/

















































































































































































































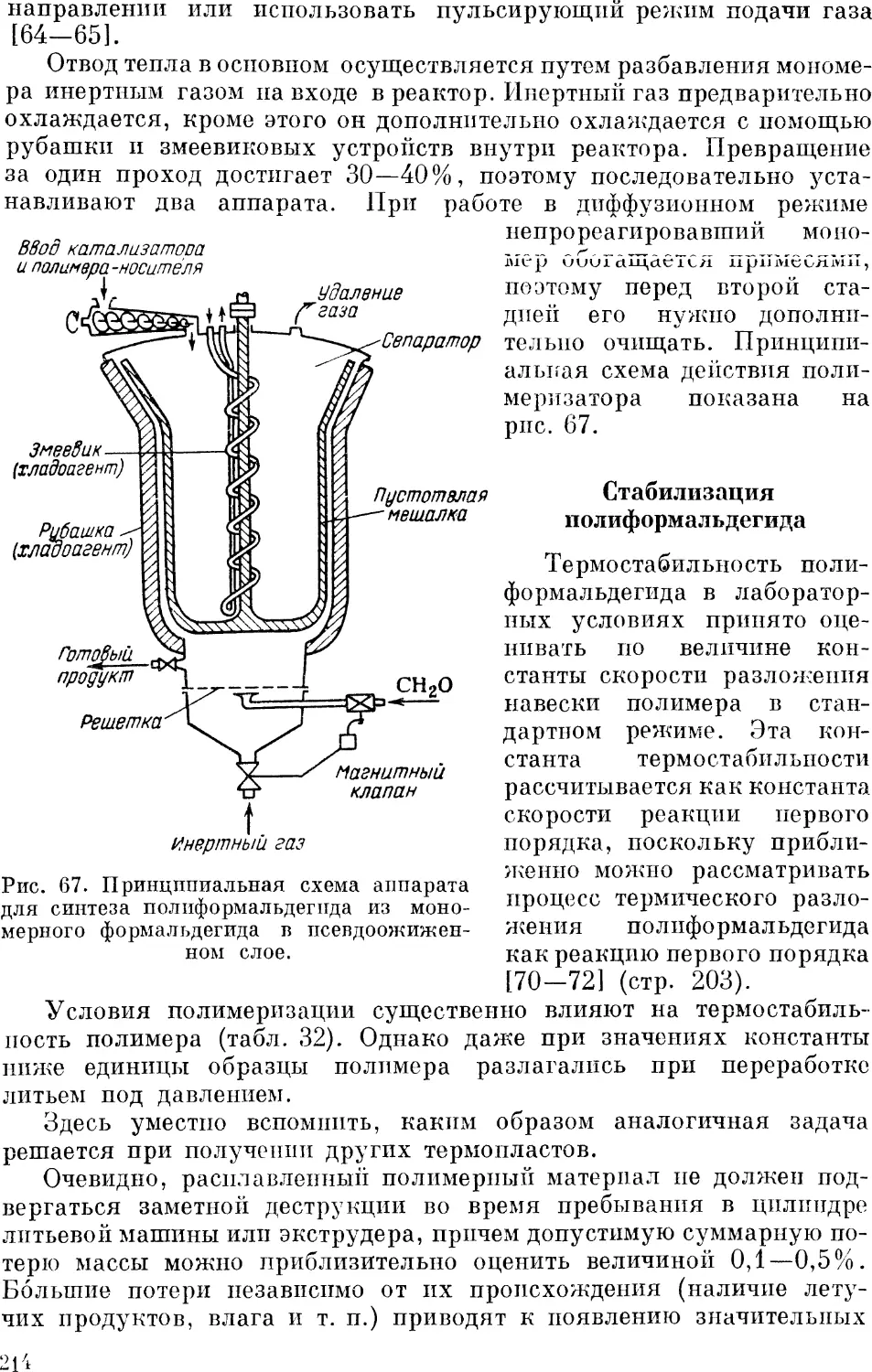





































































Текст
УДК 678.644' ¦ 141
Е 63
ЕНИКОЛОПЯН Я. С, ВОЛЬФСОН С. А.
Химия и технология полиформальдегида
Книга посвящена изложению теоретических основ
синтеза нового полимерного материала —
полиформальдегида, применяемого для производства различных
деталей в машиностроении, приборостроении и
других областях народного хозяйства.
В книге рассматриваются различные аспекты
процессов полимеризации формальдегида и его олиго-
меров, сополимеризации формальдегида с -другими
мономерами, а также принципиальные
технологические схемы синтеза гомополимеров и сополимеров
на основе формальдегида. В специальной главе
описаны свойства полиформальдегида различных
марок, методы переработки и основные области
применения этого полимера.
Книга рассчитана на работников
научно-исследовательских институтов и заводских лабораторий,
технологов, преподавателей химических вузов и
факультетов, аспирантов и студентов старших
курсов, специализирующихся в области синтеза, химии,:
физико-химии и технологии полимеров.
В книге содержится 46 таблиц, 85 рисунков и 755
библиографических ссылок.
3-14-9
9*67
С О Д Е Р Ж А Н И Е
Предисловие
Глава /. Свойства формальдегида и его олигомеров
Методы производства формальдегида
Свойства формальдегида
Растворы формальдегида
Низкомолекулярные полиоксиметилены
Параф ормальдегид .'
а-Полиоксиметилен и другие модификации полиоксиметилена
Циклические полиоксиметилены . ^
Литература
Глава И. Полимеризация формальдегида
Полимеризация в газовой фазе
Термодинамика полимеризации
Кинетика полимеризации ¦
Полимеризация в растворе
Катализаторы полимеризации
Кинетика полимеризации
Определение молекулярного веса
Радиационная полимеризация :
Полимеризация в полярных средах
Литература 62
Глава Ш. Полимеризация триоксана 65
Термодинамика полимеризации гетероциклических соединений 66
Кинетика полимеризации в жидкой фазе 70
Катализаторы и растворители 70
Индукционный период. Механизм инициирования
полимеризации 73
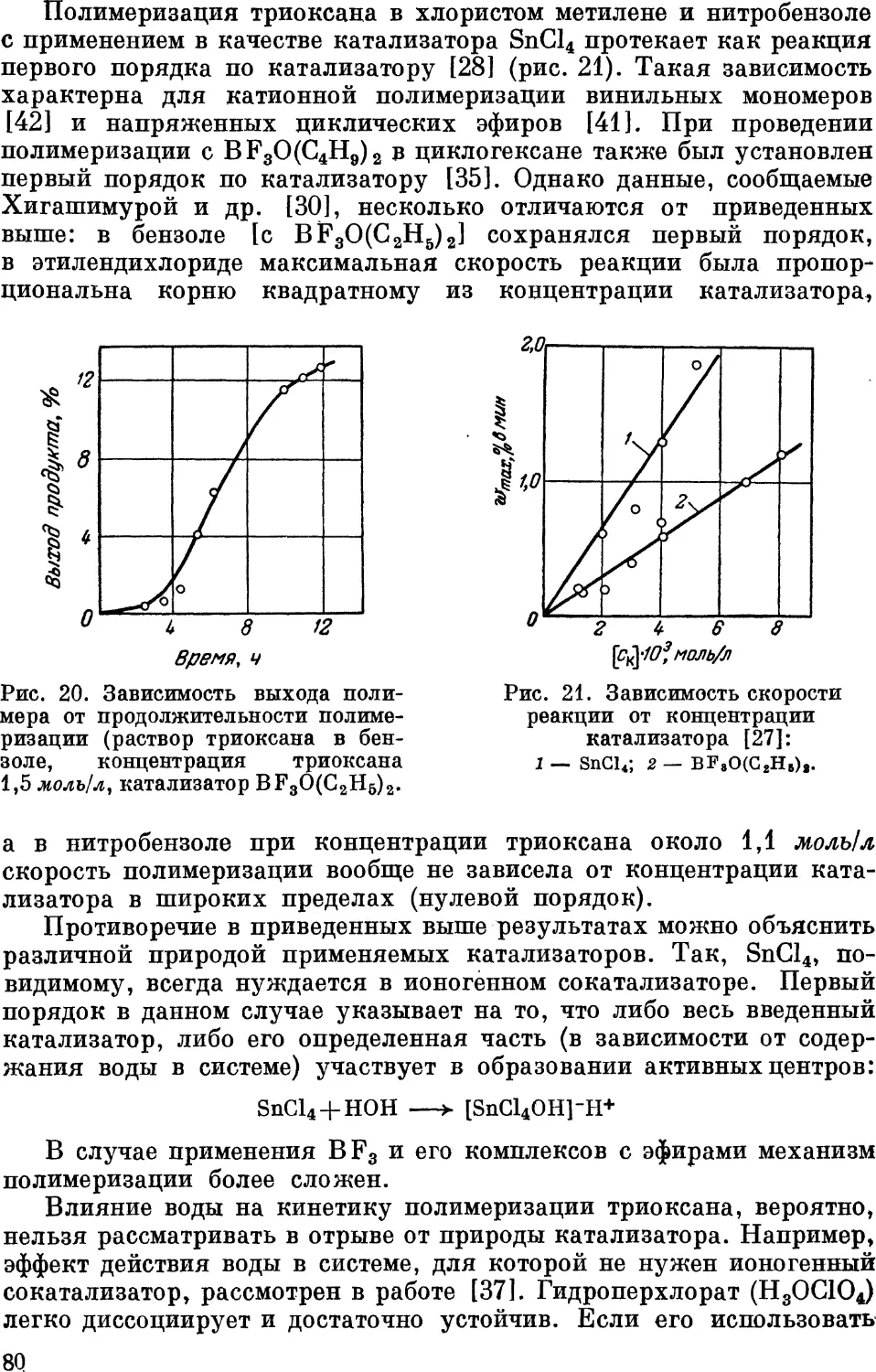
Скорость полимеризации 79
Молекулярный вес 85
Полимеризация в расплаве 89
Полимеризация в твердой фазе 91
Спонтанная полимеризация кристаллического триоксана ... 92
Кинетика полимеризации в твердой фазе 92
Литература 97
Глава IV. Деструкция и стабилизация полиформальдегида 101
Термическая деструкция ЮЗ
Химическая деструкция (ацидолиз) НО
Термоокислительная деструкция ИЗ
Стабилизация 121
Литература 132
1* 3
Глава V. Сополимеризация формальдегида (и триоксана) 136
Некоторые теоретические представления 137
Анионная сополимеризация 140
Сополимеризация с незамещенными альдегидами 140
Сополимеризация с производными альдегидов и кетенами . . . 142
Сополимеризация с циклоангидридами монооксикарбоновых
кислот 144
Сополимеризация с изоциановой кислотой и ее эфирами . . . 144
Сополимеризация с винильными мономерами 145
Катионная сополимеризация. Передача цепи с разрывом .... 146
Механизм передачи цепи с разрывом 148
Влияние передачи цепи с разрывом на молекулярно-весовое
распределение гомополимеров и сополимеров 150
Влияние передачи цепи с разрывом на состав и строение
образующихся блоксополимеров 151
Сополимеризация с окисью этилена . 153
Сополимеризация с 1,3-диоксоланом и другими гетероциклами 156
Сополимеризация с винильными мономерами 160
Радиационная и радикальная сополимеризация .... v ... 162
Литература 164
Глава VI. Структура полиформальдегида 166
Кристаллическая структура полиоксиметилена 167
К информация и некоторые свойства изолированной полиоксимети-
леновой цепи 173
Структура полиоксиметилена, полученного в различных условиях
полимеризации 180
Литература 181
Глава VII. Технология производства полиформальдегида из мономерного
формальдегида 184
Выделение мономерного формальдегида 184
Пиролиз низкомолекулярных полиоксиметиленов 186
Пиролиз гемиформалей (полуацеталей) 189
Парциальная конденсация паров формалина 192
Способы очистки мономерного формальдегида 196
Форполимеризация 197
Физические методы 200
Химические методы 202
Полимеризация мономерного формальдегида 202
Исследования режима полимеризации в гетерогенной системе 204
Аппаратурное оформление процесса полимеризации 212
Стабилизация полиформальдегида 214
Ацетилирование 216
Аппаратурное оформление стадии ацетилирования 218
Дополнительная стабилизация 220
Влияние условий реакции на свойства продукта 221
Литература 222
Глава VIII. Технология производства сополимеров триоксана (и
формальдегида) 225
Синтез и очистка триоксана 226
Гомополимеризация триоксана 230
Сополимеризация триоксана 233
Влияние условийJреакции на структуру и состав сополимера 235
Обработка сополимера 237
Стабилизация сополимера 239
Физико-химические свойства сополимера 240
Сополимеризация мономерного формальдегида 241
Соподимеризация с диметилкетеном 242
Сополимеризация на катализаторах Фриделя — Крафтса 244
Литература 244
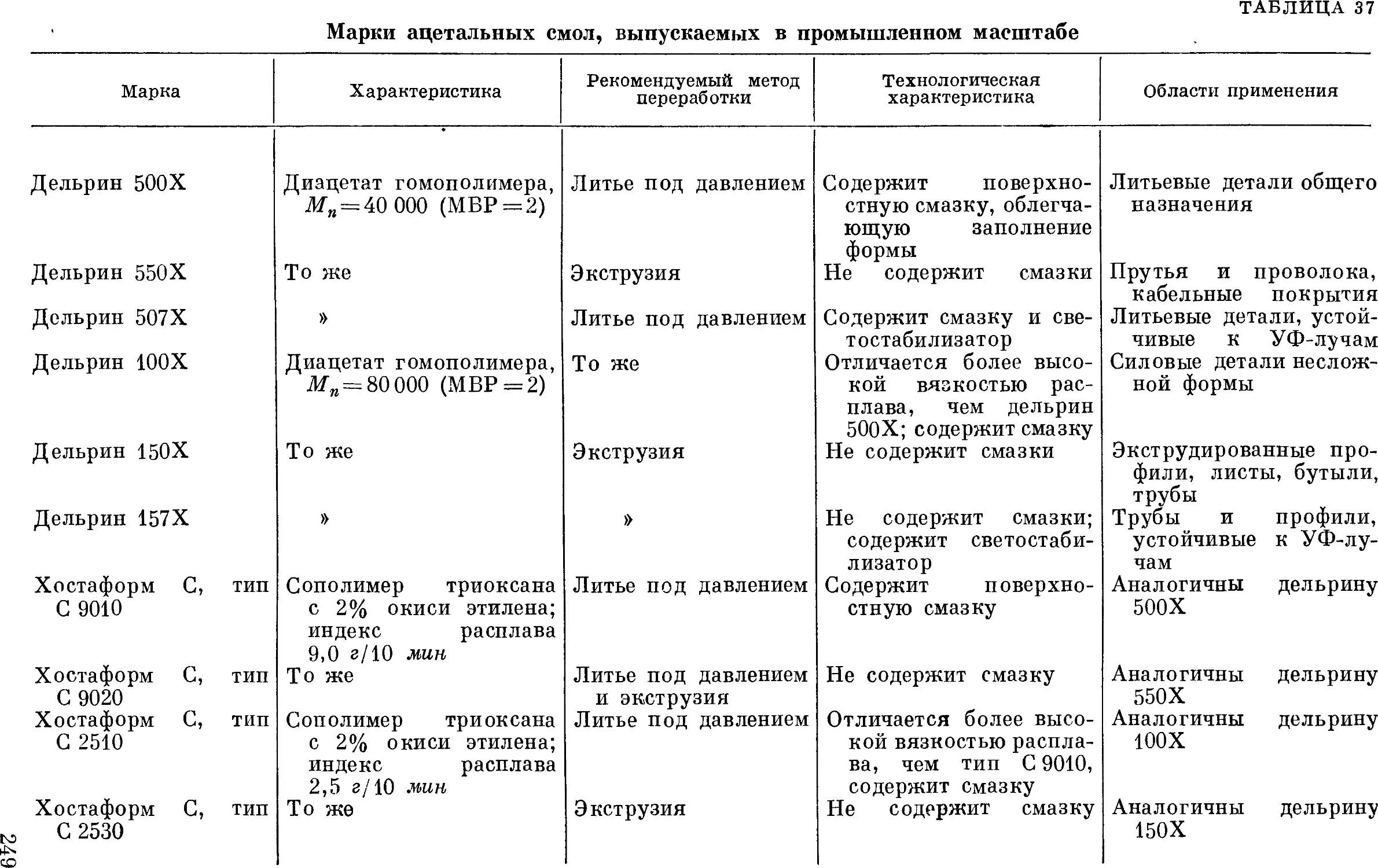
Глава IX. Свойства, переработка и применение полиформальдегида 247
Марки полиформальдегида 248
Физико-механические свойства 251
Электрические свойства 256
Химическая стойкость и физико-химические свойства 257
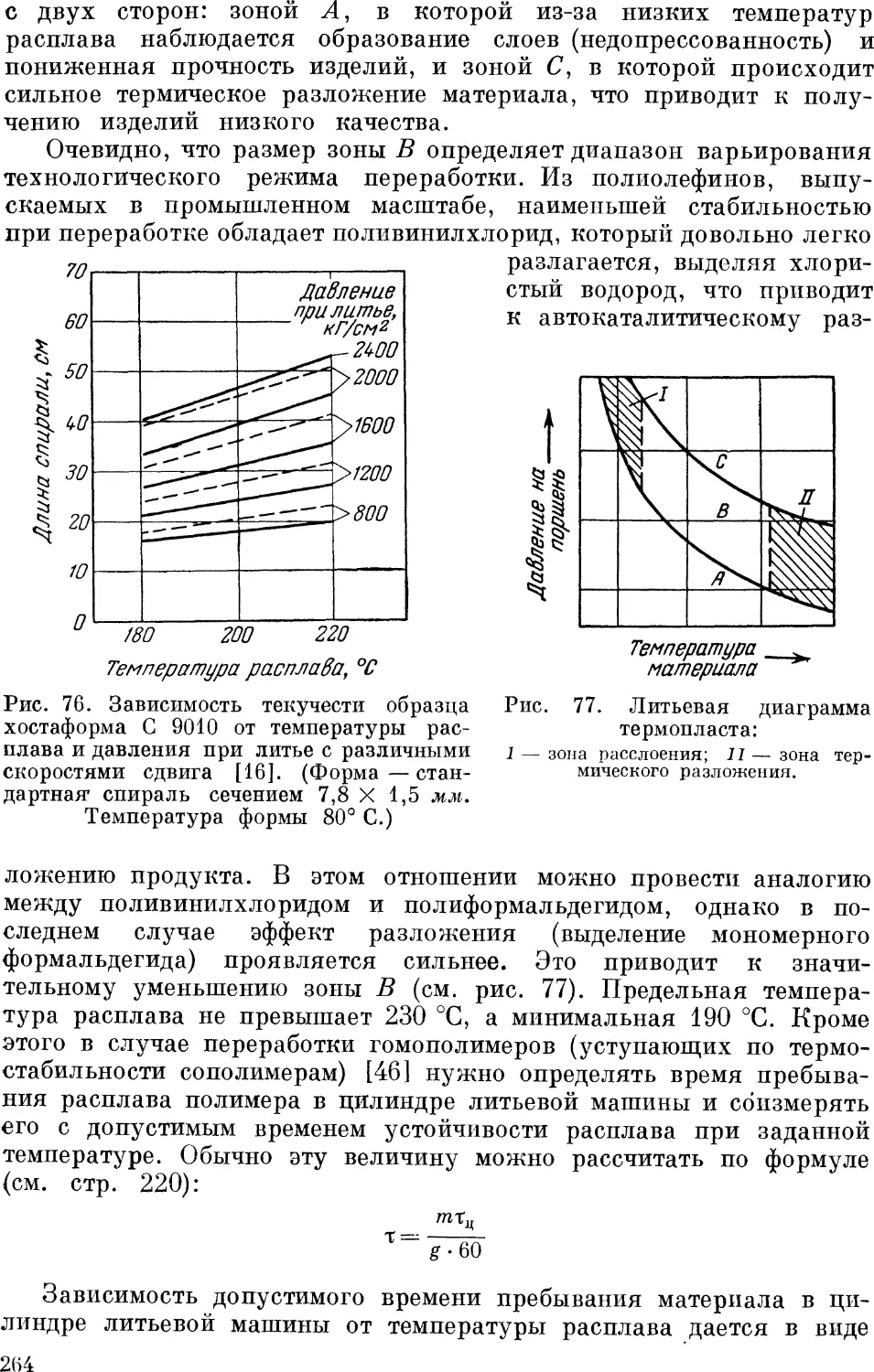
Переработка полиформальдегида 263
Литье под давлением 266
Экструзия 268
Применение полиформальдегида 269
Литература 273
Предметный указатель 275
[ПРЕДИСЛОВИЕ
Благодаря бурному развитию химии
высокомолекулярных соединений в последнее десятилетие
промышленность получила целый ряд новых полимерных материалов,
обладающих самыми разнообразными свойствами. Особо
следует отметить полиформальдегид — линейный полимер
формальдегида полиоксиметиленовой (полиацетальной)
структуры
Н Н Н
III
.. . -с—о—с—о—с—о-...
А к А
Этот термопластичный материал обладает ценным
комплексом механических свойств, которые позволяют
ему успешно конкурировать с цветными металлами и
сплавами при изготовлении различных конструкционных
деталей. Поскольку сырьевые ресурсы для производства
полиформальдегида необычайно велики (природный газ),
можно рассчитывать на низкую себестоимость готового
продукта. Все это создает предпосылки для массового
применения полиформальдегида в народном хозяйстве.
Интерес к проблеме полимеризации формальдегида
возник около 100 лет назад. В промышленности нашли
применение олигомеры формальдегида типа параформ-
альдегида, которые использовались в некоторых
химических синтезах. В 30-е годы благодаря работам школы
Штаудингера химия полиформальдегида была изучена
весьма основательно. Однако прошло еще около 30 лет
прежде чем удалось осуществить промышленное
производство высокомолекулярного полиформальдегида.
Такой разрыв во времени вызван рядом причин.
Во-первых, полимеризация формальдегида протекает по
ионному механизму, а, как известно, до середины 50-х
годов в промышленном масштабе был осуществлен всего
лишь один процесс ионной полимеризации (получение
полиизобутилена при низкой температуре). Это
объясняется тем, что ионные полимеризационные процессы
изучены недостаточно полно по сравнению со
свободнорадикальными. Кроме того, их сложно
регулировать вследствие высокой чувствительности к примесям
и больших скоростей реакции при низких температурах.
Во-вторых, полиформальдегид, в отличие от
большинства карбоцепных полимеров, обладает низкой
термостойкостью, что не дает возможности формовать
изделия из расплава полимера без предварительной его
стабилизации.
В-третьих, применение полиформальдегида (и ряда
других новых полимеров) в качестве конструкционного
материала для изготовления прецизионных изделий
потребовало нового подхода к проблеме расчета
долговременной прочности изделий из пластмасс, а также
создания новых типов перерабатывающих машин (с предпла-
стикацией материала, с вакуум-отсосом летучих
продуктов и т. п.).
В этой книге собран материал по синтезу, свойствам,
технологии и методам переработки полиформальдегида
и его производных, для которых в литературе
предложен общий термин «ацетальные полимеры». Сюда
относятся гомополимеры с различными концевыми группами,
получаемые полимеризацией формальдегида или три-
оксана, а также сополимеры на основе этих мономеров.
Полное и всестороннее освещение указанных выше
проблем едва ли возможно в настоящее время, поскольку
химия гетероцепных кислородсодержащих полимеров
именно сейчас переживает период становления. Ряд
вопросов, затронутых в этой книге, достаточно подробно
освещается в уже вышедших монографиях. Назовем,
например, монографию Уокера «Формальдегид» (Госхимиздат,
1957 г.), в которой собран наиболее полный материал
по свойствам мономерного формальдегида. Методы
переработки и вопросы применения ацетальных полимеров на
примере «дельрина» (полиоксиметилендиацетат,
вырабатываемый фирмой «Дюпон» в США) рассмотрены в книгах:
R. В. A k i n, The Acetal Resins, Reinhold Plast N. Y,
1962; M. Sit tig, Polyacetal Resins, Gulf Publ. Co.,
JVouston, 1963. Вопросы технологии синтеза
полиформальдегида, как и любого другого нового продукта, весьма
€купо освещаются в литературе, они описаны главным
образом в патентах.
Материал по главам распределен следующим образом:
в I гл. излагаются краткие сведения о свойствах
мономера и низкомолекулярных линейных и циклических
полимерах формальдегида; в гл. II и III рассмотрены
закономерности полимеризации мономерного формальдегида
и триоксана; гл. IV посвящена механизму деструкции
полиформальдегида и способам его стабилизации; в гл. V
рассмотрены вопросы, связанные с сополимеризациеи
формальдегида и триоксана; в гл. VI описана структура
полиоксиметилена, в гл. VII и VIII —некоторые вопросы
технологии синтеза гомополимеров и сополимеров поли-
ацетальной структуры. Заключительная глава (IX)
посвящена описанию свойств, методов переработки и
основных направлений применения ацетальных полимеров.
Главы I, II, VII, VIII и IX написаны С. А. Вольф-
соном, главы III, IV и V — совместно С. А. Вольфсоном
и Н. С. Ениколопяном, глава VI — Э. Ф. Олейником,
под общей научной редакцией Н. С. Ениколопяна.
Авторы выражают искреннюю благодарность всем
сотрудникам лаборатории кинетики полимеризационных
процессов ИХФ АН СССР, принявшим участие в
обсуждении отдельных глав этой книги.
И. ЕНИКОЛОПЯН, С. ВОЛЬФСОН
ГЛАВА І
СВОЙСТВА ФОРМАЛЬДЕГИДА
И ЕГО ОЛИГОМЕРОВ
Формальдегид относится к числу наиболее широко применяемых
в химии реагентов. Честь его открытия принадлежит А. М.
Бутлерову, который в 1859 г. получил формальдегид при попытке
синтеза метиленгликоля СН2(ОНJ путем гидролиза метиленацетата
[1]. Он описал свойства газообразного формальдегида, его водных
растворов, а также некоторые химические реакции формальдегида.
Бутлеров первым получил и твердый полимер формальдегида,
действуя йодистым метиленом на щавелевокислое серебро и на окибь
серебра. Он высказал предположение, что этот продукт является
полимером окиси метилена (полиоксиметиленом).
В основу современных способов производства формальдегида
положена реакция, открытая Гофманом [2] в 1868 г.: при
пропускании смеси паров метанола с воздухом через накаленную платиновую
спираль образуется формальдегид с достаточно высоким выходом.
Мировое производство формальдегида в настоящее время
составляет более 2 млн. т в год и продолжает расти. Формальдегид
находит широкое применение в промышленности и сельском хозяйстве
для самых разнообразных целей. Исключительная реакционная
способность и низкая стоимость сделали формальдегид ценным
полупродуктом для различного рода синтезов.
Ниже приводятся приближенные данные [3] о различных
областях потребления формальдегида (в %) в виде водного раствора:
Промышленность синтетических смол 72
Органический синтез 18
Текстильная и бумажная промышленность ... 2
Сельское хозяйство 2
Дубление и отверждение материалов 1
Прочие области , 5
Основным потребителем формальдегида в настоящее время
является промышленность синтетических смол: производство феноло-
формальдегидных, мочевино-формальдегидных, меламино-формаль-
дегидных и других термореактивных смол; смол, модифицированных
путем обработки формальдегидом; маслорастворимых лаков,
покрытий, клеев, слоистых пластиков и т. п.
Поскольку мономерный формальдегид из-за его высокой
реакционной способности трудно хранить и транспортировать, в технике
он обычно используется в химически связанной форме и может быть
легко выделен непосредственно в момент реакции. Наиболее часто
употребляют водный раствор — технический формалин,
содержащий 37 вес. % формальдегида. Кроме этого применяют параформ-
альдегид — низкомолекулярный твердый полимер формальдегида,
содержащий около 90% основного продукта (остальное— вода),
а также гексаметилентетрамин (CH2NN4 (уротропин). В небольших
масштабах производят а-полиоксиметилен — более
высокомолекулярный, чем параформальдегид, линейный полимер формальдегида
и триоксан — циклический тример формальдегида.
Химия формальдегида интенсивно изучается на протяжении уже
100 лет. Первая монография, посвященная проблеме формальдегида,
была опубликована в России в 1908 г. Е. И. Орловым под названием
«Формальдегид, его добывание, свойства и применение»*. Наиболее
систематическое описание свойств формальдегида принадлежит Дж.
Ф. Уокеру, монография которого «Формальдегид» выдержала в США
уже 3 издания (в 1944, 1953 и 1964 гг.). В русском переводе эта
книга вышла в 1957 г. (перевод 2-го издания)**. Производству
формальдегида посвящена, в частности, книга С. О. С к в о р ц о в а,
«Производство формалина и уротропина», Гослесотехиздат, 1947.
МЕТОДЫ ПРОИЗВОДСТВА ФОРМАЛЬДЕГИДА
Современные методы производства формальдегида развиваются
в двух направлениях: окисление метанола и прямое окисление
углеводородных газов. Производство метанола в связи с бурным
развитием нефтехимической промышленности за два последних
десятилетия претерпело серьезные изменения. Если в прошлом метанол
синтезировали из окиси углерода, которая в свою очередь получалась
из угля и кокса, то сейчас основное сырье для синтеза окиси
углерода — природный газ.
При окислении метанола формальдегид является основным
продуктом реакции, в то время как при окислении природного газа
образуется смесь кислородсодержащих продуктов (альдегидов,
спиртов, кетонов, кислот и других), из которой нужно выделять
формальдегид, выход которого за один проход составляет [8, 9] всего
3-4,5%.
Производство формальдегида из метанола до сих пор остается
главным способом его получения в промышленном масштабе.
Процесс осуществляется в двух основных вариантах: с применением
серебряного или медного катализатора и смесей метанола с воздухом,
обогащенных метанолом; с применением окисных катализаторов
на основе окисей железа и молибдена и смесей, содержащих
небольшое количество метанола. По второму варианту удается получить
растворы формальдегида, практически свободные от непрореагиро-
* Второе издание вышло в Ленинграде в 1935 г.
** У о к е р Дж. Ф., Формальдегид, Госхимиздат, 1957.
10
вавшего метанола [10, 11], что особенно важно в производстве
полиформальдегида.
В присутствии металлического катализатора основной является
реакция дегидрогенизации [12]:
СН3ОН Т""* СН2О + Н2 — 20 ккал/моль
Параллельно протекает экзотермическая реакция окисления водо*
рода:
1
2-°2 > Н2О + 58 ккал/молъ
В присутствии окисного катализатора большое значение
приобретает реакция газофазного окисления метанола:
СН3ОН + уО2 —> СН2О + Н2О + 38 ккал/молъ
Механизм этого процесса в настоящее время изучен еще
недостаточно.
Путем тщательного регулирования температурного режима и
состава парогазовой смеси (метанола и воздуха) при синтезе
формальдегида на современных промышленных установках удается
достигнуть общей конверсии на пропущенный метанол приблизительно
60—70% и конверсии на прореагировавший метанол около 80—93%
(на серебряном или'медном катализаторе). Эффективность
катализатора определяют по составу отходящих газов.
Чтобы получить продукт с высоким выходом, необходимо свести
к минимуму побочные реакции, главными из которых являются
пиролитическое разложение формальдегида и его дальнейшее
окисление:
СН2О —> СО + Н2
СН2О + -~О2 —> НСООН
CH2Of~O2—> СО + Н2О
СН2О + О2 —> СО2 + Н2О
Даже при оптимальном режиме ведения процесса выход
формальдегида ниже теоретического, и в отходящих газах помимо кислорода
и азота (из воздуха) находится некоторое количество окиси углерода,
двуокиси углерода, метана и водорода*
* При использовании окисных катализаторов водород в отходящих газах
практически отсутствует, а количество кислорода увеличивается.
11
Нише приведен состав отходящих газов (в объемн. %) процесса
производства формальдегида из метанола на серебряном
катализаторе (серебро на пемзе)*:
2
СО2
СО
СН4
О2
21-17
4,8—6
0,2—0,8
0,2—0,5
0,3
N3 (по разности) 73,5—75,7
Принципиальная схема производства формальдегида подобным
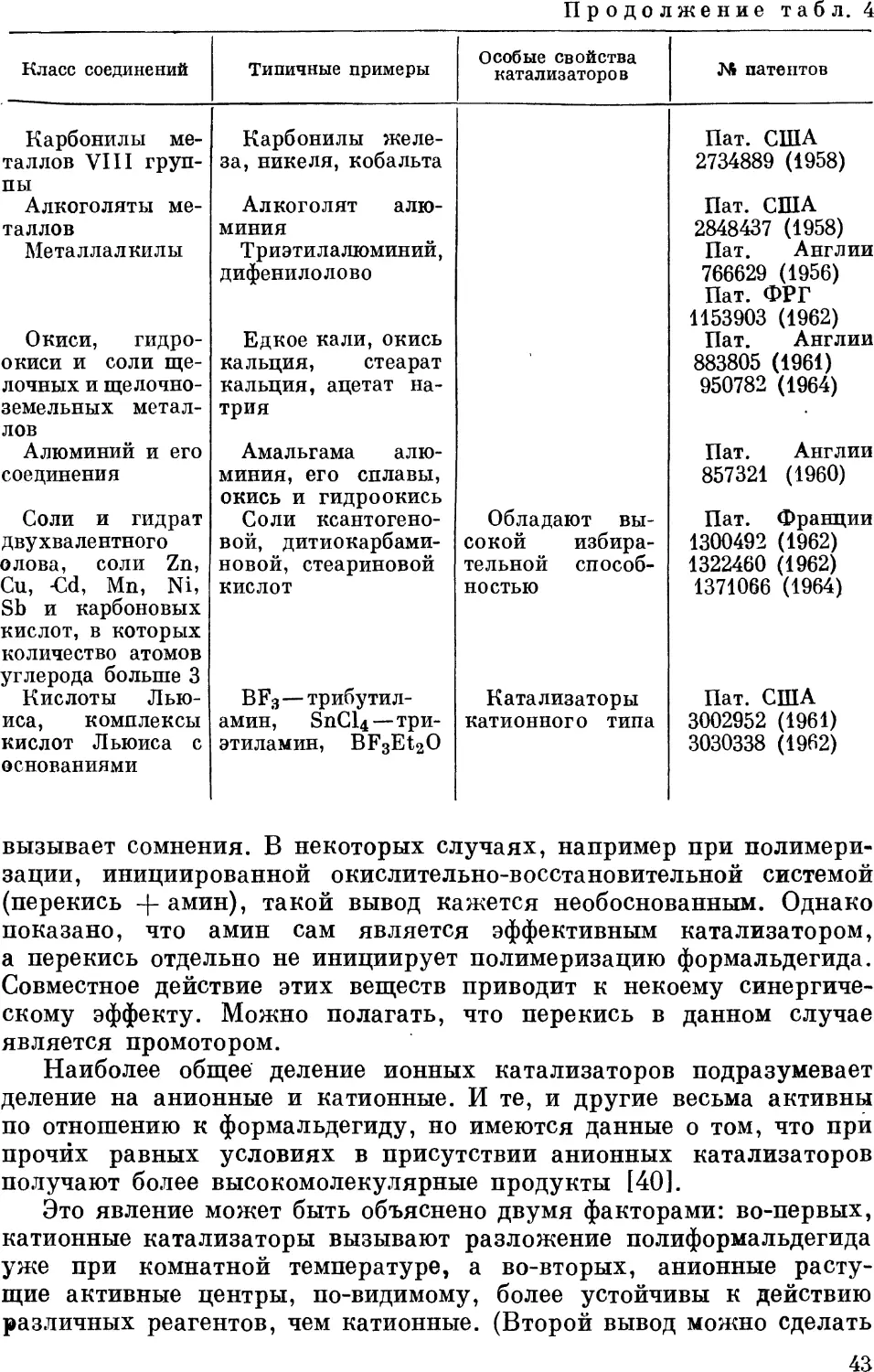
способом показана на рис. 1. Отходящие из реактора газы
поступают на улавливание в систему скрубберов, орошаемых непрерывно
Смесь
метанола
Метанол
Рис. 1. Принципиальная схема производства формальдегида путем окисления
метанола:
11 — реактор; 12 — холодильник; 13 — абсорбер I ступени; 14 — холодильник; 15 —
абсорбер II ступени; 16 — холодильник; 17 — промежуточный сборник; 18 — цистерна для
готовой продукции.
циркулирующим раствором. Из первичного скруббера выводят
раствор, содержащий 37% формальдегида и 7—12% метанола (по
весу). Стандартизация осуществляется обычно путем разбавления
раствора или добавления метанола. Более экономично ведение
процесса под давлением в несколько атмосфер, что позволяет значительно
уменьшить объем аппаратуры.
Путем регулирования технологического режима синтеза при
работе на серебряном катализаторе можно снизить содержание
метанола в формалине до 3—5%, но при этом уменьшается выход
формальдегида по сравнению с теоретическим. На некоторых заводах
дополнительное отделение метанола («обезметаноливание»)
достигается путем ректификации формалина на специальных колоннах.
Нежелательной примесью в формалине является муравьиная
кислота, для удаления которой применяют ионообменные смолы
* Состав отходящих газов изменяется при «старении» катализатора. При
этом концентрация побочных продуктов' реакции увеличивается.
12
(в тех случаях, когда ее содержание превышает допустимые
стандартом нормы).
Особенности процесса синтеза формальдегида на окисных
катализаторах до сих пор не описаны в литературе, хотя уже
функционируют промышленные установки на этих катализаторах. Основные
отличия этого метода заключаются в использовании смесей с
большим количеством воздуха и применении более мягкого
температурного режима. Содержание метанола в формалине, полученном на
установках фирмы «Монтекатини», зависит от времени использования
катализатора: на «свежем» катализаторе оно составляло 0,3—0,5%,
на «отработанном» — до 1% (от массы формальдегида).
Использование метана (природных газов) для непосредственного
окисления в формальдегид, минуя стадию получения метилового
спирта, позволяет уменьшить себестоимость формальдегида.
Принципиальные трудности, возникающие при осуществлении этого
процесса, объясняются малой реакционной способностью метана по
сравнению с формальдегидом: реакцию окисления трудно
инициировать, а начавшись, она быстро протекает до конца по схеме:
~О2—>- СН3ОН
-|-О2 —> СН2О + Н2О
|-Q2 —> СО + Н2О
СН2О —> СО + Н2
Таким образом, необходимо создать условия, облегчающие
инициирование и тормозящие скорость окисления промежуточных
продуктов. Это достигается применением газообразных и твердых
катализаторов, высоких скоростей газовой смеси, «закалкой»
(быстрым охлаждением) газов на выходе из реактора. Выход
формальдегида за один проход через реактор, как уже отмечалось, невелик,
поэтому применяется принцип многократной рециркуляции [8].
Известна работа Медведева с сотр. [9], в которой описан
смешанный каталитический процесс получения формальдегида
(катализаторы — хлористый водород и фосфаты олова, алюминия или железа).
За один проход метано-кислородной смеси через реактор получалось
до 4,5% формальдегида (считая на метан), однако процесс требовал
поддержания строгого температурного режима, что в условиях
применения твердого катализатора оказалось весьма сложным.
Наиболее перспективные результаты были получены при
использовании в качестве гомогенного катализатора окислов азота. Если
вместо воздуха для окисления метана применять кислород [13],
то выход формальдегида достигает 16%. В промышленных условиях
с применением 6—10-кратной циркуляции и окисления ,воздухом
выход формальдегида [14] составляет около 10%.
13
Формальдегид образуется в значительных количествах при
окислении высших газообразных углеводородов. В данном' случае речь
идет о процессе, аналогичном процессам нефтехимического синтеза,
так как формальдегид является только одним из основных продуктов
окисления. Высокому выходу целевого продукта способствуют более
мягкие условия окисления D00—500° С и давление 5—30 атм),
благодаря которым потери кислородсодержащих продуктов в
результате разложения незначительны.
Помимо формальдегида (выход 15—25%) из реакционной смеси
выделяют ацетальдегид, ацетон, метанол, пропиловый и бутиловый
спирты, органические кислоты и другие продукты [15—17, 79].
СВОЙСТВА ФОРМАЛЬДЕГИДА
Интерес к свойствам мономерного формальдегида резко возрос
в последнее время в связи с проведением работ по полимеризации
его в высокомолекулярный полиоксиметилен. Ранее мономерный
формальдегид вызывал интерес скорее в теоретическом, чем в
практическом отношении.
Чистый безводный формальдегид в обычных условиях
представляет собой бесцветный газ с резким запахом, который
раздражающе действует на слизистые оболочки уже при очень небольших
концентрациях [18]. Предельно допустимая концентрация 5 мг/м3.
Формальдегид кипит при —19° С и замерзает при —118° С,
превращаясь в твердое кристаллическое вещество.
При температурах выше 100° С чистый безводный газообразный
формальдегид достаточно устойчив, но при понижении температуры
легко полимеризуется, причем ничтожные примеси воды и других
полярных веществ действуют как катализаторы полимеризации.
Газообразный формальдегид горит (теплота сгорания [19] равна
4,47 ккал/г), с воздухом образует взрывчатые смеси при
концентрациях 7—72 вес. %. Температура самовоспламенения [20] —около
300° С.
Теплоемкость формальдегида (С ) была вычислена Уокером [4]
по методу, описанному Добрацем [21] и основанному на
спектроскопических измерениях. Для зависимости Ср от температуры
предложено выражение:
(G^ = 9,48 + 9,14 • Ю-»?1 —6,4- Ю-^а A)
Теплоемкость при высоком давлении вычисляли на основе
следующих вероятных значений критических величин для
формальдегида: Тщ = 141° С, PKV = 65 атм. При этом получили:
D14 \3
Значения теплоемкости, вычисленные по этой формуле [4],
приводятся в табл. 1.
ТАБЛИЦА 1
Теплоемкость газообразного формальдегида
Температура
°С
0
100
200
300
С (в кал /моль) при давлении
0 атм
9,48
10,39
11,28
12,16
0,1 атм
9,51
10,39
11,28
12,16
і атм
9,75
10,49
11,33
12,19
Теплота образования формальдегида из элементов по имеющимся
термохимическим данным вычислена Россини [22]:
С (графит) + Н2 + — О2 —> СН2О (газ)+28 ккал
Теплота сгорания мономерного формальдегида определена фон
Вартенбергом [19]:
СН2О (газ) + О2 —> Н2О
Величина свободной энергии формальдегида [5] при 25° С равна
—27 ккал/моль.
Плотность жидкого формальдегида, по данным Кекуле [29],
составляет 0,915 г/см3 при —80° С и 0,8153 г/смъ при —20° С.
Давление насыщенного пара над чистым жидким формальдегидом
определили Спенс и Уайлд [30]. Согласно их данным, эта величина (в атм}
может быть вычислена по уравнению:
1429
+ 1,75 lg T- 6,3 • 10-ЗГ +3,0177
Теплота испарения жидкого формальдегида при температуре
кипения (—19° С), рассчитанная по давлению насыщенного пара,
равна 5,57 ккал/моль.
Жидкий формальдегид можно получить конденсацией тщательно
очищенного газообразного формальдегида. Описание способов
получения жидкого формальдегида в препаративных целях приводится
рядом авторов: Кекуле [29], Траутцем и Уфером [31], Штаудинге-
ром [32], Уокером [33], Спенсом и Уайлдом[34], Тоби и Рутцом [35].
Спектры поглощения паров формальдегида в ультрафиолетовой
области изучались в работахо [23—27]. Спектр состоит из 35—40
полос о в области 3700—2500 А с максимальным поглощением при
2935 А. Спектр поглощения жидкого формальдегида (раствор в
гексане) при —70° С, по данным [24], идентичен спектру мономерного
газа, но смещен на 5 А по направлению к инфракрасной области.
Прайс [27] на основании данных по изучению спектра
поглощения в далекой ультрафиолетовой области вычислил прочность связи
С—О в молекуле формальдегида и нашел ее равной 164 ккал/молъ.
15
Главные линии инфракрасного поглощения формальдегида
приводятся ниже [28]:
Длина волны
МП
3.54
3,60
5,74
6,65
7,81
8,57
Частота
см-1
2825
2780
1744
1503
1280
1167
Групп
сн2
СН2
СО
сн2
сн2
сн2
Жидкий формальдегид смешивается в любых соотношениях со
многими неполярными органическими растворителями и в виде
раствора несколько более устойчив при хранении.
Химические свойства формальдегида во многом определяются
особенностями его структуры. Как известно, первые члены
гомологических рядов часто по своим свойствам резко отличаются от
последующих. То же самое можно сказать о формальдегиде и других кар-
бонилсодержащих соединениях, реакционная способность которых
в зависимости от природы заместителя у карбонильной связи
изменяется в следующем ряду:
Hv Rv R'v R'4
>С=О > >С=О > >С=О > >С=О
W ' НХ R"/ R"CK
Для карбонильной группы характерна поляризация я-связи,
при этом я-электроны смещаются в сторону более
электроотрицательного атома кислорода:
д+ д-
Отсутствие заместителей в молекуле формальдегида приводит
к тому, что реакционная способность как нуклеофильного, так и
электрофильного центров примерно одинакова, и формальдегид
способен реагировать с реагентами обоих типов.
Чистый формальдегид достаточно химически устойчив.
Разложение (пиролиз) на окись углерода и водород [36, 37] заметно только
при температурах выше 300° С.
Окислением в мягких условиях формальдегид можно
превратить в муравьиную кислоту [38]. В качестве промежуточных
продуктов обнаружены перекисные соединения.
Реакции взаимодействия молекул формальдегида между собой
могут протекать в четырех направлениях:
1) реакции полимеризации;
2) реакции типа альдольной конденсации;
3) реакция Канниццаро;
4) реакция Тищенко.
16
Реакция полимеризации рассматривается в следующей главе,
здесь следует лишь отметить, что она наиболее характерна для
формальдегида.
Для нормального протекания реакции альдольной конденсации
необходимо наличие а-водородных атомов. Однако в щелочной среде
в водных растворах формальдегида происходят превращения, по-
обные альдольной конденсации, в результате которых образуются
оксиальдегиды, оксикетоны и сахара. Первичным продуктом, по-
видимому, является гликолевый альдегид [39]:
2СН2О —> СН2ОН—GHO
Реакция Канниццаро заключается в восстановлении одной
молекулы формальдегида с одновременным окислением другой:
2CH2O-f Н2О —у СНзОН+НСООН
Благоприятными условиями для протекания этой реакции
является температура 50—60° С и щелочная среда. Однако было
найдено, что она протекает и в газовой фазе в присутствии следов
влаги. Добавки кислоты не влияют заметным образом на скорость
этой реакции.
В некоторых случаях формальдегид способен вступать в реакцию
Тищенко, образуя метиловый эфир муравьиной кислоты (метил-
формиат) *:
2СН2О А1(ОСНз)з-> НСООСНз
Эта реакция является побочной при пиролизе
низкомолекулярных полиоксиметиленов, при выделении триоксана из растворов
формальдегида, содержащих серную кислоту [80], и т. п.
Ниже кратко перечислены другие химические реакции
формальдегида, так как подробно они рассмотрены в монографии Уокера.
В реакциях восстановления формальдегид действует как
восстановительный агент, окисляясь сам до муравьиной кислоты (в
щелочных средах). Реакции присоединения или конденсации играют
важнейшую роль при синтезах с участием формальдегида. При этом
формальдегид легко взаимодействует с различными соединениями,
имеющими подвижные атомы водорода, образуя метилольные
производные. В качестве типичного примера можно привести реакцию
образования метиленгликоля:
HOH-f СН2О «z> НОСН2ОН
В большинстве случаев эта реакция протекает в щелочной и
нейтральной средах, в то время как в кислой происходит образование
метиленовых производных, более устойчивых, чем метилольные.
¦ Образование метилформиата может быть таюке следствием реакции
Канниццаро. В работе [81] п^кдаал^рчдро-^е^илформиат образуется при
пропускании газообразного фхм^Аьде^ида Щд^вЧ^ші низкомолекулярным поли-
оксиметиленом. Одновременно4 наблюдается:' ув^шм^ение концентрации
метанола и муравьиной даслоты.
2 Заказ 1135. . 17
Так, например, в кислой среде при взаимодействии метанола и
формальдегида образуется метилаль — вещество, которое можно считать
типичным метиленовым производным:
2СН3ОН + СН2О —>¦ СНзОСНзОСНз + НзО
Во многих случаях образованию метиленових производных
предшествует образование метилольных. Это показано, в частности,
при получении формалей при взаимодействии формальдегида со
спиртами:
CH3OH<f СН2О ^=> СН3ОСН2ОН
Метиленовые производные могут представлять собой
непредельные соединения. В этом случае метиленовая группа соединяется
с остальной частью молекулы через двойную связь:
CH2=CHR
Такие соединения играют важную роль при получении
термореактивных смол, так как за счет разрыва непредельной связи
достигается сшивка линейных макромолекул.
РАСТВОРЫ ФОРМАЛЬДЕГИДА
Растворы формальдегида разделяют на два типа: истинные
растворы, в которых формальдегид присутствует в мономерной форме,
и растворы, в которых формальдегид химически связан с
молекулами растворителя.
Растворы первого типа до последнего времени не имели
практического значения, так как при комнатной и более высоких
температурах формальдегид очень плохо растворим в неполярных
растворителях — алифатических, циклоалифатических и ароматических
углеводородах, эфирах, ацеталях, галоидпроизводных углеводородах
11 ДРУГИХ неполярных, инертных к формальдегиду соединениях.
Максимальная концентрация мономера при комнатной температуре
не превышает 1,5—3,0%. Сейчас они применяются при синтезе
полиформальдегида из мономерного формальдегида.
Как уже отмечалось, жидкий формальдегид смешивается с
неполярными растворителями при температуре ниже —19° С. В этом
случае можно получить раствор любой ксшцентрации.
Растворение газообразного формальдегида в воде и спиртах
сопровождается выделением большого количества тепла A4—
15 ккал/молъ). Исследование спектров поглощения в
ультрафиолетовой области, а также спектров комбинационного рассеяния этих
растворов показало практически полное отсутствие карбонильных
групп, т. е. негидратированного мономерного формальдегида [40,
41]. Совокупность данных по анализу свойств водных и спиртовых
растворов формальдегида позволила еще в начале этого века сделать
вывод о том, что в этих растворах формальдегид присутствует в виде
равновесной смеси моногидрата (моногемиформаля в случае спирто-
18
вого раствора) и полиоксиметиленгликолеи, или полиоксиметилен-
гемиформалей.
В водном растворе моногидрат представляет собой метиленгликоль
СН2(ОНJ. Все попытки выделить его в чистом виде остались
безуспешными, так как он легко разлагается на формальдегид и воду.
Термодинамика и равновесие в водных растворах формальдегида
изучаются вот уже в течение 80 лет. Первое подробное исследование
принадлежит Ауэрбаху и Баршаллу [42], из последних работ
необходимо отметить исследование Илисето и Беззи [43—47].
Бибер и Трюмплер [48], а затем Холл и Пайрет [49] и Илисето
и Беззи [44] доказали существование равновесия между негидрати-
рованным формальдегидом и метиленгликолем в разбавленных
водных растворах и вычислили значения константы равновесия при
различных температурах по следующему уравнению:
к = [СН2О]Ж[Н2О]
[СН2(ОНJ]ВОДН.
Приближенное значение константы равновесия при 0° С составляет
10~4, при 64° С — 30,1 • 10~4. Используя эти данные, Бибер и
Трюмплер вычислили значение молярной теплоты гидратации мономера
A4,6 ккал/молъ). Илисето приводит цифру 8,10 ккал/молъ, что с
учетом теплоты сжижения E,57 ккал/молъ) также дает величину, близкую
к 15 ккал/молъ.
Равновесие между моногидратом и полимергидратами
формальдегида, имеющими общую формулу НО(СН2О)ПН, зависит от
температуры и концентрации формальдегида в растворе. Константы
равновесия измеряли криоскопическим методом.
Скорость установления равновесия при низких температурах была
крайне мала; Илисето и Беззи [45] для установления равновесия
ждены были выдерживать растворы в течение месяца. Данные
едних молекулярных весах, полученные Илисето и Беззи
криоскопическим методом по депрессии точки замерзания растворов,
заметно отличаются от аналогичных данных, полученных Ауэрба-
хом и Баршаллом [42]. Гипотеза Ауэрбаха заключалась в том, что
в растворах с концентрацией формальдегида не выше 30% в
равновесии находятся метиленгликоль и триоксиметиленгликоль. Илисето
и Беззи с сотр. [46] показали, что имеет место гораздо более сложное
равновесие, так как в растворе присутствуют молекулы
полиоксиметиленгликолеи самой различной длины. С увеличением концентрации
формальдегида происходит перераспределение длин молекул за
счет уменьшения концентрации метиленгликоля, димеров, тримеров
и т. д.
Кинетика изменения равновесия в растворах формальдегида
изучалась Вадано с сотр. [50] с помощью интерферометрического метода.
Найдено, что константы скорости реакции деполимеризации при
разбавлении растворов подчиняются уравнению реакции первого
порядка. Концентрация водородных и гидроксильных ионов в
растворе существенным образом влияет на скорость этой реакции,
2* ._ 19
причем скорость минимальная в интервале pH от 2,6 до 4,5, а
влияние гидроксильных ионов в 107 раз сильнее, чем ионов водорода.
Теплота активации реакции деполимеризации равна
17,4 ккал/молъ. Вадано пришел к заключению, что в интервале pH,
которому соответствуют минимальные значения скорости
деполимеризации, гидраты формальдегида ведут себя как амфотерные
соединения, а в более кислых или щелочных средах образуют анионы
и катионы:
Область pH
Ниже 2,6 СН2(ОНJ Т=^ СН2—ОН + ОН-
От 2,6 до 4,5 СН2(ОНJ •+=? СН2—С) + ОН"-^Н+
Выше 4,5 СН2(ОНJ ^=± СН2(ОН)О + Н+
Такой же характер имеет диссоциация полиоксиметиленгликолей.
Термохимия растворов формальдегида изучалась Илисето [47],
который по калориметрическим данным рассчитал теплоты реакций
(а) и (б), протекающих в жидкой фазе:
НОСН2ОН^НОСН2ОН <=± НО(СН2ОJН-^Н2О + 0,87 ккал (а)
НОСН2ОН ф НО(СН2О)ЯН ^=> НО(СН2О)Я+1Н + Н2О + 0,59 ккал (б)
НИЗКОМОЛЕКУЛЯРНЫЕ ПОЛИОКСИМЕТИЛЕНЫ
Полно ксиметиленгликоли общей формулы НО(СН2О)ЯН, где
п^> 10, выпадают из водного раствора формальдегида при
температуре ниже 10° С. Метиловый спирт, добавляемый в технический
формалин, играет роль стабилизатора. Безметанольные растворы
формальдегида устойчивы только выше некоторого температурного
предела насыщения, который зависит от концентрации раствої
Ниже предела насыщения наблюдается спонтанное образовав
смеси твердых полиоксиметиленгликолей (ПОМ) различного
молекулярного веса.
Детальное изучение свойств низших полиоксиметиленгликолей
провел Штаудингер с сотр. [51], которому удалось выделить многие
из них в чистом виде. В общих чертах методика выделения
заключалась в экстракции полиоксиметиленов ацетоном из
концентрированного водного раствора формальдегида и дробном осаждении фракций
петролейным эфиром после предварительной сушки смеси безводным
сернокислым натрием.
Фракции полиоксиметиленгликолей с различной степенью
полимеризации различались температурой плавления и растворимостью.
Наиболее легкая фракция представляла собой смесь ди- и триокси-
метиленгликолей с температурой плавления 82—85° С.
Характеристики различных фракций, являющихся кристаллическими
веществами, приведены в табл. 2.
С низкомолекулярными полиоксиметиленгликолями связано
представление 6 чрезвычайно плохой термической стойкости полимеров
20
ТАБЛИЦА 2
Свойства низкомолекулярных полиоксиметиленгликолей [51]
Степень
полимеризации
2-3
4
6
7
8
9
И
12
Параформ-
альдегид
Содержание
вычислено
76,9—83,3
87,0
90,9
92,1
93,0
93,8
94,8
95,2
95—98
СШО, %
найдено
79,3
86,5
90,9
92,0
92,9
93,9
94,8
95,0
95,5
Температура
плавления
°С
82—85
95—105 (с
разложением)
—
—
115—120 (с
разложением)
—
—
—
140 (с
разложением)
Растворимость в ацетоне
Легко растворяется на
упттп TtV
Л-UJIU Д^г
То же
Растворяется на холоду
То же
Растворяется при
нагревании
То же
Растворяется при
кипении
С трудом растворяется
при кипении
Почти не растворяется
формальдегида. Штаудингер показал, что после тщательной очистки
от муравьиной кислоты путем перекристаллизации (из ацетона,
диоксана, хлороформа, пиридина и даже из воды) можно получить
вполне устойчивый продукт со слабым запахом формальдегида.
Следы кислоты и щелочи вызывали разложение полимергидратов
уже при низких температурах.
Благодаря работам Штаудингера и сотр. [52, 53] мы располагаем
обширными сведениями о химическом строении полиоксиметиленов
и их свойствах. Для последующих работ в области
высокомолекулярных полиоксиметиленов и вообще высокополимеров принципиальное
значение имело сделанное Щтаудингером заключение о влиянии
концевых групп на свойства макромолекул.
Впервые низкомолекулярные производные
полиоксиметиленгликолей были получены в самом начале века [55] действием параформ-
альдегида на уксусный ангидрид в присутствии хлористого цинка.
Впоследствии Штаудингер и Люти [56] разработали способ
получения диацетатов с более длинными цепями и путем
перекристаллизации из формамида выделили диацетаты со средней степенью
полимеризации до 35.
Ацетилирование проводили уксусным ангидридом в присутствии
пиридина в среде диоксана или ацетона. Перекристаллизованные
однородные фракции диацетатов отличались большей
термостабильностью, чем дигидраты, и разлагались при 190—200° С. Неперекри-
сталлизованный продукт, судя по содержанию формальдегида,
найденному аналитически (92,7%), имел среднюю степень
полимеризации 50, но помимо диацетатов, по-видимому, содержал ацетатгидраты
и дигидраты полиоксиметилена, что приводило к понижению его
стабильности. Он плавился с разложением при 150—170° С. При
21
кипячении в разбавленных растворах щелочи он легко разлагался,
что объясняется неустойчивостью сложноэфирных групп к
щелочному гидролизу.
Еще большей стабильностью обладали простые диметиловые
эфиры полиоксиметиленгликоля, которые были получены действием
метилового спирта на параформальдегид в присутствии серной
кислоты с последующей перекристаллизацией из различных
растворителей. Наиболее высокомолекулярные фракции (со степенью
полимеризации около 100) были получены фракционированием
Y-полиоксиметилена путем перекристаллизации из формамида, цикло-
50 100 150
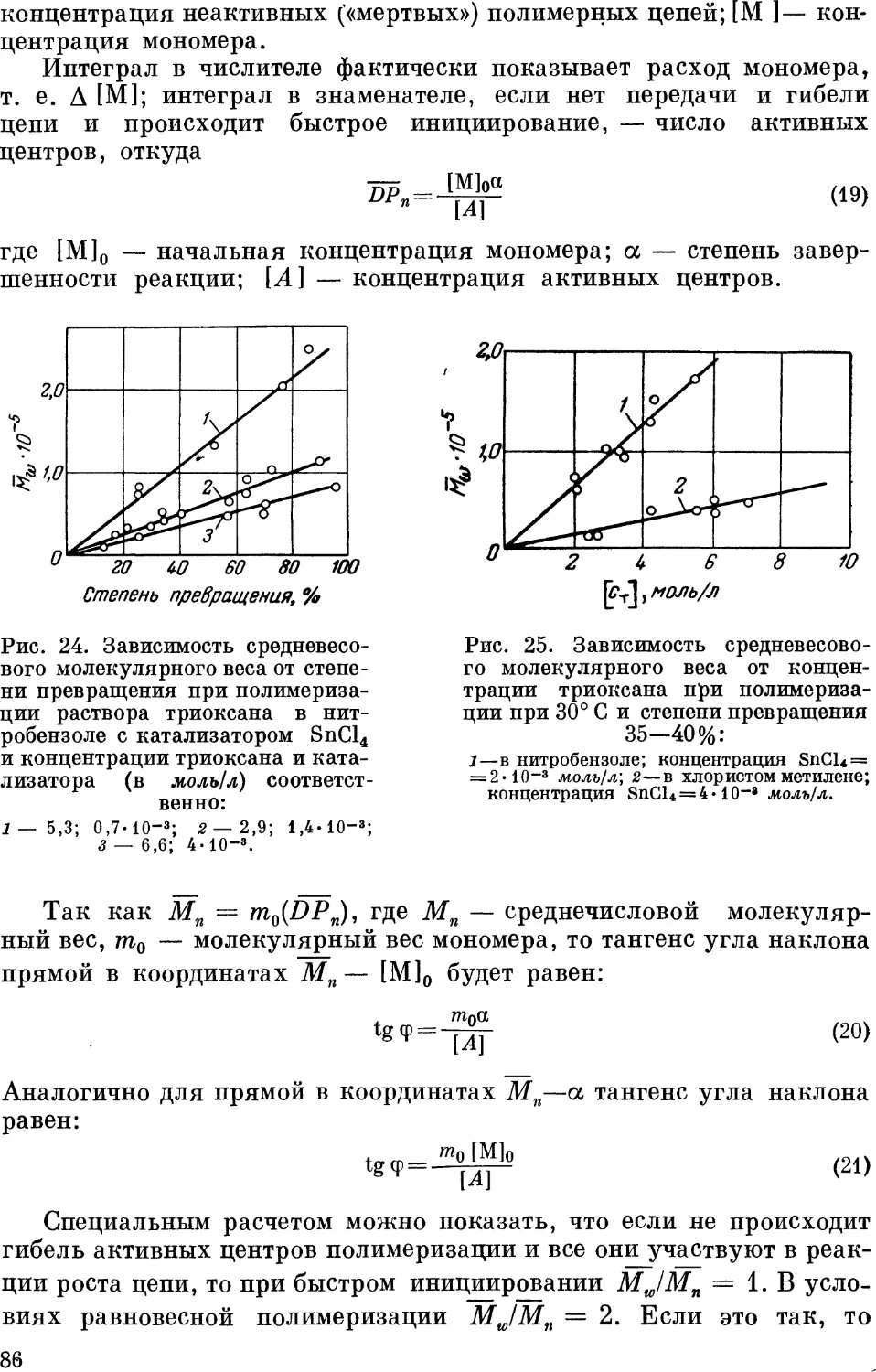
Степень полимеризации
Рис. 2. Зависимость температуры
плавления от степени полимеризации
диметилового эфира полиоксиметилена [51].
50 WO 150
Степень полимеризации
Рис. 3. Зависимость температуры
разложения от степени полимеризации
диметилового эфира
полиоксиметилена [51].
гексанола, анизола и других высококипящих растворителей при
нагревании. Фракции диметиловых эфиров получали при кипячении
со слабыми щелочами, к действию которых диметиловые эфиры
весьма устойчивы, в то время как метилэфиргидраты и дигидраты
ПОМ быстро разрушаются. Разбавленные кислоты полностью
разрушают их при кипячении, как и другие полиацетали. На
основании этих данных был сделан вывод о том, что полиоксиметиленовые
цепи устойчивы к щелочам и неустойчивы к кислотам.
Зависимость температуры плавления от степени полимеризации
диметилового эфира ПОМ показана на рис. 2. Температура
разложения наиболее высокомолекулярных фракций лежит в пределах
190-200° С.
Сравнение температур плавления диметиловых эфиров и диацета-
тов ПОМ с одинаковыми степенями полимеризации показывает, что
низкомолекулярные диметиловые эфиры ПОМ плавятся при более
высокой температуре, однако различия нивелируются при степени
полимеризации порядка 35.
Зависимость температуры разложения от степени полимеризации
диметилового эфира ПОМ показана [51] на рис. 3. По мере
увеличения степени полимеризации стойкость диметилового эфира ПОМ
22
понижается, и при степени полимеризации около 150 полимер
плавится o разложением. Растворимость диметиловых эфиров ПОМ
в горячем формамиде изменяется симбатно с кривой температур
плавления. Экстраполяция к большим степеням полимеризации дала
основание предполагать, что диметиловые эфиры ПОМ со степенью
полимеризации около 200 будут разлагаться раньше, чем наступит
растворение.
Параформальдегид
Параформальдегид представляет собой смесь полиоксиметилен-
гликолей различной степени полимеризации, содержащую 93—98%
формальдегида (остальное — химически связанная и
адсорбированная влага). Концентрация формальдегида зависит от средней степени
полимеризации и определяет такие свойства параформальдегида,
как растворимость в воде, температура плавления и т. п. По данным
Штаудингера [57], обычный технический параформальдегид
содержит молекулы со степенью полимеризации от 8 до 100. Содержание
формальдегида определяется с помощью обычных методов
количественного анализа — сульфитного или иодометрического [6].
По внешнему виду параформальдегид — бесцветное твердое
вещество, порошкообразное или в гранулах, в зависимости от степени
помола, с запахом формальдегида. Плавится с разложением в
интервале температур 120—150° С.
Параформальдегид медленно разлагается при хранении,
по-видимому, из-за присутствия следов муравьиной кислоты.
Деполимеризацию можно ускорить нагреванием или добавкой щелочного или
кислотного агента. Скорость растворения в воде, как показали Лебе-
ринг и Юнг [60], зависит от температуры, pH среды (фактически это
скорость гидролиза) и степени полимеризации.
Наиболее распространенный способ производства
параформальдегида заключается в концентрировании под вакуумом водных
растворов формальдегида до затвердевания кубового остатка, который
затем размалывается и высушивается.
Механизм образования параформальдегида до сих пор детально
не выяснен. Ранее предполагалось [7], что это типичный процесс
поликонденсации, хотя бы на первых стадиях реакции, когда
молекулы метиленгликоля взаимодействуют друг с другом. Для
объяснения эффекта увеличения молекулярного веса твердых полимерных
гликолей Штаудингер [58] предложил схему, по которой
мономерный формальдегид, образующийся при частичной деполимеризации
полимерных гликолей (в присутствии следов Муравьиной кислота),
вступает в топохимическую реакцию с твердыми полноксиметилен-
гликолями:
СН2О + НО(СН2О)ЯН —> НО(СН2О)Я+1Н
Илисето [43] высказал предположение, что если агрегация
молекул в растворе происходит по поликонденсационному механизму, то
23
в присутствии метилового спирта должны образовываться молекулы
диметилового эфира полиоксиметиленгликоля по реакции:
ИОСН3 —>- CH3O(CH2O)W+1GH3 + H2O
Однако показано, что при спонтанной полимеризации диметило-
вых эфиров не образуется. Каталитическое действие щелочей и
кислот также не согласуется с представлением о конденсационном
механизме процесса.
Анализ описанных в литературе методов получения параформаль-
дегида с различной растворимостью показывает, что существует
три принципиально различных способа синтеза смеси
низкомолекулярных твердых полиоксиметиленгликолей:
1) спонтанная полимеризация формальдегида в пересыщенном
[61, 62] растворе;
2) каталитическая полимеризация в присутствии основных или
кислотных агентов [63];
3) топохимические реакции на поверхности частичек параформ-
альдегида, которые суспендируют в растворе или обрабатывают
парогазовой смесью формальдегида и воды [64—66].
а-Полиоксиметилен и другие модификации
полиоксиметилена
При действии щелочей и кислот на концентрированные растворы
формальдегида осаждается кристаллический порошкообразный
продукт, по свойствам весьма напоминающий параформальдегид,
но отличающийся от него плохой растворимостью в воде, даже при
кипячении, и более высокой температурой плавления (плавится
с разложением при 170—180° С). Этот полимер получил название
а-полиоксиметилен [42]. Аналитическое определение концентрации
формальдегида в нем дает величину 99,4—99,8%, т. е. это смесь
полиоксиметиленгликолей со средней степенью полимеризации
выше 100.
При растворении в щелочной или кислой среде
а-полиоксиметилен, так же как и параформальдегид, гидролизуется. Оба
полимера неустойчивы при хранении и медленно^ разлагаются с
выделением мономерного формальдегида, однако скорость разложения
а-полиоксиметилена ниже.
?-Полноксиметиленом Ауэрбах и Баршалл [42] назвали продукт,
получающийся при полимеризации концентрированного водного
раствора формальдегида в присутствии серной кислоты. Этот
полимер по свойствам и молекулярному весу очень близок к а-полиокси-
метилену, однако он окклюдирует некоторое количество серной
кислоты. ?-Полиоксиметилен способен к некоторым превращениям,
обусловленным присутствием серной кислоты в его кристаллах.
Например, после длительного хранения он теряет способность
растворяться в щелочных средах. Это может быть связано с
образованием диметиловых эфиров ПОМ, устойчивых к щелочам. При нагре-
24
вании на воздухе р-полиоксиметилен легко возгоняется, не плавясь,
и превращается частично в триоксан, вероятно, в результате ацидо-
литического расщепления линейных молекул кислотой.
Если для получения ?-полиоксиметилена применять раствор
формальдегида, содержащий метиловый спирт, то вместе с ?-поли-
оксиметиленом образуется некоторое количество -у-полиоксиметилена
[42], выход которого, как показал Штаудингер, зависит от
концентрации метилового спирта в растворе [59]. Выделение
?-полиоксиметилена осуществлялось путем экстракции его из смеси раствором
сернистого натрия, в котором -у-полиоксиметилен нерастворим.
-у-Полйоксиметилен; представляет собой [54] смесь диметиловых
эфиров ПОМ со средней степенью полимеризации более 100. Его
температура плавления лежит в интервале 160—200° С (плавится
с разложением). Из всех описанных выше модификаций полиоксиме-
тилена эта наиболее устойчива. Полимер при комнатной температуре
не имеет запаха формальдегида, нерастворим в воде, горячем
ацетоне и щелочах, но гидролизуется в кислой среде.
При кипячении Y-полиоксиметилена в воде он подвергается
частичной перегруппировке. Как показал Штаудингер с сотр. [57], при
этом в цепях макромолекул образуется некоторое количество
углеводных группировок типа
—СНз-О-СН-О-СНг—О—
ОН
Получающийся продукт получил название б-полиоксиметилен.
Ни один из этих полимеров не обладает пластическими
свойствами вследствие низкого молекулярного веса. Долгое время
считалось, что при полимеризации водных или спиртовых растворов
формальдегида вообще невозможно достигнуть высоких степеней
полимеризации (порядка 1000 и более) из-за специфики
протекающих поликонденсационных процессов.
Циклические полиоксиметилены
Известны два циклических олигомера полиоксиметиленовой
структуры — триоксан и тётраоксиметилен, шести- и восьмичленные
циклические ацетали:
О
О / \
/ \ Н2С СН2
Н2С СН2 | |
II оо
ОО ||
\ / н2с сн2
V
триоксан тётраоксиметилен /
Оба они являются весьма устойчивыми химически
индивидуальными продуктами со специфическими свойствами. До последнего
' 25
времени они рассматривались как некие феномены среди полимеров
полноксиметиленовой структуры и не имели практического
значения*. В связи с открытием способа полимеризации триоксана
в высокомолекулярный линейный полиоксиметилен интерес к нему
заметно возрос.
Структурная формула триоксана выведена на основе определения
молекулярного веса и изучения его физических и химических
свойств.
Молекула триоксана представляет собой шестичленное кольцо
с атомами, не лежащими в одной плоскости. Рентгенограммы
триоксана указывают на его отчетливо выраженное кристаллическое
строение (подобно другим полиоксиметиленай). Дипольный момент
триоксана (в бензоле) равен 2,18 • 10~18 эл.-ст. ед.
Чистый триоксан — бесцветное кристаллическое вещество с
характерным запахом, не похожим не запах формальдегида. Триоксан
плавится при 61—62° С и кипит без разложения при 114—115° С.
С водой он образует азеотропную смесь, кипящую при 91Ф С и
содержащую 70% триоксана [67]. Пары триоксана образуют с воздухом
взрывчатую смесь в интервале концентраций 3,6—28,7 объемн. %.
Диэлектрическая проницаемость при 20° С, измеренная при 1,6 Мгц,
равна 3,2—3,4; диэлектрическая проницаемость расплавленного
триоксана [68] при 70° С равна 8, при 80° С — 15,6.
Триоксан хорошо растворим в воде, а также в ароматических
и циклоалифатических углеводородах, спиртах, органических
кислотах, фенолах, простых и сложных эфирах и т. п. Несколько хуже
растворимость триоксана в алифатических углеводородах.
Расплавленный триоксан сам является превосходным
растворителем для многих органических соединений и весьма схож по
свойствам с диоксаном.
Теплота сгорания и теплота образования триоксана
определялись Делепиным и Бадош [69], а также Палфреем [70]:
Теплота сгорания, ккал/молъ 120,9 118,9
Теплота образования, ккал/молъ . . . 41,3 43,4
Теплота образования триоксана из мономерного формальдегида
(тепловой эффект реакции) составляет 13—15 ккал/моль СН2О,
т. е. практически совпадает с величиной теплового эффекта реакции
полимеризации мономерного формальдегида в линейный
полиоксиметилен. Теплота испарения (при температуре кипения) равна
приблизительно 9,8 ккал/молъ триоксана.
Триоксан представляет собой весьма инертное вещество. Хорошо
очищенный продукт выдерживает нагревание до 200—250° С без
заметных следов разложения. Термическое разложение наблюдается
при температурах порядка 300° С, что является косвенным
доказательством достаточно высокой термической стойкости ацетальных
* Триоксан в ограниченных количествах использовался как растворитель
(в препаративных целях).
26
связей. Разложение триоксана в газообразном состоянии при 270—
345° С изучали Бернетт и Бэлл [71], которые определили, что это
реакция первого порядка.
Благодаря отсутствию концевых групп триоксан проявляет
высокую стойкость к щелочным агентам. Его, например, можно
кипятить и перегонять над твердой щелочью. По этой же причине
невозможно определить концентрацию триоксана по содержанию
формальдегида в нем, пользуясь сульфитным или иодометрическим
способами.
В кислотной среде триоксан, подобно всем ацеталям, гидроли-
зуется. Кинетика кислотного гидролиза в разбавленных водных
растворах подчиняется уравнению реакции первого порядка [72].
При сравнении констант скорости кислотного гидролиза при 25° С
триоксана и других ацеталей с простыми эфирами были получены
следующие результаты (в пересчете на один эквивалент кислорода
эфирной связи [73]):
\
Диэтиловый эфир 1,46 • 101
Триоксан 5,3 • 10"'
Метилаль 7,6 • 10~4
Паральдегид 2,73 • 10~3
Ацеталь 30,0
Деполимеризация триоксана с образованием формальдегида в
присутствии кислотного агента протекает как равновесная реакция.
Условия равновесия описаны в работе [74]; было найдено, что при
180° С оно полностью сдвинуто в сторону образования мономерного
формальдегида.
Впервые триоксан синтезировал Пратези [75] в 1885 г.
нагреванием параформальдегида с небольшим количеством серной кислоты
в запаянной трубке при 115° С. Ауэрбах и Баршалл [42] получили
триоксан путем пиролиза параформальдегида в токе азота.
Наиболее распространенный способ синтеза триоксана
разработан Фрэнком [76] в 1942 г. Раствор формальдегида F0—65%-ный)
перегоняют в присутствии 2% серной кислоты и затем экстрагируют
триоксан из дистиллята [72] или кристаллизуют [77]. Если вести
фракционированную перегонку, то можно отбирать фракцию, по
составу близкую к азеотропной смеси триоксан — вода и
содержащую около 60% триоксана.
Тетраоксиметилен был впервые получен Штаудингером [56]
путем деполимеризации относительно высокомолекулярного диаце-
тата полноксиметиленгликоля. Тетрамер, по-видимому, всегда
образуется в некотором количестве при получении триоксана или аци-
долитическом расщеплении полиоксиметиленов (наряду с триокса-
ном). В недавно опубликованной работе [78] приводятся данные
о зависимости выхода циклических продуктов от типа и
концентрации применявшейся кислоты.
27
Свойства тетраоксиметилена практически не изучены. Известно,
что он образует игольчатые кристаллы, плавящиеся при 112° С,
и легко возгоняется. Можно предполагать, что по свойствам он
подобен триоксану.
ЛИТЕРАТУРА
В u 11 е г о f A. M., Ann., Ill, 242 A859).
Hof mann A. W., Ann., 145, 357 A868).
У ок e p Дж. Ф., Формальдегид, Госхимиздат, 1957, стр. 484.
У ок ер Дж. Ф., Формальдегид, Госхимиздат, 1957, стр. 49.
Уокер Дж. Ф., Формальдегид, Госхимиздат, 1957, стр. 51.
У о к е р Дж. Ф., Формальдегид, Госхимиздат, 1957, стр. 420.
Уокер Дж. Ф., Формальдегид, Госхимиздат, 1957, стр. 148«
Bibb С. Н., Ind. Eng. Chem., 24, 10 A932).
Медведев С. С, Природные газы, № 4—5, 29 A932);
Медведев С. С, Робинзон Е. А., Труды физико-химического
института им. Карпова, т. 4, 1925, стр. 117.
10. Payne W. А., пат. США 2519788, 1950 г.
И. Montecatini, пат. ФРГ 1162344, 1964 г.
12. Le Blanc M., P 1 a s с h k e E.? Z. Electrochem., 17t 45 A911).
13. P a t г у М., М о n с e a u x P., Compt. rend., 223, 329 A946).
14. А и и с о н я н А. А., Гудков С. Ф., Е н и к о л о п я н Н. С,
Клейменов Н. А.,Маркевич А. М.,Н албандян А. Б.,
Сидоров А. П., Газ. пром., 6, 32 A957).
15. Ениколопян Н. С, Бельговский И. М., ЖФХ, 34, 1571
A960).
16. Bludwerth J. E., Celanese Corp., пат. США 2128908, 1938 г.
17. К irk pat rick S. D., Chem. Eng., 56, 91 A949).
18. H aggard H. W., J. Ind. Hyg., 5, 390 A923).
19. Wartenberg H,, Lerner-Steinberg В., Angew, Chem., 38,
591 A925).
20. S pence R., J. Chem. Soc,, 1936, 649.
21. D obratz С J., Ind. Eng. Chem., 33, 759 A941).
22. В і с h о w s k у F. R., Rossini F. D., Thermochemistry of the
Chemical Substances, Reinhold Publ., N. Y., 1936.
23. Purvis J. E., Me Cl ell and N. P., J. Chem. Soc, 101, 1810 A942).
24. Schou S. A., Henri V., Nature, 118, 225 A926); Z. Phys., 49,
774 A928).
25. Dieka G. H., Kistianowsky G. В., Phys. Rev., 45, 4 A934).
26. H u г z b e r g e G., Trans. Farad. Soc, 27, 378 A931).
27. P r і с e W. С, J. Chem. Phys., 3, 256 A935).
28. Randall H. M., Fowler R. G., Fuson N., Dangl J. R.,
Infrared Determination of Organic Structures, 49, N. Y., D. Van Nost-
rand Co., Inc., 1949.
29. К e k u 1 e A., Ber., 25, C), 2435 A892).
30. S p e n с e W., W і 1 d N.. J. Chem. Soc* 1935, 506.
31. T r a u t z M., U f e r E., J. prakt. Chem., 113, № 2, 105 A926).
32. Штаудингер Г., Высокомолекулярные органические соединения,
ОНТИ, Химтеорет, 1935, стр. 278.
33. W а 1 к е г J. F., J. Am. Chem. Soc, 55, 2825 A935).
34. S p e n с e W., W і 1 d W., J. Chem. Soc, 1935, 338.
35. T о b у V., R u t z E. F., J. Polymer Sei., 60, 169 A962).
36. С a 1 v e г t J. G., S t e а с u E. W., J. Chem. Phys., 19, 176 A951)*
37. H албандян А. Б., ЖФХ, 22, 1443 A948).
38. В owen E. J., Tietz E. L., J. Chem. Soc, 1930, 284.
39. E u 1 e r H., E u 1 e r A., Ber., 39, 50 A906).
40. Schou S. A., J. Chem. Phys., 26, 72 A929).
41. N і e 1 s e n* N., E b e r s E., J. Chem, Phys., 5, 823 A937).
42. Auerbach F., Barschall H., Studien uber Formaldehyd, T. I,
Berlin, 1905, S. 10.
43. I 1 і с e t о A., Ann. chim. Appl., 39, 703 A949).
44. В i S Ilieto A Chim Ind 33 429
45.
46
G.,
В es si S., її is et о A., Chim. Ind., 33, 429 A951).
Iliceto А., В e s s і S., €him. Ind., 33, 212 A951).
Iliceto A., Bessi S., Dallaporta N., Giarometti
Gazz. chim. ital., 81, 915 A951).
Iliceto A.j Gazz. chim. ital., 81, 786 A951); 84, 536 A954).
R., T r u m p 1 e r G., Helv. chim. acta, 30, 1860 A947).
"" P і г e t E. L., Ind. Eng. Chem., 41, 1277 A949).
С, Hess К., Вег., 67, 174 A934).
о _ _ __., Hochmolekularen organischen Verbindungen,
Berlin, 1932.
Staudinger H., Die Hochmolekularen organischen
Berlin, 1932, S. 225.
Staudinger H., Die Hochmolekularen organischen
Berlin, 1932, S. 240.
Staudinger H.? Die Hochmolekularen organischen
Berlin, 1932, S. 241.
D e s с u d e M., Ann. chim. phys., 29? 502 A903).
Sta'udinger H.y L u t h у М., Helv. chim. acta, 8, 41 A925).
R., Johner H., Luthy
Schweitzer O., Ann., 474,
В і є b er
49. H all M.
50. W a d a n о M., T г о g u s
51. S t au d і ng er H., Die
эз:
Verbindungen,
Verbindungen,
Verbindungen,
Staudinger H.'f Singer
Kern W., Russidis D.
A929).
58i Staudinger H., Singer R., Johner
Kern W., Russidis D., Schweitzer
O.,
H.,
o.,
Luthy
Ann., 474,
241
M.,
255
L u t h у М-*
Ann., 474, 221
2551365, 1951 г.; 2694076,
Staudinger H., Singer R., Johner H.,
Kern W., Russidis D., Schweitzer O.,
A929).
L o b e г і n g J., Y u n g K. P., Monatsh. Chem., 70, 281 A937).
Пат. США 2498206, 1950 г.; 2568016, 1951 г.
Пат. США 2568018, 1951 г. 2915560, 1959 г.
Пат. США 2369504, 1945 г.; 2519550, 1950 г.;
1954 г.
Пат. США 2992277, 1961 г.
Пат. США 2593862, 1956 г.
Англ. пат. 866089, 1961 г.; 895193, 1962 г.
Walker J. F., пат. США 2347447, 1944 г.
Walker J. F., Carl і si e P. J., Chem. Eng. News, 21, 1250 A943).
Delepine M., Badoche M., Compt. rend., 214, 717 A942).
Palfrey G. F., см. Уокер Дж. Ф., Формальдегид, Госхимиздат,
1957, стр. 172.
Burnett R., Bell R. P., Trans. Farad. Soc, 34, 420 A938).
Walker J. F., Ch adwick A. F., Ind. Eng. Chem., 39, 974 A947).
Scrabal A., Stockmair W., Schreiner H., Z. phys. Chem.,
A-169, 177 A934).
G і e f e г A.? Jacks V., К e r n W., Makromol. Chem., 74, 46 A964),
P r a t e z і L., Gazz. chim. ital., 14, 139 A885).
Franc С E., пат. США 2304080, 1942 г.
S о с о 1 Н., пат. США 2465489, 1949 г.
Kern W.jCherdron H.,Hohr L., Makromol. Chem., 52, 48 A962).
Травкин И. С, Беликов В.'П., авт. свид. 117583, 1958 г.
Hamm ick D. L., В о er e e А. В., J. Chem. Soc, 123, 288 A923).
Минин В., Файдель Г. И., Зисман Д. И., Пласт, массы,
№ 10, 53 A966).
Mortillaro L., Galiazo G., Bessi S., Gazz. chim. ital., 94,
109 A964).
ГЛАВА II
ПОЛИМЕРИЗАЦИЯ ФОРМАЛЬДЕГИДА
В 1892 г. Кекуле [1] наблюдал полимеризацию очищенного
жидкого формальдегида в твердый полимер, названный им Еи-
полиоксиметиленом. Свойства этого полимера были подробно
изучены Штаудингером [2], который на основании полученных данных
пришел к выводу, что 2?и-полиоксиметилен построен из линейных
молекул большой длины с концевыми группами, отличающимися
по своей природе от звеньев оксиметилена, входящих в состав молекул.
При нагревании образцов і?гг-полиоксиметилена, полученных
в различных условиях (в блоке и в растворе соответственно при
—80 и —20° С), они размягчались без четко выраженной точки
плавления, одновременно частично разлагаясь с выделением
мономерного формальдегида. В размягченном состоянии эти образцы
приобретали пластические свойства, их можно^было формовать,
изготовлять из них пленки, вытягивать нити и т. п.
Другие известные модификации полиоксиметиленов — а-, ?-, Y~
или б-полиоксиметилены при плавлении превращались в
низковязкие жидкости и не обладали пластическими свойствами.
Рентгенографические исследования показали, что пленки и
«стекла», полученные из .Егг-полиоксиметилена, имеют
кристаллическое строение, подобное тому, которое было найдено ранее при
исследовании низкомолекулярных полиоксиметиленов* [3]. Поскольку
рефлексы в случае і?и-полиоксиметилена оказались значительно
шире, было сделано заключение, что его кристаллиты имеют не
столь хорошо упорядоченную структуру, как а- или ?-полиоксиме-
тилен. Это было подтверждено измерением плотности различных
образцов при 20ф С пикнометрическим методом. Плотность «стекла»
2?и-полиоксиметилена, получаемого полимеризацией в блоке при
—80° С и обладающего, по-видимому, наибольшей степенью
полимеризации, равна 1,407 г/см*. Плотность у-полиоксиметилена
оказалась равной 1,467 г/смг.
Штаудингер представлял себе процесс получения .Еи-полиокси-
метилена как цепную реакцию присоединения молекул
формальдегида к активированным молекулам. Прекращение роста полимерной
цепи могло быть вызвано какой-либо другой реакцией. Детальный
механизм образования макромолекул полиоксиметилена не
выяснили, поскольку не удалось идентифицировать концевые группы и
определить степень полимеризации продукта из-за разложения
при растворении в формамиде и других растворителях.
30
Косвенным образом на основании изучения физических свойств
было найдено, что молекулярный вес 2?м-полиоксиметилена зависит
от температуры реакции. Так, например, продукт, полученный
полимеризацией в блоке при —80° С, оказался менее эластичным и более
твердым, чем продукты, полученные при —20 и -(-20° С. Продукт,
полученный при —(-100° С (реакция происходила спонтанно в газовой
фазе), обладал типичными свойствами а-полиоксиметилена. Таким
образом, можно полагать, что с увеличением температуры реакции
молекулярный вес полимера уменьшался.
Новейшие работы в области синтеза полиформальдегида,
проводимые с 50-х годов, ставили своей целью получение термически
стойкого высокомолекулярного полиоксиметилена, пригодного для
переработки в изделия. В связи с этим детально изучались механизм
и кинетика полимеризации формальдегида в различных
условиях, включая полимеризацию газообразного, жидкого и
растворенного формальдегида, а также его водных и спиртовых
растворов.
В соответствии с современными представлениями о строении
и свойствах полимерных веществ основное внимание в этих работах
уделялось выяснению следующих вопросов:
а) влияние условий проведения полимеризации на стабильность
получаемого продукта;
б) влияние условий проведения полимеризации на молекулярный
вес и молекулярно-весовое распределение продукта;
в) возможность образования различных химических структур
(разветвлений цепи, участков другой структуры в оксиметиленовой
цепи и т. п.);
г) природа концевых групп.
Кинетика полимеризации формальдегида изучалась при
проведении реакции в присутствии различных каталитических систем
и под действием ионизирующего облучения.
Несмотря на простоту строения полиоксиметилена и легкость его
получения, при проведении кинетических исследований приходится
сталкиваться со значительными экспериментальными трудностями.
По-видимому, этим в первую очередь и объясняется относительно
небольшое число работ по кинетике полимеризации формальдегида.
Анализ полимера и в настоящее время связан с известными
трудностями (вследствие плохой растворимости продукта и склонности
к деполимеризации). Кроме того, как уже отмечалось выше,
мономерный формальдегид крайне чувствителен к примесям,
присутствующим в реакционной системе.
ПОЛИМЕРИЗАЦИЯ В ГАЗОВОЙ ФАЗЕ
Газообразный формальдегид, полученный, например, пиролизом
параформальдегида или а-полиоксиметилена, спонтанно полимери-
зуется на холодных стенках сосудов, труб и т. п., образуя слей
белого твердого полимера.
31
Впервые полимеризацию безводного газообразного
формальдегида наблюдал Гофман [4]. Измерения давления газообразного
формальдегида при полимеризации на поверхности сосуда показали, что
кинетика этого процесса описывается уравнением реакции первого
порядка [5]. Было обнаружено, что абсолютно чистый формальдегид
имеет лишь незначительную склонность к полимеризации, но
небольшие количества влаги оказывают каталитическое действие. Точно
так же действуют следы кислот и щелочей.
Термодинамика полимеризации
Известно, что если формальдегид нагреть при атмосферном
давлении до 100—150° С, то полимеризация не происходит*. По
сообщению Карузерса и Норриша [71, кривая зависимости скорости
полимеризации газообразного формальдегида от температуры имеет
максимум в интервале 40—80° С. Эти же авторы нашли, что при
100° С давление формальдегида в реакционном сосуде понижается
не до нуля, а до некоторой равновесной величины.
Эти факты долгое время не находили удовлетворительного
объяснения, пока в работах Дейтона и Айвина [81 не была
сформулирована концепция равновесной полимеризации.
Они предположили, что акт роста цепи при полимеризации носит
обратимый характер:
\
Отсюда скорость реакции равна:
а[Щ
dt
kv[A][M]-kA[A] B)
где Ap[Al [M] — скорость роста цепи; А;д [А] — скорость
деполимеризации; [А].— общая концентрация активных центров; [М] —
концентрация мономера; &р и кА — константы скоростей роста и
деполимеризации.
Выражая константы скоростей реакций роста и деполимеризации
через уравнение Аррениуса, получим:
л -Е IRT . . -EJRT
где Ev и Ел — энергии активации прямой и обратной реакций
соответственно. Можно записать, что І?д — Ер с^ —АН0, где АЯ° —
изменение стандартного теплосодержания. В большинстве случаев
(когда тепловой эффект полимеризации положителен) ЕА > Ev,
Лд > А^ поскольку деполимеризация — мономолекулярная реак-
* Хранение газообразного формальдегида при повышенных температурах
нецелесообразно, так как при этом протекают побочные реакции и прежде всего
реакция Канниццаро.
32
ция, а ей соответствуют большие значения предэкспоненциального
множителя по сравнению с бимолекулярной реакцией роста.
Отсюда следует, что если даже при комнатной температуре
скорость деполимеризации незначительна, то при повышении
температуры она будет возрастать быстрее, чем скорость реакции роста цепи,
и при некоторой температуре, называемой предельной G^.), будет
равна скорости роста цепи. Очевидно, что скорость полимеризации
в этот момент равна нулю.
Тогда &р[А] [М] = &Д[Л], откуда
[М]р=-^ C)
где [М]р — равновесная концентрация мономера, К — константа
равновесия.
Предельная температура не является константой, так как
зависит, от концентрации мономера или от его термодинамической
активности, если реакция протекает в растворе. Приравнивая скорости
прямой и обратной реакций и подставляя в уравнение Аррениуса,
получим:
Т АЯ° D)
Я In—5j
Другое определение предельной температуры следует из общего
уравнения термодинамики для свободной энергии полимеризации:
Д?о=Д#о_ ГД^о E)
где АС?0, АН0 и AS0 — соответственно изменения стандартных изо-
барно-изотермического потенциала системы, энтальпии и энтропии
при полимеризации. Условию равновесия (скорость реакции равна
нулю) удовлетворяет положение, когда AG0 =0, т. е.
т АЯ° (м
Таким образом, предельную температуру можно определить как
температуру, при которой свободная энергия полимеризации равна
нулю *.
Рассмотренные выше представления о равновесной
полимеризации являются весьма приближенными. Например, свободная
энергия макромолекул, в отличие от свободной энергии
низкомолекулярных соединений, зависит не только от температуры и давления, но и
от таких факторов, как степень полимеризации, молекулярно-
весовое распределение, строение цепи (микротактичность, тип
* Зависимость Tnv от концентрации мономера выражается формулой Tnv =
AHO
¦, которая выводится из общего уравнения термодинамики не-
In IM]
равновесных процессов.
3 Заказ і 135. 33
присоединения молекул мономера и т. п.). Вблизи предельной
температуры необходимо учитывать вклад реакций инициирования и
обрыва цепи.
Влияние степени полимеризации на величину равновесной
концентрации мономера рассмотрено в работах Тобольского с сотр.
[88, 94]. Они получили следующее уравнение
1 J_
DP
[М]р = jp- G)
где DP — средняя степень полимеризации. Очевидно, что при
больших величинах DP это выражение будет аналогично уравнению C).
Влияние стереорегулярности на энтропию макромолекул
рассмотрено в работе [96]. Показано, что энтропия полимера со
статистическим распределением звеньев в цепи менее отрицательна (на
1,38 калі град-моль больше, чем энтропия кристаллического
полимера). Дэйнтон и Айвин[8] рассмотрели влияние изомеризации
мономера на термодинамику полимеризации. Шварц с сотр. [95]
исследовали влияние изменения константы скорости реакции роста в ходе
полимеризации.
Дэйнтон и Айвин рассматривали случай полимеризации, когда
полимер не растворим в реакционной среде и термодинамическая
активность мономера равна его концентрации. Байвотер [90, 91]
показал, что в более общем случае, когда полимер растворим в
реакционной среде, необходимо применять теорию растворов полимеров
Флори — Хаггенса [89].
Ениколопян с сотр. [92] изучали влияние степени полимеризации
и МБР на энтропию полимера. Они показали, что при низких
степенях полимеризации энтропия полимера зависит от DP:
| (8)
где т — масса полимера, R — газовая постоянная.
Влияние МБР для двух крайних случаев (равновесное
распределение и распределение Пуассона) дается уравнением:
Д? = =Aп7х?Ч-1) (9)
Кинетический анализ в области предельной температуры
является одним из способов экспериментального определения
термодинамических констант. Впервые он был применен к полиоксиметиленгли-
колю [9], поскольку это единственный полимер, у которого
равновесие мономер ї± твердый полимер достигается очень быстро.
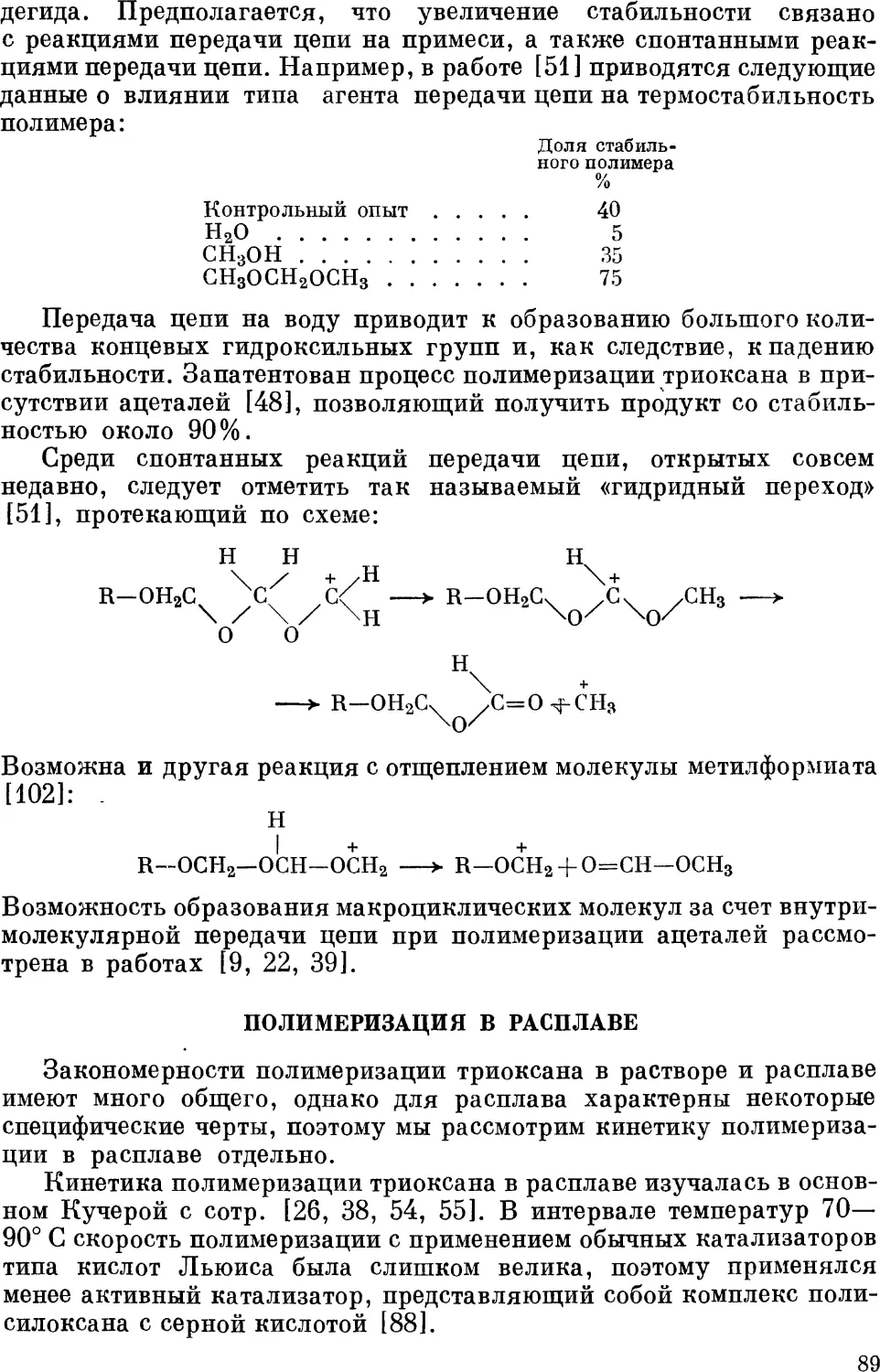
Измерения упругости паров над полиоксиметиленгликолем (рис. 4) со
степенью полимеризации порядка 100 при различных температурах
позволили рассчитать Д#г—тв. полим. по формуле
34
И A<SS_TB. полим. ПО формуле
AHO . AS*
RT + R
A1)
Вычисленные значения АН0 и A*S° составили соответственно
—A3,3 ± 1,5) ккал/молъ и —C2,5 ± 4) калі моль • град.
Предельная температура при атмосферном давлении равна 137° С.
Впоследствии эти данные
неоднократно проверялись другими
исследователями с помощью калориметрических
методов, по теплотам сгорания, а также
по зависимости равновесного давления
паров над полимером от температуры
(табл. 3).
Обращает на себя внимание
значительное расхождение величин
термодинамических констант даже при использовании
одного и того же метода измерения.
Сопоставляя величины энтальпии
полимеризации, полученные путем измерения
тангенса угла наклона кривой в координатах
In [М] =- для образцов полиЬксиме-
Z2
2,0
1,8
к*
l,b
1Л
1,2
4П
\
\
\
\
\
\
U2,7 2,8 2,9 3,0 3,f 3,2
тилена, синтезированных различными
способами, можно сделать вывод, что
равновесное давление формальдегида над
Рис. 4. Зависимость
равновесного давления паров
формальдегида над твердым
полиоксиметиленгликолем
от температуры [73].
ТАБЛИЦА 3
Термодинамические константы полимеризации формальдегида
Метод измерения
По равновесному давлению
паров над параформальдеги-
дом по данным [10]
По равновесному давлению
паров над 2?м-полиоксимети-
леном
Калориметрический * метод
Определение теплот сгорания *
По равновесному давлению
паров над а-полиоксиметиленом
По равновесному давлению
паров над орторомбическим
полимером с 100%-ной
степенью кристалличности
АЯ°
ккал/моль
— A3,3 ±1,5)
—12,24
—
—13,23
— 16,3
-17,3-18,0
-17,2
AS0
кал / моль' град
— C2,5 ±4)
- C0,66 ± 0,22)
-41,8
-41,6
—41,8
—
-43,8
¦'пр.
°С
137
126
—
—
117
—
133
Литература
9
11
12
13
14
15
16
* Образец—дельрин; AS0 для реакции образования дельрина из газообразного
формальдегида, вычисленное по опубликованным в литературе значениям энтропии
мономера E2,26 пал}моль*град [77]) и полимера A0,61 кал/моль• град [12]), равно
—42,65 кал/моль'град.
3* 35
полимером уменьшается с ростом степени кристалличности*. Это
подтверждают данные, полученные Уокером [16].
Таким образом, отсутствие полимеризации мономерного
формальдегида при температурах выше 130—150° С объясняется
термодинамическими факторами, так же как и существование равновесной
концентрации мономерного формальдегида над твердым полимером
при более низких температурах. Очевидно, что предельная
температура полимеризации формальдегида будет повышаться с увеличением
давления в реакционной системе.
Кинетика полимеризации
Впервые кинетику полимеризации газообразного формальдегида
в присутствии муравьиной кислоты изучали Карузерс и Норриш [7].
Они обнаружили, что реакция протекает на поверхности сосуда при
м температуре около 90° С.
Образовавшийся твердый полимер
имел температуру плавления 157° С и был
похож на описанный Штаудингером
продукт, полученный полимеризацией при
100° С.
Скорость реакции имела
приблизительно второй порядок по формальдегиду.
Муравьиная кислота оказалась сильным
катализатором. Полимеризация
происходила по типу разветвленной цепной
реакции. С увеличением концентрации кислоты
наблюдалось резкое возрастание скорости
реакции (рис. 5). Была предложена
следующая схема реакции:
инициирование (на охлажденной
поверхности):
ои
50
30
20
Ю
/
и Ю 20 30 ЬО
Давление НСООН, мм рт. ст.
Рис. 5. Зависимость
скорости газофазной
полимеризации СН2О от
концентрации HGOOH [7].
Нс<о
+ СН2О
<осн2он
о
рост цепи:
HCOO(CH2O)nCH2OH-f СН2О —?> HGOO(GH2O)w+iCH2OH
обрыв цепи:
НСОО(СН2О)йСН2ОНфСН2О —> HGOO(GH2O)WGH2GHO + H2O
разветвление цепи:
НСОО(СН2О)ЯСН2ОН +
Н
НО
НСОО(СН2О)ПСН2ОНС
ОН
он
* Сам по себе вклад кристаллизации в энтальпию полимеризации не может
превышать 1,78 ккал/моль (величина теплоты плавления).
36
Если [А] — концентрация активных центров, то отдельные стадии
полимеризации можно записать в виде следующих уравнений:
= *і[НСООН1[СН2О] A2)
A3)
A4)
р A5)
Предполагая, что устанавливается стационарная концентрация
активных центров, получим:
^У- = кх [HGOOH] [СН2О] — &з [Л] [СН2О] + &4 [А] [НСООН] = О
fr [НСООН] [СН,О]
й3[СН2О]«-fr [НСООН] К 0)
Общая скорость полимеризации (в случае, когда длина
полимерных цепей велика) выражается уравнением:
d [СН2О] к,к2 [НСООН] [СН,О]«
dt = ЫЛ] [СН2°]-^[СН2О]^^ [НСООН]
Это уравнение соответствовало полученным экспериментальным
данным. Вода и уксусная кислота оказывали действие, сходное с
действием муравьиной кислоты.
Предложенная авторами кинетическая схема объясняет
автоускоренный характер процесса реакцией активных центров с
муравьиной кислотой. Последняя в этом случав выступает и в роли
инициатора полимеризации, и в роли агента, ускоряющего роста цепи. Эта
схема существенно отличается от классической схемы автокатализа
в цепных реакциях. Очевидно, что максимальная скорость процесса
будет определяться (по этой схеме) концентрацией кислоты, даже
если пренебречь реакцией гибели цепи.
Позднее было изучено каталитическое действие хлористого
водорода, фтористого бора и хлорного олова на полимеризацию
газообразного формальдегида [17]. Эти катализаторы оказались еще
более активными, чем муравьиная кислота, хотя характер процесса
(включая автоускорение при увеличении концентрации
катализатора) такой же.
Подобие рассматриваемых процессов позволило предложить для
них тот же механизм полимеризации, что и для случая катализа
муравьиной кислотой. Однако механизм разветвления, предложенный
для муравьиной кислоты, не применим к реакции, катализируемой
хлористым водородом и катализаторами Фриделя — Крафтса. В
последнем случае предполагалось, что, как и при кислотном гидролизе
ацетальных связей, эти катализаторы могут расщеплять полимерную
цепь на два фрагмента, способных к дальнейшей полимеризации:
инициирование:
СН2О + НС1 —>- С1СН2ОН
37
рост цепи:
C1(CH2O)WCH2OH + CH2O —> C1(GH2O)W+1GH2OH
обрыв цепи:
G1(CH2O)WGH2OH + GH2O —>• С1(СН2О)ЯСН2СНО ^ Н2О
разветвление цепи (кинетически):
Cl(GH2O)w+mGH2OH + HGl —>¦ С1(СН2О)яСН2ОНфС1(СН2О)ш.1СН2ОН
Неустойчивость полимера к действию кислотных катализаторов
подтвердилась прямыми опытами при 100° С, когда скорость
разложения полимера увеличивалась с ростом концентрации
катализатора.
Анализ экспериментальных данных показывает, что
лимитирующим фактором в этих реакциях является диффузия. Так, например,
скорость полимеризации при применении в качестве катализаторов
фтористого бора, хлорного олова и соляной кислоты увеличивалась
при повышении концентрации катализатора и достигала некоторого
предельного значения. При этом предельные значения скоростей
полимеризации были одинаковы во всех трех случаях, но
достигались при концентрации BF3 — 0,1%, SnCl4 — 1,0% и HG1 — 5,0%.
Предложенный в работах Карузерса, Норриша и Бевингтона
механизм газофазной полимеризации формальдегида в настоящее
время мало убедителен по нескольким причинам:
1) предполагается, что инициирование происходит в газовой
фазе, хотя известно, что процесс гетерогенный и полимер всегда
образуется на стенках реакционных сосудов. Кроме этого, скорость
полимеризации пропорциональна величине поверхности;
2) вода рассматривается как неактивный компонент (при обрыве
цепи), что противоречит экспериментальным фактам*;
3) образование С—С-связей и разветвленных макромолекул,
предполагаемое при газофазной полимеризации формальдегида, не
было доказано аналитическим путем и, судя по имеющимся в
настоящее время данным, маловероятно.
Механизм инициирования и обрыва цепей при газофазной
полимеризации остается невыясненным. То же самое следует сказать
о природе активных центров. Инертность радикальных инициаторов
и в то же время каталитическое действие, оказываемое кислотами и
щелочами, позволяют предполагать, что это ионный процесс.
Открытым остается вопрос о возможности протекания «гемифор-
мальной», ступенчатой полимеризации путем взаимодействия
концевых гидроксильных групп.
Одна из причин, затрудняющих исследования механизма
газофазной полимеризации формальдегида, заключается в протекании
спонтанной полимеризации. Этому вопросу были посвящены работы
* Известно, что степень полимеризации образующегося полимера обратно
пропорциональна концентрации воды в реакционной системе.
38
Сотери [18]. Он пришел к выводу, что характер поверхности, на
которой протекает реакция, играет большую роль в механизме
полимеризации. Поэтому было трудно определить эффективный порядок
реакции и другие кинетические константы, так как отсутствовала
воспроизводимость результатов. В этом смысле лучшие результаты
получили Тоби и Рутц [19] при проведении реакции на
поверхности кварца, который перед опытами нагревали до температуры
красного каления и вакуумировали. Примеси в мономерном
формальдегиде, к сожалению, не анализировали; пригодным для опыта
считался образец жидкого формальдегида, в котором после
недельного хранения при —78° С отсутствовали следы полимера.
При полимеризации в интервале температур 0—40° С скорость
изменения давления формальдегида в системе подчинялась
эмпирическому уравнению:
(F0-F)n = kt при п>1 A8)
где Fo — начальное давление; F — давление в момент времени t.
Значения к1ч вычисленные при нескольких температурах па
уравнению
Fo-F^ht0'28^0'0* A9}
представлены ниже [19]:
Температура, °С 0 22 30 40
&1-10, моль/л 1,5 1,6 1,7 2,8
Кинетику спонтанной полимеризации формальдегида исследовал
Ениколопян с сотр. [45]. Опыты проводились в стеклянных ампулахг
нагретых до заданной температуры. Формальдегид получали
пиролизом а-полиоксиметилена при 150—180° С; его очищали, пропуская
через колонку, заполненную силикагелем, при —15° С, и
конденсировали, а затем дважды перегоняли. Скорость реакции измеряли
по падению давления мономера в реакционной системе.
Попытки регенерировать поверхность стекла (пирекс) для
получения воспроизводимых результатов окончились неудачей, так как
даже после прокаливания поверхность сохраняла повышенную
активность, инициируя полимеризацию мономера. Однако было
найдено, что если поверхность не очищать от пленки полимера, то
результаты достаточно хорошо воспроизводятся.
Добавление воды приводило к резкому возрастанию скорости
реакции, которая линейно зависела от концентрации воды. Через
некоторое время (после того как степень конверсии достигала 15—20%)
наблюдалось замедление реакции. Полимер, образующийся в
результате реакции, протекающей с большой скоростью, растворялся
в ацетоне на холоду, т. е. содержал не более 10—12 звеньев
формальдегида в цепи. Полимер, полученный медленной полимеризацией
очищенного формальдегида на поверхности с температурой от —15
до +50° С, по свойствам приближался к .Ец-полиоксиметилену.
Иная картина наблюдалась при полимеризации газообразного
формальдегида в присутствии четыреххлористого титана и
триэтиламина. Реакция протекала с большой скоростью (без
торможения) с образованием высокомолекулярного полиоксиметилена.
На основании этих данных можно предположить, что вода
принимает участие и в актах инициирования, и в актах ингибирования
процесса. Низкий молекулярный вес образующегося в ее присутствии
полиоксиметиленгликоля можно объяснить реакцией передачи цепи
на примесь (вода).
Ингибирование процесса полимеризации может происходить
в результате дезактивации активных центров, сорбированных на
поверхности, слоем низкомолекулярного полимера
(физико-статистическая дезактивация), который удаляется при обработке поверхности
б вакууме при 100° С. В этом случае полимеризация свежей порции
-очищенного мономера происходит без торможения и образуется
высокомолекулярный полиоксиметилен.
ПОЛИМЕРИЗАЦИЯ В РАСТВОРЕ
Штаудингер описал полимеризацию формальдегида в
абсолютном эфире и показал, что продукты, полученные полимеризацией
в растворе при низких температурах, аналогичны продуктам
полимеризации в массе, но имеют меньший молекулярный вес и не обладают
способностью образовывать пленки.
Добавки веществ основного (амины) и кислотного (соляная
кислота) характера приводили к ускорению реакции, что заставляло
предполагать ионный механизм полимеризации жидкого
формальдегида. Известно, что инициаторы свободнорадикального типа не
вызывают полимеризации формальдегида, но разрушают его. Это
совпадает с теоретическими представлениями о реакционной
способности карбонильной группы в альдегидах и кетонах. Средние
значения энергии диссоциации С=С- и С = О-связей, а также С—С- и С—0-
связей достаточно близки. Вместе с тем известно, что в отличие от
винильной связи в молекулах олефинов карбонильная группа сильно
поляризована. При атаке молекулы, имеющей ненасыщенные связи,
свободным радикалом конкурирующими реакциями являются
реакции замещения и присоединения к двойной связи. Реакции
замещения связаны со сравнительно большими стерическими
затруднениями (по сравнению с реакциями присоединения). В случае
олефинов скорость реакции присоединения выше за счет очень низких
значений энергии активации B—4 ккал/молъ). Соответственно
реакции замещения, имеющие более высокую энергию активации,
начинают доминировать только при повышенных температурах.
Присоединение свободного радикал я к С=О-связи происходит значительно
труднее (энергия активации 7—8 ккал/молъ). Тепловой эффект этой
реакции составляет всего 3—10 ккал/молъ. В то же время отрыв
атома водорода от молекулы формальдегида и присоединение его
к радикалу сопровождаются выделением большого количества тепла
C8 ккал/молъ) и соответственно энергия активации этой реакции
равна всего 0,5 ккал/молъ.
40
Отсюда следует, что при достаточно высоких температурах
должна наблюдаться инверсия этих процессов. Действительно^
литературе имеются соответствующие данные. Например, при
полимеризации высших альдегидов при высоком давлении, которое позволяет
сдвинуть предельную температуру *, процесс ускоряется при
введении свободных радикалов и замедляется при введении ингибиторов
[18]. Аналогичные данные получены для формальдегида при
полимеризации под давлением порядка 1000 атм и температуре 200° С.
Разработанный в последние годы способ полимеризации
формальдегида заключается в том, что очищенный от влаги и других
примесей газообразный формальдегид вводится в сосуд, в котором
находится раствор инициатора в неполярном растворителе. Процесс
проводится при комнатной температуре и интенсивном
перемешивании.
Как известно, формальдегид при температурах выше температуры
кипения (—19° С) плохо растворим в инертных растворителях.
Поэтому стационарная концентрация формальдегида во время
полимеризации не превышает 0,5—3,0% от веса растворителя. Несмотря
на это, реакция протекает с высокой скоростью и практически
количественно.
Авторы работы [20] отмечают, что полимер, полученный по
описанному выше способу, по свойствам (стабильности, прочности
и т. д.) превосходит і?м-полиоксиметилен и продукты, полученные
Штаудингером полимеризацией в растворе при низких
температурах. Для объяснения этого приводятся следующие соображения:
новый полимер имеет больший молекулярный вес; исходные
реагенты лучше очищены. Как предполагается [20, 21], рост
полимерной цепи прекращается при взаимодействии активного конца
полимерной молекулы с примесями, имеющимися в системе, например
с водой. При этом происходит передача цепи и образуется неактивная
молекула полимера и новый ион, способный присоединять молекулы
мономера [21]. Другими словами, при полимеризации
формальдегида длина кинетической цепи значительно больше длины
материальной (полимерной) цепи.
Передача цепи молекулами воды в процессе полимеризации,
инициированной анионами, может быть изображена следующей
схемой:
.. ¦ — сн2о-фн2о —>¦ ..
.-сн2он+ноно- +сн2о —> носн2о-
НОСН2О- + гсСН2О —>¦ НО(СН2О)ЯСН2О-
Таким образом, предполагается, что молекулярный вес полимера
зависит от концентрации и активности агентов передачи цепи,
присутствующих в реакционной системе.
* ГПТ) при полимеризации высших альдегидов равна приблизительно
-20° С.
41
Катализаторы полимеризации
Растворы формальдегида спонтанно полимеризуются при
хранении, причем скорость образования полимера зависит от степени
очистки раствора и от температуры хранения. До сих пор отсутствуют
сколько-нибудь надежные данные о возможности протекания
полимеризации формальдегида по радикальному механизму при
давлении, близком к атмосферному. Типичные радикальные катализаторы
{например, перекись бензоила) вызывают разложение мономера.
В то же время вещества кислотного и основного характера легко
инициируют полимеризацию формальдегида. На основании этого
считают, что полимеризация в растворе протекает по ионному
механизму.
В патентной литературе описано большое количество веществ,
катализирующих полимеризацию формальдегида в широком
интервале температур (табл. 4). Ионная природа большинства из них не
ТАБЛИЦА 4
Катализаторы полимеризации формальдегида
Класс соединений
Типичные примеры
Особые свойства
катализаторов
№ патентов
Амины
Соединения
четвертичного
аммония
Мочевина и ее
производные (кар-
бамидные
соединения)
Амиды и имиды
кислот, лактамов,
уретанов, карбо-
диимиды
Продукты
конденсации аммиака
с альдегидами
Органические
соединения
фосфора, мышьяка и
сурьмы
42
Первичные,
вторичные и третичные,
алифатические,
ароматические (три-
этиламин, N, N-ди-
метиламинсэтанол,
дифениламин,
анилин и т. п.)
Тетра-н- бути лам-
монийлаурат, диме-
тилдиалкиламмоний-
ацетат
Тиомочевина, изо-
мочевина,
полимочевина, тетраэтил-
мочевина, тиокарба-
мид, диалкиловый
эфир карбаминовой
кислоты
Диизопропилкар-
бодиимид
Гексаметилентетр-
амин
Трифенилфосфин,
трифенилстибин, три-
фениларсин
Весьма
чувствительны к
примесям
По
эффективности близки к
аминным
В некоторых
случаях помимо
каталитического
эффекта обладают
стабилизирующим эффектом
Пат. США
2768994 A956)
2734889 A956)
2828286 A958)
2841570 A958)
Пат. Англии
793673 A958)
796863 A958)
Пат. ФРГ
1176860 A964)
Пат. Англии
873673 A961)
875722 A961)
876515 A961)
Пат. Англии
934331 A963)
Пат. США
3072610 A963)
Пат. Англии
921670 A963)
Пат. США
2768994 A958)
Продолжение табл. 4
Класс соединений
Типичные примеры
Особые свойства
катализаторов
Карбонилы
металлов VIII
группы
Алкоголяты
металлов
Металлалкилы
Окиси,
гидроокиси и соли
щелочных и
щелочноземельных
металлов
Алюминий и его
соединения
Соли и гидрат
двухвалентного
олова, соли Zn,
Си, -Cd, Mn, Ni,
Sb и карбоновых
кислот, в которых
количество атомов
углерода больше 3
Кислоты
Льюиса, комплексы
кислот Льюиса с
основаниями
Карбонилы
железа, никеля, кобальта
Алкоголят
алюминия
Триэтилалюминий,
дифенилолово
Едкое кали, окись
кальция, стеарат
кальция, ацетат
натрия
Амальгама
алюминия, его сплавы,
окись и гидроокись
Соли ксантогено-
вой, дитиокарбами-
новой, стеариновой
кислот
BF3—трибу ти л-
амин, SnCl4—три-
этиламин, BF3Et20
Обладают
высокой
избирательной
способностью
Катализаторы
катионного типа
Пат. США
2734889 A958)
Пат. США
2848437 A958)
Пат. Англии
766629 A956)
Пат. ФРГ
1153903 A962)
Пат. Англии
883805 A961)
950782 A964)
Пат. Англии
857321 A960)
Пат. Франции
1300492 A962)
1322460 A962)
1371066 A964)
Пат. США
3002952 A961)
3030338 A962)
вызывает сомнения. В некоторых случаях, например при
полимеризации, инициированной окислительно-восстановительной системой
(перекись -\- амин), такой вывод кажется необоснованным. Однако
показано, что амин сам является эффективным катализатором,
а перекись отдельно не инициирует полимеризацию формальдегида.
Совместное действие этих веществ приводит к некоему синергиче-
скому эффекту. Можно полагать, что перекись в данном случае
является промотором.
Наиболее общее деление ионных катализаторов подразумевает
деление на анионные и катионные. И те, и другие весьма активны
по отношению к формальдегиду, но имеются данные о том, что при
прочих равных условиях в присутствии анионных катализаторов
получают более высокомолекулярные продукты [40].
Это явление может быть объяснено двумя факторами: во-первых,
катионные катализаторы вызывают разложение полиформальдегида
уже при комнатной температуре, а во-вторых, анионные
растущие активные центры, по-видимому, более устойчивы к действию
различных реагентов, чем катионные. (Второй вывод можно сделать
43
по аналогии с другими ионными полимеризационными процессами,
например, с полимеризацией стирола.)
Особое место занимает ряд новых катализаторов,
запатентованных фирмой «Байер» [56—58]. Это соли двухвалентного олова и
ряда металлов, не входящих в группы щелочных и
щелочноземельных металлов (цинк, кадмий, медь, свинец, марганец, железо и др.)»
и высших карбонових кислот. Специфичность действия таких
катализаторов заключается в пониженной чувствительности к воде,
метанолу, кислотам и другим примесям, которые являются
эффективными агентами передачи цепи в случае применения других
катализаторов. Даже в присутствии 1% воды или 5% метанола средне-
весовой молекулярный вес полимера был равен 100 000.
Имеющиеся в настоящее время экспериментальные данные еще
не позволяют выяснить детально механизм этого процесса. По
некоторым данным полимеры, получаемые на этих катализаторах,
отличаются очень широким молекулярно-весовым распределением.
В последнее время большой интерес вызывают также
катализаторы типа металлалкил. Предполагается, что некоторые из них
способны активировать реакцию передачи цепи на мономер, которая
протекает аналогично реакции Канниццаро. Таким образом, в
молекулах полиоксиметилена появляются формиатные и метоксильные
концевые группы. Подобным свойством обладают катализаторы
байеровского типа, алюминийалкилы и некоторые кислоты Льюиса.
Большое значение имеет выбор растворителя, который может
существенным образом влиять на кинетику и механизм
полимеризации, на дисперсность получаемого полимера и другие
технологические свойства. Растворитель должен быть нейтрален по отношению
к мономеру и катализатору. Очевидно, что этому удовлетворяют
различные классы веществ — алифатические, циклические и
ароматические углеводороды, галоидзамещенные и другие производные
углеводородов, эфиры, ангидриды и др.
Кинетика полимеризации
Кинетику полимеризации формальдегида в растворе обычно
изучают при низких температурах [41—43], что позволяет в широких
пределах варьировать концентрацию мономера. В этом случае можно
применить дилатометрический метод для снятия кинетических
характеристик, что затруднено, если полимеризацию проводить при
комнатной температуре.
Несмотря на тщательную очистку формальдегида (суммарное
содержание примесей должно быть ниже 0,01%),
удовлетворительная воспроизводимость результатов наблюдается только в сериях
опытов, проводимых с одной порцией очищенного мономера.
Считается приемлемым разброс результатов ±15%..
В качестве типичных анионных катализаторов наиболее полно
были исследованы амины [74]. Можно предполагать, что иницииро-
44
вание полимеризации формальдегида в этом случае может протекать
или за счет свободной пары электронов по схеме
r
R"
сн2 =о
(в этом случае полимеризацию ведет свободный ион), или за счет
реакции амина с химическим сокатализатором (например, водой)
и образования ионной пары:
R3N f НОН —>¦ R3NH + OH"
OH- + CH2=O —>- HO—CH2—0"
Сравнение каталитической активности аминов различного
строения и различной основности [74] показывает, что при одинаковых
основностях (ipKb) возможны значительные расхождения в выходах
полимеров при прочих равных условиях. Это как будто
свидетельствует о влиянии стерических затруднений при инициировании,
что в свою очередь можно истолковать как косвенное указание на
прямое инициирование. В то же время некоторые кинетические
данные позволяют предполагать, что химический сокатализатор при
инициировании полимеризации аминами необходим.
Рассмотрим результаты исследования кинетики полимеризации
формальдегида в растворе, опубликованные двумя группами
авторов [41—43, 45—52]. Чешские исследователи использовали раствор
СН2О в диэтиловом эфире концентрацией от 2,4 до 5 моль/л при
—58° С. Ими были взяты следующие катализаторы: тетрабутилам-
монийлаурат (ТБАЛ), дибутиламин (ДБА) и трибутиламин (ТБА).
При этом предполагалось, что ТБАЛ при ионизации образует
ионные пары, а амины обладают свободной парой электронов, т. е.
механизм действия этих катализаторов должен различаться.
Были найдены следующие общие закономерности полимеризации:
1. Зависимость скорости полимеризации от концентрации ТБАЛ
имеет нелинейный характер. Порядок реакции 0,8.
2. Зависимость скорости полимеризации от концентрации ДБА
имеет линейный характер, но существует некоторая критическая
концентрация катализатора, ниже которой реакция не идет, т. е.
часть катализатора расходуется на реакции с примесями (рис. 6).
3. Порядок реакции по мономеру был близок ко второму, но
расхождения в различных сериях опытов были весьма значительными
(от 1,7 до 2,6 в случае ДБА). Общий порядок реакции повышался
с глубиной реакции до 4,6.
Каталитическую эффективность находили как отношение
скорости реакции к концентрации катализатора. Для ряда
ТБАЛ : ТБА : ДБА она равна 4,0 : 3,5 : 1,0. Эффективная энергия
активации составляет 4,1 ккал/молъ.
45
Приведенная вязкость полимера определялась при 90 С в смеси
тетрахлорэтана с фенолом C:1). Она не зависела от типа
катализатора и его концентрации (в широких пределах). Зависимость
приведенной вязкости от конверсии во многом определялась степенью
очистки реагентов. До 40—50% конверсии молекулярный вес
незначительно увеличивался или уменьшался, но затем резко
уменьшался. Авторы не нашли удовлетворительного объяснения этому
явлению.
Для выяснения зависимости молекулярного веса от
концентрации мономера были проведены контрольные опыты с добавкой
метанола (агент передачи цепи). Если
концентрация метанола была постоянной, а
концентрация мономера увеличивалась,
то приведенная вязкость возрастала
практически линейно. Если отношение
концентраций метанола и мономера
оставалось постоянным, то приведенная
вязкость не зависела от концентрации
мономера.
Таким образом, было найдено, что
степень полимеризации
пропорциональна концентрации мономера и обратно
пропорциональна концентрации агента
передачи цепи.
Веселы и Мейзлик [44] приводят
кинетическую обработку результатов
работ [41—43] согласно следующей схеме:
зарождение цепи:
25
20
• 15
/
л
•
/°
У
2 3
Концентрация
катализатора-W6, моль/л t
Рис. 6. Зависимость
начальной скорости полимеризации
СН2О от концентрации
катализатора [47] при—59° С:
1 — ТБАЛ, концентрация СН2О
4,55 моль/л) 2— ДВА,
концентрация СН2О 4,20 моль/л.
А-СН2О-
рост цепи:
передача цепи:
повторное зарождение цепи:
х- + сн2о
.—ОСН2О-
Х-СН2СГ
Однако эта схема недостаточно хорошо согласуется с
экспериментальными данными самих авторов.
В серии работ Ениколопяна с сотр. [45—52] условия
полимеризации были заметно изменены. Мономер растворяли в толуоле,
температура реакции —30° С, раствор содержал около 80%
формальдегида. В качестве катализаторов использовали ТБАЛ, триэтиламин
(ТЭА), диэтиламиноэтанол (ДЭАЭ) и стеарат кальция (GK).
Кинетические кривые полимеризации строились путем
графического вычитания кривой спонтанной полимеризации (которая всегда
46
43
0,2
$
имела место) из соответствующей кривой каталитической
полимеризации. Таким путем для полимеризации в присутствии ТБАЛ и СК
получены воспроизводимые результаты, но для случаев
полимеризации, инициированной аминами, этот метод оказался непригодным.
Из полученных данных был сделан вывод о том, что кинетика
полимеризации формальдегида в присутствии аминов зависит от
характера и количества примесей в реакционной системе. Возможно,
что ТБАЛ и СК не требуют специального химического сокатализа-
тора, в то время как для аминов он необходим и его роль выполняют
примеси (например, вода). Исходя из начальных скоростей
полимеризации при одинаковом соотношении
концентраций мономера и
катализаторов, можно заключить, что активность
катализаторов падает в ряду: ТЭА^>
>ТБАЛ> СК.
Для случаев полимеризации в
присутствии ТБАЛ и СК найден общий
первый порядок реакции. Для ТЭА
общий порядок больше единицы.
Специальным кинетическим приемом было
показано, что первый порядок не
связан с тем, что условия реакции близки
к полимеризации в массе, где (если
полимер нерастворим в мономере, как
это имеет место в данном случае)
скорость процесса всегда пропорциональна
количеству мономера [47].
На рис. 7 показана зависимость начальной скорости
полимеризации от концентрации катализатора (СК). Как видно, скорость
реакции имеет первый порядок по концентрации катализатора. В работе
[49] показано, что эта реакция первого порядка также и по
мономеру. Эффективная энергия активации [45] — около 9 ккал/молъ.
Общий первый порядок реакции, а также первый порядок по
катализатору и мономеру может иметь место*олько в двух особых
случаях: когда не происходит гибель активных центров или когда
в системе присутствует ингибитор, расход которого приводит к
общему первому порядку. Совокупность экспериментальных данных
показывает, что при полимеризации формальдегида в присутствии
катализаторов типа ТБАЛ и СК активные центры реакции не
погибают.
Зависимость средневесового молекулярного веса (он вычислялся
по формуле [г]] = 4,44-10 Ma>6u) от степени превращения (рис. 8)
существенным образом отличалась от наблюдавшейся чешскими
исследователями, поскольку в ходе реакции происходил постоянный
рост молекулярного веса.
Рост средней степени полимеризации с глубиной превращения
может происходить в случае полимеризации «живущих» полимеров,
когда отсутствуют передача цепи и гибель активных центров.
47
u 0,2 0,4
Концентрация катализа-
тора-103, моль/я
Рис. 7. Зависимость начальной
скорости полимеризации СН2О
B3,3 моль!л) от концентрации
стеарата кальция [47] при
-30° С.
Кинетический анализ полимеризации формальдегида,инициированной
стеаратом кальция, показывает, что в этом случае увеличение
молекулярного веса объясняется другой причиной, а именно расходом
примесей (агентов передачи цепи). Усредненное значение средне-
числовой степени полимеризации описывается уравнением [63]:
Klo
Klo [і-A-а)*»/кр]
B0)
где [М]о — начальная концентрация мономера; [ск]0 — начальная
концентрация катализатора; [х]0 — начальная концентрация при-
/
/
У
о
W 20 30 W 50
Выход продукта, %
Рис. 8. Зависимость средневе-
сового молекулярного веса от
степени превращения [48]
(катализатор — стеарат кальция,
концентрация СН2О23,3 молъ/л,
температура —30° С).
k/o
/ 3 5
[ДЭДЭ]-1О\ моль/л
Рис. 9. Зависимость начальной
скорости полимеризации СН2О
от концентрации диэтиламино-
этанола [52].
меси — агента передачи цепи; а = ([М]о — [М])/ [М]о — степень
превращения; кп — константа скорости передачи цепи; /ср — константа
скорости роста
Значения DPW, вычисленные для нескольких частных случаев,
приведены в работе [53]. Строгая количественная интерпретация
полученных результатов затруднена из-за недостаточно хорошей
воспроизводимости экспериментальных данных и присутствия в
реакционной системе нескольких примесей с различными константами
передачи цепи. Однако очевидно, что, если kjkv больше единицы,
средний молекулярный вес должен расти в ходе реакции.
Полимеризация раствора формальдегида в тех же условиях
в присутствии диэтиламиноэтанола (ДЭАЭ) изучалась в работе [52].
Общий порядок реакции выше третьего, т. е., как и в случае
применения ДВА и ТЭА, полимеризация с ДЭАЭ является весьма
сложным процессом. Порядок реакции по катализатору равен
единице. Характерно наличие порогового эффекта концентрации
катализатора (рис. 9) подобно отмечавшемуся при полимеризации
48
с ДВА [42]. Следовательно, происходит частичная дезактивация
катализатора примесями.
Кинетические кривые зависимости молекулярного веса от
степени превращения для двух концентраций катализатора показаны
на рис. 10. В начале процесса наблюдается быстрый рост
молекулярного веса, затем при степени превращения около 10% происходит
резкое уменьшение молекулярного веса, который в дальнейшем
остается постоянным.
Некоторые данные о кинетике полимеризации формальдегида
в растворителях различной полярности при —78° С приводятся
в работе [54]. Авторы наблюдали
спонтанную полимеризацию
формальдегида, скорость которой увеличивалась
с повышением температуры.
Приводятся данные об активности ряда
анионных катализаторов. Наиболее
активными в неполярном растворителе (ди-
этиловый эфир, толуол) оказались
третичные фосфины (трифенилфосфин).
Амины, несмотря на большую
основность, уступали по активности фосфи-
нам в неполярных, но оказались более
активными в полярных (ацетон)
растворителях. В ряду аминов активность
уменьшалась от третичных к
первичным. Катализаторы, образующие карб-
анионы или ионные пары, типа металл-
органических соединений (бутиллитий)
или алкоголятов щелочных металлов,
обладали еще меньшей активностью.
Порядок реакции определяли
при полимеризации в присутствии три-
бутиламина. Реакция имела первый порядок по катализатору;
и мономеру. Энергия активации брутто-процесса составляла
3,9 ккал/молъ.
Сравнение экспериментальных данных с теоретическими
показывает, что начальный участок кривой зависимости молекулярного
веса полимера от степени превращения соответствует механизму
полимеризации, учитывающему одновременное существование в
реакционной системе «живущего» и «неживущего» полимеров и участие
примесей в реакциях инициирования и передачи цепи в качества
сокатализаторов. Однако снижение молекулярного веса с
увеличением выхода продукта нельзя объяснить с точки зрения предлага-:
емого механизма.
Если предположить, что в ходе процесса образуется агент
передачи цепи, причем константа скорости реакции его образований*
(кх) значительно меньше константы скорости передачи цепит то пояу-*>
ченная таким образом реакционная схема качественно описываем
10 20 > SO
Выход продукта, %
Рис. 10. Зависимость средне-
весового молекулярного веса
от степени превращения [52]
в присутствии катализатора
ДЭАЭ: :
1—2,8 х 10~4 моль/л; 2—1,1 х
х Ю-4 моль/л.
экспериментальные данные. Выражение для средней степени
полимеризации можно записать следующим образом:
Я7Г [МІР
п~~~
[ся]о l-
B1)
д
\
1 ¦_
¦на
0 0,3 0,6 0,9
Степень превращения
Рис. 11. Теоретическая кривая
зависимости средней степени
полимеризации от степени
превращения [52] (катализатор ДЭАЭ).
где q=(ku ¦ kjkl)(№]o/lcjo).
Теоретическая кривая зависимости средней степени
полимеризации от степени превращения приведена на рис. 11. Анализ
полученных данных позволяет сделать заключение, что мы еще весьма
далеки от понимания механизма полимеризации формальдегида.
Наблюдающееся противоречие в
результатах, вероятно, связано прежде
всего с неадекватностью степени
очистки реагентов в различных
опытах. Действительно, даже в
последних работах степень очистки
мономера определялась качественно (по
тенденции к самопроизвольной
полимеризации при хранении раствора).
Данные о катионной
полимеризации формальдегида в растворе еще
более скудны. До последнего
времени считалось, что из-за большой
чувствительности к примесям
катализаторы этого типа бесперспективны.
Однако успехи в области катионной
сополимеризации формальдегида
заставили пересмотреть это отношение.
Вг настоящее время опубликованы две работы в этой области [54, 75],
В первой из них приводятся качественные данные о сравнительной
активности ряда катализаторов, который включает кислоты Льюиса,
минеральные кислоты, ацетилперхлорат (особенно активный при
полимеризации триоксана) и иод. Этот ряд выглядит следующим
образом:
SnCl4 > СН0СОСЮ4 > FeCl3 > BF3Et20 > H2SO4 > I2
В работе [75] приводятся некоторые данные о кинетике
полимеризации формальдегида в растворе уайт-спирита при комнатной
температуре в присутствии хлорного олова. Учитывая, что
растворимость формальдегида в этих условиях не превышает 1% от веса
растворителя и выход полимера зависел от числа оборотов мешалки
реакционного сосуда (в котором находился раствор катализатора
в уайт-спирите и куда непрерывно подавался ток газообразного
формальдегида), можно полагать, что реакция протекала в
диффузионной области. Авторы отмечают, что молекулярный вес полимера
снижался с увеличением концентрации катализатора и ростом
температуры реакции.
50
Влияние активных добавок. Обычными примесями к
формальдегиду являются вода, метанол, муравьиная кислота, метилаль.
Специфика условий получения и очистки мономера может привести
к появлению ряда других активных примесей (особенно в
производственных условиях). Все добавки по их действию на анионную
полимеризацию формальдегида можно разделить на три класса:
1. Агенты передачи цепи (вода, метанол); анион после передачи
цепи имеет активность исходного аниона. Схему реакции можно
представить следующим образом:
. - - — СН2О--1гНОН-у . . . — СН2ОН + НО-
2. Замедлители (муравьиная и уксусная кислоты); анион после
передачи цепи менее активен.
3. Ингибиторы полимеризации (углекислый газ).
Окись углерода и кислород не оказывают никакого влияния,
так же как и хлористый водород, который, по-видимому, только
дезактивирует катализатор [43].
Относительные эффективности добавок при передаче цепи (по
влиянию на молекулярный вес) имеют следующие значения *:
Н2О : СН3ОН : СО2 : СН3СООН : НСООН = 1 : 3,1: 21,5 : 21,9 : 23,8
Среднечисловой молекулярный вес определялся по формуле
[44]:
[т]] = 5,22-10* ?°'93 B2)
Приведенную вязкость измеряли в смеси тетрахлорэтана и
фенола, как это описано выше. При помощи этого соотношения из
уравнения
мп MnJ^ [М]
(где Мп — молекулярный вес образца, полученного в контрольном
опыте) были рассчитаны значения отношений константы передачи
цепи к константе роста цепи {kjkv) для муравьиной и уксусной
кислот. Нужно отметить, что использовать эту формулу для
вычисления относительных констант передачи цепи можно только в очень
грубом приближении, так как она справедлива для мгновенной,
но не интегральной степени полимеризации, а при расчете авторы
пользовались средними значениями приведенной вязкости,
полученными из эксперимента. По-видимому, из-за этого они получили
величины относительной константы передачи цепи меньше единицы
@,36 и 0,39). Если учесть, что эффективность воды как агента передачи
цепи по данным тех же исследователей в 20 раз ниже, то для нее
относительная константа скорости передачи цепи будет порядка
* Действие добавок изучалось при полимеризации в растворе диэтилового
эфира при —59° С. Катализатор — ТБАЛ.
4* 51
0,02. Эти данные не согласуются с экспериментальными. Изучение
зависимости молекулярного веса от глубины реакции в присутствии
добавок воды [48] показывает, что относительная константа
скорости передачи цепи (kjkj на воду больше единицы.
Влияние активных добавок на полимеризацию описано также
в работах [50—51] (в качестве катализатора применяли стеарат
кальция). Реакция проводилась при комнатной температуре, причем
газообразный формальдегид вводили в верхнюю часть
реакционного сосуда, где быстро перемешивался раствор катализатора в
толуоле.
Вода и метанол не влияли на выход полимера, если их
концентрация не превышала некоторой критической величины. Средняя
степень полимеризации обратно пропорциональна концентрации
этих добавок. Отношение значений относительной эффективности
воды и метанола составляет 1 : 7,5.
Авторы высказали предположение, что увеличение
концентрации воды (или метанола) вызывает снижение концентрации
мономера в результате образования метиленгликоля (гемиформаля),
а это в свою очередь приводит к уменьшению выхода продукта:
a = Ap[M][i4* B4)
где [А] — концентрация активных центров, t — продолжительность
реакции.
При добавлении уксусной или муравьиной кислоты, а также
углекислого газа выход и молекулярный вес полимера уменьшаются.
Зависимости HDPn и 1/а от концентрации примеси в степени 1/2
имеют линейный характер. Авторы предполагают, что механизм
действия кислых добавок заключается в обрыве молекулярных
цепей путем взаимодействия с протоном, возникающим в
результате диссоциации молекулы кислоты. Предполагается, что в этом
случае в присутствии следов воды из углекислого газа образуется
угольная кислота. Относительные эффективности для ряда
СО2 : СН3СООН : НСООН выражаются как 1: 3 :14. Окись углерода не
оказывала никакого действия на кинетику полимеризации
формальдегида. Кинетика полимеризации формальдегида в присутствии стеарата
кальция изучалась также в работе [84]. Авторы исследовали
относительные эффективности действия воды, метанола и муравьиной
кислоты при комнатной температуре на молекулярный вес
полимера и получили следующие результаты:
Н2О : СН3ОН : НСООН = 1: 3: 4
В то же время по сообщению американских авторов относительные
величины констант скоростей передачи цепи для воды, метанола
и муравьиной кислоты равны соответственно 1,0; 0,50 и 20,8 при
полимеризации в присутствии анионных катализаторов типа аминов,
фосфинов, солей аммония и т. п. при комнатной температуре [55].
В качестве регуляторов длины цепи менее активных, чем вода
и метанол, предлагается использовать алифатические спирты, слож-
ьг
ные эфиры и ангидриды, амиды, имиды, галогениды, карбонаты,
силикаты, органические нитросоединения и др. [71, 72].
Влияние различных примесей на кинетику полимеризации в
присутствии SnCl4 при комнатной температуре рассмотрено в работе
[75]. Вода и метанол снижали молекулярный вес, кислоты не
оказывали никакого действия.
Определение молекулярного веса
Возможность получения относительно термически стабильного
высокомолекулярного полиоксиметилена создала предпосылки для
тщательного изучения молекулярной структуры этого полимера.
С помощью ИК-спектроскопии было показано, что полимеры имеют
полосу поглощения при 2,9 мк, что соответствует ОН-группе [22].
При этерификации полимера уксусным ангидридом в присутствии
пиридина при 139° С полоса поглощения смещается в область длин
волн 5,69 и 5,75 лік, что соответствует связиС=0в ацетатной группе.
Это указывает на то, что макромолекулы полиоксиметилена имеют
концевые гидроксильные группы.
Метоксильные концевые группы, которые образуются при
передаче цепи на метанол (в небольшом количестве имеющемся в
реакционной системе), были обнаружены аналитическим методом.
Определение вязкости раствора для контроля молекулярного
веса полимера обычно производилось в вискозиметре Кеннона —
Фенске. В качестве растворителей применяли диметилформамид
(при 150° С), л-хлорфенол (при 90—60° С), позднее были предложены
другие растворители (табл. 5).
ТАБЛИЦА 5
Растворители, применяющиеся для
Растворитель
Фенол
/г-Хлорфенол
Тетрахлорэтан
(применяется в смеси с
фенолом 3 : 1)
Бензиловый спирт
N, N-Диметилформамид
о-Дихлорбензол
Анилин
З-Фторпентанол-1
определения
полиформальдегида
Температура
кипения
°С
182
217
146
205
153
180
184
142
Температура
растворения
°С
109
98
110
132
135
164
145
110
молекулярного веса
Температура
гелеобразо
вания
°С
58
60
90
119
115
150
116
—
Литература
22
23
24
23
22
23
25
26
Выбору наиболее эффективных растворителей предшествовало
изучение взаимодействия полиформальдегида (со среднечисловым
молекулярным весом 40 000) с большим количеством веществ.
53
Так, например, в работе [28] приводятся данные анализа 406
растворителей,, испытанных для получения 1 %-ного раствора
полиформальдегида. Ни один из них не растворял полимер при температуре
ниже 60° С.
Помимо трудностей, связанных с растворением
полиформальдегида, необходимо помнить о разложении полимера в растворе. Для
уменьшения скорости разложения в растворитель обычно добавляют
1—2% антиоксиданта и акцептора муравьиной кислоты,
ускоряющей разложение (а-пинен, дифениламин и т. п.). Но и в этом случае
полиоксиметиленгликоль довольно быстро деполимеризуется (рис. 12).
Скорость деполимеризации, как
показано в работе [27], зависит
от природы полимера,
растворителя и температуры.
Попытки найти растворитель,
способный растворять
высокомолекулярный полиоксиметилен при
комнатной температуре, долгое
время были неудачными. В 1964 г.
появилось сообщение [29] о том,
что открыт новый класс
фторированных соединений (например,
гексафторацетонгидрат), которые
за счет образования водородных
связей способны растворять при
комнатной температуре
высококристаллические полимеры типа
J0 2 U 6 8
время, ч
Рис. 12. Изменение приведенной
вязкости полиоксиметиленгли-
коля (ПОМ) в зависимости от
продолжительности выдержки в
различных растворителях [27]:
1 — п-хлорфенол; 2 — диметилформ-
амид; з — тетрахлорэтан-f- фенол A : 1).
полиоксиметиленов и полиамидов.
Предельная концентрация полиформальдегида в растворе
составляет приблизительно 20% при 25° С. Раствор полиоксиметилен-
диацетата, стабилизированного 2% дифениламина, не
обнаруживает признаков разложения по крайней мере в течение суток [83].
В большинстве работ по определению среднечислового
молекулярного веса полиоксиметилена использовались растворы диацетата
полиоксиметиленгликоля в феноле [22, 30—32]. В работе [22]
молекулярный вес определяли двумя методами — осмометрическим
и аналитическим (по концентрации концевых групп).
Измерения осмотического давления проводили в растворах
фенола при 90° С в осмометре, описанном в работе [33], с капиллярами
диаметром 0,45 и 0,60 мм и целлофановой мембраной,
проницаемость которой составляла 2 • 10~б—3,5 • 10~б «г1. Величина
осмотического давления определялась статическим и компенсационным
методами в растворах различной концентрации (от 0,05 до 0,65 г
на 100 мл). Кривую зависимости осмотического давления от
концентрации экстраполировали к нулевой концентрации. Среднечис-
ловой молекулярный вес (Мп) вычисляли по общепринятой
формуле, причем значение плотности фенола при 90° С принимали рав-
54
ным 1,015 г/см3, плотность аморфного полимера 1,22 г/см3.
Величина второго вириального коэффициента А 2 находилась в пределах
1 • 10—2 • 10; молекулярный вес исследуемого полимера —
в пределах 20 • 103—98 . 103.
Концентрацию ацетильных групп определяли омылением. Для
этого полимер обрабатывали метанольным раствором хлористого
водорода и образующийся эфир омыляли едким натром.
Выделившуюся после подкисления уксусную кислоту определяли потенцио-
метрическим титрованием. Чувствительность метода проверяли на
смесях метилацетата и исходного
полимера. Она была равна 0,3 моль
ацетата на 1000 моль формальдегида
при допустимой ошибке 0,05 моль
ацетата на 1000 моль формальдегида.
Это позволило вычислять
молекулярные веса Щп) до 100 000.
Содержание метоксильных групп
измеряли модифицированным
методом Цейзеля. Чувствительность
метода позволяла в образце массой
0,3 г обнаружить метоксильные
группы при концентрации их 0,043% (со
стандартным отклонением ±0,0036).
Значения Мп, полученные при
использовании осмометрического
метода, хорошо совпали со значениями
Мп, вычисленными по результатам
определения концевых групп.
Поскольку при вычислении Мп во втором случае предполагалось, что
каждая молекула содержит только две концевые группы
A
/
0
о
/
wo
80
во
ьо
on
биго ьо во во wo
Mh fO~3}(осмометри*
чесний метод)
Рис. 13. Соотношение между
молекулярным весом ПОМ,
определенным по концентрации
концевых групп и найденным осмоме-
трическим методом [22].
п моль концевых групп/ моль
(где 30 — молекулярный вес формальдегида), то совпадение
результатов двух методов измерения (рис. 13) говорит о том, что
полиформальдегид состоит из неразветвленных линейных молекул. На это
указывает также высокая степень кристалличности, присущая
полиоксиметиленам различного молекулярного веса и с различными
концевыми группами.
В работе [22] была сделана попытка химическим путем
определить наличие в полимере ортокарбонатных и С—С-звеньев как
возможных точек разветвления, однако результат оказался
отрицательным.
Среднечисловой молекулярный вес (Мп) определяли также на
основе уравнения Хувинка [г]] = КМа, которое должно быть
справедливо для фракционированных образцов или образцов с
постоянным молекулярно-весовым распределением (МВР). В литературе
55
предлагается несколько значений констант в уравнении
Хувинка, поскольку они получены для образцов с различным МБР.
На постоянство величины МБР (MJMn) указывает линейная
зависимость lg [ц] от lg Mn, если Мп определено каким-либо
прямым методом. Майер [32, 34] использовал аналитический метод
определения концевых ацетатных групп для калибровки
модельных образцов с помощью ИК-спектрометра, что позволило в
дальнейшем определять Мп непосредственно по спектрам поглощения.
Вискозиметрические определения проводились в смеси тетра-
хлорэтана с фенолом C : 1), содержащей 2% а-пинена, при 90° С.
Концентрация раствора составляла 0,2 г/100 мл.
Характеристическую вязкость определяли по уравнению Хаггенса [38]:
: B5)
где К = 0,34.
Для образцов дельрина и ряда лабораторных образцов диацетата
ПОМ были получены линейные зависимости lg [y|] от lg Mn, что
позволило определить константы в уравнении Хувинка:
# = 2,754- Ю-* а = 0,80
Дельрин многими исследователями используется как эталон для
проверки точности метода определения Мп [25, 31, 34—37]. Для марки
500Х приводится величина 40 • 103—49 • 103. Для марки 150Х
расхождение результатов более значительно: 70 • 103—85 • 103.
МБР образцов дельрина приближается к 2,0.
Средневесовой молекулярный вес (Мю) определяли методом
светорассеяния [39]. Для серии образцов, МБР которых равен 5—7, были
получены следующие значения вискозиметрических констант:
# = 4,4-10~4 а = 0,66
Для определения вязкости (при 150° С) использовали 0,5%-ный
раствор полимера в диметилформамиде с 2% дифениламина.
Значения вязкости экстраполировали к нулевому моменту времени,
учитывая разложение полимера.
Новый растворитель — гексафторацетонгидрат> был использован
для определения вязкости и Mw методом светорассеяния в работе
[83]. Состав гидрата выражался соотношением 1 : 1,7. Растворитель
содержал 84% гексафторацетона, 1% триэтиламина и 0,5% анти-
оксиданта САО-5; d25 = 1,682 ± 0,002 г/мл, ri? = 1,3096,
вязкость при 25° С—0,06 пз. Для нефракционированных образцов
диацетата ПОМ получены следующие значения констант в
уравнении Хувинка:
> B6)
В работе [26, 93] в качестве растворителей для аналогичных
исследований применялись w-хлорфенол с 2% а-пинена при 60° С
56
и З-фторпентанол-1 при 110° С. Значения констант в уравнении
Хувинка соответственно: 4,1 . 10 и 1,33 • 10; 0,72 и 0,81. Во
всех трех случаях использовались нефракционированные образцы
с М„ • 10 от 20 до 185.
РАДИАЦИОННАЯ ПОЛИМЕРИЗАЦИЯ
В жидкой фазе при низких температурах полимеризация
формальдегида легко инициируется \~излУчением» причем процесс
можно вести в блоке или растворе [59, 60]. В последнем случае
скорость реакции и молекулярный вес ниже. Некоторые результаты
приведены в табл. 6.
ТАБЛИЦА 6
Полимеризация формальдегида в жидкое фазе при —80° С,
инициированная уизлучением [59]
Количество
мономера
г
5,0
5,2
4,2
5,4
5,4
Количество
растворителя CHjCh
г
— *
—
1,0
1,0
Доза облучения
рад/ч
0
1,96 -103
3,79 • 103
4,39-103
3,79.103
Продолжительность
облучения
14 суток
10 мин
9 мин
51 мин
53 мин
Выход
полимера
1,5
36,5
75,6
58,9
63,0
1,5
2,8
0,74
0,70
* %р 0ПРеДеляли в 0,5%-ном растворе n-хлорфенола с 2% а-пинена при 60° С.
Для выяснения механизма полимеризации проводили сополиме-
ризацию формальдегида с некоторыми винильными соединениями.
По активности винильные соединения располагаются в следующий
ряд:
изобутилен а* стирол > метилметакрилат > акрилонитрил
Был сделан вывод, что радиационная полимеризация
формальдегида в массе протекает по катионному механизму. Возможность
протекания реакции по радикальному механизму маловероятна,
так как скорость реакции намного выше, чем в случае радикальной
полимеризации стирола при комнатной температуре [61].
Как было установлено позднее [62, 63], природа растворителя
в большой мере определяет механизм процесса. Так, например,
в диэтиловом эфире полимеризация формальдегида протекает по
анионному механизму и получается полимер с максимальным
значением приведенной вязкости и минимальной степенью
кристалличности (всего 57% против 100% при полимеризации в блоке и растворе
метиленхлорид а и толуола).
Доза и интенсивность облучения влияют на скорость
полимеризации, но практически не сказываются на молекулярном весе
57
образующегося полимера. Кажущаяся энергия активации [61]
скорости реакции 4,15 ккал/молъ, энергия активации молекулярного веса —
3,50 ккал/моль.
О влиянии воды на кинетику радиационной полимеризации
имеются противоречивые данные. В работе [63] были сделаны
выводы, что вода понижает молекулярный вес полимера и увеличивает
скорость реакции (линейная зависимость). В более поздней работе
[61] влияние воды на скорость реакции не отмечается. По-новому
истолковываются также некоторые другие экспериментальные
данные. Например, природа наблюдавшегося [63] периода индукции
объясняется присутствием каких-
то неидентифицированных
примесей в формальдегиде, которые,
однако, можно удалить, отгоняя
20—30% формальдегида из
раствора, подготовленного к опыту.
Окамура [63] наблюдал второй
порядок реакции по мономеру (в то-
20
' 10
I
1-
-к—
\
,
5,52
6B?SBJM7JM
\-103
f
в 805
Рис. 14. Зависимость степени
превращения от температуры при
твердофазной полимеризации СН2О,
инициированной -у-излучением [60]
луоле). В работе [61] найден
третий порядок при степени
превращения до 5%.
Мага с сотр. [60] показал,
что энергия активации
твердофазной полимеризации
формальдегида, инициированной ^-излучением
при —196° С, имеет
отрицательное значение. Кривая зависимости степени превращения от
температуры имеет разрыв в точке плавления полимера (— 118° С)
(рис. 14). В работе [64] было найдено, что энергия активации
реакции в твердой фазе равна нулю, а в жидкой — 2,3 ккал/моль.
Пост-полимеризацию исследовали методом термического анализа.
Полимеризация предварительно облученных образцов при
нагревании протекала со взрывной скоростью. Тсуда [65] наблюдал
постполимеризацию взрывного характера даже при малых дозах
облучения. Ему удалось получить образцы с очень высоким
молекулярным весом (характеристическая вязкость, измеренная в тг-крезоле
при 135° С, была в 4—5 раз больше, чем у дельрина 500Х),
нерастворимые в тг-хлорфеноле при 100° С.
На основании этих данных автор сделал предположение, что
твердофазная полимеризация формальдегида протекает по особому
механизму. Предполагается, что под действием ионизирующего
излучения при температуре ниже —150° С формальдегид переходит
в возбужденное состояние Н2С+ — О", которое может сохраняться
длительное время без потери энергии. Полимеризация,
протекающая по ионному механизму, инициируется этими возбужденными
молекулами. Облучение при температуре —150° С инициирует
полимеризацию, протекающую по такому же механизму, как
радиационная полимеризация в жидкой фазе. Различия имеются даже во внеш-
58
нем виде получаемых полимеров. Продукт полимеризации при
—196° С — это волокно; продукт, полученный при —145° С и более
высоких температурах, — стеклообразное вещество.
ПОЛИМЕРИЗАЦИЯ В ПОЛЯРНЫХ СРЕДАХ
Долгое время считалось, что при полимеризации водного или
спиртового раствора формальдегида нельзя получить
высокомолекулярные полиоксиметилены. Известно, что при полимеризации
водных растворов формальдегида, вызываемой перенасыщением или
переохлаждением раствора, максимальная степень полимеризации
образующихся твердых продуктов не превосходит 20; добавление
кислот или щелочей и подбор специального температурного режима
позволяют добиться увеличения степени полимеризации до
200—300 (а-полиоксиметилен, у'полиоксиметилен)> н0 не выше.
Поскольку детальный
механизм процессов образования по- ^ | I >з'
лиоксиметиленов в водной среде 5[<*| 1 Ц
до сих пор не выяснен, такое §Jj| ^§j
ограничение размеров молекул Зона! |^| ЗонаП ^%\ ЗонаШ
обычно связывали с' поликон- §1 ^
денсационным характером про- §§1 I
цесса. LJ
§§1
-L-J »
В последние годы появились Концентрация СН2О
сообщения о том, что в опреде-
ленных условиях при полимери-
зации формальдегида в
полярных средах, в частности в воде
и спиртах, образуется полимер высокого молекулярного веса
C0 000—50 000). Некоторые опубликованные данные [66, 67]
заставляют предполагать, что это процесс ступенчатой полимеризации,
причем рост цепи происходит за счет присоединения молекул
мономерного формальдегида к активному центру.
Состояние формальдегида в водном растворе при постоянной
температуре и давлении показано схематически на рис. 15. Зона / —
ненасыщенный раствор формальдегида. Твердый полиоксиметилен-
гликоль НО(СН2О)ЯН, помещенный в эту зону, растворяется (де-
полимеризуется). Зона // — пересыщенный метастабильный раствор
формальдегида. Самопроизвольное образование твердых полиокси-
метиленгликолей не наблюдается. Однако если в раствор ввести
зародыши кристаллизации (твердый полиоксиметиленгликоль), то
наблюдается рост размеров частиц, сопровождающийся увеличением
степени полимеризации молекул. Зона /// — пересыщенный
нестабильный раствор формальдегида. В этой зоне наблюдается
самопроизвольное образование твердых полиоксиметиленгликолей со
степенью полимеризации порядка 10—12. Кислоты и щелочи
ускоряют этот процесс.
59
Очевидно, образование высокомолекулярного полимера возможно
только в зоне //, где реализуется равновесие:
СН2О (раствор)+ НО(СН2О)ПН (тв.) ^=± НО(СН2О)Я+1Н (тв.)
Если бы удалось непрерывно поддерживать концентрацию
раствора формальдегида, соответствующую концентрации в зоне //
(т. е. выше концентрации насыщения, но ниже концентрации начала
спонтанной кристаллизации полимера), и исключить побочные
реакции, то молекулярный вес полимера должен был неограниченно
возрастать. На самом деле этого не происходит, так как некоторое
количество низкомолекулярного полимера все же образуется.
Свойства полимера, употребляемого в качестве «затравки»
кристаллизации, могут изменяться в широких пределах. Это может
быть параформальдегид, а-полиоксиметилен или
высокомолекулярный полиоксиметиленгликоль. Важно, чтобы полимер содержал
концевые гидроксильные группы, диссоциация которых приводит
к образованию активных центров полимеризации.
Условия равновесия зависят от температуры, давления и
концентрации формальдегида в растворе, причем концентрация,
соответствующая пределу стабильности, на 4—8% превышает
концентрацию насыщения. Скорость образования высокомолекулярного
полимера зависит от температуры, концентрации формальдегида, pH
среды и типа катализатора. Увеличение температуры благоприятно
сказывается на скорости образования полимера, но в еще большей
мере ускоряет нежелательные побочные реакции (Тищенко и Канниц-
царо). Рекомендуется проводить реакцию при 60—125° С, так как
при более низких температурах скорость полимеризации
незначительна.
pH системы, по-видимому, определяется типом выбранного
катализатора. В качестве последнего рекомендуются третичные алкил-
амины, j)Kb которых менее 7. pH системы регулируется
добавлением кислоты до pH = 3,5—10. Кроме этого, известны
катализаторы — соли щелочных и щелочноземельных металлов и
минеральных или алифатических кислот. Применяются практически
насыщенные растворы этих солей, pH системы в этом случае должен быть
[68] не менее 9,3.
В ранних работах [66, 67] отмечалось, что образующийся полимер
имеет очень высокую степень кристалличности и легко ацетили-
руется (что косвенно указывает на упорядоченное расположение
концевых гидроксильных групп в кристаллах полимера).
Итальянские исследователи [68, 69] показали, что образующийся полимер
имеет метастабильную орторомбическую структуру. Механизм
перехода обычной гексагональной формы в орторомбическую во время
этого процесса не вполне ясен, так же как не ясно, какой кинетический
эффект дает образование метастабильной структуры.
Мортилларо с сотр. [68] предложили использовать в качестве
«затравки» орторомбический высокомолекулярный полимер. В этом
случае стационарное содержание орторомбической (высокомолеку-
лярной) фракции устанавливается относительно быстрее. В табл. 7
показано, как меняются свойства твердой фазы при полимеризации
метастабильного раствора формальдегида при 35° С и pH = 10,7.
В качестве катализатора использовали HCOONa, причем отношение
Н2О : HCOONa равно 1,33 (по весу).
ТАБЛИЦА 7
Изменение свойств твердой фазы при полимеризации
метастабильного раствора формальдегида [68]
Начальная
концентрация СН2О
вес. %
50
36
Свойства реакционной
системы
Соотношение жидкой
и твердой фаз (по
весу)
Содержание орторомби-
ческой фазы, %
[г|] полимера *
Соотношение жидкой
и твердой фаз (по
весу)
Содержание орторомби-
ческой фазы, %
[х\] полимера*
Время, сутки
4
2,5
5
0,16
4,2
20
0,22
14
2,5
10
0,20
3,1
55
0,37
28
2,5
30
0,28
2fi
95
0,60
42
2,5
30
0,33
2,5
95
0,68
49
2,5
60
0,34
2,5
95
0,77
56
2,5
60
0,43
69
2,5
80
0,61
Стационарный
процесс
>95
0,80
* [л] измеряли в диметилформамиде при 150° С.
Установление стационарного режима авторы связывают с
физико-статистической дезактивацией актцвных центров из-за
спонтанной кристаллизации гексагонального низкомолекулярного поли-
оксиметилена на поверхности орторомбического.
Позднее было показано [76], что образование орторомбического
полимера в этом процессе происходит только при использовании
в качестве катализатора уксуснокислого натрия и некоторых других
солей (KF, HGOOK и др.)- Необходимым условием получения
орторомбического полимера является проведение реакции при
температуре 20—35° С и определенном pH системы [85, 86]. Интересно,
что при температуре реакции 50° С наблюдается превращение
орторомбического полимера, введенного в качестве затравки, в
низкомолекулярный гексагональный полимер.
В спиртовых растворах при полимеризации в метастабильном
состоянии образуется высокомолекулярный продукт, содержащий
на концах молекул одну алкильную и одну гидроксильную группы
[87]. Это позволяет предполагать, что рост цепей происходит только
через концевые гидроксильные группы и между*собой цепи не
взаимодействуют.
61
0 полимеризации формальдегида в полярных растворителях,
не содержащих подвижные атомы водорода, известно очень мало.
В одном из патентов [79] описан процесс получения
высокомолекулярного полимера в диметилформамиде.
ЛИТЕРАТУРА
1. Kekule AM Вег., 25, 2435 A892).
2. Штаудингер Г., Высокомолекулярные органические соединения,
ОНТИ Химтеорет, 1935, стр. 256.
3. Staudinger H.? Johner Н., Singer R., Mie G.,
Hengstenberg J., Z. phys. Chem., 126, 435 A927).
4. Hof mann A. W., Ber., 1, 200 A868).
5. T r a u t z M., Ufer E.f J. prakt. Chem., 113, 105 A926).
6. S p e n с e R., J. Chem. Soc, 1933, 1193.
7. Carruthers J. E., Norrish R. G. W.s Trans. Farad. Soc, 32,
195 A936).
D а і n t о n F. S., I v і n K. J.f Guart. Rev., 12, 1 A958).
D а і n t о n F. S., I v і n K. J., Trans. Farad. Soc.l? 46, 331 A950).
Nordgren, Acta path, microbiol. scand., 40, 21 A939).
D а і n t о n F. S., I v і n K. J., Trans. Farad. Soc, 55, 61 A959).
Dainton F. S., Evans D.M., Hoare F. E., Melia Т. Р.,
Polymer, 3, 263 A962).
Parks G. S., Mosher H. P., J. Polymer Sei.* A-l, 1979 A963).
1 w a s a J.j Imoto T.? J. Chem. Soc. Japan, Pure Chem. See, 84, 29
A963).
Mortillaro L., Galiazzo G., Bezzi S., Chim. Ind. (Ital.),
45, 711 A963).
Walker J. F., Formaldehyde, 3th ed., Reinhold Publ. Corp., N. Y.,
1964, p. 180.
Bevington J. C, Norrish R. G. W., Proc. Roy. Soc, A-205,
516 A951).
S a u t e г е у R., Ann. Chim., 12-7, 36 A952).
Toby S., R u t z E. F., J. Polymer Sei., 60, 541 A962).
Schweitzer C. E., MacDonald R. N., Punderson J. O.,
J. Appl. Polymer Sei., 1, 158 A959); Химия и технология полимеров,
№ 3, 66 A960).
MacDonald R. N., пат. США 2768994, 1956 г.
Koch Т. A., L і n d v і n g P. E., J. Appl. Polymer Sei., 1, 164 A959);
Химия и технология полимеров, № 3, 73 A960).
В а г k d a 11 А. F., М а с D о n a I d R. N., пат. США 2775570, 1956 г.
Stejny J., Mayer J., Chem. Prumysl., 12, 57 A962).
Dick R., Sack H., Benott H., J. Polymer Sei., 4c A966).
Wagner H. L., Wissbkun K. F., Makromol. Chem., 81, 15 A965).
Thumler W., Plaste und Kautschuk, 12, № 10, 582 A965).
AI sup R. G., Punderson J\ O., Leverett G. F., J. Appl.
Polymer Sei., 1, 185 A959).
Chem. Eng. News, 42, № 48, 32 A964).
К озинский B.j Ф е й г и и Д., Пласт, массы, № 6, 8 A961).
М і j а к а Y., А d а s h і S., A k і m о t о M., Chem. High Pol. Japan,
21, 246 A964).
M a j er J., Makromol. Chem., 86, 253 A965).
S t a b і n J.V., Immergut E. H., J. Polymer Sei., 14, 209 A954).
Maier J., Makromol. Chem., 82, 169 A965).
Kakiuchi H., Fukuda W., J. Chem. Soc. Japan, Ind. Chem. Sec.,
66, 964 A963).
I n о u e M., T a k а у a m і g і Т., J. Polymer Sci.? 47, 498 A960).
I n о u e M., J. Polymer Sei., A-l, 2697 A963).
38. Schulz G. V., Blaschka F., J. Prakt. Chem., 158, 130 A941),
39. Б ельговский И. M., Ениколопян Н. С, СахоненкоЛ. С,
Высокомол. соед., 4, 1197 A962).
40. F u k u d а W., К а к і и с h і Н., Kogyo Kagaku Zasshi, 65, 2054 A962).
41. Mach ас ek Z., Mejzlik J., P а с I., J. Polymer Sei., 52, 309 A961),
42. M a x а ч e к 3., М є й з л и к Й., Пац Й., Высокомол. соед., 3, 1421
A961).
43. Мейзлик Й., Менчикова Й., Махачек 3., Высокомол.
соед., 4, 769 A962).
44. Веселы К., Мейзлик Й., Высокомол. соед., 5, 1425 A966).
45. Е п і к о 1 о р і а п N. S., J. Polymer Sei., 58, 1301 A962).
46. Е н и к о л о п я н Н. С, В а р д а н я н М. С, ЖВХО им. Д. И.
Менделеева, 7, № 12, 194 A962).
47. Е н и к о л о п я н Н. С, П р о ш л я к о в а Н. Ф., С а н а я И. Ф.,
Высокомолек. соед., 5, 1632 A963),.
48« Ениколопян Н. С, Прошлякова Н. Ф., С а н а я И. Ф.,
Высокомол. соед., 5, 1776 A963).
49. Ениколопян Н. С, И р ж а к В. И.,Романов Л. М.,
Высокомол. соед.* 5, 1638 A963).
5(Ь Ениколопян Н. С, Бобков а Л. П., Корсаков B.C.,
Романов Л. М., Высокомол. соед., 5, 1653 A963).
51. Ениколопян Н. С, Бобкова Л. П.? Корсаков В. С,
Романов Л. М., Высокомол. соед., 5, 1780 A963).
52. Ениколопян Н. С, Шагинян А. А.? Минин В. А.,
Кедрина Н. Ф., Высокомол. соед., 7, 1872 A965).
53. Е н и к о л о п я н Н. С, Шагинян A.A., Высокомол. соед., 7,
1866 A965).
54. Kern W., Chem. Z., 88, № 16, 623 A964).
55. Knight А. С, Du Pont, англ. пат. 796862, 1958 г.
56. Bayer F., англ. пат. 9244621, 1963 г.
57. Bayer F., англ. пат. 961942, 1963 г.
58. Bayer F., англ. пат. 943177, 1963 г.
59. Okamura S., Nakashio S., Hayashi K., Sakurada I.,
Isotopes and Radiation, 3, 242 A960).
60. С h а с h a t у С, М a g a t M., M inassi an L., J. Polymer Sei., 48,
139 A960).
61. Nakashio S., U e d a M., Takahashi K., Makromol. Chem., 83,
23 A965).
62. Okamura S., Hayashi K., Yamaoka H., NakamuraY.,
14th Annual Meeting of Japan Chem. Soc, Tokio, 1961.
63. Yamaoka H.,Hayashi K.,Okamura S., Makromol. Chem., 76,
196 A964).
64. Б a p к а л о в И. М., ГольданскийВ. И., Ениколопян Н. С,
Терехова С. Ф., Трофимова Г. М., Industrial uses of large
Radiation Sources, № 1, Vunna, 1963, p. 193.
05. T s u d a J., J. Polymer Sei., 49, 369 A961).
66. Brown N., Funck D. L., Schweitzer С. Е., пат. США
3000860, 1961 г.
67. Мог till аго L., Galiazzo G., Bezzi S., Chim. Ind., 46, № 2,
139 A964).
68. В г о w n N.. F u n с k D. L., S с h w є і t z e г С. Е., пат. США 3000861,
1961 г.
69. С a r a z z о 1 о G., Mammi M., J. Polymer Sei., 1A, 965 A963); С а-
r a z z о 1 о G., J. Polymer Sei., 1-А, 1573 A963).
70. Deutsche Gold, англ. пат. 836288, 1960 г,
71. Du Pont, англ. пат. 796863, 1958 г.
72. Stamicarbon N. V., англ. пат. 879022, 1961 г.
73. А й в и н К., Химия и технология полимеров, № 3, 78A963).
74. Кип z el Е., Giefer А., Kern W-, Makromol. Chem., 96, 17 A966).
63
Шпичинецкая Л. С, Орлова Н. В., Пласт, массы, № 8, 6
A966).
Brown N., Am. Chem. Soc, Polymer Preprints, 7 A966).
Dworjanyn L. 0., Austr. J. Chem., 13, 175 (I960).
Ж у л и н В. М., Гоникберг М. Г., Высокомол. соед., 3, 267 A961).
Пат. США 3140272, 1964 г.
Гаврилина Л. Я., Натальина Н. Н., Изв. Сибирского
отделения АН СССР, 11, 143 A962).
Bombaygh К. I., В и 11 W. Е., Anal. Chem., 9, 1237 A962).
Минин В. А., Ф а й д е л ь Г. И., 3 и с м а н Д. О., Пласт, массы,
№ 10, 53 A966).
Stockmayer W. Н., Chan L. L., J. Polymer Sei., 4, А-2, 437
A966).
Tomaszewicz M., Feigin J.f Zakrzewski L.f Przemysl.
Chem., 42, № 11, 633 A963).
Mortillaro L., G a 1 і a z z о G., Bandel A., Chim. Ind. (Milan),
46, 1143 A964).
Mortillaro L., G a 1 і a z z о G.,~ Chim. Ind. (Milan), 46, 1148 A964).
Manwiller С H., T h о m p s о n J. B.? пат. США 3193533, 1965 г.
Tobolsky A. V.,J. Polymer Sei., 25, 220 A957).
Flor у P. J., Principles of Polymer Chemistry, N. Y., Cornell Univ.
Press, 1953.
Bywater S., Trans. Faraday Soc, 51, 1267 A955).
Bywater S., Makromol. Chem., 52, 120 A962),
Д у д и H а Л. А., И p ж а к В. И., Ениколопян Н. С, ДАН
СССР 173, 1121 A967).
К о к 1 е V., В і 11 m е у е г F. W., J. Polymer Sei., ВЗ, 47 A965).
Tobolsky А. V., Eis en berg A., J. Am. Chem. Soc, 82, 289
A960).
Vrancken A., Smid D., Szwarc H., J. Am. Chem. Soc, 83,
2772 A961); Trans. Farad. Soc, 58, 2036 A962).
North A.M., Richardson D., Polymer, 5, 227 A964).
ГЛАВА III
ПОЛИМЕРИЗАЦИЯ ТРИОКСАНА
При возгонке очищенного триоксана Хеммик и Бери [1]
получили в остатке твердое вещество, названное ими е-поли-
оксимети леном. Свойства и способ получения этого вещества
заставляли предполагать, что оно имеет полиацетальную
структуру, однако его нельзя было отождествить ни с одной
известной в то время модификацией полиоксиметилена. Штаудингер [2]
высказал предположение, что это б-полиоксиметилен, т. е. продукт
частичной перегруппировки низкомолекулярного диметилового
эфира полиоксиметилена, содержащий небольшое количество
углеводных группировок.
Большое значение придавалось тому факту, что е-полиоксимети-
лен не поддавался ацетилированию. На основе этого был сделан
вывод, что он не содержит концевых гидроксильных групп *.
Позднее Колынюттер [3] наблюдал, что при возгонке ?-полиок-
симетилена (низкомолекулярного полиоксиметилена, который
окклюдирует некоторое количество серной кислоты) образуются
волокнистые кристаллы триоксана, «которые в ходе реакции претерпевают
некоторое изменение под влиянием свободного формальдегида».
В результате получаются термостойкие полиоксиметиленовые
волокна с однородной внутренней структурой.
Хотя эти факты можно истолковать как доказательство
возможности прямой полимеризации триоксана в высокомолекулярный
полиоксиметилен, такой вывод долгое время не был сделан.
Известно, что триоксан неустойчив в присутствии следов соединений
кислотного характера и уже при комнатной температуре деполимери-
зуется до мономерного формальдегида. Было естественно
предположить, что именно формальдегид, а не триоксан полимеризуется
в приведенных выше случаях.
Такая точка зрения разделялась, например, Уокером [4],
предложившим метод выделения формальдегида из триоксана путем
обработки последнего льюисовскими кислотами. Даже в 1964 г., когда
кинетика и механизм полимеризации триоксана уже интенсивно
исследовались, некоторые специалисты все еще придерживались
этой гипотезы [5].
Ацетилирование осуществляли в гетерогенной фазе.
В отличие от формальдегида триоксан способен полимеризоваться
только под действием катионных катализаторов.
Высокомолекулярные полимеры, получаемые из формальдегида и триоксана, имеют
одинаковую химическую структуру. Однако они различаются по
термодинамическим свойствам, что связано, в первую очередь,
с различиями в морфологии кристаллов. Несмотря на большой
интерес к механизму полимеризации триоксана, проявляемый в
течение последних пяти лет, следует признать, что наши знания по
данному вопросу весьма поверхностны. Это касается, в первую
очередь, термодинамики полимеризации, своеобразие которой
заключается в атермическом характере процесса раскрытия
шестичленного цикла. Кроме того, кинетика полимеризации триоксана
весьма сложна для изучения вследствие протекания целого ряда
побочных реакций.
ТЕРМОДИНАМИКА ПОЛИМЕРИЗАЦИИ
ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ
При полимеризации с раскрытием цикла изменение энтальпии
(АН) определяется величиной напряженности связей в цикле, т. е.
зависит от строения циклических соединений.
Рассматривая термодинамику полимеризации циклоалканов,
Дейнтон с сотр. [6] пришел к выводу, что при увеличении числа
членов в кольце циклоалкана значения AG0 (изменение свободной
энергии при полимеризации) должны проходить через максимум,
соответствующий именно шестичленному циклу. При этом поскольку
AG0 становится больше нуля, полимеризация напряженного цикла
термодинамически невозможна. В табл. 8 приведены величины
термодинамических констант полимеризации циклоалканов. При
раскрытии цикла и образовании полимера линейной структуры природа
химических связей не меняется. Это значит, что энтальпия
ненапряженного цикла не отличается от энтальпии полимера.
Отрицательная величина Д#° для шестичленного цикла свидетельствует о том,
что сдвиг равновесия невозможен (в данном случае понятие предельной
температуры не применимо). Однако данные, представленные
в табл. 8, вычислены полу эмпирическими способами и должны
рассматриваться как приближенные.
Замена метиленовой группы в цикле на атом кислорода (простой
циклический эфир) существенно не влияет на величины
термодинамических констант, так как межатомные расстояния и углы,
образуемые углерод-углеродными и углерод-кислородными связями,
различаются незначительно [7]:
С
С-С 1,54 A С С 109° 28'
О
С—О 1,44 A С С ИГ
66
ТАБЛИЦА 8
Термодинамические константы полимеризации [6]
для циклоалканов * общей формулы (СН2)„
п
3
4
5
6
7
8
— АН0
пиал/моль
27,0
25,1
5,2
-0,7
5,1
8,3
— AS0
кал 1 моль • град
16,5
13,2
10,2
2,5
0,7
—8,9
-AG0
икал/моль
22,1
21.Я
2,2
-1,4
4,9
11,0
* Расчет проделан для систем жидкий мономер—твердый
полимер (ж—т).
Приблизительные расчеты, выполненные Смоллом [6],
показывают, что наибольшее различие термодинамических констант
наблюдается для первых членов гомологических рядов. Так, например,
для этилена и формальдегида—АН0 равно 24,7 и 13,0 ккал/молъ
соответственно. Для циклобутана и оксациклобутана это различие
составляет уже приблизительно 3 ккал/молъ, и предполагается, что с
увеличением количества атомов в цикле оно будет нивелироваться
(табл. 9).
ТАБЛИЦА 9
Термодинамические константы полимеризации пятичленных
циклов различной структуры
Соединение
Циклопентан
Тетрагидро-
фуран
1,3-Диоксолан
Формула
н2с—сн2
1 1 "
Н2С GH.2
сн2
H2G GH2
TJ ҐА ҐЛ ТТ
rl2V-4 V-4.T12
I I
і і
GH2
-АНЖ-Т
ккал /моль
5,2
5,3
5,1—5,2
-AS0
КОЛ 1 МОЛЬ'
•град
10,2
15,8
18,6
ккал /моль
2,2
(рассчитано)
1,0
-0,44
Литература
6
8
9
Результаты термодинамических расчетов для системы триоксан
полиоксиметилен приведены в табл. 10.
5*
67
Термодинамические
Фазовое состояние
мономера *
Газообразное
Жидкое
Твердое
лн°
ккал/ моль
—3,83 **
—0,62
0,57
ТАБЛИЦА 10
константы полимеризации триоксана
AS0
кол /моль-
•град
—12,73***
—4,40
2,07
AG0
ккал /моль
—0,033
0,69
—0,046
V
301
1145
275
(нижняя предельная
температура)
* Полимер всегда образует кристаллическую фазу.
** Для расчета АЯ° использованы данные о теплоте сгорания триоксана из работы
[13], которым отдано предпочтение перед данными работы [92].
*** Величины энтропии триоксана взяты из работы [99]. Значение энтропии поли-
триоксана A0,27 кал/моль • град) взято из работы [14].
Приближенный.характер вычислений очевиден, так как в
литературе имеются противоречивые данные относительно теплот сгорания
триоксана. Результат, полученный для системы твердый мономер —
твердый полимер, предполагает существование нижней предельной
температуры. До сих пор подобный феномен известен только для двух
соединений (сера и селен), образующих восьмичленные циклы.
Действительно, твердый триоксан не полимеризуется при низких
температурах, однако этот факт находит объяснение в рамках теории
твердофазной полимеризации.
Экспериментальное определение термодинамических констант
непосредственно во время полимеризации триоксана крайне
затруднено из-за протекания побочных реакций. Прежде всего происходит
равновесная реакция образования формальдегида из триоксана,
в результате которой устанавливается равновесная (по отношению
к полимеру) концентрация формальдегида *. По аналогии с
процессами полимеризации других гегероциклов [52] в данном случае
в реакционных системах происходит накопление некоторых
циклических соединений, в частности тетраоксана [100]. Но своеобразие
этого процесса заключается в том, что выделившиеся побочные
продукты (формальдегид и тетраоксан) сами полимеризуются в данных
условиях, образуя полиоксиметилен, не отличающийся от продукта
полимеризации триоксана. Представляют интерес некоторые
калориметрические измерения, проведенные Лиизом и Баумбером [15].
Они показали, что после введения катализатора в раствор триоксана
реакция имеет эндотермический характер, причем, хотя скорость
поглощения тепла зависит от концентрации триоксана и
катализатора (наклон кривой А Б на рис. 16), общее количество
поглощенного тепла определяется только температурой реакции.
* Подобная картина имеет место и при катионной полимеризации формаль*
дегида. Показано [100], что в этом случае накапливается равновесная концен*
трация триоксана, равная 0,20 моль/л при 25е С.
68
Считая, что все тепло поглощается при образовании мономерного
формальдегида, и пренебрегая теплотой растворения формальдегида
в неполярной среде, для реакции
(СН2ОK (раствор)
ЗСИ2О (газ)
они получили значение —АН0, равное 13,7 ± 1 ккал/молъ. Если учесть,
что теплота растворения ((?), триоксана в этилендихлориде, который
являлся растворителем, составляет от —1,19 до ±0,01 ккал/молъ,
то — А#° = 13,7 +1,19я^15 ккал/молъ. Эти данные хорошо совпадают
с величиной теплоты реакции образования триоксана (см. гл. I).
С повышением температуры равновесие
реакции триоксан — формальдегид
сдвигается вправо [10, И]. Однако при
30° С предельная концентрация
формальдегида составляла приблизительно
0,01 молъ/л. Это значительно ниже
равновесной концентрации
формальдегида над полиоксиметиленом при 30° С,
так что представляется маловероятным,
чтобы образовавшийся формальдегид
являлся полимеризующимся агентом в
данных условиях.
К атермическому участку кривой на
рис. 16 мы вернемся позднее, а теперь
рассмотрим экзотермический участок.
Он совпадает по времени с периодом
видимого образования твердого
полимера в реакционной системе. Измеренный
тепловой эффект составил 2,09 ± 0,02 ккал/молъ (в пересчете
1 моль формальдегида). Полимеризацию триоксана проводили в
растворе этиленхлорида концентрацией 2,8 молъ/л при 50—60° С,
катализатор — эфират фтористого бора. Теплота кристаллизации поли-
оксиметилена [16, 17] составляет 1,76—1,78 ккал/молъ. Отсюда можно
определить напряженность цикла в молекуле триоксана, равную
приблизительно 1 ккал/молъ.
Очевидно, что полимеризация жидкого триоксана должна
характеризоваться низкими значениями предельной температуры.
Вероятность ошибки в термодинамических расчетах слишком велика.
Например, если в табл. 10 вместо теплоты полимеризации, рассчитанной
по теплотам сгорания, подставить величину, экспериментально
найденную Лиизом и Баумбером, то Гпр для одномолярного раствора
триоксана составит 173° С. Непосредственное измерение равновесных
концентраций триоксана как функции температуры еще никем не
проделано, хотя это внесло бы ясность в некоторые вопросы
термодинамики и кинетики полимеризации триоксана. Трудности подобного
эксперимента очевидны. Исследуя равновесие в системе триоксан —
полиоксиметилен, нужно учесть, что одновременно будет
Время —*-
Рис. 16. Термометрическая
кривая полимеризации
триоксана в растворе [15]:
А — ввод катализатора; АБ —
эндотермическая стадия; Б В — атер-
мическая стадия; В Г —
экзотермическая стадия.
на
реализоваться равновесие триоксан — формальдегид и формальдегид—
полиоксиметилен. Измерить равновесную концентрацию триоксана
над полимером трудно, так как распад полиоксиметилена на
формальдегид доминирует *.
В заключение нужно остановиться на гипотезе Керна и Яакса [18].
Они предположили, что схему полимеризации триоксана можно
свести к следующей системе равновесий:
триоксан —>• полиоксиметилен ^~» формальдегид
Если принять, что скорость реакции деполимеризации полимера
с образованием формальдегида выше, чем скорость полимеризации
триоксана, то, пока не установится равновесная для данной
температуры концентрация формальдегида, образование полимера не будет
происходить. Эта гипотеза как будто объясняла природу
индукционного периода, обычно наблюдаемого при полимеризации триоксана.
Однако она не объясняет, каким образом полимер может образоваться
при температуре более высокой, чем предельная температура
полимеризации формальдегида. Кроме того, было найдено, что в
некоторых случаях полимеризация может идти без выделения мономерного
формальдегида или практически без индукционного периода. Так
что, несмотря на привлекательную простоту этой гипотезы, от нее,
по-видимому, придется отказаться.
КИНЕТИКА ПОЛИМЕРИЗАЦИИ В ЖИДКОЙ ФАЗЕ
Катализаторы и растворители
Инициаторы радикальной полимеризации не действуют на
триоксан, хотя известна реакция радикального окисления триоксана
при повышенных температурах с образованием формальдегида,
воды и углекислого газа. Триоксан, как и все ацетали, весьма
устойчив к действию щелочных агентов, и напротив, разнообразные
катализаторы катионного типа, вызывающие деполимеризацию
веществ оксиметиленовой структуры, легко инициируют
полимеризацию триоксана. Сюда относятся протонсодержащие кислоты, кислоты
Льюиса, их комплексы с эфирами и другими кислородсодержащими
соединениями, аминами и т. д. (табл. 11).
Кроме этого, триоксан полимеризуется спонтанно в присутствии
следов формальдегида при кристаллизации и сублимации, а также
под действием радиационного облучения в твердой фазе.
Предполагается, что во всех случаях полимеризация протекает по катионному
механизму.
В табл. 12 приводятся данные об эффективности различных
катализаторов полимеризации триоксана в растворе [24].
* Можно ожидать, что если полимер будет находиться в растворе, то его
деполимеризация с образованием триоксана будет кинетически более выгодна.
70
Катализаторы полимеризации триоксана
ТАБЛИЦА 11
Класс катализаторов
Типичные примеры
Условия проведения
процесса
Литература
Кислоты Льюиса
Алкилсульфокислоты
(С^5)
Комплексы BF3 с
органическими
соединениями, имеющими
доноры (атомы S или О)
Комплексы BF3 с
основаниями Льюиса
(степень ионизации в
воде 1 -10-9)
Оксониевые соли
Арилдиазониевые соли
Органические кислоты
Ортоэфиры
органических кислот и кислот
Льюиса
Протонсодержащие и
сульфокислоты и их
производные (эфиры)
BF3,
А
, FeCl3, ТІСІ4,
BF3O(C2H6J
BF3-NR3, BF3-C6H5NH2
Триалкилоксонийфтор-
борат
Арилдиазонийфторборат
Щавелевая
Ортоформиат BF3
Хлорная кислота,
ацетилперхлорат
Раствор, расплав,
твердая и
газообразная фаза
Раствор, расплав,
твердая фаза
То же
Раствор
Сублимация
Раствор
Раствор, расплав
7,19,77
78
79,80
81
82
83
84
85
86,87
ТАБЛИЦА 12
Эффективность различных катализаторов при полимеризации
триоксана в растворе
Катализаторы
СН3СОСЮ4
ацетилперхлорат
FeCl3
S11CI4
ВР3О(СаНбJ
SbCl5
H2SO4
H2SO4
SbF3
ТІС14
ТІСІ4 + H2O
ТіС14
І2
AlClg
A1C13 + H2O
АІСІз + СізССООН
С13ССООН
Концентрация
катализатора
моль 1 моль
триоксана
4,5 -10-5
4,5. Ю-*
4,5 - 10-е
4,5. Ю-б
4,5 • К)
4,5-Ю-о
8 • 10
8 • 10
8.10
8.10
8 - Ю-»
8 - Ю-»
8 - Ю-«
8-Ю-3
8 • Ю-»
8 -10-8
Индукционный период
25 сек
40 сек
3 мин
18 мин
30 мин
>24 ч
2,5 мин
—
—
—
—
—
—
—
жительность
лимеризации
ч
1
1
1
1
1
24
1
1
1
1
24
24
27
27
27
>24
Конверсия
%
47
38
27
13
5
0
50
31
16
6,5
70
17
12
2
11
0
Мп
29 000
27 000
16 000
8 500
3 000
—
9 000
16 500
—
3 000
16 000
3 200
27 000
—
—
—
71
Под эффективностью катализатора обычно подразумевается его
способность обеспечить высокую конверсию мономера. Из таблицы
видно, что в большинстве случаев наблюдалась определенная связь
между конверсией, степенью полимеризации и периодом индукции:
меньшим периодам индукции соответствовала большая степень
полимеризации и высокие конверсии.
Приведенные данные имеют поисковый характер и не очень
надежны, так как выбор условий реакции и степень очистки
реагентов сильно влияют на результаты. В большинстве кинетических работ
в качестве катализаторов использовали эфират фтористого бора
(BF3*Ft20) и хлорное олово (SnCl4), которые широко применяются
при катионнои полимеризации винильных мономеров и гетероциклов
и для которых известно наибольшее количество сопоставимых
результатов.
Весьма актуальна также проблема выбора растворителя. Триок-
сан хорошо растворим в циклоалифатических и ароматических
углеводородах, а также в их галоидо- и нитропроизводных.
Значительно хуже растворимость триоксана в алифатических
углеводородах (та,бл. 13).
ТАБЛИЦА 13
Растворители, применяемые при полимеризации триоксана в растворе
Растворитель
Триоксан *
н-Гексан
н- Гептан
Циклогексан
Бензол
Хлороформ
Хлорбензол
Хлористый бутил
Хлористый метилен
Хлористый этилен
Нитробензол
Формула
(СН2ОK
С6Н14
С7Н16
С6Н12
свн6
CHClg
С6Н5С1
С4Н9С1
СН2С12
Q2H4C12
C6H5NO2
Диэлектрическая
пр они цаемость
8,0 (при 62°);
15,6 (при 80°)[26]
1,9
1,9
2,0
2,3
4,8
5,6
7,4
9,0
10,3
35,0
Дипольный
момент
0
0
0
0
0
1,15
1,56
2,11
1,55
2,0
4,0
* Полимеризация в растворе.
Растворимость триоксана зависит от температуры. В
расплавленном состоянии (выше 62° С) он смешивается с большинством
растворителей в любых соотношениях.
Растворители, содержащие подвижные атомы водорода, не могут
быть применены для полимеризации триоксана вследствие сильного
взаимодействия их с катализатором. Апротонные растворители
с высокой основностью (типа простых эфиров), которые образуют
с катализатором устойчивые оксониевые ионы, также не
пригодны.
72
Состояние триоксана в растворах до настоящего времени
практически не изучалось, между тем можно предполагать, что целый ряд
необъяснимых кинетических эффектов связан именно с
особенностями поведения триоксана в растворенном состоянии [71].
Индукционный период.
Механизм инициирования полимеризации
Кинетические кривые полимеризации триоксана в растворе
обычно имеют ярко выраженный S-образный характер (рис. 17).
Максимальной скоростью полимеризации называется скорость
* !мах , измеряемая как угол наклона касательной, проведенной
(it
в точке наиболее крутого подъема кривой в координатах конверсия —
продолжительность реакции, к оси
абсцисс. Индукционный период можно
определить как отрезок, отсекаемый
касательной на оси абсцисс. Наличие
ярко выраженного периода индукции
является важной особенностью
кинетики полимеризации триоксана в
растворе, поэтому изучению этого явления
уделяется много внимания. Часто ин-
укционный период фиксируется
визуально по начальному моменту
выпадения полимера из раствора.
Период индукции в ионной поли- Рис. 17. Кинетические кривые
меризации, точнее резкое увеличение
скорости полимеризации, часто
связывают с медленным зарождением
активных центров. Подобный случай
рассматривался Ениколопяном с сотр. [27]. Образование активных центров
можно представить как реакцию триоксана с катализатором:
полимеризации триоксана в
растворе при различных
концентрациях катализатора
tab > КЬ-
триоксан -\- катализатор
активный центр
A)
Накопление активных центров во времени для этого случая
описывается уравнением *:
[A] = [cK]0(l — e~ ° W) B)
где к0 — константа скорости инициирования; [ст] — концентрация
триоксана; [А] — концентрация активных центров; [сК]0 —
начальная концентрация катализатора.
Скорость расхода триоксана будет равна:
^1Ст] _1я г т г ЛЛ
dt
C)
* При интегрировании концентрацию мономера считаем постоянной.
73
где Je __ константа скорости роста полимерных цепей. Подставляя
B)
значение [А] из уравнения B), после интегрирования получаем:
D)
Для данного к0 [ст] и различных [ск], пользуясь уравнением
D), можно построить кинетические кривые, представленные на
рис. 17. Уравнение асимптот этих кривых будет:
(при t —>оо)
E)
20
W
/
/
Асимптоты отсекают на оси ординат
отрезок
0,2
ОА
OS
0,8
Рис. 18. Зависимость
продолжительности индукционного периода
творатриоксана в метиленхлориде,
% А и на оси абсцисс отрезок
Таким образом, индукционный
период (tt.), будучи функцией
концентрации триоксана и константы
скорости инициирования, не зависит
от концентрации катализатора.
Однако такой вывод не согласуется
с экспериментальными данными.
Заметим, что в этой реакционной схеме
инициирование рассматривается как
реакция первого порядка (концентрация триоксана считается
постоянной).
В опытах Ениколопяна, Раковой и Романова [27] наблюдалась
обратно пропорциональная зависимость периода индукции от
концентрации катализатора (SnCl4) в полярных апротонных
растворителях (рис. 18). В опытах Хигашимуры, Мики и Окамуры [30] такая же
зависимость получена для BF3O(G2H5J в этилендихлориде, а в
нитробензоле период индукции был приблизительно пропорционален
л
величине -т= . В неполярных растворителях зависимость периода
у [ск]о
индукции от концентрации катализатора [ВР3О(С2Н5J и SnClJ
носит сложный характер, однако общая тенденция сохраняется
(с увеличением концентрации катализатора период индукции
уменьшается). В работе [18] показано, что во время индукционного
периода происходит выделение мономерного формальдегида, причем
образование твердого полимера наблюдается фактически после
установления стационарной концентрации формальдегида. Эти
опыты проводились при 30° С с растворами триоксана в хлори-
74
стом метилене с катализатором BF3, а впоследствии со SnCl4 и
СН3СОСЮ4 (ацетилперхлоратом) в циклогексане [24]. Весьма
существенным является тот факт, что, судя по величинам молекулярных
весов, которые приводятся в этих работах, реакционная система
содержала относительно большие количества влаги порядка
5-Ю молъ/л. Экспериментально найденная степень полимеризации
была почти в 40 раз меньше рассчитанной на основании
предположения, что весь катализатор участвует в образовании активных
центров.
Керн и Яакс [18] объясняли природу индукционного периода,
используя представления о полимеризационно-деполимеризационном
равновесии (ПДР) при образовании полиоксиметилена (см. гл. II).
При этом предполагается, что реакция происходит по оксониево-
карбониевому механизму:
хн2~оч + хна-оч
н+о( ;сн2 —> но; )
чш2-ск \
—> НО-СН2—О—СН2-О-СН2 <¦¦> НО—СН2-О-СН2-О=СН2
В результате раскрытия цикла образуется
резонансно-стабилизованный катион, который обладает достаточной активностью для ведения
полимеризации. Образование такого катиона дает выигрыш в
энергии сопряжения, чем объясняется легкость раскрытия цикла
триоксана.
Кроме реакции роста цепи за счет взаимодействия оксоний-иона
с молекулой триоксана и последующего раскрытия цикла
+ ХН2"Оч
НО—СНа—О—СНа—О—СНа -f О NGH2
\сна—(к
сн2-оч
но—сн2—о—сна—о—сн2—о ;сн2 —> но-.(сн2оM—сн2
происходит также отщепление форма льде гидных звеньев (из-за
обратимости акта присоединения). Поскольку природа активных:
центров при полимеризации триоксана и формальдегида одинакова*
то должны действовать закономерности, рассмотренные нами для
полимеризации формальдегида в безводной среде. Отсутствие
свободного мономерного формальдегида в реакционной системе в начала
реакции приводит к тому, что равновесие
НО(СН2О)ЯСН2 ^=> HO(CH2O),_iCH2-f СН2О
сдвинуїо в сторону образования формальдегида. Отрыв молекул:
формальдегида может проходить через стадию образования оксск
7S
ниевой формы резонансно-стабилизованного катиона по следующей
схеме:
НО— СН2—О—СН2—О=СН2 —>• НО—СН2—О—СН2 + СН2О и т. д.
Если скорость отщепления формальдегида выше скорости
присоединения молекул триоксана, то образования полимера не
наблюдается до тех пор, пока не достигается предельная концентрация
мономерного формальдегида в реакционной системе и между ним
и полимером устанавливается равновесие.
Кинетический анализ этой гипотезы дается в работе [27] для
двух случаев: быстрого и медленного зарождения активных центров.
Если &д — константа скорости деполимеризации, [сф] —
концентрация СН2О и kVl — константа скорости реакции
присоединения СН2О к активному центру, то скорость накопления СН2О
в системе будет описываться уравнением:
^- = hlA]-kVi[A][4] F)
Отсюда стационарная (равновесная) концентрация СНаО в системе:
ф р
Pi
При быстром зарождении цепи [А] = [ск] и скорость образования
полимера в ходе процесса будет равна:
[Ск] ~
После интегрирования асимптота (при t —> оо) будет описываться
уравнением:
Acr = kvlcK][cT]t--?- (9)
Si
Отсюда для периода индукции получаем выражение:
х
Зависимость rt от концентрации катализатора, полученная
теоретически, подтвердилась экспериментально [27]. Зависимость
продолжительности индукционного периода от концентрации
мономера имеет более сложный характер. Большое влияние на вид кривых
накопления формальдегида во время периода индукции оказывает
тип растворителя и катализатора. Зависимость квазистационарной
концентрации формальдегида от температуры при использовании
в качестве катализаторов SnCl4n BF3-.Z?t20 приведена на рис. 19.
Эти факты ставят под сомнение гипотезу Керна и Яакса о природе
индукционного периода и прежде всего предположение о том, что
скорость отщепления звеньев СН2О от активного центра значительно
выше скорости присоединения молекул триоксана к активному
центру.
76
Прямая проверка гипотезы может быть осуществлена следующим
образом: в реакционной системе создается равновесная концентрация
мономерного формальдегида. Если наличие индукционного периода
обусловлено сдвигом равновесия полимер <± формальдегид, то
полимеризация должна начаться немедленно. Эти опыты дали
отрицательный результат [33]: добавляемый в систему мономерный
формальдегид не влиял на продолжительность индукционного периода.
Экспериментальный материал, которым мы располагаем в
настоящее время, еще не позволяет дать детальное объяснение
механизма инициирования
полимеризации триоксана. Отмечается, что
продолжительность периода
индукции является функцией
концентрации воды в реакционной
системе. Что касается
образования мономерного
формальдегида, то, по-видимому, он всегда
выделяется, если присутствуют
следы воды. В абсолютно сухих
системах индукционный период
фактически не существует (т <<
<z 1 сек) *. Недавно показано
[36], что инициирование
полимеризации триалкилоксониевой
солью (C2H5KOSbCle не
сопровождается выделением
формальдегида. Значит ли это, что инициирование протекает по какому-либо
особому механизму (без участия карбониевых ионов, например),
покажет ближайшее будущее.
Эксперименты Лииза и Баумбера [15], которые исследовали
кинетику полимеризации триоксана с помощью адиабатической
калориметрии, позволяют сделать некоторые выводы о механизме
начальной стадии полимеризации в присутствии следов воды.
На кривой изменения температуры в ходе индукционного периода
(см. рис. 16) они выделили три участка:
эндотермический, обусловленный разложением триоксана на
формальдегид до установления квазиравновесной концентрации;
атермический;
экзотермический, обусловленный выпадением твердого полимера.
Анализ этих участков термометрической кривой позволяет
сделать следующие выводы:
Количество тепла, выделившегося на первой стадии, зависит
только от температуры и не зависит от концентрации триоксана
и катализатора. Скорость разложения триоксана пропорциональна
Рис. 19. Зависимость
квазистационарной концентрации СН2О от температуры
при различных катализаторах:
1 — SnCl4; 2— BF, Et2O.
* Это справедливо для реакционной системы, не требующей ионогенного
сокатализатора.
77
квадрату концентрации триоксана и катализатора и обратно
пропорциональна концентрации воды в системе.
Показано, что на атермической стадии в основном происходит
образование растворимых олигомерных полиоксиметилен-катионов,
которое практически не сопровождается изменением энтальпии
системы. Одновременно в незначительной степени происходит
накопление формальдегида.
Кристаллизация полимера начнется, когда олигомерные катионы
достигнут критической длины, зависящей от растворителя и
температуры. При этом произойдет сдвиг равновесия в сторону
образования твердого полимера.
На основании этих данных можно представить себе следующую
качественную картину инициирования полимеризации триоксана:
1. Во время индукционного периода (рассматриваем случай,
когда содержание воды в реакционной системе относительно велико)
одновременно имеют место три равновесных реакции:
а) взаимодействие катализатора с триоксаном, приводящее к
раскрытию цикла и выделению свободного формальдегида;
б) относительно медленное взаимодействие активных центров
с триоксаном, приводящее к образованию олигомеров;
в) рост олигомеров с одновременным отщеплением формальде-
гидных звеньев и передачей цепи на воду *.
Схему реакции в этих условиях можно представить следующим
образом:
Катализатор+ Н2О диссоциация > н+ +Анион
уСН2—Оч + уСН2—Оу
н++о; хсн2 —> не/ хсн2 5=>
*~7 но—сн2—о—сн2—о— сн2
+ /сн2-оч <сн>°ь ||
но(сн2оJсн2о^ ;сн2 <—
ХсНа-о/ НО-СНз-О-СНа
НО(СН2ОMСН2 -^-> НО(СН2ОNСН26н2 —> НО(СН2ОNСН2ОН + Н+
2. Вода (помимо того, что она выполняет функцию сокатализа-
тора) в основном расходуется в реакции передачи цепи. Характер
зависимости средней длины цепи олигомеров от продолжительности
реакции определяется соотношением констант скоростей роста и
передачи цепи и концентрацией воды в системе. Чем больше послед-
* Предполагается, что в системе образуется метиленгликоль, который
участвует в реакции передачи цепи.
78
няя, тем медленнее будут расти олигомеры, и тем дольше период
индукции.
3. Образование мономерного формальдегида можно
рассматривать или как побочный процесс (если оно происходит за счет
расщепления триоксана), или как процесс, замедляющий рост олигомеров
(в результате их деполимеризации). Влияние на эти процессы
природы катализатора и растворителя пока не совсем ясно. Можно
предположить, что распад олигомеров быстрее протекает в том
случае, когда полимеризация происходит с участием ионов карбония
или вторичных ионов оксония (неустойчивых по сравнению с
третичными оксоний-ионами). Вода всегда способствует образованию
вторичных оксоний-ионов. При инициировании триалкилоксоние-
выми солями полимеризация, очевидно, протекает по оксониевому
механизму, причем деполимеризация кинетически затруднена [36]:
ХН2-0 4
/ ;СН2 5Z> CaHe-(/ )СН2 + (С2Н6JО
хсн2-ск хх
SbG+16/GH2-O4 ХН2-ОЧ
w-o^ ;сн2+о; ;сн2
(к \сн2-ск
^4
^z± С2Нб(ОСН2)й+3—О СН2
хсн2-ск
Подобная схема была предложена недавно для катионной
полимеризации тетрагидрофурана на оксониевых солях (вместо оксониево-
карбониевого механизма, предложенного Меервейном [40, 41].
Ранее она рассматривалась для оксациклобутана [43].
Уязвимое место в этой реакционной схеме — зависимость
скорости реакции от наличия в системе твердого полимера.
Действительно, показано, что добавка твердого полимера несколько ускоряет
процесс полимеризации. Но в то же время известно, что часто во
время индукционного периода происходит накопление полимера
и резкое возрастание скорости реакции наблюдается уже после
образования 3—4% полимера (от веса триоксана).
Принципиально иное объяснение заключается в том, что период
индукции связан с медленным инициированием реакции ионами гидр-
оксония. Если предполагать, что это бимолекулярная реакция второго
порядка, то обработка экспериментальных данных дает хорошие
результаты.
Скорость полимеризации
Характерная особенность полимеризации триоксана —
постепенное уменьшение скорости реакции, в результате чего кривые
зависимости конверсии от продолжительности реакции «запредели-
ваются» (рис. 20).
79
Полимеризация триоксана в хлористом метилене и нитробензоле
с применением в качестве катализатора SnCl4 протекает как реакция
первого порядка по катализатору [28] (рис. 21). Такая зависимость
характерна для катионной полимеризации винильных мономеров
[42] и напряженных циклических эфиров [41]. При проведении
полимеризации с BF3O(C4H9J в циклогексане также был установлен
первый порядок по катализатору [35]. Однако данные, сообщаемые
Хигашимурой и др. [30], несколько отличаются от приведенных
выше: в бензоле [с ВР3О(С2Н5J] сохранялся первый порядок,
в этилендихлориде максимальная скорость реакции была
пропорциональна корню квадратному из концентрации катализатора,
f
/
/
о
/
в
Время у ч
12
Рис. 20. Зависимость выхода
полимера от продолжительности
полимеризации (раствор триоксана в
бензоле, концентрация триоксана
1,5 моль/л, катализатор ВГ3О(С2НбJ.
Рис. 21. Зависимость скорости
реакции от концентрации
катализатора [27]:
1 — SnCl4; 2 — BF8O(G2H5)a.
а в нитробензоле при концентрации триоксана около 1,1 моль/л
скорость полимеризации вообще не зависела от концентрации
катализатора в широких пределах (нулевой порядок).
Противоречие в приведенных выше результатах можно объяснить
различной природой применяемых катализаторов. Так, SnCl4, по-
видимому, всегда нуждается в ионогенном сокатализаторе. Первый
порядок в данном случае указывает на то, что либо весь введенный
катализатор, либо его определенная часть (в зависимости от
содержания воды в системе) участвует в образовании активных центров:
SnCl4 + HOH —> [SnCl4OH]-H+
В случае применения BF3 и его комплексов с эфирами механизм
полимеризации более сложен.
Влияние воды на кинетику полимеризации триоксана, вероятно»
нельзя рассматривать в отрыве от природы катализатора. Например,
эффект действия воды в системе, для которой не нужен ионогенный
сокатализатор, рассмотрен в работе [37]. Гидроперхлорат (Н3ОСЮ4)
легко диссоциирует и достаточно устойчив. Если его использовать
в качестве катализатора, то, очевидно, для его активации вода
не нужна. Влияние воды изучалось при полимеризации расплава
триоксана при 70° С. Получена следующая зависимость между
максимальной скоростью реакции и концентрацией воды в системе
(рис. 22):
A1)
где wm&x — скорость реакции в присутствии воды; w0 — скорость
реакции в абсолютно безводной системе (получена экстраполяцией);
К — tg угла наклона прямой в выбранных координатах.
Из рис. 22 следует, что в абсолютно
сухой системе полимеризация будет
протекать с взрывной скоростью.
В табл. 14 приводятся данные о
влиянии воды на скорость полимеризации
в присутствии катализаторов,
нуждающихся в ионогенном сокатализаторе.
Эти данные носят весьма
противоречивый характер и, очевидно, нуждаются
в дальнейшем уточнении.
Кинетический анализ влияния воды
на скорость реакции на основе схемы,
представленной на стр. 78,
показывает, что замедление реакции нельзя
объяснить меньшей активностью
образующихся в результате атаки протона
ионов оксония:
ю3
lf°2
* 10
1,0
ч
\
\
\
\
V
\
НО(СН2О)„СН2 + НОН —> НО(СН2О)ПСН2ОН2
Рис. 22. Зависимость
скорости полимеризации
триоксана в расплаве от
концентрации воды в
реакционной системе (концентрация
катализатора НСЮ4 —
1,56 -10-6 моль/л).
Наблюдаемую экспоненциальную зависимость качественно можно
объяснить тем, что молекулы воды, образуя ассоциаты с активными
центрами, дезактивируют последние [37]. В сильно полярных средах
образование таких ассоциатов затруднено, поэтому наличие воды
мало влияет на скорость полимеризации.
По уменьшению количества воды и триоксана в ходе реакции
(определяемому аналитически) можно найти относительную
константу скорости передачи цепи на воду:
In
[сн,оЬ
A2)
Таким способом Керн, Бадер и Яакс [37] подсчитали среднее
значение к' о для случая полимеризации раствора триоксана при
6 Заказ 1135.
81
Растворитель
Циклогексан
н-Гексан
Бензол
Метиленхлорид
Этиленхлорид
Смесь н-гептана и
нитробензола
Нитробензол
Нитробензол
Влияние
Диэлектрическая
пр оницаемость
2,0
1,9
2,3
9,0
10,3
15,6*
35,0
35,0
воды на скорость поли
Катализатор
BF3O(C4H9J
BF3O(C2H5J
BF3O(CaH5J
SnCl4
-Si+HSO4-
BF3O(C2H5J
BF3O(C2H5J
* Диэлектрическая проницаемость равна таковой для расплава триоксана [26] при
** При больших добавках воды скорость полимеризации уменьшалась (экстремаль
*** Максимальная скорость наблюдалась при соотношении -~—Ц-^-я-*—\
[ск ] 2 "
25° С с катализатором Н3ОСЮ4 (к'н 0 =\2,6). Это значит, что
вода расходуется в ходе реакции (на передачу цепи) относительно
быстрее, чем триоксан.
Скорость ионной полимеризации должна зависеть от
диэлектрической проницаемости реакционной среды. Это положение впервые
было подтверждено при изучении катионнои полимеризации стирола,
при которой скорость роста цепи увеличивалась с ростом
диэлектрической проницаемости растворителя [44—47].
Экспериментальные данные, полученные в сопоставимых
условиях, показывают, что скорость полимеризации триоксана и выход
полимера увеличиваются с ростом диэлектрической проницаемости
реакционной системы [29, 34]. Однако Мики, Хигашимура и Ока-
мура [34] обнаружили неожиданный эффект: при малых
концентрациях триоксана эта зависимость имеет обратный характер.
Эта особенность полимеризации триоксана в растворе не имеет
аналогии в других катионных процессах. Наблюдаемое уменьшение
скорости реакции при низких концентрациях мономера можно
качественно объяснить на основе следующих представлений [34].
Катализатор (AB) диссоциирует на ионные пары (А+ • • • В~),
которые затем сольватируются молекулами мономера (М) или
растворителя (S):
къ^Ь
А+І-В-
:[AS]+B-
Сольватация активных центров молекулами мономера приводит
к инициированию полимеризации, причем лимитирующей стадией
может быть диссоциация молекул катализатора (скорость диссоциа-
82
ТАБЛИЦА 14
меризации триоксана в растворе
Концентрация
триоксана
моль/л
4,8
0,37
3,3
3,0
7,0
3,3
3,0
Концентрация
катализатора
моль/л* 103
1,1
9,4
10
9
16,6
1
1
Концентрация воды
моль/л» 10*
0,35-2,78
0,40-3,4
0,30-10,8
0,5—5
11,0-71,0
1,8-8,3
1-Ю
Темпе-
рату-
65
30
30
20
80
30
20
Изменение скорости
реакции с увеличением
концентрации воды
Не происходит
Увеличилась **
Экстремальная
зависимость ***
Нет
Увеличилась **
Не происходит
Нет
Литература
35
30
30
101
26
30
101
80° С.
ная зависимость).
ции зависит от полярности среды). При низких концентрациях
мономера существует конкуренция между специфической сольватацией
активных центров молекулами мономера и растворителя.
Основность триоксана невелика [23] и поэтому Jtg в некоторых полярных
средах может быть выше км.
Определение порядка реакции по мономеру привело к получению
результатов, крайне трудно поддающихся интерпретации. Некоторые
экспериментальные данные приводятся в табл. 15.
ТАБЛИЦА 15
Кинетика (порядок реакции) полимеризации триоксана
Катализатор
Растворитель
Is
BF3O(C4H9J
SnCl4
BF3O(C2H6J
SnCl4
BF3O(C2H6J
BF3O(G2H6J
BF3O(G2H5J
BF3O(C2H5J
BF3O(C2H5J
(C2H5KOSbCl6
Циклогексан
Метилендихлорид
Этилендихлорид
Нитробензол
Нитробензол
Гексан
Бензол
Хлорбензол
Метиленхлорид
Метиленхлорид
2,0
9,0
10,3
35,0
35,0
2,0
2,3
5,6
9,0
9,0
65
ЗО
ЗО
30
ЗО
55
55
55
ЗО
ЗО
1
4
3,5
2
6
6
2
2
4
4
1
1
0,5
1
0
1
1
1
1
1
35
28
33
28
33
102
102
102
102
102
6*
83
Порядок реакции по мономеру не зависит от температуры. В
работе [102] делается вывод о том, что порядок реакции зависит
исключительно от химической природы растворителя и не связан с
диэлектрическими свойствами среды. Природа противоиона не влияет на
порядок реакции. Необычно высокие значения порядков реакции
возможно объясняются специфической сольватацией мономера или
активных центров молекулами растворителя. Энергия активации
скорости реакции равна приблизительно 16 ккал/молъ и не зависит
от природы растворителя [31, 32, 34]. В то же время энергия
активации периода индукции уменьшалась симбатно с уменьшением
диэлектрической проницаемости растворителя.
Кинетические кривые конверсия — продолжительность реакции,
полученные в различных условиях, имеют одну общую
закономерность. Реакция не идет до конца. Этот эффект принято связывать
с гетерогенным характером процесса, поскольку образующийся
полимер не растворим в реакционной среде и при кристаллизации
из раствора может сорбировать активные центры.
Формальный кинетический анализ периода торможения реакции,
описываемого участком кривой в координатах конверсия — время,
приводят Оньон и Тэйлор [35] для случая полимеризации триоксана
в циклогексане при 65° С в присутствии катализатора BF3O(C4H9J.
Экспериментальные данные лучше всего описывались уравнением,
выведенным из предположения, что торможение реакции происходит
за счет ингибирования при взаимодействии активного центра с
растворителем. При этом концентрация «ингибитора» должна быть
постоянна в ходе реакции. Сделать какое-либо предположение о
химической природе этого «ингибитора» авторы статьи не смогли.
Вообще представляется сомнительным, чтобы удалось выявить
«ингибитор», который присутствует во всех растворителях.
Изменение концентрации активных центров полимеризации [А]
выражается уравнением:
--^P- = ko6vAA][S] A3)
где IS] — концентрация растворителя. Откуда
[4] = [Л]оехр(-Аобр.[?]О A4)
Уменьшение концентрации триоксана в ходе реакции составит:
откуда
К] =
Если а — доля прореагировавшего мономера (степень
конверсии) , то
1п^=-1пA-а) = ^ЦЬ=[1-вхр(-Аобр.[^]0] A6)
IctJ #обр. L°J
При t—>оо
/fD [x]n
1пA-апр.) = -г^ї A7)
Лобр. PJ
84
Рассчитанные по этому уравнению кинетические кривые
зависимости выхода от продолжительности реакции представлены
на рис. 23.
Лииз и Баумбер [49] предложили физическую трактовку явления
«занределивания». Они считают, что при быстрой кристаллизации но-
лиоксиметилена активные центры дезактивируются, попадая в глубь
кристаллов, и скорость полимеризации уменьшается. Специальные
измерения при полимеризации в присутствии больших добавок воды*
показали, что наблюдается рост
кристаллов полимера во времени,
причем зависимость размеров кристал- § 0,6
лов от продолжительности реакции
имеет линейный характер [15].
Молекулярный вес
Для триоксана было найдено, что
молекулярный вес является функцией
степени завершенности реакции,
причем в зависимости от условий
реакции вид этой функции подчас при-
ЮО 200
Время, мин
обретает весьма сложный характер. Рис. 23. Экспериментальные кри-
Зависимость молекулярного веса вые полимеризации триоксана в
от концентрации мономера и степени
завершенности реакции изучалась в
растворе этилендихлорида при
65° С и теоретические кривые
(сплошные линии) [35] при раз-
О — ІД0-І0-3 моль/л; • — 0,72-І0-3;
Д — 0,36-iO-3.
Концентрация триоксана 5,75 моль/л.
работе 150]. Полимеризацию прово- личных концентрациях катализа-
дили в хлористом метилене и нитро- тора BF3O(C4H9J:
бензоле с катализатором SnCl4.
Полученные результаты представлены
на рис. 24, 25. Как видно из этих
рисунков, между исходной концентрацией триоксана в растворе и
молекулярным весом получаемого полимера имеет место линейная
зависимость. Аналогичная зависимость наблюдалась между степенью
завершенности реакции (при постоянной исходной концентрации
триоксана и катализатора) и молекулярным весом полимера.
Подобная зависимость наблюдается и при полимеризации «живущих»
полимеров.
По определению интегральная степень полимеризации равна:
$(d[M]/dt)dt
A8)
где [R]j— концентрация растущих полимерных цепей; [Р] —
Вода позволяла замедлить скорость реакции.
85
концентрация неактивных («мертвых») полимерных цепей; [М ]—
концентрация мономера.
Интеграл в числителе фактически показывает расход мономера,
т. е. Д [М]; интеграл в знаменателе, если нет передачи и гибели
цепи и происходит быстрое инициирование, — число активных
центров, откуда
[М]0о
DPU=-
[Л]
A9)
где [М]о — начальная концентрация мономера; а — степень
завершенности реакции; [А] — концентрация активных центров.
2,0
л
>
""У
у
У
V
2
20 ЬО 60 80 100
Степень превращения, %
Рис. 24. Зависимость средневесо-
вого молекулярного веса от
степени превращения при
полимеризации раствора триоксана в
нитробензоле с катализатором SnCl4
и концентрации триоксана и
катализатора (в моль/л)
соответственно:
2 Ь 6
\сЛ у моль/л
10
Рис. 25. Зависимость средневесово-
го молекулярного веса от
концентрации триоксана при
полимеризации при 30° С и степени превращения
35—40%:
1—в нитробензоле; концентрация SnGl4 =
= 2« К)-3 молъ/л\ 2—в хлористом метилене;
концентрация SnGl4 = 4«10-a моль/'л.
1— 5,3;
0,7-Ю-3; 2— 2,9; 1,4-Ю-3;
3 — 6,6; 4-Ю-3.
Так как Мп — mo(DPw), где Мп — среднечисловой
молекулярный вес, т0 — молекулярный вес мономера, то тангенс угла наклона
прямой в координатах Мп— [М]о будет равен:
B0)
[А]
Аналогично для прямой в координатах Мп—а тангенс угла наклона
равен:
_ т0 [Mb
[Л]
B1)
Специальным расчетом можно показать, что если не происходит
гибель активных центров полимеризации и все они участвуют в
реакции роста цепи, то при быстром инициировании MJMn = 1. В
условиях равновесной полимеризации MJMn = 2. Если это так, то
86
значения средневесовых молекулярных весов, полученные
экспериментально, должны совпадать с рассчитанными по формуле A9).
Близкие значения были действительно получены при предположении,
что весь катализатор, введенный в систему, расходуется на
образование активных центров.
Из теории «живущих» полимеров следует, что молекулярный вес
полимера не зависит от температуры реакции. Однако при
полимеризации триоксана наблюдалось уменьшение молекулярного веса
при повышении температуры (Е = —9 ккал/молъ) [50]. Следовательно,
теория «живущих» полимеров в данном случае неприменима.
При равновесной полимеризации,
протекающей при температуре, близкой к
предельной, нужно считаться с уменьшением
молекулярного веса при повышении
температуры реакции (рис. 26).
Зависимость Мп от температуры
наблюдали Медведев, Розенберг и др. при
образовании «живущих» полимеров в
результате катионной полимеризации тетра-
гидрофурана [41]. Однако в этом случае
речь идет о равновесных выходах
полимера.
Большинство авторов сообщает о
нелинейном и даже экстремальном характере
зависимости молекулярного веса от
конверсии. Рассмотрим влияние примесей —
агентов передачи цепи на характер этой
зависимости. Такими примесями, обычно
сопутствующими триоксану, являются
вода, метанол, метилаль, муравьиная
кислота и др.
Степень полимеризации как функция конверсии мономера
выражается уравнением:
О 20 ьо SO 80 100
Степень пребращения, %
Рис. 26. Зависимость сред-
нечислового молекулярного
веса от степени
превращения при различных
температурах (Тг < Т2 < Г3).
Выхода, соответствующие
точкам Тг, Г2, Т3,
являются предельными
(равновесными).
da
B2)
da
где а — глубина превращения; [ск] — концентрация катализатора;
[М] — концентрация мономера; [х] — концентрация агента
передачи цепи.
Изменение концентрации мономера и агента передачи цепи во
время полимеризации равно:
dt
d[x\
dt ~
B3)
87
где [Rj] — концентрация полимерных активных центров,
состоящих из ] звеньев мономера.
Относительный расход агента передачи цепи составит:
d[M] ~" кр ' [М]~Є[М]
к
где є = -г2- — относительная константа передачи цепи.
После интегрирования получаем:
W«=Wo(l-a)e B5)
где
[M]o
При мгновенном зарождении активных центров степень
полимеризации равна:
а[М]
"т:—т.—тгг B6)
Это уравнение качественно описывает процесс полимеризации.
Дальнейшие усложнения возможны при наличии нескольких
веществ, действующих как агенты передачи цепи [20,75,89].
Так, например, экстремальный характер зависимости
молекулярного веса от конверсии можно объяснить накоплением в системе
малоактивного агента передачи цепи с относительной константой
передачи ниже единицы.
Переходя к рассмотрению действия отдельных примесей, следует
особо остановиться на влиянии воды. Очевидно, что в зависимости
от природы катализатора и способа введения воды в реакционную
систему она будет по-разному влиять на молекулярный вес полимера.
Механизм передачи цепи на воду можно представить следующим
образом [63]:
Н2О +СН2О —> НОСН2ОН
Н Н
+ I I
ОСН2+НОСН2ОН —> .-• — ОСН2-О+ —>• ОСН2-О + НОСН2
I
СН2ОН
Аналогичным образом осуществляется передача цепи на спирты и
ацетали, которые являются активными агентами передачи цепи.
Характерной особенностью полимеризации триоксана в отличие
от полимеризации формальдегида является образование относительно
термостабильного полимера. Если термическую стабильность
полимера оценивать по количеству неразложившегося остатка после
30-минутного нагревания образца r вакууме при 220° С в течение
30 мин, то доля стабильного полимера в случае полимеризации
триоксана составляет обычно 40—70% аротив 10—30% в случае формаль-
88
дегида. Предполагается, что увеличение стабильности связано
с реакциями передачи цепи на примеси, а также спонтанными
реакциями передачи цепи. Например, в работе [51] приводятся следующие
данные о влиянии типа агента передачи цепи на термостабильность
полимера:
Доля
стабильного полимера
%
Контрольный опыт 40
Н2О 5
СН3ОН 35
СН3ОСН2ОСН3 75
Передача цепи на воду приводит к образованию большого
количества концевых гидроксильных групп и, как следствие, к падению
стабильности. Запатентован процесс полимеризации триоксана в
присутствии ацеталей [48], позволяющий получить продукт со
стабильностью около 90%.
Среди спонтанных реакций передачи цепи, открытых совсем
недавно, следует отметить так называемый «гидридный переход»
[51], протекающий по схеме:
Н н Н
R-OH2C X С< —> R-OH2G4 /Сч /СН3 —>
\ / \ / \Н ХК MX
О О
—>¦ R-OH2C4 .С=О ^ СН3
Возможна и другая реакция с отщеплением молекулы метилформиата
[102]: .
н
I + +
R—ОСН2—ОСН—ОСН2 —* R—ОСН2 + О=СН—ОСН3
Возможность образования макроциклических молекул за счет
внутримолекулярной передачи цепи при полимеризации ацеталей
рассмотрена в работах [9, 22, 39].
ПОЛИМЕРИЗАЦИЯ В РАСПЛАВЕ
Закономерности полимеризации триоксана в растворе и расплаве
имеют много общего, однако для расплава характерны некоторые
специфические черты, поэтому мы рассмотрим кинетику
полимеризации в расплаве отдельно.
Кинетика полимеризации триоксана в расплаве изучалась в
основном Кучерой с сотр. [26, 38, 54, 55]. В интервале температур 70—
90° С скорость полимеризации с применением обычных катализаторов
типа кислот Льюиса была слишком велика, поэтому применялся
менее активный катализатор, представляющий собой комплекс поли-
силоксана с серной кислотой [88].
Общие закономерности полимеризации оказались аналогичными
тем, которые наблюдались при проведении процесса в растворе.
Имел место индукционный период, в течение которого происходило
накопление мономерного формальдегида [55]. Продолжительность
индукционного периода зависела от концентрации воды в системе.
Малые добавки воды играли роль сокатализатора и ускоряли
реакцию.
Авторы предполагают, что скорость взаимодействия катализахора
с мономерным формальдегидом намного выше скорости
взаимодействия катализатора с триоксаном. Отсюда следует, что после
достижения равновесной
концентрации формальдегида
инициирование реакции
происходит с большой
скоростью.
Скорость реакции в
этом случае
пропорциональна концентрации
катализатора . Зависимость
степени конверсии от
времени (рис. 27) описывается
следующими уравнениями:
индукционный период
50
200
250
100 150
Время, мин
Рис. 27. Зависимость выхода полимера от
продолжительности реакции при
полимеризации триоксана в расплаве при 85° С и
различных концентрациях катализатора
(комплекс полисилокеана с H2SO4):
1 — 1,51 ммолъ/кг; 2—0,76 ммолъ/кг;
3 — 0,54 ммолъ/кг.
[М]о — [М]
[М]0-[М]
[М]о
B7)
где 0 < t < tmn,
процесс полимеризации
т0
[Mb A2 [Mb B8)
где [M]o — число молей триоксана при t = 0; [М] — число молей
триоксана в момент времени t; /0 — число молей инициатора при
t — 0; кг — константа скорости процесса, лимитирующего общую
скорость реакции; к2 — константа скорости процесса дезактивации
инициатора; т0 — молекулярный вес триоксана (в лг/молъ); G —
вес триоксана к моменту времени t; &ф — константа скорости
реакции образования СН2О.
Кинетический анализ принятой схемы полимеризации показал,
что лимитирующей стадией является присоединение триоксана
к активному центру (А^).
Молекулярный вес полимера зависел от концентрации агентов
передачи цепи в реакционной системе и' глубины протекания
реакции. Выведено уравнение [54] для среднечисловой степени
полимеризации при условии, что агент передачи цепи расходуется во время
реакции по тому же закону, что и мономер. Полученное уравнение
90
аналогично соотношению, предложенному Тобольским для средне-
числовой степени полимеризации при радикальной полимеризации
в растворе:
[Mb«
ю
8
B9)
Степень полимеризации не зависит от концентрации выбранного
катализатора.
Экспериментальная проверка правильности этого соотношения
проводилась в серии опытов с контролируемым содержанием воды
в системе при 85° С и концентрации
катализатора 81,8-їй молъ/кг. Среднечис-
ловой молекулярный вес рассчитывался
по формуле:
— о чз
Лир. = [ЛІН-0,34 [т]]2 -0,2 (по уравнению Хаг-
генса).
Приведенная вязкость измерялась в
смеси тетрахлорэтан —фенол C : 1) при
90° С @,2 г/100 см3).
На рис. 28 показана зависимость
степени полимеризации от конверсии.
Численное значение относительной
константы передачи цепи е (—26) было
получено графической экстраполяцией
кривой, описываемой уравнением B9),
к нулевой конверсии:
А
/
у
C0)
и 10 20 30
Степень превращения, %
Рис. 28. Зависимость средней
степени полимеризации от
степени превращения при поли-
меризации триоксана в
расплаве [54]; концентрация воды в
системе:
2—27,77-10-* молъ/кг; 2 —
55,55-10-* молъ/кг; 3— 83,33-10-*
молъ/кг; 4— 111,11 • 10~4 молъ/кг.
lg A-а)
Совпадение экспериментальных [и
расчетных величин (рис. 28) довольно
хорошее.
Поскольку зависимости констант
скоростей передачи и роста цепи от температуры различны,
молекулярный вес должен также изменяться с температурой. Энергия
активации относительной константы передачи цепи равна
6,1 ккал/молъ. Ниже приведены значения е при различных
температурах:
Температура, °С 70 75 80 85
е 18 20 23 26
ПОЛИМЕРИЗАЦИЯ В ТВЕРДОЙ ФАЗЕ
Большой научный и практический интерес представляет
полимеризация мономеров в кристаллическом состоянии под действием
радиационного излучения или катализаторов. Триоксан благодаря
91
относительно высокой температуре плавления оказался весьма
удобным объектом для изучения механизма твердофазной
полимеризации. Напомним, что первые наблюдения полимеризации три-
оксана [3] были связаны с кристаллизацией и возгонкой очищенных
кристаллов триоксана.
Спонтанная полимеризация кристаллического
триоксана
Триоксан, содержащий менее 0,1% воды, склонен к
полимеризации при фазовых переходах, причем образуется чрезвычайно
высокомолекулярный полиоксиметилен. Уже одно это явление само
по себе представляло большой интерес, так как тот же триоксан
в расплаве или растворе при действии любых катализаторов образует
полимер с более низким молекулярным весом.
Было установлено, что спонтанная полимеризация свойственна
только твердому триоксану, в то время как расплав, раствор или
газообразный продукт не проявляют тенденции к самопроизвольному
образованию полимера.
Керн и Яакс [56] предположили, что катализатором
полимеризации является мономерный формальдегид. Когда из триоксана
удаляли следы формальдегида (обработкой с окисью серебра), то при
сублимации полимер не получался. Такое же действие оказывают
добавки воды, аминов и других соединений, способных
взаимодействовать с формальдегидом.
Механизм протекания этого процесса не совсем ясен. Расплав
и раствор триоксана вполне устойчивы в присутствии мономерного
формальдегида, поэтому отпадают соображения о побочных реакциях
формальдегида, приводящих к образованию муравьиной кислоты,
которая, в свою очередь, инициирует полимеризацию триоксана [57].
Вместе с тем известно, что мономерный формальдегид
взаимодействует с твердым триоксаном, вызывая полимеризацию последнего
на поверхности реакционного сосуда. Вероятно, при этом облегчается
разрыв непредельной связи в молекулах формальдегида и на
поверхности кристаллов образуются олигомерные цвиттер-ионы
О—СН2—О—СН2—О—СН2
которые «живущим» (катионным) концом могут вести
полимеризацию триоксана по катионному механизму.
Кинетика полимеризации в твердой фазе
Специфической особенностью твердофазной полимеризации
является то, что мономер находится в упорядоченном состоянии,
а это особым образом сказывается на кинетике полимеризации.
Характерные черты процессов, протекающих в твердой фазе,
заключаются в следующем [76]:
92
3,0
1,0
о
1. Времена диффузии обычно велики и значительно превышают
продолжительность реакции. Отсюда следует, что пространственное
перемещение активных центров в кристалле будет главным образом
определяться скоростью реакции роста цепи (и передачи цепи),
а не диффузией активных центров.
2. Анизотропность реакции: из-за различного геометрического
положения молекул в кристалле их активность различна.
3. Неоднородность кристаллической структуры, связанная с
наличием «дефектов». Эти дефекты мигрируют по кристаллу, причем
скорость миграции имеет тот же
порядок, что и скорость реакции.
Для инициирования полимеризации
триоксана в твердой фазе можно
использовать те же катализаторы, что и при
жидкофазной полимеризации, а также
радиационное излучение [21, 70]. В ряде
случаев каталитическую активность
могут проявлять вещества,
малоактивные в жидкой фазе, например
кристаллический иод.
Способ введения катализатора играет
весьма важную роль. Такие вещества,
как BF3 или SnCl4, можно подавать на
поверхность кристаллов из газовой
фазы или суспендировать кристаллы
триоксана в растворе этих катализаторов.
В качестве растворителя можно
использовать алифатические углеводороды,
плохо растворяющие триоксан. В
растворе распределение катализатора более
однородно [90—92].
Иод можно ввести в расплав
триоксана, а затем охладить смесь, при
этом будет достигнуто равномерное распределение иода по всему
объему мономера. В результате радиационной пост-полимеризации
также достигается равномерное распределение активных центров.
Экспериментальные данные о каталитической полимеризации
триоксана в твердой фазе приводятся в работах Окамуры с сотр.
[58, 59, 94]. Отличия от кинетики жидкофазной полимеризации
очевидны: молекулярный вес уменьшается с увеличением степени
превращения мономера (рис. 29); энергия активации молекулярного
веса имеет положительную величину и характер кривой в
координатах молекулярный вес — конверсия меняется с изменением
температуры.
Кинетический анализ экспериментальных данных, приведенных
в работах [6—62, 64—69, 76] (в качестве катализаторов применяли
SnCl4 и кристаллический иод), показал, что во всех случаях
результаты согласуются с теорией анизотропной полимеризации [76, 95].
93
10 20 30
Степень превращения, %
Рис. 29. Зависимость т|пр. от
степени превращения [58]:
1 — полимеризация триоксана в
твердой фазе,катализатор BF3Et20;
2 — то же, катализатор SnCl4; 3—
полимеризация триоксана в
растворе, катализатор BF8Et2O; 4 —
то же, катализатор SnCl4.
Формальное объяснение кинетики полимеризации в твердой
фазе основывается на предположении, что может существовать
несколько активных центров, имеющих одинаковую химическую
структуру, но различную активность. Физическую картину такого
процесса можно представить следующим образом: активный центр,
попавший в бездефектный участок кристалла, имеет высокую
константу скорости роста (/q); активный центр, находящийся в месте
какого-нибудь дефекта, проявляет значительно меньшую
активность (к2).
Запишем это положение так:
C1)
C2)
dt "
где w] и w\ — скорости образования активных центров Rx и R2\
[Ri] — концентрация активных центров, которым соответствует
константа роста кг; [R2] — концентрация активных центров,
которым соответствует константа роста к2, причем [R±] могут переходить
в [R2] (попав в дефект).
Если обозначить концентрацию дефектов [D], концентрацию
мономера [М], вероятность встречи активного центра с
дефектом а, то
[М]
Скорость перехода [і?х] в [R2] равна кга [R±].
Уравнение для выхода полимера (П) примет вид:
^f (при а « 1) C3)
При анизотропной каталитической или радиационной
пост-полимеризации к2 т^= 0. При мгновенном инициировании
1
(при кга > к2)
где [R^q и [i?2lo — начальная концентрация активных центров R±
и Д2.
Отсюда следует, что кинетическая кривая, выражающая
зависимость выхода полимера от времени, имеет два отрезка с различным
наклоном:
Во всех случаях при твердофазной полимеризации триоксана
наблюдается двухстадийный процесс, причем значение кг столь
велико, что измерить начальную скорость процесса невозможно.
Отрезок, отсекаемый кинетической кривой на оси ординат (рис. 30),
предложено называть «скачком».
При инициировании иодом [60] достигается равномерное
распределение катализатора, и величина «скачка», а также скорость
реакции пропорциональны концентрации введенного катализатора.
Такая же картина наблюдается при пост-полимеризации [69], в этом
случае пропорциональность соблюдается по отношению к дозе
предварительного облучения.
Энергия активации процесса полимеризации равна сумме энергии
активации «скачка» и энергии активации к2 (в предположении, что
[7?1]0 и Ш2]о одинаково зависят от температуры). Энергия
активации к2 приблизительно одинакова для всех случаев полимеризации
триоксана в твердой фазе C1 ±
± 3 ккал/молъ).
Инициирование
кристаллическим иодом приводит к сильной
зависимости величины «скачка»
от температуры [69] (Ejct =
= 46 ккал/молъ). Предполагается,
что это связано с большой
величиной энергии ионизации
кристаллического иода. Для SnCl4
соответствующая величина составляла
4,5 ккал/молъ [65].
В работе [59] эффект «скачка»
в начальной стадии полимеризации
практически не наблюдался из-за
сильной дефектности кристаллов
триоксана. Кажущаяся энергия
активации полимеризации
составляла 8,6 ккал/молъ. Однако если экспериментальные данные,
приведенные в работе [59], пересчитать по схеме анизотропной
полимеризации, то Е]с2 = 32 ккал/молъ.
Неожиданно высокие скорости полимеризации в твердой фазе
объясняются влиянием кристаллической решетки, которая
способствует реакции роста цепи в определенных направлениях (в
направлении оси с в кристалле триоксана). Если это так, то увеличение
дефектности кристалла должно приводить к уменьшению величины
«скачка» и скорости полимеризации. Природа дефектов в
кристаллической решетке может быть весьма различной (разрывы
поверхности, узлы, молекулы примесей и т. п.) и зависеть от размеров
кристаллов и степени их чистоты.
Наибольшие «скачок» и скорость полимеризации наблюдаются
при пост-полимеризации монокристалла. При переходе к
поликристаллам эти величины резко уменьшаются (рис. 31).
Аналогичные явления наблюдаются при полимеризации
триоксана с применением иода, когда в качестве примеси вводится
1,4-диоксан. Добавление бензола или циклогексана не влияет на
95
/ 2
Время, ч
Рис. 30. Зависимость выхода
полимера от продолжительности
полимеризации триоксана в твердой
фазе [60]:
1 — пост-полимеризация (y-из лучение);
2— катализатор SnCl4; 3 — катализатор 12.
кинетику полимеризации, так как из-за слабой энергии
взаимодействия с триоксаном эти вещества легко диффундируют из него [96].
Дефектность кристаллов может изменяться в ходе реакции,
причем чем выше температура, тем большую гибкость приобретает
кристаллическая решетка и тем легче примеси диффундируют из
кристалла. Это явление можно назвать «отжигом» дефектов.
Так, при облучении кристаллов триоксана возникают дефекты
вследствие частичного разрушения кристаллической решетки. При
температуре ниже 45° С кинетические кривые приобретают S-образ-
ную форму, причем величина индукци-
3q\ 1/II онного периода растет с увеличением
дозы предварительного облучения и
уменьшается с температурой.
1
I
W
О
А
у X
J
* зо
%20
ь
f
1
...L
3\
I
mm—о
ЬО 80
Бремя, мин
5
Время, мин
10
Рис. 31. Зависимость величины
«скачка» от дисперсности
кристаллов триоксана [64]:
1 — монокристалл; 2 — суспензия
кристаллов триоксана; з — расплав
триоксана вылит в жидкий азот;
4 — механически разрушенный
монокристалл.
Рис. 32. Влияние концентрации воды на
кинетику полимеризации триоксана в твердой
фазе [76]:
1 — очищенный образец; 2 — следы воды;
3— 5«10~6 моль Ж2О/молъ триоксана.
Подобное явление наблюдалось при инициировании
полимеризации иодом в присутствии добавок воды, метанола и левоглюкозана.
Нужно заметить, что при твердофазной полимеризации триоксана
в присутствии добавок воды удавалось получать значительно более
высокомолекулярный полимер, чем при полимеризации в растворе.
Изучение кинетики полимеризации в присутствии воды и других
перечисленных выше соединений показало, что кинетические кривые
имеют характерную S-образную форму, причем индукционный
период пропорционален количеству примеси и уменьшается
при повышении температуры (рис. 32).
На аналогию в механизме инициирования радиационной
постполимеризации и каталитической полимеризации в присутствии
примесей указывает совпадение значений энергий активации
индукционного периода [76] (—52 и —50 ккал/молъ). Накопление полимера
также приводит к увеличению концентрации дефектов вследствие
разрушения кристаллической структуры мономера. Здесь нужно
отметить, что при полимеризации монокристалла конфигурация
06
образующегося полимера полностью совпадает с конфигурацией
мономерного кристалла. Полученный таким способом полимер
отличается большой плотностью и высокой температурой плавления.
Для полимера, полученного из монокристалла, эти величины равны
соответственно 1,482 г/см3 и 198° С против 1,450 г/смэ и 186° С для
полимера, полученного из поликристалла (заметим, что и в этом
случае показатели степени упорядоченности полимера намного
превышают таковые для жидкофазной полимеризации) [59].
Если принять, что количество дефектов пропорционально
количеству заполимеризованного мономера, то предельный выход
полимера должен быть пропорционален корню квадратному из
концентрации катализатора.
Согласно этим представлениям, молекулярный вес уменьшается
в ходе реакции, а на начальных стадиях зависимость средней степени
полимеризации от выхода полимера описывается следующим
уравнением [76]:
C5)
где ?ll — концентрация дефектов при выходе П.
Если создать благоприятные условия для диффузии дефектов
из кристаллов (например, проводить пост-полимеризацию
монокристалла вблизи точки плавления), можно наблюдать постоянное
увеличение молекулярного веса [98].
Изменение молекулярно-весового распределения в процессе
анизотропной полимеризации рассмотрено в работе [97]. Теоретический
анализ вопроса показал, что в ходе процесса МБР должно
уменьшаться.
ЛИТЕРАТУРА
1, Hammick О. L., В о е г е е A. R., J. Chem. Soc, 121, 2738A922).
2.: S t a u d і n g er H., Singer R., Johner H., Luthy M.,
Kern W., Russidis D., Schweitzer 0., Ann., 474, 235
A929).
3. К о h 1 s с h u t t e r H. W., Ann., 482, 75 A930).
4. Уокер Дж. Ф., Формальдегид, Госхимиздат, 1957, стр. 176.
5. Pies с h P.H., The Chemistry of Gationic Polymerisation, Ch. 11, Per-
gamon Press, London, 1964.
6. D а і n t on F. S., D el v і n T. R. E., Small P. A., Trans. Farad.
Soc, 51, 12, 1710 A955).
7.: M ace oil, Progress in Stereochemistry, 1, 361 A954).
8. В awn G. E. H., В ell R. M., L edw і t h A., Polymer, 6, N 2, 95 A965).
9., Plesch P. H., Westermann P. H., Сообщение на симпозиуме
по макромолекулярной химии, Прага, 1965.
10. К ern W., Gi ef er А., Makromol. Chem., 74, 39 A964).
11. В аг nett R. L., В ell R. P., Trans. Farad. Soc, 34, 420 A938).
12. D el ері n e M., В ad och e M., Compt. rend., 214, 777 A942).
13. Parks G. S., M os her H. P., J. Polymer Sei, AI, 1979 A963).
14. Dainton F. S., Evans D.M., Haarse F. E., Melia T, Pa
Polymer, 3, № 3 A962).
7 Заказ 1135. 97
Leese LM В a u m b e r M. W., Polymer, 6, № 5, 269 A965).
Starkweather H. W., Boyd R. H., J. Phys. Ghem., 64, 410
A960).
I n о u e M., J. Polymer Sei., A-1, 2697 A963).
Kern W.,Jaacks V., J. Polymer Sei., 48, 399 A960).
Walker J. F., Du Pont, пат. США 230443, 1942 г.
Giefer А., Kern W., Makromol. Ghem., 74, 39 A964).
Okamura S., Hayashi K., Nakamura Y., Isotopes and
Radiation, 3, 416 (I960).
22. И в а н о в В. В., Б е р л и н Ал. Ал., Ениколопян Н. С.,
Высоком ол. соед., 9 (Б), 61 A967).
23. Iwatsuku S., Takigawa N., Okada M., Jamashita Y.T
I s h і і Y., Makromol. Chem., 82, 16 A965).
24. К er n W., J aacks V., Makromol. Ghem., 62, 1 A963).
25. Saegusa Т., Imai H., Furukawa J., Makromol. Ghem., 54,
218v A962).
26. Kucera M., Hladky E., Majerova К., Сообщение на
симпозиуме по макромолекулярной химии, Прага, 1965 г.
27. Р а к о в а Г. В., Романов Л. М., Ениколопян Н. С., Вы-
сокомол. соед., 6, 2178 A964).
28. Ракова Г. В., Авакян А. К., Романов Л. M.j
Ениколопян Н. С., ДАН СССР, 156, 1409 A964).
29. ИржакВ. Н.,Ракова Г. В., Романов Л. М.,Кравчук И. П.*
ЕниколопянН. С, Сообщение на симпозиуме по
макромолекулярной химии, Прага, 1965 г.
30. Н і g а s h і m u г а Т., M і k і Т., О к a m u r a S., Bull. Chem. Soc. Japan,
38, 2067 A965), сообщение 1.
31. H і g a s h і m u г а Т., M і k і Т., О k a m u r a S., Bull. Chem. Soc. Japan,
39, 25 A966), сообщение 2.
32. H і g a s h і m u г а Т., M і k і Т., О k a m u r a S., Bull. Chem. Soc. Japan,
39, 25 A966), сообщение 3.
33. H і g as h і m u г а Т., Miki Т., Okamura S., Bull. Chem. Soc.
Japan, 39, 25 A966), сообщение 4.
34. H і g as h і m u г а Т., Miki Т., Okamura S., Bull. Chem. Soc.
Japan, 39, 25 A966), сообщение 5.
35. Onyon P. F., Taylor K. J., European Polymer J., 1, № 2, 133
A965).
36. P о з e и б e p г Б. А.і И р ж а к В. И., Ениколопян Н. С.
Успехи химии, 35, 714 A966).
37. Kern W., Baader Н., Joacks V., Makromol. Chem., 82, 213
A965).
38. Kucera M.,Spousta M., Makromol. Chem., 82, 60 A965).
39. J aacks V., Makromol. Chem., 99, 30 A966).
40. M e erw ei n H., D elf s D., M or sch el H., Angew. Chem., 24, 927
A960).
41. Розенберг Б. А., Людвиг Е. Б., Г а н т м а х е р А.
Р.»Медведев С. С, Высокомол. соед., 6, 2035 A964).
42. Pies с h Р. Н., The Chemistry of Cationic Polymerisation, Ch. 4, Per-
gamon Press, London, 1964.
43. Rose J. В., J. Chem. Soc, 1956, 546.
44. P e p p e r D. C, Trans. Farad. Soc, 45, 397 A949).
45. P 1 e s с h P. H., The Chemistry of Cationic Polymerisation, Ch. 6, Perga-
mon Press, London, 1964.
46. I m a n і s h і Y., Higashimura Т., О k a m u r a S., Chem. High
Polymer (Japan), 18, 333 A961).
47. I man is hi Y., Nakayama H., Higashimura Т.,
Okamura S., Chem. High Polymer (Japan), 19, 154, 565 A962).
48. E и и к о л о п я н Н. С, фр. пат. 1461969, 1966 г.
49. L e e s e L,Baumber М. W., Polymer, 5, № 7, 380 A964).
Ракова Г. В...Е никол опян Н. С.,* ДАН СССР, 156, 1167 A964).
G*e r m a n Н., Fischer Е., Wessermel К., Makromol. Chem.,
90, 1 A966).
W or s fold D. J., East ham A.M., J. Am. Chem. Soc, 79, 897
A957).
Kagiga Т., Hatta M., Shimizu Т., Fukui K., J. Chem.
Soc, Japan (Ind. Chem. Soc), 66, 1890 A963).
Киша M., Spousta E., Makromol. Chem., 76, 183, 190 A964).
К и e'er a M., Spousta E., J. Polymer Sei., A-2, 3431, 3443 A964).
Kern W., J а а с k s V., Makromol. Chem., 52, 37 A962).
Wilson W., May H., Chem. Ind., 1962, 412.
Okamura S., Higashimura Т., Takeda M., Makromol.
Chem., 51, 217 A962).
59. О k a m и r a S.,Kobayashi E.,Takeda M.,Tomikawa K.,
Higashimura Т., J. Polymer Sei., 4c, № 2, 827 A963).
60. Берлин Ал. Ал.r К у з у б Л. И., Ениколопян Н. С, Высоко-
мол. соед., 8, 1200 A966).
61. Okamura S., Hayashi К., К і t a n і s h і Y., J. Polymer Sei.,
58, 925 A962).
62. Baccaredda M., В u t t a E., G і u s t у P., J. Polymer Sei., 4c,
№ 2, 953 A964).
63. Dei b ing H., Giefer A., Jaacks V., Kern W., Сообщение
на симпозиуме по макромолекулярнои химии, Прага, 1965 г.
64. Берлин Ал. Ал., К у з у б Л. И., Маркевич М. А.,
Ениколопян Н. С, Высокомол. соед., 8, 1204 A966).
65. П р у т Э. В., Е н и к о л о п я н Н. С, Высокомол. соед., 8, 1905 A966).
66. М і у a m а Н., К a m а с h і М., J. Polymer Sei., B-l, 465 A963).
67. H а у a s h і К., К і t а п і s h і Y., О k a m u r a S., Makromol. Chem.,
47, 230 A961).
68. T ak ak u г а К., Hayashi К., Okamura S., J. Polymer Sei.,
B-2, 861 A964).
69. Трофимова Г. М., Баркалов И. М., Кузьмина С. С,
Гольданский В. И., Ениколопян Н. С, ДАН СССР,
161, 882 A967).
70. О к а m u г а S., Nakashio S., Hayashi К., Sakurada L,
Isotopes and Radiation (Japan), 3, 242 A960).
71. Physicka J., Chem. Prumsl.* 14, 481 A964).
72. T а к а к u r a K., Hayashi K., Okamura S., J. Polymer Sei.,
B-2, 861 A964).
73. Takakura K., Hayashi K., Okamura S., Annual Report
of the Japonese Assotiation for Radiation Research of Polymers, 5, 139
A964).
74. Kobagashi, S і n g e r, J. Polymer Sei., B-3, 491 A965).
75. Шагинян A.A., Ениколопян H. С, Высокомол. соед., 7,
1866 A965).
76. Берлин Ал. Ал., Ениколопян Н. С, Сообщение на симпозиуме
по макромолекулярнои химии, Прага, 1965 г.
77. Celanese Corp. пат. США 2947727, 1960 г.
78. Celanese Corp., пат. США 2947727, 1960 г.
79. Celanese Corp., пат. США 2989507, 1961 г.
80. Celanese Corp., пат. США 2989506, 1961 г.
81. Celanese Corp., пат. США 2989511, 1961 г.
82. Hoechst F., англ. пат. 943684, 1963 г.
83. Hoechst F., пат. ФРГ 1330353, 1964 г.
84. Montecatini, англ. пат. 895378, 1962 г.
85. Фр. пат. 1316812, 1962 г.
86. Пат. США 3168495, 1964 г.
87. Пат. ФРГ 1152813, 1963 г.
88. Фр. пат. 1351228, 1963 г.
7* 99
89. Р а к о в а Г. В.? Шагинян А. А., ЕниколОпян Н. С, Вы-
сокомол. соед. A968).
90. А к у т ин М. С, Тихомирова Н. С, Ермолаев А. Д.,
Пласт, массы, № 12, 67 A963).
91. Baccaredda М-., Butt a E., Giusti P., J. Polymer Sei., 1С,
№ 4, 953 A963).
92. Р а 1 f г е у G. F., см. У о к е р Дж. Ф., Формальдегид, Госхимиздат, 1957^
стр. 172.
93. В а с с а г е d d a M., Butta E., Morelli F., Masetti G.f
Makromol. Chem., 93, 137 A966).
94. Okamura S., Kobayashi E., Higashimura Т.,
Makromol. Ghem., 94, 20 A966).
95. Берлин Ал. Ал., Гольданский В. И., Баркалов И. М.,
Ениколопян Н. С., ДАН СССР, 161, 373 A965).
96. П ш е ж е ц к и й В. С, К а р г и н В. А., Капансан А. Т.,
Рыбникова Л. Ф., Высокомол. соед., 6, 1442 A964).
97. Берлин Ал. Ал., Ениколопян Н. С, Высокомол. соед., 8,412
A966).
98. Т р о ф и м о в а Г. М., Баркалов И. М., К а р ю х и н а Г. А.,
Гольданский В. И., Ениколопян Н. С, Высокомол.
соед., 9 (А), 594, A967).
99. Mel і а Т. P., Bailey D.,Tyson A., J. Appl. Chem., 17, 15 A967).;
100. J а а с k s V., Makromol. Chem., 101, 33 A967).
101. Ракова Г. В., Кузнецова В. И., Харитонова Л. А.?
Ениколопян Н. С, Сообщение на 2-й конференции по строению
и реакционной способности ацеталей, Фрунзе, 1967.
102. Кравчук И. П., Ениколопян Н. С, Высокомол. соед., 9 (А),
2114 A967).
ГЛАВА IV
ДЕСТРУКЦИЯ И СТАБИЛИЗАЦИЯ
ПОЛИФОРМАЛЬДЕГИДА
При оценке стабильности полимеров принято различать стойкость
их к действию:
1) температуры (термическая деструкция);
2) кислорода (окислительная деструкция, при повышенных
температурах — термоокислительная деструкция);
3) коррозионных сред (гидролиз и ацидолиз);
4) ультрафиолетовых лучей (фотолиз или фотоокисление, если
одновременно наблюдается окислительная деструкция);
5) специальных агентов — озона, радиоактивного излучения
и др.;
6) механических нагрузок (механодеструкция).
Во многих случаях полимер или изделия, полученные из него,
подвергаются одновременному воздействию многих деструктирующих
факторов. Для изучения их влияния существуют специальные
технологические методы испытаний изделий на теплостойкость,
старение, погодоустойчивость и т. п.
Термопластичные материалы, в том числе и полиформальдегид,
должны быть достаточно термостойкими для того, чтобы выдержать
нагревание до вязко-текучего состояния во время переработки;
при этом изменение свойств материала должно быть минимальным.
Эксплуатация изделий из полимерных материалов обычно происходит
при температурах более низких, чем температура размягчения
материала.
Характерная особенность всех полиоксиметиленов заключается
в том, что при нагревании происходит отщепление молекул
мономера. Такой процесс разложения обычно называют
деполимеризацией, в отличие от деструкции — процесса разложения, не
сопровождающегося выделением летучих продуктов, который приводит
к резкому снижению молекулярного веса полимера. Оба эти процесса
могут происходить одновременно, причем в этом случае общая
картина деструкции заметно усложняется.
Было высказано предположение [4], что низкая термостойкость
полиформальдегида всецело определяется термодинамическими
причинами, а именно низкой величиной предельной температуры
полимеризации (см. гл. II). Как известно, при температуре выше
предельной (т. е. приблизительно 130° С при давлении мономера 1 атм)
101
изменение свободной энергии при полимеризации формальдегида
имеет положительную величину и равновесие в системе
HO(CH2O)WH Т"* »CH2O-f Н2О
полностью смещено в сторону образования мономера. Если учесть,
что температура плавления полиформальдегида приблизительно
на 50° выше, а вязко-текучее состояние, при котором производится
формование термопласта, реализуется при еще более высоких
температурах B00—220° С), то очевидно, что переработка
полиформальдегида происходит в термодинамически невыгодных условиях.
Соответствующие данные для некоторых термопластов приведены
в табл. 16.
ТАБЛИЦА 16
Значение предельных температур, температур плавления (размягчения)
и переработки для различных полимеров
Полимер
Полиэтилен
Полистирол
Поливинилхлорид
Полиметилметакрилат
Полиформальдегид
Предельная
температура
при
давлении 1 Ьтм
°С
407
225
312
200
130
Температура плавления
или размягчения, °С
130
80 (размягчение)
70 (размягчение)
70 (размягчение)
180
Температурный
интервал
переработки
°С
160-240
180—230
150-200
130-180
200-220
Нужно подчеркнуть, что термодинамическая неустойчивость сама
по себе не предполагает, что спонтанный переход из менее
стабильного в более стабильное состояние происходит с заметной скоростью.
Существует множество примеров, когда скорости превращения
термодинамически неустойчивых веществ исчезающе малы. Строго говоря,
большинство термопластов перерабатывается в термодинамически
невыгодных условиях, так как давление мономера над ними ниже
равновесного. а-Метилстирол и тетрагидрофуран имеют значения Гпр#
меньшие, чем формальдегид D8 и 73° С соответственно), однако
продукты полимеризации этих мономеров значительно более
термостойки, чем полиформальдегид.
Таким образом, скорость и характер деструкции полимера
зависят не только от термодинамических, но и ої кинетических факторов,
т. е. от механизма деструкции.
Проблема термостойкости полиоксиметиленов в этом аспекте
впервые была поставлена Штаудингером [1, 2], который нашел,
что деполимеризация низкомолекулярных полимеров типа пара-
формальдегида и а-полиоксиметилена обусловлена наличием у них
концевых гидроксильных групп, которые легко отщепляются при
нагревании, а также под действием основных и кислотных агентов
[3]. Замена концевых гидроксильных групп на ацетильные или мет-
102
оксильные (что достигается путем ацетилирования или метилирования
полиоксиметиленгликоля) позволяет существенно повысить
стабильность полимера. Керн и Чердрон [5] по результатам
исследования кинетики разложения диметилового эфира полиоксиметилена
со степенью полимеризации около 300 предложили пять типов
элементарных реакций, определяющих механизм деструкции
полиформальдегида:
1) деполимеризация, начинающаяся с концов цепей;
2) термическое разложение;
3) автоокислительное разложение;
4) разложение под влиянием вторичных продуктов
автоокисления;
5) гидролиз и ацидолиз.
Такая классификация элементарных реакций является в
настоящее время общепринятой.
Более подробно кинетика и механизм разложения
полиформальдегида рассмотрены в работах Ениколопяна с сотр. [6, 7].
ТЕРМИЧЕСКАЯ ДЕСТРУКЦИЯ
Сопоставление значений энергии диссоциации С--С- и С—О-свя-
зей (83,1 и 84,0 ккал/молъ соответственно) позволяет предположить,
что величины термической стойкости линейных полимеров,
содержащих эти связи, должны быть близки. Однако сравнение
термических свойств полиолефинов и полиэфиров показывает, что
различия в значениях энергии активации термического разложения,
измеренной в глубоком вакууме, весьма значительны (табл. 17).
Термическое разложение модельных соединений типа ди- или
триоксиметилена с ацетатными и бензоатными концевыми группами
изучалось [12] в интервале температур 220—230° С. Энергия
ТАБЛИЦА 17
Энергия активации термического разложения полиэфиров
и полиолефинов
Полимер
Полиметилен
Полиэтилен
Полипропилен
Полиоксиэтилен
Полиоксипропилен (атакти-
неский)
Диметиловый эфир
полиоксиметилена (степень
полимеризации 300)
Сополимер триоксана с 2%
диоксолана
Температурный
интервал
°С
400 -440
350-440
320-410
320-335
265-285
280-350
250—350
Энергия
активации
кшл/моль
76
68
61
46
20
29
46
Литература
8
9
8
10
10
5
и
103
активации этого процесса составила 29 ккал/молъ. Аналогичная
величина была получена при определении энергии активации
термического разложения триоксана [13], так что можно предполагать, что
в обоих случаях происходит разрыв молекул по ацетальным связям.
На термическую стойкость различных полимеров большое
влияние оказывает наличие «слабых» участков цепи, разветвлений,
боковых заместителей. Высококристаллические (например, изо-
тактические) полимеры обычно значительно устойчивее, чем
аморфные. Для полимеров полиацетальной структуры особое значение
имеет наличие различных примесей, способствующих протеканию
гетеролитических реакций, вследствие которых энергия активации
разложения заметно снижается.
По данным работы [14], при термической деструкции полиокси-
метиленов образуется до 98% мономерного формальдегида, что
указывает на цепной механизм этого процесса. Аналогичным
образом ведут себя полиолефины с относительно небольшими величинами
теплот и, как правило, предельных температур полимеризации
(полистирол, полиметилметакрилат, полиизобутилен, поли-а-метйл-
стирол). Исключение составляет политетрафторэтилен, теплота
полимеризации которого 46 ккал/молъ.
Для установления детального механизма деструкции большое
значение имеет определение места зарождения активных центров
деполимеризации. В зависимости от химической природы и строения
полимерной цепи активные центры могут образоваться либо на
концевых группах (зарождение по «закону концевых групп»), либо
в любой точке цепи (зарождение по «закону случая»).
Роль концевых групп в химических превращениях полимеров
впервые отметил Штаудингер [3]. Проведенное им сравнение поли-
оксиметиленов с различными концевыми группами убедительно
иллюстрирует их влияние на свойства полимеров. В дальнейшем
большое внимание было уделено полиоксиметиленам с гидроксиль-
ными концевыми группами, так как при синтезе
высокомолекулярного полиформальдегида образуются главным образом такие
продукты.
Низкая термостойкость полиоксиметиленгликолей дает основание
предполагать, что термическая деструкция (при нагревании этих
продуктов в глубоком вакууме или инертной атмосфере) происходит
по закону концевых групп. В работе [15] это доказано с помощью
строгого кинетического метода. Однозначный ответ может быть
получен при выяснении зависимости начальной скорости разложения
от средней степени полимеризации (DPn). Если эффективная
константа скорости термического разложения уменьшается с ростом DPn,
зарождение активного центра происходит по закону концевых групп
и кинетическая длина цепи меньше материальной, т. е. имеет место
гибель активных центров. Если эффективная константа скорости
увеличивается с ростом DPn1 инициирование происходит по закону
случая и гибель активных центров не происходит. При независи-
104
и,и
6fi
2,0
\
\
\
мости скорости разложения от DPn возникает неопределенность,
для устранения которой разработаны специальные методы (например,
введение в систему эффективного ингибитора). Применение
указанного метода для анализа термической деструкции
высокомолекулярного полиоксиметиленгликоля привело к заключению, что
этот полимер разлагается по закону концевых групп (рис. 33) и
эффективная константа разложения обратно пропорциональна
средней степени полимеризации. Инициирование реакции разложения
можно доказать и другим путем. Полимер ацетилировали уксусным
ангидридом, в этом случае эффективная константа разложения
уменьшалась с увеличением молекулярного веса диацетата. Отсюда
следует, что изменение природы концевых
групп приводит к изменению механизма
разложения полимера.
Если разложение макромолекул
начинается с концевых групп, то с
увеличением средней степени полимеризации
можно ожидать снижения эффективной
скорости процесса за счет уменьшения
концентрации «слабых» концевых групп
на единицу массы (или объема) полимера
[16]. Однако это справедливо только
в том случае, если деполимеризация
протекает по ступенчатому механизму.
Если же скорость инициирования
значительно меньше скорости цепной
деполимеризации (а это имеет место при
деструкции полимера выше предельной
температуры полимеризации) и в системе
нет гибели активных центров, то скорость
разложения не зависит от молекулярного
веса. Такая зависимость появится только
в том случае, если кинетическая длина цепи меньше
материальной. Мы предполагаем, что разложение полиоксиметиленгликоля
происходит именно по этой схеме.
Найдено [5], что деполимеризация полиоксиметиленгликоляг
начиная с 90° С, протекает как реакция первого порядка, путем
последовательного отщепления звеньев формальдегида от полу-
ацетального конца (или концов) макромолекулы:
... —Li?loUL(rl2UL.rl2UJtl -—>¦ ••• —L<Jtl2ULirl2Url -f- У^гіок)
I
CH2OH + CH2O f и т. д.
Приводится энергия активации разложения параформальдегида,
равная 10 ккал/молъ. Эта величина вызывает сомнения, так как
она ниже теплового эффекта полимеризации. Появление первого
порядка может быть обусловлено следующими причинами.
105
0,6
Рис. 33. Зависимость
эффективной константы скорости
термического разложения
ПОМ при 222Q С в азоте
от т] 1е (в диметилформамиде
при 150° С) [15].
Реакция термического разложения полиформальдегида
протекает в твердой фазе или расплаве, причем газообразные продукты
разложения быстро удаляются из образца. Следовательно,
концентрация полимера остается все время постоянной и порядок реакции
-0,2
-0,6
-иг
0 10
, Время, мин
а
0
-0,2
-ол
>
-0,6
-0,8
-w
-І
і і і
0 W 20 30
Время, мин
18
2
\ і
U0 50
-1,0 -
1,0
Время, мин
б
Рис. 34. Полулогарифмические
анаморфозы кинетических кривых
разложения [14] а-полиоксиметилена (а),
ПОМ (б) и диацетата ПОМ (в) при
температуре:
а) 1 — 120° С; 2 — 140° С;
4 — 160° С; 5— 170° С;
б) 1— 190° С; 2— 200° С;
5_ 150° С;
б_ 180° С.
? — 207° С.
3 — 299° С;
по потере массы должен быть первым независимо от механизма самого
процесса.
Как видно из рис. 34 (а и б), полулогарифмические анаморфозы
кинетических кривых разложения а-полиоксиметилена и
высокомолекулярного полиоксиметиленгликоля (ПОМ) представляют собой
ломаные линии, состоящие из двух прямолинейных участков.
Причину этого явления удалось объяснить на основе ранее
сделанного предположения о гибели активного центра при
деполимеризации ПОМ. Если бы образующиеся в результате гибели актив-
ного центра осколки были менее стабильны, чем исходные молекулы,
зависимость эффективной константы скорости разложения (/сэф)
от DPn нельзя было бы определить экспериментально. В этом случае
лимитирующей стадией процесса было бы инициирование реакции
разложения исходных молекул, а распад полимерной цепи
происходил бы фактически в один акт. Остается предположить, что
образующиеся осколки более стабильны, они накапливаются в системе,
а это вызывает изменение &эф во времени и появление излома на
кинетических кривых.
Для диацетата полиоксиметиленгликоля различие между
стабильностью исходных макромолекул и осколков, образующихся
в результате разрыва цепи по закону случая, невелико и отклонения
от закона первого порядка экспериментально обнаружить не
удается.
Из рис. 34 (а и б) следует, что точка перегиба на кривой
зависимости /сэф от продолжительности разложения (в зависимости
от температуры разложения) наблюдается при различных степенях
превращения. Чем выше температура, тем позднее происходит
отклонение от закона первого порядка. Этот факт говорит о том, что
реакция гибели активных центров имеет энергию активации меньшую,
чем реакция деполимеризации.
При попытках определить величину энергии активации
термической деструкции ПОМ авторы встретились со значительными
экспериментальными трудностями, так как оказалось невозможным
провести реакцию в изотермических условиях. Обычно для того чтобы
перевести реакцию разложения из диффузионной области в
кинетическую, используют следующие методы:
1. Путем уменьшения навески добиваются улучшения
теплопроводности в образце с тем, чтобы скорость реакции перестала
зависеть от массы (кинетическая область протекания процесса).
2. Путем снижения температуры эксперимента добиваются такого
положения, когда лимитирующей стадией реакции становится
собственно химический процесс. Этот метод основан на том, что скорости
химической реакции и теплопередачи по-разному зависят от
температуры (экспоненциальный закон в первом случае и степенной во
втором).
3. Добавлением инертного вещества с высокой
теплопроводностью к испытуемому образцу добиваются улучшения
теплопередачи.
Третий способ был сразу признан непригодным из-за высокой
чувствительности ПОМ к примесям, особенно к металлам и их солям,
которые обычно добавляют для повышения теплопроводности
полимера. Снижение температуры до точки плавления ПОМ оказывается
недостаточным, а ниже температуры плавления происходит резкое
уменьшение скорости разложения * за счет изменения агрегатного
состояния, т. е. второй метод также непригоден.
Этот факт был впервые отмечен еще Штаудингером [3].
107
Зависимость эффективной константы скорости разложения от
величины навески ПОМ показана на рис. 35. Авторы пришли к
выводу, что процесс разложения ПОМ протекает в переходной области,
в которой скорость реакции подчиняется закону Аррениуса, но
найденная величина энергии активации является только частью
истинной [17].
Понятие «переходная» область было введено в свое время для
некоторых химических реакций, на протекание которых
определенным образом влияет диффузия [18]. В этом случае было показано,
что экспериментально найденная величина энергии активации равна
половине истинной.
20 \ 2,0
15^-- f,SK
і
V
2
4*
0,06 0Л8 0,10
набзска, г
Рис. 35. Зависимость к*эф. от величины
навески [17]:
1 — а-полиоксиметилен при 130° С; 2 — ПОМ
при 220° С; 3 — диацетат ПОМ при 240° С.
и
ч*
п
-0>8
-1,2
-0
\
\
\
К3
\
\
о
1JB
2,0 2,2 2,U
Рис. 36. Температурная
зависимость ЛЭф# для ПОМ [17]:
1 — навеска 0,02 г; 2 — навеска 0,05 г;
з — диацетат ПОМ.
В работе [17] авторы предлагают способ, пользуясь которым
можно найти истинную энергию активации. Для ПОМ она оказалась
равной 26 шал/моль. Разложение диацетата ПОМ протекает в
кинетической области (рис. 36), и энергия активации, измеренная
непосредственно, равна 32 ккал/моль.'
Образование относительно стабильных осколков при
деполимеризации ПОМ может быть связано с реакциями передачи цепи.
Агентом передачи цепи естественно предполагать мономер, выделяющийся
при разложении. Как видно из рис. 37, давление газообразного
формальдегида действительно влияет на кинетику разложения ПОМ
независимо от температуры, навески и молекулярного веса [19].
В то же время давление формальдегида не влияет на скорость
разложения диацетата ПОМ (рис. 38).
Эти данные можно интерпретировать двояко:
повышение давления мономера приводит к изменению
предельной температуры полимеризации согласно уравнению Клаузиуса —
Клапейрона:
dP
АН
108
формальдегид вступает в реакцию передачи цепи, приводящую
к гибели активных центров деполимеризации.
Применявшиеся давления мономера слишком низки, чтобы
существенно сдвинуть равновесие мономер — полимер. Рассмотрим
второй вариант. Механизм передачи цепи можно представить следующим
образом:
Н Н Н Н
I I Нч II.
О—С—О—С • -^ )С=О —> • • • —О—С—О—С—Н >НСО
I I нх II
н н н н
При этом предполагается, что активный центр деполимеризации
имеет радикальную природу; радикал НСО распадается на окись
0,75
%30
%20
8
Время, мин
12
•Г. 0,50
0,25
\
\
-
-
OS
50
рсп2о
150
,, мм рш ст.
-3
- 2 ,
250
Рис. 37. Влияние давления мономера на
кинетику термического разложения ПОМ
[19] (навеска 0,02 г) при 210° С:
1 — 10-* лілі рт. ст.; 2 — 57 мм рт. ст.;
3—132 лш рт. ст.; 4 — 207 мм рт. ст.;
5 — 250 ли* рт. ст.
Рис. 38. Влияние давления
мономера на кинетику термического
разложения диацетата ПОМ [19]:
1 — ПОМ, навеска 0,02 г, 210° С;
2 — диацетат ПОМ, 280° С; з — а-по-
лиоксиметилен, навеска 0,07 г, 130° С.
углерода и водород, энергия активации распада составляет [20]
27 ккал/моль.
Любое вещество, имеющее прочность связи С—Н, близкую к
прочности связи в формальдегиде, и способное образовывать
малоактивный радикал, должно действовать подобным образом. В частности,
прочность С—Н-связи в толуоле близка к прочности С—Н-связи
в формальдегиде [21], поэтому толуол был использован как агент
передачи цепи при термическом разложении ПОМ. Из данных рис. 39
видно, что с увеличением давления паров толуола снижаеяся
скорость разложения ПОМ, причем влияние паров толуола на кинетику
термического разложения ПОМ аналогично влиянию
газообразного формальдегида.
Имеется ряд данных, говорящих в пользу ионного характера
активного центра деполимеризации по концевым гидроксильным
группам, например ускоряющее влияние кислот, щелочей и некоторых
109
солей на скорость разложения полимера [22, 25], низкая
энергия активации брутто-процесса, зависимость энергии активации
разложения диацетата ПОМ от химической природы катализатора
ацетилирования [19]. Гомолитическая прочность О—Н-связи
A18 ккал/молъ) достаточно высока, в то же время гетеролитический
разрыв протекает весьма легко, что также свидетельствует в пользу
ионного механизма распада. С точки
зрения ионного механизма можно
объяснить и относительную
устойчивость ПОМ в кристаллическом
состоянии, в частности высокую
стабильность смешанных
кристаллитов, включающих как молекулы
дигидрата ПОМ, так и диметилового
эфира или диацетата ПОМ, на
которую обратил внимание еще Штау-
дингер [3].
В пользу радикального меха-
100
^ 80
\ 60
I"
В
j
ft
и
кг
о-
5 Ю 15 20 25
Время, мин
Рис. 39. Влияние давления то- низма распада концевой группы
луола на кинетику термического высказались Торикаи [23] и Ней-
раЗЛОЖеНИЯ Полиформальдегида м о рп ^ ro/l Ппрттрттттир ттп
[19] B20° С, навеска 0,05 г): МаН С СОтр' lZ4b ИослеДние ПР°"
I-остаточное давление 10-^tptn. cm.; анализировали СОСТаВ ПРОДУКТОВ
2—130 мм -рт. ст.', 3—250 мм рт. ст. ТЄрМИЧЄСКОИ ДеСТруКЦИИ И обнару-
жили среди них метанол, метилфор-
миат, водород и метан. По мнению авторов, образование этих
продуктов возможно лишь в том случае, когда активным центром
являются радикалы типа
Н
Н
О—С—О-С.
І І
н н
н
I
н
или
._о-с—о—с—о.
І І
н н
причем основные реакции, протекающие в системе, — изомеризация
и распад этих радикалов.
Предположение о радикальном механизме распада кажется
весьма правдоподобным по отношению к термическому распаду
полиоксиметилена с блокированными концевыми группами. В работе
Ениколопяна и Дудиной делается предположение о совместном
существовании активных центров ионного и свободнорадикального
характера.
ХИМИЧЕСКАЯ ДЕСТРУКЦИЯ (АЦИДОЛИЗ)
При изучении термоокислительной деструкции
полиформальдегида важная роль отводится действию кислотных агентов,
поэтому, прежде чем перейти к механизму термоокислительной
деструкции, мы рассмотрим устойчивость полиацетальной цепи к действию
электрофильных агентов.
110
Механизм кислотного гидролиза низкомолекулярных ацеталей
изучен достаточно подробно [26—29]. Считается, что гидролиз
протекает по механизму SN 1 с образованием промежуточного иона
карбония. Лимитирующей стадией является распад иона карбо-
ния [27]:
OR"
OR'"
Г н ~Г
R4 /OR" Г R4
/G\ —м Ч^
Lr'/ xor''J Lr
і + *
•2H2O
J)C=O-f R'"OH + HoO+
Скорость этой реакции зависит от природы заместителей. Замена
водорода на алкил (переход от формаля к ацеталям) увеличивает
скорость разложения, введение электроноакцепторных заместителей
в а- и ?-положения, наоборот, снижает ее за счет I- или М-эффектов.
Штаудингер с сотр. [30] изучал действие кислотных и основных
агентов на полиоксиметилены с различными концевыми группами
и пришел к выводу, что основные агенты действуют
исключительно на концевые группы и не влияют на полиэфирную цепь;
кислотные агенты могут действовать по закону случая, а также
оказывать влияние на концевые группы.
Сравнительная устойчивость концевых групп к действию кислот
и щелочей изменяется в ряду:
метоксильные > алкоксильные> ацетильные > сложноэфирные ?>
?> гидроксильные
Наиболее устойчивы метоксильные и вообще простые эфирные
группы типа RO, где R — алкильный радикал. Они не гидроли-
зуются даже под действием концентрированных щелочей и среди
соединений, содержащих эфирные связи, наиболее стойки к аци-
долизу. Сложноэфирные группы легко омыляются щелочами.
Скорость разложения при ацидолизе зависит от константы диссоциации
кислоты и ее концентрации (табл. 18).
Состав продуктов разложения параформальдегида в
присутствии хлорного железа, фосфорной, я-толуолсульфоновой,
пикриновой и некоторых карбоновых кислот изучали методом газовой
хроматографии [25]. Установлено, что продуктами разложения являются
формальдегид, вода, метанол, метилмуравьиный эфир, триоксан
и тетраоксан. Можно предположить, что метиловый спирт
присутствует как примесь в мономере, вода образуется из концевых гидр-
оксильных групп, метилмуравьиный эфир выделяется по реакции
Тищенко, катализируемой кислотами. Выход циклических олиго-
меров зависит от ^Ка кислоты и возрастает с увеличением этого
показателя. Наибольший выход четырехчленного цикла A,2%)
достигался при действии пикриновой кислоты; выход триоксана при
действии FeCl3 составил 10,5% (при 150° С). Авторы предложили
111
ТАБЛИЦА 18
Зависимость скорости разложения диметилового
эфира ПОМ в атмосфере азота при 190° С от степени
диссоциации кислоты *
Кислота
Пикриновая
Салициловая
н-Оксибензойная
?-Нафтол
Гидрохинон
0,4
3,0
4,5
8,0
10,0
Доля
разложившегося за 1 ч
полимера, %
87,6
36,2
17,8
10,8
1,3
* Концентрация 10-5 ліоль/50 мг диметилового эфира ПОМ.
схему процесса, в которой разрыв происходит через стадию
образования промежуточного оксониевого иона:
О—СНа-О-СНа-О— СН2
распад по закону случая
распад по концевым
группам
н
v
х-
Н
CH2—O-CH2-O-CH2~O+
і н
+
..._с„,_о-Сн,-о-.рх^но„
—сн2—о—сн2—о=сн2
•. -О-СН2-О+СН2-О--СН2~...
Н
Образование циклических олигомеров может происходить
следующим образом:
х-
О—СН2—О-СН2-О-СН2—О—СН2
I
х-
О—CHs-O-CHg-O
СН2-О-СН2
/СН2-(\
сн2
112
Тетраоксан образуется в том случае, когда ион карбония атакует
кислород четвертого звена полиоксиметилена вместо третьего.
Кинетику ацидолитического расщепления высокомолекулярного
диацетата ПОМ под действием дихлоруксусной кислоты изучал
Мейзлик [31]. Суспензию полимера в хлористом метилене
обрабатывали кислотой при 30—33° С. Потеря массы и обратная величина
характеристической вязкости линейно возрастали во времени,
энергия активации разложения, вычисленная по потере массы, составила
A9±2,3) ккал/молъ, а вычисленная по скорости роста числа
молекул — A9,4±2,3) ккал/моль. Зависимости скоростей изменения
массы и молекулярного веса от концентрации кислоты имеют
линейный характер, причем их отношение почти не зависит от условий
проведения эксперимента (т. е. температуры и концентрации в
широком интервале величин).
Процесс ацидолиза, включающий образование промежуточного
оксониевого иона, его распад на карбониевый ион и молекулу с
концевой гидроксильной группой, протекает по схеме:
Н Н
I I +
СН2-О—СН2-0 —> СН2—
Увеличение концентрации гидроксильных концевых групп в ходе
процесса не приводит к автоускорению, поэтому можно сделать
вывод, что активным центром деполимеризации является
промежуточный ион карбония.
Японские авторы [32] исследовали кинетику разложения
высокомолекулярного ПОМ и диацетата ПОМ в феноле при 90—130° С.
Уменьшение массы полимера подчинялось уравнению реакции
первого порядка. Энергия активации разложения в обоих случаях
составляла 25 ккал/молъ, но дигидрат разлагался с большей
скоростью (с концов цепи и по закону случая). Скорость разложения
диацетата (при 118° С) изменялась в зависимости от природы
заместителя фенола в пара-положении в следующем ряду:
тг-нитрофенол ?> Аі-/?г/?е/?г-бутилфенол > тг-хлорфенол ^> фенол ]> тг-крезол
ТЕРМООКИСЛИТЕЛЬНАЯ ДЕСТРУКЦИЯ
Термическая стойкость полиоксиметилена определяется главным
образом природой концевых групп. Если рассматривать триоксан
как модель полиоксиметиленовой структуры, то можно считать, что
ацетальные связи термически устойчивы приблизительно до 270° С.
Термическое разложение полиоксиметиленгликоля со степенью
полимеризации около 1000 можно наблюдать начиная с 90° С.
Блокирование гидроксильных групп позволяет повысить термостойкость.
Заметное разложение диацетатов ПОМ происходит при 240° С,
в то время как диметиловые эфиры начинают разлагаться только при
температуре выше 270° С. Возможность образования концевых
8 Заказ 1135. ИЗ
формильных или карбонильных групп является до сих пор
проблематичной, однако им приписывается устойчивость, промежуточная
между устойчивостью гидроксильных и ацетильных групп.
После того как мы рассмотрели механизм термической деструкции
и ацидолитического расщепления полиформальдегида, следует
перейти к термоокислительной деструкции. Важность этого аспекта
проблемы очевидна, если учесть, что переработка термопластов
происходит на воздухе при повышенных температурах. А priori
можно предполагать, что полиформальдегид должен обладать низкой
стабильностью к термоокислению в расплавленном состоянии. Кроме
того, можно ожидать, что процесс окислительной деструкции будет
20 ЬО 60 80 100 120
Время у мин
Рис. 40. Кинетические кривые
термоокислительного разложения диацетата ПОМ на воздухе [34]:
2—165° С; 2— -170° С; 3—175° С; 4 — 183° С;
Л— 196° С; б —202° С; 7 — 220° С.
сопровождаться деполимеризацией, поскольку это наиболее
термодинамически выгодное направление реакции.
Для изучения термоокислительной деструкции
полиформальдегида обычно используют образцы ПОМ с блокированными
концевыми группами, которые относительно более стабщгьны.
Керн и Чердрон [5] наблюдали разложение диметилового эфира
ПОМ на воздухе при температуре выше 160° С. Реакция протекала
с автоускорением. Если учесть, что в инертной атмосфере этот же
образец был устойчив при 270° С, то разложение на воздухе можно
рассматривать как автоокислительную реакцию [33]. Керн считает,
что температура 160° С представляет собой нижний предел
разложения полиформальдегида под действием кислорода. При этой
температуре, очевидно, начинается размягчение кристаллической
решетки полиоксиметилена. В дальнейшем было показано, что
окислительная деструкция полиформальдегида характеризуется
наличием индукционного периода, во время которого выделение
мономерного формальдегида не происходит (рис. 40). При 160° С
114
величина периода индукции измеряется несколькими часами, после
чего наблюдается автокаталитическое разложение полимера. Снятие
кинетических кривых зависимости периода индукции от
температуры и экстраполяция полученных данных в область более низких
температур позволили предположить, что окислительная деструкция
полиформальдегида, сопровождающаяся разложением его с
образованием мономера, происходит начиная со 120° С. При более низких
температурах индукционный период разложения практически равен
бесконечности и действие кислорода сказывается на увеличении
хрупкости и ухудшении других механических свойств материала
(старение).
Вопросом первоочередной важности является изучение
термоокислительной деструкции полиформальдегида в диапазоне
температур 180—240° С. Методы, обычно используемые для изучения
кинетики термоокислительной деструкции карбоцепных полимеров,
в данном случае оказались непригодными (измерение поглощения
кислорода по изменению его давления), так как основным
продуктом окислительной деструкции полиформальдегида являлся
мономерный формальдегид.
В серии работ, проведенных Ениколопяном с сотр. [34—38],
кинетика термоокислительной деструкции полиформальдегида
изучалась в струевой установке по изменению массы образца и
логарифмической вязкости (в диметилформамиде при 150° С с 2%
дифениламина).
Можно предполагать, что при взаимодействии кислорода с
полимерной цепью образуются гидроперекиси
СН2—О—СН2—О—СН2—О -f О2 —>•
ООН ;
I
—> СН2—О—СН-О—СН2-0
разложение которых приводит к гемолитическому разрыву
полимерной цепи по закону случая, при котором молекулы мономера
легко отщепляются от образующегося активного радикального
центра [5, 33].
Из рис. 41 видно, что скорость разложения диметилового эфира
ПОМ на воздухе не подчиняется уравнению реакции первого порядка.
Автоускорение объясняется (по мнению авторов) ацидолизом
полимера под действием муравьиной кислоты, которая образуется в
результате окисления в мягких условиях формальдегида, выделяющегося
при деструкции. Показано [5], что если к диметиловому эфиру
добавить некоторое количество дигидрата ПОМ, то общая скорость
разложения смеси превышает расчетную величину, полученную из
раздельных измерений скоростей разложения каждого из этих веществ.
В атмосфере азота это явление не наблюдается. Влияние кислорода
особенно заметно на изменении молекулярного веса в процессе
8* 115
деструкции [341 (рис. 42). Уже при глубине превращения около 5%
в присутствии кислорода происходит резкое падение молекулярного
веса. Это характерно для разрушения полимерной цепи по закону
$ ЮО
% 80
1сГ 60
20
Ю.
\]
N
6'
<^
л
L 3
ч
300
120 180
Время, мин
Рис. 41. Кинетические кривые термоокислительного
разложения диметилового эфира ПОМ (Мп — 10 000)
на воздухе [25]:
2—120° С; 2— 180° С; 3— 190° С; 4 — 200° С; 5 — 210° С;
6 — 220° С.
случая [391. Если бы при этом получались осколки с концевыми
гидроксильными группами, то можно было бы ожидать симбатности
между изменением массы и молекулярного веса образца. Однако
1,0
s2
й—у.
20
Время, мин
60
Рис. 42. Кинетические кривые
изменения вязкости ПОМ при
термической (І) и термоокислительной B)
деструкции [34]:
# — яри 170° С; О — при 183° С;
Д — при 220° С.
20 W SO 80 WO 120 140
Время, мин
Рис. 43. Кинетические кривые
изменения вязкости диацетата ПОМ при
термической @) и термоокислительной
{1—6) деструкции [34]:
0—220° С; 7—165° С; 2— 170° С; 3 — 175° С;
4 — 183° С; 5 — 196° С; 6 — 220° С.
экспериментально найденная величина константы скорости
разложения ПОМ в кислороде всего лишь вдвое превышает величину,
полученную при разложении ПОМ в аргоне при той же температуре,
в то время как вязкость в этих же условиях изменяется в 20 раз.
116
Кинетические кривые разложения диацетата ПОМ на воздухе
(рис. 9) имеют ярко выраженный авто каталитический характер,
в то время как ПОМ разлагается без периода индукции. Кривые
изменения молекулярного веса также различаются (рис. 43).
Изучалась зависимость скорости термоокислительной деструкции
от концентрации кислорода. При 180—190° С разложение ПОМ
в инертной атмосфере протекает с большей скоростью, чем
разложение в присутствии малых количеств кислорода (ингибирующий
эффект). В случае деструкции диацетата ПОМ скорость реакции
линейно зависит от концентрации кислорода вплоть до 80%. Таким
образом, процесс окислительной деструкции в некотором интервале
концентраций имитирует первый порядок по кислороду [38].
Закономерности изменения скорости разложения ПОМ и его диацетата
достаточно хорошо описываются эмпирическим уравнением:
+
где w — скорость реакции; [О2] — концентрация кислорода; а, Ь,
с, d — эмпирические константы (Ь =^ d, b <^d, c/d^>a).
Этому уравнению можно дать следующую физическую
интерпретацию. Существуют два способа зарождения полимерных
активных центров — без участия кислорода и с его участием. В обоих
случаях гибель активных центров может происходить линейно
(самопроизвольно) и при взаимодействии с кислородом (ингибирующий
эффект).
Анализ продуктов деструкции показал, что независимо от
природы концевых групп основным продуктом реакции является
мономерный формальдегид. На 20 моль выделившегося мономера
поглощается в среднем 1 моль кислорода. Кроме того, в продуктах реакции
были найдены муравьиная кислота, водород, углекислый газ, окись
углерода, метилформиат [35, 40]. На основании этих данных можно
сделать вывод, что протекает лишь деполимеризация
полиформальдегида, инициированная кислородом, а окисление практмески
отсутствует.
Кислород инициирует разложение полиформальдегида по
схеме [35]:
СН2—О—СН2—О—СН2—О 4> О2 —>
—>¦ СНа-О-С-О—СНа—О ^НОа.
I
н
Образующийся макрорадикал может или распасться
СН2-О—С—О-СНо-О —* СН2—0~С=0^СН2-0 A)
117
или присоединить новую молекулу кислорода:
..._СН?—О—С—О-СН2—О f-O2 —>
Н
О—О-
I
—> СН2-О-~С-О-СН2-О B)
I
н
Реакция A) идет со скоростью, на 1—2 порядка превышающей
скорость реакции B). Этот вывод хорошо согласуется с
экспериментальными данными.
Некоторым подтверждением
сделанного расчета могут слу-
03с
0,6
0,2
20 ЬО
бремя, мин
ВО
20 . АО
Время, мин
б
60
\ J
fr
и
1 / rift
р
м>
Ю 20 30 40
Время, мин
в
Рис. 44. Кинетические кривые
разложения диацетата ПОМ в
различных средах [36}.
а — изменение массы при 220° С:
1— Ar; 2— Аг +0,15% HGOOH;
3—О2\ 4 — Аг +2,56% HGOOH;
5 _ Аг+2,56% HGOOH; б— О.-f
+ 2,56 HGOOH. б — изменение [тії:
1 —Аг; 2— О2; з —Аг+2,56%
HGOOH; 4 — О2+ 2,56% HGOOH.
в — изменение массы при 202° G:
1 — Ar; 2 — Аг + 2,56% HGOOH;
З — Аг, образец предварительно
обработан HGOOH; 4 — Аг +2,56%
НСООН, образец предварительно
обработан HGOOH; 5— О2; 6— О2,
образец предварительно обработан
НСООН.
жить результаты, полученные при исследовании методом ЭПР
радикалов, образующихся при у-облучении полиформальдегида [41, 42].
Авторы показали, что возникающие первоначально радикалы
типа — О—GH—О— устойчивы в вакууме при комнатной
температуре в течение нескольких суток. При 45—85° С происходит
118
wo
гибель этих радикалов по реакции второго порядка с энергией
активации A9 ±2) ккал/молъ. В присутствии кислорода порядок реакции
также второй, но константа скорости гибели радикалов выше и
зависит от давления кислорода.
Перекисные радикалы не образуются или образуются в
концентрациях, слишком малых для прямого аналитического
определения.
Было исследовано также действие муравьиной кислоты в
процессе термоокисления [36]. Поскольку наблюдающийся при
термоокислительной деструкции разрыв полимерной цепи по закону
случая связан не только с
прямым воздействием на нее
кислорода, естественно
предположить, что справедлива гипотеза
Керна о роли муравьиной
кислоты (вторичного продукта
реакции) как реагента,
вызывающего ускорение реакции
разложения .
В серии опытов муравьиная
кислота специально вводилась в
газовую фазу над полимером и
непосредственно в сам полимер.
Полученные результаты показали, что
в инертной атмосфере муравьиная
кислота вызывает ацидолиз
полимера, но ее влияние на скорость
разложения и уменьшение
молекулярного веса значительно слабее
эффекта, вызываемого
кислородом (рис. 44). Однако совместное
действие муравьиной кислоты и кислорода дает очень сильный
эффект. Иллюстрацией могут служить данные, приводимые в работе
[38J: на воздухе при комнатной температуре муравьиная кислота
взаимодействует с диацетатом ПОМ сильнее, чем в инертной
атмосфере при 202° С.
Скорость реакции разложения на воздухе увеличивается более
чем в 10 раз, период индукции, характерный для термоокисления
диацетата ПОМ, полностью снимается (рис. 45).
Эти результаты можно объяснить следующим образом: ацидолиз
полимера муравьиной кислотой приводит к образованию осколков
с концевыми гидроксильными группами.
С реакцией термического распада этих осколков
конкурирует отрыв кислородом водорода от атома, находящегося в а-поло-
жении:
W 20 ЗО
Время у мин
Рис. 45. Кинетические кривые
разложения диацетата ПОМ при 180° С
в кислороде A,2) и в кислороде
с добавкой 2,5% HGOOH C, 4).
• -0-СН2-0 -СНа-ОН
. -О-СНа-О-С-ОН + НО2 •
Н
119
В результате распада образующегося активного центра возникает
другой активный центр, способный деполимеризоваться, и новая
молекула кислоты:
О—СН2—О—С—ОН —> О—СНа-ф-НСООН
Н
Эта схема объясняет различия в характере деструкции ПОМ
и диацетата ПОМ тем, что в первом случае кислота быстрее
накапливается в результате взаимодействия кислорода с концевыми гидро-
ксильными группами. Прямым доказательством явилось измерение
скоростей накопления муравьиной кислоты в обоих случаях.
В другой работе [43] проверялась гипотеза Керна [5] о
возможности образования муравьиной кислоты за счет окисления
мономерного формальдегида непосредственно в зоне реакции. Подтверждение
этой гипотезы получено не было, так как, по-видимому, прямое
окисление формальдегида в этих условиях незначительно. Авторы
считают, что одним из вероятных путей образования муравьиной
кислоты является взаимодействие полимерной перекиси с
формальдегидом по схеме:
Н Н
I I
СН2—О—С—О—СН2 + СН2О —> СН2—О—С—О—СН2 —>
ООН О—«Н
I I
I !
о сн
I II
н о
н I
—>. СНа-О-С-О-СНа +НСООН
ОН
Поскольку эта реакция должна происходить с большой
скоростью, аналитически определимых количеств гидроперекиси в
системе может не накапливаться.
Несколько другой точки зрения на механизм
термоокислительной деструкции полиформальдегида придерживаются Нейман и Ко-
варская с сотр. [44], которые считают, что это радикально-цепной
процесс, протекающий с вырожденным разветвлением.
В пользу этого положения свидетельствуют следующие факты:
образование перекисных радикалов и гидроперекиси; S-образность
кинетических кривых окисления ПОМ; ингибирование процесса
окисления антиоксидантами радикального типа [107, 108]. Авторы
[109] использовали метод хемилюминисценции для обнаружения
перекисных радикалов, используя в качестве моделей
низкомолекулярные диметиловые эфиры ПОМ. Было показано, что люминис-
ценция появляется в результате рекомбинации перекисных радика-
120
лов, причем конечным продуктом реакции является гидроперекись.
Ее образование было подтверждено иодометрическим способом.
Отсюда можно сделать вывод, что обрыв кинетических цепей
при ингибированном окислении полиформальдегида происходит
по классической схеме:
RO2-+InH — ¦» RO2H+In.
т.
In« + RO2* —¦> Стабильные продукты
Введение добавок антиоксидантов приводит к спаду
интенсивности люминисценции. Используя известные соотношения для цепной
неразветвленной реакции [111], авторы рассчитали по полученным
экспериментальным данным значения относительных констант
скоростей реакций некоторых антиоксидантов с перекисными
радикалами (табл. 19).
ТАБЛИЦА 19
Эффективность некоторых антиоксидантов при термоокислении
диметилового эфира триоксиметиленгликоля [109]
Антиоксидант
б-трет-Еутил-м-Кфезол.
4,6-Ди-т^/тг-бутил-о-
крезол
4,4-Метилен-бис-B-ме-
тил-6-mpem -бутил
фенол)
Дифениламин
2,2-Метилен-бис-D-ме-
тяп-б-пгрет-бутилфе-
нол)
Диметилдифенилами-
нофеноксисилан
wio-7
моль/л »сек *
5,25
5,12
3,6
5,6
4,5
п**
1,8
2,5
2,4
2,0
V*p ***
л/моль» сек
0,6
2,5
5,3
4,5
3,2
8,5
Период ****
индукции
мин
18
19
148
170
190
400
Количество
разложившегося
полимера
за первые
30 мин ****
%
17
15
9
2
1
0,5
* w—скорость расходования антиоксиданта в индукционном периоде.
** п—число кинетических цепей окисления, обрываемых одной молекулой антиокси-
данта.
*** kt— константа скорости реакции антиоксиданта с перекисными радикалами; k —
константа скорости реакции рекомбинации радикалов.
**** Стабилизирующий эффект действия антиоксиданта (концентрация выше
критической) на высокомолекулярный диацетат ПОМ. Условия эксперимента: 220° С, давление
кислорода 200 мм pm. cm.
СТАБИЛИЗАЦИЯ
Стабилизация полиформальдегида предполагает подавление всех
направлений реакций разложения с тем, чтобы обеспечить
возможность переработки материала и эксплуатации изделий. Анализ
условий эксплуатации деталей, изготовленных из
полиформальдегида, позволяет сделать вывод о том, что верхний температурный
предел, при котором еще сохраняются достаточно высокие
показатели механических свойств материала, лежит около 120° С.
121
Окислительная деструкция ПОМ с блокированными концевыми
группами протекает с заметной скоростью также начиная
приблизительно со 120° С. Таким образом, блокирование концевых
гидроксильных групп ПОМ должно быть достаточным, чтобы
обеспечить необходимую термостабильность при эксплуатации изделий.
В инертной атмосфере в отсутствие следов соединений кислотного
характера диацетаты, диметиловые эфиры и другие модификации
ПОМ должны быть вполне устойчивы по крайней мере при
температурах 240—250° С, когда полимер находится в вязко-текучем
состоянии и может перерабатываться формованием. Специальные опыты
показали, что, действительно, в инертной атмосфере
полиформальдегид прекрасно формуется и изделия, полученные из него,
характеризуются хорошими механическими свойствами.
Однако конструирование специальных машин для переработки
полиформальдегида в инертной атмосфере экономически невыгодно.
Поэтому стабилизация полимера в условиях переработки оказалась
наиболее актуальной проблемой. Детальное изучение механизма
деструкции полиформальдегида создало предпосылки для решения
этой задачи. В общем виде стабилизация должна включать
следующие стадии:
1) блокирование концевых гидроксильных групп ПОМ;
2) стабилизация против действия кислорода;
3) нейтрализация следов кислоты (присутствующей в виде
примеси или образующейся в процессе деструкции);
4) связывание (поглощение) мономерного формальдегида, который
выделяется в начальный момент деструкции и может инициировать
вторичные реакции разложения или разрушать макроструктуру
изделий.
Очевидно, что термическая стойкость полиацетальной цепи будет
определять верхнюю теоретически возможную границу
стабильности полиформальдегида. Если принять, что эта граница лежит
около 270° С, то интервал между точкой начала текучести A84° С)
и верхним пределом стабильности будет достаточно велик для
осуществления переработки расплавленного материала.
Блокирование концевых групп. Природа концевых групп в
молекулах полиоксиметилена определяется механизмом передачи цепи
во время полимеризации. Если для регулирования молекулярного
веса не применяются специальные агенты передачи цепи, то абсолютное
большинство макромолекул имеет гидроксильные концевые группы.
Эти группы проявляют свойства, типичные для спиртовых групп,
и аналогичны гидроксильным группам в молекулах целлюлозы.
Наиболее простой способ модификации заключается в этерификации
концевых групп путем обработки соответствующими кислотами,
ангидридами и хлораыгидридами или другими этерифицирующими
агентами. Эти способы хорошо известны в химии целлюлозы. Однако
опыт, накопленный в результате многолетних исследований по аце-
тилированию, нитрованию, метилированию целлюлозы, не следует
переоценивать, так как полиформальдегид по устойчивости к дей-
122
ствию химических агентов существенно отличается от целлюлозы.
Поэтому, например, нецелесообразно применение хлорангидридов
для этерификации ПОМ, хотя это наиболее эффективный реагент *.
По этой же причине нецелесообразно применение кислот и кислотных
катализаторов для этерификации **. Кроме того, при стабилизации
ПОМ глубина превращения гидроксильных групп должна
приближаться к 100%, что не требуется для целлюлозы.
Штаудингер [1] осуществил ацетилирование низкомолекулярных
ПОМ путем гетерогенной обработки полимера избытком уксусного
ангидрида в среде диоксана, причем в качестве катализатора
использовался пиридин и смесь нагревалась до кипения. В этих условиях
реакция практически заканчивается за несколько минут. В патенте
фирмы «Дюпон» [45] указывается, что можно применять любой
ангидрид органической кислоты, но наиболее эффективны ангидриды
монофункциональных кислот, а среди них уксусный ангидрид.
Реакция катализируется основными агентами типа третичных
аминов, солей щелочных металлов и карбоновых кислот. Амины более
эффективны как катализаторы, но вызывают сильное
окрашивание полимера.
Степень завершенности реакции можно определить несколькими
способами:
1. Ацетилирование производят меченым уксусным ангидридом,
а затем после тщательной промывки и сушки полимера количество
ацетильных групп в нем определяют по относительной активности
выделившегося газообразного [14] СО2 после сжигания образца.
2. После ацидолиза полимера в растворе HG1 в метаноле и
омыления ацетильных групп потенциометрическим титрованием
определяют выделившуюся кислоту [45].
3. Полимер исследуют методом ИК-спектроскопии до и после
ацетилирования. Содержание гидроксильных групп определяют
по полосе поглощения при 2,88 мк, ацетильных — по полосе
поглощения при 5,69 мк. Расчет ведут по соответствующим величинам
оптических плотностей.
4. Ацетилированный полимер с добавкой 1 % подходящего амина
подвергают термической деструкции в вакууме или инертной
атмосфере выше температуры плавления A90—220° С). При этом ди-
гидраты и эфиргидраты деполимеризуются по закону концевых
групп. Можно считать, что оставшийся полимер содержит только
блокированные макромолекулы, поэтому доля стабильного
полимера будет характеризовать глубину протекания реакции
ацетилирования [46].
* При реакции гидроксила с хлораыгидридом выделяется хлористый
водород, который является сильнейшим ацидолитическим агентом по отношению
к ацетальной структуре. Все же этот способ описан в патентной литературе [57].
** Смеси ангидридов и кислот являются сильными ацетилирующими
агентами. По-видимому, можно найти оптимальный режим реакции, при котором
скорость ацетилирования будет значительно выше, чем скорость разложения,
начинающегося с конца цепей [58].
123
Этот метод анализа иллюстрируется рис. 46. В основе его лежит
использование различий в термической стойкости гидроксильных
и ацетильных концевых групп. Предложено несколько вариантов
использования этого метода, например деструкция на воздухе
с добавлением веществ, ускоряющих разложение гидроксильных
групп, и антиоксидантов, предотвращающих окисление [47].
Концентрация гидроксильных групп в высокомолекулярных
ПОМ составляет всего 0,2—0,3 моль на 100 моль формальдегидных
звеньев. Учитывая высокую
степень кристалличности полиокси-
метиленов, можно предположить,
что гетерогенная этерификация
будет весьма неэффективной из-за
диффузионных затруднений при
взаимодействии ангидрида и ката-
' лизатора с гидроксильными
группами по топохимической реакции.
Об этом свидетельствует опыт
Штаудингера по ацетилированию
низкомолекулярных ПОМ, когда
в результате действия высокой
температуры и кислоты,
образующейся по реакции
80
70
SO
so
30
W
0
1
\
/
і
/
и-
^2
3
и
\ У
т. -^
», і*
, 1
1—
ОСН2ОСН2ОН + (СН3СОJО —>
—> ОСН2ОСН2ОСОСН3 +
+ СН3СООН
полимер подвергался
значительному разложению. Тем не менее
Ю 20 30 W SO SO, 70
время, мим
Рис. 46. Анализ глубины
превращения концевых полуацетальных
групп ПОМ после ацетилирования
при остаточном давлении 10~4 мм
рт. ст. и 200° С:
1 — исходный образец ПОМ, М_п = 30 000;
2—исходный образец ПОМ, М =120 000;
3 — образец ПОМ, полученный при регу- ацЄТИЛИрОВаНИЄ ВЫСОКОМОЛекуЛяр-
лировании Мп с помощью уксусного анги- НОГО ПОМ В ГЄТЄрОГЄННОЙ Среде
дрида; 4 — диацетат ПОМ, полученный Mfil ттпптркярт с ттпртятпттттп тікггп-
ацетилированием в гетерогенной системе UDJ Протекает С ДОСТаТОЧНО ВЫСО
с ацетатом натрия в качестве катализатора. КИМИ ВЫХОДаМИ И Степенью Пре-
вращения 90—95%.
Кристаллическая структура полимера имеет большое значение при
гетерогенной этерификации. Так, например, степень превращения при
этерификации политриоксана значительно ниже, чем при этерификации
продукта полимеризации мономерного формальдегида. Орторомби-
ческий полимер, очевидно, имеет наиболее упорядоченную
структуру, в которой концевые группы расположены в основном на
поверхности кристаллитов, поэтому он легко ацетилируется в
гетерогенных условиях [48].
Кинетика ацетилирования высокомолекулярного ПОМ
рассматривается в ряде работ [16, 49, 50]. Предполагается, что
одновременно с ацетилированием протекает реакция ацидолиза полимера
и деполимеризации по концевым гидроксильным группам, причем
уменьшение выхода полимера определяется в основном последней
124
реакцией. Показано, что разложение полимера, начинающееся
с концов цепей, ускоряется действием катализаторов ацетилиро-
вания, кислот и разбавителя (если реакция проводится в
гомогенной среде). Возрастание молекулярного веса ПОМ приводит к
увеличению константы скорости ацетилирования. Очевидно, это
связано с морфологией полимера (с ростом молекулярного веса
уменьшается степень кристалличности, а гидроксильные группы в
аморфной фазе более доступны для действия реагентов).
Деполимеризацию продукта можно уменьшить, удаляя
уксусную кислоту (наиболее активный катализатор деполимеризации)
из зоны реакции или связывая ее. Для этой цели предлагается
использовать изоцианаты [51], карбодиимид [52]:
RNCO + СНзСООН —> RNHCOOCOCH3 -> RNHCOCH3
- GO*
Ацетилирование можно осуществить путем обработки ПОМ
газообразным кетеном ниже температуры плавления полимера или в
расплаве в присутствии основного катализатора (например, ацетата
щелочного металла) [53—56]. Судя по приведенным в литературе
данным, этот способ ацетилирования не дает преимуществ по
сравнению с использованием ацетангидрида.
Поскольку простые диэфиры ПОМ более стабильны, чем
сложные, предлагались различные способы получения простых эфиров
ПОМ. Нужно отметить, что ни один из них не удалось осуществить
в промышленном масштабе из-за низкого выхода полимера
вследствие разложения его под действием кислотных катализаторов,
применяемых при алкилировании.
Обработка спиртами в присутствии серной кислоты при
нагревании приводит к образованию низкомолекулярных диалкиловых
эфиров ПОМ [59—60]:
OCH2OCH2OH-f ROH -i*i§o_v OCH2OCH2OR4-H2O
Запатентован способ метилирования ПОМ в результате реакции
с диазометаном в присутствии фтористого бора или фторборной
кислоты [61], но и в этом случае разложение полимера приводит
к тому, что образуются низкомолекулярные диметиловые эфиры.
В слабокислой среде можно осуществить взаимодействие гидро-
ксильных групп ПОМ с ацеталями, кеталями, ортоэфирами или орто-
карбонатами [62—65]. Схему реакции с ацеталями можно
представить так:
OCH2OCH2OH + RO-CH2--OR —> OCH2OCH2OR + ROCH2OH
I
Реакции с кеталями и ортоэфирами протекают по схеме:
OCH2OCH2OH+(ROJC(CH3J —) OCH2OCH2OR+(CH3JCO+ROH
OCH2OCH2OH + (ROKCCH3 —у OCH2OCH2OR^CH3COOR + ROH
125
Процесе включает стадию разрыва одной из ацетальных связей
в молекуле этерифицирующего агента под действием кислотного
катализатора. Одновременно происходит расщепление гидроксиль-
ных связей, которое сопровождается быстрой деполимеризацией
образующегося полимерного иона. Низкий выход алкилированного
ПОМ определяется именно этой реакцией. Ацетальные связи в
молекулах полимера также будут рваться при электрофильной атаке.
Поэтому при этерифйкации ПОМ в присутствии кислотных
катализаторов наблюдается заметное снижение молекулярного веса
полимера *.
В качестве алкилирующих агентов можно применять вещества,
образующие стабильные концевые группы при реакции с ПОМ по
схеме ацидолитического расщепления, как это описано выше.
Такими веществами могут быть а, ?-ненасыщенные эфиры [67, 68]
типа
О
r_O-C=CH2 или v/ \
I
R'
В указанных выше случаях реакция протекает с разрывом
непредельной связи.
Для блокирования гидроксильных групп ПОМ можно
использовать также реакцию с эпоксидами [69, 71]:
R
I
ОСН2ОСН2ОН 4- CH2-CH-R —> ОСН2ОСН2ОСН2СНОН
\ /
о
Интересно отметить, что эта реакция может быть осуществлена как
в присутствии основного (пиридин), так и кислотного (кислоты
Льюиса) катализатора.
Из общих соображений следует, что выход полимера после
блокирования концевых групп в присутствии основного
катализатора должен быть выше. Описано несколько реакций такого
типа:
1. Обработка ПОМ а-хлоралкиловыми эфирами в присутствии
избытка третичного амина, играющего роль катализатора и
акцептора выделяющегося хлористого водорода [72]:
NR'
ОСН2ОСН2ОН + СН2С1—OR -¦> OCH2OCH2OCH2OR«f НС1
* Предложен способ регулирования среднего молекулярного веса полимера
путем кислотного расщепления (ацидолиза) этерифицированного полимера
в присутствии избытка алкилирующего агента [66]. В этом случае потери,
связанные с деполимеризацией, составляют около 10%.
126
2. Обработка ПОМ третичными алкилхлоридами [69] в
присутствии окиси серебра:
C^-r —>• ... -~OCH2OCH2OCR3+HC1
4
Эту реакцию проводят при низких температурах, однако
практически невозможно удалить затем окись и хлорид серебра.
3. Взаимодействие ПОМ с винильными соединениями в
присутствии щелочей или алкоголятов [69, 70]:
OCH2OCH2OH + CH2=CHR —> OGH2OCH2OCH2CH2R
4. Реакция с изоцианатами или изотиоцианатами с образованием
уретановых групп [69, 73, 74]:
OCH2OCH2OH-f ArNCO —> OCH2OCH2OCONHAr
Некоторые данные об относительной эффективности перечисленных
выше методов блокирования концевых групп ПОМ приведены
в табл. 20.
Термически стабильный полимер, который не разлагается в
вакууме при 200° С, образуется в результате ионизирующего
облучения ПОМ в вакууме при комнатной температуре [75]. Доля
стабильного полимера растет с увеличением дозы облучения, но
характеристическая вязкость уменьшается пропорционально логарифму дозы
облучения. Автор предполагает, что за счет рекомбинации
образующихся при облучении радикалов возникают концевые формокси-
или метоксигруппы.
Все рассмотренные выше методы предполагают обработку
готового ПОМ. Связанные с этим трудности очевидны. Поэтому большое
значение имеет решение вопроса о возможности проведения
полимеризации таким образом, чтобы одновременно происходило
блокирование концевых групп полимерных цепей. Теоретически это
возможно, если в системе имеется агент передачи цепи, который при
взаимодействии с активным полимерным центром образует
стабильную концевую группу и новый активный центр, также имеющий
стабильную концевую группу. Очевидно, что другие агенты
передачи цепи не должны присутствовать в реакционной системе.
Подходящими агентами передачи цепи при анионной
полимеризации формальдегида являются ангидриды и сложные эфиры [76],
при катионной полимеризации формальдегида или триоксана сюда
можно включить линейные ацетали и диэфиры олигомерных
ПОМ [33,77]. Применение этих агентов передачи цепи позволяет
получить полимер, 40—60% молекул которого имеет
блокированные концевые группы [78, 79]. Чтобы увеличить долю
блокированных молекул, необходимо иметь тщательно очищенный от
примесей мономер, так как молекулярный вес полимера обратно
пропорционален концентрации агента передачи цепи. В лабораторных
условиях могут быть получены образцы со степенью
блокирования до 85% (по весу) и достаточно высоким молекулярным весом [33].
127
ТАБЛИЦА 20
Относительная эффективность некоторых агентов,
применяющихся для блокирования гидроксильных групп ПОМ
Реагент
Уксусный
ангидрид
То же
Бензойный
ангидрид
рид
Окись пропилена
Стирол
Винилацетат
Акрилонитрил
Акролеин
Аллиловый спирт
Пропаргиловый
спирт
тг-Нитрофенил-
изоцианат
Условия реакции
20 мин, 140° С
3(
3
о
3
2
3
3
2
1
3
3
) мин, 150° С,
рас-
твор диметилфор-
мамида
ч, 100° С
ч, 50°С
ч, 125° С, под
лением
ч, 150° С, под
лением
ч, 125° С, под
лением
ч, 125° С, под
лением
ч, 125° С, под
лением
ч, 125° С, под
лением
ч, 125° С, под
лением
ч, 110° С, под
лением
дав-
дав-
дав-
дав-
Дав-
Дав-
дав-
Дав-
Катализатор
Ацетат натрия
Пиридин
Пиридин
Окись серебра
Пиридин
Ацетат натрия
Пиридин
Ацетат натрия
Ацетат натрия
Едкое кали
Пиридин
Без
катализатора
і
Выхс
мера,
95
85
88
100
95
90
90
90
85
75
30
100
Степень
завершенности
реакции *
94-96
98
69
66
65-70
62
85
70
95
94
95
9
S
со
а
Лите
69
45
69
69
69
69
69
69
69
69
69
65
* Определяется как доля полимера (в %), не разложившегося после нагревания в
течение і ч в атмосфере азота при 190 или 200° С.
Стабилизация против действия кислорода. Обычный метод
подавления окислительной деструкции высокомолекулярных
соединений заключается в подборе наиболее эффективных стабилизаторов
(антиоксидантов). Для стабилизации полиформальдегида были
испытаны различные соединения типа вторичных и третичных аминов
и фенолов, обычно применяемых для стабилизации полимерных
материалов. Предполагается, что эти соединения ведут себя как
ингибиторы радикального типа, т. е. препятствуют образованию
гидроперекисей, стабилизируют образующиеся радикалы и т. п. Амин-
ные антиоксиданты способны нейтрализовать кислоту,
накапливаемую в зоне реакции, а вторичные амины, кроме того, могут
взаимодействовать с формальдегидом, выступая в роли акцептора. Таким
образом, амины (например, дифениламин или я-фенилендиамин)
претендуют на роль универсальных стабилизаторов полиформаль-
128
дегида. Существенным недостатком антиоксидантов а минного типа
является сильное окрашивание полимера.
Фенолы не вызывают окрашивания, но они обычно менее
эффективны. Кроме того, фенолы с малыми величинами \)Ка вызывают
ацидолитическое расщепление полимера.
Кинетика ингибированного окисления полиформальдегида
рассмотрена в ряде работ Неймана и Коварской с сотр. [44, 107—109].
Ими дана оценка сравнительной эффективности соединений,
относящихся к различным классам антиоксидантов, включая
ароматические амины, диамины, аминофенолы, моно-, бис- и трис-фенолы,
серо- и фосфорсодержащие соединения и т. д. Термостабильность
полимера оценивалась по величине периода индукции на
кинетической кривой разложения и по потере массы. Условия эксперимента:
температура 220° С, давление кислорода 200 мм рт. ст. Наиболее
эффективными антиоксидантами оказались ароматические амины
и алкилированные бис-фенолы. Характерной особенностью
процесса ингибированного окисления полимеров является наличие
критической концентрации антиоксидантов, выше которой
окисление протекает как стационарный процесс, а ниже — как
автоускоренный. Подобная зависимость имеет место в случае
полиформальдегида.
Кривые зависимости числа разрывов цепи полимера (получены
на диацетате ПОМ) во время периода индукции от концентрации
ингибитора имеют автоускоренный характер. Это значит, что
ингибитор расходуется не только на реакции обрыва кинетических цепей
окисления. Как видно из табл. 19, отсутствует корреляция между
относительной константой скорости реакции взаимодействия анти-
оксиданта с радикалами и эффективностью того же антиоксиданта
при окислении полимера. Все же некоторые выводы о влиянии
строения молекул ингибитора на его активность могут быть
сделаны [110].
Неясным до сих пор остается вопрос о том, каким образом следует
оценивать эффективность стабилизирующих композиций.
Теоретически правильнее определять кинетику изменения молекулярного
веса во время термоокислительной деструкции в присутствии
различных антиоксидантов. Однако часто измерения ограничиваются
фиксированием потери массы полимера или выделившегося мономера
во время деструкции, хотя это может быть связано с побочными
процессами (недостаточно полное блокирование концевых групп,
наличие примесей кислотного характера и т. п.).
Для связывания выделяющегося при деструкции мономерного
формальдегида добавляют вещества, способные присоединять
формальдегид при повышенных температурах. Сюда относятся
полиамиды и амиды многоосновных кислот, гидразины, гидразиды,
мочевины, полимочевины и их производные [80—85]. Во многих
случаях механизм действия предлагаемых стабилизирующих
добавок не ясен; возможно, что, как и вторичные амины, они совмещают
несколько функций.
9 Заказ і 135. 129
Добавление к диацетату ПОМ перечисленных выше веществ дает
незначительный стабилизирующий эффект [86], причем наблюдается
только увеличение периода индукции при разложении (потеря
массы). Падение молекулярного веса аналогично тому, что
наблюдается на контрольном нестабилизованном образце. Была
предпринята попытка исследовать кинетику взаимодействия полиамидов
с газообразцым формальдегидом при повышенных температурах [88].
Известно, что реакция формальдегида с амидной группой приводит
к образованию метилольных или метиленовых производных.
Скорость реакции резко возрастает с повышением начального давления
формальдегида. Эффективная энергия активации брутто-процесса
составляет A6 ± 1) ккал/молъ. Общий объем поглощенного
формальдегида с повышением температуры уменьшается. С помощью
ИК-спектрометрии было показано, что при нагревании смеси
полиамида с полиформальдегидом на воздухе наблюдается постепенное
исчезновение полосы 3070 см, соответствующей колебанию
NH-группы. Вместе с тем количество поглощаемого формальдегида
не коррелируется с его количеством, выделяющимся при разложе-
ТАБЛИЦА 21
Стабилизаторы полиформальдегида
Вещество
Количество
вес. %
Предполагаемое
действие
Эффективность
Литература
Производные мети-
лен-бис-фенола,
например 4,4-бутилен-
бис-C-метил-6-иіре/п-
бутилфенол)
Производные
2,5-димеркапто-1, 3, 4
-тиодиазола
Эпоксидные смолы
Соли меди (II) и
марганца
Амиды
Ароматические ами-
носульфоны,
например га-аминофенил-
сульфон
Вторичные и
третичные
ароматические амины,
например ї^,І^-ди-р-наф-
тил-п-фенилендиамин
0,1—5
0,1-5
До 20
До 1
До 5
До 10
0,1-5
Ингибиторы
свободноради-
кальных
процессов
То же
Антистарители
Акцепторы
формальдегида
То же
Ингибиторы
свободноради-
кальных
процессов
Наиболее
эффективные среди
фенолов, не
окрашивают продукт
Не окрашивают
продукт при
окислении
Улучшают
механическую
прочность полимера
Устраняют
запах
формальдегида
Наиболее
эффективны как
антиоксиданты,
но вызывают
окрашивание
продукта
90, 91
92
93
94
95, 96
97
98
99, 100
130
ний контрольного образца. Отсюда следует, что роль полиамидов
не сводится только к связыванию свободного мономера *.
Наибольший эффект дает составление специальных
стабилизирующих композиций, включающих антиоксиданты (например,
замещенные бис-фенолы), акцепторы кислоты (амиды или амины),
акцепторы формальдегида — термостабилизаторы (полиамиды или
мочевины) [86—89].
Ниже приведены некоторые примеры стабилизирующих добавок,
предложенных для полиформальдегида (табл. 21). Следует заметить,
что не все они обладают достаточным стабилизирующим действием.
Здесь уместно напомнить, что нестабилизированный
полиформальдегид невозможно перерабатывать
из-за сильного разложения мате- * 30
риала при нагревании. Нечто по- §
добное происходит при перера- 1ц
ботке нестабилизированного поли- ^*
винил хлорида. Стабилизаторы мо- ,
гут снизить общую скорость раз- §| to
ложения, но и в этом случае пере- ^«
работка в расплавленном состоя- В
нии часто невозможна, так как
скорость разложения все еще
высока (недостаточный
стабилизирующий эффект). Фирма «Дюпон»
/
/
1
А
V
20
60
80
Время, мин
Рис. 47. Кинетические кривые
разложения различных образцов диаце-
тата ПОМ @,05 г) на воздухе при
220_Q С:
J—диацетат ПОМ, Мп = 30 000; 2—то же
с 1% А-2246 (антиоксидант); г - то же
с 1 % полиамидной смолы; 4 — то же
с 0,5% А-2246 и 1% полиамидной смолы.
впервые практически доказала, что
можно подобрать такую
стабилизирующую композицию для
высокомолекулярного диацетата ПОМ,
которая обеспечивает возможность
переработки полимера
стандартными способами (с некоторыми ограничениями по температуре и
продолжительности нагрева). На рис. 47 показаны кривые
термоокислительной деструкции различных образцов диацетата ПОМ.
Старение полиформальдегида под действием ультрафиолетовых
лучей, радиации и других факторов рассматривается в
работах [101—104]. Считается, что стабилизированный полимер в
дальнейшем сохраняет свои свойства при эксплуатации, так как
антиоксиданты полностью не расходуются при формовании. Это косвенно
подтверждается опытами по многократной переработке полимера.
На свету без добавок специальных фотостабилизаторов
происходит быстрое старение материала (табл. 22).
В качестве фотостабилизаторов, т. е. веществ, поглощающих
ультрафиолетовые лучи, для стабилизации полиформальдегида
предложены следующие соединения:
* В то же время в случае разложения гликоля ПОМ доказано, что полиамид
только снижает общую скорость разложения.
9* 131
оксибензофеноны
ОН X
I СО |
R
R'
где X — Н, ОН или галоген; R и R' — Н, галоген, алкоксигруппа;
производные циннамонитрила (а-карбометокси^-фенилциннамо-
итрил) [105]
Ar-R-CR'=CXY
где R, R' — Н, алкил, цианалкил, арил; X, Y — циано-, алкокси-
карбоксиамидо- или алкилкарбоксиамидогруппы [106];
соединения, отвечающие формуле
где R, R' — Н, алкил-, аксиламин-, циклогексил-,
или бензоатная группа.
знил-, нитро-
ТАБЛИЦА 22
Влияние фотостабилизатора на механические свойства
полиформальдегида при старении [46]
Фотостабилизатор
Без фотостабилизатора
2,2-Д ио кси-4,4-димето кси-
бензофенон B%)
Старение
месяцы
0
3
6
12
0
3
6
12
Предел
прочности
при
растяжении *
724
635
548
365
690
710
710
710
Относительное
удлинение
при
разрыве *
30
5
5
3
37
25
24
20
Удельная
ударная
вязкость **
кГ-см/см2
0,037
0,026
0,012
0,013
0,033
0,042
0,031
0,038
* Испытания по ASTM D 638—49T.
** Испытания по ASTM D 256—47Т.
ЛИТЕРАТУРА
1. Штаудингер Г., Высокомолекулярные соединения, ОНТИ, Хим-
теорет, 1935, стр. 236.
2. Штаудингер Г., Высокомолекулярные соєдинєнияі ОНТИ, Хим-
теорет, 1935, стр. 237.
3. Штаудингер Г., Высокомолекулярные соединения, ОНТИ, Хим-
теорет, 1935, стр. 250.
132
4- Фурукава Дж., Саегуса Т., Полимеризация альдегидов и
окисей, Изд. «Мир», 1965, стр. 20.
5. Kern W., Cherdron H., Makromol. Chem., 40, 101 A960).
6. Е п і k ol op у a n N. S., J. Polymer Sei., 58, 1301 A962).
7. E и и к о л о п я н Н. С, В а р д а н я н М. С, ЖВХО им. Д. И.
Менделеева, 7, 194 A962).
8. М a d о г s k у S. L., S t г a u s S., J. Res. Nat. Bur. Stand., 53, 361 A954).
9. М a d о г s к у S. L., J. Polymer Sei., 9, 133 A952).
10. М a d о г s к у S. L., S t г а и s S., J. Polymer Sei., 36, 183 A959).
11. Дудина Л. А., Кармилова Л. В., Тряпицына Е. Н.,
Ениколопян Н. С, Сообщение на симпозиуме по макромолекуляр-
ной химии, Прага, 1965 г.
12. Mejzlik J., В erg er J., Chem. Prumsl., 12, 461 A962).
13. Price L. K., Lindsay L. P., J. Am. Chem. Sei., 82, N 14, 3538
A960).
14. Д у д и н а Л. А., Ениколопян Н. С, Высокомол. соед., 4, 896
A962).
15. Д у д и н а Л. А., Диссертация, ИХФ АН СССР, 1963 г.
16. Ш в е й ц е р С. Е., Макдональд Р. Н., Пандерсон Жд. О.,
Химия и технология полимеров, № 3, 70 A960).
17. Д у д и н а Л. А., Ениколопян Н. С, Высокомол. соед., 5, 986
A963).
18. 3 е л ь д о в и ч Я. В., ЖФХ, 13, 163 A939).
19. Д у д и н а Л. А., Ениколопян Н. С, Высокомол. соед., 5, 1135
A963).
20. К 1 є і n R., S с h о е n L., J. Chem. Phys., 24, 1094 A956).
21. С е м е н о в Н. Н., О некоторых проблемах химической кинетики и
реакционной способности, Изд. АН СССР, 1958, стр. 29.
22. Рас J., Meyzlik J., V е s el у К., Chem. Prum., 12,575A962).
23. T о г і k а і S., J. Polymer Sei., A-2, 3461 A964).
24. Нейман M. В., Коварская Б. М., Блюменфельд А. Б.,
ДАН СССР, 154, 631 A964).
25. Kern W., Cherdron H.,Hohr L., Makromol. Chem., 52? 48 A962)..
26. О. D о r n a 1 J. M., L u k a s H. J., J. Am. Chem. Soc, 72, 5489 A950)*
27. Kreevoy M. M., Taft R. W., J. Am. Chem. Soc, 77, 3146, 5590
A955).
28. Яновская Л. А., Беликов В< М., Изв. АН СССР, Сер, хим.,
1363 A965).
29. Яновская Л. А., Кучеров В. Ф., Изв. АН СССР, Сер. хим.,
2189 A962).
30. Staudinger Н., Singer R., Johner H.,LuthyM., Kern W.,
Rusidis D.,Scheitzer O., Liebigs Ann. Chim., 474, 145 A929).
31. M e j z 1 і k J., Macromol. Chem., 59, 184 A963).
32. Fukuda W., Kakiuchi H., Koguo Kagaku Zasshi, 66, 387 A963).
33. Kern W., Cherdron H.,Jaacks V., Angew Chem., 73,177 A961).
34. Д у д и н а Л. А., Кармилова Л.
Высокомол. соед., 5, 1160 A963).
35. Д у д и н а Л. В., Кармилова Л.
ДАН СССР, 150, 309 A963).
36. Дудина Л. А., Агаянц Л. А.,
колопян Н. С, Высокомол. соед.,
37. Д у д и н а Л. А., Б е р л и н Ал. Ал.,
колопян Н. С, ДАН СССР, 150, 580 A963).
38. Дудина Л. А., Жарова Т. Э., Кармилова Л. В.,
Ениколопян Н. С, Высокомол. соед., 6, 1926 A964).
39. Г р а с с и Н., Химия процессов деструкции полимеров, Издатинлит,
1959.
40. Н ей м а н М. Б., А л иш о ев В. Р., К о в а р с к а я Б. М., Пласт.
массы, № 7, 11 A962).
іза
В., Е
В., Е
К а р м ]
5, 1245
Карм
н и
н и
К 0
К 0
н л о в ;
A963).
и л о в
л
л
а
а
о п
о п
Л.
Л.
я н
я н
в.,
в.,
Н
н.
Е
Е
. С.
С,
н и-
н и-
Бучаченко А. Л., Нейман М. Б., Высокомол. соед., 3, 1285
A961).
Нейман М. Б., Федосеева Т. С, Чубароча Г. В. и др.,
Высокомол. соед., 5, 1339 A963).
Дудина Л. А., Кармилова Л. В., Ениколопян Н. С,
Сб. «Химические свойства и модификация полимеров», изд. Вые. мол.
соед., 1964, стр. 228.
Nejman M. В., Aging and Stabilization of Polymers, Ch. 5, Ed. Cons.
Bur., Inc., N. Y., 1964.
Англ. пат. 770717, 1957 г.
Du Pont, англ. пат. 835841, 1960 г.
Mejzlik J.,Pac J., Macromol. Chem., 82, 226 A965).
Montekatini, фр. пат. 1364519, 1964 г.; 1364410, 1964 г.
Kosinska W., Feigin J., Zakrzewski L., Tworzuwa—
Guta, Lakiery, 5, 178 A960).
Mejzlik J., Janeckova L., Makromol. Chem., 82, 238 A965).
Bayer F., англ. пат. 868356, 1961 г.
Bayer F., Japan. Pat. Appl. Publ., 1792 A961).
Hagemeyer H. J., Ind. Eng. Chem., 41, 765 A949).
Nikon Shokubai, Kagaku Kogyo, яп. пат. І3440, 1961 г.
Compagnie de Saint-Gobain, фр. пат. 1285907, 1962 г.
Mejzlik J., Lesna M., Osecky P., Skackoval., Sadov-
s k a E., Makromol. Chem., 82, 253 A965).
Du Pont, пат. США 2964500, 1960 г.
Du Pont, пат. США 3125551, 1964 г.
Staudinger H., Singer R., Johner H., Luthy M.,
Kern W., Russidis D., Schwetger O., Ann., 474, 243 A929).
L on d erg an T. E., пат. США 2512950, 1950 г.
Societa Haliana Resine, пат. США 3061589, 1962 г.
Du Pont, англ. пат. 877256, 1961 г.
Hoechst F., пат. ФРГ 1155596, 1964 г.
Kurashiki Rayon Со Ltd, фр. пат. 1329303, 1962 г.
Du Pont, англ. пат. 868365, 1961 г.
Bayer F., англ. пат. 917016, 1963 г.
Hoechst F., фр. пат. 1286118, 1961 г.
Celanese Corp., фр. пат. 1948904, 1963 г.
Hoechst F., яп. пат. 9345, 1961 г.
Badische Anilin, пат. ФРГ, 1137214, 1963 г.
Compagnie de Saint-Cobain, фр. пат. 1348553, 1963 г.
Англ. пат. 818660, 1960 г.
Celanese Corp., англ. пат. 924925, 1963 г.
Societa Holiana Resine, англ. пат. 892178, 1962 г.; 943188, 1963 г.
Torikai S., J. Polymer Sei., A-2. 239 A964).
Du Pont, англ. пат. 796863, 1958 г.
Фр. пат. 1461969 A966).
Итал. пат. 753938 A965).
Фр. пат. 1462943 A966).
Du Pont, пат. США 2993025, 1961 г.
Bayer F., англ. пат. 927610, 1963 г.
Bayer F., пат. ФРГ 1152542, 1964 г.
Kurashiki Rayon Со Ltd, англ. пат. 913162, 1962 г.
Celanese Corp., пат. США 2936298, 1960 г.
Montecatini, фр. пат. 1356564, 1964 г.
Алишоев В. Р., Гурьянова В. В., К о в а р с к а я Л. В.,
Нейман М. Б., Высокомол. соед., 4, 1887 A962).
Д у д и ы а Л. А., Жарова Т. Э., Кармилова Л. В.,
Ениколопян Н. С, Высокомол. соед., 6, 1931 A964).
Алишоев В. Р.,Нейман М. Б.,Коварская Б. М.,Г у р ья-
н ов а В. В., Высокомол. соед., 5, 644 A963).
89. К о в а р с к а я Б. М., Нейман М. Б., Гурьянова В. В., Р о-
занцев Э. Г., Нитче О. Н., Высокомол. соед., 6, 1737 A964).
90. Du Pont, пат. США 2871220, 1959 г.
91. Du Pont, пат. США 2966476, 1960 г.
92. Badische Anilin, фр. пат. 1240264, 1960 г.
93. Kurashiki Rayon, англ. пат. 898120, 1962 г.
94. Пат. ФРГ 1098713, 1961 г.
95. Badische Anilin, пат. ФРГ 1173245, 1964 г.
Deutsche Gold, фр. пат. 1306658, 1962 г.
Stamicarbon, фр. пат. 1353368, 1964 г.
Du Pont, пат. США 3102106, 1963 г.
Du Pont, пат. США 2920059, 1960 г.
Du Pont, англ. пат. 748856, 1956 г.
Kelleher H.,Jassie L. В., J. Appl. Polymer Sei., 9, 2501 A965).
Torikae S., J. Polymer Sei., A-2, 3461 A964).
, К u r і h a r a F., J. Chem. High Polymer (Japan), 22, 539 A965).
, Англ. пат. 835841, 1960 г.
, Du Pont, англ. пат. 912033, 1962 г.
Amer. Cyanamid Co, пат. США 3079366, 1963 г.
.Блюменфельд А. Б., Нейман М. Б., Коварская Б. М.г
Высокомол. соед., 8, 1990 A966).
108. Гурьянова В. В., Коварская Б. М., Нейман М. Б.г
Нитче О. Н., Высокомол. соед., 8, 1783 A966).
109. Гурьянова В. В., Коварская Б. М., Нейман М. Б.,
Постников Л. М.,Шляпинтох В. Я., Кузнецов аГ. В,
Высокомол. соед., 7, 2175 A965).
110. Гурьянова В. В., Коварская Б. М., Нейман М. Б.*
Розанцев Э. Г.,Скрипко Л. А., Высокомол. соед., 9 (А), 2165
A967).
ГЛАВА V
СОПОЛИМЕРИЗАЦИЯ ФОРМАЛЬДЕГИДА
(И ТРИОКСАНА)
Среди всех возможных способов повышения стабильности поли-
оксиметиленов особое место заняла сополимеризация
формальдегида и триоксана с подходящими сомономерами. Термин
«подходящие сомономеры» расшифровывается в данном случае следующим
образом. Если в молекулярную цепь полиоксиметилена ввести
какие-либо звенья, стабильные в условиях деполимеризации аце-
тальной структуры, например типа —СН2—СН2— или
—СН2—СН2—СН2—, то можно ожидать, что это увеличит общую
устойчивость цепи. В блоксополимерах такие звенья,
расположенные на концах цепи, могут играть роль стабильных концевых групп.
В статистическом сополимере блоки полиоксиметиленовой
структуры будут чередоваться с блоками (или мономерными единицами)
стабильного сомономера *. Полимер такого типа образуется,
например, при сополимеризации триоксана с 1,3-диоксоланом,
содержащим звенья —СН2—СН2—, устойчивые к цепной деполимеризации:
Н2С—СН2
І І
О О
\/
сн2
В сополимере, содержащем преимущественно оксиметиленовые
звенья, большая часть концевых групп — полуацетальные:
НО—(GH2O)K-(CH2CH2O)W—(СН2О)^-(СН2СН2ОJ— . . .-(СН2О)Г-СН2ОН
Если деполимеризация инициирована по концам молекул, то
отщепление мономерных звеньев будет происходить только до встречи
* Подобным образом решается проблема улучшения термической стойкости
полиметилметакрилата, деполимеризация которого протекает по цепному сво-
^однорадикальному механизму. Введение в полимерные цепи небольших
количеств акрилокитрила уменьшает деструкцию [71].
136
активного центра с термодинамически устойчивыми блоками сомо-
номера:
. . ._O-(CH2)W-CH2CH2O-CH2O-CH2O-GH2OH
. -O-(GH2O)W-CH2CH2O-CH2O-CH2OH + GH2O
і
. -О-(СН2О)И-СН2СН2О-СН2ОН + 2СН2О
I
. . ._O—(CH2O)w-CH2CH2OH-f 3CH2O
При разрыве полимерной цепи по закону случая (например г
при ацидолизе) полимерный активный центр будет деполимеризо-
ваться также только до встречи со стабильным звеном *:
. -O-(CH2O)W-CH2CH2O-GH2-O-GH2- • • • -
. . ._O-(CH2O)W-GH2CH2O-CH2 + HO-CH2-.
. . .—О—(СН2О)Я—GH2CH2 + CH2O
Получение термически стабильных сополимеров представляет
интерес потому, что в этом случае отпадает необходимость в
блокировании концевых групп полиформальдегида путем специальной
обработки. Преимущества сополимеризации как метода синтеза
стабильного полиформальдегида не исчерпываются вопросами
термической стабильности, хотя, безусловно, это является одним из
важных моментов. Сополимеризация позволяет также в широких
пределах варьировать физико-химические свойства продуктов.**
Так, для специальных целей можно получать сополимеры
формальдегида с повышенной растворимостью, хорошей адгезией,
эластичностью, огнестойкостью и т. п.
НЕКОТОРЫЕ ТЕОРЕТИЧЕСКИЕ ПРЕДСТАВЛЕНИЯ
До середины 40-х годов сополимеры изучались только
эмпирическим путем, хотя попытки точной кинетической обработки
данных о сополимеризации делались неоднократно. В 1944 г. сразу
* Сравните с поведением блокированного гомополимера: при разрыве
полимерной цепи по закону случая она будет деполимеризоваться до конца.
** В большинстве случаев речь идет о сополимерах преимущественно аце-
тальной структуры, содержащих всего несколько процентов сомономера.
Увеличение доли сомономера приводит прежде всего к снижению степени
кристалличности [72—74].
137
нескольким исследователям, работавшим независимо друг от друга,
удалось на базе результатов радикальной сополимеризации виниль-
ных мономеров вывести уравнение, связывающее состав сополимера
с составом реакционной смеси и относительной реакционной
способностью сомономеров [1, 2, 8, 11].
При этом они исходили из следующих допущений:
а) реакционная способность радикала не зависит от его длины;
б) реакционная способность радикала зависит только от природы
концевого мономерного звена.
В этом случае в реакционной смеси, содержащей два мономера
(А и В), должны протекать следующие реакции роста:
А«+А —> А* ^11 = ^11 [А•] [А]
В. + В > В- Ы722 = А22[В.][В]
A.-fB —> В. H>i2 = Ai2[A.][B]
Если принять, что при достаточной длине молекулярной цепи
/ а\к\ d[B]\
мономеры расходуются только в реакции роста ( ~-~ и ~-- 1 ,
то отношение количеств мономеров, вошедших в сополимер, в любой
момент времени будет равно:
d [A] [А] &ii?[A-] + &2i [B-] ...
T\B\—JB\' A12[A.] t*22[B.] {)
Допуская существование стационарной концентрации для радикалов
обоих типов, получим, что w12 = w21. Сделав соответствующие
преобразования, уравнение A) можно записать в следующем виде:
1
d[B] [В] * (Aaa/A21)[B] + [A] {/)
Отношение констант кц/кі2 и k22lk21 обычно обозначают как
гг и г2, поэтому в окончательной форме уравнение
дифференциального состава бинарного сополимера (уравнение Майо — Льюиса)
примет вид:
d[B] [В] га[В] + [А]
Рассмотрим ряд предельных случаев, определяемых значениями
констант сополимеризации гх и г2. Так, если гг = г2 = 0, то
каждый мономер способен взаимодействовать только с другим
радикалом. При этом образуется сополимер, в молекулах которого
мономерные единицы обоих типов строго чередуются, как, например,
в случае сополимеризации стильбена с малеиновым ангидридом.
Если состав исходной реакционной смеси удовлетворяет условию
/1ІАЖВ] 4 т п [All-n
-" '•"• [В]"" 1-Г2
138
то из уравнения C) следует, что состав сополимера будет
соответствовать составу реакционной смеси. Во всех остальных случаях
(кроме того, когда обе константы сополимеризации равны единице)
относительная скорость расхода мономеров не равна отношению
их начальных концентраций. Кинетический анализ всех возможных
случаев соотношения констант сополимеризации приводится обычно
во всех монографиях, посвященных радикальной полимеризации
виниловых соединений.
Отметим, что уравнение C) отражает состав сополимера в
каждый конкретный момент. Для того чтобы вычислить среднее
значение состава сополимера за какой-то отрезок времени или как
функцию степени превращения, нужно перейти к интегральной форме
уравнения состава [2].
Справедливость уравнения C) подтверждена многочисленными
экспериментами в области радикальной сополимеризации [13—17].
Оно позволило разработать полуколичественную теорию
радикальной реакционной способности (схема Q — е) [12]. В дальнейшем
были исследованы и теоретически рассмотрены случаи, когда
уравнение Майо—Льюиса потребовало некоторых усложнений. К таким
случаям относится, например, протекание деполимеризации в
условиях, когда реакционная система близка к равновесию. Если
принять, что каждая из четырех приведенных выше реакций роста
обратима, то большое значение приобретает предпоследнее
мономерное звено в каждом радикале, так как оно определяет тип нового
радикала, полученного при деполимеризации. Теорию равновесной
гомополимеризации разработал Тобольский [18], ему же совместно
с Алфреем [19] удалось вывести уравнение состава и для случая
равновесной сополимеризации.
При выводе уравнения состава принималось, что рост цепи
всегда идет по бимолекулярному механизму. Однако известны
процессы, подчиняющиеся уравнению первого порядка, в которых
лимитирующей стадией является, например, изомеризация
(механизм Фонтана — Кидера) [20]. В этом случае, если все реакции
роста протекают как реакции первого порядка, уравнение Майо —
Льюиса приобретает следующий вид:
d[B)
Могут быть и более сложные случаи, если реакции роста цепи и
первого, и второго порядка.
Полная информация о строении сополимера должна включать
знание распределения молекул по длинам цепи и распределения
мономерных звеньев в макромолекулах сополимера.
Распределение мономерных звеньев в общем случае было
теоретически рассмотрено в работе [24]. Состав большинства молекул
сополимера должен быть близок к статистическому среднему составу
при достаточно большом молекулярном весе. Расчет функций
распределения по размеру и составу был выполнен для различных
139
случаев винильной полимеризации 125] на основе статистической
теории.
Имеющиеся в настоящее время данные еще не позволяют создать
столь же полную теорию ионной сополимеризации, как для
радикальных процессов. Кинетическая обработка результатов
ионной сополимеризации обычно проводится по выше приведенной
схеме.
Анализ литературы по ионной сополимеризации винильных
мономеров показывает, что в большинстве случаев результаты
хорошо описываются уравнением Майо — Льюиса [21—23].
Константы ионной сополимеризации (в отличие от радикальных
процессов) находятся в сложной зависимости от типа системы
катализатор — сокатализатор, природы растворителя и других
факторов [75].
Реакционноспособными по отношению к формальдегиду и триок-
сану мономерами могут быть альдегиды и замещенные альдегиды;
кетоны; окиси алкиленов; циклические формали; циклические
простые эфиры, циклические сложные эфиры (лактоны) и циклические
ангидриды; винильные мономеры.
По способу инициирования все реакции сополимеризации можно
разделить на анионные (с формальдегидом) и катионные (с
формальдегидом и триоксаном). Особенно интересные и
многообещающие результаты были получены при катионной сополимеризации.
В настоящее время известны сополимеры формальдегида (триок-
сана) с представителями всех перечисленных выше классов
соединений, однако далеко не все изучены в равной степени. В связи с
разработкой технологических процессов синтеза полиформальдегида
наибольший интерес вызывают сополимеры с повышенной
термостабильностью и другими ценными техническими свойствами.
АНИОННАЯ СОПОЛИМЕРИЗАЦИЯ
Из перечисленных выше сомономеров по анионной схеме
полимеризуются карбонилсодержащие соединения (альдегиды,
кетоны), окись этилена и некоторые винильные мономеры.
Сополимеризация с незамещенными
альдегидами
Формальдегид легко сополимеризуется с высшими альдегидами,
образуя линейные продукты полиацетальной структуры. К несколько
неожиданным результатам приводит сополимеризация
формальдегида с ацетальдегидом [4]. Смесь жидких мономеров полимеризо-
вали при —78° С в толуоле в присутствии различных
катализаторов типа металла л кил ов. Соотношение мономеров в
реакционной смеси (в мол. %) от 40 : 60 до 60 : 40. Результаты этой работы
приведены в табл. 23.
140
ТАБЛИЦА 23
Свойства сополимеров формальдегида и ацетальдегида
Катализатор
ыс4н9
Zn(C2H5J
трет-С^ЩО^а
А1(С2Н5JС1
А1(С2Н5JС1-ТЮ14A:1)
AI (изо-С^Щ)з
Температура
плавления
сополимера
°С
160-170
180—185
Жидкость
110 до 170
>300
300
Растворимость сополимера
в метиловом спирте
Частично растворим
То же
Растворим
»
Нерастворим
»
Свойства продуктов, полученных при использовании в качестве
катализаторов LiC4H9 и Zn (С2Н5J, не зависели от состава
исходной смеси. На основании данных ИК-спектроскопии авторы пришли
к выводу, что в этих случаях образовывалась смесь гомополимеров.
При применении в качестве катализатора бутилата натрия
получалась вязкая жидкость, представляющая собой полиэфир (по данным
ИК-спектроскопии).
Наиболее интересные результаты были получены с триизобу-
тилалюминием и комплексным катализатором: диэтилалюминий-
хлорид — хлористый титан A : 1). В обоих случаях образовывался
эластичный неплавкий сополимер, нерастворимый при обычных
температурах. Хотя точный состав его установить не удалось,
но с помощью ИКС-метода было показано, что в сополимере
присутствуют метильные и метиленовые, а также простые эфирные звенья.
Качественно также было показано, что эластичность сополимера
растет с увеличением доли ацетальдегида в полимеризационной
среде.
Возможно, что при взаимодействии формальдегида и
ацетальдегида образуется ?-оксипропионовый альдегид [26]:
=О + СН3СН=О
НОСН2СН2СН=О
Можно предполагать, что он будет реагировать с металлорганиче-
ским соединением и давать анион:
ЗНОСН2СН2СН=О + А1(ш?о-С4Н9)з —> А13+(ОСН2СН2СН=ОK + 3 изо-С4Ню
Альдегидная группа в свою очередь способна образовывать анион,
что приведет к получению разветвленного сшитого продукта.
Таким образом, несмотря на то что каждый из мономеров в
указанных реакционных условиях образует только линейный полимер
полиацетальной структуры, их сополимеризация протекает
совершенно в другом направлении.
141
В патентной литературе приводятся данные о получении
линейных сополимеров формальдегида и ацетальдегида полиацетальной
структуры [27—29]. Описан процесс, заключающийся в том, что
пары очищенного формальдегида пропускают через раствор
ацетальдегида в подходящем нейтральном растворителе при минусовых
температурах. В качестве катализатора рекомендуются амины и их
производные, например трибутиламин [27]. Соотношение
мономеров можно варьировать в любых пределах. Температура
плавления сополимера зависит от состава реакционной смеси и может
изменяться от 80 до 185° С. Структурную формулу такого
сополимера можно представить следующим образом:
•. .— сн2—о—сн2—о—сн—о—си2—о—сн—о—сн2—о—сн—о—...
СН3
По физико-химическим свойствам (стабильности, растворимости,
жесткости) он должен занимать промежуточное место между каучу-
коподобным нестойким атактическим полиацетальдегидом и
кристаллическим полиформальдегидом.
Имеются патентные данные [30] о методах сополимеризации
формальдегида с высшими альдегидами общей формулы
R—СН=О
где R может быть алкильным радикалом с количеством атомов
углерода до 10, циклоалкилом или арильным радикалом. В качестве
катализаторов рекомендуются гидриды и органические соединения
металлов II и III групп периодической таблицы.
Сополимеризация с производными
альдегидов и кетонами
Наличие электронодонорных метальных групп в а-положении
по отношению к ацетальным связям вызывает повышение
электронной плотности у связи С—О и увеличение ее чувствительности
к электрофильным агентам, вызывающим разрыв цепи. Поэтому
при введении ацетальдегидных групп в молекулярную цепь
полноксиметилена трудно рассчитывать на увеличение
стабильности получающегося сополимера по сравнению с полиоксимети-
леном.
С точки зрения повышения термической стабильности ацеталь-
ных связей более перспективным представляется проводить сополи-
меризацию формальдегида с соединениями, имеющими заместителей
с электроноакцепторными свойствами. В качестве таких сомоно-
меров в патентной литературе предложены фторальдегиды [31] и
хлораль [32].
142
Сополимер формальдегида с фторальдегидом является одним
из первых сополимеров полиоксиметилена, описанных в литературе.
Он легко образуется при пропускании паров формальдегида через
раствор дифторацетальдегида в толуоле при —35° С в присутствии
четвертичной аммониевой соли в качестве катализатора
полимеризации.
Сополимеризация с хлоралем проводилась при низкой
температуре (от —60 до —100° С) в инертном растворителе. В качестве
катализаторов применяли амины и фосфины. Сополимер содержал
от 10 до 90% хлораля. Температура плавления и температура начала
термической деструкции (после ацетилирования концевых гидр-
оксильных групп) были соответственно выше, чем у гомополимера.
Так, например, продукт, содержащий 45 вес. % хлораля, плавился
при 190° С. Сополимеры, содержащие ^ 10 вес. % хлораля, были
негорючи.
Натта с сотр. [3] осуществил анионную сополимеризацию
формальдегида с диметилкетеном
н3сх
н3с/
В зависимости от условий полимеризации [33, 34, 35] диметил-
кетен может полимеризоваться как по С—С-связи, так и по
карбонильной связи. С формальдегидом он может образовывать (в
зависимости от типа катализатора) как чередующиеся сополимеры типа
Н3С О
I II
. G-G-
СНЯ
Н3С О п
I II
—ОСН2-С-С-
СНз
—ОСН2- •
так и статистические или блоксополимеры типа
г- П ГН
• -СН2О—
II I
—С—С—СН2О—
СНо
—сн2о—сн2о-.
Равномерное распределение звеньев кетена (при добавлении
его к формальдегиду в количестве 1—3 вес. %) было достигнуто
при использовании в качестве катализатора полимеризации
третичных фосфинов. Интересно отметить, что при гомополимеризации
диметилкетена с этими катализаторами получили смесь структур
типа I и II:
Н3С О
1 II
-с-с-
1
СНз
I
ЛТТ ГЛ
II
— О—С—
II
143
При сополимеризации с формальдегидом на третичных фосфинах
всегда получалась только структура I. Строение сополимера было
доказано с помощью ИК-спектроскопии. В спектрах обнаружены
линии поглощения, характерные для эфирной и карбонильной
групп.
Введение в макромолекулу полиоксиметилена звеньев
с С—С-связью позволило, как и предполагалось, увеличить
термическую стабильность сополимера. При нагревании сополимера,
содержащего 3—4 вес. % диметилкетена, в атмосфере азота при
220° С потеря массы составляла всего 10—12%, остаток был
стабилен.
В патентной литературе описаны способы получения сополимеров
формальдегида с акролеином [35] и фурфуролом [36]. Со
полимеризацию проводили в широком интервале температур от —100 до
-f-100° С в присутствии катализаторов основного типа, например
пиридина, третичных аминов, фосфинов, стибинов. О кинетике
и механизме этих реакций пока ничего не известно, так же как
и о свойствах получаемых продуктов.
Сополимеризация с циклоангидридами
монооксикарбоновых кислот
Описана сополимеризация формальдегида в присутствии
обычных анионных катализаторов гомополимеризации (амины, фосфины,
ониевые соли) с циклическими ангидридами монооксикарбоновых
кислот (циклические гликолиды, лактиды и т. д.):
r_O-C=O
I I
О=С-О—R
Получаемые сополимеры содержат сложноэфирные группы и
неустойчивы к гидролизу [37]. Кроме того, введение объемных
заместителей в полимерную цепь приводит к резкому нарушению
регулярности строения цепи и уменьшению кристалличности.
Сополимеризация с изоциановой кислотой
и ее эфирами
Описан способ сополимеризации формальдегида с изоциановой
кислотой, имеющей реакционноспособную карбонильную группу
NH=C = O. Сополимеризацию по карбонильной связи проводят
в растворе инертного углеводорода, растворяющего оба мономера,
или в диметилформамиде при низких температурах (от —50 до
+30° С) в присутствии различных аминов или фосфинов [38].
Образующийся сополимер может содержать от 3 до 75 вес. % изоциановой
кислоты. Он отличается очень высокой температурой размягчения
(около 300° С), по-видимому, благодаря образованию сшитых струк-
144
тур и обладает высокой адгезионной способностью по отношению
к полиформальдегиду. Аналогичными свойствами обладают
сополимеры с эфирами изоциановой кислоты.
Сополимеризация с винильными мономерами
Механизм анионной сополимеризации формальдегида в
настоящее время практически не изучен. Перечисленные выше примеры
позволяют сделать вывод о том, что реакционная способность
карбонильной группы и винильной связи при анионном инициировании
существенно различаются, причем г2 (винильной связи) в
большинстве случаев близка к нулю. Исключением являются соединения
типа кетенов и, возможно, альдегидов, содержащих помимо
карбонильной группы винильные связи (акролеин, фурфурол).
Сравнительно легко можно получить блоксополимеры
формальдегида с винильными мономерами.
Известно, что некоторые «живущие» полимеры способны
инициировать полимеризацию винильных, диеновых мономеров и
циклических окисей олефинов [39—41]. Можно предполагать, что если
к «живущему» полимеру добавить безводный формальдегид, то
будет образовываться блоксополимер типа
R_GH2-O-GH2—. . •— О-СН2ОН
Получающийся сополимер состоит всего из двух блоков — карбо-
цепного, образованного при гомополимеризации винильного
мономера, и гетероцепного — ацетального. Для того чтобы получить
блоксополимер, содержащий более чем один блок карбоцеиного
мономера, необходимо найти такие условия, реакции, при
которых макроанион, имеющий алкоголятный активный конец
RCH2O—••—СН2О~, мог бы взаимодействовать с винильным
мономером и вести его полимеризацию. Была сделана попытка [42]
получить блоксополимер такого типа, в котором роль концевых
термостабильных групп выполняли бы карбоцепные блоки:
(CH2O)W-R (CH2O)W-R'H
Полимеризацию формальдегида инициировали с помощью
натрий-нафталинового комплекса, синтезированного по методу,
описанному в работе [43]. «Живущие» полимеры были получены
полимеризацией в растворе тетрагидрофурана различных мономеров:
стирола, бутадиена, изопрена. Удалось получить сополимеры,
содержащие до 15% карбоцепного сомономера. Для блокирования
второго конца цепи в реакционную систему добавляли акрилони-
трил, который хорошо полимеризуется в присутствии алкоголятов
щелочных металлов [44]. Однако данные ИК-спектроскопии и
анализ термостабильности блоксополимера показали, что
сополимеризация с акрилонитрилом не произошла.
10 Заказ 1135. 145
В то же время в одном из патентов [86] сообщается о том, что
получен блоксополимер формальдегида с винильными
мономерами, содержащий более двух блоков. Отмечается, что при
одновременном добавлении в реакционную систему, содержащую
«живущий» полимер, двух мономеров полимеризуется только один из них.
Когда он полностью исчерпается, начинается полимеризация
другого Мономера. Сополимеризация происходит при низких
температурах. Например, формальдегид взаимодействовал с «живущим»
полимером при температуре ниже 0° С; а-метилстирол ниже
—40° С, акрилонитрил ниже —50° С.
КАТИОННАЯ СОПОЛИМЕРИЗАЦИЯ.
ПЕРЕДАЧА ЦЕПИ С РАЗРЫВОМ
Катионная сополимеризация' формальдегида и триоксана
изучалась гораздо интенсивнее, чем анионная. Это объясняется прежде
бсєго тем, что при катионной полимеризации значительно
расширяется круг используемых сомономеров.
Открытие нового элементарного акта — передачи цепи с
разрывом [45, 84, 85] создало новые возможности для получения
различных типов сополимеров полиоксиметилена.
Рассмотрим механизм реакции передачи цепи с разрывом и
некоторые изменения в кинетике сополимеризации, к которым он
приводит.
При гидролизе ацеталей и простых эфиров наблюдается
образование промежуточного оксониевого комплекса, который затем
распадается с выделением нестойкого карбоний-иона и отщеплением
молекулы алкоксисоединения. Поскольку гидролиз проводится
в водной среде, карбоний-ион немедленно подвергается дальнейшему
превращению.
Экспериментально показано, что при полимеризации
формальдегида или триоксана по катионному механизму растущий
полимерный карбоний-ион может расщеплять ацетали типа диметилфор-
маля (метилаля) в безводной среде таким образом, что после разрыва
ацетальной связи одна часть молекулы присоединяется к карбоний-
иону, а другая приобретает положительный заряд и способность
продолжать полимеризацию формальдегида (триоксана). Эта
реакция используется при стабилизации гомополиформальдегида
непосредственно в ходе полимеризации (см. гл. IV) путем передачи цепи
на некоторые соединения с ацетальными и сложноэфирными связями.
Представим теперь, что полимерный катион атакует по закону
случая длинные (полимерные) цепочки, содержащие ацетальные
или какие-либо другие связи, способные рваться под действием
электрофильных агентов. В результате взаимодействия при
благоприятных условиях произойдет разрыв связи в атакуемой молекуле
с образованием новой макромолекулы и нового активного центра.
Этот вновь образовавшийся активный центр будет вести
полимеризацию мономера пока не произойдет новое столкновение между ним
146
и какой-либо другой молекулой по ацетальной связи, что приведет
к новой передаче цепи с разрывом:
. . .—СН2^О—СН2—О—R; —>• • . — СН2— О—СН2—О—R' —>
GH2 GH-2
¦I I
О О
I I
R R
—>.. . ,_сн2—O-fGH2—О—R'
СН2
О
I
R
R'_O-GH2-fO-CH2-O—R" —> и т. д
СН2
і
і
О
R'"
Количество частиц при таком взаимодействии не изменяется,
но происходит перераспределение длин и состава участвующих
в реакции молекул. Впервые на возможность такой реакции было
указано в работе [45].
Пусть в реакционную систему добавлен готовый полимер,
способный участвовать в реакции передачи цепи с разрывом. Если
предположить, что передача цепи с разрывом на добавленный
в полимеризационную систему готовый полимер происходит с
достаточно большой скоростью, то в конце концов после
многократного разрыва связей должно установиться некое равновесное
распределение блоков обоих типов в молекулах образовавшегося
сополимера.
Можно представить себе различные варианты осуществления
этого процесса с участием ацетальных структур:
1) К полимеризующемуся мономеру добавляется растворимый
в реакционной среде гомополимер. В результате реакции передачи
цепи с разрывом молекулы этого гомополимера будут дробиться
на все меньшие блоки и статистически распределяться в молекулах
растущего гомополимера.
2) Добавляемый гомополимер содержит реакционноспособные
(например, ацетальные) связи в цепи и группы, которые сами по себе
не сополимеризуются с формальдегидом (триоксаном). В результате
реакции передачи цепи с разрывом получится статистический блок-
сополимер, включающий эти группы. Гомополимер с реакционно-
способными связями можно получить, например, поликонденсацией
диолов любого строения с формальдегидом [59].
10* 147
3) Реакционноспособные связи располагаются в боковых цепях
молекул добавленного гомополимера (например, поливинил ацетата).
В этом случае образуется сшитый сополимер.
4) Можно взять два растворимых в реакционной среде готовых
гомополимера, содержащих реакционноспособные связи в главных
или боковых цепях. Добавлением соответствующего инициатора
можно вызывать реакцию передачи цепи с разрывом, которая
приведет к образованию сополимера, содержащего блоки взятых гомо-
полимеров.
Механизм передачи цепи с разрывом
Первичным актом передачи цепи с разрывом является ацидоли-
тическое расщепление полимерной цепи в безводной среде. Подобным
образом протекает полимеризация триоксана и других
циклических формалей и простых эфиров, а также передача цепи при катион-
ной полимеризации на формали, ацетали, сложные эфиры [37].
Вместо карбоний-иона может быть использован протон или
другой катион (при инициировании процесса). Суть дела от этого не
меняется.
По-видимому, во всех случаях реакция идет с образованием
промежуточного оксоний-иона:
+ уСН2—О—
. . ._о-СН2-О-СН2+:О<
ХСН3
+ >СН2—О—
Т"»' • • —О—СН2—О—СН2—О<
хсн3
Последний в силу лабильности ацетальной связи распадается
на карбоний-ион и полимерную цепь:
+ уСН-2—О—СНз
. . . —О—СН2—О—СН2—О< —у
хсн3
—>.. . .—О—СН2—О—СН2—O-fCH2—О—СН3
!
н3с
Таким образом, реакционная способность атакуемой связи
определяется основностью кислородного атома (т. е. тенденцией к
образованию оксоний-иона) и легкостью расщепления с образованием
карбоний-иона. Учитывая, что целый ряд кислородсодержащих
соединений образует оксоний-ионы, но проявляет слабую
тенденцию к расщеплению (простые эфиры, ангидриды карбоновых кислот),
Керн [46] считает, что лимитирующей является вторая стадия
реакции. Ранее аналогичный вывод был сделан в отношении
механизма ацидолиза ацеталей (см. гл. IV).
Разрыв связи кислород — углерод при превращении иона оксо-
ния в ион карбония облегчается, по мнению Керна, в результате
148
120
90
выигрыша резонансной энергии стабилизации. Например, при
передаче цепи на метила ль образуется катион, имеющий следующие
резонансные формы:
СН2—О—СН3 <—> СН2=О—СН3
С этой точки зрения передача цепи на простые эфиры типа димети-
лового или диэтилового должна быть затруднена, несмотря на их
высокую основность, так как они не образуют резонансные формы.
Керном с сотр. [46] были экспериментально найдены
относительные константы скорости передачи цепи с разрывом при
полимеризации триоксана в присутствии диме-
тилформаля. При этом
предполагалось, что в результате передачи
будут образовываться молекулы ди-
метилового эфира полиоксиметилена.
Концентрацию метоксигрупп опреде-
ляли по методу Цейзеля. От полу-
ченного полимера отделяли непроре-
агировавший диметилформаль и го-
мополимер (термическим
разложением в атмосфере азота при
190° С).
На основании уравнения Майо
в случае, когда степень
полимеризации определяется только реакцией
передачи цепи, получим:
1 __ [Агент передачи цепи]
DPn ~ п [Триоксан]
60
30
У
р
/
20 iff 60 ЗО /00
E)
Рис. 48. Влияние концентрации
диметилформаля (метилаля) на
степень полимеризации [60] при
полимеризации раствора триоксана
в нитробензоле при 25° С с
катализатором BF3Et20 @,02 ммолъ/л).
Тогда тангенс угла наклона прямой на рис. 48 дает значение
относительной константы скорости передачи цепи с разрывом для метилаля:
С = = 1 2 ± 1
кр
Данные, приведенные на рис. 48, получены при полимеризации
30 мл раствора триоксана в нитробензоле при 25° С с использованием
в качестве катализатора BF3O(G2H5J. Концентрация триоксана
100 ммолъ/л, диметилформаля 0,1 —10 ммолъ/л, катализатора
0,02 ммолъ/л, степень превращения 2—43%. При замене эфирата
фтористого бора на перхлорацетат относительная константа скорости
передачи цепи имела то же значение (в пределах ошибки измерения).
В тех же условиях определялась относительная константа
скорости передачи цепи на метилформиат (СН3ОСОН). Она оказалась
равной 0,03 (т. е. значительно ниже, чем для метилаля). Это можно
объяснить, во-первых, меньшей основностью кислорода в эфирвТой
группе метилформиата, а во-вторых, меньшей резонансной
стабилизацией образующегося формиат-катиона по сравнению с ацил-катио-
ном.
149
Константа скорости передачи цепи с разрывом при катионной
сополимеризации триоксана с полидиоксоланом, найденная Енико-
лопяном с сотр. [79], равна 1,4 (передача на полидиоксолановую
цепь), причем она в широких пределах не зависела от температуры^
что говорит о равенстве энергий активации реакций роста и передачи
цепи.
Передача цепи с разрывом может наблюдаться не только при
катионной сополимеризации. Известно, что анион может вызывать
реакцию щелочного гидролиза сложноэфирной связи (например,
омыление сложных эфиров, трансацетилирование и т. п.).
Следовательно, можно осуществить передачу цепи при анионной
полимеризации на полиэфирные молекулы, содержащие сложноэфирные
связи.
Вероятно, реакции этого типа можно наблюдать в свободно-
радикальных процессах, например на соединениях с S—S-связями.
Влияние передачи цепи е разрывом
на молекулярно-весовое распределение
гомополимеров и сополимеров
Передача цепи с разрывом не должна влиять на среднечисловой
молекулярный вес (число частиц остается постоянным). Однако
можно ожидать изменения средневесового молекулярного веса и,
соответственно, распределения по молекулярным весам. Эти вопросы
были рассмотрены в работах Ениколопяна с сотр. [47—49]. Функция
распределения имеет следующий вид:
л,=4-в-^ (в)
п
где R — общее число полимерных молекул в реакционной системе
(остающееся постоянным); п — начальная среднечисловая степень
полимеризации.
Такое же решение получено для реакционной системы, в которой
только часть молекул активна в реакции передачи цепи.
Функция распределения F) имеет такой же вид, как функция
распределения, получающаяся при поликонденсации. На рис. 49
приведены кривые числовых и весовых распределений,
соответствующих функции F), для трех значений среднечисловой степени
полимеризации п (R принято равным 104).
Для этого распределения средневесовая степень полимеризации
DPW = 2п и отношение DPJDPn = 2 (распределение Семенова —
Флори).
•Важным следствием из этого вывода является то, что независимо
от вида кривой МБР исходных гомополимеров реакция передачи
цепи с разрывом при сополимеризации должна привести к одному
и тому же распределению.
150
Специальные расчеты [48] показали, что этот вывод справедлив
и для системы олигомеров, т. е. что константа скорости реакции
передачи цепи с разрывом не зависит от молекулярного веса
атакуемого или атакующего полимера.
Рис. 49. Функции числовых (а) и весовых (б)
распределений для трех значений среднечисловой
степени полимеризации п:
1 — її*-= Ю4; 2— ^Г-= 2-Ю4; 3 — Щ = 3-Ю4.
Влияние передачи цепи с разрывом
на состав и строение образующихся
блоксополимеров
На свойства блоксополимера оказывает влияние характер
распределения звеньев сомономера в молекулярной цепи. При одинаковом
брутто-составе строение молекул сополимеров можно схематически
изобразить следующим образом:
. . ._А—А—А—А—А—В—В—В—В—В—-
-А—А—А—В—В—В—А—А—А—В—В—В— .
-А—А—А—В—А—А—А—А—А—В—А—А— •
блоксополимер
статистический
блоксополимер
статистический
сополимер
Чем больше относительная константа скорости передачи цепи
с разрывом, тем равномернее будут распределены блоки сомономера
и тем меньше будут размеры этих блоков.
Эффективность реакции передачи цепи с разрывом зависит от
активности реакционноспособных групп в молекуле добавки и их
концентрации. Положительно будут влиять также изменения
условий полимеризации, в результате которых ускоряется передача цепи по
сравнению с ростом цепи. Так как значения энергий активации
реакций передачи и роста цепи могут различаться, следовательно,
зависимости скоростей этих реакций от температуры будут различными,
и, изменяя температуру, можно регулировать относительную
константу скорости передачи цепи.
151
Рассмотрим теоретическую схему процесса сополимеризации
двух мономеров А и В при условии, что кроме четырех обычных
реакций сополимеризации (см. стр. 138) будут протекать еще четыре
элементарные реакции передачи цепи с разрывом [51]:
. . .—A.-f • •
. . .— A.+ • -
. . .—в.4- . -
. _A-B— . .
.-B-B-..
._в—А—..
. > .
. -*-•- .
••—A—A—.
..-A-B-.
. . _B—B— •
• • + •
• • + •
. . — в
. .-в
. .—A
. . .-В.+ . . .-А-А-. - . — -> . . .—в—А—. . • + • • --А. .
Наличие этих реакций, очевидно, повлияет на соотношение
концентраций активных центров А • и В •. Дифференциальное уравнение
для концентрации А- можно написать следующим образом:
.][mA-L] G)
где к12 и /с21 — константы скорости из уравнения Майо — Льюиса;
тА и тв — концентрации молекул А и В, вошедших в состав
сополимера в данный момент времени; L — концентрация блоков типа
А—А или В—В .
Константы кг, fe2, /c3, /с4 могут иметь любые значения, но вид
уравнения упрощается, если допустить, что образование активного
центра • • —В • вдвое более вероятно при атаке активным центром
• • —А« связи • • —В—В— • •, чем связи • • —А—В— • •, т. е.
2к} = к2 и по аналогии 2к3 = &4.
Конечно, такое допущение несколько ограничивает область
применения выводимых количественных соотношений, но
качественная сторона проблемы не изменяется.
Применяя условия стационарности, можно написать:
[A.]
ТВТ - к12 +к2 (тв/[В0]) (8)
Если ввести следующие обозначения: гг = кп/к12; г2 = к22/к21;
г3 = kjk21; г4 = k2lk12, п = тА/тв; г = [А0]/[В0]; а = тв/[В0],
то для небольших степеней превращения получим:
[A.]
[В.
Таким образом, если в системе протекает реакция передачи цепи
с разрывом, «мгновенный» состав сополимера уже не будет
описываться уравнением Майо — Льюиса.
152
Обозначим средние длины блоков мономеров А и В в сополимере
как sA и sB. При отсутствии передачи цепи с разрывом они равны:
A0)
B (И)
Для случая передачи цепи с разрывом, применяя введенные нами
обозначения, можно написать:
sb=~t (із)
Изменения L в результате протекания реакций передачи цепи
будут описываться системой из двух дифференциальных уравнений:
^l [L] A4)
Щ-ki [A.] [mB-L] +
.] [mA-L] A5)
Решение этой системы уравнений для стационарного состояния дает
L2 — x[mA — L][mB— Z,]=0 A6)
где х = k^kji^kg, откуда
L ^( + )
Знак -j- появляется, когда г<1 и знак — при х >> 1. Отсюда для
sA и sB получим:
A8)
A9)
Сополимеризация с окисью этилена
Открытие нового элементарного акта передачи цепи было широко
использовано для объяснения кинетики катионной сополимеризации
триоксана (и формальдегида).
153
9U
Так, известно, что катионная сополимеризация триоксана
с окисью этилена протекает следующим образом [50]:
1) в начале сополимеризации наблюдается период индукции,
значительно больший, чем при гомополимеризации триоксана в
подобных условиях;
2) продолжительность периода индукции увеличивалась с
повышением концентрации окиси этилена и уменьшалась с ростом
концентрации катализатора;
3) калориметрические измерения
показали, что изменение температуры в
течение периода индукции имеет линейный
характер (рис. 50).
Если прервать полимеризацию до
помутнения расплава в точке первого
температурного максимума путем добавления
вещества основного характера,
дезактивирующего катализатор, то можно
выделить растворимый в триоксане форполи-
мер. Таким образом, индукционный
период в значительной степени оказался
кажущимся и был связан с образованием
растворимого полимера (см. гл. III).
Ъ 82
% 76
I 70
6k
^Помут-
нвнив
/Добавление
—я?*~катализатора—
О 1
Время, мин
Рис. 50. Термометрическая
кривая начального момента
сополимеризации триоксана
с окисью этилена [50].
Методика эксперимента
Сополимеризацию расплава триоксана проводили при 70° С с 6 вес. % окиси
этилена в присутствии 0,02% BF3O(C4H9J- Процесс прерывали добавлением
третичного амина. Выход растворимого полимера составлял около 10 вес. %.
Газохроматографический анализ форполимера, образующегося в течение
индукционного периода, приводится ниже:
Превращение окиси
Состав форполимера этилена
вес.%
Растворимый полимер 54,3
1,3-Диоксолан 24,3
1,3,5-Триоксепан 8,7
1,4-Диоксан 5,1
Итого ... 92,4
Не вступившей в реакцию окиси этилена было обнаружено 4,9%.
Чтобы определить состав растворимого полимера, его гидролизовали
муравьиной кислотой и анализировали продукты гидролиза с помощью газовой
хроматографии. Были выделены олигомерные звенья, имеющие структуру поли-
оксиэтилена:
Звенья мономера —СН2—СН2—О— ... 93 вес. ч.
Звенья димера (—СЩ—СН2—О—J ... 41 вес. ч.
Звенья тримера (—СН2—СН2—О—)з ... 35 вес. ч.
Исходя из этих данных, механизм сополимеризации триоксана
с окисью этилена можно представить следующим образом:
I. Вначале идут два параллельных процесса:
154
1) Циклизация окиси этилена (одной или совместно с
формальдегидом, образующимся при ацидолизе триоксана):
2Н2С—СНа BF_to /Ч
\ / BF3Et20^ qX \q
0 ^^
1,4-диоксан
Н2С СН2+СН2О **»йТ«Ч> Н2С—СН2
\ / II
О 0 0
\ /
сн2
1,3-диоксолан
тш рч n /СН2 СН2ч
Н2С—СН2 + 2СН2О BF3Et2°^ 0Х Х0
\ / ^СНг-О-СНа^
1,3,5-триоксепан
2) Истинная сополимеризация окиси этилена с триоксаном (или
с формальдегидом). Образуется линейный сополимер со
статистическим распределением звеньев, причем, судя по данным анализа
состава, относительная константа сополимеризации окиси этилена
(гг) должна быть значительно больше, чем константа
сополимеризации триоксана (г2).
Строение образующегося статистического сополимера можно
изобразить так:
Присутствие в нем оксиметиленовых звеньев подтверждается высокой
скоростью гидролиза форполимера (полиоксиэтилен при данных
условиях деполимеризуется очень медленно).
II. Следующим этапом (после исчерпывания окиси этилена),
по-видимому, является гомополимеризация триоксана,
инициированная «живущими» концами молекул сополимера. Одновременно
имеет место передача цепи с разрывом по оксиметиленовым мостикам
форполимера. В результате блоки окиси этилена статистически
распределяются по цепи полиоксиметилена.
Что же касается образующихся на первом этапе реакции
циклических форма лей — 1,3-диоксолана и 1,3,5-триоксепана, то для них
возможно одно из двух (или одновременно оба) направления реакции:
1) Истинная сополимеризация с триоксаном по оксониевокарбо-
ниевому механизму:
H2G GH2 СІІ2—СН2
II +/
• • .-осн2осн2+ о о ¦+=+.. • •— осн2осн2о^
чсн2 сн2-(
Н2С СІІ2
+ I I
>. • .-ОСН2ОСН2ОСН2СН2ОСН2+ О О И Т. Д.
\ /
сн2
2) Гомополимеризация с последующей передачей цепи с разрывом.
155
Сополимеризация с 1,3-диоксоланом
и другими гетероциклами
Специфика этой реакции заключается в том, что сополимер можно
получить как прямой сополимеризацией триоксана с мономерным
1,3-диоксоланом, так и полимеризацией триоксана в присутствии
полидиоксолана.
Полидиоксолан
• . •— о—сн2—о—сн2—сн2—о—сн2—о—сн2—сн2— • • •
образуется при катионной полимеризации 1,3-диоксолана. Это
кристаллический продукт с температурой плавления 55° С,. хорошо
растворимый в воде и большинстве органических растворителей.
Структура молекул полидиоксолана такова, что его можно
рассматривать как чередующийся сополимер формальдегида и окиси
этилена. Ацетальные связи О—СН2—О в молекулярной цепи
полидиоксолана по устойчивости к электрофильной атаке не должны заметно
отличаться от ацетальных связей в молекуле полиоксиметилена.
Кроме того, полидиоксолан хорошо растворим в триоксане, что
облегчает проведение процесса.
Кинетика полимеризации и сополимеризации 1,3-диоксолана
изучалась в последние годы рядом автором [52—57, 77].
Предполагается, что рост цепи идет по оксониево-карбониевому механизму
с разрывом одной из ацетальных связей. Определены
термодинамические константы полимеризации 1,3-диоксолана в присутствии катион-
ных катализаторов типа кислот Льюиса [56]:Д#р_р = — Ь,2ккал/молъ;
Д5р_р = —18,6 э. е., предельная температура для одномолярного
раствора -fl° С. Из особенностей кинетики гомополимеризации
1,3-диоксолана укажем на ускорение реакции добавками ряда
кислородсодержащих веществ, например того же триоксана.
Экспериментальные данные свидетельствуют о том, что в присутствии триоксана
1,3-диоксолан полимеризуется не только со значительно большей
скоростью, но и при концентрациях ниже предельной (для данной
температуры). Последний факт не противоречит термодинамике,
поскольку в данном случае имеет место новая термодинамическая
система (диоксолан -f триоксан). Его можно объяснить тем, что
получаемый продукт является сополимером, в котором звенья 1,3-
диоксолана и формальдегида чередуются (большие блоки
1,3-диоксолана образоваться не могут, так как температура реакции выше
предельной):
• • - -О-СНа-О—СН2—СНа-О—СНа-О—СН2—О—СНа—СН2-О—СНг— * • •
звено диоксолана звено звено диоксолана
На второй стадии процесса происходит перераспределение звеньев
диоксолана за счет протекания реакции передачи цепи с разрывом.
На это указывает анализ состава сополимера, образующегося в ходе
156
процесса, и такой чувствительный индикатор, как изменение
температуры плавления сополимера с увеличением степени превращения
(рис. 51).
Доля диоксолана, вступившего в реакцию на первой стадии
процесса, зависит от концентрации инициатора и температуры процесса
и увеличивается с ростом последних. На рис. 52 показано изменение
содержания диоксолановых звеньев в сополимере с увеличением
степени превращения (хроматографическое исследование кинетики сопо-
лимеризации). Механизм этого процесса весьма сложен, так как
помимо реакции передачи цепи
с разрывом имеет место
истинная сополимеризация
полимеров.
170
•Л
***
mQ 0,2 ОЛ 0,6
Степень превращения, %
Рис. 51. Зависимость температуры
плавления сополимера триоксана
с диоксоланом от степени
превращения при полимеризации в
растворе бензола с катализатором
BF3Et20;
• — при 50° С; о — при 60° С;
X — при 70° С.
100
150 200
Время, мин
300
Рис. 52. Кинетические кривые расхода
диоксолана и триоксана при сополимери-
зации в растворе бензола с
катализатором BF3Et20 при различных
температурах:
1, Ґ — 50° С; 2, 2' — 60° С; 3, 3' — 70° С.
— кинетика расхода триоксана;
кинетика расхода диоксолана.
Параллельно изучалась кинетика сополимеризации триоксана
с полидиоксоланом [79]. В этом процессе ракция передачи цепи
с разрывом должна протекать наиболее эффективно.
Полидиоксолан растворяли в триоксане или растворе триоксана
в бензоле, затем вводили катализатор. Наблюдались большие
индукционные периоды, кроме этого, в отличие от сополимеризации
мономеров, требовался избыток катализатора.
Зависимость молекулярного веса образующегося полимера от
степени превращения, характерная для полимеризации триоксана,
наблюдалась и при сополимеризации. В последнем случае
предельные значения молекулярного веса были ниже, чем при взаимодействии
сомономеров. Это объясняется увеличением общего числа молекул
в реакционной системе.
Значение константы скорости передачи цепи с разрывом на
полидиоксолан при сополимеризации его с триоксаном определяли [79]
при 50° С. Было получено выражение для зависимости термической
стабильности сополимера (измеряемой по потере массы в вакууме
157
Сополимеризация триоксана с гетероциклическими соединениями
ТАБЛИЦА 24
Сомономеры
Окиси алкиленов
О ксацик лобутаны
Тетрагидрофуран
1,3-Диоксолан (и его
производные), где R —
это Н, СН3 или
другая группа
Структурная формула
Н2С—CHR
\ /
О
R R'
V
Н2С СН2
О
н2с—сн2
1 1
1 1
Н2С СН2
\ /
О
RHC—СН2
сн2
Катализаторы
полимеризации
Кислоты
Льюиса и их
комплексы
То же
»
»
Условия
проведения процесса
70° С; в
расплаве
В расплаве или
растворе
То же
В расплаве,
растворе или
твердой фазе
Примечание
Содержание
эпоксидных групп определялось
И КС-методом. С
окисями стирола и изобути-
лена реакция не шла
Процесс можно вести
при повышенном
давлении, а также при
пониженной температуре
Литература
50
50
50
81, 82
Циклические форма-
ли A,3-диоксан, 1,3-ди-
оксепан и т. д.)
?-Пропиолактон и
другие лактоны
Алкиленкарбонаты
R
О
CH2
R — углеводородный
радикал, содержащий
более 2 атомов углерода
CH2-(CH2)W—С=О
I о 1
11 = 1,2,3
О
/\
R С=О
v
R — ал кил,
содержащий от 2 до 20 атомов
углерода
Координационные комплексы
BF3
В расплаве
триоксана или
в твердой фазе
В расплаве или
растворе
0-100° С
Возможно получение
сополимера любого
состава
Из других лакто-
нов названы у-бутиро-
лактон, є-капролактон
При избытке
карбоната образуется
чередующийся сополимер
(карбонат неспособен к го-
мополимеризации).
Продукт омыляется
щелочью
9, 81
9, 60, 83
76
при 200 С) от степени «дробления» блоков полидиоксолана, которая
в свою очередь является функцией константы скорости передачи
цепи с разрывом [78].
Поскольку передача цепи с разрывом полиоксиметиленовой цепи
протекает и в твердой фазе (это было доказано экспериментально)
[5], то можно предполагать, что при достаточно большой
продолжительности реакции будет достигаться наиболее вероятное
распределение блоков сомономера в полимерной цепи.
Механизм сополимеризации триоксана с другими
гетероциклическими соединениями в настоящее время не изучен. В табл. 24
приводятся некоторые соединения, о возможности сополимеризации
которых с триоксаном сообщалось в литературе.
Сополимеризация с винильными мономерами
При катионной сополимеризации триоксана или формальдегида
с соединениями, содержащими винильные связи, передача цепи
с разрывом будет оказывать влияние на молекулярно-весовое
распределение и распределение звеньев по длине цепи. В этом случае
в качестве реакционноспособных групп будут выступать оксимети-
леновые блоки.
Пока мы располагаем только литературными данными об
условиях синтеза сополимеров и типе мономеров, вступающих в реакцию
(табл. 25). Кинетика этих реакций и состав сополимеров не
изучались. Исключением является работа Керна с сотр. [6] о механизме
сополимеризации триоксана со стиролом, где обсуждается вопрос
о характере присоединения атомов стирола к полимерной цепи.
Подобная задача в свое время рассматривалась для случая
радикальной и анионной сополимеризации стирола и было показано,
что присоединение идет в основном по ?-углеродному атому [58].
Причина заключается, по-видимому, в стерических затруднениях
при атаке а-углеродного атома.
При взаимодействии триоксана и стирола имеются две
возможности:
присоединение по ?-углеродному атому
. • .-О-СН2-О-СН2—СН2—СН-О-СНа-- • •
I
присоединение по сс-углеродному атому
. . .-О-СНз-О-СНз-СН
с6н5
Различие в строении цепи можно обнаружить путем ацидолиза
сополимера и анализа образующихся продуктов. Этим методом
пользуются для анализа состава сополимеров формальдегида и триоксана
с мономерами, содержащими устойчивые к ацидолизу связи, хотя,
как мы увидим в дальнейшем, интерпретация полученных результа-
460
ТАБЛИЦА 25
Сополимеры триоксана с соединениями, содержащими винильные связи
Сомономеры
Стирол (и
замещенные стиро-
лы)
Акрилонитрил
Виниловые эфиры
Изобутилен
Винилпиридин
Винилкарбазол
Инден
Структурная формула
GH=GHR
1
\/
СН2=СН
GN
GH2-CH—OR
GH2=G—GH3
CH3
CH=CH2
(\
V
H
Со
Условия проведения
процесса
От —80 до +100° С; в
растворе;
катализатор — BF3 или SnCl4
От —100 до +150° С; в
блоке, растворе или
расплаве;
катализатор — кислоты Льюиса
0—75° G; катализатор —
SnBr4
От —40 до —80° С; ка-
тализатор^— BFg
От —100 до +100° С;
катализатор —
кислоты Льюиса
То же
50—90° G;
катализатор — кислоты
Льюиса
Литература
70
67
68
69
69
69
80
тов часто затруднена из-за побочных реакций. В первом случае при
полном ацидолизе в присутствии воды должен образоваться 1-фенил-
пропандиол-1,3:
НОСН-СНа-СНзОН
Во втором случае должен соответственно образоваться 2-фенилпро-
пандиол-1,3:
НОСН2-СН-СН2ОН
I
с6н5
Экспериментальное определение строения образующихся после
гидролиза веществ оказалось более сложным, чем можно было
11 Заказ ІІ35.
161
предполагать. Прежде всего в присутствии формальдегида оба диола
превращались в изомеры 1,3-диоксана
С6Н5 Н
СН2 С
/\ /\
с6н5-нс сн2 н2с сн2
II II
0 0 0 0
\ / \ /
сн2 сн2
4-фенил-1,3-диоксан 5-фенил-1,3-диоксан
которые могли вступать в другие побочные реакции (конденсация,
сополимеризация). Все же авторам путем ацидолиза сополимера,
содержащего небольшие количества стирола, удалось получить
фенилдиоксан с достаточно высоким выходом. Продукт был выделен
путем хроматографического разделения и проанализирован ИКС-
методом.
Сопоставление полученных спектрограмм с контрольными
спектрами изомеров фенилдиоксана позволило сделать вывод о том, что
катион атакует ?-углеродный атом стирола с переносом заряда на
а-углеродный атом:
-СНа-О—СН2-О-СН2
• • .—СН2-О—СН2-О-
-f СН2=СН->
і
с6н5
—СН2—СН2—СН
с6н5
Авторы показали, что изменение диэлектрической проницаемости
среды не приводит к изменению механизма присоединения.
РАДИАЦИОННАЯ И РАДИКАЛЬНАЯ
СОПОЛИМЕРИЗАЦИЯ
Радиационная сополимеризация формальдегида. При изучении
радиационной сополимеризации формальдегида с винильными
мономерами показано [61—63], что скорость процесса уменьшается в
следующем ряду:
а-метилстирол > стирол >> метилметакрилат >
> акрилонитрил ?> окись этилена
Исследовалась [63] сополимеризация формальдегида со стиролом
при —78° С под действием дозы 10 Мрад. Контрольные опыты
показали, что при гомополймеризации формальдегида в таких же усло-
162
виях выход полимера составляет 100%, а конверсия стирола —
только 1,4%, что совпадает с литературными данными для обоих
мономеров.
Выход сополимера пропорционален содержанию формальдегида
в исходной смеси. Методом ИК-спектроскопии было показано, что
сополимер имеет всегда постоянный весовой состав: 90%
формальдегида и 10% стирола. С помощью дифференциального термического
анализа и термогравиметрии было показано, что образуется
статистический, а не привитой сополимер, как следовало бы ожидать, исходя
из того, что скорость конверсии формальдегида намного больше, чем
соответствующая величина для стирола.
Описана сополимеризация триоксана в твердой фазе с
гетероциклическими [64, 65] и винильными мономерами [63] под действием
радиации.
Радикальная сополимеризация. В литературе приводятся данные
о модификации полиоксиметилена методами механохимии. Так,
описано [66] получение привитых блоксополимеров с
разнообразными термопластичными полимерами (полиэтиленом, полистиролом,
поливинилхлоридом, поликарбонатом и т. д.). Процесс проводится
в обычном экструдере или каландре при давлении от 100 до 1500 атм
и при температуре 100—210° С в присутствии веществ, образующих
в этих условиях свободные радикалы (гидроперекись кумола,
перекись бензоила и т. д.).
В связи с этим сообщением заслуживают внимание опыты по
полимеризации формальдегида при повышенном давлении и высокой
температуре. В предыдущих главах мы уже рассматривали вопрос
о влиянии давления на предельную температуру полимеризации.
Хотя в патентной литературе часто оговаривается возможность
проведения процессов полимеризации или сополимеризации при
повышенном давлении, при этом обычно подразумевается, что такое
изменение реакционных условий не должно существенным образом
сказываться на полученных результатах.
Однако можно ожидать, что при давлении 100—1000 атм и
температурах порядка 200° С будут частично протекать реакции
радикального характера. Например, может оказаться возможным
присоединение молекул формальдегида за счет отрыва атомов водорода с
образованием С—С-связей:
. • . —СН2—О—СН2—О—СН2 —> . . .— СН2—О-СН2—О—СН.
. . ._сн2—О—СН2—О—СН. + СН2О-> .
_> . . . —СН2—О~СН2—О—СН2—СН и т. д. *
II
О
* Сополимеризация формальдегида с окисью углерода рассмотрена в
работе [10].
И* . 163
ЛИТЕРАТУРА
1. S і m h a R., В r a n s о n H., J. Chem. Phys., 12, 253 A944).
2. M а у о F. R.,Lewis F. M., J. Am. Chem. Soc, 66, 1594 A944).
3. N a t t a G., et al., Chim. e ind., 45, N 12, 1475 A963).
4. M a r к H., О g a t a N., J. Polymer Sei., A-l, N 11, 3439 A963).
5. П a x о M о в а Л. К., Ениколопян Н. С, ДАН СССР, 167 (№ 6),
1306 A966).
6. Kern W.,Horn L.,Cherdron H., Makromol. Chem., 52, 59 A962).
7. С a s t і 11 e Y. P., S t a n n e t t V., J. Polymer Sei., B-2, N 12, 1097
A964).
8. Walling С, J. Am. Chem. Soc., 71, 1930 A949).
9. J aacks V., Makromol. Chem. 101, 33 A967).
10. RagarriniM,, Modena M., GallinellaE., GevidalliG.,
J. Polymer Sei, A-2, N 12, 5203 A964).
11. A lfr ey Т., G ol df і ng er G., J. Chem. Phys., 12, 205 A944).
12. Алфрей Т., Борер К., Марк Г., Сополимеризация, Издатинлит,
1953.
13. S с h w a n Т. С, Р г і с е С. С, J. Polymer Sei., 40, 457 A959).
14. Б а г д а с а р ь я н X. С, Кинетика и катализ, 1, № 4, 503 A960).
15. О verberg er С, Tanner D., Р е а г с е Е., J. Am. Chem. Soc,
80, 4566 A958).
16. N a t t a G., J. Polymer Sei., 34, 41 A959).
17. E r r e d e Z. A., J. Am. Chem. Soc., 83, 959 A961).
18. T ob olsky A. V., J. Polymer Sei., 25, 220A957); 31, 126 A958).
19. AI fr ey Т., Tob olsky A. V., J. Polymer Sei., 38, 269 A959).
20. F о n t a n a C. M., Kidder G. A., J. Am. Chem. Soc., 70, 3745 A948).
21. Г а и T M a x e p A. P., M є д в є д є в С. С, ДАН СССР, 100, 275 A955),
22. Липатова Т. Е., Гантмахер А. Р., Медведев С. С,
ДАН СССР, 100, 925 A955).
23. Липатова Т. Е., Гантмахер А. Р., Медведев С. С,
ЖФХ, 30, 1752 (І956).
24. Goldfinger G.,Kane Т., J. Polymer Sei., 3, 462 A948).
25. Бемфорд К., Бард У., Дженкинс А., Оньон П.,-
Кинетика радикальной полимеризации, Издатинлит, 1961.
26. С т е п а н о в А. В., Щ у к о в а М., ЖРФХО, 58, 840 A926).
27. Charbonnages de France, фр. пат. 1302017, 1962 г.
28. Badishe Anilin, пат. ФРГ 1154274, 1964 г.
29. Montecatini, англ. пат. 947073, 1964 г.
30. Montecatini, англ. пат. 946832, 1964 г.
31. Du Pont, пат. США 2828287, 1958 г.
32. Courtauld Ltd., англ. пат. 902602, 1962 г.
33. Nat t a G., Mazzanti G., Pregaglia G., Binaghi H., Ma-
kromol. Chem., 44—46, 537 A961).
34. N a t t a G., Mazzanti G., Pregaglia G., Pozzi G., J.
Polymer Sei., 58, 1201 A962).
35. Deutsche Gold, пат. США 2985623, 1961 г.
36. Rechiney-Saint Gobain, англ. пат. 1000673, 1965 г.
37. Bayer F., англ. пат. 917016.
38. Du pont, пат. США, 3043803, 1962 г.
39. Szwars M., Makromol. Chem., 35, 132 A960).
40. Richards D.H., Szwars M., Trans. Farad. Soc, 55, 1644 A959).
41. Morton M., R e m b a u m А., В о s t і с k E. E., J. Polymer Sei.,
32, 530 A958).
42. N о г о К., Kavazura H., Morigama Т., Joshioka S.,
Makromol. Chem., 83, 35 A965).
43. К u w a t а Т., Bull. Chem. Soc. Japan, 33, 1091 A960).
44. A m і f є і t В., Z і 1 k h a A., J. Appl. Polymer Sei., 7, 287 A963).
45. E n і k о 1 о р і a n N. S., J. Polymer Sei., 58, 1301 A962).
46. К e r n W., J а а с k s V., В a a d e r N.. Makromol. Chem., 83, 56 A965).
1 4
Б є р л и н Ал. Ал., Иванов В. В., Е н и к о л о п я н Н. С,
Высокомол. соед., 1967 (в печати).
Иванов В. В., Шагинян А. А., Волков В.. П., Ениколо-
пян Н. С, Высокомол. соед., 7, 1833 A965).
Иванов В. В., Ш а г и н я н А. А., Е н и к о л о п я н Н& С, ДАН
СССР, 161, 154 (.1965).
Weissermel К., Fischer E., Gutweiler К.,
Hermann Н. О., Kunststoffe, 54, 410 A964).
Плечова О. А., Иванов В. В., Ениколопян Hg С, ДАН
СССР, 1967, в печати.
Kuc era М., R і gier J., Polymer, 5, 371 A964),
Кис era M., Starcuk Z., Cupak M., Hladky E., Доклад
на симпозиуме по макромолекулярной химии, Прага, 1965 г.
Iwatsuki S., Takigawa N., Okada M., Yamashita J.,
Ishii J., J. Polymer Sei., B-2, 549 A964).
Okada M., Takikawa N., Iwatsuki S., Yamashita J.,
Ishii J., Makromol. Chem., 82, 16 A965).
PI es с h P. H., Westermann P. H., Сообщение на симпозиуме
по макромолекулярной химии, Прага, 1965 г.
Okada М., Y a m ash і t a J., I s hi і J., Makromol. Chem., 80, 196
A964).
Kern W., Chem. Ztg., 88, 623 A964).
Hoechst F., фр. пат. 1343816, 1962 г.
Kern W., Jaacks V., Makromol. Chem., 83, 71 A965).
Okamura S., Nakashio S., Hayashi K., Sakurada I.,
Isotopes and Radiation, 3, 242 A959).
Hayashi K.,Yamaoka H.,Fujiwara K.,Sakamoto M.,
Mani S., Natori Т., Yioshida H., Okamura S.,
Industrial Uses of Large Radiation Sorces, IAEA, Vienna, 1963, p. 219.
С a s t і 1 1 e J. P., S t a n n e t V., J. Rolymer Sei., B-2, 1097 A964).
Skanksa Attikfabrike А. В., англ. пат. 991538, 1965 г.
Grace U. P. and Co, англ. пат. 984322, 1965 г.
X. ReMMer Н. М. В. X., англ. пат. 993434, 1965 г.
Hoechst F., пат. ФРГ 1145793, 1963 г.
Celanese Corp., пат. США 3087913, 1963 г.
Du Pont, англ. пат. 911960, 1962 г.
Brit. Ind. Plast. Ltd, фр. пат. 320029.
Melville H. W., V a 1 e n t і n e L., Trans. Farad. Soc, 46, 210 A950).
Flor у P. J., Trans. Farad. Soc, 51, 48 A955).
Fl or у P. J., J. Chem. Phys., 17, 223 A949).
J n о v e M., J. Appl. Polymer Sei., 8, 2225 A964)..
Pepper D. C, Quart. Rev., 8, 88 A954).
Пат. США 3012990, 1961 г.
В er ar din ell і F. M., Dolce I. J., Walling Sis J. Appl.
Polymer. Sei., 9, 1419 A965).
Jaacks V., Makromol. Chem., 84, 250 A965).
Иржак В. И., Ениколопян Н. С., Высокомол. соед., 8, 1486
A966).
Фр. пат. 1346554, 1962 г.
Пат. США 3027352, 1962 г.
Пат. США 3072609, 1963 г.
Пат. США 3026299, 1962 г.
Розенберг Б. А., Иржак В. И., Ениколопян Н., С,
Успехи химии, 35, 714 A966).
Ениколопян Н. С, в сб. «Химическая кинетика и цепные реакции»,
Изд. «Мир», 1966, стр. 431.
Канад. пат. 706948, 1965 г.
165
ГЛАВА VI
СТРУКТУРА ПОЛИФОРМАЛЬДЕГИДА
высокомолекулярный полиформальдегид занимает особое место
¦-^ среди всех изученных до сих пор синтетических полимеров.
Как известно, именно на примере полиформальдегида Штаудингер
доказал, что высокомолекулярные соединения построены из
линейных макромолекул, которые, в некоторых случаях, могут
упаковываться в кристаллическую решетку. Полиформальдегид был
также первым высокомолекулярным веществом, для изучения
структуры которого применили метод дифракции рентгеновских
лучей.
В конце 20-х годов Штаудингер с сотр. выполнил ряд
основополагающих работ в области исследования структуры полиоксиметиленов
[1—4]. В этих работах был заложен тот фундамент, на котором
базируются все современные представления о структуре
высокомолекулярных веществ.
Основные положения, высказанные авторами этих исследований,
сводятся к следующему:
1) высокомолекулярные соединения могут образовывать
кристаллическую решетку, причем параметры элементарной ячейки
кристалла не зависят от длины макромолекул;
2) полидисперсность по длинам макромолекул не является
препятствием для образования кристаллической решетки;
3) способность высокомолекулярных веществ к кристаллизации
определяется регулярностью строения их молекул.
В дальнейшем структура полиоксиметилена изучалась
различными авторами, использовавшими для этой цели ряд методов:
дифракцию рентгеновских лучей [5, 6], колебательную спектроскопию
[7—10], измерение дипольных моментов [И] и ядерный магнитный
резонанс [12—14, 48].
До 1961 г. была известна только одна кристаллическая
модификация полиоксиметилена (ПОМ), имеющая элементарную ячейку
гексагонального типа (гексагональный ПОМ), и поэтому подавляющее
большинство работ посвящено изучению именно этой формы
полимера. Однако в 1961 г. Беззи удалось получить новую
кристаллическую модификацию полиоксиметилена [15], имеющего ячейку орто-
ромбического типа (орторомбический ПОМ).
Структура этого полимера сейчас уже подробно описана в
литературе [16].
166
В настоящей главе рассмотрены структуры обоих
кристаллических модификаций ПОМ, однако во всех случаях, где это не
оговорено особо, под термином ПОМ подразумевается гексагональная
кристаллическая модификация полимера.
КРИСТАЛЛИЧЕСКАЯ СТРУКТУРА
ПОЛИОКСИМЕТИЛЕНА
Гексагональный ПОМ. Уже в самых ранних работах по
исследованию кристаллической структуры ПОМ [1] было показано, что
этот полимер имеет элементарную кристаллическую ячейку
гексагонального типа с постоянными решетки а = 4,46 А и с = 17,3 А.
Рис. 53. Проекция кристаллической решетки
гексагонального ПОМ на плоскость ab.
Эта структура относится к пространственной группе РЗг (cj$) или
Р32 (сі). Все многочисленные дальнейшие исследования этого
полимера подтвердили правильность полученных результатов. В то же
время более детальные исследования структуры ПОМ, выполненные
Хенгстенбергом [17, 18] и Саутером [19, 20], показали, что каждая
элементарная ячейка содержит одну макромолекулу, имеющую
форму спирали, с периодом идентичности /, который соответствует
девяти химически связанным мономерным звеньям формальдегида
(—СН2О—). Саутер предложил также молекулярную модель
кристаллического ПОМ — спираль 9/4, где девять химически связанных
мономерных звеньев делают четыре витка спирали в периоде
идентичности кристалла полимера. На рис. 53 показана проекция решетки
гексагонального полиоксиметилена на плоскость ab,
перпендикулярную осям макромолекул, а на рис. 54 — рентгенограмма этого
полимера.
Хаггинсом [21] для макромолекулы ПОМ в кристаллическом
состоянии была предложена другая молекулярная модель — 9/5.
Он предположил, что скелетные углы в цепочке ПОМ не сильно
отличаются от тетраэдрических.
167
В табл. 26 приведены структурные параметры молекулярных
конфигураций ПОМ, предложенных Саутером и Хаггинсом.
Успехи, достигнутые за последние 10—15 лет в области изучения
строения цепных полимеров [23—27], позволили вернуться к
вопросу о строении ПОМ и провести значительно более детальные
исследования его кристаллической структуры. Были получены
решающие доказательства в пользу молекулярной модели 9/5,
предложенной Хаггинсом. Как показывает теоретический анализ, веским
доказательством существования спирали типа 9/5 служит текстуррентге-
нограмма ориентированного волокна ПОМ, полученная на трубке
с медным катодом [5]. Кроме того, результаты расчетов [5, 6]
структурных амплитуд рассеяния (Fhkl) для
всех наблюдаемых на рентгенограмме
слоевых линий окончательно
разрешают вопрос о конфигурации цепи ПОМ
в пользу спирали 9/5. На рис. 55
показана модель скелета цепочки (проекция
на плоскость ab) гексагонального ПОМ
А
50° U5° 4/7° 35° 30° 25° 20° 15°
29
Рис. 54. Рентгенограмма гексагонального ПОМ (GuKa
излучение).
ТАБЛИЦА 26
Структурные параметры кристаллического ПОМ
Структурные параметры
Число витков спирали
в периоде
идентичности
Скелетные углы С С
/с\
и О О
Угол внутреннего
вращения
Расстояние атомов
скелета от оси спирали
Рас стояние между
соседними атомами
скелета, измеренное по
оси цепи
Длина связи С—О
Модель 9/5
5
110° 53'
77° 23'
0,691А
0,961A
1,43A
Модель 9/4
4
123° 9'
58° 50'
0,824A
0,961A
1,43A
168
(спираль 9/5). Цифры около кислородных и углеродных атомов
соответствуют координатам атомов С и О в долях периода идентичности.
Расстояние между любыми двумя соседними атомами С и О по оси
молекулы равно V18 периода идентичности /.
Объем элементарной ячейки ПОМ равен 344,3А, теоретически
рассчитанная плотность р = 1,505 г/см?, а плотность, измеренная
экспериментально, составляет 1,43 г/см3 [28—30].
При дальнейших исследованиях было обнаружено, что
полученные рентгенограммы не всегда достаточно хорошо совпадают друг
с другом. Это натолкнуло на мысль о возможной неточности ранее
предложенной модели. В результате тщательных рентгеновских
измерений с помощью спектрогонио-
метра и трубок, оборудованных
железным, медным и хромовым
антикатодами, были получены
величины углов 26 для всех
экспериментально наблюдаемых
рефлексов. Постоянная решетка а для
рефлексов 100 и 110 равна: 4,467 А
(CrKJ, 4,470 A (FeKJ и 4,476 А
(CuKJ. В дальнейшем в
расчетах [6] использовали величину а —
= 4,47 А в отличие от величины
а = 4,46 А, принятой ранее. Веди-
чина периода идентичности / вдоль
оси молекулы была рассчитана для
групп рефлексов, относящихся
к одной и той же слоевой линии,
и эти расчеты показали, что девять
мономерных единиц —СН2О— не
дают точно повторяющегося
элемента структуры. На основании полученных результатов был сделан
вывод, что период идентичности вдоль оси цепи гексагонального
ПОМ составляет 56,0 А и цепь содержит на этом промежутке 29
мономерных единиц, которые делают 16витков (спираль 29/16, рис. 56).
Теоретически рассчитанная плотность такой структуры
составляет р = 1,492 г/см3, а угол внутреннего вращения ф = 76°21*
вместо 77°23\ принятых ранее. Расчет структурных амплитуд
рассеяния для всех слоевых линий спирали 29/16 дал результаты,
практически не отличающиеся от полученных ранее для спирали 9/5
(с учетом различия в индексах слоевых линий).
Орторомбический ПОМ. В 1961 г. была получена новая
модификация высокомолекулярного полиоксиметилена путем
полимеризации водного раствора формальдегида в метастабильном состоянии
[15] (см. гл. II). Первоначально было обнаружено только, что
степень кристалличности этого полимера приближается к 100%, т. е.
выше, чем у высокомолекулярного гексагонального ПОМ, получаемого
169
Рис. 55. Проекция скелета
спирали 9/5 гексагонального ПОМ на
плоскость ab:
ф —атом углерода, О —- атом кислорода.
полимеризацией газообразного или жидкого формальдегида. Однако
сам по себе этот факт нельзя было считать аномальным, так
как хорошо известно, что низкомолекулярные полимеры
формальдегида, получаемые из водного раствора — параформальдегид,
а-полиоксиметилен, обладают очень высокой степенью
кристалличности. Высокомолекулярный ПОМ со степенью кристалличности,
близкой к 100%, можно получить путем радиационной
полимеризации твердого триоксана [30].
Рис. 56. Проекция скелета спирали 29/16
гексагонального ПОМ на плоскость ab:
ф — атом углерода, о — атом кислорода. (Цифры
внутри рисунка — координаты атома углерода,
внешние — координаты атома кислорода в долях периода
идентичности.)
Каразоло и Мамми подробно исследовали структуру новой
модификации ПОМ [16, 31]. На рис. 57 приведена ее рентгенограмма,
а на рис. 58 — инфракрасные спектры обычной и новой форм ПОМ.
В рентгенограмме орторомбического ПОМ четырем наиболее
интенсивным рефлексам соответствуют межплоскостные расстояния:
4,04 А; 3,83 А; 2,67 А и 2,60 А. Анализ экспериментальных данных
приводит к заключению, что кристаллическая ячейка новой формы
ПОМ имеет параметры: а — 4,77 А, Ъ = 7,65 А и с = 3,56 А. Объем
ячейки составляет |129,9 А теоретическая плотность равна
1,54 г/см3 (при условии, что элементарная ячейка содержит 4
мономерных единицы), экспериментально найденная плотность —
1,52 г/см3. Структура относится к пространственной группе i^^^.
К сожалению, орторомбический полиоксиметилен не может быть
изучен в виде ориентированной пленки или волокна, так как при
температурах выше 60° С эта структура спонтанно переходит в обычную
170
гексагональную форму. Другими словами, орторомбическая форма
является метастабильной и поэтому может быть исследована только
в том виде, в каком она
кристаллизуется в ходе полимеризации (порошок).
Учитывая некоторую
неопределенность, обусловленную исследованием
спектров порошкообразных образцов,
следует быть очень осторожным в
последующих выводах.
#7° Ь5° W° 35° 30° 25° 20° 15°
2В
Рис. 57. Рентгенограмма орторомбического ПОМ
(GuKa излучение).
Принимая во внимание большое сходство спектров
орторомбического и гексагонального ПОМ (рис. 54 и 57), Каразоло и Мамми
сделали предположение о том, что оси полимерных цепей парад-
5000 3000 2500 2000 1600 1U00 1200
юо
ЮОО 900
т-
800 см~
5000* 30002500 2000 1600 1400 1200
Рис. 58. ИК-спектры кристаллического ПОМ:
о — кристаллический гексагональный ПОМ; б — кристаллический
орторомбический ПОМ.
лельны, а решетка орторомбической формы ПОМ представляет собой
слегка искаженную решетку гексагональной формы. На рис. 59
показана проекция на плоскость ab кристаллической решетки
171
орторомбического ПОМ. Полимерные цепи в такой проекции
располагаются по четырем углам и в центре элементарной ячейки.
Следует заметить, что в гексагональной решетке ПОМ можно
выделить орторомбическую элементарную ячейку с параметрами
Рис. 59. Проекция кристаллической решетки
орторомбического ПОМ на плоскость ab.
а" = а = 4,56 А; Ь" - 2а cos 30° = 7,72 А и с' = с = 17,3 А, как
это показано на рис. 60 (ячейка выделена пунктирной линией).
Из сравнения рис. 59 и рис. 60 хорошо видно сходство решеток
гексагонального и орторомбического ПОМ.
Далее авторы предположили,
что цепь орторомбического ПОМ
содержит две мономерные единицы,
которые делают один оборот
спирали в периоде идентичности
(спираль 2/1).
Если принять, что проекция
связи С—О на ось цепи равна 7/4—
= 0,89 А, а длина связи 1,43 А,
то при равенстве скелетных углов
О С
/ \ и у/ \ их значение оказы-
G С О О
вается равным 112° 41', и проекция
цепи на плоскость,
перпендикулярную ее оси, имеет оформу
квадрата со стороной 1,12 А (рис. 60).
Дальнейшая проверка правильности предложенной структуры
определялась расчетом интенсивности (і?расЧш) * и структурных
амплитуд рассеяния (Fhkl) и сравнением их с экспериментальными
значениями.
Рис. 60. Проекция кристаллической
решетки гексагонального ПОМ на
плоскость ab.
* При расчетах интенсивностеи температурный фактор считался
изотропным и равным 2,3.
172
Полученные величины интенсивностей и амплитуд рассеяния
хорошо совпадают с экспериментальными:
„ 2 [\FhM (эксп.)| — \Ffikl (расч.)П
\Fhkl
(расч.)І
= 0,16
На рис. 61 схематически показана структура кристалла ортором-
бического ПОМ. Для достижения минимального расстояния между
атомами кислорода и углерода соседних цепей необходимо сдвинуть
цепи в элементарной ячейке друг относительно друга вдоль оси z
на величину 112. Таким образом, в кристалле орторомбического ПОМ
Рис. 61. Проекции кристаллической решетки
орторомбического ПОМ на плоскости ab, be и ас.
конфигурация каждой цепи автоматически фиксируется четырьмя
соседними цепями элементарной ячейки.
Одним из любопытных свойств структуры ПОМ (как
гексагональной, так и орторомбической) является то, что его кристаллическая
решетка построена только из изоморфных цепей (право- или левовин-
товых спиралей). Такое явление для полимеров впервые было описано
Натта [32] и очень отчетливо проявляется в случае ПОМ.
КОНФОРМАЦИЯ И НЕКОТОРЫЕ СВОЙСТВА
ИЗОЛИРОВАННОЙ ПОЛИОКСИМЕТИЛЕНОВОЙ ЦЕПИ
Все физические свойства кристаллического полиоксиметилена,
как и любого другого кристаллического полимера, определяются,
с одной стороны, структурой и свойствами самой макромолекулы,
а с другой — величиной и характером межмолекулярных сил в
173
кристалле, построенном из этих макромолекул. Чтобы связать
свойства полимерного вещества с его структурой, необходимо
выяснить особенности строения его макромолекул.
Как было показано в предыдущем разделе, макромолекула ПОМ
в кристаллическом состоянии имеет форму спирали 9/5 (либо 2/1),
т. е. любая химическая связь скелета находится в гош-положении
[33] по отношению к предыдущей паре химических связей. Таким
образом, конформацию макромолекулы ПОМ в кристаллическом
состоянии можно записать в виде [GG]n*.
Впервые вопрос об устойчивости спиральной конформации в
изолированной молекуле ПОМ был рассмотрен в работе [34]. Задача
бьща поставлена следующим образом: учитывая взаимодействие всех
валентно несвязанных ато-
10 мов цепи ПОМ, следовало
найти минимум
потенциальной энергии такой
полимерной цепи как
функцию углов вращения
вокруг связей скелета. Общую
потенциальную энергию
цепи можно записать
о
г
1
\
\
/
/
л
\
si
40° 80° 120і
160° 200і
240° 280° 320° 360°
в виде суммы
Рис. 62. Потенциальная энергия
изолированной макромолекулы как функция угла
внутреннего вращения ф.
... и т. д.
—¦ энергии
(
где Ел, Ер т
взаимодействия
(отнесенные к мономерной
единице) между заместителями, связанными с первым, вторым, третьим
и т. д. атомами скелета бесконечной цепи. Авторами
учитывались члены взаимодействия до пятого включительно, а остальные
полагались равными нулю. В качестве величин, описывающих
взаимодействие валентно несвязанных атомов типа Н Н,
С С, О- Н и т. д. как функции расстояния, выбирались
полуэмпирические потенциалы, предложенные Мезоном и Кривом
[35], Бартелом [36] и Хиршфельдером и Линнетом [37].
На рис. 62 показана кривая изменения потенциальной энергии
цепи ПОМ в зависимости от величины угла внутреннего вращения**.
Расчет приводит к выводу, что самый глубокий минимум конфор-
мационной энергии цепи достигается при значении ф приблизительна
* Каждая мономерная единица скелета ПОМ состоит из двух атомов и
поэтому конформация характеризуется двумя углами внутреннего вращения ф2
и ф2.
** Скелет цепи ПОМ состоит из атомов двух видов и, строго говоря,
потенциальная энергия является функцией двух углов фх и ф2. Однако вследствие
равенства скелетных углов эту цепочку можно рассматривать как одноатомную
и тогда Е есть функция только одного угла ф. Были проведены расчеты Е для
двухатомной цепи. Энергетическая диаграмма Е — / (фх, ф2) имеет самые
глубокие минимумы вдоль диагонали фх = ф2, что согласуется с данными,
полуденными для одного угла ф.
174
dz 67°, т. е. гош-конформация является энергетически самой выгодной
для отдельной изолированной цепи ПОМ, в отличие от цепочек
полиэтилена и политетрафторэтилена, где минимум потенциальной
энергии достигается при игр^шс-конформациях химических связей.
Некоторое расхождение вычисленной величины угла внутреннего
вращения со значением, полученным экспериментально, может быть
обусловлено как приближенным характером расчетов, связанным
с тем, что имеется недостаточно данных о потенциалах
взаимодействия атомов, так и некоторым влиянием кристаллической решетки
полимера на равновесное значение угла ф. Однако тот факт, что
цепочка ПОМ в изолированном состоянии сворачивается в спираль,
является принципиальным и не вызывает сомнений.
Вопрос о конформации изолированной цепи ПОМ с несколько
других позиций рассмотрен Флори и Марком [38]. Они рассчитали
расстояние между различными парами валентно несвязанных атомов
в цепи ПОМ в различных конформациях (трапе- и гош-) и сравнили
их с таковыми в полиэтиленовой и полидиметилсилоксановой цепях.
В табл. 27 приведены эти величины для всех трех типов полимерных
цепей.
ТАБЛИЦА 27
Расстояние между валентно несвязанными атомами в молекулах
кристаллических полимеров
Полимер
Полиоксиметилен
Полиэтилен
Полидиметилси-
локсан
Атомы или
группы атомов
СНа—О
СН2—СН2
0--0
GH2—СН2
Si—Si
О--О
Число связей
скелета
между
группами
3
( 3
1 ^
4
3
3
4
4
.4
4
Конформация
Т
G±
G^G^
G^G^
Т
G±
G±G±
G^G1^
G^G1^
G±G±
Расстояние
о
A
3,60
2,75 B,96)
3,32 C,91)
2,37 B,89)
3,89
3,01
3,65
2,69
4,62
3,69
Некоторое уменьшение расстояний между взаимодействующими
валентно несвязанными атомами и группами ПОМ по сравнению
с двумя другими полимерами не приводит к увеличению конформа-
ционной энергии цепи, так как связь С—О короче связи С—С A,43 А
и 1,54 А соответственно) и ван-дер-ваальсов радиус кислорода меньше
радиуса СН2-группы в полиэтилене. Авторы считают, что главный
выигрыш энергии гош-конформации по сравнению с ттгрбте-конформа-
цией обусловлен лондоновскими дисперсионными силами между
175
парой СН2 О (отрицательная энергия взаимодействия).
Некоторый вклад, хотя и очень незначительный, дает диполь-дипольное
взаимодействие связей С—О1+1 и С—О._г, Если дипольный момент
такой связи [39] принять равным 0,86 ?), то энергия взаимодействия
оказывается не больше 0,2 ккал/молъ.
Анализ результатов, приведенных в табл. 27, показывает, что
чередование состояний Gj-iGi* или Gj_± Gj практически невозможно
из-за сильного взаимодействия элементов цепи, разделенных
четырьмя химическими связями. Введя величины статистических весов
различных типов конформаций, можно вычислить отношение
nl/nl2 — среднеквадратичного расстояния между концами
гипотетической цепи к произведению числа связей п на квадрат длины
каждой связи. Как следует из теории поворотной изомерии линейных
полимеров [40, 41], эта величина очень чувствительна к значениям
углов внутреннего вращения. Во всех случаях были получены
величины этого отношения, превышающие таковые для цепи со
свободным внутренним вращением.
Таким образом, экспериментальные и теоретические данные
свидетельствуют об энергетическом преимуществе гогг^-конформации перед
всеми остальными конформациями изолированной цепочки ПОМ.
Из всего сказанного ясно, что при любых условиях существования
полимера большая часть его цепей будет находиться в гогг^-конформа-
ции, однако существование поворотных изомеров этой цепи отнюдь
не исключено. Так, например, для аморфного состояния полимера,
а также для расплава или раствора должна существовать по
меньшей мере еще одна устойчивая конформация цепи ПОМ, а именно
игргшс-конформация, при которой все атомы скелета макромолекулы
лежат в одной плоскости (конформация «плоский зигзаг»).
Экспериментальные доказательства существования транс-коп-
формации цепи ПОМ были получены недавно методами
колебательной спектроскопии [42, 43].
Было показано [9, 44], что инфракрасные спектры
гексагонального и орторомбического ПОМ обусловлены главным образом
взаимодействиями атомов и атомных групп внутри макромолекул, а эффекты
взаимодействия различных полимерных молекул в кристалле ПОМ
оказывают лишь слабое действие на собственные колебания цепей.
Однако замечено [10, 42], что некоторые полосы в ИК-спектрах
поглощения твердого ПОМ являются «лишними» с точки зрения
теории симметрии данной цепи **. Специальная обработка полимера
* Gf означает, что г-тая связь С—О занимает гош-положение по отношению
к предыдущей паре связей, а знаки + и — указывают на направление вращения
(вправо или влево соответственно).
** Одномерная пространственная группа цепи гексагонального ПОМ имеет
фактор-группу D A0я/9) и, в соответствии с правилами отбора, в ИК-спектре
должно наблюдаться 5 колебаний типа А 2 ( || ) и 11 дважды вырожденных
колебаний типа Ег(±_) [9]. Одномерная пространственная группа цепи
орторомбического ПОМ имеет фактор-группу D2 и активными в ИК-спектре полимера
должны быть [44] 15 колебаний типов симметрии Вг, В2 и В3.
176
I
[10, 43] для повышения содержания в нем аморфной фазы дала
возможность четко выделить такие «лишние» полосы.
На рис. 63 показаны детали ИК-спектров обычного ПОМ и ПОМ,
подвергнутого «лиофильной сушке» [10]. Во втором случае отчетливо
видны «лишние» (по сравнению с первым случаем) полосы
поглощения в области 1125 и 985 см'1. Расчеты частот и форм нормальных
колебаний для спиральной [45] и плоской цепи [46] подтвердили,
что указанные полосы обязаны своим происхождением плоской
зигзагообразной форме скелета цепи ПОМ.
Ценная информация по этому
вопросу была получена при
исследовании ИК-спектров модельных
соединений с ацетальным скелетом
типа СН3О—(СН2—О)п—СН3, где
п — 1—8. Такие соединения вплоть
до п = 7 при обычных температурах
являются жидкостями, что делает
их очень удобными объектами для |Г
изучения явления поворотной
изомерии.
В спектрах моделей [43]
наблюдаются полосы поглощения при тех
же частотах, что и «лишние» полосы
у твердого ПОМ *, которые полностью
исчезают при кристаллизации этих
веществ.
Как показано Штаудингером [1],
все олигомеры указанного типа
кристаллизуются в виде спиралей (что
также следует из анализа
колебательных спектров) и, следовательно,
дополнительные полосы обусловлены существованием (в растворе)
метастабильного состояния — а именно плоской зигзагообразной
формы. Эти данные подтверждаются теоретическими расчетами
колебательных спектров различных конформаций указанных
молекул [45]. Таким образом, получены надежные доказательства
наличия отличной от спирали конформаций цепи ПОМ,
существование которой теоретически было предсказано ранее [34].
Важнейшим свойством полимерной цепи, определяющим
наиболее характерные особенности поведения материала, является ее
гибкость. Поведение полимера при действии на него механических сил
обусловлено его кинетической гибкостью, в то же время температура
плавления связана с термодинамической гибкостью. Величины
кинетической и термодинамической гибкости полимера
характеризуются параметрами и0 и Аи — высотой потенциального барьера
800 900 1000 1100 1200 1300
Длина Волны, см'1
Рис. 63. ИК-спектры
высокомолекулярного ПОМ:
1 — подверженный «лиофильной сушке»;
2 — обычный полимер (таблетки К В г).
* Особенно отчетливо указанные явления наблюдаются [43] на модельных
соединениях с п = 2, 3 и 4.
12 заказ 113 5.
177
внутреннего вращения и разностью энергии между минимумами на
кривой зависимости потенциальной энергии молекулы от величины
угла внутреннего вращения.
Значение физических характеристик изолированной молекулы,
и в особенности величин и0 и Аи, дает возможность оценить физико-
механические свойства полимера и в ряде случаев на основании
данных о строении макромолекулы предсказать поведение материала
в различных условиях. Изучение свойств изолированной цепи ПОМ
имеет такое же значение для выяснения свойств полиацеталей, какое
имело изучение цепи полиэтилена для понимания строения и свойств
всего класса карбоцепных полимеров.
В настоящее время существует две противоположные точки
зрения относительно гибкости макромолекулы ПОМ:
1) цепь ПОМ отличается большой гибкостью, что подтверждается
исследованиями внутреннего вращения вокруг связей С — О
в малых молекулах * (вращение близко к свободному);
2) цепь ПОМ достаточно «жесткая» **, о чем свидетельствуют
имеющиеся сейчас экспериментальные данные об исследовании
физических свойств ПОМ и некоторых модельных цепных полиацеталей.
При изучении вопроса о связи термодинамической гибкости с
температурой плавления полимера [47] было отмечено, что если в цепи
парафина некоторые связи С—С заменить на связи С—О, то Тш
данного вещества резко снижается. Действительно, температуры
плавления сложных полиэфиров, как правило, лежат ниже
температур плавления соответствующих полиуглеводородов [47].
Однако как только мы переходим к сравнению цепей парафинов
с соответствующими полиацеталями, картина существенно меняется.
В табл. 28 приведены температуры плавления различных полимеров
с углеродным и ацетальным скелетом [48].
Если принять, что цепь ПОМ обладает большой гибкостью, то
высокая температура плавления должна быть обусловлена силами
межмолекулярного взаимодействия [1, 50]. Действительно, энергия
когезии на группу — О — составляет 1000 кал/моль, а на группу
—СН2—только 640 кал/моль [47], однако этим нельзя объяснить
существующей разницы температур плавления полиэтилена и ПОМ
(около 50° С).
При изучении дипольних моментов модельных полиацеталей
[И] типа СН3—О—(СН2—О)я—СН3 (где п = 1 и 2) было показано,
что наиболее устойчивой конформацией этих соединений в растворе
является гои?-конформация. Величина Аи (разность энергий между
* В настоящее время известно, что потенциальная энергия внутреннего
вращения зависит главным образом от взаимодействия валентно несвязанных
атомов [40], например от взаимодействия боковых групп большого объема.
В молекуле ПОМ отсутствуют объемные заместители и, кроме того, у атома
кислорода вообще нет никаких боковых групп, т. е. число связей у оси вращения
уменьшается от трех до одной, и если гибкость цепи ПОМ обусловлена этими
причинами, она должна быть весьма высокой.
** Во всяком случае характеризуется значениями щ и Аи большими, чем
для парафиновых углеводородов.
173
устойчивой гоіа-конформацией и менее устойчивой рфф
цией) составляет около 1,7 ккал/моль, что в три раза превышает
подобную величину для м-парафинов (приблизительно 0,6 ккал/моль).
Величина характеристического отношения Уь\/п1г (см. стр. 176)
также заметно превышает аналогичную величину для «-парафинов
[38].
По данным работы [49], величины характеристического
отношения составляют приблизительно 13 и 5,8 для температур 25 и 145° С
соответственно при
использовании модели цепи в гош- таблица 23
конформации. Эти величины Температуры плавления
свидетельствуют о жесткости некоторых полимеров
цепи ПОМ.
Еще Штаудингер [1]
обратил внимание на тот факт,
что величина ЪуЫ2 для
ПОМ (^3,4) * отличается от
таковой для других
кислородсодержащих полимеров
(например, полиэтиленгли-
коля) и несколько больше
той, которую следовало бы
ожидать для полимера со
свободным вращением
связей. Существенный интерес
представляет рассмотрение
данных, характеризующих
барьер внутреннего
вращения и0. Ввиду причин,
указанных выше, барьер
внутреннего вращения -вокруг
связи С—О в метиловом
спирте снижается втрое по
сравнению с и0 в этане A000 кал/моль и 3000 кал/моль
соответственно [40, 41].
К тем же результатам приводит теоретическое и
экспериментальное исследование диметилового эфира СН3—О—СН3[м0 (эксп.)
составляет около 2,7 ккал/моль [41] и и0 (теор.) с учетом стерического
взаимодействия CHg-групп равна приблизительно 2,5—3,0 ккал/моль].
Однако в случае молекул с ацетальным скелетом результат
неожиданно оказывается иным.
Тадокоро с сотр., как уже указывалось выше, провел
экспериментальное и теоретическое исследование колебательных спектров
ПОМ [9] и из силовой постоянной крутильных колебаний скелета
Мономерные
единицы
-СН2-Х—
-сн-х-
СНз
-СН-Д—
1
сн2
-сн-х-
(СН2J
СН3
Температура плавления, °С
X—о-
180
165*
185*
225*
х=—сн2—
137
165
125
75
* G частичным разложением.
* Эта величина получена из измерений вязкости низкомолекулярных
образцов ПОМ.
12* 179
вычислил величину и0 = 3600 кал/моль, что превышает величину
барьера внутреннего вращения в этане *.
В настоящее время еще нет решающих доказательств в пользу
«гибкой» или «жесткой» цепи ПОМ, однако их уже достаточно, чтобы
можно было усомниться в правильности обычно высказываемой
точки зрения о высокой гибкости цепи ПОМ. Если окажется, что
цепь ПОМ «жесткая», то причины, обуславливающие затрудненное
внутреннее вращение в полиацетальных структурах, имеют
принципиальный интерес.
СТРУКТУРА ПОЛИОКСИМЕТИЛЕНА, ПОЛУЧЕННОГО
В РАЗЛИЧНЫХ УСЛОВИЯХ ПОЛИМЕРИЗАЦИИ
Полимеризация триоксана с образованием ПОМ интенсивно
изучалась различными авторами [51], и полученные результаты
представляют существенный интерес. Однако целью настоящего раздела
является лишь краткое описание структурных особенностей ПОМ,
полученного в результате некоторых полимеризационных процессов,
проводимых в специфических условиях.
Особенный интерес представляет структура и некоторые свойства
ПОМ, полученного полимеризацией триоксана в кристаллическом
состоянии. При получении высокомолекулярного ПОМ
непосредственно в кристаллической решетке триоксана важную роль играет
исходная кристаллическая структура мономера. В зависимости от
совершенства исходного кристалла триоксана при полимеризации
можно получать различные структуры ПОМ — от монокристаллов
[52] с высокой степенью ориентации во всех направлениях до
волокон, ориентированных только в одном направлении, и
поликристаллов, не имеющих никакой преимущественной ориентации цепей
в пространстве.
Хаяши и Окамура [53] исследовали структуру ПОМ, полученного
радиационной полимеризацией кристаллического триоксана, и
показали, что в этом случае образуется ПОМ с гексагональной
кристаллической решеткой. Основной особенностью такого полимера
является анизотропия роста макромолекулы в различных
направлениях кристалла мономера. Оси макромолекулы образующегося
полимера ориентированы главным образом вдоль оси с кристалла
мономера, что хорошо видно на рентгенограммах.
Очевидно, что рост полимерной цепи в конденсированной фазе
регулируется структурой кристалла мономера. При полимеризации
в совершенных монокристаллах триоксана [52] были получены
монокристаллы гексагонального ПОМ, которые показывают четкую
пространственную периодичность в направлении всех
кристаллографических осей.
* При расчете авторами был принят потенциал вида
и(ф) = (уио) ^~~cos3(P)
180
Структура и прочность этих ориентированных игольчатых
кристаллов ПОМ исследовались в работе [54]. Авторы обнаружили, что
предел прочности таких образований при растяжении вдоль осей
молекул достигает 200—350 кГ/мм2, что весьма необычно для
полимерных веществ. Высокие значения прочности связаны с
особенностями строения игольчатых кристаллов ПОМ, а именно с
отсутствием складчатой структуры у ПОМ, полученного в указанных
выше условиях. Измерение дифракции рентгеновских лучей под
малыми углами подтверждает вывод об отсутствии складчатой
структуры в таких кристаллах.
В ряде случаев при полимеризации кристаллического триоксана,
инициированной у-излучением, можно заметить еще некоторые
особенности роста макромолекул ПОМ. Карозоло и др. [55]
исследовали структуру ПОМ, полученного таким
путем, и обнаружили ряд новых рефлексов
на рентгенограмме полимера. Тщательные
исследования показали, что во всех случаях
при полимеризации образуется ПОМ
гексагонального типа, но существует два типа
кристаллов, упорядоченных в пространстве
различным образом, т. е. существует «кри- Рис. 64. Ориентация
сталлический двойник» ПОМ. растущих цепей в двои-
Первый кристалл обычного типа (опи- нального ПОМ, получен-
санный ранее) образуется так, что оси макро- ного полимеризацией
молекул ПОМ совпадают с осью с кристалла триоксана в твердой
триоксана (рост происходит преимущественно Фазе*
в направлении оси с). Второй кристалл
растет в направлении W (рис. 64), образующем угол около 76° с
направлением роста первого кристалла. Все экспериментально
наблюдаемые рефлексы на рентгенограммах объясняются наложением
этих двух типов кристаллов. По данным измерений
интенсивности авторы оценили количество микрокристаллов первого и второго
типа — 54% и 46% соответственно.
Образование двойного кристалла ПОМ при твердофазной
полимеризации триоксана — это случай, который впервые встречается
в области синтетических полимеров.
Исследование механизма полимеризации различных мономеров
в твердом состоянии в настоящее время привлекает внимание многих
ученых, и можно надеяться, что изучение различных структур ПОМ,
полученных при твердофазной полимеризации триоксана, прольет
некоторый свет на многие, пока еще неясные аспекты этой
интересной проблемы физико-химии полимеров.
ЛИТЕРАТУРА
1. Штаудингер Г., Высокомолекулярные органические соединения,
ОНТИ, 1935.
2. Staudinger Н., Johner Н., Signer R., Mie G.,
Hengstenberg J.,Z. phys. Chem., 126, 425 A927).
181
3. Staudinger H., Luthy M., Helv. chim. Acta, 8, 41, 65 A925).
4. Staudinger H., Helv. chim. acta, 8, 67 A925).
5. Todokoro H., Yasumoto Т., Murahashi S., Nitta J.,
J. Polymer Sei., 44, 143, 266 A960).
6. Carazzolo G.,J. Polymer Sei., PA-1, № 55, 1573 A963).
7. Phylpotts A., Evans D., Sheppard M., Trans. Farad. Soc.,
51, 1051 A955).
8. Novak A., Whalley F., Trans. Farad. Soc., 55, 1485 A959),
9. Tadokoro H., Kobayashi M., Kavaguchi Y.,
Murahashi S., J. Chem. Phys., 38, 703 A963).
10. Олейник Э. Ф., Ениколопян Н. С., Высокомол соед. 8,1 583
A966).
11. U с h і d а Т., К и г і t a Y., К u b о М., J. Polymer Sei., 19, 365 A956)
12. У р м а н Я. Г., Слоним И. Я., К о н о в а л о в А. Г., Высокомол.
соед., 6, 1651 A964).
13. Р е t е г 1 і п А., О 1 f Н. G., J. Polymer Sei., Polymer letters, 8, 769 A964).
14. All ей J., Warren R., Chem. Ind. 15, 623 A964).
15. M 0 r t і 11 a r 0 L., Galiazzo G., Bezzi S., Ghem. Ind. (Ital.), 46,
№ 2, 139 A964),
16. G ar a z z ol 0 G., M a m m і M., J. Polymer Sei., PA-1, 965 A963).
17. H engs t enb erg J., Z. phys. Ghem., A-126, 435 A927).
18. Hengstenberg J., Ann. physik, 84, 245 A927).
19. S au t er E., Z. phys. Ghem., B-18, 417 A932).
20. S aut er E., Z. phys. Chem., B-21, 186 A933).
21. Huggins M. L., J. Ghem. Phys., 13, 37 A945).
22. Вайнштейн Б. К., Дифракция рентгеновских лучей на цепных
молекулах, Изд. АН СССР, 1932.
23. F г а п к 1 і n R. Б., К 1 u g A., Nature, 175, 379 A955); Biochim. Biophys.
Acta, 19, 403 A956).
24. Klug A., Crick F. H., Wyckoff A. W., Acta cryst., 11, 199
A958).
25. С о с h r a n W., Crick F. H. C, V a n d V., Acta cryst., 5, 581 A952).
26. Hosemann R., В age hi S. N., Acta cryst., 5, 612 A952).
27. Китайгородский А. И., Органическая кристаллохимия, Изд.
АН СССР 1955.
28. Burge R.'e., Randoll J. Т., Proc. Roy. Soc. (London), A-233,
1 A955).
29. G r 0 s s m a n W., The Chemical Structure of Proteins, Giba Found. Sim-
pos., 1953.
30. Okamura S., Hayashi K., Nishii M., J. Polymer Sei., 60,
169, 526 A962).
31. С a r a z z о 1 о G., P u t t і G., Chim. ind., 45, 7, 771 A963).
32. N a t t a G., Makromol. Chem., 35, 93 A960).
33. M у д з у с и м а С, Строение молекул и внутреннее вращение, Издат-
инлит, 1957.
34. De Santis P., Giglio E., Liguori M., Ripamouti A.,
J. Polymer Sei, A-1, 1383 A963).
35. M a s о n E., К r e e v о у M., A n і A., Chem. Soc, 77, 5808 A955).
36. В art ell L. S., J. Chem. Phys., 32, 827 A960).
37. H ir schf el d er J. О., L innett J. W., J. Chem. Phys., 18, 130
A950).
38. F 1 о г у P. J., M a r k E., Makromol. Chem., 75, 11 A964).
39. S m у t h C. P., J. Am. Chem. Soc., 60, 183 A938).
40. Волькенштейн М. В., Конфигурационная статистика
полимерных цепей, Изд. АН СССР, 1963.
41. Бирштейн Т. М., Птицы н О. Б., Конформации макромолекул,
Изд. АН СССР, 1965.
42. Олейник Э. Ф., Высокомол. соед., 6, 2103 A964).
43. Олейник Э. Ф., Ениколопян Н. С, Материалы симпозиума
по макромолекулярнои химии, Прага, 1965.
44. Zamboni V., Z e г b і G., J. Polymer Sei., C-7, 153 A964).
45. Олейник Э. Ф., Ениколопян Н. С, Оптика и спектроскопия,
1967 (в печати).
46. Олейник Э. Ф., Компанеец В. 3., Оптика и спектроскопия,
1967 (в печати).
47. Б а н н К., в сб. «Проблемы современной физики», № 12, Изд. АН СССР
1956.
48. Урман Я. Г., Слоним И. Я., Ермолаев А. Д., Высокодюл.
соед., 8 (№ 2), 251 A966).
49. S і lb ers zy с W., J. Polymer Sei., B-1, 577 A963).
50. L і n t о n W., G о о d m e n H., Di ssk, J. Appl. Polymer Sei., I, 178
A959).
51. Фурукава Дж., Саегуса Т., Полимеризация альдегидов и
окисей, Изд. «Мир», 1965.
52. J a m s on S.,Noether H., Polymer letter, 1, 51 A963).
53. Окамура С, Хаяши К., Химия и технология полимеров, Дъ 4
A965).
54. Слуцкер А. И., Громов А., Пшежецкий В., Физика
твердого тела, 6, 456 A965).
55. С arazz oll о G., Leghissa S., Mammi M., Makromol. Chem.
60, 171 A963).
ГЛАВА VII
ТЕХНОЛОГИЯ ПРОИЗВОДСТВА
ПОЛИФОРМАЛЬДЕГИДА ИЗ МОНОМЕРНОГО
ФОРМАЛЬДЕГИДА
Г"! роизводство полиформальдегида в промышленном масштабе было
' 'впервые осуществлено в 1960 г. Хотя в литературе отсутствуют
подробные данные об этом производстве, есть основания предполагать,
что оно было основано на полимеризации мономерного формальдегида
с последующей стабилизацией получаемого продукта *. Материал,
изложенный в предыдущих главах, позволяет нам выделить
основные вопросы технологии синтеза полиформальдегида. Сюда
следует отнести глубокую очистку мономерного формальдегида,
регулирование молекулярного веса в ходе полимеризации и
стабилизацию концевых полуацетальных групп в молекулах полимера.
В данной главе рассматриваются различные варианты решения
отдельных узлов технологического процесса синтеза
полиформальдегида на основе мономерного формальдегида. Большинство
излагаемых ниже вариантов синтеза, по-видимому, имеет право на
существование, а выбор между ними диктуется совокупностью
конкретных условий.
ВЫДЕЛЕНИЕ МОНОМЕРНОГО ФОРМАЛЬДЕГИДА
Первой стадией синтеза полиформальдегида является выделение
возможно более концентрированного мономерного формальдегида.
Несмотря на широкое применение формальдегида в промышленности
органического синтеза, он фактически никогда не используется
в чистом виде. В большинстве случаев по условиям реакции
допустимо применение водных растворов формальдегида, в ряде случаев
приходится использовать параформальдегид (90—95% СН2О) [1].
В связи с разработкой технологии синтеза полиформальдегида
были предложены новые способы выделения чистого мономера.
Основная трудность связана с химической неустойчивостью
формальдегида, которая проявляется в склонности к спонтанной
полимеризации и к различным реакциям превращения. Вследствие этого для
очистки формальдегида нельзя применять обычные в химической
* Фирма «Дюпон» (США).
184
технологии методы. Кроме того, высокая реакционная способность
формальдегида потребовала создания специальных устройств для
его транспортировки, хранения, перекачки, контроля температуры
.2
? Вакуум-
насосу
Формалин
Метанол,
разбавленный
раствор
формалина
и давления.
Для конструирования
аппаратуры нужно было выяснить
физико-химические
закономерности спонтанной
полимеризации формальдегида в различных
условиях. Сюда относится
изучение условий равновесия
между мономерным
формальдегидом и твердым полноксимети-
ленгликолем, влияния
различных факторов на скорость
реакции, состав образующегося
полимера и т. п.
Закономерности протекания этого
процесса оказались существенно
важными для изучения
вопросов пиролиза
низкомолекулярных полиоксиметиленов и
разработки способа очистки
мономерного формальдегида от
примесей.
Анализ влияния различных
факторов на спонтанную
полимеризацию мономерного
формальдегида позволил сделать
следующие выводы:
1. Спонтанная
полимеризация инициируется примесями
полярных веществ,
сопутствующих мономерному
формальдегиду: водой, метанолом,
муравьиной кислотой и т. д.
Уменьшение концентрации этих
примесей при прочих равных
условиях приводит к снижению
скорости спонтанной
полимеризации.
2. Температурная зависимость скорости спонтанной
полимеризации носит сложный характер из-за необычайной легкости
протекания обратной реакции (деполимеризации). Кажущаяся энергия
активации скорости реакции зависит от температуры так, что скорость
реакции приближается к нулю при температуре около 100° С
(предельная температура полимеризации) и имеет максимальное
значение при температуре около 40° С.
185
Разбавленный
раствор
формалина'
Рис. 65. [Принципиальная схема
получения мономерного формальдегида из
а-полиоксиметилена:
1 — ректификационная колонна для
укрепления и обезметаноливания формалина; 2 —
конденсатор; 3 — кипятильник; 4 — емкость
концентрированного обезметаноленного
формалина (обогреваемая); 5 — насос; б —
обогреваемые коммуникации для транспортировки
обезметаноленного формалина; 7 —
полимеризатор; 8 — мерник щелочи; 9 — мутильник;
10 — центрифуга; 11 — сборник оборотного
формалина; 12 — пневмотранспорт; 13 —
вакуум-сушилка; 14 — бункер; 15 — шнековый
питатель; 16 — центробежный деструктор.
3. Скорость спонтанной полимеризации жидкого формальдегида
(ниже —19° С) весьма велика даже при суммарном содержании
примесей в мономере менее 0,01%.
4. Предельная температура полимеризации зависит от
парциальной концентрации мономера следующим образом:
Из этого следует, что путем разбавления мономера можно
понизить предельную температуру полимеризации.
5. Инициирование газофазной полимеризации возможно только
на поверхности реакционного сосуда.
6. При спонтанной полимеризации в жидкой фазе существует
индукционный период, так как полимеризация начинается только
после образования насыщенного раствора формальдегида.
Надо отметить, что до настоящего времени не удалось подобрать
достаточно эффективных ингибиторов спонтанной полимеризации.
Для того чтобы избежать спонтанной полимеризации, стенки
трубопроводов, арматуру, аппараты и различные измерительные
устройства нагревают выше предельной температуры и мономер
пропускают с большой скоростью, что сокращает время контакта с
поверхностью. Учитывая низкий коэффициент теплопроводности
мономера, можно создать такие условия, чтобы он нагревался
незначительно.
Для получения мономерного формальдегида высокой степени
чистоты приходится использовать многостадийные схемы, поскольку
получение концентрированного мономера непосредственно при
синтезе формальдегида практически невозможно (см. гл. I). Первая
стадия заключается в поглощении мономерного формальдегида из
синтез-газа. В технике это обычно достигается путем растворения
формальдегида в воде (формалин). На следующей стадии из
формалина получают низкомолекулярные твердые полиоксиметилены (пара-
формальдегид, а-полиоксиметилен, триоксан). Кроме этого известны
способы извлечения формальдегида из реакционной смеси в виде
параформальдегида [2, 3]. Исследования, связанные с разработкой
производства полиформальдегида, привели к открытию еще двух
принципиально возможных способов выделения концентрированного
мономерного формальдегида путем образования гемиформаля
(реакцией со спиртами) и путем парциальной конденсации парогазовой
смеси формальдегида с последующим получением
концентрированного газообразного формальдегида.
Пиролиз низкомолекулярных
полиоксиметиленов
Практически весьма удобным оказалось получать
концентрированный мономерный формальдегид путем пиролиза
параформальдегида, а-полиоксиметилена или триоксана. Судя по имеющимся дан-
186
ным, на начальном этапе исследований по синтезу
полиформальдегида эти методы вызывали наибольший интерес специалистов,
занимающихся этой проблемой.
В пользу применения низкомолекулярных твердых полимеров
можно привести следующие доводы:
а) выход мономера составляет более 90% от массы исходного
вещества (ни один другой способ выделения мономерного СН2О
нельзя сравнить с рассматриваемым по этому показателю);
б) полупродукты можно хранить продолжительное время;
в) пиролиз может быть периодическим или непрерывным;
г) мономер содержит минимальное (по сравнению с другими
способами) количество примесей.
Путем пиролиза параформальдегида при 120—140° С можно
получить мономер, содержащий 90—95% основного продукта. Для
пиролиза сс-полиоксиметилена необходимы более высокие температуры
A80—200° С), при этом образуется мономерный формальдегид,
содержащий около 99% основного продукта.
Как показали исследования кинетики пиролиза а-полиоксимети-
лена, в продуктах пиролиза могут присутствовать, помимо
формальдегида, метанол, муравьиная кислота, метилаль, метилформиат,
двуокись углерода, окись углерода, вода. Зависимость состава
газовой смеси от условий, при которых проводится пиролиз, имеет очень
сложный характер, однако увеличение температуры закономерно
приводит к возрастанию суммарного количества побочных продуктов.
Триоксан в отличие от линейных полиоксиметиленов вообще не
содержит химически связанной воды. Если его кристаллы тщательно
просушены, то при пиролизе можно получить до 99,9% [8]
мономерного формальдегида^ однако скорость термической деструкции триок-
сана достаточно высока только при температурах около 300° С. Для
понижения температуры пиролиза триоксана и увеличения скорости
процесса предлагается использовать кислотные катализаторы типа
нелетучих минеральных кислот, нанесенных на твердый носитель
(кизельгур) или катионообменные смолы [4]. Условия равновесия
этого процесса рассматриваются в работе [5]. Однако в связи с
разработкой способа прямой полимеризации триоксана применимость
этого метода ограничена.
Вследствие эндотермичности процесса термодеструкции а-поли-
оксиметилена и параформальдегида и низкой теплопроводности
СН2О выделяющийся мономер имеет сравнительно невысокую
температуру. Это весьма существенно, так как при повышенных
температурах формальдегид легко вступает в реакции, которые
катализируются металлическими стенками аппаратов и смолообразными
продуктами, образующимися при пиролизе. В лабораторных установках
время поебываиия мономерного формальдегида в зоне высоких
температур невелико, что приводит к снижению количества
образующихся побочных продуктов. Так, например, содержание
муравьиной кислоты составляет около 0,005%, если пиролиз производится
в стеклянной аппаратуре, и около 0,001%, если пиролизу подвергать
187
суспензию а-полиоксиметилена в жидком инертном
теплоносителе, например минеральном масле, ди-B-этилгексил)-фталате или
вазелиновом масле. Содержание влаги в мономере зависит от
среднего молекулярного веса и степени высушивания полиоксиметилена.
При моделировании процесса пиролиза на пилотных установках
целесообразно применять шнековые и центробежные аппараты
различных конструкций (горизонтальные и вертикальные с регулируемым
временем контакта). В таких аппаратах прогрев порошка
осуществляется в тонком слое. При этом можно избежать значительного
перегрева полимера, который неминуемо приводит к увеличению
эффективности побочных реакций [9].
При расширении масштаба производства, например при переходе
от лабораторной к пилотной установке, возможны существенные
изменения состава мономерного формальдегида:
.г, - Пилотная
Лабораторная установка
установка (молибдено-
(стекло, медь) \ая сталь)
Температура теплоносителя, °С . . 140—150 180—220
Температура мономера на выходе из
аппарата, °С 40 110
Примеси в мономере, %
Н2О 0,30 0,70
СН3ОН 0,15 1,20
НСООН 0,02 0,10
СН2(ОСН,J 0,01 0,20
СНзСООН 0,01 0,08
Снизить содержание нежелательных примесей можно, применяя
жидкий нейтральный теплоноситель [6, 11] или нелетучие кислотные
катализаторы [7]. В первом случае, улучшая условия
теплопередачи, можно избежать перегрева продуктов. Кислотные добавки
увеличивают скорость деструкции полиоксиметиленов, что также
позволяет понизить температуру реакции.
При пиролизе а-полиоксиметилена на пилотной установке
увеличение продолжительности контакта формальдегида с твердым а-по-
лиоксиметиленом в зоне высоких температур (около 200° С)
приводит к увеличению содержания метанола в газовой фазе, которое не
коррелируется с изменением концентрации муравьиной кислоты.
Следовательно, можно говорить о каких-то каталитических
процессах превращения формальдегида в этих условиях.
К недостаткам способа выделения чистого мономерного
формальдегида путем пиролиза твердых полимеров можно отнести
следующие:
1) необходимость получения твердого порошкообразного
продукта по достаточно сложным многоаппаратным схемам;
2) все твердые низкомолекулярные полиоксиметилены, за
исключением триоксана, нестойки, выделяют формальдегид при хранении,
а в промышленных условиях трудно добиться полной герметичности
аппаратуры при работе с мелкодисперсными порошками;
188
3) при моделировании процессов пиролиза на промышленных
установках практически не удается подавить побочные реакции
в зоне контакта, удельный вес которых возрастает с увеличением
размеров аппаратуры.
Пиролиз гемиформалей (полуацеталей)
Впервые идея использования гемиформалей для получения
мономерного формальдегида была высказана [10] в 1958 г.
Известно, что в нейтральной и щелочной средах формальдегид,
взаимодействуя с алифатическими и циклическими спиртами
(обратимая реакция), образует сравнительно малоустойчивые соединения —
полуацетали. При нагревании эти соединения разлагаются снова
на спирт и формальдегид, чем отличаются от формалей,
образующихся из формальдегида и спирта в кислой среде:
, СН2О + С2Н5ОН <± С2Н5ОСН2ОН (нейтральная или щелочная среда)
этиловый
полуацеталь
формальдегида
СН2О^2С2Н5ОН +± С2Н5ОСН2ОС2Н5фН2О (кислая среда)
этилаль
Формали в нейтральной и щелочной средах весьма устойчивы и по
химическим свойствам сходны с простыми эфирами. Свойства
гемиформалей мало изучены. Считается, что они ведут себя подобно
растворам формальдегида в воде, например в концентрированных
спиртовых растворах монополуацетали находятся в равновесии с полиок-
симетиленполуацеталями.
Существенным является то, что гемиформали более устойчивы,
чем соответствующие гидраты формальдегида, причем температура
их разложения зависит от молекулярного веса спирта. Пользуясь
этим свойством, можно отделять гемиформали высококипящих
спиртов от гидратов формальдегида.
Принципиальная схема применения гемиформалей для
получения мономерного формальдегида включает следующие стадии:
1. Парогазовая смесь, получаемая в реакторе, где проводится
синтез формальдегида из метанола, поступает в скруббер, орошаемый
спиртом при 40—80° С. Снизу из скруббера выходит «сырой» геми-
формаль, содержащий некоторое количество воды и метанола.
2. Несконденсированный формальдегид вместе с инертными
газами поступает на дополнительное улавливание в колонны,
орошаемые водой.
3. «Сырой» гемиформаль отделяется от воды в отстойниках (если
спирт п гемиформаль нерастворимы в воде) и дополнительно
обезвоживается ректификацией в глубоком вакууме.
4. Очищенный продукт подается в испаритель, где происходит
разложение гемиформаля. Спирт отделяется от формальдегида и
возвращается в цикл.
189
По сравнению с твердыми низкомолекулярными
полноксиметиленами гемиформали как сырье для получения мономерного
формальдегида обладают рядом преимуществ: для синтеза гемиформалей
можно использовать непосредственно синтез-газ формальдегидного
производства (исключается стадия концентрирования формалина);
гемиформаль транспортабелен в нагретом состоянии, хотя и
образует твердые осадки при переохлаждении (по аналогии с
формалином) .
Очевидным недостатком приведенной схемы является большое
количество балластного спирта, который циркулирует в системе.
При прочих равных условиях количество спирта зависит от его
молекулярного веса. В табл. 29 приведены данные о концентрации
формальдегида в гемиформалях, полученных на основе спиртов
с различным молекулярным весом (при эквимолярных
соотношениях).
ТАБЛИЦА 29
Концентрация формальдегида в гемиформалях
различных спиртов
Спирт
Метанол
Этанол
Циклогексанол
Фракция спиртов С7—Сд
Пентаэритрит
Диметилолпропан
Глицерин
Температура
кипения
спирта
°С
64,7
78
161
180
262 (т. пл.)
210
290
Концентрация
СН2О
в гемиформале
вес. %
48,5
39,5
23,1
19,0
39,8
44,2
49,5
Из данных, приведенных в табл. 29, следует, что для получения
мономерного формальдегида экономически выгодно применять
многоатомные спирты [14, 15], поскольку низкомолекулярные спирты
обладают высокой летучестью. Холл и Пирет [11] установили, что
гемиформали в парах менее диссоциированы, чем метиленгликоль
(при низких температурах), причем степень диссоциации
максимальна для гемиформалей на основе пропилового спирта.
Исследования кинетики разложения гемиформалей привели к заключению,
что их температура разложения зависит от природы спирта и
концентрации формальдегида. Скорость разложения полигемиформалей
подчиняется тем же закономерностям, что и для олигомерных поли-
оксиметиленов, т. е. при температуре выше 100° С относительно
велика и практически не зависит от природы спирта. Влияние
последней наиболее сильно сказывается при пиролизе моногемифор-
малей. Чтобы полностью выделить связанный формальдегид,
необходимо нагреть гемиформаль до температуры кипения спирта.
Однако для гемиформалей на основе высоко кипящих спиртов типа
диформальпентаэритрита температура 180—190° С оказалась доста-
190
точной. Вообще температура пиролиза гемиформалей зависит от
того, каким образом осуществляется процесс деструкции. При
периодическом режиме температуру пиролиза можно постепенно повышать
для поддержания постоянной скорости выделения мономера. При
цереходе к непрерывному процессу для эффективного
использования гемиформаля необходимо поддерживать высокую температуру.
Требования к процессу пиролиза гемиформалей можно
сформулировать следующим образом:
1) разложение целесообразно вести в тонкой пленке, чтобы
уменьшить градиент температуры между пленкой и стенкой;
2) перегрев гемиформаля нежелателен, так как это приводит
к испарению больших количеств спирта;
3) унос спирта происходит за счет частичного испарения и захвата
капель жидкости газовым потоком, поэтому необходимо применять
циклоны и отбойники различных типов;
4) при отделении паров спирта в виде конденсата (путем
парциальной конденсации) контакт между конденсатом и газом
должен быть минимальным, иначе формальдегид снова растворится
в спирте;
5) для ускорения пиролиза можно использовать каталитические
добавки (особенно при пиролизе полигемиформалей).
Важным моментом является очистка гемиформалей от примесей
воды и метанола, которые растворяются в нем при синтезе. Чем
тщательнее проведена очистка, тем меньше примесей содержит
формальдегид, выделенный путем пиролиза гемиформаля. Гемиформаль,
содержащий 0,5% воды и около 20% связанного формальдегида,
фактически содержит около 2,5% воды в пересчете на формальдегид.
Для мономера, содержащего менее 0,01% воды, необходимо, чтобы
в моногемиформале на основе циклогексанола, например,
содержалось не выше 0,002% влаги.
Опыт показывает, что обезвоживание целесообразно проводить
путем двухступенчатой дистилляции в вакууме (при непрерывном
процессе очистки). В первой колонне температура куба не должна
превышать 85° С, чтобы не происходило заметного разложения
гемиформаля. Во второй колонне, куда поступает гемиформаль,
содержащий не более 1—2% влаги, температура куба доводится до 100° С
(остаточное давление в верхней части колонны около 25 мм рт. ст.).
Предложен метод удаления влаги с помощью третьего компонента,
образующего азеотропную смесь с водой (например, бензол или
циклогексан). В этом случае потери формальдегида заметно
снижаются:
Растворенные в гемиформале двуокись углерода и метилформиат
можно удалить продувкой каким-либо инертным газом.
Удаление паров спирта из парогазовой смеси после пиролиза
также представляет сложную инженерную задачу. Одним из
вариантов осуществления этой стадии является парциальная (частичная)
конденсация парогазовой смеси. Механизм этого процесса
рассмотрен ниже.
191
Сконденсировать пары спирта из парогазовой смеси трудно даже
при больших градиентах температуры. Кроме того, при
охлаждении могут протекать вторичные реакции — растворение
формальдегида в конденсате спирта, спонтанная полимеризация и т. п. Более
эффективно протекает конденсация, при которой происходит
одновременное растворение спирта в подходящем растворителе. При этом
за счет эффекта растворения парциальное давление паров спирта
в парогазовой смеси можно снизить почти до равновесного.
Подобный процесс описан [12] в патенте фирмы «Хёхст» (ФРГ).
Парогазовая смесь, полученная пиролизом гемиформаля, промывается в скруб-
берной колонне, куда противотоком подается растворитель,
который должен кипеть при температуре более низкой, чем температура
кипения спирта, хорошо растворять спирт и плохо формальдегид
и не содержать примесей, способных влиять на полимеризацию
формальдегида. В качестве растворителей можно использовать
ациклические, циклические, ароматические и замещенные углеводороды
с температурами кипения 80—160° С. Промывка осуществляется в
таких условиях, чтобы полимеризация формальдегида не
происходила (около 100° С). При достаточно высокой плотности орошения
содержание спирта в парогазовой смеси уменьшается до 0,005%.
При этом газовый поток захватывает значительные количества
растворителя, однако последний не оказывает влияния на полимеризацию.
При техническом осуществлении описанной выше схемы
получения формальдегида натолкнулись еще на одно затруднение: при
разложении гемиформаля протекают некоторые побочные реакции,
приводящие к потере спирта в основном за счет образования
линейных и циклических формалей и продуктов осмолення. Это
препятствие, конечно, не является принципиальным, так как при
тщательном контроле pH среды и очистке спирта необратимые потери можно
свести к минимуму.
Парциальная конденсация паров
формалина
Большой интерес представляет получение мономерного
формальдегида непосредственно из водного раствора. Как известно,
формальдегид в водных растворах присутствует в виде равновесной смеси
моно- и полигидратов. Парциальное давление формальдегида над
этими растворами является по существу давлением продуктов
разложения гидратов формальдегида [11].
Смесь паров воды и формальдегида обладает свойствами смеси
воды и летучего газа. Это значит, что при частичной конденсации
такой смеси содержание формальдегида в газовой фазе будет выше,
чем в конденсате. Таким путем можно из разбавленных растворов
формальдегида получить более концентрированные. Впервые
подобный способ запатентовал Уокер [13] еще в 1932 г. Парциальная
конденсация осуществлялась в охлаждаемых водой трубках Фильда
методом противотока.
192
Экспериментально было показано, что скорость растворения
формальдегида в конденсате уменьшается с понижением температуры,
и если контакт между газом и жидкостью в конденсаторе
кратковременен, то их можно вывести из соприкосновения до того,
как наступит равновесие.
На рис. 66 показана принципиальная схема парциальной
конденсации, предложенная Элдером [16]. Концентрированный
водный раствор формальдегида подается в трубчатый испаритель, где
подвергается испарению и перегревается до 110 °С. Эта температура
выбрана на основании
результатов работ Холла
и ІІирета [11], которые
показали, что при
температуре около НО °С мети-
ленгликоль полностью
диссоциирован.
Парогазовая смесь поступает в
парциальный конденсатор
I ступени,
представляющий собой вертикальную
медную трубу,
охлаждаемую водой через рубашку.
Конденсат (вода) стекает
в виде сплошной пленки
ПО Стенкам Трубы; ПО
В
Формалин
ЧІступени
Рис. 66. Принципиальная схема парциальной
конденсации:
1 — испаритель; 2 — парциальный конденсатор
I ступени; 3 — газоотделитель I ступени; 4 —
перегреватель; 5—парциальный конденсатор II ступени;
б — газоотделитель II ступени.
центру идет газ. В
конденсаторе параллельно
происходят два процесса:
конденсация воды и
растворение формальдегида
в воде. Пока жидкая
пленка покрывает стенки,
полимеризация формальдегида на них исключается, так как
сначала формальдегид должен образовать пересыщенный раствор
и только после этого начнется спонтанное образование твердого
полимера из раствора. Диаметр и длину конденсатора,
температурный режим и скорость потока подбирают таким образом,
чтобы пленка конденсата не успевала насытиться
формальдегидом. Из конденсатора парогазовая смесь и жидкий конденсат
поступают на разделение в газоотделитель. Парогазовая смесь снова
перегревается для разложения метиленгликоля и поступает в
парциальный конденсатор II ступени. По устройству и принципу
действия он аналогичен конденсатору I ступени, но отличается
меньшими размерами, так как количество влаги в парогазовой смеси
уменьшается.
Принцип действия установки парциальной конденсации, как
следует из приведенного выше описания, основан на том, что в
специально подобранных условиях скорость физического процесса
13 Заказ 113 5. 193
(конденсация воды или спирта в случае применения гемиформаля)
становится выше, чем скорость химической реакции формальдегида
с конденсатом.
Из общих соображений следует, что температура конденсата
должна быть минимальной, так как в этом случае различия в
значениях энергии активации физического и химического процессов
проявятся наиболее полно. Кроме того, скорость теплопередачи от
парогазовой смеси к стенке будет зависеть от разности температур:
¦?=/<АГ> B)
Одновременно с понижением температуры конденсата будет
уменьшаться предел насыщения раствора формальдегидом (т. е.
концентрация, определяющая начало спонтанной полимеризации).
Совместное рассмотрение эти^ трех зависимостей позволяет найти
оптимальную температуру процесса. Конечное содержание влаги (или
спирта) в газовой фазе не может быть ниже равновесного для
данной температуры конденсата.
Для достижения равновесной концентрации в газовой фазе
следует увеличивать время контакта газовой и жидкой фаз, поскольку
общее количество переданного тепла зависит от величины Д Т
(которая уменьшается в ходе процесса) и продолжительности контакта t.
Однако скорость химической реакции возрастает по мере
увеличения количества конденсата:
w = k[CR2O][R2O] C)
Отсюда следует, что конденсат нужно как можно чаще выводить из
контакта с газом. Это достигается путем применения
многоступенчатой парциальной конденсации.
Увеличение линейной скорости газового потока (турбулизация)
способствует росту коэффициента теплопередачи h, являющегося
функцией критерия Рейнольдса:
где р — плотность; v — линейная скорость; d — диаметр трубы;
(і — вязкость; а — эмпирический показатель, зависящий от
геометрических параметров системы.
Поток конденсата турбулизуется, когда, согласно эмпирической
формуле
где v0 — количество конденсата в единицу времени.
Однако симбатно с коэффициентом теплопередачи возрастает
коэффициент массопередачи, а это в пределе приводит к тому, что
процесс взаимодействия формальдегида с пленкой конденсата в
какой-то момент начинает превалировать.
194
Проведение процесса с редуцированием давления [17] позволяет
понизить мгновенную концентрацию формальдегида в зоне контакта
и увеличить тем самым выход непрореагировавшего формальдегида,
однако при этом возрастает равновесная концентрация влаги в
газовой фазе.
Регулирование режима в конденсаторах возможно эмпирически
путем перераспределения нагрузки на конденсаторы. Сложнее
регулировать режим в газоотделителе, куда поступает мономерный
формальдегид, содержащий приблизительно 1% влаги, и конденсат,
содержащий 60% формальдегида при температуре около 40 °С
(нижний предел устойчивости раствора против спонтанной
полимеризации). Как указывалось выше, спонтанная полимеризация
газообразного формальдегида невозможна при следующих
условиях:
1) давление формальдегида в системе таково, что предельная
температура полимеризации лежит ниже рабочей температуры;
2) содержание влаги в мономере таково, что конденсация при
данной рабочей температуре приводит к образованию ненасыщенного
раствора;
3) поверхность аппаратуры защищена движущейся пленкой,
в которой происходит растворение мономера, но предел насыщения
не достигается.
При конструировании газоотделителя необходимо базироваться
на этих трех принципах. В то же время температуру в
газоотделителе нельзя произвольно увеличивать, так как это приведет к
испарению конденсата.
Для увеличения устойчивости процесса можно использовать
различные методы, например автоматическое переключение на
резервный конденсатор или газоотделитель при увеличении давления
в системе.
Как показывает опыт, при автоматическом регулировании режима
возможна длительная бесперебойная работа аппаратуры.
Моделирование парциальной конденсации на большую производительность
представило бы значительную трудность, однако существует способ,
с помощью которого можно существенно облегчить задачу.
Повышения производительности можно достигнуть, например, путем увели-
чения числа конденсаторов, но при этом необходимо обеспечить
равномерную нагрузку на каждый конденсатор. Проблема
моделирования газоотделителя, однако, остается.
В табл. 30 приводятся параметры процесса, проводимого на
пилотной установке. Обращает на себя внимание факт увеличения
концентрации муравьиной кислоты после первого конденсатора.
Вероятно, это связано с протеканием реакции Канниццаро в испарителе.
Кислота может быть выведена из цикла пропусканием раствора через
подходящую ионообменную смолу при температуре, при которой
не происходит выпадение полимера из раствора. Очистка от
метанола и метилформиата достигается ректификацией раствора в
вакууме.
13* 195
ТАБЛИЦА 30
Состав парогазовой смеси и температурный режим при двухступенчатой
парциальной конденсации
Параметры процесса
Температура паров, °С
на входе
на выходе
Температура
конденсата, °С
Концентрация в
жидкой фазе, вес. %
СН2О
сн3он
НСООН
Концентрация в
газовой фазе, вес. %
Н2О (на выходе)
сн3он
НСООН
СН-ООСН
Испаритель
—
110—115
—
60-65
0,5
0,01
35-40
0,5
0,06-0,08
0,10
Парциальный
конденсатор
I ступени
110—115
60—70
40—50
33-37
0,1
0,20-0,25
10
—
—
—
Парциальный
конденсатор
II ступени
115—120
40—45
30—35
40-50
0,5
0,08—0,10
1-3
0,1
0,02
0,01
Принцип парциальной конденсации можно использовать для
получения концентрированных растворов формалина непосредственно из
парогазовой смеси, образующейся в процессе синтеза формальдегида
из метанола. Задача в данном случае обратна той, которая
рассматривалась выше, так как необходимо обогащать формальдегидом
жидкую фазу [18].
СПОСОБЫ ОЧИСТКИ МОНОМЕРНОГО
ФОРМАЛЬДЕГИДА
Глубокая * очистка мономерного формальдегида оказалась одной
из наиболее сложных технических задач при разработке технологии
синтеза полиформальдегида. В литературе предложены
многочисленные способы очистки формальдегида от примесей, однако
большинство из них трудно осуществить в промышленном масштабе
или же они не позволяют получить продукт требуемой чистоты.
Целесообразно разделить все возможные способы очистки на
следующие группы:
а) методы, основанные на форполимеризации формальдегида;
б) физические методы удаления примесей;
в) химические методы.
По-видимому, наиболее удачно эта задача была решена фирмой
«Дюпон», которая первой создала промышленное производство поли-
* Под глубокой очисткой формальдегида подразумевается очистка
концентрированного мономера до суммарного содержания примесей ниже 0,05%.
196
формальдегида, однако в литературе отсутствуют какие-либо
указания на выбранный фирмой способ очистки.
Рассмотренные нами в предыдущем разделе способы производства
формальдегида в большинстве случаев не обеспечивают получения
мономера необходимой степени чистоты. Исключением является
только метод, в котором используется триоксан.
Обычно состав мономерного формальдегида зависит от способа
его синтеза, и это учитывалось, конечно, при разработке методов
глубокой очистки, которые не являются универсальными. Данные
о составе мономерного формальдегида, полученного различными
способами, приведены в табл. 31.
Состав мономерного формальдегида
ТАБЛИЦА 31
Способ получения мономера
Пиролиз а-пблиоксимети-
лена
Пиролиз параформальде-
гида
Пиролиз гемиформалей
Парциальная конденсация
формалина
Содержание
влаги
вес. %
0,10—0,50
2,0—5,0
0,10-0,5
1,0
Концентрация
НСООН*
вес. %
0,05—0,10
0,02—0,05
0,01—0,02
0,002—0,01
* Концентрация спирта, очевидно, наибольшая при пиролизе геми-
формаля (достигает нескольких процентов от веса мономера).
Требования к чистоте мономерного формальдегида, необходимого
для получения полимера приемлемого качества, в большой степени
зависят от условий проведения полимеризации. Однако в первом
приближении на основании кинетических лабораторных
исследований можно считать, что суммарное содержание примесей не должно
превышать 0,05 вес. %.
Форполимеризация
Этот метод очистки мономерного формальдегида основан на том,
что следы воды (а также метанола и муравьиной кислоты)
инициируют полимеризацию формальдегида в жидкой и газообразной
фазе и одновременно участвуют в передаче цепи при полимеризации,
причем относительные константы скорости передачи цепи на эти
примеси больше единицы. Таким образом, примеси расходуются (на
образование концевых групп полимера) относительно быстрее, чем
мономер, и их концентрация в мономере уменьшается в ходе
реакции. Потери формальдегида на образование низкомолекулярных
полиоксиметиленгликолеи зависят от концентрации агентов передачи
цепи, температуры и условий проведения процесса. Текущая
197
концентрация агентов передачи цепи в мономере [х] во время
полимеризации будет определяться формулой:
[*] = [*]0A-д)*и/*р F)
где [х]0—начальная концентрация примеси; q — степень
превращения мономера; ки — константа скорости передачи цепи; кр —
константа скорости роста цепи.
Впервые такой метод очистки формальдегида был применен в конце
прошлого века для получения жидкого формальдегида в
лаборатории [5] и до сих пор остается общепринятым. Обычно применяют
один из двух способов проведения форполимеризации. По первому
способу формальдегид, полученный пиролизом тщательно
высушенного а-полиоксиметилена, подвергают быстрой конденсации при
низких температурах (ниже —19 °С) или растворяют в каком-либо
инертном неполярном растворителе. Образующийся твердый
полимерный продукт отфильтровывают или же отгоняют незаполимеризо-
вавшийся мономер. Повторив эту операцию несколько раз, можно
получить весьма чистый раствор формальдегида, который
относительно медленно полимеризуется при хранении.
Второй способ технически более перспективен: формальдегид
со следами влаги пропускают через стеклянные V-образные ловушки,
охлаждаемые до температуры не ниже —19 °С (чтобы не
происходила конденсация формальдегида). При этом происходит
парциальная конденсация паров воды на стенках ловушек и после насыщения
раствора формальдегидом начинается полимеризация. Размеры
ловушек, температура и скорость газового потока подбираются таким
образом, чтобы за время прохождения газа через очистительное
устройство большая часть примесей успевала вступить в реакцию.
Для расчета оптимального температурного режима этого
процесса нужно знать общее кинетическое уравнение, температурные
зависимости входящих в него параметров и тепловой эффект
реакции. Необходимы некоторые разъяснения по поводу того, что мы
называем в данном случае оптимальным режимом. Речь идет не о
достижении максимальной скорости реакции, хотя процесс очистки
в промышленных условиях должен протекать с достаточно большой
скоростью. Задача заключается в том, чтобы получить минимальный
выход твердого полимера при максимальном расходовании
примесей — агентов передачи цепи.
Текущая концентрация примеси в мономере связана с
количеством образовавшегося полимера экспоненциальной зависимостью
[уравнение F)]. Степень полимеризации образующегося продукта
равна:
G)
При данной концентрации примесей в мономере оптимизация
режима возможна только путем увеличения отношения kjkr С по-
198
вышением температуры это отношение увеличивается, а степень
полимеризации уменьшается.
При повышении температуры (приближение к предельной
температуре полимеризации) скорость расходования мономера
уменьшается из-за эффекта деполимеризации:
где [А] — концентрация активных центров.
Таким образом, следовало ожидать, что с повышением темпера-
туры степень очистки мономера будет улучшаться. Однако в этих
расчетах не учитывался акт инициирования газофазной
полимеризации формальдегида, заключающийся, по-видимому, в адсорбции
молекул воды (спирта, кислоты) на стенке и последующей ионизации.
Скорость адсорбции описывается уравнением:
где а — коэффициент адсорбции, имеющей отрицательную энергик>
активации; S — площадь поверхности стенки; [х] — концентрация
сорбируемого вещества.
С учетом этой стадии оказывается, что эффективная энергия
активации скорости расхода воды при полимеризации
газообразного мономера имеет отрицательную величину, т. е. скорость расхода
увеличивается с понижением температуры реакции.
Однако в этом случае для получения требуемой степени очистки
мономера (менее 0,05% примесей суммарно) его нужно было бы
охлаждать до температуры —A5—20) °С. Между тем анализ состава
газа на выходе из трубчатого аппарата, применявшегося в качестве
конденсатора, показал, что при температуре 25—30 °С уже
достигается заданная степень очистки от влаги и метанола, хотя потери
мономера на форполимеризацию были на порядок меньше
рассчитанных по константам передачи" цепи.
Исследования этого явления привели к заключению, что твердые
полиоксиметилены обладают высокой сорбционной способностью^
которая играет, по-видимому, основную роль в очистке мономера.
Предложено несколько вариантов использования этого эффекта:
1. Мономерный формальдегид пропускают через трубы
небольшого диаметра, охлаждаемые снаружи. Большую часть влаги удается
сконденсировать на стенке благодаря высокому коэффициенту
теплопередачи, причем она сорбируется слоем образующегося
низкомолекулярного полимера.
2. Скорость гетерогенной полимеризации формальдегида является
функцией поверхности (концентрации сорбированных активных
центров). Поэтому если охлаждать мономер в аппаратах с величиной
отношения S/V a* 0,6 см и ниже (сферическая форма), то скорость
полимеризации должна быть невелика. При этом необходимо на
входе в аппарат осуществить дросселирование газового потока.
199*
Тогда при достаточно большом времени удерживания пары влаги
успеют сконденсироваться на стенке и в газовой фазе установится
их концентрация, близкая к равновесной [19].
Техническое воплощение идей, изложенных выше, наталкивается
на большие затруднения: во время работы поверхность аппаратов
покрывается плотной коркой низкомолекулярных полимеров,
ухудшается теплообмен, возрастает сопротивление газовому потоку.
Периодически аппаратуру приходится отключать и удалять полимер
механическим путем или подвергать его термодеструкции (нагревать
через стенку или обрабатывать острым паром). Трудность подобных
операций очевидна. Были запатентованы способы непрерывного
механического удаления полимера с помощью специальных устройств
[20]. Однако использование подобной техники в крупномасштабном
производстве также сопряжено с большими трудностями.
Оказалось возможным применение для очистки формальдегида
движущейся или псевдоожиженной специальной насадки.
Подобные процессы широко используются в нефтехимическом синтезе.
Основная задача сводится к подбору соответствующей насадки,
которая должна обладать высокой сорбционной активностью к
примесям и в то же время быть инертной по отношению к мономерному
формальдегиду. Были испробованы многочисленные типы
сорбентов и, прежде всего, сам полиформальдегид. В патентной
литературе приводится описание некоторых сорбентов. Например,
алюмосиликат оказался намного эффективней, чем силикагель и алюмогель
раздельно [21].
Сообщалось также о применении молекулярных сит [22]. Расчет
соответствующей аппаратуры производится обычным способом, при
этом эмпирически подбирается такое время удерживания частиц
в контакте с мономером, чтобы они не успевали слипаться в
результате образования пленки полимера. Температурный интервал работы
насадочных колонн определяется областью наибольшей
сорбционной активности применяемого сорбента.
Физические методы
Как следует из анализа способов очистки формальдегида
/"основанных на применении частичной спонтанной полимеризации,
существенную роль в них играют физические процессы сорбции
примесей.
Следует заметить, что использование форполимеризации требует
громоздких аппаратурных схем и невыгодно экономически, так как
надо утилизировать большие количества неоднородного
низкомолекулярного полиоксиметилена. Обычно его растворяют в щелочи при
нагревании и снова возвращают в цикл, но эти операции связаны
с большими потерями формальдегида (нерастворимый остаток,
потери при растворении и перегонке). Поэтому были
исследованы варианты процесса очистки, основанные на физических
методах.
200
Чтобы избежать образования полимера, нужно вести процесс
в условиях, при которых скорость полимеризации крайне низка,
так что ощутимые количества полимера не успевают образоваться.
Предельная температура полимеризации формальдегида зависит
от концентрации мономера. Принципиально можно добиться такого
положения, чтобы формальдегид не полимеризовался при комнатной
температуре, однако равновесная концентрация в этом случае будет
слишком низка и этот путь не имеет практической ценности.
Эффективность большинства физических методов разделения
уменьшается при повышении температуры. Легче очистить
газообразный формальдегид от паров высококипящих спиртов,
парциальное давление которых имеет небольшую величину при 80—100 °С.
В этом случае достаточно пропустить парогазовую смесь через слой
инертной жидкости, нагретой до 80—100 °С, которая хорошо
растворяет спирт и плохо растворяет формальдегид [23—29]. Этот способ
можно рассматривать как один из вариантов очистки формальдегида
с применением третьего компонента. К растворителю предъявляются
обычные в этом случае требования: инертность по отношению к поли-
меризационной системе и формальдегиду.
Процесс проводится в противоточной колонне. Время фазового
контакта рассчитывается так, чтобы снизить содержание спирта
в газовой фазе до равновесного. Разделение жидкой смеси
осуществляется на ректификационной колонне. Это диктует выбор третьего
компонента — его температура кипения должна быть ниже, чем
температура кипения спирта.
При подборе веществ, способных эффективно сорбировать воду
при температурах 80—100 °С, было обнаружено селективное
действие соединений типа диалкилэфиров полиэтиленгликоля [30, 31].
Эти эфиры не взаимодействуют с формальдегидом, но сорбируют воду,
метанол, муравьиную кислоту и другие примеси из парогазовой
смеси. Концентрация примесей в газовой фазе при применении этих
сорбентов понижается до 0,10%.
Многие твердые сорбенты могут быть использованы при
повышенной температуре. Например, на кислых сорбентах, применяемых
обычно как ионообменные смолы (сульфированные полистиролы,
фенольные смолы, сшитые полиакрилаты, полифосфорные кислоты
на кизельгуре, их соли со щелочными и щелочноземельными
металлами) [32, 33], сорбцию ведут обычно при 80—125 °С, причем емкость
сорбента по воде достаточно высока. Как оказалось, нелетучие
вещества кислотного характера в небольшой степени влияют на
разложение формальдегида при повышенных температурах. Таким путем
можно снизить концентрацию воды в газовой фазе приблизительно до
0,05%. Ионообменные смолы указанного типа весьма инертны к
формальдегиду, их сорбционная емкость по мономеру всего лишь вдвое
превышает сорбционную емкость по воде.
Применение цеолитов (молекулярных сит) для сорбции примесей
натолкнулось на серьезные трудности. Цеолиты имеют щелочной
характер и ускоряют реакцию Канниццаро в газовой фазе [22].
201
Кроме того, цеолиты сорбируют значительное количество мономера
(в 3—4 раза больше, чем количество сорбированной воды). Это
происходит потому, что размеры молекул формальдегида и воды очень
близки.
Недостаток перечисленных выше методов заключается в том, что
при повышенной температуре (80—120 °С) фактически невозможно
добиться получения мономера, содержащего суммарно менее 0,05%
полярных примесей. Поэтому предлагается проводить очистку
комбинированными способами. Например, идею парциальной
конденсации можно реализовать следующим образом: мономер, содержащий
не более 0,10% примесей, подвергают парциальной конденсации
с третьим компонентом при низкой температуре @—10 °С). Чтобы
избежать полимеризации, нужно использовать вещества, хорошо
растворяющие формальдегид. Такими веществами могут быть геми-
формали спиртов с высокой температурой кипения. Это условие
необходимо, чтобы затем легко отделить пары гемиформаля от
формальдегида [34, 35]. Метод можно усовершенствовать, используя в
качестве третьего компонента смеси гемиформаля с веществами, хорошо
растворяющими воду.
Химические методы
Применение химических реагентов для связывания воды в
газообразном формальдегиде осложняется взаимодействием этих
реагентов с самим формальдегидом. В литературе имеются некоторые
сведения о химичесщіх методах обработки формальдегида водоотни-
мающими веществами. Так, например, предлагается использовать
пятиокись фосфора [36], которую добавляют непосредственно при
пиролизе сс-полиоксиметилена. Процесс нужно вести в вакууме при
температуре не выше 90 °С, чтобы предотвратить разложение с
образованием углерода и окиси углерода. Другим вариантом этого метода
[37] является пропускание формальдегида через пятиокись фосфора
при —15 °С.
Запатентован также метод, по которому пиролиз
а-полиоксиметилена производится в жидкой инертной среде, представляющей
смесь простых или сложных эфиров с углеводородами в присутствии
нелетучих кислот, растворенных в одном из двух компонентов
жидкой смеси. Рекомендуются такие кислоты, как серная, олеум,
мышьяковая, борная, себациновая, стеариновая и полифосфорная [38].
Любопытен факт, который приводится в литературе: полученный
мономер (во всех перечисленных выше случаях) не проявляет
склонность к спонтанной полимеризации при охлаждении.
ПОЛИМЕРИЗАЦИЯ МОНОМЕРНОГО ФОРМАЛЬДЕГИДА
Для разработки технологии полимеризации формальдегида важно
было определить область «перспективных» молекулярных весов
полимера, т. е. выяснить хотя бы приблизительно зависимость
физико-механических свойств от молекулярного веса. Одновременно
202
нужно было сформулировать критерии для оценки термической
стабильности полиформальдегида.
Было предложено [39] прессовать из полимера в стандартных
условиях пленку толщиной 0,08—0,18 мм, подвергать ее стареник>
при 105 °С в течение 7 суток, а затем испытывать на изгиб на 360°
(до излома). Количество полных циклов (перегиб в обе стороны)
характеризует «степень прочности» образца. Положительная оценка
давалась образцам со степенью прочности больше 100.
Для относительной оценки термической стабильности полимера
была предложена следующая методика [40, 41]. Навеску полимера
помещали в стеклянную ампулу, соединенную с атмосферой при
помощи капилляра; ампулу подвешивали к коромыслу аналитических
весов или соединяли с автоматическим прибором, регистрирующим
потерю массы, и нагревали до температуры 202 и 220 °С. По потере
массы образца можно определить скорость термического распада.
В первом приближении можно было считать, что начальная скорость
распада описывается уравнением первого порядка. Из
логарифмической зависимости количества неразложившегося полимера от
времени можно найти «константу термостабильности»:
—^St^k** hg=kTt (Ю)
где g = (g0 — gt)/g0.
Любая эмпирическая зависимость пригодна для применения
только в тех условиях, для которых она была найдена, и часто ее
использование для других условий не дает удовлетворительного
результата. Описанные выше методы не избежали подобной участи,
однако они сыграли положительную роль на первом этапе
исследований.
Основываясь на этих методах испытаний, Мак-Дональд и др.
[39, 42] пришли к выводу, что необходимо получать полимер с
логарифмической вязкостью не ниже 1,0 (измерено в тг-хлорфеноле
при 60 °С) и константой термостабильности не более 1%/лшнпри
222° С.
Кинетика полимеризации формальдегида обычно изучалась (см.
гл. II) при отрицательных температурах в растворах формальдегида
в неполярных растворителях. Основные результаты поисковых
работ можно суммировать следующим образом:
1) полимер полностью не растворим в полимеризационной среде;
2) дисперсность образующегося порошкообразного полимера
зависит от условий полимеризации и прежде всего от типа
катализатора, температуры и скорости перемешивания;
3) вязкость полимера обычно увеличивается в ходе реакции;
4) энергия активации молекулярного веса имеет отрицательное
значение;
5) приближенно можно считат.ь, что вязкость полимера обратно
пропорциональна концентрации полярных примесей (воды,
метанола, муравьиной кислоты) в реакционной системе;
20Я
6) скорость полимеризации линейно зависит от концентрации
мономера и инициатора:
-Ж=к[Ш][А]
7) тепловой эффект реакции равен приблизительно 15 ккал на
1 моль заполимеризовавшегося формальдегида.
Исходя из данных зависимости логарифма вязкости раствора
полимера от концентрации примесей в реакционной системе, можно
определить относительные константы скорости передачи цепи для
каждой примеси, имеющейся в мономере, при данной температуре.
Далее можно рассчитать концентрацию агентов передачи цепи
в реакционной системе, необходимую для получения того или
иного заданного среднечислового молекулярного веса, пользуясь
формулой:
1 ,., [Н2О] [СН3ОН] , [НСООН] [ск]
DPn~ а [М] "h п [М] "^ [М] h [М] (П)
Температурный режим полимеризации можно варьировать в
широких пределах, практически от —100 до +100 °С. Хотя в патентной
литературе предлагаются способы осуществления реакции ро всем
указанном интервале температур, наибольший интерес вызывает
полимеризация при комнатной температуре. Ведение процесса при
низких температурах потребовало бы слишком больших
энергетических затрат на поддержание изотермического режима.
Исследования режима полимеризации
в гетерогенной системе
Рассмотрим вариант осуществления процесса при положительных
температурах. В этом случае катализатор растворяется в
подходящем растворителе (алифатическом, циклическом или ароматическом
углеводороде, производных углеводородов, простых и сложных эфи-
рах и других кислородсодержащих соединениях и т. п.). Смесь быстро
перемешивается для создания максимальной поверхности контакта
с газом. Формальдегид подается непосредственно в жидкую фазу
или на поверхность раздела фаз. Хотя предельная растворимость
формальдегида в неполярных и малополярных средах при комнатной
температуре невелика (около 1 вес. %), за счет увеличения
поверхности контакта при эффективном перемешивании удается достигнуть
высоких скоростей превращения.
Таким образом, при комнатной и более высоких температурах
полимеризация происходит в гетерогенной системе (газообразный
мономер, жидкая полимеризационная среда, твердый полимер). Это
создает значительные трудности для кинетического анализа
процесса.
Для проведения кинетического анализа необходимо, чтобы
процесс протекал в кинетической области, тогда лимитирующей стадией
204
является собственно химическая реакция (полимеризация) [91].
Для перехода в кинетическую область есть несколько путей:
а) уменьшение концентрации катализатора;
б) понижение температуры реакции;
в) увеличение эффективности перемешивания.
В первом случае понижается скорость химической реакции,
поскольку она пропорциональна концентрации катализатора.
Однако уменьшать концентрацию катализатора ниже определенного
предела нецелесообразно, так как это приводит к увеличению роли
побочных процессов.
При понижении температуры скорость химической реакции и
скорость диффузии мономера в растворитель будут уменьшаться по
экспоненциальному закону, но, поскольку энергия активации во
втором случае меньше, в какой-то момент скорость полимеризации
станет лимитирующей.
Скорость диффузии пропорциональна поверхности контакта, и
увеличение интенсивности перемешивания должно способствовать
переходу процесса в кинетическую область.
Большое значение приобретает способ введения примесей в
реакционную систему. В периодическом процессе, протекающем в
гомогенной системе, соотношение концентраций мономера и примесей
непрерывно изменяется в ходе реакции (по экспоненте). Также
непрерывно изменяется средний молекулярный вес полимера в
зависимости от начальной концентрации примесей и значений констант
скорости передачи цепи [92].
При гетерогенном полунепрерывном процессе, когда
газообразный СН2 вводится в реактор в течение некоторого времени, текущая
концентрация мономера в реакционной среде будет определяться
скоростью растворения мономера и скоростью полимеризации. Если
реакция идет в кинетической области, то процессы массопередачи
должны быть равновесными и концентрация примесей в растворе,
поступающих из газа, должна определяться законом Генри. При этом
большое значение имеет начальная концентрация примесей в поли-
меризационной среде.
Мгновенное значение среднечисловой степени полимеризации
в этом случае будет выражаться уравнением *:
Jr)_
DPn [M]
к
где к'а — относительная константа передачи цеди на воду к'п = -г2- ;
[Н2О]0— начальная концентрация воды в растворе; [Н2О1Г —
концентрация воды, образующейся при конденсации из мономера
по- [закону Генри; [М] — концентрация формальдегида в
растворе.
* Принимается (для упрощения), что единственной примесью в системе
является вода.
205
В начальный момент [Н2О]0 > [Н2О]Г и образуется очень
низкомолекулярный продукт. Затем примесь «выгорает» по ходу реакции
в соответствии с уравнением:
d [H20]o ,
dt
A3)
где [А] — концентрация активных центров. В некоторый момент
концентрация воды в растворе станет равной [Н2О]Г.
Очевидно, что чем больше величина [Н2О]0 по сравнению
с [Н2О]Г, тем шире кривая молекулярно-весового распределения
получаемого полимера.
Проводя форполимеризацию подобным способом до достижения
равновесной концентрации воды в системе и затем отделяя
образовавшийся низкомолекулярный продукт, можно получить особо
чистую полимеризационную среду [43].
В непрерывном процессе, когда полимер выводится непрерывно,
концентрация воды и концентрация мономера в системе будут
постоянными (в стационарном режиме) и принципиально не должны
влиять на молекулярно-весовое распределение, которое приближается
к наиболее вероятному: -jf- = 2.
Найт [44] предложил следующую формулу для расчета средне-
числового молекулярного веса полиоксиметилена при непрерывном
режиме полимеризации (для Мп^> 10 000), протекающей в
кинетической области:
30
Мп [М] ^ [М] ^ [М]
где [хг] —молярная концентрация воды в полимеризационной среде;
[х2] — молярная концентрация метанола; [х3] — молярная
концентрация муравьиной кислоты; [М] — молярная концентрация
формальдегида; F — эмпирический поправочный фактор, учитывающий долю
непрореагировавшей примеси, которая уходит из зоны реакции
с полимером (шламом).
F=i{ 5500ІЧМ1
где [М] — концентрация формальдегида в полимеризационной среде,
выраженная в гіг раствора; Р — весовая доля твердого полимера
в шламе; w — скорость подачи (вывода) раствора в реактор в г/мин;
кп — соответствующая относительная константа передачи цепи.
Значения относительных констант скорости передачи цепи
и, соответственно, относительная эффективность примесей
(Н2О : СН3ОН : НСООН = 1 : 0,49 : 20,9) в этой формуле
отличаются от значений, полученных другими авторами (см. гл. II), что
можно отнести за счет различий в величине температурных коэф-
206
фициентов скоростей передачи цепи на воду и кислоту, с одной
стороны, и метанола — с другой.
Выражение для F можно получить путем следующих
рассуждений: при стационарном режиме в полимеризационной среде
установится некая стационарная концентрация примеси [#il:
^^ [x']0-kA4][A]-w[xi]-~$Plxi]==0 A6)
поступление расход при- уход из сорбция
примеси в ре- меси на пе- системы на поли-
акционную редачу цепи с раство- мере
систему рителем
где [х]0 — молярная концентрация воды в свежем растворителе;
[х']0 — молярная концентрация воды, полученной при
растворении мономера; кп — константа передачи цепи на воду; [А] —
концентрация активных центров; ? — коэффициент сорбции примесей
полимером.
Для учета расхода примеси можно составить следующее
уравнение:
^ *i] [Л] A7)
Уравнение материального баланса для полимера имеет следующий
вид:
--^ = ^1М][А]-и;Р = 0 A8)
откуда [А] = ^ . Подставим в выражение для передачи цепи:
wP
*«ЫМ = ^Ы A9)
Для примеси, сорбированной полимером, получаем:
!_«*» ІфЗ B0)
где [х2] — количество сорбированной примеси.
Отсюда после соответствующего преобразования получаем:
w
1 + -
Сорбция примесей из реакционной системы на твердом полимере
играет большую роль при оптимизации процесса. Если начальную
концентрацию примеси в растворе обозначить Ыо, а равновесную
концентрацию примеси после сорбции полимером [#], то
[1 С Гл.1 /ОО\
х\ ——«~ [^Jo у""/
где Ко — константа сорбционного равновесия.
207
При [х] 3> [А] мгновенная средняя степень полимеризации равна:
kv [M]
ОР--Ы B3)
Подставив в это уравнение выражение для [х], получим:
ав[М]П
B4)
Если концентрация формальдегида М постоянна, то средняя
степень полимеризации будет линейно зависеть от концентрации
твердого полимера в реакционной системе.
Отметим влияние времени удерживания растворителя в зоне
реакции (т = V/F, где т — среднее время удерживания в реакторе
идеального смешения, V — объем реактора) на величину
молекулярного веса получаемого полимера.
Общая скорость полимеризации формальдегида будет
определяться скоростями трех отдельных процессов:
Q — скорость подачи мономера; D — скорость растворения
(диффузии) мономера; R — скорость химической реакции
(полимеризации).
Кинетической области протекания процесса соответствует условие
R <^D <I Q (в более общем случае это условие можно записать так:
R <CQ и R <^D; соотношение Q и D может быть произвольным).
Введем представление о двух диффузионных областях
протекания процесса:
I диффузионная область: i? > D ^> Q.
В этом случае весь мономер вступает в реакцию и полимеризатор
работает в режиме разрежения. Относительная концентрация
компонентов в растворе равна таковой в газовой фазе. Равенство
скоростей диффузии компонентов достигается за счет различных
градиентов концентрации в поверхностной пледке.
II диффузионная область: І? ?> D <CQ.
Не весь мономер успевает вступить в реакцию. Устанавливается
равновесие между концентрациями компонентов в газовой фазе
и растворе (в зависимости от коэффициента диффузии каждого
компонента). Соответственно непрореагировавший газ может
обогащаться или обедняться примесями *.
Скорость диффузии определяется законом Фика:
dQ-D-D (ЮЛ BЬ)
— -D-Dm{d[x]) B5)
где — изменение концентрации; Dm — коэффициент диффузии;
do
~, градиент концентрации.
а[х\
* Можно рассматривать еще и III диффузионную область, когда скорость
поступления мономера из раствора к активным центрам полимеризации
лимитирует общую скорость процесса.
208
Если S — это величина поверхности раздела фаз на единицу
объема, то общая скорость диффузии будет равна:
D=SD B6>
Особый интерес представляет зависимость молекулярного веса
полимера от параметров реакции, протекающей в диффузионной
области. При стационарном режиме
•^г=Л--Ар[М][Л] = 0 B7)
Из условий стационарности вытекает также, что ¦ d = Q —D = О,
откуда Q = D. Выразим скорость подачи через концентрацию
мономера и подставим в уравнение B7). Получим, что
п=Шк B8)
где G — количество газа; t — время; [М]г — концентрация мономера
в газовой фазе.
Аналогичным образом выразим через скорость подачи
концентрацию примеси агента передачи цепи в газовой фазе:
dt
B9)
Текущая степень полимеризации, по определению, равна
отношению скорости реакции роста цепи fcp [М][Л] к сумме всех реакций
передачи и обрыва цепи 2 К \л][А].
Считая, что гибель цепи не происходит и учитывая влияние только
одного агента передачи цепи, получим, что в I диффузионной
области
(зо)
Путем интегрирования уравнения B6) (пределы интегрирования
Qx = [М] и Q2 = 0 на поверхности активных центров) получаем
выражение средней степени полимеризации для II диффузионной
области (R = D):
SDm [М] Dm [М]
DPn~ SDm [х] - Dm [х]
Экспериментально показано, что в этом случае непрореагировав-
ший мономер обогащается примесями. Это значит, что Dm%^> Dmi.
Проведение процесса в диффузионной области, где фактически
эффективные относительные константы передачи цепи становятся
равными единице (или даже меньше, как во втором случае),
представляет большой интерес с точки зрения снижения влияния примесей
14 Заказ ІІ35. 209
в мономерном формальдегиде, а также как метод регулирования
молекулярного веса полимера.
Интересно отметить, что в этом случае изменение температуры
в небольших пределах практически не скажется на величинах
молекулярного веса (энергия активации коэффициента диффузии —
низкая величина).
Проведение полимеризации в кинетической области
целесообразно с точки зрения лучшей воспроизводимости результатов,
контроля молекулярного веса и сопоставления полученных результатов
с данными лабораторных исследований.
Вместе с тем проведение процесса в диффузионной области
позволяет достигнуть максимальных скоростей реакции и нивелировать
действие эффективных агентов передачи цепи, присутствующих
в мономере. Последняя особенность диффузионного процесса наиболее
важна, если учесть, что получить мономерный СН2О высокой степени
чистоты в производственных условиях очень сложно.
Влияние природы катализатора. Большое разнообразие
катализаторов и отсутствие сколько-нибудь надежных данных о
механизме их действия приводит к тому, что подбор катализатора
производится чисто эмпирическим путем. Ниже мы попытались
перечислить основные параметры процесса, на которых может сказаться
специфика выбранного катализатора:
Основные параметры Возможный характер действия катализатора
Молекулярный вес Различная избирательность по
отношению к примесям (различные величины
относительных констант передачи
цепи)
Распределение по моле- То же
кулярным весам (МБР)
Термическая стабиль- Ухудшение стабильности (катализаторы
ность полимера катионного типа); улучшение
стабильности (азотсодержащие полимеры)
Растворимость катализа- Различный дисперсный состав полимера
тора в полимеризаци-
онной среде
Кроме перечисленных факторов катализатор, по-видимому, может
участвовать в побочных реакциях, оказывать влияние на физические
свойства полимера, изменять кинетику структурообразования и т. д.
В первом приближении можно классифицировать все
катализаторы по чувствительности к примесям полярных веществ:
1. Катализаторы катионного типа (кислоты Льюиса, их
комплексы, оксониевые соли и т. п.), которые наиболее чувствительны
к действию полярных примесей, за исключением, по-видимому,
веществ кислотного характера. Вызывают ацидолитическое
расщепление полимера. Интерес к действию этих катализаторов возник
главным образом в связи с получением термостабильных сополимеров
формальдегида [40—49].
2. Анионные катализаторы типа первичных, вторичных и
третичных аминов, фосфинов, арсипов, солей аммония, азотсодержащих
210
соединений и полимеров, карбонилов металлов VIII группы и т. д.
обладают высокой чувствительностью к действию примесей полярных
веществ. Некоторые катализаторы этого типа дают заметный
стабилизирующий эффект [50—54].
3. Соли органических кислот с числом атомов углерода в цепи
больше двух и двухвалентных металлов (олово, цинк, медь, кадмий,
никель, марганец, висмут), гидрат окиси олова, дифенил- итрифенил-
олово и др. [55, 563. Эти катализаторы занимают особое место, так
как отличаются очень низкой чувствительностью к примесям.
Например, при полимеризации в присутствии солей двухвалентного
олова мономер может содержать до 0,9% воды и до 2% метанола.
Специфика механизма действия этих катализаторов проявляется
также в том, что обычные регуляторы роста цепи по отношению к ним
неактивны, кроме того, величина кажущейся энергии активации
молекулярного веса необычно велика.
Влияние природы растворителя. Основное условие при выборе
растворителя заключается в том, чтобы он был инертен по отношению
к инициатору и мономеру. Часто приходится считаться с тем, что
хотя сам растворитель по своей природе инертен, находящиеся
в нем примеси могут взаимодействовать с инициатором или
полимерными растущими центрами.
При выборе растворителя также необходимо учитывать
технологию последующей обработки полимера. Это касается
температуры кипения, химической устойчивости, степени набухания
полимера в растворителе и других свойств. Например, если этерифика-
ция полимера осуществляется в жидкой фазе, то целесообразно
проводить ее в той же самой среде, что и полимеризацию. В связи с этим
предлагается вести полимеризацию в высококипящих растворителях
[55—57], в которых полимер хорошо набухает при нагревании, или
в таких, где он хорошо растворяется (диметилформамид, метилен-
диацетат, бутиролактон, уксусный ангидрид и т. п.). Вязкость
растворителя может существенно влиять на дисперсность
образующегося продукта.
Влияние природы регулятора роста цепи. Варьировать средний
молекулярный вес полимера можно несколькими способами.
Например, можно уменьшать степень очистки мономера или растворителя.
Наиболее приемлемым способом считается введение в полимериза-
ционную систему специальных регуляторов роста цепи.
Выбор регулятора роста цепи во многом зависит от типа
применяемого катализатора. Из общих соображений желательно
использовать вещества с относительной константой скорости передачи цепи
около единицы, чтобы не происходило «выгорания» или накопления
агентов передачи цепи в ходе реакции. Особое значение имеет
термическая стабильность образующихся в результате передачи цепи
концевых групп, так как они во многом будут определять стабильность
получаемого полимера. Представляется заманчивым за счет передачи
цепи при полимеризации полностью блокировать концевые полу-
ацетальные группы полимера [58—61]. Однако расчеты показывают,
14* 211
что в этом случае из реакционной системы должна быть полностью
удалена влага (так как средняя степень полимеризации обратно
пропорциональна полноте блокирования концевых групп,
достигаемой в результате передачи цепи). Обычно даже в лабораторных
условиях удается за счет передачи цепи блокировать не более 80%
концевых групп.
Влияние температуры. Реакции роста и передачи цепи
характеризуются различными энергиями активации и, следовательно,
различными температурными коэффициентами. Как уже отмечалось,
при работе в диффузионной области влияние температуры на
молекулярный вес незначительно, однако в кинетической области, особенно
при использовании катализаторов 1-й и 3-й группы (см. ст;\ 210),
влияние температуры на величину среднего молекулярного веса
велико.
Дисперсный состав полимера также зависит от температуры
полимеризации, а это весьма существенно сказывается на эффективности
других операций обработки полимера.
Рекомендуется вести полимеризацию в интервале температур
от —100 до +100 °С. В технологической практике обычно
поддерживают температуру полимеризации от 20 до 50 °С.
Аппаратурное оформление процесса
полимеризации
Получение однородного по свойствам полимера возможно только
при непрерывной полимеризации в стационарном режиме. Для
конструирования подходящей аппаратуры необходимо решить
следующие задачи:
1. Предотвратить спонтанную полимеризацию формальдегида
на охлажденных поверхностях, а также осаждение суспензии (шлама)
полимера в застойных зонах аппаратуры. Как было выяснено в
начале этой главы, чтобы предотвратить спонтанное образование
полимера, нужно нагреть все контактирующие с мономером устройства
выше предельной температуры полимеризации или защитить их
пленкой движущейся нейтральной жидкости. Осаждение полимера
из суспензии на стенках и различных механических устройствах
можно предотвратить, если скорость движения суспензии будет выше
критической.
Предложено критериальное уравнение для расчета критической
скорости движения суспензии v через трубу [62]:
C2)
где d — диаметр трубы; g — ускорение силы тяжести; б — средний
диаметр частиц; р — плотность жидкости; р' — средняя плотность
суспензии; а — «эффективная плотность» (ртв# — рж); \i — вязкость
жидкости.
21
В этом уравнении не учитывается фактор трения (адгезия
полимерных частиц к стенке):
где R — коэффициент трения по смоченной стенке.
Необходимый эффект достигается в аппаратах, в которых с
помощью специальных устройств (или при использовании
циркуляционных насосов) поток жидкости движется вдоль стенки. Увеличение
вязкости жидкой полимеризационной среды также способствует
повышению стабильности суспензии, однако в этом случае
увеличиваются энергетические затраты на перемешивание.
2. Осуществить равномерный вывод суспензии из реактора.
Подобная проблема встречается во многих химических процессах. Она
связана с достижением оптимальной концентрации твердой фазы
в суспензии, которая в свою очередь зависит от времени
удерживания растворителя и полимера в реакторе и определяет величину
молекулярного веса полимера.
3. Поддерживать в зоне реакции изотермические условия.
Тепловой эффект полимеризации формальдегида составляет около
15 ккал/молъ или 500 ккал/кг образуемого полимера. В реакторе
лабораторного масштаба изотермический режим можно поддержать
путем охлаждения аппарата через рубашку. Однако с увеличением
объема реактора отношение поверхности к объему уменьшается и
отвод тепла через рубашку становится недостаточным. Существует
три возможных метода поддержания изотермического режима:
а) введение дополнительных поверхностей охлаждения;
б) охлаждение поступающих в реактор компонентов;
в) введение инертных разбавителей (легкокипящих жидкостей
или инертных газов).
Первый метод, по-видимому, малоперспективен для
полимеризации формальдегида, так как поверхности охлаждения покрываются
пленкой полимера и в результате этого снижается коэффициент
теплопередачи через стенку. Другие два способа более приемлемы.
До сих пор речь шла о полимеризации формальдегида в
инертной жидкой среде. Однако можно осуществить полимеризацию в
газовой фазе [63]. Для этого используют принцип «кипящего слоя».
Особенностью процесса является то, что катализатор (аэрозоль)
наносится на поверхность раздробленных частиц полиформальдегида
и в таком виде вводится в реактор. Расчет оптимального режима
связан со следующим затруднением: при низких скоростях газового
потока время контакта и выход (степень конверсии) достаточно
велики, но частицы полимера слипаются и аппарат быстро забивается
полимером. При высоких скоростях этого явления не наблюдается,
однако степень конверсии уменьшается. Слипание можно
предотвратить, если, например, поддерживать разность температур между
газовым потоком и стенками так, чтобы она не превысила 20 °С,
а также если перемешивать ожиженный слой в горизонтальном
213
направлении или использовать пульсирующий режим подачи газа
[64-65].
Отвод тепла в основном осуществляется путем разбавления
мономера инертным газом на входе в реактор. Инертный газ предварительно
охлаждается, кроме этого он дополнительно охлаждается с помощью
рубашки и змеевиковых устройств внутри реактора. Превращение
за один проход достигает 30—40%, поэтому последовательно
устанавливают два аппарата. При работе в диффузионном режиме
непрореагировавший моно-
В8од катализатора
и полимера-носителя
Змеебик
(тладоагент)
Удаление
{'газа
¦Сепаратор
IuG*p uuui сіїцсібіОі її р II ЫбСл МП,
поэтому перед второй
стадией его нужно
дополнительно очищать.
Принципиальная схема действия
полимеризатора
рис. 67.
показана
на
Рубашка -
[хладоагент)
Готовый
продукт
Решетка
Пустот&пая
мешалка
Инертный газ
СН2О
Магнитный
клапан
Стабилизация
полиформальдегида
Термостабильность
полиформальдегида в
лабораторных условиях принято
оценивать по величине
константы скорости разложения
навески полимера в
стандартном режиме. Эта
константа термостабилышсти
рассчитывается как константа
скорости реакции первого
порядка, поскольку
приближенно можно рассматривать
процесс термического
разложения полиформальдегида
как реакцию первого порядка
[70-72] (стр. 203).
Условия полимеризации существенно влияют на термостабиль-
иость полимера (табл. 32). Однако даже при значениях константы
ниже единицы образцы полимера разлагались при переработке
литьем под давлением.
Здесь уместно вспомнить, каким образом аналогичная задача
решается при получении других термопластов.
Очевидно, расплавленный полимерный материал не должен
подвергаться заметной деструкции во время пребывания в цилиндре
литьевой машины или экструдера, причем допустимую суммарную
потерю массы можно приблизительно оценить величиной 0,1—0,5%.
Большие потери независимо от их происхождения (наличие
летучих продуктов, влага и т. п.) приводят к появлению значительных
214
Рис. 67. Принципиальная схема аппарата
для синтеза полиформальдегида из
мономерного формальдегида в псевдоожижен-
ном слое.
ТАБЛИЦА 32
Сравнительная термостабильность различных образцов полимера
Способ полимеризации
Блочный [401, при—80 °С
В толуоле [40], при
—30 °С
В толуоле, при —30 °С
В толуоле, при+25 °С
То же
»
В толуоле [69]
То же
»
В толуоле, при +25 °С
В толуоле, при +25 °С,
с добавкой уксусного
ангидрида
Логарифмическая
вязкость
(в п-хлорфе-
ноле при
60 ГС)
0,90
1,20
2,10
1,26
2,24
3,15
1,58
2,29
1,46
1,36
1,13
Константа
термостабильности
при 222 °С
%/мин
5,8
4,9
3,8
3,5
1,1
0,86
8,3
2,6
1,5
1,8
1Д
Примечание
Спонтанная
полимеризация
То же
Катализатор — стеарат
кальция
То же
»
»
Катализатор моноэта-
ноламиноолеат
Катализатор триэтанол-
аминоолеат
Катализатор тристеарат
алюминия
Полимер обработан
кипящей водой с
добавкой аммиака и затем
стабилизован добавкой
мочевины
Полимер обработан
аммиачным раствором
дефектов в изделиях и нарушению режима переработки. Отсюда
следует, что среднюю скорость разложения вообще нельзя
рассматривать как критерии перерабатываемое™ материала. Необходимое
условие заключается в том, чтобы материал вообще не разлагался
в расплавленном состоянии. Для полиолефинов принимается, что
время пребывания расплава полимера в цилиндре литьевой машины
равно приблизительно 1—2 ч при оптимальном режиме переработки.
Специальные опыты, проведенные с полиформальдегидом, пешазали,
что минимальное время при оптимальном режиме переработки
составляет 20—25 мин. Следовательно, термостабильность
полиформальдегида с точки зрения его перерабатываемое™ нужно оценивать
ке по усредненной величине скорости разложения, а по
индукционному периоду, предшествующему разложению.
Исследования кинетики термической и термоокислительной
деструкции полиоксиметиленгидратов и их производных [73—75]
позволили сделать следующие выводы:
1. Полиоксиметиленглпколи при термодеструкцпп разлагаются
с концов цепи (начиная с температуры приблизительно 90 °С),
поэтому основная задача заключается в модификации концевых групп
полимера.
215
2. Предел термической устойчивости повышается в зависимости
от природы концевых групп в следующем ряду:
гидроксильные <! формильные <С фенилуретановые < сложноэфирные ^
простые эфирные
Предел термической устойчивости полиоксиметиленовой цепи [73] —
около 270 °С.
3. Добавки веществ кислотного характера приводят к ускорению
деструкции независимо от природы концевых групп [73, 75].
4. В присутствии кислорода при температурах выше 120 °С
наблюдается автоускоренная деструкция полимера [73, 75].
Таким образом, процесс стабилизации полиформальдегида
должен включать:
а) блокирование концевых полуацетальных групп;
б) тщательное удаление кислых продуктов (введение акцептора
кислоты и акцептора формальдегида для предотвращения побочных
реакций);
в) введение подходящих антиоксидантов.
Величина индукционного периода разложения определяется
тем, насколько успешно подобраны стабилизирующие добавки при
условии полного блокирования концевых групп.
Ацетилирование
Поскольку концевые полуацетальные группы в макромолекулах
полиоксиметилена проявляют типичные для спиртовых групп
свойства, для их блокирования возможно применение реакций этерифи-
кации [76].
Сопоставление различных способов этерификации
полиоксиметилена показывает, что наиболее приемлемым в технологическом
отношении агентом является уксусный ангидрид [80—81].
Применение большинства других этерифицирующих агентов приводит к
дополнительным потерям полимера в результате деструкции или
получению продукта с недостаточно высокой термостабильностью.
Известно, что процесс ацетилирования целлюлозы в кислых
средах достаточно хорошо изучен и успешно осуществлен в
промышленном масштабе. Однако аналогию с процессом ацетилирования
полиоксиметилена нельзя проводить, так как полиацетали
неустойчивы в кислых средах. Поэтому ацетилирование проводят в
присутствии основных катализаторов в безводной среде, используя в
качестве ацетилирующего агента уксусный ангидрид.
Механизм реакции можно схематически изобразить следующим
образом:
1) RO-(GH2O)w-CH2OH
—> RO-(CH2O)n—GH2OAc+AcOH
2) RO-(GH2O)„-CH2OH —> (n-f-l)CH2O
216
3) СН20-}-Ас20 —> AcOGH2OAc
4) RO-(CH2O)W—CH20Ac^Ac0H — >
—>¦ RO—(GH2O)m—CH2OH + CH2O—(CH2O)w_m_2—CH20Ac-hAc0-
Для увеличения выхода ацетилированного продукта необходимо
свести к минимуму все реакции, кроме первой. Кинетический анализ
процесса показывает [67], что скорость реакции A) можно увеличить:,
а) путем добавления катализатора (третичный амин, соли
щелочных металлов и слабых кислот с константой диссоциации менее
1,5-Ю-4 при 25 °С);
б) повышением температуры реакции (энергия активации ацети-
лирования выше, чем энергия активации деполимеризации);
в) применением избытка уксусного ангидрида;
г) гомогенизацией реакционной смеси для облегчения
взаимодействия уксусного ангидрида с концевыми группами.
Скорость деполимеризации зависит от следующих факторов:
температуры; количества полуацетальных концевых групп в
исходном полимере; природы разбавителя (в случае гетерогенного ацети-
лирования) или растворителя полимера; природы катализатора;
концентрации уксусной кислоты, имевшейся в ангидриде и
образующейся при реакции с концевыми группами, следами воды и т. п.
Наибольшее влияние оказывает уксусная кислота, так как она
катализирует деполимеризацию и вызывает ацидолитическое
расщепление диацетата ПОМ. Для подавления этой реакции применяют
акцепторы кислоты (пиридин), тщательно очищают реагенты,
используют азеотропную перегонку кислоты из сферы реакции [87—88].
Действие деструктирующих факторов наиболее заметно на
начальной стадии реакции, во время разогрева реакционной смеси.
В ходе реакции небольшое количество мономера выделяется за счет
ацидолитического расщепления блокированных макромолекул.
Для получения термически стабильного во время переработки
продукта необходимо, чтобы степень превращения концевых гидр-
оксильных групп составила 100%. При гетерогенном способе ацети-
лирования предельная степень превращения лимитируется
особенностями кристаллического строения полиоксиметилена и его
полидисперсностью (по размеру частиц). В первом приближении можно
говорить о том, что чем больше доля аморфной фазы в полимере,
тем легче идет диффузия этерифицирующих агентов вглубь
кристаллитов и тем выше степень завершенности реакции [77]. Применяя
специальные добавки, удается довести предельную степень
превращения гидроксильных групп до 94—96%. Лучшие результаты (98%)
можно получить в гомогенных условиях, т. е. в растворе, например
в диметилформамиде или метилендиацетате [78, 79].
Природа «неблокированной» части полиоксиметилена до сих
пор не выяснена окончательно. Длительная термообработка
полиформальдегида в области начала размягчения A50—160 °С) не позволяет
достигнуть полного блокирования концевых групп, в то же время
это удается сделать при плавлении или растворении полимера.
217
Изменение молекулярного веса в процессе ацетилирования,
по-видимому, зависит в первую очередь от МВР образцов. При работе
с полидисперсными образцами (-jrf-^> 7j всегда наблюдалось
некоторое увеличение среднего молекулярного веса (см. гл. IV).
Аппаратурное оформление стадии
ацетилирования
Лабораторные исследования процесса ацетилирования показали,
что наибольший эффект дает проведение реакции в гомогенных
условиях, однако его осуществление в промышленном масштабе
связано с большими затруднениями:
а) стоимость растворителей для полиформальдегида весьма высока;
б) вследствие низкой эффективности растворителей необходимо
использовать очень разбавленные растворы полимера;
в) растворение сопровождается большими потерями полимера
в результате деструкции;
г) последующее отделение растворителя трудно осуществимо
из-за сильного взаимодействия с полимером.
Поэтому больший практический интерес представляют
газофазный и жидкофазный процессы ацетилирования, которые не связаны
с растворением полимера.
Технически возможно осуществить ацетилирование следующими
способами [68]:
1. Гетерогенная система (твердый полимер, жидкий агент этери-
фикации). Твердый полимер суспендируют в чистом уксусном
ангидриде, в котором растворен катализатор реакции. Для лучшего
смачивания полимера берется 10—20-кратный избыток ангидрида.
Суспензию быстро нагревают до температуры кипения реакционной
смеси A39 °С) при эффективном перемешивании. Пары ангидрида
конденсируются в обратном холодильнике и возвращаются в зону
реакции. В этом случае при условии тщательной очистки ангидрида
от кислоты может быть достигнут максимальный выход продукта
(порядка 95%).
Применение чистого уксусного ангидрида сопряжено с большими
затруднениями, так как его потери при регенерации весьма велики.
Поэтому обычно стремятся уменьшить его эффективную
концентрацию насколько это возможно. Исследования показали, что можно
брать полимер и ангидрид в соотношении, равном 1 : 1 и даже 1 : 0,5.
В этом случае для улучшения смачиваемости полимера используют
инертные разбавители. Поскольку полимеризация формальдегида
осуществляется в инертной жидкой среде, очевидно целесообразно
одно и то же соединение использовать и как полимеризационную
среду, и как разбавитель при ацетилировании. Тогда отпадают такие
операции, как промежуточное отделение полимера; кроме того,
можно добиться уменьшения содержания влаги в системе и
наибольшего набухания полимера.
218
2. Гетерогенная система (твердый полимер, газообразный агент
этерификации). Можно проводить этерификацию в кипящем слое,
исключив вообще применение жидкофазных разбавителей. В этом
случае парогазовая смесь азота (или другого инертного
газа-носителя), катализатора (амина) и ангидрида подается снизу на
распределительную решетку, куда непрерывно поступает
порошкообразный полимер.
Ацетилирование в жидкой фазе может быть осуществлено в
обычных автоклавах. Если ацетилирующий агент применяется в
газообразном состоянии, то можно использовать аппаратуру, работающую
по принципу псевдоожиженного слоя, или червячные устройства
различных типов.
Материал, из которого изготовляется аппаратура, должен быть
устойчив к действию паров уксусного ангидрида и уксусной кислоты.
Рекомендуется применять молибденовую сталь, эмали, стекло,
фторопласт и другие специальные некорродирующие материалы.
После ацетилирования полимер подвергается дополнительной
обработке: необходимо отделить реакционную среду, которая должна
быть регенерирована, и удалить нежелательные примеси из полимера,
в частности следы катализатора, чтобы предотвратить гидролиз
полиоксиметилендиацетата.
Ацетилированный полимер после отделения реакционной среды,
промывки и введения акцептора кислоты направляется на сушку.
Добавка акцептора кислоты (третичного ароматического амина или
амида) нужна, чтобы предотвратить ацидолитическое расщепление
полимера. Если сушка производится горячим воздухом, то
рекомендуется добавлять антиоксидант.
Для повышения эффективности операций по обработке полимера
желательно, чтобы он имел возможно меньшие размеры частиц
с максимально развитой удельной поверхностью. Анализ
полидисперсности показал, что максимум на дифференциальной кривой
распределения по размерам частиц зависит главным образом от степени
диспергирования катализатора в реакционной среде и эффективности
перемешивания. Для уменьшения размера частиц необходимо, чтобы
катализатор полностью растворялся в реакционной среде. Влияние
дисперсности частиц катализатора на свойства продукта
(полученного в уайт-спирите) показано ниже:
Стеарат каль- „, -
ция(диспер- Трибутиламин
сия) (раствор)
Средний размер частиц
полимера, мк 50 25
Удельная поверхность, м^/г . 7 20
Насыпная масса, г/см3 . . . 0,40 0,18
Удаление неблокированной части полимера можно осуществить
путем растворения полимера в среде, содержащей слабый основной
агент (для ускорения деполимеризации), или пропуская расплав
полимера через специальный экструдер, оборудованный отсосом
для улавливания летучих продуктов разложения.
219
Дополнительная стабилизация
Полностью блокированный полиоксиметилен, содержащий
акцептор муравьиной кислоты, вполне устойчив в инертной атмосфере
в расплавленном состоянии практически до 250 °С. Если навеску
такого полимера медленно нагревать в тигле при ограниченном
доступе воздуха, то при скорости нагрева ниже некоторой
критической величины (которая зависит от массы навески и геометрической
формы сосуда) можно получить расплав полимера, свойства которого
не изменяются в течение нескольких часов.
Если скорость нагрева превышает некоторую критическую
величину (для конкретных условий эксперимента), то температура начала
окислительной деструкции (около 120 °С) достигается раньше, чем
кислород успевает продиффундировать из образца, и начинается
автоускоренный процесс термоокислительной деструкции.
Для подбора наиболее эффективных стабилизирующих
композиций, включающих антиоксиданты и акцепторы формальдегида,
необходимо было выработать специальные критерии
термостабильности материала при переработке. Одним из таких критериев может
быть время от момента введения определенной навески полимера
в печь до момента разложения 1 или 0,5 вес. % материала (ткрит#).
Этот «показатель стабильности» позволяет рассчитать время
пребывания расплава в цилиндре литьевой машины как функцию
температуры:
t = yt4 C4)
t < ticpHT.
где т — время пребывания материала в цилиндре; т — объем
материала в цилиндре; g — объем впрыскиваемого материала; тц —
продолжительность одного цикла литья.
Общие требования, предъявляемые к ингредиентам, вводимым
в полимер, можно сформулировать следующим образом:
1) они должны хорошо совмещаться с полимером при плавлении;
2) не должны диффундировать из полимера (т. е. обладать низкой
летучестью);
3) не вызывать разложение полимера (не изменять окраску);
4) обладать низкой токсичностью;
5) не ухудшать реологические свойства расплава полимера.
Введение стабилизирующих добавок осуществляется обычными
в полимерной технологии способами. Вместе с этими добавками
вводят пигменты, наполнители, смазку и другие ингредиенты,
необходимые для получения композиции с заданными свойствами [88—90],
Из фенольных (не окрашивающих полимер) антиоксидантов
наиболее эффективным следует признать 4,4'-бутилиден-бис-C-ме-
тил-б^реиг-бутилфенол)» который применяется для стабилизации
полиформальдегида марки «дельрин», а также 2,2'-метилен-бис-D-ме-
тил-б-^тгрвт-бутилфенол), В качества акцептора кислоты (ингиби-
220
тора ацидолиза) в полимер вводится 0,1% дициандиамида.
Оптимальные концентрации добавок определяются обычно эмпирическим
путем. Избыток их в полимере приводит к ухудшению его физико-
механических свойств. Суммарная концентрация стабилизирующих
добавок в полиформальдегиде составляет обычно 1,5—2,0%.
Применение полиамидных смол и некоторых антиоксидантов
аминного типа для дополнительной стабилизации блокированного
полиформальдегида позволяет увеличить время удерживания
расплава полимера в цилиндре литьевой машины до 40 мин при
оптимальном температурном режиме переработки (около 205 °С). Все
же эта величина более чем в 2 раза устапает практически
допустимому времени пребывания в расплаве других термопластичных
полимеров. Поэтому переработка полиформальдегида (гомополимера)
сопряжена с некоторыми трудностями.
ВЛИЯНИЕ УСЛОВИЙ РЕАКЦИИ НА СВОЙСТВА
ПРОДУКТА
Один из этапов разработки технологии получения нового
полимера заключается в обосновании выбора характеристик стандартного
продукта. Для этого тщательно исследуются свойства полимера
и зависимость их от условий синтеза.
Рассмотрим сначала факторы, оказывающие воздействие на
свойства продуктов в процессе синтеза. Важнейшим условием
успешной переработки является однородность материала, в противном
случае было бы необходимо изменять технологию переработки
при переходе от одной партии продукта к другой. Неоднородность
материала обычно является результатом периодичности режима
синтеза (полимеризации или ацетилирования). В этом случае
достигнуть воспроизводимых результатов чрезвычайно трудно.
Непрерывный процесс полимеризации позволяет уменьшить
неоднородность по молекулярному весу. Однако для переработки
необходимы материалы с различным молекулярным весом.
Регулирование его может быть достигнуто несколькими путями. Во-первых,
за счет изменения технологического режима синтеза. Во-вторых,
путем механического смешения материала с различным
молекулярным весом. В этом случае низкомолекулярный полимер играет роль
пластификатора.
Другой способ регулирования (снижения) молекулярного веса
полиформальдегида [92, 93] заключается в расщеплении его
кислотными добавками в присутствии алкилирующих или ацетилирующих
агентов. Этот способ применим, по-видимому, к
высокомолекулярным образцам, содержащим незначительные количества
низкомолекулярных фракций.
Исследование влияния характера молекулярно-весового
распределения полиформальдегида на его реологические и
физико-механические свойства показало, что оптимальными свойствами
обладают образцы, для которых отношение MJMn близко к 2 [82—86].
221
Образцы с более широким МБР при одинаковых значениях вязкости
(измеренной в диметилформамиде) обладают более высокой
эффективной вязкостью расплава при больших скоростях сдвига. Однако
их механические свойства хуже.
Определение оптимальных значений среднего молекулярного веса
связано с исследованием реологических и механических свойств
различных образцов полимера. Так, например, относительно
низкомолекулярные образцы полиформальдегида обладают повышенной
текучестью, но довольно большой хрупкостью. Образцы с
молекулярным весом более 100 000 непригодны к переработке из-за
чрезмерно большой вязкости расплава.
Вопросам термической и термоокислительной стабильности в этой
главе было уделено достаточно места. Резюмируя сказанное выше,
можно сделать вывод, что существующие способы стабилизации
полиформальдегида позволяют получить материал с приемлемой
стабильностью в условиях переработки.
ЛИТЕРАТУРА
1. У ок ер Дж., Формальдегид, Госхимиздат, 1957, стр. 47.
2. Walker J. F., Am. Chem. Soc, 55, 2825 A935).
3. S p e n с e R., W і 1 d W., J. Chem. Soc, 506 A935).
4. Celanese Corp., пат. США 2714616, 1955 г.
5. Ciefer A., Kern W., Makromol. Chem., 74, 39 A964).
6. Goodrich B. F., пат. США 2460592, 1949 г.
7. Файдель Г. И., Вольфсон С. А., Зисман Д. О., авт. свид.
168669 A964), Бюлл. изобр. № 5, 1965.
8. Celanese Corp., пат. США 3000960, 1961 г.
9. Houilleres du Bassion du Nord, англ. пат. 888559, 1962 г.
10. Du Pont, пат. США 2848500, 1958 г.
11. Hall M. W., Piret E. L., Ind. Eng. Chem., 41, 1277 A949).
12. Hoechst F., англ. пат. 904089, 1962 г.
13. Walker J. F., пат. США 1871019, 1932 г.
14. Montekatini, англ. пат. 987457, 1965 г.
15. V. Е. В. Leuna Werke, англ. пат. 989801, 1965 г.
16. Du Pont, пат. США 2824051, 1959 г.
17. Du Pont, пат. США 2790755, 1957 г.
18. Sumitomo Chem. Co. Ltd., англ. пат. 874097, 1961 г.
19. Bayer F., англ. пат. 923460, 1963 г.
20. Ф а й д е л ь Г. И., Левшук В.Е.,Бек В. И., Баю ров В. П.,
Авт. свид. 133870, 1960 г., Бюлл. изобр. № 23, 1960.
21. Deutsche Gold, пат. США 3051755, 1962 г.
22. Файдель Г. И., Зисман Д. О., Мир с кий Л. И.,
Вольфсон С. А. и др., Авт. свид. 145237, 1961 г., Бюлл. изобр. № 3, 1961.
23. ІСІ, англ. пат. 955015, 1964 г.
24. V. Е. В. Leuna Werke, англ. пат. 953389, 1964 г.
25. Bayer F., фр. пат. 1361264, 1964 г.
26. Badishe Anilin und Soda Fabrik, пат. ФРГ 1137425, 1962 г.
27. Badishe Anilin und Soda Fabrik, пат. ФРГ 1139110, 1962 г.
28. Л е в и н А. Н., Гончаров Г. С, Р и в к и н Г. А., Пласт, массы,
№ 4, 46 A962).
29. Hoechst F., англ. пат. 904089, 1962 г.
30. Du Pont, пат. США 2780652, 1957 г.
31. Bayer F., англ. пат. 997561, 1965 г.
32. Societa Ital. Resin, англ. пат. 966895, 1964 г.
222
Societa Ital. Resin, англ. пат. 970214, 1964 г.
V. E. В. Leuna Werke, англ. пат. 980029, 1964 г.
Du Pont, пат. США 2943701, 1960 г.
Пат. ФРГ 1070611, 1960 г.
Пат. ФРГ 1104934, 1963 г.
Stamikarbon N. V., англ. пат. 980283, 1965 г.
McDonald R. W., Du Pont, пат. США 2768994, 1956 г.
Schveitzer С. Е., McDonald R. W., Punderson J. O.§-
J. Appl. Polymer Sei., 1, 158 A959).
Лужков Ю. М.и др., Пласт, массы, № 8, 60 A963).
Du Pont, пат. США 3017389, 1962 г.
Du Pont, пат. СЩА 2841570, 1958 г.
Knight А. С, англ. пат. 796862, 1956 г.
Koch Т. A., Lindvig Р. Е., J. Appl. Polymer Sei., 1, 170
A959).
Aries R., пат. США 3030338, 1962г.
Du Pont, пат. США 3002952, 1961 г.
Graseand N. R., Co.-, пат. США 2115480, 1963 г.
Deutsche Gold, фр. пат. 1271297, 1962 г.
Du Pont, англ. пат. 796862, 1958 г.
Deutsche Gold, пат. ФРГ 1139273, 1963 г.
Du Pont, пат. США 2734889, 1956 г.
Montekatini, фр. пат. 1318888, 1962 г.
V. Е. В. Leuna Werke, англ. пат. 921670, 1963 г.
Bayer F., англ. пат. 924464, 1963 г.
Bayer F., англ. пат. 943177, 1963 г.
Toyo Rayon Co. Ltd., пат. США 3140272, 1964 г.
Du Pont, пат. США 3017398, 1962 г.
Du Pont, англ. пат. 796863, 1958 г.
Stamicarbon, англ. пат. 879022, 1960 г.
Файдель Г. И., Шпичинецкая Л. С, Орлова Н. В.?
Пин Л. Д., Вольфсон С. А., Варданян М. С, Л е в-
шук М. Я., Авт. заявка 159983, 1963 г., Бюлл. изобр. № 2, 1964.
Spells К. Е., Trans. Inst. Chem. Eng., 33, 79 A955).
Bayer F., англ пат. 981376, 1965 г.
Англ. пат. 939363, 1963 г.
Фр. пат. 1291999, 1962 г.
Сотр. de Saint Gobain, фр. пат. 1285909, 1962 г.
Mejzlik Т.* Janeckova Z., Makromol. Chem., 82, 238 A965),
Du Pont, англ. пат. 770717, 1957.
Thummler W., Plast, und Kautschuk, 10, 582 A965).
Bayer F., англ. пат. 864403, 1961 г.
Kosinska W., Fejgin J., Zakrgewski Z., Tworzywa ^*
Guma, 5, 178 A960).
Mejzlik J., Lesna M., Osecky P., Srackova I., Sagov-
s k a E., Makromol. Chem., 82, 253 A965).
Kern W., Cherdron H., Makromol. Chem., 40, 101 A960).
Du Pont, пат. США 2871220, 1961 г.
E n і k о 1 о р у a n N. S., J. Polymer Sei., 58, 1301 A962).
Штаудингер Г., Высокомолекулярные органические соединения,
ОНТИ, Химтеорет, 1932.
Bu ech F., Physical Properties of Polymers, Acad. Press., p. 4, 1962..
. Du Pont, пат. США, 3050500, 1962 г.
. Montecatini, фр. пат. 1364410, 1964 г.
. Du Pont, англ. пат. 912033, 1962 г.
. Du Pont, пат. США ЗЮ2Ю6, 1963 г.
. Т і g h є В. J., О п у о n P. F., Chem. a. Ind., 27, 1217 A965).
.Mayer J., Makromol. Chem., 86, 253 A965).
. G г о h n H., Friedrich H., Сообщение на симпозиуме по макромоле-
кулярной химии, Прага, 1965.
223
Dick R., Benott H., Сообщение на симпозиуме по макромолекуляр-
ной химии, Прага, 1965.
БельговскийИ. М., ЕниколопянН. С, СахоненкоЛ. С,
Высокомол. соед., 4, 1197 A962).
Д удина Л. А., Ениколопян Н. С, Высокомол. соед., 5, 861
A963).
Дудина Л. А., Кармилова Л. В., Тряпицына Е. Н.,
Ениколопян Н. С, Сообщение на симпозиуме по макромоле-
кулярной химии, Прага, 1965.
Коварская Б. М., Нейман М. Б., Гурьянова В. В.,
Алишоев В. Р., Высокомол. соед., 4, 1887 A962).
Mejzlik J., Berger J., Chem. Prum., 12, 461 A962).
Шагинян А. А., Ениколопян Н. С, Высокомол. соед., 7,
1866 A965).
Англ. пат. 917016, 1964 г.
Grace and Co., англ. пат. 960047, 1964 rt
ГЛАВА VIII
ТЕХНОЛОГИЯ ПРОИЗВОДСТВА СОПОЛИМЕРОВ
ТРИОКСАНА (И ФОРМАЛЬДЕГИДА)
Введение в молекулярную цепь полиоксиметилена звеньев
инородной структуры, которые препятствуют цепной деполимеризации,
позволяет значительно повысить термостабильность
полиформальдегида. Поэтому в последние годы в центре внимания оказались
работы по синтезу сополимеров на основе формальдегида или триок-
сана. Специфика задачи заключается в том, что эти сополимеры
должны содержать минимально возможное количество сомономера,
равномерно распределенного по молекулярной цепи, причем
концевые группы макромолекул должны обязательно включать звенья
сомономера. При увеличении доли сомономера возрастает
стабилизующий эффект, но одновременно из-за нарушения однородности
полиоксиметиленовой структуры ухудшаются наиболее ценные
ТАБЛИЦА 33
Некоторые технологические свойства формальдегида и триоксана
Свойства
Формальдегид
Триоксан
Устойчивость при
хранении
Токсичность
Растворимость в апро-
тонных неполярных
растворителях
Устойчивость при
хранении в виде раствора
Склонность к
спонтанной полимеризации
Тепловой эффект
полимеризации
Способы очистки
Не устойчив
Токсичен (допустимая
концентрация в
воздухе не более 0,001%)
Не более 2% (при
комнатной температуре)
Не устойчив
Выражена
исключительно сильно при T<C.Tnv
Около 15 ккалъ/молъ
Обычные не пригодны
Устойчив в нейтральной
и щелочных средах в
твердом, жидком и
газообразном состоянии
Не токсичен
Хорошо растворим в
большинстве из них
Устойчив
Проявляется только у
очищенных образцов при
плавлении и
кристаллизации из расплава
Около 2 ккал/молъ
Обычные
(перекристаллизация, экстракция,
ректификация, адсорбция)
15 Зак з 11 я 5.
225
физико-химические свойства материала — кристалличность,
жесткость, твердость, понижается температура плавления.
Принципиально возможно вести сополимеризацию как на основе
формальдегида, так и на основе триоксана. Последний обладает
рядом технологических преимуществ (табл. 33), однако имеются
некоторые соображения в пользу формальдегида (например, при
использовании формальдегида отпадает надобность в получении
промежуточного продукта). В этой главе описаны принципиальные схемы
синтеза сополимеров на основе обоих мономеров.
Основные технические задачи, решение которых необходимо
для разработки технологии синтеза сополимеров триоксана, можно
сформулировать следующим образом:
1) способы синтеза и очистки триоксана;
2) выбор подходящего сомономера;
3) определение необходимой для получения продуктов с заданным
молекулярным весом степени чистоты мономеров;
4) выяснение зависимости распределения блоков сомономера
по молекулярной цепи от условий реакции;
5) разработка способов удаления полуацетальных концевых
блоков в макромолекулах сополимера (нестабильной части).
СИНТЕЗ И ОЧИСТКА ТРИОКСАНА
В основу способа получения триоксана можно положить одну
из двух известных реакций его образования:
1. Полимеризация формальдегида в водном растворе в
присутствии сильных кислот (катализатор), когда происходит равновесное
превращение линейного тримера оксиметиленгликоля в триоксан:
Н(СН2ОKОН ^± (СН2ОK + НОН
2. Ацидолитическое расщепление полиоксиметиленов. В этом
случае протекает неравновесная реакция, причем триоксан
образуется как один из продуктов деструкции. Выход триоксана зависит
от концентрации и природы кислоты.
Рассмотрим процесс образования триоксана из водного раствора
формальдегида. Равновесная концентрация триоксана в этом
растворе [55, 56] не превышает 5%. Этот факт легко объясняется
термодинамическими причинами. Тепловой эффект реакции образования
триоксана в растворе равен напряженности цикла (около
0,6 ккал/молъ), т. е. константа равновесия должна слабо зависеть от
температуры. Показано, что при 180 °С равновесие в системе
безводный триоксан — формальдегид полностью сдвинуто в сторону
образования формальдегида [1, 12]. Зависимость равновесной
концентрации триоксана от концентрации формальдегида и величин констант
равновесия от температуры приводятся в работе [55].
Условия равновесия диктуют необходимость вывода триоксана
из зоны реакции. Эту операцию можно осуществить путем
ректификации реакционной смеси (триоксан образует азеотропную смесь с во-
226
дой, содержащую 70% триоксана и кипящую при 91 °С, т. е. на
10 °С ниже, чем формалин). В этом случае процесс синтеза триоксана
проводится на ректификационной колонне, причем катализатор
(серная кислота) подается в куб. Подобный процесс [7] широко
используется для получения триоксана в лабораторных условиях. Этот
же метод был положен в основу промышленного производства
триоксана.
Реактор одновременно является кубом ректификационной
колонны. Разделительная способность колонны и скорость отбора
дистиллята (режим ректификации) определяют состав отбираемого
дистиллята, в котором содержится триоксан, формальдегид, вода,
а также примеси метанола, метилаля, муравьиной кислоты и других
компонентов.
Повышение скорости отбора дистиллята за счет более
эффективного нагрева или уменьшения флегмового числа приводит к снижению
содержания триоксана в дистилляте, однако общая
производительность аппаратуры по триоксану увеличивается. Поэтому при
расчетах оптимального режима обычно приходят к тому, что необходимо
отбирать дистиллят с меньшим, чем азеотропное, содержанием
триоксана *.
Материальный баланс для реактора, работающего в непрерывном
режиме, можно записать следующим образом:
^~ = *' [сф] [ск]-к" [ст] [cK]-w[cT] A)
где [ст] — стационарная концентрация триоксана; [сф] —
стационарная концентрация формальдегида; к' — константа скорости прямой
реакции образования триоксана; к" — константа скорости обратной
реакции (распада триоксана на формальдегид); [сК] — концентрация
катализатора; w — скорость отбора реакционной смеси.
Отсюда концентрация триоксана равна:
Скорость отбора триоксана составляет:
dQ, . . *'[Сф1
Введем величину [сф]0 — исходная концентрация формальдегида
в растворе, поступающем в реактор. Тогда
[*ф] = ['ФЪ-[*,] D)
* Скорость образования триоксана можно рассчитать, зная величины
константы равновесия [55] и константы скорости гидролиза [56] при температуре
синтеза. Так, при 100 °С К = 9,2-10-5, кГ = 3,493-10-4.
15* 227
Подставим это выражение в уравнение C):
dQT *' [сф]0 К]
E)
dt (k' \-k")[cK]+w
Скорость отвода непрореагировавшего формальдегида равна:
и ь кк —j— к і і с j. і
Проведение ректификации технически сложно из-за выделения
значительных количеств мономерного формальдегида в верхней
части колонны, что приводит к образованию полимера на
охлажденных поверхностях, ухудшению теплообмена и увеличению
гидродинамического сопротивления в колонне. Предлагается [11] вести
ректификацию при пониженном давлении с тем, чтобы уменьшить
вероятность протекания побочных реакций.
Выделение триоксана из дистиллята может быть осуществлено
путем кристаллизации или экстракцией органическими
растворителями, не смешивающимися с водой.
В другом предложенном варианте удаление триоксана
непосредственно из зоны реакции осуществляется путем экстракции его
подходящим растворителем. Формалин с растворенной в нем кислотой
циркулирует через реактор и экстракционную колонну.
Концентрация СН2О поддерживается постоянной. К недостаткам этой схемы
следует отнести низкую концентрацию триоксана в реакционной
смеси на входе в экстракционную колонну. Поэтому после отделения
экстракта последний поступает на ректификацию.
Применение серной кислоты в качестве катализатора синтеза
триоксана заставляет использовать для изготовления аппаратуры
дорогие сорта легированных сталей, специальные кислотоупорные
покрытия и т. п. Поэтому предлагается [9] пропускать формалин
через насадочную колонну, заполненную ионообменной смолой
(катионитом), играющей роль катализатора.
Определенный интерес представляет получение триоксана путем
ацидолитического расщепления полиоксиметиленов при
повышенной температуре. В этом случае триоксан нужно быстро выводить
из зоны контакта с кислотным агентом, чтобы предотвратить его
дальнейшее расщепление. В патентной литературе [8] этот способ
был описан еще в 1942 г., однако он не находил применения. Из
результатов исследования механизма ацидолитического
расщепления полиоксиметилена (параформальдегида), проведенного Керном
с сотр. [10], можно сделать вывод о том, что выход циклических
олигомеров при ацидолизе зависит от природы кислоты, ее
концентрации, температуры и времени обработки (табл. 34). По-видимому,
растворение низкомрлекулярного полиоксиметилена
(предшествующее деполимеризации) будет способствовать увеличению выхода
циклических продуктов, так как можно предполагать, что
растворенный полиоксиметилен термодинамически не устойчив по отношению
к циклическим олигомерам.
228
Анализ способа синтеза триоксана, описанного в работе [11],
дает основания предполагать, что повышенная концентрация
триоксана достигается за счет неравновесного ацидолитического разло-
ТАБЛИЦА 34
Термическое разложение параформальдегида (Мп
при 160 °С в течение 20 мин
Кислота
Трихлоруксусная
и-Толуолсульфоновая
Концентрация
кислоты
(моль/моль
СН2О)-102
5,1
1,7
5,1
Выход
триоксана
%
10,5
3,5
8,0
= 4500)
Выход
тетраокси-
метилена
%
0,04
0,09
0,15
жения параформальдегида, растворенного в избытке серной кислоты
(рис. 68). Отделение триоксана в вакууме способствует уменьшению
продолжительности контакта продукта с катализатором. Этот
процесс включает две последовательные мономолекулярные реакции:
А _?..> В Jp-> С
где В1—-целевой продукт. Необходимо, увеличивая скорость
реакции I, максимально замедлить реакцию П. Применение слишком
глубокого вакуума при дистилляции триоксана, по-видимому,
нежелательно, так как это приведет к выделению параформальдегида
в гетерогенную фазу и уменьшению выхода триоксана.
Способ последующей обработки «сырого» триоксана определяется
выбранной технологической схемой полимеризации.
Кристаллизация триоксана непосредственно из водной фазы.
Этот метод выделения триоксана осуществляется с помощью обычна
используемых в химической технологии приемов [13—15]. Посла
охлаждения до 0—5 °С и отделения твердой фазы маточник
представляет собой 10%-ный раствор триоксана в формалине. Если
дистиллят содержит большое количество растворенного формальдегида,
то при кристаллизации триоксана будет образовываться некоторое
количество низкомолекулярных линейных полиоксиметиленгликолеи,
что крайне нежелательно. Поэтому необходимо или проводить
повторную ректификацию азеотропной смеси, разбавляя ее водой (чтобы
снизить концентрацию мономерного формальдегида), или
экстрагировать триоксан из разбавленного раствора. Кристаллы триоксана
на фильтре промывают 2—3 раза ледяной водой для отделения
примесей муравьиной кислоты, метанола и формальдегида и затем
высушивают до содержания влаги около 1%. В таком виде триоксан
вполне транспортабелен и может храниться длительное время.
Экстракция триоксана из дистиллята. Для экстракции триоксана
чаще всего применяют хлористый метилен, бензол, толуол, 0-кси-
лол, хлористый бутил и др. [16]. Найдено, что для наиболее полного
229
зо
25
20
15
1
1
1
г
—-а,
ч
(98%) извлечения триоксана из дистиллята с помощью бензола
необходима трехступенчатая противоточная экстракция, в случае
применения толуола — пятиступенчатая [17, 18].
Выделение триоксана из экстракта может быть осуществлено
двумя способами: кристаллизация (в этом случае в качестве
растворителя рекомендуется толуол, так как при низких температурах
растворимость триоксана в нем ниже, чем в бензоле) и
ректификация (в этом случае предпочтительнее бензол, кипящий при более
низкой температуре).
Обезвоживание триоксана можно осуществить обычными
методами: ректификацией расплавленного триоксана или раствора с
отбором в качестве дистиллята азео-
тропной смеси триоксан — вода или
растворитель — вода, а также с
помощью различных водоотнимающих
веществ. Поскольку триоксан
неустойчив в кислой среде, эти
операции обычно проводят в присутствии
основных агентов, например твердого
едкого кали. Содержание воды в три-
оксане определяют по методу Фишера
или хроматографически [19].
Константы передачи цепи на
15 20 25 30 примеси при полимеризации три-
oe^/om оксана выше, чем при катионной
полимеризации формальдегида.
Поэтому для достижения достаточно
больших значений средней степени
полимеризации политриоксана нужно
использовать мономер более
высокой степени чистоты. В лабораторных условиях применяют
высокоэффективные химические методы очистки триоксана от
влаги, например реакцию антраценового зеркала, кипячение
над металлическим натрием, обработку триэтилалюминием, изоциа-
натом. Это позволяет получать полимеры со средней степенью
полимеризации от 4000 до 20 000 в пересчете на СН2О.
Возможность применения подобных методов в промышленном
масштабе весьма ограничена, однако, судя по имеющимся сведениям,
с помощью ректификации, очистки на сорбентах и других
технических методов очистки можно достигнуть удовлетворительных
результатов.
ГОМОПОЛИМЕРИЗАЦИЯ ТРИОКСАНА
Исследование катионной сополимеризации триоксана,
протекающей по механизму передачи цепи с разрывом, представляет
большие трудности [2—6]. На практике большое внимание было уделено
изучению гомополимеризации триоксана, поскольку сополимериза-
цию можно проводить как двухступенчатый процесс.
230
5 10
Количество
загруженного CH2O
Рис. 68. Зависимость выхода
триоксана от концентрации H2SO4
[И].
Полимеризация в твердой фазе. Можно считать, что этот процесс
протекает в диффузионной области, так как подвижность молекул
примеси в решетке кристалла ограничена. С другой стороны, в ходе
реакции по мере образования полимера кристаллы мономера
разрушаются, и это приводит к уменьшению длины образующихся
макромолекулярных цепей и среднего молекулярного веса полимера.
Зависимость среднего молекулярного веса от степени конверсии
носит сложный характер и определяется размером и дефектностью
кристаллов (см. гл. III).
В лабораторных условиях путем твердофазной полимеризации
удалось получить образцы полимера с молекулярным весом более
100 000. Однако химическое инициирование полимеризации
кристаллического триоксана представляет значительные трудности и
приводит к получению неоднородного продукта.
. Радиационная полимеризация твердого триоксана с этой точки
зрения представляет больший интерес. Сообщалось [26] о получении
таким путем полимера с очень узким молекулярно-весовым
распределением (MJMn = 1,35) и высокой степенью кристалличности. Все
же метод твердофазной радиационной полимеризации триоксана
до настоящего времени не применяется в промышленном масштабе.
Одна из причин заключается в сложности регулирования режима
сополимеризации, хотя принципиально показана возможность
получения сополимеров триоксана и в твердой фазе [20].
В технологическом отношении удобнее проводить процесс
полимеризации в суспензии кристаллов триоксана в алифатических
углеводородах или в другой среде, в которой растворяется
катализатор. Таким путем из триоксана, содержащего 0,01% влаги, можно
получить продукт с вязкостью (In т)отнJe) в 0,5%-ном растворе
диметилформамида при 150° С порядка 0,75—1,00, что соответствует
среднечисловому молекулярному весу приблизительно 80-Ю3 —
100 -103. В дальнейшем было показано, что полученный продукт
отличается большой неоднородностью и плохо поддается ацетили-
рованию. При обработке избытком уксусного ангидрида в
гомогенной среде (раствор полимера в метилендиацетате, катализатор —
пиридин) наблюдается сильное разрушение полимера и уменьшение
его вязкости; степень превращения концевых групп невелика.
Причины этого явления могут быть найдены только после
определения молекулярно-весового распределения твердофазного полимера
и изучения его химической структуры.
Известен еще один вариант проведения твердофазной
полимеризации триоксана [21]. В концентрированный раствор очищенного
триоксана при 50—75 °С вводят катализатор при интенсивном
перемешивании. После достижения 5—25%-ной степени конверсии
температуру резко понижают, так что часть непрореагировавшего
триоксана кристаллизуется и полимеризуется уже в твердой фазе.
Средний молекулярный вес продукта увеличивается, однако
полимер обладает значительной неоднородностью, что затрудняет
дальнейшую переработку и ухудшает его механические свойства.
231
Подробные исследования процесса полимеризации триоксана
с переменрым температурным режимом проведены Баккареддом,
Буттом и др. [22, 23]. Чтобы получить однородные неагрегированные
кристаллы триоксана (морфология которых, по-видимому, в
значительной степени определяет структуру получаемого полимера),
они исследовали влияние на размеры кристаллов условий
кристаллизации мономера в ходе процесса. Задача сводилась к тому, чтобы
скорость образования и роста зародышей кристаллизации не
превышала скорость полимеризации. Варьируемые параметры процесса:
скорость перемешивания, вязкость растворителя, скорость изменения
температуры и концентрация катализатора. Во всех случаях
получалась смесь полимеров, которую путем грубого фракционирования
можно было разделить на порошкообразную (по-видимому,
получаемую полимеризацией раствора) и волокнообразную (продукт
полимеризации кристаллического триоксана) фракции. Некоторые
свойства этих фракций приведены ниже:
Волокнооб-
Порошкообразная разная
r|lg в л-хлорфеноле при 60 °С ... 1,1—2,0 0,7—1,1
Температура плавления, °С .... 186—187 178—180
Степень кристалличности 97—98 90
Растворимость в уксусном ангидриде Нерастворим Растворим
Гомогенная полимеризация *. Анализ результатов поисковых
лабораторных работ показал, что целесообразно проводить
полимеризацию триоксана в растворе при возможно более низких
температурах и максимальной концентрации мономера (см. гл. III). Для
проведения процесса в растворе требуется наиболее простое
аппаратурное оформление. Это может быть, например, автоклав с
мешалкой (для периодической полимеризации). Для проведения процесса
в расплаве необходимо более сложное оборудование, поскольку
уже при 20—25%-ной степени превращения полимер образует
твердую нетранспортабельную массу. Предложены конструкции не-
прерывнодействующего полимеризатора типа роторного смесителя
Ї24], экструдера или транспортера [25].
Периодический процесс полимеризации в растворе осуществляется
следующим образом: тщательно очищенный и обезвоженный раствор
триоксана в бензоле или циклогексане концентрации 6—7 моль/л
при 50° С загружают в токе инертного газа в реактор,
представляющий собой герметичный автоклав с мешалкой и рубашкой.
Катализатор (эфират трехфтористого бора) вводят в виде раствора в том
же растворителе, что и триоксан. В качестве регуляторов роста
цепи используют спирт или другие агенты, разрушающие активные
центры. Чтобы уменьшить деполимеризацию полимера во время
реакции, можно создать небольшое избыточное давление в реакторе.
* При этом понимается, что мономер находится в гомогенной системе
(жидкой или газообразной), поскольку полиформальдегид всегда образует
твердую фазу.
232
Степень полимеризации образующегося продукта вычисляется
по формуле:
[*,] 1+jfku_A_a)Vfcp] (
LckJ
где [ст] — концентрация триоксана; [ск] — концентрация
катализатора; a — глубина превращения; [х]0 — начальная концентрация
агента передачи цепи в системе.
Регулирование молекулярного веса полимера в ходе
полимеризации можно осуществить, варьируя выход полимера или
концентрацию агента передачи цепи. Второй способ наиболее приемлем в
технологическом отношении, так как зависимость DPn = / (a) может
иметь весьма сложный характер *.
При проведении процесса полимеризации периодическим
способом наблюдается появление зависимости МВР от степени
превращения [29]. При переходе к непрерывному режиму полимеризации
молекулярный вес полимера становится функцией только
концентрации мономера и агента передачи цепи (и его эффективности) и
перестает зависеть от выхода продукта. МВР, как показано в гл. VI,
вследствие протекания реакции передачи цепи с разрывом должно
приближаться к наиболее вероятному (MJMn — 2).
Расплавленный триоксан или его концентрированный раствор
можно транспортировать по нагретым трубопроводам и хранить в
обогреваемых емкостях в атмосфере инертного газа. Чтобы
предотвратить спонтанную полимеризацию при охлаждении и
кристаллизации триоксана, рекомендуется добавлять к расплаву небольшое
количество ароматического амина, который взаимодействует с
активными центрами полимеризации.
Тепловой эффект полимеризации триоксана намного ниже, чем
формальдегида. Это облегчает отвод тепла и поддержание
изотермического режима при полимеризации. Полимеризационная система
обладает значительным тиксотропним эффектом, поэтому
реакционная смесь должна перемешиваться весьма интенсивно.
Полученный продукт не должен содержать остатков
катализатора, который вызывает ацидолитическое расщепление полимера.
Катализатор обычно нейтрализуют введением основного агента —
аммиака или подходящего амина.
СОПОЛИМЕРИЗАЦИЯ ТРИОКСАНА
Принципиальная технологическая схема синтеза сополимера
триоксана показана на рис. 69. В качестве сомономеров триоксана можно
использовать окись этилена, диоксолан, 1,3-диоксан, диоксепан,
* Если в системе имеются примеси агента передачи цепи с отношением
kn/kv < 1, то на кривой зависимости DPn от а есть максимум, после
достижения которого DPn будет уменьшаться [22, 41].
233
эпихлоргидрин и другие соединения, содержащие
углерод-углеродные и эфирные связи, способные рваться под действием
катализаторов катионного типа. Сомономер должен обладать высокой
реакционной способностью, растворяться в реакционной среде и
получаться доступным методом. С этой точки зрения окись этилена
и диоксолан оказываются наиболее перспективными сомономерами
концентрированный
формалин
Катализатор
Вода
-13
Готовая
продукция
Рис. 69. Принципиальная схема синтеза сополимера триоксана
с диоксоланом:
1 — емкость концентрированного формалина; 2 — реактор синтеза
триоксана; 3,5 — ректификационные колонны; 4 — кристаллизатор; 6 —
приемник очищенного раствора триоксана; 7 — полимеризатор; 8 — мутильник;
9, 11 — центрифуги; 10 — автоклав для термообработки сополимера;
12 — сушилка; 13 — смеситель; 14 — экструдер; 15 — гранулятор.
при сополимеризации с триоксаном. Первый — в силу своей
доступности, второй — благодаря хорошей растворимости и легкости
протекания передачи цепи с разрывом.
Методика проведения процесса сополимеризации существенно
не отличается от описанной выше методики проведения гомополи-
меризации в растворе. Правда, использование окиси этилена связано
с применением избыточного давления A—5 атм).
Из общих соображений следует, что с увеличением доли
сомономера в продукте уменьшается степень кристалличности, а это
в свою очередь приводит к ухудшению механических свойств
полимера. Чтобы в возможно большей степени сохранить высокую
упорядоченность структуры макромолекул полиформальдегида,
очевидно, следует стремиться к уменьшению доли сополимера. С другой
стороны, с возрастанием доли стабильного сомономера в
макромолекулах увеличивается стабильность сополимера.
234
Влияние условий реакции на структуру
и состав сополимера
Как было показано в гл. V, при сополимеризации триоксана
большое значение имеет реакция передачи цепи с разрывом, которая
в конечном счете приводит к перераспределению блоков сомономеров
по цепи.
Термически стабильный сополимер обычно представляет собой
остаток после термообработки в вакууме при температуре выше
точки плавления сополимера B00—220 °С). При термообработке
деполимеризуются концевые сегменты макромолекулы сополимера.
В основу количественного расчета доли концевых нестабильных
блоков положено представление о наиболее вероятном распределении
различных звеньев по цепи [27]. Зависимость доли неблокированной
части в сополимере от количества стабильных звеньев в цепи
выражается уравнением [28]:
=JM]oa_
DPn [x]
где у — весовая доля стабильной части в сополимере; [М]о —
начальная концентрация мономера; а — глубина превращения; DPn—
средняя степень полимеризации; [х] — общая концентрация
стабильных звеньев (количество сомономера).
В работе [30] подобная проблема рассматривается с позиций
классических представлений о механизме сополимеризации при
допущении, что один из сомономеров образует стабильные звенья
в макромолекулярной цепи.
Подробные результаты исследования химической структуры
сополимеров пока не опубликованы. Известно, что при
сополимеризации триоксана с окисью этилена [31, 32] (реакция проводилась
в расплаве при 65—96° С в течение 40 мин, инициатор BF3, выход 90%)
в зависимости от доли введенного сомономера образовывались
различные количества моно-, ди- и триоксиэтиленовых звеньев:
. Количество окиси этилена Количество звеньев, %
% к триоксану моно. ди. три_
0,5 92 7 1
2,0 77 19 4
6,0 57 29 14
Исследование сополимеризации триоксана с диоксоланом
показало, что на характер распределения звеньев диоксолана
оказывает большое влияние длина первоначально образующихся цепочек
сополимера, обогащенных диоксоланом, которая в свою очередь
зависит от степени очистки реагентов *.
* Данные о кинетике расхода диоксолана и триоксана в ходе реакции
см. на рис. 52.
235
Было показано, что если концентрация сомономера (диоксолана)
не превышает некоторую критическую величину, то закономерности
изменения вязкости полимера со степенью превращения остаются
такими же, как в случае гомополимеризации триоксана.
Уменьшение молекулярного веса после достижения
определенной степени превращения весьма характерно для процесса сополи-
меризации триоксана. Было высказано несколько гипотез для
объяснения наблюдаемого эффекта. Одно из объяснений формы кривых
Мп = f (а) основано на предположении, что в реакционной системе
находятся примеси с относительной константой скорости передачи
цепи меньшей, чем единица. Отсутствие гибели активных центров
полимеризации приводит к тому, что они взаимодействуют с
примесями и вследствие реакции передачи цепи с разрывом уменьшается
средняя степень полимеризации.
Знание кинетики изменения молекулярного веса и доли
стабильного полимера как функции степени превращения позволяет в этом
случае рассчитать оптимальное время проведения процесса.
Как уже отмечалось выше, реакция передачи цепи с разрывом
должна привести к наиболее вероятному молекулярно-весовому
распределению. Это экспериментально подтверждено [32] при
изучении МБР сополимеров триоксана с окисью этилена, содержащих
менее 5% углерод —- углеродных связей. Для вискозиметрических
определений использовали га-октафторпентанол-і (температура
кипения 142 °С, растворения —НО °С, константа диссоциации равна
4,5 «102). Этот растворитель был выбран благодаря низкой
кислотности; полимер не разлагался в нем в течение 12 ч при 110° С. Средне-
весовой молекулярный вес полученного сополимера определяли
методом светорассеяния, среднечисловой — с помощью осмометрии.
Характеристики некоторых образцов сополимера приведены ниже *:
Образец
№ 1 № 2 № 3 № 4
Индекс расплава при 190 °С *,
г/Ю мин 28,0 8,1 2,6 1,0
т|пр> (в гс-хлорфеноле при 60 °С,
0,01 г/дл) ** 1,03 1,33 1,71 1,99
[т]] (в октафторпёнтаноле при
110°С> 0,90 1,13 1,42 1,85
Мю.10-з 62 72 96 129
Мп • 10-3 34 40 44 56
М^. 1,8 1,8 2,2 2,3
Мп
* По методике ASTM D 1238-62T.
** В качестве антиоксиданта изпользовали а-пинен B вес. % к
растворителю).
* Образцы были получены путем сополимеризации триоксана с окисью
этилена в расплаве. Конверсия более 90%. Нестабильная часть удалялась путем
термообработки [см. фр. пат. 1253553 A961)].
236
Вычисление констант в уравнении Хувинка позволило получить
следующие соотношения между приведенной вязкостью (т)отн 1с)
в тг-хлорфеноле, характеристической вязкостью {х\\ в октофтор-
пентаноле и Mw\
4 (9)
[1I = 1,33.10-43/0,810 A0)
Индекс расплава і и Mw связаны соотношением:
» = 1,30- 1018Л?-3»55 (И)
Обработка сополимера
Образующийся в результате сополимеризации продукт
представляет собой смесь макромолекул по крайней мере трех типов.
1) молекулы блоксополимера, содержащие концевые стабильные
группы (например, оксиэтоксигруппы при сополимеризации с окисью
этилена);
2) молекулы блоксополимера с полуацетальными концевыми
группами;
3) молекулы гомополимера.
О том, что молекулы гомополимера триоксана находятся в виде
коротких цепей, косвенно свидетельствует некоторое увеличение
среднего молекулярного веса после термообработки полимера, во
время которой разрушается его нестабильная часть. Если бы все
молекулы имели одинаковый состав, то разложение некоторой
части полимера при термообработке привело бы к уменьшению
среднего молекулярного веса.
По результатам фракционирования и анализа полимеризацион-
ной среды после проведения полимеризации был сделан вывод об
отсутствии фракций сополимера, обогащенных звеньями сомономера.
Благодаря особенностям механизма сополимеризации триоксана,
на которые указывалось в гл. V, практически весь вводимый в
реакционную зону сомономер вступает в реакцию.
Полуацетальные концевые сегменты макромолекул могут
инициировать разложение при нагревании. При этом цепная
деполимеризация прекращается как только концевой группой становится
оксиэтоксигруппа (ОСН2СН2ОН) или ей подобная.
Вместо термической обработки можно использовать
дополнительную модификацию концевых полуацетальных групп путем этерифи-
кации или обработки изоцианатами [50]. В этом случае можно
избежать потерь полимера при термообработке, однако сохраняются
все технологические трудности, о которых говорилось выше.
Проблема удаления нестабильной части (полуацетальных
концевых блоков) в сополимере аналогична подобной проблеме,
обсуждавшейся в разделе, посвященном дополнительной стабилизации аце-
тилированного гомополимера. Однако важное отличие сополимеров,
237
содержащих карбоцепные звенья, заключается в том, что они
(за исключением концевых полуацетальных блоков) вполне устойчивы
к действию щелочных агентов при нагревании. Некоторая доля
концевых полуацетальных блоков разлагается относительно легко ужа
при обработке кипящей водой, содержащей небольшое количество
основного агента. Можно полагать, что эти блоки расположены
во внешней части кристаллических образований и уязвимы при
нуклеофильной атаке. Для деполимеризации концевых
полуацетальных блоков, находящихся во внутренней области кристаллитов,
методы гетерогенной обработки являются малоэффективными. Эта
часть составляет от 3 до 5% массы сополимера и может быть деполи-
меризована при растворении или плавлении продукта. Проведение
этой операции значительно облегчается тем, что сополимер более
устойчив к действию различных химических реагентов, чем гомополи-
мер. Предлагается вести термообработку в водной или спиртовой
среде при повышенном давлении и температуре, близкой к
температуре размягчения материала (около 140—150° С).
Нужный pH среды достигается введением органических или
неорганических оснований [52, 53]. В этом случае за счет
разрыхления кристаллической структуры полимера концевые полуацетальные
блоки становятся доступными для действия основного агента. Таким
путем можно добиться увеличения доли стабильной части до 97—
99%. Вместо спиртов можно применять другие агенты, например
диметилформамид, в которых полимер набухает.
Исследование физико-химических свойств сополимера триоксана
с диоксоланом показало, что техническую ценность представляют
образцы, содержащие 2—4 мол. % диоксолана. Выход стабильного
продукта в этом случае составляет 85—90%.
Более высокого выхода стабильной части можно достигнуть,
используя при сополимеризации агенты передачи цепи, образующие
два термостабильных осколка (линейные ацетали, эфиры,
ангидриды [54]). Но, как известно, введение агентов передачи цепи
приводит к снижению молекулярного веса полимера, поэтому
возможность использования этого метода находится в прямой зависимости
от степени очистки триоксана.
Термообработка сополимера может быть осуществлена в
автоклаве. Для поглощения образующегося мономерного формальдегида
обычно применяют специальные добавки (например, аммиак).
При правильном выборе режима термообработки дисперсность
продукта не изменяется. При перегреве происходит слипание частиц
или оплавление. Последующие стадии обработки сополимера
практически не отличаются от аналогичных стадий технологической
схемы синтеза гомополимера. Порошкообразный продукт подвергают
промывке, сушке и многократной гомогенизации. В него вводят
стабилизаторы, пигменты, наполнители, после чего он поступает на
экструзию и грануляцию. Удаление нестабильной части (около 5%)
производят на специальных экструдерах с вакуум-отсосом и
отношением длины червяка к диаметру (LID) более 25.
238
зо
s.
Ю
J
А
1
і h
1
21 1
V
'3
20
ВО
80
Стабилизация сополимера
После удаления неблокированной части сополимеры триоксана
с диоксоланом термически вполне устойчивы при температуре до
270° С. Стойкость к термоокислительной деструкции сложным
образом зависит от давления кислорода, концентрации и распределения
стабильных блоков в молекулах сополимера (т. е. от состава и
структуры сополимера). Были получены образцы, которые после
добавки стабилизатора выдерживали нагревание на воздухе при
220° С в течение 3—4 ч. При этом практически не наблюдалось
уменьшения молекулярного веса и не происходила деполимеризация.
Сополимер мало устойчив к
действию кислот, хотя несколько пре-
восходит в этом отношении ацети-
лированный гомополимер.
По-видимому, это объясняется тем, что
разрыв цепи по «закону случая» не
приводит к полной деполимеризации
полимерной цепи сополимера.
Сополимеры значительно пре-
восходят диацетаты гомополимера
по устойчивости к щелочным
агентам. После пребывания в 50%-ном
растворе щелочи при 140—150 °С
в течение 1 ч потеря массы
образца дельрина составляет [31] 95%,
а образца целкона (сополимера
триокрана с окисью этилена,
выпускаемого фирмой «Селанез»
в США) - всего 0,1%.
Потеря массы образца сополимера
триоксана с диоксоланом после
обработки 40%-ным водным раствором КОН в течение 12 ч при
100 °С не превышала 0,2%. Сополимеры вполне устойчивы к
действию металлов (меди, цинка, алюминия, железа и др.), в то
время как стабильность гомополимера при этом резко снижается.
Эффективность антиоксидантов, акцепторов кислоты и
формальдегида проверяли на сополимерах с различным содержанием
стабильных звеньев. Наиболее эффективными оказались антиоксиданты
аминного и фенольного типа, например антиоксидант 4010 (амин),
2,2'-метилен-бис-D-метил-6-треяг-бутилфенол), а также другие ал-
килен-бис-феыолы и циклоалифатические амины. Неожиданным
был высокий стабилизирующий эффект, достигавшийся после
обработки сополимера щелочами. Полиамиды, являющиеся
стабилизаторами для гомополимера, ие оказывают заметного
стабилизирующего эффекта на сополимеры триоксана. Показатель стабильности,
определенный в условиях переработки, для сополимеров с 2—4%
239
Время, мин
Рис. 70. Сравнительные кривые
потери массы гомополимеров
формальдегида и сополимеров при
разложении на воздухе при 220 °С:
2—дельрин 500 X; 2 — дельрин 150 X;
3 — целкон.
диоксолана был в 1,5—2 раза выше, чем для дельрина. Это значит,
что сополимер при переработке ведет себя аналогично большинству
термопластов.
Физико-химические свойства сополимера^
Превосходные механические свойства высокомолекулярных поли-
оксиметиленов связаны с их способностью кристаллизоваться в вы-
сокоупорядоченные структурные образования, что определяется в
свою очередь регулярностью строения ацетальной полимерной цепи.
Известно, что сополимеризация отрицательно влияет на
склонность полимера к кристаллизации, даже если каждый из гомополи-
меров образует кристаллические структуры [33].
Введение сомономера вызывает также понижение температуры
плавления образующегося продукта. В этом случае сомономер
действует аналогично низкомолекулярной примеси, пластификатору
или растворителю. Понижение температуры плавления сополимера,
содержащего случайно распределенные по цепи структурные звенья
А и В, описывается [34, 36] уравнением:
11 R
где Тто — температура плавления гомополимера (А); Тт —
температура плавления сополимера, который содержит долю хА мономерных
звеньев А (величина хА близка к единице, т. е. сополимер обогащен
звеньями А); Л — газовая постоянная; АН — теплота плавления,
отнесенная к одному молю А.
Необходимо отметить, что физико-химические свойства
сополимера не зависят от природы сомономера. Так, например, с помощью
дифференциального термического анализа было показано, что
температура плавления сополимеров триоксана с диоксоланом, 1,3-ди-
оксаном, диоксепаном, эпихлоргидрином линейно понижается при
увеличении концентрации сомоноь/ера, но не зависит от его природы
[35] (рис. 71).
Значения теплоты плавления полностью кристаллического
полимера (АН) были получены расчетным путем [36] и вычислены по
понижению температуры плавления при набухании [37] A490 и
1590 кал/моль соответственно).
Одновременно с понижением температуры плавления при сопо-
лимеризации наблюдается уменьшение равновесной объемной доли
кристаллической части материала. Термограммы кристаллизации,
снятые с помощью дифференциального термического анализа [34],
показывают, что с ростом доли сомономера в продукте величина
А Г, представляющая собой разность Гд (пик плавления) и Тк (пик
кристаллизации), увеличивается. AT зависит также от структуры
звена сомономера и увеличивается с ростом его длины. Введение
240
объемных боковых групп в молекулы сомономера резко понижает
степень кристалличности [45, 49].
Пластифицирующая роль сомономеров проявляется также в
уменьшении кажущейся энергии активации вязкого течения расплава
сополимера с увеличением молярной доли сомономера.
Очевидно, понижение степени кристалличности приводит к
ухудшению тех ценных физико-механических свойств, благодаря которым
полиформальдегид считается одним из лучших конструкционных
термопластов.
На основании имеющихся экспериментальных данных можно
полагать, что если добавка сомономера не превышает 5 мол. %,
то относительное ухудшение
механических свойств составит 10—15%.
Предполагается, что эта величина
будет в известной мере компенси- q?>
роваться понижением температуры |><*г 170
переработки и значительным уве- §J
личением стабильности расплава |<§
сополимера. При этом нужно иметь &|
в виду, что при переработке гомо-
полимера в промышленном масштабе 150>
средние значения показателей свойств
ухудшаться по сравнению
'О0
ч
ч
О 2 4 В 8 10
Содержание сомономера, мол.%
•с величинами, полученными в опыт- Рис- 71. Зависимость температуры
ных условиях, так как даже
незначительные изменения темпера-
плавления сополимера от типа
и содержания сомономера [34]:
О — диоксолан; • — 1,3-диоксан;
режима переработки ОКаЗЫ- Д-диоксепан; х — эпихлоргидрин.
вают сильное влияние.
Окончательное же суждение о преимуществах того или иного типа полимера
можно будет сделать только после тщательного изучения
эксплуатационных характеристик материала.
СОПОЛИМЕРИЗАЦИЯ МОНОМЕРНОГО
ФОРМАЛЬДЕГИДА
В настоящее время нет оснований предполагать, что успехи в
области сополимеризации триоксана приведут к прекращению работ
в области полимеризации мономерного формальдегида.
Одно из возможных направлений дальнейшего усовершенствования
процесса полимеризации мономерного формальдегида заключается
в переходе к синтезу сополимеров. Эта задача не встречает каких-
либо теоретических затруднений, наоборот, известным преимуществом
перед аналогичным процессом с использованием триоксана является
то, что сополимеризацию мономерного формальдегида можно проводить
на анионных катализаторах, менее чувствительных к примесям и более
стабильных, чем катионные катализаторы. О подобных разработках
сообщается в серии работ Натта с сотр. [39, 40—43]. В качеств©
стабильного сомономера они предлагают использовать диметилкетен.
16 Заказ 1135.
241
Сополимеризация с диметилкетеном
Основные задачи, решение которых необходимо для успешного
проведения сополимеризации формальдегида, аналогичны тем,
которые обсуждались при рассмотрении проблемы сополимеризации три-
оксана: концентрация стабильных звеньев в полиоксиметиленовых
цепях должна быть минимальной, чтобы сохранились механические
свойства, присущие полиоксиметилену; для достижения высокого
выхода стабильного продукта нужно равномерно распределить звенья
сомономера по молекулярной цепи.
В результате исследования реакционной способности различных
веществ в качестве сомономера для формальдегида был выбран ди-
метилкетен [40—44]." Предполагалось, что введение в оксиметиле-
новую цепь симметричного элементарного звена диметилкетена
СН3 О
I I!
—С С—
С113
приведет к увеличению стабильности сополимера (по аналогии
с другими регулярными сополимерами, имеющими структуру
сложных полиэфиров).
Принципиально возможно несколько направлений реакции с
участием диметилкетена. Так, например, полимеризация может идти
преимущественно с раскрытием двойной связи в карбонильной группе
или в одинаковой мере в обоих направлениях. Многие соединения,
являющиеся катализаторами гомополимеризации каждого из
мономеров, не эффективны как катализаторы сополимеризации, так как
способствуют образованию термически нестойких нерегулярных
сополимеров. Наилучшие результаты были получены при
использовании в качестве катализаторов третичных фосфинов и аминов.
В этом случае образуются сополимеры регулярного строения,
причем полимеризация протекает с раскрытием только непредельной
углерод—углеродной связи диметилкетена.
Диметилкетен предварительно растворяли в алифатическом,
циклоалифатическом или ароматическом углеводороде вместе с
катализатором. Затем при интенсивном перемешивании в реактор
вводили газообразный формальдегид. В других случаях сомономер
также непрерывно вводили в зону реакции. Температура реакции
не должна превышать 20 °С, так как при более высоких температурах
диметилкетен образует олигомеры и маслообразные сополимеры.
Звенья диметилкетена в сомополимере были обнаружены с
помощью ИК-спектрометрии по поглощению в областях 1738, 1165,
867 и 763 см~г, характерных для сложноэфирных групп. В то же
время отсутствие полос поглощения, приписываемых кетенным
звеньям при полимеризации по связи С — О A081 см), указывает на
однородность структуры сополимера. Содержание звеньев диметил-
242
кетена в сополимере определяли также путем ацидолитического
расщепления полимера и анализа количества образующегося альдегида.
Сополимеры, содержащие менее 10% кетена, однородны по
составу. При обработке горячим водным раствором аммиака из
них можно удалить фракцию A0—12%), которая содержит гомо-
полимеры формальдегида, цепи с одним звеном кетена и полуацеталь-
ные концевые сегменты молекул сополимера. Реакцию распада
концевых сегментов можно представить следующим образом:
О СН3
II I
R-CH2-O—G-G-(GH2-O)W-GH2OH —>
СН3
О СН3
II I
—>¦ R-CH2-O--C—С-СН2ОН + >гСН2О
сн3
Полностью отсутствовали фракции сополимера, растворимые
в кипящем хлороформе, т. е. макромолекулы, обогащенные звеньями
кетена.
Регулярность распределения элементарных звеньев кетена в
сополимере приблизительно оценивалась при изучении ИК-спектров
в области 900—905 см'1, так как полоса, характеризующая длину
оксиметиленового участка, лежит в этой области [47].
Отмечается, что при увеличении содержания сомономера
характер распределения его звеньев по цепи нарушается и
одновременно резко снижается кристалличность продукта. Если доля
кетена не превышает 5%, то степень кристалличности еще достаточно
велика, а температура плавления сополимера не ниже 165 °С.
Определение термостабильности сополимера производится
методом дифференциального термического анализа, а также путем
разложения на воздухе при 200 °С и в атмосфере азота при 220 °С
по методу, предложенному Мак-Дональдом [46]. Сополимеры
формальдегида по стабильности (измеренной по потере массы и падению
вязкости как функции времени и температуры обработки) занимали
промежуточное положение между диацетатом гомополимера и
сополимером триоксана с окисью этилена [51] (табл. 35).
По устойчивости к гидролизу и ацидолизу эти сополимеры
значительно ближе к гомополимерам, чем к сополимерам триоксана
с окисью этилена и диоксо ланом (благодаря присутствию сложно-
эфирных групп в молекулярной цепи).
Анализ физико-механических свойств сополимера формальдегида
с диметилкетеном показывает, что они изменяются в зависимости
от содержания кетена. Для получения продукта, по свойствам мало
отличающегося от гомополимера, предельное количество сомономера
не должно превышать 5%. В.этом случае степень кристалличности
продукта достигает 70% и мало зависит от скорости отжига изделий.
16* 243
ТАБЛИЦА 3
Термостабильность различных модификаций полиоксиметилєна
с логарифмической вязкостью 0,5*
Полимер
Диацетат гомополимера
Сополимер триоксана
с окисью этилена
Сополимер
формальдегида с диметилкетеном
Содержание
сомономера
%
2,5 ±1
4,0 ±1
Температура
плавления
°С
176
170
167
Потеря
массы на
воздухе при
200 °С
в течение
30 мин
%
0,8-1,4
0,2—0,5
0,5—1,0
Константа
термостабильности *• в среде
азота при 222 °С
%/мин
0,12-0,15
0,03-0,05
0,08-0,1
* Вязкость измерена в диметилформамиде при 150 °С
Сополимеризация на катализаторах
Фриделя — Крафтса
В этом направлении до последнего времени проводились только
поисковые работы. В патентной литературе описан процесс сополи-
меризации формальдегида с винильными простыми эфирами, окисями
алкиленов и азотсодержащими винильными соединениями [47, 48].
Было показано, что при использовании реакции передачи цепи
с разрывом можно синтезировать сополимеры формальдегида,
содержащие карбоцепные звенья, по термическим, физико-химическим
и механическим свойствам не уступающие аналогичным сополимерам
триоксана [52, 53].
Для реализации этого процесса в промышленном масштабе
необходимо применять эффективные способы очистки формальдегида,
так как катионные катализаторы более чувствительны к примесям
в реакционной системе, чем анионные, используемые при получении
гомополимера.
Специфика сополимеризации формальдегида (анионной и катион-
ной) проявляется в том, что стабильную часть продукта можно
отделить от нестабильной фракции путем обработки горячим
аммиачным раствором, не прибегая к специальным методам, как это
имело место в случае сополимера триоксана. Причины этого явления
еще не вполне ясны, но скорее всего они связаны с различиями в
структуре этих двух полимеров.
ЛИТЕРАТУРА
1. Walker J. F., Du Pont, пат. США 2304431, 1942 г.
2. Пат. США 2795571, 1957 г.
3. Пат. США 2947727, 947728, 1960 г.
4. Англ. пат. 895378, 1962 г.
5. Англ. пат. 878163, 1961 г.
244
6. Kern WM J а а с к s V., J. Polymer Sei., 48, 399 A960),
7. Trank С E., Du Pont, пат. США 2304080, 1942 г.
8. Пат. США 2270135, 1942 г.
9. Пат. ФРГ 1135491, 1960 г.
10. С herd r on Н.* Hohr L.,Kern W., Makromol. Chem., 52, 48
A962).
И. Натов М-* Натова Л., Кабаиванов В., Годишник хим.*
технол. ин-та (Болгария), 10, 1, 163 A963).
12. Criefer А., Kern W., Makromol. Chem., 74, 39 A964).
13. Пат. США 2465489, 1949 г.
14. Пат. ФРГ 1135491, 1962 г.
15. Натов М., Кабаиванов В. А., Михайлов М., Годишник
хим. технол. ин-та (Болгария), 7, 1-—2, 203 A961).
16. Сливкин Л. Г., О р е я к и н Д. Б., Овсянников Л. Ф., Хим.
пром., 10, 740 A965).
17. А л ь д е р с Л. Г., Жидкостная экстракция, Издатинлит, 1960.
18. Зюлковский 3., Жидкостная экстракция в химической
промышленности, 1962.
Ю.Соколов Д. Н., Головатенко Р. Т., Зав. лаб., 31, 1321
A965).
20. П а х о м о в а Л. К., Е н и к о л о п я н Н. С, ДАН СССР, 167 (№ 6),
1306 A966).
21. Bruni G. I., Celanese Corp., пат. США 2989510, 1961 г.
22. В а с с а г е d d а М., Butta Е., G і u s t у P., J. Polymer Sei., 2С$
953 A964).
23. Mar ell і F., Masetti G., Butta E., В а с с a r e~d d a M.?
J. Polymer Sei., A3, 2441 A965).
24. Celanese Corp., англ. пат. 974819, 1964 г.
25. Пат. США 2505125, 1949 г.
26. Dick R., S а с k H., Benoft H., Сообщение на симпозиуме по макро-
мол екулярной химии, Прага, 1965 г.
27. Иванов В. В., Шагинян А. А., Ениколопян Н. С, ДАН
СССР, 161, 154 A965).
28. И р ж а к В. И., И в а н о в В. В., Е н и к о л о п я н Н. С, Высоко-
мол. соед., A968).
29. Ш а г и н я н А. А.« Ениколопян Н. С, Высокомол. соед., № 11,
1866 A965).
30. J aacks V., Makromol. Chem., 84, 250 A965).
31. В er ar d і n elli F. M., Dole і Т. J., Walling С., J.: Appl,
Polymer Sei., 9, 1419 A965).
32. Wagner H. Z., Wissbkun K. F., Makromol. Chem., 8l? 15
A965).
33. Fl or у P. J., Principles of Polymer Chemistry, Cornell University Press,
N. Y., 1953, p. 565.
34. I n ou e M§, J. Appl. Polymer Sei., 8, 2225 A964).
35. Flor у P* J., Trans. Farad. Soc, 51, 848 A955).
36. Inoue M., J. Polymer Sei., 47, 498 A960).
37. Inoue М-* J. Polymer Sei., 51, 518 A961).
38. Montecatini, фр. пат. 1353226, 1964.
39. Kern W., J aacks V., Makromol. Chem., 62, 1 A969).
40. N a 11 a G., Pregaglia G., Mazzanti G., Z am bony V.f
В і s s a g h і A., Europ. Polymer J., 1, 25 A965).
41. Natta G., Mazzanti G., Pregaglia G., Binaghi M.j Ma-
kromol. Chem., 44, 537 A961).
42. Natta G., M a z z a n t і G., Pregaglia G., Pozzi G.* J.
Polymer Sei., 58, 1201 A962).
43. N a 11 a G., Mazzanti G., Pregaglia G., Binaghi M., J*
Am. Chem. Soc, 82, 5511 A960).
44. Miller R., Nield E., Turner J. A., Chem. a. Ind., 1962*
181.
245
Kitazawa Т., Tadokoko H., Chem. High Polymer Japan, 19,
203 A962).
McDonald R. N., пат. США 2768994, 1965 г.
Du Pont, англ. пат. 911960, 1962 г.
Celanese Corp., пат. США 3147234, 1964 г.
Igarashi, Mit a, Kambe, Bull. Chem. Soc. Japan, 37, № 8 A964)..
Celanese Corp., англ. пат. 986566, 1965 г.
Deutsche Gold, англ. пат. 980566, 1965 г.
Ениколопян Н. С. и др., фр. пат. 1462943 A966).
Ениколопян Н. С. и др., фр. пат. 1461969 A966).
Celanese Corp., англ. пат. 989430, 1965 г.
Ambros D., Chem. Prum., 15/40, № 6, 370 A965).
J a n d 1 J., A m b г о s D., Chem. Prum., 15/40, № 5, 278 A965),
ГЛАВА IX
СВОЙСТВА, ПЕРЕРАБОТКА И ПРИМЕНЕНИЕ
ПОЛИФОРМАЛЬДЕГИДА
настоящее время в промышленном масштабе выпускается два
материала, имеющих полиоксиметиленовую структуру: гомопо-
лимер с блокированными концевыми группами и сополимер,
содержащий небольшое количество углерод-углеродных связей (не
более 5%). В зарубежной технической литературе разновидности
полиформальдегида получили общее название «ацетальные смолы»
или просто «ацетали». Типичным примером гомополимера является
«дельрин» — материал, производимый американской фирмой
«Дюпон». Это — гомополимер, содержащий ацетильные концевые группы.
Выпускаемый в промышленности сополимер («хостаформ С»)
представляет собой сополимер триоксана и окиси этилена (производится
западногерманской фирмой «Хёхст»). Несмотря на некоторые
различия в свойствах, обусловленные меньшей степенью кристалличности
сополимера, оба материала относятся к одной группе
термопластичных полимеров. Как известно, меняя условия синтеза и вводя
различные ингредиенты, можно полимеру одной и той же химической
структуры придавать различные свойства. Полиформальдегид в этом
отношении не является исключением. Гомополимер обладает высокой
степенью кристалличности, жесткостью, твердостью, некоторой
хрупкостью. Снижая степень кристалличности путем введения
сомономера или пластифицирующих добавок, можно добиться
увеличения эластичности и повышения ударной прочности
материала. В лаборатории на основе полиформальдегида синтезированы
даже каучукоподобные материалы. Однако основное направление
применения полиформальдегида за прошедшие семь лет его
промышленного выпуска — конструирование из него различных
деталей в машиностроении, приборостроении и других отраслях
промышленности.
Поэтому наибольшую ценность представляют свойства
полиформальдегида, приближающие его к металлам.
Основные характеристики этого материала, такие, как жесткость,
прочность, твердость, хорошее сопротивление усталостным
нагрузкам, стабильность размеров и т. д., обусловлены в значительной
степени регулярностью полиоксиметиленовой структуры и лучше
всего проявляются у гомополимера. Однако сополимер, содержащий
не более 2% сомономера, отличается настолько хорошей термоста-
247
бильностью, что полностью компенсирует некоторое снижение
жесткости. Поэтому можно предполагать, что будущее принадлежит
именно сополимерам.
Помимо полиформальдегида к группе конструкционных
термопластов можно отнести полиамиды, поликарбонат, пентопласт и
полифениленоксид. Здесь термином «конструкционные термопласты»
мы обозначаем материалы, используемые главным образом для
производства силовых деталей, работающих под динамическими
нагрузками. Сравнительная характеристика этих материалов представляет
собой весьма сложную задачу, поскольку помимо технических
данных большое значение имеет экономика. Сравнительная стоимость
некоторых полимерных материалов в США в 1966 г. приведена ниже
15 ь
Полиформальдегид
Полиамиды . . .
Поликарбонат . .
Полифениленоксид
Цена, руб/т
1287
1728
1782
2277
Табл. 36 дает представление об объеме производства этих
материалов в США за последние три года.
ТАБЛИЦА 36
Объем производства конструкционных термопластов в США
Наименование
материала
Полиформальдегид
Полиамиды
Поликарбонат
1966 г.
т
27 000
30 000
9 000
Прирост 1966 г.
к 1965 г., %
58,5
• 11,1
28,5
Прирост 1965 г.
к 1964 г., %
12
20
Примечание. Полифениленоксид производится в
опытно-промышленном масштабе.
МАРКИ ПОЛИФОРМАЛЬДЕГИДА
В табл. 37 представлены характеристики некоторых марок аце-
тальных смол, выпускаемых в промышленности [9, 12—16].
Главное различие между продуктами различных марок
заключается в молекулярном весе. Высоковязкие полимеры имеют
молекулярный вес приблизительно в 2 раза больший, чем низковязкие.
Так, например, индекс расплава высоковязкого
полиформальдегида, определенный на приборе Цвика при 190 °С и нагрузке 2,16 кГ,
равен 1,8—2,5 г/10 мин; индекс расплава низковязких марок
8—15 г/10 мин. Увеличение молекулярного веса полимера приводит
к возрастанию ударной вязкости и эластичности материала. Данные,
представленные в табл. 37, отражают диапазон возможного
изменения молекулярного веса полиформальдегида. Полимер с
молекулярным весом ниже 4 • 104 обладает очень небольшой прочностью;
в то же время при молекулярном весе выше 106 текучесть материала
248
Марки ацетальных смол, выпускаемых в промышленном масштабе
ТАБЛИЦА 37
Марка
Дельрин 500Х
Дельрин 550Х
Дельрин 507Х
Дельрин 100Х
Дельрин 150Х
Дельрин 157Х
Хостаформ С, тип
С 9010
Хостаформ С, тип
С 9020
Хостаформ С, тип
С 2510
Хостаформ С, тип
С 2530
Характеристика
Диацетат гомополимера,
Мя = 40 000 (МБР = 2)
То же
»
Диацетат гомополимера,
Ми = 80 000 (МБР = 2)
То же
»
Сополимер триоксана
с 2% окиси этилена;
индекс расплава
9,0 г/Ю мин
То же
Сополимер триоксана
с 2% окиси этилена;
индекс расплава
2,5 г/10 мин
То же
Рекомендуемый метод
переработки
Литье под давлением
Экструзия
Литье под давлением
То же
Экструзия
»
Литье под давлением
Литье под давлением
и экструзия
Литье под давлением
Экструзия
Технологическая
характеристика
Содержит
поверхностную смазку,
облегчающую заполнение
формы
Не содержит смазки
Содержит смазку и све-
тостабилизатор
Отличается более
высокой вязкостью
расплава, чем дельрин
500X; содержит смазку
Не содержит смазки
Не содержит смазки;
содержит светостаби-
лизатор
Содержит
поверхностную смазку
Не содержит смазку
Отличается более
высокой вязкостью
расплава, чем тип С 9010,
содержит смазку
Не содержит смазку
Области применения
Литьевые детали общего
назначения
Прутья и проволока,
кабельные покрытия
Литьевые детали,
устойчивые к УФ-лучам
Силовые детали
несложной формы
Экструдированные
профили, листы, бутыли,
трубы
Трубы и профили,
устойчивые к
УФ-лучам
Аналогичны дельрину
500Х
Аналогичны дельрину
550Х
Аналогичны дельрину
100Х
Аналогичны дельрину
150Х
настолько низка, что его не удается перерабатывать на обычных
машинах.
Использование поверхностной смазки гранулята при литье
под давлением хорошо известно в технике переработки
термопластов. Необходимость применения смазывающих веществ вызывается
тем, что при переработке материала на плунжерных литьевых
машинах уменьшается рабочее давление вследствие преодоления
трения материала о стенки цилиндра и формы при ее заполнении.
Величина падения давления * зависит от ряда факторов, например от
длины цилиндра, конструкции узла впрыска, и может достигать
80% от прилагаемого давления [10, 11].
Введение поверхностной смазки позволяет увеличить текучесть
материала. При экструзии применение смазки нежелательно, так
как уменьшается сцепление материала с червяком.
В качестве смазывающих веществ при переработке термопластов
обычно применяют стеариновую кислоту, стеараты кальция,
кадмия, цинка, минеральные масла и воска. Количество их не должно
превышать 0,5—1,0% от массы материала, так как при увеличении
смазки резко ухудшаются механические свойства изделий.
Добавление светостабилизаторов вызвано тем, что ацетальные
смолы подвержены старению под действием УФ-лучей. Введение
светостабилизаторов (например, о-оксибензофенонов) позволяет
значительно повысить светостойкость полимера, однако они удорожают
материал и поэтому применяются t только в необходимых случаях.
Первым производителем полиформальдегида [9] стала фирма
«Дюпон» в США, организовавшая промышленное производство
в 1960 г. Два года спустя другая американская фирма — «Селанез
корпорейшн» начала промышленный выпуск сополимера триоксана
с окисью этилена (целкон). Новый продукт был разработан
совместно фирмой «Селанез» и «Хёхст» (ФРГ). В Европе он продается
под названием «хостаформ С». В США к производству ацетальных
смол (гомополимера и сополимера) приступает фирма «Теннеко ке-
миклз». Ее продукт получил фирменное название «полифайд». В
Англии планируется выпуск сополимера «алкон» (производство фирмы
«Ай-Си-Ай» по лицензии «Селанез корпорейшн»). Кроме этого еще
две фирмы объявили об организации производства ацетальных смол
в ближайшие годы. В Японии производство ацетальных смол
организуют фирмы «Сева Неопрен» (по лицензии фирмы «Дюпон»)
и «Полипластикс» («дуракон», по лицензии «Селанез корпорейшн»)
[6]. Опытно-промышленное производство ацетальных смол
организовано в Италии, Франции, а также в Болгарии («болрин») и ГДР.
В Советском Союзе производство полиформальдегида (ПФА)
начато в 1965 г. Продукт выпускается в двух модификациях. Марка
А — литьевой материал с индексом расплава 1,8 г/10 мин, который
предназначен для переработки методом литья под давлением. Из
* Эти данные были получены для полистирола, но можно полагать, что
подобная картина существует и для ацетальных смол.
250
него можно получать изделия сложной конфигурации. Марка Б
(высоковязкий полимер) предназначена для получения изделий
несложного профиля методом литья под давлением в тех случаях,
где нужна повышенная прочность.
ФИЗИКО-МЕХАНИЧЕСКИЕ СВОЙСТВА
Показатели физико-механических свойств различных марок аце-
тальных смол приведены в табл. 38. Поскольку ацетальные смолы
перерабатываются главным образом литьем под давлением, то и
образцы для испытаний обычно получают этим же методом. Хорошо
известно, что свойства испытуемого образца зависят не только от
свойств материала, но и в большой степени от условий формования.
При стандартных испытаниях по методикам ASTM, DIN и др. строго
регламентируются размеры образца и расположение литников
в форме, однако значительно труднее стандартизовать
температурный режим переработки. Имеет значение также тип применяемой
литьевой машины. Особенности режима формования сказываются
на механических свойствах изделий. Этим часто объясняется
несовпадение результатов, полученных различными исследователями
при работе с одним и тем же материалом. Поэтому данные,
приведенные в табл. 38, следует рассматривать как
приближенные.
Ацетальные смолы относятся к группе наиболее жестких
термопластов, в которую из традиционных пластиков входят полиамиды,
тройной сополимер стирола (ABC) и некоторые другие, а из новых —
поликарбонат и полифениленоксид. Предпосылкой для успешного
применения ацетальных смол в качестве конструкционного
материала является их стойкость к механическим нагрузкам и истиранию,
стабильность размеров и низкий коэффициент трения.
По сопротивлению ударным нагрузкам (удельная ударная
вязкость литьевых стандартных образцов равна 90—130 кГ - см/см2)
ацетальные смолы близки к полиамидам, но значительно более
хрупки, чем Поликарбонат (правда, это касается только однократного
нагружения образца). Эксперименты показали, что ацетальные смолы
намного превосходят поликарбонат при действии многократной
ударной нагрузки, так как обладают значительно большей усталостной
прочностью. К недостатку ацетальных смол следует отнести резкое
уменьшение удельной ударной вязкости при испытании образцов
с надрезом по Изоду. При этом величина разрушающей нагрузки
снижается более чем в 10 раз. Поэтому при конструировании деталей,
подвергающихся действию ударных нагрузок, нужно избегать
концентрации напряжений (острых углов, выступов и т. п.).
По устойчивости к истиранию ацетальные смолы уступают, по-
видимому, только полиамидам, однако превосходят последние по
стабильности размеров и влагостойкости. По этому показателю
ацетальные смолы приближаются к аморфным полимерам (полистиролу,
полиакриловым смолам). Данные об износостойкости различных
251
Физико-механические свойства ацетальных смол
ТАБЛИЦА 38
Свойства
Метод испытаний
Образец
Показатели свойств
дельрин
500Х
дельрин
150Х
хостаформ
С 9020
хостаформ
С 2520
Плотность при 20 °С,
г/см*
Индекс расплава при
190 °С, г/10 мин
Предел прочности при
растяжении при 20 °С,
кГ/см*
Предел прочности при
изгибе при 20 °С,
кГ/см*
Предел прочности при
сжатии, кГ/см*
на 10% при 23 °С
» 1% » 23 °С
Относительное
удлинение при разрыве, %
Твердость на шкале
Роквелла
Удельная ударная
вязкость на образцах
с надрезом при 20 °С,
кГ. см/см*
Гидростатический
ASTM D1238
ASTM D638-52T
Скорость растяжения
100 мм/мин
ASTMD790-49T
DIN 53452
ASTM 0695
ASTM D638-52T
ASTM D785-48T
по Изоду
ASTM D256-47T
DIN 53453
Литая пластина
толщиной 1 мм
Гранулы
Литой стандартный
брусок 25 X 3 мм
Литой стандартный
брусок 25 X 3 мм
Литая пластинка
толщиной 4 мм
Литой стандартный
брусок 25 X 3 мм
Литой стандартный
брусок 12 X 3 мм
1,425
9—13
703
1,425
1,8—2,5
710
990
1266
366
15
45
М94
7,6
12,5
1,41
680
1,41
2,5
650
850
1170 1080
1120
315
28
6,0
9,0
36
М76
10,0
Модуль упругости при
растяжении (модуль
Юнга) при 20 °С,
кГ/см*
Модуль упругости при
кручении A20 °С),
Г/*
Модуль упругости при
изгибе (модуль
жесткости) при 20 °С,
КГ/СМ1*
Коэффициент Пуассона
Водопоглощение при
20 °С, %
за 24 ч
» 90 ч
Износостойкость,
л^г/1000 циклов
под нагрузкой
1000 мг
под нагрузкой
100 мг
Коэффициент трения
(динамический)
по стали
» латуни
» алюминию
» полимеру
DIN 53371, скорость
деформации 20% /мин
ASTM D1043
ASTM D747
DIN 53472
ASTM D1044
Литая пластина
толщиной 1 мм
Литая пластина
толщиной 2 мм
Литая пластина
толщиной 1 мм
Литая пластина
толщиной 1 мм
Литая пластина
42 -103
Литая пластина
35 • 103
0,35
0,80
0,50
20
0,15
0,18
0,20
0,20
35 -103
1400
30 • 103
30 • 103
1300
28 - 103
0,35
0,80
0,50
14
полимеров приведены в табл. 39 [13], где потеря массы полиамида
в результате испытаний принята за единицу.
Как известно, все термопласты можно эксплуатировать только
в определенной области температур, где они проявляют
оптимальные эксплуатационные свойства. С одной стороны, эта область
ограничивается температурой начала размягчения материала, с другой —
появлением хрупкости. Общая для всех термопластов закономерность
проявляется в том, что при повышении температуры увеличиваются
эластичность и гибкость материала, но одновременно уменьшаются
твердость и жесткость. На рис. 72 и 73 показана зависимость
удельной ударной вязкости, предела прочности при растяжении и
относительного удлинения при разрыве образцов полиформальдегида от
-80 -ЬО О
Температура, °С
Рис. 72. Зависимость удельной
ударной вязкости (по Изоду) литьевых
образцов сополимера от
температуры.
О +50 100121
Температура, °С
Рис. 73. Зависимость предела прочности
при растяжении и относительного
удлинения при разрыве диацетата ПОМ
от температуры.
температуры. Появление хрупкости наблюдается при температуре
около —60 °С. При нагреве до 100—120 °С ацетальные смолы еще
сохраняют достаточную жесткость, однако едва ли способны
выдержать длительную эксплуатацию в подобных условиях. Верхний
предел рабочих температур для нагруженных деталей находится
около 85 °С.
ТАБЛИЦА 39
Сравнительная износостойкость полиформальдегида и других материалов ]
Материал
Полиамид «Зитель-101»
Дельрин
Полистирол
Сополимер ABC
Эфиры целлюлозы
Твердая резина
Литой алюминий
Мягкая сталь
Метод испытаний
метод
Табера
1
2-5
9—26
9
9—15
шаровая
мельница
1
4-6
15-20
10—20
10-20
10
15-20
протягивание
проволоки
1
5-6
35
15
шлифовальный станок
1
3-4
4
4—5
254
При проектировании конструкционных деталей особенно важна
устойчивость материала к длительному воздействию статических
или динамических нагрузок. Если напряжения достаточно велики,
то наблюдается изменение формы изделия во времени. Это явление
известно под названием ползучести. Характер напряжения,
длительность его воздействия и температура определяют величину
получаемых деформаций. Исследование ползучести служит основой
для оценки возможности применения деталей конструкционного
назначения из данного материала. ^
Величины допустимых
напряжений оказываются значительно
меньшими, нежели полученные при ^
кратковременных испытаниях образ- |
10 WO 1000 10000 Юлвгп
Продолжительность действия нагрузки, ч
Рис. 74. Кривые ползучести при заданной
величине деформации (сополимер триок-
сана с окисью этилена) при 22 °С:
1-0,6%; *- 1%; 5-1,5%; 4-2%.
50 100 150 200 250 300 350 U00
напряжение при растяжении, к Г/ем2
Рис. 75. Зависимость величины
модуля Юнга от величины и
длительности действия нагрузки
(хостаформ С 9010).
цов *, однако именно эти величины фигурируют в инженерных
расчетах. •
Изменение влагосодержания также влияет на ползучесть.
Благодаря хорошей влагостойкости ацетальных смол их механические
свойства относительно мало зависят от влагосодержания, в частности
ползучесть погруженного в воду образца увеличивается не более
чем на 10%.
Кривые ползучести строятся экспериментально для
определенных значений деформации по данным зависимости допустимых
напряжений при растяжении от длительности нагружения для
заданной температуры (рис. 74). Зависимость кажущейся величины модуля
упругости (модуля Юнга) от величины и длительности действия
нагрузки показана на рис. 75.
* В последнем случае говорят о максимальных значениях напряжений
или о пределе прочности.
255
При комнатной температуре и небольших нагрузках ползучесть
ацетальных смол несколько выше, чем аморфных термопластов,
однако в «тяжелых» условиях — при высокой относительной
влажности, температуре 60—90° С и значительных нагрузках —
ползучесть ацетальных смол ниже, чем у всех известных термопластов.
Предел прочности при статической нагрузке всегда выше, чем при
динамической. Это явление, характерное для любого жесткого
материала, получило название усталостной прочности. Ее величина
зависит от частоты и величины прилагаемого напряжения, а также
температуры и наличия концентраторов напряжений в образце,
т. е. от способа переработки.
Ацетальные смолы по усталостной прочности и способности
восстанавливать форму после снятия напряжения превосходят все
другие термопласты [13, 501].
Антифрикционные свойства отечественного полиформальдегида
рассмотрены в работе [8].
ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА
Ацетальные смолы обладают хорошими диэлектрическими
свойствами (табл. 40). В этом отношении они практически не отличаются
от полиамидов, хотя и уступают таким превосходным диэлектрикам,
как полистирол и полиэтилен. Основное преимущество
полиформальдегида перед полиамидами в данном случае сводится к лучшей
влагостойкости, так как в условиях 100%-ной относительной влажности
электрические свойства полиформальдегида, в отличие от
полиамидов, изменяются незначительно [23]. Диэлектрическая
проницаемость при 25 °С равна 3,3—3,8. При повышении температуры до
150 °С наблюдается небольшое снижение величины диэлектрической
Электрические свойства ацетальных смол
ТАБЛИЦА 40
Свойства
Диэлектрическая
проницаемость при 102—105 гц и
220° С
Тангенс угла
диэлектрических потерь
Удельное электрическое
сопротивление
объемное, ом • см
поверхностное, ом
Электрическая прочность,
кв/мм
Дугостойкость, сек
Метод испытания
ASTM D150
ASTM D150
ASTMD150
ASTM D150
ASTMD150
Показатели
гомополимер
3,7
0,004
6 • 1014
2•1013
49
129
(загорается)
СВОЙСТВ
сополимер
3,8
0,004
1 -1015
5 • ЮН
69,6
129
(загорается)
256
проницаемости (до 2,7). Для неполярных веществ диэлектрическая
проницаемость не должна зависеть от частоты поля. Для
полиформальдегида известно, что изменение частоты в интервале от 102
до 105 гц не влияет на диэлектрическую проницаемость и величину
тангенса угла диэлектрических потерь [48, 49, 73, 74].
ХИМИЧЕСКАЯ СТОЙКОСТЬ И ФИЗИКО-
ХИМИЧЕСКИЕ СВОЙСТВА
Полиформальдегид благодаря его полиацетальной структуре
можно рассматривать как гомолог низкомолекулярных линейных
ацеталей (формалей), отличающийся высоким молекулярным весом.
Однако известно, сколь сильно на химические свойства полиокси-
метиленов влияет природа концевых групп. Другим фактором,
имеющим большое значение, следует считать действие различных
примесей. По способу попадания в полимер их можно разделить
на две группы. Первая группа — примеси, попадающие в полимер
в процессе синтеза. Это — катализатор, химически связанный с
полимерными цепями или сорбированный при кристаллизации
полимера, загрязнения из аппаратуры, примеси к ингредиенгам
вводимым на стадиях стабилизации, окрашивания и т. д. Вторая
группа — примеси, образующиеся в процессе эксплуатации
материала в результате окисления, старения, действия УФ-излучения,
химических агентов и т. д.
Различиями в химической структуре полиоксиметиленовой цепи
можно пренебречь, так как из предыдущего изложения вытекает,
что специфическая особенность полиформальдегида заключается
в отсутствии разветвлений, двойных связей и боковых групп в
полиацетальной цепи. В то же время колебания плотности, степени
кристалличности, среднего молекулярного веса и молекулярно-весового
распределения играют важную роль.
Термическая деструкция. Этот вопрос был подробно рассмотрен
в гл. IV в связи с изложением проблемы стабилизации
полиформальдегида в целом. Здесь мы отметим только, что ацетальные смолы
в отсутствие кислорода, в частности в вакууме или в атмосфере азота,
способны в течение 6—8 ч выдерживать нагревание при 270 °С.
При этом не происходит изменения молекулярного веса полимера.
Повышение температуры до 290—300 °С приводит к разложению
материала, по-видимому, за счет гемолитического разрыва С—0-
связей в макромолекулах. Основным продуктом распада является
мономерный формальдегид. Различий в поведении между гомополи-
мером и сополимером практически не наблюдается.
Окислительная деструкция. Полиформальдегид подвергается
окислительной деструкции в присутствии кислорода уже при
температуре около 120 °С. УФ-облучение ускоряет реакцию и
инициирует ее даже при комнатной температуре. Аналогичным образом
ведут себя все другие полимерные материалы, однако специфика
полиформальдегида заключается в низкой стабильности при
17 Заказ 1135. 257
температуре переработки. Обычно разложение термопласта (появление
темно-коричневых полос, пузырей, запаха) наблюдается при
перегреве или слишком длительном цикле переработки. В случае
полиформальдегида речь идет скорее о количественном, нежели о
качественном отличии в поведении сополимера и гомополимера. При
этом сополимер ведет себя подобно большинству других
термопластов. Окисление ацетальных смол проявляется в уменьшении
молекулярного веса, снижении относительного удлинения, повышении
хрупкости. Стабилизирующие добавки в ацетальных смолах
выполняют двоякую роль: они ингибируют термоокислительную
деструкцию и препятствуют старению материала. В тех случаях, когда
материал эксплуатируется в атмосфере УФ-света, необходимы добавки
фотохимического стабилизатора.
Стойкость к действию кислот, щелочей и солей. Ацетали
устойчивы к щелочам, но легко гидролизуются под действием кислот.
Это положение справедливо по отношению к полиформальдегиду,
но следует учесть специфические свойства высокомолекулярного
кристаллического полимера. Гомополимер, блокированный
ацетильными концевыми группами (дельрин), обладает низкой
устойчивостью к действию щелочей вследствии гидролиза ацетильных групп.
Испытания в стандартизованных растворах [13] показали, что дель-
рин устойчив при температурах 20 и 60 °С в интервале pH 5—8,6.
В то же время сополимер (целкон), имеющий концевые оксиэтилено-
вые группы, устойчив к действию концентрированных щелочей при
повышенной температуре. Испытания в стандартных растворах [18]
показали, что он устойчив в интервале pH 3—13.
Сильные минеральные кислоты полностью разрушают ацеталь-
ные смолы даже при низких температурах. Такие кислоты, как
серная, соляная, азотная, действуют разрушающе уже при
концентрации раствора около 1%. Органические кислоты значительно
менее активны. Хотя точные данные отсутствуют, можно полагать,
что ацетальные смолы способны выдержать действие разбавленных
A—5%) растворов органических кислот. Действие кислот сводится
к разрыву ацетальных связей в полимерной цепи и последующей
деполимеризации осколков. Очевидно, что сополимер должен быть
более устойчив, так как деполимеризация цепей будет прекращаться
при взаимодействии активного центра с оксиалкиленовыми группами.
Галоидзамещенные полиоксиметилены обладают высокой
устойчивостью к действию кислот, однако они не выпускаются в
промышленном масштабе.
Растворы солей, а также металлическая пыль значительно
сильнее действуют на гомополимер, чем на сополимер.
Действие воды. Влагопоглощение ацетальных смол за 24 ч при
20 °С составляет около 0,2%. Эта цифра соответствует равновесному
влагосодержанию полимера при этой температуре и 50%-ной
относительной влажности воздуха. Такое влагосодержание практически
не влияет на физико-механические свойства материала. Образец
дельрина, помещенный в воду на длительное время, имел влагосо-
258
держание около 1%. При этом его физико-механические свойства
изменились незначительно. Например, после 275 суток
выдерживания в дистиллированной воде при 60 °С образец дельрина имел
модуль упругости на 18% ниже, чем у контрольного образца. Предел
прочности при растяжении не изменился. Водяной пар при
кратковременных испытаниях не оказывал заметного действия на ацеталь-
ные смолы, однако постоянная эксплуатация материала в водной
среде при температуре кипения нецелесообразна. Это относится
главным образом к гомополимеру, ацетальные концевые группы которого
склонны к гидролизу.
Сополимер должен отличаться большей устойчивостью к
действию водяных паров.
Морская вода не оказывает какого-либо заметного действия на
ацетальные смолы. Не наблюдается также отложения солей на
поверхности материала.
Действие органических жидкостей. Полиформальдегид не
растворяется в органических жидкостях при умеренных температурах.
Исключение составляют некоторые недавно синтезированные
соединения типа гексафторацетонгидрата [24]. В этом растворителе
при комнатной температуре можно получить 20%-ный раствор поди-
мера. Стабильность полимера в растворе достигается введением
стабилизатора — третичного амина. В противном случае полимер
разлагается, так как растворитель имеет кислый характер.
В табл. 41 приведены данные о стойкости ацетальных смол к
действию различных органических жидкостей. Эти данные
свидетельствуют об исключительно высокой стойкости ацетальных смол к
действию нейтральных органических жидкостей. Эта особенность
полиформальдегида привела к тому, что поиск растворителей для
физико-химического анализа полимера превратился в
самостоятельную проблему (см. гл. II). В качестве растворителей было испытано
406 веществ, относящихся к 27 различным классам соединений [36].
Ни одно из них не растворяло полимер (концентрация 1%) при
температурах ниже 60 °С. Температура гелеобразования обычно была
на 10—40 °С ниже температуры растворения. При температуре
выше 100 °С полиформальдегид хорошо растворим в фенолах,
ароматических аминах и других соединениях. Данные о поглощении
растворителей показывают, что имеется прямая связь между
количеством растворителя, поглощенного полимером, и изменением свойств.
Характеристикой растворенного вещества и растворителя,
обладающего аналогичным строением, является удельная энергия когезии
(УЭК) — количество тепла, необходимое для испарения единицы
объема вещества:
где АН — тепло, необходимое для испарения 1 моль вещества при
температуре Т\ R — термодинамическая газовая константа; V —
молярный объем растворителя при температуре Т.
17* 259
Химическая стойкость ацетильных смол
Обозначения: + устойчив*; (- / —относительно устойчив;
ТАБЛИЦА 41
неустойчив
Реагент
Ацетон
Этиловый спирт
Метиловый
спирт
Изопроиило-
вый спирт
Изоампловый
спирт
Бензин
различных сортов
74-Гексан
Керосин
Смазочное
масло
Дизельное
масло
Минеральное
масло
Мазут
Бензол
Толуол
Ксилол
Анилин
Ппрлдин
Хлорбензол
Тип полимера
Гомополи-
мер
Сополимер
Гомополи-
мер
Сополимер
Сополимер
Сополимер
Гомополи-
мер
Г ОМОН О Л11-
мер
Сополимер
Гомополи-
мер
Сополимер
Гомополи-
мер
Сополимер
Гомополи-
мер
Сополимер
Сополимер
Сополимер
Сополимер
Гомополи-
мер
Сополимер
Гомополи-
мер
Сополимер
Сополимер
Гомополимер
Гомоноли-
мер
Сополимер
Температура
°С
23
50
20
56
23
50
20
60
20
60
20
60
60
25
20
60
20
60
60
20
70
20
60
20
60
20
60
60
60
20
23
50
20
60
20
60
60
60
60
жительность
испытания
сутки
365
365
365
60
365
365
60
60
60
60
60
60
275
90
60
275
60
60
275
60
365
60
60
60
60
60
60
60
275
60
365
365
60
60
60
60
275
275
60
Изменение
предела
прочности
при
растяжении, %
—5
—7
—13
—7
-5
о
— 10
—6
— 10
-6
—6
-4
—4
От —5
ДО +1
От —7
ДО —3
—4
+4
2
0
о
+3
+4
+4
—3
+ 1
о
—о
+1
+з
—и
— 12
—7
—7
— 12
—5
-1
—1
-12
—9
—9
Изменение
массы, %
+ 1,9
+2,6
+2,9
+3,6
+2 2
+ 1,9
+1,6
+2,0
+ 1,7
+о',б
+ 1,9
+ 1^2
До +0,4
До +1,8
+0,74
+0,4
+0,7
0
+0,1
-0,2
+0,1
+0,1
+0,5
+0,5
+0,1
+0,2
+0,5
+4,0
+3,3
+2,6
+2,8
+2,1
+2,8
+ 1,2
+2,4
+8,3
+6,0
+5,0
Стойкость
+
+
+
-]_ / —
+
+
+
-j-
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+/—
+/—
+
+
+
+
+
+
—
+ '-
. J '
* Устойчивым продукт считается в том
< 3% и изменение прочности < 15%
260
случае, если изменение массы составляет
Продолжение табл. 41
Реагент
Тетралин
Тетрахлорэти-
лен
Четыреххло-
ристый
углерод
Сероуглерод
Тип
полимера
Сополимер
Сополимер
Гомополи-
мер
Сополимер
Сополимер
Температура
°С
20
60
20
60
23
50
20
60
20
жительность
испытания
сутки
60
60
60
60
365
365
60
60
Изменение
предела
прочности
при
растяжении, %
-6
л
2
-3
5
—9
—7
+ 1
Изменение
массы, %
+0,5
+3,5
+2,1
+5,3
+ 1,3
+5,7
+ 1,3
+6,3
+2,9
Стойкость
+
+/-
+
+/-
+
1 /
+
+'-
+
Для полиоксиметилена УЭК может быть вычислена по данным
элементарного звена [37]. АН для СН2О составляет 2620 кал/молъ.
Если молекулярный вес полиоксиметилена 30, а плотность 1,42 г/см3,
то УЭК будет равна 124 кал/см3. Авторы [36] наблюдали
корреляцию между величиной УЭК растворителя и набуханием полимера.
Наибольшее набухание достигалось в растворителях, УЭК которых
была близка к УЭК-полимера.
Поглощение растворителя зависит от степени кристалличности
полимера. Это было подтверждено серией опытов с образцами
различной плотности, полученными при охлаждении расплава с
различной скоростью (табл. 42).
ТАБЛИЦА 42
Зависимость степени набухания дельрина при 60° С от плотности
Показатели
Плотность
г /см3
Содержание.
фазы, вес.
Количество
полимера,
аморфной
%
адсорбиро-
ванного растворителя,
вес. %
Анилин
1,418; 1,422;
1,427; 1,438
35,0; 33,0; 30,5;
25,5
9,60; 8,66; 8,71;
6,90
Диметилформамид
1,413;
36,0;
6,35;
1,419;
34,0;
5,73;
1,422
32,0
5,62
./и-Крезол
1,418; 1,422; 1,425;
1,440
35,0; 33,0; 32,0; 25,0
16,48; 14,56; 14,60;
10,50
Увеличение плотности полимера, т. е. увеличение содержания
кристаллической фазы,, приводило к уменьшению степени набухания.
Экстраполяция кривых зависимости равновесного набухания от
плотности полимера показывает, что полимер с плотностью около
1,5 г/см3 не поглощает никаких растворителей. Для полимера
со 100%-ной степенью кристалличности расчетное значение
плотности равно 1,506.
Проницаемость. Подобно всем полимерным органическим
веществам, ацетальные смолы проницаемы для газов и паров.
26 і
Проницаемость зависит от молекулярной структуры полимера и от
химической природы диффундирующей среды. Кроме того, имеет
значение толщина материала, температура и концентрация (градиент
концентрации).
Проницаемость является функцией растворимости вещества
в полимере и скорости диффузии, т. е. ее можно выразртть формулой:
где П — проницаемость; D — диффузия и S — растворимость [38].
Зависимость константы проницаемости от температуры
выражается экспоненциальным законом:
где Еп — энергия активации проницаемости, зависящая от природы
диффундирующей среды и ее полярности; По — константа материала,
уменьшающаяся с увеличением плотности [39].
Предполагается, что проникание происходит только через
аморфные области, кристаллическую фазу можно рассматривать как
инертный наполнитель [40]. Полиформальдегид (табл. 43) обладает
низкой газопроницаемостью.
ТАБЛИЦА 43
Газопроницаемость полимеров при 30° С
Полимер
Хостаформ С *
Поливинилиденхлорид
Полиэтилентерефталат
Найлон-6
Полиэтилен
Проницаемость, мл»мм 1(см2• сек• см рт. ст.)
ЦЫО10)
для кислорода
0,3
0,053
0,22
0,38
55,0
для азота
0,06
0,009
0,05
0,10
19,0
для двуокиси
углерода
5,9
0,29
1,53
1,60
352
* Измерения проведены для пленки толщиной 80 мк.
Константа проницаемости полимерной пленки для водяных
паров в случае полиформальдегида в 10 раз больше, чем для
полиэтилена, т. е. равна приблизительно константе проницаемости полиме-
тилметакрилата и полистирола. Ацетальные смолы обладают очень
низкой константой проницаемости для органических жидкостей
и газообразных углеводородов.
Горение. Ацетальные смолы горючи, сгорают практически без
остатка. Скорость горения при стандартных испытаниях — величина
такого же порядка, как у полиэтилена и полистирола B,8 см/сек).
Токсичность. Ацетальные смолы не токсичны. При хранении
материала в закрытой таре появляется слабый запах
формальдегида, который выделяется вследствие медленного окисления, однако
262
его концентрация незначительна. Пищевые продукты не оказывают
влияния на свойства полимера при длительном хранении, хотя
горчица и горячий кофе вызывают слабое изменение окраски.
ПЕРЕРАБОТКА ПОЛИФОРМАЛЬДЕГИДА
Ацетальные смолы перерабатывают литьем под давлением,
экструзией и выдуванием. Переработка полиформальдегида (как и
других термопластов) прессованием неэффективна, так как при
этом в значительной степени уменьшается прочность материала.
Этот метод переработки применяется обычно только в лаборатории
для изготовления микрообразцов для физико-химических
испытаний.
Технологический режим переработки определяется такими
свойствами материала, как текучесть расплава, термостабильность,
скорость затвердевания и т. д.
Текучесть — величина, обратная вязкости расплава, однако
последняя не является постоянной величиной, а зависит от
температуры и скорости сдвига, так как расплавы высокомолекулярных
веществ ведут себя подобно неньютоновским жидкостям.
Реологические исследования течения расплавов
полиформальдегида имеют своей целью установить зависимость текучести
расплава от температуры, давления (напряжения сдвига) и скорости
сдвига. Существуют различные способы построения кривых течения.
Данные, получаемые с помощью экструзионных пластометров
(вискозиметров), — индексы расплава, зависимости эффективной вязкости
расплава и текучести A/т]) от температуры, напряжения и скорости
сдвига — не всегда удается моделировать к условиям
промышленной переработки. В лаборатории можно создать лишь небольшие
скорости сдвига, в то время как на стандартных литьевых машинах
они достигают 104 — 106 сек'1. Поэтому в технике часто проводят
исследования текучести материала непосредственно на стандартных
литьевых машинах с небольшим объемом загрузки материала.
Текучесть определяется по степени заполнения специальной формы.
Большое распространение получили формы в виде спирали (рис. 76)
стандартного размера, с помощью которых можно определить
оптимальный режим переработки данного материала.
При переработке полиформальдегида особое значение имеет
термостабильность расплава [45, 52]. С разложением материала
приходится считаться при переработке любого термопласта. На рис. 77
представлена схематическая литьевая диаграмма термопласта (типа
полиолефина). Нижняя кривая соответствует минимальному
давлению плунжера для данной температуры, при котором еще
происходит заполнение формы (соответствующую температуру можно
назвать температурой текучести Тт). Верхняя кривая соответствует
максимальному давлению, при котором материал начинает вытекать
из формы. Ее положение всецело определяется конструкцией
машины. Температурный интервал переработки (зона В) ограничивается
263
с двух сторон: зоной А, в которой из-за низких температур
расплава наблюдается образование слоев (недопрессованность) и
пониженная прочность изделий, и зоной С, в которой происходит
сильное термическое разложение материала, что приводит к
получению изделий низкого качества.
Очевидно, что размер зоны В определяет диапазон варьирования
технологического режима переработки. Из полиолефинов,
выпускаемых в промышленном масштабе, наименьшей стабильностью
при переработке обладает поливинилхлорид, который довольно легко
разлагается, выделяя
хлористый водород, что приводит
&о\ 1 1 "^иІ'г/"м2' к автокаталитическому раз-
V
— ¦—' ""' —
Давление
при литье,
А
-^l
'Г/СМ*
У 2000
ywoo
>/200
>800
да
20
10
0 /80 200 220
Температура расплава, °С
Рис. 76. Зависимость текучести образца
хостаформа С 9010 от температуры
расплава и давления при литье с различными
скоростями сдвига [16]. (Форма —
стандартная* спираль сечением 7,8 X 1,5 мм.
Температура формы 80° С.)
II
§1
I
Рис.
1
\
в
Температура.
материала
77. Литьевая диаграмма
термопласта:
1 — зона расслоения; 11 — зона
термического разложения.
ложению продукта. В этом отношении можно провести аналогию
между поливини л хлоридом и полиформальдегидом, однако в
последнем случае эффект разложения (выделение мономерного
формальдегида) проявляется сильнее. Это приводит к
значительному уменьшению зоны В (см. рис. 77). Предельная
температура расплава не превышает 230 °С, а минимальная 190 °С. Кроме
этого в случае переработки гомополимеров (уступающих по
термостабильности сополимерам) [46] нужно определять время
пребывания расплава полимера в цилиндре литьевой машины и соизмерять
его с допустимым временем устойчивости расплава при заданной
температуре. Обычно эту величину можно рассчитать по формуле
(см. стр. 220):
тхц
Зависимость допустимого времени пребывания материала в
цилиндре литьевой машины от температуры расплава дается в виде
264
диаграммы (рис. 78). Для того чтобы предотвратить разложение
материала, необходимо соизмерять размер изделий (объем впрыска)
и объем цилиндра литьевой машины, причем нужно стремиться,
чтобы отношение — было максимальным A : 6 или 1 : 10).
Сополимер обладает значительно большей термостабильностью,
чем гомополимер. Ограничения, связанные с продолжительностью
нагрева материала, сводятся к тому, что при 200 °С полимер не
должен находиться в цилиндре машины более й, а при 230 °С — более
30 мин.
Данные о термических свойствах полиформальдегида,
выпускаемого в промышленном масштабе, приведены в табл. 44.
ТАБЛИЦА 44
Термические свойства ацетальных смол [12—16]
Свойства
Температура
плавления
кристаллической фазы, °С
Температура
стеклования, °С
Теплостойкость
по Вика, °С
Коэффициент
теплопроводности при
20 °С,
ккал/(м • ч • град)
Удельная
теплоемкость при 20 °С,
кал/(г • град)
Коэффициент
термического линейного
расширения, град~±
Температурка начала
термоокислительной деструкции, °С
Температура начала
термической
деструкции, °С
Метод испытаний
Поляризационный микроскоп
Динамический
при 100 гц
VDE 0302
Метод двух
пластин
Дилатометр
«Leits»
Дериватограф
(по потере массы)
В вакууме
A0~3 мм pm. ст.)
Образец
Пленка
толщиной 20 мк
Пластина
(прессованная)
толщиной 4 мм
То же
10 X 100 X 100 мм
Литая пластина
Гранулы
Гранулы
Показатели свойств
гомополимер
(дельрин)
175-180
-60
167
0,27
0,35
4,5-10-o*
150
250
сополимер
(хоста-
форм С)
164-167
-60
154
0,27
0,35
1.3Х
Х10-4**
145
270
* По-видимому, в зависимости от способа измерения и температурного интервала
в литературе приводятся несколько различающиеся данные: от 4,5« 10~5 до 8,1 • 10~6 град-*.
** В литературе приводятся данные: от 0,8* 10~4 до 1,3-10~4 град-1.
Для расплавления материала к нему нужно подвести
приблизительно 100 ккал/кг (рис. 79). Скрытая теплота плавления гомополи-
мера [12] равна 39 ккал/кг. Для сополимера она несколько ниже
из-за меньшей кристалличности.
265
При внешнем подводе тепла к материалу за счет нагрева стенки
разность температур между стенкой и материалом не должна
превышать допустимых пределов, иначе в непосредственной близости
от стенки произойдет перегрев и разложение материала. Разогрев
материала происходит также за счет трения. Это явление
используется при экструзии для
поддержания изотермического режима
переработки.
Продолжительность ^
нагрева
Рис. 78. Диаграмма
продолжительность нагрева — температура
материала.
§1
50 100 150 200
Температура, °С
250
Рис. 79. Зависимость удельной
теплоемкости апетальной смолы (хостаформ С)
от температуры [18].
Литье под давлением
Литье под давлением — основной способ переработки ацетальных
смол, которым получают большую часть изделий весом от
нескольких граммов до 2 кГ. Необходимые условия переработки
полиформальдегида достаточно широко описаны в специальной литературе
и фирменных проспектах [5—7, 12—16, 18—22, 45, 52], поэтому
мы только кратко перечислим основные положения.
Тип машины. Полиформальдегид легко перерабатывается на
большинстве стандартных машин для литья термопластов.
Специфические требования, предъявляемые к оборудованию для переработки
полиформальдегида литьем под давлением, заключаются в
следующем:
1) в нагревательном цилиндре должны отсутствовать зоны застоя
материала, так как это неминуемо приводит к его разложению;
2) удельное давление должно быть равно 1000—1500 am]
3) сопло должно иметь регулируемый обогрев.
Для переработки ацетальных смол используют машины различных
типов: плунжерные, с червячным предпластикатором, червячные.
Особо следует отметить машины с червячным предпластикатором.
При переработке на таких машинах достигается однородность
литьевой массы и нагрев материала можно вести при более низкой
температуре, что важно с точки зрения стабильности материала.
Температура литья. Оптимальная температура переработки
подбирается эмпирически для каждого типа машин. Начинают пере-
266
работку обычно при высокой температуре (около 230 °С), которую
затем постепенно снижают, пока не прекратится газовыделение и
окрашивание материала. Слабый запах формальдегида может
появиться и при нормальном режиме переработки. Поэтому
рекомендуется оборудовать машины вытяжной вентиляцией.
Поскольку ацетальные смолы весьма чувствительны к изменению
температуры, необходимо, чтобы материальный цилиндр литьевой
машины был оснащен нагревателем с тщательной регулировкой и
контролем температуры (±0,5 °С). Нужно помнить также, что
термопарами замеряют температуру стенки, а не самого расплава.
Текучесть материала зависит от
температуры, поэтому обычно стремятся
работать при максимально допустимых <$
температурах. В этом случае достигаются ^
наилучшие показатели прочности изделия. ^
Зависимость вязкости расплава полиформ- |
альдегида от температуры по сравнению ^
с некоторыми другими материалами g
показана на рис. 80. Обращает на себя §
внимание тот факт, что текучесть поли- о§
формальдегида мало зависит от темпера- ^
туры.
Давление и скорость сдвига. Для
полиформальдегида характерна низкая
вязкость расплава в условиях переработки.
8
з
2-Ю2
x?—
H
^_
¦A
зЛ
~v
V
\
Рис.
177 205 232 260 288 316
Температура, °С
Зависимость вяз-
термопластов от
температуры:
1 — дельрин 150 X; 2 — поли-
метилметакрилаг; <? —полиамид
(цитель 42); і — полиэтилен
(алатон 14); 5—дельрин 500Х;
6 — полиамид (цитель 101).
Поэтому при литье полиформальдегида кости расплавов некоторых
часто применяют различные конструкции
сопел с запирающим клапаном, чтобы
предотвратить вытекание расплава при
обратном ходе плунжера (по аналогии
с переработкой полиамидов).
Давление при литье варьируют в
зависимости от сложности формы и толщины стенок изделия.
Абсолютная величина удельного давления зависит от конструкции
машины, до обычно бывает не ниже 1000 am.
При литье ацетальных смол предпочтительны высокие скорости
сдвига, однако выше определенного значения скорости сдвига
(«критическая скорость») на кривых напряжение сдвига — скорость
сдвига появляются изломы. В этом случае вытекающий из сопла
расплавленный материал становится неоднородным и качество
изделий ухудшается. Снижение скорости впрыска приводит к
уменьшению текучести материала, а также к появлению ориентационного
эффекта в изделиях.
Температура формы. Попадая в форму, расплавленный материал
расширяется и быстро охлаждается. Скорость кристаллизации
ацетальных смол очень высока [47], и кристаллизация может
произойти при недостаточном заполнении формы. Поэтому целесообразно
поддерживать температуру формы в пределах 70—140 °С. Зависимость
267
50
ЬО
зо
20
10
0
/А
?
1
текучести ацетальной смолы от температуры формы приводится
на рис. 81.
На величину усадки полимера при затвердевании и на вид
поверхности изделий большое влияние оказывают температура и
правильное оформление охлаждающих каналов.
Относительно высокая кристалличность ацетальных смол
обусловливает значительную усадку полимера при затвердевании в форме.
Величина усадки колеблется от 1,5 до 3,5%. Это свойство
ацетальных смол следует учитывать при конструировании оснастки,
особенно в тех случаях, когда предъявляются жесткие требования
в отношении предельных допусков (при изготовлении прецизионных
изделий). Поскольку величина усадки
зависит от условий переработки, формы
и размеров отливки и не поддается
точному предварительному расчету,
приходится прибегать к специальным
экспериментам. Например, в процессе опытной
переработки используют незакаленную
форму с расчетом на минимальную усадку,
размеры которой корректируют в
зависимости от полученных результатов.
Специальными опытами было показано [12,
13, 18], что усадка зависит от температуры
расплава и формы, давления впрыска,
вида литника и толщины образца.
Если форма нагрета выше 85 °С, та
после выемки отливки может происходить
дополнительная усадка в пределах 0,2—
0,3%. Для стабилизации размеров таких
изделий, а также для снятия
возможных внутренних напряжений (чтобы предотвратить коробление)
рекомендуется проводить термообработку (отжиг) изделий,
погружая их на определенное время в очищенное минеральное масло при
температуре около 150 °С. Продолжительность отжига зависит от
толщины изделий.
Заданная температура формы поддерживается обычно с помощью
электронагрева или термостата, через который циркулирует
охлаждающая жидкость.
Экструзия
Экструдеры являются необходимым звеном технологической
схемы получения ацетальных смол, так как для последующей
переработки с помощью стандартных способов нужно превратить
порошкообразный полимер в гранулят. Кроме этого экструзия позволяет
добиться хорошей гомогенности ингредиентов (стабилизаторы^
пигменты и т. п.) в расплавленном состоянии. Специфические
свойства порошкообразного полиформальдегида (наличие нестабильной
части, низкая термостабильность) заставляют применять экстру-
268
800 1200 WOO 2000 2Ш
Давление, кГ/см2
Рис. 81. Зависимость
текучести ацетальной смолы
(хостаформ С 9010) от
температуры формы [6]:
І — 70 °С; 2—85 °С: 5 —100 °С.
деры специальной конструкции. В то же время для получения гра-
нулята, поступающего в продажу, вполне пригодны одно- и двух-
червячные машины обычного типа.
Необходимым требованием, так же как и при литье под
давлением, является отсутствие, застойных зон в цилиндре и головке,
где материал может начать разлагаться. Целесообразно использовать
экструдеры с отношением длины червяка к диаметру 15—25 D,
так как в этом случае обеспечивается достаточное
пластифицирующее действие при относительно небольшом градиенте температур
между стенкой и материалом. Рекомендуются червяки такого же
типа, как и для экструзии полиамидов (с постоянным шагом).
Дозирующая зона должна быть по возможности более длинной,
а зона сжатия короткой. Степень сжатия (т. е. отношение объема
материала на выходе к объему на входе) должна составлять 3—4.
Температура переработки несколько ниже, чем при литье под
давлением: для гомополимера она составляет 180—205 °С, для
сополимеров 170—200 °С. Обычно методом экструзии перерабатывают
высоковязкие полимеры с индексом расплава 1,5—2,5 г/10 мин.
ПРИМЕНЕНИЕ ПОЛИФОРМАЛЬДЕГИДА
Анализ структуры потребления ацетальных смол показывает г
что существует явно выраженная тенденция использовать их для
замены цветных металлов и сплавов в машиностроении и других
отраслях промышленности [1—3]. В настоящее время около 90%
выпускаемого промышленностью полимера расходуется на замену
цветных металлов и стали, прежде всего металлического литья из
алюминия, цинка, бронзы, латуни и т. д. Лишь в небольшом
количестве ацетальные смолы используются для замены полиамидов и
термореактивных смол.
Литье металлов под давлением в машиностроении и других
отраслях промышленности представляет собой крупнотоннажное
производство, исчисляемое в миллионах тонн в год. Замена цветных
металлов пластмассами прежде всего выгодна экономически. Если
даже не принимать во внимание дефицитность цветных металлов,
то остаются в силе следующие соображения:
1. Стоимость деталей включает затраты на сырье, изготовление
и дополнительную обработку изделий. Хотя в настоящее время
продажная цена на цветные металлы ниже, чем на полиформальдегид,
выпускаемый в промышленном масштабе, эта разница
компенсируется значительно большими затратами на изготовление и
многостадийную обработку металлических деталей по сравнению с инжек-
ционным литьем ацетальных смол. Кроме этого следует учесть, что
удельный объем ацетальных смол значительно выше, чем у
металлов.
2. Цена на ацетальные смолы имеет определенную тенденцию
к понижению по мере усовершенствования технологии и увеличения
масштаба производства. В настоящее время мировые цены на
269
ацетальные смолы — около 1,3 руб. за 1 кг. Расчеты показывают, что
эта цена может быть снижена до 0,9 руб/кг. В Советском Союзе при
промышленном производстве стоимость полиформальдегида [42]
составит около 1,0 руб/кг. При этом следует учесть, что для
производства полиформальдегида существует практически неисчерпаемая
сырьевая база (природный газ). В то же время мировые цены на
цветные металлы подвержены достаточно сильным колебаниям с общей
тенденцией к некоторому повышению.
В качестве иллюстрации можно привести следующие данные [3,4]:
в 1964 г. в связи с повышением цен на металлы продажная цена
ацетальных смол была только в два
раза выше цены на алюминий, на
10—20% ниже цены на литьевой
цинк и составила V2 от цены на
медь. При этом следует учесть, что
плотность ацетальных смол
значительно ниже, чем плотность металлов:
Плотность
г/слі3
Сталь 7,8
Алюминий 2,7
Бронза 8,8—8,9
Латунь 8,4—8,7
Медь 8,89
Цинк 7,1
Ацетальные смолы .... 1,42
Сравнивая физико-механические
и термические свойства ацетальных
смол и металлов, можно сделать
вывод, что пластмассы значительно
уступают металлам по прочности
и теплостойкости (см. рис. 82 и
табл. 45). Между тем анализ показывает, что в различных
областях машиностроения используется большое количество
металлических деталей, работающих в сравнительно мягких условиях,
т. е. при температуре не выше 100—110 °С и нагрузках ниже
200 кГ/см2. Расчеты показывают, что в этих случаях замена
металлов на пластмассы экономически выгодна, даже если такие детали
составят всего 1% от общего потребления металлов. На самом деле
эта цифра значительно больше.
До сих пор мы говорили только об экономических преимуществах
ацетальных смол перед металлами. Следует отметить также ряд
технологических преимуществ. Высокая износостойкость, низкий
коэффициент трения, высокая усталостная прочность и стойкость
к коррозии позволяют во многих случаях получить выигрыш в
длительности эксплуатации по сравнению с другими материалами.
За 6 лет с начала промышленного выпуска ацетальных смол
их производство с 7000 т в год увеличилось приблизительно до
270
Полиметилакрилат
'дельрин
Целкон
олиамид
Поликарбонат
Рис. 82. Сравнительная диаграмма
напряжения — деформация для
некоторых материалов.
ТАБЛИЦА 4 5»
Температуры плавления и начала размягчения некоторых материалов
Материал
Медь
Бронза
Латунь
Алюминий
Магналий (сплав
Mg и AI)
Цинк
Температура
начала
размягчения
°С
1 МІМ
Температура
плавления
°С
1083
1000
900
657
610
420
Материал
Мягкий припой
Ацетальные смолы
Полиамиды
Поликарбонат
Ударопрочный
полистирол
Температура
начала
размягчения
°С
110-120
100
120
60-80
Температура
плавления
°С
180
165—180
180—220
220—230
45 000 т в год. Ожидается, что к 1970 г. мировое производство этого*
пластика достигнет 80000—100 000 т в год [3].
Структура потребления ацетальных смол различными областями
промышленности можно проиллюстрировать на примере США
(табл. 46).
ТАБЛИЦА 46
Структура потребления ацетальных смол в США
Области применения
Автомобилестроение
Конструкционные детали
Арматура
Электротехническая
промышленность
Бытовые электроприборы
Упаковка
Прочие области
1965 г.
тыс. т
3,4
3,2
2,0
1,9
1,2
0,8
3,0
%
22,2
20,9
13,1
12,4
7,9
5,2
18,3
1966 г.
тыс. т
3,9
3,5
2,3
2,3
1,4
1,0
3,6
%
21,8
19,6
12,8
12,8
7,8
5,6
19,6
Кратко остановимся на основных областях применения
ацетальсмол.
Автомобилестроение. По данным английских специалистов [44 Ї
около 25% ацетальных смол расходуется на изготовление
механических и электротехнических деталей в автомобилестроении. Сюда
относится изготовление деталей топливных насосов и трубопроводов,
подвесок, замков и ручек дверей, насоса для промывки ветровога
стекла, дворников, втулок зубчатых передач руля, приборных
щитков и т. д. [42, 75]. Методы расчетов конструкционных деталей иа
ацетальных смол рассмотрены в работах [5—8].
Машиностроение. Важным направлением в использовании
ацетальных смол стало изготовление зубчатых колес, шестерен и втулок
271
подшипников скольжения. В этих областях наиболее эффективно
проявились такие свойства полимера, как стабильность размеров,
жесткость, износостойкость, низкий статический и динамический
коэффициенты трения. Другое направление связано с изготовлением
корпусов приборов, замков, ручек, пружин, т. е. деталей, в которых
реализуется прочность и упругость материала [51, 53, 54]. Большое
значение приобретает изготовление различных армированных
деталей. Изделия из ацетальных смол хорошо поддаются механической
обработке. Их можно соединять болтами, сваривать [18]. Однако
они плохо поддаются склеиванию из-за низкой растворимости и
плохой адгезии. Описаны способы различной обработки
поверхности ацетальных смол, включая травление, металлизацию, окраску,
матирование и т. п. [18, 26, 28, 29].
В опытном масштабе выпускаются наполненные ацетальные
смолы. Наполнителем является стекловолокно и
политетрафторэтилен [3, 17]. Такой полимер обладает повышенной прочностью
и теплостойкостью. Основная область его применения —
подшипники, работающие при 80—100 °С.
Арматура. Все большее значение приобретает изготовление
из ацетальных смол корпусов и рабочих колес насосов, деталей
кранов, вентилей, задвижек, корпусов приборов и т. п. Расширенные
испытания проходят трубы, изготовленные из ацеталей [27, 30—34].
К их положительным качествам слезет отнести высокую стойкость
к динамическим нагрузкам и устойчивость к нефтепродуктам.
Электротехника. Наибольший интерес представляет
использование ацетальных смол в узлах, где необходимо сочетание хороших
электроизоляционных свойств при любой влажности с прочностью
и упругостью. Это различного типа переключатели и пружины в
телефонных и телеграфных аппаратах, электрических машинах и т. п.
[57, 59-61].
Бытовые приборы и изделия. Ацетальные смолы широко
используются в производстве бытовых электрических машин, игрушек и
других изделий широкого потребления. Приятный внешний вид
материала (естественный цвет — молочно-белый), возможность
легкого окрашивания, большая прочность создают предпосылки для
еще более широкого внедрения этого материала. Определяющим
фактором должна явиться цена на материал. Очевидно, что для
замены таких ставших традиционными материалов, как стекло,
полистирол, фенопласт, хрупкость которых причиняет известные
неудобства, ацетальные смолы должны иметь сопоставимые с ними цены
[64-70].
Другие области применения. Выдувание различных сосудов из
ацетальных смол и прежде всего контейнеров для аэрозолей
представляет большой интерес в связи с низкой газопроницаемостью
метериала и хорошими упругими свойствами. Специально для этих
целей фирма «Селаниз корпорейшн» приступила к выпуску
сополимера, содержащего три компонента, включая диэпоксид [25, 72].
Он обладает низкой вязкостью расплава и может конкурировать
272
по цене с полиэтиленом. Однако сосуды из ацетальных смол в два
раза дороже сосудов из необработанного стекла и жести [62, 63].
Кабели с покрытием из ацетальных смол могут применяться
в некоторых специальных случаях. Изготовление пленок из
ацетальных смол пока не получило большого развития [71].
Производство волокна представляет больший интерес, так как помимо
хорошей прочности (сравнимой с прочностью полиамидных
волокон) оно обладает рядом специфических свойств [34, 35].
ЛИТЕРАТУРА
1. Barrett G. F., Plastics, 25, 136 A960).
2. Mod. Plast, 40, № 5, 108 A963).
3. Mod. Plast, 47, № 5, 99 A963).
4. M і 1 1 er W. G., Brit. Plast., 37 (№ 7), 356 A964).
5. Chem. Eng. News, 44, 36 A966).
6. Brit. Plast., 39, № 9, 516 A966),
7. Akin R. В., Acetal resins, Beinhold Plast., N. Y., 1962.
8. KoxHO Ю. А., Лебединская Э. А., Мень С.М., Серги-
енко А. А., Феликсон А. М., ПІ а г и я н В. Ф.,
Полиформальдегид, Изд. «Техника», Киев, 1964.
9. L і n t о n W. H., Plast. Inst. Trans, and Journal, June 1960, 75.
10. S p e n s e r R. S., G і 1 m 0 r e G. D., Wiley R. M., J. Appl. Phys.,
21, 527 A950).
11Я E v el an d J., К ar a m H. J., Beyer G. E., Soc. Plast. End., 30
May, 1956.
12. Richardson P. N., SPE J., 16, № 2, 1324 A960).
13. С о g d e 11 J. F., H a r d e s t г у R. H., SPE J., 14, № 1, 25 A958).
14. R є і n b а с h er W. R.* Kunststoffe, 51, № 6, 321 A961).
15. Ну er H., Schmidt H., Kunststoffe, 43, № 10, 282 A964).
16. S с h m і d t H., Brit. Plast., 38, № 9, 546 A965); № Ю, 630 A965).
17. Can. Plast., 11, 42 A963).
18. Schmidt H., Plast Verarbeiter, № 10—11, 595 u. 665 A964) [Химия
и технология полимеров, № 10, 142 A965)].
19. К 1 і п е Т., Mod. Plast., 35, № 4, 153 A957).
20. Mod. Plast., 42, № 1, 104 A965).
21. M и л и ц к о в а Е. А., Викторов Е. С., Соколов А. Д.,
Костиков В. П., Пласт, массы, № 1, 23 A965).
22. Кестельман В. Н., Фельдман Л. И., КестельманН. Я.,
Пласт, массы, № 7, 65 A964).
23. Т s h і d a, Koll.-Z., 174, № 2, 162 A961).
24. Ghem. Eng. News., 42, 32 A964).
25. Weibermel K., Fischer E., Gutweiler K., Hermann H.D.,
Kunststoffe, 54, 401 A964) [Химия и технология полимеров, № 3, 92
A965)].
26. Mod. Plast., 41, № 5, 101 A964).
27. Plastics, 28, № 313, 72 A963).
28. Накамура, А р а и, Нихон Кикай Гаккайси (Япон.), 66, № 6, 843
A963).
29. В run er W., Baranano С., Mod. Plast., 39, № 4, 97 A961).
30. Oil in Ganada, 15, № 1, 37 A962) и 16, № 24, 36 A963).
31. Mod. Plast., 39, № и, 88 A962).
32. Ghem. Ing. News., 42, № 30 (I960).
33. Лосев Б. Н., Монина Н. А., Нефтяное хозяйство, N» 12, 57
A965).
34. Н all Ae L., Fibres, End. a. Ghem., І8, № 8, 257 A957).
35. Petroleum Refiner, 41, № 11, 131 A962).
18 Заказ ІІ35. 273
36. Alsup R. G., Punderson J. 0., Leverett 0. F., J. Appl.
Polymer Sei., 2, 181 A959), Химия и технология полимеров, № 3, 137
A960).
37. Dunkel M., Z. phys. Chem. (Leipzig), A128, 42 A928).
38. Pinsky J., Mod. Plast., 34, № 8, 145 A957).
39. M у е г s A. W., Rogers G. E., Stannet V., Srwarc M., Mod.
Plast., 34, № 9, 157 A957).
40. К lute С. H., Franklin P. J., J. Polymer Sei., 32, 161 A958).
41. S t a n n e t t V., S г w а г s M., J. Polymer Sei., 16, 89 A955).
42. См. ссылку 8.
43. См. ссылку 8.
44. Mod. Plast., 43, № 12 A966).
45. Гринблат В. Н., Гладышева Л. А., Лапшин В. В.,
Пласт, массы, № 8, 35 A966).
46. Косинская В., Фейгин Е., Пласт, массы, № 6, 8 A961).
47. Абрамова И* М., Ермолина А. В.т И г о н и н Л. А., К а р-
г ин В. А., ДАН СССР, 145, 1047 A962).
48. Read В. Е., W і 11 і a m s G., Polymer, 2, 239 A961).
49. Ishida Y.,MatsuoM.,Togami S., Y a m a f u j і К., T a k a'y a-
n a g і M., Kolloid-Z. und Z. Polymere, 183, 74 A962).
50. Kuchkuda R. W., M a 1 b у H. S., SPE J., 18, 907 A962).
51. H a r d e s t у R., Mach. Design, 34, № 22, 53 A962).
52. К a n о v e t z J. E., A k u t і n M.S., R о m a s h о v a A. G., Kar
p і 1 e v і t с h V» M., Chem. prumysl., 13, 209 A963).
N і s і m u r a M., Plast. Age, 6, № 11, 47 A960).
Miller R. L., Techn. Rundschau, 53, № 25, 21 A961).
С і a r 1 о n e A, N., SPE J., 18, 1367 A962).
S t a u d і n g e r J. P., Chem. and Ind., № 23, 771 A961).
Flur у К., Technika (Suisse), 11, № 16, 1161 A962).
F e jgi n J., Przem. Chem., 41, № 8, 423 A962).
К і s 11 e г J.f Kunst. — Plast., 9, № 2, 172 A962).
Lowis S., Technika, 11, № 21, 1559 A962).
61. Симидзу С, Коге Дзайре, 10, № 8, 16 A962).
€2. Patt G., Kautschuk und Gummi, 14, 281 A961).
63. Young J., Kunststoffe, 51, 445 A961).
64. Левин А. Н., Сб. «Применение полимерных материалов в
машиностроении», Машгиз, М. 1962, стр. 29.
Reicherzer R., Industrieblatt, 61, 563 A961).
Ch or і G., Plast. Age, 8, № 10, 55 A962).
К г a u b W., Maschine, 15, № 6, 25 A961),
К а б а я с и М., Коге дзайре, 10, № 8, 25 A962),
Я м а у т и А., Коге дзайре, 10, № 8, 28 A962).
S 1 а п е у S., Trau Inst. Chem. Engrs., 40, № 1 A962).
Мышленникова В. А., Л и Ц., О х р и м е н к о П. С,
Лакокрасочные материалы, № 3, 12 A962).
Mohrberg W., J. Soc. Cosmetic Chem., 12, 478 A961).
Михайлов Г. П.^Эйделнант М. П., Высокомол. соед., 2, № 10,
1552 A960).
Ishida Y., Kolloidzschr., 171, 149 A960).
D ol d er F., Technische Rundschau, № 7, 42 A962).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Автоокислительная деструкция 103
Автоускоренная деструкция 216
Агенты передачи цепи 46, 48, 49, 51,
87—89, 109, 198, 204
Адгезия 213
Активные добавки 51
Активные центры
деполимеризации 104, ИЗ, 120
деструкции 110
сополимеризации 152
Алкон 250
А ль дольная конденсация 17
Анизотропность реакций 93
Анионная сополимеризация 140 ел.
Антиоксиданти 120, 121, 128, 130,
216, 220, 236, 239
Аррениуса уравнение 32
Ацетали (ацетальные смолы) 7, 247,
248, 250
влагопоглощение 258
горение 262
проницаемость 262
свойства 251, 252, 256
термодеструкция 257
токсичность 262
химическая стойкость 258—260
циклические 25
электрические свойства 256
Ацетилирование 216 ел.
Ацидолиз 101, 103, НО ел.
полиоксиметиленов 226, 228
Болрин 250
Водопоглощение 258
Выдувание 263
Вязкость 203, 267
Газопроницаемость 262
Газофазная полимеризация
формальдегида 31 ел.
инициирование 186
катализаторы 36—39
кинетика 36 ел.
механизм 37, 38
термодинамика 32 ел., 35
Гексагональный полиоксиметилен
166, 167 ел., 180, 181
Гемиформали
очистка 191
пиролиз 197
«Гемиформальиая» полимеризация 38
Гибкость макромолекулы поли-
оксиметилена 178
«Гидридный переход» 89
Гидролиз 101, 103
Гомополимеризацрш триоксана 230,
232
радиационная 231
степень полимеризации 233
твердофазная 231
Горение 262
Давление при литье
полиформальдегида 276
Дельрин 7, 35, 247, 249, 252, 254,
258, 261, 265, 267
Деполимеризация 54, 101, 103, 137,
199
Деструкция полиформальдегида
101 ел.
механизм 103
окислительная 101, 122, 257
«Дефекты» 93
Дисперсность 203
Диффузия 93
Дуракон 250
Еи-полиоксиметилен 30, 35
«Жесткость» макромолекулы поли-
оксиметилена 178
«Живущие» полимеры 47, 49, 85,
87, 145, 178
Жидкостная полимеризация
триоксана 70 ел.
индукционный период 73 ел.
катализаторы 70 ел., 82, 83
кинетика 70, 83
механизм 73 ел.
молекулярный вес 85 ел.
растворители 70 ел., 82
скорость 79 ел.
Жидкостная полимеризация
формальдегида 57
индукционный период 186
растворители 57
275
«Закалка» газов 13
Закон Фика 208
Замедлители 51
«Запределивание» 79
Ингибиторы 51, 220
Индукционный период 73, 90, 216
Изомеризация 139
Изотермический режим 213
Инициирование полимеризации 45
триоксана 73 ел.
формальдегида 186
Ионная сополимеризация 140
Ионообменные смолы 201
Канниццаро реакция 17
Катализаторы полимеризации
триоксана 70 ел., 82, 83
формальдегида 36—39, 42—51,
60, 70 ел., 82, 83, 95, 144,
156, 158, 161, 210, 211,
215, 219, 244
Катионная сополимеризация 140,
146 ел.
Кинетика полимеризации
в «кипящем слое» 213
в растворе 92 ел.
газофазной 36 ел.
жидкофазной 70, 83
Клаузиуса — Клапейрона уравнение
108
«Константа термостабильности» 214
Константы
проницаемости 262
сополимеризации 138, 140
термодинамические,
полимеризации 35, 67, 68
Конструкционные термопласты 248
Конформации полиоксиметилена
173 ел.
Концевые группы 31, 55, 104, 257
блокирование 122 ел., 126, 128,
216
модификация 237
Коэффициенты
адсорбции 199
массопередачи 194
теплопередачи 194
Литье под давлением 249, 250, 263,
264
Люминесценция 120
Майо — Льюиса уравнение 139
Материальный баланс
полимеризации 227
Мгновенная концентрация
формальдегида 195
Метилаль 18
Механодеструкция 101
276
Молекулярно-весовое распределение
31, 34, 55, 150, 206, 210, 221,
231, 233, 236
Молекулярные сита 200, 201
Молекулярный вес 31, 53 ел., 85 ел.,
91, 202, 209, 210
полиоксиметилена 206
полиформальдегида 221
«Обезметаноливание» 12
Окислительная деструкция 101, 122,
257
стабилизация 128
Орторомбический полиоксиметилен
166, 169 ел.
«Отжиг» дефектов 96
Очистка 225
гемиформаля 191
формальдегида 196 ел.
— глубокая 196
— физические методы 196, 200 ел.
— форполимеризацией 196
— химические методы 196, 202
Параформальдегид 10, 20, 23, 187,
197, 229
Парциальная конденсация
паров спирта 191
— формалина 192, 197
Передача цепи 41, 89
с разрывом 146 ел.
— — и строение сополимеров 151
МБР 150
— — механизм 148
Перемешивание 205
Переработка полиформальдегида
102, 214, 247, 263
Пиролиз
гемиформалей (полуацеталей) 189,
197
полиоксиметиленов 186 ел., 197,
202
полиформальдегида 187, 197
триоксана 187
«Показатель стабильности» 220
Ползучесть 255
Полиамиды 248, 254, 271
Поливинилиденхлорид 262
Поливинилхлорид 102
Полидиметилсилоксан 175
Полидиоксолан 156
Поликарбонат 248, 271
Полимеризатор 214
Полимеризационно-деполимеризаци-
онное равновесие 75
Полимеризация в гетерогенной
системе 204
Полимеризация триоксана 65 ел.
в расплаве 89 ел.
Полимеризация триоксана 65 ел.
— — индукционный период 90
— — молекулярный вес 91
— — скорость 90
жидкофазная. См. жидкофазная
полимеризация триоксана
кинетика 70 ел.
твердофазная. См. твердофазная
п о л име ризация
термодинамика 66—69
Полимеризация формальдегида 202,
226
в гетерогенной системе 204
в «кипящем слое» 213
— — — катализаторы 210
— — — кинетика 203
— — — растворимость 211
— — — регуляторы роста цепи
211
— — — скорость 208
— — — температура 212
в полярных средах 59 ел.
— — — катализаторы 60
— — — pH среды 60
в растворе 40 ел.
— — активные добавки 51
— — катализаторы 42—49, 51
— — катионная 50
— — кинетика 44 ел.
— — молекулярный вес 53 ел.
— — растворители 44, 53 ел., 56
газофазная см. Газофазная
полимеризация формальдегида
жидкофазная см. жидкофазная
полимеризация
формальдегида
радиационная 57
твердофазная. См. твердофазная
полимеризация
Полиметилен 103
Полиметилметакрилат 102
Полиоксиметиленгликоли 20 ел.
термодеструкция 215
Полиоксиметилены 9, 20
гексагональный 167
низкомолекулярные линейные 20
орторомбический 166, 169 ел.
параформальдегид 23
период идентичности 167, 169
пиролиз 186 ел.
структура 167 ел., 180 ел.
термостабильность 216, 244
' циклические 25 ел.
а-Полиоксиметилен 10, 24, 59, 106,
187, 197, 202
?-, o-, у-, е-Полиоксиметилены 24,
25, 59, 65
Полиоксипропилен 103
Полиоксиэтилен 103
Полипропилен 103
Полистирол 102, 254, 271
Полифайд 250
Полифениленоксид 248
Полиформальдегид 248
газопроницаемость 262
деструкция см. Деструкция
полиформальдегида
износостойкость 254
индекс расплава 248
переработка 102, 214, 247, 263
применение 269
свойства 247, 257
стабилизация 101 ел., 214, 216
структура 6, 166 ел.
термостабильность 7
технология производства 184 ел.
химическая стойкость 257
Полиэтилен 102, 103, 175, 262
Полиэтилентерефталат 262
Пороговый эффект концентрации
катализатора 48
Пост-полимеризация 58, 95
Предел устойчивости раствора 195
Предельная температура 33, 102,
186, 195, 201
Примеси 216, 257
Проницаемость 261
Радиационная полимеризация
триоксана 91 ел., 231
формальдегида 57, 58
Радиационная сополимеризация
формальдегида 162 ел.
Радикальная сополимеризация 162,
163
Распределение
мономерных звеньев 139
по молекулярным весам 31, 34,
150, 206, 210, 221, 231, 233,
236
Семенова — Флори 150
Растворимость 203
Растворители 53, 54, 56, 57, 70 ел.,
82, 83, 211, 218
Растворы формальдегида 18 ел.
полимеризация 40 ел.
равновесие 19
Реакционная способность сомономе-
ров 138
Реакция
Канниццаро 17
Тищенко 17
Регуляторы роста цепи 211
Семенова — Флори распределение
150
Синергический эффект 43
Скорость
адсорбции 199
деполимеризации 217
277
Скорость
диффузии 209
полимеризации 73, 79 ел., 199,
204, 205, 208
сдвига 267
теплопередачи 194
Смазка 250
Сокатализаторы 45, 47, 80, 81, 90
Сополимер АБС 254
Сополимеризация триоксана 136 ел.
антиоксиданты 239
сомономеры 233
Сополимеризация формальдегида
136 ел., 241
катализаторы 141
— Фриде ля — Крафтса 244
Сополимеры триоксана
индекс расплава 236
модификация концевых групп 237
с винильными мономерами 160 ел.
с гетероциклами 156, 158
с 1,3-диоксоланом 103, 156
с окисью этилена 153
свойства 240
стабилизация 239
структура и состав 235
температуры плавления 240
теплоты плавления 240
термообработка 237, 238
технология производства 225 ел.
Сополимеры формальдегида
с акролеином. 144
с альдегидами 140
с ацетальдегидом 141
с винильными мономерами 145
с диметилк'етеном 143, 242
с изоциановой кислотой и ее
эфирами 144
с производными альдегидов и
кетонами 142
с фторальдегидом 143
с фурфуролом 144
с циклоангидридами моноокси-
карбоновых кислот 144
термостабильность 243
технология производства 225 ел.
Сорбенты 200, 201
Сорбция примесей 207
Спонтанная полимеризация 185, 212,
215, 225
триоксаиа 92
Спонтанные реакции передачи цепи
89
Стабилизаторы 130, 220
Стабилизация
и сополимеризация 136
полиформальдегида 101 ел., 216
против действия кислорода 128
сополимеров триоксана 239
Стабильность 31
278
Старение 115
Степень завершенности реакции 123
Степень полимеризации 34, 50, 57,
91, 149, 151, 198, 204, 205,
208, 209, 233
«Степень прочности» 203
Стерические препятствия при
инициировании 45
Стойкость к действию кислот,
щелочей, солей, органических
костей 258, 259
Структура полиформальдегида
166 ел.
Твердофазная полимеризация
триоксана 91 ел., 95, 181, 230*
формальдегида 58
Текучесть 263
Температуры
литья 264
начала деструкции 265
плавления 179, 265, 271
полимеризации 212
предельные 33, 102, 186, 195, 201
стеклования 265
текучести 263
формы 267
Тепловой эффект полимеризации 213,
225
Термическая деструкция 101, 103 ел...
257
параформальдегида 229
Термические свойства 265
Термодинамика полимеризации
газофазной 32 ел.
гетероциклов 66 ел.
в расплаве
в растворе
Термоокислительная деструкция 101 г
113 ел.
кинетика 114
Термостойкость 136, 203, 210, 215
полиоксиметилена 216
Тетраоксиметилен 25, 28
Тищенко реакция 17
Токсичность 262
Триоксан 10, 25, 26
выделение 229 ел.
гомополимеризация см. Гомопо-
лимеризация триоксана
и сомономеры 226, 233
и чистота мономеров 226
концевые группы 226, 236
кристаллизация из водной фазы
229
очистка 226
пиролиз 187
полимеризация см.
Полимеризация триоксана
Триоксап 10, 25, 26
производство 226
распределение блоков по цепи 226
растворимость 225
свойства 26 ел., 225
сополимеризация см. Сополиме-
ризация триоксана
сополимеры см. Сополимеры
триоксана
токсичность 225
устойчивость при хранении 225
экстракция из дистиллята 229
Удельная энергия когезии 259
Уравнение
Аррениуса 32
Клаузиуса — Клапейрона 108
Майо — Льюиса 138, 139
материального баланса 207
состава сополимера 138
Усадка 268
Фика закон 208
Фонтана — Кидера механизм 139
Формали 18
Формальдегид
выделение 184 ел.
и олигомеры 9 ел.
и с омономеры 242
ионная полимеризация 6, 38
полимеризация см.
Полимеризация формальдегида
применение 9 ел.
растворимость 225
Формальдегид
растворы см. Растворы
формальдегида
свойства 9 ел., 14 ел., 225
сополимеризация см.
Сополимеризация формальдегида
сополимеры см. Сополимеры
триоксана
технология производства 9, 10 ел.
токсичность 225
устойчивость при хранении 225
Форполимер 155
Форполимеризация 197 ел.
Фотолиз 101
Фотоокисление 101
Фотостабилизаторы 132
Химическая деструкция 101, 103,
110 ел.
Химическая стойкость 260
Хостоформ С 247, 249, 250, 252, 255,
262, 265
Целкон 250, 258
Цеолиты 201
Экструзия 249, 263, 268
Энергия активации 45, 47, 49, 58,
84, 91, 95, 96, 103—105, 108,
109, 113, 119, 130, 203
Этерификация 216 ел.
Эфиры целлюлозы 254
Эффект «скачка» 95
Николай Сергеевич Ениколопян,
Станислав Александрович Волъфсон
Химия и технология полиформальдегида
Издательство «Химия», М., 1968 г.
280 с. УДК 678.644' • 141
Редактор Г. М. Мечникова
Технический редактор 3. И. Яковлева
Художник Ю. М. Сигов
Корректоры А. А. Малкина,
С. Л. Федотова
Т-15609. Подписано в печать 25/ХИ 1967 г.
Уч.-изд. л. 18,54. Печ. л. 17,5. Бум. л. 8,75.
Формат бумаги 60x90Vie. Заказ 113 5.
Тираж 7000 экз. Цена 1 р. 31 к. Тип. бумага № 2.
Тем. план 1967 г., № 9.
Ленинградская типография № \ 4
«Красный Печатник» Главполиграфпрома
Комитета по печати при Совете Министров СССР..
Московский проспект, 91.
H.C. ЕНИКОЛОПЯН
CA.. ВОЛЬФСОН
химия
ТЕХНОЛОГИЯ
ПОЛИФОРМАЛЬДЕГИДА
ИЗДАТЕЛЬСТВО ..X ИМИЛ" МОСКВА • 1968