/












































































































































































































































































Текст
P. M. Масагутов
АЛЮМОСИЛИКАТНЫЕ
КАТАЛИЗАТОРЫ
И ИЗМЕНЕНИЕ ИХ СВОЙСТВ
ПРИ КРЕКИНГЕ
НЕФТЕПРОДУКТОВ
МОСКВА
ИЗДАТЕЛЬСТВО «ХИМИЯ» 1975
УДК 665.64.097.3:546.621'284
Масагутов Р. М.
Алюмосиликатные катализаторы н изменение их свойств
при крекинге нефтепродуктов. М., «Химия», 1975.
В книге показано влияние различных факторов на
старение и отравление алюмосиликатных катализаторов крекинга
(аморфных и цеолнтных), а также изменение показателей
процесса каталитического крекинга при дезактивации
катализаторов. Описаны различные методы предупреждения
старения катализаторов крекинга и способы предохранения их от
отравления путем очистки сырья крекинга. Изложены способы
поддержания активности катализатора на оптимальном
уровне, основанные на удалении с его поверхности отравляющих
металлов. Рассмотрены возможности повышения
эффективности процесса крекинга путем добавлення в катализатор
металлов.
Книга предназначена для инженерно-технических
работников нефтеперерабатывающих и нефтехимических заводов,
сотрудников научно-исследовательских институтов, работников
проектных организаций, обслуживающего персонала установок
каталитического крекинга и катализаторных фабрик, а также
для студентов нефтяных вузов и техникумов.
272 стр., 70 табл., 102 рис., список литературы, 389 ссылок.
31406-115
М 050@1)-75 1155
© Издательство «Химия», 1975 г.
СОДЕРЖАНИЕ
Введение 5
Глава I. Катализаторы крекинга 9
Природные катализаторы 9
Синтетические катализаторы 12
Глава II. Старение катализаторов крекинга ... 34
Изменение свойств катализатора при его старении 34
Механизм спекания алюмосиликатного катализатора 54
Кинетика процесса спекания катализатора ... 59
Влияние условий промышленной эксплуатации на
старение алюмосиликатных катализаторов . . 59
Пути улучшения свойств циркулирующего
катализатора 87
Заключение 93
Глава III. Изменение активности катализаторов в
стадии крекинга 95
Состав коксовых отложений и его зависимость от
условий крекинга 95
Влияние длительности работы катализатора на
материальный баланс крекинга 102
Влияние длительности работы катализатора на
качество продуктов крекинга 108
Влияние длительности работы катализатора на
кинетические константы 112
Проведение промышленных процессов с небольшой
длительностью работы катализатора . . . . 117
Заключение 124
Глава IV. Отравление катализаторов компонентами,
содержащимися в сырье крекинга . . . 125
Сернистые и кислородные соединения .... 125
Азотистые соединения 126
Металлоорганические соединения 134
Заключение 180
Глава V. Методы очистки сырья от компонентов,
отравляющих катализаторы крекинга . . . 181
Перегонка и термические методы очистки сырья . 181
Деасфальтнзация 182
Очистка селективными растворителями . ... 183
Адсорбционная очистка 184
Очистка серной кислотой 186
Гвдрогенизационная очистка 191
Другие методы подготовки сырья каталитического
крекинга 205
Сопоставление технико-экоиомических показателей
сернокислотного и гидрогенизационного методов
очистки сырья крекинга 207
Заключение 211
Гл а в а VI. Очистка катализатора крекинга от
отравляющих металлов 212
Механические методы 212
Химические методы 213
Применение ионообменных смол (процесс Мет-х) . 225
Процесс Демет 239
Сухая деметаллизация 243
Сопоставление технико-экономических данных
очистки сырья крекинга н деметаллизации
катализатора 253
Заключение 255
Литература 256
Предметный указатель 270
ВВЕДЕНИЕ
Процесс каталитического крекинга — один из основных катали-?
тических процессов нефтеперерабатывающей и нефтехимической
промышленности — позволяет получать не только значительные
количества высокооктанового бензина, но также газообразное и
жидкое нефтехимическое сырье. В связи с этим мощность отечест-і
венных установок каталитического крекинга в текущей пятилетке
будет увеличена вдвое при общем росте переработки нефти лишь
в 1,4—1,5 раза [1].
" Процесс каталитического крекинга применяется в
нефтеперерабатывающей промышленности почти сорок лет. При этом он
неуклонно развивается. Так, в капиталистических странах мощность
установок каталитического крекинга составляет 28,8% от
мощности установок первичной перегонки [2]. В США структура
потребления нефтепродуктов обусловливала большее развитие этого
процесса, чем в Европе. Суммарная мощность установок
каталитического крекинга в США в 1970 г. была равна 938 тыс. м3/сут, или
49,5% от мощности установок первичной перегонки (табл. 1 [3]).
Таблица 1
Мощности процессов переработки нефти в США
Годы
1960
1961
1962
1963
1964
1965
1966
1970
1975*
Атмосферная
перегонка,
тыс. мЗ/сут.
1647
1670
1684
1663
1705
1711
1705
1893
2130
Каталитический
тыс. мЗ/сут.
763
787
818
866
892
884
873
938
954
крекинг
%
46,4
47,1
48,6
52,1
52,3
51,7
51,2
49,5
44,8
• Прогноз Стеифордского института.
Процесе каталитического крекинга впервые был осуществлен
в промышленности с неподвижным катализатором. В одном и том
же реакторе проводили последовательно крекинг нефтепродуктов
и регенерацию катализатора (установка Гудри). В дальнейшем
возникли более совершенные установки с проведением реакций
крекинга и регенерации в отдельных аппаратах. Поток
катализатора непрерывно двигался через реактор и регенератор. Установки
с движущимся катализатором были оформлены в следующих двух
вариантах: 1) с движущимся плотным слоем гранулированного
катализатора (зарубежные установки термофор, гудрифлоу, гуд-
резид и отечественные установки типа 43-1 и 43-102); 2) с
кипящим слоем пылевидного катализатора (зарубежные установки
флюид, модели I, II, III и IV; ортофлоу, модели А, В, С; ЮОП и
отечественные установки типа 1-Б, 1-А, 43-103, 43-104 и ГК-3) [4].
В мировой практике наиболее распространены установки с
кипящим слоем катализатора^ Так, в США мощность установок
флюид составляет примерно 85% от суммарной мощности установок
каталитического крекинга [3].
Для каждого типа установок применяют катализаторы,
отличающиеся размерами и формой гранул. На установках Гудри
применяли гранулированный и таблетированный катализаторы
размером 4—5 мм. На установках с движущимся плотным слоем
катализатора применяли вначале таблетки размером 4—5 мм, а затем
шарики диаметром от 2 до 4 мм. Установки с кипящим слоем
пылевидного катализатора снабжали вначале размолотым
катализатором, а в дальнейшем специально приготавливали
микросферические катализаторы. Это позволило существенно снизить эрозию
аппаратуры и расход катализатора, а также улучшить
аэродинамические характеристики кипящего слоя.
На современных установках каталитического крекинга
катализатор последовательно проходит реактор, отпарную зону,
регенератор и снова поступает в реактор. В течение этого цикла в
зависимости от типа установки катализатор один или два раза
транспортируется пневмоподъемником. Условия в указанных аппаратах
разные. В реакторе катализатор при 450—500 °С контактируется
с углеводородами сырья и продуктов реакции, находящимися в
парообразном или в парожидкостном состоянии. В отпарной зоне
для удаления адсорбированных углеводородов катализатор
обрабатывают перегретым водяным паром. В регенераторе при 450—
750 °С длительное время на него действует окислительная среда
кислорода воздуха. Кроме того, на катализатор действуют
меняющиеся механические нагрузки. В реакторе, регенераторе, отпарной
секции и переточных трубах установок с движущимся плотным
слоем он истирается и находится под давлением вышележащих
слоев. В аппаратах установок с кипящим слоем и пневмоподъем-
нике с движущимся плотным слоем поверхность катализатора
подвергается усиленной эрозии вследствие многократных
столкновений с другими частицами и стенками аппаратов.
Таким образом, на установках каталитического крекинга
катализатор находится в весьма тяжелых условиях. Свежий
катализатор, догруженный в установку, довольно быстро изменяет
свои свойства [7, 8]. Прежде всего уменьшаются его
каталитическая активность и селективность. Одной из причин ухудшения
свойств катализатора является изменение его удельной
поверхности, структуры пор и других физических свойств («старение
катализатора»). Другая причина — отравление катализатора,
обусловленное изменением химических и каталитических свойств его
поверхности. Отравление катализатора может быть обратимым.
В этом случае активность катализатора после удаления
каталитических ядов полностью восстанавливается. В частности,
азотистые основания и коксовые отложения обратимо отравляют алю-
мосиликатный катализатор — при окислительной регенерации они
полностью сгорают. При необратимом отравлении каталитические
яды не удаляются на какой-либо стадии процесса и постепенно
накапливаются на поверхности катализатора. Такими ядами
являются металлы и их соединения, содержащиеся в сырье.
Накопление металлов на поверхности катализатора приводит к
увеличению образования кокса, легких газов и к уменьшению выхода
бензина. В результате существенно ухудшаются
технико-экономические показатели процесса крекинга.
Качество катализатора продолжает ухудшаться все время,
пока частица циркулирует в установке. Однако вследствие
добавления свежего катализатора для восполнения его потерь свойства
катализатора устанавливаются на каком-то уровне, который
зависит от величины догрузки, режима работы установки и качества
добавляемого свежего катализатора. Поэтому в промышленности
катализатор, находящийся в системе установки, называют
равновесным, или циркулирующим.
Катализаторы крекинга в основном представляют собой
алюмосиликатные системы с разным соотношением глинозема и
кремнезема. Кроме алюмосиликатов можно применять цирконий-
силикатные, магнийсиликатные, алюмосиликатмагниевые
катализаторы [5]. Однако последние два типа катализатора
промышленного применения не нашли [6].
Алюмосиликатные катализаторы можно приготовить из
природных глин или синтетическим путем. Природные
алюмосиликатные катализаторы применяли на первых промышленных
установках крекинга. Позднее широко распространились синтетические
алюмосиликатные катализаторы, которые пс своей структуре
делятся на аморфные и кристаллические (цеолитсодержащие). В
последнее время 90% применяемых в крекинге катализаторов
составляют кристаллические алюмосиликаты. Однако аморфные
природные и синтетические катализаторы все еще используют на
некоторых предприятиях. Кроме того, цеолитсодержащие
катализаторы обычно содержат 80—90% природных или синтетических
аморфных алюмосиликатов.
Поскольку указанные катализаторы имеют ряд общих свойств,
рассмотрим все три их типа.
Все катализаторы крекинга различаются по структуре, форме,
размерам частиц, методам приготовления, физико-химическим
свойствам, уровню активности, селективности, стабильности, но
все они обладают кислотными свойствами, что является основой
их каталитической активности. Кроме того, практически все
катализаторы крекинга содержат алюмосиликатные системы.
Поэтому в настоящей монографии термин «алюмосиликатные
катализаторы» относятся ко всем типам катализаторов крекинга, включая
природные и синтетические, свежие и равновесные, аморфные и
кристаллические (цеолитсодержащие), микросферические и
шариковые и др.
Благодаря одинаковой каталитической природе катализаторы
крекинга разных типов изменяются по одним и тем же
закономерностям. Но иногда они ведут себя по-разному. В каждом таком
случае в тексте указывается — о каком типе катализатора идет
речь.
Автор выражает искреннюю благодарность канд. техн. наук
А. С. Эйгенсону, Б. Ф. Морозову, Г. А. Бергу, С. Г. Прокопюку,
Р. А. Даниловой и С. Г. Тихановской за активное участие в
проведении исследований и обсуждении их результатов, а также
коллективу лаборатории научно-технической и патентной информации
НИИнефтехим за помощь при оформлении монографии.
Глава І
КАТАЛИЗАТОРЫ КРЕКИНГА
ПРИРОДНЫЕ КАТАЛИЗАТОРЫ
В начале развития процесса крекинга в качестве
катализаторов применяли природные глины — в исходном состоянии и после
их химической активации. Наиболее широко использовали
кристаллические глины бентонитового типа, а также активированные
минералы типа коалинита Al2O3-2SiO2-2H2O и галлуазита
А12О3 • 2SiO2 • 2Н2О • 2Н2<Э.
Основной составной частью бентонита является
монтмориллонит Al2O3-4SiO2-H2O-rtH2O. Характерная особенность
бентонитов— набухание или уменьшение объема, в зависимости от
давления водяного пара в окружающей среде. В формуле
монтмориллонита лН2О обозначает число молекул воды, вызывающее
набухание. __
В соответствии с современными представлениями, особенность
силикатных материалов — способность катиона кремния входить в
тетраэдрическую координацию с четырьмя анионами кислорода
[SiC^]4". Относительно высокая твердость и высокие температуры
плавления силикатов и объясняются прочностью связи между Si4+
и О2~. Структура силикатов создается за счет объединения в
комплексы тетраэдров [БіОЛі^с^помощью общего аниона кислорода
в одной из его вершинУДва тетраэдра могут объединиться"!? комг
плексную группу [Si2o?]6- (рис. 1). Аналогично этому три
тетраэдра могут образовать кольцеобразный комплексный анион
[БізОд]6" с тремя мостиками из ионов кислорода и шестью
отрицательными зарядами. Более сложные анионы, составленные из
четырех и шести тетраэдров, образуются таким же способом (см.
рис. 1). Тетраэдры [SiO4]4~ могут соединяться с помощью
кислородных мостиков в цепочечную структуру, две цепочки могут
связаться боковыми мостиками из кислородных ионов с образованием
пояса из двойной цепочки (рис. 2). Наконец, возможна
комбинация тетраэдров [SiO4]4~ по всем пространственным направлениям
с использованием всех ионов кислорода в качестве структурных
мостиков. В результате образуется трехмерный каркас,
напоминающий структуру пчелиных сот (рис. 3). Силикатные структуры
чрезвычайно разнообразны п многочисленны. Это разнообразие
обусловлено способностью иона алюминия соединяться с ионами
кислорода с образованием тетраэдрического комплекса [A1O4J.
В результате изоморфного замещения групп SiO4 группами А1О4
каркасы силикатов превращаются в каркасы алюмосиликатов. Так
как размеры обеих групп в пространстве почти полностью
совпадают, при замещении геометрические размеры структуры не изме-
Рис. І. Комплексы из тетраэдров Рис. 2. Цепочки и пояса из тетраэд-
[SiO^]: ров [SiO^].
Щ — кремний, О — кислород.
няются. Однако группа А1О4 вносит в единицу структуры
добавочный отрицательный заряд, так как А13+, занимая место Si4+,
насыщает только три, а не четыре свободные валентности кислорода.
Этот заряд в алюмосиликате нейтрализуется щелочным или
щелочноземельным катионом [9,
10].
Часто алюминий входит в
силикатные структуры в виде окта-
эдрической группы А1О6.
Элементарная ячейка
кристаллической структуры
монтмориллонита построена из
силикатных слоев, расположенных по обе
стороны от слоев, которые
содержат алюминий (рис. 4).
Силикатные слои состоят из
тетраэдров [SiO4]4~, вершины которых
попеременно направлены к
слоям, содержащим алюминий, и к
наружной стороне пакета. Тетраэдры, направленные к наружной
стороне, содержат гидроксильные группы. В результате
силикатный слой отвечает формуле [Si408(OH2]oo. Внутренний слой,
содержащий алюминий, составлен из октаэдров и имеет
усредненный состав [AI (О, ОНN]оо [11].
Толщина этих трехслойных пакетов равна 6,5 А. Вследствие
внутрикристаллического набухания расстояние между слоями ра-
f
Рис. 3. Трехмерный каркас из
тетраэдров [SO]
!0
стет, и толщина пакета может достигать 19,6 А. После
прокалки глины до 200—300 °С набухание увеличивается, при увеличении
температуры прокалки оно уменьшается, а при 500—550 °С глина
необратимо теряет способность к набуханию [12].
Для повышения каталитической активности монтмориллонит
обрабатывают сильными минеральными кислотами. Результат
химической активации зависит от природы глины, крепости кислоты,
температуры и длительности обработки. Активация состоит в
замене обменосиособных катинов водородом и удалении магния и
железа, а также некоторой части алюминия. Кислотная обработка
10,25і
О О ©ОН «Si oAI
Рис. 4. Структура монтмориллонита.
©OH *Si oAI
OH2O
Рис. 5. Структура галлуазита.
увеличивает удельную поверхность, объем пор и кислотность глин
[12]. Все это существенно повышает каталитическую активность
монтмориллонита. По мере ужесточения кислотной обработки
крекирующая активность глины вначале возрастает до максимума, а
затем уменьшается; максимальная активность достигается при
остаточном содержании окисей алюминия и железа 15—20%. Рент-
геноструктурный анализ показал, что при такой глубине удаления
металлов структура монтмориллонита не разрушается.
Глины типа коалинита и галлуазита также применяют в
качестве катализаторов крекинга. Эти глины состоят из двухслойной
решетки чередующихся слоев октаэдров А1(О,ОНN и тетраэдров
Si (О, ОН) 4, связанных между собой общими атомами кислорода.
Структура галлуазита, представленная на рис. 5, помогает
объяснению его свойств и, в первую очередь, отсутствия внутрикристал-
лического набухания, легкости частичного обезвоживания и
реакционной способности кремния и алюминия [11]. Эти глины приме-
11
няют в качестве катализаторов крекинга после их термической
активации или после термической активации с последующей
кислотной обработкой.
Низкая стабильность природных катализаторов крекинга
явилась причиной разработки синтетических алюмосиликатных
катализаторов, которые нашли широкое промышленное применение.
Однако дешевизна природных катализаторов не позволила
полностью от них отказаться. Появились также так называемые
полусинтетические катализаторы, полученные нанесением
синтетического алюмосиликата на частицу активированной глины. Это
позволяет использовать достоинства алюмосиликатов обоих типов.
СИНТЕТИЧЕСКИЕ КАТАЛИЗАТОРЫ
По структуре скелета синтетические алюмосиликатные
катализаторы делятся на аморфные и кристаллические. Последние
появились лишь в шестидесятых годах. Высокая активность,
селективность и стабильность способствовали быстрому распространению
кристаллических, или цеолитсодержащих, катализаторов крекинга.
Аморфные алюмосиликатные синтетические катализаторы.
Имеется много промышленных способов получения синтетических
алюмосиликатных катализаторов. Обычно аморфные
алюмосиликатные катализаторы синтезируют путем взаимодействия раство-,
ров жидкого стекла Na2O-3SiO2 и сернокислого алюминия
Al2(SO4K [12—15]. Этот синтез выражается следующим
уравнением [12, 14]:
7(Na2O -3SiO2) + A12(SO4K + 3H.2SO4 *¦ Na2O(Al„O3 -21SiO2) + 6Na2SO4 + 3H2O
При быстром смешении исходных растворов образуется
коллоидный раствор алюмосиликата натрия, который через некоторое
время превращается в гидрогель с определенной формой и
размером частиц. Для получения шарикового катализатора струйки
золя направляют в слой турбинного масла, где он разбивается на
отдельные капельки; под действием сил поверхностного натяжения
они принимают форму шариков и затвердевают. При производстве
микросферического катализатора золь распыляют в слой
трансформаторного масла сжатым воздухол;.
В дальнейшем полученные микросферы и шарики алюмосили-
катного гидрогеля подвергают термообработке, активации и
промывке. В процессе термообработки возникает структура
катализатора, обеспечивающая ему высокую механическую прочность и
необходимые диффузионные свойства. На этой стадии размер частиц
гидрогеля существенно уменьшается вследствие синерезиса —
уплотнения вещества и выделения интермицеллярной жидкости. При
обычных температурах синерезис протекает недостаточно быстро.
Для его ускорения раствор подогревают.
Полученный в предыдущих стадиях алюмосиликат натрия
Na2O(Al2O3-21SiO2), обладающий малой активностью (из-за высо-
12
го w во во
Содержание АЇ;.О3 ,°'э
WO
кого содержания натрия), активируют путем обмена ионов натрия
на ионы алюминия, происходящего при обработке катализатора
слабым раствором сернокислого алюминия. В результате
содержание натрия снижается с 5—6% до десятых и сотых процента, а
содержание алюминия увеличивается с 7—9 до 12—13%.
После активации гранулы с целью удаления вредных
соединений, главным образом ионов SO|~, промывают водой, затем
пропитывают поверхностно-активными веществами и направляют на
сушку и прокалку для удаления воды из пор геля и завершения
формирования оптимальной структуры алюмосиликатов. На стадии
сушки содержание воды снижается с 90—92 до 8—10%, а объем
частиц уменьшается в 7—8 раз. В результате прокаливания
содержание влаги в катализаторе
не превышает 1,0—1,5%
катализатор приобретает высокую
механическую прочность и
термическую стабильность.
Алюмосиликатные
синтетические катализаторы можно
получать также смешением
раздельно осажденных гелей, пропиткой
влажного или просушенного си-
ликагеля солями или
какими-либо другими соединениями
алюминия, которые при дальнейшей
обработке разлагаются с
образованием гидроокиси или окиси
алюминия.
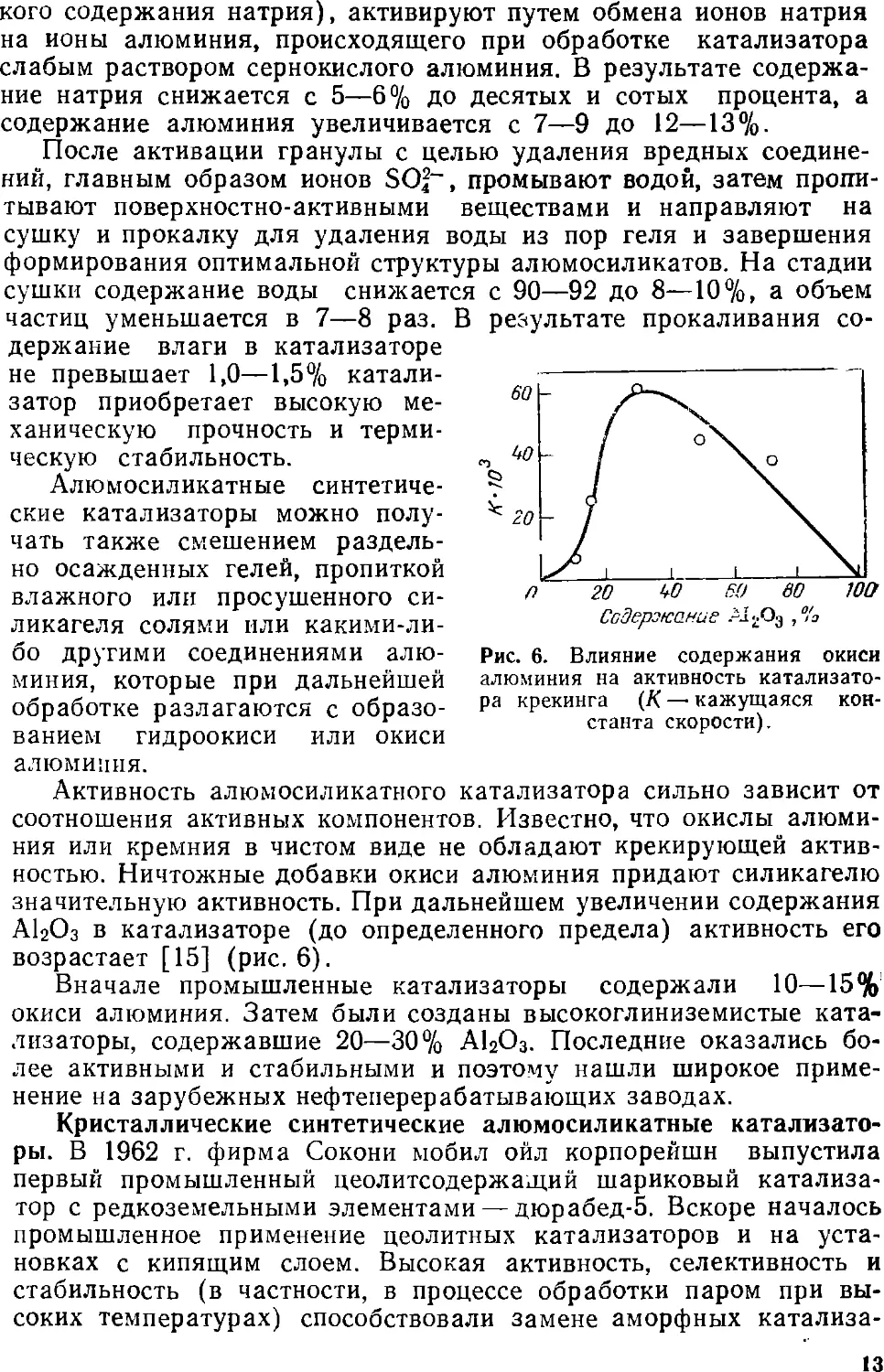
Активность алюыосиликатного катализатора сильно зависит от
соотношения активных компонентов. Известно, что окислы
алюминия или кремния в чистом виде не обладают крекирующей
активностью. Ничтожные добавки окиси алюминия придают силикагелю
значительную активность. При дальнейшем увеличении содержания
АЬОз в катализаторе (до определенного предела) активность его
возрастает [15] (рис. 6).
Вначале промышленные катализаторы содержали 10—15%
окиси алюминия. Затем были созданы высокоглиниземистые
катализаторы, содержавшие 20—30% АЬО3. Последние оказались
более активными и стабильными и поэтому нашли широкое
применение на зарубежных нефтеперерабатывающих заводах.
Кристаллические синтетические алюмосиликатные
катализаторы. В 1962 г. фирма Сокони мобил ойл корпорейшн выпустила
первый промышленный цеолитсодержащий шариковый
катализатор с редкоземельными элементами — дюрабед-5. Вскоре началось
промышленное применение цеолитных катализаторов и на
установках с кипящим слоем. Высокая активность, селективность и
стабильность (в частности, в процессе обработки паром при
высоких температурах) способствовали замене аморфных катализа-
Рис. 6. Влияние содержания окиси
алюминия на активность
катализатора крекинга (К — кажущаяся
константа скорости).
13
торов на установках каталитического крекинга цеолитсодержащи-
ми. Уже в начале 1968 г. примерно на 90% всех установок
крекинга США использовали цеолитсодержащие катализаторы [16]. По
данным 1972 г., общая годовая потребность США в катализаторах
крекинга 130 тыс. т, из них лишь 14 тыс. т не содержат
цеолитов [17].
Прежде чем подробнее характеризовать цеолитсодержащие
катализаторы, опишем свойства самих цеолитов.
Среди природных минералов существует большая группа
водных алюмосиликатов натрия, кальция и других металлов,
называемая цеолитами. Они обладают рядом примечательных свойств,
в частности способностью поглощать значительное количество во-
Рис. 7. Структура каркаса цеолита типа А (а) и фожази-
та (б).
ды. При нагревании цеолиты выделяют поглощенную воду без
разрушения структуры. Это свойство обусловило их название,
которое означает «кипящий камень» (от греческого слова цео —
кипящий, литое — камень).
При взаимодействии с водными растворами солей цеолиты
могут частично обменивать содержащиеся в их структурах катионы
на катионы, имеющиеся в растворе. Например, ионы Na+ легко
обмениваются на ионы Са2+. При обмене ионов каталитические
свойства цеолитов и размеры их внутренних пор существенно
изменяются.
Важные особенности строения цеолитов — определенные,
характерные для каждого типа минерала формы и размеры, наличие
каркаса, образованного тетраэдрами.. Основными элементами
каркаса являются полости, соединенные между собой окнами, или
каналами (рис. 7). Обычно полости имеют больший диаметр, чем
каналы (или окна). Например, диаметр полости шабазита 11,4 А,
а окна 4,9 А. В кристалле цеолита имеется огромное число
полостей. Например, в 1 г шабазита содержится 3-Ю20 полостей [9].
В каждую полость шабазита может вместиться 24 молекулы
воды. При нагреве цеолита вода удаляется и образуется ячеистая
14
структура. Удельная поверхность цеолитов достигает 700—
1000 м2/г [9].
Обезвоженные цеолиты способны избирательно адсорбировать
молекулы различных веществ, в зависимости от размеров каналов,
через которые адсорбируемые вещества проникают в полости.
Если диаметр адсорбируемого вещества больше, чем сечение канала,
то оно не может проникнуть во внутренние поры цеолита и,
следовательно, не может адсорбироваться. Так, при диаметре канала
4 А цеолит не адсорбирует углеводороды нормального строения,
диаметр молекул которых равен 4,9 А. Общая формула цеолитов:
M2/mO • Ala03-«Si02 -Ш2О
где М — катион (натрий, калий, кальций, редкоземельные
элементы и др.), обладающий валентностью т; п — коэффициент,
характеризующий тип цеолита; k — число молей воды.
Обычно тип структуры синтетического цеолита обозначают
буквами латинского алфавита А, X, Y, D, R, S, Т, L и т. д. Перед
буквой ставят символ катиона, компенсирующего отрицательный
заряд алюминия в алюмосиликате. Например, СаХ означает
цеолит типа X в кальциевой форме. Принято относить цеолиты к
различным структурным типам в зависимости от величины
коэффициента п—соотношения содержания в цеолите кремнезема к
глинозему. Значения п для цеолитов разных типов следующие:
п Тип цеолита
1,8—2,0 Цеолит А
2,3—3,0 Цеолит X
3,0—6,0 Цеолит Y
4,0—5,0 Шабазит (цеолиты D, R, S)
[18]
6,0—7,0 Эриоиит (цеолит Т) [18]
8,3—10,7 Морденит [10]
10—35,0 Цеолит L [9]
За рубежом цеолиты классифицируют несколько иначе. Перед
буквой, обозначающей тип цеолита, ставят цифру,
соответствующую максимальному диаметру молекул (в ангстремах),
поглощаемых данным цеолитом. По этой классификации цеолиту NaO
соответствует цеолит 4А, цеолиту СаА— 5А, цеолиту NaX—13Х,
цеолиту СаХ— 10Х.
Ниже приводятся размеры полостей и окон для некоторых
синтетических цеолитов [18]:
Диаметр
большой Размер окон
Цеолит полости, °.
о -А
А
LiA . . 12 4,4
NaA . . 11—12 4,0
КА 11 3,3
СаА 11 5,0
NaX (NaY) 11—13 9,0
СаХ (CaY) 11—13 8,0
Са-морденит 7 4,0
Н-морденит 7 6,6
15
Таблица 2
Классификация молекулярных сит по Барреру
Увеличение критического размера пропускаемых молекул
He, Ne, Аг, CO, H2,
O,, N2, NH3, H2O
Ca- и Ва-мордениты
(—3,8 A)
Тип 5
Кг
Хе
СН4
С2Н6
СН3ОН
CH3NH2
СН3Вг
со2, с2н2
cs2
Морденит и цеолит NaA (-4,0 A)
Тип 4
C3H8
я-С4Н10
«-с7н16
H-LltH30
С2Н6С1
С2Н6Вг
С2Н6ОН
C2H6NH2
СН2С12
СН2Вг2
CHF2C1
CHF3
(CH3JNH
СН31
Са-шабазит, цеолит СаА и гмелинит (—4,9 A)
Тип 3
Цеолит СаХ
Тип 2
CF4
CF2C1.,
CF3C12
SF„
ыэо-С4Н10
иэо-СьН12
h-CjF10
H-C,Fie
(CH3KN
(C2H6KN
C(CH3),
C(CH3)sBr
C(CH3KOH
CBri
CHC13
СНВг3 QiFgCl^
СНІ., CeHe
(СН3JСНОН CeH6CHs
(СН3JСНСі CjH^CHa),
«-C4F8 С„Н12
С4Н4О
C6H6N
Диоксан
Нафталин
Хинолин
6-Децил-1,2,
3,4-Тетра-
гидронаф-
талин,2-бу-
тил-1-гек-
силчнден
CeF„CFs
1,3,5-Триэтилбензол
1,2,3,4,5,6,7,8, 13,
13,15,16-Декагид-
рохризен
Цеолит NaX (—10 A)
Тип 1
(«-C4H0)sN
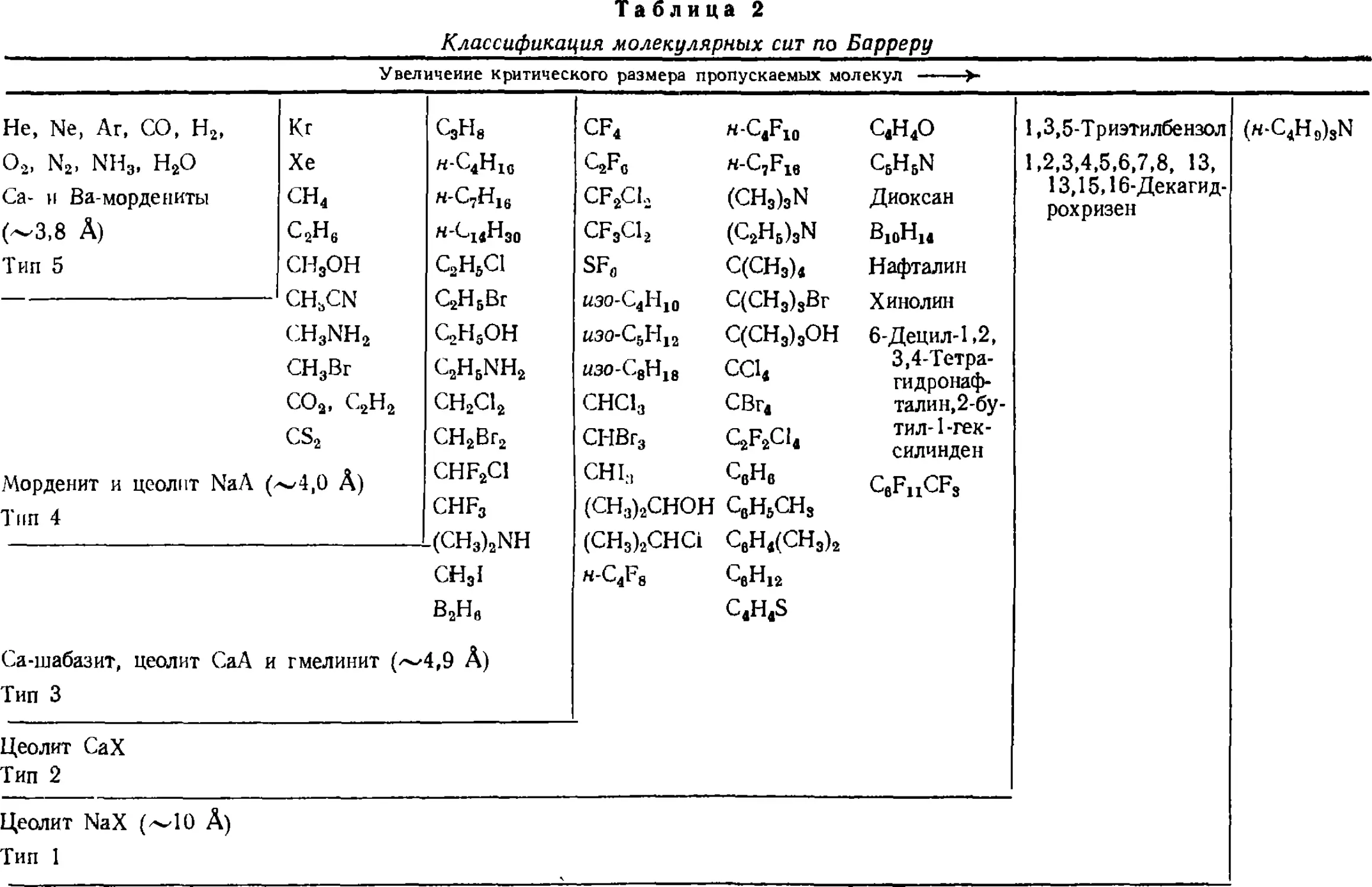
Размеры входных окон и молекул веществ обусловливают
селективную адсорбцию цеолитами или их способность служить
молекулярными ситами. Р. Баррер выделил пять типов цеолитов по
их молекулярно-ситовому действию [18] (табл. 2).
С увеличением номера типа цеолита уменьшается размер
входных отверстий в полости кристаллического каркаса. Вещества,
расположенные выше и левее черты, определяющей границу типа
цеолита, адсорбируются им, а расположенные ниже и правее, не
проникают в поры и не адсорбируются. Цеолиты адсорбируют
вещества, характерные для данного типа, и дополнительно
поглощают все те вещества, которые адсорбируются цеолитами,
имеющими более высокие номера.
У. Хирш делит цеолиты на три класса (табл. 3) [9].
Таблица 3
Вещества, молекулы которых адсорбируются
природными цеолитами различных классов
Класс
цеолита
Вещества, адсорбируемые
при комнатной илн более
низкой температуре
Вещества, медленно
адсорбируемые прн
комнатной нли более
высокой температуре
Вещества, не адсорбируемые
прн комнатной нлн более
высокой температуре
Не, Ne, Ar, H,, N2,
О2, СО, СО2, COS,
CS2, Н2О, НС1, НВг,
no, nh3, сн3он,
:h3cn, hcn, ci2,
:h3ci, сн3вг, ch3f,
^НгСід» CHoFs» CH4,
26
H2S, CH2SH
II
III
He, Ne, Аг, Н2, O2,
N2, CONH3, H2O
He, Ne, H?. O,,, N2,
H,0
Пропан и простые
нормальные углеводороды,
С2Н5ОН, C2H5NH2, C2H5F,
С2Н5С1, C2H5Br, I2 HI,
СН2Вг2, СНзІ, C2H5CN,
C2H5SH, НСО2СН3,
НСО2С2Н5, СО(СН3J,
СН3СО2, NH(CH3J,
NH(C2H5J
сн4, с2н6, сн3он,
CH3NH„ CH3CN, CH3C1,
CH3F, HCN, Cl2
Аг, НС1, NH3-.
Ароматические и
циклические углеводороды,
изопарафины и их
производные, тиофеи,
пиррол, пиридин, СНСІз,
ССЦ СНС1=СС12,
СН3СНС12, СНС12СС13.
С2Н3С13 и другие
производные брома и иода.
Вторичные алкоголи с
прямой цепью, нитрилы.
Первичные амины с
группой, присоединенной
к вторичному атому С,
четвертичные амины,
эфиры с боковыми
цепями, тиоэфиры и
вторичные амины.
Все молекулы колонн
2 и 3 класса I
То же
К I классу относятся цеолиты с размером окон 4,89—5,58 A.
Способность к адсорбции этих цеолитов неодинакова. Так,
шабазит адсорбирует нормальные парафины и не поглощает
изопарафины и ароматические углеводороды, гмелинит адсорбирует
нормальные парафины медленно. К II классу относятся цеолиты с
диаметром окон 4,0—4,89 А. Эти цеолиты (Na-морденит и др.) медленно
адсорбируют метан и этан и не адсорбируют другие
углеводороды. В то же время они быстро адсорбируют азот, кислород и дру-
2—2206
17
гие вещества с меньшими размерами молекул. К ПІ классу
относятся цеолиты с размерами каналов 3,84—4,0 А (Са- и Ва-форма
морденита). Они не адсорбируют углеводороды, но адсорбируют
аргон, азот и другие вещества, молекулы которых еще меньше.
Присущие цеолитам высокая активность, селективность,
способность противостоять обработке паром при высоких температурах
позволили использовать их для приготовления катализаторов
крекинга. Этому способствовала также их способность к легкому
обмену катионов [9, 10, 18—20], что позволяет применять цеолиты
в виде наиболее активных катионных форм.
Первые исследования каталитических свойств цеолитов,
проведенные в 1960 г., показали, что цеолиты NaX и СаХ более
активны в реакции крекинга н-декана, чем аморфный алюмосили-
катный катализатор [21], причем кальциевая форма цеолита
активнее натриевой. В присутствии цеолита NaX в продуктах
реакции отсутствовали изосоединения, но содержалось много
непредельных углеводородов, а в присутствии СаХ получались такие же
продукты, как и при наличии алюмосиликатного катализатора.
При крекинге ароматических углеводородов цеолиты
оказались менее активными, чем аморфные алюмосиликатные
катализаторы [22]. После замещения Na+ на Са2+, декатионирования и
увеличения отношения кремнезема к глинозему крекирующая
активность цеолитов типа X и Y существенно возрастает [23, 24].
Показано [23, 24], что на цеолите СаХ кумола адсорбируется в
2 раза больше, чем бензола (на аморфных алюмосиликатах их
адсорбционные коэффициенты примерно одинаковы); заметной
адсорбции пропилена на цеолитах не обнаружено, в то время как
адсорбция пропилена на аморфных алюмосиликатах тормози-т
реакцию крекинга. Эти данные в некоторой степени объясняют
более высокую активность цеолитов.
' В практике нефтеперерабатывающей промышленности в
качестве катализаторов крекинга цеолиты в чистом виде не применяют.
Их используют в виде цеолитсодержащих катализаторов [25],
представляющих собой обычные аморфные алюмосиликатные
катализаторы, в которые введено до 25% (в основном 10—20%
[17]) порошка цеолитов типа X или Y.
Аморфный алюмосиликат, который используют для введения
цеолита, часто называют матрицей. Матрица выполняет
следующие функции: 1) поддерживает цеолит в грануле катализатора;
2) уменьшает активность катализатора, поскольку при
использовании цеолита в чистом виде на существующих установках
происходит чрезмерное расщепление, сопровождающееся повышенным
газообразованием и быстрым отравлением катализатора [28].
В табл. 4 показана относительная активность чистых цеолитов
при крекинге н-гексана [6, 26, 27].
Цеолиты можно переводить в кальциевую, магниевую,
редкоземельную или в другие катионные формы, обладающие наиболее
приемлемыми каталитическими свойствами, до введения в алюмо-
18
Таблица 4
Относительная активность чистых цеолитов
Катализатор
Аморфный
Са-фожазит
ЫН4-фожазит
Re-фожазит
СаА
Са-Морденит
Н-мордеиит
МН4-морденит
SiO2
90
47,8
75,7
—
42,5
77
80,1
—
прі
і крекинге н-гексана
Содержани
А12О3
10
31,5
23,1
—
37,4
—
13,4
—¦
Na
_
7,7
0,4
0,39
7,1
1,01
0,3
0,1
;, вес. %
Са
_
12,3
—
—
13,0
—
1,54
—
Re
_
—
—
28,8
—
—
—
—
Температура
конверсии
5-20%,
°С
540
530
350
<270
580
520
300
<270
Активность
1
1,1
6400
>10 000
0,6
1,8
2500
>10000
Таблица 5
Типичные свойства цеолитных катализаторов крекинга
Показатели
Химический состав,
вес. %
SiO2
А12Оз
Сг2О3
Na
SO4
Окислы редкоземельных
элементов R2O3
Физические свойства
плотность, кг/м3
в рыхлом виде
в утрамбованном
виде
удельная
поверхность, м2/г
удельный объем пор,
см3/г
средний диаметр пор,
А
Стойкость к истиранию
по LSA — время,
требуемое для получения
50% тонкоразмолотых
частиц, с
Коэффициент диффузии
в катализатор,
(см2/с)-10-3
Дюрабед-1
свежий
9С
9,
0,
0,
0,
0
740
820
195
0,35
72
1500
15
равновесный
7
15
1
1
780
870
85
0,30
141
1500
19
Дюрабед-5
свежий
0,
0,
0,
2,
670
740
200
0,46
92
1500
26
равновесный
85
12
15
1
1
5
730
810
135
0,37
ПО
1500
26
Дюрабед-7
свежий
0
о,
о,
0,
750
840
130
0,43
132
1800
27
равновесный
51
46
15
27
1
65
760
840
115
0,39
138
2000
25
Дюрабед-8
свежий
4?
4с
0,
0
0,
1
830
920
105
0,35
135
1800
36
равновесный
,5
,8
20
1
1
20
860
960
70
0,35
200
1800
36
Таблица 6
Результаты крекинга газойли мидконтинентской нефти на пилотной установке
в присутствии цеолитных катализаторов
І Іоказателн
Активность катализатора по методу
Кат Д
Объемная скорость, ч~'
Отношение катализатора к сырью
вес. %
Температура смеси катализатора і
сырья, °С
Средняя температура в реакторе, °С
Степень превращения, объемн. %
Выход на свежее сырье, вес. %
(объемн. %)
тяжелый газойль
легкий газойль
Са +бензин (до 180 °С выкипает
90%)
всего фракции С4
сухой газ
кокс
¦¦
17
32
34
8
4
2
100
При постоянной
гсошіереші і
дгорабод-5
43
2,0
3,7
493
471
51,5
,6 A6,5)
,2 C2,0)
,2 D0,1)
,4 A2,7)
,9
,7 -
,0 101,3
степени
температуре
14
35
35
7
4
2
100
дюрабед-6
45
2,0
4,1
493
471
51,5
,8 A3,5)
,2 C5,0)
,1 D1,2)
,8 A1,8)
,8 -
3 -
0 101,5
17
32
34
8
4
2
100
дюрабі д-5
43
2,0
3,7
493
47)
51.5
,6 A6,5)
,2 C2,0)
,2 D0,1)
,4 A2,7)
,9 —
,7 -
0 101,3
При постоянном выходе кокса
дюрабед-6
47
2,0
4,1
508
482
56,0
12,4 A1,2)
33,0 C2,8)
36,7 D3,2)
9,0 A3,6)
6,2 -
2,7 —
100,0 100,8
дюрабед-7
1
4
,67
,2
482
70
4
27
48
9
7
5,
100
2
4
4
2
8
0
2
0
дюрабед-Я
1
3
,75
,9
479
70
5
22
51
10
6
3
100
,4
6
0
7
2
9
6
0
Состав легких углеводородов, вес. %
(объемп. %)
f/-C4H10
н-С4Н8
С3Н6
СзН8
сн4
н2
H2S
Октановое число бензина (до 180 °С
выкипает 90%)
моторный метод
без ТЭС
с 3 мл ТЭС
исследовательский метод
без ТЭС
с 3 мл ТЭС
дорожное октановое число
без ТЭС
с 3 мл ТЭС
1,0
4,3
3,1
8,4
1,3
2,3
0,4
0,3
0,4
0,02
0,2
4,92
79,6
86,4
88,9
96,5
86,9
94,2
A
F
D
5)
7)
5)
A2,7)
@
2)
0,9
3,9
3,0
7,8
1,3
2,1
0,4
0,4
0,4
0,03
0,2
4,83
79,3
86,0
88,9
96,0
86,7
93,8
С
F,
D,
(И
@
4)
1)
3)
,8)
,02)
1,0
4,3
3,1
8,4
1,3
2,3
0,4
о.з
0,4
0,02
0,2
4,92
79,6
86,4
88,9
96,5
86,9
94,2
A.
F,
D,
A2
5)
7)
5)
.7)
0,9
4,5
3,6
9,0
1,5
2,8
0,6
0,5
0,6
0,04
0,2
6,24
79,7
86,0
90,3
96,8
88,0
94,4
A.
G,
E,
A3
5)
0)
1)
,6)
A
G
F
A5
2,
2
7)
1)
3)
1)
7
0
A
(8
E
A5
3
1
9)
2)
5)
6)
3
8
силикат или путем обработки готового катализатора. Цеолиты
типа X и Y дают одинаковые результаты [29]. Однако
термостойкость цеолита Y несколько больше, поэтому в производстве
катализаторов крекинга его применяют более широко [17].
После первого цеолитсодержащего промышленного шарикового
катализатора крекинга — дюрабед-5 фирма Сокони мобил ойл
корпорейшн с 1965 г. начала применять дюрабед-6, который обладал
более высокой прочностью и повышенной насыпной плотностью.
Эти два катализатора содержали 2,5% редкоземельных элементов
и 0,15% окиси хрома [30]. В последнее время фирмой созданы
более активные и стабильные цеолитсодержащие катализаторы, в
частности катализатор дюрабед-8 [25].
В промышленности США наряду с синтетическими
шариковыми катализаторами применяют и цеолитсодержащие таблетиро-
ванные катализаторы на основе коалинитовых глин. К ним
относится разработанный фирмой Гудри процесс энд кемикал
корпорейшн катализатор HZ-1, который выпускают в промышленном
масштабе с 1965 г. [31]. В табл. 5 дана характеристика
гранулированных катализаторов крекинга, а в табл. 6 — результаты
каталитического крекинга в их присутствии.
Таблица 7
Характеристика микросферических катализаторов крекинга США
Показатели
Химически!': состав,
вес. %
SiO2
А12Оз
\а2О3
SO4
Fe2O3
Физические свойства
насыпная плотность,
кг/м3
удельная
поверхность, м2/г
удельный объем пор,
см3/г
Фракционный состав,
вес %, размером, [х
0—20
20—40
40—80
>80
Активность (объемн. %
конверсии) после 24 ч
дезактивации паром
I?
ІІ
II
88,1
14,9
0,05
0,27
0,046
490
440
0,9
1
2
40
57
—
84,3
14,7
0,07
0,4
0,04
380
500
0,92
1
9
64
26
72
et
и
31,0
0,06
0,4
0,08
520
335
0,6
2
15
51
32
34
Sg
N 5
Хс
30,0
0,09
0,4
0,08
520
0,6
2
17
53
28
89
¦о
N
X
36,0
0,07
0,4
0,09
550
270
0,6
3
27
57
23
89
Q О CJ
8 = *v
І5ІЗ
71,6
28,0
0,04
0,6
0,03
460
430
0,70
2
15
51
32
58
§ f г=ї
Й5ІЗ
86,8
13,0
0,03
0,3
0,03
430
—
0,77
о
15
51
32
50
22
Таблица 8
Каталитический крекинг вакуумного газойля в псевдоожиженном слое
на кристаллических алюмосиликатных катализаторах
Показатели
Дгарабед-9
515
9,8-М
7,6
7,2
40,6
31,8
6,1
6,7
62,1
82,6
86,0
94,7
98,1
2,5
1,4
6,2
4,3
0,8
518
—
8,0
6,4
37,9
34,0
6,9
6,8
59,1
82,5
85,4
94,5
97,9
2,6
1,2
6,0
3,2
0,8
XZ-25 плюс
493
1,0
—
5,40
3,90
51,4
13,0
24,6
2,9
62,4
—
—
—
3,39
0,58
2,30
3,40
0,3
XZ-36
Средняя температура в реакторе, °С
Объемная скорость, ч
Кратность циркуляции (весовая)
Выход, вес. %
сухой газ (Сз и ниже)
бутаны
дебутанизированный бензин
легкий газойль
тяжелый газойль
кокс
Степень превращения, вес. %
Октановое число дебутанизированного
бензина
моторный метод без ТЭС
то же, с 3 мл ТЭС на 1 л
исследовательский метод без ТЭС
то же, с 3 мл ТЭС на 1 л
Состав фракции С3—С4
пропилен, вес. %
пропан, вес. °/о
бутилены, объемн. %
изобутан, объемн. %
я-бутан, объемн. %
493
1,0
4,88
4,07
51,00
13,05
24,90
1,8
62,0S
3,51
0,35
2,0
4,0
0,3
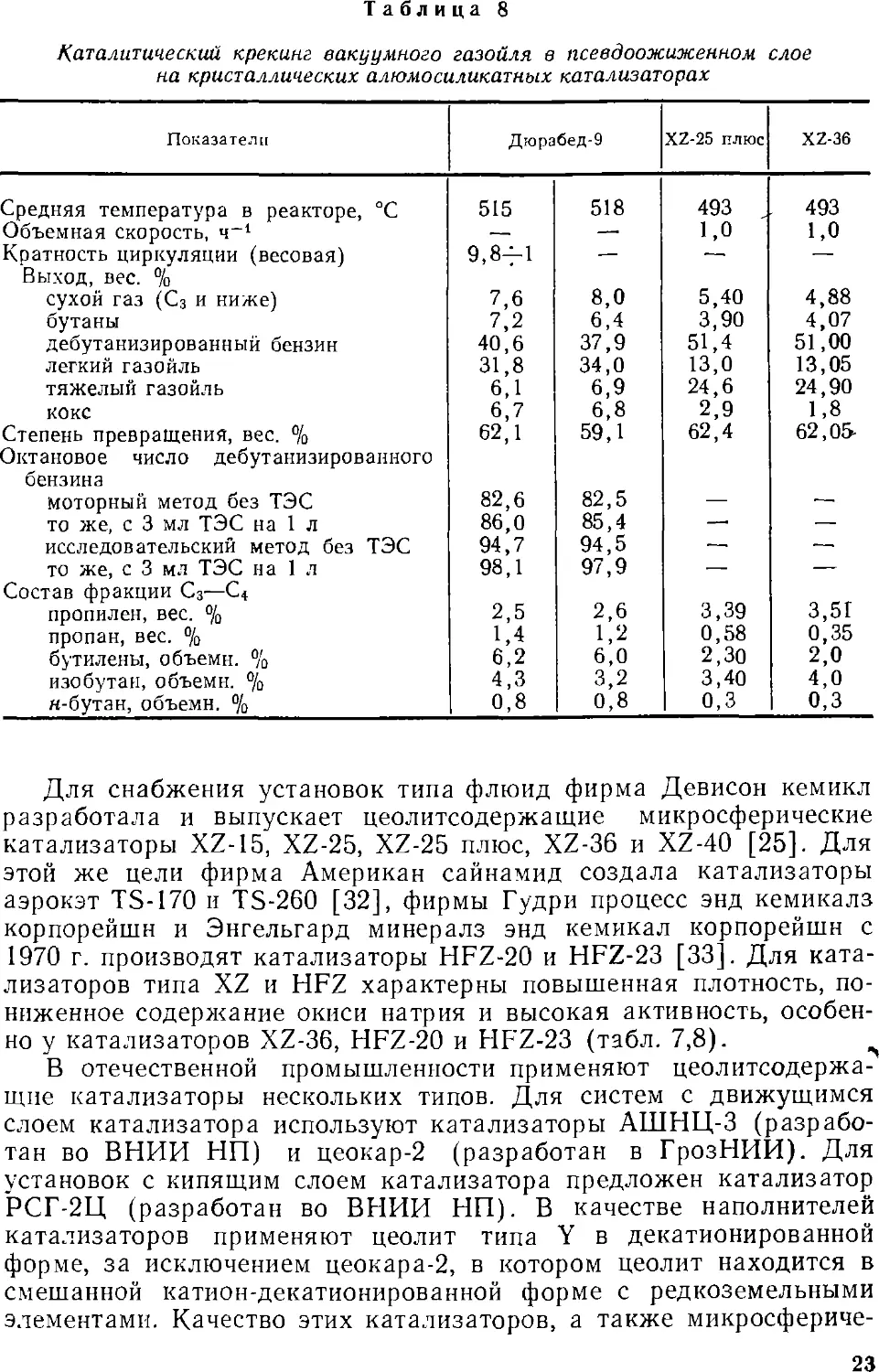
Для снабжения установок типа флюид фирма Девисон кемикл
разработала и выпускает цеолитсодержащие микросферические
катализаторы XZ-15, XZ-25, XZ-25 плюс, XZ-36 и XZ-40 [25]. Для
этой же цели фирма Американ сайнамид создала катализаторы
аэрокэт TS-170 и TS-260 [32], фирмы Гудри процесс энд кемикалз
корпорейшн и Энгельгард минералз энд кемикал корпорейшн с
1970 г. производят катализаторы HFZ-20 и HFZ-23 [33]. Для
катализаторов типа XZ и HFZ характерны повышенная плотность,
пониженное содержание окиси натрия и высокая активность,
особенно у катализаторов XZ-36, HFZ-20 и HFZ-23 (табл. 7,8). ^
В отечественной промышленности применяют
цеолитсодержащие катализаторы нескольких типов. Для систем с движущимся
слоем катализатора используют катализаторы АШНЦ-3
(разработан во ВНИИ НП) и цеокар-2 (разработан в ГрозНИИ). Для
установок с кипящим слоем катализатора предложен катализатор
РСГ-2Ц (разработан во ВНИИ НП). В качестве наполнителей
катализаторов применяют цеолит типа Y в декатионированной
форме, за исключением цеокара-2, в котором цеолит находится в
смешанной катион-декатионированной форме с редкоземельными
элементами. Качество этих катализаторов, а также микросфериче-
23
Таблица 9
Характеристика катализаторов
Показатели
Марка катализатора
Количество введенного
цеолита, вес. %
Индекс активности
Индекс стабильности
Насыпная плотность, кг/м3
Удельная поверхность, м2/г
Удельный объем пор, см3/г
Средний радиус, А
Химический состав, вес. %
А12О3
SiO2
Fe2O3
CaO
Na2O
Содержание, вес. %
никеля
ванадия
Литературный источник
Цеолитсодержащие
микросферический
РСГ-2Ц
18
47,8
51,9
790
199
0,396
39,8
12,98
84,20
0,21
1,72
0,89
0,001
0,000
[34]
шариковый
АШНЦ-З
18
48,5
51,5
703
242
0,506
41,8
10,80
87,01
0,45
0,78
0,96
0,002
0,000
[34]
ЦЕО-
КАР-2*
16
690
263
0,500
38,3
10,0
83,5
0,31
[35, 38]
Аморфные
микросферический
РСГ-2
—.
32,1
24,9
885
149
0,302
37,8
15,48
83,26
0,49
0,56
0,?1
0,032
0,013
[34]
шариковый
Стандарт-
ЩИ
32,3
25,6
785
187
0,375
40,1
10,36
87,15
0,63
1,54
0,32
0,039
0,011
[34]
Содержит 2,3% редкоземельных элементов [38].
ских и шариковых аморфных катализаторов крекинга приводится
в табл. 9.
Каталитическую активность катализаторов, разработанных во
ВНИИ НП, определяли в процессе крекинга вакуумного
дистиллята ромашкинской нефти.
Результаты каталитического крекинга ка пилотных установках
в оптимальных условиях на равновесных цеолитсодержащих и
аморфных катализаторах приведены в табл. 10—12.
В присутствии цеолитсодержащих катализаторов для
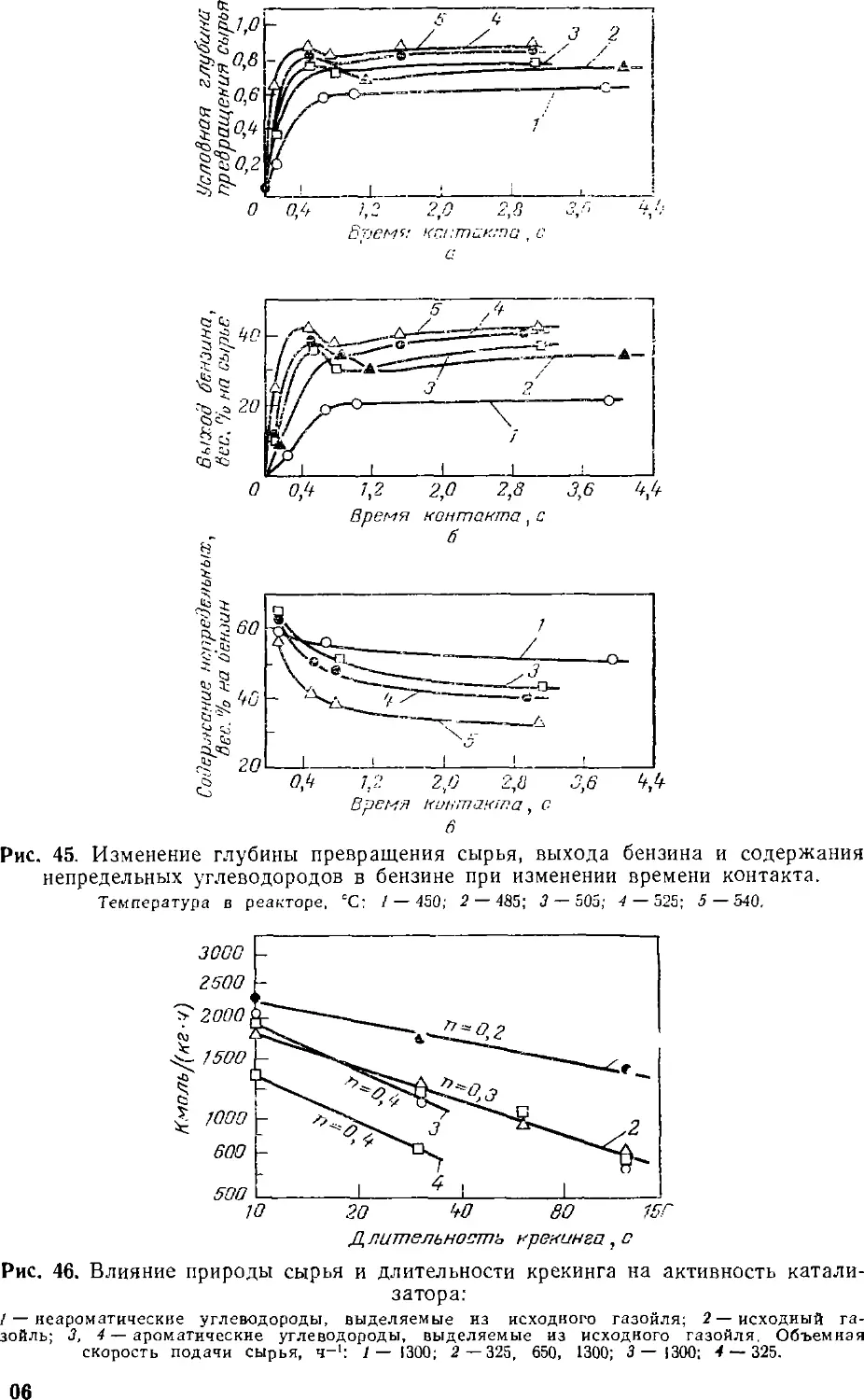
достижения высокой глубины превращения сырья, определяемой в
основном выходом бензина (табл. 10 [34]), необходим более мягкий
режим крекинга. Скорость подачи сырья следует увеличить в 2,0—
2,5 раза.
В оптимальных условиях процесса выхода бензина на
цеолитсодержащих катализаторах на 12—14 вес. % выше, чем на
стандартном катализаторе, также выше селективность их действия,
определяемая отношением выхода бензина к глубине превращения
сырья [34].
Влияние температуры на материальный баланс
каталитического крекинга фракции 400—450°С ставропольской нефти представ-
24
Таблица 10
Материальные балансы каталитического крекинга на цеолитсодержащих
и аморфных катализаторах при оптимальных режимах процесса
Показатели
Условия процесса
Температура, °С
Скорость подачи сырья, ч~'
весовая
объемная
Кратность циркуляции катализатора,
кг/кг
Выход, вес. % на сырье
Газ до С4 включительно
в том числе
H2S
н2
СН4
с2н4
С2Н6
СзН6
С3Н8
цзо-С4Н8
н-С4Н8
изо-С4Н|о
н-С4Н10
Фракция С5—195 °С
в том числе
сумма СбНю
U3O-C5H,2
н-С5Н12
Фракция 195—350 °С
Остаток выше 350 °С
Кокс
Потери
Селективность процесса, %
Катализатор
АШНЦ-3
490
—
2,8—2,0
3,0
17,5-18,8
0,74—0,78
0,03—0,04
0,47—0,81
0,88-0,90
0,43—0,62
3,96—4,18
2,52—2,82
0,75
1,01—1,09
4,96-5,40
1,48-1,41
40,6—40,8
3,47—3,02
5,72-6,56
0,54—0,87
22,0—22,3
15,1 — 13,0
3,8-4,1
1,0
47,9—46,8
стандартный
шариковый
470
—
1,0
3,0
16,5
0,61
0,61
1,34
0,68
0,94
2,26
2,85
0,94
2,02
3,53
1,17
26,0
2,62
2,68
0,42
23,8
27,9
4,8
1,0
36,1
РСГ-2Ц
470—490
3,5—5,3
—
6^7
19,4-22,0
0,72-0,84
0,04—0,02
0,50—0,73
0,64—0,94
0,52—0,60
4,70—5,04
2,12-2,27
1,08-1,64
2,50-3,36
5,45—5,33
1,03—1,23
43,3-42,8
3,35—4,98
5,88-4,41
0,69—0,60
20,0—16,8
12,0-13,0
4,3-4,4
1,0
49,6—49,2
РСГ-2
490
2,0
—
6—7
20,5
0,73
0,63
1,65
1,41
0,57
5,25
1,17
2,07
3,28
3,12
0,62
31,1
4,05
2,30
0,25
21,8
20,4
&
1,0
39,1
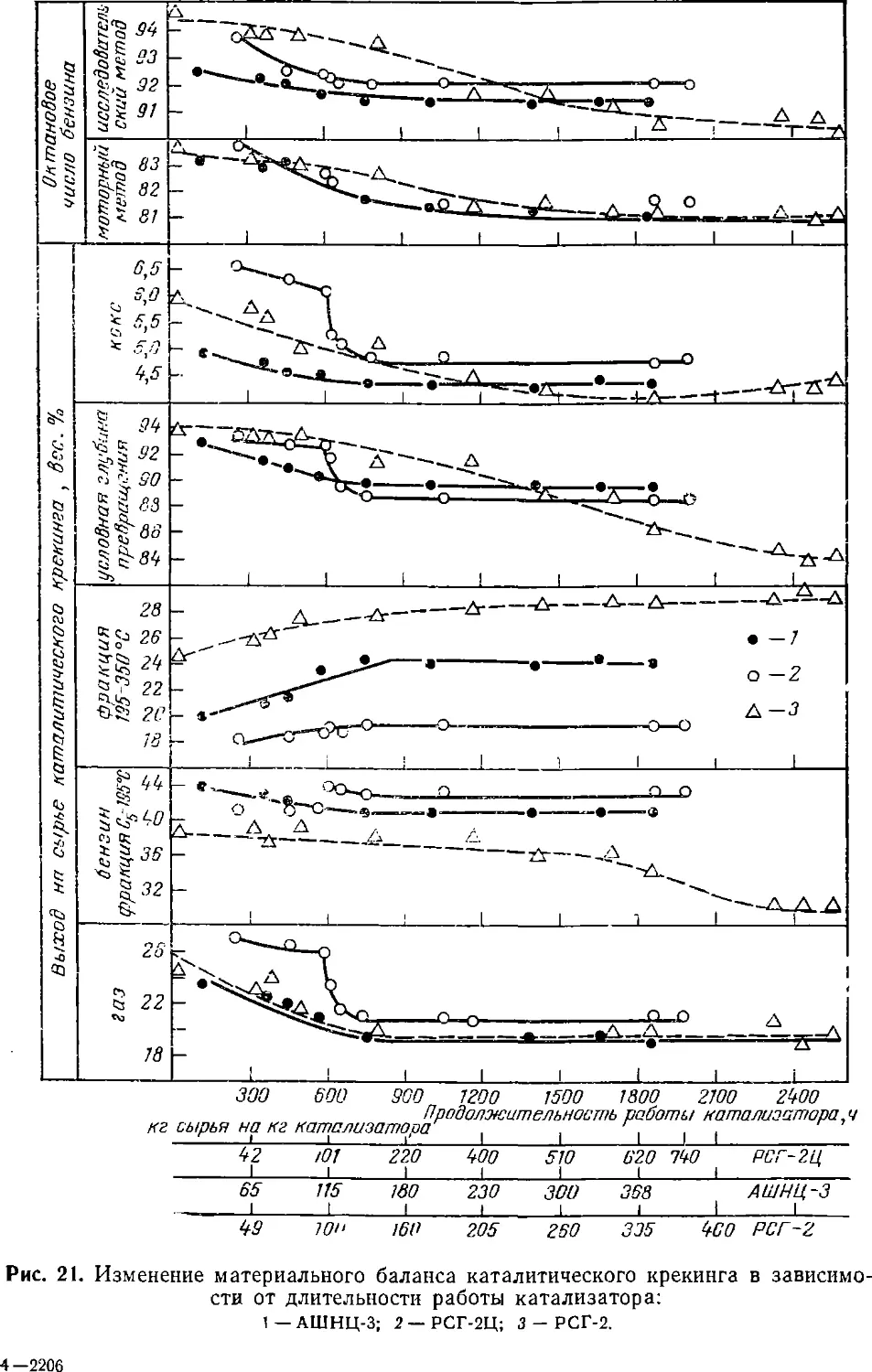
лено на рис. 8 [37J. Из рис. видно, что в случае цеолитсодержа-
щего катализатора АШНЦ-3 достигается большая глубина
превращения сырья, значительно больше выход бензиновой фракции
и несколько больше выход газа. Выходы дизельной фракции и
кокса на цеолитсодержащем катализаторе АШНЦ-3 несколько
меньше, чем на аморфном.
Уменьшение жесткости крекинга (увеличение объемной
скорости) сильно влияет на выходы кокса, бензина и на глубину
превращения сырья (рис. 9). В меньшей степени объемная скорость
влияет на выход бензина при повышенной температуре.
Наибольший выход бензина на цеолитсодержащем катализаторе АШНЦ-3
в процессе крекинга фракции 400—450 °С был достигнут при 475 °С
и объемной скорости подачи сырья 1,2 ч^1 D7,9 вес. % на сырье).
100
№
60
§-8
10
ЗО
1 ctf
$00 Ь25 450 475 500
Температура, °С
7 2 З
Объемная скорость,ч~
Рис. 8. Зависимость выхода продуктов крекинга от температуры при объемном
скорости подачи сырья 1,2 ч-1 (сплошные линии — цеолитсодержащий
катализатор АШНЦ-3, пунктирные — алюмосиликатный аморфный).
Рис. 9. Зависимость выхода продуктов крекинга от объемной скорости подачи
сырья на цеолитсодержащем катализаторе АШНЦ-3.
Температура, °С: 1 — 425; 2 — 450; 3 — 475; 4 — 500.
Аналогичные результаты получаются при крекинге тяжелой
фракции высокопарафинистой малосернистой нефти в
присутствии АШНЦ-3, ЦЕОКАР-2 и аморфного алюмосиликатного
катализатора. Крекинг на катализаторах АШНЦ-3 и ЦЕОКАР-2
характеризуется большей глубиной превращения сырья (в среднем на
9—11 вес. %), большими выходами газа (на 1,0—2,5 вес. %) и
бензина (на 10—13 вес. %), меньшими выходами кокса (на 0,4—
26
Таблица 11
Состав газов и качество бензинов, полученных в процессе каталитического крекинга
на цеолитсодержащих и аморфных катализаторах при оптимальных
режимах процесса
Показатели
Углеводородный состав
газа, вес %
До Сг включительно
Сз
с4н8
изо-С4Н|0
н-С4Н,о
Всего непредельных
» иэосоединений
Отношение изо-С4Н|о к сумме С4Н8
Показатели качества
бензинов
Октановое число в чистом виде
моторный метод
исследовательский метод
Групповой углеводородный состав,
вес. %
непредельные
в том числе изостроения
ароматические
нафтены
парафины
в том числе изостроения
Содержание серы, вес. %
Фракционный состав, °С
н. к.
10%
50%
90%
к. к.
выкипает до 100 °С (объемн. %)
АШНЦ-3
16,1 — 16,6
37,0—37,2
10,1—9,8
28,3—29,0
8,5—7,5
37,7—36,8
32,6—33,0
2,80—2,96
80,4—81,0
90,0—91,0
29,3—26,5
17,3
33,2—34,8
0,7-0,6
36,8—38,1
31,6
0,17
40
67
118
174
195
37
Катализатор
стандартный
шариковый
22,6
30,9
17,9
21,5
7,1
35,7
27,2
1,20
79,4
89,4
39,8
21,5
22,0
2,0
35,3
28,2
0,24
42
70
132
175
195
30
13
35
18
28
5
45
34
1,
81
91
28
26
0
43
0,
РСГ-2Ц
,2—14
,1 — 33
,4—22
,7—24
,3—5,
,9—50
,2—31
56—1,
,7—81
,9-92
,8—36
25,5
,7—27
,7—0,
,8—36
31,3
20—0,
41
00
112
173
195
42
,2
,2
,8
,2
6
,0
,7
06
,5
,8
,3
,7
0
,0
18
РСГ-2
24
31
26
15
3
58
25
0
82
91
55
38
28
0
15
11
0
4
,3
,1
,2
,0
,6
,3
,58
,1
,1
,0
,7
,6
,8
,6
,2
,26
42
82
130
177
195
24
0,2 вес. %) и фракции дизельного топлива (на 2—3 вес. %).
Максимальный выход бензина достигается в интервале 460—475 °С и
составляет для АШНЦ-3 46,0 вес. % и для ЦЕОКАР-2 47,5 вес. %.
Выход кокса при этом одинаков E,2 вес. % на сырье).
Для определения оптимальной температуры и весовой
скорости подачи сырья выведено уравнение регрессии [40]. Расчетом по
этому уравнению было установлено, что зависимость выхода
бензина от указанных параметров имеет экстремальный характер с
максимумом в области 470 °С и весовой скорости подачи сырья
4,8—7,3 ч-1 (рис. 10).
530
7,3 ?? :г,з іь,в і
?,scodriP. сгораешь годсчи сырья У, ¦¦"'
Рис. 10. Зависимость выхода бензина в
вес. % на сырье (цифры на кривых) от
температуры и весовой скорости подачи
сырья.
ч-ЭО
Температура, °С
Рис. 11. Изменение физико-химических
свойств бензиновой фракции в зависимости
от температуры и весовой скорости подачи
сырья.
Весовая скорость, ч~': 1 — 14,3; 2 — 9,6; 3 — 4,8.
28
При крекинге на цео-
литсодержащих
катализаторах изобутана получается
в 1,5—1,8 раза и изопента-
на в 2,0—2,5 раза больше,
чем на стандартном
катализаторе. В газе крекинга
содержится меньше
фракций до С2, отношение
выходов изобутана к сумме бу-
тиленов выше, что улучшает
состав сырья для установок
алкилирования (табл. 11)
fo4].
В бензинах крекинга,
полученных на аморфных
катализаторах, содержится
много непредельных
углеводородов, что ухудшает их
стабильность. Бензины кре-
кіінга на цеолитсодержа-
щих катализаторах
содержат меньше непредельных
углеводородов, но и они
(особенно бензины,
полученные на микросфериче-
chOM катализаторе)
требуют специальных мер для.
улучшения стабильности.
Качество продуктов,
получаемых в присутствии
катализаторов АШНЦ-3 и
ЦЕОКАР-2, практически
одинаково [35].
Изменения
физико-химических свойств бензиновой
фракции в зависимости от
условий проведения
крекинга представлены на рис. 11.
Из рис. 11 видно, что с
повышением температуры
значительно возрастает
антидетонационная стойкость
бензина и увеличивается
содержание в нем
непредельных и ароматических
углеводородов [40].
Газойлевые фракции,
Таблица 12
Характеристика легкого и тяжелого газойлей каталитического крекинга
на цеолит содержащих и аморфных катализаторах
Показатели
Легкий газойль
Цетановое число
Плотность d2?
Коэффициент преломления п2?
Пределы кипения, °С
Температура застывания, °С
Содержание серы, вес. %
Йодное число, гіг/100 г
Вязкость при 50 °С, мм2/с
Коксуемость 10%-ного остатка,
вес. %
Тяжелый газойль
Плотность d\°
Содержание серы, вес. %
Вязкость при 100 "С, мм2/с
Температура застывания, °С
Коксуемость, вес. %
Катализатор
АШНЦ-3
24—23
0,9310—0,9340
1,5498-1,5509
220—350
—20
2,3-2,4
26,1—25,3
2,6—2,5
0,18
0,9824—0,9859
2,34—2,40
7,86—7,54
+ 27
2,00—2,19
стандартный
шариковый
27
0,9111
1,5214
220—350
— 13
2,2
23,0
2,4
0,20
0,9560
1,95
7,35
+ 35
1,10
РСГ-2Ц
21
0,9461
1,5511 — 1,5541
220—350
—22
2,4—2,5
30,0—44,0
2,7-2,5
0,19
0,9810—0,9822
2,37—2,49
7,35—7,32
+ 32—+33
2,27—2,04
РСГ-2
20
0,9452
1,5495
220—350
—24
2,4
44,4
2,5
0,30
0,9710
2,20
6,83
—31
1,89
получаемые в присутствии цеолитсодержащих катализаторов,
более ароматизированы, чем получаемые в присутствии аморфных
катализаторов (табл. 12).
Глубина крекинга («80%) на аморфных катализаторах
(стандартный шариковый и РСГ-2) такая же, как и на цеолитных
катализаторах АШНЦ-3 и РСГ-2Ц, достигается при температуре
крекинга на 40 °С выше D90 против 450 °С) и объемных скоростях
подачи сырья в 3—4 раза меньших (табл. 13) [36].
Из табл. 13 видно, что выход бензина на аморфных
катализаторах меньше, чем на цеолитсодержащих. Причем эти выходы
бензина при крекинге на аморфных катализаторах являются
максимальными; для цеолитсодержащих катализаторов оптимальные
выходы достигаются при большей глубине превращения сырья.
Концентрация сероводорода в газе крекинга на цеолитном
катализаторе в 1,2—1,4 раза выше, чем в газе крекинга на
аморфных катализаторах E,0—4,9 против 3,6—3,7 вес. %). Менее
сернистыми получаются также бензины и легкие каталитические
газойли. Следовательно, реакции обессеривания на цеолитных
катализаторах протекают более глубоко.
Сообщается [38] о результатах работы промышленных
установок двухступенчатого каталитического крекинга керосино-газойле-
вой фракции на катализаторе ЦЕОКАР-2 с целью производства
авиабензина. При этом выход мотобензина — целевого продукта
первой ступени процесса (фракции, выкипающей до 240 °С) воз-
29
Таблица 13
Режимы, материальные балансы и селективность крекинга
вакуумного дистиллята ромаиікинской нефти на цеолитсодержащих
и аморфных катализаторах при глубине превращения сырья 80%
Показатели
Режим процесса
Температура, °С
Скорость подачи сырья. ч~1
Кратность циркуляции
Материальный баланс
крекинга, вес. %
Газ до Сі включительно
в том числе
н2
СН4
С2Н4
СзН6
С3Н6
СзНа
изо-С4На
н-С4На
изо-С4Н10
н-С4Ню
Фракция Сб~195°С
в том числе
сумма С5Н10
иЗО-СбН|2
н-С5Н12
Фракция 195—350 °С
Остаток выше 350 °С
Кокс
Потери
Итого . . .
Бензин:газ
Бензин: кокс
Селективность • Ю-2
Шариковые
катализаторы
АШНЦ-3
430
2,9
3,0
13,5
0,68
0,01
0,40
0,57
0,23
2,76
2,15
0,46
0,79
4,18
1,27
37,5
1,99
4,48
0,41
25,0
19,7
3,3
1,1
100
2,77
11,32
46,7
стандартный
490
1,0
3,0
23,0
0,79
0,30
2,07
1,17
1,39
3,34
3,57
1,42
2,97
4,44
1,54
26,1
3,22
2,61
0,47
21,0
23,3
5,6
1,0
100
1,13
3,50
34,0
Микросферические
катализаторы
РСГ-2Ц
450
8,0
7,5
13,2
0,65
0,01
0,21
0,36
0,24
3,06
1,04
0,88
2,19
3,72
0,84
40,9
2,43
4,27
0,28
20,1
21,5
3,3
1,0
100
3,10
12,40
52,1
РСГ-2
490
2,0
7,5
20,5
0,73
0,63
1,65
1,41
0,57
5,25
1,17
2,07
3,28
3,12
0,62-
31,1
4,05
2,30
0,25
21,8
20,4
5,2
1,0
100
1,52
6,00
39,1
рос с 43,3 до 61,8 вес. % по сравнению с выходом на аморфном
катализаторе. Одновременно отношение выходов мотобензин : газ
повысилось с 3,4 до 5,8 и отношение мотобензин : кокс с 19,7 до 23,7,
что указывает на заметно более высокую селективность в
указанном процессе катализатора ЦЕОКАР-2.
30
При замене аморфного катализатора на ЦЕОКАР-2 повысился
и выход авиакомпонента (фракция Cs—165°С) с 58,3 до 70,3%
(т. е. на 12% в расчете на сырье второй ступени) без увеличения
коксообразования. Одновременно значительно улучшились и
общие показатели процесса двухступенчатого крекинга керосино-га-
зойлевой фракции. Отбор целевого продукта (компонента
авиабензина) от керосино-газойлевой фракции составил 44,7 вместо
26,7 вес. %, т. е. возрос в 1,7 раза.
Применение катализатора ЦЕОКАР-2 способствовало
улучшению качества продуктов: уменьшилось йодное число мото- и
авиабензинов, снизилась температура выкипания 50% состава,
увеличилось содержание ароматических углеводородов в бензинах,
газойлях и тяжелой флегме. По детонационной стойкости бензины,
полученные на аморфном катализаторе и при низкой температуре
очистки D40°С) на ЦЕОКАР-2, одинаковы. Повышение
температуры очистки мотобензина на катализаторе ЦЕОКАР-2 с 440—
465 °С и одновременное использование рециркулята позволили
значительно повысить сортность авиакомпонента (с добавкой 2,6 г
ТЭС на 1 кг продукта) со 107 до 120. Расход катализатора
ЦЕОКАР-2 на установках 43-102 за период испытания в среднем
составил 0,11 вместо 0,145% для аморфного катализатора на
ступени крекинга и 0,12% вместо 0,157% на ступени очистки, т. е.
снизился на 30%. Содержание остаточного кокса на катализаторе
ЦЕОКАР-2 не превышало 0,03—0,08 вес. %.
Обобщен [39] опыт работы цеолитсодержащего катализатора
АШНЦ-3 в промышленных условиях при крекинге тяжелого
высокосернистого сырья. С целью стабилизации системы на
катализаторе АШНЦ-3 30 сут. перерабатывали легкую керосино-газойлевую
фракцию. После приобретения катализатором равновесной
активности D7 пунктов) начали перерабатывать тяжелое сырье при
следующем режиме: 445—455 °С, объемная скорость 1,7 ч~*, крат-
Таблица 14
Физико-химические свойства катализаторов
Показатели
Насыпная плотность, кг/м3
Индекс каталитической активности
Индекс каталитической стабильности
Прочность шарика, кгс
Содержание, вес. %
целых шариков
фракций более 5 мм
фракций менее 2 мм
Удельная поверхность, м2/г
Удельный объем пор, см3/г
Средний радиус пор, А
Свежий
АШНЦ-3
665
50
50
18
81
3,0
6,0
260
0,520
33
аморфный
700
37
28
19
93
3,5
3,5
365
0,555
30
Равновесный
АШНЦ-3
718
47
23
98
0,8
1,2
195
0,407
42
аморфный
770
32
—
27
94
0,4
1,1
240
0,375
32
31
ность циркуляции катализатора 1,9 т/т сырья. Максимальная
температура в регенераторе 720—740 °С обеспечивала содержание
остаточного кокса на катализаторе 0,2 вес. %.
После 12 месяцев работы установки катализатор сохранил
активные свойства (табл. 14). Выход и качество получаемых
продуктов остались на одном и том же уровне, несколько снизился
выход кокса. Расход катализатора после одного года эксплуатации
уменьшился с 2,6 до 2,2 кг/т сырья (против расхода аморфного
катализатора 3 кг/т).
Ниже приведен материальный баланс установки
каталитического крекинга на цеолитсодержащем и аморфном катализаторе:
АШНЦ-З Аморфный
Переработано сырья 100,00 100,00
Получено
стабильный бензин 31,25 21,01
легкий газойль 35,80 33,96
тяжелый газойль 14,70 28,60
сероводород 2,00 1,94
сухой газ . . . . '. 2,02 1,99
пропан-пропиленовая фракция .... 3,64 2,87
бутан-бутиленовая фракция 5,60 4,64
в том числе
бутилены 1,44 1,57
изобутан 3,26 2,34
Кокс 3,99 3,99
Потер» 1,00 1,00
Итого . . . 100,00 100,00
Из приведенных ниже данных о свойствах бензинов,
полученных на цеолитном и аморфном катализаторах, видно, что октановые
числа нестабильного бензина по моторному и по
исследовательскому методу сохраняются практически на одном уровне, вне за-,
висимости от применяемого катализатора. Содержание
непредельных в бензине, полученном на цеолитсодержащем катализаторе,
меньше, а парафинов (в том числе изостроения) больше.
АШНЦ-З Аморфный
Октановое число в чистом виде
моторный метод 78,5 79,0
исследовательский метод 84,5 84,5
Плотность rf|° 0,770 0,771
Содержание серы, вес. % 0>43 0,45
Углеводородный состав, вес. %
непредельные 22,6 26,8
в том числе
изостроения 18,1 18,5
нормального строения 4,5 7,8
ароматические 19,3 18,5
нафтены 12,6 17,7
парафины 45,5 37,0
в том числе
изостроения 40,5 32,5
нормального строения 5,0 4,5
32
Получаемые каталитические газойли — легкий (с?|° 0,9193; це-
тановое число 40,7; S 2,2 вес. %; ароматических 56 вес. %) и
тяжелый (d2Q 0,963; S 28 вес. %; ароматических 66,8 вес.
%)—более высоко ароматизированы и, следовательно, являются
исходным сырьем для производства нефтехимических продуктов.
Расчеты показали, что цеолитсодержащий катализатор
обеспечивает экономию 0,8—1,0 руб/т перерабатываемого сырья. При
ужесточении режима на пилотной установке выход бензина может
быть увеличен до 38—40 вес. %, октановое число бензина по
моторному методу повысится до 82 пунктов, отношение
непредельных к парафиновым углеводородам в бутан-бутиленовой фракции
значительно возрастает.
3—2206
Глава II
СТАРЕНИЕ КАТАЛИЗАТОРОВ КРЕКИНГА
ИЗМЕНЕНИЕ СВОЙСТВ КАТАЛИЗАТОРА
ПРИ ЕГО СТАРЕНИИ
В промышленных условиях физические и химические свойства
алюмосиликатных катализаторов резко изменяются. Одна из
причин изменения катализатора — его спекание. Скорость и степень
спекания катализаторов зависят от их химического состава, поро-
вой характеристики, температуры процесса, среды,
продолжительности обработки и др. Влияние температуры прокалки в воздухе
на удельную поверхность катализаторов показано на рис. 12 (дан-
350 500 650 800 950
Температура прокалки, °С
Рис. 12. Влияние температуры прокалки на относительную удельную
поверхность катализаторов крекинга.
Катализаторы: ;, 2, 3, 4 — синтетический аморфный алюмосиликатный; 5, 6 — природный-
7,8 — магнийспликатный.
ные взяты из работ [41—44]). До 500 °С удельная поверхность
катализаторов сохраняется постоянной. Дальнейшее повышение
температуры приводит к прогрессивному уменьшению поверхности;
при 800—1000 °С она становится равной нулю. Наименьшей
термостойкостью обладают магниисиликатные и природные
катализаторы, а наибольшей — синтетические алюмосиликаты. Разная
термостойкость алюмосиликатов, несмотря на их близкий химический
34
состав, обусловлена, по-видимому, влиянием микропримесей,
большей или меньшей неоднородностью поверхности и другими
факторами.
wo **>
0 5 10 15 20
Длительность посжалки V
25
200 'rOO Є00
Длительность прокалки , v
800
Рис. 13. Влияние продолжительности прокаливания на относительную удельную
поверхность алюмосиликатного аморфного катализатора.
Температура прокалки. °С: / — 785; 2 — 847; 3 — 900; 4 — 950.
Рис. 14. Зависимость удельной поверхности аморфного катализатора от
длительности прокалки:
/ — в воздухе при 600 °С; 2 — ъ среде водяного пара при 600 °С; 3 — нормальный интервал
для равновесного катализатора.
Скорость уменьшения удельной поверхности со временем
сильно меняется. Из рис. 13 (построенного по данным работы [41])
видно, что наиболее резко удельная поверхность уменьшается в
начале прокаливания.
600
200 ' Ш 6ОО 800
Длительность пропарки, ч
Рис. 15. Влияние длительности термопаровой обработки на удельную
поверхность алюмосиликатного катализатора:
Условия обработки; температура, °С (давление, МПа): /—576 @,03); 2 — 478 @,1); 3 — 576
@,1); 4-778 @,011); 5 - 600 @,1); 6-576 @,3); 7-778 @,08); 8-750 @,1); 9 — 778 @.1)-.
Q* 3ft
Спекание катализатора существенно ускоряется, если его
прокаливать в присутствии водяного пара. Из рис. 14 (построенного
по данным работы [45]) видно, что удельная поверхность
катализатора, прокаленного при 600 °С 400 ч в воздухе, почти в 2 раза
больше, чем пропаренного в тех же условиях.
Зависимость удельной поверхности от температуры и
длительности термопаровой обработки, или пропарки (рис. 15)
практически такая же, как и при прокалке в сухом воздухе. Только при
пропарке все изменения происходят в области более низких
температур. Из рис. 15 видно, что на результаты термопаровой
обработки большое влияние оказывает парциальное давление водяного
пара. Скорость спекания катализатора под влиянием паров воды
наиболее сильно возрастает при повышении парциального
давления с 0 до 0,1 МПа. При дальнейшем повышении давления
скорость спекания также увеличивается, но уже в меньшей степени.
Как известно, катализатор при хранении адсорбирует
значительное количество влаги, часть которой остается даже после
прокаливания. В случае прокалки катализатора при температурах
выше, чем температура предварительной осушки, остаточная влага
будет выделяться из катализатора в паровую фазу и ускорять
процесс старения катализатора, т. е. вызывать явление
«самопропарки» катализатора.
Возможность самопропарки катализатора при прокалке в
сухом воздухе была показана следующим путем [41]. Две пробы
одного и того же катализатора нагревали с 565 до 950 °С при
скорости повышения температуры соответственно 1 и 5°С/мин.
Влияние скорости нагревания на спекание катализатора показано ниже:
^Достигнутая температура ,
°С
847 900 950 Я
Медленное нагревание, 1 сС/мин
удельная поверхность, м2/г 536 455 263
удельный объем пор, см3/г 0,665 0,595 0,320
радиус пор, А 49,6 52,3 48,7
Быстрое нагревание, 5°С/мин
удельная поверхность, м2/г .... 484 384 J39
удельный объем пор, см3/г .....' 0,598 0,495 0,205
радиус пор, А 49,4 5J,5 59,0
Несмотря на большую длительность прокалки, при медленном
нагревании удельная поверхность катализатора после опыта
оказалась более высокой, чем при быстром. Влияние адсорбционной
влаги на уменьшение поверхности катализатора 'при прокалке
было показано опытами с применением вакуума для удаления
влаги. Так, если при медленном нагревании катализатора до
950 °С удельная поверхность составила 263 м2/г, то при нагревании
под вакуумом в тех же условиях она оказалась равной 430 м2/г.
При старении уменьшается не только удельная поверхность, но
и удельный объем пор; кажущаяся и насыпная плотность
катализатора возрастают.
36
В некоторых исследованиях [46] отмечается, что удельный
объем пор катализатора при прокалке уменьшается прямо
пропорционально удельной поверхности. Однако рис. 16, построенный
0,7
500
Удельная поверхность , м2/г
Рис. 16. Зависимость между удельной поверхностью и удельным объемом пор
для прокаленных катализаторов.
700 200 300 400 500 600
Удельная поверхность, мг/г ,"
Рис. 17. Зависимость между удельной поверхностью и удельным объемом пор
для катализаторов, обработанных паром:
/—теоретическая кривая для поверхностно-диффузионного спекания; 2, 3, 4. 5 — кривые,
построенные по экспериментальным данным для катализаторов, обработанных паром
соответственно при 473, 576, 678, 863 °С.
на основании литературных и наших данных, указывает на более
сложную зависимость.
Изменение удельного объема пор при спекании катализатора
под совместным действием водяного пара и температуры показано
на рис. 17. (Значение теоретической кривой, изображенной на
37
рис., см. стр. 56). В случае пропарки при температуре <650 С
удельный объем пор с уменьшением поверхности меняется
медленнее, чем при термической прокалке (см. рис. 16).
Для описания зависимости удельного объема пор V от
величины поверхности катализатора S предложены следующие
эмпирические уравнения [42].
При прокалке:
К™ = KIOS A)
При пропарке:
К™ = KinoS + Vo B)
где Кто, Ктпо, Vo — коэффициенты.
Авторы [42] подчеркивают приближенный и ограниченный
характер зависимости, выражаемый этими уравнениями. Они
применимы лишь к определенному кругу пористых тел — к
стекловидным силикагелям и алюмосиликагелям, прокаленным при
температуре не ниже 500 °С и обладающим однороднопористой
структурой. Неоднороднопористые алюмосиликаты и силикагели этой
зависимости не подчиняются [47]. При прокалке таких
катализаторов поверхность уменьшается в большей степени, чем размер
пор, и средний размер пор увеличивается.
Экспериментальные данные о изменении среднего радиуса пор
в зависимости от величины удельной поверхности для образцов
катализатора, пропаренных в лабораторных условиях и
подвергшихся старению на промышленных установках, приведены на рис.
18 [41, 48]. Характер этой зависимости сохраняется одинаковым
при всех исследованных температурах обработки; во всех случаях
пропарки в лабораторных условиях спекание катализатора
сопровождается увеличением среднего радиуса пор.
Наибольшее увеличение размера пор наблюдается при
пропарке катализатора в мягких температурных условиях. Уменьшение
поверхности катализатора в результате прокалки в промышленных
условиях сопровождается очень небольшим возрастанием
среднего радиуса пор.
Из анализа уравнений A) и B) следует, что при прокалке
катализатора в сухом воздухе величина пор не изменяется.
Действительно, если в этих уравнениях удельный объем пор V выразить
через средний радиус (гср) по известному соотношению:
21/
'ср = — C)
то будем иметь для случая прокалки:
гср = 2К-ТО = const D)
для случая пропарки:
гср = 2/(ТПо -(- ^ E)
38
Из соотношения D) видно, что при прокалке катализатора
размер радиуса пор остается постоянным, независимо от
уменьшения поверхности. При пропарке же он возрастает, так как член
2V0/S с уменьшением удельной поверхности увеличивается.
Характер изменения удельного объема пор и их радиуса для
образцов катализатора, подвергшихся спеканию в системе
промышленных установок, оказывается таким же, как и при прокалке
в сухом воздухе. Следовательно, в условиях промышленной
эксплуатации катализатора старение протекает главным образом под
влиянием высоких температур.
то
гоо зоо wo 500
Удельная поверхность tM2/z
600
Рис. 18. Изменение удельной поверхности и среднего радиуса пор катализаторов
при их спекании:
/ — по поверхностно-диффузионному механизму; 2 — при 478 и 576 °С, 0,03—0,7 МПа; 3 — при
678 "С и 0,1 МПа; 4 — при 778 °С, 0,Г МПа; 5 — при 778 "С, 0,011 МПа; 6 — при 863 °С,
0,1 МПа; 7 —в промышленных условиях.
При прокалке в сухом воздухе средний диаметр пор аморфных
алюмосиликатов и распределение объема пор по радиусам не
меняется. У пропаренных образцов появляются новые, более
крупные поры, но сохраняется и некоторое количество мелких пор [49].
Термо- и парообработка катализатора может приводить не
только к изменению структурных характеристик, но и к фазовым
превращениям материала катализатора. Алюмосиликатные гели,
содержащие до 30% окиси алюминия, являются аморфными [49].
39
Нагрев синтетических алюмосиликатных катализаторов,
содержащих 9—12% окиси алюминия, в воздухе приводит к появлению
кристаллической фазы лишь при 1100°С и выше [50, 51], т. е. в
условиях, которые вызывают полное разрушение поровой
структуры катализатора. С повышением концентрации алюминия в
катализаторе температура появления кристаллической фазы
снижается. Показано [52], что катализатор с 25% окиси алюминия после
трехчасовой прокалки при 840 X уже содержит -у-окись алюминия.
При 880°С образуется муллит. После прокалки при 1200°С
катализатор содержит а-кристобалит.
Следовательно, уменьшение активности этих катализаторов не
может быть обусловлено изменением их фазового состава. Однако
это еще не означает, что единственной причиной уменьшения
активности являются структурные изменения.
По данным работы [53], уже при прокаливании катализаторов
до 600 °С их активность заметно уменьшается, хотя структурные
изменения происходят лишь при 800 °С. После обработки
катализатора паром температура начала дезактивации катализатора
снижается примерно на 200°С при отсутствии заметных
структурных изменений. В работе [54] отмечается, что дезактивация
алюмосиликатных катализаторов при термической и термопаровой
обработке может происходить не только из-за уменьшения
величины поверхности, но и вследствие ее качественного изменения. По
мнению авторов, при спекании без водяного пара такое
качественное изменение не происходит. Об этом свидетельствует
относительное постоянство удельной обменной способности A,7—2,1-10~4мг-
экв/м2). В случае одновременного действия высокой температуры
и водяного пара удельная обменная способность катализатора
уменьшается, изменяется качественный состав продуктов реакции,
а также избирательность действия катализатора.
Причину таких изменений обменной способности поверхности
алюмосиликатного катализатора при высокотемпературной
обработке исследователи [54] видят в механизме удаления связанной
с его поверхностью воды. Предложены следующие схемы
выделения воды:
1. —Si—О—Si—ОН + НО—Si—О—Si >¦
II II
I I I I
>- —Si—О—Si—О—Si—О—Si—
I I I I
2. —Si—O—Si—
OH
J он
Выделение воды по первой схеме ведет к дальнейшему
укрупнению частиц геля, сопровождающемуся необратимым
уменьшением его поверхности. При этом качественное состояние поверхности
40
не должно измениться. Выделение воды по второй схеме не
приводит к уменьшению поверхности, но оно должно вызвать
изменение ее свойств, поскольку не только уменьшается число групп ОН
на поверхности, но и происходит замена их кислородными
мостиками. При высокотемпературном нагревании вода выделяется по
схеме, аналогичной первой. При одновременном действии
температуры и пара процесс протекает, по-видимому, одновременно по
обеим схемам. Применительно к алюмосиликатам такой процесс
можно изобразить следующей схемой [55]:
| уОН НОЧ |
—Si—О—А1< \A1_O—яі—
| ЧОН НСК |
I /ОН ' |
—Si—О—А1< НО—AI—О—Si—
Ч)Н
о
О Si—
ОН
I I I
—Si—О—AI—О—Si—
О О -f- А12О3 + ЗН2О
| I
І |
О—Si—О
Описанные изменения структуры и в ряде случаев фазового
состава материала катализатора и свойств поверхности
существенно сказываются на изменении его активности и селективности
в реакциях крекинга.
Изменение в зависимости от условий спекания активности алю-
мосиликатных катализаторов, пористой структуры, удельной
поверхности изучалось рядом авторов. В работе [56] проводили
крекинг фракции 200—400 °С ка алюмосилпкатных катализаторах с
разным диаметром пор и пришли к выводу, что поверхность пор
диаметром 5—7 А и менее не используется в реакциях крекинга
сырья из-за эффекта ультрапористости. Активность единицы
доступной поверхности катализатора оказалась приблизительно
постоянной.
Установлено [57], что у катализаторов с существенно
различающейся поверхностью (от 282 до 456 м2/г) удельная активность
поверхности сохраняется постоянной. В этой работе активность
определяли по каталитическому разложению кумола в проточной
установке при 400 °С. Скорость крекинга была пропорциональна
суммарной поверхности испытуемого образца:
W = KSVA F)
41
где W — скорость крекинга (объем газа), см3/мин; К — константа
скорости реакции, равная скорости газообразования на 1 м2
поверхности катализатора при 400 °С; S — поверхность катализатора,
м2/г; V — объем катализатора в реакторе, см3; А — насыпная
плотность, г/см3.
Так как активность единицы поверхности катализатора
постоянна, за меру каталитической активности можно принять
произведение SA (при образцах одинакового химического состава).
Приблизительное постоянство активности единицы поверхности
отмечалось и в работах [42, 58].
В работах [52, 59, 60] указывается на небольшое увеличение
активности единицы поверхности образцов, подвергнутых
термопаровой обработке; однако, по мнению большинства из авторов
данных работ, это не свидетельствует об изменении фактической
активности, а обусловлено меньшим влиянием внутренней диффузии
у более крупнопористых образцов. Исследования кинетики
крекинга кумола на образцах алюмосиликатного шарикового
катализатора [61] показали, что после прокаливания удельная
активность катализатора практически не изменяется, а после обработки
паром 24 ч при 750 °С она уменьшается очень сильно.
Относительные удельные активности для свежего, прокаленного при 900 °С и
пропаренного при 750 °С катализаторов оказались равными
соответственно 0,66, 0,60 и 0,33. Это уменьшение удельной активности
автор объясняет разрушением алюмосиликатных групп или соеди-'
нений, ответственных за каталитическую активность.
В процессе изучения влияния условий старения на активность
катализатора было установлено, что при этом изменяется и его
селективность — на образцах катализаторов, после их термической
и термопаровой обработки, при крекинге образуются более
непредельные продукты, выход кокса и газа уменьшается в большей
степени, чем выход бензина. В работе [62] такое изменение
селективности объясняется наличием на поверхности катализатора
по крайней мере двух видов активных центров. Одни из них
ответственны за реакции крекинга, и в процессе термической или
термопаровой обработки их число на единицу поверхности
катализатора не меняется. Другие катализируют реакцию
перераспределения водорода, и при спекании катализатора их свойства и
число активных центров на единицу поверхности существенно
изменяются.
Однако изменение селективности при спекании происходит и
при неизменном качественном состоянии поверхности
катализатора вследствие влияния пористой структуры. Так, ультрапоры не
участвуют в крекинге крупных молекул сырья, но могут вызывать
распад молекул бензина [56]. Влияние пористой структуры на
избирательность реакций зависит от диффузионной способности
углеводородов, а, следовательно, от времени контакта и
концентрации реагирующих веществ внутри катализатора, а также от
размера его пор [62, 63].
42
О влиянии на свойства катализатора высокотемпературной
обработки в среде других различных газов (кроме водяного пара
и воздуха) имеются лишь отрывочные сведения. Наиболее полно
влияние прокалки катализатора крекинга в среде окиси углерода,
сероводорода, двуокиси серы, аммиака, двуокиси углерода
изучалось в работе [64]. Катализатор прокаливали в проточных
условиях в интервале температур от 540 до 871СС при атмосферном
давлении. Об изменении катализатора судили по изменению
активности и селективности по отношению к коксо- и
газообразованию. Исследования показали, что газы можно разделить на 3
группы: 1) инертные (воздух, окись углерода, двуокись углерода и
серы, аммиак при температуре ниже 620 °С); 2) ускоряющие
нормальное старение (водяной пар); 3) вызывающие ненормальное
старение природного, но не синтетического катализатора
(сероводород при температуре выше 427 °С, аммиак и двуокись серы при
температуре выше 620°С).
Газы первой группы при низких температурах не влияют на
изменение активности алюмосиликатных катализаторов. Водяной
пар ускоряет уменьшение активности катализатора, но при этом
селективность его значительно не изменяется. Газы третьей
группы вызывают уменьшение активности и резкое изменение
селективности катализаторов. По мнению автора работы [64], такое
резкое изменение активности и селективности связано с
изменением состояния железа, содержащегося в природных
катализаторах, которое под влиянием сернистых соединений превращается
в активную форму и изменяет направление процесса крекинга.
Синтетический алюмосиликатный катализатор, содержащий
меньше окиси железа, ненормального старения не обнаруживает.
Инертность воздуха, двуокиси и окиси углерода и вредное
влияние паров воды на промышленных установках хорошо
иллюстрируются данными об активности синтетического алюмосиликат-
ного катализатора, прокаленного при 760 °С в течение 10 ч:
Среда Активность
Сухой воздух 48
Окись углерода 49
Двуокись углерода 48,5
Водяной пар 27
Действие высокой температуры и водяного пара на
цеолитсодержащие катализаторы крекинга совершенно иное, чем на
аморфные алюмосиликатные катализаторы. При термической и
термопаровой обработке цеолитсодержащие катализаторы
обладают высокой стабильностью. В режимах термопаровой обработки,
когда первоначальная активность аморфных катализаторов
снижается вдвое, активность некоторых цеолитсодержащих
катализаторов даже несколько повышается. Это объясняется высокой
стабильностью кристаллического каркаса цеолитного наполнителя.
Ниже приводятся максимальные температуры, при которых струк-
43
тура цеолитов сохраняется после прокалки сухим воздухом в
течение 3 ч [29]:
Цеолит Температура.
Na-фожазит, или NaX (Si/Al=l,25) ... 649
Na-фожазит, или NaY (Si/Al=2,5) .... 704
MgX 843
MgY 871
P3NaX @,85P3 + 0,15 Na) 843
P3NaY @.85P3 + 0,15 Na) 871
P3X 899
P3Y 927
NH4X 204
NH4Y 260
NH4X стабилизированный ^>980
NH4Y стабилизированный ^>980
Из этих данных видно, что цеолит типа Y более термостабилен,
чем цеолит типа X.
Значительное влияние на термостабильность цеолитов
оказывает содержание в них натрия, так как он вызывает потерю
кристалличности цеолита в результате спекания. При замене в
цеолите натрия магнием термостабильность возрастает на 170—200 °С,
при замене редкоземельными элементами — на 220—250°С. В то
же время даже термостойкость цеолитов с редкоземельными
элементами при введении в них и 15% натрия снижается на 56°С. По
данным [65], цеолит типа P3Y, содержащий 4% Na2O, менее
стабилен, чем цеолит типа РЗХ, содержащий 1% Na2O. В среде
водяного пара термическая стабильность цеолитов уменьшается. Так,
NaX полностью разрушается при 400 °С и давлении водяного пара
0,1 МПа [26]. Цеолиты типа Y обычно более устойчивы к
термопаровой обработке, чем цеолиты типа X.
Устойчивость цеолитсодержащего катализатора при
термической и термопаровой обработке изменяется в соответствии со
стабильностью цеолита. Ниже представлены данные об активности и
селективности цеолитсодержащих катализаторов с различными
цеолитами [66]:
Активность „
(выход Селективность
Образцы катализаторов бензина (отношение
вес У\ бензин:кокс)
Образец 1 (NH4X)
исходный 32,0 12,3
после пропарки 15,0 16,7
после пропарки, 12 месяцев хранения
и прокалки 14,6 13,3
Образец 2 (CaNH4X)
исходный 37,3 17,8
после 5 месяцев хранения и прокалки . 32,3 8,6
после пропарки 45,5 —
после пропарки, 7 месяцев хранения
и прокалки 29,7 7,6
после пропарки, 10 месяцев хранения
и прокалки 24,1 6,3
44
Образцы катализаторов Активность (выход Селективность
бензина, вес %) (отношение
бензинжокс)
Образец 3 (СаХ)
после пропарки 23,3 8,3
после 5 месяцев хранения и пропарки . 22,6 9,4
после пропарки, 24 месяцев хранения
и прокалки 24,5 8,2
Образец 4 (LaX)
после пропарки 32,5 11,6
после пропарки, 14 месяцев хранения
и прокалки 37,3 —
В качестве матрицы катализаторов использовали двуокись
кремния при соотношении цеолит: связующие 20 : 80. Активность
катализаторов оценивали по стандартной методике [67]. Образцы
хранили в бумажных пакетах при комнатной температуре.
Условия термопаровоздушной обработки: / = 750 °С; т = 2 ч; содержание
водяного пара в паровоздушной смеси 25%- Условия прокалки:
t = 500—550 °С; т = 4 ч.
Хранение образцов 1, 2, 3 и 4 в течение нескольких месяцев
оказало влияние на их крекирующую активность. Наиболее
высокую активность после длительного хранения сохранил образец,
содержащий лантан. Для проверки возможности восстановления
активности образец 2, хранившийся в течение 10 месяцев,
дополнительно исследовали после прокалки при 750 °С, после обработки
в среде насыщенных водяных паров н после вторичной
термопаровоздушной обработки. Во всех этих случаях активность
катализатора не восстанавливалась. Следовательно, в катализаторе во
времени происходят необратимые изменения. Активность
пропаренного образца после хранения в течение двух лет оказалась
того же порядка B4,5 вес. %)¦ Активность образца 1,
синтезированного на основе декатионированной формы цеолита типа X, была
ниже активности образца 2, содержащего катион-декатионирован-
ную форму цеолита (CaNH4X) [68].
Стабильность цеолитсодержащих катализаторов оказалась в
прямой зависимости от термической стабильности цеолитов,
использованных для приготовления катализатора. Она возрастает
в следующем порядке: NH4X<CaNH4X<CaX<LaX.
Благодаря высокой термостабильности цеолитсодержащие
катализаторы сохраняют значительную активность при жестких
режимах высокотемпературной обработки в присутствии водяного
пара. В процессе термопаровой обработки цеолитсодержащий
катализатор постепенно снижает свою активность (рис. 19), однако
даже при 870 °С глубина превращения в его присутствии такая же,
как и на обычном алюмосиликатном катализаторе, пропаренной
при 730 °С [69].
О изменении физических и химических свойств
цеолитсодержащих катализаторов крекинга под влиянием высоких температур
и водяного пара опубликовано еще мало сведений. Это
объясняется, с одной стороны, сравнительно недавним применением цеолит-
ных катализаторов в промышленности, с другой стороны,
исключительно высокой стабильностью их структуры. Основное внимание
в литературе уделяется уменьшению потерь катализатора от
истирания и разработке систем каталитического крекинга, наиболее
приспособленных для использования преимуществ цеолитных ка-
100
250
'700 7JJ SOO
Тгчл-:-рптура п
8.10 50Э
150 800 850 900
Температура пропарки, "С
Рис. 19. Глубина превращения сырья в зависимости от температуры пропарки (в
течение 4 ч) катализаторов:
/ — аморфный алюмосиликатный; 2 — каолиновый; 3 — цеолитсодержащий.
Рис. 20. Изменение удельной поверхности цеолитсодержащих катализаторов в
зависимости от температуры пропарки, проведенной в течение 4 ч:
1, 2 3 4 — образцы промышленных катализаторов; 5 — образцы катализаторов HFZ-20
и HFZ-23.
тализаторов. Из немногочисленных исследований представляют
интерес результаты работ [69, 70], в которых изучали изменение
удельной поверхности катализаторов в зависимости от
температуры их термопаровой обработки (рис. 20). Эти результаты
подтверждают высокую стабильность цеолитсодержащих
катализаторов. Их удельная поверхность уменьшается с небольшой скоростью
при повышении температуры. Особенно высокой стабильностью,
активностью и селективностью, по сравнению с существующими
цеолитсодержащими катализаторами, отличаются новые
катализаторы: HFZ-20 и HFZ-23.
Из данных табл. 15 [69], где приводятся результаты
каталитического крекинга в присутствии цеолитсодержащих
катализаторов, видно, что после пропарки селективность катализаторов
повышается.
Весьма подробно исследован характер старения некоторых
отечественных цеолитсодержащих катализаторов в работе [36].
Испытания проводили на пилотных установках с подвижным шари-
Таблица 15
Результаты каталитического крекинга в зависимости от условий
термопаровой обработки цеолитсодержащего катализатора
Условия
ература,
с
tu (j
fr- Q
774
843
871
816
816
816
816
816
пропарки
ельность,
S
ч
«=¦
4
4
4
8
8
8
8
8
їмная ско-
ь подачи
,я, 4-і
sit
1
1
1
1
2
3
4
1*
Я*
S
Я
tu
VO
58
57
46
56
51
47
45
37
X
s
tu
S
,8
,6
,1
,8
,9
,3
,2
,3
5
2
2
2
2
2
2
4
Данные,
g
03
U
о
X
,6
,9
,2
,9
,3
,0
,0
,4
вес. %
газ.
25,
14,
И,
15,
10,
8,
7,
21
полученные
2
6
5
2
9
4
2
2
сительная
ность
отно
плот
газа
1,62
1,48
1,37
1,50
1,50
1,53
1,50
1,45
по методу
в:
конв
вес.
82,5-
68,1
54,5
68,1
59,7
52,3
49,0
58,4
Кат-Д
ность
лизатора,
плот
ката
кг/л
0,86
0,89
0,93
0,90
—
—
—
—
[ьная
рхность
лизатора.
lilt
138
78
48
96
—
—
, —
95
Аморфный алюмосиликат.
ковым катализатором АШНЦ-3 и микросферическим катализато-:
ром РСГ-2Ц в кипящем слое. В качестве сырья использовали
вакуумный дистиллят ромашкинской нефти со следующими физико-
химическими свойствами:
Плотность, d|° 0,9165—0,9231
Фракционный состав, объемн. %
и. к., °С 345—352
выкипает при температуре, °С
10% ' 397—403
50% 444—456
Содержание фракций, объемн. %
до 350 °С 0,5
до 500 °С 87
Содержание, вес. %
углерода ... 85,35—85,32
водорода . . 12,75—12,73
серы ... 1,79—1,88
азота 0,11-0^,07
никеля 3—4-10 ъ
ванадия 1-10
Групповой углеводородный состав, вес. % .
парафины 26,2—26,0
нафтены 23,0—22,9
ароматические 47,4—47,2
в том числе тяжелые ароматические 15,4—15,3
смолы 3,4—3,9
Молекулярный вес 321
Коксуемость, вес. % 0,39—0,43
Вязкость кинематическая при 50°С, мм2/с . 38,71—44,33
Температура, °С
застывания 39—43
вспышки 149—153
47
Для увеличения активности и избирательности катализаторы
обрабатывали 100%-ным водяным паром при высокой
температуре. После паровой обработки активность катализаторов возросла:
шарикового — с 48,7 до 50,6, а микросферического — с 48,2 до
49,6; при этом структурная характеристика катализаторов не
изменилась, что указывает на их стойкость к действию водяного
пара.
Таблица 16
Физико-химические свойства алюмосиликатных
цеолитсодержащих и аморфных катализаторов
Показатели
Индекс
активности
стабильности
Насыпная плотность,
кг/м3
Фракционный состав,
вес. %
3—5 мм
53—220 мк
Структура
удельная
поверхность, м2/г
удельный объем пор,
см3/г
средний радиус пор,
я
л
Механические свойства
прочность на
раздавливание, кгс/шар
износ в эрлифте,
вес. %
Брак по отбору, вес. %
Индекс механического
износа, вес. %
Химический состав,
вес. %
А12О3
SiO2
Fe2O3
CaO
Na2O
Свежие катализаторы
микросферические
иг
РСГ
48,2
51,6
640
72,2
245
0,468
38,2
—
—
—
11,4
13,0
84,29
0,13
1,70
0,88
РСГ
41,8
32,5
708
—
70,7
416
0,586
28,1
—
—
—
15,0
15,54
83,52
0,18
0,58
0,18
шариковые
СО
3
48,7
51,4
689
87,7
—
264
0,532
40,0
24,1
13,3
17,0
—
10,80
87,06
0,40
0,80
0,94
Ё.
38,6
28,8
690
95,3
—
363
0,524
28,9
26,7
4,5
14,8
—
10,40
87,46
0,29
1,55
0,30
Равновесные
микросферические
я
РСГ
47,8
51,9
790
74,6
199
0,396
39,8
—
—
—
3,3
—
—
0,21
—
—¦
РСГ-
32,1
24,9
885
76,9
149
0,302
37,8
—
—
—
3,1
—.
—
0,49
—.
—
катализаторы
шариковые
СО
X
3
48,5
51,5
703
82,6
242
0,506
41,8
30,4
4,3
9,8
—
0,25
—
—
к
s
стан,
ный
3'2,5
25,6
785
80,0
187
0,375
40,1
30,8
4,0
8,3
—.
.
0,63
—
48
д. д ^_д
300
500
900 7800 1500 1800 2700 ZWO
Продолжительность работи катализатора ,ч
кг сырья на кг катализатора і і і
65
і
/Of
і
175
і
220
І
160
j
WO
1
230
\
510
300
і
его
і
368
і
VfO
1
РСГ-2Ц
АШНЦ-3
І
70"
160
205
2S0 335
РСГ-г
Рис. 21. Изменение материального баланса каталитического крекинга в
зависимости от длительности работы катализатора:
(—АШНЦ-3; 2— РСГ-2Ц; 3 - РСГ-2.
Цеолитсодержащие катализаторы сравнивали по
каталитической активности с аморфным стандартным шариковым и лучшим
из опытных образцов — микросферическим РСГ-2.
Физико-химические свойства катализаторов приведены в
табл. 16.
Стабильность катализаторов определяли при температуре
крекинга 470 °С, весовой скорости подачи сырья для
микросферического катализатора 3,0 ч~' и объемной скорости подачи сырья для
шарикового катализатора 1,0 ч~'. В таких условиях катализаторы
проработали на тяжелом сырье около 2000 ч. За этот период на
1 кг циркулирующего катализатора было пропущено сырья (в кг):
на АШНЦ-3 —569; на РСГ-2Ц и РСГ-2— соответственно 741 и
450. После стабилизации катализатора выходы продуктов
крекинга на АШНЦ-3 и РСГ-2Ц установились следующие: фракция
С5—195°С 41—43 вес. %; кокс 4,3—4,8 вес. % (рис. 21).
Благодаря улучшению в процессе работы механических свойств
катализатора его расход снизился.
Аморфный катализатор РСГ-2 оказался менее активным и
стабильным, чем цеолитный РСГ-2Ц. В процессе его испытания
выход бензина был на 12,6% ниже, чем на катализаторе РСГ-2Ц
равновесной активности (см. рис. 21).
Благодаря кристаллической структуре удельная поверхность
цеолитсодержащих катализаторов за весь период работы
снизилась незначительно — примерно в 10 раз меньше, чем активная
поверхность аморфного катализатора РСГ-2 (рис. 22). После
2000 ч работы содержание ванадия на катализаторах РСГ-2Ц и
АШНЦ-3 не превышало 0,001 вес. %; никель обнаружен не был.
При вдвое меньшем количестве пропущенного сырья на
аморфном катализаторе РСГ-2 было обнаружено в 28 раз больше
ванадия и никеля, чем на цеолитсодержащем катализаторе (см.
рис. 22).
С повышением содержания на аморфном катализаторе метал-
лоорганических соединений в газе крекинга возрастает
содержание водорода, а водородный фактор, выражающий весовое
отношение водорода к сумме метана, этана и этилена, увеличивается
по сравнению с его начальным значением в 2 раза. Водородный
фактор при крекинге на цеолитсодержащем катализаторе
практически не изменяется (см. рис. 22).
В бензинах и газах крекинга, проводимого на равновесном
цеолитном катализаторе, предельных углеводородов содержится
больше (с преобладанием углеводородов изостроения), чем в
присутствии аморфных катализаторов. Данные, полученные в конце
испытания, практически не отличались от данных начального
периода испытания. В бензинах и газах крекинга, проводимого на
аморфном катализаторе, содержание непредельных углеводородов
(по мере накопления на катализаторе металлов) непрерывно
возрастает, а изосоединений — падает (рис. 23).
50
45 -
Зі 35
г зо
1!
35
30
-Д. А
• - 1
о-2
і і__
о о
J І І І І L
o 7O0 ZOO 300 'tOO 500 BOO 100 800
Пропущено сырья , кг/кг катализатора
Рис. 22. Изменение каталитической активности, селективности и структуры
катализаторов в зависимости от количества пропущенного сырья:
/— АШНЦ-З; г — РСГ-2Ц; 3 — РСГ-2.
51
В табл. 17 показано изменение основных свойств
циркулирующего в системе цеолитного катализатора ЦЕОКАР-2 в зависимости
от продолжительности его работы [38]. В течение длительности
эксплуатации 2800 г каталитические свойства ЦЕОКАР-2 практи-
О 200 WO 600 300 WOO /200 ІЇОО /SO /SOG 2000 2200 Z'rOO 260O
Продолжительность работы кат-іяизпгоора, v
Рис. 23. Зависимость углеводородного состава продуктов крекинга от
продолжительности работы катализатора и накопления на нем ванадия и никеля:
/-АШНЦ-3; 2-РСГ-2Ц; 3 - РСГ-2.
чески не изменились. Физическая структура стабилизировалась
довольно быстро.
Аналогичные результаты были получены при промышленном
испытании на установке термофор катализаторов дюрабед-5 и
дюрабед-6 [30]. К 400 т равновесного катализатора дюрабед-5, не
содержащего соединений хрома, было добавлено по 40 т свежих
52
Таблица 17
Изменение свойств
катализатора Цеокар-2
в процессе испытания
Показатели
Насыпная плотность, кг/м3
Брак по разбору, вес. %
Выход продуктов, вес. %
бензин (н. к. —200 °С)
газ
кокс
Индекс стабильности
Физическая структура
удельная поверхность, м2/г
удельный объем пор, см3/г
средний радиус пор, А
Перед
вводом
пэрэ и
сырья
700
8,0
48,7
21,9
4,9
52
325
0,50
31
Качество катализатора после
96 ч
700
8,5
51,2
21,7
4,4
52
320
0,49
31
432 ч
710
7,6
51,2
23,4
4,5
51
280
0,48
35
624 ч
710
5,0
51,7
19,5
5,3
51
270
0,47
34
1512 ч
720
4,0
50,3
21,1
4,3
51
270
0,46
34
работы
2808 ч
720
0,7
50,3
21,2
4,3
50
270
0,46
34
катализаторов дюрабед-5 и дюрабед-6. Частицы катализатора дю-
рабед-6 содержали 0,15 вес. % Сг2О3, что позволило отделить их
от массы дюрабед-5, не содержащего соединений хрома (они име-
200
Длительность, суш
300
Рис. 24. Изменение активности
катализаторов дюрабед-6 (/) и дюрабед-5 B) в
зависимости от длительности эксплуатации.
ли другой оттенок). Во время опыта лишь дюрабед-5 использовали
для восполнения потерь. Данные испытаний (рис. 24) показали,
что дюрабед-6 более стабилен, чем дюрабед-5.
Большая часть цеолитсодержащих катализаторов более стойка
к действию водяного пара, чем обычные высокоглиноземистые
катализаторы, но и их активность при этом существенно снижается
(см. рис. 24). Поэтому многие зарубежные фирмы для
предотвращения дезактивации цеолитсодержащего катализатора не подают
водяной пар в регенератор и стремятся держать в нем
минимальные количества катализатора.
5J
МЕХАНИЗМ СПЕКАНИЯ АЛЮМОСИЛИКАТНОГО
КАТАЛИЗАТОРА
Спекание при пропарке и прокалке вызывает изменения в
пористой структуре, а также изменяет состояние вещества
катализатора. В работе [71] на основании представлений о пластинчатой
структуре алюмосиликатных катализаторов дается следующее
объяснение закономерностей изменения поверхности, удельного
объема и среднего радиуса пор в процессе спекания. При
термообработке разрушаются наружные стенки пор, и ряд пор около
наружной поверхности частицы катализатора уничтожается.
Остальная часть пор изменяется незначительно. При обработке
паром внутренние перегородки между порами разрушаются, что
приводит к увеличению размеров пор при незначительном изменении
их общего объема.
По современным представлениям, силикагели, алюмогели,
аморфные алюмосиликатные катализаторы имеют не
пластинчатое, а корпускулярное строение, т. е. состоят из сросшихся
непористых первичных частиц шаровидной формы. Первичные
частицы в зависимости от способа получения геля могут быть разных
размеров и упакованы с различной плотностью. Зазоры между
первичными частицами представляют собой поры катализатора;
эффективными диаметрами пор являются наиболее узкие места
этих зазоров (горла пор). У носителей и катализаторов диаметры
пор близки к размерам первичных частиц.
Такие представления первоначально были развиты на
основании данных по адсорбции и десорбции газов (паров); эти
процессы были проведены на спрессованных и неспрессованных
порошках из непористых шаровидных частиц, на непористых образцах
кремнезема (кварц и кварцевое стекло) и на силикагелях [72].
В дальнейшем предложенная структура ксерогелей была
многократно подтверждена с помощью электронно-микроскопических
исследований [73—75]. С точки зрения корпускулярной теории
строения скелета ксерогелей спекание катализатора при
термопаровой обработке можно представить как результат изменения
размеров, формы, взаимного расположения и связи первичных частиц,
происходящего вследствие переноса вещества этих частиц [75].
Перенос происходит в направлении уменьшения свободной
энергии дисперсной системы и приводит к сокращению поверхности, а,
следовательно, к увеличению стабильности системы.
Поверхность катализатора может уменьшаться в результате
объемной и поверхностной диффузии геля в местах срастания
первичных частиц, испарения материала одной частицы и
конденсации его на другой, а также в результате увеличения числа мест
контакта из-за перехода первичных частиц из относительно
рыхлой к более плотной упаковке. Суммарный результат структурных
изменений в катализаторе будет определяться преобладающим
механизмом спекания.
54
При термическом спекании различных дисперсных систем
большую роль играет объемная диффузия [76, 77]. При спекании
первичные частицы, которые в первоначальный момент времени
касались только в одной точке, через некоторое время будут
соприкасаться по основанию сегмента с радиусом А (рис. 25,а).
При этом одновременно уменьшается поверхность и удельный
объем пор, а также становятся меньше линейные размеры образца —
происходит его усадка. При таком механизме спекания в первые
моменты поверхность уменьшается в большей степени, чем объем
пор. Однако, в дальнейшем картина меняется. При сближении
частиц потеря суммарной поверхности постоянно уменьшается.
Исходя из этого, средний радиус пор при уменьшении удельной
поверхности должен вначале расти, а затем уменьшаться.
Рис. 25. Механизмы спекания катализатора в результате объемной (а) и
поверхностной (б) диффузии.
Водяной пар действует на поверхность, а не на всю массу
вещества первичных частиц. Поэтому он не может ускорить
термическую диффузию в объеме частиц геля, которая определяется
лишь температурой паровой обработки. В то же время при
действии пара уменьшение поверхности ускоряется, поскольку
облегчается перенос вещества путем поверхностной диффузии или
путем испарения вещества геля в одном месте и конденсации его в
другом. По поверхностно-диффузионному механизму спекание
катализатора происходит следующим образом (рис. 25,6). Вещество
меньшей из двух соприкасающихся первичных частиц движется по
ее поверхности к месту контакта обеих частиц и переходит на
большую первичную частицу. В результате этого меньшая
частица в конце концов исчезает, а более крупная частица растет.
Крупные термодинамически более стабильные частицы «поедают»
мелкие. В случае движения вещества по поверхности исходное
взаимное расположение первичных частиц сохраняется, т. е.
упорядочения упаковки геля не происходит. Поэтому внешние
геометрические размеры шарика катализатора не изменяются. Удельный
объем пор катализатора также должен оставаться постоянным, так
как независимо от размера первичных частиц общий объем
материала шариков катализатора остается прежним. В результате
уменьшения общего числа первичных единиц и увеличения их
среднего размера уменьшается поверхность единицы массы мате-
55
риала. Удельная поверхность катализатора 5 и радиус первичных
частиц R связаны уравнением:
S =
где п — число первичных частиц в 1 г катализатора.
Здесь р — истинная плотность вещества первичных частиц
катализатора.
Подставляя значение п, получим:
3 1
или
S = .Р ¦ г
так как выше нами отмечалось, что радиус пор катализатора г
равен размеру первичных частиц.
Таким образом, в случае поверхностно-диффузионного
механизма спекания должны выполняться следующие соотношения:
V = const (8)
З 1
Исследования с помощью электронного микроскопа аморфных
алюмосиликатных катализаторов и близких к ним по структуре
силикагеля, окиси алюминия и др. показали [73, 75], что в
процессе старения размеры первичных частиц действительно
изменяются. У катализатора, обработанного при 750°С паром, А.
В.Киселев с сотрудниками обнаруживали частицы диаметром 400—
700 А [75]. Диаметр частиц исходного катализатора составлял
100—200 А. У прокаленных при 900 °С образцов, как правило,
частицы имеют те же размеры, что и у исходного катализатора,
хотя иногда удается обнаружить и более крупные частицы.
В работе [73] при объяснении механизма спекания
катализатора в присутствии пара отмечается, что катализатор
дезактивируется главным образом в результате увеличения размера частиц.
Механизм термического спекания заключается в
локализованном сплавлении небольших участков внутри частицы
катализатора. Эта точка зрения ближе к механизму спекания,
предложенному в работе [71].
Зависимости изменения удельного объема пор и их радиуса от
величины поверхности при прокалке и пропарке катализаторов
позволяют судить о преобладающем механизме спекания. На рис.
16—18 кривая / соответствует поверхностно-диффузионному меха-
56
низму [уравнениям (8) и (9)]. На этих рисунках видно, что при
низких температурах пропарки экспериментальные данные
располагаются очень близко к кривой, выражающей
поверхностно-диффузионный механизм спекания. В этом случае повышение
парциального давления пара или снижение температуры не изменяют
результата, так как спекание идет практически без существенного
влияния объемной диффузии, и эти изменения могут повлиять
только на скорость процесса.
При повышении температуры все большую роль играет
объемная диффузия. Экспериментальные кривые изменения радиуса пор
и их удельного объема все дальше отстоят от кривой,
соответствующей поверхностно-диффузионному механизму спекания. В
области высоких температур механизм спекания существенно зависит
от наличия паров воды, так как при одной и той же
температуре изменение парциального давления пара влияет на
соотношение между механизмами спекания. Чем выше парциальное
давление водяного пара, тем больше роль
поверхностно-диффузионного механизма спекания. При прокалке катализаторов в сухом
воздухе поверхностная диффузия, по-видимому, полностью не
устраняется, хотя она и протекает в значительно меньшей степени,
чем в присутствии водяного пара. Суммарный результат спекания
при прокалке такой, что средний радиус пор изменяется
сравнительно мало.
При изучении механизма спекания цеолитсодержащего
катализатора необходимо учитывать наличие двух фаз: кристаллического
цеолита и аморфной алюмосиликатной матрицы. Матрица
изменяется по описанным выше закономерностям. Поскольку на
последней стадии приготовления свежего катализатора его
пропаривают в жестких условиях, удельная поверхность матрицы в
процессе крекинга уменьшается медленно. Цеолитная часть
катализатора при температурах ниже предельной, характерной для
каждого типа цеолита, не изменяется. При превышении
предельной температуры кристаллическая структура цеолитной частицы
разрушается, и частица дезактивируется.
Так же, как и аморфные катализаторы, цеолитсодержащие
катализаторы дезактивируются в основном в регенераторе.
Редкоземельные элементы катализируют сгорание кокса [78]. На
частицах цеолита он начинает гореть при температуре на ПО °С
ниже, чем температура горения кокса, отложившегося на матрице
[65].
При загорании кокса на матрице температура частицы
катализатора может повыситься до 870 °С [65]. При этом
недостаточно отпаренные частицы катализатора, содержащие повышенное
количество горючего материала, нагреваются еще сильнее.
Многократное действие высокой температуры в регенераторе приводит
к спеканию матрицы. Срок службы цеолитных промоторов
несколько больше, но постепенно они тоже дезактивируются. После
длительной работы гранулы катализатора превращаются в инерт-
57
Таблица 18
Каталитические свойства кальций-
и лантандекатионированных цеолитов
Образец
НУ
СаНУ
СаНУ
СаНУ
СаНУ
САУ
LaHy
LaHy
Lay
Содержание ионов, %
С."
20
40
62
90
95
La3+
50
90
57
Наблюдаемая константа
скорости реакции
ДО
регенерации
520
550
550
320
320
130
330
460
190
после
регенерации
5,8
6,8
46,5
320,0
220,0
130,0
230,0
290,0
190,0
ные частицы с высокой плотностью и их следует вывести из
системы.
Стабильность активности цеолитов по времени сильно зависит
от типа кристаллической решетки [79]. Исследования,
проведенные с применением импульсного
микрокаталитического метода, показали,
что фожазит имеет максимальную
активность, которая сохраняется
длительное время. Первоначальная
активность эрионита очень высока, но из-
за низкой стабильности через 5—6
импульсов она снижается примерно в
3 раза. Еще меньшей стабильностью
обладает морденит (рис. 26).
Полагают [79], что это связано с
небольшим размером пор у эрионита и мор-
денита, что благоприятствует
протеканию вторичных процессов
полимеризации и коксообразованию [79].
В табл. 18 показаны
каталитические свойства кальций- и
лантандекатионированных цеолитов до и
после их регенерации. Из данных табл. 18
видно, что первоначальная активность некоторых цеолитных
катализаторов после нескольких регенераций значительно снизилась.
Лишь при содержании более 60% на цеолит ионов Са2+ и La3+
достигается высокая стабильная активность [79].
О 5 1U 15 20 25
Число цмпульсоВ
Рис. 26. Зависимость
активности от длительности
работы фожазита (/), мордени-
та B) и эрионита C).
58
КИНЕТИКА ПРОЦЕССА СПЕКАНИЯ КАТАЛИЗАТОРА
В настоящее время выявлены многие качественные
закономерности процесса спекания катализаторов под влиянием
температуры и водяного пара. Однако кинетических уравнений,
описывающих эти закономерности, практически нет. Известна лишь одна
работа [41], в которой сделана попытка описать кинетику
спекания катализаторов. Исходя из того, что движущей силой процесса
спекания является зависящая от величины поверхности свободная
энергия, авторы этой работы принимают, что скорость спекания
пропорциональна величине поверхности в некоторой степени п:
-¦§--*¦
где т — длительность спекания, ч; 5 — удельная поверхность, м2/г.
Формально это уравнение аналогично обычному кинетическому
уравнению реакции гс-ного порядка с константой скорости
реакции k. Коэффициенты k и п для каждого конкретного случая были
подобраны экспериментально. Оказалось, что они зависят от
температуры и давления водяного пара (табл. 19). При неизменных
температуре и парциальном давлении паров воды уравнение A0)
достаточно хорошо согласуется с экспериментом.
Таблица 19
Влияние температуры и парциального давления водяного пара
на коэффициенты в кинетическом уравнении A0)
Температура,
°С
478
576
576
576
576
576
Давление
пара,
МПа
—
0,03
0,10
0,30
0,70
k
1,5 Ю-24
3,7-Ш-21
4,5 Ю-«
3,7-10-21
1,4-Ю-16
7,6-Ю-10
п
9,1
8,2
11,4
8,2
6,9
4,6
Температура,
°С
678
778
778
778
778
863
Давление
пара,
МПа
0,01
0,03
0,10
—
k
3,6- Ю-15
3,2-Ю-10
3,5 Ю5
1,4-10-13
3,2-10-1°
2,0-Ю-6
п
6,3
4,7
6,0
5,7
4,7
3,8
ВЛИЯНИЕ УСЛОВИЙ ПРОМЫШЛЕННОЙ ЭКСПЛУАТАЦИИ
НА СТАРЕНИЕ АЛЮМОСИЛИКАТНЫХ КАТАЛИЗАТОРОВ
При старении катализаторов уменьшается их удельная
поверхность и изменяются структурные характеристики — пористость,
удельный объем и средний радиус пор. В соответствии с этим
изменяется кажущаяся и насыпная плотность катализатора.
Сравнивая эти показатели у свежего и равновесного катализатора, можно
судить о степени старения последнего. Приведенные в табл. 20
данные о качестве свежего и равновесного катализатора
свидетельствуют о том, что в промышленных условиях снижение актив-
59
Таблица 20
Качество свежего и равновесного катализатора
Показатели
Активность
Удельная поверхность, м2/г
Средний радиус пор, А
Удельный объем пор, см3/г
Пористость, объемн. %
Плотность, кг/м3
истинная
кажущаяся
насыпная
Свежий катализатор
[811
34,6-38,4
386—388
23,5-24,5
0,453—0,473
51,4-52,4
2330
1080—1130
715-725
[48]
36,1—40,4
370—400
24,3—26,4
0,48—0,49
50,0—54,2
2200—2400
НІ090—1100
688—704
Рапновесный катализатор
[81]
29,0-32,0
190—114
42,5—56,0
0,400—0,320
47,8—43,0
2330
1210—1330
760—825
[48]
36,7—24,9
310—90
23,2—35,2
0,15—0,36
25,3—44,7
2100-2300
1270—1400
804—1060
[80]
24,7*—36,9
54-165
92,5—63,0
0,25—0,52
—
—
620—800
• Активность определена по методу Д+П.
ности циркулирующего катализатора в значительной степени
вызвано уменьшением его удельной поверхности. О большой роли
спекания можно судить и на основании работ [80, 81], в которых
приводится характеристика равновесного катализатора.
Из литературных данных следует, что алюмосиликатный
катализатор спекается наиболее интенсивно при переработке тяжелого
сырья. Качество циркулирующего катализатора постоянно
ухудшается. При работе установок каталитического крекинга на сырье
типа атмосферного газойля качество катализатора годами
сохраняется на высоком уровне. Так, отмечается [82], что при
переработке бензина и атмосферного газойля активность свежего
катализатора в течение первого месяца работы уменьшается на 2—3
пункта, а уже на втором месяце работы достигает равновесной
активности 34—35 пунктов. После одного года эксплуатации
активность катализатора на разных установках была от 33
до 35. Таким образом, качество катализатора практически не
меняется.
При работе на вакуумном газойле активность циркулирующего
катализатора обычно устанавливается равной 30—31 и в
некоторых случаях опускается ниже 27 пунктов. При переработке нефтей
равновесная активность катализатора сохраняется на уровне
7—21 пунктов [83]. Низкая активность катализатора обусловлена
не только его спеканием, но и значительным отравлением
металлами.
В заводских условиях качество циркулирующего катализатора
в основном контролируют по его насыпной плотности,
фракционному составу; реже определяют индекс активности. Более
детально циркулирующий катализатор систематически не исследуется.
В связи с этим представляют интерес подробные
исследования равновесного катализатора установок каталитического
крекинга типа 43-102 (табл. 21) [48]. В этой работе равновесный
катализатор, отобранный на выходе из регенератора, визуально
разбирали на «белые», полностью регенерированные частицы;
«серые», содержащие остаточный кокс в центре шариков, и
«черные» частицы, у которых не было заметно регенерированной
периферийной зоны. После дополнительной регенерации «черных»
шариков в лабораторных условиях было получено еще некоторое
количество «белых» и «серых» частиц. Оставшиеся «черные»
частицы не регенерировались даже в жестких условиях.
Как видно из табл. 21, равновесный катализатор представляет
собой смесь частиц разной степени спекания (с разной удельной
поверхностью и активностью). При этом доля низкоактивного
катализатора (активность не более 25) на блоке 3 в этот момент
составляла 63,6%, на блоке 2—10,3% и на первом блоке — 48,0%.
Наличие большого количества сильно дезактивированных частиц
(с удельной поверхностью не более 100 м2/г) свидетельствует об
эксплуатации катализатора в очень жестких условиях. Даже у
наиболее активной части катализатора («белые» частицы), доля
Таблица 21
Катализатор
Из регенератора третьего
блока
исходный
белый
серый
черный
Белый из черного
Серый из черного
Черный из черного
Из регенератора второго
блока
исходный
белый
серый
черный
Из регенератора первого
блока, исходный
Качество катализатора с
Визуальная
сортировка шариков, пес. %
белые
12,7
—
—
30,7
—
—
—
39,7
—
94,0
14,0
14,0
серые
23,7
—
—
17,6
—
—
—
50,0
—
6,0
13,0
38,0
черные
63,6
—
—
51,6
—
—
—
10,3
—
—
73,0
48,0
Насыпная
плотность
кг/мЗ
1006
822
90G
1104
904
1154
1325
877
813
803
1192
1024
установок
каталитического крекинга
Фракционный состой, вес. %
о
Л
1,5
0,4
3,4
5,5
2,0
—
0,1
1,2
0,3
2,1
0,9
—
3,0—4,0 мм
26,2
43,0
64,9
17,6
41,7
22,5
16,2
16,9
31,6
58,9
29,3
—
2,5—3,0 мм
37,3
47,3
29,6
55,3
55,3
61,5
82,0
28,0
61,8
35,8
49,6
—
2,0—2,5 мм
26,4
8,7
2,1
21,7
1,6
16,0
1,7
32,6
5,7
3,2
19,5
—
<2,0 мм
8,6
0,4
—
—
—
—
—
21,3
0,6
—
0,7
—
Индекс
активності-
29,1
36,0
37,5
24,9
34,9
18,7
0,0
32,2
—
34,5
18,5
30,7
||
115
255
290
90
142
25
0
240
187
215
35
—
Механическая
прочность, %
4,0
5,6
5,3
—
4,7
1,9
0,6
3,9
4,8
5,9
2,5
—
Средний р.
диус пор,
27,8
25,8
24,1
26,6
30,9
68,8
—
20,8
36,4
30,6
25,7
—
Плотность,
кг/m'J
кажущаяся
1620
1310
1270
1680
1500
1790
2100
1250
1270
1300
1920
—
истинная
2200
2800
2280
2150
2200
2100
2100
2250
2230
2280
2100
—
Удельный
объем пор
смз/г
0,16
0,33
0,35
0,12
0,255
0,086
0,00
0,35
0,34
0,33
0,045
—
Пористост:
27,2
43,0
44,2
21,8
21,6
14,7
0,0
44,4
47,5
42,9
8,6
—
которой составляла 12—40%, удельная поверхность была
примерно в 1,5 раза меньше, чем у свежего катализатора. Уменьшение
удельной поверхности сопровождается сильным изменением поро-
вой структуры катализатора — удельный объем пор уменьшается,
а их радиус растет. Возрастает также плотность и прочность
частиц катализатора.
Изменение пористой структуры частиц катализатора разного
размера. Нами были проанализированы образцы свежего и
равновесного катализатора с установок каталитического крекинга
различных заводов. Полученные
результаты, наши прежние
материалы [48, 84], а также
литературные данные [81] приведены в
табл. 22 и на рис. 27. Свежий
аморфный алюмосиликатный
шариковый катализатор имеет
примерно одинаковую поверхность,
объем и радиус пор для частиц
разного размера; лишь в одном
случае обнаружено уменьшение
величины удельной поверхности
при переходе от меньших
шариков к большим.
Структура частиц (шариков)
равновесного катализатора
существенно зависит от их
размеров. Чем меньше диаметр
шарика, тек меньше величина его
поверхности и объем пор.
Катализатор с установок,
перерабатывающих легкое сырье, имеет
более развитую поверхность
(кривые 6 и 7 на рис. 27), чем на
установках, перерабатывающих
вакуумные газойли (кривые 9—12).
В свежем катализаторе практически нет шариков диаметром
менее 2 мм. В равновесном катализаторе они могли образоваться
в результате спекания или истирания более крупных частиц. Во
фракциях от 2 до 4 мм содержатся частицы, получившиеся путем
истирания и спекания, а также частицы свежедобавленного
катализатора; фракция больше 4 мм состоит только из частиц,
недавно добавленных в систему.
Из рис. 27 видно, что даже самые «молодые» крупные частицы
уменьшили свою поверхность на 100—200 м2/г. При дальнейшем
уменьшении размеров частиц величина поверхности изменяется
меньше. По-видимому, в промышленных условиях спекание идет
чрезвычайно интенсивно лишь сразу после загрузки свежего
катализатора; затем скорость спекания резко замедляется.
63
10
го зо чо
Диаметр частиц,мм
Рис. 27. Зависимость удельной
поверхности катализатора от диаметра
частиц:
/—5 — свежий катализатор; 6—12 —
равновесный катализатор с промышленных
установок, перерабатывающих легкое
F 7) промежуточное (S) и тяжелое
(9—12) сырье.
Таблица 22
Пористая структура частиц разного размера,
выделенных из образцов свежего и равновесного катализатора
Номер
образца
катализатора
1
2
3
А
о
<.
Диаметр
шариков,
мм
4,0—4,2
3,6—4,0
3,3—3,6
3,0—3 3
2,6—3,0
4,0—5,0
3,0—4,0
2,5—3,0
2,0—2,5
3,0—4,0
2,5—3,0 |
2,0—2,5 1
0,5—2,0
4,0—5,0
3,0-4,0
2,0—3,0
4,0-5,0
3,8—4,0 1
3,0-3,8
2,8-3,0 J
2,3—2,8
1,9—2,3
3,0—4,0
2,5—3,0
2,0—2,5
1,0—2,0
4,0—5 0
3,8—4,0 |
3,2—3,8 /
2,0-3,0
1,0—2 0
Удельная
поврпуногть.
свежий
363
390
435
454
470
400
392
370
395
382
377
—
386
388
386
385
382
377*
—
—
—
—
404
396
410
—
М2/Г
равновесный
183
171
174
165
152
310
105
90
105
220
132 \
78 /
80
190
156
114
254
230 }
208
163 J
140
110
302
248
200
180
290
268 |
251 /
240
200
Удельный объем
пор, смЗ/г
свежий
0,416
0,416
0,423
0,431
0,434
0,500
0,490
0,490
0,490
0,489
0,478
—
0,473
0,476
0,453
0,497
0,489
0,478*
—
—
—
—
0,480
0,515
0,500
—
равновесный
0,320
0,290
0,296
0,281
0,258
0,360
0,160
0,150
0,200
0,297
0,250 1
0,200 /
—
0,400
0 374
0,320
0,405
0,376 )
0,310
0,285 J
0,240
0,200
0,390
0,290
0,264
0,233
0,377
0,320 1
0 302 J
0,305
0,300
Средний радиус
пор, А
свежий
23,0
21,4
19,5
19,0
18,5
25,0
24,5
25,0
26,4
26,3
25,4
—
24,5
24,5
23,5
25,8
26,3
25,4*
—
24,3
26,0
24,5
—
равновесный
35,0
34,0
34,0
34,0
34,0
23,2
30,5
33,3
35,2
27,0
37,8
52,0
—
42,5
48,0
56,0
32,0
32,7
29,8
35,0
34,2
36,0
25,8
23,3
26,4
25,9
26,0
23,8
24,2
25,5
30,0
Литературный
источник
[84]
[48]
[81]
Фракция 2,0—3,0 мм.
Поскольку наиболее высокая скорость спекания наблюдается
на установках, перерабатывающих тяжелое сырье (удельная
поверхность шариков одинакового размера на этих установках в
1,5—2 раза меньше, чем на установках, перерабатывающих легкое
wo
сырье), и при этом потери катализатора, как правило, в 1,5—2
раза больше, чем при переработке атмосферных дистиллятов,
скорость старения катализатора при переработке тяжелого сырья
будет еще выше.
Изменение свойств свежего и циркулирующего катализатора
по радиусу частиц. Исходными образцами для этих исследований
служили фракция 4,0—4,2 мм свежего катализатора одного из
заводов и фракции 4,0—4,2 и 3,8—4,0 мм циркулирующего
катализатора, взятые с трех установок
различных заводов.
Методом истирания из исходных
частиц диаметром 4 мм получены
частицы все уменьшающегося
размера. У свежего катализатора
удельная поверхность по глубине
частиц не меняется (рис. 28).
Циркулирующий катализатор во всех
случаях сохраняет более высокую
поверхность в центре частиц. В
образце с установки,
перерабатывающей легкое сырье, разница между
наружными и внутренними
областями частицы относительно
невелика, для остальных образцов она
значительна. Так, для одного
образца катализатора удельная
поверхность внутренней зоны на 23%
больше, чем по всему шарику. По
правилу аддитивности можло
вычислить удельную поверхность
отдельных слоев шариков. Оказывается, что для указанного
образца внешний слой шарика катализатора толщиной 0,2 мм имеет
удельную поверхность 150 м2/г, т. е. в 1,5 раза меньше, чем
удельная поверхность центральной части.
Ряд исследователей [71, 73] считали маловероятным
неодинаковое изменение катализаторов по глубине частиц. По нашим
данным, частицы в промышленных условиях спекаются неравномерно.
Чем тяжелее перерабатываемое сырье, тем больше эта
неравномерность. Однако это общий вывод, относящийся ко всей навеске,
которая состоит из большого числа частиц. Значительный интерес
представляют исследования отдельных частиц катализатора.
Известные нам методы анализа пористой структуры катализатора не
позволяют исследовать изменение ее по глубине отдельной
частицы. Некоторые качественные результаты нам удалось установить,
наблюдая за регенерацией пластинок, приготовленных из
шариков равновесного катализатора, и сравнивая их оптические
свойства на разных расстояниях от центра [85].
150
2,0 2,5 3,0 3,5
Диаметр частиц.'
Рис. 28. Зависимость удельной
поверхности алюмоснликатного
катализатора от диаметра
частиц:
/ — свежий катализатор; 2, 3, 4 —
равновесный катализатор.
5-2206
65
Пластинки толщиной 0,1—0,2 мм регенерировали в муфеле при
700 °С. Через каждые 5—10 мин регенерации микроскопом
измеряли границы части пластинки, посветлевшей в результате
регенерации. По этим данным строили кривые длительности полного
выгорания кокса с отдельных участков пластинок (рис. 29).
Из рис. 29,а видно, что «серые» пластинки весьма
неоднородны. Одни из них регенерируются довольно быстро (кривая /). По-
светление происходит почти одновременно по всей закоксованной
зоне. На других обнаруживаются зоны с разной скоростью
удаления кокса (кривая 2). Когда разница в скорости регенерации зон
1<tO
іго
100
80
60
,0
го
ft
л
vjy ;
х'
О 0,2 0,k 0,6 0,8 1,0
Расстояние от центра; доли радиуса
а б
Рис. 29. Длительность выгорания кокса с отдельных участков пластинок,
приготовленных из серых (а) и черных (б) шариков.
;, 2, 3, 4— номера образцов.
достигает еще большей величины (кривая 3), в некоторый момент
времени наблюдается закоксованное кольцо. И, наконец, у части
пластинок после первоначального уменьшения диаметра закоксо-
ванного ядра границы его стабилизируются и не изменяются
длительное время A0 ч). По-видимому, этот кокс вообще не может
быть удален с катализатора.
Пластинки, полученные из «черных» частиц, обычно
регенерируются поэтапно. Вначале освобождается от кокса одна из зон
пластинки. Эта зона светлеет одновременно. Затем длительное
время границы закоксованной части практически не меняются, пока
не посветлеет новая зона. Чаще всего первой регенерируется зона,
расположенная в центральной части пластинки, а затем зоны,
примыкающие к предыдущей (кривые /, 2 на рис. 29,6). Внешнее
периферийное кольцо обычно не светлеет даже при очень большой
длительности регенерации. Так выжигаются пластинки наиболее
часто; однако у многих пластинок из черных шариков кокс
удаляется так, как показано на кривой 3. С образованием закоксо-
66
ванных колец некоторые пластинки не светлеют даже частично,
несмотря на длительную регенерацию при температуре до 750 °С.
Таким образом, в пределах одного шарика может быть несколько
участков, которые освобождаются от кокса в разное время.
Возможно, что разная длительность выгорания кокса с
отдельных зон катализатора обусловлена разной их закоксованностью
или неодинаковыми фазико-химическими свойствами. В
дальнейшем было установлено, что концентрация кокса по глубине
частицы действительно может меняться очень сильно, и при некоторых
условиях не плавно, а скачкообразно. Разная концентрация кокса
неизбежно приводит и % разнице в длительности полного
выгорания его при регенерации, Щіастин катализатора. Но этим не
объясняется абсолютная невозможность регенерации некоторых зон
катализатора при жестких условиях даже на тонких пластинках.
Такой кокс можно представить себе лишь заплавленным в
вещество катализатора при полном спекании данного участка
частицы. Это подтверждается невозможностью регенерации основной
массы целых, не имеющих повреждений поверхности черных
шариков даже при 750 °С в среде кислорода в течение 20 ч. В то же
время центральная часть пластинок, изготовленных из таких
шариков, обычно освобождалась от кокса уже при 700 °С в воздухе
за 10—60 мин. Следовательно, черные нерегенерируемые частицы
окружены полностью спекшейся коркой, закрывающей доступ
кислорода во внутрь частиц.
Внутренние зоны катализатора, освобождающиеся от кокса в
разное время, отличаются по оптическим свойствам. На рис. 30 и
31 приведены фотографии пластинок из серых и черных частиц
в проходящем свете в разные моменты регенерации.
На фотографиях, выполненных в обычном свете (см. рис. 31),
можно различить границы некоторых зон катализатора.
Особенно четко они видны у пластинок из черных шариков. Еще лучше
их видно на фотографиях, сделанных в поляризованном свете.
Если на пути поляризованного луча нет кристаллического
вещества, то на фотографиях изменений нет. Если же на пути луча
имеется кристалл, то вследствие двойного лучепреломления на
фотографиях соответствующие участки катализатора выглядят светлыми.
По интенсивности посветления можно.приближенно судить о
концентрации кристаллического вещества. Из рис. 30, 31, видно, что
в белых частицах кристаллическая фаза отсутствует. Более
сильное посветление наблюдается в отдельных зонах
регенерированных серых пластинок. Регенерированная часть черных пластинок
также содержит несколько зон, содержащих кристаллическую
фазу, концентрация которой обычно больше, чем у зон серых
пластинок. От центра к периферии интенсивность посветления зон
обычно увеличивается вплоть до границ остаточного кокса у
серых частиц или до нерегенерируемой спекшейся корки у черных
частиц. Имеются и такие частицы, у которых светлые и темные
области чередуются.
5* 67
Рис. 30. Пластины, приготовленные из серых и белых шариков в разные
моменты регенерации:
1,3 — исходные серые лластннкн; 2, 4 — то же а конце регенерации; 2,а, 4,а — то же а
поляризованном свете; 5. 6, 7 — белые пластинки в поляризованном свете.
Рис. 31. Пластины, приготовленные из черных частиц в разные моменты
регенерации:
1—6 — в обычном свете; З.а. 6,а — в поляризованном свете.
Таблица 23
Характеристика отдельных областей катализатора
Образец катализатора
Черные шарики
после дополнительной регенерации
после размола
после размола и регенерации
черные частицы
серые частицы
Периферийный слой серых шариков
Ядра серых шариков
Посветлевшие после регенерации ядра
Непосветлевшие ядра
Удельная
поверхность,
М2/Г
0,0
5,3
16,5
40,2
54,5
15,6
149,4
36,0
Удельный
объем пор,
смЗ/г
0,0
0,0078
0,013
0,043
0,106
0,028
0,206
0,043
Средний
о
радиус пор, А
—
29,4
16
21,4
38,9
35,0
27,6
23,9
Концентрация кристаллической фазы в частицах катализатора
очень мала, и только высокая чувствительность оптических
методов позволяет ее обнаружить. Рентгеноструктурный анализ
образцов катализатора с промышленных установок показал, что они
рентгеноаморфны. Только у выбранных из равновесного
катализатора черных шариков на рентгенограмме имеются едва заметные
следы дифракционных линий.
Пластинки свежего катализатора в поляризованном свете
выглядят темными. Пластинки из частиц, прокаленных при 800 °С и
выше в течение 1 ч, несколько светлее. Следовательно,
обнаруженные нами зоны отличаются по степени спекания, причиной
которого может быть перегрев отдельных частей катализатора.
Нами предпринята попытка охарактеризовать поровую
структуру отдельных областей катализатора (табл. 23).
У черных частиц, не регенерирующихся в кислороде 20 ч при
750°С, определяли поверхность и поровую характеристику в
целом виде и после размола. Затем крошку дополнительно
регенерировали в муфеле при 750 °С 11 ч. После этого крошку
разбирали на совершенно непосветлевшую часть и частично посветлевшую.
Эти образцы также анализировали. Нулевая поверхность и
удельный объем пор черных частиц в целом виде и существенное
увеличение этих показателей после размола и дополнительной
регенерации подтверждают вывод о наличии спеченной корки и
внутренних менее измененных областей. Внутренние посветлевшие
области имеют поверхность не менее 80 м2/г, так как цифра 40,2 м2/г
получена при анализе крошки, в которой доля посветлевшей части
значительно меньше половины.
У серых частиц нам удалось отдельно проанализировать неза-
коксованную периферийную часть и закоксованное ядро
катализатора. Удельная поверхность периферийной части более чем в 3
раза превышает поверхность ядер. Таким образом, закоксованные
ядра играют незначительную роль в процессе каталитического,
крекинга. В то же время результаты анализа ядер после
регенерации и разборки свидетельствуют о том, что, обладая довольно
большой удельной поверхностью, в два с лишним раза
превышающей удельную поверхность периферийных слоев катализатора, они
могли бы эффективно участвовать в процессе.
Приведенные данные показывают, что в промышленных
условиях катализатор спекается по глубине шарика неравномерно,
поэтому структура по диаметру частицы неоднородна. Эта
неоднородность увеличивается при переходе от белых к серым и от серых
к черным шарикам.
Влияние условий работы реактора и регенератора на старение
катализатора. Для выяснения причин неравномерности изменения
в процессе крекинга пористой структуры частицы катализатора по
ее глубине был проделан ряд работ. В работе [86] показано, что
в условиях каталитического крекинга пылевидный катализатор
стареет быстрее, чем это следовало бы ожидать, учитывая только
температурные условия в аппаратах и действие водяного пара.
Для определения скорости спекания шарикового катализатора
в разных точках реактора и регенератора промышленной
установки были помещены открытые сверху контейнеры с образцами
свежего катализатора (фракция 3—4 мм). В нижней части боковых
стенок контейнеров были просверлены отверстия для прохода
газов.
В реакторе контейнеры находились 65 сут., в регенераторе —
4 месяца и 25 дней. Характеристика образцов катализатора,
находившихся в контейнерах, приводится в табл. 24.
Таблица 24
Характеристика образцов катализатора, находившихся
в контейнерах в реакторе и регенераторе промышленной установки
Образец
Исходный катализатор
Реактор
верхняя часть слоя
зона гирлянд
зона вывода
продуктов реакции
Отпарная зона
Регенератор
газовый короб
слой третьей зоны
слой пятой зоны
о 2
о х
Зля
<и _ ш а
к м о о
х ex f-
raa?s «
? о. J2
йявЧ
?iu а; са
О о шя
0,0
35,9
34,8
35,1
2,5
0,0
0,0
0,0
Насыпная
плотность,
кг/мЗ
регене-
ии
Sa
698
1020
1030
1010
716
720
739
715
ле ре-
ерации
§е
750
765
745
700
—
—
—
Удельная
поверхность,
М2/Г
регене-
ин
за
367
8
4
6
318
337
308
344
ле ре-
ерацни
g?
342
335
350
385
—
—
Удельный
объем пор,
СМЗ/г
регене-
ии
ча
0,48
0,00
0,00
0,00
0,47
0,45
0,40
0,45
ле ре-
ерацни
О X
SS
0,43
0,43
0,44
0,47
—
—
§ *
ДННЙ р'с
после
енераци
Ш О. U
О. О V
U с а
26
25
26
25
29
27
26
27
70
Из данных табл. 24 видно, что катализатор, находившийся в
реакторе, накопил большое количество кокса, который заполнил
практически весь объем пор. Поэтому удельная поверхность закок-
сованного катализатора стала очень мала.
После размола удельная поверхность образцов возросла
незначительно — до 20—25 м2/г, что свидетельствует об относительно
равномерном накоплении кокса по объему частицы. После
длительного пребывания катализатора в реакторе и регенераторе
качество образцов изменилось сравнительно мало. Их удельная
поверхность уменьшилась лишь на 20—60 м2/г, несколько уменьшился
объем пор и возросла насыпная плотность. Радиус пор изменился
мало, в основном у катализатора, находившегося в отпарной зоне
реактора.
В течение почти 5 месяцев, когда на установке проводились
опыты с контейнерами, поверхность циркулирующего катализатора
снизилась со 150 до 70 м2/г несмотря на добавление за это время
свежего катализатора примерно в 1,5 раза больше, чем
циркулировало в системе. Поверхность регенерированных образцов
катализатора, находившихся в контейнерах, во всех случаях была
более 300 м2/г. Следовательно, влиянием температуры и среды
нельзя объяснить причину существенного уменьшения удельной
поверхности циркулирующего катализатора.
Влияние закоксованности на старение катализатора. Было
исследовано влияние закоксованности катализатора на скорость его
спекания при нагреве до высоких температур в водяном паре,
азоте и воздухе. В опытах использовали свежий и закоксованный
катализатор (табл. 25).
Из табл. 25 видно, что при нагреве катализатора в водяном
паре и азоте кокс препятствует уменьшению удельной
поверхности. Это согласуется с данными работы [87], где отмечается, что
после прокалки в сероводороде 2 ч при 530 °С активность закоксо-
ванного катализатора уменьшается в меньшей степени, чем
регенерированного. Очевидно, что кокс служит барьером, затрудняющим
слияние глобул катализатора. Кроме того, он в некоторой степени
защищает поверхность катализатора от действия водяного пара.
При продувке воздуха через слой катализатора закоксованные
шарики спекаются сильнее, чем не содержащие кокса. При 700°С
(фактическая температура в регенераторе промышленных
установок) поверхность закоксованных частиц существенно уменьшается.
Однако это наблюдается лишь при достаточно большой
концентрации кокса на частице. Аналогичные опыты с катализатором, за-
коксованным до 3 и 6 вес. и/о, показали, что удельная поверхность
частиц не изменяется вплоть до 750 °С, и лишь при 800°С
наблюдается разница соответственно 15 и 31 м2/г. Увеличение скорости
спекания катализатора при выжиге в процессе регенерации
коксовых отложений обусловлено, по-видимому, значительным
разогревом его частиц. Ранее считали, что контактная масса существенно
не разогревается. Однако расчеты показали [89], что пылевидный
71
Таблица 25
Влияние закоксованности катализатора на скорость его спекания
Содержание
кокса,
вес. %
0,0
11,3
11,3
11,3
11,3
11,3
11,3
со"
3,1
3,1
6,0
6,0
12,4
12,4
12,4
темпера-
°С
750
750
750
820
820
850
700
750
800
750
800
700
750
800
Условия прокалки
среда
длительность,
ч
До прокалки
Водяной
пар
Азот
Воздух
2,0
4,0
6,0
0,5
4,0
6,0
До полной
регенерации
закоксованного
катализатора
То же
*
Удельная
поверхность
катализатора после прокалки,
незакоксоваи-
ного
380
240
218
207
229
140
330
380
380
375
380
374
380
373
373
закоксованного
—
265
250
230
266
246
360
380
380
360
378
343
355
342
250
Разница
в
поверхности,
м2/г
—
-25
—32
—23
—37
— 106
—30
0
0
+ 15
+2
+31
+25
+31
+ 125
катализатор при протекании процесса во внутридиффузионной
области может разогреться до 200—250 °С за 2 мс [88]. В работе
[89] установлено, что при регенерации алюмосиликатный
катализатор разогревается; при увеличении отложения кокса, повышении
содержания кислорода и температуры кислородсодержащего газа
разогрев усиливается. При 700 °С максимальный разогрев
катализатора 175 °С. Такое повышение температуры весьма существенно
и, по-видимому, является одной из основных причин спекания
катализатора при регенерации. О перегреве слоя катализатора при
регенерации в определенных режимах указывается также в
работе [90].
Необходимо учитывать неравномерность горения кокса во
времени. Установлено [91], что в первый момент регенерации
температура алюмосиликатного катализатора резко возрастает
вследствие быстрого окисления находящихся на поверхности кокса
активных соединений, богатых водородом. С возрастанием мольного
отношения водород: углерод и температуры наблюдается подскок
температуры, максимальная величина которого достигает 75 °С.
В дымовых газах в первые минуты выжигания кокса содержится
72
более 0,1% СО, 0,08% СО2 и водяные пары; в дальнейшем
содержание СО2 стабилизируется на уровне 0,08%, концентрация СО
падает до 0,002—0,004%; количество водяных паров резко
снижается.
О 0,2 0,4 Ofi 0,8 1,0
Радиус , доли
Рис. 32. Зависимость распределения
кокса по радиусу шарика от
температуры крекинга (а), качества сырья
(б) и содержания кокса (в).
О 0,2 0,4 0,6 0,8 1,0
j Радиус, доли
d
O,Z 0,k 0,6 0,0 1,0
Радиус, доли
После предварительной продувки закоксованного катализатора
азотом при 620 вместо 443 °С скорость окисления резко
уменьшается и температурный скачок почти полностью исчезает. Вероятно,
предварительная высокотемпературная обработка вызывает
разрушение активных поверхностных соединений.
72
Перегревы отдельных зон гранулы катализатора весьма сильно
зависят от характера распределения кокса по объему частицы.
Поэтому нами было изучено распределение кокса по диаметру частиц
методом фотометрирования тонких шлифов закоксованного
катализатора. При всех условиях крекинга периферия.шарика закоксо-
вывается сильнее, чем центр (рис. 32). Эта неравномерность
увеличивается при повышении температуры крекинга, утяжелении
сырья и уменьшении закоксованности катализатора. При
небольшом содержании кокса в катализаторе периферия в стадии
крекинга может закоксовываться в 3—5 раз сильнее, чем ядро. При
увеличении содержания кокса эта
разница быстро уменьшается и уже при
концентрации кокса 1,5% отношение
закоксованности периферии к средней
закоксованности становится
равным 1,3.
Таким образом, неравномерность
отложения кокса для сильно закоксо-
ванных частиц катализатора
относительно невелика. Картина
существенно изменяется, если катализатор
многократно участвует в стадиях
крекинга и регенерации и если при этом
новые порции кокса отлагаются в
объеме частиц, где имеются остатки
ранее отложившегося кокса.
Для установления
закономерностей накопления остаточного кокса в
центре частиц, определения его
максимально возможной концентрации рассмотрим кинетические
основы процесса выгорания кокса из пористого объема частиц.
Допустим, что кокс выгорает из объема отдельной цилиндрической
поры радиуса R (рис. 33). Принимаем, что вначале внутренняя
поверхность поры закоксована равномерно по всей длине. Через
время т после начала регенерации часть поры до глубины Х\
полностью освободилась от кокса. На участке Х\—х2, где происходит
горение, в результате окисления расходуется кислород; в точке
хг концентрация его становится равной нулю.
Кислород, подводимый к зоне горения, расходуется на реакцию
горения. Долей кислорода, расходуемого на заполнение
выжженных участков, пренебрегаем.
Поток кислорода внутрь поры:
Рис. 33. Схема сгорания кокса
в отдельной поре катализатора.
G =
(її)
где D — коэффициент диффузии кислорода; S(nR2) —площадь
поперечного сечения поры; Со — концентрация кислорода.
74
Поскольку реакция окисления кокса имеет первый порядок по
кислороду и по коксу [92, 93], количество кислорода, расходуемое
на реакцию в единицу времени на участке от Х\ до х2, будет равно:
G = S\KC0Cc-dx A2)
где Сс — концентрация кокса; К — константа скорости горения
кокса.
Принимаем, что концентрации в зоне горения кислорода и кокса
по длине поры меняются линейно (см. рис. 33).
Тогда:
--Р- A3)
dx
где Со—концентрация кислорода в устье поры; С&—начальная
концентрация кокса.
Подставив эти значения в A1) и A2) и приравнивая их,
получим:
со
Ґ о /' * \ о _?i
-s= j *.<Ці- — усс —
J
іто после интегрирования принимает следующий вид:
х ~Х-х -і/— A6)
хт-х2 х,-у ксо AЬ)
Как видно из этого уравнения, ширина зоны горения кокса хт
зависит от скорости подвода кислорода (D) и его расхода (KCq ),
т. е. от условий процесса регенерации.
Коэффициент диффузии (D) в данном случае является
коэффициентом кнудсеновской диффузии. Для условий регенерации он
может быть определен по уравнению:
D=162rcp-/7r
Константу скорости окисления кокса можно определить из
соотношения:
Т —726
lg ЛГ = 0,305+ 10,56 f
75
Таблица 26
Ширина зоны горения (в мм) при регенерации
закоксованного алюмосиликатного катализатора
Содержание
кокса на
катализаторе,
вес. %
1
3
10
30
Температура регенерации, °С
500
2,15 B,0*)
1,24
0,68
0,39
600
0,60 @,96*)
0,34
0,19
0,11
650
0,36
0,21
0,11
0,07
700
0,22 @,42*)
0,12
0,07
0,04
750
0,14
0,08
0,04
0,03
* Экспериментальные данные.
Рассчитанные нами по указанной методике значения ширины
зоны горения хт для различных условий регенерации приведены в
табл. 26. Теоретические и экспериментальные значения ширины
зоны горения оказались близки между собой.
Из данных табл. 26 следует, что ширина зоны горения при
низких температурах регенерации и небольшой степени закоксован-
ности катализатора больше, чем радиус самого шарика, равный
1,5—2 мм. Это значит, что при невысоких температурах кокс
выгорает одновременно по всему объему частицы катализатора.
В этом случае характер распределения невыгоревших остатков
кокса будет такой же, как и в начале регенерации. При высоких
температурах и большой закоксованности катализатора ширина
зоны горения в десятки и сотни раз меньше радиуса частицы (см.
табл. 26). Следовательно, в этих условиях кокс будет выгорать
послойно и остаточный кокс сконцентрируется в центре гранул.
Рассмотрим механизм накопления остаточного кокса при разных
режимах регенерации частицы катализатора, многократно
участвовавшей в процессе крекинга и подвергавшейся неполной
регенерации.
При первом цикле крекинга на частице катализатора
отложится некоторое количество кокса, которое можно определить по
уравнению Панченкова [94, 95]:
d = Ахк -f В A — e~DTk) A7)
где Сі — содержание кокса на катализаторе, вес. %;
ik—длительность цикла крекинга; А, В, D — константы.
В зависимости от условий регенерации кокс может
выжигаться в кинетической, диффузионной и переходной областях.
Рассмотрим все эти три случая.
1. В кинетической области из-за постоянства концентрации
кислорода по всему объему частицы кокс выжигается со всей
частицы с одинаковой скоростью. Поэтому даже после неполной ре-
7в
генерации кокс распределен по частице равномерно. Уравнение
регенерации в этой области имеет вид [93]:
р-то) A8)
где р — степень регенерации катализатора; Со — концентрация
кислорода; К — константа скорости окисления кокса; тр —
длительность регенерации; то — длительность накопления кислородного
комплекса; ?— коэффициент пропорциональности расходования
кислорода.
Зная длительность регенерации, найдем по этой формуле долю
кокса, оставшегося на катализаторе после первого цикла
регенерации:
СосТ! = С1 A — р)
Второму циклу крекинга подвергается катализатор, уже
содержащий кокс. Поэтому учтем время т|, соответствующее
исходной для второго цикла крекинга концентрации С0СТ1 . Тогда:
С2 = А (ті + xk) + В [ 1 - Г° « + Щ A9)
Значение т[ определяется при подстановке С0СТі в левую часть
формулы A7). Концентрация остаточного кокса после второго
цикла регенерации будет:
0>ст2 = С2 A —р)
Продолжая расчет аналогичным образом, можно найти
кривую накопления остаточного кокса в зависимости от числа циклов
крекинга и регенерации.
Чем больше концентрация остаточного кокса, тем меньше
кокса откладывается при крекинге. С другой стороны, в соответствии
с уравнением A8), степень регенерации не зависит от
первоначального содержания кокса. Чем больше закоксован катализатор,
тем больше кокса удаляется за одно и то же время регенерации.
При некотором содержании остаточного кокса должно
установиться равенство между приростом кокса при крекинге и выжигом
его при регенерации. При установившемся состоянии:
,, ?] Соет А* + В (і-є'1*')
('—Р) — с f—D(x' + тЛІ ^¦аі>
L A(T'+xk) + B[\-e °( + k>\
Это уравнение позволяет определить величину х', концентрацию
кокса после цикла крекинга С и концентрацию остаточного кокса
Сост, при установившемся состоянии.
2. Во внутридиффузионной области скорость выжига кокса
определяется диффузией кислорода в порах к зоне горения.
Поэтому кокс удаляется послойно, по мере передвижения узкой зоны
горения от периферии к центру частицы. Кинетическое уравнение ре-
77
генерации катализатора зо внутридиффузионной области при
послойном удалении кокса имеет вид [134]:
3~2p~3A~p) ] B1)
где R — радиус частицы катализатора; D3 — эффективный
коэффициент диффузии кислорода; а — коэффициент.
По этому уравнению найдем величину A—р) и определим
концентрацию остаточного кокса после первого цикла регенерации.
Остаточный кокс в этом случае сосредоточен з невыгоревшей
центральной части частицы катализатора, где его концентрация в
расчете на эту зону равна С\.
При втором цикле крекинга концентрация кокса на
регенерированной части катализатора станет равной С\, а на нерегенериро-
занной центральной части:
В случае рассмотрения второго цикла регенерации необходимо
учитывать, что кинетическое уравнение B1) справедливо при
разномерном распределении кокса по частице. Поскольку после
второго цикла крекинга концентрация кокса в центральной и
периферийной частях различна, при расчете регенерации следует
исходить из концентрации кокса С\, пока горение не достигнет
центральной части с концентрацией кокса С2. Величина р при этом
означает долю регенерированного объема частицы катализатора,
а не степени регенерации. Во зтором цикле регенерации доля
регенерированного объема оказывается равной величине р,
полученной после первой регенерации, так как при неизменной
длительности регенерации и закоксованности периферийной зоны фронт
горения успевает продзинуться на ту же величину. При этом сгорает
то же количество кокса, что и при первом цикле. Концентрация
остаточного кокса после второго цикла регенерации будет:
Сост, = Сц, A - Р) = [2Ark + В (\ - є''0**)} A - р)
а после п циклов крекинга и регенерации:
Сост = [nAxk + в (і - e~nDXk)} (I - р) B2)
Это уравнение позволяет находить концентрацию остаточного
кокса в зависимости от числа последовательных циклов закоксо-
вывания и регенерации. Из него следует, что концентрация
остаточного кокса может быть как угодно велика. Фактически после
заполнения всего свободного объема пор (достижения предельной
концентрации кокса СЩ)) в центральной части, т. е. в нерегенери-
руемой зоне частицы катализатора, дальнейшее коксоотложение
78
/ цикл
2 цикл
з цикл
Установив-^
шийся
резким
прекращается. После этого концентрация остаточного кокса
остается постоянной для данного режима:
Сост = СпрA -р) B3)
3. Переходный режим регенерации наблюдается, когда скорость
окисления кокса и диффузия кислорда в порах соизмеримы.
Скорость регенерации определяется как кинетическими, так и
диффузионными факторами. При этом условно можно считать, что на
некоторой доле объема частицы а накапливается остаточный кокс
по кинетическому механизму, а на доле объема A—а)—по внут-
ридиффузионному механизму. Концентрация остаточного кокса
для этих областей
определяется описанным
выше способом. Степень
регенерации определяется
по уравнениям для
переходного режима
регенерации [93]. Приведем
основные характерные
особенности рассмотренных
механизмов накопления
ОСТаТОЧНОГО КОКСа (рИС.
34). При кинетическом
механизме остаточный
кокс равномерно
распреб
р рр
делен по объему частицы
катализатора. По мере
накопления остаточного
кокса количество
удаляемого за цикл
регенерации кокса
увеличивается, а количество кокса,
образующегося за цикл
крекинга, уменьшается. Концентрация остаточного кокса
стабилизируется тогда, когда эти величины становятся одинаковыми.
При внутридиффузионном механизме остаточный кокс
накапливается только в центре частицы катализатора. Количество
образующегося кокса от цикла к циклу уменьшается, а количество
выгорающего кокса остается неизменным. Постоянная концентрация
остаточного кокса устанавливается тогда, когда в центре частицы
будет достигнута предельная концентрация кокса.
При переходном механизме остаточный кокс накапливается в
центре по законам внутридиффузионного механизма, а в некоторой
доле объема частицы реализуется кинетический механизм.
Количество образующегося кокса от цикла к циклу уменьшается, а
удаляемого при регенерации — возрастает, но в меньшей степени,
чем это следует из уравнения A8). Характер изменения
концентрации кокса и распределения его по частице от цикла к циклу для
Рис. 34. Схема накопления остаточного кокса
при регенерации катализатора в кинетической
(а), внутридиффузионной (б) и переходной
(в) областях:
1 — кокс, сгорающий при регенерации; 2 — остаточный
кокс.
79
трех механизмов накопления остаточного кокса схематически
показан на рис. 34.
Нами проведены лабораторные эксперименты по накоплению
остаточного кокса при различных режимах регенерации
катализатора при крекинге цетана.
На рис. 35 показана кинетика накопления остаточного кокса
при неполной регенерации
катализатора в различных
областях протекания процесса в
зависимости от числа циклов.
Там же приведены расчетные
кривые по уравнению Пан-
ченкова, которое для
использованного в опытах сырья и
катализатора при
температуре крекинга 500 °С можно
записать в виде:
С = 0,01455т + 4,52 ( 1 -е°-оШх)
Точками на рис. 35
обозначены экспериментальные
данные.
При низкой температуре
D50СС) регенерация
проходила в кинетической области
(рис. 35, а). Это
подтверждается тем, что степень
регенерации не зависит от
содержания кокса и равна 0,45 для
каждого цикла регенерации.
Экспериментальные данные
накопления остаточного кокса
несколько отличаются от
расчетных. Это происходит
потому, что после неполной
регенерации скорость коксообразова-
ния оказывается больше, чем
следует из кинетического
уравнения, вследствие разного
качества кокса, оставшегося на
катализаторе после
регенерации и свежеобразованного.
Регенерация при 700 °С осуществлялась во внутридиффузионной
области. На рис. 35, б расчетные и экспериментальные данные
близки. В соответствии с теоретическими представлениями
количество выгорающего кокса постоянно. Из рис. 35 видно, что во
внутридиффузионной области регенерации накапливается остаточный
кокс.
ю
Циклы
Рис. 35. Кинетика накопления
остаточного кокса при неполной регенерации
катализатора в кинетической (а), виутри-
диффузионной (б) и переходной (а)
областях:
/ — остаточный кокс; 2 — кокс, выгорающий
за цикл; 3— кокс, образующийся за цикл.
80
При оценке предельной концентрации остаточного кокса в
равновесном катализаторе обнаружили, что в центре частиц она
равна 25,6%. Эта концентрация близка к предельной, хотя и
несколько занижена, так как остаточный кокс у части шариков оказался
заплавленным.
Для установления возможности локальных перегревов внутри
частицы катализатора нами рассмотрено изменение теплонапря-
женности зоны горения в процессе регенерации. При этом
учитывали неравномерное распределение кокса по частице [97]. При
высоких температурах тепло сгорания кокса воспринимается очень
тонким сферическим слоем частиц, поскольку само сгорание
происходит в довольно узкой зоне. Поэтому теплонапряженность,
рассчитанная как отношение количества тепла, выделяющегося в
единицу времени, к объему зоны горения, может достигать
больших значений. Ее можно определить из выражения:
DKCI
где q — теплота сгорания кокса, отнесенная к 1 г кислорода.
Как видно из уравнения, теплонапряженность зоны горения хт
возрастает при увеличении концентрации кислорода в воздушном
дутье, температуры регенерации и начальной закоксованности
катализатора.
Расчетным путем было определено изменение величины тепло-
напряженности по мере продвижения зоны горения кокса в глубь
частицы и показано, что наиболее теплонапряженным участком
является периферия частиц и зона остаточного кокса. Это было
подтверждено прямыми экспериментами. Закоксованные частички
катализатора регенерировали при 600 X для удаления кокса с
поверхности частицы толщиной 0,5—1,0 мм. Следующий слой кокса
толщиной 0,2—0,5 мм выжигали при 700, 750 и 800 °С, а
оставшийся кокс — снова при 600 °С. Было установлено, что увеличение
концентрации кокса и повышение температуры регенерации
промежуточной зоны шарика способствуют ее селективному спеканию.
Зона спекания всегда совпадала с границами кокса, выгоревшего
при повышенной температуре.
Возможность спекания отдельных участков частицы
катализатора была показана также следующим опытом. Содержащий 12%
кокса катализатор регенерировали при 800 °С до тех пор, пока
регенерированная зона не достигла глубины примерно '/з радиуса
шариков. Затем катализатор охладили, выделили закоксованные
ядра методом мгновенного замораживания и удалили с них кокс
регенерацией при 600 °С. Удельная поверхность ядер оказалась
равной 357 м2/г, а периферийных слоев — 240 м2/г. Большая
поверхность ядер в данном случае получалась не только по причине
меньшего перегрева во время регенерации, но и вследствие
защитного действия кокса.
6—2206 81
Изменение равновесной активности катализатора в зависимости
от характера его разрушения. Активность циркулирующего
катализатора очевидно зависит от условий его разрушения и обновления.
Наиболее благоприятный случай, когда разрушается и выводится
из системы промышленной установки дезактивированная часть
катализатора, наименее благоприятный — когда теряются главным
образом активные частицы.
При изучении причин разрушения катализатора на
промышленных установках обычно наибольшее внимание уделяют узлу, в
котором частицы испытывают максимальные динамические
нагрузки,— системе пневмотранспорта. Однако даже при
нормальной работе транспорта расход катализатора может колебаться в
больших пределах. Так, при переработке тяжелого сырья он
обычно в 1,5—3 раза больше, чем в случае крекинга атмосферного
газойля. Очевидно причиной является снижение прочности частиц
под влиянием факторов технологического процесса. Прочность
шаровидных глобул катализатора определяется числом единичных
контактов этих глобул, приходящимся на единицу площади
сечения частицы катализатора, а также прочностью единичного
контакта [98]. Этим объясняется известный факт снижения прочности
алюмосиликатного катализатора при его увлажнении [99]. В
результате адсорбции воды уменьшается свободная поверхностная
энергия, в связи с чем на образование новой поверхности при
разрушении катализатора требуется затратить меньшую работу.
Особенно сильно уменьшается поверхностная энергия при
образовании монослоя адсорбированного вещества. Поэтому первые
порции воды наиболее сильно снижают прочность.
Увеличение содержания кокса приводит к увеличению
прочности частиц. Так, прочность образцов свежего катализатора,
находившихся 2 месяца в реакторе промышленной установки в
контейнерах, оказалась в 2,2—2,4 раза больше, чем у исходного неза-
коксоваиного катализатора. Остаточный кокс, находящийся в
центральной зоне частиц циркулирующего катализатора, также
способствует увеличению их прочности [100] в связи с тем, что он
увеличивает площадь контакта глобул катализатора.
Противоположное влияние отложенного кокса наблюдается при
его выжиге. При высокой температуре регенерации частицы
катализатора растрескиваются даже при отсутствии внешней нагрузки.
Так, в процессе выжига в муфельной печи при 700 °С кокса
образцов, находившихся в контейнерах в реакторе промышленной
установки, растрескивалось 40—70% шариков катализатора. Образец
свежего катализатора после семи циклов крекинга цетана и
неполной регенерации при 700°С содержал 85% треснувших
шариков. Другой образец из этой серии опытов (крекинг цетана при
500°С, регенерация при 600°С) содержал около 15% треснувших
частиц.
Растрескивание частиц катализатора может быть вызвано
попаданием на них жидкой части сырья, имеющего более низкую
82
температуру. При достаточно больших количествах жидкости
частица мгновенно охлаждается и пропитывается жидкостью. При
ее крекинге образуются пары более легких углеводородов и внутри
частицы может развиться давление, способное разорвать шарик.
Этому способствует нагрев шарика после охлаждения при его
контакте с основной массой катализатора, на которую жидкая часть
попала в меньшей степени или вообще не попадала.
Раскалывание частицы в результате внутреннего давления,
вызванного образованием большого количества паров, нами
наблюдалось на образце свежего катализатора, содержащего около 5—
7% влаги, в зоне с температурой 700°С. При этом разрушалось
до 70% катализатора. После предварительного высушивания
катализатора при 150 °С и выше растрескивание полностью
устранялось. Видимо, при переработке тяжелого сырья существенную
роль играют обе причины растрескивания: температура
регенерации и контакт частиц с жидкой фазой.
С целью определения узлов установки, вызывающих
повышенное разрушение катализатора, а также для установления
некоторых закономерностей расходования катализатора нами был
проведен специальный анализ работы одной из установок. Были
определены следующие потери катализатора: 1) выводимого из
пылевых карманов сепараторов Р-4 и Р-4А; 2) уносимого
транспортирующим агентом через трубу сепараторов Р-4 и Р-4А; 3) уносимого
дымовыми газами через трубу регенератора. Потери катализатора
при уносе его парами нефтепродуктов в колонну относительно
невелики. При хорошем состоянии установки потери катализатора
через неплотности аппаратуры также ничтожны и могут не
учитываться.
Количество катализатора, теряемого на каждом потоке,
определяли следующим образом. В период, когда установка работала
на устойчивом режиме, потоки катализатора из пылевых карманов
направляли в соответствующие мерные бачки. Затем определяли
концентрацию катализаторной пыли в газах регенерации и
транспортирующем агенте. Для этого с помощью пылесоса 10—20 мин
через фильтр пропускали газ, уходящий из труб сепараторов и
дымовой трубы. Зная объем пропущенного газа, его температуру
и количество уловленного катализатора, легко найти
концентрацию катализатора в газе.
Данные о потере катализатора, полученные этим методом и
вычисленные по изменению уровней в аппаратах в начале и конце
суток с учетом фактически догруженного на установку
катализатора, хорошо сходились. Частицы катализатора, уходящего с
установки, имеют два преимущественных размера: 0,10 и 0,35 мм
(рис. 36). Это свидетельствует о разрушении катализатора в
промышленных условиях, протекающем по двум механизмам. По
одному из них в результате истирания катализатора о стенки
аппаратов, катализаторопроводоз и при взаимном трении частиц друг о
друга образуется тонкая пыль. При этом сферическая форма шари-
6» 83
ков не изменяется; происходит лишь постепенное уменьшение их
размеров. По другому механизму, в результате разрушения (при
динамических ударах, растрескивании в процессе регенерации и др.)
получаются относительно крупные частицы. Кроме того, такие
частицы могут образовываться и в результате откалывания
поверхностных слоев гранулы при многократных ударах.
По нашим данным, доля потерь за счет истирания составляет
25—35% от общего их количества. Следовательно, преобладающая
часть расхода катализатора обусловлена полным разрушением
частиц. Кроме того, около 60% потерь приходится на ветвь
регенератора.
Одинаковые по размерам
фракции теряющегося
катализатора оказываются близкими по
величине поверхности.
Катализатор отбирали из труб
сепараторов Р-4 и Р-4А, а также из их
пылевых карманов и дымовой
трубы регенератора.
Наименьшую поверхность имеют частицы
размером менее 0,05 мм.
Увеличение их до 0,4—0,6 мм
приводит к возрастанию удельной
поверхности со 100—120 до 260—
280 м2д, а дальнейший рост
частиц вызывает некоторое ее
уменьшение. Средняя удельная
поверхность равновесного ката-
«о
э
5
1:
оЗерм
5
13
гг
9
7
5
_
-
-
Г
Г
II
У! \х
гуАг \\
\
|
0,06 O.1J O,-:, 0,2й 0,5[
Средний huc'iemp чгмілиц с/^
Рис. 36. Дифференциальные кривые
гранулометрического состава
катализатора, теряемого из реактора (/) и
регенератора B).
лизатора первого пробега была
110 м2/г, а поверхность второго
пробега—126 м2/г.
Относительно низкая поверхность мелких фракций теряющегося
катализатора свидетельствует о том, что пыль образуется
вследствие истирания всего циркулирующего катализатора. Более
крупные частицы теряющегося катализатора свидетельствуют об
образовании их за счет полного разрушения отдельных шариков. По
этому механизму разрушаются главным образом частицы свежего
догружаемого катализатора. Меньшая величина поверхности
наиболее крупных частиц теряющегося катализатора объясняется тем,
что в них имеется некоторое количество мелких шариков,
образовавшихся за счет спекания активных шариков. В среднем удельная
поверхность крошки в 2—2,2 раза больше, чем у циркулирующего
катализатора. Удельная поверхность всего теряемого катализатора
также больше, чем у циркулирующего катализатора, примерно в
1,75 раза. Следовательно, низкая активность равновесного
катализатора в значительной степени обусловлена также тем, что в
результате преимущественного разрушения активных частиц в нем
накапливаются прочные, но неактивные шарики.
84
Крупные частицы крошки и циркулирующий катализатор
содержат примерно одинаковое количество металлов. С
уменьшением размеров частиц содержание металлов в пыли резко
возрастает (рис. 37). Это обусловлено концентрированием металлов в
промышленных условиях у внешней поверхности шариков
катализатора [101, 102]. В более крупных частицах крошки,
образовавшейся за счет полного разрушения шариков, содержится металлов
столько же, сколько в равновесном катализаторе. Эти данные
согласуются с представлениями о двух механизмах разрушения
катализатора. Они также показывают, что низкое содержание
металлов в циркулирующем катализаторе установок типа 43-102, а,
следовательно, слабое его отравление обусловлено интенсивным
выносом металлов с
поверхности
катализатора в результате его
истирания.
На установках
каталитического крекинга 1-А
пылевидный катализатор
дезактивируется несколько
иначе [103]. Установку
догружают пылевидным
катализатором, составленным из
смеси свежего шарикового
катализатора, свежей ката-
лизаторной крошки и
отработанного катализатора с
установок крекинга,
использующих шариковый
катализатор. Несмотря на большое
количество отработанного
катализатора в смеси, пылевидный катализатор имеет довольно
развитую удельную поверхность — 300—400 м2/г и сравнительно
большой удельный объем пор — 0,44 см3/г. Содержание железа —
0,2 вес. %, ванадия — 0,005 вес. %, никеля — 0,003 вес. % на
катализатор.
Относительно большая активность и селективность
пылевидного катализатора объясняется тем, что по качеству указанная выше
смесь значительно лучше, чем равновесный катализатор,
циркулирующий в системе, и приближается к свежему
катализатору.
В процессе эксплуатации катализатор сильно измельчается
(табл. 27). В загружаемом и равновесном катализаторе
концентрация частиц размером 0,025—0,063 мм одинакова, что
свидетельствует об одинаковой скорости образования и дальнейшего
измельчения этой фракции катализатора. Доля частиц размером
<0,025 мм с 14 до 37% возрастает за счет уменьшения
содержания частиц размером >0,063 мм.
О 0,2 ОЛ О.6 0,8 1,0
Размер частий, , мм
Рис. 37. Влияние фракционного состава
пыли и крошки на содержание в них
металлов:
.' — железо; 2 — ванадий; 3 — никель; 4 — кальций.
85,
Таблица 27
Гранулометрический состав катализатора
в различных потоках установки 1-А
Катализатор
Догруженный в систему:
на УНПЗ
на Бакинском НПЗ
Равновесный циркулирующий
в системе*
Выделенный из шлама*
После циклона регенератора*
После электрофильтров*
Содержание
я
S
о
о
V
14
—
37
96
91,4
97,7 .
5,04 мм
Л
о
о
9
35**
9,6
4
8,5
2,3
фракций в
я
я
3
о
о"
1
о
о
16
20
13,9
—
—
—
іотоке, вес.
я
я
о
S
о
о
27
30
21,2
—
—
—
%
S
я
о
Л
34
16
18,3
—
—
—
Все данные относятся к установке 1-А УНПЗ. *" До 0,04 мм.
Катализатор, прошедший электрофильтры, состоит практически
только из самых мелких частиц. Следовательно, циклоны
регенератора и электрофильтры улавливают почти полностью частицы
размером более 0,025 мм, а
также значительное
количество более мелких частиц.
Гранулометрический анализ
катализатора, выделенного
из шлама, показал, что в
ректификационную колонну
попадает из-за неполного
осаждения в циклонах
реактора большое количество
частиц размером <0,025мм.
Исследования
показали, что физико-химические
ч
|зо
1"
-§0,4
0,025 0,05 0,075 0,10 0,125
Размер частиц,мм
Рис. 38. Зависимость удельной поверхности
катализатора (/, 2), удельного объема пор
C) и среднего размера пор D) от его
гранулометрического состава.
свойства одной и той же
фракции не зависят от
места ее отбора. С
уменьшением размера частиц удельная поверхность катализатора и объем
его пор уменьшаются по линейному закону, а средний радиус пор
увеличивается (рис. 38). Анализ пробы циркулирующего
катализатора показал, что его удельная поверхность равна 169 м2/г.
С уменьшением размера частиц катализатора содержание в
нем железа, никеля и ванадия возрастает, что видно из
следующих данных (в вес. %):
86
Размер частиц
катализатора,
мм
<0,025
0,025—0,04
0,04—0,063
0,063—0,1
0,1—0,125
Со
<0,025
0,025—0,04
0,04—0,063
Железо
Цирку
0,71
0,70
0,47
0,46
0,34
стояка з
0,85
0,77
0,46
Никель
л и р у ю щ и й
0,02
0,085
0,03
0,03
0,03
1 Л Є К Т р О ф И Л
0,023
0,014
0,01
Ванадий
0,05
0,02
0,012
0,009
0,0085
ь т р а
0,051
0,035
0,025
Выделенный и з шлама
<0,025 1,2 0,027 0,056
Большее содержание металла в катализаторе, взятом со стояка
электрофильтра и выделенного из шлама, чем в циркулирующем,
можно объяснить увеличенным содержанием з нем самых мелких
старых частиц.
Неоднородность по гранулометрическому составу оказывает
существенное влияние на активность циркулирующего
катализатора. Удаление в виде потерь с установки 1-А наиболее спекшихся
н отравленных металлом частиц катализатора способствует
эффективному обновлению катализатора и позволяет поддерживать его
высокую равновесную активность.
Катализатор
Удельная поверхность, м
Удельный объем пор, см
Средний радиус пор, А
Содержание, вес, %
железо
никель
ванадий
2/г .
3/г .
догружаемый
380
. 0,44
24
0,20
. 0,003
. 0,005
циркулирующий
170
0,32
38
0,55
0,010
0,028
выносимый
130
0,30
46
0,90
0,025
0,050
ПУТИ УЛУЧШЕНИЯ СВОЙСТВ ЦИРКУЛИРУЮЩЕГО
КАТАЛИЗАТОРА
Некоторые причины старения алюмосиликатного катализатора.
Из изложенного выше можно заключить, что основная причина
снижения активности катализатора в промышленных установках —
перегрев частиц катализатора при регенерации. Однако такое
объяснение может быть принято лишь при концентрации кокса на
активных частицах достаточно высокой для того, чтобы вызвать
спекание и разрушение катализатора при обычной температуре
регенерации.
Концентрацию кокса, который может накопиться на частицах
добавляемого свежего катализатора, мы попытались установить
на основании зависимости количества образующегося при
крекинге кокса от активности катализатора. Частицы свежего катализа-
87
тора активностью 36 пунктов помещали в один и тот же реактор
с частицами равновесного катализатора активностью 34,7; 33,5;
32,4 и 29,0 пунктов и закоксовывали их в процессе крекинга
атмосферного и вакуумного газойлей при 470 °С, объемной скорости
1,0 ч^1 и длительности 1 ч.
Из рис. 39 видно, что катализатор закоксовывается быстрее с
увеличением активности в случае крекинга вакуумного газойля.
Это согласуется с результатами работы [104], из которых следует,
что увеличение жесткости крекинга вызывает тем более сильное
увеличение коксообразования, чем выше молекулярный вес сырья.
Из рис. 39 видно также, что при крекинге легкого и тяжелого
сырья на сзежем
катализаторе кокса откладывается
ссотзетственно з 2,5 и 4,0
раза больше, чем на
катализаторе активностью
29 пунктов. В
промышленных условиях на
катализаторе, выводимом из
реактора, обычно содержится 1,8—
3% кокса. Следовательно,
при переработке тяжелого
сырья на катализаторе
активностью 29 пунктов в
добавляемых в систему
частицах свежего катализатора
на выходе из реактора
будет содержаться около 10% кокса.
Зная количество кокса, отложенного на равновесном
катализаторе и на свежих частицах, можно рассчитать степень регенерации
последних при условии, что длительность выжига обеспечивает
полное удаление кокса с равновесного катализатора. Время,
необходимое для регенерации равновесного катализатора, находят
по формуле, приведенной в работе [93].
Было рассчитано количество остаточного кокса в частицах
свежего катализатора активностью 36 пунктов, если их добавлять
в систему установок, имеющих равновесные катализаторы
активностью от 29 до 36 пунктов. Принималось, что во всех случаях
равновесный катализатор в реакторе закоксовывается до 1,8%, а
в регенераторе он полностью регенерируется. Расчеты показали,
что с увеличением разницы в активностях смешиваемых
катализаторов концентрация остаточного кокса на свежих частицах сильно
возрастает, особенно в случае переработки тяжелого сырья
(рис. 40).
В промышленных установках активность равновесного
катализатора равна примерно 33 пунктам при крекинге облегченного
сырья и 29—30 пунктам при крекинге вакуумного газойля. По
данным рис. 41, при этих условиях остаточного кокса на добавляе-
30 31 32 33
Индекс активности
Рис. 39. Зависимость относительной закок-
сованности образцов катализатора от их
активности и перерабатываемого сырья:
/ — вакуумный газойль; 2 — атмосферный газойль.
мом в систему катализаторе должно содержаться соответственно
2,8 и 10,5—12,2%.
Таким образом, расчетные и экспериментальные данные
показывают, что при переработке тяжелого сырья на частицах
свежего катализатора действительно накапливается большое количество
кокса. Учитывая это, дезактивацию катализатора в
промышленных условиях можно представить следующим образом.
При работе установки на нормальном режиме суточные потери
катализатора относительно невелики. Они восполняются догрузкой
свежего катализатора, который, циркулируя в смеси с равновесны-
31 33 35
чооть цирнулирцю'чегс
катализатора
О 0,5 1,0 1,5'
Остаточный кокс на
равновесном катализаторе, вес. %
б1
Рис. 40. Зависимость содержания остаточного кокса в свежем катализаторе от
гго активности (а) и содержания остаточного кокса в равновесном
катализаторе (б):
/ — тяжелое сырье; 2 — легкое сырье.
«и, «прирабатывается». Однако этот процесс происходит при очень
кестких условиях. Свежий катализатор сразу контактируется с
гяжелым сырьем при режиме, рассчитанном на равновесный ката-
шзатор. При этом он закоксовывается в 2—4 раза больше. В ре-
•енераторе эти частицы также попадают в жесткие условия, так
<ак при регенерации свежих, сильно закоксованных частиц выде-
іяется больше тепла и они перегреваются. Большое количество
«зкса не успевает выгореть в период регенерации, поэтому на све-
ких частицах интенсивно накапливается остаточный кокс G—
2% и более). Если такая частица вследствие неравномерности
иительности пребывания катализатора в регенераторе или по ка-
сим-либо другим причинам задержится несколько дольше в зоне
89
выжига, то начнет выгорать высококонцентрированный остаточный
кокс. В результате в этой зоне резко повышается температура и
на частице образуется кольцо спекшегося остаточного кокса.
За время работы катализатора технологический режим
переработки и качество сырья неоднократно изменяются. В связи с
этим на одной и той же частице возникает несколько сферических
зон спекания; при этом каждая последующая образуется за
пределами предыдущей, так как ранее существовавшие зоны в той или
иной степени экранируют центральную часть катализатора.
Отчасти это объясняет большую трудность избавления от остаточного
кокса в условиях промышленной установки, чем от свежеотложен-
ного. После многократных перегревов свежих частиц и
образования нескольких внутренних зон спекания их активность довольно
быстро падает почти до уровня активности циркулирующего
катализатора. Поскольку процесс стабилизации догружаемого свежего
катализатора происходит в жестких условиях, снижение
активности сопровождается большими потерями добавляемого
катализатора из-за растрескивания.
Перекоксовывание активных частиц и их перегрев нельзя
обнаружить обычными методами контроля, так как свежего
катализатора добавляют не более 2—3% от его общей массы,
находящейся на установке. Поэтому режим, характеризующий работу
циркулирующего катализатора, не отражает истинных условий
работы свежих частиц. Разница между условиями работы частиц
свежего и равновесного катализатора тем больше, чем больше
разница в их активности. При низкой активности циркулирующего
катализатора создается впечатление, что он работает в относительно
мягких условиях — на нем образуется мало кокса и для
охлаждения катализатора в регенераторе требуется небольшое количество
змеевиков. В то же время условия эксплуатации активных частиц
наиболее тяжелые, так как при низкой активности
циркулирующего катализатора в реакторе обычно несколько ужесточают режим
крекинга.
Остаточный кокс накапливается наиболее интенсивно на
крупных шариках катализатора, и они разрушаются в наибольшей
степени. Более мелкие частицы равной активности устойчивее.
Отчасти поэтому чем тяжелее перерабатываемое сырье, тем мельче
циркулирующий катализатор. Кроме того, происходит усадка
более крупных частиц, которые при этом становятся прочнее.
Следует также учесть, что равновесный катализатор обладает низкой
активностью, повышенной механической прочностью и
разрушается в основном за счет истирания, что способствует его накоплению
в системе.
На катализатор определенное действие оказывает и водяной
пар, и обычные температурные условия регенерации, а также
металлы. В присутствии металлов снижается температура начала
спекания, а при наличии тяжелых металлов в частицах они закок-
совываются гораздо интенсивнее.
90
Улучшение условий эксплуатации катализатора. Одна из
причин перекоксовывания отдельных частиц катализатора при
переработке тяжелого сырья — неравномерное распределение по слою
катализатора жидкой фазы. На некоторых промышленных
установках внедрены разработанные во ВНИИНП специальные
устройства для смешения с катализатором парожидкостного сырья.
Они не только улучшают смешение катализатора с сырьем, но и
предохраняют стенки верхней, незаполненной катализатором части,
реактора от отложений кокса.
На установках с пылевидным катализатором перекоксовывание
отдельных частиц происходит вследствие разной
продолжительности их пребывания в реакторе и регенераторе. В аппаратах с
кипящим слоем этот недостаток устраняется их секционированием.
На установках с кипящим слоем наибольшее количество кокса
накапливается на частицах, которые из реактора выносятся в
колонну с парами продуктов. В колонне они пропитываются наиболее
тяжелыми фракциями и возвращаются с потоком тяжелого
газойля и сырья в реактор, где за счет этих адсорбированных
углеводородов на них откладывается дополнительное количество кокса.
Такие частицы в наибольшей степени подвержены спеканию и
разрушению в процессе регенерации. Предотвратить это ухудшение
катализатора можно, по-видимому, путем тщательного контроля
за системой сепарации продуктов реакции от катализатора и ее
усовершенствования.
Общего улучшения условий работы катализатора можно
достичь известными методами, позволяющими снизить образование
кокса в стадии крекинга или распределить его по большей массе
катализатора. Выход кокса уменьшается (при равной глубине
превращения) повышением температуры и соответственным
уменьшением времени контакта катализатора с сырьем. Рапространен-
ным методом является также повышение кратности циркуляции
катализатора. В этом случае кокс распределяется по большой
массе катализатора. Поэтому, несмотря на некоторое увеличение
выхода кокса в % на сырье, содержание кокса в катализаторе
снижается, а следовательно, уменьшается и перегрев частиц при
регенерации. Повышение кратности циркуляции имеет и ряд других
важных преимуществ: увеличивается выход и качество продуктов
крекинга, возрастает количество тепла, вносимого с катализатором
в реактор. Это позволяет уменьшить число охлаждающих
змеевиков в регенераторе и одновременно снизить расход топлива на
предварительный нагрев сырья. Увеличение кратности циркуляции
связано с некоторыми трудностями — возрастает количество кокса,
которое необходимо выжигать в регенераторе, уменьшается
длительность пребывания катализатора в регенераторе,
увеличивается теплота сгорания кокса, из-за большого содержания в нем
водорода осложняется работа отпарной зоны реактора [105, 106].
В промышленных условиях закоксованный катализатор,
выходящий из отпарной секции реактора, всегда содержит некоторое
91
количество адсорбированных молекул сырья и промежуточных
продуктов уплотнения. Показано [88, 105], что продувкой закоксован-
ного катализатора азотом и водяным паром нельзя полностью
удалить адсорбированные промежуточные продукты; поэтому вместе
с коксом в регенератор попадает значительное количество
углеводородов, и его коксовая и тепловая нагрузки дополнительно
увеличиваются. Увеличение продолжительности отпарки не дает
больших преимуществ. Более эффективно повышение температуры от-
парной зоны до 510—540 °С. В этом случае количество горючего
материала на отработанном катализаторе уменьшится вследствие
интенсификации процесса десорбции молекул сырья и
промежуточных продуктов уплотнения, а также вследствие дококсовывания
нелетучих компонентов.
Поскольку условия регенерации оказывают большое влияние
на старение и разрушение алюмосиликатного катализатора, были
проведены специальные исследования работы регенератора [146—
150]. Оказалось, что воздух, подаваемый в аппарат, используется
неэффективно вследствие неравномерного его распределения по
сечению регенератора. Лишь 30% проходит через слой катализатора
(т. е. может быть использовано для выжига коксовых отложений
с его поверхности). Преобладающая часть воздуха из-за
несовершенства конструкции аппарата проходит вдоль стенок
регенератора и в его центральной части. В связи с этим были разработаны
конструкции устройств для равномерного распределения воздуха
по сечению регенератора и вывода дымовых газов из слоя
катализатора.
Мероприятия, обеспечивающие уменьшение образования кокса,
не могут устранить перекоксовывания наиболее активных частиц
циркулирующего катализатора. Этого можно добиться путем
сужения диапазона его активности. В настоящее время катализатор,
используемый на установках, где перерабатывают тяжелое сырье,
имеет активность от нуля до 36—37 пунктов. Полностью
спекшиеся частицы являются балластом из-за расходуемой энергии на его
циркуляцию. Так как спекшиеся частицы катализатора обладают
повышенной механической прочностью и практически в
регенераторе не растрескиваются, вывод их из системы за счет износа
осуществляется медленно, и на установке происходит накопление
таких частиц. В некоторых случаях количество их достигает 30% и
более. Поэтому возникает необходимость в избирательном
удалении такого катализатора из системы. Разработано [111, 112] два
метода выделения неактивных шариков. По одному из них
тяжелые неактивные шарики концентрируются в нижней части плотного
кипящего слоя, по другому для этой цели применяют вибрацию.
Спекание катализатора с уменьшением диаметра частиц
катализатора вдвое против нормального наблюдается также на
установке гудрифлоу [113]. Для поддержания активности
катализатора на необходимом уровне на указанной установке поставили
классификатор, из которого отводили частицы с высокой плотно-
92
¦стью, образовавшиеся в результате их высокотемпературной
дезактивации.
В некоторых случаях в систему установки, перерабатывающей
тяжелое сырье, добавляют катализатор с пониженным индексом
активности, который прошел «стабилизацию» на другой установке,
работающей на облегченном сырье. В этом случае разница между
активностью добавляемого и циркулирующего катализатора также
снижается.
В результате перекоксовывание, перегрев, старение и разруше-
,ние активных частиц, а следовательно, расход катализатора на
обеих установках уменьшается и равновесная активность его
возрастает.
Приведенный перечень мероприятий, способствующих
облегчению условий работы катализатора, не является полным. Сюда
можно отнести также увеличение объема регенератора с целью
проведения выжига кокса при более низких температурах, борьбу
с догоранием СО в слое катализатора или в непосредственной
близости от него, улучшение распределения воздуха по высоте и
сечению регенератора и др. Таким образом, существует еще много
возможностей повышения активности циркулирующего
катализатора за счет устранения причин, вызывающих его перегрев.
ЗАКЛЮЧЕНИЕ
Активность катализатора крекинга при эксплуатации на
промышленных установках снижается главным образом вследствие
уменьшения его удельной поверхности и преимущественного
износа активной части.
При циркуляции катализатор стареет значительно быстрее, чем
когда он неподвижен в аппаратах промышленной установки или
когда подвергается действию рабочей температуры и среды в
лабораторных условиях. Удельная поверхность и структура
изменяются в основном за счет объемной диффузии. Наиболее
интенсивно старение протекает в начальный период работы катализатора.
С утяжелением перерабатываемого сырья спекание катализатора
усиливается.
Особенности изменения структуры катализатора на
промышленных установках связаны с двойственным влиянием
образующегося кокса. Он замедляет спекание при нагреве катализатора в
среде инертных газов и водяного пара. При выжиге кокса спекание
ускоряется. Это связано с перегревом в процессе регенерации
частиц катализатора, по сравнению с температурой газового потока,
на 150—200 °С. Перегреваются также отдельные участки частичек
катализатора. Перегревы и защитное действие оставшегося кокса
приводят к быстрому и неравномерному спеканию частиц по
глубине.
93
На промышленных установках с движущимся шариковым
катализатором 70% износа вызвано полным разрушением шариков.
Преимущественно разрушаются активные частицы, удельная
поверхность которых в 2 раза больше, чем у средней пробы.
Повышенный расход катализатора при переработке тяжелого сырья
обусловлен растрескиванием сильно закоксованных активных
шариков в процессе регенерации.
В настоящее время имеются большие резервы увеличения
активности катализатора не только за счет повышения стабильности
и прочности свежего катализатора, но и за счет улучшения
условий его эксплуатации.
Глава III
ИЗМЕНЕНИЕ АКТИВНОСТИ КАТАЛИЗАТОРОВ
В СТАДИИ КРЕКИНГА
В процессе крекинга наряду с газообразными углеводородами,
бензином и другими целевыми продуктами образуется кокс,
который накапливается на поверхности катализатора. Вследствие
экранизации активных центров алюмосиликата коксовыми
отложениями активность катализатора быстро снижается. Эта дезактивация
является обратимой, так как после окислительной регенерации
первоначальная активность катализатора полностью
восстанавливается. Влиянию продолжительности цикла крекинга (или
длительности работы катализатора) на показатели процесса посвящены
исследования [87, 119—120]. Под терминами «продолжительность
цикла крекинга», «длительность работы катализатора» или
«продолжительность использования катализатора» понимается время
от начала до окончания контакта катализатора в реакторе с
парами сырья и реагирующей смеси. В системах с движущимся слоем
это время совпадает с продолжительностью пребывания частицы
катализатора в реакционной зоне. В этом случае длительность
работы катализатора зависит от объема реакционной зоны и
количества циркулирующего катализатора. В реакторах со
стационарным слоем катализатора длительность его работы совпадает с
продолжительностью цикла крекинга.
СОСТАВ КОКСОВЫХ ОТЛОЖЕНИЙ И ЕГО ЗАВИСИМОСТЬ
ОТ УСЛОВИИ КРЕКИНГА
Кокс представляет собой обедненные водородом смолообразные
продукты. Поскольку при закоксовывании катализатор крекинга
теряет активность, коксовые отложения обычно считаются
нежелательными. Однако в работах [124, 125] и др. показана их
положительная роль — как доноров водорода, насыщающего
непредельные продукты каталитического крекинга. Коксовые отложения
могут дать по крайней мере 50% водорода, необходимого для
насыщения непредельных продуктов крекинга [125].
Элементный состав отложений изменяется от СНО,6 До СНід
[124]. В работе [126] при крекинге индивидуальных
углеводородов получали кокс состава СН0,5-
95
Таблица 28
Коксообразование при крекинге полициклических органических соединений
Сырье
Бензол
Нафталин
Антрацен
Фенантреп
9,10-Дигидроантрацен
Хризен
Пирен
Хинолнн
Карбазол
Акридин
Дибензотиофен
Дибензофуран
Скорость
подачи
сырья,
моль/ч
[I]
3,2
12,0
5,4
11,3
11,9
12,4
10,2
16,7
12,7
9,7
13,2
12,4
11,0
5,0
11,6
13,3
18,3
О1"
Конверсия, в
0,58
7,7
6,3
13,2
29,4
28,4
16,0
20,3
8,9
17,7
15,1
15,8
6,5
21,4
28,4
4,1
4,6
Выход
на сырье
0,58
6,4
4,7
11,1
26,2
24,6
14,8
16,0
6,7
15,3
13,2
13,8
4,9
17,0
25,6
3,3**
4,0**
кокса, вес. %
на
превращенный
продукт
100,0
83,1
75,8
84,2
89,2
86,6
92,5
78,8
75,3
86,7
87,4
87,3
75,4
79,4
90,2
80,5
87,0
на
катализатор
0,06
3,8
1,9
8,7
18,7
23,0
6,8*
13,2
8,2
12,5
16,6
14,2
2,9
5,5
21,3
3,2
4,8
1
Содержание
в коксе, вес.
6,7
4,1
5,1
3,8
3,5
2,9
3,1
4,0
3,4
4,8
4,5
4,4
5,2
—
—
3,2
4,1
g
g.
Отношение
водород: угле
в коксе
0,86
0,51
0,65
0,47
0,44
0,36
0,38
0,50
0,42
0,61
0,57
0,55
0,66
—
—
0,40
0,51
Выход продуктов, вес. %
на сырье
газ
0
1,3
1,6
2,1
3,2
3,4
1,2
3,4
1,7
2,4
1,7
1,9
1,6
4,2
2,3
0,8
0,6
жидкий
продукт легче
сырья
0,4
—
0,9
0,5
0,2
0,1
0,2
0,5
—
сырье и
продукты
тяжелее сырья
99,4
92,3
93,7
86,8
70,6
71,6
84,0
79,7
91,1
82,3
84,9
84,2
93,5
78,6
71,6
95,9
95,4
Выход
газообразных
продуктов, вес. %
водород
24,7
26,0
65,4
61,2
66,0
76,0
68,2
71,5
53,5
71,5
84,4
64,7
80,2
—
12,4
меган
7Е
74
15,6
3f
34
7,2
13,4
10,1
15,2
11,5
4,3
35
7,7
87,6
тяжелее
метана
,3
,0
19,0
,8
,0
16,8
18,4
18,4
31,3
17,0
11,3
,3
12,1
—
0,7
* Скорректировано.
** Только количество углерода н водорода.
В работе [125] в результате каталитического крекинга н-окте-
на при 375 °С, 0,8 МПа, объемной скорости подачи сырья 0,9 ч~'
на алюмосиликатциркониевом катализаторе получено 12,6 вес. %
кокса на сырье; в расчете на катализатор в коксе содержалось
9,34% углерода и 1,03 % водорода, т. е. состав коксовых отложений
отвечал формуле CHi,3i- Бензолом экстрагировалось 27,6 вес. %
коксовых отложений. В свою очередь, 40% бензольного экстракта
растворялось ацетоном с получением темного вязкого масла
(молекулярного веса 202), отвечающего формуле СН[,ь Нерастворимая
в ацетоне часть экстракта представляла собой твердое вещество
красновато-коричневого цвета с т. плавл. 195—210 °С,
молекулярным ВеСОМ 396, Имеющее формулу СН0>875-
Были исследованы закономерности коксообразования и состав
коксовых отложений при крекинге различных полициклических
органических соединений. Результаты опытов, проведенных в
основном при объемной скорости подачи 10—13 моль/(л-ч)
катализатора, приведены в табл. 28.
Как следует из этих данных, полициклические соединения при
крекинге образуют в основном кокс. Для всех испытанных видов
сырья выход кокса на превращенный продукт составляет не менее
75%. Отношение водород: углерод, которое в большинстве случаев
было примерно равно или меньше, чем для коронена (для
которого это отношение равно 0,5), подтверждает существующее
мнение о том, что коксообразование включает реакции конденсации с
выделением водорода и образованием больших многоядерных
ароматических молекул.
Кокс образуется быстрее при увеличении числа колец в ряде
бензол — нафталин — антрацен, чем в ряде бензол — дифенил —
терфенил. Значительно меньше коксообразование при крекинге фе-
нантрена, чем его изомера антрацена. Четырехкольчатые
углеводороды хризен и пирен образуют меньше кокса, чем антрацен, но
все же больше, чем фенантрен.
При крекинге азотистых оснований и углеводородов, имеющих
аналогичное строение, образуется примерно одинаковое количество
кокса; при крекинге флоурена образуется кокса 8,7 вес. %, т. е.
больше, чем при крекинге гетероциклического соединения
аналогичного строения. Коксообразование возрастало в следующем
порядке: дибензтиофен<дибензфуран<;карбазол<флуорен.
При крекинге непредельных алифатических углеводородов
образующийся кокс более обогащен водородом [127]. Так, при
крекинге н-бутилена в течение 15 мин при 500 °С и объемной скорости
подачи сырья 6,8 моль/(л-ч) образуется 14,8 вес. % кокса в
расчете на сырье, содержавшее 6,3 вес. % водорода. При крекинге
полициклических углеводородов примерно в тех же условиях
водорода в коксе содержалось 2,9—5,1 вес. % (см. табл. 28).
Коксообразование на поверхности цеолитсодержащего
катализатора сильно зависит от природы сырья и возрастает в ряду:
парафины < нафтены < ароматические углеводороды. При кре-
7-2206 97
кинге ароматических углеводородов коксообразование возрастает
g увеличением числа колец в молекуле [128].
Известны случаи, когда кокса образуется лишь 1 вес. % на
сырье (например, в процессе крекинга высокопарафинистого сырья)
и 80 вес. % (в процессе крекинга полициклических ароматических
углеводородов) при одинаковой глубине конзерсии, равной 80%
?129]. При переработке смеси ароматических и парафиновых
углеводородов кокса образуется больше, чем при их раздельной
переработке [128]. Авторы этой работы установили, что выход кокса
при каталитическом крекинге в присутствии цеолитсодержащих
катализаторов находится в прямой зависимости от образования
ароматических углеводородов. Это указывает на непосредственное
участие ароматических углеводородов в процессах коксообразо-
вания.
При переходе от менее активных аморфных катализаторов к
цеолитным выход кокса уменьшается, что видно из следующих
данных:
Конверсия, объемн. %
Выход кокса, вес. % ¦
Коксовый фактор . .
Алюмосилккатнын
аморфный катализатор
с повышенным
содержанием
окиси алюминия
74,5
10,0
7,0
Цеолнт-
содержащиґі
катализатор
74,0
9,2
8,1
Поскольку кокс препятствует использованию потенциальных
возможностей катализатора, при отложениях кокса роль
термического крекинга возрастает, а каталитического — уменьшается.
При типичных степенях конверсии около половины серы,
находящейся в сырье, преобразуется в сероводород, часть ее остается
в каталитическом газойле, а остальное небольшое количество
распределяется между бензином и коксом, отлагающимся на
катализаторе. Переход серы в кокс нежелателен, так как он способствует
последующему выбросу окисей серы с отходящими газами
регенерации и, следовательно, приводит к отравлению атмосферы.
Состав коксовых отложений на катализаторах крекинга, а
также влияние условий крекинга н-гексана на состав этих отложений
наиболее подробно исследован в работе [105]. В ней указывается,
что содержание водорода в коксе при увеличении длительности
крекинга существенно снижается. Влияние продолжительности
крекинга при 520 °С, объемной скорости подачи н-гексана 0,27 ч~! и
объеме катализатора в реакторе 30 мл показано ниже [105]:
Продолжительность
крекинга,
мин
1,5
15
60
180
Образование
кокса,
мг
11,0
28,1
58,8
113,4
Состав кокса
CHi
CHfi'
CHn"o
СНп ля
98
Как следует из этих данных, кокс с увеличением
продолжительности крекинга обедняется водородом. Особенно быстро это
обеднение протекает в начале крекинга. Специальными опытами
[105] было показано, что изменение состава кокса обусловлено
лишь реакциями, протекающими при каталитическом крекинге.
Уменьшение содержания водорода в коксе сопровождается
циклизацией и конденсацией углеводородов.
Данные об изменении состава кокса в зависимости от
содержания кокса на катализаторе приводятся ниже [130]:
Расчетная формула
Количество
образующегося
кокса,
вес. %
0,643
1,51
2,71
2,96
Отношение
водород:углерод
0,100
0,074
0,074
0,052
q>89
ch0j89
CHo,625
Отмечается также [130], что при повышении температуры часть
кокса удаляется в виде легких углеводородных газов (в основном
метана) и водорода. При этом кокс еще больше обогащается
углеродом. Авторы [130] считают, что при высокой температуре
конечным продуктом такого разложения является графит, поскольку
некоторое его количество было найдено на работавших долгое время
катализаторах.
Повышение температуры в вакууме до 770—925°С приводит к
полному термическому разложению коксовых отложений,
накапливающихся на поверхности алюмохромового катализатора [131].
Аналогичные данные для алюмосилпкатного катализатора были
получены в работе [132] —после прокалки при 800—1000°С
поверхность катализатора полностью освобождается от коксовых
отложений.
Экспериментально установлено [105], что кокс обладает
ненасыщенностью. Увеличение продолжительности крекинга приводит
к уменьшению непредельности кокса. Особенно быстро это
происходит в начале цикла каталитического крекинга. В течение
первого часа йодное число кокса снижается со 110 до 60, т. е. почти
вдвое, по прошествии еще 3 ч йодные числа кокса
стабилизируются на уровне 30—35. Такин образом, кокс, полученный даже при
очень большой продолжительности крекинга, обладает некоторой
ненасыщенностью, вероятно, за счет повышенной непредельности
более поздних отложений. Было показано, что при увеличении
объемной скорости подачи сырья гюдиое число снижается
незначительно. При изменении объемной скорости подачи сырья с 0,27 до
1,74 ч~', т. е. в шесть с лишним раз, йодное число кокса
уменьшилось только на 11%. Повышение температуры крекинга с 460 до
520 °С привело к увеличению йодного числа кокса с 38 до 60 [105].
В литературе имеются весьма ограниченные сведения о составе
коксовых отложений. Эти сведения касаются в основном крекинга
индивидуальных углеводородов. В работе [133] сообщается, что
7* 90
Таблица 29
Количество и состав кокса, образовавшегося
при крекинге различного сырья
Сырье
Керосино-газойлевая
фракция туймазинской
нефти
То же, чекмагушской
нефти
Вакуумный газойль
туймазинской нефти
То же, арланской нефти
Кокс,
вес. % на сырье
1,8
2,1
4,1
4,4
Кокс на
катализаторе, вес. %
1,0
1,2
2,4
2,6
Состав кокса, вес. %
водород
3,7
3,2
3,3
3,3
углерод
95,0
94,1
95,6
94,7
сера
1,3
2,7
1,1
2,0
Содержание серы
в коксе,
% от общего
содержания его в сырье
2,1
2,1
2,4
2,6
Расчетная
формула
CHOi47SOiOO5
CHo,jiSO|Ool
^Но, 42^0,004
^42^0,008
в процессе крекинга эталонного сырья при 450 °С, объемной
скорости подачи сырья 1,2 ч~' выход кокса составлял 3,46—3,92 вес. %
на сырье и 1,77—1,98 вес. % на шариковый аморфный
катализатор, при применении таблетированного катализатора Гудри выход
кокса был 2,6 вес. % на сырье и 1,35 вес. % на катализатор.
Состав коксовых отложений был одинаков на обоих катализаторах и
колебался в пределах СН0,4—СН0,5-
Нами исследовался состав кокса при крекинге кероснно-газой-
левой фракции и вакуумного газойля, полученных из
туймазинской сернистой и арланской высокосернистой нефтей. Крекинг
проводили 30 мин на равновесном катализаторе с индексом активности
30 при 450 °С, объемной скорости подачи сырья 1,0 ч~'.
Количество и состав полученного кокса приведены в табл. 29. Из данных
этой таблицы видно, что при крекинге разных видов сырья в
одинаковых условиях содержание водорода в коксе практически не
меняется.
Отмечается [135], что содержание водорода в коксе зависит
в основном от температуры крекинга. В этой работе оно
колебалось от 2,5 до 8,0 вес. % (в среднем 4,8 вес. %) или атомарное
отношение водород : углерод было равным 0,6 [135]. В работе [136]
общее содержание водорода в коксе составляло 4—7% на
отложенный кокс. В табл. 30 приводится состав кокса, отложившегося
на катализаторах в некоторых процессах нефтепереработки [137].
Распределение серы, содержащейся в сырье, между газом,
бензином, каталитическим газойлем и коксом зависит от типа сырья
и катализатора, степени конверсии и других рабочих условий
{140]. Углубление конверсии путем уменьшения объемной скоро-
100
Таблица 30
Состав кокса, получаемого в некоторых процессах,
нефтепереработки
Катализатор
Алюмосиликатный, закоксованный при
427 °С* (среднее из 5 определений)
Окись алюминия, закоксованная при
427° С* (среднее из 3 определений)
Работавший катализатор риформинга
(среднее из 2 определений)
То же
Содержание
на катализаторе,
вес
Н
0,1116
0,0240
0,0115
0,0595
. Уо
С
2,098
0,463
0,215
1,34
Водород
в коксе.
вес. %
5,1
4,9
5,1
4,3
Атомное
отношение
Н:С
0,64
0,62
0,64
0,53
Закоксован в результате крекинга легкого восточнотексасского газойля.
сти приводит к большему обессериванию с образованием
сероводорода, чем увеличение отношения катализатор: сырье при
постоянной объемной скорости. Это означает, что степень выделения
серы в виде сероводорода зависит от продолжительности
пребывания сырья в зоне реакции. На образование сероводорода оказывает
влияние также тип катализатора. При более активном
катализаторе степень конверсии сырья возрастает, но сероводорода
образуется меньше. При постоянной степени конверсии в присутствии цео-
литсодержащего катализатора выход сероводорода ниже, чем в
случае использования аморфного катализатора в связи с
проведением процесса на активном катализаторе при более высоких
объемных скоростях [140].
Отмечается [139], что при переходе от аморфных
катализаторов к цеолитсодержащим распределение между продуктами
крекинга серы, содержащейся в сырье, изменяется мало. При этом
вследствие снижения выхода кокса выбросы серы с дымовыми
газами регенератора уменьшаются. Содержание серы в коксе
зависит от содержания ее в исходном сырье. Но доля ее в коксе (в %
от общего содержания в сырье) примерно одинакова (см.
табл. 29).
При каталитическом крекинге сернистого сырья кокс,
образующийся в количестве 5 вес. % на сырье, содержит 1,29 вес. % серы,
что составляет 5,4% от содержания ее в сырье [134].
Анализ образцов катализатора на одной из установок дал
следующие результаты [138]:
„ „ Регенернро-
Закоксованныи ванный
катализатор катализатор
Углерод, вес. % 1,2 0,61
Сера, млн.-1 115 4,00
101
Данные [140] о влиянии типа сырья и глубины конверсии на
отношение содержания серы в коксе к его содержанию в сырье
приведены на рис. 41. По мнению авторов [140], при переработке
прямогонного сырья в коксе в среднем содержится 0,7 вес. % серы
на 1 вес. % серы в сырье. Однако такое усреднение является весь-
1
•К.
Pi
Г"
рик
§ §5| 1 1 L і J і і
^ б S 50 55 tU S5 -v? 75 80
Kjudepci* , o?ie?v;. %
Рис. 41. Влияние глубины конверсии сырья
на отношение содержания серы ь коксе к
его содержанию в сырье:
/ — тяжелый каталитический газойль; 2 —
гидроочищенная высококипящая фракция западноте-
хасской кефти; 3 — высококипящая фракция
западнотечасскоп нефти (содержание серы
L',59 вес. °j); 4 — широкая фракция из мидконтк-
нентской нефти (содержание серы 0,24 вес. %);
5 — гидроочищенное высококипящее сырье.
ма приближенным. Обычно отношение содержания серы в коксе
к содержанию серы в сырье изменяется в более широком
интервале. В наших опытах это отношение колебалось от 0,3 до 1,3.
ВЛИЯНИЕ ДЛИТЕЛЬНОСТИ РАБОТЫ КАТАЛИЗАТОРА
НА МАТЕРИАЛЬНЫЙ БАЛАНС КРЕКИНГА
Изменение активности катализатора в зависимости от
длительности его контакта с сырьем изучали в лабораторных условиях в
процессе каталитического крекинга вакуумного газойля арланской
нефти в присутствии равновесного аморфного алюмоснлпкатного
катализатора, отобранного с промышленной установки.
Результаты опытов представлены на рис. 42 и 43.
Как видно из приведенных данных, с увеличением
длительности работы катализатора глубина превращения сырья, выход газа
и кокса уменьшаются. Наибольшее снижение наблюдается в
начале процесса (в первые 30 мин). Такой результат объясняется тем,
что в начальный момент вся активная поверхность катализатора
свободна и может принимать участие в осуществлении реакций,
протекающих в процессе крекинга углеводородов. Вследствие
этого крекинг углеводородов протекает глубоко. Однако со временем
образующийся в процессе кокс накапливается на поверхности
катализатора и экранирует его активные центры. В результате
уменьшается глубина крекинга, а также газо- и коксообразование.
102
Зависимость от длительности работы катализатора выхода
бензина более сложная. С момента соприкосновения паров сырья с
катализатором выход бензина начинает быстро увеличиваться. По-
0 20 40 60 60 100 120 300
О Длительность рааоты катализатора ,мин ')
Рис. 42. Влияние длительности работы катализатора на выход кокса, бензина,
газа и глубину вакуумного газойля арланской нефтн.
Объемная скорость подачи сырья 1,0 ч-1; температура, °С: / — 425; 2 — 450; 3 — 475; 4 — 500;
5 — 550.
еле 5—10 мин он достигает максимального значения, затем быстро
снижается, а после 30 мин начинает снижаться более медленно.
Такой характер изменения выхода бензина объясняется тем, что он
ЮЗ
является промежуточным продуктом, который при большой
глубине крекинга распадается на газ и кокс. При низких температурах
и небольшой длительности работы катализатора выход бензина
увеличивается вследствие распада молекул исходного сырья. При
температуре выше 450 °С (см. рис. 43) скорость расщепления
молекул бензина возрастает и образуются газ и кокс.
Таким образом, аморфный алюмосиликатный катализатор
обладает наибольшей селективностью к образованию бензина при
умеренных температурах и длительности работы до 15 мин во всем
исследованном интервале объемных скоростей @,7—2,5 ч-1). С уве-
-ї 500 550\ft5J"U 500
-1,0 ч'1
iJ%] 500 5!~>0\<f50°C 500 550\
= 1>5ч-> Ц* 2,0 V'' Ч
Рис. 43. Влияние температуры на выход бензина и газа при крекинге вакуумного
газойля арланской нефти.
Длительность работы катализатора, мин; 1 — 10; 2 — 15; 3 — 30; 4 — 120.
личением длительности работы катализатора и объемной скорости
подачи сырья, а также при понижении температуры процесса
возрастает выход газойля. Повышение температуры процесса
способствует увеличению газообразования в любые периоды работы
катализатора.
При высокой скорости образования целевых продуктов
достигаются лучшие технико-экономические показатели процесса и
требуется меньший размер аппаратуры. В связи с этим из
экспериментальных данных расчетным путем определены выходы
продуктов в вес. %/мин. Скорость образования всех продуктов в
начальный период работы катализатора исключительно высока и
объясняется высокой первоначальной активностью катализатора (рис.
104
44). После длительности работы катализатора даже 15 мин
глубина крекинга и скорость образования всех продуктов при
исследованных объемных скоростях резко уменьшается. При повышении
.41
1
1§
t
I
I
16
14
12
10
a
в
2
0
8
6
2
0
7
6
5
І З
г
і
і і і і
і і і
J L
I I I
_L
J L
J L
20 40 60 80 100 120 Z0 W ВО 80 100 120
Длительность крекинга, мин
¦0,7 ч'
-2,5 ч'
Рис. 44. Зависимость скорости образования продуктов крекинга от длительности
работы катализатора.
Температура, °С: / — 450; 2 — 500; 3 — 550.
объемной скорости глубина крекинга и скорость образования
продуктов несколько снижаются.
Скорость образования отдельных продуктов в значительной
степени зависит от температуры, особенно при работе аморфного
О ОЛ
1,2 2,0 2,8
Врем*: ксі:тпк:па , с
с
7,2 2,0 2,8
Время киншакта, о
в
,6
Рис. 45. Изменение глубины превращения сырья, выхода бензина и содержания
непредельных углеводородов в бензине при изменении времени контакта.
Температура в реакторе, СС: / — 450; 2 — 485,- 3 — 505; -/ — 525; 5 — 540,
80
Длительность ррекинга, о
Рис. 46. Влияние природы сырья и длительности крекинга на активность
катализатора:
/ — неароматическне углеводороды, выделяемые из исходного газойля; 2 — исходный
газойль; 3, 4 — ароматические углеводороды, выделяемые из исходного газойля. Объемная
скорость подачи сырья, ч-1: /—1300; 2 — 325, 650, 1300; 3—1300; 4 — 325.
06
катализатора менее .30 мин. При повышении температуры скорость
образования бензина уменьшается, а газа и кокса возрастает.
После повышения объемной скорости до 2,5 ч^1 при 450 и 500 °С
скорости образования газа, бензина и кокса несколько
выравниваются. Это подтверждает известные представления о влиянии
жесткости процесса на нежелательные вторичные реакции. Интенсивность
этих реакций возрастает с увеличением температуры (особенно при
температурах выше 500°С) и с уменьшением объемной скорости
(менее 1,0 ч^1).
В дальнейшем в ряде работ, в которых исследовалось влияние
продолжительности работы катализатора на результаты крекинга
[141—145], определялась в основном оптимальная
продолжительность контакта сырья с цеолитсодержащим катализатором. На
рис. 45 представлены данные, полученные при крекинге ромашкин-
ского вакуумного газойля в присутствии катализатора АШНЦ-3
[141, 142]. По характеру кривые на рис. 42 и 45 совершенно
аналогичны. Разница лишь в том, что при использовании цеолитсо-
держащего катализатора требуется значительно меньшее время
контакта.
Изменение активности цеолитсодержащего катализатора в
течение одного цикла сильно зависит от природы крекируемого
сырья (рис. 46). Было показано [143], что характер изменения
активности цеолитсодержащего катализатора во времени одинаков,
и экспериментальные данные для всех видов сырья хорошо
описываются уравнением k = Katn. Значения п приведены на рис. 47. При
переработке неароматических углеводородов, выделенных из
газойля, активность катализатора снижается медленно. Наиболее
быстро она снижается при крекинге ароматических углеводородов.
Это объясняется тем, что в последнем случае кокса образуется в
три раза больше, чем при крекинге неароматических
углеводородов.
В отличие от других видов сырья при крекинге ароматических
углеводородов константа скорости реакции крекинга с
понижением объемной скорости подачи сырья уменьшается, та« как при этом
возрастает скорость реакций конденсации. Поэтому при времени
контакта 10 с и объемной скорости 1300 ч^1 отложения кокса
составляют 2,3%, а при 325 ч — 3,1%.
Скорость крекинга углеводородов с одним и двумя кольцами
выше, чем парафинов такого же молекулярного веса. Но при
увеличении числа колец с 2 до 4 скорость реакций крекинга
уменьшается из-за затруднений диффузии крупных молекул ко всем
молекулам кристаллического алюмосиликата. Как видно из табл. 31, на
алюмосиликатном аморфном катализаторе константа скорости
крекинга с увеличением числа колец в молекуле возрастает, а на цео-
литсодержащем при числе колец более 2 уменьшается. Таким
образом, размеры молекул существенно ограничивают скорость
реакции на кристаллическом алюмосиликате.
107
Таблица 31
Константы скорости реакций при крекинге
различных видов сырья
Температура 482 °С, время контакта 2 мин
Реагирующий углеводород
н-Гексадекан
1,3,5-Триэтилциклогексан
1,2,6-Триметилдекалин
Пергидрофенантрен
Пергидропирен
Алюмосиликатиый катализатор
аморфный
*а
60
140
190
250
210
выход
кокса,
вес. %
на сырье
0,1
0,4
0,2
0,2
0,4
кристаллический
1000
2370
2420
953
513
ВЫХОД
кокса,
вес. %
на^сырье
1,4
2,0
0,7
1,0
1,6
Отношение
*к:*а
17
17
13
4,7
2,4
ВЛИЯНИЕ ДЛИТЕЛЬНОСТИ РАБОТЫ КАТАЛИЗАТОРА
НА КАЧЕСТВО ПРОДУКТОВ КРЕКИНГА
Улучшение химического состава продуктов каталитического
крекинга достигается в результате реакций изомеризации
углеводородного скелета, дегидрирования нафтеновых углеводородов,
реакций перераспределения водорода и др. Поскольку эти реакции
предпочтительно протекают на чистых поверхностях катализатора,
длительность работы катализатора будет оказывать влияние на
качество получаемых продуктов. Наибольшие выходы пропан-про-
пиленовой и бутан-бутиленовой фракций, изобутана и изопентана
наблюдаются при длительности работы аморфного катализатора
до 15 мин (рис. 47). По мере увеличения длительности
использования катализатора выход этих компонентов снижается. При
изменении длительности крекинга с 5 до 15 мин выход сухого газа
снижается незначительно, но заметно уменьшается количество
образующихся пропан-пропиленовой и бутан-бутиленовой фракций. Это
приводит к повышению содержания в получаемом газе водорода,
метана и этан-этиленовых углеводородов.
С увеличением объемной скорости с 0,7 до 2,5 ч~' указанные
закономерности образования компонентов газа не изменяются, а
лишь уменьшается их абсолютный выход. С повышением
температуры газообразование возрастает. При этом становится больше
компонентов сухого газа и непредельных углеводородов и
уменьшается количество углеводородов, имеющих разветвленное
строение.
Максимальный выход бутиленов и амиленов можно получить
при 550°С, объемной скорости 0,7 ч^1 и пятиминутном контакте
катализатора с парами сырья. Насыщенные углеводороды изо-
строения образуются преимущественно в течение 30 мин работы
108
аморфного катализатора (см. рис. 47). При повышении
температуры их выход уменьшается. Качество жидких продуктов процесса
также весьма сильно зависит от длительности использования
катализатора. По мере ее увеличения йодное число и содержание
серы в бензине и легком газойле возрастают. Так, в процессе кре-
' 40 80 ПО 300 40 80 12ооо
Длительность работы натализатора , мин
Рис. 47. Изменение выхода газовых компонентов от продолжительности работы
аморфного катализатора при объемной скорости подачи сырья 1,0 ч~'.
Температура, °С: / — 425; 2 — 450; 3 — 475; 4 — 50Q; 5 — 550.
кинга арланского вакуумного газойля при 450 °С и объемной
скорости подачи сырья 1,0 ч-1 в случае изменения длительности
работы катализатора с 5 до 30 мин йодное число бензина возрастает
с 29,5 до 71,4, а содержание серы — с 0,3 до 0,7%. Таким образом,
в начальный период крекинга катализатор обладает хорошей
способностью к перераспределению водорода и превращению серни-
109
Таблица 32
Групповой углеводородный состав продуктов кргкинга вакуумного газойля, вес. % на фракцию
Углеводородный состав
фракции
С5—198 °С
парафины
непредельные
ароматические
нафтены
198—350 °С
парафины
непредельные
ароматические
нафтены
>350 °С
парафины + нафтены
ароматические
смолы
0,12
19,9
58,8
13,2
8,1
28,1
28,5
23,7
19,7
49,8
47,7
2,5
При 45
0,66
21,0
56,0
17,2
5,8
22,1
33,5
31,4
13,0
42,9
54,5
2,6
"С
3,89
22,5
51,5
19,8
6,2
22,7
32,6
33,5
П,2
41,3
56,2
2,5
При 485 с
0,13
14,4
63,0
15,5
7,1
25,0
36,8
28,8
9,4
43,9
53,4
2,7
1,15
20,2
54,2
19,8
5,8
20,1
31,7
39,3
8,9
38,9
58,1
3,0
с
4,07
25,4
46,8
21,7
6,1
20,4
28,3
40,0
10,4
37,8
59,0
3,2
При 505 °
с
время контакта, с
0,11
13,6
64,7
16,0
5,7
24,6
33,6
28,3
14,5
44,0
53,4
2,6
0,79
20,0
51,1
23,0
5,9
18,3
29,3
41,2
11,2
35,6
61,2
3,2
3,11
22,0
43,8
26,0
8,2
19,5
22,8
48,6
10,1
34,4
62,1
3,5
При 524 °
0,11
13,0
64,0
16,6
6,4
26,2
32,4
26,7
14,7
43,3
54,0
2,6
0,51
16
50
26
6
18
26
46
8
33
63,
2
9
5
5
1
4
6
6
4
7
8
5
2,95
23
40
28
7
18
17
56
7
33
53,
3,
,3
6
,4
7
6
3
4
7
2
4
7
При 5-Ю °
0,10
11
57
27
3
26
28
29
15
43,
53,
3,
,6
,0
,6
8
7
1
9
3
3
0
7
0,47
25
41
29
4
15
22
51
9
33
63
3
,4
,4
,0
,2
,7
8
9
5
3
5
2
3,'J3
29
33
33
3
14
20
58
8
30,
64,
5,
,2
,4
,6
8
5
4
0
1
2
7
1
Таблица 33
Качество продуктов крекинга вакуумного газойля
го
о.
S.
с:
452
485
505
524
540
такта,
я
о
к
Ї
О.
S3 о
0,12
0,66
3,89
0,13
0,83
4,07
0,11
0,53
3,11
0,11
0,51
2,95
0,10
0,47
3,06
Фракция C'Q~ 198 °С
октановое
число,
моторный
метод
75,7
79,5
80,5
75,0
78,0
78,3
74,0
79,4
79,6
75,4
78,3
79,0
80,0
81,0
81,3
сера,
вес. %
0,62
0,51
0,34
0,77
0,46
0,31
0,68
0,33
0,32
0,75
0,36
0,30
0,44
0,30
0,31
йодное
число,
г 13/100 г
бензина
112,1
121,0
107,4
139,4
123,5
119,9
134,3
122,3
114,8
150,4
117,8
104,8
133,5
90,0
77,8
Фракция
198—350 °С
сера,
вес. %
1,7
1,7
1,9
2,2
2,6
2,5
2,5
2,6
2,4
2,1
2,7
2,9
2,0
3,1
3,1
иодиое
число,
г 13/Ю0 г
фракции
35,5
36,3
40,6
36,2
30,3
43,2
40,9
25,2
27,3
48,9
26,4
26,2
35,5
25,2
25,7
Фракция >35О °С
плотность,
кг/мз
915
910
918
912
944
945
914
947
962
910
968
983
916
1014
1040
сера,
вес. %
2,1
1,7
1,9
2,0
2,5
2,3
2,0
1,9
2,7
1,6
2,0
2,4
1,9
2,0
2,4
коксуемость
10%.
вес. %
0,43
0,33
0,43
0,54
1,70
1,34
0,83
2,34
1,86
0,29
3,23
3,65
0,80*
6,90*:
9,60*
* Коксуемость общая.
стых соединений. По мере накопления кокса на активной
поверхности катализатора протекание указанных реакций становится
менее интенсивным, и способность алюмосиликатного катализатора к
ароматизации продуктов крекинга уменьшается.
Групповой углеводородный состав и качество продуктов
каталитического крекинга ромашкинского вакуумного газойля в
присутствии цеолитсодержащего катализатора АШНЦ-3 в
зависимости от температуры процесса и времени контакта показано в
табл. 32 и 33 [141, 142].
Углеводородный состав бензина в значительной степени
зависит от температуры в реакторе и времени контакта сырья с
катализатором. Образующийся в начале процесса бензин (через 0,1—
0,13 с контакта) состоит в основном из непредельных
углеводородов (см. табл. 32). В то же время концентрация ароматических
и парафинов изостроения в бензине еще мала, и поэтому
октановое число, определенное по моторному методу, не превышает 75,5
(см. табл. 33).
При большем времени контакта октановое число бензина
возрастает в результате увеличения в нем концентрации
ароматических углеводородов и парафинов изостроения; одновременно в
бензине уменьшается концентрация непредельных углеводородов. В
исследованном диапазоне изменения времени контакта йодное число
111
бензина снижается на 15,6—55,7 единиц в зависимости от
температуры крекинга сырья. Содержание серы в бензине за первые 3—4 с
контакта уменьшается в среднем с 0,7 до 0,32 вес. %¦ При
увеличении времени контакта сырья с катализатором содержание серы
в легком каталитическом газойле несколько возрастает, а
изменение йодного числа зависит от температуры крекинга сырья (см.
табл. 33). Относительное содержание парафинов и непредельных
углеводородов во фракции 198—350 °С уменьшается в результате
реакций крекинга, циклизации и конденсации углеводородов, а
концентрация ароматических углеводородов возрастает тем
больше, чем выше температура в реакторе и чем больше
продолжительность контакта сырья с катализатором.
Состав фракции >350 °С при увеличении времени контакта и
температуры крекинга изменяется следующим образом:
содержание серы и коксуемость фракции возрастают; концентрация пара-
фино-нафтеновых углеводородов в результате их расщепления
уменьшается, а ароматических монотонно растет и достигает
64,7 вес. % (см. табл. 32 и 33).
Следовательно, как с точки зрения увеличения выхода целевых
продуктов, так и для улучшения их качества необходимо работать
при небольшой продолжительности использования катализатора.
ВЛИЯНИЕ ДЛИТЕЛЬНОСТИ РАБОТЫ КАТАЛИЗАТОРА
НА КИНЕТИЧЕСКИЕ КОНСТАНТЫ
При расчете кинетических констант нами было использовано
уравнение Г. М. Панченкова [146]:
пох = —по\а(\—х)—К B5)
где п0 — скорость подачи сырья в реактор; х — глубина
превращения сырья; К — кинетическая константа, пропорциональная
константе скорости реакции.
В начальный период работы катализатора кинетическая
константа имеет наибольшее значение, а затем быстро уменьшается и
к 30 мин работы катализатора при 450 и 500 СС составляет лишь
22—37% от первоначальной величины. При 550°С константа
снижается до 60% от ее начального значения. В последующем
кинетические константы снижаются меньше. Чем выше температура, тем
выше значение кинетической константы и тем меньше его
снижение с увеличением длительности опыта (рис. 48).
Как следует из рис. 49, с увеличением длительности работы
катализатора энергия активации процесса крекинга резко
возрастает. Низкие значения кажущейся энергии активации A0 000—
24 700 Дж/моль) соответствуют короткой длительности работы
катализатора E—10 мин) и характерны для внешнедиффузионной
области. Вероятно, в начале контакта паров с гранулами
катализатора молекулы сырья расщепляются с большей скоростью на
активной внешней поверхности и в порах, находящихся вблизи ее,
112
О ЗО 60 90 120
Длительность раиоты катализатора,
мин
Рис. 48. Зависимость константы скорости реакции от длительности работы
катализатора.
90000
80000
70000
60000
' 50000
t+0000
1
^ 30000
10000
О I-
0 ZO 40 60 80 100 120 V+0
Длительность работы катализатора, мин
Рис 49 Зависимость энергии активации процесса крекинга и предэкспоненци-
ального множителя (С) от длительности работы катализатора.
113
не проникая глубоко в мелкие поры внутри гранул. В этих
условиях подвод молекул сырья к поверхности реагирования и
продуктов реакции в свободный объем между гранулами катализатора
не должен играть решающей роли в кинетике процесса.
Таким образом, при небольшой длительности работы
катализатора молекулы сырья распадаьэтся в основном на внешней
поверхности гранул, и внутридиффузионное торможение маловероятно.
В этот период вследствие высокой активности катализатора
скорость процесса распада молекул столь велика, что транспорт
молекул из объема к поверхности и наоборот затрудняется. Это
подтверждается также литературными данными [147].
Поскольку с увеличением длительности работы катализатора
внешняя поверхность его гранул покрывается слоем коксовых
отложений и каталитическая активность этих участков резко
снижается, в дальнейшем становится необходимым проникновение не-
превраш.енных молекул сырья в более глубокие слои гранул
катализатора. Другими словами, в связи с тем, что с увеличением
длительности работы катализатора по сечению его гранулы
послойно откладывается кокс, участки с повышенной каталитической
активностью оказываются расположенными все дальше от
поверхности гранулы. Поэтому при продолжительном крекинге для
кинетики процесса все большее значение приобретает диффузия по
порам катализатора молекул сырья к активным центрам п
продуктов крекинга в свободный объем между гранулами катализатора.
Такой механизм превращения сырья и промежуточных
продуктов подтверждается данными по кажущейся энергии активации.
При увеличении длительности использования катализатора с 10 до
30 мин кажущаяся энергия активации процесса крекинга
возрастает с 18 000—24 700 до 46 000—60 000 Дж/моль, что характерно
для внешнепереходной области. Последующее увеличение
длительности процесса с 30 до 120 мин способствует росту кажущейся
энергии активации до 59 000—80 800 Дж/моль и переходу процесса
во внутридиффузионную и внутрикинетическую области. Попутно
следует отметить, что зависимость между логарифмом предэкспо-
ненциального множителя и кажущейся энергией активации хорошо
описывается уравнением линейного вида.
С увеличением глубины крекинга выход продуктов возрастает
(рис. 50). Выходы кокса и газа с углублением крекинга постоянно
увеличиваются. Несколько иной характер имеет выход бензина.
Выходы продуктов зависят от температуры процесса. С
повышением температуры крекинга увеличивается образование газа и
уменьшается образование кокса. При глубине крекинга до 20—30%
выход бензина практически не зависит от температуры в реакторе.
Однако при больших глубинах конверсии сырья увеличение
температуры вызывает существенное снижение количества
образовавшегося бензина.
Очень важно, что влияние длительности работы катализатора
на результаты крекинга ограничивается изменением глубины кон-
114
версии сырья. Поэтому данные о выходе бензина, газа и кокса,
полученные при разной длительности использования катализатора,
но при одной температуре, хорошо укладываются в пределах
соответствующих кривых. Следовательно, длительность работы ката-
го w во во
Глубина крекинга, вес, %
а
20 ЇО 60
Глубина крекинга , вес.
во
Рис. 50. Зависимость выхода бензина и кокса (а), а также газа (б) от глубины
крекинга вакуумного газойля арланской нефти.
Сплошные линии — бензин; пунктирные линии — кокс.
лизатора оказывает такое же влияние на результат процесса
каталитического крекинга, как температура, объемная скорость
подачи сырья, активность катализатора и др.
Увеличение газо- и коксообразования при возрастании
глубины крекинга обусловлено накоплением конечных продуктов ре-
fi* 116
акции. Максимум выхода бензина при глубинах крекинга 70—75%
объясняется тем, что процесс крекинга является консекутивной
реакцией, и при больших глубинах конверсии скорость
расщепления бензина преобладает над скоростью его образования.
Уменьшение выхода бензина с повышением температуры процесса при
одинаковой глубине крекинга объясняется увеличением скорости
расщепления образовавшегося
бензина.
Меньшее коксообразование
с повышением температуры
крекинга при одинаковой
глубине превращения сырья
отмечается многими авторами.
По-видимому, это вызывается
увеличением скорости
десорбции и разложения
адсорбированных углеводородов при
высоких температурах. С
повышением температуры время,
необходимое для достижения
заданной глубины крекинга,
существенно уменьшается.
Соответственно уменьшается
ширина участвующей в процессе
зоны в грануле катализатора.
Это облегчает удаление
продуктов десорбции и расщепле-
Рис. 51. Изменение выхода продуктов от
глубины каталитического крекинга в
присутствии аморфных (/, 2, 3) и це-
ния из гранул катализатора
вследствие уменьшения длины
олитсодержащих D, 5, 6) катализато- пути диффузии,
ров:
1,4 — бензин; 2, 5 — газ; 3, 6 — кокс.
В присутствии цеолитсо-
держащих катализаторов
характер зависимости выхода
продуктов от глубины крекинга не меняется (рис. 51). Эти
катализаторы отличаются от аморфных лишь по абсолютной величине
образующихся продуктов крекинга [69]. Отличительной
особенностью цеолитсодержащих катализаторов является их способность
обеспечивать значительную глубину крекинга, высокое бензинооб-
разование. Так, если аморфные катализаторы позволяют получать
максимальный выход бензина 37 объемн. % при оптимальной
конверсии примерно 50%, то цеолитсодержащие катализаторы
образуют до 60 объемн. % бензина при глубине конверсии 70—
80 вес. %. Таким образом, другой особенностью последних
является очень высокая селективность даже при больших степенях
превращения. В связи с этим при одинаковой глубине крекинга в
присутствии цеолитсодержащих катализаторов образуется
значительно меньше газа и кокса, чем при применении аморфных
алюмосиликатов.
116
ПРОВЕДЕНИЕ ПРОМЫШЛЕННЫХ ПРОЦЕССОВ
С НЕБОЛЬШОЙ ДЛИТЕЛЬНОСТЬЮ РАБОТЫ КАТАЛИЗАТОРА
Данные лабораторных исследований показывают, что
результаты каталитического крекинга сильно зависят от длительности,
работы катализатора между регенерациями. При небольшой
длительности использования катализатора резко возрастает выход,
ценных продуктов процесса: бензина, пентан-амиленовой, бутан-
бутиленовой и пропан-пропиленовой фракций, и они получаются
высокого качества. Данные о влиянии длительности работы
катализатора, полученные в промышленных условиях [149], полностью-
подтверждают результаты лабораторных исследований [150—152].
Эти данные могут быть использованы при корректировке режима
установок каталитического крекинга с целью достижения лучших,
показателей.
Во время промышленного пробега было показано, что
материальный баланс каталитического крекинга изменяется главным
образом в верхнем слое реактора высотой 900—1000 мм или при
длительности работы катализатора 9—10 мин. В этом слое выход
бензина возрастает до максимальной величины, а в остальной
части реактора он несколько уменьшается. Выходы легкого и
тяжелого газойлей резко сокращаются в верхней части реактора и
мало изменяются в остальной его части. Конечные продукты
процесса (газ и кокс) образуются по всей высоте реактора (рис. 52).
Качество катализата также изменяется, главным образом в
верхнем слое катализатора высотой не более 900 мм. При контакте
паров сырья с катализатором 10 мин плотность, коксуемость,
содержание смол и серы в катализате снижаются до минимальной
величины. В последующих слоях реактора эти показатели не
изменяются. В верхнем слое кратковременный контакт паров сырья
с катализатором, нагретым до 530—600 °С, вызывает увеличение
йодного числа дистиллята. При этом в слое высотой до 300 мм
повышается также содержание сульфируемых. В последующих слоях
содержание сульфируемых в дистиллятах снижается с 56 до 42—
43 объемн. %•
Представляет интерес образование на высоте слоя катализатора
300 мм, соответствующей длительности работы катализатора
2,64 мин, большого количества E7,4%) сухого газа (см. рис. 52).
По мере продвижения реагирующей смеси по реактору в газе
увеличивается содержание пропилена, бутиленов и бутана. После
10—15 мин работы катализатора увеличение образования
указанных компонентов прекращается, а содержание бутиленов в газе
снижается. Выход сероводорода увеличивается с возрастанием
продолжительности реагирования почти по линейному закону.
Образование большого количества сухого газа в верхней части
реактора можно объяснить глубоким крекингом сырья при контакте к
катализатором, имеющим температуру до 600°С.
117
Фракционный состав бензинов во всех пробах, отобранных из
разных точек по высоте реактора, почти одинаков. Бензин
относительно мало ароматизирован. Содержание непредельных
углеводородов снижается с 30% в верхней части реактора до 20% на вы-
50
С]
s
40
1
ЗО
20 -
10
Ў - /
я - 2
V -З
о - ч
о - 5
X - ff
Д - 7
0-е
П - 9
a -W
• - 11
УІ--А--«їа=5а.
1 Z З
Высота слоя катализатора в реакционной
Рис. 52. Выход продуктов каталитического крекинга по высоте реактора
установки 43-102:
/ — газ; 2 — бензин: 3 — легкий газойль; 4 — тяжелый газойль; 5 — (Н2+Сі + С2); 6 —
пропилен; 7 — пропан; а — бутан-бутиленовая фракция; 9 — изобутилен; Ю — бутилен;
/;—сероводород.
ходе из реактора. Содержание в бензине сернистых соединений и
парафкно-нафтеновых углеводородов изменяется незначительно.
Октановое число бензина по моторному методу при изменении
длительности работы катализатора от 2,6 до 20,5 мин возрастает с
70,0 до 72,5 и на выходе из реактора достигает 74. Низкое
значение октанового числа во время пробега объясняется содержанием
118
в сырье до 10% низкооктановой прямогонной бензиновой фракции.
Йодное число бензина снижается с 60 до 42 при увеличении
длительности работы катализатора с 2,6 до 10 мин и в дальнейшем
изменяется незначительно.
Фракционный состав легкого газойля по высоте реактора
практически остается без изменения. Йодное число его быстро
снижается с 18,4 до 8,8 в слое, обеспечивающем длительность работы
катализатора 15 мин, а на выходе из реактора оно не превышает 7.
Содержание серы в газойлевой фракции по высоте реактора
снижается с 2,87 до 2,0—2,3%. Из-за значительного содержания
ароматических углеводородов C1—50%) цетановое число газойля
было низким C6—42 пункта).
При использовании цеолитсодержащих катализаторов
отмеченные выше закономерности влияния длительности крекинга
полностью сохраняются. Следует учитывать, что они с большой скоростью
и селективно крекируют парафиновые и нафтеновые
углеводороды, а крекинг ароматических углеводородов проводят с низкой
скоростью и небольшой селективностью. Наряду с этим они
обладают высокой активностью в реакциях переноса водорода [153].
При использовании аморфных алюмосиликатов наблюдалась
значительная разница между скоростью крекинга тяжелых
газойлей и нижекипящих средних дистиллятов. Глубина крекинга
тяжелого газойля была 70—75%, а среднего дистиллята 30—35%.
В случае цеолитсодержащих катализаторов скорость
крекирования таких дистиллятов лишь слегка меньше, чем тяжелых
газойлей, Цеолитные катализаторы способны крекировать и бензиновые
углеводороды; поэтому чтобы не допускать их расщепления,
необходимо тщательно подбирать условия процесса.
Активность современных цеолитных катализаторов такова, что
для полной конверсии неароматической части сырья в
большинстве случаев требуется от 1 до 4 с [153]. На рис. 53 представлена
зависимость константы скорости реакции крекинга тяжелого
ароматизированного газойля от длительности его контакта с цеолит-
содержащим катализатором [154].
Как было показано [155], степень превращения сырья для
любых значений кратности циркуляции катализатора проходит через
максимум при длительности работы цеолитсодержащего
катализатора примерно 2 мин (рис. 54). Эти данные аналогичны данным,
полученным на аморфных алюмосиликатах (см. рис. 42).
В цеолитсодержащем катализаторе от 5 до 15% цеолита
распределено в матрице — аморфном алюмосиликате. Чтобы полнее
использовать преимущество таких катализаторов, следует
обеспечить доступ реагирующих веществ к цеолитам, который при
отложениях кокса или металлов и разрушении пористой структуры
матрицы затрудняется. Поэтому необходимо, чтобы содержание
кокса на катализаторе было минимальным, а при проектировании
предусматривать такой реакторный блок, в котором выход кокса
при выбранной глубине конверсии также был бы минимальным.
Чтобы обеспечить подвод сырья к цеолитам, необходимо
предотвратить перегрев частицы и спекание. Наличие ароматических
соединений нежелательно, так как они способствуют спеканию
частиц вследствие повышенного коксообрачования.
С переходом на работу с цеолитсодержащими катализаторами
еще более важным стало существенное снижение содержания кок-
0 12 3
Длительность, мин
Рис. 53. Зависимость
константы скорости К от
длительности реакции.
о w го зо
Длительность работы катагизатпсра
мин
Рис. 54. Зависимость степени
превращения от длительности работы цеолитсо-
держащего пылевидного катализатора
при 480 °С.
Кратность циркуляции катализатора: / — 3,00:
2—1,50; 3-0,75; 4 — 0,37; 5 — 0,18; 6 — 0,09.
са на регенерированном катализаторе. Влияние степени закоксо-
ванности катализатора на глубину превращения сырья показано
ниже:
Содержание кокса
на регенерированном
катализаторе,
вес. %
0,1
0,2
0,4
Глубина превращения
при весовой скорости
5 4—1,
объемн. %
78,5
75,1
67,8
Весопая скорость,
соответствующая
глубине превращения
75 объемн. %,
4-1
6,10
5,00
3,55
Из приведенных данных видно, что для поддержания глубины
превращения на приемлемом уровне весовая скорость подачи
сырья должна быть значительно снижена. Однако с увеличением
длительности пребывания сырья в реакторе возможен повторный
крекинг, что приводит к снижению выхода бензина и к увеличению
выхода кокса н газа [156]. По отмеченным причинам содержание
кокса на регенерированном цеолитсодержащем катализаторе в
промышленных условиях поддерживают на минимальном уровне.
Отмечается [157], что при постоянных условиях процесса и
одинаковом количестве образующегося кокса с повышением
содержания кокса на регенерированном цеолитсодержащем катализаторе
120
с 0,1 до 0,35% глубина конверсии сырья уменьшается на
2,5 объемн. %• Обследование ряда промышленных установок [158]
показало, что содержание кокса на регенерированном
катализаторе в среднем составляет 0,35%.
Для достижения минимального содержания кокса в
регенерированном катализаторе в кипящем слое большое значение имеют
температура в зоне регенерации, длительность пребывания,
давление, равномерность распределения катализатора и воздуха,
высота кипящего слоя и содержание металлов на катализаторе. Так„
в работе [159] указывается, что при температуре регенерации
649—682 °С регенерированный цеолитсодержащий катализатор
содержит от 0,05 до 0,20% кокса [159]. Считается целесообразным
поддерживать температуру в регенераторе каталитического
крекинга равной 675 °С. Это позволяет сократить расход воздуха на
выжирание кокса из-за увеличения содержания окиси углерода в
дымовых газах или же уменьшить концентрацию кокса в
регенерированном катализаторе [160].
В связи с усилением борьбы по защите атмосферы от
отравления в последнее время большое внимание уделяется составу
дымовых газов, выбрасываемых из регенератора. Обычно отношение
Таблица 34
Влияние технологии ультракат на результаты работы
трех установок каталитического крекинга флюид
Показатели
Содержание кокса на
регенерированном катализаторе, вес, %
Температура предварительного
подогрева сырья, °С
Температура в реакторе, °С
Коэффициент рециркуляции
Снижение циркуляции катализатора,
0/
70
Конверсия, объемн. % на свежее
сырье
Выход кокса, вес. % на свежее сырье
Анализ газов регенерации по Орса,
объемн, %
о2
СО2
СО
Октановое число бензина в чистом
виде
исследовательский метод
моторный метод
I
А*
0,17
318
493
1,22
75,0 <
6,8
1,9
9,9
9,3
88,9
79,5
Б*
0,03
320
509
1,13
25,6
75,0
5,0
4,1
14,2
0,0
92,0
80,2
А
0,30
274
523
1,14
79,7
7,3
0,2
10,3
11,8
93,5
81,5
і
Б
0,05
262
518
1,09
37,5
79,5
5,8
0,7
17,3
0,1
93,2
81,2
Ш
А
0,29
364
518
1,36
63,8
6,2
0,2
10,3
10,2
92,2
78,1
Б
0,03
364
518
1,16-
39,0
63,8
4,4
1,8-
16,3
0,0
92,5
78,0
* А—до применения технологии ультракат; Б—после ее применения.
121
С02 к СО находится в пределах 0,5—0,8. Для цеолитсодержащих
катализаторов характерны более низкие значения. В газах
регенерации наряду с окисью и двуокисью углерода обнаружены
также двуокись и трехокись серы. Содержание трехокиси серы
составляет от 10 до 40% от суммы окислов серы [159]. Кроме того, в
газах регенерации обнаружены сероводород, меркаптаны, серо-
окись углерода и сероуглерод, а также углеводороды (метан и
зтан). Концентрации их меняются; так, содержание сероокиси
углерода колебалось от 9 до 190 млн.~'. Из общего содержания
сернистых соединений не менее 70% составляют двух- и трехокись
серы [158].
-/ уз ,
_J і
70 ;•:,
50 SO
Конберьир, объема. %
1,0 1,5 2,0 ЗР ^0 6,0 3,0 10?
Длительности1 -ірєкинга, с
Рис. 55. Выход кокса в зависимости от глубины конверсии, типа катализатора
и реактора:
/ — аморфным катализатор; 2 — аморфный высокоглиноземистый катализатор; 3 — цеолитсо-
дсржаший катализатор из лифт-реактора; 4 — улучшенный цеолитсодержащий катализатор
ляфг реактора, 5 — цеолитсодержащий катализатор,
ультракат.
регенерированный по методу
Рис. 56. Выход бензина в зависимости от длительности крекинга:
/ — цеолі[тсоде;)жащнй :%лталнзатор; 2—аморфный. Длительность пребываЕшя катализатора:
а — в подъемнике; б — в кипящем слое.
Разработана новая технология регенерации катализатора,
называемая ультракат [161]. К ее достоинствам относятся снижение
образования кокса и выжиг большего количества кокса. В
литературе не сообщается о техническом оформлении процесса
регенерации, однако отмечается, что в процессе регенерации СО
практически полностью окисляется до СО2 в пределах регенератора.
В табл. 34 показано влияние технологии ультракат на результаты
работы трех установок каталитического крекинга флюид с
кипящим слоем катализатора.
После модификации во всех трех случаях выход кокса
уменьшился при практически прежней глубине крекинга сырья, а выход
извлекаемых жидких продуктов соответственно увеличился.
Работа установки стала более устойчивой, так как при регенерации
почти вся окись углерода окисляется и, как следствие,
предотвращается порча конструкций, расположенных по ходу движения
газов, из-за неконтролируемого их догорания. На рис. 55 показано
122
уменьшение выхода кокса по мере совершенствования технологии
каталитическего крекинга.
Улучшение показателей процесса при уменьшении
продолжительности использования катализатора в кипящем слое также
отмечалось многократно. Так, уменьшение высоты кипящего слоя
аморфного катализатора в реакторе и переход на осуществление
процесса крекинга в основном в транспортной линии позволило
увеличить глубину конверсии с 37,6 дс 42,0%, выход бензина с
18,6 до 24% и снизить образование кокса с 7,0 до 5,5%; в
результате производительность установки была повышена на 150% [162Д.
Высокоактивные цеолитсодержащие катализаторы позволяют
получать больше бензина, чем аморфные, и достигать высокой глу-|
бины конверсии в подъемнике катализатора приочень малом
времени контакта (рис. 56).
Так как основная часть конверсии сырья протекает в
подъемнике катализатора, то за рубежом появился термин лифт-реактор
За 4,4 с в лифт-реакторе достигается глубина превращения 97%
от общей ее величины, получаемой на цеолитсодержащем
катализаторе. На аморфном катализаторе за 2,9 с глубина превращения
в лифт-реакторе составляет 88% от общей глубины превращения
с дополнительным крекингом в кипящем слое в течение 9,9 с [163].
Выход бензина в результате крекинга в лифт-реакторе и в
кипящем слое при работе на цеолитсодержащем катализаторе ниже,
чем в конце лифт-реактора (см. рис. 56). На аморфном
катализаторе, несмотря на увеличение глубины превращения в кипящем
слое на 6,3 объемн. %, выход бензина возрастает только на
1 объеми. % [163].
Эти опыты подтвердили большую эффективность прямоточного
контакта в лифт-реакторе, чем в кипящем слое, вследствие
продольного перемешивания сырья и катализатора. Поэтому в
последние годы появился ряд модификаций установок каталитического
крекинга с пылевидным катализатором, основное отличие
которых— крекинг в подъедшай..трубе. В зависимости от вида
применяемого сырья и требуемой глубины конверсии имеется несколько
вариантов применения лифт-реактора. В случае переработки пря-
могокного сырья ограничиваются крекингом лишь в
лифт-реакторе. Он применяется в одном из вариантов оформления установки
флексикрекинг (рис. 57, а) [164]. Для этой системы характерны
высокие весовые скорости (от 60 до 100 ч~'), которые могут
потребоваться в случае крекинга высококачественного сырья при
умеренных глубинах превращения или при необходимости вести
высокотемпературный процесс.
Если требуется углубить крекинг или переработать труднокре-
кируемое сырье, каталитический крекинг осуществляют либо толь-*
ко в лифт-реакторе, либо в лифт-реакторе и в реакторе-сепараторе^
с небольшим слоем катализатора. Основные черты этой
модификации каталитического крекинга: высокая весовая скорость
(около 50 ч~'), минимальное перемешивание катализатора в реакторе-
12а
сепараторе. В этой системе, изменяя количество катализатора в
кипящем слое, можно весовую скорость менять в пределах 5—
50 ч-1 (рис. 57,6) [207].
Установка получается более гибкой, если раздельно подвергать
крекингу свежее и рециркулирующее сырье. Например, по схеме,
принятой в процессе флюид-тексако (рис. 58), свежее сырье
крекируют в лифт-реакторе, а более стабильное рециркулирующее
сырье перерабатывают в параллельной транспортной линии, а
затем в дополнительном реакторе с кипящим слоем катализатора
[165]. Такая схема наиболее эффективна, поскольку каждый вид
сырья перерабатывают в оптимальных условиях.
Рис. 57. Реакторный блок установки флексикрекинга:
а — крекинг только в лифт-реакторе; б — крекинг сырья в лифт-реакторе и в кипящем слое.
/ — реактор; 2 — регенератор; 3 — лифт реактор; 4 — границы кипящего слоя; 5 — сепаратор
с циклонами и секцией продувки отработанного катализатора/— сырье в реактор; //
—воздух; ///— водяной пар; IV — газы регенерации; V — продукты крекинга.
Рис. 58. Схема процесса каталитического крекинга флюид-тексако:
1 — регенератор; 2 — реактор; 3 — отпарная секция; 4 — стояки; 5 — фракционирующая
колонна. / — сырье (газойль); // — воздух; /// — дымовые газы регенерации; IV — водяной
пар на отпарку; V — циркулирующее сырье; VI — пары на прием газового компрессора;
УII — бензин неочищенный; VIII — легкий циркулирующий газойль; IX — тяжелый
циркулирующий газойль.
ЗАКЛЮЧЕНИЕ
При уменьшении длительности использования катализатора в
цикле крекинга выход и качество целевых продуктов значительно
возрастают. Намного увеличивается скорость превращения сырья.
Все это обусловлено главным образом влиянием общего
содержания кокса на катализаторе. В свою очередь, количество и состав
образовавшегося кокса сильно зависят от длительности
использования катализатора и степени экранизации активных центров
алюмосиликата.
Глава IV
ОТРАВЛЕНИЕ КАТАЛИЗАТОРОВ КОМПОНЕНТАМИ,
СОДЕРЖАЩИМИСЯ В СЫРЬЕ КРЕКИНГА
СЕРНИСТЫЕ И КИСЛОРОДНЫЕ СОЕДИНЕНИЯ
Издавна были известны вредные свойства сернистых
соединений как в процессе выработки нефтепродуктов, так и в процессе
их использования. Сернистые соединения вызывают интенсивную
коррозию аппаратуры, ухудшают условия труда обслуживающего
персонала. В связи с этим сернистые нефтепродукты необходимо
подвергать дополнительной очистке. Особенно остро стоит этот
вопрос при переработке высокосернистых нефтей. Вакуумный
газойль, полученный из таких нефтей и используемый в основном в
качестве сырья каталитического крекинга, содержит до 3,5 вес. %
серы. Поэтому изучение влияния сернистых соединений на
результаты процесса представляет несомненный интерес.
Установлено [166], что на активность синтетических алюмоси-
ликатных катализаторов сернистые соединения влияют
незначительно. В то же время активность и селективность природного
катализатора при переработке сернистого сырья снижалась. Вначале
предполагали, что это вызвано заменой гидроксильных групп
катализатора группами SH [166] или адсорбцией соединений серы
на активных центрах [167]. Однако исследования [64] показали,
что дезактивация природных катализаторов сернистыми
соединениями связана с присутствием в них железа. После разработки
методики удаления железа из глин [64] удалось получить
природный катализатор с такой же устойчивостью к действию сернистых
соединений, как и синтетический.
О влиянии кислородсодержащих соединений на активность алю-
мосиликатного катализатора известно мало [106, 168].
Объясняется это, видимо, небольшим их содержанием в сырье и
незначительным влиянием большей их части на показатели процесса. Так,
по данным работы [168], выход бензиновых фракций при
крекинге чистого сырья и сырья, содержащего до 5% фенолов, остается
постоянным. Присутствие фенолов практически не оказывает
влияния на газообразование и содержание сульфируемых в бензиновой
фракции, но отложение кокса на катализаторе и содержание
непредельных углеводородов в бензине несколько возрастает. Не
замечено также влияние кислородных соединений на регенерируе-
мость катализатора.
Кислородные соединения могут влиять на активность катали-
125
затора очень существенно, если они относятся к классу
гидроперекисей [169]. Эти соединения адсорбируются на активных
центрах катализатора и замедляют крекині. При регенерации
активность катализатора восстанавливается.
Такид: образом, при использовании синтетических
катализаторов предварительно очищать сырье от сернистых и кислородных
соединений (кроме гидроперекисей) не требуется, так как они
практически не оказывают влияния па материальный баланс ка-
72ЛІЛПЧЄСК0Г0 крекинга. D ряде случаев продукты крекинга можно
очищать дополнительно с целью улучшения их товарных свойств.
АЗОТИСТЫЕ СОЕДИНЕНИЯ
Азотистые соединения долгое время считали инертными и
безвредными примесями нефти, поэтому они мало интересовал!»
исследователей. Кроме того, вследствие их большого многообразия
и сложности, трудности выделения и количественного определения
представления о содержании в различных нефтях азота и его
соединений были весьма разноречивы. По более поздним данным
[170], азота в нефтях Советского Союза содержится 0,37—
0,015 вес. %. Особенно богаты азотом нефти Второго Баку: в
башкирских нефтях, например, содержится 0,12—0,32 вес. % азота,
в татарских — 0,18—0,31 вес. %.
В работе [172] приводится много данных о содержании азота
в днетнллятных фракциях 19 советских нефтей; с повышением
температуры кипения фракций содержание азота в них значительно
возрастает. В дальнейшем эти данные были подтверждены рядом
других исследователей [170, 171]. Они считают, что азот
концентрируется (от 85 до 100%) в остатках атмосферной перегонки.
В последние годы изучению состава азотистых соединений в
различных нефтях и дистиллятах, их свойств и их количественного
содержания было посвящено значительное количество работ. Было
установлено, что в нефти и ее дистиллятах содержатся в основном
три большие группы соединений: 1) азотистые соединения
основного характера (азотистые основания); 2) нейтральные; 3)
азотистые соединения кислого характера. Из них важное место
занимают азотистые основания. Отношение содержания их к общему
содержанию всех азотистых соединений сравнительно постоянно
и равно 0,25—0,45 [173, 174]. В работе [174] приводятся данные
о содержании азотистых соединений в 14 нефтях. В ней
указывается, что несмотря на различные количества основных азотистых
соединений (от 0,01 до 0,14) и всех азотистых соединений (от 0,04
до 0,66%), отношение содержания основного азота к общему его
количеству колеблется в пределах 0,25—0,33. Подобное
постоянство отношений (хотя и в несколько более широких пределах)
сохраняется и в узких фракциях, в том числе и в вакуумном
газойле. На основании анализа 34 образцов нефтяных дистиллятов
различного происхождения установлено [175], что дистилляты пря-
126
мой перегонки содержат меньше азотистых соединении, чем
продукты вторичных процессов. При этом продукты термического
крекинга содержат больше основных азотистых соединений, чем
соответствующие продукты каталитического крекинга. Отношение
содержания основного азота к общему азоту для сырых нефтей
равно 0,25—0,34, для дистиллятов прямой гонки — 0,31—0,67, для
продуктов каталитического крекинга — 0,25—0,67, термического
крекинга — 0,5—1,0 [174]. Результаты исследований, посвященных
распределению общего и основного азота во фракциях
вильмингтонской нефти [173], показали, что отношение содержания
основного азота к общему азоту изменяется относительно в небольшом
интервале значений (табл. 35).
Таблица 35
Содержание общего и основного азота
во фракциях вильмингтонской нефти
Фракции
<200
200—225
225—250
250—275
275—350
350—382
382—410
410—443
Остаток
Выход
фракций,
объемы. %
18,5
5,0
5,0
6,0
12,0
5,0
4,5
5,5
38,5
Азот
общий,
вес. %
0,03
0,04
0,04
0,07
0,09
0 22
0,31
0,36
1,12
Азот
основной,
вес. %
0,01
0,01
0,02
0,04
0,09
0,12
0,14
0,35
Основной
азот:общин
азот
0,25
0,25
0,28
0,44
0,40
0,39
0,39
0,31
В последнее время большой интерес был проявлен и к
нейтральным азотистым соединениям. Было доказано [174], что к
нейтральным соединениям относятся индолы, карбазолы и часть
пирролов. Повышенный интерес к этим видам азотистых
соединений объясняется тем, что они могут быть источником образования
основных азотистых соединений не только при протекании
реакций крекинга, но даже при простой перегонке нефти.
Особое место среди азотистых соединений нефти занимают
порфирины. Это высокомолекулярные комплексные соединения
азота с углеводородами, нередко содержащие ванадий или никель.
Наряду с этим около 2/3 ванадия и никеля связано с
высокомолекулярными органическими молекулами непорфиринового
характера, содержащими азот и кислород [175]. К прочим азотистым
соединениям, содержащимся в нефти, следует отнести
аминокислоты и аммонийные соли [174]. К настоящему времени выделено
и изучено около 40 индивидуальных азотистых соединений,
содержащихся в различных нефтях [174]. Опубликованы [177]
следующие данные о содержании (в вес. %) азотистых и кислородных со-
127
единений во фракциях 200—370 и 370—450 С калифорнийской
нефти:
Фракция Фракция
Ароматические соединения 200—370 °С 370—450 °С
C„H2n + zN 0,94 7,07
в том числе
индолы 0,07 0,59
карбазолы 0,28 3,38
бензокарбазолы 0,00 0,48
пнридины 0,35 0,65
хиполины 0,21 1,71
бензохинолины 0,03 0,26
C,H2n+zN2 0,00 0,12
в том числе
азаиндолы — 0,03
азакарбазолы ...... — 0,09
C„H2)l + zNO 0,15 1,14
в том числе
пиридоны 0,06 0,11
.хинолопы 0,09 1,03
C„H2„ + ZNO2 0,003 0,02
С„Н2п + гО 0,92 2,16
в том числе
бензофурапы 0,05 0,09
днбензофураны 0,48 0,69
нафтобензофураны .... 0,01 0,34
дигидробензофураны+фе-
нилкетоны 0,06 0,07
фенолы 0,32 0,97
Алифатические соединения . . 1,09 3,20
в том числе
карбоксильные кислоты . . 0,50 1,74
окиси сернистых алкилов . 0,07 1,00
другие кислородные
соединения 0,52 0,4b
Итого . . . 3,10 13,71
Изучая состав и количественное содержание азотистых
соединений, исследователи установили, что азотистые основания можно
использовать как ингибиторы коррозии, дезинфицирующие
средства, добавки для смазок, антиокислители и для других целей.
Значительно усилился интерес к азотистым соединениям после того
как было обнаружено, что они оказывают заметное влияние на
осмоление и потемнение нефтепродуктов и являются
каталитическими ядами [8, 106, 168].
В связи с существенным снижением активности катализаторов
крекинга после адсорбции или хемосорбции ими азотистых
оснований было проведено большое число исследований, посвященных
изучению механизма отравления, разработке методов
предохранения катализатора от отравления, а также изучению основ
катализа. Первыми наиболее полными работами по изучению
отравления азотистыми основаниями катализаторов крекинга являются
исследования [178, 179 и 168].
128
Таблица 36
Влияние хинолина на результаты каталитического
крекинга*
Сырье крекинга
Кумол
Кумол + 1,8 объемы. %
хинолина
Кумол**
кумола
Вь
бензола
27,6
10,9
1,9
ход, вес. %
кокса
2,3
2,7
2,8
газа
12,5
4,4
1,2
Плотность
газа dio
1,42
1,33
0,9
* Условия процесса: алюмосиликатиый катализатор фирмы Гудри
B00 мл) активностью (по методу Кат-А) 32; температура 427 °С; объемная
скорость подачи сырья 1.5 ч—1; длительность опыта 10 мин.
** Перед крекингом кумола катализатор обработан 2 мл хинолнна н
продут азотом.
Было показано [178, 179], что при каталитическом крекинге
кумола, содержащего азотистые основания, выход бензола
получается пониженным. В табл. 36 показано влияние хинолина на
результаты каталитического
крекинга кумола.
Из приведенных данных
можно заключить, что влияние
азотистых оснований значительно
сильнее при нанесении их на
катализатор до подачи сырья, чем
при введении такого же
количества азотистых соединений в
сырье. Объясняется это
различной доступностью азотистых
соединений к активным центрам
катализатора. Те же
исследователи изучали влияние различных
азотистых соединений на
дезактивацию катализатора крекинга.
По их данным (рис. 59),
зависимость между количеством яда,
адсорбированного катализатором,
и его дезактивацией в реакции
деалкилирования кумола носит
экспоненциальный характер. По
токсичности изученные
азотистые соединения располагаются
в следующем порядке: хиналь-
дин > хинолин > пиррол > пиперидин > дециламин >
анилин. Авторы работ [178—179] считают, что такой порядок
определяется не только основностью указанных соединений, но и сте-
1,0 Кб 2,0 2,5 3,0
л атслиэагпара, моль ¦ 10 *
Рис. 59. Влияние органических
азотистых соединений на
каталитический крекинг кумола:
1 — хииальдин; 2 — хинолин; 3 — пиррол;
4 — пиперидин; 5 — дециламнн; 6 — анилин.
9-2206
129
пенью их устойчивости в условиях крекинга и количеством,
остающимся на поверхности катализатора. В работе [180] на основе
изменений в распределении азотистых соединений в продуктах
крекинга подтверждается различная термическая устойчивость
разных соединений. По данным [180], с изменением температуры
процесса и кратности циркуляции катализатора изменяется и
содержание азотистых соединений на закоксованном катализаторе.
Первые исследования о влиянии азотистых соединений на
результаты каталитического крекинга нефтяных дистиллятов
Таблица 37
Влияние на результаты крекинга добавления к сырью
основных и нейтральных азотистых соединений
Показатели
Свойства сырья
Содержанке, вес. %
азотистых оснований
неосновных соединений
азота
серы
Плотность ^4°
Условия крекинга
Температура в реакторе, °С
Кокс па регенерированном
катализаторе (в среднем), вес.
Выход
Степень превращения, объемн.
Кокс. вес. %
Газ С2 и выше, м3/м3 сырья
Целевые продукты, объемн. %
фракция С2
» С4
дебутанизированный
бензин (90% отгоняется при
199 °С)
бензин иедебутанизирован-
U КГ її
НЫН
газойль
Октановое число дебутанизиро-
ванного бензина
(исследовательский метод) без ТЭС
То же с 0,8 мл/л ТЭС
Прямо-
гонный
мннасский
газойль
0,03
0,04
0,09
0,8692
529
0,10
63,2
5,0
49,3
13,9
16,8
40,6
57,4
36,8
93,5
98,3
С добавкой
иэохинелина.
0,30
0,08
0,09
0,8740
529
0,09
58,5
5,0
77,5
9,9
14,3
38,9
53,2
41,5
90,9
97,7
к
0,58
0,04
0,09
0,8778
529
0,10
51,7
5,0
77,6
7,2
10,0
35,5
45,5
48,3
88,8
96,0
С добавкой индола,
%
0,02
0,37
0,10
0,8735
529
0,12
64,2
5,0
62,6
15,5
18,8
39,6
58,4
35,8
94,8
99,6
0,02
0,65
0,10
0,8778
529
0,14
59,2
5,0
56,8
11,8
14,5
38,8
53,3
40,8
95,1
99,7
130
[168], проведенные в 1946 г., показали, что при добавлении в
сырье крекинга пиридиновых основании и хинолина выходы газа
и бензиновых фракций резко понижаются, а коксообразование в
1,5—2,0 раза больше, чем при крекинге исходного сырья.
Количество сульфируемых и непредельных углеводородов в бензине
значительно возрастает. Относительная дезактивирующая способность
сырых пиридиновых оснований из смол полукоксования углей,
пиридиновых оснований из смол коксования углей и хинолина
находятся в соотношении 1 :2,5 :20,0 при добавлении их 2 вес. % к
сырью крекинга. Хотя дезактивация катализатора азотистыми
основаниями носит обратимый характер, т. е. индекс активности
катализатора после его регенерации восстанавливается, изменения
в выходах продуктов крекинга достигают значительной величины.
Так, при переработке сырья из калифорнийской нефти выход
бензина из-за отравления катализатора азотистыми основаниями
снижается на 20% [181].
Указывается [182], что даже при незначительных добавках к
минасскому газойлю изохинолина выход продуктов
каталитического крекинга заметно уменьшается и октановое число бензина
снижается. Добавка в газойль 0,3% индола практически не влияет ни
на выход бензина и кокса, ни на качество продуктов. При
добавлении 0,65 % индола выход бензина несколько уменьшается
(табл. 37). Предполагается [182], что какая-то часть индола
превращается в азотистые основания, которые отравляют
катализатор.
ІІзучение влияния азотистых оснований на результаты
каталитического крекинга вакуумного газойля из смеси туймазинской и
ромашкинской нефтей [183] показало, что при увеличении
содержания азотистых оснований в сырье крекинга (к нему были
добавлены азотистые основания, выделенные из этого же сырья) выход
бензина и газа существенно снижается (табл. 38).
Постоянство отношения бензин : газ свидетельствует о
сохранении селективности катализатора при значительном снижении его
активности в результате отравления. Выход кокса на сырье
сохраняется на постоянном уровне.
Отравление катализатора азотистыми основаниями
сказывается на качестве продуктов крекинга. В частности, в бензинах
существенно увеличивается содержание непредельных углеводородов.
Отмечается [106] разная токсичность различных азотистых
соединений. В результате каталитического крекинга (при
атмосферном давлении, 500 °С, длительности цикла крекинга 1 ч) цетана и
декалина, содержащих 0,10 или 0,11 вес. % общего азота,
отравляющего влияния аммиака и алифатических аминов обнаружено
не было, в то время как хинолин и акридин резко снижали
глубину превращения (табл. 39). В присутствии хинолина глубина
превращения цетана и декалина снижалась примерно на
одинаковую величину. Результаты исследований показали, что
токсичность азотистых соединений возрастает с увеличением их основно-
9* 131
Таблица 38
Влияние азотистых оснований на выход и качество
продуктов каталитического крекинга
Показатели
Общее содержание азота, вес.
0/
Л
Выход продуктов, вес. % на
сьірьє
бензин до 200 °С
легкий каталитический
газойль 200—350 °С
остаток >350 °С
газ
кокс
Характеристика фракций до
ZUU ^j
плотность dl0
сульфируемые, вес. %
йодное число, г12/100 г
Отношение выходов
бензин:газ
Отношение выход
бензина:глубина разложения
Сумма светлых
нефтепродуктов, вес. %
Вакуумный газойль
трижды
обработанный
25%
H2SO4
0,07
22,6
32,6
27,7
11,9
3,5
0,7569
46,4
81,5
1,90
0,59
55,2
исходный
0,10
21,3
30,6
28,0
12,7
3,3
0,7555
47,9
81,6
1,70
0,57
51,9
с 0,5%
азотистых
оснований
0,117
19,3
—
38,0
10,0
3,3
—
—¦
1,93
0,59
—
с 2%
азотистых
оснований
0,167
15,9
28,4
43,0
8,1
3,4
0,7608
47,9
101,8
1,96
0,58
44,3
с 7,8%
азотистых
оснований
0,363
11,9
25,8
52,1
6,4
3,4
0,7680
51,9
106,8
1,86
0,55
37,7
сти и молекулярного веса. Кроме того, интенсивность отравления
катализатора зависит и от условий процесса. С увеличением
температуры крекинга отравляемость снижается.
Показана [184] линейная зависимость изменения активности
в реакциях превращения циклогексана алюмосиликатных
катализаторов от содержания на них азотистого основания. При
нанесении 6—7 моль пиридина на 100 г катализатора он полностью
дезактивируется. Токсичность пиридина, хинолина и изохинолина в
указанных условиях практически одинакова.
Определено [185] изменение константы скорости крекинга от
добавления в сырье азотистых оснований. Как видно из табл. 40,
при добавке 0,1 вес. % хинолина скорость дезактивации
катализатора несколько уменьшается. Константы скорости крекинга
газойля (k0) и образования бензина (fei) уменьшаются при этом
почти вдвое. Уменьшается и константа скорости крекинга бензина
(кг). На селективность крекинга хинолин почти не влияет.
В связи с кислотной природой активности цеолитсодержащие
катализаторы крекинга под действием азотистых соединений
изменяют свои каталитические свойства так же, как и аморфные
алюмосиликаты [79]. Однако благодаря значительной активности да-
132
Таблица 39
Отравление кремниицирконииалюминиевого катализатора крекинга
азотистыми соединениями
Добавленное азотистое соединение!
Содержание
общего
азота.
вес. %
Глубина
превращения ,
/о
Уменьшение
глубины
превращения
%
Декалин, катализатор № 1 (скорость подачи сырья
30,2 моль/кг катализатора)
Без добавки
Аммиак
Метиламин
Диамиламин
Дициклогексил амин
Пиридин
И и дол
а-Нафтиламин
Хинолин
Акридин
0,00
0,П
0,11
0,11
0,11
0,11
0,11
0,11
0,11
0,11
41,9
42,0
42,0
42,3
28,0
26,8
25,1
21,8
8,5
8,2
Декалин, катализатор № 2 (скорость подачи сырья
23,8 моль/кг катализатора)
Без добавки
Пиррол
Карбазол
Хинолин
0,10
0,10
0,10
63,9
43,2
28,1
18,1
Цетан, катализатор № 2 (скорость подачи сырья
30,2 моль/кг катализатора)
Без добавки
Хинолин
0,11
41,8
10,9
0
0
0
33
36
40
48
80
81
32
56
72
74
Таблица 40
Влияние азотистого основания на константы скорости крекинга
Показатели
Температура, °С
Скорость дезактивации
катализатора
Константа скорости
крекинга газойля /со
образования бензина /Сі
крекинга бензина /Сг
Селективность крекинга
kJk0
к2ік0
Газойль
исходный
482
32,8
28,4
24,1
2,07
0,85
0,07
538
33,9
42,7
33,9
3,59
0,79
0,08
с 0,1 вес. %
хинолина
482
29,3
13,8
10,3
0,22
0,75
0,02
538
30,5
22,5
17,2
2,21
0,78
0,10
с 0,2
вес. %
хинолина
538
27,3
18,3
12,7
1,97
0,78
0,12
же при работе на тяжелом газойле с высоким содержанием азота
глубина превращения и выход бензина в присутствии цеолитсодер-
жащих катализаторов больше. Так, на одной установке флюид в
США при содержании азота в сырье около 0,3 вес. % и переходе
его на цеолитсодержащий катализатор выход бензина увеличился
на 4,5 объемн. %, а газообразование снизилось. В другом случае
использование цеолитсодержащего катализатора при крекинге
сырья с высоким содержанием азота позволило увеличить глубину
превращения на 5%; выход бензина возрос на 4%, выход бутан-
бутиловой фракции — на 1%, а пропан-пропиленовой фракции
остался без изменения [186].
При работе на сырье, содержащем около 7-10-'% азота,
выход кокса и газа на цеолитном катализаторе был на 20% меньше,
чем на обычном аморфном, при одинаковой глубине превращения.
Но при содержании азота в сырье 3- 10~3% выход оказался
меньше только на 10%. Тем не менее работать на цеолитном
катализаторе выгодно из-за большего количества образующегося бензина
[187]. Стойкость цеолитного катализатора к действию азота
подтверждена опытами по крекингу сырья с добавкой изохинолина.
Приведенные выше результаты исследований показывают,
насколько важно контролировать содержание азотистых соединений
в сырье каталитического крекинга и принимать меры для
уменьшения их содержания.
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Содержание металлов в нефтях и нефтепродуктах. Тщательный
анализ нефтей и их дистиллятов показал наличие в них сложных
комплексов металлов с высокомолекулярными углеводородными
соединениями. Вначале изучением металлов, содержащихся в
нефтях, занимались в основном геохимики с целью обоснования
различных теорий происхождения нефти. Позднее было установлено,
что наличие металлов в нефтепродуктах приводит к резкому
увеличению коррозии оборудования и особенно лопастей
газотурбинных установок, а также является причиной резкого ухудшения
работоспособности катализатора крекинга. Это побудило
исследователей тщательно заняться изучением строения и свойств ме-
таллоорганических соединений, распределения их по различным
фракциям нефти, разработкой методик определения содержания
металлов и методов их удаления из различных нефтяных
дистиллятов и нефтей.
К настоящему времени накоплено много данных о содержании
металлов в различных нефтях. Подробные сведения о составе
золы ряда отечественных нефтей опубликованы в работе [188].
Содержание металлов в разных нефтях не одинаково. Так, в змиев-
ской нефти содержится в 40 раз больше никеля и в 1000 раз
больше ванадия, чем в полазненской нефти; содержание ванадия в
босканской нефти в 2000 раз больше, чем в полазненской.
134
0.2
0,3 1,0 /,'i 1,8
Асфальтены,вес. %
В бакинских нефтях ванадия в виде пятиокиси содержится
4-Ю-5—5-10-5%, никеля в виде окиси—следы. Несколько больше
металлов найдено в нефтях Эмбенского месторождения (ванадия
4-Ю-5—9-Ю-4; никеля 14-10~5—46-10~4%)- В нефтях восточных
месторождений металлов значительно больше: ванадия 0,010—
0,023%; никеля 0,0047—0,0064%. Основное количество металлов
связано с асфальто-смолистыми веществами, и с утяжелением
нефти их содержание резко увеличивается [189—191]. Так, в
высокосмолистых нефтях Второго Баку ванадия содержится в
200—500 раз больше, чем в малосмолистых бакинских нефтях.
После удаления из мазута 20,6% асфальто-смолистой части
содержание ванадия уменьшается на
97,2% и никеля на 36,2%.
Установлено [189], что
содержание ванадия в вакуумном
газойле прямой перегонки
зависит от природы нефти и
находится в пределах 6-10~5—
1 -10-4%, а никеля —3-Ю-5—
6 -10 -5%. В мазуте и
полумазуте содержание металлов
резко увеличивается, достигая
соответственно Ы0~4—12Х
ХЮ-3 и 3-Ю-3—42 • 10-4 % •
В дистилляте деструктивной
перегонки мазута ванадия в
4 раза, а никеля в 2 раза
больше, чем в дистиллятном
сырье прямой перегонки. Интересные попытки определить
зависимость содержания металлов от количества асфальтенов были
предприняты в работе [190]. На рис. 60 приведена одна из таких
зависимостей.
По содержанию в высокосернистых нефтях металлы^
располагаются в ряд: ванадий > никель > железо > натрий >
кальций > медь > магний > марганец [191]. В сернистой шкапов-
ской нефти ванадия содержится в 4 раза меньше, чем в
высокосернистой нефти. По концентрации в шкаповской нефти можно
выделить две группы элементов: 1) 0,002—0,004% (ванадий, натрий,
никель, кальций); 2) <0,0005% (железо, магний, медь,
марганец). В обеих нефтях содержание меди, марганца и магния
незначительно (табл. 41).
При обессоливании нефтей ванадий- и никельсодержащие
соединения арланской нефти извлекают соответственно на 16,3 и
28,5%. Более 50% остальных металлов переходит в сточные воды.
После электрообессоливания содержание железа увеличивается
более чем в 2,5 раза, что связано с коррозией оборудования.
В сточных водах, отобранных с ЭЛОУ, обнаружено (в %):
ванадия 4-Ю-6; кальция 23-Ю-4; магния 14- Ю; меди 0,3- Ю-4, нат-
135
Рис. 60. Зависимость между
содержанием серы (/), ванадия B),
никеля C) и содержанием асфальтенов.
Таблица 41
Распределение серы (в вес. %) и металлов (Ю'^-вес. %) во фракциях нефтей
Фракции,
°С
Исходная нефть
250—300
300—350
350—400
400—450
>450
Исходная нефть
200—250
250—300
300—350
350—400
>400
Исходная нефть
до ЭЛОУ
после ЭЛОУ
200—250
250—300
300—350
350—400
>400
200—350
250—360***
>350
>350***
Выход,
вес. %
100
10,0
8,8
8,1
7,6
31,9
100
7,2
3,3
7,8
8,1
50,8
100
6,2
8,3
9,6
9,7
48,9
24,1
22,9
58,6
55,0
Плотность,
0,8624
0,8124
0,8722
0,9005
0,9180
0,9985
0,8972
0,8143
0,8486
0,8766
0,9031
1,0145
0,8930
0,8913
0,8150
0,8550
0,8810
0,9055
0,9980
—
—
s
1,60
0,43
1,63
1,87
1,98
2,97
3,55
1,39
2,98
2,31
3,78
—
3,13
2,92
0,98
2,40
2,85
3,50
4,30
2,3
4,2
4,2
* Нефти получены с промыслов и обезвожены до
** Проба арланской нефти взята на одном из НПЗ
дисольван 4411.
•*• Промышленные образцы.
V
N1
Шкаповская н
44,0
0,007
0,008
0,009
0,040
120
С а у з б а ш
150,0
0,03
0,013
0,013
0,030
310
А р л а н с
180,0
150,0
0,006
0,010
0,020
0,120
300
0,012
0,008
250
280
содержания С
30,0
Следы
»
»
91
ев ск а я
50,0
Следы
»
»
0,02
90
ка я не
70,0
50,0
Следы
»
»
0,060
100
Следы
»
88
100
.1% воды.
Са
е ф т ь*
26,0
0,3
0,3
0,6
12
80
нефть*
1,0
0,4
0,4
0,4
0,4
1,7
фть**
4,0
2,0
0,5
0,5
0,3
0,5
4
0,35
0,30
3,4
4
Башкирии. Содержание хлоридов (в
Fe
3,0
0,4
0,89
4
3
9
17,0
0,4
0,6
0,9
0,8
34
13,0
32,0
0,4
0,3
0,6
1,0
65
4,4
5,0
57
70
иг/л): до
Mg
2,2
0,35
0,6
1,6
7
0,7
0,2
0,5
1,0
1,0
1,0
1,3
0,6
0,16
0,15
0,30
0,30
1,4
0,22
1,96
1,0
ЭЛОУ 3120,
Na
40,0
0,35
0,35
0,54
1,0
120
17,0
Следы
0,7
0,7
0,7
30
12,3
3,0
0,3
0,4
0,4
0,4
6,0
0,34
0,44
5,08
30,0
после ЭЛОУ
Мп
0,13
0,007
0,02
0,10
0,10
0,40
0,1
0,01
0,02
0,03
0,03
1,50
1,20
0,15
0,0032
0,006
0,090
0,80
0,30
0,018
0,014
0,38
0,30
8. Деэмульг
Си
1,3
0,01
0,02
0,10
0,40
4,0
0,7
0,04
0,04
0,035
0,040
0,19
1,7
1,0
0,043
0,060
0,090
0,120
2,0
0,068
0,05
0,6
1,0
а тор—
рия 0,1, никеля и марганца — следы. Основное количество
металлов (от 80 до 99%) при перегонке остается в тяжелых
остаточных фракциях. В дистиллятах обнаруживаются следы всех
металлов, находящихся в нефти. Во фракциях саузбашевской и арлан-
ской нефтей, выкипающих до 350 СС, содержатся следы никеля и
только во фракции 350—400 °С появляется его 2 • 10~6—6-Ю-6 вес. %.
В сернистой шкаповской нефти заметное количество никеля
обнаруживается лишь во фракции 400—450 °С.
О характере и строении соединений, в которые входят
металлы, определенных сведений не имеется. Однако большинство
исследователей считают, что металлы входят в состав порфириновых
комплексов и комплексов непорфиринового характера —
высокомолекулярных полициклических соединений, содержащих азот и
кислород [192, 193]. Сообщается [192], что в наиболее богатых
ванадием нефтяных 3—10% его содержится в виде ванадиево-пор-
фириновых комплексов. Ряд данных показывает, что ванадиево-
порфириновые комплексы нефти содержат ванадий в виде ванади-
ла VO2+. Однако не весь ванадий в виде ванадила имеет форму
порфиринового комплекса. Большая часть ванадия находится в
виде азотсодержащих комплексов. В газойлях 30—50% ванадия
содержится в виде ванадила и порфирина.
В результате исследований соединения ванадия были
разделены на 4 группы: а) летучие соединения ванадия в виде ванадила
(металло-порфириновый комплекс); б) летучие соединения, не
являющиеся ванадилом; в) нелетучие соединения ванадила; г)
нелетучие соединения не в виде ванадила.
Указывается [193], что в нефтях присутствуют по меньшей
мере три класса комплексных металлических соединений: 1) пор-
фирины металлов; 2) соединения непорфиринового характера,
легко разлагающиеся смесями ледяной уксусной и бромистоводо-
родной кислот; 3) соединения непорфиринового характера, с
трудом разлагающиеся смесями ледяной уксусной и бромистоводород-
ной кислот.
Наибольшее количество работ посвящено изучению соединений
порфирина. Молекулярный вес порфирина около 500; он хорошо
растворим в большей части углеводородов и в значительной
степени перегоняется вместе с ними. Ванадиевые комплексы менее
стабильны, чем никелевые; поэтому по мере старения нефтей
концентрация ванадиевых порфиринов снижается быстрее, чем
никелевых. Отношение ванадий: никель сравнительно
постоянно [188].
В результате сложности и многообразия изучение состава ме-
таллоорганических соединений различных нефтей приводит к
самым разнообразным представлениям. Так, в работе [194]
сообщается, что содержание порфиринов находится в тесной
зависимости от содержания в нефти серы. При этом в сернистых нефтях
порфирины на 90% представлены ванадиевыми комплексами, а в
малосернистых — никелевыми. ВанадиеЕые порфириновые ком-
137
Таблица 42
Качество алюмпсиликатного катализатора, содержащего металлы
Наносимый
металл
Без металла
То же*
Свинец
Хром
Молибден
Ванадий
Медь
Кобальт
Никель
Железо
Содержание
металла на
катализаторе,
вес. %
Отсутствует
»
0,0001
0,005
0,030
0,150
0,080
0,150
0,690
0,0001
0,002
0,010
0,040
0,060
0,600
0,650
0,0003
0,003
0,002
0,020
0,160
0,210
0,170
0,960
0,020
0,370
0,660
0,0001
0,003
0,005
0,020
0,140
0,160
0,190
0,520
0,650
3,160
0,150
0,250
0,520
1,000
Индекс
активности
36,0
36,5
39,1
39,1
36,4
32,3
34,9
32,7
24,5
38,2
38,5
34,3
35,4
33,0
18,5
16,6
38,6
38,6
38,2
35,1
25,0
21,8
21,8
12,3
25,2
14,0
11,6
30,5
28 2
25,9
21,5
16,8
16,0
16,5
10,3
9,6
10,3
30 6
26,8
21,0
18,6
Удельная
поверхность,
м2/г
371
368
361
370
369
359
366
353
340
369
362
337
334
388
347
378
349
353
358
336
336
360
356
364
370
355
351
375
343
338
363
365
385
360
358
354
340
370
362
368
371
Обменная
способность,
мг-зкя/г
общая
0,20
0,18
0,20
—
0,19
—
—
—
0,19
0,22
0,20
0,18
0,19
—
0,24
0,19
0,18
0,19
—
0 24
0,17
0,18
0,17
—
0,17
0,17
0,19
0,20
0,18
0,18
0,16
0,18
0,16
—
0,18
0,19
0,17
0,18
протонная
0,060
0,080
—
—
0,080
—
—
—
0,050
0,043
0,043
0,045
0,052
—
0,076
0,055
0,043
0,056
—
0,059
0,066
0,049
0,038
—
—
0,0-15
—
0,054
0,050
0,042
0,058
—
0,05
0,045
к ¦?
s 5
Eg
X с *
699
710
712
718
701
730
660
680
687
722
730
726
718
723
728
731
703
707
710
720
730
705
734
731
728
^22
731
706
735
716
714
718
736
710
731
719
725
710
703
701
712
s І
ju О
Н Q
Я x
U ш
о ?
? к ь
о § ізї°
100,0
100,0
109^0
109,0
101,0
90,0
97,0
91,0
68,0
106,0
107* 0
95 і 5
96,0
90,0
50,0
46,0
107'0
107',0
106,0
97,5
69^5
60 [о
60 5
ЗЗІ4
68,5
38,0
32,2
85,0
78,5
72,0
60,0
46,6
44,5
46,7
28,8
26,8
28,8
85,0
74,5
58,5
54,5
Исходный катализатор пропитан дистиллированной водой и прокален.
плексы относятся в основном к асфальто-смолистой части нефти,
а никелевые — к масляным фракциям.
В процессе изучения иракской нефти исследователи [195]
пришли к заключению, что очень незначительная часть ванадия
находится в форме ванадиево-парфириновых комплексов; практически
весь ванадий находится в асфальтенах. Около двух третей от
всего количества ванадия и никеля относят к асфальтенам и в работе
[176] на основании изучения металлоорганических соединений
нефтей Второго Баку.
Абсолютное большинство исследователей склоняется к тому,
что порфирины, содержащие металлы, представляют собой
относительно стойкие соединения, которые во время перегонки
отгоняются вместе с дистиллятом, не разрушаясь. Так, по данным [196],
около 10—15% никеля и ванадия в нефтях представлено летучими
соединениями. По данным [197], летучих соединений никеля в
исследованных нефтях было 17—65% от общего их содержания, я
ванадия 5—33%. В работе приведены данные по давлениям
насыщенных паров комплексов ванадия и никеля. Показано, что
давление насыщенных паров углеводородов в точке кипения и
насыщенных паров металлических комплексов укладывается в
сравнительно узкие пределы. О способности перегоняться группы
комплексных соединений, сопутствующих в основном асфальто-
смолистым веществам, нет данных. Однако во многих работах
указывается, что эти соединения заносятся в дистиллят в виде
капелек жидкости из-за нечеткости фракционирования.
Изменение физико-химических свойств катализатора под
действием металлов. Наши экспериментальные данные о влиянии
металлов на качество аморфного алюмосиликатного катализатора
представлены в табл. 42 и 43. Там же приводится качество
исходного образца катализатора.
Как видно из этих данных, металлы, нанесенные на алюмоси-
ликатный катализатор, не изменяют его физико-химических
свойств. Удельные поверхности, насыпные плотности, поровая
структура всех образцов катализаторов, независимо от природы
металла и его концентрации, практически остались такими же.
Обменная способность катализатора в зависимости от природы
добавляемого металла изменяется по-разному. Щелочные и
щелочноземельные металлы способствуют снижению кислотности
катализатора. Это, видимо, является следствием замещения указанными
металлами протона кислотного центра катализатора.
В отличие от щелочных металлов, никель, ванадий, железо,
хром и другие тяжелые металлы не изменяют кислотности
катализатора. Не происходит существенных изменений и в пористой
структуре. Исследователи [45, 54, 132] пришли к выводу, что при
отложении тяжелых металлов физические свойства
алюмосиликата не меняются, а образуется поверхностный слой, обладающий
совершенно иными каталитическими свойствами. В результате
металлы оказывают существенное влияние на активность катализа-
139
Таблица 43
Характеристика поровой структуры катализаторов,
содержащих металлы
Нанесенный
металл
Без металла
То же*
Никель
Кобальт
Медь
Молибден
Ванадий
Хром
Железо
Содержание
металла,
вес. %
Отсутствует
То же
0,02
0,25
0,65
0,02
0,37
0,66
0,17
0,96
0,060
0,650
0,002
0,160
0,080
0,690
0,250
0,500
1,000
Плотность, кг/мЗ
истинная
2220
2400
3320
2430
2450
2120
2320
2270
2220
2420
2310
2380
2430
2430
2210
2200
2370
2410
2180
кажущаяся
1000
1070
1030
1250
1650
1000
1030
1020
1010
1070
1000
1020
1050
1040
1050
1070
1030
1030
1010
Удельный
объем пор
смЗ/г
0,55
0,52
0,54
0,54
0,54
0,53
0,54
0,54
0,54
0,52
0,57
0,56
0,54
0,55
0,50
0,48
0,55
0,56
0,53
Средний
радиус пор,
о
А
29,60
28,20
29,85
31,40
30,50
28,70
30,40
30,70
30,40
28,60
29,40
29,60
30,10
32,70
27,30
28,20
30,40
30,40
28,60
Пористость,
%
55,00
55,40
55,60
56,70
57,00
53,00
55,50
54,90
54,40
57,70
56,70
57,00
56,85
57,30
52,50
51,50
56,50
57,40
54,50
Поверхность,
м2/Г
371
368
363
344
354
370
355
351
356
364
388
378
358
336
366
340
362
368
371
• Исходный катализатор пропитан дистиллированной водой и прокален.
тора (рис. 61). Для наглядности на рисунке активность образцов,
содержащих металлы, выражена в процентах по отношению к
исходной активности, принятой за 100. Данные, полученные в работе
[198] о зависимости активности от содержания больших количеств
нанесенного металла, не отличаются от результатов других
исследователей [45, 54, 64, 199, 202]. С увеличением количества
металла активность катализаторов уменьшается особенно резко при его
небольших содержаниях (до 0,1—0,2 вес.%). При содержании
металла больше этой величины активность уменьшается менее
значительно. Например, при ничтожно малой порции никеля 0,0001 вес.%
активность катализатора уменьшается на 5,5 пункта, или на
15 отн.%. При увеличении количества никеля до 0,02 вес.%
активность уменьшается на 40%, а при увеличении до 0,5 вес.% —
140
всего на 70 отн.%. При этом абсолютное значение индекса
10,3 пункта против 36,0 у исходного (см. табл. 42).
Поскольку все образцы, содержащие металлы, были
приготовлены и испытаны нами в одних и тех же условиях, можно
сравнить влияние различных металлов на активность катализатора. По
возрастающему отравляющему действию на катализатор
металлы располагаются в следующий ряд: хром, свинец<железо<мо-
либден, ванадий < медь, кобальт < никель.
Эксперименты, проведенные нами на катализаторах,
содержащих менее 0,01—0,02 вес.% металлов, дали интересные
результаты. Металлы, обладающие сильными дегидрирующими
свойствами, например никель, кобальт, медь, вызывают резкое уменьшение
активности катализатора. Так, при содержании на катализаторе
120 і
I 80
!
^^
! 1 1
;
-~. /
' -*• —x—-J
2
5
і і
і
^ О 0,2 0,^ 0,6 О,в 1,0
"'одєрз-і-синие мгтпаллои на катализаторе,8ес.%
Рис. 61. Зависимость активности катализатора от содержания металлов:
/ — свинец и хром; 2 — железо; 3 — молибден; 4 — ванадий; 5 — медь; 6 — кобальт;
7 — никель.
0,02 вес.% кобальта индекс активности катализатора снижается на
10,8 пункта. Некоторые металлы, например свинец, молибден,
ванадий, присутствующие на катализаторе в таких же количествах,
наоборот, вызывают увеличение активности [200, 201].
Наблюдаемое увеличение активности не является следствием мокрой
обработки (см. табл. 42 и 43). В то же время после пропитки
растворами метаванадата аммония, молибдата аммония и уксуснокислого
свинца, в зависимости от содержания нанесенного на
катализаторы металла, активность его вначале повышается, в области
концентраций 1-Ю-4—1-10~3 вес.% проходит через максимум, затем
уменьшается, достигает активности исходного катализатора и
продолжает резко уменьшаться. Максимальное наблюдаемое
увеличение активности катализатора под действием этих металлов,
по сравнению с активностью исходного образца, 2—3 пункта, что
превышает разницу между параллельными опытами.
На катализатор, а, следовательно, и на отложенные на нем
металлы во время работы действуют температура, водяной пар и
попеременно действует окислительная и восстановительная среда.
141
В этих условиях металлические отложения подвергаются
физическим и химическим изменениям: металл спекается, уменьшается
степень его дисперсности и удельной поверхности, он изолируется
в глубине пор катализатора, в результате чего активность
металла как отравляющего агента уменьшается. Чем больше возраст
отложенных металлов, тем меньше они отравляют катализатор.
Было установлено, что с увеличением в сырье содержания
никеля и ванадия глубина превращения сырья уменьшается, а при
меньшем содержании этих компонентов результаты процесса
значительно улучшаются, хотя содержание металлов на катализаторе
по-прежнему остается высоким.
О 1020309192991929816288 Гв28
VJU
IX X XI ХП
месяцы
Рис. 62. Зависимость между образованием водорода и содержанием ванадия в
сырье и на катализаторе:
1,2 — содержание ванадия в катализаторе и в сырье; 3 — образование водорода (при
нормальных условиях).
Промышленные данные [45] (рис. 62) свидетельствуют о четко
выраженной зависимости между образованием водорода и
содержанием ванадия в сырье. Между образованием водорода и
содержанием ванадия на катализаторе такой четкой зависимости не
обнаружено. Это означает, что только свежеотлагающиеся металлы
оказывают сильное влияние на активность и избирательность
катализатора. Металл, уже внедрившийся в катализатор, не влияет
на его активность. Считают, что металл изолируется в глубине пор
в результате спекания катализатора.
Одна из причин ускоренного спекания — резкое увеличение
новообразования под действием металла, что приводит к
повышению температуры регенерации. Кроме того, металлы снижают
температуру спекания поверхностей, на которых они отлагаются
[45]. Различают [203] две фазы дезактивации катализатора под
действием тяжелых металлов: 1) активный период, когда металлы
142
способствуют увеличению коксообразования и уменьшению
активности катализатора; 2) следующий за ним пассивный период.
Пассивность металла может быть вызвана спеканием
катализатора, происходящим в результате огромного выделения тепла в
зоне адсорбированного металла во время регенерации. Влияние
тяжелых металлов на спекание катализатора подтверждено
лабораторными опытами [45], в которых подвергали дезактивации два
образца катализатора, отобранных из одной и той же партии, —
незагрязненного и содержащего металлы. После одновременного
нагрева обоих образцов при 900 °С в течение 3 ч образцы имели
удельные поверхности соответственно 201 и 163 м2/г.
В работе [204] изучалось влияние попеременного действия
окислительной и восстанозительной среды на дезактивацию
металлических примесей. Катализаторы со свежими отложениями
металлов @,2 вес.°/о железа) многократно подвергали
чередующимся циклам крекинга и регенерации, затем испытывали их
способность к закоксовыванию. Было показано, что коксообразующая
способность металла заметно снижается после проведения даже
первых циклов. В результате дальнейших циклов коксообразова-
ние также снижается, но незначительно. Последующие
эксперименты показали, что для пассивации примесей металлов необходимо
именно чередование окислительной и восстановительной сред.
После действия на загрязненный катализатор 4% кислорода при
482 °С в течение 400 мин (что эквивалентно пяти циклам)
практически никакой пассивации не происходило. Заметной
пассивации металла при замене стадии крекинга восстановлением в среде
водорода не наблюдается. Увеличение продолжительности
регенерации вдвое, повышение концентрации кислорода в газе,
подаваемом в регенератор, с 4 до 21% не оказывало существенного
влияния на результаты. Очень мало влияет на пассивацию металла
и температура: повторное проведение опытов при 506 °С вместо
482 °С дало почти совпадающие результаты.
В литературе имеются некоторые данные об исследовании
влияния пропарки катализатора на степень его отравления. На
свежий катализатор наносили 0,2 вес.% нафтената железа-и
пропаривали при 566 °С перегретым паром под давлением. Параллелы
но пропаривали свежий катализатор при тех же условиях, после
чего на него обычным методом наносили 0,2 вес.% железа и затем
испытывали его активность. Было установлено, что при пропарке
существенная часть отложенных металлов поглощается
катализатором; поэтому они оказывают значительно меньшее
дезактивирующее действие, чем в случае нанесения металлов после
пропарки катализатора. Предотвращение отравления катализатора путем
воздействия на него водяного пара изучалось в работе [205].
Полученные результаты (табл. 44) иллюстрируют существенное
улучшение селективности при пропарке катализатора.
О поведении катализатора в процессе длительной эксплуатации
обычно судят по результатам испытания в условиях высокотемпе-
143
Таблица 44
Действие водяного пара* на микросферический катализатор
из трошловской глины с различным содержанием металлов
Показатели
Исходный
катализатор
ДО
обработки
после
обработки
Тот же катализатор после нанесения металла
0,06% V
до
обработки
после
обработки
0,12% V
До
обработки
после
обработки
0,095% N:
до
обработки
после
обработки
Крекинг легкогосырья на индексовой установке
Выход продуктов, вес.
бензин 21 20 14,6 20,2 11,4 13,3 13,9
газ 8,2 6,0 5,6 6,1 4,0
кокс
Относительная плотность
газа
Отношение бензин:кокс
2,7
0,83
7,8
1,0
0,98
20
2,4
0,58
6,1
1,5
1,05
13,5
3,5
0,39
3,3
3,0
1,6
0,6
8,3
6,2
4,8
0,45
2,9
Крекинг нефти на индексовой установке
Выход продуктов, вес. %
бензин
газ
кокс
Относительная плотность
газа
Отношение бензин:кокс
35,4
11,7
10,9
0,89
3,2
34,3
7,6
8,1
0,98
4,3
30,1
10,1
11,2
0,77
2,7
34,9
7,0
8,4
1,11
4,2
_
—
—
—
—
—
30,6
10,5
12,8
0,61
2,4
13,0
3,6
1,4
0,5
9,3
31,3
7,6
8,9
0,68
3,5
• Паром обрабатывали при 750 °С.
ратурной обработки в воздухе или водяном паре. Аморфный
катализатор прокаливали в муфеле при 550—900 °С в течение 4,0 ч
и пропаривали в обычной индексовой установке также в течение
4 ч при 700—900 °С и подаче пара 64 мл/ч. Полученные данные
показывают, что при термической и паровой обработке
катализатора металлы по-разному влияют на его качество.
После термообработки в присутствии металлов зависимость
качества катализатора (удельной поверхности, объема пор,
обменной способности) от температуры изменяется. Увеличение
температуры прокалки с 550 до 700 °С практически не влияет на
перечисленные выше характеристики исходного катализатора. Резкое
изменение наблюдается при повышении температуры с 850 до 900 °С.
При наличии металлов на поверхности катализатора изменение
его качества начинается уже с 800 °С. Абсолютные значения
физико-химических свойств для исходного катализатора и для
образцов, содержащих металлы, при одинаковых условиях прокалки
существенно разнятся. Так, поверхность образца, содержащего 0,66%
кобальта, при температуре прокалки 900 °С составляет всего
96 м2/г, т. е. на 52% меньше, чем удельная поверхность исходного
катализатора, прокаленного при этой же температуре. Объем пор
их уменьшился на 44%.
144
С увеличением концентрации металла на катализаторе его
удельная поверхность и объем пор уменьшаются особенно
существенно при высокой температуре прокалки (около 900°С).
Наиболее резко это наблюдается при добавлении начальной порции
металлов (до 0,1—0,2 вес.%). В дальнейшем увеличение
концентрации металлов вызывает незначительные изменения структуры
катализатора. По возрастающему действию на удельную
поверхность и объем пор металлы при всех изученных температурах
прокалки располагаются в ряд, приведенный выше (см. стр. 141).
В таком же порядке металлы располагаются и по
дезактивирующему влиянию на катализатор крекинга.
Iй
СІ
0,5 1,0 1,5 2,0 2,5
Поверхность катализатора, м^
а
О 0,5 1,0 1,5 2,0 2,5
Поверхность катализатора,м2-!'"
d
Рис. 63. Зависимость выхода бензина от поверхности пробы катализатора при
его термической (а) и паровой (б) обработке.
Катализатор: / — исходный; 2-е 0.25% железа; Л — с 0,5% железа; 4 — с 1,0% железа;
5 — с 0,25% никеля.
При термической и термопаровой обработке одновременно
изменяются насыпная плотность и удельная поверхность
катализатора. Поэтому сопоставление образцов катализатора, прошедших
обработку различной степени жесткости, затрудняется. Так как
активность катализатора оценивается при загрузке одинакового
объема A00 мл) катализатора, различные образцы лучше всего
сравнивать по величине общей поверхности пробы.
На рис. 63 показана зависимость выхода бензина от
поверхности пробы катализатора при его термической и паровой обработке.
При одной и той же поверхности пробы выход бензина на
образцах, содержащих металлы, значительно ниже. По мере
ужесточения термической обработки этот эффект усиливается. Так, при
общей поверхности пробы 2,5-104 м2 (что соответствует
температуре прокалки 550 °С, при которой катализатор не спекается) выход
бензина на образце, содержащем 0,25% никеля, уменьшился по
сравнению с выходом бензина на исходном катализаторе в 2,5
раза, а выход кокса увеличился в 3,3 раза. При прокалке до общей
10—2206
145
поверхности 2,0 -104—0,5-104 м2 выход бензина уменьшается в
3,6 раза и выход кокса увеличивается в 5,0 раз.
Таким образом при термической обработке катализатора с
нанесенным на него металлом скорость спекания катализатора
возрастает и дезактивирующие свойства металла проявляются
сильнее, что приводит к дополнительному уменьшению активности п
селективности катализатора.
При паровой обработке металлы оказывают иное действие на
качество катализатора. Они не изменяют характер влияния
температурь! пропарки на свойства катализатора. В то же время после
паровой обработки (в зависимости от ее условий, типа металла н
его концентрации) отравляющий эффект металла уменьшается или
полностью уничтожается (см. рис. 63), что хорошо согласуется
с данными других исследователей. Так, при крекинге на образце
с содержанием 0,25 вес.% железа, не обработанном иаром, выход
бензина ио сравнению с выходом бензина на исходном
катализаторе, также необработанном иаром, уменьшился в 1,3 раза, а
выход кокса увеличился в 1,5 раза. При паровой обработке обоих
катализаторов до одной и той же величины поверхности картина
совсем другая. Выход бензина при крекинге на образце с
0,25% железа при больших величинах поверхности B,0-104 м2)
несколько (в 1,2 раза) меньше, чем выход бензина на исходном
катализаторе, а ио мере ужесточения паровой обработки эти ве-
Таблица 45
Выход (в вес. % J продуктов крекинга под действием металлов
после термической обработки катализаторов до одинаковой поверхности
Катализатор
Поверхность пробы катализатора, м2.10«1
0.5
а*
6*
1.0
а
б
1,5
а
6
2,0
а
б
2,5
а
б
Исходный
С 0,25% никеля
С 0,25% железа
С 0,5% железа
С 1,0% железа
Исходный
С 0,25% никеля
С 0,25% железа
С 0,5% железа
С 1,0% железа
* а—пропарка; б—прокалка.
14,0
11,0
14,0
12,0
14,0
14,0
3,9
7,0
5,0
3,9
Б
22,2
14,0
22,0
17,0
14,0
е н з і
22,0
7,0
13,0
9,0
7,0
1 Н
29,0
17,0
26,0
20,0
17.0
29,0
9,0
18,0
13,0
10,0
33,6
18,6
28,6
21,0
18,6
34,0
12,0
22,4
16,0
14,0
36,0
Кокс
1,0
2,6
1,1
1 4
1,5
1,0
5 0
1,4
2,0
2,0
1,9
5,5
2,1
1,8
1,0
2,0
8,5
2,8
4,0
4,2
2,6
9,0
3 3
2,7
6,0
2,5
11,2
4,0
5,5
6,4
3,5
11,0
4,8
4,5
8,0
3,5
13,0
5,5
8,0
9,4
—
36,0
14,5
26,8
19,8
17,0
4,6
15,4
6,8
8,5
11,0
146
личины становятся практически равными между собой. То же
самое можно сказать и о выходе кокса. При термообработке
катализатора, содержащего металлы, качество его ухудшается
значительно больше, чем катализаторов без металла. Выход бензина при
одной и той же поверхности катализатора в случае пропаренных
образцов выше, а кокса ниже, чем в случае прокаленных
(табл. 45).
В связи с полученными результатами представляет интерес
изучение стабильности при прокалке катализаторов, промотирован-
вых микродобавками металлов. Испытуемые образцы помещали
в слой катализатора промышленного регенератора в специальном
двухсекционном жесткозакрепленном контейнере, в одну секцию
которого загружали свежий катализатор, а в другую —
катализатор, содержащий 0,002 вес.% ванадия. После девятимесячного
воздействия температуры и среды в промышленном регенераторе
удельная поверхность и обменная способность обоих образцов
практически не изменились. При этом индекс активности катализа*
тора, содержащего ванадий C5,6 пункта), оказался несколько
выше, чем свежего катализатора, взятого из контейнера C5,5
пункта). Выход кокса при крекинге эталонного сырья составлял
для обоих образцов соответственно 2,8 и 3,6 вес.%. Таким образом,
увеличение активности катализатора, наблюдаемое при нанесении
микродоз ванадия, сохраняется после его длительной прокалки и
воздействия реальной среды в условиях работы промышленного
регенератора.
Резюмируя полученные результаты по влиянию металлов, как
свежеотложенных, так и находящихся в условиях старения
катализатора, можно отметить следующее. Металлы оказывают сильное
влияние на активность катализатора, выражающееся не только в
качественном изменении его поверхности, — они способствуют
также увеличению скорости спекания при термопаровой обработке.
Изменение активности и увеличение скорости спекания зависит от
концентрации металла и его природы, а также от вида обработки.
Среди металлов, загрязняющих катализатор, наибольшую
опасность представляет никель. Он уменьшает активность и ускоряет
спекание катализатора. Ванадий до концентрации 0,02%, которая
может накопиться на шариковом катализаторе при переработку
вакуумного газойля, наоборот, увеличивает его активность. Это
влияние ванадия является стабильным и сохраняется в условиях,
характерных для промышленного регенератора.
Изменение активности и селективности алюмосиликатного
катализатора в зависимости от содержания и природы металлов.
Еще в 1947 г. было обнаружено [202], что катионы никеля и
железа, адсорбированные на алюмосиликатном катализаторе,
обладают дегидрирующей способностью. Продукты, которые получаются
при каталитическом крекинге на алюмосиликатных
катализаторах, обработанных солями никеля и железа, содержат большое
количество газа, состоящего преимущественно из водорода. Впервые
10* 147
наиболее подробно влияние тяжелых металлов на активность и
селективность алюмосиликатных катализаторов было изучено в
работе [64].
При исследовании снижения селективности у катализатора в
процессе крекинга было установлено, что одной из причин
старения катализатора является отравление металлами. Результаты
лабораторных опытов показали [64], что железо, никель, ванадий и
медь, содержащиеся в некоторых видах нефтяного сырья,
адсорбируются и накапливаются на катализаторе. Даже ничтожные
количества @,007%) этих металлов ухудшают селективность
катализаторов и снижают выход бензина. Селективность катализатора в
работе [64] оценивается коксовым и газовым фактором —
отношением выхода кокса или газа на исследуемом катализаторе к
выходу кокса или газа на исходном (стандартном) катализаторе при
одной и той же степени превращения. Ухудшение селективности
при содержании на катализаторе перечисленных выше металлов
выражается в резком повышении коксового и газового фактора.
Отравляется природный и синтетический катализатор.
Отравление катализатора крекинга тяжелыми металлами происходит и
в промышленных условиях [206]. Снижение селективности
катализатора от отравления вызывает резкое ухудшение
экономических показателей процесса. Это было показано на следующем
эксперименте. На промышленной установке каталитического крекинга
с псевдоожиженным слоем пылевидного катализатора в течение
двух с лишним лет перерабатывалось сырье трех типов с высоким
содержанием металлов; полученные результаты сравнивали с
результатами работы установки на чистом вакуумном газойле.
Содержание металлов в сырье (в вес.%) приводится ниже:
Никель Ванадий
Чистый газойль <0,05 0,06
Сырье
А 0,35 1,]0
Б 0,40 0,50
В 0,20 0,45
Анализ работы установки показал, что приблизительно
половина общего количества ванадия и никеля из сырья отложилась на
катализаторе. По мере накопления металлов на катализаторе его
селективность ухудшалась. По повышении коксового фактора с 1,0
до 3,0 относительный выход бензина в расчете на разложившееся
сырье снизился с 93 до 82%. По мнению авторов [206], при таком
снижении выхода бензина на установке, производящей 157 м3/сут
бензина, потери составят несколько тысяч долларов в сутки. Для
поддержания коксового фактора на необходимом уровне пришлось
увеличить расход катализатора. Сравнительная экономическая
оценка потерь в выходе бензина при высоком коксовом факторе
и потерь катализатора, необходимых для поддержания низкого>
коксового фактора, вскрыла необходимость уменьшения количества
148
металлов в.сырье, поступающем на каталитический крекинг, с
помощью каких-либо других процессов.
В дальнейшем в связи с широким применением в качестве
сырья крекинга тяжелых газойлей нефтепереработчики вплотную-
столкнулись с проблемой отравления катализатора и
необходимостью удаления металлов из сырья или с катализатора. При
попытках определить допустимую норму металлов в сырье крекинга и на
катализаторе оказалось, что эти нормы зависят от типа установки.
Так, было найдено, что в псевдоожиженном слое пылевидного
катализатора происходит более существенное отравление, чем в
движущемся слое шарикового катализатора. Металлы обычно
концентрируются на внешней поверхности шарика [101, 102, 207]. При
изучении распределения никеля и ванадия, отложившихся из
сырья по сечению шариков катализатора, оказалось, что около
44% всего количества никеля и 48% всего содержащегося
ванадия располагается в слое внешней поверхности гранул
катализатора глубиной 35 мк, что составляет 5% от массы гранулы. При
работе установки с циркулирующим слоем шарикового
катализатора поверхность шариков истирается, и таким образом основная
масса металлов, содержащаяся на катализаторе, выводится из
системы вместе с катализаторной пылью. Это подтверждается
следующими данными, которые были получены при истирании в
лабораторных условиях катализатора, отравленного 0,0105% никеля:
Суммарное количество пыли,
образовавшейся при истиранин Содержание никеля
зерен, % от исходной массы в полученной пыли, вес. %
0,5 0,0403
1,5 0,0408
4,5 0,0318
100 0,0105
В промышленных условиях вывод металлов из систем с
шариковым и пылевидным катализатором в результате его истирания
неодинаков. При истиранин поверхности шарикового
катализатора значительная часть накопившихся металлов выводится из
системы, и таким образом содержание металлов в катализаторе
существенно снижается. В пылевидном катализаторе металлы
практически равноверно отлагаются по всему объему частиц; поэтому
при истирании их поверхности избирательного удаления металлов
не наблюдается и содержание их выше, чем в шариковом
катализаторе.
Таким образом, допустимые нормы по содержанию металлов в
сырье для установок с движущимся шариковым катализатором
будут выше, чем для установок с псевдоожиженным слоем.
По разработанной методике нами было рассчитано количество
металлов, которое может отложиться на катализаторе при
переработке вакуумного газойля арланской нефти, содержащего
0,5-10-*% никеля; 1,0 -10-4 % ванадия и 2,4-10~4 % железа. В
случае установок с кипящим слоем пылевидного катализатора расче-
149
ты были проведены для производительности 1650 т/сут при расходе
катализатора 2 кг/т сырья и количестве катализатора в системе
500 т. Расчеты показали, что максимальное количество никеля,
которое может накопиться на пылевидном катализаторе, составит
0,025, ванадия 0,05 и железа 0,125 вес. %• Из рис. 64 видно, что
равновесие устанавливается приблизительно в течение 200—
250 сут.
200 250 О
Длительность, сут
150 200 250
Рис. 64. Концентрация металлов на циркулирующем шариковом (/, 3, 5) и,
пылевидном B, 4, 6) катализаторе в зависимости от длительности работы:
1, 2 — ванадий; 3, 4 — железо; 5, 6 — никель.
Применительно к условиям установки с шариковым
катализатором типа 43-102 расчеты были выполнены для
производительности 1200 т/сут., расхода катализатора 4,0 т/сут., количество
катализатора в системе 200 т. Максимальное количество никеля,
которое может отложиться на шариковом катализаторе при
переработке арланского вакуумного газойля, составит 0,01%, ванадия 0,02%
и железа 0,05% (см. рис. 64).
Данные о содержании металлов в сырье и в катализаторе,
полученные на промышленных установках с движущимся шариковым
катализатором, приведены ниже [207];
Содержание металла, вес. %
никеля ванадия
Установка А
Легкий газойль 0,000014 0,000005
Тяжелый газойль 0,000015 0,000025
Равновесный катализатор 0,0077 0,0020
150
Установка Б
Легкий газойль 0,000013 0,000029
Тяжелый газойль 0,000045 0,000106
Равновесный катализатор 0,0029 0,0056
Установка В
Легкий газойль 0,000007 0,000015
Тяжелый газойль 0,000138 0,00027
Равновесный катализатор 0,0051 0,0031
Определить точно количество металлов, которое вызывает
отравление, вряд ли возможно. На одной установке такой
концентрацией была 2,0-10~2—2,5 ¦ 10-2% [186], на другой — крекинг
проводили в присутствии высокоглиноземйстого катализатора беа
его отравления при содержании в катализаторе никеля до
2,5-10~2% - При содержании в системе 60% цеолитсодержащего
катализатора и концентрации в нем никеля до 4,5-10~2%
признаков отравления замечено не было.
Во время экспериментов на пилотных установках при глубине
превращения сырья 50% в результате изменения содержания
никеля на катализаторе с 2,0-10~3 до 4,25-10~2% выход кокса
возрастал с 5,7 до 9,2, водорода — с 12,3 до 68,5 м3/м3; сухого газа —
с 50 до 103 м3/м; выход бензина снижался с 32,3 до 29,2%.
Отмечается [208], что при содержании на
высокоглиноземистом катализаторе 0,0015—0,0020% меди влияние металла не
ощущалось, но при увеличении содержания меди до 0,04—0,05%
катализатор сильно дезактивировался, что сопровождалось резким
возрастанием в газе отношения водорода к метану.
Указывается [209], что в установке с псевдоожиженным слоем,
отложение на катализаторе никеля 0,010 и ванадия 0,003 вес. %
является нормальным. Ниже приведены данные о влиянии
избыточного отложения никеля и ванадия на результаты крекинга:
Нормяльное Избыточное
отложение отложение
металлов металлов
Содержание металлов на катализаторе,
вес. %
никель 0,010 0,017
ванадий 0,003 0,008
железо — 0,26
Степень превращения, % 59,8 53,8
Выход продуктов *
водород} м3/м3 сырья 4,3 27
сухой газ, вес. % 6,7 6,0
сумма С4, объемн. % 11,4 11,0
Q—221 ° объемн. % 48,9 42,9
газойль, объемн. % 40,2 46,2
кокс, вес. % 5,0 5,0
* При температуре в реакторе 493 °С.
15Г
Как это видно, с увеличением содержания никеля и на
катализаторе на 0,007% для поддержания постоянного выхода кокса при
неизменной производительности установки степень превращения
пришлось снизить на 6%. При этом образование водорода
возросло в шесть раз.
Цеолитсодержащие катализаторы более стойки к отравлению
металлами. В одном случае при работе на аморфном катализаторе
содержание ванадия было 800—1000 млн-1, а никеля 300—
400 млн.. При этом коксовый и газовый фактор был равен
0,8—1,0. С переходом на новый катализатор он снизился до 0,4—
0,8 [210]. Полагают, что чем выше активность катализатора, тем
больше допустимо на нем отложение металлов (учитывая
наличие большого числа активных центров). На одной установке,
работавшей на цеолитном катализаторе, при уменьшении количества
догружаемого катализатора активность его заметно не
понизилась, что свидетельствует о более высокой стойкости к отравлению
металлами этого катализатора, чем обычного.
Высокая температура регенерации приводит к более быстрой
дезактивации катализатора металлами.
При использовании катализаторов XZ-25 и XZ-30 образуется
меньше водорода, чем на обычном катализаторе. Это обусловлено,
по-видимому, меньшим влиянием на них металлов. Имеется опыт
регенерации цеолитного катализатора при 638—660 °С без
заметного отравления. Влияния хлоридов на действие металлов не
наблюдалось [187]. В более поздней работе [211] отмечается, что
наличие ионов хлора в сырье каталитического крекинга
способствует протеканию термического крекинга.
При подборе веществ, снижающих истирание катализатора и
эрозию оборудования, пробовали и свинец. Содержание свинца до
1-10'30/о заметно не влияло на качество катализатора. Им,еются
данные, что даже 3 ¦ 10~3 % свинца на катализаторе не влияет на
выход продуктов [187]. Это совпадает с нашими результатами.
Опубликованы [212] результаты исследований, посвященных
изменению активности шарикового и пылевидного катализатора в
условиях промышленных установок. Кривая, представляющая
изменение индекса активности равновесного шарикового аморфного
катализатора от величины его удельной поверхности, описывает
также изменение активности свежего шарикового аморфного
катализатора, у которого величина удельной поверхности менялась
в результате пропарки.
Таким образом, при сложившихся условиях эксплуатации
шариковые катализаторы теряют свою активность практически
только в связи со старением, а уменьшения активности вследствие от^
равления металлами не наблюдается. Анализы равновесных
катализаторов ряда установок, проводившиеся систематически в
течение нескольких лет, показали, что на шариковом катализаторе
содержится (в вес, %): никеля до 0,005; ванадия до 0,003;
железа 0,06—0,09; кальция — 0,25—0,34; магния 0,03—0,09. Значитель-
152
ное количество металлов уносится пылью и мелкой крошкой,
образующимися при обкалывании поверхностных слоев шариков..
Самая мелкая фракция теряющегося катализатора @,04—0,06 мм)
содержит в 5—10 раз больше металлов, чем циркулирующий
катализатор.
Кривая зависимости активности пылевидного равновесного
катализатора от удельной поверхности располагается значительно
ниже аналогичной кривой для шарикового катализатора. Это
указывает на то, что на установке с кипящем слоем катализатор
отравляется отложениями металлов, которые уменьшают его
активность, не изменяя величины удельной поверхности.
Циркулирующий пылевидный катализатор содержит металлов намного больше*
чем шариковый катализатор (никеля — 0,03—0,09; ванадия —
0,02—0,12; железа — 0,8—0,11; магния — 0,05—0,11; кальция —
0,2—0,5; натрия — 0,4—0,6 вес. %).
Проведенный анализ качества теряющегося и циркулирующего
равновесного пылевидного катализатора показал, что содержание
металлов в этих образцах различается незначительно, т. е.
эффект выноса металлов за счет истирания частиц с поверхности
невелик.
Снижение активности пылевидного катализатора в системе
крекинга на 20—30% по сравнению с активностью шарикового
катализатора, вызвано довольно низкой активностью отработанной
крошки, из которой изготовляют пылевидный катализатор, и
дополнительным его отравлением металлами при эксплуатации.
Обследования значительного числа установок флюид в США
и Канаде показали, что в период с 1960 по 1965 г. содержание
ванадия и никеля в равновесных аморфных катализаторах было на
уровне 300 и 140 млн.-1; после 1965 г., когда широкое
распространение нашли цеолитсодержащие катализаторы, содержание в них
указанных металлов возросло соответственно до 500 и 300 млн.-1.
За этот же период микроактивность катализатора увеличилась
с 55—60 (что типично для катализатора с высоким содержанием
алюминия) до 81. Эти данные также подтверждают меньшее
отравление цеолитсодержащих катализаторов металлами.
Природа металла оказывает сильное влияние на степень
отравления. На рис. 65 и 66 показано влияние некоторых металлов на
активность катализатора и на фактор коксообразования [213]. Из
рисунков видно, что разница в увеличении коксового фактора и
снижении степени превращения под действием различных металлов
весьма значительна.
Авторы [213] считают, что по отравляющему действию на
катализаторы крекинга металлы располагаются примерно в таком же
порядке, как и по отравляющему действию на катализаторы
дегидрогенизации. Большинство авторов считают, что наиболее
сильным ядом является никель. Однако эта точка зрения не является
единственной. Некоторые исследователи [64, 214] полагают, что-
такой же сильный эффект, как никель, вызывает и медь. По дан-
15а
О 0,2 0,4 0,6 0,8
Содержание металла на
катализаторе , вес. %
Рис. 65. Влияние различных металлов на
катализаторы с 30%-ной начальной
активностью по методу Д+П (дистил-
лят + потери):
/ — ванадий; 2 — кальций; 3 — натрий; 4 —
медь; 5 — цинк; б — никель; 7 — железо;
8 — хром.
О 0,2 O,U 0,6 0,3
Содержание металла
¦ на катализаторе, 8ее.%
Рис. 66. Влияние различных
металлов на фактор коксообра-
зования катализатора с
30%-ной начальной
активностью по методу Д + П:
/ — никель; 2 — цинк; 3 — хром;
4 — железо; 5 —ванадий; б — медь;
7 — натрий; 8 — кальций.
О 0,5 1,0 1,5 2,0
Содержание металлов, бес. %
а
О,
' 0 0,5 1,0 1,5 2,0
Содержание металлов, вес.%
d
Рис. 67. Влияние тяжелых металлов на активность (й) и закоксовывание (б)
катализаторов крекинга:
/ — ванадий; 2 — железо; 3 — никель; 4 — медь; 5 — свинец.
ным [54], медь оказывает большее отравляющее действие, чем
никель.
Существует мнение [45, 213], что в концентрациях менее
0,3 вес. % никель более вреден, чем другие металлы, но при более
высоких концентрациях его действие соизмеримо с отравляющим
эффектом ванадия, железа и меди. На рис. 67 приведены данные
[45] о влиянии на активность и закоксовывание катализатора
различных металлов. Авторы [45] считают, что отравляющее
действие металлов, по-видимому, снижается в таком порядке: никель>
>железо>ванадий>медь>свинец. Другие исследователи
предлагают следующий порядок: никель>медь>железо>ванадий;
они даже приводят количественные соотношеия силы воздействия
этих металлов: никель 1,0; медь 1,0; железо 0,55; ванадий 0,091
[214]. При увеличении коксового фактора количественные
соотношения несколько возрастают: железо 0,66; ванадий 0,61; при
увеличении выхода газа железо 0,66; ванадия 0,106.
В присутствии в сырье никеля выход кокса в 4,5 раза больше'
и количество бензина снижается в 7,9 раза больше, чем в
присутствии такого же количества ванадия [215]. Потеря селективности
при наличии на катализаторе никеля и меди в 10 раз больше, чем
при наличии железа [202]. Коксообразование, вызываемое
содержанием на катализаторе никеля, в 4 раза больше, чем в
присутствии железа [204]. При изучении влияния различных металлов,
на степень отравления катализатора большинство исследователей
проводили опыты с относительно большими количествами
металлов по сравнению с содержанием их на промышленном
равновесном катализаторе. Поскольку в работе [216] были использованы
данные по содержанию металлов в промышленном катализаторе,
определенные зависимости отличны от всех остальных. Уравнение,
предложенное автором этой работы для определения активности
катализатора, имеет следующий вид:
активность = 3400V — 17S5N1 — 123 OOOCu -f 27,4Fe — 60Na2O
откуда видно, что в присутствии ванадия активность
катализатора существенно увеличивается; коэффициент перед содержанием
железа также взят со знаком плюс.
Таким образом, имеющиеся данные свидетельствуют о
существенном влиянии природы отравляющего металла на степень
отравления. Видимо, из-за различия в методах отложения металлов и
испытания катализаторов единого мнения об относительной силе
отравляющих металлов нет. Теоретического объяснения влияния
типа металла также не имеется. В работе [202], правда,
делается попытка представить в общем виде возможное поведение
адсорбированных на поверхности алюмосиликатного катализатора
различных катионов. В ней изучалось влияние на каталитическую
активность натрия, калия, бария, цинка, магния, водорода,
алюминия, тория. Исходный натрийалюмосиликат пропитывали
водными растворами соответствующих солей. Общее количество рас-
15S
твора брали в большом избытке с расчетом на полное вытеснение
натрия. После обработки катализатор тщательно промывали
дистиллированной водой, сушили и прокаливали. Активность
проверяли на реакции деалкилирования смеси бутилбензолов (фракция
155—185 °С). Опыты проводили при 400 °С в течение 30 мин и
объемной скорости 1 ч~'.
Изменение каталитической активности алюмосиликатного
катализатора под влиянием адсорбированных катионов приводится
ниже [202]:
Выход фракции
Адсорбированный катион до 150 °С,
объемн. %
Натрий (исходный катализатор) 0
Калий 0
Барий 11
Цинк 22
Магний 28
Аммоний 48
Алюминий 48
Торий 42
На основании полученных данных автор [202] делает
предположение о существовании связи между валентностью катиона, его
размерами и влиянием на каталитическую активность
алюмосиликатного катализатора. Действительно, одновалентные катионы
неактивны, двухвалентные обладают некоторой активностью,
возрастающей по мере уменьшения радиуса катиона (от бария к
магнию), трех- и четырехвалентные катионы активны так же, как
и Н+.
Не всякое соединение металла отравляет катализатор.
Известно большое число патентов на различные методы реактивации
катализаторов, в результате которых отравляющий эффект металла
уменьшается путем перевода одного соединения в другое; общее
количество металла в катализаторе не изменяется. Еще в работе
[64] отмечалось, что количественные соотношения между
концентрацией металла и его влиянием зависят от распределения металла
по поверхности катализатора, от формы соединения, в котором
находится металл. Когда пробы катализатора с промышленных
установок содержали относительно высокое количество окиси железа
(она накапливалась, очевидно, вследствие коррозии и эрозии
оборудования), сконцентрированной на поверхности таблеток,
активность и селективность катализатора несколько уменьшалась.
Кроме того, в случае смеси катализатора с порошком окиси железа
зависимость селективности от количества железа была другой, чем
при пропитке катализатора водным раствором соли железа.
Проделана большая работа [217] по изучению влияния типа
соединений металлов, отложенных на катализаторе, на его
активность и стабильность. Было изучено влияние железа и его
соединений (металлического, гидрата окиси, ферросиликатов — двух-
и трехвалентного) на активные свойства двух типов синтетических
156
Таблица 46
Влияние различных соединений железа на активность
алюмосилікатного катализатора
Показатели
Исходный
катализатор
Добавки к катализатору
металлическое
железо
ферро-
силикат
трехвалентный
ферро-
силикат
двухвалентный
гидрат
окиси
железа
Катализатор I
Содержание F2O3, вес. %
Выход, вес. °/о
дистиллят
бензин <200°С
газ
кокс
Относительная плотность газа
Следы
78,5
36,7
17,4
2,3
1,42
1,03
77,0
36,1
18,9
3,8
1,30
0,96
76,1
22,8
15,9
6,8
0,87
0,97
74,1
19,3
19,8
5,6
0,9
Катализатор 1 [
Содержание Fe2O3, вес. %
дистиллят
бензин <200 °С
газ
кокс
Относительная плотность газа
Следы
61,5
36,2
27,7
6,9
1,3
0,97
62,0
34,8
26,1
6,7
1,36
1,01
63,3
30,7
26,4
9,2
1,02
0,98
61,4
31,3
26,3
—
0,91
1,09
74,3
13,7
9,7
0,45
0,92
62,3
20,4
26,4
10,3
0,90
алюмосиликатных катализаторов — с низкой и высокой
стабильностью. Из данных табл. 46 [217] видно, что добавки различных
соединений железа действуют отравляюще на катализатор, снижая
его активность. Однако степень снижения активности для
различных соединений железа при сравнительно одинаковом его
содержании не одинакова. Наибольшее отравляющее действие
свойственно гидрату окиси железа. Интересно отметить, что при
существенном изменении выхода бензина и кокса выход газа в
присутствии различных соединений железа почти не изменяется, в то
время как плотность газа уменьшается.
Из табл. 46 видно, что присутствие металлического железа на
катализаторах не вызывает существенного отравляющего
действия; все показатели у образцов с добавкой металлического железа
оставались примерно такими же, как и у чистого катализатора.
Присутствие различных соединений железа в алюмосиликатных
катализаторах отражается на их иаростабильности, причем
наиболее резкое действие наблюдается при добавке гидроокиси
железа. Так, индекс стабильности для катализатора I (см. табл. 46)
снизился на 48%, а для катализатора II — на 41%. Для
образцов с двухвалентным ферросиликатом индекс стабильности
снизился соответственно на 40 и 50%- Действие добавки
трехвалентного ферросиликата сказывается менее резко.
157
Наши данные по крекингу вакуумного газойля в присутствии
аіморфного алюмосиликатного каталізатора, содержащего
различные металлы в большой концентрации, согласуются с
результатами других исследований (рис. 68). При содержании на катализа-
тоое металлов более 0,01 вес.% выход бензина резко уменьшается.
Катализатор, содержащий 0,52 вес. % никеля, дает выход бензина
лишь 14,7% вес. %•
О 0,2
Содержание
металлов
Рис. 68. Влияние металлов на выход
бензина при крекинге вакуумного
газойля:
/ — свинец; 2 — хром; 3 — железо; 4 —
молибден (X), ванадий (Д); 5 — кобальт (О), медь
( + ), 6 — иикель.
0 0,2 0,^ U,6
Содержание металлов, вес. %
Рис. 69. Влияние металлов на выход
кокса при крекинге вакуумного
газойля;
/ — никель; 2 — медь ( + ), кобальт (О), 3 —
молибден (X), ванадий (А), свииец (V),
хром (?), 4— железо.
Все изученные металлы в испытанных пределах концентраций
вызывают увеличение коксообразования и объемного выхода газа.
Наиболее резко выход этих продуктов возрастает при содержании
на катализаторе малых количеств металла (до 0,1—0,2 вес. %).
В дальнейшем увеличение становится небольшим (см. рис. 69).
При нанесении на катализатор 0,5 вес. % никеля выход кокса
достигает 15,8%, а выход газа — 13,4 л; еыход при крекинге на
свежем катализаторе соответственно 6,6% и 4,6 л. По степени
возрастающего влияния на изменение в выходах продуктов крекинга
металлы располагаются в той же последовательности, в какой они
вызывают уменьшение активности катализатора: свинец <хром<
<железо, ванадий, молибден < медь, кобальт<никель.
Катализаторы с повышенным содержанием металлов имеют низкую
селективность, оцениваемую отношением бензин : кокс. Селективность
катализаторов, активированных микродобавками металлов,
отличается от исходной незначительно.
При небольшой концентрации некоторые металлы (ванадий,
молибден и др.) промотируют образование бензина (см. рис. 68).
Зависимость выхода бензина от содержания металла при крекинге
вакуумного газойля имеет в основном такой же характер, как и
158
зависимость выхода бензина при работе на стандартном сырье.
Оптимальная концентрация ванадия, молибдена также находится
в области 1-Ю-4—Ы0~3% от массы катализатора. При этом
выход бензина увеличивается на 2,0—2,5 вес. % на сырье. Оптимум
по содержанию свинца при крекинге вакуумного газойля
несколько шире. Даже при нанесении 0,15 вес. % свинца выход бензина
такой же, как на катализаторе, содержащем 1-10~4 вес. % свинца.
При нанесении металлов на алюмосиликатный катализатор в
газах крекинга возрастает количество водорода, сухого газа и
уменьшается количество тяжелой его части. Абсолютные значения
этих величин зависят от природы металла и его содержания на
катализаторе. Наиболее резко состав газов в указанном
направлении изменяется при содержании на катализаторах никеля,
кобальта, меди, менее всего — при содержании железа, хрома свинца.
С увеличением концентрации металла на катализаторе указанные
тенденции усиливаются. Так, при содержании на катализаторе
0,16 вес. % никеля концентрация водорода в газе 10,5 вес. %, а
сухого газа 42,0 вес. %. что соответственно в 35 и 14 раз больше,
чем в газе крекинга, полученном на исходном катализаторе. В то
же время количество пропан-пропиленовой и бутан-бутиленовой
фракции уменьшается в 1,2 раза. При содержании на
катализаторе 0,5 вес. % никеля концентрация водорода в газе увеличивается
в 50 раз, сухого газа — в 16 раз, а концентрация
пропан-пропиленовой и бутан-бутиленовой фракций уменьшается соответственно
в 1,2 и 1,4 раза.
В результате увеличения содержания в газах водорода и легких
углеводородов их плотность снижается. При содержании на алю-
моснликатном катализаторе 0,5 вес. % никеля плотность газа
уменьшилась в 3,7 раза и достигла 0,43 г/л против обычной
плотности 2,6 г/л. Весьма характерно, что при нанесении на
катализатор металла в газе увеличивается содержание олефинов. Так, про-
пан-пропиленовая и бутан-бутиленовая фракции, полученные в
процессе крекинга вакуумного газойля на катализаторе,
содержащем около 0,5 вес. % никеля, кобальта и меди, состоят
соответственно на 70— и 50% из олефинов, в то время как в обычных
условиях их содержится примерно 50 и 30%.
При добавлении к алюмосиликатному катализатору металлов,
особенно сильных дегидрирующих агентов, наряду с уменьшением
активности ухудшается и изомеризующая способность
катализатора. Так, с увеличением концентрации металлов на катализаторе
до 0,5—1,0 вес. % содержание изобутана в бутан-бутиленовой
фракции уменьшается в 1,2—1,7 раза.
Бензин крекинга, полученный на катализаторе, активированном
микродобавками, по составу практически ничем не отличается от
продуктов, полученных на исходном катализаторе. Бензины,
образовавшиеся при крекинге на катализаторах со значительными
количествами металлов, характеризуются повышенными
плотностями и большим содержанием серы и непредельных углеводородов
159
Таблица 47
Качество бензина крекинга вакуумного газойля на катализаторах,
содержащих металлы
Металл,
наносимый
на катализатор
Без металла
Свинец
Хром
Без металла
Молибден
Ванадий
Медь
Кобальт
Никель
Железо
Содержание
металла на
катализаторе,
вес. %
—
0,0001
0,003
0,03
0,15
0,08
0,15
0,69
—
0,0001
0,002
0,01
0,04
0,06
0,6
0,0003
0,003
0,002
0,02
0,16
0,18
0,96
0,02
0,37
0,66
0,0001
0,003
0,02
0,16
0,52
0,25
0,50
Характеристика бензина
плотность
0,760
0,771
0,770
0,771
0,770
0,753
0,769
0,767
0,754
0,767
0,772
0,781
0,785
0,80
0,806
0,779
0,785
0,785
0,781
0,792
0,805
0,777
0,798
0,818
0,777
0,760
0,779
0,816
0,816
0,769
0,794
содержание
серы,
вес. %
0,48
0,44
0,48
0,39
0,43
0,18
0,16
0,15
0,17
0,50
0,52
0,45
0,62
0,64
0,87
0,45
0,46
0,40
0,38
0,95
1,29
0,48
1,08
1,32
0,50
0,48
0,64
1,22
1,55
0,55
0,87
Йодное
число,
г 12/Юи г
37,7
37,7
38,9
37,1
37,5
39,4
40,7
57,8
38,5
32,0
42,9
50,1
52,0
56,0
40,0
39,0
30,7
31,5
46,0
53,4
71,6
40,8
61,0
68,0
38,1
55,0
69,0
75,1
52,6
57,2
содержание ,
непредельных
углеводородов,
вес. %
17,5
18,5
19,1
10,2
18,5
17,2
19,9
27,8
17,0
15,4
22,9
27,5
30,8
35,8
21,2
24,7
31,4
45,6
21,2
37,0
53,1
17,3
29,2
50,0
53,0
—
по сравнению с бензинами крекинга на исходном катализаторе
(табл. 47). Абсолютные значения этих величин зависят от природы
металла и его концентрации на поверхности катализатора.
Наибольшие изменения бензина в указанном направлении вызывают
160
добавки никеля, кобальта, меди, наименьшие — добавки железа,
свинца. При наличии на катализаторе 0,15 вес. % никеля
относительная плотность (d'l°) бензина увеличивается в 1,1 —1,2 раза
и достигает 0,816; содержание серы и непредельных
углеводородов возрастает на 130%.
Таким образом, металлы, нанесенные на алюмосиликатный
катализатор, не изменяя его физико-химических свойств, вызывают
резкие изменения его активности и селективности, которые очень
сильно зависят от концентрации металла и его природы. При
содержании на катализаторе исследованных ¦ металлов более
0,02 вес. % катализатор отравляется, что проявляется в
значительном уменьшении выхода бензина, увеличении выхода кокса, газа
и водорода. Наряду с общепризнанным отравляющим действием
нами было обнаружено и промотирующее действие некоторых из
этих металлов (свинца, ванадия, молибдена) в пределах
концентраций 1-Ю-4— 1 • Ю-3 вес. % от массы катализатора. Если
нанесенного металла более 0,02 вес. %, то активность катализатора
становится меньше первоначальной. Видимо, поскольку большинство
исследователей при изучении механизма отравления оперировали
относительно большими концентрациями металлов, они не могли
отметить возможность промотирования катализатора крекинга.
Об отравлении цеолитсодержащих катализаторов
металлсодержащими соединениями встречаются лишь разрозненные данные.
В частности отмечается, что эти соединения влияют на цеолитные
катализаторы в меньшей степени, чем на аморфные. Так,
сообщается [218], что при содержании от 0,027 до 0,05% никеля и от 0,04
до 0,09% ванадия заметного отравления катализатора не
наблюдается. Отмечается [219], что катализатор XZ-25 допускает
присутствие в сырье до 0,03 вес. % никеля и до 0,1 вес. % ванадия.
В работе [220] меньшее отравление цеолитных катализаторов
металлами объясняется накоплением их в основном в высококипящих
фракциях, диффузия которых в полости цеолитов затруднена.
Однако применение цеолитов полностью не устраняет
затруднений, связанных с отравлением катализаторов крекинга. Поэтому
обращается внимание на предварительную очистку сырья и
циркулирующего газойля. В частности, показана [221] высокая
экономичность гидрогенизационной очистки сырья и в случае
применения цеолитных катализаторов.
Весьма интересным является промотирование цеолитных
катализаторов различными газами. Так, активность щелочных и
щелочноземельных форм цеолитов типов А и X в реакциях крекинга
различных углеводородов значительно возрастает в присутствии
двуокиси углерода. Например, конверсия кумола на цеолите СаХ
при 468 °С и объемной скорости подачи сырья 1,0 ч~' после
добавки двуокиси углерода увеличилась с 60 до 90%. Количество
промотора составляет 0,005—25 вес. % на катализатор в статических
условиях проведения процесса и 3—200 вес. % в 1 ч — в
проточных. Промотирование цеолитов двуокисью углерода объясняют
11-220« 161
[222] обогащением их поверхности протонами в результате
образования соединений типа карбонатов. Эффект промотирования
наблюдался также при предварительной обработке двуокисью
углерода кальциевых цеолитов типа У; однако он быстро затухал. .
Промоторами цеолитов оказались сернистый ангидрид,
Сероводород, закись и окись азота и водород. Показана [223] стойкость
в.-лсококремнистых цеолитов
к действию двуокиси
углерода, сернистого
ангидрида, закиси и окиси азота и
сероводорода при высоких
температурах.
Как уже отмечалось в
гл. I, цеолитсодержащие
катализаторы крекинга
наиболее активны и стабильны
при их промотировании
редкоземельными элементами
[224, 225]. В связи с этим
они содержатся в
современных промышленных
катализаторах; наибольший
эффект достигается при
обмене катионов более 40—60%.
Влияние металлов на
новообразование. Особое
место в процессе
каталитического крекинга занимает
установление влияния
металлов, содержащихся на
катализаторе, на
образование кокса. До настоящего
времени исследователи
единодушно рассматривали
металлы как добавки,
усиливающие образование кокса.
, 0,8 /,2
Количество металла , добавленного
к катализатору, вес. %
Рис. 70. Зависимость продолжительности
отложения 2 вес. % кокса от
содержания металла в алюмосиликатном
катализаторе:
/ — литий; 2 — ііатрий; 3 — калий; 4 — цезий;
5 — бериллий; 6— магний; 7 — кальций; 8 —
стронций; 9 — свинец; 10 — ванадий; // — хром;
12 — молибден; 13 — железо; 14 — кобальт;
15 — никель; 16 — медь. _
Однако результаты наших
исследований показали, что
некоторые металлы при небольших концентрациях активируют
катализатор крекинга [200, 201]. Поэтому появилась
необходимость проведения специальных исследований по установлению
влияния металлов на коксообразование при каталитическом
крекинге.
Эксперименты проводили на установке, разработанной в
ГрозНИИ [226], которая позволяет непрерывно взвешивать
образец катализатора. Крекинг газойля артемо-малгобекской нефти
проводили при 450 °С, объемной скорости подачи сырья 1,0 ч;
продолжительность каждого опыта выбирали так, чтобы на образ-
162
це аморфного алюмосиликатного катализатора накапливалось
2 вес. % кокса.
По способности изменять величину коксообразования при
каталитическом крекинге нефтепродуктов металлы можно условно
разделить на три группы. К первой группе относятся щелочные и
щелочноземельные металлы (литий, натрий, калий, цезий, бериллий,
магний, кальций, стронций), которые подавляют
коксообразование. Из исследованных щелочных металлов наименьшее
коксообразование вызывает добавка калия и цезия (рис. 70). Время,
требуемое для отложения на катализаторе 2 вес. % кокса, с
увеличением концентрации добавляемого металла возрастает. При
добавлении щелочноземельных металлов это время возрастает не так
резко. Характерная особенность щелочноземельных металлов —
при добавлении их к катализатору в равных концентрациях
количество образующегося кокса на всех образцах практически
одинаково.
Ко второй группе относятся кобальт, медь и никель.
Добавление их к катализатору способствует существенному увеличению
выхода кокса; это зависит не только от природы металла, но и от
его содержания в образце (см. рис. 70). По сравнению с исходным
катализатором выход кокса на образцах, содержащих 0,5—
0,7 вес. % никеля, меди и кобальта, увеличивается в 3,2—3,5 раза.
Третью группу составляют ванадий, молибден, хром, свинец,
железо. При большой концентрации они вызывают увеличение
коксообразования, так же, как и металлы второй группы, но в
меньшей степени. Так, при содержании их в катализаторе 0,5—0,7%
коксообразование возрастает лишь в 1,3—1,5 раза. Влияние этих
металлов на коксообразование при крекинге примерно одинаковое.
Весьма характерная особенность металлов этой группы — при
небольшом их содержании в катализаторе образование кокса в
процессе крекинга уменьшается. Так, при концентрации ванадия
0,02—0,003 вес. % выход кокса в 1,25 раза меньше, чем в
присутствии исходного катализатора. і
При осуществлении процесса каталитического крекинга важно
не только общее содержание кокса на катализаторе. Для
эффективной работы узла регенерации большое значение имеет
характер распределения кокса по сечению гранулы. Этому вопросу
посвящены работы [85, 102]. Однако исследований о влиянии
металлов на распределение кокса по сечению шарика катализатора, нам
обнаружить не удалось. В связи с этим нами были осуществлены
специальные работы. Микрофотометрированием тонких шлифов,
приготовленных из шариков катализатора, которые содержали
различные металлы, определяли распределение кокса по диаметру
частиц катализатора. Каждый образец содержал 2 вес. % кокса.
Было установлено, что кокс по сечению шарика исходного
катализатора распределяется неравномерно. Максимальное
содержание кокса наблюдается у внешней поверхности шарика (рис. 71,
кривая /). Исследования показали, что металлы по-разному влия-
»* 163
ют на распределение кокса по сечению частицы. По своему
влиянию их можно разделить на две группы: 1) металлы,
способствующие равномерному распределению кокса (щелочные и
щелочноземельные металлы) и 2) металлы, усиливающие неравномерность
распределения кокса. Наиболее равномерно распределен кокс на
образцах, содержащих натрий, литий, калий и цезий.
з.о г—
Относительны:: ,?¦.¦¦ :•-¦¦. .¦:¦,: ....¦ :'ика
Рис. 71. Зависимость характера распределения кокса по сечению шарика
катализатора от природы и концентрации металла:
/ — исходный катализатор; 2, 3. 4 — содержание натрия соответственно 0,39; 0,84; 1,40 вес. %:
¦5, 6 7 —содержание меди 0,012; 0,09- 0,70 вес %; 8, 9, 10 — содержание хрома 0,06; 0,10;
0,80 вес. %.
Для примера на рис. 71 представлены данные о влиянии
натрия на распределение кокса по сечению частиц катализатора. Чем
больше содержится металла в катализаторе, тем равномернее
откладывается кокс по сечению частицы. Так, на образцах,
содержащих 0,5 вес. % щелочного металла, отношение содержания кокса
на периферии шарика к его содержанию в центре равно 1,24, а на
образцах, содержащих 1 вес. % металла, это соотношение
снижается до 1,07.
Кокс распределяется на образцах, содержащих бериллий,
магний, кальций, стронций, почти так же, как и на образцах
катализатора, содержащих щелочные металлы. Влияние
щелочноземельных металлов аналогично друг другу, и характер распределения
164
кокса зависит только от концентрации металла в катализаторе. На
образцах, содержащих хром, свинец, ванадий, молибден, железо,
кобальт, никель, медь, характер распределения кокса иной. На
всех образцах, содержащих эти металлы, кокс откладывается менее
равномерно по сечению гранулы, чем на исходном катализаторе.
Эта неравномерность зависит от природы металла и от его
содержания в образце. Наибольшая неравномерность отложения кокса
по сечению частицы катализатора наблюдается при добавлении
железа. В этом случае кокс в основном откладывается в
периферийных слоях частицы. В центре шарика накапливается
незначительное его количество, — почти в 5 раз меньше, чем в исходном
образце. При содержании в катализаторе 0,8 вес. % железа
отношение содержания кокса на периферии к его содержанию в центре
шарика составляет 10,0; на исходном образце это отношение
равно 1,45.
При добавлении к катализатору меди (см. рис. 71) и никеля
кокс также распределяется по сечению шарика неравномерно. Но
в этом случае концентрация кокса в центре шарика меньше, чем
в исходном образце, в 2 раза, и отношение содержания кокса на
периферии к содержанию в центре шарика при добавлении 0,5—
0,7 вес. % металла составляет 3,0—3,4.
Образцы, содержащие кобальт, молибден, хром, ванадий и
свинец, близки по характеру коксоотложений по сечению частицы к
исходному алюмосиликатному катализатору, особенно при малых
концентрациях металлов. Так, при содержании в образцах
0,8 вес. % хрома отношение содержания кокса на периферии и в
центре шарика составляет 1,85, а при содержании 0,06—0,10 вес. %
хрома кривая распределения кокса близка к кривой исходного
катализатора (см. рис. 71). Такую разницу в распределении кокса по
сечению частицы катализаторов, содержащих различные металлы,
можно объяснить их различным влиянием на коксообразование.
При добавлении к катализатору щелочных и щелочноземельных
металлов кислотность катализатора и его активность понижаются.
Из-за большего отложения металла в поверхностных слоях ката-
. литическая активность периферийных слоев катализатора
снижается больше, в то время как центральные слои сохраняют более
высокую каталитическую активность. Это приводит к
перемещению реакций крекинга в глубину частицы катализатора с
одновременным отложением кокса в центральных слоях частиц.
Поэтому кокс отлагается более равномерно по сечению шарика на
образцах, содержащих щелочные и щелочноземельные металлы, чем
на исходном катализаторе. Тяжелые же металлы не оказывают
влияния на кислотность катализатора. Накапливаясь главным
образом в периферийных слоях частицы. Поэтому кокс откладывается
на этих образцах большей частью в периферийных слоях частицы
катализатора, т. е. крайне неравномерно. По мере увеличения
содержания в катализаторе металлов неравномерность отложения
кокса возрастает. Металлы, увеличивающие коксообразование,
165
способствуют большей неравномерности отложения кокса по
сечению отдельных гранул.
Влияние металлов на регенерацию катализатора. Металлы,
накапливающиеся в процессе работы на поверхности
катализатора, должны оказывать определенное влияние и на процесс выжига
кокса. Так, на одной установке, долго работавшей на остаточном
сырье, при увеличении на катализаторе содержания никеля от
6-Ю-2 до 7-Ю-2 вес. %, а ванадия от 3,5-10~2 до 18-Ю-2 вес. %
содержание остаточного (после выжига) кокса уменьшалось с 0,4
до 0,2 вес. %. После прекращения подачи остаточного сырья и
существенного уменьшения количества металлов содержание
остаточного кокса возросло до 0,3 вес. % [186]. О влиянии
некоторых металлов на выжиг коксовых отложений с катализатора в
литературе имеются лишь отрывочные данные [78, 238—241]. Для
получения более полных данных нами были проведены
эксперименты на аппарате ГрозНИИ в кинетической E00 °С) и
диффузионной F50 °С) областях при удельном расходе воздуха 1500 ч~'.
Во всех опытах отлагалось кокса 2 вес. % на катализатор. В
кинетической области горения при добавлении в катализатор
различных металлов качественный характер регенерации катализатора
на всем ее протяжении не изменялся. Однако металлы, нанесенные
на катализатор, способствуют существенной интенсификации
выжига кокса в начальный период по сравнению со скоростью
выжига исходного катализатора.
Наибольшее ускорение выжига кокса в начале регенерации
наблюдается на образцах, содержащих хром. За первые 25 мин на
образце катализатора, содержащем 0,8 вес. % металла, сгорает
84 вес. % отложенного кокса, в то время как на исходном
катализаторе за это же время сгорает только 52 вес. % кокса. С
уменьшением содержания хрома в образце скорость выжига кокса
заметно снижается. На образцах, содержащих ванадий, медь,
молибден, доля сгоревшего кокса в начальный момент времени также
значительно выше, чем на исходном катализаторе, но несколько
меньше, чем на образцах, содержащих хром. Так, при примерно
таком же количестве металлов за первые 25 мин выгорает
только 70—74% отложенного кокса. При добавлении железа, никеля
и кобальта скорость регенерации исходного катализатора мало
изменяется. Так, при содержании 0,8 вес. % железа за первые
25 мин сгорает только 66% отложенного кокса, а на образцах,
содержащих 0,48— 0,50 вес. % никеля и кобальта, за то же время
сгорает 55% кокса; при регенерации исходного образца
катализатора сгорает 52% кокса. Свинец не влияет на регенерацию
катализатора.
В последующие моменты выжиг кокса на всех образцах
катализатора существенно замедляется. За последние 35 мин сгорает
только 20—25 вес. % кокса, а на исходном катализаторе —
35 вес. %.
При добавлении в катализатор щелочных металлов регенера-
166
ция катализатора в начальные моменты времени идет также более
интенсивно, чем исходного образца, а затем значительно
замедляется. Наиболее сильное влияние на выжиг кокса оказывает
добавление лития. За первые 25 мин на образце, содержащем
1,32 вес. % лития, сгорает 81 % кокса. Калий и цезий практически
не изменяют скорость регенерации катализатора. Так же
незначительно влияют щелочноземельные металлы.
В диффузионной области горения наибольшее влияние на
выжиг коксовых отложений оказывает добавление железа. На
образце катализатора, содержащем 0,8 вес. % железа, отложенный кокс
сгорает в два раза быстрее, чем на исходном катализаторе.
Остальные металлы в какой-то степени ускоряют выжиг кокса при
их содержании в катализаторе в больших концентрациях. Так, на
образцах, содержащих до 0,5—0,8 вес. % никеля, меди, кобальта,
хрома, молибдена и до 1,5—1,3 вес. % лития, натрия, калия,
бериллия, магния, кальция, стронция, кокс выжигается в 1,2 раза
быстрее. На образцах, содержащих микродобавки этих металлов,
скорость горения кокса такая же, как исходного образца
катализатора. Добавка свинца не влияет на скорость регенерации
катализатора.
Быстрое сгорание кокса на образце, содержащем железо,
обусловлено характером распределения кокса по сечению частицы
катализатора. На этом катализаторе кокс в основном откладывается
в периферийных солях частицы, в связи с чем средняя
необходимая глубина проникновения кислорода в зону горения
уменьшается. Это способствует улучшению регенерации катализатора в
диффузионном режиме горения. Таким образом, в диффузионной
области горения металлы, за исключением железа, почти не влияют
на скорость выжига коксовых отложений. Полученные данные
являются закономерными, так как в этой области скорость
регенерации определяется скоростью подвода кислорода к зоне горения и
отвода продуктов реакции из этой зоны, а не скоростью
протекания химической реакции.
Как показали наши эксперименты, в кинетической области в
присутствии в катализаторе крекинга различных металлов выжиг
коксовых отложений существенно ускоряется, особенно в первый
момент регенерации.
На рис. 72 показано влияние концентрации различных
металлов, содержащихся в катализаторе, на длительность сгорания
половины отложенного на катализаторе кокса. Из рисунка видно,
что при содержании металлов в катализаторе более 0,05 вес. %
характер их влияния на скорость сгорания кокса практически
одинаков. По мере увеличения содержания металла в катализаторе
регенерация его ускоряется. Наибольшее ускорение достигается
при малом их содержании в катализаторе, а с увеличением содер-
жания этот эффект становится все меньше и после достижения
некоторой максимальной концентрации металла скорость выжига
коксовых отложений перестает изменяться.
167
Эта максимальная концентрация, а также максимальное
ускорение регенерации катализатора зависят от природы металла.
С уменьшением окислительной способности металлов
максимальная концентрация возрастает. Так, если максимальная
концентрация для хрома равна 0,1 вес. %, то для ванадия она равна 0,3—
0,4%, а для молибдена и меди составляет уже примерно 0,5—
0,6 вес. %• Одновременно с уменьшением способности
катализировать окисление кокса уменьшается максимальное ускорение
регенерации катализатора. Наши данные позволяют расположить ис-
О 0,It 0,8 1,2
Количество металла, добавленного
к катализатору, Sec. %
Рис. 72. Зависимость длительности выжига 50% отложенного кокса от
количества металла, добавленного к катализатору:
' — литий; 2 —натрий; 3 — калин; 4 — цезий; 5 — бериллий; ff—магний; 7 — кальцин;
Я —стронций; 9 — свинец; 10 — ванадий; // — хром; 12 — кобальт; 13 — железо; 14 —
молибден; 15 — никель; 16 — медь.
пытанные нами металлы в следующий ряд по степени убывания
их воздействия на скорость окисления кокса в кинетической
области: хром>ванадий>литий>молибден, медь, натрий >железо>
>кобальт, никель>бериллий, магний, кальций, стронций >ка-
лий>цезий>свинец. Этот ряд не случаен. Аналогичная
последовательность была получена для тяжелых металлов по их влиянию
на окисление метана [242] и для щелочных и щелочноземельных
металлов при окислении сажи [243].
В соответствии с существующими представлениями [92, 244],
процесс окисления кокса протекает через ряд стадий. Первая
стадия — хемосорбция кислорода с образованием устойчивого
поверхностного углерод-кислородного комплекса. Вторая стадия —
разложение комплекса с образованием окиси и двуокиси углерода.
Чтобы объективно оценить влияние металлов на различные стадии
процесса регенерации на основании экспериментальных данных
были вычислены константы скоростей образования и разложения
углерод-кислородного комплекса. При этом было использовано
уравнение Г. М. Панченкова и Н. В. Голованова [92],
описывающее процесс выжига кокса в кинетической области. Численные
168
значения коэффициентов этого уравнения приведены на рис. 73
и 74.
Расчеты показывают, что характер влияния металлов на
константы скорости К\ и Ki — образования и разложения
кислородного комплекса — неодинаков.
Константа скорости образования комплекса К\ увеличивается
по мере добавления металлов и зависит от природы металла.
Максимальное влияние на величину К\ оказывают концентрации
металлов — примерно до 0,3—0,4 вес. %. При больших
концентрациях металлов константа скорости образования
углерод-кислородного комплекса изменяется незначительно (см. рис. 73). Наиболее
резко эта константа изменяется у образцов с добавками хрома;
при содержании его в катализаторе от 0,1 до 0,8% К\ становится
в 3 раза больше, чем для исходного. Среди щелочных и
щелочноземельных металлов сильнее всего на константу образования
комплекса влияет литий. В присутствии 1,3 вес. % этого металла она
возрастает в 2,5 раза. Константа скорости Ki разложения
комплекса не зависит от содержания металла в катализаторе и
определяется только его природой (см. рис. 74). Большая часть
исследованных металлов уменьшает константу скорости Кч разложения
комплекса. Так, наименьшая величина константы скорости разложения
комплекса наблюдается на образцах, содержащих хром. В этом
случае Кг в 2,4 раза меньше константы скорости разложения
исходного катализатора (см. рис. 74). Среди щелочных металлов эта
константа наиболее резко уменьшается при добавлении лития
(в L2 раза). Щелочноземельные металлы практически не влияют
на константу разложения кислородного комплекса.
Зависимости изменения константы скорости образования
комплекса и продолжительности выжига половины отложенного кокса
имеют одинаковый характер. Это показывает, что влияние
металлов на выжиг коксовых отложений заключается в ускорении
первой стадии процесса — образования углеродно-кислородного
комплекса вследствие их способности катализировать реакции
окисления.
Некоторые металлы (особенно щелочные) и их окислы могут
способствовать ускорению окисления коксовых отложений также
вследствие того, что они повышают хемосорбцию кислорода в
углероде и ослабляют связь углерод—углерод [245].
Чтобы объяснить влияние металлов на регенерацию
катализатора в кинетической области, когда кокс горит одновременно по
всей внутренней поверхности катализатора, необходимо допустить,
что кокс не откладывается по ней равномерно. Иначе металлы
оставались бы под коксовыми отложениями, и их влияние можно
было бы установить лишь в завершающей стадии регенерации.
Таким образом, наши данные являются дополнительным
подтверждением неравномерного отложения кокса на поверхности
катализатора и накопления кокса на активных центрах в виде
высокомолекулярных конгломератов [246].
169
0 0,2 0,4 0,8 1,0
Содержание металла в катализаторе,йес.%
Рис. 73. Зависимость константы скорости образования углерод-кислородного
комплекса К\ от содержания металла в катализаторе:
/ — свинец. 2 ^ванадий; 3 — хром; 4 — молибден; 5 — железо; 6 — кобальт, натрий; 7 —
никель; 8 — медь; 9 — литий; 10 — калий, цезий, бериллий, магний, кальций, стронций.
23
21
19
77
75
13
11
11,72,73,1^,75,75
70
5,9
0 0,*t 0,8
Содержание металла
в катализаторе, бес. %
7,2
Рис. 74. Изменение константы скорости разложения углерод-кислородного
комплекса Кі в зависимости от содержания металла в катализаторе (на
исходном катализаторе Кг = 0,0226):
1 — свинец; 2 — ванадий; 3—хром; 4 — молибден; 5 — железо; 6 — кобальт; 7 — никель;
8 — медь; 9 — литий; 10 — натрий; И — калин; 12— цезий; IS — бериллий; 14 — магний;
15 — кальций; 16 — стронций.
Дополнительные данные о влиянии металлов на регенерацию
катализатора были получены при помощи термографического
анализа. На термограммах всех образцов, содержащих кокс,
обнаружен эндотермический эффект при 130—150 °С, соответствующий
удалению из катализатора сорбированной воды, и экзотермический
эффект в интервале температур от 300 до 750 °С, отвечающий
сгоранию коксовых отложений. Характерным является понижение
температуры, при которой наблюдается максимальный
экзотермический эффект, обусловленное добавлением тяжелых металлов.
Это понижение достигает 80 °С и имеет наибольшее значение при
добавлении хрома и ванадия. Обнаруженное явление указывает на
то, что тяжелые металлы катализируют процесс выжига коксовых
отложений.
Механизм отравления и промотирования катализатора
металлами. В литературе существует мнение, что металлы могут влиять
на качество катализатора двояко. Такие металлы, как никель,
ванадий, железо и другие, снижают активность и избирательность
катализатора [45, 64, 202, 213]; щелочные металлы, например
натрий, уменьшают только активность катализатора не изменяя
избирательности [45].
Отравление обоих видов рассматривается в работе [45], где
изучалось влияние различных концентраций никеля, ванадия,
железа, меди, свинца и натрия на результаты крекинга и качество
катализатора. Металлы наносили на катализатор пропиткой его
водными растворами солей. Ванадий вводили в виде метаванадата
аммония, а натрий — в виде ацетата. Остальные металлы вводили
в виде нитратов. Чтобы избежать попадания в катализатор
посторонних примесей растворы солей металлов приготовляли в
двукратно дистиллированной воде, а все сосуды перед употреблением
тщательно очищали, промывали и споласкивали также двукратно
дистиллированной водой. Пропитанные образцы высушивали при
90 °С, а затем прокаливали в воздухе при 600 °С в течение 2 ч для
разложения солей металлов до окислов и полного удаления
летучих веществ. Выходы продуктов крекинга в стандартных условиях
на полученных образцах катализатора приведены в табл. 48 [45].
Там же приводятся данные о кислотности, удельной поверхности
и поровой характеристике этих образцов.
Из табл. 48 видно, что никель, ванадий, железо, медь, свинец
уменьшают активность катализатора и избирательность:
уменьшается выход бензина, увеличивается выход газа и кокса. При этом
кислотность катализатора не изменяется. Данных о влиянии
металлов на удельную поверхность и поровую характеристику
катализатора в этой работе очень мало.
Активность катализаторов обусловлена наличием на их
поверхности кислотных активных центров и, следовательно, она связана
с кислотностью. В то же время активность зависит от удельной
поверхности катализатора. Неизменность указанных факторов при
отложении тяжелых металлов на поверхности катализаторов при-
171
Таблица
Выходы продуктов крекинга на
Показатели
Активность
Выход продуктов, мл, из
100 мл сырья
бензин
газ
в том числе
водород
кокс (г)
Отношение газ:бензин
Кислотность
катализатора, мг КОН/г
Удельная поверхность,
м2/г
Удельный объем пор.
м3/г
Средний диаметр пор, А
Содержание металлов
и
37,1
19,6
2533
184,9
0,274
129,2
38,74
133,6
0,183
54,8
никель
0,047
28,8
15,4
2300
627,9
0,259
149,4
39,99
—
—
0,121
26,0
13,9
2360
818,3
0,412
169,8
37,78
122,6
0,185
58,6
1,03
17,8
8,3
2800
152Ь,8
0,607
337,3
36,69
124,4
0,187
60,1
ванадий
0,(И2
36,0
18,4
2962
450,2
0,291
161,0
38,58
—
—
0.211
26,7
13,4
3290
1503,5
0,376
245,5
—
—
—
0,847
13,0
7,0
2916
1627,1
0,585
416,6
39,29
—
—
же
0,14
34,5
18,7
2075
215,8
0,302
110,0
—
—
—
вела исследователей [45, 203] к выводу, что эти отложения
образуют новый поверхностный слой, обладающий совершенно иными
каталитическими свойствами, в результате чего выход кокса и
водорода в процессе увеличивается. Испытание силикагеля с
нанесенным на него никелем подтвердило, что никель в условиях
крекинга полностью лишен крекирующей способности, но катализирует
образование кокса и водорода (см. табл. 48). Активность же
самого катализатора снижается вследствие блокирования его
активных центров повышенным отложением кокса, вызываемым
металлами [247].
Другая точка зрения на механизм отравления катализатора
высказывается в работе [239]. Ее авторы нашли, что зависимость
степени превращения сырья от кислотности алюмосиликатного
катализатора изображается прямой А (рис. 75), уравнение которой
имеет вид: степень превращения, вес. % =34Хкислотность+11,2
(кислотность определяли по адсорбции нормального бутиламина,
а изменяли ее водной либо кислотной обработкой катализатора).
После нанесения на катализатор примесей металлов пропиткой его
водными растворами солей опять определяли кислотность
образцов и их активность (по методу Кат-А). Эта зависимость для
образцов катализатора с содержанием окислов хрома, натрия, меди и
цезия изображена на рис. 75 пунктирными линиями. Из рисунка
видно, что при нанесении на катализатор металлов зависимость
между кислотностью и степенью превращения, установленная для
172
48
полученных образцах катализатора
в катализаторе, вес. %
лезо
0,334
25,1
13,0
3025
1216
0,431
232,7
38,80
—
—
—
0,929
13,9
7 0
3200
1309
0,495
437,1
37,72
—
—
—
медь
0,06
28,3
16,0
1600
373
0,279
112,5
39,65
—
—
—
0,20
26,8
14,5
2216
538
0,326
152,8
40,13
—
—
—
2,0
16,0
7 7
2750
932
0,607
355,7
36,32
—
—
—
свинец
0,06
33,3
17,7
1987
196
0,288
112,2
—
—
—
0,20
28,4
16,3
2000
223
0,264
122,7
38,40
—
—
—
2,0
35,5
14,1
2813
1015
0,317
199.5
37,15
—
—
—
натрий
0,06
35,4
19,1
2075
172,8
0,225
108,6
36,83
—
—
—
0,20
30,9
17,9
1688
180,6
0,200
94,3
35,93
133,5
0,189
56,6
2,1
5,5
3 Я
350
12,3
0,045
92,1
15,82
103,7
0,158
60,9
Содержание
металла в силика-
геле, вес. %
0
2,1
0 73
300
0
0,03
410,9
408,3
0,295
28,6
0,12
никеля
4,1
1,48
733
202,3
0,177
495,2
383,5
0,280
29,2
0.2
натрия
1,7
0,4
240
0,029
600
269,4
0,238
35,4
исходного катализатора, не соблюдается. Для образцов
катализаторов с металлами вычисленная кислотность выше, чем
фактическая. Отсюда авторы [239] делают вывод, что при добавлении
окиси металла некоторая доля протонов удерживается от участия в
реакции. Количество этих каталитически неактивных протонов
определяли по разнице между экспериментально найденной и
вычисленной кислотностью. Найдено, что 0,5 моль окиси хрома
связывает 1 моль протонов; 1 моль окиси меди связывает 2 моль протонов
и 0,5 моль окиси натрия — 3 моль протонов. Указанные
авторы считают, что изученные добавки металлов промотируют
крекинг в направлении образования кокса, водорода и низших
углеводородов в следующей последовательности (по возрастанию
активности) : натрий<цезий<хром<медь.
Увеличение содержания натрия в катализаторе вызывает
почти полное падение активности, что приводит к резкому
уменьшению выхода бензина, газа и кокса. Относительные выходы
продуктов крекинга при этом не изменяются, а кислотность катализатора
резко падает. Удельная поверхность, объем и радиус пор
катализатора при содержании натрия на катализаторе более 0,2 вес. %
уменьшаются. По данным [203], при содержании натрия на
катализаторе более 0,5% уменьшение удельной поверхности
становится значительным. При наличии иа катализаторе 0,7% натрия
удельная поверхность катализатора уменьшалась на 10% после
20 ч пропаривания при 677 °С. После термической обработки при
173
900 °С в течение 5 ч активность катализатора, содержащего 0,7%
окиси натрия, составляла 22%, а несодержащего 60%.
Отравление катализатора натрием объясняется тем, что он
входит в структуру алюмосиликата, замещая кислотный протон. Для
нанесения натрия на катализатор применяли [45] пропитку его
ацетатом натрия. По-видимому, натрий взаимодействует с
катализатором по уравнению:
X—Н + CH3COONa
X-Na + CHoCOOH
0,9 1,0 1,1 1,2 1,3
Кислотность, мг-зк8/мг
где X — остаток структуры катализатора.
При образовании продукта замещения высвобождается
уксусная кислота; это наблюдается на холоду, но особенно заметно при
нагреве. Аналогичная реакция
замещения ранее была предложена в
работе [248]. При замещении
протона ионом натрия возможность
протонного обмена между
катализатором и карбоний-ионом
уменьшается и с увеличением
концентрации металла активность
катализатора снижается.
Наши эксперименты показали,
что, в отличие от обычных
представлений, не все металлы,
накапливающиеся на алюмосиликатном
катализаторе, дезактивируют
катализатор крекинга. Некоторые
металлы при их концентрации на
катализаторе, выраженной
тысячными и сотыми долями процента,
способствуют увеличению выхода
бензина и уменьшению образования
кокса, хотя эти же металлы при большем их содержании
отравляют катализатор крекинга.
В других каталитических процессах известно много фактов
двойственного поведения примесей катализаторов. Улучшение
активности катализатора при небольшом содержании некоторых
веществ и существенное его отравление при их большем содержании
хорошо изучено советскими учеными [249, 250, 251].
Промотирование алюмосиликатного катализатора металлами
может быть объяснено карбоний-ионной теорией, хорошо
описывающей основные закономерности процесса каталитического
крекинга [252]. В соответствии с положениями этой теории,
непредельные углеводороды в присутствии алюмосиликатного
катализатора легко образуют карбоний-ион и претерпевают быстрое
дальнейшее превращение. Однако первая стадия превращения
парафиновых п нафтеновых уїлеводородов — образование карбоний-
Рис. 75. Зависимость между
интенсивностью крекинга и
кислотностью катализатора при
добавлении к нему окиси металлов.
Стрелками показано увеличение
концентрации металла.
174
иона — является наиболее длительной стадией, определяющей
суммарную скорость всего процесса. Ускорение образования кар-
боний-иона из насыщенных углеводородов позволило бы
существенно увеличить превращение сырья. В присутствии на
поверхности катализатора некоторого количества металлов эта медленная
стадия ускоряется. Воздействие металлов на ускорение
образования карбоний-иона из парафиновых углеводородов можно
представить следующим образом. і
Металлы, обладающие дегидрирующими свойствами,
способствуют образованию непредельных углеводородов из парафиновых
углеводородов. Последние под действием кислотной части
катализатора быстро образуют карбоний-ионы, которые могут, с одной
стороны, претерпевать дальнейшее превращение с образованием
обычных продуктов крекинга, с другой — могут отнимать ионы
водорода от парафиновых углеводородов, превращая их в карбоний-
ион. Оба направления способствуют ускорению расщепления
парафиновых углеводородов. В зависимости от природы металла,
присутствующего на поверхности катализатора, и его концентрации
ускорение образования непредельных углеводородов может
привести к самым различным результатам. Если металл является
слабым дегидрирующим агентом и содержится в небольшом
количестве в катализаторе, то образуется определенное количество
непредельных углеводородов, которые инициируют крекинг
насыщенных углеводородов; тем самым увеличивается степень
превращения сырья и возрастает выход бензина.
Оптимум по содержанию дегидрирующего металла,
обусловливающий максимальную активность катализатора, обеспечивает
оптимум и по содержанию непредельных углеводородов в
реакционной смеси. Имеются работы, подтверждающие резкое
увеличение начальной скорости крекинга парафиновых углеводородов
при добавлении в них олефинов и наличие определенного
оптимума. В работе [253] показано, что в присутствии олефинового
углеводорода в 4 раза ускоряется начальная скорость крекинга
парафинового углеводорода. Существует оптимум по давлению олефи-
на, сильно зависящий от температуры кипения. Так, для 399 и
482 °С он равен соответственно 15 и 20%. С увеличением
количества олефинов скорость крекинга насыщенных углеводородов
уменьшается, так как непредельные углеводороды адсорбируются
на активных центрах катализатора с большей скоростью, чем
насыщенные углеводороды.
Получающиеся при дегидрировании непредельные
углеводороды крекируются и образуют в несколько раз больше коксовых
отложений, чем парафиновые углеводороды. Так, по данным [254],
при крекинге пентена-2 и триметилэтилена коксообразование
почти в 18 раз больше, чем при крекинге соответствующих
парафиновых углеводородов. Кокс экранирует активные центры
катализатора, и в результате активность катализатора резко уменьшается.
Видимо, поэтому для металлов с сильными дегидрирующими
175
свойствами, например никеля, мы не обнаружили максимума
активности катализатора.
По современным представлениям, коксообразование на алю-
мосиликатном катализаторе протекает путем постепенного отрыва
атомов водорода от адсорбированных на поверхности
катализатора ненасыщенных молекул [15, 252]. Карбоний-ион
взаимодействует с молекулой олефина, образуя новый ненасыщенный
карбоний-ион, после распада которого образуется диолефин, а после
повторения процесса — триолефин и т. д. Образование карбоний-
ионов зависит, в первую очередь, от кислотности катализатора.
Нейтрализация кислотного центра катализатора (например,
щелочными металлами) уменьшает возможность протонного обмена
между катализатором и карбоний-ионом, что, в свою очередь,
уменьшает коксообразование. Хотя щелочные и щелочноземельные
металлы тормозят реакцию коксообразования, степень их влияния
на образование кокса для различных металлов неодинакова.
Сильно дегидрирующие металлы (никель, медь, кобальт) даже
при ничтожном их содержании в катализаторе приводят к резкому
увеличению коксоотложения вследствие повышенного образования
непредельных углеводородов. Слабодегидрирующие металлы
(ванадий, хром, молибден, железо) при небольшом их содержании в
катализаторе (до 0,01 вес. %) образуют меньше кокса, чем
исходный катализатор. При большем содержании металла в
катализаторе коксообразование увеличивается. При содержании тяжелых
металлов в катализаторе более 0,03—0,05 вес. % характер их
влияния на изменение времени, необходимого для отложения 2%
кокса, одинаков. По уменьшению количества образующегося кокса
исследованные металлы располагаются в следующем порядке:
никель, медь>кобальт>молнбден, ванадий>железо, хром>сви-
нец>бернллий, магний, кальций, стронций>литий>натрий>ка-
лнй>цезпй. Тормозящее влияние щелочных металлов возрастает
в соответствии с увеличением их основности [257].
Энергия связи катализатора с взаимодействующими на его
поверхности атомами оказывает влияние на осуществление любых
каталитических реакций. Величины энергий связи атомов,
участвующих в реакции, с активными центрами катализатора Qa-к
являются важнейшими характеристиками системы, так как они
учитывают специфику как реагирующих молекул, так и активной
поверхности катализатора. Очевидно в частном случае
каталитического крекинга — при более прочной связи атомов углерода,
которые являются источником образования кокса, с металлами, они
будут дольше удерживаться на поверхности катализатора, и это
будет способствовать увеличению коксообразования. Таким
образом, для повышения коксообразования требуется большая энергия
связи углерода с металлом катализатора Qc-k- Действительно при
уменьшении энергии связи Qc-к углерода с металлом, введенным
в состав катализатора, время до отложения 2% кокса
увеличивается (табл. 49).
176
Таблица 49
Зависимость между энергиями связи углерода
катализаторами Qc-K и коксообразованием в процессе
каталитического крекинга
Атомный номер
элемента
4
24
26
23
42
28
29
Окись металла
или металл
ВеО
Сг2О3
Fe
v2o3
МоО3
Ni
Си
О
Д ж/моль
47 200
83 000
80 800
86 200
97 000
117 500
141000
Время до
отложения 2% кокса,
мин
26
23
23
21
20
10
10
Таким образом, зная энергию связи углерода с металлом,
введенным в катализатор, можно предсказать коксообразующее
влияние добавок. Чем выше будет энергия связи, тем интенсивнее
должно быть коксообразование в процессе каталитического
крекинга в присутствии данного катализатора.
Физико-химические и каталитические свойства вещества
определяются в конечном счете электронной структурой его атомов
(ионов). В связи с этим представляет интерес проследить влияние
металлов, добавленных к алюмосиликатному катализатору, на
коксообразование и регенерацию катализатора в зависимости от их
положения в периодической системе элементов Д. И. Менделеева.
Наши данные, представленные на рис. 76, показывают, что
действительно существует определенная зависимость изменения
коксообразования катализатора и его регенерации от положения
металла в периодической системе. Если рассмотреть элементы
4 периода, по которым мы имеем более полные данные, то видно,
что металлы, расположенные по концам периода (калий, рубидий),
способствуют уменьшению коксообразования, в то время как на
скорость выжига кокса они влияют незначительно. Металлы же,
расположенные в средней части периода (кобальт, никель, медь),
ускоряют процесс коксообразования и некоторые из них
одновременно сильно катализируют и регенерацию катализатора.
Элементы, входящие в главную подгруппу I группы, мало различаются
по характеру их влияния на скорость образования кокса. Но особо
здесь можно выделить легкие металлы, которые резко усиливают
регенерационную способность алюмосилікатного катализатора.
Влияние на скорость образования кокса и на регенерацию
катализатора элементов главной подгруппы II группы совершенно
идентично.
Учитывая, что добавленные металлы находятся на поверхности
катализатора в виде окислов, мы можем связать их действие на
коксообразование и регенерацию с такими свойствами металлов,
12-2206
177
которые, согласно существующим теоретическим представлениям
[258], непосредственно определяют каталитическую активность,
как, например, ширина запрещенной зоны, работа выхода
электрона.
w го зо ko
Атомный номер элемента
50
Рис. 76. Зависимость длительности ззкоксовывания и регенерации алюмосили-
катного катализатора от атомного номера элемента.
Пунктирная линия относится к исходному катализатору.
Как видно из приведенных на рис. 77 и 78 кривых, коксообразо-
вание увеличивается с уменьшением ширины запрещенной зоны и
с увеличением работы выхода электрона. В такой же зависимости
от этих свойств находится и длительность выжига коксовых
отложений на катализаторе (см. рис. 79).
Существование описанных выше зависимостей позволяет
предсказывать влияние металлов, нанесенных на алюмосиликатный ка-
178
тализатор, на направление каталитического крекинга. Элементы
главных подгрупп I и II групп способствуют уменьшению
образования кокса в процессе каталитического крекинга при
одновременном снижении активности катализатора в результате
нейтрализаr: 2 if В 8 10
'Нирино запрещенной зоны , эВ
Рис. 77. Зависимость времени до
отложения 2% кокса от ширины
запрещенной зоны.
60 V=
W -
I
"'s
I
10 -
- 'ca
-•
Sr
-
і
/ 2
W
3
'•Be
v ««mcA
Co
Cu
if 5
-
•• I
6
N Работа Выхода электрона, эВ
Рис. 78. Зависимость коксообразую-
щей способности металла от работы
выхода электрона.
ции кислотных центров. Воздействие металлов I группы сильнее,
чем II группы. Легкие элементы I группы существенно улучшают
регенерацию катализатора. Элементы побочных подгрупп I и
о
if 8 1Z
Ширина запрещенной
зоны, эВ
г 4
Работа выхода
электрона, зв
Рис. 79. Зависимость длительности выжига половины отложившегося кокса от
работы выхода электрона и ширины запрещенной зоны.
II групп и элементы VIII группы резко усиливают коксообразова-
ние, но улучшают выжиг коксовых отложений. Элементы III и
IV групп, по-видимому, будут незначительно влиять на
образование кокса и регенерацию катализатора. Наиболее выгодно
добавлять в алюмосиликатный катализатор металлы V и VI групп, ко-
12* Ш
торые при большой концентрации умеренно усиливают коксообра-
зование и весьма значительно улучшают регенерацию
катализатора. Особенно важно, что при небольшом содержании эти металлы
модифицируют алюмосиликатный катализатор, в результате чего
увеличивается выход бензина и улучшается качество всех
продуктов крекинга.
Существенного изменения влияния металлов на коксообразова-
ние в пределах одной группы не замечено. В то же время ясно
проявляется тенденция к уменьшению их влияния на регенерацию
катализатора в пределах одной группы при переходе к тяжелым
металлам.
На практике могут быть отклонения от предсказанного в ту или
иную сторону, поскольку в реальных условиях металл на
поверхности катализатора может находиться в виде самых различных
соединений п в разных валентных состояниях. Однако, по нашему
мнению, высказанные предположения могут быть полезны при
подборе модифицирующих добавок и при предварительной оценке
отравляющего действия на катализатор отдельных металлов.
ЗАКЛЮЧЕНИЕ
Сернистые и кислородные соединения не оказывают влияния на
качество синтетических катализаторов, однако азотистые
основания, нейтрализуя кислотные центры, очень сильно их
дезактивируют. Особенно резкое влияние на результаты процесса
каталитического крекинга оказывают металлы. В отличие от существующих
представлений, некоторые металлы (ванадий, молибден и др.) при
небольшом содержании способствуют увеличению активности
катализаторов. По влиянию на новообразование металлы делятся
на три группы: 1) увеличивающие образование кокса (никель,
медь и др.); 2) уменьшающие (щелочные и щелочноземельные
металлы) ; 3) уменьшающие коксообразование при небольшой
концентрации и усиливающие его при значительном содержании
(ванадий, молибден, хром и др.).
Металлы, содержащиеся на поверхности катализатора,
практически не влияют на скорость выжига коксовых отложений в
диффузионной области и существенно ускоряют регенерацию
катализатора в кинетической области. Исследованные нами металлы
по степени убывания их воздействия на скорость окисления кокса
в кинетической области располагаются в следующий ряд: хром>
>ванадий>литий>молибден, медь, натрий>железо>кобальт,
никель>бериллий, магний, кальций, стронций>калий>цезий>
>свинец.
Снижение выхода бензина и увеличение коксообразования при
накоплении металлов и других ядов на поверхности алюмосили-
катного катализатора существенно ухудшает
технико-экономические показатели процесса каталитического крекинга. В связи с
этим весьма важно или предварительно очистить сырье или
удалить металлы с поверхности отравленного катализатора.
180
Глава V
МЕТОДЫ ОЧИСТКИ СЫРЬЯ ОТ КОМПОНЕНТОВ,
ОТРАВЛЯЮЩИХ КАТАЛИЗАТОРЫ КРЕКИНГА
Некоторые компоненты сырья (азотистые основания,
соединения металлов, смолистые вещества и др.) отравляют алюмосили-
катные катализаторы, в связи с чем значительно ухудшаются
результаты каталитического крекинга [7, 8]. Резко уменьшается
выход целевых продуктов каталитического крекинга и,
следовательно, существенно ухудшаются экономические показатели процесса.
Существуют два метода предотвращения вредного воздействия
компонентов сырья на результаты крекинга: а) очистка сырья
крекинга с целью удаления металлов и других нежелательных
компонентов; б) удаление накопившихся металлов с поверхности
катализатора крекинга.
Разработаны разнообразные методы подготовки сырья для
процесса каталитического крекинга [8]. Использование их более
целесообразно, чем деметаллизация катализатора, так как при этом
удаляются не только металлы, но и другие нежелательные
компоненты сырья.
ПЕРЕГОНКА И ТЕРМИЧЕСКИЕ МЕТОДЫ
ОЧИСТКИ СЫРЬЯ
В процессе фракционирования нефти можно в какой-то мере
регулировать количество металлов в дистиллятах, направляемых
на каталитический крекинг. Это вполне выполнимо, поскольку
содержание металлов в дистиллятах резко увеличивается по мере
утяжеления фракций [176]. Считают, что металлы могут
попадать в состав дистиллята при вакуумной перегонке вследствие
летучести органических соединений металлов, а также из-за уноса
капель жидкости в процессе. Поэтому на характер распределения
металлов по фракциям существенное влияние оказывает
используемый метод ректификации. По влиянию условий работы
колонны и величины отбора вакуумного газойля на содержание в нем
металлов весьма показательны данные, приведенные в работе
[259]. При одинаковом отборе из туймазинской нефти A2%) на
колонне с четырьмя желобчатыми тарелками вакуумный газойль
содержал значительно меньше металлов, чем полученный на ко-
18t
лонне с двумя отбойными тарелками, что видно из следующих
данных (в %):
Колонна січетьірьмя Колонна с двумя
тарелками тарелками
Ванадий 0,076-Ю-4 О.Зб-Ю
Никель 0,036-Ю-4 0,17 Ю
Железо 0,53-Ю-4 0,82-10-*
На основании экспериментальных данных авторы этой работы
делают вывод, что при отсутствии специальных методов
подготовки сырья крекинга конец кипения отбираемой фракции в
вакуумной колонне не должен превышать 500 °С. Данные работы [260]
показывают, что промышленные вакуумные колонны позволяют
получать вакуумный газойль, содержащий не более 0,05 -10~4%
металлов. Имеющиеся литературные данные [8] подтверждают
результаты предыдущей работы. Таким образом, при
квалифицированной вакуумной перегонке качество получаемого газойля можно
существенно улучшить. Однако дальнейшее улучшение качества
сырья каталитического крекинга и увеличение его отбора без
применения специальных методов очистки невозможно.
Один из таких методов — так называемый термический метод
подготовки сырья каталитического крекинга. Имеются данные
[261], что летучесть металлоорганических соединений даже после
очень слабой термической обработки значительно снижается.
В связи с этим появилось несколько вариантов предварительной
термической обработки сырья крекинга. Сочетание легкого
крекинга с вакуумной перегонкой дает по содержанию металлов лучшие
результаты по сравнению даже с вариантом предварительной деас-
фальтизацни, не говоря уже об обычной перегонке. Авторы [261]
приходят к выводу, что лучшей при выборе схемы завода
является глубокая подготовка сырья с углубленным отбором (например,
легкий крекинг в сочетании с вакуумной перегонкой) с
последующим двухступенчатым каталитическим крекингом.
Весьма распространенный метод увеличения ресурсов сырья
каталитического крекинга — коксование [260] — не может быть
признан лучшим вследствие большого содержания в таком сырье
непредельных углеводородов. Для повышения качества сырья на
ряде заводов прибегают к циркуляции дистиллятов в процессе
коксования или к дополнительной очистке сырья крекинга, а также
циркулирующих газойлей каталитического крекинга.
ДЕАСФАЛЬТИЗАЦИЯ
Путем деасфальтизации тяжелых остатков пропаном или
бутаном можно получать большие количества высококачественного
сырья каталитического крекинга. Деасфальтизат можно
смешивать с другими видами сырья, направляемыми на каталитический
крекинг. Поскольку основное количество вредных для
катализатора компонентов содержится в высококипящих фракциях сырья,
182
эффективной является даже очистка незначительной глубины. Так,
при деасфальтизации мазута пропаном и удалении 7,3% асфаль-
тенов содержание ванадия снижается на 58%, никеля на 52%,
серы на 27,6%', азота на 25%. При углублении очистки качество ра-
фината изменяется не так существенно. При очистке с выходом
рафината 72 и 65% ванадий в обоих случаях удаляется на 96%,
никель на 94%, азот на 78—80%, сернистые соединения на 30—
33% [8]. Результаты деасфальтизации и обычной вакуумной
перегонки свидетельствуют о преимуществе деасфальтизации [196].
Удаление металлов, азотистых и сернистых соединений позволяет
существенно улучшить показатели каталитического крекинга.
Весьма подробный анализ некоторых методов подготовки сырья
был проведен в работе [261]. Эффективность методов
сравнивалась по величине коксообразования при каталитическом крекинге
и по содержанию металлов в сырье. В этой работе отмечается, что
при почти одинаковом выходе дистиллята деасфальтизация дает
сырье несколько лучшего качества, чем вакуумная разгонка. С
точки зрения содержания металлов в сырье крекинга наблюдается
явное превосходство деасфальтизации по сравнению с вакуумной
перегонкой.
При изучении зависимости степени удаления металлов от
выхода деасфальтизата [262] оказалось, что достаточно глубокое
удаление металлов при очистке достигается лишь при больших
потерях продукта с тяжелым остатком. При повышении глубины
отбора деасфальтизата с 40 до 60 объемн. % концентрация в нем
вредных металлов возрастает на 400%. В связи с этим некоторые
исследователи предприняли попытки интенсифицировать процесс
деасфальтизации [196, 262] и даже комбинировать его с другими
методами, например с фенольной и гидрогенизационной очисткой
деасфальтизатов [263].
Определенный интерес представляет метод деасфальтизации
с применением промывной жидкости [196, 262]. В качестве такой
жидкости предлагается использовать фракции, богатые
ароматическими углеводородами, экстракты, полученные из тяжелого
газойля каталитического крекинга [262], рециркулирующее масло,
кипящее выше 455 °С и включающее остатки продуктов крекинга
газойля [196], и др. Имеются также предложения [8] по осаждению
коксообразующих компонентов в процессе деасфальтизации
органическими водонерастворимыми солями.
Поскольку деасфальтизация позволяет получать
дополнительное количество качественного сырья для каталитического крекинга
из остаточных продуктов, она нашла широкое распространение в
зарубежной практике.
ОЧИСТКА СЕЛЕКТИВНЫМИ РАСТВОРИТЕЛЯМИ
Из селективных растворителей несколько большее внимание
привлек фурфурол. Имеются попытки использовать его для
очистки не только циркулирующих газойлей, но и всего сырья крекинга
183
в целом. По данным работы [8], при очистке вакуумного газойля
фурфуролом его качество существенно улучшается, но теряются
относительно большие количества газойля. Весьма тщательный
анализ процесса очистки фурфуролом как сырья, так и
циркулирующего газойля каталитического крекинга приведен в работе
[264]. Однако по этим данным, наиболее приемлемая по качеству
продуктов очистка сопряжена с относительно большими потерями
исходного сырья. Каталитический крекинг очищенного сырья
позволяет увеличить глубину разложения до 52,5% вместо 51,3 при
неочищенном сырье. Выход бензина увеличивается с 41,0 до 47,6%
при примерно одинаковом выходе кокса. В результате очистки
тяжелого циркулирующего газойля каталитического крекинга
коксуемость снижается с 1,17 до 0,02%, содержание серы — с 0,95
до 0,46, содержание металлов (ванадий и никель) — с 2,1-10~4 до
1,1-10-"%.
В литературе имеется еще несколько сообщений об
использовании фурфурола для очистки сырья крекинга [265] и
циркулирующего газойля [266]. Указывается [264], что расход фурфурола
составляет всего лишь 0,015% от сырья. В работе [266] описывается
процесс очистки циркулирующего газойля каталитического
крекинга и приводятся результаты анализа рафината и экстракта. При
использовании в качестве сырья ароматического экстракта из
циркулирующего газойля каталитического крекинга выход кокса 12%,
бензина 3%, газа 4,7%. При крекинге рафината выход этих
продуктов соответственно 1,6; 42,2 и 6%. Использование этого
процесса считается экономически оправданным.
Некоторые данные о применении очистки селективными
растворителями приводятся также в работе [267].
АДСОРБЦИОННАЯ ОЧИСТКА
В литературе имеются указания об удалении металлических
загрязнений из сырья каталитического крекинга и сырой нефти
путем адсорбционной очистки на бокситах, отбеливающей глине,
гранулированном коксе, материале, состоящем в основном из
окислов металлов, и на катализаторе крекинга.
Для удаления натрия нефтяное сырье пропускают через слой
боксита при 345—455 °С под давлением порядка 7 МПа.
Дальнейшая обработка в таких же условиях в присутствии водорода
способствует удалению ванадия [8, 268]. Контактирование сырья
каталитического крекинга, нагретого до температуры выше 200 °С,
с отбеливающей глиной приводит к адсорбции на ее поверхности
органических соединений металлов. Адсорбент после отпарки
направляется на регенерацию. В качестве контакта для очистки
тяжелого газойля от металлических загрязнений можно применять
гранулированный кокс. Процесс осуществляют при 425—455 °С
и объемной скорости подачи сырья 0,5—5,0 ч^1, обеспечивая выход
184
продукта до 90%. Содержание никеля снижается примерно на
85%, а ванадия на 80% [269].
Пропуская сырую нефть или нефтепродукты через контактный
материал, состоящий из окислов титана и алюминия или окислов
железа и алюминия или немагнитного гематита, при 400—427 °С
и 3,5—10,5 МПа, можно очистить сырье от ванадия и натрия,
которые остаются на адсорбенте,. Из остаточных нефтепродуктов
(например отбензиненной нефти) металлы удаляют при контакте с
немагнитным гематитом, имеющим частицы с поверхностью более
20 м2, при 410—470 °С, давлении 3,5—10,5 МПа, объемной
скорости подачи сырья 0,5—2 ч^1 в присутствии водорода [270]. После
фильтрации нефти через слой фосфорнокислого катализатора при
100 °С и объемной скорости подачи сырья 1,0 ч~' содержание
ванадия снизилось с 0,023 до 0,013% и никеля с 0,0053 до 0,0018%
[271]. Имеются данные [272] об удалении металлов из нефтяного
сырья, предназначенного для крекинга в псевдоожиженном слое.
Сырье каталитического крекинга (мазут или отбензиненная нефть)
контактируется с тонкоразмолотым катализатором крекинга при
150—540 °С. Длительность контакта зависит от температуры: при
260 °С — до 10 ч, при 540 °С — менее 1 мин. В то же время
превращение тяжелого сырья в низкокипящие продукты не должно
превышать 20—25%. Количество контакта должно быть от 0,1 до
10 кг/кг коксового остатка в обрабатываемом сырье. Из зоны
предварительной очистки очищенное от металлсодержащих
компонентов сырье вместе с контактом поступает в зону каталитического
крекинга с псевдоожиженным слоем катализатора, величина
частиц которого значительно больше величины частиц контакта.
Контакт вместе с катализатором после реактора поступает на
регенерацию, затем в сепараторах циклонного типа отделяется от
катализатора и возвращается в зону очистки.
Указывается [272], что металлические загрязнения и коксооб-
разующие вещества удаляются из сырья при обработке его в
трубе-реакторе при температуре более 480 °С отработанным
катализатором, выгруженным из основного реактора вследствие потери
активности.
Для удаления азотистых соединений из сырья крекинга
предлагается контактировать его с адсорбентами, такими, как бентонит,
каолинит, после превращения их в кислую форму [273]. Подача
глины составляет 0,18 м3 на 1 м3 сырья. Отработанную глину
регенерируют путем выжига отложений или обработкой селективным
растворителем, растворяющим азотистые основания (смесь
бензола и спирта). Сырье можно очищать от азотистых органических
соединений, пропуская его через частично дегидратированный цео-
литный алюмосиликат, содержащий металл [274]. В результате
контактирования получают адсорбированные азотистые
соединения и рафинат. Насыщенный адсорбент обрабатывают
специальным агентом и выделяют экстракт, содержащий десорбированные
органические азотистые соединения. Очистке сырья от азоторгани-
185.
ческих соединении на синтетическом цеолите посвящен патент
США [275].
Несмотря на большое число предложений по применению
адсорбционных методов очистки сырья крекинга, широкого
промышленного применения они еще не получили.
ОЧИСТКА СЕРНОЙ КИСЛОТОЙ
Метод облагораживания нефтепродуктов путем очистки серной
кислотой был весьма распространен. Однако он имел следующие
недостатки: большая длительность отстоя кислых гудронов, а
также отработанных щелочей и промывных вод в стадии
нейтрализации, в связи с чем (а также из-за образования относительно
стойких эмульсий) требовалось иметь большие емкости для отстоя;
коррозия аппаратуры; невозможность использования кислого
гудрона. Поэтому очистка серной кислотой была заменена другими
методами.
В последнее время основные недостатки сернокислотной
очистки были устранены. Этот метод получил новое технологическое
оформление с применением электроосадителя для отделения кис^
лого гудрона и отработанной щелочи [276, 277]. Разделение фаз
в электрическом поле позволяет резко сократить длительность
отстоя. Это дает возможность применить более эффективные методы
контактирования реагентов с нефтепродуктом и обеспечить
максимальную глубину очистки при минимальном расходе реагентов, а
также существенно уменьшить размеры аппаратов. Появился
значительный опыт по борьбе с коррозией аппаратуры. Появились и
разнообразные методы утилизации кислого гудрона [8]. Все
вышеуказанное позволило опять использовать этот метод для
подготовки сырья каталитического крекинга.
Впервые для удаления металлов, содержащихся в сырье
каталитического крекинга, была предложена промывка нефтепродуктов
кислотой концентрацией 10% при расходе ее около 30 объемы. %
[278]. Это позволило существенно снизить содержание металлов.
Были опубликованы также работы, в которых для очистки сырья
каталитического крекинга рекомендовалось применение
концентрированной кислоты [183; 279—284]. Одновременно появились
публикации об исследованиях по применению сернокислотной
очистки сырья каталитического крекинга ряда советских [285] и
зарубежных [182, 286, 287] авторов.
В работе [280] в лабораторных условиях очистке серной
кислотой концентрацией 70—95% подвергали вакуумные газойли,
полученные из сернистой туймазинской, а также из высокосернистых и
высокосмолистых чекмагушской и арланской нефтей. Расход
кислоты составлял от 0,5 до 10,0 объемн. %. Было установлено, что
с увеличением расхода и концентрации кислоты выход очищенного
продукта несколько уменьшается. Наиболее резко выход продукта
изменяется при расходе кислоты до 1—2 объемн. %, а также при
186
очистке крепкой кислотой. В результате удаления наиболее
тяжелой смолистой части сырья плотность газойля снижается, причем
особенно резко при очистке газойля кислотой концентрацией
более 90%. При расходах кислоты до 2 объемн. % коксуемость и
содержание серы быстро изменяются (рис. 80). Однако, как и
следовало ожидать, глубина обессеривания во всех исследованных
условиях не превышает 15%; очистка кислотой концентрацией 85%
практически не снижает содержания серы в вакуумном газойле.
то
1
і
ии
80
60
20
•—•
-
Г
ч
о
J
2
Ь
во
60
2
Расход
і* Б 8
кислоты , оаьемн %
10
SO 85 30
Концентрация кислоты ,
%
Рис. 80. Зависимость степени удаления азотистых и сернистых соединений и
смолистых веществ от расхода кислоты концентрацией 94,5% при сернокислотной
очистке вакуумного газойля арлаиской нефти:
/ — сера; 2 — кокс по Конрадсону; 3 — общий азот; 4 — основной азот.
Рис. 81. Влияние концентрации кислоты на степень удаления азотистых и
сернистых соединений и смолистых веществ нз вакуумного газойля арланской
нефти:
/ — основной азот, расход кислоты 5 и 10%; 2 — общий азот, расход кислоты 5 и 10%;
3, 4 — кокс по Конрадсону, расход кислоты соответственно 10 н 5%; 5 — сера, расход
кислоты 5 и 10%.
Сернокислотная очистка дает хорошие результаты по удалению
азотистых соединений. При расходах кислоты выше 1,0 объемн. %
степень удаления азотистых соединений достигает 98—99% для
основного и 78—79% для общего азота и не зависит от
концентрации кислоты (рис. 81).
При сернокислотной очистке вакуумного газойля из него
удаляется значительное количество металлов [288]. Характер
зависимости степени удаления металлов от расхода кислоты для всех
металлов одинаков (рис. 82, а). При небольших расходах кислоты
1ST
степень их удаления быстро увеличивается, а при больших
расходах практически не изменяется. Опыты, проведенные при
относительно больших расходах кислоты E, 10%), показывают, что при
О 2 4 6 в 10
Расход кислоты, ооьемн. %
-100
|
75 -
I
25 -
A
АО / V
-
R
а
-
—0а
1
2
\
і
і
—л.
д
<?0 55 5tf SS"
Кониенп,рация кислоть/, вес. %
6
Рис. 82. Зависимость степени удаления металлов от расхода (а) и
концентрации (б) кислоты:
I — никель; 2 — ванадий; 3 — железо.
изменении концентрации кислоты от 80 до 95% степень удаления
металлов не меняется (рис. 82, б). После сернокислотной очистки
групповой углеводородный состав вакуумных газойлей
существенно изменяется: снижается
содержание смолистых веществ и
полициклических ароматических
углеводородов и увеличивается
содержание ыетано-нафтеновых
углеводородов. Благодаря этому
ценность очищенного вакуумного
газойля как сырья для
каталитического крекинга возрастает.
Была проведена оценка
очищенного сырья на лабораторной
устанозке с неподвижным слоем
шарикового равновесного алюмо-
силикатного промышленного
катализатора с индексом
активности 29—30 пунктов при 450 °С и
объемной скорости подачи сырья
0,7; 1,0; 1,3 ч~'. Анализ
полученных данных показал, что для
данного вида сырья по мере
увеличения расхода и концентрации
о г г з 4 5
Расход кислоты, dec. %
Рис. 83. Влияние расхода кислоты на
выход бензина при крекинге
вакуумных газойлей:
I, 2 —вакуумный газойль туймазннскнй,
концентрация кислоты соответственно 80 и
94,9%; 3, 4 — вакуумный газойль
соответственно чекмагушскнй и арлаиский,
концентрация кислоты 94,6 и 94,5%.
188
кислоты выход бензина возрастает (рис. 83). При очистке сырья
крепкой кислотой резкое увеличение выхода бензина наблюдается
при расходе кислоты до 1,0 объемн. %.
Выход легкого газойля с увеличением расхода кислоты на
очистку несколько повышается. При очистке сырья крекинга
2 объемн. % 95%-ной кислоты выход легкого газойля при крекинге
туймазинского вакуумного газойля повышается в среднем на 1%,
а при крекинге чекмагушского газойля — на 2,5—3,0% на сырье,
60 <
\ 30
О 25 50
Концентраци
75 100
го
\
О 25 50 75 100
Концентрации кислоты , %
Рис. 84. Изменение выхода бензина (а) и кокса (б) при каталитическом
крекинге туймазинского вакуумного газойля, очищенного серной кислотой разной
концентрации:
а — расход кислоты 2,0%; объемная скорость, ч-1: / — 0,7; 2—1,0; 3—1,3; б — расход
кислоты, объемн. %: /. ¦*, 7 — 0.5; 2, 5, 8 — 2,0; 3, 6. 9 — 5.0; объемная скорость, ч-': 1,2.3 —
0,7; 4, 5. 6 — 1,0; 7, 8. 9 — 1,3.
что составляет соответственно 3 и 10 отн. %. Количество тяжелого
газойля уменьшается в среднем на 4—7% на сырье. При
увеличении глубины очистки сырья крекинга выхода газа и кокса также
уменьшаются.
На рис. 84 приводятся данные о выходе бензина и кокса при
очистке туймазинского вакуумного газойля кислотой разной
концентрации. Во всех случаях при концентрации кислоты более 80%
выход бензина возрастает и образование кокса уменьшается.
Результаты сернокислотной очистки сильно зависят от природы
перерабатываемого сырья и улучшаются в случае менее
качественных видов вакуумного газойля.
В процессе каталитического крекинга вакуумного газойля туй-
мазинской нефти, очищенного 95%-ной серной кислотой при ее
расходе 2,0 объемн. %, относительное увеличение бензинообразо-
вания составляет 107—109%, а выход кокса снижается на 12—
25%. Очистка более смолистого и сернистого сырья — вакуумного
газойля чекмагушской нефти — при тех же условиях приводит к
189
увеличению выхода бензина на 12—19% и уменьшению коксообра-
зования на 30—38 отн. % от выхода кокса при крекинге
неочищенного газойля. При сернокислотной очистке газойля арланской
нефти выход бензина увеличивается на 23 отн. %, и коксообразование
снижается на 30 отн. %.
Качество продуктов каталитического крекинга вакуумного
газойля — исходного и очищенного серной кислотой — неодинаково.
Содержание серы в бензине по мере углубления очистки вакуум-
1 Z 3 4 5 в
Расход кислоты, ?бъемн. %
а
г з
кислоты ,
б
объем н
5
%
Рис. 85. Содержание серы (а), ароматических и непредельных углеводородов
(б) в бензинах в зависимости от расхода кислоты на очистку вакуумных
газойлей:
й. 1, 2 — вакуумный газойль туймаэинский, концентрация кислоты соответственно 94,9 и
80,0%; 3 — вакуумный газойль чекмагушский. концентрация кислоты 94,6%; 4 — вакуумный
газойль арлаискнй, концентрация кислоты 94,5%; объемная скорость 1,0 ч-1. б. вакуумный
газойль чекмагушский: объемная скорость, ч—': /, 6—1,3; 2, 5 — 1,0; 3, 4 — 0,7; ¦ /', 2, 3 —
непредельные углеводороды; 4, 5, 6 — ароматические углеводороды.
ного газойля уменьшается (рис. 85, а). При крекинге очищенных
серной кислотой вакуумных газойлей туймазинской и арланской
нефти снижается содержание непредельных и увеличивается
содержание ароматических углеводородов в бензинах каталитического
крекинга (рис. 85, б). Это способствует улучшению стабильности и
октановой характеристики бензинов.
В результате сернокислотной очистки сырья несколько
облагораживается состав газа каталитического крекинга. В частности,
уменьшается содержание легких его компонентов. Так, в жирном
газе крекинга исходного арланского вакуумного газойля
содержится водорода 1,5%, метана 10,2%, в то время как в газе крекинга
того же газойля, очищенного 5%-ной H2SO4, содержится
метана 6,3%, водорода 0,94%. Наряду с этим несколько увеличива-
190
ется образование тяжелых компонентов газа. В частности, выход
бутан-бутиленовой фракции возрастает с 3,4 до 4,4 вес. % на сырье
крекинга, или на 30 отн. %. Это увеличение протекает в основном
за счет большего образования изобутана (с 1,8 до 2,5 вес. % на
сырье). Последнее обстоятельство указывает на предпочтительное
протекание вторичных реакций при крекинге очищенного сырья.
Эффективность сернокислотной очистки наиболее наглядно
видна при сопоставлении глубины превращения неочищенного и
очищенного сырья в случае одинакового выхода кокса D,5 вес. %').
Углубление крекинга в случае очищенного сырья составляет
14 отн. % для туймазинского вакуумного газойля и 49 отн. % Для
чекмагушского.
Следовательно, при очистке серной кислотой даже худшего
вида вакуумного газойля материальный баланс каталитического
крекинга улучшается. Кроме того, улучшение соотношения
выходов кокса и светлых нефтепродуктов делает такую очистку
особенно эффективной на действующих установках, где
производительность и глубина каталитического крекинга лимитируются коксовой
нагрузкой регенераторов. Установлено, что улучшение
материального баланса и качества продуктов крекинга достигается при
очистке кислотой концентрацией 95% и расходе 2 объемн. %•
Опыты по сернокислотной очистке и каталитическому крекингу
вакуумных газойлей, проведенные на пилотных установках,
полностью подтвердили лабораторные данные.
ГИДРОГЕНИЗАЦИОННАЯ ОЧИСТКА
В последние годы большое внимание уделяется подготовке
сырья каталитического крекинга методом гидроочистки. Интерес
к этому методу особенно возрос в связи с появлением ресурсов
дешевого водорода с установок каталитического риформинга.
Процесс гидроочистки является относительно громоздким и
дорогостоящим, однако он имеет следующие существенные достоинства:
резко увеличивается выход целевых продуктов крекинга;
существенно снижается отравление алюмосиликатного катализатора;
отпадает необходимость в дополнительной очистке дистиллятов
крекинга от сернистых соединений; достигается полная утилизация
серы, содержащейся в сырье; уменьшается коррозия аппаратуры;
улучшаются условия работы обслуживающего персонала и др.
Использованию гидроочистки для подготовки сырья крекинга
посвящено много работ [289—298]. Исследования проведены в
широком диапазоне режимов с применением в качестве сырья
различных фракций из разнообразных нефтей. Одним из наиболее
детальных зарубежных исследований в этой области является
работа [289], в которой приводятся результаты лабораторных,
полузаводских и промышленных экспериментов. Гидроочистку проводили
по типу процесса фирмы Шелл с низкими скоростями газа,
вследствие чего большая часть сырья, пропускаемого нисходящим пото-
191
ком через слой катализатора, остается в жидком состоянии.
Авторы этой работы считают, что таким путем жидкое сырье смывает
с катализатора вещества, снижающие срок его службы. Из
большого числа испытанных катализаторов наиболее активными для
этого процесса оказались никельвольфраммолибденовый и никель-
молибденовый на носителях. Отмечается, что при одинаковом
весовом содержании активных металлов стоимость последнего
меньше, чем обычно применяемого кобальтмолибденового
катализатора.
Гидрогенизации подвергали типичный вакуумный газойль из
западнотехасской нефти при различных режимах. Было
установлено, что во всех случаях в результате гидрогенизации качества
вакуумного газойля существенно изменяется: снижается содержание
полицнклнческих углеводородов, ухудшающих результаты
каталитического крекинга, а также содержание серы, азота и металлов.
При крекинге очищенного газойля выход кокса снижается до
65% по сравнению с выходом из неочищенного сырья при
одинаковой глубине превращения. Выход бензина повышается на 20%.
Крекинг-бензин, получаемый из гидрированного сырья, имеет
более высокие октановые числа — 86,0 против 84,1 (по моторному
методу), меньшее содержание серы @,01 вместо 0,18%). Легкий
газойль характеризуется пониженным содержанием серы @,12
против 1,5%), более высоким дизельным индексом C0 против 22) и
улучшенной стабильностью цвета. Проверка этого метода в
промышленных условиях в течение 1,5 лет показала
удовлетворительное совпадение с результатами лабораторных исследований.
Несмотря на сравнительно мягкие условия гидрирования качество
крекинг-сырья значительно повышается, о чем свидетельствует
снижение плотности, уменьшение содержания серы на 80%,
коксуемости по Конрадсону на 65%, содержания азотистых
оснований на 25%. Какого-либо влияния металлов, содержащихся в
вакуумном газойле, на катализаторы гидрирования в течение
полуторагодичной работы обнаружено не было.
Как отмечается [290], гидроочистка характеризуется
следующей степенью удаления нежелательных компонентов сырья:
Уменьшение
Компонент содержания,
%
Сера 90—98
Азот 60—85
Полициклические ароматические
углеводороды 25—70
Металлы 90—98
Кокс по Конрадсону 80— 95
Имеются и другие исследования, а также результаты
промышленных испытаний процесса подготовки сырья каталитического
крекинга методом гидроочистки [8]. Изучались дистилляты самых
различных по происхождению нефтей. При этом все выводы иссле-
192
дователей сводятся к тому, что подготовка сырья крекинга
методом гидроочистки весьма целесообразна.
Присутствие полициклических ароматических углеводородов в
сырье, направляемом на каталитический крекинг, нежелательно,
так как крекинг их протекает с трудом. Они образуют
непропорционально большое количество кокса и почти не увеличивают
выхода наиболее ценных компонентов. При гидрогенизационной
очистке, особенно при повышенном давлении, полициклические
ароматические углеводороды превращаются в моноциклические
ароматические углеводороды и цикланы. Моноциклические
ароматические углеводороды образуются с большей скоростью, чем
цикланы. Поэтому вначале концентрация моноциклических
углеводородов возрастает до равновесной. Затем равновесие нарушается,,
и содержание моноциклических ароматических углеводородов
снижается со скоростью, соответствующей скорости их насыщения
водородом [289]. -\
Вследствие высокого содержания полициклических
ароматических углеводородов гидроочистка циркулирующих газойлей
каталитического крекинга имеет большое значение, так как позволяет
получать хорошие результаты по выходу бензина и кокса при по^
следующем крекинге этих газойлей. Поэтому исследованиям,
посвященным гидроочистке циркулирующих газойлей с целью
улучшения показателей процесса каталитического крекинга, посвящено
много работ. Этот метод широко применяется в промышленности
для увеличения глубины превращения сырья крекинга [298].
Имеются также предложения по применению процесса ХДС
для гидрообессеривания сырья каталитического крекинга из
остаточных продуктов. Этот процесс можно осуществлять в двух
модификациях: 1) полное превращение остатка без выработки
тяжелого остаточного топлива; 2) гидрогенизационная обработка остатка
с целью удаления содержащихся в нем загрязнений. Первая
модификация процесса осуществляется под давлением 14—21 МПа.
Остаток превращается в высококачественное сырье для крекинга.
В этом процессе металлы и сера удаляются на 95% и более,
значительно снижается коксуемость сырья крекинга. По второму
варианту процесс ХДС проводят под давлением 3,5—70 МПа. При
этом остатки можно превратить в малосернистые топлива или
получить значительное количество высококачественного сырья для
крекинга. В этом случае остаточное топливо также становится
малосернистым.
В результате гидрообессеривания мазута кувейтской нефти при
среднем давлении процесса выход легких дистиллятов возрастает
с 16 до 24 объемн. % на сырье, а степень обессеривания
повышается с 67 до 93%- При этом остаток и дистилляты
характеризуются низким содержанием серы [299]. Наиболее важно, что гидро-
обессеривание остатков дает возможность получать из них
дополнительное количество сырья для каталитического крекинга. Так,
при гидрообессеривании 50%-ного мазута кувейтской нефти после
13-1206 193
Таблица
Характеристика гидроочищенного
о
Плотность
0,909
пес
2
о
Содержани
3,25
¦ч©
к
с.
и
а
Степень об
—
Содержание
металлов,
10-1 вес. %
иикель
0,50
1
1,10
железо
2,40
Степень
удаления
металлов, %
никель
—
1
Я
Я
—
о
а
д
Коксуемост
0,11
Степень сн
коксуемост
—
3
о *
о
3
fr" '
Сернокисло
объеми. %
13,4
Температура
0,882
0,888
0,894
0,900
0,904
1,32
1,66
1,88
2,68
2,95
59,3
49,0
42,1
18,5
9,2
0,12
0,13
0,23
0,27
—
0,35
0,37
0,70
0,75
—
0,65
0,30
0,62
0,40
—
76,0
74,0
54,0
46,0
—
68,1
66,3
36,3
31,8
—
0,03
0,05
0 06
0,08
0 08
72,7
54,5
45,4
27 3
27 3
7,7
8,6
10,4
12,0
13,0
Температура
0,871
0,875
0,88
0,894
0,891
0,47
0,74
1,13
1,98
2,49
85,6
77,2
65,2
39,1
23,4
0,032
0,03
0,11
0,20
0,31
0,09
0,10
0,40
0,70
0,87
0,87
0,20
0,52
0,47
7,40
93,6
93,9
78,0
60,0
38,0
91,8
90,8
63,6
36,4
20,9
0,003
0,02
0 04
0,06
0,07
97,2
90,9
63,6
45,4
36,4
2,0
3,0
7,2
10,4
11,8
Температура
0,863
0,865
0,874
0,883
0,890
0,22
0,36
0,76
1,38
2,02
93,2
88,9
76,6
57,5
37,8
0,
0,
0,
0,
0,
012
016
037
14
19
0,
0,
0,
0,
0,
03
034
21
43
61
0,
0,
0,
0,
0,
25
27
33
30
34
97,6
96,8
92,6
72 0
62 0
97,3
96,9
80,9
60,8
44,5
0,
0,
0,
0,
0,
001
006
02
04
06
99,0
98,1
81,8
63,6
45,4
0
1,0
4,0
9,2
10,8
Температура
0,864
0,871
0,880
0,886
0
0
1
1
,37
,65
,25
,65
88,
80,
61,
49,
6
0
6
2
0
0
0
,014
,12
,16
0,
0,
0,
025
38
56
0,
0,
1,
40
95
20
99
76
68
,2
,0
,0
97
65
49
,6
,4
,1
0,002
0,005
0,02
0,04
98,
95,
81,
63,
1
4
8
6
0
4
7
10
,8
,0
,4
,2
удаления 80% серы выход газойля (фракция 354—538°С)
составляет 60% при выходе остатка > 538 °С 23%.
В последние годы были разработаны более эффективные
катализаторы, и процесс гидрообессеривания нашел применение на ря-
194
50
вакуумного
§
Sa
Степень сни
сернокнслот
—
о
я
248
газойля
Выкипает, объеми. %,
при температуре, °С
250
—
275
2,0
300
5,5
325
16,5
350
28,0
375
44,0
475
о
-
І
№
іасті
Температурі
—
о
о
S
(Я
о
о а?
Содержание
азота, вес.
0,056
ения
га, <
5S
Степень удг
основного а;
—
та
•q
Молекулярн
303
я
д;
S
о
а.
с
я
я
1,4955
гидроочистки 350 °С
42,5
35,9
22,3
10,5
3,0
218
—
—
—
—
—
—
—
3,5
—
—
—
10
—
—
—
—
20
—
—
—
—
31,5
—
—
—
47
—
—
—
466
—
—
—
—
—
0,041
0,044
0,046
25,0
21,4
19,6
291
303
304
гидроочистки 380°С
85,1
77,6
46,3
22,3
22,9
191
204
—
—
212
2,5
2,5
—
—
—
5
6
—
—
4,5
13
11,5
—
10
23
22
—
—
17
38
36
—
—
28
57
52
—
—
45,4
463
468
468
22
19
18
18
0,020
0,020
0,027
0,030
0,030
64,4
64,4
51,7
46,4
46,4
256
290
269
311
гидроочистки 410 °С
100
92,5
60,2
31,4
19,3
—
190
—
—
—
—
—
—
14
—
—
—
—
22
—
—
—
—
32,5
—
—
—
—
49
—
—
—
65
.
—
444
—
18
18
20
18
0,015
0,024
0,031
0,030
0,036
73,3
57,1
44,6
46,4
35,7
277
277
287
гидроочистки 430°С
94,0
70,1
44,9
23,8
174
—
—
10
—
—
—
14,5
—
—
—
22
—
—
—
32
—
—
46
—
64
—
460
—
—
19
—
0,018
0,027
0,032
0,041
67,8
51.9
42,8
26,8
250
256
—
277
де нефтеперерабатывающих заводов мира при подготовке сырь^
для современных установок каталитического крекинга с
лифт-реакторами, использующих цеолитсодержащие катализаторы [300].
Наряду с процессом гидрообессеривания для подготовки сырья ка-
13* 195
талитического крекинга в США применяют и первые ступени
процессов гидрокрекинга [301].
При гидроочистке вакуумного газойля средний срок службы
катализатора за цикл 24,2 м3 сырья на 1 кг катализатора. В среДнем
катализатор выдерживает две регенерации и суммарный срок
службы его 52,5 м3 сырья на 1 кг катализатора. В случае гидро-
очнстки циркулирующего легкого каталитического газойля эти
показатели примерно в 2 раза ниже — суммарный срок службы
катализатора не превышает 28 м3 сырья на 1 кг катализатора. После
первой и второй регенерации остается 85%, а после третьей —
только 58% первоначальной активности катализатора. Но иногда
даже после пятой регенерации остается 80% первоначальной
активности катализатора [302, 303].
В Советском Союзе вопрос о целесообразности гидрогениза-
ционной подготовки сырья крекинга впервые рассматривался
применительно к дистиллятам высокосернисты.х арлан-чекмагушских
нефтей [304, 305]. Позднее этот вопрос был изучен более
детально [281, 306—309].
Ниже излагаются результаты наших исследований по
установлению оптимального режима гидроочистки сырья каталитического
крекинга, а также описаны опыты по каталитическому крекингу
сырья с различной глубиной очистки. Гидроочистку проводили на
пилотной установке с системой циркуляции водородсодержащего
газа при давлении 5 МПа, циркуляции водородсодержащего газа
(при нормальных условиях) 800 л/л сырья и объемной скорости
подачи сырья 0,5; 1,0; 2,0; 5,0; и 10,0 ч~К На каждой объемной
скорости опыты проводили при 350, 380, 410 и 430 °С. В опытах
применяли образец промышленного алюмокобальтмолибденового
катализатора, сырьем служил вакуумный газойль из арланской
нефти.
Характеристика гидроочищенного газойля приведена в табл. 50.
Влияние гидроочистки на качество вакуумных газойлей.
Степень удаления сернистых соединений в процессе гидроочистки
является одним из важнейших показателей эффективности процесса.
Изменение содержания серы в гидрогенизатах в зависимости от
объемной скорости подачи сырья и температуры гидроочистки
показано на рис. 86, а. При небольшом фиктивном времени
пребывания сырья в реакционной зоне гидроочистки содержание в нем
серы резко снижается. При дальнейшем его увеличении скорость
снижения содержания серы замедляется и наконец остается без
изменения.
Гидроочистка сопровождается весьма глубоким удалением
смолистых веществ. Характер изменения содержания сернокислотных
смол и коксуемости в гидрогенизате в зависимости от объемной
скорости подачи сырья такой же, как и изменения содержания
сернистых соединений. Глубина удаления смолистых соединений очень
велика. При температуре гидроочистки 410 °С и объемной
скорости подачи сырья 0,5 ч сернокислотные смолы удаляются пол-
196
ностью. Вследствие этого коксуемость гидрогенизатов значительно
меньше, чем сырья. Наиболее глубокое удаление коксообразующих
веществ достигается при 430 °С — даже при объемной скорости
подачи сырья 10,0 ч~' коксуемость гидрогенизата примерно в 3
раза меньше коксуемости сырья. При 380 °С такое снижение
коксуемости возможно при объемных скоростях подачи сырья менее
2,0 ч-1 (рис. 86, б). Гидроочистка при 350 °С позволяет также
значительно снижать коксуемость, но для этого требуются объемные
скорости менее 0,8 ч~'. При гидроочистке вакуумного газойля его
фракционный состав несколько облегчается. Больше всего он из-
П 05 1,0 1,5 2,0 r
Фиктивное время реагированиям-^
10 5 2 Г 0,5
Объемная скорость подачи сырья и, ч
О 0,5 1,0 1,5 2,0
Фиктивное времн^ реагирования u)-t-
10 5
0,5
Объемная скорость подачи сырья и '/"
Рис. 86. Влияние режима гидроочистки иа содержание серы (а) и коксуемость (б)
в очищенном вакуумном газойле из арлаиской нефти:
7 — 350 °С; г —380 °С; 3 — 410 °С: 4 — 430 °С.
меняется при небольших величинах фиктивного времени
реагирования. С увеличением времени реагирования изменение
фракционного состава несколько замедляется (см. табл. 50).
Гидроочистка вакуумного газойля позволяет резко уменьшить
содержание в нем металлов (рис. 87). При наиболее
распространенном режиме гидроочистки (температура 380 °С, объемная
скорость подачи сырья 1,0 ч~') металлы удаляются примерно на 90%.
Кроме того, после гидроочистки снижается содержание еще одного
яда алюмосиликатного катализатора — азота. Наиболее глубоко
(до 50—70%) удаляются соединения азота основного характера
(см. табл. 50).
При гидрооблагораживании различных нефтяных дистиллятов
наряду с изменением содержания в них серы меняются показатели
их качества (плотность, коксуемость, содержание смол, металлов
197
и др.)- Поскольку процесс гидроочистки характеризуется в
основном изменением содержания серы, была сделана попытка найти
математические зависимости изменений различных показателей
качества гидрогенизата от глубины его обессеривания [306, 310].
0.5
2,0,
°
Фиктивное время реагирования ,ш=-
ю 5 г і 05
Обьемная скорость подачи сырья v, ч'1
Фиктивное в Den я реагирования
1—1 і : і г Ґ
10 5 2 1
Объемная скорость подачи сырья
б
2.0 j
о,5
Рис. 87. Влияние режима гидроочистки на степень удаления никеля (а) и
ванадия (б) из вакуумного газойля арланской нефти:
/ — 350 °С; 2 — 380 °С; 3 — 410 °С: 4 — 430 °С.
Эмпирические формулы, выражающие зависимость изменения
качества гидрогенизата от глубины обессеривания, имеют
следующий вид:
Удаление никеля, %:
0,456 + 0,00522*
Удаление ванадия, %:
Y2= 1,055*
Снижение содержания сернокислотных смол, °/о:
Y3 = 0,0001019x3 — 0,004162x2+0,5567*
Снижение плотности, %:
B6)
B7)
B8)
Y4 = 0,05x B9)
где Y — степень изменения какого-либо из показателей качества
гидроочищенного вакуумного газойля; х — степень
обессеривания, %.
Наши экспериментальные данные, полученные при
гидроочистке вакуумных газойлей различного происхождения, а также
данные зарубежных исследователей [289, 291, 292, 293]
удовлетворительно описываются этими эмпирическими уравнениями.
198
Каталитический крекинг очищенного сырья. Опыты по
каталитическому крекингу вакуумных газойлей из чекмагушской и
арланской нефтей, подвергнутых гидроочистке до различной
глубины, осуществлялись на лабораторной установке с реактором
I
35
30
I
: го
I
С 0,5 1,0 1,4 2:0 ,
¦гиктивние бремя реагирования ui-—
! I I | L_ V
10 5 2 I 0,5
?*\.'м.у/7<? скорость подачи сырья is, </"'
- 0,5 '.'! 1,5 2,0
Фикти&нос буен? реагирования из-—
L:v
105 2
Обьемнпч скорость г.пдачи сырья
0,5
Рис. 88. Изменение выхода бензина (а), газа и кокса (б) при каталитическом
крекинге вакуумного газойля чекмагушской нефти после гидрогенизационной
очистки.
Температура крекинга 450 °С; объемная скорость подачи сырья при крекинге, ч-1:
1,4 — 0,7; 2, 5 — 1,0; 3, 6 — 1,5; /, 2, 3 — выход газа; 4, 5, 6 — выход кокса.
объемом 100 см3. Для опытов применяли аморфный катализатор,
отобранный из системы промышленной установки каталитического
крекинга, который имел индекс активности 30—32 пункта. По мере
углубления очистки сырья каталитического крекинга
увеличивается выход бензина, газа и в небольшой степени легкого газойля и
уменьшается выход кокса и тяжелого каталитического газойля.
Изменение выходов продуктов крекинга происходит наиболее
резко при небольшой глубине очистки сырья, фактически при
переходе от исходного сырья к сырью, очищенному при объемной
скорости 10,0 ч-1 (рис. 88).
При крекинге вакуумного газойля, гидроочищенного при
объемной скорости 10 ч~', выход бензина увеличивается на 22,5—
27,5 отн. % по сравнению с его выходом в случае каталитического
крекинга неочищенного вакуумного газойля, а выход кокса
снижается на 1,5—2,5 отн. %. Крекинг вакуумного газойля,
гидроочищенного при объемной скорости 0,5 ч, приводит к еще большему
увеличению выхода бензина (на 36,5—40 отн. %) и снижению
выхода кокса на 25,0—40,0 отн. %. Разница в выходах продуктов
увеличится еще больше, если их сравнивать при одинаковом
выходе кокса (табл. 51).
199
Таблица 51
Материальные балансы каталитического крекинга исходного
и гидроочищенных вакуумных газойлей из чекмагушской нефти
при одинаковом коксообразовании
D,5 вес. %)
Показатели
Расход водорода, вес. %
Выход, вес. %
бензина
легкого газойля
тяжелого газойля
газа
Потери, вес. %
Условная глубина разложения,
вес. %
Сумма светлых, вес. %
Отношение бензин:кокс
Увеличение выхода, %
бензина
легкого газойля
Исходное
сырье
0,00
21,5
24,1
39,0
9,7
1.2
35,7
45,6
4,78
100,0
100,0
Вакуумный газойль, гидроочищенный при
объемной скорости.
10,0
0,39
28,4
26,4
29,0
10,7
1,0
43,6
54,8
6,32
132,0
109,5
2,0
0,75
30,2
26,9
24,0
13,7
1.7
48,4
57,1
6,71
140,5
111,5
4-І
0,5
1,05
34,7
26,5
17,5
15,4
1,4
54,6
61,2
7,71
161,5
110,0
При повышении температуры гидроочпсткн арланского
вакуумного газойля с 380 до 410 °С выход основных продуктов крекинга
практически не изменяется, а зависит лишь от объемной скорости
подачи сырья в стадии гидроочистки. По мере углубления
очистки сырья улучшается и качество продуктов каталитического
крекинга. В бензинах значительно уменьшается содержание
сернистых соединений и непредельных углеводородов, увеличивается
содержание ароматических углеводородов. В легком каталитическом
газойле уменьшается содержание сернистых соединений,
количество сульфирующихся и йодное число.
Из сырья, гидроочищенного при объемной скорости 0,5 ч~~1,
получают в процессе каталитического крекинга (объемная скорость
1 ч-1) бензин с содержанием серы 0,07%, непредельных
углеводородов 24,0% и ароматических 13,3%. В получаемом из очищенного
сырья легком газойле содержание серы снижается с 2,87 до 0,68%,
модное число — с 11,5% ДО 6,9%. Бензин, образующийся из
исходного неочищенного сырья при таком же режиме крекинга,
содержит серы 0,55%, непредельных углеводородов 29,1 %' и
ароматических 9,4%. Следовательно, фракции бензина и легкого газойля,
полученные в процессе каталитического крекинга сырья, которое
было очищено при объемной скорости 0,5 ч~', не требуют
дополнительной очистки при производстве из этих дистиллятов товарных
продуктов.
200
В результате углубления очистки сырья улучшается также
состав газа каталитического крекинга. При крекинге вакуумных
газойлей, очищенных гидрогенизационным методом, выход пропан-
пропиленовой и бутан-бутиленовой фракции больше, чем при
крекинге исходного сырья, а выход сероводорода резко снижается.
Так, в процессе крекинга арланского вакуумного газойля,
подвергнутого гидроочистке при 380 °С и объемной скорости 0,5 ч-1,
выход (в вес. % на сырье) пропан-пропиленовой и
бутан-бутиленовой фракции соответственно 3,42 и 5,29, а при крекинге
неочищенного сырья соответственно 2,31 и 3,06. Выход сероводорода после
гидроочистки снижается почти в 25 раз.
Результаты лабораторных исследований были подтверждены во
время промышленных пробегов на Ордена Ленина Уфимском
нефтеперерабатывающем заводе и Уфимском
нефтеперерабатывающем заводе им. XXII съезда КПСС [311, 312]. Во время этих двух
пробегов в качестве сырья использовали вакуумный газойль ар-
ланской нефти. Гидроочистку проводили при 360—375 °С, объемной
скорости подачи сырья 0,83—1,0 ч~', давлении 3,0—3,5 МПа,
циркуляции водородсо держащего газа в присутствии кобальтмолибде-
нового катализатора, который использовался уже до этого в
течение 1 г. для очистки дизельного топлива. Оба пробега дали
совпадающие результаты; поэтому ниже приводятся данные пробега
лишь на первом заводе [311].
Из приведенных ниже данных видно, что несмотря на
относительно мягкие условия процесса была достигнута достаточно
высокая глубина очистки:
Вакуумный газойль
гндроочн-
неочищенный щенный
Плотность dl° 0,906 0,878
Содержание, вес. %
серы 2,75 0,48
азота 0,11 0,06
Содержание сернокислотных смол, объемн. %
% 13,5 5,0
Содержание металлов Ю-'-вес. %
ванадия 0,250 0,035
никеля 0,10 0,10
Коксуемость, вес. % 0,09 0,03
Температура застывания, °С 24,0 22,0
Фракционный состав (разгонка по
Богданову), °С
н. к 198 178
до 350 выкипает, объемн. % 29 42
90% 445 430
к. к 464 453
Расход водородсодержащего газа в период пробега составлял
1,98 вес. %, или в пересчете на чистый водород 0,7 вес. % на сырье
Во время опытного пробега стабилизационная колонна работала
недостаточно удовлетворительно. Других осложнений в работе не
наблюдалось. Весь гидроочищенный вакуумный газойль был под-
2 01
вергнут каталитическому крекингу. Результаты сопоставляли с
показателями процесса каталитического крекинга на неочищенном
вакуумном газойле, проводимого на том же режиме: объемная
скорость подачи сырья — 1,5 ч~\ температура в реакторе 460—
465 °С, весовое отношение катализатор : сырье=1,7.
В процессе каталитического крекинга очищенного сырья
образуется на 16 отн. % больше бензина, чем при крекинге
неочищенного сырья. Ниже приводится материальный баланс
каталитического крекинга (в вес. % на неочищенный газойль):
Вакуммный газойль
Выход продуктов гидроочи-
неочищенный щенный
Газы по С< включительно 13,5 10,0
в том числе
сероводород 1,39 0,20
водород 0,22 0,20
метан 1,32 0,63
сумма С2 1,84 1,26
пропан 1,63 1,40
пропилен 2,27 2,10
изобутан 2,06 1,98
н-бутан 0,74 0,60
сумма бутиленов 2,03 1,73
Бензин 19,8 23,0
Легкий газойль 46,2 48,0
Тяжелый газойль 14,6 9,8
Кокс+потери 5,9 4,0
Всего . . . 100,0 95,8
Сумма светлых 66 71
Отношение газ:бензин 0,683 0,435
При крекинге очищенного сырья получают жидкие продукты,
по качеству существенно иные, чем при крекинге неочищенного
сырья. Бензин по содержанию серы отвечает требованиям ГОСТ
на высококачественный автомобильный бензин. Октановые числа
для обоих бензинов одинаковы. В то же время приемистость к ТЭС
бензина, полученного из очищенного сырья, была несколько
повышенной.
Качество полученных жидких продуктов каталитического
крекинга приводится ниже:
Вакуумный газойль
гндро-
неочищенный очищенный
Бензин
Плотность dj° 0,761 0,752
Содержание серы, вес. % 0,71 0,10
Йодное число, г 12/100 г 65 62
Сульфируемых, объемн. % 38 33
Фракционный состав, °С
н. к 52 45
10% 86 82
50% 140 135
90% 181 178
к. к 204 195
202
Октановое чнсло по моторному методу
в чистом виде 78 78
с 0,41 г ТЭС/1 кг бензина 80 82
с 0,82 г ТЭС/1 кг бензина 81 83
Октановое чнсло по исследовательскому
методу с 0,82 г ТЭС/1 кг бензина .... 90,4 92,0
Легкий газойль
Плотность dl0 0,889 0,864
Содержание серы, вес. % 2,20 0,39
Йодное число, г 12/100 г продукта .... 15 12
Сульфируемых, объемн. % 42 35
Фракционный состав, °С
н к . 230 208
10% 250 225
50% . .... 300 285
90% .. . . 350 342
к. к 357 356
Цетановое чнсло 38,8 39,6
Тяжелый газойль
Плотность dl« 0,914 0,908
Содержание серы, вес. % 1,80 0,41
Фракционный состав, °С
н. к 210 190
10% . 335 350
50% 380 390
90% 415 422
к. к 435 435
Вязкость условная, °ВУ5о 2,20 1,83
Легкий газойль каталитического крекинга из очищенного сырья
содержит меньше серы, имеет меньшее йодное и большее
цетановое число. Он может быть использован непосредственно в
качестве компонента товарного дизельного топлива. Получаемый из
очищенного сырья тяжелый газойль также лучше по качеству; он
может быть успешно использован в качестве малосернистого
компонента котельного топлива.
Полученные результаты полностью подтвердили приведенные
выше данные лабораторных и пилотных испытаний процессов гид-
рогенизационной очистки вакуумного газойля и последующего его
каталитического крекинга и послужили основой для постоянной
работы по этому варианту промышленной установки 1-А на УНПЗ
им. XXII съезда КПСС.
Особый интерес представляет гидроочистка сырья
каталитического крекинга в связи с использованием цеолитных
катализаторов. Так, при крекинге неочищенного сырья выход бензина на цео-
литсодержащем катализаторе на 5% больше, чем на обычном. При
использовании гидроочищенного сырья и высокоактивного цеолит-
содержащего катализатора выход бензина на 10% больше [301].
203
В другой работе [313] подчеркивается, что гидроочистка газойля
легкого крекинга и коксования при работе на цеолитсодержащем
катализаторе более экономически выгодна, чем при крекинге на
аморфном катализаторе. В работе [221] также
отмечалась'целесообразность предварительной очистки сырья крекинга при работе
на цеолитсодержащих катализаторах. В связи с этим были
проведены специальные исследования [290] по подготовке сырья,
подвергаемого крекингу в присутствии высокоактивных катализаторов
крекинга [290].
В табл. 52 приведены данные о качестве сырья и результатах
крекинга гидроочищенного сырья на цеолитсодержащих
катализаторах. А — вакуумный прямогонный газойль с высоким
содержанием азота; Б — смесь из 40 объемн. % вакуумного газойля и
Таблица 52
Качество исходного сырья и результаты
каталитического крекинга гидроочищенного сырья
в присутствии цеолитсодержащего катализатора
Плотность rfjf,'!
Содержание, вес. %'•
серы
азота
Коксуемость по Конрадсону,
вес. %
Суммарное содержание никеля
и ванадия, 10~4 вес. %
Анилиновая точка, °С
Вязкость, мм2/с
при 64,4 °С
при 98,9 °С
Разгонка по АСТМ
5%
10%
50%
90%
Расход водорода (при
нормальных условиях), м3/м3 сырья
Увеличение степени конверсии
сырья прн каталитическом
крекинге при постоянном
режиме, объемн. % от
исходного сырья
Каталитический крекинг
очищенного сырья со степенью
превращения 60 объемн. %
увеличение выхода бензина,
объемн. % на сырье
уменьшение выхода кокса,
вес. °/о на сырье
А
0,9593
1,75
0,80
1,9
2,0
67,2
420
33,3
442,2
450,4
496,1
561,1
138,0
—
9
6
Б
0,9490
1,90
0,20
3,8
3,0
97,2
360
40,8
472,8
484,4
546,7
—
96,1
14
7
5
В
0,9194
0,82
0,13
0,9
—
81,7
18
5,2
376,1
378,9
398,9
482,2
108,6
18
9
Г
0,8978
1,44
0,06
0,04
0,1
74,4
9,6
3,3
286,1
295,5
367,2
467,8
73,0
12
5
3
204
60 объемн. % деасфальтизата; В — смесь 57 объемн. % газойля
каталитического крекинга, 32 объемн. % тяжелого газойля
коксования и 11 объемн. % экстракта — побочного продукта масляного
производства; Г — высокосернистый вакуумный прямогонный
газойль.
Из данных табл. 52 видно, что гидроочистка исходного сырья
приводит к существенному увеличению степени конверсии и
выхода бензина, а также к уменьшению коксообразования.
Таким образом, при гидроочистке сырья каталитического
крекинга существенно улучшается материальный баланс
каталитического крекинга и повышается качество получаемых продуктов.
Кроме того, уменьшается отравление катализатора вследствие
удаления металлов и азотистых соединений, уменьшается коррозия
аппаратуры и благодаря удалению сернистых соединений
улучшаются условия эксплуатации установки.
ДРУГИЕ МЕТОДЫ ПОДГОТОВКИ СЫРЬЯ
КАТАЛИТИЧЕСКОГО КРЕКИНГА
Наиболее радикальный метод очистки сырья —
гидроочистка — требует больших капитальных вложений и наличия
дешевого водорода. Более дешевые методы обычно менее эффективны.
Поэтому и по сей день изыскиваются новые методы очистки. Были
сделаны попытки использовать для этой цели различные кислоты,
такие, как фтористоводородная [314], иодистоводородная в смеси
с гидроароматическим углеводородом, например тетралином, что
позволяет в отдельных случаях достичь степени удаления
металлов до 50% [315]. Предлагается [316] деметаллизировать нефть
и остаточные фракции контактированием их с 1—30% жидкой,
нерастворимой в нефтепродуктах ароматической сульфокислотой при
65 °С. После второй экстракции ксилолсульфокислотой содержание
никеля снижается с 0,2-10~2 до 0,1 %-Ю-2, ванадия — с 0,4-10~2
до 0,18% • 10~3. После вторичной экстракции толуолсульфокислотой
количество никеля уменьшается до 0,4% • Ю~4, ванадия — до
0,6%-Ю-4.
Для очистки нефтяных дистиллятов от азотистых оснований
предлагается [317] применить борную кислоту и ароматические
полиоксисоединения типа глицерина, пропилэтиленгликоля,
пирокатехина. Однако этот метод проверен всего лишь на бензинах и
керосинах. Более тяжелые фракции B04—265 °С) рекомендуется
очищать от азотистых оснований бензиловым эфиром метаборной
кислоты [318].
В патенте [319] предлагается очищать сырье крекинга оксига-
лоидными производными серы или фосфора @,005—0,2% на
сырье) в присутствии 20—25 вес. % селективного растворителя,
например сернистого ангидрида. Органические азотистые
соединения образуют осадок, который удаляется с экстрактом, а очищен-
205
ная фракция — с рафинатом. После очистки газойля содержание
серы снижается с 2,19 до 1,8 вес. %, а азота — с 0,098 до 0,04%.
Выход бензина при каталитическом крекинге возрастает с 22,5 до
30,4%, выход кокса снижается с 3,2 до 1,05%.
Удаление вредных примесей в виде шлама путем обработки
сырья фтористым бором описано в патенте [320]. Шлам отделяют
при 0—60 °С в электрическом поле с разностью потенциалов не
менее 5000 В/см. Коксообразующие и металлсодержащие
компоненты хорошо удаляются также при обработке нефтепродуктов
эфиратом фтористого бора [321]. Высокая степень удаления
металлов отмечается при контактировании сырья каталитического
крекинга с NaOCl или N-бромсукцинимидом при 15—150 °С, 0,1 —
1,0 МПа и мольном отношении реагента к металлу от 1 до 5 [322].
В результате металлические загрязнения переходят в галоидные
соли, которые удаляются вместе с реагентом. Отмечается [195],
что в осажденном пропаном асфальте содержится 97,3% ванадия,
99,2% никеля и 86% железа от их количеств в остаточном топливе.
Особо следует отметить ряд патентов, предлагающих
использовать для удаления вредных для катализатора крекинга примесей
воду в чистом виде [323], воду подкисленную [324], воду в
присутствии щелочных металлов [325]. После промывки газойля 10—
20% подкисленной воды [324] при 70—100 °С зольность снижается
с 2,1 до 0,3%. Описана [325] очистка масляных фракций от
азотистых оснований обработкой водной эмульсии бисульфатом
щелочного металла или аммония при 40 °С. Имеются также указания
о возможности очистки сырья каталитического крекинга щелочью,
щелочью с последующей промывкой кислотой и водой.
Предлагается [326] удалять из нефтяных фракций смолистые
вещества путем окисления при повышенных температурах
кислородом или озоном с последующим отделением продуктов
окисления и промывкой водой. Металлпорфириновые комплексы из
нефтяных фракций удаляют растворением фракции в легких
растворителях с образованием гомогенного раствора, который
контактируют с 0,1 —1,5% озона. Затем растворитель отделяют и фракцию
промывают водой. После очистки таким путем тяжелого остатка
содержание в нем металлов снижается на 75% [326].
Сообщается [327] о целесообразности комбинирования
обработки кислородом с другими методами. Так, фракцию,
выкипающую выше 500 °С, обрабатывают в присутствии кислорода
плавленым гидратом окиси щелочного металла при 162—370 °С,
промывают водой для удаления металлов. Примеси отделяют
фильтрованием, центрифугированием или отстаиванием.
Весьма интересное предложение описано в патенте [328].
Тяжелые дистилляты частично сжигают в среде
кислородсодержащего газа с таким расчетом, чтобы около 0,5% углерода сырья
выделилось в свободном виде вместе с соединениями тяжелых
металлов. Выделенные металлы можно переводить в фосфаты и
использовать в производстве стали.
206
Особо следует отметить ряд попыток перевода летучих
комплексов металлов в нелетучие [192]. Так, при добавлении к
дистилляту, содержащему ванадий в виде порфиринового комплекса,
небольшого количества пиридина образуется нелетучий пиридинва-
надиевый комплекс. После разгонки продукта дистиллят
существенно очищается от ванадия. Весьма интересные результаты
приведены в работе [329] по изучению облучения газойля дозой 7-Ю7 Р.
Анализ показал, что количество летучих соединений ванадия и
никеля заметно снизилось, вероятно, вследствие того, что металл-
порфириновые комплексы неустойчивы к облучению. Этот факт
может быть использован для перевода летучих соединений никеля и
ванадия в нелетучие формы.
В этом разделе изложены в основном патентные сведения,
лишенные достаточно подробных данных. В литературе нет
результатов испытания по указанным методам в промышленных
условиях. Ряд методов разработан всего лишь для использования их в
лабораторных условиях. Однако сами принципы подхода к
решению вопроса очистки сырья крекинга представляют интерес.
СОПОСТАВЛЕНИЕ ТЕХНИКО-ЭКОНОМИЧЕСКИХ ПОКАЗАТЕЛЕЙ
СЕРНОКИСЛОТНОГО И ГИДРОГЕНИЗАЦИОННОГО МЕТОДОВ
ОЧИСТКИ СЫРЬЯ КРЕКИНГА
Для установления целесообразности метода подготовки сырья
каталитического крекинга необходимо наряду с технологическими
данными располагать экономическими показателями. Нами были
проведены экономические расчеты по подготовке сырья
каталитического крекинга (вакуумного газойля) сернокислотной и гидро-
генизационной очисткой. Во всех случаях на каталитический
крекинг поступало 744 000 т вакуумного газойля. По каждому
варианту выход кокса при одинаковой производительности установок
каталитического крекинга составлял 4,5 вес. %. В основном можно
отметить следующее.
Себестоимость сырья каталитического крекинга по мере
углубления его очистки увеличивается, причем наиболее резко при
переходе от неочищенного сырья к сырью, гидроочищенному при
объемной скорости 10 ч~'.
Себестоимость бензина при переработке очищенного сырья
меньше, чем неочищенного, вследствие увеличения выхода бензина.
Суммарная годовая стоимость бензина и легкого газойля
оказывается на 0,5—0,8 млн. рублей больше, чем при крекинге
неочищенного сырья. Однако продукты каталитического крекинга
неочищенного вакуумного газойля чекмагушской нефти содержат много
серы, и поэтому для получения товарных продуктов требуется их
очистка. При гидроочистке бензина и легкого газойля, полученных
в результате каталитического крекинга неочищенного сырья,
стоимость бензина и легкого газойля удорожается на 1,9 млн. руб.
207
Таблица 53
Технико-экономические показатели различных методов очистки сырья
каталитического крекинга (вакуумного газойля)
Показатели
Сырье — вакуумный газойль
неочищенный, т
Расход водорода, т
» кислоты, т
Получено, т
бензина
легкого газойля
сероводорода
Стоимость сырья, руб./т
неочищенного
очищенного
Эксплуатационные расходы,
тыс, руб,
па 1 т сырья, руб.—коп.
па 1 т бензина, руб,—коп.
Себестоимость, руб./т
бензина
легкого газойля
Затраты*, тыс. руб.
по бензину
но легкому газойлю
Суммарные .затраты*, тыс. руб.
Капиталовложения суммарные,
млн, руб,
Капиталовложения па 1 т
бензина, руб.
Неочи-
щонныи
744 000
—
—
160 000
179 200
—
4-80
6089,0
8—18
38—06
48—74
4-80
—
—
—
9,90
49,94
Неочи-
с
последующей
доочисткоЛ
дистиллятов
744 000
2173
156 546
168518
5322
4—80
7735,46
10—40
49—41
57—87
10-85
+ 821,1
+ 1080,00
+ 1901,10
11,70
55,71
Гидроочи-
щеннын,
V=]0 4-і
753 870
2040
211 800
195 400
6360
4—80
8-84
9077,90
12-20
42-96
47—54
8-84
-176,1
+ 723,60
+ 547,50
13,51
50,59
Гидроочищен-
ный,
V —10 4-і,
с последующеґ
О4НСТКОЙ
дистиллятов
753 870
5450
207 240
185400
11 750
4-80
8—84
10992,80
14—77
53—04
52—44
14-92
+ 607,90
+ 1812,30
+ 2420,70
15,32
54,95
Гндроочи-
щенный,
V=2 4-1
759 722
5698
224 690
200 130
11923
4—80
10—13
10040,80
13—49
44—68
47—85
10—13
— 148,90
+954,40
+ 805,50
13,51
47,18
Гидроочи-
щенныи,
К=0,5 4-і
780 120
8190
258 160
197 160
19 737
4—80
11—32
10859,20
14—60
42—06
44—37
11—32
—696,10
+ 1167,30
+ 471,20
13,51
44,32
Очищенный 96%-нои серной
кислотой при расходе, вес, %
2,0
785 638
—
15712,7
208 320
193 440
4-80
6—34
7034,15
9—45
33—77
48—05
4—80
—910,50
—910,50
10,80
43,01
5.0
802 588
—
40129,4
226 920
197 160
4—80
7—26
7635,27
10-26
33—65
42-59
4—80
-983,46
—983,46
10,80
40,03
10,0
852 233
—
85223,3
236 592
208 320
4—80
9—08
8754,57
11-77
37—00
46—52
4—80
— 355,30
—355,30
10,80
30,10
* Знак плюс указывает увеличение затрат по сравнению с крекингом неочищенного сырья, а знак минус—их уменьшение.
Таким образом, даже с точки зрения выработки качественных
нефтепродуктов предварительная гидроочистка сырья каталитического
крекинга является экономичной. Кроме того, экономичность
каталитического крекинга с предварительной гидроочисткой сырья
возрастет благодаря меньшему отравлению катализатора металлами,
а также уменьшению коррозии аппаратуры. Последние факторы,
ввиду отсутствия необходимых данных, не могли учитываться при
экономических расчетах.
Подготовка сырья сернокислотной очисткой была рассчитана
по данным лабораторных опытов, в которых каталитическому
крекингу подвергали исходный и очищенный чекмагушский
вакуумный газойль.
Полученные технико-экономические показатели при
сернокислотной и при гидроочистке приведены в табл. 53.
Как видно из табл. 53, эксплуатационные расходы по мере
углубления очистки возрастают. Однако в связи с увеличением
выхода бензина и легкого газойля себестоимость бензина
каталитического крекинга очищенного сырья ниже, чем бензина, полученного
из неочищенного вакуумного газойля. Из данных табл. 53 видно,
что при очистке серной кислотой концентрацией 95—96%
экономически более выгодно расходовать 2,5 вес. % кислоты на сырье.
В табл. 53 приведены размеры дополнительных
капиталовложений по обоим методам очистки. В случае сернокислотной
очистки они составляют 0,9 млн. рублей, а при подготовке сырья
гидроочисткой необходимо затратить 3,61 млн. рублей, т. е. в 4 раза
больше. Доля капитальных затрат, приходящихся на 1 т бензина,
по мере углубления очистки обоими методами уменьшается, но в
случае гидроочистки она выше, чем при сернокислотной очистке.
При оптимальных условиях очистки затраты на капитальное
строительство, приходящиеся на 1 т бензина, ниже, чем при крекинге
неочищенного сырья.
Аналогичные экономические расчеты были выполнены
зарубежными исследователями [292, 295—297]. Они также показали
несомненную экономическую эффективность предварительной гидро-
очисткн сырья крекинга. Однако для других конкретных условий
были получены иные результаты. Так, в работе [294] при
сопоставлении различных вариантов улучшения качества бензина
авторы ее пришли к выводу, что предварительная гидроочистка
сырья каталитического крекинга оправдывается только в том
случае, если в условиях данного завода нет других путей получения
бензина с требуемым октановым числом [294].
Отмечается [292], что в условиях Винневудского завода
гидроочистка сырья каталитического крекинга не является чрезмерно
выгодной с экономической точки зрения. В работе высказывается
мнение, что вряд ли нефтепереработчики будут сооружать
установки гидроочистки только с целью очистки сырья крекинга, даже
если при этом и будет достигаться некоторая экономия. Наиболее
важна гибкость этого процесса.
I4-220G 209
В работе [289] изучалась экономичность гидроочистки сырья
двухступенчатого каталитического крекинга. Расчеты показали,
что дополнительные капиталовложения, необходимые для
осуществления гидрогенизационной очистки сырья II ступени крекинга,
окупаются менее чем за два года. Кроме того, очистка сырья
II ступени крекинга позволяет получать из 1 м3 сырья
дополнительную продукцию стоимостью 3,15 долл. Эта прибыль D5—¦
60%) сохраняется приблизительно постоянной в широком
интервале степеней превращения.
Указывается [298] на предпочтительность предварительной
очистки сырья каталитического крекинга по сравнению с
гидроочисткой продуктов крекинга, несмотря на то, что в последнем
случае мощность установок гидроочистки значительно меньше.
Другими авторами [6, 165, 330] отмечается, что при крекинге в
присутствии цеолнтных катализаторов сырья, прошедшего
предварительную гидроочистку, достигается больший эффект, чем при
крекинге на обычных катализаторах. Почти все исследователи не
только подчеркивают улучшение материального баланса п
качества продуктов каталитического крекинга, но и указывают на
экономические преимущества этого метода подготовки сырья
крекинга.
Как следует из приведенных данных, и сернокислотную, и гид-
рогенизационную очистку можно применять для подготовки сырья
каталитического крекинга, но каждый из этих методов должен
быть использован только в определенной области. Так, гидрогени-
зационную очистку, позволяющую получать товарные продукты из
самых некачественных видов сырья, рекомендуется применять для
подготовки высокосернистых, высокосмолистых вакуумных
газойлей. Стоимость крекинга с предварительной гидроочисткой сырья
из таких нефтей будет выше, чем крекинга неочищенного сырья,
но сама гидроочистка будет примерно на 1,0—1,5 млн. руб.
дешевле гидроочисткп бензина и легкого газойля, полученного в случае
крекинга неочищенного сырья.
Высокоэкономичная сернокислотная очистка из-за небольшой
степени удаления сернистых соединений из сырья может быть
рекомендована только для подготовки тех его видов, из которых
получают товарные продукты, не требующие дополнительной
очистки от сернистых соединений. Следовательно, этот метод будет
эффективен при очистке низкосерннстых и среднесерийных видов
сырья.
Результаты гидроочистки вакуумных газойлей можно улучшить
путем применения более активных и стабильных катализаторов,
предназначенных для переработки тяжелых дистиллятов. В
частности, алюмоникельмолибденовый катализатор оказался более
активным, чем промышленный кобальтмолибденовын катализатор
[331—336]. Преимущества нового катализатора наиболее
сильно проявляются при переработке тяжелых нефтяных
дистиллятов.
210
ЗАКЛЮЧЕНИЕ
Специальная подготовка сырья для установок каталитического
крекинга является исключительно важной. Наиболее дешевым и
распространенным способом такой подготовки является
тщательная перегонка нефти при получении дистиллятов, предназначенных
для переработки в процессе каталитического крекинга. Нельзя
ограничиваться однократным испарением, а необходимо
использовать методы современной ректификации. Однако даже
квалифицированные методы ректификации не могут обеспечить получение
качественного сырья, особенно из нефтей с повышенным
содержанием азотистых соединений, смолистых веществ и металлов. Часто
для повышения экономичности процесса каталитического крекинга
приходится применять различные физические и химические методы
облагораживания сырья. Из них наиболее универсальным
способом является гидрогенизационная очистка; она пригодна и для
очистки сырья, и для облагораживания циркулирующего газойля.
Этот метод позволяет глубоко очищать от вредных компонентов
любые, даже наиболее неквалифицированные виды сырья. К
сожалению, гидроочистка является относительно дорогостоящим
методом, поскольку требуется значительное количество дефицитного
водорода. Тем не менее его применение для очистки некачественных
видов сырья каталитического крекинга экономически вполне
приемлемо. При подготовке сырья, содержащего немного
нежелательных компонентов, можно наряду с гидроочисткой применять
описанные выше другие, более дешевые методы очистки.
14*
Глава VI
ОЧИСТКА КАТАЛИЗАТОРА КРЕКИНГА
ОТ ОТРАВЛЯЮЩИХ МЕТАЛЛОВ
В результате отравления алюмосиликатных катализаторов
крекинга металлами значительно уменьшается выход целевых
продуктов и резко возрастает коксообразование. Описанные выше методы
предварительной подготовки сырья позволяют существенно снизить
накопление металлов на поверхности катализатора крекинга.
Однако очистка сырья от металлов при их незначительном
содержании весьма сложна. Поскольку для очистки сырья требуются
большие капиталовложения и эксплуатационные расходы, в
последние годы усилия исследователей были направлены на
разработку способов обработки катализатора с целью восстановления
его активности.
На выходящем из регенератора катализаторе металлы
находятся в виде окислов. Это было доказано на примере ванадия. В пор-
фирине ванадий находится в четырехвалентной форме (V4+). При
отложении ванадия из такого соединения на катализатор
валентность его не изменяется, что установлено по спектрам
электронного парамагнитного резонанса катализаторов крекинга,
отравленных ванадием [337]. После обработки загрязненных ванадием
катализаторов крекинга воздухом в условиях, обычно применяемых
для выжига, четырехвалентный ванадий переходит в другое
окисленное состояние, вероятно, в пятивалентное, и не
обнаруживается методом электронного парамагнитного резонанса. В связи с тем,
что активность отравленного катализатора сильно зависит от вида
соединения, в котором металл присутствует на катализаторе [217],
для восстановления первоначальной активности и селективности
отравленных катализаторов металлы следует либо совсем удалять,
либо перевести в новые, неактивные соединения.
Предложено большое число различных методов реактивации
катализаторов каталитического крекинга, отравленных
металлами. Все они основаны на физическом пли химическом действии на
катализатор. Ниже приводится их описание.
МЕХАНИЧЕСКИЕ МЕТОДЫ
Многими исследователями установлено, что металлы,
отложенные на катализаторе, сконцентрированы в основном на его
внешней поверхности. В частности найдено [101], что 44% всего никеля
212
и 58% всего ванадия находятся на наружной части гранулы
катализатора (толщиной 35 мк), которая составляет лишь 5% от его
массы. На основании этих исследований предложен ряд способов
механической обработки катализатора с целью истирания
поверхностного слоя катализатора и удаления при этом металлов. По
одному из методов [338], катализатор, содержащий металлы,
выводят из системы и обрабатывают потоком газа, содержащим
суспендированные абразивные гранулы размером 100—800 мк, т. е.
значительно меньшие по размеру, чем частицы катализатора, и
значительно большие, чем крошка, образующаяся во время истирания.
Рис. 89. Магнитный сепаратор,
используемый для удаления
металлических загрязнений с катализатора:
/ — отработанный катализатор; 2 — сетка;
3 — алюминиевый кожух; 4 — вращающийся
магнит; 5 — извлеченные металлические
частицы; 6 — очищенный катализатор.
Эти гранулы, будучи более твердыми, чем частицы катализатора,
истирают их поверхностный слой. В сепараторе образивные
гранулы отделяют от каталпзаторной пыли и используют вторично.
Предложены [339] методы магнитной обработки катализатора.
Для этой цели на нефтеперерабатывающем заводе в Мандане
фирма Американ Ойл (США) применяет специальный магнитный се~
паратор (рис. 89) [339]. Обрабатываемый катализатор поступает
через загрузочную воронку в алюминиевый кожух, внутри которого
медленно вращается магнитный барабан, извлекающий
металлические частицы. Производительность аппарата 14—15 т/сут.
ХИМИЧЕСКИЕ МЕТОДЫ
В периодической и патентной литературе встречается большое
число работ, посвященных удалению металлов с поверхности
отравленного катализатора путем обработки его кислотами:
минеральными (серная, соляная, азотная, фосфорная и др.) и
органическими (уксусная, щавелевая, винная и др.) [340—346]. После
кислотной обработки катализатор промывают водой. В случае серной
и соляной кислоты требуется тщательная промывка для полного
удаления анионов. Анионы органических кислот можно удалять
213
менее тщательно. При обработке катализатора серной кислотой
было установлено, что концентрация кислоты, длительность и
температура очистки — величины взаимосвязанные. Длительность и
температура очистки обратно пропорциональны концентрации
кислоты [345]. При высокой температуре и концентрации кислоты
период очистки будет коротким и, наоборот, при использовании
разбавленной кислоты очистка потребуется более длительная.
Обычно концентрация используемой серной кислоты колеблется от
1 до 50%, длительность очистки — от 1 до 200 ч, температура
очистки — от 15 до 121 °С. После кислотной обработки
катализатор промывают водой, высушивают, обрабатывают водяным паром
в интервале 426—815 °С не менее 2 ч. Обычно обрабатывают
водяным паром при 536—706 °С от 2 до 48 ч.
Ниже приводятся результаты работы по очистке катализаторов
трех типов серной кислотой.
Катализатор первого типа, содержащий 90—85% окиси
кремния, 9% окиси алюминия и 0,15% окиси хрома, был отравлен
0,01 вес. % никеля на промышленной крекинг-установке с
движущимся слоем катализатора при переработке калифорнийского
газойля в течение 141 сут. Этот катализатор был реактивирован
15 раз обработкой серной кислотой различной концентрации и
водяным паром. Влияние режимов очистки катализаторов на
показатели каталитического крекинга приведено в табл. 54.
Из данных табл. 54 видно, что обработка катализатора только
водяным паром лишь незначительно улучшает показатели
процесса. Обработка отравленного катализатора только кислотой
практически не дает никаких результатов. После обработки
отравленного катализатора 5%-ной серной кислотой при комнатной
температуре 100 ч и последующей обработки водяным паром 24 ч при
593 °С (опыт 3) выход кокса уменьшается с 2,5 до 1,0%, а газа —
с 6,7 до 3,3%. Выход бензина на обоих катализаторах примерно
одинаков. Полученные результаты являются стабильными — после
проведения 26 циклов колебания результатов оказались
незначительными.
При очистке катализатора 5 ч кипящей соляной кислотой с
последующей обработкой паром (опыт 4) получаются результаты,
аналогичные очистке 50%-ной серной кислотой с последующей
обработкой паром (опыт 3).
В опытах 5—15 катализатор очищали, как и в опыте 2, серной
кислотой, но разной концентрации и при разной
продолжительности и температуре очистки, затем его обрабатывали паром при
593 °С в течение 24 ч. После такой обработки катализаторов
выход кокса уменьшается на 40—45% по сравнению с
первоначальной величиной и увеличивается плотность газа.
Второй тип алюмосиликатного катализатора, содержащего 91 %
окиси кремния, 9% окиси алюминия и загрязненный около
75-10~4% никеля, был испытан на стандартной установке по
определению индекса активности (опыт 16). Катализатор дает высокий
214
Таблица 54
Влияние режимов кислотной очистки катализаторов
и обработки их водяным паром на показатели
каталитического крекинга
Условия очистки
Плотность
газа
(воздух = 1)
Выход, вес. %
Катализатор первого типа
Без очистки
Пар 100%-ный*
Серная кислота 5%-ная;
комнатная температура, 100 ч
Серная кислота 5%-ная и
1ОО7о-ный пар
Соляная кислота 20%-ная; при
ПО °С 5 ч в аппарате Соксле-
та
Серная кислота 5%-ная;
комнатная температура
8 ч
24 ч
время не указано
96 ч
Серная кислота 50%-ная;
комнатная температура
24 ч
12 ч
Серная кислота 5%-ная; 85 °С
8 ч
20 ч
Серная кислота 10%-ная; 85 °С;
4 ч
Серная кислота 50%-ная; 85 °С
1 ч
2 ч
1
,09
,25
,16
,30
,38
,42
,44
,47
,45
,36
,40
,39
,43
,41
1,37
1,30
6,7
4,6
7,7
3,3
2,6
5,2
3,6
3,7
3,2
2,9
3,6
2,7
3,2
3,1
2,5
3,3
Катализатор второго типа
2,5
1,8
2,6
1,0
0,9
1,5
1,3
1,2
1,1
1,1
1,1
1,1
1,2
1,1
1,0
1,0
Без очистки
Пар 100%-ный
Серная кислота 5%-ная и
100%-ный пар
То же и 100%-ный пар
(вторично)
1,14
1,44
1,42
1,45
Катализатор третьего
Без очистки
Пар 100%-ный
Серная кислота 5%-ная и
100%-ный пар
То же и 100%-ный пар
(вторично)
0,83
0,85
1,05
1,11
8,7
7,0
8,2
5,9
типа
8,4
4,9
5,8
4,5
3,7
2,6
2,4
1,5
5,3
2,9
3,1
2,2
• Во всех опытах водяным паром обрабатывали 24 ч при 593 °С.
** После 26 циклов.
выход кокса как до обработки водяным паром, так и после.
Катализатор, обработанный водяным паром, контактнровался с 5%-ной
серной кислотой в течение 24 ч при комнатной температуре. При
этом отношение бензина к коксу только при кислотной очистке
улучшается лишь слегка. Последующая обработка паром
существенно улучшает селективность катализатора и приближает
отношение бензина к коксу к показателям неотравленного катализатора.
Такие же результаты были получены при обработке
катализатора крекинга третьего типа (монтмориллонитовая глина,
активированная кислотой), который содержал около 224¦ 10~4% никеля.
Влияние обработки катализатора фосфорной кислотой на
показатели процесса определяли следующим образом [343].
Регенерированный катализатор обрабатывали водным раствором ортофос-
форной кислоты со стехиометрическим количеством фосфора
(около 2%) по отношению к металлическим загрязнениям. После
промывки катализатор сушили при 120—200 °С в течение 18—24 ч
и прокаливали при 450—500 °С вначале в среде инертного газа, а
затем в окислительной среде с целью перевода металлов в пиро-
и метафосфаты. Результаты очистки катализатора фосфорной
кислотой представлены ниже:
Необрабо- ,
таннын Обработанный
катализатор катализатор
Индекс активности 91 97
Фактор
коксовый 1,59 1,39
водородный 4,26 2,29
Влияние обработки соляной кислотой на показатели процесса
проверяли на природном равновесном катализаторе, отобранном
из регенератора промышленной крекинг-установки и содержавшем
2200 • 10~4% никеля и ванадия [344]. Полностью регенерированный
катализатор обрабатывали 2,3 кг соляной кислоты на 1 т
катализатора, затем паром 200 мин при 480 °С и атмосферном давлении.
Полученный катализатор испытывали в процессе крекинга в
течение 13 мин при 482 °С, 0,7 МПа до глубины превращения 50%.
Результаты крекинга после обработки катализатора соляной
кислотой представлены в табл. 55.
Результаты обработки загрязненного микросфернческого
катализатора крекинга водным раствором щавелевой кислоты,
приведенные в табл. 56, показали возможность использования этой
кислоты для удаления ванадия и никеля.
К сожалению, вместе с ванадием удаляется и часть алюминия,
что в конечном счете может привести к снижению активности
катализатора. Заметное увеличение удельной поверхности
катализатора и потери алюминия обусловили поиски некислотных
соединений, применение которых исключило бы эти недостатки. Для
проверки некоторые из этих соединений сравнивали с винной
кислотой, которая также оказалась пригодной для удаления металлов
(табл. 57). Полученные результаты подтверждают возможность
216
Таблица 55
Результаты крекинга после обработки катализатора
соляной кислотой
Показатели
Необработанный
катализатор
Катализатор,
обработанный
соляной кислотой
первый
цикл
десятый
цикл
Длительность крекинга 13 мин
Бензин, объемн. %
Сухой газ, м3/м3 сырья*
Водород, м3/м3 сырья*
Кокс, вес. %
Глубина превращения, объемн.
Сухой газ, м3/м3 сырья
Водород, м3/м3 сырья
Бензин, объемн. %
Кокс, вес. %
35,1
77,4
60,0
6,4
49,9
я 50
77,4
60,0
35,1
6,4
34,0
49,8
35,8
4,5
44,5
объемн.
60,0
43,0
36,2
4,9
33,4
55,9
38,9
4,7
46,1
%
63,1
48,7
35,3
5,6.
• При нормальных условиях.
Таблица 56
Результаты обработки катализатора щавелевой кислотой
Условия обработки
Концентрация
раствора,
%
длительность,
ч
температура,
°С
Загрязненный катализатор
0,1
1,0
5,0
12
24
48
12
24
12
24
48
28
28
28
66
66
28
28
28
Содержание металлов
на катализаторе, вес. %
ванадий
0,29
0,20
0,18
0,19
0,10
0,12
0,14
0,13
0,14
никель
0,07
0,05
0,05
0,05
0,04
0,05
0,05
0,04
0,04
алюминий
16,9
17,0
16,8
16,6
14,3
14,0
15,3
14,9
15,6
Удельная
поверхность
катализатора,
М2/Г
59
51
50
49
85
82
62
68
82
Удалено, вес. %
ванадий
_
31
38
35
66
59
32
55
52
никель
_
29
29
29
41
29
29
41
41
алюмн.
ний
_
A)*
1
2
15
17
10
12
8
• Увеличение концентрации.
удаления никеля и ванадия с помощью некислотных соединений,
не растворяющих алюминий. В то время как винная кислота
удаляет 2% алюминия, ацетилацетон и диоксан не затрагивают
алюминия, удаляя заметные количества ванадия и никеля.
217
Таблица 57
Результаты обработки катализаторов некислотными реагентами
Реагент
До обработки
После обработки
ацетилацетоном
диоксаном
винной кислотой
Содержание металлов •
на катализаторе, 10—1 вес. %
ванадий
0,29
0,19
0,14
0,17
никель
0,07
0,04
0,05
0,05
алюминий
16,9
17,3
18,1
16,5
Удельная
поверхность
катализатора,
м2/г
49
53
46
61
Удалено, вес. %
ванадий
34
52
41
никель
43
29
29
алюминий
B)*
' Увеличение концентрации.
Об обработке отравленного катализатора ацетилацетоном в
литературе встречаются разноречивые данные. Известен патент
[346], в котором предлагается метод реактивации отравленного
катализатора ацетилацетоном. Эксперименты были проведены на
катализаторе, состоящем из 90,85 вес. % окиси кремния, 9 вес. %
окиси алюминия и 0,15 вес. % окиси хрома и отравленном
0,01 вес. % никеля при работе в течение 141 сут на смеси
калифорнийских газойлей на промышленной крекинг-установке с
движущимся слоем катализатора. Обрабатывали 200 г этого
катализатора 300 мл ацетилацетона при кипении с обратным
холодильником 4 и 16 ч, после чего катализатор отделяли от ацетилацетона,
промывали, сушили и прокаливали при 537 °С. Ацетилацетоном
очищали также более загрязненный синтетический катализатор,
содержащий около 91% окиси кремния, 9% окиси алюминия и
приблизительно 255 • 10^4% никеля, а также пробу природного
катализатора, активированную кислотой, — монтмориллонитовую глину,
которая содержала около 190 -10~4% никеля. Эту пробу A00 г)
обрабатывали 130—160 мл ацетилацетона 4 ч. Затем катализатор
отделяли от ацетилацетона, сушили, прокаливали при 760 °С и
обрабатывали паром 10 ч при 650 °С. Результаты крекинга после
обработки катализатора по методу Кат-А приведены в табл. 58.
После очистки катализатора только ацетилацетоном выход
кокса и газа уменьшается незначительно. Основное улучшение
показателей достигается лишь после обработки катализатора.
Обработка катализатора только паром оказывает незначительное влияние
на выход газа, кокса и бензина. Содержание никеля после очистки
катализатора не изменяется.
Отмечается [346], что причины улучшения качества
катализатора при обработке ацетилацетоном полностью не ясны.
Предполагается [346], что ацетилацетон может образовывать с никелем,
содержащимся на катализаторе, хелатное соединение, и таким
образом никель становится неактивным в реакции образования кокса.
218
Таблица 58
Результаты крекинга после обработки катализатора
ацетилацетоном
Показатели
ЇЙ
Очистка ацетилацетоном
га
а
<о х
о о
Но
аро
4 ч
3
«1-
о а с
о<о га
с о с
бра-
о
о
к г
-с о
н о,
о та
16
ч
а;
ОСЛ
S
и
§.а
о с
Синтетический катализатор, содержащий 0,15% окиси хрома
и 0,01 % никеля
Выход
бензина, объемн. %
кокса, вес. %
газа, вес. %
Содержание никеля в
катализаторе, вес. %
Удельная поверхность
катализатора, м2/г
Отношение бензин:кокс
29,2
2,5
6,7
0,01
204
11,7
30,9
1,8
4,6
205
17,2
29,2
1,7
4,7
0,01
212
17,2
28,4*
1,0*
2,6*
28,4*
27,8
1,6
5,3
0,01
209
17,40
27,9*
1,0*
2,7*
27,9
Синтетический катализатор, содержащий 255-10~4% никеля
Выход
30,1 — 27,5
3,9 - 3,4
бензина, объемн. %
кокса, вес. %
газа, вес. %
Отношение бензин:кокс
9,1
7.7
7,8
8.1
27,5**
2,6**
6,5**
10,0**
Природный катализатор, содержащий 190-10-'% никеля
Выход
34,3
3,4
5,2
10.0
бензина, объемн. %
кокса, вес. %
газа, вес. %
Отношение бензин: кокс
32,9
4,3
8,5
7.7
29,2**
2,0**
3,3**
14,6**
• Обработка водяным паром 24 ч при 593 °С. ** Обработка водяным паром 10 ч при 650 °С.
Реакция образования хелатного соединения никель-ацетилацето-
на может протекать следующим образом:
СН3 СН3
I I
/С=О .С—ОН
Н2С< =5=* HC<f
СН3
хС-О
2HC«f
NC=O
СИ,
СНз
ЧС=О
СНз
ацетилацетон
NiO
моноенольная
форма
Н
/О
СН3
СН,
СН,
Известны также патенты, предлагающие методы реактивации
отравленных катализаторов с помощью хлор- или
фосфорсодержащих соединений (фосфор или хлор откладываются на
катализаторе в количестве, достаточном для деактивации металлических
ядов) и с последующей обработкой паром при 425—815 °С [294].
В работе предлагается [347] отравленные катализаторы крекинга
реактивировать обработкой фосфорным ангидридом или
органическими и неорганическими веществами, образующими Р2О5 при
прокаливании. Органические фосфор- или фосфорсерусодержащие
вещества (эфиры фосфорных кислот, фосфины) растворяют в
сырье, в процессе крекинга они откладываются на катализаторе,
затем органическую часть выжигают. После обработки
реактивированного катализатора водяным иаром при 540—700 °С в течение
2—48 ч активность его возрастает.
После 140 циклов (при 5—6 чередующихся циклах реакции и
регенерации за 1 сут.) катализатор следующего состава (в %):
окись кремния 90,85; окись алюминия 9,0; окись хрома 0,15 и
никель 0,01 активировали и затем исследовали его активность по
методу Кат-А. Отношение бензин : кокс и выход кокса (в %) были
соответственно равны: для отравленного катализатора 11,7 н 2,5;
после двухкратного нанесения по 0,5 вес. % Р2О5 в виде три-о-кре-
зилфосфата 14,3 и 2,0; после обработки паром 19,5; 1,4; для
катализатора, реактивированного водным раствором (NH4)jHPO4 1%
от массы катализатора в расчете на Р2О5 15,6 и 2,0; обработанного
паром 18,0 и 1,6. При такой обработке катализатора содержание
никеля не изменяется.
Предлагается [348] обрабатывать катализатор при 620 °С и
атмосферном давлении с помощью РОС13 в таком количестве,
чтобы на катализаторе отложилось не менее 0,001 вес. % фосфора
и хлора. В качестве катализатора использовали активированную
глнну, отобранную из регенератора установки. Она содержала
2255-10~4% окислов ванадия и никеля и 0,35 вес. % кокса.
Катализатор обрабатывали РОС13 сразу после регенератора и после
дополнительной регенерации. Результаты представлены в табл. 59.
Эти данные показывают эффективность такой обработки
катализатора. Еще больший эффект достигается при последующей
гидратации катализатора. При крекинге на другом образце
катализатора [348], обработанном только РОС13, выход водорода 50,2 м3/м3
сырья, бензина 36,4 объемн. %, кокса 5,0 вес. %, а при крекинге
на катализаторе, обработанном РОС13, а затем водяным паром,
соответственно 36,4; 36,8; 4,6. Авторы [348] показали, что
обработка паром при 480 СС менее эффективна, чем при 590 °С: выходы
водорода, бензина, кокса при 480 °С составляют соответственно
28,7; 37,0 и 4,0, а при 590 °С 23,6; 37,7 и 3,5. Обработка
катализатора только фосфор- или только хлорсодержащими соединениями
с последующей паровой обработкой также эффективна [346].
Имеются патенты, в которых предлагается обрабатывать
отравленный металлами катализатор раствором гидроокиси щелочного
220
Таблица 59
Результаты обработки катализатора с помощью POCU
Катализатор
Полностью
регенерированный
То же
Регенерированный на
промышленной
установке
То же
»
Обработка РОСІз
Необработанный
Обработанный
Необработанный
Обработанный
*
ірья
II
« г
60,0
19,0
13,1
58,2
36,2
40,7
¦?«
Э л
2 и d
и —s
77,4
34,7
27,1
76
53,7
59,1
а?
ІІ
я %
И о
35,1
36,9
38,0
34,8
37,2
37,3
а?
О
а
оке
6,4
4,4
3,4
5,6
4,3
4,3
металла, в частности натрия [349]. Эксперименты проводили на
алюмосиликатном катализаторе, содержащем 87-10—*% никеля и
167 • 10—4% ванадия. Катализатор находился в промышленной
установке с движущимся слоем до тех пор, пока его активность не
упала от исходной 34 до 23,4. Пробу катализатора 250 см3
вымачивали в 500 см3 водного раствора, содержащего щелочи 100 г/л, в
течение 72 ч при комнатной температуре. Затем раствор сливали и
катализатор промывали водой до полного удаления соды. После
обработки щелочью катализатор обрабатывали 50 г водного
раствора, содержащего азотнокислый аммоний концентрацией 100 г/л.
Раствор сливали через 24 ч и катализатор промывали водой.
Такую обработку катализатора азотнокислым аммонием проводили
трижды. После этого катализатор промывали водой до pH
промывной воды 7—8, сушили при 130 СС в течение 2 ч и прокаливали в
воздухе 3 ч.
В довольно многих патентах вредное действие ядов
предлагается снижать нанесением на катализатор тех или иных металлов.
Например, в катализатор крекинга, отравленный железом,
предлагается вводить бериллий [350]. Отравление катализатора
железом особенно необходимо предотвращать в случае периодических
процессов. Во время цикла крекинга сера, вносимая с сырьем,
соединяется с железом реактора и образуется сульфид железа. Во
время регенерации воздух превращает сульфид железа в окись,
которая откладывается на катализаторе и таким образом
уменьшает его активность. Кроме того, окись железа катализирует
окисление, и выжиг кокса идет в основном до СО3 вместо СО, что
вызывает значительное повышение температуры во время регенерации.
При добавлении бериллия отрицательное действие окиси
железа уменьшается. Хорошие результаты получаются пропиткой
катализатора водным раствором нитрата бериллия с последующим на-
221
гревом катализатора для превращения нитрата в бериллии. Реак-
тиванию проводили следующим образом. 10 вес. ч. катализатора,
содержащего 1,0 вес. % окиси железа, пропитали 6,5 вес. ч.
разбавленного раствора нитрата бериллия. Катализатор высушивали
при 482 °С 10 ч для превращения нитрата бериллия в бериллий.
Полученные таблетки содержали около 0,22 вес. % бериллия.
Активность этих катализаторов определяли по реакции
окисления СО в СО2. Окись углерода и избыток кислорода пропускали
над пробой катализатора, двуокись углерода адсорбировали аска-
ритом. Непрореагировавшую окись углерода окисляли над окисью
меди и образующуюся двуокись углерода также адсорбировали
аскаритом. Активность катализатора сравнивали по температуре,
необходимой для превращения 15% окиси углерода. Результаты
этих испытаний приведены ниже:
Температура, требуемая
для превращения 15% СО
Катализатор в cq2
°С
Реактивированный бериллием 475
Отравленный, нереактивированный .... 357
Неотравленный 510
Видно, что при добавлении бериллия активность окиси железа
по отношению к окислению СО в СО2 уменьшается.
Было исследовано влияние добавок бериллия на показатели
процесса. Сравнивали два образца: старый промышленный
катализатор с установки Гудри, содержавший около 0,3 вес. % железа,
и этот же катализатор, реактивированный бериллием по методике,
описанной выше. Опыты показали, что катализатор,
реактивированный бериллием, образует меньше кокса и больше жидких
продуктов реакции; кроме того, отношение СОг : СО в его присутствии
значительно уменьшается.
Авторы [350] выясняли также возможность введения бериллия
в свежий катализатор для предотвращения его отравления
железом. Пробу свежего катализатора Гудри пропитали 0,4% бериллия
описанным способом, затем прокаливали при 538 °С в течение 10 ч,
пропитали 0,5% железа и добавочно прокаливали около 10 ч при
593 °С. Другую пробу свежего катализатора пропитали 0,5%
железа и прокаливали 40 ч при 593 °С. Затем эти катализаторы
испытывали в стандартных условиях.
Влияние бериллия на свежий катализатор, содержащий
железо, следующее:
Катализатор,
_ пропитанный Катализатор,
Показатели бериллием, пропитанный
затем железом железом
Выход, вес. %
газа 5,63 7,00
кокса 5,27 5,85
жидких продуктов 84,0 83,2
кислых газов 0,136 0,020
СО2:СО 5,50 3,10
Индекс активности 21,5 23,1
222
Из приведенных данных видно, что бериллий, нанесенный на
катализатор раньше железа, не приостанавливает его отравляющего
действия. Выход кокса такой же, как и на катализаторе,
содержащем только железо, индекс активности несколько меньше, а
отношение СО2 : СО в 1,8 раза больше. Очевидно, конечное действие
катализатора определяется последовательностью нанесения на
него металлов. Это же отмечалось в работе [247]. По мнению
авторов работы, катализатор реактивируется в результате образования
шпинели BeFe2O4, которая либо имеет маленькую активность к
окислению, либо совсем ее лишена.
Другие авторы [351] предлагают для реактивации
катализаторов наносить на них медь, серебро, золото, цинк, кадмий, ртуть,
олово или металлы подгруппы III Б и IV Б периодической
системы в тонкодиспергированном состоянии (менее 0,5 вес. °/о на
катализатор).
Предлагается [352] реактивировать алюмосиликатные
катализаторы, содержащие менее 0,2 вес. % никеля (или меди, железа и
ванадия), пропиткой достаточным количеством водного раствора
соединений хрома, которые разлагаются при прокаливании с
образованием окиси хрома, а затем обрабатывать катализатор при
538—705 °С водяным паром 2—48 ч. При этом на каждый атом
никеля наносится два или больше атома хрома. Количество окиси
хрома должно быть не менее 0,5 вес. % на массу катализатора.
Для пропитки применяют водный раствор нитрата, ацетата хрома,
хромата н бихромата аммония или наносят хром в процессе
крекинга добавлением к крекируемому сырью нафтената, оксалата
или комплексного цианида хрома.
В другом патенте [353] предлагается реактивировать
катализатор, отравленный небольшим количеством никеля (менее
0,2 вес. %), пропиткой его водным раствором неорганических
солей, таких, как азотнокислый алюминий, хлористый алюминий,
сернокислый алюминий, которые при прокаливании разлагаются
с образованием окиси алюминия. Ее должно отлагаться на
катализаторе не более 5,0%- Реактивировать катализатор можно также,
контактируя его с парами хлорида алюминия. Кроме того, авторы
[353] предлагают добавлять в сырье крекинга органические
соединения алюминия, например нафтенат алюминия. Пропитанный
катализатор сушат, прокаливают и обрабатывают водяным паром
при 426—815 °С в течение 2 ч.
Катализатор, состоящий из 90,85 вес. % окиси кремния,
9 вес. % окиси алюминия и 0,15 вес. % окиси хрома и
загрязненный 345• 10~4 вес. % никеля, испытывали на активность по
стандартной методике. Затем в него вводили около 2 вес. % окиси
алюминия пропиткой водным раствором азотнокислого алюминия.
После прокалки при 538 °С и обработки водяным паром в течение
10 ч при 650 °С определяли активность катализатора по методу
Кат-А. Из табл. 60 видно, что реактивированный катализатор
имеет значительно более высокое отношение бензина к коксу, боль-
223
Таблица 60
Влияние добавления окиси алюминия на качество
катализатора крекинга
Катализатор
Отравленный
Отравленный + 2% окиси
алюминия + обработка
Отравленный
Отравленный + 0,5°/о
окиси алюминия+паровая
обработка
Отравленный + 1,0%
окиси алюминия+паровая
обработка
Отравленный + 2,0%
окиси алюминия+паровая
обработка
Содержание
никеля,
Ю-*, вес. %
345
345
350
350
350
350
Насыпная
плотность,
кг/мЗ
750
850
800
810
810
830
Относительная
плотность газг
0,84
1,55
1,15
1,45
1,46
1,44
Выход
газ
12,0
7,4
7,8
6,5
6,1
6,4
продуктов, вес. %
кокс
6,3
2,1
2,6
1,9
1,9
1,7
бенэии (к. к.
210 °С),
объемн. %
30,0
37,0
30,5
31,0
33,2
31,9
Отношение
бензин.кокс
4,8
17,6
11,7
16,3
17,6
18,8
шнй выход бензина и меньший выход кокса, чем отравленный
катализатор.
При изменении содержания окиси алюминия от 0,5 до 2 вес. %
существенной разницы в результатах крекинга не обнаружено.
После добавления к отравленному катализатору окиси алюминия и
последующей обработки паром эти результаты значительно
улучшились. При этом содержание никеля на отравленном и
реактивированном катализаторе остается одинаковым. Авторы [353]
полагают, что ирп такой обработке никель не удаляется, а переходит
из активного состояния в неактивное. Предлагается [354]
пропускать через регенерированный катализатор газ, содержащий хлорид
одного из металлов, входящих в состав катализатора. Этот процесс
следует проводить при температуре 93—650 °С, предотвращающей
конденсацию хлорида, и соотношении катализатор : хлорид от 3 : 1
до 50 : 1. После пропуска хлорида катализатор продувают
инертным газом.
Некоторые авторы [355] рекомендуют контактировать
отравленный катализатор с 0,05—1,0 хлористого хромила (СгСЬСЬ) при
470—500 °С. В патенте [356] указывается на возможность
реактивировать катализаторы крекинга добавлением 0,05—5 вес. %
окиси щелочных и щелочноземельных металлов с последующим
прокаливанием катализатора при 675—955 °С в течение 1—3 ч без
водяного пара.
Проведены обширные исследования [238, 251, 357—363] по
восстановлению активности и селективности природных и отработан-
224
ных алюмосиликатных катализаторов. Было показано, что после
добавления алюминия [357, 358], титана [238, 357], хрома [238,
359], вольфрама [358, 359] и кремния [238, 251, 359] активность
отработанного катализатора возрастает. На основании
исследования металлов и их окислов авторы разделяют добавки на две
группы [238]: 1) повышающие активность катализатора без
изменения селективности (цирконий, магний, алюминий, кремний
и др.); 2) способствующие протеканию (наряду с реакциями
крекинга) реакций дегидрогенизации, дегидроциклизации,
ароматизации (окислы хрома, молибдена, вольфрама, редкоземельных
элементов, металлы и окислы металлов VIII группы и др.).
В связи с существенным улучшением показателей
каталитического крекинга при удалении металлов с поверхности алюмоси-
ликатного катализатора ряд методов реактивации был исследован
весьма подробно. В Советском Союзе разработан процесс сухой
деметаллизации катализатора. Два метода реактивации
катализаторов нашли применение в США в промышленном масштабе.
Фирма Атлантик Рифайнер (США) разработала метод очистки
катализатора крекинга, обеспечивающий достаточно полное удаление
вредных металлических примесей. Этот процесс носит название
Мет-х. Он внедрен на нефтеперерабатывающем заводе в
Филадельфии в октябре 1961 г. Другой процесс очистки катализатора —
Демет — разработан фирмой Синклер Рифайнер и внедрен на
заводе в Вудривере (штат Иллинойс) в декабре 1961 г.
Для этих процессов характерно, что они предотвращают
накопление металлов на катализаторе и позволяют крекировать сырье
с большим содержанием металлов при пониженном удельном
расходе катализатора, повышенной производительности и лучшей
селективности процесса. Ниже приводится описание указанных
процессов деметаллизации.
ПРИМЕНЕНИЕ ИОНООБМЕННЫХ СМОЛ
(ПРОЦЕСС МЕТ-Х)
В процессе Мет-х катализатор крекинга реактивируют с
помощью ионообменных смол. При контакте с ионообменной смолой
металлические примеси хорошо удаляются с катализатора.
Влияние различных параметров очистки на активность катализатора и
его коксообразующий фактор изучалось в работе [364]. Опыты
проводили на алюмосиликатном катализаторе следующего
химического состава (в вес. %): окись алюминия 14,2; натрий 0,31;
железо 0,18; никель 0,011; ванадий 0,021. В качестве
ионообменной смолы применяли пермутит, пропущенный через сито 30 меш.
Из сухого загрязненного катализатора, смолы и воды
приготовляли суспензию @,5 г катализатора на 1 мл смолы); количество
смягченной воды брали из расчета 0,55 г катализатора на 1 мл.
воды. Ионообменную смолу обрабатывали 10%-ной серной
кислотой (из расчета 544 кг на 1 м3 смолы) с последующей промывкой
15-2206 225
водой до достижения pH 4,5. Активность катализатора определяли
по суммарному выходу в процессе крекинга жидких продуктов,
выкипающих до 204 °С. На рис. 90 показано влияние
температуры, длительности ионообменной очистки и pH среды на коксообра-
зующий фактор.
Увеличение продолжительности, повышение температуры
очистки и уменьшение pH среды способствуют уменьшению коксообра-
зования, причем наиболее эффективно в первые часы очистки.
Наиболее подходящими приняты температуры от 80 до 100 °С. При
более высокой температуре результаты очистки не улучшаются.
1.8
%1,6
о-
г
Рис. 90. Влияние
температуры, длительности
ионообменной очистки и
pH среды на коксообра-
зующий фактор:
/ — 82 °С, 1 ч; 2— 100 °С,
1ч; 3 — 82 °С, 4 Ч; 4— 100 °С,
4 ч.
Рис. 91. Влияние кислотности
среды на активность катализаторов:
/ — очищенный; 2 — неочищенный.
Показано также, что на качество ионообменной очистки влияет
вид кислоты, применяемой для обработки суспензии. Наиболее
активный катализатор получается при использовании для
ионизации суспензии лимонной кислоты.
Для проверки влияния очистки на стабильность катализатора
несколько образцов равновесного катализатора, очищенного при
100 °С в течение 4 ч при разных величинах pH, и образец
неочищенного катализатора обрабатывали паром 5 ч при 566 °С и
давлении 0,2 МПа, после чего определяли активность. Влияние
кислотности среды на активность катализаторов показано на рис. 91.
Исходный катализатор и катализатор после ионообменной обработки
испытывали также на термическую стабильность. Результаты
приведены в табл. 61.
Таблица 61
Влияние ионообменной обработки на термическую стабильность
катализатора
Катализатор
Активность*
катализатора
Д + П,
объемн. %
Поверхность,
М2/Г
Удельный
объем пор,
До термической обработки
Исходный
После ионообменной обработки**
После термичее койобр
Исходный катализатор
После ионообменной обработки**
После термической обр
Исходный
После ионообменной обработки**
41
55
а б о т к и
39
43
а ботк и
28
36
• Активность оценивается выходом бензнна+потерн.
•• При pH 3,5.
98
ПО
2 ч пр и
65
95
2 ч п р и
35
50
496°
530°
С
0,33
0,35
0,24
0,31
0,12
0,17
Из табл. 61 видно, что обработанный катализатор обладает
большей термической стабильностью, чем необработанный.
Для определения степени удаления металлов методом Мет-х
был приготовлен специальный образец катализатора, содержащий
0,1 вес. % никеля. Катализатор обладал активностью 21 и
фактором коксообразования 6,5. После ионообменной обработки 4 ч при
100 °С и pH 2,7 активность катализатора повысилась до 35, а
фактор коксообразования снизился до 1,3. Содержание никеля
уменьшилось с 0,10 до 0,03%. т. е. более чем в 3 раза.
Большой интерес представляют эксперименты [365],
проведенные на пилотных установках с целью сопоставления результатов
обычной работы крекинг-установки без очистки катализатора с
результатами работы при очистке части катализатора, взятой из
системы, в процессе Мет-х. Изучали влияние процесса Мет-х на
активность, стабильность катализатора, на его механическую
прочность, регенерационную характеристику, на обессеривающую
способность, распределение и качество получаемых продуктов.
На пилотной установке были воспроизведены режимы работы
реактора и регенератора промышленной установки. Сырье и
катализатор были отобраны также с промышленной установки.
Ежесуточно 11% катализатора (от загрузки системы) с пилотной
установки очищали в процессе Мет-х и возвращали на установку.
Качество равновесного промышленного катализатора,
использованного в данных опытах, приведено ниже:
15*
227
Активность катализатора по методу Атлантик Д + П,
объемы. % 43,6
Удельная поверхность, м2/г 100
Фактор коксообразования 2,2
Содержание металлов, вес. %
железо , 0,21
никель 0,018
ванадий 0,035
натрий 0,23
алюминий 13,5
Фракционный состав, %. мк
<40 4
<50 . 25
<60 59
<80 85
Для обеспечения удовлетворительной циркуляции
катализатора на пилотной установке от него отдували до 35% мелких
фракций. Лабораторные исследования показали, что 65%-ный остаток
имел примерно такую же активность, удельную поверхность и
такое же содержание металлов, как и исходный неотвеянный
катализатор. Систему догружали свежим синтетическим алюмосиликат-
ным катализатором, содержащим 13% окиси алюминия. Сырьем
служила смесь газойлей следующего качества:
Плотность d\l^ 0,8916
Анилиновая точка, °С 81,1
Дизельный индекс 48,5
Содержание серы, вес. % 1,1
Кокс по Рамсботтому, вес. % 0.3
Разгонка по ASTM
и. к., °С 192
выкипает при температуре, °С
10% 290
50% 387
90% 489
к. к., °С 539
Содержание металлов, 10~4%
железо 2,5
никель <0,5
ванадий <0,5
натрий 2,0
Ниже приводятся рабочие условия пилотной и промышленной
установки каталитического крекинга:
Пилотная Промышленная
установка установка
Реактор
Температура, °С 491,0 488,0
Давление, МПа 0,14 0,13
Рециркулят, объемн. % 0 40,0
Объемная скорость подачи сырья*, ч-1 . . 1,9 1,5
Отношение катализатор:сырье 5 7—10
Водяной пар*, вес. % 0 1,5
Степень превращения, объемн. %*•••• 50 45
Расход свежего катализатора, кг/м3 свеже-
Л QQ І ТЙ
го сырья 0,oo 1,оо
Расход свежего катализатора за 1 сут, %
от общего количества 1,7 1,0
228
Пилотная Промышленная
установка установка
Отпарная секция
Температура, °С 482 468
Давление, МП а 0,14 0,16
Парциальное давление пара, МПа .... 0,2 0,27
Регенератор
Температура, °С 600 600
Давление, МПа 0,14 0,08
Избыток кислорода, % 6,0 1,0
Парциальное давление пара, МПа .... 0,01 0,03
* В расчете на суммарное сырье.
Для ускорения экспериментов на пилотной установке была
увеличена объемная скорость подачи сырья. Металлов на
катализаторе с пилотной установки содержалось больше из-за меньшего
добавления свежего катализатора на единицу перерабатываемого
сырья. Для создания одинаковых условий дезактивации
температуру в отпарной секции пилотной установки поддерживали
несколько выше, а парциальное давление пара ниже, чем в
промышленных условиях (не применяли вспрыск воды).
Зависимость деметаллизации равновесного катализатора от
длительности работы установки каталитического крекинга
показана на рис. 92. По мере увеличения длительности работы
катализатора без деметаллизации содержание на нем железа, никеля и
ванадия увеличивалось. Содержание натрия слегка снижалось
из-за отсутствия вспрыска воды. По скорости накопления примесей
в катализаторе было рассчитано содержание металлов в сырье. Эти
цифры, как показано ниже, хорошо согласуются с данными
анализа:
Содержание металлов, 10—1%
рассчитано по анализу
Железо 3,0 2,5
Никель 0,4 0,5
Ванадий 0,4 0,5
Натрий 1,5 2,0
При работе установки с деметаллизацией катализатора
содержание железа, ванадия и натрия на равновесном катализаторе
намного ниже, чем при работе без деметаллизации. С увеличением
длительности работы количество этих металлов уменьшается.
Содержание никеля на катализаторе не уменьшилось, но также было
меньше, чем в основном варианте. Из рис. 92 видно, что при
очистке катализатора металлы удаляются только частично. Авторы
считают, что только небольшая часть металлов является примесями,
которые активно отравляют катализатор; металлы, остающиеся на
229
катализаторе после очистки, не дезактивируют катализатор при
его последующем использовании.
В течение опытов, проводимых без очистки катализатора,
активность равновесного катализатора повысилась (рис. 93). Авторы
объясняют это тем, что
количество добавляемого свежего
катализатора по отношению ко всей
загрузке системы было на
пилотной установке выше, чем в
промышленных условиях. Кроме
того, на пилотной установке не
разбрызгивалась вода. При
использовании процесса Мет-х
равновесная активность
катализатора на протяжении всего опыта
была выше, чем в основном
варианте. Авторы объясняют это
снижением содержания натрия
после очистки. Расчеты
показыlu
0,3
0,1
^.0,0k
It- W
-—^_w _ о
го ko во
Длительность работы, сут
Рис. 92. Зависимость деметаллизации
вают, что такое снижение
соответствует 4—5 единицам
активности. Подобная же разница
наблюдается между активностью
равновесного катализатора до и
катализатора от длительности pa- п°сле очистки. Удельная поверх-
боты:
\ — основной вариант; 2 — вариант с де-
металлизацней-
ность катализатора,
работавшего по основному варианту, такая
же, как после очистки
катализатора по методу Мет-х.
Опыты на пилотной установке показали, что в результате
удаления металлов селективность катализатора улучшается, что
выражается в уменьшении фактора коксообразования и в лучшем
распределении получаемых продуктов. Изменение фактора
коксообразования равновесных катализаторов при использовании процесса
Рис. 93. Зависимость удельной поверхности, фактора коксообразования и
активности катализатора от длительности работы и применения деметаллизация:
/ — основной вариант; 2 — вариант с деметаллизацией,
230
1*
It
є
І
і
' 2
60
20
Мет-х и без него было показано на рис. 93. Данные о выходах
отдельных продуктов при работе по обоим вариантам
представлены на рис. 94. Как это видно, при работе по
основному варианту выход кокса
с увеличением длительности
опыта увеличивается из-за
меньшего количества
добавляемого свежего катализатора
на единицу перерабатываемого
сырья @,88 кг/м3 на пилотной
установке и 1,38 кг/м3 на
промышленной установке, из
которой был отобран
равновесный катализатор). Кроме
того, сырье пилотной установки
содержало несколько больше
железа, чем среднее сырье
промышленной установки. При
работе по методу Мет-х выход
кокса с течением времени
уменьшается. Через 1 месяц ра-
боты катализатора коксовый
фактор его в системе с
очисткой раза в два меньше, чем в
системе без очистки. Данные
о выходе легких
углеводородов при работе по обоим
вариантам (при степени превра-
0 20 40 60
Длительность работы , сут
Рис. 94. Выход продуктов крекинга прн
степени превращения 50% без
рециркуляции в зависимости от длительности
работы:
/ — основной вариант; 2 — варнажт с деметал-
лнзацней.
риантам (р с рр
щения 50%) приведены ниже:
Углеводороды,
вес. %
н2
¦с,
С2Н4
с2н6
СзН6
Без очнсткн
по методу
Мет-х
0,47
1,2
0,56
1.1
3,3
1,7
С4Н8 (объемн. %) 6,2
изо-С4Н,о (объемн. %) .... 2,7
н-С4Н,о (объемн. %) .... 0,7
С очисткой
по методу
Мет-х
0,2
1,0
0,4
1,0
2,9
1,4
5,6
3,5
1,0
При использовании процесса Мет-х более чем в два раза
снизился выход водорода. Существенных изменений в октановых
числах бензинов (в чистом виде и с этиловой жидкостью) не
обнаружено. На распределение серы по продуктам крекинга этот процесс
также не влияет — содержание серы в бензине в обоих случаях
(при очистке по методу Мет-х и без нее) 0,13%, а в общем
циркулирующем газойле 1,1%.
Для выяснения влияния очистки по методу Мет-х на
истираемость катализатора во время опытов на пилотной установке изме-
231
ряли размер частиц и потерь катализатора. При сравнении с
основным вариантом разницы в образовании пыли и крошки
обнаружено не было. Кроме того, распределение размеров частиц в
пробах, отобранных из равновесного катализатора в конце обоих
пробегов, было одинаковым.
После широких исследований фирма Атлантик Рифайнинг на
своем нефтеперерабатывающем заводе в Филадельфии построила
промышленную установку Мет-х производительностью 40 т/сут, на
которой очищается часть катализатора A1% от всей загрузки),
отводимого с двух установок каталитического крекинга общей
производительностью 7600 м3/сут. Установка работает
удовлетвориРис. 95. Схема процесса Мет-х:
1 — холодильник; 2 — демннералнзатор; 3 — регенератор смолы; 4 — сепаратор; 5 — реактор;
б —фильтр; 7 — резервуар для суспензии катализатора, / — загрязненный катализатор
с установки каталитического крекинга; // — дисперсионная среда для катализатора;
/// — вода; IV— серная кислота; V — сырье каталитического крекинга; VI— очищенный
катализатор на крекинге; VII — дренаж.
тельно с октября 1961 г. Построена она была в содружестве с
фирмой Пфаудлер Пермутит, которая имеет значительный опыт в
очистке воды ионообменом.
Первоначальная схема процесса изображена на рис. 95 [366,
367]. Процесс включает четыре стадии: 1) ионный обмен; 2)
разделение катализатора и смолы: 3) выделение катализатора из
суспензии; 4) регенерация смолы. Катализатор, загрязненный
металлами, поступает самотеком в один из четырех реакторов, где он
смешивается с ионообменной смолой, с которой реагируют ионы
металлов. При этом температура, концентрация, pH раствора и
длительность реакции тщательно поддерживаются на оптимальном
уровне, необходимом для удаления металлов. Каждый реактор
представляет собой котел емкостью 15 м3 с рубашкой и
механической мешалкой. Смолу подают в виде суспензии в химически
очищенной воде. Когда реактор уже полностью загружен, катализатор
и смолу тщательно перемешивают. Реакторы работают в следую-
232
щем порядке: один загружается, в двух протекает реакция,
четвертый разгружается. По окончании реакции смесь поступает в два
сепаратора, представляющие собой вертикальные колонны. В
сепараторе катализатор и смола разделяются и поступают на
регенерацию. Катализатор отделяется от смол сначала путем отмучива-
ния (отдувки), а затем при помощи фильтрации в специальных
фильтрах, которые задерживают более крупные частицы смолы и.
пропускают мелкодисперсную катализаторную массу. После сушки
катализатор направляется в емкости для повторной загрузки в
установку каталитического крекинга.
Ионообменную смолу регенерируют обработкой серной
кислотой и последующей промывкой водой. Потери смол не превышают
0,15%/сут. Важное условие успешной эксплуатации установки —
тщательная очистка воды (фильтрация, ионный обмен и др.), так
как примеси, содержащиеся в воде, загрязняют катализатор.
На основании опыта работы этой установки запроектированы
новые усовершенствованные, почти полностью автоматизированные
установки Мет-х [368]. Изменения, внесенные в схему фирмой
Келлог, которой принадлежат патентные права на этот процесс,
позволили уменьшить габариты установки. Новая схема занимает
маленькую площадь и имеет значительно меньшую высоту, чем
высота существующей промышленной установки A4 против 56 м).
На новой установке вместо четырех реакторов будет работать
только один. Ниже приводятся данные о работе установки Мет-х,
обслуживающей две промышленные установки каталитического
крекинга:
Фактор коксообразования
равновесного катализатора с установок
каталитического крекинга
очищенного катализатора с установки
Мет-х
Содержание металлов на равновесном
катализаторе, 10~4-вес. %
ванадий
никель
железо
Удельная поверхность, м2/г
Активность (по методу Атлантик Д + П) .
Расход катализатора, кг/м3
свежий
равновесный
Содержания алюминия, вес. °/о
Отношение Н2:СН4
Кокс на катализаторе после регенерации, °/о
Температура в регенераторе, °С
Превращение сырья, л/кг кокса
Сырье
эквивалент железа, 10~4-вес. % . . .
50% выкипает при температуре, °С . .
23$
До очистки
2,00
—
1250
200
2000
130
48
1,71
0,85
14,5
3,0
0,22
649
6,8
2,0
400
После работы
установки
Мет-х
в течение
2,5 месяцев
1,45
1,00
700
120
1600
110
45
1,43
1,14
11,0
1,1
0,40
632
7,8
3,0
438
Процесе Мет-х позволяет перерабатывать не только обычный
вакуумный газойль, но и более тяжелое сырье с большим
содержанием металлов при меньшем факторе коксообразования и
газообразования, меньшем расходе катализатора и большей степени
превращенного сырья на единицу выхода кокса. Поэтому
применение процесса Мет-х дает возможность значительно расширить
ресурсы сырья каталитического крекинга.
Эффективность работы деметаллизированного катализатора
проверяли также на заводе фирмы Юнайтед Рифайнинг (США)
[369]. Пробег проводили 12 сут на установке каталитического
крекинга флюид с обычным катализатором. К катализатору
добавляли по 6 т/сут деметаллизированного катализатора,
подготовленного на установке Мет-х фирмы Атлантик Рифайнинг (США).
Производительность установки каталитического крекинга
составляла 954 м3/сут. На установке крекингу подвергали смесь легкого
газойля, парафинового дистиллята, вакуумного газойля и деас-
фальтизата, полученного после пропановой деасфальтизации
гудрона. Качество сырья приведено ниже:
Плотность d\\l 0,8939
Вакуумная разгонка, °С
и! к 232
10% 316
30% 358
50% 413
70% 466
90% 544
Содержание кокса по Рамсбогтому, % 0,5
Содержание, вес. %
серы 0,66
азота 0,10
Анилиновая точка, °С 88
Содержание металлов, Ю-'-вес. %
железо 0,6
никель 0,4
медь 0,2
Качестзо используемых на установке катализаторов приведено
в табл. 62.
Деметаллизироваиный катализатор испытывали при двух
различных режимах работы установки: при максимальной степени
превращения сырья и максимальной производительности
(табл. 63).
При увеличенной глубине превращения выход бензина
повысился на 2,2%, а выход бутан-бутиленовой фракции — на 2,5% при
практически неизменном коксообразовании. При увеличенной
производительности (на 11 % по свежему сырью больше) выход
бензина по сравнению с обычным вариантом работы увеличился на
1,3%, а бутан-бутиленовой фракции — на 2,0%; общая глубина
конверсии зозросла на 2%.
Таким образом, применение Мет-х позволяет: I) увеличить
конверсию; 2) уменьшить расход катализатора; 3) перерабатывать
234
Таблица 62
Качество катализаторов
Показатели
Относительная активность
Удельная поверхность, м2/г
Фактор коксообраэования
Фактор водородообразования
Содержание металлов на катализаторе,
10-4вес. %
никель
железо
ванадий
Деметаллн-
энрованный
катализатор с
установки
Мет-х
о,п
101
1,05
1,05
170
1080
540
Пробы катализаторов,
отобранные с установки в ходе
ДО
добавления
цеметаллн-
знрованного
катализатора
0,16
166
1,81
4,20
330
3080
450
испытаний
в момент
добавления
деметалдн-
знрованного
катализатора
0,14
137
1,19
2,80
230
1640
470
равновесный
0,12
127
1,15
2,10
200
1400
450
Таблица 63
Показатели работы установки с деметаллизированным
катализатором
Показатели
Производительность, мэ/сут
Коэффициент рециркуляции
Температура, °С
в реакторах
в регенераторе
Выход продуктов реакции
Н2, вес. %
С,, вес. %
С2, вес. %
СЭН6, объемн. %
С3Н8) объемн. %
С4Нв, объемн. %
изо-С4Нш, объемн. %
«-С4Ніо, объемн. %
бензин Cs—221 °С, объемн. %
циркулирующий продукт, объемн. %
легкий
тяжелый
кокс, вес. %
Глубина конверсии, объемн. %
До добавления
деметаллнзн-
рованного
катализатора
882
1,42
493
662
0,32
0,88
1,66
6,26
2,05
6,62
3,48
0,94
47,8
24,0
12,4
7,24
63,6
С добавлением деыеталлн-
знрованного
при
увеличенной
глубине
превращения
889
1,44
498
634
0,16
1,04
1,77
6,44
2,84
7,00
4,81
1,73
50,0
24,0
9,0
6,70
67,0
катализатора
при
увеличенной
производительности
979
1,44
498
639
0,15
1,12
1,96
6,30
2,84
6,61
4,61
1,74
49,1
24,0
10,1
6,45
65,9
Таблица 64
Разделение сырья по установкам
ПЛОТНОСТЬ (І| 5'5
Средняя температура
кипения, °С
Выкипает при
температуре >510°С, %
Эквивалент железа,
Ю-4 вес. %
Расход свежего
катализатора, кг/м3 сырья
Температура в реакторе,
Равновесная активность
катализатора по
методу Атлантик Д + П
Фактор коксообразования
Содержание металлов на
равновесном
катализаторе 10~4-вес. %
железо
никель
ванадий
медь
Основной
вариант
0,8914
374
7
2,2
1,94
488
43
1,9
2900
65
360
75
Установка
А
0,8751
359
0
1,3
1,34
484
47
1,3
3800
55
285
40
Д
-0,0193
-15
— 7
-0,9
— 0,60
—4
+4
-0,6
—
—
Основной
вариант
0,9036
379
9
2,2
1,91
488
48
1,7
1600
85
490
25
Установка Б
0,91
401
13
2,8
1,45
492
41
1,5
1400
75
405
20
А
+0,0064
+22
+4
+0,6
-0,46
+ 4
—7
—0,2
—
—
сырье с высоким содержанием металлов. Эффективность
применения процесса Мет-х зависит от специфики завода и конкретных
производственных условий.
Ниже приводится один из вариантов использования процесса
Мет-х. При наличии в схеме завода двух установок
каталитического крекинга предлагается одну из них оборудовать установкой
Мет-х [370]. На установку А подают более легкое сырье с
небольшим содержанием металлов, а на установку Б, дополненную
установкой Мет-х, направляют тяжелое сырье с высоким содержанием
металлов. Работа обеих установок при таком разделении сырья
сравнивается с результатами их работы без Мет-х на смешанном
сырье (табл. 64). Уменьшение равновесной активности
катализатора на установке Б при разделении сырья авторы объясняют
спеканием катализатора,'вызванным большим количеством
возвращаемого в реактор шлама.
В результате разделения сырья расход свежего катализатора
на обеих установках уменьшился (на установке А — за счет
облегчения сырья, а на установке Б — за счет эффективной работы
Мет-х). Результаты каталитического крекинга двух установок —
с разделением сырья и без него — представлены в табл. 65.
236
Таблица 65
Результаты каталитического крекинга при разделении сырья
Производительность по
свежему сырью, м'/сут.
Степень превращения свежего
сырья
объемн. %
м3
Кокс, вес. %
Отношение Нг:СНі
Сз, м»/сут.
Избыточные бутаны, м3/сут.
Бензин (давление насыщенных
паров по Рейду 534 мм рт.
ст.), м3/сут.
Печное топливо № 2, м3/сут.
Расход свежего катализатора,
т/сут.
Выход водорода (при
нормальных условиях), m3/ms
превращенного сырья
Р
St
3080
57,1
1760
9,4
0,80
261,0
2830
1310
1000
6,6
16,3
¦ановка
г-
и
X
3790
56,4
2140
7,9
0,50
313,0
370,0
1610
1370
5,6
11,1
д
+ 710
-0,7
+38Q
+ 52
+87
+300
+ 370
-1,0
—
ювной
нант
oS
4820
65,7
3290
10,2
0,82
417
600,0
2280
1270
10,3
19,5
гановка
и
4760
62,8
3020
9,7
0,63
388
490
2230
1410
7,8
23,0
д
-60
-2,9
—270
—
—
-29,0
-ПО
-50
+ 140
-2,5
—
Общее
Д
+ 650
-2,3
+ 110
—
+ 23
—23
+ 250
+ 510
— 3,5
—
Разделение сырья позволило увеличить общую
производительность на двух установках. Несмотря на низкую степень
конверсии на установке Б, общее превращение сырья и выход бензина
на обеих установках увеличились.
В работе [370] указывается, что при переработке на
установке Б еще более тяжелого сырья общая производительность двух
установок возрастает на 1140 м3/сут. при несколько меньшей
коксовой нагрузке регенератора. Выход бензина увеличился на
410 м3/сут. Улучшилась при таком варианте работы и
селективность катализатора, существенно уменьшился расход
катализатора (на 3,5 т/сут.).
Предлагаются и другие варианты использования установки
Мет-х [263]. В частности, указывается на возможность при
наличии ее в схеме завода перерабатывать на установках
каталитического крекинга деасфальтизат. Сочетание деасфальтизации и
Мет-х позволит существенно углубить отбор деасфальтизата — с
28 до 60 объемн. % (на вакуумный остаток).
Капиталовложения в первую промышленную установку Мет-х
производительностью 40 т/сут составляют 475 тыс. долл., а
эксплуатационные расходы 573 долл./сут, или стоимость очистки 1 т
катализатора равна 14 долл. [367]. Окупаемость установки Мет-х
меньше одного года.
237
Таблица 66
Результаты работы установок каталитического крекинга
при включении в схему установки Мет-х
Произоодительность Мет-х, т/сут.
Свежее сырье, поступающее на
установку каталитического крекинга
Производительность установки, мэ/сут.
Содержание металлов (Fe, Ni, V),
Ю-4-вес %
Расход свежего катализатора, т/сут.
Степень превращения, объемн. %
Коэффициент рециркуляции
Коксовая нагрузка, кг/ч
Получаемые продукты, мэ/сут.
сжиженный газ С3
избыточные бутаны
бензин (давление насыщенных
паров по Рейду 534 мм рт. ст.)
печное топливо
тяжелый газойль
Стоимость полученных продуктов,
долл/сут.
Эксплуатационные расходы*
установки Мет-х, долл/сут.
Эксплуатационные расходы деасфаль-
тизации, долл/сут.
Прибыль
долл/сут.
долл/год**
Капиталовложения
в установку Мет-х
в деасфальтизацию
Итого
Окупаемость, годы*
Основной
вариант 1
Вариант 2
8
Вакуумный газойль
4850
0,9
—
70
1,6
—
—
—
—
—
—
—
—
—
—
—
—
—
4850
0,9
4,6
73
1,6
17200
2,98
4,15
23,7
—23,0
0
1800
313
—
1487
490000
285000
—
285000
0,58
Вариант 3
40
Вакуумный
газойль+
деасфальтизат
6600
4,8
—
62
1,2
42,0
21,5
115
151
-262,0
2360
573
3710
18777
6200000
475000
3450000
3925000
0,63
* Включая плату за пользование патентом и из расчета 330 рабочих дней в году.
•• Из расчета 330 рабочих дней в году.
В литературе [371] рассматривается много различных
вариантов использования установок Мет-х производительностью от 5 до
80 т/сут, и они сопоставляются по технико-экономическим
показателям с обычным каталитическим крекингом. Во всех случаях
экономическая целесообразность применения Мет-х очевидна, и срок
окупаемости этих установок небольшой. ¦
В табл. 66 приводятся результаты работы типичных установок
каталитического крекинга производительностью 15 800 м3/сут. при
включении в схему установки Мет-х [368]. Рассматриваются три
варианта работы установки каталитического крекинга: 1) основ-
238
ной вариант — без очистки катализатора; 2) в схему включена
установка Мет-х производительностью 8 т/сут., что позволило
увеличить степень превращения сырья при той же коксовой
нагрузке на 3,0%; 3) в схему включены установка деасфальтизации и
установка Мет-х производительностью 40 т/сут., что позволило
подвергать каталитическому крекингу смесь вакуумного газойля и
деасфальтизата с высоким содержанием металлов. Оба участка
Мет-х окупаются сравнительно быстро.
ПРОЦЕСС ДЕМЕТ
В процессе Демет металлы удаляют с катализатора в псевдо-
ожиженном слое. Катализатор подвергают химической обработке
с целью перевода соединений металла в водорастворимые и
легколетучие формы. Процесс состоит из четырех стадий: две из них —
предварительная обработка, обеспечивающая концентрацию
металлов на поверхности катализатора и превращение их в
соединения, которым трудно диффундировать обратно в матрицу,
третья — химическая обработка с целью перевода металлов в
легколетучие и легкорастворимые в воде соединения, четвертая
стадия — промывка водой. Благодаря предварительной обработке,
повышающей концентрацию металлов на поверхности
катализатора, степень удаления их на третьей стадии существенно
увеличивается [372]. Так, ванадий из катализатора можно удалить на
40—50%. Однако чтобы не вызвать изменений в структуре
катализатора, ванадий удаляют на 25—30%. Без предварительной
обработки катализатора никель можно удалить всего на 6%, а с
предварительной обработкой — до 95%. В производственных условиях
никель удаляют на 65—70%.
Первая стадия имеет целью перевод соединений ванадия,
содержащихся в катализаторе, в пятиокись ванадия и
концентрирование последней на поверхности гранул катализатора. Для этого
катализатор обрабатывают горячим воздухом, в результате чего
соединения ванадия окисляются до пятиокиси ванадия, которая
обладает летучестью и при высоких температурах в основном
сосредотачивается на доступной внешней поверхности гранул.
Влияние температуры и длительности окисления воздухом на
количество ванадия, отложившегося на внешней поверхности
катализатора, показано на рис. 96 [373]. Как это видно, равновесие между
содержанием ванадия на поверхности катализатора и в матрице
устанавливается через 4 ч. Пятиокись ванадия можно удалить с
поверхности катализатора промывкой его разбавленным водным
раствором аммония или оставить и удалить в последующих
стадиях вместе с другими металлами.
На второй стадии в результате сульфирования при 540—700 °С
никель и железо концентрируются на поверхности. Зависимость
концентрации никеля на поверхности катализатора от
температуры и длительности сульфирования показана на рис. 97. Поверх-
23»
ность, покрытая никелем, увеличивается, если условия обработки
становятся более жесткими. Сульфирование оказывает
незначительное влияние на концентрирование ванадия на поверхности,
хотя оно может способствовать получению окислов низшей
валентности и превращать их в сульфиды или оксисульфиды.
На третьей стадии металлы, содержащиеся на поверхности,
превращаются в легколетучую или легкорастворимую в воде
форму в результате одной из нескольких операций: обработка
подкисленной водой, хлорирование хлором или хлором, содержащим
промоторы (четыреххлористый углерод, хлорид серы). Сульфиды
металлов хлорируются легко, особенно в присутствии промоторов.
20
531"С
о і г з и 5 в
Длительность, ч
Рис. 96. Зависимость количества
ванадия, отложенного на поверхности
катализатора, от температуры и
длительности окисления воздухом.
/ 2 3
Длительность , ч
Рис. 97. Зависимость количества
никеля, отложившегося на поверхности
катализатора, от температуры и
длительности сульфирования.
При этом получаются летучие хлориды ванадия и железа.
Нелетучий хлорид никеля легко удаляется водной промывкой. При
хлорировании только хлором летучих хлоридов получить не удалось.
Хлориды металлов удаляются водной промывкой хлорированного
катализатора. Для увеличения количества удаляемого водой
ванадия и железа в промывную воду добавляют хелатный агент.
Эффективность хелатного агента может быть повышена добавлением
лимонной, щавелевой и винной кислот [373, 374].
По мнению авторов, процесс хлорирования характеризуется
следующими реакциями:
А1(ОН)Э + 2СС14 *¦ А1ОС1 -f 2СОС12 -)- ЗНС1
А1(ОН)Э + 2СОС12 >- А1ОС1 + 2СО2 + ЗНС1
2А1(ОН)Э -f S2C12 4- ЗС12 v 2А1ОС1 4- 2SO2 4- 6НС1
2NiS 4- ЗС12 >- 2NiCl2 4- S2C12
2FeS 4- CC14 4- Cl2 v 2FeCl3 4- CS2
2FeS 4- 4C12 > 2FeCl3 4- S2C12
При наличии на катализаторе только одного из металлов
процесс значительно упрощается. В случае необходимости удаления
240
только никеля катализатор сульфируют, затем контактируют с
воздухом в подкисленном растворе и промывают. В случае ванадия
катализатор прокаливают на воздухе и промывают водным
раствором аммиака, который реагирует с пятиокисью ванадия с
образованием ванадата аммония:
2NH4OH + V2O5
2NH4VO3 + Н2О
Различные стадии процесса Демет освещены в некоторых
патентах [375—383], посвященных реактивации катализатора
различными газами с целью перевода металлов в легколетучие или
растворимые в воде соединения.
Изменение качества равновесного катализатора, обработанного
на лабораторной установке деметаллизации, показано в табл. 67.
Таблица 67
Изменение качества катализатора в результате
деметаллизации
Показатели
Содержание металлов, 10~4вес. %
никель
ванадий
железо
Активность по методу Д+П
Относительная активность
Газовый фактор
Коксовый фактор
Относительная плотность газа
Содержание окиси алюминия, вес. %
Удельная поверхность, м2/г
Водородный фактор
Лабораторные
данные
без
процесса
Демет і
327
4240
2700
33,5
35,5
1,56
1,18
1,09
—
138
с
процессом
Демет
114
3256
1970
37,2
43,2
1,26
0,97
1,32
—
—
58
Промышленные
данные
без
процесса
Демет 5»
260
3950
2460
32,5
33,6
1,45
1,17
1,15
26,8
136
-~
с
процессом
Демет
74
1629
2010
37,7
44,2
1,07
0,87
1,31
24,8
139
Активность катализатора после деметаллизации существенно
повысилась, уменьшились коксовый, газовый и водородный
факторы.
На основе результатов работы пилотной установки
деметаллизации была построена 10-тонная установка на заводе в Вуд-Риве-
ре (США), которая обслуживает установку каталитического
крекинга производительностью 1740 м3/сут. Процесс деметаллизации
автоматизирован и идет непрерывно.
16—2206
241
Таблица 68
Экономическая оценка процесса Демет
Показатели
Очистка катализатора, т/сут.
Производительность по сырью, м3/сут.
Содержание NiO, 10~4-вес %
в сырье
на катализаторе
Удаление катализатора из системы, т/сут.
Потери катализатора с дымовыми
газами, т/сут.
Всего добавлено свежего катализатора,
т/сут.
Конверсия, объемн. %
Взято, т/сут.
нефть
н-бутан
изобутан
Получено, м3/сут.
бензин C0% премиального)
средние дистилляты
остаточное топливо
топливо для установки
другие продукты (сжиженный газ,
смазочные материалы, асфальт
и др.)
Экономичность установки Демет по
сравнению с вариантом I, долл/сут.*
увеличение валового дохода
расход на катализатор, 315 долл/т
эксплуатационные затраты на
процесс Демет**
другие производства*
Общая стоимость деметаллизации
долл/сут.
долл/год
Капиталовложения на установку Демет,
долл/год
Окупаемость (включая амортизацию и
федеральные налоги), годы
* -|- — экономия, расход.
** Включая амортизацию и авторский гонорар
I
основной
вариант
2940
0,40
250
2,8
1,9
4,7
64,5
9500
245
110
4450
2920
886
772
717
—
—
—
—
—
—
—
—
Варианты с деметаллизацией
II
уменьшение
расхода
катализатора
5
2940
0,40
250
0
1,9
1,9
64,5
9500
245
100
4450
2920
886
772
717
—
+ 880
—280
—
600
210000
250000
1,2
ш
П+умень-
шение
содержания
никеля
17
2940
0,40
250
0
1,9
1,9
67,8
9500
245
НО
4570
2920
867
722
717
+ 500
+ 880
—560
+ 235
1055
370000
550000
1,5
IV
П+утяже-
ление
сырья
18
3140
0,40
250
0
1,9
1,9
61,8
9500
245
348
4760
2920
791
885
717
+ 3420
+ 880
— 576
—750
2975
1000000
550000
0,6
Капиталовложения и эксплуатационные
Демет следующие:
Производительность установки Демет, т/сут.
Капитальные затраты, долл/сут.* ....
Эксплуатационные затраты, долл./сут. . .
затраты на процесс
5
250 000
350
20
550 000
760
242
в том числе
реагенты 90 350
эксплуатационные затраты по
обслуживанию установки** 190 260
амортизация A0% в 1 год) 70 150
Стоимость очищенного катализатора, долл/т 14 38
* Включая 10% иа обслуживание цеха.
*• При наличии одного оператора в смеиу.
Стоимость очистки катализатора на 20-тонной установке
составляет 38 долл/т против 315—355 долл — стоимости 1 т свежего
катализатора. Экономическая оценка процесса Демет дается в
табл. 68 применительно к нефтеперерабатывающему заводу
производительностью 9500 м3/сут. [372]. Рассматривается четыре
варианта работы установки крекинга: основной (I) — без деметал-
лизации и три варианта (II, III и IV) с деметаллизацией.
Для того, чтобы поддерживать содержание никеля на
катализаторе на допустимом уровне B50-10~4%), при работе по
основному варианту необходимо удалять из системы 2,8 т/сут.
катализатора помимо восполнения потерь катализатора с дымовыми
газами 1,9 т/сут. Добавление пятитонной установки деметаллизации
позволяет поддерживать допустимое содержание NiO на
катализаторе при обычном расходе 1,9 т/сут. Окупаемость установки
составляет 1,2 г. Включение в схему установки Демет
производительностью 17 т/сут. позволит уменьшить расход катализатора до
нормальной величины и уменьшить содержание NiO на катализаторе
до 100¦ 10—4%. При этом несколько увеличится выход бензина.
Капиталовложения на установку окупаются в 1,5 г. Включение в
схему установки Демет производительностью 18 т/сут. с целью
уменьшения расхода катализатора позволит увеличить
производительность по сырью и получить дополнительное количество бензина.
В результате этого срок окупаемости установки уменьшится до
0,6 г. Как видно из приведенного технико-экономического
сопоставления, наиболее выгоден вариант работы на увеличенную
производительность.
СУХАЯ ДЕМЕТАЛЛИЗАЦИЯ
Для удаления металлов с поверхности катализатора предложен
процесс сухой деметаллизации [384]. Он отличается от известных
процессов Мет-х и Демет, внедренных в промышленности США,
тем, что металлы удаляются с поверхности катализатора путем
перевода их в легколетучие карбонилы. Известно, что карбонилы
никеля и железа образуются при контакте окиси углерода со
свободными металлами. Металлы на равновесном катализаторе после
регенерации находятся в виде окислов; поэтому перед обработкой
окисью углерода для перевода металла в свободное состояние нет
обходимо его восстановить. Алюминий и ванадий в этих условиях
16* 243
карбонилов не образуют [385]. Процесе состоит из двух стадий:
1) обработка катализатора восстановительным газом; 2)
образование карбонилов при обработке окисью углерода.
Принцип образования карбонилов широко применяется в
металлургической промышленности для извлечения никеля и
железа из руд. Закономерности и особенности этого процесса
достаточно хорошо изучены и изложены в литературе [386, 387]. Однако их
нельзя целиком перенести на процесс деметаллизации
катализатора. Прежде всего в этом процессе исходным материалом является
не монолитная руда, а катализатор с высокоразвитой
поверхностью, что создает более благоприятные условия для извлечения
металлов. В то же время концентрация их на катализаторе —
всего сотые или десятые доли процента, что ничтожно мало по
сравнению с содержанием извлекаемого металла в исходной руде
(свыше 50%) и поэтому вызывает соответствующие затруднения.
Кроме того, необходимо после деметаллизации сохранить на прежнем
уровне физико-химические свойства катализатора (содержание
окиси алюминия, удельную поверхность и др.), что может
наложить ограничения на режим процесса. В связи с этим
потребовалась разработка режима процесса по стадиям и изучение влияния
различных примесей на его результаты.
Подбор оптимального режима удаления никеля. Вначале
лабораторные опыты по подбору оптимального режима деметаллизации
катализатора проводили на искусственно отравленном образце,
содержащем только никель. Исследовалось влияние температуры,
длительности обработки, объемной скорости подачи реагирующих
газов и их чистоты в стадиях восстановления и образования
карбонилов. Температура разложения, исходя из литературных
данных, была принята равной 200 °С.
Влияние параметров режима восстановления (температуры и
длительности) оценивали по конечному результату обеих стадий,
т. е. по степени удаления никеля с катализатора. В связи с этим
опыты данной серии проводили при следующем постоянном
режиме стадии образования карбонилов: 75 °С, 2 ч, объемная скорость
подачи окиси углерода 200 ч~'. В опытах изучали влияние
температуры восстановления в интервале 200—700 °С при длительности
4 ч на двух образцах катализатора с содержанием никеля 0,14 и
0,64 вес. %, а также при длительности восстановления этих же
образцов от 1 до 6 ч и температурах 600 и 400 °С. При температуре
восстановления до 300 °С никель с катализатора не удаляется
(рис. 98). При более высокой температуре степень удаления
никеля резко возрастает п достигает 80—90% от его исходного
содержания. Дальнейшее повышение температуры, с точки зрения
сохранения качества катализатора, нецелесообразно.
Существенное влияние на результаты деметаллизации
оказывает и длительность восстановления. Причина, вызывающая
снижение степени удаления никеля при чрезмерно длительной обработке
катализатора водородом, пока не ясна.
244
Влияние параметров стадии образования карбонилов
определяли при постоянном режиме восстановления: 600 °С, 4 ч,
восстановительный газ — чистый водород. Влияние режима стадии
образования карбонилов, так же как и восстановления, оценивали по
конечному результату обеих стадий. Исследовали зависимость
степени удаления никеля от температуры, длительности обработки
окисью углерода и объемной скорости подачи окиси углерода.
Усредненные данные нескольких опытов с катализатором,
содержащим 0,14% никеля, посвященных влиянию температуры,
представлены на рис. 99. Из этих данных видно, что характер зависи-
100
300 500 100
Температура восстановлении, °С
Длительность , ч
б
Рис. 98. Влияние температуры (а) и длительности (б) восстановления на
степень удаления никеля.
«ости степени удаления никеля от температуры для различного
времени контакта катализатора с окисью углерода одинаков. Вна-
іале степень удаления никеля с увеличением температуры до 75 °С
зозрастает, достигает максимума, а затем уменьшается. Следова-
гельно, скорость реакции образования карбонила никеля имеет оп-
гимум по температуре. Очевидно, уменьшение глубины удаления
іикеля при температурах выше 75 °С обусловлено преобладанием
реакции распада карбонилов над реакцией их образования.
Опыты по влиянию длительности обработки окисью углерода
яа степень удаления никеля (см. рис. 99,6) проведены при 75 °С;
эбразец катализатора содержал 0,64% никеля. При различном
режиме восстановления характер этой зависимости одинаковый.
Основное количество никеля удаляется с поверхности катализатора
в первые 1—2 ч контакта с окисью углерода. При дальнейшей
обработке полученные результаты не улучшаются. Процесс
постепенно замедляется, а затем полностью прекращается, на наш
взгляд, из-за недостаточной степени восстановления окислов ме-
галла и из-за блокирования углеродом поверхности металла, еще
активного к реакции образования карбонилов. Наличие углерода
245
на поверхности катализатора объясняется распадом в условиях
деметаллизации окиси углерода на углерод и двуокись,
катализируемым тонкораспределенным никелем [385]. Содержание
углерода, определенное его сжиганием, достигало 0,1 вес. %.
Эксперименты показали, что повторное восстановление образца
катализатора после прекращения реакции карбонилобразования с
последующей обработкой окисью углерода позволяет извлечь
дополнительное количество никеля. При трех циклах деметаллизации с
катализатора удаляется 98,7% никеля от его исходного содержания.
2ч
I I \
ее
Из
во
? 60
* го
600 "С
SO 75 100
Температура, °С
! 2 3 4
Длительность, ч
б
Рис. 99. Влияние температуры (а) и длительности (б) обработки окисью
углерода на степень удаления никеля.
В каждом из трех последовательных циклов степень удаления
никеля примерно одинакова — около 80%, несмотря на большую
разницу в содержании металла в катализаторе перед каждой де-
металлизацией.
Влияние многократного восстановления катализатора на
степень удаления никеля следующее:
Циклы
1 2 3
Содержание никеля на катализаторе, вес. %
до деметаллизации 0,225 0,05 0,01
иосле деметаллизации 0,05 0,01 0,003
Степень удаления никеля, % от его
содержания
в загрузке цикла 78,0 80,0 70,0
в исходном катализаторе 78,0 95,5 98,7
Нами определялась оптимальная объемная скорость подачи
окиси углерода (в интервале от 10,0 до 1000,0 ч~') на катализатор
с исходным содержанием никеля 0,64 вес. % при температуре в
стадии образования карбонилов 75 °С и длительности 2 ч.
Эксперименты показали, что при объемной скорости подачи окиси
углерода 100 ч и выше степень удаления никеля практически не
изменяется и находится на уровне 80%. При объемной скорости
меньше, чем 100 ч, она резко уменьшается и при 10 ч
составляет всего 10%. Таким образом, в случае объемной скорости пода-
246
їй окиси углерода ниже 100 ч-1 результаты деметаллизации суще-
:твенно ухудшаются, видимо, вследствие затруднений диффузион-
юго порядка.
Следовательно, никель, несмотря на его небольшую
концентрацію, удаляется с поверхности алюмосиликатного катализатора
іримерно на 80% при сравнительно мягких условиях. После не-
жольких циклов деметаллизации можно удалить практически весь
шкель.
Нами были проведены исследования по изучению влияния при-
лесей в водороде и в окиси углерода на результаты деметаллиза-
іии. Наличие примесей объясняется условиями производства этих
•азов и технологией ведения самого процесса деметаллизации,
3 стадии восстановления в водород могут попадать азот и серово-
юрод. Необходимость изучения допустимого содержания азота
іиктуется в основном технологическими требованиями. Перед за-
юлнением аппаратуры водородом ее освобождают от воздуха про-
гувкой азотом. Поэтому водород будет содержать некоторое коли-
іество азота, и нужно оценить степень его влияния на показатели
іроцесса деметаллизации. При восстановлении алюмосиликатного
сатализатора водородом образуется сероводород, который загряз-
іяет водород. Поэтому необходимо знать влияние сероводорода на
:тепень удаления никеля.
Эксперименты показали, что наличие кислорода в реагирующих
¦азах, и в частности в водороде, недопустимо. Вследствие малого
соличества металла на катализаторе даже ничтожные примеси
сислорода препятствуют восстановлению окислов и, следователь-
ю, образованию карбонилов никеля.
Содержание в водороде сероводорода до 0,3 объемн. % практи-
іески не влияло на степень удаления никеля. При дальнейшем уве-
іичении количества сероводорода степень удаления никеля резко
уменьшалась, и при содержании сероводорода 5% составляла все-
о около 5—7,0%- По-видимому, азот, содержащийся в водороде,
іграет роль инертного разбавителя. Он не оказывает никакого
їлияния на удаление никеля до тех пор, пока содержание
водорода в газе превышает его стехиометрическое количество, необходи-
юе для восстановления никеля. Когда водорода в газе содержа-
юсь меньше этого количества E—10%), степень удаления никеля
іезко уменьшалась.
При разбавлении водорода окисью углерода результаты деме-
аллизации значительно ухудшаются. Если при использовании чис-
ого водорода степень удаления никеля составляет 80%, то при со-
.ержании в нем 50 объемн. % окиси углерода она не превышает
0%. Поскольку окись углерода является более слабым восстано-
ителем, очевидно в тех случаях, когда потребуется неглубокое
даление никеля (около 40—50%), для восстановления ее можно
рименять в смеси с водородом.
Наличие в окиси углерода двуокиси углерода и сероводорода
казывает сильное влияние на результаты процесса. При содержа-
247
ний з окиси углерода даже небольшого количества двуокиси (до
50 объемн. %) степень удаления никеля уменьшается на 15 отн. %,
а при увеличении содержания двуокиси до 50% процесс
практически прекращается. Что касается сероводорода, то малые его
количества (до 5 объемн. %) практически не влияют на деметаллиза-
цию катализатора. При увеличении содержания сероводорода в
окиси углерода степень удаления никеля резко уменьшается, и при
наличии в газе 50 объемн. % сероводорода процесс практически
прекращается. Азот и водород, присутствующие в окиси углерода,
по-видимому, играют роль инертных разбавителей. Разбавление
азотом и водородом не оказывает никакого влияния на степень
70 \
во
С"
і ю
10
—
- /
/
7
Длительность
/
/
І
і
?.
восог^а.
d
3 If
'облени я:
'/00 500 600
7~''!п~.;сгг'{,'ра восстсуг' ^пип °С
а
Рис. 100. Влияние температуры (й) и длительности (б) восстановления на
степень удаления железа.
удаления никеля до тех пор, пока объемная скорость подачи
окиси углерода более 100 ч~'-. Дальнейшее разбавление приводит к
резкому уменьшению степени удаления никеля.
Таким образом, реагирующие газы необходимо очищать от
кислорода (до полного его отсутствия), двуокиси углерода и
сероводорода. Допустимое содержание двуокиси в окиси углерода 1 —
1,5 объемн. %, допустимое содержание сероводорода в водороде
0,3 объемн. %, а в окиси углерода — 5 объемн. %. Эти требования
к чистоте газов являются вполне приемлемыми — очистка газа от
указанных примесей не составляет технических трудностей.
Подбор оптимального режима удаления железа и совместного
удаления никеля и железа. Опыты по удалению железа проводили
на крупнолабораторной установке деметаллизации первоначально
на искусственно отравленном катализаторе. Изучали влияние
температуры и длительности обработки на стадии восстановления и
образования карбонилов, а также влияние исходной концентрации
железа. При выяснении влияния температуры и длительности
восстановления постоянными поддерживали следующие параметры:
на стадии восстановления — давление водорода 0,5 МПа, объем-
248
100 150 200 25Г
Температпі/ра 6 cmaduii
Ірсзобания ^"р^онилоб."'
Рис. 101. Зависимость степени
удаления железа от
температуры в стадии образования
карбонилов.
пая скорость подачи водорода 250 ч~'; на стадии образования
карбонилов — температура 200 °С, длительность обработки окисью
углерода 2 ч при объемной скорости ее подачи 400 ч-1, давлении
5 МПа. Подбор оптимальной температуры и длительности на
стадии образования карбонилов проводили при постоянном режиме
восстановления F00°С, 4 ч), а также при сохранении
постоянными всех перечисленных выше параметров обеих стадий.
Температура разложения карбонилов железа на основании
предварительных экспериментов была принята равной 400 °С.
С увеличением температуры восстановления количество
железа, удаляемого с поверхности катализатора, существенно
возрастает (рис. 100,а). При температурах
выше 600 °С опыты не проводили,
чтобы не ухудшить качество
катализатора вследствие изменения его поровой
структуры. Значительное влияние на
степень удаления железа оказывает
длительность обработки катализатора
водородом (рис. 100,6). Так,
восстановление в течение 1 ч обеспечивает
лишь незначительную степень
удаления, при восстановлении от 2 до 4 ч
степень удаления железа вполне
приемлемая.
Характер зависимости от
температуры степени удаления железа на
стадии образования карбонилов примерно такой же, как и при
удалении никеля (рис. 101). Прохождение степени удаления железа
через максимум можно объяснить тем, что после достижения
определенной температуры скорость обратной реакции начинает
преобладать над скоростью прямой реакции. Визуальные
наблюдения за скоростью образования металлического зеркала
показали, что основное количество железа удаляется из катализатора в
течение первого часа.
На основании полученных результатов был выбран
оптимальный режим удаления железа: на стадии восстановления 550—
600 °С, объемная скорость подачи водорода 50 ч~', длительность
обработки водородом 3—4 ч, на стадии образования карбонилов
175—200 °С, длительность 2 ч, давление окиси углерода 5 МПа,
объемная скорость подачи окиси углерода 400 ч-1. Температура
разложения карбонилов железа 400 °С. В этих условиях были
проведены опыты по изучению влияния исходной концентрации
железа на степень его удаления. Деметаллизации подвергали
катализаторы с содержанием 0,25; 0,52 и 1,0 вес. % железа. Было показано,
что независимо от исходного содержания в катализаторе степень
удаления железа составляет 60—64%.
Установлено, что железо с поверхности алюмосиликатного ка-
гализатора удаляется хуже, чем никель. Однако достигнутая в
249
оптимальных режимах глубина извлечения железа — около*
60,0%— примерно в 3 раза выше, чем в процессах Мет-х и Демет.
Оптимальные режимы удаления железа и никеля отличаются в
основном повышенным давлением. Поскольку повышение давления
является благоприятным для образования карбонилов никеля,
было интересно оценить возможную глубину удаления железа и
никеля при их совместном присутствии и определить оптимальный
режим этого процесса. С этой целью катализатор, содержащий
0,60% железа и 0,22 вес. % никеля, подвергали деметаллизации.
Было установлено, что оптимальный режим совместного удаления
никеля и железа не отличается от оптимального режима удаления
одного железа и обеспечивает глубину извлечения никеля 95%,
а железа — 60%. Следовательно, при совместном присутствии
обоих металлов на катализаторе они удаляются так же легко, как
и в случае присутствия лишь одного из них.
Влияние условий деметаллизации на физико-химические
свойства катализатора. Очень важно, чтобы в процессе деметаллизации
физико-химические свойства катализатора остались на прежнем
уровне. Наши данные свидетельствуют о том, что свойства
катализатора (прочность, насыпная плотность, удельная поверхность,
обменная способность, содержание алюминия) после
деметаллизации при атмосферном и повышенном давлении остаются такими
же, как и у исходного, независимо от степени извлечения
металлов [389]. Так, при удалении около 80% никеля содержание
алюминия на катализаторах не изменяется. Прочность катализаторов
после деметаллизации также не изменилась. Аналогичные данные
получают и при деметаллизации с целью удаления железа.
Активность и селективность катализаторов после
деметаллизации существенно улучшаются: увеличивается выход бензина,
уменьшается выход кокса, газа (по объему) и содержание в нем
водорода. Как видно из рис. 102, кривые зависимости выхода
бензина и кокса от содержания никеля и железа на катализаторе до
и после деметаллизации совпадают. Иными словами, условия
деметаллизации (обработка водородом при высокой температуре с
последующей обработкой окисью углерода) не влияют на
активность и селективность катализатора. Это подтверждается такжє
результатами «холостого опыта», заключающегося в том, что
деметаллизации при атмосферном и повышенном давлении
подвергали свежий алюмосиликатный катализатор промышленного
производства. Показатели крекинга этого катализатора до и после
деметаллизации практически одинаковы.
Деметаллизация катализатора, отравленного при крекинге
нефтяного сырья. Все описанные выше эксперименты, посвященные
деметаллизации, проводили с катализаторами, на которые металлы
были нанесены пропиткой. Однако для полной оценки
эффективности процесса деметаллизации необходимо исследование
катализаторов с металлами, накопленными в условиях крекинга
нефтепродуктов. Нами был взят равновесный катализатор, отобранный.
250
промышленной установки. Поскольку содержание металлов на
:ем было невелико (никеля 0,005, железа 0,08 вес. %), оценить
тбпень удаления металлов с этого катализатора было трудно.
Потому параллельно деметаллизации подвергали катализатор, от-
іавленньїй при крекинге мазута на пилотной периодически дейст-
ующей установке. На этом катализаторе металлов содержалось
іримерно столько же, сколько может накопиться при переработке
іа установках вакуумного газойля арланской нефти (никеля 0,22,
келеза 0,18, ванадия 0,09 вес. %). Режим крекинга при отравле-
іии был выбран близким к промышленному. Режим деметаллиза-
15
?оІ
I
вес
1
1
1
UO
35
30
25
20
10
о
0,2 0,4
Содержание никеля нп каталиэатпоюе,вес.%
а
0 0,2 Ofi 0,6 0,3 1?
Содержание железа
на катализаторе , вес. %
Рис. 102. Влияние удаления никеля (а) и железа (б) на активность
катализатора:
( — выход бензина: О — до деметаллизацни; #—после деметаллизацни; 2 — выход кокса:
D — до деметаллизации; ¦ —после деметаллнэацнн.
дии на стадии восстановления был следующим: 600 °С; 3 ч; давле-
ше водорода 0,5 и 2,0 МПа, а на стадии образования карбонилов:
175 °С; 2 ч; давление окиси углерода 5 МПа, объемная скорость ее
юдачи 400 ч~'.
Кроме того, катализатор с пилотной установки деметаллизиро-
зали на обеих стадиях при атмосферном давлении. Усредненные
результаты опытов приведены в табл. 69.
Из данных табл. 69 видно, что с катализатора, отравленного
яа пилотной установке, никель при атмосферном давлении
удаляется значительно хуже, чем с искусственно отравленного
катализатора. Увеличение давления на стадии обработки окисью углерода
позволяет увеличить степень удаления никеля.
Физико-химические свойства обоих катализаторов в результате
цеметаллизации не изменились. Однако их активность повысилась
и значительно улучшилась селективность. Особенно резко выра-
251
лением
0,5
2,0
3
О О
О О
ОО
со со
оо
85
о о
о о
О СТ>
О1 О5
СП °°
to о
со to
оо
СЛ О5
ю to
to *¦
о to
Ос
1
сл 1
СО СО
о to
to со
о со
со со
СО ЬЗ
, . „_
55
to to
¦fe -q
га
о
аолвхзз
ленный под
ш
na к
нов к
рупнол
е
абораторпой
ч
о
ч
е поел
е деметалли
2
Е
F
ленной
установки
авної
лением
0,5
2,0
зесный
МП а
катализатор
о
о
о о о
оо о
^^ ^^ ^^
Сл СЛ СО
оо о
о о о
Ос -^1 Ос
опре-
ялось
же
005
ОоосОс
Ос О ОО
~ to
Ос СЛ |
о
оо
-о |
СЛ Оо '
сл
Ос tO
1 -
со 1 to
СЛ С1
to to to
Or -q ~q
to о •—
O5 СП -4
WA 4^
СП СО 4*
ю to to
4* ОО Со
о>о о
ос ет-~
юсл?
ca
о
сстапов
леггный под
а
О Щ
а
рупнол
е
абораторпой
п
?
Ч
о
ч
?
е поел
е деметалли
ш
аборат
¦а
о
St
а*
Ч
уста
от ж
новке
е поел
е деметаллн
из
ЇЇ
ИІ
О
о
о
оо
о
о
1
286,
о
о
,
-4
СО
о
to
Ос
сл
to
Ос
1
сл
сл
1
сл
крек
И НГЄ М
ззута на пил
о
ч
з:
атал
ш
ч
о
У
отравленны
—>е
о хз
о
SS0
о
Ос
О
S
Ос
к—
269,
о
о
,
4*
~q
to
Оо
о
to
to
о
О5
¦-1
^,
СЛ
со
ю
Образцы
катализаторов
никель
железо
ванаднп
вес.
=°
Соде
Г)
нне
мета
ь
о
Насыпная плотность
кг/мЗ
Удельная
поверхность м2/г
Обменная
способность общая,
мг-экв/г
Прочность, кгс не
1 частицу
Содержание
алюминия, вес. %
а
311
-1
О
и
о.
содержание
водорода
в газе, вес. %
X
п
¦X
2
Отношение
бензин
: иске
$
-)
эта
о
X
о
3
о
tr
жены эти изменения у катализатора, полученного на пилотной
установке. Такое сильное воздействие деметаллизации объясняется
высокой степенью удаления металлов. Данные табл. 69
показывают высокую эффективность процесса сухой деметаллизации.
Разработанным нами методом удаляется практически
полностью (на 95—98%) никель и значительная часть железа (около
60%) при сохранении физико-химических свойств катализатора и
при сохранении его стабильности. В результате показатели
крекинга на деметаллизировапиом катализаторе существенно
улучшаются. Глубина извлечения металлов данным методом значительно
больше, чем в процессах Мет-х и Демет. Кроме того, процесс
отличается простотой. Он имеет лишь две стадии; исходные
реактивы доступны, не предъявляются жесткие требования к их
чистоте, и они могут использоваться в процессе многократно;
технологическое оформление установки весьма простое.
Принципиальная схема установки сухой деметаллизации.
Производительность установки деметаллизации зависит от
допустимого содержания никеля на циркулирующем катализаторе. По
нашим расчетам, производительность установки деметаллизации для
условий установки 43-102 с шариковым катализатором составляет
10 т/сут, а с пылевидным катализатором на установке 1-А —
17,3 т/сут. Ниже описана технологическая схема установки
деметаллизации.
Часть регенерированного катализатора при 600—650 °С из
установки каталитического крекинга подается в реактор установки
деметаллизации. С целью удаления кислорода реактор продувают
горячим инертным газом, а затем водородом — для
восстановления окислов металлов. Предварительно водород очищают от
следов кислорода, двуокиси углерода и осушают. Восстановленный
катализатор охлаждают циркулирующим водородом. После
установления в реакторе температуры 175—200 °С в него подают окись
углерода, предварительно очищенную от следов кислорода и
влаги. Карбонилы металлов, образующиеся в реакторе, выносятся
потоком оеагирующего газа в разложитель. Здесь при 400 °С
карбонилы разлагаются на металл, который откладывается на насадке,
и окись углерода, которую возвращают в процесс.
Реактивированный алюмосиликатный катализатор пневмоподъемником
перегружается в регенератор установки.
СОПОСТАВЛЕНИЕ ТЕХНИКО-ЭКОНОМИЧЕСКИХ ДАННЫХ
ОЧИСТКИ СЫРЬЯ КРЕКИНГА И ДЕМЕТАЛЛИЗАЦИИ
КАТАЛИЗАТОРА
Как отмечалось выше, вопрос о преимуществе предварительной
подготовки сырья крекинга или деметаллизации катализатора
является дискуссионным. Поэтому нами были сопоставлены технико-
экономические данные обоих направлений борьбы с дезактивацией
алюмосиликатного катализатора применительно к условиям
переработки вакуумного газойля арланской нефти.
253
Таблица 70
Технико-экономические показатели различных вариантов работы установок
каталитического крекинга
Статьи расхода
Вакуумный газойль арланской нефти
Расход водорода на гидроочистку
вакуумного газойля
бензина
легкого газойля
Итого по водороду
Эксплуатационные расходы
на гидроочистку вакуумного газойля
на каталитический крекинг
на гидроочистку бензина
иа гидроочистку легкого газойля
Итого эксплуатационных расходов
Всего затрат
Себестоимость целевой продукции
в том числе
бензин
компонент дизельного топлива
масса,
тыс. т
920,0
—
1,1
2,6
—
—
920,0
219,1
315,3
—
—
534,4
219,1
315,3
Первый вариант
затраты
на единицу
продукции,
руб.
14—72
—
235—00
235—00
—
—
—
—
—
—
31-42
30—54
32-03
сумма,
тыс. руб.
13542,4
—
258,5
611,0
869,5
—
4812,0
358,2
881,6
6051,8
20 463,7
16 792,1
6691,9
10 100,2
Второй вариант
масса.
тыс. т
1360,0
12,2
—
—
1360,0
—
—
-
—
912,2
361,8
550,4
затраты
на единицу
продукции,
руб.
14—72
235—00
—
—
—
—
—
—
—
31-12
31-12
31-12
сумма.
тыс. руб.
20 019,2
2867,0
—
2867,0
3979,6
5948,6
—
9928,2
32814,4
28 391,5
11261,9
17 129,6
Рассматривалось два варианта: 1) каталитический крекинг
неочищенного газойля с деметаллизацией циркулирующего
катализатора и последующая гидроочистка продуктов крекинга; 2)
предварительная гидроочистка сырья каталитического крекинга.
Расчеты проводили применительно к типовому цеху,
состоящему из трех установок крекинга. Производительность установок
определяли при одинаковой коксовой нагрузке, равной 1800 кг/ч.
Из-за разного выхода кокса производительность установок в
каждом варианте будет значительно различаться.
Полученные технико-экономические показатели представлены
в табл. 70. Они показывают, что в случае предварительной очист*
ки сырья крекинга (второй вариант) эксплуатационные расходы
примерно в 1,6 раза выше, чем в первом варианте. Однако в связи
со значительным увеличением выхода целевой продукции
себестоимость ее во втором варианте на 30 коп. меньше. Следовательно,
экономически более целесообразно перерабатывать вакуумный
газойль арланской нефти с предварительной гидроочисткой.
Поскольку в первом варианте значительную часть эксплуатационных
расходов составляют расходы по гидроочистке продуктов крекинга,
в случае переработки малосернистого сырья более выгодные
экономические показатели будут при деметаллизации сырья.
По капиталовложениям деметаллизация является более
приемлемым вариантом. Если капиталовложения на 1 т
перерабатываемой продукции при предварительной очистке сырья крекинга
составляют 14,1 руб/т, то при деметаллизации катализатора
требуется лишь 12,5 руб/ т.
ЗАКЛЮЧЕНИЕ
Из описанных способов очистки катализатора от металлов —
ингибиторов процесса каталитического крекинга наибольший
интерес для реализации в промышленных условиях представляют
Мет-х, Демет и сухая деметаллизация. Их применение позволяет
существенно уменьшить содержание металлов на поверхности
катализатора и в результате этого добиться увеличения
производства, улучшения качества и удешевления целевых продуктов
каталитического крекинга.
ЛИТЕРАТУРА
1. Федоров В. С, «Нефтепереработка и нефтехимия», 1967,
№ 10—11, с. 1—6.
2. Мощности нефтеперерабатывающих заводов США, стран
Западной Европы и Японии. Под ред. Е. М. Зелькинда.
М., ЦНИИТЭнефтегаз, 1965. 67 с.
3. Huneke J. М., Hydrocarbon Process, 1966, v. 45, № 7,
p. 119—124; Oil a, Gas J., 1971, v. 69, № 12, p. 86—88.
4. Прокопюк С. Г., Масагутов Р. М. Промышленные
установки каталитического крекинга. М., «Химия», 1974, 173 с.
5. Зульфугаров 3. Г. Влияние условий синтеза крекирующих
катализаторов на их физико-химические свойства. Баку,
изд. АН Азерб. ССР, 1957. 224 с.
6. Смит В. М. В кн.: VII Мировой нефтяной конгресс в
Мексике. Каталитические процессы переработки нефти.
М, «Химия», 1971, с. 56—66.
7. Масагутов Р. М., Морозов Б. Ф., Данилова Р. А.
Отравление металлами и старение катализаторов крекинга. М.,
ЦНИИТЭнефтехим, 1968. 78 с.
8. Масагутов Р. М., Берг Г. А. Методы очистки сырья
каталитического крекинга. М., ЦНИИТЭнефтехим, 1966. 98 с.
9. Соколов В. А., Торочешников Н. С, Кальцев Н. В.
Молекулярные сита и их применение. М., «Химия», 1964.
156"с.
10. Жданов С. П., Егорова Е. Н. Химия цеолитов. Л.,
«Наука», 1968. 158 с.
11. Эйтель В. Физическая химия силикатов. М., Издатинлит,
1962. 1055 с.
12. Давидянц А. А., Первушкин Н. И. Производство
катализаторов крекинга и высокоактивных силпкагелей. М.,
«Химия», 1972. 168 с.
13. Райленд Л. Б. и др. В кн.: Катализ в нефтехимической и
нефтеперерабатывающей промышленности. Том 3. М.,
Гостоптехиздат, 1963, с. 1! —105.
14. Скурко Р. И., Почерникова К- А. Производство
синтетических катализаторов для нефтепереработки. М.,
Гостоптехиздат, 1963. 120 с.
15. Скарченко В. К. Алюмосиликатные катализаторы. Киев,
изд. АН УССР, 1963. 119 с.
16. Stormont D. N.. Oil a. Gas J., 1968, v. 66, № 14, p. 104—109.
17. Miller R. L., Chem. Eng., 1972, v. 79, № 5, p. 60—61.
18. Мирский Я- В., Митрофанов М. Г., Дорогочинский А. 3.
Новые адсорбенты — молекулярные сита. Грозный,
Чечено-ингушское кн. изд., 1964. 108 с.
256
19. Николина В. Я. и др. В кн.: Технология синтетических
минеральных наполнителей, адсорбентов и коагулянтов.
Том 21. Л., «Химия», 1970, с. 52—60.
20. Миначев X. М., Гаранин В. И., Исаков Я- И., «Успехи
химии», 1966, т. 35, № 12, с. 2151—2177.
21. Welsz Р. В., Frllette V. J., J. Phys. Chem., 1960, v. 64,
p. 382—383.
22. Топчиева К. В., Романовский Б. В., Хо-ши Тхоанг, ДАН
СССР, 1963, т. 149, № 3, с. 644—647.
23. Борескова Е. Г., Топчиева К. В., Пигузова Л. И.,
«Кинетика и катализ», 1964, т. 5, № 5, с. 903—909.
24. Борескова Е. Г., Лыгин В. И.; Топчиева К- В., «Кинетика
и катализ», 1964, т. 5, с. 1115—1118.
25. Литвиненко А. Г. и др. Производство и применение
катализаторов в нефтеперерабатывающей промышленности
за рубежом. М., ЦНИИТЭнефтехим, 1970. 92 с.
26. Ciaoetta F. G., Chim. е. ind., 1969, v. 51, № 11, p. 1173—
1183.
27. Miale J. N.. Chen N. У., Weisz Р. В., і. Catalysis, 1966,
v. 6, № 2, p. 278—287.
28. Pickert P. ?., Bolton A. P., Lanewalia M. A., Chem. Eng.,
1968, v. 75, № 16, p. 133—142.
29. Baker R. W., Maker P. K., Blazek I. J., Hydrocarbon
process., 1968, v. 47, № 2, p. 125—132.
30. Иствуд С, Сейлор Р., Шварц А. В кн.: Каталитические
процессы переработки нефти. М., «Химия», 1971, с. 67—75.
31. Эверет У. Э., Раймонд Н., «Инженер-нефтяник», 1969,
№ 5, с. 21—28.
32. Oil а. Gas J., 1969, v. 67, № 29, p. 76—77.
33. Gussow S., Higginson G. W., Schwint J. A., Oil a. Gas J.,
1972, v. 70, №25, p. 71—75.
34. Еркин В. Н. и др., «Химия и технология топлив и
масел», 1971, № 11, с. 4—7. ~ " ¦---¦¦¦
35. Мамаев О. А., Дорогочинский А. 3., «Химия и технология
топлив и масел», 1972, № 2, с. 7—9.
36. Еркин В. Н. и др., «Нефтепереработка н нефтехимия»,
1971, № 3, с. 16—19.
37. Мамаев О. А., Дорогочинский А. 3., «Химия и технология
топлив и масел», 1971, № 8, с. 7—10.
38. Дорогочинский А. 3. и др., «Химия и технология топлив
и масел», 1972, № Цг с. 10—13.
39. Курганов В. В. и др., «Нефтепереработка и нефтехимия:»,
1973, № 4, с. 1—3.
40. Аннамухамедов М. Б. и др., «Нефтепереработка и
нефтехимия», 1973, № 11, с. 1—3.
41. Schlaffer W. G., Morgan С. Z., Wilson J. N.. J. Phys. Chem.,
1957, v. 61, № 6, p. 714—722.
42. Киселев А. В., Никитин Ю. С, «Химия и технология той-
лив и масел», 1958, № 12, с. 27—32.
43. Топчиева К. В., Уч. зап. МГУ, 1955, т. 174, с. 75—79.
44. Масагутов Р. М. и др., «Химия и технология топлив и
масел», 1959, № 1, с. 69—71.
45. Смолл Н. Д. X., Керкалди П. X. С, Ньютон А. В кн.:
IV Международный нефтяной конгресс. Технология
переработки нефти и сланцев. Том. 4, М., Гостоптехиздат,
1956, с. 251—264.
46. Никитин Ю. С, ЖФХ, 1959, т. 33, № 11, с. 2513—2516.
47. Киселев А. В., Никитин Ю. С, «Кинетика и катализ»,
1963, т. 4, № 4, с. 648—651.
257
48. Масагутов Р. М., Берг Г. А., Мамаева К- И. В кн.:
Сернистые нефти и продукты их переработки. Вып. 4, ЛІ,
Гостоптехиздат, 1960, с. 163—173.
49. Elkin Р. В., Shuld С. G., Roess L. С, Ind. Eng. Chem.,
1945, v. 37, № 4, p. 327—331.
50. Гольдин В. А., Щекин В. В., «Труды института нефти АН
СССР», 1956, т. 8, с. 114—119.
51. Облад А. Г., Милликен М. X., Миле Г. А. В кн.: Катализ
в органической химии. Пер. с англ. А. М. Рубинштейна.
М., Издатинлит, 1953, с. 185—241.
52. Баррет У., Санчес М., Смит Дж. В кн.: Катализ. Труды
первого международного конгресса. Пер. с англ. Под ред.
А. А. Баландина и А. М. Рубинштейна. М., Издатинлит,
1960, с. 622—630.
53. Trambouze J. e. a., Compt. rend., 1959, v. 248, p. 228—230.
54. Каманин Н. М. и др. В кн.: Химия и технология
переработки нефти и газа. Вып. 4. М., Гостоптехиздат, 1959,
с. 90—101.
55. Миессеров К. Г., «Успехи химии», 1953, т. 22, № 3,
с. 279—290.
56. Агафонов А. В., Калико М. Л., ЖОХ, 1949, т. 19, № 1,
с. 39—46.
57. Добычин Д. П., Целлинская Т. Ф., ДАН СССР, 1956,
т. 109, № 2, с. 351—353.
58. Топчиева К- В. В кн.: Гетерогенный катализ в
химической промышленности. М., Госхимиздат, 1955, с. 145.
59. Манкаш Е. К-, Молчанова С. И., Щекин В. В., «Труды
института нефти АН СССР», 1956, т. 8, с. 120—130.
60. Добычин Д. П., Целлинская Т. Ф., ЖПХ, 1959, т. 32, № 3,
с. 486—494.
61. Никитин Ю. С, «Кинетика и катализ», 1963, т. 4, № 6,
с. 898—903.
62. Боресков Г. К., Дзисько В. А., Борисова М. С, ЖФХ,
1954, т. 28, № 6, с. 1055—1066.
63. Уилер Э. В кн.: Катализ. Вопросы теории и методы
исследования. Пер. с англ. Под ред. А. А. Баландина и
А. М. Рубинштейна. М., Издатинлит, 1955, с. 479—563.
64. Mills G. А., Ind. Eng. Chem., 1950, v. 42, № 1, p. 182—187.
65. Ebel R. И., Oil a. Gas J., 1968, v. 66, № 14, p. 116—118.
66. Заитова А. Я-, Андрюшенко Т. П., Волкова Л. И., «Химия
и технология топлив и масел», 1972, № 3, с. 19—22.
67. Галимов Ж- Ф-, Дубинина Г. Г., Масагутов Р. М.
Методы анализа катализаторов нефтепереработки. М., «Химия»,
1973. 190 с.
68. Топчиева К. В., Романовский Б. В., Бизре Е. В., Вестн.
МГУ, сер. хим., 1972, т. 13, № 1, с. 11 — 14.
69. Mills G. A., Ashwill R. Е., Gresham Т. L, Hydrocarbon
Process., 1967, v. 46, № 9, p. 121—126.
70. Леонтьев Е. А., Лукьянович В. М. В кн.: Методы
исследования структуры высокодисперсных и пористых тел. М.,
Изд. АН СССР, 1958, с. 19—36.
71. Рис. Г. В кн.: Катализ. Катализаторы органических
реакций. Пер. с англ. Под ред. А. А. Баландина и А. М.
Рубинштейна. М., Издатинлит, 1955, с. 37.
72. Киселев А. В., ДАН СССР, 1954, т. 98, с. 431—434.
73. Adams С. R., Voge Н. И., J. Phys. Chem., 1957, v. 61, № 6,
p. 722—727.
74. Киселев А. В. и др., Коллоид, ж., 1958, т. 20, № 11,
с. 52—58.
258
75. Киселев А. В. и др., ЖФХ, 1956, т. 30, № 10, с. 2149—
2159.
76. Пинес Б. Я., Успехи физ. наук, 1954, т. 52, с. 501—559.
77. Пинес Б. Я., Ж. тех. физики, 1946, т. 16, с. 737—743.
78. Goldstein М. S., I. and EC, Process Des. a. Develop., 1966,
v. 5, № 2, p. 189—192.
79. Топчиева К. В. и др. В кн.: Основы предвидения
каталитического действия. Том 2. М., «Наука», 1970, с. 144—151.
80. Oil а. Gas J., 1961, v. 59, № 44, p. 143—144; 1962, v. 60,
№ 12, p. 143—144; № 24, p. 149; 1963, v. 61, № 24, p. 88.
81. Курганов В. M., Караваев И. М., Гонсалес А., «Химия и
технология топлив и масел», 1965, № 5, с. 9—12.
82. Каиров И. Т., Пригорнев И. Г. Опыт работы установок
каталитического крекинга. М., ГОСИНТИ, 1957. 66 с.
83. Агафонов А. В., Алиев В. С. В кн.: Переработка
нефтяных остатков. М„ ГОСИНТИ, 1957, с. 120—152.
84. Масагутов Р. М. и др. В кн.: Методы исследования
катализаторов и каталитических реакций. Том 2.
Новосибирск, СО АН СССР, 1965, с. 202—213.
85. Морозов Б. Ф., Масагутов Р. М., Дубинина Г. Г. В кн.:
Методы исследования катализатора и каталитических
реакций. Том 2. Новосибирск. СО АН СССР, 1965, с. 284—
298.
86. Ефимова С. А. и др В кн.: Труды АэНИИ НП. Вып. 2,
Баку, АзерНЕФТНЕШР, 1958, с. 86—98.
87. Whitaker А. С, Klnzer A. D., Ind. Eng. Chem., 1955, v. 47,
№ 10, p. 2153—2157.
88. Bondi A., Miller R. S., Schlaffer W. G., I and EC, Process
Des. a. Develop., 1962, v. 1, № 3, p. 196—203.
89. Жаров Ю. М. и др., ЖФХ, 1966, т. 40, № 3, с. 537—542.
90. Sampath В. S., Huges R., Process Technol. Int., 1973, v. 18,
№ 1—2, p. 39—43; «Химия и переработка нефти», 1973,
№ 26, с. 25—31.
91. Massoth F. ?., Menon P. G., I and EC, Fundamentals,
1969, v. 8, № 3, p. 383—385.
92. Панченков Г. М., Голованов Н. В., Изв. АН СССР, ОТН,
1951, № 10, с. 1513—1521; 1952, № 3, с. 384—394; № 7,
с. 1031—1036.
93. Адельсон С. В., Заитова А. Я- В кн.: Сернистые нефти и
продукты их переработки. Вып. 6. М., Гостоптехиздат,
1963, с. 49—62.
94. Панченков Г. М., Ян Гуан-Хуа. В кн.: Кинетика и
катализ. М., Изд. АН СССР, 1960, с. 264—269.
95. Панченков Г. М., Ян Гуан-Хуа, Сяо Жин-джо. В кн.:
Кинетика и катализ. М., Изд. АН СССР, 1960, с. 255—
263.
96. Орочко Д. И. Теоретические основы ведения синтезов
жидких топлив. М.—Л., Гостоптехиздат, 1951. 460 с.
97. Масагутов Р. М., Морозов Б. Ф. В кн.: Высокосернистые
нефти и проблемы их переработки. Вып. 8. М., «Химия»,
1968, с. 104—109.
98. Ребиндер П. А., Щукин Е. Д., Марголис Л. Я- В кн.:
Научные основы подбора и производства катализаторов.
Новосибирск, СО АН СССР, 1964, с. 21—27.
99. Масагутов Р. М. В кн.: Сернистые нефти и продукты их
переработки. Вып. 3. М., Гостоптехиздат, 1960, с. 158—165.
100. Эйгенсон А. С. и др. В кн.: Переработка сернистых неф-
тей. Вып. 1. М., Гостоптехиздат, 1959, с. 145—155.
101. McEvoy I. E., Milliken T N.. Mills G. А., Ind. Eng. Chem.,
1957, v. 49, № 5, p. 865—868.
25g
102. Масагутов Р. М. и др. В кн.: Высокосернистые нефти и
проблемы их переработки. Вып. 8. М., «Химия», 1968,
с. 109—114.
103. Танатаров М. С. и др., «Химия и технология тоилив и
масел», 1972, № 5, с. 15—17.
104. Бейли У. А., Сартор А. Ф. 8 кн.: Новейшие достижения
в нефтехимии и нефтепереработке. Под ред. Дж. Дж.
Мак-Кета. Пер. с англ. Под ред. И. И. Абрамова.
Т. 5—6. М., Гостоптехиздат, 1965, с. 135—162.
105. Климе.чок Б. В., Андреев Е. А., Гордеева В. А., Изв
АН СССР, ОХН, 1956, № 5, с. 525—530.
106. Voge Н. Н., Good G. М., Greensfetder В. S. In: Third World
Petroleum Congress. The Hague, 1951, Proceeding Section
4, Leiden, E. I. Brill, 1951, p. 124—137.
107. Адельсон С. В-, Масагутов Р. М., Горлова П. Н. 3 кн.:
Переработка сернистых иефтей. Вып. 1. М.,
Гостоптехиздат, 1959, с. 120—135.
108. Масагутов Р. М-, Берг Г. А. 3 кн.: Переработка
сернистых иефтей. Зып. 1. М., Гостоптехиздат, 1959, с. !36—
144.
109. Масагутов Р. М., Галинич Е. Т., Берг Г. А., «Новости
нефтяной техники», сер. Нефтепереработка, 1960, № 10,
с. 30—33.
110. Масагутов Р. М., Берг Г. А. 3 кн.: Регенерация и
транспорт катализаторов крекинга. Уфа, Башкирское кн. изд.,
1961, с. 22—32.
111. Масагутов Р. М., Берг Г. А. Авт. свид. № 127244; Бюлл.
изобр., 1960, № 7, с. 15.
112. Масагутов Р. М. и др. Авт. свид. № 184258; Изобр., пром.
образцы, товари, знаки, 1966, № 15, с. 29.
113. Oil а. Gas J., 1969, v. 67, № 7, p. 77—81.
114. Гезалов В. А., «Нефтепереработка и нефтехимия», 1963,
№ 2, с. 13—14.
115. Френкель Я- И., Журнал экспериментальной и
теоретической физики», 1964, т. 16, № 1, с. 29—38.
116. Пинес Б. Я-, Журн. ЗХО им. Д. И. Менделеева, 1960,
т. 5, № 2, с. 173—180.
117. Панченков Г. М., Казанская А. С, Печейкин В. А.,
ЖФХ, 1960, т. 34, № 10, с. 2217—2222.
118. Пат. США 3487026 A969).
119. Курганов В. AI, Гонсалес А., «Нефтепереработка и
нефтехимия», 1964, № 9, с. 12—15.
120. Btanding F. Н., Ind. Eng. Chem., 1953, v. 45, № 6, p. 1186—
1197.
121. Voorhies A., Ind. Eng. Chem., 1945, v. 37, № 4, p. 318—322.
122. Боресков Г. К- 3 кн.: Проблемы кинетики и катализа.
Гетерогенный катализ. Зып. 6, М.—Л., изд. АН СССР,
1949, с. 404—425.
123. Агапов Г. И., Левинтер М. Е. 3 кн.: Нефтехимия,
нефтехимические процессы и нефтепереработка. Зып. 51. М.г
«Химия», 1964, с. 126—128.
124. Фрост А. В. Труды по кинетике и катализу. М, Академ-
издат, 1956. 538 с.
125. Thomas С. L., J. Am. Chem. Soc, 1944, v. 66, № 9, p. 1586—
1589.
126. Greensfelder В. С, Voge Н. Н., Good G. М., Ind. Eng.
Chem., 1945, v. 37, № 12, p. 1168—1176.
127. Appteby W. G., Gibson J. W., Good G. M., I and EC,
Process. Des a. Develop., 1962, v. 1, № 2, p. 102—110.
260
128. Hofman. L, Szalajko U., Chem. Stosow., 1973, v. 17, № 2„
p. 187—193; «Химия и переработка нефти и газа», 1973,.
№ 48, с. 12—16.
129. Oil а. Gas J., 1970, v. 68, № 9, p. 61—67.
130. Облад А. Г., Милликен Т. X., Миллс Г. А. В кн.: Химиям
углеводородов нефти. Под ред. Б. Т. Брукса и др. Пер..
с англ. Под ред. А. И. Богомолова. Том 2. Л., Гостоптех-
издат, 1958, с. 140—161.
131. Роде Т. В., Баландин А. А., ЖОХ, 1958, т. 28, № п^
с 2909 2915
132. Hall I. W., Rase H.F., I and EC, Process. Des. a. Develop.,
1963, v. 2, № 1, p. 25—30.
133. Добычин Д. П., Качур Л. А., ЖПХ, 1960, т. 33, № 7,
с 1514—1519.
134. Абаева Б. Т., Арамян Е. С. В кн.: Методы исследования
нефтей и нефтепродуктов. М., Гостоитехиздат, 1955,-
с. 264—275.
135. Hagerbaumer, Rassel Lee, Petrol. Refiner, 1947, v. 26, № 6,.
p. 123—128; 1949, v. 28, № 5, p. 102—107.
136. Dart J. C, Savage R. Т., Kirkbride С G., Chem. Eng. Progr.,
1949, v. 45, p. 102—110.
137. Hindin S. G., Lee J. K-, Weller S. W., Anal. Chem., 1957,.
v. 29, № 12, p. 1850—1852.
138. Oil a. Gas J., 1972, v. 70, № 14, p. 63—68.
139. Oil a. Gas J., 1971, v. 69, № 14, p. 68—76.
140. Wollastol E. G., Forsythe W. L, Vasalos I. A., Oil a. Gas J.,.
1971, v. 69, № 31, p. 64—69.
141. Манишлин В. В. и др., «Нефтепереработка и
нефтехимия», 1973, № 9, с. 54—58.
142. Воротников А. Г., Маншилин В. В., «Нефтепереработка
и нефтехимия», 1973, № 12, с. 14—16.
143. Nace D. М., I and EC, Process Des. a. Develop., 1970, v. 9,
№ 2, p. 203—209.
144. Потеряхин В. А. и др., «Химия и технология топлив и
масел», 1972, № 7, с. 12—13.
145. Мелик-Ахназаров Т. X. и др., «Химия и технология
топлив и масел», 1970, № 9, с. 17—21.
146. Панченков Г. М-, Лебедев В. П. Химическая кинетика к
катализ, М., изд. МГУ, 1961. 551 с.
147. Топчиева К. В., Антипина Т. В., Ли Хэ-Суянь, «Кинетика
и катализ», 1960, т. 1, № 3, с. 471—477.
148. Панченков Г. М., Жоров Ю. М. В кн.: Кинетика, катализ-
и нефтехимия. Вып. 37. М., Гостоптехиздат, 1962, с. 3—23.
149. Масагутов Р. М. и др., «Нефтепереработка и
нефтехимия», 1966, № 11, с. 5—10.
150. Антонюк Г. П. и др. В кн.: Нефтепереработка и
нефтехимия. Вып. 3. Киев, «Наукова думка», 1968, с. 228—236.
151. Масагутов Р. М., Прокопюк С. Г. В кн.: Ароматические-
углеводороды из продуктов нефтепереработки и синтезы-
на их основе. Вып. 1. М., «Химия», 1969, с. 48—54.
152. Масагутов Р. М. и др., «Нефтепереработка и иефтехи-
мия», 1968, № 9, с. 7—9.
153. Murphy ]. R., Oil a. Gas J., 1970, v. 68, № 47, p. 72—78.
154. Gustafson W. R., I and EC, Process Des. a. Develop., 1972_
v. 11, JsTs 4, p. 507—512.
155. Pachovsky R. A., Wojciechowski B. M., Can. J. Chem. Eng.,.
1971, v. 49 № 3, p. 365—369.
156. Wachtel S. J. e. a., Oil a. Gas J., 1972, v. 70, № 15, p. 104—
107.
157. Пат. США 3563911 A968).
261
158. Oil a. Gas J., 1973, v. 71, № 8, p. 80—85.
159. Oil a. Gas J., 1971, v. 69, № 12, p. 76—80.
160. Hydrocarbon Process, 1970, v. 49, № 14, p. 139—148.
161. Oil a. Gas J., 1973, v. 71, № 4, p. 32—33.
162. Левинтер M. E. и др. Реконструкция установки
каталитического крекинга с пылевидным катализатором типа
1-А. М., ЦНИИТЭнефтехим, 1970. 68 с.
163. Strother С. М., Vermilion W. L, Conner A. J., Oil a. Gas J.,
1972, v. 70, № 20, p. 102—110.
164. Pierce W. L. e. a., Hydrocarbon Process., 1972, v. 51, № 5,
p. 92-97.
165. «Инженер-нефтяник», 1969, № 5, с. 16—21.
166. Davidson R. C, Petrol. Refiner, 1947, v. 26, № 9, p. 79—88.
167. Conn A. L, Brackin С W., Ind. Eng. Chem. 1949, v. 41,
№ 8, p. 1717—1722.
168. Орочко Д. И., Фрост А. В., Щекин В. В. В кн.: Химия и
технология искусственного жидкого топлива н газа.
Вып. 6. М. — Л., Гостоптехиздат, 1954, с. 105—113.
169. Претор Ч. Д., Него Р. М. Катализ. Некоторые вопросы
теории и технологии органических реакций. М., Издатнн-
лит, 1959, с. 315—363.
170. Добрянский А. Ф. Геохимия нефти. М.—Л., Гостоптехнз-
дат, 1958. 320 с.
171. Гецеу В. В., Нефт. хоз., 1954, № 11, с. 68—70.
172. Лихушин К. П. В кн.: Сборник по вопросам
нефтепереработки АзНИИ. Баку —Москва, АЗОНТИ, 1938,
с. 117—135.
173. Richter F. Р. є. а., Ind. Eng. Chem., 1952, v. 44, № 11,
p. 2601—2605.
174. Lochte H. L., Littmann E. R. The Petroleum Acids and
Bases. New York, Constable a. Company Ltd., 1955. 368 p.
175 Ward L. F., Moore R. Т., Ball 1. S., Anal. Chem., 1953, v. 25,
p. 1070—1072.
176. Агафонов А. В. и др., «Новости нефтяной и газовой
техники», Сер. Нефтепереработка и нефтехимия, 1961, № 2,
с. 17—20.
177. Snyder L. R., Anal. Chem., 1969, v. 41, № 2, p. 314—323.
178. Mills G. A., Boedeker E. R., Oblad A. G., J. Am. Chem. Soc,
1950, v. 72, № 4, p. 1554—1560.
179. Облад А., Милликен Т., Миллс Г. В кн.: Катализ в
органической химии. Пер. с англ. Под ред. А. М.
Рубинштейна. М., Издатннлит, 1953, с. 185—241.
180. Shalt I. W., Dart J. С. In: Third World Petroleum Congress.
The Hague, 1951, Proceeding, Section 4. Leiden, E. I. Brill,
1951, p. 106—123.
181. Stenzet R. W., World Petrol., 1957, v. 28, № 2, p. 78—81.
182. Viland С. К-, Petrol. Refiner, 1958, v. 37, № 3, p. 197—200.
183. Масагутов P. M. и др., «Новости нефтяной техники», сер.
Нефтепереработка, 1958, № 10, с. 24—26.
184. Щекин В. В., Морозова О. Е., Антонова А. И., «Труды
института нефти АН СССР», 1956, т. 8, с. 100—113.
185 Voltz S. Е. е. a., I and EC, Process Des. a. Develop., 1972,
v. 11, № 2, p. 261—265.
186 Hydrocarbon Process., 1968, v. 47, № 3, p. 113—128.
187. Oil a. Gas J., 1969, v. 67, № 6, p. 86—91.
188. Гуляева А. А. В кн.: Труды института нефти АН СССР.
М., изд. АН СССР, 1952, т. 2, с. 73—83; 1954; т. 3.
с. 188—206.
262
189. Агафонов А. В. и др., «Новости нефтяной и газовой
техники», сер. Нефтепереработка и нефтехимия, 1961, № 1,
с. 18—21.
190. Edib I. A. e. a., J. Chem. and Eng. Data, 1960, v. 5, № 4„
p. 550—553.
191. Абызгильдин Ю. M. и др., «Нефтепереработка и
нефтехимия», 1970, № 10, с. 1—2.
192. Dunning Н. N. е. a., J. Chem. and Eng. Data, I960, v. 5,
№ 4, p. 546—549.
193. Howe W. W. e. a., J. Chem. and Eng. Data, 1960, v. 5, № 4V
p. 106—110.
194. Радченко О. А., ДАН СССР, 1955, т. 105, № 6, с. 1285.
195. Garner F. Н. е. a., J. Inst. Petrol., 1953, v. 39, № 353,
p. 278—293.
196. Johnson P. H., Mills E. L., Benedict D. C, Ind. Eng. Chem.,
1955, v. 47, p. 1578—1585.
197. Bieter H. e. a., J. Chem. and Eng. Data, 1960, v. 5, № 4,
p. 540—546.
198. Масагутов P. M., Данилова Р. А. В кн.: Проблемы
переработки высокосернистых нефтей. М., ЦНИИТЭнефтехим,
1966, с. 106—110.
199. Gossett Е. С, Petrol. Refiner, 1960, v. 39, № 6, p. 177—180.
200. Масагутов Р. М., Берг Г. А., Данилова Р. А., «Химия и
технология топлив и масел», 1967, № 9, с. 14—17.
201. Эйгенсон А. С. и др. Авт. свид. № 189398; Изобр., пром.
образцы. Товари, знаки, 1966, № 24, с. 17—18.
202. Битепаж Ю. А., ЖОХ, 1947, т. 17, № 2, с. 199—207.
203. Loper В. М., Debautn R. М., Petrol. Eng., 1960, v. 32, № 13,
p. 122—127.
204. Grane H. R., Connor J. E., Masologites G. P., Petrol.
Refiner, 1961, v. 40, № 5, p. 168—172.
205. Агафонов А. В., Гельмс И. Э., Рабинович Э. И., «Химия
и технология топлив и масел», 1960, № 6, с. 6—12.
206. Dyffy D. F., Hart Н. М., Chem. Progr., 1952, v. 48, № 7,.
p. 344—351.
207. Бергстром Е. В. и др. В кн.: IV Международный
нефтяной конгресс. Технология переработки нефти и сланцев.
Том 4. М., Гостоптехиздат, 1956, с. 95—ПО.
208. Oil а. Gas J„ 1972, v. 70, № 15, p. 112—116, 121.
209. Гор Е. Дж. В кн.: IV Международный нефтяной конгресс-
Технология переработки иефти и сланцев. Том 4. М.,
Гостоптехиздат, 1956, с. 139—150.
210. Суханов В. П. Каталитические процессы в
нефтепереработке. Изд. 2-е. М., «Химия», 1973. 413 с.
211. Томас Ч. Промышленные каталитические процессы и
эффективные катализаторы. Пер. с англ. Под ред. А. М.
Рубинштейна. М., «Мир», 1973. 385 с.
212. Морозов Б. Ф. и др., «Нефтепереработка и нефтехимия»,
1972, № 8, с. 3—5.
213. Connor J. Е. е. а., Ind. Eng. Chem., 1957, v. 49, № 2,
p. 276—282.
214. Kraus J. A. e. a., Chem. Eng. Progr., 1961, v. 57, № 12,.
p. 37^3.
215. Donaldson R. E., Rice Т., Murphy J. R., Ind. Eng. Chem.r
1961, v. 53, № 9, p. 721—726.
216. Nelson W. L., Oil a. Gas J., 1961, v. 59, № 10, p. 143.
217. Межлумова А. И., «Новости нефтяной и газовой
техники». Сер. Нефтепереработка и нефтехимия, 1961, № 10,
с. 10—12.
263
218. Russu R. e. a., Rev. chim., 1968, v. 19, № 3, p. 157—159.
219. Oil a. Gas J., 1967, v. 65, № 4, p. 66—73.
220. Танатаров М. А. и др., Изв. вузов. Нефть и газ, 1968,
№ 6, с. 57—60.
221. Bailey W. А., Nager M. In: Seventh World Petroleum
Congress. Mexico, 1967, Section 4. London, Elsevier Publ. Corp.,
Ltd., 1967, p. 185— І92.
227. Исаков Я. И., Миначев X. Л1, «Нефтехимия», 1970, т. 10,
№ 6, с. 805—812.
223. Миначев X. М., Исаков Я- И. Приготовление, активация
и регенерация цеолитных катализаторов. М., ЦНИИТЭ-
нефтехим, 1971, 83 с.
224. Weisz Р. В., Miale 1. N., J. Catalysis, 1965, v. 4, № 4,
p. 527—529.
225. Миначев X. M. и др. Редкие земли в катализе. М.,
«Наука», 1972. 262 с.
226. Адельсон С. В., Заитова А. Я. Сернистые нефти и
продукты их переработки. Вып. 3. М., Гостоптехиздат, 1960,
с. 171 — 180.
227. Масагутов Р. М. и др. Авт. свид. № 179867; Изобр., пром.
образцы. Товари, знаки, 1966, № 6, с. 56.
228. Massagutov R. М. е. a. In: Book of Synopses of the 7th
World Petroleum Congress. Mexico, 1966, p. 133.
229. Сартаева A. H., Бувалкина Л. А., Сокольский Д. В.
В кн.: Химия и химическая технология. Т. 2. Алма-Ата,
Министерство высшего и среднего специального
образования КазССР, «Наука», 1964, с. 217—220.
230. Паушкин Я- М., Адельсон С. В., Апостолаке И. В кн.:
Мономеры и полупродукты нефтехимического синтеза.
Вып. 72. М, «Химия», 1967, с. 14—19.
231. Топчиева К. В. и др. В кн.: V Международный нефтяной
конгресс. Переработка нефти и газа. Нефтехимия. Том 3.
М., Гостоптехиздат, 1961, с. 61—83.
232. Калико М. А., Первушина М. Н. Научные основы
подбора и производства катализаторов. Новосибирск, СО АН
СССР, 1962, с. 345—355.
233. Топчиева К В., Московская И. Ф., ДАН СССР, 1955,
т. 101, № 3, с. 517—520.
234. Адельсон С. В., Глаговещан Я-, Паушкин Я- М. В кн.:
Мономеры и полупродукты нефтехимического синтеза.
Вып. 72. М., «Химия», 1967, с. 19—22.
235. Адельсон С. В., Заитова. А. Я., «Химия и технология топ-
лив и масел», 1962, № 1, с. 25—31.
236. Добычин Д. П., Клибанова В. М., ЖФХ, 1959, т. 33, № 5,
с. 1023—1029.
237. Панченков Г. М., Лазьян Ю. И., Жоров Ю. М. В кн.:
Математическое описание и оптимизация процессов
переработки нефти и нефтехимш:. Вып. 74. М., «Химия», 1967,
с. 77—88.
238. Сокольский Д. В., Бувалкина Л. А., Вестн. АН КазССР,
1964, т. 20, № 2, с. 3—14.
239. Fischer К. А., Brandes G., "Erdol und Kohle", 1956, Bd. 9,
№ 2, S. 81—86.
240. Хенсфорд Р. В кн.: Катализ. Катализаторы органических
реакций. Пер. с англ. Под ред. А. А. Баландина и
А. М. Рубинштейна. М., Издатинлит, 1955, с. 5—36.
241. Бувалкина Л. А., Сокольский Д. В. В кн.: Катализ и
методы получения катализаторов. Том 17, Алма-Ата,
«Наука», 1967, с. 120—130.
264
242. Anderson R. В., Stein К. С, Feinen I. I., Ind. Eng. Chem.„
1961, v. 53, № 10, p. 809—812.
243. Patal S., J. Appl. Chem., 1952, v. 2, p. 306—310.
244. Франк-Каменецкий Д. А. Диффузия и теплоотдача в
химической кинетике. М., «Наука», 1967. 490 с.
245. Sato Я., Akamaisu П., "Fuel", 1954, v. 33, p. 195—202.
246. Климєнок Б. В., Шехтер А. Б., ДАН СССР, 1952, т. 83,
№ 1, с. 109—110.
247. Rothrock J. J., Blrkhimer E. R., Leum L. Л'., Ind. Eng. Chem.,
1957, v. 49, № 2, p. 272—276.
248. Баллад А. П. и др., ДАН СССР, 1951, т. 78, № 3,
с. 509—512.
249. Рогинский С. 3. 3 кн.: Гетерогенный катализ в
химической промышленности. М., Гостоптехиздат, 1955, с. 29—71..
250. Долгов Б. Н. Катализ в органической химии. Л., Гос-
химнздат, 1959. 807 с.
251. Бувалкіша Л. А., Либенсон Е. X., Сокольский Д. В., Уч.
зап. Казахского университета им. С. М. Кирова, сер. хим.,.
1956, т. 22, вып. 21, с. 57—65.
252. Гринсфельдер Б. С. В кн.: Химия углеводородов нефти.
Под ред. Б. Т. Брукса и др. Пер. с англ. Под ред.
А. И. Богомолова. Том 2. Л., Гостоптехиздат, 1958,
с. 114—139.
253. Pansing W. f., J. Phys. Chem., 1965, v. 69, № 2, p. 392—399.
254. Меликзаде M. M., Mycaee M. P., Азерб. хим. ж., I960,.
№ 4, с. 17—20.
255. Рогинский С. 3. Катализ и адсорбция на неоднородных
поверхностях. М., изд. АН СССР, 1949. 830 с.
256. Филиппов Ю. В., Лебедев В. П., ЖФХ, 1966, т. 40, № 8,
с. 1846—1853.
257. Реми Г. Курс неорганической химии. Пер. с нем. Под ред.
А. В. Новоселовой. Том 1. М., Издатинлит, 1963. 920 с.
258. Крылов О. В. Катализ неметаллами. Л., «Химия», 1967.
240 с.
259. Кондаков Д. Я., Креймер М. Л., Мамаева К- Г. В кн.:
Совершеиствование технологии и контроля производства
на Ново-Уфимском НПЗ. М., ГОСИНТИ, 1961, с. 36—42.
260. Hood R. L, Oil a. Gas J., 1960, v. 58, № 7, p. 107, 110, 112,
115.
261 Medlin W V e a Petrol. Refiner, 1958, v. 37, № 5, p. 167—
172.
262 Lawson J. ?., Peet J. P., Peet N. P., Petrol. Refiner, 1959,.
v. 38. № з, p. 177—180.
263. Atteridg P. Т., Oil a. Gas J., 1963, v. 61, № 49, p. 72—77.
264. Dickinson R. R. e. a.. Oil a. Gas J., 1960, v. 58, № 8, p. 120—
124, 126.
265. Gartner R. Т., Oil a. Gas J., 1953, v. 51, № 46, p. 157—158,
160, 161, 172.
266. Tears С F., Oil a. Gas. J., 1953, v. 52, № 6, p. 119, 121, 122,
129.
267. Мещерякова Я. Г., Казанский В. Л., Королева К. И.г
«Новости нефтяной и газовой техники», сер.
Нефтепереработка и нефтехимия, 1962, № 3, с. 5—6.
268. Пат США 2687985 A954).
269. Пат. США 2911353 A959).
270. Пат. США 29510305 A960).
271. Пат. США 2865838 A958).
272. Пат. США 2944002 A960).
273. Пат. США 2943049 A960).
274. Пат. США 2925379 A960).
26S-
275. Пат. США 2962435 A960).
-276. Митрофанов М. Г., Бондаренко Н. И., Кожерников Т. С,
«Новости нефтяной и газовой техники», сер.
Нефтепереработка и нефтехимия, 1962, № 8, с. 30—33.
277. Митрофанов М. Г., Березюк Ф. А. В кн.: Улучшение
качества и совершенствование производства смазочных
масел. М., Гостоптехиздат, 1963, с. 224—229.
278. Пат. США 2778777 A957).
279. Эйгенсон А. С. и др. В кн.: Сернистые нефти и продукты
их переработки. Вып. 3. М., Гостоптехиздат, 1960,
с. 19—33.
280. Масагутов Р. М., Берг Г. А., Эйгенсон А. С. В кн.:
Сернистые нефти и продукты их переработки. Вып. 4. М.,
Гостоптехиздат, 1960, с. 15—30.
281. Масагутов Р. М. и др., «Новости нефтяной и газовой
техники», сер. Нефтепереработка и нефтехимия, 1961, № 4,
с. 12—14.
282. Масагутов Р. М. и др., «Новости нефтяной и газовой
техники», сер. Нефтепереработка и нефтехимия, 1961, № 1,
с. 41—42.
283. Масагутов Р. М. и др. В кн.: Сернистые нефти и
продукты их переработки. Вып. 5. М., Гостоптехиздат, 1962,
с. 88—93.
284. Масагутов Р. М. и др. В кн.: Сернистые нефти и
продукты их переработки. Вып. 5. М., Гостоптехиздат, 1962,
с. 94—98.
285. Гутыря В. С. В кн.: Труды АзНИИ НП. Вып. 4, Баку,
АзерНЕФТНЕШР, 1959, с. 42—54.
286. Samuelson G. J., Woelfin W., Petrol. Refiner, 1959, v. 38,
№ 3, p. 221—223.
287. Chem. Process., 1960, v. 6, № 8, p. 42—43.
288. Масагутов P. M. и др. Высокосернистые нефти и
проблемы их переработки. Вып. 8. М., «Химия», 1968, с. 81—84.
289. Abbott М. D., Archibald R. С, Dorn R. W., Petrol. Refiner,
1958, v. 37, № 5, p. 161 — 166; Oil a. Gas J., 1958, v. 56,
№ 20, p. 144—147, 150, 152.
290. Arey W. F., Kronenberger L, Oil a. Gas J., 1969, v. 67,
№ 20, p. 131, 134—136, 138—139.
291. Eberline С. R., Wilson R. Т., Larson L. G., Ind. Eng. Chem.,
1957, v. 49, № 4, p. 661—663.
292. Coonally H. Г., Oil a. Gas J., 1959, v. 57, № 38, p. 88—92.
293. Lewis E. H., Oil a. Gas J., 1955, v. 54, № 11, p. 95—96.
294. Service W. J., Payne R. E., Askey W. E., Oil a. Gas J., 1958,
v. 56, № 15, p. 90—98.
295. Robbers 1. A., Patterson N. L, Lane W. Т., Petrol. Refiner,
1961, v. 40, № 6, p. 147—154.
296 Hellwig L. R e. a., Hydrocarbon Process, and Petrol.
Refiner, 1963, v. 42, № 5, p. 121 — 128.
297. Oil a. Gas J., 1963, v. 61, № 47, p. 92—97.
298. Lewis S. H., Oil a. Gas J., 1955, v. 54, № 11, p. 95—96.
299. Beuther H., Flinn P. A., Petrol. Refiner, 1960, v. 39, p. 143—
148.
300. Hildebrand R. E., Haling G. P., Ondish G. F., Oil a. Gas J-,
1973, v. 71, № 50, p. 112, 117, 120, 122, 124.
301. Stormont D. H., Oil a. Gas J., 1968, v. 66, № 8, p. 40—42.
302. Nelson W. L, Oil a. Gas J., 1971, v. 69, № 38, p. 102—103.
303. Nelson W. L. Oil a. Gas J., 1973, v. 71, № 29, p. 102—103.
304. Агафонов А. В. и др., «Химия и технология топлив и
масел», 1959, № 4, с. 18—24.
266
305. Эйгенсон А. С. и др. В кн.: Сернистые нефти и продукты
их переработки. Вып. 3. М., Гостоптехиздат, I960, с. 3—19-
306. Масагутов Р. М., Берг Г. А., Волкова Л. И., «Химия и
технология топлив и масел», 1961, № 8, с. 8—13.
307. Масагутов Р. М., Берг Г. А., Волкова Л. И. В кн.: За
технический прогресс в нефтепереработке. Уфа,
Башкирское кн. изд., 1961, с. 7—17.
308. Масагутов Р. М., Берг Г. А., Волкова Л. И. В кн.:
Сернистые нефти и продукты их переработки. Вып. 5. М.,
Гостоптехиздат, 1962, с. 77—88.
309. Масагутов Р. М., Берг Г. А., Волкова Л. И. В кн.: Химия
сероорганических соединений, содержащихся и нефтях и
нефтепродуктах, Вып. 5. М., Гостоптехиздат, 1963,
с. 70—76.
310. Massagutov R. М. е. a. In: Seventh World Petroleum
Congress. Section 4. Mexico, 1967. London, Elsevier Publ. Corp.,
Ltd., 1967, p. 177—183.
311. Бугай Е. А. и др., «Химия и технология топлив и масел»,.
1970, № 10, с 1—3.
312. Варфоломеев Д. Ф. и др., «Нефтепереработка и
нефтехимия», 1973, № 1, с. 1—3.
313. Англ. пат. 698332 A953).
314. Пат. ФРГ 917140 A954).
315. Пат США 3008897 A960).
316. Пат. США 2848675 A960).
317. Пат. США 2848375 A958).
318. Пат. США 2862879 A958).
319. Пат. США 2780582 A957).
320. Пат. США 2665246 A954).
321. Petrol. Process., 1956, v. 11, № 10, p. 55—57.
322. Пат. США 2944965 A960).
323. Dunning H. N.. Moore J. W., Myers А. Т., Ind. Eng. Chem.,
1954, v. 46, № 9, p. 2000—2007
324. Англ. пат. 705267 A954).
325. Голланд. пат. 96087 A960).
346. Пат. США 2934497 A960).
327. Пат. США 2990365 A961).
328. Пат. США 2920936 A960).
329. Чертков Я. Ю., Зрелое В. Н., «Нефтяник», 1958, № З,
с 13-15.
330. Масагутов P. M. и др., ДАН УзССР, 1962, № 9, с. 49—52.
331. Масагутов Р. М. и др. Авт. свид. № 162823 A961); Бюлл.
изобр. и товарных знаков, 1964, № 11, с. 13.
332. Масагутов Р. М. и др. В кн.: Химия сераорганических
соединений, содержащихся в нефтях и нефтепродуктах.
Т. 5. М., Гостоптехиздат, 1963, с. 71—83.
333. Сафаев А. С. и др., ДАН УзССР, 1965, т. 22, № 1,
с. 48—50.
334. Масагутов Р. М. и др. В кн.: Высокосернистые нефти и
проблемы их переработки. Вып. 8, М., «Химия»., 1968,
с. 122—125.
335. Масагутов Р. М. и др. В кн.: Высокосернистые нефти и
проблемы их переработки. Вып. 8. М., «Химия», 1968,.
с. 126—133.
336. Oil а. Gas J., 1970, v. 68, № 7, p. 69—75.
337. В кн.: Обзор зарубежной литературы по
нефтепереработке. Вып. 5. М., ЦНИИТЭнефтехим, 1964, с. 9—13.
338. Пат. США 2892771 A959).
339. Curfman R. L., Hydrocarbon Process, and Petrol. Refiner,
1964, v. 43, № 9, p. 160, 162.
267
340. Beuther H., Flinn R. A., I and EC, Process Des. a. Develop..
1963, v. 2, № 1, p. 53—57.
341. Пат. США 2681305 A951).
342. Пат. США 2780585 A957).
343. Пат. США 2921018 A960).
344. Пат. США 2877323 A958).
345. Пат. США 2668798 A961).
346. Пат. США 2693455 A954).
347. Пат. США 2758097 A954).
348. Пат. США 2977332 A961).
349. Пат. США 2814598 A957).
350. Пат. США 2675187 A954).
351. Пат. США 2901419 A959).
352. Пат. США 2764557 A956).
353. Пат. США 2850464 A959).
354. Пат. США 2897136 A959).
355. Пат. США 2946740 A960).
356. Пат. США 2981676 A961).
357. Сокольский Д. В., Батталова Ш. ., Изв. АН КазССР,
сер. хим., 1956, № 10, с. 69—71.
358. Сокольский Д. В., Бувалкина Л. А., Кортунова 3. Н
Вестник АН КазССР, 1959, т. 15, № 4, с. 62—72.
359. Бувалкина Л. Л., Сокольский Д. В. В кн.: Катализ в
высшей школе. Выи. 1. Часть 2. М., Изд. МГУ, 1962,
с. 314—319.
360. Бувалкина Л. А., Бижанова М. Б., Вестник АН КазССР
1963, т. 19, № 7, с. 32—40.
361. Сокольский Д. В., Бувалкина Л. А., Носкова Н. Ф., Изв.
АН КазССР, сер. хим., 1953, № 123, выи. 7, с. 39—48.
362. Бувалкина. Л. А., Даулетов Б., Каталитический синтез
мономеров. Т. 8, Алма-Ата, АН КазССР, 1962, с. 115—127.
363. Сокольский Д. В., Бувалкина Л. А., Сартаева А. Н. В кн.:
Химия v. химическая технология. Выи. 1. Алма-Ата, изд.
АН КазССР, 1963, с. 136—144.
364. Leum L. N.. Connor J. F., I and EC, Prod. Research a.
Develop., 1962, v. 1, № 3, p. 145—149.
365. Fowle M. I. e. a., Chem. Eng. Progr., 1961, v. 57, № 12,
p. 66—69.
366. Bender R. ]., Chem. Eng., 1962, v. 69, № 15, p. 66—70.
367. Stormont D. M., Oil a. Gas J., 1961, v. 59, № 50, p. 74—77.
368. Stormont D. M., Oil a. Gas J., 1963, v. 61, № 2, p. 89—91.
369. Ozawa I. K-, Porter R. F., Humble J. R., Oil a. Gas J., 1964,
v. 62, №6, p. 101, 102, 105.
370. Dilliplane R. J. e. a., Oil a. Gas J., 1963, v. 61, № 31, p. 119,
122—124, 126.
371. Atteridg P. Т., Humble J., Hydrocarbon Process, and Petrol.
Refiner, 1963, y. 42, № 4, p. 167—170.
372. Sanford R. A. e. a., Petrol. Refiner, 1962, v. 41, № 7, p. 103—
108.
373 Foster R. L. e. a., I and EC, Prod. Research a. Develop.,
1963, v. 2, № 4, p. 328—332.
374. Adams N. R., Shterba M. J., Oil. a. Gas J., 1963, v. 61,
№ 16, p. 127—129.
375. Пат. США 3150104 A964).
376. Пат. США 3150103 A964).
377. Пат. США 3150075 A964).
378. Пат. США 3178364 A965).
379. Пат. США 3168462 A965).
380. Пат. США 3151059 A961).
381. Пат. США 3168481 A965).
268
382. Пат. США 3173882 A965).
383. Пат. США 3122511 A965).
384. Эйгенсон А. С. и др. Авт. свид. № 186395; Изобр., пром.
образцы. Товари, знаки, 1966, № 19, с. 18.
385. Белозерский Н. А. Карбонилы металлов. М., Металлург-
издат, 1958. 372 с.
386. Чатт Дж., Посон Н. А., Бененци Л. М. В кн.: Химия ме-
таллоорганических соединений. Под ред. Г. Цейса. Пер.
с англ. Под ред. Н. Л. Кнунянца. М., «Мир», 1964,
с. 538—604.
387. Волков В. Л., Сыркин В. Г., Хим. пром., 1965, № 5,
с. 32—36.
388. Масагутов Р. М., Данилова Р. А., Берг Г. А.,
«Нефтепереработка и нефтехимия», 1968, № 12, с. 12—14. \
-389. Масагутов Р. М., Данилова Р. А., Берг Г. А.,
«Нефтепереработка и нефтехимия», 1969, № 4. с. 5—8.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азотистые соединения 126—134, 180
отравление катализаторов 128, 131, 132,
134
Активность катализаторов 7, 8, 13, 40—45,
48, 50, 51, 58—61, 71, 82
аморфных алюмосиликатных 13, 43, 50,
119
влияние времени контакта 102, 103, 105,
106, 108, 113, 117, 124
— кислородных и сернистых
соединений 125, 180
— металлов 138—144, 146, 147, 152, 154—
158, 161, 171, 174
— природы сырья 106
— температуры 112
индекс активности 141
метод определения Кат-А 218, 220, 223
цеолптсодержащих 43—45, 50, 51, 58,
60, 106. 107, 119
Алюмосиликатные аморфные катализаторы
7, 8, 11—13, 18, 19, 24,-27, 31, 32, 50
активность 13, 43, 50, 119
влияние времени контакта 108, 109
— ыеталлорганических соединений 50
ныделение сероводорода 101
— серы 101, 109
выход при крекинге бензина 116. 122
газовых компонентов 109
кокса 98, 108, 116, 122
— — насыщенных углеводородов 108,
109
продуктов 26, 29, 116
микросферическне 24
отравление азотистыми соединениями
132—134
— металлами 152, 153, 161
прокалка 144
пропарка 144
РСГ-2 50
способы получения 12, 13
стабильность 50
структура 51
шариковые 24, 2Г>, 27, 29, 47, 48
Бензин
выход при крекинге 25, 26, 42. 47. 49,
103—106, 118
— на аморфном катализаторе 28, 29. 50
— цеолитсодержащем катализаторе 28,
29
скорость образования 107
содержание серы 112
состав 28, 111, 112
Бентонит 9
Газ каталитического крекинга 26—29, 43
выход 26, 27, 29, 42, 47, 103—105, 108, Uli
скорость образования 107
Газойль каталитического крекинга 29, ;«
Галлуазит 9
структура 11
Глины природные 9, 11, 12
Гмелинит 16, 17
Демет 239—243
Катализаторы
активность см. Активность
катализаторов
алюмосиликатные аморфные см. Алю-
носиликатные аморфные
катализаторы
АШНЦ-3 23—32, 47—52, 107; 110, 111
выделение воды 40
выжнг кокса 82
высокоглнниземистые 13
гранулированные 6
гранулометрический состав 86, 87
дезактивация 40, 56, 57, 85, 89, 95, 129,
142, 143, 145
действие азотистых соединений см.
Азотистые соединения
— водяного пара 90
— кислородных н сернистых
соединений 125, 180
— металлов см. Металлы
длительность работы 112—114
дюрабед 19—23, 52, 53
закоксовьшание 42, 67, 69, 71—74, 76,
77, 81. 88—92, 154, 178
избирательность действия 40, 48, 171
износ 93
кристаллические, см. Цеолнтсодержа-
щие катализаторы
микросферическне 6, 24, 47, 48, 50
накопление металлов 52, 61
обменная способность 139
отработанные 224
отравление см. Отравление
катализаторов
перегрев 81, 93, 120
плотность истинная 60
— кажущаяся 59, 60
— насыпная 59, 60, 70
потери 83, 84, 89, 90
приготовление 7
природные 7, 43, 224
прокалка см. Прокалка катализаторов
{Термическая обработка)
пропарка {термопаровая обработка)
см. Пропарка катализатора (Тер-
моиаровая обработка)
прочность 82, 90, 94
пылевидный см. Пылевидный
катализатор
равновесный 59—61, 63—65, 69, 82
разрушение см. Разрушение
катализаторов
реактивация см. Реактивация
катализаторов, отравленных металлами
регенерация 66—70, 72, 76—81, 87—93, 95,
121, 122, 143
рентгеноструктурный анализ 69
РСГ-2 25, 27, 29, 30 48—52
РСГ-2Ц 23—25 29, 30 47—52
свежий 59, 60, 63—65, 69
селективность 7, 8 41 43 46 51, 85,
148. 224
270
Катализаторы
синтетические 7, 12, 13, 43, 218, 219
содержание металлов 40, 85, 86
спекание см. Спекание катализаторов
средний радиус пор 38, 39, 51, 55—57,
59, 60, 64, 69, 70
стабильность 8, 50, 94
старение см. Старение катализаторов
структура 7, 41, 42, 145
термостойкость 34
удаление неактивных частиц 92
удельная активность поверхности 41, 42,
50
— обменная способность 40
— поверхность 7, 34—39, 41, 59—61, 64,
65, 72
удельный объем пор 37—39, 42, 54, 56,
59, 60, 64, 69, 70
ЦЕОКАР-2 23—31
цеолитсодержащие см. Цеолитсодер-
жащие (Кристаллические)
катализаторы
шариковые 24, 48
эрозия 6
Коалинит 9
Кокс
выжиг 66, 67, 72, 74, 76, 77, 81, 82, 93
выход при крекинге 25, 26, 30, 42, 43,
47, 49, 91, 95—98, 103—105, 107, 114—
116, 121, 122
горение 57, 72, 75
коксообразующнй фактор 226
концентрация на катализаторе 47, 67,
70, 74, 76, 82, 87, 91, 99, 114
— регенерированном катализаторе 77,
121
накопление в реакторе и регенераторе
70, 71, 74
— на катализаторе 88, 89, 95
непредельность 99
окисление 75, 168—170
остаточный 77—82, 88—90
отложения см. Коксовые отложения при
крекинге
разложение 99
распределение по частице катализатора
73, 74, 81
содержание серы 98, 100, 101
Коксовые отложения при крекинге 58, 74
76, 91, 98
влияние азотистых соединений 130, 131
— длительности процесса 124
действие металлов 142, 143, 162—166
176, 177, 180
механизм 176
состав 100
Крекинг
в кипящем слое 124
влияние азотистых соединений 130, 131
— времени контакта 102 103 105 106
108—115, 117, 120, 124 '
— температуры 110—112, 114
выделение сероводорода 98, 101
выход бензина 25, 26, 28 29 42 47 49
50, 103—106, 114—116, 118
— бутан-бутиленовой фракции 108, 118
— газа 114—116, 118
— кокса 25, 26, 30, 42, 43, 47 49, 91,
95—98, 103—105, 107, 114—116, 121, 122
— легкого газойля 118
— насыщенных углеводородов 108, 109
непредельных углеводородов 108
— пропан-пропнленовоП фракции 108, 118
на стандартном катализаторе 28, 29
— цеолнтсодержащих катализаторах 19,
29, 47, 61
установки см. Установки крекинга
Кристаллические (Цеолитсодержащие)
катализаторы см. Цеолитсодержащие
катализаторы
Матрица 18, 46. 57, 239
Металлы 138—180
ванадий 141, 148, 149, 151, 155, 159
допустимое содержание 90, 138—143,
147—150
железо 141, 148, 157, 161
кобальт 161
коксообразование 162, 163
никель 140, 141, 147—149, 151—155, 161
промотирующее действие 161, 171, 173,
174
пропарка (термопаровая обработка)
143—146
содержание в нефтях и
нефтепродуктах 134—139
Мет-х 225—239
Молекулярные сита 17
классификация по Барреру 16
Монтмориллонит 9, 10
активность 17
структура 11
Морденит 16, 17, 19, 58
125
Отравление катализаторов 7, 125
азотистыми соединениями 128, 131
действие кислородных соединений і
металлами см. Отравление
катализаторов металлам»
Отравление катализаторов металлами 139—
180
выход бензина 146, 151, 157—159, 161,
171, 174
— бутан-бутнленовой фракции 159
— водорода 161
— газа 161, 174
— кокса 146, 151, 155, 157, 158, 161, 171
— пропан-пропнлеиовой фракции 159
дезактивация 146, 174
действие металлов см. Металлы
— прокалки 144—147
— пропарки 143—146
механизм 171—173
пылевидного 149, 150, 153
реактивация см. Реактивация
катализаторов, отравленных металлами
шарикового 149, 153
цеолитсодержащих 152, 153, 161
Очистка сырья от компонентов,
отравляющих катализаторы 181—211
адсорбционная 184, 186
ПЯ1Г\М'МНЯСТ ПРПРГП17и5 1Й1 1Я9
182
вакуумная перегонка 181,
гидрогенизационная 191
деасфальтизацня 182, 183
коксование 182
селективными растворителями 183, 184
серной кислотой 186—190
термический метод 182
Прокалка катализаторов (Термическая
обработка) 34—40, 42, 43. 71, 72, 99,
144—147
в среде газов 43
цеолитсодержащих 44, 45
Пропарка катализаторов (Термопаровая
обработка) 35—40, 42, 46, 48, 143—146
аморфных 43
в среде газов 43
действие металлов 143—146
цеолитсодержащнх 43—47
Пылевидный катализатор 85, 91, 123
активность 85, 152
отравление металлами 149
перекоксовыванне 91
селективность 85
271
Разрушение катализаторов 82—85, 90, 93, 94
истирание 84, 90
перегрев 81, 93
раскалывание 83
растрескивание 82, 90
Реактивация катализаторов, отравленных
металлами 212—255
механические методы 212, 213
применение ионообменные смол
(процесс Мет-х) 225—239
:фС::е<Х ДСМСТ 239—243
cvy;ij деметаллизация 243—253
химические методы 213—225
Силикатные материалы У
структура 9, 10
Спекание катализатором 34, 36—42, 57. 61,
63, 65, 70, 71. 81, 120. Н2. І43. Но.
147
влияние закоксоваинссти 71. 72
— металлов 142, 14І
изменение структуры 54, 55, 63
кинетика процесса 59
механизм 54—57
при регенерации 71
Старение катализаторов 34. 26. 33, 42, 43.
70. 71. 87. 93
изменение свойств 34. ^'3
спекание сіл. Спекание катализаторов
Термическая обработка см. Прокалка
катализаторов (Термическая
обработка)
Термопаровая обработка см. Пропарка
катализаторов
Установки крекинга
гудрезид 6
гудри 6
гудрифлоу 6, 92
ортофлоу 6
с движущимся слоем катализатора 6, 94
с кипящим слоем катализатора 6, 13. 91
с неподвижным катализатором 6
термофор о
ті; г. а 43-1 u
—42-102 6. 118
флекепкрекииг 123
флюид 6. 121. 122. 134, 234
Флюид-тексако 124
Фожаапт 19. 58
Цсолитсодеріхащие (Кристаллические)
катализаторы 7, 8. 13. 14, 18, 23, 103, 122,
161
активность 43—45. 50. 51, 58, 60, 106.
107. 113
влияние времени контакта 119
— сероводорода и серы 101
Рафгат Мазитович Масагутов
АЛЮМОСИЛИКАТНЫЕ
КАТАЛИЗАТОРЫ
И ИЗМЕНЕНИЕ ИХ СВОЙСТВ
ПРИ КРЕКИНГЕ
НЕФТЕПРОДУКТОВ
Редактор Бабушкина С. И.
Художественный редактор Носов Н. В.
Художник Ольшевский М. Ф.
Технический редактор Кочетова А. С.
Корректоры Го л у б е в а О. И., Хрипун ова М. С.
Т-127&3. Сдано в иаб. 22/IV 1975 г. Подп. к печ. 28/VII 1975 г.
Формат бумаги 60Х90'Л«. Бумага тип. № 2. Усл. печ. л. 17.
Уч.-изд. л. 18,90. Тираж 2000 экз. Зак. 2206. Изд. № 337. Цеиа і р. 11 к.
Издательство «Химия», 107076, Москва, ул. Стромынка, д. 13.
Московская типография № 11 Союзполиграфпрома при Государственном
комитете Совета Министров СССР по делам издательств, полиграфии
и киижиой торговли. Москва, 113105, Нагатинская ул., д. 1