/







































































































































































































































































































Текст
6П7
К344
УДК 541.183 : 66.01
Кельцев Н. В.
Основы адсорбционной техники. 2-е изд., перераб. и
доп. — М., Химия, 1984. — 592 с., ил.
Во 2-м издании (1-с вышло в 1976 г.) углублены и развиты главы о
практическом применении адсорбционных процессов, в частности для за-
щиты атмосферы и гидросферы от згщрязнений, кондиционирования и хра-
нения продукции, о разделении газов в движущемся слое адсорбента. Ад-
сорбционно-десорбционный цикл и вспомогательные стадии процесса рас-
смотрены как комплекс равновесных и кинетических закономерностей, вы-
текающих из данных об адсорбционном равновесии, структуре сорбентов,
кинетики и динамики адсорбции. Изложены основы теории адсорбции и
важнейшие процессы, реализованные на ее основе.
Предназначена для инженерно-технических н научных работников хи-
мической, нефтехимической, газовой, пищевой и других отраслей промыш-
ленности. Полезна преподавателям химико-технологических вузов.
592 с.. 73 табл., 365 рис., 766 литературных ссылок.
РЕЦЕНЗЕНТЫ:
докт, хим. наук профессор В. В. СЕРПИНСКИЙ,
докт. техн, наук профессор Т. Г. ПЛАЧЕНОВ
2801020000—166
К 050(0Т)ДТ~ 49 83
5) Издательство «Химия». 1984 г.
СОДЕРЖАНИЕ
Памяти Николая Владимировича Кельцева 9
Предисловие к первому изданию 11
Предисловие редактора 13
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
АДСОРБЦИОННОЙ ТЕХНИКИ 15
1
Развитие
адсорбционного метода
очистки и разделения
смесей
Начальные сведения о применении адсорб-
ционной очистки. Этапы становления ад-
сорбционной технологии. Первые промыш-
ленные адсорбенты (угли, силикагели). Син-
тетические цеолиты — молекулярные сита.
Современное состояние адсорбционных про-
цессов очистки, разделения и кондициони-
рования газовых и жидкостных сред. Пер-
спективы использования природных цеоли-
2
Адсорбционное
равновесие
и пористая структура
адсорбентов
Адсорбция и адсорбционные силы 22
Пористая структура адсорбентов 25
Термическое уравнение адсорбции и его
частные случаи 28
Экспериментальные методы определения
изотерм адсорбции 31
Теория мопомолекулярной адсорбции 35
Теория полимолекулярной адсорбции и оп-
ределение удельной поверхности 38
Другие методы определения удельной по-
верхности 47
Капиллярная конденсация и структура ме-
зопор 50
Ртутная пирометрия 58
Потенциальная теория адсорбции 60
Теория объемпого заполнения мпкропор 61
Уравнение Фрейндлиха 71
Уравнение Кисарова 72
Физические методы исследования адсорбци-
онного взаимодействия 74
3
Основные виды
пористых адсорбентов
Активные угли 77
Силикагели 82
Активный оксид алюминия 84
Цеолиты 85
Цеолиты в природе 87
Синтетические цеолиты общего назна-
чения 91
Синтетические кислотостойкие цеолиты 100
Смешанные адсорбенты 108
Пористые стекла 110
Природные глинистые породы 111
4
Содержание
4 Теплота адсорбции 118
Термодинамические аспекты теории объем-
Теплота адсорбции ного заполнения микропор 120
и термодинамическое
обоснование теории
объемного заполнения
микропор
5 Общие закономерности адсорбции смесей 124
„ „ Экспериментальные методы изучения ад-
Избирательрость сорбции смесей 128
адсорбции Расчет адсорбции бинарной смеси газов
(паров) на основе теории Ленгмюра 131
Расчет адсорбции бинарной смеси паров на
основе теории объемного заполнения мик-
ропор 132
Приближенное определение коэффициента
разделения 134
Теория идеального адсорбционного раство-
ра 137
6 Экспериментальное изучение кинетики ад-
, сорбции 138
Кинетика адсорбции Кинетическая кривая 141
Коэффициент диффузии 142
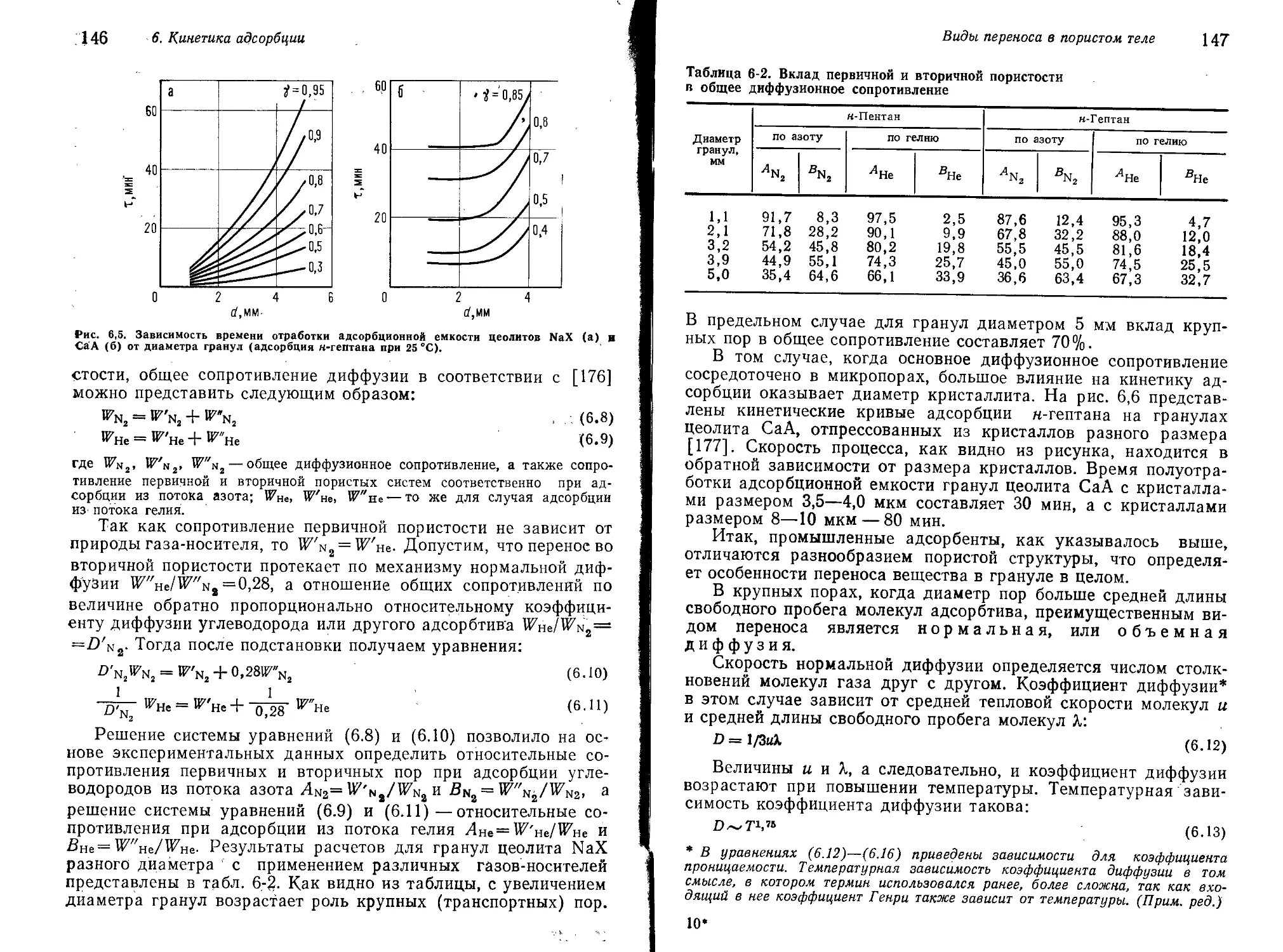
Виды переноса в пористом теле 145
7 Кинетика десорбции из зерна адсорбентов 152
,, _ Относительный метод расчета кинетики
Кинетика десорбции десорбции 154-
Выбор температурного режима ста-
дии десорбции 160
Изотермическая десорбция из слоя микро-
пористых адсорбентов 163
g Предмет динамики адсорбции 167
Экспериментальное исследование динамики
Динамика адсорбции адсорбции 170
Основные уравнения динамики адсорбции 175
Уравнение материального баланса 175
Уравнение кинетики адсорбции 178
Уравнение теплового баланса 178
Уравнения передачи тепла 170
Начальные и граничные условия 170
Математические модели динамики адсорб-
ции 180-
Динамика равновесной изотермической
адсорбции 180-
Динамика неравновесной изотермиче-
ской адсорбции 188
Динамика неизотермической адсорбции 196-
Динамика адсорбции многокомпонент-
ной смеси 201
Содержание
5
g Равновесие и динамика адсорбции
Использование метода в технике
Адсорбция
< ui6<> сорбирующихся
газов
203
211
АППАРАТУРНО-ТЕХНОЛОГИЧЕСКОЕ
ОФОРМЛЕНИЕ АДСОРБЦИОННЫХ
ПРОЦЕССОВ 215
10 Разделение газа на установках с периоди- ческими адсорберами 217
Типы адсорбционных Конструкция адсорберов в установках пе- 220 220 221 222 223 224 224 227 228 229 234 237 239 240 241 243 252 256 257
процессов и установок риодического действия Вертикальный адсорбер Горизонтальный адсорбер Кольцевой адсорбер Адсорбер с вертикальными теплообмен- ными элементами Адсорбер с прижимным устройством График работы установки периодического действия Непрерывный метод разделения смесей га- зов в движущемся слое адсорбента Принципиальная схема установки Адсорбция углеводородов в движу- щемся слое адсорбента Разделение смеси адсорбированных компонентов в хроматографической части установки Температурный режим десорбционной части Теплопередача в трубчатых нагревате- лях и холодильниках адсорбента Варианты аппаратуры Показатели непрерывного процесса Очистка водорода от примесей Выделение ацетилена из низкоконцент- рированных газов Другие варианты непрерывного адсорбци- онного метода Гидравлическое сопротивление слоя адсор- бента
11 Активные угли 262
Получение активных углей 262
Получение Типы активных углей 268
и номенклатура Силикагели 273
промышленных Получение силикагелей 273
адсорбентов Типы промышленных силикагелей 277
Приближенное определение структур-
ных характеристик силикагеля 278
Активный оксид алюминия 280
6
Содержание
Получение активного оксида алюми-
ния 280
Промышленный ассортимент 281
Синтетические цеолиты 283
Получение гранулированных цеолитов
со связующим 283
Гранулирование цеолитов без связую-
щих добавок 289
Некоторые свойства промышленных адсор-
бентов и их определение 291
J2 Характеристика влажных сред 296
Влажность газов 297
Осушка газов Влажность жидких сред 300
и органических Осушка силикагелями 301
жидкостей Осушка в статических условиях 301
Осушка в динамических условиях 303
Осушка газов с небольшим содержа-
нием влаги 306
Выбор условий регенерации силикагеля 307
Дезактивация силикагеля 307
Осушка газов под высоким давлением 309
Принципиальные схемы осушающих
установок 313
Адсорбционные установки осушки и
отбензинивания газов 315
Установки короткоцикловой безнагрев-
ной адсорбции 320
Осушка, очистка и осветление масел 324
Осушка цеолитами 324
Цеолиты как средство осушки 324
Осушка в газовой промышленности 330
Осушка в нефтехимической промыш-
ленности 351
Осушка масел 356
Осушка жидкостей в лаборатории 361
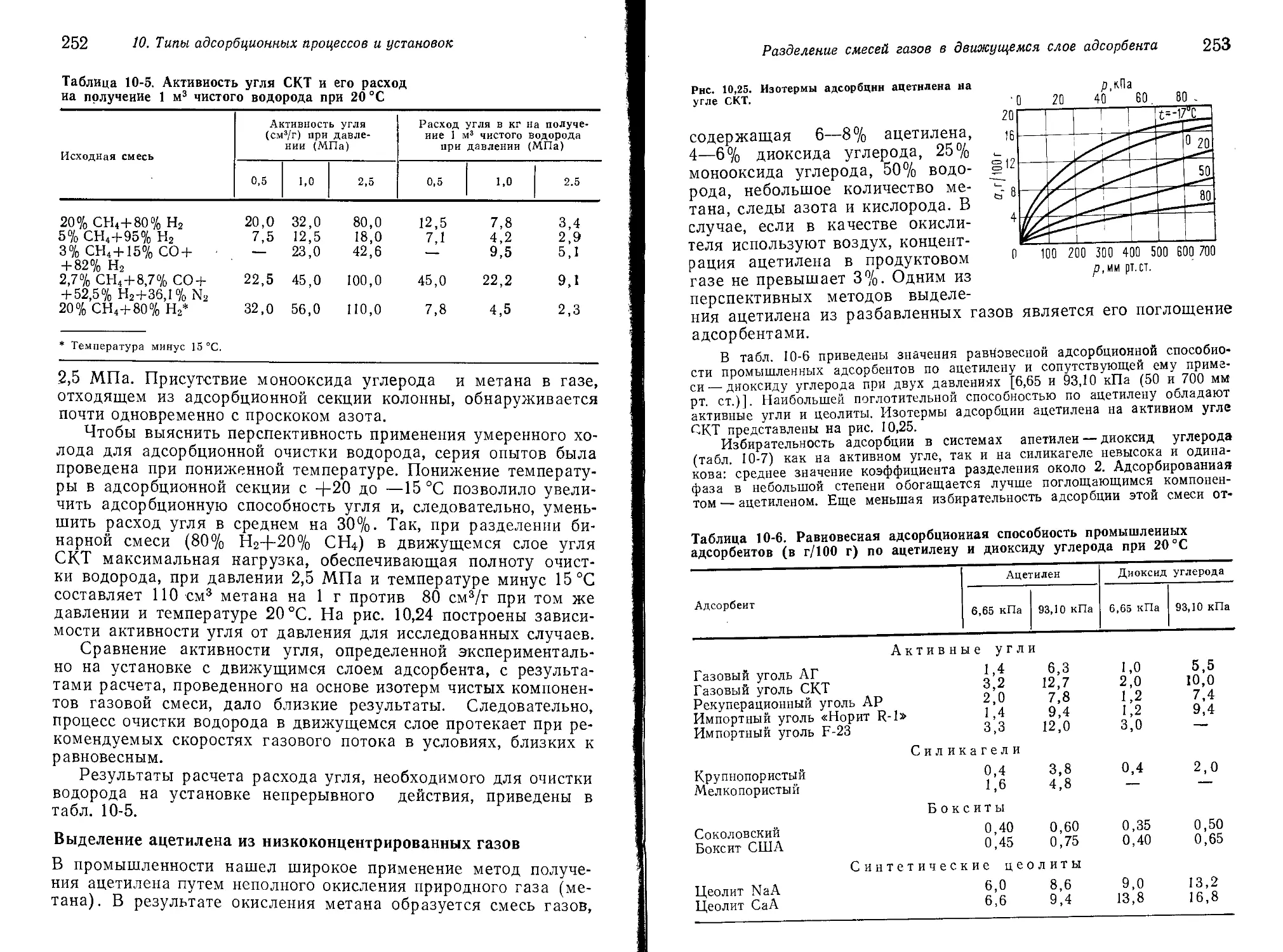
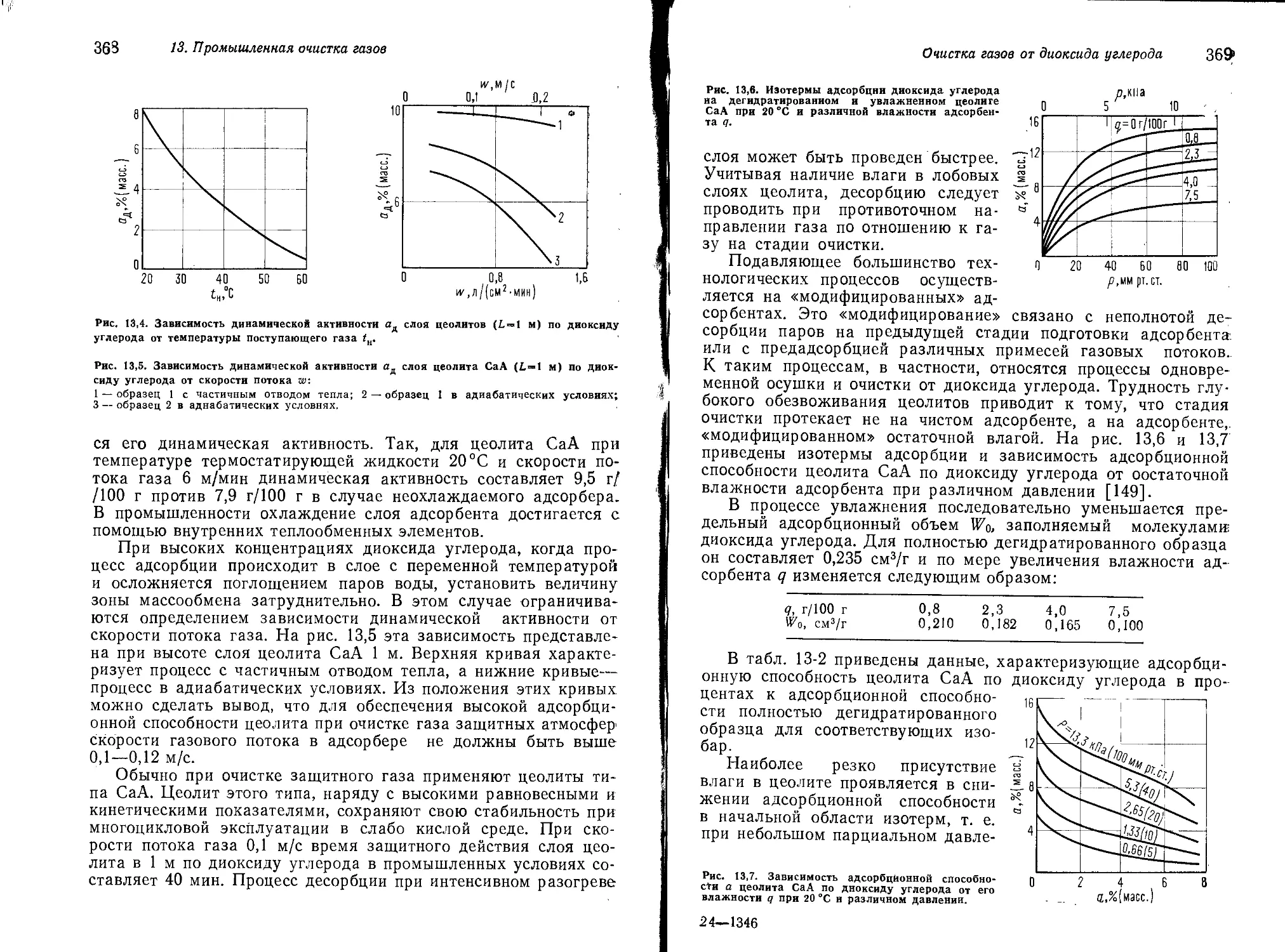
|3 Очистка газов от диоксида углерода 364
Приготовление контролируемых защит-
Промышленная ных атмосфер 366
очистка газов Очистка и осушка воздуха перед его
низкотемпературным разделением 376
Очистка природного газа от диоксида
углерода и других примесей 379
Очистка этана, пропана, водорода 381
Очистка газов от оксидов углерода с по-
мощью хемосорбентов 381
Очистка атмосферы в изолированных
системах 382
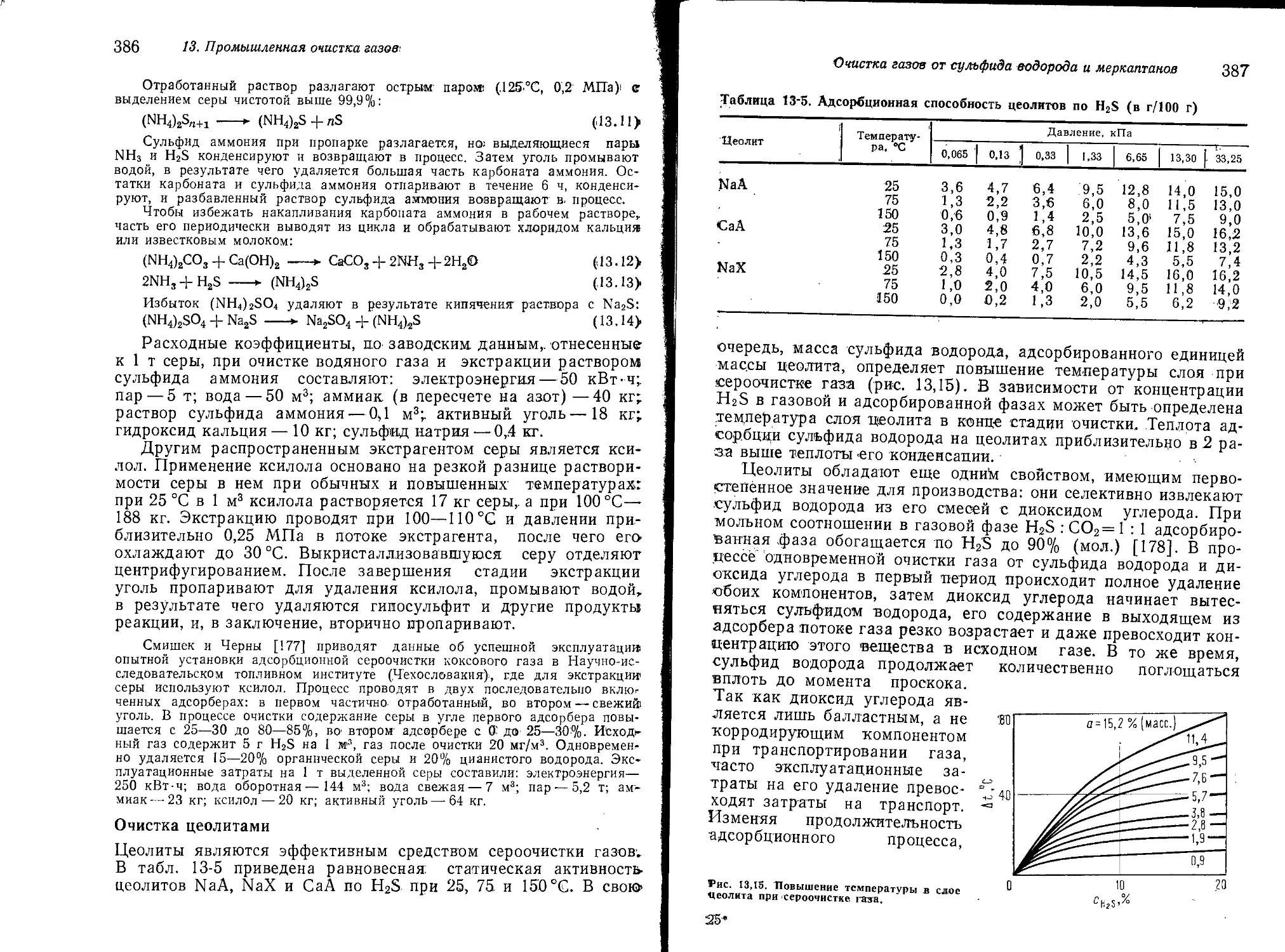
Очистка газов от сульфида водорода и
меркаптанов 383
Очистка активным углем 384
Очистка цеолитами 386
Сероочистка газов с частичным удале-
нием диоксида углерода 392
Содержание
7
Очистка газа от сероорганических сое-
динений 395
Сероочистка газов с химической реге-
нерацией адсорбента 397
Сероочистка сжиженных газов 399
Сероочистка нефтепродуктов 401
14 Рекуперация растворителей Рекуперация сульфида углерода 406 414
Защита атмосферы Рекуперация хлорорганических раствори-
от загрязнения телей 421
Очистка газов от диоксида серы 423
Методы очистки газов углями 424
Методы очистки газов цеолитами 435
Извлечение металлов из отходящих газов 438
Рекуперация семиоксида рения 438
Защита атмосферы от ртутн 439
1К Область применения метода 445
Выбор адсорбента 446
Защита гидросферы Регенерация и утилизация адсорбента 450
от загрязнения Принципиальные схемы адсорбционной
очистки воды и основные типы адсорберов 451
Технологические схемы очистки воды с
применением порошкообразного активного
угля 455
Технологические схемы очистки воды с
применением гранулированного активного
угля 457
16 Избирательность адсорбции н углях а активных 466
Использование Избирательность адсорбции на силикагелях 467
избирательности Адсорбция непредельных углеводоро-
адсорбции дов Адсорбция ароматических дов Адсорбция воды углеводоро- 467 473 478
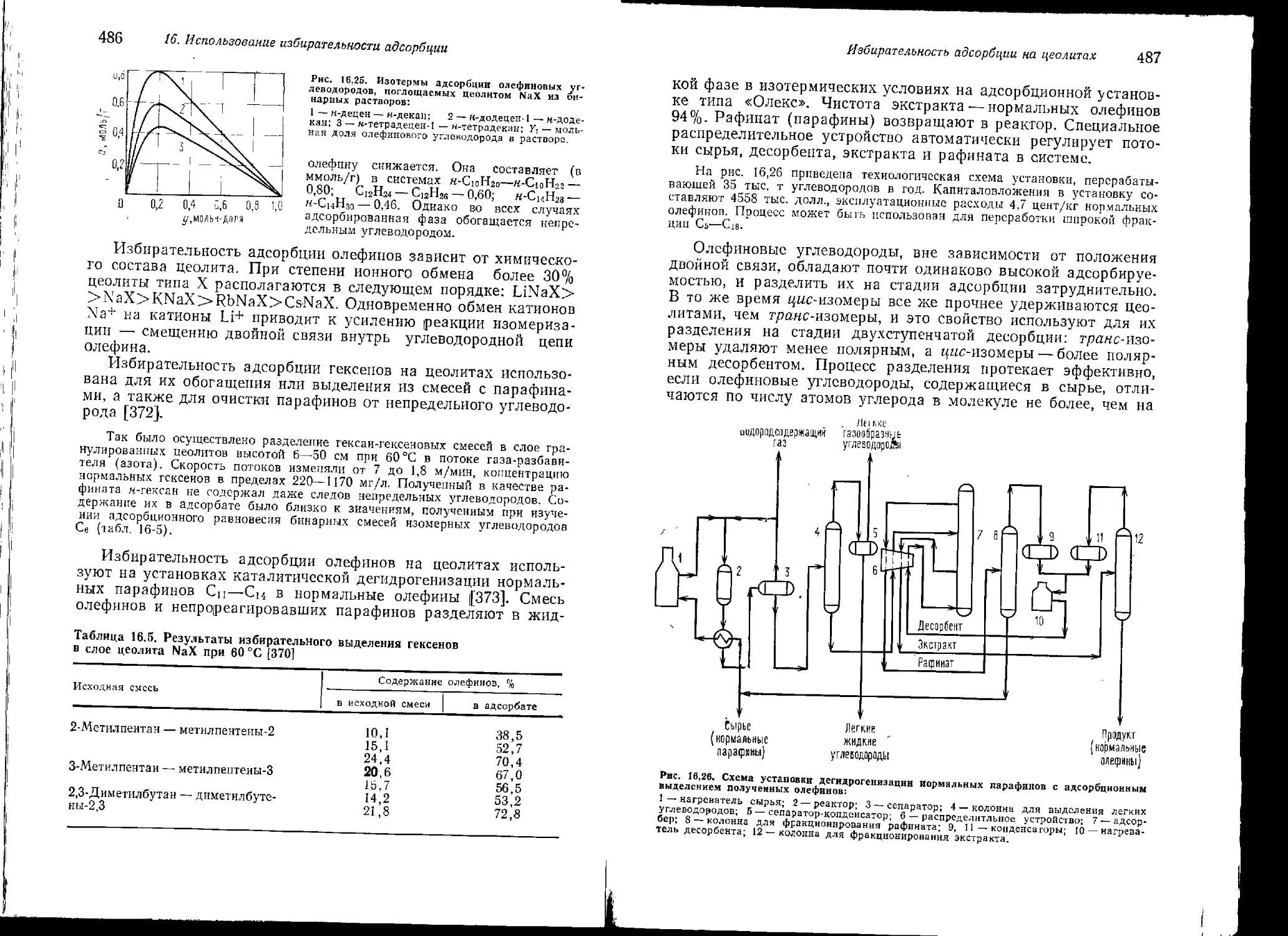
Избирательность адсорбции на цеолитах Адсорбция непредельных углеводоро- дов Адсорбция ароматических углеводоро- дов Адсорбция в системе диоксид углеро- да — сульфид водорода Адсорбция в некоторых других систе- мах 479 480 495 500 501
17 Очистка изопентана от примеси н-пентана 503
Использование Снижение токсичности выбросов автомоби- лей 505
молекулярно-ситовых Бензины прямой перегонки нефти 506
свойств цеолитов Бензины термической и каталитической
переработки нефтепродуктов 509
8
Содержание
Комбинированный процесс изомериза-
ции и денормализации углеводородных
фракций 513
Получение высокооктановых бензинов 519
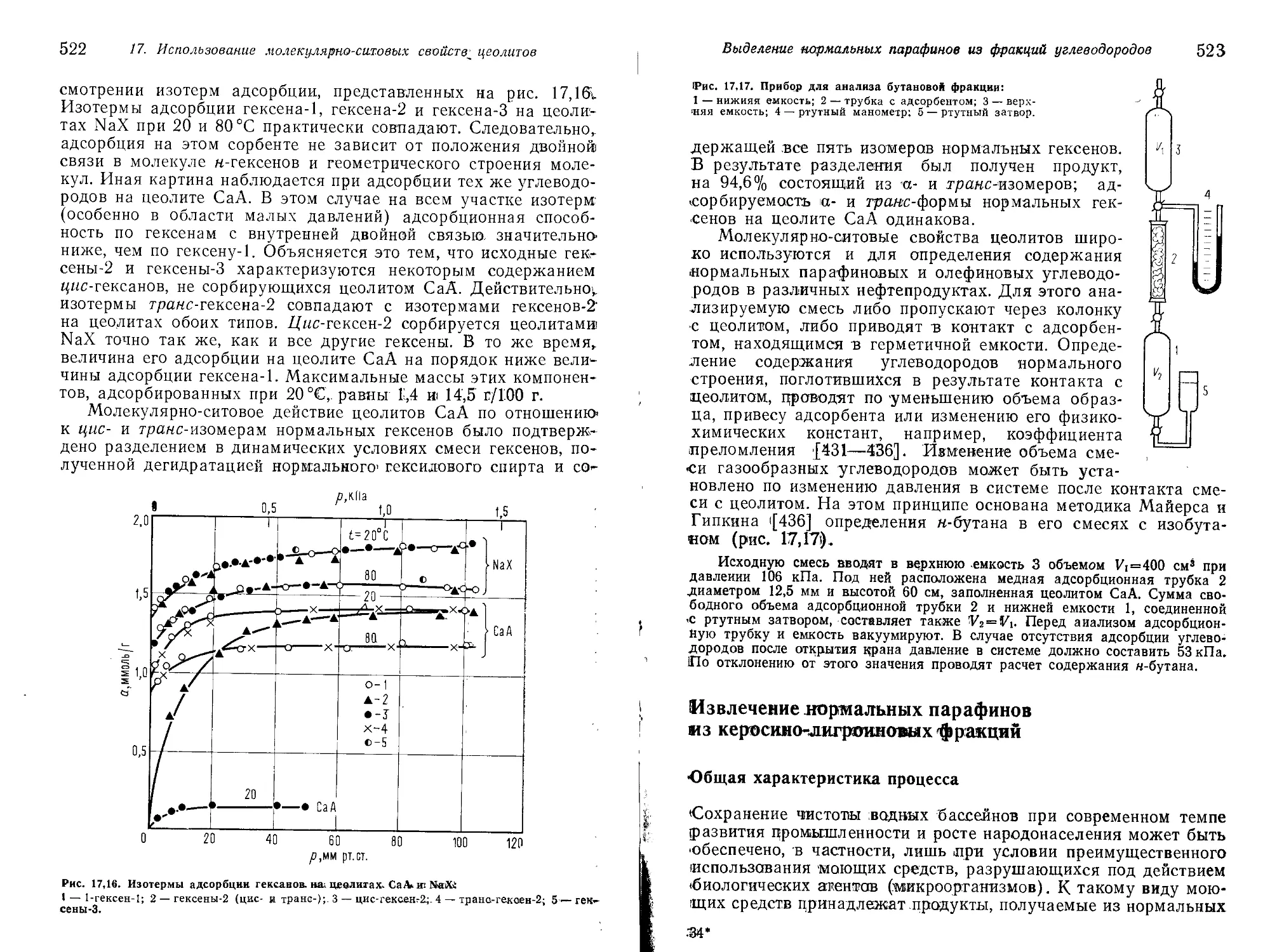
Депарафинизация в движущемся слое 521
цеолитов
Депарафинизация бензинов с примене-
нием диоксида углерода в качестве де-
сорбента 521
Выделение нормальных олефинов из смеси
с другими углеводородами 521
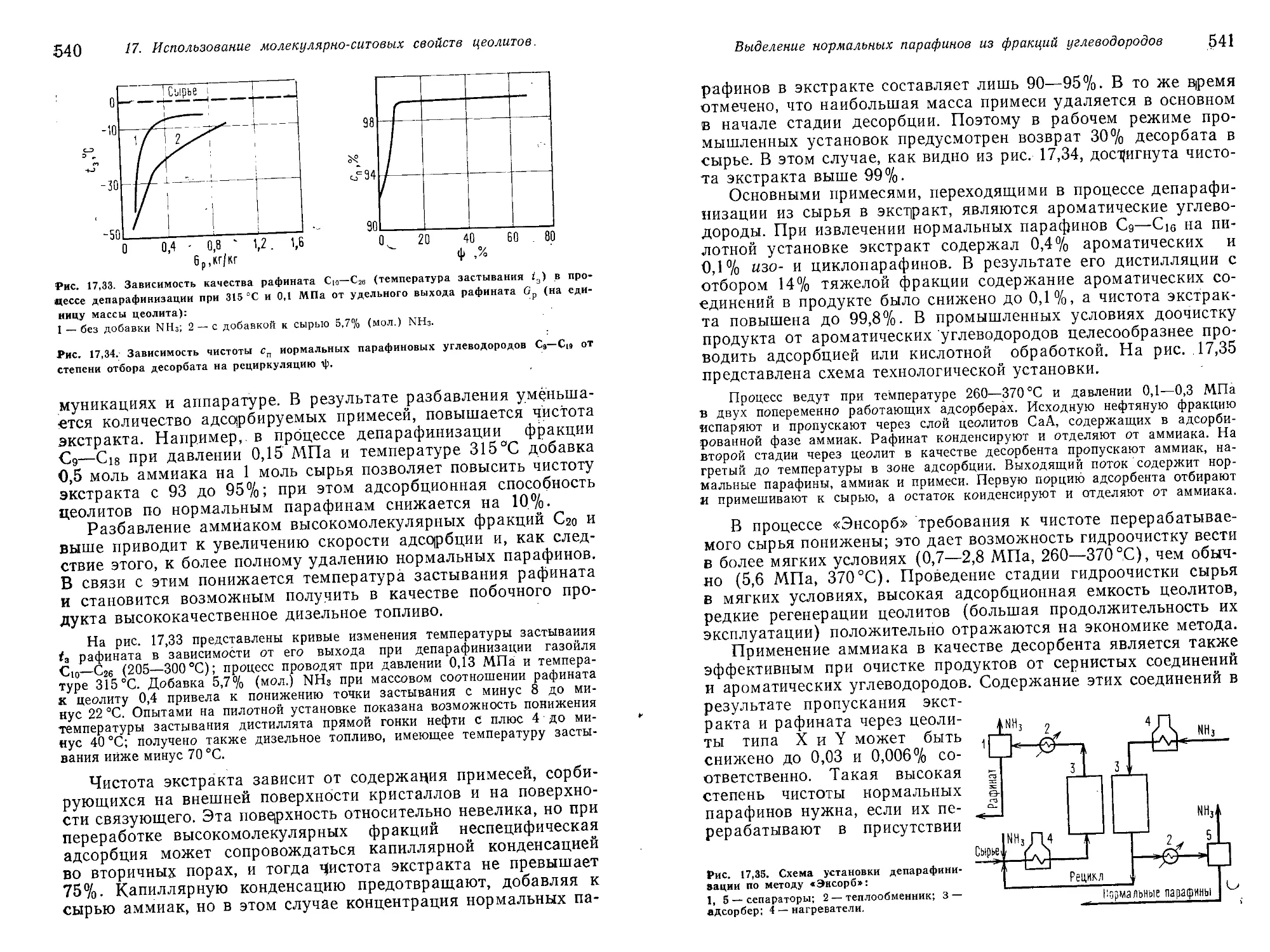
Извлечение нормальных парафинов из ке-
росино-лигроиновых фракций 523
Общая характеристика процесса 523
Процессы в паровой фазе 530
Процессы в жидкой фазе 545
Очистка аргона от кислорода 550
18 Улучшение вкусовых качеств водки, конья- 551 555 557
Применение адсорбентов для кондиционирования и хранения пищевой продукции ка, вин Осветление пива и фруктовых соков Очистка сахарных сиропов
Регулирование состава газовых сред при хранении сельскохозяйственной продукции Литература к разделу «Теоретические ос- 559
новы адсорбционной техники» Литература к разделу «Аппаратурно-техно- 567
логическое оформление адсорбционных про- цессов» 572
Заключение редактора 583
Предметный указатель 586
ПАМЯТИ НИКОЛАЯ ВЛАДИМИРОВИЧА КЕЛЬЦЕВА
В период подготовки второго издания этой книги безвременно
скончался ее автор — Николай Владимирович Кельцев.
Его смерть — тяжелая потеря для науки о сорбционных про-
цессах, в которую он внес столь существенный вклад, для хи-
мической, нефтехимической, газовой и других отраслей промыш-
ленности, которые обязаны ему внедрением многих новых про-
грессивных процессов адсорбции.
Большую утрату понесла также высшая школа, где Н. В. Кель-
цев в течение многих лет активно и талантливо участвовал в
подготовке квалифицированных инженерных кадров. Это неиз-
гладимая потеря и для всех его соратников, десятилетиями тру-
дившихся вместе с ним, для тех, с кем он делил свой энтузиазм,
свою увлеченность, свою радость от успехов советской науки и
промышленности, с которыми он был связан всю жизнь — от
студенческих лет до последнего дня.
Биография Николая Владимировича проста и типична для
всего его поколения. Он родился в 1921 г., восемнадцати лет
поступил в Московский химико-технологический институт имени
Д. И. Менделеева. Студенческие годы были прерваны войной.
В августе 1941 г. Николай Владимирович ушел добровольцем
в действующую армию. Он сражался на фронтах Отечествен-
ной войны до полного разгрома фашистской Германии и импе-
риалистической Японии, был награжден двумя боевыми орде-
нами и медалями, тяжело ранен, контужен.
После войны он завершил высшее образование и с момента
окончания МХТИ непрерывно, с постоянным успехом занимался
научной, научно-инженерной и педагогической работой в обла-
сти адсорбционных процессов. В работе его всегда отличало
резко обостренное чувство нового. Вся его деятельность — это
разработка и внедрение самых новых, самых перспективных идей
сорбционной науки и техники.
В 1951 г. Н. В. Кельцев защитил кандидатскую диссертацию,
посвященную адсорбционным процессам в движущихся слоях
адсорбента, а в 1967 г. — докторскую диссертацию о проблемах
адсорбции на едва ли не самых перспективных адсорбентах —
синтетических цеолитах.
В 1959 г. в нашей стране была создана Комиссия по цеоли-
там, преобразованная позже в Научный совет Академии наук
СССР по адсорбентам. В задачу Совета вошла координация
всех работ по синтезу, изучению и применению адсорбентов. Ни-
колай Владимирович был постоянным и активным членом сна-
чала Комиссии, а затем Научного совета. Он всегда горячо и
10
Памяти Николая Владимировича Кельцева
увлеченно участвовал в обсуждении всех планов и итогов рабо-
ты, помогая их авторам находить самые эффективные и совре-
менные решения. Поэтому не будет никаким преувеличением
сказать, что кроме его личных работ во всех достижениях со-
ветской адсорбционной науки и техники есть доля творческого
участия Николая Владимировича.
Большое значение для подготовки и повышения квалифика-
ции научных и инженерных кадров имела монография
Н. В. Кельцева «Основы адсорбционной техники». Она стала
настольной книгой для студентов, аспирантов, научных работ-
ников и инженеров, работающих в области адсорбции.
Николай Владимирович ушел из жизни, когда заканчивал
переработку своей монографии. Ко второму изданию рукопись
подготовил один из его учеников доктор технических наук
Ю. И. Шумяцкий.
Для нас — товарищей и соратников Николая Владимирови-
ча — эта книга всегда будет лучшей памятью о нем. Пусть же
и для многочисленных читателей, которые были лишены радо-
сти личного общения с ее автором, эта монография будет на-
поминанием о светлом облике крупного ученого и инженера,
талантливого педагога, хорошего и доброго человека — о Нико-
лае Владимировиче Кельцеве.
Академик М. М. ДУБИНИН
Профессор В. В. СЕРПИНСКИИ
ПРЕДИСЛОВИЕ К ПЕРВОМУ ИЗДАНИЮ
Адсорбция является универсальным методом, позволяющим
практически полностью извлечь примесь из газовой или жидкой
среды. В современной химической, газовой, нефтеперерабаты-
вающей промышленности адсорбционный метод широко исполь-
зуют для глубокой очистки и осушки технологических потоков,
улучшения качества сырья и продуктов. В технике широко при-
меняются различные адсорбенты с развитой внутренней поверх-
ностью: силикагели, алюмогели, активные угли и т. д.
Освоение метода промышленного получения адсорбентов но-
вого типа с регулярной структурой пор — цеолитов позволило
осуществить процессы разделения веществ на основе разницы
в размерах и форме молекул. С использованием цеолитов ре-
шаются некоторые важнейшие проблемы современности. К та-
ким проблемам относится в первую очередь производство био-
логически разрушающихся моющих средств. Адсорбция должна
занять ведущее место среди способов защиты биосферы от вред-
ных промышленных выбросов. В ряде случаев технологическая
очистка с помощью адсорбентов, например, очистка промышлен-
ных газов от сульфида водорода, ликвидирует выброс образую-
щегося в процессе химической переработки вредного вещества
(диоксида серы) в атмосферу, и рассматривается также как
один из методов защиты окружающей среды.
Адсорбционные процессы разносторонни. Уже при расчете
и выборе технологии основной стадии (адсорбции) инженеру
необходимо учитывать многочисленные и разнообразные аспек-
ты (равновесие, кинетика, динамика, гидравлика и т. д.). Еще
сложнее создать оптимальный вариант технологического цикла
в целом. В литературе обычно приводятся лишь разрозненные
данные о теории и показателях отдельных элементов (стадий),
что затрудняет поиск оптимального технологического режима.
Поэтому назрела настоятельная необходимость создания книги,
в которой были бы собраны воедино и обобщены результаты ис-
следований наших и зарубежных ученых. Это позволит инжене-
ру найти сведения об основных промышленных адсорбентах,
характере адсорбционного взаимодействия, важнейших техноло-
гических схемах и режимах их работы, теоретических основах
расчета отдельных стадий и процесса в целом.
Инженерные методы расчета технологических установок ба-
зируются на достижениях теории в той или иной конкретной об-
ласти науки. Большинство теоретических положений и практи-
ческих рекомендаций в книге подтверждено собственными и
литературными экспериментальными данными, которые были
12
Предисловие к первому изданию
получены на лабораторных и промышленных установках. Рабо-
та автора в лаборатории на первых стадиях протекала в кон-
такте с П. А. Теснером и А. Л. Халифом, затем с Н. С. Торо-
чешниковым; основными участниками экспериментальных работ
являлись Ю. X- Андреев, С. А. Ануров, Ю. М. Афанасьев,
С. 3. Васильев, 3. А. Жукова, О. В. Мойсейчук, А. И. Сидоров,
В. И. Смола, Л. М. Федорова, А. Ф. Старовойтова, Ю. И. Шу-
мяцкий.
Формирование взглядов советских исследователей на адсорб-
ционный процесс происходило под влиянием выдающегося уче-
ного и организатора работ в области адсорбции Героя Социали-
стического труда, академика М. М. Дубинина. Автор считает
своим долгом выразить глубокую благодарность М. М. Дубини-
ну, который взял на себя труд тщательно проанализировать сна-
чала программу, а затем, содержание книги. С разрешения
М. М. Дубинина в главах 2 и 4 мною использованы материалы
его книги «Адсорбция и пористость», касающиеся основ теории
объемного заполнения микропор и термодинамических расчетов
с использованием этой теории. Большое влияние на эволюцию
представлений о природе адсорбционных явлений оказала шко-
ла А. В. Киселева. Работы А. В. Киселева и его сотрудников
позволили понять всю сложность процессов на поверхности
раздела фаз, и их различные проявления и изменения.
Книга включает две части. В первой части адсорбционный
процесс рассмотрен как комплекс равновесных и кинетических
закономерностей адсорбционно-десорбционного цикла и вспомо-
гательных стадий. Вторая часть посвящена технологии и аппа-
ратурному оформлению, а также технико-экономическим пока-
зателям современных адсорбционных процессов очистки, осушки,
разделения газов, паров и жидкостей, в том числе в движу-
щемся слое адсорбента. Большое внимание уделено процессам,
позволяющим обезвредить промышленные выбросы, рекупериро-
вать из них ценные продукты и решить проблему защиты био-
сферы. Рассмотрены новые каталитические процессы на основе
промышленных адсорбентов.
В книге отражены наиболее крупные достижения в разви-
тии адсорбционной техники в Советском Союзе и социалистиче-
ских странах, а также в США, Франции, Англии, Италии, ФРГ.
Завершению работы над книгой способствовали все основные
специалисты в области адсорбции, которые личным советом или
через свои наиболее важные публикации внесли коррективы в
книгу. Основным автором главы 8 является Ю. И. Шумяцкий,
разделы глав 3 и 11, посвященные промышленным адсорбен-
там — силикагелям, оксиду алюминия и природным глинистым
породам, написаны совместно с Б. А. Липкиндом, Э. А. Левиц-
ким и М. А. Кердиваренко.
Н. В. КЕЛЬЦЕВ
ПРЕДИСЛОВИЕ редактора
Об адсорбции написано не так уж мало книг. Их почти исчер-
пывающий перечень приведен в библиографии к первой главе
первого издания монографии Н. В. Кельцева «Основы адсорб-
ционной техники». Но ознакомившись с ним, читатель легко
убедится, что большинство из ранее вышедших книг посвящено
различным аспектам теории адсорбции, синтезу и свойствам
адсорбентов. Адсорбция же как промышленный процесс на стра-
ницах книг освещена несравненно слабее, чем адсорбция-явле-
ние. Этот пробел и был устранен выходом в свет первого изда-
ния монографии Н. В. Кельцева.
Надо отметить, что книга Н. В. Кельцева была опубликована
чрезвычайно своевременно. Предшествующий период бурного
развития адсорбционной техники, ее внедрение в самые разно-
образные (иногда поначалу даже немыслимые) области произ-
водственной деятельности породили широкий круг остро заинте-
ресованных читателей. Именно поэтому весь тираж первого из-
дания разошелся практически мгновенно, а издательство, автор
и его коллеги получили множество писем с просьбами прислать
хотя бы один экземпляр, и не для личного пользования, а для
библиотеки завода, лаборатории. Переводы книги изданы в
Польше и ФРГ.
Но большой успех книги в первую очередь был определен
ее высоким уровнем и в целом удачным обобщением в рамках
одной монографии поистине необъятного материала. В ней, хотя
и с разной подробностью, рассмотрены три главных вопроса ад-
сорбции: теория адсорбционных явлений и процессов, свойства
и методы получения адсорбентов и, наконец, технология и аппа-
ратурное оформление промышленных адсорбционных процессов.
С наибольшей полнотой изложен третий вопрос, ибо монография
Н. В. Кельцева написана для технологов и, следовательно, пре-
имущественно о технологии. Для научного работника, инженера
или студента, входящих в проблематику, она является пособием
по всем основным вопросам адсорбции; технолог-специалист
найдет в ней «свертку» большого научного и производственного
опыта и богатый справочный материал, столь часто необходимый
ему в работе. Именно эти особенности книги Н. В. Кельцева,
не говоря уже о появлении новых и весьма актуальных прило-
жений адсорбции, обусловили целесообразность ее переиздания.
И. В. Кельцев скоропостижно скончался, в момент заверше-
ния работы над рукописью второго издания. Он внес сравни-
тельно небольшие изменения в теоретические главы книги, до-
полнил главу, посвященную адсорбентам — данными о природ-
14 Предисловие редактора
ных и некоторых нетривиальных синтетических адсорбентах —
и практически полностью переработал технологическую часть.
В новом издании книги в этой части специально отражены про-
блемы применения адсорбентов для охраны окружающей среды
(одна из глав этого направления — глава 15 — написана автором
совместно с А. В. Кельцевым), для кондиционирования и хра-
нения пищевой продукции (глава 18, написанная совместно с
М. А. Кердиваренко и В. П. Харитоновым) и др. При подготов-
ке новых глав автор, как и при написании первого варианта
книги, консультировался с академиком М. М. Дубининым и не-
однократно выражал ему свою благодарность.
При редактировании книги исключена незавершенная авто-
ром глава, посвященная теории сорбции из растворов. Глава об
адсорбентах разделена на две: в одной из них, сохраненной в
теоретической части монографии, представлены сведения о
структуре и свойствах пористых адсорбентов, а в другой, поме-
щенной в технологическом разделе, — о номенклатуре и мето-
дах производства промышленных адсорбентов. Эти изменения
находятся в соответствии с замыслами автора об общей струк-
туре книги.
Текст монографии снабжен примечаниями и комментариями,
которые, как правило, носят характер уточнений, но некоторые
из них, дополняя текст, содержат сведения, появившиеся в печа-
ти после кончины Н. В. Кельцева. Имеется, естественно, и не-
большое число примечаний, в которых отражено несогласие ре-
дактора с позицией автора.
В ходе редактирования был существенно переработан список
литературы: он дополнен новыми источниками (они в основном
указаны в примечаниях), сгруппирован не по главам, как в
первом издании, а по двум основным разделам, что дало воз-
можность исключить дублирующие ссылки; исключены также
ссылки на вторичные источники. В новый текст книги не вошел
список монографической литературы по адсорбции и трудов кон-
ференций, совещаний. Читатель, специально интересующийся
этим вопросом, как уже указывалось, найдет этот список в пер-
вом издании.
Успешная работа над рукописью была бы невозможна без
критической, объективной и доброжелательной рецензии, кото-
рую написал профессор В. В. Серпинский. Большую техниче-
скую помощь в работе над рукописью оказала вдова автора-—
3. А. Кельцева.
Ю. И. ШУМНЦКИЙ
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
АДСОРБЦИОННОЙ ТЕХНИКИ
РАЗВИТИЕ АДСОРБЦИОННОГО
МЕТОДА ОЧИСТКИ
И РАЗДЕЛЕНИЯ СМЕСЕЙ
АДСОРБЦИОННОЕ РАВНОВЕСИЕ
И ПОРИСТАЯ СТРУКТУРА
ОСНОВНЫЕ ВИДЫ
ПОРИСТЫХ адсорбентов
ТЕПЛОТА АДСОРБЦИИ
И ТЕРМОДИНАМИЧЕСКОЕ
ОБОСНОВАНИЕ ТЕОРИИ
ОБЪЕМНОГО ЗАПОЛНЕНИЯ
МИКРОПОР
ИЗБИРАТЕЛЬНОСТЬ АДСОРБЦИИ
КИНЕТИКА АДСОРБЦИИ
КИНЕТИКА ДЕСОРБЦИИ
ДИНАМИКА АДСОРБЦИИ
АДСОРБЦИЯ
СЛАБО СОРБИРУЮЩИХСЯ ГАЗОВ
Из всего многообразия проблем, которые
рассматривает теоретическая адсорбция
для инженера-практика наибольшее
значение, несомненно, имеют учение
о равновесии в адсорбционных системах,
кинетика и динамика процессов адсорбции.
Учение о равновесии, или статика
адсорбции, представляет собой
термодинамически обоснованную и в
целом хорошо разработанную совокупность
взглядов на предельное состояние системы
адсорбтив — адсорбент. Эта совокупность
первоначально складывалась применительно
к описанию адсорбции на плоской
поверхности. В дальнейшем она была
развита для адсорбции в объеме пор.
Одновременно были заложены основы
рациональной классификации пор по
размерам и адсорбентов по
преобладающему типу пор. Микро-, мезо-
и макропоры — определяющие элементы
пористой структуры адсорбентов, от
которых зависит и максимальная
адсорбционная способность, и, в известной
степени, скорость ее достижения.
Основной задачей кинетики физической
адсорбции является изучение диффузии
в зерне твердого пористого тела.
Скорость миграции вещества зависит
от условий проведения процесса и размера
пор. Сложнопористый характер реальных
адсорбентов определяет в общем случае
сложный механизм переноса адсорбтива
в них. Выявление определяющих стадий
миграции стало одним из основных
направлений исследовательских работ
в области кинетики. Другое направление
заключается в создании методов расчета
эффективного коэффициента диффузии
адсорбтива в зерне на основе параметров
пористой структуры адсорбента.
Динамика адсорбции изучает поглощение
вещества в массе гранул, имитирующих
адсорбционный аппарат. Устанавливая
основные закономерности протекания
адсорбционного процесса, она дает
представления, ориентирующие
и в разработке инженерных методов
расчета процесса, и в моделировании
его с помощью электронных
вычислительных машин.
1
РАЗВИТИЕ
АДСОРБЦИОННОГО
МЕТОДА ОЧИСТКИ
И РАЗДЕЛЕНИЯ СМЕСЕЙ
Сведения о применении твердых тел с развитой поверхностью
пришли к нам из глубокой древности. История использования
адсорбентов на первых этапах связана с углеродистыми вещест-
вами. Известно, что уже в древнем Египте уголь использовали
в лечебных целях. Во времена основоположника античной меди-
цины Гиппократа (400 лет до нашей эры) уголь являлся рас-
пространенным средством для удаления дурного запаха, исхо-
дящего от язв и ран. В 1771 г. на способность древесного угля
поглощать газы обратил внимание Шееле.
Однако развитие промышленного адсорбционного метода на-
чалось с блестящего открытия петербургского аптекаря, впослед-
ствии русского академика Ловица. В 1785 г. Ловиц обнаружил,
что древесный уголь, введенный в раствор винной кислоты,
обесцвечивает последний, поглощая органические примеси. Уче-
ный обратил внимание на универсальность угля как поглотите-
ля: растворы очищались им от разнообразных примесей. Эти
выводы были реализованы на практике спустя 9 лет. В пищевой
промышленности уголь стали применять для очистки сахарных
сиропов, в спиртоводочной промышленности — для очистки
спирта от сивушных масел, в русском флоте — для очистки
питьевой воды. Ловицем была предложена методика подготовки
угля к процессу очистки, которая заключалась в его измельче-
нии и прокаливании. Методы, разработанные этим ученым, не
потеряли своего значения до настоящего времени и широко ис-
пользуются в производстве.
Уже после смерти Ловица (1804 г.) предложенные им про-
цессы стали использоваться и в Западной Европе. В 1808 г. во
Франции начали работать сахарные заводы, на которых сироп,
полученный из свеклы, очищали древесным углем.
В 1811 г. Фигуэр продемонстрировал преимущества костного
угля перед древесным при рафинации сахара.
В 1814 г. швейцарский ученый Соссюр опубликовал резуль-
таты исследований, которые подтвердили эффективность угля
как поглотителя различных газов и паров. Он впервые отметил,
что адсорбционные процессы протекают с выделением тепла —
они экзотермичны.
I. Рчлаитие адсорбционного метода очистки и разделения смесей
17
В 1874 г. журнал Русского физико-химического общества со-
• ><>iiiit.i о работах Мельсана, который установил, что древесный
\ и.,н. поглощает равную ему массу хлора, причем в процессе
по, лощения температура в адсорбенте повышается на 30 °C.
1>|.i.'in продемонстрирована также способность угля поглощать
i.inne газы, как сульфид водорода, диоксид серы, аммиак, бро-
ни а, водорода, хлорид этила и цианид водорода. Мельсан отме-
ни, что летучие жидкости (спирты), поглощенные углем, не
вы делаются из него при температурах их кипения. Таким обра-
||>м. зародилась идея об удерживающей способности адсор-
бента.
В 1879 г. Смит сделал попытку классифицировать газы по их
адсорбируемости, приняв за единицу адсорбируемость водорода,
(>п нашел, что объемы газов, поглощенных углем, пропорцио-
нальны следующим числам: Н2=1; № = 4,25; СО = 6,03; 02 = 7,99;
СП4= 10,01; №0=12,90; СО2 = 22,05; SO2 = 36,95.
Все адсорбционные процессы основаны на избирательных
свойствах адсорбентов, поглощающих отдельные компоненты
смесей лучше, чем другие. Явление избирательности адсорбции
было использовано в 1903 г. русским ботаником М. С. Цветом.
Нм была блестяще решена задача разделения каротина (пиг-
мента, входящего в состав листьев) на пять индивидуальных
компонентов. Цвет пропустил спиртовой раствор каротина через
слой адсорбента и обнаружил образование в слое отдельных
зон с окраской, характерной для каждого компонента. Распо-
ложение зон соответствовало адсорбируемости компонентов.
Вводя растворитель, Цвет заставил мигрировать зоны по слою
и выделил компоненты в порядке возрастающего адсорбцион-
ного сродства.
Всемирную известность метод Цвета получил после опубли-
кования в 1937 г. в Вене венгерскими биохимиками Цехмейсте-
ром и Чолноки монографии «Хроматографический адсорбцион-
ный анализ». В настоящее время хроматографический анализ
занял ведущее место в химии, биохимии, медицине, биологии,
в фармацевтической и многих других отраслях промышленно-
сти для быстрой и точной идентификации вещества, его очистки
и выделения. «Метод Цвета осуществил заветную мечту хими-
ка— разделить до анализа смесь на все ее компоненты», — пи-
сали Цехмейстер и Чолноки.
В течение XIX в. непрерывно совершенствуется методика
получения углей, обесцвечивающих жидкие среды, улучшается
их качество. Однако производство углей в промышленном мас-
штабе задерживалось из-за несовершенства аппаратурного
оформления процесса. В начале XX века Острейко запатентовал
методы получения активных углей путем газовой и химической
активации. В 1911 г. в Голландии и в 1913 г. в Германии на ос-
нове этих патентов были построены заводы получения активных
2—1346
18
1. Развитие адсорбционного метода очистки и разделения смесей
углей. Одновременно лабораторные исследования создали пред-
посылки для применения углей в области очистки и разделения
газов.
Первая мировая война выдвинула перед человечеством мно-
го проблем, которые нужно было решить в максимально корот-
кие сроки. Одной из таких проблем была защита армий от от-
равляющих веществ. Отравляющие газы были применены не-
мецкими войсками 22 апреля 1915 г. на реке Ипр против
французских и английских войск, а 18 мая этого года на реке
Равка под Варшавой — против русских войск. Большое число
химиков лихорадочно принялось разрабатывать поглотители
отравляющих газов. Среди них был профессор Политехническо-
го института и одновременно заведующий Петербургской Цент-
ральной лабораторией Министерства финансов Н. Д. Зелин-
ский. Волею случая 30 лет назад, проходя стажировку в лабо-
ратории профессора Майера в Геттингеме, он синтезировал ди-
хлордиэтилсульфид, т. е. иприт, получил ожоги и, таким обра-
зом, первым испытал на себе действие этого смертоносного газа.
Именно Зелинский отказался от бесплодных попыток создать
средство защиты на основе химической реакции: химический
поглотитель мог обезвредить один конкретный газ, но не весь
арсенал отравляющих веществ, имеющийся у противника.
В результате интенсивной работы Зелинским и его сотруд-
никами был создан противогаз на основе активных углей. Его
эффективность находилась в непосредственной связи с высокой
молекулярной массой всех отравляющих веществ, которые при
атаке стелятся по земле. Вместе с тем, высокомолекулярные
вещества избирательно извлекаются из воздуха углем. Положи-
тельным качеством угля является его гидрофобность. Методика
активации угля, предложенная Зелинским, предусматривала
двухкратное прокаливание смоченного водой угля в газовых пе-
чах или ретортах при 800—900 °C. Прекрасные результаты ис-
пытаний противогаза на хлоре, фосгене, хлорпикрине, а также
простота его конструкции позволили быстро снабдить армию
защитными средствами и спасти тысячи жизней солдат и офи-
церов. Немецкая армия, располагавшая не только эксперимен-
тальными данными своих химиков, но и разведывательными
сведениями о работах русских химиков, также использовала
активный уголь как средство защиты. «Я изобрел его не для
нападения, а для защиты миллионов молодых жизней от стра-
даний и смерти» — напишет впоследствии Зелинской о своем
угольном противогазе.
Заслуга организации противохимической обороны войск при-
надлежит ученику Зелинского профессору Московского универ-
ситета Н. А. Шилову, который во время первой мировой войны
возглавлял противогазовую службу Западного фронта. В Черно-
виках при участии ближайшего помощника Шилова Л. К- Ле-
1. Развитие адсорбционного метода очистки и разделения смесей
19
пинь была организована мастерская по ремонту противогазов.
Наблюдения во фронтовой лаборатории, а также обобщение
экспериментальных работ в мирное время позволили Шилову
сформулировать законы динамики адсорбции, которые до сих
пор являются основой в инженерных расчетах адсорбционных
установок.
В настоящее время, когда вопрос жизни или смерти стоит
уже не только перед армией, но и перед всем человечеством,
обеспокоенным катастрофическим загрязнением биосферы, на-
стало время вновь обратиться за помощью к адсорбции — одно-
му из самых эффективных методов защиты окружающей среды
от загрязнений.
В США заводы по получению активных углей были пущены
в эксплуатацию в период первой мировой войны. Угли, изготов-
ленные из персиковых косточек и скорлупы орехов, предназна-
чались для противогазов. После войны (1917—1923 гг.) рост
производства активных углей стимулируется запросами про-
мышленности, в первую очередь сахарной. В этот период в ка-
честве основного сырья для активации стал служить отработан-
ный целлюлозный щелок (побочный продукт при получении бу-
маги) и бурый уголь.
Опыт противогазовой техники был использован для разра-
ботки разнообразных рекуперационных установок с неподвиж-
ным слоем активного угля. Интенсивная работа в этом направ-
лении в свое время проводилась немецкими инженерами. Улав-
ливание бензола из светильного и коксового газов, бензина —
из природных газов, растворителей — из выбросных газов рези-
новой промышленности, эфира и спирта — в производстве по-
рошков— вот далеко не полный список основных направлений
применения адсорбционного метода для рекуперации продуктов
из газовой фазы в период с 1920 по 1930 г. Десорбцию на этих
установках во всех случаях проводили водяным паром.
Установки этого типа имеют ряд существенных недостатков:
периодичность процесса, неполная отработка адсорбционной ем-
кости адсорбента, значительная площадь, занимаемая оборудо-
ванием, трудность автоматизации процессов и их управления.
Эти недостатки побудили искать новые конструктивные реше-
ния. В период с 1946 по 1955 г. в США (Берг), Советском Сою-
зе (Кельцев, Платонов), Венгрии (Бенедек, Сепеши) был раз-
работан непрерывный метод разделения газовых смесей в дви-
жущемся слое адсорбента. При этом были учтены принципы аб-
сорбционных и ректификационных установок, но четкость разде-
ления усиливалась высокими избирательными свойствами ад-
сорбента. На установках с движущимся слоем не только уда-
лось решить задачу выделения суммы компонентов из газового
потока, но и разделить их непосредственно в адсорбционной ко-
лонне, получив товарные продукты. Как правило, установки с
2*
20
1. Развитие адсорбционного метода очистки и разделения смесей
движущимся слоем рекомендуются для работы под повышен-
ным давлением (0,5—2,0 МПа), что позволяет увеличить их
пропускную способность по газу.
Для очистки больших объемов газа низкого давления, что
характерно в первую очередь для процессов обезвреживания га-
зовых выбросов в атмосферу, было предложено поглощение про-
водить в псевдоожиженном слое адсорбента (например, в псев-
доожиженном слое активного угля целесообразно очищать вен-
тиляционные газы производства химического волокна). К сожа-
лению, широкому внедрению методов с подвижными слоями ад-
сорбента препятствует отсутствие высокопрочных и достаточно
дешевых шариковых активных углей и других адсорбентов*.
Адсорбционная техника в разные периоды базировалась на
применении разных адсорбентов: до первой мировой войны —
на углеродных адсорбентах, в период между первой и второй
мировыми войнами на активных углях и силикагелях**; после
второй мировой войны не просто прогресс, но в ряде областей
техническая революция были связаны с применением синтетиче-
ских цеолитов.
Работами английского физикохимика Баррера и американ-
ского синтетика Брека были найдены пути промышленного полу-
чения уникальных алюмосиликатных адсорбентов-цеолитов, об-
ладающих не только высокой избирательностью адсорбции, но
и способностью разделять вещества, используя различия в раз-
мерах и форме молекул. На основе патентов Баррера и Брека
компания Линде (США) в 1955 г. начала в промышленном мас-
штабе изготовлять синтетические цеолиты общего назначения.
В 1959 г. при Академии Наук СССР под председательством
М. М. Дубинина была создана Комиссия по цеолитам. В ре-
зультате интенсивной работы ряда организаций, координировав-
шейся Комиссией, проблема синтеза и выпуска цеолитов в про-
мышленном масштабе в нашей стране была решена: с 1964 г.
страна обеспечена цеолитами общего назначения.
Сегодня без преувеличения можно сказать, что ассортимент
адсорбентов, изготавливаемых предприятиями химической и
нефтехимической промышленности, позволяет решить подавляю-
щее большинство задач газоочистки. Не менее эффективны ад-
сорбенты при очистке воды и иных жидких сред.
Подбором соответствующего адсорбента адсорбционному про-
цессу может быть придана как избирательность, так и универ-
сальность. Универсальными являются адсорбционные процессы,
используемые при подготовке воздуха к низкотемпературному
* Установки с кипящим слоем адсорбента производительностью до 200 тыс.
м3/ч в настоящее время успешно применяют в технике санитарной очистки
газов. (Прим. ред.).
** Промышленный синтез силикагеля был осуществлен в 20-х годах Пэтри-
ком. (Прим. ред.).
1. Развитие адсорбционного метода очистки и разделения смесей
21
разделению (извлекаемые компоненты — вода, диоксид углеро-
да, ацетилен), подготовке природного газа к транспорту (вода,
сульфид водорода, диоксид углерода), получении экзотермиче-
ской контролируемой атмосферы и т. д.
В подавляющем большинстве адсорбционных процессов для
регенерации адсорбента к нему подводят тепло. Разработанные
в последнее время установки короткоцикловой безнагревной ад-
сорбции работают без подвода тепла, благодаря чему интенсив-
ность их значительно выше интенсивности обычных установок с
политропным режимом. С помощью таких установок решают не
только проблему удаления примеси (осушка), но и задачу кон-
центрирования смеси (выделение водорода) и разделения ее на
компоненты (воздух — на азот и кислород). Установки этого
типа легко поддаются автоматизации.
Период шестидесятых — восьмидесятых годов нашего столе-
тия отличается быстрыми темпами внедрения адсорбционной
техники в медицину. Разработанный в США, Советском Союзе
и ряде других стран метод очистки крови от вредных веществ,
названный гемосорбцией, предусматривает отбор крови из ар-
терии пациента, пропускание ее через небольшую колонку со
специально приготовленным активным углем или другим сор-
бентом и возвращение в систему кровообращения пациента.
Гемосорбцию широко используют при острых отравлениях бар-
битуратами, фосфорорганическими инсектицидами, хлорирован-
ными углеводородами. Не менее важно использование гемосорб-
ции при острых и хронических заболеваниях печени: печеночной
недостаточности, желчнокаменной болезни, циррозах печени,
печеночной коме.
К гемосорбентам, естественно, предъявляются повышенные
требования, обусловленные контактом адсорбента с кровью че-
ловека. Гемосорбенты должны обладать высокой механической
прочностью, химической устойчивостью, значительной адсорб-
ционной способностью по удаляемым веществам; они должны
быть нетоксичными, стерильными, минимально травмировать
форменные элементы крови.
Большинство разработанных абсорбционных процессов бази-
руются на синтетических адсорбентах. Между тем, поверхность
земли и, безусловно, дно океана содержит огромный потенциал
природных адсорбентов. Перспективы применения природных
адсорбентов в народном хозяйстве весьма благоприятны. Одна-
ко, геологам и химикам придется еще изрядно потрудиться, что-
бы природные сорбенты стали товарными продуктами и были
внедрены в различные области промышленности. В этой связи
уместно вспомнить слова М. В. Ломоносова: «Металлы и мине-
ралы сами во двор не придут. Они требуют глаз и рук для свое-
го прииску». Но расходы на геологическую разведку адсорбен-
тов, предпринятую в широком масштабе, безусловно, окупятся,
22 2. Адсорбционное равновесие и пористая структура адсорбентов
как сторицей окупились расходы на разведку природного газа.
В последние годы, в частности, выявлены крупные залежи при-
родных цеолитов — шабазита, морденита, клиноптилолита в
Грузии, Армении, Азербайджане, Закарпатье, на Сахалине и в
ряде других районов. Концентрация цеолитов в породе достига-
ет 80 и более процентов.
На развитие адсорбционной техники в Советском Союзе по-
ложительное влияние оказали фундаментальные теоретические
обобщения, проведенные советскими учеными. Ключевыми рабо-
тами в этом направлении являются* *:
1. Теория объемного заполнения микропор, описывающая ад-
сорбционное равновесие в широкой области рабочих условий.
2. Развитие теории термодинамики адсорбции.
3. Разработка теории и основ расчета кинетики адсорбции
и десорбции.
4. Разработка теории массопереноса применительно к основ-
ным типам адсорбционных процессов.
Работы в области адсорбции в нашей стране координирует
Научный Совет Академии Наук СССР по адсорбции (председа-
тель М. М. Дубинин), созданный в 1963 г. на основе Комиссии
по цеолитам. В сфере его влияния находятся как теоретические
исследования, так и прикладные работы.
2
АДСОРБЦИОННОЕ
РАВНОВЕСИЕ
И ПОРИСТАЯ СТРУКТУРА
АДСОРБЕНТОВ
Адсорбция и адсорбционные силы
Адсорбцией называют концентрирование веществ на поверхно-
сти раздела фаз или в объеме пор твердого тела.
В процессе адсорбции участвуют, как минимум два агента:
тело, на поверхности или в объеме пор которого происходит кон-
* Библиография монографий советских и зарубежных ученых, изданных в
СССР, приведена в книге Н. В. Кельцева «Основы адсорбционной техники»
(М., Химия, 1976). В ней же представлен перечень трудов, совещаний и кон-
ференций по адсорбции, адсорбентам и адсорбционной технике (Прим. ред.).
Адсорбция и адсорбционные силы
23
центрирование поглощаемого вещества (его называют адсор-
бентом), и поглощаемое вещество. Последнее, если оно нахо-
дится в газовой или жидкой объемной фазе, т. е. в неадсорби-
рованном состоянии, называется адсорбтивом, а после того
как оно перешло в адсорбированное состояние-—адсорба-
том. Любое твердое вещество обладает поверхностью и, следо-
вательно, потенциально является адсорбентом. Однако в техни-
ке используют твердые адсорбенты с сильно развитой внутрен-
ней поверхностью (объемом пор). Развитие внутренней поверх-
ности (объема пор) в твердом теле достигается путем создания
специальных условий в процессе его синтеза или в результате
дополнительной обработки [1].
По типу сил, обусловливающих адсорбцию все адсорбцион-
ные явления можно разбить на две основные группы: физиче-
скую адсорбцию, и сорбцию, основанную на силах химиче-
ского взаимодействия, или хемосорбцию. Физическая ад-
сорбция вызывается силами молекулярного взаимодействия.
Универсальными силами молекулярного взаимодействия явля-
ются дисперсионные силы. Молекулы любого адсорбтива
обладают флуктуирующими диполями и квадруполями, вызы-
вающими «мгновенные» отклонения распределения электронной
плотности от среднего распределения. При сближении молекул
адсорбтива с атомами или молекулами адсорбента движение
флуктуирующих диполей (квадруполей) приобретает упорядо-
ченный характер, обусловливающий возникновение притяжения
между ними. Свое название дисперсионные силы получили
вследствие того, что ими вызывается и явление рассеяния света.
Дисперсионные силы не зависят от характера распределения
электронной плотности в молекулах адсорбтива и адсорбента и
поэтому взаимодействие, вызываемое ими, носит неспецифиче-
ский характер.
В ряде случаев дисперсионные силы усиливаются электро-
статическими силами — ориентационными и индукци-
онными. Ориентационные силы возникают при взаимодейст-
вии полярных молекул с поверхностью, содержащей электроста-
тические заряды (ионы, диполи), индукционные — вызываются
изменением электронной структуры молекул адсорбтива и ад-
сорбента под действием друг друга: возникновением в молеку-
лах адсорбтива дипольных моментов, наведенных зарядами ад-
сорбента, или возникновением дипольных моментов в адсорбен-
те под действием зарядов молекул адсорбтива. Взаимодействие,
вызываемое электростатическими силами, зависит от химиче-
ской природы адсорбтива и адсорбента и, следовательно, яв-
ляется специфическим. Вклад специфического взаимодействия в
общую энергию взаимодействия при адсорбции любых молекул
на электронейтральных углеродных адсорбентах практически
равен нулю, а при адсорбции полярных молекул на цеолитах,
24
2. Адсорбционное равновесие и пористая структура адсорбентов
отличающихся гетероионным характером, соизмерим с вкладом
нссиецифической составляющей.
В некоторых случаях специфическое взаимодействие может
усиливаться в результате образования водородной связи между
адсорбированной молекулой и молекулами адсорбента. Харак-
терным примером адсорбции с образованием водородной связи,
является поглощение воды и спиртов на силикагеле, поверх-
ность которого покрыта гидроксильными группами.
Адсорбция является процессом самопроизвольным и экзотер-
мическим, т. с. се протекание сопровождается выделением тепла..
В отличие от физической адсорбции при хемосорбции не
сохраняется индивидуальность адсорбтива и адсорбента. При
сближении молекул адсорбтива с поверхностью происходит пе-
рераспределение электронов взаимодействующих компонентов с.
образованием химической связи. Если физическую адсорбцию
можно сравнить с конденсацией, то хемосорбционный процесс
должен рассматриваться как химическая реакция, протекающая
на поверхности раздела фаз.
Физическую и химическую адсорбцию можно различить по
теплоте адсорбции. Теплота физической адсорбции соизмерима
с теплотой конденсации веществ и не превышает 80—-
120 кДж/моль. Теплота хемосорбции 1 моль вещества достигает
нескольких сотен килоджоулей. Хемосорбция, как правило, про-
текает с небольшой скоростью. Это обстоятельство также часто
используют для ее распознавания. Кроме того, хемосорбция мо-
жет происходить при высоких температурах, когда физическая
адсорбция пренебрежимо мала. Наконец, для хемосорбции ха-
рактерно резкое скачкообразное изменение поглотительной спо-
собности по извлекаемому компоненту при переходе от адсор-
бента одной химической природы к адсорбенту другой природы.
В отличие от физической адсорбции, при хемосорбции погло-
тительная способность может возрастать в определенном темпе-
ратурном интервале. Это явление связано с небольшой ско-
ростью хемосорбции при низких температурах.
В качестве примера на рис. 2,1 приведены температурные зависимости-
адсорбции семиоксида рения на трех адсорбентах, свойства которых будут
описаны ниже [2]. Кривые получены при концентрации Re2O? в объемной
фазе 0,02 г/м3. Как следует из графика сорбционная емкость всех исследо-
ванных адсорбентов (цеолитов) вначале резко повышается с увеличением
температуры, проходит через максимум, после чего снижается. Такое пове-
дение системы связано со сложным характером сорбции семиоксида рения,,
когда наряду с физической адсорбцией происходит образование поверхност-
ных комплексов молекулы Re2O? с элементами кристаллической решетки
адсорбента.
При хемосорбции адсорбированные молекулы не могут пере-
мещаться по поверхности адсорбента, их положение фиксиро-
вано и такая адсорбция называется локализованной. Физиче-
Пористая структура адсорбентов
25
Гис. 2,1. Зависимость адсорбционной способности цео-
.... по семиоксиду рения от температуры (концент-
рация Re2O? в газовой фазе —0,02 г/м3):
1 - NaX без связующего; 2 — NaX со связующим;
з природный морденит.
екая адсорбция может быть как лока-
лизованной так и не локализованной.
Обычно с повышением температуры
молекулы приобретают подвижность и
характер процесса изменяется: локали-
зованная адсорбция переходит в не-
локализованную.
Физическая адсорбция газов и па-
ров происходит по одним и тем же за-
кономерностям, причем переход адсорбтива из области пара
(7’<Ткр) в область газа (7’>7'кр) не сопровождается резким из-
менением адсорбируемости.
Пористая структура адсорбентов
Твердые сорбенты как правило отличаются «ажурной» внутрен-
ней структурой, включающей поры разного размера. В зависи-
мости от размеров поры подразделяют на три типа: микро-,
мезо-, и м а к р о п о р ы. Ниже дана их характеристика.
Микропоры. Наиболее мелкие поры — микропоры — имеют
размеры, соизмеримые с адсорбируемыми молекулами. По дан-
ным рентгеновского метода, их эффективные радиусы преиму-
щественно находятся в интервале от 0,5 до 1,0 нм [1, 3]. В ка-
честве верхней границы размера микропор принимают радиус
1,5 нм. Суммарный объем микропор
промышленных адсорбентов обычно не
превышает 0,5 см3/г.
Характерной чертой адсорбции в
микропорах является существенное по-
вышение энергии адсорбции по срав-
нению с адсорбцией в более крупных
порах. На рис. 2,2 изображены кривые
i;ik называемых дифференциальных
•1еплот адсорбции м-гексана для двух
углеродных адсорбентов, микропори-
стого активного угля (кривая 1) и не-
пористой сажи (кривая 2), предвари-
к'льно прокаленной при 950 °C. При
Рис. 2,2. Зависимость дифференциальной теплоты ад-
сорбции Q н-гексана активным углем (1) и графити-
роиинной сажей (2) от величины адсорбции а при
70
о,ммоль/г
а,ммоль/г
Q-10-1, кДж/ммоль
26
2. Адсорбционное равновесие и пористая структура адсорбентов
Таблица 2-J. Относительная адсорбционная способность сажи
и активных углей по бензолу при 20 °C
Давление P/Ps Сажа Активные угли с микропорами Давление p/ps Сажа Активные угли с микропорами
более крупными более мелкими более крупными более мелкими
МО"5 0,02 0,12 0,44 1-Ю-2 0,33 0,71 0,87
1 10-4 0,06 0,16 0,57 1 • ю-1 0,81 0,92 0,96
1 -10-3 0,14 0,46 0,73 0,175 1,00 1,00 1,00
одинаковой степени заполнения адсорбционного пространства*
энергия адсорбции в микропорах в среднем в 1,5 раза больше,
чем на поверхности сажи. В случае поглощения паров (или га-
зов) повышение энергии адсорбции в микропорах приводит к
резкому возрастанию адсорбционной способности в области ма-
лых равновесных давлений.
В табл. 2-1 приведены относительные значения адсорбционной способ-
ности по бензолу при 20 °C для предварительно прокаленной при 950 °C
иепористой сажи и двух образцов активных углей, обладающих различными
пористыми структурами. В этой таблице значения адсорбционной способно-
сти при заданном относительном давлении сравниваются с адсорбционной
способностью при относительном давлении p/ps = 0,175 (р —давление паров
бензола; р3 — то же, предельное при 20 °C). Для сажи это давление пример-
но отвечает покрытию поверхности сплошным мономолекулярным слоем, а
для активных углей — практическому завершению заполнения микропор.
Экспериментальные данные табл. 2-1 иллюстрируют, сколь значительно в об-
ласти низких относительных давлений возрастает количество адсорбирован-
ного пара при переходе от непористых к микропористым адсорбентам.
В таких промышленных адсорбентах, как активные угли или
синтетические цеолиты, размеры микропор соизмеримы с раз-
мерами промежутков между смежными порами, образованных
веществом адсорбента. Это приводит к тому, что все атомы и мо-
лекулы вещества адсорбента находятся во взаимодействии с
молекулами адсорбата в микропорах. Другими словами, во всем
пространстве микропор существует поле адсорбционных сил.
В этом заключается основное отличие адсорбции в микропорах
от адсорбции в более крупных порах.
Адсорбция в микропорах сводится, таким образом, к запол-
нению пространства микропор адсорбируемыми молекулами. Ос-
новным параметром пористой структуры является объем
микропор (например, для единицы массы адсорбента) и
обобщенная характеристика их размеров. Понятие «удельная
* Для данных рис. 2,2 степень заполнения адсорбционного пространства
можно определить как отношение сорбции к предельной сорбции, которая,
для угля и сажи соответственно равна 1,5 и 0,5 ммоль/г. (Прим. ред.).
Пористая структура адсорбентов
27
поверхность» для микропор не имеет геометрического смысла,
и ее определение по принятым уравнениям сводится к нахожде-
нию формальной константы этих уравнений.
Иногда наиболее крупные микропоры — радиусом от 0,7 до
1,5 нм выделяют в отдельную категорию с названием с у пер-
ми кр о поры. В супермикропорах при малых степенях запол-
нения адсорбционного объема, как считают, адсорбция может
протекать по механизму покрытия поверхности одним или даже
несколькими слоями молекул адсорбата.
Мезопоры. Эффективные радиусы более крупных пор — мезо-
нор— много больше размеров адсорбируемых молекул. Они ле-
жат в интервале от 1,5 до 100—200 нм. Стенки таких, уже отно-
сительно крупных пор, образованы очень большим числом ато-
мов или молекул вещества адсорбента; для этого случая
приобретает физический смысл понятие о поверхности раздела
фаз, т. е. о поверхности пор адсорбента. Обычно поверхность
адсорбента относят к единице его массы и называют удель-
ной поверхностью.
Для мезопор действие адсорбционных сил проявляется не во
всем их объеме, а практически только на небольшом расстоянии
от стенок. Поэтому на поверхности пор происходит мономолеку-
лярная и полимолекулярная* адсорбция паров, т. е. образова-
ние последовательных адсорбционных слоев, завершающееся
заполнением этой разновидности пор по механизму капиллярной
конденсации.
Основными параметрами мезопор являются удельная по-
верхность, объем пор и функция распределения объема пор по
размерам. Последние два параметра определяются методами
капиллярной конденсации и ртутной порометрии. В зависимости
от степени развития пор и преобладающих их радиусов удель-
ные поверхности мезопор могут заключаться в интервале от 10
до 400 м2/г. Мезопоры являются основными транспортными ар-
териями, по которым осуществляется подвод вещества к «емко-
стям» — микропорам.
Макропоры. Наконец, самые крупные поры адсорбентов —
макропоры имеют эффективные радиусы больше 100—200 нм.
11х удельная поверхность очень мала — от 0,5 до 2 м2/г, вследст-
вие чего адсорбцией на поверхности пор этого типа практически
можно пренебречь. Объем макропор у активных углей состав-
ляет от 0,2 до 0,8 см3/г. В крупных порах капиллярная конден-
сация не происходит, и единственным методом оценки их объема
и кривой распределения пор по размерам является метод ртут-
iioii порометрии. Макропоры играют роль крупных транспорт-
ных артерий в зернах адсорбентов.
" Ни шмолекулярная адсорбция происходит не в результате действия сил
<>1>1и‘нт — адсорбат, а в результате взаимодействия молекул адсорбтива с
ринг, адсорбированными молекулами адсорбата (Прим. ред.).
28
2. Адсорбционное равновесие и пористая структура адсорбентов
Все адсорбенты в соответствии с преобладающим размером
пор можно подразделить на три предельных структурных клас-
са: макропористые, мезопористые и микропористые.
Некоторые макропористые адсорбенты применяются в хрома-
тографии. К мезопористым адсорбентам принадлежит большое
число силикагелей, алюмогелей и алюмосиликатных катализа-
торов, а также многие виды природных глин, применяемых для
удаления относительно крупных молекул из различных жидких
сред (например, при очистке масел). Типичным представителем
микропористых адсорбентов являются дегидратированные кри-
сталлические алюмосиликаты — цеолиты и некоторые типы ак-
тивных углей, в частности сарановые угли.
Подавляющее большинство промышленных адсорбентов, при-
меняемых для очистки газов и рекуперации паров (активные
угли, силикагели), содержат широкую гамму пор различного
размера и относятся к смешанным структурным типам адсор-
бентов.
Термическое уравнение адсорбции
и его частные случаи
При равновесии для выбранной системы адсорбент — адсорбтив
количество поглощенного газа или пара (а) является функцией
давления поглощаемого вещества (р) и температуры (Т):
a = f(p,T) (2.1)
При описании адсорбционного равновесия количество поглощенного ве-
щества (адсорбционную способность, величину адсорбции, адсорбируемость)
выражают обычно в молях на 1 г адсорбента, граммах на 100 г чистого
адсорбента, а иногда в граммах на 1 см3 адсорбента. Естественно, соотноше-
ние между адсорбционной способностью, отнесенной к единице массы или
объема адсорбента, зависит от плотности последнего. Адсорбционную спо-
собность по относительно плохо поглощающимся газам удобно выражать в
единицах объема газа, поглощенного единицей массы адсорбента, т. е. в
см3/г.
Уравнения типа (2.1) называют термическими. Они справед-
ливы при любых температурах. В качестве характеристики ад-
сорбционных свойств твердых тел используют зависимость ад-
сорбционной способности от давления при постоянной темпера-
туре — изотерму адсорбции:
а = f (р) при Т = const (2.2)
В реальных процессах очистки и разделения веществ влия-
ние адсорбции сопутствующих веществ, а также кинетические
факторы могут вызвать необходимость внести коррективы в оп-
ределение адсорбционной способности по изотермам чистых ком-
понентов. Однако во всех случаях практического использования
Термическое уравнение адсорбции и его частные случаи
29
Рис. 2,3. Основные типы (I—V) изотерм адсорбции.
адсорбционного метода изотерма адсорбции является основной
характеристикой адсорбента и определяет выбор оптимальных
рабочих условий процесса.
Одновременно совокупность изотерм адсорбции является
источником информации о структуре адсорбента, тепловом эф-
фекте адсорбции и ряде других физико-химических и технологи-
ческих характеристик системы.
Брунауэром [4] выделены пять основных типов изотерм ад-
сорбции, которые представлены на рис. 2,3. В случае промыш-
ленных адсорбентов тип I характерен для микропористых ад-
сорбентов, практически не содержащих мезопор*. Начальные
выпуклые участки изотермы типов II и IV также связаны с
микропорами, присутствующими в сорбентах преимущественно
мезо- и микропористых, полимолекулярная адсорбция и капил-
лярная конденсация в которых определяют дальнейший ход
кривых. Начальные выгнутые участки сравнительно редко встре-
чающихся изотерм типов III и V характерны для систем адсор-
бент— адсорбат, когда взаимодействие молекул адсорбата с
адсорбентом меньше межмолекулярного взаимодействия для
молекул адсорбата, например вызванного проявлением водород-
ных связей.
Основное отличие II и III от IV и V типов заключается в
том, что объем мезопор у адсорбентов типа IV или V в резуль-
тате капиллярной конденсации заполняется адсорбатом раньше,
чем относительное давление приблизится к единице. В резуль-
тате этого на изотермах появляется верхний почти горизонталь-
ный участок.
В основе расчета любого технологического процесса, хотя он
в большинстве случаев протекает в динамических условиях, на-
ходится совокупность кривых, отражающих равновесие погло-
щаемого компонента с адсорбентом, т. е. совокупность изотерм
адсорбции. Ее определяют в широком интервале температур,
* Объяснение, которое дает основным типам изотерм адсорбции И. В. Кель-
цев, отличается от объяснения, приводимого автором классификации Бру-
науэром. Это свидетельствует о том, что адсорбция — явление в достаточной
степени сложное, и интерпретация изотерм адсорбции, исходящая только из
них самих, не является однозначной (Прим. ред.).
30
2. Адсорбционное равновесие и пористая структура адсорбентов
Рис. 2,5. Изобары адсорбции паров воды
тах:
1 — цеолит NaA; 2 — силикагель; 3 — оксид алюминия.
Рис. 2,4. Изотермы адсорбции паров воды
на цеолите СаА.
охватывающем область рабочих условий проведения как адсорб-
ции, так и десорбции. На рис. 2,4 приведена такая совокупность
изотерм адсорбции паров воды на цеолите СаА, являющаяся ис-
ходной информацией для обоснования и расчета процессов
осушки газов.
Приведенные изотермы являются типичными для адсорбции
веществ на микропористых адсорбентах. Из них видно, что ве-
личина адсорбции монотонно возрастает при увеличении дав-
ления. Рост температуры приводит к понижению адсорбцион-
ной способности. Одновременно при изменении температуры
происходит изменение формы изотермы адсорбции: она имеет
резко выпуклый характер при низких температурах, но посте-
пенно сглаживается при их повышении.
Иногда для описания адсорбционного равновесия использу-
ют зависимость адсорбционной способности от температуры при
постоянном давлении адсорбтива — изобару адсорбции*:
а == f (Т) при р = const (2.3)
На рис. 2,5 представлены изобары адсорбции паров воды на основных
типах осушителей. Сравнение изобар позволяет в рассматриваемом случае
сделать следующие практические выводы. С одной стороны, ясно, что цеоли-
* Из уравнения (2,1) вытекает еще одна частная функция — изостера
адсорбции, т. е. зависимость равновесного давления от температуры при
постоянном количестве поглощенного вещества или при постоянной степени
заполнения адсорбционного пространства:
р ~ f (Г) при а — const (2.4)
Это очень важная функция, несущая непосредственную информацию о теп-
лоте адсорбции (Прим. ред.).
Экспериментальные методы определения изотерм адсорбции 31
ил являются единственным типом адсорбентов, позволяющим проводить про-
цесс осушки при относительно высоких температурах. С другой стороны,,
регенерация цеолитов также должна протекать при сравнительно высоких
н мнературах: даже при 150—200 °C они прочно удерживают значительное-
количество влаги. ,j
Экспериментальные методы определения
изотерм адсорбции
Адсорбцию газов и паров твердыми телами исследуют стати-
ческими и динамическими методами. В статических методах ад-
сорбент помещают в атмосферу газа или пара и после установ-
ления равновесия измеряют равновесные давления, температуру
и количество поглощенного адсорбтива. Измерения адсорбцион-
ной способности производят или по привесу адсорбента (весо-
вой метод), или по разности количества адсорбтива, введенного
в измерительную ячейку и оставшегося в равновесной газовой
фазе после контакта с адсорбентом (объемный метод). Стати-
ческие методы применяются обычно при изучении адсорбции ин-
дивидуальных газов или паров с использованием вакуумной
техники. Путем вакуумирования и нагрева адсорбент освобож-
дают от ранее адсорбированных веществ.
Весовой метод. Прецизионные адсорбционные весовые установки под-
робно описаны в монографии Сарахова [5].
На рис. 2,6 показана вакуумная установка для определения адсорбцион-
ной способности с помощью весов Мак-Бэиа, широко применяющаяся в ла-
бораторной практике. Важнейшей частью этих весов является кварцевая
спиральная пружина, находящаяся в стеклянном кожухе. Пружина оканчи-
вается двумя крючками. Верхним крючком оиа через систему подвесов кре-
пится к неподвижному крючку колбы. На нижнем крючке ее подвешена ча-
Рнс. 2,6. Схема установки для исследования адсорбции весовым методом:
1 — форвакуумный насос; 2 — насос глубокого вакуума; 3 — буферные емкости и балло-
ны для хранения газов; 4 — манометр Мак-Леода; 5 -— ртутный манометр; 6 — колба с
Жидким адсорбтивом; 7—пружинные весы; 8 — термостат.
32
2. Адсорбционное равновесие и пористая структура адсорбентов
шечка с навеской адсорбента. Растяжение пружины пропорционально массе
поглощенного вещества и фиксируется по положению чашечки с помощью
отсчетного микроскопа-катетометра. Нижияя часть кожуха с пружиной поме-
щается в термостат 8.
Регенерацию образца адсорбента (удаление ранее поглощенного веще-
ства) производят его длительной откачкой при остаточном давлении поряд-
ка 1-10~3 Па-с одновременным нагревом. Максимально допустимая темпе-
ратура нагрева определяется природой адсорбента; обычно она составляет
350 °C для цеолита или угля, 200 °C — для силикагеля. Вакуум в системе
создают двумя последовательно включенными насосами: форвакуумным на-
сосом 1 и насосом глубокого вакуума 2. Для измерения давления в системе
в период регенерации предусмотрены две манометрические лампы: термо-
парная н ионизационная, соединенные с вакуумметром, например ВИТ-1.
Периодическую проверку показаний вакуумметра производят по манометру
.Мак-Леода 4. Равновесное давление газа (пара) в системе измеряется мано-
метром Мак-Леода или ртутным манометром 5, снабженным отсчетным мик-
роскопом. Точность измерения давления манометром 5 составляет около
5 Па.
Перед началом опыта с помощью катетометра отмечается положение
пустой чашечки пружинных весов. Затем иа чашечку укладывают образец
адсорбента в виде гранулы или кристаллического порошка (приблизительно
0,05 г) и производят регенерацию образцов. Длительность регенерации обыч-
.но превышает 2 ч. После этого образец термостатируют и отмечают новое
положение нижнего крючка пружины. Разность показаний катетометра при
первом и втором замерах позволяет установить навеску адсорбента.
Для измерений адсорбционной способности в установку из колбы 6 или
-баллонов 3 впускают наибольшее количество адсорбтива. После установле-
ния равновесия, о чем свидетельствует постоянство показаний весов, произ-
водят измерение равновесного давления. По разности показаний катетометра
при взвешивании чашечки с вакуумированным адсорбентом и насыщенным
.адсорбентом определяют количество адсорбированного вещества при данном
давлении. Его относят к навеске адсорбента и таким образом устанавливают
значение адсорбционной способности. При построении изотермы определяют
семейство точек, от замера к замеру изменяя давление в системе.
Объемный метод. Другая методика исследования адсорбционного
равновесия заключается в определении объема газа, поглощенного навеской
адсорбента. Принципиальная схема объемной установки представлена на
рис. 2,7. Навеску адсорбента помещают в ампулу 1 известного объема и ва-
куумируют при открытых кранах 2 и 5. После этого отсоединяют ампулу 1
краном 2. Газом-адсорбтивом заполняют емкость 3, объем которой также
известен. Затем кран 5 закрывают. Количество введенного в емкость 3 газа
определяют по уравнению состояния газа на основании показаний ртутного
манометра 4. Открывая кран 2, приводят адсорбент в соприкосновение с га-
зом-адсорбтивом. Количество поглощенного газа определяют по равновесно-
му давлению в системе с учетом остаточного содержания газа в емкостях
1 и 3, а также в коммуникациях. При работе по объемному методу кроме
•описанных выше операций часто приходится с помощью гелия измерять
-объем газового пространства, что крайне усложняет работу.
Рис. 2,7. Принципиальная схема установки ДЛ[
исследования адсорбции объемным методом:
1—ампула с адсорбентом; 2, 5 — краны; 3 — ем
кость; 4 — ртутный манометр.
Экспериментальные методы определения изотерм адсорбции
33
Таблица 2-2. Давление пара над насыщенными растворами
некоторых солей при 20 °C
Соль Давление пара Соль Давление пара
кПа мм рт. ст. кПа мм рт. ст.
СНзСООК 0,47 3,47 NaNO2 1,53 11,5
СаС12-6Н2О 0,75 5,62 NH4C1 1,83 13,8
Zn(NO3)2-6H2O 0,97 7,29 К2СгО4 2,04 15,3
Na2Cr2O7-2H2O 1,20 9,03 Na2SO3-7H2O 2,19 16,5
Таблица 2-3. Давление пара над растворами серной кислоты
при 20 “С
Концентрация кисло- ты, % Давление пара Концентрация кисло- ты, % Давление пара
Па мм рт. ст. Па мм рт. ст.
10 2300 17,24 60 490 3,71
20 2190 16,41 65 300 2,23
25 2090 15,70 70 140 1,09
30 1930 14,50 75 55 0,41
40 1490 11,29 80 16 0,12
50 970 7,37 85 7 0,05
Объемный метод позволяет изучать изотермы на крупнопористых адсор-
бентах, обладающих малой адсорбционной способностью.
Эксикаторный мет од. Этот очень простой метод определения изо-
терм сорбции применяют в заводской практике для контроля качества адсор-
бентов при их производстве. Сущность его состоит в насыщении помещенной
в бюкс навески отрегенерированного адсорбента парами адсорбтива. Если
адсорбтивом является вода, заданная концентрация ее паров в объеме экси-
катора обеспечивается либо насыщенными растворами солей, либо серной
кислотой различных концентраций. Давление паров воды над насыщенными
растворами солей приведены в табл. 2-2, над серной кислотой — в табл. 2-3.
Адсорбционная способность определяется по разности массы бюкса с адсор-
бентом до и после регенерации.
Динамические методы. Различные разновидности динамиче-
ских методов основаны на измерении концентрации адсорбтива
в потоке несорбирующегося газа-носителя на входе адсорбера
и выходе из него.
Если в качестве регистрирующего прибора применить пламенно-иониза-
ционный детектор, метод позволяет измерить количество адсорбированного
вещества от 1 10~7 г, а равновесное давление от 1,33-10~4 Па (1-10~6 мм
рт. ст.) [6].
Быстрота и достаточная точность хроматографии стимулировали разра-
ботку на ее основе различных приемов определения характеристик системы
газ — твердое тело: изотерм адсорбции, теплот адсорбции и др. Эти приемы
3—1346
34 2. Адсорбционное равновесие и пористая структура адсорбентов
можно рассматривать как разновидности динамических методов. Один из
таких методов определения изотерм основан на проявительной хроматогра-
фии.
При проявительной хроматографии в колонку, заполненную адсорбентом
и продуваемую неадсорбируемым газом-носителем, вводят образец исследуе-
мого адсорбтива. Спустя некоторое время после ввода пробы детектор, уста-
новленный на выходе газа из колонки, зарегистрирует нарастание, а затем
спад концентрации адсорбтива — хроматографический пик. Между хромато-
графической выходной кривой (пиком) и формой изотермы адсорбции имеет-
ся непосредственная связь, что используют для расчета последней*.
В каждой точке выходной кривой равновесная концентрация адсорбтива
в газовой фазе с связана с высотой h (показанием детектора) соотношением
c — kh (2.5)
Для определения k проводят калибровочный опыт. В поток газа вводят
определенное количество адсорбтива т. Через некоторое время все введен-
ное вещество выйдет из колонки, «проявившись» на ленте детектора в виде
соответствующего пика. Нетрудно показать, что
т = k (w/u) S (2-6)
где w— объемная скорость газа, см3/мин; и — скорость движения ленты са-
мописца, см/мин; S — площадь под кривой проявления, см2.
Отсюда
k = mu[wS (2.7)
Количество адсорбированного вещества а как функция концентрации
определяется из равенства
a = /(c) = -^-j (V-V0)dc (2.8)
о
где g — навеска адсорбента, г; V—объем газа-носителя, прошедшего через
колонку от момента ввода газа до появления концентрации с на выходе,
см3; Ко — свободный объем колонки, см3.
Уравнение (2.8) используют для расчета изотермы. Значение с и V
определяют на основании показателей детектора:
с = kh = (mu/wS) h (2-9)
V = (х/и) w (2.10)
где х — расстояние на ленте детектора.
После подстановки значений с и V в уравнение (2.8) получают
h
wk wk (‘ ,, ,
а f = Sh = ~ %°) dh (2-П>
u5 J
о
h
j (х — х0) dh (2.11а)
о
* При определении изотерм адсорбции процесс хроматографирования ведут
в условиях так называемого равновесного элюирования (проявления), кото-
рое определяют экспериментально в предварительных опытах, варьируя рас-
ход газа-носителя. (Прим, ред.)
Теория мономолекулярной адсорбции
35
Рис. 2,8. К расчету изотермы адсорбции по показателям детектора хроматографа h
(Ti — время, соответствующее началу впуска адсорбтива):
а — выпуклая изотерма; б — вогнутая изотерма.
Расчет изотермы проводят методом графического интегрирования. Для
этого выходную кривую разбивают горизонтальными прямыми на несколько
(например, пять) участков. В каждом участке по высоте h вычисляют рав-
новесную концентрацию с и одновременно определяют заштрихованную пло-
щадь Sh- Затем по значениям Sh для каждого участка определяют по форму-
ле (2.11) значения адсорбционной способности. Пример построения выпук-
лой и вогнутой изотермы адсорбции путем графического интегрирования хро-
матограмм приведен на рис. 2,8.
Хроматографический опыт обычно быстротечен — он продолжается часто
менее минуты. В то же время на промышленных адсорбентах адсорбционное
равновесие устанавливается иногда через несколько десятков минут, а иног-
да и часов. Поэтому хроматографический метод применим только к системам
адсорбтив — адсорбент, для которых характерно быстрое установление рав-
новесия.
Теория мономолекулярной адсорбции
Первым фундаментальным уравнением изотермы адсорбции
было уравнение Ленгмюра [7] . Оно основано на ряде допуще-
ний: адсорбция локализована и происходит на активных цент-
рах, равноценных в энергетическом отношении, на каждом ак-
тивном центре может адсорбироваться только одна молекула,
взаимодействие адсорбированных молекул друг с другом не про-
исходит. Процесс адсорбции Ленгмюр представлял как резуль-
тат динамического взаимодействия молекул адсорбтива с ак-
тивными центрами на поверхности адсорбента:
Молекула газа + Активный центр < 1 > Адсорбционный комплекс
В соответствии с кинетической теорией газов в единицу вре-
мени о поверхность, равную единице, ударяется ц молекул,
причем
р = р/(2ятКТ)Чг (2.12)
где р — давление; т — масса молекулы; К-—постоянная Больцмана;
Т — абсолютная температура.
Из числа ударяющихся молекул только часть фиксируется
на поверхности, остальные упруго отражаются в газовую фазу.
Доля неупругих столкновений от общего числа столкновений
молекул газа с поверхностью была названа коэффициентом
*
36 2. Адсорбционное равновесие и пористая структура адсорбентов
конденсации а0. Необходимо учитывать, что в процессе ад-
сорбции часть поверхности 0 уже покрыта молекулами адсор-
бата и, следовательно, активной является лишь вторая, свобод-
ная часть. Тогда скорость адсорбции, отнесенная к единице по-
верхности
Ка = «0 (1 — 0) р, (2.13)
При десорбции только те молекулы могут снова перейти в
газовую фазу, которые обладают достаточной энергией, превы-
шающей теплоту адсорбции молекул qM. Число молекул, десор-
бирующихся с единицы поверхности в единицу времени
v = Ко exp (—qvJKT) (2.14)
где Ко — энтропийный коэффициент.
Скорость десорбции с единицы поверхности
Кд = т0 (2.15)
При равновесии скорости адсорбции и десорбции равны:
а0 (1 — 0) р, = v9 (2.16)
Решая уравнение (2.16) относительно 0, получим:
9______5^__________/2 17>
° - 1 +<XoP/V ~ 1 +6р ( '
где
ст0 ехр (7м/КГ)
Ко (гшпКТ)1^
Степень заполнения поверхности можно выразить через от-
ношение адсорбционной способности при равновесном давлении
р к предельной адсорбционной способности ат, соответствующей
оккупации всех активных центров поверхности.
Уравнение изотермы адсорбции Ленгмюра
а = ambp/(\ + Ьр) (2.18)
охватывает широкий интервал давлений. В начальной области
изотермы Ьр<С1, и уравнение принимает вид
а » атЬр (2.19)
На этом участке уравнение Ленгмюра идентично уравнению
Генри для растворимости газов: адсорбционная способность
растет линейно с давлением. При большом давлении, наоборот,
Ьр^А, наступает заполнение поверхности монослоем молекул и
изотерма становится параллельной оси абсцисс:
а » ат (2.20)
Теория мономолекулярной адсорбции
37
Рис. 2,9. Изотерма адсорбции бензола на графи-
шрованной саже при 20 °C в координатах урав-
нения Ленгмюра.
Уравнение (2.18) может быть
преобразовано следующим образом:
р 1 1
а ато ат
ПЛИ
1 1 1________________1_
а ат.Ь р
(2.22)
Линейность изотермы в координатах уравнений (2.21) и
(2.22) является обязательным условием при доказательстве
правомерности применения теории мономолекулярной адсорб-
ции для рассматриваемой системы. В качестве примера на
рис. 2,9, приведена изотерма адсорбции бензола на графитиро-
ванной саже при 20 °C [8] в координатах линейной формы урав-
нения Ленгмюра. Наклон прямой и отрезок, отсекаемый прямой
на оси ординат, позволяют вычислить константы ат и Ь.
В описанном выше строгом выводе уравнения локализован-
ной адсорбции Ленгмюра величина ат, как уже указывалось,
связана с количеством центров адсорбции. Однако опыты сви-
детельствуют, что уравнение Ленгмюра справедливо и для не-
локализованной адсорбции. В этом случае ат — емкость мо-
но с л о я пропорциональна удельной поверхности адсорбента.
Для вычисления удельной поверхности по емкости монослоя ис-
пользуют уравнение
s = amyAcom (2.23)
где Na — число Авогадро, равное приблизительно 6,02-Ю23 моль-1; wm — пло-
щадь, занимаемая молекулой адсорбата в плотном слое иа поверхности ад-
сорбента (элементарная площадка).
Обычно величину ат выражают в молях на грамм твердого тела, а
(от — нм2. Тогда а (в м2/г) будет равно
s=amyAo>m-10-w (2.24)
В случае, если ат выразить в г/100 г, то получим
s = (am/M)NA(om-\0-^ (2.25)
где М — мольная масса адсорбата.
Если ат выражена в см3 (при нормальных условиях) на грамм твердого
тела, то
s = (ат/22 400) NA(Om 10~18 (2.25а)
Уравнение Ленгмюра как основа для определения поверхно-
сти может быть применено к системам, в которых процесс не
осложняется полимолекулярной адсорбцией, адсорбцией в мик-
ропорах и капиллярной конденсацией. Эти условия соблюдаются
38 2. Адсорбционное равновесие и пористая структура адсорбентов
при адсорбции газов при температурах выше критической на
непористых или крупнопористых адсорбентах. В технической
адсорбции это уравнение часто применяют в качестве равновес-
ного интерполяционного соотношения при расчете кинетики и
динамики процессов поглощения примесей из газовой среды.
Теория полимолекулярной адсорбции
и определение удельной поверхности
Большое число адсорбционных систем описывается изотермами
второго типа. Для таких изотерм характерен резкий подъем при
относительном давлении (p/ps)^Q,2, связанный, как считают,
с образованием второго и последующих слоев молекул, покры-
вающих молекулы первого слоя.
Брунауэр, Эмметт и Теллер [4] при обосновании теории по-
лимолекулярной адсорбции приняли, что, несмотря на изменение
общей модели процесса, поведение каждого адсорбированного
слоя в отдельности соответствует концепции Ленгмюра. В со-
стоянии равновесия скорость конденсации молекул на чистой
поверхности равна скорости испарения из первого слоя. Анало-
гичное равенство справедливо для скорости конденсации на
каждом предшествующем и скорости испарения в каждом дан-
ном слое. Последнее важное допущение авторов связано с ха-
рактером сил, действующих между адсорбционными слоями.
Они приняли, что эти силы подобны ван-дер-ваальсовым силам,
обусловливающим конденсацию, и, следовательно, теплота ад-
сорбции во всех слоях, кроме первого, равна теплоте конденса-
ции.
Подробный вывод уравнения полимолекулярной адсорбции,
названного по начальным буквам фамилий его авторов уравне-
нием БЭТ, приведен в ряде статей и монографий, например [4].
Поэтому здесь мы ограничимся указанием конечной формы
уравнения для плоской поверхности;
_____Ps____________ /п 9р'\
ff== (l-p/ps)[l + (C-l)P/Pd
В приложении к пористым адсорбентам уравнение БЭТ со-
блюдается в интервале относительных давлений от 0,05 до 0,35.
Его широко применяют для определения удельной поверхности
различных пористых тел.
Определение удельной поверхности обычно проводят, ис-
пользуя экспериментальную изотерму адсорбции стандартного
пара на исследуемом образце, выраженную в линейной форме
уравнения БЭТ:
P/ft = _L_ . С~1... о/п (2.27)
а (1 — p/ps) атС 1 атС P/Ps
Теория полимолекулярной адсорбции
39
Рис. 2,10. Изотермы адсорбции пропана на трех об-
разцах (1—3) силикагеля при 20 °C:
а — опытные; б —в координатах уравнения БЭТ.
На рис. 2.10а представлены изотер-
мы адсорбции пропана на силикагелях
с различной пористой структурой при
20 °C: мелкопористом (образец 3),
среднепористом (2) и крупнопористом
(1). На рис. 2,106 они же представле-
ны в координатах уравнения (2.27).
Величина ат (в моль/г) выражает ем-
кость монослоя молекул адсорбата, по-
крывающего поверхность адсорбента.
Константы ат и С находят из наклона
прямых линий [(С—1)/атС] и отрез-
ков, отсекаемых прямыми на оси орди-
нат (1/атС).
Удельную поверхность s определя-
ют из емкости монослоя, как это опи-
сано выше. Константа С непосредст-
венно связана с чистой мольной
теплотой адсорбции, соответствующей разности теплоты
адсорбции в первом слое Q[ и мольной теплоты конденсации
пара адсорбтива X:
С = exp (Qj — T)/RT
(2.28)
Последнее уравнение используют для вычисления значения
чистой теплоты адсорбции из экспериментальных данных. Зна-
чение константы С определяет вид изотерм. В случае малой чи-
стой теплоты адсорбции С<2 изотерма имеет вогнутую форму
(тип III). Когда С>2, вид изотермы соответствует S-образной
форме (тип II; см. стр. 29).
Если изотерма исследуемого вещества может быть отнесена
ко второму типу, оценка удельной поверхности может быть так-
же проведена по точке В — точке перегиба на изотерме, свиде-
тельствующей об окончании заполнения монослоя.
Соответствие расчета этими двумя методами отмечается в
случае изотерм с большой крутизной изгиба (рис. 2.11), харак-
терной для веществ с большой теплотой адсорбции. Определе-
ние точки В в случае пологих изотерм, т. е. для веществ с ма-
лой теплотой адсорбции, может привести к значительным по-
грешностям в оценке поверхности.
Обычно при измерении удельной поверхности в качестве ад-
сорбтива используют азот, и опыты проводят при —196 °C. Ве-
личина площадки молекулы азота практически на всех твердых
телах составляет 0,162 нм2, причем плотность упаковки молекул
40 2. Адсорбционное равновесие и пористая структура адсорбентов
Рис. 2,11. К определению емкости монослоя по
точке В.
в адсорбированном слое соответст-
вует их упаковке в нормальной жид-
кости.
Для определения удельной по-
верхности иногда используют и иные
адсорбтивы [9]. При этом особое
внимание уделяют их чистоте. Од-
молекул (а следовательно, и величина
нако часто ориентация
элементарной площадки) изменяется в зависимости от типа ад-
сорбента. Так, определения площади молекул «-бутана, прове-
денные различными авторами [10—12] дали очень большое рас-
хождение: от 0,32 до 0,56 нм2. Поэтому при отсутствии данных о
величине элементарной площадки на адсорбенте рассматривае-
мого типа определение удельной поверхности по нестандартным
адсорбтивам не следует рекомендовать.
Ниже приведены размеры элементарных площадок молекул
наиболее распространенных адсорбтивов, которые можно ис-
пользовать со сделанными выше оговорками:
Адсорбтив Темпера- тура опыта, °C 102 НМ2 Адсорбтив Темпера- тура опыта, °C ат* 102 нм2
Этан 20 22,7 Кислород —183 14,1
Этилен 20 22,6 Азот —196 16,2
Пропан 20 42,5 Криптон —196 19,5
н-Бутан 20 43,4 Ксенон —196 25,0
н-Пентан 20 45,0 Четыреххлорид +25 39,2
н-Гексан 20 51,5 углерода
«-Гептан 20 57,3 Бензол +20 40,0
н-Октан 20 61,0 Фреон-21 0 40,0
Аргон —196 13,7
Мак-Клиллан и Харнсбергер [13] систематизировали огром-
ный литературный материал о размерах площадок молекул
большого числа веществ на различных адсорбентах. Их данные
приведены в табл. 2-4. В этой таблице указаны также площадки
молекул (со&, сом, соп), рассчитанные тремя методами:
1) величина со& установлена на основе значения константы
уравнения Ван-дер-Ваальса b (в 102 нм2), которая определяется
по формуле Хилла:
6 = 6354(Гкр/ркр)2/з (2.29)
2) на основе геометрической модели молекулы вычислены
экстремальные значения площадки сом, которую она может за-
нять на плоской поверхности. Величина <вм значительно ниже
Теория полимолекулярной адсорбции
41
Рис. 2,12. Корреляция между молекулярными пс
площадками о,п. ’
1 '"од
действительных площадок моле- *=
кул, занимаемых на поверхности
адсорбента любой химической 3
природы. Значения со& и сом мож-
но использовать для расчета х о
только в случае, если значение
Ют неизвестно, а к точности опре-
деления удельной поверхности адсорбента предъявляются невы-
сокие требования.
3) Брунауэром и Эмметом предложен метод определения ве-
личины (Оп по плотности адсорбтива р (в г/см3) в жидком со-
стоянии:
(On = 1,091 (M/N А р)2/з (2.30)
Анализ данных таблицы позволяет установить зависимость,
по которой приближенно может быть определен размер площад-
ки молекулы на поверхности адсорбента ат по величине соп.
Эта зависимость приведена на рис. 2.12 и описывается форму-
лой:
шп — 6,16
= 0/596
где wm, о)п выражены в 102 нм2.
Существуют, однако, вещества, которые крайне нежелатель-
но использовать для оценки величины удельной поверхности ад-
сорбентов. Например, применение изотерм водяного пара может
привести к значительным ошибкам в определении. Для воды
(0т = 0,106 нм2 [14], но удельная поверхность кремнезема, опре-
деленная на основе этой величины сильно занижена. Расчет
дает 30 м2/г вместо 220 м2/г [15]. Ошибка в определении поверх-
ности по воде еще выше в случае углеродных адсорбентов [16].
Нельзя применять для оценки величины удельной поверхно-
сти кремнезема изотермы адсорбции низших алифатических
спиртов, поглощение которых сопровождается хемосорбцией [17].
Большое расхождение с истинным значением дает расчет удель-
ной поверхности углеродных адсорбентов, если в качестве ад-
сорбтива использовать аммиак (ыт = 0,13 нм2): например, по-
верхность графитированной сажи сферон-27,00, определенная по
изотермам адсорбции аммиака (/<0°С), была равна 24—
25 м2/г, а ее истинное значение составляет 84 м2/г [18].
Следует иметь в виду, что с помощью уравнения БЭТ про-
изводят определение удельной поверхности только макро- и ме-
зопористых адсорбентов, а также их смешанных типов. Присут-
ствие в адсорбенте микропор, объемно заполняющихся молеку-
лами адсорбента, приводит к искажению полученных результа-
Таблица 2-4. Размеры площадок в 102 нм2 (А2), занимаемых молекулами на поверхности
различных адсорбентов
Адсорбат Расчетные размеры площадки Опытные размеры площадки
аь % “п “т температура опыта, СС
AsH3 — мышьяковистый водород — — — 11 —78,5
CF2C12— дифтордихлорметан (фреон-12) — — — 40 —33
CFC13 — фтортрихлорметан — — — 27,6 От —78 до 33
CF4 — тетрафторид углерода 22,0 13,9 — 21,9 От —123 до —193
CHFC12— фтордихлорметан 27,2 15,4-19,2 25,7 38,8 От 0 до —78
СНС13 — хлороформ 29,5 18,3—19,7 28,5 27,9 От —78 до —50
НСО2Н — муравьиная кислота 10,6 — 19,9 13,4 38
СН4 — метан 16,5 10,6 От 15,8 (—196 °C) до 16,4 (—130 °C) 17,8+3,15 От —130 до —208
СН3ОН — метиловый спирт 22,2 12,2—14,0 18,0 21,3±5 От 0 до 29
CH3NH2 — метиламин — — 19,1 16,0 0
C12C2F4 — тетрафтор-1,2-дихлорэтан 35,0 14,8-26,0 — 38,9 От 303 до 343
C2Ffi — гексафторэтан — 13,7—21,1 — 31,7 От 223 до 253
С2Н2 — ацетилен 18,5 6,2—14,7 18,7 (—78 °C) 22,1±3,4 От —75 до —80
С2Н2С12— 1,2-днхлорэтилен — 12,4—28,6 27,8 (25 °C) 42,2 28
С2Н3С1 — хлорэтилен — 11,7—20,3 24,9 (0 °C) 36,8 0
СН3СО2Н — уксусная кислота 30,5 15,5—20,7 22,9 (25 °C) 23,8+5,7 От 20 до 38
С2Н5С1 — этилхлорид 27,2 14,1—16,6 26,0 (0 °C) 28,2±4,6 От 0 до —78
С2Н5Т — этилиодид — 17,0—17,3 28,2 (75 °C) 25,2±4,5 75
С2Н5ОН — этанол 25,7 12,8-18,8 23,1 26,5+6,45 От 0 до 25
C2H4(NH2)2— 1,2-диаминоэтан — 15,6—21,9 — 27,2+1,7 25
C3H2F6 — гексафторпропан — 20,2—28,1 — 47,5 От 303 до 343
С3Н4С12— 1,2-дихлорпропилен 30,6 22,3—24,1 28,1 (25 °C) 35,35 От 2 до 25
С3Н6 — пропилен — — — 34,5+4,5 От —3d до —00
(СН3)2СО — ацетон 31.4 20,2—23,5 27,2 (25 °C) 34 20
СоН5СО2Н — пропионовая кислота 32,4 15,8—26,7 27,4 (38 °C) 25 38
С3Ы7С1 — хлорпропан 31,6 17,3—25,2 — 42,5 25
С3Н7ОН — пропанол-1 30,8 19,4—25,3 27,2 (25 °C) 31,0 25
С3Н7ОН — пропанол-2 29,9 20,1—24,2 27,7 (25 °C) 39,3 25
C4Fin - - псцфторбутан 41,5 20,4—32,1 — 53,5 От 303 до 343
С4Н4О — фуран — 14,4—27,0 26,6 (20 °C) 40,5 23
C4I14S — тиофен 29,8 18,1—30,7 28,2 (25 °C) 33,0 25
С4Н7 — бутадиен — — — 32,9 0
СН3 (СИ) 2СН3 — гр;с-бутен-2 —: .— — 38,5 0
СН3(СН)2СН3 — транс-бутен-2 30,5 17,6—27,8 31,3 (20 °C) 35,4 0
С3Н7СО2Н — масляная кислота 33,5 20,0—32,7 31,6 (38 °C) 31,3 38
С4Н9С1 — 1-хлорбутан — 19,7—22,6 — 44,6 От 20 до 25
C4Hin — 2-метилпропап 32,0 24,5—30,3 32,3 (—11,7 °C) 50,3 От 0 до —18
С4Н9ОН — бутанол-1 32,1 17—33 31,2(25 °C) 35,6+3,4 От 20 до 25
С4Н9ОН — бутанол-2 20,0 17,6—зо;о 31,3 (25 °C) 43,3+5,5 25
СН3СН(СН3)СН2ОН — 2-метилпропанол-1 31,9 22,2—27,3 — 40,9+5,3 25
СН3С (ОН) (СН3) СН3 — 2-метилпропапол-2 33,7 22,6—30,2 — 43 25
(С2Н3) ->О — диэтиловый эфир 35,3 19,0—31,5 33,8 (20 °C) 42 20
(С9Н5) "NH — диэтиламин 36,1 26,8—31 34,2 (25 °C) 43,2 25
C4H9NH2 — н-бутиламип — 20—32 30,9 (25 °C) 32,5 25
C5H5N — пиридин — 39—44 — 38,6 20
С5Н10 — циклопентан 32,1 24,5—26,2 49,5 (0 ° С) 43,7±4,5 От 0 до 20
C5Hi0 — 1 -метилбутеп-2 36,5 22—31 — 57 От 20 до 150
(СН3)2С = СНСН3 — 2-метилбутеп-2 — — — 51,7±5,2 20
СН3(СН2)2СН = СН2— пентен-1 — 18,4—31,0 34,9 (20 °C) 49,3 20
СН3СН=СНСН2СН3 — пентен-2 •— 47,8 0
С5Н|0О2 — мстилнзобутират 40 _ 28,6—34 37,0 (20 °C) 37,7 20
СП
Адсорбат Расчетные размеры площадки Опытные размеры площадки
“6 “м “п “т температура опыта, °C
(СН3)4С — 2,2-диметилпропан (неопентан) 36,5 28—29 — 49+8 От 0 до —46
(СН3) 2СНСН2СН3 — 2-метилбутан 37 26—34 35,1 (—12 °C) 56 От 0 до 150
С5НцОН — пентанол-1 — . 19—36 — 39,2+2,8 25
Ч«кло-СбН12 — циклогексан 36,7 29,4-35 36,3 (25 °C) 44,2+8,3 От 5 до 26
ЧИО0-С5Н9СН3 — метилциклопентан — — — 47,1 20
(СНз)зССН2СНз — 2,2-диметилбутан — — — 46 20
(СН3) 2СН(СН2)2СНз — 2-метилпентаи — — — 48,5 20
(СзН7)2О — пропиловый эфир — — — 62,4 25
(С2Нз) 3N — триэтиламин 43,2 33—43 41,7 (25 °C) 43,0 320
CeFnCF3 — перфторметилциклогексан 48 37—58 — 66,5 От 303 до 343
СН3СбН5 — толуол 38,5 18—39 34,4 54±7,6 От 25 до 75
(СНз)3С(СН2)2СН3 — 2,2-диметилпентан — — — 36 65
СН3СН2СН (СН3) (СН2)2СН3 — 3-метилгексан — — 54 20
цякло-С9 Flo (CF3) 2 — ди (трифторметил) перфтор- — 36-54 — 68,8 От 303 до 343
циклогексан
C2H5CeHs — этилбензол — — — 54,7 20
(СН3)2С6Н4 — л-ксилол — — — 55,8 20
(СН3)2СвН4 — л-ксилол — — — 53,8 20
(СН3) 3ССН2СН (СН3) 2—2,2,4-т риметилпентан 56,2 20
(C4H9)2NH — ди-я-бутиламин 30—53 47,2 (25 ”С) 54,4 25
С3Н7СбН5 — пропилбензол 47,8 19—51 41 (20 °C) 61,2 20
«зо-С3Н7С6Н5 — изопропилбензол 47,8 19—51 41,2 (20 °C) 62,3 20
(СНз)зС6Н3— 1,3,5-триметилбензол — — —- 69,6 20
СН3(СН2)7СН3 — я-нонан 50,9 22—55 48,8 (26 °C) 84,4 26
(СНз)2С=СН (СН2)2с (СНз) = СНСНз - 2,6-диме- — — — 82,0 100
тилоктадиен-2,6
СНз (СН2) 8СНз — я-декан 61,0 17—61 53,5 (80 °C) 86,0 100
15 6 7 10 16,8 14,2±3,1 От —180 до —196
СО — монооксид углерода
16 4 5—11 17,0 20,3±2,1 От 35 до —78
СО2 —диоксид углерода
CS2 — дисульфид углерода 23,5 7—17 22,4 (0 °C) 23 0
15,35 От —135 до —183
Н2 — водород
Н2О — вода 23 6—8 10,5 (25 °C) 12,8±5,4 От —20 до 30
H2S — сероводород 16,5 8—10 — 21,0 —79
12 — иод — 10—17 — 27,0+10 От 20 до 25
NH3 — аммиак 15,0 7—10 12,9 15,4±2,8 От —32 до —78
N2O — диазотоксид 16,9 7—13 16,8 20,4 —78
31,4 —154
N2O2 — диазотдиоксид
23,7 От —15 до 50
N2O4 — диазоттетраоксид
10,0 —256
Ne — неон
SF6 — гексафторид серы 26,7 20,8 — 26,4 От —76 до 253
SO2 —диоксид серы 20,0 13—16 21,8±5,2 0
46
2. Адсорбционное равновесие и пористая структура адсорбентов
Таблица 2-5. Значения удельных поверхностей дегидратированных
кристаллов цеолитов [19]
Цеолит $1 по данным рентгено- структурно- го анализа, м2/г Расчет по уравнению БЭТ Расхождение ^2-100% S1
адсорбтив 52» М^/Г
СаА 1170 N2/H2O n2/h2o 770/820 34/30
NaX 1450 900/970 38/33
СаХ 1500 n2/h2o 920/910 38/39
тов по отношению к действительным величинам, хотя в опреде-
ленном интервале относительных давлений экспериментальные
данные и соответствуют линейной форме уравнения БЭТ. В слу-
чае адсорбции воды и азота на различных формах цеолитов
верхней границей этого формального соответствия является
относительное давление 0,1. Однако сравнение результатов вы-
числения удельной поверхности цеолитов с действительной гео-
метрической поверхностью, установленной на основе рентгено-
структурных данных, выявило их значительное расхождение.
Об этом убедительно свидетельствуют данные таблицы 2-5 [19].
В то же время отмечено, что отношение формальных удель-
ных поверхностей двух или серии микропористых адсорбентов,
определенных методом БЭТ, соответствует отношению их ад-
сорбционной способности, например по азоту при температуре
минус 196 °C и относительном давлении 0,1.
В рассмотренных теориях моно- и полимолекулярной адсорб-
ции было принято допущение, что молекулы адсорбата одного
слоя не взаимодействуют друг с другом. В некоторых случаях,
однако, силы взаимодействия адсорбированных молекул значи-
тельны. К таким случаям относится, в частности, адсорбция вы-
сокомолекулярных органических соединений на неполярной по-
верхности. Для описания адсорбционного равновесия при нали-
чии сильного взаимодействия адсорбат — адсорбат предложен
ряд уравнений, наиболее простым из которых является уравне-
ние Киселева:
О
+Л'„0) (2‘32)
где константа /<„ учитывает взаимодействие адсорбат — адсорбат; b — кон-
станта уравнения Ленгмюра*.
* Для локализованной ленгмюровской адсорбции на однородной поверхно-
сти Фрумкиным термодинамическим путем и Фаулером и Гуггенгеймом мо-
лекулярно-статистически получено следующее уравнение изотермы, учиты-
вающее взаимодействие адсорбат — адсорбат, разрешенное относительно р:
р = (1 — 6Г ехр (Прим, ред.)
Методы определения удельной поверхности
47
»к> уравнение описывает локализованную адсорбцию на одно-
родной поверхности в области заполнения поверхности первым
< .ноем молекул.
Другие методы определения
удельной поверхности
Метод термической десорбции. Определение удельной поверхности по
данным хроматографии может быть проведено различными способами: по
удерживаемым объемам, по размытой стороне хроматографического пика,
но результатам фронтального анализа. Для массовых определений удельных
поверхностей образцов адсорбентов или катализаторов может быть рекомен-
дован метод термической десорбции. Он основан на прямой зависимости
между расходом стандартного газа, поглощенного при низкой температуре
образцом адсорбента из потока газа-носителя (гелия), и удельной поверх-
ностью. После размораживания образца по площади хроматографического
пика судят о величине удельной поверхности. В качестве адсорбтива исполь-
зуют азот, криптон или аргон.
Простая аппаратура широко используемая в научно-исследовательских
институтах и заводских лабораториях, разработана Карнауховым и Буяно-
вой [20]. На хроматографической установке, схема которой представлена
на рис. 2,13, возможно произвести оценку двенадцати образцов в течение
6 ч.
Рис. 2,13. Схема установки для хроматографического определения удельной поверхности
методом тепловой десорбции:
1, 10 —баллоны с газами; 2, 9, 11 — расходомеры газа; 3, 4 — адсорбционные колонки;
5 — осушительная колонка; 6 — гребенка; 7—сравнительная камера катарометра; 8 —
измерительная камера катарометра; 12 — ловушка; 13 — адсорберы.
48
2. Адсорбционное равновесие и пористая структура адсорбентов
Рис. 2,14. Хроматограмма (а — стадия адсорбции;
б — стадия десорбции). На рисунке отражены ре-
зультаты параллельных определений для шест»
образцов. ,
х
адсорбентов, загруженных в
адсорберы
из балло-
Вначале шесть образцов
13—[7-образные трубки, подключают к гребенке 6 и продувают
на 1 гелием, очищенным и осушенным в колонке 5. Затем гелий и аргон из
баллонов 1 и '10 пропускают через колонки 3 и 4, где происходит осушка и
очистка газов, после чего газы смешиваются и смесь поступает в ловушку
для вымораживания воды 12, сравнительную камеру катарометра 7 и ад-
сорберы 13, погруженные в дьюар с жидким азотом.
Отходящий газ пропускают через измерительную камеру катарометра 8.
Скорость потоков устанавливают расходомерами 2, 9 и 11. После того как
катарометр зарегистрирует наступление адсорбционного равновесия, послед-
ний адсорбер «размораживают», убирая дьюар, и количество десорбиро-
ванного из образца аргона определяют по площади хроматографического
пика. Затем подобным методом анализируют следующие пять образцов.
Пример хроматограммы дан на рис. 2,14.
Среднее отклонение величины удельной поверхности, определенное хро-
матографическим способом и из изотермы, составляет 3,5%. Метод позволяет
исследовать образцы с удельной поверхностью от 0,01 до 1000 м2/г.
По экспрессности и точности метод тепловой десорбции превосходит
другие хроматографические методы. Это отчетливо видно из данных
табл. 2-6, в которой результаты определений удельной поверхности различ-
ных пористых тел методами тепловой десорбции, фронтальной и проявитель-
ной хроматографии сравниваются с оценкой по данным изотермы адсорбции,
полученным на вакуумной адсорбционной установке [21].
Экспресс-метод. Необходимость увеличивать скорость оценки удельной
поверхности образцов при массовых анализах побудила искать упрощенные
и ускоренные методы определения. Темкин [22] опытным путем установил,
что отношение количества газа, адсорбированного в заданных условиях
(давление и температура), к количеству газа, поглощенного при мономоле-
кулярном покрытии поверхности адсорбента, есть величина постоянная и
практически не зависит от природы адсорбента. Кроме того, изотермы ад-
сорбции одного адсорбтива на разных образцах подобны, и коэффициент
подобия равен отношению адсорбционных способностей этих образцов в
Таблица 2-6. Результаты определения удельной поверхности (в м2/г)
пористых тел различными методами
Адсорбент или катализатор Эффектив- ный радиус пор, нм2 Метод определения
из изотерм тепловой фронталь- проявн- десорбции ный тельный
Железомолибденовый ката- 10,00 7,5 7,5 7,1 6,5 лизатор Силикагель А 1,80 85 80 96 57 Силикагель Б 0,50 294 315 325 150 Алюмосиликатный катали- 0,20 336 336 320 135 затор
Методы определения удельной поверхности
49
одинаковых условиях опыта. Благодаря этому стало возможным оценивать
емкость монослоя по одной точке изотермы:
p/Ps) (2.33)
При оценке удельных поверхностей силикагелей и алюмогелей по одной
точке изотермы для азота в качестве стандартного относительного давления
предложено значение p/ps = Ofl.. В этом случае удельная поверхность s
(в м2/г) вычисляется по формуле:
з = 91,3а (2.34)
где адсорбционная способность а выражена в ммоль/г.
Для определения удельной поверхности образцов по этому уравнению
были разработаны простые приборы. В одном из приборов определение ос-
новано на контакте образца с воздухом при температуре жидкого кислоро-
да. Количество поглощенного газа определяют по уменьшению объема в си-
стеме, условно принимая, что адсорбируется только азот, и пренебрегая ад-
сорбцией кислорода. В другой модификации методики [33] в качестве хла-
доагента применяют жидкий азот. Для определения объема поглощенного
газа иногда используют автоматическую газовую бюретку, приспособленную
для работы в условиях вакуума [24]. Время на определение удельной по-
верхности по одной точке изотермы составляет около 15 мин.
Определение по адсорбции красителей. Для определения удельной по-
верхности крупнопористых адсорбентов иногда применяют адсорбцию из
жидкой фазы. Изотермы адсорбции из растворов строят при контактирова-
нии раствора хорошо адсорбируемого вещества в жидком растворителе с
навеской адсорбента. Массу поглощенного вещества определяют по измене-
нию концентрации адсорбтива в растворе. Наиболее часто для определения
удельной поверхности используют красители, которые обладают высокой
преимущественной адсорбцией в присутствии растворителя. В связи с этим
после образования монослоя молекул красителя растворитель практически
полностью вытесняется с поверхности. Методика определения концентрации
красителей очень проста [25].
Достаточно широко для оценки удельной поверхности используют мети-
леновую синь, изотермы адсорбции которой относятся к изотермам первого
типа, при концентрации адсорбтива 0,3% они имеют горизонтальные участ-
ки, соответствующие заполнению монослоя.
На рис. 2,15 представлена изотерма адсорбции метиленовой сини на
саже сферон из водного раствора при 20 °C [26]. По величине адсорбции,
соответствующей горизонтальному участку, легко определить удельную по-
верхность адсорбента, если известен размер площадки, приходящийся на
одну молекулу красителя. При горизонтальной ориентации на поверхности
молекула метиленовой сини должна занимать площадку 1,35 нм2, при верти-
кальной — 0,75 нм2. В действительности площадка, приходящаяся на одну
молекулу метиленовой сини, при адсорбции на углеродных поверхностях
(графите, графитированной и пеграфитированной саже) колеблется от 0,78
до 1,30 им2.
Молекула метиленовой сини имеет _______________
форму многогранника и отличается нерав- 8
номерным распределением электронной
плотности. При адсорбции на различных 7Г
силикатных поверхностях значения площа- ™
док метиленовой сини значительно отлича- — 4
ются как друг от друга, так и от указан- h.
пых выше площадок на углеродной поверх-
ности. Они составляют для силикагеля
Рис. 2,15. Изотерма адсорбции метиленовой сини ®
на саже сферон из водного раствора при 20 °C.
4—1346
БО
2. Адсорбционное равновесие и пористая структура адсорбентов
2,70, для каолинита 2,72, для кремнезема 3,27 и для монтморелонита 3,40 нм2
[26, 27].
На этих адсорбентах процесс физической адсорбции осложняется ион-
ным обменом. В связи с этим метод, основанный на адсорбции красителей,
следует применять только после установления площадки , молекулы на стан-
дартном адсорбенте идентичной химической природы. В этом случае оценку
удельной поверхности можно произвести непосредственно по соотношению
предельной адсорбционной способности исследуемого и стандартного ве-
ществ.
Попытка применить метиленовую синь для оценки удельной поверхности
вторичной пористой структуры гранулированных цеолитов не дала положи-
тельных результатов [28]. Краситель не проникает внутрь гранул, и даже
после контакта с цеолитом в течение 200 ч адсорбция ограничивается наруж-
ным слоем гранул толщиной 0,1 мм. При измельчении гранул происходит
разрушение вторичной пористой структуры. В равной степени безуспешной
•была попытка определить внешнюю поверхность кристаллов цеолитов по ад-
сорбции красителей из растворов; образование устойчивых эмульсий снижает
точность эксперимента.
Капиллярная конденсация и структура мезопор
В мезопорах промышленных адсорбентов при высоких относи-
тельных давлениях фазовый переход происходит по механизму
капиллярной конденсации. Адсорбция паров в этом случае опи-
сывается изотермами, представленными на рис. 2,16. Для таких
изотерм характерно наличие петли гистерезиса: при данном
относительном давлении точки изотерм, полученные при после-
довательном понижении давления (т. е. при десорбции), лежат
выше точек, полученных при повышении давления (т. е. при
адсорбции). Перегиб на изотермах рассматриваемого типа, со-
провождающийся расхождением ветвей гистерезиса, начинается
при относительных давлениях plps = Q,<2.—0,5. Координата точки
перегиба зависит и от вида адсорбента, и от типа адсорбтива:
для бензола при 20 °C и силикагеля КСМ она соответствует от-
носительному давлению 0,20, для воды при 20 °C — 0,40, для азо-
та при —196 °C — 0,45 и т. д.
До этой точки адсорбционная способность растет в резуль-
тате развития полимолекулярной адсорбции. Затем адсорбция
вещества начинает происходить в результате конденсации пара
в мезопорах — капиллярах, смачи-
ваемых адсорбатом. Смачивающая
жидкость образует в капилляре во-
гнутый мениск. Давление пара над
вогнутым мениском меньше давле-
ния насыщенного пара над плоской
поверхностью, вследствие чего кон-
Рис. 2,16. Изотерма адсорбции паров бензола на
мелкопористом силикагеле КСМ при 20 °C (а —
адсорбционная ветвь; б — десорбционная ветвь).
Капиллярная конденсация и структура мезопор
51.
.'нпсация наступает при plps<,\. При относительном давлении,
ранном 1, мезопоры целиком заполнены адсорбатом.
Капиллярная конденсация описывается уравнением Кельви-
на :
2 ста
lnp/ps = — —(2.35>
где pips — относительное давление пара адсорбтива; г — радиус сферического-
мениска жидкости; ст и v— поверхностное натяжение и мольный объем ад-
снрбтива в жидком состоянии.
Изотермы адсорбции в области капиллярной конденсации
(конкретно — их десорбционные ветви) используют для построе-
ния кривой распределения пор. Методика заключается в опре-
делении на основании каждой точки десорбционной ветви изо-
термы двух показателей: адсорбционной способности а (в моль-
•г-1) и относительного давления p]ps. Произведение av характе-
ризует объем пор V, заполненных жидкостью. Параллельно ПО'
уравнению (2.35) определяют эффективный радиус цилиндриче-
ских пор, в которых происходит капиллярная конденсация при
заданном значении plps-
Например, капиллярная конденсация бензола при 20 °C, согласно форму-
ле (2.35), в случае цилиндрических пор радиусом г=5 нм происходит при
относительном давлении р/щ = 0,68; в случае г=50 нм — при p/ps = 0,96;
в случае г=200 нм-—при p/ps = O,99. На основе этих расчетов строят струк-
турную кривую V=f(r), затем в каждой точке структурной кривой находят
значение производной (dV/dr или ДУ/Дг). Эти значения используют для по-
строения кривой распределения объема пор.
Анализ экспериментальных результатов о капиллярных явле-
ниях, протекающих в пористых адсорбентах и катализаторах,
не может быть выполнен без данных о геометрической форме
пор. Сами капиллярные явления, например тип гистерезисной
петли при сорбции паров, обычно не дают однозначной инфор-
мации о геометрии преобладающих пор [29]. Только независи-
мые методы, например электронная микроскопия высокого раз-
решения, позволяют в ряде случаев составить определенное
представление о геометрии пористых структур [30].
Уравнение Кельвина получено для случая, когда мениск жид-
кости имеет сферическую форму. Для адсорбентов, видимо, ха-
рактерны поры самой различной конфигурации. Каждой конфи-
гурации пор соответствует своя форма мениска. В связи с этим
результаты вычислений на основе опытов по капиллярной кон-
денсации пара отвечают не реальному адсорбенту, а эквива-
лентному модельному адсорбенту с условно принятой формой
пор, для которого десорбционная ветвь изотермы капиллярной
конденсации совпадает с соответствующей кривой для реально-
ю адсорбента. Обычно за основу принимаются эквивалентные
модельные адсорбенты с цилиндрическими порами. Вычисляв-
52
2. Адсорбционное равновесие и пористая структура адсорбентов
оценки пористой структуры
Рис. 2,17. Структурная кривая (а) и кривая рас-
пределения пор по значениям эффективных ра-
диусов ДУ/Дг (б) для силикагеля.
мые для них радиусы являются эф-
фективными величинами. Структур-
ная кривая и кривая распределения
объема пор по значениям эффектив-
ных радиусов для силикагеля пред-
ставлена на рис. 2,17.
Явление гистерезиса, проявляю-
щееся в несовпадении адсорбцион-
ной и десорбционной ветвей обяза-
но тому факту, что форма мениска
при заполнении и опорожнении пор
различна*. Карнауховым [31] про-
слежена зависимость между форма-
ми пор и мениска для основных ти-
пов адсорбентов. Эти данные приве-
дены в табл. 2-7.
Десорбция в подавляющем боль-
шинстве случаев происходит с об-
разованием сферического мениска,
вследствие чего для всех моделей
(кроме щелевидных) применимо
уравнение (2.35). Поэтому для рас-
чета кривых распределения пор по
размерам эффективных радиусов
предпочтительнее использовать де-
сорбционную ветвь изотермы.
При выборе адсорбтива с целью
следует иметь в виду, что чувстви-
тельность метода капиллярной конденсации зависит от сниже-
ния давления пара адсорбтива Ар над вогнутым мениском поры
данного размера. Величина Ар пропорциональна комплексу в
правой части формулы (2.36). Она находится в прямой зависи-
мости от поверхностного натяжения и мольного объема адсорб-
тива и в обратной от температуры опыта:
Др = К (ov/T)
(2.36)
где К. — коэффициент пропорциональности.
Наибольшими значениями комплекса от/Т отличаются четы-
реххлористый углерод, гексан и бензол, в связи с чем эти ад-
* Имеется несколько теорий гистерезиса. В большинстве из них десорбцион-
ная ветвь изотермы трактуется как истинная и рекомендуется для расчетов
пористой структуры адсорбентов (Прим. ред.).
Капиллярная конденсация и структура мезопор
53
1пГ>.иица 2-7. Форма пор и форма мениска при капиллярной конденсации
и различных пористых телах
Л «сорбент Форма пор Форма мениска
адсорбция | десорбция
Пористые стекла Цилиндрические Цилиндрический и Сферический
или близкие к ним закрытые и откры- тые поры, бутыл- кообразные поры сферический
Активные угли Шаровидные по- ры, соединенные узкими каналами (переходные и макропоры) Цилиндрический (горла), сфериче- ский (полости) Сферический
Силикагели, алю- Промежутки меж- Седловидный и седловидный
могелй, титаногели, алюмосиликатные катализаторы, са- жи, аэросил ду контактирую- щими глобулами Сферический
Хризотиласбест Поры внутри по- лых цилиндров, уложенных вдоль оси, и между ними. Цилиндрический Сферический
Монтмориллонит, окисленный гра- фит Промежутки меж- ду параллельными и наклонными плоскостями (ще- левидные поры) Цилиндрический Цилиндрический
сорбтивы чаще других применяют для оценки структуры. Одна-
ко при адсорбции крупных молекул в относительно мелких по-
рах весь объем последних может быть заполнен еще в процессе
собственно адсорбции. Тогда капиллярная конденсация не на-
ступает, а следовательно, не может быть получена эксперимен-
тальная информация, необходимая для расчета. Отмечены слу-
чаи, когда даже молекулы азота по этой причине не могли быть
использованы. Оценку структуры тонкопористых веществ сле-
дует проводить с использованием адсорбтивов, имеющих мини-
мальные размеры молекул, например, воды [32].
Выше была описана упрощенная модель капиллярной кон-
денсации. В действительности этот процесс идет, когда на стен-
ках уже имеется слой молекул адсорбата, связанных с твердой
фазой силами, характерными для физической адсорбции. Вслед-
ствие этого величина эквивалентного радиуса, вычисляемого по
формуле Кельвина, строго говоря, отвечает среднему радиусу
свободного цилиндрического пространства между адсорбцион-
ными пленками в опорожняемых (при десорбции) порах rk.
Средняя толщина адсорбционного слоя зависит от относи-
54
2. Адсорбционное равновесие и пористая структура адсорбентов
Капиллярная конденсация и структура мезопор
55
тельного давления p/ps и может быть определена на основе ре
зультатов опытов с рассматриваемым паром на непористом ад
сорбенте той же химической природы и с известной удельно!
поверхностью:
ta = ov/s = f (p/ps) (2.37
Тогда радиус опорожняемой поры:
r = а + ta
(2.38
Для построения исправленной кривой распределения объем!
пор десорбционную ветвь изотермы, изображенную в круннок
масштабе, разбивают на 10—15 участков для относительнс
близких значений десорбции Ла. Соответствующие изменение
равновесных относительных давлений для каждого этапа со-
ставляют A/i=A(p/ps). Объем опорожненных пор для каждого
этапа десорбции определяется из уравнения
Гт 2,18. Дифференциальные кривые
। .1 (расшифровка в тексте) распредс-
-н пня объема пор по значениям эф-
ф. к । явных радиусов для алюмосили-
н.ииого катализатора.
. 1>|-дняя толщина адсорбционного
- -юя для всей области гистерези-
. .1, оцененная Аултоном в /а =
1,3 нм. И наконец, кривая 1
• инечает расчету без введения по-
правок на толщину адсорбцион-
ных слоев. По внешнему виду все
кривые рис. 2,18 скорее выража-
ют распределение объемов пор
для самых разнообразных адсор-
бентов с одинаковым общим ха-
рактером пористости, нежели со-
ответствуют одному и тому же
адсорбенту.
ДУП = v
(rk)n Ч~ (Za)n
(Гк)п
(2.39'
К л-му этапу десорбции относятся следующие величины: Дап — измене-
ние величины адсорбции; (Д/а)п—изменение толщины адсорбированного
слоя на стенках, частично опорожненных в предыдущих этапах десорбции;
(rh)n — средний радиус свободного адсорбционного пространства между ад-
сорбционными пленками в опорожняемых порах и (ta)n—средняя толщинг
адсорбционной пленки. Величины, обозначенные индексом i, соответствуют
предшествующему этапу десорбции. Уточнение выражения (2.39) дано е
работе [33].
Уравнение распределения дифференциального объема пор пс
значениям эффективных радиусов для эквивалентного модель-
ного адсорбента выразится как
При оценке структуры однороднопористых адсорбентов в
первом приближении возможно рекомендовать введение посто-
янной поправки на толщину адсорбционного слоя в точке нача-
ла гистерезиса. В случае углеродных адсорбентов при 20 °C сле-
дует вычислять поправку /а (в нм) по формулам:
при адсорбции бензола
0,4715
(lgps/p-0,035)1/2
(2.42)
при адсорбции четыреххлористого углерода
0,4879
(igps/p — о.о^г^/г
(2.43)
ДУ/Дг = /(/) (2.40)
Отвечающее ему уравнение распределения дифференциаль-
ной удельной поверхности пор имеет вид
Дз 2 ДУ 2
дг = У’дг = 7"^г) (2-41)
Насколько важно учесть поправку на толщину адсорбцион-
ных слоев и правильно ее выбрать, видно из рис. 2,18, на кото-
ром представлены дифференциальные кривые распределения
пор алюмосиликатного катализатора, полученные на основе де-
сорбционной ветви капиллярной конденсации азота при —196 ’С,
по опытам Аултона [34].
Прерывистой линией обозначена кривая 2. вычисленная по уравнениям
(2.39) и (2.40). При построении кривой 2 введена постоянная поправка на
среднестатистическую толщину адсорбционного слоя 7а = 0,5 нм, отвечаю-
щую точке начала гистерезиса. Для кривой 3 принята 1акжс постоянная
Для адсорбции азота при —196 °C на кремнеземных адсор-
бентах наиболее достоверна формула де Бура:
°-4584 (2.44)
(lgps/p) /з
В работах Брукгофа и де Бура [35] сделана попытка усо-
вершенствования теории капиллярного испарения путем исполь-
ювапия более рационального уравнения Д — кривой, выражаю-
щей зависимость среднестатистической толщины адсорбционно-
го слоя t3 от равновесного относительного давления:
1п (Рь/Р) = 7П“ - ' D (Za) (2 -45)
в котором С — постоянный параметр, а корректирующий член
/)(/а) в зависимости от природы адсорбционной системы явля-
ется либо постоянной величиной, либо слабо зависящей от ta
функцией.
Да„ — (Дуп -~=г-
я (rk)t.
56 2- Адсорбционное равновесие и пористая структура адсорбентов
В дальнейшем Дубинин [36] для этой же цели предложил
использовать уравнение
1п(р,/р) = -Д- (2.46)
где К и m — параметры уравнения. Для сравнения с экспери-
ментальными данными оно было преобразовано в линейную
форму:
In In (ps/p) = 1п/С — mln/a (2.47)
Показано, что экспериментальные точки адсорбции азота на
всех изученных минеральных адсорбентах при равновесных дав-
лениях от 0,08 до 0,90 укладываются на одну прямую, отвечаю-
щую параметрам А=44,54 и т = 2,241, где величина А выра-
жена в А (10 нм). Таким образом, поправкой на изменение ве-
личины ta, приведенной ниже на примере адсорбции азота при
72 К на минеральных адсорбентах, следует пользоваться . для
расчета кривой распределения пор по радиусам в системах, ана-
логичных изученным:
pips 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,86 0,£
/а, нм 36,8 43,6 50,1 57,1 65,0 73,6 85,7 105,7 127,5 148,4
Другой, не менее важный для практики случай — анализ
данных по адсорбции бензола при 293 К на углеродных адсор-
бентах— рассмотрен в работе [37]. Параллельно была прове-
дена оценка величины t& в концепции уравнения Брукгофа и де
Бура (табл. 2-8). Как видно из таблицы, оба уравнения в ин-
тервале относительных давлений 0,08—0,8 описывают ^-кривую
с одинаковой точностью. Лишь при высоких давлениях появля-
ются расхождения, значительно возрастающие при приближении
р/Рз к 1.
Таблица 2-8. Сравнение значений (в нм) при адсорбции бензола (293 К)
на углеродных адсорбентах, вычисленные по формулам (2.45) и (2.46)
P/Ps t, вычислено по уравнению Д, % P'PS t, вычислено по уравнению Д. %
(2.45) (2.46) (2.45) (2.46)
0,08 0,4502 0,4509 0,16 0,70 1,212 1,212 0,00
0,10 0,4717 0,4723 0,13 0,80 1,536 1,543 0,45
0,20 0,5654 0,5654 0,00 0,90 2,245 2,293 2,09
0,30 0,6548 0,6542 0,02 0,94 2,939 3,088 4,64
0,40 0,7518 0,7507 0,15 0,98 5,176 6,477 20,1
0,50 0,8658 0,8644 0,10 0,99 7,369 15,801 53,4
0,60 1,0030 1,0090 0,59
Капиллярная конденсация и структура мезопор §7
11рп введении поправок на величину ta, как показано в опы-
1.|\ н<> капиллярному испарению азота при 78 К и бензола при
К из активных углей, дифференциальные кривые распреде-
пни объема мезопор по радиусам практически совпадают.
Метод капиллярной конденсации позволяет установить ха-
p.1мер распределения мезопор во всем интервале их радиусов:
• и 1,5 до 100—200 нм. Недостатком метода является длитель-
ность опыта: изучение одного образца занимает 10—15 сут.
1 ели изучение изотерм проводят весовым методом, при боль-
.... числе определений целесообразно применять установки с
несколькими пружинными весами.
Разработаны методы эталонной порометрии, позволяющие
измерять структурные и поверхностные характеристики пори-
стых тел в интервале радиусов пор от 1 до 10s нм [38]. Методы
основаны на том, что в состоянии равновесия системы, состоя-
щей из контактирующих пористых тел (метод контактной эта-
лонной порометрии), или системы пористых тел, помещенных в
одной герметичной камере (метод бесконтактной порометрии),
наблюдается равенство капиллярных потенциалов, характери-
зующих прочность связи жидкости (например, воды) с адсор-
бентом и определяющих очередность заполнения и осушения
пор. Равенство капиллярных потенциалов достигается при по-
стоянном p/ps, и для одинаково смачиваемых жидкостью твер-
дых тел отвечает заполнению капилляров одного радиуса. По-
этому оно соблюдается на всем протяжении так называемой
кривой относительного влагосодержания, которая характеризует
зависимость влагосодержания исследуемого образца vn от вла-
госодержания эталона пэ, измеренных при одинаковых p/ps.
Влагосодержапие системы пористых тел можно измерять различными
способами: испарением, конденсацией, пропиткой или отсасыванием. Наибо-
лее часто применяют испарительный способ контактной эталонной поромет-
рии. По этому способу приведенные в контакт с помощью прижимного уст-
ройства исследуемый образец и эталон, выполненные в виде дисков толщи-
ной от 0,1 до 2,0 мм, подвергаются откачке или обдувке открытой поверх-
ности образцов струей сухого воздуха. После испарения части жидкости
пористые тела быстро размещают по индивидуальным бюксам и взвешивают.
Полученную в опытах информацию используют для построе-
ния кривой относительного влагосодержания. Парные значения
и va на ней отвечают заполнению пор одинакового радиуса,
величина которых известна из независимо определенной инте-
гральной кривой распределения объема пор v3 эталона. Для тел
е разной смачиваемостью вводят поправку на угол смачивания
где индексы «и» и «э» относятся соответственно к исследуемому
образцу' и эталону.
58
2. Адсорбционное равновесие и пористая структура адсорбентов
Кривую распределения пор для эталона тщательно опред«
ляют обычными методами: по десорбционной ветви гистерезис
или методом ртутной порометрии.
Ртутная порометрия
Ртуть не смачивает поверхность пористых тел. Чтобы заполнит
ртутью объем поры, необходимо преодолеть сопротивление си
лы f, которая численно равна произведению периметра поры 1
на поверхностное натяжение жидкости о и косинус краевоп
угла смачивания 0:
/= шт cos О (2.49
Между внешним давлением р и капиллярным сопротивлени
ем в порах при равновесии существует зависимость (S — пло
щадь сечения поры):
pS=nocos0 (2.50
Для пор цилиндрической формы уравнение (2.50) приобре
тает вид
pnr2 — 2w<jcos 0 (2.51
откуда
r = 2ocos0/p (2.52
Уравнение (2.52) является исходным для расчета криво!
распределения объема пор по радиусам на основе данных ртут
ной порометрии. Каждому радиусу пор соответствует давление
при котором поры заполняются ртутью.
Средний краевой угол для системы ртуть — твердое тело со
ставляет 140° (более точно: для угля 142°, для силикагел?
145°). Если предварительно вакуумированный адсорбент поме
стить в ртуть, то минимальный радиус пор, которые заполня
ются ртутью, при атмосферном давлении составит 7-1()~4 см
Постепенно повышая давление, заполняют все более мелкие
поры. При давлении — 70 МПа заполняются поры радиусол
10 нм, при —350 МПа — 2,0 нм, —500 МПа — 1,5 нм. Таким об
разом, максимальное давление, создаваемое поромером, опреде
ляет нижнюю границу пор, которые могут быть изучены мето
дом вдавливания ртути.
В наиболее совершенных моделях аппаратуры может быть изучен,
кривая распределения пор во всем интервале мезо- и макропор. Например
поромер ПА-5, разработанный в Ленинградском технологическом институт!
под руководством проф. Плаченова, рассчитан на рабочее давление д<
500 МПа и позволяет определять объем пор в адсорбентах или катализато
рах с эффективными радиусами от 1,5 до 7500 нм. В связи со значительны
ми трудностями конструктивного характера в большинстве других поромет
рических установок верхний предел давления ограничивают 180—200 МПа
чему соответствует нижняя граница определения эффективных радиусов око
ло 3,0 нм.
Ртутная порометрия
59
Гт. 2,19. Дилатометр:
I капсюль дилатометра; 2 — крючок; 3 — перемычка;
I проволока; 5 — капилляр; 6 — головка; 7 — контакты.
Чтобы оценить объем наиболее круп-
ных пор, порометрическую установку до-
полняют порометром низкого давления,
и котором интервал измерений давлений
составляет 20—100 кПа. При этом могут
быть изучены поры с эквивалентными ра-
диусами до 4-104 нм.
При проведении опыта параллельно с
повышением и регистрацией давления в
системе определяют уменьшение объема
ртути ДУ, остающейся в свободном про-
странстве. Обычно ДУ устанавливают по
изменению электрического сопротивления
проволоки, погруженной в столбик ртути.j
Эти замеры производят с помощью дилатометра (рис. 2,19). Стеклян-
ный капсюль дилатометра I переходит в капилляр 5. Капсюль и капилляр
сообщаются через полый крючок 2. Капилляр соединен с головкой 6 из ор-
। апического стекла, на которой находятся два контакта 7. В нижней расши-
ряющейся части капилляра расположена перемычка 3, через которую пропу-
щена тонкая (0,08—0,10 мм) платиноиридиевая проволока 4. Концы прово-
локи выведены к контактам. Перед опытом в капсюль вводят навеску испы-
|усмого адсорбента, после чего нижний конец запаивают. Затем дилатометр
откачивают до остаточного давления порядка 1 • 10~2 Па, после чего капсюль
и рабочую часть капилляра заполняют ртутью, фиксируя путем измерения
сопротивления проволоки положение мениска ртути в капилляре.
Вводя в дилатометр порции воздуха, последовательно повышают в нем
давление и по изменению сопротивления проволоки определяют объем рту-
III, вошедшей в поры. Когда давление в дилатометре достигнет атмосферно-
н>, прибор подключают к поромеру высокого давления и продолжают изме-
рения.
Объем пор адсорбента, имеющих эквивалентный радиус
меньше 3 нм, определяют как разность суммарного объема пор
Уд и интегрального объема пор УПОр, вычисленного по данным
ртутной порометрии:
AV= Кд —Кпор
Значение Уд находят по результатам пикнометрических из-
мерений плотностей:
Ух = (1/Рк) — (1/Рн)
где рк и ри — кажущаяся и истинная плотность адсорбента.
На рис. 2,20 представлены структурная кривая для адсор-
бента, определенная на основе данных ртутной порометрии, и
кривая распределения пор этого адсорбента по значениям эф-
фективных радиусов, полученная дифференцированием струк-
турной кривой.
60
2. Адсорбционное равновесие и пористая структура адсорбентов
Теория объемного заполнения микропор
61
Рис. 2,20. Структурная кривая адсорбента, опре рш, 2,21.
деленная по данным ртутной порометрии (а) „ путана (1)
кривая распределения пор этого адсорбента п<
радиусам (б).
Потенциальная теория
адсорбции
В 1914—1928 гг. Поляни и его ученика
ми [39, 40] была сформулирована теория
адсорбции, которая была названа потенци-
альной. Согласно основным положения!;
этой теории, вблизи своей поверхности ад-
сорбент сильно притягивает молекулы га-
за, что и приводит к адсорбции. Силы при-
тяжения, создаваемые адсорбентом, на-
столько значительны, что могут способст-
вовать образованию на его поверхности ря-
да адсорбированных слоев. Эти слои нахо-
дятся в сжатом состоянии под действием
сил притяжения их к поверхности. Наи-
большее сжатие испытывает первый адсор-
бированный слой, несколько меньшее — вто-
рой и так до тех пор, пока плотность ад-
сорбированного вещества не понизится дс
значения ее в окружающем газе.
Рассмотрение адсорбционных сил ка1
градиента потенциала межмолекулярныз
сил взаимодействия адсорбтива и адсор-
бента приводит в случае пара к следующе-
му выражению:
& = RT\Tipslp (2.53'
получившему название адсорбционно
го потенциала. Каждому значеиик
0,1 0,2 0,3 0,4 0,5 потенциала отвечает строго определенны!
1g г(нм) ’ объем адсорбированной пленки да, которы!
при адсорбции пара может быть определен
по величине адсорбции и объему пара в сжиженном состоянии, так как ежи
маемостью жидкости, по мнению авторов теории, можно пренебречь:
w = av
(2.54)1
где а — величина адсорбции; v— объем пара адсорбтива в сжиженном (ад-
сорбированном) состоянии при температуре опыта.
Зависимость e=f(sa) носит название характеристической кри-
вой адсорбции и инвариантна к изменениям температуры. Это свойство
ее вытекает из независимости адсорбционного потенциала от температуры.
Таким образом, теория Поляни позволяет предсказать изменение величины
адсорбции при варьировании температуры, если имеется характеристическая
кривая, определенная в опытах по адсорбции, проведенных при единственной
температуре. На рис. 2,21 в качестве примера представлены характеристиче-
ские кривые адсорбции «-бутана и пропана на угле, построенные на основе
двух изотерм адсорбции 0 и 20 °C.
Теория Поляни была подвергнута серьезной критике, сущность которой
сводилась к тому, что атомные и молекулярные взаимодействия, согласно
квантовой механике, не имеют большой протяженности действия и не могут
2,21. Характеристические кривые адсорбции
) и пропана (2) на активном угле.
образования ряда последователь-
Другой недостаток теории •— отож-
обычной жидкости. Поэтому се-
пГк-СПСЧИТЬ
иi.iх слоев,
пествление свойств адсорбированной фазы со
> шшствами ' ” .
11>дня теория Поляни представляет преимуще-
( гненно исторический интерес. Однако ее су-
ществование, хорошее соответствие опытам и,
и еще большей степени, ее содержательная
критика привели к возникновению двух самых
крупных современных теорий адсорбции: полимолекулярной и объемного за-
полнения микропор.
В полимолекулярной теории, которая была рассмотрена выше, главный
недостаток потенциальной теории устранен путем замены дальнодействую-
|цих сил поверхности на взаимодействие адсорбированных молекул в смеж-
ных слоях адсорбата. Теория объемного заполнения, в значительной степей»
сихранив терминологию и аппарат теории Поляии, приписывает разные зна-
чения потенциалов микропорам различных размеров.
Теория объемного заполнения микропор
Принципиальное различие адсорбционных явлений, протекаю-
щих в микропорах и на поверхности мезо- и макропор или не-
иористых адсорбентов, требует различных теоретических под-
ходов при их описании и интерпретации. Все рассмотренные
выше теории физической адсорбции, несмотря на их кажущееся
различие, исходят из одного и того же физического образа. Этот
физический образ сводится к представлению о геометрической
поверхности раздела фаз, на которой происходит адсорбция
с образованием одного или нескольких последовательных ад-
сорбционных слоев.
Представление о микропорах [41, 42] как об областях про-
странства в твердом теле, соизмеримых по размерам с адсорби-
руемыми молекулами, позволяет утверждать, что при любой
природе адсорбционных взаимодействий (т. е. под действием
дисперсионных, электростатических или других сил), обуслов-
ливающих физическую адсорбцию, во всем пространстве микро-
пор проявляется адсорбционное поле, создаваемое твердым те-
лом. Ограниченность адсорбционного пространства микропор
предопределяет тот факт, что последовательно адсорбирующие-
ся в микропорах молекулы не образуют адсорбционных слоев.
Адсорбция в микропорах характеризуется объемным заполне-
нием адсорбционного пространства. Поэтому основным геомет-
рическим параметром, характеризующим микропористый адсор-
бент, становится объем микропор, а не их «поверхность».
Концепция объемного заполнения микропор приводит к по-
нятию о величине предельной (максимальной) ад-
сорбции а0, которая отвечает адсорбции пара при давлении,
62 2. Адсорбционное равновесие и пористая структура адсорбентов
равном давлению насыщенного пара. Величина а0, естественно,
зависит от температуры, и эта зависимость определяется тер-
мическим коэффициентом предельной адсорбции а, который
практически постоянен в широком интервале температур:
1 da0 d In «0
a = ~^7’"dF= ~ dT
(2.55)
Если предельная величина адсорбции ащ экспериментально
-определена для некоторой температуры Т\, то согласно (2.55)
предельная величина адсорбции а02 для другой температуры Т2
может быть установлена из следующего выражения:
^02 ~ я01 ехР ( а (1*2 Т])) (2.56)
Для вычисления а0 по уравнению (2.56) необходимо знать
термический коэффициент предельной адсорбции а. В работе
Николаева и Дубинина [43] был предложен метод вычисления
плотности вещества в адсорбированном состоянии (адсорбата)
для широкого интервала температур. Этим методом можно вос-
пользоваться для вычисления а. Его идея заключается в сле-
дующем.
Адсорбированное вещество в микропорах в поле адсорб-
ционных сил подобно жидкости, находящейся в сильно сжатом
состоянии. По ориентировочной оценке этому сжатию отвечает
гидростатическое давление порядка нескольких сотен атмосфер.
Для области температур, значительно ниже критических, на-
пример нормальной температуры кипения, сжимаемостью жид-
кости в объемной фазе, а следовательно, и сжимаемостью ад-
сорбата можно пренебречь. В этом случае плотность адсорбата
в первом приближении можно принять равной плотности погло-
щаемого вещества в жидком состоянии при температуре опыта
и определить по справочным данным.
По мере приближения к критической температуре плотность
жидкости в объемной фазе резко падает, а ее сжимаемость
сильно возрастает. Поэтому соотношение плотностей адсорбата
при ТКп и Ткр нельзя считать равным соотношению реальных
плотностей объемной жидкой фазы при указанных температу-
рах. Допустим, что плотность адсорбата при критической тем-
пературе соответствует максимальному сжатию, т. е. мольному
объему, выраженному константой b уравнения Ван-дер-Ваальса:
[р + (a/v2)] (v — b) = RT (2.57)
Как известно, мольный объем b отвечает учетверенному сум-
марному объему молекул и может быть вычислен по известной
формуле
& = 7?7’крЖр= 1,03-10-2 Гкр/Ркр (2.58)
Теория объемного заполнения микропор
63
(2.60)
(2.61)
(2.62)
Плотность адсорбата в микропорах при критической темпера-
туре р*кр составит:
ркр = 34/10006 (2.59)
Таким образом, отношение плотностей адсорбата при нор-
мальной температуре кипения и критической температуре мож-
но аппроксимировать отношением ро/р*кр, где р0— плотность
нормальной жидкости при а р*кр находится по формуле
(2.59). Это позволяет вычислить термический коэффициент пре-
дельной адсорбции а.
В самом деле, при постоянстве предельного адсорбционного
объема Wo, т. е. объема микропор, имеем:
для ТКП а0 = IV'oPo
для Гкр а0* = й^оРкр*
Отсюда
ао/ао* = Ро/Ркр*
Согласно (2.56) и (2.62) получаем:
_______1g («о/До’*)_____1g (Ро/Ркр*)
0,434 (Ткр-ГКп) ~ 0,434 (Гкр-Гкп)
В дальнейшем оказалось возможным в расчетах вместо ве-
личины адсорбции а0 пользоваться безразмерным параметром 0,
выражающим в данном случае степень заполнения микропор.
По определению
0 = а/ад (2.64)
В основе теории объемного заполнения микропор лежат тер-
модинамические закономерности, и поэтому при описании ад-
сорбционного равновесия используют такие термодинамические
функции как энтальпия, энтропия и энергия Гиббса. Для рас-
чета изменений этих функций в качестве стандартного состоя-
ния при рассматриваемой температуре принимается объемная
жидкая фаза, находящаяся в равновесии с ее насыщенным па-
ром при давлении ps или летучести fs.
Основной термодинамической функцией является дифферен-
циальная максимальная мольная работа адсорбции А, равная
со знаком минус изменению энергии адсорбции Гиббса AG:
А = — AG = RT In (рв/Р) (2.65)
или
А = RT In (2.66)
где р — равновесное давление; f — летучесть пара при температуре Т.
Введение в формулу летучести вместо давления позволяет
учесть неидеалыюсть газовой фазы.
64 2. Адсорбционное равновесие и пористая структура адсорбентов
Если адсорбция выражена в безразмерных единицах, то и
дифференциальную мольную работу адсорбции целесообразно
представить в форме безразмерного отношения А/Е, где Е —
характеристическая энергия адсорбции, о физическом смысле
которой будет сказано ниже. Тогда термическое уравнение ад-
сорбции можно представить в общей форме [44]:
0 = /[(А/Е),п] (2.67)
Уравнение (2.67) выражает функцию распределения микро-
пор по дифференциальной мольной работе адсорбции, причем
Е является одним из параметров этой функции. Большинство
•функций распределения в нормированной форме характеризу-
ются двумя параметрами. В формулу введен также второй па-
раметр, который условно обозначен через п.
Согласно уравнению (2.67) получаем выражение для харак-
теристической кривой:
А = Е<р(0,п) (2.68)
Если для различных паров функция <р и параметр п оста-
ются неизменными, то
А/Ао = £/£•„ = р (2.69)
т. е. характеристические кривые в координатах Л—0 являются
подобными. Другими словами, при одинаковых значениях 0 от-
ношения ординат характеристических кривых для двух паров
постоянны и равны коэффициенту подобия р. В формуле (2.69)
Ло и Ео — соответствующие величины для стандартного пара.
Из уравнения (2.68) следует, что Е = Л для некоторого зна-
чения 0° или характеристической точки, определяемых в общем
•случае из условия
<р(0°, п) = 1 (2.70)
причем при неизменности функции ф заполнение 0° будет оди-
наковым для различных паров (роль и будет рассмотрена ниже).
В связи с этим возникает возможность при известном виде
•функции ф определить характеристическую свободную энергию
адсорбции по одной точке изотермы адсорбции, соответствую-
щей заполнению 0°.
Рассматривая в термическом уравнении адсорбции (2.67)
/ как функцию распределения, мы, по существу, приняли допу-
щение о температурной инвариантности этой функции, полагая,
что ее параметры Е и п являются постоянными величинами для
рассматриваемой адсорбционной системы. Так как Е=Л для
заполнения 0°, т. е. Е является одной из точек характеристиче-
ской кривой, то допущение о температурной инвариантности
автоматически приводит к независимости характеристической
энергии адсорбции Е и, как следствие, параметра п от темпе-
ратуры.
Теория объемного заполнения микропор
65
Рассмотренное в самом общем виде представление об объем-
ном заполнении микропор позволяет на основе одной экспери-
ментальной изотермы адсорбции пара I (например, стандарт-
ного) при температуре Т\ вычислить адсорбционную способ-
ность а2 пара II при заданных параметрах: равновесном давле-
нии Р2 и температуре Т2. Для перехода от характеристической
кривой пара I к аналогичной кривой пара II необходимо знать
коэффициент подобия р. Чтобы произвести расчет, найдем рав-
новесное давление пара Ipi, соответствующее давлению пара
Пр2 для условия равенства заполнений 02 = 0ь
Согласно (2.69)
42Mi = Р21 (2.71)
Выражая дифференциальные мольные работы адсорбции
двух паров по (2.65), получим:
Г2 1g (Ps"/P2) _
Л12(Р//Р1) “Рз1
(2.72)
Откуда найдем:
1g Рг— lgР" P2i т lg(₽s7₽i) (2.73)
1 2
Из уравнения (2.64) и соотношения типа (2.60) при посто-
янстве заполнения следует
„ „ °<>2 „ Ра
й, — С1л _ — С1ч
8 1 а01 1 Р1
(2.74)
Условие температурной инвариантности характеристических
кривых выполняется в подавляющем большинстве случаев ад-
сорбции паров на микропористых адсорбентах. Принимая тем-
пературную инвариантность уравнения (2.67) и учитывая из-
вестное в математической статистике распределение Вейбула,
Дубинин и Астахов получили термическое уравнение адсорбции
(2.67) в аналитической форме [44]. Распределение заполнения
по дифференциальной мольной работе адсорбции выразится как.
0 = exp [—(А/Е)"] (2.75)
Для большого числа адсорбционных систем параметр п вы-
ражается небольшими (от 2 до 6) целыми числами.
Уравнение (2.75) позволяет перейти к уравнению характе-
ристической кривой:
А = E[ln(l/0)]V« (2.76)
Согласно (2.76) для двух различных паров при одинаковых
заполнениях 0 и условии, что показатель степени п не изменя-
ется
АДА, = Е2/Ех = ₽ (2.77)
5—1346
66 2. Адсорбционное равновесие и пористая структура адсорбентов
т. е. характеристические кривые являются подобными, и отно-
шение характеристических энергий адсорбции выражает коэф-
фициент подобия р.
Далее, из уравнения (2.76) следует, что при заполнении 0 =
= 1/е = 0,368 характеристическая энергия адсорбции Е равна
дифференциальной мольной работе адсорбции До; эта степень
заполнения получила название характеристической точки.
Выражая в уравнении (2.75) степень заполнения через вели-
чины адсорбции по (2.64), получим термическое уравнение ад-
сорбции:
а = а0 ехр [—(Д/Е)”] (2.78)
Это уравнение можно представить в линейной форме:
lga = lga0 — (0,434/Е") Лп (2.79)
В координатных осях 1g а—Ап уравнение (2.79) выражается
прямой линией, причем отрезок, отсекаемый прямой на оси ор-
динат, равен 1g а0, а угловой коэффициент прямой равен 0,434/ЕП.
Если показатель степени п известен, то из графика, выражаю-
щего линейное уравнение (2.79), легко определить предельную
величину адсорбции а0 и характеристическую энергию адсорб-
ции Е. В этом случае для каждой экспериментальной точки
изотермы (а, р) по (2.65) вычисляется соответствующее значе-
ние дифференциальной мольной работы адсорбции.
Представления об объемном заполнении микропор были раз-
виты вначале для микропористых углеродных адсорбентов или
активных углей, для которых определяющее значение в адсорб-
ционном взаимодействии имеют дисперсионные силы. Особен-
ностью большинства промышленных активных углей является
легкая доступность микропор для молекул паров веществ, мо-
лекулярные массы которых не превышают 150. Поэтому пре-
дельные значения адсорбционной способности в микропорах аа
для температур, не превышающих нормальные температуры
кипения соответствующих адсорбтивов, в удовлетворительном
приближении отвечают значениям предельного объема адсорб-
ционного пространства Wo, являющегося константой рассматри-
ваемого микропористого адсорбента [45]
Wo = а0/р = aov (2.80)
где р и v — плотности и соответственно мольные объемы нормальных жид-
костей при температурах опытов.
Для температур более высоких, чем нормальные темпера-
туры кипения, плотности адсорбатов р* и отвечающие им моль-
ные объемы v* вычисляются на основе формулы
lg Р* = lgpKn - 0,434а (Т - ТКП) (2.81)
где ркп — плотность нормальной жидкости при температуре кипения ТКП.
Теория объемного заполнения микропор
67
В этом случае
V = М/р* (2.82)
Тогда для области Т>ТКП получаем
Го = а0/р0* = <№* (2.83)
Предельный объем адсорбционного пространства Wo часто
принимается в качестве одного из параметров Термического
уравнения адсорбции взамен а0:
г /л\"1 w0 г /луч
а = аоехр I — l-g- I I =exp — l-g-l (2.84)
Для подавляющего большинства активных углей параметр
п = 2, и тогда термическое уравнение принимает вид
1Г0 Г / А VI
а = —- exp I — I -g- I (2.85)
или для общего случая адсорбции паров различных веществ
Го Г / А VI
а= ~ехР[-(ж} ] (2-86)
где р — коэффициент подобия; Wo и Ео —параметры уравнения, определяе-
мые по изотерме стандартного пара, для которого ₽=1.
Уравнение (2.86) справедливо для однородно микропористых
углей. Для адсорбентов с более широким распределением объ-
ема пор по размерам, например, углей, содержащих как микро-
поры, так и супермикропоры, можно принять, что адсорбция
в порах разных размеров происходит независимо. В таком слу-
чае уравнение адсорбции (2.86) приобретает другую форму [46]
Г01 Г ( А VI , ^02 Г (А VI
«- v, exp ^е01)]+ v* ехр [ “' ( Е02 ) ] (2,87)
В этом уравнении параметр IFoi отражает объем собственно
микропор, а параметр 1^02— объем супермикропор конкретного
адсорбента. Параметры IFoi, Eoi, Е02 могут быть определе-
ны графически даже по одной изотерме адсорбции, полученной
в широком интервале равновесных относительных давлений.
Подставляя в уравнение адсорбции (2.86) дифференциаль-
ную мольную работу адсорбции А = RT In (ps/p), получим терми-
ческое уравнение адсорбции, известное в литературе как урав-
нение Дубинина — Радушкевича:
1Г0 Г Г2 , ]
а= — exp I — В -p2“(lgPs/P)2 (2.88)
Применимость уравнения Дубинина — Радушкевича для опи-
сания адсорбционного равновесия в достаточно широком тем-
пературном интервале иллюстрируется рис. 2,22: эксперимен-
тальные точки адсорбции «-бутана при температурах от 20 до
5*
68
2. Адсорбционное равновесие и пористая структура адсорбентов
Рис. 2,22. Изотермы адсорбции
н-бутана на активном угле СКТ »
координатах уравнения Дубинина —
Радушкевича (разные точки соот-
ветствуют разным температурам).
100 °C на активном угле СКТ в координатах этого уравнения
ложатся на единую прямую.
Параметр В отражает преобладающий размер микропор. Чем
мельче микропоры в адсорбенте, тем меньше значение В и тем
круче поднимаются изотермы адсорбции газов или паров на дан-
ном адсорбенте. Параметр В непосредственно связан с характе-
ристической энергией адсорбции Е:
В= (2,3037?/Е)2 (2.89)
Между преобладающим размером микропор, характеристиче-
ской энергией стандартного вещества Ео и константой В сущест-
вует строгая зависимость. Если принять, в качестве первого при-
ближения, сферическую форму микропор активных углей со
средним радиусом г, то эта зависимость может быть прослеже-
на по значениям характеристической энергии адсорбции бензо-
ла Ео и структурной константы В для сферических пор разного
размера [46], приведенными ниже:
г, нм 0,30
Ео, кДж/моль 42,8
В-10е, град-2 0,2
0,43 0,76 0,52
30,3 17,3 8,56
0,4 1,2 5,0
В области степеней заполнения адсорбционного объема от
0,3 до 1,0 приближенный расчет адсорбционной способности
цеолитов также возможно провести по формуле Дубинина —
Радушкевича [47]. Плотности адсорбированных паров воды, ок-
сида и диоксида углерода, сульфида водорода, углеводородов с
числом атомов углерода в молекуле до 5 включительно, а так-
же других компонентов промышленных газов с небольшим раз-
мером молекул равны или близки к плотностям соответствую-
щих жидкостей при температуре опыта. В связи с регулярным
строением цеолитов предельный объем адсорбционного прост-
ранства гранулированных образцов Wo, который к тому же мо-
жет быть определен из независимых рентгенографических дан-
ных, и константа В при стандартном паре — азоте в среднем со-
ставляют:
Цеолит NaA СаА СаХ NaX
1^0, см3/г 0,205 0,223 0,235 0,235
В-10е, град-2 4,90 5,50 6,15 6,55
Теория объемного заполнения микропор
69
Таблица 2-9. Коэффициенты подобия при адсорбции веществ
микропористыми адсорбентами (стандартное вещество — азот)
Лдсорбтив Адсорбенты Адсорбтив Адсорбенты
актив- ные угли цеоли- ты актив- ные угли; цеоли- ты
Азот 1,00 1,00 Изопентан 3,20 3,10 Метан 1,52 1,34 Циклопеитан 2,80 2,80 Этан 1,70 1,68 Циклогексан 3,14 3,08 Пропаи 2,20 2,27 Метилциклогексан 3,30 3,85 н-Бутан 2,65 2,90 Бензол 3,05 4,50 н-Пентаи 3,20 3,20 Толуол 3,78 5,58 н-Гексан 3,95 3,77 Ацетилен 1,30 3,08 н-Гептаи 4,52 4,10 Метилацетилен 1,82 4,05 н-Октан 4,85 4,72 Винилацетилен 2,21 4,25 н-Нонаи 5,45 5,30 Диацетилен 2,06 4,95 н-Декаи 6,00 5,85 Вода — 2,53 Этилен 1,40 2,26 Сероводород 1,14 2,35 Пропилен 1,91 3,31 Оксид углерода 0,61 1,30 1-Бутен 2,50 3,80 Диоксид углерода 1,25 2,31 н-Гексен 3,60 5,77 Сероуглерод 2,12 2,08
Бензол не может быть использован в качестве стандартного
пара при описании равновесного состояния на цеолитах: его мо-
лекулы не проникают в адсорбционные полости значительного
числа цеолитов, а энергия взаимодействия с цеолитами, имею-
щими крупные входные окна, характеризуется большой специ-
фической составляющей, причем последняя не постоянна для
различных типов цеолитов. В связи с этим при адсорбции на
цеолитах в качестве стандартного пара используют азот. Зна-
чения коэффициентов подобия основных компонентов промыш-
ленных газов при их адсорбции на цеолитах и активных углях
(стандартное вещество — азот) приведены в табл. 2-9.
В том случае, когда адсорбция определяется действием дис-
персионных сил, коэффициенты подобия могут быть достаточно
точно оценены как отношение парахора адсорбируемых моле-
кул Р к парахору стандартного пара (при адсорбции на актив-
ных углях обычно бензола) Ро [42]:
р = Р/Р0 (2.90)
Значения парахоров не зависят от температуры и определя-
ются по соотношению
Р = Мо1/,4/р
(2.91)
где М — молекулярная масса; а — поверхностное натяжение адсорбтива;
р — плотность адсорбтива в жидком состоянии.
70
2. Адсорбционное равновесие и пористая структура адсорбентов
Парахоры являются аддитивными величинами и рассчиты-
ваются на основе данных по инкрементам парахоров отдельных
атомов Ра и связей Рс, приведенных ниже:
Элемент Связь Рг
Углерод 4,8 Двойная 23,2
Водород 17,1 Тройная 46,6
Азот 12,5 Четырехчленное кольцо 11,6
Кислород 20,0 Пятичленное кольцо 8,5
Сера 48,2 Шестичленное кольцо 6,1
В случае адсорбции на цеолитах веществ, молекулы кото-
рых отличаются неоднородным распределением электронной
плотности, под значением р подразумевают, строго говоря, эм-
пирическую константу, определяемую из опыта и позволяющую
использовать уравнения теории объемного заполнения микро-
пор для описания адсорбционного равновесия [47].
Все адсорбтивы, молекулы которых обладают дипольным мо-
ментом и квадрупольным моментом, кратными связями, при
адсорбции на цеолитах отличаются повышенными коэффициен-
тами подобия. Сравнение опытных и теоретических значений ко-
эффициента подобия показало, что для гомологических рядов
олефиновых и ароматических углеводородов соответствие тео-
рии и опыта достигается, если в качестве стандартного вещества
выбрать простейший углеводород ряда, например, этилен для
олефиновых и бензол для ароматических углеводородов [48].
Допущение о том, что плотность адсорбата соответствует
плотности ожиженного пара при температуре опыта, облегчает
практические расчеты, но имеет смысл только для p = ps и по-
этому не всегда точно выполняется. Так, были рассчитаны [49]
средние плотности адсорбата для системы вода — цеолит NaX,
как частное от деления предельной адсорбционной способности
а0 на объем адсорбционного пространства, определенный из
рентгено-структурных данных. Найдено, что плотность адсорби-
рованной воды несколько выше плотности свободной воды и кри-
вая зависимости плотности адсорбата от температуры в отличие
от кривой для обычной воды, не имеет экстремума.
Значения предельного адсорбционного объема цеолитов, вы-
численные из изотерм адсорбции высокомолекулярных углеводо-
родов, последовательно уменьшаются при увеличении числа ато-
мов углерода в молекуле, что, очевидно, связано с плотностью
упаковки молекул в полостях цеолитов. Вследствие этого при
расчете адсорбционной способности цеолитов по высокомолеку-
лярным углеводородам к значению IFO следует вводить попра-
вочные коэффициенты на плотность упаковки Ду. Для случая
Уравнение Фрейндлиха
71
адсорбции нормальных парафинов на цеолитах NaX значения
/СУ составляют: н-С6Н14— 0,88; н-С7Н16 — 0,86; н-С8Н18— 0,84, а
на цеолите СаА соответственно 0,80; 0,74; 0,70.
Для случая адсорбции под действием дисперсионных сил ко-
эффициенты подобия i-ro адсорбтива с одного стандартного ве-
щества на другое могут быть пересчитаны умножением на отно-
шение вычисленных по инкрементам парахоров веществ*:
Pi/CeHe = 0,33₽i/N2 = 0,7зрг/с3н8 (2.92)
Если требуется описать адсорбционное равновесие на цеоли-
тах более точно и в более широком интервале заполнений, сле-
дует использовать термическое уравнение адсорбции (2.78) с
конкретным значением параметра и:
для адсорбции различных веществ на цеолите с малым чис-
лом адсорбционных центров (катионов) типа морденита или
эрионита п=3;
для адсорбции крупных молекул (бензола, толуола) на цео-
литах типа X и Y м=4;
для адсорбции небольших молекул на цеолитах типа А, X и
Y значение п изменяется от 3 до 6.
Уравнение Фрейндлиха
Широкое распространение в практических расчетах по адсорб-
ции получило степенное уравнение, носящее название уравнения
Фрейндлиха:
а — ftp” (2.94)
где а — количество адсорбированного вещества; р — давление адсорбтива;
k и п — постоянные величины.
Достоинство уравнения Фрейндлиха в простоте и удовлетво-
рительном соответствии опыту в широком интервале давлений,
исключая, может быть, интервал самых низких равновесных
давлений, когда адсорбция строго описывается уравнением Ген-
ри. Обычно в расчетах используют линейную форму уравнения:
lga= IgA-f-nlgp (2.95)
* Можно предложить более универсальное правило перехода от одного стан-
дартного вещества к другому; пригодное для адсорбции, обусловленной лю-
быми силами;
₽./СН = -Л/Ь12~ (2.93)
1/C’He ₽c6h6/n2
Проиллюстрируем это правило, использовав данные табл. 2-9, для этилена.
Коэффициент подобия для адсорбции этилена на цеолитах по азоту
(мм;2 = 2,26; коэффициент подобия бензола по азоту 3ci!nb/n2=: 4,50. Следова-
тельно, коэффициент подобия этилена по бензолу на цеолитах должен быть
равен Pi/CgHg —2,26/4,50=0,49 (Прим. ред.).
72 2. Адсорбционное равновесие и пористая структура адсорбентов
Рис. 2,23. Изотермы адсорбции оксида углерода на
косточковом угле в координатах уравнения Фрейнд-
лиха.
На рис. 2,23 представлены изотер-
мы адсорбции оксида углерода на уг-
ле из скорлупы кокосового ореха по
опытам Гомфрей [50] в линейной фор-
ме уравнения Фрейндлиха. Тангенс уг-
ла наклона прямых позволяет опреде-
лить значения коэффициента и, а от-
резки, отсекаемые ими на оси ординат,
lg k. С повышением температуры зна-
чения п последовательно увеличивают-
ся, приближаясь к предельному значе-
нию, равному 1. В этом предельном случае (при низкой адсорб-
ционной способности) адсорбционное равновесие аналогично
процессу растворения и подчиняется закону Генри: адсорбция
линейно растет с давлением. Значения k с ростом температуры
уменьшаются. Для расчета адсорбционной способности по фор-
муле Фрейндлиха в справочниках обычно приводят либо гра-
фики, аналогичные представленным на рис. 2,23, либо таблич-
ные данные, как это показано для рассматриваемого случая
ниже:
Т, °C —78,3 —39,6 0 20,0 46,2
п 0,38 0,56 0,75 0,82 0,84
k 12,0 2,5 0,56 0,29 0,15
Легко показать, что для пара уравнение Фрейндлиха явля-
ется частным случаем более общего уравнения изотермы ад-
сорбции теории объемного заполнения микропор:
а = а0 exp [— (А/Е)”] (2.78)
Если п= 1
Г RT In (ps/p)
а = а0 ехр —------------g-------
0,434АТ , р
lga= lgа0 +------g----lg —
J- HS
(2.96)
(2.97)
Выражение (2.95) совпадает с уравнением (2.97), если при-
нять, что К = а0, а и = 0,434 Д7/Е.
Уравнение Кисарова
Кисаровым [51] предложено удобное для практических расче-
тов уравнение, которое в целом базируется на уравнении Ленг-
мюра, но, по мнению автора теории, учитывает силы притяжения
Уравнение Кисарова
73
между молекулами адсорбата и силы притяжения их поверх-
ностью противоположных стенок адсорбента (в случае адсорб-
ции микропористыми адсорбентами):
AB(p/Ps)ni
а ~ 1В (р/ps)n
(2.98)
Константа А в уравнении (2.98) представляет собой отношение Wn/v,
где W’n — предельный сорбционный объем без той его части, в которой мо-
жет происходить капиллярная конденсация, см3/г; v — объем одного милли-
моля в адсорбированном состоянии, см3/ммоль (при температуре выше кри-
тической вместо величины v используют константу Ван-дер-Ваальса Ь). Кон-
станта В зависит только от природы и структуры адсорбента и является
безразмерной величиной. Постоянная п равна
/г = И70 (2.99)
где k=aR — константа; Т — абсолютная температура; р — коэффициент по-
добия; постоянные п, k и а — зависят от природы и структуры адсорбента,
природы адсорбата.
Линейная форма уравнения (2.98) имеет следующий вид:
lg ("Т ~ 4“) = lg ”lg (₽s/p) (2.100)
или
/ 1 1 \ { 1 \ kT ,
,g (т - Т) = lg ( АВ J + “Г g {рМ
(2.101)
Применимость этого термического уравнения подтверждает-
ся линейностью изотерм адсорбции в координатах 1g —
_lg0s.)(CM. рис. 2,24) на различных активных углях, силика-
гелях и цеолитах в широком интервале температур и равновес-
ных относительных давлений.
При достаточно больших значениях А, т. е. при степени за-
полнения адсорбционного объема, далекой от насыщения, урав-
нение (2.98) преобразуется в уравнение Фрейндлиха
а ~ kpn
где k=ABIps.
При п=1 уравнение (2.98)
дит в уравнение Ленгмюра
АЬр
а== \ + Ьр
Рис. 2,24. Изотермы адсорбции бензола
форме уравнения Кисарова на двух обра
пористых активных углей.
74 2. Адсорбционное равновесие и пористая структура адсорбентов
Очевидно, что в этом случае А соответствует величине ат,
a b = B/ps.
Несмотря на спорность использования исходных позиций кон-
цепций Ленгмюра для описания адсорбционного равновесия на
микропористых адсорбентах, формулу Кисарова достаточно ши-
роко используют в практических расчетах.
Физические методы исследования
адсорбционного взаимодействия
Установлению характера взаимодействия адсорбат-адсорбент и состоя-
ния адсорбированных молекул способствует применение современных физи-
ческих методов. Подробное изложение техники выполнения некоторых из них
приведено в [52].
Спектроскопия в инфракрасной области впервые была применена Тере-
ниным [53] для исследования поверхностных явлений. С помощью ИК-спект-
роскопии в настоящее время решают многочисленные задачи расшифровки
механизма адсорбции [54]. Появление при адсорбции новых полос и смеще-
ние полос поглощения молекул являются основными критериями для уста-
новления не только качественных, но и количественных показателей адсорб-
ции. Особенно успешно ПК-спектроскопию применяют для оценки взаимо-
действия молекул с гидроксильными группами поверхности кремнеземных
адсорбентов [55], специфической адсорбции на катионированных цеолитах
[56] н решения ряда других задач.
Применение ИК-спектроскопии позволило Неймарку с сотр. [57] прове-
сти тщательный анализ причин, вызывающих изменение адсорбционных
свойств силикагелей при модифицировании их поверхности, и обосновать
направленный синтез силикагелей с требуемыми свойствами.
Другим широко используемым методом является исследование систем,
основанное на изменении магнитной восприимчивости. Под влиянием адсорб-
ционных сил магнитная восприимчивость системы адсорбат — адсорбент
может изменяться. В случае физической адсорбции это изменение невелико,
в случае хемосорбции — существенно. При изучении физической адсорбции
того или иного вещества магнитными методами особое внимание нужно уде-
лить подбору адсорбента, который безусловно не образует химических сое-
динений с молекулами адсорбата.
Поглощение электромагнитных волн веществом, находящимся в посто-
янном магнитном поле, которое обусловлено магнитным моментом ядер, на-
зывают ядерным магнитным резонансом (ЯМР). При этом ядра распреде-
ляются по энергетическим уровням; самое большое число ядер концентри-
руется на самом нижнем энергетическом уровне.
Если такую систему подвергнуть действию переменного магнитного поля,
плоскость вращения которого перпендикулярна к плоскости вращения по-
стоянного магнитного поля, начинается переход ядер на более высокие
энергетические уровни. При этом поглощается электромагнитная энергия.
Величина этой энергии достигает максимума при резонансе, т. е. при равен-
стве кванта разности энергий соседних энергетических уровней. Поглощение
энергии приводит к нарушению термического равновесия между сообществом
ядер и его окружением («решеткой»). Энергия ядерной системы, «темпера-
тура» которой выше «температуры» решетки, уменьшается. Наступает со-
стояние теплового равновесия. Этот процесс называют термической релакса-
цией, и его характеризуют временем спин-решеточной релаксации, т. е. вре-
менем, за которое число частиц на верхнем энергетическом уровне убывает
До величины, соответствующей тепловому равновесию.
Физические методы исследования
75
Рис. 2,25. Схема включения термопар прн терми-
ческом анализе.
Метод ЯМР позволяет выяснить ори-
ентацию молекул адсорбата на поверхно-
сти адсорбента, получить информацию о
свойствах вещества в адсорбированном со-
стоянии, подвижности молекул и решить
ряд других задач.
Электронный парамагнитный резонанс
(ЭПР), по своей природе близкий к ЯМР,
также применяют в адсорбционной техни-
ке. Основное отличие ЭПР заключается в
Инертное вещество Проба
том, что магнитные моменты электронов на три порядка превышают магнит-
ные моменты ядер. ЭПР является очень чувствительным методом обнаруже-
ния неспаренных электронов. Он успешно применен, в частности, для выяс-
нения механизма адсорбции полициклических ароматических углеводородов
(антрацена, перилена), а также их производных (дифениламина) на алюмо-
силикатных катализаторах и цеолитах.
Для решения частных задач используют также измерение диэлектриче-
ских свойств системы адсорбат—адсорбент [58], изучение спектров люми-
несценции адсорбированных веществ и некоторые другие методы [59].
Изучение адсорбентов с помощью электронной микроскопии представ-
ляет интерес лишь для процессов, протекающих в макро- и мезопорах. Одна-
ко в адсорбционной технике она может быть использована и косвенным
образом. Так, каталитический процесс превращения сероводорода протекает
на углеродной поверхности по единому механизму, независимо от того, яв-
ляется ли эта поверхность плоской и открытой или скрыта от наблюдателя
внутри пористого тела. Проведенное Лукьяновичем исследование этого про-
цесса на гладкой поверхности графита методом реплик позволило устано-
вить ее механизм: молекулы выделившейся элементной серы являются ак-
тивными центрами, на которых осаждаются вновь образующиеся молекулы;
так образуется монолит. Эксперименты, проведенные впоследствии Костри-
ковым, подтвердили, что аналогично протекает процесс выделения серы в
мезопорах активных углей.
Для получения сведений о степени неоднородности поверхности твердых
тел имеет значение метод вакуумного декорирования [60]. Твердое тело по-
мещают в вакуум, нагревают до 100—200 °C и на его поверхность методом
термического испарения наносят слой золота толщиной 0,5—1,0 нм. Затем
поверхность покрывают сплошным слоем углерода толщиной 10 нм, отделя-
ют его и- расположение захваченных частиц золота устанавливают с по-
мощью электронного микроскопа.
Важную информацию о поведении твердых тел при нагреве дает термо-
гравиметрический метод [61], позволяющий
при изменении температуры одновременно
изучать тепловые эффекты (дифференци-
ально-термический анализ) и изменение
массы (термогравиметрический анализ).
Сущность дифференциально-термиче-
ского анализа заключается в том, что об-
разцы испытуемого и термостабильного
стандартного вещества помещают в элект-
рических печь, где нагревают с постоянной
скоростью, например 5 градусов в минуту.
Температуру в образцах измеряют с по-
Рис. 2.26. Кривая дифференциально-термического
анализа.
76 2. Адсорбционное равновесие и пористая структура адсорбентов
мощью термопар. Изменение температуры в стандартном веществе выра-
жается плавной кривой. В испытуемом образце изменение температуры на
отдельных участках происходит с некоторым замедлением или опережением
по отношению к стандарту. В первом случае в веществе протекает эндо-
термический, во втором — экзотермический процесс.
В современных установках дифференциально-термического анализа пре-
дусмотрены три термопары: две помещены в стандартное вещество и одна —
в испытуемое. Схема включения термопар приведена на рис. 2,25. Одной
термопарой измеряется температура в печи. Две другие термопары включе-
ны навстречу друг к другу, и такое их расположение позволяет с помощью
чувствительного гальванометра измерить разность температур между печью
и испытуемым веществом.
В результате опыта получают термограмму, пример которой показан на
рис. 2,26. В случае, если термографическому исследованию подвергают ад-
сорбенты, эндотермический эффект соответствует удалению адсорбата из
твердой фазы, а экзотермический — изменениям в структуре адсорбента. По
величине площади эндотермического пика судят о количестве выделенного
адсорбата и температурном интервале его выделения, по экзотермическому
эффекту — о границе допустимого одноразового иагрева адсорбента, т. е.
о его термостабильности. На термограммах отмечают температуры начала,
конца и пика превращения.
Термогравиметрия базируется на непрерывном изменении массы образца
при нагреве с применением различных вариантов микровесов с автоматиче-
ской регистрацией массы образца.
Учеными ряда стран, в частности Венгерской Народной Республики,
созданы приборы, которые известны под названием дериватографов. В дери-
ватографе одновременно фиксируются четыре кривые: кривая изменения тем-
пературы (Т), кривая дифференциально-термического анализа (ДТА), кривая
изменения массы образца (ТГ) и кривая дифференциально-термогравиметри-
ческого анализа, характеризующая скорость изменения массы образца (ДТГ).
Дериватография, как разновидность термогравиметрии, стала непремен-
ным элементом исследования в первую очередь кристаллических адсорбен-
тов, например цеолитов [62]. Кроме указанных выше характеристик показа-
тели, полученные на дериватографе, позволяют рассчитать теплоты адсорб-
ции, энтальпию фазового перехода и некоторые другие величины [63]. ме-
тод был успешно применен для изучения влагоемкости и возгораемости
активных углей [64].
В тех случаях, когда термогравиметрическим испытаниям подвергаются
углеродные адсорбенты и другие углеродсодержащие вещества, опыт про-
водят в среде инертного газа (азота или аргона), что исключает появление
экзотермического эффекта при окислении углерода [65].
Активные угли
Активные угли — пористые промышленные адсорбенты, состоя-
щие в основном из углерода [66]. Их получают из различных
видов органического сырья: твердого топлива различной степени
метаморфизма (торфа, бурого и каменного угля, антрацита),
дерева и продуктов его переработки (древесного угля, опилок,
отходов бумажного производства), отходов кожевенной промыш-
ленности, материалов животного происхождения, например ко-
стей. Угли, отличающиеся высокой механической прочностью,
производят из скорлупы кокосовых и других орехов, а также из
косточек плодов.
В наиболее развитых в промышленном отношении странах активные
угли выпускают в большом количестве и в широком ассортименте. Так, в
США производят более 90 марок углей, причем по объему потребления от-
расли образуют следующий ряд (по убыванию): очистка питьевой и сточных
вод, рафинирование сахара, очистка газов и рекуперация паров, производ-
ство каучука, получение медикаментов, очистка смазочных масел, производ-
ство пластмасс, электрогальваностегия, очистка спирто-водных растворов и
вин. Ежегодное производство активных углей в США превышает 70 тыс. т.
Основным направлением применения порошкообразного угля в Японии яв-
ляется рафинирование сахара, а гранулированного угля — производство ка-
тализаторов, Потребление порошкообразного угля в Японии еще в 1965 г.
составляло 15 тыс. т, а гранулированного угля — 2 тыс. т [67].
Развивающиеся страны в качестве основы для получения активных углей
используют преимущественно местное сырье. Так, в Индии угли получают
из стеблей, скорлупы, коры и других частей тропических растений. Особен-
но высокой адсорбционной способностью обладает уголь, полученный кар-
бонизацией кожуры высушенных плодов люфы. Среди социалистических
стран, кроме Советского Союза, активные угли в значительном масштабе
производят в Польше и Югославии. В Польше источником сырья для полу-
чения активных углей под названием «карбоноль» служат березовые или
буковые древесные угли. Югославская химическая промышленность при про-
изводстве углей под названием «карбозак» также использует различные сор-
та древесины.
Активные угли как промышленные сорбенты имеют ряд осо-
бенностей, определяемых характером их поверхности и пори-
стой структуры [68]. Поверхность кристаллитов углерода элек-
78
3. Основные виды пористых адсорбентов
тронейтральна, и адсорбция на углях в основном определяется
дисперсионными силами взаимодействия.
Как правило, структура угля представлена гаммой пор всех
размеров, причем адсорбционная емкость и скорость адсорбции
компонентов промышленных газов определяются содержанием
микропор в единице массы или объема гранул*. В процессе ад-
сорбции микропоры объемно заполняются адсорбатом. Это очень
отчетливо показано Щербаковым при исследовании адсорбции
углеводородов на углях, полученных из одного исходного сырья.
Образцы углей имели близкий размер зерен (1,5—2,7 мм),
одинаковое развитие мезопористости (0,11—0,13 см3/г), но раз-
личались объемом микропор Уми (0,28—0,43 см3/г) и соответст-
венно константой предельного адсорбционного объема Wa
(0,29—0,44 см3/г).
Характеристика образцов приведена ниже:
Показатели Образцы
1 2 3 4
Плотность, г/см3 0,485 0,470 0,447 0,414
Зернение (в мм), %
2,7—2,0 33,2 34,0 31,6 56,8
2,0—1,5 63,7 62,8 63,8 42,0
1'5—1,0 3,1 3,2 4,6 1,2
Содержание, %
ЗОЛЫ 7,4 7,1 7,9 5,0
калия 0,20 0,26 0,27 0,10
серы 0,46 0,47 0,67 0,63
Объем пор, см3/г
Уми 0,28 0,32 0,33 0,43
Уме 0,11 0,13 0,12 0,11
Ума 0,28 0,31 0,35 0,35
Уа 0,67 0,76 0,80 0,80
Структурные константы Wo,
см3/г 0,29 0,33 0,35 0,44
В-10е (по бензолу) 0,44 0,54 0,54 0,67
Адсорбционная способность,
г/100 г
°с3ня 9,0 12,8 13,0 14,2
ас7н16 16,2 20,6 20,8 26,1
* В активных углях представлены две разновидности микропор: щелевидные
микропоры в кристаллитах углерода и межкристаллитные микропоры, кото-
рые в первом приближении имеют цилиндрическую форму. Характерный раз-
мер микропор угля — полуширина щели (I — в нм) — связана с характери-
стической энергией (Ео), входящей в уравнение (2.86) Дубинина — Астахова,
следующим полуэмпирическим уравнением:
I = К,’Е0
где Д=13,028—1,53-10~5 Ео^.
\ Активные угли 79
Согласно современным воззрениям (см. Дубинин М. М., Федосеев Д. В. —
1/зв. АН СССР, сер. хим., 1982, № 2, с. 246), совокупности микропор этих
Риух типов образует зоны микропористости, протяженность которых состав-
ляет 10—60 нм. Суммарный объем щелей между микропористыми зонами
является объемом мезопор для единицы массы адсорбента (Прим. ред.).
Как видно из приведенных данных, равновесная адсорбционная способ-
ность по пропану при его концентрации в газовой фазе 6% и температуре
25 °C закономерно возрастает с увеличением микропористости. Соответствую-
щий показатель по н-гептану меняется аналогичным образом. Одновременно
увеличение микропористости угля приводит к повышению скорости поглоще-
ния углеводорода, которая обратно пропорциональна так называемой высо-
те работающего слоя. На рис. 3,1 это отчетливо показано применительно к
системе н-гептан-—активный уголь. При концентрации н-гептана 1,6% (об.)
и скорости потока в адсорбере 20 м/мин высота работающего слоя с разви-
тием микропористости снижается с 9 см (образец 1) до 4,5 см (образец 4).
Улучшение равновесной и кинетической характеристик у наи-
более микропористых углей приводит к повышению эффектив-
ности работы углеродных адсорбентов в промышленных услови-
ях. При выборе типа адсорбента для промышленных установок
следует учитывать два отличительных свойства активных углей:
гидрофобность и горючесть.
Адсорбция воды на углях протекает по необычному меха-
низму. Изотермы адсорбции воды на активных углях имеют
S-образную форму (рис. 3,2). Дисперсионные силы взаимодей-
ствия молекул воды с углеродной поверхностью очень малы. На-
чальные участки изотерм воды определяются ее адсорбцией на
хемосорбированных поверхностью углей прочных кислородсо-
держащих радикалах. Эти радикалы получили название «по-
верхностных оксидов».
Поверхностные оксиды и адсорбированные на них молекулы
воды являются адсорбционными центрами, к которым за счет
водородных связей происходит присоединение других молекул
воды [69]. Число адсорбционных центров по мере повышения
давления возрастает, образуются и непрерывно увеличиваются
ассоциаты молекул воды, в результате чего адсорбционная спо-
собность резко увеличивается. В ко-
нечном итоге весь адсорбционный
объем микропор заполняется водой.
Объем поглощенной углем воды при
высоком относительном давлении
(p/ps = 0,9) близок к предельному
адсорбционному объему микропор
Рис, 3,1. Высота работающего слоя Lo при извле-
чении zt-гепгана из потока газа в слое активные
углей с возрастающим (I-М) объемом микроиор
(/«=25 °C. м/мин).
80 3. Основные виды пористых адсорбентов
ний ДТА и ТГ позволило
Рис. 3,2. Изотерма адсорбции паров воды на актив-
ном угле.
(в рассмотренном случае 0,57 сма
на 1 г). Следовательно, адсорбцион-
ная способность угля по парам во-
ды тесно связана с его микропори-
стостью. Этот вывод подтвержден
данными термогравиметрического
анализа образцов активных углей,
выдержанных во влажной воздуш-
ной'атмосфере [64]. Сравнение ли-
установить, что с увеличением микро-
пористости емкость активных углей по влаге значительно повы-
шается.
Насыщение угля влагой — процесс чрезвычайно медленный:
равновесие устанавливается в течение нескольких месяцев.
Вследствие этого во многих реальных технологических процес-
сах влажность среды практически не оказывает влияния на эф-
фективность извлечения примесей из газовой или жидкой сред.
Активный уголь — единственный гидрофобный тип промышлен-
ных адсорбентов, и это качество предопределило его широкое
использование для рекуперации паров, очистки влажных газов
и сточных вод.
Однако при низких концентрациях адсорбтива, в частности
при извлечении из газового потока микропримесей, когда про-
должительность стадии очистки велика, влажность среды в за-
метной степени снижает адсорбционную емкость угля по извле-
каемому компоненту. К такому случаю относится поглощение
сероуглерода из вентиляционных выбросов вискозного производ-
ства.
% (масс
t°c
I На рис. 3,3 построена диаграмма, на ко-
торой температурные зависимости адсорбци-
онной способности определены для каждой
абсолютной влажности исходного воздуха
производственных помещений [70]. Из рисун-
ка видно, что только изобара сероуглерода,
извлекаемого из абсолютно сухого воздуха,
имеет ниспадающую форму. Все остальные
кривые проходят через максимум. Этот мак-
симум обусловлен тем, что с одной стороны,
при повышении температуры, как и следует
по законам физической адсорбции, адсорбци-
онная способность по извлекаемому компо-
Рнс. 3,3. Зависимость адсорбционной способности ак-
тивного угля APT по сульфиду углерода (с0=1 г/м3),
извлекаемому из воздуха с различным влагосодержа-
ннем №в, от температуры процесса.
Активные угли
81-
ненту снижается, но с другой — при этом понижается относительная влаж-
ность и процесс переходит в область, где образование ассоциатов молекул
воды не происходит, т. е. в область начального участка изотермы. В этой
области влияние влаги на сорбцию примесей незначительно. Максимум посте-
пенно сдвигается с 35 (при абсолютной влажности воздуха 4 г/м3) до 50 °C
(при 18 г/м3).
Следовательно, при удалении микропримесей температурный
режим крайне продолжительной стадии очистки должен выби-
раться с учетом влажности исходного воздуха производственных
помещений. Рекомендуемые рабочие температуры в адсорбере
(или в адсорбционной секции аппарата, если процесс проводит-
ся в подвижном слое) в рассматриваемом случае составляют в
зависимости от влажности газа: 20°C (0 г/м3); 30—35°C (4—
12 г/м3); 35—50°C (12—18 г/м3).
Эти же выводы следуют из исследования поглощения паров
этанола, бензола, «-гептана и бутилацетата из воздуха, содер-
жащего пары воды [71]. Опыты показали, что влияние влаж-
ности паровоздушной среды на адсорбцию паров органических
растворителей, характеризующееся падением адсорбционной
емкости углей, проявляется, если концентрация адсорбтива ле-
жит ниже 30 г/м3.
Искусственное понижение относительной влажности воздуха
(т. е. уменьшение совместной адсорбции водяного пара), когда
это необходимо, является действенным средством повышения
адсорбционной способности активных углей при их применении
для извлечения компонентов из влажных газов.
Адсорбцию влаги на активных углях можно снизить путем модифици-
рования поверхности, например триметилхлорсиланом и диметилсульфокси-
дом. В работе [72] модификацию этими веществами проводили при 200 °C
в токе аргона в течение 1 ч, после чего образцы выдерживали 30 мин и
вакуумировали в течение 2 ч также при 200 °C. Изучение изотерм адсорб-
ции воды при 25 °C показало, что в результате такой обработки еще более
ослабляются «гидрофильные» свойства поверхности угля, снижается адсорб-
ционная способность по отношению к водяному пару, островосходящая ветвь
изотерм адсорбции перемещается в область более высоких относительных
давлений.
Отрицательной особенностью активного угля как промышлен-
ного адсорбента является его горючесть. На воздухе окисление
углей начинается при температуре выше 250 °C. Однако извест-
ны случаи пожаров на углеадсорбционных установках при более
низких температурах. Очевидно, это связано с образованием
пирофорных соединений железа типа FeS и FeaSs в результате
сероводородной коррозии аппаратуры. Загорание пирофорных
соединений железа происходит при относительно низких темпе-
ратурах, и в слое они являются очагами воспламенения всей
массы угля. Чтобы уменьшить пожароопасность к активному
углю при его получении иногда добавляют до 5% силикагеля.
Такой адсорбент называют силикарбоном.
6—1346
32 3. Основные виды пористых адсорбентов
Практически все промышленные активные угли в том или
ином количестве содержат зольные примеси. Зола и ее ингреди-
енты (минеральные примеси) являются катализаторами многих
нежелательных реакций, которые могут протекать в адсорбере.
Ниже будет показано, что на зольных углях при адсорбции
сульфида водорода в присутствии воздуха стимулируется обра-
зование серной кислоты. При повышенных температурах, ха-
рактерных для стадии десорбции (например, 250 °C), на золь-
ных углях интенсивно протекает разложение нестойких адсор-
батов. Так, значительная часть этилового спирта при 250 °C
превращается в ацетальдегид и диоксид углерода. Содержание
ацетальдегида в этаноловом конденсате после стадии десорб-
ции из малозольного угля составляет 0,025%, а из угля, содер-
жащего 30% золы, 6,2%.
Силикагели
Одним из наиболее распространенных в промышленной прак-
тике минеральных адсорбентов является силикагель, который
обладает хорошо развитой пористостью [73].
По внешнему виду силикагель представляет собой твердые
зерна: прозрачные или матовые, бесцветные или светло-корич-
невые. Выпускается силикагель в виде шариков, таблеток или
кусочков неправильной формы с зернами размером в пределах
0,1—7,0 мм. В зависимости от аппаратурного оформления реко-
мендуют следующий гранулометрический состав силикагеля:
0,1—0,25 мм —для процессов с кипящим слоем адсорбента;
•0,5—2,0 мм—для жидкофазных процессов и процессов с дви-
жущимся слоем адсорбента; 2,0—7,0 мм — для процессов в га-
зовой фазе со стационарным слоем адсорбента.
Силикагели получают на основе диоксида кремния, который,
как известно, существует в кристаллической (кварц, тридимит,
кристобалит) и аморфной формах. Силикагель по своей хими-
ческой природе является гидратированным аморфным кремне-
земом (SiO2-nH2O) — реакционноспособным соединением пере-
менного состава; превращения его протекают по механизму по-
ликонденсации [73]:
nSi(OH)4 —* Si„O2„_m + (2л — m) 11,0
Процесс поликонденсации приводит к образованию частиц
коллоидных размеров, причем эти частицы имеют форму, близ-
кую к сферической, и размер в пределах 2—20 нм (при специ-
альных методах обработки могут быть получены частицы и зна-
чительно больших размеров).
При высушивании гидрогеля кремневой кислоты структурная
сетка из связанных между собой сферических частиц сохраня-
ется. В результате увеличения числа частиц и возникновения
Силикагели
83
Рис. 3,4. Модели глобулярных систем:
а — мелкопористой (малые частицы упакованы с числом касаний 6); б — крупнопори-
стой (большие частицы упакованы с числом касаний 3).
прочных связей между ними образуется жесткий кремнекисло-
родный каркас. Поры этого каркаса рассматриваются как за-
зоры между частицами. Основные характеристики пористой
структуры определяются размером частиц и плотностью их упа-
ковки.
На рис. 3,4 представлены модели глобулярных систем, составленных
мелкими и крупными частицами с разной плотностью упаковки (числом ка-
саний) [74]. Мелкие частицы при плотной упаковке соответствуют структуре
мелкопористых силикагелей, крупные частицы прн рыхлой упаковке — струк-
туре крупнопористых силикагелей.
На химические и адсорбционные свойства силикагеля в зна-
чительной мере оказывает влияние наличие группы sSi—ОН.
ОН-группы занимают в основном вершины тетраэдров, выходя-
щие на поверхность скелета силикагеля [75].
Изменение адсорбционных свойств силикагелей может быть достигнуто
в результате химического модифицирования его поверхности — введения в
состав силикагеля амино-, сульфо- и нитрнльных групп, атомов фтора, ал-
кильных и алкенильных радикалов. Модифицирование позволяет получить
силикагели с качественно новыми свойствами. Например, хемосорбция насы-
щенных при комнатной температуре паров олефинсиланов на поверхности
аэросила привела к синтезу наполнителя полимеров с повышенными термо-
химическими и механическими свойствами; эти качества система приобрела
в результате химического взаимодействия между олефинсодержащими груп-
пами модифицированного кремнезема и макромолекулами полимера [76].
Заменой гидроксильных радикалов на атомы водорода удалось получить
водородный силикагель — гидридполисилоксан [77]. Адсорбция на нем не
сопровождается образованием водородной связи, вследствие чего он гидро-
фобен. В то же время адсорбция органических паров на гидридполисилокса-
пе происходит в тех же количествах, что и на гидроксилированном силикаге-
ле той же пористой структуры. Сочетание таких свойств, как гидрофобность,
негорючесть и достаточно высокая адсорбционная емкость по органическим
парам, позволяет рассматривать водородный силикагель как перспективный
адсорбент для целей рекуперации и обезвреживания газовых выбросов.
Силикагель — один из самых первых минеральных синтетиче-
ских адсорбентов, нашедших широкое применение в промышлен-
6*
84
3. Основные виды пористых адсорбентов
ной практике. Несмотря на то, что в последние годы бурно
развиваются адсорбционные процессы с использованием уни-
кальных кристаллических сорбентов — цеолитов, силикагель не
потерял своего промышленного значения. Более того, ожидается
существенный рост спроса на него.
Основные преимущества силикагелей: низкая температура,
требуемая для регенерации (ПО—200 °C) и, как следствие, бо-
лее низкие энергозатраты, чем при регенерации других промыш-
ленных минеральных сорбентов (оксид алюминия, цеолиты);
возможность синтеза силикагелей в широком интервале задан-
ных структурных характеристик при использовании достаточно
простых технологических приемов; низкая себестоимость при
крупнотоннажном промышленном производстве; высокая меха-
ническая прочность по отношению к истиранию и раздавли-
ванию.
Активный оксид алюминия
Другим типом неорганических адсорбентов, широко применяе-
мых в технике для осушки различных сред и для других целей,
являются активный оксид алюминия и алюмогели [78, 79].
Рост потребности в активном оксиде алюминия обусловлен
развитием таких процессов нефтепереработки, как риформинг,
гидроочистка, гидрокрекинг (в которых используются катали-
заторы, содержащие 80—99% оксида алюминия), а также ши-
роким применением его в процессах адсорбции. Достоинства ок-
сида алюминия (термическая стабильность, относительная лег-
кость получения, а также доступность сырья и др.) обеспечива-
ют возможность широкого применения его наряду с такими ад-
сорбентами, как силикагели и цеолиты.
Оксид алюминия получают прокаливанием гидрооксидов
алюминия: тригидратов (гиббсита, байерита, нордстрандита) и
моногидратов (диаспора, окристаллизованного бемита, гелеоб-
разного бемита, так называемого псевдобемита). Термическая
обработка гидрооксидов алюминия приводит к образованию
различных форм оксида алюминия. Структура оксида алюминия
зависит от типа исходного гидрооксида, остаточного содержания
воды, присутствия оксидов щелочных и щелочноземельных ме-
таллов, а также от условий термической обработки. Различают
следующие типы оксидов алюминия:
низкотемпературные оксиды (А^Оз-пНгО), в которых 0<
<п<0,6; их получают при температурах не выше 600 °C в виде
у-, р-, %- и ц-модификаций;
высокотемпературные оксиды (почти безводные), получае-
мые при 900—1000 °C в виде б-, %- и 0-модификаций;
при температурах 1000 °C и выше получают инертный а-ок-
сид алюминия (корунд).
Цеолиты
85
Промышленные сорта активного оксида алюминия обычно
содержат у-А12О3, реже %-А12О3. Эти модификации являются
разновидностями дефектной шпинельной структуры, стабилизи-
рованной небольшим количеством воды. Первичные кристалли-
ческие частицы размером 3—8 нм упакованы так, что поры, об-
разуемые ими, имеют либо щелевидную, либо бутылкообразную
форму. у-АЬОз получают прокаливанием гиббсита и бемита
(псевдобемита), а трАЬОз — прокаливанием байерита.
Многообразие областей применения активного оксида алюминия опре-
деляют необходимость производства широкой гаммы его сортов. Основные
области применения активного оксида алюминия (адсорбента) показаны
ниже:
1. Адсорбционная осушка газов. Высокая активность оксида алюминия
при взаимодействии с полярными адсорбтивамн (прежде всего, парами иоды)
обеспечивает глубокую осушку газов до точки росы минус 60 °C и ниже.
Он интенсифицирует полимеризацию непредельных углеводородов, образую-
щихся при крекинге в стадии высокотемпературной десорбции. Однако воз-
можность многократной температурной регенерации путем выжига коксовых
отложений обеспечивает долголетнюю работу адсорбента как осушителя оле-
финсодержащих потоков. Важной положительной способностью оксида алю-
миния является его водостойкость. Именно этот показатель часто опреде-
ляет выбор оксида алюминия в качестве адсорбента для осушки и перера-
ботки сред, в которых присутствует капельная влага.
2. Адсорбционная очистка масел (прежде всего трансформаторных).
Амфотерный характер оксида алюминия делает его эффективным адсорбен-
том кислот — продуктов окисления масел, накопление которых снижает ди-
электрические свойства масел.
3. Применение и статических адсорбционных системах. Активный оксид
алюминия находит применение как эффективный осушитель при консервации
приборов и оборудования, а также для таких систем, как дыхательные кла-
паны цистерн, трансформаторы и т. д.
4. Адсорбционнаи очистка газовых и жидкостных потоков от соедине-
ний, содержащих фтор-ионы. Способность оксида алюминия хемосорбировать
фтор-ионы используется для очистки вод с повышенным содержанием фтора,
улавливания паров HF из газов суперфосфатных и электролизных произ-
водств.
Цеолиты
Цеолиты — алюмосиликаты, содержащие в своем составе оксиды
щелочных и щелочноземельных металлов, — отличаются строго
регулярной структурой пор, которые в обычных температурных
условиях заполнены молекулами воды. Эта вода, названная цео-
литной, при нагреве выделяется. Цеолиты «кипят», отсюда и
произошло сочетание двух греческих корней «цео» и «лит», т. е.
«кипящие камни». Термин «цеолиты» введен в минералогию
свыше 200 лет назад шведским ученым Кронштедом. Свойства
природных цеолитов изучены и систематизированы в трудах
академиков Ферсмана [80] и Вернадского [81].
Общая химическая формула цеолитов Me2/nO-Al2O3-xSiO2-
•z/H2O (где Me — катион щелочного металла, а п — его валент-
ность). В природе в качестве катионов обычно в состав цеоли-
86
3. Основные виды пористых адсорбентов
©Рис. 3,5. Модель образования структуры содалита.
тов входят натрий, калий, кальций,
реже барий, стронций и магний.
Кристаллическая структура цеоли-
тов образована тетраэдрами SiO4 и
I А1О4. Катионы компенсируют избы-
* х^^х. точный отрицательный заряд анион-
ной части алюмосиликатного скеле-
и та неолита.
Если из цеолита удалить воду,
Т Ш [ар поры могут быть заполнены снова
JL Д- / водой или другим веществом, что и
предопределяет их использование в
i процессах осушки, очистки и разде-
ления веществ. Обратимость процес-
сов гидратации и дегидратации цеолитов была установлена в
1840 г. Дамуром. Поглощение вещества происходит в основном
в адсорбционных полостях цеолитов. Однако не все вещества
могут проникать в эти адсорбционные полости и поглощаться
в них. Это объясняется тем, что адсорбционные полости соеди-
няются друг с другом входами — окнами и строго определенно-
го размера. Проникнуть через окно могут только те молекулы,
критический диаметр которых меньше диаметра входного окна*.
Самую простую структуру имеет каркасный силикат — сода-
лит. В земной коре обнаружено пять модификаций этого веще-
ства: хлорсодалит (6NaSiA104-NaCl), гидросодалит (6NaSiA104-
•2NaOH), лазурит (4NaSiA104-2Na2S), гаюин (6NaSiA104-
2CaSO4) и нозеан (6NaSiA104-Na2S04) [82].
Содалит является строительным блоком для разнообразных форм при-
родных и синтетических цеолитов. Модель структуры содалита представлена
на рис. 3,5. Плотная упаковка кубооктаэдров (содалитовых единиц) в струк-
туре этого минерала приводит к образованию полостей, которые по форме
являются также кубооктаэдрами, следовательно, в каждой полости может
объемно поместиться дополнительный «строительный камень». Элементарную
ячейку содалита можно представить, если в куб вписать кубооктаэдр. Она
состоит, таким образом, из кубооктаэдра и восьми восьмушек соседних ку-
бооктаэдров, т. е. из двух кубооктаэдров. Входными окнами в полости со-
далита являются шестичленные кислородные кольца с диаметром свободного
прохода 0,22 нм. Из-за таких узких входов в полости содалита компоненты
промышленных газов не проникают, и применения в технике этот минерал
* Для простых молекул наглядное представление о критическом диаметре
дает диаметр наименьшей окружности, описанной вокруг молекулы. В более
сложных случаях критический диаметр определяют через диаметр кинетиче-
ский, который соответствует минимально возможному расстоянию между
покоящимися молекулами вещества. Кинетический диаметр рассчитывают по
формулам, вытекающим из квантовой химии. (Прим. ред.).
Цеолиты
87
не получил. Лишь вода адсорбируется некоторыми формами содалита, но
йог процесс протекает очень медленно: адсорбционное равновесие устанав-
ливается в течение нескольких месяцев.
Цеолиты в природе
В природе цеолиты распространены достаточно широко. Они
образовались в результате изменения вулканических туфов в
морских и континентальных бассейнах и таким образом пред-
ставляют туфогенно-осадочный тип месторождений. Катионы,
входящие в состав цеолитов, под воздействием среды могут за-
мещаться на ионы других металлов. Варьирование условий гид-
ротермального синтеза и протекание реакций катионного обме-
на в природных условиях предопределило большое разнообра-
зие типов природных цеолитов.
В зависимости от условий образования, цеолиты в природе
можно разделить на две группы: эндогенные и экзогенные. Эн-
догенные цеолиты обычно рассеяны в породах и не образуют
крупных месторождений, пригодных для использования: выде-
ление минерала из сопутствующей породы связано с очень боль-
шими затратами труда. Кроме того, обычно в таком месторож-
дении присутствует не один, а несколько типов цеолитов.
Эти обстоятельства долгое время препятствовали широкому
использованию природных цеолитов в промышленности. Однако
в последние годы в Советском Союзе и за рубежом были откры-
ты крупные экзогенные месторождения высококонцентрирован-
ных цеолитов, в первую очередь высококремнистых форм типа
морденита и клиноптилолита, в связи с чем была произведена
переоценка промышленной ценности природных цеолитов. При-
родные цеолиты быстро были внедрены в различные техноло-
гические процессы, в которых не столь существенную роль иг-
рает кристаллографическая чистота адсорбента.
Первые удачные поиски залежей природных цеолитов в нашей стране
относятся к 1945 г.: на Урале были найдены пласты осадочных пород с очень
высокой концентрацией морденита. Слои анальцима обнаружены в морских
отложениях пород вблизи Кутаиси. Крупный вклад в исследование природ-
ных цеолитов внесли грузинские ученые Твалчрелидзе и Гвахария.
Благодаря интенсивным минералогическим поискам постоянно открыва-
ются все новые типы цеолитов: например, в 1952 г. был обнаружен югава-
ралит, в 1957 г. — бикитаит и, наконец, в 1960 г. — цеолит, получивший на-
звание в честь известного ученого Полинга — полингит [83].
Наибольшее промышленное значение имеет клиноптилолит.
Крупные пласты вулканических туфов, в которых содержание высоко-
кремпислого цеолита—клиноптилолита доходит до 80—90%, открыты в Гру-
зии. Мощность туфового горизонта достигает 100—280 м. Преимущественны-
ми катионами, входящими в состав клиноптилолита, являются кальций,
натрий и калий; соотношение оксида кремния к оксиду алюминия в адсор-
бенте около 10. Термогравиметрические исследования показывают, что мак-
симум удаления воды при нагревании клиноптилолита достигается при 170 °C,
88
3. Основные виды пористых адсорбентов
предельная влагоемкость составляет 13,2% (масс.). Нагрев клиноптилолита
до температуры выше 450 °C приводит к последовательному уменьшению
способности регидратации, что связано с частичным разрушением решетки
цеолита. После нагрева до 750 °C клиноптилолит полностью теряет свои ад-
сорбционные свойства по воде.
Мощные залежи клиноптилолита найдены также в Армении, Азербайд-
жане, в Закарпатье, Средней Азии, на Камчатке, на Сахалине [84]. Промыш-
ленные месторождения высококремнистых осадочных пород открыты в США,
Японии, Восточной Африке, Италии, Франции, Югославии, на Кубе, в Чехо-
словацкой Социалистической Республике, Венгерской Народной Республике
и Народной Республике Болгарии.
Клиноптилолит был впервые описан Пирсоном и ошибочно
отнесен им к классу морденита. \
Впоследствии Шаллер, изучив оптические свойства клино-
птилолита, выделил его в самостоятельный минеральный вид
морденит-клиноптилолитовой серии. Наиболее вероятная кри-
сталлохимическая формула клиноптилолита (NaKhCaAleSiaoC^-
•24Н2О.
Рентгеновские исследования показали близость структуры
клиноптилолита и гейландита, однако эти два цеолита отлича-
ются отношением Si/Al: для гейландита оно равно 2,75—3,25,
для клиноптилолита 4,25—5,25. Кроме того эти два типа при-
родных цеолитов различаются по содержанию катионов. В кли-
ноптилолите оно соответствует (Na + K) > (Ca+Mg), в гейлан-
дите наблюдается обратная зависимость. Кислого- и термоста-
бильность клиноптилолита и гейландита также разные.
Клиноптилолит, как все цеолиты, обладает способностью эффективно
осушать газы и жидкости. Как будет показано ниже, в динамических усло-
виях слой клиноптилолитовой породы обеспечивает осушку газа до точки
росы ниже минус 40 °C. Испытания клиноптилолита в качестве средства
осушки природного газа в промышленных условиях на одном из газоперера-
батывающих заводов Советского Союза показали стабильность его работы
в процессе многоцикловой эксплуатации. Введены технические условия «Ад-
сорбенты цеолитовые природные для осушки газов и жидкостей»
(ТУ 6-12-111—78).
Перспективным является применение кислотостойких природных цеоли-
тов для целей защиты окружающей среды от ряда промышленных выбро-
сов, в частности от диоксида серы [85]. Процесс очистки отходящих про-
мышленных газов от диоксида серы с помощью клиноптилолита* и природ-
ного морденита может осуществляться в широком интервале температур
(20—150 °C).
В Японии [86] добываемый клиноптилолит используют в основном в
качестве структурообразователя почв, внося его вместе с удобрениями. Кро-
ме того, его применяют для осушки и очистки газа, разделения воздуха, в
качестве наполнителя и отбеливателя бумаги, для ликвидации запаха на
скотных дворах.
* Клиноптилолиты действительна обладают высокой начальной емкостью по
диоксиду серы. Однако в условиях циклической работы, как показано Цвет-
ковым и Шумяцким, они постепенно дезактивируются из-за практически не-
обратимой адсорбции триоксида серы — вещества, постоянно сопутствующего
диоксиду серы а образующегося из него. (Прим. ред.).
Цеолиты
89
Рис. 3,6. Цепи, образованные тетраэдрами морде-
нита.
В США разработаны методы извлече-
ния цезия из радиоактивных отходов и
удаления аммиака из сточных вод с при-
менением клиноптилолита. Его используют
на животноводческих, в частности свино-
водческих фермах. Система адсорбциоииой
доочистки сточных вод с использованием
клиноптилолита, подключенная после системы биохимической очистки, позво-
ляет практически полностью удалить аммиак: степень очистки достигает 99%.
Наряду с адсорбционными свойствами, клиноптилолит обладает высокой
каталитической активностью в некоторых процессах нефтепереработки и ор-
ганического синтеза, например при гидрировании бензола в циклогексан,
окислении этилена, деалкилировании толуола, риформинге и гидрокрекинге.
Добавление клиноптилолита в почву способствует повышению урожайно-
сти растений [87]. Введение цеолитовой минеральной добавки в корм птицы
и домашних животных увеличивает их привес [88].
Экономический анализ показал, что разработка даже бедных клинопти-
лолитовых месторождений с последующим обогащением сырья обходится в
20 раз дешевле производства синтетических цеолитов, а добыча из богатых
пород — дешевле в 100 раз.
Содержание клиноптилолита в породах определяют калори-
метрически. Этот оригинальный метод, разработанный Сендом с
сотр. [89] основан на способности дегидратированного цеолита
выделять тепло при погружении в воду.
Измельченный до размера 1 мм цеолит в тонкостенном сосуде нагрева-
ют до 350 °C. Затем сосуд охлаждают до температуры окружающей среды,
к пробе цеолита приливают воду и смесь перемешивают. При этом темпе-
ратура в сосуде повышается и через несколько десятков секунд достигает
максимума. Резкий подъем температуры из всех минералов породы харак-
терен только для цеолитов. Поэтому перепад температуры пропорционален
содержанию клиноптилолита в породе.
Структура клиноптилолита может быть существенно преоб-
разована путем различных физических и химических воздейст-
вий. Так, в результате гидротер-
мальной обработки природного кли-
ноптилолита при 100 °C и атмосфер-
ном давлении образуются цеолиты
по структуре сходные с филлипси-
том и фожазитом [90].
Другим важным типом природ-
ных цеолитов является морденит.
Он отличается высоким отношением
SiO2: А120з и является самым кис-
лотостойким цеолитом, получившим
Рис. 3,7. Проекция структуры морденита в на-
правлении главных каналов.
90
3. Основные виды пористых адсорбентов
Рис. 3,8. Адсорбционные полости шабазита.
широкое распространение в промышленно-
сти (свое название он получил от селения
Морден в Новой Шотландии, где был об-
наружен).
Природный морденит — минерал, в зна-
чительных количествах содержащийся в ру-
дах различных месторождений. В Совет-
ском Союзе он найден на Дальнем Востоке,
в Сибири, на юге Грузии, в Крыму.
Химическая формула природного морде-
нита Na2O-Al2O3-10SiO2-6H2O. В зависимо-
сти от геологических условий залегания
руд, катионы натрия могут быть частично
замещены на катионы калия, кальция или
магния. Отношение 5Ю2:А120з в мордени-
тах колеблется в интервале 8,3—10,7.
Морденит имеет тетраэдрическую струк-
туру, в которой атом кремния или алюми-
ния окружен четырьмя атомами кислорода. Тетраэдры группи-
руются в цепи, как это показано на рис. 3.6. Внутри цепей рас-
положены адсорбционные полости. Проекция структуры морде-
нита в направлении главных каналов представлена на рис. 3,7.
Природный морденит не сорбирует молекулы, критический диа-
метр которых превышает 0,4 нм.
Природные цеолиты с учетом общности их структурного
строения объединены в группы [91]. Так, для цеолитов группы
натролита характерным является наличие разветвленных цепо-
чек тетраэдров.
Структура шабазита представлена на рис. 3,8. Она построена
по ромбоэдрическому закону из шестерных колец кремнеалюмо-
кислородных тетраэдров. Шестерные кольца сдвоены и образуют
гексагональные призмы. Их сочетанием образованы адсорбци-
• -Si.Al 0-0
онные полости. Каждая полость
имеет шесть восьмиугольных окон
(а), образованных восьмерными
кольцами, и два гексагональных ок-
на (б), которые построены из сдво-
енных шестерных колец. Гексаго-
нальные окна расположены на вер-
шине и у основания каждой поло-
сти.
Размер восьмиугольных окон, по
Рнс. 3,9. Кубооктаэдрическая структура элемен-
тарной ячейки фожазнта.
F
Цеолиты 91
Рис. 3,10. Адсорбционные полости
мелинита (а) и левинита (б).
данным Мак-Бена, состав-
ляет 0,5 нм, он опреде-
ляет возможность про-
никания поглощаемых мо-_
лекул в адсорбционную -
полость. Через такое окно
проникают и поглощают-
ся молекулы воды, мети-
лового и этилового спир-
тов, муравьиной кислоты.
В то же время более
крупные молекулы бензо-
ла, ацетона, эфира шаба-
зитом не адсорбируются.
Структура филлипсита образована сочетанием четырех- и
восьмичленных колец тетраэдров. Кубооктаэдрическую структу-
ру фожазита — прообраза синтетических цеолитов типа X и Y —
характеризует рис. 3,9. Разнообразие строения природных цео-
литов отражает рис. 3,10, где представлена структура адсорб-
ционных полостей гмелинита и левинита.
Вопросы строения, образования, химии природных цеолитов
освещены в монографии Сендерова и Хитарова [83], а также
Жданова и Егоровой [91]. В табл. 3-1 приведена классификация
природных цеолитов по структурному признаку с указанием
идеализированного состава и размера окон.
Синтетические цеолиты общего назначения
Первые попытки получить цеолиты синтетическим путем были
сделаны более 100 лет назад. В 1962 г. Сент-Клер Девиль в ре-
зультате нагревания в запаянной стеклянной трубке смеси си-
ликата и алюмината калия при 200 °C получил синтетический
калиевый филлипсит. В близких условиях, при нагреве силйката
калия и алюмината натрия до 170°С был получен синтетический
шабазит. Проведенные затем многочисленные опыты, преследо-
вавшие целью синтез аналогов природных цеолитов, проводи-
лись в условиях высоких температур (250—450 °C) и давлений
(300 МПа).
Большая заслуга в синтезе цеолитов принадлежит англий-
скому физикохимику проф. Барреру. Им в 1948 г. начаты систе-
матические поиски путей синтеза, которые увенчались синтезом
морденита, шабазита, анальцима, филлипсита, фожазита, строн-
циевых и бариевых цеолитов. Работами Баррера и его сотрудни-
ков были намечены пути синтеза в условиях пониженных тем-
ператур (100°С) при нормальном давлении [92].
92 3. Основные виды пористых адсорбентов
Таблица 3-1. Классификация природных цеолитов по структурному признаку
Структурные группы и цеолиты* Идеализированный химический состав Число тет- раэдров, образую- щих «окна» Размер ОКОН, НМ
Группа анальцима
анальцим Na(AlSi2O6) -Н2О е Неизвестен
Группа натоолита
натролит N as (Al 2S13O10) • 2Н2О 8 0,26—0,39
томсонит NaCas(AlgSigOso) *6НзО 8 0,26—0,39
эдингтонит Ba(Al2Si3Oio)-3H,0 8 0,35—0,39
Группа шабазита
шабазит Ca2(Al4Si8O24) -13H2O 8 0,37—0,50
гмелинит Na2(Al2Si4OI2)-6H2O 12 0,69
эрионит (Ca, Кг, Mg)4,5-(Al9Si27O72)-27H2O 8 0,36—0,48
левинит Ca (AlsSiiOis) 6H9O 8 0,35—0,51
Группа филлип- сита
филлипсит (К, ИаЖАкЗшОэгМОНгО 8 0,28—0,48
жиемондит Ca(Al2Si2O8)-4H2O 8 0,28-0,49
Группа гейланди- та
брюстерит (Sr, Ba, Ca)2[(A102)4-(SiO2))2J- •10H2O8 8 0,23—0,50
гейландит СаД (A102)e- (Si02)25] • 24H2O 8 0,24-0,61
стильбит Na2Ca4[(AlO2)10- (SiO2)26]-28H2O 8 0,27—0,57
Группа морденита
морденит Na(AlSi5O12) -3H2O 12 8 0,67—0,70 0,29—0,57
дакиардит (НэгСа) 2 • (A14Si2o04g) 12H2O 10 8 0,37—0,67 0,36—0,48
эпистильбит Ca3[(AlO2)e-(SiO2)18].16H2O 10 8 0,32—0,53 0,37-0,44
феррьерит Na1,5Mg2[(A102)5,5- (SiO2)20,5]- 10 0,43-0,55
18H2O 8 0,34-0,48
бикиатит Li2[(AlO2)2-(SiO2)4]-2H2O 8 0,32-0,49
Группа фожазита
фожазит (NasCajso* (Al, Si) 192O384 12 0,74
ПОЛИНГИТ (Кз, Nas, Ca, Ba) 76* [(A10s)i52* • (St02)52o-700H20 8 0,39
• Кроме указанных цеолитов в группу анальцима входят вейракит, лейцит; в группу
натролита — мезонт, сколецит, гоииарднт; в группу филлипсита — гармотом; в группу
гейландита — клиноптилолит.
Структура клиноптилолита, бесспорно, важнейшего из природных цеолитов, окончательно
не установлена, поэтому в таблице ие отражена.
Параллельно Брек и другие исследователи американской
фирмы Линде нашли условия получения основных цеолитов об-
щего назначения типа NaA и NaX, широко используемых в про-
мышленности [93]. По данным Брека, за последние 16 лет син-
Цеолиты
93
Рнс. ЗД1. Структура синтетических цеолитов типов А (а) и X (б)
тезировано более 65 разных цеолитов, многие из которых не
имеют аналогов в природе. Только в США зарегистрирована
более 2000 патентов в области синтеза и применения цеолитов.
В отечественной литературе свойства синтетических цеолитон
были впервые описаны в 1957 г. [94]. С 1964 г. предприятия оте-
чественной химической промышленности начали выпускать цео-
литы общего назначения: КА, NaA, СаА, СаХ, NaX, получившие
высокую оценку как в Советском Союзе, так и за рубежом* [95].
Многие синтетические цеолиты имеют строение и геометри-
ческую структуру, аналогичные этим показателям для природ-
ных цеолитов. Аналогами фожазита являются синтетические
цеолиты типов X и Y. На рис. 3,11 представлены структуры, со-
ставленные из кубооктаэдров в цеолитах типа А и X, а на
рис. 3,12 — строение их адсорбционных полостей.
Структура цеолитов типа А состоит из больших и малых
(содалитовых) адсорбционных полостей. Химическая формула
цеолита NaA: Na2O-Al2O3-2SiO2-4,5H2O. В состав элементарной
ячейки входит одна большая и одна малая полость. Большая
полость имеет практически сферическую форму диаметром
1,14 нм [96]. Она соединена с шестью соседними большими по-
лостями восьмичленными кислородными кольцами диаметром
0,42 нм и с восемью малыми полостями шестичленными кисло-
родными кольцами диаметром 0,22 нм. Объем большой полости
составляет Kg = 0,776 нм3. В ней при полном заполнении поме-
щается 24 молекулы воды. Малая полость — также сферической
формы, ее диаметр 0,66 нм, объем Ум = 0,15 нм3.
Поскольку на каждую большую адсорбционную полость при-
ходится одна малая адсорбционная полость, доля объема боль-
ших полостей составляет 0,84. Пористость кристалла составляет
* Расшифровка обозначений цеолитов приведена на с. 96.
94
3. Основные виды пористых адсорбентов
Рнс. 3,12. Адсорбционные полости синтетических цеолитов типов А (а) и X (б)
50,2%. Входы и малые адсорбционные полости цеолитов типа
А настолько малы, что в них практически не проникают моле-
кулы адсорбируемых веществ, кроме воды. Поэтому предельный
адсорбционный объем кристаллита типа А, вычисленный из изо-
терм адсорбции паров, только в случае воды (0,325 см3/г) при-
мерно соответствует теоретическому объему пор кристаллов
(0,334 см3/г) [97]. Адсорбционный объем, заполняемый азотом,
близок к суммарному объему больших полостей цеолитов
(0,280 см3/г).
Цеолиты типа А относятся к низкокремнистым формам.
В них отношение SiOa: А12О3 не выше 2. Кислотостойкость цео-
литов последовательно повышается с увеличением этого показа-
теля. Низкокремнистые цеолиты А разрушаются в кислой среде.
В состав цеолита входят катионы, являющиеся адсорбционны-
ми центрами, которые определяют в ряде случаев форму изо-
термы адсорбции, а также ее избирательность. В цеолите NaA,
в котором таким катионом является натрий, отношение Na2O :
: А12О3 теоретически равно 1, а практически колеблется от 0,85
до 0,93. В одной элементарной ячейке находится 12 одновалент-
ных катионов: восемь катионов натрия расположены в центре
шестичленных кислородных колец, а четыре на гранях вблизи
восьмичленных кислородных колец.
Цеолиты типа X в дегидратированном виде имеют состав
Na2O-Al2O3-«SiO2. Мольное отношение SiO2.‘Al2O3 может изме-
няться от 2,2 до 3,3. Каждая большая полость имеет четыре
входа, образованных двенадцатичленными кислородными коль-
цами диаметром 0,8—0,9 нм. Вследствие этого структура цеоли-
тов такого типа более открыта и доступна для поглощаемых мо-
лекул.
Объем большой адсорбционной полости цеолита NaX лишь
незначительно отличается от соответствующей полости в цеоли-
те и равен 0,833 нм3. Малые полости имеют тот же объем, т. е.
•0,150 нм3. Комплексы больших и малых полостей составляют
элементарные ячейки. В каждой из которых содержится по
Цеолиты
9&
8 больших и малых полостей. Элементарная ячейка содержит
192 атома алюминия и кремния, а также 384 атома кислорода;,
ее объем равен 7,776 нм3. Она вмещает 256 молекул воды. Ма-
лые полости цеолитов типа X доступны для молекул азота и
других газов. Поэтому предельный адсорбционный объем кри-
сталлов типа X, вычисленный из адсорбционных измерений, бли-
зок к рассчитанному на основе геометрических размеров эле-
ментарных ячеек, т. е. 0,356 см3/г (NaX) или 0,362 см3/г (СаХ)
[98]. Этим объясняется большая адсорбционная способность-
цеолитов типа X по сравнению с цеолитами типа А, достигаемая:
при относительно высоких степенях заполнения.
В результате исследования состояния катионов Na и Са в
гидратированном фожазите установлено, что из 43 катионов,,
приходящихся на элементарную ячейку этого цеолита, лишь-
17 находятся в локализованном положении. Остальные катионы,
способны мигрировать, и состояние воды в полостях близко к
состоянию раствора электролита.
Цеолиты типа Y [99—101] также относятся к типу фожазита,.
и их структура идентична структуре цеолитов типа X. Однако в-
элементарной ячейке цеолита типа Y содержится вдвое меньше
тетраэдров АЮ4 и катионов по сравнению с цеолитом типа X.
Мольное соотношение SiO2:Al2O3 в цеолитах типа Y колеблется
от 3,1 до 6,0; они отличаются повышенной кислотостойкостью,
термостойкостью и каталитической активностью в реакциях кре-
кинга, изомеризации, алкилирования и др. Именно высокая ре-
акционная способность предопределила высокий спрос нефте-
химии на этот вид цеолитов.
В лабораторных условиях Ждановым синтезирован цеолит Е,.
относящийся к группе шабазита. Его Na-форма имеет химиче-
ский состав Na2O-Al2O3-4SiO2-6H2O. Получаемая в результате-
ионного обмена Са-форма цеолита Е отличается повышенной
адсорбционной емкостью при температуре жидкого азота и низ-
ких давлениях по компонентам воздуха. Цеолит СаЕ является
эффективным адсорбентом для криовакуумной техники.
Цеолиты — типичные микропористые адсорбенты. Поэтому
определенные по формулам БЭТ или Ленгмюра значения удель-
ной поверхности цеолитов примерно в полтора раза ниже, чем
удельной поверхности, оцененной на основе рентгеноструктурных
данных [19]. Так, из рентгеноструктурных данных следует, что
удельная поверхность цеолита СаА равна 1170 и 1450 м2/г, а
соответствующие значения, определенные из изотермы адсорб-
ции азота, значительно ниже (770 и 900 м2/г).
Цеолиты являются молекулярными ситами [102—104].
В 1945 г. было показано, что они могут быть использованы для
разделения веществ на основе не только избирательности ад-
сорбции, но и разницы в размерах и форме поглощаемых моле-
кул. Для того чтобы проникнуть в адсорбционную полость, кри-
96
3. Основные виды пористых адсорбентов
тический диаметр молекулы адсорбата должен быть меньше
размера входного окна.
Промышленностью выпускается пять типов цеолитов общего
назначения. В основу классификации, принятой в Советском
Союзе, положено двухзначное обозначение; вначале указывается
катион, преимущественно входящий в решетку цеолита (К, Na,
Са), а затем тип кристаллической решетки цеолита (А или X).
Если калиевая, магниевая или иная форма цеолита получена из
натриевой и катионный обмен проведен не полностью, цеолит
обозначают KNaA, MgNaA и т. д.
В цеолитовый каркас успешно могут быть внедрены катионы редкозе-
мельных элементов. Так, путем проведения пятикратного обмена катионов
Na+ в цеолите NaX па катионы Y3+, La3+, Na3+, Sm3+, Gd3+ получены соот-
ветствующие модифицированные цеолиты с конечной степенью обмена 50—
62% [И05]. ИК-спектры модифицированных цеолитов, снятые в интервале
400—1000 см-1, свидетельствуют о том, что при катионном обмене происхо-
дит слабая аморфизация решетки цеолита, не обнаруживаемая при рентге-
нофазовом анализе. Термостабильность модифицированных цеолитов несколь-
ко выше, чем исходных образцов.
Классификация цеолитов в США и ряде других стран предус-
матривает указание определяющего размера цеолита (округлен-
ного значения диаметра входного окна в А). Однако совпадение
классификационного номера и определяющего размера соблю-
дается только для цеолитов типа А. Ниже представлены типы
деолитов общего назначения, обозначенные по классификациям
СССР и США, и определяющие размеры цеолита:
Классификация СССР КА
Классификация США ЗА
Диаметр входного окна, 3
А (10_| нм)
NaA СаА СаХ NaK
4А 5А 10Х 13Х
4 5 8 9
Размер входных окон, определяющий молекулярно-ситовые
свойства цеолитов, зависит от расположения кислородных колец
цеолита и от числа атомов кислорода в кольце. Рис. 3,13 дает
представление о строении кислородных колец и их размерах для
основных типов природных и синтетических цеолитов.
Размер катиона, входящего в состав цеолита, и его располо-
жение также оказывает влияние на размер окна. Катион, рас-
положенный вблизи окна, сужает окно, затрудняя проход моле-
кул. При катионном обмене, в котором два катиона натрия
замещаются одним катионом кальция, входное окно расширяет-
ся; вследствие этого цеолит NaA имеет размер входного окна
0,4 нм, а цеолит СаА 0,5 нм. Однако аналогичный обмен в цео-
лите типа X приводит к некоторому сужению окна.
Классификация цеолитов в соответствии с их молекулярно-
ситовым действием была выполнена Баррером [106]. По этой
классификации цеолиты перечислены в порядке уменьшения раз-
Цеолиты
97
Рис 3,13, Строение кислородных колец в цеолитах:
I — тип А; 2 — гип X и фожазит; 3 — шабазит; 4 — гмеленит; 5 — левеннт.
мера входного окна (см. табл. 3-2). Указаны группы веществ,
сорбируемых или несорбируемых данным цеолитом. Мы не-
сколько изменили в этой классификации лишь начало, указав
в качестве цеолита с наиболее мелкими порами цеолит КА,
имеющий большое технологическое значение.
Цеолит КА при обычной температуре в значительных количествах по-
глощает только воду. Это свойство предопределило широкое применение его
для осушки нестойких веществ, склонных к реакциям полимеризации.
Цеолит NaA адсорбирует большинство компонентов промышленных га-
зов, критический размер молекул которых не превышает 0,4 нм, сульфиды
водорода и углерода, диоксид углерода, аммиак, низшие диеновые и ацети-
леновые углеводороды, метан, этан, этилеи, пропилеи и другие органические
соединения, а также неон, аргон, криптон, ксенон, кислород, азот, оксид
углерода. Последняя группа веществ в значительных количествах поглощает-
ся только при низких температурах. Пропан и органические соединения с
числом атомов углерода в молекуле более 3 не адсорбируются и таким об-
разом не подавляют адсорбцию указанных выше адсорбирующихся веществ.
Цеолит СаА адсорбирует углеводороды и спирты только нормального
строения (независимо от длины цепи), в связи с чем широко используется
в процессах разделения многокомпонентных
органических веществ на молекулярно-ситовой
основе (разделение смеси w-гексана и бензола
на цеолите СаА иллюстрирует рис. 3,14).
Кроме того, цеолитом СаА поглощаются ме-
тил- и этилмеркаптаны, органические соедине-
ния с числом атомов углерода в молекуле 2
(этиловый спирт, этиламин), диборан и др.
Среди цеолитов общего назначения типа СаА
отличается повышенной стойкостью в слабо-
кислой среде, и поэтому его используют в про-
цессах сероочистки и декарбонизации газов.
Рис. 3,14. Схема разделения смеси н-гексаиа и бен-
зола на цеолите СаА.
7—1346
Таблица 3-2. Классификация цеолитов
ю 1,3,5-триэтилбен- зол 1,2,3,4,5,6,7,8,13, 14,15,16-дека- гидрохризен
> Я
со 2 0 0
- Си CU « 2 CQ - «=( £
XX О Ч я ‘г
афталин 2 о i 7 га ’V со 03 S « “ я ь S s я с ) я О л О м s н Н Rt X О Я X И-* Нм Q -L •&\О К Си я я 2 t-H rt > . ^7 я т s ®
Я х СО U U £ ф о: я 71 r^U
§
Я ►9 вч о
я -Й X X
х" f X £ о О а я- О о О 0) к оХХХч«о$- 0J
3 s О О О w а- ь •& Ю
си
о
е» J?
„• и Й » Л' X — О
tb Ч ь s и д -? Ч. Ч Я
о ооиоооои
3
« О я" Зн *7*
со о X о X § S и- Z _г z *** Q. я гд ю г л о 5 X Ч 7 § о х « к £ зе ф и .7 о г л и-< Ч о О . t-' О
i » ~ g X л —
оо X Т! . И О О гг, <5 . О. g ? « [L, " Ч4Й^^ххх
о а» Я S О CJ О О U
и
со
сч - иГ X .04° . Л О И х"Е Й <и х и (_> О о X и
< X и г ~ - -йХ®,
и « ® я О £ X £ к
Z xzuuuXXoo
>>
о-
Я
f- ю
S О-
а> о § о ь (D О <У
X X х X
Цеолит NaA: [ сорбирует | несорбирует
Цеолит СаА: | сорбирует | несорбирует
Цеолит СаХ: [ сорбирует (несорбирует
Цеолит NaX: | сорбирует молекулы всех групп; не сорбирует (w-CiFgJsN
Цеолиты
99
Фис. 3;15. ‘Изобары адсорбции газов с различным
«рнтнческнм диаметром молекул на цеолите NaA.
МО
Цеолиты типа X имеют достаточно
широкие входные окна н адсорбируют по- m
давляющее большинство компонентов 5 5
сложных смесей: все типы углеводородов, 13 и
органические сернистые, азотистые и кис-
лородные соединения (меркаптаны, тио-
фен, фуран, хинолин, пцрнднн, диоксан и q
др.), галоидозамещенные углеводороды -200 -100 0 100
(хлороформ, четыреххлорнстый углерод, °q
фреоны, пентаборан и декарборан). Приме-
нение цеолитов СаХ и NaX основано на избирательности адсорбции, обус-
ловленной строением электронной оболочки адсорбтива, а не на молекуляр-
но-ситовых свойствах адсррбента. Прн полном замещении катиона натрия
на кальций цеолит СаХ, в отличие от цеолита NaX, не адсорбирует арома-
тические углеводороды или их производные с разветвленными радикалами,
например 1,3,5-трнэтнлбензол н метаднхлорбензол. На этом свойстве основан
метод ндентнфнкацнн цеолитов этих двух типов и установление полноты
ионного обмена прн получении цеолита СаХ.
В случае, если критический диаметр молекулы близок к
диаметру входного окна, процесс адсорбции происходит с боль-
шой энергией активации и адсорбируемая молекула должна об-
ладать определенным запасом кинетической энергии для пре-
одоления энергетического барьера. Кинетическая энергия моле-
кул повышается с ростом температуры. Одновременно повыше-
ние температуры приводит к усилению термической пульсации
решетки цеолита, что облегчает проникание молекулы в адсорб-
ционную полость. Таким образом, изменением температурного
режима можно достигнуть такого рубежа, при котором моле-
кулы адсорбтива начнут поглощаться цеолитом.
Характерным примером такого случая является адсорбция некоторых
газов на цеолите NaA прн низких температурах. Изобары сорбции криптона,
метана, азота и аргона представлены на рис. 3,15. По данным Квнтковского
и Сергиенко [107], температура начала сорбции для этих газов составляет
соответственно минус 94, 139, 155 и 165 °C. Из этого примера следует вывод
о некоторой условности приведенной выше классификации.
К-ионы создают энергетический барьер и скорость адсорбции
становится очень чувствительной к размеру молекул [108].
Ниже показано, как изменяются коэффициенты диффузии D
и энергия активации £а при поглощении газов К-модернитом
при минус 78 °C в зависимости от критического диаметра мо-
лекул dKpi
Газ Кг Аг n2 о2 н2
^1кр, НМ 0,39 0,38 0,37 0,34 0,24
D, см2/с 1,8-10-18 2,4-10-16 9,2-10-1’ 2,0-10-15 2.7-10-13
£а, кДж/мОЛЬ 41,90 35,20 20,11 18,44 10,48
7*
100
3. Основные виды пористых адсорбентов
I
/Рис. 3,16. Изменение коэффициента разделе-
нии смесн азот — метан на цеолите NaA во
времени.
Разницу в кинетике адсорбции ве-
ществ, размеры молекул которых близ-
ки к размерам входных окон, можно ис-
пользовать для их разделения. Напри-
мер, из смеси азота и метана при рав-
новесии цеолитом NaA преимуществен-
но сорбируется метан, но молекулы азо-
та, имеющие несколько меньшие разме-
ры, диффундируют внутрь кристаллита
быстрее. Благодаря этому можно подо-
брать такие условия, когда достигается
значительное обогащение адсорбирован-
ной фазы азотом.
На рис. 3,16 [109] представлено изменение коэффициента разделения
(за лучше адсорбирующийся компонент принят азот) в зависимости от вре-
мени контакта для двух температур и двух концентраций. При времени кон-
такта 4 мин и минус 79 °C значение коэффициента разделения достигает
5—10.
Критические диаметры основных компонентов промышлен-
ных газов содержатся в табл. 3-3. Высокомолекулярные соеди-
нения, указанные в табл. 3-3, могут быть применены в качестве
молекулярных щупов для распознавания типа «широкопористо-
го» цеолита. Так, цеолит NaX (диаметр пор (/=0,9 нм) адсорби-
рует трипропиламин, но не адсорбирует триперфторбутиламин.
В то же время морденит в водородной форме (d~ 1,0 нм) три-
перфторбутиламин поглощает. Цеолит NaY ((/=0,9 нм) адсор-
бирует 1,3,5-триэтилбензол, а цеолит СаХ его не поглощает.
Синтетические кислотостойкие цеолиты
К синтетическим кислотостойким цеолитам относятся морденит
(цеолит типа М), цеолит типа Y, эрионит (цеолит типа Э) и др.
Ряд кислотостойких цеолитов имеет аналоги в природе.
Морденит. Синтетические мордениты получают из гелей, в ко-
торых соотношение компонентов то же, что и в продукте: Na2O :
: А120з : SiO2 = 1 : 1 : 10. Синтез в некоторых случаях ведут при
высоких температурах (210—330 °C) и давлениях 100—200 МПа.
Морденит, полученный при высокой температуре, по молекуляр-
но-ситовым свойствам аналогичен природному. Кристаллизация
Na-морденита может быть проведена при более низкой темпера-
туре в условиях более щелочной среды [83]. Эта разновидность
морденита адсорбирует достаточно крупные молекулы типа бен-
зола. При обработке Na-морденита кислотой в результате дека-
тионирования получают водородную форму морденита Н-морде-
нит (НМ).
Другим методом получения водородной формы является за-
мещение катионов на группу NH4 с последующим термическим
I ।
Ф О О)
ф Ф Ю
Ю Ю О Ь* —« —‘ СО СП СЧ —* С) О) СП —‘ СП О О Ь- —*
Ю < Ф Ф О) Ф ф ф" ОД Ф ф ф Tj< О Ф Ф ф" ф" ф
о сп
S® ’S
3 3
CQ CQ
о о
Таблица 3-3. Критические диаметры dKp и длины* I молекул некоторых веществ (в 10 нм)
0>
со
о
вэ
3
СО
СП СП
СОФФ0>ОООО4С>ОООО4СОСО0)0)ООЬ*ЮС^
'есофсософсофсчюфююффффсо^'оо'
СО N СО Ф
ф о
СП сч со
со со со cw со
о
Sl=t
►о
и- 4 4 4
s Я S та о
SH О Н Н со
о ь- 0> 0> к
г га о. tf tf Й
< < < < < И
со ю
ОСНСНСОЬ’ФСПОО-^Ь-
СО Ю Ю Ф
ФФЮО4СЧФФО4СОС0
Длины указаны только для некоторых простейших молекул.
Критический диаметр всех нормальных парафиновых углеводородов, начиная с пропана, равен 0,49 им.
102 3. Основные виды пористых адсорбентов
разложением аммонийной формы, которое проводят прогрева-
нием цеолита в течение 1 ч при 350 °C. Свойства природных и
синтетических морденитов, аспекты их декатионирования и де-
алюминирования с целью получения водородной формы подроб-
но освещены в публикациях Беленькой, Дубинина и Криштофо-
ри [ПО]. я
Вне зависимости от типа исходной Na-формы характерный
размер структуры НМ, определяющий молекулярно-ситовые
свойства, составляет 0,66 нм, вследствие чего этот цеолит хоро-
шо поглощает низшие представители гомологических рядов уг-
леводородов (например, изо- и нормальный бутан или гексан,
циклогексан, бензол).
С другой стороны, морденит используют и для молекулярно-
ситового разделения, например метилразветвленных парафинов
(критический диаметр 0,61 нм) и высокомолекулярных арома-
тических соединений (критический диаметр более 0,8 нм).
Путем обработки «узкопористой» формы Na-морденита сна-
чала 2 н. соляной кислотой, а затем растворами NaOH и NaNO3
осуществляют ее превращение в «широкопористую» форму.
Предельный адсорбционный объем Na-морденита обоих ти-
пов составляет 0,076—0,088 см3/г, Н-морденита 0,088—
0,090 см3/г. Параметры элементарных ячеек и химический со-
став «узкопористого» и «широкопористого» Na-морденитов яв-
ляется идентичным. Очевидно, различие в молекулярно-ситовых
свойствах морденитов различных модификаций обусловлено раз-
ным расположением катионов натрия, которые в «узкопористой»
разновидности размещаются в больших двенадцатичленных ка-
налах 0,59X0,71 нм и, таким образом, сужают их эффективный
диаметр, а в «широкопористой» — локализованы в глубине ниш,
составленных двойными восьмичленными кольцами 0,39x0,47 нм
и не препятствуют прониканию крупных молекул в большие
каналы [111].
Синтетические мордениты кислотостойки и термостойки. Об-
работка 3 н. раствором НС1 в течение нескольких часов при
100—200 °C не приводит к изменению их адсорбционных свойств.
Они выдерживают пропарку при 650 °C в течение длительного
периода времени (например, 10 ч), нагрев на воздухе до 775 °C
(НМ) и 800 °C (NaM).
Водородная форма морденита обладает свойствами твердой
кислоты и является эффективным катализатором реакций кре-
кинга, изомеризации и алкилирования. С помощью Н-морденита
легко осуществляется дегидратация спиртов. Каталитическая
активность Н-морденита сохраняется при низких температурах
(около 350 °C), а также при частичной закоксованности катали-
затора.
Кислотостойкость морденитов открыла перспективу их применения для
осушки водорода и хлорида водорода, различных хлорлроизводных углево-
Цеолиты
юз.
дородов, удаления оксидов, азота из газов, отходящих с азотно-кислотных
заводов, и для решения ряда других проблем очистки и рекуперации.
В США фирма Нортон производит синтетический морденит в Na- и
Н-Лормах под фирменным названием «Зеолон» [112]. Химический состав
Na-зеолона: 71,45% SiO2; 12,05% А12О3; 0,52% FeA; 0,26% СаО; 0,31%
MgO; 7,14% Na2O; 9,15% Н2О. Соотношение в цеолите Si:Al=5:l. Товар-
ный продукт выпускается в гидратированном состоянии и перед употребле-
нием его необходимо прокалить при 600 °C. Мордениты получены также
в кальциевой и стронциевой форме.
При длительном воздействии кислой среды на кислотостой-
кие формы цеолитов происходит постепенное вымывание алю-
миния. Этот процесс получил название деалюминирования. При
деалюминировании тетраэдры (А1О4)3- замещаются на дитетра-
эдры [113]. В результате деалюминирования соотношение SiOs:
: AI2O3 может достичь 30.
В процессе декатионирования и деалюминирования мордени-
та образуются открытые структуры, из которых влага удаляется
легче, чем из цеолитов общего назначения. Вследствие этого ре-
генерация Н-морденита может быть проведена при температуре
на 25—30 °C ниже, чем цеолитов типа А [114].
Для менее кислотостойких цеолитов этот путь деалюминирования ис-
ключен. В этом случае часто прибегают к реакции, идущей через аммоний-
ную форму, обрабатывая цеолит солью аммония, например NH4C1 [115].
После этого из аммонийной формы цеолита аммиак удаляют путем нагрева
в течение 3 ч при 550 °C. Полученный продукт кипятят с ацетилацетоном.
В результате такой обработки отношение SiO2: А12О3 в цеолитах типа X и
Y возрастает с 5 до 7; при этом соответственно число атомов алюминия в
элементарной ячейке снижается с 55 до 43.
Естественно, при деалюминировании изменяется не только
состав, но и структура цеолитов. В одной из работ [116] пока-
зано, что в результате деалюминирования цеолита NaY образу-
ются вторичные поры радиусом 1,5—1,9 нм с удельным объемом
0,13 см3/г. Одновременно на 25% сокращается предельный ад-
сорбционный объем микропор — до 0,2 см3/г.
Нагревание цеолитов до температур выше 500 °C приводит
к выделению воды, входящей в решетку цеолита, т. е. к его
дегидроксилированию. При дегидроксилировании некоторых
цеолитов на каждый исходный катион выделяется одна молеку-
ла воды. Очевидно, при этом теряются гидроксил, связанный с
катионом, и протон при кислороде каркаса.
Чтобы получить «ультрастабильный» цеолит Y, обладающий повышен-
ной устойчивостью к термической и гидротермальной обработке, его дека-
тионированную форму подвергают двум дополнительным стадиям обработки.
Вначале проводят дополнительный обмен катионов на ионы аммония, воз-
действуя иа цеолит раствором соли аммония при 100 °C; в результате со-
держание Na2O снижается до 0,1%. В заключительной стадии цеолит в те-
чение 3 ч нагревают при 815 °C в присутствии паров воды; кристаллическая
структура такого цеолита сохраняется при температурах до 1000 °C. При
превращении в «ультрастойкую» форму значительно сокращается размер
элементарной ячейки. В США «ультрастабильный» цеолит известен под фир-
менным названием Z—14—US [117].
104
3. Основные виды пористых адсорбентов
Рис. 3,17. Зависимость прочности при сжатии Р гра-
нул цеолита от содержания связующего G.
При проведении процессов очистки
газов от вредных примесей следует
учитывать возможность хемосорбцион-
ного взаимодействия адсорбтива с цео-
литом. Так, исследованиями, прове-
денными в МХТИ им. Д. И. Менделеева [118], показано, что
при температурах выше 150 °C триоксид серы реагирует с Н-мор-
денитом, причем хемосорбция его практически необратима.
Кислотостойкие цеолиты, как и цеолиты общего назначения, обычно ис-
пользуют в виде гранул. Методы гранулирования морденита и других
кислотостойких цеолитов имеют свои особенности. В этом случае не только
кристаллит, т. е. сам цеолит, но и связующее, а также вся композиция
после гранулирования должны сохранять кислотостойкость. Обычные типы
связующих для этой цели непригодны; гранулы быстро разрушаются в кис-
лой среде, особенно при высоких температурах.
По одной из методик [119] гранулирование натриевой формы мордени-
та проводят золем кремневой кислоты с добавкой набухающих частиц жела-
тины. Добавка желатины обеспечивает качественную коагуляцию золя во
влажном состоянии. Гранулы при использовании желатины отличаются по-
вышенной прочностью. В процессе последующей за гранулированием стадии
прокаливания гранул морденита при температуре 500—650 °C в течение 6 ч
желатина выгорает.
На рис. 3,17 и ЗД8 представлены прочностные характеристики получен-
ных адсорбентов применительно к условиям неподвижного и движущегося
(или псевдоожиженного) слоя в зависимости от содержания связующего.
Адсорбционная емкость этих же образцов по влаге, определенная принятым
в ГДР стандартным методом, приведена на рис. 3,19. Рассмотрение этих
зависимостей позволяет заключить, что содержание аморфного диоксида
кремния в гранулах морденита с точки зрения сочетания приемлемых проч-
ностных и адсорбционных свойств должно быть в пределах 10—15% (масс.).
Гранулирование вносит определенные коррективы в характер
вторичной пористости гранул*. Результаты изучения вторичной
Таблица 3-4. Характеристика вторичных пор гранулированных
морденитов
Содержание, % Суммарный объем вторичных пор, см3/г Преимущественный радиус пор, нм
морденит глина SiO2
80 20 — 0,34 799
72,7 18 9,3 0,23 529
82,5 — 17,5 0,25 627
Зеолон-ЗОО 0,25 25
* В гранулированных цеолитах различают два вида пористости-, первичную,
сосредоточенную в самих частицах цеолита (т. е. в кристаллите), и вторич-
ную, образованную зазорами между кристаллитами и частицами связующего.
(Прим. ред.).
Цеолиты
105
Рис. 3,18. Зависимость потерь П при истирании гра-
нул цеолита NaM (размер от 1,6 до 3,2 мм) от со*
держания связующего G.
пористой структуры гранул морденита
ртутной порометрией представлены в ст
табл. 3-4. Наибольшим суммарным
объемом мезо- и макропор обладал
образец, сформованный с глиной —
0,34 см3/г, причем распределение пор
по эквивалентным радиусам охватыва-
ет интервал от 10 до 7500 нм. При ис-
пользовании в качестве связующего
золя кремневой кислоты (как при добавке желатины, так и без
нее) суммарный объем несколько уменьшается и имеет пример-
но такое же значение, как у зеолона, выпускаемого на основе
природного морденита (0,25 см3/г). Однако, преимущественный
радиус вторичных пор зеолона значительно меньше радиуса
вторичных пор приготовленных в лаборатории образцов грану-
лированных цеолитов.
Равновесная адсорбционная способность гранулированных
морденитов по парам воды и другим адсорбтивам практически
не зависит от типа связующего и методики гранулирования.
В то же время образцы, полученные с использованием золя
кремневой кислоты, отличаются замедленной кинетикой адсорб-
ции, что может быть объяснено большим числом контактов по-
рошка морденита со связующим (SiOs) и частичной блокиров-
кой входов в адсорбционные полости.
Эрионит. Другим важным типом кислотостойких цеолитов яв-
ляется эрионит. В классификации, принятой в Советском Союзе,
эрионит обозначают буквой Э, например Na3, К.Э и т. д. Его
открытие и описание относится к концу прошлого столетия. Кро-
ме катионов натрия в состав природных эрионитов могут вхо-
дить катионы калия, кальция и магния.
Соотношение SiOs: AI2O3 в эрионите меньше, чем в мордени-
те, но выше чем в фожазите: в различных модификациях оно
изменяется от 6 до 7,4. В связи с этим кислотность эрионита
является промежуточной между кислотностью цеолитов типов
X и М. Эрионит устойчив в щелочных и
кислых средах (до pH 2).
Структура адсорбционных полостей
эрионита видна из рис. 3,20. Адсорбци-
онная полость эрионита имеет вид ци-
линдра диаметром 0,63 и длиной 1,51 нм.
Каждая большая адсорбционная полость
Рис. 3,19. Зависимость предельной адсорбционной спо-
собности а* цеолита NaM от содержания связующего G.
106 3. Основные виды пористых адсорбентов
Рис. 3,20. Адсорбционная плотность эрионита.
эрионита связана с восемью сосед-
ними большими полостями шестью
восьмиугольными окнами. Кроме
того, через шестерные кольца боль-
шая полость соединена с тремя ма-
лыми полостями, а также с двумя
большими полостями, расположенными над и под ней. Эффек-
тивный диаметр пор эрионита находится в пределах 0,45—
0,54 нм; общий объем адсорбционных полостей составляет
0,21 см3/г.
В Советском Союзе месторождения природного эрионита об-
наружены в Сибири и в Грузии. Отмечены случаи образования
эрионита в вулканических отложениях и в отложениях соленых
щелочных озер (озеро Магади в Кении и озеро Чайна в Кали-
форнии) .
Синтетические эриониты получают из натриево-калиевых ге-
лей, обогащенных кремнеземом, при 100—150 °C. Условия при-
готовления эрионита искусственным путем установлены коллек-
тивами под руководством Жданова [ИЗ], Пигузовой [120] и
Николиной [121]. Адсорбционная емкость калий-натриевого
эрионита по парам воды при 25 °C составляет около 15%
(масс.). Вода полностью удаляется из него при относительно
низких температурах (150—200°C). Эрионит практически не
сорбирует бензол, а поглощение нормальных парафинов проис-
ходит с замедленной скоростью. Адсорбционная способность и
скорость поглощения нормальных парафинов могут быть повы-
шены в результате замещения катионов калия и натрия на ка-
тионы кальция и магния; равновесная адсорбционная емкость
катионообменных форм эрионита по н-гексану при 20 °C и отно-
сительном давлении 0,3 составляет для KNa3, СаНЭ и MgK3
соответственно 7,7; 10,4 и 9,2 г на 100 г адсорбента.
Под действием соляной кислоты на кристаллах эронита сна-
чала протекает процесс декатионирования, сопровождающийся
снижением содержания катионов, а затем деалюминирования,
вследствие чего соотношение компонентов ЭЮг: А1гО3 может
быть повышено до 15 без разрушения кристаллической решетки.
При декатионировании увеличивается предельный адсорбци-
онный объем эрионита, доступный для адсорбирующихся моле-
кул, а следовательно, и адсорбционная способность. Так, ад-
сорбционная способность исходного образца KNa3 по диоксиду
азота при относительном давлении 0,015 и обычной температу-
ре составляет 18, а после декатионирования — 25% (масс.).
Порог термической устойчивости К—Na-формы составляетббО—
700 °C; декатионированных форм 800 °C. Эрионит устойчив в
среде слабых кислот и в среде нитрозных газов.
Цеолиты
107
В США фирмой Линде эрионит выпускается под фирменным названием
«Цеолит А117-500». Кислотостойкий цеолит AU7-500 получают на основе эрио-
нита и шабазита. Цеолит А117-500 применяют для осушки газов, содержащих
кислые компоненты, для извлечения хлорида водорода, диоксида серы, ок-
сидов азота. Его используют при осушке водорода риформинга, содержащего
до 25-Ю-4% хлорида водорода, при осушке хлора, хлорпроизводных угле-
водородов (четыреххлористого углерода, метиленхлорида, метилхлорида и
т. п.), осушке и очистке фторпроизводных углеводородов, очистке дымовых
газов от диоксида серы, удалении хлорида водорода из водорода. Равно-
весная адсорбционная способность этого адсорбента по основным компонен-
там промышленных газов составляет:
Газ Н,0 СО, «-С4Н10 о2
t, °C 25 25 25 — 183
р, кПа 2,3 33,2 80,0 13,3
а, % (масс.) 20 13 8 17
Удаление воды и других примесей из цеолита А117-500 производят при
температурах от 150 до 315 °C; нагреватель адсорбент выше 500 °C во избе-
жание его дезактивации не рекомендуется.
Цеолит типа /. — кислотостойкий калиевый (KL) алюмосили-
кат, в котором мольное отношение SiO2: А12О3 колеблется в
пределах от 5 до 7 [122]. Он кристаллизуется из щелочных
алюмосиликатных гидрогелей, в которых отношение КгО:
: (K.2O + Na2O) выше 0,35; если это соотношение меньше 0,35 в
тех же условиях происходит кристаллизация эроинита. Химиче-
ский состав цеолита K.L: 0,88 K2O-0,12Na2O-Al2O3-6,25SiO2-
• 5,6Н2О. Цеолит K.L выдерживает длительное кипячение в уксус-
ной кислоте и нагревание в разбавленных (0,01—0,05 н.) рас-
творах соляной кислоты. Его можно применять в средах с pH
до 2—3. Кристаллическая решетка цеолита KL начинает разру-
шаться при 700 °C. Влагоемкость цеолита KL приблизительно в
2 раза ниже влагоемкости цеолита NaX.
В результате обработки цеолита KL разбавленным раство-
ром соляной кислоты получают водородную форму HKL. Влаго-
емкость цеолита HKL, в котором половина катионов щелочных
металлов заменена на Н-ионы, на 35% выше, чем у исходного
цеолита KL. Водородная форма HKL хорошо поглощает веще-
ства с относительно крупными молекулами. Предельный адсорб-
ционный объем цеолита HKL, определенный по адсорбции бен-
зола, составляет 0,16 см3/г. Декатионированные формы цеоли-
та L являются интенсивными катализаторами реакций крекинга
углеводородов, уступая по этому показателю лишь декатиониро-
ванной форме HNaY [123].
Сверхвысококремнеземные цеолиты. В последнее время раз-
работаны пути синтеза новых сверхвысококремнеземных цеоли-
тов, в которых отношение SiO2: А12О3 может составлять 100 и
выше. В США они получили фирменное обозначение ZSM [124].
Методика синтеза сверхвысококремнеземных цеолитов близка по
108
3. Основные виды пористых адсорбентов
условиям синтеза к методике получения высококремнеземных
цеолитов, однако в этом случае к исходным компонентам (крем-
незолю, алюминату аммония, свободной натриевой щелочи и
воде) добавляют вещество щелочной природы-тетралкиламмо-
ниевое (или фосфониевое) основание в виде гидроксида или
соли. В соответствии с исходным основанием образуются разные
радикалы, которые выполняют функции катионов и вместе с
катионом натрия входят в структуру конечного продукта.
Различают несколько категорий сверхвысококремнеземных
цеолитов: ZSM-5 содержит радикал [(CsHy^N]; ZSM-8 и
ZSM-12—[(C2H5)4N]; ZSM-11—[(С4Н9)Р]. Объемы органиче-
ских радикалов (0,13—0,17 нм3) намного превышают объемы
обычных катионов; например объем Na+ равен 3,94-10-3 нм3;
К+ —3,31-10~3 нм3; (NH4)+—1,4.10-2 нм3.
При синтезе цеолита ZSM в исходную смесь вводят гидро-
оксид тетрапропиламмония (C3H7)4NOH, причем соблюдаются
следующие пропорции: (ОН)-: SiO2=0,l—0,8; (R4N)+:
: [(R4N) + + Na+]=0,3—0,9; Н2О : (ОН)-= 10—300; SiO2:Al2O3 =
= 10—60. Синтез ведут при 150 °C в течение длительного време-
ни — от 1 до 20 сут. В результате получают кристаллит с разме-
ром частиц от 1 до 25 мкм и размером входного окна от 0,5
до 0,7 нм. Объем пор всех сверхкремнеземных цеолитов неве-
лик: он колеблется от 0,1 до 0,2 см3/г.
В сверхвысококремнеземных цеолитах к катионному обмену
способны только катионы натрия. Органические радикалы раз-
рушаются при нагреве до 400 °C, в результате чего поверхность
цеолита протонируется. Они являются высокоэффективными ка-
тализаторами различных превращений углеводородов: гидрокре-
кинга, изомеризации, алкилирования и т. д.
Смешанные адсорбенты
Смешанные адсорбенты не являются механической смесью раз-
личных пористых тел. Их структура, сформированная в процессе
синтеза, составлена из частиц с размерами, характерными для
коллоидальных или молекулярных систем. Пористая структура
и адсорбционные свойства смешанных адсорбентов значительно
отличаются от пористой структуры и свойств исходных адсорбен-
тов [125]. При синтезе смешанных адсорбентов проявляются та-
кие факторы как взаимное влияние компонентов при созревании
и старении, а также возможное частичное образование химиче-
ских соединений.
При добавлении к золю кремневой кислоты небольших коли-
честв золя гидрооксида металла удельная поверхность получен-
ных смешанных адсорбентов сначала резко возрастает, при оп-
ределенном соотношении достигает максимума и при дальней-
шем повышении концентрации гидроксида металла снижается
(рис. 3,21).
Смешанные адсорбенты.
109
Рис. 3,21. Зависимость удельной поверхности алюмо-
силикагеля 5 от содержания в нем оксида алюминия
СА12О3.
Это явление может быть объяснено сле-
дующим образом. В процессе формирования
структуры ксерогедя происходит рост его час-
тиц до определенных размеров, характеризую-
щих в конечном итоге удельную поверхность
гелеобразных адсорбентов [126]. При смешении
гидрооксидов металлов различной химической
природы рост частиц задерживается на грани-
це раздела вследствие взаимной стабилизации
гидрооксидов. Поэтому в смешанном ад-
сорбенте частицы вырастают до значительно
гелеобразовании отдельных компонентов.
Кроме того, рост частиц тормозится образованием комплексов на по-
верхности мицелл [127]. В случае, например, алюмосиликагелей, осажденных
в кислой среде, на поверхности мицелл кремневой кислоты образуются алю-
мосодержащие комплексы, тормозящие дальнейший рост глобул. Когда си-
стема в последующем подвергается обезвоживанию, более мелкие глобулы
образуют мелкопористую структуру, отличающуюся большей удельной по-
верхностью. При дальнейшем добавлении оксидов к кремневой кислоте ин-
тенсивность кристаллизации увеличивается, что приводит к снижению этого
показателя.
Определенные практические перспективы имеют синтезиро-
ванные цеолитоалюмогели и цеолитоалюмосиликагели, являю-
щиеся первоклассными катализаторами во многих реакциях,
а также своеобразными адсорбентами. Присутствие порошка
цеолита при созревании и формировании пористой структуры
аморфного гидрогеля приводит к изменению пористой структу-
ры аморфной части смешанного адсорбента [128]. Цеолитовый
порошок способствует разрыхлению скелета, образованию транс-
портных пор, что приводит к созданию более крупнопористой
системы. С увеличением содержания цеолита в смешанном ад-
сорбенте суммарный объем пор и объем мезопор растет
(рис. 3,22). Одновременно, естественно, увеличивается объем
микропор смешанного адсорбента. Это
отчетливо иллюстрирует табл. 3-5 [129].
Одновременно на границе раздела
аморфная фаза — цеолит возникают но-
вые активные центры, имеющие особен-
но большое значение в катализе; кислот-
ность у смешанных адсорбентов значи-
тельно выше кислотности чистых адсор-
бентов [130].
Другим классом смешанных адсорбен-
тов являются цеолитполисилоксаны (ме-
Рис. 3,22. Зависимость суммарного объема пор (1) и объ-
ема мезопор (2) V от содержания цеолита NaX в алю-
мосиликатном адсорбенте.
110 3. Основные виды пористых адсорбентов
Таблица 3-5. Распределение объема пор (см.3/г), в цеолитоалюмогеле
по эквивалентным радиусам
Содержание цеолита, % Радиус пор, 10 нм
0,29—3,1 3,1-150 / 150-690' >690
0 0,141 0,324 0,004 0,004-
6,9 0,156 0,326 0,001 0,007
16,0 0,195 0,232 0,000 0,002'
23,6 0,208 0,233 0,106 0,002
28,7 0,188 0,199 0,110 0,014'
49,1 0,257 0,158 0,124 0,253
70,1 0,268 0,086 0,132 0,545
тил-, гидрид- и винилполисилоксан). В присутствии гидрофиль-
ного порошка цеолитов скелет геля аморфной фазы при синтезе
стягивается, сжимается, макропоры уменьшаются в размерах.
Степень сжатия скелета аморфной фазы находится в зависимо-
сти не только от количества введенного цеолита, но и от при-
роды катиона, входящего в его состав, а также от размера это-
го катиона: чем больше гидратирован катион и чем меньше его-
радиус, тем сильнее сжимается гель. Переход крупных пор в
результате сжатия решетки в более мелкие в конечном итоге
приводит к повышению адсорбционной способности таких ад-
сорбентов по основным компонентам промышленных смесей.
По иному формируется структура углесиликагеля, который
был успешно синтезирован в Советском Союзе еще в тридцатых
годах [131]. После введения гидрофобного порошка угля в золь
кремневой кислоты скелет образовавшегося геля в стадии после-
дующей сушки меньше стягивается, в результате чего в смешан-
ном адсорбенте образуются более крупные поры. С другой сто-
роны, в крупные поры углеродных частиц внедряются силикат-
ные мицеллы, что приводит к уменьшению размеров пор, увели-
чению их удельной поверхности, предельного адсорбционного
объема, а следовательно, и адсорбционной способности [132].
Пористые стекла
Это пористые силикатные адсорбенты различной структуры. Их
получают в результате воздействия кислот на натриево-бороси-
ликатное стекло, в результате которого борат натрия удаляется
и остается кремнеземный скелет [133]. Пористую структуру та-
ких адсорбентов определяют состав и структура исходного
стекла, а также условия его варки. Изменение этих параметров
позволяет варьировать преимущественный размер пор в очень
широком интервале.
Природные глинистые породы
111
Рис. 3,23. Ти^ы структур в пористых стеклах
(справа приведены типичные для каждой струк-
туры изотермы адсорбции воды).
\
Микропористые и бипористые
стекла разнои\ структуры оказалось
возможным поручить изменением в
структуре исходного стекла сплош-
ной борокислородной сетки и от-
дельных включений боратной фазы.
На рис. 3,23 представлены основные
типы получаемых структур. При на-
личии в стекле только борокисло-
родной сетки после ее растворения
образуется чисто микропористая
структура. Если в стекле имеются
отдельные включения боратной фа-
зы, образуются бутылкообразные
поры с широкими полостями и очень
узкими порами. Наконец, при слия-
нии этих включений конечный про-
дукт представляет бипористую систему, состоящую из крупных
пор и отходящих от них узких микропор.
В том случае, когда в состав боратной фазы входит SiOa,
после растворения фазы кислотами образуются большие поло-
сти — макропоры; содержащийся в боратной фазе диоксид крем-
ния полимеризуется и осаждается в микропорах в виде высоко-
дисперсного гидратированного кремнезема. Образовавшийся би-
пористый конгломерат, составленный крупными порами основ-
ного кремнеземного каркаса и мелкими порами корпускулярной
структуры осадка, подвергают обработке щелочами. При этом в
первую очередь растворяется высокодисперсный кремнезем и
образуются макропористые стекла. Изменяя степень растворе-
ния стенок пор, регулируют размер и объем макропор. Радиусы
пор макропористых стекол обычно изменяются от 50 до 200 нм,
а их объемы от 0,5 до 2,0 см3/г.
Пористые стекла нашли применение для решения некоторых
задач разделения веществ, например, в медицине.
Природные глинистые породы
Среди адсорбентов значительное место занимают глинистые по-
роды, в состав которых обычно входят минералы с регулярной
структурой. Глинистые минералы по классификации Тарасевича
[134] можно разбить на три основные группы.
1. Слоистые минералы с расширяющейся ре-
шеткой. Основными представителями этой группы являются
112
3. Основные виды пористых адсорбентов
монтмориллонит и вермикулит. Они относятся к мел^опористым
сорбентам. Их структура имеет, по аналогии с гранулированны-
ми цеолитами, первичную и вторичную пористость. Первичная
пористость обусловлена кристаллическим строением минералов,
вторичные поры образованы зазорами между контактирующими
частицами. При адсорбции полярных веществ решетка первич-
ных пор расширяется, и в межпакетное пространство внедряется
один или несколько слоев адсорбата. Удельная поверхность пер-
вичных пор достигает 420—470 м2/г. Азот и углеводороды этими
порами практически не сорбируются. Преимущественный радиус
вторичных пор составляет 5—10 нм, их удельная поверхность не
превышает 60 м2/г.
2. Слоисто-ленточные минералы. Типичными
представителями их являются палыгорскит и сепиолит. Первич-
ные поры слоисто-ленточной группы представлены цеолитными
каналами 0,37X0,64 и 0,56X1,1 нм соответственно. В эти кана-
лы проникают молекулы воды, метанола, аммиака, но не угле-
водородов. Поверхность вторичных пор этих минералов доста-
точно хорошо развита, в связи с чем адсорбенты активно погло-
щают высокомолекулярные вещества, в частности, углеводо-
роды.
3. Слоистые минералы с жесткой решеткой.
Основными представителями их являются тальк, пирофиллит,
гидрослюда, каолинит. Пористость этих минералов обусловлена
зазорами между контактирующими частицами, микропоры от-
сутствуют, удельная поверхность не превышает 150 м2/г. Пло-
ские частицы слоистых минералов с жесткой решеткой уклады-
ваются преимущественно базисными плоскостями друг к другу,
т. е. в основном ориентировано. На рис. 3,24 изображена идеа-
лизированная схема расположения частиц и образования пор
между частицами в таких системах [135]. Представленная схе-
ма находится в соответствии с широким набором пор разных
размеров, обнаруженных у глуховского каолинита [136].
Структурная характеристика основных глинистых минералов
представлена в табл. 3-6.
5
Рис. 3,24. Пористая структура слоистых минералов; с жесткой структурой, состоящих
частиц одинаковой (а) и разной (б) толщины:
1 — замкнутые поры; 2 — лабиринтообразные поры; 3 —поры, образованные пластинка-
ми смежных слоев; 4 — тупокливовидньге поры; б—остроклиновндные поры.
Природные глинистые породы 113*
Таблица 3-6. Характеристика глинистых минералов
Минерал Удельная поверхность ,(в м2/г), вы- численная по адсорбции Суммарный адсорб- ционный объем, V2, смЗ/г Структура пор Отношение VMn/VX
мезопоры микропоры
VME> смЗ/г SME. М2/Г ГМЕ> Ю ям 1"МЦ, смЗ/г гми> нм
Na rt-C6H14
Монтморилло- нит пыжевский 39 36 0,37 0,05 39 45 0,32 0,48 0,86'
Вермикулит ковдорский 12 14 0,18 0,03 12 90 0,15 0,28 0,83,
Палыгорскит черкасский 224 153 0,45 0,29 153 80 0,16 0,50 0,36.
Каолинит глуховский 70 69 0,17 0,17 80 40 — — 0
В обиходе под термином «глина» понимают землистый мате-
риал с размером частиц не выше 1 мкм и высоким содержанием
глинистых минералов, который способен давать пастообразные
массы различной консистенции. В зависимости от преобладаю-
щего минерала, конкретную глинистую породу относят к тому
или иному типу. Так, монтмориллонит является основным мине-
ралом бентонитовых глин и отбеливающих земель (гумбрина,
асканита и т. д.). Отдельную группу природных глин составляют
кремнеземистые породы — диатомиты, трепел, опоки.
Глинистые материалы как адсорбенты применяют в. основном
для очистки различных жидких сред от примесей. Как правило,,
очистка жидких сред сопровождается удалением окрашенных
веществ, в результате чего продукт обесцвечивается. Отсюда
произошло название «отбеливающая земля», хотя в некоторых
современных процессах применение этих адсорбентов связано с
удалением бесцветных веществ.
Часть глинистых природных адсорбентов обладает высокой,
активностью в естественном виде и их подготовка к использова-
нию в промышленности заключается лишь в термической обра-
ботке. К этой группе относятся фуллеровы земли и флоридины.
США, гумбрины и нальчикины Советского Союза. Другая часть,
глинистых пород (бентониты, в нашей стране — асканит, ханла-
рит, гиляби) приобретают высокие адсорбционные свойства после.'
химической активации. Трепела и опоки активации поддаются
слабо. Перед применением их лишь прокаливают для удаления
адсорбционной влаги.
Природные глинистые адсорбенты являются полиминераль-
ными высокодисперсными системами со сложным химическим
составом, колеблющимся в широких пределах. Состав отбели-
вающих глин: 40—72% S1O2, 5—33% AI2O3, 1,2—15% РегО3, до
7% СаО, до 8% MgO, 4—15% оксидов щелочных и других ме-
8—1346
114
3. Основные виды пористых адсорбентов
таллов, а также вода. Состав диатомитовых земель, трепелов,
опок: 75—90% SiO2, 1,5—9% А12О3, остальное оксиды Fe, Са,
Mg, Na, К и других металлов, а также вода. i
Однозначной связи между химико-минералогическим соста-
вом и адсорбционно-отбеливающими свойствами/природных ад-
сорбентов пока не установлено. Адсорбционные й отбеливающие
•свойства природных адсорбентов и оптимальные условия их тер-
мической или химической активации определяются на основе
исследования комплекса физико-химических и адсорбционно-
структурных свойств. Конечным этапом лабораторных испыта-
ний является установление пригодности адсорбентов для кон-
кретного технологического процесса.
Наиболее распространенным видом активации природных
глин является их обработка минеральными кислотами. Чаще
всего применяют 20%-ные серную и соляную кислоты. В резуль-
тате кислотной обработки полностью или частично удаляются
•оксиды кальция, магния, железа, алюминия, других металлов.
•Одновременно с этим при химической обработке повышается
кислотность глин, происходит образование дополнительных пор,
увеличивается удельная поверхность и объем пор. Удельная
поверхность белорусских активированных глин, по данным Ко-
марова, колеблется от 20 до 100 м2/г, но может достигать
200 м2/г, средний эффективный радиус пор изменяется от 3 до
"9,5 нм.
Модифицирование поверхности и структуры глин может
быть достигнуто в результате обработки не только кислотами,
но и другими реагентами.
По наблюдениям Батталовой отбеливающая способность ак-
тивированных глин тем выше, чем больше катионообменная ем-
кость исходной глины. Высокая отбеливающая способность ак-
тивированных монтмориллонитовых глин связана с присутстви-
ем в них монтмориллонита, обладающего кислыми свойствами,
т. е. содержащего в обменном положении ионы Н+ и А13+. Уда-
ление азот- и кислородсодержащих соединений происходит
вследствие хемосорбции на кислотных центрах поверхности ад-
сорбента.
Замена катионов, входящих в состав монтмориллонита, на
катионы больших размеров приводит к «раскрытию» межслоево-
го пространства, которое становится доступным для молекул
углеводородов. Так, внедрение катионов К+, NH4+, Rb+ в ре-
шетку Пыжевского монтмориллонита увеличивает адсорбцион-
ную емкость этого минерала по н-гексану в 1,5 раза, а внедре-
ние катионов Cs+ — в 4,5 раза.
Природные адсорбенты применяют главным образом в нефтеперераба-
тывающей промышленности для очистки и регенерации смазочных, трансфор-
маторных и других специальных масел, для тонкой очистки и повышения
качества жидких топлив. Кроме того, их используют для осветления фрук-
Природные глинистые породы
Г! 5
товых соков, вин и пива, рафинирования растительных масел, очистки сало-
маса, воды. С каждым годом масштабы и области применения природных
адсорбентов расширяются.
Широкое и неуклонное увеличение масштабов применения природных
адсорбентов обусловлено тем, что обладая развитой удельной поверхностью-
и хорошими (часто специфическими), отбеливающими свойствами, они в де-
сятки раз дешевле искусственных адсорбентов. В связи с этим упрощаете»
технология их применения: из схемы часто исключают стадию регенерации.
Выявление новых месторождений природных адсорбентов к
их всестороннее исследование с целью технологического приме-
нения является актуальной проблемой большого народнохозяй-
ственного значения. Этой проблеме уделяется значительное вни-
мание. Наряду с поиском месторождений крупных промышлен-
ных масштабов, представляет несомненный интерес разведка
эффективных адсорбентов с запасами местного значения, для:
потребления их в районе добычи, что стимулирует внедрение-
новых адсорбционных технологических процессов, в том числе
для решения вопросов защиты окружающей среды.
В этом отношении показательны результаты, достигнутые-
казахскими химиками (Батталова, Ликерова). На Алма-Атин-
ском комбинате шампанских, плодово-ягодных и виноградных,
вин для их обработки успешно использована натриевая форма,
местного бентонита.
Натриевый бентонит не только осветляет, но и стабилизирует вина..
В результате контакта с бентонитом удаляется избыток как общего, так и-
белкового азота, ионов тяжелых металлов. Положительным свойством нат-
риевого бентонита является и то, что он ускоряет срок созревания и выдерж-
ки вин. Окислительно-восстановительные и другие реакции, которые медлен-
но протекают в винах во время созревания и выдержки, катализируются-
минералами и катионами, входящими в состав бентонита. Под влиянием ка-
талитических свойств бентонита вина быстро осветляются, стабилизируются,
и приобретают качества выдержанных вин.
Другим примером использования казахского натриевого бен-
тонита является мойка шкур, испытанная на Алма-Атинском:
меховом комбинате. Активированные монтмориллонитовые гли-
ны Казахстана используются и для решения других задач: до-
очистки различных масел после их обработки селективными:
растворителями, отбелки хлопкового масла, регенерации отра-
ботанного трансформаторного масла. Качество масел, очищен-
ных казахскими глинистыми адсорбентами, по цветности, кис-
лотному числу и другим показателям соответствует требовани-
ям принятых в Советском Союзе норм. Использование местных
природных ресурсов позволяет сократить ввоз в республику ад.-
сорбентов из других районов нашей страны и из-за рубежа.
8*
4
ТЕПЛОТА АДСОРБЦИИ
И ТЕРМОДИНАМИЧЕСКОЕ
ОБОСНОВАНИЕ ТЕОРИИ
ОБЪЕМНОГО
ЗАПОЛНЕНИЯ МИКРОПОР
Теплота адсорбции
Как уже отмечалось, адсорбция всегда сопровождается выде-
лением тепла. Различают дифференциальную и инте-
гральную теплоту адсорбции. Первая характеризует
тепловой эффект в интервале изменения степени заполне-
ния адсорбционного пространства, стремящемся
.к нулю. Она отнесена к единице сорбирующегося вещества и
измеряется в кДж/моль, Дж/моль и т. п.
Зависимости дифференциальной теплоты адсорбции от сте-
пени заполнения различны. Часто они носят убывающий ха-
рактер. При наличии сильного взаимодействия адсорбирован-
ных молекул друг с другом в начальной области заполнения
адсорбционной емкости наблюдается пик локального увеличе-
ния теплоты адсорбции. Нижним пределом дифференциальной
теплоты сорбции является теплота конденсации. Это
характерно для степеней заполнения, близких к предельным.
Интегрируя зависимость теплоты адсорбции от заполнения
по степени заполнения, мы получим интегральную теплоту ад-
сорбции, измеряемую в Дж, кДж и т. п. Интегральную теплоту
адсорбции, отнесенную к числу молей адсорбата, называют
средней теплотой адсорбции. Ее измеряют в тех же
единицах, что и дифференциальную теплоту адсорбции.
Иногда пользуются понятием чистой теплоты ад-
сорбции (<?), которая представляет'собой разность тепловых
эффектов фазового перехода при адсорбции Q и конденса-
ции X.
Данные о теплоте адсорбции могут быть получены непосред-
ственно при анализе сетки экспериментальных изотерм.
На основе изотерм адсорбции, полученных при разных тем-
пературах, рассчитывают изостеры, отражающие взаимосвязь
равновесных температур и давлений при постоянной степени
заполнения адсорбционного пространства. Изостеры адсорбции
бутана на промышленных адсорбентах в координатах
Igp— (1/Т) приведены на рис. 4,1. Разные прямые на этом ри-
сунке отвечают разным степеням заполнения. Изостерический
Теплота адсорбции
117
Рис. 4,1. Изостеры (а=2—12%) адсорбции «-бутана иа различных адсорбентах:
и —цеолит NaX; б —цеолит СаА; в — активный уголь.
метод расчета теплот адсорбции основан на известном уравне-
нии Клапейрона—Клаузиуса:
=_А (4
^(1/Л /a==C0nSt * 1 }
Отсюда изостерическая дифференциальная теплота адсорб-
ции определяется согласно уравнению
Как видно из графиков, изостеры адсорбции в координатах
уравнения (4.2) удовлетворительно описываются прямыми ли-
Таблица 4-1. Средние теплоты адсорбции углеводородов
и других адсорбтивов на микропористых адсорбентах (кДж/моль)
Адсорбат Теплота кон- денсации Цеолиты Активные угли Адсорбат Теплота кон- денсации Цеолиты
< га и X га Z £ о о X га О X га Z
Метан 9,01 16,76 16,76 — 18,10 Пропилеи 18,52 44,83 43,58 Этан 14,71 26,8225,98 —• 25,14 Бутилен — 58,6661,59 Пропан 18,73 36,0336,03 — 35,74 Ацетилен 16,89 45,67 — н-Бутан 22,33 44,00 41,90 39,81 41,73 Метилацетилен — 54,05 — н-Пентан 25,85 55,73 53,21 50,28 51,96 Винилацетилен — 51,12 — н-Гексан 29,12 66,20 62,85 60,34 60,55 Диацетилеи — 71,23 — н-Гептан 32,05 76,6872,91 69,14 63,69 Вода — 75,42 75,42 н-Октан 35,03 87,1582,9679,1 71,2 Диоксид 16,34 32,68 — Метилцикло- 31,63 61,59 59,50 — — углерода гексан Монооксид 6,03 15,92 — Этилен 13,53 37,71 36,87 — — углерода
118
4. Теплота адсорбции
ниями. По углам наклона изостер в соответствии с уравнением
(4.2) и рассчитывают дифференциальные теплоты адсорбции
различных веществ.
Интегрируя кривые зависимости теплоты адсорбции от за-
полнения, получим интегральные или средние теплоты адсорб-
ции. В табл. 4-1 приведены средние теплоты адсорбции различ-
ных адсорбтивов на микропористых адсорбентах по данным!
автора. Здесь учтены также результаты работы Киселева с
сотр. [137] и Астахова [138].
Рассмотрение таблицы показывает, что в случае, когда адсорбционные
силы имеют дисперсионный характер, теплоты адсорбции на микропористых
адсорбентах — цеолитах всех типов и активных углях—-близки друг к дру-
гу и приблизительно в два раза выше теплоты конденсации [138, 139]. На
крупнопористых адсорбентах это соотношение уменьшается до 1,5 [140].
Зависимость средних теплот адсорбции нормальных парафиновых углеводо-
родов от числа углеродных атомов в молекуле является линейной: с увели-
чением цепи на одну группу СН2 теплота адсорбции увеличивается иа 6—
10 кДж/м оль.
Коэффициенты в уравнении, связывающем теплоту адсорб-
ции с числом атомов углерода в молекуле нормального пара-
финового углеводорода, зависят от типа цеолита. Это видно из
данных табл. 4-2, составленной Рябухиной [141] на основе ра-
бот Баррера, Хебгуда, Ширмера, Киселева, Петерсона и их
сотрудников.
Вклад специфической составляющей взаимодействия просле-
живается при сопоставлении теплот адсорбции разных гомоло-
гических рядов. Замена одной простой связи в молекуле углево-
дорода (например, пропана) на двойную (пропилен) в случае
адсорбции на цеолитах вызывает увеличение дифференциальной
теплоты адсорбции в среднем на 8,8 кДж/моль. Увеличение на
9,2 кДж/моль отмечено при переходе от двойной связи (пропи-
лен) к тройной (метилацетилен).
Таблица 4-2. Теплоты адсорбции Q (кДж/моль) нормальных
парафиновых углеводородов п на разных цеолитах
Цеолит Углеводороды I Степень заполне- ния адсорбцион- 1 кого простран- 1 ства 0 Формула для опре- деления Q
Н-шабазит Ci—С4 0—>0 11,54-8,4 п
Са-шабазит С1-С7 0—>0 27,24-10,5 п
HL Ci—C4 0—^0 10,54-8,4 п
KL Ci—C4 0—>0 10,94-9,2 п
Na, Са, MgA Ci—Cis 0,2—0,8 22,84-6,5 п
Н-морденит C1-C4 0—^0 16,84-8,4 п
СаХ Ct-C4 0—^0 18,94-9,2 п
СаА Ci—c4 0—>0 7,14-9,6 п
NaX Ci-Ce 0,8 2,54-13,8 п
NaX Ci—C8 0,8 5,44-12,6 п
NaX C1-C6 0,1 2,54-10,1 п
Теплота адсорбции
119
Рис. 4.2. Дифференциальные теплоты адсорбции
азота иа промышленных адсорбентах (температу-
ра опыта 77 К):
1 — активный уголь СКТ; 2 — мелкопорнстый си-
ликагель; 3 — цеолит NaX; 4 — цеолит СаА.
Таким образом, влияние специ-
фического характера адсорбции на
тепловой эффект при переходе от
молекулы с простой связью к мо-
лекуле с двойной связью и далее
от молекулы с двойной связью к
молекуле с тройной при одинаковом числе атомов углерода в
молекуле углеводорода в случае цеолитов приблизительно рав-
ноценно изменению теплоты адсорбции за счет неспецифических
(дисперсионных) сил при увеличении длины молекулы на СЩ-
группу в гомологическом ряду алканов. Особенно резко прояв-
ляется специфическое взаимодействие при адсорбции на цеоли-
тах в случае молекул с двумя тройными связями. Так, теплота
адсорбции диацетилена составляет 74 кДж/моль против 43
кДж/моль для бутана и 53 кДж/моль для бутилена.
По данным Баррера [142], вклад специфической составля-
ющей, обусловленной энергией диполей и квадруполей моле-
кул, в общую теплоту адсорбции на различных типах цеолитов
для часто встречающихся адсорбтивов составляет: для азота —
от 28 до 52%; для диоксида углерода от 40 — до 75%; для ам-
миака — до 80%.
Характер влияния структуры адсорбента и химической при-
роды его поверхности отчетливо проявляется в результате рас-
чета изостерических теплот адсорбции азота, выполненного Та-
бунщиковой [143]. На рис. 4,2 представлены дифференциаль-
ные изостерические теплоты адсорбции азота на промышленных
адсорбентах при различных степенях заполнения адсорбционной
емкости.
Теплоты адсорбции азота во всех случаях уменьшаются с увеличением
степени заполнения. С другой стороны, на всем участке заполнений теплоты
адсорбции возрастают в следующей последовательности: активный уголь —
силикагель — цеолит NaX — цеолит СаА. При степени заполнения 6 = 0,5
теплоты адсорбции в указанной серии адсорбентов составляют: 9,15; 9,85;
12,30; 13,5 кДж/моль. Такое расположение адсорбентов в этом ряду объяс-
няется, по-вндимому, «нарастанием» микропорнстости при переходе от углей
и силикагелей к цеолитам и усилением адсорбционных енл за счет специфи-
ческой составляющей при адсорбции квадрупольнон молекулы азота в сили-
катной (силикагель) н катионированной алюмосиликатной (цеолит) струк-
турах.
Кроме сетки экспериментальных изотерм, данные по тепло-
там адсорбции могут быть получены из калориметрических,
хроматографических и термографических измерений. Хромато-
графический метод отличается быстротой измерений, достаточ-
120
4. Теплота адсорбции
пой точностью, стандартностью аппаратуры. В последнее вре-
мя он стал широко применяться как в теоретической, так и в
прикладной адсорбции. Область его использования, к сожале-
нию, ограничивается сравнительно малыми концентрациями ад-
сорбтива и, следовательно, областью неполной отработки ад-
сорбционной емкости.
Термодинамические аспекты теории
объемного заполнения микропор
Берингом и Серпинским [144, 145] получены термодинамиче-
ские критерии, определяющие условия выполнения основного
положения теории объемного заполнения микропор о темпера-
турной инвариантности характеристического уравнения, и урав-
нения для дифференциальных (мольных) теплоты и энтропии
адсорбции. Эти уравнения позволяют вычислять дифференци-
альные теплоты и энтропии адсорбции для различных степеней
заполнения, пользуясь параметрами уравнения адсорбции, оп-
ределенными на основе минимальной экспериментальной ин-
формации, например по одной изотерме адсорбции.
Рассмотрим общее термодинамическое соотношение, харак-
теризующее адсорбционное равновесие. Пусть при температу-
ре Т и равновесном давлении р количество адсорбированного
вещества будет а. Если принять в качестве стандартного состоя-
ния для отсчета термодинамических функций адсорбата (эн-
тальпии, энтропии, энергии Гиббса) состояние нормальной жид-
кой фазы адсорбтива при той же температуре, то максималь-
ная работа А перехода 1 моль адсорбтива из объемной фазы
на бесконечно большое количество адсорбента выразится как
Л =-ДО =In (ps/p) (4.3)
где ps — давление насыщенного пара.
Очевидно, что уравнение (4.3) и все дальнейшие уравнения
легко обобщить на случай неидеальной газовой фазы, основы-
ваясь на летучести вместо давлений.
Если условие инвариантности характеристического уравне-
ния соблюдается, то дифференциальная мольная энтропия ад-
сорбции выражается уравнением
Д5 = а (wr)r<0 <4-4)
где а — термический коэффициент предельной сорбции.
Условие (4.4) можно использовать для определения нижней
границы области температурной инвариантности. Как показано
в [145], оно соблюдается, если степень заполнения адсорбцион-
ного пространства превышает 0,15—0,20. Это значение, следо-
Термодинамика теории объемного заполнения микропор 121
вательно, и является нижней границей применимости теории
объемного заполнения микропор.
В условиях инвариантности общее выражение для диффе-
ренциальной мольной теплоты адсорбции Q таково:
f дА \
^ + А-аТ{дЫ1Г)г (4-5)
где % — теплота конденсации.
Значение А дает уравнение (4.3). Из него же определяется
производная
/ \ _ (dinhy
{dlnajT~~ [din a ]T
где h=plps — равновесное относительное давление.
С учетом (4.3) и (4.6) из (4.5) получаем:
42 = Я-7?7’1пЛ + ал[-~~-^ (4.7)
Основное уравнение теории объемного заполнения микро-
пор можно записать следующим образом:
а = а0ехр (—А/Е)п (4.8)
Разрешим его с учетом (4.3) относительно h:
In h = —(E/RT) (In а0/а)^п (4.9)
Из (4.9) путем дифференцирования находим выражение для
производной:
(£)rsi (4.10)
Зависимость значения а0 от температуры выражается урав-
нением
а0 = а0° ехр [—а (Т — Го)] (4.11)
Здесь а0° — предельная величина адсорбции, определенная при температуре
То Для исходной изотермы, служащей для нахождения характеристической
энергии адсорбции и для оценки показателя степени п.
Подставляя выражение для производной (4.10) в уравнение
для дифференциальной теплоты адсорбции (4.7), получим
Q = Л + Е [(1П ао[а)Ип + (аГ/л) (Inа./л)^п)-1] (4.12)
Уравнение (4.12) может быть записано в форме:
q _-= Q — л = Д + (a7E/n) (In (4.13)
где q — чистая дифференциальная теплота адсорбции.
122 4. Теплота адсорбции
Аналогичным образом получаем уравнение для дифферен-
циальной мольной энтропии адсорбции:
AS = —j-p-(1пав/а)<1/я>-1
(4-14)
Для характеристической точки, соответствующей степени за-
полнения объема адсорбционного пространства, равной обрат-
ной величине основания натуральных логарифмов, т. е. 0,368,
согласно формул (4.13) и (4.14) получаем:
?0 = Е[1 + (а/Л)Г] (4.15)
Д$о = —аЕ/п (4.16)
Обычно доля члена аЕТ/п в величине qQ не превышает 10—
13%. Таким образом, для характеристической точки характе-
ристическая энергия адсорбции близка к дифференциальной
теплоте адсорбции. Формула (4.16) указывает на то, что раз-
личие между qo и Е определяется энтропийным членом.
После перехода к десятичным логарифмам уравнения (4.12)
и (4.14) приобретают форму, удобную для практического при-
менения:
г 2 __аТ 1
q = Е 2, ЗО1^ (1g Oo/a)V« + —---------(1g a0/a)W«)-i (4.17)
г.ЗО'^)-!—aE
AS = - -------------(1g ао/а)0/«)-1 (4.18)
На рис. 4,3 сравниваются вычисленные по уравнению (4.17) дифферен1-
циальные теплоты адсорбции бензола (сплошные кривые) и- эксперименталь-
ные изостерические теплоты адсорбции (обозначены кружочками) для двух
образцов активных углей, изученных Дубининым и Полстяиовым [146].
В этом случае в уравнении (4.8) п=2. Верхняя кривая иллюстрирует луч-
шее соответствие между результатами вычисления и опыта, а нижняя кри-
вая — худшее. Однако и в последнем случае расхождения расчета и опыта
не превышают 5%.
На рис. 4,4 представлены чистые дифференциальные теплоты адсорбции
метана на цеолите по данным опытов Баррера и Ли [147]. Кружками обо-
Рис. 4,3. Зависимость дифференциальной теплоты адсорбции Q от массы адсорбировав—
вого бензола а для двух активных углей.
Рис. 4,4. Зависимость чистой дифференциальной теплоты адсорбции р метана на цеоли-
те L от массы адсорбированного вещества а (расшифровка точек в тексте).
Термодинамика теории объемного заполнения микропор
123
Рис. 4,6. Зависимость чистой дифференциальной теплоты адсорбции q диоксида углерода
от массы адсорбированного вещества а (расшифровка точек в тексте).
Рис. 4,5. Зависимость дифференциальной мольной энтропии адсорбции AS метана на
цеолите L от степени заполнения 0 (расшифровка точек в тексте).
значены изостерические теплоты адсорбции. Другие значки отвечают вычис-
ленным по уравнению (4.17) при п—3 чистым дифференциальным теплотам
адсорбции для различных температур (минус 30—минус 117°С), соответст-
вующих экспериментальным изостерам адсорбции. Приведенные примеры
свидетельствуют об удовлетворительном соответствии результатов вычисле-
ния и опыта.
График на рис. 4,5 изображает вычисленную по уравнению (4.18) кри-
вую зависимости дифференциальной энтропии адсорбции AS метана иа цео-
лите L от степени заполнения 0. Согласно уравнению (4.18), температура
не оказывает влияния на эту кривую, если соблюдается условие (4.4) тем-
пературной инвариантности. Поэтому вычисленные точки для температур от
—30 до —117 °C укладываются на одну н ту же кривую. Заметим, что в
рассмотренном интервале заполнений наблюдается монотонное изменение
дифференциальной энтропии адсорбции.
Другой случай соответствует адсорбции относительно не-
больших молекул с неоднородным распределением электрон-
ной плотности на цеолитах. В этом случае адсорбция молекул
происходит на двух различных энергетических уровнях: часть
молекул адсорбируется на адсорбционных центрах — катионах
цеолита, а вторая часть объемно заполняет адсорбционное про-
странство полостей цеолита, оставшееся свободным после бло-
кировки катионов молекулами адсорбтива. Хотя конечные урав-
нения для расчета энтропии и теплоты адсорбции в этом случае
имеют двучленный вид, прин-
ципиальная схема вычислений
остается прежней.
На рис. 4,6 изображена зависи-
мость чистой дифференциальной теп-
лоты адсорбции диоксида углерода
на цеолите NaX от количества ад-
сорбированного вещества а. Круж-
Рис. 4,7. Зависимость дифференциальной
мольной энтропии адсорбции AS диоксида
углерода на цеолите NaX от степени за-
полнения 6 (расшифровка точек в тексте).
и и»7
О
124 5. Избирательность адсорбции
ками обозначены экспериментальные изостерические теплоты адсорбции, оп-
ределенные по наклонам изостер. Разными значками обозначены вычислен-
ные дифференциальные теплоты адсорбции для различных температур, соот-
ветствующих экспериментальным изотермам адсорбции. Соответствие ре-
зультатов вычисления и опыта является удовлетворительным [148],
На рис, 4,7 изображена вычисленная зависимость дифференциальной
мольной энтропии адсорбции диоксида углерода на цеолите NaX от степени
заполнения 0. Обозначенные различными значками вычисленные величины
Д5, отвечающие точкам экспериментальных изотерм, практически укладыва-
ются на одну и ту же кривую. Таким образом, теория объемного заполне-
ния микропор позволяет, помимо описания адсорбционных равновесий в до-
статочно широких интервалах температур и давлений, с удовлетворительной
точностью вычислить дифференциальные (мольные) теплоты и энтропии
адсорбции. Естественно, что эти вычисления надежны только в области за-
полнений, в которых достаточно удовлетворительно соблюдаются исходные
предпосылки теории.
Термодинамические аспекты некоторых других вариантов
теории рассмотрены в работах [1, 149, 150].
5
ИЗБИРАТЕЛЬНОСТЬ
АДСОРБЦИИ
Общие закономерности адсорбции смесей
Эффективность адсорбционного разделения двух или несколь-
ких веществ и потери очищаемого компонента вследствие сов-
местной адсорбции с примесями определяются избирательными
свойствами адсорбента, количественно оцениваемыми кривыми
адсорбционного равновесия. Каждому соотношению компонен-
тов в газовой фазе соответствует определенный состав адсорби-
рованной фазы. Кривые адсорбционного равновесия отражают
мольный состав адсорбированной фазы в зависимости от моль-
ного состава газовой фазы при постоянном суммарном давле-
нии двух адсорбирующихся компонентов. В качестве меры из-
бирательности адсорбции предложено [151 —154] применять
коэффициент разделения
Кр = (5.1)
Общие закономерности адсорбции смесей
125
Рис, 5,1. Кривые адсорбционного равновесия про-
пано — бутановой смеси на активном угле АГ-2
при атмосферном давлении,
где уз и г/1 — мольные доли хуже и лучше
адсорбирующихся компонентов в газовой
фазе; х2 и Xi— мольные доли этих компо-
нентов в адсорбированной фазе.
На рис. 5,1 приведены эксперименталь-
ные данные, характеризующие зависимость
состава адсорбированной фазы от соотно-
шения пропана и бутана в газовой фазе на
активном угле АГ-2. Сравнение кривых
адсорбционного равновесия, полученных
20 и 100 °C, показывает, что в отличие от
адсорбционной способности, сильно сни-
жающейся при повышении температуры, состав адсорбированной фазы при
одинаковом составе газовой фазы изменяется незначительно даже в относи-
тельно большом интервале температур.
Коэффициент разделения не является величиной строго по-
стоянной при любых соотношениях компонентов и может быть
принят как предварительный показатель эффективности разде-
ления при некотором конкретном соотношении компонентов.
В качестве стандартного соотношения компонентов для просто-
ты предложено использовать мольное соотношение 1 :1 в газо-
вой фазе.
Коэффициент разделения несколько уменьшается с повыше-
нием температуры*. Так, средняя величина коэффициента раз-
деления для данных, приведенных на рис. 5,1, равна 4,37 при
20 °C и 3,95 при 100 °C.
Количественной характеристикой адсорбции смеси является
также зависимость адсорбционной способности от состава сме-
си в адсорбированной фазе. Анализ кривых для 20 и 100°C в
рассматриваемой системе пропан — бутан — активный уголь
(рис. 5,2), показывает, что адсорбционная способность по сум-
ме извлекаемых компонентов пщ почти линейно меняется с со-
ставом адсорбированной фазы и может быть в первом прибли-
жении рассчитана как аддитивная функция состава адсорби-
* Как показано Берингом и Серпинским, температурная зависимость коэф-
фициента разделения описывается уравнением
1пКр =
Q1-Q3
RT
где Q] и Qi — теплота адсорбции соответственно лучше и хуже адсорбирую-
щихся компонентов смеси. Таким образом, подобно константе равновесия,
коэффициент разделения экспоненциально зависит от температуры. Но ее
влияние в данном случае, естественно, проявляется слабее, так как в правой
части температурной зависимости находится не теплота адсорбции, а раз-
ность двух в общем-то близких теплот адсорбции (Прим. редф.
126 5. Избирательность адсорбции
Рис. 5,2. Зависимость адсорбционной способности
активного угля от состава смеси в адсорбирован-
ной фазе прн атмосферном давлении.
рованной фазы при постоянной сумме парциальных давлений
компонентов в системе:
== х2а2а (5.2)
где Я1° и а2° — адсорбционная способность по чистым компонентам при дав-
лении, равном сумме парциальных давлений извлекаемых компонентов.
Другой зависимостью, описывающей суммарную адсорбцию
двух компонентов, является полуэмпирическое соотношение
Льюиса, справедливое при условии постоянства суммы парци-
альных давлений этих компонентов PI2 = const:
ai/°i0 + “г/^г0 = 1 (5-3)
где Я1 и аг—адсорбционная способность по компонентам, извлекаемым из
•смеси.
Уравнение (5.3) указывает, что при постоянном общем дав-
лении компонентов адсорбция одного компонента линейно за-
висит от адсорбции другого. Применимость уравнения Льюиса
в различных системах доказывает рис. 5,3.
Рис. 5,3. График, характеризующий применимость уравнения Льюиса для различных
систем при разных адсорбентах:
уголь 1: С2Н4 — СзНв, СаНб СзНа, С4Н10 — С4Нв‘, уголь 2: СН4 — С2Н4, С2Н4 ~~ CjHe,
•С2Н4 — СзНб, СзН8 — СзНв; силикагель: СН4 — С2Н4, СаНа — СзНя, С4Н1Ф — С4Н1.
Рис. 5,4. Кривые адсорбционного равновесия в системе диоксид углерода — этилен на
.активном угле при 25 °C и давлении в пределах 7—10 кПа.
Общие закономерности адсорбции смесей
127
Увеличение давления в системе, так же как и повышение
температуры, несколько снижает избирательность поглощения
смеси. На рис. 5,4 представлены кривые адсорбционного равно-
весия этилена и диоксида углерода на активном угле при 25 °C
[155]. Адсорбция обоих компонентов происходит под действием
дисперсионных сил. На всем участке адсорбционной кривой со-
отношение энергии адсорбции СО2 и С2Н4 остается постоянным
и равным отношению поляризуемости молекул. Лучше адсор-
бирующимся компонентом является этилен. Повышение давле-
ния в системе от 7 до 40 кПа (от 50 до 300 мм рт. ст.) приво-
дит к снижению коэффициента разделения с 5 до 3,5.
Избирательность адсорбции двух компонентов не изменяет-
ся в присутствии третьего компонента. Так, коэффициент раз-
деления смеси этилена и пропана на силикагеле при 25 °C, нор-
мальном давлении и соотношении компонентов 1 : 1 составляет
2,1, а в присутствии пропилена 2,0. Соответствующие показате-
ли на активном угле равны 12,5 и 13,0.
Значение коэффициента разделения в системе может быть,
получено в результате простых арифметических действий, вы-
текающих из уравнения (5.1) на основе значений коэффициен-
тов разделения в других системах. Например, коэффициент
разделения смеси пропан — пропилен на силикагеле составляет
Лр' = 3,1, а смеси этилен — пропан КР"=2,1. Отсюда коэффици-
ент разделения смеси этилен — пропилен 7Ср/"=Лр/Лр,, = 6,5;;
опытное значение Лр=6,6.
На рис. 5,5 приведены кривые адсорбционного равновесия
для смеси этан — этилен на цеолите, силикагеле и активном уг-
ле при 20 °C. Равновесие в бинарной системе жидкость — пар
определяется свойствами этих двух компонентов. Адсорбцион-
ное равновесие во многом зависит от избирательных свойств-
третьего компонента — адсорбента. Так, в рассматриваемом
случае адсорбция на цеолите сопровождается резким обогаще-
нием адсорбированной фазы этиленом, в случае силикагеля из-
бирательность адсорбции выражена
менее отчетливо, а при поглощении
смеси активным углем наблюдается
даже инверсия избирательности: из
смеси преимущественно адсорбиру-
ется этан.
Рис. 5,5. Кривые адсорбционного равновесия сме-
си этан — этилен при 20 °C и нормальном давле-
нии на разных адсорбентах:
1 — цеолит СаА; 2 — силикагель КСМ; 3 — актив-
ный уголь СКТ.
128 5. Избирательность адсорбции
Экспериментальные методы изучения
адсорбции смесей
Для изучения адсорбции бинарных смесей применяют установ-
ки двух типов: циркуляционные и проточные. В установках пер-
вого типа адсорбция протекает в замкнутом объеме, включаю-
щем ампулу с адсорбентом, манометр и циркуляционный насос.
Для определения степени адсорбции каждого из компонентов
смеси необходима информация либо о составе газовой фазы до
адсорбции и в момент равновесия, либо о составе и содержа-
нии равновесной адсорбированной фазы. Проще определить со-
став газовой фазы, поэтому циркуляционные установки обору-
дуют газоанализаторами, либо предусматривают отбор пробы
газа на анализ.
Основной причиной ошибок при исследовании равновесной
адсорбции бинарных смесей является большая длительность
установления равновесия. Для предварительного контроля мо-
жет быть использован манометр (в циркуляционных установ-
ках), постоянство показаний которого в течение длительного
промежутка времени является необходимым признаком дости-
жения равновесия. Надежнее, однако, обеспечить непрерывный
анализ газовой фазы. Это наилучшим образом достигается в
циркуляционных установках со встроенным газоанализатором.
Детекторы должны быть точны, просты, доступны и надежны,
однако их показания часто в большой степени зависят от тем-
пературы и давления окружающей среды. Поэтому они нужда-
ются в тщательной калибровке и в термостатировании.
Адсорбция смесей, компоненты которых значительно разли-
чаются по молекулярным массам, может быть изучена на объ-
емно-весовой установке, разработанной Берингом и Серпинским.
Принципиальная схема этой установки показана на рис. 5,6.
Рис. 5,6. Схема объемно-весовой установки:
1 — бюретка; 2, 4 — первое н второе колена; 3, 6 — манометры; 5 — циркуляционный на^
сос; 7 — ячейка с адсорбентом.
Экспериментальные мет'ады
120
С помощью жидкого азота в колене 2 вымораживается определенное
количество первого компонента смеси (обычно первым вводят компонент,
адсорбирующийся хуже). Это количество затем точно измеряют по показа-
ниям газовой бюретки 1 и манометра 3, после чего также с помощью жид-
кого азота переводят в колено 4 и замораживают в нем. Аналогичным обра-
зом в установку вводят второй компонент смеси, а затем после разморажи-
вания включают циркуляционный насос 5 и дожидаются установления рав-
новесия.
Обработку результатов производят следующим образом. Предположим,
что в установку введены п01 и n02 молей первого и второго компонентов сме-
си соответственно, а после установления равновесия в газовой фазе над ад-
сорбентом осталось пг) молей непоглощенной смеси. Все эти данные рассчи-
тываются на основе известных давлений, объемов и температур по уравне-
ниям состояния газов. Количество адсорбированной смеси составляет:
в молях
«а = «а1 + «аг = пП + «оа — «П (5-4)
в граммах
Sa = Sai “F Sai = «а 1^1 «аг^2 (5.5)
где «а, «аь Иа2—суммарное количество адсорбированных компонентов, а
также количество адсорбированных первого и второго компонентов, моль;
«оь «оз — количество первого и второго компонентов, введенных в установку,
моль; пг) — суммарное количество обоих компонентов в газовой фазе после
установления равновесия, моль; g&, gai, gA2 — суммарное количество адсор-
бированных компонентов, а также количество адсорбированных первого и
второго компонентов, г.
После совместного решения этих уравнений получаем следующие рас-
четные формулы
„ «01 + «02 — «Щ — (ga/M2)
"al = 1 - (Mj/Mj (5’6>
„ ”°1 + ~~ «Ч ~ (ga/Ч) ,, _.
«а2- l-^/Afj)
Равновесные концентрации [в % (мол.)] компонентов в газовой фазе
определяются из уравнения материального баланса:
«01-”а1.1()0
1 «тп
сг= ^-^.100
(5-8)
(5.9)
Точность метода тем выше, чем больше разность между мо-
лекулярными массами компонентов смеси. Установки такого
типа достаточно точны, если молекулярные массы исследуемых
веществ разнятся более чем на 30%. Последнее обстоятельст-
во ограничивает сферу их применимости. Циркуляционные
установки как правило отличаются высокой точностью и ис-
пользуются для проведения физико-химических исследований.
Вместе с тем они сложны в изготовлении и эксплуатации.
9—1346
130 5. Избирательность адсорбции
Рис. 5,7. Схема проточной установки для изучения адсорбционного равновесия:
1 — нитрометр; 2, 16 — краны; 3 — теплообменник; 4 — адсорбционная колонка; 5 — тер-
мопара; 6 — переключатель потока; 7 — вспомогательная колонка; 8 — интерферометр;
9— манометр; 10 — автотрансформатор; 11 — смеситель; 12 — реометры для азота; 13 —
пенный расходомер; 14 — реометр для адсорбтива; 15 — дозировочный край; 17 —газо-
метр; 18 —напорная склянка.
Более простыми являются установки проточного типа. Прин-
цип их работы состоит в пропускании через слой сорбента ис-
следуемой смеси известного состава до установления равновесия
с последующим анализом адсорбированной фазы. Схема одного
из возможных вариантов проточной установки показана на
рис. 5,7.
Основной частью установки является вертикальная колонка 4 диаметром
1 см, заполненная раздробленными до фракции 0,5—4,0 мм гранулами ад-
сорбента. Колонка снабжена рубашкой для термостатирования. Термостати-
рующая жидкость (обычно вода) прокачивается через рубашку с помощью
насоса ультратермостата. Сверху на рубашку намотана нихромовая спираль
для контроля за установлением равновесия, который осуществляется с по-
мощью автотрансформатора 10.
Смесь газов, предварительно подогретая (или охлажденная) до темпе-
ратуры опыта в теплообменнике 3, поступает в колонку 4 через верхний
кран 2. Через нижний кран 2 неадсорбированные газы выводятся на анализ
для контроля за установлением равновесия, который осуществляется с по-
мощью газового интерферометра. Через сравнительную камеру интерферо-
метра пропускается некоторая смесь постоянного состава, например, воздух»
азот и т. п. Анализ выходящей из адсорбера смеси может производиться и
другими методами.
Бинарная смесь углеводородов из газометра 17 очищается* и подается
в установку с помощью крана 15. Точный расход смеси устанавливается по
пенному расходомеру 13 и контролируется по реометру 14. Расход азота
моделирующего неадсорбирующиеся компоненты смеси (метан, водород и
т. п.) измеряется и поддерживается постоянным с помощью реометров 12.
Очищающая колонка на рисунке условно не показана.
Расчет адсорбции смеси на основе теории И. Ленгмюра
131
Вспомогательная колонка 7 предназначена для установления заданных рас-
ходов обоих потоков. Нитрометр 1 предназначен для сбора поглощенных га-
зов, десорбция которых происходит при одновременном воздействии тепла
и потока диоксида углерода. Нитрометр заполнен 40%-ным раствором гидр-
оксида калия, который поглощает десорбирующий агент — диоксид углеро-
да. Анализ исходного газа и десорбата производится с помощью хрома-
тографа.
Расчет адсорбции бинарной смеси газов
(паров) на основе теории Ленгмюра
Уравнение изотермы адсорбции по Ленгмюру:
Ьр
а~ат \ + Ьр
(5.10)
где а — количество газа, адсорбированного при давлении р; ат — количест-
во газа, адсорбированного при полном молекулярном покрытии поверхности
адсорбента.
Распространяя это уравнение на случай адсорбции бинарной
смеси, Маркгем и Бентон [155] сделали допущение, что поверх-
ность адсорбента однородна, в случае адсорбции смесей на ней
в пределе может образоваться только один слой молекул, ад-
сорбция компонентов протекает в отсутствие взаимодействия
адсорбируемых молекул друг с другом.
Пусть 01 и 02 — степень заполнения поверхности соответст-
венно первым и вторым компонентами; р\ и р%— парциальные
давления компонентов; (1—01—0г)—доля свободной в задан-
ных условиях температуры и давления поверхности адсорбента;
Ki, ^(2, К.'\ И К'2 — константы скоростей реакций, характерные
для данной температуры. Тогда скорости прямого (адсорбции)
М и обратного (десорбции) v процессов для первого и второго
компонентов составят:
— KiPi (1 01 02) (5.11)
(5.12)
Л42 = К2Р2 (1-01-02) (5-13)
v2 = tf2'02 (5.14)
При равновесии, когда скорости адсорбции и десорбции пер-
вого газа равны
К1Р1(1-01-ег) = К1'01 (5.15)
Принимая и решая уравнение относительно 01;
получим:
bjPi (1 — 02)
04. 1 -р :
(5.16)
132 5. Избирательность адсорбции
Аналогичным образом для второго газа:
а __ ^2^2 О ®1)
1 + 62р2
(5.17)
После подстановки значения 02 из уравнения (5.17) в урав-
нение (5.16) имеем:
»- 1+vf+M- <5JS>
Аналогично для 02
а________Ь2р2
2 1 + A/’i + ^гРг
(5.19)
Но Qi=ai/ami и 02=«г/«т2, где ami и ат2 —предельные величины
сорбции компонентов в монослое.
Тогда
_______атА1Р1
1 1 + Ь1Р1 + ^zPz
am2^zP2
°2 ~ 1 + \P! + hP2
(5.20)
(5.21)
Уравнения (5.20) и (5.21) содержат константы, определен-
ные из изотермы чистых газов. Они указывают, что адсорбция
первого компонента снижается в присутствии второго и наобо-
рот.
При рассмотрении равновесия смеси кислорода и оксида уг-
лерода на силикагеле Маркгем и Бентон отметили хорошее со-
ответствие теории и опыта. В смесях, в которых одним из ком-
понентов являлся диоксид углерода, расхождения были более
значительными. Этот факт можно объяснить наличием значи-
тельных сил взаимодействия между адсорбированными молеку-
лами.
Теоретическое обоснование концепции Маркгема и Бентона
было подвергнуто серьезной критике. В частности, показано
[154], что ami = am2 — ctm. С учетом этой поправки формулы
(5.20) и (5.21) часто применяются для практических расчетов
адсорбции на крупнопористых адсорбентах и обеспечивают тре-
буемую точность.
Расчет адсорбции бинарной смеси паров
на основе теории объемного заполнения микропор
Согласно этой теории, изотермы адсорбции любого i-ro пара в
области высоких степеней заполнения на микропористом адсор-
бенте первого структурного типа описываются уравнением Ду-
Расчет адсорбции смеси на основе теории М. М. Дубинина 133
бинина — Радушкевича:
Fo Г ВТ2 /, , psi°
ai — Vj0 еХР I — (р.0)2 I 1g рл
(5.22)
где v°i, p°i, p°si — соответственно мольный объем, коэффициент подобия и
давление насыщенного пара i-ro компонента, адсорбируемого индивидуально
(на это указывает надстрочечный индекс — нуль).
Для дальнейших рассуждений введем следующие обозначе-
ния: х; — мольная доля i-ro компонента в поверхностном рас-
творе при равновесном парциальном давлении компонента ре,
Vi и р,- — парциальный мольный объем и коэффициент подобия
характеристической кривой для объемного раствора с тем же
составом, т. е. х(; Sa/ = ai2 — суммарная адсорбционная способ-
ность; psi2 = Xipsi + x2ps2 — сумма парциальных мачений давле-
ний насыщенных паров компонентов; vl2=X[Vi+x2v2— сумма
парциальных мольных объемов; Pi2 = *i₽i + x2p2— сумма парци-
альных коэффициентов подобия. Плотность адсорбата можно
определить по методу Николаева и Дубинина, описанному ра-
нее (см. стр. 63).
Тогда для случая адсорбции смеси паров уравнение (5.22)
может быть преобразовано:
VI Г ВУ2 /Psi VI
2^ехр[“ (2WJ (5’23)
Таким образом, для бинарной смеси:
“и = ai + а2 = —~;—=~ ехР
Vl + X2V2
(\ 2‘
4^ + *^)
ВТ2
(Х1Р1 + х2Рг)2
(5.24)
Если принять за стандартное состояние объемный раствор,
равновесный состав паровой фазы над которым имеет тот же
состав, что и над адсорбированной фазой, то легко показать,
что относительные давления обоих компонентов равны друг
другу и относительному давлению смеси паров h:
Pl/Psl = Pz/Psz ~ (Pl ~Ь Р2)/(Л>1 ~Ь Psz) = Plz/Psiz = (5.25)
Тогда уравнение (5.24) преобразуется: ’
Wa Г ВТ2 f 1 V]
“12= ч7ехр I I (5,26)
Или после логарифмирования:
1g (ад + а^) = 1g Го - 0,434ВТ2 [(1/₽12) 1g (1/й)]2
(5.27)
*134 5. Избирательность адсорбции
Рис. 5,8. Кривые адсорбции смесей хлорэтил —
диметиловый эфир (кривая 1) и диэтиловый
эфир — хлороформ (кривая 2) иа активном угле
в координатах уравнений теории объемного за-
полнения микропор (температура 50 °C).
Анализ экспериментальных данных в системах хлорэтил—ди-
этиловый эфир и диэтилэфир — хлороформ показал удовлетво-
рительное совпадение теории и опыта: логарифм заполненного
объема является линейной функцией от величины
(рис. 5,8).
Парциальные давления паров компонентов pi и Рг являются
независимыми переменными и задаются рабочими условиями
процесса. Следовательно, уравнение (5.24) содержит две пере-
менные величины di и Яг, которые необходимо определить. Для
решения поставленной задачи следует использовать дополни-
тельное уравнение, например уравнение Льюиса, которое может
быть представлено в виде
+ <5-28>
Совместным решением уравнений (5.24) и (5.28) вычисляют
значения ai и а2. Описанная выше методика расчета адсорбции
бинарной смеси разработана Берингом, Серпинским, Суриновой
{156] и была успешно использована Гроссманом и Ширмером
для предсказаний поведения пропан-пропиленовой смеси на
цеолите СаА [157].
Приближенное определение
коэффициента разделения
Анализ адсорбционного равновесия большого числа бинарных
смесей углеводородов и других компонентов промышленных га-
зов на цеолитах, силикагелях и активных углях показал, что в
большинстве систем в области высоких степеней заполнения ад-
сорбционной емкости соблюдаются следующие закономерности:
1. Коэффициент разделения сравнительно мало изменяется
(уменьшается) при повышении температуры и давления в си-
стеме.
2. Коэффициент разделения остается приблизительно посто-
янным при изменении соотношения компонентов в исходной
.смеси. '
Приближенное определение коэффициента разделения
135-
рис. 5,9. Зависимость коэффициента разделения
от соотношения коэффициентов подобия.
3. Адсорбция смеси удовлетво-
рительно подчиняется уравнению
Льюиса.
4. Избирательность адсорбции
двух компонентов не изменяется в
присутствии третьего компонента,
К сожалению, многообразие
различных сочетаний компонентов
в бинарных и более сложных сме-
сях, с которыми встречаются инже-
неры при решении конкретных задач газоразделения, не позво-
ляет дать исчерпывающий материал по адсорбционному равно-
весию, необходимый для расчета технологического процесса.
Работы теоретического плана не дают простого метода вычислен
ния меры избирательности адсорбции — коэффициента разделе-
ния. Между тем избирательные свойства адсорбентов проявля-
ются уже в результате сравнения изменений энергии компонен-
тов в процессе адсорбции; Это сравнение в теории объемного
заполнения микропор находит количественную характеристику
в виде коэффициента подобия р. Значения р установлены для
большинства компонентов промышленных газов и приведены
выше (стр. 69).
Таблица 5-1. Опытные и расчетные значения коэффициентов разделения
(давление атмосферное)
Система Адсорбент Темпера- тура опыта, VC Коэффициент раз- деления ' i
опытный расчетный
Этилен — пропилен Цеолит 25 13,0 14,4
Этан — этилен » 25 12,7 10,0
Этилен — диоксид углерода » 25 3,6 2,5
Монооксид углерода — этилен » 25 40,0 48,0
Этилен — ацетилен » 25 13,2 12,5
Пропан — пропилен » 25 17,0 19,8
н-Гексан — н-гептан » 200 2,33 2,12
н-Гептан — н-октан » 200 1,78 1,89
н-Октан — н-нонан » 200 1,50 1,60
Пропан — н-бутан Активный 25 4,37 4,80
уголь
Ацетилен — этилен » 25 1 ,40 1,70
Этилен — этан » 25 1,50 1,53
Пропилен — пропан » 25 1,10 1,13
Этилен — пропилен » 25 12,5 11,0
Метан — этилен > 25 15,4 13,2 •
Д36
5. Избирательность адсорбции
Рис. 5,10. Зависимость коэффициента разделения сме-
сей метана и его гомологов на цеолите СаА от чис-
ла атомов углерода в гомологах.
В связи с этим Кельцевым и Шу-
мяцким [158, 159] были рассмотре-
ны экспериментальные данные по рав-
новесию в 15 бинарных системах угле-
водородов и других газов (паров) на
микропористых адсорбентах. Для этих
смесей характерны особенности адсорбции, отмеченные выше.
Степень заполнения адсорбционного объема в опытах состав-
ляла не менее 30% от предельного заполнения. С применением
метода наименьших средних квадратов установлена зависимость
1g КР = —0,23 + 4,76(1— <р)
(5.29)
где ф=р2/Р1 — отношение коэффициентов подобия* хуже и лучше сорбирую-
щихся компонентов.
Соответствие экспериментальных данных расчету по этой
формуле в интервале значений (1—ф) от 0,07 до 0,40 иллюст-
рирует табл. 5-1, составленная на основе собственных и лите-
ратурных [160—162] данных (в системе более летучий компо-
нент указан первым). Экспериментальные точки, как показыва-
ет рис. 5,9, удовлетворительно ложатся на прямую, при этом
максимальное отклонение расчета от опыта составляет ±30%,
средняя ошибка равна 12%.
Метод определения коэффициента разделения по отношению
коэффициентов подобия не претендует на высокую точность. Од-
нако формула (5.29) пригодна для количественной оценки из-
бирательности адсорбции в технологических расчетах, если от-
сутствуют данные по адсорбционному равновесию смеси.
В соответствии с этой приближенной методикой оценки ко-
эффициента разделения находятся результаты опытов Ширмера
{163], изучившего адсорбцию бинарных смесей, одним из ком-
понентов которых является первый член гомологического ряда
углеводородов (например метан). На рис. 5,10 представлены
зависимости логарифма коэффициента разделения от числа уг-
леродных атомов п в молекуле парафинового углеводорода в си-
стемах метан — парафиновый углеводород на цеолите СаА при
малых степенях заполнения (0<О,1) и разных температурах.
Аналогичные прямолинейные зависимости отмечены на актив-
ном угле при давлениях выше атмосферного, т. е. при степени
* Следует подчеркнуть, что под коэффициентом подобия в данном случае
понимается определяемая из опыта величина, которая зависит от природы
адсорбента.
Теория идеального адсорбционного раствора 137
заполнения, близкой к 1. Логарифм коэффициента разделений
метан — «-бутан в этом случае составляет 2,8, 2,6 и 2,2 соот-
ветственно при 1,05, 1,9 и 3,5 МПа. Линейные зависимости от-
ражают аддитивность сил взаимодействия в этих системах. -
Теория идеального адсорбционного растврра ;
Майерс и Праусниц [164, 165] на основе подхода к термодина-
мике физической адсорбции, описанного в работе Хилла [166],
разработали описанный ниже метод расчета равновесия много-
компонентных систем.
Авторы показали, что адсорбционное равновесие смеси паров на различ-
ных адсорбентах при определенной температуре может быть предсказано с
хорошей точностью, если имеются надежные изотермы адсорбции чистых
компонентов при той же температуре.
Метод Майерса и Праусница дает хорошее соответствие с опытом, если
объемные растворы рассматриваемой смеси идеальны и подчиняются закону
Рауля. Поэтому его называют методом «идеального адсорбционного раствор
ра». К сожалению, этот метод применим только к крупнопористым адсор-
бентам. В некоторых случаях поверхностные растворы являются более иде-
альными, чем соответствующие объемные растворы. Вследствие этого на рас?
творы с небольшим .положительным отклонением от закона Рауля возможно
распространить расчет рассматриваемым методом. К такому случаю, в част-
ности, относится адсорбция смесей бензола и триметилпентана иа графити-
рованной саже [167].
Метод идеального адсорбционного раствора распространен также на
случай вычисления изотермы избыточной адсорбции компонента из бинарных
жидких растворов иа крупнопористом адсорбенте.
Адсорбция компонентов смеси является долей суммарной ад-
сорбции:
а^ Xj^t2 (5.30 )
^2 = %2^12 (5.31)
где Xi и х2—мольные доли компонентов в адсорбированной фазе; а]2—ад-
сорбционная способность по сумме компонентов.
В то же время по уравнению Льюиса:
а^сцо + а^о = I (5.32)
где а°1 и а°2—адсорбционная способность по каждому компоненту при дав-
лении, равном сумме парциальных давлений (определяется по изотермам чи-
стых компонентов).
Подстановка уравнений (5.30) и (5.31) в уравнение (5.32)
дает:
XiM0 + х,/а2° = J/ai2 ( (5.33)
Xi/a-i + (1 — М)/а2° = 1/^12 • (5;34)
Задаваясь разными значениями Xi на основе точек изотерм
чистых компонентов (cz°i и а°2), находят значения а^, а затем
по уравнениям (5.30) и (5.31) значения ai и а2 (рис. 5,11).
138 б. Кинетика адсорбции
Рнс. 5,11. Схема приближенного расчета адсорб-
ционного равновесия бинарных систем.
После этого по изотермам чистых
компонентов определяют давления
паров pi и р2, соответствующие со-
держанию компонентов в адсорби-
рованной фазе di и а2- Мольные до-
ли компонентов в газовой фазе оп-
ределяют по уравнениям:
Vi = Pi/(Pi + Р2) (5.35)
Уа=МР1 + Ра) (5.36)
Из всех значений М выбирают то, которое обеспечивает
наилучшую сходимость di2, вычисленной из (5.34), и суммой
значений о) и а2, определенных по уравнениям (5.30) и (5.31).
Кинетика адсорбции рассматривает вопросы диффузии адсор-
бата' в единичных гранулах адсорбента и скорости отработки
адсорбционной емкости этих гранул.
Экспериментальное изучение
кинетики адсорбции
Кинетику адсорбции в потоке газа изучают, используя единич-
ные гранулы адсорбента и слои толщиной в одну гранулу. На
рнс. 6,1 показана установка для изучения кинетики адсорбции
(десорбции) индивидуальных веществ в потоке газа на единич-
ных гранулах адсорбента. Установка рассчитана на работу в
интервале температур 20—400 °C.
Основной частью установки является измерительная ячейка 4, представ-
ляющая собой сорбционную стеклянную трубку, теплоизолированную в верх-
Экспериментальное изучение кинетики адсорбции
139
Рис. 6,1. Схема установки для изучения кинети-
ки адсорбции из потока газа:
1 — блок очистки; 2 — реометр; 3 — «гусек> с ад-
сорбтивом; 4 — измерительная ячейка с пружин-
ными весами; 5 — печь; 6 — блок регулирования
температуры.
В атмосферу
ней, более широкой части асбестом. Низ-
кую узкую часть сорбционной трубки по-
мещают в электрическую печь 5. Верхняя
часть сорбционной трубки снабжена при-
тертой крышкой с крючком, к которому
подвешивают чувствительный элемент •—
тонкую спираль из предварительно отож-
женной в токе водорода вольфрамовой
или молибденовой проволоки. К нижнему
концу спирали подвешивают кварцевую
штангу, к крючку которой крепят зерна
исследуемого адсорбента. Электропечь 5 за-
ключена в металлический кожух и пред-
ставляет собой керамическую трубку с рав-
номерно намотанной на ее наружной по-
верхности нихромовой проволокой, покры-
той слоем асбестовой теплоизоляции. Внут-
ри печи расположен кварцевый змеевик.
Свободное пространство внутри печи для улучшения теплопередачи заполне-
но шамотной крошкой. Печь имеет по высоте определенную зону постоян-
ной температуры, в которой размещают гранулы адсорбента во время подго-
товки к опыту.
Газ из баллона последовательно проходит блок очистки 1, реометр 2,
«гусек» с адсорбтивом 3 (в стадии адсорбции) и поступает в змеевик, распо-
ложенный внутри печи, где прогревается до температуры опыта, а затем в
нижнюю часть сорбционной трубки. Изменение положения спирали при ад-
сорбции и десорбции фиксируют катетометром через узкое окно в верхней
части сорбционной трубки.
Температуру внутри печи задают и поддерживают с помощью терморе-
гулятора. Датчиком температуры служит термопара, расположенная внутри;
печи. На входе газа в адсорбционную часть колонки имеется карман, в ко-
торый помещена вторая термопара, предназначенная для контроля прогрева
газа. Эта термопара соединена с самопишущим потенциометром. - '
Для проведения опыта одно или несколько зерен исследуемого адсорбен-
та крепят к концу кварцевой штанги, подбирая ее длину таким образом, что-
бы при колебаниях спирали под действием скоростного напора газа навеска
не выходила по вертикали из зоны постоянной температуры. Предваритель-
ную регенерацию адсорбента проводят при задаваемой и автоматически под-
держиваемой регулятором повышенной температуре (различной для разных
адсорбентов) в потоке очищенного инертного газа до стабилизации массы
адсорбента. Затем терморегулятором задают требуемую температуру и отре-
генерированный адсорбент охлаждают в токе газа до этой температуры. ВьГ-
ход установки на режим фиксируют по потенциометру.
После этого проводят процесс адсорбции: газ направляют в термостата-
руемый «гусек», где он, проходя над поверхностью жидкого адсорбтива, на-
сыщается его парами и далее через змеевиковый подогреватель поступает в
адсорбционную колонку. Так как под действием проходящего газа спираль
приходит в колебание, то во время замера газ по байпасу направляют мимо
колонки. Процесс адсорбции проводят до прекращения увеличения массы ад-
сорбента. Сразу после завершения адсорбции проводят десорбцию; газ, ми-
нуя «гусек», через змеевиковый подогреватель направляют в адсорбционную
колонку, и через определенные промежутки времени катетометром замеряют
140
6. Кинетика адсорбции
РИс. 6,2. Схема установки для изучения кинетики адсорбции слоем толщиной в одно
зерно:
1 —баллон ,с газом-носителем; 2 —моностат; 3, 4 — осушительные колонки; 5 — фильтр;
6 —реометр; 7 — термостат; 8 — адсорбционный прибор (а — сатуратор, б — смеситель);
9адсорбционная ячейка.
изменения положения спирали. Время обоих процессов (адсорбции и десорб-
ции) фиксируют по секундомеру. - - .
. „Вместо спирали в установках такого типа можно использовать, торзион-
ное весы, аналитические весы обычного .типа и т. п. ,
Весовые установки используют также для. проведения опы-
тов' по кинетике со слоем адсорбента толщиной в одной зерно.
Применение такого слоя адсорбента позволяет повысить точ-
ность измерений за счет увеличения количества адсорбента.. На
рис. 6,2 представлена схема установки, предназначенной для
изучения кинетики адсорбции в слое толщиной в одно зерно при
комнатных температурах.
. Основной частью этой установки является адсорбционный прибор 8, в
который двумя потоками поступает осушенный газ-носитель. Первый поток
проходит сатуратор, где насыщается парами жидкого адсорбтива, и посту-
пает в смеситель. Высота слоя жидкости в сатураторе, приблизительно рав-
ная 100 мм, обеспечивает практически полное насыщение первого потока.
Второй поток подают непосредственно в смеситель, минуя сатуратор-.
Из смесителя объединенный поток, содержащий пары адсорбируемого
вещества, определенной и постоянной во времени концентрации, поступает в
адсорбционную часть, где на шлифе помещают адсорбционную ячейку 9 с
навеской отрегенерированиого сорбента толщиной в одно зерно. Для равно-
мерного распределения потока по сечению адсорбционной ячейки на перфо-
рированное дно ее помещают частую металлическую сетку.
Адсорбционный сосуд термостатируют с помощью воды, циркулирующей
между ультратермостатом и термостатом 7. Соотношение обоих потоков
устанавливают по реометрам 6 с помощью кранов. Перед разделением об-
Кинетическая кривая
141
щий поток газа-носителя осушают в колонках 3 и 4, заполненных соответст-
венно силикагелем и цеолитом. Если в качестве адсорбента используют цео-
литы, то на выходе из осушающей колонки 4 газ должен иметь остаточное
содержание влаги, соответствующее точке росы не выше минус 65 °C. За-
полненный стеклянной ватой фильтр 5 предназначен для очистки потока от
механических примесей.
Для вычисления относительного давления пара адсорбтива в объединен-
ном потоке используют уравнение
_Р__________К1— /с п
где p/ps — относительное давление паров адсорбтива; р3 — давление насы-
щенного пара адсорбтива при данной температуре; ра — сумма атмосферного
давления и гидравлического сопротивления системы; Vt, V2—объемы газа-
носителя, поступающего на насыщение и разбавление соответственно.
Образец гранулированного адсорбента загружают в предварительно
взвешенную ячейку слоем толщиной в одно зерно; ячейку закрывают проточ-
ными пробками с кранами и помещают в специальную печь для регенерации
адсорбента, которую ведут в токе осушенного азота в течение 3 ч при 350 °C
(в случае использования цеолита). Затем в потоке газа образец охлаждают
до температуры опыта. После этого ячейку закрывают глухими пробками,
взвешивают и помещают в адсорбционный сосуд, который предварительно в
течение часа термостатируют при заданном расходе и составе парогазового
потока.
Адсорбционную ячейку помещают в сосуд в следующем порядке: внача-
ле в парогазовом потоке вынимают нижнюю пробку, опускают ячейку на
шлиф, быстро вынимают верхнюю пробку й включают секундомер.
Перекрывая ячейку, глухими пробками в обратном порядке, е'е периодически
взвешивают и определяют массу адсорбированного вещества, соответствую-
щую данному времени экспозиции. Основной недостаток установок такого ти-
па связан с необходимостью, прерывать опыт для взвешивания --адсорбента,
В эти паузы возможно перераспределение адсорбата-внутри гранулы, пр?
этому периоды, связанные с остановкой опыта, должны быть минимальны.мц.
Кинетическая кривая
На экспериментальных установках получают первичную инфор-
мацию о скорости адсорбции, г. е. о скорости насыщения грану-
лы адсорбатов. Обычно в качестве основной кинетической зави-
симости получают’к и н е т и чб с кую кривую,..!, e.J изменение
величины адсорбццц.во времени. . ;..........
ц = /(т) - ’ ' щ (6.2)
или степени отработки адсорбционной емкости' во времени
(рис. 6,3) ?.
у = а/а„ = <р (т) (6.3)
где а» — равновесная величина адсорбции.
Чтобы провести опыт во внутридиффузионной области и ис-
ключить влияние скорости подвода вещества к внешней поверх-
ности гранулы, предварительно проводят серию опытов при раз-
ных, последовательно увеличивающихся скоростях потока. При
142
6. Кинетика адсорбции
Рис. 6,3. Кинетические кривые адсорбции
н-гептана на цеолитах NaX разного зернения
при 25 °C.
этом в качестве рабочей выбира-
ют такую скорость потока, пре-
вышение которой уже не приво-
дит к изменению формы кинети-
ческой кривой. Обычно влияние
подвода вещества на скорость
отработки адсорбционной емкости гранул практически исклю-
чается при скоростях потока 0,25 м/с и выше [168—170].
Коэффициент диффузии
Проникание адсорбата внутрь зерна адсорбента — процесс
диффузионный и характеризуется коэффициентом диффузии.
Под коэффициентом диффузии* * D понимают количество вещест-
ва, диффундирующего в единицу времени через 1 см2 поверхно-
сти при градиенте концентрации, равном единице.
Зависимость степени отработки емкости от времени описыва-
ется сложными формулами, включающими коэффициент диф-
фузии. Однако при фиксированной степени (например, у = 0,5)
они упрощаются и легко разрешаются** относительно D:
D = (6.4)
где г —радиус гранулы; К — коэффициент, зависящий от формы гранул;
То,s—время полуотработки адсорбционной емкости, т. е. промежуток вре-
мени, прошедший от начала опыта до того момента, когда количество ад-
сорбированного вещества достигнет 50% от равновесной величины адсорбции.
Для шара /(=0,308. Для цилиндра значение К зависит от
отношения длины гранулы I к радиусу г:
l/r 1 2 4 оо
К 0,168 0,318 0,450 0,600
Численное значение коэффициента диффузии при отсутствии
молекулярно-ситовых эффектов часто лежит в интервале (1—
20) • IO-5 см2/с.
* На описанных выше установках определяют так называемый эффективный
коэффициент диффузии, который меньше коэффициента диффузии (коэффици-
ента проницаемости) на сомножитель, приблизительно равный величине, об-
ратной коэффициенту Генри. (Прим, ред.)
** Уравнение (6.4) справедливо для линейной изотермы адсорбции. При
изотермах нелинейных его используют для определения D на небольших
участках этих изотерм. Условия применимости уравнения (6.4) для расчета
эффективного коэффициента диффузии в случае нелинейных изотерм под-
робно рассмотрены в работе Адливанкиной м. А., Бочавера К. 3., Старце-
ва Л. А., ТОХТ, 1979, т. 13, № 3, с. 347—354. (Прим, ред.)
! Коэффициент диффузии !Г43
Значение D, определенное с помощью уравнения (6.4), при-
влекательного своей простотой, является в известной степени
формальной величиной, поскольку при выводе уравнения по-
лагали, что пористая среда однородна для диффундиру-
ющего газа. Между тем, гранулы любого промышленного ад-
сорбента в диффузионном отношении не являются гомогенны-
ми: в них имеются разное по характеру пористые структуры,
определяющие протекание процесса адсорбции по сложному ме-
ханизму.
Исследовалась [171] кцнётика адсорбции углеводородов на
гранулированных цеолитах' в адсорбционной ячейке слоем тол-
щиной в одно зерно. Зависимость между временем отработки
емкости для различных фиксированных степеней отработки у
и диаметром гранул аппроксимировалась степенным уравнение
ем вида
(6.5)
где xi — время от начала опыта до достижения степени отработки у,; d —
диаметр гранулы; m — константа.
Полученные в опытах значения пг для каждого углеводоро-
да находились в интервале от 1,4 до 1,6. Это означает, что обе-
щая скорость адсорбции на цеолите любого зернения опреде-
ляется диффузией как в первичной (микропоры), так и во вто-
ричной (мезо- и макропоры) структурах, и рассматривать цео-
литы как гомогенную структуру нельзя.
Аналогичный результат был получен и для гранул про-
мышленных углеродных адсорбентов. С точки зрения массопе-
редачи активные угли, подобно цеолитам, также следует рас-
сматривать как бипористую структуру [172, 173].
В работе [174] применяли тонкопористые активные угли сульфатно-ка-
лиевой активации, имеющие близкую пористую структуру, но различающие-,
ся диаметром гранул. Их структурные характеристики (см3/г): Vs=0,38—
0,41; Ума = 0,17—0,18; Уме = 0,03—0,04; VMH=0,17—0,19; №о=О,18—0,21; струк-
турная константа В прн стандартном паре—бензоле составляла (0,74—
0,80)-10-6 град-2. Исследование кинетики адсорбции паров бензола на еди-
ничных зернах проводили на проточной динамической установке с весами
Мак-Бена. Гранулы адсорбентов обрабатывали таким образом, чтобы сохра-
нялось отношение Z/d=2 (Z— длина, d— диаметр гранулы). Диаметр грану-
лы у четырех образцов составлял 1,00, 1,30, 1,88 и 3,06 мм. Концентрация
паров бензола в потоке газа-носителя (азота) равнялась 1% (об.). Скорость
газового потока составляла 0,33 м/с, при этом скорость отработки адсорб-
ционной емкости лимитировалась диффузией адсорбата в порах адсорбента,
а внешнедиффузионное торможение было исключено.
В качестве примера первичной экспериментальной информации иа
рис. 6,4 представлены кинетические кривые адсорбции бензола при 100 °C
на исследованных образцах. Время достижения степени отработки у=0,5 во
всем исследованном интервале температур для образца диаметром 3,06 мм
в среднем в 3,6 раза больше, чем для образца диаметром 1,00 мм, в то вре-
144
6. Кинетика адсорбции
с, мин
Рис. 6,4. Кинетические кривые "адсорбции бензола
при 100 °C на активных углях разного зернения
(£<>=1% об.), .
мя как отношение их диаметров составляет
3,06. Таким образом, в данном случае время
полуотработки и диаметр гранул были связа-
ны почти линейной зависимостью. Этот вывод
находится в соответствии с результатами ра-
боты [173].
При идентичности пористых струк-
тур гранул разного зернения «массо-
проводность» должна быть постоянной
и коэффициент диффузии должен
являться константой. Введем поэтому в формулу (6.4) по-
казатель пг', учитывающий негомогенность структуры. Тогда ис-
правленный коэффициент диффузии будет равен
р. =Д-г(2-т')/яТо5
(6.6)
В рассмотренном выше примере среднее значение коэффи-
циента пг' = 0,9, или т = 2—т'=1,1. Обработка эксперименталь-
ного материала по предложенному методу позволяет обсудить
зависимость коэффициента диффузии от температуры (см.
табл. 6-1). С увеличением температуры от 25 до 150°С коэффи-
циент диффузии D* увеличивается приблизительно в 2 раза. По
углу наклона прямых lg D* = f (1/Т) можно рассчитать энергии
активации (Еа) адсорбционного процесса. Для всех образцов
углей в рассмотренном примере Еа имела близкие значения и
составляла в среднем 8,4 кДж/моль.
В последующем [175] на примере системы п-ксилол —
азот — активный уголь было показано, что в транспортных по-
рах значение коэффициента диффузии близко к значению ко-
эффициента нормальной диффузии органического вещества в
газе-разбавителе.
Таблица 6-1. Исправленные коэффициенты диффузии бензола
в углях (10s D*, см2/с)
1, ’С Г, мм
0,5 0,65 0,94 1,53
25 5,67 5,90 5,35 6,50
50 7,33 7,36 6,25 7,24
100 , 10,10 10,30 9,75 10,00
150 11,55 11,41 11,59 12,27 '
Виды переноса в пористом теле 145
Виды переноса в пористом теле
Гранулированные цеолиты являются бидисперсными адсорбен-
тами. Их первая-структура — микропоры кристаллита, вто-
рая — мезопоры и макропоры, образованные при введении свя-
зующего и являющиеся транспортными каналами в грануле*.
Характер зависимости времени отработки адсорбционной ем-
кости от размера гранул цеолита резко меняется, если адсорби-
руются молекулы, близкие по размеру к размерам микропор.
Это отчетливо видно из сопоставления кривых, отражающих
кинетику адсорбции н-гептана на цеолитах NaX и СаА (рис.
6,5). Время достижения любой заданной степени отработки ад-
сорбционной емкости для гранул цеолитов СаА диаметром 2
и 3 мм практически одинаково. Некоторый рост времени отра-
ботки наблюдается лишь при переходе от трех- к четырехмил-
лиметровым гранулам. Следовательно, процесс переноса при
поглощении нормальных парафинов цеолитом СаА лимитйрует-
ся преимущественно диффузией в кристаллах цеолита. Следует
отметить, что в этом случае замена азота в качестве газа-носи-
теля на гелий не приводит к изменению скорости адсорбции:
диффузия в кристаллах слабо зависит от типа газа-носителя.
Измерение относительных коэффициентов (отношение коэф-
фициента диффузии в среде азота к коэффициенту диффузии в
среде гелия) позволило определить соотношение сопротивлений
в первичной и вторичной пористости при адсорбции углеводоро-
дов цеолитами типа X [171]. Эксперимент показал, что относи-
тельный коэффициент диффузии
^'n2 = А^/^Не (6.7>
для гранул цеолита NaX в зависимости от молекулярной мас-
сы- углеводорода изменяется от 0,67 до 0,69. Очевидно, что в
случае, если бы скорость процесса лимитировалась только диф-
фузией во вторичной пористой структуре, относительный коэф-
фициент диффузии при адсорбции должен был бы быть равен
отношению коэффициентов нормальной диффузии в азоте и в
гелии, которое для н-гептана при 25 °C равно 0,28.
Рассматривая адсорбцию на цеолитах как процесс, проте-
кающий последовательно в зонах первичной и вторичной пори-
* Основные итоги изучения кинетики адсорбции в кристаллах цеолита при-
веденье в обзоре Ruthven D. М. Separ. and Purif. Meth., 1976, v. 5, № 2,
p, 189—246. Основы количественной теорий кйнетики ' адсорбции в бипори-
стых средах вообще и в цеолитах в частности разработаны лишь в послед-
ние годы. Результаты этих работ обсуждены в статьях обзорного характера;.
Dubinin М. М. Pure and Appt. Chem., 1976, v. 48, № 4, p. 407—414; Золота-
рев П. П. В кн.: Физическая адсдрбция в микропористых адсорбентах. Расш'.
тез. докл. 5-й Всес. конф, по теор. вопр. адсорбции. Вып. 2, М., 1979, с. 23—
31; (Прим, ред.) г ; " >.
10—1346
146 6. Кинетика адсорбции
Рис. 6,5. Зависимость времени отработки адсорбционной емкости цеолитов NaX (а) и
СаА (б) от диаметра гранул (адсорбция н-гептана при 25 °C).
стости, общее сопротивление диффузии в соответствии с [176]
можно представить следующим образом:
Fn2 = F'n2 + IF'N2 = (6.8)
^Не = «7'не+В7"не (6.9)
где Wn2, IF"n2, W"n2 — общее диффузионное сопротивление, а также сопро-
тивление первичной и вторичной пористых систем соответственно при ад-
сорбции из потока азота; И^не, №'не, И7" не— то же для случая адсорбции
из потока гелия.
Так как сопротивление первичной пористости не зависит от
природы газа-носителя, то U7/N2 = U7/He. Допустим, что перенос во
вторичной пористости протекает по механизму нормальной диф-
фузии 117"Не/Й"'ма =0,28, а отношение общих сопротивлений по
величине обратно пропорционально относительному коэффици-
енту диффузии углеводорода или другого адсорбтива №не/№\2=
=Z)'n2. Тогда после подстановки получаем уравнения;
O'N2rN2 = ^'N2 + °>28r4 (6-10)
Гне = 1Г'не + -0^8- <6 •11 >
Решение системы уравнений (6.8) и (6.10) позволило на ос-
нове экспериментальных данных определить относительные со-
противления первичных и вторичных пор при адсорбции угле-
водородов из потока азота An2= Wz'n8/Wn2 и Bn2 = а
решение системы уравнений (6.9) и (6.11)—относительные со-
противления при адсорбции из потока гелия ЛНе= ^'неЖне и
Вне= W"He/WHe. Результаты расчетов для гранул цеолита NaX
разного диаметра ' с применением различных газов-носителей
представлены в табл. 6-2. Как видно из таблицы, с увеличением
диаметра гранул возрастает роль крупных (транспортных) пор.
Виды переноса в пористом теле
147
Таблица 6-2. Вклад первичной и вторичной пористости
в общее диффузионное сопротивление
Диаметр гранул, мм «-Пентан н-Гептан
по азоту по гелню по азоту по гелию
лы2 SN2 ЛНе вНе лы2 Bn2 ЛНе ВНе
1,1 91,7 8,3 97,5 2,5 87,6 12,4 95,3 4,7
2,1 71,8 28,2 90,1 9,9 67,8 32,2 88,0 12,0
3,2 54,2 45,8 80,2 19,8 55,5 45,5 81,6 18,4
3,9 44,9 55,1 74,3 25,7 45,0 55,0 74,5 25,5
5,0 35,4 64,6 66,1 33,9 36,6 63,4 67,3 32,7
В предельном случае для гранул диаметром 5 мм вклад круп-
ных пор в общее сопротивление составляет 70%.
В том случае, когда основное диффузионное сопротивление
сосредоточено в микропорах, большое влияние на кинетику ад-
сорбции оказывает диаметр кристаллита. На рис. 6,6 представ-
лены кинетические кривые адсорбции н-гептана на гранулах
цеолита СаА, отпрессованных из кристаллов разного размера
[177]. Скорость процесса, как видно из рисунка, находится в
обратной зависимости от размера кристаллов. Время полуотра-
ботки адсорбционной емкости гранул цеолита СаА с кристалла-
ми размером 3,5—4,0 мкм составляет 30 мин, а с кристаллами
размером 8—10 мкм — 80 мин.
Итак, промышленные адсорбенты, как указывалось выше,
отличаются разнообразием пористой структуры, что определя-
ет особенности переноса вещества в грануле в целом.
В крупных порах, когда диаметр пор больше средней длины
свободного пробега молекул адсорбтива, преимущественным ви-
дом переноса является нормальная, или объемная
диффузия.
Скорость нормальной диффузии определяется числом столк-
новений молекул газа друг с другом. Коэффициент диффузии*
в этом случае зависит от средней тепловой скорости молекул и
и средней длины свободного пробега молекул Л:
D = l/3uX (6.12)
Величины и и %, а следовательно, и коэффициент диффузии
возрастают при повышении температуры. Температурная зави-
симость коэффициента диффузии такова:
(6.13)
* В уравнениях (6.12)—(6.16) приведены зависимости для коэффициента
проницаемости. Температурная зависимость коэффициента диффузии в том
смысле, в котором термин использовался ранее, более сложна, так как вхо-
дящий в нее коэффициент Генри также зависит от температуры. (Прим, ред.)
10’
148 6. Кинетика адсорбции
1,0
0,5
Рис. 6,6. Кинетические кривые адсорбции к-гек*’
тана (с0=1О мг/л, /=25 °C) из потока азота и а,
прессованных таблетках цеолита СаА с кристал-
лами разных размеров.
0
т.мин
При малом размере пор, когда длина свободного пробега мо-
лекул много больше радиуса пор, фактором, определяющим
скорость диффузии, становится частота соударений со стенка-
ми пор. Такая диффузия называется молекулярной, или
кнудсеновской. При столкновении адсорбирующихся мо-
лекул с поверхностью они некоторый интервал времени фикси-
руются на активных центрах адсорбента и только после этого,
благодаря тепловому движению, удаляются в газовую фазу.
Коэффициент молекулярной диффузии определяется средней
скоростью движения молекул и и диаметром поры da:
D = l/3udn (6.14)
В то же время
и = К 8RT/nM (6.15)
Следовательно, коэффициент молекулярной диффузии и тем-
пература связаны зависимостью -
D~T9,* , (6.16)
Если поры по размерам соизмеримы с поглощаемыми моле-
кулами, адсорбционный процесс приобретает активирован-
ный характер. По аналогии с представлениями.. Аррениуса,
развитыми для химической реакции, при активированной. ад-
сорбции не все молекулы могут проникнуть в поры' и быть-там
поглощенными, а лишь те, которые обладают некоторым избы-
точным запасом энергии. Этот избыток называют энергией
активации.
Коэффициент диффузии связан с энергией активации Е& сле-
дующей зависимостью [178]:
' D = Do exp (-EJRT) (6.17)
Величина предэкспоненциального множителя Do зависит от
природы как адсорбента, так и адсорбтива. При сорбции азота
и метана на порошке цеолита NaA значения Do составляют со-
ответственно 2,3-10-9 и 58-10'9 см2/с [109]. Предадсорбция по-
лярных веществ на цеолите приводит к частичной блокировке
входных окон и, как следствие, к понижению скорости сорбции
и уменьшению величины Do [179, 180].
Виды переноса в пористом теле
149
Чем выше энергия активации, тем в большей степени изме-
няется скорость адсорбции при изменении температуры. В за-
висимости от энергии активации Еа увеличение коэффициента
диффузии при повышении температуры с 0 до 100°С происходит
в следующее число раз п:
Ег, кДж/моль 4 12 25 38 50 63
п 1,6 4,5 20 90 420 1800
Баррер и Брук [181], исследуя кинетику сорбции пропана,
л-бутана, дихлорметана и диметиламина на кристаллическом
порошке шабазита, подтвердили взаимосвязь между энергией
.активации и температурным коэффициентом кинетики адсорб-
ции. Так, для пропана энергия активации составляет
13 кДж/моль и коэффициент диффузии относительно мало из-
меняется с температурой. Для диметиламина энергия активации
равна 80 кДж/моль и коэффициент диффузии при изменении
температуры от 50 до 100°C возрастает почти в 50 раз.
Рассмотренную закономерность используют для вычисления
энергии активации на основе экспериментальных данных [182].
Для этого получают кинетические кривые адсорбции конкретно-
го вещества при двух или более температурах Т\ и Т2. Соответ-
ствующие этим температурам коэффициенты диффузии могут
быть представлены уравнениями:
P1 = Doexp(-£ai//?ri) . (6.18)
D2 = Doexp (~Ea2/RT2) (6.19)
Энергия активации практически постоянна, если интервал
(исследуемых температур 1\—Т2 не слишком вёЛиК Следова-
тельно -
Eai ~ Еа2 = Еа (6.20)
Отсюда
2% Г Еа [ 1 1 \]
Dt =ехр[~ # (л ~ та)] > (6-21)
. 01 Еа / 1 1 \ •
In Т1 — ) (6.22)
ЛТ. D,
Еа =2,3£-^-27jr- lg (6.23)
Соотношение коэффициентов диффузии может быть замене-
но на соотношение времен отработки адсорбционной емкости до
любой степени, например до 50%:
Dr Т2 (О, в)
Oj т1(0,В)
(6-24)
150
6. Кинетика адсорбции
Рис. 6,7. Зависимость энергии активации £а при
адсорбции воды цеолитами от диаметра входного
окна da.
Зависимость энергии активации
от соотношения определяющих раз-
меров пор и адсорбируемых моле-
кул отчетливо прослежена на при-
мере адсорбции воды в ряду синте-
тических цеолитов с изменяющимся
размером входного окна [183]. Из-
менение энергии активации в зави-
симости от диаметра входного окна цеолитов показано на рис.
6,7 и ниже:
Тип цеолита КА NaA СаА СаХ NaX NaY
do, нм 0,3 0,4 0,5 0,8 0,9 0,9
Еа, кДж/молъ 83,8 32,3 20,9 13,9 10,9 10,9
Энергия активации увеличивается по мере приближения
диаметра входного окна к критическому диаметру молекул ад-
сорбтива. Резкий рост энергии активации наблюдается при пе-
реходе от цеолита NaA к цеолиту КА, т. е. когда определяющий
размер пор и критический диаметр молекул (0,27 нм) стано-
вятся примерно равными величинами. Таким образом, энергию
активации можно рассматривать как молекулярно-ситовую ха-
рактеристику цеолитов [184].
Зависимость энергии активации Еа от диаметра входного ок-
на описывается уравнением
Еа Eq ехр (AdKP/d0) (6.25)
где Ео — предэкспоиеициальный множитель; А—постоянная (для воды А =
= 1,5); dKP — критический диаметр молекулы адсорбата (для воды чкр=
=0,27 нм).
Энергия активации зависит также от радиуса и размера ка-
тиона, входящего в состав цеолита. Баррером [108] она была
определена на ионообменных формах морденита. Энергия акти-
вации возрастала при переходе от Li- к iNa-форме, а затем при
переходе к К- и МН4-форме уменьшалась.
Интенсивность переноса вещества в гранулах промышлен-
ных адсорбентов усиливается благодаря миграции молекул по
поверхности пор. Этот вид переноса получил название п о-
верхностной диффузии. Движение молекул по поверх-
ности происходит скачкообразно. Если длину скачка молекулы
обозначить через А, а время пребывания молекулы в адсорби-
рованном состоянии — через т, то коэффициент поверхностной
диффузии определится из соотношения
D = Д2/4т (6.26)
Виды переноса, в пористом теле
451
В то же'время - >
т = тоехр(ЕаЖ) (6.27)
где То — период колебания атомов в структуре адсорбента, приблизительно
равный 1 10-13 с.
Тогда . • . ..........
Д«
D = ТЕ7 ехР (~£а/7?Л = Do ехр (-EJRT) (6.28)
где Do — коэффициент диффузии двухмерного газа на идеально однород-
ной поверхности, если Т—►<» или Еа = 0.
Коэффициент поверхностной диффузии связан с температу-
рой следующей зависимостью:
D~T«exp(— EJRT) (6.29)
где п может изменяться от 0,5 до 1,5.
При повышении температуры адсорбционная способность и
градиент концентрации в адсорбированной фазе уменьшаются.
Поэтому роль поверхностной диффузии в общей скорости пере-
носа вещества падает.
Интенсификация процесса поглощения примеси за счет по-
верхностной диффузии особенно отчетливо проявляется при со-
поставлении скорости адсорбции таких веществ, как вода, цео-
литами со связующим и без связующего. В последнем случае
вторичная пористая структура составлена сростками кристал-
лов цеолита и транспорт адсорбата происходит не только в объ-
еме пор адсорбента, но и по поверхности этой силикатной струк-
туры, что значительно убыстряет процесс поглощения-в целом.
Поэтому скорость поглощения воды цеолитом без связующего
выше, чем у цеолитов со связующим, в которых имеются лишь
точечные контакты между отдельными частицами. В некоторых
случаях это свойство цеолитов без связующего может быть оп-
ределяющим при выборе адсорбента для решения конкретной
задачи.
152
7. Кинетика десорбции
7
КИНЕТИКА ДЕСОРБЦИИ
Кинетика десорбции из зерна
адсорбентов
Микропористые адсорбенты, обладающие уникальными адсорб-
ционными свойствами, отличаются трудностью удаления из них
адсорбата. Для эффективного проведения десорбции из микро-
пористых адсорбентов необходимы более высокие, чем обычно,,
температуры, что повышает требования к выбору температур-
ного режима. Ошибки в выборе оптимальных температур де-
сорбции приводят к уменьшению адсорбционной емкости либо
за счет неполноты десорбции (если температура десорбции бы-
ла слишком низка), либо вследствие разрушения структуры ад-
сорбента (если температура десорбции была чрезмерно высо-
ка). В ряде случаев вопрос о внедрении новых адсорбционных
процессов в промышленность положительно решают оригиналь-
ные методы, разработанные применительно к стадии десорбции.
Иногда стадию подготовки адсорбента (в основном, стадию де-
сорбции) называют регенерацией.
Для установления оптимального температурного режима
стадии десорбции в зависимости от структуры и молекулярной
массы адсорбата Афанасьевым [139] была исследована кине-
тика десорбции нормальных парафиновых углеводородов и бен-
зола из зерна микропористых адсорбентов. Чтобы исключить
влияние внешнедиффузионного фактора, основная серия опытов
была проведена в вакууме, что отвечает одной из промышлен-
ных схем депарафинизации нефтяных фракций.
В качестве адсорбентов были использованы синтетические цеолиты СаА,
NaX, рекуперационный активный уголь и мелкопористый силикагель КСМ.
Исследование кинетики десорбции проводили на вакуумной установке с
кварцевыми весами. Подготовка образца адсорбента заключалась в вакууми-
ровании (остаточное давление »1.3-10-3 Па, или 110“! мм рт. ст.) при
350°C до постоянной массы. Подготовленный образец насыщали парами
углеводорода при температуре опыта, после чего адсорбционную ячейку с об-
разцом соединяли с вакуумной системой, в которой поддерживали давление
Кинетика десорбции из зерна адсорбентов
153
Рис. 7,1. Зависимость степени насыщенности адсор-
бента v нормальными парафиновыми углеводорода-
ми от времени экспозиции с вакуумом г — кинетиче-
ские кривые десорбции углеводородов при 150 "С на
цеолита NaX (а) и СаА (б):
1 — «-нонан; 2 — «-октан; 3 — н-гептан; 4 — «-гексан.
около 1 Па (1-10-2 мм рт. ст.). Изменение
массы гранул фиксировали катетометром че-
рез каждые 1—10 мин.
На рис. 7,1 представлены кинетические
кривые десорбции нормальных парафиновых
углеводородов С6—С9 из гранул цеолитов
NaX и СаА при 150 °C в координатах степень
насыщенности адсорбента у — время т.
При выпуклой изотерме адсорбции
скорость десорбции быстро убывает со
временем. Спустя определенное время
кинетическая кривая десорбции стано-
вится почти параллельной оси абсцисс.
Десорбция как бы прекращается, что
может быть интерпретировано как на-
личие удерживающей способности у
адсорбентов. Она может быть выраже-
на как остаточная насыщенность ад-
сорбента к моменту времени, когда
скорость десорбции становится меньше
чувствительности метода [185, 186].
Удерживающую способность ис-
пользуют при разработке рациональ-
ной структуры адсорбента и оптими-
зации режима работы адсорбционных
установок. Она зависит от свойств как адсорбента, так и адсор-
бата. С удлинением цепи углеводородов увеличивается энергия
связи молекул с поверхностью адсорбента, уменьшается
давление насыщенных паров углеводорода при данной темпера-
туре, вследствие чего удерживающая способность возрастает.
Так, при 150°С и времени экспозиции в вакууме 120 мин удер-
живающая способность на цеолите СаА соответствует остаточ-
ной адсорбции, выраженной в до- .....
лях от максимальной адсорбции для
углеводородов: н-гексан — 0,20; н-
гептан — 0,40; н-октан — 0,60; н-но-
нан — 0,77.
Тимофеевым [168] показано, что
удерживающая способность зависит
от микропористости адсорбентов:
Рис. 7,2. Кинетические кривые десорбции н-пен-
таиа в вакууме из гранул цеолитов NaX разного
размера.
154 7. Кинетика десорбции
остаточное насыщение адсорбента при десорбции, характеризу-
ющее удерживающую способность, тем больше, чем больше мик-
ропор и чем меньше их размер. В теории объемного заполнения
микропор эти два фактора характеризуются предельным адсорб-
ционным объемом Жо и структурной константой В.
, Процесс десорбции молекул из таких бидисперсщых (пер-
вичная и вторичная пористость) адсорбентов, какими являются
гранулированные цеолиты, включает следующие стадии:
; отрыв молекул адсорбата от активных центров поверхности
адсорбента; ,
диффузия молекул в первичной пористой структуре;
диффузия во вторичных порах;
: испарение с наружной поверхности гранул;
удаление десорбата из газовой фазы.
Очевидно, что в условиях десорбции в вакууме последние
две стадии протекают настолько быстро, что не могут лимити-
ровать общую скорость процесса.
Оценка вклада скорости диффузии во вторичной пористой
структуре в общую скорость процесса может быть проведена
путем сравнения скорости десорбции углеводородов из гранул
разного размера при прочих равных условиях. На рис. 7,2
представлены кинетические кривые десорбции «-пентана при
двух температурах (30 и 60 °C) из гранул цеолита NaX с раз-
мерами d = / = 4 мм и d = / = 2 мм. Из сопоставления этих кривых
т^ожно судить о величине составляющей общей скорости про-
цесса, приходящейся на диффузию во вторичных порах. Некото-
рое различие в кинетике десорбции из гранул разного размера
обнаруживается только в начальный период, соответствующий
удалению адсорбата из объема адсорбционных полостей. Прак-
тически полное совпадение кинетических кривых на втором
участке свидетельствует о том, что в условиях вакуума диффу-
зия молекул во вторичных порах не лимитирует общую скорость
десорбции.
Таким образом, в отличие от прямого процесса (адсорбция),
в котором определяющим скорость адсорбции фактором явля-
ются диффузионные процессы, при обратном процессе (десорб-
ция) решающее значение может иметь скорость отрыва
молекул от поверхности адсорбента. Как и в пря-
мом процессе, определенное влияние на скорость десорбции про-
должает оказывать диффузионное сопротивление в микропорах
(первичная пористость).
Относительный метод расчета кинетики десорбции
Если принять, что после удаления адсорбата из объема адсорб-
ционных полостей оставшиеся молекулы образуют мономоле-
кулярный слой в отсутствие взаимодействия между адсорбиро-
Кинетика десорбции из зерна адсорбентов
155
ванными молекулами, т. е. в соответствии с теорией Ленгмюра,
то скорость десорбции может быть определена из уравнения
dO/dr = —/гг0 -f- (1 — 0) (7.1)
где 0 — степень заполнения поверхности; р — давление в системе; ki н k2 —
константы скорости прямого и обратного процессов; т — время.
Для условий десорбции в вакууме р — 0 и тогда
d0/dr = — fet0 (7.2)
или, после интегрирования при начальном условии т = 0, О = Оо
In у = —fe/r (7.3)
где
У = 0т/0о = «т/й'о
В соответствии с теорией, коэффициент ki пропорционален
ехр(—E^RT}, где Ек— энергия активации процесса десорбции.
Так как
£a = Q + £a (7.4)
(где Q — теплота адсорбции; Еа — энергия активации процес-
са адсорбции), то
fei = k0 ехр [—(Q + Ea)/RT] (7 .5)
Предэкспоненциальный множитель в уравнении (7.5) зави-
сит от среднего элементарного перемещения и периода колеба-
ний атомов в решетке кристалла, т. е. связан только со свой-
ствами адсорбента. С другой стороны, энергия активации ад-
сорбционного процесса является долей теплоты адсорбции*;
£a = cQ (7.6)
Коэффициент с в случае цеолитов зависит от соотношения
между диаметром входного окна в адсорбционные полости и
критическим диаметром диффундирующей молекулы. Так. как
критический диаметр в ряду нормальных парафиновых угле-
водородов, начиная с пропана, постоянен (0,49 нм), величина с
имеет одинаковое значение для всех членов гомологического ря-
да. Поэтому уравнение (7.5) можно преобразовать;
/ Q + £а \ / Q (с + 1) \
lgy = — /г0 ехр (-—1т=—йоехр( ——jy—/Т =
/ Q \
= — ехр I cr у-1 т t (7.7)
где Ci = —(с + 1)/??.
* В общетеоретическом плане сопоставление Еа, характеризующей «долю ак-
тивных молекул», с термодинамическим параметром Q является некорректным.
Но следует учитывать, что при адсорбции это величины одного класса, так
как миграция молекул по структуре связана с отрывом их от центров поверх-
ности, который статистически и характеризуется .как теплота адсорбции.
(Прим, ред.)
156
7. Кинетика десорбции
Обработка экспериментального материала по уравнению
(7.7) выявила отклонения от предсказываемой теорией пропор-
циональности между 1g у и f(-t) в начальных участках кинети-
ческих кривых, соответствующих удалению адсорбата из объ-
ема адсорбционных полостей. В связи с этим непосредственное
использование уравнения (7.7) для определения степени десорб-
ции углеводородов затруднительно.
Однако из уравнения (7.7) следует, что десорбция двух уг-
леводородов гомологического ряда будет описываться единой
кинетической кривой, если соблюдается следующее условие:
П/Гст = Qi/QCT (7.8)
где 7; и 7СТ— температуры десорбции исследуемого вещества и вещества,
принятого в качестве стандартного; Q; и QCT — теплоты адсорбции этих же
веществ.
Соотношение (7.8) позволило разработать метод относитель-
ного расчета десорбционных кривых нормальных углеводородов
парафинового ряда по сетке десорбционных кривых стандарт-
ного вещества [187]. Расчет заключается в том, что по заданной
температуре десорбции искомого углеводорода 7\, используя
соотношение (7.8) и известные значения теплот адсорбции (при
одинаковых степенях заполнения), находят температуру стан-
дартного углеводорода 7СТ, при которой десорбционные кривые
этих углеводородов накладываются друг на друга. После этого
по сетке определяют степень десорбции искомого углеводорода
при заданном времени экспозиции в вакууме.
Рис. 7,3. Кинетические кривые десорбции я-пеитана в вакууме из гранул цеолитов NaX
(а) и СаА (б).
Кинетика десорбции из зерна адсорбентов
157
Таблица 7-1. Температура совмещения кинетических кривых десорбции
нормальных парафиновых углеводородов
Углеводо-
род
Температура совмещения, °C
С3Н8 —101 —68 —57 —53 —49 —46 —42 —39 —35 —28 —17 4-1
w-C4Hio —91 —65 —47 —32 —27 —23 —15 —10 — 7 —2 4-3 4-7
л-С5Н12 —63 —33 —13 4-5 4-Ю 4-15 4-25 4-30 4-35 4-40 +46 +50
н-С6Н14 —39 —5 +17 +38 +43 +49 +60 4-65 +71 +76 +82 +88
н-С7Н16 —15 +22 +47 +70 +75 +82 +94 +100 +106 +112 +119 +125
л-С8Н18 +9 +49 +75 +89+107 + 113 + 126+133+139+147+153 +150
н-С9Н20 4-33 +79+108+133+138+148 + 163+171+177+186+192 +200
н-С10Н22 +51 +974-127+ 155 +162 +171+187+193+202+208+217+224
я-С„Н24 +63+113+145+174+182 +191 +207+213+221 +231 +238 +247
В связи с тем, что в гомологическом ряду нормальных пара-
финовых углеводородов отношение теплоты адсорбции к тепло-
те конденсации для всех углеводородов ряда приблизительно
постоянно [188], уравнение (7.8) можно представить в виде
Т'г/Т’ст = Act (7-9)
где Л; и Лет — мольные теплоты конденсации соответственно исследуемого и
стандартного веществ.
В качестве стандартного вещества может быть использован,
например, н-пентан. Для него по экспериментальным данным
построены сетки десорбционных кривых через каждые 5 °C в ин-
тервале температур от 0 до 120 °C (рис. 7,3).
Совмещение кинетических кривых любого нормального па-
рафинового углеводорода с соответствующими кривыми стан-
дартного углеводорода осуществляется с использованием дан-
ных табл. 7-1, составленной на основе справочных значений
теплот конденсации при нормальном давлении и температуре
кипения конкретных углеводородов. Из таблицы видно, напри-
мер, что кинетические кривые десорбции углеводородов совме-
щаются с кривой десорбции н-пентана, соответствующей 30°Сг
при следующих температурах:
Углеводород С8 С4 С8 С7— С8 С9 С70 Си--
Температура, °C —39 —10 65 100 133 171 193 213
Как установлено в многочисленных опытах, максимальные
расхождения между экспериментальными и расчетными степе-
нями заполнения соответствуют относительной ошибке, не
превышающей 5%; это приблизительно равно изменению темпе-
ратуры опыта на 3—5 °C, что находится в пределах точности
термостатирования зерна адсорбента.
Хотя относительный метод расчета вначале был разработан
для случая десорбции нормальных парафиновых углеводородов
158
7. Кинетика десорбции
из цеолитов, его оказалось возможным распространить на иные
микропористые адсорбенты. Действительно, структура активных
углей и силикагелей включает поры различных размеров и
формы, но удаление адсорбата из каждой поры происходит по
тем же законам, что и в случае однородномикропористых ад-
сорбентов — цеолитов.
В работе [139] приведена сетка кинетических кривых де-
сорбции стандартного вещества — н-пентана из зерна рекупера-
ционного угля АР. Характер кинетических кривых десорбции из
активного угля отличается от характера кинетических кривых
из цеолитов. Это отличие в основном заключается в более кру-
той ниспадающей ветви в начальный период десорбции, что свя-
зано с удалением вещества из мезопор и наиболее крупных мик-
ропор. Об удовлетворительном совмещении кинетических кри-
вых десорбции различных парафиновых углеводородов из угля
свидетельствует рис. 7,4, на котором с использованием табл. 7-1
на сетку н-пентана нанесены точки экспериментальных кривых
других углеводородов. Аналогичный вид имеет сетка кинетиче-
ских кривых десорбции стандартного вещества из мелкопори-
стого силикагеля.
При десорбции углеводородов другого класса, например аро-
матических углеводородов из зерна активного угля, кинетиче-
ские кривые десорбции каждого члена ароматического ряда
практически совмещаются с кинетическими кривыми нормаль-
ного парафина с близкой молекулярной массой: бензола с н-гек-
•саном, толуола с н-гептаном и т. д. Этот факт находится в соот-
ветствии с близостью теплот адсорбции у этих адсорбтивов на
активных углях.
В большинстве реальных технологических процессов адсор-
бат подвергается десорбции в потоке газа. При достаточно вы-
соких скоростях газового потока внешний массообмен не оказы-
Рис. 7,4. Совмещение кинетических кривых десорбции различных нормальных парафи-
новых углеводородов в вакууме из гранул рекуперационного угля при различной тем-
пературе с кривыми для н-пентана. - -
Рис. 7,5. Зависимость кинетики десорбции н-пентана из гранул цеолита NaX
*=4 мм) в токе азота от скорости газа.
Кинетика десорбции из зерна адсорбентов
159
Рис. 7,6. Зависимость кинетики десорбции «-гептана из гранул цеолита СаА при ска-
рости потока азота 30 м/мин от размера гранул.
Рис. 7,7. Зависимость пороговых температур десорбции Т' нормальных парафиновых уг-
леводородов в вакууме от числа атомов углерода в молекулах углеводорода п:
1 — цеолит СаА; 2 — цеолит NaX; 3 — рекуперационный активный уголь АР; 4 — силика-
гель кем.
вает существенного влияния на скорость процесса в целом, и
кинетические кривые десорбции, полученные в опытах с продув-
кой газом, близки к кинетическим кривым в вакууме.
Зависимость степени десорбции н-пентана в потоке газа, па
данным Клушина [189], из гранул синтетического цеолита NaX
прослеживается на рис. 7,5. Анализ кривых показывает, что при
скоростях потока 24 м/мин и выше массообмен определяется
только внутридиффузионной кинетикой. Значение этой предель-
ной скорости практически не зависит от размера гранул адсор-
бента, молекулярной массы углеводорода и температуры. Близ-
кие результаты получены в работе [190]. Относительный метод,
расчета при скоростях потока выше предельной был распрост-
ранен на случай десорбции в потоке газа [191].
При десорбции в потоке газа уменьшение диаметра и высоты
зерна с 4 до 2 мм приводит к повышению скорости десорбции в
два раза. Как видно из рис. 7,6, при диаметре гранул цеолита
СаА 4 мм остаточная адсорбция //-гептана у = 0,8 достигается
при 100 °C после продувки в течение 70 мин, а при диаметре
гранул 2 мм — через 35 мин. Остаточная адсорбция у = 0,4 этого
же углеводорода при 200 °C достигается в зависимости от зер-
нения соответственно через 60 и 30 мин. Следовательно, при
десорбции в потоке газа, в отличие от вакуумной десорбции,
диффузионное сопротивление, обусловленное вторичной порис-
тостью, оказывает влияние на скорость массообмена. Анало-
гичные выводы были сделаны при анализе кинетических кривых
десорбции углеводородов из гранул активного угля и силикаге-
ля.' Однако в этих случаях отношение времени достижения оди-
наковых степеней десорбции углеводородов несколько больше,,
чем отношение линейных размеров гранул.
160
7. Кинетика десорбции
Сопоставление кинетических кривых десорбции углеводоро-
дов в потоке газа с кинетическими кривыми десорбции в ваку-
уме при прочих равных условиях показало замедление скоро-
сти десорбции в первом случае. Так, остаточная адсорбция
н-пентана у = 0,4 при его удалении из гранул цеолита СаА раз-
мером d = l = 2 мм при 120 °C в вакууме достигается через 10, а
в потоке газа — через 40 мин. Эта разница уменьшается в слу-
чае использования более мелких гранул.
Выбор температурного режима стадии десорбции
Если скорость десорбции всех углеводородов гомологического
ряда может быть отражена при определенным образом выбран-
ных температурах одной кинетической кривой, то для каждого
углеводорода существуют характерные температуры, имеющие
первостепенное значение для выбора оптимального температур-
ного режима процесса [192].
Для эффективного ведения процесса десорбции требуются
достаточно высокие скорости отвода вещества, причем скорость
переноса вещества из адсорбированной в газовую фазу опреде-
ляется главным образом температурным режимом процесса.
Минимальные температуры, ниже которых нецелесообразно про-
водить десорбцию в технологическом процессе, были названы
пороговыми (Т'). При пороговой температуре Т' поло-
вина адсорбированного вещества удаляется в вакууме в тече-
ние 10 мин.
Пороговые температуры Т' нормальных парафиновых угле-
водородов при их десорбции из зерна адсорбентов различного
типа, установленные на основе экспериментальных кинетиче-
ских десорбционных кривых, представлены на рис. 7,7 [193].
По легкости осуществления стадии
десорбции промышленные адсорбен-
ты образуют следующий ряд: сили-
кагель, активный уголь, цеолит NaX,
цеолит СаА. Чтобы в течение 10
мин удалить 50% адсорбированного
ундекана, в случае применения этих
адсорбентов требуется температура
100, 200, 260 и 340 °C соответст-
венно.
Аналогичные зависимости уста-
новлены для другой характерной
Рис. 7,8. Зависимость температур быстрой десорб-
ции Т" нормальных парафиновых углеводородов
в вакууме от числа атомов углерода в молекуле
углеводорода п:
1 — цеолит NaX; 2 — активный уголь АР; 3 — си»
ликагель КСМ.
Кинетика десорбции из зерна адсорбентов
161
температуры Т", при которой достигается быстрая десорбция.
При температуре быстрой десорбции после экспози-
ции насыщенной адсорбатом гранулы в вакууме в течение 30
мин содержание адсорбата в 100 г адсорбента снижается до
1 г. И в этом случае уголь необходимо перегревать на 100 °C,
а цеолит NaX — на 160 °C по сравнению с силикагелем, чтобы
достичь высокой степени десорбции углеводородов (рис. 7,8).
Опыты с бензолом позволили выявить влияние специфичес-
кого взаимодействия молекул этого углеводорода с гетероион-
ной поверхностью цеолитов на характер десорбции. Если по ус-
ловиям десорбции из активного угля, когда специфическое взаи-
модействие отсутствует, бензол занимает промежуточное поло-
жение между м-пентаном и «-гексаном, то в случае цеолитов
дополнительное специфическое взаимодействие усложняет де-
сорбцию бензола: из цеолитов он десорбируется труднее «-геп-
тана. Легкость регенерации силикагеля и активного угля пре-
допределяет выбор именно этих адсорбентов для большинства
адсорбционных установок с коротким циклом.
На основе экспериментальных данных было установлено, что
отношение температур Т' и Т" к нормальной температуре кипе-
ния ТКп в ряду парафиновых углеводородов остается практиче-
ски постоянным для всех исследованных адсорбентов. Этот
факт находит объяснение. Согласно правилу Трутона, для боль-
шинства веществ отношение теплоты конденсации к нормальной
температуре кипения 7КП приблизительно постоянно. Следова-
тельно, соотношение (7.9) может быть записано в следующем
виде:
Ti/TN — 7’кпг/^'кп.ст (7-10)
Все выводы по температурному режиму, сделанные на основе исследо-
вания десорбции из зерна адсорбента, действительны и для выбора условий
десорбции из слоя. Так, кинетические кривые десорбции углеводородов из
слоя активного угля в потоке газа показали, что быстрая десорбция «-бута-
на происходит пря 100—130 °C [194], что соответствует рис. 7,8.
Температурный режим технологического процесса должен выбираться в
соответствии с составом адсорбата, если он состоит из нескольких компонен-
тов. При этом в расчетах следует ориентироваться на вещество, скорость де-
сорбции которого наименьшая. По этому веществу, используя метод относи-
тельного расчета, следует определять рациональные температурные условия
десорбционного процесса.
Десорбция паров воды и диоксида углерода. Цеолиты явля-
ются эффективным и широко распространенным поглотителем
паров воды и диоксида углерода, вследствие чего выбор темпе-
ратурного режима десорбции этих компонентов из цеолитов
имеет большое практическое значение.
На рис. 7,9а приведены кинетические кривые десорбции воды из гранул
цеолита СаА размером d=/=4,4 мм в вакууме. Пороговая температура для
случая десорбции воды из цеолитов составляет, как показывает рисунок,
150 °C. Размер входных окон цеолитов СаА, СаХ н NaX достаточно велик и
11—1346
162
7. Кинетика десорбции
Ркс. 7,9. Кинетические кривые десорбции воды (а) и диоксида углерода (б) из гранул
цеолита СаА в вакууме.
не оказывает существенного влияния на скорость десорбции. Поэтому кине-
тические кривые десорбции на этих адсорбентах при одинаковых темпера-
турах близки друг к другу. Исключение составляют цеолиты КА, имеющие
размер окон 0,3 нм, близкий к критическому диаметру молекул воды
(0,27 нм). Скорость десорбции паров воды из этих цеолитов при 150°C
в 3,4 раза меньше, чем в гранулах цеолита СаХ такого же размера [114].
При десорбции вещества из кристаллического порошка мо-
лекулы адсорбата, в отличие от гранулированных цеолитов, по-
падают не в поры вторичной структуры — межкристаллитные
полости, а в свободный объем между кристаллами. Однако в
связи с тем, что основное диффузионное сопротивление с уче-
том скорости отрыва молекул от активных центров сосредото-
чено в кристаллах, кинетические кривые десорбции из порошка
имеют характер, аналогичный характеру кривых десорбции из
гранул. Скорость десорбции из порошка на всем участке опо-
рожнения адсорбционного объема приблизительно соответству-
ет скорости десорбции из гранул размером d=l=l,5 мм.
Сделана попытка определить количество воды при вакуум-
ной десорбции из цеолита NaA, которое непосредственно связа-
но с катионными активными центрами в этом цеолите. Выска-
зано предположение, что роль активных центров играют обмен-
ные катионы, причем на каждом таком центре — катионе нат-
рия адсорбируется одна молекула воды [195]. Условно процесс
дегидратации цеолита в вакууме можно разделить на две ста-
дии: быстрое удаление основного количества адсорбата при тем-
пературе до ПО °C и более медленная десорбция остаточного
его количества, удерживаемого катионами, заканчивающаяся
при 260—300 °C [196]. Степень и интенсивность дегидратации
цеолитов типа X и У практически не зависят от природы обмен-
ных катионов [197].
Диоксид углерода, как видно из рис. 7,96, десорбируется го-
раздо легче, чем вода; достаточно высокие скорости десорбции,
соответствующие пороговой температуре, достигаются уже при
0 °C. Скорости десорбции диоксида углерода одинаковы для
цеолитов типа А и X [198].
Изотермическая десорбция из слоя адсорбентов 163
Изотермическая десорбция из слоя
микропористых адсорбентов
Достаточно распространенным в технике методом десорбции
является прогрев слоя до заданной температуры внешним теп-
лоносителем с одновременной или с последующей продувкой
слоя небольшим количеством газа (или пара). В этом случае
стадия десорбции протекает практически в изотермических ус-
ловиях. Анализ процесса изотермической десорбции был прове-
ден [199] на примере десорбции этилена газообразным азотом
из слоя гранулированного цеолита NaX, размельченного до раз-
мера частиц 0,5—1,0 мм.
Изучение изотермической десорбции этилена проводили в лабораторном
адсорбере диаметром 9,5 мм с высотой слоя цеолита 46 см; загрузка состав-
ляла 16,4 г. Адсорбер был снабжен термостатирующей рубашкой, через ко-
торую циркулировала подаваемая из ультратермостата вода. Перед началом
каждого опыта цеолит регенерировали, нагревая адсорбер извне электрическим
током до 340—360 °C с одновременной продувкой азотом в течение 2,5 ч.
В этих условиях достигалось практически полное обезвоживание цеолита.
После регенерации адсорбер охлаждали до температуры опыта, а затем
через него пропускали смесь этилена с азотом до тех пор, пока не устанав-
ливалось адсорбционное равновесие, о наступлении которого судили по иден-
тичности составов входящего и выходящего газов. Десорбцию этилена про-
водили продувкой азотом; температуру при десорбции поддерживали по-
стоянной и равной той температуре, при которой проводили адсорбцию. Рас-
ход отдувочного газа азота при десорбции устанавливали реометром. Вы-
ходящую из адсорбера смесь этилена с азотом непрерывно анализировали
на хроматографе.
На рис. 7,10 приведено изменение во времени концентрации
этилена в выходящем газе при скорости продувочного газа
(азота) 0,08 м/с. Предварительное насыщение цеолита этиленом
было различным и отвечало условиям равновесия с газом, в
котором концентрация этилена изменялась от 0,8 до 14%. Гра-
фик показывает, что в любом случае на выходной кривой де-
сорбции можно отметить два характерных участка. На первом
горизонтальном участке наблюдается постоянство содержания
этилена в газе десорбции, которое соответствует концентрации
газа на предшествующей стадии насыщения. Наличие такого
участка свидетельствует о том, что между газовой фазой и оп-
ределенными слоями сорбента, включая последние по ходу дви-
жения газового потока, успевает установиться состояние, близ-
кое к равновесному. Второй участок характеризуется законо-
мерным падением содержания этилена в газе десорбции.
.Десорбционные кривые, полученные при разных исходных степе-
нях заполнения цеолита этиленом и отвечающие второму перио-
ду десорбции, достаточно точно ложатся на единую десорбци-
онную кривую. Это означает, что время отдувки до некоторой
заданной остаточной адсорбции не зависит от исходной степе-
11
164
7. Кинетика десорбции
ни насыщения цеолита.. Продолжительность первого периода
десорбции определяется графически как горизонтальный отре-
зок, проведенный из точки концентрации на оси абсцисс, соот-
ветствующей равновесной степени насыщения, до пересечения
с единой десорбционной кривой второго периода. Длина гори-
зонтального участка уменьшается с повышением степени насы-
щения цеолита адсорбатом.
Опыты, проведенные с разными скоростями продувочного
газа в интервале 0,04—0,30 м/с показали, что степень десорбции
при постоянной температуре практически не зависит от скоро-
сти потока и является функцией только количества пропу-
щенного газа. Кривые, построенные в координатах: «содержание
этилена в газе десорбции» — «удельный расход пропущенного
азота на единицу массы адсорбента», при разных скоростях
потока совпадают. Эта закономерность сохраняется как в слу-
чае высокой, так и в случае низкой исходной степени заполне-
ния цеолита адсорбатом (см. рис. 7,10).
По кривым, представленным на рис. 7,10, и значениям рав-
новесной статистической активности согласно изотермам ад-
сорбции этилена рассчитывают зависимость объема недесорби-
рованного этилена в процессе отдувки при разном удельном рас-
ходе азота. Эти данные для трех температур приведены на
рис. 7,11.
Рис. 7,10. Десорбция этилена из слоя цеолита NaX в токе азота при 25 °C:
а — скорость газа постоянна н равна 5 м/мин; б — скорость газа изменяется от 2,5 да
20 м/мин.
Изотермическая десорбция из слоя адсорбентов
165
Рис. 7,11. Зависимость объема иедесорбироваиио-
го этилена А от расхода азота Б.
Наличие размытого фронта де-
сорбции газов и паров из микропо-
ристых адсорбентов, характерное
для крутых выпуклых изотерм ад-
сорбции, позволяет применять по-
следние для вычисления выходных
десорбционных кривых [200, 201].
Если предположить, что при иссле-
дованных скоростях газового потока
равновесие между газовой и твер-
дой фазами устанавливается доста-
точно быстро, тогда десорбционные
кривые, отвечающие второму периоду, могут быть рассчитаны
из изотерм адсорбции по уравнению Викке:
V/x = Mf' (с)
(7-11)
где V—объем пропущенного отдувочного газа; х— длина адсорбера; М —
масса сорбента на единицу длины колонки; с — концентрация этилена в газе;
f'(c) — производная изотермы адсорбции по концентрации.
Для больших концентраций, кроме того, должно учиты-
ваться изменение объема газовой смеси, согласно модифициро-
ванному уравнению Викке:
У/х = Л1/'(с)(1-с|2с)2 (7-12)
где Sc — суммарная концентрация газа в объеме адсорбера, равная сумме
концентраций сорбированного газа (С2Н4) и газа-носителя (Ns).
На рис. 7,12 представлен пример расчета общей десорбци-
онной кривой этилена из цеолита NaX по изотерме адсорбции
при 25°С. Графическое дифференцирование изотермы для рас-
чета по уравнению (7.12) здесь выполнено по методике Янов-
ского [202].
Изотерма этилена разбита на ряд участков, например: 0—0,5; 0,5—1;
1—2; 2—4; 4—6% и т. д. Через точки, отвечающие местам пересечения изо-
термы с линиями, параллельными оси абсцисс, проведены касательные к изо-
терме. На оси концентраций влево от точки 0 отложен отрезок ОР, соответ-
ствующий Мх (в рассматриваемом случае 16,4 г). Из точки Р исходят лучи,
параллельные касательным к изотерме. Через каждую точку пересечения лу-
ча с осью ординат Б параллельно оси абсцисс проведена прямая до пересе-
чения с соответствующей вертикальной граничной линией. Точки, образую-
щиеся при последнем пересечении, по смыслу построения отвечают значению
V уравнения (7.11) для соответствующих концентраций. Аналогично найдены
другие точки выходной кривой.
Пунктирная линия получена с учетом поправочного множителя
(1—c/Sc)2. Экспериментальные точки достаточно точно накладываются на
166
7. Кинетика десорбции
А — концентрация этилена °/о (об.); Б — активность адсорбента, см3/г; В — расход отду-
вочного газа, л/г.
Рис. 7,12. Построение десорбционной кривой из изотермы адсорбции этилена на цеолите
NaX при 25 °C:
теоретически полученную кривую, что позволяет рекомендовать уравнения
(7.11) и (7.12) для использования при расчете десорбции из слоя микропо-
ристых адсорбентов.
Расчеты десорбционных кривых этилена, выполненные на
основе изотерм адсорбции при 25, 50, 75, 200 и 300 °C, позволили
сделать вывод, что достаточно быстрая десорбция этилена мо-
жет быть достигнута уже при 75 °C, однако большой удельный
расход отдувочного газа (1 л/г цеолита) исключает возмож-
ность получения в данном случае концентрированного этилена
в газах десорбции.
Если, согласно изотермам, принять, что в процессе адсорбции этилена
при 25 °C из смеси, содержащей 3% С2Н4, 1 г цеолитов поглощает 27 см3, а
при десорбции выделяется 90% поглощенного этилена, то в газе десорбции
при 75°C будет содержаться лишь около 4,3% этилена. С повышением тем-
пературы расход отдувочного газа резко снижается, в результате чего, на-
пример при 200 “С, концентрация этилена в газах десорбции может быть по-
вышена до 30%, а при 300 °C — до 50%.
Изложенная методика позволяет выбирать оптимальные ус-
ловия десорбции углеводородов и других адсорбатов из слоя
микропористых адсорбентов. Обратная задача—построение
изотерм адсорбции на основе кривых отдувки — позволяет по-
лучить изотермы адсорбции агрессивных газов (например, се-
роводорода), для которых использование обычной вакуумной
адсорбционной аппаратуры с ртутными элементами затрудни-
тельно.
Предмет динамики адсорбции
167
Поиску оптимального режима стадии десорбции предшеству-
ет выбор рациональной структуры применяемого адсорбента.
Адсорбент должен не только эффективно поглощать примесь того или
иного вещества, но и отдавать его при десорбции в приемлемых рабочих ус-
ловиих. Так, дли улавливании (рекуперации) легколетучих веществ, близких
по своим физико-химическим свойствам, — хлорида этила и сероуглерода —
рекомендуютсн микропористые угли с объемом микропор 0,5 см3/г и значени-
ем характеристической энергии А0 = 25,2 кДж/моль. В данном случае десорб-
ция не лимитирует адсорбционный цикл в целом. Для поглощения паров
менее летучих растворителей, например амилацетата и ксилола, наиболее
пригодны активные угли с развитой супермикропористостью при значении
£0 = 8,4—12,6 кДж/моль. Бензол по своим десорбционным свойствам занима-
ет промежуточное положение между этими двумя группами, и для его по-
глощения целесообразно использовать активные угли с промежуточными па-
раметрами микропористой структуры, т. е. при До = 16,8—21,0 кДж/моль
[203].
Предмет динамики адсорбции
В основе расчета процессов очистки и рекуперации, осуществ-
ляемых с помощью твердых адсорбентов, лежат закономерно-
сти динамики адсорбции. Динамика сорбционных процессов
рассматривает пространственно-временные распределения ком-
понентов между фазами системы (одна из которых — твердая),
возникающие при перемещении этих фаз относительно друг
друга.
Процессы адсорбции экзотермичны, обратные процессы (де-
сорбция) требуют подвода тепла. Поэтому при анализе неко-
торых задач динамики сорбции следует учитывать распределе-
ния температуры в обеих фазах системы.
Пусть концентрация адсорбата в адсорбенте до начала про-
цесса равна нулю, концентрация адсорбтива во входящем по-
токе постоянна. Введем поток в колонну и проследим за из-
менением концентрации в обеих фазах. В этот момент во всех
168
8. Динамика адсорбции
, Рис. 8,1. Формирование и перемещение фронта ад-
сорбции по гипотетическому горизонтальному адсор-
беру.
сечениях слоя, кроме лобового, кон-
центрация адсорбата равна нулю. Ад-
сорбент в лобовом слое начинает по-
глощать адсорбтив, концентрация ко-
торого в вошедшей «элементарной»
порции газа убывает. Частично исто-
щенная «элементарная» порция ад-
сорбтива вместе с потоком газа посту-
пает в следующий «элементарный»
объем слоя. В нем начинает идти пог-
лощение вещества. Далее в сорбцион-
ный процесс вовлекаются третий, чет-
вертый и т. д. «элементарные» объемы.
Рассмотрим процесс извлечения па-
ра в гипотетическом горизонтальном
адсорбере (см. рис. 8,1). В период времени ti количество посту-
пившего адсорбтива еще недостаточно для насыщения лобового
слоя адсорбента и распределение адсорбата в слое характери-
зуется кривой ть При времени т2>Т1 лобовой слой адсорбента
насыщается адсорбатом до величины равновесной адсорбцион-
ной способности.
Распределение концентраций в любой из фаз, достигнутое
ко времени т2, называют фронтом сорбции. После этого
момента кривая распределения адсорбата часто перемещается
с постоянной скоростью U, определяемой из равенства:
U = wca/av
(8.1)
где w — скорость газового потока, см/с; са — концентрация адсорбтива в га-
зе, моль/см3 газа; а?— равновесна адсорбционная способность, моль/см3 ад-
сорбента.
В период, соответствующий времени Тз, слой адсорбента
можно разделить на три зоны: полностью отработавший слой
Li, работающий слой Lo и еще не вступивший в работу
слой Ь2. В момент времени Т4 кривая распределения перемеща-
ется в концевой слой адсорбента. С этого момента, момента
«проскока», в выходящем потоке газа появляется и начина-
ет прогрессивно возрастать концентрация извлекаемого веще-
ства:
Т4 = ТПр (8.2)
где тПр — момент проскока, время защитного действия слоя
адсорбента.
Обычно, чтобы обеспечить высокую степень рекуперации па-
ра, адсорбер перед наступлением «проскока» переключают на
Предмет динамики адсорбции
169
стадию десорбции. При этом, однако, часть адсорбционной ем-
кости в работающем слое не используется; степень недо-
использования адсорбционной емкости в рабо-
тающем слое определяют коэффициентом симметрич-
ности ф, который вычисляется из соотношения площадей (см.
рис. 8,1)
Ф = Sbcd/$abcd (8-3)
Динамическая адсорбционная способность
слоя адсорбента характеризует количество вещества, поглощен-
ного до момента «проскока» и отнесенное к массе загрузки все-
го адсорбера. Отношение динамической адсорбционной способ-
ности слоя к равновесной адсорбционной способности адсор-
бента называют с те п е н ь ю использования адсорбци-
онной емкости слоя а. Следовательно
а = яд/яр (8.4)
С другой стороны, величина а определяется соотношением
высоты адсорбера L и высоты работающего слоя Lo:
а = (L — <fL0)/L (8.5)
Характер движения «проскоковой» концентрации по длине
адсорбера отражает рис. 8,2. Между временем защитного дей-
ствия слоя и длиной адсорбера существует зависимость, оп-
ределяемая формулой Шилова [204]:
xnp = kL~ т0 (8.6)
где k— коэффициент защитного действия, показывающий, какое время слой
толщиной 1 см адсорбента в условиях стационарного режима задерживает
поглощаемое вещество; То — потеря времени защитного действия, связанная
с начальным периодом формирования кривой распределения адсорбата.
Рис. 8,2. Зависимость времени защитного действия слоя (времени до «проскока» тпр)
от его длины L.
Рис. 8,3. Выходная кривая при извлечении примеси из потока газа в неподвижном слое
адсорбента.
170
8. Динамика адсорбции
Коэффициент защитного действия является величиной, об-
ратной скорости движения сорбционной волны:
k = \/U (8.7)
Величину работающего слоя Lo определяют по выходной
кривой, отражающей нарастание концентрации примеси за сло-
ем адсорбента во времени (см. рис. 8,3). Для этого можно ис-
пользовать формулу Майклса — Трейбла [205, 206]:
Дт
Lo = L тр—(1 — <р) Дт (8,8)
где Дт — разность времени между появлением равновесной тр и проскоко-
вой тПр концентраций за слоем; тр — время появления максимальной (рав-
новесной) концентрации; ф — коэффициент симметричности выходных кри-
вых или кривых распределения.
Экспериментальное исследование
динамики адсорбции
Экспериментальное исследование динамики адсорбции обычно
преследует две цели: проверку адекватности модели экспери-
менту и получение информации из выходных кривых адсорбции,
необходимой для выбора размеров и конструкции промышлен-
ных адсорберов.
Установки для изучения динамики адсорбции различаются
схемой, методами дозировки газового потока и определением
концентрации адсорбтива за слоем. Общим элементом всех ди-
намических установок является колонка, заполненная гранула-
ми адсорбента.
На рис. 8,4 показана установка для изучения процесса адсорбции диок-
сида углерода из потока воздуха при атмосферном давлении. Основным уз-
лом установки является адсорбер 3, имеющий штуцеры для отбора газа на
анализ (через гребенку 4) и ввода термопар. Температура в колонке фикси-
руется многоточечным потенциометром 1. Для регенерации адсорбента ад-
сорбер нагревают с помощью электрообмотки из нихромовой проволоки. Но-
ток газа-носителя через ротаметр 7 поступает в смеситель 5, куда из бал-
лона подается диоксид углерода. Его расход устанавливают по реометру 8.
Смесь газа-носителя (воздуха), нагнетаемого воздуходувкой 9, и диоксида
углерода поступает в адсорбер, заполненный гранулами исследуемого сор-
бента. Через штуцеры, начиная с нижнего, газ отводят на анализ, который
в данном случае проводят инфракрасным спектроскопом. В схеме установки
предусмотрена также колонка 6 с силикагелем для предварительной осушки
воздуха. Требуемая температура опыта поддерживается с помощью термо-
стата 2.
Установка иной конструкции описана в гл. 5 (см. рис. 5,7); ее в равной
степени используют для изучения динамики адсорбции и для получения рав-
новесных данных, в том числе для бинарных систем.
В выпускаемом отечественной промышленностью приборе «Сорбент»,
предназначенном для исследования динамики поглощения паров летучих рас-
творителей в широком интервале проскоковых концентраций, в качестве ре-
Экспериментальное исследование динамики адсорбции
171
гистрирующего элемента применен пламенно-ионизационный детектор [207].
Исходная концентрация адсорбтива может достигать 10—20 мг/л, минималь-
ная регистрируемая детектором концентрация равна 1-10-5 мг/л. Достоинст-
вом прибора является высокая чувствительность и точность измерений, воз-
можность автоматизации записи результатов, получение непрерывной выход-
ной кривой, надежность в работе.
При экспериментальном изучении динамики времени до про-
скока Тпр соответствует появление за слоем адсорбента мини-
мальной концентрации примеси, обнаруживаемой детектором,,
ср, времени до установления равновесия тр — появление «рав-
новесной» концентрации с2- Обычно принимают ci = 0,05 ср; С2 =
= 0,95 с0.
Использование уравнения Майклса — Трейбла для опреде-
ления Lo возможно только в случае, если фронт сорбции пере-
мещается с постоянной скоростью. Такой режим называется
режимом параллельного переноса фронта сорб-
ции. Он реализуется хотя и часто, но все же не всегда.
В работе [208] представлен краткий обзор методов опреде-
ления его реализации на основе экспериментальных выходных
кривых. Существование режима параллельного переноса мож-
но считать экспериментально доказанным, если в пределах точ-
Рис. 8,4. Схема установки для исследования динамики адсорбции диоксида углерода:
1 — потенциометр; 2 — термостат; 3 — адсорбер; 4 —гребенка; 5 — смеситель; 6 — осушаю-
щая колонка; 7 — ротаметр; 8 — реометр; 9 — воздуходувка.
172
8. Динамика адсорбции
ности опытов соблюдаются хотя бы одно из двух условий:
1) обеспечено равенство относительных концентраций адсорба-
та в газовой (у — с/со) и адсорбированной (у = а/а0) фазах си-
стемы; 2) скорости движения всех заключенных в некоторых
границах концентрационных точек адсорбционной волны равны
между собой и не изменяются во времени. Второе условие имеет
более общий характер.
На практике часто считают, что режим параллельного пере-
носа установился, если высота слоя адсорбента в 2—3 раза пре-
вышает величину работающего слоя (L^2L0).
Более совершенным критерием установления режима явля-
ется метод, изложенный в работе ([209]. Изменение концентра-
ции адсорбтива во времени на выходе из адсорбционной колон-
ки рассматривалось как функция распределения с = с(т). Эта
функция распределения описывалась 1-м и 2-м моментами рас-
пределения:
У xf (т) dx
о
^1 = ^5----------------
/ (т) dx
о
т2/ (т) dx
J f($dx
о
(8.9)
(8.10)
Очевидно, что для стадии параллельного переноса фронта
сорбции в силу постоянства его профиля вторые моменты рас-
пределения концентрации во времени не зависят от высоты
слоя, т. е. одинаковы. Если за начало отсчета времени принять
появление проскоковой концентрации за слоем адсорбента, то
будут одинаковы и первые моменты распределения. В то же
время для стадии формирования режима Mi—ст (где
М;, ст и Mi — i-й момент для стадий параллельного перемеще-
ния фронта и его формирования при увеличении высоты слоя).
На основе этих положений процесс можно считать стацио-
нарным, если моменты распределения с = с(т), полученные в
неподвижных слоях разной длины, в пределах точности измере-
ний равны между собой. Сопоставление моментов проводят по
известным статистическим методикам.
Ниже приведены значения первых ц и вторых о2 централь-
ных моментов для случая поглощения циклопропана при его
Экспериментальное исследование- динамики адсорбции 173
концентрации в газе 6% (об.) и разной высоте слоя активного
угля СКТ; скорость газового потока составляла 0,3 м/с:
L, см н а2 L, см Н О2
5,8 4,59 4,61 41,0 9,09 10,23
10,2 7,04 8,75 51,0 9,42 10,45
15,3 9,24 10,57 63,2 9,58 10,36
32,0 9,35 10,14
На основании полученных результатов можно сделать вы-
вод, что в рассматриваемой системе параллельный перенос
достигается при высоте слоя адсорбента 15 см и более.
Следует отметить, что усилия, затраченные на постановку
«чистого» опыта, обычно окупаются простотой обработки и об-
суждения полученных результатов. Поэтому метод моментов
был применен также для определения той минимальной скоро-
сти газа, при которой практически исключалось влияние внеш-
ней диффузии, для подтверждения изотермичности процесса и
для оценки влияния пристеночного эффекта.
Для использования уравнения Майклса — Трейбла необходи-
мо также, чтобы процесс адсорбции был изотермичен. При
большом же диаметре колонок затрудняется отвод выделяюще-
гося в процессе адсорбции тепла через стенки. В этом случае
выходные кривые растягиваются, и рассчитанная на их основе
высота работающего слоя обусловлена не только массообменом,
но и процессами теплопередачи.
В многочисленных системах установлено, что разогрев пото-
ка газа пренебрежимо мал, если диаметр адсорбционной ко-
лонки, снабженной снаружи водяной рубашкой, составляет не
более 16 мм. Пристенный эффект начинает проявляться, если
отношение диаметра колонки к приведенному диаметру гранул
адсорбента меньше примерно десяти. Таким образом, при диа-
метре колонки 16 мм пристенный эффект не искажает картину
массопереноса, если гранулы имеют определяющий размер* до
2,0 мм.
Высота работающего слоя является важным показателем
адсорбционного процесса, определяющим степень отработки ем-
* Диаметр промышленных гранул адсорбентов обычно лежит в интервале
от 1,5 до 5 мм. Перенос результатов, полученных с гранулами мелкого зер-
нения, на гранулы промышленных размеров, как было видно из гл. 6, пред-
ставляет значительные трудности. Поэтому опыты по «чистой» динамике ад-
сорбции, если говорить об их практической ценности, часто носят качествен-
ный характер и свидетельствуют лишь о практической осуществимости про-
цесса и его относительной эффективности. (Прим, ред.)
174 S. Динамика адсорбции
Таблица 8-1. Высота работающего слоя адсорбентов
Адсорбент Адсорбтив
С2Нв с3н8 H-C4H10 Н-С&НХ2 н-С6Н14 н-С7Н1б H-CgHig
Активный 16,0 5,0 5,8 4,0 3,1 2,9 2,5
уголь СКТ Цеолит NaX 19,0 8,0 4,0 3,6 3,8 4,8 7,4
Цеолит СаА 17,5 7,6 3,5 8,4 13,0 18,0 30,0
кости слоя адсорбента. Увеличение скорости потока, исходной
концентрации адсорбтива, температуры процесса и диаметра
зерна во всех случаях сопровождается возрастанием высоты ра-
ботающего слоя. Она увеличивается также с ухудшением ад-
сорбируемости адсорбтива. Но даже при поглощении таких от-
носительно плохо адсорбирующихся веществ как сероуглерод
(при скорости потока вентиляционного воздуха в промышлен-
ном адсорбере, загруженном рекуперационным углем APT,
0,3 м/с) высота работающего слоя не превышает 50 см. В связи
с этим высокая степень использования адсорбционной емкости
слоя угля а = ад/ар= (L—q,L0)/L порядка 75% и выше достига-
ется при высоте слоя 1,5—2 м. Большое влияние на высоту ра-
ботающего слоя оказывает скорость проникновения адсорбти-
ва в микропоры адсорбента, которая, в свою очередь, зависит
от соотношения размеров поглощаемых молекул и пор, а также
от ориентации молекул при подходе к «устью» поры.
В табл. 8-1 приведена высота работающего слоя при адсорб-
ции нормальных парафиновых углеводородов в слое микропори-
стых адсорбентов (температура 75 °C, скорость потока 0,2 м/с,
концентрация адсорбтива 0,5 об. %, зернение адсорбента 0,5—
1,0 мм). Зависимость высоты работающего слоя цеолитов от
числа атомов углерода в молекуле нормального углеводорода
(адсорбтива) проходит через минимум, соответствующий «-бу-
тану для типа СаА и н-пентану для типа NaX, в то время как в
случае активного угля эга величина монотонно убывает.
В некоторых случаях целесообразно использовать двух-
слойную шихту одного адсорбента. Поток газа проходит через
адсорбер, в замыкающей части которого содержится адсорбент
более мелкого зернения, чем в лобовой. Согласно общей теории
динамики сорбции, как будет показано ниже, при выпускной
изотерме любое начальное распределение вещества в колонне
на некотором расстоянии от точки ввода потока трансформиру-
ется во фронт постоянного профиля. Высоту замыкающего слоя
шихты подбирают так, чтобы начальное распределение веще-
ства в адсорбере, образовавшееся при прохождении лобового
Основные уравнения динамики адсорбции
175
слоя (крупные частицы) до начала проскока, успевало транс-
формироваться в распределение, отвечающее замыкающему
слою (мелкие частицы). Это дает возможность для всего ап-
парата получить адсорбционные характеристики слоя, состояще-
го из мелких гранул. Использование двухслойной шихты позво-
ляет при равных показателях процесса снизить гидравлическое
сопротивление слоя.
Основные уравнения динамики адсорбции
Для отыскания искомых функций распределения адсорбата, что
составляет задачу теоретической динамики адсорбции, исполь-
зуют два метода: феноменологический и статистический. Фе-
номенологический метод устанавливает функциональные зави-
симости между величинами, характеризующими процесс с мак-
роскопической точки зрения. При этом не ставится задача мо-
лекулярно-кинетического или микроскопического объяснения
явлений. При статистическом подходе сорбент рассматривается
как дискретная среда с произвольно расположенными сорбци-
онными центрами, через которую движется хаотический ан-
самбль адсорбируемых молекул. Процесс адсорбции характери-
зуется вероятностными функциями распределения частиц меж-
ду подвижной и твердой фазами, на основе которых находят
кривые распределения концентраций. Дальнейшее изложение
материала основано на феноменологическом подходе.
Обоснование основных уравнений динамики адсорбции и их
анализ в достаточно общей форме приведены в монографии Ра-
чинского [210]. Ниже мы рассмотрим эти уравнения в более
простой форме, приняв, в соответствии с возможностями прак-
тического расчета адсорбционных процессов, ряд упрощающих
допущений:
1. Адсорбируется лишь один компонент потока.
2. Подвижная фаза несжимаема, и концентрация адсорби-
руемого вещества в ней так мала, что можно пренебречь изме-
нениями плотности потока вследствие убыли адсорбтива.
3. Движение потока осуществляется в одном направлении
с постоянной скоростью (ш = const).
В этом случае динамика адсорбции описывается системой
следующих уравнений: уравнениями материального и теплово-
го балансов, уравнениями кинетики адсорбции и теплопередачи,
уравнением изотермы адсорбции.
Уравнение материального баланса
Ниже мы приводим вывод уравнения материального баланса,
ограничившись в соответствии со сделанными допущениями
рассмотрением одномерной задачи. Пусть х-координата соот-
176
8. Динамика адсорбции
Рнс. 8,5. К выводу уравнения материального ба-
ланса.
as
ветствует направлению перемещения
потока со скоростью w через элемент
пористого тела с площадью попереч-
ного сечения dS и толщиной dx (рис.
8,5). Скорость потока при неизотерми-
ческой адсорбции даже с учетом сде-
ланных допущений не является посто-
янной величиной, а, подобно концент-
рациям с и а, есть функция координа-
ты и времени.
Рассмотрим баланс вещества в выделенном элементарном
объеме. Количество вещества, входящего в элементарный объем
через площадь dS за время dx, составляет wc-dS-dx. Количест-
во вышедшего вещества равно: wc-dS-dx+d(wc-dS-dx). Изме-
нение количества вещества в элементарном объеме равно:
д (wc-dS-dx) dx
дх
дшс
gx -dS-dx-dx
(8.Н)
Изменение количества вещества в элементарном объеме вы-
зовет изменение концентрации вещества в адсорбенте и подвиж-
ной фазе. В адсорбенте оно будет равно (да/дх)-dx-dx-dS,
в потоке (дс/дх)-dx-dx-dS.
Отсюда общий материальный баланс в элементарном слое:
д (wc) да t дс
— ——-dS-dx-dx = ’dx-dx-dS +-^--dx-dx-dS
или
да дс д(шс) __
дх “** дх + дх
(8.12)
Учет изменения концентрации вещества в элементарном объ-
еме вследствие диффузии приводит к следующему выражению:
да дс д (wc) _ <52с
дх “** дх + дх дх2
(8.13)
где D* — коэффициент продольной диффузии, физический смысл которого
будет обсужден ниже.
Далее будем считать, что неизотермичность сорбционного
процесса в силу, например, малого коэффициента термического
расширения не приводит к изменению плотности потока. Тогда
да=const и
да , дс дс <52с
дх + "Э? + w ~дГ = Р <?х2
(8.И)
Основные уравнения динамики адсорбции
177
Такой вид уравнения материального баланса адсорбата наи-
более распространен в работах по математическому моделиро-
ванию динамики адсорбции.
Обсудим физический смысл коэффициента продоль-
ной диффузии. В ранних работах по динамике сорбции
под продольной диффузией понималась молекулярная диффузия
[211]. В 1946 г. Пшежецкий и Рубинштейн [212] указали, что
в зернистом слое может иметь место дополнительное переме-
шивание потоков, эквивалентное увеличению коэффициента мо-
лекулярной диффузии на несколько порядков. Несколько позже
Радушкевич [213] обратил внимание на то, что продольная
диффузия в зернистом слое не тождественна молекулярной диф-
фузии адсорбата, а определяется такими факторами статисти-
ческого характера, как неоднородность укладки и размера зе-
рен адсорбата, а также механическими напряжениями в слое,
вызванными действием стенок и материала. Использовав мате-
матический аппарат Викке, автор определил значения коэффи-
циентов продольного переноса для так называемой равновес-
ной задачи динамики сорбции, сущность которой будет рассмот-
рена ниже.
В настоящее время проявления продольной диффузии при
адсорбции отождествляют с нарушениями поршневой структу-
ры потока в зернистом слое [208, 214, 215]. Справедливость
этой точки зрения подтверждается, в частности, сравнением
[208] экспериментальных данных о коэффициентах продольной
диффузии, найденных Рачинским и Давыдовой [216] в опытах
с ионообменной целлюлозой, с их значениями, определенными
в специальных экспериментах при исследовании структуры по-
тока [217].
В монографии [217] приведен обзор по составляющим эле-
ментам продольного переноса. Показано, что существенное зна-
чение имеют релаксационные коэффициенты диффузии, обуслов-
ленные наличием застойных зон или неравнодоступных объемов
в зернистом слое. Замедленное изменение концентрации в этих
зонах (объемах) по сравнению с изменением ее в ядре потока
является основной причиной возникновения релаксационной со-
ставляющей продольного переноса.
Другими составляющими продольного переноса являются:
флуктуация скорости, неравномерное распределение потока по
сечению аппарата («стеночный» эффект), конвекционные пере-
мешивания и, наконец, молекулярная диффузия. Численные зна-
чения коэффициентов продольного переноса определяют экспе-
риментально по методам, изложенным в работах [217—219].
Известен ряд формул, обобщающих результаты опытов
[217, 220]. Очень удобная номограмма для определения коэффи-
циентов продольного переноса приведена в монографии Аэрова
и Тодеса [217]. _ _
12—1346
178
8. Динамика адсорбции
Уравнение кинетики адсорбции
Вопросы, связанные с кинетикой адсорбции, рассмотрены в гла-
ве 6. Кинетику нестационарного конвективного (внешнего) мас-
сообмена обычно описывают следующим уравнением:
da/dT = ₽r(c — с*) (8.15)
где а —величина адсорбции; т — время; рг — коэффициент внешнего массо-
обмена, отнесенный к единице объема адсорбента; с — текущая концентрация
адсорбтива в потоке; с*—концентрация адсорбтива на поверхности раздела
-фаз, равновесная текущей величине адсорбции.
Другой часто используемой формой кинетического уравнения
является формула, в которой движущая сила внутридиффузион-
ного процесса адсорбции записывается как разность концентра-
ций адсорбата в твердой фазе (формула Глюкауфа):
da/dx = |3Т (а* — а) (8.15а)
где рт — кинетический коэффициент; а—текущая величина адсорбции; а* —
величина адсорбции, равновесная текущей концентрации адсорбтива в потоке
на внешней поверхности гранул.
При смешаннодиффузионном переносе адсорбтива обычно
используют либо формулу (8.15) или (8.15а), понимая под р
общий коэффициент массопередачи.
Уравнение теплового баланса
Уравнение теплового баланса для элементарного объема зерни-
стого слоя по форме записывается аналогично уравнению ма-
териального баланса. Для адиабатического процесса тепловой
баланс может быть записан следующим образом:
, дТа , дТг . дТг d>(Qa±
ha дх ~ hr дх дх дх
(8.16)
где fta, hr — теплоемкость адсорбента и газа; Та, Тг — температура адсорбен-
та и газа; Q — тепловой эффект сорбдии,
Сопоставление уравнений (8.16) и (8.14) указывает, что в
уравнение теплового баланса не включено слагаемое, соответ-
ствующее продольным эффектам. В литературе [217] имеются
данные о физической интерпретации эффективного коэффициен-
та продольной теплопроводности и необходимый для его вычис-
ления справочный материал. Однако в практических расчетах
этой составляющей баланса обычно пренебрегают. В расчетах
также обычно принимают, что тепловой эффект сорбции Q яв-
ляется величиной постоянной.
Основные уравнения динамики адсорбции 179
Уравнения передачи тепла
Теплообмен между твердым телом и потоком лимитируется
или внешней теплоотдачей, или теплопроводностью гранул
। (внутренняя задача), или определяется совместно этими двумя
механизмами.
Анализ показал [221], что в реальных условиях работы зер-
нистых слоев адсорбентов и катализаторов теплообмен, в отли-
чие от массообмена, практически полностью определяется теп-
лоотдачей от ядра потока к внешней поверхности частиц твер-
дого тела. Распространение же тепла по частице проходит до-
статочно быстро В этом случае для «чисто» теплотехнической
I задачи уравнение передачи тепла можно записать следующим
I образом:
, дТ
ha^=.Kr(Tr-Ta) (8.17)
где Кт— объемный коэффициент теплоотдачи; Тг — текущая температура яд-
ра потока; 7а — текущая температура частиц твердого тела.
Для теплоотдачи, включающей тепловые эффекты адсорбции,
изменение температуры адсорбента описывается уравнением:
ha^ = Kr(Tr-Ta)+Q^- (8.18)
Для определения Кт — коэффициента теплоотдачи, отнесен-
ного к единице раздела фаз, предложены [217] критериальные
уравнения, подобные уравнениям, рекомендуемым для определе-
ния ргп. Подробный обзор работ по теплопроводности и тепло-
передаче в неподвижном зернистом слое приведен в моногра-
фии [217].
Начальные и граничные условия
Дифференциальные уравнения массо- и теплообмена в зернп-
; стом слое решаются с учетом начальных и граничных условий.
‘ Во всех рассматриваемых ниже задачах мы будем принимать
। следующие распределения концентрации и температур в на-
чальный момент времени:
т = 0; O^xsSZ.; с = 0; « = 0; Та = Тг = ТИ (8,19)
, Принятое начальное условие означает, что до начала про-
! цесса ни в газовой, ни в твердой фазах на всем протяжении ко-
лонки адсорбат не содержался.
Граничные условия при х = 0 формулируют следующим об-
разом:
г > 0; х = 0; c=c0 = const; я==«(т); Тг = Тг0 — const (8.20)
12
180
8. Динамика адсорбции
т. е. в течение всего процесса в слой поступает газовый поток
при постоянной концентрации адсорбтива и температуре.
Разумеется, условия (8.19) и (8.20) не исчерпывают всех
возможных вариантов краевых условий. Более того, практиче-
ски более важными являются задачи, в которых начальные ус-
ловия вместо (8.19) формулируются как некоторые функции
распределения с(х), а(х), Тг(х), Та(х). Решения таких задач по-
ка немногочисленны [222, 223].
Математические модели динамики адсорбции
'Описание адсорбционных процессов даже с учетом сделанных
допущений представляет большие трудности. Поэтому мы рас-
смотрим постепенно усложняющийся ряд моделей динамики
сорбции.
Динамика равновесной изотермической адсорбции
Примем, что между концентрациями адсорбтива и адсорбата в
каждый момент времени и в каждой точке слоя соблюдается
равновесное соотношение в силу, например, бесконечно большо-
го значения коэффициента массопередачи. Пусть также тепло-
выделения в слое бесконечно малы, а температуры потока и
адсорбента одинаковы. Эти допущения составляют сущность
простейшей модели динамики сорбции — равновесной изотерми-
ческой, которая представляет несомненный познавательный ин-
терес. Рассмотрение ее позволяет установить влияние вида изо-
термы сорбции на распределение адсорбата между фазами си-
стемы. Основные положения этой модели развиты Вильсоном
[224], Вейссом [225], де-Во [226], Викке [227].
В соответствии со сделанными допущениями процесс дина-
мики сорбции описывается только двумя уравнениями — урав-
нением материального баланса и уравнением изотермы адсорб-
ции:
да дс F дс д2с
—ч— -I- — -I- w — = D * д 9
дт ' дх ' дх дх2
(8.21)
а = f (с)
где a=f(c) — уравнение изотермы адсорбции.
Процесс при отсутствии продольной диффузии
Систему уравнений (8.21) можно подвергнуть дальнейшему
упрощению, предположив, что эффект продольной диффузии
пренебрежимо мал (D* = 0). В этом случае
да дс дс
дх + дт + ш дх 0
(8.22)
а = f (с)
Математические модели динамики адсорбции 181
Система (8.22) имеет два решения [210]: одно при наличии
конечного градиента концентраций в колонке (случай 1), дру-
гое при так называемом «обрывном» фронте (случай 2)*. Эти
решения записываются следующим образом:
Случай 1 Ut = j (8.23)
Случай 2 U = ,0 (8.24)
с0 Т"
где Ui — скорость движения концентрационной точки i по слою адсорбента;
f'(a)= (dafdc)i—производная изотермы адсорбции в точке i; U — скорость
движения всех точек «обрывного» фронта; с0 — концентрация адсорбтива в
потоке; ао — равновесная ей величина адсорбции.
Уравнение (8.23) носит название закона Викке, уравнение
(8.24) — закона Вильсона. Проследим действие этих законов
для разных видов изотерм сорбции при адсорбции и десорб-
ции.
Выпуклая изотерма, адсорбция. На рис. 8,6 схематично изо-
бражены изотерма адсорбции a = f(c) и кривые распределения
концентрации а = а(х) для разных промежутков времени. Пусть
начальному распределению отвечает линия то- Так как фронт
необрывный, то каждая точка этого первоначального распреде-
ления будет передвигаться по слою со своей характерной ско-
ростью, задаваемой уравнением (8.23).
Пусть движение потока по слою направлено в сторону
уменьшения концентраций. Выделим на изотерме две точки (1
и 2), перемещение которых по слою от начального положения То
будет являться предметом анализа. Для приведенного на
рис. 8,6 вида изотермы (da/dc)i> (da/dc)2, поэтому, согласно
(8.23), U!<zU2. Это означает, что расстояние между точками
1 и 2, первоначально заданное распределением т0, уменьшает-
ся— точки сближаются. Таким образом, распределение То
трансформируется в распределение ть т2 и т. п. с постепенно
увеличивающимся градиентом концентрации. При «обрывном»
фронте действие закона Викке прекращается и все точки рас-
пределения (Т5 и т. п.) распространяются с постоянной ско-
ростью U, определяемой уравнением Вильсона (8.24): U}<iU<
<U2.
Выпуклая изотерма, десорбция. В тех же условиях (вид изо-
термы, начальное распределение) изменим направление движе-
ния потока на обратное-—пусть оно совпадает с направлением
роста концентрации в слое (рис. 8,7).
* «Обрывным» в динамике адсорбции называют фронт такой гипотетической
формы, при которой концентрация адсорбтива скачкообразно изменяется
от с=са до с=0 (Прим, ред.)
182
8. Динамика адсорбции
Рис. 8,6. Выпуклая изотерма (а) и кривые распределения концентрации в слое адсор-
бента (б) при адсорбции.
Так как то при выбранном направлении движения
потока трансформирования распределения то в п, затем в т2
и т. п. проводят к увеличению расстояния между концентраци-
онными точками 1 и 2.
Равновесная изотермическая десорбция — вполне реальный
режим динамики. Ему, в частности, соответствуют данные по
десорбции этилена из слоя цеолитов NaX, рассмотренные в
гл. 7.
Если бы начальное распределение отвечало «обрывному»
фронту (подобному распределению Т5 на рис. 8,6), то действие
закона Викке не проявилось бы и фронт перемещался бы со
скоростью, предписанной уравнением (8.24), параллельно са-
мому себе.
Линейная изотерма, адсорбция и десорбция. Для линейной
изотермы уравнение (8.23) совпадает с уравнением (8.24). По-
этому независимо от направления движения потока и вида на-
чального распределения концентрационные точки перемещают-
ся по слою с постоянной и равной скоростью (17| = 17 = П2) и
любое начальное распределение сохраняет свою конфигурацию.
Вогнутая изотерма, адсорбция. Изотерма и трансформации
распределений показаны на рис. 8,8. Примем, что начальное
распределение такое же, как и для случаев, описанных выше.
Рис. 8,7. Выпуклая изотерма (а) и кривые распределения концентраций в слое адсор-
бента (б) при десорбции.
. Математические модели динамики адсорбции 183
Согласно изотерме, (da/dc}i<2 (da'idc}2, что в соответствии
с уравнением (8.23) дает Ul>U2. Таким образом, в рассматри-
ваемых условиях распределение концентраций типа То имеет
тенденцию к неограниченному «размыванию». «Обрывное» рас-
пределение концентраций, очевидно, сохраняет свою форму.
Вогнутая изотерма, десорбция. При подаче потока в направ-
лении роста концентраций первоначально размытое распреде-
ление концентраций в случае вогнутой изотермы постепенно пре-
вращается в распределение со все более и более значительным
градиентом концентраций (сжимается) и, наконец, — в «обрыв-
ный» фронт. Последний перемещается по слою с постоянной
скоростью, определяемой уравнением Вильсона.
Приведенное рассмотрение простейшей модели динамики
сорбции позволяет сделать ряд важных заключений:
1. Форма изотермы адсорбции в зависимости от начальных
условий и направления движения потока является фактором,
способствующим сжатию или расширению перемещающегося
по зернистому слою фронта сорбции.
2. Перемещение фронта сорбции по зернистому слою мо-
жет быть явлением стадийным (например, «размытое» распре-
деление преобразуется в «обрывное»), причем на каждой из
стадий проявляется действие своих характерных законов.
Процесс с учетом, продольной диффузии
В условиях рассмотренной задачи содержались предположения
о том, что скорость массообменных процессов бесконечно вели-
ка, продольная диффузия отсутствует. Рассмотрим действие
второго из этих факторов на примере системы (8.21).
Режим адсорбции при D*^=0 характеризуется тем, что од-
новременно действуют два фактора. Первый фактор — продоль-
ная диффузия, которая вызывает размывание адсорбционного
Рнс. 8,8. Вогнутая изотерма (а) и кривые распределения концентраций в слое адсорбен-
та (б) при адсорбции.
184
8. Динамика адсорбции
Рис. 8,9. Передвижение сформировавшегося фронта сорбции по слою адсорбента (а>
и выходная кривая (б).
фронта. Второй фактор связан с законом Викке и, следова-
тельно, с видом изотермы адсорбции.
Выпуклая изотерма, адсорбция. Рассмотрим случай, когда
в зернистый слой, первоначально свободный от адсорбата, вво-
дят поток при некоторой концентрации адсорбтива. В связи с
эффектом продольной диффузии первоначальное «обрывное»
распределение концентраций адсорбтива начнет размываться.
Но поведением размытого фронта управляет закон Викке. При
выпуклой изотерме адсорбции, как было показано выше, дейст-
вие закона Викке приводит к сжатию сорбционного фронта.
Естественно предположить, что при наличии двух противопо-
ложно действующих эффектов по истечении достаточно продол-
жительного времени действие их компенсируется, и фронт сорб-
ции начнет перемещаться по слою с постоянной скоростью в ре-
жиме параллельного переноса. Такая гипотеза была выдвинута
Зельдовичем и развита Тодесом [228].
Для стадии параллельного переноса фронта сорбции воз-
можно аналитическое решение системы (8.21). Оно осуществ-
ляется путем замены двух координат (х, т) одной координа-
той z, начало которой перемещается вместе с фронтом сорбции.
Связь между исходными координатами и новой координатой
дается уравнением
z = x — Ux (8.25)
где U — скорость движения сорбционной волны.
Граничные условия при
этом имеют следующий вид:
г=—оо; с = с0; а = о0;
2 = -|-оо; с = 0; о = 0;
dc/dz = 0'|
ac/dz = О J
(8.26)
Начальное условие не имеет смысла, так как рассматрива-
гтся асимптотическая стадия (г—>-оо). Решение (8.21) с уче-
Математические модели динамики адсорбции
185
том (8.25) и (8.26) приводит к выражению для скорости дви-
жения сорбционного фронта
U = wc0/(c0 + а0) (8.27)
совпадающему с уравнением Вильсона для «обрывного» рас-
пределения, и к уравнению профиля стационарного фронта:
С2
D*[(a0 + ca)/a0] Г de
г<с> = w J c-ic^fic) (8-28)
а
где ci, с2 — некоторые значения концентрационных точек на кривой распре-
деления.
Рассмотренный механизм образования стадии параллельного
переноса фронта сорбции приводит также к уравнению Шилова.
Обсудим аппарат и терминологию теории Шилова более под-
робно. На рис. 8,9а сплошной линией представлено стационар-
ное, перемещающееся параллельно самому себе распределение
концентраций в слое в фиксированный момент времени, а на
рис. 8,96 — изменение концентрации адсорбтива во времени в
постоянном сечении L неподвижного слоя.
Выберем на кривой стационарного распределения концентрации адсорб-
тива точку Ц, обеспечивающую равенство заштрихованных на рисунке пло-
щадей. Эта точка называется «центром тяжести» сорбционной
волны. Ее основное свойство таково. При подходе «центра тяжести» к не-
которому сечению слоя, например L, количество адсорбтива, введенного с по-
током в неподвижный слой, равно количеству адсорбата, содержащемуся в
слое адсорбента длиною L в состоянии равновесия. Это свойство вытекает
из равенства заштрихованных площадей. Площадь вверху — слева от «цент-
ра тяжести» — пропорциональна количеству адсорбата, которое еще в со-
стоянии поглотить слой. Площадь внизу — справа от «центра тяжести» —
пропорциональна количеству адсорбтива, не поглотившегося в слое.
Расположим в точке Ц начало координат (г=0), перемещающееся вме-
сте с сорбционной волной. Положительное направление z совпадает с поло-
жительным направлением оси х. Как указывалось, зона изменения концент-
рации от ci до с2 называется работающим слоем, илн зоной массопередачи;
ее обычно обозначают через Lo.
В некоторый момент времени концентрационная точка сь перемещавшая-
ся вместе с кривой распределения, подходит к сечению L. Положение кривой
распределения в этот момент времени обозначено на рис. 8,9а первой пунк-
тирной линией. Сечение L— это некоторая контрольная плоскость в непо-
движном слое (например, его окончание), в которой расположен детектор,
регистрирующий изменение концентрации адсорбата во времени. Запись по-
казаний детектора показана на рис. 8,96: она носит название выходной
кривой и подобно кривой распределении имеет «центр тяжести». Количе-
ство адсорбтива, пропущенного через слой к моменту появления на выходе
из слоя «центра тнжести» выходной кривой, поглошаетсн адсорбентом в со-
стоянии равновесии.
Связь между кривой распределения и выходной кривой мо-
жет быть установлена следующим образом. За промежуток вре-
186
8. Динамика адсорбции
мени Дт = Т2—ti точка с2, двигающаяся, как и все распределе-
ние, со скоростью U=wao/(ao+co), прошла расстояние Lq, т. е.
Дт = Lo/U (8.29)
где U — скорость движения фронта; t/= l/K=wo/(ao+cJ.
Фиксировать изменение концентрации во времени много
легче, чем определить распределение концентрации по длине
адсорбера. Именно в связи с этим для определения высоты ра-
ботающего слоя (зоны массопередачи) обычно используют вы-
ходные кривые.
Обсудим вопрос о выборе характерных точек (Ci и с2). В зер-
нистом слое существует непрерывный спектр концентраций и
«особыми» точками являются лишь Ci = 0 и с2 = со. Однако, как
установлено, для «особых» концентрационных точек режим па-
раллельного переноса недостижим.
С другой стороны, «центр тяжести» сорбционной волны
всегда перемещается с постоянной скоростью (это вытекает из
указанного выше его основного свойства), т. е. в режиме па-
раллельного переноса. Можно поэтому предположить, что вре-
мя, необходимое для практического установления режима па-
раллельного переноса, зависит от удаленности концентрацион-
ной точки от «центра тяжести» волны: чем дальше отстоит точ-
ка от центра тяжести, тем больший промежуток времени тре-
буется для стабилизации ее движения.
В качестве Ci можно, например, выбрать или максимально
допустимую в данном конкретном случае концентрацию адсор-
бата, или концентрацию, минимально определяемую имеющимся
анализатором. Выбранный из этих соображений уровень кон-
центрации носит название проскокового (терминология заим-
ствована из противогазового дела, где имеется в виду «проскок»
отравляющего вещества через коробку противогаза).
Время ti, отвечающее появлению за слоем адсорбента фик-
сированной длины проскоковой концентрации адсорбтива —
концентрации с\, носит название времени защитного
действия, или момента проскока тПр-
Обосновать практически разум-
ный уровень верхнего предела кон-
центраций существенно труднее
уже в силу того, что нарастание
концентраций за слоем инженера-
практика, как правило, не интере-
сует. При анализе эксперименталь-
ных результатов в качестве с2
Рис. 8,10. Время защитного действия слоя адсор-
бента при разном уровне проскоковых концент-
раций.
Математические модели динамики адсорбции
187
обычно выбирают точку, удаленную от «центра тяжести» волны
так же, как точка сР Из соображений одновременного переме-
щения точек Ci и Сч в режиме параллельного переноса их рав-
ноудаленность от «центра тяжести» следует устанавливать по
координатам г или т.
На рис. 8,10 приведены зависимости времени защитного дей-
ствия слоя от его длины для разных уровней «проскока».
Прямая, проходящая через начало координат, является трассой переме-
щения «центра тяжести» адсорбционной волны. Прямые, расположенные ни-
же ее, соответствуют проскоковым концентрациям, более низким, чем кон-
центрация в «центре тяжести». Прямые, расположенные выше трассы «цент-
ра тяжести», отвечают более высоким проскоковым концентрациям. Рисунок
в целом можно рассматривать в качестве графической иллюстрации к урав-
нению Шилова. Отсутствие режима параллельного переноса в начальный пе-
риод процесса проявляется в виде нарушений линейности зависимостей тПр—
х. На рис. 8,10 это иллюстрируют пунктирные линии, асимптотически прибли-
жающиеся к соответствующим прямым.
Режим параллельного переноса фронта сорбции и уравнение
Шилова справедливы для многих случаев адсорбции в непо-
движном слое. Для реализации этого режима необходимо су-
ществование двух эффектов, один из которых способствует сжа-
тию фронта сорбции, другой — его расширению. Природа каж-
дого из эффектов часто имеет второстепенное значение.
Выпуклая изотерма, десорбция. Эффект, компенсирующий
размывающее воздействие продольной диффузии, в данном слу-
чае отсутствует. Поэтому режим параллельного переноса не
устанавливается. Система уравнений (8.21) при необходимости
может быть решена численными методами. Приближенное опи-
сание процесса следует из уравнения Викке.
Линейная изотерма, адсорбция и десорбция. Как и в преды-
дущем случае, режим параллельного переноса не устанавлива-
ется. Из-за отсутствия эффектов сжатия фронта его протяжен-
ность непрерывно увеличивается.
Известны {229] точные решения для системы (8.21), в кото-
рой уравнение изотермы дается прямой, с начальными и гранич-
ными условиями (8.19) и (8.20), но они сложны и на практике
не используются. Достаточно простым и удобным является
уравнение следующего вида:
_у
где Л=£)*Г/(1+Г); а0/с0 — коэффициент Генри; erf (у) = (2Дя) | ехр(—y2)dy—
'о
интеграл Крампа (функция erf (г/) табулирована).
Анализ показывает, что с постоянной скоростью перемещает-
ся только «центр тяжести» кривой распределения, которому
отвечает точка с/со=О,5. Фронт в целом размывается пропор-
ционально У Нт.
188
8. Динамика адсорбции
Вогнутая изотерма. Десорбция в случае вогнутой изотермы
описывается теми же соотношениями, что адсорбция при вы-
пуклой. Аналогичным образом адсорбция при вогнутой изотер-
ме описывается так же, как десорбция при выпуклой.
Динамика неравновесной изотермической
адсорбции
Процесс при отсутствии продольной диффузии
Следуя принятой схеме, рассмотрим вначале модель динамики
неравновесной изотермической адсорбции при пренебрежимо
малом влиянии продольнодиффузионных эффектов, а затем
укажем способы их учета. Именно эта модель наиболее часто
рассматривается в литературе.
Система дифференциальных уравнений, описывающих нерав-
новесную изотермическую динамику сорбции, такова:
да/дт + дс/дт -|- w (да/дх) = О
а* = /(с)
да/дт = ср (а, с)
(8.31)
В этой системе уравнений последняя строчка представляет
собой уравнение кинетики в общей форме. В качестве началь-
ных и граничных условий примем соотношения (8.19) и (8.20).
Система (8.31), так же как и система (8.21), общего решения
не имеет. Поэтому следует рассмотреть частные случаи.
Выпуклая изотерма, адсорбция. Очевидно, в данном случае
существуют предпосылки, необходимые для возникновения ре-
жима параллельного переноса фронта сорбции на асимптоти-
ческой стадии процесса. Конечная скорость массообмена вызы-
вает растяжение фронта сорбции, выпуклая изотерма способст-
вует его сжатию. Таким образом, зависимость времени защит-
ного действия слоя от высоты его будет описываться уравнени-
ем Шилова.
Подстановка уравнения движения стационарной волны в
уравнение материального баланса и интегрирование полученно-
го выражения с учетом граничных условий приводит к следую-
щему фундаментальному соотношению асимптотической стадии
динамики неравновесной сорбции:
с/с0 = а/а0 (8.32)
Это уравнение носит название соотношения Зельдовича и
устанавливает связь между неравновесными концентрациями
адсорбата в неподвижном слое. Графическая интерпретация
этого уравнения и его соотношение с изотермой адсорбции яс-
ны из рис. 8,11.
Математические модели динамики адсорбции
189
Рис. 8,11. К расчету движущей силы адсорбционного
процесса.
Для уравнения кинетики, заданно-
го в форме (8.15), движущая сила про-
цесса при текущей концентрации ад-
сорбтива в потоке с равна: Дс = с—с*
(где с* — текущая концентрация ад-
сорбтива в потоке, равновесная теку-
щей величине адсорбции а). Для
уравнения кинетики в форме (8.15а)
движущая сила в тех же условиях со-
ставляет Аа — а*—а (где а* — величина адсорбции, равновесная:
текущей концентрации адсорбтива).
Длина зоны массопередачи при известном коэффициенте
массопередачи в случае уравнений (8.15а) и (8.15) вычисляет-
ся из следующих формул:
°2
L<> = U J рт (а* — а) (8.33).
= ш <8-34)i
Имея из опытов по динамике адсорбции экспериментально
определенные значения высоты работающих слоев, можно ре-
шить обратную задачу: рассчитать в каждом конкретном слу-
чае коэффициент массопередачи, например по уравнению
С2
r _ ЛД С dc
Рг~ Lo J с —с*
(8.35>
Интеграл в правой части этого уравнения называют чис-
лом единиц переноса и обозначают как N.
Движущую силу процесса (Дс = с—с*) определяют на раз-
личных участках интервала щ—Сг. После этого строят зависи-
мость 1/Дс=/(с) и находят значение интеграла в правой части
уравнения (2.35), т. е. число единиц переноса N.
Расчетные значения коэффициентов массопередачи, а также числа единиц
переноса N и отношения высоты работающего слоя к числу единиц переноса,
характеризующие высоту единицы переноса Н, для случая рекуперации па-
ров бензола (с0=1%об.) в слое микропористых углей типа СКТ с разным
размером гранул, по данным работы [230], приведены в табл. 8-2; скорость
потока составляла 0,33 м/с. В этих условиях процесс массопередачи лими-
190
8. Динамика адсорбции
Таблица 8-2. Характеристика массопередачи при извлечении бензола
в слое микропористых активных углей типа СКТ, различающихся
размером гранул drp (скорость потока газа 0,33 м/с)
d гр t, °C Lq, cm N H, см ₽т, с-1 d гр t, °C Lo, см N Н, см Рт, с—1
1,0 25 1,8 2,27 0,84 39,8 1,9 25 3,9 2,84 1,39 24,3
50 2,3 2,56 0,90 40,1 50 5,0 3,40 1,47 24,6
100 3,8 3,56 1,07 41,2 100 7,2 3,84 1,88 23,5
150 6,1 4,88 1,25 37,9 150 10,0 4,81 2,08 22,8
1,3 25 2,0 2,52 0,79 42,0 3,0 25 6,8 2,41 2,82 11,8
50 2,4 2,77 0,87 41,7 50 9,6 3,10 3,10 11,7
100 4,2 3,69 1,14 38,5 100 15,0 3,98 3,77 П,7
150 7,2 5,68 1,27 37,3 150 23,7 5,74 4,13 11,5
тировался исключительно внутренней диффузией. Для определения N ис-
пользовано уравнение (8.33). Поэтому
Q2
“1
da
(а* — а)
Размер гранул активного угля оказывает определяющее влияние на мас-
сообмен при адсорбции углеводородов в слое активного угля: увеличение
размера гранул в три раза приводит к снижению коэффициента массопере-
дачи приблизительно в 3,3 раза.
С повышением температуры процесса увеличивается число единиц пере-
носа (рис. 8,12) и высота единицы переноса (рис. 8,13), однако значение ко-
эффициента массопередачи остается приблизительно постоянным (рис. 8,14)
[230]. Как показывает табл. 8-3, увеличение концентрации адсорбтива в ис-
ходном газе приводит к увеличению высоты работающего слоя и числа еди-
ниц массопереноса, однако в незначительной степени сказывается на высоте
единицы переноса и, в конечном итоге, на значении коэффициента массопе-
редачи.
Пористая структура активных углей вносит определенные коррективы я
значение коэффициента массопередачи: чем больше развита микропористость,
тем выше коэффициент массопередачи при прочих равных условиях. Это об-
Рис. 8,12. Зависимость числа единиц переноса
слоем угля СКТ (с0 = 1.О% об.).
N от температуры при адсорбции бензола
Рис. 8,13. Зависимость высоты единицы переноса Н от температуры СКТ (Со=1% об.,
го=20 м/мин), при адсорбции бензола в слое углей (с0=1% об., w = 0.33 м/с).
Математические модели динамики адсорбции
191
Рис. 8,14. Зависимость коэффициента мас-
сопередачи 3 от температуры при извле-
чении примеси бензола из потока газа в
слое угля СКТ (Со»=1% об., до=20 м/мин).
50 Ч. 20 10 Фр =1,30 мм
1,00
1,62
3,06 j
273 323 373 423
ТА
стоятельство отчетливо видно из графика на рис. 8,15, где представлены зна-
чения (Зт при адсорбции бензола в слое микропористых углей с одинаковым
размером гранул (drp=l,5 мм), но различающихся константой предельного
адсорбционного объема И70.
В последние годы для решения задач по неравновесной ди-
намике сорбции стали широко использовать цифровые вычисли-
тельные машины [215, 231, 232]. Рассмотрим на примере ра-
боты [215] круг вопросов, которые можно решить с помощью
вычислительной техники.
Для большей общности обсуждения введем безразмерные
параметры: y = cjcn (безразмерная текущая концентрация ад-
сорбтива в потоке), -у = а/ао (то же, в твердой фазе) и е =
(безразмерная длина), т'= (со/ао) рт (безразмерное время) и
у* = с*1са (безразмерная равновесная концентрация). С по-
мощью указанных параметров система
да/дх + дс/дх + w (дс/дх) = 0 1
> (о.00)
да/дх = ₽г (с — с*) J
преобразуется к более простому виду:
ду/де = -(у- у*) 1 (8 37)
ду/дх’ = у — у* J
Начальные и граничные условия в новых переменных запи-
шутся следующим образом:
т' = 0; е>0; у = ехр (—е); у = 0 (8.38).
т' = 0; е = 0; у=1; у = у (т') (8.39),
Все эти преобразования несложны, и в пояснении нуждает-
ся лишь начальное условие # = ехр(—е). Экспоненциальная за-
Таблица 8-3. Характеристика массопередачи при извлечении бензола
в слое активного угля СКТ (drp= 1,9 мм) при 25 °C
и скорости потока 0,33 м/с
Со, % об. Lq, см N И, см Вт, с-1
0,5 2,99 2,10 1,42 23,3
1,0 3,90 2,84 1,39 24,3
2,0 6,25 4,78 1,31 25,5
192
8. Динамика адсорбции
висимость концентрации от длины слоя в начальный момент
времени является следствием так называемого мгновенного
проскока, распространяющегося по слою со скоростью движе-
ния потока.
В работе [215] рассмотрен класс задач, для которых равно-
весная изотерма сорбции описывается уравнением
У* = PY(1 +Р— У)-1 (8-40)
где (р^О) —параметр, определяющий форму изотермы.
При р = 0 изотерма превращается в «прямоугольную», значе-
нию р—>°о соответствует линейная изотерма.
Формула (8.40) является аналогом уравнения Ленгмюра;
•она позволяет аппроксимировать большое число равновесных
кривых. Использование безразмерных параметров и уравнения
типа (8.40) дает возможность отвлечься от конкретных значений
концентрации, скорости, времени, интенсивности массообменных
процессов и т. п. и получить решение в достаточно общей
форме.
Для решения полулинейной гиперболической системы (8.37) с учетом
(8.38) и (8.39) в работе [215] был использован метод сеток. При разработ-
ке программы иа печать вывели координаты линии у и у и сравнение у и у
в точках сетки. Это позволило рассмотреть развитие сорбционного процесса
во времени.
На рис. 8,16 представлена номограмма изменений у, у в зависимости от
х' и е при р=0,5. Движение точки у=const, как следует из рисунков, начи-
нается с момента т'=0. Точка y=const начинает движение от 8=0 в момент
времени, зависящий от р и определяемый из граничных условий. Рассмотре-
ние номограммы позволяет выделить две стадии процесса. На первой стадии
скорости движения точки у=const вдоль е меньше, чем скорость движения
точки y=t/=const. Скорости движения точек y=const и t/=const постепенно
сближаются и, начиная с некоторых значений е, х', зависящих от р и у, ста-
новятся равными друг другу с любой наперед заданной степенью точности
а=[(у—у)1у] • 100. Это, очевидно, соответствует параллельному перемеще-
нию фронта сорбции со скоростью U= wcol (До+со). Разные значения а обо-
значены на рис. 8,16 штрих-пунктирными линиями. Подобные номограммы
достаточно удобны для расчетов.
Обычно встречаются два вида задач — прямая и обратная. Прямая за-
дача соответствует определению распределений концентрации адсорбата в
зернистом слое при известном кинетическом коэффициенте (коэффициенте
массопередачи). Эта задача с помощью номограмм решается путем преобра-
зования безразмерных координат е и х' в координаты хит. Очевидно, при
этом одновременно решаются две более узкие задачи: определение выходной
кривой, т. е. зависимости с=с(т) при x=const, и соотношения между хит
для «проскоковой концентрации» [т=т(х) при с~const].
30
10
%= 0,44cm3F
273 323 373 423
7, К
Рис. 8,15. Зависимость коэффициента мас-
сопередачи Э от температуры при извле-
чении бензола в слое углей (drp=l,5 мм)
с разной константой
Математические модели динамики адсорбции
193
Рис. 8,16. Номограмма для расчета адсорбционного процесса при р=0,5.
Обратная задача, обычно встречающаяся в работах исследовательского
профиля, состоит в определении jjr по экспериментальной выходной кривой.
Эту задачу по номограммам решают методом последовательных приближе-
ний. Задавшись некоторым значением 0Г для точки r/=const, определяют е
и т', по номограмме устанавливают, соответствует ли фактическое значение у
значению, определяемому данными координатами е и т'. На основе сравнения
вносят коррективы в £г, определение повторяют и т. д. — до получения удов-
летворительного результата.
Линейная изотерма, адсорбция и десорбция. Отсутствие эф-
фектов, способствующих сжатию фронта сорбции, приводит к
его расширению, пропорциональному Ут, в силу конечного зна-
чения кинетического коэффициента.
Для уравнения кинетики в форме (8.15) в литературе при-
ведены приближенные аналитические решения. В качестве при-
мера приведем формулу Жуховицкого — Забежинского — Тихо-
нова [211]:
/0 = VLiw-V — (8.41)
где 6 = Ф-1 (1—с/О,54со); Ф-1—функция, обратная функции Крампа (табули-
рована).
Другие системы. Математическое моделирование динамики
адсорбции и решение системы соответствующих уравнений на
ЭВМ для других сочетаний изотерм сорбции с направлением
процесса осуществляется так же, как в случае адсорбции при
выпуклой изотерме. Качественные представления о протекании
процесса можно получить, если учесть, что размывающее воз-
13—1346
194
8. Динамика адсорбции
действие иеравновесности процесса на профиль распределения
концентраций по своему проявлению аналогично размываю-
щему воздействию продольной диффузии.
Процесс с учетом, продольной диффузии
Система уравнений, описывающих динамику неравновесной
изотермической адсорбции одного вещества в неподвижном зер-
нистом слое при наличии продольной диффузии, такова:
да/дт + дс/дт + w • да/дх = D* -д2с/дх2 '
a = f (с)
да/дт = ср (а, с)
(8.42)
Система (8.42) аналитического решения не имеет. Рассмот-
рим поэтому некоторые примеры.
Выпуклая изотерма, адсорбция. Этот случай является прак-
тически единственным, для которого предприняты определен-
ные попытки решения указанной задачи. На асимптотической
стадии для описания динамики адсорбции пригодно уравнение
Шилова. Из уравнений материального баланса и кинетики си-
стемы (8.42) для той же стадии можно получить [210] соотно-
шение между неравновесными концентрациями а и с. Оно имеет
следующий вид:
da _ Д* (ао + со)2 <Р с)
de w2 ’ с0 'с — (с0/а0) а \ >
Это дифференциальное уравнение является уравнением с не-
разделяющимися переменными, и для его решения приближен-
ными методами необходимо знать конкретный вид функции
<р(а, с).
Еникеева [233] показала, что при решении обратной задачи
динамики сорбции, когда известна выходная кривая адсорбции,
получаемая в опытах информация достаточна для оценки влия-
ния продольной диффузии. Уравнение, связывающее текущие
концентрации адсорбата, записывается следующим образом:
Д* («о + с0)2 de Oq_
а ~ w2 с02 dT п- с с0
(8.44)
где dc/dT — производная выходной кривой в точке с.
Так как слагаемые уравнения (8.44) положительны, то зна-
чения а = а(с) при D*^=0 будут больше, чем при D* = 0. Соотно-
шение между изотермой адсорбции, прямой Зельдовича (£)* = 0)
и линией, отвечающей уравнению (8.44), показано на рис. 8,17.
Как видно из рисунка, наличие продольной диффузии фактиче-
ски проявляется как фактор, уменьшающий движущую силу ад-
Математические модели динамики адсорбции
195
Рис. 8,17. Изотерма адсорбции и «рабочие» ли-
вни при D*=0 и D*#=0. а,
,а«
сороционного процесса и тем са-
мым снижающий эффективность
массообмена. При наличии выход-
ной кривой влияние продольной
диффузии легко оценить по уравне-
нию (8.44). Эффективный коэффи-
циент продольной диффузии может
быть определен по источникам, ука-
занным выше.
В адсорбционной лаборатории
МХТИ им. Д. И. Менделеева оценка
влияния продольной диф-
фузии была проведена более чем для десяти систем, различа-
ющихся типом адсорбента, адсорбтива и газа-носителя, агре-
гатным состоянием потока и, конечно, условиями опытов. Из
этих опытов вытекает, что на асимптотической стадии при про-
мышленно разумных скоростях потока (практически это ско-
рости, снимающие внешнедиффузионное сопротивление) размы-
вающий эффект продольной диффузии пренебрежимо мал по
сравнению с размывающим эффектом конечной скорости массо-
обменных процессов.
Влияние продольной диффузии, как отмечалось ранее, физи-
чески проявляется через снижение движущей силы процесса.
Однако формально возможен и другой подход. Можно считать
соотношение между а и с неизменным и рассматривать про-
дольную диффузию в качестве фактора, уменьшающего эффек-
тивное значение коэффициента массопередачи. Для «ленгмю-
ровской» изотермы и уравнения кинетики в форме (8.15), как
показано Тодесом и Биксоном [234, 235], такой подход приво-
дит к следующему уравнению аддитивности сопротивлений:
Д_________1_ 1 D*
Ро ~ Рг + Рт + W2
(8.45)
Использование уравнения (8.45) сводит систему (8.42) к
существенно более простой и лучше изученной системе (8.31).
Линейная изотерма. При линейной изотерме кинетическое
размывание можно рассматривать как квазидиффузионный
процесс, характеризуемый коэффициентом квазидиффузии D*K.
В рассматриваемой задаче кроме кинетики сорбции действует
продольная диффузия, имеющая эффективный коэффициент D*.
Результирующую квазидиффузионную константу можно рас-
сматривать как сумму обеих констант:
02* = DK*-|-D‘ (8.46)
13
196
8. Динамика адсорбции
Отсюда общее уравнение будет таково:
дс дс с0 д2с
дх **w дх ~ 2 а0 4- Ср дх2
(8.47)
Решение этого уравнения было приведено ранее.
Динамика неизотермической адсорбции
Рассмотренные выше математические модели динамики сорб-
ции основаны на предположении о пренебрежимо малой вели-
чине теплового эффекта 'адсорбции. Это допущение справедли-
во при небольших концентрациях адсорбтива в потоке. Однако
при повышенных концентрациях тепловой эффект может ока-
зывать существенное влияние на протекание адсорбционного
процесса и нуждается в учете. Учет тепловыделений и тепло-
обмена необходим также в тех случаях, когда температуры по-
тока и зернистого материала различны. Из неизотермических
процессов наиболее подробно изучена и описана динамика адиа-
батической адсорбции, при которой потери тепла в окружаю-
щую среду принимаются пренебрежимо малыми.
Обсудим динамику адиабатической адсорбции, предполагая
в первом приближении, что массообмен и теплообмен между
фазами системы происходят бесконечно быстро. Такой анализ
впервые был выполнен Тодесом и Лезиным [236]. Подобно рав-
новесной изотермической динамике сорбции, равновесная адиа-
батическая модель, несмотря на известную условность, позво-
ляет выявить ряд принципиальных особенностей процесса.
Основные уравнения модели могут быть записаны следую-
щим образом:
да дс дс
дх + dx +w~dT = 0
„ дТ дТ п да_ . (8.48)
Н дх +whr дх + дт ~°
а* = f (с, Т)
где Н—суммарная теплоемкость адсорбента и адсорбата.
Начальные и граничные условия пусть будут такими же, как
указано в соотношениях (8.19) и (8.20). При изотермической
сорбции, когда тепловым эффектом адсорбции можно прене-
бречь, скорость движения адсорбционного фронта составит
., С0 с0
Унзо = W - XW—-
“О “Г со ао
Из теплового баланса
ловой волны, измеренная
= whr/H
(8.49)
следует, что скорость движения теп-
ло ее «центру тяжести», будет равна
(8.50)
Математические модели динамики адсорбции
197
Рис. 8,18. Соотношение между сорбционным и тепловым фронтами (случаи а и б).
При протекании неизотермического процесса сорбционная и
тепловая волны будут двигаться взаимосогласованно. Аналити-
ческое решение системы (8.48) получено для режима парал-
лельного переноса при выпуклой изотерме адсорбции. Анализ
этого решения приводит к двум случаям:
1. Когда скорость тепловой волны (117) больше скорости дви-
жения изотермического фронта (t/изо), все выделяющееся при
адсорбции тепло отводится проходящим потоком, и адсорбция
протекает при холодном сорбенте. В этом случае теплота ад-
сорбции не оказывает влияния на процесс. По шихте распрост-
раняются две волны: тепловая со скоростью W при с = 0 и
сорбционная со скоростью U при с = с0 и а = а0. Движение этих
волн* иллюстрирует рис. 8,18а. Скорость движения сорбционной
волны и температура плато Т* определяются уравнениями
,, ,, со со
т* — ®а° U
1 Н ' W —и
(8.51)
2. Если скорость изотермической сорбционной волны (t/изо)
больше, чем скорость тепловой, то выделяющееся при поглоще-
нии тепло не успевает отводиться потоком. Это приводит к сни-
жению статической активности адсорбента в зоне поглощения.
На нагретом участке поглощается не весь адсорбат, а лишь
часть его. Поэтому по шихте распространяются две сорбцион-
ные волны, скорости движения которых задаются уравнениями
йг7* с'
= w НТ* -\-a'Q = w a' + c’ (8’52)
,, Cq -c’
U*~W Q(a0 — a')HT* ~w (c0 + a0)-(c' + a') (8‘53)
* Как показано в работе Харитонова Б. П. (Дисс., М., МВТУ им. Н. Э. Баума-
на. 1982), этот режим может наблюдаться только при нулевом начальном
заполнении адсорбента адсорбатом. (Прим, ред.)
198
8. Динамика адсорбции
При наличии изотерм сорбция (a Т*) в'се неизвест-
ные величины уравнений (8.52) и (8.53) могут быть найдены.
Графическое перемещение тепловой и адсорбционной волн по-
казано на рис. 8,186.
Сопоставление скоростей движения тепловой и изотермиче-
'ской адсорбционных волн позволяет априорно заключить, ка-
кой из режимов распространения волн реализуется в каждой
конкретной системе. Процедура такого сопоставления деталь-
но разработана Паном и Басмаджианом [237]. Они же предло-
жили достаточно удобную терминологию, которую мы будем
использовать в дальнейшем. Согласно [237], случай 1 Тодеса—
Лезина назван режимом одиночной тепловой волны, случай 2—
режимом комбинированной волны. Различают следующие уча-
стки стационарных волн: фронтальный (первая транспортная
зона), плато (средний участок) и тыловой (вторая транспорт-
ная зона).
Так как любое повышение температуры увеличивает ско-
рость движения адсорбционного фронта, то U>UU30 (где
Z7, 17ИЗО — скорости движения фронта в неизотермическом ре-
жиме и изотермических условиях). Если, далее
(8.54)
то из (8.49) и (8.50) следует, что П>17Изо>1^. Образование
одиночной волны в этих условиях невозможно, и неравенство
(8.54) является достаточным условием существования комбини-
рованной волны. Обращенное неравенство
Hlhr<aolco (8.55)
является необходимым условием существования одиночной теп-
ловой волны. Достаточное условие может быть получено из ана-
лиза соотношений для температуры плато. Для определения ее
максимального значения авторы работы [237] рекомендуют
следующее приближенное соотношение:
Т’ кс = Т„ 4- , . . (8.56)
макс и ' пг (а0/с0) + Н ’
Очевидно, достаточным условием существования одиночной
волны является более высокая скорость движения теплового
фронта, чем адсорбционного, даже при максимально возмож-
ной температуре слоя. Это соответствует неравенству <
ч* (Т*ыакс, с0) , Н
-------- >~h~ (8.57)
Со «г
Обращенное неравенство
ч* (7’*макс> со) Н
---( "акс--<т- (8.58)
Со rtT
Математические модели динамики адсорбции 1991
является необходимым условием существования комбинирован-
ной волны в области, удовлетворяющей неравенству (8.54). Ни-
же представлены различные сочетания установленных выше ус-
ловий и типов адсорбционных процессов:
Условие
(«о/со)>(////гг)
(ао/со)<(///Лг)
[а’(г*макс- со)/^о1>(///Лг) [а*(ГмаКс. «о)/со]< (Я/Лг)
Образуется одиноч- Возможны оба типа
ная тепловая волна волн
Образуется комбинированная волна
На рис. 8,19 показано влияние исходной концентрации ад-
сорбтива на тип образующихся волн. На этом рисунке пред-
ставлены две изотермы адсорбции: Т„ и Т'*Макс и луч, тангенс
угла которого равен H!hr. При исходной концентрации адсор-
бата Cq^Cqi отношение ао/со>Я//гг и a*/ca~>H]hT, что соответ-
ствует режиму одиночной тепловой волны. При концентрации
Со>с02 отношение aalct}<.H]hT и образуется комбинированная
волна. Если с01<со<со2, то, согласно приведенным данным,
возможны оба типа волн.
Следует отметить, что метод Пана—Басмаджиана пригоден
и для неизотермических процессов, в которых начальные тем-
пературы потока и слоя различны. В этом случае под Гн урав-
нения (8.56) следует понимать температуру потока на входе
в зернистый слой.
Теория равновесной адиабатической адсорбции позволяет
сделать некоторые важные практические выводы. В случае об-
разования одиночной тепловой волны процесс адсорбции имеет
тенденцию протекать независимо ог начальной температуры
слоя. Это означает, что слой будет нагрет или охлажден газом-
носителем до температуры плато к моменту подхода адсорбци-
онной волны и время защитного действия слоя адсорбента не
изменяется при изменении его начальной температуры. Следо-
вательно, в системах, удовлетворяющих условию (8.57), охлаж-
дение зернистого слоя после его термической регенерации про-
водить не обязательно. Это позволяет снизить продолжитель-
ность цикла, уменьшить расход га-
за на регенерацию и сократить ме-
таллоемкость установки. В ряде
практически важных систем, напри-
мер при осушке газов гидрофиль-
ными адсорбентами, условие (8.57)
соблюдается.
Рис. 8,19. Влияние исходной концентрации адсорб-
тива на реализацию режимов единичной и комби-
нированной волн.
200
8. Динамика адсорбции
Дальнейшее развитие теории динамики неизотермической
сорбции состояло в учете кинетики тепловых и массообменных
процессов. Лезин [238] рассмотрел модель, основанную на
предположении о бесконечно быстром протекании теплообмена
между газом и шихтой при массообмене, описываемом уравне-
нием кинетики (8.15). Основные уравнения модели записыва-
ются следующим образом:
да дс дс
~дТ+~д:г + а>~дГ^0
„ дТ дТ да 2ап
-Н~я=- — whr~^~ 4-Q—5— =—5s— Т
ат 1 дх ' * <5т R.
да
-^- = рг[с- <р(а, Г)]
(8.59)
Где ап — коэффициент теплоотдачи от стенок адсорбера в окружающую сре-
ду; R — радиус адсорбера.
Адсорбционное равновесие <р(п, Т) задавалось уравнениями
Ленгмюра и Дубинина—Радушкевича. Рассматривалось два
процесса: адсорбция и десорбция. Начальные условия при ад-
сорбции были таковы:
т = 0; с(х, 0) = а(х, 0) = Т (х, 0) = 0 (8.60)
При десорбции:
т = 0; с(х,0)=с0; а(х,0)—а0; Т(х,0)=0 (8.61)
Граничные условия при адсорбции:
х=0; т>0; с(О,т)=Со; а(0,т)=а(т); Г(0,т) = 0 (8.62)
Граничные условия при десорбции:
х = 0; т>0; с(0,т)=0; а (0, т) == а (т); Т(0,т) = 0 (8.63)
После исключения дс/дх (так как дс/дх^да/дх), перехода к
безразмерным переменным и замены дифференциальных со-
отношений вдоль характеристик конечноразностными уравне-
ниями система (8.59) была решена методом характеристик на
ЭВМ. Расчетные и экспериментальные выходные кривые, кри-
вые разогрева и охлаждения хорошо совпадали друг с другом.
В качестве примера работы, в которой учитывается кинети-
ка как массо-, так и теплообменных процессов, обсудим статью
Чи и Вазана [239]. В ней представлены модели процессов ад-
сорбции и десорбции паров воды в неподвижных слоях адсор-
бентов (силикагель и твердый носитель, пропитанный раствором
хлорида лития). В начале авторы рассматривают изотермиче-
скую модель. Основные уравнения этой модели, их приведение
к безразмерному виду принципиально не отличаются от урав-
нений и методик, использованных в работе [215]. Система урав-
Математические модели динамики адсорбции
20Г
нений была решена на ЭВМ модифицированным способом Эй-
лера и методом проб и коррекции.
Математическая модель адиабатической адсорбции включа-.
ла уравнения:
да дс дс
-dV + 'dT+w-&T = °
да
-^- = ₽г(с-с’)
д!я д! д!
+ = ° (8-64>
<5/а
~д~ = к (Тг - Та) + рг (с - с*) Q
c* = f(a,Ta)
где /а —энтальпия адсорбента; / — энтальпия потока; К — коэффициент теп-
лоотдачи, отнесенный к единице, объема зернистого слоя.
Начальные и граничные условия принципиально не отлича-
лись от краевых условий в работе Лезина [238]. Уравнения бы-
ли преобразованы путем введения безразмерных переменных е
и г'. По методикам, указанным выше, система уравнений с без-
размерными переменными была решена численными методами
на ЭВМ.
Значения коэффициентов тепло- и массообмена определяли
путем сопоставления результатов одного из опытов с резуль-
татами расчета. Полученные значения были использованы в
дальнейших расчетах. Авторы установили полную адекватность
адиабатической модели, если коэффициенты уравнений кинети-
ки определялись в реальном эксперименте.
Динамика адсорбции многокомпонентной смеси
Динамика адсорбции многокомпонентных смесей в изотермиче-
ских условиях в режиме параллельного передвижения сорбци-
онных фронтов теоретически впервые рассмотрена в работах Ду-
бинина и Явича [240]. На рис. 8,20 представлено распределение
двух адсорбатов по длине слоя адсорбента для некоторого, до-
статочно большого, фиксированного
момента времени. Слой адсорбента
ОА полностью насыщен смесью
обоих веществ. Он содержит Oi2
(первый индекс — номер рассмат-
риваемого компонента, второй —•
сопутствующего) первого компо-
Рис. 8,20. Динамика адсорбции бинарной смеси.
202
8. Динамика адсорбции
нента, находящегося в равновесии с начальной концентрацией
его в газовой фазе coi и со вторым компонентом, величина ад-
сорбции которого равна «21 при равновесной концентрации Сог-
Слой адсорбента АВ насыщен первым компонентом смеси,
но представляет собой работающий слой для второго компонен-
та. В слое ВС находится адсорбент, насыщенный только пер-
вым компонентом, а слой CD является зоной поглощения пер-
вого компонента.
Так как величина адсорбции первого компонента понижа-
ется в присутствии второго, то величина адсорбции его в обла-
сти ВС выше, чем в области ОА. Поэтому при перемещении ра-
ботающего слоя АВ вдоль длины слоя происходит частичное
вытеснение первого компонента, пропорциональное площади с
двойной штриховкой, из-за чего концентрация его на участке
ВС (с'1) выше начальной концентрации (cOi):
=== С-^ —J- occg (8.65)
где а — коэффициент вытеснения.
Поступательное перемещение работающих слоев АВ и CD
происходит со скоростями
1/3 = ^02/02! (8.66)
Ut = wc1'/a1 (8.67)
Анализ материальных балансов дает следующее выражение
для коэффициента вытеснения:
а = (oj — a^/og! (8.68)
который теоретически зависит только от равновесных факто-
ров— величин адсорбции. Отметим также, что, так как
расстояние между фронтами АВ и CD постепенно увеличивает-
ся. Представленная модель легко обобщается на случай сорб-
ции смеси с любым числом компонентов.
Решение более сложной задачи изотермической динамики
сорбции смесей, свободной от допущений о параллельном пере-
носе фронтов сорбции, было получено в работе Фролова и Лези-
на [241]. Применительно к двухкомпонентной смеси система
уравнений состояла из двух уравнений баланса и двух уравне-
ний кинетики сорбции:
да daj dct
+-5Л- + ш-5-£- = 0
<Эт ~ <5т дх
да;
-^- = Р4[сг — /(01, о2)]
(8.69)
где i— номер компонента.
В качестве уравнения равновесия использовалось уравнение
Бентона — Маркгёма.
Равновесие и динамика адсорбции
203
Начальные условия были таковы:
т = 0; с, (х, 0) = at (х, 0) = 0 (8.70)
Граничные условия:
т>0; х = 0; с; = (0, т) = coi (8.71)
Путем введения безразмерных переменных, аналогичных рас-
смотренным выше, система была преобразована в более удоб-
ную форму и, ввиду аналитической неразрешимости, была ре-
шена методом конечных разностей на ЭВМ. Концентрация ком-
понентов в каждой точке координатной сетки определялись в
результате ряда последовательных приближений.
9
АДСОРБЦИЯ СЛАБО
СОРБИРУЮЩИХСЯ ГАЗОВ
Равновесие и динамика адсорбции
Адсорбция слабо сорбирующихся газов имеет определенную
специфику. Первая особенность изотерм сорбции этих веществ
состоит в том, что их начальный участок обычно линеен и
описывается уравнением Генри. Чтобы повысить адсорбцион-
ную способность твердого пористого поглотителя по этим веще-
ствам, их адсорбцию часто проводят под высоким давлением.
Схема установки для исследования адсорбции газов под давлением при-
ведена на рис. 9,1 [242]. Основным аппаратом установки является цилиндри-
ческий адсорбер с внутренним диаметром 30 мм и длиной 1,35 м. В адсорбер
загружают око то 1000 см3 активного угля или другого адсорбента. Свобод-
ный объем адсорбера, включая объем между зернами сорбента и суммарный
объем пор, замеряют с помощью гелия. Уголь подготавливают к опыту (ре-
генерируют) продувкой водородом или гелием при атмосферном давлении
и температуре 300 °C. При регенерации адсорбер нагревают с помощью труб-
чатой печи, снабженной терморегулятором.
Для измерения изотерм адсорбции чистых газов в адсорбер из баллона
через буферную емкость накачивают газ до давления 10—20 МПа. После
того как давление в адсорбере, регистрируемое образцовым манометром, уста-
новилось, газ из адсорбера медленно выпускают; скорость выходящего газа
по реометру поддерживают постоянной — 300 см3/мин. Точный объем вышед-
204
9. Адсорбция слабо сорбирующихся газов
Рис. 9,1. Схема установки для изучения адсорбции газов под давлением:
1 — баллон с газом — адсорбтивом; 2 — буферная емкость; 3 — манометры; 4 — адсорбер;
5 — трубчатая электрическая печь; 6, 7 — реометры; 8 — газоанализатор; 9 — термостат.
шего газа определяют в газометре. Через определенные промежутки времени
отбор прекращают. Систему выдерживают, замеряют количество выделенного
газа и равновесное давление в системе. Затем отбор газа возобновляют и
т. д. до тех пор, пока не будет достигнуто атмосферное давление. Остаток
газа из адсорбера выдувают водородом или гелием при атмосферном дав-
лении.
Если V; — общий объем газа, десорбированного при снижении давления
от максимального р до атмосферного, Ё2 — объем газа, выделенного при от-
дувке водородом, Е3 — объем десорбированного газа к моменту данной оста-
новки отбора, 1/'4 — объем газа, заполняющего свободное пространство адсор-
бера при давлении р и температуре опыта t (все с учетом коэффициента
сжимаемости), тогда объем адсорбированного в данный момент газа (в см3)
определяется уравнением
^=(^ + ^-(^ + ^4) (9-1)
а изотерма выразится функцией (в см3/г)
a=E/g = f(p) (9.2)
где g — загрузка адсорбента, г.
Фомкиным и Серпинским [243] создана установка, позволя-
ющая производить измерение адсорбционной способности в ши-
роком интервале давлений (от 0,1 до 1,5-104 кПа) и температур
(от 100 до 500 К) •
Установка состоит из двух частей — стеклянной и металлической. В стек-
лянной части, принципиально не отличающейся от описанной в гл. 2 объем-
ной установки, измерения давлений производят, используя U-образный ртут-
ный манометр в комплекте с катетометром и компрессионный манометр.
Часть адсорбтива переводят с помощью жидкого азота в ампулу с адсор-
бентом, расположенную в металлической части. Затем ампулу помещают в
термостат или криостат, адсорбент нагревают до температуры замера и та-
ким образом создают соответствующее давление в системе, определяемое
грузопоршневым манометром.
Равновесие и динамика^ адсорбции 205
Рис. 9,2. Изотермы избыточной адсорбции газов
иа микропористом активном угле при 20 °C.
Результаты замера адсорбцион-
ной способности углей АГ-2 и F-23
(импортный уголь F-23 по структу-
ре близок к отечественному СК.Т)
при 20°C по метану, оксиду углеро-
да, азоту и водороду представлены
на рис. 9,2 и в табл. 9-1.
Ниспадающая ветвь изотерм от-
ражает тот факт, что при расчете
по уравнениям (9.1) и (9.2) опре-
деляется так называемая избы-
точная адсорбционная спо-
собность, соответствующая раз-
ности между общим объемом ад-
сорбированного газа и объемом
сжатого газа в свободном прост-
ранстве пор. Рост плотности газа в
свободном пространстве по мере увеличения давления и приво-
дит к снижению избыточной величины адсорбции. Таким обра-
зом, избыточная адсорбция совпадает с гиббсовым определени-
ем адсорбции и гиббсовым избытком Г. Изотермы указанного
вида неоднократно упоминались в литературе.
Полная и избыточная адсорбции связаны как
аполн — ^избРа/СРа Рг)
(9.3)
где ра — плотность газа в адсорбированном состоянии; рг — плотность газа
в газовой фазе при заданном давлении. Значения рР приводятся в справочной
литературе; величину ра можно определить по методу Дубинина — Николае-
ва, описанному в гл. 2. Этих данных достаточно, чтобы оценить полную ве-
личину адсорбции по избыточной.
Изучение равновесия хлортрифторметана, ксенона, крипто-
на [243] в широком интервале давлений и температур показа-
Таблица 9-1. Адсорбционная способность (избыточная)
активного угля АГ-2 по газам (в см3/г)
Адсорбтив Давление, МПа
1,0 2,0 3,0 4,0 5,0 6,0 7,0 8,0 9,0 10,0
Метан 37 50 53 56 54 52 49 42 37 30 Оксид углерода 30 40 46 48 49 48 48 — — — Азот 30 42 47 48 49 44 40 34 39 20 Водород 235677 6 6 5 4
206
9. Адсорбция слабо сорбирующихся газов
/зВ2,МПа
Рис. 9,3. Изотермы адсорбции метана и водорода
на активном угле при поглощении из техническо-
го водорода (Ссн4 = 15% об., /=20"С).
ло, что переход системы в закрити-
ческую область происходит без
скачков и не сопровождается изме-
нением наклона изостер. В связи
с этим можно рассчитать адсорбци-
онное равновесие при высоких дав-
лениях на основе данных, получен-
ных для области парообразного со-
стояния вещества при относительно низких давлениях и темпе-
ратурах.
Сопоставление изотерм адсорбции (см. рис. 9,2) показыва-
ет, что адсорбируемость метана, оксида углерода и азота намно-
го превышает адсорбируемость водорода; этот факт указывает
на возможность глубокой очистки водорода от названных га-
зов, часто являющихся примесями водорода. Адсорбционная
способность этих газов резко возрастает с увеличением давле-
ния до 5—6 МПа, что и определяет целесообразность исполь-
зования повышенного давления в процессе очистки.
На установке, описание которой приведено выше, может
быть изучена адсорбция не только чистых газов, но и смесей.
Для этого содержащий примеси водород из баллона при постоянном дав-
лении пропускают через адсорбер 4. Все опыты проводят с одинаковой ско-
ростью газа на выходе, устанавливаемой реометром 6. Состав газа контро-
лируют по теплопроводности газоанализатором 8, помещенным в термостат 9.
В первых порциях выходящего газа примеси как правило не обнаруживают-
ся; степень очистки близка к 100%. Затем содержание примесей нарастает и,
наконец, наступает равновесие, о чем свидетельствует тождество составов
входящего и выходящего газов. После наступления равновесия дросселиро-
ванием давления с одновременным нагревом адсорбера до 300 °C проводят
десорбцию поглощенных веществ, остаток газа из свободного объема ад-
сорбера отдувают водородом. Суммарное количество выделенных компонен-
тов за вычетом их содержания в свободном объеме адсорбера в условиях
опыта определяет состав адсорбированной фазы.
На рис. 9,3 представлены изотермы адсорбции метана и во-
дорода на угле F-23 при их поглощении из технического водо-
рода, содержащего 15% (об.) примеси — метана [244].
Из графика видно, что адсорбционная способность по метану возраста-
ет с повышением давления, достигая 40 см3/г при 2 МПа. Однако сравнение
точек изотермы чистого метана (верхняя линия) и изотермы метана в
присутствии водорода при равном парциальном давлении показывает, что
присутствие даже такого плохо сорбирующегося газа как водород значи-
тельно (в 2 раза) снижает адсорбционную способность по примеси. Анало-
гичные выводы сделаны при анализе изотерм адсорбции оксида углерода
и азота.
Одновременно в присутствии примесей снижается адсорбционная способ-
ность по водороду: при поглощении чистого водорода на угле F-23 она
Равновесие и динамика! адсорбции
207
достигает, как указывалось выше, 30 см3/г, а в присутствии примесей — лишь
4—8 см3/г.
Таким образом, при расчете глубокой очистки газа от плохо
сорбирующихся примесей при обычных температурах следует
принимать во внимание совместную адсорбцию газа-разбавите-
ля и снижение в связи с этим адсорбционной способности по
извлекаемым компонентам.
Эффективность процесса очистки газа от плохо адсорбирую-
щихся примесей может быть повышена проведением процесса
при низких температурах. Низкотемпературную очистку следует
особенно рекомендовать в тех случаях, когда примесь присут-
ствует в небольших концентрациях. Адсорбция микропримесей
в этих условиях имеет ряд дополнительных особенностей.
На рис. 9,4 приведены кривые зависимости адсорбционной способности
промышленных адсорбентов (цеолитов, активного угля, силикагеля) по мик-
ропримеси азота при температуре 77,5 К (—195,8 °C) от давления. Концент-
рация азота в газе, подлежащем очистке, составляла 0,12% (об.). Для всех
адсорбентов равновесная активность проходит через максимум при 2 МПа.
Максимум более отчетливо выражен в случае активного угля и силикагеля.
Для этих адсорбентов дальнейшее повышение давления вплоть до 15 МПа
приводит к снижению адсорбционной способности на 40% относительно мак-
симального значения. Активность цеолитов в зависимости от давления ме-
няется весьма незначительно (в пределах 6% от максимального значения) и
при высоком давлении превосходит активность угля и силикагеля. Подобное
поведение системы может быть объяснено наличием специфической составля-
ющей во взаимодействии молекулы азота, обладающей значительным квадру-
польным моментом, со щелочными и щелочноземельными катионами, входя-
щими в состав кристаллита. Для угля и силикагеля такое взаимодействие
не характерно, и в них по мере повышения давления усиливается адсорбция
водорода, а адсорбционная способность по азоту уменьшается.
Изотермы адсорбции азота на всех типах промышленных ад-
сорбентов при давлении 15 МПа и температуре 77,5 К
(—195,8°C) представлены на рис. 9,5 [245].
Рнс. 9,4. Равновесная адсорбционная способность промышленных адсорбентов по приме-
си азота (с0—0,12%), поглощаемого из водорода при Т=11 К:
I — активный уголь СКТ; 2 — цеолит NaX; 3 — цеолит СаА; 4 — мелкопорнстый силика-
гель.
Рис. 9,5. Изотермы адсорбции азота при Т=77 К, р=15 МПа иа адсорбентах:
I — активный уголь; 2 — мелкопористый силикагель; 3 — цеолит NaX; 4 — цеолит СаА.
208
9. Адсорбция слабо сорбирующихся газов
Интервал изменения концентрации азота на этих кривых составлял от,
0,06 до 0,9%. Все изотермы имеют выпуклую форму, однако крутизна изо-
терм на цеолитах выше в области малых концентраций. Поэтому активность
цеолитов в области концентраций до 0,2% больше соответствующих значений
для активного угля и силикагеля. При более высоких концентрациях азота
проявляется преимущество активного угля и силикагеля. Данные об адсорб-
ционной емкости адсорбентов по азоту для двух температур приведены
ниже:
т, к (°C) 77,5(—195,8) 67,5(—205,8)
Со, % (об.) а, см3/см3 0,08 0,19 0,47 0,91 0,08 0,19 0,47 0,91
уголь СКТ 57,6 85,8 109,0 127,0 82,3 109,0 125,5 146,0
силикагель № 6 61,2 78,8 98,5 — 81,4 100,0 115,0 126,5
цеолит NaX — 95,0 97,0 103,0 93,7 101,0 101,0 107,5
цеолит СаА 85,0 84,2 88,0 93,3 76,8 82,0 76,6 77,0
Изотермы на угле, силикагеле и цеолите NaX при темпера-
туре 67,5 К (—205,8 °C) расположены выше соответствующих
изотерм при 77,5 К (—195,8°C), т. е, активность сорбентов с
понижением температуры увеличивается. Адсорбция азота на
цеолите СаА имеет аномальный характер: при понижении тем-
пературы адсорбции с 77,5 К (—195,8°C) до 67,5 К (—205,8 °C)
наблюдается уменьшение активности. Этот факт объясняется бли-
зостью критического диаметра молекулы азота (0,4 нм) к раз-
мерам входного окна в цеолите СаА (0,5 нм), вследствие чего
адсорбция осуществляется со значительной энергией активации
и равновесие в системе не достигается.
Снижение адсорбционной емкости наблюдается и в других
системах, в которых разница между характерными размерами
молекул адсорбата и пор составляет менее 0,1 нм.
Изучение выходных кривых при очистке водорода в слое ад-
сорбентов с зернами размером 1—2 мм показало, что в случае
цеолита СаА они более пологи, чем на других адсорбентах.
Пример выходных кривых приведен на рис. 9,6. Различие в
форме выходных кривых проявляется в высотах работающего
слоя, а следовательно, и в динамической активности. На рис.
9,7 представлены зависимости высоты работающего слоя от
скорости потока, которые отвечают соотношению
L0~wn (9.4)
Коэффициент п равен: для активного угля 0,87; силикагеля 0,83; цеоли-
тов 1,0. Высота работающего слоя уменьшается в ряду: активный уголь —
силикагель — цеолит NaX в соответствии с возрастанием теплот адсорбции.
Близость диаметра молекул адсорбтива (азота) и диаметра входного окна
цеолита СаА приводит к резкому увеличению высоты работающего слоя, ко-
торый приблизительно в 2,5 раза выше работающего слоя цеолита NaX.
Замедленная диффузия адсорбата в гранулах цеолита СаА
приводит к снижению динамической активности этого адсор-
Равновесие и динамика^ адсорбции
209
Рис. 9,6. Выходные кривые при очистке водорода от примеси азота при Т—77 К, р**
==15 МПа и Со==О,12% (об.):
1 — уголь СКТ; 2 — мелкопорнстый снлнкагель; 3 — цеолит СаА; 4 — цеолит NaX.
Рис. 9,7. Зависимость высоты работающего слоя от скорости потока прн очистке водо-
рода от прнмеси азота (со=0,12% об., Т = 77 К, р = 15 МПа);
1 — цеолит СаА; 2 —активный уголь СКТ; 3 — мелкопористый снлнкагель; 4 — цеолит
NaX.
бента, которая значительно ниже соответствующего показателя
для цеолита NaX, хотя равновесные характеристики этих ад-
сорбентов близки. Данные о динамической активности слоя ад-
сорбентов (высота 300 мм, диаметр 15 мм) одинакового грану-
лометрического состава (1—2 мм) при температуре 77 К
(—195,8 °C) и постоянной производительности по расширенному
газу (1,4 м3/ч) в см3/см3 приведены ниже:
Давление, МПа 0,1 0,5 1,5 2,5 5,0 7,5 10,0 12,5 15,0
Уголь СКТ 87,6 91,3 93,4 95,0 86,0 75,2 68,8 66,4 55,1
Силикагель Ns 6 58,6 72,3 75,9 76,7 70,7 67,6 58,5 56,0 52,8
Цеолит NaX 82,6 86,2 86,6 86,5 84,0 80,5 80,5 80,5 76,4
Цеолит СаА 62,7 — — 75,0 69,7 71,2 70,0 70,2 62,5
В условиях высоких массовых скоростей, которые могут быть
применены в установках высокого давления, создаются предпо-
сылки для быстрого охлаждения отработавшего и работающего
слоев, в которых температура повысилась за счет теплоты ад-
14— 1346
210
9. Адсорбция слабо сорбирующихся газов
Рис. 9,8. Зависимость адсорбционной способности
мелкопористого силикагеля по диоксиду углерода,
поглощаемому из воздуха при минус 120 иС, от
давления.
сорбции. Вследствие этого массооб-
мен протекает в условиях, близких
к изотермическим.
Проведение адсорбционной очи-
стки газа под высоким давлением и
при низких температурах обеспечи-
вает очень высокую степень очистки. Остаточное содержание
примеси в случае применения микропористого активного угля
СК.Т и цеолитов обоих типов не превышает 5-Ю-8, а в случае
силикагеля 2-107 объемных долей.
Попытка количественно оценить вклад различных факторов
в снижение адсорбируемости примеси в присутствии газа-носи-
теля при высоком давлении была сделана Вагиным [246].
В криогенной технике используют адсорбционную очистку от диоксида
углерода при высоком давлении, причем, если в качестве адсорбента исполь-
зован силикагель, процесс проводят при низких температурах.
Кривая 2 на рис. 9,8 отражает адсорбционную способность
мелкопористого силикагеля КСМ по диоксиду углерода, погло-
щаемому из воздуха, где его концентрация составляет 0,03%
при температуре минус 120 °C в области давлений до 15 МПа.
Кривая 2 проходит значительно ниже изотермы адсорбции чи-
стого диоксида углерода (кривая 1) и имеет максимум, отвеча-
ющий давлению 2 МПа.
Известно, что при высоких давлениях присутствие газа-но-
сителя меняет химический потенциал твердого диоксида угле-
рода в соответствии с уравнением
Дц = 7?Т1пГ (9.5)
где Др, — изменение химического потенциала; F=pips — отношение давления
пара над твердым диоксидом углерода в присутствии газа-носителя к дав-
лению насыщенного пара чистого диоксида.
Изменение давления пара над твердым диоксидом углерода
под воздействием газа-носителя высокого давления было изме-
рено. Эти данные использовали для расчета кривой 3, учиты-
вающей снижение химического потенциала, или, что то же са-
мое, растворение диоксида в газе-носителе. В основу расчета
были положены следующие соображения: так как взаимодейст-
вие диоксида углерода с воздухом уменьшает летучесть в F
раз, то адсорбция диоксида углерода из смеси, в которой пар-
циальное давление примеси рсоа должно соответствовать ад-
сорбции чистого диоксида углерода при давлении pco2/F. Не-
смотря на то, что расчет передал форму экспериментальной
кривой, наличие и положение максимума, вычисленные значе-
Равновесие и динамика. адсорбции
211
ния адсорбционной способности по диоксиду углерода оказа-
лись значительно выше: в расчете не было учтено вытеснение
диоксида углерода из адсорбированной фазы воздухом.
Чтобы приблизительно учесть оба фактора, было использо-
вано уравнение адсорбции Ленгмюра, которое с учетом эффек-
та растворения в газе-носителе и адсорбционного вытеснения
для рассматриваемой системы приобретает вид:
Г (Pco2/f)
“= 1 + bco2Pco2/F + ЬвРв (9‘6)
где Г — коэффициент адсорбции СО2 при рсо2—'0; рсо, и рв — парциаль-
ные давления СО2 и воздуха. При этом pco2=i/co2p, а рв=(1—рсо2)р, где
Усо2—мольная доля СО- в газовой фазе; р — общее давление в смеси.
Константы уравнения (9.6) были определены по экспери-
ментальным данным. Соответствие расчета по этому уравнению
опытным данным прослеживается из сопоставления кри-
вых 4 и 3.
В дальнейшем на примере адсорбции примесей из гелия при
температуре 77,5 К (—195,8 °C) и давлениях до 30 МПа было
показано [247], что даже такой «неадсорбирующийся» газ, как
гелий, при высоких давлениях снижает адсорбционную способ-
ность других веществ. По мнению авторов работы, этот факт
в первую очередь связан с притяжением молекул примеси к
молекулам гелия — растворением примеси в гелии. Вытесняю-
щий эффект в изученных системах также проявлялся достаточ-
но сильно.
Использование метода в технике
В процессах низкотемпературного ожижения газов, в частности
водорода, значительную роль играет качество предварительной
очистки газов от примесей (азота и кислорода), которые от-
вердевают в ожижителе, мешая нормальной работе как ожи-
жителя, так и различных физических приборов, использующих
жидкий водород [245].
В ряде случаев, например при работе с жидководородными
пузырьковыми камерами, где недопустимо загрязнение оптиче-
ских поверхностей, требуется водород с содержанием примесей
менее 5-10 7 объемных долей. Чтобы уменьшить взрывоопас-
ность системы, применяют предварительную каталитическую
очистку водорода, которую проводят при комнатной или более
высокой температуре. Для удаления примеси азота на входе
серийного водородно-гелиевого ожижителя включены два бло-
ка очистки водорода, осуществляемой под высоким давлением
и при низкой температуре. Каждый блок имеет осушитель, теп-
лообменник и адсорбционную секцию. Максимальная произво-
14*
212 9. Адсорбция слабо сорбирующихся газов
дительность блока очистки составляет 360 м3/ч, рабочее давле-
ние 15 МПа, скорость газового потока в адсорбере 5 м/мин в
расчете на полое сечение.
Адсорбционная секция имеет шесть адсорберов, заполненных активным
углем и погруженных в ванну с жидким азотом (рис. 9,9). Изготовлены ад-
сорберы из нержавеющих труб диаметром 73X6 мм. Высота каждой секции
•600 мм. Все секции соединены последовательно, общая высота слоя адсорбен-
та составляет 4,5 м. Теплообменник и адсорберы размещены в криостате, за-
полненном жидким азотом. Тепловая изоляция криостата выполнена из пе-
нопласта. Блоки очистки работают попеременно.
В результате очистки содержание азота снижается с 5-10“4 до (1—5)
•10_7 объемных долей. Адсорберы переключают на стадию регенерации че-
рез каждые 4—8 ч. Регенерацию адсорбента веду г вакуумированием адсор-
беров до остаточного давления около 8 Па при одновременном обогреве сна-
ружи горячим азотом. Адсорбционную очистку при низкой температуре при-
меняют также для очистки баллонного водорода [248].
Адсорбционные установки, работающие под высоким давле-
нием, получили широкое распространение не только для очист-
ки водорода, но и для решения ряда других задач: осушки при-
родных газов, подготовки воздуха к низкотемпературному раз-
делению и т. д. Технология и показатели этих процессов рас-
смотрены в соответствующих разделах книги.
Рис. 9,9. Схема секции адсорбционной очистки водорода под высоким давлением:
1 — осушитель; 2 — теплообменник; 3 — адсорберы; 4 — криостат; I, II —водород; III —
греющий азот; IV — жидкий азот; V — испарившийся азот; VI —линия сброса в атмо-
сферу; VII — линия отбора пробы на газоанализатор; VIII — коммуникация к вакуум-
насосу.
Рис. 9,10. Схема установки для получения жидкого кислорода с поглощением диоксида
углерода силикагелем:
1 — детандер; 2 — теплообменник; 3 — дроссельный вентиль; 4 — нижняя колонна; 5 —
фильтр; 6 —адсорбер; 7 — конденсатор; 8 — верхняя колонна.
Использование метода в технике
213
Адсорбционный метод применяют для осушки и очистки тех-
нического водорода, разделяемого на две модификации: орто-
и параводород.
В газообразном состоянии при нормальных условиях в равновесии нахо-
дятся три части орто- и одна часть параводорода. Исходный водород, содер-
жащий 0,6% метана, 0,3% аргона и азота, 0,3% оксида углерода и 0,01 %
диоксида углерода, подвергают осушке силикагелем при давлении 4,12 МПа.
После этого его охлаждают до 89 К (—164 °C) обратным потоком водорода
и очищают от метана активным углем. В следующей стадии водород охлаж-
дают кипящим азотом до 70 К (—203 °C) и при этой температуре подверга-
ют его глубокой очистке силикагелем. Остаточное содержание примесей не
превышает 0,1 части на 1 млн. После этого компонент смеси — ортоводород
при 35 К (—238 °C) на катализаторе (оксид хрома) превращают в пара-мо-
дификацию.
Проведением процесса адсорбции при высоком давлении
(15 МПа) и температурах порядка 70—77 К (—203ч—196 °C)
эффективно решается задача глубокой очистки гелия от приме-
си азота (2°/о об.) и микропримеси неона (0,02% об.) [249].
Оптимальный режим десорбции характеризуется нагреванием
угля до минус 120 °C с последующей откачкой адсорбента в
течение 30 мин. При таком способе очистки возможно получе-
ние гелия с содержанием неона менее 1 • 10“зо/о (об.).
Очистку воздуха от диоксида углерода и взрывоопасной
микропримеси ацетилена на установке низкотемпературного
разделения проводят в холодном цикле после сжижения возду-
ха [250]. Схема установки приведена на рис. 9,10.
Сжатый и осушенный атмосферный воздух под давлением 15—20 МПа
охлаждают до 250 К (—23 °C) и разделяют на два потока. Один из них
охлаждают в теплообменнике 2 холодным азотом до 123 К (-—150°C), а
другой поток после расширения в детандере 1 до 0,6 МПа охлаждают до
ИЗ К (—160 °C), в результате чего воздух конденсируется и из жидкости
выделяется твердый диоксид углерода. Выпадение диоксида углерода из пер-
вого потока также происходит после дросселирования давления в вентиле 3.
Жидкая фаза вместе с суспензией из нижней колонны 4 поступает в фильтр
5, где с помощью пористого материала из нее выделяют суспензию диоксида
углерода. Микропримесь ацетилена и остаточное количество диоксида угле-
рода извлекают в адсорбере 6, причем их суммарная концентрация в возду-
хе после адсорбера не превышает 0,35% (в пересчете на газовую фазу).
Через трое суток адсорбер переключают на регенерацию. Очищенный жид-
кий воздух подвергают разделению в верхней (ректификационной) колонне 8
с конденсатором 7.
Рис. 9,11. Зависимость максимально допу-
стимой скорости потока в адсорберах с
активным углем от давления.
214 9. Адсорбция слабо сорбирующихся газов
Г ... рис. 9,12. Цеолитовый вакуум-насос:
I 1 — цеолит; 2 — трубка нз никелевой сетки; 3 — хладоагент.
у&д Зол Применение высоких давлений застав-
шей ляет вносить коррективы при выборе опти-
-SS оН'"1 мальной скорости потока в адсорберах.
=р: Увеличение давления вызывает замедление
я- -3 скорости диффузионных процессов при ад-
-ffi ъ': сорбции, увеличение пыления и унос ад-
Й; сорбента. Вследствие этого рекомендуется
ЗаЭйЯЕ ограничивать скорость газового потока в
адсорберах с активным углем и другими
адсорбентами в зависимости от давления,
х---как это показано на рис. 9,11 [251].
Сделана попытка использовать сниже-
ние адсорбционной способности при высоком давлении для раз-
работки изотермического процесса отбензинивания природного'
газа в периодических адсорберах, заполненных силикагелем, ак-
тивным углем или оксидом алюминия [252]. Стадию отбензини-
вания проводят при давлении, обеспечивающем максимальную
адсорбционную емкость (2—8 МПа), а стадию десорбции —
путем продувки газом при более высоком давлении. Разница
давлений в стадиях десорбции и адсорбции составляет не менее
2 МПа. Из газа десорбции после снижения давления выделяют
конденсат.
Используя низкую температуру, адсорбцию слабосорбирую-
щегося газа можно увеличить до такой степени, что адсорбент
окажется пригодным для создания в системе глубокого ва-
куума [253—256]. Адсорбционные вакуумные насосы, в отличие
от ротационных форвакуумных и диффузионных, не загрязняют
аппаратуру парами углеводородов, вследствие чего отпадает не-
обходимость установки ловушек между насосом и откачиваемым
объектом. В адсорбционных насосах применяют в основном ак-
тивные угли и цеолиты, реже силикагели. Охлаждение адсор-
бента проводят жидким азотом, водородом или гелием. Совре-
менные адсорбционные насосы обеспечивают получение вакуума
примерно до 1 • 10~7 Па.
Простейший цеолитовый насос* изображен на рис. 9,12 [253]. Цеолит
размещен в стеклянной ампуле, охлаждаемой снаружи жидким азотом или
другим сжиженным газом. Внутри ампулы с цеолитом вмонтирована тру-
бочка из никелевой сетки, образующая каналы для подвода адсорбтива.
Подготовку адсорбента (его дегазацию) проводят так же, как и в других
адсорбционных установках — вакуумированием или продувкой чистым газом
при высокой температуре. Чем хуже адсорбируется откачиваемый газ, тем
более низкие температуры требуются для охлаждения адсорбента.
* Конструкции и параметры работы адсорбционных вакуумных насосов опи-
саны в монографии А. И. Волчкевича. Высоковакуумные адсорбционные насо-
сы. М„ Машиностроение, 1973. 158 с. (Прим, ред.)
АППАРАТУРНО-ТЕХНОЛОГИЧЕСКОЕ
ОФОРМЛЕНИЕ АДСОРБЦИОННЫХ ПРОЦЕССОВ
ТИПЫ АДСОРБЦИОННЫХ
ПРОЦЕССОВ И УСТАНОВОК
ПОЛУЧЕНИЕ И НОМЕНКЛАТУРА
ПРОМЫШЛЕННЫХ
АДСОРБЕНТОВ
ОСУШКА ГАЗОВ
И ОРГАНИЧЕСКИХ ЖИДКОСТЕЙ
ПРОМЫШЛЕННАЯ ОЧИСТКА
ГАЗОВ
ЗАЩИТА АТМОСФЕРЫ
ОТ ЗАГРЯЗНЕНИЯ
ЗАЩИТА ГИДРОСФЕРЫ
ОТ ЗАГРЯЗНЕНИЯ
ИСПОЛЬЗОВАНИЕ
ИЗБИРАТЕЛЬНОСТИ
АДСОРБЦИИ
ИСПОЛЬЗОВАНИЕ
МОЛЕКУЛЯРНО-СИТОВЫХ
СВОЙСТВ ЦЕОЛИТОВ
ПРИМЕНЕНИЕ АДСОРБЕНТОВ
ДЛЯ КОНДИЦИОНИРОВАНИЯ
И ХРАНЕНИЯ ПИЩЕВОЙ
ПРОДУКЦИИ
Сегодня, вероятно, невозможно назвать
область производственной деятельности,
в которой не применялись бы адсорбенты
и адсорбционные процессы. Но все
разнообразие этих процессов основано на
сочетании сравнительно немногочисленных
приемов и легко поддается классификации.
Первым признаком такой классификации,
конечно, является отсутствие или
наличие регенерации отработанного
адсорбента. Если потребное количество
адсорбента очень невелико, или срок
службы его очень велик, или если
повторное применение адсорбента, как
в медицине, недопустимо, то используют
процессы, в которых адсорбент не
регенерируют, а заменяют.
В большинстве современных адсорбционных
процессов регенерация адсорбента
предусмотрена. Поэтому вторым
отличительным признаком является способ
ее выполнения. Таких способов,
вытекающих из физикохимии адсорбции,
всего три: повышение температуры,
понижение давления и вытеснение
адсорбата другим веществом.
Процесс, основанный на первом из них,
носит название терморегенеративного,
на втором — безнагревного, на третьем —
вытеснительного. Часто используют
комбинации этих способов, проводя
термовакуумный, термоотдувочный, либо
термовытеснительный процессы. И, наконец,
третьим существенным признаком
адсорбционного процесса является способ
его организации. По этому признаку
процессы могут быть подразделены на
периодические и непрерывные.
В установках, отвечающих первым из них,
адсорбент находится в виде
неподвижного слоя, который вначале
используют для очистки потока, а затем
подвергают регенерации. Непрерывность
обработки потока обеспечивают, применяя
параллельно работающие адсорберы.
Основными типами непрерывных
адсорбционных установок являются
колонны с движущимся или кипящим слоем
адсорбента, а также емкости с мешалкой.
По мере повышения прочности адсорбентов
области применения установок
непрерывного действия будут расширяться.
10
ТИПЫ АДСОРБЦИОННЫХ
ПРОЦЕССОВ
И УСТАНОВОК
Основным типом адсорбционных процессов остаются процессы
периодического действия, в которых адсорбер с неподвижным
слоем адсорбента после окончания стадии очистки или разде-
ления, определяемого исчерпыванием емкости адсорбента, пе-
реключается на стадию десорбции [1]. В рабочий цикл перио-
дического процесса обычно включают ряд дополнительных ста-
дий: сушку и охлаждение адсорбента, повышение и сброс дав-
ления и т. д. Широкое применение автоматизации на адсорбци-
онных установках позволило исключить ручной труд при управ-
лении процессом.
Другим перспективным методом, обладающим рядом значи-
тельных преимуществ, является очистка и разделение газов в
движущемся слое адсорбента. К сожалению, до сих пор не ре-
шена проблема получения прочных в условиях движущегося
слоя адсорбентов, в связи с чем широкая промышленная реали-
зация этого метода задерживается. В этой главе оба метода
рассмотрены на примере адсорбционных процессов отбензини-
вания природных газов.
В некоторых случаях применяется очистка и разделение га-
зов в псевдоожиженном слое адсорбента. Следует, однако, за-
метить, что уже при скоростях потока 0,25 м/с, обычных для
периодических адсорберов, адсорбционный процесс лимитиру-
ется скоростью массопереноса адсорбтива внутри гранул ад-
сорбента (внутридиффузионной кинетикой), в связи с чем пере-
вод адсорбента в псевдоожиженное состояние не сопровождает-
ся интенсификацией самого процесса поглощения примеси и
отработки адсорбционной емкости гранул. В этом основное от-
личие адсорбционных установок с псевдоожиженным слоем от
каталитических.
Преимуществом процесса в псевдоожиженном слое являет-
ся возможность его проведения при высоких скоростях потока,
которые на порядок выше, чем на установках с периодическими
адсорберами. Поэтому сделаны попытки применить этот метод
там, где нужно очистить очень большие объемы газа, а компри-
мирование газа с экономической точки зрения невыгодно. Такая
Разделение газа на установках периодического действия
217
проблема, в частности, возникла при рекуперации сероуглерода
из вентиляционных газов вискозного производства. Требования
к механической прочности адсорбента на установках с псевдо-
ожиженным слоем еще выше, чем на установках с движущимся
слоем. Адсорбционные процессы, основанные на применении
псевдоожиженного слоя, детально описаны в монографии Ро-
манкова и Лепилина [2], поэтому мы ограничимся описанием
технологического регламента лишь одной установки в разделе,
посвященном применению активных углей в промышленности
химических волокон.
Разделение газа на установках
с периодическими адсорберами
Разделение газа на установке с периодическими адсорберами
иллюстрирует описанный ниже процесс отбензинивания природ-
ного газа. Метод основан на избирательном поглощении высо-
комолекулярных углеводородов твердыми адсорбентами, обыч-
но активным углем.
На рис. 10,1 приведена сравнительная характеристика поглотительной
способности масла и активного угля по углеводородам с различным числом
атомов углерода в молекуле. Как показывает график, при извлечении про-
пана, если его концентрация в исходном газе 25 г/м3, адсорбционная способ-
ность активного угля составляет 4 г на 100 г, а поглотительная способность
масла — лишь 0,03 г на 100 г сор-
бента. Повышение давления адсорб-
ции до 1 МПа приведет к увеличе-
нию поглотительной способности мас-
ла только до 0,3 г/100 г, что ясно
иллюстрирует нецелесообразность от-
бензинивания газов с небольшим со-
держанием компонентов абсорбцион-
ным методом.
При поглощении углеводородов с
более высокой молекулярной массой
преимущества угля оказываются ме-
нее существенными, а по октану ак-
тивности угля и масла приблизитель-
но равны.
По результатам испытания на
пилотных установках сделано заклю-
чение, что активный уголь в указан-
ных условиях (Со=25 г/м3, р=
= 0,1 МПа) имеет поглотительную
способность, большую, чем абсорб-
Рис. 10,1. Поглотительная способность аб-
сорбционного масла (молекулярная масса
200) и активного угля по углеводородам,
поглощаемым нз воздуха:
1 — концентрация углеводорода 250 г/м’;
2 — то же, 100; 3 — то же, 25.
Сд С4 Сд Сд С/. Сд Cq
,^гсь1адсорбционных процессов и установок
цпонное масло, по гептану — в 6 раз, гексану — в 18 раз, пентану — в 45 раз,
бутану — в 85 раз, и по пропану — в 150 раз. Адсорбционный метод имеет
особое преимущество перед абсорбционным при извлечении легких углеводо-
родов из тощих природных газов [3].
В периодических адсорбционных установках отбензинивания
газ с определенной скоростью проходит (обычно снизу вверх)
через слой активного угля. В первый момент весь слой адсор-
бента насыщается основным компонентом углеводородной сме-
си — метаном. Затем — по мере поступления газа в периодиче-
ский адсорбер — высшие углеводороды вытесняют из нижних
слоев более легкие углеводороды, которые постепенно пере-
двигаются вверх по слою и в порядке увеличения молекулярной
массы появляются в отходящем газе.
Во избежание проскока целевого углеводорода процесс ад-
сорбции приходится заканчивать значительно раньше, чем про-
изойдет отработка адсорбционной емкости слоя адсорбента.
С целью более полного использования поглотительной способ-
ности угля часто идут на снижение степени улавливания, кото-
рая для пропана на существующих газобензиновых установках
обычно не превосходит 80% от его содержания в исходном газе.
На рис. 10,2 приведена принципиальная схема адсорбцион-
ного цеха, имеющего 12 адсорберов и предназначенного для от-
бензинивания 220 тыс. м3 природного газа в сутки.
Адсорберы объединены в 3 блока. Исходный («жирный») газ проходит
вначале через фильтры, заполненные коксом, для очистки от пыли и капель-
ной влаги и из коллектора при давлении 0,14 МПа поступает в адсорберы 1,
'‘ерегретый пар
Рис. 10,2. Принципиальная схема газобензиновой установки с периодическими адсор-
берами:
I — адсорбер на стадии отбензинивания; 2— адсорбер на стадии десорбции; 3 — адсор-
бер на стадии сушки; 4 — адсорбер на стадии охлаждения; 5 — холодильник-конденсатор;
6 — сепаратор; 7 — фракционатор уловленных углеводородов.
Разделение газа на установках периодического действия 219
находящиеся на стадии насыщения; при этом извлекаются бензиновые угле-
водороды. В качестве адсорбента применяют рекуперационный уголь APT
насыпной плотностью 520 кг/м3.
Отбензиненный газ (200 тыс. м3/сут) через коллектор поступает в Маги-
стральный газопровод, в котельную завода, а также на другие промышлен-
ные и коммунальные объекты, где используется в качестве топлива.
Адсорбционный цикл каждого адсорбера включает четыре двадцатими-
нутных стадии: адсорбции, десорбции, сушки и охлаждения. Таким образом,
'ежесуточно в цехе протекает 216 стадий, а число переключений потоков до-
стигает 1720.
После окончания стадий отбензинивания адсорбер переключают на де-
сорбцию (поз. 2). В случае углеродных адсорбентов десорбцию обычно про-
водят острым паром, поступающим из котельной под давлением 1,5 МПа
после дросселирования до давления 0,15 МПа, благодаря чему в адсорбер
•ои поступает в перегретом состоянии. Температура у-ля в конце стадии де-
сорбции составляет 120—130 °C. Направление пара обратно направлению
«жирного» газа в стадии адсорбции, т. е. сверху вниз. Благодаря этому ка-
пельки влаги, сконденсированной в первые, минуты десорбции, стекают вниз.
Кроме того, при движении пара в направлении, обратном направлению
движения «жирного» газа, выделяющиеся из верхних слоев пары легких уг-
леводородов, проходя через нижние слон, сами выполняют функции динами-
ческого агента, вытесняющего более трудно десорбируемые углеводороды,
которые концентрируются в нижних слоях адсорбера. При этом снижается
расход так называемого динамического пара (пара иа отдувку десорбирован-
ных углеводородов) и увеличивается степень десорбции.
Из адсорберов пары газового бензина и воды через сборный коллектор
направляются в конденсационную аппаратуру, состоящую из двух ступеней
(поз. 5). В конденсационной группе первой ступени, состоящей из трубча-
тых конденсаторов, при 70 °C конденсируется основная масса водяных паров
и наиболее тяжелые углеводороды, входящие в состав газового бензина.
В первой ступени для охлаждения используется вода, поступающая с гра-
дирни, а также из конденсаторов второй ступени. После конденсаторов пер-
вой ступени парогазовая смесь направляется в сепаратор первой ступени 6,
где газовый бензин отделяется от воды. В конденсационной группе второй
ступени (на схеме не показана), охлаждаемой водой, поступающей с гра-
дирни при 15—25 °C, конденсируются более легкие бензиновые углеводороды
и остаток влаги. Конденсационный бензин охлаждается в водяных теплооб-
менниках.
Остаточный газ из сепаратора направляется в газгольдер, а из газголь-
дера — на двухступенчатую компрессию: в первой ступени он компримиру-
ется примерно до 0,3, а во второй — до 1,7 МПа. После компрессии из газа
дополнительно конденсируются углеводороды — компрессионный бензин.
Смесь газовых бензинов, содержащая пропан и бутан, направляется на ста-
билизацию или фракционирование в аппарате 7 (его производительность
в рассматриваемом случае составляет 37.5 т/’сут). В результате стабилизации
получают товарные продукты: бензин с содержанием 20% бутана и сжижен-
ный газ — пропан-бутановую фракцию. На десорбцию и стабилизацию сум-
марно расходуется 160 т/сут пара, или около 6 кг на 1 кг нестабильного
бензина.
Сушку угля (поз. 3) ведут воздухом (или отбензиненным газом), нагре-
тым в трубчатых теплообменниках до 120—130 °C. В качестве греющего
агента применен пар давлением 1,5 МПа. Температура угля в конце стадии
сушки составляет от 70 до 120—130 °C. Охлаждение угля (поз. 4) ведут ат-
мосферным воздухом, подаваемым в адсорберы вентилятором производи-
тельностью 17 тыс. м3/ч. Температура угля в конце стадии охлаждения рав-
на 55—60 °C. Показателем хорошо проведенной регенерации производствен-
ники считают резкое повышение температуры в слое в первые 5 мин по-
следующей стадии отбензинивания.
220
10. Типы адсорбционных процессов и установок
Вопросы проектирования, компоновки и размещения угле-
адсорбционных установок и вспомогательного оборудования де-
тально освещены в книге Николаевского [4].
Газовый бензин, получаемый на установках периодической
адсорбции, обычно имеет высокое давление паров, что препятст-
вует его использованию как самостоятельного моторного топ-
лива. Попытки снизить давление паров бензина путем повыше-
ния температуры конденсации вызывают значительные потери
газового бензина, уловленного углем.
Рентабельно производить товарный газовый бензин непо-
средственно в периодическом адсорбере оказалось невозмож-
ным: содержание легких фракций в нем определяется условия-
ми адсорбционного равновесия, и оно тем больше, чем выше со-
отношение углеводородов (С3+С4) : С5+ в исходном газе*. На-
пример, в газах бакинских месторождений это соотношение зна-
чительно меньше, чем в газах восточных районов, и поэтому ба-
кинские углеадсорбционные газовые бензины обладают мень-
шим давлением паров.
Газовый бензин, полученный на установке с горизонтальны-
ми адсорберами при времени адсорбции 30 мин и выделенный
в холодильниках-конденсаторах, имеет высокое давление паров
(1,7 кПа) и низкую плотность (0,65 кг/см3); в составе сконден-
сированного газового бензина остается до 1—2% пропана и
16% бутанов.
Снижение до нормы содержания высших углеводородов в
сжиженном газе, полученном на установках с адсорберами пе-
риодического действия, и одновременное получение стабильного
бензина возможно только после включения в схему стабилизи-
рующих и газофракционирующих установок [5].
Конструкция адсорберов в установках
периодического действия
Вертикальный адсорбер
В адсорбционных установках периодического действия наиболь-
шее распространение нашли адсорберы, в которых отношение
высоты слоя к диаметру аппарата больше единицы. Они полу-
чили название вертикальных адсорберов. Конструкции аппара-
тов этого типа весьма разнообразны. Ниже в качестве примера
описаны адсорберы, применяемые в процессе отбензинивания.
Вертикальный адсорбер этого процесса (рис. 10,3) представляет собой
железный цилиндрический аппарат диаметром 2,1 и высотой 2,9 м. Высота
* Символом Сх+ в нефтехимии принято обозначать совокупность этого (Сх)
и всех остальных углеводородов данного гомологического ряда, следующих
за Сх. (Прим. ред).
Конструкция адсорберов
221
Рис. 10,4. Укладка подложки в адсорбере.
слоя угля в адсорберах — 2,5 м, загрузка угля в каждый адсорбер — 4 т.
Сверху адсорберы закрыты крышками, прикрепленными к цилиндрической
части болтами. Слой угля покоится на керамической плите; последняя имеет
большое число мелких отверстий, с помощью которых входящий газ равно-
мерно распределяется по сечению адсорбера. Керамическая плита опирается
на литую чугунную решетку. На керамическую плиту насыпают слой гравия
высотой 100—200 мм. Зернение гравия по высоте подложки неравномерно
(рис. 10,4): внизу располагают крупные куски размером 25—30 мм, затем
последовательно фракции 15—25, 10—15 и 5—10 мм. Гравий предохраняет
адсорбент от загрязнения конденсатом, содержащимся в «жирном» газе, а
также препятствует просыпанию мелочи и забиванию отверстий в плите.
Сверху слой угля во избежание уноса покрывают сеткой, выполненной из
проволоки толщиной 0,8 мм с отверстиями размером 2,5 мм.
В нижней части адсорбера находятся нижняя и верхняя головки с во-
семью штуцерами для ввода и вывода газов и паров на разных стадиях
процесса. Внутри адсорбера расположена труба, отводящая газ во время
охлаждения, сушки и насыщения. При десорбции водяной пар подают по
трубе в верхнюю часть адсорбера. Такая конструкция позволяет все управ-
ление адсорберами сосредоточить внизу.
Адсорберы, работающие при атмосферном давлении, обыч-
но выполняют сварными из листовой стали толщиной 10 мм. Ес-
ли в газе присутствуют агрессивные примеси, в качестве мате-
риала используют легированную или нержавеющую сталь.
Горизонтальный адсорбер
Общий вид горизонтального адсорбера представлен на рис.
10,5. Длина горизонтальных адсорберов достигает 6 м, диаметр
2 м, высота слоя адсорбента при этом равна 0,8—1,0 м. Боль-
222 Ю- Типы адсорбционных процессов и установок
Рис. 10,5. Горизонтальный адсорбер:
1 — люки для загрузки адсорбента; 2 — шту-
цер для подачн газа на стадиях адсорбции,
сушки н охлаждения; 3 — кожух; 4 — распре-
делитель водяного пара на стадии десорбции;
5 — люк для выгрузки адсорбента; 6 — штуцер
для отвода конденсата; 7 — штуцер для отво-
да паров прн десорбции; 8 — штуцер для от-
вода газа.
шое сечение горизонтальных адсорберов при малой высоте слоя
и наличие сфероидальных участков поставили под сомнение
равномерность распределения парогазовых потоков по сечению
такого аппарата.
В связи с этим при обследовании газобензиновой установки [6], обо-
рудованной горизонтальными адсорберами и предназначенной для перера-
ботки «жирного» газа, содержащего до 150—200 г/м3 углеводородов от пен-
тана и выше (или 600—650 г/м3 углеводородов от пропана и выше), один
из адсорберов был разгружен, и в разных его участках были помещены
пробы активного угля, завернутые в мешочки из металлической сетки с от-
верстиями 1 мм. После этого адсорбер был заполнен углем. В определенный
момент времени мешочки с помощью проволочек вытягивали, помещали в
герметичные пробоотборники; пробы анализировали на содержание влаги,
бензина, газообразных углеводородов.
Анализ проб угля указал на наличие в слое угля участков
с большим гидравлическим сопротивлением. Газ обтекает та-
кие участки, вследствие чего нарушается равномерность соста-
ва адсорбата в слое угля. В этих участках сорбирующиеся в
первый период углеводороды (в основном пропан и бутан) вы-
тесняются бензиновыми углеводородами медленнее, чем в дру-
гих участках. Неравномерностью распределения газового пото-
ка в горизонтальных адсорберах отчасти объясняется и труд-
ность регулирования качества продукции путем изменения вре-
мени адсорбции.
Адсорберы горизонтальной конструкции следует рекомендо-
вать только при очистке больших количеств газа в условиях,
когда к степени очистки не предъявляются слишком жесткие
требования.
Кольцевой адсорбер
В случае очистки газа от примесей, присутствующих в неболь-
ших концентрациях, иногда применяют кольцевые адсорберы.
Разрез такого адсорбера представлен на рис. 10,6.
Вся конструкция смонтирована в металлическом корпусе 1. Газ, подле-
жащий очистке, через штуцер 2 поступает во внешнюю часть адсорбера, про-
ходит в горизонтальном направлении через кольцевой слой угля, находящий-
ся между внутренней 7 и внешней 6 цилиндрическими решетками, и выво-
дится через штуцер 4. В стадиях сушки и охлаждения указанное направле-
ние потоков сохраняется. В стадии десорбции водяной пар (десорбент) по-
Конструкция адсорберов
223
Рис. 10,6. Кольцевой адсорбер:
1 — корпус; 2 — штуцер для подачн газа; 3 — штуцер
для отвода паров при десорбции; 4 — штуцер для отвода
газа и подачн пара; 5 — люк для выгрузкн адсорбента;
6, Z — решетки; 8 — люки для загрузки адсорбента.
дают через штуцер 4, а парогазовую смесь отво-
дят через штуцер 3. Загружают адсорбент через
люки 8, а выгружают через люк 5.
На одном из заводов химического волокна
кольцевой адсорбер был применен для улавлива-
ния сероуглерода из вентиляционных газов. Об-
щая высота адсорбера составляла 7,8 м, высота
слоя адсорбента 5,2 м, диаметр адсорбера 3,2 м,
внешний диаметр слоя адсорбента 2,8 м, внут-
ренний диаметр слоя адсорбента 1 м. В адсорбер
загружалось 14 т рекуперационного активного
угля АР. Содержание сероуглерода в очищаемом
воздухе колебалось от 1,5 до 1,7 г/м3, температу-
ра воздуха была около 40 °C. Проскок сульфида
углерода в выходящем из адсорбера газе фиксир
чала стадии очистки.
При значительно меньших габаритах в сравнении с верти-
кальным адсорбером, обычно применяемым на заводах вискоз-
ного волокна, кольцевой адсорбер позволил повысить произво-
дительность по газу до 40 тыс. м3/ч (в типовом вертикальном
адсорбере она не превышает 30 тыс. м3/ч) при относительно вы-
соком гидравлическом сопротивлении (300 Па).
Адсорбер с вертикальными теплообменными
элементами
В тех случаях, когда нужно ускорить процесс теплообмена в
стадиях нагрева и охлаждения или получить десорбат в кон-
центрированном виде, используют адсорберы со встроенными
теплообменными элементами. Чаще применяют адсорберы, вы-
полненные в виде трубчатого теплообменника (рис. 10,7); ад-
сорбент засыпан в трубки, а греющая или охлаждающая среда
находится в межтрубном пространстве.
Примером эффективного использования адсорберов с вер-
тикальными теплообменными элементами является очистка ар-
гона, гелия, неона от примесей кислорода и азота, осуществляе-
мая при низкой температуре (—180 °C) и давлении 0,14 МПа
[7]. Адсорбер, предназначенный для очистки в этих условиях
100 м3/ч аргона от кислорода, представлен на рис. 10,8.
Адсорбент (цеолит) расположен в трубках высотой 2 м. При общем чис-
ле трубок 51 общая загрузка цеолита в адсорбере составляет 260 кг. Ско-
рость газового потока в стадии очистки составляет 0,016 м/с. Охлаждение
цеолита производят жидким кислородом, протекающим в межтрубном прост-
ранстве адсорбера. Остаточное содержание кислорода в очищенном аргоне
составляет 1-10~3%. После отработки адсорбционной емкости цеолита его
быстро нагревают до 150 °C, подавая в межтрубное пространство азот, на-
гретый до 420 °C.
; 224
10. Типы адсорбционных процессов и установок
Рис. 10,7. Адсорбер, выполненный в виде трубча-
того теплообменника:
I, III — теплоноситель; П, IV — газ; 1 — корпус;
2, 5 — камеры; 3 — газораспределительная решет-
ка; 4 — трубчатка; 6 —карман для термопары;
7 — штуцер для отбора проб газа н измерения
температуры адсорбента.
Адсорбер с прижимным устройством
В некоторых случаях адсорберы
снабжают специальным прижим-
ным устройством для обеспечения
плотного прилегания верхней ре-
шетки с сеткой к слою адсорбента
и уменьшения его истирания.
На рис. 10,9 представлен фланцевый
адсорбер, используемый для очистки сжи-
женного воздуха от ацетилена мелкопори-
стым силикагелем в условиях низких тем-
ператур. Среднее содержание ацетилена в
жидкости перед очисткой составляет 0,2-
• 10-3 м3/м3. Десорбцию ацетилена ведут
при температуре около 0 °C и давлении
0,15 МПа продувкой слоя адсорбента га-
зом сначала при 20—25, а затем при 50—
75 °C.
При проектировании адсорбци-
онных установок с периодическими
адсорберами следует избегать подачи горячего и холодного га-
за по одним коммуникациям. Например, после переключения
адсорбера, используемого при углеадсорбционном отбензинива-
нии, со стадии сушки на стадию охлаждения в случае однопо-
точной системы подачи газа в аппарат в течение первых 5—
10 мин продолжает поступать нагретый в коммуникациях газ,
что снижает эффективность охлаждения. Аналогичный недоста-
ток отмечен и при переключении адсорбера на стадию сушки.
Четкое разграничение коммуникаций горячего и холодного газа
позволяет снизить инерционность процессов теплообмена.
С целью уменьшения потерь тепла во внешнюю среду в ста-
диях десорбции и сушки адсорберы обычно покрывают слоем
изоляции, например шлаковаты.
График работы установки периодического
действия
Переключение адсорберов периодической установки производят
в соответствии с заданным графиком работы установки. На
рис. 10,10 представлены примеры графиков для двух-, трех- и
L
График работы установки
225
Жидкий
хладоагент
Рис. 10,8. Трубчатый адсорбер для низкотемпературной очистки инертных газов:
1 — крышка; 2 — нажнмная планка; 3 —пружины; 4 — корпус; 5 — адсорбент; 6 — труб-
ка; 7 — трубная решетка; 8 — внутреннее днище; 9 — внешнее дннще.
Рис. 10,9. Адсорбер для очистки жидкости с прижимным устройством:
1 — крышка; 2—прнжнмное устройство; 3—корпус; 4 — адсорбент.
четырехадсорберной установки отбензинивания. Фигуры с раз-
ной штриховкой на этом рисунке соответствуют разным стади-
ям процесса. При двухадсорберной установке продолжитель-
ность стадии адсорбции равна сумме продолжительностей всех
других стадий, при трехадсорберной продолжительность стадии
адсорбции соответствует продолжительности стадии десорбции
или суммарной продолжительности стадий сушки и охлажден
НИЯ* И Т. И.
* Из графиков работы адсорбционной установки, или, как их еще называют,
циклограмм адсорбционного процесса, определяют необходимое число адсор-
бентов (п). Очевидно, оно должно удовлетворять неравенству
п >
где 2/ — общая продолжительность адсорбционного цикла, равная сумме
длительностей всех составляющих его стадий-, — длительность стадии ад-
сорбции.
Совершенствование адсорбционной технологии в значительной степени свя-
зано с созданием более совершенных графиков работы адсорбционной аппа-
ратуры. Здесь можно выделить две тенденции: сокращение общей продол-
жительности адсорбционного цикла, что повышает интенсивность адсорбци-
онной установки, и уменьшение числа стадий, приводящее к сокращению
числа адсорберов и, следовательно, капитальных затрат. Примером эффектив-
ности второго направления является рассматриваемый ниже двухстадийный
процесс отбензинивания. (Прим, ред.)
15—1346
226
10. Типы адсорбционных процессов и установок
Четырехадсорберная установка
Рис. 10,10. Графики работы адсорбционных установок.
Двухстадийный процесс при повышенной температуре. Кон-
денсация водяного пара в начале стадии десорбции в рекупера-
ционных установках с периодическими адсорберами значитель-
но усложняет процесс. При отбензинивании жирных попутных
нефтяных газов иногда решается задача только подготовки га-
за к транспортированию, т. е. удаление высших (бензиновых)
углеводородов, которые могут сконденсироваться в газопроводе.
Бензиновые углеводороды хорошо поглощаются углем не толь-
ко при обычных, но и при повышенных температурах, когда
конденсации пара не происходит.
Румынскими инженерами [8] разработана схема двухфаз-
ного процесса, в котором жирный газ при 90—100 °C поступает
в адсорбер с горячим углем (130—-140°C). В стадии высокотем-
пературной адсорбции происходит подсушка адсорбента. Де-
сорбцию углеводородов проводят перегретым водяным паром
при 250—280 °C. Влажность угля в процессе не превышает 5—
7% (масс.). В высокотемпературном процессе поглотительная
способность угля по бензиновым углеводородам приблизитель-
Разделение смесей газов в движущемся слое адсорбента
227
но на 50% ниже, чем в случае проведения процесса при обыч-
ных температурах. Несмотря на это, по ряду показателей (ка-
питаловложения, эксплуатационные расходы, стоимость газово-
го бензина, потребление водяного пара) новый процесс имеет
значительные преимущества перед четырехступенчатым.
Область применения двухфазного высокотемпературного
процесса не ограничена переработкой природного газа. Уста-
новки такого типа могут найти широкое распространение для
рекуперации различных растворителей и паров различных ве-
ществ.
Непрерывный метод разделения смесей газов
в движущемся слое адсорбента
Недостатки, присущие методу разделения и очистки парогазо-
вых смесей в неподвижном слое адсорбента (периодичность,
трудность автоматизации, громоздкость аппаратуры, неполная
отработка адсорбционной емкости, несовершенство стадии де-
сорбции), стимулировали поиски путей интенсификации адсорб-
ционного процесса с применением принципа непрерывности. По-
следний обеспечивается при циркуляции адсорбента в замкнутой
системе и расчленении адсорбционной колонны на локальные
зоны, в каждой из которых в оптимальных рабочих условиях
осуществляется одна из стадий процесса: адсорбция (очистка,
осушка, рекуперация компонентов газовой смеси), нагрев и де-
сорбция, охлаждение, пневмотранспорт и т. д.
Первые попытки осуществить адсорбционный метод разделения парога-
зовых смесей по непрерывной схеме относятся к тридцатым годам нашего
столетия. Но лишь в 1951 г. в США были построены 6 промышленных уста-
новок с движущимся слоем активного угля для разделения легких углево-
дородных газов [9].
Крупная установка непрерывной адсорбции была сооружена также для
выделения этилена из низкоконцентрированного газа — метановодородиой
смеси, полученной в результате перегонки нефти, и верхнего продукта деме-
танизатора. Содержание этилена в исходном газе составляло 5—7% (об.),
процесс вели при 0,6 МПа. Установка была рассчитана на максимальную
производительность 50000 м3/сут, скорость циркуляции активного угля в
•среднем составляла 8,1 т/ч, однако могла быть повышена до 14,5 т/ч.
Десорбцию проводили в трубчатом нагревателе, в межтрубное пространст-
во которого подавали конденсирующийся при 265 °C высокотемпературный
теплоноситель. Для отдувки углеводородов в колонну ниже распределитель-
ного механизма вводили острый пар (180 кг/ч). Чистота выделенного этиле-
на достигала 98%. В связи с использованием в промышленной колонне от-
носительно дешевого, но недостаточно прочного активного угля потери его
от истирания составляли 0,023% за цикл против 0,0005—0,002% за цикл иа
опытной установке, где применяли косточковый уголь.
Технико-экономическое сравнение метода непрерывной адсорбции с аб-
сорбционным методом выявило преимущество первого для ряда процессов,
в частности, для извлечения пропана из природного газа [10]. Значительное
внимание американскими инженерами было уделено повышению производи-
15*
228 10. Типы адсорбционных процессов и установок
тельности адсорбционных колони непрерывного действия и улучшению каче-
ства продуктов.
В Советском Союзе первое исследование процесса разделе-
ния газов в движущемся слое активного угля было выполнено
в Московском химико-технологическом институте им. Д. И. Мен-
делеева в 1948—1951 гг. [П]. В отечественной химической ли-
тературе непрерывный метод разделения газовых смесей в дви-
жущемся слое адсорбента подробно освещен в монографии [2].
Ряд исследований был проведен с целью расширить область
применения установок с движущимся слоем адсорбента на про-
цессы осушки промышленных газов [12], разделения углеводо-
родов с разной степенью насыщенности [13, 14], очистки водо-
рода от примесей под повышенным давлением [15], выделения
этилена из газов нефтепереработки [46, 17] и из метановодород-
ной смеси [18], выделения ацетилена из разбавленных газов
неполного окисления метана [19]. Последнее направление изу-
чалось совместно советскими и венгерскими учеными, причем в
Венгрии метод прошел опытно-промышленное испытание [20].
Процесс разделения в движущемся слое изучен также в ЧССР.
Положительные результаты получены при использовании техни-
ки движущегося слоя для рекуперации оксида азота из промыш-
ленных газов [21] и улавливания диоксида серы из отходящих
топочных газов [22].
Теории адсорбционного разделения газовых смесей в движу-
щемся слое адсорбента посвящены работы Кастена и Амундсе-
на [23], Поллока [24], Каханяка [25], Такуры [26].
Принципиальная схема установки
Принципиальная схема установки непрерывной адсорбции при-
менительно к разделению природного газа представлена на
рис. 10, 11.
Природный газ поступает в адсорбционную часть установки, где из него
в движущемся слое активного угля извлекаются высшие компоненты. На-
сыщенный углеводородами уголь под действием силы тяжести спускается в-
трубчатый нагреватель, где производится десорбция путем нагрева адсорбен-
та через стенку с подводом небольшого количества острого пара в качестве
динамического агента. При этом нс происходит увлажнения угля, и из цик-
ла полностью выпадает фаза сушкн угля, необходимая в обычных рекупе-
рационных установках периодического действия.
Пары десорбированных углеводородов поднимаются из десорбера вверх
по секции хроматографического разделения колонны, причем более тяжелый
компонент вытесняет из пор угля более легкий компонент. Благодаря этому
непрерывный метод позволяет не только выделить высшие углеводороды из
газа, но и разделить их непосредственно в адсорбционной колонне, что уст-
раняет необходимость сооружения специальных стабилизирующих или фрак-
ционирующих колонн. Активный уголь специальным питателем подают в газ-
лифт, где для транспорта угля на верх установки используют отбензиненный
природный газ. Цикл заканчивается в трубчатом холодильнике, в котором
уголь охлаждают проточной водой, прежде чем снова направить на адсорб-
цию.
Разделение смесей газов в движущемся слое адсорбента
229>
Рис. 10,11. Принципиальная схема разделения
природного газа в движущемся слое активно-
го угля.
Ценными преимуществами не-
прерывной адсорбции является пол-
ная автоматизация процесса, воз-
можность производить хроматогра-
фическое разделение смеси компо-
нентов наряду с их выделением из
газа, снижение расхода тепла на
регенерацию угля, неувлажняемого
в стадии десорбции.
Предложены различные конст-
руктивные решения процесса в дви-
жущемся слое адсорбента [27—29].
Адсорбция углеводородов
в движущемся слое адсорбента
Расчет процессов выделения угле-
водородов из потока газа в движу-
щемся слое базируется на тех же
закономерностях, что и адсорбция
в неподвижном слое адсорбента.
Однако в установках с движущим-
ся слоем адсорбента зона массооб-
мена «закреплена» относительно стенок аппарата. В простей-
шем случае — при извлечении одного компонента из потока га-
за — скорость движения адсорбента по абсолютному значению
должна быть не меньше скорости движения фронта сорбции в
периодическом адсорбере.
Слишком большая нагрузка на адсорбент, при которой коли-
чество поступающих на адсорбцию углеводородов превышает
адсорбционную емкость адсорбента, приводит к потере части
легких целевых углеводородов (при отбензинивании природного
газа — пропана). Наоборот, недогрузка адсорбента связана с
необоснованным увеличением расхода угля, повышенными по-
терями адсорбента в результате истирания и затратами тепла на
нагрев адсорбента при десорбции, что отрицательно сказывается
на технико-экономических показателях процесса. Кроме того,
при недогрузке адсорбента в секцию хроматографического раз-
деления будут поступать повышенные количества этана и даже
метана.
Скорость перемещения фронта сорбции (и в см/мин) в пе-
риодическом адсорбере может быть определена непосредственно
из экспериментальных данных по величине равновесной адсорб-
ционной способности или по уравнению Шилова, так как она
230 Ю. Типы адсорбционных процессов и установок
является величиной, обратной коэффициенту защитного дейст-
вия:
и — wc0/a0 = i/k (Ю.1)
где ао — равновесная адсорбционная способность (объем сорбируемого газа,
отнесенный к единице объема адсорбента), см3/см3; w — скорость газового
потока, отнесенная к единице сечения колонки, см3/(см2-мин); с0 — исходная
.концентрация сорбируемого компонента, см3/см3.
В установках с движущимся слоем время пребывания адсор-
бента в зоне массообмена ограничено. Поэтому адсорбент выхо-
дит из адсорбционной секции при величине адсорбции более
шизкой, чем величина равновесной активности. Поэтому для оп-
ределения скорости движения угля в промышленных установ-
ках в формулу (10.1) следует ввести поправочный коэффициент
скорости насыщения kc.H, тогда
и'= wc0/kc>sa0 (Ю.2)
Коэффициент скорости насыщения, таким образом, является
отношением теоретической скорости, вычисленной из равновес-
ных данных, к минимальной скорости движения адсорбента в
установках непрерывного действия, обеспечивающей необходи-
мую полноту извлечения компонента:
kz _н = и/и' (10.3)
Величина коэффициента скорости насыщения для ряда угле-
водородных газов, адсорбирующихся в движущемся слое актив-
ного угля, при практических расчетах может быть принята
йс. н=0,95. При известной скорости движения угля и' количест-
во циркулирующего угля (G в см3/мин) может быть определено
из соотношения
G — u'F (10.4)
где F— площадь сечения адсорбционной колонны, см2.
В действительности природные и нефтяные газы представля-
ют собой сложную смесь углеводородов, причем адсорбируе-
мость изомерных соединений на угле совпадает, а адсорбируе-
мость углеводородов с разницей числа атомов углерода в мо-
лекуле, равной единице, достаточно близка. Поэтому при ад-
сорбции целевых компонентов (п, «4-1, п+2,...) в адсорбцион-
ной секции колонны всегда будет поглощаться некоторое коли-
чество низших углеводородов (п—1, п—2, п—3,...). Количество
адсорбированных низших углеводородов определяется условия-
ми адсорбционного равновесия.
Учитывая, что адсорбционная способность по углеводород-
ным компонентам одного гомологического ряда почти линейно
меняется с составом адсорбированной фазы и может быть рас-
считана как аддитивная функция состава адсорбированной фа-
Разделение смесей газов в движущемся слое адсорбента
231
зы при постоянном общем давлении, скорость движения адсор-
бента в колонне, обеспечивающая достаточно полное извлечение
целевых компонентов, может быть рассчитана как сумма ско-
ростей по каждому компоненту в отдельности:
г 11 ИСО(П)Фп
и'а .к — ип + “n+i + “п+2 + • • • = “ь +
"см I °о(п)
^0(4+1)94+1 , шс0(П+2)Фп+2
Q0(n+l) а0(П+2)
W Г С0(4) с0(4+1) с0(П+2) "I
== Фп + "ТГ Фп+i + Фп+г + ••• (Ю.5)
^С.н"см L “0(4) °0(4+1) “0(4+2) J
где фп, фп+i, фп+2, ... — степень извлечения соответствующих углеводородов;
“о — адсорбционная способность каждого углеводорода, которая определяет-
ся из изотерм адсорбции:
an~f(Pri) (10.6а) an+i = / (Pn+i) (10.66) ап+2 — f (Рп+г) (10.6в)
йс« — коэффициент, учитывающий отклонение от закона аддитивности при
адсорбции нескольких углеводородов активным углем, который по результа-
там исследования адсорбционного равновесия углеводородов равен 0,95—
1,00*.
Количество нецелевых компонентов, одновременно извлекае-
мых по условиям адсорбционного равновесия, определяется из
уравнений:
Уп-1/Уп ~ Кр (x'n-j/Хл) (Ю.7)
Лп-1/Дп+1 — Кр (^n-i/^n+i) (Ю.8)
Уп-г/Лп+г ~ Кр (х" п-1/хп+2) (10.9)
где Хр — соответствующие коэффициенты разделения в системах первого не-
целевого компонента (п—1) с каждым из целевых компонентов; уп-\, У«,
Уп+i, уп+2, - — мольные доли компонентов в исходном газе; x'n-lt x"n-i,
х'"п-\, ... — мольные доли первого нецелевого компонента (п—1) в адсорби-
рованной фазе, определенные из соотношений адсорбционного равновесия с
каждым из целевых компонентов.
Соотношение мольных долей компонентов в исходном газе
соответствует соотношению их объемных концентраций:
Уп-1/Уп = co(n-i)/co(n) (Ю. 10)
Уп-1/Уп+1 ~ с0(4-1)/С0(4+1) ( 10. 11 )
Уп-1/Уп+2 — с0(П-1)/с0(П+2) (Ю. 12)
С другой стороны, мольное отношение компонентов в ад-
сорбированной фазе равно отношению числа молей этих ком-
* Уравнение (10.5), если исходить из его вида, основано на предположе-
нии, что все целевые компоненты смеси адсорбируются независимо. Поэто-
му наличие коэффициента йсм и его значение ниже единицы свидетельствует
лишь о некотором взаимном подавлении сорбции компонентов в смеси.
(Прим, ред.)
232
10. Типы адсорбционных процессов и установок
(10.13)
(10.14)
(10.15)
понентов, адсорбируемых в единицу времени:
х п-х!хп m n-i/mn
n-i/^n+i = m" n-i/mn+i
x“n-l/xn+2 = m"‘n-l/mn+2
Так как взаимная адсорбируемость двух компонентов оста-
нется постоянной в присутствии третьего компонента, общее ко-
.личество молей первого нецелевого (п—1)-компонента составит
„ со(л-1)тп
— т'п-г -|- т п-! 4- -|~
Со(л) лр[(гг-Щ-л]
е0(п-1)тП+1 c0(n-l)"i(n+2)
“Н ~ И” /» - а* **• (10.16)
Со(п+1) Ар с0(п+2) Ар [(п_1)_(п_|_2)]
Отсюда может быть рассчитана степень извлечения первого
нецелевого компонента; она равна
Фп-i = mn-i/l^co(n-i) (10.17)
где V— объем перерабатываемого газа в единицу времени, м3/ч; с0(„_5) —
мольная доля первого нецелевого (п—1)-компонента в исходном газе.
Тогда дополнительное увеличение скорости адсорбента, обя-
занное совместной адсорбции первого нецелевого (и—1)-компо-
нента u'n-i в см/мин составит
и’п-1 = (wco(n-l)/kc.Hao(n-i)) Фл-1 (10.18)
Аналогичным образом определяется дополнительное увеличе-
ние скорости адсорбента, обязанное совместной адсорбции вто-
рого (п—2), третьего (п—3) и т. д. нецелевого компонента:
и'п-2 ~ (ffilc0(rt-2)/^C.Ha0(^-2)) фп-2 (10.19)
Общее увеличение скорости за счет совместной адсорбции
нецелевых компонентов
^'ицк = и'п~1 + и'п-2 4~ и п-з + • • • (10.20)
Скорость адсорбента в колонне, необходимая, чтобы осу-
ществить выделение целевых компонентов до заданной степени
извлечения, определится как сумма скоростей
и' — и'цк 4~ а нцк (10.21)
На практике, чтобы упростить расчет, целесообразно огра-
ничиться углеводородами, отличающимися от целевого не бо-
лее чем на две единицы. Например, при извлечении газового
бензина (пентанов и высших углеводородов) скорость движения
активного угля следует рассчитывать с учетом совместной ад-
сорбции бутанов и пропана, допуская, что метан и этан полно-
стью вытесняются из адсорбента бензиновыми углеводородами.
Разделение смесей газов в движущемся слое адсорбента
233
Рис. 10,12. Зависимость высоты работающего слоя
при адсорбции пропана (с0«1,5%) в слое различных
активных углей от скорости потока:
I—уголь Байера; 2 — рекуперацнонный уголь; 3--
уголь АГ-2.
Кроме скорости движения адсор-
бента, параметром, необходимым для
проектирования установок с движу-
щимся слоем адсорбента, является
высота слоя в адсорбционной части
установок, гарантирующая требуемую
полноту извлечения. Как было пока-
зано Тимофеевым, высота зоны массо-
обмена в адсорбционных установках
непрерывного действия зависит от сте-
пени отработки адсорбционной емкости и сокращается с умень-
шением последней [30].
Приведенная выше методика расчета позволяет определить
скорость движения адсорбента, обеспечивающую степень отра-
ботки адсорбционной емкости, близкую к единице. В этих усло-
виях минимальная высота слоя в адсорбционной части должна
быть примерно равна высоте зоны массопередачи в периодиче-
ском адсорбере и, следовательно, может быть определена из
опытов с неподвижным слоем адсорбента (при условии одинако-
вой степени упаковки гранул).
При адсорбции смеси углеводородов высота адсорбционной
части устанавливается на основе величины зоны массообмена
по хуже сорбирующемуся целевому компоненту. Как уже ука-
зывалось, таким углеводородом при отбензинивании природных
газов является пропан.
На рис. 10,12 представлена зависимость высоты работающе-
го слоя при адсорбции пропана в слое различных активных уг-
лей от скорости потока. Некоторое различие величины работа-
ющего слоя связано, очевидно, скорее со средней величиной
зерна адсорбентов, чем с их структурой. Структуры изученных
промышленных углей АГ-2, АР-3 и Байера достаточно близки.
На основе графиков, представленных на рис. 10,12, можно сде-
лать вывод, что высота адсорбционной секции 50 см достаточна
для полного извлечения пропана и более высокомолекулярных
углеводородов в движущемся слое адсорбента (угля)*.
* Рассмотренная в тексте методика расчета секции извлечения адсорбцион-
ных колонн с движущимся слоем адсорбента, основанная на аналогии с не-
подвижным слоем, конечно, является приближенной. Более строгая поста-
новка задачи, учитывающая специфику непрерывного процесса, дана Тодесом
(в кн.: «Методы и процессы химической технологии». М.—Л., Изд. АН СССР,
1955, с. 55). Им же (см. в кн.: «Кинетика и динамика физической адсорб-
ции». М., Наука, 1973, с. 206—207) приведены решения для стационарного
режима и линейной изотермы сорбции, из которых следует, что для обеспе-
234 Ю. Типы адсорбционных процессов и установок
чения достаточно больших степеней извлечения целевых компонентов
(>9О°/о) линейная скорость продвижения угля в колонне должна в 5—10 раз
превышать скорость движения адсорбционной волны в аппарате с неподвиж-
ным слоем, а высота секции — во столько же раз превышать высоту, эквива-
лентную единице переноса массы. Аналитическое решение для линейной изо-
термы получено Себалло и др. (в кн.: «Кинетика и динамика физической
адсорбции». М., Наука, 1973, с. 250—257). В работе этих авторов изложена,
кроме того, методика численного расчета секции извлечения при произволь-
ном типе изотермы сорбции. Выходная кривая, полученная в адсорбере с не-
подвижным слоем адсорбента, была в данном случае использована только в
качестве исходной информации. (Прим, ред.)
Разделение смеси адсорбированных компонентов
в хроматографической части установки
Преимуществом метода адсорбции в движущемся слое адсор-
бента является возможность разделения гаммы выделенных из
газа углеводородов на отдельные фракции или компоненты не-
посредственно в адсорбционной колонне. Разделение происходит
в результате непрерывного массообмена между адсорбирован-
ной фазой и парогазовой вытеснительной флегмой тяжелых ком-
понентов, поднимающейся противотоком адсорбенту из десорб-
ционной части установки.
Массообмен осуществляется на основе избирательных свойств
адсорбента, характеризующихся кривой адсорбционного равно-
весия и коэффициентом разделения. При проведении процесса
отбензинивания природных газов с выделением достаточно чи-
стых пропана, бутана и газового бензина расчет секции хрома-
тографического разделения базируется на кривых адсорбцион-
ного равновесия метан — этан, этан — пропан, пропан — бутан,
бутан — пентан плюс высшие углеводороды.
При поглощении углеводородов в слое адсорбента выделяется теплота
адсорбции, вследствие чего температура адсорбента повышается и соответ-
ственно понижается его адсорбционная способность. При проектировании
газоперерабатывающих заводов обычно принимают среднюю теплоту адсорб-
ции пропана и бутана 32,5 кДж/моль, газового бензина 50,0 кДж/моль. По-
вышение температуры адсорбента определяется в каждом отдельном случае
расчетом.
Изучение температурного режима адсорбционной опытной колонны не-
прерывного действия показало, что при отбензинивании тощего природного
газа повышение температуры адсорбента в движущемся слое за счет теп-
лоты адсорбции углеводородов составляет 10—15 °C. Близкие показатели
были получены на американской углеадсорбционной колонне непрерывного
действия в Мидленде, где выделяли этилен (с0 = 5—7% об.) из метано-водо-
родной смеси; температура движущегося активного угля из косточек абри-
косов на участке адсорбционной секции повышалась с 50 до 65 °C, т. е. на
15°С.
Для достаточно полного разделения пропано-бутановой или
иной бинарной смеси углеводородов в условиях неподвижного
слоя необходимо большое число периодических операций, за-
ключающихся в обогащении адсорбированной фазы лучше сор-
Разделение смесей газов в движущемся слое адсорбента 23&
бирующимся углеводородом (например бутаном) с последую-
щим переводом адсорбата снова в газовую фазу в процессе де-
сорбции.
В первой ступени между составом адсорбированной и газовой
фаз существует соотношение
К р (Уг/У1)1 = (х2/(10.22)
Если путем десорбции перевести адсорбированные в первой
ступени углеводороды снова в газовую фазу и полученную га-
зовую смесь адсорбировать во второй ступени, то, очевидно, га-
зовая фаза во второй ступени будет иметь тот же состав, что и
адсорбированная фаза первой ступени:
(Xi/x2)i = (У1/У2)1 (10.23)
Но, кроме того
Кр Шг = (*2/*1)г (10.24)
ИЛИ •*'
Кр2 (Уа/^1)1 = (X2/Xj)2 (10.25)
Аналогично для n-й ступени уравнение принимает вид
Крп (Уг/У1)1 ~ (У2/У1)п ~ (-^aAiln+i (10.26)
или, после логарифмирования
п- 1g Лр = 1g (уМп — 1g (y2lyi)i (10.27)
Формула (10.27) рекомендуется для определения числа ад-
сорбционно-десорбционных ступеней, необходимых, чтобы по-
лучить данное изменение в составе, если известен коэффициент
разделения:
„ 1g Шп - 1g ЫУ1)1 .._ „я.
п = те (10 28>
Этого же эффекта разделения оказалось возможным достиг-
нуть при применении движущегося слоя адсорбента, насыщен-
ного разделяемой смесью, при противоточном движении паров
десорбата.
В процессе движения угля по колонне состав адсорбирован-
ной и газовой фаз непрерывно изменяется, причем изменение
состава адсорбированной фазы по сравнению с газовой в любой
точке секции хроматографического разделения компонентов оп-
ределяется так же, как и при разделении смеси путем периоди-
ческих операций адсорбции — десорбции, т. е. на основе усло-
вий адсорбционного равновесия. Таким образом, вся секция
хроматографического разделения может быть условно разбита
на ряд ступеней, в каждой из которых происходит изменение
состава, соответствующее одной адсорбционно-десорбционной
236
10. Типы адсорбционных процессов и установок
Рис. 10,13. Изменение состава газа по высоте хро-
матографической колонки (yi и t/2 — доли лучше
и хуже сорбирующегося компонента).
ступени адсорбера с неподвижным
слоем.
Если принять высоту слоя ад-
сорбента, эквивалентную одной
ступени разделения, за z, а высоту
секции хроматографического разде-
ления за LXp, тогда число ступеней,
необходимых для достижения данной степени разделения, мо-
жет быть определено из уравнения:
п — Lxq/z
(10.29)
Подставив значение п в уравнение (10.28), получим
1g Шп — 1g (y2/l/i)i = (Wz) 1g
По последней формуле может быть определена высота, эк-
вивалентная одной ступени z, если известно отношение содержа-
ния компонентов бинарной смеси в газовой фазе как функция
высоты секции хроматографического разделения, при условии,
что коэффициент разделения /Ср постоянен по всей высоте хро-
матографической секции.
В табл. 10-1 приведены данные по чистоте разделения угле-
водородов при разной высоте разделяющего слоя адсорбента.
Процентное содержание компонентов отнесено к суммарному
содержанию пропана и бутана.
По результатам этих опытов построен график в координатах
lg(f/2/f/i)i] —длина слоя Lxp (см. рис. 10,13), при-
чем за положение, соответствующее первой ступени разделения,
была принята точка отбора пропановой фракции, и за положе-
ние n-й ступени—точка отбора бутановой фракции. Первые
точки достаточно точно легли на прямую. При высоте разде-
ляющего слоя более 40 см кривая круто меняет наклон, стре-
Таблица 10-1. Чистота разделения пропано-бутановой фракции
в движущемся слое активного угля
§ Высота слоя ' разделения, 11 CM ’ > Пропановая фракция Бутановая фракция
У1 У2 У1 У2
20 86,8 13,2 12,4 86,0 1,628
30 92,5 7,5 9,5 90,5 1,969
40 92,0 8,0 3,2 96,8 2,541
70 95,6 4,4 1,4 98,0 3,184
Разделение смесей газов в движущемся слое адсорбента 237
мясь стать параллельной оси ординат. Это указывает на то,
что дальнейшее увеличение высоты колонны практически не при-
водит к улучшению степени разделения углеводородов.
Котангенс угла г наклона начального участка кривой к оси
абсцисс равен
ctgr = lg/(p/z (10.30)
откуда
z = lg/(p/ctgz (10.31)
Вычисленная по уравнению (10.31) высота движущегося
слоя активного угля, эквивалентная одной ступени разделения,
оказалась равной 10 см. Из результатов опытов с движущимся
слоем активного угля на лабораторной колонне вытекает, что
высота хроматографической секции колонны 0,8—1,0 м обеспе-
чивает получение 98%-ной бутановой и 95%-ной пропановой
фракции.
Температурный режим десорбционной части
Активный уголь находится в теплообменнике ограниченное вре-
мя (около 10 мин), в связи с чем следует выявить температур-
ные условия, при которых происходит быстрое удаление углево-
дородов, адсорбированных углем, а также определить, как эти
условия должны быть изменены при проведении процесса под
повышенным давлением. Для этого была изучена кинетика де-
сорбции бутана и фракций газового бензина: гексановой (57—
68°C) и гептановой (72—98 °C).
Выбор адсорбата был сделан применительно к условиям экс-
плуатации углеадсорбционных колонн непрерывного действия,
предназначенных для доулавливания пропана и бутана после
маслоабсорбционных отбензинивающих установок и непосредст-
венно для отбензинивания природных газов.
Анализ кинетических кривых десорбции углеводородов из
слоя активного угля высотой 10 см (рис. 10,14) позволил опре-
Таблица 10-2. Десорбция гептановой фракции из слоя активного угля АГ-2
при скорости газового потока 10 м/мин
Время, мин Степень десорбции, %
100 °C 150 °C 200 °C 210 °C
3 56,1 82,4 95,2 98,5
6 64,6 91,0 99,0 100,0
12 74,6 94,0 100,0
30 85,0 99,0 — <—
238
10. Типы адсорбционных процессов и установок
т, мин
Рис. 10,14. Кинетика десорбции бутана из активного угля (и = 10 м/мин).
Рис. 10,15. Зависимость коэффициента теплопередачи К от скорости движения угля wyr
в трубках теплообменника.
делить оптимальные температурные условия, необходимые для
быстрой десорбции каждого углеводорода: например, для пол-
ной десорбции бутана необходима температура 130 °C. При этой
температуре и скорости воздуха 10 м/мин весь бутан удаляется
через 3 мин, тогда как при понижении температуры до 100 °C
даже 30-минутная продувка угля газом не приводит к полной
регенерации адсорбента.
Минимальные температуры, обеспечивающие быстрое и пол-
ное удаление гексановой и гептановой фракций газового бензи-
на, соответственно равны 200 и 220 °C. Данные по десорбции
гептановой фракции приведены в табл. 10-2.
Насколько велико влияние динамического агента на интен-
сивность десорбции углеводородов, показал опыт без продувки
газа; в результате нагрева активного угля до 130 °C и выдер-
живания при этой температуре в течение 20 мин было удалено
лишь 55,0% бутана.
В отличие от периодических адсорбционных установок, ста-
дии адсорбции и десорбции в колоннах с движущимся слоем
адсорбента осуществляются при одинаковом давлении, которое
в большинстве случаев с целью повышения адсорбционной спо-
собности и уменьшения диаметра колонны превосходит атмос-
ферное.
Таблица 10-3/ Десорбция углеводородов из слоя активного угля АГ-2
при давлении 1,0 МПа и скорости газа 2 м/мин
Время, мнн Температура 195—200 °C Температура 230—240 °C Температу- ра 310 °C, бензин
бутан гексан гептан бутан гексан беизни
3 92,5 91,5 90,4 100 94 83,5
6 98,5 95,6 95,3 1. — — 99,6
12 100,00 97,5 97,3 — — — 100,0
Разделение смесей газов в движущемся слое адсорбента 239
. Результаты исследования десорбции под давлением приве-
дены в табл. 10-3. Они показывают, что повышение давления в
системе от 0,1 до 1,0 МПа вызывает необходимость повышения
температуры в десорбере приблизительно на 100 °C; для быстрой
и полной десорбции бутана при 1,0 МПа требуется температу-
ра 230°C, для авиационного бензина 320—330 °C. Этот вывод
был подтвержден Расуловым на примере десорбции углеводоро-
дов из слоя мелкопористых силикагелей КСМ и ШСМ и актив-
ного угля СКТ [31].
Теплопередача в трубчатых нагревателях
и холодильниках адсорбента
При проектировании установок непрерывного действия большое
внимание уделяют узлам теплообмена, в которых уголь нагре-
вают или охлаждают через стенку внешним тепло- или хладо-
агентом.
Не рассматривая детально механизм теплообмена, мы огра-
ничимся результатами экспериментального определения кон-
кретных значений общего коэффициента теплопередачи, необхо-
димых для расчета теплообменников крупных адсорбционных
установок непрерывного действия.
Определение коэффициентов теплопередачи проводили на установке, со-
стоящей из двух трубчатых теплообменных элементов: в верхнем элементе
движущийся активный уголь АГ-2 охлаждали до постоянной температуры
проточной водопроводной водой, а в нижнем — нагревали водяным паром
или горячей водой, циркулирующей с помощью насоса. Применяли трубчатые
теплообменные элементы диаметром от 16, до 50 мм и высотой от 0,5 до
4,2 м.
Заданную скорость движения активного угля обеспечивали с помощью
ложечкового дозатора, расположенного в нижней части установки. Замеры
температуры угля при его движении указали па неравномерность темпера-
турного уровня по радиусу теплообменного элемента, особенно при высоких
скоростях движения адсорбента. В связи с этим расчеты производили по
средней температуре угля, который после выхода из теплообменного элемен-
та собирали в приемник.
Чтобы приблизить эксперимент к промышленным условиям, серию опы-
тов проводили с подачей нагретого воздуха в слой активного угля, однако
существенных изменений в коэффициенты теплопередачи противоточное дви-
жение газа не внесло. Очевидно, это связано с малыми линейными скоро-
стями газового потока, соизмеримыми со скоростью движения угля (3—
6 см/с).
На рис. 10,15 представлена зависимость общего коэффициен-
та теплопередачи от скорости движения угля (®уг). Как пока-
зывает график, коэффициент теплопередачи возрастает с уве-
личением скорости угля и в области средних скоростей состав-
ляет 100—200 кДж/(м2-ч-К). Уменьшение диаметра труб теп-
лообменника приводит к некоторому повышению коэффициент;
теплопередачи.
240
10. Типы адсорбционных процессов и установок
Рис. 10,16. Температурный профиль колонны при раз-
делении природного газа.
Нагрев угля на опытной установке про-
водили в 19 вертикальных стальных трубках
диаметром 89 мм с толщиной стенки 4 мм.
Высота трубок 3 м. Трубки размещены в кор-
пусе диаметром 0,6 м. В межтрубное прост-
ранство при давлении 0,4 МПа подавали
1400 кг/ч перегретого водяного пара. Ско-
рость движения угля в трубках составляла
6—9 см/мин. Суммарная поверхность трубок
в десорбере составляла 15 м2. При количест-
ве циркулирующего угля 320 кг/ч распределе-
ние температур (в °C) в нагревателе было следующим:
Уголь на выходе из адсорбционной секции 40
Уголь в нижней части десорбера 290
Пар на входе 350
Пар на выходе 280
Пар бензиновых углеводородов на выходе из десорбера 120
В десорбер подавалось 13 кг/ч острого пара. Выход десорбируемых уг-
леводородов составлял около 5% массы циркулирующего угля, т. е. 16 кг/ч.
Расчеты показали, что общий коэффициент теплопередачи
в этих условиях составляет 50кДж/(м2-ч-К). На рис. 10,16 пред-
ставлено изменение температуры по высоте промышленной ад-
сорбционной колонны непрерывного действия при выделении
пропана и бутана из природного газа.
Варианты аппаратуры
На зарубежных установках крекинга нефтепродуктов с движу-
щимся слоем адсорбента газ в систему газлифта подается с по-
мощью циркуляционных газодувок. Это требует тщательной пы-
леочистки газа. Кроме того, ассортимент циркуляционных газо-
дувок, работающих под повышенным давлением, но при малом
перепаде давления в системе, невелик. В связи с этим были
разработаны две схемы, позволившие осуществить процесс пнев-
мотранспорта без применения газодувок [32, 33]. Их принци-
пиальные схемы представлены на рис. 10,17.
Схема «а» соответствует обычной схеме с циркуляционной газодувкой.
В схеме «б» выше холодильника угля устанавливают гидравлический затвор
из движущегося слоя активного угля. Диаметр затвора для уменьшения
объема фильтрующегося газа должен быть минимальным, но достаточным,
чтобы пропустить требующееся количество циркулирующего угля и обеспе-
чить бесперебойное (без образования пробок) движение адсорбента.
Высота затвора также должна быть выбрана достаточно точно, чтобы
процесс противоточной фильтрации газа через затвор проходил при умерен-
ных скоростях, не приводил к турбулизации и зависанию угля в затворе.
Разделение смесей газов в движущемся слое адсорбента
241
Благодаря гидравлическому сопротивлению столба угля, газ выводится с та-
релки отбора адсорбционной секции при несколько более высоком давлении
и, проходя через дозатор угля и газлифт, выполняет в нем функции рабо-
чего вещества. Из сепаратора он поступает в газопровод. Стабильность ра-
боты установки по предложенной схеме была доказана в многосуточных про-
бегах в течение 6 мес ее эксплуатации.
При расчете промышленных адсорбентов следует рекомендовать ско-
рость газа в гидрозатворе порядка 15 м/мин (в расчете на атмосферное дав-
ление). При таких и более низких скоростях не происходит зависания угля
в затворе и потери его текучести (при эксплуатации опытной установки за-
висание было отмечено, когда уровень угля в затворе понижался).
В другой предложенной схеме — «в» сырой (неотбензиненный) газ сна-
чала используют в качестве рабочего вещества для подъема адсорбента в
газлифте, а затем направляют на доизвлечение целевых компонентов в ад-
сорбционную часть установки. Такая схема также позволяет вести адсорб-
ционный процесс без циркуляционных газодувок, снизить температуру в зо-
не адсорбции, благодаря частичному насыщению угля углеводородами в газ-
лифте, сократить общую высоту установки по сравнению со схемой «б» за
счет ликвидации гидравлического затвора.
Пропускная способность адсорбционных колонн часто лими-
тируется скоростью газа в адсорбционной части: повышение
скорости газа выше критической недопустимо, так как приво-
дит к уносу адсорбента. Устройством двух или нескольких ад-
сорбционных слоев пропускная способность колонн непрерыв-
ного действия можно увеличить в соответствующее число раз.
Противоточный контакт газа со свежерегенерированным уг-
лем обеспечивают индивидуальной подачей угля в каждый ад-
сорбционный слой и регулированием скорости удаления угля из
каждого слоя с помощью специальных диафрагм, разбивающих
поток угля на несколько частей.
Показатели непрерывного процесса
Непрерывный процесс был испытан на опытной установке, пред-
назначенной для разделения углеводородов в движущемся слое
активного угля. Через установку пропускали до 3500 м3/сут при-
а б в
Рис. 10,17. Варианты схем (а—в)
адсорбционных колонн непрерывного действия.
16—1346
242 10. Типы адсорбционных процессов и установок
родного газа. Исходный газ имел следующий состав (по объ-
ему): 91% СН4, 5% С2Н6, 2% С3Н8, 1% С4Н10, 1% высших угле-
водородов.
Природный газ поступал на тарелку питания адсорбционной секции ко-
лонны, где в противотоке контактировал с движущимся слоем активного уг-
ля. Диаметр адсорбционной секции 40 см, высота 100 см. Насыщенный угле-
водородами уголь спускался через секцию хроматографического разделения
диаметром 30 см и высотой 2 м в трубчатый десорбер. Десорбер имел
19 стальных труб диаметром 89 мм и высотой 3 м, заключенных в кожух
диаметром 0,6 м. Скорость движения угля поддерживали с помощью доза-
тора шторчатого типа и регистрировали шнековым расходомером, установ-
ленным на вертикальном колене углепровода, соединяющего сепаратор угля
с холодильником. В межтрубное пространство десорбера подавали пере-
гретый водяной пар.
Уголь после выделения из газового потока в сепараторе охлаждали в
трубчатом водяном холодильнике и возвращали в (адсорбционную секцию ко-
лонны. Десорбированные углеводороды отводили в двух точках колонны: с
тарелки отбора над десорбером и с тарелки отбора над секцией хроматогра-
фического разделения. Пары бензина и бутана, отбираемые с нижней тарел-
ки, конденсировались, и конденсат собирали в сепараторе. Улучшение чисто-
ты промежуточного продукта (пропана) достигалось его пропусканием через
медленно движущийся слой отрегенерированного угля в дополнительной ко-
лонне.
Установка была оборудована контрольно-измерительными приборами,
регистрирующими температуру, перепады давления по высоте колонны, ско-
рость газовых потоков; вторичные приборы были смонтированы на централь
ном щите управления.
Большим преимуществом метода непрерывной адсорбции
является чрезвычайная гибкость процесса. В зависимости от
поставленной задачи, меняя соотношения адсорбент — газ, мож-
но добиться любой заданной степени извлечения углеводородов.
На рис. 10,18 представлена зависимость степени извлечения про-
пана от соотношения уголь — газ. График показывает, что, изме-
няя это соотношение от 1:1 до 3,5: 1, удается достичь разной
степени извлечения пропана фс„н8~от 0 до 95%.
Одновременно с увеличением степени извлечения пропана
снижается средняя емкость угля по сумме поглощенных угле-
водородов (пропан и более тяжелые углеводороды): с 5,6г/100г
в случае извлечения только бутана и газового бензина до 2,8—
3,0 г/100 г при полном извлечении углеводородов (см. рис. 10,19).
В режимах, в которых степень извлечения пропана составляла 90—95%,
было отмечено значительное поглощение этана — до 16% от потенциала,
причем поглощенный этан количественно выделялся вместе с фракцией сжи-
женного газа с тарелки отбора над секцией хроматографического разделе-
ния. В отдельных режимах соотношение адсорбент — газ было последова-
тельно увеличено до 8,6 кг/м3, при этом степень отбора этана достигла 60%.
Адсорбционные колонны непрерывного действия, рассчитан-
ные на извлечение этана и пропана или соответствующих олефи-
новых углеводородов, необходимо проектировать на давление
0,5—1,0 МПа. Эти рекомендации находятся в соответс: и с
Разделение смесей газов в движущемся слое адсорбента
243
Рис. 10.18. Зависимость степени извлечения про-
пана ф от соотношения уголь — газ N.
работой Гольберта и Платонова по
выявлению оптимального давления
в колонне непрерывного действия в
случае извлечения этилена из бед-
ных газов (концентрация этилена
около 5% об.). Сопоставлением
расходных коэффициентов стадий
адсорбции и десорбции авторы оп-
ределили оптимум с точки зрения эксплуатационных затрат —
0,5 МПа [34].
Скорости движения адсорбента целесообразно регулировать
либо по составу отходящего газа, либо по концентрации ком-
понентов в средней части адсорбционной зоны, т. е. в зоне рез-
кого падения концентрации извлекаемых компонентов. Послед-
ний вариант регулирования очень чувствителен к изменению ус-
ловий и поэтому позволяет поддерживать режим, при котором
обеспечивается высокая степень извлечения целевых компонен-
тов.
Один из режимов, в котором предусматривалось, в основном, извлече-
ние газового бензина, был реализован при соотношении адсорбент — газ, рав-
ном 0,4. В этом режиме степень извлечения бутана составляла 30—48%, бен-
зина 60—80%, в то время как пропана не более 5%. Малое содержание лег-
ких углеводородов в адсорбированной фазе повлекло резкое возрастание ад-
сорбционной способности рекуперациоиного угля — до 11—14 г/100 г.
Основными показателями для оценки процесса десорбции угле-
водородов в движущемся слое угля являются количество и со-
став недесорбированного остатка в угле ср' (в г/100 г), опреде-
ляемые на основе лабораторного анализа адсорбента, отобран-
ного в нижней части десорбера, и выход продукции В (в кг/ч),
по которому рассчитывают активность угля:
a = 5-100/G (10.32)
где G — количество циркулирующего угля, кг/ч.
Удельный расход тепла (в Дж/кг) на десорбцию углеводо-
родов составляет:
Я = (<7над + <?нугл + <7це"угл + ?пар)/^ (10.33)
где <7над — тепло на нагрев адсорбента, Дж/ч; яаугл — тепло на нагрев адсор-
бированных углеводородов, Дж/ч; <7лесУгЛ — тепло на десорбцию углеводоро-
дов, Дж/ч; ^пар — тепло, теряемое с острым паром, Дж/ч.
На рис. 10,20 даны кривые зависимости активности микропо-
ристого газового угля (а), содержания в угле недесорбирован-
ных (остаточных) углеводородов (аост) и удельного расхода теп-
ла (q) на десорбцию от конечной температуры угля, выходяще-
го из десорбера (/д).
16
244
10. Типы адсорбционных процессов и установок
Рис. 10,19. Зависимость активности рекуперацией-
ного угля а от степени извлечения пропана т).
График показывает, что оптимальной температурой, обеспе-
чивающей полную десорбцию углеводородов при давлении, близ-
ком к атмосферному, является температура 220—240 °C. Она
близка к оптимальной температуре, определенной в лаборатор-
ных опытах (для гексановой фракции газового бензина 200°C).
Разница в 20—40 °C между этими температурами объясняется
присутствием в составе газового бензина углеводородов с моле-
кулярной массой выше, чем у гексана.
Расход острого пара, обеспечивающий полную десорбцию
бензиновых углеводородов при 240 °C, может не превышать
0,5 кг в расчете на 1 кг извлекаемых углеводородов.
Понижение температуры приводит к быстрому накоплению
недесорбированных углеводородов, в связи с чем активность уг-
ля падает. Компенсация падения активности угля увеличением
его расхода приводит к росту удельного расхода тепла на де-
сорбцию. При температуре угля в десорбере ниже 120 °C на-
блюдается намокание адсорбента и нарушение его текучести,
в основном в нижней части газлифта. Несмотря на то, что в не-
прерывном процессе разделения природных газов уголь необхо-
димо перегревать приблизительно на 100 °C по сравнению с пе-
риодическим процессом, снижение расхода динамического пара
приводит к значительной экономии тепла.
Удельный расход тепла в оптимальных температурных ус-
ловиях составляет 3700—5000 Дж/кг продукции при работе на
газовом угле типа СКТ. Менее активный рекуперационный уголь
требует в тех же условиях расхо-
да тепла 5400—5500 Дж/кг. В
этих показателях не учитывают-
ся тепловые потери, которые в
условиях непрерывного процесса
и хорошей изоляции десорбцион-
ной части могут быть приняты
как 10% от общего расхода
тепла.
Рис. 10,20. Зависимость активности угля а,
остатка недесорбированных углеводородов
аост и Удельного расхода тепла на десорбцикх
<7Д от конечной температуры иагрева угля.
Разделение смесей газов в движущемся слое адсорбента
245
Рис. 10,21. Содержание углеводородов С5+ в ЮО
бензиновой фракции (1) и Сз— С* во фракции
сжиженных газов (2). ^80
60
Как уже указывалось, одним из основных преимуществ ме-
тода адсорбции в движущемся слое активного угля является
возможность разделения суммы уловленных из газового потока
углеводородов на отдельные фракции или компоненты в секции
хроматографического разделения адсорбционной колонны. Высо-
кая эффективность разделения обеспечивается правильным гид-
равлическим режимом в колонне— созданием некоторого избы-
точного давления в нижней части колонны, которое побуждает
десорбированные тяжелые фракции подниматься вверх по ко-
лонне противотоком углю и вытеснять более легкие углеводо-
роды. При отбензинивании природных газов наиболее целесо-
образным является выделение в качестве нижнего продукта —
газового бензина с требуемым содержанием бутанов и в качест-
ве промежуточного продукта — пропанобутановой фракции.
На опытной установке для регулирования процесса разделе-
ния в верхней части секции хроматографического разделения
был установлен термоэлемент, связанный с автоматическим
клапаном на выходе продукта с нижней тарелки отбора.
На рис. 10,21 показана чистота выделенных фракций газо-
вого бензина и сжиженного газа в зависимости от температуры
верха секции хроматографического разделения. Результаты изу-
чения разделения углеводородов в движущемся слое активного
угля доказали возможность регулирования выхода и качества
продуктов, отбираемых с различных точек колонны: получение
газового бензина с любым содержанием бутанов от 4 до
30 масс. % и сжиженного газа, в котором примесь высших уг-
леводородов не превышает 5—8 об. %.
Чистота промежуточного продукта не превосходит 92—95%,
так как состав газовой фазы в точке отбора определяется усло-
виями равновесия с адсорбированной фазой, в которой присут-
ствуют и тяжелые компоненты.
С другой стороны, при однократном пропускании через ад-
сорбент бинарной или многокомпонентной смеси газов в отхо-
дящем потоке можно получить хуже сорбирующийся компонент
высокой чистоты. На этом принципе достигнуто улучшение ка-
чества пропано-бутановой фракции: удаление высших углеводо-
родов из фракции сжиженного газа осуществляется в дополни-
тельной колонне с медленно движущимся слоем отрегенериро-
вэнного адсорбента.
Скорость движения угля в дополнительной колонне рассчи-
тывают, исходя из расхода перерабатываемой фракции, уело-
246
10. Типы адсорбционных процессов и установок
Таблица 10-4а. Состав входящего и отходящего газов (%)
Компонент Входящий газ Отходящий газ
режим 1 режим 2 режим 3
состав, извлече- % иие, % п/ извлече- состав, /о ние, % п, извлече- состав, % НИе, %
С, 89,25 93,23 — 92,08 — 90,34 — С, 5,15 5,99 — 4,45 — 5,08 — С3 2,47 0,48 85,5 1,45 40,5 2,18 12,1 С4 1,71 0,10 94,0 0,10 94,0 1,27 25,8 С5 1,22 0,06 95,0 0,12 90,0 0,53 56,5 СО2 0,20 0,14 — 0,00 — 0,60 — Воздух 0,00 0,00 — 1,80 — 0,00 —
вий адсорбционного равновесия и адсорбционной способности
угля по смеси углеводородов в условиях опыта. В двух режи-
мах пропано-бутановая фракция, содержащая от 8 до 13% выс-
ших углеводородов, была пропущена через небольшую допол-
нительную адсорбционную секцию с высотой слоя угля 30 см и
диаметром 10 см. Отходящий из дополнительной секции газ был
концентрированным товарным сжиженным газом с содержани-
ем высших углеводородов не выше 2%.
Изучение условий разделения многокомпонентной смеси уг-
леводородов в движущемся слое адсорбента доказало универ-
сальность метода для получения продуктов требуемого каче-
ства. В табл. 10-4 (а, б) приведены некоторые характерные ре-
зультаты разделения природного газа на опытной установке.
Меняя скорость движения угля в колонне, удается достичь лю-
бой заданной степени извлечения углеводородов, например, до
85% пропана при его содержании в исходном газе 2,5%.
Изменением количества вытеснительной флегмы, подаваемой
противотоком адсорбенту, в качестве нижнего продукта возмож-
Таблнца 10-46. Состав фракций (в %)
Компонент Бензиновая фракция Проданобутановая фракция Пропаяобутаиовая фракция после дополнительной колонны
режим 1 режим 2 режим 1 режим 2 режим 1 режим 2
С1 0,00 0,00 0,00 0,00 0,00
С2 0,05 0,00 10,30 0,00 1,50 1,50
Сз 0,30 0,84 74,70 12,72 70,57 —
с4 4,10 30,80 12,70 73,00 25,40 98,50
С5 95,55 68,36 2,30 13,40 1,53 .
со2 0,00 0,00 0,00 0,00 — —
Воздух 0,00 0,00 0,00 0,00 — —
Разделение смесей газов в движущемся слое адсорбента
247
Рис. 10,22. Потери активного рекуперационного
угля П в процессе эксплуатации.
но выделить газовый бензин с за-
данным давлением паров, опреде-
ляемым содержанием в нем бута-
нов (от 4 до 30%), и боковой про-
дукт — кондиционный сжиженный
газ (пропано-бутановую фракцию);
после пропуска бокового продукта
через небольшую дополнительную колонну с движущимся сло-
ем угля содержание примесей в пропано-бутановой фракции
снижается практически до нуля.
В условиях движения слоя адсорбента его механическая
прочность приобретает особо важное значение. Пылеобразова-
ние не только увеличивает эксплуатационные расходы в процес-
се разделения углеводородов, но и загрязняет поток отходя-
щего газа, что приводит к необходимости тщательного улавли-
вания пыли, чтобы избежать эрозии газопроводов. Накопление
пыли в системе циркулирующего угля приводит к нарушению
гидравлического режима и «текучести» угля по колонне.
Количество образовавшейся пыли определяли замером ее массы в цик-
лоне и периодическим контролем запыленности отбензиненного газа, выхо-
дящего с установки. Степень истирания угля за цикл определяли, как отно-
шение суммарной массы образовавшейся пыли к массе циркулирующего уг-
ля. На рис. 10,22 представлена кривая потерь рекуперационного активного
угля во времени. Наибольшие потери (П в %) отмечены в первый период
эксплуатации установки, когда оии достигали 0,16% в цикл. Затем потери
угля резко уменьшаются, частицы «окатываются», принимают шаровидную
или сигаровидиую форму, в системе остаются наиболее прочные зерна. Че-
рез три месяца эксплуатации потери снизились до 0,04% за цикл.
Газовый уголь, который при испытании на прочность по стандартной
методике в мельнице не имел преимуществ перед рекуперационным, оказал-
ся более прочным при испытании на опытной установке; уже через полтора
месяца эксплуатации потери этого угля снизились до 0,02% за цикл. Однако
сравнение этих показателей с показателями, полученными на установках с
движущимся слоем угля из скорлупы абрикосов и кокосовых орехов, ука-
зывают на значительно большую прочность углей, приготовленных из ко-
сточек. Потери кокосового угля на однотипных американских установках
составляют 0,001—0,002% за цикл.
Основное разрушение зерен угля на установках с движу-
щимся слоем происходит при подъеме частиц угля восходящим
потоком газа, т. е. в газлифте, причем крупные зерна, облада-
ющие большой энергией, при соударении или ударе о стенку
скорее крошатся. Этому способствуют большие скорости газо-
вого потока, которые приходится выдерживать при подъеме
крупных и тяжелых частиц (подъем рекуперационного угля идет
при минимальной скорости газа 9,5—10 м/с, а газового угля
при 6,5—7,0 м/с). Уменьшение насыпной плотности угля также
позволяет снизить скорость потока газа в газлифте. Оптиь аль-
248
10. Типы адсорбционных процессов и установок
ным размером гранул на установках с движущимся слоем сле-
дует признать 1—2 мм.
Средние размеры исходного рекуперационного угля составляли: диаметр
2,7 мм, высота 6 мм. Через сорок суток эксплуатации ситовой анализ пока-
зал следующее распределение частиц угля по размерам: более 3 мм —
22,43%; от 1 до 3 мм — 65,85%; от 0,5 до 1,0 мм — 3,14%; менее 0,5^—1,70%;
потери — 0,21 %.
Снижения потерь угля можно достичь при пневмотранспор-
те адсорбента в сплошном слое.
Широкое использование техники движущегося слоя в про-
мышленности возможно только на основе высокопрочных ад-
сорбентов, в основном шариковой формы. При испытании сфе-
рических гранул установлено, что их износ в 3—4 раза ниже
по сравнению с цилиндрическими равной прочности [35].
Стабильная работа адсорбционных установок непрерывного
действия возможна только при строгом контроле гидравличе-
ского режима. Любые отклонения от него, а также резкие из-
менения в распределении потоков приводят к нарушению ста-
бильности режима, зависанию адсорбента (в основном, в та-
релках отбора и в гидравлических затворах), потере текучести
угля, изменению качества продуктов.
Перепад давления в адсорбционной секции определяется ско-
ростью газового потока. При скорости 15 м/мин перепад давле-
ния в слое рекуперационного угля высотой 1 м составляет
1,2 кПа (плотность газа по воздуху 0,62). Перепады давления
на участке секции высотой 1 м в различных режимах приведе-
ны ниже:
№ режима 1 2 3 4
Расход газа, м3/ч 126 150 138 150
Скорость газа, м/мин 16,7 20,0 18,3 20,0
Перепад давления, кПа 1,12 1,60 1,50 1,40
Средний перепад давления на участке десорбера составлял
2,0 кПа, на участке секции хроматографического разделения —
1,5—1,6 кПа.
Перепад давления в газлифте ДРгазл определяется динами-
ческим сопротивлением газлифтной трубы ДРДИн и массы угля
gy, находящегося в газлифте сечения f:
Д^газл = ДРдин + Sylf (10.34}
Так как скорость газового потока в газлифте должна под-
бираться для конкретного угля минимальной, обеспечивающей
нормальный пневмотранспорт адсорбента, и остается во всех
режимах постоянной, то основной независимой переменной, оп-
ределяющей перепад давления, является величина gy. С другой
стороны, при постоянной скорости движения суспензии по газ-
Разделение смесей газов в движущемся слое адсорбента
249
лифту количество адсорбента, находящегося единовременно в
газлифте, прямо пропорционально часовому расходу угля G в
кг/ч. Тогда перепад давления в газлифте выражается линейной
функцией расхода угля:
ДРГазл = Д^дин + *С (10.35)
Ниже приведены результаты определения перепада давления
в газлифте при эксплуатации установки на рекуперационном
угле:
Расход угля, кг/ч 0 100 250 350 430
Давление в газлифте, кПа
внизу Pi 2,24 2,72 3,40 3,95 4,50
вверху Рг 1,20 1,20 1,25 1,30 1,30
перепад /^газл 1,04 1,52 2,15 2,65 3,20
Очистка водорода от примесей
Применение метода движущегося слоя не ограничивается разде-
лением нефтяных газов. Одним из возможных направлений раз-
вития процесса является очистка водорода от примесей (мета-
на, азота, монооксида углерода) в движущемся слое активного
угля при повышенном давлении (2—3 МПа).
Схема установки для исследования процесса очистки в движущемся
•слое адсорбента [36] принципиально не отличалась от описанной выше схе-
мы, предназначенной для разделения углеводородов. Общая высота уста-
новки составляла 6,1 м, внутренний диаметр колонны 30 мм. Водород, со-
держащий примеси, подавали в адсорбционную часть установки (высота
70 см); скорость подачи газовой смеси и соотношение между компонентами
регулировали реометрами. Очищенный водород из адсорбционной секции от-
водили сверху и степень его чистоты устанавливали по теплопроводности.
В случае очистки водорода от нескольких примесей состав выходящего газа
определяли газоанализатором. Чтобы установить распределение газа в слое
угля, в адсорбционной секции был предусмотрен отбор проб газа в про-
межуточной точке.
Насыщенный примесями адсорбент опускался в десорбционную часть
установки, причем скорость его движения регулировали специальным ло-
жечковым дозатором, расположенным ниже десорбера: угол наклона «ло-
жечки» к горизонтальной плоскости определял скорость движения угля.
Регенерировали уголь нагреванием в электрической трубчатой печи с
автоматическим регулированием температуры. В десорбционную часть пода-
вали динамический агент—водород. Регенерированный уголь через проме-
жуточные бункеры поступал в газлифт и потоком водорода транспортиро-
вался наверх установки. Состав и объем выходящих газовых потоков опре-
деляли после снижения давления до атмосферного. Скорость движения угля
контролировали с помощью частицы угля, меченной радиоактивным иодом,
путем замера времени ее прохождения между двумя счетчиками, установ-
ленными на расстоянии 1 м друг от друга; импульс радиоактивной частицы
передавался на световой сигнал.
На установках больших размеров в качестве расходомера сыпучего ма-
териала используют шнековые устройства, связанные с сигнальной лампой:
одновременно сигналы лампы свидетельствуют о непрерывном движении ад-
сорбента по колонне. При проведении опытных и лабораторных работ на
250 10. Типы адсорбционных процессов и установок
небольших установках или установках высокого давления, где шнековые
устройства оказываются непригодными, возникают большие затруднения при
установлении «текучести» адсорбента по колонне. Для этого случая в каче-
стве индикатора потока адсорбента разработано специальное звуковое уст-
ройство, регистрирующее шум движущегося адсорбента. Звуковые индика-
торы, основным элементом конструкции которых являлся камертон, были
использованы в качестве уровнемера активного угля при изучении процесса
выделения ацетилена адсорбционным методом [20].
На установках с движущимся слоем активного угля было
проведено пять серий опытов, включавших разделение бинарных
метановодородных смесей с концентрацией метана 5 и 20% (об.),
а также тройных и четверных смесей, где в качестве примесей
присутствовали метан, монооксид углерода и азот. В каждой
серии опытов давление варьировали от 0,5 до 2,5 МПа, причем
при постоянном давлении и постоянной скорости подачи газо-
вой смеси скорость движения угля в колонне повышалась от
опыта к опыту. Как уже указывалось, поглощенные примеси
десорбировали из угля при 200°C с одновременной продувкой
частью (10%) очищенного водорода. Эти условия гарантирова-
ли полную регенерацию угля и позволяли избежать его увлаж-
нения в промышленных условиях.
Горючие газы десорбции целесообразно сжигать в топочном
устройстве для использования дымовых газов в качестве теп-
лоносителя для нагрева угля [37]. Тогда углеадсорбционные
установки не требуют подвода дополнительного тепла.
В опытах, проведенных при достаточно высоких скоростях
движения угля, при любом давлении газоанализатор регистри-
ровал 100%-ную чистоту водорода, выходящего из адсорбцион-
ной секции колонны (чувствительность газоанализатора к при-
месям составляла 0,1%). Затем при определенной критической
скорости, характерной для каждого давления, нагрузка на уголь
начинала превышать его активность (1 см3 поглощенной при-
меси на 1 г адсорбента), в связи с чем чистота выходящего из
колонны водорода снижалась.
Результаты серии опытов очистки водорода от метана, со-
держание которого составляло 20% (об.), с использованием ак-
тивного угля СКТ представлены на рис. 10,23 в виде зависимо-
сти чистоты выходящего из колонны водорода от скорости дви-
жения адсорбента для разных давлений. По этим данным была
рассчитана максимальная нагрузка на адсорбент, равная сум-
марному количеству примесей, улавливаемому единицей массы
адсорбента, при отсутствии проскока. Она составляет для дав-
лений 0,5; 1,0; 2,5 МПа соответственно 20; 32 и 80 см3/г.
В следующей серии опытов была исследована очистка водо-
рода от метана, концентрация которого составляла 5% (об.),
при температуре в адсорбционной секции 20 °C и различных дав-
лениях. Максимальная нагрузка, обеспечивающая полную очи-
Разделение смесей газов в движущемся слое адсорбента
251
Рнс. 10,23. Зависимость активности адсорбента а и чистоты водорода с от расхода угля
w при очистке в движущемся слое угля СКТ (/«20 иС, Со—20% об.) при давлении 0,5,
1,0 и 2,5 МПа (соответственно I, 2 и 3).
Рис. 10,24. Активность угля по сумме примесей при различном исходном составе газа:
1—82% Н2, 15% СО, 3% СН4 (20 °C); 2-80% Н2, 20% СН4 (20 °C); 3-52,5% Н2, 8,7%
СО, 2,7% СН4, 36,1% N2 (20 °C); 4-80% Н2, 20% СН4 (-15 °C).
стку водорода в движущемся слое активного угля СКТ, состав'
ляла:
Давление, МПа 0,5 1,0 2,5
Нагрузка, см3/г 7,5 12,5 17,5
Отдельные пробы газа, отобранные из середины адсорбцион-
ной секции, показали, что при высоте движущего слоя угля
35 см и скорости потока 0,3 м/с обеспечивается полная очистка
водорода.
Высокая чистота водорода (выше 99,9%) была отмечена
также при очистке водорода от двух и трех одновременно при-
сутствующих примесей. Исходными газами служили в первом
случае модельная смесь, близкая по составу к газу, получен-
ному в результате кислородной конверсии метана (82% Н2,
15% СО, 3% СН4), и во втором — четырехкомпонентная смесь,
аналогичная по составу газам подземной газификации углей
(52,5% Н2, 36,1 % Иг, 8,7% СО, 2,7% СН4). Максимальная на-
грузка (по сумме примесей), обеспечивающая полную очистку
водорода, для тройной смеси при 1,0 МПа составляет 22 см3/г,
при 2,5 МПа — 42,6 см3/г, а для четырехкомпонентной смеси —
25, 45 и 100 см3/г при давлениях соответственно 0,5; 1,0 и
252
10. Типы адсорбционных процессов и установок
Таблица 10-5. Активность угля СКТ и его расход иа получение 1 м3 чистого водорода при 20 °C
Исходная смесь Активность угля (см3/г) при давле- нии (МПа) Расход угля в кг на получе- ние 1 м3 чистого водорода при давлении (МПа)
0,5 1,0 2,5 0,5 1,0 2.5
20% СН4+80% Н2 20,0 32,0 80,0 12,5 7,8 3,4
5% СН4+95% Н2 7,5 12,5 18,0 7,1 4,2 2,9
3% СН4+15% СО + • + 82% Н2 — 23,0 42,6 — 9,5 5,1
2,7% СН4+8,7% СО + + 52,5% Н2+36,1 % N2 22,5 45,0 100,0 45,0 22,2 9,1
20% СН4+80% Н2* * Температура минус 15 °C. 32,0 56,0 110,0 7,8 4,5 2,3
2,5 МПа. Присутствие монооксида углерода и метана в газе,
отходящем из адсорбционной секции колонны, обнаруживается
почти одновременно с проскоком азота.
Чтобы выяснить перспективность применения умеренного хо-
лода для адсорбционной очистки водорода, серия опытов была
проведена при пониженной температуре. Понижение температу-
ры в адсорбционной секции с +20 до —15 °C позволило увели-
чить адсорбционную способность угля и, следовательно, умень-
шить расход угля в среднем на 30%. Так, при разделении би-
нарной смеси (80% Нг+20% СН4) в движущемся слое угля
СКТ максимальная нагрузка, обеспечивающая полноту очист-
ки водорода, при давлении 2,5 МПа и температуре минус 15 °C
составляет ПО см3 метана на 1 г против 80 см3/г при том же
давлении и температуре 20 °C. На рис. 10,24 построены зависи-
мости активности угля от давления для исследованных случаев.
Сравнение активности угля, определенной эксперименталь-
но на установке с движущимся слоем адсорбента, с результа-
тами расчета, проведенного на основе изотерм чистых компонен-
тов газовой смеси, дало близкие результаты. Следовательно,
процесс очистки водорода в движущемся слое протекает при ре-
комендуемых скоростях газового потока в условиях, близких к
равновесным.
Результаты расчета расхода угля, необходимого для очистки
водорода на установке непрерывного действия, приведены в
табл. 10-5.
Выделение ацетилена из низкоконцентрированных газов
В промышленности нашел широкое применение метод получе-
ния ацетилена путем неполного окисления природного газа (ме-
тана). В результате окисления метана образуется смесь газов,
Разделение смесей газов в движущемся слое адсорбента
253
Рнс. 10,25. Изотермы адсорбции ацетилена на
угле СКТ.
20
содержащая 6—8% ацетилена, 16
4—6% диоксида углерода, 25%
монооксида углерода, 50% водо- 11г
рода, небольшое количество ме- 8
тана, следы азота и кислорода. В
случае, если в качестве окисли- 4
теля используют воздух, концент-
рация ацетилена в продуктовом °
газе не превышает 3%. Одним из
перспективных методов выделе-
ния ацетилена из разбавленных газов является его поглощение
адсорбентами.
В табл. 10-6 приведены значения равновесной адсорбционной способно-
сти промышленных адсорбентов по ацетилену и сопутствующей ему приме-
си— диоксиду углерода при двух давлениях [6,65 и 93,10 кПа (50 и 700 мм
рт. ст.)]. Наибольшей поглотительной способностью по ацетилену обладают
активные угли и цеолиты. Изотермы адсорбции ацетилена на активном угле
СКТ представлены на рис. 10,25.
Избирательность адсорбции в системах апетилеи — диоксид углерода
(табл. 10-7) как на активном угле, так и на силикагеле невысока и одина-
кова: среднее значение коэффициента разделения около 2. Адсорбированная
фаза в небольшой степени обогащается лучше поглощающимся компонен-
том — ацетиленом. Еще меньшая избирательность адсорбции этой смеси от-
Таблица 10-6. Равиовесиая адсорбциоииая способность промышленных
адсорбентов (в г/100 г) по ацетилену и диоксиду углерода при 20 °C
Адсорбент Ацетилен Диоксид углерода
6,65 кПа 93,10 кПа 6,65 кПа 93,10 кПа
Активные угли
Газовый уголь АГ 1,4 6,3 1,0 5,5
Газовый уголь СКТ 3,2 12,7 2,0 10,0
Рекуперационный уголь АР 2,0 7,8 1,2 7,4
Импортный уголь «Норит R-1» 1,4 9,4 1,2 9,4
Импортный уголь F-23 3,3 Силикагели 12,0 3,0
Крупнопористый 0,4 3,8 0,4 2,0
Мелкопористый 1,6 Бокситы 4,8 — ——
Соколовский 0,40 0,60 0,35 0,50
Боксит США 0,45 0,75 0,40 0,65
Синтетические цеолиты
Цеолит NaA 6,0 8,6 9,0 13,2
Цеолит СаА 6,6 9,4 13,8 16,8
254
10. Типы адсорбционных процессов и установок
Таблица 10-7. Адсорбционное равновесие в системе
диоксид углерода — ацетилен при 25 °C и нормальном давлении
Адсорбент Мольная доля в газовой фазе Мольная доля в адсорбированной фазе Л = ^СОахСгН2 __ Р ^CjH/cOj
йсо2 ус2н2 *со2 хс2н2
Силикагель 70,5 29,5 52,5 48,5 2,16
38,8 62,2 24,4 75,6 1,96
50,0 50,0 36,4 66,6 1,75
Среднее 1,96
Активный уголь 84,4 15,6 71,4 28,6 2,17
70,0 30,0 57,5 42,5 1,73
52,2 47,8 37,8 62,2 1,80
20,0 80,0 10,0 90,0 2,24
Среднее 1,96
мечена на цеолите. При соотношении компонентов в газе 1 : 1 адсорбиро-
ванная фаза содержала лишь 56% ацетилена, что соответствует коэффициен-
ту разделении Л'р=1,27. Коэффициент разделения указывает на возмож-
ность только частичного отделения от диоксида углерода в секции хрома-
тографического разделения адсорбционной колонны непрерывного действия.
Извлечение ацетилена на модельной смеси газов неполного
окисления метана воздухом с частичным отделением его от
диоксида углерода в движущемся слое адсорбента было про-
ведено на укрупненной лабораторной установке. Чтобы избе-
жать возможности воспламенения ацетилена, в качестве транс-
портного газа был использован азот. Так как по адсорбционной
и разделяющей способности в данной системе активные угли
несколько превосходят другие адсорбенты, основная серия опы-
тов была проведена с фракцией размельченного рекуперацион-
ного угля (зернение 0,1—0,2 мм).
В адсорбционной части установки при высоте движущегося
слоя угля 50 см было достигнуто полное извлечение ацетилена,
если нагрузка ацетилена на уголь не превышала 95% от рав-
новесной адсорбционной способности. Повышение температуры
угля в адсорбционной секции под действием теплоты адсорбции
равно 5 °C в случае газа, получаемого при окислении метана
воздухом, и 7—8 °C при окислении кислородом.
Большинство опытов было проведено с постоянным мольным
соотношением ацетилена и диоксида углерода 1:1. Высота хро-
матографической части составляла 120 см, диаметр 3 см, ско-
рость подачи смеси 120—200 сма/мин. Результаты опытов по-
казали, что содержание диоксида углерода в ацетиленовой
фракции составило от 1 до 10% (об.), содержание ацетилена
Разделение смесей газов в движущемся слое адсорбента
255
в промежуточной углекислотной фракции — не ниже 20% (об.).
Из-за низкой чистоты фракций на американских установках
ограничились выделением методом непрерывной адсорбции лишь
ацетилено-углекислотной фракции, из которой 98%-ный ацети-
лен извлекали в процессе последующей обработки селективным
растворителем [9].
Выделение чистого ацетилена с одновременным концентриро-
ванием его в хроматографической секции было осуществлено в
Венгерском научно-исследовательском институте нефти и при-
родного газа на опытной установке высотой 26 м; диаметр ко-
лонны в адсорбционной секции составлял 25 см, в секции хро-
матографического разделения 20 см [20]. Общая высота секций,
непосредственно предназначенных для разделения газов (ад-
сорбционной и хроматографической) была равна 5 м. Скорость
движения угля была около 15 см/мин в адсорбционной и
22 см/мин в хроматографической секции колонны. Гидравличе-
ский режим в колонне был организован таким образом, чтобы
поглощенный вначале диоксид углерода вытеснить ацетиленом
из хроматографической секции в адсорбционную и удалить его
из последней вместе с отходящим тазом.
При производительности по газу 30 м3/ч и расходе актив-
ного угля 210 кг/ч в качестве нижнего продукта получен кон-
центрированный 99%-ный ацетилен. Результаты разделения га-
зов неполного окисления метана кислородом приведены в
табл. 10-8.
Попытка венгерских инженеров осуществить десорбцию аце-
тилена только нагревом через стенку даже при невысоком дав-
лении в системе (0,15 МПа) не привела к положительным ре-
зультатам: при температуре в трубчатом нагревателе 300 °C де-
сорбция была неполной. После того как в десорбер было пода-
но 0,6—0,7 кг острого пара на 1 кг извлеченного ацетилена, или
0,7—0,8% (масс.) от массы циркулирующего угля, активность
его полностью восстанавливалась. Высота хроматографической
Таблица 10-8. Разделение газа неполного окисления метана
кислородом на опытной установке
Компонент Исходный газ Нижний продукт Верхний продукт
мЗ/ч % (Об.) мЗ/ч % (об.) мЗ/ч % (Об.)
Водород 18,90 63,30 — — 18,90 68,04 Монооксид угле- 7,64 25,50 — — 7,64 27,50 рода Диоксид углерода 1,26 4,20 0,02 0,90 1,24 4,46 Ацетилен 2,10 7,00 2,10 99,10 — — Итого 30,00 100,00 2,12 100,00 27,78 100,00
256
10. Типы адсорбционных процессов и установок
секции, в которой осуществлялось отделение диоксида углерода
от ацетилена, на опытной установке составляла 2—3 м.
В связи с тем, что теплоты адсорбции ацетилена и диоксида
углерода близки, метод регулирования количества вытеснитель-
ной флегмы в хроматографической секции по температуре слоя
в данном случае непригоден; он был заменен регулированием
разделения по данным анализа газа, отбираемого из середины
хроматографической колонны. Истирание венгерского угля со-
ставляло 0,01—0,02% за цикл, или 20—25 г/кг извлеченного
ацетилена. Активность угля при работе на модельных смесях в
течение 1 мес не снижалась, однако при наличии в газе поли-
меризующихся гомологов ацетилена в схему необходимо вклю-
чать секцию очистки от них.
Другие варианты непрерывного
адсорбционного метода
Предприняты попытки создания непрерывных адсорбционных
колонн, в которых адсорбент остается неподвижным, а зоны пе-
ремещаются по колонне в соответствии с программой управ-
ляющего устройства. Такая колонна, используемая в процессе
«молекс», описана в разделе, посвященном депарафинизации
нефтяных фракций цеолитами.
Другая конструкция изображена на рис. 10,26 [38]. Аппарат
предназначен для улавливания паров летучих растворителей из
воздуха.
Адсорбция происходит в секциях медленно вращающегося с помощью
привода 1 барабана 6. Воздух, содержащий пары растворителя, после филь-
трации (4) и охлаждения (3) вентилятором 2 нагнетается в пространство
внутри кожуха 5, проходит через слои угля в секторах и удаляется из цент-
ральной части. Вращаясь, секции поступают в десорбционную часть. Здесь
растворитель удаляют водяным паром, парогазовую смесь конденсируют
(7) и разделяют (8). Растворитель и воду удаляют через штуцеры по ком-
муникациям 9 и 10. Несмотря на компактность, установки с вращающимся
барабаном на практике применяются редко.
Очистка газа суспензиями ад-
сорбента. Высокая поглотительная
способность адсорбентов сохраняет-
ся, если последние используют в
виде суспензии в жидкой неадсор-
бирующейся среде [39]. Особенно
эффективны в таком процессе мо-
лекулярно-ситовые угли или цеоли-
Рис. 10,26. Схема установки рекуперации раство-
рителей с движущимся барабаном:
1 — привод; 2 — вентилятор; 3 — холодильник; 4 —
фильтр; 5 — кожух; 6 — барабан; 7 — конденсатор;
8 —сепаратор; 9 — штуцер для отвода раствори-
теля; 10 — штуцер для отвода воды.
Гидравлическое сопротивление слоя
257
ты с входными окнами малых размеров (0,4—0,5 нм), если они
распределены в органической жидкости, молекулы которой
столь велики, что не проникают через эти окна в адсорбцион-
ные полости. В качестве такой среды для адсорбентов с опре-
деляющим размером пористой структуры 0,4 нм может быть
взят, например, н-гексан, а при определяющем размере 0,5 нм —
изогексан.
Суспензию измельченного молекулярно-ситового угля или
цеолита NaA в н-гексане применяют для очистки природного га-
за с небольшим содержанием диоксида углерода — 0,2% (об.).
Процесс проводят в противоточной адсорбционной колонне при
давлении 4 МПа. После снижения давления до нормального рас-
твор деметанизируют. Регенерацию его осуществляют циркуля-
цией суспензии через колонну в противотоке с очищенным га-
зом.
Гидравлическое сопротивление слоя
адсорбента
Энергия, затрачиваемая на преодоление гидравлического сопро-
тивления слоя в адсорбционных установках, часто лимитирует
скорость потока в адсорбере и значительно удорожает процесс.
В настоящее время результаты многочисленных исследований
в этой области обрабатываются с помощью безразмерных пара-
метров [40]. В основу такой обработки положена зависимость
между коэффициентом сопротивления f и критерием Рейнольд-
са Re [41]:
f = <p(Re) (10.36)
причем в этом уравнении
/ = ^-ДрЩэр/(2Лб2) (10.37)
Re = d3-G/p (10.38)
где g — ускорение свободного падения; Др— перепад давления в слое; da —
эквивалентный диаметр частиц; р — плотность газа; L — длина слоя; G —
массовая скорость газа; ц— вязкость газа.
Зависимость f = cp(Re) используют для расчета перепада дав-
ления при любой вязкости, плотности, скорости газового потока,
зернения адсорбента и высоты его слоя, но при постоянном сво-
бодном объеме адсорбента, под которым даже в случае пори-
стых сред понимают объем между зернами (без учета объема
порового пространства зерен). При движении адсорбента по
колонне непрерывного действия свободный объем может не-
сколько изменяться.
По аналогии с расчетом перепада давления в круглых га-
зопроводах Блейк предложил учитывать в расчетных формулах
17—1346
258
10, Типы адсорбционных процессов и установок
IV,М/С Рис. 10,27. Гидравлическое сопротивление слоя
О 0 25 050 0 75 (1=2 м) активных углей:
— уголь Байера; б —БАУ; в — КАД-иодиый; г —
газовый уголь; д — рекуперационный гранулиро-
ванный уголь; е — рекуперационный уголь (дроб-
леный, фракция 2—3 мм).
ных адсорбентов и разных
крыт в работе [44].
величину (долю) свободного объе-
ма между зернами е [42].
В связи с широким развитием
процессов каталитического крекин-
га, каталитического риформинга,
теплообмена в слое гранулирован-
ной насадки, осуществляемых в
движущемся слое, Хаппель [43]
подробно исследовал перепад дав-
ления при прямоточном и противо-
точном пропускании воздуха через
слой движущегося катализатора
различной формы (таблетированно-
го, сферического и шарикового)
размером 0,25—4,7 мм. Автор пред-
ложил удобную функцию, хорошо
согласующуюся с опытными данны-
ми и учитывающую изменение сво-
бодного объема в неподвижном и
движущемся слоях катализатора,
связывающую модифицированный
коэффициент сопротивления fm =
=f/(l—s)3 с модифицированным
критерием Рейнольдса Rem =
= Re(l—s). Вид зависимостей f=
= <p(Re) и M = q/(Rem) примени-
тельно к разным типам промышлен-
режимах их использования был рас-
Поверхность пористого тела рассматривали как непроницаемую (в аэро-
динамическом смысле). Согласно второму допущению авторов, поверхность
считалась гладкой. Это допущение приемлемо потому, что шероховатость-
поверхности у всех промышленных гранулированных адсорбентов близка и,
следовательно, влияние шероховатости должно входить в равной степени в-
общий эмпирический коэффициент расчетных формул. Удовлетворительная
еходимость, полученная при сравнении результатов опытов с рассмотренны-
ми зависимостями, подтверждает справедливость этих допущений.
Для экспериментального исследования были выбраны про-
мышленные газовый и рекуперационный активные угли (стан-
дартный и измельченный до 2—3 мм), БАУ, КАД-иодный и
уголь Байера (производство ФРГ), а также цеолиты. В каче-
стве текучей среды использовали диоксид углерода, азот, метан.
Гидравлическое сопротивление слоя
259
и водород. На рис. 10,27 представлены кривые перепада давле-
ния в слое различных активных углей высотой 2 м. Исследо-
ванные адсорбенты имели различные размеры и различную фор-
му, в большинстве случаев цилиндрическую. Для нешаровидных
тел эквивалентный диаметр d3 определяли по удельной геомет-
рической поверхности тела Зг (поверхности единицы объема):
d3 = 6/Sr (10.39}
В свою очередь поверхность единицы объема определяется
отношением
Sr = SrP/VrP (’0.40)
где Srp и Vrp — поверхность и объем отдельной гранулы. .
Доля свободного объема для активных углей и других про-
мышленных адсорбентов в неподвижном слое составляет в сред-
нем е = 0,40 [44]. Эта величина для движущегося слоя адсор-
бента приблизительно на 10% выше, т. е. е = 0,44. Средние раз-
меры (диаметр d и высота А), поверхность Srp и объем lzrp гра-
нул, поверхность единицы объема Зг, эквивалентный диаметр
активных углей, а также цеолитов приведены в табл. 10-9.
На основе экспериментальных данных для разных систем по
формулам (10.37) и (10.38) определены значения коэффициента
-сопротивления f и критерия Рейнольдса Re, а также их моди-
фицированные формы. Зависимость f=<p(Re) представлена ниж-
ней, а зависимость fm = cp(Rem) верхней кривой на рис. 10,28.
Анализ кривых показывает, что ламинарная область в слое ад-
сорбента ограничивается числами Рейнольдса Re=10—20; это
приблизительно соответствует скоростям технологических газов,
применяемых в промышленных условиях (0,10—0,25 м/с).
Фис. 10,28. Зависимость коэффициентов сопротивления f, f от чисел Рейнольдса Re,
Фе .
'гм
Фис. 10,29. Системы в координатах /Re — Re или fmRem — Rem.
17*
260
10. Типы адсорбционных процессов и установок,
Таблица 10-9. Характеристика адсорбентов
Показатель Активные угли Цеолиты
газо- вый рекупера- ционный Байера фирмы Линде советские образцы
d, мм 1,53 2,96 4,46 1,6 3,2 2,0 4,5
h, мм 3,33 4,12 6,00 4,0 7,0 2,0 5,0
Srp=nd[/H- (d/2)], мм2 19,7 53,0 115,2 24,0 86,0 18,5 102,8
Vrp = 0,785 d2A, мм3 6,1 28,2 93,5 8,0 56,0 6,3 47,8
Sr=Srp/yrp, мм2/мм3 3,23 1,84 1,23 3,00 1,54 2,95 1,28
d3=6/ST, мм 1,86 3,26 2,88 2,00 3,90 2,04 4,69
Как показал Минский [45] при исследовании фильтрации
газа в пористых средах, между коэффициентами сопротивления
и критерием Рейнольдса существует соотношение
/=(a/Re) + * (10.41)
Указанная закономерность соблюдается и для слоя адсор-
бентов. Это четко прослеживается на рис. 10,29; зависимости в
координатах /-Re—Re и fm-Rem—ReTO аппроксимируются прямы-
ми линиями. Коэффициенты а и b определяют графически: а —
как отрезок, отсекаемый прямой на оси ординат, b — как тангенс
угла наклона прямой к оси абсцисс.
Если выразить g в м/с2, Др в кг/м2, d3 в м, р в кг/м3, L в м,
G в кг/(м2-с), ц в кг/(м-с), то после определения коэффици-
ентов формула (10.41) приобретает вид
/= (770/Re)+Ю,6 (10.42)
Аналогично
fm = (1900/Rem) + 52 (10.43)
Подставляя значения коэффициентов сопротивления и критерия
Рейнольдса, после преобразования получим
g-Ap-dgp 770р.
2lG2'--^G +10-6 <10-44)
2LG2 I 770ц \
Др = —г— ~Йг +10,6
gd3P ( 4G J
ИЛИ
2L / 770цб \
^ = l^(-^+10’6G2J
Аналогичным образом
g-^P-d3p_____ЮООц
2LG2 (1 — е)3 “ d3G (1 — е) + °2
2Z.G2 (1-е)3 Г 1900ц
Др= gd3p I d3G(l-e) +52
(10.45)
(10.46)
(10.47)
(10.48)
Гидравлическое сопротивление слоя
261
или
2L(1—е)3 Г 1900uG I
+52G2J (10-49>
Формула (10.46) предусматривает, что свободный объем ад-
сорбента составляет е = 0,4, т. е. соответствует условиям устано-
вок с неподвижным слоем. В формулу (10.49) свободный объем
входит в качестве переменной величины, т. е. она справедлива
при любой «рыхлости» упаковки, в частности и в условиях уста-
новок с движущимся слоем.
Целесообразность использования двучленных формул для
расчета гидравлического сопротивления была подтверждена на
примере прохождения воздушного потока через слой алюмоси-
ликатного катализатора [46]. Сравнения результатов вычис-
ления по метану и другим газам с опытными данными показали,
что расчет, проведенный как по двучленной формуле, так и по
графику зависимости коэффициента сопротивления от числа
Рейнольдса, дает максимальную ошибку ±7%. Изложенная ме-
тодика расчета перепада давления использована при проектиро-
вании ряда адсорбционных установок, в которых применяют ак-
тивные угли, силикагели и цеолиты.
Известны и другие формулы для расчета гидравлического сопротивле-
ния*. Так, Дубинин [47] предлагает следующие формулы для определения
перепада давления в слое активных углей:
для ламинарной области
Др = 1685ц (wL/d32) (10.50)
для переходной области
Др = 76,4р°,15р0,85/ (Тш!,33/^1,13) (10.51)
Размерность переменных в этих формулах такова: Др [бар], ц [пуаз]^
р [г/см3], L [см], w— линейная скорость газа [см/с], [см].
На основе измерений перепада давления в слое цеолитов немецкие ин-
женеры [48] предложили формулу
Др/L = / (w2p/2gd3) (10.52)
Для шаров
f = 10,9- 102/(Re')°,e4 (Ю.53)
Для цилиндров
/= 3,64-102/(Re')»,s» (10.54)
Re' = wd3/v (10.55)
где w — линейная скорость газа, м/с; v — кинематическая вязкость газа, м2/с.
* Гидравлическое сопротивление в слое адсорбента не содержит собствен-
но адсорбционной специфики, а относится к классу общих проблем зернисто-
го слоя. Поэтому более подробную информацию, включающую анализ фор-
мул, предложенных для расчета гидравлического сопротивления, читатель
найдет в монографиях [40] и, особенно, Аэрова М. Э., Тодеса О. М., Парий-
ского Д. А. Аппараты, со стационарным зернистым слоем. Л., «Химия», 1979.
176 с. (Прим, ред.)
262 11. Получение и номенклатура адсорбентов
Для процессов адсорбционной очистки в жидкой фазе перепад давления
(в Па) в слое угля высотой 1 см может быть определен по формуле
Др = 5pG/(pd3) (10.56)
Величины, входящие в формулу, выражены: ц [пуаз], G [г/(см2-с)],
[г/см3], da [см].
Как указывалось, сопротивление слоя адсорбента вносит оп-
-ределенные ограничения при выборе скорости потока. В рекупе-
рационных установках условия целесообразно подбирать таким
образом, чтобы перепад давления не превышал 5 кПа (500 мм
.вод. ст.) на 1 м слоя.
И
ПОЛУЧЕНИЕ
И НОМЕНКЛАТУРА
ПРОМЫШЛЕННЫХ
АДСОРБЕНТОВ
Активные угли
Получение активных углей
При производстве активного угля вначале исходный материал
подвергают термической обработке без доступа воздуха, в ре-
зультате которой из него удаляются летучие (влага и частично
смолы). Структура образовавшегося угля-сырца — крупнопори-
стая, он не содержит микропор и не может быть непосредствен-
но использован как промышленный адсорбент. Задача получе-
ния ажурной микропористой структуры решается в процессе
активации, которую проводят двумя основными методами: окис-
лением газом либо паром или обработкой химическими реаген-
тами.
При парогазовой активации чаще других реагентов исполь-
зуют диоксид углерода и водяной пар. Процесс в присутствии
диоксида углерода ведут при температуре около 900 °C. При
этом часть угля выгорает:
С + СО2 —2СО (11.1)
Активные угли
26S
В связи с тем, что реакция углерода с диоксидом углерода
эндотермичная, в систему требуется непрерывно подводить теп-
ло. Долю угля, выгоревшего при активации, называют сте-
пенью обгара. Наиболее микропористые угли образуются
при степени обгара 50%. В пределах степени обгара 50—75%
получают разнороднопористые активные угли с достаточно раз-
витой микро- и макроструктурой, а выше 75% —макропористые
угли.
В качестве окислителя иногда применяют водяной пар. Окис-
ление паром проводят при 850 °C:
С + Н2О -->- СО + Н2 — 130 кДж (11.2)
Параллельно протекает побочная экзотермическая реакция
СО + Н2О ► СО2 + Н2 + 42 кДж (11.3)
Реакция угля с паром катализируется оксидами и карбона-
тами щелочных металлов. Поэтому их в небольшом количестве
иногда добавляют к исходному сырью при производстве актив-
ных углей. Катализаторами процесса являются также соедине-
ния железа и меди.
Активация воздухом или другими кислородсодержащими га-
зами, протекающая по реакциям:
С + о2 ---> СО2 + 376 кДж (11.4)
2С+О2 -----► 2СО + 227 кДж (11.5)
применяется редко.
Процесс активации исходного углеродного вещества газооб-
разными реагентами сводится к следующему [49]. Исходные
карбонизированные органические материалы до температуры
активации (850—950 °C) состоят из упорядоченной части —кри-
сталлитов углерода, взаимно упакованных сравнительно нере-
гулярно, и так называемой аморфной части — высокоуглеродных
радикалов, связанных с атомами углерода призматических гра-
ней кристаллитов. В процессе активации газообразными окис-
лителями (пары воды, диоксид углерода) при температурах
850—950 °C диффузия газообразного активатора в поры угля
сопровождается химическим взаимодействием.
В первую очередь выгорает наименее плотный аморфный уг-
лерод, что приводит к образованию микропор нерегулярного
строения. При очень малых «обгарах» образуется весьма за-
метный объем микропор. При дальнейшей активации происходит
частичное или полное выгорание отдельных кристаллитов с об-
разованием основного объема микро- и супермикропор. В этой
стадии объемы образующихся микро- и супермикропор прибли-
зительно пропорциональны обгару. Параллельно завершается
развитие мезо- и макропор.
264 11. Получение и номенклатура адсорбентов
Процесс газовой активации проводят в печах трех типов.
Вращающиеся печи обогреваются теплом сгорания коксового,
генераторного газа или жидких нефтепродуктов. Сжигаемые га-
зы, пар, активируемый углеродный материал поступают в тот
конец печи, где поддерживается небольшое разрежение. Избы-
ток газа сжигается в воздухе, который вводится через форсунки,
расположенные вдоль всей длины печи. Тем самым достигается
постоянство уровня температур активации в печи. Достоинст-
вом вращающихся печей является их низкая стоимость и высо-
кая производительность. В то же время интенсивное переме-
щение твердого материала в печах этого типа приводит к пы-
.леобразованию и чрезмерным потерям от истирания.
В шахтных печах активацию ведут в слое углеродного ма-
териала. Они представляют собой систему из нескольких десят-
ков узких шахт высотой 10—20 м. Шахты выполнены из спе-
циальных фасонных кирпичей. Материал, который подвергают
активации, заполняет все пространство печи и пересыпается вниз
под действием силы тяжести. В нижней части расположены вы-
пускные отверстия, которые открываются через определенные
интервалы времени и, тем самым, регулируют скорость движе-
ния твердого материала. По вертикали каждую печь можно ус-
ловно разбить на три зоны: карбонизации, активации и охлаж-
дения.
Каждая печь состоит из двух агрегатов, которые периоди-
чески взаимно изменяют свои функции. В агрегат, который на-
ходится на фазе активации, вводят активирующий агент — пере-
гретый водяной пар, который сверху вниз проходит через си-
стему шахт. В результате эндотермичных реакций, характерных
для стадии активации, материал, нагретый в предшествующей
фазе, постепенно охлаждается до температуры 800 °C. Отхо-
дящие газы со значительным содержанием водорода и моноок-
сида углерода, в камере сгорания после смешения с воздухом
сжигаются. Продукты сгорания поступают во второй агрегат,
действующий как нагреватель. Активируемый материал, нахо-
дящийся в этом агрегате, нагревается до температуры 950 °C.
Газы, выходящие из агрегата, содержат горючие компоненты и
сжигаются при добавке воздуха, тепло горения используется
для нагрева футеровки пароперегревателя. Продукты сгорания
отводятся через дымовую трубу в атмосферу. Каждые 30 мин
агрегаты меняют свои функции.
Достоинством шахтных печей является их энергетическая са-
мообеспеченность. Тепло от внешнего источника необходимо
только в пусковой период. Однако шахтные печи имеют ряд су-
щественных недостатков: на их сооружение требуются большие
капиталовложения, они имеют низкую производительность, сте-
пень активации в разных шахтах, где скорость спускающегося
материала может быть различной, неодинакова.
Активные угли
265
Конструктивно наиболее совершенными являются печи ки-
пящего слоя, обеспечивающие получение равномерно активиро-
ванных частиц. Карбонизованный материал активируется в
псевдоожиженном (кипящем) слое смесью горячих газов и во-
дяного пара. Метод активации в кипящем слое нашел широкое
применение в производстве дешевых порошкообразных или дроб-
леных сортов активного угля. Однако при производстве грану-
лированных и порошкообразных обеспечивающих углей он пока
не используется.
Другой вид активации заключается в обработке угля соля-
ми, которые при высокой температуре выделяют газ-активатор
(например, СО2, О2): карбонатами, сульфатами, нитратами, кис-
лотами-окислителями (азотной, серной, фосфорной и др.) и го-
рячими концентрированными растворами некоторых солей. По-
следние переводят целлюлозу в раствор, при повышении темпе-
ратуры которого выделяется аморфный высокодисперсный угле-
род, образующий микропористую структуру. Угли, полученные
химическим активированием, называют в соответствии с при-
меняемым реагентом, например «уголь хлорцинковой актива-
ции».
Химическую активацию проводят при 200—650 °C. Каждый
из реагентов имеет свои недостатки и достоинства [50]. Обра-
ботку серной кислотой ведут при температуре не выше 200ЪС,
полученный после выщелачивания уголь в сухом состоянии об-
ладает незначительной адсорбционной способностью. При акти-
вации фосфорной кислотой необходимы температуры в пределах
375—500 °C; в этих условиях возникают проблемы, связанные
с коррозией аппаратуры. Активацию хлоридом цинка проводят
при 550—650 °C. Недостатком метода является некоторое за-
грязнение угля следами солей цинка. В качестве активаторов
иногда применяют хлориды магния, железа, аммония, тиоцио-
нат калия, карбонат натрия и другие вещества.
Процессы химической активации характеризуются коэффи-
циентом пропитки — отношением массы безводного акти-
ватора к массе сухого исходного углеродистого материала. Вна-
чале с увеличением коэффициента пропитки происходит разви-
тие микропористости угля, за счет чего увеличивается объем
пор. При высоких значениях этого коэффициента объем микро-
пор уменьшается, и развивается макропористость. При степени
пропитки, равной двум, уголь, полученный хлорцинковой акти-
вацией, содержит преимущественно крупные поры.
Активный уголь, полученный методом химической активации,
отличается большей однородностью структуры партии в целом,
чем структура партии угля парогазовой активации. Более вы-
сокая однородность характерна и для отдельных гранул этих
углей: их периферийные и внутренние участки однотипны.
266
11. Получение и номенклатура адсорбентов
Микропористая структура активных углей малой степени
активации, как правило, однородна: все микропоры примерно
одинаковы и имеют форму щелей. Изотермы адсорбции на этих
углях описываются одночленным уравнением теории объемного
заполнения микропор при ранге распределения п = 2, причем ве-
личина Ео в данном случае находится в обратной зависимости
от размеров щели. В сильно активированных углях имеются мик-
ропоры различного размера и описание изотерм адсорбции на
этих углях рекомендуется проводить с использованием двучлен-
ного уравнения теории объемного заполнения микропор.
Медицинские активные угли за рубежом готовят, как прави-
ло, пропиткой угля-сырца растворами сульфида и сульфата ка-
лия. После пропитки уголь подвергают активации при 900—
1000 °C. При этом протекает реакция
K2SO4 4С ---->- K2S4-4CO (11,6)
Чтобы получить уголь высокой чистоты, продукт активации
обрабатывают раствором сульфида калия, промывают соляной
и плавиковой кислотами, а затем из адсорбента при 600 °C уда-
ляют сернистые соединения в токе диоксида углерода. После
водной промывки, сепарации и сушки уголь поступает в упако-
вочное отделение.
Схема получения гранулированных активных углей методом хлорцинко-
вой активации (рис. 11,1) описана Флешером [51J. Раствор хлорида цинка
(плотность 1,8 г/см3) и пылевидный исходный материал вводят в аппарат 1
и перемешивают в течение 3 ч при 90 °C. Обычно коэффициент пропитки со-
ставляет 1,0—1,4. Учитывая коррозионность среды, внутреннюю часть аппа-
рата выполняют из бронзы. Пластичную пасту после охлаждения формуют в
машине 2, также футерованной бронзой. В зависимости от назначения размер
отверстий фильер формовочной машины позволяет получать угольные ци-
линдрики диаметром от 2 до 6 мм. Влажные гранулы сушат при температу-
ре до 180 °C во вращающейся печи 3. Длинные гранулы при этом обламыва-
ются, образуя частицы длиной 3—12 мм. Активацию проводят также во вра-
щающейся печи 4 — в противотоке с бескислородным газом при 600—700 °C.
Отходящие газы содержат пары и аэрозоль хлорида цинка, которые частич-
но рекуперируют после охлаждения газа. При активации улетучивается 30—
60% хлорида цинка, использованного для приготовления пасты.
Для того, чтобы увеличить степень улавливания хлористого цинка, по-
сле конденсационного отделения 11 устанавливают абсорбционные колонны
42, орошаемые разбавленным раствором хлорида цинка, и (или) электро-
•фильтры 13. Степень улавливания аэрозоля в электрофильтрах достигает
'98%. Уголь после активации насыщен хлоридом цинка. Его удаляют в вер-
тгикальной экстракционной колонне 5, через которую циркулирует разбав-
ленный раствор хлорида цинка, подкисленный соляной кислотой.
В нижней части аппарата остаток хлорида цинка вымывают горячей
соляной кислотой. Рекуперационный хлорид цинка возвращают в процесс.
Экстракция продолжается несколько часов. Затем угли промывают водой
в аппарате 6 до тех пор, пока кислотность и содержание хлор-ионов в про-
мывных водах не снизятся до заданного уровня. Гранулы угля сушат во
вращающейся сушилке 7 и подвергают дополнительной активации паром
в печи 8, при которой происходит не только улучшение пористой структуры
•.продукта, но и окончательно удаляется соляная кислота. Продукт рассеива-
ют и упаковывают в тару.
Активные угли
267
Схема приготовления порошкообразных углей принципиально мало отли-
чается от схемы получения гранулированных углей, но расходные показате-
ли при этом значительно ниже, что предопределяет более низкую стоимость
порошкообразных углей. При получении 1 т порошкообразных углей расхо-
дуется 300 кВт-ч электроэнергии, 2000 м3 коксового газа, 0,4—0,6 кг хлори-
да цинка. Соответствующие показатели на 1 т гранулированного угля:
1200 кВт ч, 5000 м3 и 1,0—2,2 кг.
В Институте горючих ископаемых разработан метод полу-
чения углеродных адсорбентов сферической формы из ископае-
мых углей без применения связующих веществ [52]. Сорбенты
наиболее высокого качества получены из газовых углей или из
шихт, в состав которых входили газовые и коксовые угли.
Структура активных углей сферической формы создается при
термической переработке исходного угля за счет перехода его в
пластичное состояние и создания монолитной гранулы в резуль-
тате последующих процессов полимеризации.
Технология получения таких адсорбентов сводится к следующему. Ис-
ходный уголь или смесь углей после дробления до величины 100 мкм гра-
нулируют в тарельчатом грануляторе при смачивании водой. Из фракции
с мелкими частицами гранулы получаются более плотные и прочные. Сфе-
рические гранулы размерами от 1 до 4 мм подвергают сушке и карбониза-
ции во вращающейся печи с внешним обогревом. Карбонизированные грану-
тивации:
1 — смеситель; 2 — формовочная машина; 3, 7 —сушнлки; 4, 8 — печи для активации; 5 —
экстрактор; 6 — аппарат для промывки гранул водой; 9 — классификатор; 10—тара; 11 —
конденсатор; 12 — абсорбер; 13 — электрофильтр.
/
/
268 11. Получение и номенклатура адсорбентов
лы активизируют смесью дымовых газов и водяного пара в псевдоожижен-
ном слое. Добавление при гранулировании в воду поверхностно-активного
вещества (ПАВ)—сульфитно-спиртовой барды способствовало увеличению
механической прочности «сырых» и карбонизированных гранул, что обеспе-
чивало уменьшение потерь при их транспортировании, сушке и охлаждении.
Концентрация ПАВ в растворе составляла 2,5%, расход его в расчете на
сухой уголь был равен 0,5%.
Установлено, что, применяя различный темп нагрева, можно
получить гранулы с суммарной пористостью от 0,18 до 1,3 см3/г.
При этом чем ниже темп нагрева, тем более плотные образуют-
ся гранулы. Особенно велико влияние темпа нагрева в темпера-
турной области, соответствующей переходу угля в пластичное
состояние.
Среди пор, образующихся на стадии карбонизации, преобла-
дают микропоры. Объем их невелик, размер очень мал и они
практически не доступны молекулам бензола.
Начальный период активации (степень обгара менее 20%)
характеризуется, в основном, раскрытием замкнутых пор. В этой
области объем микропор, замеренный по метанолу, существенно
превышает объем микропор, замеренный по бензолу. С увели-
чением обгара объем микропор, замеренный по этим веществам,
постепенно сближается и при 20%-ном обгаре становится прак-
тически одинаковым. Следовательно, в этот период средний эф-
фективный диаметр микропор соответствует приблизительно
0,6 нм.
Период активации, соответствующий 25—45%-ному обгару,
характеризуется равномерным развитием микро- и мезопор и
сопровождается постепенным увеличением предельного сорбци-
онного объема 1Го, структурной константы В [до (0,4—0,5) •
-10-6 при стандартном паре — бензоле] и соответственно раз-
мера микропор.
Третья область — с 45—50 до 70%-ного обжига отличается
появлением второй пористой структуры (структурная константа
В — около 8-10~6) и резким увеличением объема мезопор. При
обгаре выше 70% объем микропор существенно уменьшается и
прочность гранул падает.
При активации гранул в псевдоожиженном слое оптималь-
ными свойствами обладают образцы, полученные при 50%-ном
обгаре. Они имеют достаточно высокую прочность, объем мик-
ропор 0,4 см3/г, объем мезопор 0,09 см3/г.
Типы активных углей
По размеру и форме частиц активные угли подразделяют на
гранулированные и порошкообразные. Гранулированные угли
изготовляются обычно в форме цилиндриков диаметром 2—
5 мм, причем высота цилиндрика всегда больше диаметра. Гра-
нулированные угли применяют главным образом на установках
Активные угли
269
\
со стациойдрным слоем адсорбента при очистке и разделении
технологических потоков в газовой фазе. Чтобы увеличить ин-
тенсивность масообмена, гранулированный уголь иногда дро-
бят и после рассева получают узкие фракции, например: 0,15—
0,25; 0,25—0,55; 0,55—1,65; 1,65—2,35 и 2,35—4,70 мм. Дробле-
ные угли применяют во всех вариантах адсорбционных процес-
сов: при проведении процесса как в газовой, так и в жидкой
фазе, со стационарным, движущимся или псевдоожиженным
слоем адсорбента.
Порошкообразные угли состоят из частиц величиной менее
0,15 мм. Их используют исключительно для очистки веществ в
жидкой фазе. Некоторые усредненные физические свойства уг-
лей приведены ниже:
Плотность, г/см3
кажущаяся насыпная гранул
истинная
Теплоемкость сухого угля, кДж/(кг-К)
Теплопроводность (при 30 °C), Вт/(м-К)
0,6—0,9
1,9—2,2
0,84
0,17—0,28
В зависимости от назначения угли подразделяют на газовые,
рекуперационные и осветляющие* *. Каждый тип отличается ха-
рактерной структурой пор.
Газовые угли предназначены для улавливания относительно
плохо сорбирующихся компонентов (например, этилена) или
паров, присутствующих в газах в небольших концентрациях.
Кроме того, угли этого сорта могли быть применены и для
* В конце 60-х годов в Японии, а затем в других странах мира был освоен
синтез и налажен промышленный выпуск нового класса углеродных сорбен-
тов — молекулярно-ситовых активных углей. Их отличительной особенностью
является высокая однородность микропористой структуры, определяющие
размеры которой имеют тот же порядок, что и размеры молекул. В про-
мышленном масштабе выпускают угли с определяющим размером 0,4 и
0,5 нм, которые используют в разнообразных процессах разделения и очист-
ки сред. Имеются также сообщения о синтезе углей с размером микропор
до 0,7 нм.
Хотя фирмы-разработчики не сообщают условия получения молекулярно-си-
товых углеродных материалов, из косвенных данных все же можно заклю-
чить, что успех обеспечивается двумя факторами: 1) выбором исходного
сырья, которое, видимо, должно представлять собой однородный стереоре-
гулярный полимер и 2) малой степенью его активации. Более подробно
свойства молекулярно-ситовых углей рассмотрены в работах Астахова В. А.,
Лукина В. Д„ Машаровой Л. П. — ЖПХ, 1978, т. 51, № 8, с. 1733—1737;
Ивахнюка Г. К. и др. — В кн.: Получение, структура и свойства адсорбен-
тов, Л., «Химия», 1978, с. 12—22; Pratsch F., Juntgen И.— Verfahrenstechnik,
1978, v. 1978, № 6, s. 369—373.
Так как в последние годы стали появляться сообщения о синтезе силикаге-
лей с молекулярно-ситовыми свойствами, то ныне, можно сказать, все основ-
ные типы пористых адсорбентов (углеродные, силикатные и алюмосиликат-
ные) представлены своими молекулярно-ситовыми разновидностями. (Прим.
Рвд.)
270 11. Получение: и номенклатура адсорбентов /
/
очистки вод от примесей веществ с небольшим размером моле-
кул, в частности для дезодорации питьевой воды, извлечения
иода из буровых вод и т. д. Газовые угли должны обладать
большим объемом микропор, высокой крутизной изотермы (сле-
довательно, малой величиной структурной константы В) и уме-
ренно развитой мезопористостью, обеспечивающей достаточную
интенсивность диффузии адсорбата внутрь зерна адсорбента
(VMe не ниже 0,05 см3/г).
Представителями газовой серии в отечественной промышлен-
ности являются угли типа СКТ, полученные, как ясно из мар-
кировки, в ходе сернисто-калиевой активации. Они имеют сле-
дующие показатели: насыпная плотность 0,38—0,50 г/см3, сум-
марный объем пор Vs =0,75—1,10 см3/г, объем микропор Уми =
= 0,45—0,60 см3/г, объем мезопор 7ме = 0,10—0,28 см3/г, объем
макропор Ума = 0,16—0,25 см3/г, удельная поверхность мезопор
sMe = 60—180 м2/г, константа В=(0,65—1,00)-10-6 град-2.
Газовый уголь АГ-2 в отличие от угля СКТ имеет две мик-
ропористые структуры. Суммарный объем микропор (Уми=
= 0,30 см3/г) и суммарный объем всех пор (Vs = 0,60 см3/г) у
этого угля значительно ниже, мезопористость развита слабо
(Уме = 0,05 см3/г, sMe=33 м2/г), объем макропор (VMa = 0,25 см3/г)
соответствует этому показателю для угля СКТ. Насыпная плот-
ность равна 0,6 г/см3. Уголь АГ-2 (ГОСТ 23998—80) изготовля-
ют из каменноугольной пыли и смолы грануляцией с последую-
щей парогазовой активацией. Преимущественный размер гранул
угля 1,5—2,8 мм. В основном он предназначен для снаряжения
индивидуальных средств защиты органов дыхания, а также для
изготовления на его основе поглотителей и катализаторов.
Аналогичен по своим свойствам и технологии получения
уголь АГ-3, широко используемый для извлечения компонентов
из газовых и жидких сред (ГОСТ 20464—75). Его насыпная
плотность 0,40—0,55 г/см3. Адсорбционные свойства углей АГ-2
и АГ-3 регламентируются по времени задерживающего дейст-
вия слоя адсорбента высотой 5 см по бензолу, извлекаемому из
газового потока в стандартных условиях (ГОСТ 17218—71).
Этот показатель для угля АГ-2 должен быть не менее 45—
55 мин, для угля АГ-3—40 мин.
Уголь КАД-иодный (ТУ 16-1917—74) предназначен для по-
глощения иода из буровых минерализованных вод и для извле-
чения различных веществ из растворов. Он может использовать-
ся и для очистки газов. Уголь представляет собой зерна разме-
ром от 1 до 5 мм, полученные из каменноугольного полукокса.
По микропористой структуре он близок к углям типа АГ (VMH =
= 0,30 см3/г), но имеет значительно более развитую мезо-
(VMe = 0,15 см3/г, sMe = 110 м2/г) и макропористость (VMa =
=0,51 см3/г). Его суммарный объем пор Vs = l,00 см3/г, а
насыпная плотность 0,38 г/см3.
Активные угли
271
Рекуперационные угли. Самые мелкие поры газовых углей
доступны Для паров веществ с молекулярной массой до 150.
Поэтому с позиции собственно адсорбции их возможно исполь-
зовать и для\поглощения хорошо адсорбирующихся паров. Но
в технологических процессах определяющее значение для выбо-
ра типа угля часто приобретает стадия десорбции. Поэтому для
улавливания паров органических растворителей из воздуха и
для решения ряда аналогичных задач применяют рекуперацион-
ные угли типов АР и APT. Уголь АР, например, имеет две мик-
ропористые структуры и следующие показатели: Ух = 0,70, Уми =
= 0,33, Уме = 0,07, Ума = 0,30 см3/г; sMe = 50 м2/г; IF0i = 0,19 см3/г;
Го2 = О,18 см3/г; В1 = 0,74-10-® град-2; В2=3,42-10-6 град-2.
Современные методы промышленной газовой активации угле-
родных адсорбентов не обеспечивают однородную структуру по
толщине гранул: крупные гранулы подвергаются меньшей сте-
пени обгара в своей сердцевине. Определенную позитивную роль
при получении однородных по структуре фракций углеродных
адсорбентов может играть метод фракционирования гранул в
потоке воздуха [53]. Ниже приведены данные такого фракцио-
нирования для угля АР:
Фракция 1 2 3 4
Скорость потока, м/с 5,2 5,2 4,8 4,3
Содержание фракции, % 31,0 30,7 21,1 17,2
Насыпная плотность, г/л 528 485 405 300
Суммарная пористость, см3/г 0,60 0,78 0,97 1,33
Средний объем гранулы, мм3 7,0 6,7 5,5 5,0
Активность (равновесная) по 2,38 2,96 4,18 4,80
бензолу, ммоль/г
Объем пор, см3/г
Ими 0,211 0,252 0,372 0,393
Име 0,021 0,024 0,028 0,033
Ими 0,389 0,508 0,572 0,901
Константа уравнения
Дубинина — Радушкевича
IV'o, см3/г 0,19 0,25 0,40 0,43
В- 10е, град-2 0,88 1,10 1,50 1,70
Из представленных данных следует, что адсорбционная ем-
кость по бензолу крайних фракций различается в два раза, объ-
ем микропор в 1,85 раза, макропор в 2,3 раза.
Осветляющие угли (тип ОС) предназначены для поглощения
относительно крупных молекул или микросуспензий из жидких
сред. Они отличаются развитой мезопористостью: удельная по-
верхность мезопор составляет в среднем 150 м2/г. Осветляющие
угли получают обработкой древесного угля-сырца водяным па-
ром при температуре выше 800 °C. После активации уголь под-
вергают измельчению в порошок с размером частиц не менее
0,1 мм. Активные угли этого типа применяют для очистки пище-
272
11. Получение и номенклатура адсорбентов
вых, фармацевтических и других продуктов, а также различных
растворов. /
По действующему в Советском Союзе ГОСТ 4453—74 в за-
висимости от назначения активные осветляющие Древесные по-
рошкообразные угли изготавливают четырех видов: ОУ-А — су-
хой щелочной для очистки сиропов в сахарорафинадной про-
мышленности, воды и растворов в производствах органических
кислот, масел и жиров; ОУ-Б — влажный кислый для очистки
медицинских препаратов, растворов в крахмалопаточных произ-
водствах и на гидролизных заводах; ОУ-В — сухой щелочной
для очистки и осветления различных растворов в отраслях пи-
щевой промышленности; ОУ-Г— сухой щелочной для очистки от
высокомолекулярных смолистых и окрашивающих примесей в
органическом синтезе.
Особое внимание обращают на зольность осветляющих уг-
лей и на состав золы. Так, общее содержание соединений же-
леза (в пересчете на Fe) в угле, применяемом для фармацевти-
ческих целей, не превышает 0,05, а в других случаях 0,2%; во-
дорастворимые соединения железа в углях ОС полностью от-
сутствуют. Общая зольность — менее 6—10%, а доля водорас-
творимой золы 1—2%. Адсорбционные свойства углей ОС опре-
деляют, приводя в контакт навеску порошка с водными раство-
рами метиленового голубого и мелассы; поглотительная способ-
ность в первом случае не ниже 210—225 мг/г, во втором — 50—
100%. Массовая доля влаги в товарных углях типа А, В и Г
регламентирована: не выше 10%.
Часто осветляющие порошкообразные угли получают путем
размола не только активных дробленых и гранулированных уг-
лей, но также и отходов из них. Активный уголь КАД-молотый
(ТУ 6-16-1888—74) широко применяют, в частности, в цветной
металлургии при флотации руд полезных ископаемых, а также в
других отраслях промышленности для осветления растворов.
Порошкообразные угли не обладают вредными для организ-
ма человека свойствами, но, как всякая пыль, могут вызвать
легочные заболевания. Предельно допустимая концентрация пы-
ли активного угля в воздухе рабочих помещений 10 мг/м3. Пыль
активного угля взрывоопасна при ее концентрации в воздухе
от 114 до 400 г/м3. Температура самовоспламенения осевшей
угольной пыли в скоплении около 260 °C. При работе с порошко-
образным углем следует пользоваться противопылевым респира-
тором. Места пересыпания активного угля должны быть обору-
дованы в соответствии с нормами противопожарной безопасно-
сти (наличие вытяжной вентиляции, отсутствие открытых ис-
точников огня и т. д.).
Иногда осветляющие угли применяют в гранулированном
или дробленом виде. Основной размер гранул угля АГС-4, ис-
пользуемого для обесцвечивания сахарных сиропов, составляет
Силикагели
273
2,0—3,5 мм. Молотый древесный уголь МД используют для ре-
шения многочисленных задач обесцвечивания и осветления рас-
творов. \
Силикагели
Получение силикагелей
Известны различные методы получения силикагелей: осажде-
нием аморфного кремнезема из силикатов щелочных металлов
минеральными кислотами; смешением силикатов щелочных ме-
таллов с легко гидролизующимися солями; гидролизом галоге-
новых соединений кремния; термическим разложением кремний-
органических соединений или тетрахлорида кремния. Однако все
промышленное производство силикагелей в мире основано на
первом из этих методов.
Кремнийсодержащим сырьем для получения силикагеля яв-
ляется твердый силикат натрия (силикат-глыба), а в качестве
минеральной кислоты используют, как правило, серную кислоту:
Na2Q.3SiO2 + H.2SO4 = 3SiO2 + Н2О 4- N^SO* (11.7)
силикат-глыба аморфный кремнезем
Промышленное производство силикагелей осуществляется по
двум принципиально различным направлениям: производство
кускового и производство гранулированного (шарикового) си-
ликагеля. В производстве кускового силикагеля гелеобразую-
щие растворы сравнительно низких концентраций сливают в та-
Рис. 11,2. Схема получения гранулирован-
ных силикагелей:
1 — склад силикат-глыбы; 2 — дробилка;
3 — автоклав для разварки силикат-глыбы;
4 — емкость для жидкого стекла; 5 — ем-
кость для концентрированной серной кис-
лоты; 6 — емкость для раствора серной
кислоты; 7 — склад тригидрата оксида
алюминия; 8 — реактор для получения
сульфата алюминия; 9 — емкость для рас-
твора сульфата алюминия; 10 — насосы;
11 — ротаметры; 12 — смеситель; 13 — фор-
мовочный конус; 14—формовочная колон-
на; 15 — колонна для мокрой обработки;
16 — сборник масла; 17 — сборник сииере-
висиой жидкости; 18 — сборник для акти-
вирующего раствора; 19 —сборник для
промывной воды; 20 —сборник для транс-
портной воды; 21 — сепаратор; 22 — су-
шильный агрегат; 23 — классификатор;
24 — узел затаривания силикагеля.
18—1346
274
11. Получение, и номенклатура адсорбентов
ких отношениях, чтобы полученный золь имел кислую/реакцию и
не застудневал в короткое время. Такой золь тщательно гомоге-
низируют и оставляют в покое до образования геля. Гедь раз-
резают на куски и в кусках промывают его, а затем сушат, раз-
малывают и рассевают на нужные фракции.
Широкое распространение получили гранулированные сили-
кагели. Принципиальная технологическая схема производства
этого продукта показана на рис. 11,2.
Силикатную глыбу из склада 1 подают на измельчение в щековые дро-
билки 2, а затем в автоклаве 3 растворяют в горячей воде под избыточным
давлением 0,4—0,6 МПа при 130—140 °C. Концентрированный раствор сили-
ката натрия (жидкого стекла) разводят в емкости 4 до заданной концентра-
ции. Аморфный кремнезем из жидкого стекла осаждают серной кислотой
или подкисленным раствором сульфата алюминия. При производстве грану-
лированного силикагеля КСМ в гель в качестве упрочающей добавки вводят
4—10% А13О3. Для приготовления кислого раствора серную кислоту из ре-
зервуара 5 подают в емкость 6, где разбавляют до заданной концентрации,
или в реактор 8 для растворения тригидроксида алюминия, который дозиру-
ют в реактор 8 из бункера 7.
Концентрированный раствор сульфата алюминия разбавляют в емкости
9 до заданной концентрации. Жидкое стекло и серную кислоту или сульфат
алюминия (так называемые рабочие растворы) насосами 10 через ротамет-
ры 11 точно дозируют в смеситель 12, в котором образуется золь кремневой
кислоты. Длительность жизни золя до коагуляции составляет 4—10 с, что
достигается подбором условий его получения (концентрация рабочих рас-
творов, их температура и соотношение). Струю золя из смесителя направля-
ют на формовочный конус с желобками 13, изготовляемый обычно из орга-
нического стекла или фторпласта. Тонкие струйки золя с конуса 13 падают
на поверхность масла в формовочной колонне 14, где они разбиваются на
капли. Капли золя коагулируют в слое масла и под действием поверхност-
ного натяжения принимают форму, близкую к сферической.
Образовавшийся шариковый гидрогель выносится из формовочной ко-
лонны потоком транспортной жидкости, подаваемой из емкости 20. Гель по
наклонному перфорированному лотку транспортируют в колонну для так на-
зываемых мокрых обработок 15, а отделяющуюся на лотке транспортную
жидкость возвращают в емкость 20.
В колонне 15 протекают следующие стадии процесса: созревание (сине-
резис), активация и промывка. При созревании через гидрогель из емкости
17 прокачивают сииерезисиый (маточный) раствор, который представляет со-
бой 5—6%-ный раствор сульфата натрия. Длительность этой операции, а
также температура и pH циркулирующего раствора строго регламентируют-
ся. Затем шариковый гидрогель «активируют» — обрабатывают его разбав-
ленным раствором серной кислоты с целью удаления катионов металлов, в
первую очередь натрия. Активирующий раствор готовят в емкости 18, отту-
да насосом прокачивают через гель в колонне 15. Завершающей операцией
«мокрых обработок» является промывка геля водой для удаления сульфата
натрия и серной кислоты.
Операции «мокрых обработок» осуществляются по полунепрерывной схе-
ме, для чего на установке используют одновременно несколько колонн, чаше
всего 10—12. Примерное распределение колонн: формовка геля—1; синере-
зис— 4; активация — 2; промывка — 3; выгрузка геля—1; резерв — 1.
Через строго заданное время (длительность цикла) потоки в колоннах
переключают; обычно это время составляет 4—6 ч. Прошедший «мокрые об-
работки» шариковый гель выгружают нз колонны 15 транспортной водой, в
сепараторе 21 эта вода отделяется от шарикового геля, который поступает
иа сушку. Для сушки используют ленточные или шахтные сушилки 22, про-
Силикагели
275
цесс проводят при 130—-170 °C в атмосфере влажного воздуха. Сухой сили-
кагель рассеивают на фракции (23) и затаривают в бидоны или крафт-меш-
ки (24). х
По описанной выше технологии получают шариковый сили-
кагель с зернами размером 1,0—7,5 мм. Продукты такого фрак-
ционного состава удовлетворяют требованиям процессов со ста-
ционарным слоем адсорбента. Регулируя фракционный состав
получают более узкие фракции шарикового силикагеля (напри-
мер, 1,0—2,5; 2,0—4,0 или 3,0—5,0 мм). Это обеспечивается пу-
тем изменения числа желобков на конусе, высоты его располо-
жения над формовочной колонной или вязкости масла в ко-
лонне.
При некотором дооборудовании можно получать силикагель
с частицами размером 50—300 мкм для процессов с кипящим
слоем. Для этого отмытый гидрогель подают на растирочные
машины, а получаемую суспензию подвергают распылительной
сушке, в процессе которой образуются твердые частицы сфери-
ческой формы. Конструкции современных распылительных фор-
сунок (центробежных или пневматических) позволяют регули-
ровать фракционный состав порошкообразного продукта в до-
статочно узких пределах. Следует отметить, что при распыли-
тельной сушке не удается получить частицы крупнее 300 мкм
удовлетворительной механической прочности. Фракцию шарико-
вого силикагеля 0,3—1,0 мм пока в промышленном масштабе
не получают: до сих пор для этой цели не разработана техноло-
гия и аппаратура, удовлетворяющие технико-экономическим
требованиям.
Описанный выше технологический процесс получения грану-
лированных силикагелей позволяет в довольно широких преде-
лах регулировать их пористость [54].
Косвенной характеристикой структуры силикагелей является
насыпная плотность: у крупнопористых силикагелей она равна
0,4—0,5, у мелкопористых 0,7—0,8 г/см3. Для мелкопористых си-
ликагелей кажущаяся плотность составляет 1,1 г/см3, истинная
плотность 2,25 г/см3. Мелкопористая структура механически бо-
лее прочная, чем крупнопористая. Показатель прочности, опре-
деленный по устойчивости к.истиранию во вращающемся бара-
бане со стальными мелющими телами (см. ниже), для крупно-
пористых силикагелей находится в пределах 60—85%, а для
мелкопористых 85—95%.
В промышленной практике регулирование структуры осуще-
ствляется двумя основными методами: изменением условий со-
зревания гидрогеля (увеличение общего объема и размера пор
силикагелей происходит при повышении температуры, длитель-
ности синерезиса и pH среды) и изменением условий промывки
(при повышении температуры промывочной воды и увеличении
ее жесткости, т. е. содержания в ней солей щелочноземельных
18*
276
11. Получение, и номенклатура адсорбентов
металлов, растет общий объем и размер пор силикагеля).
Структурные характеристики силикагелей обычно Изменяют в
следующих пределах:
Объем пор, см3/г 0,3—1,2
Удельная поверхность, м2/г 300—750
Средний радиус пор, нм 1,0—7,0
Насыпная плотность, г/см3 0,4—0,9
Дополнительные возможности регулирования пористой
структуры представляются при замещении воды в отмытом гид-
рогеле различными органическими веществами и при гидро-
термальной обработке силикагелей.
Разновидностью крупнопористых структур является аэро-
сил, широко применяющийся, в частности, в качестве изоляци-
онного материала. Его получают прокаливанием кремнийорга-
нических соединений в кислородсодержащей среде, в результа-
те чего органические вещества выгорают. Аэросил состоит из
непористых сферических частиц дегидротированного кремнезе-
ма размером до 15 нм с очень рыхлой упаковкой. Свободный
объем между глобулами в 20—40 раз превышает собственный
объем частиц. Общий объем пор аэросила достигает 2,0—
2,5 см3/г, а насыпная плотность составляет всего 0,08—0,10см3/г.
Гидротермальной обработке в автоклавах при избыточных
давлениях водяного пара до 30 МПа могут подвергаться как
гидрогели, так и сухие силикагели. Такая обработка позволяет
получать силикагели с удельной поверхностью до 5 м2/г и сред-
ним радиусом пор несколько сотен нанометров. Полученные
таким образом силикагели эффективно используют в качестве
адсорбентов для хроматографического разделения, а также
носителей катализаторов. Аналогичного эффекта можно добить-
ся, прокаливая силикагели в среде водяного пара при темпера-
турах выше 650 °C.
В последние годы наблюдаются некоторые новые тенденции
в разработке технологии производства -силикагелей: получение
чистых силикагелей на основе золя кремневой кислоты; полу-
чение бидисперсных формованных силикагелей; разработка спо-
собов, позволяющих изготовлять силикагели без сброса солей в
сточные воды или обеспечивающих их эффективную утилиза-
цию; расширение ассортимента промышленных силикагелей по
характеристикам пористой структуры; разработка технологии
производства шариковых водостойких силикагелей.
Силикагели разрушаются под действием капельной влаги.
Существуют методы получения водостойких сортов силикагелей
[55, 56]. Однако водостойкие силикагели обладают пониженной
влагоемкостью, а технология их изготовления сложнее, поэтому
только обычные силикагели выпускаются промышленностью в
Силикагели
277
крупнотоннажном масштабе. Чтобы предотвратить разрушение
силикагелей при эксплуатации в тех случаях, когда возможно
проникание в адсорбер капельной влаги, в небольшом защит-
ном слое используют другие водостойкие типы промышленных
адсорбентов, например активный оксид алюминия.
Типы промышленных силикагелей
В настоящее время в СССР производятся следующие силика-
гели: кусковые и гранулированные силикагели (ГОСТ 3956—76);
силикагель — индикатор (ГОСТ 8984—58); набор силикагелей
для исследовательских работ и хроматографии по условиям, со-
гласованным с Советом по хроматографии АН СССР и «Союз-
главреактивом». В недавнем прошлом изготовлялся также сили-
кагель для бытовых холодильников (ТУ 4267—54), однако он
повсеместно заменяется шариковым цеолитом типа NaA-2MIII.
Силикагели по ГОСТ 3956—76 представляют собой твердые
стекловидные прозрачные или матовые зерна насыпной плотно-
стью в пределах 0,4—0,8 г/см3. В зависимости от характера по-
ристой структуры кусковые и гранулированные силикагели де-
лятся на крупно- и мелкопористые. Первые из них характери-
зуются средним радиусом пор 5 нм, а вторые—1 —1,5 нм. Про-
межуточную структуру (в ГОСТ не входит) составляют средне-
пористые силикагели. В зависимости от размера зерна мелко-
пористый вид кускового и крупнопористый вид гранулирован-
ного силикагеля подразделяют на 4 марки (2,8—7,0; 1,5—3,6;
0,25—2,0 и 0,2—0,5 мм), а мелкопористый вид гранулированно-
го силикагеля на две марки (2,7—7,0 и 1,0—3,6 мм). Гранули-
рованные мелкопористые силикагели в качестве упрочняющей
добавки содержат 4—10% оксида алюминия.
В зависимости от гранулометрического состава, формы ча-
стиц и характера пористости силикагели обозначают четырьмя
буквами: первая буква характеризует размер гранул, вторая
(всегда С) —силикагель, третья — размер пор, последняя — фор-
му частиц*. Так, крупный силикагель мелкопористый гранулиро-
ванный обозначают КСМГ, мелкий силикагель мелкопористый
кусковой — МСМК. Средние фракции силикагелей называют
«шихтой» и обозначают: ШСМК, ШСКГ или ШСМГ. Индика-
торный силикагель представляет собой мелкопористый силика-
гель типа ШСМГ, пропитанный солями кобальта. В зависимости
от влажности среды он изменяет цвет от светло-голубого до
розового.
Набор шариковых силикагелей разной пористости состоит из
восьми марок силикагелей с зернами размером от 1,5 до 5 мм,
* Последнюю букву часто опускают, не указе вал, таким образом, форму
гранул. (Прим, ред.)
278
11. Получение и номенклатура адсорбентов
Таблица 11-1. Набор шариковых силикагелей для исследований
по адсорбции, хроматографии и катализу
Тип силикагеля Марка (номер) силикагеля Насыпная плотность, г/см3 Характеристика пористой структуры
суммарный удельная средний объем пор, поверх- радиус см8/г ность, м2/г пор, нм
Крупнопористый 1 0,42 1,10 300 8,0—10 2 0,38—0,45 1,05—1,25 300—350 6,0—7,5 2,5 0,44—0,49 0,90—1,10 350—450 4,5—5,8 Среднепористый 3 0,50—0,55 0,76—0,85 500—600 2,6—3,5 4 0,56—0,65 0,61—0,75 550—650 1,8—2,5 Мелкопористый 5 0,65—0,75 0,46—0,60 580—680 1,5—2,0 6п 0,8 0,25—0,45 400—550 1,0—1,4 6с 0,8 0,25—0,38 600—750 0,8-1,2
различающихся величиной среднего радиуса пор в пределах от
1 до 10 нм (табл. 11-1). При использовании этого набора ис-
следователь должен иметь в виду, что все силикагели, кроме
№ 6с, в процессе изготовления прокаливали при 550—600 °C, в
результате чего их гидрофильность понижена. Поэтому прово-
дить на них работы по осушке газов и жидкостей нецелесообраз-
но. Для этих целей должен использоваться силикагель КСМ
№ 6с.
Теплоемкость силикагелей составляет 0,92 кДж/(кг-К),теп-
лопроводность при 30°C — 0,71 кДж/(м-ч-К). Средняя теплота
адсорбции паров воды на силикагелях равна приблизительно
2600 кДж/кг воды.
Приближенное определение структурных
характеристик силикагеля
Удельная поверхность. Для массовых определений удельной
поверхности силикагелей используют избирательность поглоще-
ния ими углеводородов определенного класса. Оценку удельной
поверхности производят по изменению концентрации толуола
в его смеси с изооктаном в результате контакта с навеской ад-
сорбента. Измерения производят с помощью рефрактометра.
Изотерма адсорбции толуола в изооктане проходит через мак-
симум при концентрации 40%. Благодаря резко выраженной
избирательности адсорбции ароматических углеводородов в
точке максимума в адсорбированной фазе находится только
толуол, причем его молекулы образуют на поверхности плот-
ный монослой. Количество адсорбированного толуола (в моль/г)
составляет
* V («2оисх л20кои) Рт ,,,
ат~ g-100K(l -с)М
Силикагели
279
где V.— объем исходного раствора, приливаемого к навеске адсорбента, мл;
л20исх и п20Кон — коэффициенты рефракции растворов до и после адсорбции;
g — навеска адсорбента, г; рт — плотность толуола при температуре опыта
(обычно 20°С), г/мл; /( — изменение коэффициента рефракции при измене-
нии концентрации толуола в растворе на 1% (об.), оно равно 0,00154; с —
объемная доля толуола в растворе, равная 0,4; М — молекулярная масса то-
луола.
Удельная поверхность (в м2/г) определяется из известной
формулы
5 = 10-« (11.9)
Принимая (i)m = 0,53 нм2 и подставляя в формулы (11.8) и
(11.9) другие константы рассматриваемой системы, в конечном
итоге получим
s = 47 400 (lz/g) (/г20исх —/г20КОц) (11.10)
Методика определения. Силикагель измельчают, отбирают фрак-
цию 0,25—0,5 мм и прокаливают ее в муфеле при 200 °C в течение 2 ч. За-
тем навеску адсорбента около 1 г (взвешивание производят с точностью
0,01 г) помещают в герметичную ампулу или пробирку с пришлифованной
пробкой и приливают из микробюретки точно 2 мл смеси 40% толуола и
€0% изооктана; показатель преломления исходной смеси должен быть в пре-
делах 1,4328—1,4330. Адсорбционное равновесие устанавливается через 3 ч.
Затем определяют коэффициент преломления раствора, особое внимание уде-
ляя точности термостатироваиия призм рефрактометра (20°C). Точное коли-
чество адсорбента и раствора устанавливают путем последовательного взве-
шивания пустой ампулы, ампулы с адсорбентом и раствором. Обычно с од-
ним образцом проводят параллельно 2—3 измерения.
Суммарный объем пор. Силикагели в основном относятся к
адсорбентам с развитым объемом мезопор. Мезопоры в обла-
сти высоких относительных давлений в результате капиллярной
конденсации объемно заполняются адсорбатом. Поэтому сум-
марный объем пор может быть определен эксикаторным мето-
дом по адсорбции паров бензола при относительном давлении,
близком к единице. Для того чтобы несколько понизить давле-
ние паров бензола и избежать возможности его конденсации на
внешней поверхности адсорбента, к бензолу добавляют немного
н-бутилового спирта, для которого характерно малое давление
насыщенного пара. Суммарный объем (в см3/г) пор определяют
по привесу адсорбента после установления адсорбционного рав-
новесия:
Ух = Д§/£Рб (Н.П)
где Ag— привес адсорбента за счет поглощения бензола, г; g — навеска ад-
сорбента, г; ре — плотность бензола при 20 °C, равная 0,876 г/мл.
Метод определения. В бюкс с крышкой вводят 0,2—0,5 г сухого
адсорбента (фракция 0,25—0,50 мм). Пустой бюкс и бюкс с навеской взве-
шивают на аналитических весах с точностью до 0,0001 г. Затем крышка с
бюкса снимается, и его помещают на фарфоровую подставку эксикатора,
нижняя часть которого заполнена бензолом с добавкой около 0,5% (об.)
280 11. Получение и номенклатура адсорбентов
спирта. Бюкс взвешивают через каждые 8—24 ч, пока масса не достигнет
постоянного значения. После этого по формуле рассчитывают суммарный
объем пор.
Средний радиус пор (в нм) определяют на основе двух по-
казателей адсорбента: суммарного объема пор Vs (в см3/г) и
удельной поверхности s (в м2/г):
Например, если суммарный объем пор Vs =0,45 см3/г, а
удельная поверхность s = 300 м2/г, средний радиус пор
2-103.0,45
гср — 300 ~ 3 нм
Активный оксид алюминия
Получение активного оксида алюминия
Все технологические схемы производства активного оксида алю-
миния, реализованные в настоящее время отечественной про-
мышленностью, основаны на получении гидроксида алюминия
(«гидрата глинозема»). Схема такого процесса включает рас-
творение «гидрата глинозема» в кислотах (H2SO4, HNO3) или
в щелочи (NaOH) с последующим гидролизом при нейтрализа-
ции соответственно основанием или кислотой. Процесс пере-
осаждения гидроксида алюминия связан с большими затрата-
ми кислот или оснований (2—4 т/т оксида алюминия), кото-
рые практически не регенерируются. Исключение составляет
нитратная технология (растворение «гидрата глинозема» в
азотной кислоте и нейтрализация аммиаком), которая в слу-
чае привязки к заводу, имеющему производство аммиачной се-
литры, позволяет практически полностью использовать затра-
ченные компоненты.
В производстве адсорбентов ряда типов целесообразно ис-
пользовать схемы, основанные на формировании смесей пере-
осажденного гидрооксида алюминия (или других связующих)
и «гидрата глинозема», что существенно удешевит адсорбенты
без ухудшения их качества. Это направление должно стать ос-
новным в производстве рядовых сортов осушителей.
Иногда оксид алюминия готовят в шариковой форме, и тогда техноло-
гия его приготовления напоминает технологию приготовления шариковых
силикагелей. В качестве исходного сырья применяют гидрооксид алюминия,
который отмывают от примесей. После добавки к сырью 0,15—0,20 моль со-
ляной кислоты на 1 моль АЬО.-, образуется однородная пластичная и по-
движная пульпа. Формирование шариков происходит после прохождения
пульпы через фильеры и свободного падения в слое керосина. Затем обра-
зовавшиеся капли проходят через слой аммиачного раствора, где происходит
Активный оксид алюминия
281
нейтрализация основной соли алюминия и закрепление полимерной структу-
ры адсорбента. В шарике остается хлорид алюминия (0,2 моль на 1 моль
А12О3). Его удаляют при прокаливании шариков.
В качестве примесей шариковый оксид алюминия, изготав-
ливаемый в Советском Союзе, Японии и Франции содержит 0,1—
0,6% Na2O; 0,1—1,0% SiO2; 0,1—0,4% Fe2O3. Средний диаметр
частиц 3—4 мм, насыпная плотность 0,6—0,9 г/см3, суммарный
объем пор 0,4—0,7 см3/г, средний радиус пор 3—4 нм.
Стадии формирования гранул, сушки и прокаливания явля-
ются общими для любых схем производства активного оксида
алюминия. Технология и аппаратурное оформление этих стадий
процесса нуждаются в коренном усовершенствовании.
Одним из возможных решений комплекса задач по созданию
аппаратурно-технологического оформления этих стадий процес-
са является разработанная технология производства гаммы сор-
тов активного оксида алюминия «Сфераль». Единая технологи-
ческая линия включает непрерывно действующие узлы жидкост-
ного формования, безградиентной инфракрасной сушки, интен-
сивного прокаливания и аэродинамической сепарации сфериче-
ских гранул. Схема рассчитана на выпуск широкого ряда сор-
тов продукта, различающегося формой и размером гранул (от
0,5 до 4 мм) и пористой структурой. Регулирование пористой
структуры (удельной поверхности, суммарного объема и рас-
пределения пор по эффективным радиусам) обеспечивается вве-
дением в жидкотекучие формуемые массы добавок порошков
гидрооксидов алюминия или малых добавок органических ве-
ществ (спиртов, кислот, аминов, полимеров и т. д.).
Промышленный ассортимент
Химический состав активного оксида алюминия — адсорбента
обычно жестко не регламентируется. Такие примеси, как оксид
кремния, не изменяют характеристик продукта, даже если их
содержание составляет несколько процентов. В то же время при-
сутствие железа ухудшает товарный вид продукта, и потому его
содержание обычно невелико (менее 0,2%). Существенное зна-
чение имеет содержание солей натрия: присутствие щелочи по-
нижает термическую стабильность адсорбентов и кислотность
их поверхности (последнее отражается на качестве адсорбента,
применяемого при осушке воздуха и очистке его от непредель-
ных углеводородов в производстве жидкого кислорода).
В мировой практике отмечается тенденция к переходу на
сферические гранулы активного оксида алюминия. Рекомендуе-
мый размер гранул адсорбента непосредственно связан с линей-
ной скоростью потока в аппарате. Если адсорбент работает при
высоком давлении и небольших линейных скоростях, изменение
диаметра гранул не оказывает существенного влияния на поте-
282
11. Получение и номенклатура адсорбентов
Таблица 11-2. Некоторые характеристики активного оксида алюминия,
в СССР (гранулы цилиндрической формы)
Марка ГОСТ (ТУ) Размер гранул (d\ 1), мм Насыпная плотность, г/л Удельная поверх- ность, М2/Г Суммарный объем* пор, см3/г Преобла- дающий радиус пор*, нм
А-1 ГОСТ 8136—56 4—6; 400—550 180—220 0,8—1,0 7,0 и 300 5—25 А-2 ГОСТ 8136—56 5—25 550—750 — — — А-15 ГОСТ 8136—56 2,6—3,0; 600±50 200 0,7 6,0—10,0 3—7 А-64 ГОСТ 8136—56 1,5—2,0; 600±50 200 0,7 6,0—10,0 3—7 А ТУ 38-101-190—71 4,5; 3,5 610—630 200—170 — — В ТУ 38-101-190—71 2,8; 3,5 610—630 200—170 — — АС-64 ВТУ 1-42—66 4—5; 600—700 200 0,6 — 4—7
* Показатели, характеризующие пористую структуру в действующих ГОСТ (ТУ)
измерений ряда образцов.
рю напора; в этом случае обычно применяют гранулы диамет-
ром 2—4 мм. Для систем, работающих при невысоком давлении
и значительных линейных скоростях, рекомендуются гранулы
диаметром 6 мм и более.
Для глубокой осушки технологических потоков обычно при-
меняют мелкопористый оксид алюминия с удельной поверхно-
стью 350—400 м2/г. В этих системах активность адсорбента про-
порциональна удельной поверхности. Количества мезопор
(0,1 см3/г) вполне достаточно, чтобы обеспечить интенсивный
транспорт адсорбата внутрь гранул оксида алюминия. Наличие
крупных пор (радиус десятки и сотни нанометров) весьма по-
лезно в адсорбентах, используемых для очистки и осушки ма-
сел. В статических системах адсорбции оптимальный радиус
пор выбирается в соответствии с заданной равновесной относи-
тельной концентрацией адсорбтива в окружающей среде. Поми-
мо соображений эффективности работы адсорбента, приходит-
ся учитывать и такой фактор, как относительно низкая устой-
чивость мелкопористого оксида алюминия к капельной влаге.
Увеличение водостойкости оксида достигается обычно за счет
создания бидисперсной пористой структуры, в которой представ-
лены крупные поры размером не менее 50 нм.
Некоторые характеристики отечественных типов оксида алю-
миния приведены в табл. 11-2. Как видно из таблицы, все вы-
пускаемые промышленностью сорта представляют собой грану-
лы цилиндрической формы с удельной поверхностью около
200 м2/г. Основным типом адсорбента является оксид алюми-
ния типа А-1, хотя из-за небольшой удельной поверхности при-
Синтетические цеолиты
283
выпускаемого
Содержание плимесей, %,
не болк'е
FejOa NajO SiOa
о,1 — —
0,1 —
0,02 0,03 —
0,02 0,02
0,15—0,20 0,1 1,0—1,5
0,35—0,45 0,3—0,4 0,03—0,15
0,02 0,1 1,0
ее нормированы. Приводятся результаты
менение такого адсорбента для
осушки технологических потоков
недостаточно эффективно.
В табл. 11-3 приводятся дан-
ные о ряде сортов оксида алю-
миния, известных на мировом
рынке.
При использовании оксида
алюминия в качестве осушителя
следует иметь в виду его ката-
литическую активность. Так, при
дегидратации спиртов, в частно-
сти метанола, с помощью а-А120з,
где алюминий находится в ок-
таэдрической координации, в
стадии термодесорбции происхо-
дит разложение спирта [57]. Ос-
новными продуктами при этом
_____________________________ являются оксид углерода и водо-
род. В малых количествах обра-
зуется метан, диметиловый эфир, вода, формальдегид и этилен.
Обнаружены также следы непредельных углеводородов С3—С5.
Синтетические цеолиты
Получение гранулированных цеолитов со связующим
Принципиальная схема получения гранулированных цеолитов со
связующим представлена на рис. 11.3. Сырьем для получения
натриевой формы цеолитов являются силикат-глыба, гидрооксид
алюминия и гидрооксид натрия (едкий натр). Гидрооксид алю-
миния растворяют в кипящем растворе едкого натра, разбав-
ляют и таким образом получают рабочий раствор алюмината
натрия. Силикат-глыбу разваривают острым паром, после чего
раствор жидкого стекла разбавляют водой для получения тре-
буемой концентрации.
Смесь растворов силиката и алюмината натрия поступает в смеситель 1,
где при интенсивном перемешивании образуется гидрогель, который направ-
ляют в кристаллизатор 2. При получении цеолита типа А температура в
кристаллизаторе составляет 80—90 °C, а время пребывания 6 ч; при полу-
чении цеолита типа X соответствующие показатели равны 95—100 °C и 12 ч.
Нагрев геля в кристаллизаторе ведут глухим или острым паром. Последний
применяют при большой производительности кристаллизатора.
Образовавшиеся кристаллы отделяют от маточного раствора в фильтр-
прессе 3, где одновременно происходит отмывка избытка щелочи водой. Ма-
точный раствор иногда повторно используют для получения рабочих раство-
ров. Кристаллы могут быть выделены из растворов также центрифугирова-
Таблица 11-3. Характеристика ряда марок активного оксида алюминия,
Фирма Марка Форма и размер гранул, мм Насыпная плотность, г/л Удельная поверх- ность, м2/г Суммарный объем пор, смЗ/г
Гудрн Хюлле, ФРГ Н0401 Н0415 Цилиндры: 1,6; 2,4; 3,2; 4,0; 2,8 То же 900 800 200 160 0,42 0,45
Н0416 » » 880 300 0,44
Н0417 » » 830 300 0,47
Н0403 Цилиндры: 2,0—4,0; 3,7—7,0 816—840 300—350 0,5-0,65
БАСФ, ФРГ А Цилиндры, кольца, шарики: 3—10,1—20 500-800 200—300 0,4—1,0
Пешине- Сент- Гобэн, Франция GCS-300 GDS-300 Цилиндры: 1,2—2,0 То же 700 700 300 300 0,55 0,55
SCM-250 Шарики: 2—6 650 250 0,67
Харшоу, США А1-0104Т Таблетки: 3,2 815 90 0,28— 0,33
А1-1404Т То же 815 190 0,42— 0,51
Кайзер Алюминиум, США КА-201 Шарики: 0,6—6,0 720 380 0,51
Рейнольдс Алюминиум, США RA-320 RA-1 RA-3 Гранулы или поро- шок тонкого помола 760—840 210—240 0,2—0,25
Алкоа, США F-l, F-3, F-7 Гранулы: 7—12 Порошок: 0,05 Гранулы: 2,4—6,0 830 875 210 210 —.
Р-20, F-21 Н-151 Гранулы: 0,17—0,075 Шарики: 3,0—6,0 1080 810 210 390 —
выпускаемого за рубежом
Средний преобладаю- Содержание примесей, % (масс.), не более
щий радиус пор, им Fe2O3 Na2O SiO2 Назначение продукта
8,5 — 0,4 0,05 Носитель катализаторов
11,0 — 0,7 0,05 То же
6,0 — 0,6 0,05 » »
6,0 — 0,1 0,05 Адсорбент, носитель катализато- ров
— — 0,5 0,05 Осушитель
— 0,1 0,1 0,1 Катализатор и носитель катализа- торов
7,5 0,01 0,02 0,02 Носитель
7,5 0,045 0,09 0,15 То же
11,0 0,025 0,06 0,02 » »
7,0 5,0 — — — Осушитель газов и жидкостей, катализатор, носитель, средство для обесфторивання алкилатов, нейтрализации смазочных масел
5,0 0,02 0,3 0,2; TiO2— 0,02 Осушитель
Cl— 0,001 Носитель
— 0,02 0,5 0,1 TiO2- 0,002 Осушитель газов и жидкостей, носитель катализаторов
— 0,08 0,9 0,09 Осушитель
— Осушитель, носитель катализато- ра, катализатор
— 0,08 0,9 0,09 Насадка при хроматографии
— 0,13 1,6 2,2 Осушитель (под высоким давле- нием)
286
11. Получение и номенклатура адсорбентов
Рис. 11,3. Схема получения гранулированных цеолитов со связующими добавками:
I — смеситель; 2 — кристаллизатор; 3, 11 —фильтры; 4 — бегуны; 5 — увлажнитель; 6 —
гранулятор; 7 —сушилка; 8 — сито; 9 — печь; 10 —реактор.
нисм. Частицы имеют размер от 1 до 15 мкм и за редким исключением не
могут применяться как адсорбенты технологических процессов. Поэтому в
качестве связующего в бегунах 4 к ним добавляют 15—20% пластичной коа-
линнтовой нли бентонитовой глины, обладающей высокой проницаемостью.
Время замеса составляет 30—40 мин.
Выходящая из бегунов паста имеет влажность около 35%, что обеспе-
чивает консистенцию, которая необходима для грануляции. В некоторых слу-
чаях пасту дополнительно увлажняют в аппарате 5. В таблеточной машине 6
получают гранулы требуемого размера, обычно от 2 до 4 мм. Гранулирова-
ние может быть проведено на машинах различного типа.
Гранулы подвергают сушке при 120—150 °C в аппарате ленточного 7 или
иного типа. В барабанном вращающемся сите 8 от высушенных гранул от-
сеивают мелочь н возвращают ее на повторный замес. Для придания грану-
лам термической и механической прочности их прокаливают во вращающейся
или шахтной печи 9 при 575—650 СС в течение 6—24 ч. В печи гранулы дви-
жутся противотоком к дымовому газу, получаемому в результате сжигания
бессернистого природного газа. Готовые гранулированные цеолиты упако-
вывают в герметичную тару.
При получении кальциевой формы цеолита кристаллит с фильтра 3 на-
правляют в реактор 10, где происходит катионный обмен в среде раствора
хлорида кальция. Кальциевый кристаллит выделяют на фильтре 11, после
чего гранулируют по описанной выше схеме'с.
Основное различие в технологическом регламенте при полу-
чении цеолитов типов А и X заключается не только в темпера-
турном режиме стадии кристаллизации, но и в условиях приго-
товления гидрогеля. Гидрогель цеолита NaX должен отличать-
ся относительно высоким содержанием оксида кремния и ще-
лочи. Мольное отношение SiO2: А12О3 в гидрогеле перед кри-
сталлизацией составляет при получении цеолита А 1,7—1,9; при
получении цеолита X 2,4—4,0. Мольное соотношение Na2O : А120з
соответственно составляет 2,3—2,5 и 2,7—5,2.
* В последние годы с целью получения более прочных гранул ионный обмен
часто проводят, используя формованные гранулы, а не порошок. (Прим, ред.)
Синтетические цеолиты
287
В процессе получения цеолита типа X особое внимание уде-
ляют мерам, исключающим параллельное образование цеолита
другого типа — филлипсита. С этой целью, в частности, гидро-
гель при кристаллизации не подвергают перемешиванию.
В табл. 11-4 приведены показатели качества гранулирован-
ных цеолитов, изготовляемых в СССР. Плотность гидратирован-
ного кристалла равна 2,00 г/см3, обезвоженного 1,55 г/см3, на-
сыпная плотность кристаллита 0,525 г/см3. Удельная теплоем-
кость ср гранул изменяется в зависимости от температуры:
/, °C 20 40 250
ср, Дж/(кг-К) 830 850 1000
Наряду с теплоемкостью, одним из основных показателей, на
которых базируются расчеты стадий регенерации и охлаждения,
является теплопроводность. Теплопроводность также несколько
изменяется с ростом температуры. Зависимость коэффициента
теплопроводности цеолитов СаА и NaX, установленная методом
стационарного потока в сферической прослойке, приведена на
рис. 11,4 [58]. На теплопроводность адсорбента оказывает влия-
ние теплопроводность среды и адсорбированного вещества. Так,
при 100 °C теплопроводность цеолита СаА в атмосфере воздуха
составляет 0,67, а в атмосфере СОг только 0,50 кДж/(м-ч-К).
Существенные коррективы в значении эффективного коэффици-
ента теплопроводности может внести конвекция тепла десорба-
том, выделяющимся при нагреве из твердой фазы. Теплопровод-
ность дегидратированных кристаллов при обычной температуре
составляет 2,1 кДж/(м-ч-К).
Как указано, при гранулировании в цеолит добавляют свя-
зующее, которое создает дополнительную, вторичную пористость.
В зависимости от процесса, в котором применяют цеолит, к ка-
честву связующего предъявляются различные требования. Ад-
Таблица 11-4. Характеристика гранулированных цеолитов
Показатель Тип цеолита
А X
Насыпная плотность, г/см3 0,68—0,78 0,62—0,72
Диаметр зерна цеолита (цилиндры, таблет- 2—4 2—4
ки, шарики), мм Предел прочности при сжатии, МПа Динамическая влагоемкость, мг/см3 8—1,5 6—12
90—140 100—160
Динамическая емкость по парам бензола, мг/см2 — 60—90
288
11. Получение и номенклатура адсорбентов
сорбция на поверхности связующего не является специфической
и обычно стремятся, чтобы удельная поверхность связующего не
превышала 10 м2/г. Часто в качестве связующего применяют гли-
ны. В тех случаях, когда переработке подвергаются легко поли-
меризующиеся продукты, связующее должно быть каталитиче-
ски неактивным; к связующим таких типов относят крымский
кил и цемент.
На рис. 11,5 приведены дифференциальные кривые распре-
деления объема пор по радиусам для двух образцов гранулиро-
ванных цеолитов по данным ртутной порометрии [59]. Преоб-
ладающий радиус вторичных пор для этих экспериментальных
образцов, сформованных в шнековой машине, составляет 340—
540 нм. Второй небольшой пик отвечает порам радиусом 70 нм.
При увеличении давления прессования максимумы кривых рас-
пределения пор смещаются в сторону меньших радиусов. В про-
мышленных образцах цеолитов [60] кривая распределения пор
характеризуется пиком, отвечающим эффективному радиусу
230 нм; 50% объема вторичной пористой структуры приходится
на поры в пределах эффективных радиусов пор от 160 до 355 нм,
46% объема пор имеют радиус меньше 160 нм и только 6% —
выше 355 нм.
Введение связующего уменьшает концентрацию активных
ячеек — частиц кристаллита в объеме адсорбента, в связи с чем
уменьшается адсорбционная способность. На рис. 11,6 представ-
лена зависимость равновесной адсорбционной емкости цео-
лита NaA по этилену при 20°C и его концентрации в газе
2,5% от содержания кристаллита в гранулах [61]. Как видно
из рисунка, активность цеолита закономерно убывает с увели-
чением доли связующего. При чрезмерном добавлении связую-
щего ухудшаются кинетические характеристики процессов.
Обычно добавка связующего к цеолиту составляет 18—20%.
Вычисление предельного адсорбционного объема отечествен-
ных и американских гранулированных цеолитов, проведенное на
Ц г
Рис. 11,4. Зависимость коэффициента теплопроводности Л цеолитов СаА и NaX от тем-
пературы t.
Рис. 11,5. Дифференциальные кривые распределения объема вторичных пор двух образ-
цов гранулированных цеолитов по радиусам (г в нм).
Синтетические цеолиты
289
Рис. 11,6. Зависимость равновесной адсорбцион-
ной способности цеолита NaA по этилену от со-
держания связующего ссв (/-20 °C, с0-2,5% об.).
основе обработки результатов ад-
сорбционных измерений, позволило
установить средние значения пре-
дельного адсорбционного объема
основных типов цеолитов (в см3 на
1 г): NaA —0,205; СаА —0,223;
СаХ —0,235; NaX —0,238. Эти по-
казатели на 30% ниже предельных
объемов пор кристаллитов [62].
Большое внимание уделяется разработке методов получения
цеолитов повышенной прочности, имеющих сферическую форму
и пригодных для использования в установках не только со ста-
ционарным, но и с движущимся слоем адсорбента. Одно из на-
правлений предусматривает метод закатки. При изготовлении
шариковых цеолитов этим методом цеолитовый порошок и свя-
зующее смешивают, увлажняют и в тарельчатом грануляторе
окатывают в шарики заданного размера. Шарики влажностью
15—18% по обычной методике подвергают сушке и прокалива-
нию. В качестве связующего используют алюминат натрия
(который разлагается при обработке дымовыми газами в оксид
алюминия), гексаметилентетрамин, бакелит и бакелитовый лак,
цемент, известь. В последнем случае цеолит формуется в гра-
нулы с негашеной известью, а механическая прочность гранул
обеспечивается обработкой их водяным паром при давлении
0,8—1,6 МПа в течение 12 ч.
Другим направлением является гранулирование из гелеоб-
разной смеси цеолита и жидкого стекла, которая, опускаясь че-
рез слой масла, принимает форму шаров, а затем коагулирует
в растворе хлорида натрия. По одной из методик цеолитовый
порошок перед подачей в органическую жидкость смешивают
с кремнезолем и с тонкоизмельченным оксидом магния.
Гранулирование цеолитов без связующих добавок
Введение связующего уменьшает равновесную способность ад-
сорбента, а в ряде случаев приводит и к ухудшению кинетиче-
ских свойств цеолита. В связи с этим возникла идея получения
цеолитов, в которых кристаллы соединяются в агломераты, что
исключает необходимость применения связующего.
Кристаллизацию цеолита при этом проводят при более вы-
сокой концентрации реагирующих компонентов, чем обычно. Вы-
сококонцентрированные алюмосиликатные гидрогели готовят
формованием в гранулы мелкодисперсного сухого кремнезема и
19-1346
290 И- Получение и номенклатура адсорбентов
концентрированного раствора алюмосиликата натрия. Аморфные
гранулы сушат, прокаливают и отсеивают* от пыли, как и обыч-
но. После этого проводят кристаллизацию во вращающихся ап-
паратах, через которые циркулирует горячий щелочной раствор
алюмината натрия. Вращение кристаллизаторов уменьшает
опасность слипания и срастания гранул. Температура при кри-
сталлизации 95—100 °C. Затем гранулы промывают водой до тех
пор, пока pH отходящей воды не снизится до 11,5. После того,
как из гранул удален избыток воды, их сушат воздухом или ды-
мовыми газами и упаковывают.
Если в цеолиты вместо натрия необходимо ввести другой
катион, в кристаллизатор после промывки гранул от щелочи по-
дают раствор соответствующей соли. Когда реакция обмена за-
кончена, избыток соли отмывают и дальнейшая технология со-
ответствует обычной. В качестве сырья для получения цеолитов
без связующего используют и природные материалы. В Совет-
ском Союзе в промышленном масштабе реализован способ по-
лучения шарикового цеолита NaA без связующего на основе
природного сырья — каолина Г64].
Принципиальная технологическая схема получения этого цеолита пред-
ставлена на рис. 11,7. Комовый каолин подвергается аморфизации во вра-
щающейся печи 1, куда из топки 3 поступают дымовые газы. Образующийся
при этом метакаолин измельчают в шаровой мельнице 2. Соотношение SiO2:
: А120з в метакаолине соответствует соотношению этих компонентов в цео-
литах типа А. Порошкообразный метакаолин смешивают с раствором гид-
рооксида натрия и образующуюся расту формуют в сферические гранулы с
помощью машины 4. Гранулы твердеют при высушивании в аппарате 5, а
затем подвергаются кристаллизации в аппарате 6 при циркуляции через их
слой горячего щелочного раствора, подаваемого насосом 8 из емкости 7.
В связи с разработкой процессов выделения нормальных па-
рафинов в кипящем слое был осуществлен синтез, а затем опыт-
Каолин
Рис. 11,7. Схема получения цеолита NaA из каолина:
1 — вращающаяся печь; 2 — шаровая мельница; 3 — топка; 4 — формовочная машина; 5 —
сушилка; 6 — кристаллизатор; 7 — емкость щелочного раствора; 8—насос.
Некоторые свойства промышленных адсорбентов 291
Таблица 11-5. Свойства гранулированных цеолитов NaA,
не содержащих связующих веществ [20]
Показатель Цеолиты, не содержащие связующих веществ Цеолит, содержа- щий 20% глины
из синте- тического сырья из глухо- вецкого каолина из каолина с добавкой кристал- ' лита
Плотность, г/см3 0,73 0,70
насыпная 0,67 0,74
кажущаяся 1,08 1,16 1,16 1,10
Адсорбционная емкость по парам ВОДЫ при /=20°С И Ротн = 0,1, % (масс.) Динамическая активность по парам воды при осушке воздуха до точки росы минус 70 °C, г/100 г 25,0 22,0 23,0 20,0
при высоте слоя 30 см 18,7 17,7 20,0 14,0
при высоте слоя 50 см . . 19,7 20,8. — 17,0
Удельная теплота смачивания водой, Дж/г 377 377 1 377 310
Предел прочности при сжатии, МПа 12 30 20 6
ное производство микрОсферического цеолита типа MgA, не Со-
держащего связующих веществ. Размер микросфер не превыша-
ет 500 мкм. Технология производства заключается в следующем.
Мелкодисперсную каолиновую суспензию подвергают распыли-
тельной сушке с целью формования ее в микросферические гра-
нулы, а затем прокалке для аморфйзации каолина. Затем про-
водят кристаллизацию, ионный обмен и промывку гранул.
Как показывает табл. 11-5, цеолиты без связующего по фи-
зическим свойствам близки к обычным цеолитам, но превосхо-
дят их по механической прочности. Наличие активной поверхно-
сти вторичных пор, которая образована гранями кристаллита,
позволяет значительно увеличить скорость транспорта молекул
адсорбата к микропорам. Благодаря этому цеолиты без связу-
ющего часто применяют в жидкофазных технологических про-
цессах, в первую очередь при осушке жидкостей.
Некоторые свойства промышленных адсорбентов
и их определение
Специфические свойства пористых тел, определяющие их способ-
ность быть адсорбентами, рассмотрены в гл. 2 и 3. В этом раз-
деле обсуждены неспёцифйческие свойства пористых тел (адсор-
бентов, катализаторов, носителей), используемые при их произ-
водственной оценке.
19’
292 И- Получение и номенклатура адсорбентов
Плотность адсорбентов. Для характеристики плотности пористых тел
Применяют три показателя: истинная, кажущаяся и насыпная,
или гравиметрическая плотность.
Истинная плотность ри характеризует массу единицы объема вещества,
из которого состоит адсорбент. Обычно ее определяют, помещая навеску ад-
сорбента в пикнометр и заполняя его той или иной жидкостью. Объем веще-
ства, из которого состоит адсорбент, определяют по разности объема пустого
пикнометра и пикнометра с навеской адсорбента. Объем пикнометра с навес-
кой пропорционален весу жидкости в нем.
Другой метод определения истинной плотности заключается в заполне-
нии герметичной колбы, содержащей навеску адсорбента, практически ие сор-
бирующимся газом — гелием. Разность между объемом газа в пустой колбе
и объемом газа в колбе, содержащей навеску адсорбента, соответствует объ-
ему вещества, из которого состоит адсорбент.
Кажущаяся плотность рк соответствует массе гранулы адсорбента, от-
несенной к ее объему. Объем гранул складывается из объема вещества ад-
сорбента и объема пор. Кажущуюся плотность достаточно крупных гранул
(больше 1 мм) определяют в пикнометре (так же, как истинную плотность),
но предварительно на поверхность гранул наносят тонкую пленку парафина,
препятствующую прониканию жидкости в поры. Кроме этого метода для оп-
ределения кажущейся плотности используют свойство ртути не смачивать
поверхность адсорбентов и не заполнять пор, в которых находится воздух.
Определение проводят в мерной ячейке с фиксированным объемом. Ее запол-
няют ртутью: сначала пустую, а затем — после внесения навески адсорбента.
Разность в объемах ртути, пошедшей на заполнение ячейки в первом и вто-
ром случаях, соответствует суммарному объему зерен адсорбента.
Насыпная плотность рг характеризует массу единицы объема слоя ад-
сорбента. Она практически не зависит от размера зерен адсорбента. Насып-
ную (гравиметрическую) плотность определяют в мерном цилиндре емкостью
V от 100 до 500 см3. Цилиндр при встряхивании заполняют адсорбентом до
метки, а затем взвешивают, устанавливая массу адсорбента g. Результаты
определения насыпной плотности (Pr=g/Iz) в значительной степени зависят
от интенсивности встряхивания, и поэтому условия уплотнения слоя регла-
ментируются.
Применительно к цеолитам определение проводят следующим образом.
Насыщенный в эксикаторе парами воды адсорбент тремя порциями по 30—
35 см3 каждая засыпают в стеклянный цилиндр емкостью 100 мл, установ-
ленный на вибростолике (рис. 11,8). Каждую порцию вводят приблизительно
в течение 15 с, после чего в течение 1 мин слой под действием вибрации
уплотняется. После введения третьей порции слой подвергают вибрации еще
2 мин, затем вибратор включают и адсорбент досыпают до метки 100 мл.
Затем гранулы из цилиндра высыпают в предварительно взвешенную чистую
колбу с пробкой и взвешивают с точностью до 0,1 г. Отношение массы ад-
сорбента к его объему (100 см3) дает насыпную плотность насыщенного во-
дой адсорбента. Чтобы учесть содержание влаги в адсорбенте, иавеску в 2—
3 г помещают в фарфоровый тигель и сушат до постоянной массы. Зная по-
терю массы, а следовательно, влажность цеолита, легко внести поправку в
расчет на влажность и, таким образом, определить насыпную плотность де-
гратированного адсорбента.
Пористость слоя и гранул. Под пористостью слоя есл понимают долю
объема слоя, не занятую собственно гранулами адсорбента. По этим пусто-
там проходит поток газа или жидкости в процессе адсорбции. Значение
пористости слоя зависит от формы гранул адсорбента и характера их упа-
ковки. Пористость слоя определяет один из основных показателей адсорбе-
ра — его гидравлическое сопротивление и учитывается во всех формулах, ко-
торые применяют для расчета. Она может быть определена по соотношению
кажущейся и насыпной плотности. Очевидно, что
рг=(1—есл)Рк (11.13)
Некоторые свойства промышленных адсорбентов 293
Из этого соотношения легко получить выражение для пористости слоя:'
еСл = 1 — (Рг/Рк) . ' (11.14)1
Пористость гранул и ее характер определяют равновесные и особенно
кинетические свойства адсорбента. Под пористостью гранул (его) понимают
долю пустот в грануле адсорбента. Ее определяют по соотношению кажу-
щейся и истинной плотности:
®гр = 1 — (Рк/Ри) (11.15)
Эквивалентный диаметр гранул и поверхность единицы объема. Гидрав-
лическое сопротивление слоя адсорбента при прохождении через него потока
газа или жидкости, как было показано выше, зависит от определяющего
размера гранул. В случае шаровидных гранул таким определяющим разме-
ром является диаметр d, в случае цилиндрических или иных гранул — экви-
валентный диаметр d3. Для нешаровидных тел эквивалентный диаметр мо-
жет быть определен по удельной геометрической поверхности тела Sb
(внешней поверхности единицы объема):
4» = 6/S0 (11.16)
В свою очередь, поверхность единицы объема определяется как отноше-
ние поверхности гранулы згр к ее объему огр:
= згр/пгр (11.17)
Гранулометрический состав. Он характеризуется содержанием фракций,
задерживаемых ситами с ячейками номинального размера. Рассев можег
быть машинным или ручным. Перед рассевом пробу адсорбента (25 г) вы-
держивают на воздухе в течение 1—2 ч. Остатки на ситах, а также фрак-
цию, прошедшую через нижнее сито, собирают в стаканчики с крышками в
взвешивают с точностью до 0,2 г. Сумма масс всех фракций не должна от-
Рис. 11,8. Прибор для определения гравиметрической плотности.
Рис. 11,9. Пресс для определения предела механической прочности гранулированных ад-
сорбентов при сжатии.
294
11. Получение и номенклатура адсорбентов
личаться от массы первоначальной более чем на 1 г. Гранулометрический
состав выражается в массовых процентах*.
Механическая прочность промышленных адсорбентов. Адсорбент в про-
мышленных адсорберах с неподвижным слоем подвергается измельчению под
действием статических и динамических нагрузок. К статическим нагрузкам
относится в первую очередь давление слоя адсорбента, расположенного выше
гранул в рассматриваемом сечении аппарата. Динамические нагрузки воз-
никают вследствие изменения температур и давлений в аппарате, при его
разгрузке и т. д.
Определение механической прочности синтетических цеолитов — предела
Прочности при сжатии — производят на прессе специальной конструкции,
представленном на рис. 11,9. Из пробы адсорбента (например, цеолита), под-
лежащей анализу, отбирают 26 гранул (таблеток) с относительно равными
торцевыми поверхностями. Эти таблетки прокаливают в муфеле при 350—
400 °C в течение 3 ч. Затем их охлаждают в герметичном бюксе и поочеред-
но подвергают испытанию. Для этого таблетку пинцетом ставят в центр
столика прибора, затем вращением маховичка производят постепенный иа-
жим на таблетку. Одновременно следят за показанием двухстрелочного ма-
нометра. При раздавливании одна стрелка манометра падает на нуль, вто-
рая регистрирует давление раздавливания. Раздавливающее усилие опреде-
ляется умножением давления раздавливания на константу прибора — пло-
щадь плунжера. Прочность адсорбента характеризуют индексом механической
прочности. Его получают делением значения раздавливающего усилия на
площадь минимального поперечного сечения таблетки. Среднее значение ин-
декса вычисляют как среднеарифметическое из измерения прочности 20 таб-
леток (три верхних и три нижних результата отбрасывают). Прочность гра-
нул цеолита на раздавливание (предел прочности при сжатии) составляет
4—5 МПа, но может достигать 140 МПа [65].
Определение механической прочности гранулированных адсорбентов при-
менительно к динамическим нагрузкам производят в приборе, представлен-
ном на рис. 11,10 и состоящем из трех горизонтальных барабанов, вращаю-
щихся с частотой 1,2 с-1 (75 об/мин). В каждый барабан загружают на-
веску, соответствующую объему 50 см3 адсорбента. Внутренний диаметр ап-
паратов 80 мм, длина каждого из иих также 80 мм. В каждый барабан по-
мещен истирающий элемент — стальной стержень массой 1,2 кг; его диаметр
равен 50 мм, длина 78 мм. Перед опытом адсорбент отсеивают от мелочи.
Если размер частиц адсорбента больше 1,5 мм, применяют сита с отверстия-
ми размером 1 мм. Время испытания в аппарате 3 мин. После измельчения
образец снова отсеивают от мелочи на том же сите и по разности массы
образца до и после опыта устанавливают прочность. Для характеристики
прочности применяют среднее из двух определений, причем расхождение
между ними не должно превышать 2%.
* Средний диаметр зерна отдельной ситовой фракции может быть определен
по формуле:
з/ 2d 2 л 2
. _ 1/ 1 2
йгср- У dl + d2
где di и d2 — диаметры отверстий смежных сит, применяемых при ситовом
анализе.
Средний диаметр всей партии по данным ситового анализа дается фор-
мулой
l/dcp = t^i/^icp)
где dict, и Xi — средний размер зерна в отдельной ситовой фракции и ее-мас-
совая доля. (Прим, ред.)
Некоторые свойства промыш '-'чных адсорбентов
295
Рис. 11,10. Прибор МИС-60-8 для определения механической прочности адсорбентов в
ОЯрЯОЯНв!
1 винт для регулирования положения прибора; 2 — червячный редуктор; 3 — муфта;
4 электродвигатель; 5 — барабаны; 6 — задняя бабка; 7—крышка; 8 — плита; 9 — план-
шайба. '
Рис. 11,11. Прибор для определения прочности адсорбента при испытании в эрлифте:
1 — баллон с газом; 2 — реометр; 3 — ячейка эрлифта.
- В процессе очистки или разделения газов в движущемся слое основное
истирание адсорбента происходит при пневмотранспорте, т. е. в газлифте
(или эрлифте). Описанные методики определения прочности ие отражают
условия в таких установках, поэтому целесообразно использовать метод ис-
пытания в эрлифте, обычно применяемый для оценки прочности катализато-
ров [66]. Пробу адсорбента 5> г (gi). после отсева от пыли и мелочи загру-
жают в прибор, схема которого представлена иа рис. 11,11. Ячейка эрлифта 3
имеет стеклянную трубку диаметром 15 мм и длиной 500 мм, расширенную
в нижней части; коаксиально к трубке расположен стеклянный кожух, диа-
метр которого 40 мм, а высота 530 мм. Снизу кожух вставлен в конус из
стали, оканчивающийся соплом диаметром 2 мм. На расстоянии 20 мм от
верхнего конца кожуха расположена ударная пластина. Частицы адсорбен-
та, уносимые вверх по трубе азотом, поступающим в нижнюю часть ячейки
из баллона 1 через сопло, ударяются о пластинку н по кольцевому прост-
ранству между трубкой и кожухом возвращаются вниз. Скорость азота
устанавливается по реометру 2; ее целесообразно поддерживать минималь-
ной, но обеспечивающей нормальную циркуляцию адсорбента, например
2,5 м/с. Время циркуляции составляет 15 мин. После опыта адсорбент вы-
гружают, просеивают через сито с отверстиями диаметром 1 мм, взвеши-
вают (g2), и по убыли массы определяют индекс прочности (в %);
n=(g2/gi)-100 (11.18)
Пылеобразование. В процессах транспортирования и загрузки адсорбен-
та часть гранул разрушается, образуя пыль. В технологических адсорбци-
онных процессах эта пыль, а также пыль, непрерывно возникающая при
смене температур и давлений непосредственно в адсорбционном цикле, пе-
реходит в аэрозольное состояние, что осложняет работу установок и арма-
туры. В настоящее время существует ряд методов и приборов, позволяющих
характеризовать дисперсный состав и концентрацию пыли в движущейся га-
зовой среде [67]. Одним из наиболее быстрых и удобных средств дисперсно-
го анализа запыленных газовых сред является метод фотометрии, использо-
ванный в фотоэлектрических счетчиках аэрозольных частиц. Он позволяет
подсчитать число частиц пыли размером от 0,3 до 10 мкм в единице объема
пропущенного через слой адсорбента газа.
Испытания различных промышленных адсорбентов с помощью этого при-
бора показали [67, 68], что интенсивность пыления максимальна в первые.
296 12.. Осдшкагазов и органических жидкостей
минуты прохождения газового потока через адсорбент. Доля пылн, уноси*
мой на начальной стадии продувки слоя, составляет 30—50% общей массы
частиц, выносимых нз слоя при длительной его эксплуатации. С целью со-
кращения выноса частиц пыли в циклическом адсорбционном процессе мо-
жет быть рекомендована операция короткой продувки слоя после загрузки
.его в аппарат или отмывки от пылн перед загрузкой.
Содержание влаги. В углеродных адсорбентах (активных углях) содер-
жание влаги определяют по убыли массы образца прн его высушивании до
постоянной массы. Навеску образца около 1 г в предварительно высушен-
ном и взвешенном вместе с крышкой стаканчике помещают в сушильный
шкаф н высушивают при 105—ПО °C в течение 1 ч. После этого стаканчик
.с пробой охлаждают в эксикаторе и снова взвешивают. Параллельно про-
изводят измерение на двух навесках. Точность взвешивания 0,0002 г.
Содержание влаги (в %) определяют из соотношения
^[fe-^/tex-^i-ioo
где gi — масса стаканчика с крышкой н навеской до высушивания; g2—то
же, после высушивания; g— масса пустого стаканчика с крышкой.
Расхождение между результатами двух параллельных определений не
должно превышать 0,3% прн содержании влаги в пробе от 1 до 10% и 0,4%
при содержании влаги 10—25%. Окончательный результат вычисляют как
среднее арифметическое двух определении с точностью до 0,1%.
Зольность. Зольность углеродных адсорбентов определяют следующим
образом. Две параллельных пробы адсорбента (1 г) в предварительно прока-
ленных и взвешенных тиглях помещают на под муфеля, температура в ко-
тором не превышает 300 °C. Затем образцы прокаливают прн 800±25°С в
течение 2 ч. После прокаливания тнглн с зольным остатком извлекают из
муфеля, охлаждают сначала на воздухе в течение 5 мин, а затем в эксика-
торе н взвешивают. Содержание золы А (в %) рассчитывают по формуле
4 = [100/(100-ЦТ)] 100
где gt — масса пробы исходного угля; gi — масса зольного остатка; W — со-
держание влаги в испытуемой пробе.
12
ОСУШКА ГАЗОВ
И ОРГАНИЧЕСКИХ
ЖИДКОСТЕЙ
Характеристика влажных сред
Одним из основных назначений силикагелей и цеолитов явля-
ется осушка газовых и жидких сред [69]. В связи с этим це-
лесообразно рассмотреть основные закономерности, характер-
ные для влажных сред.
Характеристика влажных сред
297
Рис. 12,1. Соотношение между абсолютной и относительной <р влажностью воздуха
при разной температуре.
Влажность газов
Влага снижает калорийность горючих газов, образует ледяные
пробки, закупоривающие газопроводы и нарушающие режим
работы технологических установок. Особое затруднение вызы-
вают кристаллогидраты углеводородов — снегообразные *гвер-
дые соединения, образующиеся при плюсовых температурах в
условиях повышенных давлений. Огромные трудности возника-
ют в зимнее время при использовании влажного воздуха в конт-
рольно-измерительных приборах. Наличие влаги в технологиче-
ских потоках часто приводит к ухудшению качества продукции
и к отравлению катализаторов. Большинство промышленных га-
зов подвергают осушке различными способами, из которых наи-
большую степень осушки обеспечивает адсорбционный метод.
Содержание влаги (в г) в единице объема газа (в м3) назы-
вают абсолютной влажностью. Каждой температуре
соответствует строго определенное предельное содержание во-
дяных паров в 1 м3 газа. Газ насыщен водяными парами, если
парциальное давление равно давлению насыщенного пара воды
при заданной температуре. С ростом температуры повышается
давление насыщенного пара и, соответственно, предельное со-
держание его в единице объема газа.
В большинстве случаев в технике имеют дело с газами, в ко-
торых содержание влаги меньше этого верхнего предела. Отно-
шение действительного содержания влаги к предельно возмож-
ному при заданной температуре, выраженное в процентах, на-
зывают относительной влажностью (<р). На рис. 12,1
приведено абсолютное влагосодержание воздуха в зависимости
от температуры при различной относительной влажности.
298
12. Осушка газов и органических жидкостей
Рис. 12,2. Прибор для определения влажности
газа пи точке росы:
1 — измерительная ячейка; 2 — зеркальце; 3 —
медный стержень; 4 — сосуд Дьюара; 5 — ста-
кан со льдом; 6 — регистрирующий прибор.
Относительная влажность может быть рассчитана также как
отношение парциального давления р к давлению насыщенного
пара при заданной температуре ps:
<Р = Pl Ре
Давление насыщенного пара воды при давлениях до 5—
10 МПа слабо зависит от общего давления газа. Поэтому ком-
примирование газа, сопровождающееся уменьшением его объ-
ема, приводит к конденсации части влаги. Компримирование ча-
сто используют на практике для частичного удаления влаги из
газа.
, Конденсацию пара можно вызвать и другим путем: понижая
температуру и вместе с тем—давление насыщенного пара. Ес-
ли при охлаждении достигнута температура, при которой пар-
циальное давление и давление насыщенного пара становятся
равными, т. е, относительная влажность равна 100%. начинает
образовываться туман. Такую температуру называют точкой
рос ы. Между абсолютной влажностью и точкой росы имеется
строгая зависимость: чем суше газ, тем ниже точка росы.
В, табл. 12-1 приведены данные, позволяющие производить
пересчет различных единиц выражения влагосодержания газа.
Здесь же указаны значения влажности еще в одной часто ис-
пользуемой единице — «пропромилле», или частей на миллион*.
Долгое время основными приборами для определения влажно-
сти газов были гигрометры, основанные на определении темпе-
ратуры начала конденсации влаги, т. е. точки росы.
Такой прибор представлен на рис. 12,2. Газ, влажность которого нужно
измерить, с постоянной скоростью, пропускают через стеклянную измери-
тельную ячейку 1. В ячейке помещено полированное металлическое зеркаль-
це 2, в которое впаяна точечная термопара. Зеркальце соединено с медным
стержнем 3, нижний конец которого опущен в дьюар с жидким азотом 4.
Холодный спай термопары выведен на лед в сосуде 5. Температура зеркаль-
ца фиксируется с помощью регистрирующего прибора 6.
Стержень погружают в жидкий азот — температура зеркальца постепен-
но понижается, достигая с течением времени значения, соответствующего
точке росы. В этот момент зеркальце слегка «запотевает» из-за осаждения
* Одна «пропромилле» (далее в тексте обозначается «част, на 1 млн») равна
1О~*°/о. Итак, 10 част, на млн. (объемных) — это 10-10~* об. %, или 10 см3
примеси в 1 м3 газа. Аналогичным образом выражается концентрация в мас-
совых «пропромилле». (Прим, ред.)
Характеристика влажных сред
299
на нем капелек влаги. Момент «запотевания» фиксируют как точку росы.
Чтобы унифицировать замеры, иногда приборы снабжают фотоэлементом,
наблюдения за зеркальцем ведут с помощью луп и т. п. В некоторых вари-
антах охлаждение зеркальца проводят, используя отрицательный тейловой
эффект, сопровождающий дросселирование газа высокого давления; воздуха
или диоксида углерода. После замера стержень вынимают из дьюара и зер-
кальце размораживается.
Недостатками метода являются субъективность замеров.,(раз-
ные люди неодновременно замечают выпавший туман) и, необ-
ходимость использования веществ, не всегда доступных: жидко-
го азота, газов высокого давления. Кроме того, при низких со-
держаниях влаги точность определения момента появления очень
тонкого конденсата уменьшается, поэтому проводить замеры
этим методом при точке росы ниже минус 70 °C не рекоменду-
ется.
В последнее время начали широко внедряться автоматиче-
ские приборы, в которых поток газа пропускают над пленкой
гигроскопичного вещества — пятиоксида фосфора, нанесенной
на тонкую платиновую спираль, которым эта влага практически
полностью поглощается. В толще пленки непрерывно происходит
электролитическое разложение влаги. Количество кулонов, по-
шедших на этот процесс, и служит мерой влажности поглотите-
ля, а следовательно, и газа. Такие приборы получили название
Таблица 12-1. Соотношение между различными единицами выражения
влагосодержания воздуха (расчетные значения)
Точка росы, СС Парциальное давление НаО, Р Абсолютная влажность
Па мм рт. ст. мг/мв част, на 1 млн.
об. масс.
— 100 1,32-10-4 9,9-10-» 1,04-10-» 1,3-Ю-з - • ЗМ?.# 8,1- IO-4
—90 9,50-IO-» 7,00-Ю"5 7,35-10-2 9,2-Ю-2 5,7-10-2
—80 5,30-10-2 4,00-10-4 4,20-10-1 5,26-10-1 3,27-JO-i
—70 2,50-10-1 1,94-10-» 2,4-0-10» 2,55-10» 1,58-10'
—60 1,07-10° 8,00-10-» 8,47-10» 1,06-101 6,59г10»
—50 3,93-10» 2,95-10-2 3,15-101 3,94-101 2,46-10«
—40 1,28-101 9,66-10-2 1,03-Ю2 1,27-Ю2 7,9) -Ю» 2,34-102
—30 3,80-101 2,85-10-1 3,00-102 3,76-102
—20 1,03-10» 7,76-10-1 8,15-Ю2 1,02-102 6,35-102
—10 2,58-102 1,95-10» 2,05-10» 2,56-10» 1,59-10»
0 6,07-102 4,57-10» 4,82-10» 6,00-10» 3774-40»
+5 8,75-102 6,60-10» 6,82-10» 8,14-10» 5,60 10»
+ 10 1,22-10» 9,21-10» 9,40-10» 1,18-Ю4 7;з8-10»
4-15 1,70-10» 1,28-101 1,30-104 1,60-10» 9,96-10»
4-20 2,33-10» 1,75-101 1,70-Ю4 2,15-Ю4 1,34-104
4-25 3,14-10» 2,38-101 2,3-104 2,88-104 1,79-Ю4
+30 4,21-10» 3,18-101 3,0-Ю4 3,80-104 2,37-104
4-40 7,55-10» 5,49-101 5,10-Ю4
4-50 1,22-104 9,20-101 8,30-104 — —
300
12. Осушка газов и органических жидкостей
кулонометрических. В Советском Союзе они выпускаются Ир-
кутским заводом «Эталон» по разработкам Ангарского филиала
ОКБА.
Шкала кулонометрических влагомеров отградуирована в
част, на 1 млн. (объемных). Для увеличения точности влаго-
меры обычно имеют несколько шкал. У влагомера «Байкал»,
например, их девять, что позволяет вести измерение влажности
в пределах от 1 • 10_ 1 до 1-Ю3 част, на 1 млн. Влагомер «Лена»
предназначен для поверки рабочих параметров других влаго-
меров и точного определения содержания влаги в газах при
проведении прецезионных научно-исследовательских работ. Диа-
пазон прибора 10~3—104 част, на 1 млн., или по точке росы:
от —102 до 4-18 °C.
Влажность жидких сред
Влага в жидкостях приносит не меньший вред, чем в газах.
В одних случаях она понижает эксплуатационные свойства про-
дукта (трансформаторное масло), в других — его реакционную
сйособйость (сырье в процессах алкилирования), в третьих —
просто при охлаждении выделяется на стенках трубопроводов
и емкостей, вызывая коррозию.
Предельное содержание влаги в жидких средах ограничива-
ется растворимостью. На рис. 12,3 представлены графики, от-
ражающие растворимость воды в углеводородах и различных
Рис. 12,3. Растворимость воды в углеводородах.
Осушка силикагелями
301
нефтяных фракциях в зависимости от температуры. Из этих
графиков видно, что растворимость воды в органических жид-
костях при обычной температуре не превышает 1% (мол.). Рас-
творимость воды уменьшается с понижением температуры.
Достоверность результатов изучения процесса осушки и воз-
можность непрерывного контроля качества осушки во многом
зависят от точности и экспрессности определения влажности
жидких сред. Методы анализа влажности жидкостей подробно
описаны в специальной литературе [70]. Анализ публикаций,
посвященных различным методам определения микроконцент-
раций воды за последние 10 лет, показал, что 30% всех работ
приходится на долю химического метода с применением реакти-
ва Фишера*.
Достаточно широко применяется гидрокальциевый, спектро-
абсорбционный, емкостный и хроматографический методы опре-
деления влаги в жидких средах.
Осушка силикагелями
Одним из видов широко применяемых для осушки газов адсор-
бентов являются силикагели.
Структура силикагеля оказывает решающее влияние на эф-
фективность его использования в адсорбционном процессе. Ад-
сорбционные процессы и конкретные требования к структурным
характеристикам силикагелей достаточно разнообразны, однако,
исследователь значительно упростит решение задачи, если
будет четко представлять себе основные закономерности, харак-
теризующие взаимосвязь структуры силикагелей и их адсорб-
ционных свойств.
На рис. 12,4 представлены изотермы адсорбции паров воды
на среднепористом и мелкопористом силикагеле со средним эф-
фективным радиусом пор соответственно 2 и 1 нм. Различия в
видах изотерм по-разному проявляются при осушке среды в
статических и динамических условиях.
Осушка в статических условиях
Осушка в статических условиях достаточно широко распростра-
нена в промышленной практике: при консервации оборудования,
поддержании заданной влажности в замкнутых объемах, запол-
нении затворов и дыхательных клапанов емкостей. Количество
* В книге Малкина Л. Ш., Колина В. Л. (Осушка и очистка малых холо-
дильных машин. М., «Легкая и пищевая промышленность», 1982. 148 с.)
описан полуавтоматический показывающий лабораторный титратор «Влага»,
выпускаемый в промышленных масштабах. Его действие основано на тит-
ровании воды иодом, электрогенерированным в ходе самого анализа из
реактива Фишера по ТУ 6-09-1487—76. (Прим, ред.)
302
12. Осушка газов и органических жидкостей
Рис. 12,4* Изотермы адсорбции паров воды на мелкопористом (2) и среднепористом (1)
силикагелях при 20 °C.
Рис. 12,5. Скорость насыщения крупно- (№ 2), средне- (№ 4) и мелкопористого (№ 8>
силикагелей в статических условиях.
загружаемого адсорбента в этом случае определяется в основ-
ном равновесной адсорбционной емкостью*.
При относительном влагосодержании среды до 55—60% мак-
симальной адсорбционной способностью по парам воды облада-
ет мелкопористый силикагель, причем преимущество его перед
средне- и крупнопористыми силикагелями тем больше, чем ниже
влагосодержание осушаемой среды. В интервале относительных
влагосодержаний 60—90% преимущество перед силикагелями
других структурных типов имеет среднепористый силикагель,,
особенно значительное при влагосодержаниях 70—80%. Пре-
имущества крупнопористого силикагеля реализуются лишь при
очень высоких относительных влагосодержаниях среды — бо-
лее 90%).
Насыщение силикагелей влагой в статических условиях-—
процесс медленный (рис. 12,5). По времени насыщения влагой
при комнатной температуре силикагели располагаются в следу-
ющем порядке: мелкопористый — среднепористый — крупнопо-
* В статических условиях адсорбенты,' как правило, используют для поддер-
жания влажности в частично герметичном объеме. Сухой адсорбент (сили-
кагель) в мешочках из ткани более или менее равномерно распределяют по
контролируемому объему. Общее количество адсорбента зависит от прони-
цаемости и размеров оболочки, поступления влаги от хранимых изделий.
Оно, конечно, тем больше, чем выше заданная продолжительность хранения
и обратно пропорционально равновесной адсорбционной емкости. Последнюю
определяют по изотерме адсорбции, соответствующей температуре хранения,,
и предельно допустимой для изделий данного вида относительной влажности,
среды. (Прим, ред.)
Осушка силикагелями
303
ристый. Длительность полного насыщения у этих силикагелей
составляет 40, 50 и 75 ч, а обработка адсорбционной емкости на
50% достигается за 8, 10 и 22 ч соответственно.
Адсорбционная емкость мелкопористых силикагелей в зави-
симости от температуры и парциального давления водяного па-
ра может быть определена из равновесных данных, приведен-
ных на рис. 12,6. Следует отметить, что при постоянной отно-
сительной влажности гаЗй влагоемкость силикагеля приблизи-
тельно постоянна, вне зависимости от температуры, при кото-
рой происходит процесс поглощения.
Осушка в динамических условиях
В подавляющем большинстве случаев осушку газов производят
в динамических условиях. " ' '
Изотермы адсорбции паров воды на силикагелях состоят из
выпуклых и вогнутых участков. В таких случаях при адсорбции
параллельный перенос в слое происходит только тогда, когда
влажность осушаемой,о.^газа невелика (для ,мелкопористых си-
ликагелей, например, при относительном давлении паров воды
не выше 0,2—0,3). При более высоких концентрациях паров во-
ды стационарный фронт образуется только для начального уча-
стка изотермы: с=0 до*'с=с'о (где с'о—концентрация адсорбти-
ва в точке перегиба 'изотермы). Участок фронта, заключенного
в эти пределы, движется с постоянной скоростью [71]:
U = wc04a0’ (12.1)
где с'а, а'о — концентрация воды в газовой и твердой фазах в точке пере-
гиба.
Рис!. 12,6. Зависимость адсорбционной способности мелкопористого силикагеля от тем*
пературы при различном парциальном давлении водяных паров.
304
12. Осушка газов и органических жидкостей
P/Pt
Рис. 12,7. Механизм осушки газа в слое силика-
геля: '
А — изотерма адсорбции; Б — кривые распределе-,
иия адсорбата в слое адсорбента.
Остальной участок фронта рас*
пределен по всей, охваченной ад-
сорбцией длине слоя адсорбента.
Такой механизм иллюстрирует рис.
12,7. Чем медленнее движется псев-
достационарный фронт, тем больше
время до проскока Тщ> и, следова-
тельно, тем выше динамическая активность:
тпр = "д' (£ — ф'ТоЭ = (L — ф'^о')
(12.2)
где Д—[высота слоя адсорбента; L'o — высота работающего слоя, определен-,
иая для интервала концентраций от 0 до с'о", ф' — фактор симметричности
для этого же участка выходной кривой.
Следовательно, динамическая активность при прочих рав-
ных условиях находится в прямой зависимости от отношения.
а'^/с'о, которое (см. рис. 12,4) наиболее высоко для мелкопори-
стых адсорбентов. Поэтому осушку газов в динамических усло-
виях, в любом случае следует вести с применением мелкопори-
стых силикагелей.
Это положение было подтверждено экспериментально. Осуш-
ку проводили в широком интервале относительных влагосодер-
жаний и скоростей осушаемого воздушного потока (20 °C) в слое
высотой 0,5 м. В качестве адсорбентов были использованы круп-
нопористый (№ 2), среднепористый (№ 4) и мелкопористый
,м/с
Рис. 12,8. Зависимость динамической активности слоя (£=1) силикагелей с разной пори-
стой структурой от скорости потока:
а — осушка до проскоковой концентрации,, соответствующей точке росы минус 40 ®С; б —
то же, 0 °C.
Осушка силикагелями
305
Рис. 12,9. Зависимость динамической активности слоя (Л=1 м) силикагелей с разной по-
ристой структурой от относительного влагосодержания воздуха:
а — осушка до проскоковой концентрации, соответствующей точке росы минус 60 °C; б —
то же, минус 40 СС; в —то же, 0°С.
(№ 6) силикагели. В опытах, результаты которых представле-
ны на рис. 12,8, осушался воздух с постоянным относительным;
влагосодержанием 75—80% (абсолютное влагосодержание 13—
14 мг/л), а скорость потока изменялась в пределах 0,03—
0,20 м/с. В следующей серии опытов (рис. 12,9) осушку прово-
дили при постоянной скорости потока, равной 0,09 м/с, а отно-
сительное влагосодержание воздуха изменяли в пределах 10—
85%.
Как видно из графиков, при постоянном относительном вла-
госодержании воздуха (75—80%) во всем интервале изученных,
и имеющих практическое значение скоростей осушаемого пото-
ка динамическая емкость силикагелей находится в обратной за-
висимости от среднего радиуса их пор. И в условиях глубокой:
(до точки росы минус 40°C), и в условиях грубой (до точки,
росы 0°C) осушки мелкопористый силикагель имеет значитель-
ные преимущества перед остальны-
ми типами силикагелей. Эта зако-
номерность не изменяется при осу-
шке воздуха с любым влагосодер-
жанием.
Динамическая активность слоя
силикагеля по влаге зависит от
размера зерна. Оценка влияния
размера зерна на динамическую
емкость мелкопористого силикагеля
была проведена Липкиндом в адиа-
батическом адсорбере с высотой
слоя адсорбента 0,3 м. Осушали
Рис. 12,10. Влияние размера зерна на динамиче-
скую активность слоя (/.=0,3 м) силикагеля КСМ.
20—1346
306
12. Осушка газов и органических жидкостей
Рве. 12,11. Зависимость динамической активности
мелкопорвстого силикагеля от температуры осу-
шаемого воздуха.
при 20 °C воздух с относительным
влагосодержанием 75—80%. Ско-
рость потока составляла 0,10 и 0,15
м/с. За момент «проскока» была
принята точка росы выходящего га-
за— минус 60 °C. Результаты опы-
тов представлены на рис. 12,10. Увеличение размера зерна в 4
раза (от 1 до 4 мм) приводит к снижению динамической ак-
тивности слоя силикагеля приблизительно в 2 раза.
Повышение температуры в значительной степени -снижает
эффективность осушки и, в первую очередь, динамическую ак-
тивность. На рис. 12,11 эта зависимость дана для слоя силика-
геля КСМ высотой 0,5 м. Перед поступлением в адсорбер воздух
насыщали парами воды до относительной влажности 80%,
а затем нагревали до заданной температуры. .Скорость потока
в этой серии опытов составляла 0,15 м/с, за проскоковую кон-
центрацию влаги, как и в предыдущей серии, была принята
точка росы минус 60°C. При температуре осушаемого воздуха
70 °C динамическая активность составляла лишь 4 г/100 г, т. е.
была в 4 раза ниже, чем при 10 °C.
Указанные особенности процесса сорбции паров воды сили-
кагелями позволяют утверждать, Что степень использования ад-
сорбционной способности а у этого адсорбента не может пре-
вышать величины, даваемой уравнением ......
Up'cg / L —
аоСо' \ L
(12.3)
где йо — адсорбционная способность, равновесная с исходным содержанием
паров воды в газовой фазе (с0).
Результаты расчета по формуле (12.3) показали, что даже
при бесконечно больших слоях силикагеля степень использо-
вания его адсорбционной емкости при осушке влажного (<р =
= 0,95) газа не превышает 75%.
Осушка газов с небольшим содержанием влаги
Мелкопористые силикагели при низких температурах могут ис-
пользоваться и для доосушки газов с низким содержанием
влаги. На рис. 12,12 представлены изотермы адсорбции паров
воды на мелкопористом силикагеле в полулогарифмической си-
стеме координат [72]. При 25 РС, концентрации 200 част; на
Осушка силикагелями
307
Рис. 12,12. Изотермы адсорбции паров воды иа
мелкопористом силикагеле в области малых кон-
центраций.
1 млн. (1g с=2,3), или 0,02% (об.)
активность мелкопористого силика-
геля составляет 4 г/100 г.
Выбор условий регенерации
силикагеля
Степень осушки газа зависит от ус-
ловий обезвоживания силикагеля в
предшествующей стадии регенера-
ции. Глубина осушки зависит, в первую очередь, от температу-
ры регенерации, а при низких температурах — от влажности:
продувочного газа W (рис. 12,13). Кривые, соответствующие раз-
личной влажности продувочного газа (от 1000 до 21500 част, на
1 млн.), носят экспоненциальный характер и остаточное влаго-
содержание по мере повышения температуры регенерации
асимптотически стремится к некоторому предельному значению-
(10 част, на 1 млн.). Это значение концентрации влаги в паро-
вой фазе, видимо, отвечает полному удалению физически ад-
сорбированной воды из пор силикагеля. При температурах ре-
генерации выше 160°C полнота регенерации силикагеля дости-
гается вне зависимости от влажности продувочного газа.
Степень осушки газов силикагелями определяется на основе
диаграммы, построенной с учетом изотерм адсорбции (рис. 12,14).
Приведем пример использования диаграммы. Пусть при регенерации си-
ликагель был нагрет до 135 °C в результате продувки влажным газом, в ко-
тором концентрация влаги, составляет 1000 част, на 1 млн. Проводим перпен-
дикуляр с= 1 • 103 част, на 1 млн. до пересечения с изотермой 135 °C. Содер-
жание влаги в адсорбенте после регенера-
ции определяем на оси ординат в соответ-
ствии с положением точки пересечения.
Оно равно около 1% (масс.). В свою оче-
редь, точки пересечения линии постоянного
влагосодержания силикагеля с кривыми 25
и 50 °C позволяют получить на оси абсцисс
значения остаточного содержания влаги в
газе на стадии осушки: 11 част, на 1 млн.
при 25 °C и 28 част, на 1 млн. при 50 °C.
Дезактивация силикагеля
Термическая обработка. При нагре-
вании насыщенного водой силика-
геля до 200 °C происходит удаление
Рис. 12,13. Зависимость глубины осушки газа мел-
копористым силикагелем К СМ-5 (остаточной влаж-
ности с) от условий регенерации (t, W).
20*
308
12, Осушка газов и органических жидкостей
Рис. 12,14. Диаграмма для определения остаточного влагосодержания в газе после осуш-
ки мелкопористым силикагелем с учетом условий регенерации (температуры и влажно-
сти продувочного газа).
физически адсорбированной воды и восстановление его адсорб-
ционных свойств. В случае дальнейшего повышения температу-
ры начинается выделение воды за счет ОН-групп поверхности.
Разрушение ОН-групп на поверхности силикагеля приводит к
ухудшению его адсорбционных свойств как осушителя.
Влияние температуры термообработки на динамическую ак-
тивность слоя мелкопористых силикагелей по парам воды было
изучено при скорости потока влажного (<р = 9О°/о) газа 0,15 м/с.
Высота слоя адсорбента составляла 100 см. В качестве адсор-
бентов-осушителей были применены шариковый силикагель
(средний радиус пор 1,0 нм) и гранулированный силикагель
(1,4 нм). Результаты исследований приведены в табл. 12-2. Пос-
Таблица 12-2. Динамическая активность (в мг/см3)
<мелкопористых силикагелей (проскоковая концентрация влаги —60
и —40 °C по точке росы)
Температура термообработки, •°C Радиус пор 1,0 нм Радиус пор 1,4 нм
—60 °C —40 °C -60'С —40 °C
100 0 3,2 0 1,0
150 0 34,0 29,0 39,0
200 81,0 95,0 60,0 66,0
250 75,0 93,0 58,0 63,0
300 50,0 67,0 38,0 51,0
400 25,0 30,0 20,0 28,0
500 0,0 20,0 21,0 26,0
600 0,0 8,0 12,0 16,0
Осушка силикагелями
309
ле дегидратации при 200 °C динамическая активность обоих об-
разцов силикагелей была максимальной.
Дальнейшее повышение температуры термообработки при-
водило к значительному снижению динамической активности.
Дегидроксилирование (отщепление структурной влаги) в ре-
зультате чрезмерного нагрева отражается на адсорбционных
свойствах силикагелей не только по парам воды, но и по дру-
гим веществам, молекулы которых характеризуются наличием
дипольного или квадрупольного моментов, например по диокси-
ду углерода [73]. Дегидроксилирование приводит также к из-
менению избирательности адсорбции смесей. Исследование ад-
сорбции смесей аргон — кислород и азот — оксид углерода по-
казало [74], что преимущественное поглощение кислорода и ок-
сида углерода менее резко выражено на дегидроксилированных
образцах.
Закоксовывание. При осушке газов, содержащих полимери-
зующиеся компоненты (например, продуктов крекинга нефти,
пиролиза этана и пропана), силикагель также, как и другие не-
органические адсорбенты, в процессе эксплуатации теряет ак-
тивность в результате отложения углеродистого осадка.
Обычно осадок выжигают в потоке кислородсодержащего
таза. Но закоксовывание можно и предотвратить. Для этого в
поток осушенного газа предложено вводить ароматический или
ароматизированный растворитель: бензол, толуол, ксилол, кре-
кинг-бензин. При термической десорбции растворитель вместе с
растворенным осадком удаляется с газом десорбции, выделя-
ется из газового потока сепарацией и возвращается в процесс
[75].
На рис. 12,15 представлена схема установки. Газ, содержащий полиме-
ризующиеся компоненты, проходит первичный сепаратор для разделения
жидкой и газообразной фаз и адсорбер, где осушается. При этом в газопро-
вод вводят растворитель. Избыток растворителя вместе с растворенным осад-
ком удаляется из первичного сепаратора снизу. Периодически адсорбер пе-
реключают иа регенерацию. При регенера-
ции горючий газ поступает в адсорбер,
проходит снизу вверх слой адсорбента и
направляется во вторичный сепаратор, где
мз него выделяется растворитель, который
возвращается в процесс.
Осушка газов под высоким
давлением
Ряд современных химических про-
цессов проводят при высоком дав-
лении. Содержание водяных паров
Фис, 12,15. Схема установки осушки с добавкой
растворителя образующегося кокса:
I, 2 — сепараторы; 3 —адсорбер.
Растворитель
310 12. Осушка газов и органических жидкостей
Рис. 12,16. Влагосодержание природно-
го газа Wr в равновесии с жидкой фа-
зой.
в газах, находящихся в рав-
новесии с жидкой водой,
при высоких давлениях дол-
жно рассчитываться с уче-
том сжимаемости. Обычно
для этих целей пользуются
графиками. Такие графики
для природного газа пред-
ставлены на рис. 12,16; они
позволяют определить вла-
госодержание в газе при
насыщении для температур
от минус 60 до 350 °C и
давлений от 0,1 до 70 МПа.
При повышении давле-
ния избыток влаги конден-
сируется, абсолютная влаж-
ность, отнесенная к единице объема газа при нормальных ус-
ловиях, снижается, вследствие чего уменьшается удельный рас-
ход адсорбента на осушку.
При исследовании равновесной и динамической характеристик процесса
осушки газов особое внимание должно быть уделено поддержанию постоян-
ного относительного содержания паров воды в исходном газе. Это- можно
обеспечить. следующим образом [76]: будем дросселировать газ, сжатый до
давления рОбщ i н полностью насыщенный парами воды, от давления рОбщ i
до рОбщ 2. Тогда относительное влагосодержание в расширенном газе соста-
вит р/р,=рОбщ i/робщ 1. Меняя Робщ г, можно провести опыты при разных
р/ра, на одном и том же общем давлении рОбщ 2.
Рис. К, 17. Схема установки для изучения процесса осушки газов под высоким давле-
нием:
1 —магнитный пускатель; 2 — компрессор; 3 — маслоотделитель; 4 — реле; 5 — маномет-
ры; 6 —буферная емкость; 7, 10—фильтры; 8 —контактный манометр; 9 — регулятор;
11 — адсорбер; 12 — ротаметр.
Осушка силикагелями
311
рис. 12,18. Пример многоступенчатой выходной
(Кривой.
Схема установки для изучения ха-
рактеристик процесса сушки под высо-
ким давлением приведена на рис. 12,17.
Воздух из компрессора 1 под давлением
Робщ । проходит через маслоотделитель'
3 и фильтр 7, где очищается от капель-
ной влаги и масла. Давление воздуха
поддерживают постоянным с помощью
электрбконтактного манометра 8. Далее воздух дросселируют до рабочего
давления рОбщ 2 и с постоянной скоростью подают в адсорбер высокого дав-
ления 11 —• термостатированный сосуд из нержавеющей стали.. Приблизитель-
ные размеры адсорбера: диаметр 10 мм, высота 150 мм.
Расход газа и его рабочее давление рОбщ •>. регулируют пневматическим
прибором мембранного типа 9. Изменения концентрации водяных паров за
слоем адсорбента в течение всего опыта непрерывно регистрируются куло-
нометрическим измерителем влажности. В процессе регенерации слой адсор-
бента продувают сухим азотом (влажность по точке росы минус 70 °C). Тем-
пературу регенерации выбирают в зависимости от типа адсорбента; в случае
силикагеля она составляет 200 °C.
После насыщения адсорбента при определенном парциальном давлении
водяных паров опыт продолжают при более высоком содержании паров
воды в воздухе, которое достигают, изменив ровщ i. Непрерывная регистра-
ция изменения концентрации водяных паров за слоем адсорбента позволяет
построить многоступенчатую выходную кривую (рис. 12,18), интегрированием
которой определяют равновесную активность адсорбента при различных со-
держаниях водяных паров.
На рис. 12,19 приведены изотермы адсорбции паров воды,
поглощаемых силикагелем из воздуха при высоким давлении.
С повышением давления крутизна изотерм ’ последовательно
уменьшается. Величина адсорбционной способности при посто-
Рис. 12,20. Зависимость адсорбционной способности мелкопористОгр силикагеля К С М-6
и цеолита NaA по парам воды от давления в системе при относительной влажности
<р*е0,95. ...
312 12. Осушка газов и органических жидкостей
янной относительной влажности с повышением давления растет,,
проходит через максимум (а; 6,0 МПа), после чего снижается,
(рис. 12,20). Этот максимум отмечен и у других типов твердых;
осушителей, например цеолитов.
Степень осушки при проведении процесса под высоким дав-
лением соответствует точке росы ниже минус 70 °C (после дрос-
селирования).
Если процесс осушки проводят под повышенным давлени-
ем, а влажность газа регистрируют прибором по точке росы по-
сле снижения давления до атмосферного, то найти действитель-
ную точку росы газа в условиях повышенного давления можно,,
пользуясь номограммой (рис. 12,21).
Рис. 12,21. Номограмма для определения точки росы газа под повышенным давлением
по показателям прибора, работающего иод атмосферным давлением»
. Осушка силикагелями
313
Она построена на допущении, что концентрация паров воды в газе, вы-
раженная в объемных процентах или част, на 1 млн. (об.), вне зависимости
от давления остается постоянной. Пусть процесс осушкн проводят примерно
прн 1 МПа, а точка росы, отмеченная прибором прн нормальном давлении,
составляет минус 48 °C. Соединяя значения на шкалах «Точка росы» (минус
48 °C) и «Давление» (О,Г МПа) иа пересечении со шкалой «Влажность», по-
лучим значение 40 част, на 1 млн. Проведя теперь через эту точку примую,
исходящую из 1 МПа на осн давления, на шкале «Точка росы» получим
—27 °C, что и соответствует влажности под давлением.
Принципиальные схемы осушающих установок
В большинстве адсорбционных осушающих установок насыщен-
ный водой адсорбент регенерируют горячим газом. Если пра-
вильно установлена температура слоя в конце регенерации, тер-
мический метод обеспечивает высокую степень десорбции. Для
(регенерации применяют либо часть осушаемого (или осушен-
ного) газа, либо инертный газ со стороны.
В соответствии с системой регенерации установки осушки
газа подразделяют на установки с открытым и закрытым цик-
лом. В установках с открытым циклом газ на стадии регенера-
ции однократно проходит через охлаждаемый и (или) нагре-
ваемый адсорбер, после чего удаляется из системы или при-
мешивается к исходному газу. В установках другого типа —
<с закрытым циклом—десорбцию влаги проводят при пропуска-
нии через адсорбент горячего газа, циркулирующего в замкну-
том контуре с помощью газодувки. В технологической нитке
циркуляции газ охлаждают, при этом из него выделяется вла-
га, затем его снова нагревают и возвращают в адсорбер, нахо-
дящийся на стадии регенерации.
Основные принципиальные схемы осушки газа рассмотрены в
работе [77].
В схеме с открытым циклом (рис. 12,22) осушку газа ведут в адсорбе-
ре 2, часть исходного газа отбирают и пропускают последовательно через
адсорбер 4, находящийся на стадии охлаждения, а затем после нагрева —
через адсорбер 5, в котором производится десорбция влаги. После охлажде-
ния газа регенерации и выделения из него в сепараторе 1 влаги поток при-
мешивают к исходному газу.
В другой схеме (рис. 12,23) охлаждение адсорбера 2 ведут частью осу-
шенного газа. Выходящий из адсорбера 4 поток соединяют с основным по-
током осушенного газа. Схема потоков и оборудование на стадии нагрева
адсорбера 5 не отличаются от описанных выше. Для установок с открытым
циклом характерны сравнительно высокие степени осушки.
На рис. 12,24 представлена схема с закрытым циклом на стадии реге-
нерации (адсорбер 4). Часть осушенного газа используют для охлаждения
адсорбера 2. Капиталовложения и эксплуатационные расходы в схемах с за-
крытым циклом выше, но работают они очень стабильно. Обычно система
переключения клапанов на осушающих установках автоматизирована и ис-
пользование ручного труда сведено к минимуму.
Продолжительность рабочего цикла определяет число ад-
сорберов, которые должны быть включены в схему, чтобы обес-
314 12. Осушка газов и органических жидкостей
Рис. 12,22. Схема установки осушки газа с открытым циклом при последовательности
стадий охлаждение — нагрев:
1 — сепаратор; 2 — адсорбер на стадии осушки; 3 — холодильник; 4 — десорбер на стадии
охлаждения; 5 — адсорбер на стадии регенерации; 6 — теплообменник; 7 — нагреватель.
Рис. 12,23. Схема установки осушки газа с открытым циклом при охлаждении осушен-
ным газом:
1 — сепаратор; 2 — адсорбер на стадии осушки; 3 — холодильник; 4 — адсорбер на стадии
охлаждения; 5 — адсорбер на стадии регенерации; 6 — теплообменники; 7 — нагреватель.
печить непрерывность работы осушающей установки. Если про-
должительность стадии осушки 6 и более часов, число адсор-
беров в установке ограничивается двумя. Для более коротких
циклов необходимо, как было рассмотрено выше, включать три
адсорбера: один на стадии осушки, второй на стадии нагрева,
третий на стадии охлаждения.
В стадии десорбции влаги из силикагеля необходимо сле-
дить, чтобы температура в адсорбере была не выше 250 °C. По-
этому воздух или другой газ перед подачей в адсорбер нагре-
вают лишь до 150—250 °C. Как ясно из рис. 12,25, десорбция!
основной массы влаги из силикагеля происходит в течение пер-
вых 4 ч, причем температура выхо-
дящего газа остается постоянной
(55°С). Затем следует достаточно
резкий подъем температуры. Реге-
нерацию заканчищют, когда темпе-
ратура выходящего воздуха достиг-
нет 100 °C.
Затраты тепла на регенерацию
при условии хорошей изоляции ад-
Рис. 12,24. Схема установки осушки газа с закры-
тым циклом греющего газа:
1 — адсорбер на стадии осушки; 2 —- адсорбер на
стадии охлаждения; 3 — теплообменники; 4 — ад*
сорбер на стадии регенерации; 5 — холодильник;
6 — нагреватель; 7 — сепаратор; 8 — газодувка.
Осушка силикагелями
315
Рис. 12,25. Изменение температуры выходящего [ЭД
«з адсорбера воздуха при регенерации силика- _
геля. 120
< 80
сорбера составляют 6,7—8,3 МДж/ 40
/кг десорбированной влаги. Силика-
гель охлаждают сухим или влаж- 0
ным газом. В последнем случае ну- 7
жно обратить внимание на то, что-
бы направление потоков газа на стадиях охлаждения и осушки
совпадали.
Достижение высокой и стабильной степени осушки возмож-
но только при достаточной продолжительности и непрерывной
эксплуатации адсорбционной установки. В период остановки
влага вследствие диффузии накапливается в основной аппара-
туре и коммуникациях, что значительно ухудшает «сухость»
газа после повторного пуска. Особенно сильное увлажнение ап-
паратуры отмечается после периодических гидравлических испы-
таний, которые проводят с помощью воды.
При проектировании адсорбционной осушающей установки
нужно по возможности избегать излишнего «мертвого» объема
аппаратов, тупиковых участков, стремиться к сокращению ком-
муникаций, располагать адсорберы вблизи мест потребления су-
хого газа. Для изготовления адсорберов, других основных ап-
паратов и трубопроводов сухого газа лучшим материалом явля-
ется нержавеющая сталь.
Адсорбционные установки осушки
и отбензинивания газов
В последние годы в Советском Союзе и за рубежом для осушки
и отбензинивания газов газоконденсатных месторождений интен-
сивно разрабатываются адсорбционные процессы с коротким
циклом.
Принято считать, что первая установка такого типа построе-
на и пущена в эксплуатацию в США в 1957 г. компанией «Шелл
Ойл» [78]. В качестве адсорбента, как правило, в короткоцик-
ловых установках используют силикагель. Вначале силикагеле-
вые установки применяли только для осушки природного газа.
Однако в процессе эксплуатации было установлено, что наряду
с влагой происходит выделение углеводородов бензинового ря-
да, и осушающие установки стали переоборудовать в установ-
ки, совмещающие функции осушки и отбензинивания. При этом
возросло число компонентов, подлежащих извлечению, поэто-
му в условиях постоянных габаритов и загрузки адсорберов
должна быть сокращена продолжительность стадии периоди-
316
12. Осушка газов и органических жидкостей
ческого процесса. В известной мере этому факту короткоцикло-
вый процесс осушки и отбензинивания природного газа обязан
своим названием.
Короткоцикловые установки предназначены в основном для
переработки относительно небольших объемов газа: до 150—
700 тыс. м3/сут. Расчеты показывают, что верхний предел их
экономичного применения не превышает производительности
2 млн-м3/сут; для переработки больших объемов газа более эф-
фективно применять низкотемпературное разделение. Так как
диаметр адсорбера, работающего под давлением 5—7 МПа, не-
рационально увеличивать более, чем до 1,5—2,0 м, то в случае
относительно высокой производительности (например 2 млн.
м3/сут) применяют дублирование аппаратуры. Принципиальная
схема короткоцикловой двухадсорберной установки с открытым
циклом приведена на рис. 12,26.
Исходный газ проходит через сепаратор 2, где происходит отделение ка-
пельной фазы, и затем подвергается осушке и отбензиниванию в одном из
двух силикагелевых адсорберов 4 или 7. Выходящий из адсорбера газ после
теплообменника 5 поступает в газопровод. В это время силикагель во втором
адсорбере подвергается регенерации. Для этого часть газа отбирают от ос-
новного потока, нагревают в теплообменнике 5 до температуры около 250—
Рис. 12,26. Схема установки осушкн и отбензинивания природного газа:
I — нагреватель; 2 — первичный сепаратор; 3 — сепаратор; 4, 7—адсорберы; 5 — тепло-
обменник; 6 — воздушный холодильник.
Осушка силикагелями
317
300 °C и пропускают через адсорбер до тех пор, пока температура слоя не
достигнет 200 °C. В процессе нагрева происходит десорбция поглощенных
компонентов, которые вместе с отдувочным газом выходят снизу адсорбера
и охлаждаются до температуры, близкой к температуре окружающей среды.
Разделение смеси на влагу, конденсат углеводородов и газ идет в трех-
фазном сепараторе 3. Выходящий из сепаратора газ примешивают к основ-
ному потоку сырого газа, поступающего на установку короткоцикловой ад-
сорбции. После того как температура слоя достигнет 200 °C, газ направляют-
в адсорбер, находящийся на стадии регенерации, минуя нагреватель. В ре-
зультате пропуска холодного газа температура слоя силикагеля понижается
до 65 °C. После этого адсорберы 4 и 7 взаимно меняют назначение. Жидкие
углеводороды, выделившиеся в трехфазном сепараторе, стабилизируют. Об-
разующиеся при этом газы используют в качестве топлива, а конденсат от-
водят в хранилище, откуда он может отбираться на дальнейшее разделение
и переработку.
В двухадсорберной установке с закрытым циклом десорбцию и охлажде-
ние адсорбента ведут газом, выделяющимся в трехфазном сепараторе и цир-
кулирующем в замкнутом цикле с помощью компрессора. Срок окупаемости
короткоцикловых установок оценивается в 1,5 г.
Таким образом, схемы короткоцикловых установок очень-
просты. Продолжительность стадий адсорбции, десорбции и:
охлаждения составляет 30—60 мин и зависит от производитель-
ности установки по перерабатываемому газу, массы адсорбента-
и содержания в газе извлекаемых компонентов. Наиболее су-
щественным отличием адсорбционной стадии силикагелевых
короткоцикловых установок от давно известных и изученных:
углеадсорбционных установок периодического действия, функ-
ционировавших, например на газовых месторождениях Запад-
ной Украины, являются высокие массовые скорости газового
потока при проведении процесса адсорбции под высоким давле-
нием.
Так как эффективная и полная конденсация легких углево-
дородов в сепараторе может быть осуществлена только в при-
сутствии тяжелых компонентов, для уменьшения эффекта хро-
матографического разделения углеводородов в стадии десорб-
ции рекомендуется направление потока газа, противоположное,
направлению потока в стадии отбензинивания.
В одной из работ [79] приведены состав исходного газа и-
составы фракций, получающихся при эксплуатации короткоцик-
ловых установок. Анализ литературных данных об эксплуата-
ции установок, работающих по закрытому циклу, показывает,,
что короткоцикловые отбензинивающие установки обеспечивают
практически полное извлечение гексана и более высокомолеку-
лярных углеводородов, в меньшей степени извлекаются пен-
таны; пропан-бутановая фракция практически остается в сухом
газе. Суммарная степень извлечения фракции пентана и более
высокомолекулярных углеводородов достигает 70% и зависит
от состава исходного газа и длительности цикла, возрастая, как
правило, по мере его уменьшения. Одновременно с отбензини-
ванием происходит осушка газа.
318
12. Осушка газов и органических жидкостей
Рис. 12,27. Зависимость степени извлечения бута-
на и газового бензина ф' иа установке осушки и
отбензинивания природного газа с применением
активного угля (1) и силикагеля (2) от содержа-
ния адсорбированного компонента а.
Так как стадию извлечения в ко-
роткоцикловом процессе проводят
при высоком давлении (4—6 МПа),
адсорбционная емкость, независимо
от типа применяемого адсорбента и
крутизны изотерм адсорбции угле-
водородов, отрабатывается практи-
чески полностью, вследствие чего
степени извлечения углеводородов
от пентана и выше в адсорбере, за-
полненном, как силикагелем, так и
активным углем, близки. Преиму-
щество активного угля проявляется только в отношении пропа-
но-бутановой фракции. Это отчетливо видно из рис. 12,27, где
•степень извлечения углеводородов в промышленном адсорбере,
заполненном либо активным углем, либо силикателем, пред-
ставлена как функция количества адсорбированных углеводоро-
дов в жидком состоянии [80].
При нагрузке на адсорбент 3 л/кг степень извлечения бензи-
новых углеводородов в силикагелевом и в угольном адсорберах
практически одинакова (90—92%), в то время как извлечение
бутана углем в три раза выше, чем силикагелем. В связи с этим
для повышения степени извлечения пропано-бутановой фрак-
ции предложены схемы с одновременным использованием сили-
кагеля и активного угля [81].
Предприняты многочисленные попытки усовершенствовать
короткоцикловый процесс [82]. В одном из вариантов осушки
и отбензинивания природных газов методом периодической ад-
сорбции с тремя стадиями (адсорбция, десорбция, охлаждение)
достигнуто увеличение степени извлечения высших углеводо-
родов и значительно улучшен тепловой баланс. Для этого пе-
ред концом стадии адсорбции к перерабатываемому газу добав-
ляют поток горячего отбензиненного газа регенерации после то-
го, как из него в сепараторе выделены влага и углеводородный
конденсат. Благодаря этому уже в стадии адсорбции начина-
ется прогрев лобового слоя адсорбента (схема установки этого
типа приведена на рис. 12,28).
Сырой газ отбензинивают и осушают в адсорбере 1, после чего исполь-
зуют для охлаждения адсорбента в адсорбере 3. После охлаждения в тепло-
обменнике 5 холодным газом регенерации температура отбензиненного газа
перед поступлением в газопровод или подземное хранилище не превышаем
<55 °C. Регенерацию адсорбента в адсорбере 2 ведут газом, циркулирующим
Осушка силикагелями
319
с помощью компрессора 7. Конденсат из газа выделяют в сепараторе 9 по-
сле охлаждения в теплообменнике 6. Влагу и конденсированные углеводо-
роды удаляют из сборника 10. Часть жидких продуктов для улучшения эф-
фективности десорбции насосом 8 закачивают в циркуляционный газ. После
небольшого црдргрева в теплообменнике 5 циркуляционную смесь подогре-
вают в солядцй ванне 4 до 290—370 °C. Температура расплава 427 °C; тем-
пература выходящего из адсорбера потока в конце стадии десорбции 205—
220 °C.
Когда емкость адсорбента в адсорбере 1 близка к полной отработке, вы-
ходящий из адсорбера 2 Газ отключают от циркуляционной системы н при-
мешивают к потоку сырого газа перед его поступлением в адсорбер. Темпе-
ратура смешанного потока может изменяться в пределах 90—200 °C. благо-
даря этому уже на стадии адсорбции начинается прогрев лобового слоя ад-
сорбента в адсорбере 1. Пропуск газа прекращают, прежде чем начинается
десорбция углеводородов. После этого адсорбер 1 переключают на стадии
десорбции и охлаждения. Степень удаления адсорбата в конце стадии де-
сорбции достигает 95%, Одновременно с переключением потока, выходящего"
из адсорбера 2, прекращают закачку конденсата в циркуляционный газ; по-
следний по байпасу, минуя ванну 4 продолжает поступать в холодном со-
стоянии сверху в сдой адсорбента, охлаждает верхние слои и выравнивает
температуру по высоте адсорбера.
В короткоцикловых адсорбционных установках осушки и от-
бензинивания природных газов на стадии регенерации обычно
применяют нагретый природный газ. Недостатком метода явля-
ется несовершенство системы конденсации высших углеводоро-
дов из потока десорбента. Но в одном из вариантов [78] на-
грев адсорбента, насыщенного высшими углеводородами (С4+),.
проводят потоком пропана. Высшие углеводороды конденсируют
из пропана после его охлаждения.
Рис. 12,28. Схема усовершенствованной трехадсорберной установки осушки и отбензини-
вания природного газа:
1—3 — адсорберы; 4 — соляная ванна; 5 — теплообменник; 6—холодильник; 7— компрес-
сор; 8 — иасос; 9 —сепаратор; Ю — сборник конденсата.
320 12. Осушка газов и органических жидкостей
Установки короткоцикловой безнагревной
адсорбции
В случае, если изотерма адсорбтива на адсорбенте не предель-
но крута, открывается возможность для проведения стадии ре-
генерации без подвода тепла — только путем снижения давле-
ния и (или) продувки газа частью очищенного потока. Этот
принцип регенерации положен в основу короткоцикловых без-
нагревных адсорбционных установок, получивших широкое при-
менение в первую очередь для осушки газа. Принцип коротко-
цикловой безнагревной адсорбции на примере осушки воздуха
поясняет рис. 12,29 [84].
Поступающий сжатый воздух проходит через клапан 1а в адсорбер 2а,
где осушается в результате контакта с отрегенерированным адсорбентом.
Основная часть осушенного воздуха (сухой прямой поток) проходит через
обратный клапан За и выводится из установки потребителю. Другая часть
осушенного воздуха (сухой обратный поток) дросселируется с помощью вен-
тиля 4 до давления более низкого, чем давление при адсорбции (обычно до
атмосферного), проходит другой адсорбер, регенерируя находящийся в нем
адсорбент, и через клапан 56 в виде влажного обратного потока сбрасывает-
ся в атмосферу.
Спустя небольшой промежуток времени, не превышающий, как правило,
"нескольких минут, пневмо- и электроуправляемые клапаны серии «а» пере-
крываются, одновременно открываются клапаны серии «б» и адсорберы вза-
имно меняют назначение. Работу адсорбера в одной из стадий (адсорбции
или десорбции) называют полуциклом. Длительность цикла в установках рас-
сматриваемого типа равна удвоенной длительности любого из полуциклов.
Короткоцикловые установки безнагревной адсорбции отлича-
ются компактностью, вследствие чего их легко выполнять пере-
движными. Если осушке подвергать не воздух, а ценный газ,
потерь последнего можно избежать, компримируя обратный
поток и присоединяя его к газу, который направляется на осуш-
ку. Отсутствие затрат тепла на нагрев адсорбента является од-
ной из причин, обеспечивающих высокую экономическую эффек-
тивность установок рассматриваемого
типа. В то же время необходимо учи-
тывать, что непрерывная эксплуата-
ция короткоцикловых установок воз-
можна только при условии надежной
и быстродействующей системы управ-
ления потоками.
Установки короткоцикловой безна-
гревной осушки только спустя значи-
тельное время после пуска начинают
давать качественный продукт. На рис.
Рис. 12,29. Схема установки короткоцикловой без*
нагревной осушки:
1, 5 — автоматические клапаны; 2 — адсорберы; 3 —
обратные клапаны; 4 — дросселирующий вентиль.
Осушка силикагелями
321
Ряс. 12,30. Изменение влажности осушенного газа
в период выхода короткоцикловой безнагревной
адсорбционной установки на режим.
12,30 показана кривая выхода уста-
новки на режим. Как видно из гра-
фика, только через 5 сут после пу-
ска влагосодержание осушенного
воздуха стабилизировалось на уров-
не 1 част, на 1 млн. при влагосо-
держании исходного воздуха 4000
част, на 1 млн. Длительность выхо-
да установки на режим может быть, однако, существенно умень-
шена, если на десорбцию подавать весь воздух, вышедший из
адсорбера-осушителя.
В дальнейшем в схему был внесен ряд ценных усовершен-
ствований, рассмотренных в обзорах [85, 86]. Предложено, на-
пример, для уменьшения расхода воздуха, идущего на регене-
рацию, а также для более плавного подъема давления в адсор-
бере при его переключении со стадии регенерации на стадию
адсорбции, использовать дополнительную емкость. На рис. 12,31
показан один из вариантов подключения этой емкости.
Промежуточная емкость в стадии адсорбции имеет давле-
ние, равное давлению в адсорбере. Газ, заполняющий ее, слу-
жит вначале для регенерации слоя, а затем для плавного подъ-
ема давления при переходе от стадии регенерации к стадии ад-
сорбции. Плавный подъем давления уменьшает истирание ад-
сорбента вследствие гидравлических толчков и способствует уве-
личению степени осушки. Установка, изображенная на рис. 12,31,
послужила прототипом одноадсорберных установок безнагрев-
ной осушки.
Пределы производительности короткоцикловых установок
очень широки: от 2 до 3800 м3/ч. Газ осушают при давлении
0,4—1,0 МПа. Степень осушки сжатого газа соответствует точ-
ке росы от минус 40 до минус 90 °C и увеличивается с пониже-
нием температуры исходного воздуха и
ростом давления.
Применение установок короткоцикло-
вой безнагревной адсорбции не ограни-
чивается областью осушки газов. Их
успешно применяют, например, для очи-
стки циркулирующего водорода рифор-
минга [87]. В одном из вариантов про-
цесса очистку производят в двух адсор-
берах, в нижней части которых разме-
щен крупнопористый силикагель, в верх-
Рис. 12,31. Схема установки короткоцикловой безнагрев-
ной очистки с одним адсорбером (1) и рессивером (2).
21—1346
322 12. Осушка газов и органических жидкостей
ней — активный уголь. Силикагель эффективно и обратимо ад-
сорбирует тяжелые углеводороды, а активный уголь — углево-
дороды Ci—С4. При противоточной регенерации десорбируемые
из угля легкие углеводороды способствуют более полной отдув-
ке тяжелых углеводородов, поглощенных силикагелем. Чистота
полученного водорода выше 99%.
В более сложной схеме [88] очистку водорода предложено
проводить при давлении 1,3 МПа в девяти адсорберах: три ад-
сорбера в каждой из трех серий. Первые адсорберы заполнены
силикагелем, последующие — активным углем. В одной из се-
рий происходит адсорбция, в другой регенерация, в третьей
подготовка к переводу с регенерации на адсорбцию. При этом
с выходом 50% получают водород, содержащий менее 0,005%
примесей, а также дополнительно до 25% водорода чистотой
99,9%. В связи со сложностью установки управление переключе-
нием клапанов производят по программе, заложенной в вычис-
лительную машину.
Применение короткоцикловых установок для очистки и осуш-
ки продуктов сжигания природного газа с целью получения за-
щитных контролируемых атмосфер рассмотрено в разделе, по-
священном вопросам декарбонизации на цеолитах. Короткоцик-
ловые безнагревные установки применяют не только для осуш-
ки и очистки газов, но и для разделения двух- или трехкомпо-
нентных газовых смесей. Примером разделения двухкомпонент-
ных смесей является получение обогащенного кислородом воз-
духа с использованием в качестве адсорбера цеолита СаА или
NaX [89]. Цеолиты избирательно поглощают азот из воздуха.
Обогащенный кислородом воздух (30—75% О2) получают в ви-
де первичного потока. Процесс проводят при давлении 0,2—
0,6 МПа; длительность полуцикла составляет от 40 с до 2,5 мин.
Примерно половина выходящего из адсорбера потока расходу-
ется на регенерацию адсорбента. Одновременно с обогащением
происходит осушка газа и очистка его от диоксида углерода*.
* Перечисленные примеры лишь в малой степени отражают возможности
метода. Он применяется также для получения высокочистого водорода (до
99,999°/0), концентрирования гелия в гелийносных газах, очистки синтез-газа
в производстве аммиака, очистки метана от гомологов и др. Отметим, что-
все эти процессы осуществляются при комнатных температурах и часто на-
столько уникальны, что не имеют иных промышленных альтернатив.
Короткоцикловые безнагревные процессы реализуются по двум главным схе-
мам: фирмы «Эссо» и фирмы «Юнион Карбайд». В основном тексте описана
первая схема, в которой регенерацию адсорбента проводят частью произве-
денного продуктового газа. В установках «Юнион Карбайд» для регенерации
применяют газ, заполняющий свободный объем продуцирующего адсорбера.
Его отбирают, завершив в этом адсорбере стадию адсорбции, и направляют
в параллельный аппарат, регенерируемый при атмосферном давлении. Из-за
наличия переходных стадий (сброс давления, подъем давления) и ради эко-
номии неадсорбирующегося продуктового газа число адсорберов в установках
разделения равно 3—4. достигая иногда 10.
Осушка силикагелями
323
О возможностях безнагревного процесса говорят следующие цифры: круп-
нейшие установки по получению кислорода этим методом имеют производи-
тельность до 40 т]сут, производя газ с 90% кислорода; установки для очист-
ки синтез-газа производительностью 200 тыс. мДч удаляют пары воды, диок-
сид углерода, монооксид углерода и метан за одну стадию с полнотой, удов-
летворяющей такой тонкий каталитический процесс, каким является синтез
аммиака. Длительность цикла в некоторых процессах сокращена до 6 с.
(Драм, ред.)
Методика инженерного расчета короткоцикловых безнагрев-
ных установок осушки сжатого воздуха разработана в МХТИ
им. Д. И. Менделеева [90]. Основное уравнение для расчета
записывается следующим образом:
lg (с/со) = (12.4)
где с и Со — конечное и исходное влагосодержания воздуха; тг — время пре-
бывания газа в адсорбере, с; тпц — длительность полуцикла адсорбции (де-
сорбции), с; d — средний диаметр гранул адсорбента, м; ko — константа, не
зависящая от исходной концентрации влаги в потоке; ее значение определя-
ется температурой, давлением при адсорбции и коэффициентом избытка об-
ратного потока (соотношение объемных расходов воздуха при десорбции и
адсорбции).
Для осушки воздуха силикагелем при 0,4 МПа и 30 °C зна-
чения константы kQ в зависимости от коэффициента избытка
обратного потока К приведены ниже:
К 1,05 1,2 1,5 1,75 2,00 2,8
ko, м-c-0’6 9,7 13,8 18,9 23,2 25,9 34,0
Технико-экономический анализ короткоцикловой безнагрев-
ной осушки воздуха силикагелями показал, что приведенные за-
траты на единицу воздуха, осушенного этим методом, несколько
ниже, чем в обычном процессе. Основным преимуществом корот-
коцикловой адсорбции является более высокая производитель-
ность установок при сопоставимых габаритах и отсутствии вспо-
могательных устройств для нагрева регенерирующего воздуха*.
* Приводим перечень важнейших работ, в которых изложена теория корот-
коцикловых процессов очистки и разделения газовых потоков: Шумяц-
кий Ю. И., Бочавер К. 3., Кельцев Н. В., Торочешников Н. С. — ТОХТ, 1970,
т. 4, № 4, с. 496—502; Бочавер К. 3., Торочешников Н. С., Шумяцкий Ю. И.—
В сб.: «Нефтепереработка и нефтехимия». М., 1972, с. 210—226; Беккер Б. И.,
Торочешников Н. С., Шумяцкий Ю. И. — ТОХТ, 1973, т. 7, № 2, с. 279—282
и № 3, с. 359—364; Устинов Е. А., Акулов А. К. — ЖПХ, 1979, т. 52, № 10,
с. 2291—2295; № 11, с. 2532—2536; 1981, т. 54, № 8, с. 1890—1894;
Gelbin D., Bunke G. — «Chem. Eng. J.», 1979, v. 17, № 3, p. 191—199; Усти-
нов E. A. — В сб.: «Получение, структура и свойства адсорбентов». Л., 1980,
с. 141—151; Акулов Д. К., Устинов Е. А., Головко Г. А., Игнатов Ю. Я.—
ЖПХ, 1980, т. 53, № 9, с. 2007—2011; Laszlo Aranji et al.—Magj. Kern, lapja,
1981, v. 36, № 1, p. 39—44; Нагаев В. В. и др. — ТОХТ, 1980, т. 14, № 6,
с. 819—824 и 1981, т. 15, № 1, с. 31—35. (Прим, ред.)
21’
324
12. Осушка газов и органических жидкостей
Рис. 12,32. Влияние структуры силикагеля (сред-
него радиуса пор гср) на его адсорбционную спо-
собность при очистке тяжелого масла.
Осушка, очистка и осветление масел
Очистка, осветление и регенерация
масел являются, наряду с осушкой,
одной из наиболее значительных
областей применения силикагелей
как адсорбентов [91]. Для этих
процессов большое значение имеет выбор оптимальной струк-
туры силикагеля. Влияние пористой структуры силикагелей на
эффективность процесса продемонстрирована на примере глу-
бокой очистки тяжелого масла — деасфальтированного гудрона
туймазинской нефти.
Деасфальтированный гудрон, растворенный в алкилате в отношении 1 :
: 3, пропускали через навеску адсорбента 10 г, помещенную в лабораторный
адсорбер диаметром 25 мм. Расход гудрона 12 см3/ч, температура в адсор-
бере соответствовала комнатной. Активность адсорбента оценивали по выхо-
ду продукта с заданным коэффициентом рефракции, а осветляющую способ-
ность — по выходу бесцветного рафината. Результаты опытов отражает рис.
12,32. Активность испытуемого силикагеля а выражена в относительных про-
центах к активности эталонного сорбента — стандартного шарикового катали-
затора крекинга.
Из приведенных данных вытекает, что оптимальными ад-
сорбционными свойствами обладают силикагели со средним ра-
диусом пор 2,5—3,0 нм.
Осушка цеолитами
Цеолиты как средство осушки
Цеолиты — уникальное средство осушки. Адсорбция воды на
цеолитах имеет ряд характерных особенностей. На рис. 12,33
представлены изотермы адсорбции паров воды на цеолитах об-
Таблица 12-3. Адсорбционная способность (в г/100 г)
гранулированных цеолитов NaA при малых давлениях
Давление Температура, °C
Па мм рт. ст. 0 25 50 100 150 200 300 350
10-3 10-3 0 0 00 000 0 10-2 ю-4 2,0 1,6 0,4 0 0 0 0 0 10-1 IO"3 4,8 3,2 2,0 1,0 0,4 0 0 0 10° IO-2 14,0 6,0 3,8 3,0 2,2 1,8 1,0 0,2 Ю1 10-1 18,0 15,0 8,0 3,6 3,2 2,0 1,8 0,4 Ю2 10° 18,0 18,0 16,0 6,0 3,6 3,0 2,4 1,0
CcytuKa цеолитами 325
р,шл рт.ст.
Рис. 12,33. Изотермы адсорбции паров воды
NaA (б).
р,мм рт.ст.
на гранулированных цеолитах NaX (а) н
щего назначения различных типов. Ранее приводились изотер-
мы на цеолите СаА (см. стр. 30). Для всех цеолитов характе-
рен очень крутой подъем изотерм в области малых концентраций
паров воды. Адсорбционная способность цеолитов при обычных
температурах уже при давлении 130—260 Па (1—2 мм рт. ст.)
близка к адсорбционной способности при максимальном насы-
щении. Даже в области очень малых давлений (табл. 12-3) цео-
лит NaA так же как и другие формы цеолитов, проявляет зна-
чительную адсорбционную способность по воде. Изотермы ад-
сорбции паров воды на цеолите СаА при малых давлениях пред-
ставлены на рис. 12,34.
Преимущество цеолитов всех типов при низких давлениях
адсорбтива перед другими адсорбентами отчетливо видно из
данных табл. 12-4, где адсорбционная способность цеолита NaX
сопоставлена с адсорбционной способностью силикагеля и окси-
да алюминия [92]. Это сравнение указывает на перспективность
использования цеолитов для осушки газов с невысоким содер-
жанием водяных паров.
Рис. 12,34. Изотермы адсорбции паров воды в области малых давлений на цеолите СаА.
326 12. Осушка газов и органических жидкостей
Таблица 12-4. Адсорбционная способность (в г/100 г)
промышленных адсорбентов по парам воды при 25 °C
Адсорбент Давление, Па (мм рт. ст.)
10-1 (10-3) 100 (10-2) 101 (Ю-1) 102 (100) юз (101)
Цеолит NaX 3,5 9,0 18,0 20,0 25,0
Оксид алюминия 1,5 2,0 3,0 5,0 14,0
Силикагель мелкопори- стый 0,2 0,4 1,2 5,0 25,0
Второй отличительной особенностью адсорбции паров воды
на цеолитах является то, что адсорбционная способность велика
даже при повышенных температурах. При 100 °C и давлении па-
ров воды 10 кПа для цеолитов она достигает 15—16 г/100 г;
даже при 200°C она еще значительна (4 г/100 г). В этой обла-
сти температур адсорбционная способность силикагелей и окси-
да алюминия практически равна нулю [92]. Возможность прове-
дения процесса осушки при высоких температурах особенно
важна в тех промышленных установках, где технологический
газ, выводимый из зоны высоких температур, должен быть воз-
вращен снова в реакционную зону.
Замена обычных сорбентов цеолитами позволяет в ряде слу-
чаев (например, при осушке природного газа на промыслах и
газоперерабатывающих заводах) избежать стадии охлаждения
осушаемого газа, что приводит к значительному сокращению
энергозатрат и упрощению схемы. Поскольку адсорбционная
способность цеолитов при повышенных температурах существен-
на, тепло, выделяющееся в процессе поглощения паров воды, не
оказывает значительного влияния на активность сорбента. По-
этому конструкция адсорберов с цеолитами может быть пре-
дельно проста (без охлаждающих змеевиков внутри аппарата),
и процесс осушки проведен в адиабатических условиях. Малая
чувствительность адсорбционной способности цеолитов к темпе-
ратуре позволяет уменьшить время охлаждения адсорбента пос-
ле его регенерации, в результате чего рабочий цикл осушающей
установки сокращается, и, следовательно, увеличивается ее про-
изводительность.
Цеолиты отличаются очень большой скоростью поглощения
влаги. Высокая степень осушки поддерживается практически в
течение всей стадии. Повышение влагосодержания в конце ста-
дии наступает не постепенно, как это наблюдается в случае при-
менения других твердых поглотителей, а резко и быстро. Вслед-
ствие такого характера выходных кривых удается почти полно-
стью отработать адсорбционную емкость слоя. Динамическая
Осушка цеолитами
327
Рис. 12,35. Зависимость остаточной влажности
цеолита Wa от условий регенерации (точка росы
продувочного газа изменяется в пределах от 20
до минус 40 °C).
активность даже относительно не-
большого слоя цеолитов близка к
равновесной статической активно-
сти. Разница между ётими величи-
нами в цеолитовых адсорберах не
превышает 10—15%.
Благодаря высокой скорости по-
глощения воды адсорбция проходит
в работающем слое небольшой вы-
соты (обычно меньше 10 см). Сле-
довательно, появляется возможность
конструировать более компакт-
ные установки. Цеолиты позволяют
скоростях газового потока и тем самым значительно интенсифи-
цировать процесс. Так, изменение скорости газа от 4 до 15
м/мин (что соответствует скоростям, применяемым в промыш-
ленных адсорберах) практически не приводит к снижению ак-
тивности. Даже при скорости, приближающейся к 20—25 м/мин,
слой сорбента высотой 60 см гарантирует осушку газа до точки
росы ниже минус 65 °C при адсорбционной способности 15 г/100
Г. В осушающих установках с цеолитами вполне допустимы ско-
рости до 30 м/мин, причем предел устанавливается скорее уно-
сом адсорбента и гидравлическим сопротивлением, чем непо-
средственно сорбционным процессом.
Цеолиты обеспечивают самую
высокую степень осушки: точка ро-
сы осушенного газа может дости-
гать минус 80 °C и даже ниже. Цео-
литы жадно поглощают влагу, но
они трудно отдают ее при регене-
рации. В промышленных адсорбе-
рах регенерацию адсорбента, как
правило, проводят продувкой слоя
горячим газом. Остаточное содер-
жание влаги в цеолите после реге-
нерации определяется температурой
слоя в конце стадии и влажностью
продувочного газа. На рис. 12,35
приведены кривые, характеризую-
щие остаточную влажность цеолита
Рис. 12,36. Зависимость степени осушки газа (точ-
ка росы /р) от условий регенерации (остаточная
влажность Ц7а) и температуры иа стадии осушки.
вести осушку при высоких
328
12. Осушка газов и органических жидкостей
в случае продувки газом, имеющим точку росы 20,0; минус 20,0
и минус 40,0 °C [92].
Условия регенерации цеолитов обычно выбирают по графи-
кам, представленным на рис. 12,36 [93]. Они позволяют опре-
делить остаточную влажность сорбента после продувки газом
с заданным влагосодержанием, а затем по остаточной влажно-
сти установить минимальную точку росы, которая может быть
получена в последующей стадии. Так, после продувки при 200 °C
газом, имеющим точку росы минус 10 °C, остаточная влажность
цеолитов составляет 3% (масс.). Такой сорбент после охлажде-
ния до 40 °C обеспечит осушку, соответствующую точке росы
минус 70 °C. Если регенерация проведена не полностью, и оста-
точная влажность составляет 10% (например, при 150°С га-
зом, имеющим точку росы 20 °C), точка росы осушенного газа
понизится только до минус 40 °C.
Скорость процесса регенерации цеолитов в промышленном
адсорбере определяется в основном скоростью разогрева ад-
сорбента, причем выделение основного количества влаги при
заданной конечной температуре происходит за относительно ко-
роткий промежуток времени. В связи с этим расчет процесса ре-
генерации должен сводиться к теплотехническому расчету нагре-
ва гранулированного адсорбента тем или иным теплоносителем
до заданной температуры с учетом количества влаги, удаляемой
в процессе регенерации*. Условия регенерации (температура,
влажность продувочного газа) выбирают в соответствии с рав-
новесными графиками, представленными на рис. 12,37, в зави-
симости от производственных условий.
Слой сорбента на установках осушки цеолитами может быть
нагрет не только продувкой горячим газом, но и с помощью
вмонтированных внутрь аппарата греющих труб, электрических
нагревательных элементов и т. п. Однако и в случае нагрева че-
рез стенку для полноты регенерации и отдувки паров влаги из
свободного объема адсорбера необходима продувка сорбента,
например, достаточно сухим азотом или воздухом.
На рис. 12,37 приведена схема газовых потоков в рассматри-
ваемых двух вариантах. При регенерации цеолитов горячим азо-
том продувку целесообразно вести в направлении, обратном на-
правлению газового потока в стадии осушки, во избежание сни-
жения степени осушки в первый период после подключения ад-
сорбера на рабочую стадию. Это может произойти, поскольку
* Основное количество воды из цеолита, действительно, удаляется сравни-
тельно быстро, но десорбция остаточных количеств ее связана с весьма про-
должительным выдерживанием адсорбента при повышенных температурах.
Так как часто удаление именно этой прочносвязанной воды определяет эф-
фективность цеолитов в разнообразных процессах, продолжительность их
термической регенерации приходится определять в специальных опытах.
(Прим, ред.)
Осушка цеолитами
329
Рис. 12,37. Варианты (а, б) регенерации цеолитов,-
1 — нагреватель; 2 — адсорбер.
остаточная влага в период регене-
рации концентрируется в последних
слоях сорбента по направлению
движения газа.
Наоборот, при равномерном обо-
греве с помощью, например, элект-
рообмотки и продувке холодным ре-
генерирующим газом применять
противоток нежелательно, так как
в этом случае наиболее ответствен-
и, следовательно, регене-
ный лобовой слой сорбента охлажден
рируется неполностью. Этот слой при осушке становится кон-
цевым, и газ, проходя через него, увлажняется. При прямотоке
же в этом случае недостаточно осушенный газ будет доосушен
последующими слоями.
Термическая устойчивость цеолитов может быть установлена
на основе данных термогравиметрического анализа, который
позволяет произвести оценку порога термической стабильности
одновременно с определением влагоемкости образца и некото-
рых других характеристик. В частности, на венгерском прибо-
ре— дериватографе одновременно фиксируется четыре перемен-
ные, изменяющиеся во времени: температура образца (Т), раз-
ность температур образца и эталона (ДТА), масса (ТГ) и ско-
рость ее изменения (ДТГ).
На рис. 12,38 представлены кривые термогравиметрического
анализа гранулированного цеолита NaA. Кривые показывают,
что максимум дифференциального эндотермического теплового
эффекта, а также резкая потеря массы за счет десорбции влаги
из цеолитов наблюдаются в области 270—290 °C. Практически
полное удаление влаги из цеолитов в отсутствии продувки дости-
гается при 500—600°C. Хотя при однократном нагреве экзотер-
мические эффекты на термограммах свидетельствуют о наруше-
нии кристаллической структуры искусственных цеолитов только
при 850 °C, высшим температурным пределом, при котором в
процессе продолжительной эксплуатации не отмечается сниже-
ния сорбционных свойств цеолитов, следует считать 550—
600 °C.
При гидратации и дегидратации размеры гранул цеолитов
несколько изменяются [94]. По наблюдениям польских инжене-
ров [95], в процессе многоцикловой эксплуатации механическая
прочность гранул цеолитов, используемых в качестве осушителя,
понижается, если содержание влаги в цеолитах превышает 15%.
Обобщая опыт применения цеолитов для осушки газов, мож-
но сделать вывод, что они обладают рядом преимуществ перед
h
333 12. Осушка газов и органических жидкостей
Рис. 12,38. Кривые термогравиметрического анализа
гранулированного цеолита.
другими осушителями: высокой погло-
тительной способностью в области ма-
лых парциальных давлений паров во-
ды, возможностью осушки при высо-
ких температурах, высокой и стабиль-
ной степенью осушки, достаточно хо-
рошими механическими свойствами.
Учитывая относительную трудность
регенерации цеолитов, основную осуш-
ку газов (например, природных) со
значительным содержанием водяных
паров иногда целесообразно прово-
дить обычным, более дешевым и лег-
че регенерируемым жидким или твер-
дым сорбентом, а глубокую доосуш-
ку — цеолитами.
Осушка в газовой промышленности
Газовая промышленность является крупнейшим потребителем
цеолитов. В значительной мере широкому использованию цеоли-
тов в этой области способствовали повышенные требования к
степени осушки газа, которые были введены в связи с внедре-
нием криогенных систем для разделения углеводородных газов.
При разделении природный газ охлаждают до минус 160 °C, на
гелиевых заводах — до минус 170 °C, Ни гликолевый метод, ни
применение обычных твердых осушителей не гарантирует глу-
бины осушки, обеспечивающей продолжительную непрерывную
эксплуатацию аппаратуры разделения в этих условиях. Решение
задачи стало возможным только после применения цеолитов.
Особое значение цеолиты приобретают в связи с открытием
мощных газовых месторождений на Крайнем Севере, где в ус-
ловиях вечной мерзлоты заглубление газопроводов не представ-
ляется возможным и газ должен транспортироваться на поверх-
ности, подвергаясь интенсивному охлаждению. Успешное реше-
ние проблемы наземного транспорта в холодном климате воз-
можно также при условии глубокой, надежной осушки транс-
портируемого газа.
Нами была проведена серия опытов на Ставропольском при-
родном газе с некоторым содержанием высших углеводородов.
Полученные результаты показали хорошую сходимость с данны-
ми по осушке воздуха на том же адсорбенте (цеолит NaA). Так,
при 50 °C и скорости газового потока 5 м/мин была достигнута
степень осушки, соответствующая точке росы ниже минус 70 °C
при активности сорбента 17 г/100 г. Селективность адсорбции
Осушка цеолитами
331
цеолитов по отношению к парам воды настолько ярко выраже-
на, что присутствие ряда компонентов практически не влияет
на характер извлечения влаги. Высшие же углеводороды не
проникают в мелкую структуру пор цеолитов NaA. Тем самым
исключается дезактивация, которая наблюдается на обычных
твердых осушителях. Поэтому срок службы цеолитов NaA зна-
чительно выше, чем обычных адсорбентов.
В то же время при осушке природного газа цеолитами типа
NaX с входными окнами более крупных размеров отмечается
некоторое уменьшение поглотительной способности и скорости
сорбции паров воды вследствие предадсорбции высокомолеку-
лярных углеводородов. В связи с этим в подавляющем большин-
стве случаев для осушки природных и других промышленных
газов применяют цеолит NaA.
Сравнительная оценка эффективности применения цеолитов
и оксида алюминия была проведена на польском природном га-
зе ;[96]. Для испытаний в схему были включены два адсорбера
емкостью по 10 л каждый. В один адсорбер было загружено
8 кг оксида алюминия, в другой 7 кг цеолита NaA. Процесс
осушки вели одновременно в параллельно включенных адсорбе-
рах при одинаковых скоростях, давлениях и температурах. Де-
сорбцию проводили после нагрева адсорбента в течение 4—5 ч
внешним теплоносителем с одновременной продувкой неболь-
шого количества горячего воздуха.
Осушке были подвергнуты отбензиненный на углеадсорбци-
онной установке природный газ и три сырых газа из различных
скважин, один из которых содержал 0,3% кислых примесей:
диоксида углерода и сульфида водорода. В цеолитовом адсорбе-
ре совместно с водой поглощалось в 10 раз меньше бензина, чем
в адсорбере с оксидом алюминия. Бензин, десорбированный из
цеолита, имел более высокие границы выкипания: на инертной
поверхности вторичных пор цеолита NaA происходит концентри-
рование только высокомолекулярных углеводородов. Цеолит во
всех случаях обеспечивал большую глубину осушки при более
высоко^ влагоемкости. Объем осушенного до точки росы минус
5 °C газа составлял 30 тыс. м3/т для оксида алюминия и
100 тыс. м3/т для цеолита.
Испытание процесса осушки природного газа цеолитами NaA
(без связующего), выполненное в промысловых условиях <[97]
на установке производительностью по газу 2000 м3/ч при давле-
нии 5 МПа и температуре исходного насыщенного водой газа
23—30 °C, позволило установить, что точка росы осушенного га-
за составляла около минус 50 °C, а влагоемкость цеолита (13—
15) г/100 г. Для регенерации был использован сухой газ (точ-
ка росы минус 35°C), при температуре (на входе в адсорбер)
300 °C под давлением 5 МПа; удельный расход газа регенера-
ции был равен 2,7—2,8 м3/ч на 1 л цеолита, а продолжитель-
332
12. Осушка газов и органических жидкостей
ность регенерации 13—14 ч. В этих условиях остаточная влаж-
ность цеолита была в пределах 3,4—3,7 г/100 г.
На адсорбционную установку газ может поступать с различ-
ной относительной влажностью. На Миннибаевском газопере-
рабатывающем заводе газ подвергают предварительной осушке
и очистке смесью диэтиленгликоля (ДЭГ) и моноэтанол амин а
(МЭА); в этом случае после абсорбционного контактора точка
росы газа около минус 10 °C. Нередко на газоперерабатываю-
щих заводах применяют двухслойную шихту: основное количе-
ство влаги удаляется в первом слое — силикагеле, остаток вла-
ги поглощается вторым слоем —цеолитом.
Целесообразность использования того или иного типа адсор-
бента либо шихты находится в зависимости от условий прове-
дения процесса, в первую очередь от влажности поступающего
на адсорбционную установку газа и температурного режима
осушки. Чтобы выяснить эту зависимость в МХТИ им. Д. И. Мен-
делеева были проведены испытания на стендовой установке
[98].
Скорость потока газа составляла 0,25 м/с. Первая серия опытов была
проведена на шариковом силикагеле. К.М.С-6 с диаметром сфер 2 мм, вто-
рая— на синтетическом цеолите NaA, гранулы которого имели диаметр и
высоту тоже 2 мм. Осушали газ различного влагосодержання (—10, —1,
+ 10 °C по точке росы) при трех различных температурах слоя 25, 50 и
75 °C. Момент «проскока» фиксировали, когда точка росы выходящего из ад-
сорбера газа достигала минус 40 °C.
Показатели процесса осушки газа в слое мелкопористого силикагеля
КСМ-6 и цеолита NaA при высоте слоя адсорбента 24 см представлены в
табл. 12-5.
Равновесная адсорбционная способность силикагеля, как и
следовало ожидать, только в одном случае превосходит соответ-
ствующий показатель для цеолита: при осушке газа с высоким
влагосодержанием (10 °C по точке росы) при относительно низ-
ких температурах (25 °C).
Таблица 12-5. Осушка газа силикагелем и цеолитом
Влагосодер- жаиие исход- ного газа по точке ро- сы, °C Темпера- тура осушки, °C Силикагель Цеолит
.Д’ г/100 г пр> г/100 г 10, см U, см/мии аД’ г/100 г аР’ г/100 г Lo, см и, см/мии
10 25 8,6 21,1 51,2 0,172 13,6 20,0 5,4 0,128 50 4,8 15,2 32,8 0,267 12,9 19,8 7,7 0,140 75 3,6 14,3 51,2 0,400 12,1 19,4 14,6 0,147 — 1 25 6,8 13,0 17,5 0,120 13,6 16,5 4,3 0,065 50 5,1 11,4 24,3 0,155 13,2 16,2 5,8 0,067 75 1,8 7,2 46,5 0,343 10,6 15,4 10,2 0,082 —10 25 5,9 9,4 10,3 0,055 12,0 14,4 4,2 0,033 50 4,8 7,5 12,4 0,084 11,7 14,1 4,8 0,035 75 1,7 4,6 26,7 0,178 8,4 12,3 9,9 - 0,045
Осушка цеолитами 333
Таблица 12-6. Время защитного действия (в мин)
При 25 °C | При 75 “С
влагосодержаиие исходного газа по точке росы, °C
15 10 —1 -10 15 10 -1 -10
Доля цеоли-
та NaA
в шихте, %
100 121 168 353 680 90 150 275 480
75 136 195 330 602 74 105 237 339
67 139 198 317 587 60 100 191 313
55 128 199 298 554 50 90 158 280
47 115 186 — — — — — —
25 93 150 200 450 29 63 80 137
0 42 102 145 322 14 42 45 100
Результаты расчета динамики процесса осушки газа сили-
кагелем, приведенные ранее, показали, что даже при бесконеч-
но большом слое силикагеля степень использования его адсорб-
ционной емкости при осушке газа с высокой относительной
влажностью не может превышать 75%. Вследствие этого во всех
режимах показатели осушки газа слоем цеолита предпочтитель-
ней, чем при использовании слоя силикагеля равной высоты:
большая динамическая активность ад, меньшая высота рабо-
тающего слоя Lo, меньшая скорость движения фронта равных
концентраций U.
В следующей серии опытов в адсорбер была загружена ком-
бинированная шихта: мелкопористый силикагель — цеолит NaA,
причем при постоянной общей высоте в адсорбере (24 см) со-
отношение слоев компонентов изменяли. Влагосодержаиие ис-
ходного газа варьировали по точке росы от 15 до —10 °C, тем-
пературу в слое при осушке поддерживали 25 и 75 °C. Резуль-
таты такой осушки показывают, что время защитного действия
слоя шихты в зависимости от условий опыта составляет от 14
до 690 мин (см. табл. 12-6).
На рис. 12,39 представлены выходные кривые при осушке
газа в слое силикагеля, цеолита NaA и их шихты в соотношении
1 : 1 при начальной температуре слоя 25 °C и исходном влагосо-
держании газа (по точке росы)
10 °C. В этих условиях реализу-
ется высокая влагоемкость сили-
кагеля в области капиллярной
конденсации, слой цеолита «под-
бирает» поступающую из сили-
Рис. 12,39. Выходные кривые при осушке газа
в слое силикагеля (1), цеолита NaA (2) и
двухслойной шихты силикагель — цеолит в со-
отношении 1 : 1 {3).
334
12. Осушка газов и органических жидкостей
Рис. 12,40. Выходная кривая (/р) и изменение тем-
пературы в замыкающем слое при осушке газа
цеолитом.
кагелевого слоя влагу, трансформи-
рует выходную кривую в форму, ха-
рактерную для случая осушки газа
цеолитами. Поэтому заметно повы-
шается динамическая активность
слоя и время защитного действия.
Большая скорость поглощения влаги цеолитом по сравнению
с силикагелем проявляется в более крутом характере выходных
кривых и в меньшей разнице между временем до установления
равновесия тР и до «проскока» тпр. Моменту «проскока» влаги
предшествует резкое повышение температуры в замыкающих
слоях адсорбента, которое зависит от типа адсорбента и условий
опыта (исходной влажности газа, температуры слоя в начале
стадии осушки и температуры газового потока).
Как видно из рис. 12,40, при осушке цеолитом NaA газа, име-
ющего точку росы 10 °C, при 75 °C максимальный разогрев слоя
составляет 15 °C; затем замыкающий слой достаточно быстро
охлаждается и спустя приблизительно 30 мин после «всплеска»
температура стабилизируется на исходном уровне. Эти наблю-
дения позволяют рассматривать процесс осушки газа в слое мик-
ропористых сорбентов (особенно цеолитов) как протекающий в
изотермических условиях. Повышение температуры в замыкаю-
щем слое часто используют для контроля режима осушки: в мо-
мент «всплеска» температуры адсорбер автоматически переклю-
чается на стадию регенерации.
На рис. 12,41 приведены кривые, отражающие изменение
времени защитного действия слоя (24 см) тпр комбинированной
шихты с разным содержанием цеолита NaA при осушке газа с
о
относительно высоким содержанием па-
ров воды (точка росы исходного газа
10°C). Как видно из рисунка, примене-
ние двухслойной шихты на адсорбцион-
ных установках с точки зрения времени
защитного действия слоя, динамической
активности и ряда других показателей
целесообразно только в случае высокого
содержания влаги в газе (10°С и выше
по точке росы), если процесс проводит-
ся при относительно низких температу-
рах (приблизительно 25 °C). Во всех
Рис. 12,41. Зависимость времени защитного действия
слоя комбинированной шихты от содержания в ней цео-
лита.
Осушка цеолитами.
335
Рис. 12,42. Схема установки осушки природного
газа цеолитами на промысле:
1 — адсорбер на стадии осушки; 2 — нагреватель
газа регенерации; 3 — адсорбер иа стадии регене-
рации; 4 — теплообменник; 5 — сепаратор влаги.
других условиях время защитного
действия слоя понижается с умень-
шением доли цеолита в шихте.
Осушка природного газа на
промыслах. Природный газ, выхо-
дящий из скважин, насыщен водя-
ными парами, и в холодное время
Осушенный газ
при транспортировании его к газо-
сборному пункту неизбежно выпадение кристаллогидратов.
Чтобы избежать их образования, раньше в газопровод впрыски-
вали гликоль или вели обогрев газопровода на участке до га-
зосборного пункта. Иногда использовали осушку газа силикаге-
лем. В настоящее время для этих целей разработан процесс с
использованием цеолитов [99].
При составлении технологического регламента установок та-
кого типа было учтено, что цеолиты осушают газ при повышен-
ных температурах, и, следовательно, не обязательно специально
их охлаждать после стадии регенерации. Сжатый природный газ
характеризуется относительно небольшим содержанием паров
воды. Если такой газ поступает в горячий адсорбер, тепловой
(холодный) фронт опережает адсорбционный фронт, и осушае-
мый газ одновременно выполняет функцию хладоагента, что
дает возможность работать по двухфазному циклу.
На одной из таких установок (рис. 12,42) осушку природного газа, на-
сыщенного парами воды в условиях пласта (давление 6 МПа, температура
32 °C), проводят в двух адсорберах, каждый из которых содержит 340 кг
цеолитов. Производительность установки составляет 380 тыс. м3 газа в сут-
ки. Стадия осушки продолжается 4 ч. После этого адсорбер переключают на
стадию регенерации. Продолжительность стадии регенерации также состав-
ляет 4 ч, причем в течение всей регенерации через адсорбент продувают го-
рячий газ. Для регенерации используют 400 м3 газа в 1 ч, т. е. 3,3% всего
осушаемого газа.
Горячий газ регенерации, выходящий из адсорбера, в теплообменниках
охлаждают осушенным газом, затем из газа регенерации выделяют сконден-
сированную влагу, после чего этот поток примешивают к осушенном газу.
Влажность объединенного потока составляет 0,05—0,06 г/м3 (точка росы ми-
нус 46°C). После стадии регенерации адсорбер переключают непосредствен-
но на стадию осушки. В течение первых 30 с влага цеолитом не поглощается,
и из адсорбера выходит газ, влажность которого практически соответствует
влажности исходного газа. Через 2 мин после переключения адсорбера тем-
пературы газа на входе в адсорбер и на выходе из него сравниваются, влаго-
содержание газа быстро уменьшается и через 10 мин составляет 0,005 г/м3
(точка росы минус 65 °C).
Промысловые цеолитовые установки отличаются простотой
управления процессом; его ведут с помощью четырех двухходо-
835 12. Осушка газов и органических жидкостей
вых или двух трехходовых задвижек. Замену цеолитов произво-
дят через 2—4 г.
Осушка газа на газобензиновых заводах. На газобензиновых
заводах природный газ из большого числа скважин собирают
и подготавливают к дальнейшему транспортированию по маги-
стральным газопроводам потребителю. Здесь осушку газа воз-
можно провести более квалифицированно, чем в промысловых
условиях.
Первая установка осушки природного газа цеолитами на газобензино-
вом заводе была пущена в эксплуатацию в 1958 г. и обеспечивала осушку
в соответствии с требованиями (точка росы минус 40°C). В последующем
требования к степени осушки были повышены. На одном из крупных газо-
бензиновых заводов США 30 млн. м3 природного газа в сутки осушают с
помощью цеолитов при давлении около 4 МПа н средней температуре 27 °C.
Содержание влаги в исходном газе составляет 0,18 г/м3, что соответствует
относительной влажности 25%.
Блок осушки состоит из четырех адсорберов диаметром 3,6 м, высота
слоя цеолитов также 3,6 м. В адсорберы в общей сложности загружено
112 т цеолита NaA. В поток осушаемого газа параллельно включены три
адсорбера. Длительность стадии осушки в каждом адсорбере составляет
36 ч. Стадия регенерации включает нагрев (8 ч) и охлаждение (4 ч). Расход
газа на регенерацию равен 360 тыс. м3/сут. Общий расход тепла 5,28-
•109 Дж/ч. Направление потоков в стадии осушки и регенерации — обратное.
Перепад давления при прохождении газового потока через слой адсорбента
в стадии осушки равен 50 кПа.
Если на газобензиновом заводе предусмотрено относительно
неглубокое извлечение этана, точка росы газа не должна быть
выше минус 40 °C. На таких заводах интенсивно строятся новые
и реконструируются на основе синтетических цеолитов старые
адсорбционные установки.
В некоторых случаях на современных газобензиновых заво-
дах степень извлечения этана предусмотрено довести до 85—
90%, пропана до 99%, бутанов практически до 100%. Столь глу-
бокое извлечение возможно после охлаждения газа до темпера-
туры минус 90 °C. На таких заводах осушку газа ведут цеоли-
тами до точки росы минус 85 °C [100]. Одновременно цеолита-
ми осушают и некоторые промежуточные потоки, например,
верхний продукт деэтанизатора.
В качестве примера удачной реконструкции установок осуш-
ки на базе цеолитов можно привести опыт одного газобензино-
вого американского завода, перерабатывающего 5 млн-м3 при-
родного газа в сутки {101].
Газ имел следующий состав (по объему): 82,5% СН4; 10,8% С2Н6; 4,2%
СзНв; 1,2% С4Н10; 0,3% С2Н!2+, а также примеси N2 и СО2. Сначала на
установке извлекался только пропан и более высокомолекулярные углеводо-
роды. Затем было решено дополнительно извлекать этан.
Осушку газа вели при давлении более 2 МПа и температуре около 40 °C
силикагелем. Установка имела два адсорбера диаметром 3,6 м с высотой
слоя адсорбента 6 м. Загрузка силикагеля в каждый адсорбер составляла
40 т. Линейная скорость потока в адсорберах была 15 м/мин. Время стадий
Осушка цеолитами
337
Рис. 12,43. Профиль температур; в слое
цеолита (1СЛ) и выходные кривые (1р) при
осушке газа.
осушки и регенерации по проектным данным должно было составлять 8 ч.
Исходный газ содержал 1 г/м3 воды. После осушки его влажность в пуско-
вой период снизилась до значения, соответствующего точке росы минус
40 °C.
Одиако при длительной эксплуатации установки с силикагелем возникли
большие трудности. Режим выдерживался, если поступающий газ имел тем-
пературу ниже 35 °C. При повышении температуры продолжительность ста-
дии осушки приходилось сокращать до 6 ч. Перевод установки иа цеолиты
позволил добиться стабильной работы с длительностью стадии осушки 12 ч.
Почти в 20 раз сократилось количество совместно поглощаемых углеводоро-
дов. Одновременно была решена задача полного улавливания сульфида во-
дорода и меркаптанов, которые до этого вследствие неполноты предвари-
тельной очистки вызывали сильную коррозию аппаратуры.
В настоящее время на установке перерабатывают газ, имеющий точку
росы 4°C и содержащий 5-Ю-4 г H2S и 3-10—3 г меркаптанов на 1 м3.
В связи с относительно невысокими требованиями к степени осушки конеч-
ная температура адсорбента при регенерации сначала составляла только
205 °C. Учитывая присутствие сернистых соединений в дальнейшем ее повы-
сили до 260 °C.
Как указывалось выше, в адсорберах с цеолитами, особенно
при высоком давлении, зона теплообмена движется по слою зна-
чительно быстрее, чем зона массопередачи. Поэтому только в
первый очень непродолжительный период после начала стадии
осушки, когда слой еще горячий, оба фронта частично совпа-
дают. Затем процесс осушки происходит практически уже охлаж-
денным цеолитом.
На рис. 12,43 представлен профиль температур в последней
по ходу газа точке слоя адсорбента /сл и выходные кривые осуш-
ки газа при атмосферном давлении после его прохождения че-
рез небольшие слои (10 и 20 см) природного клиноптилолита,
разогретого в процессе предыдущей регенерации до 175 °C
[102]. Влагосодержание исходного газа соответствовало точке
росы +10 °C; скорость потока составляла 0,25 м/с. Как видно
из графиков, в первые минуты поток поступающего газа охлаж-
дает слой адсорбента. Затем, в результате выделения теплоты
адсорбции, происходит небольшой разогрев, не влияющий на про-
цесс адсорбции. Даже при малых высотах слоя точка росы
осушенного в этих условиях газа была менее минус 50 °C; про-
скок влаги наступал уже после того, как слой охладится.
Осушка газов без включения в цикл стадии охлаждения цео-
литов использована в Канаде при подготовке природного газа
на крупном газоперерабатывающем заводе ![103].
22—1346
338
12. Осушка газов и органических жидкостей
Впервые такой процесс был разработан и применен фирмой «Малони» с
целью снижения стоимости осушки, упрощения всего цикла и сокращения
расхода энергии, В нем полностью исключена стадия охлаждения, обычно
следующая за стадией регенерации цеолитов. Благодаря этому заметно
уменьшено число холодильников, задвижек и средств контроля. Другие преи-
мущества вытекают из использования меньших слоев цеолита, меньшего рас-
хода энергии и простоты операций. По сравнению с обычной трехадсорбер-
ной схемой эксплуатационные расходы составляют лишь половину, а капи-
тальные вложения — одну третью часть. В течение продолжительной про-
мышленной эксплуатации отрицательных эффектов не обнаружено.
Установка осушкн включает первичный сепаратор 1, три адсорбера 6—
В, нагреватель газа регенерации 5, конденсатор-сепаратор 2, теплообменники
газ — газ 3 и 4, задвижки и средства контроля (рис. 12,44).
В установке использована углеродистая сталь легированная марганцем
для обеспечения ковкости и достаточных прочностных свойств и для сниже-
ния коррозии аппаратуры.
Исходный газ (1,2 млн. м3/сут) поступает на установку при давлении
7 МПа и температуре 30 °C; он может быть либо уже очищен, либо содер-
жать кислые компоненты и воду. Вся сконденсированная влага выделяется
в сепараторе 1, а газ используется для охлаждения газа регенерации в теп-
лообменниках 3 и 4. Затем исходный газ пропускают через два параллельно
включенных адсорбера 6 и 7, после чего он поступает в газопровод.
Цеолит регенерируют частью непрерывно отбираемого исходного газа,
который нагревают в теплообменниках газ — газ 3 и 4 отходящим газом ре-
генерации, а затем в соляном нагревателе о до 260 СС.
Каждый адсорбер находится на стадии осушки две трети рабочего вре-
мени, а на стадии регенерации — одну треть времени; переключение автома-
тическое. Регенерацию проводят непрерывно. Остаточное влагосодержаиие осу-
шенного при таком режиме газа поддерживают приблизительно постоянным.
Охлажденный газ регенерации примешивают к исходному газу после про-
хождения через конденсатор 2.
Температура газа, поступающего в газопровод, поддерживается не вы-
ше 60 °C (такая температура возникает в период подключения горячего ад-
сорбера в поток путем перераспределения потоков в двух параллельных ад-
сорберах 6 и 7).
Рис. 12,44. Схема адсорбционной установки без стадии охлаждения адсорбента на газо-
перерабатывающем заводе:
1 первичный сепаратор; 2 — конденсатор; 3, 4 — теплообменники; 5 — нагреватель газа
регенерации; 6—8 — адсорберы.
Осушка цеолитами
339
Таблица 12-7. Относительные затраты на осушку газа
Виды затрат Тип установки
без охлаж- дения обычная двухадсор- берная обычная трехадсор- бериая гликолевая
Капиталовложения 100 116 143 45
Стоимость химикатов 100 108 188 303
Эксплуатационные за- траты 100 139 222 207
При расчете экономики процесса учитывали, что природный
газ обычно осушают на двух или трехадсорберных установках
с цеолитами или на гликолевой установке. По сравнению с рас-
сматриваемым процессом эти традиционные варианты требуют
больших капитальных затрат и эксплуатационных расходов,
расхода химикалиев и в случае использования гликолей вызы-
вают значительно большее загрязнение окружающей среды.
Капитальные затраты, стоимость химикалиев и эксплуатацион-
ные расходы для различных вариантов процесса представлены
в табл. 12-7.
Показатели определены для установки указанной выше про-
изводительности, осушающей газ и очищающей его от кислых
компонентов (содержание сульфида водорода в исходном газе
около 22,5%).
Анализ показал, что объем слоя адсорбента на обычных цео-
литовых установках на 50—100% больше, чем на установках,
работающих без охлаждения адсорбента. Время цикла на трех-
адсорберной установке в два раза больше, чем на новой уста-
новке. Расход газа регенерации на двухадсорберной обычной
установке в два раза выше, чем на рассматриваемой установке.
Преимуществом адсорбционной осушки по сравнению с гликоле-
вой является более высокая степень удаления влаги. Кроме то-
го, применение гликолевых установок для осушки газа с кон-
центрацией сульфида водорода выше 1% нецелесообразно в
связи с загрязнением атмосферы. Потери гликоля даже при на-
личии хорошего антивспенивателя составляют около 1 кг на
10 тыс. м3 газа, или около 70 л/сут. Стоимость этих потерь зна-
чительно выше стоимости цеолитов.
Потери серы на стандартной гликолевой установке, вследст-
вие растворения сульфида водорода в адсорбенте оцениваются в
11 т/сут. При трехлетием сроке службы цеолитов период оку-
паемости установки нового типа составляет 2,5 г.
Осушка природного газа, закачиваемого в пласт. Эксплуата-
тацию газоконденсатных пластов можно вести двумя способа-
ми: либо газ направляют непосредственно потребителю, либо,
22*
340 12. Осушка газов и органических жидкостей
после извлечения из него тяжелых углеводородов (конденсата),
газ для поддержания давления в пласте снова закачивается в
пласт. В обоих случаях газ осушают, но при закачке газа в пласт
выявляется одна технологическая деталь: для уменьшения за-
трат на компримирование, осушку и отбензинивание целесооб-
разно проводить при высоком давлении.
Известна установка [104], на которой природный газ перед закачкой в
пласт осушают при давлении 15 МПа. Характерной особенностью перераба-
тываемого газа является большое содержание в нем кислых примесей: 26%
H2S и 5% СО2. Осушку производят кислотостойким цеолитом. В адсорбци-
онные полости этого цеолита проникают только молекулы Н2О, H2S, СО2,
СН< и С2Н6, в связи с чем была устранена проблема закоксовывания адсор-
бента в результате крекинга высокомолекулярных углеводородов в порах.
.Благодаря высокому адсорбционному сродству вода вытесняет из пор все
другие компоненты, и, регулируя длительность стадии осушки, можно до-
биться, чтобы цеолит поглощал из потока газа только воду.
Схема установки представлена на рис. 12,45. Газ иа устье скважины
имеет давление 21 МПа и температуру 55—75 °C. В результате снижения
давления до 15,5 МПа и пропускания газа через воздушный теплообменник 1
из него выделяется более половины влаги. Сконденсированную влагу удаля-
ют в первичном сепараторе 2. Порог образования кристаллогидратов на
установке составляет 32 °C, поэтому охлаждение газа проводят только до
40 °C. Установка имеет два адсорбера 3 и 4. В то время как адсорбер 3
находится иа стадии осушки, адсорбент в адсорбере 4 подвергают регенера-
ции. Давление в адсорберах при осушке и регенерации одинаково.
На стадии регенерации через адсорбент пропускают часть природного
газа, нагретого в соляной ванне 5 до 260 °C. Температура рассола в ванне
достигает 370 °C. Ванна автоматически отключается, когда начинается охлаж-
дение адсорбента. Температура адсорбента в конце стадии охлаждения 55 °C.
Продукты, выделяющиеся при регенерации, охлаждают в воздушном теп-
лообменнике 6, сконденсированную воду отделяют в сепараторе 7, после
Рис. 12,45. Схема установки осушки природного газа под высоким давлением:
1, 6 — воздушные холодильники; 2 — первичный сепаратор; 3 —адсорбер иа стадии осуш-
ки; 4 — адсорбер на стадии регенерации; 5 —солевая ванна; 7, 8 — сепараторы; 9 —
яасос.
Осушка цеолитами
341
Таблица 12-8. Показатели установок осушки
природного газа цеолитом AW-500
Производительность, млн. м3/сут
Показатели 0,28 0,56 1,00 1,35»
Высота слоя адсорбента, м 3,6 3,6 3,8 3,1
Диаметр адсорбера, м 0,4 0,64 0,82 1,20
Загрузка одного адсорбера, кг 450 900 1750 2250
Скорость потока в стадии осушки, 7,8 6,8 7,0 5,1
м/мин
* Трехадсорберная установка.
чего газ возвращают на осушку. Воду из сепараторов 2 и 7 подвергают
дегазации в сепараторе 8 при давлении 0,13 МПа и температуре 35—40 °C,
после чего насосом 9 закачивают в пласт. Газ из сепаратора 8 используют
в качестве топлива. Переключение адсорберов производят каждые 3 ч.
В настоящее время эксплуатируются девять установок тако-
го типа, на которых обеспечивается требуемая степень осушки
(точка росы минус 30°C). Основные показатели адсорберов
двухадсорберной установки в зависимости от производительно-
сти приведены в табл. 12-8.
При существующей системе регенерации (нагрев газа реге-
нерации в соляной ванне) на рассматриваемой установке не до-
стигается полного обезвоживания цеолита. Вследствие этого ди-
намическая активность даже свежего адсорбента по парам во-
ды не превышает 9 г/100 г. В процессе многоцикловой эксплуа-
тации активность снижается до 65% первоначального значения.
Лабораторный контроль указал на медленное накопление в цео-
лите углерода и серы. Их удаляют в результате периодических
длительных регенераций при повышенных температурах. Кроме
того, отмечено попадание в адсорбер хлорида натрия из соляной
ванны и сернистых соединений железа, образующихся в резуль-
тате коррозии аппаратуры.
Текущий ремонт адсорберов связан со сбросом и последую-
щим повышением давления в системе, когда возникают чрезмер-
но высокие скорости потока, вызывающие движение слоя и исти-
рание адсорбента. В связи с этим в верхней части адсорбера
гранулы цеолита AW-500 диаметром 1,6 мм заменяют на более
крупные — диаметром 3,2 мм. Срок службы адсорбента оцени-
вается в 2 г.
Осушка природного газа на криогенных установках. В газо-
вой промышленности криогенный метод применяют для решения
двух дадач: сжижения природного газа и извлечения из него
гелия.
342 12. Осушка газов и органических жидкостей
Низкие температуры в холодильном цикле (до минус 170 °C)
требуют практически полного удаления паров воды из газа. Эту
задачу удалось решить только благодаря применению цеолитов.
На криогенных установках за момент проскока влаги принима-
ют 1 часть на 1 млн. Имеются данные, что остаточное содержа-
ние воды при осушке газа цеолитами составляет 0,1 части на
1 млн. Это соответствует точке росы минус 100 °C. К сожале-
нию, приборами, регулирующими более низкие концентрации па-
ров воды, не располагает ни отечественная, ни зарубежная тех-
ника.
Испытания показали, что глубокая осушка газа цеолитами
полностью исключает возможность обмерзания оборудования, а
следовательно, и нарушения непрерывности эксплуатации крио-
генных установок. В течение 5 лет испытательного срока на
криогенных установках, оборудованных блоками осушки цеоли-
тами, не было отмечено ни одного случая прекращения техно-
логического потока из-за образования ледяных или кристалло-
гидратных пробок.
Как правило, при производительности осушающих устано-
вок до 3,5 млн. м3/сут применяют блоки, состоящие из двух ад-
сорберов с периодом стадии осушки не менее 8 ч. В этом случае
система автоматизации не обязательна. При большой произво-
дительности целесообразно распределять поток сырого газа че-
рез два или более параллельно включенных адсорбера, в то вре-
мя как один адсорбер из блока находится на стадии регенера-
ции. В этом случае за минимальное время осушки также прини-
мают 8 ч, а время регенерации равно 4 ч. На регенерацию пода-
ется сухой газ под давлением, равным давлению на входе в уста-
новку осушки или более низким, в зависимости от условий ре-
компрессии. На таких установках всегда предусматривают авто-
матизацию процесса.
Условия эксплуатации блоков осушки на криогенных уста-
новках приведены ниже:
Производительность, млн. м3/сут 0,3—40
Содержание паров воды, г/м3 От 0,05
до насыщения
Давление, МПа 1—10
Температура, °C 10—45
Остаточное содержание паров воды
в % 1-10-в
по точке росы, °C —100
Конечная температура нагрева цеолита при 290
регенерации, °C
Чтобы обеспечить низкое влагосодержание газа, в период
пуска адсорбционной аппаратуры необходимо тщательно просу-
шить всю систему.
Осушка цеолитами
343
Установки сжижения природного газа проектируются и
строятся на различную производительность. При использовании
выделенного на установке сжиженного газа для покрытия ме-
стных пиковых нагрузок производительность установок колеб-
лется в широких пределах 5—500 тыс. м3/сут, при его примене-
нии для транспорта в сжиженном виде — от 3 до 10 млн. м3/сут
[105].
В США практически на всех эксплуатирующихся установках
сжижения природного газа вне зависимости от продолжитель-
ности применяют цеолиты. При высоком содержании влаги в пе-
рерабатываемом газе применяют комбинированную шихту сили-
кагель — цеолит.
Одна из американских установок сжижения природного газа имеет мощ-
ность сжижающей аппаратуры 130 тыс. м3/сут и мощность испарительной ап-
паратуры 2 млн. м3/сут [106]. Емкостный парк рассчитан на 46 тыс. м3 сжи-
женного метана. В схеме установки используется комбинация каскадного н
турбоэкспандерного цикла. Осушку поступающего в холодильный цикл газа
производят при 4 МПа в адсорберах, в которых основная масса влаги погло-
щается силикагелем, а глубокая доосушка производится синтетическими цео-
литами. В холодильном цикле предусмотрены также цеолитовые адсорберы.
Газ проходит через них при минус 98 °C; при этом содержание СО2 снижа-
ется до 50 част, на 1 млн., a H2S и меркаптана (одоранта) ниже 1 части
на млн.
Производительность установок осушки природного газа влажностью
0,05—1,5 г/м3 (рис. 12,46), перерабатываемого на гелиевых заводах, колеб-
лется от 8 до 30 млн. м3/сут.
Как правило, адсорберы на гелиевых заводах имеют диаметр 2,4—3,6 м,
причем одновременно на стадии осушки находятся от трех до шести аппа-
ратов; природный газ проходит через них параллельными потоками. Дли-
тельность полного цикла (адсорбция и регенерация) колеблется в пределах
8—96 ч в зависимости от размеров аппаратов и содержания влаги в исход-
ном газе. Цеолиты при регенерации доводят до температуры 200—315 °C, для
нагрева используют сухой газ с блока ожижения.
На тех установках, где прежде в качестве адсорбента ис-
пользовали оксид алюминия, объем существующих адсорберов
заполняют цеолитами только частично. Уменьшение высоты слоя
адсорбента приводит к снижению перепада давления; в адсорбе-
рах с оксидом алюминия он составлял обычно 0,1 МПа, в цеоли-
товых адсорберах только 0,03—0,05 МПа. Снижение перепада
давления примерно на каждые 700 Па приводит к годовой эко-
номии 5000 долл, применительно к
установке производительностью 30
млн. м3 газа в сутки.
Загрузка цеолитов в один осу-
шающий блок на гелиевых заводах
Рис. 12,46. Схема установки осушки газа на ге-
лиевом заводе:
1 — адсорбер на стадии осушки; 2 — адсорбер на
стадии регенерации; 3 — нагреватель; 4 — блок
сжижения; 5 — холодильник; 6 — сепаратор.
344
12. Осушка газов и органических жидкостей
Рис. 12,47. Зависимость предельной адсорбционной способ-
ности породы по парам воды ср при 20 °C от содержания
клиноптилолита с.
США достигает 550 т. В Канаде при про-
изводстве гелия цеолиты устанавливают в
качестве «защитного» слоя после обычных
осушающих средств — для понижения точ-
ки росы газа.
В Польше закончено строительство за-
чистого гелия и 96%-ного метана. Исход-
ный природный газ содержит 0,4% Не и 46% N2 [107]. На за-
воде предполагается производить 4 млн. м3 метана и около 13
тыс. м3 гелия в сутки. Перед разделением газ очищают от ди-
оксида углерода аминами и осушают цеолитами.
Использование цеолитов в качестве осушителя природного
газа при низких температурах, естественно, вызвало вопрос, не
могут ли в их порах в этих условиях образоваться кристаллогид-
раты углеводородов, что сильно осложнило бы эксплуатацию
установок. Экспериментальное исследование давления паров во-
ды над цеолитами ![108] позволило, однако, сделать вывод, что
образование кристаллогидратов при давлениях 3,5—5 МПа тео-
ретически возможно только во вторичной пористой структуре и
при температурах ниже 150 К (минус 123°C).
Осушка углеводородных газов природными цеолитами. В свя-
зи с открытием месторождений природных цеолитов в различ-
ных регионах Советского Союза — от Закарпатья до Дальнего
Востока и Сахалина — установки подготовки углеводородных
газов к транспортированию от месторождений до химических
заводов стали оснащать адсорбционными установками.
Адсорбционные свойства по парам воды любой породы, со-
держащей клиноптилолит (или морденит), зависят от предель-
ного объема адсорбционного пространства (практически — от
объема микропор), отнесенного к единице массы или объема ад-
сорбента. Предельный адсорбционный объем находится в пря-
мой зависимости от содержания клиноптилолита в породе.
Как показано на рисунке 12,47, значения предельной сорб-
ционной способности по парам воды линейно возрастают (от
0,043 до 0,131 г/г) с ростом содержания цеолита в породе 1[ 109].
Экстраполяцией опытных данных в область более высокого
содержания цеолита получена ожидаемая величина предельной
сорбционной способности 100%-ного природного цеолита (кли-
ноптилолита), равная 0,142 г/г. В целом зависимость предельной
сорбционной способности от концентрации клиноптилолита в по-
роде описывается линейным уравнением
ар= 0,00142с (12.5)
Сравнительная характеристика эффективности четырех ти-
пов адсорбентов, опыты с которыми проводили на стендовой.
Осушка цеолитами
345
Таблица 12-9. Сравнение эффективности адсорбентов — осушителей
Влажность исходного газа по точке росы, °C Температура при осушке, °C Время защитного действия слоя адсорбента, мин
цеолнт NaA силикагель кем шихта силикагель— цеолит (3:1) порода с содержанием клиноптило- лита 90%
+10 25 168 102 150 100
50 160 57 — 82
75 150 42 63 75
—1 25 353 145 200 190
50 343 128 — 150
75 275 45 80 133
—10 25 680 322 450 505
50 664 263 — 405
75 480 100 137 305
установке, дана в табл. 12-9. Слой адсорбента при осушке газа
принимали 24 см, скорость потока 0,25 м/с, размер гранул или
зерен адсорбента 2—3 мм.
В большинстве режимов время защитного действия слоя кли-
ноптилолита значительно превосходит соответствующий показа-
тель для силикагеля и комбинированной шихты из силикагеля и
синтетического цеолита NaA. Несколько меньшая влагоемкость
природных цеолитов по сравнению с синтетическими не должна
влиять на их широкое внедрение в газовую промышленность.
Во всех случаях в адсорберах, загруженных цеолитовой породой,
достигается хорошая глубина осушки (ниже минус 40 °C по
точке росы).
В ноябре 1974 г. на Миннибаевском газоперерабатывающем
заводе им. Ленинского комсомола один промышленный адсор-
бер в адсорбционном блоке общей производительностью
1,5 млн. м3/сут был переведен на работу с загрузкой клинопти-
лолитом [НО].
До этого осушку вели в шести двухслойных адсорберах диаметром 2 м
с высотой слоя адсорбента 6 м. В каждый адсорбер загружали 9 т сили-
кагеля и 2 т цеолита NaA. Предварительно газ осушали до точки росы ми-
нус 10 °C, а также очищали от СО2 и H2S в абсорбере, орошаемом смешан-
ным поглотителем (диэтиленгликоль и моноэтаноламин).
В один из адсорберов вместо двухслойного адсорбента было загружено
18 т клиноптилолитовой породы месторождения Дзегви Грузинской ССР.
В течение всей стадии осушки при давлении 4 МПа точка росы выходящего
из адсорбера газа была постоянной и равной минус 60, температура в слое
при осушке составляла 25—30 °C.
После регистрации проскока влаги (этому моменту соответствовала точ-
ка росы минус 55 °C) адсорберы переключали на стадию регенерации, кото-
рую проводили при давлении 6 МПа продувкой горячим газом (250—280 °C)
в течение 6 ч. Расход газа на регенерацию составлял 10% всего осушенного
газа. После стадии регенерации клиноптилолит при 200—220 °C поступал на
вхлаждение сухим газом —22°C, V=6 тыс. м3/г, т=2 ч).
346 12. Осушка газов и органических жидкостей
Спустя год из промышленного адсорбера отобрали пробы
клиноптилолита. На вакуумной адсорбционной установке были
получены изотермы адсорбции паров воды в интервале темпе-
ратур 25—150 °C на свежем и выгруженном из адсорбера об-
разцах. Снижение адсорбционной способности клиноптилолита в
период эксплуатации составило менее 20%. Так, при 25 °C и
2,3 кПа равновесная адсорбционная способность свежего клино-
птилолита составляла 12,6 г/100 г, а после эксплуатации
10,5 г/100 г. Это приблизительно соответствует снижению актив-
ности синтетического цеолита NaA в процессе многоцикловой
эксплуатации при осушке природного газа в условиях газопере-
рабатывающего завода.
Физико-химические и механические показатели адсорбента,
его термо- и кислотостойкость почти не изменились. Испытания
на стенде показали, что эффективность осушки (ее глубина) на
свежем и «работавшем» в промышленных условиях клинопти-
лолите почти идентична. Следовательно, адсорбционные уста-
новки, осушающие нефтяной и природный газ, следует проекти-
ровать, принимая время эксплуатации загрузки 4 года и более*.
Динамическая активность слоя породы по парам воды долж-
на определяться исходя из содержания клиноптилолита в породе
(табл 12-10).
В технических условиях на природные цеолитные адсорбен-
ты предусмотрено деление пород на два класса. Первый класс
имеет нижнюю границу концентрации цеолита 75—80%, второй
50—60%. Степень осушки газа, зависящая от содержания цео-
лита в породе, приведена ниже:
Содержание цеолита, % 30 50 75 90
Точка росы, °C —40 —45 —50 —60
Отсюда видно, что при использовании даже низкоконцент-
рированных пород обеспечивается глубина осушки, удовлетворя-
ющая требованиям, предъявляемым к природному газу, посту-
пающему в магистральный газопровод.
Другим показателем качества является зернение адсорбен-
та. Размер зерна не должен превышать 5—10 мм. Условия ре-
генерации: температура поступающего на регенерацию газа
350—400 °C, температура адсорбента в конце регенерации 280—
290 °C. Расход продувочного газа и расход газа на охлаждение
в каждом конкретном случае определяют на основе материаль-
ного и теплового баланса процесса. Ориентировочно расход про-
дувочного газа на 1 кг цеолита составит 1,5 м3, а расход газа
на охлаждение 0,75 м3. Продолжительность стадии охлаждения
* Успешная эксплуатация клиноптилолита Дзегви на описанной установке
продолжается уже восьмой год. (Прим, ред.)
Осушка цеолитами
347
Таблица 12-10. Динамическая активность (в г/100 г)
породы цеолита по парам воды
Влажность исходного газа по точ- ке росы, °C Начальная температура адсорбции, *С Концентрации цеолита в породе, %
30 50 75 90
+ю 25 1,8 2,5 6,6 8,1
50 1,4 2,2 6,1 6,7
75 1,1 1,5 5,1 6,2
—1 25 1,9 1,8 4,7 7,5
50 1,4 1,7 3,8 5,9
75 1,1 1,5 3,1 5,3
—10 25 2,0 1,9 5,3 6,1
50 1,6 1,8 4,2 5,7
75 1,1 1,7 2,9 5,2
(если она необходима) 2—3 ч. Предельно допустимую скорость
потока выбирают исходя из гидравлического сопротивления ки-
нетики сорбции и пыления сорбента. Зависимость скорости от
давления в системе представлена ниже:
р, МПа 0,1 1,0 2,0 3,0 4,0 5,0 7,0
Г, м/с 0,30 0,23 0,16 0,14 0,10 0,08 0,05
Успешные промышленные испытания позволяют обосновать
целесообразность широкого использования природных цеолитов
в газовой отрасли. Можно рекомендовать на их основе создание
промысловых установок подготовки газов к транспортированию,
адсорбционных блоков на ГПЗ, гелиевых производствах, уста-
новках по закачке газа в пласт.
Осушка сжиженных углеводородных газов. Основным источ-
ником получения сжиженных газов являются природные и по-
путные нефтяные газы, а также газы, получаемые при стабили-
зации и переработке нефти. Значительным резервом увеличения
производства сжиженных газов является углубление отбора про-
пан-бумановой фракции из попутного нефтяного газа. Интен-
сивное использование сжиженных газов вызывает необходимость
строительства большого парка хранилищ и продуктопроводов.
При эксплуатации последних значительные трудности возника-
ют из-за присутствия влаги в сжиженных газах и других неф-
тепродуктах. Растворимость воды в них при обычных темпера-
турах не превышает 0,04% (мол.). Несмотря на это, ее присут-
ствие может вызвать значительные осложнения. Перед тем как
подать сжиженный газ в трубопровод, его необходимо тщатель-
но осушить. Применение твердых осушителей позволяет при не-
348 12. Осушка газов и органических жидкостей
сложном оборудовании получить высокую степень осушки сжи-
женных газов.
Лабораторные опыты [111] позволили сопоставить результа-
ты осушки пропана и бутана в паровой и жидких фазах в слое
цеолита NaA. Остаточное содержание влаги после прохождения
через цеолит в обоих случаях составляло менее 20 част, на
1 млн., а динамическая активность (15—19) г/100 г; динамиче-
ская активность слоя оксида алюминия значительно ниже 3—
4 г/100 г ;[112]. Основным преимуществом проведения осушки
в жидкой фазе является возможность работы при значительно
больших массовых скоростях. Время защитного действия слоя
цеолита NaA высотой 8 см в процессе жидкофазной осушки
пропана, проводимой при линейной скорости потока 0,Q04 м/с,
составляет 24 ч, а слоя оксида алюминия равной высоты — все-
го 6 ч. Испытаниями адсорбера с цеолитами, установленного на
потоке пропана, была показана возможность глубокой осушки
одним килограммом цеолита до 8 т пропана.
Установка осушки пропана (исходная влажность 300 част, на 1 млн.}
в блоке современного газоперерабатывающего завода производительностью
50 т/сут состоит из двух попеременно работающих адсорберов с общей на-
грузкой адсорбента 100—150 кг. Стадия осушки длится 12 ч. Регенерацию
проводят горячим сухим природным газом до температуры слоя на выходе
из адсорбера 250—350 °C. При скорости газового потока 30—60 м/мин реге-
нерация может быть закончена в течение 1—2 ч.
Часто осушку сжиженных газов совмещают с их сероочист-
кой. Поэтому более подробно показатели и схемы таких уста-
новок рассмотрены в гл. 13.
На современных газонаполнительных станциях сжиженный
газ перед закачкой в баллоны подвергают фильтрации и адсорб-
ционной осушке, что позволяет значительно улучшить качество
сжиженного топлива [113].
Осушка абсорбента на газоперерабатывающих заводах. По-
ложительные результаты, полученные при осушке цеолитами лег-
ких углеводородов в сжиженном состоянии, позволили распро-
странить метод на осушку ряда органических жидкостей и уг-
леводородных фракций, применяемых, в частности, в качестве
абсорбентов на газоперерабатывающих заводах [114]. Их осуш-
ка оказалась необходимой в связи с переводом абсорбционных
установок на низкотемпературный режим.
Осушку абсорбента производят с помощью адсорбционной
установки с цеолитами NaA, включенной в технологическую
нитку на пути абсорбента из десорбера в абсорбционную колон-
ну (рис. 12,48). В Шеридане (штат Техас, США) были проведе-
ны первые испытания установки такого типа; остаточное содер-
жание влаги в абсорбенте после осушки цеолитами равнялось
10—15 част, на 1 млн. Загрузка цеолитов в установку составля-
Осушка цеолитами
349
Рис. 12,48. Схема абсорбционной установки с блоком осушки тощего абсорбента цеоли-
тами:
1 — абсорбер; 2 — холодильник; 3 — блок осушки; 4 — нагреватель; 5 —сепаратор; 6 — от-
парная колонна.
Рис. 12,49. Схема блока осушки, тощего абсорбента;
1 — адсорбер на стадии осушки; 2 —адсорбер на стадии регенерации; 3 — нагреватель^
4 — холодильник; 5 — сепаратор.
ла 13,5 т. После пяти лет эксплуатации снижения активности ад-
сорбента замечено не было.
Схема блока осушки абсорбента представлена на рис. 12,49. Он состоит
из двух адсорберов: один находится на стадии осушки, другой—на стадии
регенерации. Рабочий цикл включает следующие операции: заполнение аб-
сорбентом, осушка, спуск жидкой фазы, нагрев и охлаждение. Нагрев и
охлаждение ведут природным газом. Продолжительность цикла — 8—24 ч.
Осушка цеолитами позволяет понизить температуру в аб-
сорбционной колонне до минус 20 °C и обеспечить переход на
низкомолекулярные абсорбенты, обладающие более высокой по-
глотительной способностью по углеводородам. Присутствующие
в абсорбенте добавки и ингибиторы коррозии в конце стадии
осушки полностью вытесняются из цеолита адсорбируемой во-
дой и, таким образом, не дезактивируют цеолит. По аналогично-
му регламенту работают несколько промышленных установок,
предназначенных для осушки конденсатов перед их низкотем-
пературным разделением.
Применение метода позволяет шире использовать отпарку во-
дяным паром, обеспечивающую большую степень удаления уг-
леводородов на стадии десорбции и, как следствие этого, более
высокую степень улавливания на стадии отбензинивания.
Комбинированная схема осушки природного газа. Несмотря
на широкое использование цеолитов, осушка газов диэтиленгли-
колем или триэтиленгликолем продолжает доминировать в га-
зовой промышленности. Гликолевые установки компактны, ав-
томатизированы, удобны в эксплуатации. Степень осушки газа
после гликолевой колонны зависит от влагосодержания глико-
ля, поступающего в абсорбер. Минимальную точку росы осушен-
ного газа в зависимости от температуры контакта и влажности
350 12. Осушка газов и органических жидкостей
диэтиленгликоля определяют по графикам, представленным на
рис. 12,50.
При температуре контакта 25 °C понижение точки росы в про-
цессе осушки газа триэтиленгликолем в зависимости от кон-
центрации поглотителя составляет:
Концентрация, % 98 99 99,5 99,9
Понижение точки росы, °C 36 45 55 70
К сожалению, получить абсолютно безводный гликоль рек-
тификацией не удается. Высокие температуры при регенерации
применять нельзя вследствие его разложения; температуры на-
чала разложения ди- и триэтиленгликоля составляют соответст-
венно 177 и 204 °C.
Проведение регенерации гликоля в вакууме или с продув-
кой инертным сухим газом, хотя и позволяют снизить влагосо-
держание гликоля, но недостаточно. Между тем, как показыва-
ет рис. 12,50, если в абсорбционную колонну подать гликоль с
содержанием воды 0,1 %, можно обеспечить степень осушки, со-
ответствующую точке росы минус 40 °C. Поглотитель такого ка-
чества получают, пропуская гликоль, из которого основная мас-
са воды удалена в регенераторе, через адсорбер с цеолитом
(рис. 12,51) [115].
При осушке таких вязких сред, как гликоли, лучшие пока-
затели достигаются, если процесс проводить при повышенной
температуре, например, 70 °C
[116]. В результате обработки
триэтиленгликоля, содержаще-
го 2% воды, в слое цеолита
NaA высотой 2,5 м при скоро-
сти 0,02 м/мин достигается
снижение влажности до
0,025%; влагоемкость цеолита
при этом составляет 12 г/100 г.
Определенные затруднения
вызывает стадия восстановле-
ния адсорбционных свойств
цеолита после осушки глико-
ля. При обычной термогазовой
регенерации оставшийся в ад-
сорбенте гликоль разлагается,
а продукты разложения при-
Рис. 12,50. Точки росы газа /р, равновесные
с водными растворами диэтиленгликоля
(ДЭГ), при различных температурах кон-
такта (пунктиром показаны зависимости,
полученные на гликоле, осушенном цеоли-
тами).
Осушка цеолитами
351
Рис. 12*51. Схема установки осушки газа ди-
этиленгликолем (ДЭГ), обезвоженным цеоли-
том:
] — абсорбционная колонна; 2 — регенератор;
3 — адсорбер с цеолитом.
Природный
2 3
90%-ный Т
99,9%-ный ДЭГ
водят к необратимой дезактивации адсорбента. Чтобы устра-
нить это негативное явление, было предложено регенерацию
производить в две стадии: сначала непродолжительной продув-
кой (15—20 мин) газом со скоростью 0,2—0,3 м/с, а затем про-
мывкой конденсатом водяного пара, выводимого сверху из ко-
лонны — регенератора гликоля. В первой стадии удаляется ос-
новная масса гликоля, во второй—его остатки.
Только после этого в цикл включается этап обезвоживания
цеолита горячим газом [117]. В одном из вариантов в первой
стадии промывки к воде добавляют поверхностно-активное ве-
щество, например алкил сульфон атное моющее средство [118].
Осушка в нефтехимической промышленности
Продукты нефтепереработки и нефтехимии отличаются от при-
родного сырья высоким содержанием непредельных углеводоро-
дов, осушка и очистка которых имеет определенную специфику.
Осушка непредельных углеводородов. При осушке непредель-
ных углеводородов необходимо учитывать каталитическую ак-
тивность цеолитов в реакции полимеризации. Каталитические
свойства цеолитов связаны, с одной стороны, с наличием в кри-
сталлитах активных кислотных центров, а с другой — с катали-
тическими свойствами связующего. Среди цеолитов общего на-
значения кристаллит типа NaA обладает наименьшей катали-
тической активностью >[119]. Путем подбора каталитически
инертного связующего был приготовлен цеолит NaA-З, который
используют, в частности, для осушки пропилена в жидкой фа-
зе. Процесс был опробован на пилотной установке производи-
тельностью 1 т/сут )[120].
Установка состояла из двух адсорберов диаметром 10 см, в каждый из
которых было загружено по 3,7 кг цеолита NaA-З в виде гранул размером
d = l=4 мм. Высота слоя цеолитов в адсорберах составляла 0,8 м. Влагосо-
держание исходной пропано-пропиленовой фракции (85%-ный пропилен)
изменялось от 120 до 330 мг/кг, что соответствует точке росы пропилена
после его испарения от минус 21 до минус 28 °C. Осушку проводили при
давлении 1,2—1,4 МПа, температуре 15—20 °C, скорости пропилена 13—
20 см/мин.
После окончания стадии осушки пропилен сливали, а его остатки отду-
вали при комнатной температуре в течение 2 ч азотом. Затем, ие прекращая
продувку азотом, ступенчато поднимали температуру в адсорбере до 300 °C.
Динамическая активность цеолита при указанной температуре в стадии ре-
генерации составляла (14—18) г/100 г, точка росы осушенного пропилена
352 12. Осушка газов и органических жидкостей
после испарения достигала минус 70 °C, количество пропилена, осушенного
одним килограммом цеолита, превышало 1000 кг.
При подаче на цеолиты потока жидких олефинов, например
пропилена, температура слоя резко повышается, что увеличива-
ет вероятность полимеризации. Чтобы избежать этого, в некото-
рых случаях цеолиты насыщают олефинами, предварительно
пропуская через адсорбер инертный газ, содержащий 2% олефи-
нов [121]. За этой стадией в рабочем цикле следуют: осушка,
вытеснение водой остатков олефинов, нагрев и охлаждение.
При осушке непредельных углеводородов цеолитами NaA
предпочтительно предусматривать одно и то же направление
потока газа на стадиях осушки и регенерации. В этом случае
углеводороды, сорбирующиеся в первый период, постепенно
вытесняются водой из слоя цеолитов и уносятся с потоком осу-
шенного газа. После наступления момента проскока адсорбер
переключают на стадию регенерации горячим газом. Оконча-
тельное вытеснение углеводородов адсорбирующейся водой из
замыкающего слоя цеолитов заканчивается на стадии регенера-
ции при относительно низких температурах, и таким образом
снижается до минимума дезактивация адсорбента.
В системе с тремя адсорберами газ осушают в двух последо-
вательно включенных аппаратах. Первый по ходу газа адсорбер
переключают на стадию регенерации после того, как цеолит на-
сыщается водой до состояния равновесия и все углеводороды
вытесняются из адсорбента. В этот момент поток газа направ-
ляют во второй и последовательно подключенный к нему тре-
тий адсорбер со свежеотрегенерированным адсорбентом. Темпе-
ратуру слоя в конце регенерации рекомендуется поддерживать
на уровне 230—315 °C.
Использование для регенерации цеолитов циркулирующего
азота в значительной степени удорожает процесс. Поэтому на
некоторых заводах (например, на нефтеперерабатывающем за-
воде Маркус Нук компании Сан Ойл, США) цеолиты регене-
рируют потоком природного газа, нагревая адсорбент до 230—
260 °C [122]. Охлаждение адсорбента до комнатной температу-
ры проводят также природным газом. Но в природном газе со-
держится обычно до 1% диоксида углерода, который частично
поглощается цеолитом NaA, а после переключения адсорбера
на стадию осушки загрязняет выходящий из адсорбера пропи-
лен. Концентрация диоксида углерода в первых порциях осу-
шенного пропилена составляет 400 част, на 1 млн. Эту часть
пропилена приходится возвращать в блок очистки. Только после
пропуска 40—50 л жидкого пропилена через 1 кг цеолита со-
держание диоксида углерода снижается до нормы (рис. 12,52).
Загрязнение пропилена было ликвидировано в результате за-
мены цеолита NaA на цеолит КА, который не поглощает диок-
сид углерода. Уже после пропуска 7 л пропилена через 1 кг цео-
Осушка цеолитами 353
Рис. 12,52. Зависимость содержания диоксида уг-
лерода Cqq в пропилене, осушенном цеолитом
NaA от удельного количества переработанного
пропилена ’•
1 — однократный пропуск; 2 — циркуляция.
лита КА содержание диоксида уг-
лерода в углеводороде снижается
до 1 част, на 1 млн. Динамическая
активность цеолита КА по воде со-
ставляет 10 г/100 г. Цеолиты КА
широко применяют для осушки по-
тока пропилена и других техноло-
гических потоков, содержащих не-
предельные углеводороды. Их при-
менение предусматривают не толь-
ко при строительстве новых, но и при реконструкции действу-
ющих установок осушки, на которых раньше использовали си-
ликагель или алюмогель.
Показателен опыт реконструкции установки осушки крекинг-газов на од-
ном из заводов США [123]. В результате замены оксида алюминия на цео-
лит КА дезактивация адсорбента резко снизилась, а продолжительность экс-
плуатации одной загрузки возросла с 0,5—1 года до 4 лет. После реконст-
рукции количество адсорбента в адсорбере сократилось в три раза, соответ-
ственно уменьшились гидравлическое сопротивление слоя, расход регенера-
ционного газа и тепла, необходимого для проведения стадии десорбции.
Несмотря на относительно высокую стоимость цеолитов, затраты на осушку
уменьшились в полтора раза.
Цеолит КА широко применяется для осушки разнообразных
сред, склонных к разложению и полимеризации: этилена, бути-
лена, бутадиена, стирола и его смеси с «-гексаном, амиленов,
винилацетата, изопрена, дихлорметана, хлороформа, галлоидсо-
держащих хладоагентов и др. [124]. Широкое применение цео-
литы КА получили для осушки этилена и пропилена, отбирае-
мых из подземных хранилищ, газов крекинга, подвергаемых низ-
котемпературному разделению, а также сырья для многочислен-
ных процессов полимеризации и алкилирования. К таким про-
цессам относятся получение изопренового и некоторых других
видов каучука, получение полипропилена и полиэтилена, а так-
же описанное ниже алкилирование с использованием в качестве
катализатора фтористоводородной или серной кислоты.
Осушка пропилена с одновременной очисткой от изопропило-
вого спирта. В ряде случаев технологический поток необходимо
не только осушить, но глубоко очистить от нежелательных при-
месей, например пропилен, используемый для получения поли-
пропилена. В процессе полимеризации на стадии промывки по-
лимера от катализаторного комплекса непрореагировавший про-
пилен загрязняется изопропиловым спиртом. Отработанный про-
пилен составляет значительную часть от общего объема сырья,
23—1346
35 4 12. Осушка газов и органических жидкостей
поступающего на полимеризацию. Возвращение его в цикл яв-
ляется важной задачей. Но для этого нужно очистить пропилен
от спирта. Остаточное содержание спирта не должно превышать
3 част, на 1 млн.
Использование в процессе полимеризации сырья с повышен-
ным содержанием спирта вызывает увеличение расхода катали-
затора и может привести к полному прекращению реакции. Кро-
ме того, при полимеризации загрязненного сырья снижается вы-
ход стереорегулярного полимера. До последнего времени в про-
мышленности очистку пропилена проводили с помощью ректи-
фикации с большим флегмовым числом на двух колоннах, име-
ющих суммарно не менее 100 фактических тарелок. Процесс
ректификации недостаточно стабилен, наблюдаются случаи «про-
скока» спирта, что приводит к повышению его содержания в
регенерированном пропилене.
По комбинированной схеме [125], представленной на рис. 12,53, пред-
варительную очистку пропилена ведут в двух ректификационных колоннах 1
и 2. Примесь спирта в исходном пропилене достигает 250 част, иа 1 млн.,
после первой колонны оно снижается до 100 част, на 1 млн., а после второй
до 20 част, на 1 млн. Глубокую очистку проводят цеолитом NaX в двух ад-
сорберах 9 и 10. Остаточное содержание спирта в чистом продукте не пре-
вышает 3 част, на 1 млн. Одновременно со спиртом из пропилена практиче-
ски полностью удаляется вода. Суммарная адсорбционная емкость цеолита
по спирту и воде составляла (14—17) г/100 г. При снижении влажности
исходного сырья повышается емкость цеолита по спирту и снижается норма
адсорбента на очистку сырья: 1 кг цеолита очищает до 10 000 кг пропилена.
Промышленные испытания комбинипованной ректификацион-
но-адсорбционной установки подтвердили ее эффективность.
8 — теплообменники; 9, 10 — адсорберы.
Осушка цеолитами
355
Рис. 12,54. Зависимость динамической активности слоя различных цеолитов при осуш-
ке газа, содержащего пары соляной кислоты, от количества, прошедшей кислоты gHC.
на 1 кг цеолита:
1 — GaY; 2 — NaX; 3 — KNa3; 4 — NaA.
Рис. 12,55. Зависимость динамической активности дд слоя цеолита NaX прн осушке газа,
содержащего пары соляной кислоты, от количества прошедшей кислоты gHCI на 1 кг
цеолита при различной температуре на стадии регенерации tp.
Осушка газов риформинга. При осушке циркуляционного во-
дородсодержащего газа риформинга выбор типа адсорбента сле-
дует проводить с учетом того, что в газе присутствуют микропри-
меси соляной кислоты. В многоцикловых опытах ;[126] были
оценены эксплуатационные свойства цеолитов различных типов
при осушке газа, содержащего 100—150 мг/м3 8%-ной соляной
кислоты.
Опыты проводили при 25 °C и объемной скорости 2500 ч_|. За момент
«проскока» принимали повышение точки росы осушенного газа до минус
50 °C. Десорбцию вели подъемом температуры слоя цеолита до 350 °C с од-
новременной продувкой сухим (точка росы минус 28 °C) азотом в течение
2—4 ч. Соляная кислота преимущественно и количественно сорбируется все-
ми типами цеолитов.
В процессе испытаний выяснилось, что активность цеолитов
общего назначения, а также CaY резко падает от цикла к циклу
(рис. 12,54). Исключение составляет кислотостойкий цеолит —
эрионит КЙаЭ. Его динамическая активность после 18 циклов
соответствовала активности свежего образца (9,2 г/100 г). Сте-
пень разрушения цеолитов общего назначения, например NaX,
снижается, если нагрев цеолита на стадии регенерации прово-
дить до температуры ниже 250 °C (рис. 12,55). Но следует ука-
зать, что полная десорбция соляной кислоты из цеолитов обще-
го назначения достигается только при температуре 450 °C, а из
эрионита — при 250—300 °C. В США для осушки циркуляцион-
ного водорода на установках риформинга используют кислото-
стойкий цеолит AW-500.
Осушка сырья на установках алкилирования. На заводах ал-
килирования в качестве катализаторов используют плавиковую
или серную кислоту. Эффективность и экономика процесса алки-
лирования во многом зависят от влажности катализатора, кото-
рая, в свою очередь, определяется влагосодержанием перераба-
23*
356
12. Осушка газов и органических жидкостей
В блоке HF-алкилирования
Рис. 12,56. Схема установки адсорбционной
осушки сырья на заводах алкилирования.
тываемого сырья. Содержание
влаги в исходном сырье обыч-
но составляет 100—450 част,
на 1 млн. Если алкилирование
проводят при пониженной тем-
пературе (например, минус
7°C), некоторая доля влаги
удаляется в результате вымо-
раживания. Задача глубокой
осушки сырья решена с по-
мощью цеолитов [127].
на одном из нефтеперерабаты-
вающих заводов США после ввода установки с цеолитами влаж-
ность сырья была уменьшена до 15 част, на 1 млн. и соответст-
венно влажность катализатора упала с 2—3 до 0,4—1,3 част.
на 1 млн. В этих условиях значительно снизились коррозия ап-
паратуры и расход кислоты. Уменьшилось также образование
фторорганических соединений, алкилирование стало возможным
проводить при более низкой температуре, что привело к улучше-
нию октановой характеристики алкилата. Принципиальные схе-
мы установок осушки сырья на заводах алкилирования пред-
ставлены на рис. 12,56.
На установке HF-алкилирования изобутан и олефин осушают раздель-
но в двух адсорберах. Применительно к процессу НгБОгалкилирования ком-
поненты сырья смешивают и совместно осушают в одном адсорбере, а вто-
рой адсорбер используют для осушки циркуляционного изобутана. Капитало-
вложения в установку осушки составляют 1,3—1,6% капиталовложений в
установку алкилирования в целом, а эксплуатационные расходы 1,0—1,3°/о
соответствующего показателя для всей установки алкилирования. В даль-
нейшем на установках алкилирования намечено использовать цеолиты также
для тонкой очистки сырья от серы и бутадиена.
Осушка масел
Осушка рабочей среды фреоновых холодильных масел. Надеж-
ность и долговечность герметичных холодильных машин во
многом зависит от чистоты хладоагентов («хладонов») и сма-
зочных масел. До 80% образующихся во фреоновых холодиль-
ных машинах загрязнений, вызывающих коррозию системы и, в
конечном итоге, перегорание встроенных электродвигателей, свя-
зано с присутствием влаги.
В системах, использующих фреоны, концентрация воды в
хладоагенте и в масле не должна превышать 10—60 част, на
1 млн. В отечественной холодильной технике часто используют
синтетическое масло (ХФ-22С-16) и минеральное масло
(ХФ-12-14). Растворимость воды в этих маслах различна: при
Осушка цеолитами
357
Рис. 12,57. Изотермы 20 °C (1—3) и 60 °C (4) ад-
сорбции воды из синтетического фреонового хо-
лодильного масла ХФ-22С-16 на цеолитах:
1 — КА; 2 — NaA со связующим; 3, 4 — NaA без
связующего.
обычных температурах в случае
минерального масла она не превы-
шает 100 част, на 1 млн., а в случае
синтетического масла достигает
1300 част, на 1 млн.
В связи с малым влагосодержа-
нием минерального масла адсорбционная способность цеолита
линейно зависит от концентрации воды в нем. При содержании
влаги 50 част, на 1 млн. и температуре 20°C адсорбционная
способность цеолита NaA составляет 3,5 ммоль/г [128]. Для
гигроскопичного синтетического масла, как видно из рис. 12,57,
прямая пропорциональность наблюдается только в области ма-
лого содержания воды в жидкой фазе. Предельная степень за-
полнения адсорбционного объема полостей цеолита не превыша-
ет 20% в случае минерального масла и 50% в случае синтети-
ческого масла.
Адсорбционная способность зависит от типа цеолита и убы-
вает в следующей последовательности: NaA (без связующего)
>NaA>KA.
Роль характера поверхности вторичных пор проявляется в
кинетических свойствах цеолитов [129]. При осушке масла цео-
литами без связующего степень осушки, время защитного дей-
ствия и динамическая активность значительно возрастают. По-
вышение температуры увеличивает скорость поглощения воды.
Рис. 12,58. Выходные кривые при осушке масла ХФ-22-16 цеолитом NaA без связующего
(скорость потока 210-3 м/с, фракция цеолита 1,5—3 мм).
358
12. Осушка газов и органических жидкостей
связующим и без него связано
онного потока. В случае, если
Рис. 12,59. Зависимость высоты работаю-
щего слоя (Lo) и динамической активности
ад от температуры при адсорбции воды
из масла ХФ-22-16 цеолитом NaA без свя-
зующего (скорость потока 210-3 м/с, высо-
та слоя 70 см).
Это наглядно видно из рас-
смотрения выходных кривых
процесса, представленных на
рис. 12,58.
Различие в скоростях ад-
сорбции воды цеолитами со
с разным характером диффузи-
поверхность транспортных пор
имеет «разломы», миграция вещества к микропорам, сосредо-
точенным в кристаллах цеолитов, определяется только диффу-
зией в объеме пор. В цеолитах без связующего ускорение
транспортирования адсорбата (влаги) обусловлено дополни-
тельной поверхностной диффузией его молекул по полностью
связанной поверхности вторичных пор, образованной сростками
кристаллов цеолитов.
Высокая скорость поглощения влаги позволила при исполь-
зовании цеолита без связующего ограничиться высотой слоя в
адсорбционных патронах 6—12 см. При скорости потока
0,15 м/мин и температуре 60 °C минимальная концентрация во-
ды в синтетическом масле на выходе из адсорбера составляет
15 част, на 1 млн., а динамическая активность слоя цеолита
(фракция 1,0—1,5 мм) высотой 0,7 м достигает 13 г/100 г. Пока-
затели осушки минерального масла в тех же условиях: мини-
мальная концентрация воды 5 част, на 1 млн., динамическая ак-
тивность 3 г/100 г.
На рис. 12,59 приведена зависимость высоты работающего
слоя и динамической активности при осушке синтетического мас-
ла цеолитом NaA без связующего от температуры. Наименьшая
высота работающего слоя и соответственно наибольшая динами-
ческая активность наблюдаются при 60 °C. При температуре
выше 60 °C равновесная адсорбционная емкость понижается, вы-
сота работающего слоя увеличивается, что приводит к умень-
шению динамической активности слоя цеолитов. Глубина осуш-
ки холодильных масел определяется тщательностью проведения
предшествующей стадии регенерации цеолитов. Для холодиль-
ных установок регенерацию цеолита рекомендуется проводить
сухим продувочным газом (воздухом или азотом) при темпе-
ратуре 400—450 °C в течение 3,5—4 ч.
Многократное использование цеолитов при осушке фреоновых
холодильных масел обусловило необходимость удаления масла
с внешней поверхности и из вторичной пористой структуры пе-
ред десорбцией. В противном случае масло закоксовывается и
Осушка цеолитами
359
процесс десорбции сопровождается выделением продуктов сго-
рания масла. В связи с этим разработан способ отмывки масла
фреоном, исключающий потери масла при регенерации. Схема;
установки по отмывке замасленного цеолита приведена на.
рис .12,60 [130, 131].
Отработанный при адсорбции цеолит помещают в патрон 1; жидкий фре-
он поступает в патрон из ресивера 8, проходит через слой цеолита снизу
вверх, вымывает масло из цеолита и далее дросселируется в регулирующем
вентиле 4. В испарителе 6 фреон выкипает при обогреве наружным воздухом
и далее отделяется от маслофреоновой флегмы в сепараторе 3, всасывается
компрессором 12 и нагнетается в маслоотделитель 10 для удаления капель-
ного масла, унесенного из компрессора, конденсируется (7) и стекает в ре-
сивер 8.
По окончании отмывки вентиль на ресивере 8 закрывают, а систему
(патрон 1—испаритель 6 — сепаратор 3) вакуумируют. Затем патрон 1 со-
единяют с воздухонагревательной установкой. Для освобождения испарителя
6 и сепаратора 3 от масла предусмотрен вентиль 5. Масло, унесенное из
компрессора, по мере накопления в маслоотделителе возвращается обратно
в картер через поплавковый вентиль на линии 13. С целью уменьшения энер-
гозатрат и понижения давления конденсатор охлаждают воздухом, предва-
рительно охлажденным в испарителе.
При эксплуатации фреоновых холодильных машин в их ра-
бочей среде образуются органические кислоты, которые вызы-
вают, наряду с другими причинами, перегорание встроенного в
систему электродвигателя. Для обеспечения долговременной и
надежной эксплуатации машин предельно допустимые концент-
рации по кислотам не должны превышать кислотного числа
Рис. 12,60. Схема установки отмывки цеолита от масла фреоном:
1 — патрон с адсорбентом; 2, 9 — манометры; 3 —сепаратор; 4 — поплавковый регулирую-
щий вентиль; 5 — линия спуска масла; 6 — испаритель; 7 — конденсатор; 8 — ресивер; 10 —
маслоотделитель: 11 —байпас; 12 — компрессор; 13 —линия возврата масла в компрессор.
360 /2. Осушка газов и органических жидкостей
0,05—0,6 мг КОН на 1 г; в то же время кислотное число масла
в поступающих на ремонт агрегатах достигает 1,8 мг КОН на
1 г. Это является основной причиной выхода из строя холодиль-
ных машин. Если по технической характеристике моторесурс
выпускаемых машин должен составлять 10—15 лет, то по дан-
ным на 1970 г. около 10% их выходило из строя в первом или
втором году эксплуатации, причем 70% поступало в ремонт
из-за перегорания электродвигателя.
В последнее время глубокую осушку и очистку от органиче-
-ских кислот рабочей среды ведут в патронах с высокопрочным
шариковым сорбентом NaA-2KT, сочетающим свойства цеолита
NaA (осушка) и активного оксида алюминия (очистка). Его вы-
пускает Салаватский химический комбинат и используют для
оснащения как фреоновых холодильных машин, так и домаш-
них холодильников. Внедрение метода адсорбционной очистки и
осушки при изготовлении, ремонте и эксплуатации фреоновых
холодильных машин позволило в три раза повысить срок их
службы и дало существенный экономический эффект*.
Осушка трансформаторного масла. Эти масла широко приме-
няют для изоляции в трансформаторах. Качество масла зависит
от его пробойного напряжения, а на этот показатель в большой
степени оказывает влияние влага, присутствующая в масле.
Раньше способы осушки трансформаторных масел базировались
в основном на нагревании их до температуры кипения воды.
Иногда осушку масел проводили под вакуумом. Все варианты
дистилляционного метода страдают существенными недостат-
ками.
Уже первые лабораторные испытания процесса осушки
трансформаторного масла цеолитами позволили установить пер-
спективность адсорбционного метода [132]. Контактирование
масла с цеолитами проводится при обычных температурах и
вследствие этого не вызывает окислительных процессов, про-
текающих при нагреве. Указанные преимущества и простота
аппаратурного оформления адсорбционного процесса позволили
приступить к широкому использованию осушки трансформатор-
ного масла на предприятиях Советского Союза. Благодаря ини-
циативе сотрудников «Оргрэса» созданы установки по промыш-
ленной осушке трансформаторного масла цеолитами, которые,
пройдя испытания, получили положительную оценку и рекомен-
дованы к широкому использованию [133].
* Адсорбционная осушка и очистка применительно к малым холодильным
машинам используется в трех направлениях: при подготовке веществ, исполь-
зуемых в рабочих средах машин, при изготовлении и ремонте агрегатов и
при их эксплуатации. Цеолит NaA-2KT, в частности, предназначен для экс-
плуатируемых холодильников. Специфика применения адсорбентов в каж-
дом конкретном случае детально рассмотрена в книге Малкина Л. Ш., Ко-
лина В. Л. Осушка и очистка малых холодильных машин. Л., «Легкая и
пищевая промышленность», 1982. 150 с. (Прим, ред.)
Осушка цеолитами
361
Первая передвижная промышленная установка для осушки трансфор-
маторного масла цеолитами в нашей стране была изготовлена и пущена в
эксплуатацию в 1965 г. [134]. В настоящее время все монтажные управления
используют адсорбционную осушку масла.
Типовая установка размером 4X2X2,2 монтируется в прицепном авто-
фургоне грузоподъемностью 4,5 т. Она имеет четыре адсорбера с загрузкой
50 кг цеолитов NaA в каждом, маслоиасос производительностью 3000 л/ч,
электроподогреватель масла, электронагреватель воздуха при регенерации
(до 400—450 °C) мощностью 50 кВт.
Осушка жидкостей в лаборатории
Обезвоженные органические жидкости широко применяют в ла-
бораторном эксперименте. Например, реагенты с регламентиро-
ванной влажностью используют в хроматографии при опреде-
лении воды для составления калибровочных смесей; при иссле-
довании свойств катализаторов (определение кислотности) ит. д.
Для лабораторной осушки жидких сред разработана установка проточно-
го типа, схема которой представлена иа рис. 12,61 [135]. Основными эле-
ментами установки являются сырьевая емкость 14, дозатор сырья 12,
адсорбционная колонка 10 и прибор для определения микроколичеств воды
в осушенном продукте 3. Рабочая часть дозатора, обеспечивающего посто-
янную скорость подачи сырья, изготовляется из медицинских шприцев 13
емкостью 5 мл каждый. Для сглаживания пульсации при подаче продукта
в дозатор два плунжера-насоса сдвинуты по фазе иа половину периода.
Привод насоса осуществляется от электродвигателя РД-09 через систему
шестерен и кривошипно-шатунный механизм.
Адсорбционная колонка выполнена из стекла, имеет вверху и внизу
шлифы; высота колонки 1000 мм, внутренний диаметр 12 мм. С целью мно-
гократного использования адсорбента в адсорбционно-десорбционных циклах
колонка оборудована системой регенерации; нагрев адсорбента ведут с по-
мощью печи 11. Пробы осушенного продукта собирают в приемники 5 и 6,
после чего направляют в анализатор влажности.
Прибор 3 основан на использовании метода Фишера и состоит из стек-
лянной ячейки диаметром 50 мм и высотой 130 мм, магнитной мешалки 8
и пневматической микробюретки иа 10 мл с ценой деления 0,02 мл. В ячей-
ке имеется ввод для иглы от медицинского шприца 2; с помощью иглы в
ячейку вводят растворитель или выводят из нее избыточную жидкость. Та-
кая конструкция обеспечивает надежную защиту от проникания внутрь
ячейки влаги из атмосферы. Для той же цели приемники и другие части
оборудованы осушительными патронами (1, 4, 7, 9, 15), заполненными цео-
литами. Точность определения влаги оценивается в 3—5%. При измерении
микроколичеств воды следует применять реактив Фишера, имеющий титр по
воде 1,2—1,4 мг/л. Проба органического вещества может оттитровываться
реактивом Фишера до эквивалентной точки методом потенциометрического
титрования или путем сравнения с окраской «свидетеля».
Если анализируют вещества, плохо растворяющиеся в метиловом спир-
те (бензин, ксилолы и т. п.), в качестве растворителя и для титрования ре-
активом рекомендуется использовать смесн сухого хлороформа или пиридина
с абсолютированным метиловым спиртом, а также смесь сухого тетрахлор-
углерода и абсолютированного метилового спирта в объемном отношении
4 : 1.
В качестве адсорбента для осушки применяют фракцию 1—2 мм цео-
лита NaA, полученную дроблением гранул. Загрузка цеолита составляет
60—65 г. Перед загрузкой цеолит прокаливают в муфельной печи при 450—
362
12. Осушка газов и органических жидкостей
500 °C в течение 4 ч. В последующем регенерацию проводят непосредствен-
но перед стадией осушки в самой установке.
Перед пуском установки пробоотборную систему (5, 6) и анализатор 3
в течение 2—3 ч тщательно продувают сухим инертным газом с точкой ро-
сы не выше минус 60 °C. Осушку жидких сред проводят при скоростях по-
тока 3—5 см/мин для хорошо растворяющих воду продуктов (например,
спиртов) и 7—10 см/мин для веществ, ограниченно растворяющих воду (на-
пример, ксилолов). При выборе температуры на стадии осушки следует
руководствоваться температурой кипения и вязкостью осушаемых жидко-
стей. Напомним, что в некоторых случаях при повышенной температуре
осушка идет более эффективно.
Осушенный продукт поступает в отградуированный приемник 5, из ко-
торого основную часть продукта направляют в лабораторный реактор или
сборник. Одновременно часть продукта отбирают в приемник 6 емкостью
около 10 мл, а из него — в анализатор влажности 3.
Рис. 12,61. Схема лабораторной установки для осушки жидких сред:
I, 4, 7, 9, 15 — осушительные патроны; 2 — медицинский шприц; 3 — прибор для опреде-
ления влажности продукта; 5, 6 — приемники; 8 — магнитная мешалка; 10 — адсорбцион-
ная колонна; 11 —печь; 12 — дозатор; 13 — шприцы; 14 — сырьевая емкость.
Промышленная очистка газов
363
Перед регенерацией адсорбента производят отдувку осушаемой жидко-
сти, находящейся в свободном объеме колонки, а также адсорбированной
на внешней поверхности гранул и во вторичных порах цеолита. Регенерацию
проводят при 300 °C с одновременной продувкой инертным газом; влагосо-
держание продувочного газа соответствует точке росы не выше минус 30 °C.
13
ПРОМЫШЛЕННАЯ
ОЧИСТКА ГАЗОВ
Подавляющее большинство газов, применяющихся в тех или
иных химических процессах, содержат примеси. Нередко эти
примеси являются каталитическими ядами, веществами, корро-
дирующими аппаратуру и приводящими к нарушениям рабочего
режима. Вследствие этого такие газы подвергают промышлен-
ной (технологической) очистке, причем требуемая степень уда-
ления примесей выбирается в каждом конкретном случае от-
дельно [136]. Значительную роль, особенно в последнее время,
для решения задачи промышленной очистки газов, приобрели
адсорбционные методы. Примерами удачного использования ад-
сорбентов в этом направлении могут служить процессы удале-
ния диоксида углерода, сульфида водорода и сероорганических
соединений из природных и других газов.
Ярким примером негативного воздействия каталитических ядов является
снижение активности никелевого катализатора в присутствии сульфида во-
дорода и других сернистых соединений при получении водорода для синтеза
аммиака. Как известно, этот процесс основан на взаимодействии метана, а
также его гомологов с водяным паром, кислородом и диоксидом углерода
при температурах от 600 до 1100 °C по следующим реакциям:
СН4 + Н2О = СО + ЗН2 — 206,4 кДж (13.1)
СО-|-Н2О = СО2 + Н2-|-41,0 кДж (13.2)
СН4 + СО2 = 2СО + 2Н2 — 248,3 кДж (13.3)
СН4 + 1/2О2 = СО-|-2Н2 + 35,6 кДж (13.4)
В Советском Союзе для осуществления этого процесса используют ка-
тализатор, содержащий от 5 до 10% оксида никеля на носителе из оксида
алюминия, выполненном в форме таблеток или колец диаметром 8—15 мм.
Предварительно оксид никеля восстанавливают до активной формы — ме-
таллического никеля — в токе водорода.
364
13. Промышленная очистка газов
В результате взаимодействия с сульфидом водорода активные центры
никеля переходят в моносульфид никеля NiS и другие сульфиды с общей
формулой NixS. При этом отравление никелевого катализатора наступает
при настолько малых концентрациях сульфида водорода, что они намного
ниже равновесного давления над сульфидом никеля. Вследствие этого ис-
ходный газ , применяемый в этом процессе, требуется очень тщательно очи-
стить— до остаточного содержания общей серы 1 мг в 1 м3.
Процессы очистки газов от диоксида углерода, сульфида во-
дорода, сероорганических соединений, аммиака основаны на вы-
сокой избирательности адсорбции этих компонентов технологи-
ческих газов промышленными адсорбентами.
Очистка газов от диоксида углерода
При сравнительно большом парциальном давлении диоксида
углерода и низких температурах для извлечения этого компо-
нента из газа может быть рекомендовано использование микро-
пористого активного угля типа СКТ. Примером благоприятных
условий для такого процесса является тонкая очистка газа от
диоксида углерода в производстве аммиака. Адсорбцию прово-
дят при температуре минус 50 °C и повышенном давлении, а де-
сорбцию— в потоке инертного газа при температуре «20 °C и
давлении, близком к атмосферному >[137]. В табл. 13-1 приведе-
ны данные по равновесной адсорбционной способности актив-
ного угля и цеолитов по диоксиду углерода при обычной и по-
ниженной температуре. Введение некоторых веществ, например
иода, в структуру активных углей, позволяет значительно повы-
сить адсорбционную способность по диоксиду углерода [138].
В случае, если адсорбент используется при обычных температу-
рах, преимущества цеолитов неоспоримы. Молекулы диоксида
углерода достаточно малы: критический диаметр молекул со-
ставляет около 0,33 нм, что позволяет им проникнуть во внут-
Таблица 13-1. Равновесная адсорбционная способность
промышленных адсорбентов по диоксиду углерода (в г/100 г)
Парциаль- ное давле- ние, кПа Температура 20 °C Температура минус 42 °C
СКТ NaA СаА СаХ NaX СКТ СаА
0,01 - 0,7 1,0 - - 0,2 7,5
0,1 0,1 2,8 3,5 0,6 0,7 0,6 16,5
0,6 0,3 7,7 8,6 1,5 2,7 3,0 18,0
1,0 0,6 9,6 И,2 2,2 4,2 5,0 19,0
4,0 1,6 П,4 15,2 3,2 6,7 9,9 20,0
6,6 2,3 12,2 16,7 3,5 7,7 13,2 20,6
10,0 3,1 12,4 17,4 3,8 8,3 16,5 20,8
13,0 3,9 12,8 17,8 4,0 9,0 19,8 21,0
26,0 — 14,9 18,8 4,9 11,0 26,0 21,8
Очистка газов от диоксида углерода
365
реннюю структуру подавляющего большинства цеолитов. Бла-
годаря несимметричному распределению электронной плотно-
сти, молекула диоксида углерода обладает значительным квад-
рупольным моментом.
Взаимодействие диоксида углерода с каркасом цеолита имеет сложный
характер. ИК-спектроскопические исследования [139] показали, что во всех
случаях значительное место отводится физически связанным молекулам ди-
оксида углерода, которые, легко удаляются вакуумированием. Адсорбция ди-
оксида углерода усиливается ион-дипольным взаимодействием молекулы с
катионами, входящими в состав цеолита. Наконец, в некоторых случаях (на-
пример, на цеолитах NaX, LiX, КХ, RbX, MgX) при определенных условиях,
особенно в присутствии воды, возможно образование карбонатных структур
[ИО].
Энергия адсорбции диоксида углерода на цеолитах значительно увели-
чивается в результате взаимодействия квадруполь — катион, а также до-
норно-акцепторных взаимодействий с привлечением неподеленных электрон-
ных пар атомов кислорода и вакантных орбит катионов.
Катионы являются специфическими активными центрами для
молекул диоксида углерода [141]. Величина адсорбции при не-
больших заполнениях находится в прямой зависимости от ко-
личества катионов, приходящихся на единицу массы дегидрати-
рованных цеолитов. При декатионировании цеолитов их актив-
ность по диоксиду углерода в начальной области изотерм сни-
жается, в пределе приближаясь к активности пористого стекла.
Для сравнения следует указать, что при 6,65 кПа и обычной
температуре активность пористого стекла по диоксиду углерода
в 6 раз ниже активности цеолитов. При больших заполнениях
влияние катионной плотности уменьшается и решающее значе-
ние приобретает величина предельного адсорбционного объема
цеолитов.
Специфическое взаимодействие
квадруполя молекул диоксида уг-
лерода с катионами щелочно-зе-
мельных металлов, входящими в
состав кристаллической решетки
цеолитов, проявляется в высокой
избирательности адсорбции этой
примеси из хуже сорбирующейся
газовой среды, что было использо-
вано для разработки ряда новых и
реконструкции старых схем очист-
ки газов: получения защитных ат-
мосфер в металлургической про-
мышленности, подготовки воздуха
Рис. 13,1. Изотермы адсорбции диоксида углерода
на цеолит СаА.
ptмм рт.ст.
366
13. Промышленная очистка газов
к низкотемпературному разделению, очистки этилена перед его
переработкой в полиэтилен и других процессов.
Изотермы адсорбции диоксида углерода на цеолите СаА в
интервале температур от минус 70 до плюс 80 °C представлены
на рис. 13,1. Изотермы указывают, что цеолиты обладают вы-
сокой поглотительной способностью по отношению к диоксиду
углерода, которая при 20 °C достигает 15 г/100 г. Изотермы кру-
то поднимаются в начальной области, что характеризует цеоли-
ты как эффективное средство глубокой очистки. Средние изо-
стерические теплоты адсорбции диоксида углерода на цеолитах
определенного типа составляют: СаА — 36,0; СаХ — 32,5; NaX —
33,0 кДж/моль.
Приготовление контролируемых защитных атмосфер
Совместная адсорбция диоксида углерода и воды на цеолитах.
В результате сгорания природного газа или жидких углеводо-
родов в воздухе получают азотную или азотоводородную ат-
мосферу, содержащую до 12% диоксида углерода и насыщен-
ную парами воды. Полное сжигание углеводородного газа (на-
пример метана) протекает по реакции
СН4 + 2О2 = СО2 + 2Н2О (13.5)
Если в качестве окислителя применить воздух, то основным
«непрореагировавшим» компонентом будет инертный азот со
значительным содержанием диоксида углерода и воды. Для
применения в качестве защитной контролируемой атмосферы
при термической обработке металлов газ должен быть очищен
от примесей, способных к взаимодействию с компонентами рас-
плавленного металла.
Абсорбционные методы очистки контролируемых атмосфер
от диоксида углерода рентабельны в установках большой про-
изводительности, например на металлургических заводах. Как
показывают техно-экономические расчеты, в установках малой и
средней производительности для одновременной очистки от ди-
оксида углерода и осушки экзотермической контролируемой ат-
мосферы предпочтение следует отдать синтетическим цеолитам.
Сведения об использовании цеолитов, преимущественно типа
СаА, для промышленной осушки и очистки защитных атмосфер
приведены в работах ;[ 142—148].
При одновременном поглощении паров воды и диоксида уг-
лерода влага адсорбируется в лобовых слоях, постепенно вытес-
няя из последующих слоев адсорбированный вначале диоксид.
Расчет цеолитового адсорбера для одновременного удаления па-
ров воды и диоксида углерода следует производить независимо-
по каждому компоненту.
Очистка газов от диоксида углерода
367
Рис. 13,2. Распределение адсорбированных
компонентов в слое цеолитов:
а — начало очистки; б — середина стадии
очистки; в — конец очистки; г — начало десорб-
ции; д — конец десорбции.
В стадии десорбции вначале
и достаточно быстро удаляется
диоксид углерода. Выделение
влаги начинается только после
прогрева слоя цеолита. Распре-
деление адсорбированных компо-
нентов в стадиях очистки и де-
сорбции характеризует рис. 13,2.
Температурный режим адсор-
бера в адиабатических условиях
при исходной концентрации ди-
оксида углерода 10% (об.) и
скорости потока 6 м/мин пред-
Очмстка
Поток газа —*
' Десорбция
•«— Поток газа
ставлены на рис. 13,3. Макси-
мальный разогрев в любом сече-
нии слоя, независимо от его уда-
ленности от лобового, составляет
65 °C. К моменту проскока боль-
шая часть слоя цеолитов имеет
температуру выше 40°C. Пере-
нос тепла газовым потоком, на-
гретым в лобовых слоях, прохо-
дит так, что тепловой фронт опе-
режает фронт сорбции.
На рис. 13,4 представлена за-
висимость динамической актив-
ности слоя цеолита СаА от на-
чальнои температуры слоя. В интервале температур 20—60 °C
динамическая активность по диоксиду углерода снижается с 8
до 1 г/100 г. Поэтому в промышленных установках данного ти-
па температура цеолита перед
превышать 25 °C [148].
Поместив адсорбер в жид-
костной термостат, можно от-
вести часть выделяющейся
теплоты адсорбции воды и ди-
оксида углерода. При этом
снижается разогрев слоя (в
среднем на 15 °C) и повышает-
Рис. 13,3. Изменение температуры слоя
цеолита при поглощении диоксида углеро-
да из потока газа (со»1О% об.), / «20 °C).
стадией адсорбции не должна
х,мин
368
13. Промышленная очистка газов
Рис. 13,4* Зависимость динамической активности ад слоя цеолитов (L — 1 м) по диоксиду
углерода от температуры поступающего газа
Рис. 13,5. Зависимость динамической активности ал слоя цеолита СаА (L—1 м) по диок-
сиду углерода от скорости потока w:
1 — образец 1 с частичным отводом тепла; 2 — образец 1 в адиабатических условиях;
3 — образец 2 в адиабатических условиях.
ся его динамическая активность. Так, для цеолита СаА при
температуре термостатирующей жидкости 20°C и скорости по-
тока газа 6 м/мин динамическая активность составляет 9,5 г/
/100 г против 7,9 г/100 г в случае неохлаждаемого адсорбера.
В промышленности охлаждение слоя адсорбента достигается с
помощью внутренних теплообменных элементов.
При высоких концентрациях диоксида углерода, когда про-
цесс адсорбции происходит в слое с переменной температурой
и осложняется поглощением паров воды, установить величину
зоны массообмена затруднительно. В этом случае ограничива-
ются определением зависимости динамической активности от
скорости потока газа. На рис. 13,5 эта зависимость представле-
на при высоте слоя цеолита СаА 1 м. Верхняя кривая характе-
ризует процесс с частичным отводом тепла, а нижние кривые—
процесс в адиабатических условиях. Из положения этих кривых
можно сделать вывод, что для обеспечения высокой адсорбци-
онной способности цеолита при очистке газа защитных атмосфер
скорости газового потока в адсорбере не должны быть выше
0,1—0,12 м/с.
Обычно при очистке защитного газа применяют цеолиты ти-
па СаА. Цеолит этого типа, наряду с высокими равновесными и
кинетическими показателями, сохраняют свою стабильность при
многоцикловой эксплуатации в слабо кислой среде. При ско-
рости потока газа 0,1 м/с время защитного действия слоя цео-
лита в 1 м по диоксиду углерода в промышленных условиях со-
ставляет 40 мин. Процесс десорбции при интенсивном разогреве
Очистка газов от диоксида углерода
369
Рис. 13,6. Изотермы адсорбции диоксида углерода
на дегидратированном и увлажненном цеолите
СаА при 20 °C и различной влажности адсорбен-
та q.
слоя может быть проведен быстрее.
Учитывая наличие влаги в лобовых
слоях цеолита, десорбцию следует
проводить при противоточном на-
правлении газа по отношению к га-
зу на стадии очистки.
Подавляющее большинство тех-
нологических процессов осуществ- /?,ммрт. ст.
ляется на «модифицированных» ад-
сорбентах. Это «модифицирование» связано с неполнотой де-
сорбции паров на предыдущей стадии подготовки адсорбента
или с предадсорбцией различных примесей газовых потоков.
К таким процессам, в частности, относятся процессы одновре-
менной осушки и очистки от диоксида углерода. Трудность глу-
бокого обезвоживания цеолитов приводит к тому, что стадия
очистки протекает не на чистом адсорбенте, а на адсорбенте,,
«модифицированном» остаточной влагой. На рис. 13,6 и 13,7
приведены изотермы адсорбции и зависимость адсорбционной
способности цеолита СаА по диоксиду углерода от оостаточной
влажности адсорбента при различном давлении [149].
В процессе увлажнения последовательно уменьшается пре-
дельный адсорбционный объем Wo, заполняемый молекулами,
диоксида углерода. Для полностью дегидратированного образца
он составляет 0,235 см3/г и по мере увеличения влажности ад-
сорбента q изменяется следующим образом:
q, г/100 г 0,8 2,3 4,0 7,5
№о, см3/г 0,210 0,182 0,165 0,100
В табл. 13-2 приведены данные, характеризующие адсорбци-
онную способность цеолита СаА по диоксиду углерода в про-
центах к адсорбционной способно-
сти полностью дегидратированного
образца для соответствующих изо-
бар.
Наиболее резко присутствие
влаги в цеолите проявляется в сни-
жении адсорбционной способности
в начальной области изотерм, т. е.
при небольшом парциальном давле-
Рис. 13,7. Зависимость адсорбционной способно-
сти а цеолита СаА по диоксиду углерода от его
влажности q при 20 °C н различном давлении.
24—1346
370 13. Промышленная очистка газов
Таблица 13-2. Адсорбционная способность цеолита СаА по СО2 в %
к адсорбционной способности полностью обезвоженного цеолита
Парциаль- ное давле- ние СО2, Па Влажность цеолита, г/100 г
1 2 3 4 5 6 Г
650 74 58 44 32 24 20 16
1300 76 63 54 47 40 32 26
2500 78 68 60 52 44 38 32
5000 85 75 67 61 54 48 41
13000 86 76 70 66 60 53 47
нии диоксида углерода в перерабатываемом газе. Так, при дав-
лении 0,65 кПа при влажности 2—3 г/100 г адсорбционная спо-
собность цеолита снижается вдвое. Если подвергать очистке
концентрированный газ (например, если давление диоксида уг-
лерода составляет 13,3 кПа), то падение адсорбционной спо-
собности в два раза происходит при влажности 6,5 г/100 г. Ус-
ловия обезвоживания цеолитов выбираются с учетом парциаль-
ного давления диоксида углерода в перерабатываемом газе.
Содержание влаги в цеолите после регенерации определяет
динамическую активность на стадии очистки. На рис. 13,8 пред-
ставлена зависимость динамической активности цеолита СаА на
•опытной установке получения экзотермической атмосферы в за-
висимости от качества предыдущей регенерации. Чтобы в про-
мышленных условиях достичь динамической активности цеоли-
тов по диоксиду углерода 4,5—5,0 г/100 г, температура слоя в
конце регенерации должна быть не менее 250 °C.
Процессы очистки экзогаза с терморегенерацией цеолита.
Известны разные конструкции установки для получения защит-
ной атмосферы из природного газа. Схема опытно-промышлен-
ной установки, в которой нагрев цеолитов в стадии десорбции
производится потоком горячего воздуха, представлена на
рис. 13,9 [150]. '
Природный газ сжигают в газогенераторе 1 с небольшим недостатком
воздуха. Продукты неполного сгорания, содержащие СО2, после охлаждения
в воздушном теплообменнике 3, в водяном холодильнике 4 и выделения вла-
ги поступают в один из трех адсорберов (5,
6 или 7), заполненных цеолитами СаА, где
происходит осушка газа и очистка его от
СО2. Диаметр адсорберов 688 мм, высота
1070 мм, количество цеолитов в каждом ад-
сорбере 272 кг.
Регенерацию цеолита ведут воздухом,
подаваемым вентилятором 2 и нагретым в
теплообменнике 3. Расход воздуха 350 м3/ч,
Рис. 13,8. Зависимость динамической активности
ад слоя цеолита СаА (L«l м) от температуры
слоя t в конце предыдущей стадии иагрева.
Очистка газов от диоксида углерода
371
температура 315 °C. Цеолиты на стадии десорбции нагреваются до температу-
ры не ниже 235 СС. Охлаждают цеолиты чистым газом, циркулирующим через,
систему и холодильник 8 с помощью газодувки 9, причем в начальный период
охлаждения (6 мин) газ после продувки через цеолиты выбрасывают в атмо-
сферу, а затем он циркулирует в замкнутом контуре с постоянной подпиткой
свежим газом (10% от производительности установки).
Потребность получает 57 м3/ч защитной атмосферы, содер-
жащей менее 0,005% О2 и имеющей точку росы минус 60 °C.
Содержание диоксида углерода в готовом газе колеблется от-
0,1 до 0,001%. Продолжительность каждой стадии составляет
1 ч.
Необходимость более тонкой очистки газа от кислорода по-
требовала полной изоляции цеолитов от регенерирующего воз-
духа. Для этого нагрев цеолитов в некоторых установках ведут
от внешнего теплоносителя через стенку. Установки, основан-
ные на таком принципе, обеспечивают получение газа с кон-
центрацией кислорода менее 0,0005%. Однако это приводит к
увеличению габаритов адсорберов, расхода и температуры воз-
духа.
Так, промышленная установка фирмы Линдберг (США), схема которой
принципиально не отличается от описанной выше (но с адсорбером, выпол-
ненным в виде теплообменника), при той же производительности и том же
количестве цеолитов имеет адсорберы диаметром 1010 мм и высотой 1670 мм,
а расход воздуха, подаваемого при регенерации в межтрубиое пространство,
составляет 1350 м3/ч. Температура иагрева цеолитов 230 °C. При этом гото-
Рис. 13,9. Схема установки для приготовления очищенной экзотермической атмосферы:
с регенерацией цеолитов горячим воздухом:
I — газогенератор; 2 — вентилятор; 3 — газовоздушный теплообменник; 4, 8 — водяные хо-
лодильники; 5 — адсорбер на стадии охлаждения; 6 — адсорбер иа стадии очистки; 7 —
адсорбер иа стадии регенерации; 9 — циркуляционная газодувка.
24
372
13. Промышленная очистка газов
Узел смешения ' Узел сжигания Узел конверсии СО Узел охлаждения
вая экзотермическая защитная атмосфера содержит менее 0,01% СОг и име-
ет точку росы минус 58 °C. Фирма выпускает установки производительностью
от 28 до 280 м3/ч.
О выпускаемых в зарубежных странах промышленных уста-
новках, предназначенных для получения очищенной экзотерми-
ческой контролируемой атмосферы с использованием цеолитов,
наиболее полные данные имеются по установкам типа «Нитро-
мат» фирмы Айхелин (ФРГ). На рис. 13,10 представлена техно-
логическая схема установки производительностью 100 м3/ч.
Установка состоит из узлов смешения, сжигания, конверсии оксида уг-
лерода, охлаждения и очистки от диоксида углерода и паров воды. Исход-
ное газообразное топливо и. воздух поступают в узел смешения, откуда га-
зовоздушную смесь подают в узел сжигания, который состоит из камеры
сжигания 1 и газовоздушного теплообменника 2, где продукты неполного
сгорания исходного газа отдают тепло циркулирующему в системе воздуху,
предназначенному для регенерации цеолитов. Недостаток тепла компенсиру-
ется электронагревателем 3, установленным на воздухопроводе. После узла
сжигания предварительно охлажденные продукты неполного сгорания по-
ступают в конвертор оксида углерода 4, где процесс ведут водяным паром
при 330—380 °C. Так как конверсию проводят без предварительной очистки
газа от диоксида углерода, процесс идет до достижения равновесного отно-
шения СО : СОг, т. е. превращение СО в СОг идет не полностью. Из узла
конверсии газ поступает в водяной холодильник 5, охлаждается до 20 °C,
а затем направляется в узел очистки от диоксида углерода и водяных паров.
Основу узла очистки составляют три адсорбера 6. СО2 и Н2О поглощают
в трубчатых адсорберах, заполненных цеолитами. Для отвода тепла, обра-
зующегося при адсорбции диоксида углерода и водяных паров, в воздушных
рубашках адсорберов циркулирует холодный воздух, подаваемый вентиля-
тором. Из узла очистки готовая продукция поступает к потребителю.
Регенерацию цеолитов ведут их нагревом до 300 °C с помощью цирку-
лирующего в системе горячего воздуха.
Очистка газов от диоксида углерода
373
Рис. 13,10. Схема установки «Нитромат» с
трубчатыми адсорберами производительностью
100 м3/ч:
1 — камера сжигания; 2 — газовоздушный теп-
лообменник; 3 — электронагреватель; 4 — кон-
вертор; 5 — водяной холодильник; 6 — адсорбе-
ры; 7 — вентиляторы холодного воздуха; 8 —
циркуляционный вентилятор.
Оригинальное совмещение
циркуляции воздуха с использо-
ванием тепла продуктов сгора-
ния и с электрическим подогре-
вом воздуха сокращает расход
электроэнергии на подогрев ре-
генерирующего воздуха, а соот-
ветственно, и стоимость процес-
са. Производительность устано-
вок «Нитромат» составляет от
50 до 200 м3/ч осушенной и очи-
щенной экзотермической контро-
лируемой атмосферы.
Очистку продуктов сжигания природного газа на установке
голландской фирмы Смит ведут в трех адсорберах, заполнен-
ных цеолитами, в три стадии: адсорбция, регенерация и охлаж-
дение цеолитов. Управление блоком очистки может осуществ-
ляться вручную или автоматически. Влажность готового газа
соответствует точке росы минус 70 °C. Содержание кислорода в
газе не превышает 0,001%.
Своеобразно решен метод осушки и очистки от СО2 экзотер-
мической контролируемой атмосферы в установках «Интергаз-
генератор» фирмы Эбнер (Австрия). Процесс очистки проходит
г две стадии: абсорбция водой под давлением и адсорбция син-
тетическими цеолитами. Фирма Эбнер выпускает установки на
10—250 м3/ч очищенного экзогаза, содержащего 3—5% Н2, 3%
СО, менее 0,01 % СО2, менее 0,0003% О2 и азот (остальное);
точка росы газа минус 55°C. Из-за очень низкого коэффициен-
та избытка воздуха при сжигании природного газа в экзогазе
присутствует водород.
Безнагревные процессы очистки экзогаза. В «Экзотермиче-
ском генераторе атмосферы» фирмы Бирлег (Англия) процесс
ведут по методу короткоцикловой безнагревной адсорбции под
давлением (рис. 13,11).
Продукты неполного сгорания получают в газогенераторе 1, охлажда-
ют и сжимают в одноступенчатом компрессоре 3 до давления 0,56 МПа.
После доохлаждения в водяном холодильнике 4, удаления капельной влаги
и масла в сепараторе 5 газ поступает на очистку в один из двух цилинд-
рических адсорберов с цеолитами 6. Для предотвращения колебаний давления
перед компрессором и после адсорберов установлены буферные баки —
ресиверы 2. Очищенный газ может подаваться к потребителю с максималь-
ным давлением 3,5 МПа. Регенерацию ведут без нагрева — продувкой цеоли-
374 13. Промышленная очистка газов
та чистым газом при атмосферном давлении в направлении, обратном потоку'
основного газа. Продолжительность регенерации (как и адсорбции) 10 мин.
Производительность установки 158 м3/ч. Содержание СО2 в очищенном газе-
0,1—0,4% (в зависимости от исходного содержания СО2, т. е. от коэффици-
ента расхода воздуха при сжигании). Содержание О2 в газе не превышает.
0,0002%. Система управления процессом отличается достаточной простотой
и надежностью в эксплуатации.
Короткоцикловая безнагревная установка, демонстрировав-,
шаяся на международной выставке АСНЕМА—70, обеспечивает
получение из природного газа очень чистого азота, предназна-
ченного для применения в производстве ферритов и полиамидов.
Немецкие установки, работающие по принципу короткоцикло-
вой безнагревной адсорбции, рассчитаны на производительность
до 300 м3 защитного газа в час [151]. Адсорбцию ведут при дав-
лении 0,6 или 1,5 МПа, переключение производят через 5—-
15 мин.
Рис. 13,11. Схема короткоцикловой безнагревной установки фирмы Бнрлег для получе-
ния защитной атмосферы:
1 — газогенератор; 2 — ресиверы; 3 — компрессор; 4 — холодильник; 5 — сепаратор; 6—•
адсорберы; 7 — расходомер.
Очистка газов от диоксида углерода
375
Фирма Чугай—Ро (Япония) выпускает установки типа
«NX-генератор» производительностью 7—140 м3/ч очищенного
экзогаза. Состав готовой контролируемой атмосферы: менее
0,1% СО2; менее 0,5% СО; 0,5—2,0% Н2; остальное N2. Влаж-
ность соответствует точке росы минус 40 °C и ниже. Очистку от
диоксида углерода и водяных паров проводят с помощью син-
тетических цеолитов при атмосферном давлении. В отличие от
рассмотренных выше установок цеолиты регенерируют путем
вакуумирования адсорбента. Узел очистки состоит из двух ад-
сорберов, ресиверов и двух вакуумных насосов с пусковым обо-
рудованием.
Адсорберы представляют собой цилиндрические вертикаль-
ные аппараты. Процесс ведут в две стадии: адсорбция и регене-
рация. Постоянство расхода газа на стадии очистки достигается
с помощью ресивера, соединенного с адсорбером. Переключение
адсорберов со стадии адсорбции на регенерацию и обратно осу-
ществляется с помощью автоматических соленоидных задви-
жек. Снижение давления в процессе регенерации обеспечивается
вакуумными насосами с трехфазными электродвигателями мощ-
ностью 22 кВт.
Такие варианты очистки защитных экзотермических атмос-
фер исключают необходимость стадии охлаждения, так как к
моменту окончания стадии регенерации температура адсорбен-
та понижается до необходимого уровня. Кроме того, на выхлопе
из вакуумного насоса собирают десорбат достаточно высокой
концентрации. Некоторое увеличение температуры на стадии
адсорбции, хотя и влечет за собой снижение емкости адсорбента,
однако приводит к улучшению условий регенерации и тем са-
мым— к уменьшению содержания диоксида углерода в выходя-
щем газе. Результаты опытов подтвердили это предположение
,[152]. В последнее время удельный вес установок с вакуумной
регенерацией в мировой практике значительно возрос.
В Советском Союзе прошла испытания и рекомендована в
серийное производство установка ЭКВ-0-М2 производительно-
стью до 15 м3/ч [153]. Технологическая схема узла очистки пред-
ставлена на рис. 13,12.
Охлажденные до 20—25°C продукты горения поступают в трубки, за-
полненные цеолитами, где происходит поглощение диоксида углерода и во-
дяных паров. Для нагрева цеолитов во время регенерации используют подо-
гретый в калорифере 1 воздух, который с помощью вентилятора прокачи-
вается через межтрубное пространство адсорбера. Охлаждение слоя ведут
также через стенку воздухом, подаваемым вентилятором 4 через охлади-
тель-скруббер 3.
Во время регенерации через слой продувается небольшой поток очищен-
ного экзогаза, который отбирают нз линии готового продукта. Переключе-
ние адсорберов проводят автоматически системой электромагнитных клапа-
нов. Очищенный от СО2 и осушенный экзогаз направляют потребителю.
Длина, ширина и высота установки соответственно составляют 4050,
1500 и 3725 мм; ее масса 2650 кг. В каждом адсорбере находится по 91 тру-
376
13. Промышленная очистка газов
Рис. 13,12. Схема установки ЭКВ-О-М2 конструкции ВНИИЭТО производительностью до-
15 м3/ч:
I — калорифер; 2, 4 — вентиляторы; 3 — скруббер-промыватель; 5 — адсорберы; I — газ на.
очистку; II и III — вход н слив воды; IV — отбор газа на анализ; V — на свечу; VI —
экзогаз потребителю.
бе (внутренний диаметр 33 мм). Трубы заполнены цеолитом СаА в вида
гранул диаметром 3,2 мм. Загрузка цеолита в каждом адсорбере 90 кг, вы-
сота слоя 1,5 м. На стадии очистки в межтрубное пространство подают
820 м3/ч холодного воздуха. Экзогаз при испытаниях содержал 10% СО2.
При расходе исходного газа 14 м3/ч и скорости потока в трубках адсорбера
0,3 л/(см2-мин) проскок диоксида углерода был зарегистрирован через
3 ч 10 мин.
Нагрев цеолита ведут воздухом при температуре на входе в адсорбер
460 °C; его расход составляет 200 м3/ч. Прогрев слоя цеолита до 200—
340 °C заканчивается за 3 ч, а охлаждение — за 2 ч.
Кроме этих установок в нашей стране выпущена серия уста-
новок производительностью 125 м3/ч очищенной экзотермической
атмосферы [154]. Установки ЭК-125-0М2 имеют конфигурацию
пластинчатого теплообменника, в котором плоский слой цеолита
нагревают и охлаждают через стенку. Установки внедрены на
предприятиях электротехнической, радиоэлектронной, машино-
строительной, автомобилестроительной и других отраслей про-
мышленности*.
Очистка и осушка воздуха перед его
низкотемпературным разделением
Двухступенчатый метод подготовки воздуха к разделению при
низких температурах, в котором стадии очистки и осушки раз-
* В СССР также разработаны установки производительностью до 500 мг1ч.
Удаление диоксида углерода и паров воды в них производят в слое цеоли-
тов NaX при атмосферном давлении, регенерацию — вакуумированием слоя
с одновременной небольшой продувкой его очищенным газом. (Прим. ред.).
Очистка газов от диоксида углерода
377
граничены, имеет значительные недостатки. В качестве погло-
тителя диоксида углерода обычно применяют раствор щелочи.
К числу основных недостатков традиционного метода следует
отнести невысокую и нестабильную во времени степень очистки
(остаточное содержание диоксида углерода 10—15 част, на
1 млн.), громоздкость аппаратуры, значительный расход щело-
чи. Силикагель и алюмогель рекомендуют для очистки воздуха
только при низких температурах — от —50 до —80 °C, что
усложняет аппаратуру и схему процесса.
В установках низкотемпературного- разделения воздуха при-
меняются давления до 20 МПа. В этих условиях для определе-
ния равновесных и кинетических параметров при расчете ад-
сорбционного процесса использование данных, полученных в
обычных условиях, приводит к значительным погрешностям.
В табл. 13-3 представлена зависимость динамической актив-
ности цеолита СаА по диоксиду углерода от давления при вы-
соте слоя 0,9 м и температуре 20 °C; размер зерна цеолита 2—
3 мм [155]. Данные таблицы указывают, что динамическая ак-
тивность при постоянной производительности проходит через
максимум, соответствующий давлению 10 МПа. Скорость га-
зового потока значительно влияет на величину динамической ак-
тивности. При высоких скоростях активность слоя цеолитов в
NaX значительно выше, чем цеолитов СаА, что связано с более
медленной диффузией молекул диоксида углерода через вход-
ные окна кристаллита СаА. Скорость адсорбции, от которой за-
висит величина работающего слоя, значительно выше у цеоли-
тов NaX; именно диффузионный фактор определил использова-
ние цеолитов этого типа в промышленных установках подготов-
ки воздуха.
Содержание диоксида углерода в воздухе после очистки цео-
литами составляет менее 3 част, на 1 млн. Одновременно с очи-
сткой происходит его осушка. Точка росы осушенного газа после
дросселирования давления до атмосферного ниже минус 75 °C.
Результаты опытов свидетельствуют о перспективности примене-
ния цеолитов для совмещенного метода очистки воздуха от ди-
оксида углерода и его глубокой осушки.
Таблица 13-3. Зависимость динамической активности слоя
цеолита СаА по диоксиду углерода (г/100 г) от давления
Скорость газа (в расчете на рас- ширенный), м/мии Давление, МПа
2,0 5,0 10,0 15,0 20,0 25,0
100 2,0 2,5 3,0 2,8 2,8 2,7
200 1,8 2,0 2,6 2,0 1,9 1,8
378 13. Промышленная очистка газов
Таблица 13-4. Характеристика промышленных блоков очистки и осушки воздуха
Производи- тельность, м3/ч Давление в систе- ме, МПа Размеры абсорбе- ров, мм Потребляе- мая мощность, кВт Число адсорберов
макс. МИН. диаметр высота
120 20,0 10,0 325 1380 2
400 20,0 10,0 377 1970 22,5 2
1000 7,0 3,5 530 3000 22,5 2
1000 20,0 10,0 465 3790 37,5 2
2400 7,0 3,5 750 3750 37,5 2
2400 20,0 11,0 465 3790 — 4
В 1962 г. впервые на крупном кислородном заводе использован метод
тонкой осушки н очистки воздуха перед его низкотемпературным разделе-
нием, разработанный фирмой Юнион Карбайд Канада [156]. Одновременно
с влагой и диоксидом углерода из газа практически полностью удаляется
ацетилен. Количество перерабатываемого газа составляет 85 м3/мин. Период
стадии адсорбции равен 8 ч. Регенерацию ведут продувкой горячим азотом
противотоком к газу в периоде адсорбции. Расход азота составляет около
8,5 м3/мин. Эксплуатационные затраты примерно в 5 раз ниже, чем при ще-
лочной очистке.
В Советском Союзе была разработана схема и технологиче-
ский регламент опытно-промышленной трехадсорберной установ-
ки подготовки воздуха к низкотемпературному разделению. Ад-
сорберами служили баллоны высокого давления емкостью 500 л.
Регенерацию проводили в результате нагрева цеолитов до
150 °C теплом отходящего азота, а охлаждение — тем же азо-
том при обычной температуре (20 °C).
Экономический расчет показал, что адсорбционная очистка
с применением цеолитов при полной автоматизации управления
на 20% дешевле, чем щелочная очистка, хотя энергетические
затраты на адсорбционный метод несколько выше, главным об-
разом за счет значительных затрат на регенерацию.
При замене оксида алюминия на цеолиты NaX в стандарт-
ных блоках осушки воздуха с адсорберами внутренним диа-
метром 0, 33 м получен сухой воздух, удовлетворяющий требо-
ваниям по содержанию диоксида углерода (не выше 0,001% об.);
одновременно, как указывалось, происходит полное удаление
ацетилена {157].
Испытание метода адсорбционной очистки воздуха в опытно-
промышленном масштабе проведено на стендовых установках.
Результаты испытаний были заложены в основу проектов типо-
вых адсорбционных блоков для промышленных установок раз-
деления воздуха производительностью от 120 до 2400 м3/ч.
Скорость потока в адсорберах составляет 0,5—5 м/мин (в расчете на
сжатый газ); продолжительность стадии очистки 12 ч, нагрева и охлаждения
(вместе)—6 ч. Регенерацию проводят азотом прн 350 °C; минимальная тем-
Очистка газов от диоксида углерода
379
пература в слое цеолита к концу стадии регенерации 220 °C. Скорость пото-
ка азота на стадиях нагрева и охлаждения 20—40 м/мин. Активность цео-
литов по диоксиду углерода составляет 14 см3/г при 10 °C и 7 см3/г при
30 °C. В табл. 13-4 приведены показатели блоков (температура воздуха на
входе 10 °C).
Очистка природного газа от диоксида углерода
и других примесей
Цеолиты способны одновременно удалять основные примеси
природного газа, вследствие чего их устанавливают перед по-
ступлением газа на установку низкотемпературного разделения
[92]. После очистки исходного газа в дальнейшем отпадает по-
требность в очистке полученных продуктов: этанопропановая
смесь не содержит СО2, пропан получается в виде сухого и не-
коррозионноактивного продукта, в газовом бензине отсутствует
сера. Очень низкая точка росы газа (ниже минус 70 °C) и пол-
ное удаление других вымораживающихся компонентов позволя-
ет понизить температуру при разделении. На рис. 13,13 приве-
дена схема установки подготовки природного газа к низкотем-
пературному разделению производительностью 5,6 млн-м3/сут.
Газ содержит 1% СО2, 0,1 г Н2О на 1 м3 и 0,05 г H2S на I м3. Его про-
пускают при температуре 25 °C и давлении 5,1 МПа через два параллельно
включенных адсорбера 1 и 2 диаметром 3 м с высотой слоя 6 м. Выходящий
из адсорберов газ содержит менее 2-10~3% СО2, 0,1% серы и имеет точку
росы ниже минус 70 °C. Регенерацию адсорбента в адсорберах 3 и 4 ведут
частью сухого газа, получаемого в блоке низкотемпературного разделения 8.
Расход газа на регенерацию составляет 1,4 млн. м3/сут. Между адсорберами
3 и 4 установлен нагреватель газа 5. Газ регенерации после охлаждения в
Рис. 13,13. Схема установки очистки природного газа от диоксида углерода и других при-
месей на заводах низкотемпературного разделения:
1, 2 — адсорберы на стадии очистки; 3 — адсорбер на стадии охлаждения; 4 — адсорбер
на стадии нагрева; 5 — теплообменник; 6 — холодильник; 7 — сепаратор; 8 — блок низко-
температурного разделения.
Рис. 13,14. Схема установки очистки природного газа от диоксида углерода перед его
сжижением:
1 — адсорбер на стадии очистки; 2 — адсорбер на стадии охлаждения; 3 — адсорбер на
стадии нагрева; 4 — нагреватель; 5 — холодильник; 6 — сепаратор.
380 13. Промышленная очистка газов
холодильнике 6 и отделения влаги в сепараторе 7 примешивают к магист-
ральному газу.
Метод одновременного удаления примесей из исходного газа
является экономически более выгодным, чем очистка и осушка
полученных продуктов.
В процессе извлечения кислых компонентов (сульфид водо-
рода и диоксид углерода) из природного газа продолжитель-
ность эксплуатации одной загрузки составляет от 2 до 5 лет.
В США находятся в эксплуатации 12 установок, на которых очи-
щают свыше 30 млн. м3/сут. Очистка цеолитами не имеет конку-
ренции во всех случаях, когда требуется очень глубокая степень
очистки, а в исходном газе содержание диоксида углерода не-
высоко (менее 1,5% об.).
В некоторых случаях применяют двухступенчатый метод:
удаление диоксида углерода в абсорбционных колоннах с по-
следующей осушкой цеолитами NaA. В этом случае продолжи-
тельность эксплуатации загрузки увеличивается до 3—7 лет.
Скорость газового потока в цеолитовых адсорберах целесообраз-
но поддерживать в пределах 0,1—0,3 м/с [158].
Адсорбционные блоки с цеолитами устанавливают перед по-
ступлением природного газа на установки его сжижения. Их ос-
новное назначение — удаление диоксида углерода. Решить эту
задачу оказалось легче, чем задачу сероочистки газов, так как
при декарбонизации легче решается проблема реализации га-
зов, выделяющихся при регенерации. Адсорбционные установ-
ки декарбонизации природного газа рентабельны вплоть до про-
изводительности 200 тыс. м3/сут. По другим источникам, верх-
ний предел производительности адсорбционных установок на
порядок больше [159]. На рис. 13,14 представлена схема уста-
новки для сжижения 280 тыс. м3 природного газа в сутки.
Исходный газ содержит 1% СО2 н 0,1 г Н2О на 1 м3. После пропуска
через адсорбер 1 содержание примесей снижается до 20 и 0,1 част, на 1 млн.
по СО2 и Н2О соответственно; одновременно происходит очистка газа от сле-
дов сернистых соединений. Часть очищенного газа (140 тыс. м3/сут.) исполь-
зуют для регенерации. При этом газ последовательно проходит через охлаж-
даемый адсорбер 2, а затем после нагрева (4) через нагреваемый адсор-
бер 3.
Основное различие в схемах адсорбционных блоков на установках низ-
котемпературного разделения и сжижения природного газа заключается в
том, что в последнем случае газ регенерации не может быть примешан к по-
току очищенного газа. Из газа регенерации после его охлаждения в холо-
дильнике 5 в сепараторе 6 выделяют основное количество влаги. Затем газ
с помощью циркуляционной газодувки 7 возвращают в газовую сеть.
В США имеется шесть заводов сжижения природного газа с цеолитовыми
блоками. В Канаде (г. Ричмонд) также имеется установка, где предусмот-
рено применение цеолитов для удаления примесей (СО2, H2S, Н2О) из при-
родного газа перед его сжижением [160].
Широкое применение нашли цеолиты на заводах получения
гелия. Содержание паров воды в природном газе, поступающем
Очистка газов от диоксида углерода
381
на установку выделения гелия, должно соответствовать точке
росы не выше минус 75 °C, а содержание диоксида углерода —
быть не выше 50 част, на 1 млн. На большинстве современных
гелиевых заводов многоступенчатый процесс подготовки при-
родного газа к низкотемпературному разделению (очистка с по-
мощью раствора моноэтаноламина и щелочи с последующей
осушкой силикагелем или алюмогелем) заменен на одноступен-
чатый с применением цеолитов, обеспечивающий содержание
диоксида углерода ниже 1 част, на млн.
Для повышения поглотительной способности цеолитов и
улучшения степени очистки гелия или другого инертного газа
от микропримесей диоксида углерода, а также паров воды,,
в очищаемый газ предложено добавлять небольшое количество
третьего компонента [161]. Этот компонент образует с молеку-
лами примесей химические соединения, обладающие низкой ле-
тучестью и прочно удерживаемые адсорбентом. Так, при добав-
ке в поток очищаемого газа аммиака в результате реакций об-
разуются карбонаты NH4HCO3 и (МН^СОз. Отработанный
цеолит нагревают сначала до 100 °C. При этом карбонаты раз-
лагаются. Затем продукты разложения десорбируют в резуль-
тате вакуумирования при 350 °C.
Очистка этана, пропана, водорода
Диоксид углерода избирательно адсорбируется не только из ме-
тана, но и из его высших гомологов. В связи с этим процесс
очистки углеводородных газов от диоксида углерода распрост-
ранен на этан, пропан и этанопропановую фракцию [162]. На
типовой установке один адсорбер (или два включенных после-
довательно) находится на стадии очистки, один на десорбции
и один на охлаждении. Для регенерации используют часть очи-
щенного продукта, природный газ или иной технологический по-
ток. В США находятся в эксплуатации по крайней мере три та-
ких установки.
Применение цеолитов позволило резко улучшить чистоту
водорода, получаемого каталитической конверсией углеводоро-
дов с паром. Ранее в результате применения щелочной очистки
с последующей ступенью метанирования получали водород чи-
стотой 98,0—99,9%. Использование одноступенчатого адсорб-
ционного метода для очистки от диоксида углерода обеспечило-
получение водорода чистотой до 99,99% [163].
Очистка газов от оксидов углерода
с помощью хемосорбентов
При очистке влажных газов от диоксида углерода цеолит в;
первую очередь поглощает влагу и это в некоторых случаях
приводит к значительному удорожанию процесса. Избиратель-
382
13. Промышленная очистка газов
вое поглощение диоксида углерода может быть достигнуто с
помощью хемосорбентов.
Улучшение равновесных и кинетических свойств хемосорбен-
тов по диоксиду углерода достигается в результате комбиниро-
вания химических ингредиентов и модифицирования поверхно-
сти химическими реагентами. К таким адсорбентам относятся
пористые гранулы, в состав которых входит оксид магния (80%)
.и гидроксид щелочного металла: натрия, калия, рубидия и цезия
(20%) [164]. Добавление гидроксида калия к оксиду магния
приводит к увеличению адсорбционной емкости поглотителя в
2 раза и росту скорости адсорбции в 10 раз.
Получают такой сорбент путем смешения соли 4MgCOs-
•Mg(OH)2-5H2O с водой, в которой растворено некоторое ко-
личество гидроксида. Пасту выдерживают в течение 0,5—1 ч,
'формуют в виде гранул диаметром 3 мм, сушат и подвергают
термической обработке при 500 °C. При этом MgCOs превра-
щается в MgO и адсорбент становится пористым. Затем грану-
лы обрабатывают насыщенным паром, в результате чего их
плотность увеличивается вдвое. Такой адсорбент, в отличие от
цеолитов, влагу не поглощает. Регенерацию гранул ведут тер-
мическим методом.
Оксид углерода относится к плохо адсорбируемым вещест-
вам. Для его удаления из газов рекомендуется либо проводить
адсорбционный процесс при пониженных температурах, либо
применять высокоселективные поглотители. Такой сорбент, поз-
воляющий извлечь оксид углерода из смеси с азотом, согласно
патенту <[165], получают в результате термической обработки
японских природных материалов при 350—700 °C. Основными
компонентами исходных материалов являются оксиды кремния,
алюминия и вода. Отношение оксида алюминия к оксиду крем-
ния в сорбенте составляет 1:10. Сорбент содержит также 10%
(в сумме) оксидов калия, натрия и кальция*.
Очистка атмосферы в изолированных системах
Удаление продуктов жизнедеятельности, в первую очередь вла-
ги и диоксида углерода, в современных изолированных обитае-
мых системах (например, космических кораблях) может быть
проведено по-разному. Одним из эффективных методов, обеспе-
чивающих поддержание концентрации диоксида углерода в изо-
лированной системе на уровне не выше предельной (0,5%) яв-
ляется адсорбция на цеолитах [166—168].
* Высокая емкость по монооксиду углерода материала такого состава, если
только в него не введены активирующие добавки, сомнительна. (Прим, ред.)
г
Очистка, газов от сульфида водорода и меркаптанов 383
Очистка газов от сульфида водорода
и меркаптанов
Сульфид водорода — сильный нервно-паралитический яд. Очист-
ка газа от него и сероорганических соединений осуществляется
в двух направлениях [169]: 1) санитарная очистка отходящих
производственных газов и вентиляционных выбросов; 2) очист-
ка природных, коксовых и других промышленных газов, ис-
пользуемых в качестве сырья для синтеза, а также газов, обра-
зующихся на разных ступенях технологической нитки химиче-
ских и нефтехимических производств. Во втором случае реша-
ются параллельно две задачи. С одной стороны, вредный для
каталитических процессов яд — сульфид водорода выводится из-
схемы и, как правило, преобразуется в товарные продукты: се-
ру, диоксид серы, серную кислоту и т. п. С другой стороны, уда-
ление сульфида водорода и сероорганических соединений снижа-
ет или даже ликвидирует выброс в атмосферу диоксида серы,,
образующегося на окислительных стадиях химического процесса.
Все горючие газы, содержащие сульфид водорода, должны
подвергаться очистке от него. К таким газам, кроме природного
и коксового, относятся все газы нефтепереработки (крекинга, ри-
форминга, гидроочистки и т. д.), сланцепереработки, генератор-
ный газ.
Газы различаются по содержанию сульфида водорода. При-
родные газы, имеющие наибольшее значение для народного хо-
зяйства, могут быть совершенно бессернистыми, но могут содер-
жать до 5 и даже до 20% сульфида водорода. С другой сторо-
ны, имеются газы с очень низким содержанием H2S, но с очень-
большим общим выбросом этого компонента. Так, в вентиляци-
онных выбросах вискозного производства содержится всего
0,01—0,1% H2S, но очистка такого газа обязательна: выброс вен-
тиляционного воздуха в атмосферу только на одном заводе до-
стигает многих миллионов м3/ч.
Требования к степени очистки газа от сульфида водорода
различаются в зависимости от назначения газа. В СССР для
природного и других газов, которые готовят к транспортирова-
нию по магистральным газопроводам и к использованию для
бытовых целей, предельно допустимое содержание сульфида во-
дорода составляет 0,02 г/м3, в газе для производства обычной
мартеновской стали допускается 2—3 г/м3, а для химического
синтеза, в зависимости от процесса, от 1 до 50 мг/м3. Сульфид
водорода, уловленный из газов, в зависимости от метода очист-
ки, выделяется в виде элементной серы или концентрированного
Н25-газа, который перерабатывают каталитически в серную кис-
лоту и элементную серу. По технико-экономическим соображе-
ниям H2S целесообразнее перерабатывать в элементную серу, ко-
торая является дефицитным продуктом. Из всего производства
384
13. Промышленная очистка газов
•серы в капиталистических странах на долю серы, извлекаемой из
газов, приходится около 40%.
Содержание сульфида водорода в воздухе регламентируется
предельно допустимыми концентрациями: в рабочей зоне —
10 мг/м3, а в присутствии углеводородов — 3 мг/м3. Максималь-
но разовая и среднесуточная предельно допустимая концентра-
ция в атмосферном воздухе равна 0,008 мг/м3.
Ючистка активным углем
Активный уголь не только поглощает сульфид водорода из га-
зовой среды, но и катализирует реакцию окисления поглощен-
ного H2S в адсорбированной фазе кислородом до элементной
«серы:
2HaS + О2 -► S2 + 2Н2О + 440 кДж (13.6)
Многочисленные промышленные газы, в первую очередь кок-
совый, подвергают сероочистке активным углем [170, 171]. При
этом адсорбционно-каталитические свойства углей используют,
чтобы не только очистить газ, но и получить товарный продукт —
серу. Иногда процесс сероочистки комбинируют с улавливанием
углеводородов. Если очищают бескислородный газ, требуется
добавка к нему кислорода из расчета, чтобы выходящий газ со-
держал не более 0,1% кислорода.
Следует отметить, что отложение серы в виде кристаллов
происходит в основном в мезопорах и от развития мезопористо-
сти зависит адсорбционная емкость по сульфиду водорода. По
.данным польских исследователей [172], наиболее интенсивно
процесс очистки протекает в порах диаметром от 3,5 до 8,0 нм.
.Для улавливания сульфида водорода рекомендуется, например,
венгерский уголь нуксит LBS II, имеющий суммарный объем
пор 0,8 см3/г и следующее распределение пор по размерам:
Радиус пор, нм До 1,1 1,1—2,0 2,0—10,0 10,0—100,0 Выше 100,0
=Объем пор, см3/г 0,15 0,10 0,15 0,24 0,21
Размер гранул угля нуксита от 2 до 6 мм, насыпная плот-
ность 390 кг/м3, адсорбционная емкость по бензолу 20 г/100 г.
Окисление H2S в присутствии гомогенного катализатора —
аммиака происходит в течение нескольких секунд. Сероемкость
угля при насыщении достигает 100 г/100 г. При выборе типа
угля для улавливания сульфида водорода следует также иметь
в виду, что пропитка адсорбента некоторыми реагентами, в ча-
стности иодом, значительно повышает сероемкость активного
угля и ускоряет процесс образования серы [173—175]. Иодиро-
ванные угли стали применять на практике [176].
Очистка газов от сульфида водорода и меркаптанов 385
Трудность использования активного угля для решения зада-
чи сероочистки долгое время заключалась в том, что при улав-
ливании H2S углем в присутствии кислорода основная реакция
преобразования адсорбата в серу сопровождается побочным
сильно экзотермическим процессом образования серной кислоты:
H2S + 2О2 ---► + 790 кДж (13.7)
Удельный вес побочной реакции, протекающей в порах актив-
ных углей обычных типов, настолько значителен, что при высо-
кой концентрации H2S в очищаемом воздухе слой сильно разо-
гревается и возникает опасность возгорания. Интенсивность об-
разования серной кислоты зависит от содержания в угле метал-
лов, и в первую очередь — железа. При их высоком содержании
более одной трети H2S превращается в H2SO4.
Образующуюся серную кислоту нейтрализуют раствором со-
ды с последующим вымыванием продуктов нейтрализации во-
дой. Впоследствии стали широко применять нейтрализацию сер-
ной кислоты газообразным аммиаком. Образующиеся аммоний-
ные соли вымываются водой легче, чем соли натрия, в связи с
чем расход воды при нейтрализации аммиаком в 4 раза меньше.
Аммиак можно вводить в систему периодически на стадии регенерации
(охлаждения) адсорбента или непрерывно примешивать к очищаемому газу.
В этом случае наряду с основной реакцией образования серы в результате
окисления сульфида водорода на поверхности угля протекают побочные ре-
акции, в результате которых образуются сульфат и карбонат аммония:
2NH3 + H2S + 2О2 --► (NH4)2SO4 (13.8)
2NH3 + СО2 + Н2О --> (NH4)2CO3 (13.9)
Вследствие высокой экзотермичности процесса каталитиче-
ского окисления верхним пределом концентрации H2S в очищае-
мом газе, при которой еще целесообразно применять активный
уголь, принято считать 5 г/м3. Однако известны случаи адсорб-
ционной очистки газов, в которых концентрация сероводорода
достигает 10—13 г/м3. Температура угля на установках с такой
высокой концентрацией сероводорода, эксплуатирующихся в Че-
хословакии и Венгрии, повышается до 70—100 °C.
Для регенерации насыщенного серой угля обычно применя-
ют водный раствор сульфида аммония, который при взаимодей-
ствии с серой превращается в многосернистый аммоний:
(NH4)2S + nS --- (NH4)
A+l (13.10)
Исходный раствор получают насыщением 10—20%-иого раствора аммиа-
ка сульфидом водорода; в 1 л раствора содержится до 150 г H2S. Экст-
ракцию серы проводят в несколько ступеней: сначала используют сильно на-
сыщенные серой растворы, затем слабо насыщенные н только в конце све-
жий раствор. Содержание серы в насыщенном растворе сернистого аммония
достигает 240 г/л.
25—1346
386
13. Промышленная очистка газов:
Отработанный раствор разлагают острым паром (,12Ja-°C, 0,2 МГГа)| с
выделением серы чистотой выше 99,9%:
(NH4)2S„+1 -> (NH4)2S + nS (.13.11 >
Сульфид аммония при пропарке разлагается, но; выделяющиеся пары
NH3 и H2S конденсируют и возвращают в процесс. Затем уголь промывают
водой, в результате чего удаляется большая часть карбоната аммония. Ос-
татки карбоната и сульфида аммония отпаривают в течение 6 ч, конденси-
руют, и разбавленный раствор сульфида аммония возвращают в. процесс.
Чтобы избежать накапливания карбопата аммония в рабочем растворе,,
часть его периодически выводят из цикла и обрабатывают хлоридом кальция
или известковым молоком:
(NH4)2CO3 + Са(ОН)2 -► СаСО3 -% 2NH, + 2НаО (13.12>
2NH3+H2S ----► (NH4)2S (,13.13>
Избыток (NH^JzSO^ удаляют в результате кипячения- раствора с Na2S:
(NH4)2SO4 + Na2S --> Na2SO4 + (NH4)2S (13.14>
Расходные коэффициенты, по заводским, данным,, отнесенные
к 1 т серы, при очистке водяного газа и экстракции раствором
сульфида аммония составляют: электроэнергия — 50 кВт-ч;,
пар — 5 т; вода — 50 м3; аммиак (в пересчете на азот) —40 кг;
раствор сульфида аммония—0,1 м3; активный, уголь—18 кг;
гидроксид кальция — 10 кг; сульфид натрия—0,4 кг.
Другим распространенным экстрагентом серы является кси-
лол. Применение ксилола основано на резкой разнице раствори-
мости серы в нем при обычных и повышенных температурах:
при 25 °C в 1 м3 ксилола растворяется 17 кг серы,, а при 100 °C—
188 кг. Экстракцию проводят при 100—НО °C и давлении при-
близительно 0,25 МПа в потоке экстрагента, после чего его
охлаждают до 30 °C. Выкристаллизовавшуюся серу отделяют
центрифугированием. После завершения стадии экстракции
уголь пропаривают для удаления ксилола, промывают водой,
в результате чего удаляются гипосульфит и другие продукты
реакции, и, в заключение, вторично пропаривают.
Смишек и Черны [177] приводят данные об успешной эксплуатации,
опытной установки адсорбционной сероочистки коксового газа в Научно-ис-
следовательском топливном институте (Чехословакия), где для экстракции
серы используют ксилол. Процесс проводят в двух последовательно вклюг
ченных адсорберах: в первом частично отработанный, во втором — свежий
уголь. В процессе очистки содержание серы в угле первого адсорбера повы-
шается с 25—30 до 80—85%, во- втором адсорбере с 0.' до 25—30%. Исход-
ный газ содержит 5 г H2S на 1 м3, газ после очистки 20 мг/м3. Одновремен,
но удаляется 15—20% органической серы и 20% цианистого водорода. Экс-
плуатационные затраты на 1 т выделенной серы составили: электроэнергия—
250 кВт-ч; вода оборотная—144 м3; вода свежая — 7 м3; пар — 5,2 т; ам-
миак — 23 кг; ксилол — 20 кг; активный уголь — 64 кг.
Очистка цеолитами
Цеолиты являются эффективным средством сероочистки газов-.
В табл. 13-5 приведена равновесная статическая активность,
цеолитов NaA, NaX и СаА по H2S при 25, 75 и 150 °C. В свок>
Очистка газов от сульфида водорода и меркаптанов
387
Таблица 13-5. Адсорбционная способность цеолитов по H2S (в г/100 г)
Цеолит Температу- ра, Ю Давление, кПа
0,065 ' 0,13 ’ 0,33 1,33 6,65 13,30 33,25
NaA 25 3,6 4,7 6,4 9,5 12,8 14,0 15,0
75 1,3 2,2 3,6 6,0 8,0 11,5 13,0
150 0,6 0,9 1,4 2,5 5,0‘ 7,5 9,0
СаА 25 3,0 4,8 6,8 10,0 13,6 15,0 16,2
75 1,3 1,7 2,7 7,2 9,6 11,8 13,2
150 0,3 0,4 0,7 2,2 4,3 5,5 7,4
NaX 25 2,8 4,0 7,5 10,5 14,5 16,0 16,2
75 1,0 2,0 4,0 6,0 9,5 11,8 14,0
150 0,0 0,2 1,3 2,0 5,5 6,2 9,2
очередь, масса сульфида водорода, адсорбированного единицей
массы цеолита, определяет повышение температуры слоя при
«сероочистке газа (рис. 13,15). В зависимости от концентрации
H2S в газовой и адсорбированной фазах может быть определена
температура слоя цеолита в конце стадии очистки. Теплота ад-
сорбции сульфида водорода на цеолитах приблизительцо в 2 ра-
за выше теплоты его конденсации.
Цеолиты обладают еще одни'м свойством, имеющим перво-
степенное значение для производства: они селективно извлекают
сульфид водорода из его смесей с диоксидом углерода. При
мольном соотношении в газовой фазе H2S : СО2= 1 : 1 адсорбиро-
ванная .фаза обогащается по H2S до 90% (мол.) [178]. В про-
цессе одновременной очистки газа от сульфида водорода и ди-
оксида углерода в первый период происходит полное удаление
обоих компонентов, затем диоксид углерода начинает вытес-
няться сульфидом водорода, его содержание в выходящем из
адсорбера потоке газа резко возрастает и даже превосходит кон-
центрацию этого вещества в исходном газе. В то же время,
сульфид водорода продолжает количественно поглощаться
вплоть до момента проскока.
Так как диоксид углерода яв-
ляется лишь балластным, а не
корродирующим компонентом
при транспортировании газа,
часто эксплуатационные за-
траты на его удаление превос- °“-
ходят затраты на транспорт. *=
Изменяя продолжительность
адсорбционного процесса,
Уис. 13,15. Повышение температуры в слое о/
Неолита при сероочистке газа.
388
13. Промышленная очистка газов
можно получить любую заданную степень извлечения диоксида
углерода.
В противоположность адсорбционному методу, обычный ме-
тод жидкостной очистки газа эталонамином не обладает изби-
рательностью по кислым компонентам и предусматривает по-
глощение в равной степени как сульфида водорода, так и диок-
сида углерода. Поэтому экономическое преимущество сероочист-
ки газа цеолитами особенно проявляется, если в исходном газе
соотношение CO2:H2S>3. При переработке газа с высоким со-
держанием кислых компонентов на базе газов десорбции может
быть организовано производство серы и твердого диоксида уг-
лерода. Особенно перспективным является применение цеолито-
вой сероочистки в тех случаях, когда содержание сернистых
соединений в части газа строго не регламентируется. Тогда эту
часть газа используют для регенерации |[179]..
На рис. 13,16 приведена схема установки очистки природ-
ного газа от сероводорода и меркаптанов в неподвижном слое
синтетических цеолитов [180].
Установка состоит из четырех адсорберов. В схеме с открытым циклом
сырой природный газ после отделения конденсата в первичном сепараторе 1
и пыли в фильтре 2 последовательно проходит через' адсорберы 3 и 4, где
подвергается сероочистке, и поступает в газопровод. В адсорбере 4 проис-
ходит доулавливание сульфида водорода. Его переключают на место адсор-
бера 3 после того, как тот переведут на стадию регенерации. Часть очищен-
ного газа отбирают и используют сначала для охлаждения цеолита в ад-
сорбере 5, а затем, после нагрева,— для регенерации цеолита в адсорбере 6.
Из газа регенерации в сепараторе 9" отделяют воду и. жидкие углеводоро*-
ды, обычно содержащиеся в природном газе. После этого газ используют в
качестве топлива на местные нужды, например на установках синтеза аммиа-
ка. Таким образом, в каждом адсорбере последовательно проводят следую-
щие стадии процесса: регенерацию, охлаждение; доулавливание, сероочистку.
Рис. 13,16. Схема установки сероочистки природного газа:
I, g — сепараторы; 2 — фильтр; 3—адсорбер на стадии очистки; 4—адсорбер на стадии
доулавливания; 5—адсорбер на стадии охлаждения; 6—адсорбер на стадии регенерации^
7—теплообменник; 8—холодильник; 10—подогреватели
Очистка газов от сульфида водорода и меркаптанов
389
Сульфиды, образующиеся в газопроводе, также поглощаются цеолитами
Для тех случаев, когда объем газа регенерации превышает
потребность в топочном газе, разработана схема с замкнутым
циклом, по которой из газа регенерации в абсорбере, орошае-
мом водой или другими растворителями, выделяют основную
массу сульфида водорода, после чего его примешивают к по-
току сырого газа, с которым он проходит повторную очистку.
Десорбцию сульфида из воды (или растворителя) проводят сни-
жением давления. При очистке газа от меркаптанов в схему с
замкнутым циклом включают дополнительный адсорбер, оро-
шаемый органическим растворителем; регенерацию органиче-
ского растворителя проводят в десорбере при повышенной тем-
пературе.
Стоимость установки такого типа, разработанной компанией Энжинирс
энд Фабрикейторс (EFCO) на производительность по газу 2,8 млн. м3/сут,
включая стоимость 53 т цеолитов, составляет 1 млн. долл, без учета стоимо-
сти осушающей установки, которая необходима в случае применения жид-
костного скруббера. Технико-экономические данные приведены применительно
к газу, содержащему 6% СО2 и 0,5—0,7 г H2S и меркаптанов в 1 м3. По
крайней мере две установки типа EFCO находятся в эксплуатации.
Извлечение сернистых соединений из газового потока проис-
ходит с высокой скоростью, что позволяет для снижения пере-
пада давления использовать цилиндрические горизонтальные ад-
сорберы. Установка сероочистки природного газа с горизон-
тальными адсорберами сооружена компанией Эль Пасо Нейче-
рел гез в штате Нью Мексико [182].
На установке при давлении 5,2 МПа и температуре 35—40 °C перера-
батывается 5,6 млн. м3 природного газа в сутки. В 1 м3 исходного газа со-
держится 0,05 г сернистых соединений и 0,1 г воды. Регенерацию цеолита
проводят частью очищенного газа при 315 °C. Выходящий газ содержит
сульфида водорода 1,4-10~4 г/м3, что находится в рамках требований к ма-
гистральному газу. Аналогичная установка меньшей производительности
(1,4 млн: м3/сут) сооружена другой фирмой [183].
Что1ы снизить расход, газа на десорбцию, иногда очистку проводят в ад-
сорберах с внутренним подводом тепла; при таком оформлении процесса
расход газа на регенерацию составляет 2—5% всего пропущенного газа.
При высоком содержании H2S адсорбционный процесс мо-
жет быть проведен в изотермических условиях (регенерация
снижением давления в адсорбере). На небольшой канадской
установке изотермическим методом перерабатывали газ, содер-
жащий 33% H2S и 12% СО2. Остаточное содержание сульфида
водорода после очистки составило менее 2-10”2 г/м3, что удов-
летворяет требованиям, предъявляемым к магистральному газу.
Газ, выделяющийся из адсорбера при снижении давления, со-
держал менее 5%) углеводородов и являлся исходным сырьем
для получения серы [184].
390 13. Промышленная очистка газов.
В Советском Союзе открыты крупнейшие месторождения при-
родных газов с высоким содержанием сернистых соединений.
Задача состоит в том, чтобы эти соединения превратить из, ис-
точника загрязнений в товарный продукт. На цеолитах с опреде-
ляющим размером пор не менее 0,46 нм, например типа NaX,
может протекать каталитическая реакция Клауса — превраще-
ние уловленного сульфида в серу [185—186].
Исследования. [187] показали, что при концентрации H2S
в газе от 3 до 15%, температуре 300—320 °C и объемной скоро-
сти потока до 5000 ч-1 степень превращения достигает 90%. Со-
поставимый выход серы при использовании традиционного ка-
тализатора— боксита достигается при гораздо меньших, объ-
емных скоростях: 500—1000 ч-1. Цеолиты типа X и У отлича-
ются высокой стабильностью: в течение полугода эксплуатации
загруженного образца заметного падения его активности не на-
блюдалось. Цеолиты NaA как катализаторы в реакции Клауса
применены быть не могут.
Долгое время отсутствовал метод сероочистки газов, добы-
ваемых в скважинах, непосредственно в полевых условиях. Но в
1969 г. трестом «Саратовнефтегаз» на Урицком газовом про-
мысле была создана опытно-промышленная установка сероочи-
стки природного газа производительностью 200 тыс. м3/сут [188].
Содержание сульфида водорода в исходном газе составляло
1 г/м3, после очистки 2 г/100 м3.
: Установка состоит из трех вертикальных адсорберов высотой 7 и и' диа-
метром 0,8 м. Объем цеолитов, загруженных в каждый адсорбер, составляет
2,5 м3. Очистку газа ведут при давлении 5 МПа и линейной скорости 0,1—
0,2 м/с. В качестве адсорбента применяют цеолит СаА. Продолжительность
стадии очистки 6 ч. Десорбцию проводят частью (8—10%) перерабатывае-
мого газа, нагретого до 315 °C. После эксплуатации в течение двух лет цео-
литы сохранили достаточно высокую адсорбционную емкость. Одновременно
с удалением сернистых соединений обеспечивается глубокая осушка газа.
В дальнейшем в промысловых условиях на передвижной
опытной адсорбционной установке был изучен процесс сероочи-
стки газа месторождения Гучурлы Туркменской ССР и Север-
ный Мубарек Узбекской ССР [189]. Содержание H2S в исход-
ном газе составляло 0,3—0,4%, диоксида углерода 1,0—1,35%.
В первичном сепараторе из газа в результате снижения давления до
6 МПа выделяли капельную влагу и жидкие углеводороды. После этого газ
пропускали снизу вверх через один из трех адсорберов; здесь из него уда-
ляли влагу, сульфид водорода и частично диоксид углерода. Очищенный и
осушенный газ поступал потребителю. Адсорберы имели многополочиую кон-
струкцию, высота слоя на каждой полке составляла 30—40 см. Скорость газа
в стадии адсорбции была 0,2—0,3 м/с, температуру газа изменили от минус
1 до 12 °C. Точка росы осушенного газа снижалась до минус 45—50 °C, содер-
жание сернистых соединений в отходящем газе не превышало 2—5 мг/м3.
Десорбцию поглощенных веществ и регенерацию адсорбентов вели нагре-
тым до температуры 350 °C газом при давлении 5,8—6,0 МПа.
Очистка газов от сульфида водорода и меркаптанов 391
Присутствие в очищаемом газе диоксида углерода в диапа-
зоне парциальных давлений вплоть до 3-104 Па не оказывает
заметного влияния на равновесную адсорбционную способность
цеолита NaX по меркаптанам, однако приводит к некоторому
увеличению работающего слоя, а следовательно, к снижению ди-
намической активности и сокращению времени защитного дейст-
вия слоя адсорбента ф!90].
Большинство цеолитовых установок для сероочистки природ-
ного газа рассчитано на переработку 150—300 тыс. м3/сут. Они
работают при давлении в газопроводе 1,7—5,0 МПа и обеспечи-
вают степень очистки до остаточного содержания сульфида во-
дорода и меркаптанов 2-10~3 г/м3 и ниже. При низких исходных
концентрациях H2S (50 част, на 1 млн. и ниже) ло кинетиче-
ским показателям предпочтительнее применять цеолиты СаА без
связующего. При более высоких концентрациях (800 част, на
1 млн.) показатели очистки газа цеолитами без связующего и
со связующим равноценны [191]. Данные по промышленной и
опытно-промышленной реализации процесса сероочистки газов
и его некоторые показатели приведены в работах [192, 193].
При удалении нескольких компонентов из газов температур-
ный режим стадии регенерации приходится выбирать по хуже
десорбируемому компоненту. В случае удаления воды, сульфи-
да водорода и диоксида углерода таким ключевым для расчета
стадии регенерации компонентом является вода, которая, как
указывалось выше, из цеолитов удаляется только при 300—
350 °C.
Проведение процесса удаления серии компонентов в несколь-
ко стадий усложняет схему и условия управления процессом, но
в некоторых случаях может привести к значительному сниже-
нию эксплуатационных затрат. Так, в рассматриваемой системе
в первой ступени охлаждением газа до -ф5 °C и обработкой его
в слое цеолитов точку росы снижают до минус 60 °C и ниже.
После этого сероочистку и декарбонизацию сухого газа прово-
дят йо второй ступени также цеолитами. При этом температу-
ра регенерации может быть ограничена 100—150 °C [194].
В тех случаях, когда концентрация диоксида углерода вели-
ка, а сульфид водорода находится в виде микропримеси, темпе-
ратуру стадии нецелесообразно задавать по хуже десорбируе-
мому сульфиду водорода. Поэтому разработана система, состоя-
щая, как минимум из трех адсорберов {195]. Два адсорбера
работают с коротким циклом: в одном из них проводят очистку
при повышенном давлении (например, 1 МПа), во втором—де-
сорбцию путем снижения давления с последующим вакуумиро-
ванием или продувкой частью очищенного газа. Продолжитель-
ность этих стадий 30 мин.
Без'нагревная регенерация не обеспечивает удаления других
примесей; они накапливаются, постепенно дезактивируя адсор-
392
13. Промышленная очистка газов
бент. Поэтому третий адсорбер находится на стадии «горячей»
десорбции. В этой стадии адсорбент разогревают до 200—
350 °C. Разогрев можно проводить потоком нагретого очищен-
ного газа или через стенку в результате теплообмена с внеш-
ним теплоносителем. В последнем случае адсорберы оборудова-
ны теплообменными элементами (змеевиками, трубчатками и
т. п.). Продолжительность стадии «горячей» десорбции — 8 ч.
Интервал, через который каждый адсорбер переключают на
стадию «горячей» десорбции, составляет 16 ч. В таком процессе,
по сравнению с обычными, достигается снижение капитальных
затрат и эксплуатационных расходов, особенно за счет эконо-
мии расхода тепла.
Сероочистка газов с частичным удалением
диоксида углерода
Основные трудности в адсорбционном методе сероочистки газов
связаны с утилизацией газов регенерации. Эта проблема удачно
решается в случае, если из исходного газа со значительным со-
держанием диоксида углерода удалить только часть его.
Предельное содержание С02 в природном газе по нормам США состав-
ляет 3%. В этом случае может быть рекомендована комбинированная схема,
представленная на рис. 13,17. Две установки такого типа производительно-
стью 5—6 млн. м3 природного газа в сутки каждая находятся в эксплуата-
ции [196]. Природный газ, содержащий 6% СО2, 1 г/м3 Н2О и 0,2—2,0 г/м3
H2S, разделяют на два примерно равных потока. Одну часть газа пропуска-
ют через адсорбер 1 с цеолитом до момента проскока H2S; таким образом, в
слое цеолита практически полностью удаляются Н2О и H2S. Вторую часть
очищают от H2S и СО2 в моноэтаноламиновом адсорбере 6, а затем осу-
шают гликолем в колонне 7. Выходящий из этого абсорбера газ содержит
0,05 г/м3 H2S, 1 г/м3 Н2О и практически полностью очищен от СО2.
После смешения очищенных потоков газ поступает в газопровод. При
этом содержание примесей в нем составляет 3% СО2, 0,05 г/м3 H2S и
0,5 г/м3 Н2О. Регенерацию (десорбцию и охлаждение) цеолита в адсорбере
2 проводят продувкой частью подо-
гретого газа (3), очищенного адсорб-
ционным методом (280 тыс. м3/сут).
Газы десорбции после охлаждения
(4) и выделения влаги в сепарато-
ре 5 примешивают к потоку газа,
поступающего в аминовый абсорбер.
Для переработки газов, содержащих
кислые компоненты, часто использу-
ют кислотостойкие цеолиты, напри-
мер AW-500. После 3000 регенераций
Рис. 13,17. Схема установки сероочистки
природного газа с частичным удалением
диоксида углерода:
1 — адсорбер на стадии очистки; 2 — адсор-
бер иа стадии регенерации; 3 — нагрева-
ГаЗ тель; 4 — холодильник; 5 —сепаратор; 6 —
этаноламиновый адсорбер; 7 — этиленгли-
колевый адсорбер.
Очистка газов от сульфида водорода и меркаптанов
393
Рис. 13>18. выходные кривые при сероочистке при-
родного газа цеолитами.
активность кислотостойкого цеолита, в промышленных условиях еще состав-
ляла 65% первоначальной [197].
Выше было указано, что селективность цеолитов при погло-
щении смеси сульфида водорода и диоксида углерода является
одним из преимуществ этого адсорбционного метода. Однако
нужно учитывать, что при очистке малосернистых газов с отно-
сительно высоким содержанием диоксида углерода за стадией
адсорбции H2S следует реакция образования серооксида углеро-
да, определяющая общую скорость процесса и катализируемая
адсорбентом:
H2S-|-CO2----»-COS+H2O (13.15)
Серооксид углерода удерживается цеолитом слабо, вследст-
вие чего в очищенном газе он появляется раньше проскока суль-
фида водорода. Это отчетливо видно из выходных кривых, при-
веденных на рис. 13,18.
Серооксид углерода не приводит к коррозии металла: его со-
держание ограничивают лишь нормы на общую серу в магист-
ральном газе. Но в случае переработки природного газа в не-
которых химических процессах, например при синтезе аммиака,
этот компонент является каталитическим ядом. Кроме того, если
из природного газа выделяют пропано-бутановую фракцию,
концентрация серооксида углерода в жидких углеводородах мо-
жет оказаться чрезмерно большой, так как его температура ки-
пения на 8 °C ниже температуры кипения пропана. Наоборот,
если содержание серооксида углерода в газе допустимо, реак-
ция (13.15) желательна; в этом случае сокращается количество
адсорбируемого сульфида водорода, увеличивается время за-
щитного действия слоя или может быть уменьшена загрузка
слоя.
Равновесие в системе серооксид углерода — диоксид углеро-
да рассмотрено в работе [198]. Схема экспериментальной уста-
новки, на которой производилась очистка природного газа, со-
держащего от 30 до 160 част, на 1 млн. H2S и от 1 до 3% СОг,
представлена на рис. 13,19.
Исходный газ содержит 0,05% кислорода. Чтобы избежать быстрого на-
копления серы в порах адсорбента, в процессе регенерации в газ добавляют
50% избытка оксида углерода по сравнению со стехиометрией реакции
СО + VA ^=t СО2 (13.16)
394
13. Промышленная очистка газов
По этой схеме в реакторе 1 кислород удаляют из исходного газа при
120 °C на катализаторе, содержащем 30% меди. Концентрацию сжиженных
газов регулируют, добавляя смесь пропана, бутана и пентана. Смесь суль-
фида водорода и диоксида углерода дозируют в поток газа, выходящего из
реактора, из баллонов 2 и 3. Затем газовую смесь компримируют до давле-
ния 6,2 МПа двухступенчатым дифференциальным компрессором 4. Темпе-
ратуру входящего в адсорбционный блок газа (ог 4 до 25 °C) контролиру-
ют в теплообменнике 5, охлаждаемом пропаном.
Цеолиты загружают в два адсорбера 7 и 8 высотой 2,4 м каждый с
внутренним диаметром 3,25 см, включенных последовательно; общая высота
слоя 4,8 м. Температуру в адсорбере измеряют термопарами на входе, в се-
редине и на выходе из каждого адсорбера. При регенерации используют
электрический подогреватель 6; в этот период адсорберы также нагревают
электрической обмоткой до заданной температуры. Хроматограф непрерывно
замеряет концентрацию диоксида углерода и пропана во входящем и выхо-
дящем газе. Концентрацию сульфида водорода замеряют газоанализатором
£ чувствительностью 10 част, на млн.
Чтобы определить концентрацию серооксида углерода, образец газа
пропускают через небольшую гидрогенизационную колонку, где он превра-
щается в сульфид водорода. Повышение концентрации H2S пересчитывают
на содержание серооксида. Влажность газа определяют по точке росы.
Перед загрузкой цеолита адсорбер продувают азотом, чтобы снизить до
минимума контакт с воздухом. Расход газа в стадии адсорбции 27 м3/ч.
'Часть очищенного газа отбирают в емкость 9, чтобы использовать на стадии
’^регенерации, остальную часть сжигают.
Цеолиты регенерируют нагревом до 315—350 °C. В течение 2 ч слой цео-
лита разогревают, после чего в течение 2—4 ч ведут отдувку. Противоток
азота обеспечивает десорбцию поглощенных веществ. Содержание влаги в
газе (по точке росы) после адсорбера на стадии регенерации снижалось от
атмосферного до соответствующего температуре —40 °C и ниже. Затем газ
охлаждают в течение 1 ч, пропуская из емкости в том же направлении очи-
щенный газ, как и на стадии очистки.
Результаты опытов показали, что по каталитической актив-
ности цеолиты можно разбить на три группы. Самую низкую
Рис. 13,19. Схема опытной установки для исследования процесса удаления мнкропримеси
сульфида водорода ирн высоком содержании диоксида углерода-.
1 —реактор; 2, 3 — баллоны с H2S и СО2; 4 — компрессор; 5 — теплообменник; 6 —нагре-
ватель газа регенерации; 7, 8 — адсорберы; 9 — емкость.
Очистка газов от сульфида водорода U меркаптанов
395
Рис. 13,20. Зависимость степени превращения
сульфида водорода от ого концентра-
ции в газе Cj_j
конверсию дают цеолиты типа
СаА. Промежуточной активно-
стью обладают цеолиты NaA, Са-
мую высокую активность имеют
цеолиты NaX. На рис. 13,20 пред-
ставлена зависимость степени
конверсии сульфида водорода в
слое цеолита NaA от концентра-
ции этого компонента в исходном
газе, содержащем 2,2% ди-
оксида углерода.
Очистка газа от сероорганических соединений
Если в природном газе кроме сульфида водорода присутствуют
сероорганические соединения, например этилмеркаптан, для се-
роочистки целесообразно применить цеолит NaX [199].
Цеолитовые адсорберы для очистки газа от меркаптанов
размещают после аминовых установок. Вследствие высокой ад-
сорбционной способности цеолитов по меркаптанам экономиче-
ские показатели адсорбционных установок благоприятны. При
переработке 2,25 млн. м3/сут, газа, содержащего 1 г/м3 меркап-
танов установка имеет 2 адсорбера с загрузкой 13,5 т цеолита
в каждом. Расход газа на регенерацию составляет 80 тыс. м3/сут.
Значения адсорбционной способности (а) цеолитов СаА и NaX
по изоамилмеркаптану (р— его парциальное давление) приве-
дены ниже:
р, Па 0,67 1,62 2,79 319 638 931 1330 2005
а, г/100 г СаА — 0,06 — 0,12 0,28 0,38 0,69 1,65
NaX 1,8 4,2 12,4 19,2 19,8 20,1 20,9 21,9
При проектировании сероочистных установок и выборе ус-
ловий регенерации цеолитов следует учитывать, что сероорганй*
ческйе соединения обладают значительно более низкой термиче-
ской стабильностью, чем углеводороды [200]. Наименее термо-
стабильны меркаптаны, разложение которых начинается уже
при 200 °C:
2RSH ---> RSR4-H2S (13.17)
Дисульфиды разлагаются при 350—400 °C:
CHaSSCH3 ► СН2=СН2 4- S 4- H2S (13.18)
396 13. Промышленная очистка газов
Исследованием ИК-спектров тиофена, тиофана и тиофенола, адсорбиро.
ванных цеолитами типа X в натриевой и кальциевой формах, установлено,
что адсорбционное взаимодействие молекул сероорганическнх соединений
Идет через атом серы с катионами цеолита и через атом водорода SH-rpyn-
Пы с атомами кислорода решетки [201]. Не исключена при этом возможность
образования меркаптидов. Прн адсорбции соединений ароматического харак-
тера (например, тиофенола) их молекулы взаимодействуют также с поверх-
ностью цеолита через систему л-электронов ароматического кольца. Обна-
ружена возможность образования на поверхности соединений, включающих
«окисленный атом серы, а также карбонатно-карбоксильных соединений и их
«серосодержащих аналогов. На цеолите в кальциевой форме разложение
.молекул' сероорганических соединений происходит более интенсивно.
Цеолиты успешно применены для очистки оренбургского га-
;за, поступающего на Волжский автомобильный завод, от мер-
каптанов [202]. В цехах термической и химической обработки
деталей, высокотемпературной пайки и спекания металлокера-
мических изделий в качестве контролируемой среды использу-
’.ют эндотермическую атмосферу, получаемую сжиганием природ-
ного газа в присутствии катализатора на никелевой основе
ТИАП-25. По условиям процесса используемый природный газ
должен содержать не более 10 мг/м3 сернистых соединений.
В то же время, оренбургский газ имел в 1 м3 25—40 мг суль-
фида водорода, 8—15 мг сероокиси углерода, 250—470 мг ме-
тил-, этил-, пропил-, бутилмеркаптанов (все в пересчете на
серу).
Цеолитовая установка, которая обеспечила очистку газа, состоит из
двух попеременно работающих адсорберов диаметром 0,2 м с высотой слоя
цеолита NaX 0,95 м производительностью 45 м3/ч с электрическим нагре-
вом при регенерации. Загрузка адсорбента в каждый адсорбер 17 кг. Линей-
ную скорость потока в аппарате поддерживают 0,37 м/с. Процесс очистки
ведут при температуре 30—40 °C и давлении, близком к атмосферному.
После появления проскока сернистых соединений в очищенном газе
(10 мг/м3) проводят регенерацию внешним прогревом слоя с помощью элект-
рического тока до 300—350 °C при одновременной продувке азотом
(10 м3/ч). Удаление сернистых соединений происходит примерно за 4 ч, при-
чем наиболее интенсивно десорбция идет при 190—200 °C. В этих условиях
приблизительно 50% уловленных меркаптанов конвертируется до сероводо-
рода. Динамическая способность цеолитов по сумме сернистых соединений
до проскока сероокснда углерода и сульфида водорода после 5—10 циклов
стабилизировалась на уровне 2 г/100 г, а до проскока меркаптанов — на
уровне 3 г/100 г. Следует учитывать, что исходный газ отличался значитель-
ным содержанием высокомолекулярных углеводородов, которые могли сов-
местно адсорбироваться и тем самым снижать адсорбционную способность
по сернистым соединениям: 1,4% пропана, 1,0% бутана н более тяжелых уг-
леводородов.
Включение адсорбционной установки в технологическую нит-
ку завода позволило в 3 раза повысить срок службы катализа-
тора, улучшить надежность автоматического регулирования со-
става экзогаза и атмосферы в печах, снизить расход электро-
энергии, а также объем работ по ремонту и обслуживанию обо-
рудования.
Очистка газов от сульфида водорода и меркаптанов
397
СерОочистка газов с химической регенерацией
адсорбента
Цеолиты являются не только прекрасными адсорбентами, но
и катализаторами некоторых химических процессов. Так, была
установлена высокая каталитическая активность цеолитов по
отношению к реакции Клауса — Ченса:
2HjS + SO2 ---► 1,5S24-2H2O (13.19)
Некоторые сорта цеолитов при комнатной температуре сти-
мулируют также реакцию Клауса:
•2HaS + О2 ---> S2 + 2Н^О (13.20)
Предпринята попытка использовать эти свойства цеолитов
для разработки новой технологической схемы серооочистки га-
зов с химической регенерацией, в которой газы, образующиеся
при сгорании серы, используют для окисления адсорбированно-
го сульфида водорода и получения легко конденсируемых па-
ров серы [203]. При химической регенерации тепло реакции не-
посредственно используют для повышения температуры слоя.
В процессе регенерации необходимо избегать чрезмерного окис-
ления сульфида водорода, что уменьшает выход серы и приво-
дит к опасному для структуры цеолитов перегреву.
На рис. 13,21 представлена принципиальная схема трехад-
сорберной установки сероочистки газа с химической регенера-
цией адсорбентов [204].
Природный газ под давлением 3,5 МПа проходит сверху вниз через
слой адсорбента в адсорбере 1. Очищенный газ может быть использован для
дополнительного охлаждения адсорбера 2. Одновременно адсорбер 2 выпол-
няет функцию второй ступени, обеспечивая увеличение степени удаления
сульфида водорода.
Для того, чтобы провести следующую стадию — десорбцию при возмож-
но более низком давлении, после насыщения цеолита сульфидом водорода
давление в адсорбере понижают до 0,2 МПа. Газ для регенерации получа-
ют сжиганием жидкой серы в горелке 8. Продукты сжигания после охлаж-
дения до требуемой температуры в теплообменнике пропускают в направле-
нии сверху вниз через адсорбер 3. В результате экзотермического теплового
эффекта реакции между сульфидом водорода и диоксидом серы температу-
ра слоя цеолитов повышается на 220—250 °C. Выходящую снизу из адсор-
бера 3 при 290 °C парогазовую смесь охлаждают в теплообменнике 5. Скон-
денсированную серу выделяют из газового потока в сепараторе 7 прн 140 °C.
Приблизительно одну треть серы в жидком состоянии насосом 9 подают на
сжигание.
В составе сбросных газов из сепаратора находятся иепрореагировавшие
SO2, H2S и N. Перед началом стадии охлаждения для удаления этих газов
адсорбер 2 продувают природным газом. Адсорбент охлаждают очищенным
газом при высоких скоростях потока, обеспечивающих быстрое протекание
процесса. После охлаждения адсорбер подключают к линии очищаемого га-
за и давление повышают до рабочего. Все необходимое тепло получают в
результате сжигания серы, а избыточное тепло используют в котле-утили-
заторе.
398
13. Промышленная очистка газов
Рис. 13,21. Схема установки сероочистки газа с химической регенерацией цеолита:
1 — адсорбер на стадии очистки; 2 — адсорбер на стадии охлаждения; 3 — адсорбер на
стадии регенерации; 4, 6 — холодильники; 5 — конденсатор серы; 7 — сепаратор; 8 — горел-
ка; 9 — иасос для серы; I — исходный газ; II — очищенный газ; III —диоксид серы;
IV—воздух; V —жидкая сера; VI — пары серы; VII —товарная сера; VIII — газ в га-
зопровод.
Если содержание сульфида водорода в исходном газе невелико и полу-,
чение серы на его основе нецелесообразно, то для регенерации может быть
использован воздух. На опытной установке, состоящей из трех адсорберов
диаметром 20 см и высотой 2,4 м с загрузкой цеолитов 45 кг в каждом, при
переработке газа с содержанием от 2 до 20% H2S активность цеолита па
H2S составляла 4,7—5,2 г/100 г. Продолжительность стадий: адсорбции 15—'
25 мни, сброса давления 5—15 мии, химической регенерации 30 мин, охлаж-
дения 20—25 мин, повышения давления 5—10 мин.
Процесс сероочистки газа с химической регенерацией при промышленной
реализации встретил серьезные трудности*. Поэтому в дальнейшем было пред-
ложено конструктивное решение, предусматривающее расчленение зон ад-
сорбции и катализа [205]. Аппарат, в котором протекает процесс, разделен
на две зоны: вверху расположена большая, адсорбционная зона, внизу —
меньшая, каталитическая. Обе зоны заполнены цеолитом.
Извлечение сульфида водорода из газового потока происходит в адсорб-
ционной зоне прн 15—150 °C; одновременно газ осушается. После того, как
адсорбент насытится сульфидом водорода и влагой, газовый поток пере-
ключают на следующий адсорбер. Затем потоком инертного горячего газ®
последовательно разогревают каталитическую зону до 190—230 °C, а адсорб-
* В литературе отсутствуют данные о практическом использовании процесса,,
представленного на рис. 13,21. Его лабораторная проверка, проведенная'
в МХТИ им. Д. И. Менделеева и НИИОгазе, показала, что при реализации-
процесса возникают принципиально непреодолимые трудности. Они связаны
с тем, что тепло, выделяющееся в верхних слоях адсорбера 3 в результате-
взаимодействия сульфида водорода и диоксида серы, нагревает поток газа-
носителя, который, поступая в нижележащие слои адсорбента, термические
десорбирует сульфид водорода. Поэтому реакция начинается, но быстро за-
тухает. (Прим, ред.)
\ 'Очистка .газов от сульфида водорода и меркаптанов 399
щионную зону до 200—31.5 °C, при этом особое внимание обращают на то,
чтобы не было локальных перегревов цеолита выше порога его термической
стабильности, т. е. 540 °C.
Десорбированный в потоке инертного газа сульфид водорода проходит
через реакционную зону, в которую в этот период подают воздух или кис-
лородсодержащий газ, нагретый до 150—260 °C. Содержание кислорода
должно быть достаточным для полной конверсии H2S. Чтобы в реакционной
зоне получить газовую смесь заданного постоянного состава, температуру
1газа, вводимого в адсорбционную зону, последовательно повышают. Пары
серы и воды конденсируют.
После окончания стадии десорбции слой адсорбента в адсорбционной зо-
«е охлаждают до 120—150 °C продувкой воздухом, а затем до 15—40 °C по-
током сырого газа. В последнем периоде охлаждение адсорбента совмеща-
ется с сероочисткой газа, причем к концу стадии охлаждения отрабатыва-
ется приблизительно одна треть сорбционной емкости цеолита. Аналогичный
процесс н аппаратура предложены для получения серы нз сернистых жидких
нефтепродуктов.
При проектировании и расчете адсорбционных установок,
предназначенных для сероочистки природных и попутных нефтя-
ных газов, необходимо учитывать влияние высших углеводоро-
дов, подавляющих адсорбцию H?S. Установлено [206], что в
обычных температурных условиях сульфид водорода и н-бутан
цеолитом СаА адсорбируются в соизмеримых количествах.
Природные цеолиты (клиноптилолит и морденит) по адсорб-
ционной способности (по H2S) значительно уступают синтетиче-
ским цеолитам [207], а сероорганические соединения, молекулы
которых сравнительно велики, ими практически не сорбируются.
Однако кислотная обработка 0,25 н. раствором НС1 приближает
их активность по сульфиду водорода к активности цеолитов ти-
па А [208] и делает возможным проникание молекул низкомо-
лекулярных меркаптанов в пористую структуру. Вследствие
этого природные цеолиты могут рассматриваться как перспек-
тивные адсорбенты для сероочистки природных и промышлен-
ных газов.
Сероочистка сжиженных газов
Высокая адсорбционная способность и избирательность ад-
сорбции сернистых соединений на цеолитах сохраняются и при
проведении очистки в жидкой фазе. Это свойство цеолитов ши-
роко используют для осушки и очистки ряда продуктов, выде-
ляемых на газобензиновых заводах и используемых в качестве
сырья для органического синтеза и жидкого топлива.
В 1959 г. был осуществлен пуск первой промышленной установки осуш-
ки и очистки цеолитами пропана. Уже в 1962 г. в США в эксплуатации бы-
ло более 50 цеолитовых установок осушки и сероочистки легких углеводород-
ных жидкостей, .а вскоре их число увеличилось до 75 [209]. В двух третях
из ннх простой заменой твердого адсорбента-осушителя (силикагеля и алю-
могеля) на цеолиты были совмещены функции очистки и осушки в одном
аппарате н нз схемы исключена стадия щелочной очистки. Более 50% пропа-
ща, вырабатываемого на газобензиновых заводах США, пропускают через
адсорберы с цеолитами [210].
400 13. Промышленная очистка газов /
Адсорбционная способность цеолитов по сернистым соедине-
ниям значительна даже при очень низком их содержании в жид-
кой фазе. Так, при концентрации H2S в пропане 0,6-10-2 г/м3
равновесная адсорбционная способность цеолитов составляет
4,5 г/100 г, а при концентрации 2,3 г/м3— 13,5 г/100 г [211].
Установка очистки пропана (10 м3/ч) состоит из двух адсорберов диа-
метром 0,5 м с высотой слоя 2,5 м и загрузкой цеолитов NaX 340 кг в каж-
дом; диаметр гранул цеолита 0,08 мм. Состав (мольный) исходного газа:
8% С2Н6, 91% CsHs, 1% C4Hio (примеси: 0,006 г/м3 H2S и 0,01 г/м3 СО2).
Адсорберы работают попеременно: когда в одном проводится очистка, вто-
рой последовательно проходит три стадии — удаления жидкой фазы, десорб-
ции и охлаждения. Таким образом, рабочий цикл включает четыре стадии
[212):
1. Очистку пропана проводят при обычной температуре и давлении, ис-
ключающем возможность местного испарения (около 1,5 МПа), в течение
16 ч. На разных установках пропускная способность изменяется от 0,75 до
6,0 кг на 1 см2 сечения адсорбера в 1 ч.
2. Удаление жидкой фазы ведут путем вытеснения газом высокого дан-
ления (например, метаном), подаваемым в верхнюю часть адсорбера. Затем
давление в системе снижают до 0,4—0,8 МПа и испаряющийся пропан после
компрессии возвращают на очистку. Продолжительность операции 15 мин.
3. Десорбцию сернистых соединений проводят при том же давлении и
температуре 200—290°C продувкой горячим сухим газом (обычно метаном),
не содержащим сернистых соединений и кислорода, в течение 4 ч (реакция
кислорода с сернистыми соединениями может вызвать образование серы,
отлагающейся в порах и дезактивирующей адсорбент). Разогрев слоя др
заданной температуры заканчивается через 1,5 ч. Удельный расход газа
на регенерацию составляет 16 м3/кг цеолита. Полнота десорбции сульфида
водорода достигается при 200 °C, низших меркаптанов — при 290 °C.
4. Для охлаждения пропана используют очищенный пропан с содержа-
нием серы менее 2,5-10~2 г/м3. Продолжительность охлаждения 0,5—1,0 ч,
расход пропана 1,7—2,5 л/кг цеолита.
Несколько иной регламент работы установки адсорбционной
очистки пропана описан в работе [213]. Период очистки длится
8 ч. Перед регенерацией пропан из слоя цеолита удаляют газом
высокого давления. Регенерацию ведут газом низкого давлений
(0,35—0,4 МПа), нагретым до 200—260°C, в течение 6 ч. Сжи-
гание газа регенерации покрывает потребность установки в топ-
ливе. В заключение адсорбер в течение 1 ч охлаждают очищен-
ным пропаном.
С целью увеличения пропускной способности установок се-
роочистки пропана и уменьшения истирания адсорбента вслед-
ствие пульсации потока на всех -стадиях сохраняется направле-
ние движения сверху вниз. Схема двухадсорберной установки
для очистки пропана представлена на рис. 13,22. Схема рабо-
ты клапанов управления на установке осушки сжиженных га-
зов описана в работе [214].
После 8-месячной эксплуатации отложения углеродистых со-
единений в цеолите не превышали 1 г/100 г, а сернистых соеди-
нений 0,02 г/100 г. Понижение активности в конце испытатель-
Очистка газов от сульфида водорода и меркаптанов
401
Рис. 13,22. Схема двухадсорберной уста-
новки очистки пропана цеолитами:
1, 2 — адсорберы; 3 — нагреватель; 4 — ем-
кость чистого пропана; 5 — теплообменник.
кого периода не превышало
10%, потерь цеолита не было
отмечено.
В литературе имеются со-
общения об успешной экс-
плуатации двухадсорберной
компактной цеолитовой уста-
новки. Установка перерабаты-
вает 20 м3/ч пропана; содержание сернистых соединений в про-
пане 1,425 г/м3. Регенерацию проводят при 260—315°C. Про-
должительность адсорбции и регенерации по 8 ч [215]. Оста-
точное содержание сульфида водорода в жидком пропане после
цеолитовой очистки не превышает 7-10-3 г/м3 [216].
Строящиеся в настоящее время установки, как правило, рас-
считаны на загрузку 900 кг цеолита NaX в каждый адсорбер.
При определении эксплуатационных затрат планируют замену
адсорбера через каждые 2—4 года. Установки для очистки бу-
тана имеют аналогичные конструкции и показатели.
Уже с 1967 г. все основные компании Канады начали широко применять
цеолиты для очистки сжиженных газов. Очистка пропана и бутана успешно,
решена с помощью цеолитов NaX на крупных канадских установках /217].
Диаметр гранул использованных цеолитов составляет 1,5 мм. Каждая уста-
новка имеет производительность около 3 тыс. м3 сжиженных газов в сутки
(диаметр адсорберов 0,9—1,2 м, высота 2—3,6 м). В исходном пропане-
содержание H2S достигает 1,5%. Продукты практически полностью очища-
ются от сульфида водорода, метил- и этилмеркаптанов; степень удаления-
сероокиси углерода зависит от длительности цикла, в начале стадии очистки-
удаление практически полное. Контроль за качеством продукции осуществля-
ется микроколориметрическим детектором (COS, меркаптаны) и портатив-
ным абсорбционно-потенциометрическим прибором (общая сера).
Первая установка сероочистки сжиженных газов в Англии пущена в
эксплуатацию в 1966 г. На ней очищают 11 т пропана и 22 т бутана в сутки.
Установки имеются во Франции, Австралии и других странах.
Сероочистка нефтепродуктов
Метод сероочистки цеолитами применим не только к сжижен-
ным углеводородам, но и к более тяжелым нефтяным продук-
там: газовым бензинам, лигроинам и т. п. В пропане, наряду с
сульфидом водорода присутствуют метилмеркаптаны, в бута-
не— метил- и этилмеркаптаны, в бензинах п лигроинах — раз-
личные сернистые соединения. При очистке высокомолекулярных
фракций следует иметь в виду, что по избирательности адсорб-
ции поглощаемые компоненты в порядке убывания адсорбцион-
ного сродства образуют ряд: вода — меркаптаны — сульфид во-
дорода — диоксид углерода [218].
26—1346
402 13. Промышленная очистка газов
Обычно нефтяные продукты получают в результате ректифи-
кации, и поэтому каждому погону сопутствуют определенные
(по температуре кипения) сернистые соединения. С увеличени-
ем молекулярной массы углеводорода повышается его совмест-
ная сорбция с сернистыми соединениями, понижается избира-
тельность адсорбции сернистых соединений и соответственно ди-
намическая активность цеолитов.
Чем уже границы кипения фракции, тем выше адсорбцион-
ная способность цеолитов по сернистым соединениям, загрязня-
ющим погон. Меркаптанную серу эффективно удаляют из всех
нефтяных продуктов с достаточно узкими границами кипения,
сероуглерод — из пропана и бутана, серооксид углерода при вы-
соком содержании диоксида углерода не удаляется.
При выборе типа цеолита следует иметь в виду, что цеолиты
СаА имеют такой размер пор, что с их помощью проблема пол-
ного извлечения из бензинов сульфида водорода и меркаптанов
может быть решена; при этом частично удаляются сульфиды и
.дисульфиды. Циклические сернистые соединения удаляются
только цеолитами типа X. Применение цеолита NaX позволило,
.например, полностью обессерить бензин туймазинской нефти
(конец кипения 205°C); исходное содержание сернистых соеди-
нений в нем составляло 0,05% [219].
Описаны [220] лабораторные опыты по сероочистке бензинов цеолитами.
В небольшом адсорбере (диаметр 5 см, высота слоя цеолита NaX 1,2 м,
крупность его частиц 0,8 мм) сероочистке был подвергнут деэтанизирован-
ный газовый бензин, выкипающий в пределах 30—120 °C. Содержание сер-
нистых соединений в бензине составляло: 8,36-10~2% общей серы; 5,81 •
• 10~2% меркаптанной серы; 5-10~4% элементной серы; 1-10-3% дисуль-
фидов; 1-10“30/о сульфида водорода. В составе нафтено-парафинистого бен-
:зина было 3,2% трудно десорбируемых ароматических углеводородов, в ос-
новном бензола.
Подготовку (обезвоживание) цеолита вели продувкой сухим азотом при
340 °C в течение 4 ч. Сероочистку бензина проводили при 38 °C, давлении
0,45 МПа и объемной скорости 0,95 ч~*. Проскок меркаптаииой серы
5 част, на 1 млн. отмечен после пропуска 3,5 кг бензина через 1 кг цеолита;
при этом содержание общей серы в выходящем из адсорбера бензине со-
ставляло 11 част, на 1 млн.
Выходные кривые при очистке бензина представлены на рис. 13,23. При
влажности цеолитов 8% динамическая активность по сернистым соединени-
ям снижается на 50% (пунктирная кривая). Одновременно с сернистыми
соединениями происходит (в первом периоде полное, а в последующем —
частичное) удаление ароматических углеводородов, вследствие чего увели-
чивается приемистость бензина к тетраэтилсвинцу (ТЭС). При пропускании
0,8 кг бензина на 1 кг цеолита в выходящем потоке практически отсутству-
ют ароматические углеводороды, после пропуска 2 кг/кг их концентрация
составляет 0,4%, затем резко возрастает до 2,8% и в последующем достига-
ет содержания их в исходном бензине. Приемистость бензина к ТЭС после
лропуска 9 кг бензина на 1 кг цеолита повысилась с 10—15 до 19—20 пунк-
тов.
Десорбцию проводили по мере повышения температуры слоя до 370 °C
в течение 2 ч с последующей продувкой сухим газом в течение 5 ч. Дезакти-
вации сорбента в результате десорбции отмечено ие было.
Очистка газов от сульфида водорода и меркаптанов
403>*.
Рис. 13,23. Зависимость содержания сернистых соеди-
нений cs в бензине, выходящем из адсорбера с цео-
литами от пропущенного количества.
1 — общая сера; 2 — меркаптанная сера; 3 — то же,
на влажном цеолите.
При промышленном осуществле-
нии процесса сероочистки применяе-
мый для десорбции горячий газ не
должен содержать высших компонен-
тов (гликолей, абсорбционного мас-
ла), дезактивирующих адсорбент. Газ
или жидкость, применяемые для ох-
лаждения, должны быть сухими и не
содержать сернистых соединений, аро-
матических и высокомолекулярных уг-
леводородов.
Во избежание падения активности
у, кг/кг
цеолитов, в рабочем цикле адсорбционной установки преду-
сматривается стадия окислительной регенерации кислородсо-
держащим газом. Концентрацию кислорода в газе выбирают с
таким расчетом, чтобы температура в слое не превышала поро-
га термической стойкости цеолитов.
Первая промышленная установка очистки деэтанизирован-
ного газового бензина с содержанием серы от 0,09 до 0,23% име-
ла производительность 1135 м3/сут.
Установка состояла из двух адсорберов (диаметр 1,5 м, высота 15 м),.
загруженных цеолитом NaX с диаметром зерен 3 мм; загрузка цеолита в
оба адсорбера составляла 33 т. Сероочистку бензина вели одновременно с
его осушкой, причем в конце процесса цеолит в среднем содержал 0,33 г/
/100 г воды и 10,3 г/100 г сернистых соединений. При содержании в бензине
0,1% S продолжительность стадии очистки составляла 15 ч, при 0,22% S —
10,5 ч. Рабочие условия процесса: температура 38 °C, давление 2,5 МПа.
В последующем адсорбционный метод очистки газовых бен-
зинов был использован на двух промышленных установках. Про-
изводительность каждой из них составляла около 1400 т бен-
зина в сутки. Содержание серы в виде меркаптанов и дисуль-
фидов в сыром бензине было 0,1%. Установки имели по два вер-
тикальных адсорбера диаметром 1,5 м и высотой 5,1 м. Регене-
рацию проводили газом, нагретым до 315 °C; его расход со-
ставлял 10 тыс. м3/ч. Капиталовложения иа такую установку со-
ставляют 300 тыс. долл.
Экономика процесса зависит в первую очередь от цены цео-
лита и срока его службы. Предварительные расчеты американ-
ских инженеров показали, что сероочистка цеолитами по эконо-
мичности соответствует содовой очистке при цене цеолита
1,2 долл/кг и сроке службы 1 год или цене 3,3 долл/кг и сроке-
службы 2,8 года [221].
26
404 13. Промышленная очистка газов
Регенерация адсорбента включает следующие ступени: слив
бензина из адсорбера; продувку адсорбента сверху вниз холод-
ным инертным или природным газом для удаления из свободно-
го объема неадсорбированных примесей; продувку горячим
инертным или природным газом снизу вверх с последующим
пропуском парогазовой смеси через конденсатор; продолжитель-
ную продувку колонны снизу вверх горячим газом при 290—
370 °C, минуя конденсатор; охлаждение адсорбента в результа-
те пропускания сверху вниз бензина, очищенного от сернистых
соединений.
В случае, если использование сернистых соединений не пре-
дусмотрено, регенерация может заключаться в их выжиге при
температурах слоя адсорбента ниже порога термической ста-
бильности цеолитов. Периодическая окислительная регенерация
может быть предусмотрена и в других случаях, когда актив-
ность цеолита при эксплуатации падает.
Стадии осушки и сероочистки жидкостей иногда бывает це-
лесообразно расчленить. В этом случае жидкость пропускают
сначала через слой цеолита NaA, в котором поглощается вода
и низкомолекулярные кислые компоненты, а затем через слой
цеолита NaX, где происходит извлечение высокомолекулярных
сернистых соединений [222]. Особенно высокие требования по
содержанию серы предъявляются к углеводородам, применяе-
мым в ряде процессов их каталитической переработки, полиме-
ризации и т. п. Избирательность адсорбции сернистых соедине-
ний используется также для оценки качества образцов цеоли-
тов [223].
Более подробное описание регламентов процессов очистки
углеводородов приведено в ряде обзоров, например [224].
14
ЗАЩИТА АТМОСФЕРЫ
ОТ ЗАГРЯЗНЕНИЯ
Защита окружающей среды от загрязнений вредными выброса-
ми промышленности и транспорта является глобальной пробле-
мой человечества. Вопрос загрязнения биосферы нельзя рас-
сматривать в отрыве от факторов интенсивной индустриализа-
ции, стремительного роста народонаселения планеты и концент-
рации жителей в крупных промышленных центрах. В этих ус-
ловиях перед учеными и инженерами стоит важнейшая задача
создания безотходных производств, что исключит загрязнение
основных компонентов биосферы: атмосферы, гидросферы и ли-
тосферы*.
Создание безотходных производств обеспечивается не толь-
ко коренным изменением технологии, но и созданием новых,
а также реконструкцией существующих очистных установок.
Большую роль в обезвреживании отходящих промышленных
газов и сточных вод, рекуперации из них компонентов с даль-
нейшим превращением их в ценные товарные продукты или с
возвратом в процесс должны играть адсорбционные методы.
Санитарная очистка газов представляет собой комплекс тех-
нических решений, препятствующих чрезмерному загрязнению
атмосферного воздуха. Следует подчеркнуть, что адсорбционный
метод позволяет решить задачи глубокой очистки технологиче-
ских и отходящих промышленных газов, содержащих разнооб-
разные вредные вещества. Если правильно выбраны технологи-
ческий регламент, схема и аппаратура процесса, примесь мо-
жет быть удалена адсорбционным методом практически полно-
стью: это дает возможность обеспечить концентрацию примесей
в воздушной среде значительно ниже предельно допустимой
* В задачи безотходной технологии входит не только защита биосферы от
загрязнений, но и рациональное использование сырья и энергии. Все эти за-
дачи взаимосвязаны, так как естественно, что, например, утилизация компо-
нентов выбросов не только предотвращает загрязнение окружающей среды,
но и сокращает расход природного сырья, идущего на получение этих ком-
понентов. Аналогичным образом, использование вторичных энергоресурсов
уменьшает расход топлива и исключает тепловое «загрязнение» окружающей
среды. (Прим, ред.)
406 ' 14. Защита атмосферы от загрязнения ~ ........
концентрации (ПДК). Аналогичное заключение может быть сде-
лано по применению адсорбционного метода для очистки сточ-
ных вод*.
Рекуперация растворителей
В рекуперационных установках этого назначения осуществляют
улавливание и возврат в технологический процесс растворите-
лей из производственных или вентиляционных газов. Улавлива-
ние растворителей приносит не только большой технико-эконо-.
мический эффект, но и имеет огромное социальное значение,
оздоровляя условия жизни человека и обеспечивая охрану
окружающей среды. Несмотря на меры, принимаемые в Совет-
ском Союзе, потери растворителей и их выбросы в атмосферу r
настоящее время оцениваются в 600—800 тыс. т в год. Наибо-
лее остро стоит проблема улавливания ацетона, бензина, бензо-
ла, толуола, ксилола, метилэтилкетона, простейших спиртов,,
нормальных парафинов Се и С?, сероуглерода, диэтилового и
изопропилового эфиров, уайт-спирита, хлорпроизводных углево-
дородов (хлороформа, дихлроэтана, хлорбензола, метиленхло-
рида).
В большинстве случаев концентрация растворителя в отходя-
щих газах низкая (несколько граммов в 1 м3), в связи с чем
возрастает роль адсорбционного метода рекуперации. На реку-
перационных установках в качестве адсорбента применяют
только активный уголь благодаря его гидрофобности. Кроме
того, рекуперационные угли обладают высокой адсорбционной
способностью по парам органических веществ, небольшой удер-
живающей способностью; они достаточно прочны, не требуют
* В соответствии с законом «Об охране атмосферного воздуха» (июнь?
1980 г.) для промышленного предприятия СССР основным критерием чистоты
выбросов является не ПДК., которое фиксирует концентрацию в приземном
слое атмосферы, а предельно допустимые выбросы (ПДВ). Они устанавли-
ваются нормативно на уровне, при котором выбросы загрязняющих веществ
«от конкретного и всех других источников в данном районе с учетом пер-
спективы его развития не приведут к превышению нормативов ПДК загряз-
няющих веществ в атмосферном воздухе». ПДК выражают в мг/м3, ПДВ —
в мг на единицу вырабатываемой продукции. Соотношения между ПДВ и
концентрацией загрязняющей примеси в атмосфере, верхнее допустимое зна-
чение которой и есть ПДК, в достаточной степени сложны. Они зависят от
мощности предприятия, наличия других источников выбросов в данном райо-
не, оборудованности предприятий средствами рассева выбросов, атмосферных
условий и т. п. .
; Но с учетом всех этих факторов нормативы ПДВ, если их выразить в со-
поставимых единицах, значительно выше, чем ПДК. Например, ПДК на диок-
сид. серы равен 10 мг/м3, а нормы ПДВ, соответствующие 90%-ой очистке
газовых выбросов от этого вещества, будут составлять примерно 500 мг/м3.
Поэтому такое достоинство адсорбционных процессов, как большая глубина,
очистки, в этом их приложении часто не имеет решающего значения. (Прим,
ред.)
Рекуперация растворителей
407
значительных затрат энергии на преодоление гидравлического
сопротивления слоя адсорбента при прохождении потока газа.
•Окупаемость (рентабельность) адсорбционных установок зави-
сит от концентрации растворителя. Нижний порог рентабельно-
сти (по концентрации с) адсорбционных установок с неподвиж-
ным слоем активного угля может быть установлен на основе
приведенных ниже данных [225, 226]:
Растворитель С, Г/мЗ Растворитель с, г/мЗ
Бутилацетат 1,5 Ксилол, метил- 2,1
Трихлорэтилен, 1,8 ацетат, этил-
этанол ацетат
Бензин, бензол, 2,0 Ацетон 3,0
метил енхлорид, Тетрахлоругле- 4,5
толуол род Сероуглерод 6,0
Степень извлечения растворителя на рекуперационных уста-
новках обычно составляет 95—99%, а остаточное содержание
растворителя в отходящем из адсорбера газе не превышает
•0,5 г/м3.
Как правило, проектным разработкам рекуперационных уста-
новок предшествует стадия лабораторных исследований. Изуче-
ние адсорбции растворителей в динамических условиях можно
Рис. 14,1. Схема лабораторной установки для изучения рекуперации паров растворителя
из газа;
1 — баллон с азотом; 2 — осушитель; 3 — реометры; 4 — дополнительные осушнтелн; 5 —
испаритель («гусек»): 6 — смеситель; 7 — терд/остат, 8 — трехходовые краны; 9 — тепло-
обменник; 10 — термопара; 11 — адсорбционная колонка; 12 — трансформатор; 13 — потен-
циометр; 14 — интерферометр.
408
14. Защита атмосферы от загрязнения
осуществить на установке, схема которой представлена на
рис. 14,1.
Основной частью установки является вертикальная термостатированная
стеклянная колонка 11, заполненная гранулами активного угля; ее диаметр
25 мм, высота 0,5 м. Перед началом каждого опыта проводят регенерацию
адсорбента путем нагревания колонки (с помощью нихромовой обмотки) до
200—250 °C с одновременной продувкой газообразным азотом, подаваемым
из баллона 1 после осушки в колонке с цеолитом 2. Температурный режим
при регенерации регулируют автотрансформатором 12 по показаниям по-
тенциометра 13, соединенного с термопарой 10. Затем слой адсорбента
охлаждают до заданной температуры опыта. Для этого колонку снабжают
рубашкой, через которую циркулирует вода, подаваемая из термостата 7..
При температуре опыта выше 80 °C колонку термостатируют с помощью ни-
хромовой обмотки. Затем создают требуемую концентрацию адсорбтива в;
потоке газа, насыщая часть газа парами растворителя в «гуське» 5, нахо-
дящемся в термостате. Измеренные реометрами 3 и дополнительно осушен-
ные в трубках 4 потоки газа-носителя (азота) и растворителя после смеше-
ния в смесителе 6 до начала каждого опыта направляют в байпасную колон-
ку, гидравлическое сопротивление которой равно сопротивлению рабочей
колонки, затем через трехходовой кран 8 выбрасывают в атмосферу.
После стабилизации заданной скорости газового потока и концентрации'
адсорбтива поворотом трехходового крана газовый поток направляют в ра-
бочую колонку 11. Благодаря равенству сопротивлений рабочей и байпас-
ной колонок при этом переключении не происходит нарушения гидродинами-
ческого режима и газ, поступающий на адсорбцию, с начала опыта прохо-
дит через слой адсорбента с постоянной скоростью, имея постоянный состав.
Газовую смесь перед поступлением в рабочую колонку предварительно по-
догревают до температуры опыта в теплообменнике 9. Участок коммуника-
ции между выходом из теплообменника и входом в колонку тщательн®
теплоизолирован асбестовым шнуром. Выходящий из колонки газ проходит
через рабочую камеру интерферометра 14, через сравнительную камеру ко-
торого пропускают азот. Концентрацию растворителя за слоем адсорбента
непрерывно регистрируют с точностью 0,01 % (об.). Периодически с помощью
переключения трехходовых кранов 8 интерферометром контролируют кон-
центрацию адсорбтива в исходном газе.
Изучение выходных кривых дает информацию о динамиче-
ской и равновесной адсорбционной способности, величине рабо-
тающего слоя, времени защитного действия слоя и т. д.
Схема простейшей адсорбционной рекуперационной установ-
ки представлена на рис. 14,2. В принципе технологический ре-
жим и аппаратура установки не отличаются от описанных в
предшествующих главах установок периодической адсорбции.
При высоких скоростях потока, чтобы избежать уноса адсор-
бента, слой прикрывают сверху сеткой или ведут процесс при
направлении потока сверху вниз. Чтобы адсорбционная способ-
ность была достаточно высокой, в некоторых случаях исходный
газ перед подачей на адсорбционную установку охлаждают.
Современные рекуперационные установки полностью авто-
матизированы. Контроль и регулирование концентрации рас-
творителя за слоем адсорбента ведут непрерывно с помощью
высокочувствительного прибора, связанного с гидравлической
или пневматической системой переключения. Десорбцию про-
г
Рекуперация растворителей 409
Фис. 14,2. Схема рекуперациоииой установки:
2 — воздуходувки; 3 — нагреватель; 4 — адсорбер; 5—конденсатор; 6 — холодильник;
7 — сепаратор.
водят перегретым паром, пропускаемым противотоком к направ-
лению газа на стадии адсорбции. Пары воды и растворителя
•охлаждают, конденсируют и разделяют непосредственно в се-
параторе или в дополнительной фракционирующей колонне.
После стадии десорбции уголь сушат и охлаждают потоком
атмосферного воздуха.
Одна из первых систем автоматизации управления процессом рекупера-
ции была испытана в Киеве на установке извлечения бензина [227]. Управ-
ление осуществлялось с центрального щита посредством двух командных
электрических приборов (КЭП), блока реле, газоанализатора на бензин и
универсальных переключателей, с помощью которых открывались задвижки
на трубопроводах, включались электромагнитные пускатели электродвигате-
лей, вентиляторов и насосов, КЭП и газоанализатор.
Газоанализатор подает сигнал о переводе первого адсорбера иа стадию
десорбции за пять минут до достижения предварительно выбранной концен-
трации растворителя в отходящем газе. Система предусматривает также
автоматическое регулирование и контроль температуры в адсорберах, дав-
ления пара, поступающего в адсорберы, температуры конденсата на входе
в сепаратор, давления паровоздушной смеси после вентиляторов, масла и
воды после насосов. Световая сигнализация позволяет быстро определить
место неисправности в случае ее возникновения. При аварии установка ав-
томатически отключается. Наряду с этим предусмотрена возможность руч-
ного управления установкой.
Наиболее распространенными материалами при изготовле-
нии рекуперационных установок являются обычная сталь, спец-
сталь, чугун, медь, оцинкованное железо, латунь, алюминий.
Адсорберы для извлечения галогенорганических соединений ча-
410 14. Защита атмосферы от загрязнения
сто выполняют из титана*. В ряде случаев предусмотрена про-
тивокоррозионная защита. Так, на установках рекуперации
хлорэтила стенки адсорбера покрыты слоем диабазовой плит-
ки и слоем графита. Колосниковая решетка с металлической
сеткой, на которой размещается активный уголь, заменена гра-
фитовой плитой толщиной 50 мм с отверстиями 12—14 мм. На
плите расположены слои стекловаты, колец Рашига и стеклово-
локна.
Расходные коэффициенты на 1 т уловленного растворителя
при его концентрации 10 г/м3 приведены ниже:
Пар (0,3—0,5 МПа), т 2—3,5
Охлаждающая вода (15 °C), м3 30—50
Электроэнергия, кВт-ч 100—250
Активный уголь, кг 0,5—1,0
Иногда длительность работы одной загрузки адсорбента
рассчитывают на 10 000 адсорбционно-десорбционных циклов.
Окупаемость рекуперационных установок обычно оценивают в
1—2 года. Эксплуатационные расходы зависят от концентрации
растворителя в воздухе. Расход энергии обратно пропорциона-
лен содержанию растворителя, расход пара и воды изменяются
в меньшей степени. При отборе паров растворителей с рабочих,
мест и проектировании системы вентиляции следует стремиться,
чтобы концентрация адсорбтива в газе была по возможности
максимальной, однако меньше 0,5 от нижней границы взрывае-
мости (НТВ). Пределы взрываемости показаны в табл. 14-1.
Для поддержания необходимой концентрации разработаны
регуляторы, обеспечивающие безопасность производства. В не-
которых случаях смесь разбавляют чистым воздухом.
В последнее время в ряде случаев адсорбционные процессы
проводят при повышенных температурах. Такой метод особен-
но перспективен в тех случаях, когда отходящие газы горячие,
например в производстве ряда пластмассовых изделий. Кон-
денсация водяного пара в начале стадии десорбции на рекупе-
рационных установках, на которых стадию адсорбции проводят
при обычных температурах, значительно осложняет процесс,
требуя включения в цикл дополнительных стадий сушки и
охлаждения активного угля. При повышенной температуре весь
цикл можно осуществить без увлажнения адсорбента, в две
фазы — адсорбции и десорбции.
Исследования процесса рекуперации бензола и толуола [228,
229] показали, что оптимальный температурный режим двух-
фазного процесса, исключающий увлажнение активного угля, —
* При выборе материала учитывают коррозионные свойства среды, адсорбти-
ва и продуктов гидролиза адсорбтива. (Прим, ред.)
Таблица 14-1. Пределы взрываемости веществ в смеси с воздухом
Компонент Нижний предел Верхний предел
% (об.) Г/мЗ % (об.) ’ Г/мЗ
«-Амилацетат 1,1 *>9 изо-Амилацетат т’о ®4 8 Амиловые спирты 1,2 44 — Дммияк ' 19,0 10v Zo, и 1 УО Ацетилен 2,4 26 78,0 844 AnewH с 3<0 68 10’8 247 Бензол 14 45 6,7 217 л-Бу?ан 1 9 46 9,0' 217 = .шо-Бутан ]-8 43 8,4 203 «-Бутилацетат 1,7 82 7,6 367 .ызо-Бутилацетат 2,4 116 10,5 50/ «-Бутиловый спирт 1,3 46 11,3 8 «зо-Бутиловый спирт !’! о -,Тл СО ВоД°Р°Д t’9 44 74 265 гркрдц 1,2/ 43 /, 4 ZOO ДиокСан 2,0 73 22,5 824 Дихлорэтилен 5,6 226 13,0 524 Жлол 1,1 48 6,0 265 .и-Ксилол 1,1 48 7,0 309 л-Ксилол 1,1 48 7,0 309 Метан 5,2 35 14,0 93 Метилацетат 3,1 95 15,6 480 Метиловый спирт 6,7 89 36,5 486 Метилпропилкетон 1,5 54 8,1 290 Метилэтилкетон 1,8 54 9,5 285- Монооксид углерода 12,5 145 74,2 ооЗ «-Пентан 1,4 42 7,8 234 .ызо-Пентан 1,4 42 7,6 228 Пиридин 1,8 49 12,4 407 Пропан 2,1 38 9,1 167 «-Пропиловый спирт 2,1 52 13,5 337 ызо-Пропиловый спирт 2,0 50 12,0 300 Сероуглерод 1,3 41 44,5 1408 Сероводород 4,3 61 45,5 645 Толуол 1,5 57 — Уксусная кислота 5,4 135 — — Хлористый этил 3,8 102 15,4 413 Этан 3,0 37 12,5 156 Этилен 3,1 36 32,0 373 Этиловый спирт 4,3 82 18,9 362 Этиловый эфир 1,9 58 48,5 1494 Этиловый эфир муравьиной 2,7 83 13,5 416 кислоты Этиловый эфир пропионовой 1,9 80 11,1 471 кислоты
412
14. Защита атмосферы от загрязнения
Рис. 14,3. Зависимость содержания толуола в слое
активного угля а от числа циклов п (концентра-
ция толуола в газе 0,4% об., температура процес-
са 80 °C):
1 — после адсорбции; 2 — после десорбции.
это 60—80°C. Высота работающего
слоя микропористых углей при по-
вышенной температуре не превыша-
ет 30 см, и, следовательно, процесс
рекуперации с высокой степенью
использования адсорбционной ем-
кости слоя (выше 0,8) возможно проводить в обычной адсорб-
ционной аппаратуре. При десорбции перегретым водяным па-
ром, имеющим на входе в адсорбер температуру 150°С, двух-
фазная установка после 5 циклов (рис. 14,3) выходит на ста-
бильный режим. При этом в случае извлечения толуола из газа
при скорости потока 0,1 м/с полезная динамическая адсорбци-
онная емкость слоя активного угля высотой 0,3 м составляет
5 г/100 г. В этих условиях примерно такое же количество толуо-
ла остается недесорбированным из угля.
За критерий оптимальности в первом приближении может
быть принята разность стоимости рекуперированного толуола в
пара, отнесенная к 1000 м3 очищенного газа. Зависимость кри-
терия оптимальности К от температуры процесса представлена
на рис. 14,4. При температуре ниже 60 °C уменьшение адсорб-
ционной способности вызывается увлажнением адсорбента в.
ходе паровой регенерации, выше 80 °C — снижением емкости
адсорбента по поглощаемому веществу. Эти факторы определя-
ют оптимальный температурный уровень двухфазного процесса.
Двухфазный метод рекуперации толуола и изопропилового
спирта из отходящих газов успешно применяется в США на
предприятиях по производству фильтровальной бумаги [230].
Система включает два адсорбера высотой 12 м и диаметром 3 м.
В одном адсорбере осуществляется стадия адсорбции, в дру-
гом— стадия десорбции растворителей. Выделенные раствори-
тели повторно используют в технологических процессах.
Проведение процесса рекуперации на частично увлажненном
угле часто позволяет устранить
опасность воспламенения угля
или его дезактивацию, если из-
влекаются реакционноспособные
вещества (например, оксид мези-
тила, циклогексанон) [231].
Рнс. 14,4. Зависимость критерия оптимально-
сти К от температуры очищаемого газа (кон-
центрация адсорбтива — толуола — 0,6% об.).
Рекуперация растворителей
413
Вначале рекуперацию оксида мезитила проводили при 30 °C и линейной
скорости паровоздушной смеси 0,3 м/с в четырехфазиом цикле. Десорбцию
вели водяным паром при 105 °C в течение 40 мии при скорости потока
0,1 м/с, а сушку угля — воздухом при 80 °C. Были отмечены случаи возгора-
ния угля, стимулируемые в железной аппаратуре. После 25 циклов актив-
ность рекуперационного угля по оксиду мезитила снизилась в 4 раза.
При улавливании оксида мезитила с использованием двухфазного цикла
адсорбция — регенерация иа увлажненном угле не отмечено ни одного слу-
чая воспламенения. Испытания рекуперации смеси растворителей при цикли-
ческой работе показали, что динамическая активность угля АР-А по этому
компоненту снижается с 15 г/100 г в первом цикле до 11 после пяти цик-
лов, после чего стабилизируется в процессе многоцикловой эксплуатации.
На Нефтекамском комбинате искусственных кож установка
рекуперации смеси растворителей была сдана в эксплуатацию
лишь после перевода ее четырехфазного цикла (по проекту) на
двухфазный, что исключило возможность воспламенения угля с
циклогексаноном.
В некоторых случаях простая пропарка острым паром не
приводит к восстановлению адсорбционных свойств активного
угля. Например, в производстве синтетических полиэфирных во-
локон получение диметилтерефталата ведут окислением «-кси-
лола кислородом воздуха в присутствии кобальтовых солей
жирных кислот. Очистку отходящего в атмосферу воздуха от
многочисленных и разнообразных органических веществ про-
водят в адсорберах с активным углем. Однако адсорбент бы-
стро отравляется такими высококипящими соединениями, как
диметилтерефталат, терефталевая кислота и другие вещества,
которые обычными средствами десорбции не удаляются из пор
угля. Это приводит к резкому уменьшению адсорбционной спо-
собности, особенно по таким относительно плохо поглощаемым:
из газового потока углем продуктам реакции как метанол и
формальдегид.
Для решения проблемы успешно могут быть использованы:
комбинированные способы регенерации адсорбентов. Так, ад-
сорбционные свойства активного угля в данном производстве
полностью восстанавливаются после обработки его 10%-ным!
раствором КОН с последующей продувкой паром в течение 1 Ч'
при 120—130 °C и давлении 0,5 МПа и сушкой горячим возду-
хом при 100 °C [232].
Производительность рекуперационных установок, введенных
в эксплуатацию, находящихся в стадиях строительства или
освоения в Советском Союзе, колеблется от 10 до 150 тыс. м3
газа в час, а концентрация в нем адсорбтива изменяется от
0,5 до 20 г/м3 [233]. Их используют в различных отраслях про-
мышленности: пищевой, полиграфической, резинотехнической,
химической (пластмассы, лаки, краски), асбестотехнической, де-
ревообрабатывающей, текстильной, фотохимической, машино-
строительной.
«414 14. Защита атмосферы от загрязнения
' В ряде случаев, если десорбат и конденсат водяного лара
взаимно растворимы друг в друге, выделение растворителя про-
изводят на дополнительных ректификационных колоннах. Так,
при рекуперации этилацетата и диметилформамида из газовых
выбросов отделочных машин на Луцком заводе синтетических
кож на ректификацию поступает как органическая фаза десор-
бата — этилацетат с содержанием воды 3,2%, так и водная фаза
•с суммарной концентрацией в ней смеси этилацетата и форм-
амида'9%.
Интенсивность и рентабельность работы рекуперационных
'установок во многом зависит от правильно выбранного графика
цикла. Так, перевод установки рекуперации бензина производи-
тельностью 20 тыс. м3/ч на новый цикл: адсорбция 90 мин (вме-
сто 120 мин), десорбция и сушка по 45 мин (вместо 60 мин) —
позволил (повысить степень рекуперации этого растворителя с 74
до 85% со значительным положительным экономическим, эф-
фектом.
Г •
Рекуперация сульфида углерода
Выбросы сульфида углерода в атмосферу непосредственно свя-
заны с развитием промышленности химических волокон. Напри-
мер, комбинат химического волокна, выпускающий 50 т корд-
ного, 120 т штанельного и 20 т целлофанового волокна, выбрасы-
вает в атмосферу в сутки 35 млн. м3 вентиляционного воздуха.
Суточные потери растворителей с этим воздухом составляют
10 т H2S и 56 т CS2. Такие значительные выбросы вызывают
(необходимость очистки вентиляционного воздуха для обеспече-
ния нормальных санитарно-гигиенических условий окружающей
•среды, а также из экономических соображений, так как CS2 и
(продукт его окисления — сера являются ценным сырьем для про-
мышленности.
Во всех случаях <CS2 улавливают с помощью активного уг-
ля. Большую часть H2S в некоторых схемах удаляют жидкими
поглотителями. Трудность использования активного угля для
очистки от о'боих компонентов долгое время заключалась в том,
что при улавливании сульфида водорода углем в присутствии
кислорода основная реакция преобразования адсорбата в серу
(14.1) сопровождается побочным сильно экзотермическим про-
цессом образования серной кислоты (14,2):
H2S-p/2O2 * S-|-H2O4-220 кДж (14.1)
H2S 4- 2Ог -► H2SO4 4- 790 кДж (14.2)
Удельный вес побочной реакции, протекающей в порах ак-
тивных углей обычных типов, настолько значителен, что при вы-
сокой концентрации ’H2S в очищаемом воздухе слой угля силь-
но разогревается:и возникает опасность возгорания CS2.
Рекуперация сульфида углерода
415
Рис. 14,5. Зависимость степени конверсии сульфи-
да водорода в серную кислоту ан250д от с0"
держания солей поливалентных металлов в актив-
ном угле см.
Однако абсорбционная стадия
не решает проблему полного из-
влечения H2S; его концентрация в
газе после абсорбера составляет
20—50 мг/м3. Кроме того, на актив-
ном угле в небольшом масштабе
протекает реакция гидролиза CS2 с
образованием H2S:
CS2 + 2Н2О ----» СО2 + 2H2S (14.3)
Интенсивность реакции образования серной кислоты (14.2)*
зависит от содержания в угле металлов и в первую очередь же-
леза. Как показывает рис. 14,5, при высоком содержании тяже-
лых металлов более одной трети сульфида водорода превра-
щается в серную кислоту. При наличии специальных сортов без-
зольных активных углей возможно перейти к внедрению совме-
щенного метода очистки вентиляционных газов от H2S и CS2
[234—236]. Такой процесс может быть аппаратурно оформлею
различно. Очистку можно вести в одном адсорбере одним ти-
пом угля, в одном адсорбере с двумя слоями различных углей
и в двух последовательно включенных адсорберах с углем од-
ного или разных типов.
Образовавшуюся серную кислоту раньше- нейтрализовали;'
раствором соды с последующим вымыванием продуктов нейтра-
лизации водой. Впоследствии стали широко применять нейтра-
лизацию серной кислоты газообразным аммиаком. Образовав-
шиеся аммонийные соли вымываются водой легче, чем соли1
натрия, в связи с чем расход воды при нейтрализации аммиаком
в 4 раза меньше.
Аммиак можно вводить в систему периодически на стадии-
регенерации (охлаждения) адсорбента или непрерывно в не-
большом количестве примешивать к очищаемому газу. На
рис. 14,6 приведены кривые падения активности угля по CS2 в
процессе многоцикловой эксплуатации. Нижняя кривая показы-
Рис. 14,6. Зависимость активности угля по
сульфиду углерода при многоцикловой экс-
плуатации:
1 — непрерывная добавка NH3 в исходный воз-
дух; 2 — периодическая добавка NH3 на стадии
охлаждения; 3 — без добавки NH3.
-416 14- Защита атмосферы от загрязнения
вает, насколько быстро падает активность угля без добавки
аммиака. Падение активности резко замедляется при непрерыв-
ном добавлении аммиака в очищаемый газ (верхняя пунктир-
ная линия). Периодическая нейтрализация кислоты аммиаком
на стадиях охлаждения угля каждый раз приводит к частичному
восстановлению адсорбционных свойств угля. После того как
активность угля упадет на 20—25%, адсорбер переключают на
стадию промывки водой.
Отложение элементной серы в порах активного угля приво-
дит к снижению предельного адсорбционного объема, равно-
весной и динамической активности. При этом увеличивается
структурная константа В, что указывает на уменьшение числа
микропор, доступных для адсорбируемых молекул. Изменение
формы изотерм в процессе забивки пор угля серой приводит к
увеличению числа единиц переноса и, как следствие этого, к
возрастанию длины зоны массообмена. Теплота адсорбции
сульфида углерода газовыми и рекуперационными углями в
среднем составляет 31,5 кДж/моль. Изменение структурных
констант активного угля APT, высоты работающего слоя (Lo)
и динамической активности (ад) слоя угля высотой 0,5 м по
>сульфиду углерода при различном содержании в угле элемент-
ной серы (Cs) показано ниже:
CS, % 0 4,4 6,4 7,4 10,4 16,4 18,4 20,4
Го, см3/г 0,418 0,320 —_ — 0,210 0,152 0,143 0,103
В-10-6, град-2 3,54 4,13 — — 7,63 8,00 8,30 8,87
Lo, см 5,10 6,96 9,30 10,00 —_ — 17,60 18,30
«Д, % 4,05 2,53 1,95 1,78 — —— 0,38 0,23
Как показали данные ртутной порометрии, сера в виде плот-
ных «линз» откладывается в макро- и мезопорах, блокируя вхо-
ды в микропоры. Этим объясняется сравнительная легкость ре-
генерации сернистых углей. Регенерацию осерненного активного
угля проводят промывкой растворителем, обычно сульфидом
углерода.
Схема совмещенного метода очистки вентиляционных выбро-
сов, разработанная западногерманской фирмой Пинч-Бамаг,
приведена на рис. 14,7 |[237].
Воздух, содержащий примеси, с помощью воздуходувки 9 пропускают
через один или несколько параллельно включенных адсорберов 8. К воздуху
.примешивают аммиак. Сульфид водорода окисляется в лобовом слое угля,
при этом в порах угля отлагается сера. Одновременно происходит физиче-
ская адсорбция сульфида углерода. Очищенный воздух выбрасывают в ат-
мосферу через трубу. Содержание примесей в 1 м3 очищенного воздуха со-
ставляет 10—20 мг CSa и 1—2 мг H2S. На стадии очистки концентрацию
'CS2 в очищенном газе непрерывно измеряют газоанализатором, и в момент
«проскока» поток воздуха с помощью исполнительного механизма автомати-
•чески переключается на адсорбер с отрегенерированным углем, а адсорбер 8
^переключается на стадию регенерации.
Рекуперация сульфида углерода
417
Рис. 14,7. Схема установки очистки вентиляционного воздуха от сульфида водорода и
рекуперации сульфида углерода:
1 — испаритель сульфида углерода; 2, 5, 7 — емкости; 3, 11 — конденсаторы; 4, 12 —хо-
лодильники; 6, 14 — сепараторы; 8 — адсорберы; 9, 13 — воздуходувки; 10 — подогрева-
тель.
Смесь сульфида углерода с воздухом взрывоопасна уже при 80 °C,
и поэтому перед стадией десорбции адсорбер продувают инертным газом
до тех пор, пока концентрация кислорода не упадет ниже границы взрывае-
мости сульфида углерода. При этом снижается также возможность окисле-
ния CS2 на последующей стадии десорбции, которую проводят паром. Смесь
паров CS2 и Н2О конденсируют (И) и дополнительно охлаждают до 25—
30 °C (12). В сепараторе 14 жидкая фаза разделяется ввиду разницы в
плотностях. Нижний сероуглеродный слой (CS2) является высококачествен-
ным товарным продуктом; воду направляют в канализацию. В конце стадии
десорбции практически весь сульфид углерода удаляется из угля.
Сушку и охлаждение угля проводят атмосферным воздухом, подаваемым
в адсорбер воздуходувкой 13. На стадии сушки воздух подогревают до
120 °C в паровом нагревателе 10. На стадии охлаждения подачу пара в на-
греватель прекращают или воздух пропускают по байпасу мимо нагревателя.
Когда температура в адсорбере снизится до заданного уровня, продувку хо-
лодного воздуха прекращают. Адсорбер с закрытыми вентилями оставляют
в резерве до тех пор, пока очередной адсорбер не насытится сульфидом уг-
лерода.
Периодически адсорбер отключают от основного рабочего цикла и отмы-
вают сернистые соединения из угля. Сначала водой удаляют аммонийные
соли, а затем сульфидом углерода из емкости 7 — элементную серу. Выходя-
щий из адсорбера сульфид углерода, содержащий растворенную серу, соби-
рают в емкости 5, затем испаряют (1), конденсируют (3), охлаждают (4) и
после отделения от воды в сепараторе 6 собирают в емкости 7. Серу рас-
плавляют и в жидком состоянии выводят из испарителя 1. Содержание эле-
ментной серы в сульфиде углерода, выходящем из испарителя, не превышает
200 мг/т.
Установки рекуперации сульфида углерода в высшей степе-
ни пожароопасны, и их управление полу- или полностью авто-
матизировано. Особое внимание уделено температурному режи-
му и точному регулированию технологических потоков. При об-
наружении неполадок включается звуковой и световой сигналы
тревоги, и установка отключается.
В табл. 14-2 приведены расходные коэффициенты на рекупе-
рацию 1 т сульфида углерода для двух концентраций примесей.
27—1346
418
14. Защита атмосферы от загрязнения
Таблица 14-2. Расходные коэффициенты
на 1 т рекуперированного сульфида углерода
Показатели Совмещенный метод, кон- центрация примесей в 1 м3 Абсорбция НзЭ+ад- сорбция CSsJ кон- центрация примесей в 1 м3: 12 г CS2, 2 г H2S
4 Г CS2, 1,5 г H2S 12 г CS2, 2 г H2S
Пар, т 7,9 4,2 4,9
Вода, т 107 59 240
Электроэнергия, кВт-ч ИЗО 395 460
Аммиак, кг 23 10 0,5
Активный уголь, кг 5,7 3,1 1,5
Защитный газ, м3 90 50 30
Сжатый воздух, м3 1 1 750
Кроме того, приведены данные для случая, когда CS2 предва-
рительно удаляют абсорбцией. При их расчете приняты следу-
ющие температуры: вентиляционного воздуха 25 °C, охлаждаю-
щей воды 15 °C, насыщенного пара 130 °C и перепад давления
в системе 2 кПа.
Применять совмещенный метод очистки следует при кон-
центрации H2S не выше 3 г/м3. В других случаях перед адсорб-
ционным блоком следует размещать абсорбционную колонну
для удаления сульфида водорода.
На Рязанском комбинате искусственного волокна для очистки вентиля-
ционных выбросов сооружена установка производительностью 180 тыс. м3/ч»
на которой извлечение CS2 проводят в адсорберах с неподвижным слоем
активного угля (диаметр 5,6 м, высота слоя угля 1,6 м). Линейные скоро-
сти газового потока в адсорберах достигают 0,35 м/с. Содержание CS2 на
входе в адсорбер 4—5 г/м3, степень очистки воздуха 95—98%. В целом про-
цесс очистки ведут непрерывно. Одновременно в одних адсорберах идет по-
глощение H2S, в других — десорбция, сушка и охлаждение угля. Переклю-
чение аппаратов с одной стадии на другую ведут автоматически по заданной
программе. Установки с неподвижным слоем активного угля надежны в
эксплуатации благодаря специальной системе автоматизации.
На рис. 14,8 приведена схема управления процессом рекуперации суль-
фида углерода [238, 239].
Автоматическое управление проводится многоцелевыми (расположенными
в операторной) командными приборами МКП, которые выполняют програм-
му вместе с реле времени РВ, контролирующими продолжительность отдель-
ных операций. Программа управления задается ключом КВУ. В схеме пре-
дусмотрено также дистанционное управление ключами КВД и КДУ с пульта
при сохранении всех блокировок безопасности, действующих при автоматиче-
ском управлении.
Через релейную схему сигнал о подаче команды поступает в блок возду-
хораспределителей БВ, где преобразуется из электрического в пневматиче-
ский (давление 0,6 МПа). Последний передается в блок управления высоко-
го давления БУВД, непосредственно управляющий исполнительными меха-
низмами запорных клапанов адсорберов.
Рабочий цикл адсорбера включает восемь операций: 1) адсорбция,
2) перекрытие всех клапанов, 3) продувка азотом, 4) десорбция, 5) сниже-
Рекуперация сульфида углерода
419
пне давления, 6) осушка, 7) охлаждение, 8) открытие клапана очищенного
воздуха. Суммарная продолжительность операций 2—5 соответствует про-
должительности стадий 7—8. Переключение адсорберов на регенерацию и
переход от одной операции к другой осуществляются по заданному времени;
стадии десорбции, сушки и охлаждения управляются по времени и по тем-
пературе в адсорбере. Для переключения адсорбера температура на стадии
десорбции должна достичь 100 °C, а на стадии охлаждения снизиться до
40 °C. Если температура в десорбере превысит 150 °C, произойдет его от-
ключение от всех технологических линий.
Формирование пневматического сигнала о процессе в адсорбере проис-
ходит в блоке сигнализации положения клапанов БСПК, затем сигнал в
Дистанционное
управление
Автоматическое управление
Рис. 14,8. Схема управления процессом рекуперации сульфида углерода:
I — блок управления высокого давления БУВД; II — блок сигнализации положения кла-
панов БВПК; 1—10 — адсорберы.
27
420
14. Защита атмосферы от загрязнения
блоке преобразователей БП трансформируется в электрический и регистри-
руется на ленте самопишущего прибора через блок записи БЗ.
Сведения о любых отклонениях от нормального режима поступают в опе-
раторную в форме светозвуковых сигналов.
При очистке газов от микропримесей органических или серо-
органических соединений небольшое содержание влаги в угле
незначительно сказывается на адсорбционной способности по
извлекаемому компоненту. На рис. 14,9 представлены зависимо-
сти равновесной и динамической активности слоя угля APT вы-
сотой 1 м при концентрации CS2 в воздухе 1 г/м3, скорости по-
тока 0,25 м/с, относительной влажности 40% и температуре
35°C. Эти кривые показывают, что резкое падение активности
угля по CS2 происходит при влажности адсорбента выше 6—
8 г/100 г.
Изучение влияния относительной влажности газовоздушной
смеси и содержания влаги в сорбенте позволяет в значительной
мере аргументировать переход от четырехфазного процесса
адсорбции сульфида углерода в неподвижном слое адсорбента
к двухфазному, из которого полностью исключены стадии суш-
ки и охлаждения угля. Как показали промышленные испытания
на Калининском комбинате химического волокна, при некото-
ром изменении конструкции аппаратуры (применение адсорбе-
ров с паровой рубашкой) после стадии десорбции газовоздуш-
ная смесь, имеющая относительную влажность 50—60%, при
40—60°C может быть подана в слой активного угля, прошедше-
го только стадию десорбции перегретым паром. При этом про-
цесс очистки протекает достаточно эффективно. Фронт тепловой
волны опережает передвижение фронта сорбции, уголь охлаж-
дается непосредственно на стадии очистки, одновременно про-
исходит его подсушка до требуемого уровня влажности, т. е.
до 6—8 г/100 г.
Применение двухфазного режима работы позволяет увели-
чить производительность адсорбера на 20—25%, снизить рас-
ход пара, электроэнергии, эксплуатационные затраты в целом.
Как указывалось, наиболее эф-
фективно процесс очистки от суль-
фида водорода протекает в мезопо-
рах. В то же время, для улавлива-
ния CS2 целесообразно использо-
вать микропористые угли. Поэтому
в одном из вариантов процесса вен-
тиляционный воздух производств.
Рис. 14,9. Зависимость равиовесиой (1) и динами-
ческой (2) активности а угля APT по сульфиду
углерода от влажности адсорбента ТГа.
Рекуперация хлорорганических растворителей 421
очищают двумя типами активных углей: сульфид водорода —
углем с развитой мезопористостью, сульфид углерода — микро-
пористым углем.
Установки с периодическими адсорберами требуют большой
площади для размещения, и их производительность ограничи-
вается 300—400 тыс. м3 вентиляционного воздуха в час. Стрем-
ление увеличить пропускную способность углеадсорбционных
установок стимулировало разработку процессов с кипящим
слоем адсорбента. Скорости потока в этом случае на порядок
выше скоростей в адсорберах со стационарным слоем.
В 1956 г. в Англии фирмой Куртольдс была создана промышленная уста-
новка, перерабатывающая 1 млн. м3| вентиляционных выбросов в час в ки-
пящем слое активного угля — норита. От сульфида углерода воздух очища-
ют в адсорбционной колонне диаметром 16 м на трех перфорированных пол-
ках; высота слоя адсорбента в спокойном состоянии на полке составляет
50 мм, в кипящем 70—80 мм. Газовоздушная смесь с содержанием CS2
0,4—0,6 г/м3 поступает в нижнюю часть колонны. Противотоком газу непре-
рывно подают уголь, который «стекает» с полки на полку через специаль-
ные переточные устройства, постепенно насыщаясь. Насыщенный уголь по-
ступает в отпарную колонну, где в движущемся слое происходит десорбция
сульфида углерода, и адсорбент подсушивается. После сушки уголь вновь
возвращают на адсорбцию.
При циркуляции в цикле адсорбция — десорбция в активном угле про-
исходит накопление сернистых соединений в результате окисления кислоро-
дом воздуха сульфида водорода, оставшегося после первой ступени очистки
жидкостного типа. Для восстановления адсорбционных свойств угля часть
его непрерывно выводят па глубокую регенерацию, которую проводят пере-
гретым паром при 450 °C.
Промышленное освоение установок с кипящим слоем на-
толкнулось на ряд трудностей. Основной их недостаток — зна-
чительные потери активного угля вследствие истирания. Крупно-
масштабные установки с кипящим слоем могут быть пущены в
эксплуатацию только при наличии шариковых адсорбентов вы-
сокой механической прочности. Кроме того, установки с кипя-
щим слоем сложны в эксплуатации, аппаратура подвергается
интенсивной эрозии и коррозии, часто выходит из строя вслед-
ствие циркуляции в системе угля, содержащего серную кислоту.
Рекуперация хлорорганических растворителей
Работа современных предприятий химической чистки системы
бытового обслуживания сопряжена с загрязнением окружающей
среды хлорорганическими соединениями, используемыми в тех-
нологических процессах в качестве растворителей. Одна из ос-
новных причин таких выбросов обусловлена существующей
технологией заключительных стадий процессов химической
чистки — сушки обработанных предметов и проветривания.
Операция сушки в машинах химической чистки основана на удалении
растворителя из этих аппаратов путем циркуляции паровоздушной смеси
422 14. Защита атмосферы от загрязнения
через замкнутую систему, включающую калорифер, моющий барабан и кон-
денсатор, с выделением в последнем растворителя. Эта операция продолжа-
ется до тех пор, пока содержание растворителя в обрабатываемых предме-
тах и циркулирующей паровоздушной смеси не достигает определенного
уровня, при котором дальнейший вывод растворителя из системы путем
конденсации нецелесообразен. Остаточный растворитель (1—3% от массы
обрабатываемых предметов) удаляется из внутреннего объема моечного ба-
рабана и транспортных магистралей во время операции проветривания.
В качестве агентов химической чистки в основном используют дорого-
стоящие и дефицитные хлорорганические растворители — перхлорэтилен и
трихлорэтилен. Кроме того, действующие санитарные нормы предопределяют
необходимость обезвреживания поступающих в атмосферу газовых выбросов
предприятий химической чистки: концентрация вредных веществ в атмосфе-
ре не должна превышать максимально-разовую предельно допустимую кон-
центрацию. Поэтому паровоздушные потоки операции проветривания обыч-
но направляют в углеадсорбционные аппараты моечных машин с целью
выделения содержащихся в газовой фазе хлорорганических растворителей с
последующим возвращением их в производство. На некоторых предприятиях
такие операции проводят в общецеховых адсорбционных установках, обслу-
живающих группу машин химической чистки.
Таблица 14-3. Адсорбция паров перхлорэтилена слоем
активного угля высотой 36 см
Тип угля и диаметр гранул Показатели
t, °C Со. % (об.) Тпр. МИИ Тр, мнн Lq, см
Газовый, микропористый, 50 0,50 296 353 6,0 3,05 мм 0,91 126 177 12,0 1,17 86 126 12,6 То же, 1,88 мм 20 0,57 350 392 4,7 0,76 208 245 5,2 1,50 81 116 13,2 50 0,54 320 358 4,1 0,79 173 196 5,2 1,25 79 108 13,5 75 0,43 282 335 6,2 1,03 133 153 7,0 1,33 66 89 10,7 100 0,56 242 298 7,5 1,15 97 115 6,5 1,33 63 86 11,7 То же, 1,05 мм 50 0,49 335 376 4,1 0,84 165 189 5,0 1,24 89 109 7,3 Рекуперационный, 3,0 мм 20 0,50 165 233 13,0 1,02 60 99 15,6 1,26 44 85 22,6 50 0,52 151 196 9,7 1,00 50 85 15,6 1,25 40 75 22,1 75 0,50 115 155 10,7 1,02 45 79 19,7
Очистка газов от диоксида серы.
423
Динамика сорбции паров перхлорэтилена из его низкокон-
центрированных смесей с воздухом изучена [240] на трех об-
разцах газовых углей различного зернения с примерно одина-
ковой пористой структурой: Ws =0,40 см3/г; 1Уми=0,18 см3/г;
1^ме = 0,04 см3/г; 1^ма=0,18 см3/г, а также в слое рекупераци-
онного угля, наиболее широко использовавшегося в качестве
адсорбента при чистке паровоздушных смесей на предприятиях
химической чистки.
Исследование вели на лабораторной установке в адсорбцион-
ной колонке диаметром 1,4 см при температуре 20—100 °C й
концентрации перхлорэтилена в паровоздушных потоках 0,5—
1,5% (об.). Паровоздушную смесь подавали в слой адсорбента
со скоростью 0,25 м/с. Табл. 14-3 характеризует результаты
этого исследования. Из нее видно преимущество использования
микропористых активных углей на предприятиях химической
чистки. Высокая степень отработки адсорбционной емкости слоя
может быть достигнута при его высоте 0,5—1,0 м.
Очистка газов от диоксида серы
Диоксид серы по масштабу выброса превосходит все другие
загрязнители атмосферы. Он образуется при сгорании серы,
сульфида водорода, а также при нагревании различных суль-
фидов в токе воздуха или кислорода. Сера, содержащаяся в
топливе и рудах, при их сжигании или термической переработ-
ке превращается в этот агрессивный газ. Предварительная се-
роочистка топлива (например, многосернистого угля) и руд не
может в полной мере обеспечить ликвидацию выбросов диок-
сида серы в атмосферу. Основным источником загрязнения ат-
мосферы диоксидом серы являются теплоэлектростанции. Кон-
центрация SO2 в дымовых газах теплоэлектростанций зависит
в основном от содержания серы в топливе. Так, при сжигании
мазута с содержанием серы 3% концентрация диоксида серы в
дымовых газах составляет 0,4% (об.), а при содержании серы
3,5% — 0,45% (об.).
Загрязняя атмосферу, диоксид серы наносит вред окружаю-
щей среде (человеку, животному и растительному миру). Улав-
ливание диоксида серы необходимо, но не только для с сани-
тарно-гигиенической точки зрения, а и по технико-экономичес-
ким соображениям. Показателен процесс, достигнутый в цветной
металлургии, где диоксид серы улавливают из части конвертор-
ных газов, ранее выбрасываемых в атмосферу. Выпуск по-
лучаемой из него серной кислоты составляет 30% общего ее
производства в Советском Союзе, причем она в три раза де-
шевле кислоты, получаемой обычными методами в химической
промышленности.
-424
14. Защита атмосферы от загрязнения
Химическая промышленность, хотя по общему выбросу и за-
нимает среди других отраслей лишь шестое место, в районе
местоположения сернокислотных заводов интенсивно загряз-
няет атмосферу. В отходящих газах сернокислотного производ-
ства содержится 0,2—0,3% SO2, а также SO3 и аэрозоль
H2SO4. Улавливание аэрозоля производят в абсорбционных
башнях и электрофильтрах.
Для обезвреживания газов, содержащих диоксид серы, пред-
ложено большое число методов, но ни один из них не может
быть признан идеальным. Ниже будут описаны основные мето-
ды адсорбционной очистки отходящих газов сернокислотного
производства, тепловых электростанций и других промышлен-
ных объектов.
Методы очистки газов углями
Адсорбенты являются эффективным средством для рекупера-
ции и обезвреживания диоксида серы, причем их потенциаль-
ные возможности в этом направлении выявлены еще далеко не
полностью. Для улавливания SO2 в основном применяют угле-
родные пористые вещества £241].
Если улавливание производить из потока газа, не содержа-
щего кислород, поглощение диоксида серы происходит по зако-
нам физической адсорбции, и при десорбции активность адсор-
бента полностью восстанавливается. Изотермы адсорбции диок-
сида серы на активном угле характеризует рис. 14,10. Теплота
адсорбции диоксида серы в среднем составляет (в кДж/моль):
на силикагеле — 23, на графите — 30, на активном угле — 44,
на активных полукоксах — до 42 [242]. Однако реальные техно-
логические и вентиляционные газы в подавляющем большинст-
ве случаев — кислородсодержащие.
Активный уголь и другие углеродные адсорбенты являются
катализаторами превращения поглощенного диоксида серы в
триоксид [243]:
SO2 + V2O2 --► SO3 (14.4)
В присутствии воды триоксид серы, в свою очередь, обра-
зует серную кислоту:
SO3-j-II2O --- H2SO4 (14.5)
Серная кислота в избытке воды разбавляется. Концентра-
ция образовавшейся серной кислоты зависит от условий прове-
дения процесса и влажности очищаемого газа.
Вне зависимости от концентрации диоксида серы в исходном
газе, в адсорбированной фазе содержится около 50% (масс.)
этого компонента, остальная часть приходится на продукты его
Очистка газов от диоксида серы
425
Рис. 14,10. Изотермы адсорбции диоксида серы на
активном угле СКТ.
окисления и гидролиза. Это соотно-
шение изменяется только в зави-
симости от температуры и влажно-
сти среды.
Таким образом, адсорбат содер-
жит три категории веществ: физи-
чески адсорбируемый диоксид серы,
обратимо удаляемый из твердой
фазы вакуумированием или продув-
кой газа при температуре десорб-
ции, например 100 °C; необратимо
адсорбированную серную кислоту,
не выделяющуюся при температу-
ре до 190 °C, но удаляемую в ре-
зультате промывки водой; серни-
стые соединения, прочно связанные
с углеродом (около 0,5 г/100 г адсорбента), которые не удаля-
ются при промывке водой, но можно провести их экстракцию
перекисью водорода. Соотношение между обратимо и необрати-1
мо адсорбированным диоксидом серы зависит от температуры
адсорбции [244].
При высоких температурах возрастает скорость превраще-
ния диоксида серы в серную кислоту, в результате чего харак-
тер связи молекул адсорбата с адсорбентом преимущественно
химический, т. е. необратимый. При низких температурах массы
обратимо и необратимо поглощенных примесей становятся со-
измеримыми.
Скорость экстракции серной кислоты зависит от структуры
активных углей. Чрезмерное развитие микропористости угля
вызывает необходимость увеличения периода экстракции и рас-
хода промывной воды. Поэтому иногда понижают микропо-
ристость углей, применяемых для извлечения диоксида серы,
путем их дополнительной активации при температурах выше
700 °C.
Для промышленных установок адсорбционного извлечения
диоксида серы в неподвижном слое адсорбента следует реко-
мендовать механически прочные активные угли с небольшим
гидравлическим сопротивлением, т. е. в виде крупных гранул.
Метод «Хитачи» [245—248]. Метод «Хитачи» разработан в
Японии. Испытания его прошли последовательно на двух уста-
новках: сначала — на опытной производительностью 6 тыс. м3/ч,
обслуживающей два котла тепловой электростанции, а затем—1
на опытно-промышленной производительностью 150 тыс. м3/ч,)'
где очищался газ шести котлов. Опытная установка проработа-'
426
14. Защита атмосферы от загрязнения
ла в течение 3500 ч. Очистку проводили при температуре выше
100°C, степень очистки превышала 90%. Опытно-промышленная
установка работала в течение года. Технологическая схема про-
цесса «Хитачи» представлена на рис. 14,11.
Отходящие газы из котельной 1, прежде чем поступить в дымовую тру-
бу 5. проходят через пылеуловитель 2 и несколько параллельно включенных
адсорберов с активным углем 4. Гидравлическое сопротивление слоя адсор-
бента (500 Па) преодолевается с помощью газодувки 3.
Один из адсорберов (4а) находится на стадии промывки. В первый пе-
риод промывку ведут достаточно концентрированной серной кислотой, затем
последовательно — кислотой средней, низкой концентрации и, наконец, во-
дой. Затем раствор проходит через маслоотделитель 7 и собирается в емко-
сти 6. Жидкость, полученная при промывке, является 20%-ной серной кисло-
той. Ее упаривают в блоке 8 до концентрации 70%, а затем после охлаж-
дения (9) и центрифугирования (10) собирают в емкостях И и 12, откуда
она поступает потребителю как товарный продукт.
После промывки влажность угля достигает 20—25%, и его сушат по
одному из двух вариантов. По первому варианту, как показано на схеме,
используют часть загрязненного газа, который при сушке частично очищают
и затем примешивают к основному потоку очищенного газа, выбрасываемого
в трубу. По второму варианту часть газа после газодувки 3 пропускают
через адсорбер, находящийся на стадии сушки, а затем примешивают к по-
току газа, который направляется в адсорберы на очистку. В этом случае
в схеме следует предусмотреть установку дополнительной газодувки. Если
сушку проводить газом, образующимся в результате сжигания тяжелого
жидкого топлива, температура газа на выходе из адсорбера до завершения
Рис. 14,11. Схема установки очистки дымовых газов активным углем по методу «Хи-
тачи»:
1 — котельная; 2 — пылеуловители; 3 — газодувка; 4 — адсорберы иа стадии очистки; 4а —
адсорбер на стадии промывки; 46 — адсорбер на стадии сушки угля; 5 — дымовая тру-
ба; 6 — резервуары с разбавленной кислотой и водой; 7 — маслоотделитель; 8--концент-
ратор кислоты; 9 — холодильник; 10 — центрифуга; 11 — емкость для кислоты; 12 —сбор-
ник кислоты; 13 — насос для подачи концентрированной кислоты иа склад.
Очистка газов от диоксида серы
427
Рис. 14,12. Схема установки очистки газа
в горизонтальных адсорберах при пони-
женной температуре по методу «Лурги»:
I — скруббер Вентури; 2 — сепаратор; 3 —
адсорбер; 4 сборник кислоты; 5 — цирку-
ляционный насос.
стадии устанавливается на уровне
50—60 °C. После завершения испаре-
ния влаги температура выходящего
газа быстро растет до уровня темпе-
ратуры газа, поступающего на очи-
стку.
Установка производительностью
150 тыс. м3/ч была предназначена
для очистки газа от агрегата мощностью 55 МВт, работающего
с содержанием серы 1,8%. Она состояла из 5 адсорберов высотой
и диаметром 5 м. Приблизительная длительность стадий: очистки — 30 ч,
промывки — 20 ч, сушки — 10 ч.
на
мазуте
11,2 м
Процесс «Лурги» [249]. В отличие от японских установок в
установках фирмы Лурги (ФРГ) газ, содержащий водяные пары
и кислород, пропускают через горизонтальный адсорбер с ак-
тивным углем при температурах ниже 100 °C (рис. 14,12).
Образовавшаяся серная кислота периодически отмывается разбрызгивае-
мой сверху водой. Выделяющуюся 10—15%-ную кислоту из сборника 4 цир-
куляционным насосом 5 закачивают в скруббер Вентури 1. В скруббере
происходит увлажнение и охлаждение газа до требуемой температуры, а за
счет его тепла серная кислота упаривается до концентрации 30—70% и вы-
деляется в сепараторе 2.
Испытание процесса на опытной установке производительностью
1000 м3/ч доказало возможность очистки газа, образующегося при сжигании
сеонистого мазута и содержащего 0,1% SO2, со степенью очистки 98—99%.
По этому методу в ФРГ эксплуатировалась установка для очистки
50 тыс. м3/ч отходящих газов производства серной кислоты, содержащих
0,18% SO2. Проектная степень очистки составляла 90%.
Процесс «Штратман» [250—252] проводится также в адсор-
берах с неподвижным слоем активного угля, но уголь непре-
рывно орошается водой. В адсорбере поддерживают температу-
ру в пределах 65—80 °C. При более низких температурах реак-
ция окисления диоксида серы затухает, при более высоких наб-
людается интенсивное испарение влаги, в связи с чем возрас-
тают потери тепла. Температура в адсорбере зависит от темпе-
ратуры поступающего газа (при температуре орошающей воды
30 °C):
Температура газа, °C 200 300 500 700 1000
Температура в адсорбере. °C 60 65 70 75 80
Обычно газы поступают на очистку при температуре ниже
200 °C, и для поддержания заданного режима в адсорбер до-
полнительно вводят пар. На установке для очистки дымовых
428 14. Защита атмосферы от загрязнения
газов (120°С) ТЭЦ мощностью 1000 МВт расход добавочного/
пара может колебаться от 400 (задана температура в адсорбе-
ре 65°C) до 1600 т/ч (задана температура 80°C). Концентра-
ция полученной кислоты не превышает 8%.
Концентрацию кислоты можно повысить, проводя очистку в
две ступени. На первой ступени газ в противотоке очищается
разбавленной серной кислотой, поступающей со второй ступени.
Очистку на второй ступени проводят в адсорбере, орошаемом
водой, причем направление движения газа и воды прямоточное.
Расходные коэффициенты на установке очистки дымовых газов
ТЭЦ мощностью 1000 МВт в расчете на часовую производи-
тельность приведены ниже:
Активный уголь, т 0,3 Вода, т 200
Электроэнергия, КВт.ч 4000 Пар (добавочный), т >200
Основным недостатком метода является большой расход
пара. Кроме того, увлажнение очищенного газа и снижение
его температуры перед выбросом приводит к ухудшению усло-
вий рассеивания в атмосфере. Для того чтобы улучшить рассеи-
вание, приходится подогревать очищенный газ или очищать
только часть выбрасываемого газа; эту часть перед подачей в
трубу смешивают с неочищенным газом, имеющим более высо-
кую температуру. Естественно, последнее решение связано с
понижением общей степени улавливания диоксида серы.
Метод «Райнлюфт» [253, 254]. По методу «Райнлюфт»
(ФРГ) регенерацию угля проводят не в результате экстракции
серной кислоты, а ее испарением путем нагрева угля до 400 °C.
При этом все другие сернистые соединения, поглощенные углем,
превращаются в диоксид серы в соответствии с реакциями
2SO3 4-С >2SO24-CO2 (14.6)
2SO3 + S -->- 3SO2 (14.7)
Таким образом, углерод и сера, образовавшаяся в адсорб-
ционно-десорбционном цикле, являются восстановителями три-
оксида серы в диоксид.
Побочные реакции, например образование других сернистых
соединений (H2S, CS2, COS), при этих температурах не проте-
кают. В то же время, образовавшийся диоксид серы легко уда-
ляется из адсорбированной фазы в небольшом потоке продувоч-
ного газа. Его концентрация в выходящей парогазовой смеси,
состоящей из азота, диоксида углерода и водяных паров, дости-
гает 40—55%. При циклической работе расход активного угля
вследствие «угара» (химических реакций) составляет 0,2 кг на
1 кг уловленного диоксида серы. Процесс очистки проводят в
движущемся слое адсорбента (рис. 14,13).
Очистка газов от диоксида серы
429
Рис. 14,13. Схема установки очистки газов в движущемся слое адсорбента по методу
«Райнлюфт»:
1 — зона каталитической конверсии диоксида серы; 2 — вентилятор; 3 — теплообменник;
4 — зона поглощения непрореагировавшего диоксида серы; 5 — десорбционная зона; 6 —
холодильник; 7 (а—в) — блоки переработки в товарные продукты.
Исходный газ проходит через две зоны с движущимся слоем: в первой
зоне 1 при температурах до 150 °C происходит каталитическая конверсия
диоксида серы. После этого поток газа выводят из адсорбционной колонны
и вентилятором 2 подают в теплообменник 3, где охлаждают воздухом, ко-
торый идет на сжигание топлива (одновременно воздух подогревается), а за-
тем во вторую адсорбционную зону 4. Температура газа на входе в эту зону
составляет 100—150 °C. Образовавшаяся серная кислота полностью остается
в циркулирующем угле, и поэтому теплообменник 3 изготовляют из нелеги-
рованной стали. Во второй ступени происходит улавливание непрореагиро-
вавшего диоксида серы. Степень очистки газа можно изменять в результате
изменения соотношения адсорбент — газ. Она может достигать 99,9%.
Насыщенный диоксидом серы и серной кислотой уголь спускается в де-
сорбционную зону 5. Здесь он нагревается потоком циркуляционного газа,
нагретого до 400 °C. Часть газа непрерывно отводят из десорбера и охлаж-
дают в теплообменнике 6; здесь из газа выпадает и выводится из системы
пыль и конденсат, содержащий ионы хлора и фтора. Диоксид серы
(50%-ный) в смеси с азотом и диоксидом углерода поступает в блок 7 на
переработку в товарные продукты: сжиженный диоксид серы, серную кис-
лоту или элементную серу. Серу получают по методу Клауса с использова-
нием в качестве восстановителя газа, который поступает с нефтеперераба-
тывающих, коксовых и других установок и содержит сульфид водорода. Ре-
генерированный уголь выводят из десорбера снизу, отсеивают от пыли и воз-
вращают в цикл.
Метод «Райнлюфт» позволяет перерабатывать газы, содер-
жащие в 1 м3 до 2 г пыли, золы, сажи и других твердых час-
тиц. Пыль задерживается адсорбентом, а затем выводится из
цикла при отсеве мелочи. Выходящий из адсорбционной секции
очищенный газ содержит в 1 м3 менее 150 мг твердых частиц.
430 14. Защита атмосферы от загрязнения
Если в газе присутствуют оксиды азота, они полностью по-
глощаются углем, а затем в десорбере восстанавливаются до
элементного азота. Сложнее обеспечить улавливание галогенов
и их соединений. В некоторых газах концентрация хлора, хло-
рида и фторида водорода достигает 2 г/м3. Эти компоненты пол-
ностью поглощаются адсорбентом, но затем загрязняют десор-
бированный диоксид серы, что нужно учитывать при переработ-
ке последнего в товарный продукт.
Сернистые соединения могут присутствовать в перерабаты-
ваемом газе не только в виде оксидов, но и в виде CS2, COS4.
H2S и паров серы. В небольшом количестве эти сернистые со-
единения не мешают технологическому процессу, но при их из-
бытке отмечается чрезмерное заполнение пор элементной се-
рой, что приводит к затруднениям при эксплуатации установки.
Если в газе присутствуют смолистые вещества и углеводо-
роды, иногда дурнопахнущие, иногда канцерогенные, они также
полностью поглощаются адсорбентом, а в десорбере под дей-
ствием серной кислоты и высоких температур превращаются в
кокс. Поэтому ни очищенный газ, ни рекуперированный диоксид
серы этих вредных веществ не содержат.
При проектировании газоочистных установок необходимо
разрабатывать систему утилизации образующейся угольной пы-
ли и сточных вод. На 1 т уловленного диоксида серы получают
около 100 кг угольной мелочи. Эту мелочь можно не только
сжигать в пылеугольных топках, но и найти ей более рацио-
нальное использование.
В адсорбере, десорбере, вентиляторах, системе управления
процессом, транспортерах адсорбента серная кислота и влага
не выделяются, и угроза коррозии отсутствует. Поэтому эти
узлы изготовляют из нелегированной стали. В узле очистки и
охлаждения газа, содержащего диоксид серы, а также в блоках
переработки его в товарные продукты должно быть предусмот-
рено использование кислотостойких материалов: легированных
сталей, керамики, свинца и т. д.
Производительность типовой установки с движущимся слоем
адсорбента составляет 180 тыс. м3/ч. Такая установка занимает
площадь 15X20 м, а ее высота равна 55 м. Если нужно перера-
батывать большие объемы газа, строят несколько параллельно
работающих установок. Эксплуатационные затраты при очист-
ке методом «Райнлюфт» дымовых газов ТЭЦ мощностью
1000 МВт, работающей на жидком топливе с содержанием серы
2,5%, при степени очистки дымовых газов 90% составляют (в
час):
Адсорбент, т 2 Охлаждающая вода, т 400
Электроэнергия, кВт-ч 6 Мазут, т 1
Очистка газов от диоксида серы
431
Рис. 14,14. Изотермы адсорбции диоксида серы
на полукоксе, полученном из бурого угля Каиско-
Ачинского бассейна Ирша-Бородинского место-
рождения при температуре пиролиза 600 °C.
Высокий расход активного угля
на установках с движущимся сло-
ем заставил провести цикл работ
по изысканию эффективных, но бо-
лее дешевых твердых поглотителей
[255]. В результате этих работ бы-
ло показано, что стоимость очистки
может быть значительно снижена,
если вместо дорогого активного уг-
ля применять другие углеродсодер-
жащие адсорбенты: адсорбционный
кокс, полукоксы из бурого и ка-
менного угля, коксы из торфа и т. д. [256].
Исследования показали, что активный адсорбент для диок-
сида серы может быть получен на основе бурых углей Совет-
ского Союза. Так, полукокс, образующийся при температуре
пиролиза 600—650 °C из бурого угля Ирша-Бородинского место-
рождения, при 100 °C имеет адсорбционную способность по
диоксиду серы 4,2 г/100 г, если концентрация адсорбтива в
инертном газе составляет 1% (об.). Его удельная поверхность
равна 420 м2/г, он имеет развитую м,икропористость. Дифферен-
циальные кривые распределения пор по данным ртутной поро-
метрии указывают на наличие двух максимумов: в области ме-
зо- (эффективный радиус 30,0 нм) и макропор (эффективный
радиус 10 000 нм). Изотермы адсорбции диоксида серы на
этом полукоксе представлены на рис. 14,14.
В процессе эксплуатации адсорбционная способность полу-
коксов по диоксиду серы и продуктам его конверсии не только
не уменьшается, но сначала значи-
тельно возрастает. Это явление са-
моактивации связано с «угаром»
углерода под воздействием оксидов
серы при высокой температуре и
изменением его пористой структу-
ры — развитием микропористости.
На рис. 14,15 приведены кривые из-
менения активности полукоксов от
числа циклов при различной тем-
Рис. 14,15. Изменение активности а полукоксов по
SOa в процессе многоцикловой эксплуатации при
различной температуре десорбции (п — число цик-
лов).
432
14. Защита атмосферы от загрязнения
Рис. 14,16. Схема установки очистки газа по методу «Бергбау — Форшунг»:
ас термической регенерацией; 1 — адсорбер; 2 — транспортер; 3, 4 — бункеры; 5 — де-
сорбер; 6—сепаратор; 7 — линия пневмотранспорта; 8 — холодильник; б — с экстракцион-
ной регенерацией; 1 — адсорбер; 2 — экстрактор; 3 — линия пневмотранспорта; 4 — на-
клонная решетка; 5 — емкость для воды; 6 — стояк.
пературе десорбции. При 350 °C первоначальная активность со-
ставляла 14 г/100 г, а к 30-му циклу она увеличилась до
20 г/100 г. В дальнейшем происходит, как обычно при активации,
разрушение структуры микропор и активность падает: после
60 циклов исходный и бывший в эксплуатации образцы по по-
глотительной способности становятся равноценными.
Вследствие значительной разницы в стоимости полукоксов
и активных углей и с учетом явления самоактивации установки
с движущимся слоем адсорбента целесообразно эксплуатиро-
вать на полукоксах [257]. Активность полукоксов может быть
повышена при введении в адсорбент катализаторов — железа,
кобальта, ванадия или их соединений [258]. Потеря адсорбци-
онного кокса, полученного из каменного угля, от механического
истирания на установках с движущимся слоем составляют
0,005% за цикл [259].
Процесс «Бергбау-Форшунг» (ФРГ) разработан [260] в двух
вариантах: с термической (рис. 14,16а) и экстракционной
(рис. 14,166) стадиями регенерации.
Ниже описан первый вариант процесса. Его проводят в движущемся
слое активного угля или полукокса, причем адсорбер имеет прямоугольное
сечение; на стадии очистки применено перпендикулярное направление движе-
ния газа и адсорбента. Использование адсорбента с частицами крупных раз-
Очистка газов от диоксида серы
433
меров (до 9 мм) позволяет проводить процесс при малом гидравлическом
сопротивлении и перерабатывать сильно запыленные газы. Гидравлическое
сопротивление в адсорбционной секции в зависимости от запыленности пока-
зано ниже:
Запыленность, г/м3 0,2 1,5 2,0 3,0
Сопротивление, Па 400 700 1000 1300
Очистку проводят при температуре около 130 °C, время пребывания угля
в адсорбере 26 ч. Насыщенный серной кислотой уголь питателем подается
в транспортер, поднимается на верх установки в бункер и оттуда непрерыв-
но поступает в десорбер, где он смешивается с горячим теплоносителем —
песком и нагревается. Десорбированный диоксид серы удаляется сверху, пос-
ле чего регенерированный адсорбент в сепараторе отделяется от мелких
частиц песка, охлаждается в теплообменнике и транспортером возвращается
в цикл.
Процесс «Вестфако» (США). В этом процессе [261—264]
очистку дымовых газов ТЭЦ или отходящих газов из печи до-
жигания на установках Клауса производят в адсорбционной
секции — в псевдоожиженном слое высокопрочного активного
угля (см. рис. 14,17).
Для поддержания оптимального температурного режима (65—150 °C) в
секцию впрыскивают воду. В результате реакций диоксид серы в адсорби-
рованной фазе в присутствии кислорода и воды превращается в серную кис-
лоту.
В последующем активный уголь «перетекает» в ниже расположенные
секции, где также находится в псевдоожиженном состоянии. В регенераторе
серы насыщенный серной кислотой уголь контактирует с сульфидом водоро-
да, который адсорбируется и в порах угля реагирует с серной кислотой с об-
разованием серы, остающейся в адсорбенте (знаком* обозначено, что ве-
щество находится в адсорбированной фазе):
H2S ---> (H?S)* (14.8)
(H2SO4)* + 3(H2S)* --> (4S)* + 4H2O (14.9)
Часть (около 25%) элементной серы выделяют из угля в результате на-
грева адсорбента до 550 °C в десорбере серы с последующей конденсацией
паров серы:
(4S)’ Sf + (3S)* (14.10)
Полная регенерация угля достигается в секции получения сульфида во-
дорода, где остаток серы при 550 °C реагирует с газом-восстановителем (на-
пример, водородом):
(3S)*+3H2 -----> 3H2S (14.11)
Водород для этой реакции получают в газогенераторе твердого топлива
в результате конверсии природного газа или нефти. Из секции генерации
сульфида водорода образовавшийся газ подают в секцию генерации серы,
чтобы обеспечить протекание реакции получения серы. Суммарная реакция
процесса имеет вид:
SO2 -% i/2O2 + ЗН2 --► S-j-3H2O (14.12)
28—1346
434
14. Защита атмосферы от загрязнения
Рис. 14,17. Блок-схема установки ад-
сорбционно-каталитической очистки га*
за в псевдоожиженном слое адсорбента
по методу «Вестфако».
Регенерированный уголь пнев-
мотранспортом или механическим
подъемником из секции генерации
сульфида водорода подают в зо-
ну адсорбции, и цикл повторяет-
ся. Таким образом, отличитель-
ной особенностью очистки дымо-
вых газов ТЭЦ и некоторых дру-
гих газов от диоксида серы по
методу «Вестфако» является по-
лучение непосредственно в про-
цессе очистки товарной серы, од-
нако при потреблении значитель-
ного количества водорода.
Адсорбционная защита
кабин крановщиков. На
предприятиях цветной ме-
таллургии передвижные
краны работают в атмосфе-
ре воздуха, загрязненного
диоксидом серы, содержа-
ние которого достигает 2 г/м3. Рабочие, обслуживающие краны,
находятся в течение длительного времени в загрязненной атмо-
сфере. Задача защиты обслуживающего персонала от диокси-
да серы в Советском Союзе решена герметизацией кабин кра-
новщиков и оборудованием их системами очистки и кондицио-
нирования воздуха [265].
Блок очистки (рис. 14,18) состоит из трех секций: в первой секции
улавливают пыль из воздуха производственных помещений, во второй —-
поглощают диоксид серы активным углем СКТ, третья секция предназначена
для улавливания пыли, которая может образоваться при разрушении частиц
угля. Производительность блока очистки составляет 100 м3/ч. Очищенный
воздух вентилятором 4 подают в увлажнитель 5, а затем либо в кондицио-
нер 6, либо непосредственно в кабину 7. В кабину поступает 1400 м3/ч кон-
диционированного воздуха.
При испытании системы на Балхашском горнометаллургическом комби-
нате содержание диоксида серы и пыли в цехе составляло соответственно
100 и 50 мг/м3. В кабине крана концентрация диоксида серы была не выше
3 мг/м3, а пыль отсутствовала. Температура воздуха в помещении цеха ко-
лебалась от 14 до 32 °C, в кабине она устойчиво поддерживалась на уровне
Рис. 14,18. Схема установки очистки и кондицио-
нирования воздуха в кабинах крановщиков:
1, 3 — фильтры; 2 — адсорбер; 4 — вентилятор;
увлажнитель; 6 — конденсатор; 7—кабина.
Очистка газов от диоксида серы
435
22—24 °C. Относительная влажность воздуха в цехе была около 35%, в ка-
бине 60%. Кондиционер смонтирован на мосту крана непосредственно над
кабиной. Газоочистная установка с увлажнителем и вентилятором, а также
вся аппаратура системы управления размещены в кабине крана. При разра-
ботке конструкций особое внимание обращено на доступность всех элемен-
тов оборудования для обслуживания и профилактического и капитального
ремонта.
Фильтры с активным углем применяют и для улавливания
других вредных компонентов из промышленных и вентиляцион-
ных газов, причем для увеличения эффективности очистки уголь
пропитывают различными реагентами, которые подбирают в за-
висимости от типа поглощаемой примеси: бикарбонатом ка-
лия— для удаления диоксида серы, солями цинка — для удале-
ния аммиака и сульфида водорода, солями меди — для удале-
ния цианида водорода, иодом—для удаления ртути и сульфида
водорода.
Методы очистки газов цеолитами
Большие надежды возлагаются на использование кислотостой-
ких цеолитов для защиты окружающей чреды от ряда вредных
промышленных выбросов, в частности, содержащих диоксид
серы. К перспективным адсорбентам следует отнести природные
цеолиты — клиноптилолит и морденит, а также их синтетичес-
кие аналоги [266, 267].
Основные различия в химическом составе природных цеоли-
тов разных месторождений относятся к содержанию обменных
катионов. В зависимости от геологических условий залегания
цеолитов катионы Na+ в них могут быть в той или иной степени
замещены на катионы К+, Са2+ или Mg2+. Отношение (Са2+-Ь
+Mg2++K+)/(Ca2+4~Mg2++K++Ma+) в клиноптилолитовых
породах Советского Союза колеблется от 0,52 до 0,94.
В табл. 14-4 приведены данные по адсорбционной способно-
сти природных цеолитов различных месторождений при погло-
щении диоксида серы из бескислородной газовой смеси, содер-
жащей 0,5% (об.) SO2; опыты проводили при 25°С. Анализ
данных таблицы показывает, что на адсорбционные свойства
цеолитов (клиноптилолита) оказывает влияние не только содер-
жание его в породе (только этот фактор определяет адсорбци-
онную способность по парам воды), но и химический состав:
адсорбционная способность по диоксиду серы увеличивается
пропорционально приведенному выше соотношению катионов.
Для извлечения диоксида серы рекомендуется применять
цеолитовые породы с содержанием клиноптилолита 50—60% и
выше, преимущественно в кальциево-магниевой форме. Особен-
но перспективно их применение на промышленных предприя-
тиях, расположенных вблизи мест добычи цеолитов.
28*
436
14. Защита атмосферы от загрязнения
Таблица 14-4. Адсорбционная способность природных
клиноптилолитовых пород по SO2 (Со=О,5%; 1=25 °C)
Месторож- дение Содержа- ние клино- птило- лита, % (масс.) Отно- шение SiOg Содержание оксидов, % (масс.) Отношение Са2'*Ч- +Mg2++K+ _ Ca2+-[-Mg2‘4- +K+-(-Na + XI00, % Адсорб- ционная способ- ность по SO3, % (масс.)
СаО MgO Na2O К2О
Л1гО3
Сокирницкое 75 10,0 Закарпатье 3,50 0,83 0,62 3,06 92,1 6,3
Берестянское 60 9,75 3,10 0,83 2,42 1,68 70,0 2,4
Ноемберен- 70 9,-5 Закавказье 4,02- 1,25 0,74 1,31 90;0- 4,1
ское Суворовское 65 9,3 2,70 Приморье 0,89 0,83 2,11 87,3 4,8
Милоградов- 60 10,0 2,22 0,70 0,37 2,56 93,7 5,3
ское Люточское 70 9,2 1,54 Сахалин 0,66 3,24 1,38 52,2 4,3
Корсаков- 60 8,9 3,36 1,24 1,42 1,02 79,5 3,7
ское Гейзерное 80 9,2 3,0 Камчатка 50,99 1,28 2,45 83,5 6,0
Быстрая и полная десорбция диоксида серы происходит при
350—400°C*. Содержание диоксида серы в газах десорбции за-
висит главным образом от адсорбционной емкости цеолита, рас-
хода продувочного газа и скорости прогрева слоя цеолита
[268].
Адсорбционный процесс очистки отходящих газов синтетическими цеоли-
тами прошел испытание на двухадсорберной установке в г. Огайо (США)
[269].
Установка получения серной кислоты традиционна и включает три основ-
ные ступени. Вначале диоксид серы получают сжиганием серы, сульфида
водорода или из сернокислотных остатков переработки нефти. Затем его ка-
талитически окисляют в триоксид серы на ванадиевом катализаторе, после
этого поглощают серной кислотой с получением концентрированной серной
кислоты. Концентрация диоксида серы в газе, поступающем в контактное
отделение, составляет 8,5%.
* Приведенные в тексте утверждения верны только для бескислородного
газа. Мы уже указывали, что природные клиноптилолиты катализируют ре-
акцию окисления диоксида серы в триоксид и хемосорбируют последний.
Лишь глубоко декатионированные цеолиты из-за того, что центрами хемо-
сорбции триоксида серы являются гидратированные обменные катионы, три-
оксид, возможно, не хемосорбируют и регенерируются при сравнительно низ-
ких температурах. (Прим, ред.)
F
Очистка газов от диоксида серы 437
Рис. 14,19. Схема очистки отходящих газов
сернокислотного производства цеолитами:
1 — адсорбер иа стадии очистки; 2 — ад-
сорбер на стадии регенерации; 3 —топка;
4 — газодувка.
_аи2-содержащая, газ
8 производство H2SO
Отходящий газ
из производ-
ства H2S0
Чистый газ на выброс
Окисление диоксида серы проте-
кает не полностью и некоторое коли-
чество этого непрореагировавшего
компонента выбрасывается с потоком
азота в атмосферу. В зависимости от
схемы процесса, числа ступеней кон-
тактирования и свойств ванадиевого
катализатора на большинстве установок концентрация диоксида серы в отхо-
дящих газах составляет от 0,2 до 0,4%. Отходящий газ содержит также не-
большое количество триоксида серы, аэрозоля и паров серной кислоты. Кроме
того, в результате сгорания углеводородов и азотистых соединений, присут-
ствующих в одном из видов сырья — кислом гудроне, образуется диоксид уг-
лерода и следы оксидов азота.
Схема обезвреживания газа в неподвижном слое кислото-
стойких цеолитов разработана фирмой Юнион карбайд кор-
порейшен и названа «Пьюрасив S».
Адсорбционный блок (рис. 14,19) вмонтирован в линию отходящих газов
сернокислотного завода. Максимальное количество отходящих газов состав-
ляет 300 м3/мин, предельное содержание в них диоксида серы 0,35%. Газ
после адсорбционной очистки содержит около 2-10“3% диоксида серы, 1-
10-;;% триоксида серы, 1-10~3% оксидов азота, 1-10“2% влаги, 2-10“2%
серной кислоты в виде пара и аэрозоля, 7,5% диоксида углерода, 10% кис-
лорода, 0,2-10“4% других минеральных кислот (соляной, плавиковой и др.),
0,5-10-''% органических сернистых соединений, 0,5-10“4% галогенов, остаток
составляет азот.
Через два месяца после пуска в строй установка была переведена на
режим испытаний. В этот период расход газа варьировали в пределах 235—
280 м3/мин, а концентрацию диоксида серы — от 0,25 до 0,5% (об.). Степень
извлечения диоксида серы при этом составляла 99%, а его концентрация
в отходящем газе (1,5—2,5)-10“3% (об.). Это соответствует поступлению в
атмосферу менее 0,2 кг диоксида серы на 1 т полученной серной кислоты.
Адсорбционная установка работала при средней температуре 95 °C и
давлении 0,12 МПа. Переключение адсорберов производили, когда концен-
трация диоксида серы за слоем адсорбента достигала 0,01%; концентрация
аэрозоля серной кислоты за слоем адсорбента была менее 3 мг/м3. Регене-
рацию цеолита проводили продувкой слоя горячим воздухом. Газы регенера-
ции возвращали в контактное отделение. Содержание диоксида серы в газе
регенерации, поступающем на сернокислотную установку, в процессе эксплуа-
тации постепенно возросло с 0,3 до 4%.
Этот рецикл оказывал лишь незначительное влияние на условия эксплуа-
тации самой сернокислотной установки. Первые два слоя ванадиевого ката-
лизатора вследствие рецикла дали повышение температуры на 10 °C, сле-
дующие слои на рецикл не реагировали. Концентрация кислоты после пуска
адсорбционного блока сначала повысилась (с 99,1 до 99,3%), но затем снова
стабилизировалась на уровне 99,1%. Основное оборудование было выполнено
в варианте «на салазках». Всюду, за исключением труб холодильника посту-
пающего газа, использовали углеродистую сталь. Основное оборудование ад-
сорбционной установки было типичным для сернокислотного производства—•
аппараты с неподвижным слоем насадки, газодувки, теплообменники и огне-
вые нагреватели.
428
14. Защита атмосферы от загрязнения
Площадь для размещения установки равнялась 120 м2. Период сооруже-
ния составлял 3 мес. Установка управлялась системой задатчиков времени,
обычно применяемых на адсорбционных установках. Оператор сернокислот-
ной установки обслуживал и адсорбционную установку; дополнительного
лабораторного оборудования не потребовалось.
Замену цеолитов на установке намечено производить каждые два года.
Эксплуатационные расходы зависят от количества перерабатываемого газа
и находятся иа уровне 1,0—2,3 долл, на 1 т кислоты.
Извлечение металлов из отходящих газов
Рекуперация семиоксида рения
С целью комплексного использования ценных компонентов, со-
держащихся в отходящих газах металлургических производств,
стадии механической очистки газов может предшествовать ста-
дия рекуперации из них паров или летучих соединений метал-
лов. Так можно улавливать, например, семиоксид ренря.
Как известно, при термической обработке медных и медно-молибдеиовых
концентратов, содержащих рений, последний иа 60—90% переходит в газо-
вую фазу и в виде семиоксида выбрасывается в атмосферу с отходящими
газами. В настоящее время применяются в основном «мокрые» способы его
извлечения, в которых реиий извлекается посредством абсорбции водой, вод-
ными растворами кислот или другими жидкими абсорбентами. Но «мокрые»
методы извлечения целесообразны лишь при переработке относительно не-
больших количеств отходящих газов, так как в противном случае образуются
значительные объемы кислых сточных вод. Поэтому для извлечения рения
из газов отражательных печей и печей электроплавки, которые в настоящее
время выбрасывают в атмосферу, большой интерес представляют «сухие»
способы.
Исследована [270] адсорбционная способность по семиоксиду рения син-
тетических цеолитов NaX и природного морденита Закарпатского месторож-
дения в широком интервале температур и исходных концентраций извлекае-
мого компонента. В экспериментах использовали гранулированный материал
с размером частиц 2—3 мм; их обрабатывали в слое толщиной в одно зерно
потоком, скорость которого поддерживали иа уровне 0,5 м/с. Модельная
газовая смесь содержала 2% влаги, 2% диоксида серы, 10% диоксида угле-
рода, что соответствует составу реальных отходящих газов цветной метал-
лургии.
В табл. 14-5 приведены значения сорбционной емкости исследованных
цеолитов. Из таблицы следует, что наибольшей адсорбционной способностью
Таблица 14-5. Сорбционная емкость цеолитов
по семиокиси рения (в г/100 г)
Цеолит Температу- ра, °C Концентрация Re2O7, г/м3
0,005 0,010 0,020 | 0, 050
NaX без связующего 250 0,40 0,90 1,40 2,30
350 0,59 1,45 2,30 4,10
NaX со связующим 250 0,32 0,60 1,00 1,70
350 0,65 1,05 1,80 2,90
Природный морденит 250 0,20 0,40 0,68 1,05
350 0,09 0,15 0,30 0,55
Извлечение металлов из отходящих газов
439
:по семиоксиду рения из исследованных цеолитов обладает NaX без связую-
щего. Сорбционная способность по ИегО? природного морденита значительно
ниже емкости синтетических цеолитов, что объясняется недоступностью по-
ристой структуры кристаллов для проникания в адсорбционные полости мо-
лекул семиоксида рения, критический диаметр которых (около 0,65 нм) боль-
ше характерного размера структуры морденита, определяющего молекулярно-
ситовые свойства последнего. В этом случае поглощение молекул ReaOj про-
.исходит, очевидно, в крупных порах, образованных породой. Однако деше-
визна и доступность природных цеолитов не исключают возможность их
применения для достижения поставленной цели.
Значительное увеличение сорбционной способности «широкопористых»
цеолитов по семиоксиду рения при повышении температуры процесса вплоть
до 350 °C, как уже было указано ранее, связано с хемосорбционным харак-
тером поглощения этого вещества в первичной пористой структуре адсор-
бента.
Водное выщелачивание насыщенных рением цеолитов в течение 2—3 ч
приводит к полному переходу семиоксида рения в раствор, из которого цен-
ный компонент извлекают известными методами: экстракцией, сорбцией иа
ионообменных смолах и т. п. Процесс рекуперации рения из отходящих га-
зов может быть осуществлен в различных вариантах: в неподвижном, дви-
жущемся и псевдоожиженном слое адсорбента.
Защита атмосферы от ртути
i
I Одним из наиболее важных нерешенных вопросов защиты ок-
ружающей среды от загрязнений является обезвреживание бы-
стро увеличивающегося выброса в атмосферу с отходящими га-
зами различных соединений ртути.
Состав отходящих промышленных газов, в частности, содер-
жание в них ртути, может колебаться в самых широких преде-
j лах. Изменение состава наблюдается не только на разных заво-
дах, но и на одном и том же металлургическом агрегате. Это
связано с условиями протекания процессов обжига, пылеулав-
ливания и т. и. В значительной степени при прочих равных ус-
J ловиях на состав газов оказывает влияние разбавление техно-
ь логических потоков за счет подсосов в систему. При обжиге
?, бедных ртутных руд потери с технологическими газами увели-
чиваются обратно пропорционально содержанию ртути в руде:
Содержание ртути в руде, % 0,5 0,2 0,1
Потери ртути, % 0,56 1,4 2,8
Необходимо учитывать также потери ртути с вентиляцион-
ными газами, количество которых можно оценивать в 15—20%
от объема технологических газов. Общие потери ртути с техно-
логическими и вентиляционными газами составляют примерно
40 г/т руды.
Проблема очистки таких газов приобрела особое значение
не только из-за увеличения безвозвратных потерь ртути, но и в
связи с необходимостью защиты воздушного бассейна вокруг
440
14. Защита атмосферы от загрязнения
ртутных предприятий. Вопросами обезвреживания и очистки
технологических и вентиляционных газов от соединений ртути
занимаются уже давно. Разработанные способы очистки газов
от ртути основаны, главным образом, на контакте их с мате-
риалами, способными удерживать ртуть в химически связанном
или сорбированном состоянии. Такой контакт обеспечивается
либо при пропускании газов через слой твердого поглотителя,
либо путем орошения потока газов раствором или суспензией
реагента.
При выборе поглотители необходимо учитывать, что очистке
подвергаются большие объемы газов (до 1000 м3/т руды), по-
этому поглотитель должен не только хорошо улавливать ртуть,,
но и быть достаточно дешевым и недефицитным. Он должен
обеспечивать глубокую очистку; ртуть относится к высоко ток-
сичным веществам, и среднесуточная предельно допустимая кон-
центрация ее в воздухе населенных пунктов по санитарным
нормам Советского Союза составляет 0,0003 мг/м3.
Наиболее испытанными способами очистки отходящих газов ртутных за-
водов являются мокрые способы с применением хлорида кальция (хлорной
извести), газообразного хлора, азотной и серной кислот. Основные показате-
ли некоторых из реализованных процессов приведены ниже:
Показатели Са(С1О)2 С1а HNO3 H2SO4
Расход газа, м3/ч 9000 25000 1800 55000
Скорость газов, м/с 0,43 0,4 — —
Содержание ртути, мг/м3 7,12 1,0—9,0 23,2 40—80
Степень очистки, % 79,4 95 98,0 99
Расход реагентов, кг/кг активный хлор 70 1,2 —
аммиак — 0,2 — —
кислота — — 5,0 —
Существенный недостаток этих способов — использование материалов,
которые либо сами являются токсичными (хлорная известь, хлор), либо со-
держат строго лимитируемые санитарными нормами вредные вещества, либо
образуют токсичные соединения. Известно, что почти все растворимые ртут-
ные соли являются ядами, и при использовании мокрых способов появляется
проблема очистки сточных вод. Именно вследствие загрязнения воды, а через
нее окружающей среды, более перспективными следует считать сухие спо-
собы.
Сухие способы очистки основаны на улавливании ртути из
газов соответствующими адсорбентами. Сущность их заключа-
ется в пропускании газовой смеси через адсорбционную колон-
ну, в которой происходит избирательное улавливание ртути,
В последнее время получила развитие адсорбционная очистка,
газов от паров ртути с применением углеродных и кремнийсо-
держащих адсорбентов, адсорбентов на основе соединений ме-
таллов, а также различных ионообменных смол. Многие из этих
Извлечение металлов из отходящих газов
441
сорбентов обладают высокими показателями термостойкости,
химической устойчивости, хорошими деформационно-прочност-
ными характеристиками и т. п.
Для очистки газов от ртути активный уголь обычно подвер-
гают предварительной обработке химическими реагентами.
В одной из работ [271] представлены результаты испытания
активного угля, содержащего 3—5% хлорирующего агента (от
массы угля), в процессе поглощения ртути из отходящих газов
химического производства.
Поглотитель испытывали на небольшом адсорбере производительностью
по газу 0,28 м3/ч, состоящем из двух колонок диаметром 22 мм, заполнен-
ных активным углем. Общая высота слоя угля 1 м (высота в первой секции
10 мм). Скорость газа 0,2 м/с, температура сорбции 18—25 °C. Установлено,
что при начальной концентрации ртути в газе 0,48—19,44 мг/м3 в течение
234 ч работы установки первый по ходу газа слой обеспечивал степень
очистки от 92,8% (в начале испытаний) до 55,9% (в конце испытаний). Ем-
кость активного угля по парам ртути составила 8,8% (масс.). Проскок паров
ртути через весь слой не был обнаружен до конца испытаний.
Равновесная адсорбционная способность активного угля при
содержании ртути в исходном воздухе 0,11; 0,94; 3,5 и 8,2 мг/м3
составляет соответственно 4,4; 5,3; 8,8 и 14,3 г/100 г [272]. Оче-
видно, что в порах угля физическая адсорбция паров ртути
сопровождается химической реакцией, в результате которой об-
разуются менее летучие и более прочно удерживаемые углем
соединения: сулема (HgCl2) и каломель (Hg2Cl2).
Адсорбционный метод успешно применен для очистки отхо-
дящих газов производств винилхлорида, хлора и каустической
соды.
Ртутьсодержащие отходящие газы (до 600 м3/ч) в производстве винил-
хлорида образуются на стадии приготовления катализатора гидрохлорирова-
ния ацетилена. По химическому составу они представляют собой загрязнен-
ную смесь воздуха и азота. Для удаления из газа пыли, сулемы и кислых
примесей его промывают 20%-ым водным раствором щелочи. После этого
газ, выходящий из скрубберов при 20—28 °C, подогревают на 5—7 °C для
предотвращения конденсации паров воды и пропускают через адсорбер диа-
метром 1,8 м с высотой слоя сорбента 0,5 м.
Концентрация ртути в поступающем на адсорбцию газе в течение де-
вятимесячной эксплуатации установки колебалась в пределах 0,5—9,8 мг/м3,
составляя в среднем 1,8 мг/м3. Концентрация ртути на выходе газа из ад-
сорбера составляла 0,001—0,003 мг/м3,, что находится в пределах санитар-
ных требований. Степень очистки при этом превышала 99,8%.
В последнее время разработан [273] один из вариантов хе-
моуглеадсорбционного способа, заключающийся в фильтрова-
нии газа через слой рекуперационного активного угля, модифи-
цированного хлоридом водорода. Активный уголь пропитывают
непосредственно в процессе очистки газа путем кратковремен-
ного введения в газовый поток перед адсорбером небольших
количеств хлорида водорода, полностью поглощаемого адсор-
бентом. Хлорид водорода подают из баллонов или в виде паров
соляной кислоты.
442
14. Защита атмосферы от загрязнения
В условиях электролампового завода метод был испытан на установке
производительностью по вентиляционному воздуху 90 тыс. м3/ч. Высота слоя-
сорбента в адсорбере составляла 0,13 м, скорость газового потока 0,4 м/с.
При 0,1 мг/м3 паров ртути па входе степень очистки составляла 98%.
Для очистки печных газов производства ртути, содержащих туман ртути,
пыль (100—300 мг/м3), диоксид серы (2—3 г/м3) и насыщенных парами-
ртути, разработан и внедрен в промышленность так называемый галогенид-
ный способ. Опытно-промышленная установка, работающая по этому спосо-
бу, выполнена из сплава титана, смонтирована на одном из ртутных комби-
натов и имеет производительность 12 тыс. м3/ч. Поступающий на установку
газ, в котором концентрация ртути составляет 35 г/м3, предварительно очи-
щают от пыли и туманообразпой ртути в абсорбере распиливающего типа,,
орошаемом водой, а затем уже очищают от ртути в абсорбере с псевдо-
ожиженным слоем шаровой насадки, орошаемым 2%-ным водным раствором
хлорида натрия с катализирующей добавкой (2%) порошкообразного ак-
тивного угля. В циркулирующем растворе первой ступени газоочистки на-
капливаются (до определенной концентрации) кислые примеси. В отстойнике
скапливается шлам, содержащий 2—7% ртути, который направляют на из-
влечение металла в печи.
Во второй ступени основная масса ртути реагирует с хлоридом натрия
и превращается в частицы каломели, взвешенные в циркулирующем растворе
вместе с частицами угля. По мере накопления в растворе каломели ртутный
шлам периодически выводят из циркулирующего раствора (содержание рту-
ти в нем доходит до 60% от массы сухого шлама) в аппарат, где обраба-
тывают щелочью для перевода каломели в оксиды ртути. После этого шлам
также направляют в печи, где извлекается металл. Степень очистки превы-
шает 90%, ее себестоимость составляет 50 коп. на 1000 м3 газа. Экономиче-
ский эффект за счет возврата уловленной ртути в производство равен
24 тыс. руб. в год.
С помощью активных углей успешно решается задача улав-
ливания ртутьсодержащих органических соединений.
На заводе средней мощности по производству ртутного протравителя
семян сельскохозяйственных культур — гранозана в атмосферу выбрасыва-
ется 150 тыс. м3/ч воздуха, содержащего высокотоксичные пары ртутьсодер-
жащих органических соединений, главным образом диэтилртути (QHsbHgp
Концентрация этого компонента составляет несколько миллиграммов в 1 м3.
Равновесная адсорбционная способность отечественных рекуперационных и
газовых углей по диэтилртути находится в пределах от 27 до 39% (масс.).
На промышленной установке адсорбционной очистки отходящего газа
производства гранозана был предусмотрен [274] двухслойный адсорбер с об-
щей высотой слоя адсорбента 0,6 м. После полного насыщения первого по
ходу газа слоя и появления проскока паров ртутьсодержащих органических
соединений за вторым слоем первый слой выгружают и заменяют свежим уг-
лем, а очищаемый воздух пропускают в противоположном направлении.
Продолжительность стадии очистки достигала нескольких месяцев.
Обычный метод десорбции водяным паром в рассматриваемом случае
применить затруднительно вследствие загрязнения конденсата соединения-
ми ртути, от которых необходимо снова освобождаться. Поэтому регенера-
цию вели в отдельном аппарате, где отработанный активный уголь движется
под действием силы тяжести, нагреваясь до 400—500 °C в слабом токе инерт-
ного газа. В этих условиях соединения ртути' разлагаются до металлической
ртути, которая легко конденсируется: температура кипения ртути 356 °C,
а диэтилртути только 156 °C. Адсорбент после десорбции из него соединений
ртути возвращают в процесс.
За два года эксплуатации установки было уловлено около 3,5 т ртуть-
содержащих органических соединений при почти полной очистке воздуха.
Извлечение металлов из отходящих газов
443
Достаточно эффективные сорбенты для улавливния паров
ртути получены пропиткой активных углей минеральными кис-
лотами [275].
Адсорбент получают из водных растворов кислот при соотношении
уголь — раствор 1:1с последующим отделением угля от раствора фильтра-
цией и высушиванием угля на воздухе. Содержание кислот в угле опреде-
ляется их концентрацией в пропиточных растворах. Концентрация паров рту-
ти на входе в колонку с сорбентом составляла 4,8—7,8 мг/м3. Результаты
очистки воздуха слоем активного угля высотой 26 см, модифицированного
кислотами, при скорости потока 0,4 м/с приведены ниже:
Кислота H,SO4 HNO„ НС1
Скисл, % 2,2 12,3 0,9 3,8 1,2 2,1
Тпр, Ч 2375 4896 1070 1971 1339 1745
бд, % 10,60 25,28 4,85 9,40 8,88 10,10
Предложено также очищать воздух от паров ртути активны-
ми углями, пропитанными хлоридом железа [276].
Испытание процесса проводили при 20 °C, относительной влажности воз-
духа 50 и 90%, начальной концентрации ртути 10 мг/м3. Высоту слоя ад-
сорбента изменяли от 5 до 30 мм, линейная скорость составляла 0,08 м/с.
Сорбент готовили на основе активных углей марок АГ и АР пропиткой их
раствором хлорида железа различной концентрации. Установлена прямая за-
висимость динамической активности слоя сорбента от содержания хлорида
железа в активном угле вплоть до значения 7,4 г/100 г:
Содержание FeCl3, % (масс.) 1,5 4,7 7,4 11,0
Динамическая активность, % (масс.) 0,7 4,1 6,7 6,6
Пои дальнейшем увеличении добавки FeCl3 до 12—13 г/100 г адсорб-
ционная способность не возрастает. Изменение относительной влажности га-
зовой смеси в пределах 50—90% практически не влияет на поглотительную
способность импрегнированного хлоридом железа активного угля.
Процесс адсорбции паров ртути углеродными адсорбентами
был испытан также без применения химических реагентов
[277]. Технологические газы переработки сульфидных ртутных
руд кроме соединений ртути, представленных в виде металличе-
ской, оксидной и сульфидной форм, содержат пары воды, оксц-
ды серы, кислород, диоксид углерода и азот.
При очистке сухой газовоздушной смеси активные центры
(микропоры) блокируются в первую очередь диоксидом серы.
Поэтому пары ртути из газовой смеси извлекаются плохо. Сте-
пень очистки сухой газовой смеси невелика. Однако в присутст-
вии паров воды в адсорбированной фазе с высокой скоростью
протекает реакция SO2+H2O + 1/2O2 = H2SO4. Образовавшаяся
кислота химически связывает ртуть и ее соединейия, обеспечи-
вая высокую степень очистки от этих токсичных компонентов.
Испытание метода проводили на производственной установке для очистки
сбросных газов печей кипящего слоя. Производительность установки по газу
444
14. Защита атмосферы от загрязнения
1000 м3/ч. Основной частью установки является адсорбер сечением 0,98 X
Х1,16 и высотой 2 м с пятью полками. Так как исходный газ в системе
конденсации ртути насыщен парами воды, для предотвращения конденсации,
паров воды его подогревали. Установка снабжена системой контроля основ-
ных параметров процесса: скорости, расхода, температуры и влажности га-
зов, концентрации паров ртути и оксидов серы иа входе в систему и иа вы-
ходе из нее.
Состав отходящих газов печей КС (по объему): 14—18% О2; 3—4% СО2;
72—78% Na. Концентрация других компонентов составляла (г/м3): SO2—
от 4 до 8; SO,г1—от 0,02 до 0,1; H2S— от 0 до 2; Н2О — от 10 до 50;
пыль — от 0,2 до 1,0. Ртуть в газах была представлена следующими форма-
ми (%): парообразная ртуть — от 40 до 65; аэрозоль металлический рту-
ти— от 30 до 50, аэрозоль сульфидной ртути — от 1 до 4; аэрозоль суль-
фатной и оксидной ртути — от 0,5 до 2. Температура газа колебалась от 25
до 40 °C.
На первую полку по ходу газов загружали огарок в виде частиц 10—
20 мм с высотой слоя до 20 см. Слой огарка улавливал пыль, а также кап-
ли воды. На каждые последующие полки загружали по 70 кг активного
угля марки СКТ с размером частиц 1,5—3,0 мм; высота слоя иа каждой пол-
ке была 10 см. Очистку проводили при следующих условиях:
Температура газа, °C 60—80
Содержание ртути в газе, мг/м3
минимальное 0,32
максимальное 7,65
среднее 2,75
Содержание диоксида серы (среднее), г/м3 0,7
Относительная влажность, °/о 85
В течение первых пяти суток пары ртути полностью поглощались ак-
тивным углем. Концентрация ртути в газе, соответствующая нормам, после
второго слоя была зарегистрирована лишь спустя 30 сут непрерывной ра-
боты.
Вакуумтермическая десорбция (остаточное давление в сис-
теме менее 0,05 МПа) при 450 °C в течение 1 ч обеспечивает
восстановление адсорбционной способности активного угля.
Влияние остаточного давления в системе на степень десорбции
невелико: при остаточном давлении 0,07 и 0,05 МПа степень
десорбции ртути составляет соответственно 93 и 96,7%.
ЗАЩИТА ГИДРОСФЕРЫ
ОТ ЗАГРЯЗНЕНИЯ
Все более широкое применение находит адсорбционный метод
для очистки вод. Положительными факторами адсорбционной
обработки воды являются: высокая степень очистки, отсутствие
отходов и загрязнений на самой установке, стабильность степе-
ни очистки при неожиданных залповых выбросах загрязнений,,
экономичность, связанная с многократностью использования
сорбента*.
Область применения метода
Адсорбенты способны удалять из воды разнообразные по харак-
теру загрязнения: механические примеси, эмульгированные и
коллоидные вещества, молекулы и ионы. Однако использование
адсорбции для удаления гетерогенных примесей экономически
не оправдано и не практикуется. Блок адсорбционной очистки,,
как правило, включают в схему на заключительной стадии
обезвреживания воды, когда из нее отстаиванием, фильтрацией,,
коагуляцией уже удалена основная масса взвешенных частиц,
эмульгированных смол и масел и вода освобождена от крупных
мицелл коллоидных систем.
Адсорбционная очистка эффективна во всем диапазоне кон-
центраций примеси в воде, однако более всего ее преимущества
сказываются на фоне других методов очистки при низких кон-
центрациях загрязнений. Данные рис. 15,1 наглядно демонстри-
руют это. Сравнение адсорбционного (линия 1) и экстракцион-
ного (линия 2) обесфеноливания воды показывает, что при кон-
центрации фенола в воде до 0,01% выгоднее проводить очистку
воды адсорбентом, а при концентрации фенольной воды более
0,01 % —экстрагентом. Аналогичные результаты дает сравнение
* Адсорбция жидких сред вообще и сорбционная очистка сточных вод в
частности имеют ряд особенностей, отличающих их от адсорбции газов и-
паров. Учитывая это, а также то значение, которое сорбционная техника при-
обрела для охраны гидросферы, приводим перечень новых монографий по-
этому вопросу: А. М. Когановский и др. Адсорбционная технология очистки
сточных вод. — К., Техшка, 1981; Смирнов А. Д. Сорбционная очистка воды —
Л., Химия, 1982. Основное внимание в этих книгах уделено инженерному:
расчету процессов и конструкциям сорбционной аппаратуры. (Прим, ред.)
446
15. Защита гидросферы от загрязнения
Рис. 15,1. Поглотительная способность активного
угля (1) и экстрагента — бутилацетата (2) по фе-
нолу, извлекаемому из водной среды.
адсорбционной очистки с другими
наиболее распространенными мето-
дами очистки воды: реагентной
нейтрализацией, дистилляцией, био-
химической очисткой [278]. Эконо-
мические исследования также под-
тверждают все возрастающую кон-
курентноспособность адсорбционной очистки в сравнении с тра-
диционными методами (табл. 15-1).
Таким образом, теоретически и экономически целесообраз-
ным является применение адсорбции для удаления из воды ма-
лых (остаточных) количеств молекулярно растворенных ве-
ществ и ионов. Основные области применения адсорбционных
процессов в очистке воды — технология подготовки питьевой
воды и доочистка сточных вод.
Выбор адсорбента
При адсорбции из растворов происходит поглощение молекул
загрязнения и среды. Для очистки водных растворов характер-
на конкуренция двух видов межмолекулярных взаимодействий:
гидратация молекул адсорбтива, т. е. взаимодействие их с моле-
кулами воды в растворе, и взаимодействие молекул адсорбтива
с адсорбентом.
Разность энергий этих двух процессов и представляет собой
энергию, с которой извлеченное из раствора вещество удержи-
вается частицами погруженного в раствор адсорбента. Если по-
верхность адсорбента имеет электрически заряженные центры,
то наиболее сильное адсорбционное взаимодействие наблюда-
ется в тех случаях, когда в структуре молекул имеются кратные
связи, образующиеся с участием л-электронов. Поэтому арома-
тические соединения на таких сорбентах сорбируются лучше
Таблица 15-1. Экономические показатели очистки сточных вод
различными методами в СССР [279, 280]
Метод очистки Удельные капи- тальные затра- ты, руб/м3 Себестоимость очистки, руб/ма Приведенные затраты, руб/м*
Биохимический 0,42—2,00 0,09—1,20 0,14—1,44
Нейтрализация 0,08—0,20 0,35—0,65 0,26—0,67
Электрохимический 1,94—5,56 0,41—2,53 0,64—3,60
Дистилляция и сжигание 4,60—9,50 0,76—6,30 1,31—7,44
Адсорбционный 1,50—2,75 0,65—1,46 0,80—1,70
Выбор адсорбента
447
алифатических. С другой стороны, наличие электрического заря-
да, ориентирующего вокруг себя диполи воды, ухудшает адсор-
бцию, поэтому гидратированные ионы адсорбтива сорбируются
значительно хуже, чем недиссоциированные молекулы того же
вещества. Адсорбция ухудшается также при увеличении в мо-
лекулах адсорбтива числа гидроксильных групп, обладающих
большой энергией гидратации.
Конкуренция процессов гидратации и адсорбции лежит в.
основе разграничения сорбентов для удаления из воды органи-
ческих и неорганических веществ. Для сорбции органических
веществ применяют углеродные пористые материалы — актив-
ные угли, дробленые материалы различного органического про-
исхождения: уголь, кокс, топливные шлаки, сорбенты на основе
целлюлозы и резины, синтетические полимеры [281—286]. По-
лярные гидрофильные материалы — иониты, глины, силикагели,
алюмогель, цеолиты, оксиды и гидроксиды для адсорбции орга-
нических веществ малопригодны, так как энергия взаимодей-
ствия их с молекулами воды равна энергии сорбции молекул
органических загрязнений или превышает ее. Эти гидрофильные-
материалы используют для удаления из воды неорганических
соединений, присутствующих в ней, как правило, в ионной фор-
ме [287—292].
Наиболее распространенным адсорбентом для очистки воды
являются активные угли. С их помощью возможно практически
полное удаление из растворов почти всех органических соеди-
нений, а при определенных условиях и эффективная очистка во-
ды от некоторых токсических ионов неорганических веществ.
При подборе активных углей для очистки вод и организации1
процесса прежде всего обращают внимание на pH раствора в-
связи с тем, что активные угли адсорбируют из воды вещества
в молекулярной форме и практически не извлекают гидратиро-
ванные ионы. Поэтому в присутствии слабых электролитов (на-
пример, фенолов, пиридиновых оснований, спиртов и органиче-
ских кислот) необходимо проводить очистку и группировать по-
токи так, чтобы процесс проходил при оптимальных значениях
pH. Эти значения определяются из выражений:
рНоПгкисл рКдисс — 3
(15.1>
рНоптоси рКдисс + 3
где рНопткиол И рНоптосн- оптимальные показатели для разбавленной кис-
лоты и основания соответственно; рКдисс—десятичный логарифм от обрат-
ной величины константы электролитической диссоциации извлекаемого веще-
ства. Оптимальное значение pH при адсорбции фенола составляет, например,.
рН<8, а анилина рН>8 [293].
С конкуренцией процессов гидратации и адсорбции связано
и другое известное и важное правило организации обезврежива-
*t*to
io. защита ги росферы от загрязнения
ния сточных вод активными углями. Считается, что более рас-
творимое вещество адсорбируется на углях хуже, чем малорас-
творимое. Так, показана’[294] обратная зависимость между рас-
творимостью органических кислот и их адсорбцией на активном
угле БАУ. Однако правило это не универсально, и многочислен-
ные исключения затрудняют его использование.
Свойства извлекаемых веществ оказывают большое влияние
на их адсорбцию. Проявляется это влияние в избирательности
действия активных углей по отношению к различным адсорбти-
вам. Подавляющее большинство литературных данных указы-
вает на трудность предсказания и объяснения избирательности
адсорбции, а следовательно, и выбора оптимальных адсорбентов
для очистки сточных вод [294—297].
Для реальных сточных вод наиболее приемлем термодинами-
ческий подход к решению проблемы избирательности адсорбции
и выбора адсорбента. Находясь в сточной воде, загрязнения
адсорбируются на угле с различной силой, которую можно оце-
нить уменьшением свободной энергии ДТадс (в кДж/моль) при
адсорбции вещества в стандартных условиях (из бесконечно
разбавленного раствора).
Чем выше Д/Чдс, тем сильнее вещество адсорбируется из
водного раствора и вытесняет с поверхности адсорбента все
вещества с меньшим значением AFaRC.
Показано, что при А/7адс = 17 кДж/моль константа адсорб-
ционного равновесия невелика и для извлечения таких веществ
из раствора требуется большой расход активного угля. Поэтому
адсорбционные методы очистки следует применять для воды с
высоким содержанием соединений, отличающихся повышенны-
ми значениями AFaac (не ниже 20 кДж/моль).
Величина АТадС мало зависит от типа активного угля и об-
ладает свойством аддитивности. Это позволяет вычислять ее
значение по ранее определенным инкрементам dAFanc для от-
дельных структурных элементов и функциональных групп. Для
промышленных углей основных типов определены значения
^AFaac различных структурных элементов ароматических соеди-
нений [298]. Используя значения АРаяс и dAFaac, расчетным
путем можно оценивать эффективность и избирательность
адсорбции. К сожалению, такой подход иногда сталкивается с
большими трудностями [299], а изредка оказывается просто не-
удачным [296].
В настоящее время общепризнанным считается, что при
обезвреживании сточных вод активными углями их группиров-
ку и предварительную обработку следует вести таким образом,
чтобы в адсорбер поступала вода, загрязненная компонентами,
имеющими близкие значения AFaac.
Важным фактором, обусловливающим избирательность сорб-
ции, является химическая природа поверхности сорбента. Так,
Выбор адсорбента
44?
Рис. 15,2. Выходные кривые при очистке воды от
фенола:
I — адсорбция из бинарного раствора фенол — во-
да; 2 — адсорбция из сточных вод коксохимиче-
ского производства (V— количество профильтро-
ванного через слой раствора).
на поверхности активных углей всегда присутствуют оксиды,
имеющие различный характер — основные и кислотные. Много-
численными исследованиями адсорбции паров доказано, что
присутствие этих оксидов не влияет на физическую адсорбцию
неполярных молекул, но увеличивает адсорбцию полярных ве-
ществ. Что же касается адсорбции из водных растворов, то
здесь сведения противоречивы: с одной стороны, указывается,
что сорбция нейтральных и цислых веществ снижается на ак-
тивных углях с кислым характером поверхностных окислов, а с
другой стороны, — именно такие угли рекомендуют для извлече-
ния слабых электролитов из растворов [300].
Принципы подбора активного угля для проведения адсорб-
ции из жидкой фазы в зависимости от характера его пористой
структуры пока изучены недостаточно. Считается, что для уда-
ления из сточных вод веществ с небольшими молекулами струк-
тура активных углей не должна значительно отличаться от
структуры углей, применяемых для адсорбции газов и паров,
т. е. необходима развитая м,икропористость [301]. В то же вре-
мя, до сих пор не решен вопрос о минимально необходимом
объеме транспортных пор и возможности применения активных
углей с очень мелкими порами.
Для очистки воды от молекулярно растворенных органиче-
ских веществ могут применяться отечественные активные угли
типа ОУ, КАД-иодный, БАУ, ДАК, СКТ, АР, АГ и другие.
Их характеристика приведена ранее в гл. И. Влияние парамет-
ров пористой структуры этих адсорбентов на эффективность
очистки сточных вод от некоторых органических соединений
подробно изучено в работе [302]. Установлено, что эффектив-
ность очистки сточных вод от растворенных ароматических сое-
динений определяется объемом микропор активного угля; для
углей с бидисперсной макроструктурой, т. е. имеющих поры с
эффективным радиусом до 0,7 нм (собственно микропоры) и
поры радиусом 0,7—1,6 нм (супермикропоры) характерно явле-
ние вытеснительной десорбции. Выходная кривая на рис. 15,2
иллюстрирует это явление. Появление участка кривой для 'Кон-
центрации примеси выше исходной (с0) объясняется десорбцией
29—1346
450
15. Защита гидросферы от загрязнения
данного вещества из пор активного угля лучше сорбируемыми
компонентами очищаемого стока*.
Вытеснительная десорбция органических соединений, как
показано в работе [302], происходит из супермикропор сорбен-
та, в связи с чем динамическая активность угля по фенолам и
другим молекулярно растворенным органическим веществам
(ад) пропорциональна предельному объему адсорбционного
пространства микропор угля (1Г0= F01+F02), а равновесная
статическая активность (ар)—объему адсорбционного прост-
ранства собственно микропор (lFOi), т. е.
ад — V» и Чр~А^11го1 (15.2)
где Г01 и Ж, 2 — объемы микропор и супермикропор в угле.
Для адсорбции из растворов крупных молекул рекоменду-
ются активные угли с развитой макро-, мезо- и супермикропо-
ристостью [300, 302]. Необходимо отметить, что при проведении
процесса очистки воды с регенерацией адсорбента пористую
структуру активного угля чаще подбирают, ориентируясь на
оптимизацию именно стадии регенерации. Поэтому в отечест-
венной практике очистки воды чаще других применяют актив-
ный уголь типа КАД-иодный [303, 304].
Регенерация и утилизация адсорбента
При извлечении загрязнений из сточных вод важное место за-
нимает проблема регенерации отработанного адсорбента. Как
правило, именно стадия регенерации определяет экономику
процесса адсорбционной очистки воды в целом (доля затрат
на регенерацию в общей стоимости очистки составляет от 40 до-
85 %). Если стоимость сорбента ниже затрат на его регенера-
цию, и разница затрат не компенсируется стоимостью извлечен-
ных при очистке продуктов, регенерацию считают нецелесооб-
разной и отработанный адсорбент утилизируют (например,,
сжигают).
Из методов регенерации наиболее широко используется хи-
мическая (обработка кислотами, щелочами или окислителями^
удаление загрязнений в результате экстракции, отгонки с водя-
ным паром, отдувки нейтральным газом-носителем), а также
термическая регенерация. Последнюю проводят продуктами го-
рения газа и водяным паром при 650—900 °C. Только химиче-
скую регенерацию используют в процессах, связанных с приме-
нением адсорбентов-ионообменников; самые разнообразные ме-
тоды применяют для восстановления поглотительных свойств
* Возникновение выходных кривых типа кривой 2 на рис. 15,2 связано с об-
щими закономерностями динамики адсорбции смеси в неподвижном слое-
(Прим, ред.)
Принципиальные схемы адсорбционной очистки воды
451
активных углей и других углеродистых сорбентов. Дополни-
тельные сведения о регенерации адсорбентов, используемых для
очистки сточных вод, приведены ниже при рассмотрении кон-
кретных процессов адсорбционной очистки воды.
Принципиальные схемы адсорбционной очистки воды
и основные типы адсорберов
Разнообразие состава различных сточных вод и условий их
очистки, большое число факторов, влияющих на адсорбцию,
отсутствие единого мнения о принципах выбора наилучших
адсорбентов и правилах подготовки воды к адсорбционной очи-
стке порождают большое разнообразие в организации процесса,
в его технологическом оформлении. Достаточно полно основные
принципы схемы адсорбционной очистки воды рассмотрены в
работе [305].
Адсорбционную очистку воды можно проводить порошкооб-
разными или зернеными адсорбентами в периодическом или не-
прерывном процессе. В настоящее время наиболее широко рас-
пространена очистка воды порошкоообразными адсорбентами
(особенно питьевой воды активными углями).
Схема очистки в этом случае проста: адсорбент (5—10 г/л)
засыпают в аппарат с очищаемой водой и образующуюся сус-
пензию перемешивают 30—40 мин. Затем адсорбент отфильтро-
вывают от очищенной жидкости. Процесс может быть органи-
зован как в периодическом, так и в непрерывном режиме; от-
работанный адсорбент либо регенерируют, либо утилизируют,
например, как топливо. Основные варианты схем жидкофазной
контактной очистки порошкообразным углем представлены на
<рис. 15,3.
По простейшему одноступенчатому варианту А воду смешивают со све-
жим углем, выдерживают при перемешивании в смесителе, после чего очи-
щенную воду и отработанный уголь разделяют. По двухступенчатому проти-
воточному варианту Б вначале воду обрабатывают уже частично отрабо-
танным углем, а тонкую очистку проводят свежим углем. Таким образом,
вода движется с первой ступени иа вторую, а уголь, наоборот, со второй сту-
пени иа первую. Конечно, степень очистки и отработки адсорбционной емко-
сти в двухступенчатом варианте выше. Иногда число ступеней увеличивают
до трех. По варианту В отработку воды проводят в каждой из двух после-
довательных ступеней свежим углем.
При использовании порошкообразного угля трудно осуще-
ствить его регенерацию, что приводит к значительному расходу
адсорбента. На очистном сооружении с пропускной способ-
ностью 50 тыс. м3/сут (по воде) годовой расход порошкообраз-
ного угля достигает 200 т. Стоимость адсорбционной обработки
1 м3 питьевой воды таким методом оценивается в 0,3 коп.
Быстрота достижения условий, близких к равновесным, де-
шевизна порошковых сорбентов и отсутствие в большинстве
29*
452
15. Защита гидросферы от загрязнения
уголь вода
ВАРИАНТ Б
Исходная Свежий Свежий
Отработанный Отработанный
уголь уголь
Вариант В<
Рис. 15,3. Варианты (А—В) схемы контактной очистки воды порошкообразным актив*
иым углем:
1 — контактор; 2 — сепаратор.
случаев стадии регенерации, простота оборудования и техноло-
гической схемы обусловили широкое использование порошко-
вых сорбентов в практике водоочистки. Однако статистика ми-
рового производства адсорбентов указывает на более высокие
темпы роста производства гранулированных поглотителей по
сравнению с порошковыми [306].
Известно, что теоретически процесс очистки воды фильтро-
ванием через слой гранулированного сорбента всегда выгод-
нее, чем очистка порошковым сорбентом [307]. Это связано с
тем, что в первом случае насыщение угля происходит при на-
чальной, а во втором —при остаточной концентрации загряз-
нения. В настоящее время все шире проектируют и строят очи-
стные станции, работающие на гранулированных сорбентах, а
порошки применяют, как правило, малыми дозами для предва-
рительной очистки от основного количества примесей.
Адсорбцию на зерненом поглотителе можно проводить пе-
риодически и непрерывно. При периодическом процессе адсор-
бер, заполненный неподвижным слоем поглотителя, отключает-
ся от потока после отработки слоя, разгружается и вновь за-
Принципиальные схемы адсорбционной очистки воды
453 ’
полняется свежим или регенерированным сорбентом для нового'
цикла. Типичный адсорбер с неподвижным слоем зерненогсг
сорбента показан на рис. 15,4.
Как правило, в схемах с неподвижным слоем сорбента предусмотрено'
несколько адсорберов, в каждом из которых в один и тот же момент време-
ни протекает определенная стадия процесса. При такой схеме адсорберы
работают периодически, а очистка воды происходит непрерывно. Взвешенные
вещества, задержанные в верхних слоях адсорбента, периодически уда-
ляют при промывке водой, проходящей через слой активного угля снизу
вверх. Перед этой стадией обычно проводят так называемую поверхностную
промывку от мелочи.
Модификация этого способа организации процесса отражена в ориги-
нальной схеме адсорбера с «пульсирующим слоем», согласно которой из ад-
сорбера периодически удаляется лишь отработанная часть слоя сорбента при
одновременном добавлении в него свежего регенерированного поглотителя.
Такая конструкция позволяет иметь лишь один адсорбер и резко сокращает
непроизводительные затраты времени на отключение адсорбера и повторный
пуск его в эксплуатацию (на продувку, сушку, разогрев слоя и т. п.) [308].
Адсорберы с «пульсирующим слоем» используются фирмой Органо (Япо-
ния).
Непрерывный процесс адсорбции проводят в схемах с дви-
жущимся или псевдоожиженным слоем сорбента, в которых
может быть осуществлено различное направление движения
потоков адсорбента и исходного раствора, а стадия регенера-
ции может проводиться как в том же самом, так и в отдельном
аппарате [309].
Значительным недостатком в работе адсорберов с неподвиж-
ным слоем зерненого поглотителя является их невысокая про-
изводительность, так как скорость потока воды должна поддер-
живаться на уровне 2—6 м/ч. На
порядок большую производи-
тельность имеют адсорберы с
псевдоожиженным слоем сорбен-
та, получающие в последнее вре-
мя все большее распростране-
ние. Разработаны различные кон-
струкции таких адсорберов. Две
Из них, созданные под руководст-
вом Кульского, показаны на рис.
15,5 ([310].
Рис. 15,4. Адсорбер с неподвижным слоем ак-
тивного угля:
I — корпус аппарата; 2— адсорбер; 3 — люк
для загрузки угля; 4 — штуцер подачи воды на
очистку; 5 — штуцер подачи воды для поверх-
ностной промывки загрузки; 6 — решетка; 7 —
штуцер для отвода очищенной воды и подачи
промывной воды; 8 — люк для выгрузки от-
работанного адсорбента.
454
15. Защита гидросферы от загрязнения
Рис. 15,5. Конструкция адсорберов с псевдоожиженным слоем активного угля:
а — одноярусный адсорбер: 1--выносной уплотнитель; 2 — центральная труба; 3 - дыр-
чатая кольцевая труба; 4— распределительные решетки; 5 — диффузор; б — двухъярусный
адсорбер; 1 — эжектор; 2 — распределительная решетка; 3 — камера первого яруса; 4 —
горловина; 5 — камера второго яруса; 6 — кольцевой желоб; 7 — центральная труба; 8 —
переливная труба; 9 — труба для эжектирования отработанной воды; 10 — регулирующий
клапан; 11 — выносной уплотнитель.
Установка с одноярусным взвешенным слоем (вид а) активного угля ос-
нащена выносным углеуплотнителем 1 с принудительным отсосом адсорбен-
та, который подают вместе со струей воды, поступающей на очистку по
центральной трубе 2 через диффузор 5. Пройдя распределительные решет-
ки 4, вода вступает в контакт со взвешенным слоем угля; очищенную воду
отводят из верхней части установки через дырчатую затопленную кольцевую
трубу 3.
В двухъярусном противоточном адсорбере (вид б) суспензию активного
угля через центральную трубу 7 подают под распределительную решетку вто-
рого яруса адсорбера, где смешивают с частично обработанной водой, выхо-
дящей из камеры первого яруса 3 через горловину 4. Над решеткой в каме-
ре 5 активный уголь образует взвешенный слой. Очищенную воду отводят из
верхней части адсорбера с помощью кольцевого желоба 6. Загрязненная вода
поступает на очистку в нижнюю часть адсорбера через эжектор 1, который
подсасывает избыток активного угля из камеры второго яруса по переливной
трубе 8.
Вода, смешанная в эжекторе с уже частично насыщенным углем, про-
ходит распределительную решетку 2 и образует нижний слой в камере пер-
вого яруса 3. Очистку воды в этом слое проводят за счет разности кон-
центраций загрязнений в необработанной и очищенной воде. Избыток взве-
шенного слоя отработанного в нижнем ярусе угля попадает в выносной угле-
Очистка воды порошкообразным активным углем
455
уплотнитель 11. Процесс усиливается эжектированисм обработанной воды из
уплотнителя через трубу 9, снабженную регулирующим вентилем 13, в пере-
ливную линию. Из углеуплотннтеля отработанный сорбент направляют на ре-
генерацию.
Применение адсорберов такой конструкции положительно зарекомендо-
вало себя в технологии кондиционирования воды для удаления растворен-
ных органических, в том числе дурнопахнущих загрязнений.
В адсорберах с псевдоожиженным слоем поглотителя мож-
но использовать сорбенты с размером частиц 0,2—0,5 мм, что
значительно сокращает время достижения адсорбционного рав-
новесия. Кроме того, при использовании адсорберов псевдоожи-
женного слоя допустимо подавать на очистку воду со значи-
тельным содержанием взвешенных частиц (неподвижные слои
адсорбента забиваются при концентрации взвеси 10—20 мг/л).
Недостатком схемы с псевдоожижением является быстрый из-
нос аппаратуры и частичек адсорбента из-за эрозии.
Технологические схемы очистки воды
с применением порошкообразного активного угля
Кондиционирование воды на станциях водоподготовки. Для
приготовления питьевой воды на заключительных стадиях водо-
подготовки часто применяют порошкообразный уголь. Одна из
схем приведена на рис. 15,6.
Питьевую воду забирают из водохранилища с помощью сооружения 6 и
перекачивают потребителям насосными станциями первого (7) и второго
(12) подъемов. Суспензию активного угля приготовляют в баке 3. С помощью
Рис. 15,6. Схема углевания при водоподготовке:
* — воздухозаборник; 2 — компрессор; 3 —резервуар для приготовления угольной суспен-
зии; 4 —эрлифт; 5 —дозатор; 6 — водозаборное устройство; 7—насосная станция перво-
го подъема: 8 — контактор угля с водой; 9 —отстойник; 10 — фильтр; И — резервуар чи-
стой воды; 12 — насосная станция второго подъема.
456
15. Защита гидросферы от загрязнения
воздуха, забираемого из атмосферы устройством 1 и сжимаемого компрессо-
ром 2, суспензия по эрлифту 4 дозатором 5 направляется в линию транспор-
тирования питьевой воды. Место ввода активного угля в системе подготов-
ки питьевой воды должно обеспечивать условия достаточной диффузии за-
грязнений к поверхности угля и необходимое для очистки время контакта.
С этой точки зрения наиболее целесообразным местом ввода является на-
сосная станция первого подъема 7. После контакта с углем в резервуаре 8
адсорбент выделяют из воды в отстойнике 9 и фильтре 10. Из резервуара
чистой воды 11 вода поступает в насосную станцию второго подъема. В неко-
торых случаях дополнительную долю угля вводят в воду перед ее подачей
на фильтр 10 [304].
Доочистка воды после биохимической обработки. Техноло-
гия процесса адсорбционной очистки сточных вод этого произ-
водства порошкообразным углем полностью еще не отработана:
ведутся исследования, направленные на создание оптимальных
условий контакта угля со сточными водами и последующего их
разделения, а также регенерации отработанного угля. Пилотные
установки доочистки биохимически обработанных вод имеются
в ряде стран [311]. Схема одной из них (на 55 м3/сут) показана
на рис. 15,7.
Для повышения эффективности осаждения угля рекомендовано введение
в контактные резервуары 2 и 7 минеральных компонентов в сочетании с
полиэлектролитами. Во избежание снижения адсорбционной способности угля
коагулянты вводят после осуществления контакта воды с углем в аппара-
тах 1 и 6. При дозе активного угля 25 мг/л, продолжительности контакта
воды с углем 15 мин и последующем введении хлорированного раствора
сульфата железа показатель ХПК сточной воды после двух ступеней снижа-
ется с 50 до 10 мг/л. Содержание ионов трехвалентного железа в обрабаты-
ваемой воде составляет 20—30 мг/л, содержание алкилбензосульфопатов в
Рис. 15,7. Схема двухступенчатой очистки сточных вод порошкообразным углем:
1 — контактор с углем первой ступени; 2 — реакционная камера первой ступени; 3 — от-
стойник первой ступени; 4 — дозатор полиэлектролита; 5 — дозатор свежего угля; 6 —
контактор с углем второй ступени; 7— реакционная камера второй ступени; 8 — отстой-
ник второй ступени; 9 —песчаный фильтр.
Очистка воды гранулированным активным углем
457
воде не превышает 0,2 мг/л. Порошкообразный уголь из экономических со-
ображений предложено применять при исходном содержании растворенных
загрязнений не более 30 мг/л (по ХПК)*.
В настоящее время отрабатываются вопросы регенерации порошкового
угля в печах с фонтанирующим и с псевдоожиженным слоями [312].
Исследования в области применения порошковых активных
углей для очистки сточных вод широко ведутся в настоящее
время. Предложены схемы для одновременного удаления из
воды органических загрязнений и ионов тяжелых металлов
[313], для разложения эмульсий [314], очистки воды от краси-
телей и фторированных углеводородов [312]. Последняя схема
внедрена на одном из заводов фирмы Дюпон (США). Пыле-
видный уголь (50—300 мг/л) вводят на второй ступени биохи-
мической очистки. Стоимость установки 200 тыс. долл. [315].
Технологические схемы очистки воды с применением
гранулированного активного угля
Темпы роста числа установок очистки сточных вод с исполь-
зованием гранулированного активного угля неуклонно возрас-
тают во всех экономически развитых странах, практически во
всех отраслях промышленности. Сведения о действующих в
США сорбционных установках, по данным немецких авторов
[316], приведены в табл. 15-2. Регенерацию активного угля в
четырех случаях проводят в многоподовых печах (в 3-м слу-
чае— промывкой 3%-ным раствором щелочи).
Ниже приводятся конкретные схемы сорбционной очистки
воды гранулированным углем.
Адсорбционная доочистка бытовых сточных вод. Потенци-
альные возможности применения активного угля для очистки
бытовых сточных вод оцениваются выше, чем для промышлен-
ных.
По мнению сторонников применения активного угля, фильтрация через
него бытойых сточных вод после обычной (первичной и вторичной) биооб-
работки дает такую чистую воду, что она пригодная для питья. Стоимость
такой воды в среднем 2,6—4,5 цента за 1 м3 (3—4 коп/м3), включая все три
стадии обработки [317]. '
Очистка бытовых вод гранулированным углем используется на ряде во-
доочистных станций США. На очистной станции в г. Помоне (рис. 15,8) од-
новременно работают четыре последовательно расположенных закрытых
угольных фильтра 5—8 с неподвижной загрузкой угля; фильтр 4 — резерв-
ный; он необходим для подмены при выходе иа регенерацию фильтра 5.
Время контакта сточных вод с адсорбентом около 50 мин, линейная скорость
* Химическое потребление кислорода (ХПК)—величина, характеризую-
щая общее содержание в воде восстановителей (неорганических и органиче-
ских), реагирующих с сильными окислителями. Практически ХПК определя-
ется количеством кислорода, эквивалентным бихромату калия, расходуемому
на окисление органического и неорганического вещества1 В' стандартных ^ус-
ловиях.
458
]5. Защита гидросферы от загрязнения
фильтрации 17—24,5 м/ч. Фильтры снабжены устройством для поверхностной
промывки. Фильтр 5 промывают один раз в сутки в течение 30—40 мин,
остальные - по мерс необходимости. Расход воды на одну промывку состав-
ляет около 19 м3.
Установка в Помоне в настоящее время очищает около 38 тыс. м3 воды
в сутки [318|. В результате очистки сточных вод ХПК снижается с 60 до
7 мг/л (сорбционная емкость активного угля по ХПК — 0,26 кг/кг). Регене-
рацию активного угля проводят прокаливанием в мпогоподовой печи, при
этом потери угля составляют около 10%.
В настоящее время в США вводится в строй ряд новых установок
очистки бытовых и смеси бытовых и промышленных сточных вод с исполь-
зованием стадии очистки активным углем [317]. В частности, запатентован
так называемый Z-M-процесс, по которому биологическая обработка сточных
вод полностью заменена физико-химической с применением адсорбции грану-
лированным углем [319]. Эффективность адсорбции по этому способу допол-
нительно увеличена в результате гидролиза молекул большой молекулярной
массы (от 2 тыс. до 2—3 млн.), так как продукты гидролиза лучше сорби-
руются активным углем. Гидролиз ведут при добавлении Са(ОН)2 или H2SOt
с перемешиванием и последующим отстаиванием (рис. 15,9).
Сточную воду после отделения взвешенных частиц на сите 1 направляют
в реакторы-отстойники 2 и 3. Выделяющийся в них при взаимодействии с
известью шлам собирают в накопителе 7. Известь сепарируют на вакуум-
фильтре 13, обжигают в печи 14, охлаждают в аппарате 15, отделяют от
золы на сите 16 и через склад извести возвращают в процесс. Определенную
долю свежей извести добавляют к рециркуляту.
Сточную воду после освобождения от твердых веществ окончательно
очищают в адсорбере с активным углем 5 и направляют потребителю. Пе-
Таблица 15-2. Характеристика некоторых действующих в США
установок очистки сточных вод
Сточные воды Расход сточных вод, мЗ/ч Содержание загряз- нений, мг/л Размеры адсорбера, м Время контакта с ак- тивным углем, мин Загрузка активного угля, м3 Стоимость обработки 1 м3 воды
в исходной воде | в очищенной воде
Воды красиль- ного произвол- 80 200 — 0=2,85 40 56,6 0,15 марки (4,5 коп)
ства Фенол содержа- щие воды (за- вод 1) Фенолсодержа- щие воды (за- вод 2) Воды, загряз- ненные нефтью и маслами 88 50—200 1 0=2,4 //=10,5 280* 60 0,70 марки (20,9 коп)
30 400—2500 1 0=3,15 75 37 0,47 марки (14 коп)
660 350 (по ХПК) 30 (по ХПК) 57 53 0,03 марки (0,9 коп)
Полиолсодер- 16 дкащие воды * В двух последовательно 700 2 0=1,2 //=4,5 включенных адсорберах. 20 0,45 марки (13,5 коп)
Очистка воды гранулированным активным углем
459
Рис. 15,8. Схема установки очистки сточных вод в неподвижном слое гранулированного
угля: 1
1 — хлоратор сточных вод, поступающих из вторичных отстойников; 2 — резервуар био-
логически очищенных сточных вод; 3— резервуар промывной воды; 4 — резервуарный ад-
сорбер; 5—8 — последовательно включенные адсорберы; 9 —сборник очищенной воды;
10 —бункер для обезвоживания отработанного угля; Н — шестиподовая печь для реге-
нерации угля; 12 —резервуар для охлаждения регенерированного угля.
риодически уголь из адсорбера 5 выгружают, переводят в емкость 8, обезво-
живают в системе 10, регенерируют в печи 11 и из бака для охлаждения 12
возвращают в адсорбер 5. В этот период очистку сточной воды проводят в
резервном адсорбере 9.
При очистке по Z-M-процессу сточных вод (38 тыс. м3/сут) ВПК* воды
снижается с 250 до 5 мг/л, ХПК — с 500 до 10 мг/л, содержание механиче-
ских примесей — с 250 до 5 мг/л и общее содержание соединений фосфора —
с 10 до 0,3 мг/л. По расчетам фирмы, такая установка будет стоить иа
50 % меньше, чем обычная установка с полной биохимической обработкой,
эксплуатационные расходы снизятся на 40%.
Адсорбционная доочистка сточных вод в коксохимической
промышленности. Одна из первых в мире схем очистки сточных
вод гранулированным углем была применена в Англии для
полного обезвреживания 775 м3/сут сточной воды аммиачного
отделения коксохимического завода следующего состава
* Биологическая потребность в кислороде (БПЦ)— степень загрязнения при-
родных и сточных вод органическими веществами, определяемая количеством
кислорода, который используется на биохимическое окисление этих веществ.
Расход кислорода на нитрификацию в этот показатель не включается.
БПК определяют по разности между содержанием кислорода до и после ин-
кубации пробы в стандартных условиях. Цифра в показателе БПК обозначает
время инкубации (в сут), например Б ПРИ, БПК-М и т. д.
460
15. Защита гидросферы от загрязнения
(мг/л): взвешенные вещества — 2000; фенолы — 600—1800; ро-
даниды— 150—450; тиосульфаты (в пересчете на серу)—40—
210; хлориды — 4000—5500; высшие фенолы — 250—450 [320].
Окисляемость воды ХПК — 4000 мг Ог/л, pH — 9—10.
В схеме очистки (рис. 15,10) предусмотрены четыре стадии:
1) отстаивание и охлаждение, 2) фильтрование через песчаные
фильтры, 3) удаление тиоцианатов и тиосульфатов ионитами,
4) удаление фенолов и других примесей активным углем.
На первой и второй стадиях воду последовательно охлаждают с 90 °C
до 30 °C, концентрация взвешенных веществ в ней уменьшается с 2000 до
5 мг/л. На стадии ионного обмена воду подкисляют до pH 5—6, а затем
с помощью анионита Диацидит-Е в хлоридной форме удаляют из нее тио-
цианаты и тиосульфаты. Это делается из-за того, что присутствие данных
веществ и некоторых других неорганических ионов снижает активность углей
в процессе удаления органических веществ. Регенерацию смолы на третьей
стадии проводят последовательной обработкой аммиаком и хлоридом водо-
рода. Воду после ионитной очистки нейтрализуют известью, а на четвертой
стадии удаляют фенолы и другие органические вещества активным углем.
Имеется три группы по два последовательно включенных адсорбера;
одна из групп работает на стадии адсорбции, вторая — на стадии регенера-
ции угля горячим бензолом и третья — на стадии перегрузки угля. Раствор
фенолов в бензоле направляют в смолоперегонный цех завода для ректифи-
кации. Очищенная вода содержит менее 10 мг/л фенолов, около 70 мг/л выс-
Рис. 15,9. Схема комбинированной физико-химической очистки сточных вод:
I _ сито’ 2, 3 — реакторы-отстойники; 4 — фильтр; 5, 9 — адсорберы; 6— склад извести;
7 — шламон’акопитель; 8 — хранилище активного угля; 10 — система обезвоживания угля;
11 — печь регенерации; 12 —бак для охлаждения угля; 13 — вакуумфильтр; 14 — печь для
обжига извести; 15 — охладитель извести; 16 — отстойник; 17 — сборник золы.
Очистка воды гранулированным активным углем
461
Рис. 15,10. Схема доочистки сточных вод коксохимического завода (Англия):
1 — отстойник; 2 — оросительный холодильник; 3 — сборник охлажденной воды; 4 — пес-
чаные фильтры; 5 — бак промывной воды для фильтров; 6 — хранилище аммиака для
регенерации ионитов; 7 — ионообменные фильтры; 8 — хранилище сточной воды; 9 — ем-
кость для раствора после регенерации ионитов; 10 —адсорберы с активным углем; 11 —
сборник очищенной воды; 12 — аппарат для регенерации бензола после промывки адсор-
беров.
ших фенолов, ее окисляемость (ХПК) менее 100 мг О2 на 1 л. Эффектив-
ность очистки снижается из-за падения адсорбционной емкости угля от цик-
ла к циклу, поэтому через 16 фильтроциклов уголь подвергают термообра-
ботке для удаления недесорбируемых бензолом смол и высших фенолов.
Расход реагентов на очистку следующий: 3,38 кг/м3 НС1; 0,74 кг/м3 NH3;
2,02 кг/м3 Са(ОН)2; 0,58 л/м3 С6Н6. Кроме того, приходится восполнять по-
тери ионообменной смолы и активного угля. Реализация извлеченных фено-
лов компенсирует лишь 5% эксплуатационных расходов.
Более прогрессивной и экономически эффективной является
схема, предложенная для очистки сточных вод коксохимическо-
го производства фирмой Кэлгон корпорейшн и внедренная на
одном из металлургических заводов США [320].
Состав воды подаваемой на очистку, следующий (мг/л):
взвешенные вещества — 50; общий органический углерод* —
* Общий органический углерод — полное содержание органических соедине-
ний в воде — определяют по методу «сожжения» Бэкмана и корреспондируют
с показателем ХПК.
462
15. Защита гидросферы от загрязнения
Рис. 5,11. Схема доочистки сточных вод коксохимического завода (США):
1 — емкость реагентов для регулирования pH; 2 — емкость для коагулянтов; 3 —отстой-
ник; 4 — фильтр; 5 — загрузочный бункер; 9 — адсорбер; 7 — сборник отработанного угля;
8 — шнековый обезвоживатель; 9 — печь регенерации; 16 — охладитель угля; 11—сборник
регенерированного угля; 12 —емкость для сточной воды; 13 —емкость для соединений
меди; 14 — газгольдер с воздухом или кислородом; 15 — реактор для разложения циани-
дов.
2100; растворимый органический углерод* — 1900; фенолы —
2235; цианиды — 0,01; аммиак — 4000; роданиды — 700; pH 7,2.
Процесс очистки проводят в три стадии по технологической схеме, пока-
занной иа рис. 15,11. На первой стадии ведут осветление воды коагуляцией
взвешенных частиц с помощью FeCl? и синтетического органического полиме-
ра с последующим фильтрованием через слой гранулированного неактивиро-
ванного угля (позиции 1—4). На дайной стадии очистки происходит умень-
шение концентрации взвешенных веществ до 5 мг/л, а содержание общего и
растворенного углерода выравнивается, достигая 1750 мг/л.
Вторая стадия — адсорбционная очистка воды от органических веществ
гранулированным активным углем (позиции 5,6) в четырех адсорберах. Каче-
ство воды, прошедшей две стадии очистки (в мг/л): органический углерод —
менее 156; фенолы — менее 0,1; окисляемость — не более 1260, вода является
бесцветной.
На третьей стадии (позиции 12—15) из воды полностью удаляются циа-
ниды в результате химической реакции с соединениями меди. Ионы меди
связывают CN-ионы в комплекс, который сорбируется активным углем в
реакторе. Для полного окисления CN-ионов в систему вводят воздух или
кислород. В схеме предусмотрена технологическая нитка для регенерации
угля (позиции 7—11).
* Растворимый органический углерод — содержание растворимых в воде
органических соединений. Определяется по методу Бэкмана в профильтро-
ванной пробе воды.
Очистка воды гранулированным активным углем
463
Отмечается, что стоимость очистки воды по данному способу
является более низкой, чем при обесфеноливании другими ме-
тодами. Снижение концентрации фенола с 2000 до 0,1 мг/л об-
ходится примерно в 45 центов/м3 (около 40 коп/м3), причем
содержание растворимых органических веществ мало влияет на
процесс, а сточные воды очищаются более полно, чем при обыч-
ной биоочистке. Достоинством установки является ее компакт-
ность: при производительности 1500 м3/сут она занимает пло-
щадь 540 м2.
Адсорбционная очистка сточных вод в производстве химиче-
ских волокон. На рис. 15,12 показана схема промышленной уста-
новки, созданной на Киевском комбинате химического волокна
для очистки сточных вод производства сульфида углерода .[321],
содержащих не только CS2, ио и H2S.
Принцип работы установки состоит в следующем. Сточную воду после
отстаивания в приемных горизонтальных баках 1 с помощью насоса 2 про-
фильтровывают через три адсорбера 3, соединенных последовательно и загру-
женных активным углем, после чего отводят в канализацию. Четвертый ад-
сорбер (на рис. 15,12 не показан) находится на регенерации или в резерве.
В каждом новом цикле первым по ходу воды включают адсорбер, который
был вторым в предыдущем цикле. При этом сульфид углерода из сточной
воды удаляется полностью, а содержание сульфида водорода понижается.
Вода движется в адсорберах снизу вверх со скоростью 1,5—2 м/ч. Уве-
личение скорости потока выше 5 м/ч приводит к значительному уменьшению
времени защитного действия слоя активного угля. Сорбционная емкость ад-
сорбента находится в пределах 15—30%. Масса адсорбента, загружаемая в
один адсорбер, 900—1300 кг.
Извлечение сульфида углерода из угля проводят паром низкого давления
(0,05—0,02 МПа). Пар движется в адсорбере сверху вниз, его расход колеб-
лется от двух- до пятикратного количества от массы извлеченного из угля
сульфида углерода. Смесь паров пропускают через холодильник 4, разделяют
сконденсированную фазу на воду и сульфид углерода в сепараторе 8, после
чего воду в холодильнике 4 примешивают к очищенной воде.
Рис. 15.12. Схема адсорбционной очистки сточных вод производства сульфида углерода:
j__сборник сточных вод; 2 — насосы; 3 — адсорберы; 4 — холодильник; 5 — сборник; ъ —
хлоратор; 7 — баллон с хлором; 8 — сепаратор.
464 15, Защита гидросферы от загрязнения
Коагулянт
Для более полной очистки воды от сульфида водорода ее после охлаж-
дения хлорируют в аппарате 6, куда хлор подают из баллонов (3 кг на 1 кг
H2S). После этого воду отводят в канализацию.
Производственная установка на Киевском комбинате хими-
ческих волокон позволила не только полностью удалять суль-
фид углерода из сточных вод, но и возвращать в производство
до 7,5 т CS2 ежемесячно (около 90 т/год). Себестоимость одного
килограмма возвращаемого сульфида —12 коп. Таким образом,
только от утилизации сульфида углерода экономический эф-
фект составляет около 10 тыс. руб. в год.
Сорбционная очистка стоков предприятий химического во-
локна все шире практикуется в СССР [322] и в ряде зарубеж-
ных стран [323].
Адсорбционная очистка сточных вод в химической промыш-
ленности. Обработка воды гранулированным активным углем
наиболее широко практикуется в химической промышленности,
и в особенности там, где сточные воды содержат биологически
трудноразрушаемые вещества. Описаны схемы удаления грану-
лированным углем поверхностно-активных веществ [324], кра-
сителей [325], нефтепродуктов [326, 327], хлорорганических про-
изводных [328] и др.
; На рис. 15,13 показана технологическая схема адсорбцион-
ной, деструктивной очистки сточных вод Рубежанского химиче-
ского комбината [329].
Кислые и щелочные сточные воды смешивают в усреднительном бассей-
не I, где pH поддерживают в пределах 4—5 для обеспечения выделения
гидробкеида железа. При этом из стоков выделяется значительное количество
Очистка воды гранулированным активным углем
465
Рис. 15,13. Принципиальная схема станции
адсорбционной очистки сточных вод Рубе-
жанского химкомбината:
1 — накопитель-отстойник; 2 — насос; 3 —
смеситель; 4, 5 — мерники извести и коагу-
лянта; 6, 10 — осветители; 7—смеситель
воды с активным углем; 8—бункер-доза-
тор активного угля; 9 —адсорбер; 11 —
песчаный фильтр; 12 — транспортер (обез-
воживание угля); 13 — печь регенерации.
органических соединений кислого ха-
рактера. Высокоразвитая поверх-
ность гндрооксида железа способст-
вует выделению коллоидов. После
отделения выпавшего шлама (6—
10 кг/м3 в расчете на сухое вещест-
во) ХПК стока снижается на 25—
30%, а интенсивность окраски — в
2—3 раза.
После отстаивания стоки перека-
чивают насосом 2, нейтрализуют в
аппарате 3 известковым молоком
(4) и освобождают от выпавшего
осадка в осветлителях 6 или верти-
кальных отстойниках. В смеситель 3
подают также коагулянт из мерника 5. Оптимальная скорость восходящего
потока в осветлителях, обеспечивающая работу взвешенного слоя — 0,2—
0,3 мм/с, т. е. значительно меньше принятой в практике водоподготовки, так
как осадки обладают малой прочностью и легко пептизируются.
В результате осветления нейтральной смеси сточных вод из нее удаляют
не только хлопья гидрооксидов металлов, но и адсорбированные на них кра-
сители и жидкие смолы. Обработка в осветлителях, как правило, обеспечи-
вает удаление от 30 до 60% органических веществ, содержащихся в стоках
(по ХПК), и значительное снижение интенсивности окраски воды. Оставшие-
ся в воде загрязнения в основном состоят из молекулярно растворенных ор-
ганических соединений ароматического и алифатического рядов и минераль-
ных солей (сульфатов и хлоридов натрия, аммония, калия и др.).
Из осветлителя эту воду направляют в смеситель 7, куда из бункера 8
дозируют активированный антрацит — высокопрочный углеродистый сорбент
микропористой структуры. Полученную водноугольную суспензию насосом
перекачивают в коническое днище одноярусного адсорбера 9 с псевдоожи-
женным слоем. Очищенную в адсорбере воду возвращают на повторное от-
стаивание для освобождения от пыли, выносимой из аппарата. В качестве
коагулянта во втором осветлителе 10 используют сульфат железа (до
100 мг/л). Воду, прошедшую через второй осветлитель, профильтровывают
через песчаный фильтр И. После такой обработки стоки не содержат приме-
сей ароматических соединений, а содержат преимуществеиио биологически
легкоокисляемые алифатические соединения (БПКб 300—500 мг/л).
Непрерывную термическую регенерацию (позиции 12 и 13) сорбента про-
водят одновременно с активацией дробленого антрацита в двухзонной печи
псевдоожиженного слоя.
Эксплуатационные затраты на адсорбционную очистку по
приведенной схеме 10000 м3/сут сточных вод и извлечение из
них 8—10 т/сут органических веществ составляют 60—70 коп
за 1 м3 стока, в том числе затраты на активированный антра-
цит— 20—25 коп [330]. Схемы, аналогичные рассмотренной,
запроектированы также для очистки сточных вод на некоторых
других химкомбинатах.
30—1346
16
ИСПОЛЬЗОВАНИЕ
ИЗБИРАТЕЛЬНОСТИ
АДСОРБЦИИ
Подавляющее большинство процессов разделения бинарных или
более сложных систем проводят, используя избирательные
свойства адсорбента по отдельным компонентам, составляющих
смесь. Выбор адсорбента оптимального типа в каждом конкрет-
ном случае во многом облегчает задачу разделения. Общие
закономерности адсорбции смесей рассмотрены в гл. 5.
Избирательность адсорбции иа активных углях
Активные угли принадлежат к адсорбентам электронейтралыю-
го типа, и адсорбция на них определяется дисперсионными си-
лами взаимодействия между твердой поверхностью и поглощае-
мыми молекулами. Как правило, активными углями избира-
тельно адсорбируются вещества, молекулы которых обладают
высокой поляризуемостью и большой массой.
Коэффициенты разделения (определение см. на стр. 124)
углеводородов на активном угле при атмосферном давлении и
температуре 20 °C, по данным Льюиса, следующие: для С2Н2—
С2Н4 Кр=1,4; С2Н4—С2Н6 (1,5); С3Н6 —С3Н8 (1,1); С2Н6—
—С3Н8(3,6); С2Н4—С3Н6(12,5); СН4—С2Н4 (15,4); С2Н4—
—С3Н8(13,6) [331]. Первым указан хуже адсорбирующийся
компонент.
Многие методы промышленной очистки газов, защиты ат-
мосферы от загрязнений основаны на избирательных свойст-
вах активных углей и рассмотрены раньше. В настоящем разде-
ле остановимся лишь на одном примере использования избира-
тельности адсорбции на угле [332]. Ранее (стр. 124) описан
совмещенный метод очистки вентиляционных отходящих газов
производств химических волокон от сульфидов водорода и угле-
рода. Если газ, содержащий эти компоненты и кислород, посту-
пает в адсорбер с активным углем, в первом слое угля проис-
ходит реакция окисления H2S с образованием элементной серы,
в то гремя как последующие слои выполняют функции улавли-
вания CS2 по механизму чисто физической адсорбции.
В бескислородной среде лучше адсорбирующимся компо-
нентом является сульфид углерода: его молекулярная масса
Избирательность адсорбции на активных углях
467
равна 76, в то время как молекулярная масса сульфида водо-
рода только 34. Технологические газы некоторых отраслей про-
мышленности, связанных с получением и применением сульфида
углерода (производство тетрахлорида углерода, собственно
сульфида углерода, ускорителей вулканизации) содержат в
чрезвычайно высоких концентрациях CS2 и H2S. Примерный
состав такцх газов после установки масляной адсорбции сле-
дующий (в об. %): CS2—12,8; H2S—49,3; COS—13,5; СО2—8,7;
СО—5,6; N2—8,1; Н2О—2,0.
При пропускании этой смеси через слой активного угля про-
скок сульфида водорода наступает практически немедленно, в
то время как период защитного действия даже небольшого слоя
угля по сульфиду углерода при скорости потока 0,03 м/с изме-
ряется часами.
Процесс извлечения CS2 из бескислородных смесей проводят в верти-
кальных адсорберах, снабженных внутренними змеевиками, в которых цирку-
лирует вода, охлаждающая слой адсорбента. Благодаря этому удается, не-
смотря на высокий экзотермический эффект при переработке высококонцен-
трированной газовой смеси, поддерживать температуру угля на уровне
40 °C. За проскоковую концентрацию принимают 3% CS2. Продолжитель-
ность стадии адсорбции колеблется от 6 до 48 ч. Десорбцию ведут острым
насыщенным водяным паром в течение 2—3 ч при давлении, близком к атмо-
сферному; расход пара составляет 2,5 кг/кг CS2. Заключительную стадию —
охлаждение угля проводят в течение 6—10 ч пропускаемой через змеевики
водой. Внедрение в промышленность адсорбционного метода рекуперации
позволило получать ежегодно дополнительно около 7 тыс. т сульфида угле-
рода.
Избирательность адсорбции на силикагелях
Адсорбция непредельных углеводородов
Силикагели относятся к адсорбентам анионного типа. В этом
случае на адсорбционную способность и характер адсорбции
большое влияние оказывает химическая природа адсорбтива:
специфическое взаимодействие адсорбированных молекул нак-
ладывается на общий «фон» взаимодействий, обусловленных
дисперсионными силами.
Известно, что углеводороды олефинового ряда обладают
большим давлением паров, чем парафиновые углеводороды с
тем же числом атомов углерода в молекуле, и при разделении
таких смесей ректификацией в нижнем продукте происходит
обогащение парафиновыми углеводородами. При адсорбции
углеводородов разных гомологических рядов на силикагелях
преимущественно адсорбируются углеводрроды, имеющие двой-
ные и тройные связи в молекуле. В случае силикагелей такие
вещества являются менее летучими компонентами |[333].
Различие в поведении углеводородов разной степени насыщения на углях
и силикагелях проявляется уже при рассмотрении изотерм адсорбции. На
30*
468
16. Использование избирательности адсорбции
рис. 16,1 представлены изотермы адсорбции пропана и пропилена на актив-
ном угле и силикагеле при 25 °C. Силикагель обладает значительно меньшей
адсорбционной способностью, особенно по пропану, но различие в адсорби-
руемости на нем выражено гораздо более отчетливо. В соответствии с этим
кривые адсорбционного равновесия на силикагеле, в отличие от угля
(рис. 16,2), указывают на значительное обогащение адсорбированной фазы
непредельным углеводородом и, следовательно, на возможность разделения
этих компонентов на силикагеле.
Разница в теплотах адсорбции является количественным выражением до-
полнительных специфических сил адсорбции. На поверхности углеродных ад-
сорбентов, например графитированной сажи, на которой отсутствуют поляр-
ные группы и ионы, теплоты адсорбции пропилена и пропана близки. При
степени заполнения адсорбционной емкости 0 = 0,5 они равны соответственно
26,0 и 27,2 кДж/моль [334]. На силикагеле теплота адсорбции пропилена
в тех же условиях в 1,5 раза выше теплоты адсорбции пропана (30,6 и
20,9 кДж/моль) [335].
Изменение температуры в интервале от —20 до 25 °C приводит к несу-
щественному изменению избирательности адсорбции пропан-пропиленовой
смеси на силикагеле: коэффициент разделения находится в пределах 3,1—
3,8 [333]. Алюмогель обладает несколько более низкой избирательностью. При
25 °C и атмосферном давлении коэффициент разделения на алюмогелях раз-
личных типов колеблется между значениями 2,20 и 2,86. Преимущественная
лс3н„
Рис. 16,1. Изотермы адсорбции при 25 °C в системах:
I — СзН8 на активном угле; 2 - С3Н8 на активном угле; 3 — СзНа на силикагеле- 4 —
СзН, на силикагеле.
Рис. 16,2. Адсорбционная способность активного угля (1) и силикагеля (2) по смеси
С8Нв — С3Н8, кривые адсорбционного равновесия этой смеси иа силикагеле (3) и актив-
ном угле (4) при температуре 25 °C и нормальном давлении.
Избирательность адсорбции на силикагелях
469
Рис. 16,3. Кривые адсорбционного равновесия смеси С2Н, — С2Н6 на силикагеле при раз-
личном давлении:
1 — 0,1 МПа; 2 — 0,26 МПа; 3 — 0,78 МПа; 4 — 1,92 МПа; 5 — равновесие на мелкопори-
стом угле (для сравнения).
Рис. 16,4. Кривая адсорбционного равновесия смеси изобутана и 1-бутена на силикагеле
при 25 °C и нормальном давлении.
адсорбция олефинового углеводорода на силикагеле отмечена также в систе-
мах этан — этилен (рис. 16,3) и изобутан — бутилен (рис. 16,4).
Избирательность адсорбции при атмосферном давлении, достаточная для
разделения углеводородов, которые различаются по степени насыщенности,
сохраняется и при повышенном давлении. Это подтверждает кривая адсорб-
ционного равновесия этаи-этиленовой смеси иа силикагеле при 25 °C и дав-
лении около 0,5 МПа, представленная на рис. 16,5. По данным Льюиса с
сотр., повышение давления в системе этан — этилен — мелкопористый сили-
кагель от 0,03 до 1,9 МПа приводит к снижению коэффициента разделения
сЗ до 1,6.
Ацетилен как углеводород с тройной связью в молекуле адсорбируется
преимущественно из смеси с этиленом. Это ясно из сравнения изотерм ад-
сорбции этих двух углеводородов (рис. 16,6) и кривых адсорбционного рав-
новесия (рис. 16,7). При поглощении смеси активным углем отмечено незна-
чительное обогащение адсорбированной фазы этиленом.
На рис. 16,8 представлены изотермы адсорбции этилена, пропана и про-
пилена на силикагеле. Адсорбция двух компонентов, у которых наличие
двойной связи в молекуле проявляется в
тельной энергии адсорбции, приводит к
тому, что избирательность адсорбции в
системе этилен — пропилен как на анион-
ном адсорбенте — силикагеле, так и на
электронейтральиом активном угле при-
близительно одинакова (рис. 16,9). В то
же время, увеличение энергии адсорбции
этилена иа силикагеле за счет специфиче-
ской составляющей по сравнению с энер-
гией адсорбции пропана, обязанной дей-
ствию только дисперсионных сил, приво-
дит к снижению избирательности адсорб-
Рис. 16,5. Кривые адсорбционного равновесия сме-
си С2Н4—С2Н8 на мелкопористом силикагеле (1)
и рекуперационном активном угле (2) при 20 °C,
0,5 МПа.
приблизительно равной дополни-
470
16. Использование избирательности адсорбции
Рис. 16,6. Изотермы адсорбции при 25 °C:
1 — С2Н4 на активном угле; 2 — С2Н2 на
С2Н4 на силикагеле.
активном угле; 3 — С2Н2 на силикагеле; 4 —
Рис. 16,7. Адсорбционная способность активного угля (1) и силикагеля (2) по смеси
С2Н2-С2Н4, кривые адсорбционного равновесия этой смесн на силикагеле (3) и актив-
ном угле (4) при 25 °C и нормальном давлении.
ции этой смеси на силикагеле по сравнению с избирательностью адсорбции
на активном угле (рис. 16,10 и 16,11).
Избирательность адсорбции непредельных углеводородов на силикагеле
используется для выделения этих компонентов из смесей с углеводородами
других гомологических рядов [336]. Так, в движущемся слое силикагеля АСМ
была подвергнута разделению на две фракции смесь следующего состава:
11,6% изо-CiHs; 22,1% н-СТЬ; 63,1% н-СгН.о д. кзо-С/ДТ^,; 3,2% С5Н124-
+ С5Н10. После разделения содержание непредельных углеводородов в ниж-
ней фракции составляло 97—98%, а в верхней фракции — только 7—10%.
Возможность разделения этан-этиленовой и пропан-пропиле-
новой смесей в движущемся слое мелкопористых силикагелей
Таблица 16-1. Адсорбционное равновесие смеси ацетилен — диоксид
углерода на силикагеле ШГСГ при 25 °C и нормальном давлении
Мольная доля в газовой фазе, % Мольная доля в адсорбированной фазе, % Коэффициент разделения /Ср
у2(со2) | ^2(COs) | X) (СзНз)
70,5 29,5 52,5 48,5 2,16
50,0 50,0 36,4 66,6 1,75
38,8 62,2 24,4 75,6 1,96
Избирательность адсорбции на силикагелях
471
также доказана экспериментально. Основные трудности при
проведении процесса разделения связаны с полимеризацией
олефинов в секции десорбции и, следовательно, закоксовыва-
нием адсорбента.
При выборе типа адсорбента для выделения ацетилена из
промышленных газовых смесей необходимо учитывать совмест-
ную адсорбцию других компонентов и в первую очередь диок-
сида углерода. Молекулы как ацетилена, так и диоксида угле-
рода обладают значительными квадрупольными моментами, а
следовательно, и дополнительными специфическими составляю-
щими энергии адсорбции. При пропускании сме^и ацетилена и
диоксида углерода через слой шарикового силикагеля с добав-
кой глинозема ШГСГ (удельная поверхность 400 м2/г) первые
порции выходящего газа содержали только хуже адсорбирую-
щийся компонент — диоксид углерода. Результаты анализа
адсорбированной фазы после установления равновесия приведе-
ны в табл. 16-1. Так как по адсорбционной способности актив-
ные угли в отношении углеводородов значительно превосходят
силикагели, а по разделяющей способности в системе ацети-
лен — диоксид углерода угли и силикагели равноценны, приме-
нение активных углей для разделения рассматриваемой смеси
предпочтительнее.
В системах углеводородов одной степени насыщенности, но разной моле-
кулярной массы на силикагеле, так же, как и на других адсорбентах, пре-
имущественно поглощается более высокомолекулярный углерод. Так, коэф-
Рис. 16,8. Изотермы адсорбции углеводородов на силикагеле:
а — С2Н4; б — СзЩ; в — СзНв-
4/Z
I . Использование избирательности адсорбции
Рис. 16,9. Кривые адсорбционного равновесия смеси С2Н(—- С5Н6 (давление 0,1 МПа):
1 на активном угле (25 °C); 2—4 — на силикагеле соответственно при 0 (2), 25 (3) и*
40 °C (4).
Рис. 16,10. Кривые адсорбционного равновесия смеси С2Н4—С3Н8:
М — на активном угле при 25 °C и давлении соответственно 0.1; 0.26 и 0,74 МПа; 4 —
на силикагеле при 25 °C н 0,1 МПа.
фициент разделения у силикагеля в системе этилен — пропилен при 0°С и
атмосферном давлении составляет 7,7; при 25 °C — 6,6; при 40 °C — 6,1
(рис. 16,12). Коэффициент разделения в системе углеводородов алканового
ряда — этана и пропана — при 25 °C и нормальном давлении составляет на
силикагеле 4,8, на активном угле 3,6.
Избирательность адсорбции в бинарных системах углеводородов разной
массы и степени насыщенности на силикагеле при атмосферном давлении
показана ниже;
Система
Кх.а
t, °C
к,
СН4—С2Н4
СИ;
25
19,0
С 2Н4 CgHg С2Н4 С3Н8 С2Н4—С3Н8
С2Н4 С2Н4 С2Н4
О 25 40
2,1 2,0 1,9
В системе этилен — пропан наличие дополнительной специфической со-
ставляющей взаимодействия этилена с адсорбентом проявляется в резком
снижении избирательности по сравнению с
избирательностью на активном угле, хотя
в обоих случаях хуже адсорбирующимся
компонентом является этилен. Коэффици-
ент разделения при нормальном давлении
на угле составляет 13,6, иа силикагеле —
только 2,1. В системе метай — этилен, на-
оборот, преимущественная адсорбций эти-
лена усиливается при контакте углеводо-
родов с силикатной поверхностью, Коэф-
фициент разделения в одинаковых услови-
ях на силикагеле составляет 19,0, а иа уг-
ле 15,4.
Рис. 16,11. Адсорбционная способность силикагеля
по смеси С2Н4 — CSHB при нормальном давлении. •
Избирательность адсорбции на цеолитах
473
Рис. 16,12. Кривые адсорбционного равновесия
(а) и адсорбционная способность (б) в системе
С2Н4 — С3Н8 — силикагель при нормальном давле-
нии.
Адсорбция ароматических
углеводородов
Ароматические углеводороды изби-
рательно адсорбируются силикаге-
лем из смесей с углеводородами
других классов. На рис. 16,13 пред-
ставлены кривые адсорбционного
равновесия бинарных смесей бензо-
ла и толуола с нормальными пара-
финами и циклопарафинами С6—Cs
при 150°C [337]. Кривые указыва-
ют, что адсорбируемость бензола
значительно превышает адсорбиру-?-.
емость «-гексана и циклогексана f
(т. е. углеводородов с близкой мо- R.
лекулярной массой) и приблизи-
тсльно соответствует адсорбируемо-
сти «-октана. Избирательность по-
глощения ароматических углеводо-
родов сохраняется и при проведе-
нии процесса в жидкой фазе. Изби-
рательность адсорбции углеводоро-
дов на силикагелях широко исполь-
зуют для анализа многокомпонентных
смесей [338, 339].
Впервые попытка использовать силикагель для промышлен-
ного разделения углеводородов была предпринята в 1946 г.
[340]. Промышленный процесс выделения ароматических угле-
водородов из различных нефтяных фракций в стационарном
слое силикагеля был разработан фирмой Сан-Ойл и известен
под названием «аросорб». На рис. 16,14 приведена схема этого
процесса, предназначенного для выделения бензола и толуола
[341]-
Деароматизацию проводят при обычных рабочих условиях в жидкой
фазе, пропуская сырье из емкости 1 через слой силикагеля, загруженного в
адсорбер 3. Продолжительность стадии 30 мин. Неадсорбированные парафи-
новые и циклопарафиновые углеводороды поступают в ректификационную
колонну 4, где отделяются легкие углеводороды—бутан или пентан, исполь-
зуемые в следующей стадии «промывки» адсорбента. Продолжительность ста-
дии промывки 10 мин. Нижний продукт ректифицируют в колонне 6. Де-
сорбцию ароматических углеводородов проводят потоком ксилолов, которые
затем отделяют от примеси парафиновых углеводородов в колонне 5, а от
ароматического адсорбата — в колонне 7. Продолжительность стадии де-
474
16. Использование избирательности адсорбции
Рис. 16,13. Кривые адсорбционного равновесия смесей бензола (а) и толуола (б) с па-
рафиновыми углеводородами на мелкопористом силикагеле при 150 С и нормальном
давлении:
1 — циклогексан; 2 — н-гсксаи; 3 — гептан; 4 — н-октан.
сорбции 40 мин. В течение первых 25 мин смесь десорбата и десорбента вы-
ходит из десорбера с некоторым содержанием парафиновых углеводородов.
Смесь бензола и толуола фракционируют в колонне 8. Десорбент циркулирует
в замкнутом цикле.
При концентрации ароматических углеводородов в исходной смеси 25%.
адсорбционная способность силикагеля составляет около 20 г/100 г. В дина-
мических условиях эта величина на 20—30% меньше. Чистота выделенных,
ароматических углеводородов достигает 97%, степень извлечения аромати-
ки— 95%. Скорость перемещения сырья в адсорбционной колонне состав-
ляет 1-10~3 м/с. Расход промывочной жидкости равен 370 м\ а десорбента
1250 м3 на 1000 м3 сырья.
В процессе используют вертикальные цилиндрические адсорберы высотой
4,5—6,0 и диаметром 1,3 м. Загрузка силикагеля в такой адсорбер составляет
около 15 т. При жидкофазной деароматизации применяют мелкую фракцию
адсорбента с размером частиц 0,07—0,6 мм. Перед подачей в адсорбер с
силикагелем все потоки подвергают осушке до остаточной влажности не выше-
20 част, на 1 млн.; при такой остаточной влажности адсорбционная способ-
ность силикагеля по ароматическим углеводородам за счет совместной ад-
сорбции воды снижается не более, чем на 15%. Ядами для силикагеля.
уменьшающими длительность его эксплуатации, являются азотистые, серни-
стые и кислородные соединения, а также непредельные углеводороды, при-
сутствующие в сырье. Последние
опасны, так как при их полимери-
зации происходит закоксовывание ад-
сорбента. Для технико-экономиче-
ских расчетов продолжительность
эксплуатации одной загрузки сили-
кагеля принимают равной 1 год.
Рис. 16,14. Схема установки деароматиза-
ции нефтяиых фракций в стационарном
слое силикагеля:
I — емкость сырья; 2 —осушители потоков;
3 — адсорбер; 4—8 — ректификационные ко-
лонны; I — ароматические углеводороды;
II — широкая фракция неароматических
углеводородов; III —фракция С4—С5; IV —
неароматические углеводороды Сб+; V —
бензол; VI — толуол; VII — десорбент
(ксилолы).
Избирательность адсорбции на силикагелях
475
Кроме ксилолов, используемых в процессе «аросорб», в ка-
честве десорбента могут применяться и другие вещества, если
они обладают достаточно высокой адсорбируемостью и легко
отделяются от десорбата. Эффективным десорбентом тяжелых
ароматических углеводородов являются ароматизированные уг-
леводородные фракции с интервалом кипения более низким, чем
интервал кипения сырья [342, 343]. Так, при деароматизации
фракции продукта гидроформинга прямогонного лигроина 150—
205°C с содержанием ароматических углеводородов до 50% де-
сорбентом может служить фракция 71—88°C, содержащая 60%
бензола. Десорбент и десорбат в этом случае легко разделя-
ются фракционированием.
В одном из вариантов процесса [344] деароматизацию про-
водят под давлением. Затем силикагель промывают пропаном
или бутаном для удаления неароматических углеводородов и
проводят десорбцию. На стадии десорбции в аппарат подают
жидкий, но нагретый низкокипящий водорастворимый десорбент
(ацетон, метанол, ацетальдегид). После окончания стадии де-
сорбции давление в адсорбере снижают до атмосферного, де-
сорбент испаряется, его пары конденсируют. Выделение арома-
тических углеводородов из основной массы десорбента проводят
добавлением воды к смеси; при этом происходит образование
двух фаз — ароматической и водной. Десорбент сушат и возвра-
щают в цикл.
При деароматизации легких нефтяных фракций десорбция
может быть проведена обычным методом: нагревом до 300 °C
при одновременной продувке нейтральным газом (водородом,
азотом, оксидом углерода, углеводородами Ci—Сз) [345]. На-
оборот, если разделению подвергаются тяжелые фракции, в ка-
честве десорбента применяют эффективные смеси типа смеси
дихлорэтилена, бензола и пентана. Такая смесь, в частности,
рекомендована в качестве десорбента при деароматизации па-
рафинистого смазочного масла, которую проводят с целью по-
вышения эксплуатационных свойств масла — понижения темпе-
ратуры застывания и улучшения индекса вязкости [346]. Учиты-
вая высокую вязкость масла, перед подачей в адсорбер его раз-
бавляют низкомолекулярным парафиновым углеводородом, на-
пример гептаном.
Другим конструктивным решением является проведение про-
цесса деароматизации в подвижном слое адсорбента [347, 348].
Как правило, адсорбент находится в виде взвеси в легких пара-
финовых углеводородах или в газойле. Масса силикагеля во
взвеси в 10—15 раз превышает массу ароматических углеводо-
родов, подлежащих извлечению. Соотношение всех углеводоро-
дов и силикагеля колеблется от 2 : 1 до 1:1.
В Советском Союзе разработан технологический процесс
деароматизации фракции нормальных парафинов в движущемся
476
16. Использование избирательности адсорбции
Отработанный _ Воздух
1Г 7
слое алюмосиликатного адсорбента с преимущественным разме-
ром зерен 0,25—0,8 мм [349, 350]. Промышленная установка
введена в эксплуатацию на одном из нефтеперерабатывающих
заводов в 1969 г. Схема установки приведена на рис. 16,15.
В адсорбер 5 подается раствор сырья в бензиновой фракции 80—120 °C.
Сырье выкипает в интервале 250—360 °C. В нем содержится 2—3% арома-
тических соединений и 0,12—0,14% серы. Массовое отношение подаваемого
адсорбента к сырью составляет 1 : 2, кратность разбавления сырья раствори-
телем (по объему) — 1:1. Температура в адсорбционной зоне 40—45 °C.
Одновременно с ароматическими углеводородами из сырья удаляются
сернистые соединения, смолы; при этом происходит полное обесцвечивание
раствора. В промывной секции адсорбционной колонны в результате проти-
воточного контакта с тем же растворителем проводят десорбцию ароматиче-
ских углеводородов. Концентрат выделенных ароматических соединений яв-
ляется побочным продуктом процесса. Отработанный адсорбент содержит
2,5—3,5% смолистых веществ.
Сушку пульпы адсорбента проводят в аппарате 6 кипящего слоя при
145—160 °C. Перегретый пар подают как непосредственно в слой адсорбента,
так и во встроенные змеевики. В процессе окислительной регенерации, кото-
рую проводят в аппарате кипящего слоя 3 при 600—650 °C, содержание кок-
са в адсорбенте снижается до 0,05—0,1%. Избыточное давление в нижней
части регенератора равно 0,03—0,04 МПа. Адсорбционная способность алю-
Избирательность адсорбции на силикагелях
477
Исходные сырье
Зкырам аромати-
ческих углеводородов
Рис. 16,15. Технологическая схема
установки деароматизации жидких па-
рафинов в движущемся слое силикат-
ного адсорбента:
1 — подогреватель; 2 — холодиЛ>ник;
3 — регенератор; 4 — сепаратор; 5—ад-
сорбер; 6 —сушилка адсорбента; 7 —
теплообменники; 8 —емкости; 9 — водо-
отделитель; 10 — отпарная колонна для
очищенных парафинов; Ц—то же, для-
экстракта ароматических углеводоро-
дов; 12 — газлифты.
мосиликата после регенерации со-
ставляет 75—80% от адсорбцион-
ной способности свежего адсор-
бента.
Перед возвращением в адсор-
бер регенерированный адсорбент
охлаждают в кипящем слое (2)
воздухом при 40—45 °C. Хладо-
агентом в кипящем слое, кроме
воздуха, служит вода, проходя-
щая внутри встроенных теплооб-
менников. Скорость потока в газ-
лифте при пневмотранспорте ад-
сорбента составляет 7—10 м/с.
Качество очищенных продуктов характеризуют следующие данные:
Показатели Продукт 1 Продукт 2
Плотность р420 Температура, °C 0,732 0,785
застывания 24 24
вспышки Содержание, % 128 130
комплексообразующих компонен- тов 96 96
ароматических углеводородов 0,40 0,47
серы 0,01 0,03
воды .— —
механических примесей — .—.
Кислотное число (по КОН), мг/г — 0,002
Показатель преломления 1,4332 1,4338
Пределы выкипания, °C 268—360 279—356
Цвет Бесцветный Бесцветный
При получении бензолсульфонатов из керосинового дистил-
лята (215—245 °C) содержание ароматических углеводородов
-----u,juupuie ьности адсорбции
матнзированную керосиновую
Рнс. 16,16. Изотермы адсорбции нафталиновых (1)
н бензольных (II) углеводородов при 20 °C из
растворов в керосине:
1 — бензол; 2 — а-метйлнафталнн; 3 — |3-метилнаф-
талии; 4 — нафталин; 5 — бензол; л' п объемные
ар
доли углеводородов.
не должно превышать 1,5% • Аро-
матические углеводороды сырья
(до 20%) представлены двумя
группами, отличающимися по ад-
сорбируемости: алкилбензолы и ал-
килнафталины. Для их извлечения
используют силикагели [351, 352].
На рис. 16,16 представлены изотермы
адсорбции двух ароматических групп на
силикагеле. В качестве растворителя при
определении изотерм использовали деаро-
фракцию (215—245 °C). Изотермы адсорбции
этих групп пересекаются при концентрации ароматических углеводородов
3% (об.). В области концентраций от 0 до 3%, (об.) изотерма адсорбции
ароматических углеводородов ряда бензола проходит выше, чем углеводо-
родов ряда нафталина. Испытания показали, что при жидкофазной деаро-
матизации скорости потока в колонне могут достигать 1,8 см/мин.
В процессе деароматизации керосина в качестве побочного продукта по-
лучают 70%-ный концентрат ароматических углеводородов, который может
рассматриваться как товарный продукт. Одновременно удаляется 90% сер-
нистых соединений, а содержание нафталинов понижается приблизительно на
30%. Относительная сорбируемость сернистых соединений подобна адсорби-
руемости бициклических ароматических соединений. Расчеты показали, что
стоимость очистки керосина адсорбционным методом на 40% ниже, чем клас-
сическими методами: серной кислотой и олеумом. Положительными фактора-
ми при адсорбционной очистке является отсутствие отходов и коррозии ап-
паратуры.
Разделение ароматизированных нефтепродуктов может быть
проведено и на стадии десорбции. Так, для разделения тяжелой
нефтяной фракции, типа масляного дистиллята, содержащего
парафиновые, нафтеновые, моноциклические и конденсирован-
ные полициклические углеводороды, рекомендован метод изби-
рательной десорбции [353].
Сначала все углеводороды из жидкой фазы при обычных условиях ад-
сорбируют силикагелем. Затем проводят последовательное вытеснение адсор-
бата четырьмя растворителями с прогрессивно повышающимся адсорбцион-
ным сродством. Алканы Сг,—Сю избирательно десорбируют парафиновые и
нафтеновые углеводороды, алкены С5—Сю (например, диизобутилен)—моно-
циклические ароматические углеводороды, низкомолекулярные ароматические
углеводороды (бензол, толуол)—полициклические ароматические углеводо-
роды, растворитель — кислород-, азот- и серосодержащие соединения.
Адсорбция воды
Силикагели — традиционное средство осушки. Однако избира-
тельность адсорбции воды в присутствии других хорошо сорби-
рующихся компонентов не так резко выражена, как в случае
Избирательность адсорбции на силикагелях
479
цеолитов, и совместную адсорбцию этих примесей необходимо
учитывать при расчете установок осушки. С такой проблемой
инженеры сталкиваются, например при проектировании устано-
вок осушки природных и попутных нефтяных газов со значи-
тельным содержанием высокомолекулярных углеводородов.
На рис. 16,17 представлены кривые адсорбционного равновесия в систе-
ме водяной пар — н-гексан на силикагеле при объемном соотношении компо-
нентов в газовой фазе 1 ; 1 и суммарной концентрации извлекаемых компо-
нентов 0,5% (об.) [354]. Избирательность адсорбции влаги значительна, но
уменьшается с повышением температуры. Коэффициент разделения состав-
ляет: при 20 °C — 32; при 40 °C—16; при 60 °C — 7. Присутствие углеводо-
рода при обычных температурах снижает адсорбционную емкость силикагеля
по воде на 10—20%. Повышение температуры приводит к резкому снижению
влагоемкости силикагеля. В этих условиях освобождается значительное число
вакантных адсорбционных центров на поверхности силикагеля, уменьшается
конкуренция между разнородными молекулами, в связи с чем присутствие-
воды в газе оказывает более слабое влияние на активность силикагеля по па-
рам углеводорода.
Избирательность адсорбции на цеолитах
Цеолиты являются эффективным средством для разделения
многокомпонентных смесей углеводородов. Процессы разделе-
ния основаны на двух свойствах цеолитов: молекулярно-сито-
вом действии и резко выраженной избирательной адсорбции
ненасыщенных углеводородов и полярных молекул. В этом раз-
деле рассмотрены процессы, основанные на избирательности.
Рис. 16,17. Кривые адсорбционного равновесия в системе водяной пар — н-гексан при
соотношении компонентов в газовой фазе 1 : 1 н суммарной концентрации извлекаемых,
компонентов 0,5% (об.).
Рис. 16,18. Изотермы адсорбции этилена на различных адсорбентах при 20 °C:
1 — активный уголь СКТ; 2 — цеолит СаА; 3 —- цеолит NaA; 4 —активный уголь АГ-2;
5 — мелкопористый силикагель.
480
16. Использование избирательности, адсорбции
Адсорбция непредельных углеводородов
Чем ярче выражена ненасыщенность углеводородов, тем выше
избирательность их поглощения из смесей. По возрастающей
селективности адсорбции газообразные углеводороды образуют
следующий ряд: парафины < олефины < ацетиленовые углево-
дороды. Избирательность адсорбции непредельных углеводо-
родов связана с усилением адсорбционных сил за счет специфи-
ческого взаимодействия л-связей углеводорода с гетероионной
решеткой цеолита, накладывающегося на общий фон неспеци-
фического, дисперсионного взаимодействия, характерного для
адсорбции углеводородов всех типов.
Для изотерм адсорбции непредельных углеводородов на цео-
литах свойственно резкое возрастание адсорбционной способно-
сти в начальной области. В этой области изотермы адсорбции
олефинов на цеолитах поднимаются гораздо круче, чем изотер-
мы адсорбции этих углеводородов на мелкопористых углях.
Система этан — этилен. Изотермы адсорбции этилена на
различных адсорбентах представлены на рис. 16,18 [61].
Сравнительная характеристика адсорбционной способности различных
адсорбентов показывает, что при низких парциальных давлениях в отно-
шении адсорбции углеводородов ряда этилена цеолиты превосходят другие
адсорбенты. Приведены [3551 данные по адсорбционному равновесию этан-
этиленовой смеси иа искусственных цеолитах типа NaA при нормальном
давлении и трех температурах. Сопоставление этих данных с коэффициента-
ми разделения, полученными для систем этан — этилен на других адсорбен-
тах, выявляет преимущества цеолитов в отношении избирательности адсорб-
ции олефинов. Средний коэффициент разделения составляет 16,3 при 0 °C и
13,7 при 20 °C, в то время как для силикагелей при обычных температурах
он, как было указано выше, равен всего 2—3. В случае адсорбции смеси
этих углеводородов (1:1) на цеолите СаА прн 25 °C и нормальном давле-
нии коэффициент разделения составил 12,7.
Адсорбционная способность цеолита по смеси этана и этилена при за-
данном общем давлении системы может быть с достаточной для техниче-
ских расчетов точностью выражена как линейная функция от мольного со-
става смеси в адсорбированном состоянии (рис. 16,19).
Высокая поглотительная способность цеолитов по низшим
олефинам в области малых парциальных давлений и концентра-
ций, резко выраженная избирательность при выделении этого
класса углеводородов из Смесей с углеводородами других гомо-
логических рядов, отсутствие адсорбции высших углеводородов
(на цеолитах типа А) создали предпосылки для использования
цеолитов с целью извлечения низших олефинов из ряда нефте-
заводских потоков {356]. Получение этилена высокой чистоты
в случае применения активных углей возможно лишь при от-
сутствии в исходной смеси этана и других парафиновых угле-
водородов [357].
Влажность цеолита значительно снижает его адсорбционную способность
по низшим олефинам, и поэтому при подготовке к стадии адсорбции он
Избирательность адсорбции на цеолитах
481
Рис. 16,19. Адсорбционная способность цеолита
СаА по смеси С2Н, — С2Н8 прн 20 С и нормальном
давлении.
должен быть тщательно обезвожен. На рис. 16,20 представлена кривая, от-
ражающая влияние остаточной влажности гранулированного цеолита СаА
на адсорбционную способность по этилену при 25 °C и 2,7 кПа. График по-
казывает, что для эффективного проведения процесса остаточная влажность
цеолита не должна превышать 1%.
Избирательность адсорбции цеолитов в отношении этилена и их адсорб-
ционную способность в начальной области изотермы можно повысить в ре-
зультате ионного обмена. Это изменение свойств находится в непосредствен-
ной связи с энергией взаимодействия л-электронов этилена с катионами ад-
сорбента [358]. Взаимодействие адсорбат — адсорбент особенно велико в се-
ребряных формах цеолитов. На рис. 16,21 приведена изотерма адсорбции
этилена на цеолите AgX при 35 °C и обмене катиона Na+ на катион Ag+
[359].
При разработке технологических процессов извлечения оле-
финовых углеводородов нужно учитывать каталитическую ак-
тивность последних по отношению к реакциям полимеризации,
проявляющуюся в условиях повышенных давлений и темпера-
тур.
Например, в результате нагревания пропилена до 200 °C при давлении
в несколько десятков атмосфер в автоклаве был обнаружен жидкий продукт,
содержащий углеводороды от С4 до Cis, в основном разветвленные парафи-
ны Се, С9 и С12 [360, 361]. Интенсивность полимеризации убывает в следую-
щем порядке: изо-С4Н8>СзНв>СгН*.
Каталитическая активность цеолитов по
отношению к реакции полимеризации оле-
финов связана с наличием в цеолитах кис-
лых активных центров на основе А1гОз.
Скорость реакции полимеризации пропиле-
на уменьшается в следующей последова-
тельности: CaX>NaX>CaA>NaA>KA.
Очень высокой каталитической активностью
обладает цеолит HY. В его присутствии
реакции полимеризации, дегидрирования и
циклизации олефинов с образованием аро-
матических колец начинают протекать уже
при 150 °C [362].
В связи с этим в тех технологи-
ческих процессах, где в состав ад-
сорбата входят олефины, особое
внимание следует обратить на про-
Рис. 16,20. Зависимость адсорбционной способно-
сти а цеолита СаА по этилену от влажности ад-
сорбента lFa при 20 °C и 26 кПа.
31—1346
482 16. Использование избирательности адсорбции
ведение процесса десорбции в возможно более мягких условиях
с постепенным повышением температуры. Правильно подобран-
ные условия на стадии десорбции обеспечат поддержание ак-
тивности цеолитов при многоцикловой эксплуатации. Так, в
процессе выделения олефинов из метано-водородной фракции
при давлении 0,1—0,7 МПа и температуре 12—15 °C была уста-
новлена стабильность активности цеолита СаА с глуховской гли-
ной в качестве связующего в течение 25 циклов [363].
Исходная фракция имела следующий состав (по объему): 29,20% Н2;.
61,30% СН4; 5,50% С2Н4; 0,34% С2Н6; 1,60% С3Н6; 0,8% С4Н8; 0,16% С4Н6;
1,11% С2Н2; 0,10% СО2; 0,2 мг/л H2S; 0,2 мг/л Н2О. Десорбцию поглощен-
ных углеводородов при этом проводили, продувая слой газообразным азотом
или этановой фракцией (содержание этаиа 92—96%) с малой скоростью
при постепенном повышении температуры путем теплообмена с внешним теп-
лоносителем. Скорость подъема температуры составляла 3—4 °C в 1 мин.
При извлечении таких целевых компонентов, как этилен, из
потока газа особую трудность представляет технологическое
решение стадии десорбции, в результате которой извлеченный
компонент мог бы быть выделен в концентрированном виде.
В связи с этим в некоторых случаях может быть использована
схема, в которой в качестве отдувочного газа применяют газ,,
образующий с этиленом реакционную смесь, например моноок-
сид углерода [364].
Так, в случае извлечения этилена из разбавленных газов нефтеперера-
ботки с последующим получением пропионового альдегида и пропионовой
кислоты в реакциях оксосинтеза десорбцию целесообразно проводить нагре-
ванием адсорбента до 200 °C с продувкой слоя требуемым количеством мо-
нооксида углерода или смесью монооксида углерода с водородом. Опыты,
проведенные в лабораторном адсорбере, показали возможность получения
в этом случае реакционной смеси с содержанием 30% этилена, используемой
для оксосинтеза.
В производстве чистого этилена цеолиты используют также
следующим образом [365]. Для очистки этилена от примеси,
ацетилена применяют орошение ацетоном. Пары ацетона улав-
ливают, пропуская этилен через слой цеолитов. Очистку прово-
дят при повышенном давлении (0,7 МПа). В этих условиях кон-
центрация ацетона снижается с 1 • 10~3% до нуля. Динамическая
активность составляет 2 г/100 г.
Система пропан — пропилен. Избирательность адсорбции
непредельных углеводородов на цеолитах сохраняется и в более
высокомолекулярных смесях, в частности в смеси пропана и
пропилена [366]. Действие дополнительных специфических сил
адсорбции количественно проявляется в разнице теплот адсорб-
ции. В табл. 16-2 приведены данные о теплотах адсорбции цро-
пилена и пропана на основных типах адсорбентов. На углерод-
ной поверхности теплоты адсорбции пропана и пропилена близ-
ки, на силикагеле и цеолитах отмечена значительная разница в
теплотах адсорбции.
Избирательность адсорбции на цеолитах
483
Таблица 16-2. Теплоты адсорбции пропана и пропилена
на различных адсорбентах (в кДж/моль)
Углеводород Графитиро- ванная сажа, 6 = 0,5 Силикагель, 6 = 0,5 Цеолиты
СаА, 6=0,5 по данным хроматографии (0->О)
СаА | NaX
Пропилен 26,0 31,0 50,8 57,6 46,2
Пропан 27,2 21,0 42,3 39,5 32,7
Разница —1,2 10 8,5 18,1 12,5
В соответствии с разницей в теплотах адсорбции находится избиратель-
ность адсорбентов по пропан-пропиленовой смеси. При температуре 25 °C и
нормальном давлении коэффициент разделения составляет иа активном угле
1,1 [367], на мелкопористом силикагеле 3,5 [25], на цеолите СаА 17,0 [61,368].
На рис. 16,22 представлены кривые адсорбционного равновесия пропан-про-
пиленовой смеси на модифицированном цеолите СаА, а также для сравнения
на других типах адсорбентов. Катионный обмен, как уже указано, является
действенным средством изменения избирательности адсорбции. На рис. 16,23
прослеживается, как усиливается преимущественная адсорбция пропилена по
мере увеличения степени замещения натрия на кальций, а затем кальция на
серебро в цеолите типа А.
Высокая избирательность адсорбции пропилена на цеолитах
позволила адсорбционный метод расценивать как конкурентно-
способный с другими известными методами разделения пропан-
пропиленовой смеси: ректификацией, экстракцией растворами
солей металлов, абсорбцией органическими растворителями и
серной кислотой. Разработаны методы промышленного разделе-
ния пропана и пропилена на цеолитах.
Перспективным является разделение пропан-пропиленовой смеси в дви-
жущемся слое сферического цеолита. В ряде работ разделению в движущемся
!Рис. 16.21. Изотерма адсорбции этилена на цеолите AgX при 35 °C.
Рис. 16,22. Кривые адсорбционного равновесия в системе СзНа—СзН6 при 25 °C и нор-
мальном давлении:
1 — цеолит СаА; 2 — мелкопористый силикагель; 3 — активный уголь.
ЗР
484
16. Использование избирательности адсорбции
Рис. 16,23. Участки кривых адсорбционного равновесия в системе С3Н8— С3Нв иа катион*
замещенных формах цеолитов*.
1 —CaNaA (25% Са); 2 — СаА (51% Са); 3 — СаА (5% Ag); 4 — СаА (15% Ag).
Рис. 16,24. Кривые адсорбционного равновесия гексана-гексеновых смесей на адсорбен-
тах:
1 — СаХ; 2 — NaX; 3 — СаА; 4 — рекуперацнонный активный уголь.
слое цеолита NaX подвергались бинарные смеси, в которых содержание не-
предельного углеводорода изменялось от 19,3 до 80,6% (об.). Во всех случаях
степень извлечения пропилена (от его содержания в сырье) достигала 99%,
а чистота после десорбции 99,5%. Удельный расход цеолита, в зависимости
от исходной концентрации пропилена, колебался от 21 до 45 г иа 1 л извле-
ченного углеводорода. Рекомендуемая скорость газового потока в адсорб-
ционной секции колонны непрерывного действия равна 3 см/с. Десорбцию
проводят при 200—210 °C. В качестве динамического агента может быть ис-
пользован диоксид углерода. На адсорбционных установках с движущимся
слоем цеолита могут быть эффективно решены и другие задачи нефтехимии,
например выделение нормальных бутиленов из С4-фракции продукта термо-
крекиига с использованием цеолита СаА или MgA.
Системы с высокомолекулярными олефиновыми углеводоро-
дами. При повышении молекулярной массы углеводородов из-
бирательность адсорбции в системе олефин — парафин с равным
числом атомов углерода в молекуле снижается, но остается дос-
Таблица 16-3. Избирательность смеси я-гексан — гексен-1
на фожазите [369]
Состав газовой фазы (мальн. доля) Состав адсорбированной фазы (мольн. доля) Коэффициент разделения
*свн12 zCeH14
0,22 0,78 0,38 0,62 2,2
0,45 0,55 0,70 0,30 2,8
0,46 0,54 0,64 0,36 2,4
0,46 0,54 0,66 0,32 2,3
0,71 0,29 0,88 0,12 3,1
Избирательность адсорбции на цеолитах
485
Таблица 16-4. Избирательность адсорбции углеводородов
С6 на фожазите
Система* Мольные доли хуже ад- сорбирующегося компонента Коэффици- ент разде- ления
У2
«-Гексан — гексен-1 0,544 0,336 2,4
1-Гексан — гексадиен-1 0,560 0,267 3,5
1,5-Гексадиеи — бензол 0,552 0,170 6,0
н-Гексаи —• гексадиен-1,5 0,586 0,181 6,4
1-Гексен — бензол 0,554 0,072 16
н-Гексан — бензол 0,572 0,055 23
* Первым указан хуже адсорбирующийся компонент.
таточной для осуществления разделения смеси. В табл. 16-3
приведены данные по совместной адсорбции бинарной смеси
«-гексан — гексен-1. Из смеси преимущественно поглощается
последний, причем коэффициент разделения в зависимости от
состава смеси изменяется от 2,2 до 3,1 (97аС; 59,8 кПа). В то
же время олефиновые углеводороды являются хуже адсорби-
рующимся компонентом в смесях с диеновыми и ароматически-
ми углеводородами. Это отчетливо прослеживается на основе
данных таблицы 16-4 (условия те же).
На рис. 16,24 представлены кривые адсорбционного равновесия «-гек-
саи — н-гексеновых смесей [370]. Коэффициент разделения на цеолите СаА со-
ставляет 5, иа цеолите NaX—10; его значение практически не зависит от
положения двойной связи в молекуле олефина. При контакте с цеолитами в
гексенах отмечается значительная изомеризация (перемещение двойной свя-
зи). Изомеризация изогексенов иа цеолитах NaX протекает уже при ком-
натной температуре, а при повышенных температурах (60—100 °C) выделен-
ный цеолитами продукт содержит смесь изомеров с различным положением
двойной связи в молекуле. Только 2,2-диметилбутеи остается практически не-
изменным. Более стабильными являются гексены нормального строения.
В процессе выделения гексеиа-1 при 100°С цеолитом NaX лишь 1—5% пере-
ходит в цис- и транс-изомеры.
Каталитическая активность цеолитов СаА и CdA еще выше. Выделение
гексена-1 цеолитом СаА при 100 °C сопровождается его превращением в гек-
сены с внутренней двойной связью.
Избирательность адсорбции олефиновых углеводородов в
более высокомолекулярных системах продемонстрирована в ра-
ботах ![371]. На рис. 16,25 представлены изотермы адсорбции
нормальных олефиновых углеводородов с числом атомов угле-
рода от 10 до 14, поглощаемых из бинарных растворов в па-
рафиновом углеводороде с одинаковым числом углеродных ато-
мов при 100 °C на цеолите NaX.
С увеличением молекулярной массы избирательность адсорбции олефи-
нов несколько снижается, концентрация парафина в адсорбированной фазе
увеличивается, в связи с чем максимальная адсорбционная способность по
486 16. Использование избирательности адсорбции
Рис. 16,25. Изотермы адсорбции олефиновых уг-
леводородов, поглощаемых цеолитом NaX из би-
нарных растворов:
1 — н-децен — н-декап; 2 — к-додецен-1 — н-доде-
кан: 3— к-тетрадецен-1 — н-тетрадекан; У7 — моль-
ная доля олефинового углеводорода в растворе.
олефину снижается. Она составляет (в
ммоль/г) в системах я-СюНго—я-СюНгг —
0,80; С12Н24 — С12Н26 — 0,60; я-СцНгв —
«-С14Н30 — 0,46. Одиако во всех случаях
адсорбированная фаза обогащается непре-
дельным углеводородом.
Избирательность адсорбции олефинов зависит от химическо-
го состава цеолита. При степени ионного обмена более 30%
цеолиты типа X располагаются в следующем порядке: LiNaX>
>NaX>KNaX>RbNaX>CsNaX. Одновременно обмен катионов
Na+ на катионы Li+ приводит к усилению реакции изомериза-
ции — смещению двойной связи внутрь углеводородной цепи
олефина.
Избирательность адсорбции гексенов на цеолитах использо-
вана для их обогащения или выделения из смесей с парафина-
ми, а также для очистки парафинов от непредельного углеводо-
рода [372].
Так было осуществлено разделение гексаи-гексеновых смесей в слое гра-
нулированных цеолитов высотой 6—50 см при 60 °C в потоке газа-разбави-
теля (азота). Скорость потоков изменяли от 7 до 1,8 м/мин, котщентращпо
нормальных гсксеиов в пределах 220—1170 мг/л. Полученный в качестве ра-
фината н-гексан не содержал даже следов непредельных углеводородов. Со-
держание их в адсорбате было близко к значениям, полученным при изуче-
нии адсорбционного равновесия бинарных смесей изомерных углеводородов
Се (табл. 16-5).
Избирательность адсорбции олефинов на цеолитах исполь-
зуют на установках каталитической дегидрогенизации нормаль-
ных парафинов Си—Си в нормальные олефииы |[373]. Смесь
олефинов и непрореагировавших парафинов разделяют в жид-
Таблица 16.5. Результаты избирательного выделения гексенов
в слое цеолита NaX при 60 °C [370]
Содержание олефинов, %
Исходная смесь
в исходной смеси | в адсорбате
2-Метилпеитан — метилпентены-2 10,1 38,5
15,1 52,7
24,4 70,4
З-Метилпентаи — метилпептеиы-З 20,6 67,0
15,7 56,5
2,3-Диметилбутан — диметилбуте- 14,2 53,2
ны-2,3 21,8 72,8
Избирательность адсорбции на цеолитах
487
кой фазе в изотермических условиях на адсорбционной установ-
ке типа «Олекс». Чистота экстракта-—нормальных олефинов
94%. Рафинат (парафины) возвращают в реактор. Специальное
распределительное устройство автоматически регулирует пото-
ки сырья, десорбепта, экстракта и рафината в системе.
На рис. 16,26 приведена технологическая схема установки, перерабаты-
вающей 35 тыс. т углеводородов в год. Капиталовложения в установку со-
ставляют 4558 тыс. долл., эксплуатационные расходы 4,7 цеит/кг нормальных
олефинов. Процесс может быть использован для переработки широкой фрак-
ции Cs—Cis.
Олефиновые углеводороды, вне зависимости от положения
двойной связи, обладают почти одинаково высокой адсорбируе-
мостью, и разделить их на стадии адсорбции затруднительно.
В то же время ^ис-изомеры все же прочнее удерживаются цео-
литами, чем транс-изомеры, и это свойство используют для их
разделения на стадии двухступенчатой десорбции: транс-изо-
меры удаляют менее полярным, а цпе-изомеры — более поляр-
ным десорбентом. Процесс разделения протекает эффективно,
если олефиновые углеводороды, содержащиеся в сырье, отли-
чаются по числу атомов углерода в молекуле не более, чем на
Рис. 10,26. Схема установки дегидрогенизации нормальных парафинов с адсорбционным
выделением полученных олефинов:
1—нагреватель сырья; 2 — реактор; 3 — сепаратор; 4 — колонна для выделения легких
углеводородов; 5 — сепаратор-копденсатор; 6 — распределитльпое устройство; 7 — адсор-
бер; 8 — колонна для фракционирования рафината; 9, II — конденсаторы; 10 — нагрева-
тель десорбента; 12 — колонна для фракционирования экстракта.
488 16. Использование избирательности адсорбции
2—4. Поэтому широкие фракции, в которых имеются олефииы
Сб—Cis, перед подачей на адсорбционную установку предвари-
тельно фракционируют.
Процесс разделения может быть проведен как в жидкой, так и в паровой
фазах, в широком интервале температур (О—100°С) и давлений (0.1—
7,0 МПа). В качестве адсорбента применяют цеолиты AgX или AgY; кроме
того, могут быть использованы Си-, Au-, Zn-, Cd-, Hg-формы цеолитов типа
X и У. По этой методике удачно решена задача разделения транс- и цис-геп-
тенов в жидкой фазе при обычной температуре. Десорбцию вначале проводят
чистым пентаном, а затем к пентану добавляют возрастающие количества
этилового эфира. Выделение транс-гептена-3 происходит при концентрации
эфира 20%, а цис-гептена-3 — в чистом эфире.
Система этилен — диоксид углерода. В смеси с диоксидом
углерода этилен при адсорбции на цеолитах является хуже
адсорбирующимся компонентом. Это свойство используется для
его очистки в производстве полиэтилена. Обычно получающийся
на нефтехимических заводах этилен содержит до 0,1—0,3% СОг,
что намного превышает нормы, допустимые при Полимеризации.
Изменение содержания в этилене примесей — диоксида углерода в пре-
делах 50—250 см3/м8 и общей серы в пределах 0,8—2.4 мг/м3 — практически
ие оказывает влияния на такие свойства полиэтилена, как прочность, отно-
сительное удлинение при разрыве, морозостойкость н диэлектрическая прони-
цаемость. С другой стороны, диэлектрические потери (tg6) заметно зависят
от содержания примесей. На рис. 16,27 прослежено влияние примесей в эти-
лене на тангенс угла диэлектрических потерь полиэтилена, полученного при
185—190 СС и давлении 12—13 МПа. Первая серия испытаний проводилась
прн постоянном содержании серы (0,9—1,1 мг/м3), вторая серия — при по-
стоянном содержании диоксида углерода (0,012% об.). Полиэтилен, соответ-
ствующий лучшим мировым стандартам (tg 6=2-10-4) может быть получен
при содержании диоксида углерода не выше 30—50 см3/м3 и общей серы ие
выше 0,5 мг/м3.
В табл. 16-6 приведены данные по совместной адсорбции
этилена и диоксида углерода на цеолите СаА i[61J. Коэффи-
циент разделепя при 25 °C, атмосферном давлении и соотноше-
нии компонентов в газовой фазе
1 : 1 составляет 3,75. Высокая энер-
гия адсорбции при поглощении ди-
оксида углерода вызывается взаи-
модействием квадруполя молекулы
с активными центрами — катионами
цеолита [374].
Избирательность адсорбции ди-
оксида углерода была использова-
на для разработки адсорбционного
процесса очистки этилена.
Рис. 16,27. Влияние примесей (а — диоксида угле-
рода. б--сернистых соединений — общей серы),
присутствующих в этилене, на тангенс угла ди-
электрических потерь полиэтилена.
Избирательность адсорбции на цеолитах
489
Таблица 16-6. Адсорбционное равновесие смеси этилен — днокснд углерода
иа цеолите СаА
Состав газовой фазы Состав адсо]эбироввниой фазы Коэффициент разделения
"с2н4 "СОг ХС2Н, *С02
0,224 0,776 0,120 0,880 2,12
0,505 0,495 0.215 0,785 3,75
0,693 0,307 0,323 0,677 4,64
0,758 0,242 0,485 0,515 3,33
Испытания, проведенные на Уфимском заводе синтетического каучука
[375], показали, что при давлении 2,1-—2,3 МПа цеолиты всех испытанных
типов (NaA, СаА, СаХ, NaX) гарантируют полное удаление диоксида угле-
рода из этилена; его содержание в исходном газе колебалось от 0,01 до
0,03%. Адсорбционная способность цеолитов по диоксиду углерода при этом
составляет 1,0—1,2 г/100 г. Оптимальная скорость газового потока (сжатого
газа) 0,6—3,0 м/мин.
Наиболее эффективно очистка проходит в слое цеолитов СаА. Одновре-
менно с удалением диоксида углерода обеспечивается тонкая сероочистка и
осушка газа: содержание серы снижается с 3 до 0,5 мг/м3, точка росы с ми-
нус 56 — минус 64 до минус 69 — минус 75 °C. Десорбцию проводили после
дросселирования давления до атмосферного продувкой азотом или метано-
водородиой смесью при 230—300 °C в течение 2 ч при расходе газа на 1 л
цеолита 250—300 л/ч. Потери этилена в результате совместной адсорбции с
диоксидом углерода составляют 0,74% всего пропущенного этилена. Пони-
жение температуры слоя цеолита при очистке с Зб до 3°С приводит к по-
вышению динамической активности в б раз. За проскоковую концентрацию
принято содержание диоксида углерода 0,005%.
В США процесс очистки этилена от диоксида углерода про-
шел испытания па пилотной установке, состоящей из двух ад-
сорберов диаметром 0,3 м при высоте слоя цеолита СаА 1,14 м
[376].
Рабочие условия процесса: температура 25 °C, давление 1,75 МПа. Де-
сорбцию проводили нагревом цеолита через стенки до 290—300 °C посредст-
вом вмонтированных трубок при одновременном пропускании 14 м3 нагрето-
го метана при давлении 0,17 МПа. Концентрация диоксида углерода в очи-
щенном газе была менее 1 част, на 1 млн. Мольное соотношение этилена и
диоксида углерода в адсорбированной фазе 1,7 : 1. Попытка проводить нагрев
слоя цеолита очищенным этиленом привела к дезактивации адсорбента вслед-
ствие отложения углеродистого осадка, в результате чею продолжительность
стадии очистки до проскока быстро снизилась с 18 до 8 ч.
На основе результатов этих испытаний создана промышленная установка
производительностью по этилену 50 тыс. м3/сут; установка имеет два адсор-
бера (£>=1,5 м, //=10 м) с загрузкой 6,8 т цеолита СаА в каждом. Этилен
очищает в течение 12 ч, при этом поглощается 140 м3 СОг. После снижения
давления температуру слоя цеолитов повышают до 240 °C, через слой в те-
чение 3 ч продувают 140 м3 метана. Затем адсорбер переключают на стадию
охлаждения и, когда температура снизится до 60 °C, от остатка метана осво-
бождаются в медленном токе очищенного этилена. После повышения дав-
ления адсорбер снова переключают иа стадию очистки. Одновременно с
очисткой происходит осушка этжлеиа до точки росы минус 73 °C н удаление
490
16. Использование избирательности адсорбции
некоторых микропримесей. В ряде случаев осушку этилена цеолитами про-
водят как самостоятельный процесс [377].
Цикл процесса очистки этилена от диоксида углерода показан ниже:
Стадия Объемная скорость, м!/(м3-ч) Продолжн- 1 сльность. мин
Адсорбция 210 720
Снижение давления — 15
Десорбция 25—30 180
Охлаждение Без продувки 180
Продувка этиленом Очень малая 10
Повышение давления — 20
На существующих установках возможно осуществить совме-
щение стадий очистки и осушки этилена в одном аппарате
путем замены твердого осушителя — активного оксида алюми-
ния цеолитом i[378j.
Это проверено в опытах на модельном адсорбере с загрузкой 300 см3
цеолита СаА. Параллельно с цеолитовым адсорбером был подключен алюмо-
гелевый. Регенерацию цеолитов проводили в тех же условиях, в каких
обычно проводят иа заводе регенерацию оксида алюминия: температура
185 °C, давление 0,5—0,7 МПа, объемная скорость продувочного газа
600 ч-1, его точка росы минус 40—минус 50 °C, продолжительность 4 ч.
В последующей стадии осушки и очистки этилен пропускали с объемной
скоростью 200 ч-1 при температуре 10—14 °C, давлении 2,2—2,3 МПа. Про-
должительность очистки до проскока составила 44 ч. Несмотря иа мягкие ус-
ловия подготовки адсорбента, цеолит па степени осушки ие уступал алюмо-
гелю; в обоих случаях точка росы снижалась с минус 60 — минус 65 до ми-
нус 73 —минус 74 °C. Одновременно с парами воды в слое цеолита удаля-
лись диоксид углерода и сернистые соединения. После пропуска 1000 объемов
этилена через 1 объем цеолита за 5 ч содержание СОг снизилось с 0,02 до
0,003% (об.), серы — с 4 до 0,5 мг/м3.
Для реконструируемых заводов, на которых до сих пор ие
была предусмотрена тонкая очистка этилена (пирогаза), пред-
ложена схема, представленная на рис. 16,28.
Этилен после гидрирования примеси
ацетилена поступает в один из адсорбе-
ров 1, откуда после осушки и очистки на-
правляется в блок разделения. В колон-
не 2 из него удаляется метаио-водородиая
фракция, затем в колонке 3 — тяжелые уг-
леводороды. Из колонны 3 очищенный эти-
лен направляется иа полимеризацию. Мета-
Рис. 16,28. Схема тонкой очистки этилена:
1 — адсорберы; 2, 3 — ректификационные колонны;
I — метановодородная смесь из цеха разделения
на десорбцию; II —этилен на тонкую очистку:
111 —газы десорбции на очистку нли на факел;
IV — метановодородная смесь на десорбцию или
факел; V — этилен па ректификацию; VI — чистый
этилен; VII — более тяжелые углеводороды.
Избирательность адсорбции, на цеолитах
491
но-водородная фракция нагревается в теплообменнике и используется в ка-
честве продувочного газа для регенерации отработанного адсорбента. Газы
регенерации возвращают в цех предварительной очистки или используют в
качестве топлива.
Система «-бутиловый спирт — «-кротиловый спирт. Избира-
тельность адсорбции по отношению к веществам с кратной
связью сохраняется и в гомологическом ряду спиртов [379].
Адсорбционное равновесие смеси к-бутилового п н-кротилового спиртов
(р-мстилкарбииолы) иа цеолитах исследовано При 150 °C иа проточной уста-
новке. Для этого смесь спиртов с содержанием н-кротилового спирта 1, 2, 3
5, 8 и 10% (масс.) в виде пара пропускали через термостатируемый слой цео-
лита до установления равновесия, наступление которого фиксировали по
идентичности составов входящей и отходящей смесей. Поглощенную смесь
десорбировали в токе азота, конденсировали и определяли ее состав
Результаты изучения адсорбционного равновесия в системе
«-бутиловый — «-кротиловый спирт на цеолитах NaX и СаА
показали, что «-кротиловый спирт избирательно сорбируется
цеолитами обоих типов. Среднее значение коэффициента разде-
ления при нормальном давлении п температуре 150°С (газовая
фаза) на цеолите NaX составляет 7,35. При содержании в ис-
ходной смеси 8% «-кротилового спирта концентрация последне-
го в десорбате, извлеченном из цеолита NaX, составляет 40%.
Рафинат представлял собой химически чистый бутиловый спирт.
Свойства цеолитов в рассмотренной системе могут быть ис-
пользованы в промышленности синтетического каучука.
Системы этилен — ацетилен, монооксид и диоксид углерода—
ацетилен. Очень отчетливо выражена избирательность адсорб-
ции на цеолитах в отношении углеводородов с тройной связью
в молекуле, в частности ацетилена. Высокие селективные свой-
ства цеолитов по отношению к ацетиленовым углеводородам
обусловлены наличием двух видов
взимодействия: ацетилено-катиоп-
иого и ацетилено-анионного [380].
Как показывает рис. 16,29, изотермы
адсорбции ацетилена на цеолитах в обыч-
ных производственных температурных ус-
ловиях (20 °C) уже при давлении 3,3 кПа
достигают значений, близких к максималь-
ной адсорбции. Поэтому адсорбционная
способность цеолитов при небольших кон-
центрациях ацетилена в 2—3 раза выше
адсорбционной способности самых мелко-
пористых активных углей. С повышением
температуры разница в их адсорбционной
способности еще больше.
Рис. 16,29. Изотермы адсорбции ацетилена на
цеолите СаА (пунктиром указаны изотермы при
20 °C на активном угле СКТ и мелкопористом си-
ликагеле мсм>.
р,мм риг.
492
16. Использование избирательности адсорбции
Рис. 16,30. Кривая адсорбционного равновесия си-
стемы CiH<—СцНг на цеолите СаА при 20 "С и
нормальном давлении.
В смеси с этиленом ацетилен преиму-
щественно адсорбируется на цеолитах всех
типов. На рис. 16,30 приведена кривая ад-
сорбционного равновесия системы этилен—
ацетилен на цеолше СаА. В результате
пропускания смеси, содержащей 70% эти-
лена и 30% ацетилена, через слой цеолита
СаА при 20 °C и нормальном давлении пос-
1 ле установления состояния равновесия
обогащение ацетиленом в адсорбированной
фазе достигает 85%. Такому соотношению
компонентов в газовой н адсорбированной
фазах отвечает коэффициент разделения
ть цеолитов по смеси этилен-—ацетилен ив-
13,2. Адсорбционная способиосч
ляется лилейной функцвей состава адсорбированной фазы (рис. 16,31).
Изучение адсорбционных свойств цеолитов по ацетилену
выявило их значительное преимущество перед активными угля-
ми и силикагелями, особенно при низких концентрациях ацети-
лена в газе [381].
При проведении десорбции углеводородов и подготовки цео-
литов к последующей стадии разделения особое внимание дол-
жно быть уделено тщательности их обезвоживания. Остаточная
влажность сильно снижает адсорбционную способность цеоли-
тов (рис. 16,32). Условия обезвоживания могут быть определе-
ны по графикам, приведенным в разделе 12.
Рис. 16,32. Зависимость адсорбционной способности а цеолита СаА по ацетилену от оста-
точной влажности при 20 “С и различном давлении.
Избирательность адсорбции на цеолитах
493
у?,ММ рт.ст.
Рис, 1в,33. Изотермы адсорбции метилацетилена при 20 °C на различных адсорбентах:
1 —активный уголь RKD-IV; 2 — активный уголь СКТ; 3 — Цеолит NaX; 4 — цеолит СаА
(фирмы Линде); 5 — цеолит СаА (СССР); 6 — ректификационный активный уголь; 7 —
цеолит NaA; а — мелкопористый силикагель.
Ацетилен преимущественно сорбируется цеолитами из сме-
сей с большинством компонентов промышленных газов. Так,
коэффициент разделения в системе монооксид углерода — аце-
тилен — цеолит СаА превышает 40. Исключение составляет сис-
тема диоксид углерода — ацетилен. В адсорбированной фазе
при адсорбции смеси диоксид углерода — ацетилен отмечено
слабое повышение концентрации ацетилена по сравнению с га-
зовой фазой (Др— 1,3).
Методом ионного обмена возможно повысить адсорбционную
емкость по ацетилену. Так, с использованием марганецзамещеи-
ных цеолитов типа X на Руставском химическом комбинате
был разработан метод комплексной осушки и очистки от ацети-
лена воздуха перед его низкотемпературным разделением {382].
Система ацетилен — гомологи ацетилена. В процессе конвер-
сии углеводородов наряду с ацетиленом образуется значитель-
ное количество его высших гомологов. Так, при электрокрекин-
ге метана доля высших гомологов достигает 15—20% образо-
вавшихся ацетиленовых соединений.
Одним из перспективных методов очистки ацетилена от выс-
ших гомологов является адсорбционный метод, однако до пос-
леднего времени для этой цели использовали только активные
угли. Исследования, проведенные Мякиненковым [383], выявили
высокие адсорбционные свойства Цеолитов по высшим гомоло-
гам, которые дополнительно к одной тройной связи в молекуле
имеют метильную группу (мети л ацетилен), двойную связь (мо-
494 16. Использование избирательности адсорбции
Рис. 16,34. Кривые адсорбционного равновесия
в системе ацетилен — метилацетнлен при 2(J ’G
на различных адсорбентах:
1 — цеолит NaX; 2 —цеолит СаА: 3 —актив-
ный уголь RKD-IV; 4—рекуперационный ак-
тивный уголь.
иовинилацетилеп) или дополни-
тельную тройную связь (диацети-
лен).
На рис. 16,33 приведены изо-
термы адсорбции метплацетиле-
на иа цеолитах, активных углях
и силикагелях. Тот же характер
изотерм сохраняется для моио-
винилацетилена и диацетилена.
Благодаря крутой форме изотерм адсорбционная способность-
цеолитов уже при 266 Па близка к предельной. Адсорбционная
способность активных углей в этой области давлений не превы-
шает 10% предельной, а силикагеля совсем ничтожна.
Избирательность адсорбции гомологов ацетилена на цеолитах выше, чем
иа активных углях. Это отражается в форме кривых адсорбционного равно-
весия в системе ацетилен — гомологи ацетилена. В качестве примера иа
рис. 16,34 представлены кривые для бинарной системы ацетилен—иетклаце-
тилеи.
Избирательность адсорбции гомологов ацетилена в смеси с ацетиленом,
аа исключением случая адсорбции иа цеолите NaA, убывает в следующем
порядке: диацетилеи — моновииилацетилен — метилацетнлен. При повышении
температуры коэффициент разделения адсорбентов несколько снижается. Во
всех случаях избирательность адсорбции иа цеолитах СаА и NaX выше изби-
рательности адсорбции иа активных углях. Из активных углей наибольшей
избирательной способностью обладает микропористый уголь RKD-IV, приме-
няемый за рубежом для очистки ацетилена от высших гомологов, и ряд со-
ветских опытных образцов.
Разделение многокомпонентной смеси ацетиленовых углеводородов в
слое адсорбента происходит по законам вытеснительной хроматографии. Вна-
чале из адсорбера выходит инертный, газ, затем последовательно ацетилен,
метилацетнлен, моиовинилацетилеи и диацетилен. При адсорбции ацетилено-
вых углеводородов из потока газа в слое адсорбента генерируется большое
количество тепла, вызывающего повышение температуры, а тем самым сни-
жение динамической активности адсорбента. Значения средних теплот ад-
сорбции гомологов ацетилена во всем интервале заполнений, вычисленные из
взостер адсорбции углеводородов, составляют 46,2—71,4 кДж/моль при ад-
сорбции на цеолитах и 21,0—29,4 кДж/моль при адсорбции иа углях.
Особое значение для решения вопроса о возможности исполь-
зования цеолитов для разделения ацетиленовых углеводородов-
приобретает разработка условий десорбции ацетиленовых угле-
водородов в возможно более мягких температурных условиях.
При повышении температуры активность адсорбентов падает
вследствие полимеризации гомологов ацетилена й выделения
углеродистого осадка в порах. Интенсивно полимеризация про-
текает во внутренней структуре цеолитов. В случае использова-
ния активных углей катализаторами полимеризации являются
Избирательность адсорбции на цеолитах
495
отдельные ингредиенты золы. Для промышленного разделения
ацетиленовых углеводородов разработаны методы десорбции,
которые снижали бы до минимума возможность отложения по-
лимерных соединений в порах адсорбента.
Один из таких методов [384] заключается в продувке адсорбента влаж-
ным (<р = 0,6—0,9) газом, нагретым до 60—80 °C. Влага адсорбируется и
вытесняет из цеолита поглощенные ирнмеси, которые переходят в газовую
фазу н уносятся с потоком. Нагретый газ может содержать большие массы
вытеснителя — паров воды: до 0,15 кг/м1 при 60 °C и 0,55 кг/м3 при 80 °C.
Такой метод десорбции был проверен на лабораторном адсорбере, загружен-
ном цеолитами СаА н NaX.
Через цеолит пропускали смесь следующего состава: 0,5% метнлацети-
лена, 0,5% моновниилацетилена, 0,5% днацетилена г: остальное — азот. При
обычной термической адсорбции активность цеолитов падает через несколько
циклов и уже к 10-му циклу составляет 25% первоначальной. При десорбции
влажным воздухом активность вплоть до 100-го цикла находилась на уровне
70% первоначальной. Применение продувки слоя цеолитов азотом, содержа-
щим 2% кислорода, при 500°C полностью восстанавливало активность. Метод
особенно перспективен, если адсорбат ие представляет ценности как само-
стоятельный продукт и его выделение из газа десорбции не обязательно.
Адсорбция ароматических углеводородов
Ароматические углеводороды имеют высокие критические диа-
метры молекул (выше 0,6 им), вследствие чего не сорбируются
цеодитами СаА, поэтому их выделение из смеси с углеводорода-
ми других классов (например, с нормальными парафинами) мо-
жет быть осуществлено на основе молекулярио-ситового эффек-
та. В этом случае ароматические углеводороды, молекулы ко-
торых ие могут проникнуть через окна цеолитов в адсорбцион-
ные полости, остаются в фильтрате, а адсорбированная фаза
содержит концентрат нормальных парафинов.
Напротив, в тех случаях, когда входные окна достаточно ши-
роки, чтобы сделать возможным вход молекул в адсорбционные
полости, цеолиты проявляются резко выраженную избиратель-
ность по отношению к ароматическим углеводородам. Это свой-
ство используют как для промышленных, так и для аналитиче-
ских целей.
Особое значение адсорбцион-
ный метод выделения ароматиче-
ских углеводородов имеет для
смеси углеводородов с близкими
физико-химическими коистаита-
Гис. 16,35. Изотермы адсорбции бензола из его
разбавленных растворов в циклогексане на
различных адсорбентах при 25 С:
1 — активный уголь СКТ; 2 — цеолит NaX; 3 —
цеолит СаХ; 4 — активный уголь БАУ; 5 —
С-уголь; в — мелколористый силикагель; 7 —
цеолит NaA; 6—крупнопористый силикагель.
496
16. Использование избирательности адсорбции
^С6Н(
цеолите NaX (1)
при 80 °C и нор-
Рис. 16,36. Кривые адсорбционного равновесия си-
стемы циклогексан — бензол на ......v
U мелкопористом силикагеле (2)
мальном давлении.
и циклогек-
ми, например бензола
сана. Вследствие близости темпера-
тур кипения бензола и циклогекса-
на разделение пх простои ректифи-
кацией невозможно, и должны быть
применены специальные методы.
На рис. 16,35 представлены изо-
термы адсорбции бензола из его
разбавленных растворов в цикло-
гексане на различных адсорбентах при 25 °C [385].
В отличие от изотерм для силикатных адсорбентов, изотермы адсорбции
бензола на цеолитах NaX и СаХ поднимаются очень круто и адсорбционная
способность цеолитов близка к адсорбционной способности наиболее мелко-
пористых активных углей СКТ. Аналогичный характер имеют изотермы ад-
сорбции бензола в паровой фазе; при температуре 20ъС и давлении 266 Па
адсорбционная способность цеолита СаХ (на 100 г) достигает 15 г, цеолита
NaX—17,5 г. Максимальное количество сорбированного бензола при 5,3 кПа
составляет соответственно 20 и 23 г. Изотермы адсорбции циклогексана на
цеолитах имеют более пологий характер и лежат значительно ниже значений
адсорбционной способности по бензолу при равном давлении. Так, при 20 °C
и 266 Па адсорбционная способность цеолита NaX по циклогексану состав-
ляет лишь 5 г/100 г.
Повышение температуры в адсорбере до температуры кипения бензола
(80 °C) снижает адсорбционную способность по бензолу только иа 20—25%;
даже при 150 °C цеолиты продолжают поглощать до 13 г/100 г бензола.
Избирательность адсорбции цеолитов и силикагелей в систе-
ме бензол — циклогексан была изучена в работе [386]. Кривая
адсорбционного равновесия, представленная на рис. 16,36, ука-
Таблнца 16-7. Избирательность адсорбции в системе
н-гексан — бензол на цеолите типа Y
Температу- ра, ’С Давление. кПа Мольная доля лучше адсорби- рующегося компонента — бензола Коэффициент разделения
*С,Н,
69 26 0,26 0,94 44
78 25 0,28 0,91 26
97 24 0,29 0,90 23
97 59 0,014 0,81 18
97 60 0,11 0,86 52
97 59 0,29 0,95 47
97 60 0,43 0,94 23
97 60 0,74 0,97 13
Избирательность адсорбции на цеолитах
497
зывает на ярко выраженную избирательность адсорбции бензо-
ла на цеолитах. Так, при содержании в паровой фазе 10% бен-
зола обогащение адсорбированной фазы бензолом достигает-
84%. В соответственных условиях адсорбированная силикаге-
лем фаза содержит только 33% бензола. Вычисленные отсюда,
коэффициенты разделения составляют для цеолита 47, а для.
силикагеля только 5.
В табл. 16-7 приведены данные по совместной адсорбции,
смеси н-гексан — бензол на цеолите типа Y. Коэффициент раз-
деления в этой системе в зависимости от условий опыта изме-
няется от 13 до 52.
Показатели очистки циклогексана в динамических условиях были изуче-
ны Лузяниным в лабораторном тсрмостатируемом адсорбере (D=l см. Я=
25 см), загруженном цеолитами NaX с размером частиц 0.5—1,0 мм. Опыты-
проводили при двух температурных режимах: 20°C (жидкая фаза) и 80 °C
(паровая фаза) иа трех смесях с различной концентрацией бензола: 0,5, 1,0
и 5%. В очищенном продукте спектрографически бензола обнаружено не-
было, следовательно, степень чистоты циклогексана превышала 99,999%.
Цеолиты типа X предложено применять при получении рас-
творителя пищевого масла [387]. Подлежащий очистке полупро-
дукт представляет собой смесь неароматических углеводородов;
с числом атомов углерода в молекуле 6—8. Его промывают во-
дой до содержания сульфолана менее 0,5% и очищают от при-
месей ароматических углеводородов в слое цеолита с диамет-
ром входных окон в адсорбционные полости более 0,7 нм. Усло-
вия деароматизации могут изменяться в широких пределах:
температура 10—95°С, давление 0,1—3,5 МПа, объемная ско-
рость 0,05—2,0 ч-1.
Адсорбционная установка состоит из двух попеременно работающих ад-
сорберов. Продукты, выделяющиеся при регенерации адсорбента, пропускают
через абсорбционную колонну для улавливания сульфолана в блоке экстрак-
ции и через колонну улавливания бензола в блоке дистилляции.
Широко применяется метод адсорбционной деароматизации
для получения высококачественного реактивного топлива [388]..
При этом особое внимание _ 2
уделяется подбору эффектив- | ууууС]__________»[ |_Рзф™т......
ного десорбепта. В качестве 1 ,_____, Дес«р8вп
последнего обычно применяют |*Н---Iе (ин,)
аммиак. Чтобы сократить рас- 3
ход тепла на десорбцию, в тех-
нологических схемах обеспе-
чивают теплообмен между по-
токами. Одна из таких схем
представлена на рис. 16,37.
Рис. 16,37. Схема установки деароматиза-
ции реактивного топлива:
1 — нагреватель; 2, 3 — адсорберы; 4 — рек-
тификационная колонна; Б — хододжльнкк.
Экстракт ароматических
углеводородов
-498 76. Использование избирательности адсорбции
Реактивное топливо с пределами кипения 150—290 °C нагревают в теп-
лообменнике 1, испаряют и в парах пропускают через слой цеолита NaX
в адсорбере 2, где избирательно поглощают ароматические углеводороды. Из
адсорбера выходит термически стабильное реактивное топливо с хорошим
люминометрическим числом. Десорбцию проводят, вводя аммиак в насыщен-
ный ароматическими углеводородами цеолит в адсорбере 3. Первую порцию
(5—20%) десорбата, которая близка по составу к исходному сырью, возвра-
щают в качестве рсциркулята в процесс. Последующие порции десорбата
являются концентратом ароматических углеводородов, и их выводят из си-
стемы.
Рсциркулят вводят в нижнюю часть тарельчатой колонны 4, выполняю-
щей роль не только сепаратора аммиака, ио и своеобразного теплообменника.
Сверху в колонну подают свежее холодное сырье. В результате теплообмена
пары десорбированных углеводородов конденсируются, а сырье при этом на-
гревается до 80—П0сС. Если свежего сырья для охлаждения десорбата не-
достаточно, часть выходящих из колонны углеводородных газов охлаждают
в теплообменнике 5 и примешивают к сырью. Аммиак отводят из верхней
'части колонны. Подогретую таким образом смесь углеводородов направляют
«а деароматизацию. Колонна заменяет систему из теплообменника и сепара-
тора. Схема может быть использована и в других процессах адсорбционного
^разделения, в частности при депарафинизации нефтяных фракций цеолитами.
Применительно к деароматизации нефтяных фракций разра-
ботана схема, в которой стадии адсорбции и десорбции разби-
вают на два периода [389].
В первом периоде стадии адсорбции процесс ведут до проскока лучше
.адсорбирующегося компонента, получая рафинат высокой чистоты. Во вто-
ром периоде получают фракцию рафината, загрязненную извлекаемым ком-
понентом. Затем адсорбер переключают на стадию вытеснительной десорб-
ции. В первый период стадии десорбции с помощью вытеснителя из адсорби-
рованной фазы удаляют адсорбат. Период заканчивается до момента появ-
ления вытеснителя в выходящем потоке. Затем во второй период десорбцию
.вытеснителя проводят в потоке нечистой фракпии рафината, полученной во
втором периоде стадии адсорбции.
Выходящую из адсорбера смесь разделяют иа вытеснитель и углеводо-
роды. Процесс заканчивают, когда содержание вытеснителя в выходящем по-
токе достигнет 6% всего вытеснителя, поданного в адсорбер. Для того, что-
чбы процесс разделения был непрерывным, устанавливают не менее двух ад-
сорберов. В качестве адсорбента применяют синтетические цеолиты типа X,
в качестве вытеснителя, как уже говорилось, — аммиак.
Очистку нормального гексана от примеси бензола (8%) про-
водят в периодическом адсорбере высотой 1,35 м, заполненном
цеолитом СаХ, при атмосферном давлении и температуре 340°C.
Схема установки приведена на рис. 16,38.
Сырье (смесь СбН9 и w-CeHis) нагревают в печи 1 и пропускают через
адсорбер 2 с цеолитом СаХ. Бензол адсорбируется цеолитами. В первый пе-
риод отбирают чистый н-гексан и собирают его в емкости 6. Во второй пе-
риод адсорбционной стадии загрязненную бензолом фракцию направляют в
«емкость 5. Затем адсорбер переключают на стадию десорбции. В первый пе-
риод стадии десорбеит — аммиак из сепаратора 7 пропускают через слой
цеолита в адсорбере 2 в направлении, обратном направлению потока в ста-
дии адсорбции.
Первые порции выходящей смеси углеводородов по составу близки к ис-
ходной смеси и их из емкости 8 возвращают иа разделение. Затем в сепара-
тор направляют смесь бензола ц аммиака. Давление в сепараторе поддержи-
Избирательность адсорбции на цеолитах
499
вают таким, чтобы аммиак в последующем подавался в адсорбер без ком-
прессора. Период заканчивается прежде, чем появится аммиак в выходящем-
потоке. Во второй период смесь бензола н н-гексана из емкости 5 с помощью
иасоса 4 подают в печь 3, где она нагревается и испаряется в адсорбер 2.
Выходящий поток разделяют в сепараторе па аммиак и обогащенный бен-
зином продукт.
Аналогичный принцип может быть также реализован в движущемся слое-
адсорбента. Схема потоков в этом случае приведена в нижнем правом углу
рис. 16,38.
Ароматические углеводороды с разным числом ароматичес-
ких колец в молекуле по-разпому адсорбируются на цеолитах*
СаХ и NaX; это свойство Майер и Шмайенгер использовали в.
Смесь' C6Ht4+ СВН6
Рис. 16,38. Схемы установки и процесса адсорбционной деароматизации нефтяных фрак-
днй:
1. 3 — печи; 2 — адсорбер; 4 —насос; 5. б, 3 — емкости; 7 —сепаратор.
500 16. Использование избирательности адсорбции
аналитических целях для изучения состава высококипящих неф-
тяных фракций [390].
В технике избирательность адсорбции углеводородов цеоли-
тами типа X используют для глубокой очистки различных рас-
творителей, доочистки нормальных парафинов, выделенных из
нефтяных фракций цеолитом СаА, получения высококачествен-
ного дизельного топлива для сверхзвукового авиационного тран-
спорта, концентрирования бензола и толуола из углеводородных
смесей [391].
Для выделения ароматических углеводородов из смсссй с
углеводородами других гомологических рядов могут применять-
ся также цеолиты типа Y, предпочтительно в катнонированной
форме [392].
Адсорбция в системе диоксид углерода — сульфид водорода
Цеолиты являются уникальными сорбентами, избирательно по-
глощающими сульфид водорода из смеси с диоксидом углеро-
да. Кривая адсорбционного равновесия в системе диоксид угле-
рода — сульфид водорода на цеолите типа СаА представлена
на рис. 16,39. При соотношении компонентов в газовой фазе
1 : 1 адсорбированная фаза в обычных условиях в случае цеоли-
та СаА содержит только 12% диоксида углерода. Коэффициент
разделения в системе диоксид углерода — сульфид водорода —
цеолит СаА составляет 7—9. В связи с этим стало возможным
полностью удалить из газа вредные примеси (влагу, сульфид
водорода) и до любой заданной степени балластную примесь —
диоксид углерода. Особенно большое значение цеолитовые уста-
новки приобретают для очистки природных и промышленных га-
зов с соотношением компонентов СО2: H2S>3. Подробно вопрос
сероочистки газов рассмотрен в гл. 13.
При расчете адсорбционных установок, предназначенных для сероочистки
природных и попутных нефтяных газов, необходимо учитывать влияние выс-
ших углеводородов, подавляющих адсорбцию сульфида водорода. Исследова-
ние адсорбционного равновесия в системе сероводород — н-бутаи, проведен-
ное на цеолитах NaX, показало, что адсорбированная фаза обогащается
«-бутаном. Чем выше температура, тем меньше концентрация H2S в адсор-
бированной фазе. Соответственно увеличивается коэффициент разделения
£393]:
t, °C 0 26 53 75
КР 1,56 2,36 4,30 6,20
Снижение относительного содержания сульфида водорода в адсорбиро-
ванной фазе при повышении температуры отмечено также в системе про-
тай— сульфид водорода — цеолит. Следовательно, применение низких темпе-
ратур при сероочистке газов предпочтительнее не только для повышения ад-
сорбционной способности по извлекаемым примесям, ио н для улучшения
избирательных свойств адсорбента.
Избирательность адсорбции на цеолитах
501
Рис. 16,39. Кривая адсорбционного равновесия си-
схемы диоксид углерода — сульфид водорода на
цеолите СаА при 25 °C и нормальном давлении. '
Метод сероочистки цеолитами
применим также к жидким нефте- g
продуктам: пропан-бутановой фрак- =>>
ции, бензинам, лигроинам и т. п.
В пропане, наряду с сульфидом водо- q ?
рода, присутствует метилмеркаптан, в бута-
не—метил- и этилмеркаптаны, в бензи-
нах— другие сильфиды н полисульфиды. д'
Обычно нефтяные продукты получают в
результате ректификации, поэтому каждо-
му потону сопутствуют определенные, со-
ответствующие по выкипаемости, сернистые соединения. С увеличением ин-
тервала температуры выкипания фракции понижается избирательность ад-
сорбции сернистых соединений. Наоборот, чем уже границы кипения фрак-
ции, тем выше избирательность цеолитов по сернистым соединениям, тем
эффективнее процесс очистки.
j Адсорбция в некоторых других системах
I В состав дымовых газов обычно входят азот, кислород, диоксид
* углерода и диоксид серы. Адсорбируемость диоксидов углерода
и серы на цеолитах намного превосходит адсорбируемость кис-
лорода и азота. На рис. 16,40 представлены изотермы адсорб-
ции диоксида серы на Н-морден,ите [394]. Присутствие азота
незначительно сказывается на адсорбционной способности по
диоксиду серы. В то же время, в присутствии диоксида углерода
в газе адсорбционная способность значительно снижается.
р ,< Па
Рис. 16,40. Изотермы адсорбции диоксида серы на н-мордеиите:
1 — чистый диоксид серы при 0 °C; 2 — то же, из смеси с азотом; 3 — то же, из смеси
с диоксидом углерода; 4 — чистый диоксид серы при 100 °C; 5 — то же, нз смеси с азо-
лом; б — то же, из смеси с диоксидом углерода.
Гис. 16,41. Кривые адсорбционного равновесия систем диоксид углерода — диоксид серь»
м азот — диоксид серы при нормальном давлении на я-мордените.
502
16. Использование избирательности адсорбции
щается диоксид углерода. С
Рис. 16,42. Кривые адсорбционного равновесия
в системе диоксид азота — хлор на различных
адсорбентах:
1, 2, 3 — цеолит НКЭ соответственно при 25, 50
и 75 °C; 4 — активные угли СКТ и АР при
25 °C; 5 — цеолит NalO при 25 °C.
Температурный уровень ока-
зывает решающее влияние на из-
бирательность поглощения диок-
сида серы в присутствии диок-
сида углерода. Как показывают
кривые адсорбционного равнове-
сия (рис. 16,41), при низких тем-
пературах из такой смеси морде-
нитом преимущественно погло-
повышением температуры проис-
ходит инверсия избирательности, из смеси начинает избира-
тельно поглощаться диоксид серы, причем при 100 °C коэффи-
циент разделения близок к 9*. Одновременно при высоких тем-
пературах в большей мере подавляется совместная адсорбция
азота и кислорода.
Высокая избирательность адсорбции диоксида серы на мор-
дените, стойкость цеолита этого типа позволяют оптимистиче-
ски оценить возможности использования в некоторых случаях
кислотостойких форм цеолитов для очистки дымовых газов.
Одним из методов получения нитритов калия и натрия явля-
ется конверсия соответствующих хлоридов концентрированной
азотной кислотой, жидкими или газообразными оксидами азота.
Отходящие газы в этом производстве содержат хлор, диоксид
азота и хлорнитрозил. Разделение смеси указанных компонен-
тов может быть решено адсорбцией.
На рис. 16,42 представлены кривые адсорбционного равно-
весия в системе диоксид азота — хлор на различных адсорбен-
тах (395]. Особенно высокой избирательностью адсорбции обла-
дает водородная форма калиевого эрионита НКЭ. Значение
коэффициента разделения смеси NO2—С12 при 25 °C в исследо-
ванном диапазоне соотношений компонентов колеблются от 35
до 120; для газовых углей они составляют только 6—11.
Усиление избирательности адсорбции на цеолитах связано
со специфическим взаимодействием полярных молекул NO2 с
частично протонизированными атомами водорода поверхностных
гидроксильных групп адсорбента, в то время как адсорбция
аполярных молекул хлора ограничивается лишь дисперсионны-
* Причины, изменения селективности цеолитов в этой системе не ясны. Воз-
можно, что при повышенных температурах происходит хемосорбция триокси-
да серы, что приводит к увеличению общего количества поглощенных соеди-
нений серы. (Прим, ред.)
Очистка изопентана от примеси н-пентана
503
ми силами адсорбат — адсорбент. Также избирательно погло-
щаются полярные молекулы нитрозила. Все компоненты рас-
сматриваемой системы десорбируются из цеолита при 200 °C,
степень десорбции составляет 95—98%.
Кислотостойкий цеолит — эрионит не разрушается в атмо-
сфере оксидов и хлороксидов азота при многоцикловой эксплуа-
тации. Использование избирательности адсорбции диоксида азо-
та из смеси с хлором на кислотостойких цеолитах не ограничи-
вается рассматриваемым случаем, а может служить и для
решения ряда других технологических задач.
Рассмотренные примеры применения адсорбентов не охва-
тывают всю область адсорбционного разделения веществ. Мно-
гочисленные случаи использования селективности адсорбции на
активных углях, силикагелях, цеолитах, изменения ее путем
ионного обмена и другими методами приводятся в отечествен-
ных и зарубежных обзорных работах.
17
ИСПОЛЬЗОВАНИЕ
МОЛЕКУЛЯРНО-СИТОВЫХ
СВОЙСТВ ЦЕОЛИТОВ
Однородная кристаллическая структура природных и синтети-
ческих цеолитов, наличие входных окон строго определенного
размера дают возможность использовать их для разделения ве-
ществ на основе разницы в размерах и форме молекул. Особое
значение новые адсорбенты приобрели для разделения смесей,
компоненты которых близки по физико-химическим константам
(например, по температуре кипения и замерзания, плотности),
когда обычные методы не могут обеспечить требуемую степень
разделения.
Очистка изопентана от примеси «-пентана
В качестве примера использования молекулярно-ситовых свойств
цеолитов для лабораторной и технологической практики может
.служить разделение изо- и «-пентана [396—399].
504
17. Использование молекулярно-ситовых свойств цеолитов
Рис. 17,1. Изотермы адсорбции к-пентана иа
цеолите СаА.
В составе газовых бензинов некото-
рых месторождений Советского Союза
содержится до 25% изопентана, кото-
рый после выделения может быть ис-
пользован в качестве сырья для произ-
водства изопренового каучука и других
химических продуктов. Обеспечить чет-
кое разделение изо- и я-пентана обыч-
ными методами ректификации в про-
мышленных установках трудно вслед-
ствие близости температур кипения этих
углеводородов (соответственно 28 и
36 °C). Кальциевая форма цеолитов типа
А с размером входных окон 0,5 нм яви-
лась эффективным средством для целей
тонкой очистки изопентана (критиче-
ский диаметр молекул 0,56 нм) от при-
месей н-пентана (критический диаметр
молекул 0,49 нм).
Изотермы адсорбции н-пентана, представленные на рис. 17.1,
имеют обычный характер изотерм физической адсорбции. В то
же время, адсорбция изопентана, которая связана с поглоще-
нием на внешней поверхности адсорбента и во вторичной пори-
стой структуре, при температурах 20—60 °C не превышает
1 г/100 г. Адсорбционная способность цеолита СаА в этих тем-
пературных пределах уже при 1,33 кПа достигает величины
(9—10 г/100 г), близкой к максимальной активности (11 —12 г
на 100 г). В тех же условиях изопентана сорбируется менее
0,3 г/100 г. При давлении 39,9 кПа адсорбционная способность по
изопентану не превышает 0,8 г/100 г. Подобно изопентану, на
цеолите слабо адсорбируются и другие углеводороды с крити-
ческим диаметром молекулы выше 0,5 нм. По данным i[400],
100 г гранулированного цеолита СаА при 25 °C сорбируют 0,02—
0,04 г 2-метилбутана, 0,50—0,55 г 2-метилпентана, 1,2—1,8 г
метилциклогексана, 1,00—1,87 г толуола; при 100 °C — 0,45—
0,53 г метилциклогексана, 0,13—
0,52 г толуола, 0,10—0,39 г цикло-
гексана.
Эти данные характеризуют одно
из важнейших свойств цеолитов —
разделять смеси углеводородов с
различной структурой молекул.
Рис. 17t2. Октановое число этилированных нор-
мальных парафиновых углеводородов (по иссле-
довательскому методу) OZa.
Снижение токсичности выбросов автомобилей 505
Уже сравнение изотерм изо- и н-пентана на цеолитах СаА ука-
зывает на возможность их четкого разделения в адсорбционном
процессе.
Снижение токсичности выбросов автомобилей
В автомобилестроении укрепилась тенденция создания машин
с мощными двигателями, работающими при высокой степени
сжатия. Для таких двигателей необходим высококачественный
бензин. До последнего времени повышение качества (октано-
вого числа) прямогонных бензинов достигалось добавкой этило-
вой жидкости, содержащей тетраэтилсвинец. При горении топ-
лива это соединение разлагается, и пары свинца выделяются в
атмосферу с выхлопными газами, резко повышая их токсич-
ность.
Перспективный путь получения моторных топлив (или их
компонентов) высокого качества и с пониженной токсичностью
заключается в удалении низкооктановых парафиновых углево-
дородов путем молекулярно-ситового разделения бензиновых
фракций на цеолитах типа СаА.
В состав авиационных и автомобильных бензинов входят углеводороды
различных классов: ароматические, нафтеновые, парафиновые и олефиновые.
Самыми низкими октановыми числами обладают нормальные парафины, при-
чем с увеличением молекулярной массы их антидетонационные свойства рез-
ко ухудшаются. Из нормальных углеводородов только н-бутан имеет анти-
детонационную характеристику, соответствующую современным бензинам. На
рис. 17.2 приведен график, характеризующий октановые числа этилирован-
ных нормальных парафинов* в зависимости от числа атомов углерода в мо-
лекуле.
Удаление нормальных парафиновых углеводородов (парафи-
нов) из бензинов обычными методами, например фракциониро-
ванием, является чрезвычайно трудной проблемой вследствие
близости их температур кипения и температур кипения других
углеводородов.
Процесс депарафинизации на цеолите СаА, основан на его
молекулярно-ситовом действии. Этот цеолит адсорбирует все
нормальные парафины с критическим диаметром молекул
0,49 нм, и не поглощает ароматические, нафтеновые и изопара-
финовые углеводороды, которые не могут проникнуть в адсорб-
ционные полости через входные «окна» цеолита.
При адсорбции низших нормальных парафиновых углеводородов проис-
ходит заполнение всего объема адсорбционных полостей. С повышением чис-
ла атомов углерода в молекуле нормального парафина уменьшается число
молекул, адсорбируемых в одной полости цеолита СаА: при 25 °C макси-
мальное число молекул н-CiHio, приходящихся на одну полость, составит 5,
* Этилированное топливо, этилированный углеводород и т. п. — продукты,
содержащие добавки тетраэтилсвинца. (Прим, ред.)
506
17. Использование молекулярно-ситовых свойств цеолитов
Рис. 17,3. Зависимость числа молекул нормаль-
ных парафиновых углеводородов п иа одну по-
лость цеолита при насыщении от числа атомов-
углерода в молекуле углеводорода пс-
«-С10Н22 — только 2, а для высокомолекулярных парафинов с числом атомов
углерода больше 15 приближается к 1 (рис. 17,3) [401]. С повышением тем-
пературы число молекул углеводорода, адсорбирующихся в одной полости,
уменьшается.
Скорость поглощения нормальных парафиновых углеводородов при про-
ведении адсорбции в паровой фазе зависит от основных параметров процес-
са: температуры, давления, скорости паров, концентрации извлекаемого ком-
понента, его молекулярной массы. Высота работающего слоя при выделении
индивидуальных углеводородов: я-гексана из фракции 60—80 °C и я-гептана
из фракции 96—120 °C при скорости потока 20 см/с составляет около 10 см.
Значения высоты работающего слоя для рассматриваемого случая [402] пред-
ставлены на рис. 17.4.
Бензины прямой перегонки нефти
Самое высокое содержание нормальных парафиновых углеводо-
родов приходится на газовые бензины и бензины прямой пере-
гонки. В табл. 17-1 приведены результаты выделения нормаль-
ных парафинов из бензинов различного происхождения (403,
404].
Повышение октанового числа прямогонных бензинов в ре-
зультате пропускания через слой цеолита СаА может достигать
25 пунктов. По мере увеличения молекулярной массы нормаль-
ных парафинов их содержание в бензинах прямой перегонки и
в газовых бензинах уменьшается, но одновременно в связи с
уменьшением октанового числа увеличивается их отрицательное
действие на моторные свойства бензина. Вследствие этого повы-
шение октанового числа выделенных фракций прямогонного
бензина на всех участках разгонки остается приблизительно по-
стоянным. Об этом свидетельствуют данные табл. 17-2.
В Советском Союзе проведе-
ны опыты по депарафинизации
бензинов различных месторожде-
ний. Так, Митрофанов и Мир-
ский [405] подвергли адсорбци-
онному разделению в слое цео-
Рнс. 17,4. Зависимость высоты работающего
слоя £о от скорости потока при извлечении
«-гептана (1) и «-гексана (2) нз бензиновых
фракций в слое промышленного цеолита СаА
при температуре 180 °C н концентрации угле-
водорода 22% (об.).
Снижение токсичности выбросов автомобилей 507
Таблица 17-1. Повышение октанового числа бензинов в чистом виде (1)
и с добавкой 0,8 мл/л ТЭС (2) в результате
депарафинизации цеолитом СаА
Бензин До адсорб- ции После ад- сорбции Повышение окта- нового числа
1 1 2 1 2 1 1 2
Широкая фракция платфор- 87,1 97,1 95,3 101,1 8,2 4,0
минг-бензина Бензин термического крекинга 76,0 88,5 89,1 96,1 13,1 7,6
Тяжелый бензин каталитиче- 88,0 94,0 94,4 98,8 6,4 4,8
ского крекинга Легкий бензин каталитического 93,8 99,2 95,7 100,6 1,9 1,4
крекинга Авиаалкилат каталитического 91,1 104,4 33,6 — 0,5 —
крекинга Полимер-бензин из пропилена 93,0 98,0 93,6 98,6 0,6 0,6
Легкий прямогонный бензин 69,0 87,1 83,7 96,6 14,7 9,5
Тяжелый прямогонный бензин 44,1 66,4 70,6 — 26,5 —
литов СаА, синтезированных ими, прямогонные бензины гроз-
ненских нефтей. В результате удаления нормальных парафино-
вых углеводородов октановое число прямогонных бензинов по-
вышалось на 11—20 пунктов. Содержание нормальных пара-
финов в десорбате превышало 95%.
Положительные результаты получены при депарафинизации
бензинов прямой гонки башкирской [406], долинской [407], та-
тарской [408] нефтей. Адсорбционная депарафинизация бензи-
нов, выделенных из конденсатов, не имеющих самостоятельного
использования вследствие низкой октановой характеристики,
позволяет получить компоненты автомобильных бензинов А-72
и А-76 [409].
Таблица 17-2. Изменение октанового- числа фракций легкого бензина
прямой гонки в результате разделения иа цеолите СаА [403]
Фракция (интервал кипения), °C Выход фракции, % (об.) Октановое число Фракция (интервал кипения), °C Выход фракции, % (об.) Октановое число
по иссле- дователь- скому методу с ТЭС повы- шение по иссле- дователь- скому методу с ТЭС повы- шение
4—30 22 101,5 10,2 60—65 11 96,2 9,8
30—39 И 96,6 6,5 65—74 11 99,0 10,2
39—50 12 98,0 8,0 74—80 4 94,4 4,7
50—55 12 96,6 9,0 80—100 6 92,2 Н,7
55—60 11 97,0 12,0
508 17. Использование молекулярно-ситовых свойств цеолитов
Исследование процесса выделения нормальных парафинов из двух фрак-
ций бензинов прямой гонки башкирской нефти выполнено в стеклянных ад-
сорберах диаметром от 26 до 38 мм с высотой слоя цеолитов от 0,87 до
1,66 м [406]. Адсорбцию проводили при 155 °C, десорбцию — при 250—300 °C
и небольшом разрежении. Удаление нормальных гексана, гептана, октана и
нонана из фракций 70—150 °C повышает октановое число на 17,5 пунктов
(с 48 до 65,5), а при удалении я-гексана и н-гептана из фракции 69—94 °C
повышение октанового числа достигает 21,5 пункта (с 54,5 до 76,0). Концен-
трация нормальных парафинов в адсорбате составляла 97—98%.
Удаление нормальных парафиновых углеводородов из фракций с точкой
начала кипения 62—85 °C, выделенных из бензина долинской нефти, прове-
дено на пилотной адсорбционной установке [407]. Термостатируемый адсор-
бер (D=67 мм, А = 4 м) был заполнен цеолитом СаА. Скорость паров состав-
ляла 2,1—2,4 м/мин, температура 150—200 °C, давление 0,5—0,6 МПа. Де-
сорбцию проводили сбросом давления с последующим вакуумированием. Эта
стадия определяла продолжительность цикла в целом и в оптимальном слу-
чае составляла 20 мин.
Равновесная адсорбционная емкость цеолита по нормальным парафинам
С4—Се, входящим в состав фракции, при 150 °C составляла 5,1, а при
200 °C — 4,6 г/100 г; в циклическом процессе адсорбционная емкость снижа-
лась в течение первых циклов на 20%, после чего стабилизировалась. Со-
держание парафинов С4—С6 в исходной фракции составляло 43,2%, а после
пропускания через цеолит — 22,2%. Состав продукта после депарафинизации
был близок к составу продукта изомеризации фракции при 380 °C. Октановое
число фракции по моторному методу при адсорбционной депарафинизации
повышено с 72,4 до 84,0. Адсорбат содержал: 9,1% Н-С4Н10, 52% Я-С5Н12,
20,7% н-СбНц.
Не менее успешно проведена депарафинизация бензиновых
фракций из татарских нефтей. Повышение октанового числа
после пропускания через цеолит СаА составило 15—17 пунктов
[408].
Таким же способом были удалены нормальные парафины из
бензиновых фракций карадагского конденсата (60—105, 105—
140, 140—200 и 60—200 °C) в результате их пропускания через
слой цеолита СаА при температуре на 25—30 °C выше темпера-
туры конца кипения фракции и подаче сырья с объемной ско-
ростью 0,5 ч-1 до проскока нормальных парафинов [409]. В ре-
зультате повышения октанового числа на 12,4—16,5 пунктов
бензиновые фракции 60—150 °C, 105—140 °C и широкая фракция
60—200 °C приобретают антидетонационную характеристи-
ку, соответствующую автомобильным бензинам.
Выделение из адсорбата узких фракций, соответствующих по
температурам кипения индивидуальным парафинам, открыло
путь к получению последних на базе природного сырья, осу-
ществляемому одновременно с получением высокооктанового
бензина. О достаточно высокой чистоте выделенных нормаль-
ных парафинов свидетельствует совпадение их физико-химиче-
ских констант с табличными значениями констант углеводоро-
дов [405].
Результаты процесса извлечения нормальных парафинов из
бензиновых фракций карадагского конденсата приведены ниже:
Снижение токсичности выбросов автомобилей
509
Фракция, °C 60—105 105—140 140—200 60—200
Содержание парафинов, % 50,5 53,5
перед адсорбцией — —
после адсорбции 29,5 40,9 — —-
изменение Октановое число 21,0 13,6 — —
перед адсорбцией 66,8 50,0 33,4 50,9 Т
после адсорбции 78,4 66,2 49,6 67,4*-
изменение 12,4 16,2 16,2 16,5
Выход депарафинизирован- ного бензина, % 74,5 79,0 — 75,0
* С добавкой ТЭС октановое число 77,9.
При проектировании адсорбционных установок депарафини-
зации бензинов внимание должно быть уделено выбору опти-
мальной скорости паров в адсорбере. Этот показатель опреде?»
ляет как габариты аппарата, так и время цикла. Ниже приведе-
ны данные по продолжительности стадии разделения смеси'
н-гептан—толуол при 200 °C и динамической активности слоя
цеолита СаА высотой 1 м в зависимости от скорости подач»
смеси {410]:
Расход смеси, см3/ч 104 207 410
Скорость подачи смеси, см/ч 15,0 29,6 58,6
Скорость потока паров, м/мин 0,07 0,14 0,28
Объемная скорость, ч-1 0,15 0,30 0,60
Продолжительность стадии адсорб- 75 29 10
ции, мин Динамическая активность, г/100 г 5,6 5,3 2,9
Повышение скорости подачи смеси с 15 до 58,6 см/ч сни-
жает динамическую активность с 5,6 до 2,9 г/100 г. Таким ско-
ростям подачи смеси соответствуют скорости потока паров уг-
леводородов 0,07 и 0,28 л/(см2-мин), или 0,7—2,8 м/мин.
Бензины термической и каталитической
переработки нефтепродуктов
В бензинах каталитического риформинга общее содержание па-
рафинов, независимо от режима процесса, составляет около
10%. После удаления нормальных парафинов цеолитом СаА.
продукт обладает высокими антидетонационными свойствами,,
что способствует применению цеолитов для переработки рифор-
минг-бензинов. Повышение октанового числа бензина платфор-
минга достигает 8 пунктов, каталитического крекинга 6—7 пунк-
тов, термического крекинга 13 пунктов. Бензины, полученные
процессах алкилирования и полимеризации низших олефинов,
W 9?
510 17. Использование молекулярно-ситовых свойств цеолитов
Таблица 17-3. Изменение октанового числа фракций продукта
каталитического риформинга после обработки в слое цеолитов СаА
Фракция (интервал [ кипения), °C Выход фракции, % (об.) Октановое число Фракция (интервал кипения), °C Выход фракции, % (об.) Октановое число
по иссле- дователь- скому методу с ТЭС повы- шение по иссле- дователь- скому методу с ТЭС повы- шение
20—55 11 101,0 5,7 105—120 11 101,0 3,4
55—72 11 96,4 7,1 120-128 10 102,6 1,2
71—85 10 91,0 6,7 128—155 10 103,5 0,7
85—88 10 101,0 6,7 155—175 7 104,0 0,3
88—105 10 100,0 3,8 175—230 10 106,0 1,2
виду малого содержания нормальных парафинов подвергать ад-
сорбционному разделению нецелесообразно.
Эффект повышения октанового числа во фракциях бензинов
;риформинга, выкипающих при разных температурах, неодина-
ков, в отличие от переработки прямогонных бензинов.
Данные табл. 17-3 показывают, что наибольший эффект повышения окта-
гнового числа наблюдается для фракций, выкипающих до 88 °C, со значитель-
ным содержанием и-пентана и н-гексана. Повышение октанового числа сильно
.ароматизированных фракций, выкипающих при температуре выше 120 °C, не
превышает 0,3—1,2 пункта. В то же время октановое число высококипящих
этилированных фракций (выше 100 °C) соответствует требованиям, предъ-
являемым к высококачественному моторному топливу. Поэтому на тех уста-
новках, где продукт каталитического риформинга подлежит разделению на
легкую и тяжелую части, адсорбционной депарафинизации целесообразно
подвергать только легкую часть, выкипающую при температуре до 120 °C.
.Это позволяет на 40% сократить расход цеолита при приблизительно рав-
ном качестве продукта. Капитальные затраты на установки адсорбционной
депарафинизации бензина окупаются за 1/$ года.
При поглощении углеводородов в адсорбере протекает про-
цесс вытеснительной адсорбции углеводородов: углеводород с
'-большей молекулярной массой постепенно вытесняет углеводо-
род с меньшей молекулярной массой, в результате чего адсор-
бированная фаза обогащается тяжелыми углеводородами. При
разделении пентан-гексановой фракции первым в выходящем
потоке изопарафиновых углеводородов появляется н-пентан.
Этот момент (момент проскока) фиксируется с помощью хрома-
тографического анализа, который ведут непрерывно, после чего
адсорбер отключают от линии подачи сырья. Момент проскока
может быть определен также по повышению температуры в кон-
•цевом участке слоя. Если в сырье присутствует примесь н-бута-
ша, его появление в выходящем продукте предшествует появле-
нию н-пентана. Выходные кривые нормальных парафинов в
^процессе адсорбционного разделения пентан-гексановой фракции
'бензина с небольшим содержанием бутанов представлены на
фис. 17.5.
Снижение токсичности выбросов автомобилей
5111
Рис. 17,5. Выходные кривые нормальных парафи-
новых углеводородов при адсорбционном разде-
лении пентан-гексаиовой фракции с примесью
я-бутана (N — относительная концентрация нор-
мальных парафиновых углеводородов в выходя-
щем из адсорбера потоке).
Лабораторные опыты, извлечения н-
гептана (30%) из смеси с толуолом (70%)
в изотермических условиях, в стеклянном
адсорбере (Z) = 3 см, L=1 м), заполнен-
ном гранулированным цеолитом СаА
(rfrp=l,6 мм, /ггр = 3 мм) с добавкой 20%
инертного связующего, показали, что рав-
новесная адсорбционная способность при
200 °C составляет 8,6 г/100 г [410].
Большинство адсорбционных процессов денормализации уг-
леводородных фракций проводят в три стадии, образующие
цикл: адсорбция нормальных парафинов в полостях цеолитов
СаА — продувка слоя для удаления вещества из свободного
объема аппарата — десорбция нормальных парафинов. Четкость
разделения, а следовательно и чистота выделенных фракций вы-
ше при проведении процесса в паровой фазе, что и определил»'
более широкое развитие различных парофазных процессов.
На первых установках депарафинизации ориентировались на использова-
ние в качестве десорбента тех веществ, которые легко отделяются в после-
дующем от десорбата. Это направление не потеряло своего значения и до
настоящего времени. Однако анализ изотерм различных нормальных пара-
финовых углеводородов на цеолите СаА (см. рис. 17,6) показал, что, по.
крайней мере, бензиновые углеводороды Cs — С9 могут быть успешно десорби-
рованы в результате обычного снижения давления.
В промышленных условиях вполне достижимо разрежение,,
соответствующее остаточному давлению в системе (1,3 кПа).
В связи с этим фирма Бритиш Петролеум видоизменила свой’
вариант денормализации с вытеснительной десорбцией MS-1 на
вариант с десорбцией под вакуумом MS-2. Процесс MS-2 — изо-
термический, трехступенчатый, циклический, осуществляемый в
паровой фазе (411].
Процесс основан на разной адсорбци-
онной способности цеолита при разных
давлениях. Рабочий интервал давлений
разбит на три ступени, (см. рис. 17,7). Па-
рообразное сырье пропускают через слой
цеолитов при давлении выше атмосферно-
го. Нормальные парафины, присутствую-
щие в сырье избирательно адсорбируются
до равновесного насыщения в конкретных
условиях процесса.
Адсорбированное количество нормаль-
ных парафинов приближенно вычисляют
как разницу в равновесной адсорбционной
Рис. 17,6. Изотермы адсорбции нормальных пара-
финовых углеводородов на цеолите СаА при
400 °C.
$12
17. Использование молекулярно-ситовых свойств цеолитов
сти адсорбента. Выделяется также
Рис. 17,7. Равновесие изменения адсорбцион-
ной способности иа ступенях процесса MS-2.
способности при давлении на стадии ад-
сорбции и при давлении на стадии пред-
шествующей десорбции: a=A!+A2.
На стадии адсорбции из адсорбера
выходит денормализованный продукт.
После окончания этой стадии давление
в аппарате снижают, и при этом уда-
ляются вещества, заполняющие свобод-
ный объем и поглощенные на поверхно-
некоторое количество десорбированных
нормальных парафинов, определяемое разностью равновесной адсорбционной
способностью цеолита при давлении на стадии адсорбции и на второй (про-
межуточной) стадии Ар Десорбированные нормальные парафины выполняют
функции динамического агента и способствуют удалению углеводородов дру-
.гих классов. Продукт, выходящий во время этой стадии из адсорбера, приме-
шивают в качестве рецикла к сырью, чтобы повысить общую степень извле-
чения. Аналогичным образом поступают на второй ступени.
Наконец давление в аппарате доводят до самого низкого уровня. При
этом нормальные парафины из адсорбционных полостей удаляют в соответ-
ствии с разницей между равновесной адсорбционной способностью на про-
межуточной стадии и стадии десорбции.
Схема установки M.S-2 приведена на рис. 17,8. При выборе конструкции
адсорберов особое внимание уделяют правильному и равномерному распре-
делению потоков при переключении их со стадии на стадию. Цеолиты под-
лежат замене через 10 лет эксплуатации.
На адсорбционной установке MS-2 в Руре денормализации подвергали
шрямогонную фракцию, выкипающую в пределах 96—153 °C и содержащую
29,3% нормальных парафинов С?—Сю. Другие показатели фракции: удель-
ная плотность при 15 °C 0,745 г/см3; содержание общей серы 8, влаги
4 част, на 1 млн.; бромное число 0,24 г Вг на 100 г; содержание ароматиче-
ских углеводородов 11,2%- Содержание нормальных парафинов в денормали-
зованном продукте составило 5%, в десорбате 99,5.%.
В результате разделения пентан-гексановой фракции с общим содержани-
ем нормальных парафинов 17,6% на пилотной установке MS-2 получен про-
чие. 17,8. Схема установки MS-2:
Л, 8 — емкости; 2 — теплообменник; 3 — нагреватель сырья; 4 — адсорбер; 5 — конденса-
тор продукта второй стадии; 6 — конденсатор продукта стадии десорбции; 7 — холодиль-
щик; 8 — сепаратор.
Снижение токсичности выбросов автомобилей
513
дукт, содержащий всего 1 % нормальных парафинов; его октановое число по
исследовательскому методу (в чистом виде) повысилось с 82,9 до 88,4 пунк-
тов. Концентрация парафинов в десорбате составила 96%.
Капиталовложения на установку MS-2 производительностью 200 тыс.т/год
99%-ной денормализованной фракции С5—С8 (одновременно получают
50 тыс.т/год побочного продукта-—нормальных парафинов чистотой 96%)
составляют (включая стоимость цеолитов) 2,7 млн. долл. Ежегодные эксплуа-
тационные затраты на пополнение цеолитов и химикалии оценены в
13 тыс. долл. Расходные коэффициенты на 1 кг денормализованного продук-
та: топливо 0,52 кВт-ч, электроэнергия 0,02 кВт-ч; пар 0,001 кг, охлаждаю-
щая вода 0,1 л.
Комбинированный процесс изомеризации
и денормализации углеводородных фракций
В промышленности разработаны непрерывные процессы выделе-
ния нормальных парафиновых углеводородов обработкой угле-
водородных смесей в слое цеолита СаА. Иногда из исходного
сырья вначале на фракционирующих колоннах выделяют узкую
фракцию, которую в дальнейшем подвергают адсорбционной де-
парафинизации. Особенно эффективны установки с блоками
изомеризации выделенных нормальных парафиновых углеводо-
родов. Один из таких процессов осуществляется в изотермиче-
ских условиях в аппаратуре, изготовленной из обычных мате-
риалов, с применением неподвижного слоя цеолита.
Схема опытной установки изомеризации, предназначенной
для выделения нормальных парафинов из пентан-гексановой
фракции, представлена на рис. 17,9 [412, 413].
Производительность установки по сырью составляет 1000 м3/сут. Смесь
углеводородов СбН12—CeHu подвергают каталитической гидроизомеризации
Рис. 17,9. Схема установки гидроизомеризации с адсорбционным разделением продуктов:
1 — подогреватель; 2 — адсорбер на стадии разделения; 3 — адсорбер на стадии десорб-
ции; 4 — вакуум-насос; 5 — подогреватель; 6 — реактор; 7 — сепаратор; 8 — колонна ста-
билизации; 9—оборудование для регенерации; 10 — адсорбер на стадии регенерации.
33—1346
514
17. Использование молекулярно-ситовых свойств цеолитов
Снижение токсичности выбросов автомобилей
515
при 250—375 °C в присутствии платинового катализатора. Нормальные па-
рафиновые углеводороды выделяют из сырья в паровой фазе. Для этого
сырье нагревают при давлении 0,52 МПа в подогревателе 1, испаряют и про-
пускают при 300 °C через адсорбер 2, заполненный цеолитами СаА. Загруз-
ка цеолита в каждый адсорбер составляет 3,6 т.
Вследствие выделения в процессе адсорбции тепла температура в слое
цеолитов повышается приблизительно на 30 °C (с 300 до 330 °C). Теплота
адсорбции парафиновых углеводородов нормального строения на цеолитах
составляет 714—1050 кДж/кг [414]. Процесс прекращают сразу после появ-
ления «-пентана в потоке рафината. После этого адсорбер переключают иа
стадию десорбции. Десорбцию проводят в две стадии: сначала давление в
адсорбере 3 снижают почти до атмосферного (0,12 МПа), а затем систему
откачивают с помощью шестицилиндрового вакуум-насоса 4 до остаточного
давления 6,65 кПа. Диаметр рабочего цилиндра насоса составляет 96,5 см.
Давление на линии всасывания компрессора равно 4,65 кПа.
Такой метод десорбции позволяет обеспечить быструю смену операций,
сократить время цикла до 10 мин при длительности рабочей стадии 5 мин.
Распределение продолжительности остальных операций таково: снижение
давления 20 с, перекрывание клапанов 6 с, вакуумирование 4 мин 18 с, пере-
крывание клапанов 6 с, создание давления 10 с (итого 5 мин).
На установках используют быстродействующие клапаны, которые по кон-
струкции аналогичны клапанам, применяемым на установках дегидрирования
«-бутана. Остаточное содержание н-пентана в адсорбированной фазе при тем-
пературе 300 °C и давлении 6,65 кПа не превышает 1 г/100 г.
Десорбированные нормальные парафины смешивают с водородом, нагре-
вают в подогревателе 5 и изомеризуют в реакторе 6. Жидкие углеводороды,
выделенные в сепараторе 7, после стабилизации в колонне 8 смешивают с ис-
ходным сырьем и возвращают в адсорбер 2. Горючие газы из стабилизацион-
ной колонны удаляют сверху.
Процесс депарафинизации бензинов с вакуумной десорбцией подробно
описан в литературе [415—417].
В процессе эксплуатации активность цеолитов, особенно при переработке
фракций, содержащих высокомолекулярные углеводороды, снижается вслед-
ствие отложения углеродистого осадка. Для восстановления активности ка-
тализатора в систему включают третий адсорбер 10, подключенный к блоку
регенерации 9, как это показано на рис. 17,9. Выжиг «кокса» проводят ана-
логично восстановлению платинового катализатора процессов гидроформин-
га — продувкой кислородсодержащим газом при температурах ниже порога
термической стабильности цеолита. На первых установках депарафинизации
бензиновых фракций регенерацию цеолита было предусмотрено проводить
через каждые 14 сут эксплуатации в 3 стадии: 1) нагрев цеолита циркуля-
ционным инертным газом до 400 °C (15 ч); 2) выжиг углеродистых образо-
ваний (42 ч); 3) охлаждение цеолита циркуляционным газом до температу-
ры 300 °C (13 ч).
Смесь, поступающая на установку (31,7 т/ч), состоит из двух частей:
17,8 т/ч исходной пентан-гексановой фракции, содержащей 45% нормальных
парафиновых углеводородов, и 13,9 т/ч продукта изомеризации, содержащего
34% нормальных парафиновых углеводородов. Концентрация нормальных па-
рафиновых углеводородов в смеси, таким образом, составляет 40%. После
пропускания через слой цеолитов поток содержит менее одного процента нор-
мальных парафиновых углеводородов, в основном пентана.
Концентрация нормальных парафиновых углеводородов в адсорбате со-
ставляет 86%; в равновесных условиях она достигает 95%. На установке
изомеризации выделяется 213 м3 горючего газа, основными компонентами
которого являются водород, метан, этан и пропан. Выход газа составляет
12 м3 на 1 т перерабатываемого сырья. С другой стороны, в процессе
изомеризации затрачивается на каждую тонну сырья 10 м3 водородсодержа-
щего газа с концентрацией водорода 86%. Адсорбционная способность цео-
лита СаА по нормальным парафиновым углеводородам С5—С6 при темпе-
ратуре 300 °C и давлении 0,52 МПа составляет 6 г/100 г.
Сопоставление экономических показателей двух вариантов
переработки пентан-гексановой фракции (изомеризация +
фракционирование и изомеризация-)-адсорбция) показало
преимущества варианта с применением синтетических цеолитов
[418].
Расчет проведен для установки производительностью по сырью 400 т/сут.
В связи с тем, что в случае адсорбционного удаления нормальных парафинов
потребность в четком фракционировании углеводородов отпадает, блок фрак-
ционирования имеет меньшие габариты, благодаря чему капитальные затра-
ты на установку разделения, несмотря на необходимость сооружения до-
полнительного адсорбционного блока, снижаются на 5%. Одновременно,
вследствие полного вывода из цикла нормальных парафинов, капитальные
затраты на блок изомеризации уменьшаются на 17%. По статье эксплуата-
ционных расходов второй вариант позволяет осуществить экономию на 25%
в основном за счет экономии пара. Дополнительным источником экономии
служит увеличение на 10% выхода продукта изомеризации при небольшом в
стоимостном выражении понижении выхода горючего газа.
Октановое число этилированного продукта в случае разде-
ления углеводородов с применением цеолитов составляет 97,6,
что на 1,9 пункта выше, чем при чистом фракционировании.
Стоимость повышения октанового числа 1 м3 продукта на 1
пункт в случае адсорбционного разделения снижается на 32%.
Следует отметить, что затраты на первоначальную загрузку цео-
литов составляют 9% всей суммы капитальных затрат при стои-
мости адсорбента 2,2 долл, за 1 кг.
Если при разделении системы с небольшим числом компо-
нентов экономические показатели процессов фракционирования
и адсорбционного разделения сопоставимы, то по мере услож-
нения системы и увеличения в ее составе числа углеводородов
преимущество адсорбционного метода становится более ощути-
мым, а в ряде случаев он является единственно приемлемым
для технического осуществления.
В настоящее время метод изомеризации нормальных бута-
на, пентана и гексана получил широкое распространение не
только в США, но и в Европе.
Так, в 1969 г. крупная промышленная установка изомеризации пентана
и гексана по методу «Пенекс» пущена в эксплуатацию в г. Манитце (Ита-
лия). Преимущества установок, оборудованных адсорбционными блоками,
позволяют надеяться на широкое применение цеолитов для целей выделения
и возвращения на изомеризацию непрореагировавших нормальных парафинов
[419].
В большинстве зарубежных установок депарафинизацию бензиновых
фракций проводят в паровой фазе в изотермическом режиме при 300—315 °C
по методу «Изосив», в котором, как указано выше, десорбцию проводят за
счет снижения давления [420]. Ряд установок «Изосив» 'перерабатывают лег-
кое углеводородное сырье [421]. В Вильмингтрне (Калифорния, США)’ про-
изводят растворитель — нормальный гексан, а депарафинизированный продукт
используют в качестве добавки-ж моторному топливу.. В: ФРГ иа нсфтсперс-
33*
516 17. Использование молекулярно-ситовых свойств цеолитов
рабатывающем заводе «Фризла» (г. Эмден) основным продуктом депарафи-
низации является рафинат, содержащий углеводороды С5—С? и используе-
мый как компонент высокооктанового бензина; экстракт применяют в качест-
ве растворителя.
В Японии промышленная установка построена в г. Иоккайши [422]. Она
предназначена для разделения бензиновой фракции С5—С7 с получением нор-
мальных парафиновых углеводородов, используемых в качестве растворителя,
а также изопарафиновых и циклических углеводородов.
Десорбция низкомолекулярных нормальных парафиновых
углеводородов из слоя цеолитов может быть проведена не толь-
ко вакуумированием, но и иным путем. В процессе «Тексако»
стадию десорбции ведут продувкой парами легких углеводоро-
дов, легко разделяемых с десорбатом простым фракционирова-
нием [423]. Обычно в качестве динамического агента применяют
изопентан.
Схема установки, рассчитанной на переработку 44 м3, или около 30 т/ч
продукта изомеризации гексановой фракции с содержанием 22% «-гексана,
представлена на рис. 17,10. Установка состоит из двух вертикальных адсор-
беров с загрузкой 54 т цеолита СаА в каждом. Диаметр адсорберов 2,45 м,
их высота 21,35 м, высота слоя цеолита 17,0 м. Продолжительность цикла
составляет 20 мин. Процесс разделения проводят при 260 °C, давлении
0,8 МПа и скорости паров углеводородов в адсорбере 7 м/мин. Динамиче-
ская активность слоя цеолита в этих условиях около 4 г/100 г.
После снижения давления до 0,24 МПа при температуре 305 °C прово-
дят десорбцию нормальных парафиновых углеводородов потоком изопента-
на, нагретого до 430 °C, который пропускают через слой со скоростью
21 м/мин. Расход изопентана составляет 31,8 м3/ч, или 0,28 объема на
1 объем адсорбента в 1 ч. Таким образом, удельный расход изопентана со-
ставляет 0,72 объема на 1 объем сырья, или 3,3 объема на 1 объем десорбиро-
ванных нормальных парафиновых углеводородов. На установке получают
34,4 м3/ч изомерных и 9,7 м3/ч нормальных углеводородов. Смесь углеводо-
родов, выделяющихся при снижении давления (10,3% сырья), может быть
Рис. 17,10. Схема установки адсорбционного разделения продукта изомеризации по мето-
ду «Тексако:
1 — адсорбер иа стадии разделения; 2 — адсорбер иа стадии десорбции; 3 — фракциони-
рующая колоииа; I — сырье; II — продукт; III — изопентан; IV — десорбент и десорбат;
V— десорбированные нормальные парафиновые углеводороды.
Снижение токсичности выбросов автомобилей 517
возвращена на повторное разделение; в этом случае производительность про-
цесса снижается до 0,44 объема сырья на 1 объем цеолита в час.
На установке производительностью 1600 л гексановой фракции в 1 ч
стабильное разделение углеводородов проводят в течение 2000 циклов.
В среднем чистота изогексана составляла 98,5%, я-гексана 94,7%.
Изопентан является недостаточно эффективным десорбентом, вследствие
чего процесс приходится проводить в неизотермическом режиме. Отсюда вы-
текают основные недостатки метода — необходимость большого расхода цир-
кулирующего изопентана, достаточно сложное оборудование и управление
процессом разделения. Иногда в качестве десорбента применяют я-бутап или
я-пентан.
Стоимость разделения гексановой фракции при производительности уста-
новки «Тексако» 1060 м3 сырья в сутки оценена в 1,72 долл./м3. При расчете
калькуляции цена 1 кг цеолита принята в 2,2 долл., а срок его службы —
1 год.
Проблема уменьшения загрязненности биосферы свинцом
при одновременно высоком качестве моторного топлива успешно
решается в комбинированных установках «тотального процесса
изомеризации» (TJP), в котором сочетаются метод изомериза-
ции «Хайзомер» фирмы Шелл и денормализации продукта
«Изосив» фирмы Юнион Карбайд [424]. Процесс TJP эксплуа-
тируется на трех нефтеперерабатывающих заводах: в Японии,
ФРГ, Швейцарии. Цеолиты в этом процессе используют не толь-
ко как адсорбент, но и как компонент катализатора реакций
изомеризации.
Комбинированный процесс обеспечивает повышение октано-
вого числа на 15—20 пунктов, причем «чувствительность» про-
дукта (разница в октановом числе по исследовательскому и
моторному методу) невелика: около 3 пунктов против 10 пунк-
тов у продукта каталитического риформинга. Схема процесса
приведена на рис. 17,11.
Процесс проводят при постоянных давлении (1,4—3,5 МПа) и темпера-
туре (205—370 °C) по одному нз двух вариантов в зависимости от того, ка-
кую цель преследуют: обеспечение максимального выхода продукта или до-
стижение его максимального октанового числа.
В первом случае сырье, нагретое в теплообменнике 1, подвергают денор-
мализации в адсорбере 2 с цеолитом СаА. Неадсорбированиые углеводороды
освобождают от легких, газообразных компонентов в стабилизаторе 5 и ис-
пользуют их как компонент бензина. Десорбат подвергают изомеризации в
реакторе 3, который заполнен цеолитовым катализатором, содержащим бла-
городный металл. Такой катализатор более устойчив в присутствии паров
воды и сернистых ядов, чем обычные низкотемпературные катализаторы, ак-
тивированные галоидами. Если стадию десорбции проводят не вакуумом, а в
потоке десорбента, последний вместе с десорбатом пропускают через реак-
тор, конденсируют в холодильнике 4, отделяют в сепараторе 7, нагревают
в теплообменнике 6 и возвращают в адсорбер, находящийся на стадии де-
сорбции. Конденсат, содержащий непрореагировавшие нормальные парафины
Ci+, в качестве рецикла добавляют к сырью, направляемому на денормали-
зацию.
По второму варианту, когда все сырье вначале изомеризуют, низкоокта-
новые гептаны частично подвергают крекингу; при этом увеличивается соот-
ношение углеводородов Cs : Св, следовательно и октановое число; однако
вследствие реакций крекинга снижается общий выход продукта.
.518 17. Использование молекулярно-ситовых свойств цеолитов Г
Рис. 17,11. Схема комплексной установки переработки легкой бензиновой фракции:
1, 6 — нагреватели; 2—секция адсорбционного разделения; 3 — секция каталитической
изомеризации; 4 — конденсатор; 5 — фракционирующая колонна; 6 — теплообменник; 7 —
секция выделения десорбента.
Рис. 17,12. Схема установки получения высококачественного моторного топлива:
1 — фракционирующая колонна; 2 — реактор гидроизомеризации остатка; 4, 5 — адсорбе-
ры с цеолитом СаА; 6 — сборник высокооктанового продукта.
Десорбат нормальных
парафинов '
парафинов
б[
При переработке пеитан-гексановой фракции по комбинированному мето-
ду в промышленных условиях октановое число в чистом виде по исследова-
тельскому методу повышается с 72,2 до 88,5 пунктов, а по моторному методу
с 68,7 до 85,6 пунктов.
В ГрозНИИ [425] предложен процесс, в котором стадии изо-
меризации на платиноцеолитном катализаторе (платина, нане-
сенная ионным обменом на цеолит типа Y) и денормализации
продукта изомеризации с помощью цеолитов типа А тесно увя-
заны между собой: обе стадии проводят в паровой фазе при
одинаковых давлениях (3 МПа) и температурах (280—350°C),
причем в качестве десорбента используют водородсодержащий
газ из блока изомеризации. В свою очередь, водород в реактор
изомеризации поступает из цеха риформинга. Цеолитовый ката-
лизатор не требует добавки промоторов, а также глубокой очи-
стки сырья и водородсодержащего газа от примесей. Горячий
продукт десорбции в паровой фазе вместе с водородом и рецир-
кулятом возвращаются в блок изомеризации. Окончательный
продукт содержит не более 1,5% нормальных парафинов.
Принципиальная технологическая схема следующая. Сырье нагревают в
теплообменнике и печи, смешивают с горячим десорбционным потоком водо-
рода и нормальных парафиновых углеводородов из десорбера и подают в
реактор изомеризации. Затем проводят адсорбцию нормальных парафинов
цеолитами с последующим охлаждением и конденсацией денормализованного
продукта в специальном блоке. После стабилизации продукта во фракцио-
нирующей колонне получают высококачественный компонент бензина.
Для обеспечения непрерывности процесса адсорберы периодически (через
5—10 мин) переключают со стадии адсорбции на стадию десорбции. Регене-
рацию адсорбента и катализатора проводят одни раз в год. Выход компонен-
та моторного топлива достигает 97% от его содержания в сырье. Октановое
число смеси углеводородов Cs—Св в результате комбинированного процесса
Снижение токсичности выбросов автомобилей
519
повышается приблизительно иа 20 пунктов и по исследовательскому методу
в чистом виде достигает 87,4, а с добавкой ТЭС 102,7 пунктов. При перера-
ботке Самотлорской и ромашкинской нефтей на заводе производительностью
12 млн. т сырой нефти в год возможно направить 450—520 тыс. т пентан-
гексановой фракции на изомеризацию с адсорбционным выделением нормаль-
ных парафинов.
Капиталовложения в процесс и себестоимость 1 т получен-
ного продукта сопоставимы с соответствующими показателями
промышленных установок только изомеризации пентан-гексано-
вой фракции. Комбинированный процесс отвечает требованиям
безотходной технологии: в нем практически отсутствуют выбро-
сы в атмосферу и сточные воды, а на стадии изомеризации
вредные промоторы не применяют.
Получение высокооктановых бензинов
Адсорбционные цеолитовые установки применяют в комбиниро-
ванной схеме, позволяющей получить бензины с очень высоким
октановым числом. На рис. 17,12 представлена схема такого
процесса [426].
Бензин прямой гонки фракционированием разделяют на пентан-гексано-
вую фракцию и тяжелую фракцию, содержащую углеводороды, начиная с
гептана. Повышение октанового числа пентан-гексановой фракции осу-
ществляют в процессе каталитической гидроизомеризации, а остатка — в про-
цессе гидроароматизации (платформинга). Нормальные парафиновые угле-
водороды, не подвергшиеся конверсии, выделяют на цеолитовой установке
и возвращают в процесс. Смесь фракций, из которых они удалены, является
высококачественным моторным топливом.
Переработка бензина по комбинированной схеме имеет опре-
деленные преимущества по сравнению с обычной схемой пере-
работки бензинов, заключающейся в платформинге фракции С7
и выше и последующем смешении продукта с фракцией Cs—Се.
Максимальное октановое число этилированных бензинов, по-
лучаемых в наиболее жестких условиях платформинга, состав-
ляет 102 пункта; продукт, получаемый по комбинированной схе-
ме, может иметь октановое число до 105 пунктов. На рис. 17,13
выход бензина представлен как функция заданного октанового
числа конечного продукта. При этом включение в схему блока
разделения на цеолитах позволяет проводить риформинг в более
Рис. 17,13. Зависимость выхода бензина Р от за-
данного октанового числа по исследовательскому
методу OZn (с ТЭС):
1 — переработка по схеме иа рис. 17,12; 2 — про-
дукт риформинга фракции С?+ после смешения с
углеводородами Cg—Сб, выделенными фракциони*
роваинем.
520 17. Использование молекулярно-ситовых свойств цеолитов
Рис. 17,14. Зависимость октанового числа этилиро-
ванного продукта OZH от жесткости условий про-
цесса риформинга (содержания ароматических уг-
леводородов в бензине сар):
1 — переработка по схеме на 17,12; 2 — продукт
риформинга фракции С?4- после смешения с угле-
водородами С5—Сб, выделенными фракционирова-
нием.
мягких условиях и тем самым по-
высить выход жидкого продукта.
Преимущество комбинированной
схемы возрастает по мере повыше-
ния требований, предъявляемых к
качеству конечного продукта.
каталитического риформинга в бо-
Кроме того, проведение
лее мягких условиях приводит к снижению содержания арома-
тических углеводородов. На рис. 17,14 представлена зависимость
октанового числа этилированного продукта от содержания аро-
матических углеводородов. Переработка по комбинированной
схеме позволяет при заданном октановом числе продукта сни-
зить содержание в нем ароматических углеводородов на 5—
7%. Уменьшение содержания ароматических углеводородов в
бензине приводит к улучшению его качества, в частности к сни-
жению «чувствительности» (разницы в октановых числах по ис-
следовательскому и моторному методу). Повышение содержания
в бензине ароматических углеводородов на 10% вызывает уве-
личение «чувствительности» в среднем на 1,5 пункта.
Рис. 17,15. Схема установки депарафинизации углеводородных фракций в движущемся
слое мелкосферического цеолита:
1 — адсорбер; 2 — воздуходувка; 3 — воздухоподогреватель; 4 — зона отдувки; 5, 9 — хо-
лодильники; 6, 10—сепараторы; 7 — нагреватель циркуляционного газа; 8 — десорбер;
11 — компрессор.
Выделение нормальных олефинов из смесей
521
Депарафинизация в движущемся слое цеолитов
Преимущества непрерывных процессов побуждают искать новые
технологические решения. Одним из них является проведение
процесса депарафинизации в жидкой фазе углеводородных
фракций в движущемся слое мелкосферического цеолита [427].
Схема установки представлена на рис. 17,15.
Поглощение нормальных парафиновых углеводородов проводят в колон-
не 1 в жидкой фазе в противотоке со слоем адсорбента, медленно спускаю-
щегося под действием силы тяжести. Насыщенные цеолиты поступают в зону
отдувки. Здесь, в потоке нагретого (3) до 95 °C воздуха, нагнетаемого воз-
духодувкой 2, с внешней поверхности гранул цеолита и из вторичной пори-
стой структуры удаляют углеводороды, не принадлежащие к классу нор-
мальных парафинов. Эти углеводороды после охлаждения (5) выделяют в
сепараторе 6 и возвращают в адсорбционную колонну 1.
Десорбцию нормальных парафиновых углеводородов из цеолита прово-
дят в десорбере 8, где температуру слоя с помощью циркуляционного газа
повышают до 300 °C. Температура газа на выходе из нагревателя 7 состав-
ляет 325 °C. Перемещение цеолита из зоны в зону обеспечивают с помощью
механических транспортеров и газлифта, оборудованного компрессором 11.
Депарафинизация бензинов с применением
диоксида углерода в качестве десорбента
В Советском Союзе (ГрозНИИ) разработан [428] процесс депа-
рафинизации бензиновых фракций, в котором десорбцию про-
водят в потоке диоксида углерода. Стадии адсорбции и десорб-
ции ведут в изотермическом режиме при 140—180 °C и 0,5—
0,9 МПа. Кратность циркулирующего диоксида углерода состав-
ляет от 2,5 до 2,9 частей на 1 часть выделяемых н-парафиновых
углеводородов. Адсорбционная способность цеолита (до насы-
щения) по н-гексану составляет 5%, по н-гептану — только
3,1 г/100 г; снижение адсорбционной емкости в последнем слу-
чае связано с неполным вытеснением углеводорода диоксидом
углерода на стадии десорбции.
Выделение нормальных олефинов из смеси
с другими углеводородами
Молекулярно-ситовые свойства цеолитов СаА в равной степени
могут быть использованы для выделения нормальных олефино-
вых углеводородов из смесей с углеводородами других типов
[429]. При этом нужно, однако, учитывать протекание реакций
изомеризации и полимеризации олефинов.
При поглощении высших нормальных олефиновых углеводо-
родов, например гексенов, цеолиты СаА обеспечивают молеку-
лярно-ситовое разделение изомеров, различающихся геометри-
ческим строением [430]. Этот вывод можно сделать при рас-
34—1346
522 -/7. Использование молекулярно-ситовых свойству цеолитов
смотрении изотерм адсорбции, представленных на рис. 17,161.
Изотермы адсорбции гексена-1, гексена-2 и гексена-3 на цеоли-
тах NaX при 20 и 80 °C практически совпадают. Следовательно,
адсорбция на этом сорбенте не зависит от положения двойной
связи в молекуле н-гексенов и геометрического строения моле-
кул. Иная картина наблюдается при адсорбции тех же углеводо-
родов на цеолите СаА. В этом случае на всем участке изотерм:
(особенно в области малых давлений) адсорбционная способ-
ность по гексенам с внутренней двойной связью, значительно
ниже, чем по гексену-1. Объясняется это тем, что исходные гек-
сены-2 и гексены-3 характеризуются некоторым содержанием
г{ас-гексанов, не сорбирующихся цеолитом СаА. Действительно^
изотермы тра«с-гексена-2 совпадают с изотермами гексенов-2
на цеолитах обоих типов. Дис-гексен-2 сорбируется цеолитами
NaX точно так же, как и все другие гексены. В то же время,
величина его адсорбции на цеолите СаА на порядок ниже велщ
чины адсорбции гексена-1. Максимальные массы этих компонен-
тов, адсорбированных при 20 °C, равны 11,4 hi 14,5 г/100 г.
Молекулярно-ситовое действие цеолитов СаА по отношению»
к цис- и транс-изомерам нормальных гексенов было подтверж-
дено разделением в динамических условиях смеси гексенов, по-
лученной дегидратацией нормального1 гексилового спирта и со-
Рис. 17,16. Изотермы адсорбции гексанов, на. цеолитах. СаА. и-N-aX;
1 — 1-гексен-1; 2 — гексены-2 (цис- и транс-);. 3 — цис-гексенг2;. 4 — трано-гекоен-2; 5— гек-
сены-3.
Выделение нормальных парафинов из фракций углеводородов
523
1Рис. 17,17. Прибор для анализа бутановой фракции:
1 — нижияя емкость; 2 — трубка с адсорбентом; 3 — верх-
няя емкость; 4 — ртутный манометр; 5 — ртутный затвор,
держащей все пять изомеров нормальных гексенов.
В результате разделения был получен продукт,
на 94,6% состоящий из а- и транс-изомеров; ад-
сорбируемость а- и транс-формы нормальных гек-
сенов на цеолите СаА одинакова.
Молекулярно-ситовые свойства цеолитов широ-
ко используются и для определения содержания
нормальных парафиновых и олефиновых углеводо-
родов в различных нефтепродуктах. Для этого ана-
лизируемую смесь либо пропускают через колонку
с цеолитом, либо приводят в контакт с адсорбен-
том, находящимся в герметичной емкости. Опреде-
ление содержания углеводородов нормального
строения, поглотившихся в результате контакта с
цеолитам, проводят по -уменьшению объема образ-
ца, привесу адсорбента или изменению его физико-
химических констант, например, коэффициента
преломления {431—436]. Ивменени-е объема сме-
си газообразных углеводородов может быть уста-
новлено по изменению давления в системе после контакта сме-
си с цеолитом. На этом принципе основана методика Майерса и
Гипкина {436] определения н-бутана в его смесях с изобута-
ном (рис. 17,17i).
Исходную смесь вводят в верхнюю -емкость 3 объемом Vi=400 см® при
давлении 106 кПа. Под ней расположена медная адсорбционная трубка 2
диаметром 12,5 мм и высотой 60 см, заполненная цеолитом СаА. Сумма сво-
бодного объема адсорбционной трубки 2 и нижней емкости 1, соединенной
,с ртутным затвором, составляет также V2=Vi. Перед анализом адсорбцион-
ную трубку и емкость вакуумируют. В случае отсутствия адсорбции углево-
дородов после открытия крана давление в системе должно составить 53 кПа.
По отклонению от этого значения проводят расчет содержания н-бутана.
Извлечение нормальных парафинов
из керосино-лигроиновых фракций
•Общая характеристика процесса
^Сохранение чистотел водных бассейнов при современном темпе
развития промышленности и росте народонаселения может быть
•обеспечено, в частности, лишь при условии преимущественного
использования моющих средств, разрушающихся под действием
'биологических агентов (микроорганизмов). К такому виду мою-
щих средств принадлежат продукты, получаемые из нормальных
.34
524 17. Использование молекулярно-ситовых свойств цеолитов'
парафиновых углеводородов от С10Н22 до Ci8H38, т. е. компонен-
ты легких и тяжелых керосинов. Моющие средства, полученные
из нормальных парафинов, разлагаются микроорганизмами с
очень большой скоростью. Содержание таких веществ’ в речной,
воде после 4 сут снижается с 5 до 2 мг/л, а после 15 сут прак-
тически до нуля [437]. Особенно быстро разлагаются моющие
средства, полученные из чистых нормальных парафиновых угле-
водородов, в которых общее содержание примесей не превышает
1%, а ароматических углеводородов — не более 0,05%.
Молекулярно-ситовые свойства цеолитов СаА могут быть,
использованы для увеличения концентрации нормальных пара-
финовых углеводородов (например, С[7—С24) во фракциях, вы-
деленных в традиционном процессе карбамидной депарафиниза-
ции масел. По одной из методик [438], для этого 5%.-ный рас-
твор фракций в изооктане контактируют в автоклаве с цеолитом
СаА в течение 2 ч. Десорбцию проводят с помощью вытес-
нителя н-гептана при 320 °C в течение 2 ч. В случае концентри-
рования высокомолекулярных фракций (375—400°C) на стадии
десорбции применяют смесь н-гептана и бензола.
Чистота нормальных парафиновых углеводородов, получен-
ных с использованием цеолитов, значительно превышает чисто-
ту парафинов, выделенных другими методами. Возросшая по-
требность в моющих средствах, разрушающихся под действием
микроорганизмов, и повышенные требования к чистоте сырья
побудили распространить адсорбционный метод на непосред-
ственное выделение нормальных парафинов из керосиновых’
фракций.
Другим не менее важным направлением переработки чистых
парафиновых фракций является микробиологическое производ-
ство белковых веществ. Из всех классов углеводородов микро-
организмами усваиваются только нормальные парафины. Па
аминокислотному составу белки, полученные из нормальных
парафинов, не уступают белкам животного происхождения и
могут снизить дефицит кормового белка. В зарубежных странах
производство белково-витаминных концентратов превысила
3,5 млн т/ год [439], а производство нормальных парафинов
Сю—Сю с помощью цеолитов 1,5 млн т/год [440]. Оно освоена
на 24 промышленных установках и осуществляется по методам:
«Изосив», «Техсако», «Бритиш Петролеум»,. «Энсорб», «Парекс»,.
«Молекс».
Нормальные парафины Сю-—С20 широко применяют также
для получения разбавителей каучука и ингибиторов воспламе-
нения (полихлорпарафинов), пластификаторов поливинилхло-
рида, разнообразных спиртов и олефинов. Нормальные парафи-
ны от Сю до Сю используют для производства н-алкилбензол-
сульфонатов, н-алкенилсульфонатов, этоксилированных н-алкил-
фенолов, эфиров, аминов, меркаптанов и т. д.
Выделение нормальных парафинов из фракций углеводородов 525
Первая установка извлечения парафинов в паровой фазе по методу
«Изосив», пуск которой осуществлен в декабре 1961 г. в г. Силсби (Техас,
США), вначале была предназначена для выделения нормальных углеводоро-
дов С5Н12—С12Н26, но спустя 5 мес после начала эксплуатации была пере-
ведена на керосиновое сырье. Установка перерабатывает 160 м3 керосина в
сутки. Затем в ноябре 1963 г. была пущена установка в г. Встсоие (Кали-
форния, США), вырабатывающая 56 м3/сут нормальных парафинов, а в
1964 г. —крупный завод в Техасе, где получают сырье для производства
фенилпроизводных нормальных алканов путем алкилирования бензола с по-
следующей переработкой полупродукта в моющие средства (441, 442].
Начиная с 1962 г. отмечен быстрый рост ввода в эксплуатацию промыш-
ленных установок выделения нормальных парафинов. Крупные исследования
и разработки новых технологических схем проведены в социалистических
странах: Советском Союзе (процесс ГрозНИИ), ГДР (процесс «Парекс»),
Польше (процесс «Интенекс»), Чехословакии (процесс «Вуруп»). В число
стран, располагающих установками адсорбционной депарафинизации, вхо-
дят: США, Англия, ФРГ, Италия, Япония, Испания, Голландия.
Все процессы различаются в основном методом десорбции.
Только один процесс проводится в жидкой фазе—• «Молекс». Он
был предложен для выделения нормальных парафинов С6—Сд
из бензинов, но ни одна промышленная установка не была по-
строена для этих целей. Процесс был реализован в промышлен-
ности для выделения нормальных парафинов Си—Cis из керо-
синов [443].
Выбор метода десорбции. Стадия десорбции является опреде-
ляющей для экономики процесса адсорбционного разделения и
общей продолжительности цикла. Ниже рассмотрены некоторые
методы десорбции, применяемые для удаления углеводородов
из цеолитов.
Повышение температуры слоя при десорб-
ции уменьшает адсорбционную емкость цеолита по углеводоро-
дам и тем самым стимулирует перевод адсорбата из адсорбиро-
ванной фазы в газовую. Это иллюстрирует изобара адсорбции
н-додекана при нормальном давлении, представленная на
рис. 17,18. Нагрев цеолитов можно проводить горячими потока-
ми инертного газа, нормальных парафинов или углеводородных
фракций [444]. Однако в связи с достаточно сложным аппара-
турным оформлением, низкой интенсивностью, большими затра-
тами тепла и холода метод проведения десорбции высокомолеку-
лярных углеводородов путем нагрева не получил распростране-
ния.
Снижениедавления, как метод десорбции, широко при-
меняется на практике. Оно позволяет проводить разделение
практически в изотермических условиях. Особенно эффективен
этот метод, если он дополнен вакуумированием. Применительно
к разделению высокомолекулярных углеводородов, однако, его
использование наталкивалось .на значительные трудности. Как
показывает рис. 17,19, применение умеренного вакуума, освоен-
ного, в промышленности, позволяет сравнительно полно удалить
из адсорбированной фазы в изотермических условиях только
526 /7- Использование молекулярно-ситовых свойств цеолитов
Рис. 17,18. Изобара адсорбции н-додекаиа на цео-
лите СаА при давлении 0,1 МПа.
бензиновые углеводороды (н-гек-
сан). Лишь создание новой техники
получения высокого вакуума, осно-
ванной на конденсации и охлажде-
нии самого десорбата, позволило в
последнее время распространить
метод вакуумной десорбции на процесс молекулярно-ситового
разделения высокомолекулярных фракций нефти.
Вытеснение адсорбата. Адсорбируемость углеводо-
родов на цеолитах, как и на других адсорбентах, возрастает с
увеличением молекулярной массы. Это позволяет с высокой
скоростью провести вытеснение адсорбированных углеводородов
из твердой фазы другими углеводородами с большой молекуляр-
ной массой. Однако при промышленном применении метод
встретил непреодолимую трудность: не удается эффективно ре-
шить проблему удаления десорбента перед повторной стадией
депарафинизации. Нормальные парафины, содержащиеся в
сырье, очень медленно замещают высшие углеводороды в
адсорбционных полостях цеолитов, а неполнота удаления пос-
ледних приводит к значительному снижению адсорбционной
емкости.
Поэтому в процессах разделения высокомолекулярных угле-
водородов используют метод де со р бц и и хуже адсорби-
рующимся веществом. В этом случае десорбент одновре-
менно выполняет функции динамического агента и вытеснителя.
Понижая парциальное давление адсорбата и одновременно
сорбируясь на цеолитах, десорбент постепенно вытесняет угле-
водороды, поглощенные из сырья. Углеводороды десорбента лег-
ко вытесняются на последующие стадии депарафинизации.
В связи с тем, что и стадия адсорбции, и стадия десорбции про-
Рис. 17, 19. Изотермы адсорбции н-гексана (1) и н-додекаиа (2) на цеолите СаА при
315 “С.
Выделение нормальных парафинов из фракций углеводородов
527
Рис. 17,20. Зависимость времени, необхо-
димого для десорбции тд, от разницы числа
атомов углерода в молекулах адсорбата и
десорбента Дпс.
водятся, когда адсорбент уже насыщен углеводородами, и в хо-
де десорбции происходят только замещение, тепловые эффекты
оказываются незначительными, и условия процесса можно счи-
тать изотермическими.
При таком способе десорбции обеспечивается полнота уда-
ления адсорбата и достигается высокая активность адсорбента
на стадии разделения. В качестве десорбента применяют нор-
мальные парафины, нормальные олефины [445, 446], различные
углеводородные фракции [447, 448]. Кроме углеводородов, для
десорбции используют вещества, обладающие значительной до-
полнительной специфической энергией адсорбции: диоксид угле-
рода, аммиак [449], сульфид водорода [450] и воду [405]. Пос-
ледние два компонента не нашли широкого применения в про-
мышленности: сульфид водорода из-за своей агрессивности, а
вода вследствие разрушающего действия на структуру цеолита
в условиях высоких температур и высоких концентраций водя-
ного пара [451]. В качестве десорбента могут быть применены
также SO2, CH3NH2, С2Н5С1, C2H5F [452].
Обязательным требованием, предъявляемым к десорбенту,
является возможность легкого его разделения как с фильтратом,
так и с экстрактом методами обычной ректификации или даже
сепарации. Процесс депарафинизации, как уже отмечалось,
обычно проводят в условиях, близких к изотермическим. Недос-
татком способа является большая кратность циркуляции де-
сорбента.
Обычно в качестве десорбента используют нормальные пара-
фины [453]. Десорбцию проводят углеводородом, температура
кипения которого ниже темпе-
528 17. Использование молекулярно-ситовых свойств цеолитов
Рис. 17,22. Зависимость времени, необходимого
для десорбции тд нормальных парафиновых угле-
водородов, выделенных из лигроина (150—205 °C)
в потоке пеитана от температуры /.
родных атомов в молекулах нормальных парафинов сырья и
десорбента, тем быстрее происходит десорбция. На рис. 17,20
представлена зависимость времени десорбции нормальных па-
рафинов, выделенных из фракции 150—205 °C, от этого показа-
теля. При сопоставимых временах в случае, например, десорб-
ции н-нонана из слоя цеолита СаА при температуре 250°C, ско-
рости потока десорбента 8 см/с и массовом соотношении десор-
бент— адсорбат 5:1 степень десорбции составляет 100% при
использовании н-гексана и только 63% при использовании тех-
нической пропан-бутановой фракции [454].
Выбор температуры и давления в процессе депарафинизации.
Если процесс проводят в паровой фазе, имеется возможность
варьирования температуры в достаточно широком интервале.
При повышенных температурах увеличивается скорость адсорб-
ции нормальных парафинов, но понижается равновесная
адсорбционная емкость цеолита; одновременно возрастает кок-
сообразование. Совокупность кинетических и равновесных пока-
зателей определяет оптимальную температуру, при которой ди-
намическая активность достигает максимума.
Положение оптимума зависит от длины цепи в молекуле
парафина и в случае депарафинизации керосиновых фракций
соответствует примерно 330°C (рис. 17,21). Большинство паро-
фазных процессов проводят при температурах, которые несколь-
ко ниже этого оптимума. Осуществление процесса в паровой
фазе значительно интенсифицирует стадию десорбции, так как
при этом меньше сорбируется нецелевых углеводородов и они
легче удаляются при продувке. Повышение температуры уско-
ряет десорбцию поглощенных веществ. На рис. 17,22 представ-
лена зависимость времени десорбции нормальных парафинов,
выделенных из фракции 150—205°C, от температуры. При вы-
боре оптимального режима процесса следует обеспечить такие
условия, чтобы углеводороды находились в паровой фазе и в
то же время не подвергались интенсивному крекингу.
Выбор расхода десорбента. Увеличение расхода десорбента
приводит к повышению степени десорбции, полезной адсорбци-
онной емкобти на стадии депарафинизации и соответственно к
снижению капиталовложений в адсорбционную установку.
С другой стороны, при этом увеличиваются капиталовложения
на разделение десорбента и десорбата. Выявление оптимума
Выделение нормальных парафинов из фракций углеводородов
529
Рис. 17,23. Зависимость времени тд, необходимого для десорбции нормальных парафи-*
новых углеводородов с интервалом кипения 175—260 °C, от объемной скорости десорбента
(н-гептана) Н.
расхода десорбента проводят на основе технико-экономического
анализа.
На рис. 17,23 показано время десорбции нормальных пара-
финов, выделенных из керосина фракцци 177—260 °C в зависи-
мости от часовой объемной скорости десорбента (н-гептана).
В установках депарафинизации на стадиях адсорбции и де-
сорбции обычно применяют противоток. Тогда десорбированные
из лобовых слоев легкие углеводороды служат динамическим
агентом при отдувке тяжелых компонентов, находящихся в по-
следних слоях адсорбера.
Восстановление адсорбционной способности цеолитов. Сни-
жение адсорбционной способности цеолитов определяется комп-
лексом сложных химических реакций, в основе которых лежит
крекинг нормальных парафиновых углеводородов. Образую-
щиеся при крекинге олефины подвергаются ароматизации и
полимеризации с образованием твердых углеродсодержащих
соединений. Попутно может протекать процесс изомеризации.
В свою очередь, ароматические углеводороды и продукты изо-
меризации, критический размер молекул которых больше раз-
мера входных окон цеолита СаА, оказываются «запертыми» в
адсорбционных полостях и продолжают постепенно подвергать-
ся крекингу и полимеризации.
Дезактивация цеолитов происходит также вследствие накоп-
ления в адсорбированной фазе загрязнений, прочно удерживае-
мых в адсорбционных полостях и не удаляемых при десорбции.
К таким загрязнениям относятся сернистые соединения и поляр-
ные вещества, имеющие в своем составе гидроксильные, карбо-
нильные, нитро- и аминогруппы.
Восстановление адсорбционных свойств цеолитов (выжиг кокса и за-
грязнений) обычно проводят в потоке кислородсодержащего газа. При вы-
боре концентрации кислорода в газе, подаваемом в адсорбер на стадии ре-
генерации, необходимо учитывать, что повышение температуры цеолита ft-
результате выжига зависит от содержания углерода в адсорбенте. Кривые,,
характеризующие нагрев цеолита при выжиге па 140 и 250 °C, продета плены
на рис. 17,24. Температура цеолита при выжиге не должна превышать поро-
га его термической устойчивости.
Предприняты попытки снизить каталитическую (крекирую-
щую) активность синтетических цеолитов типа СаА в резуль-
530
17. Использование молекулярно-ситовых свойств цеолитов
Рис. 17,24. Зависимость концентрации кислорода
с0^ в газе регенерации, обеспечивающей задан-
ное повышение температуры в слое цеолита (Д£ =
= 140 и 250 °C), от закоксоваииости gc (в г. атом
С иа 100 г цеолита).
тате замещения катионов кальция на катионы лантана или трех-
валентных металлов группы лантанидов |[455, 456]. Интенсив-
ность коксообразования на лантанзамещенных формах цеолитов
снижается приблизительно на 30%.
В процессе депарафинизации активность цеолитов снижается быстрее,
если в сырье содержатся микропримеси соединений железа в виде мельчай-
шей взвеси, образующейся в результате коррозии и эрозии трубопроводов и
аппаратуры. Поэтому предложено [457] для увеличения срока службы цео-
литов исходную фракцию углеводородов перед подачей в адсорбер пропус-
кать через целлюлозный фильтр с порами диаметром не выше 0,45 мкм.
г Другим источником дезактивации цеолита являются серни-
стые соединения. При разделении сырья, содержащего серни-
стые соединения, последние переходят как в рафинат, так и в
экстракт, загрязняя их. Сернистые соединения очень прочно
удерживаются цеолитами, не удаляются на стадии десорбции,
накапливаются и дезактивируют адсорбент. Одновременно они
являются промоторами реакций образования кокса.
Во избежание накопления олефинов в циркулирующем де-
сорбенте выше нормы может быть проведено их гидрирование.
Для этого к десорбенту примешивают водород. В качестве ка-
тализатора гидрирования используют никель, оксиды кобальта
и молибдена на носителе. Процесс проводят при температуре
до 360 °C, давлении до 0,42 МПа, объемной скорости 0,1—20 ч-1,
расходе водорода 0,2—0,4 м3/л углеводородов. При этом гидро-
очистке подвергаются также сернистые соединения.
Но радикальными методами борьбы с накоплением вредных
соединений в цеолитах продолжают оставаться предваритель-
ная сероочистка сырья и выжиг загрязнений из цеолита одно-
временно с выжигом кокса.
Процессы в паровой фазе
Процесс «Изосив»— это наиболее распространенный парофаз-
ный процесс извлечения парафиновых фракций. Он разработан
фирмой Юнион Карбайд. Его применяют для выделения нор-
мальных углеводородов с числом атомов углерода в молекуле
от 5 до 22. Для различного сырья и в зависимости от требова-
ний к чистоте рафината и экстракта разработаны разнообраз-
Выделение нормальных парафинов из фракций углеводородов 531
ные варианты процесса {[458—463]. Технологическая схема уста-
новки «Изосив» представлена на рис. 17,25.
Сырье насосом подают в теплообменник 1, где нагревают и испаряют,
а затем пропускают через первый слой Цеолита СаА в адсорбере 2. После
того, как первый слой насытится нормальными парафинами, поток переклю-
чают на второй слой. В зависимости от температуры кипения выделенных
парафинов и, соответственно, трудности их удаления из адсорбированной
фазы на стадии десорбции используют циркуляционный газ или десорбент.
Десорбент подогревают до требуемой температуры в теплообменнике 6,
пропускают через второй слой цеолитов и вместе с десорбированными нор-
мальными парафинами направляют в блок фракционирования 7. При фрак-
ционировании в качестве иижнего продукта выделяют концентрат нормаль-
ных парафинов, а в качестве верхнего —десорбент, возвращаемый в цикл.
Свежий и рециркулирующий десорбент смешивают в емкости 5. Аналогич-
ным образом в блоке 4 производят разделение рафината и десорбента, ос-
тавшегоси в цеолите после стадии десорбции и выделяющегося в поток
рафината на стадии разделения. Отлагающийся на адсорбенте кокс периоди-
чески выжигают с помощью системы регенерации 3.
Промышленные испытания показали, что содержание приме-
сей в сырье может составлять: до 0,03% серы, вода (вплоть до
полного насыщения углеводородов), азотистые соединения (не-
много) и следы металлов.
Повышение концентрации нормальных парафинов в экстрак-
те достигают включением между адсорбцией и десорбцией ста-
Рис. 17,25. Схема установки депарафинизации методом «Изосивэ: J
1, 6 — теплообменники; 2 — адсорбер; 3 — система регенерации; 4, 7 — блока фрйкЦиойВ’
рования; 5 — емкость.
532
17. Использование молекулярно-ситовых свойств цеолитов
Рис. 17,26. Температура начала капилляр
ной конденсации fKK и точка росы fp га-
। зойля:
1 — £кк чистого газойля; 2 — то же, с Д(Н
бавкой 30% н-гексана; 3 — fp чистого газой*
ля; 4 — то же, с добавкой 30% «-гексана
дии отдувки примесей в потоке инертного газа (464]. Благодаря
введению стадии отдувки на Рурском нефтеперерабатывающем!
заводе достигнута концентрация нормальных парафинов С13—
С17 в экстракте до 99,5% [465]. Установка включает 5 адсорбе-
ров, один из которых периодически подключают на регенера-
цию. Десорбцию проводят пропусканием н-пентана через слой
адсорбента, после чего н-пентан и экстракт разделяют обычным
(фракционированием.
<С целью извлечения нормальных парафинов из газойлевых
фракций метод «Изосив» был усовершенствован. Депарафини-
зацию газойля стали проводить в слое цеолита СаА в паровой
фазе при 370 °C и 0,28 МПа в присутствии н-гексана, добавляе-
мого для того, чтобы избежать капиллярной конденсации высо-
комолекулярных углеводородов во вторичной пористой струк-
туре.
На рис. 17,26 приведены кривые температур начала капил-
лярной конденсации и точки росы газойля (чистого и с добав-
кой 30% гексана) в зависимости от давления в системе. При
давлении 0,20 МПа температура капиллярной конденсации га-
зойля равна 390°C, точка росы 345 °C, в присутствии 30% гек-
сана характерные температуры соответственно равны 370 и
330 °C. Схема усовершенствованной установки представлена на
рис. 17,27.
Установка состоит из нагревателя сырья 1, адсорберов 3 н 5, в’ которых
осуществляется рабочий цикл, блоков отделения гексана от рафината 6
и экстракта 8. Адсорбер 2 подключен на стадию регенерации, для чего пре-
дусмотрена система 4. После насыщения цеолита нормальными парафинами
адсорбер продувают потоком гексана, в результате чего удаляются примеси
из свободного объема, вторичной пористой структуры и с внешней поверх-
ности кристаллов. Затем гексан используют в качестве десорбента. Смесь
десорбированных высокомолекулярных парафинов и гексана разделяют в
ректификационной колонне блока 8.
На последующей стадии разделения смесь сырья и гексана поступает в
слой, уже насыщенный гексаном. В процессе разделения гексан постепенно
вытесняется высокомолекулярными нормальными парафинами сырья. Смесь
рафината и гексана совместно со смесью углеводородов и гексана со стадии
продувки поступает в ректификационную колонну блока 6, где рафииат от-
деляют от гексаиа. Гексан, выделенный в блоках 6 и 8 из сборника 7, воз-
вращают в процесс. Десорбцию из экономических соображений целесообраз-
но проводить не полностью. Периодически, ведут выжиг образовавшегося
кокса смесью воздуха и азота.
Выделение нормальных парафинов из фракций углеводородов
533
Для поддержания высокой адсорбционной емкости и скоро-
сти массообмена, а также исключения интенсивной дезактива-
ции цеолита в результате крекинга углеводородов температуру
процесса при переработке керосина поддерживают в пределах
.315—335 °C, газойля—дд 400 °C. При оптимизации процесса
расход десорбента выбирают в зависимости от состава и каче-
ства перерабатываемого сырья. Стоимость производства нор-
мальных парафинов линейно возрастает с увеличением средней
молекулярной массы перерабатываемой фракции [466].
Процесс «Бритиш петролеум». Процесс, разработанный фир-
мой Бритиш петролеум, осуществляется в изотермических и
изобарических условиях. В этом процессе сырье перед подачей
в адсорбер во всех вариантах разбавляют. Часто разбавителем
является десорбент. В качестве сырья используют керосины,
содержащие нормальные парафиновые углеводороды Сю—Сю и
фракцию газойля (Си—Сю). После стадии адсорбции пары
сырья из цеолита отдувают азотом. Десорбцию проводят, как
-правило, «-пентаном или другим углеводородом.
Рекомендуется соотношение продолжительности стадий ад-
сорбции, отдувки и десорбции поддерживать равным 1:1:2, на-
пример 6, 6 и 12 мин. Максимально допустимое содержание сер-
нистых соединений в сырье оценено в 0,1 %. В системе предус-
мотрен четвертый адсорбер на стадии регенерации. Регенера-
цию проводят при 350—400 °C через каждые 100—400 ч эксплуа-
тации установки.
В варианте процесса «Бритиш петролеум» |[467] депарафи-
низацию нефтяных фракций, выкипающих до 350 °C, проводят
Рис. 17,27. Схема установки депарафинизации по усовершенствованному методу «Изо-
4 — теплообменник; 2, 3, 5 — адсорберы; 4 — система регенерации; 6 — блок фракциони-
рования десорбента и рафината; 7 — емкость; 8 — блок разделения десорбента н экс-
тракта.
534
17. Использование молекулярно-ситовых свойств* цеолитов
Рис. 17,28. Схема установки депарафинизации методом «Бритиш петролеум»:
I, 4 — теплообменники; 2 — адсорбер; 3 — система регенерации; 5, 6 — блоки фракциоии^
рования. *
в изотермических и изобарических условиях при температуре
от 300 до 450 °C и давлении до 1,5 МПа. Соотношение продол-
жительности стадий адсорбции, продувки и десорбции состав-
ляет 1 : 1 : п, где п>3. Продувку ведут азотом или углеводорода-
ми метанового ряда с числом атомов углерода в молекуле от В
до 5. В качестве эффективного десорбента используют н-бутан
или н-пентан.
Первая промышленная установка по методу «Бритиш Петро-
леум» производительностью 3 тыс.т/год нормальных парафинов!
Сю—Сю введена в строй на Рурском нефтеперерабатывающем
заводе в Дислакене (ФРГ) [468]. Вторая установка, производи-
тельность которой достигает 50 тыс. т/год, построена в Англии.
Чистота нормальных парафинов, получаемых из керосина и га-
зойля по этому методу, достигает 99,5%. Технологическая схема
процесса представлена на рис. 17,28 [469].
Сырье, разбавленное десорбентом, нагревают, испаряют и смесь пропус-
кают в паровой фазе через один из адсорберов 2, заполненных цеолитами.
Рафинат отделяют от десорбента в блоке разделения 5, после чего десорбент
возвращают в процесс. В это время из второго адсорбера поглощенные нор-
мальные парафины удаляют в потоке нагретого (4) десорбента. Смесь экст-
ракта и десорбента разделяют в блоке 6 и десорбент возвращают в процесс.
Таким образом, в схеме один компонент выполняет функции и разбави-
теля и десорбента, причем его циркуляцию проводят в двух раздельных
замкнутых контурах. На установке предусмотрена система регенерации цео-
литов 3. В промышленных адсорбционных установках запроектировано 5 ад-
сорберов: один находится на стадии адсорбции, один — на стадии продувки,
два — на стадии десорбции и один периодически переключают на регеиера*
дию.
выделение нормальных парафинов из фракций углеводородов 535
Освоение техники вакуумной десорбции позволило эту (тип
MS = 1) и некоторые другие установки перевести на новый ре-
жим (тип MS-2, см. стр. 511).
Во избежание накапливания сернистых соединений в адсор-
бенте сырье рекомендуется подвергать гидроочистке до оста-
точного содержания общей серы менее 1 -10 2%. Замену цеоли-
тов производят каждые 5 лет. Расходные показатели на 1 кг
выделенных парафинов составляют: топливо — 4220 кДж, элект-
роэнергия— 0,1 кВт-ч, пар—0,25 кг, охлаждающая вода — 36 л.
Процесс со снижением давления обеспечивает получение нор-
мальных парафинов (выше 99%), что особенно важно при их
использовании для производства белка.
Денормализованный побочный продукт имеет низкую темпе-
ратуру застывания, и его рекомендуется использовать в каче-
стве среднего дистиллята в северных широтах. Процесс денор-
мализации высокомолекулярных нефтяных фракций с вакуум-
ной десорбцией эффективно протекает на советских промышлен-
ных цеолитах типа СаА и MgA [470].
Применение разбавителя позволяет полностью испарить ис-
ходную смесь углеводородов, добиться равномерного распреде-
ления потоки по слою, повысить скорость адсорбции, уменьшить
капиллярную конденсацию во вторичной пористой структуре и
сократить количество углеродистых отложений. Все это повы-
шает качество продуктов.
Процесс «Тексако селектив финишинг» (ТСФ) предназначен
для депарафинизации тяжелых нефтяных фракций. Десорбцию
в нем проводят углеводородами с меньшей молекулярной мас-
сой, чем масса углеводородов в сырье [471].
По одному из вариантов процесса ТСФ [472] выделение нор-
мальных углеводородов Сю—Сго производят при повышенной
температуре 315 °C и давлении 0,2 МПа в паровой фазе в не-
подвижном слое цеолитов СаА. Рабочий цикл состоит из сле-
дующих стадий: адсорбция при указанном выше давлении, сни-
жение давления до атмосферного, продувка слоя цеолита при
этом давлении, повышение давления до 0,28 МПа и, наконец,
снижение давления до первоначального. Процесс короткоцикло-
вый: продолжительность стадии адсорбции 13—15 мин, продув-
ки— 1—6 мин, десорбции — 25 мин, изменения давления —
0,3—0,5 мин.
В качестве продувочного агента и десорбента используют
фракцию, содержащую в основном нормальный парафин с чис-
лом атомов углерода в молекуле на 1—3 меньше, чем в моле-
куле самого низшего парафина сырья. Объемная скорость паров
углеводородов на стадии продувки составляет от 50 до 1000 ч~\
расход десорбента от 0,1 до 10 об/об. цеолита. Направление па-
ров на стадии адсорбции, с одной стороны, и стадиях продувки
а десорбции, с другой, противоточное.
536
17. Использование молекулярно-ситовых свойств, цеолитов
По этой методике разделили керосиновую фракцию с границами кипения
172—260 °C, предварительно подвергнутую гидроочистке и содержащую
16,2% нормальных парафиновых углеводородов Сю—Сю. Фракцию вводили,
в нижнюю часть адсорбера при температуре 315 °C и давлении 0,2 МПа.'
Расход сырья составлял 1,1 м3/(м3-ч). Адсорбер имел диаметр 7,5 см, высо-
ту 1,1 м, объем 5,3 л. Он был загружен 4630 г цеолита типа СаА с диамет-
ром гранул 1,6 мм.
Сверху из адсорбера отбирали продукт, содержащий 76,8% углеводоро-
дов Сю—Сю, ие принадлежащих к классу нормальных парафинов, 1,6% нор-
мальных парафинов Сю—С15 и 21,6% десорбента. Скорость отбора состав-
ляла 6140 см3/ч. Продукт подвергали фракционированию. Выход фракции'
Сю—Сю, ие содержащей нормальных парафинов, составил 84,4% (на исход-
ное сырье). Через 8,5 мин после начала процесса сорбции цеолит полностью
был насыщен нормальными углеводородами. Однако смеси продолжали про-
пускать еще в течение 13 мин. При этом 10% нормальных парафинов вы-
ходило с верхним продуктом. Затем пропускание керосина прекращали, и
давление в адсорбере в течение 0,5 мин снижали до атмосферного. Адсорбент
продували парами фракции, содержащей 84,3% я-С7Н16 и 15,7% изо- и цик-
лопарафииов, выкипающих от 95 до 114 °C. Объемная скорость паров иа
стадии продувки составляла 170 ч-1; направление потоков на стадиях ад-
сорбции и продувки было противоточным. Продувку закончили после того,
как пропустили 2,6 объемов пара иа 1 объем цеолита.
Выходящий из адсорбера продукт содержит 19,9%. углеводородов, ад-
сорбированных на внешней поверхности кристаллов и, во вторичной пористой,
структуре, 45,6% фракции, используемой для продувки, и 34,5% (масс.) нор-
мальных парафинов. Скорость отбора продукта 5560 см3/ч. После охлажден
ния до 32 °C продукт примешивали к сырью, направляемому иа адсорбцию..
Продолжительность стадии продувки 0,9 мии. Затем с помощью той же
фракции давление в системе повышали в течение 0,3 мин до 0,18 МПа и при
этом давлении проводили десорбцию в потоке паров фракции в течение
24,6 мии. Направление потоков иа стадиях продувки и десорбции совпадало.
Скорость подачи десорбента (на жидкое состояние), составляла 0,6 об./об.
цеолита в час.
Продукт содержит нормальные парафины Сю—Сю (выход 15,6% иа ис-
ходное сырье) и десорбеит (выход 77,8% от общего расхода адсорбента,
включая стадию продувки). После разделения десорбент возвращают в цикл.
Степень десорбции нормальных парафинов 80%. После этого давление в си-
стеме снижали и цикл повторяли. Выход нормальных парафинов в процессе
в целом составил 90,0% по отношению к их содержанию в сырье. Концен-
трация нормальных парафинов Сю—Сю в продукте 99,6%.
Первая промышленная установка была пущена в эксплуатацию в 1965 г.
в Тринидаде. На ией получают более 70 тыс. т нормальных парафинов в год.
Их чистота превышает 99%. В основном это углеводороды Сю—Сю, извле-
каемые из керосиновых и газойлевых фракций и применяемые в качестве-
детергентов и пластификаторов, а также в качестве сырья для получения
спиртов и других химических производных, топлива, добавок к смазочным-
маслам, растворителей.
Рабочий цикл состоит из трех стадий: адсорбции, продувки и десорбции.
На стадии адсорбции сырье в паровой фазе пропускают через слой цеолитов,
в результате чего из него извлекают нормальные парафины. После адсорбции
следует короткая стадия продувки, для удаления углеводородов других клас-
сов. Наконец, в результате стадии десорбции получают концентрат нормаль-
ных парафинов.
При разработке процесса учитывали, что адсорбент должен не только
эффективно вытеснять адсорбат, но и сам достаточно легко удаляться в по-
следующем. При депарафинизации достаточно широкой газойлевой фракции
Сю—Сго в паровой фазе этим условиям удовлетворяет бинарная смесь, со-
стоящая из легкого и тяжелого компонентов. Легкий компонент содержит в:
Выделение нормальных парафинов из фракций углеводородов 537
Свежий легкий
десорбент
парафины
Рис, 17,29. Схема установки депарафинизации с двухступенчатой десорбцией:
1—4 — адсорберы; 5—7 — фракционирующие колонны; 8 — сборник.
молекуле иа 5—7 атомов углерода, а тяжелый на 1—3 атома углерода мень-
ше, чем наиболее легкий углеводород сырья. В качестве такого десорбеита-
рекомендуют, в частности, использовать смесь н-гептаиа и н-ундекана.
В зависимости от состава адсорбата и условий проведения
процесса десорбент может быть обогащен легким [473] или тя-
желым [474] компонентом. При этом в цикл включают стадию*
продувки тем же десорбентом, которую проводят при давлении
более низком, чем давление на стадиях адсорбции и десорбции.
Для депарафинизации фракции, содержащей углеводороды
Сю—С24, разработан процесс с двухступенчатой десорбцией
[475]. Депарафинизацию проводят при температуре 300—370°C
и давлении 0,13—0,65 МПа, а стадию десорбции при давлении
на 0,01—0,14 МПа более высоком, чем давление на стадии ад-
сорбции. В цикл включена стадия продувки слоя цеолитов азо-
том, которую проводят при атмосферном давлении.
На первой ступени десорбции через цеолит пропускают по-
ток первого десорбента нормальных парафинов, соответствую-
щих первым четырем компонентам адсорбата (например, Сю—
С13), в результате чего происходит обогащение адсорбированной
фазы этим десорбентом. Затем первый десорбент удаляют в по-
токе второго десорбента — более легких парафинов, молекулы
которых содержат па 1—4 атома углерода меньше, чем самый
легкий компонент сырья (например, С7—С8). Для такой слож-
ной схемы рекомендуют выбирать сырье с незначительным со-
35—1346
538 17- Использование молекулярно-ситовых свойств цеолитов
Рис. 17,30. Зависимость выхода десорбата G'
(нормальных парафиновых углеводородов Ст—С9)
от расхода десорбеита G'R при 315 °C и 0,1 МПа
(значения G' и G'? отнесены к единице массы цео-
лита).
держанием сернистых и азотных со-
единений, олефинов, ароматических
углеводородов и легких парафинов
Ci—Сд. Технологическая схема ус-
тановки представлена на рис. 17,29.
Сырье снизу поступает в адсорбер 1, где цеолит СаА избирательно по-
глощает нормальные парафины. Рафинат, выходящий из адсорбера 1, со-
держит легкий (второй) десорбент, оставшийся в адсорбированной фазе после
предыдущей стадии. Во фракционаторе колонны 5 выделяют продукт, не со-
держащий нормальных парафинов, который направляют в хранилище, и
легкий десорбент, возвращаемый в цикл.
В это время цеолиты в адсорбере 2 подвергают продувке легким десор-
бентом. Перед этим давление в адсорбере после стадии адсорбции снижают
до атмосферного, открывая вентиль на выходе. Направление потока на ста-
дии продувки — обратное направлению на стадии адсорбции.
Выходящий из адсорбера 3 поток содержит углеводороды сырья, часть
тяжелого адсорбента и часть легкого десорбента, оставшегося в адсорбиро-
ванной фазе после предыдущей стадии. Эту смесь направляют в фракцио-
натор 7, где разделяют на продукт — нормальные парафины, легкий адсор-
бент и тяжелый десорбент. Тяжелый десорбент непосредственно возвращают
в цикл, а легкий подвергают дополнительной очистке от тяжелых углеводо-
родов во фракционаторе 6, после чего также возвращают в цикл. Тяжелый
десорбент перед стадией разделения отдувают из цеолита потоком легкого
десорбента в адсорбере 4.
Процесс «Энсорб». Эффективным десорбентом высокомоле-
кулярных углеводородов является аммиак [476, 477). Он был
применен в процессе «Энсорб». С помощью аммиака из цеолита
СаА могут удаляться поглощенные нормальные парафины
вплоть до Сзз- Преимущество использования аммиака по срав-
нению с азотом отчетливо видно при рассмотрении рис. 17,30.
Оно позволяет достичь практически полного удаления нормаль-
ных парафинов из адсорбированной
фазы; в этих же условиях степень
десорбции углеводородов продувкой
азотом не превышает 35%.
С увеличением молекулярной
массы сырья расход аммиака повы-
шается. Как показывает рис. 17,31,
при 315 °C достаточно полное уда-
Рис. 17,31. Зависимость степени десорбции ад нор-
мальных парафиновых углеводородов от удельного
расхода аммиака (иа единицу массы цео-
10‘22 5 1СГ12 5 10“ 2 5 10' лита): 3
Е„и,кг|м 1 — C,Hie — С3Н20 при 315°С; 2 — С9Н20 — СцНз,
N"3 ПРИ 315 °C; 3 — С1вНз2 — Сзз - Н6> При 385 °C.
Выделение нормальных парафинов из фракций углеводородов 539
Рис. 17,32. Зависимость отношения содержания
нормальных парафиновых углеводородов в рафи-
нате 6р к их содержанию в сырье от количе-
ства рафината Gp, отбираемого из 1 кг цеолита
(разные точки соответствуют опытам, проведен-
ным в присутствии аммиака и без него).
ление парафинов С7—Сд достигается при расходе аммиака 0,05*
кг/кг цеолита, а нормальных парафинов Сд—Cis — при расходе-
аммиака 0,2 кг/кг цеолита. В то же время в случае переработ-
ки фракции, содержащей нормальные парафины С!5—Сзз, даже-
при 385°С и расходе аммиака 1 кг/кг цеолита степень десорб-
ции не превышает 80%.
Оценивая эксплуатационные свойства аммиака как десор-
бента, следует отметить следующие его положительные качест-
ва: он легко отделяется от десорбата, термически стабилен, не
образует продуктов разложения, дезактивирующих адсорбент,,
и, наконец, вследствие щелочного характера подавляет катали-
тическую активность цеолитов по отношению к реакции крекинг-
га тяжелых углеводородов. Вследствие этого, хотя на установ-
ках «Энсорб» и предусмотрен блок регенерации цеолитов, в ра-
бочем графике намечено проводить регенерацию один раз в год..
Кроме того, аммиак достаточно легко вытесняется из цеолитов,
нормальными парафинами Сю+, что позволяет проводить про-
цесс при постоянном давлении, варьируя лишь соотношение ам-
миака и углеводородов.
На рис. 17,32 представлены данные о динамике адсорбции нормальных
парафинов Сю—Сю из фракции, в которой их содержание составляет 20%.
Опыты были выполнены на «чистом» адсорбенте и адсорбенте, содержавшем;
аммиак. Как видно нз рисунка, характеристики процесса не зависят от того;,
присутствовал ли аммиак в адсорбированной фазе перед началом стадии!
депарафинизации или нет. В этих опытах при использовании аммиака в ка-
честве десорбента достигнута степень извлечения парафинов 99%.
Эффективность процесса снижается при переработке низко-
молекулярных фракций; в этом случае предварительная адсорб-
ция аммиака значительно уменьшает степень извлечения нор-
мальных парафинов. Так, для нормальных парафинов Cs и Сд.
степень извлечения не превышает 95%. Чтобы повысить адсорб-
ционную емкость цеолитов при извлечении н-гептана и более-
низкомолекулярных парафинов, в цикл включают стадию отдув-
ки аммиака потоком рафината, не содержащего нормальных:
парафинов. В некоторых случаях аммиак добавляют к сырью:
Разбавление аммиаком позволяет провести процесс извлечения
даже высокомолекулярных парафинов типа Сго—Сз0 в паровой;
фазе при обычном давлении, избежать их конденсации в ком7
35!
540
17. Использование молекулярно-ситовых свойств цеолитов.
Рис. 17,33. Зависимость качества рафината Сю—См (температура застывания i ) в про-
цессе депарафинизации при 315 °C и 0,1 МПа от удельного выхода рафината Gp (на еди-
ницу массы цеолита):
1 — без добавки NH3; 2 —с добавкой к сырью 5,7% (мол.) NH3.
Рис. 17,34. Зависимость чистоты сП нормальных парафиновых углеводородов С»—Сю от
степени отбора десорбата на рециркуляцию ф.
муникациях и аппаратуре. В результате разбавления уменьша-
ется количество адсорбируемых примесей, повышается чистота
экстракта. Например, в процессе депарафинизации фракции
Сэ—Си при давлении 0,15 МПа и температуре 315 °C добавка
0,5 моль аммиака на 1 моль сырья позволяет повысить чистоту
экстракта с 93 до 95%; при этом адсорбционная способность
цеолитов по нормальным парафинам снижается на 10%.
Разбавление аммиаком высокомолекулярных фракций С20 и
выше приводит к увеличению скорости адсорбции и, как след-
ствие этого, к более полному удалению нормальных парафинов.
В связи с этим понижается температура застывания рафината
и становится возможным получить в качестве побочного про-
дукта высококачественное дизельное топливо.
На рис. 17,33 представлены кривые изменения температуры застывания
t3 рафината в зависимости от его выхода при депарафинизации газойля
Сю—С26 (205—300°C); процесс проводят при давлении 0,13 МПа и темпера-
туре 315 °C. Добавка 5,7% (мол.) NH3 при массовом соотношении рафината
х цеолиту 0,4 привела к понижению точки застывания с минус 8 до ми-
нус 22 °C. Опытами на пилотной установке показана возможность понижения
температуры застывания дистиллята прямой гонки нефти С плюс 4 до ми-
нус 40 °C; получено также дизельное топливо, имеющее температуру засты-
вания ниже минус 70 °C.
Чистота экстракта зависит от содержания примесей, сорби-
рующихся на внешней поверхности кристаллов и на поверхно-
сти связующего. Эта поверхность относительно невелика, но при
переработке высокомолекулярных фракций неспецифическая
адсорбция может сопровождаться капиллярной конденсацией
во вторичных порах, и тогда Чистота экстракта не превышает
75%. Капиллярную конденсацию предотвращают, добавляя к
сырью аммиак, но в этом случае концентрация нормальных па-
Выделение нормальных парафинов из фракций углеводородов 541
рафиков в экстракте составляет лишь 90—95%. В то же время
отмечено, что наибольшая масса примеси удаляется в основном
в начале стадии десорбции. Поэтому в рабочем режиме про-
мышленных установок предусмотрен возврат 30% десорбата в
сырье. В этом случае, как видно из рис. 17,34, достигнута чисто-
та экстракта выше 99%.
Основными примесями, переходящими в процессе депарафи-
низации из сырья в экстракт, являются ароматические углево-
дороды. При извлечении нормальных парафинов Сэ—CJS на пи-
лотной установке экстракт содержал 0,4% ароматических и
0,1% изо- и циклопарафинов. В результате его дистилляции с
отбором 14% тяжелой фракции содержание ароматических со-
единений в продукте было снижено до 0,1%, а чистота экстрак-
та повышена до 99,8%. В промышленных условиях доочистку
продукта от ароматических углеводородов целесообразнее про-
водить адсорбцией или кислотной обработкой. На рис. 17,35
представлена схема технологической установки.
Процесс ведут при температуре 260—370 °C и давлении 0,1—0,3 МПа
в двух попеременно работающих адсорберах. Исходную нефтяную фракцию
испаряют и пропускают через слой цеолитов СаА, содержащих в адсорби-
рованной фазе аммиак. Рафинат конденсируют и отделяют от аммиака. На
второй стадии через цеолит в качестве десорбента пропускают аммиак, на-
гретый до температуры в зоне адсорбции. Выходящий поток содержит нор-
мальные парафины, аммиак и примеси. Первую порцию адсорбента отбирают
и примешивают к сырью, а остаток конденсируют и отделяют от аммиака.
В процессе «Энсорб» требования к чистоте перерабатывае-
мого сырья понижены; это дает возможность гидроочистку вести
в более мягких условиях (0,7—2,8 МПа, 260—370°C), чем обыч-
но (5,6 МПа, 370°C). Проведение стадии гидроочистки сырья
в мягких условиях, высокая адсорбционная емкость цеолитов,
редкие регенерации цеолитов (большая продолжительность их
эксплуатации) положительно отражаются на экономике метода.
Применение аммиака в качестве десорбента является также
эффективным при очистке продуктов от сернистых соединений
и ароматических углеводородов. Содержание этих соединений в
результате пропускания экст-
ракта и рафината через цеоли-
ты типа X и Y может быть
снижено до 0,03 и 0,006% со-
ответственно. Такая высокая
степень чистоты нормальных
парафинов нужна, если их пе-
рерабатывают в присутствии
Рис. 17,35. Схема установки депарафини-
зации по методу «Энсорб»:
1, 5 — сепараторы; 2 — теплообменник; 3 —
адсорбер: 4 — нагреватели.
Нормальные парафины
542 17. Использование молекулярно-ситовых свойств цеолитов
высокочувствительных катализаторов. Кроме того, удаление аро-
матических и сернистых соединений позволяет получить высоко-
стабильное дизельное топливо, применяемое для сверхзвуковых
самолетов, и растворители высокой чистоты.
При очистке дизельного топлива вместе с ароматическими
углеводородами удаляется часть нафтенов с большим количест-
вом колец, что приводит к дополнительному повышению люми-
нометрического показателя.
Так, при пропускании дизельного топлива, выкипающего в пределах 160—
264 °C, через цеолит NaX содержание в топливе ароматических углеводородов
снижено с 18,6 до 1,75%, трехкольчатых нафтенов с 1,5 до 0,8%, серы —
с 0,08 до 0,005% • При этом люминометрический показатель повышен с 47
до 95; в случае удаления только ароматических углеводородов он должен
был составлять 81,7.
Процесс «Парекс» разработан в ГДР (478]. После испыта-
ний процесса на полупромышленной установке в г. Лёйна, где
разделению подвергались фракции, выделенные из ромашкинс-
кой нефти, было принято решение соорудить крупную установку
на заводе в г. Шведт на Одере. В качестве сырья здесь исполь-
зуют погоны нефти 190—230 °C и 230—320 °C с содержанием
нормальных парафинов 22%. Отличительной чертой процесса
«Парекс» является то, что депарафинизацию проводят в паро-
вой фазе и в среде водорода. Водород не только создает иде-
альные условия с точки зрения кинетики адсорбции, но и гид-
рирует содержащиеся в сырье непредельные углеводороды. Для
ускорения реакции гидрирования вначале в адсорбент типа
СаА, который в ГДР носит фирменное название «Зеосорб-5А»,
добавляли специальные промоторы. Затем была синтезирована
новая модификация «Зеосорб-5АМ», удовлетворительно рабо-
тающая и без гидрирующих промоторов. Технологическая схема
процесса представлена на рис. 17,36.
Рис. 17,36. Схема установки депарафинизации по методу «Парексэ:
1, 2 — адсорберы с цеолитом NaA; 3 — теплообменник; 4—6 — нагреватели; 7, 10 —адсор-
беры с цеолитом СаА; 11 — блок разделения.
Выделение нормальных парафинов из фракций углеводородов
543
Фис. 17,37. Схема установки депарафинизации с вспрыском сырья в адсорбер:
1— печь; 2, 5 — теплообменники; 3, 4 — адсорберы; 6 —сепаратор; 7 — газодувка.
Сырье осушают в адсорбере с цеолитом 1, подогревают в теплообменни-
ке 3 и смешивают с водородом, осушенным цеолитами в адсорбере 2. Затем
углеводороды нагревают до требуемой температуры, испаряют в нагревате-
ле 4 и в потоке водорода пропускают через адсорбер 7, где происходит де-
парафинизация. В это время адсорбер 8 находится на стадии продувки
нагретым водородом (5), а адсорберы 9 и 10 — на стадии десорбции. Десор-
бент перед подачей в адсорберы осушают и испаряют в нагревателе 6.
В блоке разделения 11 получают в чистом виде рафинат, экстракт, водород
и десорбент.
Вначале в качестве десорбента в процессе «Парекс» применяли н-пентан,
в последующем метод усовершенствован. В последнем варианте в качестве
десорбента используют аммиак. Содержание примесей в сырье регламенти-
ровано: не более 0,01% серы, не более 2% олефинов. Расходные коэффици-
енты на 1 т выделенных парафинов: 0,09 т жидкого топлива, 80 м3 охлаж-
дающей воды, 394 кВт-ч электроэнергии, 6 кг десорбента, 30 м3 водород-
содержащего газа, 0,35 кг цеолита.
Программа производства биологически разрушающихся мою-
щих средств и белковых веществ в Советском Союзе предус-
матривает широкое использование техники «Парекс». Чистота
нормальных парафинов, выделенных этим методом из различ-
ного исходного сырья, составляет 98—99%.
Разбавление сырья газом предусмотрено и в процессе, раз-
работанном голландской фирмой Петролеум матчепей Батафта.
Принципиальная схема процесса представлена на рис.. 17,37.
544
17. Использование молекулярно-ситовых свойств цеолитов
Углеводородное сырье нагревают в теплообменниках 2, а затем в печи 1.
до 300—500 °C, после чего вводят сверху в вертикальный адсорбер 3, запол-
ненный цеолитом СаА с частицами размером 1,6—6,4 мм. Процесс депарафи-
низации проводят при давлении 0,1—1,0 МПа. Для повышения скорости ад-
сорбции и соответственно степени извлечения нормальных парафинов в верх-
нюю часть адсорбера подают нагретый инертный газ (азот, водород, диоксид
углерода) или легкие углеводороды (метан, этан, пропан). При выборе тем-
пературного режима учитывают, что выше 450 °C начинается крекинг угле-
водородов. Соотношение высоты адсорбера к диаметру составляет 10: 1.
Чтобы уменьшить стекание сырья по стенкам адсорбера, внутри адсорбера
смонтированы специальные конические элементы.
Продукт после депарафинизации охлаждают в теплообменнике 5, выде-
ляют из газового потока в сепараторе 6, после чего инертный газ с помощью
газодувки 7 возвращают в процесс. Поглощенные нормальные парафины и
часть примесей выжигают в адсорбере 4 воздухом.
Если нормальные парафины представляют ценность, вместо воздуха по
той же коммуникации подают соответствующий десорбент. Метод применяют
в частности, для получения смазочных масел с низким содержанием нор-
мальных парафинов (менее 2%).
Процесс Гроз НИ И. Успешный синтез микросферических цео-
литов позволил осуществить процесс депарафинизации по не-
прерывной схеме. На рис. 17.38 приведена схема депарафиниза-
ции в псевдоожиженном слое микросферического цеолита MgA,
разработанная в СССР [479]. Процесс проводят в изотермиче-
ских условиях. Размер частиц адсорбента составляет от 20 до
100 мкм.
Исходное сырье насосом подают в трубчатую печь 1. Здесь оно испаря-
ется и нагревается до требуемой температуры. Депарафинизацию проводят
в адсорбере 7. Рафинат выходит из адсорбера с некоторым содержанием де-
сорбента, проникающего в адсорбер вместе с цеолитом из десорбционной
зоны. Рафинат и десорбент разделяют в ректификационной колонне 2, обо-
рудованной нагревателем, холодильником и емкостью для флегмы. Насыщен-
Рис. 17,38. Схема установки депарафинизации в псевдоожиженном слое цеолита:
1—.трубчатая печь; 2, 10 — ректификационные колонны; 3 —точки анализа фракций;
1 —сборник; 5, 9 — печи; 7 —адсорбер; 8 —десорбер.
Выделение нормальных парафинов из фракций углеводородов 545
ный микросферический цеолит непрерывно поступает в десорбер 8, где погло-
щенные нормальные парафины отдувают в потоке десорбента. Смесь нор-
мальных парафинов и десорбента поступает в ректификационную колонну 10.
Верхний продукт колонны, десорбент, нагревают в печи 9, после чего возвра-
.щают в цикл для повторного использования. Нормальные парафины, отби-
раемые из колонны снизу, охлаждают, конденсируют и направляют потре-
бителю.
Технологическая схема упрощается, если в качестве десорбента применить
вещества неуглеводородного характера, отделяемые от экстракта и рафината
простой сепарацией, и даже, возможно, водяной пар. (Цеолит MgA по мо-
лекулярно-ситовым свойствам аналогичен цеолиту СаА, но более устойчив
в среде водяного пара.)
На опытной установке ГрозНИИ производительностью 50 л
нормальных парафинов в сутки установлен оптимальный режим
процесса переработки керосиновых и дизельных фракций [480]:
температура адсорбции и десорбции 400°C, соотношение адсор-
бент: сырье=10:20, расход водяного пара 4% от массы цирку-
лирующего цеолита. Часть цеолита (5%) непрерывно отводится
на регенерацию, проводимую при 380—450°C.
При переработке сырья с температурой конца кипения 365 °C
и содержанием серы до 0,8% получены нормальные парафины
чистотой 97,5—98,7%, содержащие от 0,3 до 1,1% ароматиче-
ских углеводородов и примесь серы (0,01—0,03%). Степень из-
влечения нормальных парафинов составляла 75—85%.
Непрерывный процесс выделения нормальных парафинов ис-
пытан на опытно-промышленной установке производительностью
по сырью 150 т/сут. Масса загруженного цеолита 40 т, а интен-
сивность его циркуляции между адсорбером и десорбером
160 т/ч. Разделению подвергали неочищенные прямогонные ке-
росиновую (180—270°C) и дизельную (180—320 °C) фракции
ставропольской нефти. За 3,5 мес. непрерывной работы на уста-
новке выработано 2,3 тыс. т нормальных парафинов с примесью
ароматических углеводородов от 0,5 до 1,3%. После олеумной
очистки содержание примеси ароматических углеводородов бы-
ло снижено до 0,1%. Средняя степень извлечения нормальных
парафинов составляла 62—64%.
Процессы в жидкой фазе
Процесс «Молекс». Процессом депарафинизации в жидкой фазе,
получившим широкое распространение, является процесс «Мо-
лекс», разработанный фирмой Юниверсал ойл прадактс. Основ-
ным преимуществом является его высокая производительность.
В условиях пилотной установки жидкофазной депарафинизации был под-
вергнут [481] керосин, имеющий следующие показатели: плотность при 15,5 °C
4=0,84, интервал выкипаемости 200—270 °C, содержание углеводородов: па-
рафинов 86,4% (из них нормальных парафинов 21,6%), олефинов 0,7%, аро-
матических углеводородов 12,9%; люминесцентный показатель 5, максималь-
ная высота некоптящего пламени 26,6 мм, температура замерзания ми-
546
17. Использование молекулярно-ситовых свойств цеолитов
Рис. 17,39. Схема установки жидкофазного-
разделения компонентов в движущемся
слое адсорбента:
I, II, III и IV — адсорбционная, первая
хроматографическая, десорбционная и вто-
рая хроматографическая зоны аппарата.
нус 35 °C. После десорбции был по-
лучен 96,6 %-ный концентрат нор-
мальных парафинов с люминесцент-
ным показателем 141, максимальной
высотой некоптящего пламени 50 мм,
температурой замерзания ми-
нус 10 °C. Десорбат включал сле-
дующие нормальные парафины: 0,1 %.
С10; 30,3% С,,; 23,5% С!2; 18,5%.
С,3; 13,0% С14; 8,2% Ci5; 0,6% С16;
0,4% Ci?. Близкие результаты полу-
чены при адсорбционной депарафинизации легкого газойля, выкипающего в
интервале температур 213—324 °C. Содержание нормальных парафинов в-
десорбате в этом случае достигало 98,6%. В этих опытах отмечена зависи-
мость между чистотой и выходом нормальных парафинов.
Идеальным решением для извлечения нормальных парафи-
нов в жидкой фазе могло бы явиться осуществление процесса в
движущемся слое цеолита. Принципиальная схема такого гипо--
тетического жидкофазного процесса разделения компонентов А
и В (А обладает лучшей адсорбируемостью) представлена на
рис. 17,39; в правой части рисунка показано изменение состава
жидкой фазы по высоте колонны.
Вертикальная колонна по высоте разбита на 4 зоны, каждая из которых
выполняет самостоятельные функции. В низ зоны I вводят сырье (компо>-
ненты А+В); в низ зоны 111 — десорбент Д. В адсорбционной зоне I из
восходящего жидкого потока сырья в условиях противоточного массообмена
с движущимся слоем адсорбента избирательно извлекается компонент А.
Адсорбент, поступающий сверху в адсорбционную зону, содержит в адсорби-
рованном состоянии компоненты В и Д. Отбираемый нз колонны сверху
рафинат содержит, кроме хуже сорбируемого компонента В, десорбент Д,
который частично вытесняется из адсорбированной фазы компонентом А,
Из зоны I адсорбент, содержащий в адсорбированной фазе компоненты А
и В, поступает в первую зону вытеснительной хроматографии II. По мере
того, как адсорбент спускается по зоне II, компонент В постепенно вытес-
няется из адсорбента восходящим жидким потоком А и Д. Жидкая фаза в
нижней части зоны II содержит только компоненты А и Д.
Зона III — десорбционная, в ней компонент А полностью вытесняется из
адсорбента десорбентом Д. Из десорбционной зоны сверху отводят экст-
ракт— компонент А в смеси с десорбентом Д. В зону IV — вторую зону вы-
теснительной хроматографии — снизу вводят смесь компонентов В и Д, в ре-
зультате чего компонент Д частично вымывается из адсорбента компонен-
том В, образуя восходящий поток вытеснительной флегмы Д, поступающей
в зону III. Десорбент из выделенных в колонне продуктов—рафината и
экстракта — удаляют в результате простой ректификации.
Но процесс в движущемся слое в промышленном масштабе
может быть осуществлен лишь при наличии шариковых и мик-
росферических цеолитов, по механическим свойствам не усту-
Выделение нормальных парафинов из фракций углеводородов
547
пающим наиболее прочным активным углям. Реализация непре-
рывного процесса адсорбционного разделения на крупномас-
штабных установках связана с рядом серьезных технических
трудностей. Вследствие этого была разработана схема разделе-
ния, имитирующая технику движущегося слоя. По этой схеме
(процесс «Молекс») движение слоя заменено периодическим
переключением потоков, поступающих в вертикальный адсорбер,
заполненный цеолитами СаА, и отводимых из него.
Упрощенная схема процесса депарафинизации керосиновых
фракций, реализованная в масштабе опытной установки, пред-
ставлена на рис. 17,40 [482].
Фракцию прямой перегонки нефти, содержащую преимущественно нор-
мальные парафины Си—Си, подвергают каталитической гидроочистке, нагре-
вают и пропускают через верхнюю часть адсорбера I; из адсорбера сверху
отбирают рафинат — керосни, из которого удалены нормальные парафины.
В нижней части адсорбера проходит десорбция нормальных парафинов в по-
токе нагретого десорбента — углеводородных фракций, содержащих нор-
мальные С?—Cg.
Десорбент отделяют от рафината и экстракта в ректификационных колон-
нах 3 и 5. Периодически с помощью распределительного устройства 2 пере-
ключают потоки и смещают зоны в адсорбере.
Промышленные установки «Молекс» (рнс. 17,41) имеют более сложную
конструкцию [483]. Слой адсорбента разбит на 12 зон, каждую из которых
с помощью системы коммуникаций и распределительного устройства подклю-
чают к соответствующему технологическому потоку. Согласно схеме, в дан-
ный момент времени «активными» являются коммуникации 3, 6, 9, 12, все
остальные коммуникации заглушены. Когда позиция распределительного уст-
ройства сместится на один индекс, десорбент начнет поступать в адсорбер
Рис. 17,40. Принципиальная схема установки выделения нормальных парафиновых угле-
водородов по методу «Молекс»:
1 — адсорбер: 2 — распределитель потоков; 3 — фракционатор рафината; 4 — сборники; 5—-
фракционатор нормальных парафинов; 6—печи.
548
17. Использование молекулярно-ситовых свойств цеолитов
по коммуникациям 4 вместо коммуникации 3. Соответственно сместятся и
другие позиции, например удаление рафината из адсорбера будет осущест-
вляться из позиции 1, вместо позиции 12.
При разделении керосиновой фракции, содержащей 35% нормальных па-
рафинов (преимущественно Си—С14) иа промышленной установке «Молекс»
получен экстракт, в котором концентрация нормальных парафинов составляет
96,1%. Состав примесей приблизительно таков; 50% ароматических, 25% изо-
парафиновых и 25% циклопарафиновых углеводородов'. -Рафинат используют
в качестве дизельного топлива.
Капиталовложения в установку производительностью по сырью 400 м3/сут
в ценах 1965 г. составляют 1466 тыс. долл., производительностью
1600 м3/сут —2750 тыс. долл.; стоимость выделенных нормальных парафинов
соответственно 2,1 и 1,5 цент/л при их концентрации в исходном сырье 20%..
Эксплуатационные расходы равны соответственно 28 и 19 долл./т нормаль-
ных парафинов. Степень извлечения нормальных парафинов в технико-эконо-
мических расчетах принята 85%, концентрация н-парафинов в экстракте 96%.
[484].
В связи с тем, что процесс «Молекс» проводят при относи-
тельно низких температурах, сернистые и азотистые соединения,
непредельные углеводороды, если они присутствуют в сырье,
необратимо сорбируются цеолитами, снижая их адсорбционную
емкость. Поэтому сырье, поступающее в адсорбционную уста-
новку «Молекс», подвергают тщательной гидроочистке. В этих
условиях длительность эксплуатации цеолитов оценивается в
2 года.
Выделение нормальных парафинов из фракций углеводородов 5491
Рис. 17,42. Зависимость адсорбционной способно-
сти а по кислороду, аргону и азоту при минус
196 °C от содержания катиона кальция сСа при
ионном обмене натрия на кальций в шабазите.
управления разделением
Точное регулирование процесса
в жидкофазном способе обеспечи-
вают, применяя автоматический
анализатор, позволяющий каждые
1—2 мин определять содержание
нормальных парафинов в техноло-
гических потоках с точностью 1%. Для
используют также малогабаритные вычислительные машины,
контролирующие 24 параметра процесса.
Выделение пара-ксилола. Конструктивное решение процесса
«Молекс» использовано для выделения пара-ксилола из смеси
с другими изомерами (485, 486]. Его проводят при температурах
120—175 °C и умеренном давлении.
Смесь ксилолов пропускают через слой цеолита, при этом извлекают
весь пара-ксилол и небольшую долю других изомеров. Примеси отмывают
частью выделенного пара-ксилола. Затем его вытесняют из цеолита угле-
водородным десорбентом. Десорбат и десорбент разделяют дистилляцией и:
последний возвращают в цикл.
Степень извлечения пара-ксилола достигает 100%, причем продукт по-
лучают очень высокой чистоты. Для сравнения укажем, что метод фракцион-
ной кристаллизации в одну ступень обеспечивает извлечение лишь 60% пара-
ксилола. Для разделения используют специальный адсорбент, разработанный
фирмой Ричард Нойзил и, видимо, относящийся к типу ВаХ.
Получение реактивного топлива. Цеолиты СаА успешно при-
меняются для получения высококачественного реактивного топ-
лива [487]. С этой целью керосин подвергают двухступенчатой
гидрогенизации в следующих условиях: температура 121—
455 °C, давление от 2,2 до 14,1 МПа, объемная скорость жидкого
сырья 0,2—6,0 ч-1, расход водорода от 0,26 до 1,4 м3/л углево-
дородов. В реактор загружен кобальт-молибденовый, кобальт-
вольфрамовый, никель-молибденовый или молибден-вольфрамо-
вый катализатор на носителе — оксиде алюминия или алюмоси-
ликате. Адсорбцию проводят
примерно при тех же темпера-
туре и давлении; объемную
скорость изменяют в пределах
1-—6 ч-1. После удаления нор-
мальных парафинов цеолитами
Рис. 17,43. Схема установки очистки арго-
на от кислорода цеолитами:
1, 2 — адсорберы на стадии очистки; 3--
адсорбср на стадии регенерации; 4 — газо-
дувка; 5 — нагреватель.
550
17. Использование молекулярно-ситовых, свойств цеолитов
СаА продукт обладает отличными показателями по вязкости и
температуре застывания.
Очистка аргона от кислорода
Молекулярно-ситовые свойства цеолитов используют для разде-
ления не только углеводородов, но и других веществ, например
для разделения аргона и кислорода.
Катионы, входящие в состав цеолитов, имеют слишком ма-
лые размеры, чтобы оказать влияние на свободный объем внут-
рикристаллической структуры. Однако они могут размещаться
таким образом, что замедлят или даже полностью прекратят
диффузию поглощаемых молекул внутрь адсорбционной полости.
На рис. 17,42 показано, как изменяется низкотемпературная
-сорбция газов шабазитом в процессе обмена катионов Na+ на
катионы Са+.
Варьирование размера катиона позволяет в некоторых_слу-
чаях осуществить тонкое разделение веществ на основе разницы
в размерах адсорбируемых молекул. Так, при минус 183 °C ле-
(винит [488] и цеолит NaA [489] не сорбируют аргон, но сорби-
руют кислород. Установки адсорбционной очистки аргона от
кислорода производительностью от 40 до 180 м3/ч имеется в
СССР и США [490]. Схема установки представлена на
рис. 17,43.
Загрязненный аргон пропускают последовательно через два адсорбера
1 и 2 с встроенными змеевиками, охлаждаемыми кипящим внутри них жид-
ким воздухом. Адсорбер 3 находится иа стадии регенерации. Для регенерации
используют сухой азот с установки разделения воздуха, подогретый до 90 °C.
Нагрев цеолита заканчивают, когда температура в слое достигнет 20 °C. Пос-
ле этого слой охлаждают жидким воздухом с продувкой небольшого коли-
чества неадсорбирующегося гелия. В конце продувки содержание азота в ге-
лии не должно превышать 0,1% (об.). Примесь кислорода в аргоне после
.адсорбционной очистки не превышает 2 част, на 1 млн.
18
ПРИМЕНЕНИЕ
АДСОРБЕНТОВ ДЛЯ
КОНДИЦИОНИРОВАНИЯ
И ХРАНЕНИЯ
ПИЩЕВОЙ ПРОДУКЦИИ
Для кондиционирования и хранения пищевой продукции исполь-
зуют адсорбенты различных типов: активные угли, природные
глины и другие. К этим адсорбентам, особенно к тем из них,
которые непосредственно контактируют с продуктами, предъяв-
ляются требования биологической безвредности: они должны
быть прочными, не токсичными и не канцерогенными.
Улучшение вкусовых качеств водки, коньяка, вин
Водкой называют напиток, получаемый смешением этилового-
спирта и специально подготовленной воды. Перед смешением
с водой спирт-сырец подвергают ректификации с отбором трех
основных фракций: тяжелой (сивушные масла), легкой (низко-
кипящие альдегиды и другие кислородсодержащие органичес-
кие соединения) и средней — собственно спирт. Однако в спир-
те-ректификате всегда остаются примеси, снижающие его вкусо-
вые качества. Продукт, полученный после смешения, в ликеро-
водочном производстве называют сортировкой.
Типы водок отличаются друг от друга характером неболь-
ших вкусовых добавок (сахара, меда, лимонной кислоты, окса-
лата натрия, перманганата калия и др.), но во всех технологи-
ческих схемах производства этого продукта предусмотрена обя-
зательная стадия обработки сортировки активным углем [491,
492]. В водочном производстве используют активные угли расти-
тельного происхождения (березовый, БАУ, буковый). Они от-
личаются низким содержанием золы (менее 8%), низкой насып-
ной плотностью (0,26 г/см3), умеренным развитием микропор-
(0,23 см3/г) и небольшим объемом мезопор (0,08 см3/г). В схе-
мах с неподвижным слоем угля размер гранул угля БАУ сос-
тавляет 1—4 мм, в схемах с псевдоожиженным слоем размер*
зерен должен быть 0,2—0,4 мм.
В сортировке, так же как и в спирте-ректификате, содержат-
ся примеси альдегидов, кетонов, сложных эфиров, карбоновых,
кислот и высокомолекулярных веществ (сивушные масла).
Уголь не только извлекает эти компоненты, но и способствует
-552
18. Применение адсорбентов для обработки пищевой продукции
их каталитическому превращению, протекающему в адсорбиро-
ванной фазе: окислению непредельных соединений, спиртов и
альдегидов, этерификации кислот и омылению сложных эфиров
[493]. Кислород для реакций окисления поступает из водно-
спиртовых растворов, где он содержится в растворенном состоя-
нии. При этом этиловый спирт сам играет активную роль.
Наиболее типичными реакциями являются следующие:
Окисление
Фурфурол
НС—сн
II
НС
с—с
(18.1)
Пирослизевая кислота
СН3СН2ОН + 1/2О2 --» СН3С-^ + Н2О
\н
Этиловый спирт Уксусный
альдегид
zO о
CH3cf + 1/А —сн3—С"
хн \он
(18.2)
(18.3)
Уксусный
альдегид
Уксусная
кислота
Этерификация
/О
CH3C(f + СН3СН2ОН
\ОН
СН3С—ОС2Н5 + н2о
(18.4)
о
•Омыление
/О
CH3CH2OC(f + Н2О
\СН3
Этидацетат
/О
СН3СН2ОН + СН3С"
(18.5)
Каждый эфир обладает специфическим ароматом. Например, уксусно-
этиловый эфир обладает фруктовым запахом, уксусноизоамиловый эфир —
запахом грушевой эссенции, изовалерианоизоамиловый эфир — запахом яб-
лочной эссенции, н-масляноэтиловый эфир — запахом ананасной эссенции.
После контакта сортировки с активным углем образуются новые эфиры —
каприловой, пеларгоновой, каприоновой и других высокомолекулярных кис-
лот [494]. Сложные эфиры маскируют оставшиеся неизвлеченными неприятные
примеси и улучшают вкусовые качества водки.
Эффективность адсорбционной очистки во многом определя-
-ется пористой структурой активного угля, при этом решающая
роль принадлежит микропорам. Для использования в промыш-
ленных условиях рекомендованы угли с предельным объемом
адсорбционного пространства 0,3 см3/г. Так как для использо-
вания в пищевой промышленности особенно большое значение
Улучшение вкусовых качеств водки, коньяка, вин
553
Рис. 18,1. Схема установки очистки водки в неподвижном слое активного угля:
1 — песочные фильтры; 2 — адсорберы с активным углем; 3 — керамические фильтры;.
4 — холодильник; 5 —сборник отгона.
приобретает механическая прочность адсорбента, такие актив-
ные угли целесообразно получать при степени обгара до 30%.
Размеры микропор определяют скорость каталитических реак-
ций, протекающих в адсорбированной фазе. С этой точки зре-
ния наиболее перспективны угли с размером пор 0,8—1,3 нм
[495]. Окисленность поверхности угля отрицательно влияет на
процесс [496].
Сахароза, вводимая вместе с сахаром в состав сортировок
при получении водок «Экстра» и «Столичная», не поглощается
активным углем из спиртово-водочных растворов крепостью
выше 30%. В то же время уголь частично извлекает глюкозу
и фруктозу, которые присутствуют в меде, добавляемом к сор-
тировкам водки «Российская». В этом случае добавку следует
вводить после пропускания сортировки через слой активного
угля. Схема установки очистки сортировки в неподвижном слое
активного угля представлена на рис. 18,1.
Сортировку пропускают через песочные фильтры I, пв.ч последеmiricjii.iio-
включенных адсорбера 2 и после очистки от уют.пой пыли и песочных или
керамических фильтрах 3 направляют в сборные чаны. Адсорберы в виде
цилиндров выполнены из листов нержавеющей стали толщиной 3 мм. Их
диаметр 0,7 м, высота 4,2 м. Масса угля и одном адсорбере составляет от
250 до 300 кг. Направление потока в нем па стадии очистки — снизу вверх.
Предельно допустимые скорости подачи сортировок на один адсорбер са
свежим или регенерированным углем в зависимости от сорта водки измеия-
554
18. Применение адсорбентов для обработки пищевой продукции
ются в пределах* 30—60 дал/ч. Длительность стадии очистки соответствует
пропусканию через адсорбер от 15 тыс. до 100 тыс. дал.
Адсорбер переключают на регенерацию 3—4 раза в год, когда органо-
лептический и химический анализ зафиксируют снижение качества водки до
предельно допустимого уровня. Регенерацию отработанного активного угля
проводят сухим водяным паром при 115 °C, который пропускают через ад-
сорберы сверху вниз. В результате регенерации из одного адсорбера получа-
ют от 50 до 60 дал спиртового отгона крепостью 55—60%, который конден-
сируют в холодильнике 4, собирают в бачке 5 и направляют на ректифика-
цию. Продолжительность стадии регенерации составляет 3—4 ч, расход пара —
4 кг на 1 кг угля. Часть отгона, не содержащего спирта, отправляют в ка-
нализацию. После регенерации уголь охлаждают в токе воздуха до 50 °C,
при этом из него удаляется поверхностная влага, и он насыщается кислоро-
дом. Расход угля в результате его дезактивации, механического разрушения
и уноса оценивается в 1,2 кг на 1000 дал сортировки.
Иногда регенерацию угля проводят в централизованном порядке—про-
каливанием во вращающихся печах при 800—850 °C. Потери угля при прока-
ливании достигают 20%, но угли практически полностью восстанавливают
свои адсорбционные и каталитические свойства.
На заводах производительностью менее 700 тыс. дал/год сор-
тировку очищают периодически контактным методом ([497].
Активный уголь отсевают от мелких частиц (до 0,5 мм), засы-
пают в чан с сортировкой из расчета 2 кг на 1000 дал водки и
суспензию перемешивают в течение 30 мин сжатым воздухом,
который из чана выводят в атмосферу через спиртоловушку.
После этого систему подвергают двухступенчатому отстаиванию
и осветленную часть окончательно освобождают от частиц угля
в двух последовательно включенных фланцевых рамных филь-
трах с поверхностью фильтрации 2 м2, а затем на фильтр-прес-
•се.
На Московском ликеро-водочном заводе эксплуатируется не-
прерывно действующая установка очистки сортировки в псевдо-
ожиженном слое мелкозернистого активного угля производи-
тельностью 500 дал/ч.
Предварительно профильтрованную в песочных фильтрах сортировку очи-
щают в четырех последовательно включенных адсорберах — цилиндрах диа-
метром 0,7 м и высотой 3,5 м с коническим днищем. Адсорберы предвари-
тельно на 55—60% по объему загружают активным углем. Сортировку по-
дают в адсорбер через кольцевую перфорированную трубу, расположенную
в конической части, в направлении снизу вверх. При скорости потока
5 л/(м2-с) уголь «взрыхляется», и занимаемый им объем увеличивается в
1,5 раза. В условиях турбулентного режима при небольшом размере частиц
адсорбента массообмен между жидкой и твердой фазами происходит очень
интенсивно. В верхней части адсорбера расположен сепаратор угля диаметром
1,2 м и высотой 0,4 м. Благодаря снижению скорости потока и изменению
направления его движения (от центра к периферии) унесенные сортировкой
частицы угля вновь возвращаются в адсорбер. На крышке сепаратора распо-
ложен люк для загрузки аппарата активным углем и штуцер для отвода
очищенной сортировки. Уголь из адсорбера выгружают через люки, располо-
* Дал — декалитр — принятая в виноделии единица измерения объема жид-
кости. (Прим. ред.).
Осветление пива и фруктовых соков
555
женные в нижней его части. От угольной пыли сортировку очищают на двух
ступенях фильтрации.
Система регенерации предусматривает прохождение потока водяного
пара через адсорбер сверху вниз, и принципиально не отличается от описан-
ной выше в схеме для условий неподвижного слоя адсорбента.
Вкусовые качества коньяка улучшают, добавляя 5 г пыле-
видного активного угля на 1 л [498]. В результате контактиро-
вания с адсорбентом из коньяка удаляются кислоты, фурфурол
и таннин. Уголь не поглощает эфиры, ацетальдегиды и высшие
спирты. При большей дозе угля можно достичь частичного уда-
ления микропримеси сивушного масла.
При улучшении качества вин технологи встречаются с тремя
задачами: обесцвечивание вина без изменения его «букета»,
удаление привкуса и запаха без изменения цвета, обесцвечира-
ние одновременно с изменением «букета». Порошкообразный
уголь сначала смешивают с небольшим количеством вина (или
виноградного сусла) и суспензию закачивают в чан, в котором
находится вино (или сусло). Для интенсификации процесса
адсорбции в чан снизу подают воздух, который барботирует че-
рез слой вина. Может быть предусмотрено и механическое пе-
ремешивание. Время контакта составляет несколько часов. Доза
угля, в зависимости от задачи и типа вина, составляет от 100
до 1200 г на гектолитр. Частицы угля удаляют в результате
отстаивания. Если при отстаивании мутность полностью не ис-
чезает, вино пропускают через фильтр.
Осветление пива и фруктовых соков
Прозрачное и стойкое пиво получают после специальной обра-
ботки таннином, водорастворимыми синтетическими полимера-
ми, ферментами, активным углем или силикагелем. Порошко-
образный уголь вводят в пиво перед его розливом для улучше-
ния цвета и запаха (удаления фенолов) 1[499]. Доза угля сос-
тавляет 20—25 г на гектолитр. Кроме того, активный уголь при-
меняют для очистки воздуха, диоксида углерода, а также воды.
В СССР, ФРГ, Англии, США широко используют различные
методы осветления пива бентонитовыми глинами. На одном из
крупнейших пивоваренных заводов бентонит вводят в танк с
пивом и оставляют на 5 суток при 0°С. Химически инертные
вещества, обладающие невысокой щелочностью — бентониты хо-
рошо набухают в водной среде. Отрицательно заряженные час-
тицы бентонитовых глин, образующих суспензию в пине, связы-
вают положительные ионы металлов, присутствующих в нем,
и сорбируют коллоидные частицы протеинов, которые в кислой
рреде пива заряжены также положительно. Добавка 0,2—0,4 г/л
бентонитовых глин Средней Азии и Казахстана, размельченных
до частиц размером 0,25 мм, к жигулевскому пиву с последую-
55 6 18- Применение адсорбентов для обработки пищевой продукции
Рис. 18,2. Кинетика осветления виноградного сокэ
молдавским бентонитом при разных дозах адсорбен-
та сад Гординешты-А (мутность пробы М выражена
в единицах шкалы фотоэлектрического колориметра).
щей выдержкой суспензии в течение
7 сут приводит к осветлению пива
[500]. Цветность, кислотность и вкус
пива при этом не изменяются.
Фруктовые соки содержат в своем:
составе сложную смесь белковых, пек-
тиновых, дубильных, красящих ве-
ществ в молекулярном, коллоидном и
граничащем с коллоидным — более
грубодисперсном состояниях. Эти при-
меси вызывают мутность сока. Освет-
ляют фруктовые соки также бентонитами (контактным спосо-
бом). Традиционная технология осветления включает три ос-
новных этапа: контактирование сока с бентонитами при интен-
сивном перемешивании, отстаивание сока для отделения отра-
ботанного бентонита, содержащего адсорбированные примеси,
и окончательное осветление, осуществляемое методом фильтра-
ции.
Процесс осветления, очевидно, основан не только на явлении
физической адсорбции, но и дополняется другими видами взаи-
модействия между твердой и жидкой фазами, например ионным
обменом.
Кердиваренко {501] в результате многочисленных исследова-
ний адсорбционных методов очистки соков, вин и других про-
дуктов обратил внимание на кинетический аспект проблемы кон-
тактной очистки. На рис. 18,2 приведены кинетические кривые
осветления виноградного сока молдавским бентонитом (суспен-
зию перемешивали при частоте вращения мешалки 16 с-1, или
1000 об/мин). Из рассмотрения кривых становится ясным, что
снижение мутности (осветление) происходит при очень крат-
ковременном контакте (менее 1 мин). Эти результаты указали
на возможность интенсификации процессов контактной очистки
путем включения в схему мешалок быстрого контактирования
очищаемой жидкой среды с адсорбентом и центрифуг для вы-
деления отработанной суспензии [502]. Таким образом, продол-
жительность адсорбционной очистки сократилась до десятков
секунд, а процесса осветления в целом — до десятков минут,
вместо многих часов и суток по прежней технологии 1[503]. Од-
новременно сократились габариты установок: из схемы удалены
крупные отстойники отработанной суспензии.
Схема прошла промышленные испытания и внедряется на
соковых и винодельческих заводах Молдавии [504]. Доза бенто-
нитов в 5 г/л обеспечивает полное осветление соков. Содержа-
Очистка сахарных сиропов
557
ние белковых веществ в виноградном и яблочном соках снижа-
ется на 40%, пектиновых веществ — на 20—25%. Адсорбция
•белковых и других крупных молекул происходит преимущест-
венно на внешней поверхности частиц бентонита: на Г; м2 по-
верхности раздела фаз адсорбируется приблизительно 1 мг бел-
ковых веществ. При равновесной конечной концентрации бел-
ков в соке 100 мг/л одним граммом бентонита поглощается
10—20 мг белковых веществ*.
Очистка сахарных сиропов
Использование твердых адсорбентов для очистки сахарных си-
ропов началось в 1812 г. на базе костяного угля.
Костяной уголь получали в результате термической обработки (750—
‘950 °C) предварительно измельченных костей, подвергнутых экстракции орга-
ническими растворителями. Основной составной частью костяных углей явля-
ется фосфат кальция (до 80%), в его структуру вкраплены частицы мелко-
дисперсного угля (около 20%), однако именно эти вкрапления и определяют
обесцвечивающие свойства костяного угля. В то же время, фосфатные соеди-
нения являются эффективным средством для поглощения солей. С начала
XX в. костяной уголь в сахарной промышленности стал вытесняться активным
углем.
Обесцвечивание сиропов с помощью адсорбента является
последней стадией очистки сахара. Веществами, которые ока-
зывают наиболее интенсивное воздействие на цвет сахара, явля-
ются меланоидины — азотистые вещества, образовавшиеся при
восстановлении сахара аминосоединениями, особенно их колло-
идные частицы с молекулярной массой от 8000 до 15000. Одно-
временно с коллоидными частицами из сиропа углем поглоща-
ются и другие примеси, в том числе неорганического происхож-
дения (зола). В связи с этим при эксплуатации на обесцвечи-
вающих установках изменяется химический состав активного
угля: в нем понижается содержание углерода, но увеличивается
зольность, содержание азота, а также водорода и кислорода.
Принципиальная схема двухступенчатой очистки сахарного си-
ропа представлена на рис. 18,3.
Воду и сахар смешивают в обогреваемом аппарате I, где сахар расплав-
ляется и образуется сироп. Обесцвечивание сначала проводят в контакторе 2,
в который подают уголь, частично отработанный во второй ступени. Расход
угля составляет 5—10 г на 1 л сиропа, время контакт;! 30 мин. Скорость
адсорбции примесей из водных растворов в значительной мере зависит от
температуры. При равном времени контакта с пылевидным активным углем
(иапример, 20 мин) остаточное содержание примесей в сахарном сиропе при
80 °C в 2 раза, а при 70 °C в 3,5 раза больше, чем при 90 °C.
Отработанный уголь в виде суспензии насосом подают на фильтр-
пресс 3. В процессе фильтрования толщина слоя угля на фильтрующем эле-
* Адсорбенты, в частности порошкообразный кристаллит цеолита NaA, ис-
пользуют также для концентрирования соков. (Прим, ред.)
558
18. Применение адсорбентов для обработки пищевой продукции
Рис. 18,3. Схема установки двухступенчатой очист-
ки сахарного сиропа:
1 — смеситель; 2.1- контакторы; 3,5 — фильтры,
менте увеличивается приблизительно до-
30 мм, в связи с чем сопротивление систе-
мы возрастает от 0,2 до 0,4 МПа. Затем
уголь смывают 5—10-кратным избытком
воды и выводят из системы. Сахарный си-
роп подвергают «углеванию» во второй
ступени (контактор 4 и фильтр 5), Сте-
пень обесцвечивания достигает 99%.
Регенерацию порошкообразных углей
проводят лишь на крупных заводах. Для
удаления золы угли сначала кипятят в
1—4%-ной соляной кислоте, а для удаления
поглощенных красящих веществ в 1%-ном
растворе NaOH. Затем, после сушки угля,
в печах при 500—600 °C подвергают кре-
кингу органические вещества, поглощенные
углем. При этом, естественно, поры угля
частично блокируются образовавшимся уг-
леродом, и адсорбционная способность угля
снижается на 10—20%. Потери порошко-
образного угля при регенерации составля-
ют 5—10%. Расход угля от 0,5 до 1% мас-
сы сахара-рафипада.
Типичный порошковый уголь для обесцвечивания — деколар,
изготовляемый в Венгрии путем химической активации, имеет
развитую систему микро- и макропор. Распределение пор имеет
следующий характер (преимущественный размер частиц угля
50—100 мкм):
Размер пор, нм До 1,1 1,1—2,0 2,0—10,0 10,0—100,0 Выше 100,0
Объем пор, см%г 0,12 0,39 0,52 0,69 До 0,78
Кроме сахарной промышленности, угли такого типа приме-
няют для обесцвечивания напитков (коньяков, вин, фруктовых
соков), лекарств и наркотиков (ацетанилида, алкалоидов, ко-
феина, салициловой кислоты), неорганических кислот (борной,
фосфорной), органических кислот (лимонной, молочной, уксус-
ной) и других органических веществ (ацетойа, спиртов, глице-
рина), компонентов средств чистки (тетрахлоруглерода, бензи-
на, эфирных масел), жиров и масел (например, воска), смол
(агара, желатины, пектина) и неорганических веществ (буры,
сульфатов натрия и цинка, хлорида свинца). В ряде случаев
одновременно с обесцвечиванием продуктов происходит удале-
ние запаха, привкуса, коллоидных и иных примесей. Угли этого
типа эффективны и в процессах водоочистки.
Для очистки сахарных сиропов применяют и установки с
гранулированным активным углем. В этом случае расход угля
Очистка сахарных сиропов
559
на порядок меньше—не выше 0,1% массы сахара-рафинада.
Очистку проводят при 80°C, пропуская сироп через слой адсор-
бента сверху вниз. Адсорберы высотой 8—10 м и диаметром 1 м
работают при скорости потока сиропа 1,5—2,5 м/ч. Время кон-
такта сиропа с адсорбентом 3—6 ч, длительность работы загру-
женной массы (загрузки) угля 15—30 сут. Отработанный уголь
выгружают из колонны, отмывают от поглощенных неорганиче-
ских примесей, сушат, подвергают термической обработке при
1000—1100 °C в слабоокислительной атмосфере и активируют
паром [505].
По другой методике гранулированный отработанный уголь
обрабатывают горячей водой, затем при 80 °C 0,5 н. NaOH, от-
мывают избыток щелочи водой и обрабатывают 0,33 н. НС1 при
50°C до тех пор, пока выходящая жидкая фаза не покажет
слабощелочную реакцию. С числом циклов регенерации, прове-
денной этим методом, поглотительная способность угля посте-
пенно снижается, стабилизируясь после 40 циклов на уровне
65% активности свежего угля.
В Советском Союзе, США, Англии, Японии и в ряде других
стран разработаны и применяются непрерывные методы обес-
цвечивания сахарных сиропов в движущемся слое активного
угля [506]. Установки, построенные на Яготинском и Черкасском
сахарных заводах, эксплуатируются на угле АГС-3. Высота зо-
ны контактирования в адсорберах составляет 7,5 м, диаметр
1 м. Производительность такой установки равна 3—3,5 м3/ч.
Скорость потока сиропа — около 3,5 м/ч. Характерной особен-
ностью этих установок является то, что применяемые скорости
обеспечивают расширение слоя адсорбента, но еще не переводят
его в псевдоожиженное состояние.
Отработанный уголь выводят из нижней части колонны, про-
мывают восходящим потоком воды, по гидротранспортеру на-
правляют сначала в циклон, а затем во вращающуюся печь,
где при 930 °C он регенерируется. По гидротранспортеру уголь
поступает сверху в адсорбер, но предварительно его освобож-
дают от капельной влаги на сите с размером ячеек 2,5 мм.
Регулирование состава газовых сред при хранении
сельскохозяйственной продукции
Адсорбционные способы регулирования газового состава искус-
ственных атмосфер применяют в сельском хозяйстве, торговле
и пищевой промышленности при длительном хранении скоро-
портящихся продуктов сельскохозяйственного производства:
плодов, овощей, картофеля, ягод, винограда, кормов, семян
([507—510].
Метод хранения плодоовощной продукции в регулируемых газовых сре-
дах (РГС) представляет собой усовершенствованный метод холодильного
560
18. Применение адсорбентов для обработки пищевой продукции
.хранения, при котором плоды и овощи сохраняют в герметизированных хо-
лодильных камерах при постоянных оптимальных значениях температуры, от-
носительной влажности, пониженной концентрации кислорода (в 7—10 раз
меньшей, чем в атмосферном воздухе) и повышенной концентрации диоксида
углерода. Регулируемые газовые среды плодоовощехранилищ отличаются по
•составу от обычного воздуха только соотношением основных компонентов —
азота, кислорода и диоксида углерода. Содержание других веществ в атмо-
сфере холодильных камер не должно превышать предельно допустимых кон-
центраций.
Научное обоснование метода дал в 1914 г. профессор Ф. В. Церевитинов
в статье «Влияние углекислого газа на сохранение фруктов». В процессе хра-
нения плодов и овощей происходит постепенное дозревание и старение, что
сопровождается изменением физического и химического состава тканей, поте-
рей массы за счет распада углеводов (сахаров, крахмала), уменьшением за-
паса органических кислот, дубильных веществ, витаминов, изменением вкуса,
окраски, размягчением тканей. При этом плоды поглощают кислород из окру-
жающей среды, выделяют диоксид углерода, водяные пары, этилен, органиче-
ские летучие вещества. Чем интенсивнее протекают биохимические процессы
внутри плодов, тем скорее наступает их старение, снижается качество, разви-
ваются микробиологические и физиологические заболевания.
На интенсивность процессов жизнедеятельности можно влиять различ-
ными способами: замедлить процессы дозревания можно снижением темпера-
туры хранения (не ниже криоскопической), уменьшением концентрации кис-
лорода и увеличением концентрации диоксида углерода в определенных пре-
делах. Интенсивность протекания биохимических процессов при хранении
плодов и овощей в РГС уменьшается примерно в три раза по сравнению
с интенсивностью их протекания в обычной атмосфере при одинаковой тем-
пературе.
Это приводит к продлению сроков хранения на несколько месяцев, сни-
жению потерь от микробиологических и физиологических повреждений в 2—
3 раза, лучшему сохранению качества, витаминов и других полезных ве-
ществ. Если учесть, что потери плодоовощной продукции составляют значи-
тельную часть урожая, то можно сделать вывод: применение РГС даст
такую добавку к реализуемой продукции, которая соизмерима со среднего-
довым приростом ее производства.
Эффект совместного влияния низких температур, низких концентраций
кислорода, повышенных концентраций диоксида углерода и повышенной от-
носительной влажности зависит от конкретных значений этих параметров,
вида продукции, ее состава, срока хранения. Рекомендуемые значения кон-
центраций активных компонентов (кислорода и углекислого газа) требуется
поддерживать с погрешностью, не превышающей ±0,5%. В табл. 18-1 приве-
дены примеры рекомендуемых к применению параметров регулируемых газо-
Таблица 18-1. Примеры применяемых газовых сред плодоовощехранилищ
Плоды, ОВОЩИ Состав газовых сред, % °C Плоды, овощи Состав газовых сред, % °C ..
со2 | о2 n2 соа i оа n2
Груши 5 2 93 0 Салат 2 3 95 1
2 3 95 0 Помидоры 2—3 4—5 92—94 1
Яблоки 3 3 94 4 Ягоды 10 3 87 0
0 2 98 4 Цветы 2 1 97 0
Картофель 5 1-5 3 1—3 92 93—97 3 10 Грибы 3 2 95 0—2
Хранение сельскохозяйственной продукции
561
вых сред по материалам Международной организации по стандартизации
(ИСО) Международного института холода и других источников.
Оптимальные газовые среды в плодоовощехранилищах создаются сразу
же после загрузки и герметизации холодильных камер различными способа-
ми. Самый простой из них — естественная модификация атмосферы за счет
дыхания плодов. В этом случае требуемая концентрация кислорода дости-
гается за две-три недели, а концентрация диоксида углерода повышается до
предельно допустимых величин через несколько суток.
В расчетах принимают среднюю интенсивность выделения СОг плодами
(яблоками) в пределах от 3 до 5 г/(т-ч): дыхательный коэффициент, опре-
деляемый как отношение объема выделившегося диоксида углерода к объему
поглощенного кислорода, находится в пределах 1,0—1,1. Емкость холодильных
камер с регулируемыми газовыми средами составляет 100—300 т (до 1000 т),
плотность загрузки равна 0,2 т/м3. Приведенные цифры и опыт эксплуатации
показывает, что скорость повышения концентрации диоксида углерода в не-
регулируемых герметичных плодоовощехранилищах составляет 0,5—1,0% в
сутки.
При естественном способе создания оптимальной газовой
среды (пассивном способе создания РГС) необходимо через
двое-трое суток после герметизации холодильных камер уда-
лять избыток СО2 из атмосферы хранилища, что может быть
сделано с помощью адсорбционной установки.
На рис. 18,4 представлена принципиальная схема адсорбци-
онной установки марки ТМ9, выпускаемой фирмой Эйр Ликвид
Рис. 18,4. Принципиальная схема адсорбционной установки типа ТМ9:
1, 2, 6—9—клапаны; 3 — нагреватель; 4, 11 — цеолнтовые адсорберы; 5 — регенеративный
теплообменник; 10 — увлажнитель; 12, 13 — вентиляторы.
Рис. 18,5. Принципиальная схема газогенераторной установки типа УРГС-2:
1, 4, 14 — газодувки; 2, 6 — четырехходовые клапаны; 3, 8 — адсорберы с активным уг-
лем; 5, 15трехходовые клапаны; 7 — исполнительный механизм; 9 — контактный холо-
дильник; 10 — сборник конденсата; И — емкость для хранения газов; 12 — камера сгора-
ния; 13 — устройство подготовки газов.
36—1346
562 18. Применение адсорбентов для обработки пищевой продукции
(Франция) для удаления диоксида углерода из атмосферы хо-
лодильных камер с регулируемой газовой средой.
Установка включает в себя два, заполненных цеолитами адсорбера, ра-
ботающих попеременно в режимах очистки газовой среды и регенерации.
В режиме очистки газовую среду подают из хранилища вентилятором 13
через клапан 9 в адсорбер 4, где очищают от диоксида углерода и возвра-
щают в хранилище через клапан 1 и увлажнитель 10. Степень увлажнения
контролируется специальным регулятором. Продолжительность режима очист-
ки равна 1,5 ч.
Режим регенерации (продолжительность 1,5 ч) состоит из двух
фаз: нагревания цеолита и его охлаждения, каждая продолжительностью
0,75 ч. В фазе нагревания цеолита атмосферный воздух вентилятором 12 че-
рез клапан 6 подают в регенеративный теплообменник 5, затем в нагреватель 3
и через клапан 2 направляют в адсорбер 11, из которого воздух и продукты
десорбции удаляют через клапаны 8 и 7.
В фазе охлаждения цеолита клапаны 6 и 7 переключают. Вентилятор 12
подает воздух через клапаны 7 и 8 в адсорбер 11, вследствие чего цеолит
охлаждается. Подогретый в адсорбере воздух проходит клапан 2, нагрева-
тель 3 и регенеративный теплообменник, где он отдает тепло аккумулирую-
щей насадке.
Технические характеристики цеолитной установки ТМ9 приведены ниже:
Производительность по удаляемому СОг, кг/ч
Ссо2-=3%
ССО2'=4%
Ссо2=5%
Мощность, кВт
Расход воды, м3/ч
Масса, кг
Габаритные размеры, мм
Емкость обслуживаемого холодильника при концентра-
ции диоксида углерода от 3 до 5%, т
9
12
15
60
2
5000
3700x2350x2950
3000—5000
Пассивный способ создания оптимальной газовой среды да-
леко не всегда является приемлемым. Для многих видов и сор-
тов плодоовощной продукции требуется создавать оптимальные
газовые среды за короткий период времени (несколько суток).
Создание оптимального газового состава среды с применением
технических средств быстрого удаления кислорода из атмосфе-
ры холодильных камер получило название активного способа
формирования газовой среды.
Одним из видов оборудования, реализующего активный спо-
соб формирования газовых сред, являются газогенераторные
установки проточного и рециркуляционного типов с адсорбци-
онными аппаратами очистки. На рис. 18,5 приведена принци-
пиальная схема газогенераторной установки типа УРГС-2.
Установка состоит из газогенератора ГНС и адсорбционных аппаратов
очистки типа АО. Газогенератор предназначен для получения газовой среды
с низким содержанием кислорода путем сжигания в потоке атмосферного
воздуха природного или сжиженного газа. Продукты сгорания в качестве
основных компонентов содержат азот, диоксид углерода (до 13%), водяные
Хранение сельскохозяйственной продукции
563
пары в количестве, соответствующем стопроцентной относительной влажности
при 20—25 °C, следы кислорода (0,2—0,5%).
При работе газогенератора газ из емкости 11 через устройство подго-
товки газа 13 направляют в камеру сгорания 12, в которую воздуходувкой 14
в требуемом количестве нагнетают атмосферный воздух. Газовоздушная смесь
сгорает на кусковом катализаторе при температуре, близкой к 2000 °C. Про-
дукты сгорания охлаждают проточной водой до 50—75 °C и через конденса-
тосборник 10 направляют в контактный холодильник 9, откуда они выходят
при 20—25 °C. Сконденсированные водяные пары отделяют от продуктов сго-
рания в конденсатосборнике 10.
В режиме создания газовой среды продукты сгорания через трехходовые
клапаны 5 и 15 направляют на очистку от диоксида углерода, а затем в
холодильную камеру для вытеснения из нее атмосферного воздуха.
Адсорбционный аппарат очистки состоит из заполненных активным уг-
лем адсорберов 3 и 8, работающих попеременно с циклом 5—10 мин. Управ-
ление процессом производят с помощью клапанов 2, 4, переключающих по-
токи вентиляторов 1 и 4. Регенерацию активного угля проводят атмосфер-
ным воздухом; расход воздуха через адсорбер в режиме регенерации при-
мерно в три раза превышает расход газовой среды. Продукты десорбции
выбрасывают в атмосферу. Продолжительность режимов очистки и регенера-
ции одинакова. Переключение адсорберов производят с помощью синхрон-
ного переключения четырехходовых клапанов 2 и 4 с общим приводом от
электрического исполнительного механизма 7. Проточный газогенератор УРГС
работает в режиме создания газовой среды по разомкнутому контуру: газо-
генератор— хранилище — атмосфера. Установка УРГС-2Б производитель-
ностью 120 м3/ч обеспечивает создание оптимальной газовой среды в загру-
женной холодильной камере емкостью 100 т плодов не более, чем за 10—12 ч.
Газогенераторная установка РГГС-400 относится к газогене-
раторам рециркуляционного типа и создает в холодильной каме-
ре оптимальную газовую среду при работе по замкнутому кон-
туру: хранилище — газогенератор — хранилище. Принцип дей-
ствия рециркуляционного газогенератора основан на каталити-
ческом беспламенном окислении пропана или природного газа
кислородом, содержащимся в атмосфере хранилища. Схема га-
зогенератора РГГС-400 приведена на рис. 18,6.
В режиме создания газовой среды после герметизации холодильной ка-
меры 1 воздух из камеры отбирают вентилятором 2 через вентиль 14, подо-
гревают в газовом подогревателе 3 до температуры 600—650 °C и смешивают
с необходимым количеством пропана или природного газа, поступающего
через вентиль 13. Реакцию каталитического окисления проводят в проточном
реакторе 4, продукты катализа охлаждают в теплообменнике 5 водой и через
-четырехходовый клапан 6 подают в один из переключаемых адсорберов 7
или 10, заполненных активным углем. Очищенную от диоксида углерода газо-
вую среду через клапан 8 направляют в увлажнитель 9, где она охлаждается
и увлажняется, а затем через вентиль 15 возвращают в холодильную камеру.
Активный уголь регенерируют атмосферным воздухом, продуваемым вен-
тилятором 11 через подогреватель 12. Продукты десорбции выбрасывают в
атмосферу. Производительность рециркуляционного газогенератора РГГС-400
‘400 м3/ч, расход пропана 2,4 м3/ч или природного газа 6 м3/ч, расход охлаж-
дающей воды 0,8 м3/ч, мощность 3 кВт, масса 3700 кг, габаритные размеры
3800 X 2600X1960 мм.
При активном способе формирования газовой среды опти-
мальный состав атмосферы в хранилищах практически любой
36*
Е64
18. Применение адсорбентов для обработки, пищевой продукции
Рис. 18,6. Принципиальная схема газогенераторной установки типа РГГС:
1 — холодильная камера; 2, II — вентиляторы; 3 — подогреватель; 4 — проточный реактор-
5 — холодильники; 6, 8 — четырехходовые клапаны; 7, 10 — адсорберы, заполненные ак-
тивным углем; 12 — подогреватель; 13—17 — вентили.
емкости можно создать не более, чем за трое суток. В общей
длительности хранения, составляющей 6—8 мес, этот важный
период занимает по времени относительно малую долю. После
достижения требуемых параметров переходят на режим регули-
рования состава газовой среды, сущность которого сводится к
поддержанию концентрации кислорода и диоксида углерода в
узких заданных пределах.
Опыт эксплуатации промышленных хранилищ с регулируе-
мой газовой средой показал, что при современном уровне гер-
метизации холодильников концентрация кислорода в загружен-
ной камере не возрастает, чаще всего она убывает на доли
процента за сутки. Количество диоксида углерода, выделяемого'
плодами, соответствует приведенным выше цифрам, и для ка-
меры емкостью 200 т достигает 12 м3/сут. Для поддержания по-
стоянного газового состава внутри хранилища необходимо не-
прерывно или периодически удалять избыточный диоксид угле-
рода, а также другие вредные вещества, выделяемые плодами
в процессе хранения.
Эффективным и экономичным способом решения этой зада-
чи является использование адсорбционных аппаратов очистки,
в частности адсорбционных установок типа рассмотренной выше
установки ТМ9. Предусмотрена возможность использования ад-
сорбционных аппаратов очистки, входящих в состав газогене-
раторных установок, для рециркуляционной очистки атмосферы
хранилищ от диоксида углерода.
Хранение сельскохозяйственной продукции
565
В таком режиме установка УРГС-2 работает следующим образом (см.
рис. 18,4). Газогенератор выключен, вентилятором отбирают газовую среду
из холодильной камеры и направляют ее в один из адсорберов 3 или 8; очи-
щенную от диоксида углерода среду возвращают в камеру через четыреххо-
довый клапан 2. Регенерацию активного угля проводят описанным выше
способом.
Рециркуляционный газогенератор РГГС-400 работает в режиме регули-
рования газовой среды также по замкнутому контуру (см. рис. 18,5), но при
отключенном подогревателе 3 и закрытых вентилях подачи газа 13 и воды 17.
Применение адсорбционных установок с цеолитовыми адсор-
берами обеспечивает надежное регулирование газового состава
атмосферы плодоовощехранилищ во всем требуемом диапазоне
концентраций кислорода и диоксида углерода в соответствии
с табл. 18-1 и технологическими рекомендациями. Недостатком
цеолитовых установок являются высокий расход электроэнер-
гии и относительно сложное аппаратурное оформление.
Углеадсорбционные установки значительно проще, дешевле,
отличаются малыми энергозатратами. Однако адсорбционная
емкость активных углей по диоксиду углерода невелика, что
порождает немало технических трудностей в силу следующих
обстоятельств. В режиме очистки в адсорбер поступает газовая
среда при температуре, близкой к О °C, с относительной влаж-
ностью 95—97%, что приводит к охлаждению и увлажнению
угля. Увлажнение угля, а следовательно, и снижение адсорбци-
онной емкости по СО2, может продолжаться и в режимах реге-
нерации, особенно при высоких температуре и влагосодержании
атмосферного воздуха, так как при движении сквозь слой хо-
лодного активного угля температура воздуха понижается, а
относительная влажность возрастает.
Пониженная адсорбционная емкость активного угля в ре-
альных условиях эксплуатации обусловливает очень короткие
циклы работы адсорбера (3—10 мин). Высокая частота пере-
ключений приводит к большому переносу атмосферного воздуха
внутрь холодильной камеры и недопустимому росту концентра-
ции кислорода. Эти недостатки существующих углеадсорбцион-
ных установок могут быть существенно уменьшены в результа-
те усовершенствования технологической схемы, оптимизации ре-
жимных параметров и конструкции адсорбера.
Применение адсорбционных установок на промышленных
хранилищах с регулируемой газовой средой впервые в нашей
стране было осуществлено на фруктовом холодильнике в совхозе
имени Джандосова (г. Алма-Ата), и на хранилище винограда
в Крымском совхозе-заводе «Коктебель». В настоящее время
адсорбционные установки используются на хранилищах с регу-
лируемой газовой средой в Казахстане, на Украине, в Белорус-
сии, РСФСР. Адсорбционные установки включены в комплект
оборудования, предусмотренного для типовых проектов вновь
сооружаемых холодильников.
ЛИТЕРАТУРА К РАЗДЕЛУ
«ТЕОРЕТИЧЕСКИЕ ОСНОВЫ АДСОРБЦИОННОЙ
ТЕХНИКИ»
1. Дубинин М. М. Адсорбция и пористость. М., Изд-во Воен. акад. хим. за-
щиты, 1972. 127 с. 2. Кельцев Н. В., Толстикова М. Г. — Труды МХТИ им.
Д. И. Менделеева, 1979, вып. 109, с. 10—12. 3. Dubinin М. М., Plavnik G. М.—
Carbon, 1968, v. 6, № 2, р. 183—192. 4. Бруиауэр С. Адсорбция газов и паров/
/Пер. с англ. М., Издатинлит, 1948. 781 с. 5. Сарахов А. И. Весы в физико-хими-
ческих исследованиях. М., Наука, 1968. 229 с. 6. Березин Г. И., Наситов М. М.—
ЖФХ, 1979, т. 53, № 9, с. 2402—2403. 7. Langmuir J. — J. Amer. Chem. Soc.,
1918, v. 40, № 9, p. 1361—1403. 8. Киселев A. В. — В кн.; Курс физической
химии, т. 1/Под ред. Я. И. Герасимова. 2-е изд. М., Химия, 1969, с. 412—509.
9. Nishimura Motoshi — J. Chem. Soc. Japan, Chem. a. Ind. Chem., 1979, v. 52,
№ 1, p. 10—15. 10. Davis R. T., De Witt T. W., Emmett P. H. — J. Phys. Coll.
Chem., 1947, v. 51, № 6, p. 1232—1248.
11. Emmett P. H., Cimes M. — J. Phys. Coll. Chem., 1947, v. 51, № 6,
p. 1248—1262; № 6, p. 1329—1341. 12. Эльтеков Ю. A. — В кн.: Адсорбция и
пористость. М., Наука, 1976, с. 86—87. 13. McCiellan A. L., Harnsberger Н. F. •—
J. Coll. Interface Sci., 1967, v. 23, № 4, p. 577—599. 14. Livingston H. K. — J.
Amer. Soc., 1944, v. 66, № 4, p. 569—573. 15. Young G. J. — J. Coll. Sci., 1958,
v. 13, № 1, p. 67—85. 16. Young G. J. e. a. — J. Phys. Chem., 1954, v. 58, № 4,
p. 313—315. 17. Kiselev A. V. — In: The Structure and Properties of Porous Ma-
terials. London, Butterworth, 1958. 195 p. 18. Dell R. M., Beebe R. A. — J. Phys.
Chem., 1955, v. 59, № 8, p. 754—762; Canad. J. Chem., 1957, v. 36, № 12,
p. 1542—1544. 19. Дубинин M. M., Жуковская E. Г., Мурдмаа К. O. — ЖФХ,
1963, t. 37, № 2, c. 426—432. 20. Буянова H. E., Гудкова Г. Б., Карнау-
хов А. П. — Кинетика и катализ, 1967, т. 8, № 2, с. 428—432.
21. Дзисько В. А., Карнаухов А. П., Тарасова Д. В. Физико-химические
основы синтеза окисных катализаторов. Новосибирск, Наука, 1978. 384 с.
22. Темкин М. И. —ЖФХ, 1955, т. 29, № 9, с. 1610—1613. 23. Клячко-Гур-
вич А. Л. —Изв. АН СССР. ОХН, 1961, № 10, с. 1884—1886. 24. Берг Л. Г.,
Абдурахманов Р. А. — ЖФХ, 1970, т. 44, № 8, с. 527—529. 25. Ewing W.,
Liu F. W. — J. Coll. Sci., 1953, v. 8, № 2, p. 204—213. 26. Books C. S. — Koll.
Z. Polimere, 1964, Bd. 199, № 1, S. 30—36. 27. Пошкус Д. П., Козлаус-
кас P. M. — Коллоидн. ж., 1965, т. 27, № 5, с. 738—764. 28. Березкина О. Ф.,
Сарахов А. И., Дубинин М. М. — ЖФХ, 1970, т. 44, № 5, с. 1237—1241. 29. Ду-
бинин М. М., Катаева Л. И. — Изв. АН СССР. Сер. хим., 1977, № 3, с. 516—
522. 30. Карнаухов! А. П. — Кинетика и катализ, 1971, т. 12, № 4, с. 1025—
1033, 1235—1242.
31. Карнаухов А. П. — Кинетика и катализ, 1967, т. 8, № 1, с. 172—181.
32. Джигит О. М., Киселев А. В., Неймарк И. Е. — ЖФХ, 1954, т. 28 № 10,
с. 1804—1811. 33. Deiiimore D„ Heal G. R. — J. Coll. Interface Sci., 1970, v. 33,
№ 4, p. 508—519. 34. Oulton T. D. — J. Phys. Coll. Chem., 1948, v. 52 № 8,
p. 1296—1341. 35. Broekhoff J. C„ de Boer J. H. — J. Catal, 1967, v. 9, № 8,
p. 15—18; 1968, v. 10, № 2, p. 153—165; № 4, 368—376, 377—390, 391—400.
36. Дубинин M. M. — Изв. АН СССР. Сер. хим., 1980, № 1, c. 22—26. 37. Ду-
бинин M. M., Катаева Л. И. — Изв. АН СССР. Сер. хим., 1980, №3, с. 498—502.
38. Вольфкович Ю. М. — Коллоидн. ж., 1979, т. 41, № 4, с. 640—647. 39. Pola-
nyi М. — Verh. Deutsche Phys. Ges., 1914, Bd. 16, S. 1012. 40. Polanyi M.—
Trans. Faraday Soc., 1932, v. 28, p. 316—333.
41. Заверииа E. Д,, Дубинин M. M — ЖФХ, 1939, t. 13, № 2, c. 151—162;
Дубинин M. M. Юбилейный сборник АН СССР, ч. 1. М., изд. АН СССР, 1947,
с. 562—581. 42. Дубинин М. М., Заверина Е. Д. — ЖФХ, 1950, т. 24, № 10,
Литература
567
с. 1262—1272. 43. Николаев К. М., Дубинин М. М.— Изв. АН СССР. ОХН,
1958, № 10, с. 1165—1174. 44. Дубинин М. М., Астахов В. А. — Изв. АН СССР.
Сер. хим., 1971, № 1, с. 5—17. 45. Дубинин М. М. — ЖФХ, 1965, т. 39, № 6,
с. 1305—1317. 46. Дубинин М. М.— Изв. АН СССР. Сер. хим., 1979, № 8,
с. 1691—1696. 47. Дубинин М. М., Жукова 3. А., Кельцев Н. В. —В кн.: Синте-
тические цеолиты. М., Изд-во АН СССР, 1962, с. 7—'17. 48. Шумяцкий Ю. И.,
Кельцев Н. В., Торочешииков Н. С. — Труды МХТИ им. Д. И. Менделеева,
1965, вып. 48, с. 242. 49. Рахмуков Б. X. и др. — Изв. АН СССР. Сер. хим.,
1979, № 11, с. 2419—2422. 50. Homfrau J. F. — Z. Phys. Chem., 1910, Bd. 74,
№ 1, S. 129—130.
51. Кисаров В. M.— ЖФХ, 1969, т. 43, № 4, с. 1037—1039. 52. Эксперимен-
тальные методы в адсорбции и молекулярной хроматографии/Под ред.
А. В. Киселева и В. П. Древинга. М., Изд-во МГУ, 1973. 448 с. 53. Тере-
нин А. Н. —ЖФХ, 1940, т. 14, № 9—10, с. 1362—1369. 54. Киселев А. В., Лы-
гии В. И. Инфракрасные спектры поверхностных соединений и адсорбирован-
ных веществ. М., Наука, 1972. 459 с. 55. Филимонов В. Н. — В кн.: Основные
проблемы физической адсорбции. М., Наука, 1970, с. 116—131. 56. Кисе-
лев А. В., Лыгии В. И. — Там же, с. 132—150. 57. Неймарк И. Е. — Там же,
с. 151—165. 58. Жиленков И. В.— Там же, с. 235—240. 59. Кость М. В. и др.—
Там же, с. 172—173. 60. Лукьянович В. М., Трофимов В. И. — Там же, с. 287—
295.
61. Новикова О. С. Канд. дне. М., Ин-т общей и неорг. хим. им. Н. С. Кур-
накова, 1956. 62. Батиашвили Т. В. Термографическое исследование цеолитов
среднеэоцеоновых вулканогенных толщ Грузии. Тбилиси, Мецниерба, 1979, 79 с.
63. Пилоян Г. О., Новикова О. С. — Изв. АН СССР. Сер. Неорг. хим., 1966, №2,
с. 1298—1301. 64. Кельцев Н. В., Клушии В. Н., Соловьев В. Н. — Труды МХТИ
им. Д. И. Менделеева, 1970, вып. 65, с. 92—94. 65. Гаврилов М. 3., Ермолен-
ко И. Н. — Изв. АН БССР. Сер. хим. наук, 1973, № 3, с. 32—36; 1976, № 1,
с. 23—28; 1977, № 5, с. 22—26. 66. Колышкии Д. А., Михайлова К. К. Актив-
ные угли. Л., Химия, 1972. 57 с. 67. Эгути Е. — Катаку коге, 1966, т. 17, № 11,
с. 1053—1058. 68. Dubinin М. М. — Chem. Phys. Carbon, 1966, v. 2, № 4, p. 51—
122. 69. Dubinin M. M., Zaverina E. D„ Serpinsky V. V. — J. Chem. Soc., 1955,
№ 6, p. 1760—1766. 70. Костриков В. И., Кельцев Н. В., Панова Л. Н. — В кн.:
Углеродные адсорбенты и их применение в промышленности, ч. 2. Пермь, 1969,
с. 12—21.
71. Паторжииский И. В., Серпиоиова Е. Н. — Труды МХТИ им. Д. И. Мен-
делеева, 1970, вып. 65, с. 79—82. 72. Wojcik G., Karpinsky К. — Chem. stosow.,
1978, v. 22, № 2, р. 351—360. 73. Неймарк И. Е., Штейифайи Р. Ю. Силикагель,
его свойства, получение и применение. Киев, Наукова думка, 1973. 200 с.
74. Карнаухов А. П. — Кинетика и катализ, 1962, т. 3, № 4, с. 583—598.
75. Кольцов С. И., Алесковский В. Б. Силикагель, его строение и химические
свойства. Л., Госхимиздат, 1963. 96 с. 76. Бурушкииа Т. Н. Канд. дис. Киев,
Ин-т физ. хим. им. Л. В. Писаржевокого, 1968. 77. Будкевич Г. Б., Слиияко-
ва И. Б., Неймарк И. Е. — ДАН СССР, 1964, т. 154, с. 692—694; Коллоидн. ж.,
1965, т. 27, № 5, с. 758—764; 1966, т. 28, № 1, с. 21—26; Изв. АН СССР. Сер.
хим., 1968, № 3, с. 467—474. 78. Белоцерковский Г. М., Савченко В. И. — Изв.
АН СССР. Сер. хим., 1966, № 2, с. 32—42. 79. Шморгуиеико Н. С., Левиц-
кий Э. А. — Изв. АН СССР. Сер. хим., 1966, № 2, с. 43—49. 80. Ферсмаи А. Е.
Материалы к исследованию цеолитов в России. Избранные труды, т. 1. М.,
Изд-во АН СССР, 1952. 863 с.
81. Вернадский В. М., Курбатов С. М. Земные силикаты, алюмосиликаты
и их аналоги. Л.-М, ОНТИ СССР НКТП, 1937. 378 с. 82. Виичелл А. Н. Опти-
ческие свойства искусственных минералов/Пер. с англ. М., Мир, 1967. 526 с.
83. Сеидеров Э. Э., Хитаров Н. И. Цеолиты, их синтез и условия образования
568
Литература
в природе. М., Наука, 1970. 283 с. 84. Михайлов А. С. — Советская геология,
1975, № 9, с. 70—78. 85. Кельцев Н. В. — Жури. ВХО им. Д. И. Менделеева, 1979,
т. 24, № 1, с. 54—58. 86. Челищев Н. Ф., Береиштейи Б. Г., Смола В. И. Ис-
пользование природных цеолитов. М., Изд-во ВИЭМС, 1977. 53 с. 87. Mump-
ton F. А. — Ind. Minerals, 1973, № 73, p. 30—36. 88. Челищев H. Ф., Челище-
ва Р. В. — Вести, сельскохозяйственной науки, 1978, № 2, с. 126—131. 89. Cul-
faz A., Kiesling С. A., Sand L. В. — Amer. Miner., 1973, v. 58, р. 1044—1047.
90. Пенчев В. и др. — Изв. хим. Блг. АН, 1979, т. 12, № 2, с. 317—324.
91. Жданов С. П., Егоров Е. Н. Химия цеолитов. Л., Наука, 1968. 158 с.
92. Barrer R. М., Marschall D. J. — J. Chem. Soc., 1964, № 7, 2296—2305.
93. Breck D. W., Eversole W. G., Milton R. M. — J. Amer. Chem. Soc., 1956,
v.78, № 10, p. 2338—2339. 94. Кельцев H. B. — Газовая пром., 1957, № 9, с. 38—
40. 95. Оиусайтис Б. А. — Вестник АН СССР, 1965, № 1, с. 114—118. 96. Bar-
rer R. М., Meier W. М. — Trans. Faraday Soc., 1958, v. 54, № 7, p. 1074—1085.
97. Breck D. W. e. a.—J. Amer. Chem. Soc., 1956, v. 78, № 23, p. 5972—5977.
98. Дубинин M. M., Жуковская E. Г., Мурдмаа К. О. — Изв. АН СССР. ОХН,
1962, № 5, с. 760—769. 99. Пигузова Л. И. Высококремиеземистые цеолиты и
их применение в нефтепереработке и нефтехимии. М., Химия, 1974. 156 с.
100. Ростовцева И. В., Аиякова Р. М., Орехов В. В. — Вопросы химии и хим.
технол. нефти и газа, № 1, Грозный, Изд-во Чечено-Ингуш. уи-та, 1978, с. НО—
112.
101. Липкиид Б. А. — В кн.: Адсорбенты, их получение, свойства и приме-
нение. М., Наука, 1971, с. 51—59. 102. Соколов В. А., Торочешников Н. С.,
Кельцев Н. В. Молекулярные сита и их применение. М., Химия, 1964. 156 с.
103. Брек Д. Цеолитовые молекулярные сита/Пер. с англ. М., Мир, 1976. 702 с.
104. Grubner О., Jiru Р., Ralek М. Molekularsiebe. VEB Deutscher Verlag der
Wissenschaften. Berlin, 1968. 176 S. 105. Zielinski S., Sarbak Z. — Chem. stosow.,
1980, v. 24, № 1, p. 43—50. 106. Barrer R. M. — Brennstoff, 1954, Bd. 35, № 2,
S. 325—328; Brit. Chem. Eng., 1959, v. 4, № 5, p. 267—269. 107. Квитков-
ский Л. H., Сергиенко С. P. —ДАН СССР, 1962, т. 147, № 6, с. 1399—1401.
108. Barrer R. M. — Trans, Faraday Soc., 1949, v. 45, № 4, p. 358—373. 109. Hab-
good H. F. — Canad. J. Chem., 1958, v. 36, № 10, p. 1384—1397. ПО, Белень-
кая И. M., Дубинин Н. М., Криштофери И. И. — Изв. АН СССР. Сер. хим.,
1967, № 10, с. 2164—2171; 1968, № 10, с. 2184—2190; 1971, № 7, с. 1391—1397;
в кн.: Адсорбенты, их получение, свойства и применение. Л., Наука, 1971,
с. 118—121.
111. Мишин И. В. и др. — Изв. АН СССР. Сер. хим., 1972, № 2, с. 2370—
2373; 1973, № 5, с. 1017—1021, с. 1136—1138. 112. Chem. a. Eng. News, 1962,
v. 40, № 11, р. 52. 113. Жданов С. П., Новиков Б. Г.— Изв. АН БССР. Сер.
хим. наук, 1966, № 1, с. 44—58. 114. Федорова Л. М., Кельцев Н. В., Щерба-
ков В. А. — Труды МХТИ им. Д. И. Менделеева, 1970, вып. 65, с. 88—91.
115. Пат. 3640681, 1972 г. (США). 116. Lohse U. е. а. — Z. anorg. u. Allgem.
Chem., 1980, Bd. 460, № 1, р. 179—180: 117. Пат. 329319, 1966 г. (США).
118. Мииасян Ш. О., Шумяцкий Ю. И., Кельцев Н. В. — ЖФХ, 1970, т. 46,
№ 12, с, 3136—3139. 119. Bettke W. е. a. —Chem. Technik, 1978, № 12, S. 637—
640. 120. Пигузова Л. И. и др.— Изв. АН БССР. Сер. хим. наук, 1966, Хе 1,
с. 59—64.
121. Николина В. Я., Киыш Л. И., Соколова П. А. — Там же, с. 65—69.
122. Никелина В. Я. и др. — Изв. АН БССР. Сер. хим. наук, 1967, № 4, с. 38—
42. 123. Ширииская Л. П. и др. — Изв. АН БССР, Сер. хим. наук, 1973, № 1,
с. 5—8. 124. Пигузова Л. И. Новые сверхвысококремнеземиые цеолиты и их
применение в нефтепереработке. М., ЦНИИТЭнефтехим, 1977. 76 с. 125. Ней-
марк И. Е. — В кн.: Адсорбенты, их получение, свойства и применение. Л.,
Наука, 1978, с. 16—21. 126. Киселев А, В., Никитин Ю. С. — Хим. и технол.
Литература
569
топлив и масел, 1958, № 12, с. 27—32. 127. Высоцкий 3. 3., Неймарк И. Е.
Укр. хим. ж., 1954, т. 20, № 5, с. 513—-522. 128. Шейифайи Р. Ю., Стась О. П.,
Неймарк И. Е. — ЖПХ, 1972, т. 46, с. 2192—2195. 129. Пиоитовская М. А. и др. —
В кн.: Адсорбенты, их получение, свойства и применение. Л., Наука, 1971,
с 73—77 130. Бобович Ф. М. и др. — Жури. теор. и эксперим. хим., 1975, № И,
с. 121—123.
131. Шилов Н. А., Дубинин М. М., Торопов С. А. — ЖРФХО, 1929, т. 51,
с. 1765—1768. 132. Высоцкий 3. 3., Неймарк И. Е. — Укр. хим., ж., 1956, т. 22,
№ 3, с. 485—488. 133. Жданов С. П. — В кн.: Адсорбция и пористость. М., Нау-
ка, 1976, с. 21—26. 134. Тарасевич Ю. И. Докт. дис. Киев, Ин-т коллоидн. хи-
мии и химии воды АН УССР, 1972. 135. Карнаухов А. П. — В кн.: Моделирова-
ние пористых материалов. Новосибирск, Ин-т катализа СО АН СССР, 1976,
с. 5—30. 136. Тарасевич Ю. И., Овчаренко Ф. Д. Адсорбция на глинистых ма-
териалах. Киев, Наукова думка, 1975. 351 с. 137. Киселев А. В., Храпова Е. В.,
Щербакова К. Д. — Нефтехимия, 1962, т. 2, № 6, с. 877—883. 138. Аста-
хов В. А. — Докт. дис. Л., ЛТИ им. Ленсовета, 1972. 139. Афанасьев Ю. М.
Канд. дис. М., МХТИ им. Д. И. Менделеева, 1967. 140. Biachell Н. С., Lan-
ge К. R. —J. Phys. Chem., 1956, v. 60, № 3, р. 307—308.
141. Рябухина Л. Г. Канд. дис. М., МГУ им. М. В. Ломоносова, 1973.
142. Barrer R. М. — J. Coll. Interface Sci., 1966, v. 21, № 4, p. 415—434; Chem.
a. Ind., 1968, № 36, p. 1203—1212. 143. Табуищикова О. К. Канд. дис. М.,
МХТИ им. Д. И. Менделеева, 1971. 144. Беринг Б. П., Серпииский В. В. — ДАН
СССР, 1963, т. 148, № 6, с. 1331—1334. 145. Беринг Б. П„ Серпииский В. В.—
Изв. АН СССР. Сер. хим., 1971, № 4, с. 847—849. 146. Дубинин М. М„ Пол-
стяное Е. Ф. — Изв. АН СССР. Сер. хим., 1966, № 9, с. 1507—1513. 147. Bar-
rer R. М., Lee J. А. — Surface Sci., 1968, v. 12, № 2, р. 354—368. 148. Аста-
хов В. А., Дубинин М. М. — Изв. АН СССР. Сер. хим., 1971, № 1, с. 17—20.
149. Мумииов С. 3., Беринг Б. П., Серпииский В. В. — Изв. АН СССР. Сер.
хим., 1966, № 1, с. 43—55. 150. Беринг Б. П., Махашвили Н. И., Серпии-
ский В. В. —Изв. АН СССР. Сер. хим., 1969, № 9, с. 2039—2041.
151. Lewis W. е. а. — Ind. Eng. Chem., 1950, v. 42, № 7, p. 1319—1326.
152. Кельцев H. В. Канд. дис. М., МХТИ им. Д. И. Менделеева, 1951. 153. Чер-
нышев А. Б., Кельцев Н. В., Халиф А. Л. — ДАН СССР, 1952, т. 82, № I,
с. 75—77. 154. Беринг Б. П., Серпииский В. В. —ЖФХ, 1952, т. 26, № 2, с. 253—
269. 155. Marham Е. С., Benton A. F. — J. Amer. Chem. Soc., 1931, v. 53, № 2,
p. 497—507. 156. Беринг Б. П., Серпииский В. В., Суринова С. И. — Изв. АН
СССР. Сер. хим., 1965, № 5, с. 769—776. 157. Grossman A., Schirmer W.—
Chem. Technik, 1968, Bd. 20, № 1, S. 34—38. 158. Кельцев H. В. Докт. дис. М.,
МХТИ им. Д. И. Менделеева, 1967. 159. Кельцев Н. В., Шумяцкий Ю. И. —
ЖФХ, 1970, т. 44, № 5, с. 1327—1328. 160. Кельцев Н. В. — В кн.: Физическая
адсорбция из многокомпонентных фаз. М., Наука, 1972, с. 30—38.
161. Гриненко С. Б. — В кн.: Нефтехимия, Киев, Наукова думка, 1964,
с. 63—67. 162. Гаинко Н. К-, Дорогочииский А. 3. — Хим. и технол. топлив и
масел, 1965, № 11, с. 28—32. 163. Ширмер В. — В кн.: Физическая адсорбция
из многокомпонентных фаз. М., Наука, 1972, с. 61—63. 164. Kinday A. J.,
Myers A. L. — AIChE J., 1966, v. 12, № 5, р. 981—986. 165. Myers A. L. — Ind.
Eng. Chem., 1968, v. 60, № 5, p. 45—49. 166. Hill T. L. — J. Chem. Phys., 1949,
v. 17, № 6, p. 520—535. 167. Беринг Б. П., Серпииский В. В., Суринова С. И.—
Изв. АН СССР. Сер. хим., 1973, № 1, с. 3—15. 168. Тимофеев Д. П. Кинетика
адсорбции. М., Изд-во АН СССР, 1963. 252 с. 169. Тимофеев Д. П., Алексе-
ева Н. И. —ДАН СССР, 1966, т. 166, № 4, с. 917—919. 170. Коваленко Л. А.,
Кельцев Н. В. — Труды МХТИ им. Д. И. Менделеева, 1967, вып. 56, с. 137—
139.
570
Литература
171. Кельцев Н. В., Коваленко Л. А., Шумяцкий Ю. И. — ТОХТ, 1967, т. 1,
№ 5 с. 648—653. 172. Золотарев П. П., Дубинин М. М. — ДАН СССР, 1973,
т. 210, № 1, с. 136—139. 173. Кельцев Н. В. и Др. — Труды МХТИ им. Д. И. Мен-
делеева 1970, вып. 65, с. 86—87. 174. Воштова Г., Кельцев Н. В., Му-
хин В. М. — ЖФХ 1978, т. 52, № 7, с. 1765—1767. 175. Бобок Д, Мухин В. М.,
Кельцев Н. В. — ЖФХ, 1979, т. 53, № 12, с. 3139—3143. 176. Тимофеев Д. П.
и др. — В кн.: Синтетические цеолиты. М, Изд-во АН СССР, 1962, с. 24—30.
177. Коваленко Л. А. Канд. дис. М, МХТИ им. Д. И. Менделеева, 1968.
178. Barrer R. М. — Trans. Faraday Soc., 1941, v. 37, № 10, p. 590—600.
179. Barrer R. M., Cobins A. B. — Trans. Faraday Soc., 1953, v. 49, № 7, p. 807—
815. 180. Barrer R. M., Reeg J. V. — Trans. Faraday Soc., 1954, v. 50, № 8,
p. 853—863.
181. Barrer R. M., Brook D. — Trans Faraday Soc., 1953, v. 49, № 9, p. 1049—
1059. 182. Тимофеев Д. П., Ерашко И. T. — Изв. АН СССР, ОХН, 1961, № 7,
с 1192—1197 183. Коваленко Л. А., Шумяцкий Ю. И,, Кельцев Н. В. — ТОХТ,
1968, т. 2 № 6 с. 868—874. 184. Дубинин М. М. — ДАН СССР, 1964, т. 159,
№ 1, с. 166—169. 185. Товбин М. В., Гринберг А. Д. — ЖФХ, 1952, т. 26, № 2,
с. 1755—1765. 186. Кожевников А. Г., Мусакин Г. А. — В кн.: Получение, струк-
тура и свойства сорбентов. Л., Наука, 1978, с. 88—94. 187. Кельцев Н. В., Афа-
насьев Ю. М, Шумяцкий Ю. И. —ЖФХ, 1968, т. 42, № 6, с. 1480—1483.
188. Barrer R. М, Sutherland J. W. — Proc. Roy. Soc., 1956, A237, № 1211,
p. 439—463. 189. Клушин В. H. Канд. дис. М, МХТИ им. Д. И. Менделеева,
1968. 190. Тимофеев Д. П., Кабанова О. Н. — Изв. АН СССР. Сер. хим., 1966,
№ 4, с. 642—648.
191. Кельцев Н. В., Клушин В. Н., Торочешников Н. С. — Изв. вузов. Сер.
хим. и химич. технол., 1970, т. 13, № 7, с. 975—978. 192. Афанасьев Ю. М., Жу-
кова 3. А., Кельцев Н. В. — В кн.: Цеолиты, их синтез, свойства и применение.
М.-Л, Наука, 1965, с. 352—355. 193. Афанасьев Ю. М., Кельцев Н. В., Шумяц-
кий Ю. И.— Газовая пром., 1969, № 12, с. 32—35. 194. Кельцев Н. В., Ха-
лиф А. Л., Ходанович И. Е. — В кн.: Переработка и транспорт природных га-
зов. М, Гостоптехиздат, 1953, с. 136—141. 195. Астахов В. А., Меерсон Л. А.,
Клюшкова Г. С.— Изв, АН БССР, Сер, хим, наук, 1978, № 3, с, 118—121.
196. Продан Е. А. и др. — Изв. АН БССР. Сер. хим. наук, 1978, №3, с. 8—12.
197. Бобович Ф. М. и др. —ЖПХ, 1979, т. 62, № 8, с. 1860—1864. 198. Doel-
le Н. J, Riekert L. — Angew. Chem., 1979, Bd. 91, № 4, S. 309—316. 199. Кель-
цев H. В., Торочешников Н. С., Шумяцкий Ю. И. — В кн.: Цеолиты, их синтез
свойства и применение. М.-Л., Наука, 1965, с. 356—360. 200. Шай Г. Теорети-
ческие основы хроматографии газов. Пер. с нем. М, Издатннлит, 1963. 382 с.
201. Рогинский С. 3., Яновский М. И. —ЖФХ, 1950, т. 24, № 2, с. 137—
143. 202. Яновский М. И. —ЖПХ, 1951, т. 24, № 6, с. 661—666. 203. Дуби-
нин М. М. — В кн.: Адсорбенты, их получение, свойства и применение. Л., Нау-
ка, 1978, с. 4—9. 204. Шилов Н. А., Лепинь Л. Н., Вознесенский С. А.—
ЖРФХО, 1929, т. 61, № 7, 1107—1123. 205. Michaels A. S. — Ind. Eng. Chem,
1952, v. 44, № 8, р. 1922—1930. 206. Treiball R. E. Mass-Transfer Operation.
Mc-Graw Hillbook, N.-Y. — London, 1955. 666 p. 207. Дубинин M. M. — Зав.
лаб, 1973, № 4, с. 487—489. 208. Торочешников Н. С, Кельцев Н. В, Шумяц-
кий Ю. И. — В ки.: Кинетика и динамика физической адсорбции. М, Наука,
1973, с. ПО—116. 209. Щербаков В. Л.—Там же, с. 150—151. 210. Рачин-
ский В. В. Введение в общую теорию динамики сорбции и хроматографии. М,
Наука, 1964. 135 с.
211. Жуховицкий А. А, Забежинский Я. Л, Тихонов А. Н. — ЖФХ, 1945,
т. 19, № 6, с. 253—261. 212. Пшежецкий С. Я., Рубинштейн Р. Н. — ЖФХ,
1946, т. 20, № 12, с. 1421—1434. 213. Радушкевич Л. В. —ДАН СССР, 1947,
Литература
571
т. 57, № 5, с. 471—474. 214. Радеке К. X., Буйке Г., Гелбин Д. — В кн.: Кине-
тика и динамика физической адсорбции. М., Наука, 1973, с. 132—139. 215. Ко-
лин В. Л. и др.— Там же, с. 98—100. 216. Рачинский В. В., Давыдов Е. Г.—
ЖФХ, 1966, т. 40, № 6, с. 1190—1195. 217. Аэров М. Э., Тодес О. М. Гидрав-
лические и тепловые основы работы аппаратов со стационарным и кипящим
зернистым слоем. Л., Химия, 1968. 510 с. 218. Левеншпиль О. Инженерное
оформление химических процессов. М., Химия, 1969. 621 с. 219. Тодес О. М.,
Лезин Ю. С., Шейнина Л. С. — ТОХТ, 1968, т. 2, № 1, с. 103—115. 220. Ed-
wards М. F., Richardson J. Р. — Chem. Eng. Sci., 1968, v. 23, № 2, p. 109—
123.
221. Карберри Л. В. — В кн.: Пористая структура катализаторов и процес-
сы переноса в гетерогенном катализе. Новосибирск, Наука, 1970, с. 39—45.
222. Crickmore Р. J., Wojciechowski В. W. — J. Chem. Soc. Faraday Trans., 1977,
v. 73, № 8, part 1, p. 1216—1233. 223. Нагаев В. В. и др. — ТОХТ, 1981, т. 14,
№ 1 с 31—35; К» 6, с. 819—824. 224. Wilson J. — J. Amer. Chem. Soc., 1940,
v. 62, № 6, p. 1583—1591. 225. Weiss J. — J. Chem. Soc., 1943, v. 145, № 7,
p. 293—303. 226. De Vault D. — J. Amer. Chem. Soc., 1943, v. 65, № 4, p. 535—
540. 227. Wicke E — Koll-Z., 1939, Bd. 86, № 3, S. 295—313. 228. Тодес О. M.—
ЖПХ, 1945, т. 18, Кв 11—12, с. 591—608. 229. Баррер Р. Диффузия в твердых
слоях/Пер. с аигл. М., Издатинлит, 1948. 508 с. 230. Мухин В. М. Канд. дис.
М., МХТИ им. Д. И. Менделеева, 1979.
231. Дубинин М. М., Лезин Ю. С.— В кн.: Алгоритмизация расчета процес-
сов и аппаратов химических производств, вып. 5. Киев, Наукова думка, 1969,
с. 3—8. 232. Лезин Ю. С. — ЖФХ, 1968, т. 42, № 7, с. 1762—1767. 233. Енике-
ева Н. И. Каид. дис. Казань, КХТИ им. С. М. Кирова, 1969. 234. Тодес О. М.,
Биксон Я. М. — ДАН СССР, 1950, т. 75, № 5, с. 727—730. 235. Биксон Я. М. —
ЖФХ, 1953, т. 27, № 10, с. 1530—1538. 236. Тодес О. М„ Лезин Ю. С. — ДАН
СССР, 1956, т. 106, № 2, с. 307—310. 237. Pan С. J., Bastnadjan D. D. — Chem.
Eng. Sci., 1970, v. 25, № 11, p. 1653—1664. 238. Лезин Ю. С. —ЖФХ, 1968,
т. 42, № 7, с. 1762—1767. 239. Chi С. W., Wasan D. Т. — AIChE J., 1970, v. 16,
№ 11, р. 23—31. 240. Дубинин М. М., Явим М. — ЖПХ, 1936, т. 9, № 7, с. 1191—
1203.
241. Фролов В. Ф., Лезин Ю. С. — В кн.: Кинетика и динамика физической ад-
сорбции. М., Наука, 1973, с. 264—271. 242. Жукова 3. А., Кельцев Н. В.—
В ки.: Переработка природного газа, вып. 6. М., ГНТИ нефтяной и горно-топ-
ливной литературы, 1959, с. 154—160. 243. Фомкин А. А., Серпинский В. В.—
Изв. АН СССР. Сер. хим., 1974, № 9, с. 2108—2110; с. 2111—2114. 244. Жуко-
ва 3. А. Канд. дис. М., МХТИ им. Д. И. Менделеева, 1962. 245. Белоногов А. В.,
Табунщикова О. К.— Хим. и нефт. машиностроение, 1969, № 12, с. 15—16
246. Вагин Е. В. — ДАН СССР, 1969, т. 189, № 3, с. 566—567. 247. Вагин Е. В.,
Тулянская И. И. — ЖФХ, 1979, т. 53, №3, с. 685—688. 248. Никифоров В. П.
и др. —ЖФХ, 1970, т. 44, № 2, с. 535—537. 249. Никитина И. Е. и др. — Газо-
вая пром., 1979, № 4, с. 46—49. 250. Орехов И. И., Обрезков В. Д. — В кн.:
Холод в процессах химической технологии. Л., Изд-во ЛГУ, 1980, с. 175—181.
251. Chem, Eng. Progress, 1966, v. 62, № 4, p. 11. 252. Пат. 3325971, 1967 г.
(США). 253. Сорбционные процессы в вакууме/Под ред. К- И. Мызиикова. М.,
Атомиздат, 1965. 312 с. 254. Федорова М. Ф. Исследование физической адсорб-
ции и ее практическое применение. Харьков, Изд-во АН УССР (ФТИ), 1966.
91 с. 255. Вопросы атомной науки и техники. Сер. Низкотемпературная адсорб-
ция и криогенный вакуум. Харьков, ХФТИ, 1971, вып. 1. 164 с.; 1972, вып. 2.
39 с. 256. Коган В. С., Бреславец К. Г. — В кн.: Адсорбция и пористость. Труды
4 Всесоюз. конф, по теоретич. проблемам адсорбции/Под ред. М. М. Дубинина
и В. В. Серпинского. М., Наука, 1976, с. 270—272.
572 Литература
ЛИТЕРАТУРА К РАЗДЕЛУ «АППАРАТУРНО-
ТЕХНОЛОГИЧЕСКОЕ ОФОРМЛЕНИЕ АДСОРБЦИОННЫХ
ПРОЦЕССОВ»
1. Серпионова Е. Н. Промышленная адсорбция газов и паров. М., Высшая
школа, 1969. 414 с. 2. Романков П. Г., Лепилин В. Н. Непрерывная адсорбция
газов и паров. Л., Химия, 1968. 228 с. 3. Халиф А. Л., Кельцев Н. В. — В кн.:
Вопросы добычи, транспорта и переработки попутных газов. М., Гостоптехиз-
дат, 1951, с. 295—303. 4. Николаевский К. М. Проектирование рекуперации ле-
тучих растворителей с адсорбентами периодического действия. М., Оборонгиз,
1961. 238 с. 5. Грицев Н. Д. Углеадсорбционный метод получения бензина из
газа. М.-Л., Гостоптехиздат, 1950. 85 с. 6. Кельцев Н. В., Халиф А. Л. — Газо-
вая пром., 1958, № 7, с. 44—47. 7. Орехов И. И., Обрезков В. Д. — В кн.: Хо-
лод в процессах химической технологии. Л., Изд-во ЛГУ, 1980, с. 175—181.
8. Constaninescn Р. — Petrol, si gaze, 1968, v. 17, № 5, p. 323—325. 9. Berg C. —
Trans. Amer. Inst. Chem. Eng., 1946, v. 42, № 4, p. 665—680; Gas, 1947, v. 23,
№ 1, p. 32—37; Petrol. Eng., 1947, v. 18, № 5, p. 115—118; Petrol. Ref., 1949,
v. 28, № 11, p. 113—130; Chem. Eng. Progr., 1951, v. 47, № 11, p. 585—591;
Petrol. Ref., 1951, v. 30, № 9, p. 241. 10. Holt В. M. — Petrol. Proc., 1949, v. 4,
№ 6, p. 681—682.
И. Кельцев H. B. — Канд, дис., M., МХТИ им. Д. И. Менделеева, 1951.
12. Кельцев Н. В.—'Газовая пром., 1956, № И, с. 32—36. 13. Кельцев Н. В.—
Хим. и технол. топлива, '1956, № 6, с. 38—43. 14. Григорьян X. А., Алиев 3. Э.,
Кулиев А. М. — В кн.: Труды по исследованию нефтей и нефтепродуктов, вып. 3.
Баку, 1958, с. 357—378. 15. Жукова 3. А., Кельцев Н. В. — В кн.: Разделение
и анализ углеводородных газов. М., Изд-во АН СССР, 1963, с. 134—141. 16. Голь-
берт К. А., Георгиев Ц. О. — Там же, с. 101—113. 17. Гольберт К. А., Плато-
нов В. М., Павлова Л. Ф. — В кн.: Химическая переработка нефтяных углеводо-
родов. М., Изд-во АН СССР, 1956, с. 231—141. 18. Пат. 2651666, 1953 г. (США).
19. Кельцев Н. В., Халиф А. Л. — Хим. технол. топлива, 1956, № 12, с. 17—
22. 20. Benedek Р., Szepesy L. — Erdol u. Kohle, 1956, № 9, S. 593—596.
21. Пат. 3502724, 1967 г. (США). 22. Jurtgen Н., Knoblauch К., Kruel М.—
Chem. Ing. Techn., 1970, Bd. 42, № 2, S. 77—81. 23. Kasten P. R., Amund-
sen N. R. — Ind. Eng. Chem., 1950, v. 42, № 7, p. 1341—1346. 24. Pollok K- —
Ind. Eng. Chem., 1958, v. 50, № 1, p. 4—7. 25. Kachanak S. — Chem. zvesti,
1960, v. 14, № 1, p. 85—90; 1961, v. 15, № 8, p. 585—589; 1962, v. 16, № 6,
p. 417—430. 26. tlsida Tacura — Plant, a. Process, 1968, v. 10, № 2, p. 11—25.
27. Пат. 1492396, 1967 г. (Франция). 28. Пат. 1525959, 1968 г. (Франция). 29.
Пат. 509319, 1955 г. (Канада). 30. Тимофеев Д. П. — ЖПХ, 1963, т. 36, № 5,
с. 1021—1028.
31. Расулов Н. М. — Изв. АН БССР. Сер. хим. наук, 1966, № 1, с. 128—
133. 32. А. с. 109363 (СССР). 33. А. с. 109590 (СССР). 34. Гольберт К. А., Пла-
тонов В. М. — В кн.: Химическая переработка нефтяных углеводородов. М.,
Изд-во АН СССР, 1956, с. 218—230. 35. Поляков Л. М., Винокуров Э. И.—
Кинетика и катализ, 1979, т. 20, № 5, с. 1311—1314. 36. Кельцев Н. В., Жуко-
ва 3. А. —Труды ВНИИгаз, 1961, вып. 12(20), с. 143—149. 37. А. с. 146286
(СССР). 38. Bryan A. D. — Brit. Chem. Eng. 1961, v. 6, №2, p. 114—116. 39. Ас-
тахов В. А. и др, —ЖПХ, 1970, т. 43, № 12, с. 2655—2658; 1971, т. 44, № 2,
с. 319—322. 40. Аэров М. Э., Тодес О. М. Гидравлические и тепловые основы
работы аппаратов со стационарным и кипящим зернистым слоем. Л., Химия,
1968. 510 с.
Литература
573
41. Cholton Т. Н., Colburn А. Р. — Ind. Eng. Chem., 1931, v. 23, p. 913—
919. 42. Blake F. C. — Trans. Amer. Inst. Chem. Eng., 1921, v. 14, p. 415—420.
43. Happel J. — Ind. Eng. Chem., 1949, v. 41, № 6, p. 1161 —1174. 44. Кель-
цев H. В., Халиф A. JI. — Газовая пром., 1957, № 12, с. 31—35 45. Мин-
ский Е. М. —ДАН СССР, 1951, т. 78, № 3, с. 409—412. 46. Курганов В. М.,
Гонсалес С., Караваев Н. М. — Хим. и технол. топлив и масел, 1965, № 8
с. 4—7. 47. Дубинин М. М. — ЖПХ, 1941, т. 14, № 7—8, с. 906—913. 48. Mittels-
transs М., Fuertig Н., Weber М. — Chem. Technik, 1969, Bd. 21, № 2, S. 90—96.
49. Дубинин M. M. — Изв. АН СССР. ОХН, 1961, № 5, с. 750—756. 50. Forn-
wald Н. J., а. е. — Brit. Chem. Eng., 1963, v. 8, № 8, p. 546—550.
51. Flaser M.— In: Smisek M., Cerny S. Active Carbon. Amsterdam — Lon-
don-New York, Elsevier Publ. Co., 1970, p. 34—42. 52. Костомарова M. A.,
Суринова С. И. — В кн.: Адсорбенты, их получение, свойства и применение. Л.,
Наука, 1978, с. 54—56. 53. Виноградов С. Л., Ковальская А. П. — Пром, и са-
нит. очистка газов, 1980, № 2, с. 20—21. 54. Неймарк И. Е. — Усп. химии, 1956,
т. 25, № 6, с. 748—769. 55. Белоцерковский Г. М. и др. — ЖПХ, 1969 т. 42
№ 12, с. 2749—2756. 56. Пат. 3579464, 1971 г. (США). 57. Трохимец А. И. и
др. — Изв. АН СССР. Сер. хим., 1979, № 3, с. 51—55. 58. Васильев С. 3. и др. —
Электрометрия, 1970, вып. 97, с. 20—23. 59. Коваленко Л. А., Шумяцкий Ю. И.,
Кельцев Н. В. — ТОХТ, 1968, т. 2, № 6, с. 869—874. 60. Вишнякова М. И.
и др. —Изв. АН СССР. ОХН, 1961, № 3, с. 396—406.
61. Шумяцкий Ю. И. Канд. дис. М., МХТИ им. Д. И. Менделеева, 1965.
62. Дубинин М. М., Жуковская Е. Г„ Мурдмаа К. О. — Изв, АН СССР. ОХН,
1962, № 6, с. 960—968. 63. Мирский Я. В. Докт. дис. Л., ЛТИ им. Ленсовета,
1969; Изв. АН БССР. Сер. хим. наук, 1966, № 1, с. 114—118. 64. Мегедь Н. Ф.
и др. — В кн.: Адсорбенты, их получение и применение. Л., Наука, 1978, с. 27—
31. 65. Щукин Е. Д., Бессонов А. И., Паранский С. А. Механические испытания
катализаторов и сорбентов. М., Наука, 1971. 55 с. 66. Кельцев Н. В. — В кн.:
Получение, структура и свойства сорбентов. М., Госхимиздат, 1959, с. 39—51.
67. Клименко А. П. Методы и приборы для измерения концентрации пыли. М.,
Химия, 1978. 208 с. 68. Кельцев Н. В., Клушин В. Н., Артемова Е. Н. — Труды
МХТИ им. Д. И. Менделеева, 1979, вып. 109, с. 121—123. 69. Сидоров А. И.,
Шумяцкий Ю. И. Адсорбционная осушка газов. М., МХТИ им. Д. И. Менделе-
ева, 1972. 104 с. 70. Ничуговский Г. Ф. Определение влажности химических ве-
ществ. Л., Химия, 1977. 200 с.
71. Кельцев Н. В. и др. — В кн.: Адсорбенты, их получение, свойства и
применение. Л., Наука, 1971, с. 187—189. 72. Шолахов А. и др. — Труды МХТИ
им. Д. И. Менделеева, 1970, вып. 65, с. 76—78. 73. Bhat S. G. Т., Naraj-
ап К- S.— Indian J. Chem., 1978, v. А 16, № 4, р. 294—299. 74. Lemkoff N. О.,
Sing К. S. W. —J. Coll. Interface Sci., 1977, v. 61, № 2, p. 227—232. 75. Пат.
3606730, 1971 г. (США). 76. Назаров Б. Г., Торочешников Н. С., Кель-
цев Н. В. — Труды МХТИ им. Д. И. Менделеева, 1967, вып. 56, с. 126—129.
77. Mittelstrass М., Fuertig Н., Weber М. — Chem. Technik, 1969, Bd. 21, № 2,
S. 90—96. 78. Oil a. Gas J., 1958, v. 56, № 44, p. 74—77. 79. Drake O. S., Daw-
son E.R.— J. Petrol. Techn., 1960, v. 12, № 7, p. 12—21. 80. Dow W. M. — Pet-
rol. Ref., 1957, v. 36, № 6, p. 141—145.
81. Беликовский А. С., Расулов A. M. — Газовая пром., 1966, № 3, с. 50—
52. 82. Пат. 3477206, 1969 г. (США). 83. Пат. 3514396, 1970 г. (США). 84. Пат.
2944627, 1960 г. (США). 85. Громова К. И. и др. — Хим. пром, за рубежом,
1967, № 7(67), с. 60—75. 86. Лишневский М. И. Безнагревные установки для
осушки сжатого воздуха. М., ЦНИИТЭнефтехим, 1969. 53 с. 87. Chem. Eng.,
1963, v. 70, № 23, р. 225—226. 88. Пат. 3225518, 1965 г. (США). 89. Knob-
lauch К-, Reichenberger J.— GWF — Gas/Erdgas, 1975, Bd. 116, № 9, S. 382—
386. 90. Ермаков В. И. и др. — Хим. и нефт. маш., 1972, №9, с. 18—19.
91. Kuliev R., Samedova F. — Schmierst. u. Schmierungtechn., 1969, № 36,
S. 33. 92. Thomas T. L., Clark E. L. — Proc. 46th Ann. Convent Natur. Gas Pro-
574
Литература
cessors Assoc., Houston, Texas, 1967. Techn. pap., p. 51—60. 93. Hales G. E.—
Hydrocarb. Processing, 1971, v. 50, № 6, p. 151 —154. 94. Кононюк В. Ф., Capa-
хов А. И., Дубинин М. М.— Изв. АН СССР. Сер. хим., 1972, № 8, с. 1691—
1697; Изв. АН БССР. Сер. хим. наук, 1972, № 5, с. 45—49. 95. Siemienlewska Т.,
Tomkov К., Woszek В. — Przem. chem., 1972, v. 51, № 4, р. 231—233. 96. Cha-
jes W. — Vortr. geochem. u. chem. — phys. Probl. Erdol-Ergas. Erkund u. For-
ders, 1968, Bd. 3, S. 176—186. 97. Лакеев В. П. и др. — Газовая пром., 1968,.
№ 11, с. 49—51; 1969, № 5, с. 44—50. 98. Кельцев Н. В. и др. — Нефтепромыс-
ловое дело, 1978, № 10, с. 33—36. 99. Kulperger R. J. — Oilweek, 1968, v. 19,.
№ 7, р. 25. 100. Erdol u. Kohle-Erdgas — Petrochem., 1970, Bd. 23, № 6, S. 369.
101. Kessock A., Fris J. P. — Hydrocarb. Processing a. Petrol. Ref., 1965,.
v. 44, № 4, p. 123—124. 102. Кельцев H. В., Жукова 3. А., Ниорадзе С. C.—
В кн.: Клиноптилолит. Тбилиси, Мецниерба, т. 5, 1977, с. 199—206. 103. Pal-
mer G. Н. — Hydrocarb. Processing, 1977, v. 56, № 4, p. 103—106. 104. Kray-
chy P. N., Hasuda A.— Oil a. Gas J., 1966, v. 64, №32, p. 66—68. 105. Criog. a.
Ind. Gases, 1970, v. 5, №8, p. 33—38. 106. Hale D. — Amer. Gas J., 1969, v. 196,.
№ 14, p. 24—26. 107. Nitrogen, 1972, №79, p. 40. 108. Щербакова П. P.,
Бык С. Ш.— Газовая пром., 1971, № 6, с. 41—44. 109. Кельцев Н. В., Моисей-
чук О. В., Плужников Г. С. — Нефтепромысловое дело, 1979, № 8, с. 48—51..
ПО. Кельцев Н. В. — В кн.: Адсорбенты, их получение, свойства и применение.
Л., Наука, 1978, с. 184—186.
111. Кельцев Н. В., Титова Ю. К-, Торочешников Н. С. — Труды МХТИ им.
Д. И. Менделеева, 1964, т. 47, с. 61—68. 112. Вялкина Г. С. и др. — В кн.:
Цеолиты, их свойства, синтез и применение. М.-Л„ Наука, 1965, с. 303—304.
113. L. Р, — Gas, 1969, v. 29, № 12, р. 36—39. 114. Collins J. J., Raidt R. A.—
Oil a. Gas J., 1969, v. 60, № 10, p. 100—102. 115. Пат. 3151959, 1964 г. (США).
116. Плужников Г. С., Баженов Ю. М., Кельцев Н. В. — Нефтепромысловое
дело, 1976, № 11, с. 33—34. 117. Плужников Г. С. и др. — Нефтепромысловое
дело, 1978, № 12, с. 38—40. 118. Плужников Г. С. Канд. дис. М., МХТИ им.
Д. И. Менделеева, 1979. 119. Norton С. J. — Chem. a. Ind., 1962, № 6, р. 258—
259. 120. Юзефович В. И. и др. — Хим. пром., 1967, № 4, с. 23—24.
121. Stormont D. Н. — Oil a. Gas J., 1963, v. 61, № 10, р. 64—66; Gas Li-
quefatti, 1963, v. 9, № 4, p. 26—30. 122. Connel J. A. — Hydrocarb. Processing
a. Petrol. Ref., 1963, v. 42, № 12, p. 97—98. 123. Chem. Eng. Progress, 1966,
v. 62, № 5, p. 238. 124- Hales G. E. — Chem. Eng. Progress, 1971, v. 67, № 11,
p. 49—53. 125. Гельме И. Э. и др. — Хим. и технол. топлив и масел, 1971, № 1,
с. 13—15. 126. Гельме И. Э., Юзефович В. И., Юдинсон Р. Н. — Хим. и технол.
топлив и масел, 1967, № 9, с. 17—19. 127. Collins J. J. — Oil a. Gas J., 1962,
v. 60, № 27, р. 97—99. 128. Малкин Л. Ш. и др. — Хим. и технол. топлив и ма-
сел, 1970, № 9, с. 22—23. 129.Малкин Л. Ш. и др. — Хим. и технол. топлив и
масел, 1970, №12, с. 11—13. 130. Малкин Л. Ш. и др. Очистка и осушка ра-
бочей среды фреоновых холодильных машин синтетическими адсорбентами. М.,
ЦНИИТЭИлечпищепром, 1972. 44 с.
131. Малкин Л. Ш-, Колин В. Л., Кельцев Н. В., Самойленко В. И.—
В кн.: Адсорбенты, их получение, свойства и применение. Л., Наука,
1971, с. 213—215. 132. Кельцев Н. В., Назаров Б. Г., Торочешни-
ков Н. С. — Хим. и технол. топлив и масел, 1962, № 4, с. 21—24. 133. Соко-
лов Б. И. — Энергетик, 1970, № 12, с. 24—25. 134. Маневич Л. О. Обработка
трансформаторного масла. М., Энергия, 1975. 72 с. 135. Юзефович В. И. и др. —
Хим. и технол. топлив и масел, 1972, № I, с. 59—61. 136. Очистка промышлен-
ных газов. М., Химия, 1977. 488 с. 137. Зельвенский Я. Д., Харьковская Е. Н.—
Хим пром., 1960, № 4, с. 393—402. 138. Морозова А. А. и др.— Изв. АН
БССР. Сер. хим. наук, 1978, № 2, с. 36—40. 139. Bertsch L., Habgood Н. W. —
Литература
575
J. Phys. Chem., 1963, v. 67, № 8, p. 1621—1628. 140. Ward J. W., Hab-
good H. W. —J. Phys. Chem., 1966, v. 70, № 4, p. 1178—1182.
141. Хвощев С. С. и др. — ЖФХ, 1968, т. 42, № 1, с. 171—176. 142. Kurz G. —
Chem. Rdsch. (Schweiz), 1972, Bd. 25, № 38, S. 1175—1178. 143. Маергойз И. И.,
Летрук А. П. Контролируемые атмосферы в электрических печах. М., Энергия,
1971. НО с. 144. Васильев С. 3. и др.— Газовая пром., 1971, № 2, с. 40—43;
Газовое дело, 1971, № 2, с. 38—41; Электротермия, 1969, № 80, с. 39—41; 1970,
№ 97, с. 20—23; 1971, № 112, с. 21—24. 145. Сидоров А. И. Канд. дис. М.,
МХТИ им. Д. И. Менделеева, 1965. 146. Барк С. Е. и др. — В кн.: Синтетические
цеолиты. М., Изд-во АН СССР, 1962, с. 276—283. 147. Schutzgas Faschenbuch.
Essen, Vulkan-Verl., 1974, 142 S. 148. Васильев С. 3., Маергойз И. И., Пушка-
рев Л. И. Установки экзогаза. М., Энергия, 1977. 83 с. 149. Жукова 3. А.
и др. — Журн. ВХО им. Д. И. Менделеева, 1969, т. 14, № 3, с. 352—353.
150. Эстрин Б. М. Производство и применение контролируемых атмосфер. М.,
Металлургия, 1973. 392 с.
151. Sarnes R. — Blech, Rohre, Profile, 1972, Bd. 18, № 2, S. 65—69.
152. Колесников В. П. и др. — Труды МХТИ им. Д. И. Менделеева, 1974, т. 79,
с. 29—30. 153. Березовская И. М. и др. — Электротермия, 1969, № 82, с. 22—
24. 154. Васильев С. 3. и др. — В кн.: Адсорбенты, их получение, свойства и
применение. Л., Наука, 1978, с. 190—194. 155. Торочешников Н. С., Кель-
цев Н. В., Сидоров А. И. — Труды МХТИ им. Д. И. Менделеева, 1964, т. 47,
с. 69—74; 1966, т. 51, с. 99—104; в кн.: Цеолиты, их синтез, свойства и приме-
нение. М.-Л., Наука, 1965, с. 240—249. 156. Chem. Processing, 1963, v. 47, №3,
р. 51—54. 157. Стефаненко П. М. и др. — Кислородная промышленность. Техн,
и экономия, информация, 1970, № 3, с. 37—40. 158. Hedden К-, Mark F. Е.,
Rao В. R. — Chem.-Ing.-Techn., 1978, Bd. 50, № 4, p. 311; Ger. Chem. Eng., 1978,
v. 1, № 5, p. 305—312. 159. Thomas T. L., Clark E. L. — Oil a. Gas J., 1967,
v. 65, № 1, p. 112—115. 160. Кельцев H. B. — В кн.: Физическая адсорбция из
многокомпонентных фаз. М., Наука, 1972, с. 30—38.
161. Пат. 1270258, 1972 г. (Англия). 162. Perry С. R. — Oil a. Gas J., 1977,
v. 75, № 75, р. 76—79. 163. Haase Н. — Chem. Anlagen Verfahren, 1970, № 9,
S. 42—45. 164. Пат. 3557011, 1971 г. (США). 165. Пат. 3772852, 1973 г. (США).
166. Воронин Г. И., Поливода А. И. Жизнеобеспечение экипажей космических
кораблей. М., Машиностроение, 1967. 211 с. 167. Stengel R. F. — Design News,
1969, v. 24, № 7, р. 36—37. 168. Walker Р. L., Chelef M. —Carbon, 1967, v. 7,
№ 1, p. 7—11. 169. Кельцев H. В. Очистка отходящих газов промышленности
от вредных примесей. М., МХТИ им. Д. И. Менделеева, 1978. 48 с. 170. Зель-
венский Я. Д., Волков А. Е. — Труды ГИАП, 1953, вып. 1, с. 131—158.
171. Егоров Н. Н. и др. Очистка от серы коксовального и других горючих га-
зов. М., Металлургиздат, 1960. 341 с. 172. Swinnarrski A., Siedlewski J. — Che-
mia Stosawana, 1961, v. 5, № 2, p. 211—224. 173. Пат. 724911, 1962 г. (ФРГ).
174. Пат. 1087272, 1965 г. (Англия). 175. Пат. 1388453, 1965 г. (Франция).
176. Fischer F., Kraus Н. — Chem.-Ing.-Techn., 1964, Bd. 36, № 9, S. 963—968.
177. Smisek M., Cerny S. Active carbon. Manufacture, Properties a. Applications.
Amsterdam — London — New York, Elsevier Publ., 1970. 479 p. 178. Fails J. C.,
Harris W. D. — Oil a. Gas J., 1960, v. 58, № 28, p. 86—90. 179. Kunkel L. V.,
Chobotuck J. W. — Petrol. Int., 1974, v. 21, № 5, p. 48—55. 180. Boston F. C.—
Hydrocarb. Processing a. Petrol. Rep., 1964, v. 43, № 8, p. 141—145.
181. Petrol. Ref., 1957, v. 36, № 7, p. 136—140. 182. Conviser S. A. —Oil a.
Gas J., 1965, v. 63, № 49, p. 130—135. 183. Kessock A., Fris J. P. — Hydrocarb.
Processing a. Petrol. Ref., 1965, v. 44, № 4, p. 123—124. 184. Haines H. W.,
Wielingen G. A., Palmer G. H. — Petrol. Ref., 1961, v. 40, № 4, p. 123—126.
185. Пат. 3144307, I960 г. (США). 186. Пат. 3186789, 1961 г. (США). 187. Ад-
576
Литература
ливаикина М. А. и др.— Труды МХТИ им. Д. И. Менделеева, 1967, т. 56„
с. 160—164; 1969, т. 60, с. 123—126; 127—129. 188. Адинсков Б. П., Спи-
рин И. А. — В кн.-. Использование газа в народном хозяйстве, вып. 10. Саратов,.
Саратовский ун-т, 1973, с. 315—321. 189. Кулиев А. М. и др. — Газовая пром.,
1974, № 7, с. 39—41. 190. Набутовский 3. А.— Газовая пром., 1977, № 4,
с. 18—20.
191. Chi Chang W., Lee Hanju — AIChE. Symp. Ser., 1973, v. 69, № 1/4,
p. 95—101. 192. Торочешников H. С., Соколов В. А., Кельцев H. В. Молекуляр-
ные сита и нх применение. М., Химия, 1964. 156 с. 193. Еникеева Н. И. и др.—
Труды МХТИ им. Д. И. Менделеева, 1966, т. 51, с. 93—98; труды КХТИ им.
С. М. Кирова (Казань), 1969, т. 40, ч. 1, с. 288—294. 194. Пат. 972284, 1964 г.
(Англия). 195. Пат. 3594983, 1971 г. (США). 196. Dudzik Z„ Bilska М. — Przem.
chem., 1974, v. 53, № 12, S. 727—730. 197. Kulperger E. J. —Oilweek, 1968,
v. 19, № 7, p. 25—28. 198. Фролов Г. С. и др. — Пром, и санитарная очистка
газов, 1977, № 3, с. 16—17. 199. Харьковская Е. И., Зельвенский Я. Д., Про-
нина Р. Н. — Хим. пром., 1966, № 4, с. 268—272. 200. Мазгаров А. М., Фахри-
ев А. Ф., Неячилов А. В. — Хим. и технол. топлив и масел, 1978, № 8, с. 3—8.
201. Вольцов А. А. и др. — Кинетика и катализ, 1979, т. 20, № 1, с. 253—
256. 202. Афанасьев Ю. М. и др. — Пром, и санитарная очистка газов, 1977,
№ 6, с. 16—18. 203. Пат. 3144307, 1964 г. (США). 204. Petrol. Ref., 1961, v. 40,
№ 11, р. 191—192. 205. Пат. 3154383, 1964 г. (США). 206. Жукова 3. А., Кель-
цев Н. В. —ЖФХ, 1970, т. 44, № 8, с. 2067—2068. 207. Дияров И. Н. и др.—
В кн.; Природные цеолиты. Тбилиси, Мецниерба, 1979, с. 269—273. 208. Грабов-
ский Ю. П., Меркулов Е. А., Цыбулевский А. М. — Там же, с. 186—189.
209. Hydrocarb. Processing a. Petrol. Ref., 1964, v. 43, № 9, p. 198—199. 210.
Lirpatrick S. D. — Chem. Eng., 1961, v. 68, № 23, p. 192—196.
211. Clark E. L. e. a. — Chem. Eng. Progress, 1960, p. 192—196. 212. Mess-
mer J. K., Brunner O. — Erdol u. Khole, 1962, Bd. 15, Ns 3, S. 185—186. 213. Ber-
ti B., Brunner O., Clark E. L. — Gas liguefatti, 1965, v. 11, № 117, p. 14—15.
214. Clark E. L. — Oil a. Gas J., 1962, v. 60, № 46, p. 178—179. 215. Debaty M. —
Bull. Ass. Frans. Technieciens Petrole, 1959, № 135, p. 591—621. 216. Ed-
wards J. F. Brit. Chem. Eng., 1961, v. 6, № 2, p. 112—114. 217. Kowalchuk A.—
J. Petrol. Technol., 1972, May, v. 24, p. 540—544. 218. Schoofs R. J. — Oil a. Gas
Internal., 1967, v. 7, № 7, p. 30—35. 219. Галич П. H., Куприянова Л. A. — Нефт.
и газ. пром., 1968, Научно-произв. сб. 5, с. 47—48. 220. Henke А. М., Stanf-
fer Н. С., Karr N. L. — Oil a. Gas J., 1962, v. 60, № 18, p. 78—80.
221. Uhl W. C. —World Petrol., 1961, v. 32, № 4, p. 32—34. 222. Scher-
wood P. W. — Petroleum, 1963, v. 26, № 6, p. 247—252; Erdol u. Kohle, 1963,
Bd. 16, № 8, S. 843—846; Techniques du Petrol., 1963, v. 18, № 204, p. 23—28..
223. Виноградова В. С., Кофман Л. С. — В кн.: Синтетические цеолиты. М.,
Изд-во АН СССР, 1962, с. 99—102. 224. Кофман Л. С., Виноградова В. С.,
Ефимова Г. И. — Вести, технич. экономич. информации Гос. комитета Совмина
СССР по химии, 1961, № 4, с. 10—16. 225. Кисаров В. М. — Журн. ВХО им.
Д. И. Менделеева, 1969, т. 14, № 4, с. 388—393. 226. Кисаров В. М. Современ-
ное состояние техники рекуперации летучих органических растворителей. М.,
ЦИНТИхимиефтемаш, 1976. 48 с. 227. Лазарев Р. Я. и др. — Каучук и рези-
на, 1962, № 8, с. 45. 228. Кельцев Н. В., Мухии В. М. — Пром, и санитарная
очистка газов, 1978, № 4, с. 18—19. 229. Кельцев Н. В. и др.— Труды МХТИ
им. Д. И. Менделеева, 1979, вып. 9, с. 12—22. 230. Chem. Processing, 1977, v. 40,
Ns 13, p. 95.
231. Суханова T. А., Субботин А. И., Ковальская А. П— Пром, и санитар-
ная очистка газов, 1980, № 2, с. 19—20. 232. Якубеня Н. А. и др. — ЖПХ,
1978, т. 51, № 8, с. 1779—1783. 233. Кисаров В. М., Субботин А. И. — Пром, и
Литература
577
санитарная очистка газов, 1980, № 2, с. 18—19. 234. Костриков В. И., Кель-
цев Н. В., Нивин П. И.—Хим. волокна, 1968, № 2, с. 45—47. 235. Костри-
ков В. И., Кельцев Н. В., Панова Л. Н. — В кн.: Углеродные адсор-
бенты и их применение в промышленности, ч. 2. Пермь, 1969, с. 12—21.
236. Кельцев Н. В. и др. — Хим. волокна, 1970, № 1, с. 37—38. 237. Бубно-
ва Г. П., Кострикова В. И. — Журн. ВХО им. Д. И. Менделеева, 1969, т. 14,
с. 399.—405. 238. Ким В. П. и др. — Хим. волокна, 1971, № 5, с. 66—68. 239. Чер-
вяков В. А. — Хим. волокна, 1973, № 2, с. 55—57. 240. Кельцев Н. В. и др.—
Труды МХТИ им. Д. И. Менделеева, 1981, вып. 119, с. 12—17; 39—46.
241. Смола В. И., Кельцев Н. В. Защита атмосферы от двуокиси серы. М.„
Металлургия, 1976, с. 44—128. 242. Смола В. И. Канд. дис. М., Гинцветмет,.
1972. 243. Давтяи О. К., Овчинникова Е. Н. — ДАН СССР, 1955, т. 104, № 6,
с. 857—860. 244. Michiharu Seki, Kyoko Jomamoto. Manuscript for ACS Houston
Meeting Tokyo, February 1970. 245. Оно T. Сангё кочай, 1969, т. 5, № 6, с. 326—
329. 246. Sleek А. V, —Chem. Eng., 1967, v. 47, № 25, p. 168—192. 247. Bro-
cke W. — Staub-Reinhalt-Luft, 1968, Bd. 28, № 3, S. 101—107. 248. Chem. Eng.,
1970, v. 77, p. 71—73. 249. Вилесов H. Г., Костюковская А. А. Очистка выброс-
ных газов. Киев, Техника, 1971. 194 с. 250. Johswitch F. — Brennstoff-Warme-
Kraft, 1965, Bd. 17, № 5, S. 985—990.
251. Федышин Б. M. и др. — Хим. пром., 1977, № 8, с. 597—598. 252. Фе-
дышин Б. М. Канд. дис. М., МХТИ им. Д. И. Менделеева, 1981. 253. Jung-
ten Н., Karweil J. — Erdol u. Kohle, 1962, Bd. 15, № 12, S. 985—990. 254. Ban-
ning K. — Brennstoff-Warme-Kraft, 1968, Bd. 20, № 12, S. 588. 255. Spregler G. —
Staub, 1963, v. 23, № 3, S. 171—175. 256. Смола В. И., Рахлин Е. С., Кель-
цев Н. В. — В кн.: Пылеулавливание и очистка газов в цветной металлургии.
М., Металлургия, 1970, с. 142—151. 257. Brennstoff-Warme-Kraft, 1971, Bd. 23,.
№ 3, S. 91—97. 258. Fischer F., Krau H. — Chem.-Ing.-Techn., 1964, Bd. 36, №9,.
S. 963—966. 259. Jungten H., Knoblach K-, Krue M. — Chem.-Ing.-Techn., 1970,
Bd. 42, № 2, S. 77—81. 260. Machine Design, 1972, v. 43, № 19, p. 32.
261. Peper Trade J., 1971, v. 155, № 14, p. 36. 262. Pupl. a. Peper, 1971,.
v. 72, № 3, p. 120—122. 263. Oil a. Gas J., 1971, v. 69, № 14, p. 64—65. 264. To-
les G. E. — Chem. Processing, 1971, v. 17, № 12, p. 13—14. 265. Смола В. И.,.
Рахлин Е. С., Кельцев Н. В. — Водоснабжение и санитарная техника, 1972,.
№ 4, с. 33—36. 266. Кельцев Н. В. — Журн. ВХО им. Д. И. Менделеева, 1979,
т. 24, № 1, с. 54—58. 267. Бермаи И. Ф. и др. — В кн.: Природные цеолиты. Тби-
лиси, Мецниерба, 1979, с. 55—61. 268. Берман И. Ф., Кельцев Н. В. — Пром. и
санитарная очистка газов, 1978, № 4, с. 15—16. 269. Collins J. J. е. а. — Chem.
Eng. Progr., 1974, v. 70, № 6, р. 58—62. 270. Кельцев Н. В., Толстикова М. Г.,
Бермаи И. Ф. — Пром, и санитарная очистка газов, 1978, № 4, с. 16.
271. Степанов А. С., Фадеев А. И. — Хим. пром., 1972, № 8, с. 71—73.
272. Степанов А. С., Фадеев А. И. — Пром, и санитарная очистка газов, 1976,
№ 3, с. 22—23. 273. Степанов А. С., Фадеев А. И., Львов Г. М. — Пром, и са-
нитарная очистка газов, 1979, № 4, с. 16—17. 274. Степанов А. С., Зем-
сков И. Ф. — Хим. пром., 1970, № 1, с. 43—45. 275. Bergh V. К.-Н. — Z. Chem.,
1977, Bd. 17, № 3, S. 610—617. 276. Haani M., Kuivala A. — Kemia-Kemi, 1972,
v. 29, p. 577—580. 277. Бермаи И. Ф., Смола В. И., Кельцев Н. В. — Труды
МХТИ им. Д. И. Менделеева, 1977, вып. 93, с. 30—35. 278. Lyman W. J. Car-
bon adsorption. «Unit Oper. Treat. Hazardous Ind. Waster». Perk Ridge, N. Y.,
1978, p. 97—137. 279. Кудеико H. E. — Труды ВНИИводгео, 1973, вып. 40, ч. 2,
с. 74—78. 280. Степанова Н. В., Мерсова Н. А. — Хим. пром, за рубежом, 1972,
вып. 9(117), с. 22—35.
- 281. Wu V.—Water Poll. Contr. Developm. Countries. Proc. Int. Conf., Bang-
kok, 1978, p. 573—578, 282. Этиигер И. Л., Шларакова Э. И. — Вести, маши-
37—1346
-578 Литература
«остр., 1979, № 8, с. 52—54. 283. А. с. 595258 (СССР). 284. Исхаков X. А., Сол-
датчеико А. Н., Сапожникова Ф. X. — Химия твердого топлива, 1977, № 4,
с. 131—133. 285. Родионов А. И., Скорняков В. В., Беляева Н. М.— Цветная
металлургия, 1978, № 3, с. 26—28. 286. Kunin R. — Polym. Eng. a. Sci., 1977,
v. 17, № 1, р. 58—62. 287. Mignonsin Е. Р., Guillaume М. — Tijiadschr. water-
vootz. en. afvalwaterbchandel., 1975, Bd. 8, № 18, S. 368—373. 288. Герасимен-
ко Б. Д. — Изв. Сев.-Кавказ, научн. центра высш, школы. Сер. техн, науки,
1977, № 3, с. 81—82. 289. Пат. 4019982, 1975 г. (СШН). 290. Пат. 53—57190,
1968 г. (Япония).
291. Галиева Ю. Г. и др. — В кн.: Гигиенические аспекты охраны окружаю-
щей среды. М., изд. Акад. мед. наук. СССР и ин-та общ. и коммун, гигиены им.
А. И. Сысина, 1974, вып. 2, с.86—88. 292. Wasser, Luft u. Bctr., 1978, Bd. 22,
.№ 11, S. 576—586. 293. Когановский A. M. и др. Очистка промышленных сточ-
ных вод. Киев, Техника, 1974. 257 с. 294. Сорбция из растворов высокополи-
мерами и углями. Минск, Изд-во Белорусского ун-та, 1961. 152 с. 295. Кири-
ченко В. А., Левченко Т. М.„ Когановский А. М. — В кн.: Адсорбция и адсор-
бенты. Киев. Иаукова думка, 1972, вып. 1, с. 32—37. 296. Gomella С. — In: Tech-
niques et sciences municipales et reveu d’lau, 1970, v. 65, p. 383—389. 297. Ро-
зин A. T. Канд. дис. Минск, Ин-т торфа АН БССР, 1968. 298. Ровинская Т. М.,
Когановский А. М. — Коллоидн. ж., 1962, т. 24, № 1, с. 67—73. 299. Стад-
.иик А. М. —Труды ВНИИводгео, 1974, вып. 47, с. 28—39. 300. Колышкин Д. А.,
Михайлова К- К. Активные угли. Справочник. Л., Химия, 1972. 57 с.
301. Дубинин М. М., Онусайтис Б. А. — В кн.: Углеводородные адсорбенты
и их применение в промышленности, ч. 1. Пермь, 1969, с. 3—26. 302. Кель-
цев А. В. Канд. дис. М., МХТИ им. Д. И. Менделеева, 1977. 303. Гринберг А. М.
Юбесфеноливание сточных вод коксохимических заводов. М., Металлургия, 1968.
.212 с. 304. Шевченко М. А. Физико-химическое обоснование процессов обесцве-
чивания и дезодорации воды. Киев, Наукова думка, 1973. 152 с. 305. Глушан-
ков С. Л., Корин А. М., Ковалева Э. А. — В кн.: Углеродные адсорбенты и их
.применение в промышленности, ч. 2. Пермь, 1969, с. 99—113. 306. Сугияма Т.—
Когату кейдзай, 1970, т. 17, № 5, с. 458—465. 307. Giglio А. — Chim. е. indust-
ria, 1972, v. 54, № 1, р. 59—62. 308. Пат. 4105549, 1978 г. (США). 309. Пат.
4085043, 1978 г. (США). 310. Кульский Л. А. Теоретические основы и техно-
логия кондиционирования воды. Киев, Наукова думка, 1980. 564 с.
311. Лукиных Н. А., Липман Б. Л., Криштул В. П. Методы доочистки сточ-
ных вод. М., Стройиздат, 1974. 96 с. 312. Browing J. Е. — Chem. Eng., 1972,
-у. 79, № 4, р. 36—40. 313. Technocrat, 1976, v. 9, № 2, р. 85—90. 314. Новое
•оборудование для очистки газов, сточных вод и переработки шламов, выпускае-
мое фирмами ФРГ. М., Черметинформация, Экспресс-информация, вып. 1, 1979,
с. 3. 315. Grulich S. е. а. — AIChE Sump., 1973, v. 69, № 127, р. 134—137.
316. Kaempf Н. Т. е. а. — Wasser, Luft u. Bert., 1972, v. 16, № 6, S. 169—172.
317. Ролев M. Я., Большакова В. А., Шимко И. Г. — В кн.: Новые методы очи-
стки сточных вод в производстве искусственных волокон. М., НИИТЭхим, 1974,
с. 33—69. 318. Wasser, Luft u. Bert., 1972, Bd. 16, № 11, S. 397—401. 319. Пат.
3635817, 1972 г. (США). 320. Van Stone J. L. — Iron a. Steel Eng., 1972, v. 48,
-№ 4, p. 63—66.
321. Фишмаи Г. И., Литвак А. А. Водоснабжение и очистка сточных вод
предприятий химических волокон. М., Химия, 1971. 161 с. 322. Стемпков-
ская Л. А. — В кн.: Исследования в области промышленного применения сор-
бентов. М„ Изд-во АН СССР, 1961, с. 127—134. 323. Roy С„ Volesky В. — Са-
rnad. Text. J., 1977, v. 94, № 8, p. 47—63. 324. Соколов В. П. — Хим. пром.,
1969, № 4, с. 20—22. 325. Krepke J. — Chemiefasern, 1972, Bd. 22, № 4, S. 302—
308. 326. Пат. 51898, 1970 г. (Румыния). 327. Dejohn Р. В., Adams A. D.—
Hydrocarb. Processing, 1975, v. 5, № 10, sec. 1, p. 104—111. 328. Lamben A. E.,
Литература
57»
Sharp D H. — Mantifact. Chemist., 1960, № 31, p. 198—201. 329. В кн.: Очистка
производственных сточных вод. Л., Химия, 1967, с. 187—240. 330. Семе-
нов Л. В., Давыдов В. А. Экономика химической переработки угля. М., Метал-
лургия, 1972. 97 с.
331. Lewis W. К. е. а. — J. Amer. Chem. Soc., 1950, v. 72, № 3, p. 1153—
1157. 332. Левит P. M., Белоцерковский Г. M.— В кн.: Углеродные адсорбенты
и их применение в промышленности, ч. 2. Пермь, 1969, с. 31—43. 333. Le-
wis W. К. е. a. —Ind. Eng. Chem., 1950, v. 42, № 7, p. 1319—1326. 334. Бе-
зус А. Г., Древинг В. П.,Клячко-Гурвия А. Л. — Коллоидн. ж., 1961, т. 23,
№ 3, с. 241—247. 335. Гольберт К. А., Платонов В. М., Павлова Л. Ф.—
В кн.: Химическая переработка нефтяных углеводородов. М., Изд-во АН СССР,
1956, с. 231—241. 336. А. с. 104741 (СССР). 337. Веремчук М. К. Дис. Львов,
Львовский политехи, ин-т, 1972. 338. Mair В. J. е. а. — Ind. Eng. Chem., 1950,.
v. 42, № 7, р. 1279—1286. 339. Rossini F. D. e. a. — Proc. Amer. Petrol. Inst.,
1942, v. 23, № 3, p. 7—10. 340. Пат. 2398101, 1946 г. (США).
341. Harper J. J., Olsen J. L., Shuman F. R. — Chem. Eng. Progr., 1952,.
v. 48, № 6, p. 276—280. 342. Пат. 718219, 1954 г. (Англия). 343. Пат. 2854495,
1958 г. (США). 344. Пат. 2819326, 1958 г. (США). 345. Пат. 143882, 1962 г..
(Венгрия). 346. Пат. 2773803, 1956 г. (США). 347. Пат. 2768942, 1956 г. (США).
348. Пат. 2768942, 1956 г. (США).349. А. с. 147711 (СССР). 350. Левинсон С. 3.
и др. — Хим. и технол. топлив и масел, 1971, № 8, с. 17—20.
351. Urbanscek S. — Konf. Chem. u. Chem. Verarb. Erdol u. Erdgas, Buda-
pest, 1965. Budapest, 1968, S. 944—950. 352. Chachulski J., Urbanscek S. — Erdol
u. Kohle, 1966, Bd. 19, № 5, S. 351—354. 353. Пат. 3150079, 1964 г. (США).
354. Мосейчук О. В., Кельцев Н. В. — Труды МХТИ им. Д. И. Менделеева, 1973,.
вып. 73, с. 13—14. 355. Кельцев Н. В. — Газовая пром., 1960, № 9, с. 49—53.
356. Collins J. J. — Chem. Eng. 1959, v. 66, № 17, p. 128—130. 357. Георгиев Ц. O'.
Канд. дис. М., МХТИ им. Д. И. Менделеева, 1957. 358. Carter J. L. е. а. — J.
Phys. Chem., 1966, v. 70, № 4, р. 1126—1136. 359. Jates D. S. C. — J. Phys..
Chem., 1966, v. 70, № 11, p. 3693—3696. 360. Norton C. J. —Chem. a. Ind.,.
1962, № 6, p. 258—259.
361. Chem. Age, 1962, v. 87, № 2235, p. 785. 362. Eberly P. E. — J. Phys..
Chem., 1967, v. 71, №6, p. 1717—1722. 363. Кельцев H. В. и др. — Нефтепере-
работка и нефтехимия, 1969, № 9, с. 33—34. 364. А. с. 192178 (СССР)..
365. Scherwood Р. W. — Brennstoff-Chemie, 1959, Bd. 40, № 11, S. 1773—1778..
366. Кельцев Н. В. — Газовая пром., 1959, № 9, с. 38—40. 367. Szepesy L.,.
Jlles V. — Acta chim. Acad. Sci. Hung., 1963, v. 35, № 3, p. 245—254. 368. Гаии-
ko H. K-, Дорогочииский A. 3. — Хим. и технол. топлив и масел, 1965, № II,
с. 28—32. 369. Eberly Р. Е., Cimberlin С. N., Beker L. Е. — J. Appl. Chem., 1967,
v. 17, № 2, р. 44—48. 370. Петряева Г. С. Канд. дис. М., Ин-т органич. химии-:
им. Н. Д. Зелинского, 1968.
371. Березутский В. М. и др. — Нефтепереработка и нефтехимия, 1979,.
№ 12, с. 36—38; 1980, № 2, с. 25—26; с. 34—36. 372. Петряева Г. С., Тимофе-
ева Е. А., Шуйкии Н. А. —ДАН СССР, 1967, т. 172, № 2, с. 361—363. 373. Brit.
Chem. Eng., 1970, v. 15, № 3, p. 301—303. 374. Жданов С. П., Хвощев С. С.,
Самулевич Н. Н. Синтетические цеолиты. М., Химия, 1981. 260 с. 375..
Вольф М. Б. и др.—-Хим. и технол. топлив и масел, 1963, № 8, с. 29—30.
376. Canad. Chem. Proc., 1958, v. 42, № 7, p. 55—62. 377. Далин M. А. и др. —
Хим. пром., 1962, № 2, с. 77—79. 378. Вольф М. Б. и др. — Пластич. массы.
1966, № 11, с. 26—28. 379. Виноградская М. В., Кельцев Н. В., Львов С. В.—
ЖФХ, 1970, т. 44, № 1, с. 238—240. 380. Barren R. М., Steart W. J. — Proc.
Roy. Soc., 1959, v. A249, № 1259, p. 464—483.
37*
-580
Литература
381. Джарылпакова Ж. А., Торочешников Н. С., Кельцев Н. В, —Труды
МХТИ им. Д. И. Менделеева, 1961, т. 35, с. 1'58—161; Журн. ВХО им.
_Д. И. Менделеева, 1960, т. 5, № 6, с. 710—712. 382. Сихарулидзе Н. Г. — Азот-
ная пром., 1969, № 1, с. 79—83. 383. Кельцев Н. В., Мякиненков В. И., Торо-
чешников Н. С. — Хим. пром., 1964, № 11, с. 813—817; журн. ВХО им. Д. И. Мен-
делеева, 1962, т. 7, № 6, с. 694—695; труды МХТИ им. Д. И. Менделеева,
1963, т. 41, с. 186—190; в кн.: Разделение и анализ углеводородов. М., Изд-во
АН СССР, 1963, с. 122—133. 384. А. с. 173726 (СССР). 385. Зельвенский Я. Д,
Кочетков В. Л. — Нефтехимия, 1963, т. 3, № 2, с. 285—295. 386. Лузянин Б. П.,
Кельцев Н. В., Торочешников Н. С. — Труды МХТИ им. Д. И. Менделеева,
1963, т. 41, с. 191 — 196. 387. Пат. 3457166, 1969 г. (США). 388. Пат. 330684,
1967 г. (США). 389. Пат. 3476822, 1969 г. (США). 390. Mair В. J., Schmaien-
ger М. — Analyt. Chem., 1958, v. 30, № 2, p. 276—279.
391. Квитковский Л. H. Докт. дис. Львов, Львовский политехи, ин-т, 1975.
392. Satterfield Ch. N., Cheng Ch. S. — AIChE J., 1972, v. 18, № 4, p. 720—723.
393. Еникеева H. И. Канд. дис. Казань, КХТИ, 1969. 394. Joubert J., Zwie-
ber J. — .Advanc. Chem., 1971, Ser. 102, p. 209—216. 395. Кофер P. Г., Дани-
лов H. Ф., Пойлов В. 3. — В кн.: Адсорбенты, их получение, свойства и приме-
нение. Л., Наука, 1978, с. 204—206. 396. Кельцев Н. В., Старовойтова А. Ф.—
Газовая пром., 1961, № 8, с. 34—37. 397. Кельцев Н. В., Старовойтова А. Ф.,
Торочешников Н. С. — В кн.: Синтетические цеолиты. М., Изд-во АН СССР,
1962, с. 239—244. 398. А. с. 157343 (СССР). 399. Виноградова В. С., Коф-
ман Л. С. — В кн.: Синтетические цеолиты. М., Изд-во АН СССР, 1962, с. 99—•
102. 400. Nelson К. Н., Grimes М., Heinrich В. J. — Analyt. Chem., 1957, v. 29,
-№ 7, р. 1026—1029.
401. Peterson D. L., Redlich O. — J. Chem. Eng. Data, 1962, v. 7, № 4,
S. 70—75. 402. Мирский Я. В., Дорогочинский А. 3., Мегедь Н. Ф. — Нефте-
переработка и нефтехимия, 1976, № 4, с. 22—26. 403. Franz W. F. е. а. — Oil а.
“Gas J., 1959, v. 57, № 15, р. 116—124. 404. Хим. и техиол. топлив и масел,
1962, № 9, с. 70—71. 405. Митрофанов М. Г., Мирский Я. В. — В кн.: Синте-
тические цеолиты. М., Изд-во АН СССР, 1962, с. 236—238. 406. Вольф М. Б.,
Алексеева Р. В. — В кн.: Синтетические цеолиты. М., Изд-во АН СССР, 1962,
с. 233—235. 407. Балинский И. С., Квитковский Л. Н. — Нефт. и газ. пром.,
1970, № 3(51), с. 37—39. 408. Телешова М. Н., В ига люк Р. В. — Нефтеперера-
ботка и нефтехимия, 1964, № 1, с. 13—15. 409. Алиев В. С. и др. — Азерб. хим.
ж., 1963, № 5, с. 13—16. 410. Defives D., Buson Р. — Gen. chim., 1961, v. 68,
№ 3, p. 64—73.
411. Greebbell J. — Oil a. Gas J., 1975, v. 73, № 15, p. 85—101. 412. Gries-
mer G. J., Rhodes H. B., Kijonaga K- — Petrol. Ref., 1960, v. 39, № 6, p. 125—
132; Erdol u. Kohle, 1960, Bd. 13, № 9, S. 650—656. 413. Oil a. Gas J., 1959,
v. 57, № 27, p. 58. 414. Мирский Я. В., Митрофанов М. Г. — В кн.: Синтетиче-
ские цеолиты. М., Изд-во АН СССР, 1962, с. 94—98. 415. Broughton D. В., Car-
son D. В. — Oil a. Gas J., 1959, v. 57, № 15, р. 112—115. 416. Samuels S. С.,
Ray G. C., Reiche A. D. — Ind. Eng. Chem., 1959, v. 51, № 1, p. 73—76. 417. Sher-
wood P. W. — Brennstoff-Chemie, 1959, Bd. 40, № 11, p. 354—358. 418. Chem.
'Eng. News, 1959, v. 37, № 13, p. 39. 419. Bour G., Schwoerer С. P., Asse-
lin С. P. — Oil a. Gas J, 1970, v. 10, № 11, p. 101—105; Oil a. Gas J., 1970,
v. 68, № 43, p. 57—61. 420. Avery W. F., Lee M. N. J. — Oil a. Gas J., 1962,
v. 60, № 23, p.121—123;Erd61 u. Kohle, 1962, Bd. 15, № 5, S. 356—358; Chim.
e. Ind., 1962, v. 88, № 4, p. 417—420; Petrole Information, 1962, № 331, p. 83—
84.
421. Oil a. Gas J., 1967, v. 65, № 12, p. 160. 422. Lindner K-— Seifen-Fette-
Wachse, 1968, v. 94, № 4, p. 81—84; № 5, p. 110—112. 423. Oil a. Gas L., 1959,
v. 57, № 13, p. 52. 424. Jammartino N. R. — Chem. Eng., 1975, v. 82, № 9, p, 62—
Литература
581
63. 425. Дорогочинская В. А. и др. — Хим. и технол. топлив и масел, 1981,
№ 1, с. 39—40. 426. Carson D. В., Broughton D. В. — Petrol. Ref., 1959, v. 38,
№ 4, р. 130—134. 427. Ziegenhain W. С. — Petrol. Eng., 1957, v. 29, № 9,
Sect. 6, p. 6—12. 428. Мирский Я. В., Митрофанов М. Г., Дорогочинский А. 3.
Молекулярные сита. Грозный, Чечено-Инг. нзд-во, 1964. 107 с. 429. Anders Кч
Konnecke Н. G., Pulz A. Konf. Chem. a. chem. Verarb. Erdols u. Erdgas. Buda-
pest, 1968, S. 671—685. 430. Петряева Г. С., Кельцев Н. В., Тимовеева Е. А.—
Изв. АН СССР. Сер. хим., 1967, № 8, с. 1860—1862.
431. Larson L. Р., Becker Н. С. — Analyt. Chem., 1960, v. 32, № 9, р. 1215—
1216. 432. Квитковский Л. Н., Грушецкая Е. В. — Хим. и технол. топлив и ма-
сел, 1962, № 3, с. 61—64. 433. Павлова С. Н., Дриацкая 3. В., Мхчиян М. А. —•
Хим. и технол. топлив и масел, 1962, № 3, с. 58—60. 434. Schwartz R. D.,
Brasseaux D. J. — Analyt. Chem., 1957, v. 29, № 7, p. 1022—1036. 435. Адыко-
ва T. T., Рябова Н. Д., Усманова Д. А. — Узб. хим. ж., 1962, № 2, с. 77—79.
436. Myers Н. S., Hipkin Н. G. — Petrol. Ref., 1956, v. 35, № 7, р. 175—177.
437. Swisher R. D. — Chem. Eng., Progr., 1964, v. 60, № 12, p. 41. 438. Фалько-
вич M. И., Черножуков H. И., Эльтеков Ю. A. — Хим. и технол. топлив и масел,
1969, № 12, с. 11—15. 439. Chem. Ind., 1972, v. 24, № 1, р. 14. 440. Chem. а.
Eng. News, 1975, v. 53, № 40, p. 18—19.
441. Oil a. Gas J., 1963. v. 61, № 39, p. 93—94. 442. Chem. Eng., 1963,
v. 70, № 19, p. 69. 443. Ponder T. C. Hydrocarb. Processing, 1969, v. 48, № 10,
p. 141 — 144. 444. Пат. 2996558, 1961 г. (США). 445. Пат. 898059, 1962 г. (Анг-
лия). 446. Пат. 2920038, 1960 г. (США). 447. Пат. 2935476, 1960 г. (США).
448. Пат. 2938864, 1961 г. (США). 449. Пат. 3070542, 1961 г. (США). 450. Пат.
2974179, 1961 г. (США).
451. Галич П. Н. и др.—Нефтехимия, 1962, № 2, с. 193—195, 452. Пат.
268616, 1966 г. (Австралия). 453. Cooper D. Е. е. а. — Europ. Chem. News, 1966,
v. 10, № 254, Suppl., p. 23. 454. Галич П. H„ Коновальчиков О. Д., Дуров-
ская Г. П. — В кн.: Нефтепереработка и нефтехимия. Киев, Наукова думка,
1965, с. 121—124. 455. Пат. 69593, 1969 г. (ГДР). 456. Топчиева К. В., Хо Ши
Тхоанг —ДАН СССР, 1971, т. 198, № 1, с. 141—144. 457. Пат. 995937, 1965 г.
(Англия). 458. La Plante L. J. — Oil a. Gas J., 1970, v. 68, № 6, p. 55—57.
459. Brit. Chem. Eng., 1962, v. 7, № 9, p. 649. 460. Avery H'. F., Lee M. N. —
Petrol. Management, 1962, v. 34, № 6, p. 239.
461. Ind. Eng. Chem. Intern. Ed., 1962, v. 54, № 5, p. 12—13. 462. Chem.
Eng. News, 1962, v. 40, № 17, p. 59—60. 463. Oil a. Gas J., 1962, v. 60, № 15,
p. 92. 464. Stormont D. H. — Oil a. Gas J., 1963, v. 61, № 10, p. 64. 465. Brit.
Chem. Eng., 1965, v. 10, № 2, p. 85. 466. La Plante L. Ja—Oil a. Gas Int., 1970,
v. 10, № 8, p. 99—104. 467. Пат. 996398, 1965 г. (Англия). 468. Chem. Age,
1965, v. 94, № 2403, p. 139. 469. Hydrocarb. Processing, 1968, v. 47, № 9, p. 229.
470. Америк M. Б., Панченков Г. M., Скобло В. А. — Хим. и технол. топлив и
масел, 1978, № 9, с. 16—ЗВ.
471. Cooper D. Е. е. а. — Chem. Eng. Progr., 1966, v. 62, № 4, p. 69—73.
472. Пат. 3523075, 1970 г. (США). 473. Пат. 3539502, 1970 г. (США). 474. Пат.
3539501, 1970 г. (США). 475. Пат. 3395397, 1966 г. (США). 476. Chem. Eng.,
1964, v. 71, № 9, р. 73—76. 477. Asher W. J. e. a. — Hydrocarb. Processing, 1969,
v. 48, № 1, p. 134—138. 478. Wehner K-, Welker — In: Siedel G. Chem. Technik,
1970, Bd. 22, Sonderheft, S. 44—49. 479. Мирский Я. В. и др. Синтетические
цеолиты и их применение в нефтепереработке и нефтехимии. М., ЦНИИТЭнеф-
техим, 1967. 90 с. 480. Мирский Я- В. и др. — В кн.: Адсорбенты, их получение,
свойства и применение. Л., Наука, 1978, с. 16—18.
481. Broughton D. В. —Chem. Eng. Progr., 1968, v. 64, № 8, p. 60—65
482. Sterba M. J.— Petrol. Ref., 1965, v. 44, № 6, p. 151—153. 483. Пат. 1172724,
582 Литература
1969 г. (Англия). 484. World Petrol., 1965, v. 36, № 9, р. 14. 485. Brough-
ton О. В. е. а. — Chem. Eng. Progr., 1970, v. 66, № 9, p. 70—75. 486. Wolf F.
e. a. —Chem. Technik, 1975, Bd. 27, № 12, S. 739—743. 487. Пат. 3527693,
1970 г. (США). 488. Novak G. J.— Ind. Eng. Chem., 1959, v. 51, № 10, p. 36A—
38A. 489. Barrer R. M. — Chem. a. Ind., 1968, № 36, p. 1203—1213. 490. Голов-
ко Г. А. Установки для производства инертных газов. М., Машиностроение,
1974. 350 с.
491. Бачурин П. Я., Смирнов В. А. Технология ликеро-водочного производ-
ства. М., Пищевая промышленность, 1975, с. 91—106. 492. Справочник техноло-
га ликеро-водочного производства/Под ред. В. Л. Яровенко. М., Пищевая про-
мышленность, 1976, с. 175—180. 493. Славуцкая Н. И. — Ферментация и спир-
товая пром., 1968, № 6, с. 14—17. 494. Ошмян Г. Л. — В кн.: Углеродные ад-
сорбенты и их применение в промышленности, ч. 2. Пермь, 1969, с. 126—135.
495. Макеева А. Н. Канд. дис. М., ВНИИПрБ, 1980. 496. Исаева Т. Г., Ушако-
ва Г. И., Макеева А. Н.— Ферментация и спиртовая пром., 1972, № 5, с. 35—
37. 497. Смирнов В. А. и др. — Ферментация и спиртовая пром., 1966, № 5,
с. 10—14. 498. Tolbert N. Е., Amerine М. А. — Ind. a. Eng. Chem., 1943, v. 35,
№ 10, р. 1078—1082. 499. Kaiser Н. — Brauwelt, 1957, Bd. 97, S. 1203—1205.
500. Смолина Л. Б., Сафиулина Ф. Ш., Дайман И. К. — В кн.: Адсорбционные
свойства природных синтетических сорбентов. Ташкент, Фан, 1969, с. 147—150.
501. Кердиваренко М. А. и др. — В кн.: Адсорбенты, их получение, свойст-
ва и применение. Л., Наука, 1978, с. 225—227. 502. Кердиваренко М. А. Мол-
давские природные адсорбенты и технология их применения. Кишинев, Карта
молдованяскэ, 1975. 190 с. 503. А. с. 483429 (СССР). 504. Кердиваренко М. А. —
Консервная и овоЩесушнльная пром., 1976, № 6, с. 13—15. 505. Карташев А. К.
и др. — Сахарная пром., 1964, т. 38, № 11, с. 821—825. 506. Кравец Я. О.—
В кн.: Углеродные адсорбенты и их применение в промышленности, ч. 2. Пермь,
1969, с. 136—146. 507. Харитонов В. П. Адсорбция и кондиционирование на
холодильниках для плодов и овощей. М., Пищевая промышленность, 1978.
192 с. 508. Метлицкий Л. В. и др. Хравнение плодов в регулируемой газовой
среде. М., Экономика, 1972. 183 с. 509. Журин А. А., Харитонов В. П. Оборудо-
вание плодоовощехранилищ с регулируемой газовой средой. М., ЦБНТИ
МИНТОРГ СССР, 19.72, вып. 1. 79 с. 510. Колесник А. А., Осенова Е. X., Фе-
доров М. А. Хранение плодов в регулируемой атмосфере. М., Колос, 1973.
120 с.
ЗАКЛЮЧЕНИЕ РЕДАКТОРА
Книга Н. В. Кельцева завершена. За ее пределами оказались
лишь некоторые специфические адсорбционные процессы. По-
этому даже самый придирчивый читатель должен согласиться —
книга отражает современный уровень развития адсорбционного
метода. Она дает достаточно полное представление о типах про-
мышленных адсорбентов, организации адсорбционных процессов
и применении метода в промышленности.
Пенной особенностью книги является и то, что в ней, хотя и
в неявной форме, содержится прогноз общего развития адсорб-
ционной техники в предстоящие годы. Автор констатирует тен-
денции к сокращению длительности адсорбционного цикла (и,
следовательно, габаритов адсорберов), уменьшению числа ста-
дий и расширенному применению природных адсорбентов вза-
мен синтетических. Эти тенденции обусловлены двумя причина-
ми: общей (экономия материальных ресурсов) и специфической
(временный дефицит некоторых промышленных адсорбентов).
Долгосрочный характер действия первой из них несомненен;
действие второй, конечно, будет преодолено. Поэтому можно
предполагать, что в будущем совершенствование адсорбцион-
ных процессов, наряду с экономией материалов, будет опреде-
ляться соображениями экономии энергии. Необходимость учета
этого требования с возрастающей настойчивостью предъявляет-
ся ко всем областям техники, и мало вероятно, что адсорбция —
процесс очень энергоемкий —• составит исключение.
Для адсорбции требования сбережения энергии означают
уменьшение расхода тепла на регенерацию адсорбента, исполь-
зование низкопотенциального и отбросного тепла, рекуперацию
тепла, внесенного в слой, и проведение регенерации в непико-
вые периоды потребления энергии.
Самые большие резервы повышения энергетической эффек-
тивности адсорбции, конечно, связаны с созданием новых адсор-
бентов. В наиболее распространенных в настоящее время уста-
новках с периодически работающими адсорберами доля условно
полезного тепла (под которым понимается теплота адсорбции
(десорбируемых примесей) не превышает ЗО°!о. Остальная вводи-
мая в процесс энергия расходуется на нагрев адсорбента и кон-
струкции, потери с газами, покидающими адсорбер при регене-
рации, и потери в окружающую среду за счет лучеиспускания.
Очевидно, что при прочих равных условиях долю полезной энер-
гии можно увеличить, если применять адсорбенты с более высо-
кой адсорбционной способностью.
584
Заключение редактора
Другой способ повышения эффективности заключается в пе-
реходе от процессов периодических к процессам непрерывным.
Выполнение отдельных операций, соответствующих стадиям пе-
риодического процесса, в специальных аппаратах исключает
расход энергии на нагрев конструкции и позволяет рекупериро-
вать тепло ряда потоков. Однако возможность создания непре-
рывных процессов, как неоднократно подчеркивалось
Н. В. Дельцевым, тоже связана с необходимостью применения
адсорбентов, обладающих определенной совокупностью свойств
и, в первую очередь, высокой механической прочностью. Можно
предполагать, что решение проблемы энергосбережения будет
способствовать и расширению областей применения непрерыв-
ных процессов, и производству специальных адсорбентов для
них.
Утилизация отбросного тепла промышленности станет воз-
можной, если техника будет располагать адсорбентами, регене-
рируемыми при 50—150°С.
Таким образом, требования экономии энергии стимулируют
работы по созданию адсорбентов, обладающих высокой ем-
костью, большой механической прочностью и низкой температу-
рой регенерации. Сочетание этих свойств в одном адсорбенте
обеспечить трудно, но улучшение даже отдельного свойства
конкретного адсорбента уже способно привести к существенно-
му прогрессу в адсорбционной технологии.
Экономия энергии особенно важна для типовых установок:
осушки воздуха, рекуперации растворителей, производства за-
щитных атмосфер. Эта категория адсорбционных установок бу-
дет развиваться как в направлении увеличенного производства
традиционных объектов, так и в результате расширения номен-
клатуры (ближайший кандидат в типовые процессы — это ад-
сорбционное разделение воздуха). Из-за широкой сферы приме-
нения в типовых установках используют самые доступные и со-
вершенные виды энергии: электроэнергию, природный газ, пар.
Эффективное применение энергии в них может быть обеспечено,
если регенерацию адсорбента проводить в периоды сокращенно-
го потребления энергии — в ночное время.
Обсудим на примере самых распространенных типовых уста-
новок осушки воздуха, к каким изменениям технологии может
привести это мероприятие. В установках рассматриваемого типа
адсорбент (силикагель) через каждые 8 ч подвергают регенера-
ции, которую осуществляют воздухом, нагретым с помощью
электричества. Требование энергосбережения в данном случае
означает увеличение цикла до 24 ч. Для установок традиционно-
го типа повышение длительности цикла равносильно увеличению
размеров аппаратуры, расхода адсорбента и металла. Требова-
ние сбережения энергии, таким образом, вступает в противоре-
чие с требованиями сбережения материалов.
Заключение редактора
585
Трудно однозначно сказать, какой путь изберет технический
прогресс для разрешения этого противоречия. Возможен, на-
пример, такой вариант, как создание одноадсорберных устано-
вок, что оправдано для предприятий с одно- и двухсменной ра-
ботой. Осушенный газ получают в течение рабочих смен, а реге-
нерацию проводят ночью. Количество адсорбента в данном слу-
чае не больше, чем в ныне выпускаемых установках с восьмью-
часовым циклом.
Взаимосвязь энергетики и адсорбции имеет и другую сторо-
ну— это использование адсорбентов и адсорбционных процес-
сов в решении собственных проблем энергетики. В основном
тексте книги Н. В. Кельцев подробно осветил вопросы примене-
ния адсорбции для очистки отходящих топочных газов тепло-
электростанций от диоксида серы и при подготовке природного
газа к дальнейшему транспортированию. Менее известно, что в
настоящее время проводятся интенсивные исследовательские
работы по созданию на основе адсорбентов хранилищ энергии.
Такие хранилища — тепловые насосы предназначены для пре-
образования солнечной радиации и отбросного тепла электро-
энергетики и промышленных предприятий в энергию, используе-
мую для обогрева жилых домов, административных зданий и
производственных сооружений в зимний период и для охлажде-
ния воздуха в них летом. Отметим, что около ЗО°/о энергии, про-
изводимой в промышленно развитых странах, расходуются на
эти цели.
В хранилищах энергии полезным свойством адсорбента яв-
ляется его способность выделять тепло при адсорбции. За счет
энергии указанных выше недефицитных источников адсорбент
регенерируют. Емкость с адсорбентом герметизируют, получая в
сущности аккумулятор тепла. Разрядка этого аккумулятора
происходит при контакте от регенерированного адсорбента с во-
дой или ее парами. Емкость такого источника энергии достигает
250—700 кД.ж/м3 адсорбента. Продолжительность разрядки ре-
гулируют скоростью ввода воды (паров воды). В качестве рабо-
чих материалов хранилищ испытаны силикагель, активный оксид
алюминия и цеолиты. Специально для хранилищ синтезированы
новые адсорбенты-. Sorbead R и W. Проведены успешные испы-
тания хранилищ емкостью 1 и 6 м3 с адсорбентом типа Sorbe-
ad-R (более подробные сведения об этом новом и интересном
применении адсорбции см. в Papers Psented of the International
Conference on Energy Storage, vv. 1—2, Brighton, U. K-, 1981,
551 p.).
Таковы в кратком изложении основные тенденции развития
адсорбционного метода, сущность и приложения которого со-
ставляют основной предмет книги Н. В. Кельцева.
Ю. И. ШУМЯЦКИЙ
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адсорбаты
влагосодержание 296
диаметр молекул критический (ки-
нетический) 86
мольный объем 62 сл.
парахор 69, 70
плотность 133
подобия коэффициент 29, 135 сл.
площадки молекул элементарные,
размер 42 сл.
Адсорбенты
адсорбционная (ое)
взаимодействие 74 сл.
емкость 169 сл., 303
полость 90 сл.
пространство 67, 68
равновесие 22 сл.
способность см. Адсорбционная
способность
активность динамическая слоя 377
активные угли см. Угли активные
активный оксид алюминия см. Ок-
сид алюминия активный
виды 77 сл.
влагоемкость 79 сл., 287
влагосодержание 57, 296
высота работающего слоя 79,
173 сл., 209, 233, 358
гидрофобность 79
глинистые породы природные
111 сл.
гранулометрический состав 293, 294
дезактивация 307 сл.
емкость монослоя 37, 39, 40
зольность 78, 296
классификация 92, 98
макропористые 27, 28
мезопористые 27, 50 сл.
микропористые 25 сл., 78 сл.
молекулярно-ситовые свойства
503 сл.
плотность 78, 287, 291 сл.
поверхность 27, 37 сл., 47 сл., 109,
113, 259, 279, ,283, 291 сл.
пористость 25 сл., 52 сл., 78, 109,
284, 292
природные
глинистые породы 111 сл.
цеолиты 87 сл.
прочность 287, 288, 291, 293 сл.
регенерация 307, 327 сл., 370 397,
450, 451
Адсорбенты
силикагель см. Силикагель
смешанные 108 сл.
стекла пористые ПО, 111
степень использования адсорбцион-
ной емкости 169
— насыщения 153
теплоемкость 287
теплопроводность 287, 288
удерживающая способность 153
цеолиты см. Цеолиты
Адсорберы
вертикальные 220, 221
с движущимся слоем адсорбента
229
кольцевой 222, 223
периодического действия 217 сл.
с прижимным устройством 224, 225
со стационарным слоем 453
с теплообменными элементами 223,
224
трубчатые 223, 225
Адсорбиты 23, 40, 41, 69
Адсорбционная емкость 145, 291, 438,
484
Адсорбционная способность (актив-
ность, емкость)
активного оксида алюминия 326
активных углей 26, 78, 80, 126, 217,
244, 252, 264, 364, 468, 470
сажи 26, 70, 468
силикагеля 253, 302, 303, 306, 311,
326, 468, 470, 472
уравнения 306
цеолитов см. Цеолиты, адсорбцион-
ная способность
шабозита 549
Адсорбционный потенциал 60
Адсорбция
движущая сила 189
динамика 167 сл., 203 сл.
математические модели 180 сл.
уравнения 170 сл.
число единиц переноса 189
дисперсионные силы 23, 24
избирательность 126 сл., 466 сл.
изобары см. Изобары адсорбции
изостеры 30
изотермическая
неравновесная 188 сл.
равновесная 180 сл.
изотермы см. Изотермы адсорбции
Предметный указатель 587
Адсорберы
кинетика 138 сл.
кривые равновесия см. Кривые ад-
сорбционного равновесия
мономолекулярная 35 сл.
неизотермическая 196 сл.
полимолекулярная 38 сл.
предельная (максимальная) 61 сл.
применение см. Осушка, Очистка,
Разделение
равновесие см. Кривые адсорбцион-
ного равновесия
расчет 34, 35, 129 сл., 137, 138,
154 сл.
скорость 36
теория
идеального адсорбционного рас-
твора 137 сл.
мономолекулярная Ленгмюра
объемного заполнения микропор
Дубинина 22, 61 сл., 132 сл.
полимолекулярная Браунауэра —
Эммета — Теллера /БЭТ/ 38 сл.
теплота
дифференциальная 121
интегральная 121 сл.
мольная 39
средняя 121 сл.
уравнение(я)
БЭТ 38, 39
динамики 170 сл.
термическое 28 сл.
характеристическая кривая 60 сл.
энергия мольная 64, 68
энтальпия 120
энтропия 120 сл.
Браунауэра — Эммета — Теллера
(БЭТ) уравнение 38, 39
де Бура формула 55
Ван-дер-Ваальса уравнение 62
Взрывоопасность смесей веществ с
воздухом 411
Викке уравнение 165
Вильсона уравнение 181
Влагоемкость 287
Влагосодержание
адсорбатов 296
воздуха 237, 298
газов 297 сл., 310
цеолитов 296
Время защитного действия слоя 168
Гидравлическое сопротивление слоя
адсорбента 257 сл.
Глинистые породы 111 сл.
Гмелинит 91
Графит 53
Давление паров над растворами 33
Десорбция
в вакууме 153 сл.
гептана 159
диоксида углерода 161 сл.
кинетика 152 сл., 238
парафинов нормальных 157 сл.,
паров воды 161 сл.
пентана 158
пороговая температура 159
расчет кинетики 154 сл.
скорость 36
совмещение кинетических кривых
157, 158
температурный режим 160 сл.
углеводородов 153
Дилатометр 59
Дифференциально-термический анализ
75, 76
Диффузия
активированная 148, 149
зависимость от пористости 143, 144
-----температуры 144
коэффициенты 142 сл.
молекулярная (кнудсеновская) 148
нормальная (объемная) 147
поверхностная 150, 151
продольная 180 сл.
скорость 36
Дубинина теория 61 сл.
Дубинина уравнение 56
Жуховицкого — Забешинского — Ти-
хонова (ЖЗТ) уравнение 193
Зельдовича уравнение 188
Извлечение
ароматических углеводородов
473 сл., 495 сл.
бензола 73, 190
бутана 68
непредельных углеводородов 467 сл.,
480 сл.
— парафинов, процессы
«Бритиш петролеум» 533 сл.
«ГрозНИИ» 544 сл,
«Изосив» 530 сл.
«Молекс» 545 сл.
«Перекс» 542 сл.
«Токсако селектив финшииг»
535 сл.
«Энсобр» 538
параксирола 549
588 Предметный указатель
Извлечение
пропана 243
ртути 239 сл.
Изобара(ы) адсорбции
бензола 73
бутана 68
газов на цеолите 99
додекана на цеолите 526
паров воды 30
пропана 39
Изобара десорбции 526
Изостеры адсорбции 30
Изотерма (ы) адсорбции
азота на разных адсорбентах 207
ароматических углеводородов из
керосина 478
ацетилена на разных адсорбентах
37, 470, 478, 495
вогнутая 182, 183
водорода из технического водорода
на активном угле 206
воды 111, 357
выпуклая 181, 184 сл., 194, 195
газов иа микропористом активном
угле 205, 206
гексенов иа цеолите типа X 522
диоксида серы
на активном угле 425
— мордените 501
— полукоксе 431
диоксида углерода на цеолите СаА
365, 369
линейная 182, 187, 193, 195
метана из водорода активным углем
206
метилацетилена на разных адсор-
бентах 493
метиловой сини на саже «сферон»
49
нафтеновых углеводородов на раз-
ных адсорбентах 478, 511
оксида углерода на косточковом уг-
ле 72
олефиновых углеводородов 486
паров бензола на силикагеле К.СМ
50
— воды
иа активном угле 80
при малых давлениях 325
н-пентана на цеолите СаА 504
пропана на разных адсорбентах 468
пропилена иа разных адсорбентах
468
на силикагелях 302, 303, 307, 311
— цеолитах гранулированных 325
-----СаА 30
типы основные 29
этилена на разных адсорбентах 470,
479, 483
Капиллярная конденсация 27 сл.,
51 сл.
Катализаторы (поверхность, порис-
тость) 48, 53, 55
Кельвина уравнение 51
Кинетика
адсорбции 132 сл.
десорбции 152 сл., 238
Кисарова уравнение 72 сл.
Киселева уравнение 46
Клауса— Ченса реакция 397
Клиноптилолит 87 сл., 436
Коэффициент (ы)
диффузии 99, 142 сл.
зависимость от температуры 144,
149
относительный 146
поверхностный 150
продольной 176 сл.
эффективный 142
конденсации 35, 36
массопередачи 191
подобия при адсорбции 69, 135 сл,
предельной адсорбции 62 сл.
пропитки 265
разделения 100, 124 сл., 134 сл.
расходные см. Расходные коэффи-
циенты
скорости насыщения 230 сл.
сопротивления слоя адсорбента
259 сл.
теплопередачи 239, 240
теплопроводности 287, 288
Кривые адсорбционного равновесия в
системах
ацетилен — метилацетилен на раз-
ных адсорбентах 494
бензол — парафиновые углеводоро-
ды на силикагеле 474
водяной пар — и-гексан на разных
адсорбентах 479
диоксид серы — диоксид углерода
на мордените 501
диоксид углерода — сульфид водо-
рода иа цеолите СаА 501
пропан — пропилен на разных ад-
сорбентах 483
толуол — парафиновые углеводоро-
ды на силикагеле 474
циклогексан — бензол на цеолитах
496
этилен — пропан на силикагеле 473
этилен — пропилен на активном уг-
ле и силикагеле 469, 472
Ленгмюра уравнение 35 сл., 131 сл.,
211
Льюиса уравнение 126
Предметный указатель 589*
Майклса—Трейбла формула 170
Массопередача 189 сл.
Монтомориллонит 53
Морденит 24, 25, 89, 90, 100, 102 сл.
Оксид алюминия активный 30, 84, 85
адсорбционная способность 326
ассортимент 85, 281 сл.
влагоемкость 33
получение 84, 280, 281
Октановое число, повышение 519 сл.
Осветление
масел 324
пива 555 сл.
фруктовых соков 555 сл.
Осушка
абсорбента цеолитами иа газобеи-
зиновых заводах 336 сл., 348,
349
воздуха 297, 298, 376 сл.
выходная кривая 311, 333, 334, 337
газов
под высоким давлением 309 сл.
с небольшим содержанием влаги
силикагелем 306, 307
природных 330 сл., 349 сл.
риформинга цеолитами 355
углеводородных 344 сл.
в газовой промышленности 330 сл.
— динамических условиях 303 сл.
жидкости в лаборатории 361 сл.
масел 324, 356 сл., 360
механизм 304 сл.
нейтральных углеводородов цеоли-
тами 351 сл.
природного газа 330 сл., 349 сл.
под давлением 340
закачиваемого в пласт, цеолитами
339 сл.
на криогенных установках цеоли-
тами 341 сл.
— промыслах цеолитами 335
пропилена 353 сл.
в статических условиях 301 сл.
сырья иа установках алкилирова-
ния, цеолитами 355, 356
углеводородных газов 344 сл.
Очистка
аргона от кислорода 55
воздуха от диоксида углерода 366
водорода от примесей 249 сл., 381
воды
после биохимической обработки
456, 457
бытовых стоков 457 сл.
граиулироваиным углем в непо-
движном слое 459
коксохимических производств
। 459 сл.
Очистка
воды
порошкообразным углем, двухсту-
пенчатая 456
производств химических волокон-
463, 464
угольной суспензией 455 сл.
физико-химическая, комбиниро-
ванная 460
воздуха 376 сл.
выбросов автомобилей 505 сл.
газов
от диоксида серы 383 сл., 423 сл.
— диоксида углерода 464,.
479 сл., 492 сл.
— меркаптанов 383 сл.
— оксида углерода 381 сл.
— сероорганических соединений
395, 396
изопентана от «-пентана 503 сл.
масел 324 t
нефтепродуктов 401 сл.
отходящих газов сернокислотного-»
производства 437
пропилена от изопропилового спир-
та 353 сл.
сточных вод 445 сл.
углями, методы (процессы)
«Бергбау — Форшуиг» 432, 433
«Вестфако» 433, 434
«Лурги» 427
«Рейилюфт» 428 сл.
«Хитачи» 425 сл.
«Штратман» 427, 428
цеолитами 435 сл.
экзогаза 370 сл.
этана от диоксида углерода цеоли-
тами 381
Парахор 69, 70
Плотность
активного оксида алюминия 283
активных углей 78, 276
цеолитов 287, 291 сл.
Поверхность удельная
активных углей 276, 278, 279
силикагеля 48
цеолитов 46, 293
Поглотительная способность адсор-
бентов см. Адсорбционная спо-
собность
Пористые стекла 53, НО, 111
Порометрия ртутная 57 сл.
Прочность адсорбентов 287, 288, 291»
293 сл.
Разделение
газов неполного окисления метаиаа
255
;590 Предметный указатель
^Разделение
газовых смесей
азот — диоксид серы 501
ацетилен — гомологи ацетилена
493 сл.
«-бутиловый спирт — «-кротило-
вый спирт 491
«-гексан — гексен-1 484
диоксид углерода — диоксид серы
501
диоксид углерода — сульфит во-
дорода 500, 501
пропан — пропилен 482 сл.
этан этилен 480 сл.
этилен — ацетилен 491 сл.
этилен — диоксид углерода 488 сл.
Растворение воды в углеводородах
300
Расчет 34, 35, 137, 138
адсорбции бинарных смесей 129,
131 сл.
кинетики адсорбции 148 сл.
— десорбции 154 сл.
Рекуперация
адсорбентов 307, 327 сл., 370, 397,
450, 451
растворителей 256, 406 сл.
сульфида углерода 414 сл.
хлорорганических 421 сл.
рения семиоксида 438, 439
силикагеля 307
'Сажа графитированная 25, 26, 37, 49
Сероочистка газов
активным углем 384 сл.
с регенерацией адсорбента 397 сл.
сжиженных 399 сл.
с удалением диоксида углерода
392 сл.
цеолитами 386 сл.
Силикагель (и) 30, 39, 82 сл., 301 сл.
адсорбционная способность 207, 253,
302, 303, 326, 468, 470, 472
активность динамическая 304 сл.
гранулирование 273 сл.
гранулометрический состав 82
дезактивация 307 сл.
избирательность адсорбции 467 сл.
модифицированные 83
поверхность удельная 48
получение 82
структура 83
•Содалит 86
"Теплоемкость
активных углей 269
силикагелей 278
цеолитов 287
Угли активные
адсорбционная способность 26, 78,
80, 126, 217, 244, 252 264, 364,
468, 470
активность 416
влагоемкость 79 сл.
выпуск за рубежом 77
высота рабочего слоя 79
газовые 269, 270
гидрофобность 79 сл.
горючесть 82
зернистость 78
избирательность адсорбции 466
классификация 277 сл.
осветляющие 271, 272
пропитка 265
плотность 78, 276
поверхность удельная 276, 278, 279
получение 262 сл.
поры
объем 78, 276, 278, 279, 384
радиус средний 276, 384
структура 78
форма 53
потери 247, 248
рекуперационные 271
теплоемкость 269
типы 268 сл.
Уравнение (формула) см. также имен-
ные уравнения
адсорбции термическое 28 сл., 64
адсорбционного равновесия 46
адсорбционной способности адсор-
бентов 306
БЭТ 38, 39
влажности относительной 298
десорбции 54
динамики адсорбции 170 сл.
дифференциальной мольной работы
адсорбции 63
диффузии 146 сл.
ЖЗТ 188
изобары адсорбции 30
изостеры адсорбции 28
кинетики адсорбции 178 сл.
материального баланса динамики
адсорбции 175 сл.
мольного объема 62
мольной работы адсорбции 63,
120 сл.
— теплоты адсорбции 121 сл.
объема пор 59
перепада давления в газлифте 249
предельного объема адсорбционного
пространства
предельной адсорбции
распределения объема поверхности
пор 54, 55
теплового баланса 178, сл.
Уравнение
термическое адсорбции 28 сл., 64
удельной поверхности 278, 279
характеристической кривой адсорб-
ции 65
числа адсорбционно-десорбционных
ступеней 235
Установки адсорбционные
адсорбции углеводородов 229 сл.
выделения нормальных парафинов
по методу «Молекс» 547
газогенераторной 564
газобензиновой с периодическими
адсорберами 218
с движущимся слоем адсорбента
229 сл., 477
деароматизации
жидких парафинов 477
нефтяных фракций 499
реактивного топлива 497
депарафинизации
с двухступенчатой десорбцией
537
по методу «Бритиш петролеум»
534
•----«ГрозНИИ» 544
— — «Перекс» 542
----- «Техсако селектив фин-
шинг» 535
-----«Энсобр» 541
в псевдоожиженном слое 544
обезвоживания жидких сред 362
объемно-весовой лабораторной 128
осушки газа
на алкилирования заводах 356
воздуха 320 сл.
на гелиевом заводе 343
природного 313 сл., 316 сл., 336
на промыслах 336
отбензинивания 315 сл.
очистки
аргона 549
атмосферы защитного газа 366
водорода под давлением 212,
249 сл.
вод сточных после биохимической
обработки 456
-----бытовых 457 сл.
— — коксохимических произ-
водств 461, 462
-----кондиционирующей 455
----углем 455 сл., 459
-----физико-химическая комби-
нированная 460
водки 553
воздуха 417
газов 388, 392 сл., 426, 427, 429,
432, 434, 437
сахарных сиропов 558
этилена 490
получения защитных атмосфер 371
проточной установки для изучения
адсорбционного равновесия 130
Установки
разделения
природного газа 229
продуктов изомеризации 516
рекуперации 407 сл.
сероочистки 381 сл., 383 сл., 392 сл.,.
395, 396, 401 сл.
типы 217 сл.
экзотермической атмосферы 371 сл.
Фрейндлиха уравнение 71, 72
Хемосорбция 23, 24
Хилла формула 40
Цеолиты
адсорбционная способность
СаА 30, 46, 207 сл., 253, 364, 369,.
387, 481
СаХ 46, 253, 289
NaA 30, 253, 289, 324, 364, 387
NaM 105
NaX 25, 105, 109, 207, 326, 364
адсорбционный объем 369
активность динамическая 291, 368:
влагоемкость 283, 296
высота работающего слоя
гранулированные 104, 105, 293
емкость по бензолу 287
зернистость 287, 293 сл.
кислотостойкие 100 сл., 107
классификация
природных 92
синтетических 96 сл.
коэффициенты подобия (афинности)1
69
марки зарубежные 283, 284
молекулярно-ситовые свойства)
503 сл.
общего назначения 91 сл.
плотность 287, 291 сл.
поверхность 46, 293
получение
без связующих 289 сл.
гранулированных со связующим-
283, 286 сл.
пористость 288, 292
природные 87 сл.
прочность 104, 287, 288, 291, 293 сл.
регенерация 327 сл.
сверхвысококремнеземистые 107, 108;:
синтетические 91 сл.
структура 88 сл., 93 сл.
теплоемкость 287
теплопроводность 287, 288
термическая устойчивость 102, 103
формулы химические 85, 86, 92
Шабозит 90, 91
Шилова формула 169
Энергия активации 99, 148 сл.
Энтальпия 120 сл.
Энтропия 120
Эропит 105 сл.
Николай Владимирович Кельцев
ОСНОВЫ
АДСОРБЦИОННОЙ
ТЕХНИКИ
Редакторы Ю. И. Шумяцкий,
М. В. М ин икс
Художественный редактор Н. Н. Носов
Технический редактор В. В. Хазикова
Графики А. И. Перфирьев,
Е. В. Перфирьева
Корректоры М. В. Черняховская,
Т. Н. Реброва
Макет, переплет и оформление
художников Ю. Г. Ворончихина
и С. С. Водчица
ИБ № 1181
Сдано в наб. 10.06.83. Подп. в печ. 14.11.83.
Т. 19928. Формат бумаги 60X90716. Бумага тип.
№ 1. Гарн. литературная. Печать высокая. Усл.
печ. л. 37. Усл. кр.-отт. 37,25. Уч.-изд. л. 42,78.
Гнраж 5000 экз. Заказ № 1346. Цена 2 р. 60 к.
Изд. № 2049.
Ордена «Знак Почета» издательство «Химия»,
107076, Москва, Стромынка, д. 13.
Московская типография № И Союзполиграфпрома
при Государственном комитете СССР по делам
издательств, полиграфии и книжной торговли.
113105, Москва, Нагатинская ул., д. 1.