/










































































































































































































































































































Текст
О.А.Сотина
В. А. Захаров /
/Амперометрическое титрование
X'J
УДК 543.257.5
Сонгина О. А., Захаров В. А.
Амперометрическое титрование. — 3-е изд., перераб. — М.: Химия, 1979. 304 с., ил.
В книге изложены теоретические основы метода амперометрического титрования. В третьем издании (второе издание вышло в 1967 г.) приведены новейшие данные о роли кинетических факторов при последовательно протекающих реакциях, о зависимости редокс потенциалов реагирующих систем от состава и концентрации среды. На новых теоретических положениях основан важнейший раздел о выборе потенциала индикаторного электрода; уделено внимание выбору материала индикаторных электродов. Описаны методики определения более 60 элементов.
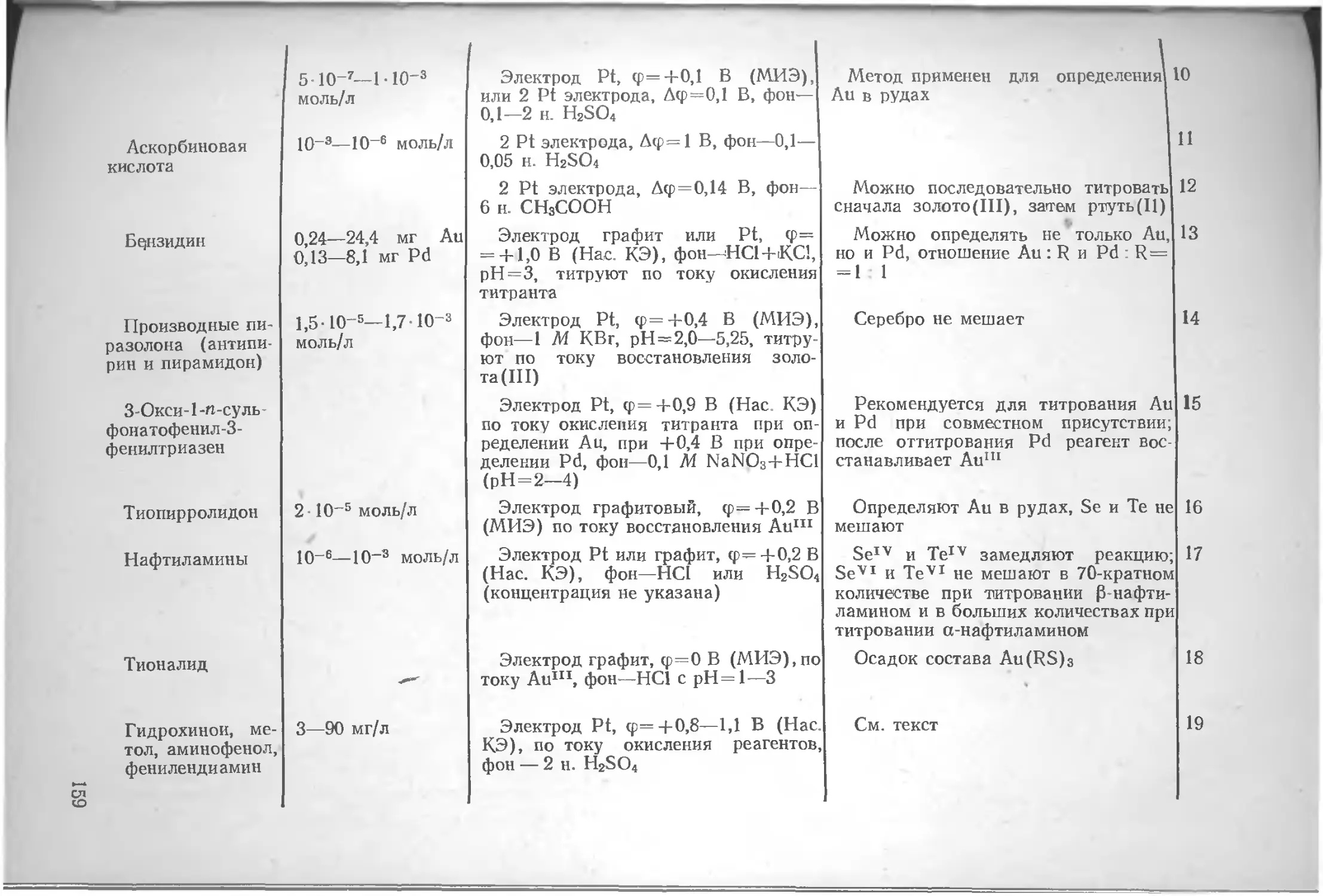
Книга предназначена для химиков-аналитиков производственных и научно-исследовательских лабораторий. Она представляет несомненный интерес для студентов, аспирантов и преподавателей химических вузов.
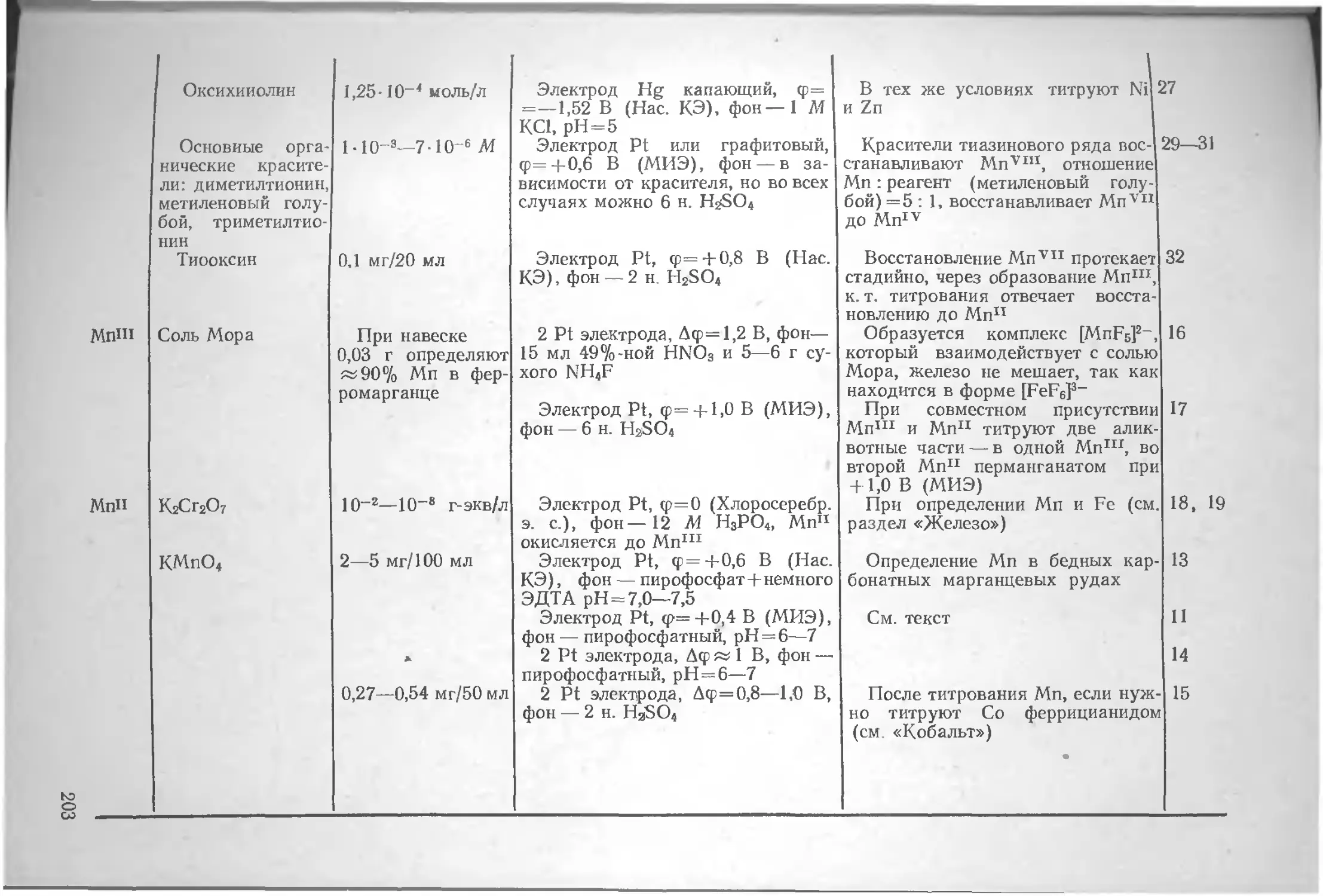
304 с., 45 табл., 48 рис., 1507 литературных ссылок.
20506-16
С 050(01)-79 16-79-1804000000.
© Издательство «Химия», 1979 г.
содержание
Предисловие 7
Введение. . . . . 8
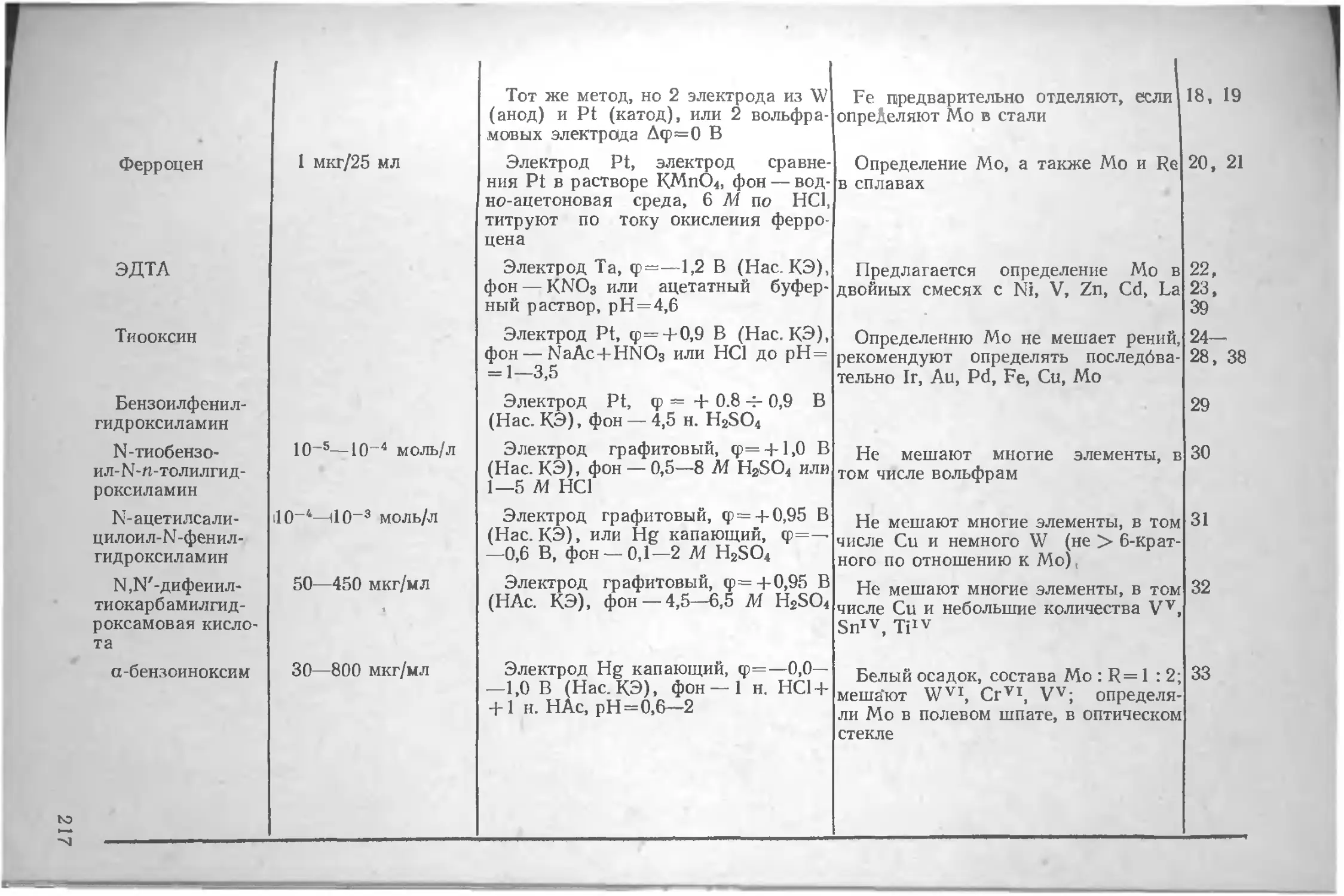
Глава I. Общие сведения об амперометрическом титровании.............................................. 9
Основные принципы.............................. 9
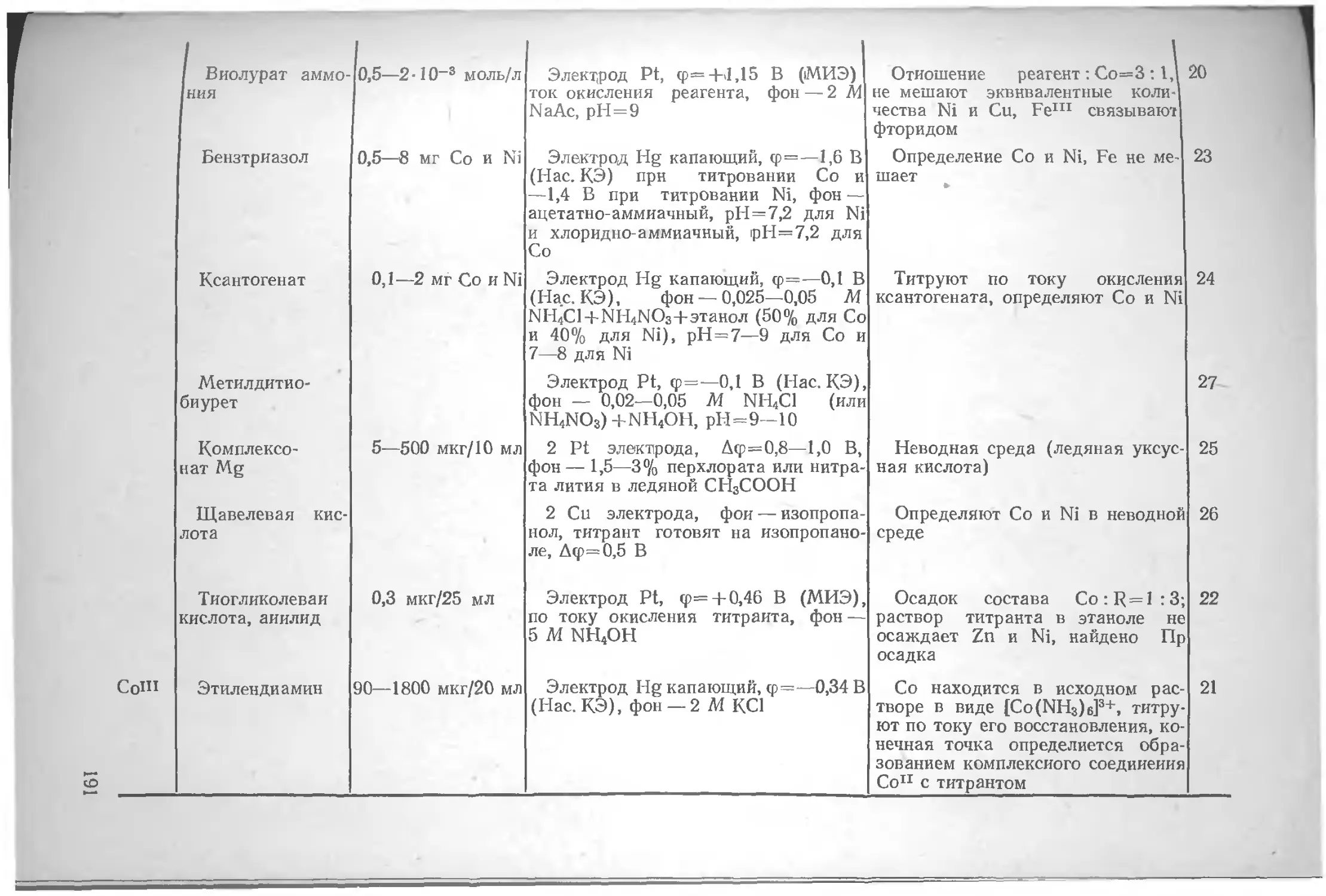
Роль и место амперометрического титрования в современной аналитической химии 14
Литература .. . 17
Глава II. Общие вопросы теории амперометрического титрования.............................................19
Предельный ток на ртутном и твердых электродах 19
Факторы, влияющие на диффузионный ток 22
Материал индикаторного электрода 23
Некоторые особенности твердых электродов 30
Изменение поверхности электрода за счет адсорбционных явлений . . 30
Изменение поверхности электрода за счет электродного процесса . ...........................34
Литература ................ . . 35
Глава III. Амперометрическое титрование с одним индикаторным электродом.................................38
Установки для амперометрического титрования 38
Выбор потенциала индикаторного электрода . 46
Типы электродных реакций ... 47
Катодное восстановление окислителей и анодное окисление восстановителей .... 47
Восстановление ионов металлов . 53
3
Анодное окисление органических реактивов ... 54
Восстановление и окисление малорастворимых веществ . 56
Методы определения конечной точки....................58
Литература . 51
Глава IV. Амперометрическое титрование с двумя индикаторными электродами...............................64
Принцип метода............................. ... 65
Выбор напряжения для титрования . 69
Теоретические основы метода ...... 73
Аппаратура и область применения метода 78
Литература...........................................81
Глава V. Типы реакций в амперометрическом титровании ..................................................82
Реакции осаждения................................... 82
Реакции комплексообразования ..... 83
Реакции окисления — восстановления...................85
Литература 97
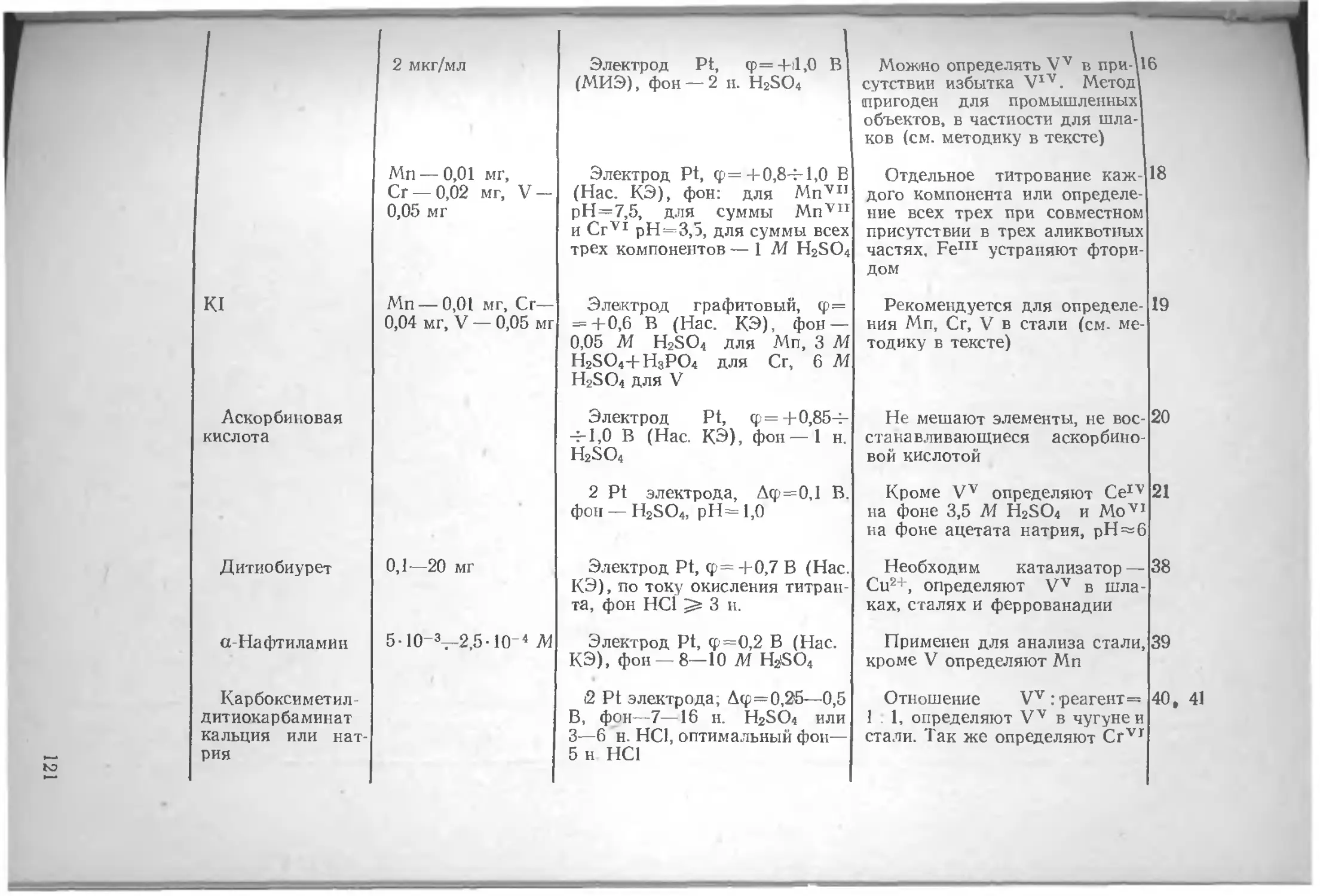
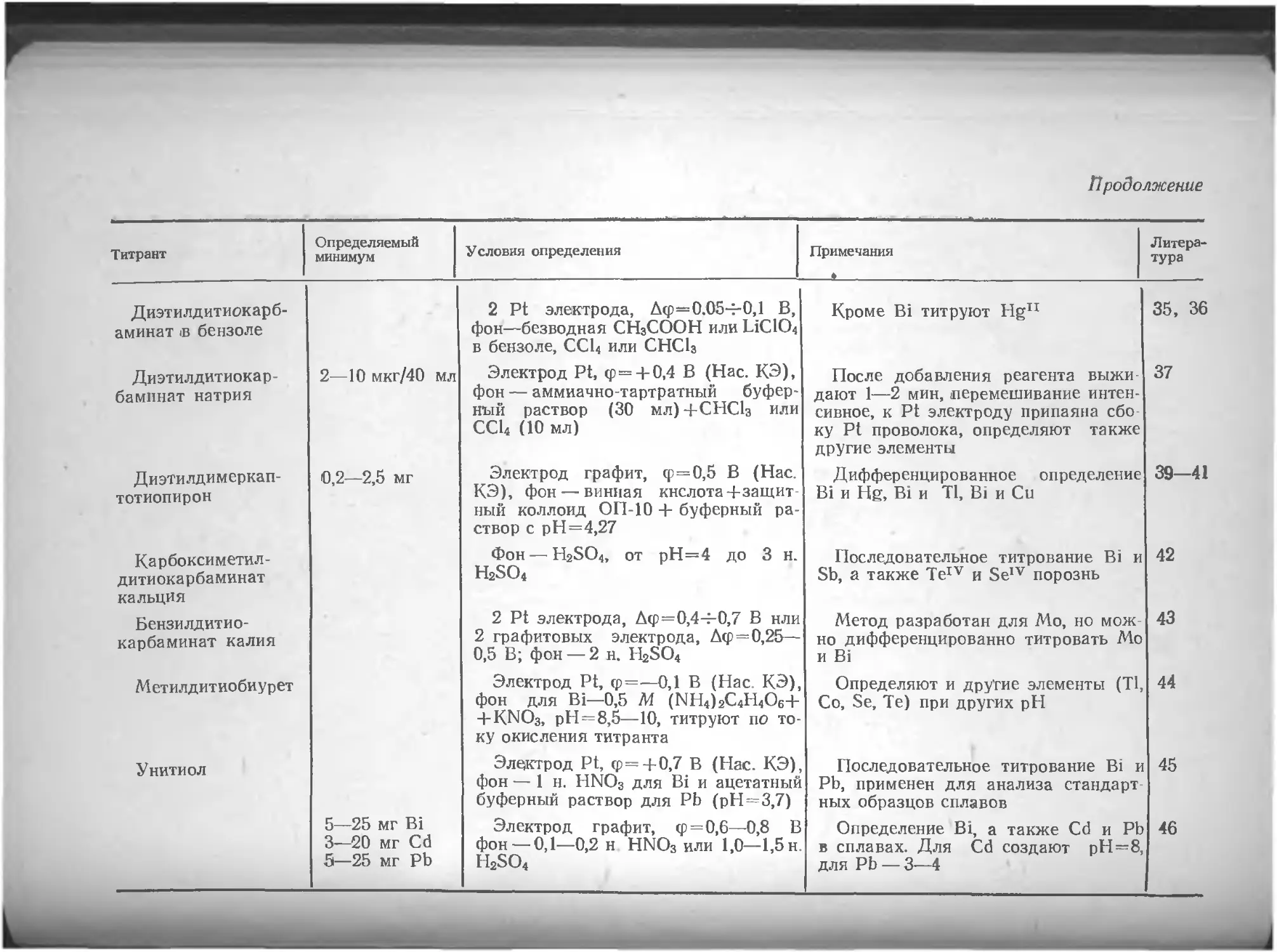
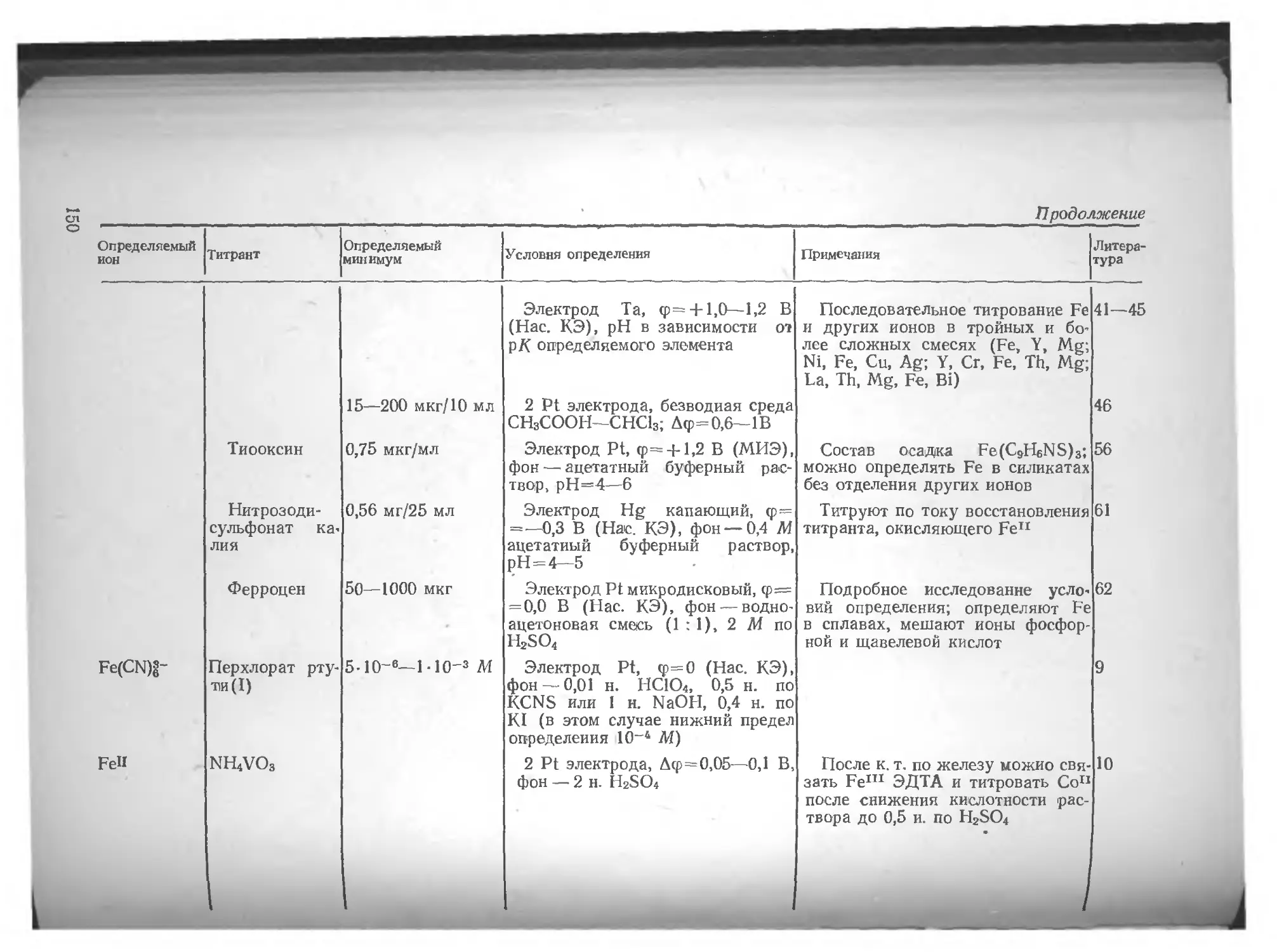
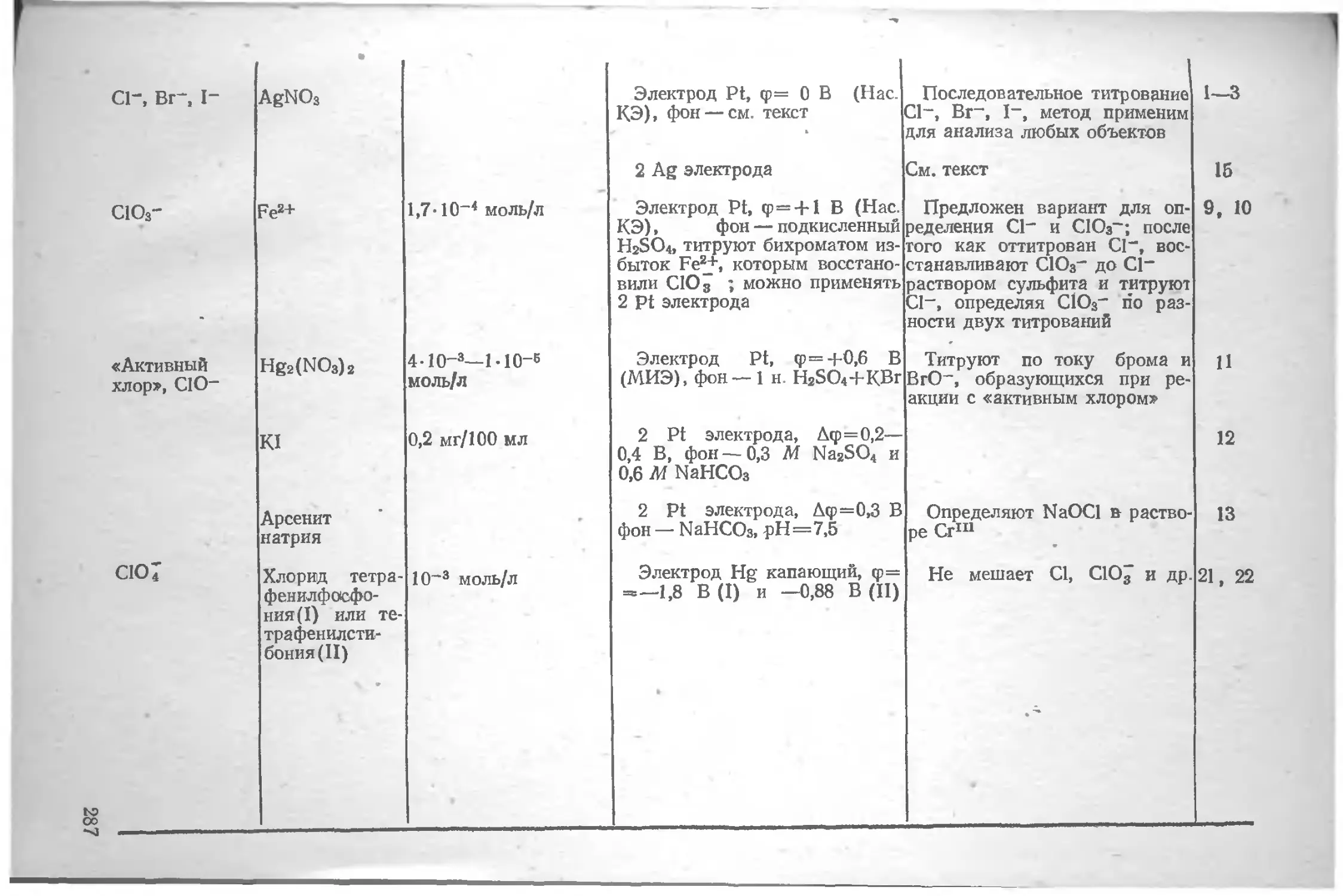
Глава VL Определение различных элементов методом амперометрического титрования , . по
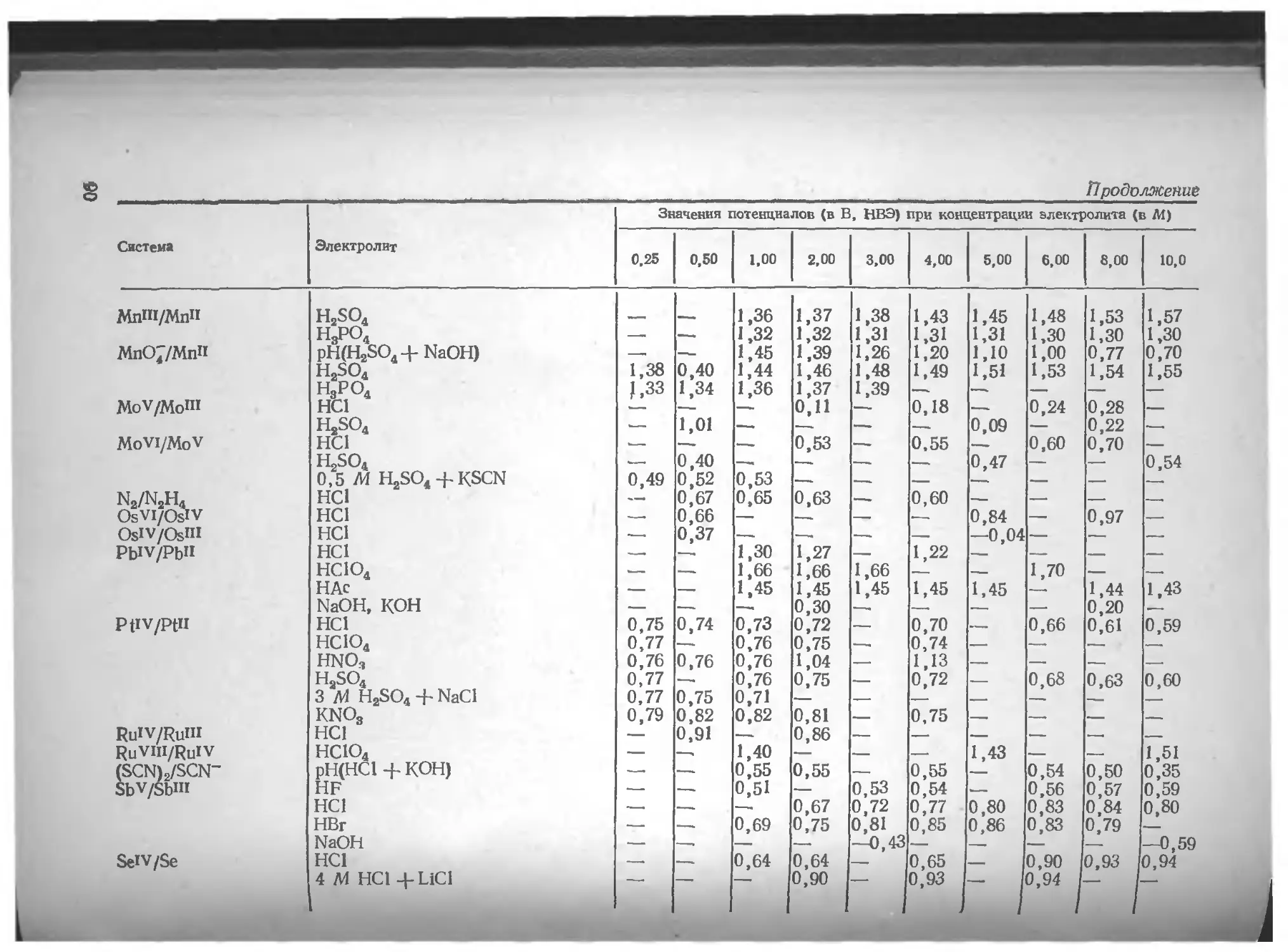
Азот .... юо Вольфрам . 135
Литература 106 Литература 136
Алюминий . . 106 Галлий 137
Литература . 107 Литература 140
Барий . . . . . . но Г афний 141
Литература ш
Бериллий . . . 111 Литература 142
Литература 112 Германий 142
Бор . . . . 112 Литература 143
Литература 113 Железо 143
Бром . . . . . . . 113 Литература 153
Литература 113
Золото 154
Ванадий 114
Литература 125 Литература 162
Висмут 127 Индий . . 163
Литература 134 Литература . . . 167
4
Иод . . . . • Литература 167 170 Осмий Литература 228 228
Иридий . . 171 Палладий .... 228
Литература . . . 173 Литература 232
Иттрий . „ - . . . 174 Платина ... 233
Литература . . . 174 Литература 235
Кадмий .... . . . 175 Плутоний н нептуний 235
Литература . . . 179 Литература 235
Калий .... . . . 180 Рений ... . 235
Литература 181 Литература 236
Кальций .... . . . 181 Родий ... 236
Литература . . . 185 Литература 237
Кислород .... . . . 186 Ртуть . . . . 237
Литература . . . 188 Литература 241
Кобальт .... . . . 188 Рутений 242
Литература . . . 189 Литература 243
Кремний . . . 192 Свинец 243
Литература . . . 193 Литература 244
Лантан, редкоземельные элементы 193 Селен ...... 245
Литература 198 Литература 249
Литий .... . . . 199 Сера ... 250
Литература ... 199 Литература 255
Магний .... ... 199 Серебро . 256
Литература . . . 200 Литература 260
Марганец .... . . . 200 Скандий 261
Литература . . . 204 Литература 262
Медь . . 205 Стронций . 262
Литература . . . 212 Литература 262
Молибден Литература Мышьяк .... . . . 214 . . 218 . . . 219 Сурьма Литература Таллий .... 262 264 265
Литература . . 222 Литература 269
Натрий .... . . . 223 Теллур 269
Литература . . . 224 Литература 272
Никель .... . . . 224 Титан ... 273
Литература . . 225 Литература 274
Ниобий ... . . 225 Торий 274
Литература . . . 226 Литература 276
Олово .... . . . 226 Уран . . ... 276
Литература . . . 227 Литература 278
5
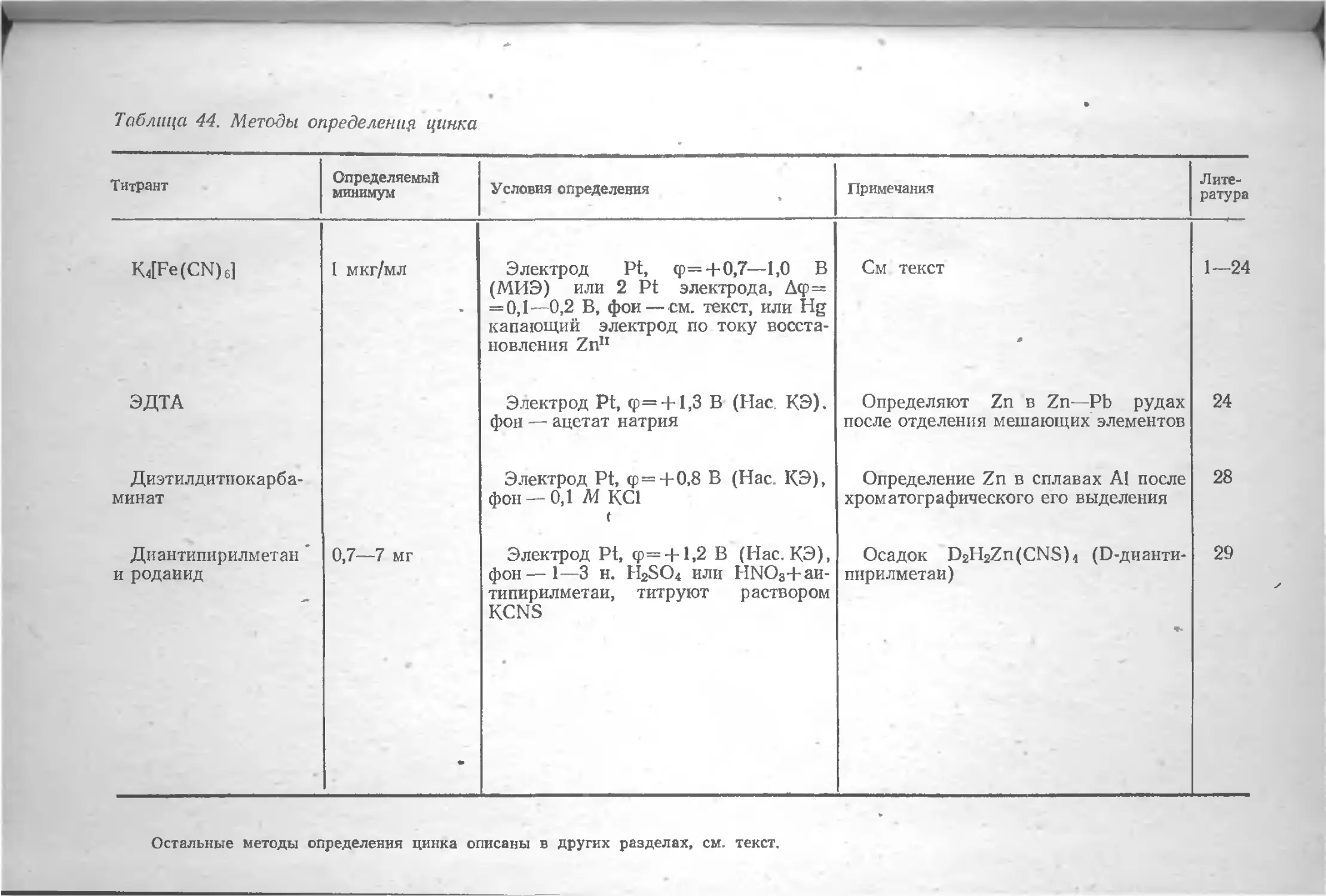
Фосфор 279 Церий 295
Литература 281 Литература 295
Фтор 282 Цинк 296
Литература 283 Литература 300
Хлор . . . . . 284 Цирконий 301
Литература . . . 289 Литература 303
Хром ... . . . 290
Литература .... 294
ПРЕДИСЛОВИЕ
В 1967 г. вышло второе издание книги О. А. Сонгиной «Амперометрическое (полярометрическое) титрование». В настоящее время область применения амперометрического метода значительно расширилась вследствие введения новых реактивов и новых приемов работы. Пополнились также сведения о применении различных твердых электродов и их поведении при катодных и анодных процессах в различных средах и о влиянии этих процессов на ход кривых амперометрического титрования. Эти данные важны потому, что твердые электроды все больше вытесняют ртутный капающий электрод из практики амперометрического титрования: методам с применением ртутных электродов посвящено в настоящее время лишь около 15% всех публикуемых в мировой печати работ по амперометрии.
В связи с этим назрела необходимость значительно переработать издание 1967 года, пополнить его новыми сведениями теоретического и практического характера и изъять некоторые устаревшие данные. Предлагаемая читателям монография является по существу новой книгой, в которой авторы стремились охватить новейшие работы в области теории и практики амперометрического титрования. Вместе с тем, поскольку за последнее десятилетие появилось очень много различных работ по амперометрическому титрованию, авторы вынуждены были ограничиться приведением в практической части книги лишь тех методик определения элементов, которые хорошо оправдали себя на практике или отличаются новизной и оригинальностью.
В книге широко использованы результаты работы ее авторов и их сотрудников и учеников. Эти работы выполнялись в 1950— 1958 гг. на кафедре аналитической химии Казахского университета (зав. кафедрой профессор, академик АН КазССР М. Т. Козловский), а с 1959 года — на кафедре химии редких элементов того же университета, руководимой одним из авторов книги. Авторы благодарят всех своих товарищей по работе, принимавших участие в подготовке настоящей монографии к печати.
С огромной признательностью отмечают авторы исключительно большой труд, выполненный кандидатом химических наук Г. Б. Бектуровой по подбору литературы для гл. VI и по оформлению рукописи.
Все указания на недостатки, несомненно имеющиеся в этой книге, будут приняты авторами с большой благодарностью.
АВТОРЫ
7
ВВЕДЕНИЕ
Во «Введении» к первому и второму изданиям монографии «Амперометрическое (полярометрическое) титрование» было подробно рассмотрено происхождение названия метода и описана критика, развернувшаяся в 40—50 годах в связи с тем, что авторы метода— Я. С. Гейровский и его сотрудники — не соглашались с термином «амперометрическое» титрование, предложенным И. М. Кольтгофом в 1939 г. Чешские химики считали необходимым сохранить за предложенным ими в конце двадцатых годов методом такое название, которое отражало бы неразрывную связь этого метода с его источником — полярографией, например «полярометрическое титрование», подобно тому, как титриметрический вариант потенциометрии называют потенциометрическим титрованием. Однако предложенный И. М. Кольтгофом термин «амперометрическое», отражающий природу измеряемой величины, т. е. тока, получил всеобщее признание. Автор I и II изданий этой монографии, отдавая должное приоритету и точке зрения чешских химиков, все же пользовался термином «амперометрическое» титрование. Сейчас этот термин узаконен готовящейся к утверждению ЙЮПАК номенклатурой терминов, применяемых в электрохимических методах анализа.
В быстром признании термина «амперометрическое титрование» сыграло роль еще одно обстоятельство, а именно все больший отход амперометрического титрования от полярографии в том смысле, что вместо ртутного капающего электрода— основного инструмента полярографии — теперь для индикации конечной точки применяют главным образом не ртутный, а различные твердые электроды, а та область электрохимического анализа, которая раньше называлась полярографией, сейчас является частным случаем более широкого понятия, характеризуемого термином «вольт-амперометрия». В соответствии с этим полярограммы, снимаемые не на ртутном, а на твердых электродах, правильнее называть вольт-амперными кривыми. Авторы настоящего, третьего, издания «Амперометрического титрования» по мере возможности придерживались этого правила.
ГЛАВА I
Общие сведения об амперометрическом титровании
Основные принципы
Амперометрическое титрование представляет собой титриметриче-ский метод анализа, в котором для индикации конечной точки используют ток, возникающий на электроде за счет разряда иоиов, участвующих в титровании, или продуктов их взаимодействия. Амперометрическое (полярометрическое) титрование представляет собой модификацию полярографического метода анализа, основанного, как известно, на пропорциональности между диффузионным током и концентрацией вещества, участвующего в электрохимическом процессе на электроде и обусловливающего наблюдаемый диффузионный ток.
Непосредственную связь между амперометрическим титрованием и полярографическим методом удобнее всего показать графически. Представим себе, что в ряде растворов.соли цинка определена концентрация цинка полярографическим методом; соответствующие полярограммы представлены на рис. 1,а. Полярографические кривые выражают зависимость между силой тока и приложенным напряжением; как видно из рис. 1,а, сила тока сначала постепенно возрастает при увеличении приложенного напряжения, затем кривая переходит в горизонтальный участок (параллельный оси абсцисс). Это означает, что дальнейшее изменение потенциала электрода уже не может вызвать увеличения силы тока. В таком случае принято называть наблюдаемый ток предельным или, в определенных условиях, — диффузионным, если лимитирующей стадией электродного процесса является диффузия ионов к электроду*.
Высота «площадки» диффузионного тока (волны) зависит от содержания цинка в исследуемом растворе: кривая 1 соответствует наибольшему содержанию цинка, кривая 6 — наименьшему. Исходя из этих полярограмм, можно построить другой график, на оси ординат которого по-прежнему откладывают силу тока, а на оси абсцисс — содержание цинка; если перенести высоту волны, обозначенную на рис. 1,а цифрами 1, 2, 3, 4, 5, 6, на график, изображенный на рис. 1,6, то получается ряд точек, расположенных
Подробнее о явлении предельного тока см. гл. II.
9
Рис 1, Графическое изображение связи между полярографическим и полярометрическим (амперометрческим) титрованием:
а — полярограммы растворов соли цинка различной концентрации; б — изменение силы тока в растворах солн цинка при титровании
на прямой линии, выражающей собой уравнение, лежащее в основе полярографии*:
Id = KC
Прямую, подобную приведенной на рис. 1,6, можно получить экспериментально: совершенно очевидно, что если к раствору, содержащему, например, 3,2 мг цинка (случай, изображенный на рис. 1,а, кривая 1), будем добавлять какой-либо осадитель и тем самым постепенно понижать концентрацию цинка в растворе, предварительно установив потенциал электрода так, чтобы он находился в пределах площадки предельного тока, например —1,65 В (Нас. КЭ)**, то понижение концентрации ионов цинка в растворе вызовет соответствующее уменьшение силы тока, и весь процесс будет совершенно аналогичен процессу последовательного снятия полярограмм ряда растворов солей цинка различной концентрации.
Таким образом, для осуществления амперометрического титрования необходимо установить на индикаторном электроде по
* См. гл. I.
** В книге приняты следующие сокращения: НВЭ — нормальный водородный электрод сравнения; Нас. КЭ — насыщенный каломельный электрод сравнения; МИЭ — меркур-иодидный электрод сравнения.
10
тенциал, отвечающий области предельного тока того вещества, кот, рое участвует в электродном процессе и концентрация которого изменяется в процессе титрования. Для этой цели применяют реакции осаждения (например, титрование ионов цинка ферроцианидом калия), реакции окисления-—восстановления (например, титрование ванадат-иона солью Мора) и реакции комплексообразования (например, титрование ионов кальция раствором ЭДТА).
Важной особенностью амперометрического титрования, выгодно отличающей его от полярографическких определений, является следующее: если для полярографического определения необходимо, чтобы сам определяемый ион давал электродную реакцию, т. е. восстанавливался (или окислялся) на электроде, то для амперометрического титрования это совершенно не обязательно. Достаточно, чтобы на электроде мог восстанавливаться или окисляться хотя бы один из двух участвующих в титровании реагентов или продукт этой реакции. Если электродную реакцию дает титруемое вещество, то с самого начала титрования появится ток (при соответственно подобранном напряжении), который во время титрования будет постепенно понижаться вследствие связывания этого вещества титрующим реагентом в осадок, в малодиссоцииро-ванное соединение или переведения его в другую степень окисления, уже не дающую электродной реакции при данном потенциале В таком случае кривая титрования будет иметь вид, изображенный на рис. 2, а.
Если же требуется определить (оттитровать) вещество, которое не может восстанавливаться или окисляться на электроде вообще или при заданном потенциале электрода, то следует подбирать такой реактив, который сам обладал бы этой способностью. В этом случае кривая титрования будет иметь зеокально обратный ход. Пока добавляемый реактив расходуется на образование осадка или на иную реакцию с определяемым веществом, сила тока будет оставаться практически без изменения; лишь после конечной точки, когда в титруемом растворе появится избыток ионов титранта и начнется их восстановление (или окисление), сила тока будет повышаться, такая кривая изображена на рис. 2, б.
Возможны случаи, когда оба участвующие в реакции титрования— титруемое и титрующее — вещества способны восстанавливаться (или окисляться) при заданном потенциале электрода. В этом случае сила тока вначале будет уменьшаться, а после конечной точки — возрастать; данный случай представляет собой как бы комбинацию обоих рассмотренных выше примеров. Кривая титрования при этом имеет форму, представленную на рис. в. Если же одно из веществ при данном потенциале будет восстанавливаться. а другое окисляться, то сила тока после конечной точки изменит свое направление: стрелка (или «зайчик») гальванометра начнет отклоняться в противоположную сторону (по отношению к нулю гальванометра). В таком случае кривая
11
титрования пересечет ось абсцисс — форма ее будет подобна кривой, показанной на рис. 2, г; в данном примере титруемое вещество восстанавливается, а титрующее окисляется; если, наоборот, титруемое будет окисляться, а титрующее восстанавливаться, то кривая будет иметь зеркально обратный ход.
Если в титруемом растворе присутствуют разные ионы, способные давать соединения с одним и тем же титрующим реагентом, причем один из этих ионов может участвовать в электродной реакции при данном потенциале, а другой —нет, то можно получить кривую, изображенную на рис. 2, д при условии, что титрант также дает электродную реакцию. Такие кривые называют иногда дифференцированными*. Классическим примером является титрование ионов свинца и бария раствором бихромата: ионы свинца обусловливают начальный ток; по мере титрования ток уменьшается и, достигая минимального значения, остается постоянным во время титрования ионов бария; после конечной точки ток снова возрастает вследствие восстановления бихромата (имеется в виду ртутный капельный электрод).
Наконец, возможны и такие случаи амперометрического титрования, при которых измеряется только ток продукта химической
* Встречающееся иногда выражение «дифференциальные» нельзя признать правильным.
12
пеакции, протекающей в растворе. Например, при титровании мышьяка (V) раствором иодида калия в определенных условиях возможно только электродное восстановление свободного иода, образующегося в результате реакции:
AsOf" 21” 2Н+ * AsO|” -р 1г -р НгО
В этом случае сила тока будет возрастать по мере титрования, достигнет максимума в точке эквивалентности и после этого останется постоянной, так как дальнейшее выделение свободного иода прекратится ((кривая титрования представлена на рис. 2, е).
Приведенные примеры не исчерпывают все возможные типы кривых амперометрического титрования. Эти примеры относятся к наиболее распространенным. Некоторые специальные типы кривых титрования будут рассмотрены ниже.
Амперометрическое титрование можно проводить даже в том случае, если ни одно из веществ, участвующих в реакции, и ни один из продуктов реакции не дает электродной реакции. В этом случае титрование возможно по так называемому индикаторному методу, предложенному Рингбомом и Вилькманом [1]. Этот метод заключается в следующем: если требуется определить ион, не дающий электродной реакции, при помощи иона, также не способного ни восстанавливаться, ни окисляться на электроде, то к исследуемому раствору добавляют небольшое количество такого вещества, которое было бы способно давать электродную реакцию и, кроме того, взаимодействовало бы с тем же реактивом, но лишь после того, как закончится реакция с определяемым ионом. Примером может служить определение алюминия (III), при помощи титрования раствором фторида калия в присутствии индикатора— железа(III) [2—5]. Алюминий, бериллий и цирконий образуют более прочные фториды, чем железо(III), и поэтому реагируют с фторид-ионом в первую очередь; когда же они будут практически полностью связаны фторидом, последний начнет реагировать с железом (III). При этом ток, обусловленный присутствием железа (III), начнет уменьшаться, и кривая титрования будет иметь форму, изображенную на рис. 3. Четкость подобной кривой титрования определяется разностью прочности комплексных соединений фторид-иона с железом (III) и алюминием (Ш).
Существует еще один вариант амперометрического титрования, при котором используют способность некоторых индикаторных электродов (ртутного, алюминиевого, медного) электрохимически окисляться в присутствии титруемого или титрующего вещества при определенном потенциале [6, 7, 8]. Например, при титровании ионов алюминия раствором фторида натрия с алюминиевым индикаторным электродом [7] до полного связывания алюминия (III) во фторидный комплекс ток в цепи отсутствует, когда же в растворе появится избыток фторида, начинается окисление элек-'Да, и ток в цепи возрастает, т. е. получается кривая, изображенная на рис. 2,6. Наоборот, если индикаторный электрод окис-
13
Рис. 3. Кривая амперометрического титрования по индикаторному методу.
ляется в присутствии определяемого вещества, получается кривая, показанная на рис. 2, а.
При любом варианте амперометрического титрования конечная точка устанавливается по изменению тока, протекающего в электрохимической цепи, включающей два электрода. Если электроды разделены, т. е. находятся в разных сосудах, соединенных солевым мостиком, то электрод, находящийся в титруемом растворе, называется индикаторным или поляризуемым электродом, а второй — неполяризуемым, или электродом сравнения. В этом случае метод называют «амперометрическое титрование с одним индикаторным (или поляризованным) электродом» (см. гл. III)—обычное «классическое» амперометрическое титрование. Если же в титруемом растворе находятся оба электрода, то метод называют «амперометрическое титрование с двумя индикаторными (или поляризованными) электродами» (см. гл. IV).
Роль и место амперометрического титрования в современной аналитической химии
Амперометрическое титрование обладает одной важной особенностью: электродная реакция, которая служит для индикации конечной точки титрования, зависит от того, какой потенциал будет установлен на индикаторном электроде в данном растворе; химическая же реакция между титруемым и титрующим веществами будет протекать совершенно независимо от того, какой материал выбран для индикаторного электрода и какой установлен на нем потенциал, если, конечно, компоненты раствора не взаимодействуют с материалом электрода (например, ртуть может непосредственно окисляться перманганатом, особенно в кислой среде). Поэтому, выбирая тот или иной потенциал индикаторного электрода, можно добиться селективной электродной реакции и, следовательно, наблюдать за изменением силы тока при анализе данного вещества в присутствии различных других веществ.
Возможности амперометрического титрования весьма широки также вследствие того, что его можно проводить не только с ртутным капельным, но и с различными твердыми электродами. Особенно удобен вращающийся платиновый электрод, позволяющий использовать реакции восстановления (протекающие при высоких положительных значениях потенциала и невыполнимые
14
вследствие этого на ртутном электроде) и разнообразные «анодные» реакции. Преимущество анодных методов заключается в том, что при положительных значениях потенциала исключается восстановление ионов многих металлов, могущих присутствовать в пастворе, и устраняется влияние растворенного кислорода, котовый также не может восстанавливаться при потенциалах более положительных, чем +0,65 В (НВЭ).
Характерной чертой амперометрического титрования является то что оно дает возможность определять различные вещества в весьма разбавленных растворах. При амперометрическом титровании, кроме того, исключаются цветные индикаторы, а следовательно, устраняются и ошибки, часто связанные с их применением. Амперометрическое титрование, подобно потенциометрическому и кондуктометрическому, позволяет проводить определение в мутных и окрашенных растворах. При этом нужно иметь в виду, что чувствительность амперометрического метода значительно выше потенциометрического, так как при больших разбавлениях
скачок потенциала уже не может достигнуть заметного значения.
В связи с тем, что для амперометрического титрования могут быть использованы самые разнообразные химические реакции (осаждения, окисления — восстановления, комплексообразования и иногда нейтрализации), можно подобрать тот или иной реактив для определения большинства элементов периодической системы. В этом отношении перспективы амперометрического титрования расширяются благодаря введению в практику аналитической химии различных органических реактивов. Преимущества органических реактивов в отношении их чувствительности и избирательности действия общеизвестны. Многие органические реактивы, широко применяемые в аналитической практике, например оксихило-лин, диметилглиоксим, о.-бензомноксим (купрон) и ряд других, способны восстанавливаться в определенных условиях на ртутном капельном электроде, другие же, как, например, купферон или тиокарбамид, окисляются на платиновом электроде. Очень большую роль в настоящее время играют в амперометрическом титровании различные комплексоны, значительно увеличившие возможность определения иоков электроотрицательных элементов — кальция, магния, редкоземельных элементов и т. д.
Практическое применение амперометрическое титрование находит в самых различных областях аналитической химии: в анализе минерального сырья и продуктов его переработки, природных вод и промышленных растворов, в анализе почв (определение микроэлементов), фармацевтических препаратов, различных органических соединений и т. д. Иногда встречаются определенные Нудности при разработке метода амперометрического определения того или иного вещества, особенно в присутствии других веществ (примесей), которые могут осложнить ход определения, таких случаях приходится изучать электрохимическое поведение примесей в различных условиях, изыскивать способы устране
15
ния влияния этих примесей и находить способы избирательного определения данного элемента без отделения других. Не следует сразу же отбрасывать опыт как неудачный, если результат его не совпадает с предположениями, из которых исходили при постановке данного опыта. При амперометрических исследованиях, в частности, необходимо выяснять причины, объясняющие наблюдаемое поведение электрода (часто говорят о «неправильном ходе кривых титрования»), причем следует иметь в виду, что электрод никогда не «ошибается», а точно отражает те условия, в которые его поставил экспериментатор.
Вообще характерной особенностью амперометрического титрования является и тот факт, что оно может быть использовано, помимо практического применения для аналитических целей, и как метод исследования. При этом следует иметь в виду, что индикаторный электрод является весьма чувствительным инструментом и отражает те явления, которые обусловлены составом раствора, материалом электрода, состоянием его поверхности и потенциалом. Так как амперометрическое титрование сочетает электрохимические явления, происходящие на электроде, с чисто химическими, протекающими в объеме титруемого раствора, то оно представляет собой особо ценный метод исследования, позволяющий изучать состав образующихся осадков, их растворимость, кинетику образования, процессы комплексообразования и т. д.
Особо следует упомянуть о применении амперометрического метода для определения конечной точки при кулонометрическом^ титровании, быстро развивающемся в последние годы. Эти случаи4 рассмотрены в периодических обзорах по амперометрическому титрованию, публикуемых в журнале Analytical Chemistry.
Вообще по амперометрическому титрованию имеется в настоящее время обширная литература. В руководствах [9—23] по электрохимическим методам анализа содержатся краткие разделы, посвященные амперометрическому титрованию. Имеется и несколько монографий: два издания настоящей книги, вышедшие в 1957 и 1967 г. [24, 27] (последнее издание переведено на польский язык [28]1); монография [25] и книга Дж. Стока [26]. Общее число статей, опубликованных с 1940 г., приближается к 3000 (только с 1966 по 1976 г. обзорными статьями Дж. Стока [29] охвачено более 2000 работ). Ежегодно публикуется 300—400 статей, причем количество их ежегодно возрастает, хотя и незначительно, но неуклонно, что свидетельствует о неослабевающем интересе к этому методу. Вклад советских исследователей в общее число работ составляет в среднем около 35%, достигая в отдельные годы почти 50% (в 1972 г. 49,5%).
Следует, однако, признать, что амперометрическое титрование применяют еще не так широко, как оно того заслуживает, по-ви-димому, потому, что широкому распространению электрохимических методов анализа иногда мешает все еще недостаточное знакомство сотрудников лабораторий с теоретическими основами этих
16
етодов а иногда и некоторая недооценка чисто химической сто-поны вопроса. Не следует думать, что инструментальный метод может полностью освободить аналитика от необходимости соответствующим образом подготавливать пробу для анализа, выбивать среду, учитывать влияние присутствующих в растворе веществ и т. д. Наоборот, именно умение «химически мыслить» обеспечивает успех инструментального метода в наиболее сложных
случаях анализа, когда, с одной стороны, приходится учитывать состав и свойства исследуемого объекта и, с другой,— выбирать наиболее рациональный метод анализа.
ЛИТЕРАТУРА
1 Ringbom A., Wilkman В. — Acta chem. scand., 1949, v. 3, № 1, p. 22—26.
2 Усатенко Ю. И., Беклешова Г. Е. — Укр. хим. ж., 1954, т. 20, № 4, с. 690— ’ 695.
3 Усатенко Ю. И., Беклешова Г. Е.—Зав. лаб., 1953, т. 19, № 2, с. 147—150; ' № 8, с. 892—895.
4 Усатенко Ю. И., Уварова К. А. — Укр. хим. ж., 1963, т. 29, № 2, с. 193— 196.
5 . Chariot G., Tremillon В. — Elektroanalyt. Chem., 1962, v. 3, № 1, p. 1—6.
6 . Петухова А. И., Торопова В. Ф.— Изв. вузов. Химия и хим. технол., 1971, т. 14, № 5, с. 685—690.
7 . Човнык Н. Г., Алемаскина Г. А —В кн.: Физико-химические методы анализа металлов, сплавов и электролитных ванн. М., 1975, с. 56—58.
8 . Boef G., Freese F., Kramer M., Pope H.—Taianta, 1970, v. 17, № 10, p. 1006— 1009.
9. Крюкова T. А., Синякова С. И., Арефьева Г. В. Полярографический анализ. М., Госхимиздат, 1959, с. 772.
10. Виноградова Е. Н., Галлай 3. А., Финогенова 3. М. Методы полярографического и амперометрического анализа. М., изд-во МГУ, 1963, с. 299.
11. Гейровский Я., Кута Я. Основы полярографии. — Пер. с чешского/Под ред. С. Т. Майрановского. М., Мир, 1965, с. 55.
12. Кольтгоф И. М., Лингейн Дж. Дж. Полярография. — Пер. с англ./Под ред. А. П. Виноградова. М., Госхимиздат, 1948.
13. Stackelberg М. V. Polarographische Arbeitsmethoden. Berlin, W. de Gruyter u. Co., 1950.
14. Zyka J. Organic reagents in Polarometric (Amperometric) Titration, Progress in Polarography. New York, Interscience, 1962.
15. Chariot G., Bezier D. Methodes modernes d’analyse quantitative minerale, Paris, 1955.
16 Шарло Г. Методы аналитической химии. — Пер. с франц/Под ред. Ю Ю. Лурье. М., Химия, 1965, с. 975.
17. Lingane J. J. Electroana’ytical Chemistry, New York, 1958.
18. Chariot G., Badoz-Lambling J., Tremillon B. Les Reactions clcctrochimiqucs. Paris, Masson, 1959.
1® Делахей П. Новые приборы и Издатинлит, 1957.
методы в электрохимии. — Пер. с англ. М.,
20.
19инг Г- В. Инструментальные М.. Госхимиздат, 1960, 509 с.
Руководство по аналитической химии. М ,- М ио 1975.' 468 а 2—1694
методы химического анализа. — Пер. с англ.
21.
БИБЛИО ТЕ К Л омского
Государстзенвого
ч ।
17
22. Лопатин Б. В. Теоретические основы электрохимических методов анализа. М., Высшая школа, 1975. 295 с.
23. Рейшахрит Л. С. Электрохимические методы анализа. Л., изд-во ЛГУ, 1970 194 с.
24. Сонгина О. А. Амперометрическое титрование в анализе минерального сырья. М., Госгеолтехиздат, 1957, 211 с.
25. Dolezal J., Zyka J. Polarometricke titrace. Praha, Statni Naklad. Techn. liter 1961, 150 p.
26. Stock J. T. Amperometric Titrations. New York, Interscience, 1965, 730 p.
27. Сонгина О. А. Амперометрическое (полярометрическое) титрование. М., Химия, 1967. 387 с.
28. Songina О. A. Miareczekowanie amperometryczne. Warszawa, wyd. Naukovo-techn., 1972, 487 c.
29. Stock I. T. — Analytical Chem., 1966, v. 38, № 5, 452—460; 1968, v. 40, № 5, 392R—401R; 1970, v. 42, № 5, R276—R284; 1972, v. 44, № 5, 1R—9R; 1974, v. 46, № 5, 1R—8R; 1976, v. 48, № 5, 1R—9R.
Г
ГЛАВА II
Общие вопросы теории амперометрического титрования
Предельный ток на ртутном и твердых электродах
В основе метода амперометрического титрования лежит измерение предельного тока при постоянном потенциале. В зависимости от лимитирующей стадии электродного процесса ток может быть диффузионным, адсорбционным, кинетическим или каталитическим [1—3]. Когда предельный ток (гпр) является диффузионным Щ), т. е. когда скорость электродного процесса определяется скоростью поступления (диффузии) ионов к электроду, id определяется в общем случае выражением:
id=SnFDC — (II. 1)
6
где S — поверхность электрода; п — число электронов, участвующих в реакции; F — число Фарадея; D — коэффициент диффузии; С — концентрация разряжающегося вещества в растворе; 6 — толщина диффузионного слоя.
Толщина диффузионного слоя зависит от типа и конструкции электрода, перемешивания раствора, коэффициента диффузии и некоторых других факторов [1—3].
В амперометрическом титровании применяют два типа электродов— ртутный капающий и, чаще, твердые. При работе с обычным ртутным капающим электродом уравнение (II. 1) принимает вид известного уравнения Ильковича.
Id = 605л£> (II. 2)
где m — масса ртути, вытекающей за 1 с; t — период капания.
Произведение m2'3t1/e называют характеристикой капилляра.
Для вращающегося с достаточно большой скоростью (более 15 см/с) ртутного капающего электрода диффузионный ток подчиняется уравнению [1]
id = (II.3)
где и скорость движения раствора у поверхности электрода.
Для твердых вращающихся электродов или стационарных в перемешиваемом растворе уравнение (П.1) принимает вид
ld = KSnFD^yrQC (II. 4)
воат^ к°нстанта, зависящая от конструкции электрода; со — угловая скорость Р щения электрода или движения жидкости; v — кинематическая вязкость рас
2*
19
твора; р и q — показатели степени, принимающие в зависимости от конструкции электрода значения от 0 до 1 [5, 6].
Так, на вращающемся дисковом электроде при ламинарном течении раствора диффузионный ток определяется уравнением [5]
id = 0,62SnFD,,!’<o'f^~'lsC (II.5)
Для цилиндрического (проволочного) электрода [10]
id = (II. 6)
где d — диаметр электрода.
Наряду с вращающимися электродами в амперометрическом титровании применяют, особенно при работе с малыми объемами (1—2 мл) раствора, вибрирующие электроды [6]. Диффузионный ток на таком электроде зависит от частоты и амплитуды колебания [7].
Все рассмотренные выше для диффузионного тока уравнения можно представить в виде
id = KC (II. 7)
т. е. ток прямо пропорционален концентрации электроактивного вещества в растворе или, как принято говорить в электрохимии, порядок скорости электродной реакции по концентрации разряжающегося вещества равен единице (показатель степени для С равен 1).
На практике приходится иметь дело не только с предельньуя диффузионным, но и другими токами, обусловленными той или иной лимитирующей стадией электродного процесса. В этом случае порядок скорости электродной реакции по концентрации разряжающегося вещества может отличаться от единицы и уравнение для тока примет вид
td = KC“ (II 8)
Показатель степени а зависит от природы тока. Для твердых электродов в случае диффузионного тока а=1, адсорбционного— а=0,5, смешанного диффузионно-адсорбционного-—0,5<а<1, кинетического, смешанного диффузионно-кинетического и каталитического— 0<а^1 [1—3, 5].
Следует отметить, что режим электродного процесса для одной и той же электрохимической реакции может изменяться с изменением условий опыта, так как скорость собственно электрохимического акта определяется потенциалом электрода [2], а скорость подвода вещества к электроду определяется толщиной приэлект-родного слоя [5]. Поэтому, изменяя условия опыта (потенциал электрода, интенсивность перемешивания раствопа, концентрацию разряжающегося вещества и т. д.), можно перевести электродный процесс из одного режима в другой. С точки зрения амперометрического титрования наиболее благоприятным режимом является
20
рис 4 Кривые амперометрического титрования для /=КС“ при а=1 (кривая /) и а<! (кривая 2).
диффузионный, поскольку в этом случае порядок электродной реакции по деполяризатору равен единице, т. е. соблюдается равен-ств0 1=КС, и восходящая (нисходящая) ветвь амперометрической кривой имеет линейный ход.
Если же порядок реакции по разряжающемуся веществу меньше единицы, то на кривой титроза ния появляются «загибы» (рис. 4).
Загибы возникают не только при дробном порядке электрохимической реакции. Другой причиной этого явления может быть самопроизвольное изменение (во время титрования) наложенного на индикаторный электрод потенциала за счет изменения падения напряжения (//?) в растворе.
Потенциал индикаторного электрода (анода и катода) в соответствии с законом Ома для проводников второго рода опреде-
ляется выражением
'Ран, инд — -вн + 'р кат — IR
ИЛИ
'Ркат, инд — 'Ран ^вн-р IR.
(П.9)
Обычно сопротивление (R) электролитической ячейки при амперометрическом титровании не превышает 2000 Ом, а сила тока (I) составляет единицы микроампер. В этом случае IR не превышает сотых долей вольта, и потенциал индикаторного (поляризующего ся) электрода практически соответствует приложенному внешнему напряжению относительно второго неполяризующегося электрода (электрода сравнения). Если же во время титрования по каким-либо причинам сильно увеличится сопротивление (выделение на индикаторном электроде плохо проводящих ток веществ — оксидов, гидроксидов, органических соединений из раствора и т. д.) или _ок достигает нескольких десятков микроампер (высокая концентрация деполяризатора, большая поверхность индикаторного электрода и др.), то IR составит уже десятые доли вольта, и истинное значение потенциала индикаторного электрода не будет соответствовать установленному внешнему напряжению. Вследст-ие этого электродная реакция во время титрования может перей-из^ диффузионного режима в смешанный диффузионно-кинети-конКИИ ИЛИ кинетический, и ток будет уже зависеть не только от центрации разряжающегося вещества, но и от потенциала элек
21
трода [2]. Это также приводит к тому, что на кривых титрования появляются загибы.
Предел концентрации деполяризатора, до которого ход кривой титрования отвечает уравнению 1=КС, зависит от размера индикаторного электрода. В частности, для проволочного электрода длиной 4—5 мм (диаметр 0,5 мм) этот предел составляет в среднем НО-3 мэль/л [8].
Факторы, влияющие на диффузионный ток
Уравнения (II. 1—11.12) показывают, как и от каких параметров зависит диффузионный ток. Некоторые из этих параметров, в свою очередь, могут зависеть от других факторов, которые в явном виде не входят в эти уравнения. Это относится, в частности, к коэффициенту диффузии. Коэффициент диффузии зависит от многих факторов: природы и размера диффундирующего иона или молекулы, заряда иона, вязкости раствора, температуры, природы, концентрации и состава фонового раствора. Количественный учет влияния всех этих факторов на коэффициент диффузии далеко не всегда возможен.
Коэффициент диффузии обратно пропорционален вязкости раствора и, следовательно, диффузионный ток будет тем больше, чем меньше вязкость раствора, при прочих одинаковых условиях.
Вязкость раствора, в свою очередь, зависит от температуры, с увеличением которой вязкость уменьшается, и поэтому коэффициент диффузии будет увеличиваться с повышением температуры.
На коэффициент диффузии и, следовательно, на диффузионный ток, существенное влияние оказывают природа, концентрация и состав фонового раствора. Влияние этих факторов обусловливается различными причинами: изменением формы существования разряжающегося иона (например, ванадий в нейтральных растворах существует в виде иона VO", в умереннокислых — в виде VO2, а в сильнокислых — в виде VO3+); изменением степени гидратации иона; явлением комплексообразования, при котором в зависимости от концентрации комплексообразующего иона и лиганда могут образовываться различные по составу комплексы [например, висмут (III) в солянокислой среде образует BiCl2+, BiCis', BiCl3, BiC17, BiCll"!! BiCIg-]. В случае комплексообразования фон влияет не только на коэффициент диффузии, но и на потенциал восстановления или окисления данного иона. Поэтому при подготовке раствора к титрованию, начиная с разложения пробы и при всех последующих операциях, необходимо учитывать, какой при этом создается солевой состав раствора и как он может сказаться на состоянии определяемого иона в этом растворе.
22
Материал индикаторного электрода
При амперометрическом титровании можно пользоваться индикаторными электродами из различных металлов, сплавов и других материалов, обладающих достаточной электропроводностью и химической индифферентностью по отношению к фоновому электролиту и компонентам титруемого раствора. В качестве индикаторных электродов чаще всего применяют платиновый и графитовый электроды (80% всех работ, посвященных амперометрическому титрованию), меньше — ртутные (около 15%)- Выбор материала электрода определяется в первую очередь тем, какой элемент оп
ределяется и какой электрохимической реакцией предполагается воспользоваться для индикации конечной точки титрования.
Ртутный электрод. Для ртутного электрода, как известно, ха-
рактерно высокое перенапряжение для выделения водорода, которое при средних плотностях тока составляет около 1,1 В* [2]. Вследствие этого водород на ртути выделяется при потенциалах около—1,1 В в кислых растворах, при —1,5 В — в нейтральных и при —1,9 В — в щелочных. Поэтому ртутные электроды (капающий, амальгамированные и др.) обычно применяют в тех слу-
чаях, когда хотят воспользоваться током восстановления какого-либо электроотрицательного металла (например, цинка, кадмия, бария и т. д.) или органического соединения.
Ртуть не является электрохимически индифферентной и легко растворяется анодно при невысоких положительных потенциалах. Поэтому возможности ртутного электрода для осуществления процессов электроокисления веществ весьма ограничены. Потенциал электрорастворения ртути определяется природой аниона фона, и поэтому рабочая область потенциалов ртутного электрода зависит как от кислотности, так и о природы используемого раствора (табл. 1). - г г
Платиновый электрод. На платиновом электроде перенапряжение для выделения водорода при средних плотностях тока практически отсутствует [2] и водород выделяется при потенциале, близком к равновесному системы Н2/Н+, т. е. около О В в кислом
Таблица 1. Рабочая область потенциалов ртутного электрода
Ф°Н. 1 н. раствор Потенциалы. В (НВЭ) Фон, 1 н. раствор Потенциалы. В (НВЭ)
н3р°4 H2SO4 HNO, НС1О4 НС] 4 +0,64—1,1 K2SO4 +0,64—1,5
+0,64—1,1 KNO3 +0,84—1,5
+0,84—1,1 ксю4 -т-0,84— 1,5
+0,84—1,1 КС1 +0,34—1,5
+0,34 1,1 кон +0,14—1,9
В гл. II потенциалы приведены относительно НВЭ
23
Таблица 2. Рабочая область потенциалов платинового электрода
Фон, 1 н. раствор Потенциалы, В (НВЭ) Фон, 1 н. раствор Потенциалы, В (НВЭ)
н3ро4 + 1,74-0,0 K2SO4 + 1,34- —0,4
H2SO4 +1,74-0,0 KNOa + 1,34- —0,4
HNO3 +1,7-з0,0 KC'O4 + 1,34- —0,4
НС1О4 + 1,74-0,0 КС1 + 1,24- —0,4
НС1 +1,34-0,0 кон +0,94- —0,8
растворе, —0,4 В в нейтральном и —0,8 В в щелочном. Поэтому на платиновом электроде возможно электровосстановление лишь немногих электроотрицательных ионов, например свинца (II) (Еп——0,12 В) и таллия(1) (Ео=—0,33 В).
Но, в отличие от ртути, платина анодно практически не растворяется и на ней весьма значительно перенапряжение для выделения кислорода. Вследствие этого платиновый электрод можно поляризовать до очень высоких положительных потенциалов (+1,7— +2,0 В) и тем самым осуществлять анодное окисление различных неорганических и органических восстановителей, а также катодное восстановление электроположительных ионов металлов (серебро, золото, иридий и т. д.), и сильных окислителей (перманганат, бихромат и т. д.). Передел анодных потенциалов, до которых возможна поляризация платинового электпода, определяется про„-цессом выделения молекулярного кислорода при данной кислотности раствора за счет разряда молекул воды или анодным процессом окисления аниона фона (например, в хлоридной среде— электроокисление хлорид-иона). Поэтому рабочая область потенциалов платинового электрода, как и ртутного, зависит от кислотности и природы фонового раствора (табл. 2).
Кроме того, для платины характерны некоторые специфические особенности, связанные с поляризацией ее в водных растворах. При поляризации платина претерпевает изменения, обусловленные хемосорбцией водорода и кислорода, а также образованием окис-лов. Такие изменения могут оказать влияние на электрохимическое поведение участников титрования и тем самым сказаться на самом титровании.
Состоянию поверхности платинового электрода в водных растворах посвящены специальные исследования [9—15]. Установлено, что при потенциалах +0,4—0,0 В (кислые растворы), т. е. когда еще не выделяется молекулярный водород, на поверхности платинового электрода адсорбируется атомный водород за счет разряда ионов гидроксония или молекул воды
Н3О+ (или Н2О) + е~ ч -X Надс + Н2О (или ОН-) (И. 10)
Адсорбированный водород может ингибировать протекание электродных реакций. Так, предварительно адсорбированный при
24
катодной поляризации водород обусловливает уменьшение тока восстановления перманганата [16], бихромата [17], железа(Ш) г 18] молекулярного кислорода [19, 20], а также тока окисления гидразина [21]. Поэтому при амперометрическом титровании следует избегать предварительной поляризации платинового электрода при потенциалах выделения водорода. Очистить поверхность платины от адсорбированного водорода можно путем промывания ее раствором вещества, способного восстанавливаться водородом [железо(III), церий(IV) и т. д.], или путем поляризации платины при потенциалах 0,5—0,6 (НВЭ).
Адсорбция кислорода и образование оксидных пленок на платине происходят при менее положительных потенциалах, чем выделение молекулярного кислорода [9—15, 22—30], за счет разряда молекул воды или гидроксил-иона
Н2О(или 2ОН~)—2й~ —> Оадс + ОН- (или Н2О) (II. И)
и за счет химического взаимоде.йствия адсорбированного кислорода с платиной. Адсорбция кислорода и образование оксидов платины при ее анодной поляризации в кислых растворах (например, 1 М H2SO4) начинается при +0,65 В (НВЭ) и с увеличением pH на единицу смещается в катодную сторону на 0.06 В. С увеличением анодного потенциала и продолжительности поляризации общее количество адсорбированного кислорода на поверхности платины возрастает, причем одновременно с этим происходит упрочнение связи атомов кислорода с платиной [10, 12, 24, 25, 28, 29, 30]. Относительно предельного количества адсорбирующегося кислорода, формы его существования и состава оксидной пленки высказываются различные мнения. Так, согласно работам [23, 24], при потенциалах 0,7—1,2 В образуются низшие оксиды платины РЮ, а при более положительных потенциалах — PtO(O)aB.c- По данным работы [26], при анодной поляризации платины образуются фазовые окоиды РЮ и РЮ2-«Н2О.
Адсорбция кислорода и образование оксидных пленок на платине происходят и при ее соприкосновении с молекулярным кислородом [22] или при ее выдерживании в растворе сильного окислителя: бихромата, церия (IV) и т. д. [26, 31]. Если же, наоборот, окисленную платину выдержать в растворе восстановителя, например мышьяка(III) или железа(II), то происходит снятие оксидной пленки [26, 31]. Удалить оксидные пленки и адсорбированный кислород с поверхности платины можно также электрохимически. Для этого нужно выдержать электр эд в кислом растворе фона при потенциалах 0,5—0,6 В (НВЭ) до тех пор, пока сила тока не станет равной нулю.
Хемосорбированный кислород, находящийся в атомном состоянии или в виде радикала ОН, играет активную роль в процессах электрохимического окисления, протекающих с изменением кислородного баланса в окисляемом веществе, причем взаимодействие окисляющегося вещества с адсорбированным кислородом (ради
25
калом ОН) определяет скорость электродной реакции в целом. В общем случае процесс окисления веществ, протекающих с из-нением кислородного баланса, можно представить схемой:
Pt+nH2O — 2ие- х=> Pt(O)„, адс 4-2иН+ (11.12)
или
Pt+ пН2О — пег ..ь. Pt(OH)n> адс+ «Н+
Pt(O) и, аде + АО^- —> Pt+АОДО- (11.13)
или
Pt(OH)адС + АО*" —> Pt + AO^+”’-+«№
Например, для окисления хлорид-иона до хлората принимается следующий механизм [32]:
Pt-p «Н20 —Pt(O)n4-2«H+
(П. И) pt(O)„+ci- —> pt(O)„_3+cio;
Подобные схемы предлагаются также для электрохимического выделения кислорода и анодного образования озона [24—33], электроокисления иодида и иода до йодата [34], сульфида и сульфита до сульфата [23, 35], урана (IV) [36], арсенита [37], тиогликоля-та [38] и многих других неорганических и органических веществ. Одним из основных признаков, указывающих на участие адсорбированного кислорода в анодном процессе, является понижение высоты волны окисления деполяризатора с уменьшением скоросттр. снятия полярограммы, что обусловливается увеличением прочности связи адсорбированного кислорода с платиной во времени, и как следствие этого — уменьшением скорости реакции (11.13).
Если адсорбированный кислород играет активную роль в процессах электроокисления, то поверхностные оксиды оказывают, наоборот, ингибирующее влияние на многие анодные процессы: окисление перекиси водорода [12], железа(II) [39], урана(IV) Г36], хлорида [40], иода [34], иодида [34], сульфита [35], арсенита [37], тиокарбамида [41], различных серусодержащих органических реагентов [38, 42, 43], спиртов [9, 44, 45], альдегидов [43] и ряда других веществ. Особенно сильное торможение поверхностные оксиды платины оказывают, как правило, на скорость реакций, протекающих с изменением кислородного баланса в окисляемом веществе или сопровождающихся деструкцией деполяризатора. В этих случаях на полярограммах отмечается появление максимумов и спадов силы тока при высоких анодных потенциалах, а между поляризационными кривыми, снятыми в различном направлении, имеется большой гистерезис. Наглядным примером могут служить вольт-амперные кривые арсенита и тиокарбамида (рис. 5).
Таким образом, адсорбированный кислород и оксиды платины, а также адсорбированный водород оказывают значительное влияние на диффузионный ток разряжающегося вещества. В кис-
26
D„r 5 Вольт-амперные кривые арсенита (Л 2) Р Тиокарбамида (3, 4) на фоне 1 М серной глоты снятые в различных направлениях поляризации платинового электрода.
лых растворах на поверхности платинового электрода при потенциалах j 7—0,7 В (НВЭ) находится адсорбированный кислород и оксиды платины, при 0,4—0 В — адсорбированный водород, и лишь при потенциалах 0,7— 04В поверхность платины свободна от этих веществ.
При переходе от кислых к щелоч-
ным растворам область потенциалов адсорбции кислорода и водорода сдвигается в сторону более отрицательных значений соответственно сдвигу всей вольт-амперной кривой фона с изменением его
кислотности.
Графитовый электрод. В настоящее время в амперометрическом титровании широко применяют твердые электроды из материалов на основе углерода (стеклоуглерод, углеситал) и из графита [47—52]. Стеклоуглерод и углеситал — имеют незначительную поверхностную пористость (2—3%) и практически газонепроницаемы. Для графита, например так называемого «спектрального» марки осч, поверхностная пористость достигает 25—30% [53], и он легко газопроницаем. Поэтому используют электроды не из чистого графита, а из графита, предварительно пропитанного воском, парафином, клеем БФ, эпоксидной смолой, полиэтиленом или некоторыми другими веществами — импрегнаторами [47—52]. Для пропитанного графита поверхностная пористость составляет 5—8%, и он, так же как и стеклоуглерод, газонепроницаем.
Пропитка графита импрегнаторами практически не сказывается на предельном токе деполяризатора, значительно снижает остаточный (емкостный) ток, улучшает воспроизводимость результатов и значительно расширяет пределы потенциалов поляризации графитового электрода (табл. 3). Различие в пределах рабочей ласти потенциалов пропитанного и непропитанного графитовых электродов связано с емкостным током и наличием в порах непропитанного графита кислорода воздуха, способного электровосста-навливаться. Поэтому при пропитке электрода необходимо стре-иться к тому, чтобы воздух, наполняющий поры графита, полно-тью из них вышел.
Графитовый электрод особенно перспективен, поскольку по-ДйпХН?сть его ferK0 может быть обновлена путем зачистки наж-вос'НОИ бумагой, а некоторые посторонние процессы, в частности становление растворенного в электролите молекулярного кис
27
лорода, протекают на графитовых электродах при потенциалах на 0,3—0,4 В отрицательнее, чем на платиновом электроде. Это обстоятельство с точки зрения амперометрического титрования является положительным, поскольку в случае титрования с графитовым электродом по катодному принципу при невысоких значениях положительных потенциалов растворенный кислород не будет обусловливать начальный и остаточный ток.
Графитовый электрод, по сравнению с платиновым, обладает весьма малой адсорбционной способностью по отношению к водороду и кислороду. Так, согласно работе [55], даже при длительной катодной поляризации водород не адсорбируется на электродах из спектрального, реакторного и пиролитического графита. На активном угле, по данным Фрумкина [56], количество адсорбированного водорода не превышает 2% от монослоя. Весьма незначительно адсорбируется водород на стеклоуглероде и углесита-ле. Поэтому процесс разряда деполяризаторов практически не зависит от адсорбированного водорода.
Адсорбция кислорода на графитовых электродах происходит при потенциалах положительнее 0,3—0,4 В [57, 58]. Однако заполнение поверхности хемосорбированным кислородом до потенциалов выделения молекулярного кислорода, т. е. до 1,4—1,6 В, составляет не более 2—3% от монослоя. Это обстоятельство сказывается на электрохимическом поведении деполяризаторов, окисление которых сопровождается изменением кислородного баланса в окисляющемся веществе. Окисление таких деполяризаторов-ца графитовом электроде в рабочей области его потенциалов не происходит. Наглядным примером могут служить, например, вольт-амперные кривые арсенита и иодида. На рис. 6 приведены эти вольт-амперные кривые на графитовом и платиновом электродах. Как видно, иодид на платиновом электроде (кривая 1) дает две волны: первая при потенциалах 0,6—1,1 В соответствует окислению иодид-иона до иода, а вторая волна при потенциалах более 1,1 В — окислению иодид-иона до йодата. В случае же графитового электрода иодид-ион окисляется только до иода (кривая 2). Волну окисления арсенита до арсената удается получить лишь на платиновом электроде (кривая 3). Отсутствие второй волны иоди-
Т об лица 3. Рабочая область потенциалов различных графитовых электродов
Фон, 1 н. раствор Графит непропитанныЙ 4 Графит импрегнированный Стеклоуглерод [53] Углеснтал (54)
h2so4 HNOS НС1О4 НС1 Na2SO4 КС1 кон +++++++ о о о о о о о OJ 00 Ъо ОО СО СО 00 •I- -I- -1- •!• 1 1 1 1 1 1 1 О О О О О О О Ф* ND ND 1— »— 1— •— + 1,44—0,6 +1,44—0,6 +1,44—0,6 +1,14- _о, 6 +1,44—0,8 +1,14—0,8 +0,84—1,0 + 1 + 1 1 1 + о J"- J"- -4 4^ СП •I- I I I -° -° <— со со + 1,34—0,6
28
6 Вольт-амперные кривые иодида и арсе-нита на фоне 1 М H2SO4:
иодида на платиновом электроде; 2 — иодида на 1 алитовом электроде; 3 — арсенита на платиновом г₽ „тсоде- 4 — арсенита на графитовом электроде; дЛ6 фона на графитовом электроде.
*¥>,в(нвэ) и
1аН,М^
да и волны арсенита на графитовом электроде связано с тем, что электроокисление иодида до йодата и арсенита до арсената протекает с участием адсорбированного на электроде кислорода, а графит при анодной поляризации адсорбирует, как уже отмечалось, весьма незначительное количество кислорода, которое не может внести за
метного вклада в анодное окисление иодида до йодата и арсенита до арсената на этом электроде. Этими двумя примерами не исчерпываются, разумеется, все случаи электроокисления веществ, идущие с изменением кислородного баланса.
Танталовый и карбидотанталовый электроды. Танталовый и карблдотанталовый электроды успешно применяет Ташкентская школа химиков-аналитиков [59—63]. Танталовый электрод представляет собой танталовую проволоку (длина 10—15 мм, диаметр до 1 мм), вмонтированную в стеклянную трубку при помощи воска.
Карбидотанталовый электрод изготавливают тоже из танталовой проволоки, которую специально карбидизируют [63]; отрезок танталовой проволоки помещают в узкий паз, пропиленный вдоль тонкого графитового стержня, слегка запрессовывают смесью порошкообразного графита с сажей и накаливают стержень электрическим током в высоком вакууме до температуры 1500 °C в течение 1—2 мин. Рабочая область потенциалов этих электродов почти такая же, как платинового, их применяют при титровании по току окисления реагента.
Золотой электрод. Золото как материал индикаторного элект-в амперометрическом титровании используют пока редко [Ь4 65]. Рабочая область потенциалов золотого электрода несколько больше, чем платинового. Так, в растворе 1 М H2SO4 эта область для золотого электрода составляет +1,8-;—0,1 В, тогда ак для платинового +1,74-0,0 В (НВЭ), что обусловлено различ-1м перенапряжением для выделения водорода и кислорода на их электродах. Золото, в отличие от платины, анодно растворя-ся, особенно в присутствии комплексообразователей — галогенидонов, цианид-ионов, серусодержащих органических реагентов и Р-, что необходимо учитывать при работе с этим электродом.
кис 3 ЗОЛОТОм электроде, как и на платиновом, хемосорбируется лород и образуются поверхностные оксиды [66—70]. Адсор
29
бированный кислород принимает участие в анодных процессах протекающих с изменением кислородного баланса в окисляющемся веществе, а оксиды золота ингибируют эти процессы [68—74]. Поэтому вольт-амперометфическое поведение веществ, окисляющихся с изменением кислородного баланса, на золотом электроде подобно поведению их на платиновом.
Алюминиевый электрод. Алюминиевый электрод чаще всего применяют при определении фторид-ионов и при использовании фторида в качестве титрующего реактива, например для определения алюминия [75—-77]. Однако в этом случае индикаторный электрод не является инертным — он анодно депопляризуется в присутствии ионов фтора, в результате чего появляется анодный ток, пропорциональный концентрации фторид-ионов.
Другие электроды. Кроме упомянутых электродов, в литературе имеются указания на применение индикаторных электродов из палладия [77, 79], вольфрама [65, 80], висмута [77, 81], олова [77, 82], серебра [80, 83], свинца [80], молибдена [80], меди [80, 83, 84], ниобий-цирконий-титанового сплава [85], а также оксидов свинца(IV) [86, 87], таллия(III) [87, 88], марганца (IV) [86, 88], висмута(Ш) [86, 88] и некоторых других оксидов [86—88]. Электроды из.этих металлов чаще всего применяют в качестве катода, а оксидгые электроды в качестве анода. Часто эти электроды используют в паре с платиновым при амперометрическом титровании с двумя индикаторными электродами.
Некоторые особенности твердых электродов
Поверхность твердых электродов, в противоположность ртутному капающему, не обновляется во время титрования, и поэтому возможны ее изменения, связанные с адсорбцией компонентов раствора, выделением нерастворимых продуктов электродной реакции и другими явлениями. Изменение состояния поверхности электрода, в свою очередь, может повлиять на электрохимическое поведение участников титрования и, как следствие этого, на ход амперометрических кривых. При этом влияние может быть столь значительным, что оно приводит к полному прекращению разряда деполяризатора.
ИЗМЕНЕНИЕ ПОВЕРХНОСТИ ЭЛЕКТРОДА
ЗА СЧЕТ АДСОРБЦИОННЫХ ЯВЛЕНИИ
Адсорбция ионов и молекул. На твердых электродах могут адсорбироваться присутствующие в растворе*ионы и нейтральные молекулы: галогенид-, фосфат-ионы, таллий(I), кадмий(II), спирты и другие неорганические и органические вещества [14, 15, 42, 89— 92]. Адсорбционная способность зависит от ряда факторов, в первую очередь от природы адсорбата, кислотности раствора и потенциала электрода. Например, галогениды по прочности связи и по
30
дл1ша 4 Потенциалы начала окисления -железа (II) и восстановления железа (Ш) пРи различных концентрациях иодида на фоне 1 М H2SOt
Отношение молярных концентраций Fe2+:I Потенциал начала окисления Fe11, В (НВЭ) Отношение молярных концентраций Fe3+; I Потенциал начала восстановления Fe®, В (НВЭ)
1:0,0 0,6 1:0,0 0,7
1:0,05 1:0,1 1:0,2 0,8 0,9 1,0 1:0,02 0,5
1:0,4 1,1 1:0,1 0,4
1:1,5 1,2 1:0,2 0,3
адсорбируемости располагаются в ряд: F~<Cl_<Br~<:I_, их адсорбируемость при переходе от кислых к щелочным растворам, а также при высоких анодных или катодных потенциалах значительно уменьшается (вплоть до полной десорбции).
Наличие в электролите посторонних адсорбирующихся ионов и молекул обусловливает обычно торможение электродных процессов, что выражается в той или иной мере сдвига потенциала окисляющегося вещества в сторону более положительных, а восстанавливающегося— в сторону более отрицательных значений (табл. 4), и в уменьшении тока (скорости разряда) потенциал-определяющих веществ. Влияние адсорбированных веществ проявляется тем сильнее, чем большей адсорбционной способностью оно обладает и чем больше его концентрация в электролите. Так, в случае галогенидов эффект влияния на те или иные электродные процессы усиливается в ряду: С1_<Вг_<Г_.
Особенно существенное влияние на электрохимическое поведение веществ оказывают иодид-ионы, причем эффект влияния наблюдается не только в присутствии этого галогенида в растворе, но и в том случае, если он в электролите отсутствует, а вольт-амперные кривые снимаются на «иодированном» электроде, т. е. на электроде, на котором предварительно были адсорбированы ионы иода. На таком электроде, как и при наличии в растворе свободных иодид-ионов, восстановление, например железа(III), происходит цри менее положительных потенциалах, а его ток уменьшается (рис. 7). По мере того как увеличивается время иодирования электрода (количество адсорбированного иодида), этот эффект усиливается настолько, что процесс разряда ферри-ионов почти прекращается (рис. 7, кривая 4). Аналогичную картину можно наблюдать также в случае полярографирования на иодированном электроде растворенного кислорода [93], арсенита I13'J и других веществ.
ио С°веРшенно очевидно, что если адсорбированные на электроде элеИД И°НЬ1 (Равно как и Другие ионы и молекулы) влияют на ТИтКТрохимическое поведение тех или иных присутствующих в руемом растворе веществ, то это, в свою очередь, может при-
31
Рис. 7. Вольт-амперные кривые восстановления железа(Ш) (5-10'4 М) на платиновом электроде, иодированном в 0,1 М рас. творе KI. Продолжительность иодирования-1 — 0.0; 2— 5; 3—15; 4—30 мин; 5 — фон (1 серная кислота на иодированном электроде)-электрод сравнения — НВЭ. ' *
вести к осложнениям при амперо-метрических определениях. Мы часто наблюдали, что платиновый электрод, на котором проводилось титрование по току окисления иодида
или восстановления иода, оказывался совершенно непригодным для работы при определении других веществ [железа (III), церия(IV), бихромата и т, д.].
Удалить адсорбированный иод с электрода можно нескольки
ми способами, в частности: прокаливанием платины в пламени газовой горелки, выдерживанием электрода при потенциалах около 1,5—1,7 В (НВЭ) в сернокислом растворе или промыванием 1— 2 М раствором едкой щелочи.
При очистке электрода от иода следует иметь в виду, что иод не только адсорбируется на поверхности, но и проникает в глубь платины. Поэтому операцию десорбции следует проводить не менее 3—5 раз через 20—30 мин или оставлять электрод в растворе
щелочи на ночь.
Адсорбирующиеся вещества могут оказывать и положительное влияние. Так, адсорбция тиокарбамида облегчает процесс электровосстановления платины (IV) (рис. 8). В данном случае адсорбированный тиокарбамид играет роль своеобразного «мостика», спо_-собствующего переносу электронов от электрода к иону PtCll [94].
Адсорбция продуктов химической реакции. При амперометрическом титровании иногда возникают осложнения, связанные с пассивацией электрода адсорбирующимися продуктами химической реакции. Такое явление, например, наблюдается при титровании цинка [95—97] и некоторых других металлов [95, 98] в присутствии алюминия ферроцианидом по току окисления последнего на платиновом электроде при потенциале 1,0 В (НВЭ) на хлоридно-аммиачном фоне и в слабокислой (0,01 М H2SO4)
среде.
Амперометрическая кривая в этом случае характеризуется или очень пологим ходом после конечной точки, или вообще отсутствием конечной точки. Подобное влияние алюминия наблюдается также при титровании циркония купфероном [96, 99]. Это осложнение хода амперометрической кривой обусловлено экранированием поверхности электрода гидроксидом алюминия, образующим-
результате гидролитических явлений в растворе. Устранить СЯо осложнение можно путем связывания алюминия в цитратный roc; Q71 или тартратный [100] комплексы, а также путем проведения титрования при потенциалах 1,5—1,6 В (НВЭ) [96, 101]. Ппи этих потенциалах на платиновом электроде одновременно протекают два процесса — окисление ферроцианида и окисление воды с выделением молекулярного кислорода, вследствие чего поиэлектродный слой подкисляется и адсорбцрованный на электроде гидроксид алюминия растворяется.
Р Другим примером осложнения хода амперометрической кривой являются случаи титрования никеля (II) [8], свинца (II) [8], скандия (Ш) и ионов некоторых РЗЭ [102] феррицианидом калия по току его восстановления на платиновом или графитовом электродах.
Получающиеся в этом случае кривые вообще не имеют перегиба, соответствующего конечной точке, т. е. наблюдается явление пассивации электрода по отношению к процессу разряда фер-рицианид-иона. Это явление обусловлено следующей причиной: в растворе, с самого начала титрования, из-за некоторой растворимости феррицианида никеля (свинца, РЗЭ), присутствует какое-то количество свободных ионов феррицианида, которые восстанавливаются на электроде до ферроцианид-ионов. Появляющиеся в приэлектродном слое ферроцианид-ионы образуют с ионами никеля (свинца, РЗЭ) менее растворимые (в отличие от феррицианидов) соединения, которые плотно обволакивают электрод и тем самым блокируют его поверхность.
Пассивация электрода за счет блокировки его осадком может происходить и в том случае, когда этот осадок является продуктом взаимодействия участников титрования. В частности, такое явление наблюдается при титровании ионов цинка, марганца, кобальта раствором феррицианида калия [8] по току его восстановления на платиновом электроде, а также при титровании ванадия (IV), мышьяка (III) и вольфрамата [103, 104] в растворах с рН=4—7 тиооксином по току его окисления на платиновом или • графитовом электродах. Наблюдаемые при этом кривые титрования вообще не имеют перегиба.
Рис. 8. Вольт-амперные кривые восстановления платины (IV) (1.5Х10-4 ) на платиновом электроде, пред-т^вЛЬг1° выДеРжанном в 0,1 М растворе тиокарбамида:
[Продолжительность выдерживания: 1 — кислота) мин: 3 — Фон (0.5 М соляная
3—1694
33
ИЗМЕНЕНИЕ ПОВЕРХНОСТИ ЭЛЕКТРОДА ЗА СЧЕТ ЭЛЕКТРОДНОГО ПРОЦЕССА
Выделение металлов. Количество металла, выделяющегося на электроде за время титрования, настолько мало, что его не принимают во внимание. Однако оно вполне достаточно, чтобы полностью покрыть поверхность твердого электрода хотя бы тончайшим слоем. При этом происходит, по существу, самопроизвольная замена одного материала индикаторного электрода другим. Этот факт, как ни странно, далеко не всегда учитывается исследователями, считающими, что они работают, например, с платиновым электродом, тогда как он покрыт другим металлом, который может вести себя иначе, чем платина, и оказывать влияние на электродный процесс. Подобный случай наблюдается [105], например, при аргентометрическом определении хлорид-ионов в растворах, содержащих медь (II), если титрование хлорида проводят без наложения внешнего напряжения с применением меркур-иодидного электрода сравнения, т. е. при потенциале +0,02 В (НВЭ). При этом происходит процесс восстановления ионов меди(II), и кривые имеют настолько искаженный ход, что определение хлорида оказывается вообще невозможным. Причина этого явления заключается в том, что на свежеосажденной меди резко увеличивается скорость электровосстановления взвеси осадка хлорида серебра [105]. Если же титрование проводят при потенциале +0,4 В, при котором восстановление меди (II) и взвеси осадйа хлорида серебра исключено, то определение хлорид-иона в медных электролитах идет беспрепятственно.
С другой стороны, выделяющийся металл может способствовать проведению амперометрического титрования. Например, титрование солями или солей свинца по току восстановления ионов РЬ2+ на платиновом электроде при потенциале —0,5 В (НВЭ) становится возможным в слабокислой среде потому, что электрод покрывается металлическим свинцом, на котором резко повышается перенапряжение выделения водорода. На чистой же платине при этом потенциале основная реакция состоит в выделении водорода.
Выделение оксидов металлов. На поверхности электрода наряду с металлами могут осаждаться их оксиды. Осадки оксидов металлов могут образовываться в результате анодного или катодного процесса, а именно:
а) анодный процесс — окисление ионов низшей валентности до высшей: РЬ2+—*-РЬОг; Мп2+—*-МпОг; Т1+—*-Т12О3.
б) катодный процесс — восстановление ионов высшей валентности до низшей: СггО?"—>Сг2О3; МпО?—>МпО2.
В обоих случаях выделяющиеся на электроде оксиды, обладая малой электропроводностью, повышают сопротивление на границе электрод — раствор, в результате чего обмен электронами между электродом и разряжающимися ионами затрудняется, и сила
34
тока заметно снижается. Это может вызвать недостаточно четкий ход кривой титрования.
Снять осадок оксида металлов с поверхности электрода мож-путем промывания азотной кислотой или растворами других ешеств, взаимодействующих с оксидом. Например, для удаления диоксида марганца можно воспользоваться раствором щавелевой кислоты или ее соли.
ЛИТЕРАТУРА
1 Гейровский Я-, Кута Я- Основы полярографии. — Пер. с чешского/Под ред. ’ С Г Майр айовского. М., Мир, 1965.
2 Дамаскин Б. Б., Петрий О. А. Введение в электрохимическую кинетику. М, Высшая школа, 1975.
3 Галюс 3. Теоретические основы электрохимического анализа. — Пер. с поль-ского/Под ред. Б. Я- Каплана. М., Мир, 1974
4 Эпштейн И. М. — Электрохимия, 1966, т. 2, № 6, с. 734.
5 Плесков Ю. В., Филиновский В. Ю. Вращающийся дисковый электрод. М., Наука, i972.
6 . Adams Р. N. Electrochemistry at solid elektrodes. N. Y., 1969.
7 . Графов Б. M. — Электрохимия, 1967, т. 3, № 8, с. 935.
8' Сонгина О. А., Захаров В. А. — Изв. АН КазССР. Сер. хим., 1961, вып. 1 ’ (19), с. 52.
9. Хазова О. А., Васильев Ю. Б., Багоцкий В. С. — Электрохимия, 1965, т. 1, ’ № 1, с. 84.
10. Will F. С., Knorr С. А. — Z. Elektrochem., 1960, v. 64, № 2, р. 258.
11. Breiter М. W., Knorr С. A., Volk М. Z. — Z. Elektrochem., 1955, v. 59, № 4, р. 681.
12. Bold W., Breiter M. — Elektrochim. Acta, 1961, v. 5, № 2, p. 145.
13. Breiter M. W.— J. Electroanal. Chem., 1964, v. 8, № 3, p. 449.
14. Сокольский Д. В., Закумбаева Г. Д. Адсорбция и катализ на металлах VIII группы в растворах. Алма-Ата, Наука, 1973.
15. Захаров В. А., Сонгина О. А., Бессарабова И. М. — Изв. АН КазССР, Сер. хим., 1968, № 5, с. 28.
16. Сонгина О. А., Рождественская 3. Б.—Ж. анал.хим., 1956, т. 2, № 6, с. 717.
17. Сонгина О. А. Амперометрическое титрование. М., Химия, 1967.
18. Захаров В. А., Сонгина О. А. — Изв. АН КазССР. Сер. хим., 1964, вып. 4, с. 3.
>9- Оше А. Тихомирова В. И., Багоцкий В. С. — Электрохимия, 1965, т. 1,
20. Захаров В. А., Сонгина О. А. — Изв. АН КазССР. Сер. хим., 1964, вып. 2, с. 10.
21- Р- И., Захаров В. А., Сонгина О. А. — Изв. АН КазССР. Сер. хим., 1969. вып- 61 с- 20 * * * * * * * * 29-
Нестерова В. И., Фрумкин А. И.—Ж- физ. хим., 1952, т. 26, № 8, с. 1178.
о? Борисова Т. И., Веселовский В. И,—Ж. физ. хим., 1953, т. 27, № 8, с. 1195.
с°б37гаЛ6 И" Веселовский В- И~ДАН ссср> 1956, т- Ш. № 3,
И'’ ТихомиРова В- Багоцкий В. С.—Электрохимия,
27^'мп S°h Г'<?' BinSane J. I. — J. Amer. Chem. Soc., 1957, v. 79, № 10, p. 4901.
2R PoihL A BanSer S. H. — J. Electrochem. Soc., 1964, v. Ill, № 2, p. 438.
M, л erg<S' W-> Enke C. G., Bricker С. E. — J. Electrochem. Soc., 1963, v. 110, 29r-4’,p-826
3KL Man» Tn Woods R — J- Electroanal. Chem., 1969, v. 20, № 1, p. 73.
31 In еТп' * ПетРий О. A. — Электрохимия, 1967, т. 3, № 7, с. 901.
ln сгг Д-Апа1. Chem., 1961, v. 33, № 4, р. 934.
3*
35
32. Елина Л. М., Борисова Т. И., Филиппова Т. С.—Ж. физ. хим., 1956, т. Зо № 6, с. 1283.
33. Раков А. А., Потапова Г. Ф„ Веселовский В. И. — Электрохимия, 1970 т. 6, № 11, с. 1730.
34. Захаров В. А., Сангина О. А.—Ж. физ. хим., 1962, т. 36, № 6, с. 1226.
35. Кузьмина Н. Н., Сангина О. А. — Изв. вузов. Химия и хим. технол., 1963, т. 6, № 2, с. 201.
36. Розенталь К. И., Веселовский В. И,—Ж. физ. хим., 1958, т. 32, № 6 с. 1341.
37. Захаров В. А., Сангина О. А.—Ж. физ. хим., 1964, т. 38, № 3, с. 766.
38. Бессарабова И. М., Захаров В. А., Сангина О. А. — В кн.: Химия и химическая технология. Алма-Ата, 1970, вып. 1, с. 24.
39. Кабанова О. Л.—Ж. физ. хим., 1961, т. 35, № 11, с. 2465.
40. Тедорадзе Г. А.—Ж. физ. хим., 1959, т. 33, № 1, с. 129.
41. Кузьмина Н. Н., Сангина О. А. — Изв. вузов. Химия и хим. техиол., 1961, т. 4, № 6, с. 928.
42. Захаров В. А., Сангина О. А., Оспанов X. К- — Изв. АН КазССР. Сер. хим., 1968, № 6, с. 21.
43. Бессарабова И. М. и др. — Изв. вузов. Химия и хим. технол., 1969, т. 12, № 9, с. 1202.
44. Бескоровайная С. С. и др. — Электрохимия, 1966, т. 2, № 8, с. 932.
45. Фрумкин А. Н„ Подловченко Б. И. — ДАН СССР, 1963, т. 150, № 2, с. 349.
46. Богдановский Г. А., Шлыгин А. И. — Ж. физ. хим., 1959, т. 33, № 8, с. 1769.
47. Рождественская 3. Б., Сангина О. А., Бараков В. Г. — Зав. лаб., 1963, т. 29, № 1, с. 30.
48. Галлай 3. А. и др. — Ж. анал. хим., 1964, т. 19, № 12, с. 1464.
49. Вариков В. Г., Сангина О. А., Климка Е. В. — Зав. лаб., 1969, т. 35, № II, с. 1312.
50. Галлай 3. А. и др. — Ж. анал. хим., 1975, т. 30, № 10, с. 1947; 1975, т. 30 Ns 12, с. 2346.
51. Чуйко Т. В., Аришкевич А. М„ Тулюпа Ф. М.—Ж. анал. хим., 1976, т. 31, Ns 4, с. 671.
52. Брайнина X. 3. Инверсионная вольт-амперометрия твердых фаз. М., Химия, 1972.
53. Евсеева М. А., Ашпур В. В., Машкович Л. А. и др. — Зав. лаб., 1973, т. 30, № 11, с. 1314.
54. Кабанова О. А., Гончаров Ю. А. — Ж. анал. хим., 1973, т. 28, Ns 9, с. ' 665.
55. Drossbach F., Schuls J. — Electrochim. acta, 1961, v. 9, Ns 7, p. 1391.
56. Пономаренко E. А., Фрумкин A. H., Бурштейн P. X. — Изв. АН СССР. Сер. хим., 1963, Ns 9, c. 1549.
57. Фрумкин А. И., Пономаренко E. А., Бурштейн P. X.—ДАН СССР, 1963, т. 149, Ns 5, с. 1123.
58. Тарасевич М. Р. и др. — Электрохимия, 1971, т. 7, Ns 4, с. 586.
59. Хадеев В. А., Глазунова Л. А. —Узб. хим. ж., 1959, Ns 3, с. 24.
60. Жданов А. К., Ахмедов Г. —Узб. хим. ж., 1969, Ns 4, с. 29.
61. Жданов А. К, Акентьева Н. А., Корытко П. К — ДАН Узб. ССР, 1972, Ns 7, с. 33.
62. Геворгян А. М„ Талипов Ш. Т., Хадеев В. А.—Узб. хим. ж., 1973, Ns 4.
63. Хадеев В. А., Геворгян А. М. — ДАН Узб. ССР, 1976, № 7, с. 47.
64. Helbing N. — Z. analyt. Chem., 1969, v. 245, Ns 2, p. 359.
65. Галлай 3. А., Марьяне вская Г. Я.—Ж- аиал. хим., 1963, т. 18, Ns 8, с 924.
66. Will F. G„ Knorr С. А. — Z. Elektrochem., 1960, v. 64, Ns 1, р. 270.
67. Hoare I. P. — Electrochim. Acta, 1966, v. 11, Ns 1, p. 203.
68. Мичри А. А., Пшеничников А. Г., Бурштейн P. X. — Электрохимия, 1972, т. 8, Ns 3, c. 364.
69. Винников Ю. Я-, Шелепин В. А., Веселовский В. И. — Электрохимия, 1972, т. 8, Ns 9, с. 1384.
36
70 Захаров В. А., Кальницкая Л. П., Сонгина О. А,— Изв. АН КазССР. Сер. ,, ЧахаоовВ А и др2—Электрохимия, 1971, т. 7, № 8, с. 1215.
711 В А., Сонгина О. А., Кальницкая Л. П. — Электрохимия, 1971,
72. 702
74 ^Тюпикова’о. Г., Миллер Н. Б., Веселовский В. Я. — Электрохимия, 1972, т 8 № 4, с. 618.
. \nruta I. Pradac I. — J. Electroanal. Chem., 1966, v. 17, № 1, p. 177.
74 Kolthoff 7- M., Sambucetti C. J. — Anal. chim. acta, 1959, v. 21, № 1, p. 17;
1959 v. 21, № 2, p. 155.
7fi Човнык H. Г., Алемаскина Г. A. — В кн.: Физико-химические методы ана-лиза металлов, сплавов и электролитических ваин. М., 1975.
77 Gacil F. F., Jovanotic Li. S., Canic V. D. — Z. anal. Chem., 1974, v. 272, ' № 2, p. 117. »
78 Flvino P. I., Smith D. I. — Anal. Chem., 1960, v. 32, № 11, p. 1849.
79 Harzdorf C. — Z. anal. Chem., 1969, v. 244, № 6, p. 368.
80, Филенка А. И.—Ж. анал. хим., 1967, т. 22, № 1, с. 161.
81 Козулина М. М„ Лепин Я. К-, Сонгина О. А. — В кн.: Химия и химическая технология. Алма-Ата, 1975, вып. 17, с. 122.
82. Gaal F. F-, Leovac V. М., Avramovic В. D. — Z. anal. Chem., 1973, v. 266, № 5, p. 355.
83. Масалович В. M., Лагунова А. — Труды Уральского научно-исследовательского химического ин-та, 1971, вып. 26, с. 58.
84. Борк В. А., Швыркова Л. А., Файзулаев О.—Ж. анал. хим., 1974, т. 29, № 9, с. 1844.
85. Жданов А. К, Бархударьян А. А.—Ж. анал. хим., 1975, т. 30, № 10, с. 2645.
86. Kainz G., Muller Н. A., Sontag G. — Z. anal. Chem., 1971, v. 256, № 5, p. 345.
87. Sontag G., Kainz G., Margaritella P. — Z. anal. Chem., 1975, v. 273, № 4, p. 263.
88. Kainz G., Sontag G. — Z. anal. Chem., 1974, v. 269, № 4, p.'267.
89. Дамаскин Б. Б., Петрий О. А., Батраков В. В. Адсорбция органических соединений на электродах. М., Наука, 1968.
90. Балашова Н. А. — Электрохимия, 1967, т. 3, № 6, с. 750.
91. Казаринов В. Е. — Электрохимия, 1966, т. 2, № 12, с. 1389.
92. Парцхалава Дж., Васильев Ю. Б., Багоцкий В. С. — Электрохимия, 1970, т. 6, № 1, с. 110.
93. Захаров В. А., Сонгина О. А.—Ж. физ. хим., 1963, т. 37, № 7, с. 1450.
94. Гавва Н. Ф„ Захаров В. А., Сонгина О. А. — Изв. АН КазССР. Сер. хим., 1976, № 1, с. 67.
95. Сонгина О. А., Войлошникова А. П„ Козловский М. Т.—Зав. лаб., 1952, т. 18, № 4, с. 390.
96. Усвяцов А. А., Сонгина О. А. — Зав. лаб., 1965, т. 31, № 6, с. 661.
97. Сонгина О. А., Савицкая И. С., Рождественская 3. Б. — Изв. вузов. Химия и хим. технол., 1969, т. 12, № 7, с. 858.
98. Сонгина О. А., Войлошникова А. П., Козловский М. Т. — Изв. АН КазССР. 0Q ~еР- хим., 1953. вып. 6, с. 69
inn „сатенко Ю. И., Беклешова Г. Е, — Зав. лаб., 1957, т. 23, № 1, с. 12. iw. Дюлгерова А. С., Сонгина О. А., Захаров В. А.— Изв. АН КазССР. Сер. 101 ^ИМ” 19711 № 31 с' 66-
’ Ж)н?ина °. А., Бессарабова И. М., Усвяцов А. А.— Зав. лаб. 1972, т. 38, £ 21.
1021 J°Ki/weea г Т., Сонгина О. А., Захаров В. А. — Изв. АН КазССР. Сер. 103 гт ’ 9711 № 21 с- 79-
"а™ова И. М„ Сонгина О. А, —Изв. АН КазССР. Сер. хим., 1967, № 2,
вы^З0^ ^98^ U — В КН" Сборник работ по химии. Алма-Ата, 1973, 105‘ 3ахаР°в В. А. и др,—Ж. анал. хим., 1969, т. 24, № 9, с. 1401.
37
ГЛАВА Ш
Амперометрическое титрование с одним индикаторным электродом
Установки для амперометрического титрования
Принципиальные схемы установок для амперометрического титрования с одним индикаторным электродом приведены на рис. 9 и 10 и подробно описаны в предыдущем издании этой книги [1]. Конструктивное оформление установок может быть весьма различным— от простых «открытых» схем, собираемых из обычных измерительных приборов, до специальных компактных установок, например типа АУ [2] и УАТ-ЦНИИЧМ [3, 4]. Амперометрическое титрование можно выполнять, конечно, также на любом по-лярографе. Так, имеются сведения о применении классических полярографов ПВ-5 [5] и LP-55 [6], а также осциллографического [7], переменнотокового [8, 9] и импульсного [10] поляро-гоасров.
Особое внимание должно быть уделено индикаторному электроду. При работе с ртутным капающим электродом желательно подбирать характеристику капилляра так, чтобы число капель в минуту было не менее 60, т. е. подбирать быстро капающий электрод. Такие электроды обеспечивают меньшие колебания зеркальца (или стрелки) гальванометра, что значительно облегчает отсчет силы тока после каждого прибавления титрующего раствора.
Что касается твердых электродов, то конструкция их может быть весьма разнообразной как в отношении размера и формы рабочей части электрода, так и в отношении осуществления его вращения.
На рис. 11 показаны часто применяемые формы электродов, представляющих собой платиновую проволоку диаметром 0,5 мм, впаянную в стеклянную трубку так, что наружный (рабочий) конец проволоки имеет длину 4—5 мм. Рабочая длина платиновой проволоки имеет большое значение. В некоторых случаях при определении малых количеств вещества можно прибегать к большим электродам, длиной до 10 мм.
Для титрования и особенно для снятия вольт-амперных кривых часто применяют также электрод в виде вращающегося диска. На рис. 11, в приведена простейшая конструкция такого электрода, который представляет собой торец платиновой проволоки диа^-метром 1—3 мм, впаянной в стеклянную трубку и зашлифованной заподлицо с ее концом. Хотя электрод этого типа не имеет «кры-
38
Рис. 9. Схема амперометрической установки:
1 — индикаторный электрод; 2 — шкив и передача к мотору, осуществляющему вращение платинового индикаторного электрода; 3 — сосуд для титрование 4__промежуточный сосуд; 5 — электрод сравнения; 6 — гальванометр с
шунтом; 7—вольтметр; 8 — реостат; 9— источник тока; 10, 11 — контакты, при помощи которых можно замыкать электроды непосредственно на гальванометр или присоединять источник тока с реостатом и вольтметром.
лышек», он все же обеспечивает достаточно быстрое перемешивание титруемого раствора, если частота вращения электрода достигает 600 об/мин. Для интенсивного перемешивания раствора при небольших частотах вращения (200—300 об/мин) к концу стеклянной трубки припаивают два «крылышка» или используют элект-< род, изображенный на рис. 11, а. Для изготовления электрода такого типа используют обычно фторопласт; расширенный конец фторопластовой трубки срезают в форме эллипса, в центре кото-
рого просверливают сквозное отверстие и в него запрессовывают платину в виде проволоки или «гвоздя», торцовая сторона (или шляпка) которого служит рабочей поверхностью электрода.
Аналогичную конструкцию имеют графитовые электроды.
• Ю. Схема амперометрической установки ДВУМЯ вращающимися электродами — ^икаториым и электродом сравнения: ния Э”Тм<т^1ИЗерь1’ 5 — шкивы передачи враще-’лектвопы- Здам; 3 ~ платиновые вращающиеся помете п’ * соединительный мост; 5 — гальва-в~ HCTO4n.JfyHT°M: 6~ вольтметр; 7 —реостат; ЭлектромотораТ0Ка’ 9~двойной ведущий шкив
39
Рис. 11. Индикаторные электроды проволочные (а, б) и дисковые (в, г):
1 — платина; 2 — стекло; 3 — фторопласт
Особое внимание необходимо обращать на качество впаивания проволоки в стеклянную трубку во избежание появления тончайших трещин, нарушающих работу электрода [1].
Большое значение имеет способ очистки твердых электродов после работы и хранение их. При выделении на электроде металлов или окислов или при адсорбции различных веществ электроды следует очистить тем или иным способом: промывание водой и обтирание фильтровальной бумагой, электролитическая очистка поверхности [1].
Довольно много внимания уделяется тому, как лучше хранить электрод в промежутках между работой: на воздухе, в воде или в каком-либо электролите. Обычно способ хранения электрода не оказывает существенного влияния на характер кривых титрования , Можно хранить его просто на воздухе (после предварительной промывки по окончании работы) или погружать в дистиллированную воду. Не следует оставлять электроды на воздухе в том случае, если они работали в условиях, которые способствуют образованию оксидных пленок или других адсорбционных слоев. При хранении на воздухе может происходить окисление или «старение» этих пленок, растворимость их значительно уменьшается, и при возобновлении работы получаются меньшие значения силы тока. Подобные явления очень заметны при работе с растворами ферроцианида: в этом случае на поверхности электрода образуется налет турнбуллевой сини, пассивирующий электрод настолько, что последний уже на следующий день почти полностью теряет чувствительность, если был оставлен на ночь просто на воздухе. При хранении электрода в кислоте подобная пассивация не наблюдается.
Электроды сравнения. Основным требованием, предъявляемым к электроду сравнения при полярографических и амперометрических определениях, является его неполяризуемость: потенциал электрода сравнения должен оставаться практически постоянным
40
юбом наложенном напряжении. Для этого площадь его дол-ПрИ "быть неизмеримо больше площади индикаторного электрода. Роли площадь электрода сравнения мала, то плотность тока, про-ЬСЛ шего в цепи, может оказаться настолько большой, что потен-Х°ал электрода изменится — электрод поляризуется. Поляризация Цпектрода сравнения приведет к изменению потенциала индикаторного электрода, поскольку гран (или фкат) в уравнении
~ Van — '?кат+ 7/?
VJKe не будет постоянной.
У При амперометрическом титровании поляризация электрода сравнения может привести к тому, что на кривых титрования сократятся прямолинейные участки (отвечающие основному уравнению id=KC), и определение конечной точки станет затруднительным.
В табл. 5 приведены составы и потенциалы наиболее часто применяемых электродов сравнения.
Для пересчета потенциала одного электрода на другой удобно пользоваться следующим графическим приемом. На рис. 12 приведена «ось потенциалов»; потенциалы, положительные по отношению к нормальному водородному электроду, располагаются слева от него (т. е. слева от нуля); потенциалы, отрицательны по отношению к водородному электроду, — справа от него (т. е. справа от нуля).
Из этого рисунка следует, например, что если потенциал цинковой пластинки, погруженной в 1 н. раствор соли цинка, равен —0,76 В относительно стандартного водородного электрода, то относительно насыщенного каломельного он будет равен: —0,76— (+0,25)=—1,01 В.
При расчете потенциал насыщенного каломельного электрода всегда вычитают, так как за нуль принят уже не водородный электрод, а каломельный, по отношению к которому водородный электрод является более отрицательным. Если за нуль принять потенциал меркур-сульфатного электрода, то потенциал той же цинко-
+0,43
-----------7/4--------
--------------1iD1.
+0,25 О
-+-м-----1-------1—f-
-0,7В
в, в
Меркур-
Цинк в 1н. растворе ZnSO4
~ Каломельный Нормальный
сУль<ратныи.^ насыщенный воворовный
электродов* с” ПОтенциалов>> для пересчета потенциалов различных
41
Таблица 5. Состав и потенциалы некоторых электродов сравнения
Электрод Состав электрода Потенциал по отношению к нормальному водородному электроду (прн 20 °C), в
Нормальный водородный (НВЭ; Pt, Н2|Н+|| Платинированная платиновая пластинка в 1 М растворе серной кислоты, насыщенном водородом 0
Меркур-иодидный (МИЭ) Металлическая ртуть, 4,2 г иоди- +0,02
Hg|HgI2, KI, kciii Каломельные (КЭ) Hg|Hg2, CI2, KCIII да калия и 1,3 г иодида ртути (II) на 100 мл насыщенного раствора хлорида калия Металлическая ртуть, паста из металлической ртути и каломели, раствор хлорида калия:
0,1 н. (0,1 НКЭ) 0,1 н. +0,337
1,0 н. (НКЭ) 1,0 н. +0,284
насыщенный (Нас. КЭ) насыщенный +0,247
Меркур-сульфатный HglHg2SO4, H2SO4|| (М-сульф. Э) Металлическая ртуть, паста из металлической ртути и сульфата ртути (I), 2 н. серная кислота +0,682
Хлор-серебряный Ag AgCI, KCIII (ХСЭ) Металлическое серебро, покрытое пленкой хлорида серебра, в 1 н. растворе хлорида калия +0,290
вой пластинки в тех же условиях будет равен: —0,76—(+0,68) = =—1,44 В.
Если, наоборот, указан потенциал относительно того или иного 3J ектрода сравнения, то по отношению к стандартному водородному электроду он будет определен как сумма заданного потенциала и потенциала электрода сравнения, поскольку электрод сравнения является положительным по отношению к стандартному водородному электроду. Например, если титрование проводят при потенциале —0,6 В по отношению к насыщенному каломельному электроду (см. рис. 12), то это значит, что потенциал индикаторного электрода составляет по отношению к стандартному водородному электроду: —0,6+ (+0,25) =—0,35 В.
Если титрование проводят при +0,15 В относительно насыщенного каломельного электрода, то относительно стандартного водородного этот потенциал будет составлять:+0,15+(+0,25) = = +0,40 В.
Если указан потенциал меркур-сульфатного электрода и требуется пересчитать его на каломельный, то, как это вытекает из того же рис. 12, этот потенциал составит: +0,68—(+0,25) =+0,43 В
В первых работах по полярографии электродом сравнения (обычно анодом) служил слой металлической ртути, наливаемой на дно рабочей ячейки. Простота такого устройства сравнения не может компенсировать все присущие ему недостатки: прежде всего, потенциал этого электрода меняется при переходе от одного
42
к другому, что не позволяет сравнивать между собой раСгпьтаты полученные в различных случаях; кроме того, донная Резу может так или иначе реагировать с титруемым или титрую-рту раствором (например, с перманганатом, тиосоединениями ™ д ) искажая ход кривой титрования.
И Предложены и другие типы электродов сравнения: жидкие амальгамы [П, 13], металлические стержни в агаровой массе, со-пеожащей соли того же металла [13], или просто платиновая пластинка размером 1 см2, помещенная в электролитическую ячейку, в которой проводят титрование Следует, однако, заметить, что хотя этот электрод прост по устройству, работать с ним неудобно, так как помещение анода в один сосуд с катодом вызывает осложнения, о которых упоминалось в связи с донным ртутным электродом, а именно: потенциал электрода сравнения будет зависеть от солевого состава анализируемого раствора, который к тому же может существенно изменяться во время титрования.
Эти недостатки можно устранить, если платиновый электрод сравнения сделать выносным. Электроды такого типа в практику амперометрического титрования были введены Михальским с сотр. [14—16]. Он предложил применять электроды сравнения, представляющие собой растворы нитрата ртути (II), перхлората церия, смеси растворов иода и иодида, насыщенный раствор иоди-да таллия (I), в которые погружена неподвижная платиновая проволока. К сожалению, в работах Михальского отсутствуют данные о потенциалах указанных электродов сравнения, что, по существу, не дает возможности учесть, при каких потенциалах индикаторного электрода следует проводить то или иное титрование. Следует также отметить, что при применении ненасыщенных растворов и неподвижного электрода не исключена возможность его поляризации вследствие изменения концентрации соли у поверхности платинового электрода сравнения.
Однако этого явления можно избежать, применяя вращающийся платиновый электрод в насыщенном растворе окислителя или восстановителя [2—4, 17—19]. На рис. 10 приведена схема простого устройства, обеспечивающею вращение обоих электродов— индикаторного и электрода сравнения. Размеры обоих электродов Должны быть примерно одинаковые, но еще лучше, если электрод сравнения будет иметь больший размер, чем индикаторный. Растворы можно подобрать так, что получается непрерывный ряд электродов с различными потенциалами. Так как потенциал окислительно-восстановительных систем часто зависит от концентрации кислоты, то, выбирая соответствующие окислители и кислотность раствора, можно создать электроды сравнения с любым заданным потенциалом, в пределах от +1,6 до +0,1 В (НВЭ), как это видно на рис. 13.
Необходимо учитывать, что в зависимости от природы выбран-либ° веи*ества электрод сравнения будет работать либо как анод, ° как катод. Если в электроде сравнения находится зкисли-
43
Концентрация щелочи, М Концентрация киелотЫ] М Катоды н28О4--К3[1'и(ск)(]|4
|f 1,,Р04-^К.2СГ.>07 | 5
| h2so4^nh4vo3^toh \Z
|H2S04«-KMn04^-KOH 11
+e,b js jk- ‘ 1,2 ' jo ' d,e ' o,b ' dfi ' 0,2 1 b
|H2S04^-K4[Fe(CN)6]| ff
FqSQ4-^-H3PoJg
-^h/u!8
Дноьы I-^ho2!^
Рис. 13 Зависимость потенциала платиновых электродов сравнения в насыщенных растворах различных солей от кислотности электролита:
1 — перманганат в серной кислоте и едком кали; 2 — ванадат в серной кислоте и едком кали; 3 — бихромат в фосфорной кислоте; 4 — феррицианид в серной кислоте; 5 — ферроцианид в серной кислоте; 6 — железо (II) в фосфорной кислоте; 7—хлорид олова (П) в соляной кислоте; 8 — иодид калия в серной кислоте. В нижней части рисунка приводятся «ось потенциалов» и составы растворов, позволяющих получать желаемый потенциал электрода сравнения, выступающего в роли катода или анода; электрод сравнения — НВЭ.
тель, например перманганат, то электрод будет функционировать как катод при любом потенциале; для того чтобы электрод срав-> нения работал как анод, необходимо воспользоваться восстановителем, например иодидом или ферроцианидом калия.
Описанные выше электроды сравнения [2—4, 14—19] специально рассчитаны на титрование без наложения внешнего напряжения. Титрование без внешнего напряжения удобно с чисто практической стороны, так как оно позволяет упростить схему установ-
44
электрическая цепь в этом случае замыкается между индика-ИИ: iM электродом и электродом сравнения непосредственно че-Т°РНгальванометр. Тем самым из схемы амперометрической установки исключаются внешний источник постоянного тока и все приборы, связанные с ним.
Приготовление таких электродов сравнения чрезвычайно прото- в раствор кислоты, концентрация которой должна отвечать желаемому потенциалу при использовании данного окислителя, вносят этот окислитель в виде сухой соли в таком количестве, чтобы образовался насыщенный раствор. Потенциал устанавливается быстро и отвечает значениям, приведенным на рис. 13, с точностью до сотых долей вольта. Готовят электроды сравнения непосредственно перед титрованием. Ими можно пользоваться в течение всего рабочего дня.
Другие приборы. Для измерения силы тока применяют микроамперметры постоянного тока с чувствительностью 10-6— 10-9 A/деление, например, милливольтмикроамперметры типа М-193, М-194, М-198 и М-1200, гальванометры типа М-195 и др. Наиболее удобны для работы гальванометры М-194 и М-1200, которые снабжены шунтом и позволяют работать в широком интервале силы тока.
Для измерения напряжения можно пользоваться любым вольтметром постоянного тока, позволяющим контролировать приложенное напряжение с точностью 0,01 В. Наложение необходимого потенциала на индикаторный электрод осуществляют при помощи реостата (см. рис. 9 и 10) переменного сопротивления на 1000— 1500 Ом.
В качестве источника тока обычно достаточно иметь два щелочных аккумулятора на 1,2 В каждый или один кислотный. Можно также с успехом применять соответствующие сухие батареи, в том числе батарейки для фонаоиков и транзисторов.
При работе с вращающимися электродами перемешивание раствора осуществляется вращением самого электрода. Электрод вращают обычно со скоростью 200—600 оборот/мин. Более быстрое вращение, хотя и рекомендуется некоторыми авторами, не всегда желательно, поскольку оно приводит к техническим трудностям, например образованию глубокой воронки, разбрызгиванию и т. д. Для вращения электрода используют малогабаритные электромоторы переменного тока с постоянным числом оборотов, например моторы типа ДАП и ЭДГ. При работе со стационарными электродами раствор перемешивают обычно магнитной мешалкой.
Соединительные мосты (см. рис. 9 и 10) представляют собой стеклянные трубки, заполненные насыщенным раствором индифферентного электролита (КС1, KNO3, K2SO4) или чистым агар-*-ром. Однако применения агар-агара следует избегать, поскольку На агаРе могут адсорбироваться ионы исследуемого раствора с Ли Даже осадок при титровании по методу осаждения); при по-едующих титрованиях с тем же мостиком эти ионы могут ока
45
зать весьма нежелательное влияние на ход определения. Кроме того, агар-агар относится к числу поверхностно-активных веществ и следы его в растворе могут изменить состояние поверхности индикаторного электрода. Поэтому мы считаем более надежным ц удобным совершенно исключить агар-агар из практики амперометрического титрования и пользоваться простыми мостиками, концы (или даже один конец) которых закрыты маленькими пробками из фильтровальной бумаги (беззольные фильтры).
Длина соединительных мостиков не должна быть слишком большой; необходимо помнить, что увеличение длины и уменьшение диаметра мостика вызовет увеличение сопротивления системы, чего следует избегать при полярографических и амперометрических определениях.
В качестве электролизеров (сосудов) для титрования лучше всего использовать стеклянные стаканы емкостью 50—100 мл. Что касается бюреток, то обычно применяют стандартные микробюретки емкостью 1,2 или 5 мл. При этом желательно оттягивать кончик бюретки так, чтобы размер капли раствора был небольшим (не 20 капель на 1 мл, как обычно, а 80—100). В качестве бюретки иногда применяют медицинские шприцы [21], поршень которых приводится в движение от мотора.
Выбор потенциала индикаторного электрода
Титрование следует проводить при таком потенциале индикатор—, ного электрода, который соответствовал бы области диффузионного тока иона, дающего электродную реакцию (см. гл. I). Это значит, что разработка методов амперометрического титрования должна начинаться со снятия полярограммы (вольт-амперной кривой) соответствующего иона. Полярограмму снимают на том электроде, на котором предполагается в дальнейшем проводить титрование, и в той же среде*.
Правильность выбора потенциала можно легко проверить следующим образом: в стакан для титрования помещают раствор, который должен служить фоном при предполагаемом титровании; в бюретку помещают раствор, содержащий тот ион, который должен давать электродную реакцию. На индикаторном электроде устанавливают выбранный потенциал. При титровании фона этим раствором никакой химической реакции в растворе не происходит, но концентрация электровосстанавливающегося (окисляющегося) иона постепенно возрастает и пропорционально ей возрастает диф-
—т,
* Если титрование предполагается проводить с ртутным капающим электродом, то можно воспользоваться таблицами потенциалов полуволн (Ej/2), публикуемых в руководствах ло полярографии [21-—24] и в справочных изданиях [25, 26], учитывая, что в них приведены значения потенциалов полуволны, а не потенциалы, относящиеся к области диффузионного тока. Поэтому потенциал индикаторного электрода следует устанавливать на 0,1—0,3 В более отрицательным, чем указанный в таблице Ёу2-
46
пый ток. Если потенциал выбран правильно, то прямоли-фузион * Кр’ИВой титрования сохранится в широких пределах НеИНентраций. Если же потенциал не соответствует области диф-КОН^онногс тока, то на кривой титрования более или менее быст-*УЗИоявится «загиб», свидетельствующий о том, что прямолиней-ЕяяПзависимость id=KC не выдерживается.
Н Вольт-амперные кривые во многих случаях могут быть исполь-ваны и для полярографического определения на твердых элект-Зодах соответствующего элемента, однако подробное рассмотрение работ в этой области выходит за рамки данной монографии*. Тем не менее необходимо особенно подчеркнуть, что изучение по-1ЯП, -рамм, полученных при работе с твердыми электродами, имеет очень большое значение для правильного понимания некоторых процессов, наблюдаемых при амперометрическом титровании.
Типы электродных реакций
Выбор потенциала индикаторного электрода определяет возможность прохождения той или иной реакции на твердом электроде, в частности:
1) катодное восстановление окислителей и анодное окисление восстановителей, не сопровождающееся выделением осадков на электроде;
2) восстановление ионов металлов, сопровождающееся выделением осадка на электроде;
3) анодное окисление органических реактивов, используемых в амперометрическом титровании;
4) восстановление и окисление малорастворимых веществ.
Следует подчеркнуть, что при выборе потенциала необходимо считаться с так называемыми реальными потенциалами (Ер), таблица значений которых приведена в гл. V.
КАТОДНОЕ ВОССТАНОВЛЕНИЕ ОКИСЛИТЕЛЕН И АНОДНОЕ ОКИСЛЕНИЕ ВОССТАНОВИТЕЛЕЙ
В качестве титрующего раствора при амперометрических определениях часто пользуются ферри- или ферроцианидом калия. Феррицианид калия Кз[Ее(СК)б] может восстанавливаться-на платиновом электроде, а ферроцианид К4[Ре(СМ)б] окисляться. Если в растворе присутствуют одновременно ионы ферри- и ферроци-внчда, то получается непрерывная катодно-анодная волна (рис. пересекающая ось абсцисс при потенциале, который можно считать равновесным для данной системы в данных условиях (по-тенциал в отсутствие тока). Такие системы, дающие непрерывную тв ^екотоРое представление о состоянии работ в области полярографии на с«ий Ю злектродах и о литературе по этому вопросу дает книга Делимар-] 9/q Скобец Е. М. Полярография на твердых электродах. Киев, Техника,
47
Рис. 14. Полярограммы эквимоляр, ной смеси ферри- и ферроцианида калия (0,001 Л4) на платиновом элек| троде на фонах 1 М нитрата калия (/) и 2 М сериой кислоты (2).
катодно-анодную кривую, относят к так называемым «обратимым» системам. Равновесный потенциал системы фер-ри-/ферроцианид зависит от кислотности раствора, с увеличением которой этот потенциал возрастает, по-видимому, вследствие изменения соотношения окисленной и восстанов-
ленной форм за счет образования различных по прочности «кислых» ионов типа [HnFe(CN)6](4-n)_ и [HnFe(GN)6](S-n)_. Поэтому с увеличением концентрации кислоты полярограмма системы [Fe(CN)6]3~/[Fe(CN)6]4~ и область предельного тока ионов ферри- и ферроцианида смещается в сторону более положительных потенциалов (см. рис. 14, табл. 6).
Большое практическое значение имеют системы, образованные свободным галогеном и его ионами. На рис. 15 приведены вольт-амперные кривые трех систем: С1г/2С1_, Вг2/2Вг" и I2/2I" на фоне 1 М серной кислоты. Если в растворе присутствуют одновремен
но ионы всех трех галогенов, то можно предвидеть, что, например, при потенциале +1,0 В в электродной реакции будет участвовать только тот ион, который дает анодную волну при более отрицательных потенциалах, чем +1,0 В, т. е. только иодид-ион. Ес-
ли же установить потенциал индикаторного электрода равным
Таблица 6. Полярографические характеристики системы ферри-!ферроцианид на платиновом электроде при различной кислотности раствора
Фон Равновесный потенциал, В (НВЭ) Область потенциалов предельного тока ионов, В (НВЭ)
[Fe(CN)6]S~ [Fe(CN)6]<-
1 М KNO3 0,50 0,404-—0,3 0,604-1,3
0,2 М H2S04 0,63 0,504-0,0 0,754-1,5
0,5 Л4 H2S04 0,68 0,604-0,0 0,804-1,5
1,0 М H2SO* 0,75 0,654-0,0 0,854-1,5
2,0 Л4 H2SO4 0,80 0,704-0,0 0,904-1,6
4,0 М H2SO, 0,86 0,754-0,1 1,04-1,6
6,0 Л4 H2SO4 0,95 0,854-0,1 1,14-1,6
48
„„„ 15 Полярограммы систем галоген/ Угалогеиид-ион на платиновом электроде fa фоне 1 М серной кислоты:
2-система CU/2C1-; 3- система ВП/2ВГ-’; 4- система W2I-.
I 1 з в, ТО одновременно с иодид-иотом на электроде будет окисляться и бромид-ион, а при потенциале 4-1,6 В —все три иона. Следует заметить, что анодное окисление галогенид-ионов очень сильно зависит от среды. Если при хлоридном фоне среда остается нейтральной или слабокислой и концентрация хлорид-ионов не превышает 0,2—0,3 М, то хло-рид-ионы не будут окисляться и, титрованию; если же кислотность
следовательно, не могут мешать среды достигает примерно рН=1
и концентрация возрастает, то с
возможным окислением хлорид-
иона уже нельзя не считаться.
Особо следует отметить анодное поведение иодид-иона. Окис-
ление иодид-иона на платиновом электроде протекает в две стадии (см. рис. 15, кривая 4): сначала до иода (первая анодная волна) и далее до йодата (вторая анодная волна). В процессе окисления иодида до йодата принимает участие хемосорбированный кислород (см. гл. II), и поэтому на второй волне вместо площадки предельного тока имеется максимум. Аналогично поведение иодид-иона и на золотом электроде (табл. 7), для которого также характерна хемосорбция кислорода. В случае же графитового электрода, на котором кислород практически не адсорбируется, вторая волна не наблюдается (см. рис. 6, кривая 2). Поэтому если по каким-либо причинам титрование проводят при высоком анодном потенциале (более +1,1 В) и желательно, чтобы окисление иодид-иона шло только до иода, следует воспользоваться графитовым электродом.
На рис. 15 видно также, что свободные галогены восстанавливаются на платиновом электроде. В связи с тем, что растворы хлора и брома мало устойчивы, их обычно не применяют при количественных определениях, но при некоторых реакциях, в результате которых образуются галогены, следует считаться с возможным их восстановлением на электроде. Например, при титровании броматом в конечной точке титрования начинается выделение брома.
Иод способен на платиновом электроде как восстанавливаться, ак и окисляться (табл. 7). Ток окисления иода обычно не исполь-уют в амперометрическом титровании, но это обстоятельство сле-У т учитывать в случае титрования при высоких анодных потен-в-иалах (более +1,1 В), так как процесс окисления иода может
Таблица 7. Сравнительная характеристика полярографического поведения иодида и иода на различных электродах (фон — 1 М серная кислота)
Параметр-< Платиновый электрод Графитовый электрод
иодид иод иодид ИОД
первая анодная волна вторая анодная волна анодная волна катодная волна анодная волна катодная волна
Потенциал начала волны (НВЭ), В Потенциал ДЧг, В Область предельного тока, В Продукт электродной реакции 0,66 0,73 0,85— 1,20 Иод 1,20 Р,35 Макс, при 1,4 Иодат 1,12 1,30 Макс, при 1,4 Иодат 0,80 0,71 0,6—0,0 Иодид 0,66 0,74 0,85—1,50 Иод 0,82 0,73 0,65— 0,0 Иодид
Таблица 8. Некоторые характеристики полярограмм 0,001 М растворов ванадата, бихромата и перманганата на фоне серной кислоты различной концентрации на платиновом электроде
Ванадат Бихромат Перманганат
Концентра- Потенциал, В (НВЭ) Потенциал, В (НВЭ) Потенциал, В (НВЭ)
ция серной начала начала начала
кислоты, и области области полу- области
полуволны диффузионно- полуволны диффузионно- волны диффузи-
го тока го тока тока
0,01 До начала выделения До начала выделения +1,26 + 1,0
0,10 водорода восстановление водорода восстановление +1,26 + 1.0
0,20 не происходит не происходит +1,26 +1,0
Сила тока возрастает
по сравнению с чистым фоном, но волны еще нет
2 Сила тока несколько Небольшой перегиб +1,26 +1,о
возрастает по сравнению с чистым фоном, но вол- при + 0,7
ны еще нет
4 +0,82 +0,4 + 1,26 + 1,о
6 +0,96 +0,6 +1,26 + 1,0
8 +1,06 +0,8 +1,30 +1,о
1С Небольшой перегиб +1,08 +0,8 + 1,43 +1,1
При +0,4 + 1,22 +1,0 + 1,2
12 +0,94 +0,5 +1.40
16 +1,12 +0,7 + 1,34 + 1,1 При дальнейшем увеличении концен-
трации кислоты
раствор перманга-
ната обесцвечива-
ется
20 + 1,26 + 1,1 При дальнейшем уве-
24 + 1,29 + 1,2 личении концентрации
30 + 1,38 + 1,3 кислоты результаты плохо воспроизводимы
Оильнекислая среда Слабокислая среда 1кат
Iнагл
Vv-*-Vwu Fe3t*Fe2t
Y=+0,3B
Объем титранта б
^кат
Объем титранта а
?кат
&
РиС 16. Полярограммы растворов, Гпяержащих ионы ванадия (V) и Sa(II и Ш), и схематическое изображение кривых титрования ванадия (V) раствором соли Мора в различных условиях: , „Осстановление ванадия (V) в силь-7 „Та опеле- 2 — ТО же, в слабокис-нокнслой ср Д и серная кислота); 3 — катодно-анодная кривая смеси раствор poB_Fe]Li£±—--------------
отразиться на ходе амперометрической кривой. Исключить процесс окисления иода при высоких анодных потенциалах можно, воспользовавшись графитовым электродом.
Кислородсодержащие анионы — перманганат, бихромат и ванадат, имеющие большое применение в практике амперометрического титрования, восстанавливаются на электроде необратимо. Это значит, что если в растворе присутствуют одновременно окисленная и восстановленная формы, например Сг2Оа7- и Сгш, то непрерывной катодно-анодной волны, подобной тем, которые были изображены выше (см. рис. 14 и 15), получить нельзя. На катодном поведении кислородсодержащих ионов на твердом электроде особенно сильно сказывается кислотность среды (табл. 8).
На рис. 16 сопоставлены вольт-амперные кривые для системы Feni/Fn и ванадат-иона. Предположим, что титрования ванадия (V) железом (II) проводят в сильно-сРеде при потенциа-ДисМ1^’^ В" В этом слУчае Внач ЗИ°ННЫЙ Т0К МОГУТ Дать ионы и ванадия(V) и железа(II).
d але, еще до титрования, сила тока будет обусловлена присут-4*
t ¥= + О,ВВ
Объем титранта
Iкат
V=+1,2B
1ан
Объем титранта е
Объем титранта В
7кат
1ан
Объем титранта д
51
ствием ионов ванадия (V). По мере титрования количество последних будет убывать, но вместо них будут появляться ионы FeIn, которые тоже могут восстанавливаться. Следовательно, сила тока практически не будет изменяться (рис. 16,а).
Если же при том же потенциале +0,3 В проводить титрование не в оильнокислой, а в слабокислой среде, например в 0,2 н. или 1 н. серной кислоте, то ванадат не сможет восстанавливаться на электроде, поэтому сила тока будет зависеть только от ионов FeIH, которые по мере титрования накапливаются в растворе. Сила тока, сначала практически равная нулю, постепенно повышается, и кривая титрования принимает вид, изображенный на рис. 16,6. Теперь представим себе, что среда снова сильнокислая, но установлен потенциал +0,6 В. В таком случае железо(III) фактически уже не сможет восстанавливаться на электроде (рис. 16, а), тогда как ионы ванадата при этом потенциале в сильнокислой среде дают диффузионный ток. Следовательно, кривая титрования примет вид, изображенный на рис. 16, в. В слабокислой среде при потенциале +0,6 В сила тока во время титрования вообще не изменяется (рис. 16, а), так как при этом потенциале ток железами) отсутствует, а для электрохимического восстановления ванадата нужна более кислая среда.
При потенциалах более положительных, чем +0,6 В, например при +1,2 В, в сильнокислой среде на платиновом электроде возможно и восстановление ванадия(V), и окисление железа(II). Поэтому в данном случае до конечной точки титрования наблюдается катодный ток ванадата, который, однако, сравнительно незначителен, так как потенциал +1,2 В соответствует не области диффузионного тока, а начальной части волны восстановления ванадата (кривая 1, рис. 16, а). После точки эквивалентности, когда в титруемом растворе появятся избыточные ионы железа (II), наблюдается анодный ток их окисления и кривая титрования примет вчд, изображенный на рис. 16,6. Если уменьшить кислотность раствора, то электрохимическое восстановление ванадия (V), как указано выше, совсем не наблюдается. Тогда при титровании при потенциале +'1,2 В сначала нет никакого тока; лишь после конечной точки за счет избыточных ионов Fe11 возникает анодный ток. Кривая титрования будет иметь вид, изображенный на рис. 16, е-Этот тип титрования находит широкое применение при определении различных окислителей солью Мора (см. гл. VI).
Приведенные примеры показывают, что при амперометрическом титровании по методу окисления — восстановления при одной и той же химической реакции в растворе на электроде могут восстанавливаться или окисляться, в зависимости от среды и потенциала индикаторного электрода, не только ионы титрующего и титруемого раствора, но и вещества, образующиеся в результате химической реакции между ними. Из изложенного вытекает, что, меняя кислотность среды и потенциал индикаторного электрода, можно получить самые разнообразные формы кривых титрования.
52
С другой стороны, если кривая титрования не имеет той формы, С орую предполагали получить, устанавливая тот или иной потен-к°аЛ индикаторного электрода, то это означает, что либо среда оказалась неподходящей, либо в растворе присутствовали другие юны так или иначе участвующие в процессе титрования (либо на электроде, либо непосредственно в растворе).
ВОССТАНОВЛЕНИЕ ИОНОВ МЕТАЛЛОВ
На твердых электродах могут быть восстановлены до металла катионы всех электроположительных металлов, а также некоторых электроотрицательных, в частности свинца, обладающего сравнительно невысоким отрицательным потенциалом наряду с высоким (почти как у ртути) перенапряжением для выделения водорода.
На рис. 17 приведены вольт-амперные кривые восстановления ионов меди(II), серебра (I) и золота (III) на платиновом электроде на фоне 1 М азотной кислоты. Такие же полярограммы этих ионов получаются на графитовом и золотом электродах. Из сопоставления кривых рис. 17 вытекает практически важное следствие: если потенциал индикаторного электрода будет установлен около 4-0,1 В, то все три иона — и медь(П), и серебро(1), и золото (III)—смогут восстанавливаться; если же установить потенциал 4-0,4 В, то ионы меди(II) восстанавливаться не смогут, тогда как ионы серебра (I) и золота(III) будут давать ток восстановления. Этот факт позволяет проводить титрования, связанные с восстановлением ионов серебра (или золота), в присутствии меди без какого-либо влияния с ее стороны (см. гл. II). Из рис. 17 следует, что при потенциале +0,8 В будет исключено уже и восстановление иона серебра, а золото при этом потенциале будет восстанавливаться. Следовательно, проводя титрование при 4-0,8 В, можно совершенно исключить влияние ионов серебра, меди и других металлов, менее электроположительных, чем золото (III).
Приведенные на рис. 17 вольт-амперные кривые были получены при постепенном изменении потенциала индикаторного электрода от положительных к отрицательным значениям. Если же эти кривые снимать в обратном направлении, то при потенциалах несколько менее положительных, чем потенциал восстановления ме-Ди(П), серебра(1) или золота (III), будет наблюдаться анодный пик окисления металлической меди (серебра или золота), которая выделилась на электроде во время снятия вольт-амперной кривой.
мели Уп П°ляРогРаммы растворов сульфата стоил ’ нитРата серебра (2) и золотохлори-киспл °Р°ДН0Й кислоты (3) на фоне азотной °ты. Электрод сравнения — НВЭ.
hanr
53
Следует отметить, что ионы некоторых металлов могут окисляться на платиновом электроде: например, свинец(П) до свинца (IV) с образованием окисной пленки РЬО2, ТР до Т120з, Мп11 до МпО2. Ток окисления ионов металлов редко используют в амперометрическом титровании, но с образованием оксидных пленок приходится иногда считаться.
АНОДНОЕ ОКИСЛЕНИЕ ОРГАНИЧЕСКИХ РЕАКТИВОВ
В настоящее время широкое практическое применение в качестве титрующих реагентов при амперометрическом определении тех или иных элементов находят [24, 27, 30, 31] различные органические вещества (см. гл. VI). Ограничимся лишь несколькими примерами, показывающими, какие факторы необходимо учитывать при выборе потенциала индикаторного электрода в случае титрования по току органического титранта.
К органическим веществам, наиболее часто применяемым в амперометрическом титровании, относятся серусодержащие соединения [30, 31], поскольку они дают возможность использовать различные типы химических реакций между определяемым и титрующим веществами. Типичными представителями этих соединений
Рис. 18. Вольт-амперные кривые тиокарбамида на фоне 1 М серной кислоты, снятые в анодном (сплошные линии) и катодном (пунктирные линии) направлении. Материал электрода:
1 — платина; 2 — золото; 3— графит.
Рис. 19. Вольт-амперные кривые тиооксина на фоне 1 М серной кислоты, снятые в анодном (сплошные линии) и в катодном (пунктирные линии) направлении. Материал электрода:
1 — платина; 2 — золото; 3 — графит.
54
м0Гут служить, например, тиокарбамид и тиооксин (8-меркапто-ХИННаИрис- 18 и 19 приведены вольт-амперные кривые тиокарб-
и а и тиооксина на различных твердых электродах на фоне 1 М ^рной кислоты. Анодное окисление этих веществ (равно как и Многих других серусодержащих) начинается, независимо от мате-пиала твердого электрода, практически при потенциалах 0,5— Об В (НВЭ) [32—40]. Однако характер вольт-амперных кривых тиокарбамида сильно зависит от природы материала электрода и от направления снятия полярограммы и резко отличается от характера вольт-амперных кривых тиооксина. При снятии вольт-амперных кривых тиокарбамида на платиновом и золотом электродах в направлении увеличения положительной поляризации электрода (рис. 18, сплошные кривые 1 и 2) наблюдается две волны, причем на второй волне имеется более или менее резко выраженный при потенциале около 1,3—1,4 В максимум, характерный для
тех случаев, когда окисление вещества протекает с участием хемосорбированного кислорода (см. гл. II). Первая волна отражает окисление тиокарбамида до карбамидодисульфида, а вторая-— дальнейшее окисление тиокарбамида до различных кислородсодержащих продуктов, в том числе до сульфат-иона [33, 37, 41]. В случае же снятия полярограммы тиокарбамида на графитовом электроде (см. рис. 18, кривая 3), для которого хемосорбция кислорода не характерна (см. гл. II), наблюдается только одна волна, отражающая окисление тиокарбамида до карбамидоди сульфида [38, 41].
При снятии вольт-амперных кривых тиокарбамида в направлении уменьшения положительной поляризации платинового или золотого электрода полярограммы имеют несколько иной вид: максимум при 1,3—1,4. В исчезает, но зато появляется другой максимум при потенциале 1,0—1,1 В (см. рис. 18, пунктирные кривые / и 2), причем между прямым и обратным ходом поляризационных кривых наблюдается значительная петля гистерезиса. Что касается графитового электрода, то его «обратная» полярограмма практически совпадает с вольт-амперной кривой, полученной в направлении справа налево (см. рис. 18, кривая 3).
Тиооксин, в отличие от тиокарбамида, дает независимо от природы, материала твердого электрода одну анодную волну, ход которой не зависит от направления снятия полярограммы (см. рис. 19). Это связано с тем, что тиооксин окисляется [32, 34, 37, 39, ’Ч только до его дисульфида (8,8'-дихинолилдисульфид).
Вообще следует отметить, что если анодное окисление того или ВДого серусодержащего вещества проходит только до его дисульфида, то вольт-амперометрическое поведение этого вещества прак-
1Чески не зависит от природы твердого электрода и от направ-ения снятия полярограммы и титровать-можно при любом потен-иале, соответствующем площадке анодного тока.
5Ь
ВОССТАНОВЛЕНИЕ И ОКИСЛЕНИЕ
МАЛОРАСТВОРИМЫХ ВЕЩЕСТВ
На индикаторном электроде могут разряжаться не только растворенные в электролите вещества, но и взвеси малорастворимых соединений: галогенидов [40, 42—51]1, сульфидов [45, 50, 51], сульфатов [50, 51], хроматов [45], молибдатов [52, 53], вольфраматов [54, 55] и некоторых других соединений [45, 46, 50, 51]. Ток восстановления или окисления малорастворимых соединений может быть использован для индикации конечной точки титрования. Так, в работе [56] осуществлено амперометрическое определение иодид-иона в хлоридсодержащих растворах нитратом серебра по току восстановления взвеси AgCl, образующейся после завершения реакции иодид-иона с серебром (см. гл. VI, раздел «Иод»),
Возможность разряда осадков на индикаторном электроде необходимо учитывать при осадительных титрованиях, так как образующиеся осадки могут оказывать весьма существенное влияние на ход кривой титрования. Примером может служить титрование
Рис. 20. Полярограммы сульфата ртути(П) (кривая 4), иодида калия (кривая 3) и взвеси иодида ртути (II) (кривая 2) на фоне 1 М серной кислоты (кривая 1). Индикаторный электрод — платина, электрод сравнения — МИЭ.
Рис. 21. Кривые титрования ртути(П) (0,5 мг) 0 01 н. раствором иодида калия по току окисления иодид-иона при потенциалах:
1 — от +0,8 до +1,0 В; 2 Н,2 В; 3 И,4 В; 4—4-1,5 В, Электрод сравнения — МИЭ.
56
99 Кривые титрования ртути (II) РиС" \ о 01 н раствором иодида калия ПО току восстановления ионов ртути при ПОТо™+0 5адо +0.3 В; 2 —+0.2 В; 3-+0.1 В. /эпХо1()с5ра—-НВЭ.
двухвалентной ртути иодидом калия [42]- На рис. 20 сопоставлены вольт-амперные кривые ионов двухвалентной ртути (кривая 4), иодид-иона (кривая 3) и взвеси иодида ртути(Ш) (кривая 2). Предположим, что титрование ртути(II) иодидом калия проводят при потенциалах от +0,8 до +0,1 В, т. е. когда единственным электродным процессом может быть только анодное окисление иодид-ионов. В этом случае кривая титрования имеет четко выраженную форму (рис. 21, кривая /). Если же титрование проводить при более положительных
потенциалах, чем +1,0 В, при которых уже происходит окисление
иодида ртути(II) (см. рис. 20, кривая 2), то ход амперометрической кривой становится несколько иным: сила тока до конечной
точки титрования возрастает по мере накопления в растворе иодида ртути (II), причем тем сильнее, чем более положителен потенциал электрода (см. рис. 21, кривые 2—4). При этом наряду с изменением хода кривых титрования происходит смещение конечной точки титрования в сторону большего расхода титрующего реактива, что связано с повторным взаимодействием ионов ртути (II), освобождающихся при анодном окислении Hgl2, с добавляемыми иодид-ионами.
Более сложный вид кривых наблюдается при титровании ртути (II) иодидом калия по катодному принципу. Исходя из рис. 20, следовало ожидать, что если титрование проводить при потенциалах от -}-0,5 до 4-0,3 В, то кривая титрования должна иметь форму а. Однако в действительности на кривой появляется анодный участок с максимумом (рис. 22, кривая 1). Причина этого явления кроется в том, что при указанных потенциалах на поверхности платинового электрода [до того, как вся ртуть(П) оказывается связанной в осадок] выделяется металлическая ртуть, которая затем в присутствии избытка свободных иодид-ионов электрохимически окисляется по реакции
2Hg+ 21- — 2ё- = Hg3I2 (Ео = —0,04В) и тем самым обусловливает появление анодного тока.
57
Если титрование по катодному процессу проводить при еще менее положительных значениях потенциала индикаторного электрода, чем +0,3 В, то в этом случае, вследствие одновременного восстановления ионов ртути (II) в растворе и в осадке иодида ртути (II) (количество первых уменьшается в процессе титрования, а количество второго, наоборот, увеличивается), кривые титрования имеют настолько «размытый» вид (рис. 22, кривые 2 и 3), что определение конечной точки не представляется возможным.
Методы определения конечной точки
При амперометрическом титровании конечную точку определяют обычно графически: на оси ординат откладывают силу тока (обычно в делениях шкалы гальванометра), по оси абсцисс — расход титранта, а конечную точку определяют как точку пеоесечения двух касательных к обеим ветвям кривой титрования.
Если на кривой титрования имеются «загибы» (рис. 23), причины которых рассмотрены в гл. II, то при определении конечной точки их не следует принимать во внимание. Конечную точку находят только по прямолинейным участкам кривой титрования.
При титровании по методу осаждения форма кривых титрования в значительной степени зависит от растворимости образующегося осадка: чем выше растворимость осадка, тем более размытой получается кривая титрования.
Иногда при таком ходе кривой может возникнуть вопрос, какую часть ее следует учитывать при определении положения конечной точки. Например, на рис. 24 приведена кривая, на которой можно наметить два прямолинейных участка: а—b и с—d. Если за конечную точку принять пересечение горизонтальной части кривой с отрезком а—Ь, то конечная точка будет соответствовать значительно меньшему объему титранта, чем нужно по расчету. Правильным будет второй прием, так как избытку титранта соответствуют точки именно на отрезке с—d, а отрезок а—Ъ соответствует еще процессу образования осадка. Если возникает сомнение в том, какой участок кривой принимать во внимание, то сле-
58
Рис 24. Определение положения конечной точки при титровании веществ образующих недостаточно малорастворимые осадки.
Рис 25. Определение положения конечной точки титрования по метолу Лангера и Стивенсона.
дует оттитровать стандартный раствор соответствующего иона (на соответствующем фоне) и по количеству взятого для титрования вещества определить способ установления конечной точки.
К числу заметно растворимых осадков относятся осадки, с которыми проводили первые амперометрические титрования, а именно осадки сульфата свинца и сульфата бария. В связи с этим первые работы по амперометрическому (полярометрическому} титрованию и, в частности, работа В. Майера [57] содержат математический анализ кривых титрования, приводящий к выводу о том, что кривая титрования по методу осаждения представляет собой гиперболу, а точка эквивалентности — точку пересечения обеих асимптот гиперболы.
Лангер и Стивенсон [58] показали возможность графического нахождения конечной точки даже в тех случаях, когда кривые титрования настолько размыты, что обычный метод касательных уже неприложим. В таких случаях рекомендуется для кривых а и b принять ось абсцисс за одну из асимптот гиперболы и, воспользовавшись геометрической теоремой (отрезки секущей, заключенные между асимптотами и ветвями гиперболы, равны между собой), найти сначала вторую асимптоту, а затем точку ее пересечения с первой асимптотой, т. е. конечную точку титрования. На рис. 25 в качестве примера показан этот способ определения конечной точки для кривой формы «а»: через кривую титрования проводят две секущие (прямые 1 и 2), на них от восходящей ветви кривой откладывают отрезки cd и рт, равные соответственно 0& и eh, и через точки d и т проводят прямую до пересечения с осью абсцисс. Точка пересечения соответствует конечной точке титрования.
Известны другие методы определения конечной точки [59—73]. Нднако использование этих методов при проведении серийных работ затруднительно.
59
Рис. 26. График, поясняющий расчетный метод определения конечной точки по Д. П. Щербову.
Рис. 27. Зависимость силы тока от разбавления раствора при титровании:
/ — экспериментальная кривая: 2 — кривая после пересчета силы тока с учетом разбавления раствора.
Д. П. Щербов [74], стремясь к экономии времени при серийных определениях, предложил применять вместо графического метода нахождения конечной точки простой расчетный метод, заключающийся в следующем: если титрование проводят по восходящей ветви кривой (рис. 26), т. е. кривой формы «б», то конечную точку рассчитывают по формуле:
а(гц — л,,) х — А----------
Щ---П
где х — объем титранта, соответствующий конечной точке титрования; А — объ-ем титранта, соответствующий показанию tt\ гальванометра, а — объем титранта, соответствующий разности показаний гальванометра П\—п; по — начальное показание гальванометра.
Непременным условием применения расчетного метода является следующее: можно использовать только прямолинейные участки кривых титрования.
При графическом нахождении конечной точки в случае значительного увеличения объема раствора во время титрования рекомендуется обычно вводить поправку на разбавление раствора, пересчитывая экспериментальные значения силы тока по уравнению:
Уо + У . гпересч — ,, гэксп
'о
где-Vo — исходный объем раствора; V — добавленный объем титранта.
Если применять в качестве титрующего и титруемого растворы примерно равной концентрации, то влияние разбавления на силу 60
заметно скажется на форме кривой титрования, как это вид-Т°К например, на рис. 27, на котором приведены эксперимевталь-Н°я (Л и «пересчитанная» (2) кривые титрования нитрата серебра н*м калия. Следует отметить, что особенно резко влияние Избавления сказывается при титровании по току титруемого иона, т е когда кривые титрования имеют форму а. Такой именно злу-чай и приведен на рис. 27.
Все затруднения, связанные с влиянием разбавления, легко устраняются, если применять для титрования растворы, коицгнтра-ция которых примерно в 10—20 раз больше концентрации определяемого иона.
ЛИТЕРАТУРА
1 Сонгина О. А. Амперометрическое титрование. М., Химия, 1967.
2 Усвяцов А. А., Исаев Э. И. Амперометрические установки и техника амперометрического титрования. М„ НИИХМ, 1968.
3. Соломатин В. Т., Ржавичев С. П. — Зав. лаб., 1976, т. 42, № 5, с. 529—530.
4. Соломатин В. Т., Балусов В. А., Ржавичев С. П. — В кн.: Методы определения неметаллов и других вредных примесей в промышленных материалах. М., Металлургия, 1977, с. 149—153.
5 Лебедева М. И., Исаева Б. И. — Труды Тамбовского ин-та хим. машиностр., 1969, № 3, с. 113—147.
6 Рора Gr., Cruceru D., Baiulescu Gh., Greff С. — Rev. Chim. (RPR), 1965, v. 16, № 7, p. 321—322.
7. Габович A. M. — Труды Кишиневского с.-х. ии-та, 1968, т. 43, с. 125—131.
8. Gupta S. L., Sharma S. К-—J. Indian Chem. Soc., 1965, v. 42, № 6, p. 381— 383.
9. Gupta S. L., Sharma S. K-, laitly I. N. — Indian J. Chem., 1966, v. 4, № 4, p. 166—167.
10. Myers D. J., Osteryoung J. — Anal. Chem., 1974, v. 46, № 3, p. 356—359.
11. Макарьева С. П„ Беззубик 3. Г., Проскурин M. А.—Зав. лаб., 1947, т 13, № 12, с. 1347—1349.
12. Kolthoff I. М., Sambucetti С. I. — Anal. chim. acta, 1960, v. 22, № 2, p. 253— 259.
13. Чирков С. К.—Зав. лаб., 1956, т. 22, № 8, с. 896—900.
14. Michalski Е., ledrzejewski W. — Roczn. Chem., 1955, v. 29, № 3, p. 862—868.
15. Michalski E., Pawluk V. — Chem. Analit., 1961, v. 6, № 3, p. 480—485.
16. Michalski E. — Chem. Analit., 1958, v. 3, № 3, p. 423—429.
17. Сангина О. А., Усвяцов A. A.-—Зав. лаб., 1964, т. 30, № 11, с. 1419—1450.
18. Усвяцов А. А., Сонгина О. А.—Зав. лаб., 1965, т. 31, № 6, с. 663—666.
19. Усвяцов А. А., Сонгина О. А. — В кн.: Современные методы химического и спектрального анализа материалов. М., Металлургия, 1967, с. 241—245.
1960°KOJMCKIZ^ & С' Клименко Ю. В. Ваиадатометрия. М., Металлургиздат,
21. Gonzalez В., Taylor J. К.—Trans. Elektrochem. Soc., 1948, v. 92, № 2, p. 437— 442.
22. Крюкова T. А., Синякова С. И., Арефьева Т. В. Полярографический анализ.
2ч Госхимиздат, 1959. 772 с.
о. I ейровский Я., Кута Я. Основы полярографии. — Пер. с чешского/Под ред.
24 Р Майрановс.тго М., Мир, 1С65 559 с.
гейшахрит Л. С. Электрохимические методы анализа. Л., нзд-во ЛГУ, 1970, 194 с.
25 Лурье Ю Ю. Справочник по аналитической химии. М., Химия, 1971. 454 с. ’ "Т>авочник химика/Под ред. Никольского Б. П. Т. III. М. — Л., Химия, 1964. 1168 с.
Сонгина О. А. —Зав. лаб., 1965, т. 31, № 10, с. 1163—1172.
61
28. Галлай 3. А., Шеина Н. М.— В кн.: Успехи аналитической химии. М Hav ка, 1974, с. 278—280. Л'
29. Сонгина О. А., Захаров В. А. — В кн.: Успехи аналитической химии. М Наука, 1974, с. 273—277.
30. Сонгина О. А., Бессарабова И. М. — Зав. лаб., 1973, т. 34, № 6, с. 641—655
31. Бессарабова И. М., Сонгина О. А. — Ж. анал. хим., 1974, т. 29, № 5, с. 998— 1002.
32. Бессарабова И. М., Захаров В. А., Сонгина О. А.—В кн.: Химия и химическая технология. Алма-Ата, 1970, вып. 1, с. 24—30.
33. Кузьмина Н. Н., Сонгина О. А.—Изв. вузов. Химия и хим. техиол., 1961 т. 4, № 6, с. £ 28 -935
34. Захаров В. А., Сонгина О. А., Бессарабова И. М.— Изв. АН КазССР Сер. хим., 1968, № 5, с. 28—32.
35? Усатенко Ю. И., Шумская А. И. — Зав. лаб., 1960, т. 26, № 2, с. 148—151.
36. Маслий А. И., Лепенко Г. К, Бек Р. Ю.—Изв. Сиб. отд. АН СССР, 1976, вып. 4, № 9, с. 14—20.
37. Захаров В. А. и др.—Электрохимия, 1971, т. 8, № 8, с. 1215—1217.
38. Захаров В. А. и др. — Электрохимия, 1973, т. 9, № 1, с. 58—59.
39. Супрунович В. И., Усатенко Ю. И.—Труды Днепропетровского хим. ехиол. ин-та. Химия и хим. технол., 1962, вып. 16, ч. I, с. 123—129.
40. Захаров В. А. и др.—Ж- анал. хим., 1969, т. 24, № 9, с. 1401—1403.
41. Захаров В. А. Исследование в области амперометрического титрования с твердыми (платиновый, золотой, графитовый) электродами. Автореф. докт. дисс. Алма-Ата, 1974. 62 с.
42. Сонгина О. А., Захаров В. А. — Зав. лаб. 1962, т. 28, № 8, с. 908—910.
43. Kolthoff 1. М., Stock J. Т. — Anal. Chem., 1955, v. 80, № 5, р. 860—864.
44. Stock I. T., Turner W. R.— Chem. and Ind., 1961, № 42, p. 1710.
45. Micka K. — Chem. listy, 1961, v. 55, № 3, p. 852—890.
46. Сонгина О. А., Даушева M. P. — Электрохимия, 1965, т. 1, № 12, с. 1464— Ь68.
47. Даушева М. Р., Сонгина О. А., Жданов С. И. — Электрохимия, 1970, т. 6,.. № 2, с. 285—288.
48 Даушева М. Р., Сонгина О. А. — Электрохимия, 1972, т. 8, № 1, с. 97—100.
49. Захаров В. А., Сонгина О. А.—Изв. АН КазССР. Сер. хим., 1962, вып. 2 (20), с. 55—58.
50. Гладышев В. П., Киреева Е. П. — Электрохимия, 1972, т. 8, № 6, с. 838; 1972, т. 8, № 7, с. 977—981.
51. Даушева М. Р., Сонгина О. А.—Усп. хим., 1973, т. 42, № 2, с 323—342.
52. Мамбетказиев Е. А., Сонгина О. А., Захаров В. А. — В ки.: Химия и химическая технология. Алма-Ата, 1967, вып. 6, с. 130—134.
53. Сонгина О. А., Захаров В. А., Мамбетказиев Е. А.—Ж- анал. хим., 1968, т. 23, № 3, с. 453—455.
54. Мамбетказиев Е. А., Захаров В. А. — В кн.: Химия и химическая технология. Алма-Ата, вып. 7—8, с. 62—65.
55. Сонгина О. А., Захаров В. А., Мамбетказиев Е. А. — Изв. АН КазССР. Сер.
* хим., 1968, № 6, с. 31—35.
56. Вариков В. Г., Сонгина О. А., Клименко Е. В.—Зав. лаб., 1969, т. 35, №11, с. 1312—1313.
57. Mayer VI. — Z. Elektrochem., 1936, Bd. 42, № 1, S. 121—125.
58. Langer A., Stevenson D. — Ind. Eng. Chem., Anal. Ed., 1942, v. 14, № 2, p. 770—775.
59. Хадеев В. А. Некоторые вопросы теории амперометрического метода титрования, основанного иа реакциях осаждения. Ереван, изд-во Ереванск ун-та, 1957. 180 с.
60. Хадеев В. А.—Узб. хим. ж., 1959, № 2, с. 27—32. |
61. Adamek Р., Dolezal I., Zyka I. — Coll. Czech. Chem. Comm., 1963, v. 28, № 8. p. 2131—2137.
62. Goldman I. A., Meites L. — Anal. chim. acta, 1964, v. 30, № 1, p. 280—284.
63. Liteanu C., Gormos D. — Acta Chim. Hungar., 1961, v. 27, № 1, p. 9—15.
62
Lett., 1976, № 4—5, Ташкентского ун-та, 5; 1969, т. 24, № 3,
€4 Goi^man J. Л.— J. Electroanal. Chem. and Interfac. Electrochem., 1968, v. 19, 65 Mer^oj^A^Lukkari O., Lukkari tf. —Finn. Chem.
66 Xfldee*8#- А-Узб. хим. ж., 1969, № 3, с. 3; Труды 1Q72 вып. 49, с. 67.
А7 Марьянов Б. М.— Ж- анал. хим., 1967, т. 22, № 1, с.
67. Л1аРьД332. 1972> т 27> № п, с. 2099-2106.
68 Марьянов Б. М., Богданов В. С.—Ж. анал. хим., 1975, т. 30, № 3, с. 459—
ко Сидоренко В. И., Гордиенко В. И.— Ж- анал. хим., 1969, т. 24, Ns 3, с. 315; Изв вузов. Химия и хим. технол., 1969, т. 12, Ns 9, с. 1189—1193.
70 Гордиенко В. И., Коваленко П. И., Иванова 3. И. — Изв. вузов. Химия и хим технол., 1965, т. 8, Ns 4, с. 549—554.
71 Гордиенко В И., Сидоренко В. И. — Изв. вузов. Химия и хим. технол., 1969, т 12, Ns 3, с. 250—254.
79 Гордиенко В. И., Михайлюк Ю. И.—Ж. анал. хим., 1970, т. 25, Ns 12, с 2267—2272; 1972, т. 27, № 6, с. 1069—1073; 1974, т. 29, Ns 2, с. 210—213.
73 Гордиенко В. И., Худякова Л. П., Сидоренко В. И,— Ж. анал. хим. 1974, ‘ т. 29, Ns 8, с. 1467—1473.
74 . Щербов Д. П.—Зав. лаб., 1952, т. 18, Ns 11, с. 1310—1313.
ГЛАВА IV
Амперометрическое титрование с двумя индикаторными электродами
История возникновения электрохимического метода анализа, описываемого в настоящей главе, несколько необычна. Метод был, по существу, два раза открыт и оба раза почти забыт. Первый раз его описал Саломон [1] в 1897—1898 гг. Саломон погрузил два посеребренных серебряных электрода в раствор нитрата калия, содержавший немного нитрата серебра, и установил, что сила тока, возникающего в цепи, зависит от приложенного напряжения и от концентрации нитрата серебра; в отсутствие нитрата серебра при небольшом напряжении ток через цепь не проходил. Саломон предположил, что сделанное им наблюдение можно использовать для количественного определения серебра и других металлов, титруя раствор соответствующим реактивом до прекращения тока. Он подчеркивал перспективы этого метода, который, по его мнению. должен обладать большой точностью, и рекомендовал заняться подробнее его разработкой. Однако лишь в 1905 г. появилась работа Нернста и Мерриама [2], которые провели кислотно-основное титрование, пользуясь двумя палладиевыми игольчатыми электродами, погруженными в раствор нитрата калия, содержащий небольшое количество соляной кислоты, и наблюдая за изменением силы тока при титровании этого раствора щелочью при определенном наложенном напряжении.
Затем, уже в 1926 г., Фоулк и Боуден [3] случайно обнаружили, что если в раствор иода погрузить два платиновых электрода и наложить на них очень небольшое напряжение (10—15 мВ), то в цепи возникает ток, уменьшающийся по мере титрования раствора иода тиосульфатом и полностью исчезающий в конечной точке. В связи с последним Фоулк и Боуден назвали «свой» метод (как видно, он вполне аналогичен методу, предложенному Саломоном) методом «dead-stop and point», т. е. методом «мертвой конечной точки».
Однако и после опубликования работы Фоулка и Боудена этот метод практически не находил применения, по-видимому, вследствие того, что сущность его не получила надлежащего толкования, и развитие метода могло идти только эмпирическим путем. Сами авторы метода «dead-stop» объясняли резкое изменение силы тока в конечной точке поляризацией электродов в результате адсорбции
64
nvKTOB электролиза: водорода на катоде и кислорода на аноде. т₽°ько в 1942 г. Бетгер и Форхе [4] совершенно справедливо повили под сомнение объяснение Фоулка и Боудена, считая, что Сого незначительного напряжения, при котором проводят титрова-Т ге недостаточно для электродного выделения водорода и кисло-н’ ’ Однако этот взгляд, несмотря на его очевидную правильность не сразу нашел признание.
Прежде чем перейти к описанию этого метода необходимо остановиться на терминологии. Кроме названия «мертвая конечная точка» в нашей переводной литературе можно часто встретить самые разнообразные выражения — «метод заторможенной конечной точки», «метод титрования до полной остановки», «метод резкой конечной точки», равно как и часто встречающийся не только в иностранных, но и в отечественных статьях термин «биамперометрическое титрование». Все эти названия носят описательный характер, не отражая сущности процесса. Гузман и РапканпО [5] называют его «деполярометрическим», что также нельзя признать удачным. Вполне правильное название было предложено Кольтгофом [6]: «амперометрическое титрование с двумя индикаторными электродами». Этого названия придерживаются и авторы настоящей монографии, считая его верным по существу самого процесса, лежащего в основе метода*.
Принцип метода
Если погрузить в один и тот же раствор одновременно два электрода одинакового размера и наложить на них постоянное напряжение, то, в отличие от обычного амперометрического титрования с одним индикаторным электродом, поляризоваться будут оба электрода, и при соответственно подобранных реагентах во время титрования будет изменяться ток, являющийся функцией наложенного напряжения и зависящий также от концентрации ионов, участвующих в титровании. Ход процесса титрования лучше всего можно проследить, пользуясь вольт-амперными кривыми (рис. 28). Например, если на электроды, погруженные в подкисленный раствор соли железа (II), не содержащей Fe111, наложить небольшое постоянное напряжение — около 0,05 В (рис. 28, а, пунктирные линии), то ток в цепи не возникает, так как для протекания электролиза в растворе сульфата железа (II) нужно приложить значительно большее напряжение (напряжение разложения в этом случае составляет примерно 1,0 В). Если же добавить какои-либо окислитель, например бихромат, то эквивалентное ему количество Ее2+-ионов будет окислено до Ре3+-ионов. Система е+/Fe2+ является, как известно, обратимой системой — это зна-
а ® 1 -Давно изданной ШРАК терминологии по электрохимическим методам менять^нР511110 УказЬ1вается’ что термин «биамперометрическое титрование» при-5-1694 ' «
Рис. 28. Схематические полярограммы для различной степени оттитрованное™ железа (II) (а, б, в, г, д) окислителем и кривая титрования (е) с двумя индикаторными электродами.
чит, что для осуществления этой электродной реакции достаточно наложить на электроды минимальное напряжение, поскольку потенциалы катодного восстановления Fe3+ и анодного окисления Fe2+ практически одинаковы. Следовательно, при напряжении порядка 0,05 В электродный процесс сможет начаться — в цепи появится ток (рис. 28, б); на катоде начнется восстановление Fe3+, на аноде — окисление Fe2+. Измерительный прибор зарегистрирует соответствующую силу тока. По мере титрования ток будет возрастать и достигнет максимума при оттитровывании 50% Fe2+, т. е. когда концентрации ионов железа обеих степеней окисления будут равны (рис. 28, в). По мере дальнейшего титрования сила тока начнет убывать, так как концентрация Fe2+ будет все время уменьшаться (рис. 28, г), и в конечной точке электролиз прекратится вследствие исчезновения этого иона, обеспечивавшего анодный процесс при данном напряжении. Система возвращается, по существу, в исходное положение, с той разницей, что вместо ионов Fe2+ в растворе находятся сейчас ионы Fe3+ Таким обра-
М
достигается тот момент, который Фоулк и Боуден называли 3°“к^ой точкой» (рис. 28, д). Кривая такого титрования имеет «рКеркьп <.-о₽6> (ркс. 28, е).
‘ rpj3 STOro примера видно, что явление торможения в конечной очке нельзя объяснить поляризационным эффектом, основанным на адсорбции газа на поверхности электрода, как полагали Фоулк и Боуден. Причина появления или исчезновения тока в конечной точке заключается в наличии или отсутствии соответствующих «пар» ионов, способных обеспечивать катодный и анодный процессы при данном напряжении. Чем больше «обратимость» системы, обеспечивающей процесс титрования, тем меньшее напряжение' требуется налагать на электроды. Наоборот, при работе с необратимыми системами налагаемое напряжение должно быть соответственно больше. Практически титрование с двумя электродами возможно в любом случае, если приложенное напряжение превышает разность .катодного и анодного потенциалов, устанавливающихся в условиях титрования.
Так же, как и при титровании с одним индикаторным электродом, необходимо следить за тем, чтобы сопротивление цепи было минимальным (для обеспечения резкости конечной точки).
Таким образом, основными условиями амперометрического титрования с двумя электродами являются следующие:
V = const; <ран и <pKaTTtconst;
i=f(C, V); R— очень мало
Характер зависимости i, т. е. личины, от С и V будет подробно
Во избежание недоразумений, возникающих при описании метода амперометрического титрования с двумя индикаторными электродами, рассмотрим другой метод, с которым его часто путают. Это метод потенциометрического титрования под током. Принцип его заключается в следующем: в
измеряемой при титровании ве-рассмотрен ниже.
I
Рис. 29. Схематическое изображение изменения силы тока и потенциала индикаторного электрода согласно полярограммам в процессе титрования различными электрохимическими методами: !!аз₽ез а~^ — амперометрическое титрование a'—h™ индикат°Рным электродом; ра-рез мя и амперометрическое титрование с дву-. d~ ™дикгорными электродами; разрез с— ком- °™нДиометрическое титрование под то-пииё^.,„„Ь ° ~ Ф — классическое потенциометрическое титрование.
5*
67
Таблица 9. Условия выполнения различных методов электрохимического анализа
Метод Измеряемая величина Постоянные и переменные величины и функциональная зависимость
Ток 1 Напряжение Потенциалы Сопротивление R Графическое изображение соответствующих электрохимических процессов (см. рис. 29)
катода <ркат анода <раи
Полярография Ток id=f(C,E) С = const Переменное Переменный Постоянный По возможности малое Полярограммы, отражающие зависимость силы тока от концентрации
Амперометрическое титрование с одним индикаторным электродом id = f(C) Постоянное Постоянный То же То же Изменение предельного тока в процессе тит- рования по разрезу ab
Амперометрическое титрование с двумя индикаторными электродами X- i — V) » Переменный Переменный » Изменение тока и потенциала одного индикаторного электрода в процессе титрования по разрезу а'Ь
Потенциометрическое титрование (классическое) в отсутствие тока Потенциал i = 0 Нет Фкат “ f(O Постоянный Постоянный фан = ДО Очень большое Изменению потенциала индикаторного электрода в процессе титрования отвечают точки пересечения полярограмм с осью потенциалов
Потенциометрическое титрование под током с одним поляризованным электродом То же i == const, но очень мал (несколько микроам-пер)' Постоянное Фкат = Л Постоянный Постоянный Фаи = ДО i) » Изменению потенциала одного индикаторного электрода в процессе титрования отвечают точки пересечения полярограмм с линией cd
Потенцио метр ическое титрование под током с двумя поляризованными электродами Дф То же Дф = фан—Фкат = ДЩ )) » Изменению потенциала индикаторного электрода в процессе титрования отвечают точки пересечения полярограмм с линией cd*
* Изменение потенциала второго индикаторного электрода на рис 29 не показано
J
тоУемый раствор погружают два электрода и пропускают через ТИ д небольшой (несколько микроампер) ток; при этом в цепь це0дЯт очень большое сопротивление. Поэтому ток в цепи будет В ал- включая в цепь потенциометр, можно измерять ДЕ, т. е. разность потенциалов катода и анода. Следовательно, этот метод — потенциометрический, в противоположность методу «dead-stop», и котором измеряемой величиной является сила тока, достигающая больших значений вследствие отсутствия сопротивления в цепи.
Наглядное представление о взаимосвязи полярографического, амперометрического и потенциометрического методов дает схема, приведенная на рис. 29, на которой изображены кривые в координатах сила тока — потенциал, т. е. поляризационные или вольт-амперные кривые. Эти кривые лежат в основе следующих электрохимических методов анализа: полярографии; амперометрического титрования — с одним и с двумя индикаторными электродами; потенциометрического титрования—обычного в отсутствие тока и под током, с одним и с двумя поляризованными электродами.
Для выполнения определения по тому или иному методу необходимо соблюдать условия, приведенные в табл. 9.
Выбор напряжения для титрования
Для выбора налагаемого на электроды напряжения следует исходить из вольт-амперных кривых веществ, участвующих в титровании.
Поясним это на примере титрования ферроцианида перманганатом на фоне 1 н. серной кислоты. Ферроцианид является восстановителем по отношению к перманганату, причем система феррицианид— ферроцианид относится к числу обратимых, а перманганат восстанавливается необратимо. На рис. 30 приведены полярограммы ферроцианида, феррицианида, перманганата и фона (серной кислоты). Для того чтобы начался электролиз раствора серной кислоты, т. е. чтобы происходил разряд Н+-ионов на катоде и выделение кислорода на аноде, необходимо, как известно, наложить напряжение около 1,7 В. Это хорошо видно и на кривой 1 рис. 30. Если же в растворе будут находиться ферроцианид-ионы, способные окисляться на платиновом аноде (кривая 2), то анодный процесс начнется при значительно менее положительном потенциале, чем выделение кислорода, — разность потенциалов между анодным и катодным процессом уменьшится до 0,4 В (катодный процесс — по-прежнему восстановление ионов водорода, так как других ионов, способных восстанавливаться на катоде, в растворе нет). Если же наложить еще меньшее напряжение, то ток в цепи будет отсутствовать до тех пор, пока не начнется титрование окислителем. Как только будут добавлены первые капли перманганата, в растворе появятся ионы феррицианида, спссоб-
69
Рис. 30. Полярограммы компонентов, присутствующих при титровании ферроцианида перманганатом:
1— I и. серная кислота (фон); 2 — 0,001 М раствор ферроцианида калия; 3— 0,001 М раствор феррицианида калия; 4— 0,001 и раствор перманганата калия. Электрод сравнения — МИЭ.
ные восстанавливаться на катоде (кривая 3) и образующие с ферроцианидом хорошо обратимую систему (пунктирная линия между кривыми 2 и 3). Поэтому даже при наложенном напряжении около 5—10 мВ в цепи появится ток за счет работы этой обратимой пары — анодное окисление ферроцианида и катодное восстановление феррицианида. Кривая титрования будет идти так, как это показано на рис. 28-получится характерный горб, максимум которого приходится на 50% оттитрованное™ ферроцианида.
На рис. 31 приведена экспериментальная кривая, полученная при титровании ферроцианида перманганатом при напряжении 0,04 В. Высота горба зависит от концентрации титруемого иона, но для определения конечной точки важен только непосредственно примыкающий к ней участок кривой. Поэтому во многих работах, посвященных описываемому методу, приводятся только эти «конечные» участки кривой титрования.
Ход кривой после конечной точки будет также зависеть от наложенного напряжения. Если оно составляет 0,04 В, то, как это видно на рис. 31, ток в цепи будет практически отсутствовать, так как в растворе уже нет иона ферроцианида, обеспечивавшего анодный процесс. Конечный участок кривой титрования достаточно резок, поэтому отсутствие тока после конечной точки не мешает ее определению. Ток после конечной точки появится только в том случае, если на электроды будет наложено значительно большее напряжение, обеспечивающее образование новой «пары»: восстановление избытка перманганата на катоде (кривая 4, рис. 30) и выделение кислорода (кривая 1, рис. 30) на аноде. Как видно из сопоставления соответствующих вольт-амперных кривых, для этого требуется наложить напряжение около 0,5 В.
Предположим теперь, что все титрование проводят при напряжении 0,5 В. В этом случае с самого начала уже появится ток, так как такое напряжение достаточно для того, чтобы начался электролиз: на аноде пойдет окисление ферроцианида, на като-
70
— восстановление Н+ (кривые 2 и 1, рис. 30). На рис. 32 при-Де ены конечные участки кривых титрования ферроцианида пер--веДганатом, полученных при различных значениях наложенного ма^сяжения. Как видно, напряжение не влияет на положение ко-^чной точки, но с увеличением его конечный участок кривой делается прямолинейным на большем протяжении. На практике, од-ако не всегда целесообразно увеличивать напряжение, стремясь к получению таких резко выраженных участков, так как при большом напряжении в процесс электролиза могут быть вовлечены другие компоненты раствора, а это может привести к искажению фор-
мы кривой титрования.
В рассмотренном примере участвовали одна обратимая и одна необратимая система. Титрование возможно и в том случае, если обе системы обратимы. Примером может служить титрование ферроцианида раствором иода на фоне нитрата калия. Равновесный потенциал системы 12/21~ составляет на этом фоне +0,58 В, а системы [Fe(CN)6]3-/[Fe(CN)6]4- равен +0,45 В. Следовательно, иод на фоне нитрата калия является окислителем по отношению к ферроцианиду (в кислых растворах, наоборот,
феррицианид окисляет иодид, так как реальный потенциал системы [Fe(CN)6]3~/Fe(CN)6]4^ смещается в кислых растворах в сторону положительных значений до +0,76 В). Если титрование проводят раствором иода, а в титруемом растворе находится ферроцианид, кривая титрования при небольшом напряжении (5— 10 мВ) имеет до точки эквивалентности такой же ход, как в случае титрования ферроцианида перманганатом. Но после конечной точки ток резко возрастает, так как пара 12/21— идеально обрати-
ма и минимального напряжения достаточно для обеспечения ка-
тодного и анодного процессов — восстановления иода до иодида и окисления иодида до иода (рис. 33, кривая 2). Если наложенное напряжение заметно превышает минимально необходимое, например если оно составляет 0,2 В, то ход кривой до точки эквивалентности меняется за счет участия и второй обратимой системы 12/21~ (рис. 33, кривая 1)\ подъем тока обусловлен в данном случае возрастанием концентрации феррицианида в процессе титрования, уменьшения же тока после 50% оттитрованности ферроцианида не происходит, так как взамен ионов ферроцианида, убывающих при титровании, в растворе появляются иодид-ионы, концентрация которых возрастает во время титрования (иодид-ионы вво-
састпл^’ кривая титрования ферроцианида
нии 004°В пеРманганата калия при напряже-
71
1
2 J МЛ
Рис. 32. Кривые титрования ферроцианида раствором перманганата калия вблизи точки эквивалентности при различных напряжениях на фойе 1 н. серной кислоты.
дятся также с раствором иода). После конечной точки, при появлении избыточного иода, ток возрастает резко, как и в случае кривой 2, так как начинает работать пара 1г/21_.
Если же необходимо осуществить такое титрование, при котором не образуется ни одной обратимой системы, то можно добавить в раствор то или иное вещество в качестве своеобразного индикатора. Это вещество должно образовывать обратимую систему с одним из участвующих в титровании веществ, например титрование цинка ферроцианидом калия по методу осаждения можно либо вести при очень большом напряжении — около 1 В, либо добавить в раствор соли цинка несколько капель раствора феррицианида и титровать при 0,04 В. В первом случае ток после конечной точки появится за счет пары ферроцианид (анодный процесс)—выделенный водород (катодный процесс), во втором — вследствие возникновения хорошо обратимой пары [Fe(CN6]3~/ f(Fe(CN)6]4~. Второй способ предпочтительнее,
гак как при малом напряжении, как уже говори-
лось выше, практически устраняется возможность протекания других электродных реакций.
В табл. 10 приведено несколько примеров, показывающих зависимость электродных процессов и формы кривых титрования от наложенного напряжения [7]. Как видно из этих примеров, определение конечной точки становится невозможным, если электро-
химические реакции до и после точки эквивалентности одни и те же^
В качестве возможного катодного процесса в этой таблице в некоторых случаях принят процесс восстановления ионов Н+; следует, однако, иметь в виду, что при соответствующих условиях (состав фона, температура) роль катодного агента может играть также
Рис. 33. Кривые титрования ферроцианида раствором иода при разных напряжениях:
1 — 0,05 В; 2 — 0.3 В.
72
ястворенный кислород. Замена одного процесса другим не отра-Р ся на форме кривой титрования, но приведет к тому, что элек-ЗИолиз будет протекать при меньшем напряжении, чем в случае восстановления водорода.
Теоретические основы метода
Теорией этого метода начали интересоваться главным образом в конце 40-х и в 50-х годах. Многие исследователи занимались выяснением влияния различных факторов на ход кривых титрования и предлагали математическую трактовку кривых [8—12].
Для практических целей важно уравнение, приводимое в книге Делахея [13]. Делахей, опираясь на работы других исследователей, приводит следующее уравнение для силы тока, протекающего в цепи при титровании обратимой системы необратимой:
2 \ dy)i=Q
где Uc — наложенное напряжение.
После ряда подстановок соответствующих величин и некоторых преобразований это выражение принимает вид:
nF i=-^P(l-WCUc
где Р— коэффициент пропорциональности, зависящий от природы вещества, обусловливающего электродную реакцию (предполагается, что Р анодного процесса равно Р катодного); X —степень оттитрованности; С — концентрация.
Из этого уравнения следует, что ток проходит через максимум, когда Х=0,5. Отношение i к iMaKC в каждой точке титрования можно рассчитать по уравнению:
i
= 2(1-*) ‘-макс
Кривая титрования, построенная по этому уравнению*, полностью совпадает с экспериментальной кривой на рис. 31.
При выводе этого уравнения принимали, что оба электрода имеют одинаковый размер и что, следовательно, плотность тока на обоих электродах одинакова. При разных размерах электродов ход кривой может.существенно измениться, притом тем больше, чем больше разность между поверхностями обоих электродов [И]. Уменьшение размера, одного из электродов приводит к смещению максимума и к уменьшению силы тока. Такой ход кривых объясняется следующим. В начале титрования мала концентра-ЦИя ионов феррицианида^ образующихся из ферроцианида, при ю * Когда Т ЙД расчет ведут из соотношения iMaKc/i; когда Ъ > 0,5, то берут ОШенИе с/^махс-
73
Таблица 10 Зависимость электродных процессов и формы кривых титрования от
Вещества, участвующие в титровании Фон Наложенное напряжение, В Возможные ***
До конечной
процессы (катодный/анодный)
Ферроцианид- Серная кнс- 0,02—0,04 [FefCN)eP-/[Fe(CN)er-
перманганат лота
0,5 [Fe(CN)e]3-, H+/[Fe(CN)ep-
>1.6 [Fe(CN)el3-. H+/H2O, [Fe(CN)ep-
Ферроцианид- Нитрат ка- 0,02—0,04 [Fte(CN)e]3-/[Fe(CN)er-
нод ЛИЯ
0,3 [Fe(CN)e]3-/[Fe(CN)e]4-, Г
>1,6 [Fe(CN)e]3-, H2O/H2O, [Fe(CN)e]4-
Цинк-ферро- Серная кис- 0,02—0,04 Электролиз
цианид в при- лота-(-сульфат
сутствии ферри- аммония
цианида >1.2
Цн нк-ферро- То же <0,4 Электролиз
цианид вез фер-
рицианида
>0,4 Электролиз
* Под «степенью обратимости», выраженной в вольтах.
понимается протяженность
титровании его перманганатом. Ток в цепи зависит, как известно, от концентрации вещества, присутствующего в меньшем количе-
стве, следовательно, именно ионы феррицианида, т. е. катодный процесс, лимитируют ток на протяжении первой половины титрования. При этом плотность тока на обоих электродах одинакова, если они имеют одинаковый размер. Во второй половине титрования процесс лимитируется уже анодным процессом — ионами ферроцианида. Известно, что плотность тока зависит от размера электрода, следовательно, если катод будет маленьким, а анод большим, то плотность тока на них будет неодинакова, поэтому максимум, отвечающий середине титрования при электродах разной поверхности, сдвинется в рассматриваемом примере в сто-
74
напряжения
—"^ктродные процессы . Форма кривой титрования
точки после конечной точки
степень обратимости*, В процессы (катодный/анодный) степень обратимости•, В
0 (обратима) Электролиз не щ, [ет /
0,4 MnOJ7H2O 0,4
1,6 [Fe(CN)e]3-, Н+/Н2О 1,6 Конечную точку
определить нельзя
^/0 (обрати- VI" ~0 (обратима)
ма)
0,2 V [Fe(CN)e]3-/r 0,2 /
1,6 [Fe(CN)e]3~, I2, H2O/H2O, Г 1,6 Конечную точку
определить нельзя
яе идет [Fe(CN)e]3-/[Fe(CN)ep- ~0 (обратима)
1,2 [Fe(CN)e]3-/[Fe(CN)ep- 1.2 То же
не идет Электролиз не идет Конечную точку
определить нельзя
не идет H+/[Fe(CN)eP- 0,4
участка вольт-амперной кривой, совпадающего с осью абсцисс.
рону анода. Если же, наоборот, катод будет большим, а анод маленьким, то более резкое возрастание тока будет происходить в начале титрования, так как ионы феррицианида имеют возможность восстанавливаться на большом катоде с большой скоростью. Вторая же половина титрования пройдет при меньшем токе, так как малый размер анода будет ограничивать скорость окисления ферроцианида — максимум сместится в сторону катодного процесса.
Обычно при амперометрическом титровании с двумя индикаторными электродами пользуются неподвижными электродами, перемешивая раствор мешалкой. Но можно сделать оба электрода вращающимися. Если оба электрода имеют одинаковый раз
75
мер и вращаются с одинаковой скоростью, то кривая останется симметричной, но ток возрастает. Это совершенно понятно, так как вращение электрода, как это показано в гл. II и III, приводит к уменьшению толщины диффузионного слоя и, следовательно, к возрастанию тока. Если же придать вращение только одному из электродов, то ход кривой измелится так же, как при изменении размера одного из электродов — максимум сместится в сторону процесса, протекающего на вращающемся электроде. Влияние вращения электродов на ход кривой титрования подробно изучено А. И. Филенко [15—17], показавшим хорошее совпадение экспериментальных кривых с расчетными, построенными по уравнению Делахея, видоизмененному с учетом неравномерности подачи электроактивного вещества к электродам.
Некоторое 'изменение положения максимума и «сглаживание» правой ветви кривой при титровании Fe11 раствором бихромата объясняется различием в коэффициентах переноса обоих ионов [18].
Однако, какова бы ни была причина смещения максимума, это смещение само по себе не влияет на положение конечной точки титрования, но может сказаться на резкости конечного участка кривой титрования. Поэтому при амперометрическом титровании с двумя электродами желательно применять электроды одинакового размера или применять электрод большего размера для того процесса, которым определяется конечная точка титрования, или придавать вращение соответствующему электроду.
Асимметрия кривой титрования может быть вызвана и наложением большого напряжения, изменяющего характер электродных процессов. Рассмотрим случай, возникающий при титровании ферроцианида перманганатом с двумя электродами одинакового размера при напряжении 0,6—0,7 В. В этом случае появляется большой начальный ток, обусловленный анодным окислением ферроцианида, присутствующего в растворе; катодный процесс обеспечивается выделением водорода (см. рис. 30). Во время титрования в растворе появляются ионы феррицианида, которые восстанавливаются на катоде одновременно с ионами водорода. В результате ток возрастает и проходит через максимум (рис. 34, кривая 1), но максимум оказывается смещенным влево. Иначе говоря, в данном случаё увеличение концентрации феррицианид-ионов вызывает такое же смещение максимума, как и увеличение размера катода по сравнению с анодом.
Если же при напряжении 0,6 В (в этом же случае — при титровании ферроцианида перманганатом) работать с электродами разного размера, то ход кривых приобретает совершенно другой характер. Так, если при большом анбде применить катод меньшего размера, то начальный ток будет значительно меньше, а максимум резко смещается вправо (см. рис. 34, кривая 2), причем ход анодного участка кривой полностью совпадает с кривой 1, чего и следовало ожидать, поскольку размер анода в обоих слу-
76
34 Кривые титрования ферроцианида распопом перманганата калия при различном соотношении размеров электродов, напряжение 0,60 В: “ „ба электрода — пластинки размером 1 см»; 2 — j — ovo катод _ Проволока длиной 10 мм; 3 — ка-®од^- 1 см3, анод — проволока длиной 10 мм.
чаях одинаков. Однако на всем протяжении титрования сила тока не достигает большого значения, так как она лимитируется малым размером катода. При большом катоде и малом аноде, наоборот, сила тока будет лимитироваться анодным током, а весь ход кривой (см. рис. 34, кривая 3) будет аналогичен кривой обычного амперометрического титрования с одним электродом, так как второй электрод представляет собой в данном случае практически неполяризую-щийся водородный электрод.
Существует [19] еще одна причина асимметрии кривых титрования — необратимость титруемой системы. Расчетным путем доказано, что если «доля по-
тенциала», определяющая процесс анодного окисления и катодного восстановления, неодинакова, то максимум должен смещаться в ту или другую сторону. Это положение иллюстрирует разобранный выше пример титрования при 0,6 В с электродами одинакового размера, поскольку при этом кроме обратимой системы феррицианид— ферроцианид на электродах работает и необратимая пара ферроцианид — ион водорода, и «доля потенциала», приходящаяся на катодный и анодный процессы, не может быть абсолютно одинаковой.
Важной особенностью амперометрического титрования с двумя индикаторными электродами является тот факт, что при работе с обратимыми системами можно увеличивать размеры электродов, не опасаясь потерь определяемого вещества в результате электролиза. Дело в том, что если оба электрода находятся в од- [ом растворе и если на одном из них идет катодный процесс, то на другом пойдет соответствующий анодный процесс, например при электролизе систем [Fe(CN)6]3-/[Fe(CN)6]4~ или 12/21—, которые взаимно компенсируются, вследствие чего необратимая потеря вещества в результате электролиза исключается. Об этом Упоминали Кольтгоф [12] и Киз [8]; впоследствии этот факт был Установлен экспериментально [7]. Так как увеличение размера электрода приводит к более резко выраженной конечной точке (вследствие увеличения силы тока), таким приемом пользуются а практике при работе с разбавленными растворами. Однако не
77
следует забывать, что этот прием допустим лишь при наличии обратимой системы в титруемом растворе и при малом наложенном напряжении, обеспечивающем только работу этой системы. Если же наложить большое напряжение, при котором в электродную реакцию будут вовлечены ионы Н+ или ОН~, то, как было показано выше, кривая титрования становится асимметричной—-«равновесие» между катодным и анодным процессами нарушится, и потери вещества в результате электролиза окажутся возможными.
Аппаратура и область применения метода
Метод амперометрического титрования^ с двумя индикаторными электродами заслуживает большого внимания и широкого распространения. Он очень прост по аппаратурному оформлению, достаточно точен и чувствителен. На рис. 35 приведена схема установки, не требующая специальных пояснений (бюретка не показана во избежание загромождения рисунка). Характеристика всех приборов такая же, как при титровании с одним электродом (гл. III). Обычно пользуются неподвижными электродами, перемешивая раствор пропеллерной мешалкой, как показано на рисунке, или магнитной мешалкой. Для варианта с одним или двумя вращающимися электродами применяют те же устройства, что и для вращения электрода при обычном титровании. В выпуске «ГосИНТИ» [20] описана схема установки с двумя электродами, один из которых, с изогнутым почти под прямым углом концом, вращается и перемешивает раствор. Предложена также полностью автоматизированная установка, описанная, к сожалению, в малораспространенном и труднодоступном итальянском издании [21].
Электроды делают обычно из платиновой проволоки, подобно описанным в гл. III, или из листовой платины размером около 1 см2. Удобны в работе и графитовые электроды (см. гл. III). Применяют также висмутовые [22], серебряные [23], вольфрамовые [24], карбидотанталовые [25] и некоторые другие электроды. Интересны ртутные электроды [26], позволяющие пользоваться электродными реакциями, протекающими в отрицательной области потенциалов благодаря высокому перенапряжению водорода на ртути. Ртутные электроды обладают еще одним свойством: ртуть, как известно, легко окисляется электрохимически в присутствии комплексообразователей, вследствие чего создается практически идеальная обратимая система анод — катод. Это позволяет определять различные неорганические и органические вещества, титруя их раствором соли ртути(II).
Вообще в некоторых случаях целесообразно применять наряду с платиновым катодом анод из металла, легко анодно растворимого при налагаемом на электроды напряжении. Например, Киз [27] применял ртутный анод для титрования галогенидов и се-78
35 Схема амперометрической установки с двумя индикаторными электро-f ^платиновые индикаторные электроды; 2 — 1 3 — гальванометр; 4 — вольтметр;
“^реостат; б —источник тока.
ребра: анодное окисление металла электрода заменяет'в этих случаях индикаторный электрод. При определении сахара, основанном на титровании избытка реактива Фелинга стандартным раствором сахдра, применяют медный анод: окисление меди
обеспечивает анодный ток, вос-
становление ионов меди'—катодный [28, 29]. Гоффар, Мишель и
Питанс [30] пользовались серебряным анодом при титровании марганца (II) перманганатом: растворение серебра составляло анодный процесс, восстановление перманганата — катодный.
Однако растворение анода не всегда благоприятно. Отрица
тельное влияние этого процесса на титрование перманганата, бихромата и ванадата солью Мора описано в работе [31]: изучение зависимости налагаемой э. д. с. и тока от природы материала
одного из электродов (второй электрод во всех случаях — платина) показало, что анодное растворение электродов из вольфрама, молибдена, меди, серебра, нихрома и нержавеющей стали проис-
ходит при менее положительных потенциалах, чем анодное окисление Fe11, вследствие чего кривая титрования «смазывается». Применять эти же металлы в качестве катода не только можно, но даже желательно, так как соответственно подобранный материал катода позволяет налагать меньшее напряжение и тем самым уменьшать влияние могущих присутствовать в растворе примесей, способных к электродной реакции [31]. Дело в том, что соответственно подобранная «пара» расширяет возможности метода за счет различия в перенапряжении выделения водорода, ионизации кислорода и других электродных реакций на том или ином материале за счет электропроводности металла или его окислов, могущих отлагаться на электроде, и т. д. Примером служит исследование титрования молибдена (VI) на различных электродах (платина — платина, вольфрам — вольфрам и вольфрам — платина), показавшее, что применение пары вольфрам — платина позволяет вдвое снизить напряжение, налагаемое на электроды, по сравнению с двумя платиновыми электродами 1^4].
Область применения метода амперометрического титрования с Двумя электродами весьма обширна. Особенно часто этот метод применяют для определения воды по методу Фишера в самых раз-
79
Саль Мора, пл KMnQj, мл
Рис. 36. Схематические кривые последовательного титрования нескольких ионов с двумя индикаторным электродами:
1 — титрование урана(Ш) и железа(П) перманганатом при 0,1 В; 2 —титрование перманганата и ванадата солью Мора в 2 и. растворе серной кислоты при 0,5 в-3 — титрование ванадия (V и IV) сначала солью Мора' затем перманганатом при 0,4 В.
нообразных объектах— неорганических и органических. В этом методе используют окислительно-восстановительную реакцию 12/21_, которая, как было показано выше, относится к числу почти идеально обратимых систем и потому особенно пригодна для амперометрического титрования с двумя электродами. При определении воды в неводных растворах очень
важно то, что при титровании с двумя электродами исключается применение электрода сравнения и солевого мостика. По существу этот метод может быть применен почти во всех случаях, для которых рекомендовано «классическое» титрование с одним индикаторным электродом.
Следует упомянуть и о том, что при титровании с двумя электродами можно определять последовательно несколько ионов, как и при обычном амперометрическом титровании, но с более резкими перегибами кривой в точках эквивалентности. На рис. 36 приведены схематические кривые последовательного титрования нескольких ионов в растворе [32—34]. Разница в ходе кривых 1 и 3 после конечной точки титрования объясняется разным значением приложенного напряжения: в первом случае (кривая 1) напряжение 0,1 В недостаточно для обеспечения тока после конечной точки.
Помимо непосредственного применения для собственно амперометрического определения различных веществ, амперометрический метод с двумя электродами широко применяют в кулонометрическом титровании, поскольку он позволяет с большой точностью определять конечную точку титрования. В недавно опубликованном обзоре Т. К. Хамракулова [35] даны ссылки на случаи определения различных веществ при помощи этого метода. Применение «биамперометрии» и кулонометрическом титровании описано также в книге А. П. Зозули [36] и в обзорах Дж. Стока по амперометрическому титрованию (см. гл. I). Методу с двумя индикаторными электродами посвящены также обзорные статьи чешских исследователей [37—39]. Применение метода для конкретных случаев описывается в гл. VI в числе различных методик, рекомендованных для определения отдельных элементов.
80
ЛИТЕРАТУРА
<? inmnn Е __Z phys. Chem., 1897, Bd. 24, S. 55; 1898, Bd. 25, S. 366.
9 NernstW, Merriam E.-Z. phys. Chem., 1905, Bd. 53, S. 235.
O Sew Bowden A. T.—J. Am. Chem. Soc., 1926, v. 48, p. 2045.
3 T RnttaerW, Forche Я. — Mikrochemie, 1942, Bd.. 30, S. 138.
f Guzman E, Rancano A. — An. Exp. Quim., 1934, v. 32, p. 899.
В Кол^оф Й. M. -Усп. хим., 1955, т. 24, с. 673.
7 Савицкая И. С. Некоторые особенности метода амперометрического титро-вания с двумя индикаторными электродами. Канд. дисс. КазГУ, Алма-Ата,
8 Kies Н.— Anal. chim. acta, 1952, v. 6, p. 90; 1954, v. 10, p. 161, 575; 1958, v 18, p. 14.
9 Duyckaerts G. — Anal. chim. acta, 1953, v. 8, p. 57.
10 Kolthoff 1. M., Nightingale J. — Anal. chim. acta, 1957, v. 17, p. 329.
11 Bradbury J. H.— Trans. Faraday Soc., 1953, v. 49, p. 304; 1954, v. 50, p. 959.
12 Кольтгоф Й. M.—Усп. хим., 1955, т. 24, с. 673.
13 * Делахей П. Новые приборы и методы в электрохимии. — Пер. с англ./Под ред. Б. В. Эршлера. М., Издатинлит, 1957.
14 Сонгина О. А., Савицкая И. С.—Зав. лаб., 1959, т. 25, с. 1028—1032; 1961, ’ т. 27, с. 1068—1072.
15. Филенка А. И.—Ж- анал. хим., 1966, т. 21, № 12, с. 1495—1497.
16 Филенка А. И. — В кн.: Химия и химическая технология. Том 1. Алма-Ата, ’ 1964, с. 114—120.
17. Филенко А. И., Кужель А. М. — Ж- анал. хим., 1968, т. 23, № 7, с. 1036.
18. Rayon J.— Compt. rend., 1965, v. 261, № 20, p. 4083—4085.
19. Tockstein A., Romers K. — Chem. Listy, 1958, v. 52, p. 1479—1482.
20. Филенко А. И. — Передовой науч.-техн. и произ. опыт, М., ГосИНТИ, 1966, № 2,-66—1225/138.
21. De Robertis A., Bellomo A., d'Arrigo С., de Marco D. — Inst. Chim. Anal. Univ. Messina, Italy, 1973, v. 63 (11—12), p. 855—859; Chem. Abstr., 1975, № 2, 21379j.
22. Козулина M. M., Лепин fl. K-, Сонгина О. A. — В кн.: Химия и химическая технология, Алма-Ата, 1975, вып. 17, с. 122—127.
23. Масалович В. М., Лагунова Н. Л. — Труды Уральского НИИхимии, 1971, вып. 26, с. 28—62.
24. Николаева Е. Р. и др. — Ж. анал. хим., 1970, т. 26, № 1, с. 119—122; Вестник МГУ, 1968, № 2, с. 135—139.
25. Мухамеджанова Д., Хадеева В>. А., Талипов Ш. Т.—ДАН УзбССР, 1973, Ks 11, с. 31—32.
26. Петухова А. И., Торопова В. Ф. — Изв. вузов. Химия и хим. технол, 1970, т. 13, №8, с. 1101—1103; 1974, т. 17, № 8, с. 1253—1254.
27. Kies Н. — Anal. chim. acta, 1955, v. 12, p. 180.
28. Bell E., Graham J. — Intern. Sugar J., 1950, v. 52, p. 90.
29. Coalstad S.—J. Chem. Soc. India, 1946, v. 65, p. 230.
qi Goffart G., Michel G., Pitance Th.—Anal. chim. acta, 1947, v. 1, p. 393.
qo Филенко А. И.— Ж. анал. хим., 1967, т. 22, № 1, с. 161—164.
чч Klinkhamer J. — Rec. trav. chim., 1954, v. 73, p. 1009.
чд -Руденская Л. С., Сонгина О. A.—Зав. лаб., 1960, т. 26, с. 1102.
ос" ~ДвиЦкая И. С., Сонгина О. А. — Зав. лаб., 1963, т. 29, с. 41.
□К миракулое Т. А.— Зав. лаб., 1975, т. 41, № 9, с. 1041—1066.
47 \Я3Уля А. П. Кулонометрический анализ. Л., Химия, 1969.
^tuiik К., Vydra F. — Chem. Listy, 1966, v. 60, № 5, p. 628—645.
эд Миш K., Vydra P. — Chem. Zvesti, 1967, v. 61, № 21, p. 274—304.
• btulik K-, Vydra F. — J. Electroanal. Chem. and Interfacial El.-Chem., 1968, v- 16, № 3, p. 385—395.
6—1694
81
ГЛАВА у
Типы реакций в амперометрическом титровании
Реакции осаждения
При определениях по методу осаждения первостепенное значение имеет растворимость образующегося соединения, так как от нее зависит полнота взаимодействия участников титрования и чувствительность метода. Для осуществления титрований по методу осаждения необходимо, чтобы растворимость была меньше 10-4 моль/л. Если же осадки обладают заметной растворимостью (порядка 10~3 моль/л), то для понижения ее титрования проводят в присутствии спирта или ацетона.
Необходимо также считаться и со скоростью образования осадка: обычно чем выше растворимость осадка, тем меньше скорость его образования. Если осадок образуется не мгновенно, то в первый момент после добавления титрующего раствора ток возрастает пропорционально количеству добавленного реактива и -затем постепенно уменьшается по мере того, как происходит связывание добавленного реактива и образование осадка. В таких случаях не следует регистрировать первый, «бросковый» ток, а
Таблица 11. Состав ферроцианида цинка, образующегося на различных фонах
Ферроцианид цинка Состав фона Литература
K2Zn3[Fe(CN)e]2 0,05—1,0 М H2SO4, НС1, НАс [5, 9]
(NH4)2Zn3[Fe(CN)6]2 1Л4 NH4C1 + НС1 до pH = 2 — 3 0,5 M HTart + NH4OH до pH = 7 — 8 0,5 M HTart + 0,75 M NH4Ac + NH4OH до pH = 7 — 8 [10] [11] , ]11, 12]
Zn2[Fe(CN)6] 1,5 —2,0 M HAc, NH4Ac 0,5 —0,3 M NH4OH 0,2 M NH4OH+ 0,2 M K2SO4, NaAc 0,1 — 0,3 M HCit + NHjOH до pH = 8 [5, 13] [5] |5] [14]
[NHj2Zn[Fe(CN)6] 1 — 4 M (NH4j2SO4 0,75 M NH4Ac-f- HAc до pH = 4— 5 0,1 — 0,5 M HCit + NH4OH до pH = 2 — 5 0,5 M HTart + NH4OH до pH = 4 — 5,5 0,5 M HTart + 0,75 M NH4Ac-f- NH4ON до pH =4 —5,5 [1, H] ill! [11! 121
82
бходимо выждать, пока между раствором и осадком не уста-Не°° ся равновесие, после чего сила тока примет постоянное зна-Н°Вие Кривая титрования имеет в таких случаях вид, изображен-ЧеН“ на рис. 37. Верхняя кривая соответствует значениям «брос-НЬШого» тока, нижняя — силе тока после выжидания. Обычно выедать приходится не больше 0,5 мин. Только в исключительных Жучаях, при титровании растворов высокой ионной силы, в кото-пьгх образование осадка может быть сильно замедлено, приходится выжидать по 2—3 мин, особенно в начале титрования, когда концентрация одного из ионов очень мала по сравнению с концентрацией другого. Подобное длительное выжидание необходимо, например, при определении микроколичеств цинка анодным ферроцианидным методом без отделения примесей [1].
F При титровании по методу осаждения необходимо считаться еще с тем, что состав образующегося соединения может зависеть от природы и кислотности фонового электролита. Так, 8-меркап-тохинолин (тиооксин) с золотом (III) в нейтральных и слабокислых (рН = 3—6) растворах образует осадок Au(C9H6NS)3, а на фоне 1 М раствора НС1 — HAu(C9H6NS)2C12 [2]. Этот же реагент с палладием (II) образует при рН=4—7 осадок Pd(C9E6NS)2, а в 0,1 н. растворе НС1 — Pd2(C9H6NS)3Cl [3].
Особенно характерно образование осадков различного состава для ферроцианидов [4]. Так, при титровании цинка(II) раствором ферроцианида калия образуются соединения, в которых в зависимости от природы и состава фона отношение Zn2+ и Fe(CN)e- может составлять 1 :1,2 : 1, 3 : 2 (табл. 11).
При титровании ионов редкоземельных элементов раствором ферроцианида калия образуются осадки четырех типов [15—18]: Ln4[Fe(CN)6]3,MLn[Fe(CN)6],MLn5[Fe(CN)6]4HM4Ln8[Fe(CN)6]7 (М — щелочной металл, Ln — лантаноид). Состав ферроцианидов РЗЭ обусловливается природой лантаноида и природой катиона щелочного металла фона, а также кислотностью раствора (табл. 12). Образование ферроцианидов РЗЭ различного состава связано, вероятно, с ионными радиусами соответствующих РЗЭ и щелочных металлов. Для лантаноидов с радиусом, большим 0,113 нм (элементы цериевой подгруппы), независимо от размера катиона щелочного металла фона, характерно об-
Рис. 37. Кривые титрования кальция рас-„ °Р°м Ферроцианида калия при замедлен-собразовании осадка:
с₽азУ после добавления титрующего раствора; 2 — через 30 с.
6*
83
разование соединения MLn[Fe(CN)6]. Лантаноиды с радиусом меньше 0,113 нм (иттриевая подгруппа) образуют соли указанных выше четырех типов, причем чем меньше размер лантаноида иттриевой подгруппы и больше размер катиона щелочного металла фона, тем в большей мере происходит внедрение щелочного металла в осадок ферроцианида (рис. 38).
Таким образом, при титровании по методу осаждения необходимо не только считаться с растворимостью образующегося соединения, .но и учитывать возможность влияния природы и кислотности титруемого раствора на состав образующихся осадков.
Реакции комплексообразования
Реакции образования устойчивых воднорастворимых комплексных соединений часто используют в амперометрическом титровании для определения различных элементов (см. гл. VI). В качестве комплексообразующего реагента-титранта наиболее широкое применение получил ЭДТА, который с ионами многих металлов образует прочные комплексы постоянного состава с отношением кэмплексообразователя к лиганду 1 : 1. Образование и устойчивость комплексонатов зависят от природы катиона металла и кислотности раствора [19]. Поэтому, варьируя pH титруемого раствора, можно осуществлять амперометрическое определение одних элементов в присутствии других, а также раздельное определение нескольких ионов при совместном их присутствии. Например, можно последовательно оттитровать раствором ЭДТА смесь ионов висмута, цинка и магния: сначала при рН = 1—2 титруют
Состав осадка
Sc(nH=3), La, Pr, Sm, Eu
- MLn[Fe(CN)6]
47.47-1
Y, Yb
• -M4Ln8[Fe(CN)e]y
0,47-
ScfpH=5J
Na+
Li
ф ь,
47,7-
MLn5[Fe(CN)e]4
-•Ln4[Fe(CN)6]3
K+NH4
0,08 0,10 0,12 0,U 0,1 В R,HM
Pnc 38. Зависимость состава ферроцианидов РЗЭ от ионного радиуса катиона фона.
84
,щ\ затем, понизив кислотность раствора до рН=4,7— БлСМУитоуют цинк(И), после этого еще понижают кислотность до 5h-Tq_-10 и титруют магний (II) [20]. Подобный прием примера пои анализе смесей таллий (I) — медь (II), таллий(1)-кад-Htft(ID —магний(II), ртуть(II)—висмут(Ш)—цинк(П) и ряда других [21—25].
W Из других комплексообразующих реагентов все большее при-енение находят различные органические серусодержащие соединения [26, 27]. Характерной особенностью этих реагентов являет-ня т0> что образованию растворимых комплексов часто предшествуют образование осадка или протекание окислительно-восстановительного взаимодействия. Так, при титровании ртути(II) унитиолом [28, 29] сначала выпадает белый осадок, который затем растворяется с образованием комплексного соединения, в котором молярное отношение ртути (II) к унитиолу равно 1:1. При титровании теллура (IV) тиооксином [30] происходит восстановление теллура(IV) до теллура(II) и последний образует комплекс [Те(С9НбП§)4]2+- Подобным образом протекает взаимодействие платины (IV) с тиокарбамидом в азотнокислой среде [31]: пла-THHa(IV) восстанавливается до платины(II), а платина(II) образует комплекс [Pt(SCN2H4)4]2+.
При титровании по методу комплексообразования необходимо учитывать возможность влияния природы и концентрации фона на протекание интересующей аналитика реакции, так как в растворах ионы металлов не существуют в «свободном» состоянии и находятся всегда в виде тех или иных комплексных ионов, в том числе в виде аквакомплексов. Если анионы фона способны образовывать с определяемым ионом металла достаточно прочный комплекс, то титрование ионов этого металла комплексообразующим реагентом может не дать положительного результата. Например, титрование ионов PtClg" и PtCl4“ тиокарбамидом [31] реализуется без каких-либо осложнений на фоне азотной кислоты, но не удается на солянокислом фоне вследствие упрочнения комплексных ионов за счет значительного избытка хлорид-иона. Другой пример: при титровании палладия(II) в солянокислой среде роданидом калия [32] образуется при концентрации НС1 меньше 0,2 М смешанный хлорид-роданидный комплекс [Pd(CNS)2Cl2]2-, а при концентрации НС1 больше 0,2 Л-1— одновременно комплексы [Pd(CNS)2Cl2]p- и [Pd(CNS)Cl3]2-. Эти примеры наглядно показывают, что при титровании по методу комплексообразования, как и осаждения, необходимо принимать во внимание возможность влияния тех или иных факторов на пол-н°ту и стехиометрию реакции.
Реакции окисления — восстановления
При титровании по методу окисления — восстановления также неходкие, чтобы взаимодействие участников титрования протека-количественно и быстро. О .возможности и направлении проте-
85
Таблица 12. Состав ферроцианидов РЗЭ, образующихся на различных фонах
Фон Sc (0,83 A)* Y (1,06 A) La (1,22 A)
LiNO3 Li+ —0,78 А LiSc[Fe(CN)B] (pH = 2,2—4,4) Sc4[Fe(CN)B]3 (pH = 6,0—6,0) Li4YB[Fe(CN)B]7 (pH = 3,2—6,5) LiLa[ Fe(CN)6] (pH = 3,5—6,0)
NaNO, Na+ —0,98 А NaSc[Fe(CN)e] (pH = 2,2—3,5) Sc4[Fe(CN)B]3 (pH = 4,0—6,0) Na4Ye[Fe(CN)B]7 (pH = 3,2—7,0) NaLa[Fe(CN)B] (pH = 3,5—6,1)
KNO3 К+—1,33 А KSc[Fe(CN)e] (pH = 2,0—3,5) Sc4[Fe(CN)6]3 (pH = 4,0—6,0) KY[Fe(CN)6] (pH = 2,5—6,0) KLa[Fe(CN)B] (pH = 3,5—5,8)
F!bNO3 Rb+ — 1,49 A RbSc[Fe(CN)B] (pH = 1,4—6,3) RbY[Fe(CN)B] (pH = 1,7—6,0) RbLa[Fe(CN)BJ (pH = 2,3—6,5)
CsNO3 Cs+ — 1,65 A CsSc[Fe(CN)6] (pH = 1,4—6,3) CsY[Fe(CN)6] (pH= 1,7—6,0) CsLa[Fe(CN)B] (pH = 2,3—6,5)
nh4no3 NH| — 1,43 A NH4Sc[Fe(CN)B] (pH = 2,0—3,5) Sc4[Fe(CN)„] (pH = 4,0—6,0) NH4Y [Fe(CN)B] (pH = 2,5—6,0) NH4La[Fe(CN)B] (pH = 3,5—5,9)
* Ионные радиусы по Гольдшмидту.
кания таких реакций обычно судят, исходя из значений стандартных (нормальных) потенциалов. Стандартные потенциалы характеризуют окислительно-восстановительные системы в идеальных условиях, т. е. при активности потенциалопределяющих ионов равной единице, отсутствии посторонних ионов и т. д. Практически такие условия неосуществимы, наоборот —в реальных условиях приходится иметь дело с растворами, содержащими те или иные ионы, которые могут оказывать влияние на значение окислительно-восстановительного потенциала данной системы. Особенно сильно на окислительно-восстановительные потенциалы влияют посторонние вещества, способные выступать в качестве лигандов или изменять форму существования потенциалопределяющих ионов. Так, стандартный потенциал системы Au3+/Au+ равен 1,41 В, а реальный потенциал этой системы в 1 и 4 М растворах НС1 составляет 1,03 и 0,87 В. Стандартный потенциал системы VO2+/V3+ равен 0,34 В, а реальный потенциал этой системы в 1 М растворе Н3РО4 — 0,52 В. Поэтому характер протекания окислительно-восстановительных реакций может оказаться иным, чем этого следовало бы ожидать, исходя из значений стандартных потенциалов. Так, исходя из стандартных потенциалов систем Fe(CN) б'/ Fe(CN)e" (£'о=О,36 В) и 1г/1~ (£о = 0,54 В), казалось бы, что Fe(CN)6- нельзя применить для титрования иодид-иона. Однако
86
и при различных pH [15—18]
О Рг (1,16 А) Sm (1,13 A) Eu (1,13 A) Gd (1,11 A) Yb (1,00 A)
LiPr[Fe(CN)e] (pH =3,0—6,4) LiSm[Fe(CN)B] (pH = 2,3—6,5) LiEu[Fe(CN)B] (pH = 2,2—6,5) Gd4[Fe(CN)B]3 (pH = 3,2—6,6) Li4Ybe[Fe(CN)B]r (pH = 2,3—6,5)
NaPr[Fe(CN)B] (pH = 4,5—6,0) NaSm[Fe(CN)6] (pH = 2,5—6,0) NaEu[Fe(CN)B] (pH = 2,6—6,4) Gd4[Fe(CN)B]3 (pH = 4,5—6,0) Na4Ybs[Fe(CN)B]r (pH =3,8—7,0)
KPr[Fe(CN)6] (pH = 2,9-6,2) KSm[Fe(CN)6] (pH = 2,3—6,2) KEu[Fe(CN)B] (pH = 2,3—6,1) KGd6[Fe(CN)B]4 (pH = 2,3—6,0) KYb[Fe(CN)B] (pH = 2,0—6,7)
RbPr[Fe(CN)B] (pH = 1,6—6,2) CsPr[Fe(CN)B] (pH = 1.6—6,3) RbSm[Fe(CN)B] (pH = 1,6—6,7) CsSm[Fe(CN)B] (pH = 1,6—6,3) RbEu[Fe(CN)B] (pH = 1,6—6,6) CsEu[Fe(CN)B] (pH= 1,6—6,6) RbGd6[Fe(CN)B]4 (pH = 1,5—6,6) CsGd6[Fe(CN)B]4 (pH = 1,4—6,8) RbYb[Fe(CN)B] (pH = 1,4—6,2) CsYb[Fe(CN)B] (pH = 1,4-6,0)
NH4Pr[Fe(CN)B] (pH = 2,9—6,5) NH4Sm[Fe(CN)B] (pH = 2,4—6,2) NH4Eu[Fe(CN)B] (pH = 2,2—6,0) NH4GdB[Fe(CN)„]4 (pH = 2,4—6,3) NH4Yb[Fe(CN)„[ (pH = 2,0—6,0)
такое титрование возможно, если его проводить на фоне 8 М серной кислоты [33], так как в растворе 8 М серной кислоты потенциал системы ферри—ферроцианид (0,98 В) намного положительнее потенциала системы иод —иодид (0,39 В) [34].
Для характеристики окислительно-восстановительной системы в реальных условиях пользуются понятием «реальный (формальный) потенциал» (Ер или Е01), который соответствует потенциалу. установившемуся в данном конкретном растворе при равенстве исходных концентраций окисленной и восстановленной форм потенциалопределяющих ионов, без учета каких-либо поправок на процессы комплексообразования, гидролиза и др. Реальные потенциалы окислительно-восстановительных систем с аналитической точки зрения более ценны, чем стандартные потенциалы, так как истинное поведение системы определяется не стандартными, а Реальными потенциалами, и именно последние позволяют предвидеть протекание окислительно-восстановительных реакций в конкретных условиях опыта. Реальные потенциалы различных окислительно-восстановительных систем, имеющих значение для аналитической практики, приведены в табл. 13. Как видно из этой >лицы, реальные потенциалы той или иной системы зависят от кислотности и природы электролита и изменяются в широких пРеделах.
87
Оо
00
Таблица 13. Реальные потенциалы окислительно-восстановительных систем [34]
Система Электролит Значения потенциалов (в В, НВЭ) при концентрации электролита (в М)
0,25 0,50 1,00 2,00 3,00 4,00 5,00 6,00 8,00 10,0
AsV/Asiii pH(NaCl) . 0,52 0,50 0,48 0,44 0,31
HF — — .—. -—. — — — 0,54 0,57 0,61
HC1 — — 0,58 0,61 0,63 0,65 0,67 0,70 0,82 —
HBr — — 0,59 0,62 0, 3 0,66 0,68 0,72 0,82
нею. —. — 0,62 0,64 0,65 0,67 0,69 0,73 0,84 —
H2so4 — — 0,58 0,59 0,63 0,65 0,67 0,69 0,78 0,86
NaOH — — —0,07 — —0,09 — —0,16 — — —
AuVAu HC1 1,13 1,12 1,Н 1,07 1,05 1,02 — — — —
Аиш/Au KC1 0,97 0,95 0,92 0,90 0,89 0,88 — — — —
HC1 0,96 0,93 0,90 0,88 0,86 0,85 0,84 0,83 0,80 0,78
HBr 0,85 — 0,82 — 0,77 -—. — — — —
h2so4 1,10 1,09 1,08 1,05 1,02 1,01 1,00 — — —
NaOH 0,60 0,55 0,50 0,47 0,44 -—. — —. — —
Auin/Aui HC1 — — 1,03 0,98 0,90 0,87 — — — —
Br2/Br HC1 1,20 1,17 1,13 1,08 1,06 1,05 1,04' 1,03 1,00 0,94
H2SO4 1,12 1,12 1,11 1,10 1,07 1,05 1,03 1,00 0,98 0,96
BrO-/Br~ H2SO4 — — 0,66 — 0,70 — — — —
NaOH — — 0,69 -—. 0,66 — 0,65 — -—. .—
ВгОз/Вг" HC1 1,39 1,38 1,33 1,27 1,23 1,20 1,18 -—. 1,12 —
H2SO4 1,12 1,20 1,28 1,34 1,40 1,45 1,50 . — —
BrO7/Br° HC1 — 1,25 1,30 1,30 1,29 1,27 1,25 1,24 1,22 —
H2so4 1,20 1,27 1,33 1,38 1,43 — — .— —— —
CelV/CeHl HC1 1,26 — 1,28 — — — — — — —
HNO3 — — 1,61 1,62 —. 1,61 — — 1,56
HC1O4 — —. 1,70 1,71 — 1,75 — -— 1,87
H2so4 1,44 1,44 1,44 1,43 — 1,36 — 1,28 — 1,24
ci2/ci- HC1 1,40 1,35 1,31 1,29 1,25 1,22 1,17 1,14 1,10 1,08
H2SO4 — — 1,27 1,25 1,23 1,21 1,20 — — —
Coin/Coii HN03 — , — 1,83 — 1,84 1,85 — — — —
Crin/Crii HC1 —0,40 — —0,38 — —» -—. — — — —
Cr2O|-/Crin pH(KNO3) — — 1,Ю 0,95 0,86 0,80 0,68 0,61 0,48 0,35
HC1 — — 1,06 1,07 1,08 1,10 1.12 1,14 1J5 1 1,20
00
Cuil/Cul
FeHi/Fell
[Fe(CNe]3-/[Fe(CN)J*-
Hg^+/Hg
Hg2+/Hg
I2/r
IC17/IS 104/10:7 Iriv/irin
МпШ/МпП
HC1O4
H2SO4
HSPO4
NaOH
pH(l M KC1)
KC1
HC1
0,5 M H2SO4+ KSCN
pH (ацетатный и баратный буферы)
CHSCOOH (pH = 2)
CHSCOOH (pH = 3)
HCI
1 M HC1 + HSPO4
H2SO4
0,5 M H2so4 + HF
0,5 M H2SO4 + NaF
0,015 M h2so4+kscn
0,5 M H2SO4+H4P2O7
H3PO4
pH(KNO3)
KC1
HCI
HgSOe
NaOH
KSCN
KSCN
pH(NaCl)
HCI
H2SO4
HCI
pH
KC1, NaCl
LiCl
HCI
H2SO4
pH(0,3!M^Na4P2O, 4- H2SO4)
2^M NH4F+H2SO4, pH
0,61
0,74
0,63
0,39
0,60
0,48
0,63
0,63
0,45
0,20
0,20
0,62
0,97
1,19 1,22 1,36 1,47 1 1,55
1,08 1,10 1,14 1.17 1,21 1,25 1 1,28 1,34 1,38
1,02 1,07 l.H 1,15 1,18 1,22 1,32 1,45
—0,12 .— —0,09 — —0,08 —. — —
0,13 0,12 0,06 0,06 0,06 0,03 0,01 0,00 0,15
0,14 0,12 — -— —. — — ——
0,43 0,45 0,50 0,56 0,58 0,60 0,61 0,61 0,60
0,59 0,57 — — -—. — -— — —
— 0,80 0,79 0,71 0,60 0,43 0,22 —0,15 —0,35
0,72 0,69 0,66 0,63 — 0,58 -—. —— ——
0,58 0,54 0,50 0,47 -—. 0,43 — — —
0,71 0,70 0,68 0,67 0,66 0,64 0,62 0,56 0,53
0,61 0,57 0,52 0,50 — — —. — —
0,71 0,70 0,69 0,68 0,67 0,66 — — —
0,58 0,55 0,50 0,47 0,45 0,44 0,42 — —
0,25
0,57 0,55 — — — — — — —-
0,44 0,42 —. — — — -—. -— —
0,62 0,59 0,56 0,54 -—. 0,53 —- 0,52 —
0,65 0,56 0,52 0,47 0,45 0,45 0,44 0,44 0,44
0,45 0,46 0,48 0,50 0,51 — — — —
0,65 0,70 0,74 0,79 0,82 _—. — — —
0,68 0,73 0,79 0,84 0,88 0,93 0,97 1,04 —.
0,46 0,48 0,49 0,50 0,51 0,52 — — —
0,15 0,12 — —« — — -— — ‘
0,15 0,12 — —- — — — -—. —
— 0,65 0,65 0,65 0,65 0,65 0,65 0,65 —
0,61 0,60 0,59 0,58 0,56 0,54 0,51 0,48 0,45
0,60 0,58 0,54 0,51 0,47 0,43 0,41 0,39 0,37
— 1,08 1,07 1,05 1,03 1,01 0,97 0,87 —
.— 1,26 1,18 1,11 1,06 1,02 0,98 0,94 0,76
0,98 0,99 1,01 1,02 1,03 — — — —
—. 0,99 1,00 —- 1,02 — 1,04 1,05 1,06
— 0,96 0 98 -—. 1,02 — 1,03 1,03 1,04
1,02 1,02 1,02 — 1,03 — 1,04 1,06 1,08
— — — 0,96 0,79 0,66 0,51 0,43 —
— — — 0,83 0,76 0,72 — — —
s
Система Электролит
МпШ/МпП H2SO4 HsPO4
Mno;/Mn« pH(H2SO,+ NaOH) H2SO4 HSPO4
MoV/Moin HC1 H2SO4
MoVi/MoV HC1 H2so4 0,5 M H2SO, + KSCN
n2/n2h4 OsVi/OslV Osiv/Osin Pbiv/PbH HC1 HC1 HC1 HC1 HC1O4 НАС NaOH, KOH
Ptiv/Ptn HC1 HC1O4 HNO, HaSO4 3 M H2SO4 + NaCl KNO3
Ruiv/RuHi RuViH/RulV (SCN)2/SCN-SbV/Sbin HC1 HC1O4 pH(HCl + KOH) HF HC1 HBr NaOH
Seiv/Se HC1 4 M HC1 4-LiCl
Продолжение
Значения потенциалов (в В, НВЭ) при концентрации электролита (в Л!)
0.25 0,50 1,00 2,00 3,00 4,00 5,00 6,00 8,00 10,0
— 1,36 1,37 1,38 1,43 1,45 1,48 1,53 1,57
— 1,32 1,32 1.31 1,31 1,31 1,30 1,30 1,30
— — 1,45 1,39 1,26 1,20 1.Ю 1,00 0,77 0,70
1,38 0,40 1,44 1,46 1,48 1,49 1,51 1,53 1,54 1,55
,,33 1,34 1,36 1,37 1,39 — — — ——
I— — ——- о.п —. 0,18 0,24 0,28 —
I— 1,01 — — — 0,09 — 0,22 —.
I— — —— 0,53 — 0,55 0,60 0,70 —
— 0,40 —— —* —* —, 0,47 — — 0,54
0,49 0,52 0,53 —„ — —. — — —.
0,67 0,65 0,63 0,60 — — — ——
— 0,66 — —ь —— — 0,84 — 0,97 —.
—— 0,37 —* •— — —0,04 .—
1,30 1,27 — 1,22 — — — —
1,66 1,66 1,66 .— 1,70 — —.
— — 1,45 1,45 1,45 1,45 1,45 — 1,44 1,43
— —* 0,30 . .—. -—. 0,20 .—.
0,75 0,74 0,73 0,72 — 0,70 — 0,66 0,61 0,59
0,77 0,76 0,75 —_ 0,74 -—. — — —
0,76 0,76 0,76 1,04 — 1,13 — — — —
0,77 '— 0,76 0,75 — 0,72 .— 0,68 0,63 0,60
0,77 0,75 0,71 — -—. — —. — — —.
0,79 0,82 0,82 0,81 — 0,75 — — — —
— 0,91 - 0,86 — — — — —
— —. 1,40 — — — 1,43 — — 1,51
— 0,55 0,55 — 0,55 — 0,54 0,50 0,35
—- — 0,51 — 0,53 0,54 ш— 0,56 0,57 0,59
-—. —* — 0,67 0,72 0,77 0,80 0,83 0,84 0,80
— 0,69 0,75 0,81 0,85 0,86 0,83 0,79 —
— —. —. —. —0,43 — —. — — —0,59
—. — 0,64 0,64 —. 0,65 — 0,90 0,93 0,94
— — —• 0,90 — 0,93 — 0,94
16
н
§
Д о 5 < сл сл 5. г ~ <
о 2 Д Д Я Я ^яРР
м
W О
2 .3
X, X X S X X XPS хрхх IXX-2.--XX х-х^х ХХьэ
>хР РР Р^Р С/) HJM О Г? * сл Эсл^сл & р 5 з хР Я сл сл <Ррр я + я w4 СЛ 0 К»
+ «р + ++ X ° +
Z XX р
S 2 3 Я I *я х^ •п g
ч Ъ а (Л О
(/> р
КЭ
Система Электролит
VIV/VHI Н3С„Н6О7 (pH 1,5) nh4f H2SO4 HSPO4 H2C4H4Oe pH (4 М H2CeHeO.( -j- NaOH)
WVi/WV HCI 0,33 M HSPO4+HC1
Ди су льфид/тиооксин pH HCI, H2SO4
Дисульфид/дитиобиу-рет HCI H2SO4
Тетратионат/тиосуль-фат HCI H2so4 NaOH NaHCO3
Хиноп/гидрохинон' H2SO4 NaOH
Продолжение
Значения потенциалов (в В. НВЭ) при концентрации электролита (в М)
0,25 0.50 1,00 2,00 3,00 4.00 5.00 6.00 8,00 10.0
— — 0,24 0,24 0,25 0,25 0,26 0,27 —. —
— 0,35 0,32 0,29 0,27 — — — — —
— 0,36 0,35 0,34 0,33 0,32 0,32 — — —
— 0,44 0,52 0,62 — — — 0,75 — —
— 0,33 0,35 — — — —• — —
— — — 0,28 0,28 0,27 0,20 0,15 0,05 —
— — — — — — — 0,17 0,20 0,24
— 0,31 0,23 0,18 0,17 — 0,16 — — —
— — 0.31 0,28 0,24 0,20 0,18 0,16 0,05 —
0,38 — 0,43 — — — — •— — —
0,33 — 0,29 0,27 0,27 0,26 0,26 0,26 — —
— 0,38 0,37 0,35 0,37 — — — —
0,06 0,06 0,08 0,08 0,08 0,08 0,07 0,06 — —
0,0 0,06 0,07 0,08 0,09 0,12 о.н — — —
—0,01 —0,02 —0,03 —0,04 —0,06 — — — — —
0,02 0,03 0,04 — — — — — — —
— 0,70 — — — — 0,77 — •— •—
— — 0,13 — — — — — — 0,20
Когда пользуются реальными потенциалами, то уравнение Нернста для любой окислительно-восстановительной системы, в том числе описываемых реакцией
МОУ—-J-2хН+4-=₽=*= M(y+"~2v)-+хН2О
имеет вид:
где Ер — реальный потенциал, В; Сок и Свое — концентрация окисленной и восстановленной форм потенциалопределяющих ионов, М.
Для одной и той же окислительно-восстановительной системы значение £р может зависеть от многих факторов (природы электролита, его концентрации и т. д.), но в одних и тех же условиях значение £р постоянно.
При расчете полноты протекания реакции, пользуясь реальными потенциалами, исходят из следующего. Представим окислительно-восстановительную реакцию в виде:
fcOKt -f- dBoc2 < -» /BoCj -|- йОк2 (2)
По достижении равнсзесия
Р . £7' (Сок,)* 6 , RT (Сок,)*
Р(1> 1 пЕ1П (СВ0С1)/ — ср(2) ' 1 in J nF
ДЛИ
£р(1> 7?р{2) — Д£р — RT , In nF (Qta)fe (СВ0с,)7 (CbocJ* (СОК1)6 ( ’
Поскольку Okj реагирует с Вос2 в отношении b: d, а Вощ и
Ок2 образуются в отношении f: k, то в точке эквивалентности
бвоса — b Gout (4)
Сою — , Своп
(5)
Подставляя выражения (4) и (5) в уравнение (3), получаем для точки эквивалентности
лр _ (fe//)*(CBoc.)w
р nF (d/b)a(COKl)d+b
где СвОс4 — концентрация Воск Сощ—концентрация непрореагировавшего Окь Принимая во внимание, что
б*Ок1 — ^Ок, ^Ок, (7)
Свос1=-уСЬК1 (8)
С от исходная концентрация Оки СОК1—количество прореагировавшего
и подставляя соотношения (7) и (8) в уравнение (6), получим PT (kif)k[(//b)C'OKy+f
Д£р- nF ln (d/b)d C'0^d+b (9>
Выражая Сощ относительно CoKt в процентах (Р=СоК1Х ХМО/Сощ), что соответствует полноте протекания реакции (2), и заменяя натуральный логарифм десятичным, получим
0,059 (k/f)k (fP/b) ft+/(Q)K1/100) k+f-d-b
Л£р = n lg (d/b}d(lOQ — P)d+b (10)
Уравнение (10) отражает взаимосвязь разности реальных потенциалов участников окислительно-восстановительного взаимодействия с полнотой протекания реакции и позволяет рассчитать как полноту протекания реакции при том или ином значении Д£р, так и минимальное значение Д£р, необходимое для протекания реакции с заданной полнотой.
Анализ уравнения (10) для интервала Со^ от 0,1 до 10~6 М, т. е для содержаний, обычно определяемых редокс-титриметри-ческими методами, показывает, что наибольшая разность реальных потенциалов при одинаковой полноте протекания окислительно-восстановительной реакции требуется в том случае, когда все параметры реакции’ (b, d, f, k, п) равны единице, и что для практически количественного (Р=99°/о) протекания какой-либо окислительно-восстановительной реакции достаточна разность реальных потенциалов, равная 0,24 В.
Разность потенциалов Д£р является необходимым, однако еще недостаточным условием для осуществления редокс-титриметри-ческих определений, поскольку окислительно-восстановительные потенциалы характеризуют термодинамическое состояние систем и направленность реакции, но не ее кинетику. Очень часто окислительно-восстановительные реакции, несмотря на термодинамическую выгодность, идут настолько медленно, что скорость реакции оказывается лимитирующим фактором при таких определениях. Поэтому при решении вопроса о возможности использования той или иной окислительно-восстановительной реакции в аналитических целях необходимо учитывать не только реальные потенциалы, но и кинетические характеристики системы.
Кинетика реакций обусловливается их механизмом, который для окислительно-восстановительных реакций обычно довольно сложен, и может существенно зависеть от природы и кислотности электролита, наличия в растворе посторонних ионов и от других факторов [35—38]. Варьируя условия протекания реакции, можно добиться увеличения ее скорости настолько, что она не будет лимитировать практическое 'применение данной реакции. Варьированием условий можно достигнуть также и того, что скорость реакции одного из компонентов раствора станет доста
94
точно отличающейся от скорости реакций остальных, и тем самым окажется возможным его избирательное определение без отделения от других веществ с близкими химическими свойствами. Так, на различии скорости основано раздельное амперометрическое определение мышьяка(III) и сурьмы(III) при совместном их присутствии при помощи бихромата калия [39]: мышь-як(Ш) быстро окисляется бихроматом в растворах H2SO4 лишь более 2 М, а сурьма (III)—при любой кислотности, и поэтому в слабокислой среде (0,1—0,5 М H2SO4) оттитровывается одна сурьма (III), а в сильнокислой среде (3—4 М H2SO4)—сумма мышьяка (III) и сурьмы (III). Другой пример: окислительно-восстановительное взаимодействие иодид-иона с перманганатом протекает быстро и количественно при кислотности более 0,03 М H2SO4, с хроматом — более 2 М H2SO4 и с ванадатом — более 5 М Нг8О4, что позволяет осуществить амперометрическое титрование перманганата в присутствии хромата и ванадата, а также хромата в присутствии ванадата. Таким образом, можно раздельно определить эти три окислителя с помощью иодида калия при совместном их присутствии [40].
Реакции окисления — восстановления подразделяются на реакции, протекающие по так называемому «внешнесферному» и «внут-рисферному» механизму или типу [35—38]. Скорость реакций с внешнесферным механизмом лимитируется скоростью переноса электрона, а с внутрисферным 'механизмом — скоростью переноса атома или группы атомов между реагирующими веществами. Для реакций, протекающих по внешнесферному механизму, Маркусом развита теория [35, 38], согласно которой скорость окислительно-восстановительной реакции определяется соотношением
К = (МК2К3/)1/2 (11)
где К — константа скорости окислительно-восстановительной реакции, Ki и Кг— константа скорости восстановления окислителя и окисления восстановителя; Кз — стандартная константа равновесия окислительно-восстановительной реакции; f — некоторый член, определяемый выражением
(1пКз)2 41n(MK2/Z2)
(12)
где Z — частота столкновения двух незаряженных частиц (принимается равной 10“ м-ьс-1).
Поскольку в уравнение (11) входит константа равновесия (Kz), для которой справедливо
lgK3 =
nF&E°
2,ЗЕТ
(13)
то из теории Маркуса вытекает, что скорость окислительно-восстановительных реакций с внешнесферным механизмом увеличивается с увеличением разности стандартных потенциалов систем, участвующих в реакции.
95
Рис. 39. Зависимость разности реальных потенциалов редокс-систем Fe(CN)e~/Fe(CN) (сплошные линии) и скорости взаимодействия золота (III) с ферроцианидом (пунктирные линии) от природы и концентрации фона.
Рис. 40. Зависимость разности реальных потенциалов редокс-систем (1 — MnVI1/Mn11—IrIV/Irln; 2 — Crv>/ Criii—Iriv/IriH; 3 —vv/V’V—Iriv/ Пг1П) и скорости окисления ири-дия(Ш) перманганатом (1а), биохроматом (2а) и ванадатом (За) от концентрации серной кислоты.
Рис. 41. Зависимость логарифма скорости взаимодействия золота (Ш) с ферроцианидом в среде КС1 (1), НС1 (2) и H2SO4 (3) от разности реальных потенциалов.
Авторы работы [38], анализируя данные о кинетике ряда окислительно-восстановительных реакций, приходят к заключению, что скорость возрастает с увеличением разности стандартных потенциалов независимо от механизма.
В работах [41—45] на примере различных окислительно-восстановительных реакций исследован характер изменения скорости реакции и разности реальных потенциалов взаимодействующих систем в зависимости от концентрации фонового электролита, а также характер зависимости скорости от разности реальных по-
96
Рис. 42. Зависимость логарифма скорости взаимодействия иридия(III) с перманганатом (1), бихроматом (2) и ванадатом (3) в среде H2SO4 от разности реальных потенциалов соответствующих окислительно-восстановительных систем.
тенциалов систем, участвующих в реакции. В этих работах показано, что с изменением концентрации фона скорость (®) той или
иной окислительно-восстанови-
тельной реакции и разность реальных потенциалов (A£p) редокс-систем в этой реакции изменяются симбатно (рис. 39, 40), а между lg w и AfP какой-либо конкретной реакции в каком-либо конкретном фоне имеется обычно прямая зависимость (рис. 41, 42). Это свидетельствует о том, что в электролите данной природы скорость окислительно-восстановительной реакции является функцией раз-
ности реальных потенциалов реагирующих редокс-систем: скорость тем больше, чем больше эта разность. Зависимость скорости от разности реальных потенциалов, исходя из экспериментальных данных [41—45], можно представить в виде
lgw = Д-р ВЛЕр
(14)
где Д и В — некоторые коэффициенты, зависящие от природы окислительно-восстановительной реакции, природы фона и, возможно, некоторых других факторов.
Таким образом, пользуясь реальными потенциалами окислительно-восстановительных систем, можно не только судить о возможности и направлении протекания окислительно-восстановительной реакции в конкретных условиях опыта, но и предвидеть, при каких условиях в электролите данной природы окислительно-ьосстановительное взаимодействие будет проходить быстрее или, наоборот, медленнее, т. е. предвидеть наиболее благоприятные условия амперометрического титрования.
ЛИТЕРАТУРА
1. Войлошникова А. П., Козловский М. Т., Сонгина О. А.—Зав. лаб., 1957, т. 23, № 3, с. 273—276.
2. Усатенко Ю. И., Супрунович В. И. — Изв. АН ЛатвССР. Сер. хим., 1963, Ns 2, с. 181—185.
3. Усатенко Ю. И., Супрунович В. И. — Изв. АН ЛатвССР. Сер. хим., 1963, № 1, с. 18—30
4. Тананаев И. В. и др.—Химия ферроцианидов. М., Наука, 1971, с. 320.
5. Сонгина О. А. Войлошникова А. П„ Козловский М. Т.—Зав. лаб., 1951, т. 17, № 1, с. 3—15; 1952, т. 18, № 3, с. 390—394.
6. Бутенко Г. А., Рынская Е. С. — Ж. анал. хим., 1950, т. 5, № 2, с. 145—149.
7. Woodson A. L., Johnson В. Н., Cooper S. R. — Anal. Chem., 1952, v. 24, № 4, p. 1198—1203.
7—1694
9?
8. Агасян П. К- — Вестник МГУ. Сер. физ., хим., астроном., 1959, № 6, с. 156— 159.
9. Тананаев И. В., Корольков А. П. — Изв. АН СССР. Сер. неорган. матер 1965, т. 1, № 10, с. 1577—1583.
10. Parks Т., Smith О., Radding S. — Anal. chim. acta, 1954, v. 10, № 2, p. 485— 489.
11. Дюлгерова А. С., Сонгина О. А., Захаров В. A. — В кн.: Прикладная и теоретическая химия. Алма-Ата, 1971, вып. 3, с. 36—40.
12. Дюлгерова А. С., Сонгина О. А., Захаров В. А. — Изв. АН КазССР. Сер. хим., 1971, № 3, с. 66—68.
13. Сонгина О. А., Котлярская В. 3., Войлошникова А. П. — Изв. АН КазССР. Сер. хим, 1955, вып. 8, с. 77—81.
14. Солнцев Н. И., Таль Э. М.—-Зав. лаб., 1953, т. 19, № 10, с. 1027—1031.
15. Токушева Г. Т., Захаров В. А., Сонгина О. А.— В кн.: Химия и химическая технология. Алма-Ата, 1968, вып. 7—8, с. 217—220.
16. Сонгина О. А., Захаров В. А., Токушева Г Т.—Ж. анал. хим., 1970, т. 25, № 1, с 6^- -67.
17. Токушева Г. Т„ Захаров В. А. — В кн.: Прикладная и теоретическая химия. Алма-Ата, 1974, вып. 5, с. 50—54.
18. Захаров В. А., Сонгина О. А., Токушева Г. Т.— В кн.: Сборник работ по химии. Алма-Ата, 1973, вып. 3, с. 206—211.
19. Шварценбах Г., Флашка Г. Комплексонометрическое титрование. — Пер. с нем. М., Химия, 1970, с. 360.
20. Жданов А. К-, Мархабаев И. А., Ленченко Т. А. — Изв. вузов. Химия и хим. технол., 1971, т. 14, № 3, с. 355.
21. Жданов А. К-, Капица Н. В., Ахмедов Г. — Труды Ташкентского ун-та. 1967, вып. 288, с. 31—34; с. 35—38; с. 39—41.
22. Жданов А. К-, Акентьева Н. А., Капица Н. В.—Узб. хим. ж., 1967, № 5, с. 18—20, № 6, с. 12—14.
.23 . ‘Жданов А. К, Капица Н. В., Акентьева Н. А.—Ж. анал. хим., 1977, т. 26, № 4, с. 835—837.
24. Жданов А. К-, Муминов Н. П.—Ж. анал. хим., 1974, т. 29, № 7, с. 1424— 1426.
25. Жданов А. К., Мархабаев И. А.—Узб. хим. ж., 1972, Ns 3, с. 25—27.
26. Сонгина О. А., Бессарабова И. М.— Зав. лаб., 1973, т. 39, № 6, с. 641— 655.
27. Бессарабова И. М„ Сонгина О. А. — Ж. анал. хим., 1974, т. 29, Ns 5, с. 998— 1002.
2^, Усатенко Ю. И., Климкович Е. А., Лошкарев Ю. М. — Укр. хим. ж., 1961, т. 27, Ns 9, с. 823.
29. Сонгина О. А., Оспанов X. К-, Рождественская 3. Б.—Ж- анал. хим., 1965, . т. 20, № 1, с. 55—59.
30. Сонгина О. А. и др. — Ж. анал. хим., 1972, т. 27, № 6, с. 1121—1124.
31. Гавва Н. Ф., Захаров В. А., Сонгина О. А.—Ж. анал. хим., 1976, т. 31, № 7, с. 1334—1337.
32. Захаров В. А., Айтхожаева Т. А.—Зав. лаб;, 1975, т. 41, Ns 2, с. 164—165.
33. Сонгина О. А. и др. — Зав. лаб., 1973, т. 39, № 8, с. 920—921.
34. Захаров В. А., Сонгина О. А., Бектурова Г. Б.—Ж- анал. хим., 1976, т. 31, Ns lil, с. 2212—2221.
35. Марк Г., Рехниц Г. Кинетика в аналитической химии. — Пер. с англ./Под ред. К- Б. Яцимирского. М., Мир, 1972, с. 368.
36. Терни Т. Механизмы реакций окисления—восстановления. — Пер. с англ./ Под ред. А. И. Бусева. М., Мир, 1968.
37. Сокольский Д. В., Дорфман Я. А. Катализ лигандами окислительно-восстановительных реакций в водных растворах. Алма-Ата, Наука, 1972.
38. Басоло Ф., Пирсон Р. Механизмы неорганических реакций. — Пер. с англ./ Под ред. А. Н. Ермакова. М., Мир, 1971, с. 592.
39. Чокина Н. Ю., Сонгина О. А., Захаров В. А.—Ж. анал. хим., 1976, т. 31, № 4, с. 720.
40. Сонгина О. А. и др.—Ж. анал. хим., 1974, т. 29, № 8, с. 1594—1598.
58
Чахапов В А Исследование в области амперометрического титрования с 41' ™Рпдыми (платиновый, золотой, графитовый) электродами. Автореф. докт. дисс Алма-Ата, 1974, с. 62.
42 Тойбаев Э. И., Сонгина О. А., Захаров В. А. — В кн.: Прикладная и теоретическая химия. Алма-Ата, 1975, вып. 7, с. 113 117.
43 Бектурова Г. Б., Захаров В. А. — В кн.: XI Менделеевский съезд по общей и прикладной химии. Аналитическая химия. М., Наука, 1975, с. 34.
44 Сонгина О. А. и др. — Изв. АН КазССР. Сер. хим., 1976, № 2, с. 75—77.
45 Захаров В А., Сонгина О. А., Айтхожаева Т. А.—Ж. анал. хим., 1977, Т- 32, № 9, с. 1786—1789.
7*
ГЛАВА VI
Определение различных элементов методом амперометрического титрования
В данной главе рассмотрены методы амперометрического определения элементов и почти для каждого элемента даны таблицы, в которых сгруппированы реагенты, применяемые для определения той или иной формы данного элемента, и дана краткая характеристика условий определения. Эти сведения приведены в том виде, в каком они опубликованы авторами методов, поэтому не во всех случаях есть указания на состав фона, pH раствора, потенциал электрода и т. д. С целью экономии места в таблицах приняты обычные условные сокращения, названия сопутствующих элементов даны символами, без указания степени окисления. В последней графе таблицы приведены ссылки на литературный источник.
В тексте, сопровождающем таблицы, кратко описан принцип метода, на котором основано определение той или иной ионной формы элемента, критическая оценка некоторых методов, а для наиболее оригинальных или зарекомендовавших себя при практическом применении изложен подробный «ход определения». Таким образом, читатель может выбрать по таблицам тот или иной метод и, получив в тексте общую сравнительную оценку отдельных методов, либо воспользоваться приводимой в тексте методикой, либо обратиться к соответствующему литературному источнику.
За последние годы усилилась тенденция к определению с помощью одного и того же реагента нескольких (иногда до 5—6) элементов, ионы которых совместно присутствуют в анализируемом растворе. В таких случаях, во избежание многократных повторений, возможность определения каждого из этих элементов обсуждается в том разделе, к которому относится главнейший элемент смеси или первый в алфавитном порядке. Например, часто определяют ванадий, марганец и хром в одном растворе: в разделе «Ванадий» описано определение всех трех элементов, а в разделах «Марганец» и «Хром» даны ссылки на раздел «Ванадий».
Азот
Амперометрический метод дает возможность определять азот _в форме различных его соединений — в виде аммиака, МОз", NO2, гидразина, гидроксиламина и т. д. Поскольку в некоторых случа-
100
ях при разложении органических веществ используют образование цианидов, то определение последних дано в настоящем разделе как метод определения азота.
Обычно три определении «органического» азота соответствующие вещества разлагают по методу Кьельдаля и затем титруют образовавшийся аммиак или соли аммония каким-либо окислителем по току его восстановления [1—4]. Впервые амперометрический вариант этого метода был предложен Кольтгофом с сотрудниками [1].
Другие методы, предложенные позднее [2—4], представляют собой по существу лишь варианты метода Кольтгофа (табл. 14).
Нитратный азот, например в нитрующих смесях, можно определять и более простым методом, без предварительного восстановления до аммиака, а именно непосредственно титровать нитрат-ион солью Мора в сильнокислой среде (H2SO4) [5]. Этот метод рекомендуют и другие исследователи [6, 7], считающие его более быстрым, чем методы, основанные на предварительном восстановлении нитрита до аммиака, и особенно ценным для определения малых количеств азота в виде нитрата. Предложено также [8] определение нитрат-иона при помощи ацетата свинца в неводной среде.
Для амперометрического определения нитрит-иона применяют различные окислители, например перманганат, сулыЬаминовую кислоту или церий(IV) [9, 10]. Этот метод, особенно в перманганатном варианте, по сравнению с другими методами [11—14] наиболее прост и удобен.
В последнее время много внимания уделяют определению гидразина путем окисления его различными окислителями и титрования по току окислителя. Чаще всего рекомендуется перманганат [15—18] в различных вариантах. Методы, описанные в работах [15—16], аналогичны, с той только разницей, что в одном случае окислителем является иод, а в другом — бром, образующиеся при взаимодействии перманганата с вводимыми в титруемый раствор бромидом или иодидом калия. Для связывания восстановленной формы марганца (по-видимому, Мпш) добавляют фосфаты. Из других окислителей, рекомендуемых для определения гидразина [19—22], привлекает внимание трикарбонатокобальти-ат-гексаминокобальт(Ш), «универсальный» реактив, обладающий высоким окислительным потенциалом [21] и потому реагирующий со многими восстановителями. Однако в определенных условиях возможно селективное определение того или иного вещества, например гидразина или гидроксиламина.
Для определения азида рекомендовано два метода [24, 25], из которых, по-видимому, предпочтение заслуживает метод, описанный в работе [25].
Цианиды титруют только раствором нитрата серебра [26—30], но^в различных вариантах, подробности которых указаны в таол. 14 в графе «Примечания».
101
~ Таблица 14. Методы определения азота
Определяемое вещество Титрант Определяемый минимум Условия определения Примечания Литература
NH3 NaClO 2Pt электрода, Д<р=0,15 В, боратный буфер, pH - 8,3 Определение NH3 в газах 2
NaBrO 4-Ю-6— 6- ю-« М Электрод Pt, <р=+0,2— +0,15 5 (Нас. КЭ), фон— Na2SO4, NaHCO3, pH «9 Определение в различных объектах, в том числе органических после разложения по Кьельдалю 1, 3
NaClO+KBr 2 Pt электрода Электролитическое восстановление N03 До NH3 4
no; Соль Мора 6 мкг/мл 2 Pt электрода, Л<р=100 мВ, фон — H2SO4 (75%-пая) Охлаждение льдом титруемого раствора, температура не выше 60 °C. Удалять растворен ный кислород. Определение в нитрующих смесях и других объектах 5. 6, 7
Ацетат свинца 20 мг Электрод Hg капающий, анод—ртутное дно, <р =—1,0 В, фон—0,1 М безводный Li Ас в безводном СНзСООН 8
no; КМпО4, сульфаминовая кислота, CeIV 5-Ю-5 М Электрод Pt, <р= + 1,05 В (Нас КЭ), фон—0,05 М H2SO4 Реакция с сульфаминовой кислотой медленна и пригодна для более высоких концентраций. чем с КМпО4; CeIV реагирует почти так же, как К<МпО4 9, 10
Тиокарбамид, обратное титрование КЫОз Электрод Pt, <( - +0,4 В ('МИЭ), фон — 5М H2SO4 Обратное титрование избыт ка тиокарбамида. Рекомендовано для определения NO2_ в серной кислоте 11
As111 Электрод Pt, <р = +0,1 В (Нас. КЭ), фон—NaHCO3, рН=8 NO2~ восстанавливают тио карбамидом в кислой среде до N2; удаляют избыток тиокар- 12
Аскорбиновая кислота Электрод Pt, ф= + 1,0 В, фон—5 н H2SO4
NO7 и NO- KI 2 Pt электрода, А<р=0,1 В
N2H4 КМпО4 или Вг2 Электрод Pt, электрод сравнения Pt в растворе KI, без наложения напряжения, кислая среда+КВг
КМпО4 или 12 0,026 мг/40 мл Электрод Pt, <р=—0,1 В (Нас. КЭ), фон—0,2—0,3 М NaHCO3+l,3 мг К1+0,5—0,6 г Na3PO4
КМпО4 0,12 мг/20 мл Pt электрод, <р= + 1,0 В, фон—lNa2P2O7+H2SO4, рН=6— 7, водно- ацетоновая среда
КМпО4 3,7—5,9 мкг Электрод Pt, <р=0 (Нас. КЭ), фон—0,01—0,06 н. H2SO4+ +0,013—0,15 н. CuSO4+0,8— 1,5% NaF
NaNO2 0,025 мг/40 мл Электрод Pt, по току окисле ния гидразина, фон—3,5—5,5 H2SO4
бамида раствором NaBrO, избыток ВгО- титруют As111
Титруют аскорбиновую кис- 13 лоту исследуемым раствором нитрита. Пользуются отсчетами броскового тока. Титр аскорбиновой кислоты устанавливают по стандартному раствору KNO2
МОз~ — сначала восстанавли- 14 вают до NO2 в Cd-редукторе
Можно определять различные 15 производные гидразина; Вг2— продукт окисления Вг~ перманганатом
12— продукт окисления I-16 перманганатом; вместо КМпО4 можно применять Т1111
На фоне 2М HCl+КВг мож- 17 но определять диметилгидра-зин
NaF для связывания в ком- 18 плекс восстановленного марганца (III)
Гидразин дает 2 анодных 19 волны на Pt электроде, 2,5 М H2SO4, при +0,5 и +1,0 В (Нас. КЭ).
Титруют по этому току, после к. т. по току окисления NO2
Продолжение
Определяемое вещество Титрант Определяемый минимум Условия определения Примечания Литература
ICN или BrCN 3—140 мг Электрод Pt, <р=+0,15 В (Нас. КЭ), рН=4,6—5,0 при титровании раствором ICN и 4,8— 5,3 при титровании раствором BrCN Тем же способом определяют аллилксантат при pH=5,0—5,4 и тиокарбамид на фоне 1 н. H2SO4 20
Трикарбонатоко-бальтнат-гексам-минкобальт (III) 2 вращающихся Pt электрода, Д<р=0,1 В, фон—1 н. H2SO4 +0,5 г КВг Реактив предложен для определения многих элементов в разных средах (см. ниже — гидроксиламин) 21
КВгОз Г 0,03—3,0 мг Электрод Pt, Нас. КЭ, фон— 2 н. H2SO4 или HCl+KBr Метод превосходит по «точности» визуальные варианты с индикаторами 22
nh2oh Трикарбонатоко-б альтиат-гексам -минкобальт (III) 2 вращающихся Pt электрода; Д<р=0,1—0,2 В, фон — 0,5— 10 н H2SO4, или один Pt электрод при 0,9 В (Нас. КЭ) Определение только по способу замещения: добавляют Fe2(SO4)3 (2,5 мл 4%-ного раствора на объем 40 мл) и 2,5 мл H2SO4 (1:2), кипятят, охлаждают и титруют Fel1 (см. раздел «Железо») 21
CeIV, рекомендовано также для определения N2H4, N3 и NO7 Электрод Pt, <р=0 (Нас. КЭ), фон — 5 н. НСЮ4 Рекомендовано прямое титрование NH2OH н азид иона, обратное титрование гидразина и нитрат-иона — избыток Celv титруют раствором оксалата (Примечание авторов книги: метод вряд ли практически удобен) 23
NaNs Cu(NO3)2 2,6—13 мг Электрод Hg капающий, <p= =—0,4 В (Нас. КЭ), фон—ацетат аммония +0,1% желатины Мешают ионы Ag+, Hg+, Pb2+,|24
галогениды, CN-, .->CN_, В20з2-, As3+, SM+, Bi3+
к 0,3—65 мг Электрод Pt, <р=0 (Нас. МИЭ), фон — KNO3, pH=6—9 Титруют избыток I2 тиосульфатом 25
CN~ AgNO3 1,4—-1500 мг/л Электрод Pt, <р=0 (МИЭ), фон — KNO3 Азот переводят в CN- сплав 1 лением вещества с металлическим калием Рекомендовано для органических веществ, вод коксохимического производства 26—30
CN~ CNCT (и Cl") AgNO3 10-3—10-2 м CN" 5-10-3— 2-Ю"2 M CNO" Электрод Pt, <р=0 (Нас. КЭ), фон — 0,1 М KNO3 Последовательное титрование: сначала CN~, затем С1“ (с добавлением желатины), затем в присутствии СН3ОН, при 5 °C — CNO- 28
CN" SCN-CNO" AgNO3 10-3—10-2 M CN~и SCN-5.10-3— 1 • IO-2 M CNO- Электрод Pt, ф=0 (Нас. КЭ), фон — KNOS или Ba (NO3)2 Сначала титруют сумму CN~ и SCN-, затем CNO~; отдельно SCN-, маскируя CN- формальдегидом, избыток его разрушают HNO3, устанавливают рН<3 и титруют SCN- 29
ЛИТЕРАТУРА
1. Kolthoff I. M., Stricks W., Morren L.— Analyst, 1953, v. 78, p. 405.
2. Broz L., Malek J.— Chem. Listy, 1976, v. 70, № 11, p. 1208—1213.
3. Ватутина Г. M., Михайленко А. С. — Химия твердого топлива, 1974, № 1, с. 121—122.
4. Kleinova I., Eadie К-, Re sac Z. — Anal. Chem., 1964, v. 199, № 1, p. 35—39.
5. Williams A. F., Brooks J. Proceedings of the International Symposium on Microchemistry. Birmingham, 1960, p. 430; РЖХим, 1960, 77166.
6. Sandi S., Flanquart G. — Chim. analyt., 1957, v. 30, Kg 1, p. 20—23.
7. Enyedi B., Bencz G., Bagyinszki F. — Mag. Kern. Lap., 1961, v. 16, № 1, p. 131—134.
8. Крешков А. П., Борк В. А., Швыркова Л. А. и др.—Зав. лаб., 1966, т. 32, № 1, с. 10—12.
9. Stock I. Т., Bjork G. — Microchem. J., 1962, v. 6, № 2, p. 219.
10. Stock J. T., Bjork G.— Taianta, 1964, v. 11, № 2, p. 315—318.
11. Кузьмина FL H., Срослова С. И. — В кн.: Методы контроля химического состава неорганических и органических соединений, Куйбышев, Средне-Волжское ЦБТИ, 1966, с. 30—38.
12. Chatterjee S. S., Banerjee В. И. — Technology. (India), 1971, v. 8, № 3—4, р. 285—286; РЖХнм, 1973, 4Г129.
13. Ахмедов Г., Жданов А. К-, Джамалова 3. О. — Узб. хим. ж., 1970, № 5, с .29—31.
14. Mahr М., Pungor Е. — Hung. Sci. Instrum., 1975, № 34, 31—34; РЖХим., 1976, 10Г118.
15. Усвяцов А. А. и др.—Зав. лаб., 1971,. т. 37, № 2, с. 150—154.
16. Жданов А. К, Ахмедов Г. — Изв. вузов. Химия и хим. технол., 1970, т. 13, № 12, с. 1720—1721.
17. Усвяцов А. А. и др.-—Ж. анал. хим., 1974, т. 29, № 1, с. 170—172.
18. Hommam А. М. — Z. anal. Chem., 1975, Bd. 276, № 4, S. 303—306.
19. Жданов А. К, Ятрудакис С. М., Ильина В. Г.—Узб. хим. ж., 1966, № 3, с. 25—28.
20. Ram С. Р., Raj К. С., Ram Р. — Curr. Sci. (India), 1973, v. 42, № 14, p. 501— 503; РЖХим, 1974, ЗГ107.
21. Хадеев В. А., Мухамеджанова Д.—Зав. лаб., 1970, т. 36, № 12, с. 1443— 1446.
22. Sant В. R., Mukherji А. К- — Anal. chim. acta, 1959, v. 20, № 5, p. 476—480.
23. Dziegiec I., Ignaczak M. — Soc. Sci. lodz. Acta chim., 1971, v. 16, p. 169— 175; РЖХим, 1972, 5Г113.
24. Rao A. L., Puri В. К — Z. Anal. Chem., 1969, Bd. 248, № 1—2, S. 33—34.
25. Ikeda Sanae, Metokata lunko. — J. Chem. Soc. Jap., 1975, № 5, 932—934; РЖХим, 1975, 22Г89.
26. Чумаченко M. И., Мухамедшина Р. А. — В кн.: Проблемы аналитической химии. Т. 1. М., Наука, 1970, с. 164—172.
27. Коган Л. А. и др. — Кокс и химия, 1970, № 3, с. 42—45.
28. Муся Соиорио, Нисимура Такао.—Japan Analyst, 1965, v. 14, № 9, р. 795— 803; РЖХим, 1966, 13Г94.
29. Муся Соитиро, Икэда Санаи. — Нихон кагаку дзасси, 1968, v. 89, № 8, р. 767—771; РЖХим, 1969, 11Г107.
30. Икэда Санаи, Муся Соитиро. — Когё кагаку дзасси, 1969, v. 72, № 10, р. 2221—2223, РЖХим, 1970. 8Г141.
Алюминии
Хлюминий (III) не восстанавливается ни на ртутном капельном, ни тем более на платиновом электроде. Поэтому амперометрическое определение его возможно только при помощи реагента, способного давать электродную реакцию на индикаторном электроде, или при помощи электрохимического индикатора (см. гл. I).
Оксихинолин образует с алюминием весьма малорастворимый осадок и восстанавливается на ртутном капельном электроде. Титрование алюминия уксуснокислым раствором оксихинолина было впервые предложено А. М. Занько [1] и осуществлено впоследствии в различных вариантах [2, 3]. Однако гораздо большее значение приобрели производные ЭДТК, в частности ЭДТА [4— 9]. Известно несколько вариантов этого титрования. Например, обратное титрование избытка ЭДТА раствором хлорида железа (III) с платиновым электродом, 'без наложения напряжения при потенциале Нас. КЭ [4]. Этот метод может быть использован для определения алюминия в присутствии магния, так как при pH = 5, при котором рекомендуется титровать, константы нестойкости обоих комплексонатов сильно различаются.
В монографии Стока [5] описан интересный пример определения алюминия, висмута, кальция и магния в смеси их солей.
Другой вариант [6] основан на обратном титровании избытка ЭДТА раствором ванадия (VO2+) на фоне ацетатно-буферной смеси с рН~4 по току окисления ванадия на платиновом электроде при +0,6 В (Нас. КЭ). Интересно, что этот метод может быть применен в присутствии фторидов, поскольку прочность комплексных соединений алюминия с ЭДТА выше, чем его фторидных соединений. В присутствии циркония, который также может быть определен этим методом, титруется сумма обоих элементов.
Предложены новые реактивы [10, 11], позволяющие определять алюминий в присутствии некоторых примесей. В частности, купферон [11] позволяет определять алюминий в присутствии кальция и магния.
Успешно определяют алюминий в присутствии амперометрического индикатора (см. гл. I). Метод осуществлен в различных вариантах [12—18] и применен для определения алюминия в промышленных объектах, например в бронзах [17] ив высоколегированных сплавах на основе никеля [16]. Следует, однако, иметь в виду, что титрование алюминия фторидом, лежащие в основе этого метода, рекомендовано также для определения бериллия, циркония, тория (см. ниже), которые могут, следовательно, мешать определению алюминия.
Известно также определение алюминия при помощи титрования его фторидом натрия без введения индикатора, но на алюминиевом электроде [19—20]. Метод применен для анализа электро-
107
— Таблица 15. Методы определения алюминия (А11п)
СО Титрант Определяемый минимум Условия определения Примечания Литература
Оксихинолин Hg капающий электрод, Нас. КЭ, по току восстановления реактива Рекомендован для определения А1 в природных объектах и в дюралюми-не 1, 2, 3
Комплексоны ЭДТА Электрод Pt, ф=0 (Нас. КЭ), или +0,6 В (Нас. КЭ), фон — ацетатный буферный раствор рН~4—5 Рекомендуется обратное титрование избытка ЭДТА раствором FeCl3 или ванадила. Возможно раздельное определение А1 и Mg, а также последовательное определение Al, Bi, Са и Mg 4, 5
0,1—10 мг/40 мл Электрод карбидо-танталовый, <р= + 0,86 В относительно Pt катода, фон — тартратный буферный раствор, pH=4,5 Косвенный вариант предполагает титрование избытка ЭДТА раствором соли цинка 6
2 Pt электрода, вращающихся, Д<р= = 0—1,2 В, ацетатный буфер, рН= =2—4 Титруют не только А11П, но и Feni 7
2 электрода: один Pt, второй оксидный (Pt, покрытая слоем — Т12О3, N12O3, СО2О3, МпО2, PtO2) Титруют кроме А1 и другие ионы, пользуясь обратным титрованием — ЭДТА титруют определяемым ионом 9
1,2-Диаминциклогексантетр ауксус-ная кислота 2 Pt электрода, Д<р= 1 В, фон — рН=1,5—2,0 (после разложения пробы HNO3); после к. т. вводят СН2С1СОС)Н до рН=В,5—4,0, нагревают до 50—80 °C Определение А1 и Fe при совместном присутствии. Рекомендуется для природных и искусственных силикатов Не мешает Mg и многовалентные катионы 8
.Купферон
Электрод Hg капающий, <р=—>1,4 В, фон — ацетатный буферный раствор, pH=4,6
Са и Mg не мешают
10
Ализарин S
1,25 -10~3, 5-10_3 моль/л
Электрод графитовый, фон — 0,5 М Na2SO4+H2SC>4 рН=4,5—4,8 для определения А1,.рН=3 для CeIV, рН= =4,3 для Сп2+ и Fe®+
Комбинацией pH н прямого и обратного титрования можно определять А1, Се, Си и Fe
NaF
Индикаторный метод с добавлением Fe111 или электрод Pi, <р=0,1 В (Нас. КЭ), 2 Pt электрода, Д<р=0,5 В
Рекомендуется добавлять спирт для 12—17
понижения растворимости осадка A1F3; определение А1 в различных силикатах, сплавах и т. д.
10-4—10-2 моль/л
Электрод алюминиевый, <р=0,0 (Нас. КЭ), фон—ацетатный буферный раствор в 0,5 М KNO3+45% спирта, pH=3—4
При появлении избытка ионов фто- 19, 20
ра ток возрастает за счет растворения материала электрода
Th(C104)4 в присутствии F-
Индикаторный метод с Fe111, элек-
трод Pd, <р= +0,3 В (НВЭ)
Используют различную прочность фторидных комплексов А1 и Th
18
ЛИТЕРАТУРА
1. Занько А. М. — ДАН СССР, 1940, № 6, с. 27—29.
2. Мухина 3. С.—Зав. лаб., 1951, т. 17, № 3, с. 289—292.
3. Жданов А. К-, Кожевникова Н. В. — Зав. лаб., 1952, т. 18, № 4, с. 529—531.
4 Бабенышев В. М., Кузнецова О. М.—Ж. анал. хим., 1960, т. 15, № 4 с. 568—570.
5. Stock J. Т. Amperometric Titrations. New York, Interscience, 1965.
6. Хадеев В. A. — РЖХим, 1974, 10Г57, депон.
7. Vydra F., Stulik К- — J. Electroanal. Chem., 1968, v. 16, № 3, p. 375—383.
8. Vydra F., Stulik K- — Acta geol. et geogr. Univ. Comenianae geol., 1968, № 15, p. 87—89; РЖХим, 1969, 5Г161.
9. Kainz G„ Sontag G. — Z. Anal. Chem., 1974, Bd. 269, № 4, S. 267—275.
10. Lohonyai N., Szabo-Akos Zs.— Period, polytechn. Chem. Engng., 1968, v. 12, № 3, p. 217—222; РЖХим, 1969, 21Г94.
И. Аргова T. В. — Вести. ЛГУ, 1969, № 22, с. 164—165.
12. Усатенко Ю. И., Беклешова Г. Е.—Зав. лаб., 1953, т. 19, № 2, с. 147—150.
13. Беркович М. Т., Сирина А. М., Лагунова Н. Л.—Труды Уральского НИИхим., вып. 11, 1964, с. 26.
Т4. Varma А. — Bull. Chem. Soc. Japan, 1962, v. 35, p. 1444; РЖХим, 1963, 15Г74.
15. Усатенко Ю. И., Беклешова Г. Е.—Зав. лаб , 1954, т. 20, № 3, с. 266—269.
16 Шемякин Ф. М., Беляков Н. И. — Зав. лаб., 1954, т. 20, № 5, с. 552—555.
17. Усатенко Ю. И., Беклешова Г. Е., Гренберг Е. И. и др.—Зав. лаб., 1955, т. 21, № 1, с. 26—29.
18 Breckenridge I. Н., Harris W. Е. — Anal. Letters, 1969, v. 2, №9, р. 473.
19. Алемаскина Г. А., Човнык Н. Г. — В кн.: Методы контроля химического состава неорганических и органических соединений. Куйбышев, Средне-Волжское ЦБТИ,. 1966, с. 47—50.
20. Човнык Н. Г., Алемаскина Г. А.—В кн.: Физико-химические методы анализа металлов, сплавов и электролитических ванн. Б. М. 1975, с. 56—58; РЖХим, 1975, 22Г132.
Барий
Восстановлением ионов бария на ртутном капающем электроде на фоне хлорида тетраметил аммония воспользовались Гейровский и Березицкий [1, 2] для определения ионов бария при помощи титрования его сульфатом лития. Эта работа была вообще первой работой по «полярографическому», как назвал его Гейровский, титрованию. Сульфат лития применяют {3] для определения бария и сейчас, но ведут титрование на фоне этанольно-водного раствора бромида тетраэтиламмония, этим достигается более высокая чувствительность определения вследствие уменьшения рас-^ творимости осадка и улучшения условий восстановления ионов бария на ртутном капающем электроде при 2 В (Нас. КЭ). Метод применим для определения «следовых» количеств бария в радиоактивных растворах.
В пятидесятых годах успехом пользовался хроматный метод [4—6], позволяющий определять барий в рудах в любых количествах, притом в присутствии кальция и стронция.
Образование труднорастворимого карбоната бария также использовано для амперометрического его титрования в присутствии ионов таллия(I) в качестве индикатора [7].
НО
В настоящее время получают распространение различные ва-пианты определения бария при помощи ЭДТК и различных ее Епоизводных [8-11]. Метод в общем привлекателен своей простотой и возможностью работы с платиновыми электродами, но необходимо иметь в виду, что те же реактивы дают комплексные соединения с ионами других элементов (кальция, магния, стронция из группы щелочно-земельных и др.) и поэтому при определении бария приходится соблюдать очень точные условия опыта. В качестве индикаторных применяют платиновый электрод [8, 9] при методе с двумя электродами, вращающийся ртутный электрод [Ю]. В работе [11] изучено титрование бария в присутствии свинца(II), определены рК комплексных соединений ионов бария и других щелочно-земельных элементов с ЭДТА и с другими комплексонами.
ЛИТЕРАТУРА
1. Heyrovsky J., Berezicky S. — Coll. Czech. Comm,, 1929, v. 1, p. 10.
2. Heyrovsky J., Berezicky S. — Chem. News, 1929, v. 138, p. 195.
3. Ziitel E., Miller F. J., Thomson P. F. — Anal. Chem., 1959, v. 31, p. 1351.
4. Васильев A. M., Попель A. A. — В кн.: Труды Комиссии по аналитической химии АН СССР. IV (VII), 1952, с. 126.
5. Калмыкова Е. И. — Зав. 'лаб., 1954, т. 20, № 3, с. 397—399.
6. Ballczo W., Schenk W. — Mikrochim. Acta, 1954, p. 163.
7. Вешева Л. В., Добрянская A. M., Рейшахрит Л. С. — Вестник ЛГУ. 1967, № 164 с. 179—182.
8. Vorlicek J., Vydra F. — Coll. Czech. Chem. Comm., 1966, v. 31, № 6, p. 2510.
9. Srivastava H. R. — J. Indian Chem. Soc., 1976, v. 53, № 2, p. 206—207.
10. Boef G., Freese F., Kramer M., Poppe H. — Taianta, 1970, v. 17, № 10, p. 1006—1009.
11. Sierra F., Perez R., Martinez L., Polo Comacho J. L. — Quim. anal, (pura у apl.), 1975, v. 29, № 6, p. 368—373.
Бериллий
Для бериллия, так же как и для алюминия, предложен индикаторный метод титрования 'фторидом калия [1]. Ход определения такой же, как в случае алюминия, однако в связи с более высокой растворимостью фторида бериллия содержание этанола в растворе необходимо увеличить до 70%, причем кислотность рас-
твора должна соблюдаться особенно строго (pH = 2,1—2,3). Для установления кислотности применяют метиловый оранжевый, как описано для определения алюминия, но используют «свидетель», состоящий из 35 мл 1%-ной соляной кислоты, содержащей 3,2 мл 0,5 н. раствора хлорида кобальта в 1%-ной соляной кислоте и 9,3 мл такого же раствора хлорида железа. Состав осадка соответствует формуле Na2BeF4. Примеси, не мешающие определению алюминия, не мешают и при определении бериллия, но сам алюминии титруется вместе с бериллием, причем расход фторида не соответствует сумме находящихся в растворе бериллия и алюми-ия. Это исключает возможность совместного определения ионов оих металлов и определение одного из них по разности.
111
В растворах же, не содержащих алюминия, амперометрическое определение бериллия может, по-видимому, с успехом заменить длительные гравиметрические методы.
В серии работ, депонированных в ВИНИТИ [2—5], предлагается определять бериллий прямым [2, 3] и косвенным [4, 5] методами при помощи ацетилацетона [2, 4], сульфосалицилата натрия [3] и ферроцианида [5].
Один из современных комплексонов — урамил-И,И-диуксусная кислота — образует с ионом бериллия прочное комплексное соединение [6]' при pH = 5—6, при котором ЭДТА с бериллием не реагирует. Это позволило разработать методику определения бериллия в сплавах бериллий — магний — алюминий — цинк (содержание бериллия 1,64 и 0,97%): ЭДТА связывает все ионы кроме бериллия, который и оттитровывают урамил-М-Ы-диуксусной кислотой по току ее окисления [6].
ЛИТЕРАТУРА
1. Усатенко Ю. И., Беклешова Г. Е.—Зав. лаб., 1953, т. 19, № 7, с. 892 -895. 2. Хадеева Л. А. — РЖХим, 1974, 18763, депон.
3 Хадеева Л. А., Талипов Ш. Т.—РЖХим., 1974, 18Г64, депон.
4. Хадеева Л. А., Джиянбаева Р. X. — РЖХим., 1974, 8Г134, депон.
5. Хадеева Л. А., Джиянбаева Р X. — РЖХим., 1974, 9Г47, депон.
6. Галлай 3. А., Зверяева И. В.—Ж. аналит. хим., 1971, т. 26, № 12, с. 2340— 2342.
Бор
Самый простой метод определения бора [1, 2, 3] заключается в титровании борной кислоты в присутствии маннита или инвертного сахара (как и при обычном титриметрическом определении борной кислоты) раствором едкого натра с платиновым платинированным электродом при потенциале +0,55 В (Нас. КЭ). Появление анодного тока в точке эквивалентности связано с окислением ОН_-ионов.
Японские исследователи предложили прямое титрование борной кислоты фруктозой на фоне хлорида лития [4] или хлорида кальция [5]. Метод довольно сложен, так как требует удаления растворенного кислорода. Титруют по току восстановления фруктозы, Ei/2 которой на ртутном капающем электроде составляет —1,7 В.
Предложены также методы определения других форм бора в растворе — тетрафенилбората [6], борогидрида [7] и смеси борной кислоты с бурой или серной кислотой [8].
Предложено титровать борную кислоту раствором хлорида кальция [9]. Условия титрования несколько необычны: оба электрода, опущенные в титруемый раствор, — медные, один из них (катод) амальгамирован. Кривая титрования имеет три излома,
112
отвечающих, по предположению авторов метода, последовательному оттитровыванию ионов водорода борной кислоты. Общий ход кривой аналогичен кривой кондуктометрического титрования, что дает авторам метода основание для объяснения хода кривой изменением электропроводности раствора.
ЛИТЕРАТУРА
1 Боговина В. И., Селиванов В. Г. — Зав. лаб., 1958, т. 24, № 12, с 1200.
2 Родионова М. А., Мун А. И.—Труды ин-та хим. наук АН КазССР. 1969, ' т. 25, с. 68—69.
3. ^л.едов Г., Жданов А. К-—Учен. зап. Душанбинского гос. пед. ин-та, 1976, вып. 99, с. 118—121.
4. Ishibashi М^ Fujinaga Т., Nagai Т. — Bull. Inst. Chem., Res., Kyoto Univ., 1958, v. 36, p. 134; Stock J. T. Amperometric Titrations, New York, Interscience, 1965, p. 365.
5 Ishizuka T., Sunahara H., Kato E. Бунсеки кагаку, Jap. Analyst (яп.), 1971, ’ v. 20, № 7, p. 818—823; РЖХим, 1972, 4Г228.
6 Ikeda Sanae, Hirata Junko. — Нихон кагаку кайси; J. Chem. Soc. Jap. Chem. ’ and Ind. Chem., 1973, № 5, p. 953—957; РЖХи..., 1972, 1Г63.
7. Liberti A., Lazzari P. — Ric. Sci., 1956, v. 26, p. 825; Stock J. Amperometric Titrations, New York, Interscience, 1965, p. 612.
8. Ким Хен Нак, Цой Ku Ок. — Чосон минчучуи инмин конхваквон тхонбо, 1965, № 4, с. 26—32; РЖХим, 1966, 23731.
9. Борк В. А., Сальникова К- С.—Ж. анал. хим., 1974, т. 23, № 6, с. 901—-907.
Бром
Для определения бромид-иона предложено несколько вариантов аргенто- или меркуриметрического методов. Поскольку все эти варианты предусматривают одновременное определение других галогенов, притом при преобладающем содержании хлорид-иона, описание этих методов дано ниже, в разделе «Хлор». От этих «классических» методов отличается метод определения бромид-иона с применением медного амальгамированного электрода в среде безводной уксусной кислоты [1]. Бромид (и хлорид, если титруют смесь) осаждают титрующим раствором — ацетатом кадмия в виде бромида (или хлорида) кадмия, нерастворимого в уксусной кислоте. Раствор ацетата кадмия предложен также для титрования бромида на фоне уксусной кислоты, но с платиновым электродом [3]. Гипобромит-ион, в числе прочих окислителей, предложено титровать раствором перекиси водорода [3] или раствором соли ванадия (IV) [4] в присутствии бромита ВгОг; для определения последнего его восстанавливают раствором фенола. Определение ведут в щелочных растворах брома. Практическое приложение этих методов не описано.
ЛИТЕРАТУРА
Б1-, Борк В. А., Сальникова К С.—Ж. анал. хим., 1967, т. 22, 2 № 9, с. 1392-1397.
ии^К &едухина Г. П. — Труды Московского химико-технологического
-та им. Д. И. Менделеева, 1967, вып. 54, с. 119—121.
8-1694 , . о
3. Nucci L., Raspi G. — Ric. Sci., 1969, v. 39, № 4—6, p. 400—404.
4. Сахаров А. А., Сальникова P. Д.—Учен. зап. Петрозаводского ун-та, 1966/ /1967, т. 14, № 3, с. 96—102.
Ванадий
Определение ванадия(V) основано на реакциях окисления — восстановления, причем наиболее распространенным является метод амперометрического титрования ванадия (V) солью Мора по току окисления последнего на платиновом вращающемся электроде. Этот метод был предложен И. П. Алимариным и Т. К. Кузнецовым и вслед за ними Г. А. Бутенко и Е. С. Рынской для определения ванадия, хрома и марганца в легированных сталях [1]-
Затем вышла работа И. П. Алимарина и Б. И. Фрид [2] по приложению этого же метода к микроопределению ванадия и хрома в минералах, рудах и горных породах и ультрамикровариант— титрование 2,5-1 СП8 г ванадия (V) в объеме 0,3 мл на предметном столике микроскопа при помощи вибрирующего платинового электрода [3].
Амперометрическое определение ванадия (V) с помощью соли Мора имеет существенные достоинства: оно может быть применено в присутствии многих других ионов — меди, никеля, свинца, цинка и других элементов, позволяет определять не только ванадий, но три компонента (ванадий, хром, марганец) в одной навеске, дает результаты высокой точности, пригодно для определения весьма малых количеств ванадия, хрома и марганца (десятые и сотые доли миллиграмма в титруемом объеме раствора), может быть выполнено в мутных и окрашенных растворах.
Предложено несколько вариантов этого метода для определения ванадия в различных объектах: в металлическом ванадии, в хромите, в урансодержащих веществах. По-прежнему много внимания уделяют этому методу при анализе легированных сталей. Предложен этот метод и для определения ванадия и хрома в си-лико-алюминиевых катализаторах крекинга нефти, причем вместо обычного'в таких случаях селективного окисления хрома(III) его восстанавливают до трехвалентного при помощи азида натрия; хром(III) не мешает титрованию ванадия солью Мора.
Ниже дан «классический» ход определения ванадия (V) солью Мора в стали и в рудах [2].
Ход анализа. Навеску стали разлагают 30—40 мл серной кислоты (1:5), добавляют азотную кислоту, выпаривают до появления паров серной кислоты, разбавляют водой до 100—120 мл. Масса навески зависит от содержания ванадия и хрома; при содержании их меньше 1% — 1 г, до 6% —0,5 г, выше 6% — 0,2 г. При анализе руд и горных пород сплавляют навеску с пнросульфатом; если проба содержит много кремнекислоты, то желательно сначала удалить ее фтористоводородной и серной кислотами и остаток сплавить с пиросульфатом.
Пиросульфитный сплав растворяют в серной кислоте с таким расчетом, чтобы концентрация ее составляла от 0,1 до 0,5 н. Если определению подлежит 114
ько ванадий, то его окисляют на холоду перманганатом (температура долж-Т°быть выше 20 °C), добавляя последний до появления слабо-розовой окраски Няствора Избыток перманганата удаляют 0,2%-ным раствором нитрата натрия, я избыток последнего — карбамидом (0,2—0,5 г). Титруют 0,1 (или 0,01) М раствором соли Мора при потенциале +1,0 (Нас.КЭ)—кривая формы б.
Нитрит-ион, добавляемый для удаления избытка перманганата, является восстановителем по отношению к ванадию (V) и, следовательно, может влиять на результаты титрования ванадия(V) раствором Fe11. Специальное исследование кинетики этой реакции [4] показало, что она зависит от концентрации нитрита и серной кислоты и что поэтому лучше вообще обходиться без добавления нитрита, очень осторожно добавляя перманганат, не допуская его избытка. Если же добавляют нитрит натрия, то его количество контролируют по показанию гальванометра, прекращая добавление в момент минимального показания.
Если одновременно определяют и ванадий и хром, то окисление их проводят персульфатом в присутствии нитрата серебра (кислотность раствора не должна быть выше 2 н.), удаляют избыток персульфата кипячением. Для устранения влияния марганца (VII), образовавшегося в результате окисления марганца(II) в пробе, добавляют 5 мл 5%-ного раствора хлорида натрия или несколько капель концентрированной соляной кислоты, кипятят 3 мин, охлаждают и титруют солью Мора сумму ванадия и хрома. Затем окисляют восстановленный при титровании ванадий перманганатом на холоду, как указано выше, и титруют только ванадий(V), так как хром(III) в этих условиях не окисляется. Содержание хрома определяют по разности.
Если определяют не только ванадий и хром, но и марганец, то окисление проводят сначала персульфатом в присутствии нитрата серебра, как указано выше (хлорид в этом случае добавлять не следует), и титруют охлажденный раствор солью Мора, определяя таким образом сумму всех трех металлов. Затем снова окисляют персульфатом, далее поступают, как указано выше, и титруют сумму хрома(VI) и ванадия^). После этого окисляют ванадий (IV) перманганатом и титруют только ванадий (V). Хром и марганец определяют по разности.
Железо(III) и хром(III) не оказывают влияния на точность определения ванадия. Часто рекомендуемая для связывания железа (Ш) фосфорная кислота, необходимая при визуальном титровании, .при амперометрическом варианте не нужна. Наоборот, при содержании фосфорной кислоты в титруемом растворе свыше 3 Л! наклон кривой после конечной точки становится пологим вследствие увеличения вязкости раствора, а это затрудняет установление конечной точки титрования [6].
Реакция между ванадием(V) и железом (II) протекает в рас-ворах серной кислоты быстро и строго стехиометрично, не за-нася от формы существования ванадия^) в растворе (катион-’ ПОливанадаты). Это позволяет пользоваться так называемым
8*
115
«теоретическим титром» раствора соли Мора, устанавливаемым по стандартным растворам перманганата [7]. Описываемый метод титрования ванадия, хрома и марганца раствором соли Мора был применен для анализа сталей в упрощенном варианте — без наложения напряжения на платиновый электрод при перманганатном электроде сравнения [8].
Во многих случаях при определении ванадия (V) с помощью соли Мора применяют два индикаторных электрода, поскольку система Fe3+/Fe2+ хорошо обратима (см. гл. IV). Работ в этом направлении известно много [1, 9—13, 14], но различаются они только некоторыми деталями в подготовке пробы, в кислотности раствора, сущность же метода одна и та же.
Титриметрическое определение ванадия (V) раствором соли Мора давно пользуется большой популярностью, обычно его выполняют в визуальном варианте с фенилатраниловой кислотой в качестве индикатора. Недавно было поставлено специальное исследование [22]—сравнили три варианта этого метода: визуальный, потенциометрический и амперометрический. Последний оказался наилучшим по ряду локазателей.
Вместо соли Мора можно применять ферроцианид калия [15, 16], являющийся восстановителем ванадия (V) при pH =С4. Лучшей средой для этой реакции следует признать серную кислоту с рН~1, в которой реакция протекает практически мгновенно, при повышении pH скорость реакции убывает и при pH = 4 реакция превращается, так как реальные потенциалы обеих систем становятся равными [15, 17].
Методика определения ванадия (V) с помощью ферроцианида калия очень проста [15, 17]:
0,1—0,5 г пробы (например, шлака) растворяют в 5—7 мл HF, слегка нагревают, прибавляют еще столько же HF и 10 мл H2SO4 (1 : 4), нагревают до появления белых паров; по охлаждении разбавляют водой, прибавляют 1—2 мл азотной кислоты, упаривают до появления белых паров и переносят в мерную колбу емкостью 250 мл. Для полного окисления ванадия добавляют раствор перманганата до появления желтой окраски раствора. Аликвотную часть 5—10 мл разбавляют до 20 мл 1 М раствором H2SO4 и титруют 0,01 М раствором ферроцианида калия при потенциале +1,0 В (МИЭ) по току окисления ферроцианида (кривая формы б).
Путем подбора pH на основании изучения зависимости реальных потенциалов и скорости реакции от кислотности среды выбраны условия, позволяющие титровать ферроцианидом марганец (VII), хром(VI) и ванадий(V) без повторного окисления и восстановления компонентов [18]. Достаточно создавать определенную среду при титровании каждого из трех элементов: марганец (VII) быстро и количественно реагирует с ферроцианидом при любых значениях pH, хром(VI)—при рН^З,5, а ванадий(V) только в кислой среде. Поэтому в одной аликвотной части при pH = 7,5 оттитровывают марганец, в другой при pH = 3,5 — марганец и хром, в третьей — ионы всех трех элементов на фоне 1 н. серной кислоты.
1 16
На этом же принципе — изучении влияния состава и pH фона значение окислительно-восстановительного потенциала реаги-Нующих систем и на скорость их взаимодействия — основан метод определения ванадия, хрома и марганца в высшей степени окисления при помощи иодида калия [19]. Восстановление марганца (VII) протекает быстро и количественно, уже начиная с концентрации серной кислоты 0,05—0,1 М; хром(У1) требует более высокой кислотности, порядка 4 М, и для ускорения реакции добавления Н3РО4, связывающей образующийся хром(III) в комплексное соединение; для восстановления ванадия (V) с достаточной для титрования скоростью необходима 6—8 М серная кислота. Следовательно, определению марганца (VII) не будут мешать ни хром(VI), ни ванадий(V), хром(VI) будет титроваться совместно с марганцем (VII), а ванадий(V)—совместно с марганцем (VII) и хромом (VI) при высокой кислотности раствора.
Ход анализа. Навеску (0,1—0,2 г) анализируемой стали разлагают смесью кислот, состоящей из 25—30 мл H2SO4 (1 :4), 5—10 мл концентрированной НзРО,- и 1—2 мл HNO3 (1 : 1). Раствор упаривают до выделения густых паров серной кислоты, остаток разбавляют водой до 80—100 мл, добавляют 1—1,5 мл 1%-ного раствора AgNOs, нагревают до кипения и прибавляют порциями по 0,1 г твердой (NH4)2S2O8 до появления малинового окрашивания, после чего нагревают на песочной бане до полного разрушения избытка персульфата (до прекращения выделения пузырьков кислорода). Раствор охлаждают, переносят в-мерную колбу емкостью 200—250 мл, приливают 0,5—1 мл 0,5%-ного раствора КС1 (для связывания ионов серебра), разбавляют до метки водой и отбирают три одинаковых аликвотных части раствора по 5—10 мл. В одну из них приливают 0,1 М H2SO4 до объема 20—25 мл, добавляют 1—1,5 г твердого K2SO4 и титруют марганец(VII) амперометрически 0,01 н. раствором KI при потенциале графитового электрода +0,6 В (Нас.КЭ). К другой аликвотной части раствора приливают 4 М H2SO4 до объема 20—25 мл, 1—2 мл концентрированной НзРО4 и оттитровывают сумму марганец(УП) и хром (VI). К третьей аликвотной части приливают 8 М H2SO4 до объема 20—25 мл, добавляют 0,3—0,65 г твердого KF для связывания железа(Ш) во фторидный комплекс, так как оно при высокой кислотности фона может частично реагировать с иодидом, и оттитровывают сумму Mnvn, CrVI и Vv. По разности находят количество CrVI, Vv.
Для определения ванадия (V) можно применить и другие восстановители, в частности аскорбиновую кислоту [20, 21].
Методы осаждения для амперометрического определения ванадия (V) фактически не применяют. Известна всего одна работа [23], рекомендующая осаждать ванадий (V) в виде ортованадата меди, А^тод вряд ли найдет практическое применение.
Ванадий (IV) можно определять по методу окисления или восстановления и по методу осаждения. В качестве окислителя рекомендованы растворы церия(IV) на фоне серной кислоты [24], марганца(III) [25] и иод [26] (табл. 19). Ниже приведена методика определения ванадия(IV) [24].
30—4(?Д анализа> Навеску феррохрома 0,5—1,0 г растворяют при нагревании в До п МЛ сеРн°й кислоты (I : 4), добавляют немного азотной кислоты, кипятят бавл°ЯВЛеНИЯ „паРов- Раствор переносят в мерную колбу емкостью 100 мл, раз-новым°Т вод°й Д° метки. Аликвотную часть раствора (25 мл) титруют с плати-0,004М вРащающимся электродом при потенциалах от +0,8 до +0,9 В (Нас.КЭ) н. раствором сульфата церия(IV). Содержание ванадия в феррохроме со-
117
ставляло от 0,11 до 0,20%. Ошибка определения не превышала тысячных долей процента.
Определяют ванадий(IV) и при помощи окисления его раствором тетраацетата свинца (IV) с ртутным капающим [27] или с двумя платиновыми электродами [28].
Для определения ванадия(IV) по методу восстановления рекомендуются растворы ванадия(II) [29] или титана (Ш) [30]. Последние два метода могут оказаться полезными в некоторых специальных случаях, применение же их в широком масштабе при массовых определениях в производственных лабораториях затруднительно вследствие неудобства работы с легко окисляющимися растворами ванадия(II) и титана(III).
Ферроцен [31] также восстанавливает ванадий(IV) до ванадия (III), но фон должен быть водно-спиртовым, содержащим обязательно фосфорную, серную или соляную кислоту. Спирт может быть любой — метиловый, пропиловый, можно пользоваться и муравьиной и уксусной кислотами, ацетонитрилом, ацетоном. Раствор ферроцена готовят на этиловом спирте. Титруют по току окисления ферроцена на платиновом или графитовом электроде. Определению мешает железо(III), которое надо удалять экстракцией. Это обстоятельство, равно как и необходимость пользоваться органическими растворителями, значительно снижает практическую ценность метода для производственных лабораторий.
Для окислительно-восстановительного титрования ванадия (IV) в присутствии ванадия (V) рекомендован метод с двумя платиновыми индикаторными электредами: сначала титруют ванадий (V) солью Мора, затем ванадий(IV)—перманганатом, вычитая из общего количества ванадия (IV) ту его часть, которая отвечает восстановлению ванадия(V) на первом этапе титрования [32]. Этот метод был применен для определения ванадия (V) и (IV) в ванадиевых катализаторах [33].
Несколько отличен от других метод амперометрического титрования ванадия раствором тиооксина [34, 35]. Особенность его заключается в следующем: при титровании в кислых растворах и в растворах, pH которых не превышает 4, ванадий(V) восстанавливается тиооксином и может быть беспрепятственно оттитрован (кривая титрования имеет форму б) в присутствии ванадия (IV), который в этих условиях с тиооксином не реагирует. Точка эквивалентности отвечает мольному отношению ванадия к тиооксину, равному 1:1. Если же вести титрование при pH больше 5, то восстановление ванадия (V) не происходит, а выпадает осадок зеленого цвета, имеющий состав VOsCgHeSN (т. е. мольное отношение ванадия к тиооксину составляет 1:1, как и в предыдущем случае). Титрование идет также по кривой формы б, но в этом случае ванадий (IV) титрованию мешает, так как он также осаждается тиооксином при указанном pH, образуя осадок неопреде' ленного состава. Практическая методика, йроверенная на стандартных образцах, приведена ниже.
118
„ анализа. Навеску ферросплава около 0,1 г растворяют в 40 мл серной Л 1 (1'4) при нагревании. Полученный раствор переносят в мерную колбу КПСЛ°тью Л| 250 мл, доводят до метки водой и отбирают аликвотную часть ччК°л в коническую колбу или стакан емкостью 50—100 мл; добавляют по кап-2° м0 1 н раствор перманганата [для дополнительного окисления ванадия(ГУ)] ЛЯМпоявлёния неисчезающей в течение 1 мин розовой окраски. Для удаления Д°бытка перманганата прибавляют несколько капель соляной кислоты (1:1) и И пятят около 10 мин для удаления хлора. По охлаждении добавляют около (П г сухого фторида аммония [для связывания железа (III)] и титруют 0,1 М раствором тиоксина при потенциале +1,1 В (МИЭ). Кривая титрования фор-Р а. ] мл 0,1 М раствора тиооксина отвечает 5,095 мг ванадия.
Этот метод разработан для определения больших количеств ванадия — порядка 40—50%; однако его можно применять и для любых других содержаний, соответственно уменьшив концентрацию титрующего раствора и увеличив навеску. Наименьшее количество ванадия, поддающегося определению при помощи тиооксина,— 60 мкг в 20 мл раствора.
Взаимодействие ванадия (V) и (IV) с тиооксином было изучено также на фоне 2 н. ацетата натрия [36]. При титровании на этом фоне осадок (состава 1 :2) образует не только ванадий (IV), но и ванадий (V), состав этого осадка 1:1. На кислом фоне ванадий (V) может быть последовательно оттитрован в присутствии железа и меди или молибдена [37].
Для определения ванадия (V) применяют также, помимо тиооксина, другие органические реактивы: дитиобиурет [38], требующий введения ионов меди(II) в качестве катализатора; а-наф-тиламин [39], позволяющий определять до 104 М ванадия^); карбоксиметилдитиокарбаминаты кальция и натрия [40, 41]. Вообще органические реактивы рекомендуются для определения ванадия в разных степенях окисления. Так, можно воспользоваться ЭДТА [42]: константы нестойкости комплексонатов ванадия (III) и ванадия(IV) различаются почти на порядок (в 7 раз), и это позволяет последовательно титровать при рН = 3—4 сначала ванадий(III), а затем- ванадий(IV). Для определения ванадия в степени окисления III, IV и V рекомендуется бензгидроксамовая кислота [43], а также эриохромцианин и галлоцианин, образующие комплексные соединения с ванадием(III) и (IV) [44]. Ванадий (III) образует комплексное соединение с тироном [45], и его можно определять, титруя раствор на фоне ацетатного буферного раствора по току восстановления ванадия(III) на ртутном капающем электроде; ванадий (IV) при этом не мешает. Практиче ски не мешает ванадий(IV) и при титровании ванадия(III) раствором бихромата на фоне 1 н. серной кислоты [46]: ванадий(Ш) реагирует с бихроматом почти мгновенно, тогда как ванадии (IV)— очень медленно и не количественно, даже при повыше-ии температуры. Полного окисления ванадия (IV) можно достиг-по ftv Фоне смеси 1 н. серной кислоты и 3 н. фосфорной кислоты ^ри 80 С. Поэтому при комнатной температуре в 1 н. серной кис-Одн ванадий(Н1) можно определять в присутствии ванадия(IV).
ако, если содержание ванадия (IV) превышает содержание ва-
119
J3 Таблица 16. Методы определения ванадия
Определяемое вещество Титрант Определяемый минимум
W Соль Мора (Fe11)
/ Микрограммы
K4Fe(CN)6 Сг—0,1—3,5 мг, Мп — ОД—2,3 мг, V—0,04—2 мг
Условия определения Примечания Питера -гура .
Электрод Pt, <р= + 1,0 В (Нас. КЭ), фон—H2SO4 Известно несколько вариантов этого метода, позволяющих определять V в присутствии Fe, Сг, Мп и других элементов или V, Сг, Мп при совместном присутствии 1, 3
Электрод Pt, электрод сравнения — раствор перманганата в H2SO4, <р=0 Вариант того же метода, предложен для определения V. Сг, Мп в стали 8
2 электрода Pt, Д<р=0,6 В, фон — H2SO4+H3PO4 Определение только ванадия или хрома и ванадия в двух аликвотных частях: сумма Сг+ 4-V и только V 9, 10
2 электрода: один Pt, второй из различных металлов; Лер зависит от металла второго электрода Определение V, Сг, Мп можно осуществлять, заменяя один Pt электрод электродом из W, Mo, Си, Ag и других металлов только в качестве катодов 11
2 Pt электрода, Д<р=0,6— 0,9 В, фон— 3- 6 н. H2SO4 Предложен для определения V, Сг и Мп в почвах, сплавах и силикатах 12, 13
2 Pt электрода, Д<р=0,1 В, фон—0,2 М H2SO4 для V, 2,5 М H2SO4 для Се Титрование Vv и CeIV при совместном присутствии 14
Электрод Pt, <р= 4-1,1 В (Нас г\Э), фрн- H2SO4: 2 н для CrVI, 0,5 1н. для MnVI1, 4—5 н. для V'* Метод разработан на чистых растворах, для отдельного определения V, Сг и Мп порознь 1 15
2 мкг/мл Электрод Pt, q>= -4-11,0 В Можно определять Vv в при-|16
(МИЭ), фон —2 н. H2SO4 сутствии избытка V. Метод! пригоден для промышленных] объектов, в частности для шла-| ков (см. методику в тексте)
Мп — 0,01 мг, Сг — 0,02 мг, V-0,05 мг Электрод Pt, ср= 4-0,84-1,0 В (Нас. КЭ), фон: для MnVI! рН=7,5, для суммы Mnvn и CrVI рН=3,5, для суммы всех трех компонентов — 1 М H2SO4 Отдельное титрование каждого компонента или определение всех трех при совместном присутствии в трех аликвотных частях, Fe111 устраняют фторидом 18
KI Мп — 0,01 мг, Сг— 0,04 мг, V — 0,05 мг Электрод графитовый, ф= = 4-0,6 В (Нас. КЭ), фон — 0,05 М H2SO4 для Мп, 3 М H2SO44-H3PO4 для Сг, 6 М H2SO4 для V Рекомендуется для определения Мп, Сг, V в стали (см. методику в тексте) 19
Аскорбиновая кислота Электрод Pt, ср= 4- 0,854-4-1,0 В (Нас. КЭ), фон—1 н. H2SO4 Не мешают элементы, не восстанавливающиеся аскорбино вой кислотой 20
2 Pt электрода, Д(р=0,1 В. фон — H2SO4, рН= 1,0 Кроме Vv определяют CeIV на фоне 3,5 М H2SO4 и Movl на фоне ацетата натрия, рН~6 21
Дитиобиурет 0,1—20 мг Электрод Pt, (р= 4-0,7 В (Нас. КЭ), по току окисления титранта, фон НС1 3 н. Необходим катализатор — Си2+, определяют Vv в шлаках, сталях и феррованадии 38
а-Нафтиламин 5-Ю-3—2,5-10-4 М Электрод Pt, Ф=0,2 В (Нас. КЭ), фон —8—10 М H2SO4 Применен для анализа стали, кроме V определяют Мп 39
Карбоксиметил -дитиокарбаминат кальция или натрия <2 Pt электрода; Д<р=0,25—0,5 В, фпн -7- 16 н. H2SO4 или 3—6 н. НС1, оптимальный фон— 5 н НС1 Отношение Vv:'peareHT= 1 1, определяют Vv в чугуне и стали. Так же определяют CrVI 40, 41
Продолжение
Определяемое вещество Титрант Определяемый минимум Условия определения Примечания Литера тура
Тиооксин Электрод Pt, (р= + 1,0 В (Нас. КЭ), фон — H2SO4, НС1, HCl+iNaAc. Определение Vv и, в зависимости от среды, также Fe, Си или Мо по трем изломам на кривой титрования 37
CuSO4 0,2 М Электрод Hg капающий, q>=—0,2 В, фон—0,1 М KNO3+ +желатина и 25% спирта, 60 °C Осаждение ортованадата меди 23
Viv Марганец (III) Электрод Pt, ср =+0,8—0,9 В (МИЭ), фон —5 М H2SO4 В присутствии Fe11 сначала титруется именно Fe11, затем VIV, форма кривой анодно-катодная-—по току окисления Fe11 и восстановления избытка Мп111 25
12 2 Pt электрода, Лс[.=0,02Ь В, фон, — аммиачный буферный раствор, pH=9—10 26
PbAci" 5-10-6 моль/л 2 Pt электрода, А(р=50 мВ, фон— 1—6 н. НС1 Определение ванадия и Fe в алюмованадиевом сплаве, бронзе и латуни 28
Ферроцен 50 мкг/30 мл Электрод Pt или графитовый, перманганатный щелочной электрод сравнения (ф = +0,72 В) без наложения напряжения, по току титранта, фон — водно-органический, содержащий НС1 или H2SO4, Н3РО4 Мешают Fe3+, MoVI (см. текст) 31
yv и Vv I Соаь Мора и 1 КМпО4
yin ТиООКСЩ! Бензгидрокса-мовая кислота Тирон 60 мкг/20 мл 0,1—2 мг
VIII и to СлЗ Viv К2СГ2О7 0,13—1,7 мг/20 мл
2 Pt электрода, Дф=0,4 В, фон — 1 М H2SO4
2 Pt электрода, Хер = 0,4 В, фон — 1 М H2SO4
Сначала титруют солью Мора 32 Vv, затем VIV перманганатом (см. текст и кривую титрования на рис. 36)
Приложение метода [29] к ва- 33 надиевым катализаторам, рекомендовано также определение VIV в присутствии V111 (см текст)
Электрод Pt, ф = + 1,1 В (МИЭ), фон — сернокислый
Электрод Pt, ф= + 1 В (Нас КЭ), фон —2 н. NaAc *
Электрод Hg капающий, при титровании по току восстановления титранта или графитовый при титровании по току окисления
В зависимости от среды и pH возможно определение Vv в присутствии VIV (см. методику)
34
Ванадий (V) и (IV) осаждаются тиооксином на фоне 2 н. NaAc. Титрование по току окисления тиооксина
С титрантом реагируют: Vv— два соединения состава 1 :1 и 1 2 с рК~10~9 и 10~16; V111 и VIV по одному соединению состава 1 : 1 с рК« 10~10 и 10~8
43
соответственно
Электрод Hg капающий. Ф = —1,1 В (Нас. КЭ), по току восстановления V111, фон — ацетатный буферный раствор. рН = 4
Электрод Pt, ф = + 0,3 В (МИЭ), фон—1 н H2SO4
Viv Vin
не мешает определению 45
Титрование V111 в присут- 46
ствии пятикратного количества Viv
Продолжение
Определяемое вещество Титрант Определяемый минимум Условия определения Примечания Литература
VIII и VIV К2СГ2О7 для V111 CrSO4 для VIV 0,27—2,7 мг Электрод графит (Нас. КЭ). ср =+0,3 В при титровании V111 в токе азота на фоне 1 н. HaSO4 раствором К2СГ2О7, <р= = +0,4 В при титровании V1V тоже в токе азота на фоне 6 н. H2SO4 Определение V111 и VIV в стеклах и шпинелях 47
КМпО4 0,5 мг/20 мл Электрод Pt, ср =+0,5 (МИЭ), фон — 4—5 М НгВО4 Отдельное и последовательное титрование V111 и VIV, вместо Нг5О4 можно применять HCI, КС1, KNO3 48
ЭДТА Электрод Pt вибрирующий, ср= +0,9 В (Нас. КЭ), фон — H2SO4, pH=3—4 Два перегиба на кривой титрования, отвечающие к. т. ванадия (III) и ванадия (IV) 42
yin, VIv, vv Эриохромцианин R Галлоцианин 1,14-IO-5 М Электрод Pt, ср =+0,64 В (Нас. КЭ), фон-0,4 М КС1, рН=2,4 V111 образует 2 комплексных соединения с обоими реагентами состава 1:1 и 1:2. Аналогично реагирует Ti111 44
р 43 Кривая титрования смеси ванадия(III) F /IV) 0,1 н. раствором перманганата калия на фоне 5 М H2SO4.
I
25
15
0,1
Q8 0,5 0,7 мл
надия(Ш) более чем в 5 раз, ошибка может стать заметной вследствие частичного взаимодействия ванадия (IV) с бихроматом, особенно при медленном титровании.
Известен вариант этого метода [47]: ванадий(III) титруют бихроматом на фоне 1 и. серной кислоты, как описано выше, а ванадий (IV) титруют
новителем —раствором сульфата хрома (II), дий(Ш) не взаимодействует. Авторы этого метода рекомендуют пропускать через титруемый раствор азот во избежание окисления хрома(II), хранить который надо, разумеется, тоже в особых ус-
не окислителем, а восста-с которым вана-
ловиях.
Беспрепятственно титруются ванадий(III) и (IV) перманганатом, причем форма кривой титрования зависит от состава и pH фона [48], при этом с одинаковым успехом можно применять серную, соляную кислоту, растворы хлорида или нитрата калия. На фоне 5 М серной кислоты на кривой титрования отчетливо получаются две конечные точки (рис. 43), отвечающие последовательному оттитровыванию сначала ванадия (III), затем ванадия (IV). Подъем тока между первой и второй конечными точками объясняется катодным восстановлением образующегося при окислении ванадия(IV) ванадия (V), поскольку титрование веДут при +0,5 В (МИЭ), т. е. при потенциале, отвечающем появлению катодных токов ванадия (V).
ЛИТЕРАТУРА
1. Сонгина О. А. Амперометрическое (полярометрическое) титрование. М., Химия, 1067, с. 387.
2. Алимарин И. П., Фрид Б. И. — Зав. лаб., 1952, т. 18, с. 1300—1303.
3. Петрикова М. И., Алимарин И. П.—Ж. анал. хим., 1957, т. 12, с. 462—464.
4. Сонгина О. А., Бектурова Г. Б. — Зав. лаб., 1969, т. 35, № 12, с. 1433—1435.
5. Kekedy L., Makkay Er. — Studia Univ. Babeg—Bolyai, 1966, v. 11, № 1, P- 21—31; РЖХнм, 1967, 8Г151.
Ь. Сонгина О. А., Бектурова Г. Б. — Труды ВсесоюзНИИ стандарт, образцов и спектр, эталонов, 1968, № 4, с. 119—122.
'• Сонгина О. А. и др.—Труды ВсесоюзНИИ стандарт, образцов и спектр, эталонов, 1968, № 4, с. 112—119.
» Иванова И. Сонгина О. А, —Зав. лаб., 1970, т. 36, № 5, с. 540-541.
in ^иленко А- Я—Укр. хим. ж., 1967, т. 33, с. 632—635.
’ ;^ленко А- И-—Изв. вузов. Химия и хим. технол., 1965, т. 8, № 3, с. 397—
11 401’
’ А. И.—Ж. аналит. хим., 1967, т. 22, № 1, с. 161—164; 1964, т. 19,
№ 6, с. 709—713.
125
12. Жданов А. К., Маслова В., Ятрудакис С. М. — РЖХим, 4973, 1'1'Г65. дел.
13. Астафьев В. В., Загинайченко Н. И. — В кн.: Методы анализа руд Кольского полуострова. Апатиты, 1970, с. 137—142.
14. Singh D., Varma А. — J. Scient. Res. Banares Hindu Univ., 1964—1965, v. 15 № 1, p. 68—72; РЖХим, 1966, 10Г60.
15. Жданов А. K-> Ахмедов Г. — Научные труды Ташкентского ун-та. 1972, вып. 419, с. 43—49.
16. Сонгина О. А., Бектурова Г. Б., Захаров В. А. — В кн.: Химия и химическая технология. Алма-Ата, 1968, вып. 7—8, с. 221—225.
17. Бектурова Г. Б. и др.—Ж. анал. хим., 1973, т. 28, № 11, с. 2211—2213.
18. Захаров В. А., Сонгина О. А., Бектурова Г. Б. — Ж. анал. хим., 1976, т. 31, № 11 с. 2212_____2221.
19. Сонгина О. А. и др. — Ж. анал. хим., 1974, т. 29, № 8, с. 1594—1598.
20. Галлай 3. А., Типцова В. Г., Пешкова В. М.—Ж- анал. хнм., 1957, т. 12, № 3, с. 469—472.
21. Singh D., Bhatnagar V. — Israel J. Chem., 1967, v. 5, № 1, p. 29—33; РЖХим., • 1967, 22Г55.
22. Быков И. E. — Труды ин-та химии Уральского научного центра АН СССР, 1974, вып. 27, с. 152.
23. Gupta С. М., Joshi М. Р. — J. prakt. Chem., 1968, Bd. 38, № 5—6, S. 358— 361.
24. Алимарин И. П., Терин С. И. — Зав. лаб., 1955, т. 21, № 7, с. 777—780.
25. Сонгина О. А., Кемелева Н. Г. — В кн.: Прикладная и теоретическая химия. Л...ма-Ата, МВО КазССР, 1971, вып. 3, с. 41—43.
26. Singh D., Varma А. — J. Scient. Res. Banares Hindu Univ., 1964—1965, v. 15, № 1, p. 131; РЖХим., 1966, 10Г89.
27. Deshmukh G. S., Choudhury V. N. — Indian J. Chem., 1974, v. 12, № 5, p 540—541; РЖХим, 1975, 6Г57.
28. Агасян П. К., Сираканян М. А.—Ж. анал. хим., 1970, т. 25, № 9, с. 1677— 1681.
29. Галлай 3. А., Марьяновская Т. Я.—Ж. анал. хим., 1963, т. 18, № 8, с. 924— 927.
30. Галлай 3. А., Шеина Н. М.—Ж- анал. хим., 1961, т. 16, № 7, с. 706—710.
31. Соломатин В. Т., Артемова Т. Н. — Зав. лаб., 1974, т. 40, № 9, с. 1044— 1046; Ж- анал. хим., 1977, т. 32, № 9, с. 1742—1747.
32. Сонгина О. А., Савицкая И. С.— Зав. лаб., 1963, т. 29, № 3, с. 401—404.
33. Елизарова Г. П., Кузнецова А. С.—Ж. анал. хим., 1968, т. 23, № 1, с. 50—
54.
34. Павлова И. М., Абетова 3. К- — В кн.: Химия и химическая технология. Алма-Ата, 1966, вып. 3—4, с. 298—302.
35. Павлова И. М., Сонгина О. А.—Изв. АН КазССР. Сер. хим., 1967, № 2, с. 33—36.
36. Усатенко Ю. И. и др. — В кн.: Химическая технология. Респуб. межвузовский научно-техн, сборник. Харьков, 1971, вып. 17, с. 183—190.
37. Супрунович В. И., Усатенко Ю. И., Величко В. В.—Зав. лаб., 1970, т. 36, № 6, с. 652—656.
38. Сухоручкина А. С., Усатенко Ю. И. — Зав. лаб., 1969, т. 35, № 6, с. 647— 650.
39. Вартанян С. В., Тараян В. М. — Зав. лаб., 1975, т. 41, № 1, с. 15—17.
40. Галушко С. В., Усатенко Ю. И. — В кн.: Новые методы контроля материалов на остаточные элементы и микропримеси. М., 1975, с. 139—143.
41. Галушко С. В., Усатенко Ю. И. — В кн.: Органические реактивы в анализе, Саратов, Сарат. ун-т, 1975, с. 75—79.
42. Церковницкая И. А., Кустова Н. А.—Вестник ЛГУ. Сер. физ. и хим., Л., № 16, вып. 3, с. 148—149.
43. Григорьева М. Ф., Слесарь Н. И., Церковницкая А. И. и др. — Изв. вузов. Хим. и хим. технол., 1977, т. 20, № 1, с. 27—30.
44. Церковницкая И. А., Перевощикова В. В.—Ж. анал. хим., 1973, т. 28, № 1> с. 81—85.
126
45 Церковницкая И. А., Григорьева М. Ф. — Ж- анал. хим., 1966, т. 21, № И, 46 Сонгина О А., Б^ктурова Г. В., Захаров В. Л. и др.—Ж- анал. хим., 1972, т 27 № 12, с 2392—,2894.
47 Гпигорьева В. М., Захаренко В. М„ Церковницкая И. Л.—Зав. лаб., 1976, ’ т 42, № 6, с. 660—661.
48 Бектурова Г. Б., Сонгина О. А., Захаров В. А.—Зав. лаб., 1972, т. 38, № 5, с. 143—145.
Висмут
Висмут относится к электроположительным элементам, и ионы его могут восстанавливаться как на ртутном капающем, так и на платиновом электроде. Для определения висмута можно использовать различные реактивы, образующие с ним малорастворимые осадки, в частности фоофат-ион (см. раздел «Фосфор»), Есть данные о том, что можно титровать висмут(III) феррицианидом калия на ртутном капающем электроде [1]. Однако практическое значение этого метода невелико, так как определению будут мешать все ионы, образующие осадки с феррицианидом.
Иодид калия, также образующий практически нерастворимое соединение с висмутом (III) в отсутствие избытка иодида калия, не может быть рекомендован, так как состав осадка меняется в зависимости от кислотности раствора и содержания висмута [2, 3]. Значительно лучше результаты получаются при сочетании иодида калия с некоторыми органическими веществами. Так, можно применять [4] оксихинолин в комбинации с иодидом калия, воспользовавшись тем, что иодид висмута образует с оксихидолином в кислой среде малорастворимое соединение — иодвисмутит оксихинолина C9H7ON • HBiU. На этом же принципе основано титрование висмута(Ш) изохинолином—(образуется осадок состава C9H8N-HBiI4) [5] и а-капролактамом [6].
Вообще для определения висмута применяют в основном органические реактивы. Пирогаллол образует с висмутом(III) практически нерастворимый осадок и позволяет определять висмут в присутствии свинца [7], однако применение его сопряжено с рядом трудностей и практического применения не получило. Особенно много внимания уделяется определению висмута при помощи ЭДТА и ее производных. Метод был разработан Пршибилом и Матыской [8, 9] в 1950—1951 гг. и выполняется сейчас в различных вариантах.
Пршибил и Матыска титруют висмут (III) раствором ЭДТА, используя восстановление висмута(III) на ртутном капающем электроде. При рН=1—2 комплексонаты других металлов не образуются, поэтому добавляемый при титровании ЭДТА связывается только с висмутом, по току которого проводят титрование при потенциале около —0,18 В (Нас. КЭ). Мешать этому определению могут ионы, восстанавливающиеся при том же потенциа-сурьма, железо, олово; их связывают в лимонно- или вин
127
нокислый комплекс, сдвигая потенциал их восстановления в более отрицательную область. По данным Пршибила и Матыска, висмут можно таким образом определять в присутствии всех других катионов. Это позволяет применять титрование комплексоном для определения следов висмута в металлическом свинце.
Ход анализа. Навеску свинца 50 г (при содержании тысячных долей процента висмута) растворяют в концентрированной азотной кислоте; прибавляют столько тартрата или цитрата натрия, чтобы концентрация его в растворе составила 0,3—0,5 М (8—12 г тартрата или 10—15 г цитрата натрия на 100 мл раствора); переводят в мерную колбу емкостью 100 мл, добавляя аммиак с таким расчетом, чтобы pH раствора оказался равным примерно 2. Титруют аликвотную часть раствора 0,02 М раствором ЭДТА при напряжения от —0,18 до 0,22 В (Нас.КЭ).
Метод позволяет определять 0,004% висмута в свинце.
Можно титровать висмут (III) раствором ЭДТА и по току окисления последнего на платиновом электроде, поскольку он легко окисляется анодно как в щелочной, так и в кислой среде [10]. Чешские химики титруют висмут раствором ЭДТА на установке с двумя платиновыми электродами [11—13] на фоне азотной кислоты. Рекомендуемая ими методика определения висмута в силикатах и другом минеральном сырье сводится к следующему [13]: 0,5—2 г тонко истертой пробы обрабатывают HF и HNO3 с таким расчетом, чтобы конечная концентрация азотной кислоты в титруемом объеме (80—100 мл) была 0,5 М и титруют ЭДТА с двумя платиновыми электродами (ДЕ=1,5 В). Титрованию не мешают многие ионы, мешают индий(III), таллий (III) и цирконий (IV). Минимальное определяемое количество висмута — 10 мкг.
Рекомендуется и более сложный вариант [14], включающий осаждение висмута(Ш) 2%-ным раствором диэтилдитиокарбамата натрия, экстракцию осадка хлороформом с последующим выпариванием экстракта с азотной кислотой (1:1). Титруют при рН = = 0,8—1,2 также с помощью двух платиновых электродов.
Если требуется определить одновременно висмут и другие элементы, присутствующие в пробе, то можно также воспользоваться ЭДТА [15—28]. Во всех случаях сначала оттитровывают висмут(Ш) на азотнокислом фоне, затем в том же растворе, соответственно меняя pH, титруют тот или иной ион (табл. 17). Во многих случаях применяют танталовый электрод [15, 17—27]. Конечно, все эти титрования можно выполнять и на обычном платиновом вращающемся электроде. Еще ранее [28] был предложен аналогичный метод с применением ртутного капающего электрода для определения висмута (III), свинца (II) и кальция (II) при одновременном присутствии их в титруемом растворе: сначала титруют висмут при pH=2 по току его восстановления [—0,25 В (Нас. КЭ)], затем в том же растворе устанавливают рН=4 и титруют свинец(И) при — 0,55 В (Нас. КЭ) по току его восстановления и, наконец, при pH=8 и при потенциале +0,05 В
128
(Нас КЭ) титруют кальций(II); после конечной точки появляется анодный ток окисления ртути в присутствии ЭДТА.
Ход анализа. Раствор, содержащий от 50 до 60 мг висмута, столько же свинца и от И до 22 мг кальция, разбавляют до 50 мл водой и устанавливают jq=2 при помощи твердой хлоруксусной кислоты. Добавляют ра_твор желатина с таким расчетом, чтобы концентрация ее составляла около 0,05%; раствор не должен содержать хлоридов. Титруют 0,1 М раствором ЭДТА, приготовленным, из перекристаллизованного препарата, при —0,25 В (Нас. КЭ). Когда висмут будет оттитрован (сила тока перестанет понижаться), добавляют кристаллический ацетат натрия для создания рН=4, окончательно устанавливают pH при помощи раствора аммиака (1:1). Продолжают титрование при —0,55 В по току восстановления свинца. Когда титрование свинца закончится устанавливают рН=8 при помощи раствора аммиака (1:1) и титруют при +0,05 В: ток сначала остается постоянным, когда же весь кальций будет оттитрован, появится ток окисления ртути в присутствии ЭДТА.
Установлена также возможность определения висмута (III) наряду с ионами других элементов (медь, индий, цинк, свинец, галлий, германий, кадмий, скандий) при помощи ЭДТА в безводной уксусной кислоте и в смеси ее с хлороформом или другими органическими растворителями [29—32], а в некоторых случаях при необходимости определения малых количеств висмута рекомендуется экстрагировать его хлороформом в виде иодида.
Своеобразный метод титрования висмута (и ионов других элементов) ЭДТА предложен в работе [16]. Титруют с двумя платиновыми электродами, но один из них (анод) предварительно покрывают (электролитически) тончайшей пленкой РЬО2. Внешнее напряжение не налагается, но после конечной точки титрования ток резко возрастает за счет взаимодействия избытка ЭДТА с РЬО2. Авторы работы полагают, что ЭДТА окисляется оксидом, а восстановленный при этом ион оксида окисляется анодно. К сожалению, работа чисто эмпирическая, в ней нет никаких соображений относительно редокс-потенциалов, рА комплексона-тов и роли pH среды. Указано лишь, что висмут титруют с указанными выше электродами при рН=1—2, при котором можно также титровать Fe3+, Th4+, Zr^, Т13+, а при pH=4,5 — ряд других ионов (двухзарядные ионы ртути, кадмия, цинка, свинца и трехзарядные— церия, лантана, скандия), но уже с другими оксидными пленками на электроде — Т12О3 или Со2Оз. Оксид свинца при этом pH, как замечают авторы статьи, «менее чувствителен», чем при рН=1—2. Не обсуждая здесь механизм всего процесса, укажем лишь, что такой способ титрования излишне сложен, поскольку ЭДТА, как известно, хорошо окисляется анодно на обычном платиновом или другом твердом электроде.
Предложено еще несколько органических реактивов, позволяющих определять висмут как отдельно, так и в присутствии ионов Других элементов, например этилксантогенат калия [33]. Висмут (III) образует с этим реактивом нерастворимое соединение с отношением висмута к реагенту, равным 1 :3. Этилксантогенат окисляется на платиновом электроде, так что титрование можно! вести по току его окисления; в присутствии свинца (II) образует-9 1694 loo»
Таблица 17. Методы определения висмута (Bi111)
Титрант Определяемый минимум Условия определения Примечания Литература
Изохинолин + KI 4—20 мг Электрод Hg капающий, ср = 0,3 В (Нас. КЭ), фон — 2 М НСООН Изохинолин добавляют в титруемый раствор, титруют раствором KI прн удалении О2 током азота 5
8-капролактам 8—150 мг Электрод Pt, ср=0,45 В, (Нас. КЭ), фон — 0,5 М HNOs Принцип тот же, что и выше. В раствор добавляют 15—20 мл 10 %-кого раствора реагента и титруют раствором KI 6
ЭДТА 0,004% Электрод Hg капающий, <р= =-0,18-4-0,22 В (Нас. КЭ), фон — тартрат или цитрат Na, pH ^2 Определяют Bi в свинце 8, 9
Электрод Pt, ср = +0,9 В (Нас. КЭ), фон — нитратный или сульфатный, рН = 1—2 Определяют Bi в легкоплавких сплавах 10
5—100 мг 2 Pt электрода, Дср=1,5 В, фон — 0,4 М HNO3 {Определяют Bi в галените 11—13
0,2—36 мг 2 Pt электрода, <р=1,4 В, фон — HNO3, рН=0,8—1,2 В присутствии Zn, Tl, In, Мо, Си, Fe осаждают Bi раствором диэтилди-тиокарбамината Na, экстрагируют СНС13 несколько раз, выпаривают с HNOs 14
1—2 мг 2 Pt электрода, Дср=0,5—1,0 В, фон — HNOs для Bi, хлорацетат Na до рН=1,2—2 для Fe Совместное титрование сначала Bi, затем Fe111 15
Электрод Pt, покрытая пленкой МпО2, PtO2 или Bi2O5 (анод), катод— платина, фон — рН=1—2 Кроме Bi111 при рН=1—2 титруются Fe111, Tl111, Thlv, ZrIV, при рН=4—5 — ряд других элементов (см. текст) 16
0,05—9 мг Электрод Та, ср= + 1,2 В (Нас. КЭ), фон — 0,2 н. HNOs Можно определять не только Bi, но и другие элементы, а также последо- 17—26
4 мкг/мл
Электрод Та или из сплава Nb — Zr — Ti, <р= + 1,0—1,1 В (Нас. КЭ)
5—10 мкг
Электрод Hg капающий, <р=0,25 В (Нас. КЭ), фон — рН=2 для Bi; <р = =--0,55 В, рН=4 для РЬ; <р=+О,ОЬ В, рН=8 для Са
2 Pt электрода, А<р =0,54-0,7 В или карбидотанталовый электрод, <р= = +0,9—1,1 В (Нас. КЭ), титруют по току окисления ЭДТА
вательно титровать Bi на фоне HNO3, затем в том же растворе, но при другом pH Cd, Pb, Zn, Ni, Hg, Си, Mg, Ag, Th
Определение Bi в присутствии La и 27 Th; определение этих элементов, а также Mg, Fe, Са, Be, Ag в других условиях
Мешает С1_ 28
0,02—1 мг
Этилксантогенат калия
Ы-бис(Р-аминоэтил) дикарбаминовая кислота
СО
2 Pt или карбидотанталовых электрода, Aq>=i0,8 В или один Pt электрод, tp=+0,95 В (Нас. КЭ), фон — CH3COOH+CHCI3
Электрод Pt, q>= +0,85 В (Нас. КЭ), фон—НС1+1—2 мл 0,1 н. нитрата свинца
Электрод Hg капающий, tp=0,2 В (Нас. КЭ), фон—рН=0,02—0,15
Определяемый катион Bi3+ (и др.) экстрагируют в виде дитизонатов или диэтилдитиокарбаминатов, к экстракту (10—12 мл) добавляют 25—30 мл НаО, несколько капель 2 н. HNO3, 0,2 мл 1 Al раствора AgNOs, перемешивают магнитной мешалкой 2— 3 мин и устанавливают pH водной фазы 1,5—3 (для Bi); титруют 0,01— 0,0001 М раствором ЭДТА, выжидая по 0,5—3 мин
Показана возможность прямого определения Bi, In, Ga, Sc и последовательного титрования двух металл-ионов в том же растворе
Определяют Bi в легкоплавких сплавах (см. методику)
Титруют при других pH также Cd, Т1, РЬ и Ni; Cd, Т1 —при рН=9—10; Pb—9,5, Ni—7,5—8,5
38
29—32
33
34
Продолжение
Титрант Определяемый минимум Условия определения Примечания 4 Литература
Диэтилдитиокарб-аминат в бензоле 2 Pt электрода, А<р=0.05-"-0,1 В, фон—безводная СНзСООН или L1CIO4 в бензоле, ССЦ или СНС1з Кроме Bi титруют Hg11 35, 36
Диэтилдитиокар-бампнат натрия 2—10 мкг/40 мл Электрод Pt, <р=4-0,4 В (Нас. КЭ), фон — аммиачно-тартратный буферный раствор (30 мл) +CHCI3 или CCU (10 мл) После добавления реагента выжидают 1—2 мин, перемешивание интенсивное, к Pt электроду припаяна сбо ку Pt проволока, определяют также другие элементы 37
Диэтилдимеркап-тотиопирон 0,2—2,5 мг Электрод графит, <р=0,5 В (Нас. КЭ), фон — винная кислота 4-защитный коллоид ОП-Ю 4- буферный раствор с pH=4,27 Дифференцированное определение Bi и Hg, Bi и Tl, Bi и Си 39- -41
Карбоксиметил-дитиокарбаминат кальция Бензилдитио-карбаминат калия Фон — H2SO4, от рН=4 до 3 н. H2SO4 2 Pt электрода, А<р=0,44-0,7 В или 2 графитовых электрода, А<р=0,25— 0,5 В; фон — 2 н. H2SO4 Последовательное титрование Bi и Sb, а также TeIV и Seiv порознь Метод разработан для Мо, но можно дифференцированно титровать Мо и Bi 42 43
Метилдитиобиурет Электрод Pt, tp=—0,1 В (Нас КЭ), фон для Bi—0,5 М (NH4)2C4H4O64-4-KNO3, pH=8,5—10, титруют по току окисления титранта Определяют и другие элементы (Т1, Со, Se, Те) при других pH 44
Унитиол Электрод Pt, fp=4-0,7 В (Нас. КЭ), фон — 1 н. HNO3 для Bi и ацетатный буферный раствор для РЬ (рН=3,7) Последовательное титрование Bi и РЬ, применен для анализа стандартных образцов сплавов 45
5—25 мг 3—20 мг 5- 25 мг Bi Cd Pb Электрод графит, <р=0,6—0,8 В фон — 0,1—0,2 н HNO3 или 1,0—1,5н. H2SO4 Определение Bi, а также Cd и РЬ в сплавах. Для Cd создают рН=8, для РЬ — 3—4 46
ся его этилксантогенатный комплекс, который способен также окисляться на платиновом электроде. Так как осадок этилксанто-гената висмута выпадает в первую очередь, то образование свинцового комплекса может служить для индикации конечной течки. Этот метод предложен для определения висмута в легкоплавких сплавах.
Ход анализа. Навеску сплава около 0,2 г растворяют в смеси 15 мл соляной (1:1) и 5 мл азотной (плотностью 1,4 г/см3) кислот; добавляют 40—50 мл воды и кипятят 1—2 мин. По охлаждении нейтрализуют аммиаком до появления мути и добавляют 2 н. соляную кислоту (около 25 мл). Раствор переводят в мерную колбу емкостью 250 мл и доводят до метки водой. К аликвотной части раствора (5 или 10 мл) добавляют 40 мл 0,1 н. соляной кислоты и 1—2 мл 0,1 н. раствора нитрата свинца, после чего титруют раствором этнлксантогената калия при +0,85 В (Нас. КЭ)
В числе ряда других элементов висмут можно определять с помощью Г\),Ь1-бис(р-аминоэтил)дитиокарбаминовой кислоты [34]; серия работ посвящена изучению титрования висмута(III) ди-этилдитиокарбаминатом [35—37], реактивом не только на висмут, но и на Cd, Zn, Ni, Ag, In, Pb, Си. Комплексные диэтилдитио-карбаминаты этих элементов экстрагируют, если нужно, хлороформом и четыреххлористым углеродом, титрование осуществляют в среде уксусная кислота — хлороформ бензольным раствором диэтилдитиокарбамината свинца [35]. Метод интересен, но сложен и вряд ли пригоден для массовых определений вследствие необходимости пользоваться органическими растворителями, особенно бензолом. Другсй вариант этого метода заключается в том, что диэтилдитиокарбаминатные (или дитизонатные) комплексы
различных элементов, в том числе висмута, сначала экстрагируют органическими растворителями и затем их титруют ЭДТА [38].
В серии работ [39—41] рассмотрено титрование висмута ди-этилдимеркаптотиопироном, реагирующим также с медью(II), ртутью (II) и таллием (III). При соблюдении определенных условий возможно последовательное титрование меди и висмута [40], ртути, висмута и таллия [41]. Карбоксиметилдитиокарбаминат кальция позволяет титровать висмут(III), сурьму(III), селен(IV) и теллур(IV) и определять последовательно висмут и сурьму в одном и том же растворе [42]. Применяют также бензилдитио-карбаминат калия, предложенный в основном для определения молибдена (VI) [43], но позволяющий раздельно определять и молибден и висмут, метилдитиобиурет [44] и унитиол [45, 46], позволяющий раздельно определять в азотнокислой среде висмут, свинец и кадмий. Метод прост и удобен. Методика определения висмута и свинца следующая: 0,5 г сплава, содержащего определяемые металлы (например, 45—55% висмута, 25—35% свинца и *5 20% олова), растворяют при нагревании в 5—10 мл концентрированной азотной кислоты, кипятят 1—2 мин для удаления оксидов азота, отделяют оловянную кислоту выпариванием с азотной кислотой, фильтруют, фильтрат разбавляют водой до мл. К 1 5 мл полученного раствора прибавляют концентри
133
рованную азотную кислоту до ее конечной концентрации в растворе 1 н. и титруют висмут раствором унитиола. Нейтрализуют этот раствор едким натром по универсальной бумаге, прибавляют 5 мл ацетатного буферного раствора (pH=3,72), 1 мл 1 %-него раствора поверхностно-активного вещества ОП-10 и титруют РЬ2+ тем же раствором унитиола. Продолжительность анализа 15—20 мин. Метод применен для определения висмута в легкоплавких сплавах.
ЛИТЕРАТУРА
1. Бабенышев В. М., Шелкановцева А. Я-, Кузнецова О. М. — В кн.: Сборник научных трудов Куйбышевского индустриального института, 1957, № 7, с. 37—41.
2. Сонгина О. А., Войлошникова А. П., Козловский М. Т. — Изв. АН КазССР. Сер. хим., 1949, вып. 3, с. 32—34.
3. Сонгина О. А. — В кн.: Труды комиссии по аналитической химии АН СССР. 1952, IV (VII), с. 116—120.
4. Жданов А. К., Хадеев В. А., Муртазинова Г. Ф. — Зав. лаб., 1955, т. 21, № 5, с. 518—520.
5. Rao A. L., Puri В. К- — Z. anal. Chem., 1969, v. 246, № 5, р. 322—326.
6. Бу сев А. И. и др. —Ж. анал. хим., 1966, т. 21, № 4, с. 422—426.
7. Мусина Т. К-, Сонгина О. А.— Изв. АН КазССР. Сер. хим., 1957, вып. И, с.^36—40.
8. Pribil R., Matyska В. — Chem. Listy, 1950, t. 44, s. 305.
9. Pribil R., Matyska B. — Coll. Czech. Chem. Comm., 1951, v. 16, p. 139.
10. Усатенко Ю. И., Виткина M. А.—Зав. лаб. 1960, т. 26, № 6, с. 542—544.*
И. Vydra F., Vorlicek J. — Taianta, 1966, v. 13, № 4, p. 603—607.
12. Vorlicek J., Petak P. — Microchem. J., 1967, v. 12, № 4, p. 466—471.
13. Vorlicek J. — Acta geol. et geogr. Univ. Comenianae. Geol., 1968, № 15, p. 121—125.
14. Vorlicek J., Petak P., Sulcek Z.— Z. anal. Chem., 1968, Bd. 240, № 5, S. 317— 320.
15. Kemula W., Hulanicki A., Jedral W.—Chem. analit. (Polska), 1969, v. 14, № 2, p. 225—234.
16. Kainz G., Muller H. A., Sontag G, — Z. anal. Chem., 1971, Bd. 256, № 5, S. 345—348.
17. Жданов A. K-, Ятрудакис С. M., Андреева T. И.—Научные труды Ташкентского ун-та, 1964, вып. 264, с. 73—78.
18. Жданов А. К, Капица Н. В., Ахмедов Г. — Труды Ташкентского ун-та, 1967, вып. 288, с. 26—30; с. 31—34.
19. Жданов А. К-, Копица И. В., Ахмедов Г.—Труды Ташкентского ун-та, 1967, вып. 288, с. 35—38; с. 39—41.
20. Жданов А. К- и др. — Узб. хим. ж., 1969, № 2, с. 17—19.
21. Жданов А. К-, Капица Н. В., Акентьева И. А. — Ж. анал. хим., 1971, т. 26, № 4, с. 835—837.
22. Жданов А. К-, Мархабаев И., Ленченко Т. А.—Ж. прикл. химии, 1971, т. 44, № 2, с. 456—457.
23. Жданов А. К-, Мархабаев И., Ленченко Т. А. — Изв. вузов. Химии и хим. техн., 1971, т. 14, № 3, с. 355—357.
24. Жданов А. К., Мархабаев И. — Журн. ВХО, 1971, т. 16, № 5, с. 585.
25. Жданов А. К., Капица Н. В., Ахмедов Г. — Труды Ташкентского ун-та, 1967, вып. 288, с. 31—34.
26. Жданов А. К, Капица Н. В., Ахмедов Г. — Труды Ташкентского ун-та, 1967, вып. 288, с. 39—41.
134
27 Мархабаев И. А., Жданов А. К.—Ж. анал. хим., 1973, т. 28, № 1, с. 168— 170
9R Reilleu Ch., Scribner W., Temple C. — Anal. Chem., 1956, v. 28, p. 450.
94 Геворгян A M.- Талипов Ш. T.. Хадеев В. А.— Ташкент, 1978, депон. ВИНИТИ 28/IX—73, № 6822—73.
30 Геворгян А. М., Талипов Ш. Т., Хадеев В. А. — Узб. хим. ж., 1973, № 4, ’ с. 23—25.
31 Мухамеджанова Д., Хадеев В. А., Талипов Ш. Т.—ДАН УзбССР, 1974, ’ № 7, с. 37—38.
32 Геворгян А. М. и др. — В кн.: XI Менделеевский съезд по общей и прикладной химии. Рефераты докладов и сообщений. № 5. М., Наука, 1975, с, 32
33 Усатенко Ю. И., Тулюпа Ф. М.—Укр. хим. ж., 1963, т. 29, с. 747—751.
34 Chattopadhyay S. S. — Indian J. Chem., 1970, v. 8, № 12, p. 1142—1143; ’ РЖХим., 1971, 14Г71.
35. Геворгян A. M., Хадеев В. А., Постылев В. С. — Научные труды Ташкентского уи-та, 1976, № 513, с. 28—30.
36. Геворгян А. М. и др. —РЖХнм., 1975, 10751, депон. ВИНИТИ.
37 Мухамеджанова Д., Талипов Ш. Т., Хадеев В. А.—Узб хим. ж., 1970, Ns 3, ’ с. 8—10.
38 Мухамеджанова Д., Хадеев В. А., Талипов Ш. Т.—ДАН УзбССР, 1973, ’ Ns 11, с. 31—32.
39. Аришкевич М. А., Ахметшин А. Г., Усатенко Ю. И. — Зав. лаб., 1967, т. 33, № 6, с. 692—695.
40. Ахметшин А. Г., Усатенко Ю. И., Аришкевич А. М. — В кн.: Комплексообразование, межмолекулярное взаимодействие и соосаждение в некоторых системах. Днепропетровск, 1970, с. 130—135; РЖХим., 1971, 23Г67.
41 Ахметшин А. Г-, Усатенко Ю. И., Аришкевич А. И. Там же, с. 136—143; РЖХим., 1971, 23Г68.
42. Павличенко В. А., Усатенко Ю. И., Тулюпа Ф. М. — В кн.: Химическая технология. Республ. межвед. научно-техн. сб. 1968, вып. 13, с. 88—91.
43. Галушко С. В., Усатенко Ю. И. — Зав. лаб., 1974, т. 40, № 7, с. 776—778.
44. Deshmukh G. S., Nandi R. К. — Current Science (India), 1969, v. 38, Ns 21, p. 512—513.
45. Усатенко Ю. И., Даниленко E. Ф. — Зав. лаб., т. 36, № 8, с. 915—916.
46. Даниленко Е. Ф., Усатенко Ю. И. — В кн.: Химическая технология. Респуб. межвед. научно-техн. сб. 1971, вып. 18, с. 164—.167.
Вольфрам
Для амперометрического титрования вольфрама (VI) предложены в основном методы осаждения, подробно описанные в книге [1]. В качестве осадителей рекомендованы соли свинца(II), таллия (I), меди(II), ртути(I), а также оксихинолин. Однако все эти реактивы образуют соединения со многими другими элементами, в том числе с молибденом, часто сопутствующим вольфраму, поэтому их применение ограничено, по существу, только теми случаями, когда вольфрам отделен от всех мешающих примесей. Кроме того, растворимость вольфраматов недостаточно мала, приходится работать в водно-спиртовой среде и строго регулировать кислотность растворов. Поэтому амперометрическое титрование ольфрама не получило практического применения. В последние Г9^ц1П°ЯВИЛась’ однако> серия работ болгарских исследователей пя fvn РекомендУюЩих титровать не вольфрам(VI), а вольф-Р M(v) или вольфрам(III) .после восстановления вольфрама^!)
» 135
той или иной амальгамой*. Во всех случаях титрантом служит раствор феррицианида калия, являющегося окислителем по отношению к восстановленным формам вольфрама в кислой среде. В работе [2] указывается, что в определенных условиях можно определять таким образом не только вольфрам, но и молибден и сумму обоих элементов (см. также раздел «Молибден»),
Для определения вольфрама в гетерополисоединениях типа кремневольфрамовой и фосфорновольфрамовой кислоты рекомендован диантипирилметан (ДАМ), образующий с вольфрамат-ио-ном осадок сложного состава: (ДАМ.)5-Н6\У12О39-хН2О, в котором отношение ДАМ:\У=5:12, а с кремневольфрамовой кислотой (flAM)3H4[Si(W3O10)]4-xh2O, т. е. ДАМ:\У = 3:1 [7, 8].
Ход анализа. Навеску 0,1—0,2 г растворяют в 5 н. серной кислоте, добавляя по каплям азотную кислоту и нагревая для растворения остатка. Добавляют 50 мл веды, затем по каплям 20 %-дай раствор едкого натра до pH=9 и переводят в мерную колбу емкостью на 200 мл. Для титрования отбирают 5—10 мл раствора и добавляют понемногу 10 н. серную кислоту до рН=1,5—2, затем 2— 3 мл 0,004 М раствора силиката натрия и нагревают на водяной бане 5—6 мин, после чего вводят серную кислоту до концентрации ее 1 н. Титруют сернокислым раствором ДАМ при потенциале +1,4 В (МИЭ). Титр ДАМ устанавливают по стандартному образцу стали.
Попытка определять вольфрам(VI) с помощью раствора нитрата серебра в расчете на образование осадка вольфрамата серебра [9] не дала положительного результата. Оказалось, что кривая титрования искажена вследствие катодного восстановления осадка вольфрамата серебра, выпадающего при титровании [10].
Определение вольфрама (VI) по методу окисления — восстановления затруднено невысоким стандартным потенциалом системы WVI/WV. Поэтому для амперометрического определения приходится прибегать к очень сильным восстановителям, например к хрому(П) [11], что связано с определенными неудобствами вследствие того, что растворы хрома(II) неустойчивы при хранении.
ЛИТЕРАТУРА
1. Сонгина О. А. Амперометрическое титрование. М., Химия, 1967.
2. Кулев И., Добрева Я. — Годишн. Висш. хим.-техн. нн-т, Бургас, 1972—1970, т. 7, с. 53—61 (болг.); РЖХим., 1972, 5Г71.
3 Кулев И., Гаванаров И., Станев Д. — Рудодобив (болг.), 1973, т. 28, № 4, с. 23 -29; РЖХим., 1973, 23Г12Э.
4. Кулев И., Видева М. — Z. Chemie, 1976, Bd. 16, № 10, S. 411—412.
5. Kulev I., Widewa M. — In: 2nd Nat. Conf, on Anal. Chem. with Internat. Participation, Golden Sands, Varna, 1976, p. 188.
6. Кулев И., Цайков У. — Годишн. Висш. хим.-техн. ин-т, Бургас, 1972—1970, т. 7, с. 63—66 (болг.); РЖХим, 1974, 5Г113.
7. Луговой С. В., Рязанов И. П. — В кн.: Сборник научных трудов Магнитогорского горно-металлургического ин-та. 1971, вып. 87, с. 64—67; РЖХим, 1971, 24Г135.
* Восстановление вольфрама (VI) амальгамами описано в книге Н. А. Та-нанаева «Объемный анализ», ОНТИ, 1934.
136
8 Луговой С. В., Паклерова Л. Д.—Ж. анал. хим., 1974, т. 29, № 3, с. 1748— 1753.
9 Сонгина О. А., Котлярская В. 3., Войлошникова А. П. — Изв. АН КазССР. Серия хим., 1955, вып. 8, с. 77—80.
10 Сонгина О. А., Захаров В. А., Мамбетказиев Е. А. — Изв. АН КазССР. Се-’ рия хим., 1968, № 6, с. 31—35.
11 Пешкова В. М., Галлай 3. А., Алексеева Н. Н. — В кн.: Химия редких элементов. М., изд-во АН СССР, 1957, вып. 3, с. 119—130.
Галлий
Для амперометрического определения галлия можно применять только реакции осаждения и комплексообразования, так как окислительно-восстановительные реакции для него, как известно, нехарактерны. К наименее растворимым соединениям галлия относится его ферроцианид, образованием которого воспользовался еще Лекок де Буабодран при выделении галлия из кислых растворов. Состав ферроцианида галлия установлен И. В. Тананае-вым и Н. В. Баусовой [1], разработавшими также амперометрический метод определения по току окисления ферроцианида на платиновом электроде. Конечная точка отвечает молярному отношению галлий : ферроцианид = 4 : 3, т. е. осадок имеет состав Оа4[Ре(СМ)б]з- Растворимость ферроцианида галлия очень мала— можно определять до 10 мкг галлия в 20 мл. Алюминий, который почти всегда сопутствует галлию в растворах, не мешает титрованию, хотя при относительно больших количествах алюминия ток окисления ферроцианида заметно понижается, вследствие чего определение конечной точки становится менее отчетливым. Влияние ,алюминия было замечено и при других титрованиях и может быть устранено титрованием с таким электродом сравнения, потенциал которого лишь не намного отличался бы от потенциала окисления ферроцианида, например с перманганатным электродом (см. гл. V).
Феттер и Свайнхарт [2] титруют галлий(III) также ферроцианидом, но с двумя индикаторными электродами при напряжении 0,23 В и при 50 °C. Напряжение выбрано чисто эмпирически, причем авторы [2], по-видимому, титруют без добавления феррицианида (см. гл. VI) и поэтому вынуждены применять высокую температуру. Вряд ли этот метод удобен. Во всяком случае ему следует предпочесть очень простой и вполне надежный метод Та-нанаева и Баусовой.
Подобно титану(IV), цирконию(IV) и скандию(III), галлий (III) образует малорастворимое комплексное соединение с М-бензоилфенилгидроксиламином [3, 4] и с купфероном [5]. Оба реактива окисляются на платиновом электроде. При титровании купфероном надо восстанавливать железо(III) до железа(II), затем экстрагировать галлий бутилацетатом из 6 н. соляной кислоты и реэкстрагировать его водой, подкисленной серной кисло-т°и до рН = 3—4 и содержащей некоторое количество хлорида
137
£5 Таблица 18. Методы определения галлия (Ga111)
0° _ Титрант Определяемый минимум Условия определения Примечания Литература
K4Fe(CN)6] 10 мкг/20 мл Электрод Pt, <р= +0,8 В (Нас. КЭ) См. текст 1
2 Pt электрода, А<р=0,23 В, 50 °C См. текст 2
N-бензоилфенил-гидроксиламин Электрод Pt или графит, <р= = + 1,1 В (Нас. КЭ), фон —рН= = 2,4—3 (НС1 + тартрат калия) Этот же титрант рекомендуется для определения Sc, Ti и Zr 3, 4
Купферон Электрод Pt, вибрирующий; <р = = + 1,2 В (НВЭ), фон —0,001—0,1 н. H2SO4 См. текст 5
ЭДТА Нижний предел 7 мкг/мл Электрод Та, tp= +1,2 В ,(Нас. КЭ), фон — тартратный буферный раствор, pH=5,0 Нё мешают большой избыток ионов щелочных и щелочноземельных элементов и обычные анионы 7
Электрод Та, <р= + 1,2 В (Нас. КЭ), фон — HNO3 (рН=2—2,5) для In и Ga, рН=5 и <р= 1,0 В для Cd и In + Cd Сначала титруют сумму Ga и In, затем вводят раствор NaAc до рН= = 5, титруют Cd; в другой аликвотной части титруют сумму In и Cd, определяют Ga и In по разности 8
1—30 мкг/мл 2 Pt электрода, А<р=1 В, фон — 40%-ный бензол, 0,2 М СН3СООН В тех же условиях можно титровать индий(Ш) 10
1—2 мкг/мл в экстракте 2 Pt электрода; А<р=0,7 В, фон — хлороформный экстракт ацетилацето-ната+25—30 мл воды и HNO3 до рН= =2—3; добавляют отмеренный избыток ЭДТА Титруют ЭДТА ацетилацетонаты Ga и других элементов (Си, In, Th), экстрагированные раствором ацетилаце-тона в хлороформе; для Ga — обратное титрование избытка комплексона раствором Bi3+ 11
0,35 мкг (10-6 М) Электрод ртутный вращающийся, tp=+0,35 В (Нас. КЭ), фон—рН=2, хлориая кислота
Электрод Au, амальгамированный, избыток ЭДТА титруют раствором соли цинка, <р=—0,8 (Нас. КЭ). Подчеркивается высокая точность амперометрического метода
Тиооксин 2,5 мкг/мл Электрод Pt, <р= +1,0 В (НВЭ) по току окисления тиооксина, фон — ацетатный буферный раствор, pH=4—7
0,1 мкг/мл Электрод Pt, фон — ацетатный буферный раствор, pH=3,7—6,7 или 2 н. NaAc и 1 н. KNO3, <р= + 1,0 В (Нас. КЭ)
Урамил-|М,М-диук-сусная кислота 1,4.10“5А4 Электрод графит, <р= +0,74-0,8 В (Нас. КЭ), фон — H2SO4, рН=2—3
N-Циннамоилфе-иилгидроксиламин и N-3-стирилакрило-фенилгидроксиламин 0,3—42 мкг/мл Электрод графит, <р= +0,9 В (Нас КЭ) или ртутный капающий, <р=—1,1 В, фон — бифталатный или аммиачно-ацетатный заствор, рН=2.2—3,8. Титруют на Pt по току окисления титранта, на Hg — по току его восстановления
Оксиарильные кислоты 0,8—12мг/20мл Электрод графит, ср=0,5—0,8 В (Нас КЭ), фри — 0,1 М NH4OH
Обратное титрование избытка ЭДТА1 12 раствором Bi3+ концентрации 5-10~4 М| титруют по анодному току ЭДТА на ртутиом электроде
Метод рекомендован Национальным бюро стандартов для установления стехиометрии арсенида галлия; отношение Ga : As найдено равным 0,999994+0,000065
Состав осадка Ga(OH) (C9H6HS)2, т. е. отношение; Ga : тиооксина = 1 2, можно определить Ga, In, Tl при совместном присутствии на разном фоне
Состав осадка на ацетатном фоне 1 : 2, на фоне KNO3—1 : 1, предложено последовательное титрование Ga и других элементов (см. текст)
Можно титровать также индий при рН=1—2
Состав комплексного соединения R:Ga = 3:l, мешают Fe111, TiIV и Pb11, Fe111 восстанавливают аскорбиновой кислотой
Обратное титрование избытка реактива раствором NiSO4, можно определять также In (от 2,3 мкг до 23 мг/20мл)
13
14
15
16
17, 18
19
натрия. Алюминий не мешает определению в том случае, если его количество не превышает 50-кратный избыток по отношению к галлию.
ЭДТА, впервые примененный для титрования галлия в присутствии фосфора еще в 1959 г. [6], оказался хорошим реагентом и для определения галлия в присутствии других элементов [7—13]. Рекомендованы различные электроды (платина, тантал, ртуть, амальгамированное золото) и различные фоны в зависимости от рЛ комплексонатов определяемых элементов. Титрованию во всех случаях мешает алюминий(III), его маскируют фторидом.
Применяют для определения галлия и 8-меркаптохинолин (тиооксин), образующий с галлием труднорастворимый осадок [14]. Методика определения галлия чрезвычайно проста: навеску пробы (например, таллиевого концентрата) 0,5—1,0 г растворяют в 20—25 мл серной кислоты (1:1), переводят в мерную колбу емкостью 100 мл, доводят водой до метки, отбирают 5 мл, доводят объем до 20 мл ацетатным буферным раствором с pH = 6 и титруют 0,01 А4 раствором тиооксина при +1,0 В (НВЭ). После каждого титрования налипший на электрод осадок удаляют промыванием электрода в азотной кислоте (1:1) и протиранием фильтровальной бумагой. Одно из важных преимуществ тиооксина заключается в том, что он не реагирует с алюминием, часто сопутствующим галлию в природных и промышленных объектах.
Если совместно с галлием (III) присутствуют индий(III) и таллий (Ш), можно определять каждый из этих ионов без предварительного их разделения [14]: таллий (Ш) титруют на фоне 1 М серной кислоты (см. раздел «Таллий»), индий — в другой аликвотной части на фоне тартратного буферного раствора с pH = 4,5 (см. раздел «Индий»), а галлий определяют по разности после суммарного титрования его и индия на ацетатном фоне, как указано выше.
Авторы работы [15] подробно изучили зависимость состава осадка от состава и pH среды и рекомендовали последовательное титрование галлия и ионов других элементов тиооксином в ряде двойных и тройных смесей.
ЛИТЕРАТУРА
1. Тананаев И. В., Баусова Н. В. — Химия редких элементов. М., изд-во АН СССР, 1957, вып. 3, с. 41.
2. Fetter N. R., Swinehart Р. F. — Anal, chem., 1956, v. 28, p. 122.
3. Галлай 3. А., Алимарин И. П.—Ж. анал. хим., 1963, т. 18, № 7, с. 1442.
4. Галлай 3. А., Шеина И. А., Алимарин И. П. — Ж. анал. хим., 1965, т. 20, с. 1093—1096.
5. Церковницкая И. А., Калинин А. И., Морачевский Ю. В. — Зав. лаб. 1960, т. 25, с. 797.
6. Чудинова Н. Н.—Ж- анал. хим. 1959, т. 14, № 3, с. 636.
7. Жданов А. К., Акентьева Н. А. —Узб. хим. ж., 1969, № 6, с. 3—4.
8. Жданов А. Ц., Акентьева -И. А., Лукьяненко И. А. — ДАН УзбССР, 1970, № 2, с. 28—29.
140
о Геворгян A M., Талипов Ш. Т., Хадеев В. А. —депон. в ВИНИТИ ' 26/IX—73 г., № 6822—73 Депон.
Ю Геворгян А. М„ Талипов Ш. Т., Хадеев В. В.—депон. в ВИНИТИ РЖХим.. 1973, 23Г101.
И Михамеджанова Д., Хадеев В. А., Талипов Ш. Т. — ДАН Узб ССР, 1974, № 7, с. 37—38.
19 Boef G. Oostervink R.—Anal. chim. acta, 1976, v. 87, № 2, p. 515—517.
13 Marinenko G., Durst R. A.— Natl. Bur. Stand. USA, Techn. Note 583, 18, 29, ’ 31, 1973.
14 Захаров В. А и др.—Ж. анал. хим., 1970, т. 25, № 5, с. 879—884.
15 Супрунович В. И., Усатенко Ю. И., Величко В. В. — Укр. хим. ж., 1973, т. 39, ’ № 5, с. 488—493.
16 Бусев А. И., Галлай 3. А., Зверяева И. В. — Зав. лаб., 1974, т. 40, № 5» ’ с. 514—517.
17 . Галлай 3. А. и др.—Ж. анал. хим., 1975, т. 30, № 12, с. 2346—2349 18. Галлай 3. А. — Ж. анал. хим., 1977, т. 32, № 9, с. 1722—1725.
19 Бусев А. И., Галлай 3. А., Зверяева И. В. — Ж. анал. хим., 1977, т. 32, № 9, ’ с. 1927—1730.
Гафний
Гафний является полным аналогом циркония и в природных соединениях практически не встречается отдельно от него. Пока еще не предложено ни одной специфической реакции для гафния, и определять его приходится теми же методами, что и цирконий, несколько видоизменяя условия опыта. Амперометрическое определение гафния в растворах, не содержащих циркония, возможно при помощи купферона [1] и при помощи тартразина и флавази-на '[2]. Титрование проводят по току восстановления купферона на ртутном капающем электроде при —0,65 В (Нас. КЭ) на фоне 1 н. серной кислоты [1]. Титрование можно проводить в присутствии фторидов и фосфатов. По-видимому, можно заменить ртутный электрод платиновым, проводя титрование по току окисления купферона при +0,8 (Нас. КЭ), тогда отпадает необходимость удаления растворенного кислорода воздуха, которое необходимо при работе с ртутным электродом. Авторы работы [1] продувают раствор током азота после каждого добавления купферона. Состав осадка купфероната гафния отвечает формуле-НПКупф)4.
Второй метод заключается в титровании тартразином или фла-вазином, также образующими осадки с гафнием (и с цирконием). Рекомендуется титровать при рН=1—1,5 с ртутным капающим электродом при 1,8 В (Нас. КЭ) и 70 °C. Кислород необходимо удалять. Хотя этот метод предложен [2] для определения малых количеств гафния (от 0,3 до 2 мг), что является положительной, его стороной, он все же, надо полагать, весьма неудобен для практического использования.
Простой и удобный метод определения гафния заключается в-титровании его раствором ЭДТА в сильнокислой среде (серная ислота, pH = 0,4—0,8), в которой большинство катионов не обра-ует комплексонатов [3]. Титруют на установке с танталовым
I4S
электродом при +1,2 В (Нас. КЭ) по току окисления титранта. Определяемое количество гафния — от 1 до 40 мг в титруемом объеме [3].
ЛИТЕРАТУРА
1. Elving Ph., Olson Е. — Anal. Chem., 1955, v. 27, p. 1817.
2. Popa Gr., Baiulescu Gh., Moldoveanu S. — Rev. chim. Acad. RPR, 1963, v. 8, p. 287.
3. Акентьева H. А., Жданов A. K-—Журн. BXO, 1971, t. 16, № I, c. 118.
Германий
Для амперометрического определения германия (IV) предложено пока три типа реакций: образование комплексных соединений с о-дифенолами и родственными им соединениями [1, 2], титрование гетерополикислоты нитроном [3] и осаждение в виде герма-ната магния [4].
Титрование пирокатехином [1] проводят с ртутным капающим электродом по току восстановления германия (IV) при —1,7 В (Нас. КЭ) на 4юне боратно-ацетатного буферного раствора с рН=7—9. Кривая титрования имеет форму а, перелом наступает при молярном отношении германия к пирокатехину 1 :3. Однако авторы подчеркивают, что это титрование дает хорошие результаты только в том случае, если содержание германия не слишком мало — не менее 4-10~3М. Пирокатехин взаимодействует не только с германием, но и с другими элементами, поэтому германий приходится предварительно отделять экстракцией четыреххлористым углеродом.
Ход анализа. Навеску 2—4 г при содержании германия до 1 % или 0,2—1 г при содержании более 1 % разлагают, как обычно, кислотами или сплавлением с перекисью натрия. Во втором случае после выщелачивания сплава обязательно добавляют гидроксиламин для разрушения перекиси водорода. Раствор, объем которого должен составлять 15—30 мл, переносят в делительную воронку, добавляют трехкратный объем концентрированной соляной кислоты и 50 мл четыреххлористого углерода. Затем смесь охлаждают и экстрагируют в течение 2 мин. Для второй экстракции берут 40 мл СС1«, после чего органический слой промывают 10 мл 10 н. соляной кислоты.
Из объединенных экстрактов реэкстрагируют германий (IV) водой в два приема—сначала 10 мл, затем 5 мл. Водный раствор, содержащий германий(1У), переносят в мерную колбу емкостью 25 мл, доводят pH до 8 буферным раствором и, если требуется, несколькими каплями 1 н. раствора едкого иатра по фенолфталеину. После этого окисляют мышьяк(Ш) (если вместе с германием присутствует мышьяк, то он экстрагируется тоже и должен быть окислен до Asv, чтобы ие мешать титрованию GeIV пирокатехином) 0,01 и. раствором иода в присутствии двух капель 1%-ного раствора крахмала и объем раствора доводят до метки буферным раствором. Переносят в электролизер 10 мл раствора и после пятиминутного продувания водородом титруют 0,1 М раствором пирокатехина из микробюретки. Титр раствора пирокатехина устанавливают по стандартному раствору германия тем же методом на том же фоне.
Описанный метод был проверен его авторами на пробах, содержащих 0,25—2,8% германия наряду со значительными количе
142
ствами меди, цинка, свинца, мышьяка и других элементов. Среднюю ошибку определения авторы метода оценивают приблизительно в 4%.
Вместо пирокатехина можно титровать раствор галловой кислотой в солянокислой среде, содержащей сульфат аммония [2].
Как известно, германий (IV) образует, подобно кремнию (IV), фосфору (V) и мышьяку (V), гетерополикислоты; германомолибде-новая гетерополикислота восстанавливается на ртутном капающем электроде и может быть осаждена [3] нитроном в гликолевом буферном растворе с pH = 2—2,5.
Очень прост метод осаждения германия (IV) раствором сульфата магния [4]. Титрование ведут по току восстановления ионов германия на ртутном капающем электроде при —1,6 В (Нас. КЭ) на хлоридно-аммиачном фоне. Составу осадка отвечает формула MgjGeO^ Титровать можно от 0,05 мг германия и выше в объеме около 15 мл. Анионы кремневой, мышьяковистой и борной кислот не мешают титрованию. Интересно отметить, что это титрование может быть выполнено и в присутствии катионов, образующих осадки с сульфатом, поскольку сульфат-ион не играет никакой роли в процессе титрования. Мешать титрованию может мышьяк (V), который, как известно, также осаждается солями магния.
Новых реактивов для определения германия за последние годы не предложено, но возобновлен в электрохимическом варианте способ определения германия (II) при помощи иода после восстановления германия (IV). Авторы статьи [5] подробно изучили условия восстановления германия(IV) гипофосфитом натрия и последующее титрование германия(II) раствором иода в иодиде калия. После восстановления германия (IV) в специальной ячейке, соединенной с сосудом для титрования, титруют с одним платиновым электродом или с двумя электродами (в первом случае при потенциале 0—1-0,3 В (Нас. КЭ), во втором при Е=50— 200 мВ) на фоне фосфатного буферного раствора с рН~1. Можно титровать также раствором йодата калия. Нижний предел определения— от 0,0032 до 3,2 мг в 40 мл раствора. Конечная точка определяется по появлению тока восстановления титранта.
ЛИТЕРАТУРА
1. Зелянская А. И., Сташкова Н. В. — Ж. анал. хим., 1961, т. 16, № 2, с. 430. 2. Nair A., Ibrahim S. — Current Sci., 1956, v. 25, p. 10.
3. Hahn H., Wagenknecht E. — Z. anal. Chem., 1961, Bd. 182, S. 343.
4. Драницкая P. M., Иашвили Э. В. — Ж. анал. хим., 1964, т. 19, № 5, с. 1031. 5. Агасян П. К., Рамла М,— Ж. анал. хим., 1973, т. 28, № 5, с. 1011—1014.
Железо
Амперометрическое титрование ионов железа осуществляется различными методами, которые можно подразделить на следующие группы: а) методы, основанные на восстановлении Fe3+;
143
б) методы, основанные на окислении Fe2+; в) методы, основанные на комплексообразовании ионов железа; г) методы осаждения.
а) Для прямого титрования железа(III) еще в 50-х годах была предложена аскорбиновая кислота [1—3]. Титровали с одним и с двумя платиновыми электродами [1, 2] и с ртутным капающим — в этом случае использовали ток восстановления железа (HI) [3]. Однако сравнительно легкая окисляемость аскорбиновой кислоты не способствовала широкому распространению этого метода. По той же причине не вошли в практику и методы титрования раствором хрома (II) [4] и раствором олова (II) [5].
Железо(III) успешно титруется нитратом ртути(I) в присутствии роданида калия. Этот метод был разработан В. М. Тараян в 1958 г. как обычный титриметрический метод — так называемый «меркурометрический» — и впоследствии применен для амперометрического титрования железа(III) [6]. Присутствие роданида необходимо для понижения окислительно-восстановительного потенциала системы Hg2+/Hg+ для того, чтобы обеспечить восстановление железа (Ш). Аналогичный метод, но не с нитратом, а с перхлоратом ртути(I) разработан для определения очень малых количеств железа (Ш) и других ионов [7, 8], в частности феррицианида [9].
б) Более распространены различные варианты титрования железа (II) после восстановления железа(III) тем или иным восстановителем. Окислителями служат обычные вещества, применяемые и при визуальном титровании — ванадат [10, 11], бихромат [12—18], перманганат [19—21]. Амперометрический вариант позволяет устранить ряд затруднений, связанных с индикацией конечной точки при визуальном ее определении (например, при общеизвестном определении железа по методу Циммермана — Рейн-гардта) [21]. Амперометрическое установление конечной точки полностью устраняет ошибки, часто возникающие при бихромат-ном методе определения железа(II) с индикатором дифениламиносульфонатом натрия в пробах, содержащих ванадий.
Растворы церия(IV) относятся к числу сильных окислителей (Ес системы Celv/CenI= +1,44 В), их также успешно применяют для титрования железа (II) [17, 19, 22—24].
Растворы церия (IV) отличаются большой устойчивостью, их титр не меняется в течение времени, и поэтому они особенно удобны с практической точки зрения. В. И. Боговина, В. П. Новак . и В. Ф. Мальцев [24] применили 0,002 н. раствор сульфата церия (IV) для определения железа (II) в электролитах, содержащих серную и щавелевую кислоты (так называемые «оксалатные ванны»).
Ход анализа. Отбирают 10 мл отфильтрованного электролита в мерную колбу емкостью 100 мл и доводят до метки дистиллированной водой. К аликвотной части 5—10 мл добавляют 1 мл серной кислоты (1:5), разбавляют водой до 20—25 мл и титруют при +0,6 В (Нас. КЭ) 0,002 н. раствором церия(IV), титр а 44
которого устанавливают также амперометрически по стандартному раствору железа (П).
Железо (II) в рудах также можно определять амперометрическим методом [25, 26] на основе известного метода А. В. Шеина, по которому пробу руды разлагают смесью серной и фосфорной кислот в присутствии оксида ванадия (V), являющегося окислителем железа (II). Избыток оксида ванадия титруют солью Мора.
Ход анализа. В стакан емкостью 100 мл, слегка влажный внутри (1—1,5 мл воды), вносят точную навеску (0,08 г) оксида ванадия(V), 20 мл смеси серной и фосфорной кислот (200 мл серной кислоты плотностью 1,84 г/см3 и 100 мл фосфорной кислоты плотностью 1,7 г/см3); закрывают стакан часовым стеклом и ставят на сильно нагретую плитку для растворения оксида ванадия, которое происходит очень быстро. Вместо сухого оксида ванадия можно использовать заранее приготовленный раствор оксида ванадия в смеси кислот. Для приготовления такого раствора поступают следующим образом: 4 г V2O5 растворяют при умеренном нагревании в 100 мл смеси серной и фосфорной кислот и затем доводят раствор водой в мерной колбе до 1 л. Для разложения навески 0,25 г берут 20 мл этого раствора, упаривают до начала кристаллизации, после чего добавляют 10 мл смеси кислот, вновь нагревают до растворения, вносят навеску и снова нагревают до полного растворения. Если проба была тонко растерта, то полное растворение ее происходит за 5—15 мин в зависимости от состава*.
Совершенно прозрачный раствор после полного охлаждения осторожно разбавляют водой, переносят в мерную колбу емкостью 100—200 мл и доливают до метки воду. В таких же точно условиях проводят холостой опыт с раствором, содержащим то же количество оксида ванадия и ту же смесь кислот. Длительного нагревания при этом следует избегать, так как могут выпасть малорастворимые основные соли.
Вместо титрования избытка ванадия (V) при определении железа (II) в хроматах описанным методом можно титровать непосредственно ванадий (IV), образовавшийся при окислении железа (II). Однако обычное титрование перманганатом в присутствии зеленовато-фиолетовой окраски ионов хрома(III) затруднительно. Амперометрический же вариант этого титрования дает достаточно точные результаты. Титруют при +0,60 В (МИЭ) по току восстановления избытка 'перманганата. Преимуществом прямого титрования ванадия(IV) является то, что в таком случае отпадает необходимость во взятии точной навески оксида ванадия(V) для окисления железа (II).
Описанный метод, разработанный для хромитов, может быть применен для железа (II) в любом другом минерале, в некоторых горных породах и в промышленных продуктах (огнеупорных материалах и т. д.).
в) Определение железа при помощи реакций комплексообразования сводится в основном к титрованию железа(III) ЭДТА. Оно осуществляется в различных вариантах. С ртутным капаю-
Как известно, железо(II) в хромитах очень устойчиво и ие окисляется иа воздухе в процессе растирания пробы; для других же пород очень тонкое растирание, во избежание окисления железа (II), не рекомендуется.
10—1694 145
щим электродом [27] титрование железа(III) можно проводить раствором ЭДТА при потенциалах от +0,020 до +0,100 В (Нас. КЭ) при рН=3—4,5 в присутствии ионов щелочноземельных металлов, вольфрама(V), молибдена(VI), марганца(II) (при не очень большом его содержании) и различных анионов, за исключением хлорид-иона, который может, как известно, образовывать с ионами ртути комплексные или малорастворимые соединения, если ртуть находится под достаточно «анодным» потенциалом для ее окисления. Поэтому в присутствии хлоридов титрование следует проводить при потенциалах менее положительных, чем указанный выше потенциал, рекомендуемый для комплексонометрического определения железа (III) в отсутствие хлорид-иона.
При работе с платиновым электродом используют ток окисления ЭДТА при +0,9 В (Нас. КЭ) в солянокислом растворе [28j, так что кривая титрования имеет форму б.
Применяют также танталовые [29] и графитовые [30] электроды. Некоторые исследователи применяют метод с двумя электродами— платиновыми [31—35], графитовыми [35] и рутутными [36].
Последний метод [36] разработан для определения не только железа, но и других элементов.
Описаны методы определения при помощи ЭДТА железа (III) в рудах [37], в машинных маслах [38], в кислотах [33] и в других объектах. Много внимания уделяется определению железа (Ш) в присутствии других ионов. Так, по данным работы [35], можно определять наряду с железом другие ионы, основываясь на различной устойчивости их комплексонатов и соответст-венно*регулируя pH раствора.
Если в растворе одновременно присутствуют железо (III) и железо (II), то пользуются тем, что комплексонат железа (III) несравненно более устойчив, чем комплексонат железа (II), рК обоих комплексонатов составляют соответственно 25,1 и 14,3 в растворе нитрата калия [39]. Кроме того, устойчивость комплексонатов зависит от pH раствора. Поэтому можно титровать сначала железо(III) при рН^5 (ацетатный буферный раствор), а после конечной точки изменить pH до 7 и титровать железо(II) тем же раствором ЭДТА. Однако вторая конечная точка, вследствие относительно малой устойчивости комплексоната железа (II), недостаточно отчетлива, поэтому авторы описываемого метода рекомендуют следующий прием. Пссле того как оттитровано железо (Ш). добавляют избыток ЭДТА, достаточный для связывания всего железа(II), устанавливают рН«7 при помощи раствора щелочи и титруют раствором феррицианида. В присутствии ЭДТА окислительно-восстановительный потенциал системы Feni/Fen сильно понижается (до +0,136 В), поэтому феррицианид оказывается окислителем по отношению к железу(II), связанному в комплекс с ЭДТА (£р системы [Fe(CN)6]3+[Fe(CN)6]4- в данной среде равен +0,45 В).
146
1
Для определения железа и алюминия в природных и искусственных силикатах предложена следующая методика [40].
Ход анализа. Мелкоизмельченную пробу сплавляют с 5-кратным количеством Na СОз или К2СО3, плав выщелачивают азотной кислотой и удаляют SiO2. Аликвотную часть полученного раствора нейтрализуют до рН=1,5—2,0, разбавляют до 100 мл и при перемешивании титруют 0,005 М раствором 1,2-диаминциклогек-сантетрауксусной кислоты (раствор 1) при напряжении на индикаторных платиновых электродах (поверхностью 0,5 см2) 0,6—1 В (в зависимости от чувствительности применяемого гальванометра). По достижении точки эквивалентности вводят монохлорацетатно-аммиачный раствор до рН=3,5—4,0, нагревают до 50— 80 °C и при перемешивании титруют 0,05 М раствором 1 при напряжении на индикаторных электродах 0,8—1 В. При анализе шлаков (10,14% Fe и 9,24% А1) и бокситов (11,68% Fe, 22,65% Al, 0,14% С, 2,21 Ti и 2,70% SiO2) ошибка определения А1 и Fe<l%.
Комплексонометрическое определение железа в двойных и более сложных смесях с рядом других элементов было разработано ташкентскими химиками [41—45] в основном с танталовым электродом. Последовательное определение элементов достигается изменением pH раствора в соответствии с рК комплексонатов соответствующих элементов. При необходимости можно титровать железо (II) ЭДТА также в среде хлороформа и безводной уксусной кислоты [46].
Относительно применения других комплексообразующих реагентов для амперометрического определения железа сведений почти нет, за исключением оксалата калия, который был предложен [47] для совместного титрования кальция и железа, и уни-тиола, образующего комплексные соединения с железом (II) [48].
г) Определение железа по методу осаждения может быть выполнено при помощи различных реактивов, образующих с железом малорастворимые соединения. Поскольку железо(III) восстанавливается как па платиновом, так и на ртутном электроде, титрование можно проводить и такими реагентами, которые сами не дают электродной реакции, например раствором двузамещенного фосфата натрия [49] или щелочи [50]; можно применять также оксихинолин [51, 52], купферон [53, 54], а-нитрозо-р-наф-тол [55] и тиооксин (меркаптохинолин) [56], позволяющий определять железо (III) не только отдельно, но и попутно с другими элементами — медью, палладием, иридием [57, 58].
Однако оцределение железа по методу осаждения не имеет преимуществ перед описанными выше методами, так как селективность действия осадителей недостаточно высока и, кроме того, состав осадка сильно зависит от условий осаждения.
Несколько особняком стоят работы по титрованию перхлората железа(II) Fe(C104)2 в ледяной уксусной кислоте различными окислителями — раствором хромового ангидрида в уксусной кислоте [59], монохлоридом иода в присутствии ацетата натрия или бромом [60]. Все три метода выполняют с двумя платиновыми электродами. Область применения этих методов, очевидно, невелика, но в некоторых случаях они могут оказаться полезными.
*°* 147
Таблица 19. Методы определения железа
Определяемый ион Титрант Определяемый минимум Условия определения Примечания Литература
Fein Аскорбиновая кислота Электрод Pt (титрование по току окисления титранта) или ртутный капающий (титрование по току восстановления Fein) 1—3
Sn” Нитрат ртути (I) Перхлорат ртути (I) 4-10-5 М Fe 3 '10- 5 М Си Нижний предел io-5 М 2 угольных электрода, Л<р=О,1 В, фон — 0,5 н. НС1, 65—75 °C Электрод Pt, <р= +0,3 В (МИЭ), фон — 0,5—5 и. H2SO4+роданид калия Электрод Pt, tp=+O,l В (Нас. КЭ), фон —0,2 н. KCNS+0.2 н НС1О4 (1 : 1), титруют по току восстановления Fe111 Применено при анализе железных руд, определена константа скорости реакции Fe3++Sn2+ Определение железа в силикатах и минеральной воде, можно определять также Си и Fe: в одной аликвотной части сумму, в другой — Си, маскируя Fe пирофосфатом 5 6 7
Предел 5 1 О'5—10~3 М 2 Pt электрода, Л<р от 50 до 200 мВ, фон —0,5 М KCNS+iO.OIl М НС1О для КзГ'Ре1(СМ)б], ОД М KCNS ,-0,1 М НСЮ4 для Fe3+, 0,02 М Ki+0,05 М H2SO4 для 12, 0,3 М KCN S ; 0,02 М *3+0,02 М НС1О4 для Си11, 0,01 -М KI + 0,2 М НС1О4 для CeIV Определяют Fe111 и Кз[Ре(СМ)6], а также Си11, CeIV и 12 8
ЭДТА Электрод ртутный, <p=0,02-F0,l В (Нас. КЭ), фон —рН=3—4,5 Электрод Pt, <р=+0,9 В (Нас КЭ), фон — солянокислый См. методику в тексте Титрование по току окисления ЭДТА 27 28
1 мкг/мл Электрод Та, (₽= 1,2 В (Нас. КЭ), фон — кислый, но не больше 0,1 н. < 29
От 2 до 70% Fe в рудах и минералах 2 Pt электрода, Дср=1 В, фон— НС1, pH =1,6—2,0 После разложения и удаления SiO2 окисляют Fe11 до Fe111 перекисью водорода, разлагают ее кипячением; считают, что метод более точен, чем обычные перманганатный и хроматный методы 31
1—100 мг 2 Pt электрода, Дер =1,0 В, фон— pH = 1,5—2,0, 60—70 °C Определение Fe111 в различных кислотах 33, 34
2 Pt или графитовых электрода, Д(р=|14-1,4 В, фон — НС1, pH регулируют соответственно рК образующегося комплексоната Определяют Fe111, при регулировка pH возможно селективное определение других иоиов 35
2 ртутных электрода, Дср=40 мВ, фон для Fe111—любой Кроме Fe111 можно определите и другие ионы (Bi, Pb, Ni, Zn, Cd) избыток ЭДТА титруют раствороь нитрата ртути (II) >36
2 Pt электрода, Дср=1 В, фон — рН=1,5—2,0, 60—70 °C Определяют Fe в рудах и дру гих материалах -37
0,1—0,025 мг Электрод Pt, (р=—0,2 В (Нас КЭ), фон—изопропиловый спирт подкисленный H2SO4 до 0,005 М, удаляют О2 продуванием СО2 Предложено определять Fe вма шинном и других маслах, мешаю Сг111, Си11 и TiIV -38
2 Pt электрода, фон — pH к 5 (ацетатный буферный раствор) для Fe111 и рН=7 для Fe11; Д(р= =0,6—1 В Определение Fe111 в присутст вии Fe11 (см. методику в тексте 39
/ 2 Pt электроца, Дср=0,6—1 В фон — для Fe рН=1,5—2,0; для А pH=3,5—4,0, 50- 80 °C Определение Fe и А1 в природ ных и искусственных силиката, (см. методику в тексте) -40
Продолжение
Определяемый ион Титрант Определяемый минимум Условия определения Примечания Литература
Электрод Та, <р= + 1,0—1,2 В (Нас. КЭ), pH в зависимости от рК определяемого элемента Последовательное титрование Fe и других ионов в тройных и более сложных смесях (Fe, Y, Mg; Ni, Fe, Cu, Ag; Y, Сг, Fe, Th, Mg; La, Th, Mg, Fe, Bi) 41—45
15—200 мкг/10 мл 2 Pt электрода, безводная среда СНзСООН—СНС13; Дф=0,6—1В 46
Тиооксин 0,75 мкг/мл Электрод Pt, <р= + 1,2 В (МИЭ), фон — ацетатный буферный раствор, pH—4—6 Состав осадка Fe(C9H6NS)3; можно определять Fe в силикатах без отделения других ионов 56
Нитрозодисульфонат калия 0,56 мг/25 мл Электрод Hg капающий, <р= =—0,3 В (Нас. КЭ), фон — 0,4 М ацетатный буферный раствор, pH=4—5 Титруют по току восстановления титранта, окисляющего Fe11 61
Ферроцен 50—1000 мкг Электрод Pt микродисковый, ф= = 0,0 В (Нас. КЭ), фон — водно-ацетоновая смесь (1:1), 2 М по 112SO4 Подробное исследование условий определения; определяют Fe в сплавах, мешают ионы фосфорной и щавелевой кислот 62
Fe(CN)g- Перхлорат ртути (I) 5-IO-8—1-Ю-3 М Электрод Pt, ф=0 (Нас. КЭ), фон — 0,01 н. НС1О4, 0,5 н. по KCNS или I н. NaOH, 0,4 н. по KI (в этом случае нижний предел определения 10~4 М) 9
Fell NH4VO3 1 . 2 Pt электрода, Д(р=0,05—0,1 В, фон — 2 н. H2SO4 После к. т. по железу можно связать Fe111 ЭДТА и титровать Со11 после снижения кислотности раствора до 0,5 и. по II2SO4 10
КдСгзОу
КМпО4
6—77% Fe при навесках 3—20 мг
10-6 М
сл
Электрод Pt, <р= + 1,0 В (Нас. КЭ), фон — HC1+H2SO4, предварительное восстановление Fe111 в Bi-редукторе
2 Pt электрода, Дер=0,05 В, 60—75 °C, фон—0,3—0,6 н. НС1
Электрод Pt, <р= + 1,0 В (Нас. КЭ), фон—1 М НС1
2 Pt электрода, Дер=0,05—0,1 В, фон — 2 н. H2SO4, Feln восстанавливают металлическим цинком
Электрод Pt, ер= + 1,0 В (Нас. КЭ), фон — 2 н. H2SO4, предварительное восстановление Fe111 в висмутовом редукторе
2 Pt электрода, Дер = 0,1 В, фон— 12 М Н3РО4, продувают N2
В присутствии V можно оттитро- 11 вать его после определения железа: окисляют VIV до Vv и титруют солью Мора
Предложен для определения 12 Fe11 и присутствии Fe111 Fe111 титруют тиосульфатом при 60—70 °C
Предложен для титрования Fe 13 в бронзах, поэтому перед определением Fe удаляют Си, восстанавливая ее до металла на цинковой стружке
После определения Fe11 титруют 14, Со11, добавив ЭДТА для понижения £р Со111/Со11
Метод рекомендован в микрова- 16 рианте для определения Fe в рудах
Наблюдаются два минимума на кривой титрования: первый соответствует к. т. определения Fe11, второй — Мп11, окисляющегося до Мп111
18
15
2 Pt электрода, А<р=0,05 В, фон— Титруют Fe11 и смесь Fe11 с VIV,
1 н. H2SO4 а также Fe11 в присутствии окса-
лата; в этом случае титрант— CeIV на фоне 5 М H2SO4
Электрод Pt, электрод сравне- Кроме Fe титруют различные ния — ХСЭ, <р=0 В, фон—0,5 М другие вещества
H2SO4
19
20
Продолжение
Определяемый иои Титрант * Определяемый минимум Условия определения Примечания Литература
Электрод Pt, <р= +1,1 В (МИЭ) или +0,5 В при титровании по току восстановления КМпО4, фон— солянокислый Вариант общеизвестного метода Фольгарда—Вольфа [восстановление железа (III) SnCl2]; амперометрическим методом исследованы все стадии процесса 21
Ce(SO4)2 Электрод Pt, <р = +1,25 В (Нас. КЭ), фон — сернокислый, титруют по току восстановления титранта Электрод Pt, <р=+0,9 В (Нас. КЭ), фон — сернокислый Электрод Pt, tp=0,6 В (Нас. КЭ), фон — сернокислый+щавеле-вая кислота («оксалатные ванны») В тонких ферритовых пленках определяют Fe, Мп, Zn и Со; Fe определяют без предварительного отделения, после восстановления в Bi-редукторе Если присутствует VIV, то после оттитровывания Fe11 титруют VIV тем же реактивом, но при ср=0,5В (по току восстановления CeIV) После к. т. ток не возрастает, так как присутствует оксалат-ион и CeIV продолжает расходоваться на его окисление 22 23 24
Соль Мора КМпО4 или 2 мкг/мл Электрод Pt, ср=0,9 В (МИЭ), фон—H2SO4-f Н3РО4 Определение Fe11 в рудах, после окисления Fe11 при сплавлении с V2O6 титруют раствором соли Мора избыток Vv или образовавшийся VIV раствором КМпО4 (см. методику в тексте) 25, 26
Унитиол Электрод Pt, ср =+0,6 В (МИЭ), фон — хлорйдно-аммиачный Титруется Fe11 — образуется комплексное соединение 48
В последние годы были применены еще два реактива для титрования железа(III): нитрозодисульфонат калия [61] и ферроцен [62], восстанавливающий железо (III) в водно-ацетоновой среде.
ЛИТЕРАТУРА
1. Усатенко Ю. И., Беклешова Г. Е. — Зав. лаб., 1954, т. 20, с. 266.
2. Singh D., Varma А. — Z. anal. Chem., 1961, Bd. 183, S. 172.
3' Erdey L., Ear say A. — Acta chim. Hungar, 1956, v. 9. p. 43.
4. Галлай 3. A.—Ж- анал. хим., 1961, т. 16, № 4, с. 63.
5 Бидиловский М. Я-, Усатенко Ю. И., Семенова Г. А. — Ж- анал. хим., 1970, ' т. 25, № 12, с. 23:5—2400.
6 Тараян В. М., Шапошникова Г. Н., Анарян Г. С.—Арм. хим. журн., 1970, ’ т. 26, Кв 1, с. 25—29.
7. Stock J. Т., Heath Р. — Analyst, 1965, v. 90, № 1072, р. 403.
8. Merrer R. Е, Stock ]. Т. — Anal. chim. acta, 1971, v. 53, № 2, p. 233—238.
9. Stock I. T., Merrer R. J. — Analyst, 1967, v. 92, № 1091, p. 98—102.
10 Singh D., Bhatnagar U. — J. Indian Chem. Soc., 1967, v. 44, № 3, p. 237— 238; РЖХим., 1967, 24Г95.
11 Ратовская А. А. и dp. — Труды Уфимского нефтяного ин-та, 1974, вып. 16, с. 212—214; РЖХим., 1975, 10Г94.
12. Будиловский М. Я-, Усатенко Ю. И. — Зав. лаб., 1970, т. 36, № 7, с. 788— 792.
13. Усатенко Ю. И. и др. — Зав. лаб., 1955, т. 22, Кв 1, с. 26—29.
14. Singh D., Bhatnagar U.—J. Scient. Res. Banares Hindu Univ., 1966—67, v. 17, № 2, p. 29—31; РЖХим., 1968, ЗГ96.
15. Singh D., Prasad В. B., Bhatnagar U. — J. Indian Chem. Soc., 1967, v. 44, № 3, p. 200—202; РЖХим., 1967, 24Г96.
16. Алимарин И. П., Фрид Б. И. — Зав. лаб., 1952, т. 18, с. 1300—1303.
17. Хадеев В. Л. — РЖХим., 1976, ЗГ111, деп. ВИНИТИ 14/Х—75 г., № 2976— 75 депон.
18. Singh D., Bhatnagar U. — Allgem. u. prakt. Chem., 1967, Bd. 18, № 4, S. 107— 108.
19. Singh D., Bhatnagar U. — J. Scient. Res. Banares Hindu Univ., 1964—1965, v. 15, Kb 2, p. 276—282.
20. Singh D., Sharma Shee’.a — Indian J. Chem., 1970, v. 8, № 2, p. 192; РЖХим., 1971, 22Г42.
21. Сонгина О. А., Ходасевич С. А.—Ж. анал. хим., 1961, т. 16. Кв 3, с. 516— 520.
22. Зенкевич О. В. — В кн.: Материалы II Межотраслевого совещания по методам получения и анализа ферритовых материалов и сырья для них. Ч. 2. М„ 1969, с. 39—44; РЖХим., 1969, 19Г69.
23. Алимарин И. П., Терин С. И. —Зав. лаб., 1955, т. 21, Кв 7, с. 777.
24. Боговина В. И., Новак В. П., Мальцев В. Ф. — Зав. лаб., 1963, т. 29, Кв 6, с. 654.
25. Хондрахина Е. Г., Егорова Л. Г., Сонгина О. А. — Изв. АН КазССР. Сер. хим., 1957, вып. 11, с. 45.
26. Кондрахина Е. Г., Сонгина О. А. — В кн.: Труды ин-та строительства и стройматериалов АН КазССР, 1958, т. 1, с. 149.
оо Matyska В. — Coll. Czech. Chem. Comm., 1951, v. 16, p. 139.
28. Усатенко Ю. И., Виткина М. А. — Зав. лаб., 1958, т. 24, № 10, с. 1058.
29. Жданов А. К., Умарова М. М. — В кн.: Некоторые вопросы химической технологии и физико-химического анализа. Ташкент, АН УзбССР, 1963.
*’0- F’’ Р. — J. Electroanalyt. Chem., 1970, v. 24, Кв 2—3, р. 379—
31. Martiny Е., Siresko V. — Acta geol. et geogr. Univ. Comenianae geol., 1968, № 15, p. 91—97; РЖХим., 1969, 5Г160.
153
32. Vydra F., Stulik K- — J. Electroanalyt. Chem., 1968, v. 16, № 3, p. 375—383.
33. Vorlicek Vydra F. — Microchem. J., 1965, v. 9, № 1, p. 1—8; РЖХим., 1966^57149.
34. Vorlicek J., Vydra F. — Coll. Czech. Chem., Comm., 1965, v. 30, № 12, p. 4272—4279.
35. Vydra F., Vorlicek I. — Taianta, 1964. v. 14, p. 665.
36. Петухова А. И., Торопова В. Ф.—Изв. вузов. Химия и хим. технол., 1974, т. 17, № 8, с. 1253—1254.
37. Будиловский М. Я-, Семенова Г. А., Усатенко Ю. И. — В кн.: Химическая технология. Респ. межвед. темат. научно-техн, сб., 1971, вып. 24, с. 28—32; РЖХим., 1972, 12Г124.
38. Гордиенко В. И., Сидоренко В. Н., Стеколыцикова И. Н. — Химия и технол. топлив и масел, 1969, № 11, с. 57—58.
39. Hall С., Flanigan D. А. — Anal. Chem., 1961, v. 33, р. 1495.
40. Vydra F., Stulik К- — Acta geol. et geogr. Univ. Comenianae Geol., 1968, Ns 15, p. 87—89; РЖХим., 1969, 5Г161.
41. Акентьева H. А., Жданов А. К., Закиров Б. Г. — ДАН УзбССР, 1970, № 4, с. 15—16.
42. Жданов А. К., Мархабаев И. А.—Журн. ВХО, 1973, т. 18, № 3, с. 350— 351.
43. Жданов А. К.., Бархударьян А. А.—ДАН УзбССР, 1976, № 22, с. 56—58.
44. Жданов А. К., Мархабаев И. А.—Узб. хим. ж., 1972, № 3, с. 25—27.
45. Мархабаев И. А., Жданов А. К-—Ж- анал. хим., 1973, т. 28, № 1, с. 168— 170.
46. Геворгян А. М., Талипов Ш. Т., Хадеев В. А.—РЖХим., 1973, 21Г91, депон. ВИНИТИ.
47. Усатенко Ю. И., Виткина М. А. —Укр. хим. ж., 1957, т. 23, с. 788.
48. Оспанов X. К.., Рождественская 3. Б. — Изв. вузов. Химия и хим. технол., 1965, вып. 3—4, с. 216.
49. Теодорович И. Л., Абрамов М. К.— ДАН УзбССР, 1958, № 1, с. 29.
50. Kamecki /., Slabon М. — Roczn. Chem., 1955, v. 29, p. 107.
51. Sandberg B. — Svensk Kem. Tiding., 1946, v. 58, p. 197; C. A., 1949, 349.
52. Berraz G., Delgado O., Vassallo M. — Rev. Fac. Ing. Quim. Univ. Nacion. Litoral, Santa Fe, Argent., 1958, v. 27, p. 65; Stock J. Amperometric Titrations. New York, 1965, p. 419.
53. Kolthoff I. M., Liberti A. —Analyst, 1949, v. 74, p. 635.
54. Canela A., Garcia Ladona A. M. — Quim. and pura у apl., 1974, v. 28, № 3, p. 144—146; РЖХим., 1975 2Г43.
55. Wilson Lovelady H. — Anal. Chem., 1955, v. 27, p. 1231.
56. Бессарабова И. M., Захаров В. А., Сонгина О. А. — Зав. лаб., 1968, № 8, т 34 с 936_____938
57. Усатенко Ю. И., Супрунович В. И.—ДАН СССР, 1963, № 153, с. 622.
58. Усатенко Ю. И., Супрунович В. И. — Зав. лаб., 1964, т. 30, № 6, с. 622.
59. Hinsvark О. N., Stone К. G. — Anal. Chem., 1955, v. 27, р. 371.
60. Piccardi G., Cellini P.—Anal. chim. acta, 1963, v. 29, p. 107.
61. Торопова В. Ф„ Дегтярева В. Н. — Изв. вузов. Химия и хим. технол., 1973, т. 17, № 2, с. 175—178.
62. Соломатин В. Т., Артемова Т. Н.—Зав. лаб., 1974, т. 40, № 8, с. 916—919.
Золото
Амперометрическое определение золота может заменить (во всяком случае частично) длительный и трудоемкий пробирный метод, поэтому за последнее время амперометрическому определению золота уделяется все больше и больше внимания.
В 1955 г. Н. А. Езерская [1] разработала два варианта амперометрического определения золота (Ш). Первый вариант осио-
154
ван на образовании малорастворимого осадка золота с меркаптобензотиазолом, второй — на титровании раствором тиосульфата натрия, с которым золото образует достаточно прочный комплекс. Титрование ведут без наложения напряжения относительно Нас." КЭ по току восстановления золота(Ш). Этот метод несколько менее чувствителен, он позволяет определять до 0,08 мг золота в 15 мл раствора (концентрация золота порядка 10-Е М). При меньших количествах ошибка определения достигает 50— 60%. По-видимому, это связано с тем, что по более поздним данным [2] состав продуктов окисления тиосульфата зависит от концентрации золота(III) в растворе, от кислотности раствора, скорости приливания реактива и некоторых других факторов, специально изученных в работе [2]. В 1976 г. к этому же методу вновь обратились индийские исследователи, предложившие определять золото в присутствии урана [3].
В настоящее время разработка амперометрических методов определения золота продолжается в трех направлениях: использование окислительно-восстановительного взаимодействия золота (П1) с различными восстановителями, образование растворимых комплексных соединений золота и осаждение золота (III) се-русодержащими органическими реагентами. Для титрования по методу восстановления золота(Ш) применяют следующие вещества: иодид калия [4], сульфат гидразина [5], ферроцианид [6, 7], соль Мора [8]. Соль Мора дает возможность определять золото в черновой меди без отделения золота от меди. Однако реак7 ция между золотом (III) и железом (II) в солянокислой среде проходит медленно. Поэтому лучше восстанавливать золото (III) избытком соли Мора и титровать не вошедшие в реакцию ионы Fe2+ раствором бихромата калия по току окисления Fe2+ на платиновом электроде при +1,2 В (МИЭ). Метод позволяет определять десятые доли миллиграмма золота в титруемом объеме, например в черновой меди. Хорошие результаты дает восстановление золо-та(П1) растворами солей ртути(1)—ее нитратом или перхлоратом. Разработано несколько вариантов этого метода [9, 10] с одним или двумя электродами.
Предложено также несколько органических восстановителей для титрования золота(III), например аскорбиновая кислота [П, 12], бензидин [13], производные пиразолона [14] и другие соединения [15—18]. Различные амины, а также гидрохинон и метол были испытаны еще в 1962 г. [19]. Эти вещества окисляются на платиновом электроде при потенциале от +0,8 до +1,1 В (Нас. КЭ) на фоне 2 н. серной кислоты, так что титровать можно по току окисления титрующего раствора. Золото(III) при потенциале + 1,0 В, при котором ведут титрование, не восстанавливается на электроде, так что кривая титрования имеет форму б. Ио-Hbi элементов, часто сопутствующих золоту,— селена, теллура, палладия, иридия, рубидия, рутения — ни на электроде при + 1,0 В, ни в растворе этими восстановителями не восстанавли
155
ваются и не мешают определению золота. Метод позволяет определять от 3 до 90 мг/л золота в промышленных продуктах.
В некоторых случаях восстановившееся при титровании золото (III) в виде золота (I) образует комплексные соединения с восстановителем. Таким восстановителем оказался тиокарбамид [20]. По данным авторов этой работы, точка эквивалентности при титровании на фоне азотной и серной кислот различной концентрации (от 0,001 н. до 12 н.) приходится на отношение золота к тискарбамиду 1:3. Титруют по току окисления тиокарбамида при + 0,65 В (Нас. КЭП Присутствие хлоридов в количествах, превышающих 0,1 М, мешает титрованию. При титровании на фоне с рН=4,5 конечная точка отвечает отношению золото: тиокар б-амид=2:3 [21]. Однако подробное исследование процесса комплексообразования золота(I) с тиокарбамидом [22] показало, что на кислом фоне расход тиокарбамида на титрование золота (III) соответствует 1:3; это значит, что сначала идет восстановление золота (Ш) до золота (I), на что расходуется 2 моль тиокарбамида, а затем 1 моль тиокарбамида связывает образовавшееся золото (I). Титрование золота тиокарбамидом было применено [22, 23] для определения золота в различных концентратах, рудах, шламах электролиза меди и т. д.
Ход анализа. Навеску 1—5 г (в зависимости от ожидаемого содержания золота) разлагают при нагревании 30—40 мл царской водки, затем выпаривают до объема 5—7 мл. Если проба не полностью разложилась, то добавляют еще 5—10 мл азотной кислоты (концентрированной) и окислители: 1—2 г хлората калия, 2—4 г нитрата аммония или немного жидкого брома. После полного разложения пробу упаривают с 15—20 мл соляной кислоты (1:1) до половины объема (для удаления основной массы окислов азота). Затем приливают 5— 7 мл концентрированной соляной кислоты, 10—15 мл воды и кипятят 15— 20 мин. После охлаждения переносят раствор вместе с осадком в мерную колбу емкостью 50 или 100 мл и доводят до метки водой. Переносят 20 мл раствора в стакан емкостью 50 мл н титруют при +0,7 В (Нас.КЭ) 0,002—0,0002 М (в зависимости от содержания золота) раствором тиокарбамида. После добавления каждой порции тиокарбамида выжидают 30 с перед снятием показаний гальванометра. Кривая имеет форму б.
Раствор тиокарбамида готовят растворением в воде точной навески дважды перекристаллизованного продажного препарата; 1 мл 0,001 М раствора тиокарбамида соответствует 0,0657 мг золота.
При анализе сульфидных руд и концентратов пробу следует сначала обработать 15—20 мл соляной кислоты (1: 1) при нагревании до полного удаления сероводорода и лишь после этого разлагать ее, как указано выше. Если же в пробе содержится большое количество двуокиси кремния, то для полного разложения пробы ее необходимо обработать смесью серной и фтористоводородной кислот и затем разлагать, как указано выше.
Если одновременно с золотом требуется определить палладий, то пробу разлагают, как указано выше, из мерной колбы отбирают две аликвотные части и титруют в одной из них сумму золо-та(Ш) и палладия(П) при +0,8 В (Нас. КЭ), а в другой сначала восстанавливают золото(III) солью Мора (несколькими кристал
156
ликами) при нагревании [золото(III) восстановится в этом случае до металлического] и титруют оставшийся в растворе палладий (П) раствором тиокарбамида. Содержание золота определяют по разности.
Так же как тиэкарбамид (восстановление с последующим комплексообразованием) реагирует с золотом и рубеанозодород-ная кислота [24].
Ряд органических реагентов дает комплексные соединения непосредственно с золотом (III), например димеркаптотиопироны [25, 26], производные дитиокарбаминовой кислоты [27, 26 с., 28], висмутолы [29].
Для титрования золота (III) по методу осаждения применяют в основном серусодержащие реагенты, образующие с золотом трудно растворимые соединения. Тиооксин (меркаптэхинолин) [30] и его производные [31, 32], а также фенилдитиобиурет [33] дают осадки, состав которых зависит от pH и состава среды, осадки постоянного состава образуются с тионалидом [17], с тиосалициловой кислотой [34], бензолсульфанилтиобензамидом [27, 23 с.], и унитиолом [36]. Унитиол— димеркаптопропанол-сульфонат натрия — восстанавливает золото(III) до золота(I), ко-/SAu (где торое осаждается унитиолом в виде соединения Rz
\SAu
R— условное обозначение группы CH2CHCH2SO3Na). На восстановление 1 моль золота(III) до золота(I) расходуется 1 моль унитиола:
/SH /S—s4
2R< + 2ДцЗ+ -->R >R-p2Au++4H-
XSH -4e~ XS—Sz
На связывание золота (I) расходуется еще 1 моль унитиола; таким образом, соотношение золота и унитиола в конечной точке титрования составляет 2:3. Следует^ иметь в виду, что в избытке реактива образовавшийся осадок унитиолата золота растворяется с образованием комплексного соединения, окрашивающего раствор в золотисто-желтый цвет. Конечная точка титрования определяется, однако, очень резко. Титрование ведут по току окисления унитиола на платиновом электроде при +0,8 В (Нас. КЭ), форма кривой б. В присутствии меди (II), если ее не больше чем ЮО-кратное количество по отношению к золоту, возрастание тока после конечной точки происходит особенно резко, так как образу-ющиееся при взаимодействии меди(II) с избытком унитиола комплексное соединение меди(1) окисляется на электроде с большей скоростью, чем чистый унитиол, т. е. медь играет в данном случае роль индикатора. Если меди в титруемом растворе нет, то рекомендуется прибавлять несколько капель раствора сульфата меди (около 10 мг считая на медь) к титруемому раствору. Остальные элементы, чаще всего сопутствующие золоту (свинец, цинк,
157
Таблица 20 Методы определения золота (Auln)
Титрант Определяемый минимум Условия определения Примечания Литература
Меркаптобенз-тигазол 0,01—2,0 мг Электрод Pt, tp=O В (Нас КЭ) по току восстановления золога(1П), фон — щелочной, спиртовой раствор титранта Железо (III) связывают фторидом, медь (II) мешает, так как осаждается реактивом 1
Тиосульфат натрия До 0,08 мг Электрод Pt, <р=0 В (Нас. КЭ), по току восстановления золота (III) Железо(Ш) связывают фторидом, медь(II) не мешает, если установить <р= 4-0,2 В (Нас. КЭ) 1
Электрод Pt, ср= 4-0,2 В (Нас. КЭ), фон—0,1 н. H2SO4 Определяют не только Au, но и последовательно Au и U 3
KI 2,4-10-3—6,3-IO-6 моль/л Электрод Pt, <р=+0,5—0,2 В (МИЭ) по току восстановления золота (III) или 4-4,0 В по току окисления I-, фон—рН=2,5—4,0 При титровании по току восстанов ления золота (III) реакция заканчивается при отношении AuIH:I_= = 1:2; при титровании по току окисления I- образуется Aul, т. е. отношение Au111:1~= 1 : 3 4
K4Fe(CN)6 10-4—10-6 моль/л Электрод Pt, ср= 4-0,4 В (МИЭ) по току восстановления золота (III) или при 4-1,0 В по току окисления реагента, фон — KNO3, NH4NO3, Na2SO4 Мешают ионы, реагирующие с фер роцианидом 6. 7
Соль Мора Десятые доли миллиграмма Электрод Pt, tp= 4-1,2 В (МИЭ), фон—НС1, разбавленная. Титруют избыток соли Мора бихроматом Рекомендовано для определения золота в черновой меди 8
Hg2(NO3)2 или. Hg2 (CIO4) 2 L0'5—10-3 моль/л Электрод графитовый, tp= 4-0,4 В (|МИЭ), фон — 0,1—0,5 М HCI, титре юз по катодному току золота(III) Железо(1П) устраняют пирофосфатом при рН=3, или восстанавливают золото(1П) до Au (мет.), растворяют в царской водке, выпаривают добавляют НС1 и Н2О2, кипятят, разбавляют водой и титруют 9
5-Ю-7— ЫО'3 моль/л Электрод Pt, tp=+O,l В (МИЭ), или 2 Pt электрода, Atp=O,l В, фон— 0,1—2 н. H2SO4 Метод применен для определения Au в рудах 10
Аскорбиновая кислота 10-3—10-6 моль/л 2 Pt электрода, А<р= 1 В, фон—0,1— 0 05 н. H2SO4 2 Pt электрода, Аф = 0,14 В, фон— 6 н. СНзСООН Можно последовательно титровать сначала золото(Ш), затем ртуть(П) 11 12
Бензидин 0,24—24,4 мг Au 0,13—8,1 мг Pd Электрод графит или Pt, ф= = + 1,0 В (Нас. КЭ), фон-НС1+КС!, рН=3, титруют по току окисления титранта Можно определять не только Au, но и Pd, отношение Au : R и Pd : R = = 1 1 13
Производные пи-разолона (антипирин и пирамидон) 1,5-IO-S—1,7-IO-3 моль/л Электрод Pt, tp=+0,4 В (МИЭ), фон—1 /И КВг, pH=2,0—5,25, титруют по току восстановления золо-та(Ш) Серебро не мешает 14
З-Окси-1-п-суль-фонатофенил-3-фенилтриазен Электрод Pt, ср=+0,9 В (Нас. КЭ) по току окисления титранта при определении Au, при +0,4 В при определении Pd, фон—0,1 /И NaNO3+HCl (pH=2—4) Рекомендуется для титрования Au и Pd при совместном присутствии; после оттитрования Pd реагент восстанавливает Аиш 15
Тиопирролидон 2-10~5 моль/л Электрод графитовый, <р=+0,2 В (МИЭ) по току восстановления Au111 Определяют Au в рудах, Se и Те не мешают 16
Нафтиламины 10~6—10~3 моль/л Электрод Pt или графит, <р= +0,2 В (Нас. КЭ), фон—HCI или H2SO4 (концентрация не указана) SeIV и TeIV замедляют реакцию; SeVI и TeVI не мешают в 70-кратном количестве при титровании Р-нафти-ламином и в больших количествах при титровании а-нафтиламином 17
Тионалид Электрод графит, ср=О В (МИЭ),по току Au111, фон—НС1 с рН=1—3 Осадок состава Au(RS)3 18
Гидрохинон, метол, аминофенол, фенилендиамин СП о 3—90 мг/л Электрод Pt, <р=+0,8—1,1 В (Нас КЭ), по току окисления реагентов фон — 2 н. H2SO4 См. текст 19
Продолжение
Титрант Определяемый минимум Условия определения Примечания Литература
Тиокарбамид Электрод Pt, <р= +0,65 В (Нас. КЭ), фон — 0,001—12 н. IINO3 или H2SO4 Отношение Au : тиокарбамид = 1 : 3 20
Электрод Pt, фон — HCl+NazCOa до pH=4,5 Отношение Au : тиокарбамид=2 : 3 21
Электрод Pt, <р= + 0,7 В (Нас. КЭ), фон — НС! Отношение Au :тиокарбамид= 1 :3 22, 23
Рубеановодо-родная кислота РЮ-3—3-10"6 Электрод Pt, <р=+0,2 (МИЭ) при титровании по току восстановления Aum или +0,8 В по току окисления титранта Отношение Au : титрант= 1 : 1 24
3-этил-2,6-ди-меркапто-1,4-тио-пирон; З-метил-5-фе-нил-2,6-димеркапто-1,4-тиопирон и 3-пиридил-2,6-ди-меркапто-1,4-тио-пирон 5-Ю-4—5-Ю-2 мг/мл Au- -50—600 мкг Электрод графитовый, ср=+0,7 В (Нас. КЭ), фон —0,1—8 714 НС1 или другие кислые фоны, а также ацетатный буферный раствор с рН=4 Отношение Au : титрант при рН = = 2,5 — 2 : 3, при pH=il — 1:2; можно раздельно титровать бинарные смеси— Au+Se(Te), Au+Ag (Си, Hg, РЬ), последовательное титрование Au—Ag—Си « 25, 26
Производные ди-тиокарбаминовой кислоты 0,02—2,0 мг Электрод графитовый, (( = +0,7 В (Нас. КЭ), фон—1 М НС1 Применено для определения Au в гальванических ваннах, в черновой меди, в кислых растворах отношение Аи:титрант=1 :2 при рН=8—11 27
11—1694
2-Цианэтиловый эфир бензол суль-фонилдитиокарба-миновой кислоты
5-меркапто-З-(нафтил-2)-1,3,4-тиадиазолтион-2 0,0025—2,5 мг/мл
Тиооксин (8-мер-каптохинолин) и его производные 0,3 мг/л
Тиосалициловая кислота 3-Ю-4—6-Ю-6 моль/л Au
Бензолсульфо-нилтиобензамид
0,009 мг
Унитиол
2 мкг/мл
Электрод графитовый, <р= + 1,0 В (Нас. КЭ), фон —0,1 М НС1О4 Hg11 и Pd11 титруются суммарно с Au, отношение Au : титрант=1 :2 28
Электрод графитовый, <р=0 В (Нас. КЭ) по току восстановления Аиш, фон — pH—5,3- -6,0 Отношение Au:титрант =1 :2 при указанном pH, с изменением pH состав комплекса меняется 29
Электрод Pt, <р=+0,9 В (Нас. КЭ), фон — 0,5 н. H2SO4 Состав осадка зависит от кислотности среды, лучшие результаты дает 5-бромзамещениое соединение 30-
Электрод графитовый, без наложения напряжения (МИЭ), фон — 0,1 М KNO3 для Ag, <р= +0,25 В, pH =11 для Au Отношение Au : реактив= 1 :3 для Au и 2 : 1 для Ag; другие катионы маскируют ЭДТА; определению Au и Ag мешают SeIV, Те1 , Т1+, MoVI 34
Электрод графитовый, <р=+0,9— 1 В (Нас. К-Э), фон — ацетатный буферный раствор, рН = 3; титруют по току окисления титранта Отношение Au : титр ант = 1 : 3, мешают Hg и Ag; применен для определения Au в иридиевых сплавах 35
Электрод Pt, <р=+0,8 В (Нас. КЭ), по току окисления титранта, фон — 0,5—4 М НС1 Отношение Au : унитиол=2 :3 36
•32
о
серебро, висмут), не мешают определению. Метод был проверен на золотых концентратах с содержанием золота от 170 до 900г/т.
Ход анализа. Навеску 1—5 г в зависимости от ожидаемого содержания золота, помещают в коническую колбу емкостью 250 мл и нагревают 5—10 мин с 15—20 мл соляной кислоты (1 : 1) до удаления сероводорода. Затем прибавляют 15 мл азотной кислоты плотностью 1,40 г/см3 и упаривают до объема Ь— 7 мл, если после этого остается темный королек серы, то повторно добавляют 2—3 мл азотной кислоты и выпаривают, добавляя если нужно, окислители — хлорат калия, нитрат аммония. Прибавляют 0,1—0,2 г хлорида натрия и раствор дважды упаривают с 7—10 мл соляной кислоты до половины объема (выпаривать досуха нельзя во избежание частичного разложения хлоридов золота). Затем приливают 6—7 мл соляной кислоты плотностью 1,19 г/см3 и 10—15 мл воды и кипятят до растворения солей, охлаждают, переводят в мерную колбу емкостью 50 или 100 мл и доводят до метки водой.
Переносят 25 мл раствора в стакан для титрования, добавляют сухой фторид аммония до обесцвечивания раствора (или до светло-голубого окрашивания в присутствии меди). Если от прибавления фторида аммония появится муть или осадок, это не повлияет на результат определения золота. Титруют при +0,8 В /Нас.КЭ) 0,002—0,001 М раствором унитиола, выжидая 30 с после каждого добавления реактива. Перед каждым титрованием электрод промывают водой и протирают фильтровальной бумагой.
Продолжительность определения золота в одной пробе составляет 80— 90 мин. Определяемый минимум — 50 мкг золота в 25 мл титруемого раствора, 1 мл 0,001 Л1 раствора унитиола соответствует 0,131 мг золота.
ЛИТЕРАТУРА
1 Езерская Н. А. — Изв. сектора платины АН СССР, 1955, вып., 32, с. 38.
2. Сонгина О. А., Пащенко А. И. — Изв. вузов. Химия и хим. технол., 1969, № 12, с. 558- 562.
3. Рапсе у Р. N., Singh М. М., Upadhaya Y. D. — Ind. J. Chem., Sec. A, 1976, v. 14, № 4, p. 288—289; РЖХим., 1977, 4Г11.
4. Тараян В. M., Шапошникова Г. Н.—Учен, зап Ереванского ун-та. Ест. н., 1968, № 3 (109), с. 123—126.
5. Бардин М. Б., Ляликов Ю. С., Темянко В. С. Анализ благородных металлов. М. Изд-во АН СССР, 1959, с. 80.
6. Пащенко А. И., Сонгина О. А.—Ж. анал. хим., 1964, т. 19, с. 303—306.
7. Захаров В. А., Сонгина О. А., Кальницкая Л. П.—Ж- анал. хим., 1971,
т. 26, № 3, с. 482—486.
8. Пащенко А. И., Сонгина О. А. — Зав. лаб., 1964, т. 30, № 10; с. 1064—1066.
9. Тараян В. М., Шапошникова Г. Н., Ачарян Г. С. — Зав. лаб., 1968, т. 34,
№ 5, с. 522—523.
10. Тараян В. М., Шапошникова Г. Н., Ачарян Г. С.—Арм. хим. ж., 1974, т. 27, № 4, с. 350—352.
11. Varma А.—J. Scient. Res. Banares Hindu Univ., v. 12, № 31, p. 1961—1962.
12. Singh D., Bhatnagar U. — Ind. J. Chem., 1967, v. 5, № 4, p. 164—165; РЖХим., 1967, 24Г57.
13. Вешева Л. В., Рейшахрит Л. С., Щербакова С. М. — В кн.: Применение органических реагентов в аналитической химии. Л., изд-во ЛГУ, 1969, с. 218— 220.
14. Луговой С. В., Рязанов И. П„ Темянко В. С. — Изв. вузов. Химия и хим. техн., 1967, т. 10, № 1, с. 23—27.
15. Темянко В. С. — В кн.: Сборник трудов Магнитогорского горно-металлургического ин-та. 1971, вып. 87, с. 34—36.
16. Тараян В. М., Саркисян А. А., Шапошникова Г. Н. — Арм. хим. ж., 1975, т. 28, № 6, с. 461—466.
17. Вартанян С. В., Тараян В М. — Арм. хим. ж., 1973, т. 26, № 3, с. 211—214.
,162
18 Шапошникова Г. Н„ Тараян В. М. — Зав. лаб., 1972, т. 38, № 10, с. 1188—
19 Рейшахрит Л. С., Сухобокова И. С.—Учен. зап. ЛГУ. Сер. хим., 1960, № 19, ‘ 150___154; Теория и практика полярографического анализа. Материалы
II совещ. по полярографии, Кишинев, Штиинца, 1962, с. 332.
20 Шумская А. И., Усатенко Ю. И. —В кн.: Научные труды Днепропетровского хим.-технол. ин-та. 1961, вып. 12, ч. 2, с. 173.
21 Deshmukh G. S., Tatwawadi S. V., Surendranath M. — Current Science, 1963, ' v. 32, p. 69.
22 Пащенко А. И., Сонгина О. A.—Зав. лаб., 1965, т. 31, № 12, с. 1312—1314.
23 Пащенко А. И., Сонгина О. А., Каргина П. И. — Зав. лаб., 1965, т. 31, № 11, ' с. 1312—1314.
24 Шапошникова Г. Н., Галфаян Н. Т., Тараян В. М.—Арм. хим. ж., 1975, ’ т. 28, № 2, с. 94— ’00.
25. Чуйко Т. В., Аришкевич А. М., Тулюпа Ф. М. — Ж- анал. хим., 1976, т. 31, № 4, с. 671—675.
26 Волошина В. В., Усатенко Ю. И., Аришкевич А. М.—Зав лаб., 1970, т. 36, ' Кв 5, с. 530—534.
27. Усатенко Ю. И., Тулюпа Ф. М„ Гарус 3. Ф. — В кн.: Химическая технология. Респ. межвед. научно-техн, сб 1968, вып. 14, с. 26—28.
28. Гарус 3. Ф. и др. — В кн.: Вопросы химии и химической технологии. Респ. межвед. темат. научно-техн, сб., 1972, вып. 26, с. 49—54.
29 Бусев А. И., Симонова Л. Н., Арутюнян А. А. — Ж. анал. хим., 1972, т. 27, Кв 6, с. 1209—1212.
30 Усатенко Ю. И., Супрунович В. Н. — Изв. АН ЛатвССР. Сер. хим., 1963, Кв 2, с. 181—184.
31. Берзиня В. К, Янсон Э. Ю., Седола В. А.—Учен. зап. Латв, ун-та, 1967, т. 88, с. 75—82.
32. Берзиня В. К, Янсон Э. Ю„ Зинченко Р. В. — Учен. зап. Латв, ун-та, 1967, т. 88, с. 89—92.
33. Усатенко Ю. И., Сухоручкина А. С.—Ж. анал. хим., 1963, т. 18, Кв 7, с. 1447—1449.
34. Тараян В. М., Погосян А. Н. — Изв. вузов. Химия и хим. технол., 1969, т. 12, Кв 6, с. 728- 732.
35. Тараян В. М., Саркисян А. А. — Арм. хим. ж., 1966, т. 19, № 12, с. 932— 937.
36. Сонгина О. А., Оспанов X. К, Рождественская 3. Б —Зав. лаб., 1964, т. 30, № 7, с. 664—668.
Индий
Известно три основных метода определения индия: осаждение ферроцианидом калия и тиооксином и образование очень прочного комплекса с ЭДТА.
Титрование ферроцианидом [1] проводят с ртутным капающим электродом при потенциале —0,75 В (Нас. КЭ) в солянокислой среде (0,1 М) шо току восстановления индия(III). Состав осадка соответствует формуле 1п4[Ре(СМ)6]з- Метод позволяет определять от 2 до 80 мкг индия. Если индий (III) присутствует вместе с Другими катионами, осаждаемыми ферроцианидом, то необходимо предварительно отделить его.
Совершенно очевидно, что это же определение можно выполнять с платиновым электродом, пользуясь током окисления ферроцианида, подобно тому, как определяют аналог индия — галлий (см. выше) и многие другие элементы.
~ Таблица 21. Методика определения индия (1пт) ---- -
Титрант Определяемое количество Условия определения Примечания Литература
K4Fe(CN)6] 2—80 мг Электрод Hg капающий, <р= =—0,75 В (Нас. КЭ), фон — 60%-ная уксусная кислота Fe111 связывают фторидом 1
Тиооксин 1 мкг/мл Электрод Pt, <р= +0,86 В (Нас. КЭ), фон — боратный или ацетатный буферный раствор, pH ^8 Титруют по току окисления тиооксина 2
1 мкг/мл Электрод Pt, <р= + 1,0В (НВЭ), фон — ацетатный буферный раствор, рН=4—7 Можно определять индий в присутствии Т1 и Ga (см. текст) 3
ЭДТА Электрод Hg капающий, <р=—0,7 В (Нас. КЭ), фон —рН=1—2 См. методику в тексте Г 4—6
8 • 10-6 моль/л Электрод Hg капающий, <р=—0,5 В (Нас. КЭ), фон —NaBr, pH = 1—1,5 Можно титровать раздельно In и Cd, In и РЬ 7
0,1 мкг/мл Электрод Та, <р= +1,25 В (Нас. КЭ) Мешающие примеси отделяют на катионите КУ-2 8
2,5—50 мг/100 мл 2 Pt или графитовых электрода Д<р=1,5 В, фон —0,1 М HNO3, рН= = 1,5—3,5 Определяют индий в сплавах Cd— Sb—In и Zn—Sb—In 9
1 Электрод Та, <р= + 1,2 В (Нас. КЭ) фон —HNO3, pH =1,5 Последовательное титрование In и Zn In и Pb, In, Ga, Cd (в искусственных смесях) 10, И
0,06—20 мг/40 мл Электрод Та, <р= +1,2 В (Нас. КЭ), фон — HNO3, pH=2—3 для титрования индия, затем добавляют сухой ацетат калия до pH=4,5—5,5 для Cd или РЬ Последовательное титрование In, Cd, РЬ в сплавах 12, 13
50—1000 мкг Электрод Pt, ф= +1,0—13 В (Нас. КЭ) или 2 Pt или Та электрода, Д<р=0,8—(1,0 В, фон — 10%-ный раствор безводного ацетата натрия в ледяной уксусной кислоте + хлороформный экстракт дитизоната или диэтилдитиокарбамата определяемого элемента Титруют йосле нагревания исследуемого раствора с несколькими каплями 0,5 М раствора СгО3 в ледяной уксусной кислоте для разрушения экстрагента. Раствор ЭДТА—в 11%-иой Na Ас в ледяной уксусной кислоте. Определяют In, Zn, Си, Bi 14—16
Те же условия без экстракции Титруют те же элементы 17
20—400 мкг/10 мл Те же, условия, но к фону добавляется бензол, хлороформ, СС1« Титрование In, Ga Sc и других металлов 18, 19
10—20 мкг/10 мл Электрод Та, <р= 1,5—3 В (Нас. КЭ), фон — экстракт иодидных комплексов индия в ацетилацетоне в присутствии AgNO3, рН= 1,5—3 Проверено на чистых растворах (см. также разделы «Галлий» и «Висмут») 21
Урамил-N — N-диуксусная кислота w i 6,4-10-5 моль/л Электрод Pt, или графит, <р= = + 0,8 В (Нас. КЭ), фон — H2SO4, рН=1—2 См. также раздел «Галлий» 22
Комплексонат магния в растворе ледяной уксусной кислоты + КАс СП 46—230 мкг Электрод Та, <р=+0,95—1,0 В (Нас. КЭ), фон —ледяная уксусная кислота + КАс или LiClO4; в присутствии 12, <р=0,75—0,8 В (концентрация 12=5-10-4—1-10-® моль/л) Определение In и Си; можно титровать индий в виде иодидного комплекса 20
Тиооксин (8-меркаптооксихинолин) осаждает индий (III) в ацетатной, боратной, янтарнокислой среде при pH=3,5—8 в молярном отношении 1:3 [2] и позволяет определять индий(Ш) в присутствии таллия(III) и галлия(III) [3]. Индий (III) титруют и на фоне тартратного буферного раствора с pH=4,5, в котором галлий (П1) и таллий (III) не реагируют с тиооксином. Галлий (Ш) можно определять на ацетатном фоне (см. раздел «Галлий»), а таллий (Ш)-в 1 н. серной кислоте, в которой он восстанавливается тиооксином до таллия(I). В ацетатной среде титруются индий и галлий совместно.
ЭДТА был предложен для определения индия(Ш) в 1956 г. [4]. В настоящее время описаны варианты этого метода [4—13]. Для определения индия в продуктах металлургического производства и в сфалеритовых концентратах с малым содержанием индия предложена следующая методика.
Ход анализа. При определении индия в сфалеритах навеску 2 г растворяют в фарфоровой чашке в 15—20 мл бромистоводородной кислоты плотностью 1,4 г/см3. После прекращения бурной реакции раствор упаривают на водяной бане до 2—3 мл, добавляют 1 мл концентрированной азотной кислоты и выпаривают досуха. К сухому остатку добавляют 5 мл бромистоводородной кислоты, 1 мл азотной кислоты и снова выпаривают на водяной бане досуха (при этой операции удаляются мышьяк и олово). Остаток растворяют в 10—15 мл 5 н. раствора бромистоводородной кислоты, добавляют около 0,1 г иодида калия и затем постепенно, при помешивании, вводят тиосульфат натрия до обесцвечивания жидкости и еще несколько кристаллов [эта операция имеет целью восстановление железа(Ш) и сурьмы(V) и выделение молибдена(У1) в виде сульфида].
Обесцвеченный раствор фильтруют через сухой фильтр диаметром 5 см, осадок на фильтре промывают 2—3 раза 5н. раствором бромистоводородной кислоты порциями по 2—3 мл. Фильтрат вместе с промывными водами собирают в делительную воронку емкостью 50—60 мл, добавляют равный объем бутилацетата и, закрыв пробкой, встряхивают 1 мин. После разделения нижний слой сливают и отбрасывают, к экстракту добавляют 3 мл 5 н. бромистоводородной кислоты, несколько кристалликов тиосульфата натрия и энергично встряхивают воронку. Нижний слой опять сливают, вторично промывают экстракт раствором бромистоводородной кислоты. Из слоя органического растворителя индий реэкст-рагируют водой, водный раствор переводят в стакан и еще раз реэкстрагируют индий водой. Объединенные растворы упаривают на песочной бане примерно до 5 мл, добавляют 1 мл 4%-ного раствора аскорбиновой кислоты для восстановления железа (III) и 0,1-0,4 мл 5 %-кого раствора тиокарбамида для связывания следов мед1.'(П). Нейтрализуют разбавленным раствором аммиака по тропеолину ОО и прибавляют 15 мл буферного раствора (50 мл 0,2 М раствора КС1 и 97 мл 0,2 н. соляной кислоты в объеме 200 мл). Титруют из микробюретки емкостью 1 мл 0,005 М раствором ЭДТА, прибавляя его по 0,2 мл. 1 мл титранта соответствует 0,574 мг индия.
Кривая титрования имеет форму «а». Метод позволяет определять сотые доли процента индия в пробах.
Для некоторых особых случаев можно рекомендовать методики определения индия при помощи ЭДТА в неводных растворах [14—22].
Для определения индия можно использовать также диэтилдитиокарбамат натрия, 8-оксихинолин, фосфат натрия. Однако наилучшие результаты, по мнению Долежала и Зыки [23], специаль
166
но исследовавших вез эти методы, дают методы с применением ЭДТА и ферроцианида калия.
ЛИТЕРАТУРА
1 Nimer N., Натт R., Lee G. — Anal. Chem., 1950, v. 22, р. 790.
2 Усатенко К). И., Супрунович В. И. — Изв. АН ЛатвССР. Сер. хим., 1963, № 5, с. 557.
3 Захаров В. А., Сонгина О. А., Бессарабова И. М. и др. — Ж. анал. хим., 1970, т. 25, № 5, с. 879—884.
4. Treindl L. — Chem. Listy, 1956, v. 50, (80), p. 534.
5. Владимирова В. M.—Зав. лаб., 1957, т. 23, № 12, с. 1286.
6. Цывина Б. С., Владимирова В. М.—Зав. лаб., 1958, т. 24, №3, с. 278.
7. Натт R., Furse С. — Anal. Chem., 1962, v. 34, р. 219.
8. Хадеев В. А., Базаров И. — Узб. хим. ж., 1962, т. 6, с. 47.
9. Vydra F., Vorlicek J. Taianta, 1966, v. 13, № 3, p. 349—443.
10 Жданов А. K-, Акентьева H. А., Лукьяненко И. Л.—ДАН УзбССР, 1970, ' № 12, с. 28—29.
11 Жданов А. К-, Акентьева Н. А., Корытко П. А-—'ДАН УзбССР, 1972, № 7, с. 33—34.
12. Жданов А. К., Капица Н. В. —ДМА УзбССР, 1969, № 1, с. 29—30.
13. Жданов А. К-, Капица Н. В. —ДАН УзбССР, 1969, № 1, с. 26—27.
14. Геворгян А. М. и др. — В кн.: XI Менделеевский Съезд по обшей и прикладной химии. М., Наука, 1975, № 5, с. 32.
15. Геворгян А. М-, Талипов Ш. Т„ Хадеев В. А. — Зав. лаб., 1976, т. 42, № 6, с. 646—647.
16. Мухамеджанова Д., Хадеев В. А., Талипов Ш. Т.—ДАН УзбССР, 1973, № 11, с. 31—32.
17. Геворгян А. М., Талипов Ш. Т., Хадеев В. А. — Узб. хим. ж., 1973, № 4, с. 23—25.
18. Геворгян А. М., Талипов Ш. Т„ Хадеев В. А. — РЖХим, 1974, 4Г49, депон. ВИНИТИ.
19. Геворгян А. М., Талипов Ш. Т., Хадеев В. А. — РЖХим, 1973, 23Г101, депон. ВИНИТИ.
20. Геворгян А. М. и др. —Узб. хим. ж., 1977, № 2, с. 80—82.
21. Мухамеджанова Д., Хадеев В. А., Талипов Ш. Т.—ДАН УзбССР, 1974, № 7, с. 37—38.
22. Бусев А. И., Галлай 3. А., Зверяева И. В.—Зав. лаб., 1974, т. 40, № 5, с. 514—517.
23. Dolezal J., Zyk& J. — Coll. Czech. Chem. Comm., 1961, v. 26, p. 1464.
Иод
Для определения иодид-ионов могут быть использованы как методы осаждения его. например в виде иодида серебра, так и окислительно-восстановительные реакции. Во всех случаях применяют платиновый вращающийся или графитовый электрод.
При титровании нитратом серебра образуется иодид серебра, обладающий очень малым произведением растворимости 10-16Л1. с то обстоятельство дает возможность определять иодид-ион не только в сильно разбавленных растворах, но и в присутствии других галогенидов, в частности хлорида, так как вследствие малой растворимости иодида серебра для его выделения можно применять аммиачные растворы, из которых хлорид серебра, как известно, не выпадает. Методики определения иодид-иона с помо
167
щью нитрата серебра в присутствии хлорид- и бромид-ионов приведены в разделе «Хлор». Можно титровать иодид-ион также солями ртути, поскольку осадки иодида ртути(I) и ртути(II) также мало растворимы [1—5].
. Несколько отличается от других методика спределения иодида на фоне подавляющих количеств хлорид-иона [6]. При титровании раствором нитрата серебра иодид-ион осаждается в первую очередь, причем ток в цепи отсутствует (титруют по току восстановления серебра). Когда иодид-ион оттитрован, начинается реакция между ионами серебра(I) и хлорид-ионами, образуется осадок AgCl, который восстанавливается на электроде [7]. Ток возрастает, конечная точка обозначается очень резко. Хлорид-ион в этом случае не определяют, так как концентрация его в растворе очень велика, не менее 1 М.
Иодид-ион окисляется на платиновом электроде до иода, и, в зависимости от условий, даже до положительно_ заряженного иона 1+ и кислородсодержащих анионов Ю~, Юз и т. д. [8—10]. На диффузионном токе окисления иодида основано «анодное» определение серебра(I) и ртути(II) (см. соответствующие разделы). Этот же анодный ток может быть использован и для определения самого иодида.
При одновременном присутствии в растворе ионов 1“ и Юз оба иона могут быть оттитрованы последовательно раствором нитрата серебра [11, 12].
Определение иодида удобно проводить при помощи двух платиновых электродов. При титровании по методу осаждения раствором нитрата серебра к титруемому раствору добавляют немного свободного иода — получается хорошо обратимая пара 12/21_, на кривой титрования очень резко обозначается конечная точка. Напряжение накладывают небольшое, 0,025 В [13]. При таком же напряжении можно титровать иодид раствором йодата калия, причем в раствор добавляют несколько миллилитров хлороформа для растворения выделяющегося цри титрования иода [14]. На двух платиновых электродах титруют иодид окислителями (перйодатом, нодатом, броматом) в присутствии цианид-иона [15]. При изучении взаимодействия бихромата с различными восстановителями [16] было установлено, что иодид-ион может быть оттитрован бихроматом в присутствии любых количеств бромида и хлорида, поскольку иодид окисляется бихроматом в первую очередь, вследствие большой разности реальных потенциалов соответствующих редокс-систем.
Среди методов определения иода при помощи восстановителей в первую очередь надо назвать тиосульфатный метод. Титрование тиосульфатом применяют как для определения самого иода, так и для других определений, основанных на реакции между иодидом и веществом, вытесняющим иодид из его соединений, в частности для определения меди (II), железа (1П), мышьяка (V) и т. д. Эти методы описаны в соответствующих разделах. Следует под-
168
Таблица 22. Методы определения иода
Определяемый ион Титрант Определяемое количество Условия определения Примечания т итера- УРа
г Hg(NO3)2 Нижний предел 10-4 моль/125 мл ♦ 3 J0~s моль/л 2,5-10-3— 4-10-5 моль/л Электрод Pd, f=0,l мкА, фон — 0,1 М HNO3 в 80%-ном этаноле. Титрование потенциометрическое с наложением тока, но может быть осуществлено в амперометрическом варианте Два Hg электрода, Дф=40 мВ, фон —0,1 М H2SO4+0,5 М K2SO4 Электрод Pt, <p=+d,3 В (МИЭ), фон — 0,1 М H2SO4 Применение палладиевого электрода не оправдано hi обходимостью, титрование успешно пойдет и с Pt электродом; так же титруют С1~ и Вг_ Кроме I- определяют С1_ и Вг~; смесь С1~ и Вг~ можно титровать без разделения прн отношении С1_ : Вг~ не выше 7 : 1 Можно последовательно титровать I-, Вг~, С1~ при соотношении их в пределах 10: 1—1 : 10 2 3
К2СгаО7 3 мкг/мл Электрод Pt, <р= + 1,0 В (Нас.КЭ), фон — 4 н. H2SO4 Не мешают 1000—2000-кратные количества С1~ н Вг_ соответственно 16
AgNOa 0,01—1,5 мг/20 мл в 1 М NaCl Катод графитовый, анод графитовый, покрытый серебром (электролитически), анолит — KNO3, католит — исследуемый раствор, содержащий 1 моль/л NaCl, Дф»0,4 В Рекомендуется для титрования I-в минеральных водах и других солевых растворах 6
I-, 10- AgNOs 0,001—0,01 моль/л I-, 0,002—0,01 моль/л 10 з Электрод Pt, <р=0 В (Нас.КЭ), фон — KNO3, ацетат Na или аммония для определения 1~, затем вводят 0,02% желатины и 30—40% этанола — 1О3 Электрод Pt, ф=0 В (Нас.КЭ), фон — нейтральная среда; для определения IO з понижают температуру и добавляют 40% этанола Последовательное титрование I-, IO7 основано на разнице в растворимости осадков Agl и AglOs Титрование автоматическое, последовательное определение I- и 1О3 11 12
I. HgC104 5-10~s—10-3 моль/л Электрод Pt, ’ф=0 В (Нас. КЭ) или 2 Pt электрода, Дф=50—200 мВ фон —0,02 М KI+0,05 М HgSj4 4, 5
Na2S2C)g Любые количества Электрод Pt, титруют по току восстановления иода, ф=+0,2 В (НВЭ) или 2 Pt электрода, Дф=0,025 В Пример определения малых коли честв иода в морской воде приведег в тексте 17. 18 1
черкнуть, что амперометрический метод определения иода более точен, чем обычный титриметрический метод с применением крахмала. Специальное исследование [17] показало, что амперометрическое титрование иода тиосульфатом 'позволяет определять от 20 до 40 мкг иода в 50—200 мл раствора с большей точностью, чем другие методы электрометрического титрования. Реакция иода с тиосульфатом стала «классической» в методе с двумя индикаторными электродами, так как образующаяся .при титровании обратимая пара 12/1~ обеспечивает резкую конечную точку.
В качестве примера приведем методику определения соединений иода в морской воде [18], .позволяющую определять тиосульфатным методом до 10-7 М иода.
Ход анализа. Анализируют 100 мл морской воды, подкисленной 10 мл 25%-ной уксусной кислоты, после предварительного удаления кислорода воздуха током азота. Перед титрованием добавляют 1 мл 0,0002 н. раствора тиосульфата и 2—3 кристаллика иодида калия. Присутствующий в воде иодат окисляет иодид, при этом выделяется эквивалентное количество иода, который вступает в реакцию с добавленным раствором тиосульфата. Избыток тиосульфата титруют раствором йодата калия, концентрация которого должна быть точно 0,000500 н. (растворы такого разбавления готовят из стандартного 0,0500 н. раствора, приготовляемого путем растворения 1,9496 г йодата калия в 1 л воды). Раствор тиосульфата приготовляют ежедневно из исходного 0,1 н. раствора, который готовят растворением 25 г ПагБгОз-БНгО в 1 л воды с добавлением около 1 мл сероуглерода для стабилизации раствора. Дистиллированная вода должна быть свежепрокипяченной. Разбавляют растворы также свежепрокипяченной водой.
В другой порции пробы окисляют иод до йодата бромной водой, при этом необходимо следить за тщательным удалением избытка брома, для чего раствор упаривают на водяной б а пр до половины объема, после чего разбавляют водой до прежнего объема. Далее продолжают анализ, как указано выше. Параллельно проводят холостой опыт.
Содержание иода и йодата находят по разности результатов титрования двух’ проб воды.
Метод позволяет определять десятые доли микрограмма иода в титруемом объеме воды.
Для определения иода можно применять и другие восстановители — гидразин [19], мышьяковистую [19], фениларсоновую [20] и аскорбиновую [21] кислоты, однако особых преимуществ по сравнению с тиосульфатом эти титранты не имеют.
Известен также весьма сложный метод определения иода при помощи железа (II) в присутствии ЭДТА [22], требующий инертной атмосферы при титровании, и метод титрования раствором трикарбонатокобальтиатогексамминкобальта (III), позволяющим определять многие ионы, в том числе и иодид-ион, цо не отличающимся большой устойчивостью [23].
ЛИТЕРАТУРА
1. Hanzdorf С. — Z. anal. Chem., 1969, v. 244, № 6, р. 368—371.
2. Петухова А. И., Торопова В. Ф.— Изв. вузов. Химия и хим. технол., 1971, т. 14, № 5, с. 685—690.
3. Шапошникова'Г. Н., Тараян В. М.—Арм. хим. ж,., 1976, т. 29, № 1, с. 22— 25.
170
л Stock J. T. — Analyst, 1966, v. 91, № 1081, p. 280—282.
5 Merrer R. Л Stock J. T. — Anal. chim. acta, 1971, v. 53, № 2, p. 233—238.
6 Вариков В Г., Сонгина О. А., Климко Е. В.— Зав. лаб., 1969, т. 35, № 11, ’ с. 1312—1313.
7 Захаров В. А., Сонгина О. А., Чултурова В. Ш. и др.—Ж. анал. хим., 1969, ‘ т. 24, № 7, с. 1401—1404.
8 Kolthoff I. М., Jordan J. — Anal. Chem., 1953, v. 25, p. 1833—1836.
9 Сонгина О. A.—Зав. лаб., 1955, т. 21, № 7, с. 665—667.
Ш Захаров В. А., Сонгина О. А.—Ж- физ. хим., 1962, т. 36, с. 1226—1229.
И Икэда С.—Anal. Instrum. (Japan), 1972, v. 10, № 1, p. 14—19; РЖХим., ’ 1972, 15Г101.
12 Мися Соитиру, Икэда Санаэ, Нисида Гиро — Бунсеки Кагаку, 1968, т. 17, ’ № 3, с. 332—338; РЖХим., 1969, 1Г103.
13. Philibert Н.—Medecine tropicale, 1955, v. 15, р. 356—359.
14. Stone К. О. — Anal. Chem., 1954, v. 15, p. 396—399.
15. Ries H. L. — Anal. chim. acta, 1958, v. 18, p. 14—17.
16 Миляева И. M„ Сонгина О. A.—Ж- анал. хим., 1969, т. 24, № 12, с. 1794— 1799
17. Knowles G., Lowden G. — Analyst, 1953, v. 78, p. 159—162.
18. Barkley R. A., Thompson T. G. — Anal. Chem., 1960, v. 32, p. 154—157.
19. Panvar K- S., Mathur N. K-, Rao S. P. — Anal. chim. acta, 1961, v. 24, p. 541— 544.
20. Kramer H., Moore W., Ballinger D. — Anal. Chem., 1952, v. 24, p. 1892—1895.
21. Галлай 3. А., Типцова В. Г., Пешкова В. М. — Вестник МГУ. Сер. мат., мех., астрой., физ. и хим., 1952, № 13, с. 209—212.
22. Sierra F., Sanchez-Pedreno С., Perez Ruiz Т., Martinez Losano С. — An. quim. Real. Soc. esp. fis. у quim., 1972, v. 68, № 7—8, p. 999—1004.
23. Хадеев В. А., Мухамеджанова Д.—Зав. лаб., 1970, т. 36, № 12, с. 1443— 1446.
Иридий
Для амперометрического определения иридия применяют редокс-методы, основанные на восстановлении иридия (IV) до иридия (III) или на окислении иридия(III) до иридия(IV). Кроме трех восстановителей — гидрохинона, аскорбиновой кислоты и тиооксина, подробно описанных ранее [1—3],—применяют и другие восстановители, а титрование тиооксином' [4, 5] распространено на определение иридия в сложных смесях. При титровании иридия (IV) ди-этанолдитиокарбаминатом [6] не мешают ионы платиновых металлов [палладий (II), платина (IV), родий (III)], а также медь(Ц) и ртуть(II).
Подробно изучено титрование иридия (IV) обычными неорганическими восстановителями — иодидом калия [7], ферроцианидом калия и солью Мора [8], а также иридия(III) окислителями — перманганатом [9, 10], бихроматом и ванадатом [10]. Измерение реальных потенциалов системы иридий(IV)/иридий(III) в различных средах показало зависимость этой величины от состава и кислотности среды, а такие же измерения потенциалов ре-Докс-систем восстановителей и окислителей, наряду с изучением кинетики соответствующих реакций между иридием и реагентом, позволили выбрать наиболее выгодные условия определения ири-
ДРазличной среде. Поскольку обе системы, т. е. система иридии (IV)/иридий (III) и редокс-система указанных выше реаген-
171
Таблица 23. Методы определения иридия
Определяемый ион Титрант Определяемый минимум Условия определения Примечания Литература
Iriv Гидрохинон или аскорбиновая кислота 2 мкг/мл Электрод Pt, (р= +0,44-0,5 В (Нас. КЭ), фон — НС1+N аС1, pH= = 1,5 Титруют по току восстановления иридия(1У), Pt, Pd, Си, Ni, Se, Те не мешают 1
Тиооксин ♦ 5—1000 мкг Электрод Pt, ср=+0,9 В (Нас. КЭ), в присутствии Pd11, фон — 0,5 М НС1 Электрод Pt, ф=+0,8+1,1 В (Нас. КЭ), фон — рН=0,9—3,5 2 Pt электрода, фон — 0,1—2 н. НС1, Д<р=0,1 В Титруют по току окисления 1г111, образующегося при восстановлении IrIV тиооксином, Pd11 играет роль индикатора и титруется сам Дифференцированное определение IrIV + PdII+MoVI и IrIV + + Cun+iMoVI Титруют IrIV, а также FeIH, Tl111, кроме того — другие ионы на другом фоне 2, 3 4 5
Диэтилдитио-карбаминат 0,002—0,2 мг/40 мл Электрод графитовый, <р=+0,2+ +0,4 В (Нас. КЭ), фон — буферный раствор с pH=3,5—4 Не мешают Fe111, Pd11, PtIV, Rh™, Hg11 и Си11 6
Иодид калия 1 мкг/мл Электрод Pt, q>=+0,8 В (НВЭ), фон — 1 М KNO3, или 1 М НС1, или 1 М H2SO4 He мешают Pt, Ru, Rh, на фоне НС1 не мешает также Pd 7
K4Fe(CNa)] или соль Мора 1 мкг/мл Электрод Pt, q>=+0,8 В (НВЭ), фон — 1 М H^SO4 или 1 М НО Не мешают Pt, Rh, Pd, ионы Fe111 связывают KF 8. 9
Irin КМпО4 1,5 мкг/мл Электрод Pt, tp= + l,l В (НВЭ), фон — 0,5—1 М H2SO4 He мешают Pt, Pd, Rh, Ru 10
К2СГ2О7 1 мкг/мл Электрод Pt, <р=-+1,0 В (НВЭ), фон — 8 М Нй5О4 He мешают Pt, Pd, Rh 10
:NH4VO3 2 мкг/мл Электрод Pt, <р= + 1,0 В (НВЭ), фон — 8 М H2SO4 He мешают Pt, Pd, Rh 10
тов дают вольтамперные кривые с катодными и анодными участками, то форма кривых титрования может быть различной, в зависимости от потенциалов, при которых проводят титрование. В табл. 23 указаны те потенциалы, при которых кривая титрования имеет резкий перелом в конечной точке при перемене направления тока.
Практически удобнее других перманганатный метод: реакция протекает быстро, при любой кислотности среды, тогда как бихромат, и особенно ванадат, реагируют гораздо медленнее и требуют для успешного титрования кислотности порядка 8 М серной кислоты. По сравнению с иодидным методом [7] для иридия (IV) перманганатный метод имеет то преимущество, что он позволяет определять иридий(III) в присутствии преобладающего количества палладия(П) и рутения(IV).
При титровании иридия (IV) восстановителями методика подготовки пробы к анализу сводится к следующему.
Навеску пробы (например, катализатора) 0,1—0,2 г разлагают 30—40 мл царской водки, выпаривают до влажных солей, добавляют 10—15 мл соляной кислоты (1:1) и снова выпаривают до состояния влажных солей После охлаждения приливают 20—30 мл воды, добавляют 3—5 мл 30%-ного раствора перекиси водорода для полного окисления иридия(III) до иридия(IV), кипятят до полного' разложения перекиси водорода, переносят в мерную колбу емкостью 100 мл и доводят до метки водой. К аликвотной части 2—5 мл добавляют тот или иной фон, в зависимости от выбранного титранта (см табл. 23).
Если титруют иридий(III) окислителями, то рекомендуется следующая подготовка пробы.
Навеску 0,1—0,5 г сплавляют с 5—6-кратным количеством перекиси натрия при 700—800 °C в корундовом или фарфоровом тигле 2 ч, выщелачивают соляной кислотой (1:1) и упаривают до состояния влажных солей. Добавляют 10— 15 мл воды, переносят, ие отфильтровывая кремневую кислоту, в мерную колбу емкостью 100 мл, доводят объем до метки водой, отбирают аликвотную часть (5—10 мл), приливают раствор фона (1 М сериую кислоту, если титруют перманганатом, или 10 М, если титруют бихроматом) до объема 20—25 мл. Чтобы выяснить, полностью ли восстановлен иридий (IV), налагают на платиновый электрод потенциал +0,8 В (НВЭ), т. е. потенциал, при котором иридий(ГУ) электрохимически восстанавливается до иридия(Ш), и, если при1 этом наблюдается катодный ток, то добавляют 0,2—0,5 г фторида натрия гь вводят по каплям 0,001 М раствор соли Мопа до полного исчезновения катодного тока. Затем устанавливают потенциал + 1,0+1,1 В (НВЭ) и титрую? перманганатом или бихроматом по теку их восстановления. До конечной точки наблюдается небольшой анодный ток окисления иридия(Ш), после конечной точки — катодный ток восстановления реагента.
Показано, что, пользуясь амперометрической индикацией конечной точки, можно определять малые количества иридия (IV) при кулонометрическом титровании его электрогенерированными ионами меди(1) [Ц] и титана (III) [12].
ЛИТЕРАТУРА
1- Пшеницын Н. К., Езерская И. А.—Ж- анал. хим., 1959, т. 14, № 1, с. 81. с 1963К° ’ СУпРУнович В- В- — Изв. АН ЛатвССР. Сер. хим. № 1,
173;
3. Усатенко Ю. И., Супрунович В. И.—ДАН СССР, 1963, № 153, с. 622.
4. Супрунович В. И., Усатенко Ю. И.—Ж- анал. хим., 1965, т. 20, № 7, с. 800—806.
5. Супрунович В. И., Усатенко Ю. И., Ямнова Т. П. — В кн.: Вопросы химической технологии. Респ. межвед. темат. научно-техн, сб., 1974, вып. 34, с. 18—25; РЖХим., 1974, 21Г26.
6. Шаронина Р. М., Усатенко Ю. И., Тулюпа Ф. М. — В кн.: Новые методы химического анализа материалов. Ns 2. М., 1971, с. 24—27.
7. Захаров В. А., Сонгина О. А., Айтхожаева Т. А.—Ж. анал. хим., 1975, т. 30, Ns 7, с. 1430—1432.
8. Айтхожаева Т. А., Сонгина О. А., Захаров В. А. — В кн.: Прикладная и теоретическая химия. Вып. 6. Алма-Ата, 1975, с. 8—14.
9. Айтхожаева Т. А., Захаров В. А.— Зав. лаб., 1976, т. 42, № 9, с. 1042—1043. 10. Захаров В. А., Сонгина О. А., Айтхожаева Т. А.—Ж. анал. хим., 1977, т. 32, Ns 9, с. 1786—1789.
11. Стенина В. И., Агасян П. К.—Ж- анал. хим., 1966, т. 21, Ns 10, с. 1223.
12. Стенина В. И., Агасян П. К., Беренцвейг Г. А.—Ж- анал. хим., 1967, т. 22, № 1, с. 91.
Иттрий
До недавнего времени был известен только один метод амперометрического определения иттрия (III): титрование его купфероном с применением двух индикаторных платиновых электродов [1]. Разумеется, это титрование может быть выполнено и с одним индикаторным электродом по току окисления купферона на платиновом электроде при +0,8 В (Нас. КЭ) или по току восстановления его на ртутном капающем электроде при —0,7 В (Нас. КЭ). Определение при псмощи купферона не селективно, поскольку купферон реагирует с другими ионами
В разделе «Лантаноиды» описано определение иттрия(III) с помощью ферроцианида калия [2, 5]. Широкие возможности комплексонометрического титрования позволили применять ЭДТА и для определения иттрия. При помощи ЭДТА определяют иттрий как отдельно [6], так и в присутствии скандия(III) и некоторых ионов группы РЗЭ [2, 7] и других ионов, регулируя pH раствора. Известен также метод комплексонометрического определения иттрия, скандия и некоторых лантаноидов на фоне безводной уксусной кислоты [3] и применение для определения этих же ионов в водной среде другого комплексона — триаминпентауксусной кислоты [4].
В статьях [5, 8] рекомендуются другие реактивы для определения иттрия (III) [и скандия (III)]—N-циннамоилфенилгидро-ксиламин и N-циннамоил-п-толил-гидроксиламин, образующие с этими ионами осадки состава 1 : 3. Титруют по току окисления титранта на графитовом электроде.
ЛИТЕРАТУРА
1. Василенко В. Д. —Укр. хим. ж., 1960, т. 26, с. 767.
2. Жданов А. К., Мархабаев И. А. —Узб. хим. ж., 1972, Ns 3, с. 25—27.
3. Геворгян А. М., Талипов Ш. Т., Хадеев В. А. — Научные труды Ташкентского ун-та, 1973, вып. 435, с. 63.
174
. хадеев В. А. — РЖХим., 1977, 10Г79, депон. в ВИНИТИ.
Д Шеина Н М., Галлай 3. А., Шведене Н. В.—Ж анал. хим., 1974, т. 29, № 10, с. 1924-1928.
6 Жданов А. К., Мархабаев И. А.—Ж. аналит. хим., 1972, т. 27, № 6, с. 1090— 1094.
7 Жданов А. К-, Мархабаев И. А.—Жури. ВХО, 1973, т. 18, № 3, с. 350—351. я Галлай 3. А., Шеина Н. М., Шведене Н. В. и др.—Ж- анал. хим., 1975, т. 30, № 10, с. 1947—1950.
Кадмий
Для амперометрического определения кадмия известно довольно много реагентов, перечень которых значительно пополнился в последние годы, главным образом за счет органических соединений.
Наиболее известным реагентом является ферроцианид калия. Определения кадмия с помощью ферроцианида проводят по току окисления ферроцианида на платиновом электроде [I] при потенциалах от +0,8 до +1,1 В (МИЭ). Кадмий(П) образует с ферроцианидом в аммиачной среде простую соль Со2[Ре(СЫ)б]> а в ацетатной — двойную, состава K6Cd^[Fe(CN6)]4- Растворимость осадка ферроцианида кадмия заметно выше, чем соответствующих осадков цинковых солей. Так, предельной концентрацией, при которой удается определить кадмий, является 1 • 10-3 М (112 мг/л) в ацетатной и 2,5-10-4 М (28 мг/л) в аммиачной среде. В аммиачно-ацетатной среде растворимость осадка ферроцианида еще выше: из 4 • 10—3 М (450 мг/л) раствора осадок уже не выпадает. Это имеет большое значение при определении цинка (см. разд. «Цинк»), но неблагоприятно для определения кадмия: в присутствии цинка кадмий определять нельзя (даже суммарно).
Таким же образом можно проводить титрование ферроцианидом при помощи ртутного капающего электрода по току восстановления кадмия (II) [2—4].
В 1 М соляной кислоте или хлориде калия ферроцианид осаждает [5, 6] только цинк, кадмий же остается в растворе. Это явление использовано в простом амперометрическом методе раздельного определения [6] обоих элементов. Титрование проводят с Двумя платиновыми электродами при напряжении 0,05 В.
Ход анализа. Навеску пробы (губчатый кадмий) около 0,5—0,8 г растворяют в 10%-ной соляной кислоте, фильтруют, выпаривают с 0,5 мл концентрированной азотной кислоты, к осгдку добавляют 50 мл 6 н. соляной кислоты, экстрагируют железо(II) эфиром и после отделения эфирного слоя разбавляют раствор водой в мерной колбе емкостью 250 мл. В стакане для титрования 50 мл этого раствора разбавляют 10 мл воды, добавляют 5 капель 0,6%-ного раствора феррицианида калия и титруют 0,05 М раствором ферроцианида калия; возрастание тока указывает на полное связывание цинка. Затем в этот же раствор вводят 9 г сухого сульфата аммония, выжидают некоторое время, пока сила тока не упадет до первоначальной и вновь титруют тем же раствором ферроцианида до вторичного возрастания тока — осаждается кадмий. Этот метод, как указывают его авторы [6], дает удовлетворительные результаты при содержании 10% цннка и 62—70% кадмия.
пН~кесто сУльФата аммония раствор можно нейтрализовать аммиаком до-
175
7 аблица 24. Методы определения кадмия (Cd11)
Титрант Определяемый минимум Условия определения Примечания Литература
K4Fe(CN)6 28 мг/л Электрод Pt, <р=+0,84-1,1 В (МИЭ), фон — аммиачный или ацетатный См. текст 1
KI Электрод Hg капающий, <р=—1,0 В (Нас.КЭ), фон—1,2 М Na2SO4+nH-рамидон, рН=3 Медь цементируют на железной проволоке 8
KSCN Электрод Hg капающий, ср=—0,85 В (Нас. КЭ), фон—0,5М CH3COONH4+ + 0,01% желатины+изохинолин, рН= =5,5 Мешают Hg, Ag и галогениды влияние Ni, Zn, Со устраняют изменением pH 12
Натриевая соль трифосфорной кислоты Электрод Hg, <р=—0,8 В (Нас. КЭ), фон — 1 М КЬЮз Для титр< вания РЬ11 ср=—0,6 В, для Fe11, Со11 и Ni11 ср=—1,4 В; отношение M:R для Cd=l:2, для остальных 1 : 1 13
ЭДТА 1 мкг/мл Электрод из сплава Nb — Zr — Ti; ср =+0,8 В (Нас.КЭ), фон — для Cd и Zn, рН=4,5—5, для Ni рН=9—10 Одновременное определение Cd и Ni (или Zn и Ni); сначала титруют Ni диметилглиоксимом, затем, изменяя pH, кадмий ЭДТА 15
Электрод Та, <р= +1,14-1,2 В (Нас.КЭ), фон — для Cd, Zn и РЬ ацетат натрия, для Hg — 0,25 М HNOa Титрование бинарных смесей Hg— —Cd(Zn, РЬ); сначала титруют Hg в кислом растворе Cd (или Zn или РЬ) — в ацетатном 16
Электрод Та, ср= + 1,2 В (Нас.КЭ), фон-—pH=0,4—1 для Т1, pH=4—4,5 для Cd Такой же прием для определения Т1, затем Cd и Ag (иодидом калия) 17
12—1694
6 мкг/мл Cd
3 мкг/мл Zn и Мп
Электрод Hg, ср=—0,8 В для Cd, —1,6 В для Zn и —1,75 В для Мп (Нас. КЭ), фон—0,2 М раствор окса1-лата с pH=6,0 для Cd и Zn, pH=8 для Мп
Метод совместного определении Cd, Zn и Мп
KBr
М-бис(Р-амино-этил)дитиокарбоновая кислота
Дитиотриокси-метиламиномета-нат
Унитиол
K2CS3
Метилдитиобиурет
Тиооксин
Хинальдинат натрия
Электрод Pt, ср= + 1,3 В (Нас. КЭ), фон — безводная уксусная кислота с добавлением сухих солей Мешают Pb, Ag, In; неводная среда 25
Электрод Pt, <р= +0,1 В (Нас. КЭ), фон —0,05—0,2 М C2H4O4+KNO3+ + NH4OH, рН=7,5—9,0 26
0,2—0,26 мг Cd и Си Электрод Hg капающий, <р= +0,2 В (Нас. КЭ), фон — тартрат аммония F +KNO3, pH=8,0 Определение Cd и Си, в присутствии Си кривая титрования имеет 2 излома (см. текст и рис. 44) 27
Электрод Pt, <р = + 0,6+0,8 В (Нас. КЭ), фон — бориоборатный буферный раствор, pH = 8—9 28
Электрод Hg капающий, (р=—0,4 В (Нас. КЭ), фон—0,05—0,2 М С2Н4О4+ + KNO3+NH4OH, рН=7,5—9 Можно титровать этим же реагентом также Си, РЬ и Zn 29
Электрод Hg капающий, <р=—0,2 В (Нас. КЭ), для Cd и Ni, +0,05 В для Си, фон — 0,05 М тартрат аммония+ + KNO3+NH4OH, pH « 9 Определение смесей Си — Cd и Си—Ni по току окисления реагента и восстановления М2+ 30
0,5 мкг/л—1 мг/л Cd и Си Электрод Pt, <р=+0,9 В (Нас.КЭ), фон — хлоридно-аммиачный, рН= 1—3 Можно титровать смеси Си — Cd и Си—Cd—MoVI;.. Fe111, маскируют ЭДТА 32
2,9—6,4 мг Электрод Hg капающий, ср= + 0,9+ +1,1 В (Нас. КЭ), фон —0,5 М NaNO3, рН=6,2—7,2 35
Подобно висмуту, кадмий образует малорастворимые осадки с иодидом калия в присутствии органических веществ. Это позволило разработать метод определения кадмия в сплавах, заключающийся в титровании его иодидом калия в присутствии пирамидона при рН=2—5 [7]. Сравнительно недавно этот метод был применен для анализа кадмиевой губки и сплавов '[8].
Титрование роданидом калия, предложенное ранее на фоне пиридина [9—11], проводят в присутствии изохинолина [12]. В обоих случаях титруют с ртутным капающим электродом при —0,85 В (Нас. КЭ) по току восстановления Cd11. Метод [12] позволяет определять не только кадмий, но и медь и никель при совместном присутствии.
Четвертый неорганический реагент — натриевая соль трифос-форной кислоты — предложена также для определения кадмия в присутствии кобальта (II), никеля (II), железа (III) и свин-ца(П) [13].
Еще в 1962 г. [14] было предложено совместное титрование никеля и кадмия: сначала осаждают никель(II) избытком раствора диметилглиоксима и, не отфильтровывая осадка, титруют кадмий (II) раствором ЭДТА, после чего титруют избыток диметилглиоксима стандартным раствором соли никеля. Определение проводят на хлоридно-аммиачном фоне с капающим ртутным электродом по току восстановления обоих ионов.
Этот же принцип использован для определения кадмия в присутствии никеля и цинка [15]. Серия работ по определению кадмия в различных смесях с другими ионами выполнена на танталовом электроде по току окисления комплексона при +1,2 В (Нас. КЭ). Часть этих работ описана в разделе «Висмут» и в разделе «Индий» и поэтому, во избежание повторений, в данном разделе не рассмотрена. На том же электроде титруют кадмий в присутствии ртути (II), цинка и свинца (И) [16] и таллия (I) и се-ребра(1) [17].
Для комплексонометрического определения кадмия в присутствии цинка и марганца(П) рекомендуется [18] ртутный капающий электрод, а определение кадмия в присутствии свинца(II) и цинка выполнено методом осциллографической полярографии также на ртутном электроде [19]. Рекомендуется ряд других комплексонов [20—23].
При необходимости можно определять кадмий и ионы других элементов при помощи раствора ЭДТА в неводной среде [24]. Титруют кадмий и в ледяной уксусной кислоте бромидом калия [25]. Экстракционные методы, описанные в разд. «Висмут», рассчитаны и на определение кадмия.
Известно довольно много реактивов, содержащих се'ру в том или ином состоянии и успешно применяемых для определения кадмия. Например дитиокарбаминовая кислота, описанная в разделе «Висмут», рекомендована и для кадмия; она же в другом варианте [26] применена для определения кадмия в присутствии
178
рис 44 Схематическая кривая последовательного титрования меди(II) н кадмия(II) оаствором дитиотриоксиметиламиномета-ната (К).
мл
меди и никеля. Для последовательного титрования кадмия и меди применен дитиотриоксиметил амино-метанат калия [27]. Реагент сначала взаимодействует с медью(II), причем на капающем ртутном электроде регистрируется убывающий ток ее восстановления, затем титру-
ется кадмий, а после конечной точки появляется при том же потенциале (—0,2 В Нас. КЭ) ток окисления реагента. Кривая титрования имеет, следовательно, форму, изображенную на рис. 44.
Кроме этих сложных серусодержащих соединений для определения кадмия применяют унитиол [28] (см. «Висмут»), тиокарбонат калия [29], дающий анодную волну на ртутном электроде, метилдитиобиурет [30]1 — также анодно окисляющийся, тиоацетамид [31], позволяющий титровать кадмий в присутствии 100-кратного количества цинка, тиооксин [32, 33]. Из не содержащих серу органических соединений можно упомянуть еще антрахиноновую кислоту [34] и хинальдинат натрия [35].
В заключение следует отметить, что приведенными выше примерами не исчерпываются все описанные в литературе способы определения кадмия. Упоминания об определении кадмия можно найти в других методиках, посвященных определению элементов, вместе с которыми может находиться кадмий.
ЛИТЕРАТУРА
1. Сонгина О. А., Войлошникова А. И., Козловский М. Т.—Зав. лаб., 1951, т. 17, № 1, с. 3—7.
2. Човнык И. Г. и др. —-Зав. лаб., 1949, т. 15, № 5, с. 517—520.
3. Ramaiah И. A., Agarwal S. К.., Gupta S. L. — Naturwis., 1956, Bd. 43, S. 197, 325.
4. Saraswat H. C. — Proc. Ind. Acad. Set, 1960, v. 51A, p. 34.
5. Агасян П. R. — Зав. лаб., 1958, т. 24, № 5, с. 532—535.
6. Гао Сяо-Ся, Чжунан Вэнь-дэ — РЖХим., 1959, реф. 19095.
7. Жданов А. К-, Хадеев В. А., Макриикая Е. Е.—Зав. лаб., 1956, т. 22, № 12, с. 1286.
8. Ziemba S. — Chem. Anal., 1970, v. 15, № 4, p. 829—832.
9. Попель А. А., Марунина A. T. — В кн.: Теория и практика полярографического анализа. Материалы I Всесоюзного совещания по полярографии. Ки-10 лгИНеВ’ ^тнннца' 1962, с. 139—144.
Марунина А. Т. — В кн.: Труды Казанского химико-технологического ин-та, 1962, вып. 30, с. 193—196.
и. Deshmukh G. S., Rao A. L. — Z. anal. Chem., 1963, Bd. 194, S. 101.
lz Rao A. L., Puri B. K. — Z. anal. Chem., 1969, Bd. 247, S. 18—20.
12»
179
13. Марунина А. Т„ Попель А. А.— Учен. зап. Казанского ун-та, 1965, т. 124 № 3, с. 203—207.
14. Новаковская Э. Г. — Зав. лаб., 1962, т. 28, № 1, с. 28.
15. Жданов А. К, Бархударьян А. А. — Узб. хим. ж., 1974, № 5, с. 15.
16. Жданов А. К-, Капица И. В.—Узб. хим. ж., 1968, № 1, с. 29—31.
17. Жданов A. K-j Акентьева Н. А., Капица Н. В.—Узб. хим. ж., 1967, № 6, с. 12—14.
18. Нодзаки Тору, Косиба Кундзи, Накаи Синъити, Цукуру Тадаси, Иосиму-ра — Бунсеки Кагаку, 1969, т. 18, № 11, с. 1394—1396; РЖХим., 1970, 12Г71.
19. Габович А. М.— Труды Кишиневского с.-х. ин-та, 1966, № 43, с. 125—131.
20. Kodama Matsuo — Bull. Chem. Soc. Jap., 1975, v. 48, № 12, p. 3598—3602; РЖХим., 1976, 13Г89.
21. Arthur P., Hunt В. R. — Anal. Chem., 1967, v. 39, № 1, p. 95—97.
22. Хадеев В. A. — РЖХим., 1977, 10Г79, депон.
23. Flaschka H., Speights R.— Anal. chim. acta, 1963, v. 28, p. 433.
24. Геворгян A. M., Талипов Ш. T„ Хадеев В. A. — РЖХим., 1973, 16Г64, де-понир. ВИНИТИ, 28.-11.-73, № 5548—73, депон.
25. Крешков А. П., Борк В. А., Федухина Г. П. — Зав. лаб., 1968, т. 34, № 3, с. 265—268.
26. Deshmukh G. S., Chattopadhyay S. S. — Indian J. Appl. Chem., 1972, v. 35, № 4—6, p. 115—116; РЖХим., 1975, ЗГ74.
27. Chaudhuri H., Chatterjee S. S., Mahanti H. S. — Technology, (India), 1974, v. 11, № 1, p. 58—60; РЖХим., 1974, 13Г46.
28. Даниленко E. Ф., Усатенко Ю. И. — В кн.: Вопросы химии и химической технологии. Респ. межвед. темат. научно-техн. сб. 1973, вып. 28, с. 56—60.
29. Deshmukh G. S., Garde Р. — Bull. Soc. Japan, 1967, v. 40, № 6, p. 1543— 1545; РЖХим., 1968, ЗГ20.
30. Deshmukh G. S., Nandi R. K- — Z. anal. Chem., 1969, Bd. 248, № 3—4, S. 170—172.
31. Przysczewska M. — Acta chim. Hungar., 1962, v. 34, p. 135.
32. Куликовская Ж. Б., Супрунович В. И. — В кн.: Методы определения неметаллических и других вредных примесей в промышленных материалах. М., Металлургия, 1977, с. 133—137.
33. Усатенко Ю. И., Супрунович В. И. — Изв. АН ЛатвССР. Сер. хим., ,1963, № 5, с. 549.
34. Костромин А. И., Апаршева М. И. — Зав. лаб., 1956, т. 22, № 3, с. 544.
35. Negoin D, Gagescu D. — Studii sci. Cercetati chim. Acad. RSR, 1965, v. 13, № 12, p. 1257—1260; РЖХим., 1967, 2Г59.
Калий
В литературе [1] подробно описаны три метода амперометрического определения калия: титрование солями дипикриламина с ртутным капающим электродом, анодный ферроцианидный метод с платиновым электродом и тетрафенилборатный метод. В настоящее время, в связи с быстрым и широким распространением фотометрических методов определения щелочных металлов, дальнейшая разработка методов амперометрического титрования шла, в основном, в направлении усовершенствования тетрафенилборатного метода.
Тетрафенилборат Na[B(C6H5)4] образует с калием труднорастворимые осадки. Известны методы обратного титрования избытка реагента солями серебра, ртути, таллия (I) с двумя платиновыми электродами или с одним ртутным капающим [1]. Практически гораздо удобнее и проще прямое титрование, известное
180
в нескольких вариантах [2—10]. Упомянем лишь об одном, основанном на анодном окислении реагента на графитовом электроде Г9]. Тетрафенилборат дает при окислении две волны, отвечающие двум стадиям его окисления [10]. Для титрования рекомендуется устанавливать потенциал -j-0,55 В (Нас. КЭ), соответствующий площадке предельного тока первой волны.
Этот метод, безусловно, очень прост и удобен в выполнении. Авторы метода применили его для определения 0,25—8,5% калия в различных материалах — глинах, стеклах, природных солях, огнеупорных изделиях и т. д., причем метод был проверен на стандартных образцах Национального бюро стандартов США.
Ход анализа. Навеску, смоченную водой, обрабатывают в платиновой чашке смесью серной и фтористоводородной кислот [на 1 г навески 5 мл серной кислоты (1:1) и около 10 мл 48%-ной фтористоводородной кислоты]. Если в пробе присутствует много карбонатов, то сначала осторожно, во избежание потерь, обрабатывают навеску соляной кислотой. Выпаривают досуха на песочной бане, и, если нужно, повторяют разложение серной и фтористоводородной кислотами. К сухому остатку добавляют соляную кислоту (1 :20), фильтруют, осадок промывают соляной кислотой (1 :20). Затем добавляют по каплям концентрированный раствор едкого натра до рН=3 по универсальному индикатору. После этого титруют при потенциале +0,55 В (Нас. КЭ) раствором тетрафенилбората, титр которого устанавливают по стандартному раствору соли калия. Кривая титрования несколько закруглена вблизи конечной точки, но она определяется легко вследствие резкого возрастания тока окисления избытка тетрафенилбората.
Следует упомянуть о двух методах определения калия в неводной среде. В одном из них [11] калий осаждают в виде сульфата в изопропиловом спирте, в другом [12]—титруют щавелевой кислотой в той же среде (изопропиловый спирт).
ЛИТЕРАТУРА
1. Сонгина О. А. Амперометрическое титрование. М., Химия, 1967, с. 227—231.
2. Geyer R., Bormann Н. D. — Wiss. Z. Techn. Hochschule Chem., Leuna-Merseburg, 1965, Bd. 7, № 3, S. 243—246; РЖХим., 1966, 13Г63.
3. Stock J. T., Bowerman D. R.— Microchem. J., 1972, v. 17, Ns 3, p. 253—257.
4. Siska E.'—Elmiszervizsg. Kozl. (венгр.), 1972, v. 18, № 3, p. 147—151; РЖХим., 1973, 4Г76.
5. Ikeda Sanae, Hirata Junko. — Л Chem. Soc. Jap., Chem. and Ind. Chem., 1973, № 5, p. 953—957; РЖХим., 1974, 1Г63.
6. Басов В. H.. Агасян П. К., Базуева Т. А.—Ж. анал. хим., 1976, т. 31, № 10, с. 2046—2048.
7. Siska Е., Pungor Е. — Fres. Z. anal. Chem., 1971, Bd. 257, S. 7—10.
8. Церковницкая И. А., Дубровина Л. Т. — Вестник ЛГУ, 1973, 4, с. 128. 9. Smith Е., Jamieson D., Elving Р.—Anal. Chem., 1960, v. 32, 1253—1255. 10. Turner W. R., Elving P. — Anal. Chem., 1965, v. 37, p. 207—210.
И. Сальникова К- С., Кузьмичев О. В.—Ж. анал. хим., 1971, т. 26, № 10, с. 1924—1927.
12. Борк В. А., Швыркова Л. А., Ким Л. Б.—Ж. анал. хим, 1971, т. 26, с. 269—272.
Кальций
Амперометрическое титрование кальция можно осуществлять по реакции осаждения и по реакции комплексообразования с ЭДТА и его аналогами.
181
Таблица 25. Методы определения кальция
Определяемый ион Титрант Определяемый минимум Условия определения Примечания Литература
Са2+ K4Fe(CN)6] Электрод Pt, ср—+0,8—1,1 В (МИЭ), фон •— ацетат аммония + +60% спирта (по объему) См. текст 1
ЭДТА Электрод Hg капающий, <р= —1,5 В (Нас. КЭ), фон—NH4NO3+ + Zn(NO3)2, 2п2+-амперометриче-ский индикатор Концентрация Zn(NO3)2 — 6 мг в 50 мл титруемого раствора, магний не мешает, если его содержание не превышает содержания Са больше чем в 5 раз 6, 7
Электрод Hg капающий, <р= —1,55 В (Нас.КЭ), фон — 97 мл СНзОН+3 мл NH4OH+ZnO|-(индикатор) Определение Са в нитрофосфате и удобрениях, содержащих Ca(NO3)2, титруют экстрагированный Са2+, фоном служит экстрагент 9
«5 мкг 2 Pt электрода, Д<р=1,5 В, фон—«0,3 м кон Не мешает 15-кратный избыток магния, определяют Са в воде 12
0,1—0,02 мкг 2 Pt игольчатых электрода диаметром 0,01 мм, Д<р=1,0+1,1 В, фон — 0,2—0,02 М КОН Анализируемый объем 0,1— 0,5 мл, работают под микроскопом 13
Са2+ Mg2+ ЭДТА Са 0,1 мг Mg 0,4 мг Электрод Hg капающий, <р= = 0,55 В (Нас. КЭ), фон—NH4OH+ + NH4NO3, pH «9 для суммы Ca+Mg, для Mg— 12,5 (боратный буферный раствор) Индикатор — раствор TINO3 (см. текст) 10
Ca2++iSr2+ и Ba2+
15 мкг 2 электрода: Pt (катод) и амаль- Определение Са и Mg в воде и 14, 15
* Десятые и сотые доли миллиграмма в 50 мл гамированное Ag (анод), Д<р= = 130 мВ, для Са, 210 мВ для Mg, pH соответственно 11,75 и 9,6; /=37°С, амперометрический индикатор Hg(NO3)2 Электрод Та, <р = + 0,65 В (Нас. КЭ) для 2Ca+Mg, по току окисления ЭДТА, для Са <р= + 0,44-0,86 В по току окисления индикатора — TliNO3; фон — аммиачный буферный раствор, рН= =9,5—11 молоке; в статье 115] предложены 2 электрода из одного и того же материала — Pt или графит Отдельное определение Са и Mg или Ca+Mg, избирательное определение Са в присутствии Mg с индикатором Т1+ по току его окисления; применили для анализа доломита 16
5 мкг — 5 мг в 40—100 мл 2 Pt электрода, Д<р=1,4 В, фон — 0,5 н. NaOH Рекомендуется для определения суммы Mg и Са в воде; рН=9,6 (боратный буферный раствор) 17
ЭДТА и ЭГТА 0,03 мкг/мл Электрод Hg неподвижный (см. текст) Определение Са и Mg в биологических субстратах (см. текст) 11
ЭДТА 0,12— 1,4 мг 2 Pt электрода, Д<р= 1,0 В для Са и Ва, 0,9 для Mg, фон — рН= =2,5—3 для Са, рН=1,5—2 для Mg 18
ЭДТА для Mg. Диамино диэтиленгликолевый эфир тетрауксусной кислоты для Са Электрод РЬО2, <р = + 1,2 В (Нас. КЭ), фон —0,1 М NH4OH, рН=11 Электрод Т12О3, фон — NH4C1+ + NH4OH, рН=10 Титруют по току окисления титрантов 25 26
ЭДТА 1—20 4—44 3—20 мг Са мг Sr мг Ва 2 Pt электрода, Д<р=1,6 В, фон — 0,5 М КОН Во время титрования раствор разогревается до 60—70 °C 19
СО
Рис. 45. Кривые титрования кальция рас-1ан гвором ферроцианида калия в отсутствие
, (/) и в присутствии (2) хлорида аммония.
7 •/
/ / Для осаждения кальция предло-
л. ? / жено несколько реагентов — ферро-
/ цианид, оксалат, фторид-ион. Фер-
/ Д роцианид калия окисляется на пла-
/ , типовом электроде, позволяя вести
Д ' 1 Йл титрование по анодному току. Со-
т.з став осадка соответствует формуле
CaK2[Fe(CN)6].
Для понижения растворимости осадка рекомендуется добавлять около 60% спирта (по объему) и вводить в раствор ионы аммония [1], замещающие ионы калия в осадке и способствующие уменьшению его растворимости. Если же в растворе присутствуют ионы натрия, то присутствие ионов аммония обязательно, так как они препятствуют образованию более растворимого кальций — натрий ферроцианида. Например, если в 20 мл титруемого объема содержится 70 мг натрия (Na+), то кривая титрования имеет совершенно неправильный ход (рис. 45, кривая /), указывающий на образование растворимых соединений; если при таком же содержании натрия в растворе присутствует 18 мг аммония (NHJ), то кривая титрования имеет нормальный вид (рис. 45, кривая 2) и результат титрования оказывается весьма точным. Магний и алюминий мешают определению кальция ферроцианидом, так как, с одной стороны, они также осаждаются ферроцианидом в присутствии спирта, с другой — в присутствии алюминия наблюдается понижение тока окисления ферроцианида, а при больших содержаниях — увеличение растворимости осадка ферроцианида кальция.
Титрование кальция следует проводить в нейтральных или уксуснокислых растворах (концентрация уксусной кислоты не оказывает влияния на ход титрования). Наличие минеральных кислот (соляной и азотной), даже разбавленных, недопустимо, так как и в их присутствии осадок ферроцианида кальция растворяется. Недопустимо присутствие не только серной кислоты, но и сульфатов; как известно, кальций может частично выпасть в осадок в виде сульфата.
Этот же принцип был использован позднее Михальским и Галус [2] для определения относительно больших количеств кальция (0,2 н. растворы). Они титровали раствором ферроцианида аммония либо обычным амперометрическим методом на платиновом электроде без наложения внешнего напряжения с меркур-сульфатным электродом сравнения, либо с двумя индикаторными электродами при напряжении 0,4 В. В присутствии магния результаты завышаются — авторы считают, что сумма кальция и магния может быть оттитрована вполне удовлетворительно (например, в известняках сумма была определена с ошибкой 0,5%).
При титровании кальция оксалатом можно использовать окисление его на платиновом электроде [3] или вводить медь(П) в качестве амперометрического индикатора [4]. Однако наибольшее внимание уделяется титрованию кальция раствором ЭДТА и некоторыми его аналогами. Этот метод, основанный на образовании очень прочного растворимого комплексного соединения кальция,
184
осуществляется в различных вариантах и на различных электродах. При работе с ртутным электродом [6—11] приходится прибегать к амперометрическим индикаторам, восстанавливающимся на ртути и образующим с ЭДТА менее прочные соединения, чем кальций. При работе с платиновыми электродами можно использовать ток окисления ЭДТА [12—19].
Много внимания уделяется и практически важной задаче — определению кальция ь присутствии магния и совместному определению обоих элементов [10, И, 14, 15, 16, 17], а также определению кальция в присутствии стронция и бария [18, 19]. Указания к этим методам приведены в табл. 25.
Описаны также методы определения кальция в более сложных смесях — в присутствии цинка и меди (II) [20], висмута (III) и сурьмы(Ш) [21], ртути(П) и цинка(П) [22], молибдена(VI), цинка(II) и магния (II) [23]. Работа [24] специально посвящена подробному изучению взаимодействия ионов всех четырех щелочноземельных элементов (Са, Mg, Sr, Ва) с ЭДТА и его аналогами — этиленгликольбисаминоэтиловый эфир тетрауксусной кислоты ЭГТА и диэтилентриаминпентауксусной кислоты ДТРА. Приводятся сравнительные данные о прочности соединений, образуемых четырьмя ионами со всеми тремя комплексонами и указывается, что на фоне нитратно-аммиачного буферного раствора с рН=10 можно определять кальций в присутствии 1000-кратного избытка магния, если титровать раствором ЭГТА. Порядок определяемых величин (всеми тремя реагентами) 10-6 М.
Рекомендуется также применять электроды из диоксида свинца [25] и триоксида таллия [26]. В обоих случаях титруют по току окисления ЭДТА и второго реагента — диаминодиэтиленгликоля тетрауксусной кислоты. Эти реагенты легко окисляются и на платиновом электроде, так что замена платины оксидными электродами вряд ли имеет смысл.
Ионы кальция и остальных щелочноземельных элементов могут быть оттитрованы раствором щавелевой кислоты при помощи двух медных электродов на фоне изопропилового спирта [27].
ЛИТЕРАТУРА
1. Сонгина О. А., Войлошникова А. П., Козловский М. Т.—Изв. АН КазССР. Сер. хим., 1953, вып. 6, с. 69—73.
2. Michalski Е., Gaius Z. — Chemia Analitycna, 1958, v. 3, p. 431—434.
3. Усатенко Ю. И., Виткина М. А.—Укр. хим. ж., 1957, т. 23, с. 788—791.
4. Усатенко Ю. И., Виткина М. А. — В кн.: Трвды Комиссии по аналитической химии АН СССР. 1956, VII/X, с. 155.
5. Smith W. М.— Chem. Weekbl., 1960, v. 56, р. 25.
6. Pribil R., Vicenova Е. — Chem. Listy, 1952, v. 46, p. 535.
7. Laitinen H., Sympson R.— Anal. Chem., 1954, v. 26, p. 566.
°- Richardson M. L.— Taianta, 1953, v. 10, № 1, p. 103.
9. Bhatnagar R. M., Roy A. K. — Technology (India), 1965, v. 2, № 3, p. 130— 136; РЖХим., 1966, 19Г163.
10. Торопова В. Ф., Батыршина Ф. М. — В кн.: Теория и практика полярографического анализа. Кишинев, Штиинца, 1962, с. 352.
185
11. Monnier D., Roueche A.— Helv. chim. Acta, 1964, v. 47, p. 103.
12. Vorlicek J., Fara M., Vydra F. — Microchem. J., 1967, v. 12, № 3, p. 409—411-РЖХим., 1968, 7Г51.
13. Алимарин И. П., Петракова М. Н.—Ж. анал. хим., 1969, т. 24, № 8, с. 1138— 1140.
14. Sethu R. D. е. а.—Analyst, 1964, т. 89, с. 608—612.
15. Vydra F., Vorlicek J. — Taianta, 1965, v. 12, № 6, p. 647—650.
16. Хадеева В. А., Ахмеджанова'С. A.—Научн. труды Ташкентского ун-та, 1967, jsbin. 284, с. 3—9.
17. Vorlicek J., Fara M., Vydra F. — Z. anal. Chem., 1968, Bd. 241, № 5, S. 314— 318.
18. Srivastava H. R.—J. Indian Chem. Soc., 1976, v. 53, № 2, p. 206—207; РЖХим., 1976, 19Г103.
19. Vorlicek J., Vydra F. — Coll. Czech. Chem., Comm., 1966, v. 31, № 6, p. 2510— 2517.
20. Campbell R., Reilley Ch.—Taianta, 1962, v. 9, № 2, p. 153.
21. Reilley Ch., Scribner W„ Temple C. — Anal. Chem., 1956, v. 28, p. 460.
22. Sierra F. e. a. — An. quim. Real. Soc. esp. fis. у quim., 1972, v. 68, № 9—10, p. 1091—1096; РЖХим., 1973, 4Г52.
23. Соколова А. А., Мухина 3. C. — В кн.: Физико-химические методы анализа металлов и сплавов. М., 1969, с. 94—95.
24. Boef G. е. a. —Taianta, 1970, v. 17, № 10, р. 1006—1009.
25. Huber С. О., Dahnke К, Hinz F. — Anal. Chem., 1971, v. 43, № 1, p. 152— 154.
26. Kainz G., Sontag G., Schaller F. — Mikroctiim. Acta, 1975, v. 2, № 4—5, p. 381—388; РЖХим., 1976, 9Г218. *
27. Борк В. А., Швыркова Л. А., Ким Л. Б.—Ж. анал. хим., 1970, т. 25, № 6, с. 1079—1084.
Кислород
Растворенный кислород (кислород воздуха, всегда насыщающего исследуемые растворы) может восстанавливаться на индикаторных электродах (ртутном и платиновом) и тем самым мешать определению других веществ. Однако это же обстоятельство может быть использовано и для определения самого кислорода, поскольку диффузионный ток восстановления кислорода пропорционален его концентрации в исследуемом растворе. Процессу восстановления кислорода посвящено много исследований, на основании которых разработаны методы главным образом для автоматического контроля содержания кислорода в жидкостях: различных природных водах и рассолах, биологических растворах, воде аквариумов и т. д. Поскольку в основе этих методов лежат полярографические приемы, т. е. непосредственное измерение высоты волны восстановления кислорода, а не титрование его каким-либо раствором, то подробного описания этих методов в настоящей монографии не приводится.
Для амперометрического определения кислорода предложены следующие методы: восстановление кислорода до ОН~-ионов на ртутном электроде и последующее титрование [1] соляной кислотой при —1,8 В (Нас. КЭ), а также амперометрический вариант определения кислорода по классическому методу Винклера, т. е.
186
титрование выделяющегося иода тиосульфатом и определение по току восстановления иода. Этому методу определения кислорода посвящено большое число работ, различающихся подробностями технического оформления и аппаратурой. Очень удобно пользоваться в этом случае двумя платиновыми электродами (см. гл. IV).
Метод с двумя электродами принят, например, в США для определения кислорода в воде, питающей котельные установки. Нижний, предел определения оценивается [2] в 0,4 мкг кислорода на 1 л воды.
В монографии Стока [3] приведено около 10 работ, посвященных определению кислорода по методу Винклера с амперометрическим окончанием. Не так давно появилось описание прибора для автоматического контроля содержания озона в воздухе тем же методом — титрованием тиосульфатом иода, выделяющегося в результате окисления иодида озоном [5].
Методика амперометрического титрования выделяющегося иода при помощи двух электродов весьма проста. Для выполнения этого титрования достаточно руководствоваться указаниями, приведенными в гл. IV и V.
Тот же метод с двумя платиновыми электродами рекомендован и для определения озона в различных производственных растворах оригинальным методом, позволяющим «консервировать» озон в виде пирофосфатного комплекса марганца(III).
Ход анализа. В исследуемый раствор, подкисленный серной кислотой до рН<1, вводят пирофосфат и марганец(П) (сульфат), окисляющийся озоном до марганца(Ш), образующего, как известно, прочный пирофосфатный комплекс в сернокислой среде. Этот комплекс затем титруют гидрохиноном при напряжении на двух платиновых электродах 0,3 В. Кривая титрования имеет V-образную форму, так как сначала ток обусловлен ионами марганца(III), обеспечивающими катодный процесс, а после конечной точки ток резко возрастает вследствие работы пары хинон — гидрохинон.
Этот метод позволяет определять 2,4 мкг озона в 50 мл раствора, т. е. в 10 раз меньше, чем при визуальном варианте того же метода. Метод разработан применительно к промышленным запросам в одном из ведущих исследовательских институтов [6].
Определение перекиси водорода сводится, по существу, к определению кислорода. Перекись окисляют сильными окислителями, в частности перманганатом, и проводят определение по току восстановления кислорода на платиновом электроде [7—9].
В заключение следует упомянуть о том, что амперометрический метод был косвенно применен для определения кислорода, растворенного в металлической меди. Образец металла нагревают в токе водорода, поглощая образующуюся воду метанолом, и титруют ее затем амперометрически реактивом Фишера, обычно применяемом для определения воды в различных веществах [4]. По данным, приводимым авторами статьи [4], метод позволяет опреде-чть 0,0005% кислорода в меди.
187
ЛИТЕРАТУРА
1. Ketnula W., Siekirski S. — Coll. Czech. Chem. Comm., 1950, v. 15, p. 1069.
2. Janssen C., Smith G. B.— Analyt. chim. acta, 1957, v. 16, p. 276.
3. Stock J. Amperometric Titrations. New York, Interscience, 1965.
4. Fischer W., Bastius H., Mehlhorn R. — Acta chim. Hungar., 1962, v. 43, p. 167. 5. Steinberger E. H., Goldwater F. — J. Phys. E. Sci. Instrum., 1972, v. 5, p. 373. 6. Штреккер Г. П., Романова Л. Ф., Иванова Г. И. — В кн.: Очистка сточных и оборотных вод предприятий цветной металлургии. Труды Казмеханобра, Алма-Ата, 1970, № 4, с. 315—318.
7. Жданов A. R., Ятрудакис С. М. — Узб. хим. ж., 1966, № 1, с. 12—17.
8. Жданов А. К., Ятрудакис С. М.—Узб. хим. ж., 1968, № 2, с. 16—18.
9. Michalski Е., Pawluk N. — Chem. analit (Polska), 1966, v. 11, № 5, p. 917— 922.
Кобальт
Основным реактивом для амперометрического определения кобальта в виде кобальта (II) являлся феррицианид калия, предложенный еще в 1958 году [1]. Метод с использованием этого реактива представляет собой амперометрический вариант известного потенциометрического определения кобальта, основанного на реакции окисления кобальта (II) феррицианидом калия в аммиачной среде и подробно описанного в ряде руководств, в частности в книге С. Ю. Файнберга и Н. А. Филипповой [2]. Титрование проводят при —0,2 В (Нас. КЭ) с платиновым вращающимся электродом по току восстановления избытка феррицианида.
Амперометрический метод по сравнению с потенциометрическим имеет то преимущество, что конец титрования обозначается более резко и позволяет определять меньшие количества кобальта, чем потенциометрический. Подробная методика разложения пробы и подготовки раствора к титрованию изложена в упомянутой книге [2].
Этот метод может быть успешно применен для определения кобальта не только в сплавах, но и в различных других объектах, например в фармацевтических препаратах [3].
В последние годы разработано несколько вариантов феррицианидного метода: титрование с двумя электродами [4, 5, 6], предложена практически ценная методика разложения пробы, обеспечивающая предварительное окисление Мп11 до Мпш, не мешающего определению кобальта [6], изучено влияние состава среды на ход титрования [7].
Недавно появилась серия работ [8, 9], в которой предлагается предварительно переводить Со11 в комплексонат ЭДТА или диэти-лентриаминпентаацетат с целью еще большего понижения редокс-потенциала системы СоП1/Соп.
В пробах сложного состава также определяют кобальт с помощью феррицианида, например, при анализе ферритных пленок и других материалов [9]. Кобальт титруют на хлоридно-аммиач-ном фоне без предварительного отделения от ионов других элементов. В качестве окислителей для титрования Со11 применяют би
188
хромат [10], или ванадат [11], или CeIV [12], предварительно связав кобальт(II) ЭДТА. Из органических реагентов предложены рубеановодородная [13] и антраниловая [14] кислоты, ниооксим [15] и а-нитрозо-р-нафтол [16, 18]. В настоящее время этот перечень дополнен хлорамином Т [19], виолуратом аммония [20], этилендиамином [21] и тиогликолевой кислотой [22].
Если кобальт(II) требуется определить в присутствии железа(III), то прибегают к методам, описанным в разделе «Железо». В присутствии никеля(II) можно определять оба иона с помощью раствора бензтриазола [23] или ксанто-гената [24]. Сложную смесь ионов Со, Т1, Bi, Se, Те (см. раздел «Висмут») анализируют при помощи метилдитиобиурета по реакции осаждения. Вообще же реакции осаждения для кобальта мало избирательны и вряд ли могут иметь практическое значение.
Появились также работы, предлагающие определять кобальт в неводной среде — на фоне ледяной уксусной кислоты [25] и в изопропаноле [26].
ЛИТЕРАТУРА
1. Торопова В. Ф., Стрекалова О. С. — Зав. лаб., 1958, т. 28, № 5, с. 524.
2. Файнберг С. Ю., Филиппова И. А. — Анализ руд цветных металлов. М., Ме-таллургиздат, 1963.
3. Абрамов М. К-—Аптечное дело, 1961, т. 10, с. 19.
4. Кужель А. М„ Филенко А. Н„ Сурмий А. М.—Изв. вузов. Химия и хим. технол., 1974, т. 17, № 5, с. 760—761.
5. Кужель А. М., Сурмий А. М., Филенко А. И.—Ж- анал. хим., 1973. т. 28, № 11, с. 2251—2254.
6. Дятел С. Я.— Зав. лаб., 1976, т. 42, № 9, с. 1059.
7. Malik W., От //.—Taianta, 1967, v. 14, № 11, р. 1341—1343.
8. Sierra F., Hernandez-Artiga M. P., Hernandez-Canavate J. — An. quim. Real.
Soc. esp. fis. у quim., 1976, v. 72, № 5, p. 460—467; РЖХим., 1977, ЗГ128.
9. Sierra H. M. T., Hernandez J., Sierra F. — An. quim. Real. Soc. esp. fis. у quim., 1975, v. 71, № 3, p. 310—313; РЖХим., 1976, 21Г118.
10. Зенкевич О. В. — В кн.: Материалы II Межотраслевого совещания по методам получения и анализа феррит, материалов и сырья для них. Ч. 2. М., 1969, с. 39—44.
11. Varma А. — J. Sci. Ind. Res., 1962, v. 21, p. 142; J. Sci. Res. Banaras Hindu Univ., 1961—1962, v. 12, p. 275.
12. Singh D., Varma A. — Coll. Czech. Chem. Comm., 1963, v. 28, p. 524.
13. Alschwang H. — Bull. Soc. Sci. natur. (franq.), 1941, 455.
14. Жданов А. К., Цейтлин P. И., Якубов A. M.—Зав. лаб., 1955, т. 21, № 1, с. 7.
15. Calzolari С., Furlani С. — Ann. Triest. Cur. univ. Trieste, 1953, v. 2, p. 22.
16. Kolihoff I. M_, Langer A. — J. Am. Chem. Soc., 1940, v. 62, p. 3172.
17. Okac A., Gracova L. — Chem. Listy, 1953, v. 47, p. 367.
18. Szmidt K-, Weber J. — Prace Inst. Meeh., 1957, v. 6, p. 71.
19. Singh D., Prasad В. B., Bhatnagar U. — J. Ind. Chem. Soc., 1965, v. 42, № 8, p. 560—562; РЖХим., 1966, 7Г95.
20. Ершова Л. В., Темянко В. С. — В кн.: Сборник научных трудов Магнитогорского горнометаллургического ин-та. 1971, вып. 87, с. 67—70.
21. Hofbauerova Н., Mocak J.t Bustin D. I. — Chem. Zvesti, 1974, v. 28, № 5, p. 609—613.
22. Бусев А. И., Шестидесятая H. Л.—Ж- аналит. хим., 1971, т. 26, № 2, с. 338—341.
о/ ^oddar s- N„ Rag К. —J. Ind. Chem. Soc., 1973, v. 50, № 6, p. 391—393.
24. Deshmukh G. S., Saraswathi K-— Indian J. Chem 1969, v. 7, № 8, p. 827;
РЖХим., 1969, 9Г104.
189
Таблица 26. Методы определения кобальта
Определяемый ион Титрант Определяемый минимум Условия определения Примечания Литера тура
Со» K3[Fe(CN)6] Электрод Pt, ф=—0,2 В (Нас. КЭ), фон — хлоридно-аммиачный 2 Pt электрода, Д<р=0,4 В для Со, 0,8 для Мп, фон — аммиачный буферный раствор, рН=10—11 для Со, для Мп 2 н. H2SO4 См. текст Определение кобальта |4] и кобальта и марганца [5]. Сначала титруют Мп11, потом Со в том же растворе, изменив pH. Метод применен для определения Мп и Со в стали и сплавах 1 4, 5
2 Pt электрода, Д(р=0,1 В, фон — аммиачный Титр раствора KsIFe(CN)6] устанавливают по фосфорно-кислому раствору электролитического кобальта 6
2—30 мг/мл Электрод Hg капающий (Нас. КЭ), на аммиачно-цитратном фоне (I) е pH=9,8, <р=—0,3 В и на глициновом фоне (II) с рН=8, <р=—0,55 В На фоне (I) не мешают Си, Ni, Сг111 и Vv, мешают Се111, Fe111; на фоне (II) не мешают 10-кратные количества Се111 и Сг111. мешают следы Си11 и Vv 7
1 или 2 Pt электрода, <р=—0,15 В (Нас.КЭ), Дф=0,15 В, фон—ацетатный, рН=3—5 Со11 предварительно связывают диэтилентриаминпентаацетатом, не мешают Мп и Ni 8
1 или 2 Pt электрода, Дф = 0,25 В, <р=0 (Нас. КЭ), фон — ацетатно-ам-миачный, pH аг 3,5 Со11 переводят в комплексона? ЭДТА путем добавления комплексон ата Zn 9
Хлорамин 2 Pt электрода, Д<р=0,1 В, фон — pH=2—6,5 Со11 связывают предварительно ЭДТА 1 19
Виолурат аммония 0,5—2-10—3 моль/л Электрод Pt, <р=-Н1,15 В (МИЭ) 1 ток окисления реагента, фон — 2 М NaAc, pH=9 Отношение реагент : Со=3 :1, не мешают эквивалентные количества Ni и Си, Fe111 связывают фторидом 20
Бензтриазол 0,5—8 мг Со и Ni Электрод Hg капающий, <р=—1,6 В (Нас. КЭ) при титровании Со и —1,4 В при титровании Ni, фон — ацетатно-аммиачный, pH=7,2 для Ni и хлоридно аммиачный, ipH=7,2 для Со Определение Со и Ni, Fe не мешает 23
Ксантогенат 0,1—2 мг Со и Ni Электрод Hg капающий, <р=—0,1 В (Нас. КЭ), фон — 0,025—0,05 М ЬПКО+МКНОз+этанол (50% для Со и 40% для Ni), рН=7—9 для Со и 7—8 для Ni Титруют по току окисления ксантогената, определяют Со и Ni 24
Метилдитиобиурет Электрод Pt, ф=—0,1 В (Нас. КЭ), фон — 0,02—0,05 М NH4C1 (или NH4NOs) +NH4OH, рН=9—10 27
Комплексонат Mg 5—500 мкг/10 мл 2 Pt электрода, Дф=0,8—1,0 В, фон— 1,5—3% перхлората или нитрата лития в ледяной СН3СООН Неводная среда (ледяная уксус ная кислота) 25
Щавелевая кислота 2 Си электрода, фои — изопропанол, титрант готовят на изопропаноле, Дф=0,5 В Определяют Со и Ni в неводног среде 26
Тиогликолеваи кислота, аиилид 0,3 мкг/25 мл Электрод Pt, ф= + 0,46 В (МИЭ), по току окисления титраита, фон — 5 М NH4OH Осадок состава Со : R = 1 : 3 раствор титранта в этаноле не осаждает Zn и Ni, найдено П{ осадка 22
Coi” Этилендиамин 90—1800 мкг/20 мл Электрод Hg капающий, ф=—0,34 В (Нас. КЭ), фон — 2 М КС1 Со находится в исходном рас творе в виде (Co(NH3)e]3+, титру ют по току его восстановления, ко вечная точка определяется обра зованием комплексного соединен!» Со11 с титрантом 21
25. Геворгян А. М., Костылев В. С., Молодожен Е. В. и др. — РЖХим., 1977, 8Г104, депон.
26. Борк В. А., Швыркова Л. А., Ким Л. Б. — Труды Тамбовского ин-та химического машиностр., 1970, вып. 4, с. 112—116.
27. Deshmukh G. S., Nandi R. К- — Current Sci. (India), 1969, v. 38, № 21, p. 512—513; РЖХим., 1970, 13Г101.
Кремний
.Для определения кремния еще в пятидесятых годах было предложено титрование солями свинца(II) [1—3]. Однако эти методы были недостаточно обоснованы и разработаны [4, с. 243]. Более точные рекомендации были даны в статье [15], авторы которой предложили определять кремний в органических соединениях (после разложения вещества) титрованием раствором свинца(II) на ртутном капающем электроде. Кремний в этом случае находится в растворе в виде БЮз-. Титрование раствором нитрата свинца предложено и для тех случаев, когда кремний присутствует в виде аниона кремнефтористоводородной кислоты, например в гальванических ваннах хромирования [6]. Электролит, в котором присутствует серная кислота и хромат, разбавляют водой и осаждают SOF и СгОГ сухим карбонатом свинца, перемешивая раствор, до обесцвечивания. Избыток РЬ11 удаляют с помощью раствора карбоната натрия, фильтруют через пористый тигель, осадок промывают водой. К фильтрату добавляют 10 мл 0,1 М раствора КС1 и 2 мл 0,1 М соляной кислоты; продувают воздухом для удаления СО2, разбавляют водой до объема около 100 мл, устанавливают раствором едкого калия pH л: 6,5 (по слабо-зеленой окраске бромтимолового синего) и титруют 0,1 М раствором нитрата свинца при —1,0 В (Нас. КЭ) по току восстановления свинца(II) на ртутном капающем электроде. Содержание фтора не должно быть больше 6 мг.
Предложено также титровать кремний в виде кремнемолибденовой кислоты, восстанавливающейся на ртутном капающем электроде [7]. Титруют в этом случае раствором нитрона. Но можно проводить определение и с двумя платиновыми электродами раствором железа (II) [8, 9]. Этот метод применен для анализа алюмосиликатов, циркона и других минералов, содержащих от 3 до 95% кремния [9].
Кремнемолибденовую кислоту титруют также раствором диан-типирилметана [10, 11]. В работе [12] рекомендуется переводить кремний в кремневольфрамовую кислоту и титровать также диан-типирилметаном, образующим с ней весьма труднорастворимое соединение. Метод особенно удобен для определения кремния в ферровольфраме. Избыток вольфрамат-ионов связывают фторидом аммония.
Ход анализа. Навеску ферровольфрама 0,1—0,2 г сплавляют с 3—5 г перекиси натрия в никелевом тигле. Охлажденный сплав выщелачивают 50—100 мл воды, кипятят до разложения перекиси натрия, переводят в мерную колбу емкостью 100—200 мл и доводят до метки водой. К аликвотной части 5—10 мл добавляют постепенно 10 н. серную кислоту до рН=1,5—2 и нагревают на водя-
192
ной бане 3_5 мин. Добавляют еще кислоты приблизительно до 1 н. ее концент-
папии затем в горячий раствор вводят 0,2—0,4 мл 0,5 М раствора фторида аммония’ и нагревают 1—2 мин. После охлаждения титруют раствором диантипи-пилметана (титр его устанавливают по стандартному образцу ферровольфрама), раствор прилипают порциями через каждые 15 с. Потенциал платинового электрода + 1,4 В (МИЭ), отношение реагент : кремиевольфрамовая кислота =3 : 1.
ЛИТЕРАТУРА
1. Беркович М. Т. —Зав. лаб., 1950, т. 16, № 6, с. 558.
2. Беркович М. Т. — Z. anal. Chem., 1952, Bd. 137, S. 461.
3. Беркович M. Т., Васильева Л. П.—Зав. лаб., !952, т. 18, № 2, с. 179—181.
4. Сонгина О. А. Амперометрическое титрование. М., Химия, 1967.
5’ Чумаченко М. И., Бурлака В. П. — Изв. АН СССР. ОХН, 1963, № 1, с 5. 6 Ramachandran S., Natarajan S. — J. Electroanal. Chem., 1966, v. 11, № 3, ’ p. 230—233.
7. Hahn H., Wagenknecht E.— Z. anal. Chem., 1961, Bd. 182, S. 343.
8. Burnel D., Malaprade L. — Chim. analyt., 1967, v. 49, № 5, p. 253.
9. Liodec N., Debras-Guedon J. — Bull. Soc. Fran§. ceram., 1968, № 81, p. 15—20. 10. Луговой С. В., Рязанов И. П. — Зав. лаб., 1967, т. 33, № 6, с. 688—692. 11. Луговой С. В., Паклерова Л. Д. — Ж анал. хим., 1974, т. 26, № 9, с. 1748. 12. Луговой С. В., Паклерова Л. Д. — Зав. лаб., 1975, т. 4Ji>№ 6, с. 646—648.
Лантан, редкоземельные элементы
Химические свойства РЗЭ весьма близки, что очень затрудняет их селективное определение при одновременном присутствии ионов нескольких редкоземельных элементов в растворе. Описываемые ниже методы могут быть применены либо для определения одного из РЗЭ в отсутствие всех остальных, либо для определения общего количества всех присутствующих в растворе РЗЭ. Эти методы применимы только для определения лантаноидов(III). Церий, обладающий устойчивой валентностью не только 3 + , но и 4+, рассмотрен в специальном разделе «Церий», так как в состоянии 4-1-он является сильным окислителем и определение его основывается на иных принципах, чем определение лантаноидов (III).
Для титрования по методу осаждения могут быть применены различные реактивы, образующие малорастворимые соединения с лантаноидами. Одним из таких реактивов является ферроцианид калия. Титрование лантаноидов ферроцианидом калия было известно в двух вариантах — с двумя платиновыми электродами [1], и с ртутным капающим электродом [2]. В последнем случае титруют по току восстановления лантана (III) (метод описан именно Для лантана в чистых растворах) при —1,85 В (Нас. КЭ). Для понижения растворимости осадка добавляют около 30% этилового спирта. Первый метод, с платиновыми электродами, несомненно удобнее второго. Если ввести в титруемый раствор несколько капель раствора феррицианида калия, то конечная точка титрования обозначится очень резко, так как при избытке ферроцианида начнет «работать» обратимая пара [Fe(CN)ft]3-/[Fe(CN)6]4~ (см.
13—1694
193.
Таблица 27. Состав осадков некоторых ферроцианидов РЗЭ в зависимости от состава, концентрации
и pH фона (содержание каждого лантаноида — 1 мг[25 мл)
Элемент Фон (MlNOa) Концентрация фона, M pH фона Состав осадка
La, Pr, Sm, LiNO3 l ,0 3,5—6 MiM[Fe(CN)6]
Eu (М) KNO3 1,0 3,5—6 M|M [Fe(CN)6
nh4no3 1,0 3,5—6 M,M Fe(CN)c
RbNO3 0,5 2,0—6 Fe(CN)6
CsNO3 0,5 2,0—6 M^lFefCNje]
LiNO3 0,5 3,2—6,6 Gd4[Fe(CN)6]
Gd NaNO3 0,5 4,5—6,0 Gd4[Fe(CN)6]
K(NH4)NO3 0,5 2,3—6,3 M,Gds[Fe(CN)6]4
Rb(Cs)NO3 0,5 1,4—6,6 M1Gd5[Fe(CN)6]4
LiNO3 1 ,o 2,3—6,3 Li4Ybs [Fe (CN) б] 7
Yb NaNO3 1.0 3,8—7,0 Na4Yb8[Fe(CN)6]7
K(nh4)no3 1,0 2,0—6,7 M|Yb[Fe(CN)6]
Rb(Cs)NO3 0,5 1,4—6,7 M,Yb[Fe(CN)6J
LiNO3 1,0 2,2—4,4 LiSc[Fe(CN)6]
Sc Na(K, NH4)NO3 1,0 5,0—6,0 Sc4[Fe(CN)6]3
Rb(Cs)NO3 0,5 4,0—6,0 Sc4[Fe(CN)e]3
Rb(Cs)NO3 0,5 1,4—6,3 M,Sc[Fc(CN)6]
LiNO3 1,0 3,2—6,5 Li4Y8[Fe(CN)6]7
Y NaNO3 0,5 3,2—7,0 Na4Y8[Fe(CN)6]7
K(NH4)NO3 1,0 2,5—6,0 M,Y[Fe(CN)6]
Rb(Cs)NO3 0,5 1,7—6,0 M|Y[Fe(CN)6]
Из работ И. В. Тананаева [3] известно, что состав осадка ферроцианидов лантаноидов зависит от того, какой катион входит в состав ферроцианида •— калий, натрий, литий и т. д. Специальное исследование, о котором упоминалось в гл. V '[4—7] показало, что в условиях амперометрического титрования состав осадка определяется также составом, концентрацией и pH фона. На этом основании разработаны методики амперометрического определения La, Pr, Sm, Eu, Gd, Yb, а также Sc и Y на фоне, гарантирующем получение осадка определенного состава [4—7]. В табл. 27 указаны фоны, их концентрация, pH и состав соответствующих осадков. Важно отметить, что скандий можно титровать в присутствии лантана, пользуясь тем, что осадок ферроцианида скандия образуется на фоне 1 М раствора нитрата натрия, тогда как лантан в этом случае не осаждается. Значения pH, приведенные в табл. 27, «крайние», т. е. при более низких значениях осадок выпадает не количественно, кривая титрования может иметь характерный для таких случаев «горб», а при более высоких значениях pH начинается выпадение осадков гидроксидов или основных солей лантаноидов.
По методу осаждения лантаноиды титруют также купфероном [8], фторидом калия [9], молибдат- [10], и вольфрамат-ионами
194
щ___13] Показано, что состав осадка, в частности самария(Н1),
зависит от pH среды: при рН=6—7 осаждается нормальный вольфрамат Sm2O3-3WO3, при рН=5,5--6—Sm2O3-7WO3, при пН=4—5—Sm2O3-9WO3. Для определения самария рекомендуйся рН=6—7 [13].
Известно, что лантаноиды осаждаются щавелевой кислотой. Эта реакция применена и для амперометрического титрования отдельных лантаноидов или их суммы по току восстановления щавелевой кислоты на ртутном электроде [14] или по току ее окисления на оксидных электродах [15].
ЭДТА позволяет определять некоторые лантаноиды как отдельно, так и суммарно. Есть данные о возможности определять отдельные лантаноиды в присутствии других элементов, например лантан в присутствии тория i[16], самарий (III) в присутствии тория, железа и магния [17] и др. ,[18—22]. Конечно при работе с ЭДТА необходимо помнить, что разница в р/< комплексонатов лантаноидов относительно невелика и что многие другие элементы могут реагировать с ЭДТА в очень близких условиях.
Применяют и другие комплексоны, например триамииопентауксусную кислоту для определения лантана, неодима, гадолиния, иттрия наряду с кадмием [23] (см. также «Кадмий», ссылка [22]), ализаринкомплексон [24], тринатрие-вую соль днэтилентриами (пентауксусиой кислоты [251. С помощью последней можно титровать европий(Ш) в присутствии определенных количеств лантана, церня(Ш) и неодима при рН~5, на ртутном капающем электроде по току вос-сгановления европия(III) при —0,85 В (Нас. КЭ); при этом европий(III) восстанавливается до европия(П).
Предложено также титровать лантаноиды наряду с иттрием в среде безводной уксусной кислоты уксуснокислым раствором ЭДТА (см. раздел «Иттрий», ссылка [5]).
Приводится описание несколько необычного способа амперометрического титрования лантаноидов на фоне расплавленного нитрата калия при 360 °C [26]. Титрантом служит расплав нитрата калия, содержащий вольфрамат или молибдат калия. Конечная точка соответствует образованию двойных вольфраматов или молибдатов лантаноидов.
В заключение следует упомянуть об определении празеодима (IV) в смеси оксидов лантанидов. По данным Амброжия и Гольцева [27], празеодим (IV) можно определять косвенным методом— по реакции взаимодействия его с марганцем(II). Празеодим (IV) окисляет марганец(П) до перманганата, который титруют (визуально) раствором оксалата аммония. Тщательное исследование этой реакции показало [29], что окисление марганца(II) проходит не до марганца (VII), а до марганца(III), причем, если вести эту реакцию в присутствии пирофосфата или фосфорной кислоты, то образуется устойчивый комплекс марганца(III), который также титруется оксалатом, поскольку стандартный окислительно-восстановительный потенциал системы Мп3+/Мп2+ имеет Т° ^теттзначение> что и потенциал системы МпОл/Мп2-1-. Марга-нец(Ш) в виде фосфатного комплекса также способен восстанав-
8 Таблица 28. Методы определения лантаноидов
Определяемый ион Гитрант Определяемый минимум Условия определения Примечания Литература
Lain, Prill, Smin, ЕиШ, Gdin, YblH, ScHl, Yin K4Fe(CN)6 3—10 мкг/мл Электрод Pt, гр=+0,6 В (Нас. КЭ), фой — см. текст См. текст 4—7
Lain, Cein, РгШ, NdlH Купферон 2 мкг/мл Электрод Pt, <р= + 1,0 В (Нас. КЭ) или 2 Pt электрода, Д(р= = 1,2 В, фон — рН=4—6, для коагуляции осадка 3—5 г NaCl Выжидать 1 мин, после каждого добавления титранта состав осадка — Ln : Cup= 1 : 3 8
Lain Na2MoO4 Нижний предел 10-s моль/л Электрод Hg капающий, <р= = + 1,5 В (Нас. КЭ), фон — кислый. если по току восстановления MoOf-, нейтральный —- по току восстановления La111, pH=5—6 Через раствор пропускают азот, состав осадка La2O3-3MoO3 10
Lain, Smin NasWO4 10-4 моль/л Электрод Hg капающий, <р= =—1,5 В (Нас. КЭ), фон — рН= =5—6, 50% спирта С изменением pH меняется состав осадка (см. текст) 12, 13
Любой лантаноид Щавелевая кислота 0,1—5 мг/10 мл Электрод Hg, <р=—1,3 В (Нас. КЭ), фон—0,1 М LiCl+лимон-ная кислота, рН=5—7 14
Lain, СеШ, ThlV Нижний преде.т 10-4 моль/л Электрод РЬО2 или МпО2, Т12О3 PtO2, (р= +1,3 В, фон — рН=2—3 Подробно изучена зависимость хода реакции на электродах oi pH; оксалат на электроде не реа гирует, только кислота 15
Lain, ThlV
Smin, Lain, Ybin, Luin, Cein, Tmin
Lain, Cein, Thiv
Prill, NdlH, Frill, Yin
PrO2, TbO2
ЭДТА
Ализарин-комплексон
K2WO4, К2МОО4, KVO3
Сульфат марганца и щавелевая кислота
10—200 мг
3—5 мкг/мл
50—250 мкг La, 6—1300 мкг Се, 100—2300 мкг Th
Электрод Hg, <р=—<1,25 В (Нас. КЭ) по току восстановления NO3, появляющемуся иа фоне 1 М раствора NaCl в присутствии тритона Х=100, рН=3
Мешают щелочноземельные металлы, аммоний и многие другие ионы
16
Электрод Та, <р=—1,0—1,2 В, Титруют отдельно лантаноиды, 17—22
фон — в основном ацетатный бу- бинарные и более сложные ком-
ферный раствор с различными позиции лантаноидов, а также лан-
pH; или 2 Pt электрода, Д<р=0.5— -4-0,8 В [18] таноидов с другими ионами
Электрод Pt, ср=+0,8-•—1,0 В (Нас. КЭ), фон — КЫОз+НЬЮ3 или ацетатный; для Се и La рН= =4,5—6,5; для Th—4—4,5 Так же определяют медь на фо не с рН=3,5—10 24
Электрод Pt, <р=—1,0 В, титруют в расплаве KNO3 расплавом KNO3, содержащим реагенты Образуются двойные соли К—№-Ln 26
Электрод Pt, <р= + 0,4 В (МИЭ), фон — H4SO4+H3PO4 (см. текст) См. текст 29
ливаться на платиновом электроде, поэтому можно проводить титрование раствором оксалата или щавелевой кислоты при +0,4 В (МИЭ) по току восстановления марганца(III).
Ход анализа. Навеску оксида празеодима(IV) или смеси его с оксидами других лантаноидов растворяют при нагревании в серной кислоте (1:3) в присутствии сульфата марганца и пирофосфата натрия. Сульфат марганца можно вводить в виде раствора, содержащего примерно 50 мг/мл марганца (общий расход примерно 5 мл при навеске оксида празеодима около 0,05 г), и пирофосфат— в виде насыщенного раствора (5—6 мл). По охлаждении образовавшийся марганец(Ш) титруют 0,1 н. раствором щавелевой кислоты из микробюретки емкостью 1—2 мл. При расчете исходят из того, что празеодим(IV) и марга-нец(П) реагируют в соотношении 1:1.
Совершенно так же протекает реакция окисления марганца(II) и железа(II) оксидом тербия ТЬ4О7 [11, 28].
ЛИТЕРАТУРА
1. Василенко В. Д.— В кн.: Сборник докладов на совещании по химическим методам контроля. Днепропетров. книжн. издат., 1960, с. 167.
2. Gour I. N., Zutshi К. — J. Electroanal. Chem., 1963, v. 5, p. 208.
3. Тананаев И. В., Сейфер Г. Б., Харитонов Ю. Я. и др. Химия ферроцианидов. М., Наука, 1971.
4. Сонгина О. А., Захаров В. А., Токушева Т.—Ж. анал. хим., 1970, т. 25, № 1, с. 64—67.
5. Токушева Г. Т., Захаров В. А., Сонгина О. А.—В кн.: Химия и химическая технология. Алма-Ата, вып. 7—8, с. 217—220.
6. Токушева Г. Т„ Захаров В. А. — Прикладная и теоретическая химия. Алма-Ата, 1974, вып. 5, с. 50—54.
7. Захаров В. А., Токушева Г. Т. — В кн. Сборник работ по химии КазГУ. Алма-Ата, 1973 вып. 3, с. 206—209.
8. Василенко В. Ю. — В кн.: Теория и практика полярографического анализа. Кишинев, Штиинца, 1962, с. 24.
9. Singh D., Varma А.—J. Sci. Res. Banares Hindu Univ., 1961, v. 112, p. 254. 10. Saxena R. S., Mittal M. L. — Taianta, 1964, v. 11, p. 649.
i 11. Сангина О. А., Кемелева H. Г. — В кн.: Методы определения и анализа редкоземельных элементов III—IV группы периодической системы. Свердловск, 1975, с. 19.
12. Gupta С. М.—Bull. Chem. Soc. Jap., 1965, v. 38, № 8, p. 1401; РЖХим., 1966, 5Г78.
13. Gupta С. M., Joshi M. P.— J. Ind. Chem. Soc., 1967, v. 44, № 11, p. 943— 946; РЖХим., 1968, 11Г19.
14. Комлев О. И., Марина В. С., Головчин Г. А.—ДАН УРСР (укр. яз ), 1971, Б, № 1, с. 42—43.
15. Sontag G., Kainz G., Margaritella P. — Z. anal. Chem., 1975, Bd. 273, № 4, S. 263--270.
16. Mukherji A. К—J. Electroanal. Chem., 1967, v. 13, № 4, p. 425—432.
17. Жданов А. K„ Мархабаев И. А., Муминов И. П.—Журн. ВХО, 1975, т. 20, № 2 с. 238_____239.
18. Мархабаев И. А., Жданов А. К. — ДАН УзбССР, 1973, № 10, с. 34—35.
19. Жданов А. К, Мархабаев И. А.—Изв. вузов. Химия и хим. техн., 1973, т. 16, № 9, с. 1331—1333.
20. Мархабаев И. А., Жданов А. К.—Ж. анал. хим., 1973, т. 28, № 1, с. 168— 170.
21. Жданов А. К, Муминов И. П.—Ж. анал. хим., 1974, т. 29, № 7, 1424— 1426.
22. Мархабаев И. А., Жданов А. К. — Научн. труды Ташкентского ун-та, 1974, вып. 462, с. 89—91.
198
94 Хадеев В. А. — РЖХим., 1977, 10Г79, депон. ВИНИТИ.
24 Хадеева Л. А. — РЖХим., 1976, ЗГЗЗ, депон. ВИНИТИ.
25 Kobaiashi М., Iwase А. — Yamagata Daigaku Kiyo Shizen Kagaku, 1960, v. 5, P- ЗОИ Stock J. T. Amperometric titrations. New York, 1960, p. 397.
26 . Вальцев В. К.. Куприянова А. К- — Изв. Сиб. отд. АН СССР. Сер. хим., 1969, вып. 2, с. 51—53.
27 . Амброжай М. Н., Гольцев А. М. — Науч. докл. высшей школы, Химия и хим. технол., 1958, № 3, с. 491.
28 . Сонгина О. А., Кемелева И. Г. — В кн.: Исследования в области химии Р. 3. Э. Саратов, изд-во Сарат. ун-та, 1975, с. 92.
29 Сонгина О. А., Кемелева Н. Г., Пихтовникова А. К—Зав. лаб., 1968, т. 34, ’ № 1, с. 10—12.
Литий
Химические и электрохимические свойства лития таковы, что амперометрическое титрование его затруднено. Он почти не образует малорастворимых солей, образованием которых можно было бы воспользоваться для прямого титрования, не образует также комплексных соединений и имеет сильно отрицательное значение стандартного потенциала. Поэтому пока известно только два способа определения лития амперометрическим методом: косвенное определение, заключающееся в осаждении лития уранил ацет атом цинка, отделении и растворении осадка с последующим титрованием цинка раствором ферроцианида калия на фоне тартратно-ацетатного буферного раствора с рН=7,5—8 в водно-этанольной среде. Титруют при потенциале +0,8 В (Нас. КЭ) на платиновом электроде. Количество определяемого лития — от 1 до 3 мг. Мешает определению уран(VI). Метод опробован на литийсодержащих материалах [1]. Второй способ — титрование в среде изопропилового спирта раствором щавелевой кислоты. Электроды — медный амальгамированный катод и медный анод, Дф=1,0 В. Нижний предел определения 1-10~4 моль/л. Метод разработан для последовательного определения калия (см. «Калий»), натрия и лития, причем авторы статьи '[2] замечают, что оксалат лития образуется в последнюю очередь и что в отсутствие калия и натрия литий практически не титруется.
ЛИТЕРАТУРА
1. Кулев И. И., Стенев Д. С., Бакалов В. Д. — Химия и индустрия (болг.), 1974, т. 46, № 10, с. 445.
2’ В- М Швыркова Л. А., Ким Л. Б.—Ж. анал. хим., 1971, т. 26, № 2, с. 269.
Магний
Рингбом и Вилькман [1], авторы индикаторного метода в амперометрическом титровании, попытались титровать магний раствором фторида калия, применив в качестве индикатора железо(III).
днако прямое титрование, вследствие относительно высокой ра
199
створимости фторида магния, не приводило к получению четкой конечной точки, поэтому авторы [1] предпочли добавлять к исследуемому раствору избыток фторида и титровать его раствором соли алюминия с тем же индикатором — железом(III).
Титрование магния оксихинолином известно в нескольких вариантах [2—6]. Однако лучших результатов можно ожидать от титрования магния растворами ЭДТА. Соответствующие методики описаны в разделе «Кальций», поскольку в основном титрование раствором ЭДТА разработано именно для раздельного определения магния и кальция. ЭДТА применен также для определения магния в присутствии висмута(III) (см. раздел «Висмут» ссылки [22, 23]) и в присутствии скандия и празеодима(III) [7].
Магний образует также прочное комплексное соединение с красителем пентахром виолет SW. Это позволяет определять магний при рН=11 путем титрования раствора красителя анализируемым раствором с ртутным капающим электродом при —0,6 В (Нас. КЭ). Метод применен для определения магния в известняках. Влияние кальция подавляют этилендиамином [8].
ЛИТЕРАТУРА
1. Ringbom A., Wilkman В.— Acta chem. Scandin., 1949, v. 3, p. 22.
2. Zyka J. — Ceska Farmacie, 1955, v. 4, p. 301.
3. Jordan D. E., Collis C. F. — Anal. Chem., 1958, v. 30, p. 1991.
4. Berraz G., Delgado O., Vassallo M — Rev. Fac. Ing. Quim. Univ. Nation. Litoral, Santa Fe, Argentina, 1958, v. 27, p. 65.
5. Powers R. M., Day R. A., Underwood A. L.—Anal. Chem., 1958, v. 30, p. 254.
6. Ishibashi M., Fuijinaga T. — Bull. Chem. Soc. Japan, 1950, v. 23, p. 229; Sbor-nik I mezinarodniho polarografickeho sjezdu, cast 1, Praha, 1951, s. 115.
7. Мархабаев И. А., Жданов A. К. — Изв. вузов. Химия и хим. технол., 1973, т. 16, № 4, с. 520—522.
8. Dean I., Bryan И. А. — Anal. chim. acta, 1957, v. 16, p. 180.
Марганец
Марганец относится к элементам с переменной валентностью, поэтому для его определения могут быть использованы окислительно-восстановительные методы. Еще в 1950 г. было предложено титровать марганец (VII) солью .Мора [1, 2]. Титрованию не мешают железо, кобальт, никель и другие обычные компоненты минерального сырья.
Ход анализа [3, 4]. Навеску стали растворяют в смеси 20—25 мл серной кислоты ('1:5) и 5—10 мл фосфорной кислоты (плотностью 1,7 г/см3). Затем прибавляют немного азотной кислоты для полного окисления Fe2+, упаривают до появления паров, разбавляют водой до 75—100 мл, добавляют 1,0—il,5 мл 1 %-ного раствора нитрата серебра и нагревают раствор до кипения. Прибавляют 15—20 мл 20%-ного раствора персульфата аммония и кипятят 2—3 мин до появления малиновой окраски (перманганат-ион). Затем переносят стакан с раствором на песочную баню и при слабом нагревании разлагают персульфат аммония (до прекращения выделения пузырьков кислорода). Жидкость охлаждают и титруют сумму марганца(VII), ванадия(У) и xpoMa(VI) 0,1 н. раствором соли
200
Мора. Затем вновь окисляют восстановившиеся при титровании ионы — марга-нец(П), хром(III) и ванадий(1У)—раствором персульфата при нагревании до появления малиновой окраски, после чего добавляют 2—3 мл 50%-ного раствора хлорида натрия и кипятят раствор до исчезновения малиновой окраски (селективное восстановление перманганата). Охлаждают раствор и титруют сумму хрома(У1) и ванадия(V) раствором соли Мора. Добавляют 0,1 н. раствор перманганата до появления малиновой окраски, т. е. селективно окисляют вана-flHfi(IV), разрушают избыток перманганата, добавляя по каплям 3%-ный раствор нитрата натрия, и тотчас же вводят 0,2—0,3 г тиокарбамида для связывания избытка нитрата. Затем титруют раствором соли Мора ванадий(V). Хром и марганец определяют по разности. Титрование проводят с двумя индикаторными электродами при напряжении около 0,1 В. Можно титровать и с одним индикаторным электродом [1], но титрование с. двумя электродами (см. гл. IV) несколько проще в техническом отношении и очень удобно в практике производственных лабораторий [4].
Методы определения марганца в присутствии ванадия и хрома описаны в разделе «Ванадий». Марганец(УП) в этих случаях восстанавливают солью Мора [6], ферроцианидом калия [7], иодидом калия [8]. Титрование ио дидом калия не дает воспроизводимых результатов [8], однако подробное исследование скорости реакции между Mnvn и I- показало i[9], что этот метод дает вполне надежные результаты на фоне 0,1 М серной кислоты. При этой концентрации кислоты Mnvn восстанавливается до Мп11. При концентрации же кислоты 7,5 н. восстановление может идти до Мп111, чем и может объясняться невоспроиз-водимость результатов.
Определение марганца (VII) и хрома (VI) в присутствии ванадия (V) осуществляют при помощи бромида калия [10], поскольку скорость реакции ванадия (V) с бромидом во много раз меньше, чем скорость реакции между бромидом и перманганатом и бихроматом, даже в 10 н. серной кислоте. Хорошо идет также реакция окисления Мп11 до Мп111 перманганатом при pH «6—7, причем Мп111 связывают пирофосфатом в прочное комплексное соединение. Подробное изучение этой реакции [11], позволило установить, что наиболее удобным для титрования является потенциал платинового электрода +0,4 В (МИЭ), при котором полностью исключается как анодный ток окисления Мп11, так и катодный ток образующегося при титровании Мп111, вследствие чего кривые титрования получаются весьма отчетливыми (формы б), так как возрастание тока обусловлено только избытком перманганата. Состав пирофосфатного комплекса отвечает формуле М^НгРгО?)!". Метод проверен на стандартных образцах марганцевой руды (№ 1-а, 44-а, смеси стандартного образца № 44-а и стандартного образца хромовой руды № 132). В присутствии хрома добавляют во время разложения пробы около 10 мл фосфорной кислоты плотностью 1,7 г/см3 и выжидают 15—20 мин, чтобы хром(III) связался в комплекс, не взаимодействующий с перманганатом i[12].
Окисление Мп11 перманганатом применено также для определения марганца в бедных рудах '[13], при анализе стали [14—16] И при совместном присутствии Мп11 и Мп1и [17].
201
Таблица 29. Методы определения марганца
Определяемый ион Титрант Определяемый минимум Условия определения Примечания Литература
№07 Соль Мора K4[iFe(CN)6] KI KBr N a2S2O3 H2O2 NaNO2 Тиосалициловая кислота Тиокарбамид 5 мкг/мл 0,01 мг/20 мл Мп—0,025 мг/20 мл Сг—0,042 мг/20 мл 0,02—3 мг/40 мл 0,01—3 мг От 0,2 мг и больше (4-10~3— 2-Ю-4 М) 4-Ю-З—2,4-10-4 моль/л Электрод Pt или 2 Pt электрода, Дф=0, 1 В с одним Pt электродом <р= +0,1 В (Нас. КЭ), фон—15 мл 9 и. H^SO4+5 мл 5 н. HNO3+H2O до 100 мл Электрод Pt, <р=+0,3—1,0 В (Нас. КЭ), фон — ацетат натрия + + 0,1 М— 1 М NaOH до рН=7,5 Электрод графитовый, <р=0,6 В (Нас. КЭ), фон —0,04 М H2SO4 Электрод Pt, <р= + 1,2 В (Нас. КЭ), фон — для Мп 6—8 М, а для Сг 10 М H2SO4 2 Pt электрода, Дф=0,6—0,8 В, фон — 50 мл 4 н. H2SO4+4 мл 18 н. Н3РО4 Электрод Pt, <р= + 1,2 В (Нас. КЭ), фон —HNO3 или H^SO4 Электрод Pt, ф= + 1,1 В (Нас. КЭ), фон — 2,5 и. H2SO4 Электрод Pt, ф= + 1,2 В (МИЭ), фон — 2 н. H2SO4 Электрод Pt, ф= + 1,2 В (МИЭ), фон — 8 н. H2SO4 Методика в тексте Определение Мп возможно в присутствии Сг и V (см. раздел «Ванадий») Определение Мп в присутствии V и Сг (см. раздел «Ванадий») Определение Мп и Сг в присутствии ванадия Не мешают Сг, Ni, V; определяют Мп в сталях; окисляют Мп11 до MnVI1 не в присутствии AgNO3 как обычно, а смесью сульфатов Со и Ni Кроме Мп можно определять хром (VI) и церий (IV) Mnvu сначала восстанавливается до Мп111 — первая к.т., затем Мп111 переходит в Мп11 без изменения тока; после второй к.т. окис ляется титрант; расчет делают по второй к. т. отношение Мп : титрант = 1:1 То же, что при титровании тиосалициловой кислотой, отношение Мп : реагент = 1 • 2, проверено на Мп рудах 1—4,6 7 9 10 20 21 22, 23 24 25, 26
Оксихииолин Основные органические красители: диметилтионин, метиленовый голубой, триметилтио- 1 1,25-10-4 моль/л 1-Ю3—7-Ю6 М Электрод Hg капающий, <р= =—1,52 В (Нас. КЭ), фон—1 М КС1, pH = 5 Электрод Pt или графитовый, ф= +0,6 В (МИЭ), фон — в зависимости от красителя, но во всех случаях можно 6 н. H^SO4 В тех же условиях титруют Ni и Zn Красители тиазинового ряда восстанавливают MnvnI, отношение Мп: реагент (метиленовый голубой) = 5 : 1, восстанавливает Mnvn до MnIV 27 29—31
МШИ Мп» Тиооксин Соль Мора К2С12О7 КМпО4 0,1 мг/20 мл При навеске 0,03 г определяют «90% Мп в ферромарганце 10-г—10~8 г-экв/л 2—5 мг/100 мл А 0.27—0,54 мг/50 мл Электрод Pt, ф=+0,8 В (Нас. КЭ), фон — 2 н. H2SO4 2 Pt электрода, Дф=1,2 В, фон— 15 мл 49%-ной HNO3 и 5—6 г сухого NH4F Электрод Pt, ф= +1,0 В (МИЭ), фон — 6 н. H2SO4 Электрод Pt, ф=0 (Хлоросеребр. э. с.), фон— 12 М НзРО4, Мп11 окисляется до Мп111 Электрод Pt, ф=+0,6 В (Нас. КЭ), фон — пирофосфат+немного ЭДТА pH=7,0—7,5 Электрод Pt, ф=+0.4 В (МИЭ), фон — пирофосфатный, pH = 6—7 2 Pt электрода, Дф « 1 В, фон — пирофосфатный, pH=6—7 2 Pt электрода, Дф=0,8- -1,0 В, фон — 2 н. Н28О4 Восстановление MnVI1 протекает стадийно, через образование Мп111, к. т. титрования отвечает восстановлению до Мп11 Образуется комплекс [MnF5]2_ который взаимодействует с солью Мора, железо не мешает, так как находится в форме [FeF6]3- При совместном присутствии Мп111 и Мп11 титруют две аликвотные части — в одной Мп111, вс второй Мп11 перманганатом при + 1,0 В (МИЭ) При определении Мп и Fe (см раздел «Железо») Определение Мп в бедных кар бонатных марганцевых рудах См. текст 11осле титрования Мп, если нуж но титруют Со фсррицианидок (см. «Кобальт») • 32 16 17 18, 19 13 11 14 15
В амперометрическом варианте можно осуществить и известный метод Фольгарда — Вольфа [6], т. е. титровать Мп11 перманганатом, рН=5,5. При этом варианте устраняются все недостатки обычного визуального варианта этого метода.
Возможно окисление Мп11 бихроматом в кислой среде, в присутствии больших количеств фосфорной кислоты [18—19].
Из восстановителей, рекомендованных для титрования перманганата, кроме упомянутых выше, можно упомянуть еще тиосульфат натрия [20], перекись водорода [21] и нитрит натрия [22, 23]’.
Для определения марганца применяют также некоторые органические реактивы [24—28], в том числе органические красители [29—31], тиооксин [32].
Определение марганца в присутствии других элементов описано в разделах «Ванадий», «Железо», «Кадмий».
Реакции осаждения для определения марганца практически не применяют. Рекомендованное ранее [6] осаждение Мп11 ферроцианидом калия не имеет преимуществ перед редокс-реакция-ми, в частности перед восстановлением Mnvn тем же ферроцианидом калия.
ЛИТЕРАТУРА
1. Бутенко Г. А., Беклешова Г. Е. — Зав. лаб., 1950, т. 16, № 7, с. 650.
2. Гренберг Е. И., Генис М. Я. —Зав. лаб., 1950, т. 16, № 10, с. 1002.
3. Филенко А. И. —Зав. лаб., 1963, т. 29, № 12, с. 1423.
4. Филенко А. И.—Ж. анал. хим., 1964, т. 19, № 4, с. 709.
5. Brann В. F., Clapp М. H. — J. Am. Chem. Soc., 1929, v. 51, р. 39.
6. Сонгина О. А. Амперометрическое титрование. М., Химия, 1967.
7. Бектурова Г. Б. и др.—Ж анал. хим., 1973, т. 28, № 11, с. 2211—2213.
8. Жданов А. К., Кондратова В. Ф., Акентьева И. А.— Узб. хим. ж., 1969, № 1, с. 13—15.
9. Сонгина О. А. и др.—Ж. анал. хим., 1974, т. 29, № 8, с. 1594—1598.
10. Сонгина О. А. и др. — Изв. АН КазССР. Сер. хим., 1976, № 2, с'. 75—77. И. Сонгина О. А., Даушева М. Р., Ходасевич С. А.—Ж- анал. хим., 1962, т. 17, № 5, с. 966—969.
12. Даушева М. Р., Сонгина О. А. — Зав. лаб., 1966, т. 32, № 10, с. 910—912.
13. Дюлгерова А. С., Ку лев И. И. — Химия и индустрия (НРБ), 1973, т. 45, № 6, с. 261—263.
14. Филенко А. И., Кужель А. М. — В кн.: Химическая технология. Респ. межвед. научно-техн. сб. 1968, вып. 14, с. 29—33.
15. Кужель А. М., Сурмий А. М„ Филенко А. И. — Ж. анал. хим., 1973, т. 28 № 11, с. 2251—2254.
16. Доренко Г. И., Филенко А. И.—Зав. лаб., 1967, т. 33, № 7, с. 795—797.
17. Сонгина О. А., Кемелева И. Г. — В кн.: Прикладная и теоретическая химия. Алма-Ата, 1974, вып. 5, с. 196—200.
18. Singh D., Sheela S. — Current Sci. (India), 1972, v. 41, № 9, p. 329—330; РЖХим., 1972, 20Г60.
19. Singh D., Bhatnagar V.—Allgem. und prakt. Chemie, 1967, v. 18, № 4, p. 107—108.
20. Бузинова 3. H., Филенко А. И.—Укр. хим. ж., 1971, т. 37, Кв 6, с. 603— 605.
21. Жданов А. К-, Ятрудакис С. М. — Узб. хим. ж., 1965, Кв 5, с. 18—24.
22. Жданов А. К, Ятрудакис С. М. — Зав. лаб., 1966, т. 32, Кв 11, с. 1336.
23. Жданов А. К., Ятрудакис С. М. — Узб. хим. ж., 1966, № 2, с. 17—21.
204
24 Бессарабова И. М„ Сонгина О. А.— Изв. вузов. Химия и хим. технол., 1976, ’ т 19 № 8, с. 1207—1209.
25 Бессарабова И. М„ Сонгина О. А. Изв. АН КазССР. Сер. хим., 1976, № 6, ' с -51.
26 Бессарабова И. М., Кемелева Н. Б., Бектурова Г. Б. — В кн.: Химия и хи-мическая технология. Алма-Ата, 1971, вып. 19, с. 164—170.
27 Gupta S. L., Sharma S. К., Jaitly I. N., Indian. J. Chem., 1966, v. 4, № 4, ’ p 166—167; РЖХим., 1967, 1Г62.
28 Чикрызова E. Г., Вассерштейн Ш. Е., Пронина И. В.—Ж- анал. хим., 1970, ’ т. -25, № 8, с. 1568—11274.
29 Овсепян Е. И., Махакян Л. А.—Арм. хим. ж., 1975, т. 28, № 14, с. 894— ’ 897.
30 Овсепян Е. Н., Махакян Л. А. — Учен. зап. естеств. наук, Армения, 1976, ’ № I (131), с. 71—74.
31. Овсепян Е. И., Махакян Л. А. —Арм. хим. ж., 1974, т. 27, с. 1004.
32 Бессарабова И. М„ Сонгина Q-. А., Захаров В. А.—Ж- анал. хим., 1975, ' т. 30, № 4, с. 755—758.
Медь
В предыдущем издании этой книги [1] упоминалось о том, что для амперометрического определения меди предложено около 40 реактивов, а общее число работ превышало 100. В настоящее время число титрантов приблизилось к 80, главным образом за счет органических соединений. Далеко не все реагенты, предложенные ранее, продолжают «жить» в настоящее время. С другой стороны среди титрантов, предложенных в 60—70 годах, также имеются такие, которые вряд ли найдут применение в будущем по тем или иным причинам (неизбирательность, труднодоступ-ность, отсутствие каких-либо преимуществ по сравнению с ранее предложенными более простыми титрантами и т. д.). Поэтому ниже будут приведены из прежних методов только те, которые не утратили своего значения, а из новых — те, которые имеют перспективы дальнейшего развития. Остальные методы можно найти в предыдущем издании этой книги и в обзорах Дж. Стока, публикуемых каждые два года в журнале «Analytical Chemistry».
Из давно известных методов следует упомянуть о методе, проверенном на анализе никелевых штейнов и других объектов, содержащих медь наряду с большими количествами никеля и железа [2]. Медь(II) образует с салицилальдоксимом (C7H7O2N) в уксуснокислом растворе светлый зеленовато-желтый осадок состава Cu(C7H6O2N)2; реактив специфичен для меди, никель(II) ко-бальт(Ц) и железо(П) им не осаждаются. Однако железо(Ш) мешает титрованию тем, что восстанавливается при тех же потенциалах, что и медь, по току восстановления которой проводят титрование; поэтому железо (III) связывают фторидом, добавляя его в количестве, достаточном для выпадения осадков фторида железа.
Ход анализа. Навеску штейна 0,25 г (содержание меди от 6 до 12%) рас-воряют в царской водке, выпаривают досуха на песочной бане, прибавляют мл соляной кислоты плотностью 1,19 г/см3 и 60 мл воды. Осторожно (чтобы выпал гидроксид железа) добавляют аммиак до слабокислой реакции; нагре-
205
вают раствор почти до кипения и осаждают железо также горячим 4%-ным раствором фторида натрия (35 мл). После охлаждения добавляют 5 мл уксусной кислоты (1:1) и титруют раствором салицилальдоксима. Титрант готовят следующим образом: 10 г салицилальдоксима растворяют в 50 мл спирта и приливают этот раствор по каплям к 950 мл воды, нагретой до 80 СС, взбалтывают, после исчезновения белой суспензии фильтруют. Титр устанавливают амперометрически по стандартному раствору меди (II), приготовленному из чистой электролитной меди. ,
Такой же ход анализа применяют и при определении меди в анодном никеле с той разницей, что навеску увеличивают до 0,5 г (содержание меди около 4%), а объем титруемого раствора и количество раствора фторида натрия уменьшают вдвое. Анодный никель разла! ают азотной кислотой—15 мл (3:2).
Из других органических реактивов, ранее рекомендованных для определения меди, следует упомянуть об а-бензоиноксиме (купроне), специфическом реактиве на медь {3—6]. Он интересен тем, что дает осадки с медью(II) в аммиачной среде, а в кислой — осаждает молибден(VI) и вольфрам (VI). Эта реакция, не лишенная практического интереса, ждет применения в амперометрическом варианте.
Рубеановодородная (амиддитиощавелевая) кислота [7—9] интересна тем, что позволяет определять медь(II) и никель(II) раздельно при их совместном присутствии в растворе. С медью она образует черный осадок в аммиачной или слабокислой среде. Реакция считается весьма чувствительной и может быть применена для определения меди в некоторых промышленных объектах — в алюминиевых сплавах, легированных сталях и латуни.
Серусодержащие органические соединения широко применяют для определения меди. Тиоацетамид осаждает медь(1) в виде Cu2S в аммиачно- или нитрато-уротропиновой среде [10—12], причем определению не мешают многие ионы. Метилдитиобиурет позволяет определять медь в присутствии свинца (II) [13] и кадмия (см. «Кадмий»). Тиооксин (8-меркаптохинолин) давно применяемый для определения меди(II) [14, 15], предложен для ее определения, в частности в присутствии ртути(II), серебра(I), палладия (II), цинка, молибдена(VI) и кадмия [16], для титрования ее в присутствии ванадия, молибдена и т. д. [17, 18] (см. также «Ванадий»), Производные тиооксина — 5-бром- и 4-метилпроиз-водные позволяют титровать медь и молибден на разных фонах [18].
Диэтилдитиокарбамат был предложен для титрования меди еще в 1950—52 гг. [19, 20]. Впоследствии этот реактив неоднократно применяли в разных вариантах [21—23]. Комплексное соединение свинца (II) с дитиокарбаматом менее прочно, чем такое же соединение меди, поэтому присутствие свинца не только не мешает определению меди, но улучшает резкость конечной точки благодаря тому, что диэтилдитиокарбамат свинца окисляется на электроде с большей скоростью, чем сам диэтилдитиокарбамат [21]. Испытаны в качестве реагентов для меди также различные производные диэтилдитиокарбамата: диметилдитиокарбамат [24], пиперидиндитиокарбамат i[25], диалкилдитиокарбамидогидразин 206
Г26]. Сравнительно недавно опубликованы сведения {27, 28] о титровании меди и свинца при их одновременном присутствии Г4-быс-('₽-аминоэтил)дитиокарбаминовой кислотой. Изучены также реакции меди с димеркаптотиопироновой кислотой и ее производными [29, 30]; некоторые реакции описаны в разделе «Висмут».
Из других титрантов для меди можно упомянуть органические тиосоединения [31—34] и некоторые не содержащие серу соединения, например бензтриазол [35], бензимидазол [36], диаминоглиоксим [37] и пикрат натрия [38].
ЭДТА и другие комплексоны этого типа продолжают играть важную роль в амперометрическом определении меди [39—45] Есть рекомендации для определения меди в присутствии магния [45]1, цинка [46], серебра и таллия [47]. Определение меди при помощи комплексонов в присутствии других элементов описано в соответствующих разделах («Висмут», «Железо», «Индий», «Лантан», «Кадмий», «Палладий», «Ртуть», «Молибден», «Серебро»).
Из неорганических реактивов следует в первую очередь упомянуть о ферроцианиде калия [48—55]. Реакцию с этим реактивом изучали на разных фонах, с разными электродами, по катодному току меди(II) на ртутном и по анодному току ферроцианида на платиновом электроде. К сожалению, ферроцианидный метод не мог получить широкого распространения потому, что он применим лишь для анализа растворов, не содержащих других катионов, способных осаждаться ферроцианидом.
Довольно известна реакция осаждения меди в виде Си1 роданидом калия [56—59] и образование осадков с селенитом '[60] и теллуритом натрия [61]. Осаждают медь также тиокарбонатом калия, рекомендованным и для определения других элементов (см. «Кадмий»),
К окислительно-восстановительным методам определения меди относится восстановление меди(II) солями ртути(I) [62] в присутствии роданида (см. «Железо») и амперометрический вариант известного йодометрического метода [63]. Иод, выделяющийся при взаимодействии меди (II) с иодид-ионами в подкисленном растворе, легко восстанавливается на платиновом электроде и может быть оттитрован раствором тиосульфата натрия. Этот метод позволяет определять любые количества меди.
Ход анализа. Исследуемый раствор подкисляют серной кислотой (до 1,0 н, по серной кислоте), погружают в него платиновый электрод, замыкают цепь без наложения внешнего напряжения (МИЭ), добавляют 0,5—1,0 г кристаллического иодида калия. При этом наблюдается резкое падение, до нуля, начального тока — тока восстановления меди (II), соответствующего ее концентрации в растворе. Такое резкое падение силы тока объясняется практически полным связыванием меди(П) в иодид После этого начинается выделение иода; сила тока возрастает и достигает максимума, отвечающего концентрации иода в растворе.
Титрование тиосульфатом следует начинать тогда, когда стрелка гальванометра займет устойчивое положение. По мере восстановления иода тиосульфатом концентрация иода падает и, если раствор не содержит других ионов, способных восстанавливаться в данных условиях, ток к концу реакции снижается до нуля, конечная точка лежит на пересечении кривой титрования с осью абсцисс.
207
g Таблица 30. Методы определения меди (Си11)
ОС — Титрант Определяемый минимум Условия определения Примечания Литература
С ал ицил альдоксим Электрод Pt или Hg капающий, <р=0 В (МИЭ), или —0,25 В (Нас. КЭ), при Pt электроде; при Hg электроде <р=—0,52 В (Нас. КЭ) Определение Си в штейнах, в анодном никеле и других объектах 2
а-Бензоиноксим (купрон) Сотые доли мг в 20 мл Электрод Hg капающий, <р=—0,25 В (Нас. КЭ), — по току восстановления меди(П), фон — 0,03 н. NH4OH Можно определять Си в присутствии Мо 6
Рубеановодородная кислота Электрод Hg капающий, <р = —0,1 В (Нас. КЭ), фон—аммиачный или слабокислый, раствор реактива — спиртовой Можно последовательно определять Си и Ni 7, 8
Тиоацетамид 10~4—10“3 моль/л В 1NH4OH; в кислой чреде 6-10-5—il • 10"? моль/л Электрод Hg капающий, <р=—0,6 В (Нас. КЭ), фон -NH4OH+NH2OH+ +H2SO4 или ККОз+уротропин, рН= =2—6; на таком фоне <р=—0,25-4 4-0,4 В (Нас. КЭ) Осадок Cu2S, Си предварительно восстанавливают гидроксиламином, не мешает Zn и другие металлы 10—12
Мети лдитиоб иур ет 0,13—0,65 мл Си и 1,66—0,4 мг РЬ в 25 мл Электрод Hg капающий, <р=—0,2 В (Нас. КЭ), фон — тартратно-аммоний-ный+МН4ОН до pH«9, продувают раствор азотом Последовательное титрование Си11 и РЬ11 — сначала по току восстановления Си11, затем, пока титруется РЬ, ток не меняется, после к.т. — ток окисления реактива 13
Тиооксии 14 мкг/20 мл 2 Pt электрода, Дф=0,1Ч-0,15 В фон — 0,1—2 н. НС1 или 0,1 н H2SO4 Определение Си, Fe, Мо, титруют последовательно Fe111 и Си11 17
5-бром- и 4-ме-тил-8-меркапто-хинолин (тиооксин) Электрод Pt, <р=+0,9 В (Нас. КЭ), фон — 1 н. H2SO4 при титровании 5-Вг-производным, КС1 + НС1 (рН=2) Можно титровать также MoVI на фоне H3PO4+NaOH (рН=2) 18
14—1694
Диэтилдитио-карбаминат и его производные при титровании 4-метил-производ-ным Электрод Hg капающий или Pt по току определяемого иона или по току окисления реактива, фоны различные См. текст 19—26
\-бис(Р-амиио-этил) дитиокарба-минован кислота Электрод Hg капающий или Pt, <р=+0,1 В (Нас. КЭ) для Pt, -0,2 В для Hg электрода, фон с pH=7,5—9, тартрат аммония + KNO3 Совместное титрование смеси Си+ +Cd+Ni (см. «Кадмий») и Cu+Pb, в последнем случае электрод Hg 27, 28
Димеркаптотиопирон и его производные 3—600 мкг/40 мл при титровании реактивом МФ Электрод графитовый, <р= + 0,8 В (Нас. КЭ) фон — ацетатный буферный раствор, рН=4,2 Изучено 22 производных, отношение Си: R в к.т. зависит от pH фона, при pH ~ 4 отношение 1 : 3, названо 6 лучших производных, в частности МФ-метилфенилпроизводные 30
2-меркапто-бензоксазол 1—4 мг Си 0,5—4 мг Ag 1—4 мг Pd Электрод Hg капающий, <р=—0,3 В (Нас.КЭ), фон — ацетатный буферный раствор желатина, pH = 4—6, реакция с Си2+ идет в 2 ступени, на кривой 2 перелома, к.т. отвечает второму перелому Мешает Hg1, Hg11 и PtIV 31, 32
Меркаптотриазол 4-Ю-5—4-Ю-7 моль/л Электрод графитовый. ф=0 В (МИЭ) фон — pH от 5 до 4 н. H2SO4 Осадок состава C2HN3SCu 33
Висмутол (димеркаптотиотиазол) 0,02 мг в 30 мл Электрод Pt, <р= +0,9 В (Нас. КЭ) фон — ацетатный или яитарно-борат-ный буферный раствор при pH от 5 до 1 н. НС1 Осадок состава Cu:R=l : 1, Fe111 связывают фторидом; метод проверен на алюминиевых сплавах с содержа нием Си» 4% 34
Диаминоглиоксим 10-2—5-10“4 моль/л Электрод Hg капающий, <р = —0,76 В (Нас. КЭ), фон — аммиачный буферный раствор Осадок состава Си : R = 1 : 2 • 37
Продолжение
Титрант Определяемый минимум Условия определения Примечания Лите ра-тура
Пикрат натрия 0,016—0,9 мг/мл Электрод Hg капающий, <р=—0,4 В (Нас.КЭ), фои —3 М NH,OH + + 1 капля 10 М раствора NaOH + + 1 мл 0,2%-иого раствора желатина Образуется осадок состава 1 :2, т.е. Cu(NH3)4- (pier) 2 38
ЭДТА до 5 -10—8 г Электрод Pt, вибрирующий <р= + +0,2 В (Нас.КЭ), по току Си11, фон —аммиачный буферный раствор, рН=9 В этой же работе описана возможность определения таких же количеств Си с помощью купферона по току его окисления при +0,9 В 39
да 2 Pt электрода, Д<р=1,0—1,6 В, фон — pH=6 до 3 н. HNO3 Мешают Со, In, Fe111, Pb, Hg, Ni, Tl, Th, Zn и CNS- 40, 4
10-7—<10~6 моль/л Электрод Hg капающий, пульс-по-лярографический метод 42
Триэтилентетрамин и ЭДТА Си 1 мкг/50 мл Mg 2 мкг/50 мл Электрод Hg, вращающийся (Pt проволока, покрытая ртутью, предварительно позолоченная), фон ацетатный буферный раствор, рН=5—6 Метод рекомендован для Си с три-этиленамином и для Mg с ЭДТА 43, 44
ЭДТА Электрод Та, <р=—0,1 В (Нас. КЭ) по току Си11 или при + 1,2 В по току ЭДТА, фон — с рН=2 для Си11, для Zn — 2 М NaAc + 1 М НАс Возможно последовательное титрование Си и Zn, меняя кислотность раствора 45
K4[Fe(CN6)]
0,03 мг/20 мл
KCNS
Электроды и фоны могут быть разные, в зависимости от объекта анализа См. текст 48—55
Электрод Hg капающий, (р=—0,5 В (Нас. КЭ), фон — NH4Ac + НАс + + NH2OH Си11 предварительно восстанавливают гидроксиламином до Си1 56—59
HgC104
KI +тиосульфат
Ферроцен
0,05—15 мг
10-5—JO-4 моль/л
Электрод Pt, ср=—0 В по току С11, фон — 0,3 М + 0,02 М НСЮ4+0,06 М KI (Нас. КЭ) KGNS + KI добавляют для подавления реакции между Си2+ и CNS~ с образованием (CNS)2, могущем быть при избытке KCNS в растворе [(CNS)2 окисляет 1~] 62
Электрод Pt, (р = 0 В фон — H^SO4 ~ 1 н. (МИЭ), См. текст 63—68
Электрод Pt, электрод сравнения Pt в КМпО4, фон — НС1О44 ацетонитрил (1:2) 72
Определению меди этим методом не мешают ни цинк, ни свинец, ни серебро, ни другие элементы, сопутствующие меди в различных природных объектах или промышленных продуктах, так как все эти элементы либо вообще не реагируют с иодидом калия, либо образуют малорастворимые иодиды, и, поскольку иодид калия добавляют в избытке, они выпадают в осадок и не мешают дальнейшему ходу анализа. Кай и при обычном визуальном титрометрическом методе, определению меди мешает железо(III), которое Необходимо перед титрованием связать в достаточно прочное комплексное соединение [64] с фторидом натрия: к исследуемому раствору добавляют сначала раствор ацетата натрия (насыщенный) до тех пор, пока не появится красная окраска ацетата железа, после чего вводят 30%-ный раствор фторида натрия до исчезновения этой окраски и дополнительно еще 4—5 мл этого же раствора фторида. При больших содержаниях железа обычно выпадает осадок фторида железа, который не мешает титрованию.
Йодометрический метод в амперометрическом варианте следует рекомендовать для определения меди в окрашенных растворах, например в присутствии большого количества никеля или хрома(III) и др. Этот метод оказался пригодным для определения микроколичеств меди в различных природных объектах — рудах, почве, а также в некоторых шлаках и других материалах [65—68].
Вместо тиосульфата некоторые исследователи считают возможным применять гидразин [69].
Для титрования меди в неводной среде рекомендованы разные титранты — ЭДТА [70], диэтилдитиокарбаминат свинца [71], ферроцен [72]. Кроме того, во многих работах описано определение меди в неводной среде попутно с другими элементами (см. разделы «Висмут» и «Серебро»),
ЛИТЕРАТУРА
1. Сонгина О. А. Амперометрическое титрование. М., Химия, 1967.
2. Матвеев Н. И., Креймер С. Е., Свишева В. И., Головина В. А. — Зав. лаб., 1953, т. 19, № 10, с. 1019—1023.
3. Langer А. — Ind. Eng. Chem. Anal. Ed., 1942, v. 14, p. 283.
4. Стрекалова О. С. Амперометрический метод анализа высококоэрцитивных магнитных сплавов. Канд. дисс. Казанский гос. ун-т, 1956.
5. Ishibaski М., Fujinaga Т. — Sbornik mezinarodniho polarografickcgo sjezdu, cast 1, Praha, 1951, s. 115.
6. Мусина T. К- Применение некоторых органических соединений для амперометрического титрования Канд. дисс. АН КазССР, Алма-Ата, 1952.
7. Левитман X. Я-, Резникова Р. И. — Изв АН БССР, 1953, с. 3.
8. Левитман X. Я-, Кривчик 3. А. —Зав. лаб., 1955, т. 21, № 4, с. 397.
9. Жданов А. К, Хадеева В. А., Вякозина О. К- — Зав. лаб., 1955, т. 21, №10, с. 913; 1959, т. 25, № 11, с. 1036.
10. Pryszczewska М., Krzeszowska Е. — Zvest. nauk. Politechn. szczecin. 1967, № 88, S. 31—36.
11. Pryszczewska M., Krzeczowska E. — Taianta, 1971, v. 18, № 6, p. 638—642.
12. Pryszczewska M., Krzeczowska E. — Zvest. nauk Pszczec., 1971, № 127, S. 21—22.
212
13. Deshmukh О. S., Nandi R. К-— Chem. and Ind., 1969, № 20, p. 655.
14 Усатенко Ю. И., Супрунович В. И. — В кн.: Новые методы анализа на металлургических и металлообрабатывающих заводах. М., Металлургия, 1964, с. 89.
15 Супрунович В. И., Усатенко В. И., Куликовская Ж. Б.—Ж. анал. хим., ’ 1970, т. 25, № 10, с. 1890--1893.
16 Усатенко Ю. И., Супрунович В. И., Ямнова Т. П. — Зав. лаб.,1973, т. 39, ' № 10, с. 175—180.
17. Супрунович В. И., Усатенко Ю. И. — Труды Днепропетровского химико-металлургического ин-та, 1962, вып. 16, с. 123—126.
18 Берэиня В. К, Янсон Э. Ю., Приедите Р. А.—Учен. зап. Латвийского ун-та, 1970, № 117, с. 138—141.
19г Okac A., Brejskova Е.—Anal. Chem., 1952, v. 24, р. 914.
20. Calzolari С. — Leybolds polarographische Berichte, 1950, v. 1, Ref. 267.
21. Усатенко Ю. И., Тулюпа Ф. М.— Зав. лаб., 1958, т. 24, № 12, с. 1327.
22. Лотарева В. И. — Ж- анал. химии, 1964, т. 19, № 1, с. 185.
23. Stricks W., Chakravarti S. К — Anal. Chem., 1962, t. 34, p. 508.
24. Усатенко Ю. И., Уварова К. А. — Труды Днепропетровского химико-технологического ин-та, 1959, вып. 12, с. 17.
25. Усатенко Ю. И., Уварова К- А. — Зав. лаб., 1960, т. 26, № 10, с. 1098.
26. Jacobsen Е„ Hansteen С.—Anal. chim. acta, 1963, t. 28, p. 249
27. Deshmukh G. S., Chattopadhaya S. S. — Indian J. Appl. Chem., 1972, v. 35, Ks 4—6, p. 113—114; РЖХим., 1975, ЗГ75.
28. Deshmukh G. S., Chattopadhaya S. S. — Indian J. Appl. Chem., 1972, v. 35, № 4—6, p. 115—116; РЖХим., 1975, ЗГ74.
29. Усатенко Ю. И., Аришкевич А. М. Авт. свид. СССР, 145051, 26/П—1962; РЖХим., 1962, 23Д154.
30. Шидловская А. И., Аришкевич А. М, Усатенко Ю. И. — В кн.: Химическая технология Респуб. темат. межведом, научно-техн, сб., 1971, вып. 18, с. 155— 159.
31. Вега В. С., Chakrabartty М. М. — Anal. chim. acta, 1965, v. 33, № 5, p. 564— 566.
32. Bera В. C., Chakrabartty M. M.— Chem. and Ind., 1966, № 11, p. 458; РЖХим,. 1966, 19Г135.
33. Овсепян E. H., Сираканян M. A.-—Арм. хим. ж., 1968, т. 21, № 4, с. 936— 940.
34. Беклешова Г. Е., Виткина М. А. — В кн.: Химическая технология. Респ. межвед. научно-техн, сб., 1966, вып. 5, с. 57—63.
35. Poddar S. N., Roy К. — Indian J. Appl. Chem., 1970, v. 33, № 3, p. 183— 188; РЖХим., 1971, 22Г98.
36. Poddar S. N. e. a. —J. Ind. Chem. Soc., 1974, v. 51, № 11, p. 974; РЖХим., 1975, № 18—19.
37. Mdnok F., Varhelyi C, Benko A. — Stud. Univ. Babes-Bolyai, ser. Chem., 1971, v. 16, № 1, p. 75—78.
38. Morales A., Gonzalez F. — J. Electroanal. Chem., 1970, v. 25, № 1, p. 151— 154.
39. Helbig W. — Z. anal. Chem., 1969, Bd. 246, № 3, S. 169—173.
40. Vorlicek J., Vydra F. — Taianta, 1965, v. 12, № 7, p. 671—676.
fl' ^ог^се^ Б, Vydra F. — Microchem. J., 1965, v. 9, № 2, p. 152—156.
42. Myers D. J., Osteryoung J. — Anal. Chem., 1974, v. 46, № 3, p. 356—359.
43. Freese F., Jasper H.' J., Boef G. —Taianta, 1970, v. 17, № 10, p. 945—953.
44. Хадеева Л. A. — РЖХим., 1977, 8Г76, деп.
45. Freese F., Boef G. — Taianta, 1966, v. 13, № 6, p. 865—866.
46. Жданов А. К, Андреева T. И. — Научн. труды Ташкентского ун-та, 1964, вып. 264, с. 69—72.
47. Жданов А. К.., Акентьева Н. А., Капица Н. В.—Узб. хим. ж., 1967, № 5, с. 18—20.
ло’ У°внык Н. Г., Кузьмина Н. Н. — Ж- анал. хим., 1949, т. 4, № 1, с. 96.
4У. Човнык Н. Г. и dp.— Зав. лаб., 1949, т. 15, № 6, с. 517.
213
50. Kalvoda R., Zyka J. —Chem. Listy, 1951, v. 45, p. 462.
51. Човнык H. Г., Клейбс Г. A. —Ж. анал. хим., 1948, т. 3, № 2, с. 303.
52. Riccoboni L., Goldschmied Р. — Proc. Intern. Congr. Pure Appl. Chem. IUPAC, 11th, London, № 1; 1947,-p. 199.
53. Kalvoda R., Zyka J. — Col. Czech. Chem. Comm., 1950, v. 15, p. 630.
54. Сонгина О. А., Войлошникова А. П., Козловский M. T.—Зав., лаб., 1951, т. 17, № 1, с. 3.
55. Хадеев В. А., Околова Я- И. — Труды Среднеазиатского гос. ун-та. Хим. науки, 1958, т. 10, с. 23.
56 Матвеев Н. И., Креймер С. Е. — В кн.: Сборник технической информации. Гипроникель. Л., 1954, № 21.
57. Хадеев В. А., Жданов А. К —Узб. хим. ж., 1958, № 3, с. 57.
58. Марунина А. Т.—Зав. лаб., 1962, т. 28, № 1, с. 25.
59. Попель А. А., Марунина А. Т. — В кн.: Теория и практика полярографического анализа. Кишинев, Штиинца, с. 139.
60. Deshmukh G. S., Asthana О. Р. — J. Sci. Res. Banares Hindu Univ., 1959, v. 10, p. 41.
61. Deshmukh G. S., Rao V. S. — Indian J. Chem., 1967, v. 5, № 1, p. 37—38. 62. Stock J. T.— Analyst, 1966, v. 91, № 1078, p. 27—32.
63. Сонгина О. А., Войлошникова А. П., Козловский M. T. — Изв. АН КазССР. Сер. хим., 1951, вып. 5, с. 3.
64 Сонгина О. А. — Зав. лаб., 1953, т. 19, № 9, с. 887.
65. Войлошникова А. П., Козловский М. Т., Сонгина О. А.—Зав. лаб. 1957, т. 23, № 3, с. 273.
66. Езерская Н. А. Применение полярографического и амперометрического методов для анализа некоторых золотосодержащих продуктов. Канд, дисс., ИОНХ АН СССР, 1955.
67. Кондрахина Е. Г. — Огнеупоры, 1961, т. 26, с. 381.
68. Singh D., Agarwala V. S. — Current Sci., 1961, v. 30, p. 12.
69. Panwar K. S., Mathur N. K-, Rao S. P. — Anal. chim. acta, 1961, v. 24, p. 541.
70. Геворгян A. M., Талипов Ш. T., Хадеева В. A.—РЖХим., 1974,'7Г64, депон. 71. Геворгян А. М. и др. — РЖХим., 1976, ЗГ121, депон.
72. Соломатин В. Т., Бусев А. И., Артемова Т. Н. — В кн.: Стандартные образцы в черной металлургии. № 2, М., Металлургия, 1973, № 2, с. 104—ПО.
Молибден
Определение молибдена (VI) можно осуществлять по методу осаждения солями свинца [1—9], как и в обычном гравиметрическом анализе. Разработан своеобразный вариант этого метода [10], основанный на каталитических свойствах молибдена: если титровать раствором ацетата свинца комплексное соединение молибдена (VI) и перекиси водорода на ртутном капающем электроде, то катодный ток его восстановления сначала возрастает, достигая максимума в конечной точке, когда практически весь молибден связан в осадок, а остаточный ток определяется произведением растворимости молибдата свинца в данном растворе. Поэтому при любой концентрации молибдата максимум находится на одной и той же высоте, равно как и остаточный ток. Начальный же ток и положение максимума относительно оси абсцисс зависят от содержания молибдена в растворе. Кривые титрования изображены на рис. 46.
Известен и другой каталитический метод определения Movl, основанный на каталитическом эффекте молибдата в реакции
214
Рис. 46. Кривые титрования перекисных’ комплексов молибдена (VI) 5 2-Ю-8 М раствором ацетата свинца на фсГне ацетатного буферного раствора (рН=4,5).
между перекисью водорода и иодидом калия [11]. Определение выполняют на двух платиновых электродах, Д<р = = 0,05 В, среда сернокислая.
Молибден (VI) можно титровать солями ртути(I) [12] или ферроцианидом калия [13]. Можно сначала молибден^!) восстановить гидразином до молибдена (V) и титро-
вать молибден (V) феррицианидом; по мере титрования выпадает осадок ферроцианида молибдена(VI) в результате взаимодействия продуктов реакции в растворе [14].
Вообще реакции восстановления для молибдена (VI) мало характерны, поскольку окислительно-восстановительный потенциал системы Movl/Mov относительно невысок. Поэтому их осуществление требует таких сильных восстановителей как хром (II) или ванадий(II) [15—17]. Разработан также вариант этого метода с двумя электродами из разных металлов [18, 19], причем наилучшими оказались пары вольфрам (анод)—платина (катод) и вольфрам — вольфрам, работающие при минимальной наложенной э. д. с. (0,1 В). В водно-органических средах можно титровать молибден^!) ферроценом [20, 21].
ЭДТА применен для титрования молибдена(VI) в двойных смесях с различными элементами [22, 23]. Для определения молибдена успешно применяют 8-меркаптохинолин (тиооксин) [24— 28], рений(VII) [27] и другие ионы [28] не мешают определению.
Привлекают внимание и другие органические реагенты [29 33].
Нанограммовые содержания молибдена предложено определять на основе каталитической реакции окисления иодид-иона перекисью водорода в присутствии молибдена (VI) [34, 35]. Определение выполняют приемами микротехники, с объемом раствора около 2 мл, два платиновых вибрирующих электрода. Титрования как такового нет, измеряют ток, обусловленный выделяющимся иодом, определяя его количество по калибровочному графику. Аналогичный метод, но не в микроварианте, был предложен еще
Для определения молибдена в присутствии вольфрама рекомендуется метод, описанный в разд. «Вольфрам» [37].
215
9И
Таблица 31. Методы определения Молибдена (МоО%~)
Титрант Определяемый минимум Условия определения Примечания Литература
Ацетат или нитрат РЬ11 Электроды различные, Hg капающий, или Pt, титруют по току восстановления РЬ2+ на ацетатном или нитратном фоне при рН=4—5 Применяют для определения Мо в стали, сплавах и т. д. 1—7
0,5—2,5 г/л Электрод Hg капающий, tp=—0,8 В (Нас. КЭ), фон—0,1 М K'N03+0,01% желатины, рН=3—4,8 8
Электрод Pt, q>= + l,l В (Нас.КЭ), фон—ацетатный буферный раствор, рН=5 Титруют по току окисления РЬ11 до PbIV 9
10-6 моль/л Электрод Hg капающий, <р=+0,1 В (МИЭ), фон — ацетатный буферный раствор, рН=4,5 Титруют раствором РЬАс2 по каталитическому току восстановления Н2О2 (см. текст) 10
HgC104 2 • 10-3— 10-6 моль/л 2 Pt электрода, Д<р=0,3—0,6 В, фон—1 н. H2SO4+2% KCNS Мешают Си11 и Fe111, реагирующие с Hg1 12
K3[Fe(CN)6] 0,01% Мо в горных породах ) 2 Pt электрода, Аф = 0,05—0,1 В, фон — 4 М НС1 Предварительно восстанавливают MoVI гидразином; по мере титрования выпадает осадок ферроцианида MoVI 14
Электрод Pt, <р=—0,1 В (Нас.КЭ), фон — 4 1!. НС1 + лимонная кислота Восстановление MoVI до Мо111 амальгамой Cd (см. текст); можно определять и W, и сумму Mo + W 37
K4[Fe(CN)6] Pt электрод, <р=0,8 В, фон — 2МНС1 ,NH4C1 13
Сг11 или V11 Электрод Pt, ф=+0,6 В (Нас. КЭ), по току окисления восстановителя или с Hg капающим электродом при —0,8 В по току восстановления MoVI Можно осуществить дифференцированное титрование Мо и W 15—17
Ферроцен
ЭДТА
Тиооксин
Бензоилфенил-гидроксиламин
N-тиобензоил-N-П-ТОЛИЛГИД-роксиламин
N-ацетилсали-цилои л-N-фенилгидроксиламин
N.N'-дифеиил-тиокарб амилгид-роксамовая кислота
а-бензоиноксим
1 мкг/25 мл
10~5—10~4 моль/л
110-4—110-8 моль/л
50—450 мкг/мл
Тот же метод, но 2 электрода из W (анод) и Pt (катод), или 2 вольфрамовых электрода Аф=0 В
Электрод Pt, электрод сравнения Pt в растворе КМпО4, фон — вод-но-ацетоновая среда, 6 М по НС1, титруют по току окисления ферроцена
Электрод Та, (р =—1,2 В (Нас. КЭ), фон — KNO3 или ацетатный буферный раствор, рН = 4,6
Электрод Pt, tp=+0,9 В (Нас. КЭ), фон — NaAc+HNO3 или НС1 до рН= = 1—3,5
Электрод Pt, ф = + 0.8 4- 0,9 В (Нас. КЭ), фон — 4,5 н. H2SO4
Электрод графитовый, tp= + l,0 В (Нас. КЭ), фон — 0,5—8 М H2SO4 или 1—5 М НС1
Электрод графитовый, (р= + 0,95 В (Нас. КЭ), или Hg капающий, (р=— —0,6 В, фон — 0,1—2 М H2SO4
Электрод графитовый, <р=+0,95 В (НАс. КЭ), фон —4,5—6,5 М H2SO4
30—800 мкг/мл
Электрод Hg капающий, <p=—0,0— —1,0 В (Нас. КЭ), фон — 1 н. НС1 + + 1 и. НАс, pH=0,6—2
Fe предварительно отделяют, если 18, 19 определяют Мо в стали
Определение Мо, а также Мо и Re 20, 21 в сплавах
Определению Mo не мешает рений, 24— рекомендуют определять последбва- 28, 38 тельно Ir, Au, Pd, Fe, Си, Мо
Предлагается определение Мо в 22, двойных смесях с Ni, V, Zn, Cd, La 23, 39
29
Не мешают многие элементы, в 30 том числе вольфрам
Не мешают многие элементы, в том 31 числе Си и немного W (не > 6-кратного по отношению к Мо),
Не мешают многие элементы, в том 32 числе Си и небольшие количества Vv, SnIV, TiIV
Белый осадок, состава Мо : R= 1 : 2; меша’ют Wvl, Crvi, Vv; определяли Мо в полевом шпате, в оптическом стекле
Описан способ определения молибдена в присутствии циркония и цинка с помощью ЭДТА [39] (см. также «Железо») в присутствии меди и висмута с помощью бензоилдитиокарбамината калия (см. «Висмут»), в присутствии церия(III) и ванадия(V)—с помощью аскорбиновой кислоты (см. «Ванадий») и бензоилфенил-гидроксиламин в присутствии цинка, кальция и магния (см. «Кальций»),
ЛИТЕРАТУРА
1. Thanheiser G., Willems J. — Arch. Eisenhiittenwesen, 1939, Bd. 13, S. 73.
2. Мухина 3. C. —Зав. лаб., 1948, т. 14, № 12, с. 1194.
3. Aylward G N.—Anal. chim. acta, 1956, v. 14, p. 386.
4. Торопова В. Ф, Срубинская Г. 3.—Зав. лаб., 1953, т. 19, № 1, с. 42.
5. Чирков С. К-, Студенская Л. С.—Зав. лаб., 1959, т. 25, № 11, с. 1034.
6. Бабко А. К.-, Волкова А. И-. —Зав. л^б., 1958, т., 24, № 2, с. 135.
7. Saxena R. S., Gupta G. М. — Naturwis., 1960, v. 47, p. 201.
8. Saxena R. S., Jain M. C. — Indian J. Chem., 1968, v. 6, № 4, p. 224—225; РЖХим., 1968, 22Г112.
9. Байменова A. T. — В кн.: Химия и химическая технология, Алма-Ата, 1966, вып. 5, с. 115—117.
10. Шарипова Н. С., Сонгина О. А.—Ж. анал. хим., 1973, 28, Кв 12, с. 2348— 2351.
11. Jedrzejewski W. — Chem. Anal. (Polska), 1960, v. 5, № 2, p. 207.
12. Тараян В. M., Ачарян Г. С., Шапошникова Г. Н.—Арм. хим. ж., 1974, т. 27, № 4, с. 279—282.
13. Дегтерев И. М. — Зав. лаб., 1955, т. 21, № 10, с. 917—918.
14. Церковницкая И. А., Кустова Н. А. — В кн.: Вопросы аналитической химии минеральных веществ, Л., ЛГУ, 1966, с. 28—30.
15. Пешкова В. М., Галлай 3. А., Алексеева Н. Н.— В кн.: Химия редких элементов. Изд-во ИОНХ АН СССР, 1957, № 3, с. 119.
16. Галлай 3. А. — Научные доклады высшей школы. Хим. и хим. технол., изд. МВ и ССО СССР, 1958, вып. 3, с. 498.
17. Гусев С. И., Николаева Э. М.—Ж. анал. хим., 1964, т. 19, № 4, с. 715.
18. Николаева Е. Р., Агасян П. К., Таренова К. X. — Вестник МГУ ун-та, 1968, № 2, с. 135—139.
19. Николаева Е. Р. и др.—Ж- аналит. хим., 1970, т. 25, № 1, с. 119—1.22
20. Соломатин В. Т., Яковлев П. Я., Артемова Т. Н., Лапшина Л. А. Ж. анал. хим., 1973, т. 28, № 11, с. 2197—2201.
21. Соломатин В. Т. и др.—В кн.: Стандартные образцы в черной металлургии, М., Металлургия, 1975, № 4, с. 108—113.
22. Жданов A. К., Умурзакова И. А.—В кн.: Некоторые вопросы химической технологии и физико-химического анализа. М., изд-во МГУ, 1964, с. 69.
23. Жданов А. К., Бархударян А. А. — Узб. хим. ж., 1976, № 6, с. 6—9.
24. Супрунович В. И., Усатенко Ю. И. — Зав. лаб., 1965, т. 31, Кв 3, с. 262.
25. Супрунович В. И., Усатенко Ю. И.-*-Ж анал. хим., 1965, т. 20, Кв 4, с. 800.
26. Боговина В. И., Новак В. П„ Мальцев В. Ф.—Ж- анал хим., 1965, т. 20, Кв 10, с. 951.
27. Павлова И. М., Сонгина Q А. — В кн.: Химия и химическая технология. Алма-Ата, МВ и С СО КазССР, 1966, Кв 3—4, с. 218-.
28. Супрунович В. И., Усатенко Ю. И. — В кн.: Химическая технология. Респ. межвед. научно-техн. сб. 1966, вып. 4, с. 27—33.
29. Соколова А. А., Мухина 3. С. — Зав. лаб., 1968, т. 34, Кв 9, с. 1060—1061.
30. Галлай 3. А. и др.—Ж. анал. хим., 1976, т. 31, Кв 5, с. 921—924; авт. свид. № 524119, РЖХим., 1977, 14Г185.
31. Галлай 3. А. и др.—Ж. анал. хим., 1975, т. 30, Кв 6, с. 1148—1152.
32. Галлай 3. А. и др. — Вестник МГУ. Химия, 1974, т. 15, Кв 5, с. 582—584.
218
33 Церковницкая И. А., Шипунова Л. Г. — В кн.: Применение органических реагентов в аналитической химии. Л., ЛГУ, 1969, с. 173—179.
34 Шафран И. Г., Розенблюм В. П., Штейнберг Г. А. — В кн.: Труды Все-союзНИИ химических реактивов и особо чистых химических веществ. 1969, вып. 31, с. 171—180.
35. Розенблюм В. П„ Шафран И. Г., Штейнберг Г. А. — В кн.: Методы аналитической химии реактивов и препаратов, 1971, вып. 18, с. 175.
36. Шарипов Р. К-, Сонгина О. А. — Зав. лаб., 1963, т. 29, № 12, с. 1293—1296.
37 Ку лев И., Ибраева Я. — Годишн. Висш. хим. техн, ин т (болг.), Бургас, 1972(1970), т. 7, с. 53—64.
38. Супрунович В. И., Усатенко Ю. И., Ямнова Т. П. — В кн.: Вопросы химии и химической технологии. Респ. межвед. тематич. сб. вып. 34, Харьков.
39 Жданов А. К-, Акентьева И. А., Закиров Б. Г. — ДАН УзбССР, 1971, № 2, ' с. 37—38.
Мышьяк
Для определения мышьяка применяют платиновый вращающийся электрод, титранты — растворы соответствующих окислителей при титровании мышьяка(III) или восстановителей при определении мышьяка (V). Прекрасные результаты дает титрование мышьяка (III) раствором бромата калия [1—5]. Титрование обычно проводят в солянокислой среде, но можно пользоваться и сернокислой, если вводить в раствор некоторое количество хлорида или бромида (натрия или калия). В конечной точке очень резко возрастает сила тока вследствие того, что избыточный бромат окисляет хлорид- или бромид-ионы до свободного галогена, который и дает ток восстановления на платиновом индикаторном электроде.
Для окислительного титрования мышьяка(III) предложено еще несколько реактивов: перманганат [6, 7], церий (IV) [8—11], сви-нец(1У) в виде ацетата [12], иод [13, 14, 16], перйодат калия [16], бихромат калия [17, 18]. Димеркаптотиопирон и его различные производные осаждают мышьяк(Ш) и мышьяк(У) [19, 20], так же как и производные антипирина ,[21] и несколько необычный реактив — пентасульфид фосфора [22], осаждающий мышьяк(III). Мышьяк(V) можно титровать и восстановителями, например тиооксином [23] или иодидом калия [4, 24] так же, как это описано в разд. «Медь».
Мышьяку почти всегда сопутствует сурьма. Обычно приходится тем или иным способом разделять их (например, отгонять мышьяк), но некоторые амперометрические методы позволяют определять мышьяк и сурьму с помощью одного и того же реактива в одном и том же растворе. К этим методам относятся, в частности, методы титрования иодом [18] и бихроматом [13].
Метод раздельного йодометрического определения Asin и Sb,n заключается в следующем: в одной аликвотной части раствора титруют сумму мышьяка и сурьмы „иодом в кислой среде; к другой аликвотной части добавляют бихромат, который быстро окисляет сурьму (III.) в бикарбонатиой среде и лишь медленно реагирует с мышьяком(III), что позволяет оттитровать мышьяк раствором иода, не затрагивая сурьму. Содержание сурьмы находят по разности двух титрова-
219
{g Таблица 32. Методы определения мышьяка
Определяемый ион Титрант Определяемый минимум Условия определения Примечания Литература
Asin КВгОз Электрод Pt, <р = 0,1—0,6 В (Нас. КЭ), фон — H2SO4 + немного сухого КВг В этих же условиях титруется Sb111, вместо бромата можно применить иодат калия 1—5
KMnO4+NH4F 0,9—2,4 мкг/л Электрод Pt, <р« +0,2 В (Нас. КЭ), фон —0,1—0,6 н. H2SO4+«sl% NHJ1’ 6
КМпО4 5 мкг/20 мл Электрод Pt, <р=+0,4 В (Нас. КЭ), фон — 0,1—2 М НС1 Определяют As111 в кеках и спеках металлургического производства, так же определяют SbIir 7
Ce(SO4)s Электрод Pt (или Та), <р=0,14-0,4 В (Нас. КЭ), фон — 0,25 н. KC1+KI или IC1 или OsO4 в качестве катализатора Кроме Ce(SO4)2 применяют также КМпО4, KIO3, Т1111, добавляя во всех случаях KI 8—10
РЬ(Ас)4 3-10~5 моль/л Электрод Hg капающий, <р=—0,8 В (Нас. КЭ), по току восстановления As111 и PbIV, фон — 3 н. H2SO4 12
кю4 2 Pt электрода, Д<р = 0,05—0,1 В на фоне NaHCO3+KI или 0,1—0,2 В на фоне 4—5 Л4 НС1 + несколько капель 0,1 н. раствора IC1 Так же определяют Sb111 на фоне 0,5 н. H2SO4-( КВг 16
P2SB 3,3—ilOO мкг/мл Электрод Pt, <р = 0 В (Нас. КЭ), фон—ацетатный буферный раствор, pH=4,5 Влияние мешающих элементов устраняют ЭДТА, определяли As в (NH4)2SO4, 0 поведении Sb не упоминается 22
3-карбонато-кобальтиат гек-саминкобальта 2 Pt электрода, Дер = 0,02—0,1 В, фон — 1 н. H2SO4+ 0,3—0,5 г КВг Определяют многие другие элементы в частности Sb (см. разд. «Азот» и «Висмут») 28
AsV Диантипирил-метан 0,05 мкг/мл Электрод Pt, <р=4-1,4 В (МИЭ), фон — H2SO4 или HNO3, оптимальный фон 0,02 М NH4NO3, 0,8 н. H2SO4, 0,02 М (NH4)2Mo04 Титруют Asv в виде As — Мо — гетерополикислоты, образуется труднорастворимый осадок; не мешают Ge, Si, V и др. Влияние Sb не оговаривается 21
Тиооксии 2 Pt электрода, Д<р=0,05—0,1 В, фон — 1—2 н. НС1 или H2SO4 Восстановление Asv до As111, предлагается серия последовательных титрований различных элементов, 0 Sb не упоминается 23
AsV, As»! Asin, Sbin KI Димеркаптотиопирон н его аналоги I2 и KI I2 Ce(SO4)2 К2Сг20у Доли миллиграмма в 20 мл 1 мкг/мл 1 мкг/мл 10~4 моль/л 370 мкг As 570 мкг Sb Электрод Pt, <р= + 0,2 В (МИЭ), фон— НС1~ 10 М Электрод графитовый, <р=+0,6 В (Нас. КЭ), фон — 0,1—5 н. НС1 или 1—20 н. H2S0„ Электрод Pt, <р= + 0,1 В (Нас. КЭ), фон — бикарбонатный для As111, титруют раствором иода, для Asv—9— 10 М НС1, титруют раствором KI Электрод Pt, <р=+0,1 В (Нас. КЭ), фон — бикарбонатиая среда 2 Pt электрода, Д<р=О, 1 В, фон — 4—5 М H2SO4+OsCl4 (катализатор) Электрод Pt, <р=0,25 В (Нас. КЭ), фон — 0,5—8,0 н. H2SO4 или 2,5—8 н. НС1 Мешают Си11, Fe111 Asv и As111 образуют труднорастворимые соединения в отношении Asv : R= 1 : 5 и As111: R=3 : 2 Титруют отдельно As111 и Asv в двух аликвотных частях раствора, применено для анализа руд Раздельное определение As и Sb (см. текст) Необходимо отделение As111 от Sb111 Необходимо отделение As111 от Sb111 24 19, 20 14 18 И 17
AsV, Sbin K2Cr2O? Тиооксин 2 мкг/мл 10 мкг/мл As 2 мкг/мл Sb Электрод Pt, <р= +0,2 В (Нас. КЭ), фон —для As 3 М H2SO4, для Sb 0,5 М H2SO4 Электрод Pt, <р=+0,8 В (Нас. КЭ), фон — для As 2 М H2SO4, для Sb — ацетатный буферный раствор, рН=5 Раздельное определение As и Sb при совместном присутствии (см. текст) Отношение As : тиооксин =12, a Sb : тиооксин = 1 : 3; дифференцированное титрование в одном растворе (см. текст) 13 25
Asin, Sbin, Snll to to I2 1—6 мкг/мл Электрод Pt, <р = + 0,05—0,2 В (Ag/AgCl) или 2 Pt электрода, фон — метанол 0,5 М по НС1 для определения Sn11 или 3,5 М по NaAc для As111 и Sb111; ацетонитрил или этанол — во всех случаях 2 М по NaAc, изопропанол — 4 М по NaAc Определяют As111, Sb111 и Sn11 в отдельности или при совместном присутствии, титрование с 2 Pt электродами дает наилучший результат 29
На основании изучения реальных потенциалов взаимодействующих систем и скорости реакций в растворах серной кислоты различной концентрации, был разработан метод, позволяющий определять с помощью бихромата мышьяк(III) и сурьму(III) в двух аликвотных частях одного и того же раствора при разной кислотности: в одной аликвотной части определяют сумму мышьяка и сурьмы, в другой — только сурьму [13]. Содержание мышьяка определяют по разности. Метод применен для анализа черновой меди и мышьяковистого кека.
Ход определения. Навеску пробы (0,5—1,0 г) разлагают 30—40 мл смеси H2SO4 и HNO3, упаривают до появления паров серной кислоты, охлаждают, добавляют 15—20 мл воды и 1—1,5 г сульфата гидразина для перевода мышьяка и сурьмы из высшей степени окисления в трехвалентное состояние. Раствор упаривают до объема 5—4 мл, что необходимо для разрушения избыточного количества гидразина, переносят в мерную колбу емкостью 100 мл, доводят до метки водой и отбирают две аликвотные части по 5 мл. К одной аликвотной части добавляют воду до объема 20 мл, а к другой — 3 At K2SO4 до такого же объема и амперометрически титруют 0,02 н. раствором бихромата калия обе аликвотные части при потенциале платинового индикаторного электрода +0.2 В (Нас. КЭ).
В первом случае оттитровывают сурьму(III), а во втором — сумму сурь-мы(Ш) и мышьяка(Ш). Содержание мышьяка находят по разности результатов двух титрований.
Определение мышьяка (V) и сурьмы(III) при совместном присутствии можно осуществить также при помощи тиооксина [25], пользуясь тем, что в кислой среде мышьяк (V) восстанавливается тиооксином, а сурьма(III) образует с тиооксином комплексное соединение (отношение сурьма: тиооксин=1 :2). Это позволяет дифференцированно титровать оба элемента, получая кривую титрования с двумя перегибами: первый соответствует окончанию реакции тиооксина с сурьмой (III), второй — восстановлению мышьяка^). Фон — 2 н. серная кислота, <р=-[-0,8 В (Нас. КЭ) по току окисления тиооксина.
В других работах [26—29] рассмотрено применение ВгС1 и BrI, а также трикарбонатокобальтиата гексамминкобальта(Ш) для определения ионов нескольких элементов, в том числе мышьяка и сурьмы. Для определения мышьяка и сурьмы (а также олова) в неводной среде предложен йодометрический метод [29].
В предыдущем издании этой книги были упомянуты некоторые другие реагенты [30], однако методы с использованием этих реагентов не получили дальнейшего развития и в настоящем издании не рассматриваются.
ЛИТЕРАТУРА
1. Laitinen Н., Kolthoff I. М. — Phys. Chem., 1941, v. 45, р. 1079.
2. Kobrow a М. — Chem. Listy, 1954, v. 48, p. 1039.
3. Kolthoff J. M., Stricks W., Morrett L. — Analyst, 1953, v. 78, p. 405.
4. Konopik N., Szlaczka K.— Oesterr. Chem. Z., 1951, Bd. 52, Helt 11, S. 205.
5. Kalvoda R., Zyka J. — Chem. Listy, 1951, v. 45, p. 461.
6. Issa I. M., Chandour M. A., Hammam A. M. — J. Indian Chem. Soc., 1974, v. 51, № 10, p. 872—874.
222
7 Чокина Н. Ю., Сонгина О. А., Захаров В. А. — В ки.: Прикладная и теоре-тическая химия. Алма-Ата, МВ и ССО КазССР, вып. 6, с. 18—22.
8 Ахмедов Г., Жданов А. К-—Узб. хим. 1971, № 2, с. 9—11.
9 Gopala Rao G., Gandikota M., Viswanath S. G. — Anal. chim. acta, 1976, ' v. 87, № 2, p. 511—514.
10. Adamek P., Dolezal J., Zyk.a J. — Microchem. J., 1964, № 1, s. 1—5.
11" Singh D., Prasad В. B., Bhatnagqr U. — J. Sci. Res. Banares Hindu Univ., ’ 1966—1967, v. >7, № 1, p. 70—74; РЖХим., 1967, 13Г151.
12. Yang N. S., Tien С. С. Kao H. H. — In: Amperometric titrations. New York, Interscience, 1965, p. 520.
13 Чокина H. Ю., Сонгина О. А., Захаров В. A.—Ж- анал. хим., 1-976, т. 31, ’ № 4, с. 720—726.
14 Захаров В. А., Сонгина О. А., Драгавцева Н. А.—Зав. лаб., 1960, т. 26, ' № 5, с. 537.
15. Кольтгоф И. М.— Зав. лаб., 1945, т, 11, № 7, с. 626 -632.
16 Хадеев В. А., Мухамеджанова Д. — В кн.: Труды Ташкентского ун-та. 1968, ’ вып. 323, с. 112—121.
17 Ва/ганд В., Николак В., АнтонщевиЬ Р. — Гласник Хем. друшт. Београд, 1975, т. 60, № 5—6, с. 347—351; РЖХим., 1976, 12Г160.
18 Захаров В. А., Войлошникова А. П., Сонгина О. А. — Зав. лаб., 1962, № 1, с. 27—31.
19. Усатенко Ю. И., Аришкевич А. М., Дьяченко Л. Ф.— Зав. лаб., 1967, т. 33, № 5, с. 550—551.
20. Дьяченко Л. Ф., Усатенко Ю. И., Аришкевич А. М. — В кн.: Химическая технология. Рэсп. межвед. научно-техн, сб., 1968, вып. 11, с. 119—124.
21. Луговой С. В., Одуд 3. Ж-—Ж. анал. хим., 1972, т. 27, № 9, с. 1754— 1758.
22. Chatterjee S. S., Banerjee В. К- — Technology (India), 1971, v. 8, №3—4, р. 245—248; РЖХим., 1973, 4Г131.
23. Ямнова Т. М., Супрунович В. И., Усатенко Ю. И. — В кн.: Вопросы химии и химической технологии. Респуб. межвед. научно-техн, сб., 1976, вып. 44, с. 71—76.
24. Сонгина О. А., Войлошникова А. П. — Зав. лаб., 1958, т. 24, № 12, с. 1331— 1336.
25. Захаров В. А., Сонгина О. А., Бессарабова И. М. и др. — В кн.: Сборник работ по химии. Алма-Ата, 1973, № 3, с. 198—205.
26. Корнева Л. Э., Генгринович А. И., Муртазаев А. М.—ДАН УзбССР, 1965, № 9, с. 28—31.
27. Нарзулаев С. Н., Генгринович А. И., Муртазаев А. М.—ДАН УзбССР, 1965, № 5, с. 23—25.
28. Хадеева В. А., Мухамеджанова Д. — Зав. лаб., 1970, 36, № 12, с. 1443— 1446.
29. Рамадан А. А., Агасян П. К-, Петров С. И.—Ж. анал. хим., 1975, т. 30, № 1, с. 89—94.
30. Сонгина О. А. Амперометрическое титрование. М., Химия, 1967.
Натрий
Амперометрическое определение натрия встречает те же трудности, что и определение лития. Известна пока всего одна работа L1], в которой предложено определять натрий в морской воде после осаждения натрия цинкуранилацетатом, растворения осадка и титрования цинка раствором ферроцианида, т. е. такой же метод, как и для лития (см. «Литий»), Можно рекомендовать также как и в случае лития, титрование щавелевой кислотой в среде изопропанола с двумя медными электродами («Литий»).
223
ЛИТЕРАТУРА
1. Ку лев И. И., Стенев Д. С., Бакалов В. Д. Химия и индустрия (болг.) 1974 г., т. 46, № 2, с. 65.
Никель
Новых реагентов для амперометрического титрования никеля, по сравнению с теми, которые были описаны в предыдущем издании настоящей книги [1] почти не появилось. Большая часть новых публикаций посвящена ранее предложенным реактивам — диметилдиоксиму и другим соединениям этого типа [2], и некоторым другим органическим соединениям, применяемым для определения не только никеля, но и других элементов. С помощью диметил-глиоксима определяют никель в никель-молибденовых сплавах [3], а также в сложных смесях, остальные компоненты которых определяют комплексонометрически. Эти работы описаны в разделах «Кадмий» (см. ссылку [15]) и «Железо» (см. ссылку >[43]). Никель во всех случаях титруют при рН~9. Предложены новые оксимы — о-оксиацетофеноноксим [4] и дигидрооксиацетофено-ксим [5].
Диметилглиоксим позволяет определять никель в присутствии кадмия в активной массе щелочных аккумуляторов [6]. Сначала осаждают никель титрованным раствором диметилглиоксима, проверяя полноту осаждения (диметилглиоксим должен быть взят в избытке), затем, не отфильтровывая осадок, титруют кадмий раствором ЭДТА по току вссстановления кадмия на ртутном капающем электроде при —1,2 В (Нас. КЭ). Когда титрование кадмия закончено, добавляют в раствор несколько капель раствора соли кадмия для того, чтобы избыток ЭДТА не мог связываться с никелем, солью которого титруют избыток диметилглиоксима. Это титрование проводят по току восстановления избыточного никеля при том же потенциале; pH раствора должен быть около 10 (pH устанавливают при помощи аммиака и хлорида аммония). Автор метода подчеркивает, что для титрования лучше применять не обычный ЭДТА, т. е. двузамещенную натриевую соль, а четырехзамещенную, во избежание появления в растворе ионов водорода. Описанный метод был применен для анализа массы щелочных аккумуляторов при соотношении никеля и кадмия 1 : 30, а также для анализа ванн, используемых для приготовления пластин аккумуляторов.
Ферроцианид калия, рекомендованный ранее многими исследователями |[1], применен для анализа сплавов на никелевой и железной основе [7].
Определение никеля с помощью ЭДТА [8] было применено также для последовательного определения никеля и кобальта [9].
Для титрования никеля, меди, кобальта и цинка применяют также натриевую соль хиноксалин-2-карбоновой кислоты. Осадки 224
I
с ионами никеля и кобальта образуются быстро, а ионы кобальта и цинка титруют «обратно», добавляя избыток реактива, так как осаждение происходит медленно [10].
Остальные работы, выходившие в последние годы, касаются в основном определения никеля в различных смесях с другими ионами и описаны в соответствующих разделах: «Кобальт», «Висмут», «Кадмий».
ЛИТЕРАТУРА
1. Сонгина О. А. Амперометрическое титрование. М., Химия, 1967.
2. Пешкова В. М., Галлай 3. А.—Ж- анал. хим., 1952, т. 5, № 3, с. 286.
3. Байменова А. Т., Суворова О. А. — В кн.: Химия и химическая технология. Алма-Ата, 1969, вып. 9, с. 175—179.
4 Poddar S. N., Ray К. — Indian J. Appl. Chem., 1970, v. 33, Ns 4, p. 225—227; РЖХим., 1971, 22Г131.
5 Gupta B. D., Malik W. U. — Indian J. Appl. Chem., 1969, v. 32, № 6, p. 348— 350; эЖХим., 1970, 24Г11.
6. Новаковская Э. Г. —Зав. лаб., 1962, т. 28, № 1, с. 28.
7. Романова В. С., Шишкина И. И., Кремлева Г. Д. — Труды Уральского НИИ черных металлов, 1970, № 9, с. 30—35.
8. Жданов А. К., Ахмедов Г. —Узб. хим. ж., 1969, Ns 4, с. 29—31.
9. Жданов А. К., Бархударьян А. А.—Ж- анал. хим., 1975, т. 30, № 10, с. 2045—2047.
10. Dutt N. К., Sanyal G. S. — Z. anal. Chem., 1972, Bd. 258, Ns 2, S. 107—110.
Ниобий
Для определения ниобия (V) и тантала (V) известен метод, основанный на образовании комплексного соединения с пирокатехином. Последний, так же как и другие фенолы, окисляется на платиновом электроде, давая площадку диффузионного тока в пределах потенциалов от +0,5 до +0,7 В (Нас. КЭ). Авторы работы [1, 2] воспользовались этим обстоятельством для титрования ниобия и тантала при различных pH: ниобий — при pH « 3, тантал при pH лг 8. Оба элемента могут присутствовать в растворе в виде оксалатных комплексов. Титан (IV), который очень часто сопутствует ниобию и танталу в минеральном сырье, тоже реагирует с пирокатехином, поэтому его маскируют раствором ЭДТА.
Ход анализа. Навеску руды (лопарит) разлагают серной кислотой в присутствии сульфата аммония при медленном нагревании, остаток растворяют в 5%-ном растворе оксалата аммония и переводят раствор в мерную колбу емкостью 100 мл. Затем отбирают три аликвотные части и титруют ниобий и тантал 0,002 М раствором пирокатехина в одной аликвотной части при pH « 8, вводя около 0,5 мл 0,01 н. раствора ЭДТА для связывания титана(1У); в другой аликвотной части точно так же титруют тантал, но при рНягЗ; в третьей аликвотной части титруют при рН~3, не добавляя ЭДТА, общее количество тантала и титана, после чего рассчитывают содержание титана.
Метод позволяет определять от 12 мкг и выше ниобия или тантала при отношении их к титану в пределах 1 : 30.
Этот метод был применен для определения ниобия в присутствии вольфрама в сплавах и минералах.
15—1694 225
Попытка титровать ниобий раствором купферона [4] не дала положительных результатов, так как состав осадка непостоянен, но впоследствии было показано [5], что титровать ниобий купфероном и неокупфероном все же можно, применяя в качестве фона серную кислоту с добавлением хлорида натрия в качестве коагулянта образующегося осадка. Неокупфероном можно титровать на фоне 1—2 н. соляной кислоты.
Предложено определение ниобия с помощью ксиленолового оранжевого [6], основанное на образовании комплексного соединения состава 1:1. Метод применен для определения ниобия в лопаритовом концентрате.
Серия работ [7—9] посвящена применению N-бензоилоксима и его аналогов для определения ниобия в присутствии многих не мешающих титрованию элементов. В Статье [10] приведены обобщающие сведения о применении этих реагентов для амперометрического определения ниобия.
ЛИТЕРАТУРА
1. Церковницкая И. А., Комолова И. Г. — В кн.: Методы количественного определения элементов. Л., изд-во ЛГУ, 1964, с. 77.
2. Комолова И. Г., Церковницкая И. А. — Зав. лаб., 1964, т. 30, № 12, с. 1329.
3. Церковницкая И. А., Комолова И. Г. — В кн.: Вопросы аналитической химии минеральных веществ. Л., изд-во ЛГУ, 1966, с. 21—27.
4. Elving Р., Olson Е. — Anal. Chem., 1956, т. 28, р. 338.
5. Усатенко Ю. И., Тихонова А. Н.— Зав. лаб., 1967, т. 33, № 8, с. 938—942.
6. Церковницкая И. А., Боровая Н> С., Анкудинова М. М. — В кн.: Применение органических реагентов в аналитической химии. Л., изд-во ЛГУ, 1969, с. 180—183.
7. Галлай 3. А., Шеина Н. М., Нифонтова Н. В.—Ж- анал. хим., 1968, т. 23, № 6, с. 942—944.
8. Галлай 3. А., Шеина Н. М„ Нифонтова Н. В.— Ж- анал. хим., 1970, т. 25, № 4, с. 737—741.
9. Галлай 3. А., Нифонтова Н. В., Литвак Н. Б. и др.—Ж- анал. хим., 1970, т. 25, № 7, с. 1337—1341.
10. Галлай 3. А., Шеина И. М.— В ки.: Успехи аналитической химии. М., Наука, 1974, с. 278—280.
Олово
Обычный широко распространенный йодометрический метод определения олова(II) может быть очень хорошо осуществлен в амперометрическом варианте. И. П. Алимарин и С. И. Терин [1] применяют для титрования олова (II) не только растворы иода, но и церий (IV) и перманганат. Титрование проводят при —0,2 В (Нас. КЭ) или без наложения внешнего напряжения (МИЭ) по току восстановления окислителя.
Ход анализа. От раствора олова(П), находящегося в мерной колбе, отбирают аликвотную часть 5 мл, помещают в стакан для титрования, вводят 3 мл концентрированной соляной кислоты и восстанавливают олово(П) металлическим цинком или алюминием. Затем слабо подогревают для растворения губчатого олова, охлаждают при пропускании двуокиси углерода, предварительно разбавив раствор до 25—-30 мл так, чтобы концентрация соляной кислоты в растворе составляла 3,5—4% (1 и.), и титруют окислителем.
226
Недавно йодометрический метод был вновь предложен для определения олова (II), мышьяка(III) и сурьмы(III) в неводной среде, содержащей в случае титрования олова соляную кислоту [2]. Метод описан в разделе «Мышьяк».
Вообще для определения олова(II) можно применить многие из реактивов, предложенных для титрования мышьяка(III) и сурьмы(Ш): бромат [3], иодат [4], хлорамин [5], иодхлорид [6], бихромат [7], разумеется в отсутствие растворенного кислорода воздуха. За последние годы было предложено несколько новых реактивов для определения олова в сложных смесях. Эти методы описаны в разделах «Марганец» и «Висмут». Для определения олова(II) в присутствии титана (III) предложена метиленовая синяя, восстанавливающаяся на платиновом электроде [8, 9].
В щелочной среде, в присутствии ЭДТА олово (II) дает на платиновом электроде хорошо выраженную [10] анодную волну с площадкой диффузионного тока в пределах потенциалов от 0 до —0,3 В (НВЭ). Этим фактом можно воспользоваться для титрования сурьмы (III) или мышьяка (III) раствором ртути (И) в присутствии олова (II): получается отчетливая «дифференциальная» анодно-катодная кривая титрования с хорошо выраженными конечными точками для олова(II), которое титруется в первую очередь, и для сурьмы (или мышьяка). Конечный подъем тока обусловливается восстановлением избыточной ртути (И).
Олово (IV) можно титровать хлоридом тетрафениларсония и нитрофениларсоновой кислотой [11], купфероном [12] и таннином [13]. Все эти методы выполняют на ртутном капающем электроде при продувании азотом и вряд ли они удобны для практических целей.
ЛИТЕРАТУРА
1. Алимарин И. П., Терин С. И. — Зав. лаб., 1954, т. 20, № 1, с. 18.
2. Рамадан А. А., Агасян П. К-, Петров С. И. — Ж. анал. хим 1975, т. 30, № 1, с. 89—94.
3. Subramanian И. — J. Sci. Ind. Res. (India), 1957, v. 16B, p. 516.
4. Fiorani M., Botnbi G., Freddi R. — Ricerca scient., 1963, parte 2, sez. A, 3, № 5, p. 749.
5. Хадеев В. А., Жданов А. К., Речкина Л. Г. — Узб. хим. ж., 1960, № 6, с. 28.
6. Генгринович А. И., Корнева Л. Е., Муртазаев А. М. — Труды Ташкентского фармацевтического института, 1960, т. 2, с. 355.
7. Deshmukh G. S., Murty S. V. — Z. anal. Chem., 1964, Bd. 198, s. 328.
8. Кузьмина H. H., Никулаева T. А., Ярцев M. Г. — В кн.: Методы контроля химического состава неорганических и органических соединений. Куйбышев,
9. Кузьмина Н. Н., Никулаева Т. А., Ярцев М. Г. — Изв. вузов. Химия и хим. технол., 1966, т. 9, № 5, с. 709—712.
10. Войлошникова А. И., Сонгина О. А.— Зав. лаб., 1964, т. 30, № 1, с. 18. io !• М., Johnson R. — J. Electrochem. Soc., 1951, v. 98, p. 138, 231.
i,atPamura S., Rein I., Booman G. — Anal. Chem., 1959, v. 31, p. 1868. Щ. Subramanian N. — J. Sci. Ind. Res. (India), 1957, 16B, p. 518.
15*
227
Осмий
В литературе описано только титрование осмия (VI) диэтанолди-тпокарбаминатом натрия на платиновом электроде при потенциале +0,8—1,1 В (Нас. КЭ) на фоне 1 н. серной кислоты; PtIV и Rh111 не мешают определению осмия [1]. Вторая работа посвящена амперометрическому определению осмия(VIII) без титрования: измеряют ток, обусловленный появлением иода при реакции окисления иодида перекисью водорода, катализируемой осмием(VIII). Фон — ацетатный буферный раствор с рН=3—3,5. Потенциал платинового электрода устанавливают 0,2—0,3 В (Нас. КЭ). Можно определять 10-5 мкг/мл осмия(VIII), рутений (IV) мешает, если содержание его больше чем в 10 раз превышает содержание осмия [2]. Возможно также определение осмия по его каталитическому эффекту на реакцию бромата калия с мышьяком(III) [3].
ЛИТЕРАТУРА
1. Швыдка А. Ф. и др. — В кн.: Анализ и технология благородных металлов. ' М„ Металлургия, 1971, с. 103—106.
2. Алексеева И. И. и др. — Ж- анал. химии, 1972, т. 27, № 8, с. 1566—1568.
3. Алексеева И. И. и др. — Ж. анал. хим.,_1974, т. 29, № 5, с. 1017—1019.
Палладий
Амперометрическое определение палладия(II) основано главным образом на реакции осаждения. Реагентов для этой цели предложено очень много. Один из самых простых и доступных методов — осаждение палладия(II) иодидом калия [1], с которым палладий (II) так же, как и серебро, дает осадок, практически нерастворимый в воде, но сильно отличающийся по растворимости в аммиаке: константы устойчивости аммиачных комплексов палладия (II) и серебра (I) различаются больше чем на 20 порядков. Отсюда следует, что из аммиачной среды в осадок будет выпадать только иодид серебра, а палладий останется в растворе. Золото (III) не может мешать при этом титровании, равно как не мешают ему и ионы цветных металлов, даже в 100—1000-кратном избытке (см. описание иодидного метода определения серебра в разд. «Серебро»),
Ход анализа. Навеску пробы 1—5 г (в зависимости от содержания палладия) помещают в коническую колбу емкостью 250 мл, смачивают водой и разлагают 20—40 мл концентрированной азотной кислоты при нагревании. Если после разложения пробы остаются еще темные частицы, то добавляют 2—3 г нитрата аммония и продолжают нагревание до полного обесцвечивания остатка. Упаривают до минимального объема, охлаждают и вновь упаривают с 10—20 мл серной кислоты (1:1) до появления паров, которым дают выделяться в течение 5—'10 мин (это необходимо для полного удален 1я окислов азота). Затем колбу охлаждают, обмывают стенки 15—20 мл воды и кипятят 10—15 мин для полного удаления нитрозилсерной кислоты. Вновь охлаждают и осторожно нейтрализуют 10—20 мл концентрированного раствора аммиака. По охлаждении раствор переносят в мерную колбу емкостью 100 мл и доводят до метки водой. Отбира-
228
ют две аликвотные части, устанавливают pH в одной части около 2—3, в дру-rOg_около 5, контролируя среду по универсальной индикаторной бумаге.
В аликвотной части с pH=2—3 титруют сумму серебра и палладия, в другой — только серебро, определяя палладий по разности. Конечно, если серебра в пробе не содержится, то титрование палладия можно проводить при рН^4 в одной аликвотной части. Титруют 3,005—0,0001 н. раствором иодида калия при +1,0 В (МИЭ).
Если в пробе содержится много палладия (больше 100 г/т), то при длительном нагревании с серной кислотой часть палладия (II) может восстановиться до металлического, поэтому упаривание надо проводить осторожно, только до появления белых паров, после чего дважды повторить упаривание с добавлением воды. Этот простой метод определения палладия проверен на промышленных образцах и дает вполне удовлетворительные результаты. '
Рекомендуется также осаждать палладий(II) иодидом калия в присутствии изохинолина или пиридина i[2, 3] в расчете на образование двойных солей типа РбгЯгЬ. причем вместо иодида калия можно применять азид натрия или роданид калия, получая соответствующие двойные соли того же типа.
Большую роль играют различные оксимы, с которыми палладий (II) так же, как и его аналог никель(II), образует весьма прочные соединения [4—9]. ,
К числу реагентов-осадителей относятся также бенздиазол [10], бензтриазол [И], 2-оксифенилбензоксазол [12] и меркаптодиметилтиазол [13], который по мнению предложивших его авторов значительно лучше, чем меркаптобензтиазол '[14]. Вообще серу-содержащие органические реагенты часто рекомендуют для титрования палладия(II), например висмутол [15], дитиокарбаматы [16—18], тионалид [19, 20], димеркаптотиопироны [21, 22], мер-каптобензоксазол (см. «Медь»),
Палладий(II) хорошо титруется тиокарбамидом [23—26], однако этому титрованию мешают золото(III) и серебро(I), вследствие чего авторы работы [23] прибегают к экстракции золота (III) эфиром из солянокислого раствора, а серебро выделяют в виде хлорида.
Титруется палладий(П) и меркаптохинолином (тиооксином) [27, 28], который часто рекомендуют для анализа сложных смесей, в состав которых входит и палладий (см. «Иридий», «Медь», «Молибден»).
Недавно для определения палладия(II) по методу осаждения применены два неорганических реагента — ферроцианид калия и роданид калия [29, 30]. Ферроцианид [29] в нитратной или сернокислой среде образует осадок Pd2[Fe(CN)6] с ПР=2-10-15. В солянокислой среде образуется растворимый комплекс смешанного состава [Pd2Fe(CN)6C12]2- с рК=4-10~34. Ионы других платиновых металлов (Pt, Rh, Ru, Ir) не мешают определению, если их содержание превышает содержание палладия не более чем в и раз, а влияние элементов с высоким окислительным потенциа-
229
Таблица 33. Методы определения палладия (PdM)
Титрант Определяемый минимум Условия определения Примечания Литература
KI 0,25 мкг/мл Электрод Pt; <р= 4-1,0 В (МИЭ), фон — 0,1 М H2SO4 или 0,05 7И НС1 Одновременно можно определить Ag (см. текст) 1
KI + изохинолин или пиридин 4—10 мкг/мл ' Электрод Hg капающий, <р=0 В (Нас. КЭ), фон — ацетатный буферный раствор, рН=4—6 Мешают Си, Ag, Аи, РЬ при изохинолине, при пиридине не мешают платиновые металлы 2, 3
Различные окснмы 10 мкг/мл Электрод Hg капающий, <р=0 В (Нас. КЭ),* или Pt, <р= 4-0,25 В (Нас. КЭ), фоны —0,2 М НС1, 0,1 М NaNOs, ацетатный буферный раствор с pH=5 4—9
2-оксифенонбензокса’ зол 10 мкг/мл Электрод Hg капающий, <р=0 В (Нас. КЭ), фон — ацетатный буферный раствор, рН=5—6 Мешают Ir, Pt, Си, Hg 12
2-меркапто-4-5-ди-метил-тиазол 0,02 мкг/мл Электрод Pt или графитовый; <р= 4-0,8—1,0 В (Нас. КЭ), фон — различный, но лучше ацетатный буферный раствор с pH»7 Мешает Hg11, если ее в 30 раз больше, чем Pd, и сильные окислители 13
Дитиокарбаматы 2 мкг/мл Электрод Pt, <р=4-0,8 В (Нас. КЭ), фон — 1 М НС1 или H2SO4 Мешают Ir, Pt, Au, Си, Sb 16—18
Тионалид 2 мкг/мл Электрод Pt, <р=4-1,1 В (Нас. КЭ), фон — 0,5 М H2SO4 Мешает 50-кратный избыток Си, As, Sb. Au, Se 19—20
Димеркаптотиопироны 1 мкг/мл
Тиокарбамид и его производные 2 мкг/мл
Тиооксин и его производные [ мкг/мл
K4[Fe(CN)6] 4 мкг/мл
KCNS 3 мкг/мл
Унитиол 0,4 мкг/мл
Бензидин 4 мкг/мл
Триазен (диазоаминобензол)
NaClO
2 мкг/мл
20 мкг/мл
Электрод графитовый, <р= + 0,6 В На фоне 10 н. НО не мешает 21—22
(Нас. КЭ), фон—1—10 н. НС1 или H2SO4 Электрод Pt, ср= +1,0 В (Нас. КЭ), фон — 1 М H2SO4 5-кратный избыток Au, Pt, Ir, Ag, Hg, Mo, Fe 23—26
Электрод Pt, <р = +1,0 В (Нас. КЭ), фон —• НО, H2SO4, K1NO3 При pH<2 состав осадка Pd(Thio)3, при pH>2 — Pd(Thio)2 27, 28
Электрод Pt, ср= +0,9 В (Нас. КЭ), фон —0,1—0,5 М H2SO4 Платиновые металлы не мешают, применен для анализа катализаторов 29
Электрод Pt, ср= + 0,88 (Пас. КЭ), фон —0,5—1 М H2SO4 Не мешает 10-кратпый избыток Ptlv, Rh111, RuIV, Irlv, Fe связывают в фторидный комплекс 30
Электрод Pt, ср=+0,8 В (Нас. КЭ), фон — 2 М НО Мешают Au, Ag, Fe, Си 31
Электрод Pt или графитовый, д>= + 1,0 В (Нас. КЭ), фон — КС1 + + НО рН~3 Не мешают Pt, Se, Fe, Си, Ni 32
Электрод Pt, ср= + 0,4 В (Нас. КЭ), фон — 0,1 М NaNO3+HCl, рН=2—4 Не мешает Аи111 33
Электрод Pt, ср= + 0,5 В (Нас. КЭ), фон —0,5 М NaCI + 0,5 М NaNO3, pH=2—3 Окисление Pd11 до PdIV 34
лом устраняется при помощи нескольких капель соли Мора до исчезновения катодного тока перед титрованием.
Роданид калия, подобно ферроцианиду, образует с Pd11 осадки в нитратной и сернокислой среде и комплексные соединения в солянокислой. Для практических целей предпочтительнее сернокислая среда [30]. Оба реактива успешно применены'для определения палладия в катализаторах.
Комплексные соединения, образование которых можно использовать для титрования палладия (II), дают также унитиол [31], бензидин ([32], триазен [33] и З-окси-1-п-сульфонатофенил-З-фе-нилтриазен, уже упоминавшийся в разд. «Золото».
Окислительно-восстановительные реакции для палладия мало характерны. Пока известен только один амперометрический метод— окисление палладия (II) раствором NaClO в присутствии азида натрия и хлорида натрия в кислой среде [34].
ЛИТЕРАТУРА
1. Сонгина О. А., Пащенко А. И., Маслова П. И.—Зав. лаб., 1965, т. 31, №6, с. 661—663.
2. Rao A. L„ Puri В. K. — Z. anal. Chem., 1972, Bd. 258, S. 286—289.
3. Рао А. Л., Пури Б. К. — Ж.^анал. хим., 1973, т. 28, № 2, с. 185—186.
4. Tomicek О., Chihalik J., Dolezal J., Simon V., Zyka J. — Chem Listy, 1952, v. 46, p. 710—714.
5. Бардин M. Б., Ляликов Ю. C.—Ж- анал. хим., 1957, т. 12, № 3, с. 390—394.
6 Бардин М. Б., Мелека Н. Д.—Учен. зап. Кишиневского ун-та, 1957, т. 27, № 1, с. 87—90.
7. Ивонина О. М.—Ж. анал. хим., 1964, т. 18, № 5, с. 344—р47
8. Bark L. S., Lain М. — Anal. chim. acta, 1970, v. 49, p. 349—351.
9. Krishna R. V., Brahmaji R. S., Appala R. N- — Proc. Ind. Acad., 1975, A—81, N, 1, p. 32—37; РЖХим., 1975, 20Г94.
10. Цветашвили В. HI., Хавтаси H. С., Гаприндашвили В. Н. — Сообщ. АН ГрузССР, 1972, № 65, с. 317—320.
11. Wilson R., Wilson L.— Anal. Chem., 1956, v. 28, p. 93—97.
12. Wilson R., Baye L., James J. — Z. anal. Chem., 1959, Bd. 171, S. 103—106.
13. Баркалов В. С. и др. —Зав. лаб., 1978, т. 44, № 1, с. 1—3.
14. Majumdar А. К., Chakrabarty М. М. — Anal. chim. acta, 1959, v. 20, p 286— 390.
15. Bera В. C., Chakrabarty M. M. — Anal. chim. acta, 1965, v. 33, p. 564—569; Talar.ta, 1966, v. 13, p. 1186—1188.
16. Усатенко Ю. И., Уварова К. А. — Труды Днепропетровского химико-технологического ин-та, 1961, № 12, с. 177—180; Укр. хим. ж., 1963, т. 29, № 2, с. 193—195.
17. Усатенко Ю. И., Тулюпа Ф. М., Ткачева Л. М. — В кн.: Современные методы химической технологии и контроля производства. Ростов-на-Дону, изд-во Ростовского ун-та, 1968, с. 67.
18. Усатенко Ю. И., Тулюпа Ф. М., Ткачева Л. Н. — В кн.: Химическая технология. Харьковский ун-т, 1971, № 18, с. 150—153.
19. Усатенко Ю. И., Заморская Т. В. — Зав. лаб., 1963, т. 29, № 3, с. 291— 294.
20. Усатенко Ю. И., Заморская Т. В. — В кн.: Новые методы анализа иа металлургических и металлообрабатывающих заводах. М., Металлургия, 1964, с. 124.
21. Аришкевич А. М. и др. — Зав. лаб., 1970, т. 36, № 3, с. 265—268.
22. Аришкевич А. М., Усатенко Ю. И. — В кн.: Анализ и технология благородных металлов. М._ Металлургия, 1971, с. 197.
232
23 Усатенко Ю. И., Толубара А. И.—Зав. лаб., 1965,. т. 31, № 1, с. 34—36.
24 Бардин М. Б., Баландина М. С., Тодорова Г. И, —Ж. анал. хим., 1964, т. 19, ' № 11, с. 1228—1232.
25 Усатенко Ю. И., Сухоручкина А. С. — В кн.: Химическая технология. Харьковский ун-т, 1962, вып. 17, с. 178 181.
26. Оробинская В. А., Иванова В. П., Евтифьева Г. К.— Научн. труды СибНИпроект. ин-та цветной металлургии. Красноярск, 1971, вып. 4, с. 7—11.
27 Усатенко Ю. И., Супрунович В. И. — Изв. АН ЛатвССР. Сер. хим., 1963, № 1, с. 18—30; ДАН СССР, 1963, т. 153, с. 622—625.
28 Берзиня В. К., Янсон Э. Ю., Седола В А. — Учен. зап. Латвийского ун-та. ’ Сер. хим., 1967, № 88, с. 75—80.
29 Захаров В. А., Сонгина О. А., Айтхожаева Т. А.—Ж- анал. хим, 1974, ' т. 29, № 6, с. 113и—1133.
30 Захаров В. А., Айтхожаева Т. А. — Зав. лаб., 1975, т. 41, № 2, с. 164—165. 31. Сонгина О. А. и др. —Ж. анал. хим., 1967, 22, № 6, с. 1170—1174.
32. Вещева Л. В., Рейшахрит Л. С., Щербакова С. И. — В кн.: Применение органических реагентов в аналитической химии. Л., ЛГУ, 1969, с. 218—220.
33. Темянко В. С. — В кн.: Сборник научных трудов Магнитогорского горно-металлургического ин-та, 1971, № 87, с. 34—38.
34. Clem R., Hoffman Е. — Anal. Chem., 1968, v. 30, p. 945—950.
Платина
Амперометрическое титрование платины до самых последних лет было практически не разработано, несмотря на то, что платина (IV) и платина(II) способны вступать в реакции окисления — восстановления с различными реагентами. В 70-х годах появились работы, в которых описаны методы определения платины, основанные именно на этой реакции: восстановление платины (IV) иодидом калия [1] и тиокарбамидом [2—4], окисление платины (II) сульфатом церия (IV) |Д 6] и перманганатом [7]. Показана также возможность определения платины(II) по методу комплексообразования при помощи тиокарбамида [4].
В дополнение к данным табл. 34 следует отметить, что иодо-метрическому определению платины (IV) не мешает значительный избыток многих ионов: Rh111, 1г,п, Ru1”, Asv, Sbv, А1Ш, ThIV, SeIV, TeIV, CrVI, Vv, Ni11, Fe”, In111, а также равные количества Ru. Мешают Pd11, Fe111, IrIv. Однако мешающее влияние палладия (II) и железа (III) легко устраняется путем перевода палладия в осадок диметилглиоксимата, а железа — во фторидный комплекс. Влияние иридия (IV) устраняется путем восстановления его до иридия (III) солью Мора.
Ход определения платины (IV). Навеску (0,1—0,5 г) анализируемого материала растворяют в царской водке, упаривают дважды с концентрированной соляной кислотой до объема 5—10 мл. Переносят раствор в мерную колбу емкостью 100 мл и доводят водой до метки. Отбирают аликвотную часть раствора 5 мл, приливают воду до объема 20 мл и прибавляют 0,5 г твердого фторида натрия. Затем помещают раствор в амперометрическую установку, задают потенциал платиновому электроду +0,25 В (Нас. КЭ) и добавляют по каплям 0,001 М раствор соли Мора до исчезновения катодного тока восстановления иридия (IV) и других окислителей. Если катодный ток отсутствует, то операцию добавления соли Мора не проводят. Изменяют потенциал с +0,25 В на —0,25 В (Нас КЭ)—потенциал восстановления иода, прибавляют 1 г твердого ацетата натрия, добиваясь полного его растворения, добавляют 3 г KI и, спустя 2— о мин, титруют выделившийся иод 0,005 и. раствором тиосульфата натрия.
233
Таблица 34. Методы определения платины
Определяемый ион Титрант Определяемый минимум Условия определения Примечания Литература
PtIV KI и Na2S2O3 0,5 мкг/мл Электрод Pt, ср=—0,25 В (Нас. КЭ), фон — ацетатный буферный раствор с рН=6—8; берут 500—1000-кратный избыток KI по отношению к Ptlv Йодометрическое определение ptci Г +6P--^Pti42- +i2+eci- 12+ + 2S20i-—<-S4O в +21~ применено для анализа сплавов и катализаторов 1
Тиокарбамид 4 мкг/мл 10 мкг/мл Электрод Pt, q>=+0,9 В (Нас. КЭ), фон — 3 М KNO3 Электрод Pt, ср = + 0,6 В (Нас. КЭ), фон — 2 М HNO3 Тнтруют сульфатный комплекс PtIV, который в данной среде реагирует с тиокарбамидом в отношении 1 : 5; при этом PtIV восстанавливается до Pt11, a Pt11 образует комплекс [РЦЭОЙгН^зОН]*, применено для анализа катализаторов Хлоридный или сульфатный комплекс PtIV взаимодействует в данной среде с тиокарбамидом в отношении 1 : 6, при этом Pt ^восстанавливается до Pt11, a Pt11 образует комплекс [Pt(SCiN2H4)4]2+ 2 3,4
Ptn Тиокарбамид 10 мкг/мл Электрод Pt, ср =+0,6 В (Нас. КЭ), фон — 2 М HNO3 PtCl 1~ взаимодействует с тнокарба-мидом в отношении 1:4с образованием комплекса [Pt(SCN2H4)4]2+ 4
00(804)2 2 мкг/мл 2 Pt электрода, Дср=0,1 В, фон—-0,1—0,5 М НС1 или H2SO4, температура 80 °C Pt11 получают восстановлением PtIV хлоридом меди (I), при титровании CeIV сначала реагирует с избытком Си1, а затем с Pt11, вследствие чего на кривой получаются две к. т.; применено для аналцза сплавов и катализаторов 5,6
КМпО4 0-'э мкг/мл Электрод Pt, ср =+0,65 В (Н.В.Э), фон — 0,1—2 М НС1 или H2SO4 Pt11 получают восстановлением PtIV хлоридом меди (I), избыток Си1 удаляют нагреванием; применено для анализа сплавов и катализате ров 7
ЛИТЕРАТУРА
1 Захаров В. А., Гаева И. Ф., Сонгина О. А.—Ж. анал. хим., 1976, т. 31, №4, ' с 746—750.
2 Гавва Н. Ф., Сонгина О. А., Захаров В. А.—Ж. анал. хим., 1975, т. 30, №9, ' с 1780—1783.
3 Гавва Н Ф„ Захаров В. А., Сонгина О. А. — Изв. АН ЕазССР. Сер. хим., 1976, № 1, с. 67—70.
4 Гавва И. Ф., Захаров В. А., Сонгина О. А.—Ж. анал. хим., 1976, т. 31, №7, с. 1334 -1337.
5 . Hulanicki A., ledral W., Rubel S. — Analyst, 1972, v. 97, № 1154, p. 340—345.
6 . Hulanicki A., Jedral W- — Chim. Anal., 1974, v. 19, № 4, p. 765—769.
7 Гавва H. Ф., Сонгина О. А., Захаров В. A.—Зав. лаб., 1976, т. 42, № 9, ' с. 1040—1041.
Плутоний и нептуний
Плутоний имеет переменную валентность, что дает возможность применять для его амперометрического определения методы окисления — восстановления. На платиновом электроде можно титровать плутоний(VI) в виде РиО1+ раствором сульфата железа (II), причем можно использовать как ток окисления Fe2+ после конечной точки [1], так и ток восстановления [2] РиОг+ до Рп4+. В первом случае титруют при +0,6 В относительно М.СЭ (мер-кур-сульфатного электрода сравнения), во втором — при 4-1,0 В (Нас. КЭ). Оба метода осуществляют в микро- или полумикроварианте с применением весовой бюретки и специальной аппаратуры.
Этот же принцип — восстановление NpVI и PuVI железом (II) положен в основу кулонометрического титрования этих элементов электрогенерированными ионами Fe2+ с амперометрической индикацией конечной точки [3, 4] на фоне фосфорной и серной кислот.
ЛИТЕРАТУРА
1. Seils С., jr., Meyer R., Larsen R. — Anal. Chem., 1963, v. 35, p. 1673.
2. Helbig №. —Z. anal. Chem., 1961, Bd. 182, S. 15.
3. Моисеев И. В., и др. — Радиохимия, 1976, т. 18, № 1, с. 128—136.
4. Симакин Г А. и др.—Ж. анал. хим., 1974, т. 29, № 8, с. 1585—1589.
Рений
Амперометрическое определение рения встречает большие трудности, связанные с тем, что все соли, образуемые перренат-ионом, хорошо растворимы (даже наименее растворимая таллиевая соль — перренат таллия — растворяется 1,6 г на 1л воды при 20 °C) и что окислительно-восстановительные потенциалы всех систем, образуемых рением (VII), равно как рением в других валентных состояниях, имеют относительно низкие значения (не выше +0,5 В) и поэтому соединения рения не могут быть восстановлены обычными восстановителями, применяемыми в визуальном, титриметриче-ском анализе [соли железа (II), аскорбиновая кислота и т. д.].
235
Известна серия работ, рекомендующих титровать рений(VII) очень сильными восстановителями — хромом(II) [1], титаном(III) [2], ванадием(II) [3], а также железом (II) в присутствии фосфорной и серной кислот, настолько понижающих реальный потенциал системы Fe3+/Fe2+ вследствие комплексообразования, что восстановление рения (VII) становится возможным [4].
Предложен новый реагент для титрования рения (VII) в водноорганической среде — ферроцен [5], восстанавливающий рений (VII) до рения(V). Изучение кинетики реакции ферроцена с p3HHeM(VII), молибденом (VI) и вольфрамом (VI) позволило разработать методику определения рения(VII) в присутствии молибдена^!) и вольфрама (VI) [6] на фоне смеси ацетонитрила с серной кислотой. О возможности определения рения в сумме с молибденом и последующем определении только молибдена упомянуто в разделе «Молибден».
На восстановлении рения(VII) основан метод определения его а- или р-нафтиламином: рений (VII) сначала восстанавливается реактивом до рения (V), который образует с нафтиламином комплексное соединение [7].
ЛИТЕРАТУРА
1. Галлай 3. А., Рубинская Т. Я., Федорова А. В. — Ж. аиал. хим., 1966, т. 21, № 5, с. 584—589.
2. Галлай 3. А., Рубинская Т. Я-—Ж- аналит. хим., 1966, т. 21, №8, с. 961—964.
3. Рубинская Т. Я-, Жусид Л. Б., Галлай 3. А.—Ж. анал. хим., 1967, т. 22, № 10, с. 1519—1522.
4. Галлай 3. А., Рубинская Т. Я- — Ж. анал. хим., 1967, т. 22, № 9, с. 1378— 1381.
5. Соломатин В. Т., Артемова Т. И.—Ж. анал. хим., 1974, т. 29, № 3, с. 489— 492.
6. Ржавичев С. П., Каплан Б. Я., Соломатин В. Т. — Зав. лаб., 1978, т. 44, №1, с. 4—7.
7. Тараян В. М., Вартанян С. В., Шапошникова Г. И. — ДАН АрмССР, 1973, т. 56, 2, с. 92—95.
Родий
Сведений об амперометрическом определении родия пока еще очень мало. В щелочной среде (1—5 М NaOH) родий (III) в форме Rh(OH)6‘ окисляется гипобромитом натрия до родия (IV). Титровать можно как по току окисления родия (III) при +0,4 В (Нас. КЭ), так и при +0,1 В по току восстановления образующегося родия (IV). Форма кривой титрования, соответственно, «а», или «г». Нижний предел определения Ы0-4 г-ион/л. Из ионов платиновых металлов мешают иридий (III) и осмий (VI) [1].
Другой метод определения родия (III) заключается в окислении его избытком церия (IV) и обратном титровании церия (IV) солью Мора или тиооксином с одним или двумя платиновыми электродами при +0,8—1,0 В (Нас. КЭ) или при Д<р—0,05—0,25В
236
соответственно. Восстановление церия(IV) родием (III) происходит только при кипячении на фоне 4 М серной кислоты в течение Ю—15 мин. Нижний предел определения родия — 10 мкг в 20— 50 мл. Мешают следы иридия в любой степени окисления [2].
Третий метод [3] основан на осаждении родия (III) солями бария в щелочном растворе. Состав осадка отвечает отношению Rh : Ва=2 : 3, формула его Ва3[Ш1(ОН)б]2- Титруют на платиновом электроде при <р = +0,4 В (Нас. КЭ), на фоне 0,5—2 М NaOH, по току окисления родия (III). Определяемое количество родия 0,2—4 мг. Титруют раствором хлорида или нитрата бария.
ЛИТЕРАТУРА
1 Бардин М. Б„ Хоанг Тхо Тин — Ж., анал. хим., 1975, т. 30, № 8, с. 1771— ‘ 1774.
2 Величко В. В., Магульский Б. И., Усатенко Ю. И. — Зав. лаб., 1976, т. 42, № 8, с. 923—924.
3 . Бардин М. Б., Хоанг Тхо Тин — Координ. химия, 1977, т. 3, № 2, с. 247—251.
Ртуть
Для определения ртути наиболее простым, легкодоступным и удобным реактивом является иодид калия, образующий с ртутью(II) очень мало растворимый осадок Hgl2. При большом избытке иодид-ионов этот осадок, как известно, растворяется с образованием комплексного соединения [HgH]-2. Растворимость иодида ртути в воде очень мала (ПР=4-10~29), что позволяет определять ртуть(II) при весьма малых содержаниях ее в титруемом растворе — порядка 10-6 моль/л. При столь малых содержаниях ртути во время титрования незаметно (визуально) образование осадка или появление опалесценции. Осадок становится заметным только тогда, когда содержание ртути в, 25 мл раствора достигает примерно 0,2 мг.
Если правильно выбрать потенциал электрода при титровании, как указано в гл. V, то можно совершенно исключить влияние разряда взвеси осадка иодида ртути на платиновом электроде, электродные реакции других ионов и влияние комплексообразования избытка иодида с образовавшимся осадком иодида ртути [1—5].
Мешает определению ртути, вернее осаждается иодидом вместе с ртутью (II), серебро, поскольку растворимость иодида серебра имеет порядок, близкий к растворимости иодида ртути (см. «Серебро»), Поэтому в присутствии серебра рекомендуется предварительно отделять ртуть отгонкой или разлагать пробу царской водкой в расчете на то, что хлорид серебра, относящийся к числу малорастворимых соединений, не растворится при последующей обработке пробы серной кислотой.
Применяя кислотное разложение проб, необходимо иметь в виду возможные потери ртути за счет улетучивания ее в виде хло-
237
g Таблица 35. Методы определения ртути
со _______________________________________
Определяемый ион Титрант Определяемый минимум Условия определения Примечания Литература
Hgii KI 1 мкг/мл Электрод Pt, ср = + 0,8 В (МИЭ), фон — 0,5 н. H2SO4 См. текст 1—6
ЭДТА Электрод Та, ср = + 1,2 В (Нас, КЭ), фон — 0,1—0,5 н. H2SO4 или HNO3 Можно последовательно титровать Hg11 и Ag1 9
Аскорбиновая кислота — 2 Pt электрода, Дср = 0,14 В, фон—6н. СН3СООН Последовательно титруют Au111 и Hg11 10
Тиокарбамид • Электрод Hg капающий, ср = —0,3 В (Нас. КЭ), по току восстановления Hg11, или Pt электрод, ср= + 1,0 В (Нас. КЭ), по току окисления титранта, фон — H2SO4 ИЛИ HNO3 Можно последовательно титровать Hg11 и Ag1 и, 12—14
Тиооксин и его производные Электрод Pt, ср= + 1,0 В (Нас. КЭ), фон — 2—6 н. H2SO4 Отношение Hgn:THooKCHH= 1:2, комплексное соединение, окисляющееся на Pt электроде, возможно раздельное определение Hg11 и Hg1 18—22
Тиосалнциловая кислота 0,4 мг/ 30 мл Электрод Pt, ср== + 1,0 В (Нас. КЭ), фон — 4—6 н. H2SC>4 Применен для анализа руд 23
Тиоацетамид Ю-5_1о-з моль/л Электрод Hg капающий, q>=—0,7 В (Нас. КЭ), фон — NaOH+4-амидо-сульфобензоат натрия 24
Ксантогенат Электрод Pt, ср=—0,2 В (Нас. КЭ), фон — аммиачно-тартратный, рН=9, ток восстановления Hg11 и окисления титранта В присутствии РЬ11 сначала осаждается Hg11, затем РЬ11 дифференцированное титрование 25—26
1 Тиосемикарбазид 0,005 моль/л Электрод Pt, ср=1,0 В (Нас. КЭ), фон — 1 М HNO3 или H2SO4 Отношение Hg : титрант =1:2 можно титровать также Ag на фоне 1 М HNO3 28
Диэтнлднтио-карбаминат Электрод Pt, <р=+0,8—0,9 В (Нас. КЭ), фон—3—6 М HNO3 или H2SO4+ + 2 мл 0,1 М C11SO4 (индикатор) Мешает Ag 29
Пиперазин и гндроксиэтилпи-перазиндитиокар-баминат 0,2—4 мг/50 мл Электрод Pt, ср= +0,8 В (Нас. КЭ), фон — рН=2,8—9,1 Мешает Ag, его устраняют фторидом, возможно дифференцированное титрование Hg11 и Си11 при рНягб ’ 30
Бензолсульфо-нилтиобенз амидин 0,03 мг/25 мл Электрод графитовый, ср=+0,9— —1,0 В (Нас. КЭ), фон — ацетатный буферный раствор, pH » 4 Осадок состава HgRs, метод применен для анализа ртутных руд 31
2-меркаптобен-зоксазол 0,4—3,5 мг Электрод Pt, ср=О В (Нас. КЭ), фон — ацетатный буферный раствор. pH=5—4 Образуются 2 комплексных соединения с соотношением Hg:pea-гент= 1:1 и 1:2 32
Дитизон 5—50 мкг Электрод Pt, ср=+0,2 В (Нас. КЭ), прямое экстракционное титрование раствором дитизона в CHCI3 или СсНв Метод довольно сложен, можно определять, кроме Hg1 и Hg11, также Ag и Cd 33
Hgi КЮз Ю-5— 10-4 моль/л 2 Pt электрода, Дгр= 100 мВ, фон— 1 М НС1 для Hg1, осадок Hg2CI2 растворяется во время титрования Титруют так же Sb111 и ТР, для Sb1» — фон 3 М НС1, для Т11—5 М НС1, Дф= 150мВ 8
HgH и Hgi Тиопиперидон Миллиграммы в титруемом объеме Электрод графитовый, ср=О В (МИЭ), по току Hg или +1,5 В по току окисления реактива, фон—рН = = 1,0 Отношение Hgn:R=l:2, белый осадок разлагается с выделением HgS, H2S и пиперидона 27
Унитиол Электрод Pt, <р= + 0,8 В (Нас. КЭ), фон — 1 М II2SO4 См текст 15—I7
рида. Однако, учитывая, что эта возгонка становится ощутимой лишь при температуре выше 160 °C, можно вести разложение пробы как описано ниже, поскольку потери ртути в этих условиях, как было установлено специальными опытами [1], не выходят за пределы расхождения, допускаемых обычными нормами. Метод был успешно применен для определения ртути в рудах, содержащих от 2,2 до 0,08% ртути.
Ход анализа. Навеску руды 0,3—1 г помещают в коническую колбу емкостью 100 мл, смачивают водой, приливают 5 мл царской водки и разлагают при умеренном нагревании (температура песочной бани около 100 =С). По окончании разложения жидкость охлаждают, приливают около 10 мл серной кислоты плотностью 1,84 г/см3 и выпаривают до 2—3 мл, избегая перегрева выше 300 °C. Не следует затягивать эту операцию дольше, чем нужно. К охлажденному раствору осторожно добавляют 5 мл воды и выпаривают до появления белых паров. Эта операция необходима для удаления нитрозилсерной кислоты, мешающей последующему титрованию иодидом калия. После охлаждения раствора его разбавляют 10 мл воды и добавляют немного сухого карбамида для удаления остатков окислов азота. Затем раствор вместе с осадком переносят в стакан для титрования, доводят водой до 20—25 мл и титруют раствором иодида калия при + 0,85 В (МИЭ) или +0,6 В (Нас. КЭ). При содержании ртути в рудах 0,1% и меньше титрование следует проводить 0,01 М раствором иодида калия, а при большем содержании—0,1 М.
Можно титровать и с двумя индикаторными электродами [6]. Недавно иодидный метод был вновь предложен [7] для определения ртути(II), палладия (II), серебра. Ртуть(I) титруют йодатом калия [8], этот же реагент позволяет титровать сурьму (III) и таллий (I).
Число же органических реактивов, примененных для определения ртути, очень велико. Ограничимся упоминанием лишь наиболее интересных из тех, которые предложены в последние годы.
В основном применяют серусодержащие реагенты, из других органических реактивоз упоминаются только два — ЭДТА [9] (см. также «Кальций») и аскорбиновая кислота, позволяющая последовательно титровать ртуть(II) и золото(III) [10].
Из большого числа серусодержащих соединений различного типа в первую очередь следует назвать тиокарбамид [11-—14] и унитиол [15-17], дающий с ртутью(II) сначала белый осадок, который постепенно растворяется с образованием комплексного соединения, в котором соотношение ртути (II) и унитиола равно 1 : 1. Ртуть(I) восстанавливается унитиолом до металла [16].
Обе валентные формы ртути можно титровать унитиолом не только порознь, но и при совместном присутствии их в растворе при потенциале -[-0,8 В (Нас. КЭ) на фоне примерно 1 М серной кислоты (электрод платиновый). Сначала, в момент замыкания цепи, наблюдается ток, значение которого зависит от количества ртути (I), поскольку этот ток обусловливается анодным окислением ртути(I) до ртути(II). При титровании в первую очередь образуется унитиолат ртути (II), поэтому ток остается постоянным (или незначительно повышается по причинам, подробно рассмотренным в работе [16]). Когда реакция унитиола с ртутью(II)
240
закончится, начинается взаимодействие унитиола с ртутью(I) — появляется муть от выпадающей в осадок металлической ртути, ток понижается вплоть до второй [16] конечной точки, которая обозначается так же как и первая конечная точка, очень резко. Молярное отношение ртути (I) и унитиола равно 2: 1.
Ход анализа. Н (веску руды 0,2—0,5 г помещают в коническую колбу емкостью 250 мл, смачивают водой, приливают 10 мл серной кислоты плотностью 1 84 г/см3, слабо нагревают 5—10 мин, затем добавляют 1—1,5 мл азотной кислоты плотностью 1,40 г/см3 и по окончании бурной реакции выпаривают осторожно, как указано выше (при описании иодидного метода определения ртути в рудах), до появления белого дыма. Для окончательного удаления окислов азота кипятят раствор после разбавления его 20—30 мл воды. Охлаждают, переводят раствор с осадком в мерную колбу емкостью 100 мл, доводят водой до метки. К аликвотной части 25 мл добавляют 2—3 капли насыщенного раствора пирофосфата натрия или очень немного сухого фторида аммония для связывания железа(III) и титруют 0,01 или 0,001 М раствором унитиола. Нижний предел определения — 2 мкг/мл. Цинк и свинец не мешают определению, медь не мешает, если содержание ее не превышает содержание ртути более чем в 500 раз.
Много внимания уделено тиооксину и его производным [18— 22]. Применяют также тиосалициловую кислоту [23], тиоацетат [24], различные ксантогенаты [25, 26], тиопиперидон [27], тиосемикарбазид [28], диэтилдитиокарбаминат и его аналоги [29, 30], бензосульфанилтиобензамидин [31], 2-меркаптобензоксазол [32] и 3,5-диэтил-2,6-димеркапто-1,4-тиопирон, уже упоминавшийся в разделе «Висмут».
В неводных средах ртуть титруют раствором дитизона в хлороформе или бензоле [33], бензольным раствором диэтилдитиокар-бамината свинца на фоне ледяной уксусной кислоты (см. «Висмут») или диэтилдитиокарбаминатом натрия в смешанных водноорганических растворах (см. «Висмут»), При титровании ртути(II) рекомендуется в качестве органического компонента хлороформ или четыреххлористый углерод, в качестве высаливателя — нитрат натрия.
ЛИТЕРАТУРА
1. Захаров В. А., Сонгина О. А., Терземан Л. Н. — Зав. лаб., 1960, т. 26, с. 787.
2. Сонгина О. А., Войлошникова А. П., Козловский М. Т. — Изв. АН КазССР. Сер. хим., 1949, вып 3, с 81.
3. Сонгина О. А.—Труды комиссии по аналитической химии АН СССР, 1952, IV (VII), с. 116.
4. Сонгина О. А., Захаров В. А. —За . лаб 1952, т. 28, № 9, с. 9СЗ
5 Захаров В. А., Сонгина О. А. — Изв. АН КазССР. Сер хим., 1961, вып. 2 (20), с. 53.
6. Kies S.~Z. anal. Chem., 1963, Bd. 195, S. 249; 1964, Bd. 194, S. 104.
7. Siska A. —Hung. Sci. Instrum., 1975, № 33, p. 13—17; РЖХим., 1976, 10Г34.
8. Freddi R„ Botnbi G. G., Fiorani M. — Z. anal. Chem., 1966, Bd. 222, N 4, p. 369—381.
9. Хадеев В. А., Базарбаев A. 7.—Узб. хнм. ж., 1960, № 5, с. 38—41.
^ingh D., Bhatnagar V. — Indian J. Chem., 1967, v. 5, № 4, p. 164—165;
РЖХим., 1967, 24Г57.
11 Торопова В. Ф. — Ж. неорган. хим., 1956, т. 1, с. 930.
16—1694 941
12. Шумская А. И., Усатенко Ю. И. — Труды Днепропетровского химико-технологического ин-та, 1959, вып. 12, ч. 1, с. 11.
13. Усатенко Ю. И., Шумская А. И. — Зав. лаб., 1960, т. 26, № 2, с. 149.
14. Усатенко К>. И., Шумская А. И. — Труды Днепропетровского химнко-техно-логического ин-та, 1961, вып. 12, ч. 2, с. 165.
15. Усатенко Ю. И., Климкович Е. А., Лошкарев Ю. М.—Укр. хим. ж., 1961, т. 27, с. 823.
16. Сонгина О. А., Оспанов X. К, Рождественская 3. Б.—Ж- анал. хим., 1965, т. 20, № 1, с. 55—58.
17. Оспанов X. К-, Рождественская 3. Б.—Ж- анал. хим., 1965, т. 20, № 3, с. 258—261.
18. Усатенко Ю. И., Супрунович В. И., Куликовская Ж. Б.—Ж- анал. хим., 1970, т. 25, № 10. с. 1890—1893.
19. Супрунович В. И., Куликовская Ж- Б., Усатенко Ю. И. — В кн.: Химическая технология. Респ. межвед. научно-техн. сб. 1971, вып. 17, с. 191—197.
20. Куликовская Ж- Б., Супрунович В. И., Усатенко Ю. И. — В кн.: Химическая технология. Респ. межвед. научно-техн. сб. 1974, вып. 36, с. 64—65.
21. Берзиня В. К, Янсон Э. Ю., Седола В. А. — Учен. зап. Латвийского ун-та, 1967, с. 88. \
22. Берзиня В. К, Янсон Э. Ю., Тейбе А. Я- — Изв. АН ЛатвСС!5; Сер. хим., 1971, № 5, с. 560—562.
23. Крюкова Л. В., Усатенко Ю. И. — Зав. лаб., 1975, т. 41, № 4, с. 387—389.
24. Facsko G., Nemes М.— Rev. roum. chim., 1974, v. 19, № 1, p. 141—144-РЖХим., 1974, 16Г99.
25. Усатенко Ю. И., Тулюпа Ф. М. — Укр. хим. ж., 1963, т. 29, с. 747.
26. Deshmukh G. S., Saraswathi К- — Indian J. Chem., 1965, v. 3, № 11, p. 489— 490; РЖХим., 1966, 13Г59.
27. Тараян В. M., Саркисян А. А., Мушегян А. В.—Арм. хим. ж., 1971, т. 24, № 9, с. 769—776.
28. Норенко Г. М. и др. — В кн.: Вопросы химии и химической технологии. Респ. межвед. тематич. научно-техн. сб. 1974, вып. 34, с. 39—43.
29. Усатенко Ю. И., Тулюпа Ф. М., Ткачева Л. М.— В кн.: Химическая технология. Респ. межвед. научно-техн. сб. 1971, вып. 18, с. 150—154.
30. Тулюпа Ф. М., Ткачева Л. М., Усатенко Ю. И. — В кн.: Химическая технология. Респ. межвед. научно-техн. сб. 1967, вып. 7, с. 87—93.
31. Усатенко Ю. И., Ситало Л. И.—Ж. анал. хим., 1974, т. 29, № 1, с. 35—39. 32. Вега В. С., Chakrabartty М. М., Bag S. Р., Mallik К. Б.—Taianta, 1966, v. 13, № 3, р. 528—531.
33. Мухамеджанова Д., Талипов Ш. Т., Хадеев В. А. — Узб. хим. ж., 1970, №4, с. 12—14.
Рутений
Известен амперометрический метод определения рутения(IV), основанный на его титровании в виде «черной соли» KzRuCk растворами гидрохинона или аскорбиновой кислоты, восстанавливающими рутений(IV) до рутения(II). Титруют при +0,5 В (Нас. КЭ) по току восстановления рутения (IV) на платиновом вращающемся электроде на фоне соляной кислоты (1:1). В присутствии золота (III), которое также восстанавливается на электроде при указанном потенциале, ток повышается, но это не мешает определению конечной точки, если количество золота не слишком велико — не больше 15—20-кратного по отношению к рутению. Если золота больше, то оно одновременно с рутением восстанавливается гидрохиноном. При относительно малых количествах золота эта реакция цезаметна, так как она протекает значительно медленнее,
242
чем восстановление рутения (IV). Определению рутения этим методом мешают железо(II) (анодный ток окисления, компенсирующий катодный ток рутения) и иридий (IV). Метод применим для определения 0,02—2,0 мг рутения.
Разработан метод определения рутения(IV) по его каталитическому действию на реакцию окисления иодида калия перекисью водорода [3, 4]. Амперометрического титрования как такового нет, измеряют ток восстановления выделяющегося иода (так же, как при каталитическом определении осмия, см. выше). Потенциал платинового индикаторного электрода +0,3 В (Нас. КЭ), фон — ацетатный буферный раствор, рН = 5,4. Определяемые концентрации рутения — 5-10~4 — 4-10—2 моль/л. Минимальная концентрация рутения, определяемая этим методом, на два порядка ниже, чем при спектрофотометрической регистрации.
Предложено также использовать другую каталитическую реакцию— окисление ферроина перйодатом калия [5]. Каталитический эффект зависит от той формы, в виде которой рутений(IV) находится в растворе. В статье [5] приведены результаты исследования этой реакции и даны соответствующие методики.
ЛИТЕРАТУРА
1. Пшеницын Н. К-, Езерская Н. А. — Ж- анал. хим., 1961, т. 16, № 2, с. 196.
2. Пшеницын Н. К-, Езерская Н. А. — В кн.: Теория и практика полярографического анализа. Кишинев, Штиинца, 1962, с. 145.
3. Алексеева И. И. и др.—Ж- неорган. хим., 1971, т. 16, № 6, с. 1654—1658. 4. Алексеева И. И. и др.—Ж. анал. хим., 1973, т. 28, № 7, с. 1368—1371.
5. Алексеева И. И. и др. — Ж- анал. хим., 1974, т. 29, № 9, с. 1859—1861.
Свинец
В 1939 г. была опубликована [1] одна из первых работ по амперометрическому («полярометрическому», как тогда его называли) титрованию вообще, а именно амперометрическое титрование свинца (II) раствором бихромата калия. С тех пор этот метод неоднократно изучали и совершенствовали [2]. Ход определения аналогичен описанному выше в разделе «Барий». /Титрование можно проводить с ртутным капающим электродом без наложения внешнего напряжения;'в этом случае кривая будет иметь форму б, так как свинец(П) при потенциале Нас. КЭ не восстанавливается, а бихромат восстанавливается. Можно титровать и при —1,0 В, в таком случае кривая имеет форму в, так как восстанавливаться будут и бихромат, и свинец. В качестве фона можно использовать растворы нитрата калия или аммония, ацетатный раствор или даже слабоазотнокислые растворы (не выше 0,3 Л4). В последнем случае необходимо помнить, что осадок хромата свинца растворим в минеральных кислотах.
При титровании менее 200 мг/л свинца надо иметь в виду следующее обстоятельство, на которое обычно не обращают внима-,6* 243
ния: если для растворения осадка сульфата свинца по ходу анализа применяли ацетат аммония, то в случае малых количеств свинца при последующем титровании бихроматом осадок может не выпасть. Это связано с тем, что в большом избытке ацетата хромат свинца растворяется, как это было подтверждено рядом опытов с разными количествами свинца и ацетата. Поэтому практически необходимо следить за тем, чтобы концентрация ацетата аммония в титруемом растворе не превышала 0,5 М (т. е. около 4%).
Свинец(II) образует мало растворимый осадок с ферроцианидом калия [3], что особенно удобно в тех случаях, когда наряду со свинцом в пробе присутствует барий(П), так как он не осаждается ферроцианидом. Сурьма(III) также не образует осадков с ферроцианидом и потому не мешает определению свинца. Вис-мут(Ш), железо(П), цинк, кадмий, медь(П) будут титроваться вместе со свинцом, поэтому при анализе полиметаллических руд необходимо прибегать к обычному выделению свинца в виде сульфата. Ферроцианидный метод позволяет определять свинец при разбавлениях до 100 мг/л (т. е. до 2 мг в объеме 20 мл).
Предложены для определения свинца и другие неорганические реагенты, например раствор пентасульфида фосфора в щелочи [4], позволяющий определять свинец в присутствии ртути и серебра, трифосфорная кислота (см. «Кадмий»), теллурат . натрия (см. «Медь»).
Однако больше всего рекомендовано различных органических реактивов [2], ЭДТА и ряд серусодержащих соединений [5]. Некоторые из них уже упоминались при описании определения других элементов. Так, ЭДТА позволяет определять свинец в присутствии висмута (см. «Висмут»), индия(Ш) («Индий»), Диаминоциклогексанонтетр ауксусная кислота (ЦДТА) предложена для совместного определения свинца и таллия (I) [6], а также свинца и кадмия (см. «Кадмий»), Рекомендованы два производных ди-меркатотиопирона [7], диэтилдитиокарбаминат [8], пиперидинди-тиокарбаминат [9]; другие карбаминаты применены для совместного определения свинца и рути(П) [10], свинца и висмута (см. «Висмут»), свинца и меди (см. «Медь»). Ксантогенаты, в частности этилксантогенат [11], рекомендованы для определения свинца в присутствии ртути (см. «Ртуть-»); унитиол — для свинца и висмута (см. «Висмут»), В последние годы предложен новый реактив — дитиотриоксиметиламинометанат калия '[12], позволяющий определять свинец в присутствии ртути и серебра.
ЛИТЕРАТУРА
1. Neuberger А.—7. anal. Chem., 1939, Bel. 116, S. 1.
2. Сонгина О. А. Амперометрическое титрование. М., Химия, 1967.
3. Сонгина О. А., Войлошникова А. П., Козловский М. Т. — Зав. лаб., 1951, т. 17, № 1, с. 3—6.
4. Chaudhuri И., Chatterjee S. S., Mahanti Н. S. — J. Indian Chem. Soc., 1974, v. 51, № 9, p. 836—837; РЖХим., 1975, 12Г97.
244
5. Сонгина О. А., Бессарабова И. М. — Зав. лаб., 1973, т. 39, № 6, с. 641—655. 6* Gaur J. N., Jain D. S. — Acta chim. Acad, scient. Hung., 1967, v. 51, № 2, ' p. 171—174.
7 Аришкевич A. M., Алыбина А. И. — Зав. лаб., 1967, т. 33, № 11, с. 1367— 1369.
8 Усатенко Ю. И., Тулюпа Ф. М.—Ж. неорган. хим., 1959, т. 4, № 12, с. 2495—2497.
9 Черноморченко Л. И., Гржегоржевский А. С., Бутенко Г. А. — Зав. лаб., 1976, т. 42, № 4, с. 401—402.
10. Stricks XV., Chakrawarti S. — Anal. Chem., 1962, v. 34, p. 508—510.
11. Усатенко Ю. И., Тулюпа Ф. М. — Укр. хим. ж., 1963, т. 29, с. 747—750.
12 Deshmukh G. S„ Chaudhuri И., Mahanti Н. S. — Bull. Chem. Soc. Jap., 1975, v. 48, № 3, p. 1089—1090; РЖХим., 1975, 20Г81.
Селен
Для определения селена известно несколько амперометрических методов, основанных на осаждении иона 5еОз" различными реагентами и на способности селена(IV) вступать в реакции окисления— восстановления. При осадительном титровании применяют в качестве титрантов нитрат ртути(1) [1], нитрат серебра [2], ацетат свинца(II) [3].ДЭти методы малочувствительны, определению селена (IV) мешают многие анионы.
При определениях по методу окисления — восстановления широко используют реакцию селена (IV) с иодид-ионом [4—9]. Возможно как прямое титрование селена(IV) раствором иодида калия1 [4, 5], так и титрование иода, выделившегося в результате взаимодействия селена (IV) с иодидом, тем или иным восстановителем: гидразином [6, 7], тиосульфатом [7], аскорбиновой кислотой [8, 9]. Йодометрический вариант положен в основу метода раздельного определения селена(IV) и теллура(IV) при совместном их присутствии [7]. В этом методе использована различная реакционная способность иода по отношению к теллуру и селену в гидрокарбонатной среде, -в которой теллур быстро и количественно окисляется иодом, а селен с иодом практически не реагирует. Поэтому титрованием выделившегося иода в кислой среде определяют сумму селена и теллура, а в гидрокарбонатной среде — только селен.
_ Ход анализа [7]. Навеску пробы 0,2-—1 г разлагают концентрированной азотной кислотой при слабом нагревании и упаривают раствор до 5—8 мл. Если в анализируемом объекте содержится медь(II), приливают 40—50 мл воды, цементируют на свинцовую пластинку медь, добавляют 15—20 мл H2SO4 (1:1) и упаривают до выделения паров серного ангидрида. Если же медь отсутствует, то серную кислоту добавляют сразу после упаривания азотнокислого раствора. Пробу охлаждают, приливают 30—40 мл воды, переносят в мерную колбу емкостью 100 мл и доводят до метки водой.
Отбирают две аликвотные части по 5—10 мл, приливают 4 М раствор КС1 до объема 20—25 мл, вводят 0,1—0,2 г твердого фторида аммония для связывания железа(III) и 0,5—1 г твердого KI. Накрывают раствор часовым стеклом и выжидают 10 мин до полного выделения селена, теллура и иода. В одной аликвотной части титруют выделившийся иод раствором тиосульфата, а в другом после нейтрализации твердым гидрокарбонатом натрия титруют иод раствором гидразина. При титровании иода тиосульфатом оттитровывается сумма
245
246
Таблица 36. Методы определения селена
Определяемый иои Титрант Определяемый минимум Условия определения Примечания Литература
Seiv Hg2(NO3)2 10 мкг/мл Электрод Pt, <р= +0,1 В (Нас. КЭ), фон — 0,005 М H2SC>4 Образуется осадбк Hg2SeO3 1
AgNO3 20 мкг/мл Электрод Pt, <р=0 В (Нас. КЭ), фон — KNO3+NaOH, рН=10 Образуется осадок Ag2SeO3 2
Pb(CH3COO)2 14 мг Электрод Hg капающий, ср=0,1 В (Нас. КЭ), фон —KNO3 (pH=3—6) Образуется осадок PbSeO3 3
KI 1 мкг/мл Электрод Pt, <р=+0,8 В (Нас. КЭ), фон — 6-—8 М НС1 Восстановление Selv до элементного, не мешает TeIV 4,5
2 мкг/мл 2 Pt электрода или пара Pt—графит, Дср = 0,1 В, фон -— 6 М НС1 Восстановление Selv до элементного, не мешает TeIV 5
1 мкг/мл Электрод Pt, <р=+0,1 В (Нас. КЭ) SeIV восстанавливают KI до Se° в солянокислой среде и выделившийся иод титруют в гидрокарбо-натной среде гидразином; не мешают TeIV, Asv, Sbv, Pb, Zn, Ag, Tl, In; применено для анализа продуктов цветной металлургии 6,7
Электрод Pt, ср=0,04-0,2 В (Нас. КЭ), фон —0,05 М КС1+НС1 до рН=1 К SeIV добавляют пилуторократ-ный избыток KI и выделившийся иод титруют аскорбиновой кислотой, не мешает Те IV 8,9
Гидразин 1 мкг/мл Электрод Pt, ср = +0,2 В (МИЭ) SeIV восстанавливают в солянокислой среде гидразином и избыток гидразина титруют в гидро-карбонатной среде иодом, не ме-шаютТе1У, Asv, Sbv, Pb, Cd, Zn, Tl, In 6
Аскорбиновая кислота
Тиокарбамид
10 мкг/мл
2 мкг/мл
Электрод Hg капающий, <р = —0,05 В (Нас. КЭ), фон —0,01—0,1 н. НС1 или H2SO4 (рН=1—2)
Электрод Pt, <р = + 0,8 В (Нас. КЭ), фон — 6—8 М НС1
Димеркаптотио-пироньт
1 мкг/мл
Электрод графитовый, <р=+0,4— 0,8 В (Нас. КЭ), фон —6—8 М НС1 илн H2SO4
Дитиокарбами-наты 1 мкг/мл
Дитпобиурет
Ксантогенат
Дитиотриоксиметил аминометан
Унитпол 0,4 мкг/мл
Тиооксин 1 мкг/мл
Электрод Pt, <р= + 0,7—0,8 В (Нас. КЭ), фон — рН=3 до 1 М НС1
Электрод Pt, ф= +0,1 В (НВЭ), фон — ацетатный буферный раствор, pH=4—5
Электрод Hg капающий, ср —0,5 В (Нас. КЭ), фон — 1—6 M НС1
Электрод Hg капающий, ср=—0,2 В (Нас. КЭ), фон — смесь 0,1 М KNO3 и 0,1 М тартрат аммония рН=7,5
Электрод графитовый, ср=+0,6 В (Нас. КЭ), фон — 0,1—4 М НС1 или H2SO4
Электрод Pt, <р= + 1,0 В (Нас. КЭ), фон — 1—4 М H2SO4
Тиосульфат
Электрод Pt, ср = —0,2 В (Нас. КЭ), фон 0,1 М НС1
Selv и SeVI восстанавливают при 60 ° С до Se°, не мешают TeIV, Си, Pb, Fe
SeIV восстанавливают при 60 °C тиокарбампдом и избыток тиокарбамида титруют бихроматом калия; применено для анализа серы
В зависимости от производного дпмеркаптотиопирона и природы фона образуются осадки при соотношении Selv:R=l:2; 1:3; 1:4; применено для анализа сплавов, руд, шламов
Образуется комплекс с отношением SeIV:R = l:4
Соотношение lSeIV:R=l:4
10
11
12—16
17—20
21
Образуется осадок с отношением 22
Se,v R=l:4, мешает Те IV
Образуется осадок с отношением 23
Selv:R = l:4, мешает TeIV
SeIV восстанавливается до элементного, не мешают Telv, Vv, Asv, Ag и др.
Selv и TeTV титруются на фоне 1 М H2SO4 дифференцированно, SeIV восстанавливается до элементного, применено для анализа пылей, концентратов, стали
Образуется комплекс, в котором селен двухвалентен; берут избыток тиосульфата, который оттитровы-вают иодом
24
25, 26
27
Продолжение
Определяемый ион Титрант Определяемый минимум Условия определения 1 Примечания^ I Литература
Seiv, РЬ(СН3СОО)4 КМпО4 NaOBr KI 1 мкг/мл Электрод Hg капающий, <р=—0,3 В (Нас. КЭ), фон — 1 М НС1 Электрод Pt, <р= + 0,3 В (Нас. КЭ), фон —8—10 М H2SO4 Электрод Pt, ф=+0,3 В (Нас. КЭ), фон — 0,6 М NаНСО3 Электрод Pt, <р= +0,1 В (Нас. КЭ) SeIV окисляется до SeVI SIV окисляется до SeVI, мешает TeIV SeIV окисляется до SeVI К SIV и TeIV добавляют KI и 28 29 30 7
TelV З-метил-5-фе- 1 мкг/мл Электрод Pt, <р=0,0—0,2 В (Нас. КЭ), фон —0,05 М КС1+НС1, рН=1 Электрод графитовый, ф= + 0,7 В выделившийся иод титруют: в одной аликвотной части в гидрокар-бонатной среде гидразином, определяя селен, в другой аликвотной части в кислой среде тиосульфатом определяют сумму селена и теллура; применено для анализа кеков, пыли, селеновых концентратов К Selv и TeIV добавляют KI до 0,05 М и титруют выделившийся 12 аскорбиновой кислотой, определяя Se, затем после к.т.т. добавляют KI до 0,12 М и титруют комплекс [Те!6]2- аскорбиновой кислотой; применено для анализа стали Se1’ и Telv титруют дифферен- 8,9 15, 16
нил-2,6-димер-капто-1 ",4-тиопи-рон Диэтилдитио-карбаминат 1 мкг/мл (Нас. КЭ), фон — 8—9 М НС1 Электрод Pt, ф = 0,7—0,8 В (Нас. КЭ) цированно, применено для анализа полупроводников В одной аликвотной части при рН=5—6 титруют Telv, в другой— на фоне 0,1 М НС1 титруют сумму
селена и теллура, а при титровании гидразином — только иод, отвечающий реакции с селеном. Титруют с платиновым индикаторным электродом при потенциале 4-0,1 В (Нас. КЭ). Метод применен для анализа продуктов свинцово-цинкоього производства.
Из других восстановителей используют гидразин [6] и аскорбиновую кислоту [10], которые позволяют определять селен (IV) в присутствии теллур а (IV).
Для определения селена (IV) широкое применение нашли различные серусодержащие вещества [11—27], которые в зависимости от природы реагента и кислотности фона могут или восстанавливать селен(IV), или образовывать осадки и комплексные соединения. Некоторые серусодержащие реагенты (например, 3-пири-дил-2,6-димеркапто-1,4-тиопирон, диэтилдитиокарбаминат, унитиол, тиооксин) обладают различной реакционной способностью по отношению к селену (IV) и теллуру (IV), что позволяет осуществить как титрование селена в присутствии теллура [12, 14, 20, 24], так и раздельное определение этих ионов при их совместном присутствии >[15—18, 25, 26]. Для раздельного определения селена (IV) и теллура(IV) удобно дифференцированное титрование их тиооксином [25].
Ход анализа. Навеску 0,2—1 г разлагают 15—20 мл концентрированной азотной кислоты, упаривают до ’/з объема, приливают 10—15 мл воды, цементируют на свинцовую пластину медь, ртуть, серебро и другие электроположительные металлы (медь(И), ртуть(П) и сереброД) мешают определению селена и теллура). Затем добавляют 15—20 мл H2SO4 (1 : 1), упаривают до выделения белых паров, охлаждают, переносят в мерную колбу емкостью 100 мл и доводят до метки водой. Отбирают аликвотную часть раствора 5—10 мл, приливают 1 М серную кислоту до объема 20—25 мл и титруют 0,01 М раствором тиооксина при потенциале платинового электрода 4-1,0 В (Нас. КЭ). В начале тиооксин реагирует с теллуром (IV) по реакции:
TeIV+ eCgHeNSH = [TefCJLNS) 4]2+ + C8H6NS — SNH6C9
а затем с селеном (IV), восстанавливая его до элементного состояния. Комплекс [Te(CsH6NS)]2+ электрохимически активен, и поэтому при титровании ток в цепи сначала возрастает, а после завершения этой реакции тиооксин взаимодействует с селеном (IV) и ток в цепи остается постоянным до появления избытка тноок-сина.
Предложены также методы титрования селена(IV), основанные на окислении его ацетатом свинца (IV) [28], перманганатом [29], гипобромитом [30].
ЛИТЕРАТУРА
1. Hubicki XV., Dabrowska М. — Ann. Univ. М. Curie-Sklodowska, Lublin, Polonia, 1951, AA6, p. 161—163.
2. Ikeda S., Molonaka J. — J. Chem. Soc. Jap., Chem. and Ind. Chem., 1975, v. 8, p. 1337—1341.
3 Savencu S., Bordea A., Moroi Gh„ Rosmarin. M., Matel F. — Bui. Inst, poli-techn. Jasi, 1966, v. 12, Hs 3—4, p. 157—160.
4. Сонгина О. А., Войлошникова А. П. — Зав. лаб., 1958, т. 24, № 11, с. 1331— 1336.
249
5. Агасян Л. Б., Николаева Е. Р„ Агасян И. К- — Ж- анал. хим., 1966, т. 21, № 12, с. 1470—1474.
6. Захаров В. А., Сонгина О. А., Клюева Р. И.—Зав. лаб., 1969, т. 35, № 11, с. 1309—1311.
7. Захаров В. А., Сонгина О. А., Клюева В. И.—Зав. лаб., 1972, т. 38, № 9, с 1066—1067.
8. Иванова 3. И., Игнатенко Е. Г., Тарасова В. А.—Зав. лаб., 1973, т. 39, № 10, с. 1180—1182.
9. Иванова 3. И., Игнатенко Е. Г., Тарасова В. А.—Ж- аналит. хнм., 1973, т. 28, № 10, с. 1980—1984.
10. Simon V., Grim V. — Chem. Listy, 1954, v. 48, № 8, p. 1774—1778.
11. Кузьмина И. И., Сонгина О. A.—Ж. анал. хим., 1962, т. 17, № 4, с. 495— 499.
12. Аришкевич А. М., Усатенко Ю. И., Волошина В. В.—Ж. анал. хим., 1969, т. 24, № 7, с 1069—1076.
13. Сурмий А. М., Аришкевич А. М., Усатенко Ю. И —Труды комиссии по аналитической химии АН СССР, 1968, вып. 16, с. 114—119.
14. Аришкевич А. М„ Волошина В. В., Дьяченко Л. Ф. — В кн : Определение микропримесей. М., 1968, № 2, с. 67—71.
15. Волошина В. В., Усатенко Ю. И., Аришкевич А. М. — Зав. лаб., 1970, т. 36, № 5, с. 530—534.
16. Аришкевич А. М., Волошина В. В., Усатенко Ю. И.—Укр. хим. ж., 1970, т. 36, № 5, с. 499—504.
17. Усатенко Ю. И., Тулюпа Ф. М., Баркалов В. С. — Зав. лаб., 1966, т. 32, № 7, с. 787—790.
18. Тулюпа Ф. М., Баркалов В. С., Усатенко Ю. И.—Ж- анал. хим., 1967, т. 22, № 2, с. 399—402.
19. Павличенко В. А., Усатенко Ю. И., Тулюпа Ф. М. — В кн.: Химическая технология. Респ. мвжведомств. научно-техн. сб. 1968, вып. 13, с. 88—91.
20. Chatterjee S. S., Banerjee В. К- — Technology (India), 1971, v. 8, №3—4, р. 242—244.
21. Deshmukh G. S., Nandi R. A.— Current Sci. (India), 1969, v. 38, № 21, p. 512—513.
22. Deshmukh G. S., Sankara Rao V. S. — Current Sci. (India), 1968, v. 37, № 20, p. 584—585.
23. Chaudhuri H., Chatterjee S. S., Mehanti M. S. — Technology (India), 1973, v. 10, № 3—4, p. 264—266.
24. Даниленко E. Ф. и dp.—В кн.: Вопросы химии н химической технологии. Респ. межвед. темат. научно-тех. сб. 1974, вып. 33, с. 77.
25. Сонгина О. А. и др.— Ж. аналит. хим., 1972, т. 27, М? 6, с. 1121—1124.
26. Супрунович В. И., Величко В. В., Коган Д. С. — В кн.: Методы определения неметаллических и других вредных примесей в промышленных материалах. М„ 1977, с. 137—142.
27. Jensen R. — Chim. analyt. (Paris), 1959, v. 41, № 1, p. 304—307.
28. Deshmukh G. S., Chaudhuri V. N. — Z. anal. Chem., 1974, Bd. 272, № 2, S. 125.
29. Тойбаев Э. И., Сонгина О. А., Захаров В. А. — Изв. АН КазССР. Сер. хим., 1977, № 2, с. 55—59.
30. Deshmukh G. S., Bapat М., Balkrishnan Е., Eshwar М. — Naturwiss., 1958 Bd. 45, № 1, S. 129—132.
Сера
Определение серы в виде сульфата было первой полярометрической работой, выполненной в 1929 г. Гейровским и Березицким. Описанию этого титрования посвящено много работ, в которых рассмотрены главным образом условия титрования разных объек
250
тов. Почти все исследователи рекомендуют титровать в присутствии какого-либо органического растворителя, понижающего растворимость сульфата свинца, — этилового или метилового спирта, других высших спиртов, ацетона [1—91. Вместо солей свинца можно применять для титрования сульфат-иона растворы солей бария, но в этом случае прибегают к обратному титрованию избытка хлорида бария раствором бихромата калия [10—12], или раствором ЭДТА [13].
Для определения серы в виде сульфит-иона (8Оз~ ) рекомендуется окислительное титрование раствором иода [14, 15], иодхлори-да [16], иодбромида [17], бромхлорида [18] и хлорамина Т [19, 20]. Можно титровать сульфит-ион раствором ртути(II) [21], в этом случае образуется комплекс Hg(SO3)2".
Тиосульфат ион (82Оз“) обычно определяют титрованием раствором иода [14, 22—24]. В качестве титрантов предложены также иодхлорид [25], иодбромид [17], иодат калия [26], бихромат [26], сульфат. церия [23] и другие окислители. Тиосульфат-ион можно титровать солями ртути(П) [21—27].
Для определения серы (S0) предложены методы [28—30], заключающиеся в переводе ее в среде 50%-ного изопропанола в форму CNS~ при помощи KCN, маскировки избытка KCN формальдегидом и титровании CNS- раствором нитрата серебра.
Сульфидную серу (82--ион) определяют при помощи ацетата кадмия i[31, 32], нитрата ртути (II) [33], сульфата цинкаД34] и нитрата серебра [35], с которыми сульфид-ион образует малорастворимые осадки в нейтральных и щелочных растворах. Определение проводят или путем прямого титрования 82--иона раствором соли металла [31, 33—35], или путем осаждения 82--иона избытком титрованного раствора ацетата кадмия и обратного титрования его раствором ферроцианида калия [32]. Сульфидную серу можно окислять бромной водой до сульфат-иона и последний титровать раствором нитрата свинца [36]. Йодометрический метод определения сульфида применен для анализа малых количеств сероводорода в воздухе [37].
Серу в виде роданид-иона (CNS) определяют чаще при помощи нитрата серебра [36, 38—40], при этом титрование обычно проводят в водно-спиртовой среде для уменьшения растворимости осадка AgCNS. Можно определять CNS_-hoh титрованием окислителями: иодом, иодхлоридом, перманганатом калия [41].
Для определения политионатов применяют в качестве реагента гипохлорит натрия [42].
Были предприняты также попытки одновременного определения сульфидов, полисульфидов и тиосульфата при совместном присутствии их в растворе [43].
В заключение следует упомянуть, что для определения тионных ( = C=S) и тиольных ( = С—SH) групп в различных соединениях могут быть применены те вещества, титрование которых возможно серу со держащими соединениями этого типа [44].
251
Таблица 37. Методы определения серы
Определяемый ион Титрант Определяемый минимум Условия определения Примечания Литература
SO1- Pb(NO3)2 0,1 мг Электрод Hg капающий, <р=—1,2 В (Нас. КЭ), фон —KNO3, рН=3—6 Для уменьшения растворимости образующегося сульфата свинца титрование проводят в водно-спиртовых средах 1—7
Pb(NO3)2 или РЬ(СН3СОО)2 0,1 мг Электрод Pt, <р— —0,6 В (МИЭ) или +1,5 В (МИЭ), фон — 0,4 М KNO3 или 4—5% СН3СООН Титруют в водно-спиртовой среде, применено для анализа электролита цианистого кадмирования 8, 9
ВаС12 0,1 мг Электрод Hg капающий, <р=0 В (Нас. КЭ), фон — рН=5,3—7,3 Обратное титрование избытка Ва2+ раствором бихромата или ЭДТА 10—13
so2- 12 0,05 мкг Электрод Pt, <р=0 В (Нас. КЭ) или 2 Pt электрода, Д<р=20 мВ, фон — ацетатный или гидрокарбонатный раствор, pH=4—7 Применено для анализа желатины 14, 15
IC1 0,1 мг Электрод Pt, <р=0 В (Нас. КЭ), фон — концентрированная СН3СООН или 0,1 М НС1 Мешает S2O|~ 16
IBr 0,05 мг Электрод Pt, <р=0 В (Нас. КЭ), фон — 0,2 М НВт Мешает S2O|~ 17
ВгС1 0,05 мг Электрод Pt, <р=0 В (Нас. КЭ), фон — 1 М НС1 18
Хлорамин Т 5-10_ 4 моль/л Электрод Pt, <р=0 В (Нас. КЭ), фон — ацетатный буферный раствор, pH=4—6 19—20
ад- h 10-6 моль/л Электрод Pt, <р=0,1 В (Нас. КЭ), фон — раствор с рН=4—8 Возможно раздельное определение S2O|“ и SO1~: в одной аликвотной части титруют сумму этих ионов, а в другой после связыва- 14, 22, 23, 24
IC1 0,05 мг Электрод Pt, <р=0,1 В (Нас. КЭ), фон —0,1—1 М НС1 ния SO3- с помощью СН2О титруют S2O|_ 25
IBr 0,05 мг Электрод Pt, <p=0 В (Нас. КЭ), фон — 0,2 М НВг Мешает SO3~ 17
KIQs, Три, КВгОз, K2Cr2O7, KMnOi 0,1 мг Электрод Pt, <р=—0,1 В (Нас. КЭ) Титрование проводят в присутствии KI как катализатора; титруют растворами КЮ3 на фоне 0,075 н. H2SO4, Tl111 на фоне 0,125 н. H2SO4, КВгО3 на фойе 1 н. НС1, 'КгСггО? на фоне 3,75 н. H2SO4, КМпО4 на фоне 0,75 н. H2SO4 26
Ce(ISO4)2 0,05 мг Электрод Pt, <р=0,2 В (Нас. КЭ), фон — 0,25 н. H^SO4 Титруют в присутствии KI как катализатора 23
HgCl2 Hg(.NO3)2 5-10“5 моль/л IO-5 моль/л Электрод Pt, <р=0 В (Нас. КЭ), фон — раствор с pH=7—8 2 Hg капающих электрода, Д<р= = 50 мВ, фон — КС1 и Na2B4O? Образуется комплекс Hg(S2O3)22-, возможно раздельное определение S2O|_ и SO|~, в одной аликвотной части титруют сумму этих ионов, а в другой после связывания SO3_ с помощью СН2О титруют S2O|~ 21 27
s° KCN AgNO3 Электрод Pt, <р=0,06 В (Нас. КЭ), фон — 1 М KNO3 Элементную серу переводят при помощи KCN в CNS- и титруют раствором AgNO3 28 30
S2- Cd(CH3COO)2 2 Pt электрода, Д<р=0,6 В, или один Pt электрод, <р = 4-0,8 В (Нас. КЭ) В методе с двумя электродами Pt — прямое титрование, в методе с 1 электродом—избыток Cd2+ от-титровывают раствором K4[Fe(CN)6] 31, 32
Продолжение
Определяемый ион Титрант Определяемый минимум Условия определения Примечания Литература
Hg(NO3)2 0,1 мг 2 Hg капающих электрода, Д<р= = 40 мВ, фон — 2 М КОН Не мешают 62О|_, SO|~ 33
ZnSO« 2 Pt электрода, Д<р=0,8 В или Д<р = = 1,2 В, фон — 1 М NaOH Применено для анализа, сульфидно-щелочных растворов сурьмы и ртути 34
AgNO3 Электрод Pt, <р=0 В (Нас. КЭ), фон — 0,2 М NH4OH 35
Pb(NO3)2 Электрод Hg капающий, <р=—1,0 В (Нас. КЭ), фон — раствор с рН=4—5 S2- окисляют бромной водой до SO|_ , Который титруют раствором соли свинца (И) 36
Is 10 мкг 2 Pt электрода, Д<р = 0,1 В S2- окисляют иодом и избыток иода титруют тиосульфатом 37
CNS" AgNO3 10-4 моль/л Электрод Pt, <р=0 В (Нас. КЭ), фон — 0,01—0,03 М HNO3 или 0,1 М KNO3+0,01 М NH4OH Для уменьшения растворимости AgCNS титруют в присутствии 20—30% спирта 36, 38—40
I2> IC1, KMnO4 0,5 мкг/мл Электрод.Pt, <р=—0,1 В (Нас. КЭ), фон — 0,3 М NaHCO3 при титровании раствором 12 или IC1, 0,05 М H2SO« при титровании раствором КМпО4 41 ч
Поли-тионаты NaOCl 2 Pt электрода, Д<р=50 мВ, фон — фосфатный буферный раствор с рН = = 6 42
ЛИТЕРАТУРА
1 Jshibashi М., Fujinaga Т.— Bull. Chem. Soc. Japan, 1952, v. 23, p. 142.
2 Gordon В. E., Urner J?. — Anal. Chem., 1953, v. 25, p. 897.
3 Крисс E. E„ Якубсон С. И., Геллер Б. A. — Зав. лаб., 1954, т. 20, № 9, ’ с. 824.
4 Бутенко Г. А., Пиндас В. К- —Зав. лаб., 1946, т. 9, № 7, с. 634.
5 Васильев В. А., Герцова С. А.— Зав. лаб., 1949, т. 15, № 12, с. 1414.
6^ Белая В. С., Пученкина И. И., Коршунов И. А.— Зав. лаб., 1945, т. 11, № 7, с. 644.
7. Алемаскина Г. А., Векслина В. А., Рутберг Л. Г. Амперометрическое опре-' деление сульфатов, хроматов, фосфатов, фторидов. Куйбышев, Книжное изд-во, 1963.
8. Усвяцов А. А., Сонгина О. А. — Зав. л аб., 1965, т. 31, № 7, с. 663.
9 Кузьмина Н. Н. и др.—Изв. вузов. Химия и хим. технол., 1972, т. 15, № 7, ’ с. 996—1000.
10 Wars chaw sky В., Shook Т. Е., Shanta Е. J. — Anal. Chem., 1954, v. 26, № 6, ’ р. 1051—1055.
11 Скобец В. Д., Печак Е. Е., Скобец Е. М.—Укр. хим. ж., 1976, т. 42, № 6, ’ с. 640—644.
12 Терентьева Е. А., Бернацкая М. В.—Ж- аналит. хим., 1964, т. 19, № 7, ’ с. 876—879.
13 Мураками Сатоми, Хаякава Хисас — Bull. Fac. Educat. Kobe Univ., 1968, 1968, v. 40, p. 11; РЖХим., 1970, 8Г136.
14. Ахмедов Г.—< Научи, труды Ташкентского ун-та, 1970, вып. 379, с. 182—188.
15. Matschiner Н., Siemroth I., Wolff R.— Mikrochim. acta, 1970, v. 2, p. 321— 326.
16. Генгринович А. И., Корнева Л. E., Муртазаев A. M. — Труды Ташкентского . фармацевтического ин-та, 1960, т. 2, с. 355—360.
17. Назруллаев С. Н., Генгринович А. И., Муртазаев А. М.—ДАН УзбССР, 1965, № 5, с. 23—25.
18. Корнева Л. Э., Генгринович А. И., Муртазаев А. М. — ДАН УзбССР, 1965, № 9, с. 28—317.
19. Хадеев В. А., Жданов А. К-, Речкина Л. Г.—Узб. хим. ж., 1960, № 6, с. 28—31.
20. Matsuda Toshio. — Rev. Polarogr., 1975, v. 21, № 1—6, p. 135.
21. Yamasaki Sumio, Ohura Hiroki, Nakamori Issei. — Bunseki Kagaku, 1975, v. 24, № 12, p. 764—771; РЖХим., 1976, 14Г125.
22. Kiss S. A. — Z. anal. Chem., 1965, Bd. 207, S. 184—188.
23. Жданов A. K-, Ахмедов Г., Лукьянова T. В.-—Узб. хим. ж., 1969, № 5, с. 12—14.
24. Robin У., Pithon F., Watanabe R., Combes R. — C. r. Acad. Sci., 1977, v. 284, № 1, p. 33—35.
25. Жданов А. К. и dp. —Узб. хим. ж , 1961, № 2, с. 44—47.
26. Жданов А. К-, Ахмедов Г. — Узб. хим. ж., 1971, № 5, с. 6—8.
27. Петухова А. И., Торопова В. Ф.—Изв. вузов. Химия и хим. технол., 1970, т. 13, № 8, с. 1101—1103.
28. Chatterjee Р. К., Banerjee D., Sirear А. К. — J. Scient. and Ind. Res., 1960, BC19, № 10, B403; РЖХим., 1961, 12Г137.
29. Икэда Саназ, Муся Соитиро— Japan Analyst, 1966, v. 15, № 8, р. 871; РЖХим., 1967, 12Г127.
30. Икэда Санаэ, Муся Соитиро — J. Chem. Soc. Japan. Ind. Chem, Sec., 1970, v. 73, № 2, p. 296—299, A14; РЖХим., 1970, 18Г100.
3*- Kiss S. —Taianta, 1961, v. 8, N 6, p. 726—729.
32. Ленская В. H., Терехова Р. //. — Изв. вузов. Химия и хим. технол., 1962, т. 5, № 6, с. 726—729.
33. Петухова А. И., Торопова В. Ф.— Изв. вузов. Химия и хим. технол., 1972, т. 15, № 3, с. 341—344.
34. Севрюков Н. Н., Корякин И. В —Зав. лаб., 1969, т. 35, № 9, с. 1036—1039.
255
35. Икэда Санаэ, Муся Соитиро—J. Chem. Soc. Japan. Ind. Chem. Sec., 1970 v. 73, № 2, p. 299—302, A15; РЖХим., 1970, 18Г62.
36. Evans И B., Mori S. — Anal. Chem., 1968, v. 40, Ns 1, p. 217—218.
37. Levin L., Swann W. B.—Taianta, 1958, v. 1, N 3, p. 276—280.
38. Banerjee D., Gupta T. P. — Indian J. Chem., 1966, v. 4, № 2, 91; РЖХим., 1967, 18Г104.
39. Икэда Санаэ, Нисида Гиро — Japan Analyst, 1966, v. 15, № 6, p. 610—613; РЖХим., 1967, 5Г110.
40. Ikeda S., Hirata J. — J. Chem. Soc. Japan., Chem. and Ind. chem., 1973, v. 76, № 2, p. 425—427; РЖХим., 1973, 17Г132.
41. Жданов А. К-, Ахмедов Г., Розенблит М. М. — Изв. вузов. Химия и хим. технол., 1972, т. 15, № 4, с. 503—505.
42. Масалович В. М., Вессерман Л. И., Пирская Т. X. — Труды Уральского научно-исследовательского химического ин-та, 1971, вып. 26, с. 46—51.
43. Kiss S. — Z. anal. Chem., 1962, Bd. 188, S. 341—344.
44. Сонгина О. А., Бессарабова И. М. — Зав. лаб., 1973, т. 39, № 6, с. 641—655.
Серебро
Для определения серебра было предложено в основном два реактива — хлорид и иодид калия или натрия, образующие, как известно, малорастворимые осадки с серебром (I). Так как растворимость иодида серебра составляет 1,2—10-8 М при 25 °C, а растворимость хлорида серебра в тысячу раз больше (1,2-10-5 М), то предпочтение следует отдать иодиду калия (натрия).
Серебро титруют обычно с платиновым вращающимся электродом. С ртутным капающим электродом титровать серебро затруднительно, так как ртуть непосредственно взаимодействует с ионами серебра, восстанавливая их до металла.
Известно два варианта титрования иодидом калия — катодный [1—5], основанный на восстановлении ионов серебра (кривая имеет форму а), и анодный [4], основанный на окислении избыточных исдид-ионов (кривая формы б).
При титровании катодным методом определение можно проводить без наложения внешнего напряжения (МИЭ). Однако, во избежание восстановления других ионов (Си11, В1ш и др.), лучше титровать при +0,4 В (МИЭ). Влияние железа(III) устраняют фторидом калия. Катодный метод применен [5] для определения серебра в различных рудах, сплавах, шламах, цианистых электролитах и т. д.
Титрование по анодному принципу, т. е. по току окисления избыточных ионов иодида [4], проводят при потенциале +1,0 В (МИЭ). При этом потенциале не восстанавливается ни один из обычных катионов, могущих присутствовать в анализируемом растворе вместе с серебром, в том числе и железо(III).
Для того, чтобы получать устойчивые, хорошо воспроизводимые результаты титрования надо соблюдать некоторые предосторожности. Температура раствора перед титрованием не должна превышать 20—22 °C, так как растворимость осадка иодида серебра возрастает с увеличением температуры. После каждого определения электрод следует протирать фильтровальной бумагой, а в
256
конце рабочего дня погружать в раствор тиосульфата. Титрование лучше проводить при pH ^2—2,5, так как такая кислотность раствора способствует возрастанию тока окисления избыточного иодида. Анодный метод можно с уверенностью применять для определения серебра в любых объектах, в частности в минеральном сырье и, что особенно важно, в черновой меди.
Для определения серебра в рудах и концентратах предложена следующая методика.*
Ход анализа. Навеску пробы 1—5 г (в зависимости от ожидаемого содержания серебра) помещают в коническую колбу емкостью 250 мл, смачивают водой и разлагают 20—40 мл концентрированной азотной кислоты при нагревании (царской водкой пользоваться нельзя — выпадает осадок хлорида серебра!). Если после разложения еще остаются темные частицы пробы, то добавляют 2— 3 г нитрата аммония и нагревают до полного обесцвечивания пробы. Упаривают до минимального объема, охлаждают и прибавляют 10—20 мл серной кислоты (1 : 1), нагревают до появления белых паров, которым дают выделяться в течение 5—10 мин. Затем раствор охлаждают, осторожно обмывают стенки колбы 15—20 мл воды и кипятят раствор 10—15 мин для полного разрушения нитро-зилсерной кислоты (эта операция очень важна, так как в присутствии следов окислов азота титровать иодидом калия нельзя). Охлаждают, осторожно нейтрализуют аммиаком основную массу кислоты (примерно 10—15 мл концентрированного раствора аммиака). Охлажденный раствор вместе с осадком переносят в мерную колбу и доводят до метки водой. Обычно используют мерную колбу емкостью 100 мл. Отбирают аликвотную часть в стакан для титрования и доводят pH аммиаком до 2—3 по универсальной индикаторной бумаге. Охлаждают раствор в стакане до 18—20 °C проточной водой и титруют серебро 0,005— 0,0001 н. раствором иодида калия при потенциале +1,0 В (МИЭ). Силу тока измеряют через 30 с после каждого добавления иодида.
Если проба содержит больше 15—20% двуокиси кремния, то пробу разлагают несколько иначе: после нагревания с серной кислотой осторожно приливают 10 мл концентрированной фтористоводородной кислоты и упаривают снова до появления паров; эту операцию повторяют трижды, после чего выпаривают с 10—15 мл воды н далее поступают так, как описано выше.
Анодный иодидный метод позволяет определять любые количества серебра в рудах и концентратах; нижний предел для руд — 5 г/т.
Для определения серебра в черновой меди применяют следующую методику [6].
Ход анализа. Навеску черновой меди 1 г (при содержании серебра 2000— 2500 г/т) смачивают водой, приливают 10 мл азотной кислоты плотностью 1,40 г/см3 и после окончания бурной реакции упаривают до минимального объема. Охлаждают, добавляют 10—15 мл серной кислоты (1:1) и нагревают до появления белых паров и полного обесцвечивания жидкости. После охлаждения осторожно приливают 20—30 мл воды и кипятят несколько минут (для полного разрушения нитрозилсерной кислоты). Полученный раствор охлаждают, переносят в мерную колбу емкостью 100 мл и доводят водой до метки. Для титрования отбирают 10 мл, осторожно нейтрализуют концентрированным раствором аммиака до появления осадка основных солей меди, т. е. до pH=2—3. Выпавшие соли растворяют, добавляя 2—3 капли серной кислоты (1 : 1), охлаждают раствор до комнатной температуры (20—22 °C) и титруют 0,005 н. раствором иодида калия из микробюретки с ценой деления 0,01 мл при потенциале платинового электрода +1,0 В (МИЭ).
* Методика разработана А. И. Пащенко на кафедре химии редких элементов Казахского государственного университета и проверена на промышленных объектах.
17—1694
257
g Таблица 38. Методы определения серебра (Ag1)
Титрант Определяемый минимум Условия определения Примечания Литература
KI K4Fe(CN)6] 0,02- -0,03 мг/л (ка-тодн.), 1 мкг/20 мл, 5 г/т (анодн.) Электрод Pt, <р= + 0,44-0,45 В (ка-тодн.) (МИЭ), <р= + 1,0 В (анодн.), фон — сернокислый, рН=2—S Электрод Hg капающий, <р=— 0,3+ +0,5 В (Нас. КЭ), фон —0,1 М KNO3 См. текст 1—11 13
4.Ю-3—Ь Ю-в моль/л Электрод Pt, <р= +0,8+1,4 В (МИЭ), фон —0.1 н. H2SO4, HNO3, KNO3 Титруют по току окисления ферроцианида, определяли Ag в отработанной фотопленке 14
Аскорбиновая кислота 2 Pt электрода, для Т1111 Д<р= =0,1 В, фон — 0,1—1 н. H2SO4; для Ag1 Д<р=0,05 В, фон— (NH4)2SO4, pH=6,5—7,0 Определение Ag1 и Т1111 15
Ферроцен 0,12 мкг/мл Электрод углеситалловый, электрод сравнения КМпО4 в щелочном растворе, фон — 2—4 н. H2SO4 или НС1О4 Ag1 восстанавливается до Ag металлического, мешают все ионы, способные восстанавливаться ферроцв’ ном 16
Тиокарбамид Электрод Pt, <р=+0,2 В (Нас. КЭ) или 1,0 В, т. е по току восстановления Ag1 или окисления титранта, фон — 0,1—2 н. H2SO4 См. текст 17
Т иосалицилов ая кислота Нижний предел 0,01 мг/30 мл Электрод Pt,<p= +1,0 В (Нас. КЭ), фон — кислый, рН= 1 Определяли Ag в сплаве с медью и в шламах 18, 27
Унитиол Нижний предел 50 мг/25 мл Электрод Pt, <р =4-0,8 В (Нас. КЭ), фон — 0,001—0,5 н. H2SO4 Осадок состава R:Ag= 1:2 при рН< <3, при рН>3 осадок растворяется; в присутствии Pd образуется смешанный Ag—Pd комплекс с унитиолом 19
Ксантогенат Электрод Pt, <р= 4-0,05 В (Нас. КЭ), фон — аммиачно-тартратный буферный раствор4 KNO3, рН=7—9 Ag1 титруют по току его восстановления, можно титровать РЬ11 по току окисления реагента 22
Димеркаптотиопироны Электрод графитовый, <р= 4-0,5 В (Нас. КЭ), фон — 0,5—3 и. НС1 или 0,1—15 и. H2SO4, можно и щелочной фон, pH =10 В кислой среде можно последова тельно титровать Ag, Au, Sb, Tl, Те As 23
iN бис-(р-амино-этил) -дитиокарбоновая кислота Электрод Pt, <р=0 В (Нас. КЭ), фон — аммиачно-тартратный, буферный раствор, pH=8—9 Ток окисления реагента 24
Диэтилдитио-карбаминат свинца 2 Pt электрода, Д<р=0,05—0,1 В, фон — безводная уксусная кислота или перхлорат лития в бензоле Не мешают Си, Bi, Ni, Cd, Pb 26
Дитизон 20—150 мкг/10 мл серебра 5—50 мкг/10 мл меди 2 Pt электрода, Д<р«0,1 В, фон — безводная уксусная кислота или уксусная кислота 4-СНС13 в присутствии LiClO4, НСЮ4, CH3COONa Титруют Ag и Си; вместо Pt элек тродов пробовали Ag и Си электродь 27
к> Си
Описаны различные варианты иодидного метода с применением двух платиновых или ртутных электродов [7—10], а в статье [11] рекомендовано определять ртуть и серебро, титруя сначала ртуть(II) раствором ЭДТА, а затем серебро иодидом [серебро(I) не образует устойчивых комплексов с ЭДТА].
В разделе «Палладий» описан метод определения палладия (II) и серебра (I) с помощью иодида при совместном присутствии обоих ионов, а в статье [12] предложено, во избежание «палладиро-нания» электрода, добавлять в титруемый раствор пиридин.
Серебро титруют также раствором ферроцианида калия [13, 14], роданида, нитропруссида [13], аскорбиновой кислотой [15], ферроценом [16], который восстанавливает серебро(I) до металла. К сожалению этому титрованию мешают все ионы, способные восстанавливаться ферроценом.
Большое распространение получили для определения серебра различные серусодержащие реагенты, в том числе тиокарбамид. Изучение реакции серебра(I) с тиокарбамидом показало ,[17], что образуется несколько растворимых соединений и одно нераствори-.мое, в котором соотношение серебра и тиокарбамида равно 1 : 1 (0,1—2 н. серная кислота).
Серебро (I) образует труднорастворимый осадок с тиосалициловой кислотой [18, 19], метод описан также в разделе «Золото». Унитиол предложен для определения серебра(I) и палладия (II), причем установлено, что при совместном присутствии обоих ионов образуется смешанное палладиево-серебряное соединение с унитиолом, состав которого зависит от исходного соотношения обоих элементов [20]. Изучено взаимодействие серебра(I) с тиопиперидоном [21], ксантогенатом [22], димеркаптотиопиронами [23], различными производными этилдитиокарбоновой кислоты [24] и с другими сложными соединениями [25].
Бензольные растворы диэтилдитиокарбамината свинца применены для титрования серебра в безводной среде как отдельно [26], так и в присутствии других элементов (см. «Висмут»), В неводной среде титруют серебро также дитизоном [27] и тиооксином (см. «Медь»)' и его производными (см. «Золото») в присутствии многих элементов.
ЛИТЕРАТУРА
1. Сонгина О. А., Войлошникова А. П., Козловский М. Т. — Изв. АН КазССР. Сер. хим., 1949, вып. 3, с. 81.
2. Сонгина О. А., Войлошникова А. П., Козловский М. Т. — Изв.‘ АН КазССР. Сер. хим., 1951, вып. 4, с. 80.
3. Сонгина О. А. — Труды комиссии по аналитической химии АН СССР. IV (VII), 1952, с. 116.
4. Сонгина О. А. — Зав. лаб., 1955, т. 21, № 7, с. 665.
5. Езерская Н. А. Применение полярографического и амперометрического методов для анализа некоторых золотосодержащих продуктов. Канд. дисс. ИОНХ АН СССР, 1955.
6. Мищенко А. И., Сонгина О. А. —Зав. лаб., 1963, т. 29, № 2, с. 162.
7 Абрамов М. К- — Аптечное дело, 1962, № 11, с. 44.
1260
8 Kiss S. — Z. anal. Chem., 1963, Bd. 195, S. 249.
9 Kies H. L. — Anal. chim. acta, 1955, v. 12, p. 280.
10 Накасима Рёдзо, Маруяма Такаёси, Фу рукава Масамити— Ногая когё гид
' зюцу сикнасё хококу, 1968, v. 17, № 9—10, р. 241—244; РЖХим., 1970, 4Г183.
11. Хадеев В. А., Базарбаев А. Т.—Узб. хим. ж., 1960, № 5, с. 38—41.
12^ Торопов Ю. А. — В кн.: Методы анализа руд Кольского полуострова. Апатиты, 1970, с. 143—144.
13. Kalvoda R., Zyka J.— Chem. Listy, 1951, v. 45, p. 82.
14. Шапошникова Г. H.— Арм. хим. ж., 1976, т. 29, № 2, с. 148—151.
15 Singh D., Bhatnagar V.— Bull. Chem. Soc. Japan, 1966, v. 39, № 6, p. 1101— ‘ 1103; РЖХим., 1967, 5Г60.
16. Соломатин В. T. и др. — Ж. анал. хим., т. 32, № 5, с. 948—953.
17 Кузьмина Н. А., Сонгина О А.—Ж. анал. хим., 1963, т. 18, № 2, с. 323— 326.
18. Крюкова Л. В., Усатенко Ю. И. — В кн.: Вопросы химии и химической технологии. Респ. межвед. темат. научно-техн. сб. 1975, вып. 38, с. 63—66.
19 Тараян В. М., Погосян А. Н.— Изв. вузов. Химия и хим. технол., 1969, т. 12, № 6, с. 728—732.
20. Сонгина О. А. и др. —Ж. анал. хим., 1967, т. 22, № 8, с. 1170—1174.
21 Тараян В. М., Саркисян А. А.', Мушегян А. В. — Арм. хим. ж., 1968, т. 21, № 8, с. 662—668.
22. Deshmukh G. S., Saraswathi К. — Bull. Chem. Soc. Japan, 1967, v. 40, № 6„ p. 1545—1546; РЖХим., 1968, 5Г65.
23. Аришкевич A. M., Кроик А. А., Усатенко Ю. И. — В кн.: Химическая технология. Респ. межвед. темат. научно-техн, сб., 1971, вып. 18, с. 160—163.
24. Chattopadhyay S. S. — Indian J. Chem., 1971, v. 9, № 7, p. 720—721; РЖХим.* 1972, 5Г76.
25. Deshmukh G. S., Chaudhuri H., Mahanti H. S. — Indian J. Chem., 1975, v. 13, p. 82—83; РЖХим., 1976, 2Г97.
26. Геворгян A. M. и dp. — РЖХим., 1975, 20Г65, депон.
27. Геворгян A. M. и др. —Узб. хим. ж., 1976, № 4, с. 12—13.
Скандий
Для определения скандия рекомендован бензоилфенилгидроксила-мин, который применяют и для определения галлия (III) (см.. «Галлий»), титана(IV) и циркония(IV). Скандий можно определять при помощи этого реактива, титруя его избыток либо раствором соли цинка с ртутным капающим электродом, либо раствором перманганата с платиновым электродом. По данным авторов метода можно определять 0,5 мг скандия в присутствии 100-кратных количеств неодима, лантана и иттрия.
Рекомендуется также N-циннамоилфенилгидроксиламин как только для скандия [1], так и для скандия и иттрия (см. «Иттрий»), и N-циннамоил-п-толилгидроксиламин для ионов обоих элементов (см. «Иттрий»). Титруют в обоих случаях с графитовым электродом при +0,7 В (Нас. КЭ), на фоне с рН=4,5—7. Состав комплексных соединений отвечает соотношению скандий: реагент = 1 :3. Нижний предел определения скандия 0,003 мг.
Можно титровать скандий и раствором ЭДТА, но, как указывалось в разделе «Лантаноиды», метод мало селективен.
Методы, связанные с применением ЭДТА, описаны в разделах «Иттрий», «Лантаноиды», «Магний» и «Индий».
261
Определение скандия с помощью ферроцианида [2] описано в разделе «Лантаноиды», где указана зависимость состава осадка ферроцианида скандия от состава, концентрации и pH фона и от природы щелочного металла.
ЛИТЕРАТУРА
1. Шеина И. М. и др.—Anal, lett, 1974, v. 7, № 2, р. 125—132.
2. Сонгина О. А., Захаров В. А., Токушева Г. Т.—Ж. анал. хим., 1970, т. 25, № 1, с. 64—67.
Стронций
Стронций был одним из первых элементов, определенных амперометрически: еще в 1929 г. Гейровский и Березицкий титровали стронций «полярографически» на ртутном капающем электроде растворами карбоната и гидрокарбоната натрия [1]. Лишь через много, лет был предложен другой метод определения стронция: в отсутствие ионов бария, кальция, магния и многих других стронций может быть оттитрован [2] раствором ЭДТА на платиновом электроде по току окисления избытка ЭДТА при +0,85 В (Нас. КЭ) на фоне нитратно-аммиачного раствора с рН=8—10. Затем были разработаны методики определения стронция в присутствии ионов других щелочноземельных металлов с помощью титрантов, описанных в соответствующих разделах — например, «Кальций».
ЛИТЕРАТУРА
1 Heyrovsky J., Beresicky S. — Coll. Chem. Comm., 1929, v. 1, p. 10; Chem. News, 1929, N 138, p. 195.
2 . Рейшахрит JI. С., Пустошкина M. П., Тихонова 3. И.—Вестник ЛГУ, 1964, № 4, с. 122.
Сурьма
Для определения сурьмы предложено несколько новых реактивов— диэтилдитиобиурет для Sbv [1], амилдимеркаптотиопирон [2], 2,6-димеркапто-3,5-диэтил-1,4-тиопирон [3] и ЭДТА [4] для Sb111. Сурьму (V) можно титровать ферроценом, восстанавливая ее до сурьмы(III) [5]. Интересно, что этому титрованию не мешает мышьяк(V), но мешают медь(II) и железо(II), хотя по значениям редокс-потенциалов этих двух элементов можно было ожидать, что они восстанавливать сурьму (V) не будут. Однако наибольшее развитие получили методы, основанные на окислении сурьмы(III) различными окислителями, в первую очередь броматный и йодатный методы (см. «Мышьяк»), Считается, что броматный метод —лучший из всех, предложенных для определения <урьмы [6]. Его успешно применяют для определения сурьмы в различных сплавах [7, 8]. Его можно осуществлять и с двумя ин-
262
Таблица 39. Методы определения сурьмы
Определяемый ион Титрант Определяемый минимум Условия определения Примечания Литература
SblH КВгОз Димеркапто-3,5-диэтил-1,4-тиопирон 5—800 мкг/25 мл Электрод Pt, <р=+0,4 В (МИЭ), фон — 2 М НС1 или H2SO4+NaCl; можно с 2 Pt электродами, Д<р=0,015 В Электрод графитовый, <р= + + 0,7 В (Нас. КЭ), фон для Sb— —H2SO4+HCI, для Se и Те НС1 или H2SO4 Мешает As Отношение Sb:R=l:3, титруют тем же реагентом Se и Те, отношение (Se, Te:/R= 1:13 на фоне НС1 и 1:4 па фоне H2SO4) 6—10 2
Ам и лди меркаптотиопирон 10—600 мкг/40 мл Электрод графитовый, <р=+0,7 В (Нас. КЭ), фон —3 н. НС1 Осадок состава Sb(HR)3; Си и As осаждаются вместе с Sb, определяли Sb в Sn—Pb припоях 3
ЭДТА и аминциклогексан тетра-уксусная кислота 0,02—4 мкг/мл Электрод Hg капающий, <р=— —0,55—0,7 В (Нас. КЭ), фон pH = 3—4 Не мешают Fe11 и As111, влияние Fe Iu, La111, In111, Си устраняют тартратом, мешают Bi, SnIV, Zn 4
Sbv Ферроцен Дитиобиурет 2 мкг/20 мл Электрод Pt, вспомогательный электрод К(МпО4 в растворе NaOH, фон — 6 н. НС1 +ацетон (1 1 или 1 : 1,5) Электрод графитовый, <р= + +0,55 В (Нас. КЭ), фон —6 н НС1 Мешают Си11 и Fe11 (см. текст), определяют Sb в сплавах Определение Sb в сплавах, влияние As не оговаривается 5 1
Sbin, Asin КЮз 1 мкг/мл Электрод Pt, <р=0 В (Нас. КЭ), фон — 1 М H2SO4 для As+Sb; для Sb111—1 М H2SO4+KI Раздельное определение Sb и As, см. в тексте 11
К2СГ2О7 2 мкг/мл Электрод Pt, <р=+0,2 В (Нас КЭ), фон —H2SO4 Раздельное определение Sb111 и As111 при совместном присутствии: сумма Sb111 и As111 на фоне 3 М H2SO4, а на фоне 0,5 М H2SO4, титруется только Sb111 13
SbSj-, SbSJ- Сульфат цинка 2 Pt или графитовых электрода, Д<р=1,2 В, фон — 1 М NaOH Сульфат цинка количественно осаждает сурьму в виде Sb2Ss, 1 мл 0,1 н. раствора ZnSO4 соответствует 0,0406 г Sb 12
дикаторными электродами [9] при напряжении +0,015 В. Вместо бромата можно применять гипобромит [10], титруя в гидрокарбонатной среде при +0,3 В на платиновом электроде.
Разработан вариант йодатного метода, позволяющий раздельно титровать мышьяк(Ш) и сурьму(Ш) при их совместном присутствии [11], основанный на различной скорости окисления мышьяка (Ш) и сурьмы(III) йодатом в отсутствие и в присутствии иодида калия. Ниже приведена методика, позволяющая определять мышьяк(Ш) и сурьму(Ш) при их отношении 1:10—5:1.
Ход анализа. Навзску пробы 0,2—1,0 г разлагают 20—30 мл смеси H2SO4 и HNO3, упаривают до появления паров серной кислоты, охлаждают, добавляют 15—20 мл воды и 1—1,5 г сульфата гидразина для перевода мышьяка и сурьмы в трехвалентное состояние. Раствор упаривают до объема 4—5 мл, что необходимо для разрушения избыточного количества гидразина, охлаждают, приливают 10—15 мл воды, добавляют для предотвращения гидролиза сурьмы винную кислоту или ее соль в количестве, превышающем навеску в 4—6 раз, переносят в мерную колбу емкостью 50—100 мл, доводят до метки водой и отбирают две аликвотные части по 5 мл. К одной аликвотной части добавляют 1 М H2SO4 до объема 20—25 мл, титруют 0,02 н. йодатом калия при <р=0 В (Нас. КЭ) сумму мышьяка и сурьмы. К другой аликвотной части добавляют 1 М H2SO4 до объема 20—25 мл и 0,5—1,0 мл 1 %-кого раствора KI и титруют тем же раствором йодата калия сурьму. Содержание мышьяка находят по разности результатов двух титрований.
Для раздельного определения сурьмы(III) и мышьяка(III) при совместном присутствии можно применять бихромат калия [12]. Применяют и другие окислители: церий (IV), перйодат калия, иод, хлористый и подпетый бром, соединения кобальта(III), перманганат, а также тиооксин, описанные в разделе «Мышьяк». В разделе «Ванадий» описано титрование сурьмы(III) раствором ацетата свинца на ртутном капающем электроде.
Для определения сурьмы в растворах тиосолей предложено титрование раствором сульфата цинка [13], описанное подробно в разделе «Сера».
ЛИТЕРАТУРА
1. Сухоручкина А. С. и др. — В кн.: Вопросы химии и химической технологии. Респ. межвед. темат. научно-техн. сб. 1974, вып. 32, с. 44.
2. Сурмий А. М., Аршикевич А. М., Усатенко Ю. И. — Труды Комиссии по аналитической химии АН СССР, 1968, т. 16, с. 114—118.
3. Сурмий А. М., Усатенко Ю. И., Аршикевич А. М. — Зав. лаб., 1967, т. 33, № 8, с. 937—939.
4. Rubel S., Bern J. — Chem. anal. [PRL], 1972, v. 17, № 2, p. 279—284.
5. Соломатин В. T., Яковлев П. Я-, Ржавичев С. П.—Ж. анал. хим., 1974, т. 29, № 10, с. 1981—1985.
6. Konopik N., Szlaczka К. — Oesters. Chem. Ztg., 1951, Bd. 52, S. 205.
7. Garate M., Astudillo M., Burriel-Marti F. — Anales Real. Soc. Espan. Fis. Quim. (Madrid), 1960, v. B56, p. 775; Stock J. Amperometric Titrations. New York, 1965, p. 556.
8. Bradshaw G. — Analyst, 1963, v. 88, p. 599.
9. Иованович M., Драгоевич M. — Гласник хим. Друшт.. 1962, т. 27, с. 95; РЖХим., 1963, 21Г99.
10. Deshmukh G. S., Eshwar М. С. — Current Sci. (India), 1960, v. 29, p. 388.
11. Захаров В. А., Чокина H. Ю., Сонгина О. А.—Ж. анал. хим., 1979, т. 34, № 2, с. 314—319.
264
12 Чокина Н. Ю., Сонгина О. А., Захаров В. А.—Ж. анал. хим., 1976, т. 31, ' Ks 4, с. 720—723.
13 Севрюков Н. Н„ Корякин И. В. — Зав. лаб., 1969, т. 35, № 9, № 10, с. 1036— ' 1039.
Таллий
Таллий(I) во многом сходен с серебром(I) и свинцом(II), но в отличие от них сравнительно легко может быть окислен до трехвалентной формы, в которой он проявляет уже иные свойства. В связи с этим при определении таллия можно использовать как реакции осаждения, так и реакции окисления — восстановления.
Растворимость иодида таллия(I) составляет 2,4-10~4 М, т. е. сравнительно высока. Поэтому при титровании таллия иодидом калия [1, 2] обычно получаются заниженные результаты, несмотря на довольно отчетливый ход кривой титрования. Для понижения растворимости осадка добавляют спирт [3] или ацетон [4, 5], но все же нижний предел остается на уровне 4 мг/20 мл. Титровать можно с платиновым электродом по току окисления иодида [2—3] или на ртутном капающем [4, 5] по току восстановления таллия (I) при пропускании азота. Нельзя забывать, что серебро(I) и свинец(II) также осаждаются иодидом. Таллий (I) осаждается также тетрафенилборатом [6, 7], тиоацетамидом [8, 9], рубеано-водородной кислотой [10]. В шестидесятых годах были предложены и другие реактивы для определения таллия(I), не нашедшие однако практического применения [11].
Более надежными являются окислительно-восстановительные реакции. Их можно осуществлять в двух вариантах — титровать таллий (Ш) восстановителями или таллий(1) окислителями. Окислительно-восстановительная система T1III/T1I обладает высоким стандартным потенциалом (£= + 1,28 В) и является, следовательно, окислителем по отношению ко многим ионам, в частности по отношению к бромид-иону. Реакция между таллием (III) и бромид-ионами может быть использована для амперометрического определения таллия с вращающимся платиновым микроэлектродом [12]. Титровать можно либо по току восстановления таллия(III), либо по току окисления бромид-иона, т. е. при потенциалах электрода от +1,3 до +1,4 В. Кривая титрования имеет форму б. Однако этот метод требует предварительного окисления таллия (I) до таллия (III) ц связан с некоторыми дополнительными условиями [Н], поэтому он менее удобен, чем прямое титрование таллия(I) сильными окислителями, например перманганатом в кислой среде, содержащей хлорид [13], который связывает образующийся таллий (III) в комплексное соединение и тем самым понижает окислительно-восстановительный потенциал системы Т1Ш/Т11.
Осадок хлорида таллия (I), выпадающий сначала, не мешает, так как по мере титрования перманганатом он полностью растворяется вследствие окисления Т11 до Т11П. Титр перманганата при
265
Таблица 40. Методы определения таллия
Определяемый ион Титраит Определяемый минимум Условия определения Примечания Литература
T1I К1 4 мг/20 мл Электрод Pt, <р= + 1,0 В (МИЭ), можно и при—0,6 В по току восстановления Т11П, при +1,0 В более точные результаты; фон — 0,1 М NH4NO3 или KNO3 См. текст- 1—3
Электрод Hg капающий. <р = =—0,6-0,7 В .(Нас. КЭ), фон— 3 н ККОз+10% ацетона Пропускают азот 4,5
Тиоацетамид 2-Ю-4—1,5-10-3 М Электрод Hg капающий, <р= =—0,8 В (Нас. КЭ), фон — аммиачный «7,5 М (в присутствии Zn—12 М + твердый гидразинсульфат « 2,5%) Пропускают водород, 1000-кратный избыток Zn не мешает 8,9
Рубеаново-дородная кислота 1,5—3,5 мг/20 мл Электрод Hg капающий, <р= ——1,0 В (Нас. КЭ) фон—аммиачный буферный раствор 10
КВг 0,1 мг/20 мл и более Электрод Pt, <р= + 1,3—1,4 В (МЦЭ), фон —2 н. H2SO4 См. текст 12
Celv 2 Pt электрода, Д<р=0,2 В, фон— 1—4 н. HCl+MnSO4 или IC1 в качестве катализатора В этих же условиях можно титровать ТР бихроматом, но на фоне 10 н. H^SO4 15
КМпО4 Электрод Pt, <р=+0,75 В(МИЭ), фон — 1 н. НС1 См. текст 13
10—200 мкг/20 мл Электрод Pt, <р=—0,36В (МИЭ), фон — 3 н. Na2CO3 В к. т. отношение Т1:МпО4- = =3:2 (см. текст), не мешает Cd и РЬ 20
K3[Fe(CN)6] 0,025 мг/мл Электрод Pt, <р=—0,05 В (Нас. КЭ), фон — сильно щелочной4-+следы осмиевой кислоты в качестве катализатора Окисляющийся Т1 выпадает в оса-1 док в виде Т1 (ОН) 3 16
КЮз, KBrO3 1 0,1—0,6 мг 2 Pt электрода, или Pt и графитовый, Д<р=0,1 В, фон — 2 н. НС1 или 2 н. H2SO44-NaCl, добавляют КВг или IC1, или смесь H2SO44-4-Н3РО4 17—19
Tim Ферроцен Электрод Pt, электрод сравнения щелочной раствор КМпО4, фон— 1 и. H2SO44-ледяная СН3СООН (этиловый спирт или ацетон) Подробное исследование взаимодействия ферроцерона с Т1ш, изучено влияние других ионов 21
Тиокарбамид 1 мкг/40 мл Электрод Pt, <р= + 1,0 В (Нас. КЭ), фон — HNO3, рН=1—1,4 22, 23
Димеркаптотиопироны 10- -200 мкг/30 мл Электрод графитовый, <р= = 4-0,6—0,7 В или 4-1,0 В (Нас. КЭ), фон — различный, рекомендуют 0,1—2 н. HCI4-CH3COOH 0,2 н. или ацетатный буферный раствор, рН»4 Можно дифференцированно титровать Tl, Ag, Hg и TeIV 24, 25
Унитиол 25 мкг/25 мл Электрод Pt, <р=4-1,0 В (НВЭ), фон — сернокислый Мешают TeIV и SeIV 26
Бензолсульфо-нилтиобензамид 0,5—0,004 мг Электрод Pt, <р=4-0,9—1,1 В (Нас. КЭ), фон — HNO3, рН=1 Осадок состава TIR3 27
Тиооксин и его производные Электрод Pt, <р=4-1,2 В (Нас. К.Э), Фон — сернокислый, рН= 1—2 Можно последовательно титровать Т1 и Си, Т1 и Hg [28] и определять Т1 в присутствии Ga и 1п [29] (см. текст) 28, 29
ЭДТА Электрод Та, <р=4-1,0-4-1,2 В (Нас. КЭ), фон —pH 0,9—1,0, для Zn pH=4,5—5 Титруют ТРч и «пары» Т1—Zr, Tl—Zn, Tl—Pb, Tl—Zn—Mg 30
этом определении следует устанавливать по стандартному раствору соли таллия (I).
При титровании таллия(1) растворами солей церия(1У) также применяют солянокислую среду [14, 15].
На том же принципе — понижении стандартного потенциала системы Tlra/TP— основано титрование таллия(1) раствором феррицианида калия в сильнощелочной среде [16]; феррицианид может окислять таллий (I) потому, что образующийся Т1ш связывается в практически нерастворимый Т1(ОН)з.
Известен метод титрования таллия(I) ра-створами йодата или бромата калия в солянокислой среде [17, 18], а также на фоне смеси серной и фосфорной кислот [19]. Можно титровать таллий (I) раствором соли церия(ГУ) в присутствии сульфата марганца в качестве катализатора [15].
В гидрокарбонатной среде таллий(I) окисляется перманганатом [20] благодаря тому, что потенциал системы Tl'H/Tl1 резко снижается — таллий (III) выпадает в осадок; раствор при титровании вследствие этого мутнеет, приобретая коричневатую окраску за счет двуокиси марганца, образующейся при восстановлении перманганата таллием (I).
Для определения таллия (III) можно использовать реакции всех трех типов — осаждения, комплексообразования, окисления— восстановления. Показано [21], что ферроцен восстанавливает таллий (П1) полностью до таллия(I) на фоне уксусной и серной кислоты; предложен соответствующий метод амперометрического определения таллия(Ш). Восстанавливается таллий(Ш) также тиокарбамидом [22, 23], димеркаптотиопиронами [24, 25], образующими в зависимости от условий комплексные соединения с таллием. Титруется таллий(III) унитиолом [26]. Бензолсульфонил-тиобензамид [27] осаждает таллий(Ш) в виде ТШз, а производные тиооксина [28] реагируют с таллием (III) различно, в зависимости от заместителя в молекуле реактива (8-меркаптохинолина); 5-Вг-меркаптохинолин в кислой среде образует осадок состава ТЩз, а 4-метилмеркаптохинолин восстанавливает таллий(III) до таллия(I). Этому реагенту отдают предпочтение [28]. Тиооксин также восстанавливает таллий(III) в кислой среде, что использовано [28] для определения таллия при совместном присутствии его с галлием и индием (см. «Галлий» и «Индий»),
Вообще работ, посвященных определению таллия(III) в присутствии других элементов, довольно много. Таллий(III) титруют раствором ЭДТА в присутствии циркония, свинца,„.цинка, магния [30]. При помощи того же ЭДТА или его аналогов определяют таллий в присутствии серебра и меди (см. «Медь»), кадмия и серебра (см. «Кадмий»), висмута (см. «Висмут»), свинца (см. «Свинец») и т. д. Диэтилдимеркаптотиопирон позволяет определять таллий в присутствии ртути и висмута (см. «Висмут»),
Таллий(I) титруют в присутствии сурьмы(III) и ванадия(V) раствором ацетата свинца(IV) (см. «Ванадий»),
268
ЛИТЕРАТУРА
1. Сонгина О А., Войлошникова А. П., Козловский М. Т. — Изв. АН КазССР. Сер. хим., 1949, вып. 3, с. 81.
2 Сонгина О. А. — Труды комиссии по аналитической химии АН СССР, IV ' (VII), 1952, с. 116.
3. Michalski Е., Stapor W.— Chem. analytycz. (Polska), 1958, v. 3, p. 441.
4. Kalvoda S., Zyka J. — Coll. Chech. Chem. Comm., 1950, v. 15, p. 630.
5. Kalvoda P., Zyka J. — Chem. Listy, 1951, v. 45, p. 82.
6. Williams R. B., Frye H.—Anal. Chem., 1964, v. 36, p. 945—948.
7. Siska E. — Magy. Kern. Foly, 1975, v. 81, p. 201—203.
8. Pryszczewska M., Iwanowska L. — Zvesz. nauk.. Politechn. Szczecin, 1967, № 88, p. 25—30.
9. Iwanowska L. — Zvesz. nauk., PSZCZEC, 1972, № 141, p. 61—68.
10. Левитман X. Я-, Лаптева Л. E. — В кн.: Сборник статей Белорусского политехнического ин-та, 1967, № 7, с. 31—33.
11. Сонгина О. А. Амперометрическое титрование, М Химия, 1967.
12. Сонгина О. А., Войлошникова А. П. — Зав. лаб., 1956, т. 22, № 1, с. 19—23.
13. Сонгина О. А., Кемелева Н. Г. — Металлург, и хим. пром-сть Казахстана, 1962, № 4, с. 76—80.
14. Singh D., Agarvala V.—J. Sci. Ind., 1962, v. 21, p. 212.
15. Singh D., Bhatnagar U. — J. Scient. Res. Banares Hindu Univ., 1964—65, v. 15, № 2, p. 263—268; РЖХим., 1967, 1Г72.
16. Хадеев В. А., Мялковская С. Г. — В кн.: Некоторые вопросы химической технологии и физико-химического анализа. Ташкент, АН УзбССР, 1963, с. 143.
17. Хадеев В. А., Халикова Ф. X., Королева В. И. — Научн. труды Ташкентского ун-та, 1964, вып. 264, с. 3—17.
18. Prasad В. В., Khandelwal G. D.-^ Isr. J. Chem., 1972, v. 10, № 5, p. 981; РЖХим., 1973, 10Г67.
19. Хадеев В. А., Мухамеджанова Д. М.—Научн. труды Ташкентского ун-та, 1964, вып. 264, с. 18—26.
20. Сонгина О. А., Кемелева И. Г., Сушкова Л. М. — В кн.: Приящадная и теоретическая химия. Алма-Ата, 1974, вып. 5, с. 190—195.
21. Соломатин В. Т., Ржавичев С. П. —Ж. анал. хим., 1976, т. 31, № 12, с 2345_____2352
22. Усатенко Ю. И., Куценко Л. М. — Зав. лаб., 1966, т. 32, № 4, с. 398—400.
23. Куценко Л. М., Усатенко Ю. И. — Химическая технология, изд. Харьковского ун-та, 1971, вып. 20, с. 30—33.
24. Дьяченко Л. Ф., Усатенко Ю. И. — Вопросы хим. и хим. технол., 1974, вып. 34, с. 25.
25. Куценко Л. М., Аришкевич А. М„ Усатенко Ю. И. — Зав. лаб., 1969, т. 35, № 4, с. 404—407.
26. Сонгина О. А., Оспанов X. К., Китайгородская В. Я-—Ж- анал. хим., 1970, т. 25, № 3, с. 482—484.
27. Усатенко Ю. И., Куценко Л. М.—Ж. анал. хим., 1968, т. 23, № 6, с. 858— 862.
28. Берзиня В. К-, Я неон Э. Ю., Тейбе А. Я- — Изв. АН ЛатвССР, 1971, № 1, с. 108—109.
29. Захаров В. А. и др. —Ж- анал. хим., 1970, т. 25, № 5, с. 879—884.
30. Жданов А. К-, Акентьева И. А., Капица Н. В. — В кн.: Труды Ташкентского ун-та, 1968, вып. 323, с. 157—159.
Теллур
Амперометрические методы определения теллура в основном аналогичны методам определения селена, поскольку эти элементы сходны по химическим свойствам и поэтому обычно титруются совместно. Однако некоторые отличия в свойствах теллура и селена
269
Lq Таблица 41. Методы определения теллура (TeIV)
Титрант Определяемый минимум Условия определения Примечания Литература
12 1 мкг/мл Электрод Pt, <р= +0,1 В (Нас. КЭ), фон — гидрокарбонатная среда TeIV восстанавливают цинковой пылью до Те0, окисляют иодом и избыток иода оттитровывают гидразином; не мешает Se; применено для анализа кеков и селеновых концентратов 1
KI 1 мкг/мл Электрод Pt, <р= +0,1 В (Нас. КЭ), фон — 1 М НС1 Электрод Pt, <р=0 В (Нас. КЭ), фон — 0,05 М КС1+НС1 до рН=1 • TeIV восстанавливают KI, и выделившийся нод оттитровывают тиосульфатом; возможно раздельное определение TeIV и SeIV при совместном присутствии (см. раздел «Селен») К TeIV добавляют KI до концентрации 0,1 н. и образовавшийся комплекс [Tele]2- оттитровывают аскорбиновой кислотой, возможно раздельное определение TeIV из SeIV, применено для анализа стали 2 3,4
Тиооксин Ксантогенат 2 мкг/мл Электрод Pt, <р=+1,0 В (Нас. КЭ), фон 1—8 М H2SO4 [5, 6], или <р= +0,8 В (Нас. КЭ), фон ацетатный буферный раствор, pH=3,7 [6] Электрод Hg капающий, <р=—0,5 В (Нас. КЭ), фон — 4—5 М НС1 В растворах H2SO4 менее 4 М TeIV восстанавливается до Те11, который образует комплекс [Te(C9H6NS)4]2+; в растворах H2SO4 более 4 М TeIV восстанавливается до элементного; в ацетатном буферном растворе образуется осадок с отношением TeIV:R= = 1:4; возможно дифференцированное титрование TeIV и Se,v при совместном присутствии (см. «Селен») Образуется осадок с отношением TeIV : R = 1 : 4, мешает SeIV 5,6 7
Дитиобиуреты 1 мкг/мл Электрод Pt, <р=—0,1 В (Нас. КЭ), При рН=9,5—10 образуется осадок 1 8,9
фон — боратный буферный раствор с рН=9,5—10 [8] или <р=0,6 В (Нас. КЭ) и фон —6 Al НС1 [91 с отношением TeIV : R = 1:4, в 6 Al НС1 TeIV восстанавливается до элементного соотношения TeIV : R= = 1:2, мешает селен (IV)
Димеркаптотиопироны 2 мкг/мл Электрод графитовый, <р= + 0,4— —0,86 В (Нас. КЭ), фон—1—10 н. НС1 ил HjSO4 В зависимости от природы тиопирона и природы фона TeIV взаимодействует с реагентом в отношении 1:2; 1:3; 1:4; возможно дифференцированное титрование при помощи 3-метил-5-фенил-2,6-димеркапто-1.4-тиопиро-на; применено для анализа полупроводниковых материалов 10—12
Дитиокарбами-наты 1 мкг/мл Электрод Pt, <р=+0,6—1-0,8 В (Нас. КЭ), фон — ацетатный буфер-ный раствор, pH=4—6 Образуется комплекс при отноше-чии TeIV : R=l:4, возможно раздельное определение TeIV и SeIV :TeIV титруют при pH=5—6; а сумму TeIV и SeIV при рН^З.6 с использованием свинца (II) в качестве электрометрического индикатора 13—15
Дитиотриокси-метиламиномета-нат Электрод Hg капающий, <р=—0,2 В (Нас. КЭ), фои — смесь тартрата аммония и KNO3 (общая концентрация 0,4 М, pH=7,5) Образуется осадок с отношением TeIV :R=1:4, мешает SeIV 16
КМпО4 1 мкг/мл Электрод Hg капающий [17] или Pt [18. 19], <р=—0,8 В (Нас. КЭ) [17]+ +0,4 В (МИЭ [18], или +0,3 В (Нас. КЭ) [19], фон —0,1—1 М NaOH [17, 18] или 0,1—6 М Hj|SO4 [19] TeIV окисляется до TeVI, МпО4~ в щелочной среде восстанавливается до МпО2, в кислой до Мп111; в щелочной и слабокислой среде не мешает SeIV; применено для анализа кека, пыли 17—19
К2СГ2О7 Электрод Pt, <р=-1 Г,15 В (Нас. КЭ), фон — 0,5—1 М H^SO4 TeIV окисляют бих>оматом и избыток бихромата титруют солью Мора, не мешает SeIV, Си, Pb, Ag, Bi, Sbv, Asv 20
го
позволяют осуществить раздельное их определение при совместном присутствии. Так, на различии в кинетике окисления элементных теллура и селена иодом в гидрокарбонатной среде основано амперометрическое определение теллура в присутствии селена [1]. Это определение проводят следующим образом.
Ход анализа. Навеску пробы 0,1—1 г разлагают 30—40 мл концентрированной азотной кислоты при слабом нагревании, упаривают раствор до 5—8 мл, приливают 15—20 мл серной кислоты (1:1) и нагревают до выделения паров серного ангидрида. Затем раствор охлаждают, обмывают стенки колбы 15— 20 мл дистиллированной воды, кипятят и вновь охлаждают. Содержимое колбы переносят в мерную колбу емкостью 100—200 мл и доводят до метки водой. Отбирают аликвотную часть 5—10 мл в стакан для титрования, добавляют 2— 3 капли 1 % -ного ацетата свинца и при слабом нагревании прибавляют небольшими порциями цинковую пыль для восстановления теллура(1У). Добавлять цинковук пыль следует очень осторожно, добиваясь того, чтобы она полностью прореагировала в растворе. После полного осаждения теллура и растворения избытка цинковой пыли добавляют 0,1—0,2 г фторнда аммония, выжидают 30— 40 мин пока железо(П) не перейдет в железо(Ш), доводят объем раствора в стакане до 20 мл, приливают отмеренное количество 0,01 н. раствора иода, нейтрализуют раствор твердым гидрокарбонатом натрия и оттитровывают избыток иода раствором гидразина или тиосульфата. В гидрокарбонатном растворе теллур быстро взаимодействует с иодом, тогда как селен — очень медленно. Титрование проводят по току восстановления иода при потенциале платинового электрода + 0,1 В (Нас. КЭ). Метод применен для анализа кека и селенового концентрата.
Кроме иода и иодида калия [2—4] для определения теллура рекомендуются серусодержащие органические реактивы, образующие с TeIV комплексные соединения [5, 6, 10—-15], осадки [7, 9, [16], а также окислители — перманганат |[ 17—19] и бихромат [20].
Методы раздельного определения теллура и селена при их совместном присутствии рассмотрены в разделе «Селен».
ЛИТЕРАТУРА
1. Клюева Р. И. и др. — Зав. лаб., 1970, т. 36, № 7, с. 792—794.
2. Захаров В. А., Сонгина О. А., Клюева Р. И.—Зав. лаб., 1972, т. 38, № 9, с. 1066—1067.
3. Иванова 3. И., Игнатенко Е. Г., Тарасова В. А. — Ж. анал. хим., 1973, т. 28, № 10, с. 1980—1984.
4. Иванова 3. И., Игнатенко Е. Г., Тарасова В. А.— Зав. лаб., 1973, т. 39, № 10, с 1180—1182.
5. Сонгина О. А. и др. — Ж- аиал. хим., 1972, т. 27, № 6, с. 1121—1124.
6. Супрунович В. И., Величко В. В., Коган Д. С. — В кн.: Методы определения неметаллических и других вредных примесей в промышленных материалах. М„ 1977, с. 137—142.
7. Deshmukh G. S., Sankara Rao V. S. — Current Sci. (India), 1968, v. 37, №20, p. 584—585.
8. Deshmukh G. S., Nandi R. K. — Current Sci. (India), 1969, v. 38, № 21, p. 512—513.
9. Сухоручкина А. С., Постников В. А., Усатенко Ю. И. — В кн.: Вопросы химии и химической технологии. Респ. межвед темат. научно-техн. сб. 1972, вып. 26, с. 44—49.
10. Сурмий А. М., Аришкевич А. М., Усатенко Ю. И.—Труды комиссии по аналитической химии АН СССР, 1968, вып. 16, с. 114—118.
11. Волошина В. В., Усатенко К). И., Аришкевич А. М.—Зав. лаб., 1970, т. 36, № 5, с. 530—534.
272
12 Аришкевич А. М., Волошина В. В., Усатенко Ю. Я.—Укр. хим. ж., 1970, т. 36, № 5, с. 499—504.
13. Усатенко Ю. И., Тулюпа Ф. М., Баркалов В. С.—Зав. лаб., 1966, т. 23, №7, с. 787—790.
14 Тулюпа Ф. М., Баркалов В. С., Усатенко.Ю. И. — Ж. анал. хим., 1967, т. 22, Кв 2, с. 399—402.
15. Павличенко В. А., Усатенко Ю. И., Тулюпа Ф. М. — В кн.: Химическая технология. Респ. межвед. научно-техн. сб. '968, вып. 13, с. 88—91.
16. Chaudhuri Н., Chatterjee S. S., Mahanti И. S. — Technology (India), 1973, v. 10, Кв 3—4, р. 264—266.
17. Issa I. М., Issa R. M., Allam M. G.— Anal. chim. acta,' 1960, v. 23, Ks 1, p. 196—200.
18. Сонгина О. А., Тойбаев Б. К. — Изв. АН КазССР. Сер. хим., 1962, чып. 2 (22), с. 53—57.
19. Тойбаев Э. И., Сонгина О. А., Захаров В. А.-—Изв. АН КазССР, Сер. хим., 1977, Кв 2, с. 55- 59
20. Boszai Z., Hegedus Z.— Mag. Kem. Fol., 1952, v. 58, Ks 3, p. 331—339.
Титан
Для определения титана может быть использована его переменная валентность — восстановленный титан(Ш) титруют различными окислителями или комплексообразователями. Восстанавливают титан (IV) в кадмиевом редукторе [1] или электролитически [2]. После восстановления в редукторе раствор титруют бихроматом или ванадатом аммония [1]. Так как титан(III) легко окисляется кислородом воздуха, то все определение (и восстановление титана, и последующее титрование) проводят в присутствии сульфата аммония примерно в 2 н. растворе по серной кислоте. В таких условиях титан (И1) связывается в сульфатный комплекс и окисляё'гся медленно. После электролитического восстановления [2] титан (III) титруют комплексообразователями — эриохромцианином или галлоцианином, так же, как ванадий(Ш) (см. «Ванадий»), Раствор продувают азотом.
Для титрования титана(III) предложена также метиленовая синяя (см. «Олово»).
В разделе «Железо» упоминается титрование титана (III) окислителями после восстановления титана (IV) двухвалентным хромом.
Для непосредственного титрования титана (IV) восстановителями рекомендован раствор ванадия (II) при обязательном удалении кислорода [3].
Более удобно титрование титана (IV) по методу осаждения, для чего используют купферон [4—6], образующий с титаном (IV) осадок состава TiR4, и неокупферон [7], образующий с титаном еще менее растворимый осадок, чем купферон. Осадки образует также бензоилфенилгидроксиламин [8] и N-бензоил-о-тиолгидрок-силамин [9], осаждающий также и цирконий (купферон также реагирует с цирконием).
Титан (IV) образует достаточно устойчивые комплексонаты, что позволяет титровать его раствором ЭДТА с ртутным капающим
18—1694
273
электродом, используя ток восстановления титана (IV) при <р = =—0,22 В (Нас. КЭ) [10]. Рекомендуется обратное титрование избытка ЭДТА [11]. ।
ЛИТЕРАТУРА
1. Пешкова В. М., Галлай 3. А. — Вестник МГУ, 1954, т. 9, № 10, вып. 7, с. 73.
2. Церковницкая И. А., Перевощикова В. В. — Ж. анал. хим., 1973, т. 28, № 1, с. 82—85.
3. Галлай 3. А., Марьяновская Т. Я.—Ж. анал. хим., 1963, т. 18, №8, с. 924— 928.
4. Усатенко Ю. И., Беклешова Г. Е.—Зав. лаб., 1955, т. 21, № 8, с. 779.
5. Усатенко Ю. И. и др. — Зав. лаб., 1956, т. 22, № 6, с. 528.
6. Elving Ph., Olson Е. — Anal. Chem., 1955, v. 27, p. 1817—1820. ,
Галлай 3. А. и dp. —Ж. анал. хим., 1964, т. 19, № 8, с. 1464.
8. Галлай 3. А., Алимарин И. П.—Ж- анал. хим., 1963, т. 18, № 8, с. 1442.
9. Галлай 3. А. и др.—Ж. анал. хим., 1970, т. 25, № 10, с. 1851—1854.
10. Takao X., Муса С. — Japan Analyst, 1961, v. 10, р. 160; РЖХим., 1961, р. 1863.
11. Goldstein G., Manning D. L., Zittel H. E. — Anal. Chem., 1963, v. 35, p. 17—32.
Торий '
Амперометрическое определение тория возможно только по методу осаждения или комплексообразования. Лучше всего разработано титрование раствором молибдата аммония, которое позволяет определять торий(1У) в присутствии урана (VI) [1], ионов редкоземельных элементов [2] (анализ монацитового песка), и в других объектах [3, 4]. Титрование фторидом [5] представляет собой «обратный» метод определения фторида при помощи солей тория. Можно также титровать торий (IV) по методу осаждения селенитом натрия [6], тороном [7], перйодатом [8], купфероном [9]. Известен метод титрования тория (IV) ферроцианидом калия или щавелевой кислотой [10] и осаждение в виде ThO2-WO2 [11], однако в этих случаях мешают ионы, реагирующие с ферроцианидом и вольфраматом.
Все остальные методы амперометрического определения тория связаны с применением ЭДТА, с которым торий образует весьма прочное комплексное соединение (р/С=23,2). Известно несколько вариантов этого метода. В 1960 г. было предложено [12] прямое титрование тория (IV) раствором ЭДТА с двумя платиновыми электродами при внешнем напряжении 0,4—0,5 В при pH ^5.
Ход анализа. Монацитовый песок (5 г) разлагают серной кислотой, как обычно, и отделяют торий вместе с редкоземельными элементами при помощи щавелевой кислоты. Прокаленный осадок растворяют в соляной кислоте (1 : 1), добавляют перекись водорода для восстановления церия (IV) и празеодима(IV), переносят раствор в мерную колбу емкостью 100 мл и доводят объем до метки водой. Берут аликвотные части по 1—3 мл, создают pH раствора 2—3 (при помощи аммиака), доводят объем до 10—20 мл водой и титруют 0,06 М раствором ЭДТА.
274
Рис. 47. Кривая титрования тория (IV) раствором ЭДТА в присутствии индикатора — железа (II) и железа (П1).
При другом способе с двумя электродами [13] используют соли железа (Ш) как амперометрический
индикатор
Ход анализа. В сосуд для титрования вносят сначала ацетатный буферный раствор, на фоне которого ведут титрование, затем вводят 0,02 мл 0,1 М раствора сульфата Железа(III) и 0,06 мл 0,03 М раствора ЭДТА для переведения почти всего железа(III) в комплексное соединение. Затем добавляют 0,2 мл 0,1 М раствора соли Мора и налагают напряжение ЦО мВ. При таком напряжении ток в системе не появляется, так как разность потенциалов восстановление комплексоната железа(Ш) и окисления ионов железа(П) больше, чем приложенное напряжение, и процесс электролиза начаться не может. Начинают титрование 0,05 М раствором ЭДТА, причем после добавления нескольких его ка-пел1 сила тока резко возрастает (начиная с точки б на рис. 47), потому что ЭДТА связал остатки Fe3+ и начал взаимодействовать с Fe2+. В результате появляется «пара»: комплексонат железа(Ш)—комплексонат железа(П), которая вполне обеспечивает катодно-анодный процесс при напряжении 110 мВ. В это время вводят аликвотную часть исследуемого раствора (содержание тория в нем должно составлять приблизительно 1—10 мг), после чего ток резко падает: торий «отнимает» ЭДТА у железа(П), анодная составляющая пары исчезает, ток превращается (точка г). Затем начинают титровать торий(IV) ЭДТА до резкого возрастания тока после конечной точки (точка <Э); избыток ЭДТА вновь связывает железо (II) и, следовательно, вновь появляется анодная составляющая электродного процесса.
Для приготовления исследуемого раствора навеску разлагают азотной кислотой (1:1) при нагревании, выделяют, как обычно, свинец в виде сульфата, отфильтровывают осадок, фильтрат и промывные воды упаривают до объема приблизительно 15 мл, добавляют кристаллический гидроксиламин (для связывания урана, если он присутствует в пробе) и нагревают. Затем устанавливают рН«1, переводят раствор В мерную колбу емкостью 25 мл и доводят до метки водой. Аликвотную часть титруют, как описано выше.
Этот оригинальный метод был применен для определения тория в ураните [13].
В качестве амперометрического индикатора применяют также соли меди(П) '[14] и перманганат калия [15]. При титровании без индикатора налагают очень большое напряжение, порядка 1,5 В [16]. Известны также методы титрования при помощи ЭДТА с одним индикаторным электродом [17, 18]. Авторы работы [19] вернулись к методу Флашке [20], т. е. к методу, заключающемуся в вытеснении ионами тория ионов другого элемента из его комплексоната, менее прочного, чем комплексонат тория, в частности ионов свинца [20] и висмута [19].
Об определении тория при помощи ЭДТА см. также в разделах «Лантан», «Индий» и «Висмут».
18* 275
ЛИТЕРАТУРА
1. Gordon L., Stine R.—Anal. Chem., 1953, v. 25, p. 192.
2. Gordon L., Vanselow C., Willard H.—Anal. Chem., 1949, v. 21, p. 1323.
3. Burastero i., Martres R. W. — Anal. Chem., 1962, v. 34, p. 378.
4. Saxena R. S., Mittal ML. — Naturwis., 1963, v. 50, p. 373.
5. Sundaresan M., Karkhanavala M. — Current Sci. India, 1954, v. 3, p. 258.
6. Deshmukh G. S., Asthana О. P. — Z. anal. Chem., 1962, Bd. 187, S. 81.
7. Церковницкая И. A., E. Жу-цзю— Вестник ЛГУ; 1963, № 16, с. 135.
8. Алимарин И. П., Ченг Я. А., Пуздоренкова И. В. — Вестник МГУ, Сер. мат., механ., астрон., физ. и хим., 1958, № 13, с. 201.
9. Василенко В. Д., Островская Э. Б,-—Ж- анал. хим., 1961, т. 16, № 3, с. 433. 10. Хадеев В. А., Масалова Л. С. — Труды Ташкентского ун-та, 1967, вып. 288, с. 3—11.
11. Gupta С. М. — Bull. Chem. Soc. Japan, 1966, v.' 39, № 4, p. 837—838; РЖХим., 1966, 22Г77. .
12. Палей П. H., Удальцова Н. И. — Труды Комиссии по аналитической химии АН СССР, XI, 1960, с. 299.
13. Церковницкая И. А., Е. Жу-цзю— Вестиик ЛГУ, 1963, № 22, с. 168.
14. Dean 3. R., Harris W. Е. — Anal. Letters, 1969 v. 2, № 2, p. 93—97.
15. Хадеев В. А., Маслова Л. С.— Труды Ташкентского ун-та, 1967, вып. 288, с. 12—17.
16. Vydra F., Vorlicek J. — Coll. Czech. Chem. Comm., 1966, v. 31, № 1, p. 51—57. 17. Freese F., Oostervink R., Boef G. — Taianta, 1971, v. 18, № 10, p. 1064—1066. 18. Жданов А. К., Умарова H. M.—Труды Ташкентского ун-та, 1964, вып. 264, с. 40—42.
19. Хадеев В. А., Кочергина С. А.—Узб. хим. ж., 1967, № 6, с. 15—18.
20. Flaschka Н., Barakat М. F. — Z. anal. Chem., 1957, Bd. 156, S. 321.
Уран
Определение урана может быть основано йак на реакциях осаждения, так и на окислительно-восстановительных процессах. Очень подробно исследовано [1, 2] осаждение урана (VI) ферроцианидом калия и разработан соответствующий метод [2, 3]. Рекомендована также фениларсоновая кислота [4], восстанавливающаяся, как и уранил-ион, на ртутном капающем электроде. Известны другие методы титрования уранила, основанные на осаждении: осаждение теллуратом натрия [5], 2, 3, 4-триоксихальконом [6]1, ванадатом натрия [7].
Уран (VI) можно титровать по методу окисления-восстановления двумя способами: прямое титрование урана (VI) восстановителями и титрование урана (IV). Для прямого титрования нужны сильные восстановители, так как нормальный потенциал системы уран(У1)/уран(1У) составляет всего +0,334 В. К таким восстановителям относятся титан(Ш) [8] и хром(П) [9], предложенные также для титрования ванадия(V) (см. «Ванадий»), и соль Мора в сильной фосфорно-кислой среде, снижающей редокс-потенциал системы ферри-ферро [10].
Для предварительного восстановления урана (VI) применяют редукторы висмутовый, кадмиевый, цинковый (так называемый редуктор Джонса) или электролитический метод восстановления. Последний способ предпочтительнее потому, что в раствор соли урана не вносятся посторонние ионы. В качестве окислителей при
276
меняют перманганат [11, 12], церий(ГУ) [11, 13], железо (III) [10, 13, 14], ванадат аммония [13, 15, 16]. В зависимости от способа восстановления уран может находиться в растворе либо только в виде урана (IV), либо вместе с ураном (III)., как, например, в случае применения редуктора Джонса.
Уран(III) и уран(IV) титруют [13] отдельно раствором сульфата железа (III) при —0,05 В (Нас. КЭ) с платиновым электродом на фоне 0,3 н. серной кислоты, при этом до первой конечной точки наблюдается анодный ток — по-видимому, анодное окисление урана (III)—затем во время титрования урана (IV) ток остается практически постоянным и резко возрастает' после конечной точки вследствие восстановления Fe3+. Авторы работы [14] подчеркивают, что успех этого титрования зависит от того, насколько полно будет удален растворенный кислород, который окисляет не только уран(III), но в той или иной степени и уран(IV), особенно если кислотность раствора понижается. Титровать можно от 5 до 200 мг урана с относительной погрешностью 0,1—0,4%. Титрованию мешают ванадий (V) и молибден^У!), так как при восстановлении урана^!) они также восстанавливаются, а затем окисляются желе-зом’(Ш) вместе с восстановленным ураном.
Титровать уран(IV) можно с одним и с двумя индикаторными электродами. Сравнительное изучение различных электрометрических способов определения конечной точки показало, что метод с двумя электродами дает наилучшие результаты [13, 16].
В разделе «Железо» описан метод восстановления железа (III) и других элементов, в том числе урана (VI) солями хрома (II) и последующего титрования урана (IV) растворами бихромата калия или сульфата церия [17]. ,
Для определения урана применяют также реакции комплексообразования. Изучено титрование урана (VI) [18] раствором ЭДТА по методу с двумя индикаторными -электродами или с одним по току окисления избытка ЭДТА. Использован [19] также «индикаторный» метод: индикатором служат ионы ртути(II), обусловливающие начальный ток и достаточно резкую конечную точку после того, как оттитрован весь уран (IV). Этот метод основан на большой разнице в прочности комплексонатов урана(IV) и рту-th(V)—рА равно соответственно 26,1 и 21,8. Определять можно доли микрограмма урана, титруя разбавленными растворами ЭДТА вплоть до 10-6 7И.
Ход анализа. К 25—50 мл анализируемого раствора, содержащего 10-3— Ю-® г урана(У1), добавляют серною кислоту до рН=2 по универсальному индикатору. Добавляют 1—1,5 мл раствора формамидинсульфоновой кислоты (1 г на 150 мл 0,25 н. серной кислоты) для восстановления урана(У1) до ураиа(1У) и выпаривают на плитке примерно до 5 мл. Разбавляют в эдой вдвое, охлаждают до комнатной температуры, доводят объем примерно до 25 мл 0,2 М раствором нитрата аммония. Затем вводят 0,3—0,5 мл 0,0001 М раствора нитрата ртути (II) и титруют раствором ЭДТА соответствующей концентрации при потенциале графитового электрода +0,2 В (МИЭ). Концентрация раствора ЭДТА выбирается в соответствии с предполагаемым содержанием урана: при содер-’ жании урана более 1 мг в титруемом объеме применяют 10-2 М раствор
277
ЭДТА, при 10—‘100 м.<г используют 10~3 М, при ,1—10 мкг применяют 10~4 М и при содержании урана менее 1 мкг—10~s или 10~6 М растворы. Применение графитового электрода с относительно большой рабочей поверхностью (торец стержня для спектрального анализа) позволило повысить чувствительность определенья до указанных выше пределов.
Рекомендовано также прямое титрование урана (VI) раствором ЭДТА в присутствии других элементов [20].
’Аскорбиновая кислота, будучи по отношению к урану(VI) недостаточно сильным восстановителем, реагирует с ним в отрешении 1:1, образуя комплексное соединение, что позволяет применять ее'для прямого титрования урана (VI) [21].
Известно также осаждение урана(VI) аммиаком в виде диураната аммония (NH4)2U2O7 с последующим растворением осадка [22] в насыщенном растворе гидрокарбоната натрия, с добавкой 0,5 г бромида калия и титрованием раствором гипохлорита кальция Са(ОС1)2 с платиновым вращающимся электродом без наложения внешнего напряжения (Нас. КЭ).
Метод интересен тем, что позволяет определять уран (VI) в присутствии некоторых других ионов, в частности титана (IV) и бериллия (П). Только в случае, если эти катионы присутствуют в очень больших количествах, они могут затруднить отмывание ионов аммония от осадка, поскольку они также выпадут в осадок. Мешает титрованию ванадий (V), который частично осаждается в виде метаванадата аммония; последний растворяется в гидрокарбонате и является причиной завышения результатов.
Уран (VI) можно также титровать [23] в неводной среде при помощи органииеских реактивов с ртутным капающим электродом.
ЛИТЕРАТУРА
1. Клейбс Г. А.—Ж. неорган. хим., 1956, т, 1, с. 2178, 2198; 1958, т. 3, с. 2621.
2. Сочеванов В. Г„ Шмакова И. В., Волкова Г. А. — Ж. неорган. хим., 1957, т. |2, с. 20-9, Ж- анал. хим., 1960, т. 15, № 1, с. 77.
3. Almagro V. — At,, quim. Real. Soc. esp. fis. у quim., 1969, v. 65, №4, p. 405'— 410; РЖХим., 1972. 4Г106.
4. Rolthoff I. M., Tohnson R. A.— J. Electrochem. Soc., 1951, v. 98, p. 138—142.
5. Mittal M. L., Saxena R. S. — Indian J. Appl. Chem., 1967, v. 30, № 3—4, p. 75—79- РЖХим., 1968, 20Г23.
6. Syamasundar K. — Cflrrent Sci. (Indian), 1967, v. 36, №22, p. 6’07-^608; РЖХИ1 , 1968, 14Г87.
7. Mittal M L., Saxena R. S. — Experientia, 1965, v. 21, № 8, p. 481—482; РЖХим., 1966, 5Г89.
8. Галлай 3. А., Шеина И. В. — Ж. анал. хим., 1961, т. 16, № 4, с. 706.
9. Галлаи 3. А., Каланчу к Г. Е.—Ж. анал. хим., 1961, т. 16, № 1, с 63
10. Garcia Е Е., Botbol М., Casabe I. — Inform. Comis. пас. energia atom., 19C8, № 241, 11 p. (исп.); РЖХим., 1968, 20Г118.
11. Silvermann И. P., Skoog D. A. — Anal. Chem., 1963, v. 35, p. 131—133.
12. Singh D., Sharma Sh. — Ind. J. Chem., 1970, v. 8, № 2, p. 192—195; РЖХим., 1970, 22Г42.
13. Удалыюва H. И. — Ж- анал. хим., 1962, т. 17, № 5, с. 476.
278 »
14 Sympson R. F., Larsen R. P., Meyer R. J., Oldham R. D. — Anal. Chem., ’ 1965, v. 37, p. 58.
15 . Ескевич В. Ф., Комарова Л. А.—Ж- анал. хим., 1960, т. 15, № 1, с. 84.
16 Агасян П. К., Николаева Е. С., Демина Л. А. — Зав. лаб., 1964, т. 30, № 12, ‘ с. 1434.
17 . Хадеев В. А. — РЖХим., 1976, ЗГ111, депон. в ВИНИТИ.
18 . Палей П. И., Удальцова Н. И. — Ж- анал. хим., 1960, т. 5, № 11, с. 2211.
19 Рождественская 3. Б, Сонгина О. А., Вариков В. Г. — Зав. лаб., 1963, т. 29, ’ № 1, с. 30.
20 Хадеев В. А., Зоренко Л. П. — Труды Ташкентского ун-та, 1973, вып. 435, с. 20—23.
21 Singh D., Varma А. — J. Sci. Res. Banaras Hindu Univ., 1962—1963, v. 13, p. 313.
22 . Sekerka I., Vorlicek J. — Chem. Listy, 1953, v. 47, p. 572.
23 . Церковницкая И. А., Быховцева T. T.—Ж. анал. хим., 1964, т. 196, Ks 3, с. 574.
Фосфор
Длительность и трудоемкость гравиметрического определения фосфора пос'лужили причиной многочисленных попыток разработать амперометрический метод его определения. Для этой цели рекомендовали соли свинца [1, 2] и железа [3, 41, однако эти методы не получили практического применения, по всей вероятности в связи с тем, что состав осадка недостаточно постоянен и сильно зависит от pH раствора. При титровании солями свинца, кроме того, мешают сульфаты и хлориды. Гипофосфит (анион фосфорноватой кислоты) осаждают в виде PbsPsOe в водно-спиртовой среде (10— 25% спирта по объему). Этим же способом титровали фосфат-ион и при анализе фосфорно-никелевых сплавов [5]. Метод титрования солями железа(III) недавно был вновь применен для определения фосфат-ионов [6]. Титруют фосфаты также раствором ванадила [7], нитратом ртути(I) [8] и уранил-ацетатом, образующим осадок состава KUO2PO4, отличающийся малой растворимостью и постоянством состава [9—11].
Малорастворимые осадки с фосфат-ионом дает, как известно, *- висмут(Ш). Перхлорат висмута(Ш) был предложен для титрования фосфата при помощи ртутного капающего электрода [12]. Впоследствии вместо ртутного был применен платиновый электрод [13] и показана возможность титровать не только перхлоратом, но и нитратом висмута (III). Состав осадка зависит от кислотности раствора, титрование следует проводить при pH=g:l,5, т. е. при сине-зеленой окраске метилового фиолетового. В работах [14, 15] предложен, как наилучший, фон состава азотная кислота+нитрат-1-гл икоколь. Подробное изучение процесса взаимодействия фосфата с висмутом(III) при различной кислотности раствора [16, 17] показало, что наиболее близкие к теоретическим результаты получаются при рН=1,1; при рН<0,9 начинается растворение осадка, при рН>1,4 — образование основных солей. Было установлено также, что большое влияние на ход титрования имеют соли железа(III) [16] вследствие образования не только фосфатных
279
комплексов с самим железом(III), но и сложных комплексов железо— висмут — фосфат. Наличие железа(Ш) поэтому замедляет осаждение фосфата висмута, а при недостаточной кислотности вообще парализует его- выпадение. Вследствие этого железо(III) следует перед титрованием восстановить до железа (И) аскорбиновой кислотой. Хлорид- и сульфат-ионы также не должны присутствовать в растворе, поэтому навеску удобрений (аммофос, суперфосфат, двойной суперфосфат и др.) разлагают азотной кислотой, удаляя сульфат-ион осаждением нитратом бария. Ионы фтора, часто присутствующие в природных фосфатах, не мешают [17].
Титрование нитратом висмута хорошо идет с висмутовым индикаторным электродом, поскольку поверхность этого электрода не изменяет свою природу во время титрования, тогда как на плати-' новом электроде выделяется металлический висмут, что приводит к изменению хода некоторых электродных процессов [18]. Пригодность метода подтверждена результатами анализа минеральных удобрений [13, 17].
Для определения фосфора в триполифосфате предложен метод титрования его раствором триэтилендиаминкобальта(Ш) по току его восстановления на ртутном капающем электроде [19], а определять пирофосфат в присутствии орто- и триполифосфата можно при помощи нитрата кадмия в присутствии этанола для понижения растворимости осадка {20].
Для определения фосфора в виде гексафторфосфата предложено титрование раствором хлорида тетрафениларсония [21] на хло-ридно-аммиачном фоне с pH=9 по току вэсстановления реактива с ртутным капающим электродом при —1,7 В (Нас. КЭ). Можно такжЪ воспользоваться для определения фосфора (V) образованием фосфорно-молибденовой кислоты [22], однако этот метод очень сложен и сопровождается экстракцией, многократными промываниями и фильтрованиями, причем мышьяк надо удалять возгонкой, а ванадий (V) восстанавливать до низшей валентности, чтобы он не участвовал в образовании гетерополикислоты. По нашему мнению, такой способ вряд ли может получить практическое применение не только вследствие исключительной громоздкости, но и потому, что точность его весьма сомнительна.
Несколько проще современные варианты этого метода [23, 24]. В одном из них [23] молибден (VI), прямо титруют ферроценом на водно-ацетоно-изобутанольном фоне, 6 М по НС1. Однако образованием фосфорно-молибденовой гетерополикислоты можно воспользоваться и иначе, а именно непосредственно титровать фос-фат-ион в присутствии молибдата аммония раствором диантипи-рилметана (ДАМ), соблюдая определенную кислотность фона [25].
Мышьяк (V) мешает этому титрованию, так как его гетерополикислота образуется в тех же условиях, но влияния кремния можно избежать, придерживаясь указанной в методике кислотности раствора.
280
Ход анализа. Навеску стали 1 г растворяют в 30 мл азотной кислоты (1 : 1), прибавляют 5 мл 4%-ного раствора перманганата и кипятят до выпадения бурого осадка МпО(ОН)2. Не прекращая нагревания, прибавляют по каплям 10%-ный раствор сульфата железа(П) до полного растворения осадка. Большого избытка сульфата железа(П) следует избегать. После растворения осадка раствор кипятят еще 2—3 мин, охлаждают, переводят в мерную колбу емкостью 100 мл и доводят до метки 3—4 и. серной кислотой. Отбирают аликвотную часть 10 мл, прибавляют к ней 5—10 мл 5%-ного водного раствора молибдата аммония и титруют раствором ДАМ. Реактив прибавляют через каждые ЗС с. Тятр раствора ДАМ устанавливают по стандартному образцу.
Аналогичный способ применяют [25] и для определения кремния (см. «Кремний»).
ЛИТЕРАТУРА
1. Ляликов Ю. С.—Ж. анал. хим., 1946, т. 1, № 1, с. 147.
2. Торопова В. Ф., Яковлева Г. С.—Ж. анал. хим., 1946, т. 1, № 2, с. 290.
3. Сайкина М. К-, Торопова В. Ф. — Труды комиссии по аналитической химии АН СССР, 1952, IV (VII), с. 141. .
4. Новак В. Н., Богдвина В. И., Мальцев В. Ф. — Зав. лаб., 1965, т. 31, А" 2, с. 159.
5. Байменова А. Т. — В кн.: Материалы XXII научи, конф, проф.-преп. состава Казахстанского педагогического ин-та им. Абая, Алма-Ата, 1969, с. 176.
6. Bixler J. W., Colwell L. F. — Anal. chim. acta, 1976, v. 85, № 1, p. 185—188.
7. Золотавин В. Л., Кузнецова В. К. — Зав. лаб., 1955, т. 21, № 11, 'с. 1255— 1258.
8. Румянцева Л. С., Теодорович И. Л.—ДАН УзбССР, 1967, № 11, с. 35—36. 9. Cogbill Е., White Susana С. — Analyt. Chem., 1955, v. 27, p. 1955.
10. Kolthoff I. M., Cohn G. — Ind. Eng. Chem., Anal. Ed., 1942, v. 14, p. 1942.
11. Човнык H. Г., Векслина В. A.— В кн.: Методы контроля химического состава неорганических и органических соединений. Куйбышев, 1966, с. 59—67.
12. Neuberger А.—Z. anal. Chem., 1939, Bd. 116, S. 1.
13. Сонгина О. А., Войлошникова А. П., Козловский М. Г.— Изв. АН КазССР. Сер. хим., 1963, вып. 5, с. 3—13.
14. Теодорович И. Л., Румянцева Л. С.—Зав. лаб., 1966, т. 32, № 11, с. 1134.
15. Румянцева Л. С. — В кн.: Физико-химические исследования глинистых. минералов и силикатных материалов. Ташкент, ФАН, 1970, с. ИЗ—114.
16. Лепин Я. К, Козулина М. М., Сонгина О. А. — В кн.: Химия и химическая технология. Алма-Ата, 1972, вып. 13, с 152—156.
17. Лепин Я. К, Козулина М. М., Сонгина О. А.—Зав. лаб., 1973, т. 39, № 3, с. 283—284.
18. Козулина М. М., Лепин Я. К., Сонгина О. А. — В ки.: Химия и химическая технология. Алма-Ата, 1975, вып. 17,. с. 122—127.
19. Омаркулова Г. О., Сонгина О. А., Фрезе Н. А. — Зав.,лаб., 1970, т. 36, № 1, с. 20—21.
20. Сонгина О. А., Близнюк В. М., Омаркулова Г. О.—Зав. лаб., 1971, т. 37, № 3, с. 271—273.
21. Affsprung Н. Е., Atcher V. S. — Analyt. Chem., 1963, v. 35, p. 976.
22. Chlebovsky T. — Hutn. Listy, 1958, v. 13, p. 252.
23. Соломатин В. T., Артемова Т. И., Яковлева П. Я- — Ж. анал. хим., 1974, т. 29, № 3, с. 525—528.
24. Шафран И. Г., Розенблюм В. П. — Труды ВсесоюзНИИ хим. реактивов и особо чистых веществ, 1971, т. 33, с. 179.
25. Луговой С. В., Рязанов И. П. — Зав. лаб., 1967, т. 33, № 6, с. 638—692.
281
Фтор
Амперометрическое определение фтора основано на малой растворимости фторида тория [1—6]. Титруют нитратом тория с ртутным капающим электродом при —1,7 В (Нас. КЭ), причем интересно то, что измеряемая после конечной точки сила тока обусловлена, по всей вероятности, не ионами тория, а нитрат-ионами, которые могут восстанавливаться на ртутном капающем электроде только в присутствии ионов многовалентных элементов и, в частности, тория >[7]. Можно сначала добавить к титруемому раствору соли железа (III) и затем титровать с палладиевым электродом раствором соли тория(IV), который вытесняет железо(III) из его комплекса с фторидом. По мере титрования освобождающееся железо (III) восстанавливается на электроде, ток возрастает до конечной точки, после чего остается постоянным [6].
Титрование растворами солей тория можно проводить также с двумя электродами, в частности с парой алюминий — платина [5] или с двумя алюминиевыми [8] без наложения напряжения (см. ниже).
Соли свинца также образуют трудно растворимые осадки с фторидом [9—11].
Описан способ определения фтора, основанный на его осаждении хлоридом кальция с последующим титрованием избытка кальция ферроцианидом [12].
Подробно изучено [13—19] титрование фторида солями железа (III). Результат титрования в значительной степени зависит от ряда взаимосвязанных условий: образования феррифторидных комплексов разного состава при различном pH и составе среды, зависимости хода титрования от температуры и от наличия примесей; титрованию мешает алюминий и многие другие катионы. Однако авторы метода [16. 17] использовали его для определения фторида в травильных ваннах.
Ход анализа. В мерную колбу емкостью 100 мл помещают 5 мл электролита и доводят до метки водой. Берут аликвотную часть (5 мл), .добавляют 5 мл этилового спирта, вводят 1—2 г чистого кристаллического хлорида натрия в устанавливают рН=5—6 по универсальному индикатору 2%-ной азотиой кислотой или 2%-ным раствором едкого натра. Общий объем титруемого раствора должен составлять примерно 20 мл. Титруют из микробюретки раствором хлорида железа(Ш), содержащим +01 г железа(Ш) в 1 л раствора. Потенциал платинового электрода +0,5 В (НВЭ).
Описанный метод применяют для определения свободных фто-рид-ионов. Так как в травильных .ваннах могут присутствовать и связанные ионы фтора, в частности фториды железа(Ш), то сначала осаждают гидроксиды раствором едкого натра из нагретого до 50—70 °C раствора, и в фильтрате определяют фторид-ион, как указано выше.
Для определения фтора применяют также алюминиёвый элект род [17, 20] Кольтгоф и Самбучетти [21] подробно изучили осо
282
бенности работы этого электрода и установили, что при потенциале —0,75 В (Нас. КЭ) или при работе без наложения внешнего напряжения, но с электродом сравнения, имеющим потенциал такого же порядка (амальгама кадмия, Е=—0,77 В), ток окисления алюминия в присутствии фторида пропорционален концентрации последнего. Это явление было использовано для амперометрического титрования [22].
В растворе, содержащем фторид-ион в концентрации порядка 10~3—10~4 М, сначала измеряют ток окисления на вращающемся алюминиевом электроде при —0,75 В (Нас КЭ) или при указанном выше электроде сравнения; раствор должен иметь pH «4 (ацетатный буфер) и содержать примерно 50% спирта и некоторое количество цитрата калия или натрия (концентрация нитрата щелочного металла должна быть примерно 0,5 Л1) для того, чтобы образующееся соединение (Na2AlF6 или KsA'Fe) имело постоянный состав. Пропускают азот для удаления растворенного кислорода и затем тнтруют 0,01 М раствором штрата алюминия, продувая раствор азотом после каждого добавления реактива. Кривая титрования имеет форму а.
Указанные выше условия следует соблюдать строго, иначе кривая получается размытой и конечная точка трудно определима, так как по ходу титрования могут образовываться комплексные фториды алюминия другого состава. Все факторы, обусловливающие успешное осуществление этого метода, очень подробно обсуждены в литературе [21, 22, 23].
Бейерманн [24] также определяет фторид при помощи алюминиевого электрода, но основывает это определение на несколько ином принципе. Он измеряет прирост тока между алюминиевым и платиновым (или тоже алюминиевым) электродами при введении фторид-ионов в раствор фона. Содержание фтора находят по калибровочному графику.
Можно упомянуть также о титровании [25] фторида растворами перхлората или хлорида урана (IV). Метод основан на образовании комплексного соединения типа Нг^СЬБг]. Титруют на фоне Формиатного буферного раствора при pH=3,5 с капающим ртутным электродом при +0,1 В (Нас. КЭ) по анодному току урана (VI), кривая титрования имеет форму б. Титровать следует в отсутствие кислорода воздуха во избежание окисления урана (IV). Определять можно от 0,04 до 2,2 мг фторида.
ЛИТЕРАТУРА
1. Langer А. — Ind. Eng. Chem., Anal. Ed., 1940, v. 12, p. 511.
2. Лузина Г. С. —Зав. лаб., 1949, т. 15, № 12, с. 1412.
3. d’Amore G., Faraone G.— Ann. chim., Roma, 1957, v. 47, p. 142
4. Benesi H. A. e. a. — Amperometric Titratios, New York, 1965, p. 305.
5. JohannessonL. K- — Chem a.. Ind., London, 1957, p. 480.
6. Harris W. E. — Anal. Chem., 1958, v. 30, p. 1000.
7. Гейровский Я. Техника полярографического исследования. М., Издатинлит,
8. Gaal F. F., Jovanotic L. S., Canic V. S. — Z. anal. Chem., 1974, Bd. 272, Гв 2, S. 117—120.
9. Талипов Ш. T., Хадеев В. А.— Зав. лаб., 1953, т. 19, № 11, с. 1145.
283
10. Haul R., Griess W. — Z. anorg. allg. Chem., 1949, Bd. 259, S. 42.
11. Petrow H., Nash L. — Anal. Chem., 1950, v. 22, p. 1274.
12. Сонгина О. А., Войлошникова А. П., Козловский M. T.— Изв. АН КазССР. Сер. хим., 1953, вып. 6, с. 69.
13. Herder 1. J., van Pinxteren J. A. C. — Pharm. Weekblad, 1958, v. 93, p. 1013. 14. Aarts E. M.— Pharm. Weekblad, 1961, v. 96, p. 373.
15. Kadic K, Resac Z.— Chem. Listy, 1955, v. 49, p. 570.
16. Мальцев В. Ф., Новак В. П. —Зав. лаб., 1959, т. 25, № 11, с. 1296.
17. Мальцев В. Ф. — В кн.: Производство труб. Вып. 12. М., Металлургия, 1964, с. 129.
18. Новак В П., Резник Б. Е., Мальцев В. Ф.—Ж. анал. хим., 1967, т. 22, №4, с 608—640.
19. Кузьмина Н. Н„ Рунтов В. И., Ярцев М. Г. — В кн.: Методы контроля хи мического состава неорганических и органических соединений. Куйбышев, 1966, с. 15—19.
20. Baker В. В., Morrison J. D.— Anal. Chem., 1955, v. 27, p. 1307.
21. Kolthoff I. M, Sambucetti C. J.— Anal. chim. acta, 1960, v. 22, p. 253.
22. Kolthoff I. M., Meehan E. J., Sambucetti С. I.—Anal. chim. acta, 1960, v. 22, p. 351.
23. Алемаскина Г. А., Човнык H. Г. — Труды Куйбышевского медицинского ин-та, XXIII, 1966, с. 28.
24 Веуегтмп К —Z. anal. Chem., 1963, Bd. 194, S. 1.
25. McEwen D. DeVries Th. — Anal. Chem., 1959, v. 31, p. 1347.
Хлор
«Классической» реакцией на хлорид-ион является осаждение его в виде хлорида серебра. Эта же реакция лежит в основе амперометрического определения хлорида, а также бромида и иодида. Титрование проводят с платиновым вращающимся электродом, чаще всего без наложения внешнего напряжения с меркур-иодидным или насыщенным каломельным электродом сравнения.
Как известно, произведения растворимости хлорида, бромида и иодида серебра равны соответственно 10-10, 10~13, 10-16. Большая разница между этими величинами позволяет определять амперометрически все три галогенида последовательно, если они совместно присутствует в растворе. Для того чтобы переломы на кривой титрования были более отчетливы, в процессе титрования изменяют среду. Сначала добавляют аммиак — около 2 мл концентрированного раствора аммиака на 50 мл титруемого раствора. В аммиачной среде бромид и особенно хлорид серебра, как известно, растворимы, тогда как иодид серебра в аммиаке практически не растворяется; таким образом, устраняется одновременное осаждение других галогенидов серебра [1]. Вследствие малой растворимости иодида серебра во время титрования наблюдается лишь очень незначительный остаточный ток (этот ток пропорционален концентрации серебра над осадком, т. е. определяется произведением растворимости иодида серебра). Когда весь иодид-ион оттит-руется, сила тока начнет возрастать за счрт образования осадка бромида серебра, произведение растворимости которого на трипорядка выше произведения растворимости иодида. В этот момент титрование приостанавливают и нейтрализуют раствор азотной
284
' Рис. 48. Кривая последовательного титрования смеси ионов иодида, бромида и хлорида раствором нитрата серебра.
хлорид. Роль желатины со-
кислотой. В нейтральном или слабокислом растворе хорошо осаждается бромид серебра. После того как бромид будет оттитрован, в раствор вводят желатину и продолжают титрование; теперь уже титруется стоит в том, чтобы препятствовать непосредственному восстановлению серебра из его хлорида [2, 3].
На рис. 48 приведена ступенчатая кривая титрования всех трех галогенидов при совместном их присутствии. Четкость перехода от иодида к бромиду почти всегда вполне удовлетворительная, но переход от бромида к хлориду выражен менее четко, особенно если содержание хлора превышает содержание брома. Известно, что галогениды серебра, и особенно бромиды и хлориды, образуют смешанные кристаллы. Если же бромид отсутствует, то раздельное титрование иодида и хлорида проходит очень четко. Этот метод может быть использован для определения иодид- и хлорид-ионов в природных и иных водах, солях и промышленных растворах.
При титровании галогенидов раствором нитрата серебра следует помнить, что растворимость галогенидов серебра и, в частности, хлорида серебра очень сильно зависит от температуры. Растворимость AgCl меняется с повышением температуры следующим образом [2]: •
Температура, °C . ....................... 5 15 -25 35 48
Произведение растворимости, (ПР-1010) . 0,26 0,71 1,78 4,14 8,97
На основании специально поставленных опытов [2] рекомендуется титровать малые количества хлорид-иона при температуре не выше +5 °C (цри дальнейшем понижении температуры растворимость меняется уже незначительно). Эти указания имеют большое практическое значение, особенно для лабораторий, расположенных в южных районах. Нам в нашей практике (г. Алма-Ата) не раз приходилось наблюдать резкое ухудшение результатов определения хлорид-иона в жаркое время года, когда «комнатная» температура достигает 30—32 °C.
Аргентометрический метод дает возможность успешно определять хлорид-ион в медных электролитах и вообще в присутствии меди (II) [4] при потенциале платинового электрода +0,4В (МИЭ), т. е. в условиях, исключающих электродное восстановле-
285
Jg Таблица 42. Методы определения хлора
Определяемый ион Титрант Определяемый минимум Условия определения Примечания Литература
ci- AgNO3 Электрод Pt, ф= + 0,4 В (МИЭ), фон определяется составом анализируемого раствора, например медного электролита Ф=+0,4 В (МИЭ) устанавливается во избежание восста- новления меди и осадка AgCl 4, 28
- Электрод Pt, <р= 0 В независимо от типа э._ектрода сравнения (Нас. КЭ или МИЭ), фон — практически любой ' Определение С1“ во всевозможных материалах 5—9
МО-'-ЬЮ-' моль/л Электроды Ag и AgCl, Aq>= =30—70 мВ, фон — 0,025 н. HNOS+CHSOH • 26
Соли Cd 2 Си электрода, катод амальгамирован; Дф от —0,6 до —2,0 В, фон —ледяная уксусная кислота Помимо С1“ можно определять различные анионы, в частности фосфаты, оксалаты и бораты 24, 25
Hg(NO3)2 •> Электрод Pt, ф=0,1—0,2 В (Нас. КЭ), фон — ацетат натрия-)-10—15% изопропилового спирта Определяют С1~ в виде LiCl 27
С1-, I- AgNO3 Электрод Pt, покрытый Ag, Ф=0 В (Нас. КЭ), фон — 0,004 М KNOs См. текст 14
ci-, Вг~, I- AgNO3
сю3- Fe2+ 1,7- IO-4 моль/л
«Активный хлор», C1O~ Hg2(NO3)2 4-10-3—ЫО-6 моль/л
KI 0,2 мг/100 мл
Арсенит натрия
cioz Хлорид тетра-фенилФосфо-ния(1) или те-трафенилсти-бония(П) 10~® моль/л
Электрод Pt, (р= О В КЭ), фон — см. текст
(Нас.
Последовательное титрование С1~, Вг~, I-, метод применим для анализа любых объ< ктов
1—3
2 Ag электрода
См. текст
(Нас.
Электрод Pt, <р= + 1 В
КЭ), фон — подкисленный H2SO4, титруют бихроматом избыток Fe^j", которым восстановили СЮ з ; можно применять 2 Pt электрода
Предложен вариант для определения С1~ и С10з~; после того как оттитрован С1~, восстанавливают СЮз~ до С1_ раствором сульфита и титруют С1_, определяя С1О3~ по разности двух титрований
Электрод Pt, q>=+0,6 В (МИЭ), фон — 1 и. H2SO4+KBr
Титруют по току брома и ВгО~, образующихся при реакции с «активным хлором»
2 Pt электрода, Лер=0,2— 0,4 В, фон—0,3 М Na2SO4 и 0,6 М N аНСОз
2 Pt электрода, Л(р=0,3 В фон — N аНСОз, pH=7,5
Определяют NaOCl в растворе Сг111
15
9, 10
11
12
13
Электрод Hg капающий, <р= =—1,8 В (I) и —0,88 В (II)
Не мешает С1, СЮд и др.
21, 22
ние меди (II). В противном случае кривая титрования искажается, так как с самого начала появляется ток восстановления меди (II) и осадка AgCl, вследствие чего после конечной точки ток восстановления ионов серебра заметен уже недостаточно резко.
Известно много методов определения хлорид-ионов в различных объектах [5—8]. Предложено определять хлорид-ион с С1С"з при совместном присутствии, причем хлорид титруют аргентометрически, а СЮз восстанавливают железом (II) и титруют избыток его бихроматом [9, 10].
Для определения СЮ- разработаны способы титрования растворами солей ртути (I) с добавкой бромида калия [11], иодида калия [12] и арсенита [13].
Из ранее предложенных методов определения хлорида и иодида интересен вариант аргентометрического определения иодида и хлорида при их совместном присутствии [14]. Иодид титруют на фоне 0,004 М раствора нитрата калия по его анодному току с посеребренным платиновым электродом без внешнего напряжения (Нас. КЭ). В конечной точке ток практически становится равным нулю и не изменяется на всем протяжении титрования хлорида, после чего резко возрастает, так как в этих же условиях на электроде восстанавливаются ионы серебра, появляющиеся в растворе после конечной точки титрования. Таким образом, на кривой имеется анодная и катодная ветви с резкими переломами в конечных точках для иодида и хлорида.
Для последовательного титрования иодида, бромида и хлорида может быть применен метод с двумя серебряными электродами [15] при напряжении 0,01 В. Анодный процесс обеспечивается в этом случае растворением серебра, а катодный — электровосста-новленйем серебра (I) из его галогенидов. Поскольку растворимость галогенидов серебра различается на несколько порядков, то на кривой титрования наблюдается три резких пика, отвечающих последовательному восстановлению иодида, бромида и хлорида серебра, а минимумы на кривой титрования отвечают конечным точкам для каждого галогенида.
Для определения свободного хлора предложено несколько методов, основанных на измерении тока восстановления газообразного хлора на твердых электродах. При этом возможно как полярографическое определение, т. е. непосредственное измерение высоты волны восстановления хлора, так и амперометрическое титрование тем или иным восстановителем. Примером первого типа определений является метод, заключающийся в автоматической записи силы тока восстановления газообразного хлора на вращающемся серебряном катоде [16]. Анализируемый воздух пропускают с определенной скоростью через прибор; газообразный хлор при этом растворяется в электролите (1О°/о-нь1Й раствор хлорида кальция, 0,1 н. по соляной кислоте) и восстанавливается на индикаторном электроде; вторым электродом (анодом) служит амальгама кадмия. Разность потенциалов между обоими электродами состав
288
ляет около 0,7 В. Этот метод позволяет определять от 0,05 до 1,5% хлора в анализируемом газе.
Амперометрическое определение свободного хлора в различных водах осуществляют главным образом с помощью фениларсоновой кислоты в присутствии иодида калия [17]. Предложены специальные упрощенные приборы для такого рода определений [18].
Для определения хлора в воде [19] и водных растворах органических веществ [20] применяют обычный метод — выделение иода из иодида калия. Выделившийся иод титруют непосредственно тиосульфатом [20] или добавляют тиосульфат в избытке и титруют его раствором йодата калия [19]. Для этих титрований очень удобно пользоваться методом с двумя индикаторными электродами.
Если нужно определить хлор в виде перхлората (СЮ 4), то можно титровать его растворами хлорида тетрафенилфосфония [21], или сульфата тетрафенилстибония [22]. В обоих случаях применяют ртутный капающий электрод и титруют по току восстановления реактивов: в первом случае при —1,8 В и во втором случае пои —0,88 В относительно Нас. КЭ. В противоположность другим методам определения СЮ4, данный метод позволяет определять перхлор ат-ион в присутствии хлорида, хлората и других анионов. Определять можно концентрации перхлората порядка 10-3Л4.
Для определения хлорида и бромида в безводной среде, в частности в безводной уксусной .кислоте, используют образование хлорида и бромида кадмия, малорастворимых в уксусной кислоте [23]. Титруют раствором нитрата кадмия с ртутным капающим электродом при —1,2 В относительно донной ртути, или с двумя медными электродами [24, 25] (см. также «Бор»), В метаноле титруют хлорид аргентометрически [26], а определение при помощи солей ртути (I) возможно в изопропиловом спирте [27]. Соли ртути (I) вообще рекомендуют для титрования галогенидов не только в безводных растворах. Соответствующие методы описаны ранее в разделе «Иод».
ЛИТЕРАТУРА
1. Laitinen И., Jennings W., Parks Т. — Ind. Eng. Chem., Anal. Ed., 1946, v. 18, p. 355, 358.
2. Kolthoff I. M., Kuroda P. — Analyt. Chem., 1951, v. 23, p. 1306.
3. Kolthoff I. M., Stock J. — Analyst, 1955, p. 860.
4. Сонгина О. A.—Зав. лаб., 1958, т. 24, № 3, с. 273—277.
5. Сонгина О. А.—Амперометрическое титрование. М., Химия, 1967.
6. Пиксарев А. Ю., Хедреярв X. X., Ваарманн А. Я-—Труды Таллинского политехнического ин-та, 1974, № 359, с. 55—61.
7. Мухамедшина Р. А., Чумаченко М. И.—Узб. хим. ж., 1966, № 3, с. 22—24.
8. Масалович В. М. и др. —Зав. лаб., 1971, т. 37, № 5, с. 525—527.
9. Икэда Санаэ, Муся Соитиро — Нихон кагаку ' дзасси, 1969, v, 90, №2, р. 180—186; РЖХим., 1970, 23Г111.
10. Масалович В. М„ Пирская Т. X., Сычева Т. В.—Труды Уральского научно-исследовательского химического ии-та, 1973, вып. 30, с. 77—82.
19—1694
289
11. Тараян В. М„ Ачарян Г. С., Дарбинян Г. А. — Арм. хим. ж., 1970, т. 23, № 1, с 27—30.
12. Масалович В. М. и др.—Ж- аналит. хим., 1970, т. 25, № 10, с. 1995—1998.
13. Масалович В. М., Пирская Т. X. — Труды Уральского научно-исследовательского химического ин-та, 1971, вып. 26, с. 52—58.
14. Nijland М. М., Eggels Р. Н. —Chem. Weekblad., 1958, v. 54, р. 289.
15. Masten М. L., Stone G. К. — Analyt. Chem., 1954, v. 26, p. 1076.
16. Макарова С. П., Беззубик 3. Г., 7роскурнина 5f. А,'—Зав. лаб., 1947, т. 13, № 12, с. 1347.
17. Marks Н., Williams D., Glasgow G. — J. Amer. Water Assos., 1951, v. 23, p. 201.
18. Mahan M. — Water Sewage Works, 1949, v. 96, p. 171.
19. Holluta J., Meissner H. — X. analyt. Chem., 1956, v. 152, p. 112.
20. Bet R. P., Yates K.— J. Chem. Soc. (London), 1962, p. 3285.
21. Нэдзу X.—Japan Analyst, 1961, v. 'JO, p. 571; РЖХим., 1963, 7Д49.
22. Morris M. — nnalyt. Chem., 19f>5, v. 37, p. 977.
23. Грешков А. П. и dp.—Ж- анал. хим., 1965, т. 20, № 4, с. 704.
24. Борк В. А, Сальникова Г. С.—Ж- анал. хим., 1968, т. 23, № 6, с. 901—907.
25. Грешков А. П., Борк В. А., Сальникова К. С.—Ж. анал. хим., 1967, т. 22, № 9, с. 1392—1397.
26. Heitmankova 1., Heitmanek М. — Coll. Czech. Chem. Comm., 1969, v. 34, № 2, p. 504—511.
27. Сидоренко В. И., Гордиенко В. И., Гизатуллина К. И.-— В кн.: Доклады Нефтехимической секции Башкирского респ. правления ВХО. Уфа, 1968, вып. 4, с. 94—101.
28. Захаров В. А., Мамбетказиев Е. А., Чултрова В. Ш. — Ж- анал. хим., 1969, т 24, № 9, с. 1401—1404.
Хром
Для амперометрического определения хрома до недавнего времени использовали только восстановление хрома (VI) тем или иным восстановителем. В настоящее время разработано несколько методов определения хрома в форме хром (III), а именно титрование раствором ЭДТА [1, 2] и диаминциклогексантетрауксусной кислотой [3], а также прямое окисление хрома (III) перманганатом в аммиачно-ацетатном растворе [4]. Для определения хрома (III) в материалах, содержащих одновременно хром (VI), рекомендовано окислять хром (III) избытком гипохлорита натрия, обладающего высоким окислительным потенциалом (Е° = +1,63 В) при рН=3— 6 и титровать избыток гипохлорита в гидрокарбонатной среде раствором иодида калия [5]. В хроматных растворах, содержащих хром (III), также титруют его в сильно щелочном растворе при нагревании дс 50 °C раствором феррицианида калия [6].
Несколько необычный способ, названный «псевдотитрсванием» [7], заключается в том, что при отрицательном потенциале ртутного электрода образуется хром(II), катализирующий реакцию образования комплексоната хрома при титровании ЭДТА.
Соль Мора по-прежнему Остается прекрасным реактивом для титрования хрома (VI). Соответствующих методик известно много [8—22], но принцип метода везде един и тот же: титрование на платиновом (или другом твердом электроде) по току окисления железа(II). Описанные в литературе способы различаются только
290
методами разложения пробы, зависящими от ее состава, материалом электрода (платина, графит, тантал) и числом электродов — один или 2 электрода. Очень часто определение хрома (VI) совмещается с определением ванадия (V) и марганца (VII). Эти случаи описаны в соответствующих разделах «Ванадий» и Марганец».
Приводим две конкретных методики определения хрома(VI), разработанные для анализа различных материалов и, соответственно, различающихся способом разложения навески и подготовки раствора к титрованию.
Ход анализа. Определение хрома в черных металлах [11]. Навеску 2 г металла растворяют при нагревании в смеси 25 мл серной (1.5) и 5 мл фосфорной кислот. После растворения нгзески окисляют железо (II) азотной кислотой, упаривают до появления паров, охлаждают, прибавляют 50 мл воды, 5 мл 1 %-ного раствора нитрата серебра, нагревают до кипения и окисляют хром(Ш) и могущий присутствовать в пробе марганец(И) 10 Мл 10%-ного раствора персульфата аммония. Избыток персульфата разлагают кйпячением, а марганцевую кислоту восстанавливают хлоридом натрия (5 мл 5%-ного раствора). После охлаждения титруют раствором соли Мора, концентрация которого определяется количеством хрома (VI) в титруемом растворе. Можно титровать либо весь раствор, либо, переведя его в мерную колбу, титровать Только аликвотную часть (в зависимости от содержания хрома и от взятой навески). В этом же растворе можно определять и ванадий (V), как указано в соответствующем разделе.
Описанным методом определяют от 0,03 до 0,15% хрома в различных чугунах, сталях и в стандартном образце стали № 20-Г. Метод считается наилучшим (по сравнению с колориметрическим или обычным титриметрическим) методом определения хрома.
Определение хрома в хромитах и хромомагнезитовых огнеупорных материалах (с одновременным определением общего содержания железа [15]. Навеску 0,2—0,25 г сплавляют со смесью перекиси натрия и карбоната натрия. Плав выщелачивают горячей водой и гидроксид железа отфильтровывают через бумажный фильтр. Фильтрат сохраняют для определения хрома. После промывания 1%-ным раствором карбоната натрия осадок растворяют в горячей 2 н. серной кислоте. Железо(Ш) восстанавливают висмутом и титруют аликвотную часть 0,1 или 0,01 н. раствором бихромата калия (в зависимости от содержания железа).
Для определения хрома (VI) фильтрат после предварительного кипячения в течение 20 мин для разрушения перекиси натрия нейтрализуют серной кислотой и добавляют избыток ее с таким расчетом, чтобы после доведения раствора в мерной колбе до определенного объема концентрация кислоты составляла 0,1 н. Аликвотную часть титруют 0,1 н. или 0,05 н. раствором соли Мора.
Титрование хрома и железа в хромитах и в хромомагнезитовых огнеупорных материалах протекает быстро и очень четко. Выбирая соответствующую навеску и разбавление и применяя Титрующие растворы различной концентрации, можно определять самые различные концентрации хрома (и железа), например от 0,05% до 15% Сг2О3 в магнезитовых кирпичах, бывших в работе, и от 35% до 55% Сг2Оз в хромитах.
Помимо соли Мора титровать хром (VI) можно другими восстановителями: ферроцианидом калия (см. «Ванадий») и иодидом калия (см. «Ванадий» и «Марганец»).
19*
291
Jg Таблица 43. Методы определения хрома
to Определяемый ион Титрант Л Определяемый минимум Условия определения Примечания Литература
Сг!1[ ЭДТА 0,25 мкг Электрод Hg вращающийся, <р=—0,33 В (Нас. КЭ), избыток ЭДТА титруют раствором Bi(NO3)3 при рН=2. Реакцию Сг111 с ЭДТА осуществляют при нагревании 1
Электрод графитовый, <р=+ 1,1 В (Нас. КЭ), фон — NaAc+HAc, pH=5, избыток ЭДТА титруют раствором CuSO4 Реакцию Сг111 с ЭДТА осуществляют при длительном нагревании до 100 °C, введение Na2CO3 или NaAc ускоряет реакцию, не мешает СгО4 2
Диаминциклогексантетр а-уксусная кислота Электрод Hg капающий, <р=— —1,0—1,1 В (Нас. КЭ), фон-0,1 М НС1О4, KNO3 и КС1, рН= =3,4—3,6 3
КМпО4 X. Электрод Pt, электрод сравнения выносной тоже Pt, Д<р=О В, фон —- ацетатно-аммиачный буферный раствор * 4
NaClO 2—32% Сг 2 Pt электрода, Д<р—0,1 В, фон— гидрокарбонатный, pH « 8, титруют избыток NaClO раствором KI См. текст 5
• • -*
293
Ks[FeCN)e]
ЭДТА
CrVI Соль Мора Fe11
Арсенит натрия 1-10-3—7-Ю"6 М
• •
Карбокснметил-тиокарбамипат калщия 4-32%
2 Pt электрода, Д<р=0,05 В, фон — сильно щелочной, 20 н. NaOH, при 50 °C
Электрод Hg капающий, <р = —1,075 В (Нас. КЭ)
Электрод Pt, <р=+0,6 В (Нас. КЭ), фон — в основном сернокислый, pH зависит от состава титруемого раствора
Электрод Pt, <р= +0,4 В (МИЭ), по току восстановления CrVI или при +1,1 В по току окисления арсенита, фон — 20%-ная НС1 или H2SO4
2 Pt электрода, Д<р=0,5 В, амперометрический индикатор — Vv, фон — 8—18 н. H2SO4
Метод предложен для определения Сг1П в присутствии политионатов, которые предварительно окисляют гипохлоритом «
См. текст
Метод пригоден для любых хромсодержащих материалов (см текст)
Метод применен для определения хрома в кирпиче и цементе
Отношение Сг : R=1 : 0,75 (в присутствии ванадия)
6
7
8—22
24
25
'Один из сравнительно давно известных методов [23]—титрование перекисью водорода — описан в разделе «Марганец», но вряд ли перекись водорода может иметь какие-либо преимущества перед другими более надежными восстановителями.
Из новых методов определения хрома (VI) следует упомянуть о восстановлении его раствором арсенита [24] и об определении хрома (VI) титрованием карбоксилметилдитиокарбаминатом Г25] (см. «Ванадий»),
Бейерман [26] применил для определения бихромата не амперометрическое титрование, а прямую амперометрию: он измерял изменение силы тока, протекающего между двумя электродами, на чистом фоне и при внесении исследуемого раствора. Аналогичный метод описан в разделе «Фтор» и может быть применен также, по данным его автора [26], для определения элементов, обладающих высоким окислительным потенциалом. Метод позволяет определять субмикрограммовые количества вещества.
ЛИТЕРАТУРА
1. Den Boef G., Oostervink R.— Anal. chim. acta, 1974, v. 72, № 2, p. 434—436.
2. Цоодол Ш., Алимарин И. П„ Галлай 3. А.— Изв. АН СССР. Сер. хим., 1968, № 8, р. 1726—1729.
3. Chiacchierini Е.—Ann. chim., 1966, v. 56, № И, р. 1405—1425; РЖХим., 1967, 14Г65.
4. Michalski Е'., Pawluk N. — Soc. scient. Lodz, acta chim., 1966, t. 11, s. 39—43.
5. Пирская T. X., Масалович В. M., Агасян П. К.—Зав. лаб.’, 1976, т. 42, № 7, с. 786—787.
6. Масалович В. М., Вассерман Л. И, Пирская Т. X. — В кн.: Сборник трудов УНИХИМ, вып. 26, с. 46—50.
7. Bustin D. J., Mocak J.—Taianta, 1970, v. 20, № 11, p. 1185—1190; p. 1191— 1197.
8. Алимарин И. П., Кузнецов Т. К- — Рэфераты докладе и Всесоюзн. совещания по электрохимическим методам анализа, изд-во АН СССР, 1949.
9. Алимарин И. П., Фрид Б. Й. —Зав. лаб., 1952, т. 18, № 12, с. 1300.
10. Бутенко Г. А., Беклешова Г. Е. — Зав. лаб., 1950, т. 16, № 7, с. 650.
11. Гренберг Е,- И., Генис М. Я-—Зав. лаб., 1952, т. 18, № 2, с. 174; 1950, т. 16, № 11, с. 1002.
12 Кольтгоф Й. М,- Зав. лаб., 4945, т. Ы, № 7, с. 626.
13. Kolthoff I. М., May D.— Ind. Eng. Chem., Anal. Ed., 1946, v. 18, p. 208.
14. Parks Th., Agazzi E. — Anal. Chem., 1950, v. 22, p. 1179.
15. Кондрахина E. Г., Егорова JI. Г., Сонгина О. A. — Изв. АН КазССР, Сер. хим., 1957, вып. I (II), с. 45.
16. Кондрахина Е. Г., Сонгина О. А. — Труды ин-та строительства и стройматериалов АН КазССР, 1958, т. 1, с. 149.
17. Hiett Т., Kobetz Р. — Analyt. Chem., 1956, v. 28, р. 1495.
18. Филенко А. И.—Ж- анал. хим., 1964, т. 19, Ns 4, с. '709; Зав. лаб., 1963, т. 29, Ns 12, с. 1423; 1966, т. 32, Ns 3, с. 287—290.
19. Apple R. F., Zittel Н. Е. —Anal. Chem., 1964, v. 36, р. 983.
20. Brhacek L., Golonka A., Kurzova K. — Hutn. Listy, 1960, v. 15, p. 679.
21. Арефьева T. В., Пац P. Г. — В кн.: Сборник научных трудов Гинцветмета. 1958, Ns 14, с. 74.
22. Гинзбург В. И. — Зав. лаб., 1961, т. 27, Ns 12, с. 1337.
23. Ятрудакис С. М., Жданов А. К. — Узб. хим. ж., 1964, Ns 5, с. 23.
24. Шапошникова Г. Н.—Арм хим. ж., 1975, т. 28, Ns 8, с. 667—668.
25. Усатенко Ю. И., Галушко С. В. — Зав. лаб., 1975, т. 41, Ns 3, с. 270—272.
26. Beyermann К- — Z. anal. Chem., 1962, Bd. 191, S. 346—349.
294
Церий
Церий (III) может быть оттитрован любым из реактивов, предложенных для определения лантаноидов, либо в отдельности, если другие лантаноиды не поисутствуют в растворе, либо суммарно с ними. Однако методы окисления — восстановления для церия более характерны и это выгодно отличает его от остальных лантаноидов. Обычно используют восстановление церия (IV), вследствие высокого стандартного потенциала системы CeIV/Cein, но можно и окислять церий(Ш), снижая потенциал тем или иным способом. Например, предложено титровать церий(III) феррицианидом калия в растворе, насыщенном карбонатом калия [1, 2]. В такой среде потенциал системы CeIV/CeHI настолько сдвигается в отрицательную сторону, что феррицианид калия оказывается окислителем по отношению к церию (III). Если титровать с платиновым электродом без наложения напряжения (Нас. КЭ), то получается смешанная катодно-анодная кривая: сначала наблюдается анодный ток окисления карбонатного комплекса церия (III), убывающий по мере титрования, а после конечной точки — катодный ток восстановления феррицианида.
Вместо феррицианида применяют также перманганат [3]. В серии работ [4—7] очень подробно изучено окисление церия (III) перманганатом в пирофосфатной среде, также способствующей снижению редокс-потенщщла вследствие образования комплексных соединений церия (IV). Разработана методика разложения проб чугуна, стали и других сплавов, способы подготовки раствора к титрованию и т. д. [5—7].
Для титрования церия (IV) по методу восстановления предложены аскорбиновая кислота [8—10] (см. также «Ванадий»), щавелевая кислота [Н, 12], соль Мора [11, 13], (см. также «Ванадий»), перхлорат и нитрат ртути(1) [14, 15], арсенит натрия [16], перекись водорода [17], нафтиламин [18], цистеин [19], метиленовая голубая [20], гидрохинон [21]. В разделе «Марганец» упоминается титрование церия (IV) нитритом натрия. Купферон, применяемый для осаждения церия (III), также является восстановителем по отношению к церию (IV) и может быть применен для его определения [И]- в водно-органической среде церий (IV) может быть оттитрован ферроценом [22].
ЛИТЕРАТУРА
1. Leonard G., Rally Н., Ните D.—Anal. chim. acta, 1957, v. 16, p. 185—188.
2. Жданов А. К-, Курочкина H. А. —Узб. хим. ж., 1961, № 3, с. 15—17.
3. Жданов А. К, Демидова Н. В.—Труды Ташкентского ун-та, 1964, вып. 264, с. 43—47. t
4. Henriet D., Gelis P. — Chim. analyt., 1968, v. 50, № 10, p. 519—530.
5. Боговина В. И., Усатенко Ю. И., Мальцев В. Ф. — Зав. лаб., 1966, т. 32, № 4, с. 412—413.
6. Боговина В И., Усатенко Ю. И., Мальцев В. Ф. — В кн.: Производство труб. М., Металлургия, 1970, вып. 24, с. 259—263.
295
7. Боговина В И., Усатенко Ю. И., Мальцев В. Ф.— Ж- анал. хим., 1968, т. 23, № 8, с. 1152—1157.
8. Галлай 3. А., Типцова В. Г., Пешкова В. М. — Ж. анал. хим., 1957, т. 12, № 3, с. 469.
9. Singh D., Varma. А.— Z. anal. Chem., 1961, Bd. 183, S. 172.
10. Жданов А. К., Рязанов T. М. — Узб. хим. ж., 1962, № 4, с. 18.
II. Василенко В. Д., Мажаровская Г. С. — В кн.: Редкоземельные элементы. М., изд-во АН СССР, 1963, с. 322.
12. Сонгина О. А., Кемелева Н. Г., Устимов А. М. — Жури. ВХО, 1964. т. 9, с. 697.
13. Боговина В. И., Усатенко Ю. И., Мальцев В. Ф. — Зав. лаб., 1969, т. 35, № 3, с 260—265.
14. Merrer R. J., Stock Л —Analyst, 1971, v. 96, № 1142, р. 361—363.
15. Ачарян Г. С., Тараян В. М.—Арм. хим. ж., 1972, т. 25, № 9, с. 753—757.
16. Жданов А. К., Ахмедов Г. — Ж- прикл. хим., 1971, т. 44, № 3, с. 660—662.
17. Жданов А. К., Ятрудакис С. М.—Труды Ташкентского ун-та, 1967, вып. 288, * с. 18—25.
18. Вартанян С. В., Тараян В. М.—Арм. хим. ж., 1974, т. 27, № 6, с. 469—473.
19. Шапошникова Г. Н., Галфаян Н. Г.—Арм. хим. ж., 1974, т 27, № 5, с. 373—376.
20. Проценко В- А.—Ж. неорг. хим., 1967, т. 31, № 3, с. 824.
21. Никасима Р., Фурукава М., Сасаки С., Сибата С. — Repts. Govt. Ind. Res. Inst., Nagoya, 1968, v. 17, № 9—10, c. 235—240; РЖХим., 1969, 4Г98.
22. Соломатин В. T„ Яковлев П. Я-, Харламова Л. И. — В кн.: Новые методы испытания металлов. М., Металлургия, 1976, № 3, с. 20—23.
Цинк
Наиболее надежным, хорошо оправдавшим себя на практике методом определения цинка, является анодный ферроцианидный метод [1—17], основанный на образовании весьма малорастворимого осадка. Феррицианид калия также образует осадок с цинком, притом простого и постоянного состава Zn3[Fe(CN)6]2, но растворимость этого осадка выше, чем ферроцианидного, и поэтому амперометрическое определение цинка с помощью феррицианида не зошло в практику. Ферроцианид калия образует с цинком осадки различного состава: из обычных растворов цинковых солей выпадает осадок K2Zn3['Fe(CN)6]2, из комплексных соединений, в частности из аммиачных растворов, выпадает простая соль Zn2[Fe(CN)6].
Титрование ферроцианидом можно выполнять с ртутным капающим электродом по току восстановления цинка [14—17] или с платиновым вращающимся электродом по току окисления избытка ферроцианида [1-12]. Последний вариант ферроцианидного метода имеет все преимущества анодных амперометрических методов. Разработке этого метода определения цинка в различных видах минерального сырья и промышленных объектах предшествовало подробное выяснение влияния различных элементов, сопутствующих цинку, и выбор среды, позволяющей проводить титрование без отделения цинка от этих элементов [1].
Подбирая соответствующие условия, можно определять цинк в присутствии больших количеств кальция, магния, свинца(II) (при сульфатном фоне свинец в большей своей части окажется в
296
осадке), меди(II) (до соотношения меди и цинка 1:1); кадмия (до соотношения кадмия и цинка 1 : 10), алюминия и железа. Такая возможность достигается подбором фона, способствующего связыванию мешающих ионов в комплексные соединения или выпадению их в осадок. Так, в ацетатно-аммиачной среде .медь(II) .и кадмий удерживаются в виде комплексных соединений, а цинк, обладающий наименьшей по сравнению с другими ионами растворимостью ферроцианидного соединения, выпадает в осадок. Некоторые авторы [18] считают возможным титровать цинк в присутствии даже 100-кратного количества кадмия. Железо(III) в аммиачной среде выпадает в осадок и не мешает титрованию, если его содержание не слишком велико, так .как в ином случае цинк может адсорбироваться осадком гидроксида железа. Поэтому при высоких содержаниях железа (около 10% и выше) следует прибегать к добавлению лимонной кислоты [5], связывающей его в достаточно прочный комплекс, из которого ферроцианид не осаждав ет железо. Добавление лимонной кислоты [6] также ослабляет влияние алюминия, которое вообще довольно заметно при всех титрованиях с платиновым электродом [алюминий(III) пассивирует электрод вследствие образования тончайшей пленки гидроксида, появляющейся в результате гидролиза солей алюминия].
В присутствии лимонной кислоты несколько уменьшается также влияние Марганца (II), который сильно мешает проведению титрования цинка по двум причинам. Как было указано в разделе «Марганец», он может не только давать осадки с ферроцианидом, но и окисляться на платиновом электроде- при тех потенциалах, при которых проводят титрование (от +0,7 до +1,0 В, МИЭ). При повышенных содержаниях марганца его приходится предварительно окислять (например, персульфатом) и обязательно отфильтровывать осадок, так как осадок оксида марганца (IV), тем более свежеосажденный, является сильным окислителем и непосредственно взаимодействует с ферроцианидом, искажая кривую титрования и вызывая повышенный расход титрующего раствора.
Во избежание помех со стороны некоторых элементов (свинец, кобальт, кадмий, марганец и медь) предложено маскировать их ионы диаминопиклогексантетрауксусной кислотой [19], титруя цинк ферроцианидом с двумя платиновыми электродами (с добавкой феррицианида, см. гл. IV).
В кислой среде, даже в чистых растворах, нельзя получить воспроизводимых результатов при определении малых количеств цин-‘ ка (порядка 1 мг и меньше в 20 мл раствора), вследствие того, что растворимость осадка ферроцианида цинка в кислой среде достаточно высока [6]. При нейтрализации серной кислоты аммиаком до pH «5 получаются вполне удовлетворительные результаты. Вообще присутствие ионов аммония в исследуемом растворе улучшает результаты титрования вследствие того, что замещение ионов калия ионами аммония в двойных ферроцианидах приводит к значительному уменьшению растворимости осадка. Подобное же
297
благоприятное действие ионов аммония описано выше в разделе «Кальций». Однако, по мнению авторов [20] i, состав осадка стабилизируется и при добавлении к раствору во время титрования некоторого количества сульфата калия.
В присутствии спирта (50%) растворимость осадка ферроцианида цинка еще больше понижается, что позволяет определить [6] до 6 мкг цинка в 20 мл раствора. Титрование проводят в таких случаях 0,0002 М раствором ферроцианида калия.
Без добавления спирта можно определять также достаточно малые количества цинка •—до 30 мкг в 20 мл. При титровании таких количеств цинка не образуется не только осадка, но даже опалесценции— растворы остаются прозрачными, и тем не менее конечная точка титрования обозначается вполне отчетливо.
Ниже приводится методика определения цинка анодным ферроцианидным методом в промышленных растворах (электролите и др.).
Ход анализа. Отбирают пипеткой определенное количество электролита или иного раствора, содержащего цинк, и разбавляют водой в мерной колбе, емкость которой выбирают соответственно содержанию цинка в исходном растворе; например, при содержании цинка 140 г/л- (цинковый промышленный электролит) отбирают 20 мл и разбавляют до 1 л. Отбирают 50 мл этого раствора и разбавляют водой до 100 мя. Переносят 10 мл вновь полученного раствора в стакан для титрования, добавляют 5 мл 3 М раствора ацетата аммония (можно и ацетата натрия, ио ацетат аммония следует предпочесть, так как ионы аммония понижают растворимость осадка, а ионы натрия, наоборот, могут ее повысить). Затем добавляют около 5 мл 2%-ного раствора аммиака до появления слабого запаха. Объективно это количество аммиака должно быть достаточным для того, чтобы растворилась выпавшая муть. Можно нейтрализовать и по фенолфталеину до появления слабо-розовой окраски, т. е. до рН=8. Избыток аммиака вреден. Титруют при потенциалах от +0,7 до +0,1 В 0,1 М раствором ферроцианида калия с платиновым электродом по току окисления избыточного Лерроцианида. Кривая, имеет форму б. Состав осадка отвечает в этом случае простой соли с формулой Zn2[Fe(CN)e], по которой и рассчитывают содержание цинка.
* При определении цинка в рудах, концентратах, полупродуктах металлургической их переработки, в почвах и других объектах навеску разлагают тем или иным способом, а титрование проводят так же, как указано выше. Если нужно, то титровать можно и лри рНягЗ—6, но в таких случаях надо добавлять соли аммония, так как осадок состава (NII4)2Zn[Fe(CN)6] значительно менее растворим.
В работах, выполненных совместно с болгарскими исследователями [21—23], подробно рассмотрены условия определения микро-граммовых содержаний цинка в присутствии винной кислоты, связывающей в комплексные соединения железо(Ш) и алюминий, и влияние смешанных аммиачно-тартратно-ацетатных фонов на поведение меди (II), олова(II) и кадмия при определении цинка.
Много внимания уделено определению цинка при помощи ЭДТА или его аналогов. В работе [24] указано, что титрование ЭДТА (также как и ферроцианидом) дает результаты более точ-
298
Таблица 44. Методы определения цинка
Титрант Определяемый минимум Условия определения Примечания Литература
K4[Fe(CN)6] 1 мкг/мл Электрод Pt, <р=+0,7—1,0 В (МИЭ) или 2 Pt электрода, Д<р= = 0,1—0,2 В, фои — см. текст, или Hg капающий электрод по току восстановления Zn11 См текст 1—24
ЭДТА Электрод Pt, <р= +1,3 В (Нас. КЭ). фон — ацетат натрия Определяют Zn в Zn-—РЬ рудах после отделения мешающих элементов 24
Диэтилдитиокарба-минат Электрод Pt, <р=+0,8 В (Нас. КЭ), фон — 0,1 М КС1 ( Определение Zn в сплавах А1 после хроматографического его выделения 28
Диантипирилметан ’ и роданид 0,7—7 мг Электрод Pt, <р= +1,2 В (Нас. КЭ), фон—1—3 н. H2SO4 или НМОз+аи-типирилметаи, титруют раствором KCNS Осадок D2H2Zn(CNS)4 (D-дианти-пнрилметаи) *- 29
Остальные методы определения цинка описаны в других разделах, см. текст.
яые, чем полярографический метод определения цинка. Практически все работы, посвященные титрованию цинка раствором ЭДТА, касаются не только цинка, но и других элементов. Сведения об этих работах даны в разделах «Медь», «Индий», «Висмут», «Кадмий», «Лантаноиды», «Кальций», «Молибден». Ранее предложенные методы титрования раствором ЭДТА описаны в предыдущем издании этой книги [25], равно как и некоторые другие реактивы, о дальнейшем применении которых сведений в литературе нет.
Диэтилдитиокарбаминат, о применении которого упоминалось ранее '[26, 27], позволяет определять цинк при помощи ртутного капающего [27] или платинового [26, 28] электродов, но реактив не избирателен по отношению к цинку, поэтому его приходится отделять при помощи ионообменной колонки [28].
Титрование цинка оксихинолином описано в разделе «Марганец», дитиотриоксиметиламинометаном в разделе «Серебро», тиооксином в разделах «Ртуть» и «Кальций». Титруют цинк также диантипирилметаном [29] и, подобно меди(II), свинцу(II) и кадмию— тиокарбонатом калия (см. «Кадмий»).
В серии работ, посвященных титрованию в неводных растворах и экстракционным методам с последующим ' амперометрическим титрованием, в числе других элементов упоминается и цинк (см. разделы «Висмут», «Индий», «Кадмий»).
В болгарской печати появился обзор, посвященный амперометрическому определению малых количеств цинка различными реагентами [21].
ЛИТЕРАТУРА
1. Сонгина О. А., Войлошникова А. П., Козловский М. Т.—Зав. лаб., 1951, т. 17, № 1, с. 3
2. Бутенко Г. А., Рынская Е. С.—Ж- анал. хим., 1950, т. 5, № 1, с. 145.
3. Сонгина О. А.—Труды комиссии по аналитической химии АН СССР, 1952, IV (VII), с. 116.
4. Солнцев Н. И., Чудина Р. И. — В кн.: Сборник научных трудов Гинцветме-%а. 1958, № 14, с.. 103.
5. Солнцев Н. И., Таль Э. М. — Зав. лаб., 1953, т. 19, № 11, с. 1027.
6. Войлошникова A. TL, Козловский М. Т., Сонгина О. А.—Зав. лаб., 1957, т. 23, № 3, с. 273.
7. Гао Сяо-Ся, Юнь-Вань — Acta chim. sinica, 1955, v. 21, p. 253.
8. Parks Th., Smith O., Rodding Sh. — Anal. chim. acta,. 1954, v. 10, p. 485.
9. Boszai J. — Acta chim. Hungar., 1956, v. 9, p. 195.
10. Kies H. L., Hien T. S. — Z. anal. Chem., 1955, Bd. 148, S. 91.
11. Kao S. S., Chuang W. T. — Chem. Abs., 1959, v. 53, c. 973.
12. Сонгина О. А., Савицкая И. С. — Зав. лаб., 1965, т. 31, № 3, с. 259.
13. Усвяцов А. А., Сонгина О. А. —Зав. лаб., 1965, т. 31, № 7, с. 663.
14. Khosla В., Gaur Н —J. Indian Chem. Soc., 1953, v. 30, p. 637.
15. Nimer E., Hamm R., Lee G. — Anal. Chem., 1950, v. 22, p. 790.
16. Spalenka M. — Coll. Czech. Chem. Comm., 1939, v. 11, p. 146.
17. Woodson A., Johnson B., Cooper S. — Anal. Chem., 1951, v. 23, p. 1712.
18. Sierra F., Sanchez-Pedreno C., Perez T., Lozano С. M. — Inform, quim. anal., 1969, v. 23, № 1, p. 11—16; РЖХим., 1970, 20Г68.
19. Sierra F„ Martinez U. C., Perez R. T., Sancnez-Pedreno C. — Inform, quim ami., 1972, v. 26, № 5, p. 209—216; РЖХим., 1973, 8Г57.
300
20 Олыианова К. М., Човнык И. Г., Шепеленко Л. Н.—Ж- анал. хим., 1970, т. 25, № 11, с. 2115—2117.
21 Дюлгерова А. С., Сонгина О. А., Ку лев И. И. — Химия и индустрия, София, 1972, т. 44, № 7, с. 320—322
22. Дюлгерова А. С., Сонгина О. А., Захаров Б А. — Изв. АН КазССР. Сер. хим., 1971, вып. 3, с. 66—67.
23. Дюлгерова А. С., Захаров В. А. — В кн.: Прикладная и теоретическая химия. Алма-Ата, 1971, в'ып. 3, с. 18—20.
24. Ziemba St. — Rudy i metale niezel., 1969, t. 14, № 3, s. 154—158.
25. Сонгина О. А. Амперометрическое титрование. M., Химия. 1967
26. Усатенко Ю. И., Тулюпа Ф. М. —Зав. лаб., 1960, т. 25, № 8, с. 783.
27. Stricks W., Chakrabarti S. К- — Anal. Chem., 1962, v. 34, p. 508.
28. Шепеленко Л. Г.—Труды ин-та хроматографии Воронежского ун-та, 1968, вып. 2, с. 141—142.
29. Луговой С. В., Рязанова И. П., Темянко В. Г. — Зав. лаб., 1966, т. 32, № 6, с. 663—666.
Цирконий
В амперометрическом титровании циркония большая роль принадлежит купферону [1, 2]. Это титрование выполняют с ртутным капающим электродом по току восстановления купферона при —0,84 В (Нас. КЭ). Оно может быть осуществлено в присутствии многих других катионов и анионов, в частности фторидов и фосфатов. Фториды часто присутствуют в растворах циркония (IV) поскольку металлический цирконий переводят в раствор обычно при помощи фтористоводородной кислоты, а фосфат применяют для отделения циркония и гафния от тория [1]. Титрованию мешают только железо (П1), ванадий (V) и большие количества олова (IV). Остальные катионы не мешают. Гафний, если он присутствует в пробе, проходит через все операции вместе с цирконием. Уран(VI) и ниобий (V) Определяют в аликвотных частях первоначального раствора полярографически. Купфероновый метод был применен для определения циркония в магниевых сплавах, в которых определяют «общий», кислотно-растворимый и кислотно-нераствооимый цирконий, применяя различные способы разложения навески сплава [1].
Интересна способность циркония переходить в осадок купферо-ната даже в том случае, если он связан фосф ат-ионом и находится в растворе в виде взвеси фосфата циркония. Такой же способностью обладает фосфат гафния. Это позволяет амперометрически титровать не растворы циркония, а взвеси его фосфата, что оказывается очень удобным в тех случаях, когда необходимо, например, определить цирконий в растворах, содержащих, кроме циркония, уран и ниобий [1].
Ход анализа. К известному объему раствора добавляют концентрированную сериую кислоту, доводя объем раствора до 10 мл, если присутствуют оксалаты (в том случае, если исследуемый раствор предварительно обрабатывали щавелевой кислотой для извлечения редкоземельных элементов), прибавляют 25 мл концентрированной азотной кислоты. Упаривают до появления обильных паров. Охлаждают, переводят в мерную колбу емкостью 100 мл, доводят до метки ВОДОЙ. 1
301
Таблица 45. Методы определения цирконя (Zriv)
Титрант Определяемый минимум Условия определения Примечания Литература
Купферон, нео-купферон Электрод Hg капающий, <£= -0,84 В (Нас. КЭ), восстановление купферона, фон— 1—2 М H2SO4 Осадок Zr(Cupf)4, см текст 1, 2
- Электрод Pt, <р= 4-0,6 В (Час. КЭ). или 2 Pt электрода, Д<р=0,4 —0,5 В или с Pt электродом сравнения в растворе КМпО4, фон — сернокислый СП СО СП 4*
ЭЦТА 2 Pt электрода, Д<р=0,6—0,7 В, амперометрический индикатор AgNOa или KsIFe(CN)6], или Та электрод, <р= + 1,2 В (Нас. КЭ), фон — 0,5— 1,0 н. H2SO4 илй электрод Hg капающий, <р=—0,2 Ь (Нас. КЭ) при обратном титровании избытка ЭДТА нитратом висмута рН~2 - СО СП
Ортооксифенил-и ми н о ди уксуси а я кислота Электрод Pt «ли графитовый, <р= = 4-0,8 В (Нас. ДЭ), фон — KNOs4-4-НС1, pH =2 Отношение Zr : R=1 : 1, комплекс ZrR более прочен, чем комплекс Fe3+ и других ионов 10
Азопнразолои 0,2—0,4 мг Электрод Hg капающий, <р=—0,7 В (донная ртуть), фон — НС1О4 (3:2) Осадок состава Zr : 'R= 1 : 2 11,12
N-фуроилфенил-гидроксиламин 0,5—3,5 мг/10 мл Электрод графитовый, <р=4-1,0 В (Нас. КЭ), фон — 0,1 н. H2SO4 или буферный растворе рН=2—2,2 Отношение Zr: R = 1:4, пр именяют для сплавов состава 13
N-бензоил-о-то-ли лги др оксил амин 0,4—5 мг/10 мл Электрод графитовый, <р= 1 В (Нас. КЭ), фон — 0,5—2 М H2SO44-сухой NaCl для коагуляции осадка Отношение Zr : R= 1 : 4, реактив растворяют в смеси спирта с водой, мешают Ti, Mo, W, Hf 14
Аликвотную часть, содержащую около 0,1 ммоль циркония (около 10 мг), юносят в центрифужную пробирку емкостью 125 мл, добавляют 50 мл 1' «' ной сепной кислоты, 3 мл 30%-ной перекиси водородами 25 мл 10%-ной оной кислоты насыщенной однозамещенным фосфатом аммония NH4H2POt тимепю 25 г фосфата на 100 мл кислоты). Перемешивают и центрифугируют затем несколько раз промывают фосфат циркония, центрифугируя его с паствопом содержащим 50 мл указанного выше раствора фосфата в серной кислоте 50 мл 10%-ной серной кислоты в 3 мл 30%-ной перекиси водорода. После промывания взбалтывают осадок-с 50 мл 10%-ной серной кислоты и тит-nvror купфероном. Можно также сначала отфильтровать осадок фосфата циркония через небольшой быстрофильтрующий бумажный фильтр и после промывания погрузить осадок с фильтром в 10%-ную серную кислоту, измельчить фильтровальную бумагу мешалкой и титровать образовавшуюся суспензию купфероном.
Титрование купфероном можно выполнять и с платиновым электродом по току окисления купферона [3, 4]. Вместо купферона рименяют неокупферон [5]. Купфероновый метод недавно был вновь применен для определения циркония с помощью двух платиновых электродов [6]. Для титрования циркония применяют ЭДТА [6—9], анализ проводят как с ртутным капающим, так и с твердыми электродами, причем известны случаи, когда цирконий определяют при одновременном присутствии других элементов (см. «Галлий», «Железо», «Висмут»), Ортооксифенилиминодиук-сусная кислота также служит для титрования циркония [10], равно как азопиразолон [11, 12], ферроилфенилгидроксиламин [13] и N-бензоил-о-толилгидроксиламин [14].
Возможно амперометрическое определение циркония с помощью ксиленолового оранжевого [15].
ЛИТЕРАТУРА
1. Elving Ph., Olson Е. — Anal. Chem., 1955, v. 27, p. 1817; 1956, v. 28, p. 251, 338.
2. Olson E., Elving Ph. — Anal. Chem. 1954, v. 26, p. 1747.
3. Усатенко Ю. А., Беклешова Г. — Зав. лаб., 1957, т. 23, К° 12, с. 1406.
1. Усвяцов А. А., Сонгина О. А. — Зав. лаб., 1965, т. 31, № 7, с. 663.
5. Галлай 3. А. и др. —Ж. анал. хим., 1964, т. 19, № 7, с. 1464.
6. Хадеев В. А.— РЖХим., 1977, 8Г90, депон. в ВИНИТИ.
7. Владимирова В. М.— Зав. лаб., 1956, т. 22, № 6, с. 529.
8. Goldstein G„ Manning D., Zittel H. — Anal. Chem., 1962, v. 34, p. 358.
9. Хадеев В. А., Квашнина Ф. Ф.— Изв. вузов. Химия и хим. технол., 1960, т. 3, с. 251.
10. Виткина М. А., Беклешова Г. Е. — В кн.: Химическая технология. Респ. межвед. научно-техи. сб., 1969, вып. 15, с. 100.
11. Рора Gr., Cruceru D., Baiulescu Gh., Greff C. — Rev. Chim., (RPR), 1965, v. 16, № 7, p. 321.
12. Popa Gr., Cruceru D., Dreff C. — An. Univ. Bucuresti, Chim., 1971, v. 20, № 1, p. 65—70.
13. Галлай 3. А. и др. — Вестник МГУ. Химия, 1971, т. 12, № 1, с. 94—97 14. Галлай 3. А. и др. — Ж. анал. хим., 1970, т. 25, № 10, с. 1851—1854.
15. Боровая Н. С., Церковницкая И. А. — В кн.: Проблемы современной аналитической химии. Л., изд-во ЛГУ, 1976, вып. 1, с. 95—105.
303
Ольга Альфредовна Сонгина Владимир Андреевич Захаров
А мае р о метрическое
титрование
Редактор В. Л. Абрамова
Технический редактор В. М. Скитина
Художественный редактор Н. В. Носов Корректоры М. А. Ивлиева, Л. В. Лазуткина
ИБ 845 * «
Сдано в наб. 16.07.79. Подп. в печ. 5.12.79. Т—18799.
Формат бумаги бОХЭО1/^ Бумага тип. № 3. Гарн. литературная. Печать высокая. Усл. печ. л. 19 Уч.-изд л. 20,96. Тираж 7800 экз. Заказ № 1694. Цена 1 р. 70 к. / Изд. № 1702.
Ордена «Знак Почета» издательство «Химия». 107076. Москва, Стромынка, 13.
Московская типография № 11 Союзполиграфпрома при Государственном комитете СССР по делам издательств, полиграфии и книжной торговли.
Москва, 113105, Нагатинская ул., д. 1.