/








































































































































































































































































































Текст
В.Е.СОРОКО, С.В.ВЕЧНАЯ, Н.Н.ПОПОВА
Основы
химической
технологии
для техникумов
В.Е.СОРОКО, С. В. ВЕЧНАЯ, Н.Н. ПОПОВА
7
3
I
11
Основы
' химической
технологии
11
I
(з
I
6
a "os о.
Под редакцией
профессора д-ра. техн, наук
В. Е. Сорока
Допущен Министерством
химической промышленности СССР
в качестве учебника
для учащихся техникумов
химических
•1. 1|[П|1
к МОГЯЛРВ
ВИ6ЛК5ТГКА
нко- екнолсгнчвыого
техникуме
Инв.
Ленинград "ХИМИЯ"
Ленинградское отделение
1986
1
1
5
I,
г
1
9
6П7.1
С 655
УДК 66.01 (075.8)
Рецензенты: 1. Зам. директора Щелковского химико-
механического техникума Сосонко В. Е.
2. Преподаватель Щелковского химико-ме-
ханического техникума Наумова А. М.
УДК 66.01 (075.8)
Сороко В. Ё., Вечная С. В., Попова Н. Н.
Основы химической технологии: Учеб, для технику-
мов. — Л.: Химия, 1986. — 296 с. ил.
Обобщены основные закономерности ведения технологических процес-
сов. Даны рекомендации по рациональному аппаратурно-технологическому
оформлению химических производств. Изложены принципы работы основных
типов оборудования, приведены их расчеты, режимы эксплуатации, показа-
ны области применения. Рассмотрены типовые химико-технологические про-
цессы. Особенностью изложения является связь теоретических основ с при-
кладными вопросами.
Для учащихся техникумов, готовящих специалистов химической, нефте-
химической, нефтеперерабатывающей, пищевой, фармацевтической и смеж-
ных отраслей промышленности.
Табл. 3. Ил. 208.
2801000000-096
С ' '51 (OW
© Издательство „Химия", 1986
ОГЛАВЛЕНИЕ
Предисловие ........................................... 7
Введение ............................................................. 9
Глава 1. Химическая промышленность в СССР.......................... 11
1 1. Значение химической промышленности в народном хозяйстве ..... И
(1.2. История развития химической промышленности.......................Н
1.3. Основные направления развития химической техники и технологии . . 13
Глава 2. Сырьевая и энергетическая база химической промышленности ... 16
2.1. Сырье химической промышленности \.............................. 16
-1. 2.1.1. Виды и запасы сырья........................................16
2.1.2. Принципы обогащения сырья.............. ... . . 18
2.2. Вода и воздух в химической промышленности........................21
2.2.1. Вода в химической промышленности . ........................21
2.2.2 Воздух в химической промышленности...........................2о
2.3. Энергетика химической промышленности........................ ... 26
2.3.1. Основные источники энергии..................................27
2.3.2 Энергетическая программа СССР и химическая промышленность . . 29
2.3.3. Экономия энергии в химической промышленности . ............29
Глава 3. Основные характеристики химико-техиологических процессов ... 32
3.1 Классификация химико-технологических процессов....................33
-3.1.1. Уровни анализа, описания й расчета ХТП......................34
3.2. Основные показатели химико-технологического процесса.............35
3.2.1. Степень превращения.........................................36
3.2.2. Выход продукта..............................................36
3.2.3. Скорость XIП.............................................. 37
3.2.4. Избирательность ............................................40
3.2.5. Расходные коэффициенты......................................41
3.3. Материальный баланс..............................................42
3 4. Тепловой баланс................................................42
3.5. Термодинамические характеристики химических процессов............>4
3.5.1. Задачи термодинамического анализа...........................44
3.5.2. Константы равновесия и расчет равновесных степеней превращения . 45
Глава 4. Закономерности гомогенных процессов..........................49
4.1. Общая характеристика гомогенных химических процессов............49
4.2. Влияние концентрации на скорость реакции и степень превращения . . 50
4.2.1. Простые необратимые и обратимые реакции . . 51
4.2.2. Влияние концентрации на избирательность.....................52
4 2.3. Методы увеличения концентраций.......................... 53
4.3. Температура как фактор повышения скорости процесса и управ тения
выходом продукта и избирательностью..............
4.3.1. Интенсификация необратимых процессов . ... ........
4.3.2. Влияние температуры на скорость и степень превращения при прот
кании обратимых реакций........................................
1*
4.3.3. Влияние температуры на скорость, избирательность и выход продукта
при протекании сложных реакции . . . .... 58
4.4. Влияние давления на скорость газофазных процессов..................59
4.5. Применение катализаторов в гомогенных системах (гомогенный катализ) 61
4.5.1 Значение и области применения катализа..........................61
4.5.2. Сущность и виды катализа .... ... 62
4.5.3. Скорость гомогенного каталитического процесса . . .63
4.6. Изменение основных показателей гомогенных процессов во времени . . 64
4.7. Принципы расчета оптимальных параметров проведения процессов ... 67
Глава 5. Закономерности проведения гетерогенных химико-технологических
процессов . . 71
5.1. Общая характеристика гетерогенных ХТП .............................71
5.2. Процессы, протекающие во внешнедиффмзионной области 73
5.3 Виутрнднффузионная область протекания процессов . ...........76
5.4. Кинетическая область протекания процессов . 79
5.5. Особенности протекания гетерогенно-каталитических процессов . ... 80
5.5.1. Основные требования к катализаторам .... . 81
5.5.2. Свойства твердых катализаторов и их производство . ... 82
5.5.3 Области протекания гетерогенно-каталитических процессов. Влияние
диффузионных торможений на избирательность ... 83
5.6. Моделирование — основной метод расчета гетерогенных ХТП . 88
5.6.1. Понятие о моделировании н моделях..............................88
5.6 2. Характеристика и моделирование процессов в системе газ (жид-
кость) — твердое .................................................. 89
5.6.3. Характеристика и моделирование процессов в системе газ (жид-
кость) — жидкость ....................................................93
5.6.4. Процессы взаимодействия между твердыми фазами н в многофазных
системах ............. ....................................... . . 95
Глава 6. Химические реакторы, закономерности их работы и конструкции 95
6.1. Основные характеристики потоков и их влияние на протекание ХТП . . 97
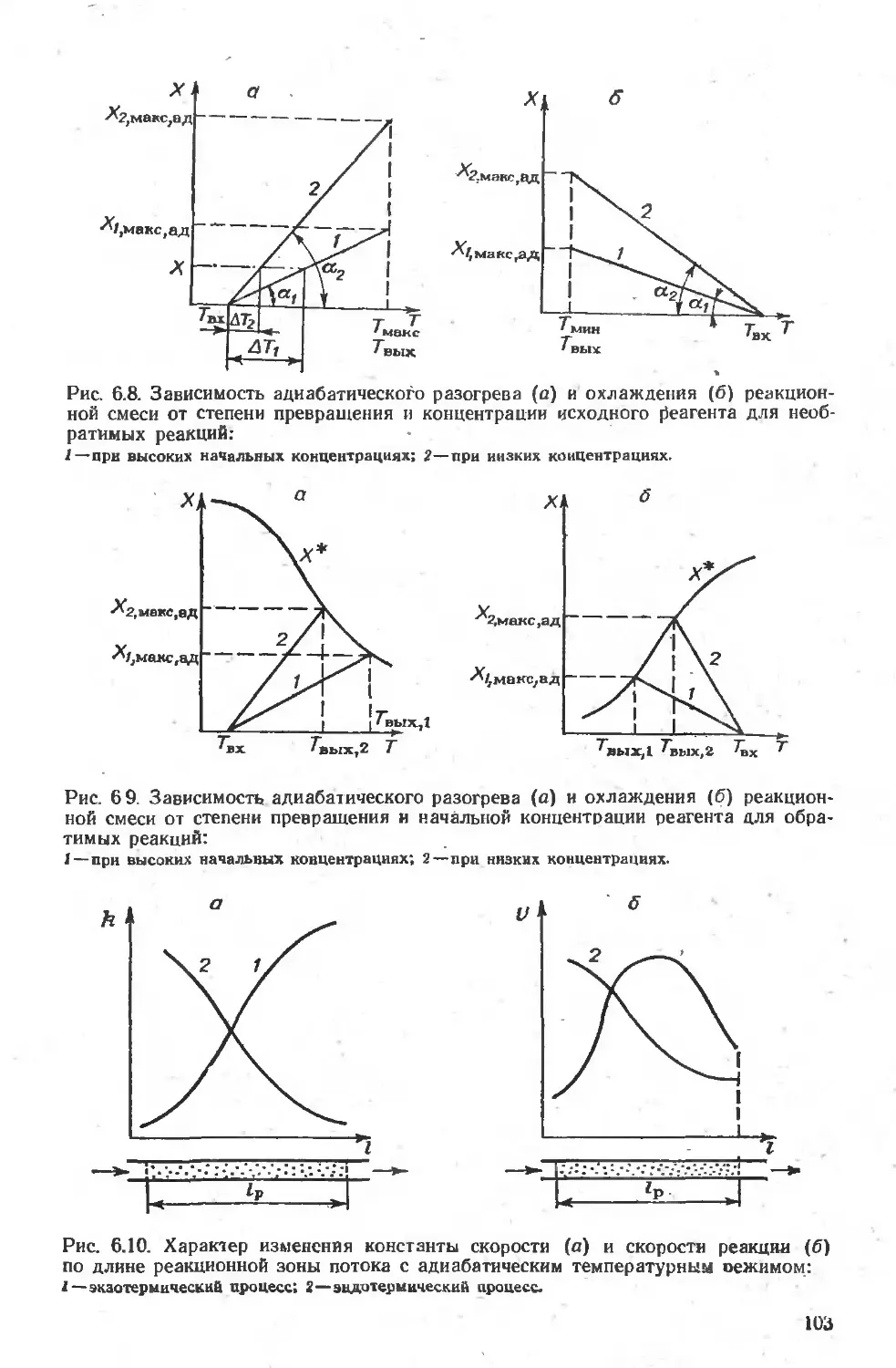
6.2. Протекание ХТП в потоке идеального вытеснения ...... .98
6.2.1. Общая характеристика и температурные режимы . . ... 98
6.2.2. Изотермический температурный режим ... . .................99
6.2.3. Адиабатический температурный режим . . ............ ... 102
6.2.4. Политермическнй температурный режим...........................104
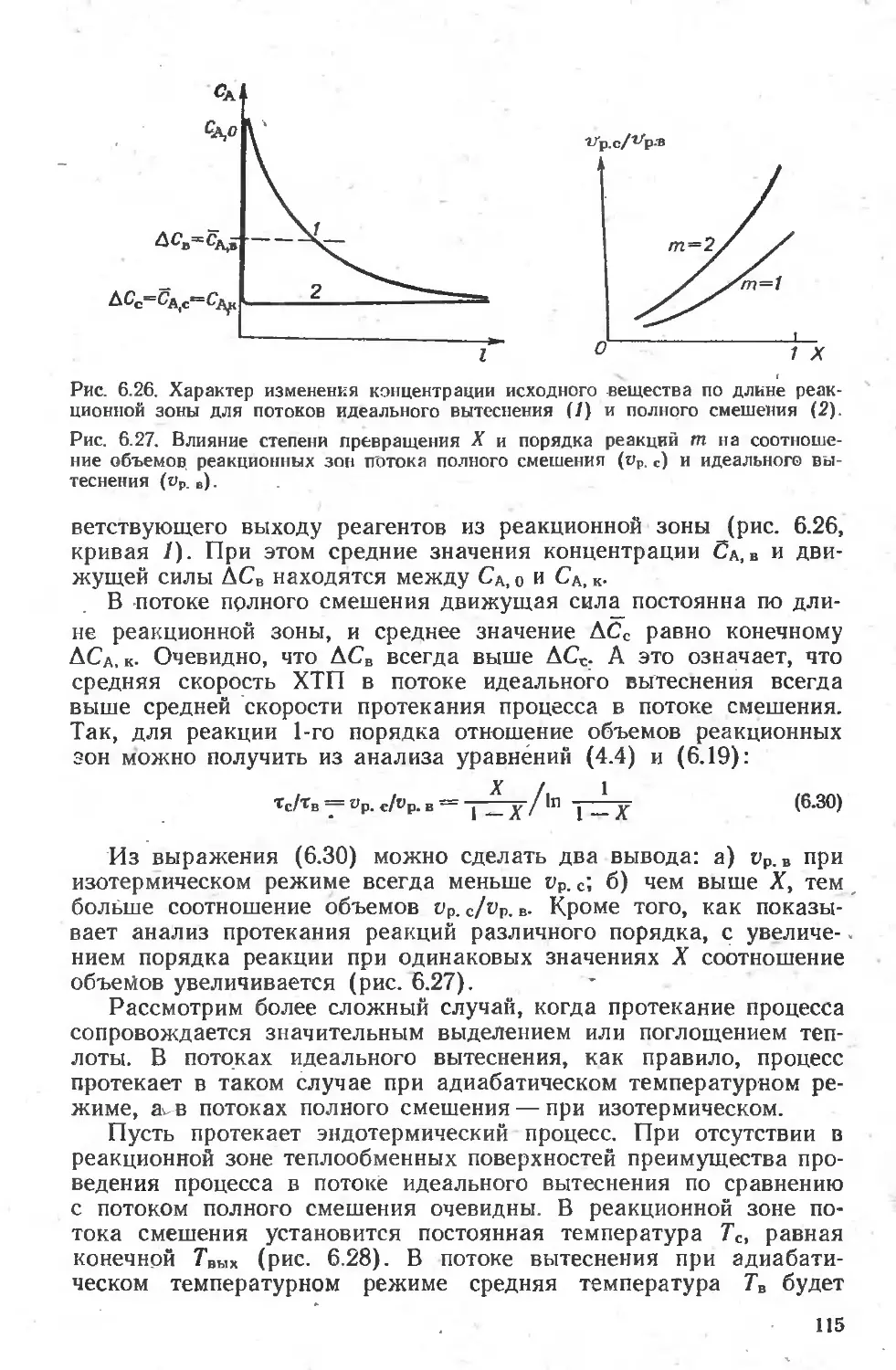
6.3. Протекание ХТП в потоке полного смешения . . ... . 107
6.3.1. Общая характеристика режима.................................. 107
6.3.2. Закономерности протекания ХТП в потоке без теплообмена .... 108
6.3.3. Теплообмен с окружающей средой как фактор интенсификации ХТП
в потоке ... . .......... ............................... НО
6.3.4. Секционирование реакционной зоны потока. . ...................142
6.4. Сопоставление протекания ХТП в идеальных потоках...................И4
6.4.1 Простые обратимые и необратимые реакции .114
6.4.2. Влияние типа потока на избирательность .... . .... 116
6.5. Протекание ХТП в неидеальных потоках . . . . . 119
6.5.1. Отклонения реальных потоков от идеальных ... ...........119
6.5.2. Модели неидеальных потоков................................. . 120
6.6. Классификация и основные показатели работы химических реакторов . 120
6.7. Реакторы для гомогенных процессов.................................122
6.7.1. Система жидкость - жидкость (Ж—Ж) .... 122
6.7.2. Система газ — газ (Г—Г).......................................123
6.8. Реакторы для некаталитических гетерогенных процессов . .... 124
6.8.1. Система газ — жидкость (Г—Ж)................................ 124
6.8.2. Система жидкость — твердое вещество (Ж—Т) . . ..... 127
6.83. Система газ—твердое вещество (Г—Т) и газ — твердое вещество —
жидкость (Г—Т—Ж).....................................................128
4
(5.9. Аппаратурное оформление гетерогенно-каталитических процессов . . . 135
6.9.1. Контактные аппараты с фильтрующими слоями катализатора (ФО . 135
6.9.2. Контактные аппараты с кипящими слоями катализатора (КС) . . .141
6.9.3. Контактные аппараты с движущимся слоем катализатора . . 143
6.10. Анализ работы многоступенчатых каталитических реакторов..........144
6.10.1. Контактные аппараты ФС с промежуточным теплообменом . . 145
6.10.2. Контактные аппараты КС с промежуточным теплообменом 147
6.10 3. Контактные аппараты КС с внутренним теплообменом 148
6.11. Температурная устойчивость реакторов. ...................150
Глава 7. Химико-технологические системы (ХТС) . . ............... 152
7.1. Общая характеристика химико-технологических систем................152
7.2. Способы отображения структуры химико-технслогических систем . . 153
7.3. Основные типы связей между элементами ХТС 155
7.3.1. Последовательная связь ... ................... . . 155
7.3.2. Последовательно-обводная технологическая связь............. . 156
7.3.3. Параллельная технологическая связь........................... 157
7.3.4. Обратная (рецнклическая) технологическая связь . 158
7.3.5. Перекрестная технологическая связь .... .......... 160
7.4. Понятие о математической модели ХТС . - 160
7.5. Задачи, решаемые при исследовании ХТС.............................160
7.6. Проектирование ХТС............................................... 160
7.6.1. Основные этапы создания производств и стадии проектирования . . 130
7.6.2. Предпроектная разработка ............ . . . . 162
7.6.3. Разработка проекта.................................. . 164
7.6.4. Составление рабочей документации............................. 165
7.6.5. Послепроектная стадия....................................... 166
7.6.6. Методы проектирования ........................................166
Глава 8. Принципы экологической технологии . . . . 166
8.1. Основные направления охраны окружающей среды от промышленных
выбросов...............................................................166
8.2. Очистка газообразных промышленных выбросов........................169
8.2.1. Очистка газообразных выбросов от взвешенных частиц (аэрозолей) . 170
8 2.2. Очистка газов от газо- или парообразных примесей . - 172
8.3. Очистка сточных промышленных вод . . ...................... 173
8.4. Предотвращение теплового загрязнения . . .................... 176
Глава 9. Производство серной кислоты . ...................... 177
9.1. Общие сведения................................................... 177
9.2. Способы производства .... 177
9.3. Сырье для производства серной кнстоты . . . 178
9.4. Производство сернистого газа..................................... 178
9.4.1. Сжигание серы . ..... 178
9 4.2. Обжиг колчедана . . . . 180
9.5. Окисление оксида серы(1У).........................................183
9.6 Абсорбция оксида серы (VI)........................................188
9.7. Химико-технологические системы производства серной кислоты . 190
9.8. Основные направления интенсификации сернокислотны’: производств . . 192
Глава 10. Производство аммиака . ...... 193
10.1. Общие сведения об аммиаке. Значение соединений азота . 193
10.2. Способы производства.......................................... 194
10.3. Сырье для синтеза . . 194
10.3.1. Получение азота .......................... . ... 195
10.3.2. Получение водорода..........................................195
б
10.4 Физико-химические основы синтеза аммиака........................ 199
10.5. Реакторы для синтеза............................................202
10.6. Химико-технологические системы синтеза аммиака . . 203
10.7 Основные направления совершенствования производства аммиака . . . 205
Глава 11. Производство азотной кислоты............................... 205
11.1. Общие сведения................................................ 205
11.2. Способ получения .............. . . 206
11.3. Сырье для производства..........................................206
11.4. Физико-химические основы производства. Аппаратурное оформление . . 207
11.4.1. Контактное окисление аммиака ........207
11.4.2. Окисление оксида азота(П) . . ... 210
11.4.3. Абсорбция оксида азота(1А/) водой . . . . . 211
11.5. Химико-технологические системы производства кислоты.............212
11.6. Основные направления совершенствования производства.............214
Глава 12. Химическая переработка топлив ..............................215
12.1. Классификация топлив............................................215
12.2. Переработка твердого топлива....................................216
12.2.1. Виды, свойства, состав и методы переработки твердого топлива 216
12.2.2 Коксование каменного угля . . 219
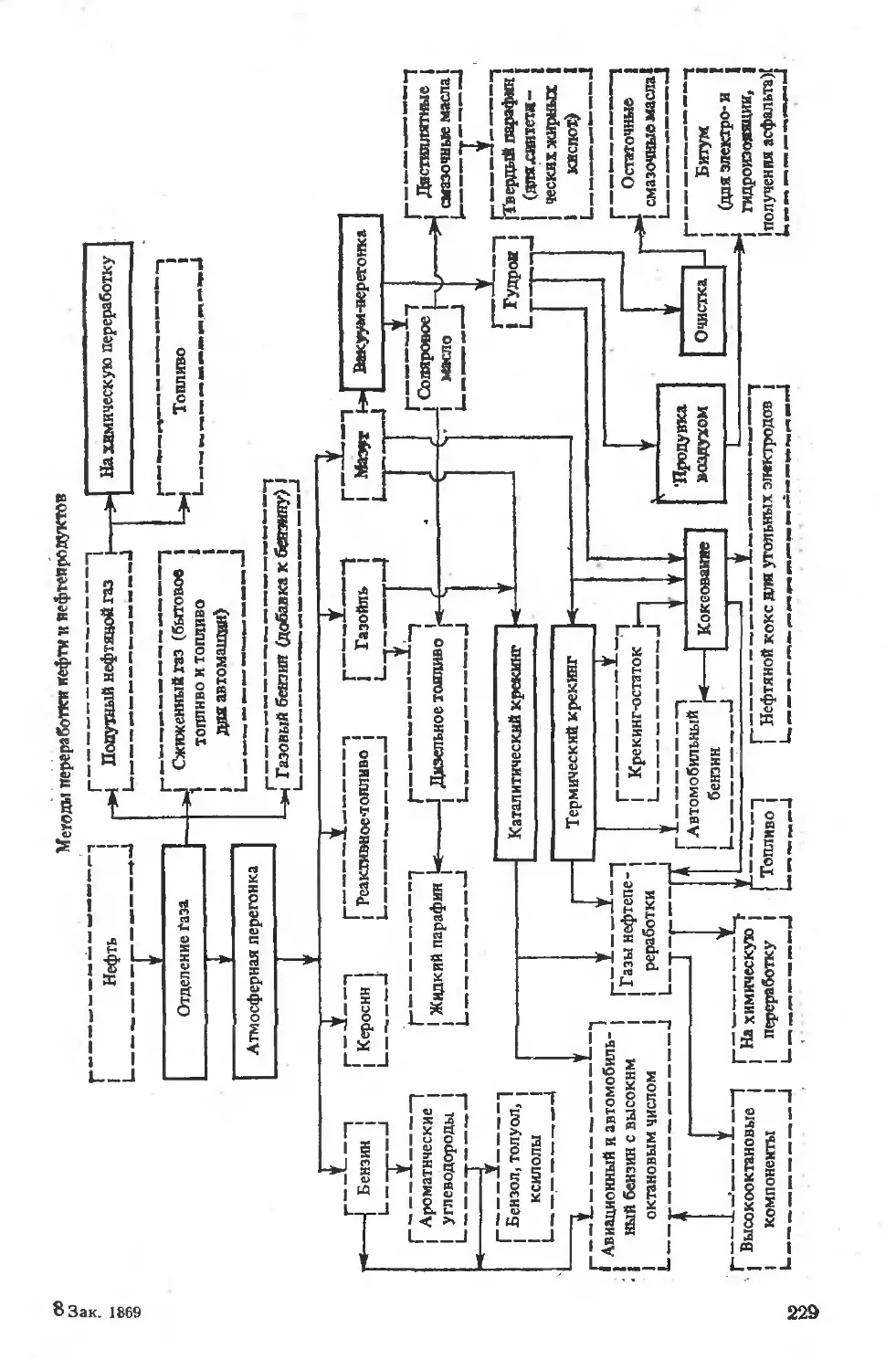
12.3. Переработка нефти и газообразных топлив . .... 22ч
12.3.1. Состав нефти и нефтепродуктов............................. 224
12.3.2. Методы переработки нефти....................................227
12-3,3. Перегонка нефти.............................................'30
12.3 4. Химические методы переработки нефти ... .............234
12.3.5. Методы разделения газообразных топлив.......................242
Глава 13. Органический синтез.........................................246
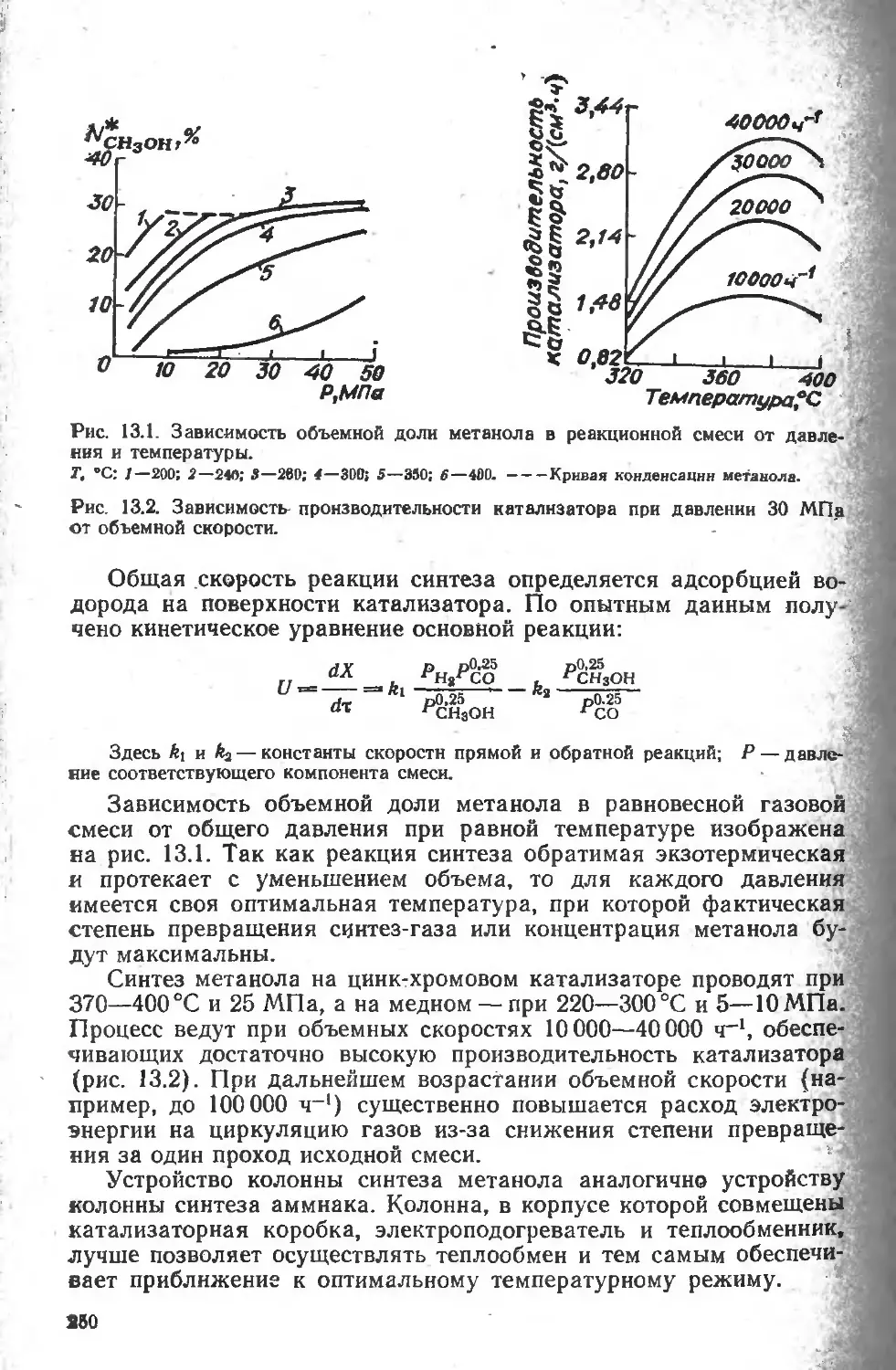
13.1. Характеристика сырья и основных процессов.......................246
13.2. Производство метанола........................................... 249-
13.3. Производство формальдегида............ .........................252
13.4. , Производство ацетилена . ............... . .... 257
13.5 Производство уксусной кислоты...................................260
13.6. Получение этанола...............................................263
Глава 14. Технология высокомолекулярных соединений....................268
14.1. Методы получения высокомолекулярных соединений..................268
14 2 Производство синтетических смол и пластмасс......................274
14.2 1/ Полнмеризационные смолы и пластмассы на их основе..........274
14.2.2. Поликонденсационные смолы и пластмассы на их основе . ... 285
Литература............................................................288
Предметный указатель..................................................289
ПРЕДИСЛОВИЕ
Химическая наука и промышленность играют большую роль
в создании и развитии материально-технической базы коммуниз-
ма. В соответствии с «Основными направлениями экономического
и социального развития СССР на 1986—1990 годы и на период
до 2000 года» и решениями XXVII съезда КПСС во все более широ-
ких масштабах будет проводиться химизация народного хозяйства.
Число химических производств неуклонно возрастает, а освоенные
производства претерпевают существенные изменения. Одновремен-
но происходит все возрастающая их типизация, при которой
в различных производствах усиливается применение аналогичных
технологических приемов интенсификации процессов, управления
ими. улучшения качества целевых продуктов.
Основное внимание г настоящем учебном пособии, как и предус-
мотрено программой курса «Основы химической технологии», уде-
лено изучению общих закономерностей протекания химико-техно-
логических процессов, основных приемов рациональной эксплуата-
ции реакционных аппаратов; типовым методам интенсификации
технологических процессов; оптимальной организации производства
в целом. С целью облегчения усвоения теоретическою материала
основные закономерности химической технологии проанализиро-
ваны на примере сравнительно небольшого числа производств,
имеющих наибольшее народнохозяйственное значение.
Поскольку теория процессов тепло- и массопередачи изложена
в курсе «Процессы и аппараты химической технологии», содержа-
ние данного учебника ограничено научными и практическими осно-
вами химико-технологических процессов, являющихся главными
в химических производствах. Курс «Основы химической техноло-
гии» имеет также тесную связь с такими дисциплинами, как «Физи-
ческая химия», «Охрана труда»х «Охрана окружающей среды»,
специальными предметами. Авторы старались избегать дублирова-
ния материала с этими курсами, хотя не всегда это оказывалось
возможным из-за естественного стремления сохранить единство
предмета
Авторы старались учесть специфику среднего технического
образования и обобщить обширный материал в виде узловых воп-
росов теоретических основ химической технологии, которые состав-
ляют обязательный минимум знаний в этой области.
Стремление изложить материал в соответствии с современным
уровнем развития химической технологии как науки привело
к некоторому расширению ряда тем учебной программы. Этот ма-
териал, набранный петитом, должен помочь учащимся составить
наиболее полное и правильное представление о закономерностях
7
протекания химико-технологических процессов и рекомендуется
для самостоятельного углубтенного изучения.
Учитывая, что материал должен быть доступен учащимся, все
обобщения даны преимущественно в виде графиков или несложных
математических корреляций в соответствии с программой и уровнем
знаний, получаемых учащимися по математике.
Введение и главы 1, 8, 10, И написаны С. В. Вечной, глава 2 —
С. В. Вечной совместно с В. Е. Сороко, главы 3—7 и 9 — В. Е. Со-
роко, главы 12—14 — Н. Н. Поповой.
Авторы приносят благодарность участвовавшим в написании
учебника В. А. Коновалову (разд. 3.3, 3.4) и Н. В. Кузичкину
(разд. 7.1, 7.2, 7.4, 7.5) и выражают признательность профессору
И. П. Мухлеиову и рецензентам В. Е. Сосонко и А. М. Наумовой
за их ценные замечания, которые учтены при окончательной подго-
товке рукописи к изданию.
ВВЕДЕНИЕ
Термин «технология» происходит от двух греческих слов: «тех-
нос» — искусство или ремесло и «логос» — наука. Следовательно,
технология — это наука о ремеслах. Современный уровень разви-
тия промышленности вкладывает новое содержание в слово техно-
логия Технология — это наука о наиболее экономичных спосо-
бах и процессах получения сырья, полупродуктов, продуктов или
изделий.
Способ получения — это совокупность всех операций,
которые проходит, например, сырье при получении из него продук-
та. Операция может быть осуществлена в одном или нескольких
аппаратах. В химических аппаратах, как правило, одновременно
протекают несколько процессов: гидравлические, тепловые, диф-
фузионные и чисто химические (основные).
Технологию делят на механическую и химическую.
Механическая технология изучает процессы, в кото-
рых изменяется форма или внешний вид и физические свойства
материала без изменения состава и внутренней структуры веще-
ства. Так, из металлов и пластмасс штамповкой и резанием изго-
товчяют детали машин и аппаратов.
Химическая технология изучает процессы, в которых
происходят глубокие изменения состава, свойств и внутреннего
строения веществ. Например, в результате переработки природ-
ных газов, нефти, углей получают удобрения, пластические массы,
растворители, красители, химические волокна и другие продукты,
имеющие совершенно иные свойства, строение и состав, чем исход-
ные вещества.
Выделение химической технологии в отдельную область знаний
началось в первой половине XIX в. Именно в это время в Россий-
ской Академии наук была утверждена кафедра химической техно-
логии (1803 г.). Окончательно химическая технология оформилась
в самостоятельную научную дисциплину в первом десятилетии
XX в., когда было разработано учение об основных процессах в ап-
паратах и общих закономерностях химико-технологических про-
цессов.
Деление технологии на механическую и химическую в ряде слу-
чаев условно, так как изменение вида и формы материала может
сопровождаться химическими превращениями. Так, при литье
пластмасс происходят и химические превращения. Химические про-
цессы, в свою очередь, во всех производствах сопровождаются ме-
ханическими.
Химическую технологию подразделяют на технологию неоргани-
ческих веществ (включает производства неорганических продуктов:
о
Леральных кислот, солей, щелочей, силикатов и др.) и техноло-
,<ю органических веществ (такие производства, как переработка
углей, нефти, горючих газов, получение органических кислот, спир-
тов, красителей, пластмасс, химических волокон и других продук-
тов). В настоящее время стираются грани между органическими и
неорганическими производствами, так как они имеют общие прин-
ципы и закономерности, подчас и общее аппаратурное оформление.
Химическая технология — наука синтетическая, базирующаяся
на закономерностях общей, органической и физической химии, ме-
ханики, экономики и других наук. Используя достижения различ-
ных наук, а также производственный опыт, химическая технология
изучает совокупность физических и химических процессов, основ-
ные закономерности построения оптимальных химико-технологиче-
ских процессов и разрабатывает типовые методы их интенсифика-
ции и общие принципы построения химических производств. Хими-
ческая технология является научной базой химической, нефтехими-
ческой, металлургической, целлюлозно-бумажной, пищевой про-
мышленности и многих других отраслей.
В нашей стране основы химической технологии как науки зало-
жили русские ученые и инженеры А. К. Крупский, И. А. Тищенко,
Д. И. Менделеев, Д. П. Коновалов, Л. Ф. Фокин.
Изучение основ химической технологии имеет важное значение
в современных условиях развития химической промышленности,
идущей по пути увеличения масштабов производства, улучшения
его технического оснащения, создания сложных технологических
процессов, комбинирования и кооперирования различных произ-
водств. Чтобы ориентироваться в производственных условиях,
овладеть сложной технологией и управлять высокоразвитой техни-
кой, обслуживающему персоналу нужно полностью знать все усло-
вия производства, и главным становится умение проникать в сущ-
ность химико-технологических процессов, находить и поддерживать
оптимальные режимы их ведения, экономно расходовать материа-
льные ресурсы и получать продукцию высокого качества.
ГЛАВА 1
ХИМИЧЕСКАЯ ПРОМЫШЛЕННОСТЬ В СССР
1.1. ЗНАЧЕНИЕ ХИМИЧЕСКОЙ ПРОМЫШЛЕННОСТИ
В НАРОДНОМ ХОЗЯЙСТВЕ
Химическая промышленность СССР является одной из важней-
ших отраслей тяжелой индустрии, во многом определяющей науч-
но-технический прогресс в народном хозяйстве^ Трудно найти
отрасль народного хозяйства, в которой бы не применялись способы
и средства химической технологии и то огромное количество про-
дуктов, которое поставляет химическая промышленность.
На базе продуктов химической промышленности получили мощ-
ное развитие такие современные отрасли, как металлургическая,
машиностроение, автомобильный и авиационный транспорт, произ-
водство строительных материалов и товаров народного потреб-
ления.
Химическая промышленность является материальной базой
химизации народного хозяйства СССР.
Химизацией называют внедрение химических методов, про-
цессов и материалов в народное хозяйство. Химизация сельского
хозяйства имеет важное значение в решении Продовольственной
программ^ СССР. В настоящее время невозможно добиться полу-
чения большого количества высококачественных продуктов сель-
ского хозяйства без применения минеральных удобрений, ядохими-
катов (средств борьбы с вредителями и сорняками), консервантов
и искусственных кормов. Применение химических способов очистки
вредных выбросов различных производств (например, металлур-
гических, энергетических, целлюлозно-бумажных, текстильных)
способствует решению проблемы охраны окружающей среды. Мно-
гие продукты химической промышленности имеют широкое приме-
нение в технике, быту и коммунальном хозяйстве. Химизация имеет
также важное значение для повышения эффективности произ-
водства.
1.2. ИСТОРИЯ РАЗВИТИЯ ХИМИЧЕСКОЙ ПРОМЫШЛЕННОСТИ
» .
3 Г Химическая промышленность нашей страны в своем развитии
прошла сложный пут^ В дореволюционный период по уровню раз-
вития она во много раз отставала от химической промышленности
США и стран Западной Европы. Сырье и 70 % средств производ-
ства в основномдэбеспечивались за счет импорта. Не использовался
и такой благоприятный фактор, как высокий уровень русской хими-
ческой наукихБольшой вклад в развитие химической науки внесли
выдающиеся русские ученые М. В. Ломоносов, Н. Н. Зинин,
Н Н. Бекетов, А. М Бутлеров, Д. И. Менделеев, В. В. Марковни-
ков, А. Е. Фаворский, Д. П. Коновалов и дрУ‘
11
1913 г. Россия располагала 524 химическими предприятиями,
преимущественно мелкими, с общей численностью рабочих
около 80 тыс. По производству серной кислоты, например, Россия
в 14 раз отставала от США, а объем производства суперфос-
фата составлял всего лишь 2 % от объема его производства
в CUIjQ
Во время гражданской войны и иностранной интервенции мно-
гие предприятия были разрушенье
После установления Советской власти правительство во главе
с В. И. Лениным рассматривало развитие химической промышлен
ностп как одну из важных задач.£Планом ГОЭЛРО (1920 г.) пре-
дусматривались опережающие темпы развития химической про-
мышленности по сравнению с другими ведущими отраслями народ-
ного хозяйства.
>^В конце восстановительного периода (1926 г.) около 90 % всей
fхимической промышленности концентрировалось в основном
в Европейской части нашей страны. Объем химической про-
дукции в 1926/27 гг. почти в 5,5 раза превысил ее объем в
1922/23 rrj
В первой пятилетке (1928—32 гг.) построены крупные предприя-
тия по производству синтетического аммиака, азотных, калийных
и фосфатных удобрений, карбида кальция, химических волокон,
а также реконструированы и расширены предприятия, построенные
ранее. Пуск в эксплуатацию комбината «Апатит» на базе Хибин-
ского месторождения позволил не только отказаться от импорта
фосфатного сырья, но и начать его экспорт. Настоящим триумфом
советской науки был пуск (1932 г.) первого в мире завода синтети-
ческого каучука, производимого по методу С. В. Лебедева. Это поз-
волило резко сократить импорт натурального каучука.
К 1940 г. в СССР создана мощная азотная промышлен-
ность, промышленность основной химии и ряда других подот-
раслей.
ГЬольшой урон химической промышленности нанесла Великая
Отечественная война/ Объем химической промышленности резко
снизился (например, серной кислоты на 77 %, аммиака на 50%).
^|Ъольшая часть заводов была разрушена, а часть их должна была
4 перебазироваться на Востоку
—I В послевоенный период химическая промышленность быстро
восстанавливалась^ создавались новые заводы на основе более
совершенной техники и технологии. {Уже в 1950 г. производство
химической продукции превысило уровень 1940 г. в среднем
в 1,8 разг.
Партия и правительство нашей страны постоянно придают боль-
шое значение развитию химической промышленности, что нашло
значительное отражение в решениях XXI—XXVI съездов КПСС
и пленумов ЦК КПСС, а также в постановлениях Советского пра-
вительства, направленных на дальнейшую химизацию народного
хозяйства страны^ В результате интенсивного наращивания мощно-
стей химическая промышленность СССР по объему' продукции
12 ,
занимает второе место после США, а по некоторым видам продук-
ции— первое место в мире/ (например, по выпуску минеральных
удобрений). -
1.3. ОСНОВНЫЕ НАПРАВЛЕНИЯ РАЗВИТИЯ
ХИМИЧЕСКОЙ ТЕХНИКИ И ТЕХНОЛОГИИ
Главная цель развития химической технологии—повышение
производительности труда, улучшение качества продукции и сни-
жение ее себестоимости. Эта цель может быть достигнута путем
совершенствования техники и технологии в следующих основных
взаимосвязанных направлениях: увеличение мощности производ-
ства; комплексное использование сырья; разработка энергосбере-
гающих производств; создание безотходных производств; внедрение
механизации трудоемких работ, автоматизации и автоматиче-
ских систем управления технологическими процессами; замена
периодических процессов непрерывными.
1. Увеличение мощности производств осуществляется тремя пу-
тями: использование параллельных технологических линий, увети-
чение размеров аппаратов и интенсификация работы аппаратов.
Использование параллельных технологиче-
ских линий, т. е. установка нескольких однотипных аппаратов
параллельно, позволяет осуществлять монтажные работы, пуск
в эксплуатацию, а также последующий ремонт оборудования без
остановки всего производства.
Увеличение размеров аппаратов повышает их про-
изводительность *, а следовательно, и производительность труда, а
также снижает себестоимость продукции. Например замена двух
небольших аппаратов на один большего размера уменьшает удель-
ный расход конструкционного материала и сокращает удельные
производственные площади, занимаемые оборудованием.
Однако рост мощности производства путем увеличения разме-
ров аппаратов имеет свои ограничения. Так, при сйльном увеличе-
нии размеров аппаратов усложняется их конструкция, затрудняется
обеспечение ремонтных работ и появляются другие негативные
факторы.
Интенсификация* работы аппаратов позволяет по-
высит ь их производительность без увеличения размеров. Достигает-
ся она двумя путями: V -
улучшением конструкции аппаратов (например, применение
форсунок для распыления жидкости, скоростных мешалок и т. д.);
совершенствованием технологических процессов в аппаратах
данного типа (изменение температуры и давления, применение
катализаторов, внедрение механизации и автоматизации и т. д.).
Эти два пути тесно связаны между собой. Улучшение констр) к-
ции аппарата повышает интенсивность технологического процесса.
* Сущность понятий «производительность» и «интенсивность» и формулы
для их расчета приведены в главе 6 [см. уравнения (6.43) и (6.44)].
1
13
За последние двадцать лет значительно возросла единичная
мощность аппаратов. Так, в производстве серной кислоты и
аммиака —в 30 раз, что позволяет получать до 450 тыс. т/год
продукции с сдной технологической линии.
2. Комплексное использование сырья. Это направление имеет
существенное значение для экономии сырья, снижения себестои-
мости продукции и решения экологической проблемы. Подробно
о комплексном использовании сырья см. разд. 2.1.1.
3. Разработка энергосберегающих производств дает возмож-
ность экономить природные энергоресурсы (каменный уголь, нефть,
природный газ и др.) и ведет к снижению себестоимости продук-
ции. С этой целью в химической технологии широко используют
теплоту экзотермических реакций для подогрева исходных реаги-
рующих веществ или для получения товарного водяного пара. При-
мерами могут служить производства аммиака, серной и уксусной
кислот, альдегидов и др. Столь же большое значение имеет рацио-
нальное использование теплоты сжигания топлива для проведения
эндотермических процессов, а также энергии на транспортировку
газов и жидкостей. Для уменьшения тепловых потерь в окру-
жающую среду применяют изоляцию аппаратов и трубопро-
водов.
4. Создание безотходных производств — важный фактор в реше-
нии экологической проблемы и снижении себестоимости продукции.
Существует несколько путей создания безотходных производств:
организация производств по замкнутому циклу
(циркуляционных), сущность которых состоит в многократном
возвращении непрореагировавших веществ в один и тот же аппарат
до полного их превращения в конечный продукт; такое произ-
водство не имеет отходов, загрязняющих окружающую среду. Это
наиболее рациональный путь решения экологической проблемы,
а повышение степени превращения сырья уменьшает его расход
и снижает себестоимость продукции. Примером таких производств
служат производства аммиака, метанола и др.
кооперирование химических производств с дру-
гими (например, металлургическими) позволяет использовать
выбрасываемые ранее отходы этих производств в качестве сырья
для других отраслей промышленности. Это в значительной мере
способствует охране окружающей среды и снижает себестоимость
продукции в результате экономии природных материалов;
применение методов очистки отходящих газов и сточ-
ных вод с использованием специальных очистных аппаратов. Пока
это самый простой и доступный метод защиты окружающей среды,
но в некоторых случаях при этом повышается себестоимость
продукции и не всегда обеспечивается достаточная степень очистки
вредных выбросов.
В настоящее время комбинируют все три направления.
5. Механизация трудоемких работ—замена физического труда
^^ловека машинным. Механизация, как правило, повышает
14
4
Рис. 1.1. Схема непрерывного процесса.
производительность труда за счет интенсификации работы аппара-
тов и сокращения штата обслуживающего персонала.
Автоматизация — применение приборов, позволяющих осу-
ществлять производственные процессы без участия человека, а
лишь под его контролем. Автоматизация — высшая степень механи-
зации, которая позволяет резко увеличить производительность тру-
да, улучшить условия труда и качество продукции. Приборы авто-
матического контроля дают возможность поддерживать на задан-
ном уровне параметры технологических процессов (температуру,
давление, расход реагентов и вспомогательных материалов и т. п.).
Для управления процессами наиболее эффективным является ис-
пользование автоматизированных систем управления технологиче-
скими процессами (АСУТП) с широким применением электронно-
вычислительных машин (ЭВМ).
6. Замена периодических процессов непрерывными интенсифи-
цирует процессы, способствует увеличению производительности
труда, улучшает качество продукции и условия труда.
Периодические процессы характеризуются тем, что
порция сырья загружается в аппарат, в .течение определенного
времени проходит в нем ряд стадий обработки, и после этого
полученный продукт выгружается. Периодические процессы имеют
ряд существенных недостатков: аппарат простаивает в период
загрузки и выгрузки; параметры технологического режима изме-
няются во времени (например, температуру после загрузки сырья
постепенно повышают, а перед разгрузкой—понижают), это ухуд-
шает качество продукта; продолжительность всего процесса
больше, чем непрерывного; энергетические затраты выше из-за
потерь теплоты в периоды загрузки и выгрузки; периодические
процессы трудно механизировать и автоматизировать.
В непрерывных процессах поступление сырья и вы-
грузка продукции происходят непрерывно (рис. 1.1). В этих про-
цессах отсутствуют недостатки, характерные для периодических
процессов. Непрерывный процесс отличается стабильностью пара-
метров технологического режима (температура, давление, концен-
трации веществ и т. п.), что, как правило, улучшает качество
продукта и позволяет автоматизировать процесс.
15
ГЛАВA 2
СЫРЬЕВАЯ И ЭНЕРГЕТИЧЕСКАЯ БАЗА
ХИМИЧЕСКОЙ ПРОМЫШЛЕННОСТИ
2.1. СЫРЬЕ ХИМИЧЕСКОЙ ПРОМЫШЛЕННОСТИ
Сырьем называют исходные материалы, используемые в про-
изводстве промышленных продуктов. Сырье химической промыш-
ленности отличается большим разнообразием. Выбор того или
иного вида сырья в значительной степени определяет технологию
производства, себестоимость и качество получаемого продукта.
В качестве сырья могут быть применены природные материалы,
полупродукты и отходы производства.
Полупродукты (полуфабрикаты) — это сырье, кото-
рое подвергалось промышленной переработке.
О т х о д ы •— побочные продукты, которые не используются на
данном предприятии, но могут служить сырьем для производства
продуктов на других предприятиях. Например, полупродуктами
в производстве метанола являются Н2 и СО, в производстве
H2SO4 — SO2. В производстве цветных металлов из природных руд
диоксид серы SO2 является отходом, содержащимся в отходящих
газах, и может быть использован в качестве сырья для получе-
ния H2SO4.
Сырье, прошедшее ряд стадий обработки (обогащение, измель-
чение, разделение на фракции и т. д.) с целью подготовки его
к применению, называют технологическим.
Как исходное сырье, так и готовые продукты должны отвечать
определенным требованиям, соответствующим стандарту (ГОСТ,
ОСТ). Сырье перед применением всегда подвергают качественному
и количественному анализу на содержание основного вещества
и примесей
2.1.1. Виды и запасы сырья
Виды сырья, используемого в химической промышленности, раз-
нообразны. Сырье можно классифицировать по происхождению —
минеральное, растительное и животное; по химическому составу —
неорганическое и органическое; по агрегатному состоянию—твер-
дое, жидкое и газообразное.
Минеральное сырье делят на рудное (металлическое),
нерудное (неметаллическое) и горючее (органическое). Рудное
минеральное сырье, Состоящее из природных минералов, исполь-
зуют для получения металлов. В основном это оксиды и сульфиды
металлов (Fe3O4, Fe2O3, CU2S, CuS, ZnS и др.).
Нерудное минеральное сырье- разнообразно по химическому
аоставу и применяется либо в естественном состоянии (песок,
глина, асбест, слюда и др.), лйбо поступает на химическую пе-
16
реработку (фосфориты, апатиты, природные калийные соли
и др.) для получения сульфатов, фосфатов, карбонатов, хлори-
дов и др.
Горючие минеральные ископаемые — торф, бурые и каменные
угли, сланцы, а также нефть и природный газ — относят к органи-
ческим соединениям и используют в качестве источников сырья или
энергии.
В химической промышленности широко применяют такие доступ-
ные и дешевые источники сырья, как воздух и вода (см. разд. 2.2).
Растительное и животное сырье по своему назначе-
нию разделяют на пищевое (например, масла, жиры, молоко)
и техническое (древесина, хлопок, кожа, шерсть и т. д.).
Сырьевая база химической промышленности СССР располагает
всеми необходимыми видами сырья. СССР занимает первое место
в мире по запасам железных руд, калийных солей, сульфата нат-
рия, поваренной соли и др. В СССР находится свыше половины
мировых запасов угля и торфа. Однако мировые запасы нефти,
газа и некоторых других видов сырья истощаются и являются
невозобновимыми.
По оценкам различных ученых мировых запасов большинства
видов минерального Сырья хватит лишь на несколько десятков лет.
Так, срок жизни запасов нефти и природного газа при современ-
ном уровне потребления составляет 30—50 лет, медных, свинцовых,
вольфрамовых и цинковых руд — 20—45 лет, алюминиевых и мар-
ганцевых руд—80—120 лет, железных и хромовых руд — 200—500
лет. Более благополучно положение с углем, запасов которого, по-
видимому, хватит на 300—1000 лет.
Основными направлениями в решении сырьевой проблемы
являются: 1) использование более дешевого сырья; 2) применение
отходов как вторичных материальных ресурсов; 3) применение
концентрированного сырья; 4) комплексное использование сырья;
5) замена пищевого сырья непищевым.
Использование более дешевого сырья имеет важ-
ное значение как для решения сырьевой проблемы, так и для сни-
жения себестоимости продукта. Стоимость сырья составляет
60—70 %\ себестоимости продукта. Снижение затрат на сырье
достигается разными способами. Прежде всего стремятся исполь-
зовать местное сырье, тем самым исключая затраты на его перевоз-
ку Важное значение имеет применение легкодобываемого сырья,
как более дешевого. Так, себестоимость нефти в 3,5 раза, а природ-
ного газа—в 12 раз меньше, чем угля, добытого шахтным мето-
дом. Дешевый и доступный природный газ и продукты нефтепере-
работки обеспечивают снижение себестоимости таких продуктов,
как пластмассы, синтетические волокна, каучук, моющие сред-
ства и др.
В качестве дешевого сырья расширяются возед
Wjuiiyn И ВТходо-. .
как о¥падЖ?"захряты ia добычу и
ериЬДНБ Я « 0 •
пользования полуп]
ных производств, та
переработку природных ма'
)ЖНОСТИ и с -
j р а злич-
х« иа<й ехяолсгЕчесхого
17
Применение концентрированного сырья означает
применение сырья, в котором велико содержание (концентрация)
полезных компонентов. Использование такого сырья упрощает
и удешевляет его транспортировку и переработку, позволяет прово-
дить реакции с большей скоростью, большим выходом и высоким
качеством продукта. Для увеличения концентрации сырья чаще
всего его подвергают обогащению на так называемых обогатитель-
ных фабриках или в процессе получения из него продуктов.
Комплексное использование сырья — это исполь-
зование всех составных частей сырья для производства различных
продуктов и материалов. В этом случае нет отходов производства:
все, что содержится в сырье, используется. Примерами могут слу-
жить: переработка нефти, угля, природного газа, воздуха, поварен-
ной соли, серусодержащих руд, фосфоритов и апатитов. При этом
сокращается расход сырья й снижается себестоимость продуктов.
Так, при коксовании угля, кроме целевого продукта — металлурги-
ческого кокса — получают коксовый газ и смолу, переработка кото-
рых дает сотни ценных веществ: ароматические углеводороды,
фенолы, пиридин, аммиак, водород, этилен и др. Применение ука-
занных веществе в качестве продуктов народного хозяйства привело
к снижению себестоимости кокса.
Комплексное использование сырья тесно связано с таким важ-
ным направлением развития химической техники, как комбинирова-
ние предприятий — наиболее прогрессивной и экономичной формой
организации химического производства. Особенно это важно для
вновь организуемых производств на базе использования отходов
производств. При этом на 60—70 % сокращаются капиталовложе-
ния на общезаводское хозяйство (складирование материалов, тран-
спорт и т. д.), снижается себестоимость продукции. При комплекс-
ном использовании сырья решается также и экологическая проб-
лема, так как отсутствие отходов сохраняет чистоту окружающей
среды и исключает расход на строительство очистных сооружений.
Замена пищевого сырья непищевым важна для решения
Продовольственной программы, поскольку позволяет высвободить
пишевые продукты для нужд населения. Себестоимость продуктов,
полученных из непищевого сырья, ниже, чем из пищевого. Расти-
тельное и животное пищевое сырье уже в основном вытеснено син-
тетическим.
Выбор сырья играет значительную роль в организации производ-
ства, в выборе наиболее экономичного способа производства,
в решении вопросов экономного и рационального использования
природных ресурсов и решении проблемы охраны окружающей
среды.
2.1.2. Принципы обогащения сырья
Природное сырье, как правило, кроме основного компонента,
содержит примеси. Поэтому сырье подвергают обогащению с целью
повышения в нем концентрации основного компонента. Значение
18
Рис. 2.1. Принципиальная схема мокрого гравитационного обогащения:
1 — 3—осадительные камеры.
применения концентрированного сырья описывалось выше (см.
разд. 2.1.1).
Обогащение сырья чаще производят в местах добычи- сырья
на обогатительных фабриках для уменьшения транспортных расхо-
дов на перевозку его к месту переработки.
Методы обогащения твердых материалов основаны на различии
физических и химических свойств составляющих компонентов
сырья: плотности, твердости, растворимости, температур плавления
и возгонки, электрической проводимости, магнитной проницае-
мости, смачиваемости отдельными жидкостями и т. п.
Ниже приведены обычно используемые методы обогащения.
Рассеивание (грохочение) основано на различной
крупности зерен входящих в состав сырья минералов.
При дроблении менее прочные (хрупкие) минералы образуют
более мелкие зерна. После просеивания смеси через сита с отвер-
стиями разных размеров происходит разделение на фракции, обо-
гащенные определенным минералом. Применяемые для рассеива-
ния аппараты называют грохотами. Их снабжают механизмами
для сотрясения, вибрации или качания.
Гравитационное обогащение (мокрое и сухое) осно-
вано на различии скоростей падения частиц, имеющих разную
плотность или крупность, в потоке жидкости или газа или на дей-
ствии центробежной силы. Чаще всего проводят мокрое обогаще-
ние в потоке воды (рис. 2.1). Измельченное сырье смешивают
с водой в баке с мешалкой и в виде суспензии (взвесь твердого
материала в жидкости) подают в'осадительные камеры 1—3. Так
как ширина камер постепенно увеличивается, происходит замедле-
ние скорости потока, что облегчает осаждение твердых частиц.
Самые легкие и мелкие частицы (обычно пустая порода) уносятся
из камер потоком воды.
Сухое гравитационное обогащение применяют для сортировки
материала после измельчения. Схема воздушного сепаратора цен-,
тробежного типа представлена на рис. 2.2. Тонкоизмельченный
материал подается на тарелку 2 и разбрасывается по сечению
внутреннего конуса 3. Мелкие частицы увлекаются вверх потоком
воздуха, создаваемым вентилятором, выбрасываются в наружный
19
Измельченный материал
Мелкие частице/
Рис. 2.2. Схема воздушного сепаратора:
/—крылатка вентилятора; 2—вращающаяся тарелка; 3—вну-
тренний конус; 4 —наружный конус.
конус 4, опускаются по его стенкам вниз и выводятся в виде мелких
верен. Крупные частицы падают вниз и выводятся из внутреннего
цилиндра. Воздух циркулирует в сепараторе.
Электромагнитное и электростатическое обо-
гащение основано на различии магнитной проницаемости или
электрической проводимости компонентов сырья. Эти способы при-
меняют для разделения магнитовосприимчивых частей от немаг-
нитных и электропроводящих от диэлектриков. Разделение осуще-
ствляют в электромагнитных и электростатических сепараторах,
имеющих сходный принцип действия. Так, в электромагнитном
сепараторе (рис. 2.3) в барабан 2 ленточного транспортера вмон-
тирован электромагнит 3. При прохождении измельченного сырья
магнитные частицы задерживаются на ленте 1, пока лента не вый-
дет из поля действия магнита, а затем падают в соответствующий
бункер; немагнитные частицы попадают в бункер для немагнитной
фракции.
Флотация — широко распространенный способ обогащения,
основанный на различной смачиваемости зерен отдельных минера-
лов водой. Частицы несмачиваемого (гидрофобного) минерала 1
будут как бы вдавливаться в жидкость (рис. 2.4), но, не преодолев
сил поверхностного натяжения, оставаться на ее поверхности,
тогда как частицы смачиваемого (гидрофильного) материала 2
Рис. 2.4. Влияние смачивания.
Рис. 2.3. Схема электромагнитного сепаратора:
I—лента транспортера; 2—барабан транспортера; 3— электро-
магнит; 4, 5—бункеры. ~
20
Рис. 2.5. Флотационная машина с воз-
душным перемешиванием:
/ — циркуляционная камера; 2—коллектор воз-
духа*. 3—желоб для концентрата; 4—воздушные
трубки.
обволакиваются пленкой жидкости и, преодолев силы поверхност-
ного натяжения жидкости, опускаются на дно аппарата. Частицы
несмачмваемого материала снимаются с поверхности жидкости,
и таким образом происходит разделение руды на фракции. Для
ускорения флотации создают условия неодинаковой смачиваемости
водой отдельных компонентов породы, для чего применяют различ-
ные химические соединения — флотационные реагенты. Они изби-
рательно усиливают или ослабляют смачиваемость воды, а также
прилипаемосгь к пузырькам воздуха взвешенных минеральных
частиц. Для флотации применяют флотационные машины двух
типов: камерные с механическим перемешиванием суспензии с воз-
духом и корытные с пневматическим (воздушным) перемешива-
нием (рис. 2.5).
Термическое обогащение твердого сырья основано
иа различии в плавкости компонентов сырья. Например, нагрева-
нием серусодержащей породы отделяют легкоплавкую жидкую
серу от пустой породы, состоящей из более тугоплавких известня-
ков, гипса и др.
Жидкие растворы различных веществ концентрируют различ-
ными методами: выпариванием растворителя, до н ас ы-
щением раствора полезным компонентом, вымо-
раживанием, осаждением примесей или выделе-
нием их в газовую фазу.
Газовые смеси разделяют разными методами: конденса-
цией, ректификацией, абсорбцией и адсорбцией,
растворением. Так, из коксового газа газообразный водород
выделяют при сжижении всех остальных компонентов, а сероводо-
род растворяют в растворе этаноламина с последующим нагрева-
нием раствора и выделением H2S в газовую фазу.
2.2. ВОДА И ВОЗДУХ В ХИМИЧЕСКОЙ ПРОМЫШЛЕННОСТИ
Воду и воздух широко используют в химической промышлен-
ности благодаря их доступности, дешевизне и ряду ценных свойств.
2.2.1. Вода в химической промышленности
Вода обладает универсальными свойствами, благодаря чему
применяется в качестве сырья, химического реагента, растворителя,
тепло- и хладоносителя, в некоторых случаях как катализатор.
21
Например, из воды получают водород, применяемый в производ-
стве спиртов, анилина, моющих средств и т. п. Вода служит реаген-
том для получения кислот, щелочей и оснований, различных орга-
нических продуктов — спиртов, уксусного альдегида, фенола, в ре-
акциях гидратации- и гидролиза. Воду применяют в качестве
растворителя твердых, жидких и газообразных веществ. Как тепло-
носитель и хладоагент вода используется для проведения экзо- и
эндотермических процессов. Горячая вода и водяной пар имеют
значительные преимущества перед другими теплоносителями: высо-
кую теплоемкость и термическую стойкость, простоту регулирова-
ния температуры в зависимости от давления и пр.
В целях экономии воды применяют так называемую оборотную
воду, т. е. многократно использованную и возвращенную в произ-
водственный цикл. Для экономии воды как тепло- и хладоносителя
ее заменяют воздухом.
Природные воды подразделяют на атмосферные, поверхностные
и подземные.
Атмосферная вода — дождевые и снеговые, осадки — ха-
рактеризуется небольшим содержанием примесей в виде растворен-
ных газов. В этой воде почти полностью отсутствуют растворенные
соли.
Поверхностные воды — реки, озера и моря — отличаются
разнообразным составом примесей, содержат газы, соли, основания,
кислоты. Наибольшее содержание минеральных примесей в мор-
ской воде (солесодержание более 10 г/кг).
Подземные воды — колодезные, ключевые, артезианские —
характеризуются различным составом растворенных солей, который
зависит от состава и структуры почв и горных пород. В подзем-
ных водах обычно отсутствуют примеси органического проис-
хождения.
Морская вода и вода буровых скважин является богатым источ-
ником сырья химической промышленности.
Основные показатели качества воды: жесткость, общее солесо-
держание^ прозрачность, окисляемость и реакция воды.
Жесткость воды бывает временная, постоянная и общая.
Временная (устранимая) жесткость обусловлена наличием в воде
гидрокарбонатов кальция и магния. Эти соли сравнительно легко
удаляются из воды при кипячении: Са(НСО3)2 —► СаСО3 + Н2О +
-Ь СО2.
Постоянная жесткость обусловлена присутствием в воде хлори-
дов, сульфатов, нитратов кальция и магния. Эти соли при кипяче-
нии из воды не удаляются. Временная и постоянная жесткость
дают в сумме общую жесткость, измеряемую количеством ионов
кальция или магния, содержащихся в виде солей в 1 кг воды.
Общее со песодержание, или сухой остаток, — это
масса вещества, остающаяся после испарения воды и высушивания
полученного остатка при 105—110°С в виде минеральных и органи-
ческих примесей.
22
Прозрачность измеряется толщиной слоя воды, через кото-
рый можно различить визуально или при помощи фотоэлемента
изображение стандартных знаков.
Окисляемость воды характеризуется наличием в ней орга-
нических примесей и выражается массой кислорода, расходуемого
на окисление веществ, содержащихся в 1 кг воды.
Реакция воды — кислотность и щелочность воды опреде-
ляются концентрацией ионов водорода или значением pH. Реакция
природных вод близка к нейтральной; pH колеблется в пределах
6.8—7,3.
Потребляемые воды в зависимости от назначения условно под-
разделяют на промышленные и питьевые.
Питьевые воды в первую очередь освобождаются от бак-
терий; к ним предъявляют особые требования в отношении вкуса,
цвета, запаха. Эти требования установлены соответствующим стан-
дартом.
Промышленные зоды не должны содержать большого
количества растворенных солей и механических примесей.^ Макси-
мально допустимее содержание примесей регламентируется соот-
ветствующими ГОСТами, в зависимости от качества получаемых
продуктов. Грубодисперсные, механические взвеси засоряют трубо-
проводы и аппараты, уменьшая их производительность, образуют
пробки, которые могут вызвать аварию.
Огромный вред приносят растворенные в воде соли и газы, вы-
зывающие образование накипи и поверхностное разрушение метал-
лов вследствие коррозии. Накипь ухудшает теплообмен, приводит
к перегреву труб, что влечет за собой преждевременный их износ.
В текстильной промышленности соли, растворенные в воде, приво-
дят к большому перерасходу мыла в процессах мойки. Потери
мыла в жесткой воде достигают 80 %.
Природную воду, поступающую в произвэдстзо, подвергают
очистке различными методами в зависимости от характера приме-
сей и требований, предъявляемых к воде данным производством.
Промышленная водоподготовка представляет собой комплекс
операций, обеспечивающих очистку воды от механических приме-
сей, растворенных солей и газов. Основными операциями явля-
ются: очистка от взвешенных примесей отстаиванием и фильтра-
цией, умягчение и обессоливание воды, дегазация, обеззаражи-
вание.
Отстаивание от крупных частиц осуществляют в непрерыв-
но действующих отстойниках большой емкости. От мелких частиц
освобождаются фильтрацией. Для осаждения коллоидных
частиц (мельчайших глинистых частиц и белковых веществ) их под-
вергают коагуляции путем введения коагулянтов — сульфатов или
двойных солей алюминия (алюмокалиевые квасцы). Ион-коагулянт
должен иметь заряд, противоположный заряду коллоидной части-
цы, чтобы он мог адсорбировать на поверхности заряженной
частицы; это приводит к слипанию (коагуляции) отдельных частиц
и образованию осадка.
2»
Умягчение и обессоливание воды — это основные
процессы водоподготовки, которые состоят в удалении солей каль-
ция, магния и других металлов. Различают физические, физико-
химические и химические способы умягчения воды.
Физические способы — термический (кипячение), дистилляция
и вымораживание. Термическим способом удаляют соли времен-
ной жесткости. Дистиллированную воду, не содержащую солей,
получают перегонкой на специальных дистилляционных установках.
Она необходима для приготовления химически чистых реактивов,
лекарственных препаратов, в лабораторной практике и т. п. Вымо-
раживание основано на различии температур кристаллизации воды
и примеси.
Физико-химические способы — электрохимические, основанные
на применении электродиализа, электроосмоса, электрокоагуля-
ции, и ионитовые (ионообменные), получившие широкое распро-
странение. '
Ионообменные способы основаны на удалении из воды ионов
кальция и магния при помощи ионитов (труднорастворимых твер-
дых веществ), способных обменивать свои ионы на ионы, содержа-
щиеся в воде. Различают процессы катионного и анионного обмена,
соответственно иониты называют катионитами и анионитами.
В основе катионного процесса умягчения лежат реакции обмена
ионов натрия и водорода китионитов на ионы Са2+ и Mg2+. Обмен
ионов натрия называется Na-катионированием, а ионов водорода —
Н-катионированием:
Na2 [Кат] + Са(НСО3)2 Са [Кат] + 2NaHCO3
Na2 [Кат] + MgSO4 Mg [Кат] + Na2SO4
H2 [Кат] + MgCl2 ч=* Mg [Кат] + 2НС1
Н [Кат] + NaCl Na [Кат] + НС1
Приведенные реакции показывают, что ионообменным спосо-
бом достигается полное обессоливание воды (умягчение и обессо-
ливание).
Реакции ионообмена обратимы, и для восстановления обменной
способности ионитов проводят процесс регенерации. При помощи
растворов поваренной соли осуществляют регенерацию Na-катиони-
тов, при помощи минеральных кислот — Н-катионитов.
Са [Кат] + 2NaCl Na2 [Кат] + СаС12
Na [Кат] + НС1 Н [Кат] + NaCl
Примером анионного обмена может служить реакция обмена
анионов ОН- по уравнению:
[Ан]ОН + НС1 =г=>= [Ан]С1 + Н2О
Регенерацию анионита проводят при помощи растворов
щелочей:
[AH]Cl+NaOH [Ан]ОН + NaCl
Па рис. 2.6 представлена схема обессоливания воды с приме-
нением ионообмена (катионирование и аннонирование). Вода сна-
24
Рис. 2.6. Схема установки для обессоливания воды с применением ионообмена:
1 — катионнтовый фильтр; 2—анноннтовый фильтр; 3—дегазатор; 4—сборник воды.
чала освобождается от ионов кальция, магния и натрия в Н-катио-
нитовом фильтре 1 (рис. 2.7), где на слое крупного кварцевого
песка расположены зерна катионита. Затем вода поступает в анио-
нитовый фильтр 2 для удаления анионов и далее в дегазатор Зг
где происходит выделение растворенных в воде СО2, О2.
Из электрохимических способов значительный интерес представ-
ляет электрокоагуляция. Сущность ее состоит в получении электро-
химическим путем гидроксида алюминия, обладающего высокой
сорбционной способностью по отношению к вредным примесям.
Процесс осуществляют в электролизерах. К достоинствам способа
электрокоагуляции относят: высокую сорбционную способность
электрохимического А1(ОН)з, возможность механизации и автома-
тизации процесса, малые габариты очистных сооружений.
Химические способы умягчения воды заключаются в обработке
ее растворами некоторых химических соединений с целью связыва-
ния ионов Са2+, Mg2+ и других в нерастворимые и легко удаляе-
мые соединения.
Дегазация — это удаление из воды растворенных газов
СО2, О2. Газы, содержащиеся в воде, представляют опасность из-за
образования воздушных пробок, которые могут привести к наруше-
нию технологического процесса. Растворенные газы могут вызывать
коррозию аппаратов и трубопроводов. Дегазацию проводят хими-
ческим и физическим способами. Например, для удаления СО2
воду пропускают .через фильтр, заполненный гашеной известью,
или добавляют к воде известковое молоко: СО2 + Са (ОН )2 =
= СаСОз| + Н2О. Для удаления О2 применяют фильтр, заполнен-
ный железными опилками, стружками.
Рис. 2.7. Катионнтовый фильтр
1 — катионит, 2—песок (SlOj).
25
Физические способы дегазации сводятся к частичному удалению
газов из воды при ее нагревании паром или нагревании воды
в вакууме.
Обеззараживание воды, используемой для бытовых нужд,
производят с целью уничтожения болезнетворных бактерий и окис-
ления органических примесей. Обеззараживание осуществляют
хлорированием (газообразным хлором), а также хлорной известью
и гипохлоритом кальция.
2.2.2. Воздух в химической промышленности
Воздух в химической промышленности применяют в качестве
сырья, реагента, а также для энергетических целей. Его широкое
применение обусловлено химическим составом. Сухой чистый воз-
дух содержит, % (по объему): N2—78,10, О2— 20,93, Аг — 0,94,
СО2 — 0,03, незначительные количества Не, Ne, Кг, Хе, Н2, СН4,
Оз, NO.
Кислород воздуха часто применяют в качестве окислителя:
при обжиге сульфидных руд в производстве цветных металлов, при
обжиге серного колчедана в производстве серной кислоты, при
неполном окислении углеводородов в производстве спиртов, альде-
гидов, кислот и др.
В качестве сырья воздух используют для получения азота, кис-
лорода и инертных газов. Азот служит сырьем для получения
аммиака, который, в свою очередь, широко применяют для произ-
водства всех азотсодержащих солей. Кислород, выделенный из воз-
духа, в больших количествах расходуют для кислородной плавки
металлов, в доменном процессе и т. п.
Энергетическое применение воздуха связано прежде всего
с использованием кислорода как окислителя для получения тепло-
вой энергии при сжигании различных топлив. Воздух является
хладо- и теплоносителем в теплообменных процессах. В пневмати-
ческих барботажных смесителях сжатый воздух используют для
перемешивания жидкостей и суспензии, в форсунках — для распы-
ления жидкостей в реакторах и топках.
2.3. ЭНЕРГЕТИКА ХИМИЧЕСКОЙ ПРОМЫШЛЕННОСТИ
Энергетическая обеспеченность — необходимое условие разви-
тия и технического прогресса любой промышленности. Химико-
технологические процессы протекают или с выделением (экзо-
термические процессы), или с поглощением (эндотер-
мические процессы) теплоты. В первом случае необходимо
рационально использовать выделяющуюся энергию и тем самым
обеспечивать повышение экономичности производства. Часто теп-
лота экзотермических процессов не только компенсирует затраты
энергии на осуществление основных и вспомогательных операций,
но и используется другими производствами. При проведении эндо-
26
термических процессов важно правильно выбрать источник энергии
и организовать подвод теплоты в зону реакции. Раздел науки,
занимающийся изучением оптимального сочетания химической тех-
нологии и ее энергетики, в том числе и особенностей работы энер-
гетического оборудования, называют энерготехнологией.
Химико-технологические системы, обеспечивающие требуемый
выпуск продукции при рациональном использовании энергии, назы-
вают энерготехнологическими. Показателем эффектив-
ности использования энергии является ее расход на единицу полу-
чаемой продукции. Расход энергии измеряют в единицах энергии
(кДж, кВт-ч) или определяют массу топлива, затраченного на про-
изводство единицы массы или объема продукта (кВт-ч/т, кДж/кг,
кг/м3 и т. п.).
В последние годы эффективность производства продуктов
в химической промышленности и других отраслях характеризуют
так называемым топливным эквивалентом, под которым
понимают совокупность сырьевых и энергетических затрат, соот-
ветствующую по теплотворной способности эквивалентному коли-
честву топлива (например, нефти). Для условного топлива, тепло-
творная способность которого принята равной 29,3-10® кДж/т, топ-
ливные эквиваленты ряда химических продуктов (в тоннах услов-
ного топлива на тонну продукции) составляют: этилен — 4,1; про-
пилен— 5,1; стирол — 9,7; дивинил — 10,3; изопрен— 12,1; полиэти-
лен — 5,5; аммиак— 18; фосфор — 35.
2.3.1. Основные источники энергии
Отечественные химические и нефтехимические производства
весьма энергоемки. Эти производства, выпуская около 7 % промыш-
ленной продукции, потребляют около 20 % энергии, расходуемой
всей промышленностью.
В зависимости от особенностей химико-технологических процес-
сов химические производства используют различные энергоресурсы:
топливо (уголь, мазут, природный газ), электрическую энергию
и пар (получаемый с ТЭЦ и вырабатываемый на самом предприя-
тии из различных видов топлива или при использовании теплоты
экзотермических процессов). Соотношение важнейших потребляе-
мых энергоресурсов в химических и нефтеперерабатывающих про-
изводствах СССР приведено ниже:
_ В иефтепере-
В химических рабатывающих
Вилы виергоресурсов произведет- произввдст-
вех, % вах,
Топливо всех видов при 31,5 38,7
прямом использовании
Электрическая энергия 3.8,5 15,2
Пар, горячая вода 30,0 46,1
в том числе, получа- 22,0 38
емые со стороны
Как видно из этих данных, в химических производствах более
2/з энергетических ресурсов приходится на электрическую энер-
гию и энергию пара.
27
Электрическую энергию применяют для проведения электротер-
мических (нагрев, плавление, разложение, синтез при высоких тем-
пературах и т. д.) и электрохимических (электролиз растворов
и расплавов) процессов. На всех химических предприятиях значи-
тельные затраты электрической энергии приходятся на обеспечение
работы электродвигателей. Средний расход электрической энергии
на производство некоторых химических продуктов приведен ниже:
Продукт
Расход электроэнергии,
кВт-ч/т
Алюминий
Фосфор
Карбид кальция
Аммиак
Хлор
Серная кислота
Аммиачная селитра
Суперфосфат
18 000—20 000
13 000—20 000
3 000—3 500
3 000—3 500
2 300—3 500
60—100
7—15
2—10
Оснрвное количество электрической энергии (около 78 % ) полу-
чают от тепловых электростанций. При этом расходуется примерно
20—22 % добываемого топлива. Доля угля, потребляемого на выра-
ботку электроэнергии, составляет около 38 %, мазута — 30 %, при-
родного газа — 27 %. Оценка запасов этих источников представлена
в разд. 2.1.1.
Примерно 12 % электрической энергии поставляется гидроэлек-
тростанциями, а 8 % приходится на долю атомных электростанций.
Запасы урановых руд, на которых базируется ядерная энергетика,
по своим энергетическим возможностям существенно превышают
потенциальную энергию разведанных запасов органического топ-
лива. В будущем все шире будут использоваться для выработки
электрической энергии возобновляемые источники — энергия солн-
ца, ветра, приливов, геотермальные воды.
Тепловая энергия в виде пара или горячей воды поступает
от ТЭЦ, работающих на угле, мазуте или природном газе, а также
от котельных установок, использующих теплоту продуктов реакций,
главным образом экзотермических процессов.
Горячую воду и пар низкого давления (до 0,6 МПа) в химиче-
ской промышленности применяют для процессов нагрева, плавле-
ния, сушки, выпарки, дистилляции и т. п. Пар высокого давления
(более 4,0 МПа) во многих случаях непосредственно используют
для привода компрессоров, обеспечения работы насосов и т. д.
Средний расход пара (т/т) на производство некоторых химических
продуктов составляет: винилацетата — 7, едкого натра — 6, суль-
фата аммония — 5.
Существенную часть топлива используют непосредственно для
обеспечения проведения технологических процессов, в частности
для целей пламенного обогрева в процессах сушки, термообра-
ботки, выпарки и т. п. Большое значение в этом плане имеет
природный газ, который не только служит для энергетических
нужд, но и является химическим сырьем.
2.3.2. Энергетическая программа СССР
и химическая промышленность
Энергетика народного хозяйства, в частности химической про-
мышленности, развивается в соответствии с Энергетической про-
граммой СССР. Программа определяет главные направления
и важнейшие мероприятия по расширению энергетической базы
и совершенствованию топливно-энергетического комплекса страны
до 2000 года,. Среди мероприятий, намеченных Энергетической
программой, многие имеют прямое отношение к химической про-
мышленности и должны быть реализованы при участии химической
науки и химических производств.
, Основные положения Энергетической программы СССР пред-
усматривают:
активную энергосберегающую политику на базе ускоренного
научно-технического прогресса во всех звеньях народного хозяйства,
всемерную экономию топлива и энергии, обеспечение на этой ос-
нове значительного снижения энергоемкости всех видов продукции.
Химическая и нефтехимическая промышленность, производя около
7% промышленной продукции, расходуют 13% всех топливно-энер-
гетических ресурсов, поэтому разработка и внедрение менее энер-
гоемких процессов в этих отраслях особенно актуальны;.
обеспечение роста ресурсов моторных топлив за счет увеличения
объема и глубины переработки нефти и организации производства
синтетических моторных топлив из газа и горючих сланцев. Это
ставит перед химической наукой и химической промышленностью
неотложные задачи разработки новых катализаторов для перера-
ботки нефти, технологии переработки новых видов .топлив, эконо-
мически эффективных методов производства жидких топлив из
угля;
форсированное развитие ядерной энергетики для производства
электрической и тепловой энергии и освобождение на этой основе
значительного количества органического топлива. Здесь перед хи-
миками стоит задача создать комплексные технологические
системы покрытия энергетических расходов при осуществлении
эндотермических реакций с помощью ядерной энергии.
* Химические производства потребляют большое количество те-
плоты (свыше 1000 млн. ГДж). Наибольшее потребление энергии
в химической промышленности приходится на долю азотной, хлор-
ной, содовой отраслей, на производство химволокон и фосфора.
Основная задачи технологов — это правильный выбор и .осуществ-
ление различных энергосберегающих мероприятий.
2.3.3. Экономия энергии в химической промышленности
Основные принципы энергосберегающей технологии в химиче-
ских производствах сводятся к совершенствованию технологии,
улучшению использования энергоресурсов, в организации энерго-
сберегающей политики.
29
Решение задачи совершенствования технологии с целью энерго-
сбережения можно разделить на следующие конкретные направле-
ния: выбор оптимального вида сырья; применение более эффектив-
ных катализаторов; использование менее энергоемких методов
выделения готовой продукции; применение энерготехнологического
комбинирования и энергосберегающего оборудования и улучшение
его эксплуатации.
Влияние качества с ы р ь-я на расход энергии заклю-
чается в том, что в химической промышленности один и тот же
конечный продукт может быть получен из различных видов сырья,
а каждая схема производства характеризуется своим расходом
энергоресуреов. Так, при получении аммиака на основе газифика-
ции полукокса расход электроэнергии составлял 1780 кВт-ч на
1 т азота, а при использовании современных агрегатов, работаю-
щих на природном газе, эта величина не превышает 100 кВт-ч.
Применяемое сырье должно обеспечивать наименьшее число стадий
переработки в конечный продукт при высокой степени использова-
ния сырья и теплоты реакций.
Повышение активности к а т а л и з а т о р а, его избира-
тельности и стабильности также приводйт к понижению энерго-
затрат. Так, в каталитическом риформинге повышение активности
катализатора на 1% обеспечивает снижение энергозатрат на 2,9%.
Переход на новые, более активные и стабильные катализаторы
в производстве ароматических углеводородов снижает удельные
энергозатраты на 1 т продукции на 0,26 т условного топлива.
Выделение и очистка готовой продукции пред-
ставляют одну из наиболее энергоемких стадий химических про-
изводств. Например, на долю ректификации приходится около
28% суммарных энергозатрат в химической и нефтеперерабатыва-
ющей промышленности. Замена ректификации менее энергоем-
кими методами разделения, такими, как экстракция, адсорбция,
мембранное разделение, может существенно снизить энергозат-
раты.
Энерготехнологическое комбинирование может
выполняться на уровне процессов (проведение экзо- и эндотермиче-
ских процессов в одном объеме), технологических установок (теп-
лота продуктов одной установки используется для обеспечения
работы другой установки) и крупных производств (сочетаются
энерговыделяющие производства с энергопотребляющими). Так,
использование для нагрева шихты теплоты отходящих газов фос-
форных печей снижает расход электроэнергии на 15—20%, кокса —
на 25%.
Совершенствование использования энергоресурсов в существую-
щих химических производствах предусматривает улучшение ис-
пользования первичных и утилизацию вторичных энергоресурсов
(ВЭР). ВЭР — это энергетический потенциал конечных, побочных
и промежуточных продуктов, а также отходов, образующихся в тех-
нологических агрегатах, который может быть использован для
энергоснабжения других агрегатов. Утилизация ВЭР представляет
30
Продукты реакции
Рис. 2.8. Принцип рекуперативного использования теплоты продуктов реакции или
отходящих газов:
I —теплообменник; 2—реакционный аппарат.
Рис. 2.9. Схема работы регенератора:
1.3— регенераторы: 2—насадка регенератора; 4—заслонка.
важнейшую задачу для химических производств, так как в ВЭР
пеоеходит значительная часть потребляемой электроэнергии, топ-
лива и тепловой энергии пара. Только в азотной промышленности
ВЭР превышают 15 млн. т условного топлива в год.
В простых случаях теплота продуктов реакции или отходящих
газов может быть использована для предварительного нагрева ма-
териалов, поступающих в реакционные аппараты. Принципиальная
схема использования теплоты продуктов реакции представлена на
рис. 2.8. В этом примере теплообмен между горячими продуктами
и холодными исходными реагентами происходит через стенки тру-
бок теплообменника.. Такие теплоиспользующие аппараты назы-
вают -рекуператорами.
Теплоту продуктов реакции высокотемпературных процессов
используют также с помощью регенераторов — периодически дей-
ствующих камер, заполненных насадкой из жаропрочного матери-
ала. Схема работы регенератора показана на рис. 2.9. Газы с высо-
кой температурой проходят через насадку, нагревая ее. После
этого через нее пропускают холодные газы, которые нагреваются,
отбирая теплоту от насадки. Для создания непрерывного процесса
устанавливают два и более регенератора с автоматическим управ-
лением заслонками, перекрывающими движение горячих или холод-
ных газов.
Основным направлением применения высокотемпературных
ВЭР, особенно при наличии больших количеств, является их ис-
пользование в котлах-утилизаторах для получения водяного пара.
В настоящее время в химической и нефтеперерабатывающей про-
мышленности работает более 1000 котлов-утилизаторов, в которых
вырабатывается около 100 млн. ГДж теплоты.
В процессах, протекающих при высоких давлениях, для сниже-
ния расхода электрической энергии, преобразуемой в механиче-
скую, стремятся использовать энергию сжатых газов, находящихся
под давлением. На рис. 2.10 показана схема энергосберегающего
31
Рис. 2.10. Схема агрегата «электродвигатель — насос — турбина»:
1—реакционный аппарат; 2—электродвигатель; 3—турбина; 4 — иаСос.
Рис. 2.11. Использование системы утилизации горючих ВЭР с включением газовой
турбины: '
1 — топка; 2—газовая турбина; 3—вал; 4—воздушный компрессор; 5 — парогенератор.
агрегата «электродвигатель — насос — турбина». Газ под давле-
нием поступает в нижнюю часть реакционного аппарата 1 и кон-
тактирует с орошающей его жидкостью. Выходящая снизу жид-
кость п< ддавлением поступает на турбину 3 и вращает ее рабочее
колесо. Турбина вращает рабочие колеса многоступенчатого насоса
4, которые находятся на одном валу с колесом турбины. Насос
подает жидкость на орошение турбины. Потери энергии компенси-
руются питанием электрической энергией двигателя. По такой же
схеме может использоваться и потенциальная энергия сжатого
газа, выходящего из реактора.
Большой интерес представляет применение систем, вырабатыва-
ющих за счет использования ВЭР горючих отходящих газов одно-
временно электроэнергию, пар и сжатый воздух. Пример такой
схемы утилизации приведен на рис. 2.11.
ГЛАВА 3
ОСНОВНЫЕ ХАРАКТЕРИСТИКИ
ХИМИКО-ТЕХНОЛОГИЧЕСКИХ ПРОЦЕССОВ
В современной химической промышленности используют де-
сятки тысяч способов переработки сырья. Каждый способ состоит
из нескольких операций, выполняемых последовательно или парал-
лельно в соответствующих машинах и аппаратах. В общем случае
каждая операция представляет собой сочетание нескольких эле-
ментарных физических, физико-химических и химических процес-
сов. Совокупность элементарных процессов, включающих химиче-
скую реакцию, называют химико-технологическим про-
цессом (ХТП). Химико-технологический процесс, как правило,
протекает в несколько последовательных стадий: 1) подвод реаген-
тов в зону реакции; 2) химическая реакция; 3) отвод из зоны
реакции полученных продуктов.
82
Все многообразие современных способов химической перера-
ботки сырья в продукты потребления и средства производства
можно описать через относительно ограниченное число химико-тех-
нологических процессов. Для выделения химико-технологических
процессов, характеризуемых одинаковыми закономерностями про-
текания, все ХТП классифицируют по нескольким признакам. Для
удобства использования классификации инженерно-техническими
работниками число признаков должно быть невелико и отвечать
практическим задачам химической технологии, к которым, в част-
ности, относится выбор и управление технологическим режимом.
Технологическим режимом называют совокупность ос-
новных факторов (параметров), влияющих на интенсивность ра-
боты аппаратов и качество получаемого продукта.
3.1. КЛАССИФИКАЦИЯ ХИМИКО-ТЕХНОЛОГИЧЕСКИХ ПРОЦЕССОВ
В химической технологии за основу классификации химико-тех-
нологических процессов обычно принимают их физико-химические
особенности, обеспечивающие правильный выбор основных факто-
ров интенсификации производств. В этом плане наиболее важной
является характеристика агрегатного состояния реагирующих ве-
ществ. По такому признаку различают гомогенные (одно-
родные) процессы, когда все взаимодействующие вещества
находятся в одном из трех возможных агрегатных (фазовых)
состояний, и гетерогенные (неоднородные) процессы,
в которых реагирующие вещества Первоначально находятся в раз-
личных агрегатных состояниях.
Если реагенты перед вступлением в реакцию хорошо переме-
шать, то скорость гомогенных процессов определяется скоростью
непосредственно химического превращения веществ. В гетероген-
ных процессах химические реакции обычно сопровождаются чисто
физическими промежуточными стадиями, которые определяют или
влияют на наблюдаемую скорость процесса. В простейшем случае
при протекании гетерогенного химико-технологического процесса
можно выделить два элементарных процесса — диффузию веществ,
находящихся в одной фазе, к поверхности раздела фаз или от нее
и химическую реакцию внутри одной из фаз. В зависимости от
того, какой из элементарных процессов — диффузия или реакция —
определяет скорость ХТП, последние разделяют по области проте-
кания. Например, если определяющее значение на скорость ХТП
оказывает скорость диффузии, то говорят о химико-технологических
процессах, протекающих в дйффузионной области; если
скорость ХТП определяется скоростью химической реакции, то про-
цесс протекает в кинетической области. Методы и приемы
интенсификации ХТП, протекающих в диффузионной и кинетиче-
ской областях, совершенно различны. Знание области протекания
процесса особенно важно для анализа гетерогенно-каталитических
процессов и управления ими.
2 Зак. 1869
33
Для правильного выбора теплового режима проведения ХТП
весьма существенно, с каким тепловым эффектом протекает реак-
ция. Поэтому ХТП разделяют на экзотермические (с выделе-
нием теплоты) и эндотермические (с поглощением теплоты).
Для выбора рациональных температур проведения ХТП необхо-
дима информация об обратимости химической реакции. По этому
признаку выделяют обратимые и практически необра-
тимые ХТП, а также процессы со сложными параллельными или
последовательными реакциями.
Перечисленные признаки — фазовое состояние реагирующих ве-
ществ, характеристики теплового эффекта и обратимости реакции,
а также области протекания процессов — являются основными,
позволяющими выделять однотипные ХТП с одинаковыми законо-
мерностями управления технологическим режимом.
3.1.1. Уровни анализа, описания и расчета ХТП
Сложный химико-технологический процесс представляет собой
совокупность элементарных физических, физико-химических и хи-
мических процессов. Число элементарных процессов, их характе-
ристики и последовательность определяют структуру ХТП в целом.
В подавляющем большинстве практических случаев структура хи-
мико-технологических процессов сложна и очень трудно правильно
выбрать технологический режим их проведения. Действительно,
каждая реакция (а при проведении ХТП могут протекать одновре-
менно несколько реакций) характеризуется своим типом влияния
основных параметров — температуры, давления, концентрации.
Кроме того, реакции сопровождаются массообменом, выделе-
нием или поглощением теплоты и соответствующим теплообменом.
Как будет показано в последующих разделах, на протекание ХТП
сильно влияет-характер движения потоков реагирующих веществ,
конструктивные особенности реакторов и другие факторы. Изуче-
ние ХТП с целью выбора рациональных технологических режимов
и управления ими проводят не в сложной совокупности элементар-
ных процессов, а по частям (уровням). Уровень — это простые
составляющие протекающего сложного процесса. Именно уровне-
вый подход дает возможность наиболее правильного и быстрого
выбора технологических режимов сложных ХТП и управления ими.
Таким образом, анализ и описание ХТП проводят последовательно
с учетом уровня протекания процесса.
Молекулярный уровень предполагает описание процес-
сов как молекулярное взаимодействие реагирующих веществ. При-
нимают, что реагирующие вещества хорошо перемешаны и их мо-
лекулы могут беспрепятственно вступать во взаимодействие друг
с другом. Протекание ХТП описывается закономерностями кине-
тики химических реакций. При описании, анализе и расчете проте-
кания химической реакции как элемента ХТП используют извест-
ные кинетические закономерности без вскрытия механизма реакции.
Поэтому говорят, что протекание процесса проводят на языке
34
формальной кинетики. В простейшем случае эти закономерности
непосредственно можно использовать для описания гомогенных
процессов, когда реагенты, вступающие в реакцию, хорошо предва-
рительно перемешаны, а также для гетерогенных процессов, про-
текающих в кинетической области в аппаратах периодического
действия.
На уровне малого объема описывается протекание гете-
рогенных процессов для малого объема взаимодействующих фаз,
например для частицы твердого материала, реагирующего с газом
или жидкостью, зерна катализатора, пузырька газа, поднимающе-
гося в жидкости, капли жидкости, омываемой газом и др. Необхо-
димость рассмотрения процессов на этом уровне связана с тем, что
для анализа и расчета гетерогенных процессов знания закономер-
ностей протекания только химических реакций в большинстве слу-
чаев недостаточно. Эти закономерности необходимо дополнить за-
кономерностями притекания физических процессов переноса массы
и теплоты. Совместное протекание химических реакций и процес-
сов переноса теплоты и массы описывается закономерностями мак-
рокинетики.
Уровень потока рассматривает протекание процесса на
совокупности твердых частиц, капель жидкости, зерен катализатора
и других разновидностях «малого объема», находящихся в потоке
реагирующих веществ. Учитываются эффекты, связанные с ха-
рактером движения потоков, изменением температур и концен-
траций в различных участках реакционного объема. Для гетеро-
генных процессов закономерности их протекания в малом объ-
еме дополняются закономерностями изменения концентрации и
температур по длине реакционной зоны, через которую проходит
поток.
Уровень реактора учитывает конструктивные особенности
реакционных зон, их число, взаимное расположение, соотношение
технологических показателей процесса при прохождении потока
из одной реакционной зоны в другую.
Уровень химико-технологической системы (ХТС)
учитывает взаимные связи между реакторами, теплообменниками,
смесителями и другими аппаратами, используемыми для перера-
ботки сырья в конечные продукты.
Последующий материал изложен с учетам рассмотренных уров-
ней анализа протекания ХТП.
3.2. ОСНОВНЫЕ ПОКАЗАТЕЛИ
ХИМИКО-ТЕХНОЛОГИЧЕСКОГО ПРОЦЕССА
Степень совершенства технологического режима количественно
оценивают с помощью показателей ХТП. К основным показателям
относят: степень превращения исходных реагентов, выход про-
дукта, скорость превращения, избирательность (селективность)
процесса, расходные коэффициенты,
,?*
35
3,2.1. Степень превращения
Совершенство процесса с позиции полного использования сырья
оценивают с помощью степени превращения (конверсии).
Степень превращения — это отношение количества
(массы, объема) одного из реагентов, вступившего в реакцию,
к начальному количеству (массе, объему) этого реагента. Напри-
мер, для реакции
аА + ЬВ = dD
степень превращения веществ X рассчитывают по соотноше-
J ниям:
ХА = (тА,а- тл)/тА. О = («А. О - «а)/«А. О = (° А. О “ МЛ’Л. О
~ (отв. о ~ тв)/та, о “ (пв. о "в)/"в. о“ (°в. о г’в)/°в. о
Здесь тА, о, тв, о, иА, о, Пв, о, va, о, Св. о — масса, число молей и объем реа-
гентов А и В в начале процесса; тА, тв, пв сА, чв — то же в конце про-
цесса.
Если реагенты А и В взяты в стехиометрическом соотношении,
то ХА = Хв. Степень превращения реагента, взятого с избытком от
стехиометрического соотношения, всегда ниже, чем для реагента,
поступающего в недостатке. Если <%в — коэффициент избытка В по
отношению к стехиометрии, то
ХА — ав-^в (3.2)
Если реакция протекает без изменения объема v, то концентра-
ция (С, моль/м3) реагента или его объемная доля (со, %) в начале
и в конце процесса равны:
СА. О = «А. о/° СА = «а/»-' “а. О “ (о . о/°) • 100
“а = №)-10° (3.3)
Для реакций, протекающих с изменением объема
ХА = (СА. О — Са)/(СА. О ~ 8АСа) (3-4)
Здесь еА — относительное изменение объема реакционной смеси при полном
превращении реагента А.
Для процессор межфазной массопередачи (испарение, конден-
сация, абсорбция, десорбция и др.) степень превращения называют
степенью межфазного перехода, например степенью
абсорбции, степенью десорбции, степенью кон-
денсации ит. п.
3.2.2. Выход продукта
Для сложных (параллельных, последовательных и др.) реакций
наряду со степенью превращения исходных веществ используют
понятие «выход продуктов».
Выход продукта Ф — это отношение фактически получен-
ного продукта Пф(или Шф} к его максимальному количеству пмак0
36
(или /иМакс), рассчитанному по стехиометрическому уравнению
реакции при степени превращения исходных реагентов, равной 1:
Фф = П4/Пмакс = 'П.1/,Пиакг (3-5)
Если в реакции участвуют два или более веществ, например
А + В -> D, то различают выходцелевого продукта Dno исходному
веществу А(ФС(Д)) и по веществу В (Фо(в>)- Если А и В> содер-
жатся в исходной смеси не в стехиометрическом соотношении, то
Фо(А) =#= Фб(В).
Для простых реакций (т. е. в отсутствие параллельных
и последовательных превращений) выход продукта числен-
но равен степени превращения исходныхвеществ.
Для простых необратимых реакций максимальный фактический
выход Фф. макс равен 1. Для обратимых реакций исходное вещество
превращается в продукт не полностью, и если в формулу (3.5)
вместо «ф подставить количество продукта, полученное в условиях
равновесия «*, то Фф. „акс < 1. Этот выход называют равновесным
ф*_-
Фж = пЧп = Ф* (3.6)
ф. макс макс ' ’
Для характеристики степени приближения обратимого процесса
к равновесию на практике используют так называемый выход от
теоретического или выход от равновесного, который
равен отношению количества фактически полученного продукта
«ф к количеству его, которое получилось бы, если процесс пришел
в состояние равновесия п*:
фт = пф/"*= фф/ф* (3-7)
Выход продукта выражают в долях единицы или в процентах.
Этот показатель во многом отражает степень совершенства техно-
логического процесса, затраты сырья на единицу продукта и эконо-
мические показатели производства.
3.2.3. Скорость ХТП
Чтобы определить время, необходимое для достижения задан-
ного выхода продукта или заданной степени превращения исход-
ного реагента, требуется знать скорость ХТП. Она определяет ин-
тенсивность протекания процессов.
Скорость хими к о-технол о г ичес к о г о процесса
выражают изменением количества какого-либо компонента, прореа-
гировавшего или образовавшегося в результате химической реак-
ции в единицу времени в единице реакционного объема v (для го-
могенных ХТП) или на единице поверхности S раздела фаз (для
гетерогенных ХТП).
Если при протекании процесса не происходит обмен веществом
с окружающей средой, средняя скорость' расходования исходного
вещества или образования продукта за интервал времени
37
Дт =' та — Ti в общем случае равна:
U = (п2 — П1)/[о (т2 — Т1)1 = tm/(v • Дт) (3.8)
Здесь nt, пг — число молей вещества в моменты времени tj и т2 соответ-
ственно; v — средний объем реагирующей смеси.
Средняя скорость равна интенсивности протекания процесса.
При Дт = О получают мгновенную, или истинную,
скорость, соответствующую конкретному моменту времени.
Мгновенная скорость ХТП выражается соотношением:
и = lim т— = ± 7— (3.9)
v • Дт v ат
Необходимо подчеркнуть, что пользоваться этим уравнением
можно при соблюдении двух условий: а) реакционный объем оста-
ется постоянным; б) объем можно считать закрытым.
Закрытым, или замкнутым, объемом считают такие
условия, когда реагирующая система не обменивается веществом
с окружающей средой. Как будет показано в последующих разде-
лах, это относится к процессам, протекающим в реакторах перио-
дического действия.
Если скорость определяют по Образующемуся продукту реак-
ции, то выражение (3.9) используют со знаком плюс, если по ис-
ходному веществу — со знаком минус. Следует помнить, что ско-
рость ХТП всегда положительна.
На практике часто пользуются более простым уравнением, при-
годным для ХТП, протекающих при постоянном объе ме. Учитывая,
чтс rit/v = С,, т. е. равно концентрации вещества, при постоянном
объеме имеем:
dC.
<ЗЛ0>
Для процессов с газообразными веществами скорость удобно
выражать через изменение парциальных давлений Pt реагирующих
компонентов смеси:
dP.
и=±-зг <ЗЛ1>
- Иногда скорость выражают через изменение степени прев-
ращения:
dX.
u" = ~dT <ЗЛ2)
Взаимосвязь между значениями U, U', U" описывается соотношением
V = U'KRT) = С{ 0U" (3.13)
Здесь R — универсальная газовая постоянная, равна 8,314 Дж/(моль-К);
Т — температура, К; Ci, о — начальная концентрация i-то компонента.
Для гетерогенных ХТП, когда реакция протекает на границе
раздела фаз, скорость часто относят к единице межфазной поверх-
38
ности S:
1 dn{
(3.14)
Для гетерогенно-каталитических процессов скорость реакции
удобно выражать в расчете на единицу массы тк катализатора:
1 dn,
= ±-----
тк dx
(3.15)
Численные значения скорости ХТП, выраженной относительно различных
реагентов, будут зависеть от соотношения соответствующих коэффициентов
в стехиометрическом уравнении. Например, для реакции
аА + ЬВ + ... -> dD + fF + ...
численные значения скорости процесса, выраженной через изменения концентра-
ций реагентов А, В, ...-, D, F ..., будут различны, т. е.:
dCA
dx
dCft dCD dCp
dx
(3.16)
Для однозначного определения скорости достаточно знать скорость образо-
вания (или расходования) какого-либо реагента, например, А:
Л?д
U=>----
dx
(3.17)
Это значение скорости связано со скоростями по другим реагентам соотно-
шением:
dC• a dCR a dC'i a dCp
и---------= ••• --тг~ (3.18)
dx b dx d dx f dx
Если скорость выражать как
U. =-----j— (для любого Z) (3.19)
* vt ах
(где Vi — стехиометрический коэффициент при соответствующем реагенте), то
она будет Иметь одинаковое значение, независимо от того, к какому из реаген-
тов ее отнести. Такое значение скорости называют инвариантным.
Скорость ХТП в общем случае зависит от скоростей физических
и химических процессов, которые в свою очередь определяются
гидродинамическими параметрами движения реагентов, условиями
перемешивания, диффузии, концентрациями, давлением, темпера-
турой и другими факторами, оказывающими влияние на массо- и
теплоперенос.
В самом общем виде формула для расчета скорости процесса
может быть представлена в виде:
t/ = -^-=K-AC (3.20)
ах
Здесь К — коэффициент скорости процесса; АС — движущая сила процесса.
Коэффициент скорости процесса зависит от области протека-
ния процесса и является функцией констант скоростей прямой
лМ» обратной (kt) и побочных (Ani, kai) реакций, коэффициентов
39
диффузии исходных реагентов к поверхности раздела фаз
(£>i, £)г, ...) и продуктов реакций от поверхности раздела
(о;, о;....):
42. ьп,.4п!. .... D,,D2, .... dJ, о;...) <З.И1>
Обычно зная область протекания процесса, технологи выбирают
основные параметры, наиболее сильно влияющие на скорость про-
цесса. В этом случае выражение для расчета коэффициента ско-
• рости процесса упрощается. Так, для гомогенных процессов или
гетерогенных, протекающих в кинетической области, К зависит
лишь от констант скоростей основной и побочных реакций:
А = /(*., k2, ka, ...) (3.22)
Для гетерогенных процессов, протекающих в диффузионной
области, определяющее влияние на скорость оказывают коэффици-
енты диффузии газа или жидкости внутри более плотной фазы (на-
пример, в порах твердого вещества):
К = f (Ц, D2. .... d'.d'^ ...) (3.23)
Движущая сила процесса в общем случае является сложной
функцией текущих (Сь С2,....) и равновесных (CJ, концен-
траций реагирующих веществ, константы равновесия Кр и других
параметров. Вид функции зависит от типа реакции, области проте-
кания процесса. В простейшем случае протекания необратимой
реакции первого порядка А->В в кинетической области
АС = СА (3.24)
Уравнение (3.20) в этом случае принимает вид:
l/=KAC = Jfe1CA (3.25)
3.2.4. Избирательность
Избирательность (селективность) — важнейшая
характеристика процессов, в которых наряду с основной реакцией
образования целевого продукта протекают побочные параллельные
или последовательные реакции с образованием нежелательных
или менее ценных продуктов. Различают конечную суммарную
(интегральную) избирательность и мгновенную (дифференциаль-
ную).
Суммарная избирательность фсум определяется как отношение
количества исходного вещества, превратившегося в целевой про-
дукт, к общему количеству прореагировавшего исходного веще-
ства. Например, для параллельных реакций аА->ЬВ и aA-»-dD,
целевым продуктом которых является вещество В, суммарная из-
бирательность фВ, сум выражается в виде:
<₽в, сум = (а/ь) "в/(иА, о ~ «а) = nBl[(a/b) nti + (a/d) nD] (3.26)
Связь между степенью превращения Ха, выходом продукта Фв<а)
и суммарной избирательностью фВ>Сум определяется из выражения
40
(3.26) и соотношений (3.27) и (3.28):
ха = («а. о — пк)!пк. о (3.27)
ФВ (А) = (а/Ь) «в/«А. О <3-28)
^А’Рв, сум = ФВ (А) (3.29)
Мгновенная избирательность измеряется отношением скорости
образования целевого продукта к скорости потребления исходного
реагента на образование всех продуктов реакции. Иными словами,
мгновенная избирательность равна отношению скорости основной
реакции к общей скорости процесса, выраженной через количества
исходного реагента:
Ч)В, мгн = ив/иА (3.30)
Фв, мгн имеет смысл мгновенного выхода целевого продукта.
Суммарная избирательность равна сумме всех мгновенных выхо-
дов. Иногда мгновенную избирательность оценивают отношением
скоростей образования целевого и побочного продуктов:
3.2.5. Расходные коэффициенты
Расходные коэффициенты (РК) характеризуют затраты сырья
на производство.
Расходный коэффициент — это отношение количества
сырья, затраченного при проведении ХТП, к количеству получен-
ного целевого продукта. Различают теоретические и практические
расходные коэффициенты. Теоретический расходный коэффициент
рассчитывают по стехиометрическому уравнению основной реак-
ции с учетом содержания исходного реагента в сырье. Например,
для реакции аА—>-ЬВ теоретический расходный коэффициент т]т для
получения продукта В определяют по соотношению:
Т]т = аМ^ЦЬМцЫ^ с) (3.32)
Здесь Ma, Mb — молекулярные массы реагента А и продукта В; <ва, с — мас-
совая доля компонента А в сырье, %.
При расчете практических расходных коэффициентов учитывают
степень превращения исходного реагента Ха, выход продукта
Фв(А), избирательность фв, сум:
, Чп = 'Пт/Иа’Рв, сум) = ЧТ/ФВ (А) (3.33)
Практический расходный коэффициент всегда выше теоретиче-
ского. Задача технологов — уменьшить отношение т)п/т)т без увели-
чения себестоимости продукта. А это достигается путем повыше-
ния степени превращения, избирательности и как следствие — вы-
хода продукта.
41
3.3. МАТЕРИАЛЬНЫЙ БАЛАНС
, Основой технологических расчетов являются расчеты материаль-
ных потоков при протекании ХТП. Знание материальных потоков
необходимо для проведения конструктивных расчетов производ-
ственного оборудования и коммуникаций, оценки экономической
эффективности процесса. Самым распространенным видом мате-
риальных расчетов является' составление материальных балансов
Материальные балансы составляют на основе закона сохранения
массы вещества с учетом стехиометрических соотношений. Мате-
риальный баланс означает, что общая масса (X т<Х) всех входя-
щих в аппарат (или цех) материалов (приход) равна общей-массе
(£т/ых) выходящих веществ (расход):
р р
<3,34)
£ = 1 / = 1
Число реагентов может изменяться от 1 до р. Расчет обычно
выполняют в единицах массы (кг, т), реже — в единицах количе-
ства вещества (моль, кмоль).
На производстве обычно составляют материальные балансы
для реактора, цеха,завода, отрасли.
Различают балансы на единицу времени и единицу продукции.
Материальный баланс на единицу времени
составляют чаще всего для непрерывных процессов с целью расчета
размеров аппаратов, диаметров трубопроводов, а также для опре-
деления входных и выходных значений концентраций реагирующих
компонентов.
Материальный балансна единицу продукции
составляют как для непрерывных, так и для периодических процес-
сов с целью определения теоретических и практических расходных
коэффициентов.
Связь между балансами на единицу времени и на единицу про-
дукции определяется производительностью.
Материальные балансы отделения, цеха, производства, завода
рассчитывают для определения габаритов складских помещений
для сырья и готовой продукции, расчета трубопроводов и промежу-
точных емкостей. Одновременно балансы могут быть использованы
для сравнения эффективности работы аппаратов, отделений, цехов
одинаковой мощности на различных завода^.
Баланс отрасли позволяет оценивать возможности сырьевой
базы для функционирования производств и проанализировать
структуру потребления получаемых продуктов.
Материальный баланс чаще всего представляют в виде таблиц,
которые включают приходную и расходную части.
3.4. ТЕПЛОВ 5Й БАЛАНС
По данным ¥йтериального баланса с учетом тепловых эффек-
тов реакций и физических превращений, подвода теплоты извне
или отвода ее составляют тепловой баланс, позволяющий опреде-
-м
лить потребность в топливе, температуру в зоне реакции, площадь
теплообменных поверхностей и другие параметры.
Основой теплового баланса является закон сохранения энергии,
в соответствии с которым в замкнутой системе сумма всех видов
энергии постоянна. Для расчета затрат энергии на производство
может быть составлен энергетический баланс, учитывающий рас-
ходы электроэнергии, технологического пара и воды на получение
продукта. В общем случае уравнение теплового баланса можно
представить в виде: '
Овх + Оф + Ор + Онагр = <Ux + Оф + Q? + <2ОХЛ (3.35)
Здесь Qb» и Qbux — соответственно количество теплоты, вносимое в аппарат
или выносимое из него жидкими или газообразными веществами; Оф и Оф —
теплота физических процессов, происходящих с выделением (') и поглощением
(") теплоты, Qp и Qp— теплота экзотермических (') и эндотермических (")
химических реакций; QBari! и 0ОХЛ — соответственно количество теплоты, подво-
димое в аппарат извне для нагрева реакционной смеси и отводимое через холо-
дильники для поддержания заданного температурного режима
На производстве тепловой баланс чаще всего составляют для
аппарата с целью определения технологических параметров и усло-
вий для обеспечения заданного температурного режима.
Величины QBX и QBMx рассчитывают по уравнениям:
рвх
Obx=Z«Wbx.« <3‘36>
рвых
^вых = т/с//вых. I ^3’37^
Здесь mt, т,- — масса (объем, количестзо) исходных реагентов и продуктов
реакции, прошедших через аппарат в едингцу времени кг/с (м3/с, кмоль/с);
с,, с, — средняя удельная теплоемкость компонентов, кДж/(кг-К) [кДж/(м3-К),
кДж (кмоль-К)]; 7’вх. I, Гвых. I — температура входного и выходного потоков. К;
Рвх, рвых — чис ю компонентов, участвующих в процессе
Теплоты фазовых переходов реагентов в ходе процесса и
Оф, которые в зависимости от знака теплового эффекта фазо-
вого перехода учитываются в приходной (кристаллизация, субли-
мация, конденсация) или расходной (испарение, плавление) частях
баланса, рассчитывают по уравнению:
п
<3-38)
Здесь Шф1 — масса (объем, количество) i-реагеита, участвующего в фазовом
переходе,. кг/с(м3/с, кмоль/с); 9ф; — тепловой эффект фазового перехода /-компо-
нента, кДж/кг (кДж/м3; кДж/кмоль).
Теплоту реакций определяют по уравнению:
<*₽=-£ «w* <з-39)
. i—1
43
Здесь mpi — масса (объем, количество) /-реагента, вступившего в реакцию
или образовавшегося в ходе реакции, кг/с(м3/с, кмоль/с); — тепловой эффект
реакции, кДж/кг(кДж/м3, кДж/кмоль).
Расчет Q„arp и QOxj. ведут по основному уравнению теплопере-
дачи, согласно которому количество переданной теплоты Q равно:
Q = KTF АТ (3.40)
Здесь Кл — коэффициент теплопередачи, Вт/(м2-К); F—площадь поверхно-
сти теплообмена, м2; А7 — разность- температур теплоносителя и реакционной
зоны (движущая сила процесса теплопередачи).
Из уравнения теплового баланса можно рассчитать любой из
параметров при известных остальных. Технолога чаще всего инте-
ресуют Твх, Тк, F.
Тепловой баланс представляют в виде таблиц, имеющих при-
ходные и расходные статьи тепловых потоков.
3.5. ТЕРМОДИНАМИЧЕСКИЕ ХАРАКТЕРИСТИКИ
ХИМИЧЕСКИХ ПРОЦЕССОВ
3.5.1. Задачи термодинамического анализа
Роль термодинамического анализа в химической технологии
определяется решением следующих задач:
1. Установлением принципиальной возможности самопроизволь-
ного протекания предполагаемого химического процесса.
2. Определением энергетических эффектов химико-технологиче-
ских процессов, необходимых для составления энергетических
балансов и расчета затрат энергии на проведение ХТП.
3. Расчетом максимально возможных степеней превращения
и выходов продуктов при проведении обратимых процессов.
4. Определением диапазона температур и давлений, при кото-
рых рационально осуществление ХТП.
Из курса физической химии известно, что условием принци-
пиальной осуществимости протекания химической
реакции в прямом направлении без затраты работы является
уменьшение свободной энтальпии системы.
Количественно возможность протекания реакции при той или
иной температуре оценивается на основе тепловых эффектов реак-
ции ДН и изменения энергии Гиббса Дб. Как правило, чем более
экзотермична реакция при данной температуре, тем вероятнее ее
протекание. На практике используют стандартные значения Дб°
для условий Р = 101 кПа, Т — 298 К. Эти значения приведены в
справочной литературе. Опыт показывает, что если Абс,Э8 <
< — 40 кДж/моль, то предполагаемая реакция возможна, а если
Дб°98 > 40 кДж/моль — реакция невозможна в любых условиях.
Энергетические эффекты при протекании химико-тех-
нологических процессов оценивают на основе теплот образования
и сгорания веществ. Для многих веществ теплоты образования
44
и сгорания известны и сведены в таблицы. Их называют стандарт-
ными тепловыми эффектами и обозначают С помощью этих
таблиц путем комбинации известных значений ДЯ°98 можно полу-
чить Д/7 практически для любых реакций, не прибегая к экспе-
рименту.
Относительно оценки максимально возможных
степеней превращения Хмакс следует отметить, что для
необратимых процессов Хмакс = 1.
Г л у б и н у п р оте к а н и я обратимыхХТП и диапа-
зон рабочих температур и давлений можно пред-
сказать на основе данных о равновесии химической реакции. Все
обратимые ХТП стремятся к равновесию, при котором скорости
прямого и обратного процессов уравниваются и соотношение кон-
центраций, определяющее равновесную степень превращения X* =
= Хмакс, остается неизменным, пока не изменятся внешние условия.
3.5.2. Константы равновесия
и расчет равновесных степеней превращения
Для количественной оценки состояния равновесия используют
значения констант равновесия, равновесных концентраций реаген-
тов С*, равновесных степеней превращения X* и выходов про-
дукта Ф*. В общем случае для реакции
аА+6В+ ... «=> rfD + eE+ ...
константы равновесия можно выразить через равновесные концен-
трации реагентов (Cj):
Кс = (C*d)4 (4)е • • -/[(сА)а (С’в)1 (3.41)
Для газов константа равновесия часто выражается через рав-
новесные парциальные давления реагентов (Р*)'
КР = (P^d (РЁ)е • /[(РА)° (Р’в)"1 • (3.42)
При технологических расчетах константы равновесия иногда
выражают через мольные доли реагентов в состоянии равновесия
W):
-/Ща (AQ1 - (3.43)
Следует помнить, что для реакций, протекающих с изменением
числа молей, численные значения констант равновесия зависят от
формы записи стехиометрического уравнения реакции. Например,
реакцию окисления оксида серы (IV) можно записать в виде:
SO2 + 0,5О2 SO3 или 2SO2 + O2 «=* 2SO3.
В первом случае:
Кс\ = <-‘so3/[cso2 (С02)0'5] (3 44)
Во втором:
^С2 = (cs63)7l(cso2);! С о2] (3.45)
45
Kci и Ксг имеют разные размерности и Кс\ = л/Ксч- Аналогично
для указанных реакций Кр\ Кр2 и Kn\ K.N2- В общем случае
Кс#= К₽=/= Kn. Связь между ними устанавливается на основе урав-
нения Менделеева — Клапейрона и закона Дальтона. Уравнение
Менделеева Клапейрона позволяет выразить связь между пар-
циальным давлением i-ro компонента газовой смеси (Р,) и его мо-
лярной концентрацией (С,):
Р и = п^Т (3.46)
Cz = n,/t> = />,/(/? D (3.47)
Здесь ni и v — число молей вещества и объем реакционной смесн.
Связь между парциальным давлением (-компонента (Pi), общим
давлением смеси (ЁОбщ) и мольными долями (М,) устанавливается
соотношениями:
Р0бш=£Р/ (3-48)
г-1
= (3-49)
Таким образом, Кс, Кр и Кн связаны между собой соотно-
шениями:
^ = Kc(/?r)Zv' (3.50)
У V/
*Р=К^общ (3-51)
*c = tfwpS^(*r)-EV' (3-52)
Здесь vi — алгебраическая сумма стехиометрических коэффициентов при
реагентах и при продуктах реакции. При этом коэффициенты при продуктах
Реакции — положительные, а при исходных реагентах принимаются со знаком
минус.
Значения констант равновесия для различных реакций приве-
дены в справочных пособиях.
Численные значения Кс и Кр не зависят ни от концентрации
реагентов и их соотношений, наличия или отсутствия примесей или
разбавителей, ни от давления (если газы идеальные, а давление
не очень велико). Константы химического равновесия зависят лишь
от природы реагентов и температуры. Связь межд) константой
равновесия и температурой описывается уравнением Вант-Гоффа:
d In Кр ЬЯ
dT
Если известно значение константы равновесия Kpi при темпе-
ратуре It и требуется рассчитать значение константы КР2 при тем-
пературе Т2, то используют соотношение:
lg (КР1/КР2) = [ЛЯ/(2,3/?)] (1/Г2 - 1/Г,) (3.54)
При этом принимается, что в диапазоне изменения температуры
от 7\ до Т2 тепловой эффект не изменяется.
(3.53)
46
Рис, 3.1. Влияние температуры на равновесную степень превращения при проте-
кании обратимых экзотермических (/) и эндотермических (2) процессов.
Рис. 3.2. Влияние давления на равновесную степень превращения при протекании
реакций с уменьшением (?) и увеличением (2) объема.
Константы’равновесия в той или иной форме входят в состав
многих кинетических уравнений. Значения констант равновесия
используют главным образом для расчета составов равновесных
смесей и определения максимально возможных степеней превра-
щения Лмакс — X* или выходов продукта Ф*.
Если численные значения констант равновесия зависят только
от природы реагентов и температуры, то на равновесные составы,
степени превращения и выходы продуктов влияет не только тем-
пература, но и давление, концентрация исходных веществ, нали-
чие примесей, разбавителей. Качественно, влияние этих параметров
определяет известный принцип Ле Шателье, согласно которому
в системе, выведенной внешним воздействием из состояния равно-
весия, происходят изменения, направленные к ослаблению воздей-
ствий, выводящих систему из равновесия.
Так, для экзотермических процессов повышение температуры
должно привести к изменениям в системе, которые обеспечили бы
уменьшение тепловыделения. Этому отвечает уменьшение равно-
весной степени превращения исходных веществ, т. е. сдвиг равно-
весия влево, в сторону образования исходных веществ. Эндотер-
мическим процессам отвечает обратная картина (рис. 3.1). Для
газофазных процессов, протекающих с повышением объема, увели-
чение давления будет приводить к уменьшению равновесной сте-
пени превращения или сдвигу равновесия влево. Если в процессе
превращения число молей реагирующей смеси уменьшается, то
увеличение давления должно вызвать такое изменение в равновес-
ной смеси, которое бы компенсировало внешнее воздействие, т. е.
должна увеличиться степень. превращения, что приведет к умень-
шению числа молей смеси и падению давления (рис 3.2).
В рассмотренных случаях степень смещения равновесия зави-
сит от абсолютного значения теплового эффекта реакции и от
относительного изменения объема реакционной смеси. Чем больше
&Н, тем значительней влияние температуры. Чем больше относи-
тельное изменение объема реакционной смеси еА (см. разд. 3.2.1),
тем сильнее сказывается увеличение давления иа сдвиг равно-
весия.
47
Рис. 3.3. Влияние избытка кислорода на равно-
весную степень окисления оксида серы(IV).
В соответствии с принципом Ле-Шателье введение в равновес-
ную систему дополнительного количества какого-либо компонента
сдвигает равновесие в направлении уменьшения его концентрации.
Поэтому избыток одного или нескольких исходных веществ спо-
собствует увеличению степени превращения других веществ, сме-
щая равновесие вправо. Дополнительное введение продуктов реак-
ции вызывает смещение равновесия влево. Удаление продуктов ре-
акции из смеси позволяет более глубоко проводить химическое пре-
вращение. Эти обстоятельства широко используют в промышлен-
ности. Так, при окислении оксида серы (IV) в производстве серной
кислоты реакцию проводят с избытком кислорода, добиваясь бо-
лее полного окисления SO2 (рис. 3.3.), а между двумя последова-
тельными стадиями окисления выводят путем абсорбции из реак-
ционной смеси оксид серы (VI), что позволяет еще более повысить
степень превращения оксида серы(IV).
Введение в равновесную смесь инертного газа при постоянном
давлении приводит к уменьшению парциальных давлений реаген-
тов и, таким образом, эффект разбавления подобен эффекту умень-
шения общего давления в системе.
Для количественной оценки влияния температуры, давления,
концентраций, коэффициента избытка одного из реагентов и кон-
центрации разбавителя используют соотношения между констан-
тами равновесия, равновесным составом и равновесной степенью
превращения. Эти соотношения для каждого конкретного случая
получают путем решения системы уравнений, задаваемых констан-
тами равновесия, и уравнений стехиометрического баланса. Вид
формул, связывающих константы равновесия, равновесный состав
и равповесную степень превращения, зависит от типа реакции.
Как правило, мольные доли компонентов равновесной смеси свя-
зывают с числом молей исходного вещества, прореагировавшего
к моменту равновесия, или со степенью превращения исходного
вещества Хр. Число молей каждого из реагирующих веществ
в исходной смеси может быть произвольным. Обычно при расчетах
исходят из 1 моль основного реагента и определяют число молей
других веществ.
Рассмотрим, например, реакцию, протекающую в газовой фазе:
А + 2В D
Константу равновесия, выраженную через равновесные пар-
циальные давления Р*,, можно записать в виде:
Кр=Ро/[Р1(^в)2] (3.55)
48
Парциальное давление любого компонента по закону Даль-
тона равно:
р/ = («7£к/)робщ = л'/роб1ц1 . (356)
РА = N А^общ’ ^В = NBPo6m< PD = NDPo6m
Здесь ni — число молей i-компонента в равновесной смеси; nt — общее число
молей всех компонентов, составляющих равновесную смесь; N{ — мольная доля
i-компонента равновесной смеси.
После подстановки парциальных давлений, выраженных в соот-
ношениях (3.56), в уравнение (3.55) получим:
Кр = ^о/[<(л/*в)2Рабщ] (3-57)
Примем, что число молей в исходной смеси гад, о= 1, гав, о = 2,
nD 0 = 0. В равновесной смеси га’Л=1 — у, га* = 2(1 — у), n*D = y
(у — число молей исходного вещества, прореагировавшего к мо-
менту равновесия). Суммарное число молей в равновесной смеси
У, n*i — 3 — 2у. Состав равновесной смеси, определяемой мольными
долями компонентов в ней:
4 = (1 - «/)/(3 - 2f/), 4 = 2(1 -у)/{3-2у), NtD = y/(.3-2y) (3.58)
Уравнение, связывающее известное значение константы равно-
весия с искомым значением у, для данной реакции принимает вид:
К'р = у (3 - 2f/)7[4 (1 - у)3 4бщ] х (3.59)
Определив из уравнения (3.59) значение у и подставив его
в (3.58), можно рассчитать состав равновесной смеси.
Поскольку при расчете на 1 моль основного компонента А ко-
личество превратившегося вещества А численно равно степени его
превращения, т. е. у = Хк, то соотношение (3.59) фактически свя-
зывает Кр и Ха:
Хр = 4 (3 - 24)7(4 (1 - 4)3 робщ] (3.60)
Аналогично получают уравнения, связывающие константы рав-
новесия и равновесные степени превращения для различных моль-
ных соотношений реагентов.
ГЛАВА 4
ЗАКОНОМЕРНОСТИ ГОМОГЕННЫХ ПРОЦЕССОВ
4.1. ОБЩАЯ ХАРАКТЕРИСТИКА ГОМОГЕННЫХ
ХИМИЧЕСКИХ ПРОЦЕССОВ
Гомогенные процессы в промышленности в основном осуществ-
ляются в газообразной или жидкой фазе. В качестве примера го-
могенных процессов, протекающих в газообразной фазе, в техно-
логии неорганических веществ можно привести сжигание серово-
49
дорода или паров серы для производства оксиды серы(IV), окис
ление оксида азота(II) до оксида азота(IV) в производстве азот-
ной кислоты, окисление оксида серы (IV) при каталитическом дей-
ствии газообразных оксидов азота, частично протекающее при
нитрозном способе производства серной кислоты, и ряд других.
Наиболее широко гомогенные процессы в газовой фазе представ-
лены в технологии органических веществ. Примерами служат про-
цессы термического хлорирования некоторых парафинов в произ-
водстве растворителей, парофазное хлорирование бензола и суль-
фохлорирование углеводородов при получении моющих средств,
синтез хлороводорода, бензола, ацетилена и др. В технологии не-
органических веществ в качестве примера гомогенных процессов
в системе Ж — Ж можно привести производство сульфата аммо-
ния путем взаимодействия аммиачной воды и серной кислоты:
2NH4OH + H2SO4 = (NH4)2SO4 + 2Н2О. По такому же принципу
протекают некоторые обменные реакции в растворах солей. В тех-
нологии органических веществ гомогенно в системе Ж — Ж про-
водят процессы полимеризации винилацетата, получают этилен-
гликоль, простые и смешанные эфиры из спиртов и др.
При проведении гомогенных процессов химические реакции
протекают во всем объеме реакционной смеси, и если реагенты
в исходной смеси достаточно хорошо перемешаны, то диффузион-
ные процессы не лимитируют общей скорости процесса, которая
определяется, как правило, кинетикой химического превращения.
Закономерности гомогенных процессов применимы и для анализа
гетерогенных процессов, протекающих в кинетической области.
В этой главе рассмотрено влияние концентраций реагирующих
веществ, температуры, давления на основные показатели ХТП,
проводимого в замкнутом объеме или, как будет показано ниже,
в реакторах периодического действия. Для таких уровней прове-
дение гомогенных процессов описывается закономерностями ки-
нетики химических реакций, т. е. закономерностями процессов,
протекающих на молекулярном уровне.
Для более общего случая анализа работы реакторов непрерыв-
ного действия необходимо дополнительно знать законо. 1ерности
протекания процессов в потоках, которые будут рассмотрены
в гл. 6.
4.2. ВЛИЯНИЕ КОНЦЕНТРАЦИИ НА СКОРОСТЬ РЕАКЦИИ
И СТЕПЕНЬ ПРЕВРАЩЕНИЯ
Математическую формулу, связывающую скорость с концент-
рациями, называют кинетическим уравнением реак-
ции. Вид кинетического уравнения определяет порядок реак-
ции, который равен сумме показателей степеней при концентра-
циях реагентов в уравнении. Влияние концентраций реагирующих
веществ на скорость определяется законом действия масс, являю-
щимся основным законом химической кинетики.
50
Для реакции
аА + 6 В + . • • = Продукты
кинетическое уравнение можно представить в виде:
' dC. ,
U = —f~ = kCACR ... (4.1)
ат
Порядок реакции т равен:
т = а' + Ь' + • • • (4.2)
Порядок реакций в большинстве случаев не совпадает с моле-
кулярностью реакций т' = а -ф b -ф ... что объясняется сложным
механизмом таких реакций. Показатель степени при концентрации
какого-либо компонента в кинетическом уравнении (4.1) называют
порядком реакции относительно этого компо-
нента.
4.2.1. Простые необратимые и обратимые реакции
Наиболее простые случаи гомогенных реакций описываются
кинетическими уравнениями 1- и 2-го порядков:
т=1 ---;— = kC 1-й порядок
ас (4,3)
т = 2 ---= kC2 2-й порядок
Как следует из уравнений, зависимость скорости ре-
акции от концентрации сильно изменяется при
возрастании порядка реакции. Так, при увеличении те-
кущей концентрации реагентов в 2 раза скорость реакции 1-го по-
рядка увеличивается в 2 раза, а реакции 2-го порядка — в 4 раза.
После интегрирования кинетических уравнений и последующих алгебраиче-
ских преобразований получают формулы, описывающие взаимосвязь начальной
и конечной концентраций исходных реагентов, времени, необходимого для дости-
жения заданной степени превращения или выхода продукта. Для простых не-
обратимых реакций 1-го порядка эти формулы имеют вид:
т = (\/k) In (СА д/Сд) = (1/fe) In [1/(1 - х)1 (4.4)
СА = СА 0 exp (— kt) (4.5)
ХА = 1 — exp (— kt) (4.6)
Для технолога важно знать влияние начальной концентрации
реагентов на среднюю скорость процесса. Именно этот факт опре-
деляет конечную производительность установки. Учитывая урав-
нение (4.4), несложно показать, что для реакции 1-го порядка
U = (СА, о - СА)/т = kCA 0Ха/1п [1/(1 - ХА)[ (4 7)
т. е. средняя скорость U линейно увеличивается с ростом СА, о при
заданном постоянном значении степени окисления ХА. Влияние
концентрации на скорость обратимых реакций аналогично, хотя
51
Рис. 4.1. Влияние повышения концен-
трации Св и хоэффициента избытка
ав вещества В на степень превраще-
ния вещества А и выход продукта D
по веществу А в реакции А + В D.
выражение движущей силы процесса ДС, в которую входит на-
чальная концентрация, имеет существенно более сложный вид.
Как известно, для простых необратимых и обратимых реакций
выход продукта по какому-либо веществу численно равен степени
превращения этого вещества. Если в реакции участвуют два ве-
щества А и В, то увеличение концентрации одного из них (напри-
мер, В) приводит к повышению выхода продукта (например, D)
по другому веществу Фо<а)- Этот прием широко распространен
в инженерной практике, когда необходимо более полно использо-
вать дорогостоящее, дефицитное или вредное для окружающей
среды вещество А. Увеличивая избыток другого — дешевого или
безвредного — вещества В путем увеличения его концентрации Св,
повышают степень превращения дорогого вещества ХА и выход
продукта по этому веществу Фо<д) (рис. 4.1). При этом верхний
предел выхода Фо(д)мвкс для необратимых реакций равен 1, а для
обратимых — равновесному выходу.
4.2.2. Влияние концентрации на избирательность
На практике чаще всего приходится управлять сложными ре-
акциями, к которым относятся обратимые, параллельные и после-
довательные. В таких реакциях протекает не одна-единственная,
а две и более реакций с участием одних и тех же реагирующих
веществ.
Влияние концентрации исходных веществ на протекание гомо-
генных процессов наиболее существенно для параллельных и по-
следовательных реакций с разными Порядками основной и побоч-
ной реакций.
Рассмотрим параллельные реакции, при которых одно веще-
ство А участвует в двух реакциях, давая различные продукты В
и D, причем основная реакция с получением целевого продукта
описывается уравнением 2-го порядка, а побочная—уравнением
1 го порядка:
2Д В, А D
В этом случае скорость образования целевого продукта В опи-
сывается уравнением:
dCR „
= = = k, [CKt 0(1 - ХА)]2 (4.8)
52
Скорость образования побочного продукта D:
dCn
(jD = -^ = k2C2 = fe2CA fi (1 - ЛА) (4.9)
Избирательность, выраженная через соотношение скоростей
Uв и Ud, равна:
Фв. мгн = Wo = ( W С А. О (1 - *А) (4.10)
Из соотношения (4 10) видно, что увеличение начальной кон-
центрации вещества А прямо пропорционально повышает избира-
тельность и увеличивает выход продукта В [см. уравнение (3.29)].
К аналогичному выводу можно прийти, если проанализировать
кинетику последовательных реакций. Например, в простейшем слу-
чае двух последовательных необратимых реакций, в которых ки-
нетика получения основного продукта В описывается уравнением
2-го порядка, а побочного продукта D — уравнением 1-го порядка,
соотношение количеств В и D растет пропорционально начальной
концентрации исходного реагента А. Соответственно увеличивается
и избирательность.
4.2.3. Методы увеличения концентрации
Для повышения исходных концентраций реагентов в гомоген-
ных системах применяют следующие методы:
Для газов: сорбция реагентов из смесей с последующей де-
сорбцией в более концентрированном виде; сжатие; сжижение;
растворение газов для проведения реакции в растворе.
Для жидкостей: выпаривание; вымораживание; сорбция
и последующая десорбция необходимых компонентов в более кон-
центрированном виде; дополнительный ввод реагентов в раствор
с последующим растворением твердых или смешением жидких
концентратов. j
4.3. ТЕМПЕРАТУРА КАК ФАКТОР ПОВЫШЕНИЯ
СКОРОСТИ ПРОЦЕССА И УПРАВЛЕНИЯ ВЫХОДОМ ПРОДУКТА
И ИЗБИРАТЕЛЬНОСТЬЮ
В химической практике одним из самых распространенных при-
емов увеличения скорости реакций является нагревание. Для не-
обратимых гомогенных процессов увеличение скорости реакций
с ростом температуры обусловлено только повышением константы
скорости реакции. В обратимых процессах изменение температуры
сказывается и на значении движущей силы процесса через изме-
нение константы равновесия и равновесных концентраций реаген-
тов, входящих в состав математического выражения АС. Для про-
цессов с параллельными и последовательными реакциями увели-
чение температуры существенно может влиять на избирательность
при одновременном ускорении основной и побочных реакций.
53
4.3.1. Интенсификация необратимых процессов
Согласно простому эмпирическому правилу (правило В ант-
Гоффа) при повышении температуры на ЮК скорость хими-
ческой реакции увеличивается в 2—4 раза. Такое правило дает
весьма приближенную оценку влияния температуры. Скорость
необратимых реакций прямо пропорциональна константе скорости,
связь которой с температурой чаще всего может быть описана
уравнением Аррениуса:
k == k0 exp [- E,'(RT)] (4.11)
Здесь k0 — предэкспоненциальный множитель (предэкспонента); Е— энергия
активации, т. е. избыточное количество энергии по сравнению со средним уров-
нем энергии молекул исходных веществ, необходимая для их участия в хими-
ческой реакции; R — универсальная газовая постоянная.
Значения /го и Е технологи обычно находят в справочной ли-
тературе. Если известно значение fei для одной температуры Т\,
то, зная Е, можно рассчитать k2 для любой другой температуры
Т2, если в заданном температурном диапазоне Е = const. Для
етого используют соотношение:
In = (E/R) [(Г2 - ГОЛГ.Гг)! (.4.12)
Формальный смысл Е: скорость реакции тем сильнее зависит
от температуры, чем больше энергия активации.
Поскольку движущая сила гомогенных процессов с необрати-
мыми реакциями не зависит от температуры, характер изменения
скорости реакций определяется изменением значений k.
Следует помнить, что указанный характер влияния темпера-
туры на скорость необратимых реакций не зависит от их тепло-
вого эффекта. Таким образом, повышение температуры увеличи-
вает скорость как экзотермических, так и эндотермических необ-
ратимых реакций. Их скорость прямо пропорциональна константе
.скорости, изменение которой описывается уравнением (4.11).
Отметим, что и средняя скорость процесса С, которая опреде-
ляет выпуск продукции в единицу времени, или производитель-
ность, также прямо пропорциональна изменению константы ско-
рости, если при изменении температуры проведения процесса тре-
буется поддерживать неизменной заданную степень превраще-
ния X. Это следует из анализа изменения времени т реакции
с ростом Т в выражении средней скорости процесса. Например,
для реакции 1-го порядка имеем:
и ~ (са, оса)/т = £а. о^л/^'у ,п 1 _ =
= Ak = AkB exp [— Е/ (RT)] (4.13)
Здесь А — постоянная.
В производственной практике технолога интересует, как изме-
няется О (или производительность) при увеличении температуры
проведения процесса при неизменном заданном времени реакции
та (или при постоянной скорости подачи реагентов и неизменном
64
Рис. 4.2. Характер изменения средней скорости не-
обратимого химического процесса в зависимости ст
температуры.
объеме реактора). Для получения такой зависимости в выраже-
нии средней скорости надо проанализировать изменение конечной
концентрации исходного реагента СА:
^=(са.о-са)Л = (са,оЛ)П -exp(-frr)] (4.14)
При этом зависимость k от Т описывается уравнением (4.11).
Характерный график изменения V для такого случая показан
на рис. 4.2. Так как 1 —ехр(—kt) = Ха, то изменение ХА с ростом
температуры также описывается S-образной кривой.
Для каждого производственного процесса существуют ограни-
чения повышения температуры, обусловленные или термостой-
костью конструкционных материалов, из которых изготовлены ре-
акторы, или затратами энергии на поддержание высокой темпера-
туры. Кроме этого, повышение температуры выше некоторого пре-
дела Гмакс может привести к возникновению побочных реакций
и, как следствие, к сильному снижению выхода продукта. Поэтому
процессы проводят при определенных ограничениях по темпера-
турному режиму.
4.3.2. Влияние температуры на скорость
и степень превращения при протекании обратимых реакций
В отличие от необратимых регкцкй влияние температуры на
скорость обратимых реакций имеет более сложный характер, так
как скорость прямой и скорость обратной реакций характеризуются
разными энергиями активации, а значит и разной «чувствитель-
ностью» к изменению Т. Для обратимой экзотермической реакции
Е2 > fi, для эндотермической — энергия активации прямой реак-
ции (f|) выше, чем обратной (Ея). Для простых обратимых реак-
ций типа А В ± q скорость реакции можно записать в виде:
U = klCA-k2CB (4.15)
С учетом соотношений
^а = 6^а и (1 — ХА). СВ=СА ОХА, fej = k0 exp [— £'1/(/?Г)], KQ = kJk2
г
уравнение (4.15) преобразуется:
U = k0 exp [- EJIRT)] 0 [1 - ХА (1 + 1 /Кс)] (4.16)
55
Рис. 4.3. Влияние температуры на значения кон-
станты скорости Л и константы равновесия Кс-
б
Рис. 4.4. Характерные зависимости скорости об-
ратимой экзотермической реакции от температуры
и степени превращения < Хг < Х3 < л<).
Влияние температуры на скорость проявляется через изменение
константы скорости и константы равновесия Кс- Другими словами,
если представить уравнение (4.3) в виде U = kKC, то можно ска-
зать, что при изменении температуры изменяется и константа ско-
рости реакции k и движущая сила процесса:
дс = са,о['-*а(1 + 1/*с)] (4.17)
В случае экзотермической реакции влияние температуры на
кинетический фактор (константа скорости) и термодинамический
фактор (константа равновесия) противоречиво: с ростом Т увели-
чивается k и уменьшается Кс (рис. 4.3). Это приводит к экстре-
мальной зависимости изменения скорости процесса от темпера-
туры. Если в уравнение (4.16) подставить соответствующие зна-
чения йоь Сд. о. то можно построить график зависимости U от Т
для определенного значения ХА (рис. 4.4, а). Если таким же об-
разом построить графики для других значений XA(Xi, Х2, -Хз,
то можно показать, что с увеличением степени превращения ско-
рость реакции снижается, а температуры, соответствующие мак-
симальным скоростям, Топт, ь Т^опт,2. (оптимальные темпера-
туры) уменьшаются (рис. 4.4,6).
Линию MN, соединяющую максимальные значения скоростей,
называют линией оптимальных температур. Она пока-
зывает, как надо изменять температуру по мере увеличения сте-
пени превращения, чтобы процесс шел с максимальной скоростью.
Значения ХА при некотором постоянном т с ростом Т вначале уве-
бв
a
б
Рис. 4.5. Характерные зависимости степени превращения от температуры (Т| >
> г2 > т3 > т4).
личиваются, достигают максимума, а затем снижаются, так как
процесс ограничен равновесной степенью превращения Ха
(рис. 4.5, а).. Кривые Х(7) для различных значений т показаны на
рис. 4.5, б. Анализируя графические зависимости, представленные
на рис. 4.5, б, можно сделать следующий вывод: для проведения
обратимых экзотермических процессов с максимальной скоростью
температуру необходимо уменьшать с ростом степени превра-
щения.
При проведении процессов с обратимой эндотермической
реакцией влияние температуры на константу скорости и движу-
щую силу процесса однозначно: с ростом Т увеличивается и k,
и Кс, входящая в состав движущей силы в уравнении (4.16).
С помощью уравнения (4.16) несложно получить графическую за-
висимость скорости реакции от температуры для разных степеней
превращения (рис. 4.6). А зависимость Х,\ и пропорциональной ей
средней скорости процесса U = Ca,oX\/t от Т при постоянстве
заданного времени контакта изображена на рис. 4.7.
Рис. 4.6. Изменение скорости обратимой эндотермической реакции в зависимости
от температуры и степени превращения (X) < Х2 < Х3 < Х4).
Рис. 4.7. Влияние температуры на степень превращения для обратимой эндотер-
мической реакции А В — q (ti >• т2 > т3 > т4).
57
Общая закономерность влияния температуры на скорость обра-
тимых реакций заключается в гом, что при малых степенях пре-
вращения и низких температурах преобладает фактор повышения
константы скорости прямой реакции, а при высоких температурах
сказываются термодинамические ограничения, затормаживающие
рост эндотермических процессов и уменьшающие значения степеней
превращения в экзотермических процессах.
4.3.3. Влияние температуры на скорость, избирательность
и выход продукта при протекании сложных реакций
Рассмотрим параллельные реакции А —В и А —D,
где В — целевой продукт, D — побочный. Пусть скорости образо-
вания этих продуктов описываются уравнениями:
£В = = klCA = km exp [- E'/tRT)]
de (4Л8)
= -rfT = k2C* = *02 «Р [- CA2
Избирательность по продукту В будет пропорциональна отно-
шению скоростей образования целевого и побочного продуктов:
Фв ~ UR/UD^ [*01С£/(й0,Са)] е*Р К£2 - WUm] <4.19)
Для выявления характера влияния температуры на избиратель-
ность принимают все величины, входящие в уравнение (4.19), по-
стоянными и тогда
ФВ ~ ехр [ДЕ/(/?Г)] (4.20)
Если Энергия активации побочной реакции выше, чем основ-
ной, т. е. ДЕ > 0, то для повышения избирательности необходимо
уменьшать температуру. Однако, если при этом сохраняется по-
стоянным время реакции т, неизбежно уменьшается скорость об-
разования продукта В, степень превращения исходного вещества
А и, как правило, понижается выход целевого продукта (рис. 4.8, а).
Рис. 4.8. Влияние температуры на избирательность, степень превращения и выход
целевого продукта для параллельных реакций:
а—б—Е\>Е}.
58
Таким образом, задача технолога в этой ситуации найти рацио-
нальную степень снижения температуры, обеспечивающую повы-
шение избирательности при допустимом снижении производитель-
ности и выхода продукта.
Более просто решается задача повышения избирательности
при Ei > £2 (рис. 4.8,6). В этом случае увеличение температуры
повышает избирательность, степень превращения Ха и, как след-
ствие, выход Фв и производительность.
Рассмотрим последовательную реакциюА —* В —* D,
причем В — целевой продукт. Скорости образования целевого и
побочного продуктов описываются соотношениями:
dCB
^В = = — ft2CB (4.21)
С/О = Й2СВ (4-22)
Принимая, что избирательность по продукту В пропорцио-
нальна отношению Ub/Ud, можно записать:
Фв ^в/^D= ^1^а/^2^В I (4.23)
При постоянстве С а и Св избирательность будет тем выше, чем
больше отношение k\jki, т. е.
Фв ~ 4k-i = *oi ехР [~ Ei/UU">]/{kot ехР [~ ЩМ]} = ЛехР [Л*/(ЛП] (4-24)
Здесь А — постоянная.
Как и при рассмотрении параллельных реакций, несложно сде-
лать вывод, что при Д£ > 0 для повышения избирательности це-
лесообразно уменьшать температуру, а при Д£ < 0— ее увеличи-
вать. В последнем случае растет также выход целевого продукта,
его количество, получаемое в единицу времени, степень превра-
щения исходного вещества.
4.4. ВЛИЯНИЕ ДАВЛЕНИЯ НА СКОРОСТЬ
ГАЗОФАЗНЫХ ПРОЦЕССОВ
Влияние давления на скорость реакций и другие показатели
гомогенных процессов наиболее сильно проявляется, когда реаги-
рующие вещества находятся в газообразном состоянии. Для не-
обратимых реакций влияние давления сказывается одно-
значно: с увеличением давления пропорционально растут концен-
трации реагирующих веществ, а следовательно, и движущая сила,
и скорость процесса в целом.' Например, для реакции аА + ЬВ->
скорость можно описать уравнением:
и = kC*CbB = /? [PfJ(RT)]a' [PB/(RT)]b' = k'PaA Р* (4.25)
В соответствии с законом парциальных давлений Ра = АдДобщ,
Р в — А вР общ-
59
Рис. 4.9. Влияние давления Р на скорость газофаз-
ных необратимых реакций различного порядка т.
Следовательно, скорость реакции можно представить, как функ-
цию давления Роещ, при котором проводят процесс:
и = лХА'в>общ' = *'л^бщ (4.26)
Таким образом, с увеличением давления Робщ скорость гомо-
генного процесса растет пропорционально, давлению в степени,
равной порядку реакции. Чем выше порядок реакции, тем сильнее
влияние давлёния на скорость (рис. 4.9).
Для обратимых реакций влияние давления на движу-
щую силу процесса имеет более сложный характер, так как с уве-
личением Р растут не только концентрации реагирующих веществ,
но изменяются параметры равновесного состояния, входящие в со-
став выражения движущей силы. Это могут быть равновесные кон-
центрации или равновесная степень превращения.
Для пояснения сказанного примем, что движущая сила про-
цесса пропорциональна разности фактической и равновесной кон-
центраций реагента А для реакции аА + ЬВ dD, т. е.
ДСд ~ сд - (4.27)
Если реакция протекает с уменьшением объема реагирующей
смеси (а -ф b > d), то с увеличением Робщ растет СА и снижается
Сд. Движущая сила однозначно связана с изменением Робщ, и ско-
рость реакции возрастает с увеличением давления, при котором
протекает реакция. По сравнению с необратимыми реакциями
того же порядка влияние роста давления на увеличение скорости
обратимых реакций, протекающих с уменьшением объема, как пра-
вило, более существенно.
Для реакции, протекающей с увеличением объема (а -ф b < d),
зависимость скорости реакции от давления имеет максимум
(рис. 4.10). Оптимальное давление, при котором скорость реакции
максимальна, зависит от степени приближения реагирующей сме-
си к равновесию. С увеличением степени превращения оптималь-
ное давление уменьшается.
Аналогично изменяются степень превращения исходных ре-
агентов и выход продукта (рис. 4.11). Линия MN~ соединяющая
максимумы на кривых ХА, Фо<а) — f(Робщ), называется линией
оптимальных давлений. Положение линии оптимальных
давлений в координатах X — Р показывает, что по мере увеличе-
ния степени превращения необходимо уменьшать или общее, или
60
Рис. 4.10. Влияние давления на скорость обра-
тимой реакции, протекающей с увеличением
объема (Xt < Х2).
Рис. 4.11. Влияние давления на степень превра-
щения и выход продукта для обратимых реак-
ций, протекающих с увеличением объема (Xi >
> т2 > Т3>.
парциальные давления реагентов. На практике для уменьшения
парциальных давлений реакционную смесь по мере увеличения
степени превращения разбавляют инертным газом или паром.
При проведении параллельных или последователь-
ных реакций влияние давления эквивалентно изменению кон-
центраций реагирующих веществ. Например, повышение давления
во всех случаях увеличивает скорость образования целевых и по-
бочных продуктов. Если порядок основной реакции выше, чем по-
бочной, то увеличивается и выход целевого продукта, и избира-
тельность.
Для гомогенных процессов с жидкой фазой влия-
ние давления сказывается на изменении константы скорости реак-
ции. Но это проявляется лишь при высоких давлениях. Напри-
мер, полимеризацию этилена осуществляют при давлении свыше
100 МПа.
4.5. ПРИМЕНЕНИЕ КАТАЛИЗАТОРОВ В ГОМОГЕННЫХ СИСТЕМАХ
(ГОМОГЕННЫЙ КАТАЛИЗ)
4.5.1. Значение и области применения катализа
Наиболее эффективным способом увеличения скорости хими-
ческих процессов является использование катализаторов. Химико-
технологический процесс с химической реакцией, осуществляю-
щейся в присутствии катализатора, называют каталитиче-
61
с к и м. Роль каталитических процессов в химической технологии
неуклонно возрастает: около 90 % новых производств, освоенных
за последнее время химической промышленностью, включают ка-
талитические процессы. Использование катализаторов значительно
облегчает практическое осуществление многих химических реак-
ций. Их применяют как для получения важнейших неорганических
продуктов основной химической промышленности (водорода, ам-
миака, серной и азотной кислот и_др.), так и в технологии орга-
нических веществ. С помощью катализаторов осуществляют мно-
гие процессы органического синтеза — окисление, гидрирование,
дегидрирование, гидратацию, дегидратацию, полимеризацию, по-
ликонденсацию и др. Каталитическими являются многие процессы
при переработке нефтепродуктов: крекинг, риформинг, изомериза-
ция, ароматизация и алкилирование углеводородов.
4.5.2. Сущность и виды катализа
Катализом называют изменение скорости химических реак-
ций в результате воздействия веществ — катализаторов, которые
участвуют в реакции, вступая в промежуточное химическое взаи-
модействие с реагентами, но восстанавливают свой химический
состав по окончании каталитического акта.
Катализаторы могут находиться в газообразном, жидком и
твердом состояниях. По фазовому состоянию реагентов и катали-
затора каталитические процессы разделяют на две основные груп-
пы: гомогенные и гетерогенные.
Сущность ускоряющего действия катализаторов связана глав-
ным образом с изменением реакционного пути, на котором обра-
зование промежуточных соединений требует меньших энергетиче-
ских затрат, т. е. снижается энергия активации. Вследствие этого
увеличиваются количества промежуточных соединений, образую-
щихся и распадающихся в единицу времени. Особенностью многих
катализаторов является способность ускорять преим^тцественно
какую-либо одну или группу сходных реакций. Подбирая соответ-
ствующие катализаторы, можно из большого числа термодина-
мически возможных реакций выбрать те, которые приводят к об-
разованию требуемых продуктов. Иначе говоря, катализатор мо-
жет изменять селективность ХТП.
Действие катализаторов не влияет на равновесие, а лишь уско-
ряет достижение равновесия при данной температуре. Сущность
катализа одинакова для всех его видов, в частности для гомоген-
ного и гетерогенного.
Все каталитические реакции по типу взаимодействия между
реагентами и катализатором делят на окислительно-восстанови-
тельные и кислотно-основные.
Окислительно-восстановительный механизм
катализа связан с переносом электронов между реагентами и ка-
тализатором, который облегчает электронные переходы в реаги-
рующих молекулах. По этому принципу происходят процессы
62
окисления, дегидрирования, конверсии углеводородов, синтез ам-
миака, метанола и др.
Механизм кислотно-основного катализа заключа-
ется в переносе положительно заряженного иона, например про-
тона, или отрицательного иона, например гидроксила, между ката-
лизатором и реагентами, в результате чего происходят внутримо-
лекулярные превращения, облегчающие взаимодействия исходных
веществ. По такому типу протекают реакции гидратации, дегидра-
тации, гидролиза, этерификации, поликонденсации в растворах
и др.
Значительное число каталитических процессов в жидкостях и
газах протекает по цепному механизму, в котором катали-
затор служит инициатором. Ускорение достигается в результате
появления в процессе самой реакции богатых энергией частиц —
свободных радикалов. По радикально-цепному механизму проте-
кают реакции окисления алканов, в частности метана в формаль-
дегид, а также реакции хлорирования, полимеризации и др.
4.5.3. Скорость гомогенного каталитического процесса
Формально влияние катализаторов на кинетику процессов учи-
тывается изменением предэкспоненциального множителя и энер-
гии активации, входящих в уравнение Аррениуса. Рассмотрим
мономолекулярную гомогенно-каталитическую реакцию (К — ка-
тализатор) :
Каталитический процесс протекает в две стадии:
fel ь
А + К^АК-Л-В+К
kz
Обычно промежуточное соединение возникает в результате
быстрой обратимой реакции. При этом устанавливается равнове-
сие, которое можно охарактеризовать уравнением:
felCA (СК, О ~САК) = fe2CAK (4 -28)
Здесь Ск, о— начальная концентрация катализатора; Сак—.концентрация
промежуточного соединения; (Ск, о — Сак) — концентрация не связанного ката-
лизатора.
Поскольку 2-я стадия является лимитирующей, то она и опре-
деляет скорость процесса:
i/Ct,
^=-dr = ^CAK (4.29)
Из уравнения (4.28) следует, что концентрация промежуточ-
ного соединения равна
Сак = Л1САСК,о/(й2 + Л1Са) (4-30)
63
Скорость гомогенно-каталитического процесса оказывается
прямо пропорциональна начальной концентрации катализатора:
dCR
+ (4.31)
Влияние температуры, концентрации реагентов и давления на
скорость гомогенно-каталитических реакций аналогично вышепри-
веденным общим закономерностям протекания гомогенных про-
цессов. Основной недостаток гомогенного катализа — сложность
полного отделения катализатора от продуктов реакции. Вслед-
ствие этого велики неизбежные потери катализатора и загрязнение
им продуктов реакции.
4.6. ИЗМЕНЕНИЕ ОСНОВНЫХ ПОКАЗАТЕЛЕЙ
ГОМОГЕННЫХ ПРОЦЕССОВ ВО ВРЕМЕНИ
Продолжительность протекания ХТП — один из важных факто-
ров, определяющих значения основных показателей процессов, а
через них и многие показатели работы реакторов, в частности их
производительность, экологическую безопасность, а также эконо-
мические показатели производства в целом.
Изменение показателей во времени наиболее наглядно для
простых необратимых и обратимых процессов. При
проведении необратимых реакций (рис. 4.12, а, б) эти показатели
количественно можно рассчитать для любого отрезка времени, ис-
ходя из анализа кинетических уравнений.
Отличие характера изменения этих зависимостей для обрати-
мых реакций заключается в том, что концентрации реагентов и
степень превращения при больших т стремятся к своим равновес-
ным значениям (рис. 4.12,в,г).
Из приведенных графиков видно, что для того, чтобы довести
реакции до конца (ХА = 1 для необратимых реакций и Ха —
для обратимых), требуется очень большое время (теоретически
т = оо). При таком времени скорость процесса практически рав-
на 0. На практике процесс не доводят до конца, и технолог на-
ходит рациональную продолжительность проведения процесса трац,
при которой обеспечивается приемлемая степень превращения, а
процесс проходит с достаточно высокой скоростью. Выбор трац
главным образом проводят на основе экономических расчетов.
При проведении параллельных реакций характер изме-
нения концентраций исходных веществ во времени принципиально
не отличается от вышеприведенных графиков. Однако в этом слу-
чае мы получаем два или более продуктов, скорость изменения
концентраций которых различна и зависит, как отмечалось выше,
от соотношения констант скоростей основной ЛОС[, и побочной
k„ реакций и их порядка (рис. 4.13). Для реакций одинакового
порядка избирательность по мере увеличения продолжительности
проведения процесса остается постоянной, а ее численное значение
64
Рис. 4.12. Характер изменения концентраций реагентов С, степени превращения
X и скорости процесса U в зависимости от продолжительности т его протекания
для реакции А-> В (а, б) и As±B(e, г).
определяется соотношением —а = АОЕн/&п (рис. 4.14). С учетом
того, что степень превращения ХА растет во времени, а избиратель-
ность не изменяется, выход целевого продукта увеличивается и
Рис. 4.13. Изменение концентрации исходных реагентов и продуктов параллель
k\ kl
«ых необратимых реакций первого порядка А —> В и А —> D (ki > k2}.
Рис. 4.14. Характер изменения степени превращения и избирательности от г для
*, % *1 *2
реакций А —> В, А —> D и А —> В, А —> D (ki/k2>- kjk<^.
3 Зак. 186®
66
Рис. 4.15. Характер изменения избирательности во
времени при протекании параллельных реакций
разного порядка:
1 — порядок основной реакции ниже, чем побочной; 2—по-
рядок основной реакции выше, чем побочной. В — целевой
продукт.
при очень большом времени становится численно равным суммар-
ной избирательности (в пределе при т = оо, Фв(д> = <рв). Если про-
текают параллельные реакции разного порядка, то избиратель-
ность изменяется по мере увеличения продолжительности про-
цесса.
Например, если реакция образования целевого продукта В
имеет 2-й порядок, а побочная реакция— 1-й, то
Фв ~^В/^ = *1Са/(*2Са) = W*2==«CA. О (1 ~Ха) (4.32)
Из уравнения (4.32) следует, что по мере увеличения Х& изби-
рательность уменьшается. С учетом роста ХА с увеличением т сле-
дует вывод и об уменьшении избирательности во времени.
Если основная реакция описывается уравнением болёе низкого
порядка, чем побочная, то избирательность растет по мере увели-
чения продолжительности процесса (рис. 4.15).
Еще более сложно во времени изменяются показатели ХТП
при протекании последовательных реакций. Рассмотрим
наиболее простой случай, когда реакция происходит в соответ-
ствии с уравнением А —► В —*D. Если обе необратимые реак-
ции — 1-го порядка и объем реакционной системы постоянен, то
скорости расходования исходного вещества А и образования це-
левого В и побочного D продуктов можно описать следующими
уравнениями: dCA dt ==К1СА (4.33)
rfCR dx “° ~ *2^В - (4.34)
dC0 u в ь г (435)
dr = ^в
При этом
Ч А — А. 0 — £ В — (4.36)
После преобразования и решения системы уравнений (4.33)—
(4.36) получают выражения, описывающие изменения концентра-
ций реагентов во времени, время тОПт, за которое концентрация
целевого продукта достигает максимального значения, и само зна-
чение максимальной концентрации В, Св, макс. Характерное изме-
66
Рис. 4.16 Характер изменения концентраций ре-гентов во времени при протека-
Aj
нии последовательных реакций А —» В —> D.
Рис. 4.17. Изменение степени превращения Ха. избирательности <рв, выхода про-
Л, Л1
дукта Фв во времени при протекании последовательных реакций А —> В —► D
нение концентраций во времени показано на рис. 4.16. Значения
Св. макс, топт и соотношение скоростей изменения концентраций за-
висят от начальной концентрации Сд, о и констант скоростей
и k2, однако вид кривых сохраняется для широкого диапазона
изменения указанных параметров. Огеврдно, что степень превра-
щения вещества А увеличивается во времени (рис. 4.17). Избира-
тельность же, пропорциональная отношению скоростей образова»
ния продукта В и расходования А, будет максимальна в первый
момент времени от начала реакции, а затем падает. Выход целе-
вого продукта В, как и его концентрация, описывается кривой,
имеющей максимум при т = тОПт.
Таким образом, в случае последовательных реакций необхо-
димо знать оптимальную продолжительность протекания процесса,
обеспечивающую максимальный выход целевого продукта.
4.7. ПРИНЦИПЫ РАСЧЕТА ОПТИМАЛЬНЫХ ПАРАМЕТРОВ
ПРОВЕДЕНИЯ ПРОЦЕССОВ
Оптимальным значением какого-либо параметра процесса при-
нято считать такое, при котором конечные показатели процесса
являются наилучшими. Чаще всего оптимизируют лишь по одному
показателю, который называют критерием оптимизации.
Наиболее обоснованными являются экономические крите-
рии оптимизации, так как они отражают конечный результат
производства. Например, к такому показателю можно отнести се-
бестоимость продукта. Однако учесть влияние всех параметров
технологического режима на экономические показатели и рассчи-
тать наилучший режим очень сложно. Поэтому в качестве крите-
рия оптимизации часто используют технологические по-
казатели, характеризующие эффективность проведения процес-
сов и в конечном итоге косвенно обусловливающие экономические
8*
67
Рис. 4.18. Влияние степени превращения на себестоимость (Се) продукта как
экономический критерий оптимизации.
Рис. 4.19. Влияние температуры на степень превращения как технологический
Критерий оптимизации.
показатели. К таким критериям относят скорость процесса, свя-
занную с производительностью системы, выход продукта или сте-
пень превращения сырья и др. Технологические критерии наиболее
удобны при оптимизации проведения отдельного процесса или ра-
боты того или иного реактора. Оптимизирующими фак-
торами называют параметры технологического режима, с по-
мощью которых технолог может управлять процессом. При
оптимизации гомогенных процессов такими факторами являются
соотношение концентраций реагентов, температура, давление, про-
должительность протекания процесса и др.
Технологические и экономические критерии оптимизации часто
взаимосвязаны. Рассмотрим, например, производство продукта В
из исходного реагента А:
А В +
Пусть экономическим критерием оптимизации является себе-
стоимость продукта В. При малых степенях превращения А(Хд->0)
себестоимость В будет велика из-за высоких затрат на сырье
(если непрореагировавший компонент А не используется). При
высоких значениях Хд->Хд необходимо обеспечить большую про-
должительность протекания реакции т. В пределе для достижения
равновесной степени превращения Хд т-*оо. Это неизбежно при-
водит к высоким затратам на создание реакторов большой емко-
сти и их обслуживание, и себестоимость продукта В будет опять
высокой. Из этих рассуждений очевидно, что существует оптималь-
ная степень превращения ХдОПт<Ха, при которой себестои-
мость В будет минимальна (рис. 4.18). В этом случае Хд является
оптимизирующим параметром. Для наиболее экономичного дости-
жения Ха опт необходимо правильно выбрать температуру прове-
дения процесса ТОПт, при которой процесс будет протекать с мак-
симальной скоростью U. Скорость процесса становится критерием
оптимизации, а оптимизирующим параметром — температура
(рис. 4.19).
68
Критерий оптимизации, который записывается в виде функции
параметров, воздействующих на процесс, называют целевой
функцией. В число таких параметров входят и оптимизирую-
щие факторы. Математически задача оптимизации — это
определение (или расчет) таких значений оптимизирующих фак-
торов, при которых целевая функция достигает экстремума (мак-
симума или минимума). Такие значения оптимизирующих факто-
ров называют оптимальными. Существует несколько методов
определения оптимальных значений параметров. Простейшим из
них является аналитический, принцип которого заключается в том,
что после необходимых преобразований целевую функцию диф-
ференцируют по оптимизирующему параметру и находят его зна-
чение из условия равенства производной нулю.
Если целевая функция представлена в виде F = (ух, у2, у2, ...), то анали-
тический'метод определения оптимальных значений оптимизирующих параметров
Ух, Уъ Уз, ... заключается в решении системы;
dF
дух
0;
dF
ду2
= 0;
”-0
ду3
(4.37)
Решениями системы будут значения ух, опт, У2, опт, У». опт, • • , КОТОрЫе В СО-
вокупности определяют оптимальный режим технологического процесса.
Рассмотрим наиболее простой случай определения оптималь-
ной температуры проведения необратимой реакции 1-го порядка:
А —► В. За критерий оптимизации примем скорость процесса U.
Тогда целевую функцию можно записать в виде:
F =С7 = Аоехр[-£/(ЛГ)]СА (4.38)
Как очевидно, целевая функция с ростом температуры растет
неограниченно, и экстремум, как таковой, отсутствует (производ-
ная скорости по температуре ни при каких значениях последней
не равна нулю). В этом случае оптимальное значение температуры
определяется ограничениями по максимально допустимой темпе-
ратуре Тмакс, рассмотренными выше. То есть
Т'опт = Т’макс (4.39)
В качестве второго примера приведем решение более сложной задачи —
определение оптимальной температуры обратимой экзотермической реакции:
k,
A^tB
fe,
Пусть прямая и обратная реакции являются реакциями 1-го порядка по
соответствующим реагентам (А и В). Критерием оптимизации будем считать
скорость реакции. Целевая функция запишется в виде:
F = U = fe01 exp [- Ei/(RT)] Ск - k02 exp [- E^RT)] CB (4.40)
Входящие в уравнение (4.40) концентрации СА и Св не являются оптимизи-
рующими, так как они независимы; их изменения происходят в результате реак-
ции, и мы не можем управлять процессом, изменяя СА и Св. Единственный
независимый параметр, влияющий на целевую функцию, — это температура. Для
ее расчета в соответствии с условием (4г39) запишем частную производную
69
скорости по температуре-при фиксированных значениях С л и Св:
> = {*01 «р [- СА - *02 ехР [- ЦМ] св} = О (4.41)
После дифференцирования н преобразований получаем формулу для расчета
оптимальных темперит' р:
г“" - - £,)/(« In )-(£,- £,)/(« I. <«)
Подставляя значения Гопт для соответствующих концентраций в уравнение
(4.42) строят линию оптимальных температур в координатах V — Т.
Из уравнения (4.42) следует, что при Лл-*-0 ТОпт -»-со. Поэтому для малых
степеней превращения вводят ограничения. ТОПт = Т„акс.
Как отмечалось выше, начальная концентрация реагирующих
веществ существенно влияет на скорость процесса и другие пока-
затели. Для простых необратимых и обратимых реакций техно-
логи стремятся увеличить начальную концентрацию реагента. Од-
нако, когда смесь состоит из двух реагентов, повышение концен-
трации одного из них неизбежно вызовет снижение концентрации
другого. При предельной концентрации (0 и 100%) любого из
компонентов скорбеть реакции равна нулю. Значит между этими
пределами должны существовать оптимальные концентрации или
оптимальное соотношение концентраций реагентов, при котором
скорость процесса становится максимальной.
Рассмотрим пример определения оптимального соотношения концентраций
водороду и азота для синтеза аммиака. За критерий оптимизации примем ско-
рост: > реакции синтеза NHs. Она описывается уравнением:
I/ = *Л2 Ш^Нз)0'5 - *2 (^Нз W5 (4-43)
Здесь PHj, PN2, PNHb — парциальные давления водорода, азота и аммиака
соответственно.
Для упрощения задачи рассмотрим случай, когда реакция протекает вдали
от равновесия, т. е. когда скоростью обратной реакции И влиянием продуктов
реакции можно пренебречь. Тогда целевая функция
F = t/ = fe1PN/>iS2 (4.44)
Обозначив через JVHs мольную долю Н2 в смеси и учитывая, что РНа ==
= ^н/’общ- РЫг •= (1 — Лгн2) робщ> скорость ре (кции запишем в виде уравнения:
С/ = А1(1-1УН2)^ = *1да2-№н52) (4.46) 1
Продифференцировав целевую функцию по определим оптимальное
значение 1VH2, соответствующее максимальной скорости
*1-2М£ onT = V 1.52V&1 ОПТ(1-6/з*н2) = 0 (4-46) , 'J
откуда Nh2, опт = 3/з; для азота: (1 — NHa, опт) =« 2/s-
Следовательно, оптимальное соотношение концентраций азота и водорода
для рассматриваемого случай составляет:
(VH3)onT = 2/3
При про: канни реакции вблизи равновесия необходимо учитывать и ско-
рость обратной реакции, Выполнив анализ уравнения (4.43), подобный изложен- |
70
ному выше, получим, что максимальная скорость реакции синтеза будет обеспе-
чиваться при соотношении Na : Hz = 1:3.
Аналогично решаются задачи нахождения оптимального вре-
мени для последовательных реакций, при котором обеспечивается
максимальный выход целевого продукта, и оптимального давле-
ния при проведении газофазных реакций, протекающих с увели-
чением объема.
Рассмотренные основные закономерности протекания гомоген-
ных процессов применимы и для анализа гетерогенных процессов,
протекающих в условиях отсутствия диффузионных торможений,
т. е. щля кинетической области. В то же время в промышленных
условиях большинство гетерогенных процессов протекает при су-
щественном влиянии процессов массопереноса (диффузионных
процессов) на общую (наблюдаемую) скорость ХТП. Основные
закономерности протекания гетерогенных процессов, влияние па-
раметров технологического режима на скорость массопереноса и
на общую скорость процесса рассмотрены в следующей главе. Сле-
дует отметить, что эти закономерности, как и приведенные выше,
справедливы для ХТП, проводимых в замкнутом объеме, в отли-
чие от процессов, в которых реакция осуществляется в потоке.
ГЛАВА 5
ЗАКОНОМЕРНОСТИ ПРОВЕДЕНИЯ ГЕТЕРОГЕННЫХ
ХИМИКО-ТЕХНОЛОГИЧЕСКИХ ПРОЦЕССОВ
5.1. ОБЩАЯ ХАРАКТЕРИСТИКА ГЕТЕРОГЕННЫХ ХТП
При протекании большинства промышленных ХТП реагенты на-
ходятся в разных фазах. Такие процессы называют гетероген-
ными. В химической технологии наиболее распространены гетеро-
генные процессы, когда реагирующие вещества находятся в следу-
ющих сочетаниях фазовых состояний: газ — жидкость (Г — Ж),
газ — твердое вещество (Г — Т), жидкость — твердое вещество
(Ж — Т), твердое вещество — твердое вещество (Т — Т), газ —
жидкость — твердое вещество (Г — Ж —Т). В качестве примеров
промышленных гетерогенных процессов в указанных системах
можно привести:
производство оксидов металлов и оксида серы (IV) путем об-
жига сульфидов в кислородсодержащей атмосфере
2FeS (тв.) + 30 (г.) = 2FeO (тв.) + 2SO2 (г.)
4FeS2 (тв.) + 11О2 (г.) = 8SO2 (г.) + 2Fe2O3 (тв.)
сжигание твердых углеродсодержащих материалов с образова-
нием оксидов углерода, а также получение водяного газа при взаи-
модействии углерода с водяным паром
С (тв.) + Н20 (г.) == СО (г.) + Н2 (г.)
71
получение цианамида кальция при взаимодействии карбида
кальция и атмосферного азота
СаС2 (тв.) + N2 (г.) = CaCN2 (г.) + С (тв.)
I
производство сероуглерода синтезом из элементов
С (тв.) + 2S (ж.) = CS2 (г.)
Как правило, "при проведении гетерогенных ХТП химическая
реакция протекает на поверхности контакта фаз, к которой предва-
рительно должны быть подведены реагенты. Таким образом, в от-
личие от гомогенных процессов на скорость гетерогенных процессов
влияет не только скорость химической реакции, но и скорость под-
вода реагентов к поверхности контакта фаз и скорость отвода про-
дуктов реакции от нее. Условная поверхность контакта фаз нахо-
дится внутри одной из фаз, и она в общем случае не-совпадает
с поверхностью раздела фаз. Глубина проникновения реагентов
внутрь одной из фаз к поверхности контакта зависит от соотноше-
ния скоростей диффузии и реакции. При очень низкой скорости
реакции процесс может протекать во всем объеме одной из фаз.
Влияние физических процессов переноса вещества (массопередачи
между фазами) тем больше, чем выше скорость химической реак-
ции. Стадию, скорость которой существенно меньше возможных
скоростей других стадий, называют лимитирующей.:При нали-
чии лимитирующей стадии наиболее эффективны те воздействия на
процесс, которые изменяют скорость именно лимитирующей стадии.
В свою очередь, массопередача сама может протекать в не-
сколько последовательных стадий. Так, для гетерогенного процесса
с участием пористого твердого вещества (например, кокса) и газа
(например, кислорода) можно выделить несколько элементарных
•стадий: 1) подведение путем диффузии реагирующих веществ (О2)
из потока к внешней поверхности твердого тела; 2) диффузия га-
зообразных реагентов (Ог) в порах зерна твердого тела к его внут-
ренней поверхности; 3) собственно химическая реакция (с образо-
ванием оксидов углерода); 4) диффузия продуктов реакции (СО,
СО2) из внутренних областей твердого вещества к внешней повер-
хности; 5) диффузия продуктов реакции с внешней поверхности
в поток.
Любая из этих стадий при определенных условиях может ока-
заться лимитирующей.4 Скорость процесса в целом и закономер-
ности его протекания определяются скоростью лимитирующей ста-
дии. Если лимитирующей является стадия 1 или 5, то говорят, что
процесс протекает во внешнедиффузионной области, т. е.
общая скорость ХТП определяется скоростью диффузии реагентов
или продуктов через пограничный слой газа или жидкости к внеш-
ней поверхности твердого вещества. Если скорость ХТП лимитирует
2- или 4-я стадии — процесс протекает во внутр и диффузио н-
ной области, когда лимитирующей стадией является химиче-
ская реакция, — в кинетической области. При несуществен-
72
ном различии скоростей смежных стадий процесс протекает в со-
ответствующих переходных областях.
^Лимитирование важно учитывать как при описании процессов,
так и при выборе наиболее эффективных приемов их интенсифика-
ции и управления химико-технологическими процессами. Эти при-
емы различны для разных областей протекания ХТП. Для эффек-
тивного воздействия на процесс технологу необходимо определить
лимитирующую стадию, т. е. выявить область протекания ХТП,
а затем воздействовать именно на лимитирующую стадию. При
протекании процессов как в крайних, так и в переходных областях
изменение скорости одной стадии приводит к изменению скоростей
и других стадий. При этом возможен переход процесса из одной
области протекания в другую.
В общем случае скорость гетерогекного ХТП выражается урав-
нением, в котором в том или ином виде учитывается поверхность
контакта фаз. Например, если скорость относят к единице объема
реакционного пространства v или к единице массы катализатора тк
то ее значение будет пропорционально поверхности контакта фаз,
приходящейся на единицу объема (Гуд, м2/м3) или массы (Руд,
м2/кг):
и = KFyJt ДС; U' = K'Fyд ДС (5.2)
Коэффициент скорости К и движущай сила ДС процесса зависят
от области протекания ХТП.
/ Если гетерогенный процесс протекает в кинетической области,
то скорость процесса, лимитируемая скоростью химических реак-
ций, описывается кинетическим уравнением реакции. При этом за-
кономерности гетерогенного ХТП, протекающего в кинетической
области, аналогичны закономерностям гомогенных химических про-
цессов (см. главу 4).
5.2. ПРОЦЕССЫ,
ПРОТЕКАЮЩИЕ ВО ВНЕШНЕДИФФУЗИОННОЙ ОБЛАСТИ
При протекании процессов во внешнедиффузионной области ли-
митирующей стадией является массопередача реагентов из потока
к наружной поверхности раздела фаз. В этом случае наблюдаемая
скорость процесса определяется скоростью переноса реагентов из
потока к наружной поверхности или отвода продуктов от наруж-
ной поверхности в поток.
Диффузия газа или жидкости в пористом теле и скорость хи-
мической реакции в состоянии обеспечить взаимодействие между
значительно большими массами реагентов, чем это возможно в рас-
сматриваемых условиях. В таком случае говорят о наличии
внешнедиффузионного торможения, препятствующего
73
максимальному использованию возможностей химической реакции.
Увеличение разности между потенциально возможной скоростью
реакции и фактически наблюдаемой скоростью процесса свиде-
тельствует о повышении диффузионного торможения.
Перенос вещества осуществляется в результате молекуляр-
ной диффузии, связанной с тепловым движением молекул, и путем
конвекции, характеризуемой движением масс газа или жид-
кости вследствие механического воздействия. Конвекция и моле-
кулярный перенос — два независимых параллельно действующих
фактора. Молекулярный перенос является определяющим в тонком
пограничном слое, в котором конвективный перенос практически
отсутствует. В то же время усиление конвекции, например путем
перемешивания, уменьшает толщину пограничной пленки и тем са-
мым облегчает доступ реагентов к поверхности раздела фаз. Вклад
молекулярной диффузии в большинстве практических случаев не-
велик и не превышает 10 %.
Скорость молекулярной диффузии Uu можно оценить по формуле:
Здесь F — внешняя поверхность раздела фаз; DM—коэффициент молекуляр-
ной диффузии, - б — толшнна пограничного слоя; АС — рйзнееГь концентраций
реагента в потоке и на поверхности.
Для газа движущую силу обычно выражают как разность давлений реа-
гента в потоке и у поверхности.
Коэффициент молекулярной диффузии зависит от природы и физических
свойств компонентов смеси, от температуры Т и давления Р:
DH = aTu-44‘1-8' (5.4)
, Du = bP~l (5.5)
Здесь а, Ь — постоянные.
Скорость конвективного переноса UK определяется по формуле:
UK = ₽ АС (5.6)
Здесь Р — коэффициент массопередачи, рассчитываемый по эмпирическим
уравнениям,.
При совместном вкладе молекулярной и конвективной диффузии для оценки
скорости переноса вещества к поверхности раздела фаз (С/») используют значе-
ния эффективных коэффициентов диффузии (Йэ), которые рассчитывают также
по эмпирическим критериальным уравнениям.
dn
Суммарный диффузионный поток в единице реакционного объема
в этом случае будет равен:
_^_=6/эДуд=-^ДСГуд (5.7)
Основные признаки протекания процесса во внешнедиффузион-
ной области: а) сильное влияние линейной скорости потока или
интенсивности перемешивания на наблюдаемую скорость процесса
при постоянстве времени контакта фаз; б) слабая зависимость
влияния температурыхна скорость процесса, обусловленная низким
значением энергии активации; в) наблюдаемый порядок реакции
74
Рис. 5.1. Влияние перемешивания на ско-
рость и степень превращения для внещне-
циффузионной области протекания прогесса.
/—внешне диффузионная область; /7—внутри
диффузионная или кинетическая область.
не превышает 1, независимо от истинного порядка реакции; г) на-
личие градиента температур между потоком и внешней поверх-
ностью твердого вещества при проведении реакций с тепловым
эффектом. При этом для газофазных экзо- и эндотермических про-
цессов с большим тепловым эффектом эта разность может дости-
гать десятков градусов.
Первый фактор используют для определения внешнедиффузион-
ной области протекания процесса. Для этого изменяют интенсив-
нисть перемешивания /п или скорость потока и измеряют скорость
процесса или степень превращения исходного вещества (рис. 5.1).
Зона 1 сильного влияния перемешивания характерна для протека-
ния процесса во внешнедиффузионной области. Неизменность U или
X в зоне И указывает, что скорость подвода реагентов к поверх-
ности раздела фаз не лимитирует скорости процесса и он проходит
или во внутридиффузионной или в кинетической областях.
Основным фактором интенсификации процесса, проходящего во
внешнедиффузионной области, является повышение турбулизации
в зоне раздела фаз. Это достигается или повышением интенсивно-
сти перемешивания или увеличением линейной скорости движения
жидкой или газовой фаз.
Менее существенно повышение температуры, которая сказы-
вается лишь на молекулярной составляющей диффузии. Следует
учитывать, что с ростом температуры процесс еще более углуб-
ляется во внешнедиффузионную область, так как скорость хими-
ческой реакции возрастает быстрее скорости молекулярной диф-
фузии. Таким образом, несмотря на некоторое увеличение наблю-
даемой скорости процесса с повышением Т, внешнедифЛузионные
торможения растут, и этот способ интенсификации не является ра-
циональным.
Скорость процесса увеличивается прямо пропорционально
удельной внешней поверхности раздела фаз. При проведении гете-
рогенного процесса с системами Г—Т или Ж—Т внешнюю поверх-
ность увеличивают в основном за счет измельчения твердой фазы.
Однако с уменьшением размера частиц твердой фазы могут сни-
жаться значения коэффициента массопередачи, и в каждом конк-
ретном случае существует оптимальный размер пастиц.
В системах Г — Ж поверхность раздела фаз развивают путем
организации движения жидкости в виде тонких пленок, распыления
жидкой фазы на мелкие капли, диспергирования газа в объеме
жидкости методом барботирования, диспергирования жидкости
75
и газа с использованием кинетической энергии газовых струй или
за счет механического перемешивания реагирующих фаз. Следует
отметить, что в последнем случае с увеличением поверхности одно-
временно растет и коэффициент массопередачи и этот прием интен-
сификации является наиболее рациональным.
При протекании процесса во внешнедиффузионной области дви-
жущей силой в уравнении скорости процесса (5.2) является раз-
ность концентраций реагентов в потоке С,о и на внешней поверх-
ности C/F. При этом увеличение С,о способствует увеличению на-
блюдаемой скорости процесса.
5.Э. ВНУТРИДИФФУЗИОННАЯ ОБЛАСТЬ ПРОТЕКАНИЯ ПРОЦЕССОВ
Протекание процессов во внутридиффузионной области наибо-
лее часто наблюдается в системе Г—Т или Ж—Т, если твердое
вещество является пористым. Лимитирующей стадией в этом слу-
чае будет диффузия реагентов или продуктов реакции в порах.
Наблюдаемая скорость процесса определяется или скоростью до-
ставки реагентов от внешней поверхности к внутренней, на которой
осуществляется химическая реакция, или скоростью диффузии про-
дуктов реакции из объема пористого тела к его внешней поверх-
ности. Поскольку скорость химической реакции в рассматриваемом
случае ограничивается возможностями доставки реагентов к внут-
ренней поверхности, то говорят о внутридиффузионном
торможении процесса. Если внешнедиффузионные тормо-
жения снижаются с уменьшением толщины пограничного слоя, то
внутридиффузионные торможения при неизменных условиях прове-
дения процесса ослабляются с уменьшением длины пор. ч
В отличие от внешнедиффузионной области протекания про-
цесса перенос вещества в порах осуществляется исключительно за
счет диффузии молекул.
Механизм движения молекул в порах зависит от соотношения диаметра пор
d„ и средней длины свободного пробега молекул X. Если dn существенно превы-
шает X (например, в 10 раз и более), то молекулы сталкиваются друг с другом
гораздо чаще, чем со стенками поры, и последние не оказывают существенного
влияния на скорость молекулярной диффузии. В этом случае нет разницы между
диффузией в поре и диффузией в объеме неподвижного газа или жидкости.
Скорость переноса вещества вдоль поры описывается уравнениями (5.2)—(5 4)
Например, при атмосферном давлении (X ~ 10-7 м) по такому механизму осу-
ществляется диффузия в порах с диаметром больше 10_® м. Чем выше давление,
тем меньше может быть диаметр пор, при котором действием стенок пор на мас-
соперенос можно пренебречь. При малом диаметре пор (например, 10-8 м) и
низких давлениях, когда средняя длина свободного пробега молекул превышает
dn, молекулы чаще ударяются о стенки, чем сталкиваются друг с другом, и за
счет отражения от стенок препятствуют продвижению молекул вдоль пор. За-
кономерности диффузии в таких условиях проанализированы Кнудсеном, и коэф-
фициенты диффузии при X >> d„ называют кнудсеновскимн (Дкн). Для кнудсе-
новской диффузии характерна независимость DKH от давления, слабая зависи-
мость от температуры и прямая пропорциональность диаметру пор:
Окн — fdn у/т
(6.8)
76
Рнс. 5.2. Зависимость коэффициентов внутренней
диффузии от диаметра пор.
Здесь f — коэффициент, зависящий от молекулярной массы диффундирую-
щего газа.
Таким образом, коэффициент кнудсеновской диффузии пропорционален диа-
метру пор и не зависит от давления, в то время как коэффициент молекулярной
диффузии не зависит от dn и обратно пропорционален давлению. Из-за столкно-
вений молекул со стенками пор скорость кнудсеновской диффузии меньше ско-
рости объемной молекулярной.
Характер изменения коэффициентов диффузии в широком диапазоне изме-
нения диаметра пор показан на рис. 5.2.
В зоне I при d„ X массоперенос осуществляется объемной молекулярной
диффузией. В зоне II при dn С X имеет место кнудсеновская диффузия. В зоне III
диаметр пор в несколько раз превышает диаметр молекул du, и из-за действия
силового поля внутренней поверхности столкновения молекул со стенками пор
уменьшаются и соответственно наблюдается некоторый рост D с уменьшением d„.
Зона IV соответствует диаметру пор, соизмеримому с диаметром молекул. В этой
зоне при заданном диаметре пор коэффициент диффузии сильно зависит от диа-
метра молекул, вследствие чего такое явление в химической технологии исполь-
зуют для разделения веществ на молекулярных ситах (цеолиты, некоторые сорта
пористых стекол, активированных углей).
Характерные признаки протекания процессов во внутридиффу-
зионной области: а) сильная зависимость скорости процесса от
размера частиц d4 твердого материала, связанная с ослаблением
внутридиффузионных торможений с уменьшением d4 и соответ-
ственно средней длины пор; б) незначительное влияние темпера-
туры на скорость процесса, особенно при мелкопористой структуре
твердого вещества, диффузия в котором протекает по механизму
Кнудсена; в) изменение порядка реакции и энергии активации при
изменении условий протекания процесса (Т,Р,С ...); г) наличие
градиента температур внутри пористого твердого материала. Раз-
ность температур между внутренней и внешней поверхностью мо-
жет превышать 100 градусов при высоком тепловом эффекте и низ-
кой теплопроводности материала пористого тела.
Первый фактор используют для определения внутридиффузион-
ной области протекания процесса. С этой целью измеряют скорость
процесса или степень превращения реагентов в зависимости от раз-
мера частиц твердой фазы при постоянном времени контакта фаз
(рис. 5.3). С уменьшением d4 снижаются внутридиффузионные тор-
можения, и вследствие этого растет скорость (зона I). Практиче-
ская неизменность U и X при уменьшении размера частиц ниже
некоторого значения (зона //) свидетельствует о переходе процесса
в кинетическую область.
Фактор снижения диффузионных торможений с уменьшением
размера частиц твердой фазы является основным при интенсифи-
кации процессов, протекающих во внутридиффузионной области.
77
Рнс. 5.3. Влияние размера частиц твердой
фазы на скорость процесса и степень пре-
вращения.
Другой прием интенсификации — повышение давления Р. С ро-
стом Р увеличивается движущая сила процесса диффузии (ДС) в
уравнении (5.2).
При низких давлениях в тонкопористых материалах основную роль в пере-
носе массы играет кнудсеновская диффузия. Так Как коэффициент кнудсеновской
диффузии не зависит от давления, а движущая сила диффузии растет с увели-
чением Р, то наблюдается рост скорости диффузии и соответственно скорости
процесса (зона I на рис. 5.4). При более высоких давлениях (зона II) происхо-
дит частичный переход кнудсеновской диффузии в объемную молекулярную.
Рост скорости диффузии и процесса замедляется из-за уменьшения коэффициента
молекулярной диффузии. Область III характерна для объемной молекулярной
диффузии, когда снижение не компенсируется ростом ДС с увеличением дав-
ления.
В тех случаях, когда возможно управление пористой структурой
твердой фазы, выгодно до некоторого предела увеличивать сред-
ний диаметр пор da (рис. 5.5, зона /). Снижение скорости процесса
с дальнейшим увеличением d„ связано с уменьшением удельной по-
верхности контакта фаз (зона II).
Увеличение удельной поверхности, как следует из уравнения
(5.2), приводит к прямо пропорциональному росту скорости процес-
са только в том случае, если при развитии поверхности путем уве-
личения числа пор и уменьшения их размеров не происходит пере-
ход объемной молекулярной диффузии в кнудсеновскую (рис. 5.6.
зона/). Если размеры пор уменьшаются до значений, соответствую-
сти:
/—кнудсеновская диффузия; //—смешанная диффузия; III—объемная молекулярная диффу-
зия.
Рис. 5.5. Влияние размера пор на скорость процесса во внутридиффузионной об-
ласти:
1 — переход кнудсеновской диффузии в объемную молекулярную: II— уменьшение удельной
поверхности твердой фазы.
Рис. 5.6. Влияние удельной внутренней поверхности на скорость процесса во вну-
тридиффузионной области.
78
щих кнудсеновской диффузии, то рост скорости замедляется вслед-
ствие увеличения диффузионных торможений (зона II). При даль-
нейшем уменьшении dn увеличение удельной внутренней поверх-
ности (зона III) не компенсирует уменьшения скорости диффузии.
Увеличение скорости процесса bq внутридиффузионной • области
следует также ожидать при повышении начальной концентрации
реагентов газовой или жидкой фаз, так как при этом возрастает
движущая сила процесса диффузии.
S.4. КИНЕТИЧЕСКАЯ ОБЛАСТЬ ПРОТЕКАНИЯ ПРОЦЕССОВ
7 При протекании процессов в кинетической области стадия хи-
мической реакции является лимитирующей, и ее скорость опреде-
ляет скорость процесса в целом. Диффузионные торможения в этом
случае отсутствуют, и скорость процесса рассчитывают, используя
методы истинной или формальной кинетики, по кинетическим урав-
нениям, рассмотренным в главе 4. *
Основные признаки протекания процессов в кинетической об-
ласти: а) сильная зависимость скорости процесса от температуры/
описываемая уравнением Аррениуса (4.11). При этом энергия ак-
тивации, как правило, велика и при уменьшении температуры
в большинстве случаев практически не изменяется;-' б) независи-
мость скорости процесса от линейной скорости газа или жидкости
(при сохранении неизменным времени кснтакта) и интенсивности ~
перемешивания (при проведении процесса в замкнутом объеме),
а для высокопористых материалов — и от размера частиц.
Для определения кинетической области протекания процесса
проводят измерения скорости процесса или степени превращения
исходных веществ при переменных значениях линейной скорости
потока, интенсивности перемешивания, размера пористых частиц
(см. рис. 5.1, 5.3). Неизменность U или X при изменении указан-
ных параметров свидетельствует о протекании процесса в кинет и-
ческой области.
/Основные приемы интенсификации основаны на правильном
выборе температуры, давления, соотношения концентраций реаген-
тов и других параметров, анализ влияния которых на скорость хи-
мических реакций выполнен в главе 4. Эти приемы направлены на
увеличение константы скорости реакции и движущей силы процес-
са^ В отличие от гомогенных процессов, дополнительным средством
интенсификации гетерогенных процессов, протекающих в кинетиче-
ской области, является развитие удельной поверхности контакта
фаз.
Приемы увеличения поверхности для систем Г — Ж, Г (Ж) — Т
(непористое) при кинетической области протекания процесса прин-
ципиально не отличаются от описанных выше (см. разд. 5.2) спо-
собов для внешнедиффузионной области.
Для пористых твердых материалов для системы Г(Ж)—Т
основным фактором развития внутренней поверхности является
79
уменьшение диаметра пор при увеличении их числа. В отличие от
диффузионных областей скорость процессов, протекающих в кине-
тической области, всегда прямо пропорциональна удельной поверх-
ности материала. Отклонение от прямолинейной зависимости ука-
зывает на переход процесса во внутридиффузионную область.
5.5. ОСОБЕННОСТИ ПРОТЕКАНИЯ
ГЕТЕРОГЕННО-КАТАЛИТИЧЕСКИХ ПРОЦЕССОВ
Гетерогенный катализ играет решающую роль во многих отрас-
лях химической и нефтехимической промышленности. Поэтому тех-
нолог постоянно встречается с проблемами гетерогенного катализа.
При гетерогенном катализе катализатор и реагенты находятся
в разных фазах, и реакция протекает на поверхности раздела этих
фаз. Наибольшее практическое значение имеют каталитические
процессы при взаимодействии газообразных или жидких реагентов
с твердыми катализаторами.
В общем виде скорость реакции на твердом катализаторе опи-
сывают уравнением:
и==k°ехр [_ E,{RT}] ‘лс
(5-9)
Как отмечалось выше (см. разд. 4.3), при высоких температу-
рах увеличивается скорость реакции, но некоторые продукты при
этом могут разлагаться. При проведении обратимых экзотермиче-
ских реакций с ростом температуры уменьшается равновесное со-
держание продукта. Применение катализаторов позволяет прово-
дить реакции при пониженных температурах, обеспечивая при
этом достаточно высокую скорость и степень превращения (рис. 5.7).
В качестве примеров гетерогенно-каталитических процессов
можно привести: крекинг углеводородов на алюмосиликатных ка-
тализаторах; синтез спиртов гидратацией олефинов на магний-
силикатных катализаторах; получение бутадиена из этанола; окис-
ление о-ксилола или нафталина во фталевый ангидрид и оксида
серы(IV) в оксид серы(VI) на ванадиевых катализаторах; окисле-
ние аммиака в оксиды азота на платино-родиевых катализаторах;
этилена в оксид этилена и метанола в формальдегид на серебря-
ных катализаторах; пропилена в акролеин на медьсодержащих
катализаторах; отверждение жиров на никелевых катализаторах;
Рис. 5.7. Зависимость степени превращения
от тег.пеоатуры для каталитической (/) и
некаталитической (2) обратимой экзотерми-
ческой реакции.
&)
конверсия оксида углерода и синтез метанола на цинкхромоксид-
ных катализаторах; синтез аммиака на железосодержащих ката-
лизаторах; полимеризация этилена на хром молибденоксидных ка-
тализаторах и др.
Примерами жидкофазного катализа служат: получение бензаль-
дегида из метилбензола в растворе сульфата меди; производство
ацетальдегида из ацетона на суспензированном сульфате ртути;
синтез высших спиртов из простых спиртов, осуществляемый на
коллоидных платиновых или палладиевых катализаторах.
5.5.1. Основные требования к катализаторам
Выбор катализатора для того или иного процесса определяется
в основном технологическими и экономическими соображениями.
Катализаторы должны иметь высокую активность, избирательность,
низкую температуру зажигания, быть стабильными в работе, мало-
чувствительными к перегреву, к действию контактных ядов, иметь
достаточную механическую прочность.
При сравнении различных катализаторов для одного процесса
используют понятие «активность» катализатора.
Активность катализатора — это мера ускоряющего
действия его на данную реакцию. Наиболее часто активность вы-
ражают разностью или соотношением скоростей каталитической
и некаталитической реакций, степенью превращения исходного реа-
гента или выходом целевого продукта при определенном техноло-
гическом режиме. Иногда активность представляют соотношением
констант скоростей каталитической и некаталитической реакций
или отношением энергий активации.
Избирательность (селективность) действия катали-
заторов важна для большинства каталитических процессов органи-
ческой технологии, в которых термодинамически возможен ряд
параллельных и последовательных реакций. Избирательность неко-
торых катализаторов позволяет сильно ускорять только одну реак-
цию из ряда возможных, проводить процесс при такой температуре,
при которой подавляются другие реакции.
Температура зажигания — минимальная температура,
при которой катализатор имеет активность, достаточную для авто-
термической работы в промышленных услозиях. Понижение темпе-
ратуры зажигания обеспечивает снижение энергетических затрат
на предварительный нагрев реагентов и облегчает пуск аппаратов
и производств в целом.
Термостойкость катализаторов определяет возможность
стабильной работы при высоких температурах. Высокая термо-
стойкость особенно важна при проведении сильно экзотермических
процессов. При высоких температурах могут происходить процессы
образования неактивных кристаллов, процессы спекания, приводя-
щие к уменьшению внутренней поверхности контакта.
Стойкость катализатора к действию контактных
ядов — важнейшая характеристика, определяющая возможность
81
отравления катализатора. Отравление катализатора—это частичная
или полная потеря активности под действием небольшого коли-
чества веществ, называемых контактными ядами. Потеря актив-
ности происходит вследствие выключения части активной внутрен-
ней поверхности. При отравлении катализаторов различают истин-
ное отравление и блокировку поверхности. Истинное отравление
наступает при химическом взаимодействии яда с катализатором
с образованием каталитически неактивного соединения. Снижение
активности из-за блокировки связано с механическим экранирова-
нием поверхности катализатора твердыми веществами, образую-
щимися при катализе. Отравление бывает обратимым и необрати-
мым. При обратимом отравлении активность восстанавливается
после исключения яда из исходной смеси. При необратимом от-
равлении первоначальная активность не восстанавливается.
Прочность катализатора должна обеспечивать его эксплуа-
тацию в течение длительного времени (до нескольких лет). Он не
должен разрушаться при загрузке, выгрузке, транспортировке по
трубопроводам и т. д. t
5.5.2. Свойства твердых катализаторов и их производство
При разработке катализаторов технологи стремятся обеспечить
высокую активность путем увеличения как каталитической актив-
ности единицы поверхности, так и самой удельной поверхности.
Поиышение активности единицы поверхности достигается путем
правильного выбора состава катализаторов или, как их часто на-
зывают, контактных масс. Контактные массы обыно не являются
индивидуальными веществами, а представляют смесь собственно
каталитически активных соединении с актива горами, тем или иным
способом нанесенную на носители.
Активаторы, или промоторы — это вещества, повышаю-
щие активность основного вещества-катализатора. При этом сами
активаторы могут не обладать каталитическими свойствами. Ме-
ханизм активирующего действия промотором сложен и разнообра-
зен Иногда активатор увеличивает поверхность основного активного
вещества или число Активных центров на единице поверхности.
В других случаях активация происходит вследствие взаимодей-
ствия промотора с основным активным веществом и образования
продуктов с более высокой активностью. Промоторы могут также
способствовать повышению стойкости контактных масс или слу-
жить защитой от воздействия контактных ядов.
Носителями или трегерами называют инертные веще-
ства, на которые наносят каталитически активные соединения или
промоторы. Как правило, это пористые вещества с достаточно вы-
сокой термостойкостью. Иногда они выполняют функции и актива-
торов. Примерами носителей служат: пемза, каолин, асбест, сили-
кагели, керамика, пористый уголь, различные соли. Следует отме-
тить, что если скорость химической реакции очень высокая, то
бессмысленно применять пористые носители. Например, реакция
82
окисления оксида серы (IV) на оксиднованадиевых катализато-
рах— умеренно быстрая, и ее проводят на пористых катализато-
рах. Э'га же реакция или реакция окисления аммиака до оксидов
азота ца платиновом катализаторе — очень быстрые. Практически
всегда они заканчиваются на внешней поверхности. Поэтому ис-
пользуют непористые катализаторы (и соответствующие носители),
выполненные в виде сеток.
В пористых катализаторах высокая удельная поверхность обес-
печивается пористой структурой носителя, характеризуемой долей
объема пор от общего объема частицы (пористость), размерами
пор и их числом.
Основные методы изготовления катализаторов:
1. Осаждение гидроксидов или карбонатов из растворов их со-
лей с последующим формованием и термообработкой контактной
массы (осажденные катализаторы).
2. Смешение порошкообразных Каталитически активных ве-
ществ, промоторов, носителей и связующих с последующим прес-
сованием и термообработкой (смешанные катализаторы).
3. Сплавление нескольких оксидов с последующим восстанов-
лением металлов из оксидов (плавленые катализаторы). Иногда
после сплавления одно из веществ удаляют путем выщелачивания
(скелетные катализаторы).
4. Пропитка пористого носителя раствором, содержащим соли
активных элементов, с их последующей сушкой и термообработкой
(нанесенные катализаторы).
Катализаторы выпускают в виде таблеток, колец, шариков,
цилиндрических гранул, сеток и др.
5.5.3. Области протекания гетерогенно-каталит» ческих процессов.
Влияние диффузионных торможений на избирательность
Каталитические процессы как частный случай гетерогенных ХТП в зависи-
мости от технологических условий могут протекать в диффузионных и кинетиче-
ских областях.
Признаки и закономерности протекания каталитических процессов во внешне-
диффузионной области, а также приемы интенсификации аналогичны рассмо-
тренным в разд. 5.2.
Большинство промышленных процессов со сложными параллельными и по-
следовательными химическими реакциями являются каталитическими. В зависи-
мости от области протекания процесса диффузионные торможения могут суще-
ственно влиять иа избирательность. Умение технолога учитывать это влияние
предопределяет рациональность того или иного варианта проведения химико-
технологического процесса.
Следует помнить, что внешнедиффузионные торможения не оказывают влия-
ния на избирательность при параллельных реакциях и сильно сказываются на
избирательности при протекании последовательных реакций. Так, для последова-
тельных реакций 1-го порядка А—-> В—>• D (В — целевой продукт), проте-
кающих на непористом катализаторе, избирательность <р, пропорциональная от-
ношению скоростей образования продуктов В и D, зависит от отношения коэф-
фициента массопередачи ₽ к константе скорости второй реакции
<pB~t/B/t/D = ₽/A2 (5.Ю)
83
Рис. 5.8. Характер изменения концентрации СА исходных реагентов и скорости
реакции U по длине поры / при протекании процесса во внутриднффузионной об-
ласти:
о—при низкой скорости диффузии и высокой скорости реакции; б—при высокой скорости
диффузии и низкой скорости реакции.
В кинетической области р/Й2->°°, и в начале процесса <р = 1. Вследствие
малой скорости реакции В -> D (низкие значения k2) и интенсивного отвода
продукта В от поверхности (из-за высоких значений Р) продукт D практически
ие образуется до тех пор, пока не произойдет накопления вещества В в потоке.
В этом случае будет наблюдаться высокая избирательность.
Если же процесс протекает во внешнеднффузионной области (P/fe->-0),
продукт В из-за низкой скорости массопередачи не успевает диффундировать
в поток и практически целиком вступает в реакцию с образованием побочного
продукта D. Избирательность в этом случае будет близка к нулю. Таким обра-
зом, в случае протекания процесса во внешнедиффузионной области увеличение
интенсивности массопереноса путем повышения линейной скорости потока или
перемешивания (см. разд. 5.2) ие только интенсифицирует процесс в целом, но
и повышает избирательность.
Прн протекании гетерогенно-каталитического процесса с использованием по-
ристых катализаторов во внутридиффузионной области влияние диффузионных
торможений определяется так называемой степенью использования
внутренней поверхности, численно равной отношению фактической скорости про-
цесса (7ф,кт к скорости процесса в отсутствие диффузионных торможений нли,
иначе, к максимальной скорости, соответствующей кинетической области t/MaKC:
4 = %^/^ (5.Н)
Влияние диффузионных торможений на скорость процесса при протекании
реакции 1-го порядка А В поясняет рис. 5.8. Если скорость химической реак-
ции велика по сравнению со скоростью диффузии (например, кнудсеновская
диффузия в порах высокоактивного катализатора), то концентрация Са быстро
уменьшается по мере приближения к центру зерна (рнс. 5.8, а) и соответственно
падает скорость реакции, пропорциональная концентрации вещества А. Средняя
концентрация исходного реагента Са и средняя скорость реакции U будут много
ниже максимальных, соответствующих процессу в начале поры.
Если увеличить скорость диффузии (например, путем увеличения диаметра
пор до значения, соответствующего переходу кнудсеновской диффузии в моле-
кулярную), то снижение СА по длине поры замедлится (рис. 5.8,6), средняя
скорость возрастет и увеличится эффективность использования внутренней по-
верхности поры 1).
С постом активности катализатора при неизменной скорости диффузии па-
дение концентрации по длине поры будет круче и степень использования поверх-
ности уменьшится, несмотря на увеличение скорости процесса в целом.
Если среднюю длину поры уменьшить (например вдвое, измельчив зерна нли
гранулы катализатора), то средняя концентрация вещества А в каждой из двух
коротких пор повысится, наблюдаемая скорость процесса О увеличится, возра-
стет и г] (рис. 5.9).
84
Рис. 5.9. Характер изменения концентрации и скорости реакции при изменении
длины поры (измельчении пористого катализатора):
6-Z=/p/2.
Степень использования внутренней поверхности катализатора можно рас-
сматривать как относительную глубину проникновения реагентов, на которой
поверхность поры используется полностью. Тогда наблюдаемая фактическая ско-
рость реакции т-го порядка будет равна:
£/факт = П*Ст (5.12)
Таким образом, степень использования поверхности растет с уменьшением
диаметра частиц катализатора d4 (уменьшается длина пор), с понижением кон-
станты скорости химической реакции k и концентрации реагентов на внешней
поверхности катализатора Ci, г (k и Ci. f определяют скорость реакции), а также
с увеличением коэффициента диффузии реагентог D.
т) ~ (l/d4) д/С/(*С^‘) (5-13)
Подставляя выражение (5.13) в уравнение (5.12), несложно показать, что
при сильном внутридиффузионном торможении порядок реакции уменьшается
от т до (т-|-1)/2. Одновременно снижается и измеряемая энергия активации,
которая при низких значениях 1] может составлять лишь половину истинного
значения энергии активации реакции.
При протекании сложных реакций под влиянием внутридиффузионных тор-
можений может сильно изменяться избирательность. Рассмотрим параллельные
реакции: А —► В и А —► D. Влияние внутренней диффузии на избирательность
проявляется при условии, что порядки основной и побочной реакций различны.
Как следует из выражения (5.13), значение г) больше для реакции низкого по-
рядка, чем для реакции высокого порядка.-Следовательно, под влиянием диффу-
зионных торможений реакция низкого порядка получит преимущество, и если
целевой продукт получается по этой реакции, то избирательность возрастет.
Если В является целевым продуктом и kt — константа скорости реакции
1-го порядка, а йа — константа скорости реакции 2-го порядка, то избиратель-
ность можно увеличить, используя катализатор с крупными зернами, но мелкими
порами, в которых протекает кнудсеновская диффузия.
При протекании последовательных реакций А -*• В -► D содержание промежу-
точного продукта В в порах тем выше, чем больше диффузионные торможения.
При этом промежуточный продукт быстро начинает превращаться в конечный
продукт D. Если В является целевым продуктом, то для увеличения избиратель-
ности следует уменьшать внутридиффузионные торможения. Это достигается
увеличением размера пор катализатора (при кнудсеновской диффузии) и неко-
торым искусственным снижением активности катализатора, напрймер путем ча-
стичного его отравления.
Умение управлять избирательностью путем изменения диффузионных тормо-
жений еще не решает полностью проблему повышения выхода целевого продукта.
Главная задача в этом направлении — разработка новых катализаторов, обла-
85
Рис. 5.10. Влияние температуры на
скорость диффузии и реакции и
область протекания процесса:
1'— скорость химической реакции; 2—ско-
рость диффузии внутри поры: 3—ско-
рость конвективной диффузии; (о) — на-
блюдаемая скорость процесса. 7—кипе
тическая область; II — внутридиффу-
зиоиная область; III — внеш не диффу-
зной на я область.
дающих способностью резко увеличивать избирательность. Во многих процессах
органического синтеза цель применения катализаторов состоит не столько в по-
вышении обшей скорости реакций, как в увеличении именно избирательности.
Только благодаря разработке и применению катализаторов, многократно увели-
чивающих скорость реакций в требуемом направлении, удалось в последние
десятилетия создать новые высокоинтенсивные промышленные способы получения
ценных продуктов органического синтеза.
Рассмотренные выше закономерности протекания процессов во внешнедиф-
фузиониой, внутридиффузионной и кинетической областях отражают предельные
случаи, когда одна из соответствующих стадий является лимитирующей и ее
скорость определяет скорость процесса в целом. Изменение параметров техноло-
гического режима (температуры, давления, концентраций, линейной Скорости по-
тока, интенсивности перемешивания, размеров частиц твердой фазы, йх пористой
структуры,' длительности взаимодействия фаз и др.) может привести к переходу
процесса из одной области протекания в другую.
В этом отношении характерно'влияние температуры на протекание гетероген-
ного процесса в системе Г — Т (пористый катализатор). При низких температу-
рах, как правило, процесс лимитируется скоростью химической реакции. Скорость
подвода вещества к внешней и внутренней поверхности раздела фаз (кривые 3
и 2 в зоне / на рис. 5.10) может существенно превосходить скорость химической
реакции (кривая I в зоне /). С ростом температуры константа скорости химиче-
ской реакции растет по экспоненциальному закону с показателем степени от 0,5
(для кнудсеновской диффузии) до 1,8 (для молекулярной диффузии). Коэффи-
циент конвективной диффузии и ее скорость при подводе реагентов к внешней
поверхности катализатора практически ие зависят от температуры (линия 3).
Таким образом, темп роста константы скорости реакции существенно выше, чем
увеличение коэффициентов диффузии. А это означает, что при некоторой темпе-
ратуре стадия внутренней диффузии станет более медленной по сравнению с хи-
мической реакцией и процесс перейдет постепенно во виутридиффузионную
область, а его скорость будет ограничиваться скоростью внутренней диффузии
(кривая 2 в зоне //).
При дальнейшем повышении температуры процесс может лимитироваться
стадией подвода реагентов к внешней поверхности (линия 3 в зоне III), т. е.
перейдет во внешнедиффузионную область. Увеличение скорости процесса, на-
пример при переводе процесса из кинетической области во внешнедиффузионную,
не означает, что последняя выгоднее, для процесса. Измельчив, например, ката-
лизатор, можно опять перейти в кинетическую область и еще более увеличить
скорость процесса. Оптимальные параметры, как и рациональная область 'проте-
кания процесса, определяются совокупностью технологически::, энергетических и
экономических условий.
При переводе процесса из одной области протекания в другую технолог
должен руководствоваться правилом, согласно которому необходимо стре-
миться к переводу процесса в кинетическую область пу-
тем такого воздействия на лимитирующую стадию, кото-
рое не снижало бы скорость химической реакции. При этом
критерием выбора оптимальных параметров технологического режима должны
являться экономические факторы.
86
Рнс. 5.11. Влияние коэффициентов массопередачи ₽ (а) и коэффициентов скоро-
сти реакции k (6} на концентрацию реагента на поверхности раздела фаз для
переходной области протекания процесса (Pi < Рг < Рз; kt < k? < k3\ б — тол-
щина пограничного слоя):
а—ft^const; б—0=const.
Области протекания ХТП при параметрах технологического режима, соот-
ветствующих переходу процесса из одной области в другую, называют пере-
ходными или промежуточными. При протекании ХТП в таких об-
ластях скорость процесса или лимитируется двумя смежными стадиями, или ни
одна из стадий не является лимитирующей и эффективность процесса может
повышаться путем воздействия на любую из них.
В качестве простейшего примера рассмотрим протекание необратимой реак-
ции 1-го порядка при взаимодействии газа с непористым твердым материалом.
При обтекании частицы такого материала потоком газа с концентрацией реа-
гирующего вещества Ct. о концентрация его у поверхности будет меньше из-за
протекания химической реакции и составит Ct, f. В результате появления разности
концентраций ДСг = С), о— Ct. г (движущая сила массообмена) возникает поток
массы через пограничный слой. Скорость диффузии через единицу поверхности
ил и скорость реакции Ur, отнесенная к единице поверхности, могут быть опи-
саны уравнениями:
(5.14)
UP = kCi.P
(5.15)
Так как на поверхности раздела фаз в реакцию вступает столько газа,
сколько успело продиффундировать к ней, то Ua — Up. Приравняв правые части
уравнений (5.14) и (5.15), получаем выражение для расчета неизвестной кон-
центрации реагента на границе раздела фаз:
С//г = рС(0/(й + р) (5.16)
Подставив уравнение (5.16) в (5.14) или (5.15), получим выражение ско-
рости процесса для рассматриваемой переходной области:
С/ = Л₽С/0/(fe + ₽) = KCi 0 (5.17)
Здесь К — коэффициент скорости процесса, зависящий и от скорости реак-
ции и от скорости диффузии.
Из формулы (5.17), видно что всегда К < k, т. е. наличие диффузионной
стадии тормозит процесс.
При протекании процесса в переходной области, когда значения Р и k срав-
нимы, изменение константы скорости реакции k и коэффициента массопередачи р
влияет на концентрацию реагента у поверхности раздела фаз, а через нее и на
скорость диффузии (рис. 5.11).
87
Из уравнения (5 17) следует, что при р fe
U = kCt0 (5.18)
т. е. процесс лимитируется скоростью химической реакции и протекает в кинети-
ческой области. При р k
0 (5.19)
т. е. процесс лимитируется стадией подвода реагентов к поверхности раздела
фаз н проходит во вгэшнедиффузионной области.
При определении лимитирующей стадйи можно сравнивать численные зна-
чения коэффициентов массопередачи и скорости реакции (при условии одинако-
вой размерности). При сравнимых значениях р и k любое изменение параметров
режима, влияющих на коэффициенты массопередачи или константу скорости
реакции, приводит к изменению наблюдаемой скорости процесса.
5.6. МОДЕЛИРОВАНИЕ — ОСНОВНОЙ МЕТОД РАСЧЕТА
ГЕТЕРОГЕННЫХ ХТП
5.6.1. Понятие о моделировании и моделях
Объект, обладающий определенными реальными свойствами,
изменяющимися в зависимости от условий его существования, на-
зывают оригиналом. Если оригинал достаточно сложен, то
его непосредственное исследование в большинстве случаев неэко-
номично, трудоемко, т. е. требует больших материальных и вре-
менных затрат. Поэтому свойства сложного оригинала чаще всего
изучают на его модели, а результаты исследования модели после
их обработки переносят на оригинал. Создание модели, воспроиз-
водящей изучаемые особенности структуры и поведения ориги-
нала, и последующее исследование этой модели с распростране-
нием результатов на оригинал называют моделированием.
В прикладных науках моделирование проводят с использованием
материальных моделей. Материальные модели разделяют на фи-
зические и математические. При физическом моделирова-
нии процессы в оригинале и физической модели не отличаются
по физической природе. Основное отличие между оригиналом и мо-
делью — их размеры. Опытные данные, полученные при исследо-
вании физической модели, представляют в виде уравнений, содер-
жащих критерии подобия (Рейнольдса, Архимеда, Фруда, Пекле,
Прандтля, Нуссельта и др.), и безразмерных соотношений геомет-
рических и физических величин. Физическое моделирование сво-
дится к воспроизведению равенства определяющих критериев
подобия в модели и оригинале в пределах изменения основных
параметров процесса, которые исследованы на модельных уста-
новках. В подавляющем большинстве случаев ХТП настолько
сложны, что соблюдение подобия модели и оригинала, заключаю-
щееся в одновременной идентичности многих критериев подобия,
практически невозможно. Кроме этого, современные ХТП иногда
не поддаются изучению в «чистом» эксперименте. Не всегда
имеется экспериментальная база и возможности выделения боль-
шого числа квалифицированных кадров.
88
Поэтому в настоящее время расчет ХТП проводят, как пра-
вило, путем математического моделирования. Центральной пробле-
мой при разработке технологом математической модели является
создание мысленной модели оригинала, включающей протекание
всех основных процессов. Мысленную схему протекания процессов,
выраженную языком математики, называют математической
моделью. Математическая модель может быть представлена
в виде систем уравнений, неравенств, графиков, таблиц, с помощью
которых описывается поведение и основные свойства оригинала.
Термины «математическая модель» и «математическое описание»
часто употребляют как синонимы. Составление математического
описания процесса в совокупности с решением полученных урав-
нений на ЭВМ называют математическим моделирова-
нием.
В соответствии с рассмотренными выше (см. разд. 3.1) уров-
нями анализа протекания химико-технологического процесса ма-
тематическая модель ХТП строится путем последовательного пе-
рехода описания процесса от низшего уровня его протекания
(молекулярный уровень) до высшего (уровень цеха).
Если описание процесса на молекулярном уровне при изве-
стном механизме химической реакции в большинстве случаев
может быть ограничено подбором соответствующего числа кинети-
ческих уравнений, то анализ кинетических закономерностей проте-
кания гетерогенного ХТП на уровне малого объема (зернах, кап-
лях, пузырьках) требует дополнительного учета переноса вещества
и теплоты между фазами. Возможность достаточно точного описа-
ния кинетических закономерностей протекания гетерогенных ХТП
прежде всего зависит от того, как технолог мысленно представ-
ляет протекание основных его стадий в их взаимосвязи или, иначе
говоря, насколько мысленная схема или модель протекания про-
цесса отражает его реальные существенные стороны.
Таким образом, эффективность расчета гетерогенного ХТП
в условиях одновременного протекания химической реакции и
массо- и теплопереноса возможна лини на основе математического
описания мысленных представлений об основных стадиях про-
цесса.
В последующих разделах рассмотрены наиболее простые и ти-
пичные представления и модели, используемые для описания, ана-
лиза, расчета гетерогенных ХТП и управления ими для различных
систем взаимодействующих фаз.
5.6.2. Характеристика и моделирование процессов
в системе газ (жидкость) — твердое
К ХТП с участием газообразных и твердых реагентов (Г — Т)
относят: адсорбцию газов твердыми поглотителями, катализ на
твердых катализаторах, пиролиз и горение твердого топлива, об-
жиг твердых материалов, их термическую диссоциацию, коррозион-
ные процессы и т. Д.
89
Процессы е участием твердых и жидких реагентов (Т — Ж)'
включает: растворение и плавление твердых веществ, кристалли-
зацию их из растворов и расплавов, экстрагирование и выщела-
чивание, адсорбцию растворенных в жидкости веществ твердыми
поглотителями и обратный процесс — десорбцию, ионообмен, ка-
тализ в жидкой фазе на твердых катализаторах и т. п.
Кинетические зависимости такого рода превращений с учетом
химических реакций и диффузии газообразного реагента в зону
реакции рассмотрим на примере обжига твердого непористого ма-
териала.
Обжиг — это высокотемпературный ХТП с участием твердых
и газообразных реагентов. Типичным примером может служить
окислительный обжиг колчедана и других сульфидных руд с хи-
мическим превращением типа:
А (г.) + В (тв.) = С (тв.) + D (г.)
Процесс начинается обычно в активных точках (центрах) на-
ружной поверхности кристалла, в которых зарождается новая
твердая фаза, как правило, пористая. На этом первом этапе осу-
ществляется диффузия газообразного исходного вещества А через
газойую ламинарную пленку, окружающую зерно, к поверхности
раздела фаз, химическая реакция и диффузия продуктов D в газо-
вый поток.
По мере образования новой твердой фазы, химически инертной
к воздействию реагента А, появляется внутридиффузионное тор-
можение, связанное с диффузией реагента А (или продукта D)
к поверхности непрореагировавшего вещества В (или от нее). При
этом граница раздела фаз, на которой протекает химическая реак-
ция, перемещается во времени к центру частицы.
Аналогичный механизм можно наблюдать (или представить)
и при растворении или выщелачивании. Например, при разложе-
нии непористых материалов кислотами можно также выделить
отмеченные в предыдущем примере этдпы. В частности, при выще-
лачивании растворимой части минералов скорость замедляется
в результате появления внутридиффузионных торможений при
диффузии оастворителя через поры нерастворимой части породы
к поверхности минерала. И в этом случае граница раздела фаз или
радиус сферы, на которой протекает реакция, перемещается к
центру частицы. -
Такова простейшая мысленная модель последовательности
протекания основных этапов рассматриваемого процесса. Ее на-
зывают моделью частицы с невзаимодействующим
ядром. Рис. 5.12 поясняет сущность такой модели. Характер
изменения концентрации газообразных веществ в зависимости от
лимитирующей стадии и области протекания процесса показан на
рис. 5.13.
Математическое описание процесса сводится к составлению
системы уравнений скорости подвода газообразных веществ к
внешней поверхности зерна, скорости диффузии реагентов через
' 90
Рис. 5.12. Основные этапы протекания гетерогенного ХТП согласно модели ча-
стицы с невзаимодействующим ядром:
а—начальный этап протекания процесса прн низкой степени превращения; б—этап, характе»
рнзующнйся интенсивным образованием слоя пористого продукта; в — высокая степень превра-
щения. /-^Ламинарная пленка газа; 2—слой исходного твердого вещества В; 3—слой порг •
стого продукта С; 4—граница раздела фаз; —> —диффузионный поток исходного реагента А;
<----• - диффузионный поток продукта D; Яо^начальныЙ радиус Частины; Яя—радиус непро’
реагировавшего ядра.
пористый слой продукта С, скорости химической реакции на по-
верхности раздела твердых фаз. Последующее решение такой
системы уравнений при соответствующих граничных условиях поз-
воляет получить зависимость, описывающую кинетику протекания
процесса.
Для процесса горения твердого непористого топлива, когда
твердый продукт может не образовываться, также используют
модели частицы с невзаимодействующим ядром. Однако отсут-
ствие внутридиффузионных торможений существенно упрощает
математическое описание.
/?0 RH0 НяЯ0
Рис. 5.13. Характер изменения концентрации газообразных реагентов нри проте-
кании процесса по модели частицы с невзаимодействующим ядром.
Области Протекания процесса: а—внещнедиффузш нчая, б—внутриднффузионная; в—кинети-
ческая. /—Ламинарная пленка газа; "—слой исходного твердого вещества В; 3—слой пори-
стого продукта С.
91
Рис. 5.14. Основные этапы протекания гетерогенного некаталитического ХТП по
квазигомогенной модели:
Степень превращения вещества А: а—низкая; б—средняя; в—высокая.
Рассмотрим другой случай взаимодействия в системе Г — Т,
когда твердое вещество является пористым. Такими процессами
могут быть: горение кокса, восстановление металлов из пористых -
гранул окислов (в металлургии, при получении катализатора син-
теза аммиака и др.), каталитические процессы на твердых порис-
тых катализаторах, процессы отравления таких катализаторов
и т. д.
Использование модели частицы с невзаимодействующим ядром
для анализа таких процессов недопустимо, так как она не отра-
жает в данном случае особенностей их протекания. Действительно,
при наличии крупных пор, достаточно высокой скорости диффузии
реагентов внутри твердых частиц и малой скорость химической
реакции последняя будет протекать практически на всей внутрен-
ней поверхности. В этом случае используют так называемую к в а -
зигомогенную м од ель, согласно которой газ проникает
внутрь частицы и взаимодействует с ее веществом во всем объеме
до тех пор, пока частица находится в зоне реакции или пока она
не превратится в продукты реакции. Типичным примером может
служить отравление зерен катализатора. Квазигомогенная модель
наиболее хорошо отражает явления при малой скорости химиче-
ской реакции на поверхности крупнопористых тел. Рис. 5.14 по-
ясняет сущность квазигомогенной модели для некаталитического
процесса: А(г.)+В(тв.) = D(tb.).
При проведении каталитических процессов в зависимости от
внутридиффузионных торможений изменяется форма профиля
концентраций по диаметру частицы катализатора (рис. 5.15).
Расчет сводится к составлению системы уравнений, описываю-
щих диффузионные потоки вещества к внешней поверхности, вну-
три зерна катализатора и скорость химической реакции по длине
пор. Решение системы уравнений позволяет получить характерис-
тики профиля концентраций по диаметру зерна и определить сте-
пень использования внутренней поверхности, знание которой не-
обходимо для расчета скорости процесса при известном кинетиче-
ском уравнении.
92
Рис. 5.15. Распределение концентрации газообразных реагентов внутри шаровой
пористой гранулы катализатора по квазигомогенной модели для реакции А->В.
Области протекания процесса: а—внешнедиффузионная; б —внутри диффузионная; в—кинети-
ческая. 1 — Ламинарная пленка газа; 2—катализатор.
Как уже отмечалось, от правильного выбора мысленной модели
процесса, которая отражала бы основные этапы его протекания,
зависит возможность использования полученной математической
модели для расчета различных показателей и управления химико-
технологическим процессом. В значительной степени успех зави-
сит от квалификации технолога.
5.6.3. Характеристика и моделирование процессов
в системе газ (жидкость) — жидкость
ХТП с взаимодействием газообразных и жидких компонентов
очень разнообразны. К ним относят: адсорбцию газов жидкостями,
в том числе сопровождающуюся химической реакцией; десорбцию
газов; перегонку жидких смесей (дистилляцию, ректификацию);
пиролиз жидкостей с испарением продуктов пиролиза; хлорирова-
ние углеводородов газообразным хлором; производство амида нат-
рия путем взаимодействия аммиака с расплавом натрия и др.
Процессы, сопровождающиеся химической реакцией при взаи-
модействии несмешивающихся жидкостей, используют во многих
производствах органического синтеза. Например, к ним относятся
процессы сульфирования и нитрования, заключающиеся в обра-
ботке жидких углеводородов растворами серной, хлорсульфоно-
вой или азотной кислот.
Механизм взаимодействия фаз в системах Г—Ж и Ж — Ж
(несмешивающьеся) имеет много общего. Для простоты ограни-
чимся анализом системы Г — Ж. При переносе такого анализа на
систему Ж — Ж условно надо считать газовую фазу как вторую
жидкость, изменив лишь терминологию.
Пусть газовая смесь, содержащая вещест во А, взаимодействует
с жидкостью, содержащей вещество В. При этом А и В должны
химически прореагировать между собой:
А (г.) + В (ж.) = D (ж.)
93
Рис. 5.16. Распределение концентра! чи реаген-
тов и продуктов реакции А(г.)+В(ж.) =О(ж.)
по толщине слоя жидкости согласно пленочной
модели:
I—турбулентный поток газа; II — пограничный слой
газа; III— поверхность раздела фаз; IV — пограничный
слой жидкости; V — турбулентный поток жидкости;
---- ---концентрация А;-------концентрация В,
------------------------------концентрация D.
В рассматриваемой системе реакции протекают в основном
в жидкой фазе. В общем случае химической реакции предшествует
стадия доставки газообразных реагентов диффузией их в газовой
смеси к поверхности раздела фаз и растворение в жидкости. Та-
кова простейшая мысленная модель протекания основных этапов
процесса. Для количественного описания таких процессов широко
используют пленочную модель, согласно которой по обе стороны
межфазной поверхности Г (Ж)—Ж существует ламинарная по-
граничная пленка. Скорость диффузии через пленку прямо про-
порциональна разности концентраций по обе стороны пленки
и обратно пропорциональна ее толщине. Предполагается, что вне
пределов пограничной пленки изменение концентраций отсутствует,
а на поверхности контакта фаз устанавливается равновесие между
концентрацией поглощаемого компонента в жидкой и газовой
фазе.
На рис. 5.16 показан характер изменеиня .концентраций реагентов А и В и
продукта D в пограничных пленках согласно пленочной модели для случая,
когда скорость диффузии соизмерима со скоростью реакции. Концентрация В
в жидкости равна Св, о, компонеста А — отсутствует. Вследствие этого он диф-
фундирует через пограничный слой газа II, поверхность раздела фаз III, погра-
ничный слой жидкости IV и в нем вступает в химическую реакцию с В, образуя
продукт D. При этом концентрация А уменьшается до нуля к моменту оконча-
ния реакции на расстоянии а от поверхности раздела фаз. Компонент В диффун-
дирует сквозь жидкчй погргничный слой в направлении поверхности раздела
фаз Е~о концентрация уменьшается и на расстоянии b становится равна нулю.
Продукт D накапливается в слое толщиной а до некоторой концентрации и
диффундирует в поток жидкости. '
При увеличении скорости химической реакции и уменьшении скорости" диф-
фузии п-»-0. При малой скорости реакции и высокой скорости диффузии реак-
ция заканчивается в зоне турбулентного потока жидкости (зона V).
Расчет процесса сводится к составлению и решению системы уравнений ско-
рости диффузии реагентов А и В через пограничные слои и скорости химической
реакции при соответствующих граничных условиях.
Если процесс абсорбции сопровождается протеканием химиче-
ской реакции, то последняя сильно интенсифицирует процесс мас-
сслередачи. Это связано с увеличением движущей силы процесса
диффузии, обусловленным исчезновением реагирующих веществ
84
вследствие вступления их в реакцию. Например, если коэффи-
циент массопередачи при абсорбции СО2 водой принять-за 1, то
при абсорбции СО2 водными растворами щелочей, химически со-
единяющих СО2, массопередача интенсифицируется в 50—75 раз.
5.6.4. Процессы взаимодействия
между твердыми фазами и в многофазных системах
Взаимодействие только между твердыми фазами наиболее
характерно для процессов спекания. Спекание — это процесс
получения агрегатов частиц из мелкозернистых, порошкообразных
или пылевидных материалов при высоких температурах. Реакции
между твердыми материалами часто проходят с участием газовой ,
или жидкой фаз, образующихся во время реакции. При выборе
модели, пригодной для описания таких процессов, учитывают по-
явление этих фаз, последующие стадии их диффузии и химическую
реакцию.
Установлено, что появление газовой и жидкой фаз способствует
ускорению взаимодействия между твердыми материалами. По-
этому такие процессы относят к многофазным, и скорость их опре-
деляется скоростью одной из перечисленных элементарных стадий.
В качестве типичного примера многофазного ХГП можно при-
вести карбонизацию аммиачно-содового раствора в производстве
соды. В этом процессе при взаимодействии газовой и жидкой фаз
образуется твердая фаза (гидрокарбонат натрия). При выборе
модели и расчете подобных процессов необходимо вводить упро-
щения, оставляя в модели лишь основные факторы, определяющие
поведение системы.
ГЛАВА 6
ХИМИЧЕСКИЕ РЕАКТОРЫ,
ЗАКОНОМЕРНОСТИ ИХ РАБОТЫ И КОНСТРУКЦИИ
Аппараты, в которых протекает ХТП, называют химиче-
скими реакторами. В каждом реакторе можно выделить
реакционную зону (реакционный объем), в которой непосредствен-
но осуществляется химический процесс и через которую проходят
потоки реагентов. ХТП в реакторах могут протекать в замкнутом
реакционном объеме при отсутствии сквозных потоков или в потоке,
проходящем через реакционную зону.
Протекание ХТП в замкнутом реакционном объеме, т. е. когда
отсутствует массообмеи с окружающей средой или с другими
аппаратами, подчиняется закономерностям, рассмотренным в гла-
вах 4 и 5. При протекании ХТП в таких реакторах параметры и по-
казатели технологического режима (температура, давление, концен-
трации, степень превращения, выход продукта, избирательность,
Рис. ,6.1. Параметры технологического режима при протекании процесса в за-
мкнутом объеме.
Рис. 6.2. Характер изменения степени превращения и концентрации исходных реа-
гентов во времени при протекании ХТП в замкнутом объеме:
--------без перемешивания;--------с перемешиванием.
скорость и др.) постоянны во всем объеме и изменяются лишь
во времени (рис. 6.1)^ Если процесс протекает при постоян-
ной температуре и давлении, то для расчета показателей ХТП
используют, например, уравнения (4.4) — (4.6) с учетом поправок
на диффузионные, торможения, рассмотренных в главе 5. Если тем-
пература и давление в реакционном объеме изменяются во вре-
мени, то при использовании отмеченных уравнений учитывается
характер изменения ТиР
Следует отметить, что в замкнутом реакционном объеме всегда
имеется циркуляционное движение реагентов (циркуляционные
потоки), обусловленное конвективным массообменом. Но характер
изменения показателей технологического режима не зависит от
интенсивности циркуляции и перемешивания. Например, для гете-
рогенных процессов в случае отсутствия перемешивания перенос
вещества к поверхности контакта фаз будет определяться моле-
кулярной диффузией, и кривые изменения во времени концентрации
и степени превращения (рис. 6.2) могут иметь лишь более поло-
гий вид по сравнению с процессом, протекающим в условиях
интенсивного перемешивания. Если скорость химической реакции
меньше скорости подвода реагентов за счет молекулярной диф-
фузии, то применение перемешивания вообще не окажет влияния
на изменение показателей процесса во времени. Как отмечалось
в главе 5, это характерно для протекания процессов в кинетиче-
ской области.
Подавляющее большинство ХТП проводят в сквозных потоках,
проходящих через реакционные зоны аппаратов. Эти потоки раз-
личаются по интенсивности перемешивания исходных реагентов
и продуктов реакции, по характеру изменения температуры по
длине реакционной зоны. Закономерности работы таких реакторов
определяются характеристиками этих потоков.
96
6.1. ОСНОВНЫЕ ХАРАКТЕРИСТИКИ ПОТОКОВ
И ИХ ВЛИЯНИЕ НА ПРОТЕКАНИЕ ХТП
Для анализа и управления реакторами, через которые прохо-
дят потоки реагентов, технологу необходимо учитывать влияние
характеристик таких потоков на показатели ХТП. К основным
гидродинамическим характеристикам потока, оказывающим влия-
ние на протекание ХТП, относят линейную скорость и перемеши-
вание, к термодинамическим — изменение температуры по длине
реакционной зоны, через которую проходит поток и др. При даль-
нейшем изложении материала мы не будем делать различия
между потоками газа, жидкости, зернистых твердых материалов
или их смесями. Для простоты их называют потоками жидкости,
а под термином «частица» подразумевают часть потока, движе-
ние которого в какой-либо момент можно рассматривать как дви-
жение единого целого. В зависимости от гидродинамических ха-
рактеристик и структуры потока продолжительность пребывания
таких частиц (зерен, капель, малых объемов жидкости или газа)
в зоне реакции будет различна, процесс, протекающий в каждой
такой частице, будет иметь различную степень завершенности.
Это, в свою очередь, окажет отрицательное влияние на средние
выходные показатели протекания ХТП в реакторах.
Фактор линейной скорости характеризуется двумя па-
раметрами — средним значением и неоднородностью поля скоро-
стей. Соответственно влияние линейной скорости на протекание
ХТП в потоке рассматривают с двух сторон: 1) с повышением
средней скорости уменьшаются внешнедиффузионные торможения
и, следовательно, интенсифицируется процесс; 2) с увеличением
неоднородности распределения скоростей по сечению потока рас-
тет неравномерность времени пребывания реагирующих веществ
(частиц) в реакционной зоне.
Перемешивание в потоке может быть продольным и радиаль-
ным. Продольное перемешивание обусловливает смеше-
ние частиц, только что вошедших в реакционную зону, с части-
цами, давно в ней находящимися. В большинстве случаев продоль-
ное перемешивание ухудшает показатели ХТП из-за снижения
движущей силы. Радиальное перемешивание интенсифи-
цирует массообмен между соседними участками в поперечном се-
чении потока и уменьшает отрицательное влияние неоднородности
поля линейных скоростей.
Продольное перемешивание и неоднородность поля скоростей
приводят к одному и тому же отрицательному явлению — к нерав-
номерности времени пребывания частиц в зоне реакции, через ко-
торую проходит поток.
Среднее время пребывания частиц рассчитывают по формуле:
t = t>p/V (6.1)
Здесь — объем реакционной зоны; V — объемный расход жидкости (газа,
смеси).
4 Зак 1869
97
Рис. 6.3. Иллюстрация влияния неравномерно-
сти времени пребывания реагентов в реакцион-
ной зоне на степень превращения.
При одном и том же среднем времени т различные степени неравномерности
времени пребывания частиц в реакционной зоне приведут к различным значе-
ниям показателей ХТП. Это связано с тем, что уменьшение степени превращения
в частицах из-за покидания ими реакционной зоны раноше времени т не ком-
пенсируется увеличением X за счет задержки в реакционной зоне других частиц
дольше среднего времени пребывания. Рис. 6.3 поясняет сказанное. Пусть одни
частицы покинут реакционную зону раньше т на отрезок времени Ат, а другие
частицы останутся в зоне реакций на Ат дольше. Из-за меньшего времени пре-
бывв.чия частиц степень их превращения уменьшится на AXi, а степень превра-
щения задержавшихся частиц увеличится на АХг- Как следует из графика,
AXt > ДХ2. Чем больше отклонение времен пребывания от среднего значения т
тем больше разность AXi — ДХ2. Таким образом, чем больше неравномерность
времени пребывания, те11 сильнее отрицателен >е воздействие этой неравномерно-
сти на показатели ХТП
Для учета влияния структуры потока, неоднородности поля ско-
ростей, перемешивания на протекание ХТП в реакторах технолог
должен построить упрощенные мысленные модели потоков. Такая
модель должна отражать наиболее существенные особенности по-
тока н давать возможность описывать влияние этих особенностей
на протекание ХТП, прогнозируя тем самым его поведение.
Технологи часто используют для анализа ХТП две простейшие
максимально упрощенные модели потоков, называемых идеаль-
ными— модель потока с идеальным вытеснением
реагентов и модель потока с идеальным (полным) j?
смешением. Соответственно реакторы, через которые проходят
идеальные потоки, называют реакторами идеального вытеснения
и полного смешения. Эти модели, несмотря на свою простоту, во
многих случаях достаточно точно описывают поведение ХТП в
реальных потоках, проходящих через промышленные реакторы.
6.2. ПРОТЕКАНИЕ ХТП В ПОТОКЕ ИДЕАЛЬНОГО ВЫТЕСНЕНИЯ
6.2.1. Общая характеристика и температурные режимы
При прохождении потока идеального вытеснения через реак
ционную зону все частицы движутся с одной -скоростью, не пере-
мешиваясь друг с другом. Если выделить в потоке малый объем,
занимающий все поперечное сечение, то этот объем будет дви-
гаться, как поршень (рис. 6.4).
Вследствие одинаковой скорости w всех частиц время прохож-
дения ими любого отрезка по длине реакционной зоны и время
96
Рис. 6.4. Поток с идеальным вытеснением.
пребывания в ней одинаковы. В данном случае истинное время
пребывания каждой частицы равно среднему времени пребывания,
рассчитанному по формуле (6.1).
В потоке идеального вытеснения исходные реагенты не смеши-
ваются с продуктами реакции, в каждом поперечном сечении по-
тока концентрации их выровнены, но плавно изменяются по длине
или высоте реакционной зоны. Аналогично изменяется скорость
процесса.
Количественная оценка изменения концентрации, степени пре-
вращения, выхода продукта, избирательности, скорости и других
показателей ХТП при протекании его в потоке с идеальным вы-
теснением зависит от характера изменения температуры по длине
реакционной зоны. Различают три температурных режима реак-
ционной зоны — изотермический, адиабатический и политерми-
ческий.
Изотермический температурный режим характе-
ризуется постоянством температуры по всей длине (высоте) реак-
ционной зоны, через которую проходит поток.
Адиабатический температурный режим характе-
ризуется изменением температуры по длине реакционной зоны
вследствие тепловых эффектов процесса при отсутствии теплооб-
мена с окружающей средой.
Политермический температурный режим харак-
теризуется изменением температуры по длине реакционной зоны
вследствие совместного влияния тепловых эффектов и теплообмена
с окружающей средой.
6.2.2. Изотермический температурный режим
Изотермический температурный режим в потоке идеального
вытеснения наблюдается в случае протекания ХТП без теплового
эффекта или когда скорость тепловыделения (теплопоглощения)
мала, а теплопроводность среды в реакционной зоне высока. На-
пример, , это может быть гетерогенный процесс, протекающий
с низкой скоростью, при наличии в реакционной зоне металличе-
ского катализатора с высокой теплопроводностью.
Если в потоке идеального вытеснения выделить малый объем,
то при движении его вместе с потоком ни одна частица не выйдет
из этого объема, так как все они имеют одинаковую скорость. Та-
кой объем можно рассматривать как замкнутый, и скорость реак-
ции в нем будет описываться уравнениями типа:
U,=—± = kb.C
i dt
(6.2)
4*
99
А так как время пребывания всех частиц в потоке одинаково,
то закономерности протекания ХТП в замкнутом объеме и в по-
токе идеального вытеснения можно описать одинаковыми форму-
лами, например (4.4) — (4.7), (4.13), (4.14), применимыми для
анализа и управления кимико-технологическими процессами, про-
текающими при изотермическом температурном режиме. Необхо-
димое время пребывания реагентов в реакционной зоне рассчи-
тывают по уравнению:
СА
С 4Са
т= \ (6.3)
ед о
Выражение (6.3) называют характеристическим урав-
нением реактора идеального вытеснения. При этом
фактор времени должен быть связан с увеличением длины пути,
пройденного реагентами в реакционной зоне /р, или ее текущей
длиной (высотой) I
т = l/w (6.4)
Здесь w — линейная скорость потока.
Например, для реакции 1-го порядка уравнение (4.5) для реак- |
ционной зоны с потоком идеального вытеснения можно предста-
вить в виде:
СА = СА 0 exp (— kl/w) (6.5)
При I — 1Р
CA-=CA,O^P(-klplW) <6-6)
Для расчета степени превращения используют уравнение (4.6),
представленное в виде:
X = 1 — ехр (— kl/w) (6.7)
При I = 1Р получают конечную степень превращения Хк:
XK=l — exp (— klp/w) . (6.8)
Скорость реакции по длине реакционной зоны изменяется
ь соответствии с выражением:
U = kCA — kCA оехр( — kl/w) (6.9)
При выбранной линейной скорости потока w, задаваясь раз-
личными значениями I, можно построить график изменения кон-
центрации Са, степени превращения X и скорости реакции U по
длине реакционной зоны (рис. 6.5).
В каждом поперечном сечении реакционной зоны потока ука-
занные параметры (С, U, X, фв) и другие постоянны во времени.
При этом увеличение длины реакционной зоны от 1Р до (/р -)- М)
не оказывает влияния на характер изменения и численные значе-
ния параметров на участке от I = 0 до /Р (рис. 6.6) .
100
При протекании сложных реакций (А -> В D) характер изменения показа-
телей ХТП по длине реакционной зоны аналогичен их изменению во времени при
протекании процессов в замкнутом объеме (рис, 6.7). Если В является целевым
продуктом, то правильный выбор оптимальной длины реакционной зоны /р. Опт
определяет возможность достижения максимальной концентрации и выхода це-
левого продукта. Для определения lf, ОПт можно по формуле (4.48) рассчитать
Топт- Тогда
^р, опт = И’Топт (6.10)
При протекании в потоке гетерогенных ХТП следует учиты-
вать влияние массопередачи на наблюдаемую скорость. При этом
для условий идеального вытеснения и изотермического темпера-
Рис. 6.7. Характер изменения кон-
центраций реагентов по длине ре-
акционной зоны потока идеального
вытеснения для последовательной
реакции А -> В -> D.
101
турного режима в реакционной зоне закономерности протекания
ХТП на одной частице полностью отражают закономерности про-
текания ХТП в потоке. Это вытекает из того, что все частицы
имеют одно и то же время пребывания в реакционной зоне.
-6.2.3. Адиабатический температурный режим
Адиабатический температурный режим наблюдается при отсут-
ствии в реакционной зоне теплообменников и хорошей изоляции ее
от окружающей среды. Если при протекании ХТП не происходит
фазовых переходов, связанных с плавлением, испарением, конден-
сацией и другими физическими процессами, то изменение тепло-
содержания реагирующей смеси будет обусловлено лишь тепловым
эффектом реакции и степенью превращения исходных вещесгв.
Пусть протекает экзотермическая реакция А->В + ^. При не-
изменном значении теплового эффекта реакции q количество выде-
лившейся теплоты Qp будет прямо пропорционально количеству
прореагировавших веществ или степени превращения Ха, т. е.
Qp ~ ХА. За счет этого количества теплоты будет происходить
разогрев реагирующей смеси. При неизменной теплоемкости смеси
изменение температуры АТ будет прямо пропорционально QP, т. е.
Т ~ Qp. Из этого можно сделать вывод, что изменение темпера-
туры АТ при протекании ХТП в адиабатических условиях прямо
пропорционально степени превращения реагирующих веществ. Та-
кая зависимость между АТ и X описывается уравнением адиабати-
ческого процесса:
ДУ = ±ххА (6.11)
Здесь X — коэффициент пропорциональности, который называют коэффициен-
том адиабатического разогрева (охлаждения); зависит от начальной концентра-
ции реагирующего вещества Сд, о, теплового эффекта реакции q и теплоемкости
реакционной смеси с:-
1 X ~ СА (6-12)
I
При технологическом расчете какого-либо конкретного про-
цесса изменение X обусловлено изменением начальной коицентра-
' ции реагирующих веществ, а зависимость адиабатического изме-
нения температуры от степени превращения в координатах X — Т
можно представить в виде прямой, проходящей под углом а к оси
Т (рис. 6.8). При этом, чем выше СА, о, тем меньше угол а, тем
больше изменение температуры при одинаковой степени превра-
щения.
Для экзотермических необратимых реакций максимальная сте-
пень превращения Хмакс. ад в потоке с адиабатическим температур-
ным режимом ограничивается лишь предельно допустимым разо-
гревом реакционной смеси (см. разд. 4.3). Для эндотермических
необратимых реакций ХмаКс. ад ограничивается минимально допус-
тимым охлаждением реакционной смеси до Тмин, ниже которой
процесс «затухает». При протекании обратимых экзо- и эндотер-
102
Рис. 6.8. Зависимость адиабатического разогрева (а) и охлаждения (б) реакцион-
ной смеси от степени превращения и концентрации исходного реагента для необ-
ратимых реакций:
1—при соких начальных концентрациях; 2—при низких концентрациях.
Рис. 6.10. Характер изменения константы скорости (а) и скорости реакции (б)
по длине реакционной зоны потока с адиабатическим температурным оежигтом:
/—экзотермический процесс; 2—эндотермический процесс.
Рис. 6 9. Зависимость адиабатического разогрева (а) и охлаждения (б) реакцион-
ной смеси от степени превращения и начальной концентрации реагента для обра-
тимых реакций:
1—при высоких начальных концентрациях; 2—при низких концентрациях.
103
Рис. 6.11. Характер изменения степени
превращения X и температуры Т по дли-
не реакционной зоны потока с адиаба-
тическим температурным режимом для
экзотермических ХТП.
мических реакций ХмаКс. ад ограничивается, как правило, достиже-
нием равновесия (рис. 6.9).
Характер изменения концентраций и степеней превращения по
длине реакционной зоны потока с адиабатическим температурным
режимом аналогичен изотермическим потокам. Однако при рас-
чете необходимо учитывать изменение константы скорости и ско-
рости реакции по длине реакционной зоны (рис. 6.10). При этом
следует помнить, что согласно уравнению (6.11) изменение темпе-
ратуры прямо пропорционально изменению степени превраще-
ния. Это означает, что кривые, описывающие изменение Т и X по
длине реакционной зоны, имеют одинаковый характер (рис. 6.11).
Расчет длины реакционной зоны, необходимой для обеспечения
заданной степени превращения Хк или заданной конечной кон-
центрации Ск, проводят интегрированием кинетического уравнения.
Например, для реакции А->В:
dCA
f/=----(C) = k^C
Сл к
г dCA ск dX
= j ~ = Сд- ° j Тле
СА, 0 °
(6.13)
(6.14)
При этом k изменяется по длине реакционной зоны в соответ-
ствии с изменением Т. Необходимая длина реакционной зоны со-
ставит:
/р = rw (6.15)
6.2.4. Политермический температурный режим
Политермический температурный режим возможен лишь при
наличии в реакционной зоне теплообменной поверхности. Проведе-
ние ХТП в этом режиме позволяет существенно повысить степень
превращения, выход продукта, скорость процесса и другие показа-
тели. Например, при проведении необратимых экзотермических
процессов, отводя необходимое количество теплоты из зоны реак-
ции, можно обеспечить достижение очень высоких степеней пре-
вращения, не опасаясь превышения максимально допустимых тем-
ператур Тмакс- При этом, если температура потока на входе в реак-
104
Рис. 6.12. Характер изменения температуры по длине реакционной зоны при поли-
термическом режиме для экзотермического необратимого процесса.
Рис. 6.13. Диаграмма X— Т для необратимого экзотермического процесса в по-
токе идеального вытеснения с политермчческим температурным режимом.
ционную зону Твх существенно ниже Гмакс, то на первом участке
реакционной зоны процесс может протекать адиабатически до до-
стижения Гмакс, а затем — с отводом теплоты при постоянной тем-
пературе, равной Тмакс (рис. 6.12). Характеристику протекания
процесса в таких условиях дополняет диаграмма Х — Т (рис. 6.13).
Еще более эффективно влияние политермического режима на
протекание ХТП с обратимой экзотермической реакцией. Изменяя
интенсивность отвода теплоты по длине реакционной зоны, можно
не только существенно увеличить максимально достижимую сте-
пень превращения исходных реагентов и выход продукта, но
и обеспечить условия протекания ХТП с максимальной скоростью.
При этом изменение температуры в зависимости от достигаемых
степеней превращения должно соответствэвать линии оптимальных
температур (см. разд. 4.3). На рис. 6.14 показан характер изме-
нения температуры в зависимости от достигаемой степени превра-
щения. Если температура потока на входе в реакционную зону
Тех существенно ниже ТмаКс, то на первом участке реакционной
зоны целесообразнее проводить процесс при адиабатическом тем-
пературном режиме, а после достижения максимальной скорости
Рис. 6.14. Диаграмма X — Т для обрати-
мого экзотермического процесса, проте-
кающего и потоке идеального вытеснения
с политермическим температурным режи-
мом:
1 — линия адиабатического разогрева; 2—линии
оптимальных температур; 3— фактическое изме-
нение температуры в зоне расположения тепло-
обменных поверхностей; 4—линия равновесного
состояния.
105
Рис. 6.15. Характер изменения температуры по длине реакционной зоны потока
идеального вытеснена с политермическим температурным режимом (обратимая
экзотермическая реакция):
1—линия адиабатического разо. рева. 2—линия оптимальных температур; 3—фактическое
изменение температуры.
Рис. 6.16. Характер изменения скорост, экзотермического обратимого процесса по
длине реакционной зоны потока идеального вытеснения:
1 — адиабатический температурный режим; 2—линия максимальных скоростей, соответствующих
оптимальным температурам; 3 —по литер мнческий температурный режим.
уточка пересечения адиабаты 1 с линией 2 оптимальных темпе-
ратур) необходимо обеспечить такой отвод теплоты, чтобы тем-
пература изменялась в соответствии с кривой 2. На практике
обеспечить точное проведение процесса по оптимальной темпера-
турной зависимости не удается. Поэтому фактическое изменение
температуры (кривая 3) может несколько отличаться от теорети-
ческого (кривая 2).
Изменение температуры по длине реакционной зоны для рас-
сматриваемого случая показано на рис. 6.15.
На рис. 6.16 представлен характер изменения скорости экзо-
термического обратимого процесса по длине реакционной зоны
для случая, когда ТВх существенно ниже Тмакс-
Если температура потока на входе в реакционную зону Твх
близка к Тмакс, то преимущества политермического температурного
режима становятся еще более очевидными. В этом случае воз-
можно проводить процесс при оптимальном температурном ре-
жиме. Основные показатели процесса будут близки к предельным
теоретически возможным. Характер их изменения соответствует
кривым 2 на рис. 6.14—6.16.
Технологи "должны стремиться проводить ХТП в потоках иде-
ального вытеснения с оптимальным температурным режимом,
однако не всегда удается преодолеть технические трудности, свя-
занные с обеспечением требуемой интенсивности отвода или под-
вода теплоты по длине реакционной зоны реактора.
6.3. ПРОТЕКАНИЕ ХТП В ПОТОКЕ ПОЛНОГО СМЕШЕНИЯ
6.3.1. Общая характеристика режима
При прохождении потока полного смешения частицы, посту
лающие в реакционную зону, мгновенно смешиваются с частицами,
уже находящимися в этой зоне, т. е. равномерно распределяются
по-всей длине и во всем объеме реакционной зоны. В результате
во всех точках реакционной зоны мгновенно выравниваются все
параметры, характеризующие ХТП — концентрации исходных ве-
ществ и продуктов реакции, степени превращения и выходы про-
дуктов, скорость, избирательность и др. Вследствие полного пере-
мешивания выравнивается и температура во всем объеме реак-
ционной зоны. Таким образом, ХТП в потоке полного смешения
может протекать только при изотермическом температурном ре-
жиме, независимо от значений теплового эффекта, концентрации
и степени превращения исходных веществ.
На входе в реакционную зону концентрации, степени превра-
щения, скорость реакции, температура изменяются скачкообразно
(рис. 6.17) вследств'ие мгновенного смешения компонентов, содер-
жащихся в потоке до входа в реакционную зону, с компонентами,
находящимися в реакционной зоне. Время пребывания частиц
в реакционной зоне потока полного смешения распределено нерав-
номерно. При среднем времени пребывания f — up/V часть час-
тиц покинет реакционную зону раньше т, другие частицы задер-
жатся дольше т.. Смешение исходных реагентов с продуктами
реакции и неравномерность времени пребывания частиц в реак-
ционной зоне приводят к уменьшению движущей силы процесса
по сравнению с проведением его в потоке идеального вытеснения.
В отличие от потока идеального вытеснения в любой части
потока полного смешения происходит обмен веществом с другими
частями. Замкнутых объемов в потоке полного смешения не
имеется. Поэтому уравнения, используемые для расчета ХТП, про-
ходящих в замкнутых объемах, для
потоков полного смешения неприме-
нимы.
Для вывода уравнения, характе-
ризующего протекание процесса в
потоке полного смешения, составим
материальный баланс по любому
исходному веществу, например А.
Пусть за единицу времени в реак-
ционную зону войдет смесь объемом
Рис. 6.17. Характер изменения концентра
цнй С, степеней превращения X, скорости
процесса U и температуры Т в реакционной
зоне потока полного смешения (реакция:
А-*В + <?),
107
V. Если концентрация вещества А в исходной смеси равна Сд, 0,
то в реакционную зону поступит количестоо вещества А, равное
УСа,0- Если реакция протекает без изменения объема, то реакцион-
ную зону в единицу времени будет покидать вещество А с концен-
трацией Сд в количестве КСд. Из определения скорости V про-
цесса следует:
(VC^-VCJ/v^U (6.16)
U > 0, если в процессе реакции вещество А образуется, и
U < 0, если вещество А расходуется.
С учетом того, что V/vp= \/х, уравнение (6.16) преобразуется
к виду:
^ = (Са.о-са)/^ (6.17)
Так как СА = СА,о(1—Ад), то уравнение (6.17) можно пред-
ставить в виде:
(6.18)
Для реакции l-ro порядка:
^ = Аа/(1-Аа) (6.19)
Уравнения (6.17) и (6.18) называют характеристическими урав-
нениями ХТП в потоке полного смешения или просто характе-
ристическим уравнением реактора полного сме-
шения. По этим уравнениям рассчитывают среднее время пре-
бывания реагентов в реакционной зоне ее объем цр и длину 1Р,
необходимую для достижения заданных концентраций Сд или сте-
пеней превращения Ад. При этом связь между f, оР и /р выра-
жается соотношением:
т = ор/У — lp/w (6.20)
При-протекании сложных реакций типа A->B->D для дости-
жения максимального выхода продукта В необходимо, как и для
потоков идеального вытеснения, правильно выбрать длину реак-
ционной зоны потока полного смешения.
6.3.2. Закономерности протекания ХТП в потоке
без теплообмена
При отсутствии теплообмена с окружающей средой изменение
энтальпии (теплосодержания) реакционной смеси определяется
лишь тепловыми эффектами реакций, протекающих в потоке. Как
отмечалось выше, в реакционной зоне потоков полного смешения
всегда устанавливается изотермический температурный режим
независимо от значений теплового эффекта, концентраций исход-
ных веществ и степеней превращения. Признак же адиабатического
протекания процесса — прямая пропорциональность между изме-
нением температуры Т и степени превращения А — проявляется
в характере изменения конечных (выходных) степеней превраще-
108
Рис. 6.18. Влияние длины реакционной зоны на
температуру для потока полного смешения:
а—экзотермический ХТП; б —эндотермический ХТП.
ния и температур. Например, для про-
стых необратимых реакций А->-В + <7 с
изменением длины реакционной зоны бу-
дут изменяться достигаемые степени пре-
вращения ХА, а следовательно, и темпе-
ратура смеси в реакционной зоне (рис.
6.18). При этом значения Т для каждого
значения /р остаются постоянными Ха-
рактеристику протекания ХТП для рас-
сматриваемых условий допо'лняет диа-
грамма X—Т (рис. 6.19).
Вертикальные прямые, параллельные
оси X, характеризуют изотермичность
температурного режима потока полного
смешения, а пунктирные наклонные пря-
мые, соединяющие значения конечных
степеней превращения, — адиабатические условия проведения ХТП
с позиции теплового режима. При этом угол наклона а, как и для
потока идеального вытеснения с адиабатическим тепловым режи-
мом, зависит от начальной концентрации исходного вещества.
Для расчета адиабатического разогрева или охлаждения при
достижении заданной степени превращения X используют уравне-
ние адиабаты (6.11). Следует отметить, что при проведении гетеро-
генных каталитических процессов в системе Г — Т температура
потока полного смешения на входе в реакционную зону Твх, в от-
личие от потока идеального вытеснения, может быть ниже темпе-
ратуры зажигания катализатора. Это связано с перемешиванием
частиц катализатора, благодаря которому охлажденные на входе
Рис. 6.19. Диаграмма X— Т протекания экзотермического (а) и эндотермического
(б) процесса в потоке полного смешения
109
Рис. 6.20. Влияние Ты на разогрев реагирующей
смеси и достигаемую степень превращения при
проведении обратимого экзотермического процесса
в потоке полного смешения без теплообмена с
внешней средой:
1 — Xp=f(r); ?—линия оптимальных температур; 3—изо-
термы.
в реакционную зону частицы сменяются горячими частицами, по-
ступающими из центральных областей реакционной зоны. Значе-
ние Твх для потоков полного смешения во многих случаях можно
рассчитать исходя из теплового баланса реакционной зоны.
Для проведения необратимых экзотермических реакций пре-
имущество потоков полного смешения заключается в возможности
поддержания максимальной температуры по всей длине реакцион-
ной зоны. Это обеспечивает поотекание ХТП при максимальном
значении константы скорости.
Для обратимых экзотермических реакций благодаря выравни-
ванию концентраций по длине реакционной зоны и изотермичности
температурного режима становится возможным поддержание оп-
тимального значения температуры, соответствующего достигае-
мой степени превращения. Изменяя в допустимых пределах Тах,
можно всегда выбрать такую температуру реакционной зоны, ко-
торая будет являться оптимальной для соот ютствующей дости-
гаемой степени превращения (рис. 6.20).
Проведение обратимых экзотермических ХТП при оптимальной
температуре в условиях отсутствия участков плавного разогрева
реакционной смеси и перегрева ее выше ТОПт положительно ска-
зывается на общей скорости процесса. В большинстве случаев эти
факторы компенсируют снижение движущей силы процесса при
протекании его в потоке полного смешения по сравнению с пото-
ком идеального вытеснения.
6.3.3. Теплообмен с окружающей средой
как фактор интенсификации ХТП в потоке
При отсутствии теплообмена с окружающей средой достигае-
мые степени превращения при протекании ХТП с большим тепло-
вым эффектом ограничиваются предельным разогревом или охлаж-
дением реакционной смеси. Отвод или подвод теплоты в реак-
ционную зону потока полного смешения позволяет существенно
увеличить степень превращения вещества таких ХТП.
НО
Рис. 6.21. Изменение температуры реакционной зоны потока полного смешения
с ростом интенсивности отвода теплоты для экзотермического ХТП:
/—без отвода теплоты; 2—средняя интенсивность отвода теплоты; 3 —высокая интенсивность
отвода теплоты.
Рис. 6.22. Диаграмма X — Т для экзотермического обратимого ХТП в потоке пол-
ного смешения при различной интенсивности отвода теплоты:
1— без отвода теплоты; 2—средняя интенсивность отвода теплоты; 3—высокая интенсивность
отвода теплоты: 4— линия оптимальных температур.
Если при адиабатических условиях проведения экзотермиче-
ских ХТП технолог стремится -максимально снизить Твх, увеличить
допустимый разогрев и за счет этого повысить степень превраще-
ния, то при отвод&'Теплоты из реакционной зоны потока полного
смешения принципиально возможно обеспечить любую степень
превращения независимо от Твл.
Чем большее количество теплоты будет отводиться из реакци-
онной зоны, тем ниже устанавливается температура по длине ре-
акционной зоны (рис. 6.21). При этом для обратимой экзотерми-
ческой реакции становится возможным не только поддержание
оптимальной температуры, но и повышение достигаемой степени
превращения вследствие снижения ТОВт при увеличении интенсив-
ности теплоотвода (6.22).
При протекании эндотермических процессов необходимо под-
водить теплоту в реакционную зону. Чем больше будет подведено
теплоты, тем меньше будет падение температуры (рис. 6.2'3). При
этом, если на входе в реакционную зону температура потока равна
максимально допустимой (Твх = Тыакс), то при определенном под-
воде теплоты вообще может не наблюдаться падения температуры
(линия 3). В случае, если по технологическим условиям Твх
меньше Ткакс, то при достаточно интенсивном подводе теплоты
температура реакционной зоны может быть выше Твл (линия 4)
и ее целесообразно поддерживать близкой к Тмакс. При поддержа-
нии в реакционной зоне максимально допустимой температуры
процесс протекает при максимальном значении константы скоро-
сти и обеспечивается возможность более эффективного использо-
вания теплоты смеси, выходящей из реакционной зоны.
Ш
Рис. 6.24. Диаграмма X — Т для обратимого эндотермического ХТП в потоке
полного смешения при различной интенсивности подвода теплоты:
1 — беа теплоподвода; 2—средняя интенсивность; 3—высокая интенсивность; 4—высокоинтеи-
снвный теплоподвод при ?вх ниже Тмакс; 5—линия равновесных степеней превращения.
Рис. 6.23. Изменение температуры реакционной зоны потока полного смешения
с ростом интенсивности подвода теплоты для эндотермического ХТП:
1—без подвода теплоты; 2—средняя интенсивность; 3—высокая интенсивность; 4—высоко-
интенсивный теплоподвод при Твх ниже ТМаКС-
Кроме этого, для обратимых реакций появляется возможность
достижения максимальных степеней превращения (рис. 6.24).
Таким образом, осуществление теплообмена в реакционной
зоне потоков полного смешения при сохранении всех преимуществ
изотермического температурного режима позволяет улучшить
основные показатели как экзотермических, так и эндотермиче-
ских ХТП.
6.3.4. Секционирование реакционной зоны потока
В реакционной зоне потока полного смешения концентрации
реагентов мгновенно снижаются до конечных значений и, если
необходимо обеспечить высокую степень превращения (т. е. низ-
кие конечные концентрации исходных реагентов), то скорость
Процесса будет очень низкой и потребуется реакционная зона боль-
шого объема. В этом случае технологи используют эффект разде-
ления (секционирования) реакционной зоны на несколько участ-
ков. Каждый такой участок (ступень) работает как самостоятель-
ная реакционная зона или отдельный реактор. Таким образом
технолог создает систему последовательно соединенных реакцион-
ных зон (каскад реакционных зон, каскад реакторов). В каскаде
состав реакционной смеси мгновенно изменяется при переходе из
одной реакционной зоны в другую. При этом в каждой реакцион-
ной зоне, как это характерно для потоков полного смешения, тех-
нологические параметры процесса постоянны по всему объему.
112
Рис. 6.25. Влияние условий секционирования
потока полного смешения на изменение кон-
центрации и суммарную длину реакционных
зон:
I — несекционироваиная зона*. 2—каскад из 2-хреак-
ционных зон; 3—каскад из 3-х реакционных зон.
Проведение ХТП в каскаде реак-
торов позволяет существенно увели-
чить среднюю скорость превраще-
ния, так как в первых ступенях
каскада поддерживается достаточ-
но высокая концентрация реагентов
и лишь последние работают при
низких скоростях, обусловленных
малой остаточной концентрацией исходных реагентов. Для дости-
жения одинаковых степеней превращения потребуется меньший
суммарный объем реакционных зон в секционированном потоке
по сравнению с объемом несекционированной одиночной реакцион-
ной зоны.
Эффективность секционирования зависит от требуемой степени
превращения и числа ступеней в каскаде. Сущность и эффектив-
ность секционирования для реакции А -> В поясняет рис. 6.25.
С увеличением числа ступеней каскада характеристики проте-
кания ХТП в потоке полного смешения приближаются к характе-
ристикам протекания процессов в потоке идеального вытеснения.
При числе ступеней больше 6, как правило, расчет каскада можно
проводить по модели потока идеального вытеснения.
При расчете каскада реакционных зон исходят из материального баланса,
составленного для каждой ступени каскада Для необратимой реакции 1-го по-
рядка (А->В), протекающей в кинетической области, уравнение материального
баланса 1-й ступени имеет вид:
СА 0У = СА । V + vpkCk । = СА t (V + vpk) (6.21)
Из уравнения (6.21) следует:
СА. 1 = СА. о/(‘ + kvp/V) = СА1 0 /(1 + kx) (6.22)
Аналогично получают уравнение материального баланса для 2-й и любой
n-й ступени каскада:
СА. Л = СА.21/ + МСА,2 (6 23)
СА. «^“^Afn+H^ + ^p^Aln+l) (6.24)
Соответственно концентрации исходного реагента, выходящего из 2-й и п-й
ступени, можно рассчитать по формулам:
СА,2 = СА,о/(1+^)2 (6.25)
CA.„ = CA,o/(l + fei)n (6-26)
Степень превращения ХА связана с начальной концентрацией вещества СА, о
и его конечной концентрацией СА = СА, п:
*A = (CA.o-CA)/CAt0 (6.27)
113
Из выражений (6,26) и (6.27) можно получить связь между заданным зна-
<ением ХА и необходимым числом ступеней каскада:
ХА = I - 1/(1 + (6.28)
Уравнения (6.21) — (6.28) выведены для условий разделения реакционной
зоны потока на равные объемы v и постоянной температуры в каждой ступени
каскада. Уравнение (6.28) используют для проверочных расчетов работы кас-
када, если известно число ступеней и объем реакционной зоны каждой ступени.
Для анализа протекания ХТП, описываемого более сложным кинетическим
уравнением, технолог часто использует различные графические методы расчета
каскада. t .
Секционирование реакционной зоны потока полного смешения
позволяет также при необходимости поддерживать в каждой сту-
пени каскада различные температуры. Например, экзотермические
эбратимые процессы можно проводить при температуре, оптималь-
ной для каждой ступени. Анализ проведения ХТП при таких усло-
виях будет рассмотрен а разд. 6.10.2 и 6.10.3.
6.4. СОПОСТАВЛЕНИЕ ПРОТЕКАНИЯ ХТП В ИДЕАЛЬНЫХ ПОТОКАХ
6.4.1. Простые обратимые .и необратимые реакции
Сравнение протекания ХТП в различных потоках проводят по
достигаемой степени превращения при одинаковых объемах реак-
ционных зон, по необходимым объемам реакционных зон при за-
данной степени превращения, по избирательности и другим харак-
теристикам. При сравнении принимают одинаковыми начальные
условия протекания ХТП (Твх, концентрации, давления, размеры
частиц и др.), хотя каждому виду потока присущи свои рацио-
нальные значения входных параметров проведения процессов. Та-
ким образом, проводимое ниже сравнение является в определенной
мере условным.
Рассмотрим протекание простых реакций при изотермическом
температурном режиме. Сравнение проведем по объемам реакци-
онных зон потоков идеального вытеснения ХР. в и потоков смеше-
ния Up. с, необходимых для обеспечения одной и той же степени
превращения.
Для гомогенных ХТП скорость процесса описывается уравне-
ниями типа:
О' —/г АС (6.2?
При изотермическом температурном режиме константы скоро-
сти реакций, проводимых в потоке идеального вытеснения и пол-
ного смешения, равны между собой, и поэтому основное различие
протекания процесса обусловлено характером изменения движу-
щей силы АС. Для реакции А->-В ДС = СА. В потоке идеального
вытеснения Сь плавно -изменяется от максимального значения в
начале реакционной зоны Сд, 0 до конечного значения СА, к, соот-
114
Рис. 6.26. Характер изменения концентрации исходного вещества по длине реак-
ционной зоны для потоков идеального вытеснения (1) и полного смешения (2).
Рис. 6.27. Влияние степени превращения X и порядка реакций т на соотноше-
ние объемов реакционных зон потока полного смешения (ор. с) и идеального вы-
теснения (Ор. в)
ветствующего выходу реагентов из реакционной зоны (рис. 6.26,
кривая 1). При этом средние значения концентрации Са, в и дви-
жущей силы ДСВ находятся между СА, о и СА, к-
В потоке прлного смешения движущая сила постоянна по дли-
не реакционной зоны, и среднее значение ДСс равно конечному
ДСА, к- Очевидно, что ДСВ всегда выше ДСс. А это означает, что
средняя скорость ХТП в потоке идеального вытеснения всегда
выше средней скорости протекания процесса в потоке смешения.
Так, для реакции 1-го порядка отношение объемов реакционных
зон можно получить из анализа уравнений (4.4) и (6.19):
X / 1
Тс/Тв == Цр. с/^р. в = । - ~ х ! I - л (6.30)
Из выражения (6.30) можно сделать два вывода: а) ир. в при
изотермическом режиме всегда меньше ир. с; б) чем выше X, тем
больше соотношение ооъемов ир. c/vv. в. Кроме того, как показы-
вает анализ протекания реакций различного порядка, с увеличе-
нием порядка реакции при одинаковых значениях X соотношение
объемов увеличивается (рис. 6.27).
Рассмотрим более сложный случай, когда протекание процесса
сопровождается значительным выделением или поглощением теп-
лоты. В потоках идеального вытеснения, как правило, процесс
протекает в таком случае при адиабатическом температурном ре-
жиме, а.в потоках полного смешения — при изотермическом.
Пусть протекает эндотермический процесс. При отсутствии в
реакционной зоне теплообменных поверхностей преимущества про-
ведения процесса в потоке идеального вытеснения по сравнению
с потоком полного смешения очевидны. В реакционной зоне по-
тока смешения установится постоянная температура Тс, равная
конечной ТВых (рис. 6.28). В потоке вытеснения при адиабати-
ческом температурном режиме средняя температура Тв будет
115
Рис. 6.28. Характер изменения температуры по длине реакционной зоны потоков
идеального вытеснения (/) и полного смешения (2) при проведении эндотермиче-
ских процессов без теплообмена.
Рнс. 6.29. Характер изменения температуры по длине реакционной зоны потоков
идеального вытеснения (/) и полного смешения (2) при проведении экзотерми-
ческих процессов без теплообмена
находиться в интервале между Твх и Твых. При этом ТВ>ТС. Следо-
вательно, и значения константы скорости в потоке идеального вы-
теснения kB будут выше, чем для потока полного смешения fec,
т. е. kL > £с. Поскольку АСВ > ДСс, несложно показать, что
UJUc = kB/kc = > 1 (6.31)
т. е. средняя скорость процесса в потоке вытеснения UB будет
всегда выше средней скорости ХТП, осуществляемого в потоке
полного смешения Ос-
Если в реакционную зону потока полного смешения подводится
теплота, то, как показано в разд. 6.3.3, Тс может быть близка
к Тмакс и существенно превышать Тв. В этом случае необходимо
сравнивать отношение движущих сил АСв/АСс (которое больше 1)
и отношение средних значений констант скоростей kB/kB (которое
меньше 1).
Если протекает экзотермический процесс, то средняя темпера-
тура 7С и среднее значение константы скорости fec в реакционной
зоне потока полного смешения во всех случаях выше соответствую-
щих значений 7В и kB (рис. 6.29) и для выбора рациональных
условий проведения процесса необходимо сопоставление соотно-
шений АСв/АСс и kc/kB. В большинстве случаев при высоких зна-
чениях тепловых эффектов, начальных концентраций исходных
веществ и степеней превращения АСв/АСс < kc/kB, и ХТП рацио-
нально проводить в потоках смешения с изотермическим темпера-
турным режимом. При этом наиболее очевидно их преимущества
проявляются при отводе теплоты из реакционной зоны (см.
разд. 6.3.3).
6.4.2. Влияние типа потока на избирательность
Эффективность проведения ХТП со сложными реакциями во многом опреде-
ляется типом потока, в котором протекает процесс. Технолог должен выбрать
такие условия проведения процесса, при которых обеспечивалась бы возмож-
116
ность получения максимального количества целевого продукта и минимум по-
бочных продуктов или отходов.
fc k.
Для параллельных реакций А —> В А —D избирательность по целево-
му продукту В пропорциональна соотношению UbIUd, т. е.
Ч>В ~ Ur/Ud (6.32)
Пусть скорость образования целевого продукта В описывается уравнением
t/B=^T = felCA <6-33>
а скорость образования побочного продукта D — уравнением
dCn
t/D = ^=fe2C^ (6.34)
Тогда
= (6.35)
Анализ уравнения (6.35) показывает, что на избирательность могут влиять
многие параметры технологического режима. Например, отношение kjkz зависит
от типа катализатора и размера его частиц, температуры, давления. Сильно
влияет на избирательность и концентрация исходного реагента, поскольку при
X = const Са ~ Сь, о.
Рассмотрим сначала протекание процесса при изотермическом температурном
режиме, т. е. когда тепловым эффектом можно пренебречь. В этом случае
<рв ~ Сдт, Дт = т, — т2 (6.36)
Если порядок реакции получения целевого продукта В больше, чем порядок
реакции образования побочного продукта D, т. е. если Длг>0, то избирательность
повышается с ростом СА. В этом случае целесообразно перерабатывать смесь с
повышенной начальной концентрацией вещества А. Процесс выгодно проводить
в потоке идеального вытеснения, так как средняя концентрация исходного ве-
щества Ga, в по длине реакционной зоны такого потока при одинаковых степенях
превращения будет выше концентрации А в реакционной зоне потока полного
смешения Сд с (см. рис. 6.26).
Когда Дт <0. <рв ~ \/С^т. В этом случае целесообразно работать с низ-
кими начальными концентрациями А и проводить ХТП в потоках полного сме-
шения.
Если процесс сопровождается большим тепловым эффектом, а теплопровод-
ность реакционной смеси низка, то в реакционной зоне потока идеального вы-
теснения реализуется адиабатический температурный режим. В реакционной зоне
потока полного смешения, как отмечалось выше, температурный режим всегда
изотермический. В этих случаях при выборе гидродинамических условий прове-
дения ХТП кроме концентрационного фактора необходимо учитывать и темпе-
ратурный фактор, т. е. характер изменения температуры по длине реакционной
зоны. Температурный фактор влияет на соотношение ki/kz в уравнении (6.35).
Выразив зависимость kt и kz от температуры через уравнение Аррениуса, получим:
kijk2 = й01 exp [— EiAflDI/ffco? exp [— E2HRT)} } =
= k' exp [(£2 - E.)/(/?T)] = k' exp 'AE/IET)] (6.37)
Здесь С, и Ег— энергии активации образования целевого и побочного про-
дуктов; k‘ — коэффициент, зависящий от механизма реакции и природы катали-
затора.
Пусть протекает экзотермический ХТП в потоках идеального вытеснения и
полного смешения при отсутствии теплообмена в реакционных зонах. Характер
изменения температуры по длине реакционной зоны в сравниваемых потоках при
117
одинаковой температуре реакционной смеси на входе Тех и одной и той же сте-
пени превращения вещества А показан на рис. 6.29.
Если Е, < Е2 и От) > т2, процесс выгодно проводить в потоке идеального
вытеснения, в котором устанавливается пониженная температура и достаточно
высокая концентрация вещества А, что должно обеспечить повышенную изби-
рательность.
Когда Ei > Е2, но Ш) > т2, при проведении ХТП в любом из рассматривае-
мых потоков наблюдают противоречивое влияние концентрационного и темпера-
турного факторов на избирательность. Так, концентрационный режим для пото-
ков идеального вытеснения благоприятно воздействует на избирательность (обес-
печивается высокая средняя концентрация вещества А), но адиабатический тем-
пературный режим не всегда обеспечивает требуемую высокую среднюю темпера-
туру по длине реакционной зоны.
Аналогичная ситуация возникает, когда Et < Е2, но mi < т2. В этих случаях
необходимо дополнительно количественно сравнить влияние концентрационного
и температурного факторов на избирательность, а затем уже выбрать тип пото-
ка. в котором целесообразно проводить процесс.
Если необходимо обеспечить высокую избирательность при протекании эндо-
термического процесса, то следует помнить, что по длине реакционной зоны по-
тока полного смешения устанавливается температура Тс более низкая, чем сред-
няя температура реакционной зоны потока идеального вытеснения Тв (см.
рис, 6.28). В этом случае можно рекомендовать проведение ХТП в потоке пол-
ного смешения, если 9i < Е2, а т, < т2. При Е, > Е2 и т, > т2 ХТП выгодно
проводить в потоках идеального вытеснения.
Варианты, когда Е, > Е2, но Ш] < т2 или когда Ei < Е2, > т2, требуют
выполнения дополнительного численного анализа, при котором сопоставтяется
влияние концентрационного и температурного факторов на избирательность
Избирательность последовательных реакций зависит также от соотношения
скоростей их протекания, а выбор типа потока, в котором рационально прово-
дить ХТП, обусловлен характером изменения концентраций и температуры по
длине реакционной зоны.
Для последовательных реакций типа А —В —> D кинетические уравнения
имеют вид:
dCA
—dr=k'C* (6-38)
dCB
t/B=-d7’ = ^CA-^CB <6-39)
nD = -^- = fe2CB (6.40)
Соотношение скоростей образования целевого В и побочного D продуктов,
пропорциональное избирательности, равно:
, 4>в ~ V D = А ^2^в)/(^2^в) = ^1^а/(^2^в) ’ (6.41)
Из соотношения (6.41) следует, что избирательность тем эспьше, чем выше
концентрация исходного реагента С* и отношение k\jk2. Поэтому, исходя из кон-
центрационного фактора, ХТП предпочтительно проводить в потоке идеального
вытеснения, средняя концентрация вещества А в котором выше, чем в реакцион-
ной зоне потока полного смешения (см. рис. 6.26). Это всегда справедливо, если
тепловые эффекты невелики и процесс протекает при изотермическом темпера-
турном режиме.
Для ХТП, протекающего со значительным тепловым эффектом, необходимо
проанализировать и влияние температурного режима на избирательность. Как и
при анализе параллельных реакций, для последовательных реакций можно запи-
сать:
ki/k2 =feOi exp I— Eil(RT)\l{kOz exp [— E2/(RTY\ } =
= k exp [ДЕ/(ЯГ)]; ДЕ = E2 - Ei (6.42)
118
Если Ei > Е2, т. е. ДЕ < 0, то проведение процесса при повышенной тем-
пературе способствует увеличению избирательности Для потока идеального вы-
теснения это будет наблюдаться при протекании эндотермических процессов (см.
рис. 6.28).
При Е] < Е2 необходимо стремиться к проведению ХТП при пониженных тем-
пературах. В этом случае эффективность проведения процессов в потоке идеаль-
ного вытеснения будет максимально проявляться для экзотермических реакций
(см. рис. 6.29).
Во всех других вариантах эффективность проведения ХТП в реакционной
зоне потоков идеального вытеснения будет несколько снижаться из-за отрица-
тельного воздействия на избирательность адиабатического температурного ре-
жима. Для таких случаев более эффективным может оказаться проведение ХТП
в секционированном потоке полного смешения.
6.5. ПРОТЕКАНИЕ ХТП В НЕИДЕАЛЬНЫХ ПОТОКАХ
6.5.1. Отклонения реальных потоков от идеальных
Реальные потоки, с которыми технологам приходится сталки-
ваться на практике, могут существенно отличаться по своим ха-
рактеристикам от идеальных. При расчете ХТП в таких потоках
необходимо введение поправок на степень неидеальности потока.
Наиболее существенными и часто встречающимися отклоне-
ниями реальных потоков вытеснения от идеальных являются два:
неравномерность скорости движения частиц в поперечном сечении
потока и наличие, как правило, незначительного продольного пе-
ремешивания.
Отклонения реальных потоков смешения от идеальных моделей
связаны с наличием застойных зон, с проскоком существенной ча-
сти реагирующих компонентов без химического взаимодействия
(байпасная часть потока), с недостаточной интенсивностью пере-
мешивания, не обеспечивающей при высоких скоростях процесса
полного выравнивания концентраций и температур.
В некоторых случаях наблюдаемые отклонения поведения ре-
альных потоков существенно не сказываются на выходных пока-
зателях ХТП, рассчитываемых по наиболее подходящим для кон-
кретных условий моделям идеальных потоков. Так, для ХТП, про-
текающего с невысокой скоростью в аппаратах с кипящими
слоями, даже при не очень интенсивном перемешивании наблюда-
ется постоянство концентраций и температур по длине реакционной
зоны потока, а показатели процесса достаточно точно рассчитыва-
ются по модели потока полного смешения.
Режим в потоках большого диаметра при движении жидкости
или газа через неподвижный слой зернистого материала (напри-
мер, катализатора), несмотря на понижение линейной скорости
течения у стенок, можно отнести к идеальному вытеснению. Про-
текание ХТП в потоке газа или жидкости, движущихся в длинных
трубках небольшого диаметра, также может быть описано с ис-
пользованием модели идеального вытеснения, особенно если ско-
рость химической реакции невелика.
119
6.5.2. Модели неидеальных потоков
Для описания процессов, проводимых в неидеальных потоках,
используют различные физические представления (модели). В на-
стоящее время для описания структуры неидеальных потоков при-
меняют ячеечную и диффузионную модели. Согласно ячеечной мо-
дели реакционная зона условно разбивается на ряд секций (ячеек),
в каждой из которых поток описывается моделью полного смеше-
ния. Суммарный объем всех ячеек равен объему реакционной
зоны реального потока. Степень отклонения от идеальности харак-
теризуется экспериментально найденным числом ячеек п, которое
может изменяться от единицы до бесконечности. Фактически яче-
ечная модель — это аналог каскада реакционных зон потока сме-
шения (см. разд. 6.3.4). При п->-1 режим в потоке приближается
к режиму полного смешения, при п->°о — к режиму идеального
вытеснения. Зная из экспериментальных данных конечную концен-
трацию вещества, выходящего из реакционной зоны потока СА,
или конечную степень превращения X, можно описать процесс
в реальном потоке с помощью ячеек полного смешения, подбирая
соответствующее их число п. При этом можно использовать урав-
нения (6.26) и (6.28).
6.6. КЛАССИФИКАЦИЯ И ОСНОВНЫЕ ПОКАЗАТЕЛИ
РАБОТЫ ХИМИЧЕСКИХ РЕАКТОРОВ
В промышленности используют тысячи различных химических
реакторов, характеризующихся конструктивными особенностями,
режимами протекания процессов. Независимо от типа реактора
можно выделить три основных конструктивных элемента: устрой-
ства для ввода исходных реагентов и вывода продуктов реакции,
устройства для смешения или распределения потоков в реакцион-
ной зоне и теплообменные элементы. Учитывая, что конструкция
реакторов должна обеспечивать заданный гидродинамический и
температурный режим потока, именно характерные свойства по-
тока должны быть положены в основу классификации реакторов.
К характерным признакам классификации, охватывающим все
многообразие применяемых в химической промышленности реакто-
ров, относят следующие факторы: временной, характеризую-
щий изменение параметров технологического режима во времени;
гидродинамический, характеризующий режим движения и
структуру потока реагентов, проходящих через реактор; темпе-
ратурный, определяющий характер изменения температуры по
длине реакционной зоны. По временному фактору все реакторы
разделяют на непрерывнодействующие и реакторы пе-
риодического действия. Для описания и анализа работы
непрерывнодействующих реакторов используют закономерности
протекания ХТП в потоках (см. разд. 6.1—6.3). Описание работы
реакторов периодического действия проводят на основе законо-
120
мерностей протекания ХТП в замкнутом объеме (см. гл. 4, 5).
По гидродинамическому режиму реакторы непрерывного действия
разделяют на реакторы вытеснения и реакторы сме-
шения, а по температурному признаку — на адиабатиче-
ские, изотермические и политермические. Конструк-
тивные особенности реакторов определяются также фазовым со-
стоянием реагирующих компонентов и термодинамическими пара-
метрами протекания ХТП, т. е. температурой и давлением.
Основные показатели работы реакторов условно разделяют на
технологические, энергетические, эксплуатационные и экономи-
ческие.
К технологическим характеристикам работы реак-
торов можно отнести показатели ХТП, протекающих в том или
ином реакторе (см. разд. 3.2). Кроме того, работу реактора ха-
рактеризуют производительность и интенсивное гь.
Производительностью' П (моль/с,'кг/с, м3/с) называют коли-
чество G (массу т, объем и) выработанного продукта или пере-
работанного сырья за единицу времени:
П = G/т - (6.43)
Повышение производительности может быть достигнуто увели-
чением размера реакторов или скорости протекания ХТП. Макси-
мальную производительность часто называют мощностью реак-
тора. Увеличение мощности реакторов является одним из основ-
ных направлений развития химической промышленности, так как
позволяет улучшить экономические показатели работы произ-
водств.
Интенсивность работы реактора / )моль/(с-м3), кг/(с-м3),
м3/(с-м3)] выражают как отношение производительности П
к объему реактора цР:
( = П/Vp = G/(tcp) (6.44)
Интенсивность можно повысить улучшением конструкции реак-
торов и изменением режимбв протекания ХТП. Поскольку интен-
сивность работы реакторов фактически отражает среднюю ско-
рость протекания ХТП4 Для увеличения I используют все приемы
повышения скорости процессов, изложенные в главах 4, 5 и
разд. 6.3.3, 6.3.4, 6.4.2.
Энергетические показатели работы реакторов харак-
теризуют затраты энергии на преодоление гидравлических сопро-
тивлений потоком реагирующих веществ и на их перемешивание,
а также эффективность использования теплоты, подводимой в ре-
актор для проведения эндотермических процессов или выделяю-
щейся при протекании экзотермических процессов.
К эксплуатационным характеристикам относят
легкость управления и обеспечения устойчивого режима и без-
опасности работы реактора. Эти характеристики зависят от кон-
структивного совершенства реактора, которое определяет также
и его ремонтоспособность.
121
Экономические характеристики реактора определяют
стоимость его изготовления и монтажа, а также затраты на про-
ведение ремонтных работ.
Основные требования к реакторам сводятся к повышению ин-
тенсивности его работы, снижению энергетических затрат на транс-
портировку реагентов через реактор, уменьшению потерь теплоты
при проведении эндотермических процессов, увеличению степени
использования - теплоты экзотермических реакций, обеспечению
устойчивости технологических режимов протекания ХТП и без-
опасности работы, снижению стоимости изготовления реакторов
и их ремонта.
6.7. РЕАКТОРЫ ДЛЯ ГОМОГЕННЫХ ПРОЦЕССОВ
Промышленные реакторы для гомогенных процессов занимают
промежуточное положение между реакторами идеального вытес-
нения и полного смешения.
Если исходные реагенты перед поступлением в реактор хорошо
перемешаны, то ХТП протекает в кинетической области и пере-
мешивание внутри реакционной зоны может быть нежелательным
фактором.
Часто исходные реагенты подают в реактор отдельными пото-
ками без предварительного смешения. Перемешивание потоков
происходит непосредственно в реакторе, и от интенсивности пере-
мешивания зависит не только скорость ХТП, но и взрывобезопас-
ность.
Следует учитывать, что перемешивание во всех случаях интен-
сифицирует теплообмен.
6.7.1. Система жидкость — жидкость [Ж — Ж)
Реакторы для проведения гомогенных процессов с участием
жидких реагентов могут быть периодического и непрерывного дей-
ствия. Как правило, они представляют собой камеру, снабженную
каким-либо перемешивающим устройством.
Рис. 6 30. Каскад реакторов смешения.
Рис 6.31 Колонный реактор с насадкой:
/—корпус; 2—насадка из колец; 3—колосниковая решетка; /—разбрызгивающее устройство.
122
Рис. 6.32. Трубчатый реак-
тор:
1—внутренняя труба; 2—тепло-
обменная рубашка; 3 —колено.
Режим движения жидкости в реакционной зоне реактора не-
прерывного действия близок к полному смешению. Для повыше-
ния скорости процесса и увеличения степени превращения реак-
торы устанавливают в виде каскада (рис. 6.30).
К реакторам вытеснения относят колонные и трубчатые реак-
торы. Колонные реакторы (рис. 6.31) выполнены в форме
высокого цилиндра, внутри которого установлены перегородки
или решетки, обеспечивающие незначительное* перемешивание.
Интенсивность перемешивания в этих реакторах невелика, и его
роль сведена лишь к выравниванию температур и увеличению ско-
рости теплообмена.
Трубчатые реакторы (рис. 6.32) выполнены из длинных
труб, соединенных последовательно в секции. Для обеспечения
теплообмена трубы снабжены рубашками, в которых циркули-
рует тепловой агент. Длина труб определяется необходимым вре-
менем контакта. Так, непрерывнодействующий проточный реактор
для гидролиза дихлорэтана имеет длину около 1 км. Несмотря на
турбулентное течение жидкости внутри трубы, в таких реакторах
режим движения реагирующих компонентов соответствует вытес-
нению.
6.7.2. Система газ — газ (Г — I]
Для газофазных процессов применяют камерные и трубчатые
реакторы только непрерывного действия. Смешение реагентов
осуществляют с помощью инжекторов, центробежных смесителей
и др. Характерная конструкция реактора камерного типа с
инжекторным смешением показана на рис. 6.33.
Такой реактор применяют, например, для
синтеза хлороводорода. Смешение исходных
потоков хлора и водорода осуществляется в
камере 1, в которой режим движения газовой
смеси соответствует смешению. Непосред-
ственно синтез проводят в камере 2. Анало-
гичные реакторы используют для термоокис-
лительного крекинга метана и других газофаз-
ных процессов.
уйе/тют
Рис. 6.33. Реактор для хлорирования метана:
1—смесительная камера; 2—реакционная камера.
123
Рис. 6.34. Камерный реактор с центробежным перемешиванием газовой смеси.
Рис. 6.35. Кожухотрубный реактор с теплообменом:
1 — кожух с двумя днищами; 2—трубиые решетки; 3—теплообменные трубки.
Для улучшения перемешивания потоки могут поступать в реак-
ционную зону тангенциально. Схема такого камерного реактора
с центробежным перемешиванием реагентов изобра-
жена на рис. 6.34. Режим работы близок к полному смешению.
В реакторе поддерживается изотермический температурный режим.
В режиме вытеснения работают трубчатые реакторы. На
рис. 6.35 представлен трубчатый реактор с теплообменом между
двумя газами. По температурному режиму он политермический.
В промышленности часто используют трубчатые реакторы с ру-
башкой для жидкостного нагрева или охлаждения.
6.8. РЕАКТОРЫ ДЛЯ НЕКАТАЛИТИЧЕСКИХ
ГЕТЕРОГЕННЫХ ПРОЦЕССОВ
Конструкции реакторов для гетерогенных процессов, как пра-
вило, сложнее, чем для гомогенных ХТП. Это связано с необхо-
димостью обеспечения интенсивной массопередачи и создания воз-
можно большей поверхности контакта реагирующих фаз. В от-
личие от гомогенных процессов в гетерогенных системах каждая
фаза может двигаться через реактор в своем режиме. Например,
одна фаза может проходить через реакционную зону в виде по-
тока смешения, а другая — в режиме вытеснения. Кроме того, сами
потоки могут быть различно ориентированы относительно друг
друга: прямоток, противоток или перекрестный ток. От конструк-
ции реактора зависит возможность проведения ХТП в наиболее
выгодной области — диффузионной или кинетической.
6.8.1. Система газ — жидкость (Г — Ж)
Химические процессы между газом и жидкостью протекают •
при производстве фосфорной, кремнефтористоводородной кислот,
нитрата, фосфата и сульфата аммония, гидрокарбоната натрия и
других солей, во многих производствах органических продуктов.
124
Для проведения таких ХТП часто используют типовые аппараты,
применяемые также и для осуществления физических массообмен-
ных процессов: абсорбции, десорбции, ректификации, теплообмена
и др. К таким аппаратам относят различные типы колонных аппа-
ратов: пленочные, барботажные, разбрызгивающие, пенные.
В основном это реакторы непрерывного действия, хотя некоторые
конструкции (например, барботажные, пенные) могут использо-
ваться в режиме полупер иод ического действия с непрерывным пи-
танием по газовой фазе. Все они выполнены в виде колонн, внут-
реннее устройство которых предназначено для развития поверх-
ности контакта фаз и ее обновления в процессе взаимодействия
реагентов.
Например, в пленочных реакторах жидкая фаза дви-
жется тонкой пленкой по поверхности насадочных тел. В качестве
насадок используют керамические .и металлические кольца, шары,
спирали, системы плоскопараллельных пластин и др. (см.
рис. 6.31). Условно принимают, что жидкость течет по всей поверх-
ности насадок.'Газ движется прямотоком или противотоком. Дви-
жение как жидкости, так и газа соответствует режиму вытеснения.
При протекании ХТП с большим тепловым эффектом, как пра-
вило, устанавливается политермический температурный режим.
Благодаря большому свободному сечению насадки гидравлическое
сопротивление реакторов невелико.
Колонные реакторы насадочного и трубчатого типа широко
используют для проведения адсорбционно-десорбционных процес-
сов в производстве синтетического каучука, азотной кислоты, при
переработке коксового газа, в некоторых установках органиче-
ского синтеза и др.
Общим недостатком таких реакторов является низкая интен-
сивность, обусловленная относительно малой удельной поверх-
ностью контакта фаз и несущественным обновлением этой поверх-
ности. Большие ограничения интенсификации связаны с невозмож-
ностью работы при повышенных скоростях газа (как правило, не
более 1,5 м/с), так как это приводит к срыву пленки, зависанию
жидкости и значительному ее уносу.
В барботажных реакторах (рис. 6.36) газовая фаза
диспергируется и проходит через жидкость в виде мелких пузы-
рей. Большое число пузырьков в единице.объема жидкости обес-
печивает необходимую поверхность контакта фаз. Газовая фаза
движется в режиме вытеснения, жидкая фаза при высоком газо-
содержании — в режиме смешения. Для увеличения движущей
силы процесса и степени превращения в колонне устанавливают
до нескольких десятков колпачковых или ситчатых тарелок, на
каждой из которых образуется барботажный слой, т. е. реакцион-
ную зону секционируют. При этом газовая и жидкая фазы на
каждой тарелке движутся перекрестно. В то же время по высоте
колонны соблюдается принцип противоточного движения фаз.
Характерным примером использования колонных барботаж-
ных реакторов являются процессы концентрирования кислот
125
Рис, 6.36. Схема устройства барботажного реактора с колпачковыми (а) и сит-
чатыми (б) решетками:
1—колпачковая решетка; 2—переливная труба; 3—ситчатая решетка; 4—сливной порог.
Рис. 6".37. Полая башня с разбрызгиванием жидкости:
1 — корпус; 2—распылители жидкости.
в производстве соды. Их применяют также в нефтехимических
производствах, в технологии органических веществ.
Будучи по сравнению с пленочными реакторами более слож-
ными по конструкции барботажные реакторы могут работать при
более высоких скоростях газовой фазы и обеспечивать большую
интенсивность.
В колонных реакторах разбрызгивающего типа
развитие поверхности контакта фаз достигается путем дисперги-
рования жидкой фазы с помощью механических или пневматиче-
ских форсунок. Различают полые форсуночные и скоростные пря-
моточные реакторы.
В полых колонных реакторах (рис. 6.37) газовый поток дви-
жется вертикально вверх и встречает на своем пути распыленную
жидкость. Высокая скорость газового потока (до 5 м/с) обеспе-
чивает снятие внешнедиффузионных торможений. При неизмен-
ной тонкости распыления поверхность контакта фаз пропорцио-
нальна плотности орошения — объему жидкой фазы, подаваемой
в единицу времени на единицу площади поперечного сечения
реактора. Поэтому при низких плотностях орошения [менее
10 м3/(м2-ч)] эти реакторы работают неудовлетворительно. Обыч-
но плотность орошения составляет 30—45 м3/(м2-ч). Ввиду боль-
шого брызгоуноса на выходе газового потока из реактора уста-
навливают брызгоотделитель. Из-за неравномерности распределе-
ния факела распыления по объему реакционной зоны интенсив-
ность полых реакторов невелика.
В скоростных реакторах (рис. 6.38) организовано прямоточное
' движение газа (Г) и жидкости (Ж) Распыление жидкой фазы
126
Рис. 6.38. Скоростной прямоточный реактор.
Рис. 6.39. Пенный однополочный (а) и многополочный (б) аппарат:'
1 — решетка: 2—приемная короСкг , 3—сливной порог: 4—коробка для разрушения пены. ,
осуществляется за счет кинетической энергии газового потока,
движущегося с большой скоростью (более 50 м/с). Вся распылен-
ная жидкость уносится с газовой фазой и отделяется в сепараци-
онных устройствах.
Конструкции пенных реакторов и колонных барботажных
в основном аналогичны. Главное отличие состоит в структуре
двухфазной системы, находящейся на распредепительной решетке
(рис. 6.39). Пенный слой образуется при такой скорости газа, ко-
гда силы трения газа о жидкость уравновешивают массу послед-
ней. В результате возникает взвешенный слой подвижной пены
в виде быстродвижущихся пленок, капель и струй жидкости, пе-
ремешанной с пузырьками и струями газа. Такая двухфазная си-
стема имеет очень большую и при этом интенсивно обновляю-
щуюся поверхность контакта фаз. Подобный режим обеспечивает
наиболее интенсивное взаимодействие газовой и жидкой фаз. Ско-
рость газа в сечении реактора" составляет от 1 до 3,5 м/с.
Движение жидкости соответстг ует режиму смешения, а движе-
ние газа — ближе к режиму вытеснения. Для анализа протекания
ХТП используют различные ячеечные, и диффузионные модели.
Для системы Г — Ж используют и трубчатые реакторы (см.
рис. 6.32), в которых обычно проводят высокотемпературные про
цессы пиролиза органических веществ. Их применяют также для
проведения абсорбционно-десорбционнйх процессов. Режим дви-
жения фаз — вытеснение, температурный режим — политермиче-
ский.
6.8.2. Система жидкость — твердое вещество [Ж — Т)
Реакторы для некаталитических ХТП в гетерогенных системах
Ж — Т находят широкое применение в неорганических производ-
ствах. К ним относят: - производство фторида алюминия, фосфатов
натрия, суперфосфата, экстракционной фосфорной кислоты и др.
127
Рис. 6.40. Вертикальный реактор для получения фторида алюминия.
Рис. 6.41. Двухвальный шнековый реактор:
1—кожух; 2—вал; 3—лопасти.
Как правило, для проведения процессов используют реакторы пе-
риодического действия или непрерывного действия с механическим
перемешивающим устройством. Характерным примером может
служить вертикальный реактор с лопастной мешалкой,
используемый для получения фторида алюминия из H2SiFe и
А1(ОН)3 (рис. 6.40). Это реактор периодического действия. Пере-
мешивание необходимо для уменьшения внешнедиффузионных
торможений. Требуемая температура (90—95 °C) поддерживается
подачей острого пара непосредственно в реакционную зону.
Для переработки смесей с высоким содержанием твердой
фазы используют реактор с двухвальными шнеками
(рис. 6.41). В корытообразном кожухе 1 такого реактора разме-
щено два вала 2 с лопастями 3, расположенными по винтовой ли-
нии. Валы вращаются навстречу друг другу и обеспечивают об-
новление поверхности контакта фаз и перемещение материала от
штуцера загрузки к штуцеру выгрузки. Основное перемешивание
смеси происходит в поперечном направлении, поэтому режим дви-
жения фаз через реакционную зону принимают соответствующим
вытеснению. В подобных реакторах, например, получают моно-
кальцийфосфат действием фосфорной кислоты на известняк, а
также проводят нейтрализацию суперфосфата.
6.8.3. Система газ — твердое вещество (Г — Т|
и газ — твердое вещество — жидкость (Г — Т — Ж)
Наиболее типичными ХТП с участием газообразных и твердых
реагентов (Г — Т) являются пиролиз твердого топлива, различные
виды обжига твердых материалов, реакции газообразных компо-
нентов на твердых катализаторах и др.
Большинство некаталитических ХТП в системе Г — Т проте-
кает при температуре выше 700 К. Химические реакторы, предна-
значенные для осуществления такого рода процессов, имеют ха-
рактерные особенности и называются печами. Например, в печах
осуществляют прокалку карбонатов (СаСОз, MgCO3 и др.) и
128
гидроксидов [А1(ОН)з] при производстве СОг и оксидов, окисли-
тельный обжиг сульфидных руд (FeS2, CuS) при производстве
оксидов металлов и оксида серы (IV), синтез соединений из эле-
ментов, например оксидов азота из азота и кислорода и др. При
этом в ряде случаев вследствие высоких температур возможен
переход твердой фазы в жидкую, например из-за плавления час-
тиц. Таким образом, получается третья фаза и процесс протекает
в системе Г — Т — Ж.
Исключительное разнообразие проводимых в промышленности
высокотемпературных процессов привело к большому числу раз-
личных типов печей. Промышленные печи классифицируют по
энергетическим, технологическим и конструктивны^ признакам.
Энергетические признаки включают характеристики источника
тепловой энергии и способа нагрева реакционной смеси. По источ-
никам энергии различают: 1) топливные печи, использующие раз-
личные виды топлива; 2) электрические печи; 3) реакционные
печи, в которых теплота выделяется в результате химической ре-
акции ХТП.
По способу нагрева различают: 1) печи прямого нагрева, в ко-
торых источник энергии находится непосредственно в реакционной
зоне; 2) печи косвенного нагрева, в которых теплота от источника
тепловой энергии передается в реакционную зону через стенки
реактора.
Технологические признаки связаны с особенностями отрасли
производства, гидродинамическим и температурным режимами
в реакционной зоне. у
По конструктивным признакам выделяют шахтные, полочные,
распылительные печи, печи кипящего слоя, барабанные вращаю-
щиеся, камерные, туннельные, ванные, трубчатые и др.
Ниже рассмотрены основные конструкции печей с учетом энер-
гетических и технологических признаков.
Шахтные печи — это реакторы непрерывного действия, ре-
акционная зона которых выполнена в виде вертикальной шахты
круглого или прямоугольного сечения (рис. 6.42). Шахта запол-
нена кусковым материалом (шихтой). Исходный твердый мате-
риал загружается сверху и медленно опускается по мере отвода
твердых продуктов реакции в нижней части шахты. Газ подают
снизу, а газообразные продукты реакции выводятся из верхней
части шахты. Таким образом, газовая и твердая фазы движутся
противотоком в режиме, близком к идеальному вытеснению.
Шахтные печи — это топливные печи прямого нагрева. Теп-
лота выделяется в самом нагреваемом материале или в резуль-
тате окисления твердого топлива (кокса), входящего в состав
шихты, или вследствие горения природного газа, вводимого в ре-
акционную зону.
По температурному режиму шахтные печи политермичны. По
высоте печи можно выделить несколько зон. В верхней зоне про-
исходит подсушка и нагрев твердого материала за счет теплоты
образования газообразных продуктов, движущихся противотоком.
Б Зак. 1869
129
^Исходный твердый материал
/оо |о'о\н Гязообразные продукты
/о О к о V ” реакции
Го°/100а]
вМ
г0°|О° I
\° О 00
к?о 1©О /
V о-^°/
Г° о°/
\°0 О°/
Исходный \“°с!оо
1~лЛ о I
газ
Т Рвер0ые продукты
х реакции
Рис. 6.42. Шахтная печь.
Рис. 6.43. Печь обжига пылевидного колчедана.
В средней зоне протекают непосредственно химические реакции.
В нижней зоне происходит охлаждение твердых продуктов реак-
ции и нагрев исходной газовой смеси.
Шахтные печи широко распространены в промышленности и
применяются для проведения процессов обжига известняка, суль-
фидных руд, выплавки чугуна, газификации твердого топлива, по-
лукоксования углей, торфа и др. Это одни из самых мощных пе-
чей. Например, мощность доменных печей достигает 10 000 т/сут.
Высота печи может превышать 50 м. Удельная производитель-
ность— до 100 кг/(м3-ч). Диаметр печи для обжига известняка
и мела — до 7 м, высота — более 20 м, производительность — до
300 т извести в сутки, удельная производительность — до
50 кг/(м3-ч).
В подавляющем большинстве случаев процессы переработки
пористых материалов в шахтных печах протекают во внутридиф-
фузионной области. Это связано с тем, что для обеспечения необ-
ходимой равномерности распределения газа по сечению шахты и
для снижения гидравлического сопротивления размер исходных
частиц твердого материала не должен быть меньше 10 мм. По-
этому в шахтных печах трудно использовать два важных фактора *
интенсификации — развитие поверхности контакта фаз и перевод
процессов в кинетическую область путем уменьшения размера
частиц твердой фазы.
Распылительные печи работают по принципу распыле-
ния тонкоизмельченного твердого материала в потоке газовой
фазы при прямоточном движении реагентов. В основном их ис-
пользуют для процессов обжига, протекающих с выделением теп-
лоты. По энергетическим признакам распылительные печи отно-
сят к реакционным печам прямого нагрева. На рис. 6.43 показана
печь для обжига колчедана. Газовый поток с распыленным колче-
130
даном (размер частиц менее 100 мкм) поднимается в верхнюю
зону печи, где смешивается с дополнительной частью воздуха. Об-
разовавшиеся газообразные продукты реакции выводятся сверху,
а огарок (твердый остаток) падает в нижнюю часть печи и уда-
ляется.
Режим движения фаз соответствует смешению. Температурный
режим — изотермический. Использование тонкодисперсной твердой
фазы обеспечивает уменьшение и даже снятие внутридиффузион-
ных торможений, что обусловливает высокую скорость процесса
на каждой твердой частице. Однако относительно низкая концен-
трация твердой фазы в потоке (доля объема, занятая твердой фа-
зой, не превышает 10%) не позволяет эффективно использовать
объем реакционной зоны. Интенсивность работы печей обжига
пылевидного колчедана составляет около 30—40 кг/(м3-ч). Велик
унос твердой фазы с потоком выходящего газа. Отсутствие тепло-
обменных элементов внутри печи ограничивает возможность ин-
тенсификации обжига путем повышения концентрации реагирую-
щих веществ (в частности, за счет обогащения воздуха техниче-
ским кислородом). Адиабатический разогрев при протекании ХТП
достигает 500—800 °C.
Пени с кипящим (взвешенным, псевдоожижен-
ным) слоем обладают наибЪлее высокой интенсивностью, так
как в них можно использовать все основные факторы интенсифи-
кации. Кипящий слой (КС) образуется при пропускании снизу
вверх через твердый зернистый материал газового потока с опре-
деленной скоростью. Минимальной линейной скорости, называемой
скоростью взвешивания шв, соответс~вует равенство силы трения
газа о поверхность частиц в плотном слое силе их тяжести. При
этой скорости частицы начинают хаотически двигаться, не поки-
дая в то же время слоя. Слой становится похожим на кипящую
жидкость и приобретает свойство текучести.
Кипящий слой обладает следующими основными свойствами,
которые предопределили широкое применение реакторов КС:
1) независимость гидравлического сопротивления от размера
частиц твердой фазы, линейной скорости и запыленности газового
потока;
2) исключительно высокая эффективная теплопроводность,
обеспечивающая изотермический температурный режим и возмож-
ность интенсивно отводить или пбдводить теплоту с помощью теп-
лообменных элементов, расположенных непосредственно в реак-
ционной зоне.
Благодаря этим свойствам протекание ХТП в реакторах с ки-
пящим слоем может быть максимально интенсифицировано за
счет предельного развития наружной поверхности контакта фаз
для Непористых частиц, полного использования внутренней по-
верхности для пористых частиц и создания наиболее благоприят-
ного температурного режима.
Возможность подвода или отвода теплоты непосредственно
из реакционной зоны обеспечивает поддержание рациональной
6*
131
высокой температуры по всей высоте слоя и, следовательно, высо-
ких значений, констант скоростей.
На рис. 6.44 показана печь КС для обжига колчедана. Это
реакционная печь прямого нагрева. Интенсивность печи примерно
в 10 раз выше, чем у механических полочных, и в 2 раза выше
по сравнению с печами для обжига пылевидного материала. Теп-
лота реакции горения достаточно полно используется для выра-
ботки пара, направляемого на турбогенераторы.
Режим движения твердой фазы соответствует смешению даже
при скоростях газа, близких к началу псевдоожижения. По газо-
вой фазе режим отвечает промежуточному положению между сме-
шением и вытеснением. При высоких скоростях газового потока,
близких к скорости уноса шу, режим соответствует смешению.
Температурный режим при интенсивном псевдоожижении —
всегда изотермический.
Основной недостаток печей КС — высокая запыленность газо-
вого потока (для печей обжига колчедана содержание пыли — до
200 г/м3). Печи КС широко используют для обжига сульфидных
руд цветных металлов, колчедана, извести, для сжигания и гази-
фикации твердого топлива, термической обработки фосфоритов и
антрацита, сжигания шламов и др.
Барабанные вращающиеся печи представляют собой
горизонтальный или наклоненный под небольшим углом (до 7°)
вращающийся цилиндр (барабан), внутри которого перемещается
обрабатываемый твердый материал. Частота вращения барабана
0,5—10 об/мин. Вращение барабана обеспечивает перемешивание
твердой фазы в радиальном направлении, что способствует обнов-
лению поверхности контакта фаз. Твердый материал и газовый
поток могут двигаться прямотоком или противотоком. Режим дви-
жения фаз по длине реактора соответствует вытеснению.
По энергетическому признаку барабанные печи — топливные,
в основном прямого нагрева. Обогреваются при непосредственном
контакте твердого материала с горячими топочными газами. Ино-
гда применяют барабанные печи с косвенным нагревом через
стенку реактора. Такие печи используют, например, при производ-
стве плавиковой кислоты, минеральных пигментов, кальциниро-
ванной соды и др.
По температурному режиму барабанные печи — политермиче-
ские. Как и печи КС, барабанные печи позволяют перерабатывать
мелкодисперсный материал при минимальных диффузионных тор-
можениях.
На рис. 6.45 представлена вращающаяся печь для производ-
ства цементного клинкера. Исходная шихта непрерывно загружа-
ется в верхнюю часть печи и при вращении барабана постепенно
перемещается к нижней части, где выгружается готовый продукт —
клинкер. Необходимая для спекания теплота обеспечивается сжи-
ганием топлива, которое подается в печь с нижнего конца. Таким
образом, газы и твердый материал движутся противоточно. При
этом твердая фаза проходит зоны сушки, нагрева, кальцинации
132
7'
Шихта
— - Топочные
—*“ Тазы.
Топливо
Воздухе"—. -
Продукт
Рис. 6.44. Печь КС для обжига колчедана:
I —кипящий слой; 2—сепарационная зона; 3—теп чообмеииые элементы; 4—газораспредели-
тельная решетка.
Рис. 6.45. Барабанная вращающаяся печь.
Рис. 6.46, Характер распределения температуры по длине вращающейся цемент-
ной печн.
(900—1200°С), спекания (до 1450°С) и охлаждения. Характер
распределения температуры по длине печи показан на рис. 6.46.
Производительность наиболее мощных печей 120—300 т в час.
Благодаря простоте устройства, легкости обслуживания, управ-
ления, устойчивости в работе и универсальности применения ба-
рабанные печи получили широкое распространение в производстве
силикатных материалов, глинозема, соды, щелочей, солей и мно-
гих других.
Камерные печи представляют собой камеры с хорошей
теплоизоляцией, с прямым или косвенным нагревом. Печи косвен-
ного нагрева иногда называют муфелями.
Камерные печи наиболее просты по устройству. Конструкции
их различаются по форме камеры, периодичности или непрерыв-
ности работы. Наибольшее распространение имеют печи полуне-
прерывного действия, в которых твердая фаза загружается и вы-
гружается периодически, а газ проходит через реакционную зону
в непрерывном режиме. По характеристикам источника теплоты
различают топливные и реакционные -печи. Реакционные печи ис-
пользуют для сжигания фосфора, серы, топливные часто объеди-
няют в батареи и применяют в крупнотоннажных производствах
при коксовании углей, обжиге керамики и др.
Режим движения газа в печах непрерывного и полупериодиче-
ского действия — вытеснение. В большинстве случаев температур-
ный режим близок к изотермическому.
Основной недостаток печей — низкая интенсивность и произво-
дительность, а также невысокая-степень использования остаточной
теплоты выходящих дымовых газов,
133
Для интенсификации реакционных камерных печей газовый
поток вводят с большой скоростью по тангенциальным каналам.
Такие печи называют циклонными. В них газовый поток враща-
ется вокруг продольной оси аппарата, что интенсифицирует сме-
шение, протекание диффузионных процессов, тем самым увеличи-
вается скорость ХТП. Циклонные печи применяют для сжигания
топлива, обесфторивания природных фосфатов и в ряде других
процессов.
Достоинство циклонных печей — высокая интенсивность, высо-
кая полнота сжигания, низкое аэродинамическое сопротивление.
Режим движения газа в циклонных печах больше соответствует
смешению.
- Туннельные печи выполнены в виде длинного канала
(туннеля), футерованного огнеупорным кирпичом. Внутри туннеля
движутся вагонетки с полками, на которых находится кусковой
материал. Газовый поток движется противотоком. По энергети-
ческим признакам туннельные печи являются топливными, пря-
мого нагрева. Температурный режим — политермический. По дли-
не печи имеется несколько температурных зон — подогрева, об-
жига, охлаждения. Режим движения фаз лучше всего описывается
моделью вытеснения с проскоком части непрореагировавшего
газа.
Туннельные печи легки в упразлении, работают в непрерывном
режиме, обладают высокой производительностью, просты и на-
дежны в эксплуатации. Их широко применяют для обжига огне-
упоров, керамических изделий, для полукоксования сланца, про-
изводства алюмосиликатных катализаторов и др.
Характерная схема туннельной печи для производства кера-
мических изделий представлена на рис. 6.47. Длина печи больше
160 м. В зону обжига подают газообразное топливо, которое обес-
печивает температуру до 1500 °C.
Ванные печи — это печи, в которых твердый материал пла-
вится и затем подвергается обработке газами в жидком состоя-
нии. Теплота передается материалу или конвективными потоками
сжигаемого газа, или путем лучеиспускания от раскаленного свода
и стен, нагреваемых газовыми горелками.
Разновидностью ванных печей являются электрические печи,
в которых высокая температура (до 3500 °C) создается в резуль-
тате преобразования электрической энергии в тепловую. По спо-
собу преобразования электрические печи разделяют на печи со-
противления, дуговые и индукционные.
J—горелки; 3—вагонетки о изделиями.
134
При электрическом нагреве исключается загрязнение обраба-
тываемого вещества продуктами горения топлива. В электриче-
ских печах получают карбид и цианамид кальция, карбиды крем-
ния и бора, электрокорунд, фосфор, сероуглерод, графит, стали,
цветные металлы и др.
6.9. АППАРАТУРНОЕ ОФОРМЛЕНИЕ
ГЕТЕРОГЕННО-КАТАЛИТИЧЕСКИХ ПРОЦЕССОВ
Реакторы, в которых осуществляются каталитические процес-
сы, называют контактными аппаратами. Устройство кон-
тактных аппаратов весьма разнообразно и зависит от способа
контакта между катализатором и реагирующими веществами (го-
могенный и гетерогенный катализ), характеристик потока взаимо-
действующих веществ (реакторы вытеснения и смешения), спо-
соба осуществления теплообмена (реакторы с теплообменом не-
посредственно в реакционной зоне и реакторы с выносными
теплообменниками), требуемого температурного режима в реак-
ционной зоне (реакторы с политермическим, адиабатическим или
изотермическим температурным режимом).
Большинство контактных аппаратов использую'7' для проведе-
ния процессов между газообразными реагентами на твердых ка-
тализаторах. Их можно разделить на три основные группы: кон-
тактные аппараты с фильтрующими (неподвижными) слоями ката-
лизатора, контактные аппараты с кипящими (псевдоожиженными)
слоями катализатора и контактные аппараты с движущимся ка-
тализатором.
6.9.1. Контактные аппараты
с фильтрующими слоями катализатора (ФС)
В контактных аппаратах с фильтрующим слоем газовый поток
проходит через слой неподвижного катализатора в режиме вытес-
нения. Различают шахтные, радиальные, полочные и трубчатые
контактные аппараты с фильтрующим слоем (ФС).
Контактный аппарат ФС шахтного типа является
наиболее простой и распространенной конструкцией. Принципи-
альные схемы таких аппаратов показаны на рис. 6.48. Газовый
поток поступает в реактор сверху или снизу и проходит через
неподвижный слой катализатора (рис. 6.48,а), расположенного
на решетке. Катализатор имеет форму шариков, таблеток, цилин-
дрических гранул или выполнен в виде сетки. Поскольку гидрав-
лическое .сопротивление аппарата возрастает с уменьшением раз-
мера частиц катализатора, то, как правило, используют крупные
гранулы катализатора с размером не менее 4—5 мм. Температура
газа на входе в реакционную зону должна обеспечить автотермич-
ное протекание процесса, а объем катализатора должен быть до-
статочен для достижения заданной степени превращения.
135
Рис. 6.48. Контактные аппараты шахтного типа:
а—с крупно зернистым катализатором; б—поверхностного контакта с сетками.
Рис. 6.49. Каскад реакторов фильтрующего слоя с промежуточными теплообмен-
никами:
1—3 — контактные аппараты; 4—6—теплообменники.
Если активность катализатора и скорость реакции высоки, то
используют катализатор в виде сеток, расположенных друг' над
другом в несколько слоев (рис. 6.48,6). Время контакта газа
с поверхностью сеток составляет тысячные доли секунды. Реакция
протекает практически на внешней поверхности катализатора. По-
этому такие реакторы называют контактными аппаратами
поверхностного'контакта. Их применяют для окисления
аммиака на платино-родиевых сетках, для окисления метанола
на медных или серебряных сетках и т. п.
Если катализатор малоактивный и скорость реакции низка, то
высота слоя катализатора может достигать нескольких метров.
Температурный режим в реакционной зоне — адиабатический.
Обычно шахтные контактные аппараты для таких процессов при-
меняют в тех случаях, когда тепловой эффект реакции невелик и
равновесная степень превращения, соответствующая температуре
газа на выходе из слоя катализатора, близка к заданной или для
реакций с низкой концентрацией реагентов, например при катали-
тической очистке газов окислением или гидрировании примесей.
Простота конструкции контактных аппаратов шахтного типа
не компенсирует невозможность обеспечения оптимального темпе-
ратурного режима при проведении экзотермических реакций с
большим тепловым эффектом. Для этих случаев используют кас-
кад шахтных контактных аппаратов с промежуточными внешними
теплообменниками (рис. 6.49), в которых осуществляют охлажде-
ние газа после каждого контактного аппарата. Вследствие этого
параметры процесса (концентрации, парциальные давления, сте
пени превращения) удаляются от равновесных значений и увели-
чивается движущая сила процесса на входе в каждый последую-
щий реактор. Хладагентом, как правило, служит исходный хо-
лодный газ, который, проходя'через теплообменники, нагревается
перед входом в 1-й реактор до необходимой температуры.
136
Рис. 6.51. Схема радиального контактного аппарата.
Рис. 6.50. Каскад реакторов фильтрующего слоя с промежуточными смесителями
потоков.
1—3—контактные аппараты; 4, 5—смесители холодных и горячих потоков.
Иногда температурный режим поддерживают, смешивая после
каждого реактора горячие и холодные потоки (рис. 6.50).
Радиальные контактные аппараты представляют
разновидность шахтных. В них газовый поток движется горизон-
тально через слой катализаюра, расположенного в кольцеобраз-
ной перфорированной катализаторной коробке (рис. 6.51). При
атом, как правило, газовый потод поступает в центре, а отводится
из периферийной зоны катализаторной коробки. Режим движения
газовой фазы — вытеснение. При проведении процессов с большим
тепловым эффектом реализуется адиабатический температурный
режим.
В полочных контактных аппаратах теплообменники
расположены между слоями катализатора. В контактных аппара-
тах большой мощности устраиваются выносные теплообменники,
а полки с катализатором, расположенные в одном корпусе, раз-
делены друг от друга горизонтальными перегородками.
Теплообмен осуществляется с помощью газовых, водяных труб-
чатых или змеевиковых теплообменников, а также введением
между стадиями контактирования одного из реагентов. Принципи-
альные схемы полочных контактных аппаратов для проведения
экзотермических гетерогенно-каталитических процессов с различ-
ным технологическим и конструктивным решением промежуточ-
ного отвода теплоты представлены на рис. 6.52.
В полочных контактных аппаратах с трубчатыми теплообмен-
никами теплообмен происходит по принципу противотока. Самый
холодный газ охлаждает реакционную смесь перед последней
полкой и, проходя последовательно через теплообменники, нагре-
вается до температуры «зажигания» катализатора. Охлажденные
продукты реакции поступают на разделение.
Если по тепловому балансу оказывается, что количество тепло-
ты, выделяющееся в результате реакции, существенно превышает
количество теплоты, необходимое для нагревания реакционной
137
В Реагенты
В первый теплообменник
Из первого теплообменника
Во второй теплооБменник
Из второго теплообменника
Рис. 6.52. Полочные контактные аппараты с встроенными (а) и выносными (б)
газовыми теплообменниками:
1 — слон катализатора; 2—решетки; 3—теплообменники; 4—перегородки.
Рис. 6.53. Контактный аппарат с вспрыскиванием воды между слоями катализа-
тора:
I—полки с катализатором; 2—форсунка; 3—насадка испарителя.
Рис. 6.54. Трубчатый контактный аппарат с катализатором в трубках и обогревом
топочными газами:
Г—огнеупорная кладка; 2—трубки с каталиаатором.
смеси, то отвод теплоты проводят с помощью водяных, паро-
вых теплообменников, входящих в состав установки по исполь-
зованию втощшных энергетических ресурсов. Иногда тепловым
агентом служит воздух, который после нагрева до высокой тем-
пературы можно подавать на газовую турбину.
В некоторых каталитических процессах промежуточное охлаж-
дение осуществляют между слоями катализатора дополнительным
введением исходной смеси, отличающейся по температуре от про-
реагировавшей смеси (см. рис. 6.50). Этот прием часто используют
для охлаждения газа после 1-й полки контактных аппаратов для
окисления оксида серы(IV). Иногда дополнительно вводят только
один из реагентов. Примером может служить каталитическая кон-
138
Рис. 6.55. Трубчатый контактный аппарат с катализатором в трубках и тепло-
обменом между реагентами и продуктами:
1—корпус: 2—трубка с катализатором; 3—трубная решетка.
Рис. 6.56. Фактическое (1) и оптимальное (2) распределение температур по
длине катализаторной трубки.
версия оксида углерода(П) с водяным паром: СО + Н2О = СО2+
+ Н2 + q. Для увеличения выхода водорода и более полного ис-
пользования оксида углерода(II) и одновременно для снижения
температуры газ после 1-й полки с катализатором охлаждают и
насыщают водяным паром, получаемым при испарении воды в ис-
парителе (рис. 6.53). Вода подается в форсунки и испаряется на
полке с инертной насадкой.
В полочных контактных аппаратах каталитическую реакцию и
теплообменные процессы проводят раздельно, последовательно.
Режим движения газа в полочных аппаратах, как и в шахтных,
соответствует вытеснению. Температурный режим при протекании
ХТП с высоким тепловым эффектом, как правило, адиабати-
ческий.
В трубчатых контактных аппаратах теплообмен осу-
ществляется непосредственно в реакционной зоне. В простейшем
случае такие аппараты устроены по принципу трубчатых тепло-
обменников. При этом в трубках находится катализатор, а в меж-
трубном пространстве — теплоноситель (рис. 6.54). Реагирующая
смесь проходит внутри катализаторных трубок навстречу теплоно-
сителю. В качестве примера можно привести контактные аппараты
для проведения эндотермического процесса дегидрирования бу-
тана, каталитической конверсии метана и др.
В трубчатых контактных аппаратах для проведения экзотер-
мических ХТП трубки охлаждаются холодным исходным газом,
который после нагрева поступает сверху в катализаторные трубки
(рис. 6.55). Продукты реакции отводят снизу.
Для создания оптимального профиля температуры по длине
трубок необходим наиболее интенсивный отвод теплоты в верхней
части и снижение интенсивности теплообмена по мере уменьшения
скорости процесса, т. е. в нижней части трубок интенсивность теп-
лообмена должна быть меньше, чем в верхней. В рассмотренных
139
Рис. 6.57. Диаграмма X— Т для экзотермической реакции в трубчатом контакт-
ном аппарате:
1—равновесная кривая; 2—линия оптимальных температур; 3—фактический профиль темпе-
ратуры.
Рис. 6.58. Элемент аппарата с двойными теплообменными трубками:
1—катализатор; 2—внутренняя трубка; 3—решетка; 4—внешняя трубка; 5—кольцевое про-
странство.
Реагенты
конструкциях это обеспечить невозможно, так как наименьшая
движущая сила процесса теплопередачи создается в верхней части
реактора, где достигается максимальная температура теплоноси-
теля при минимальной температуре слоя катализатора. Пример-
ное распределение температуры по длине катализаторной трубки
в таком аппарате показано на рис. 6.56. Характерный вид диа-
граммы X—Т для рассматриваемых контактных аппаратов, в ко-
торых протекает экзотермический ХТП, представлен на рис. 6.57.
Существенное отклонение фактического температурного режима
от оптимального отрицательно сказывается на интенсивности ра-
боты контактных аппаратов.
Лучшее приближение к оптимальному температурному про-
филю достигается в контактных аппаратах с двойными теплооб-
менными трубками (рис. 6.58). В такой конструкции обеспечива-
ется большее соответствие между скоростью реакции (она мак-
симальна в верхней зоне слоя катализатора) и интенсивностью
теплообмена. Охлаждение газа, движущегося в кольцевом зазоре,
более холодным исходным газом, поступающим в центральную
трубку, увеличивает движущую силу процесса теплопередачи в
верхней зоне.
Недостатки трубчатых контактных аппаратов связаны с нерав-
номерностью температур по горизонтальному сечению слоев ка-
тализатора и малой интенсивностью отвода теплоты. Все контакт-
ные аппараты с фильтрующими слоями катализатора обладают
следующими недостатками, затрудняющими интенсификацию ХТП,
особенно при работе с концентрированными смесями при высоких
тепловых эффектах реакций:
1. Сложность использования мелкозернистых катализаторов,
позволяющих проводить процессы в кинетической области. Это
140
связано с высоким гидравлическим сопротивлением слоев с такими
катализаторами. Степень использования внутренней поверхности
крупнозернистых катализаторов при размере частиц 4—8 мм, как
правило, не превышает 60—70 %.
2. Низкая пылепропускная способность фильтрующих слоев,
приводящая к необходимости тонкой очистки газа от механиче-
ских частиц. Вследствие засорения пылью, вносимой газовым по-
током или образующейся в результате разрушения гранул катали-
затора, нарушается равномерность распределения газа по сече-
нию контактного аппарата, повышается его гидравлическое сопро-
тивление в процессе эксплуатации.
3. Низкая теплопроводность катализатора, не обеспечиваю-
щая интенсивного отвода (или подвода) теплоты из реакционной
зоны и равномерности распределения температуры по диаметру
и высоте слоя катализатора.
4. Сложность непрерывного отвода катализатора для регенера-
ции, которая необходима для многих процессов в Технологии ор-
ганических веществ.
Из-за этих недостатков контактные аппараты с фильтрующими
слоями для многих процессов постепенно заменяются контактными
аппаратами с кипящими слоями катализатора.
6.9.2. Контактные аппараты
с кипящими слоями катализатора (КС)
Контактные аппараты с кипящими слоями катализатора (КС)
находят все более широкое применение. Они обеспечивают проте-
кание каталитических процессов при изотермическом температур-
ном режиме даже при высоких тепловых эффектах реакции. Не-
зависимость гидравлического сопротивления кипящих слоев от
размера частиц и линейной скорости газа дает возможность при-
менения мелкозернистых катализаторов. Это позволяет эффек-
тивно проводить процессы в кинётической .области при полном
использовании внутренней поверхности катализаторов. рТысокая
теплопроводность кипящего слоя^ обусловленная подвижностью ча-
стиц, создает благоприятные условия для отвода или подвода теп-
лоты непосредственно в слое катализатора, без опасения вызвать
локальные затухания или перегрев контактной массы. При этом
вследствие высоких значений коэффициентов теплопередачи от
кипящего слоя, к тепловому агенту обеспечив ается наиболее эф-
фективный теплообмен и соответственно уменьшаются размеры
теплообменных узлоц?
^^Через кипящий слой свободно проходят все мелкие частицы,
находящиеся в газовом потоке, а пыль, образующаяся при разру-
шении гранул катализатора, выносится потоком. Практически
неограниченная пылепропускная способность кипящих слоев
обеспечивает возможность переработки запыленных газовых сме-
сей, снижает требования к работе пылеулавливающей аппа-
ратуры.
141
Рис. 6.59. Многополочный контактный аппарат с кипящими слоями катализатора:
1—распределительные решетки; 2—слои катализатора; 3—теплообменные элементы; 4— газо-
распределительный конус.
Рис. 6.60. Характер изменения температуры и константы скорости по высоте кон-
тактных аппаратов с фильтрующим (/) и кипящим (2) слоями.
^Свойство текучести кипящего слоя позволяет осуществлять не-
прерывный отвод катализатора для регенерации] Именно это свой-
ство обусловило первое промышленное применение контактных
аппаратов КС при каталитическом крекинге нефтепродуктов.
Контактные аппараты с кипящими слоями по режиму движе-
ния и степени перемешивания твердой фазы относятся к реакто-
рам смешения. Режим движения газовой фазы больше соответ-
ствует модели вытеснения с проскоком части газа.
Принципиальная схема полочного контактного аппарата с ки-
пящими слоями .катализатора для экзотермических процессов
представлена на рис. 6.59. Газовая смесь проходит последова-
тельно снизу вверх через несколько слоев катализатора 2, находя-
щихся в режиме псевдоожижения. Отвод теплоты проводят с по-
мощью теплообменных элементов 3, расположенных непосред-
ственно в кипящих слоях. Конструкция теплообменных элементов
проста, а общая поверхность теплообмена меньше, чем в аппа-
ратах с фильтрующими слоями, благодаря более высоким коэф-
фициентам теплоотдачи от кипящего слоя к поверхности эле-
ментов.
Преимущества контактных аппаратов КС по сравнению с реак-
торами фильтрующего слоя наиболее сильно проявляются при про-
ведении ХТП с большим тепловым эффектом и особенно, если кон-
центрация реагирующих элементов в газовой смеси велика. Это
преимущество обусловлено возможностью осуществления необхо-
димого теплообмена непосредственно в слоях катализатора. Для
контактных аппаратов КС, работающих без теплообменных эле-
ментов, вследствие экзотермичности температурного режима тем-
пература 7\с, константы скорости экзотермической реакции /гкс
сущестзенно выше средних значений температуры 7фС, константы
142
Рис. 6.61. Влияние температуры потока на
входе в контактные аппараты и температур-
ного режима на степень превращения:
/—фильтрующий слой; 2—кипящий слой; 3— равно-
весная кривая.
скорости йфС процесса, осуществляемого в адиабатическом филь-
трующем слое (рис. 6.60).
При проведении экзотермических процессов газовую смесь
можно подавать в контактный аппарат КС при меньших темпера-
турах, чем для контактных аппаратов с фильтрующим слоем. Это
позволяет повысить максимальную степень превращения по срав-
нению с реакторами ФС (рис., 6.61).
Основной недостаток контактных аппаратов с кипящими слоя-
ми катализатора- связан с проскоком части непрореагировавшего
газа, проходящей через слой катализатора в виде пузырей. При-
менение специальных насадок, помещаемых в кипящий слой,
уменьшает это вредное явление. Другой недостаток аппаратов
КС — необходимость использования износоустойчивых катализато-
ров, так как обычные контактные массы быстро истираются и уно-
сятся с газовым потоком.
6.9.3. контактные аппараты
с движущимся слоем катализатора
Контактные аппараты с движущимся слоем катализатора ис-
пользуют в тех случаях, когда требуется непрерывная циркуляция
катализатора между реактором и регенератором, например в уста-
новках каталитического крекинга. Контактная масса в таких аппа-
ратах может двигаться в двух режимах; а) в виде взвеси мелко-
зернистого катализатора в восходящем потоке газа (рис. 6.62);
б) плотным слоем крупнозернистого катализатора, опускающегося
под действием силы тяжести в аппарате шахтного типа при прямо-
точном или противоточном движении газа (рис. 6.63).
В контактном аппарате с потоком взвеси катализатора (см.
рис. 6.62) скорость газовой смеси превышает скорость уноса час-
тиц катализатора. В результате измельченный катализатор выно-
сится потоком из верхней части контактного аппарата 2, отделя-
ется в сепараторе 3 от продуктов реакции и поступает в регене-
ратор 5. В регенераторе в режиме кипящего слоя осуществляется
регенерация катализатора, заключающаяся в выжигании возду-
хом углеродных веществ из пор. Регенерированный катализатор
перетекает через стояк *, смешивается с парами сырья и эжекто-
ром подается в контактный аппарат. Пары через реактор прохо-
дят в режиме вытеснения.
143
Рис. 6.62. Реакторы каталитического крекинга с потоком взвеси пылевидного ка-
тализатора:
/ — инжектор; 2—контактный аппарат с движущимся катализатором; 3—сепаратор: 4—отпар-
ная емкость; 5—регенератор с кипящим слоем; 6— стояк.
Рис. 6.63. Контактный узел с плотным движущимся слоем катализатора:
/—контактный аппарат; 2—регенератор.
-
В аппарате с плотным движущимся слоем (см. рис. 6.63) ка-
тализатор медленно опускается при противоточном движении с
исходными парами и продуктами реакции. После отвода продук-
тов крекинга на разделение катализатор проходит через регенера- ,
тор 2 и далее в контактный аппарат 1.
Режим движения газа и катализатора соответствует вытесне- ,
нию. Температурный режим близок-к адиабатическому.
'1
6.10. АНАЛИЗ РАБОТЫ МНОГОСТУПЕНЧАТЫХ
КАТАЛИТИЧЕСКИХ РЕАКТОРОВ
В настоящее время наиболее распространены полочные кон-
тактные аппараты с фильтрующими или кипящими слоями ката-
лизатора. Аппараты с фильтрующим слоем работают в режиме
вытеснения. В них практически отсутствует продольное перемеши-
вание, уменьшающее движущую силу процесса. Однако адиаба-
тический температурный режим, реализуемый в таких аппаратах,
обусловливает необходимость большого числа ступеней контакти-
рования для достижения высоких степеней превращения при про-
ведении процессов с большим тепловым эффектом.
Аппараты с кипящими слоями катализатора работают при
изотермическом температурном режиме и обеспечивают большее
приближение температур к оптимальным.
144
При реализации режима смешения, что наблюдается при ин-
тенсивном псевдоожижении слоев катализатора малой высоты,
возможно проведение ХТП при строго оптимальной температуре.
Это связано с тем, что по всей высоте кипящего слоя устанавли-
вается одинаковая концентрация продуктов, степень превращения
и соответствующая им одна оптимальная температура.
6.10.1. Контактные аппараты ФС
с промежуточным теплообменом
К основным параметрам технологического режима работы кон-
тактных аппаратов относят: температуру Т, константу скорости
процесса k, концентрации реагирующих веществ и продуктов
реакции С, степень Превоащения X, движущую силу ДС и ско-
рость U процесса. Рассмотрим изменение этих параметров по вы-
соте полочного контактного аппарата с фильтрующими слоями
катализатора и теплообменниками, встроенными между полками
(см. рис. 6.52,а).
Сделаем упрощение, согласно которому контактный аппарат
должен обеспечить достижение заданной степени превращения
при минимальном числе полок. Это означает, что температура ре-
акционной смеси на входе в каждую полку Твх должна быть
близка температуре зажигания катализатора Тваж, а степень пре-
вращения, достигаемая на каждой полке, должна быть близка
равновесной.
Для экзотермического процесса
А4- В D + <?
диаграмма X—Т, соответствующая указанному ограничению,
представлена на рис. 6.64.
Характер изменения основных параметров технологического
режима по высоте контактного аппарата при движении реагирую-
щей смеси снизу вверх показан на рис. 6.65. По мере прохожде-
ния газовой смеси через слой катализатора температура ее повы-
шается вследствие выделения теплоты (рис. 6.65, с). Наиболее
интенсивный рост наблюдается на начальных участках каждого
слоя и замедляется по мере приближения степени превращения
к равновесной. Важно отметить, что конечная температура смеси
на выходе из каждого последующего слоя ниже выходной темпе-
ратуры каждого предыдущего слоя. Это следует из характера из-
менения равновесной кривей 1 на диаграмме X — Т (см. рис. 6.64).
Аналогично средняя температура каждого последующего слоя
должна быть ниже средней температуры каждого предыдущего
слоя. Характер изменения средней температуры должен соответ-
ствовать линии оптимальных температур (кривая 2 на рис 6.64).
С ростом температуры увеличивается и константа скорости
процесса k (см. рис. 6.65,6). Количественное изменение k описы-
вается уравнением Аррениуса.
145
Рис. 6.64. Диаграмма X — Т для контактного аппарата с фильтрующими слоями
катализатора (экзотермический обратимый процесс):
1—равновесная кривая; 2—линия оптимальных температур; 3—линия адиабатического разо-
грева; 4—линия охлаждения реакционной смеси в промежуточных теплообменниках.
Рис. 6.65. Характер изменения параметров технологического режима по высоте
контактного аппарата с фильтрующими слоями (экзотермический обратимый про-
цесс) :
J—слои катализатора; 2—теплообменники.
На рис. 6.65, в представлен характер изменения по высоте ре-
актора концентрации СА и степени превращения ХА исходного ве-
щества А, а на рис. 6.65, г — текущей CD и равновесной СЬ кон-
центрации продукта D. При этом следует понимать, что измене-
ния значения СЬ определяются лишь изменениями температуры
по высоте реактора. Максимальные значения СЬ соответствуют
областям реактора, имеющим минимальную температуру (т. е. на-
чальным участкам каждого слоя). В соответствии с принципом
Ле Шателье минимальная равновесная концентрация продукта
будет отвечать участкам с максимальной температурой. Это соот-
ветствует участку выхода газовой смеси из 1-го слоя.
Как указано выше, движущая сила процесса АС в общем слу-
чае является сложной функцией текущих и равновесных концен-
траций и характеризует степень приближения процесса к равно-
весию или полному превращению одного из веществ. Можно услов-
но принять, что АС пропорциональна разности между равновесной
и текущей концентрацией продукта D. Характер изменения дви-
жущей силы процесса по высоте реактора показан на рис. 6.65, д.
В начале каждого слоя движущая сила максимальна и умень-
шается до нуля в конце каждого слоя при достижении равнове-
сия. Таким образом, роль теплообменников, отводящих избыточ-
ную теплоту, сводится к повышению движущей силы процесса.
Скорость процесса при неизменной удельной поверхности кон-
такта газа и катализатора пропорциональна произведению кон-
станты скорости и движущей силы. Так как по высоте слоя k
нелинейно увеличивается, а АС — уменьшается, то изменение ско-
рости процесса имеет экстремальный характер (см. рис. 6,65, е).
На каждой полке максимальные значения Смакс наблюдаются в
146
Рис. 6.66. Диаграмма X — Т для контактного аппарата с фильтрующими слоями
катализатора (эндотермический обратимый процесс):
1—равновесная кривая; 2—линия адиабатического разогрева; 3—линия нагрева смеси в тепло-
обменниках.
Рис. 6.67. Характер изменения параметров технологического режима по высоте
контактного аппарата с фильтрующими слоями (эндотермический обратимый про-
цесс) :
1—слои катализатора; 2—теплообменники.
центральных областях слоев катализатора. В начале каждого слоя
скорость низка из-за малого значения k, а в конце каждого слоя —
из-за приближения к равновесию, когда ДС->0.
Для эндотермического каталитического процесса
D < В + F — q
диаграмма X — Т при указанных выше ограничениях представлена
на рис. 6.66, а характер изменения основных параметров техноло-
гического режима по высоте реактора — на рис. 6.67.
Следует отметить, что для эндотермических обратимых реак-
ций, протекающих в адиабатическом реакторе вытеснения, и кон-
станта скорости k и движущая сила АС уменьшаются по высоте
слоя катализатора. Следовательно, в отличие от экзотермических
процессов, скорость U нелинейно уменьшается по высоте каждого
слоя катализатора. Средние же значения скорости, как и для эк-
зотермических процессов, снижаются по мере прохождения каж-
дого последующего слоя.
6.10.2. Контактные аппараты КС
с промежуточным теплообменом
Реакторы КС с промежуточным теплообменом для проведения
экзотермических процессов применяют в основном в тех случаях,
когда теплоту реакции используют лишь для нагрева исходной
газовой смеси. Схема рассматриваемых контактных аппаратов
сильно не отличается от представленных на рис. 6.52. Перемеши-
вание газовой смеси при прохождении через псевдоожиженный
слой катализатора более существенно, чем при движении газа в
режиме фильтрации. Тем не менее режим движения газа через
кипящий слой достаточно большой высоты (например, более 1 м),
147
Рис. 6.68. Диаграмма X—Т для контактного аппарата с кипящими слоями (экзо-
термический процесс):
а—режим вытеснения (высокие слои); б—режим смешения (низкие слои); 1 — линия изменения
степени превращения при изотермическом температурном режиме; 2—линии охлаждения смеси
в теплообменниках и нагрева на входе в слой; 8—линия оптимальных температур; 4—равно-
весная кривая; 5—протекание процесса прн изотермическом температурном режиме в усло-
виях полного смешения.
Рис. 6.69. Характер изменения движущей силы АС (д) и скорости U (б) экзотер-
мического процесса по высоте реактора КС (режим движения газа — смешение):
1 — слои катализатора; 2— теплообменники.
как показывают многочисленные эксперименты, больше соответ-
ствует вытеснению. В низких слоях, высота которых не превышает
0,1 м, турбулизация потока в прирешеточной зоне обеспечивает
режим, близкий к идеальному смешению.
Характерные диаграммы X— Т протекания экзотермического
процесса в 3-полочных контактных аппаратах с высокими и низ-
кими кипящими слоями представлены на рис. 6.68. При движении
газа в режиме вытеснения через изотермический слой катализа
тора невозможно осуществление процесса при оптимальной тем-
пературе для всего слоя, так как степень превращения по высоте
реакционной зоны изменяется. Обычно температура равна опти- ’
мальной, соответствующей конечной степени превращения на вы-
ходе из каждого кипящего слоя (рис. 6.68,а).
Для низких слоев с интенсивным псевдоожижением процесс
можно вести строго при оптимальной температуре, соответствую-
щей достигаемой в каждом слое степени превращения
(рис. 6.68,6). Это становится возможным благодаря постоянству
по высоте реакционной зоны и температуры и степени превраще-
ния или концентрации реагентов. Протекание процесса изобража-
ется в координатах X — Т точками 5, лежащими на линии опти-
мальных температур.
Характер изменения основных параметров процесса по высоте
реактора при движении газа в режиме смешения представлен
на рис. 6.69. Вследствие постоянства фактических и равновесных
концентраций движущая сила и скорость процесса тоже посто- j
янны по высоте реакционной зоны и несколько уменьшаются по
мере прохождения каждого последующего кипящего слоя.
6.10.3. Контактные аппараты КС с внутренним теплообменом
При большом избытке теплоты становится целесообразным
размещать теплообменные элементы непосредственно в кипящем
слое катализатора. При этом достигается совмещение технологи- ’
148
Рис. 6.70. Диаграмма X — Т протекания экзотермического процесса в реакторе КС
с внутренним теплообменом:
1—линии изменения X при изотермическом режиме; 2—линии мгновенного изменения темпе-
ратуры смеси иа входе в кипящий слой; S—линия оптимальных температур; 4—равновесная
кривая.
Рис. 6.71. Характер изменения показателей технологического режима по высоте
реактора КС с внутренним теплообменом:
1 — слои катвлизатора; 2—теплообменники.
ческих и энергетических функций при максимальной компактности
контактного узла. Схематично такой контактный аппарат пред-
ставлен на рис. 6.59. В большинстве случаев высота кипящего
слоя существенно превышает высоту прирешеточной зоны и, сле-
довательно, режим движения газовой смеси больше соответствует
вытеснению. Пэ высоте кипящего слоя наблюдается градиент кон-
центраций реагирующих веществ.
Диаграмма X — Т протекания экзотермического процесса в та-
ком реакторе представлена на рис. 6.70.
Температура смеси на входе в слой катализатора Твх не зави-
сит от температуры кипящего слоя. Она может быть и ниже
температуры зажигания катализатора Тзаж и выше максимально до-
пустимой температуры Тмакс, при которой эксплуатация катализа-
тора невозможна. Изменяя количество отводимой из слоя тепло-
ты, можно легко поддерживать заданную температуру. Например,
если ТвХ < Тр, то часть выделившейся теплоты будет расходо-
ваться на нагрев входящей газовой смеси до рабочей температуры
кипящего слоя Тр и нагрузка на теплообменники уменьшается.
Если Твх> Тр или Твх > Т'макс, то теплообменники должны не
только обеспечить отвод теплоты реакции, но и охлаждение газо-
вой смеси до температуры кипящего слоя. Теплообменники в этом
случае будут иметь большую поверхность.
Отсутствие промежуточного теплообмена предопределяет ха-
рактерный вид линии охлаждения на диаграмме X— Tt обуслов-
ленный тем, что смесь поступает на каждую полку с температу-
рой, более высокой, чем температура кипящего слоя.
Характер изменения показателей процесса для рассматривае-
мого типа аппаратов показан на рис. 6.71. Как видно на этом
рисунке, соотношения параметров для различных полок контакт-
ного аппарата сохраняются независимо от изменения степени
149
превращения по мере прохождения газовой смеси через слои ката-
лизатора. Эти соотношения определяются характером зависимости
оптимальной температуры от степени превращения (т. е. линией
оптимальных температур на рис. 6.70) и не зависят от количества
выделившейся теплоты. В то же время количество теплоты, отво-
димое теплообменными элементами из реакционной зоны, будет
зависеть от соотношения степеней превращения реагента, дости-
гаемых в каждом кипящем слое катализатора.
6.11. ТЕМПЕРАТУРНАЯ УСТОЙЧИВОСТЬ РЕАКТОРОВ
Стационарное протекание ХТП соответствует условиям, при которых все тех-
нологические показатели в любой точке реакционной зоны не изменяются во вре-
мени. При проведении экзотермического процесса в адиабитическом реакторе тем-
пература и константа скорости по мере увеличения степени превращения растут
из-за тепловыделения, а движущая сила уменьшается в результате расходования
реагентов и снижения их концентрации. Вследствие этого для простой необрати-
мой реакции зависимость скорости процесса от температуры выражается кривой,
имеющей S-образкую форму (см. разд. 4.3.1). Поскольку скорость выделения теп-
лоты Qp пропорциональна скорости процесса, то зависимость Qp от температуры
также описывается S-образной кривой.
В рассматриваемом адиабатическом реакторе выделяющаяся теплота расхо-
дуется только на нагрев реагирующей смеси. QP зависит от константы скорости
и теплового эффекта реакции, концентрации Сд и скорости подачи реагентов VCM.
Эти параметры определяют положение кривой в координатах Qp—Т.
Для адиабатических условий скорость отвода теплоты из реактора Qotb равна
скорости отвода теплоты с потоком жидкости или газа, выходящих из реактора,
т. е.:
Qotb = (7р 7ВХ) (6.45)
Здесь р, с — плотндсть и удельная теплоемкость смеси.
Для простоты можно принять, что Кем, рис одинаковы как на входе, так и
на выходе. Уравнение (6.45) выражает линейную зависимость Qотв от темпера-
туры, которая графически в координатах Q— Т может быть представлена пря-
мой линией (рис. 6.72). Угол наклона ее зависит от Кмрс, а ось Т пересекается
при Т = Тех. В зависимости от Км, р, с и ТВх изменяются характеристики тепло-
отвода и положение прямой линии в координатах Q — Т.
При стационарном состоянии (т. е. при не изменяющихся во времени показа-
телях технологического процесса)
Qp = Qotb (6.46)
При совмещении кривая тепловыделения и прямая теплоотвода могут пере-
секаться в одной или нескольких точках (рис. 6.73). Точки пересечения a, b, с, d
соответствуют уравнению (6.46), т. е. условию стационарного протекания про-
цесса. Но эти стационарные состояния могут быть устойчивыми и неустойчивыми.
Если процесс протекает устойчиво, то при отклонении его от данного состояния
(возмущении) он самопроизвольно возвращается в начальное состояние. При не-
устойчивом стационарном состоянии любое возмущение приводит к переходу
процесса в другое устойчивое состояние. При работе аппаратов неизбежны слу-
чайные колебания параметров технологического режима, в том числе темпера-
туры.
Рассмотрим сначала стационарное состояние, соответствующее температуре
Г| (точка а'). Если по каким-либо причинам произойдет повышение температуры
на ДГ, то увеличится и скорость тепловыделения (из-за увеличения скорости ре-
акции) и скорость теплоотвода (из-за роста разности между 7Р и 7ВХ). Но ско-
рость теплоотвода, как видно из графика, будет выше скорости тепловыделения;
после снятия возмущения реактор начнет охлаждаться и процесс возвратится' в
150
Рис. 6.72. Зависимость скорости отвода теплоты от температуры для различных
условий протекания процесса в реакторе смешения:
1, 3—при VCM 2—при ГСМ1 2 (VCM ,> VCM> 2).
Рис. 6.73. Температурные зависимости скорости теплоотвода (/—3) и тепловыде-
ления (4) при проведении необратимых экзотермических процессов.
Рис. 6.74. Температурные зависимости скорости тепловыделения (7) и теплоот-
вода (2 и 3) при проведении обратимых экзотермических процессов.
первоначальное стационарное состояние при Г]. При случайном снижении темпе-
ратуры на Д7 скорость тепловыделения окажется выше скорости теплоотвода, и
температура будет повышаться до тех пор, пока ие достигнет Т\. Таким образом,
это состояние является устойчивым. Однако интенсивность работы реактора будет
низка, так как при низких температурах скорость процесса мала.
Стационарное состояние при температуре Гз, отвечающее точке Ь, является
неустойчивым, поскольку при выведении процесса из состояния равновесия откло-
нения будут расти и процесс перейдет в другое стационарное состояние, соответ-
ствующее точке с при увеличении температуры.
Стационарные состояния при температурах Г3 и Г4 являются устойчивыми.
Прямая теплоотвода идет круче кривой тепловыделения. Поэтому при температуре
Тз + ДТ (или Г4 + ДТ) скорость отвода теплоты окажется выше скорости ее вы-
деления. Значит после снятия возмущения (случайного или искусственного) реак-
тор охлаждается до перехода к стационарному состоянию, соответствующему тем-
пературе Гз (или Г4).
Режим процесса, отвечающий стационарному состоянию в области высоких
температур, является наиболее благоприятным, так как скорость процесса высока.
На практике возможна такая ситуация, когда работа при высокой темпера-
туре невыгодна из-за низкой избирательности и рациональная температура дол-
жна быть близка к Т2. Для обеспечения устойчивости работы при этой темпера-
туре необходимо увеличить интенсивность теплоотвода (например путем увеличе-
ния скорости подачи реагентов или введением в реакционную зону теплообменных
элементов) и изменить температуру входа.
При достаточно больших их значениях прямая теплоотвода 2' будет круче
прямой 2 и можно обеспечить устойчивое протекание процесса при высокой изби-
рательности (см. рис. 6.73).
Общее условие обеспечения устойчивости состоит в том, что прямая тепло-
отвода должна идти круче кривой тепловыделения в точке их пересечения. Фор-
мула, описывающая это условие, может быть представлена в виде;
^отв
dT
dQf
~dT
(6.47)
При проведении обратимого экзотермического процесса
деления от температуры имеет более сложный вид (рис.
зависимость тепловы-
6.74). Стационарные
151
состояния при температурах Т>, Та и Т< (точки а, с, d) соответствуют устойчи-
вым режимам, а при температуре Та (точка Ь) — неустойчивому.
Анализ температурной устойчивости работы реакторов является ответствен-
ным этапом при разработке технологических схем, выборе оборудования и опреде-
лении рациональных условий проведения ХТП.
ГЛАВА 7
ХИМИКО-ТЕХНОЛОГИЧЕСКИЕ СИСТЕМЫ {ХТС)
7.1. ОБЩАЯ ХАРАКТЕРИСТИКА
ХИМИКО-ТЕХНОЛОГИЧЕСКИХ СИСТЕМ
Химико-технологический процесс осуществляют с целью преоб-
разования сырья в товарные продукты. Систему аппаратов с раз-
личным функциональным назначением, взаимосвязанных мате-
риальными и энергетическими потоками и действующих как
единое целое с целью выпуска товарной продукции заданного каче-
ства, называют химико-технологической системой
(ХТС). Число аппаратов, последовательность их участия в произ-
водственном процессе, направления материальных и тепловых
потоков между аппаратами характеризуют структуру ХТС. Струк-
туру ХТС образуют входящие в ее состав элементы и связи между
ними. Элементами химико-технологической системы являются
отдельные аппараты. Связи между элементами выступают в виде
трубопроводов, по которым передаются материальные и тепловые
потоки.
Каждый элемент ХТС выполняет определенные функции по
преобразованию параметров входящих в него потоков в пара-
метры выходящих потоков. Следовательно, каждый элемент ХТС
можно рассматривать в этом смысле как преобразователь (или
технологический оператор), качественно и (или) количественно
преобразующий параметры входных материальных и энергетиче-
ских потоков Уь Уг, ..., Уя в физические параметры выходных
материальных и энергетических потоков Z\, Z2, ..., Zn (рис. 7.1).
Так, реактор конверсии оксида углерода представляет собой тех-
нологический оператор, осуществляющий как качественное [в со-
став выходного технологического потока входят новые химические
компоненты: оксид углерода(IV) и водород], так и количествен-
ное (увеличение температуры и изменение количественного состава
выходного потока) преобразование физических параметров вход-
ного технологического потока, включающего оксид углерода (II)
и водяной пар.
Типовые технологические операторы — это элементы химиче-
ского превращения, теплообмена, массообмена, смесители и дели-
тели потоков, устройства для повышения и понижения давления,
изменения агрегатного состояния вещества. Условные изображе-
ния этих элементов приведены на рис. 7.2.
152
a i S t в г
Рис. 7.1. Элемент ХТС как технологический оператор.
Рис. 7.2. Условные обозначения типовых технологических операторов:
а — элемент химического превращения; б—элемент массообмзна; в —смеситель потоков;
г — распределитель потоков; д—элемент теплообмена; е — элемент сжатия или расширения;
ж—элемент изменения фазового состояния вещества.
Математически в общем виде технологический оператор можно
представить следующим образом:
Y = F (X, U) (7.1)
Здесь У — обобщенная характеристика параметров выходных технологиче-
ских потоков; X— обобщенная характеристика параметров входных технологиче-
ских потоков; U — обобщенная характеристика конструкционных и технологиче-
ских параметров аппаратов (объем реактора, поверхность теплообмена в тепло-
обменнике, флегмовое число в ректификационной колонне и т. д.).
Параметрами состояния технологических по-
токов являются расход, температура, концентраций компонентов,
давление и другие характеристики. Величину, равную числу пара-
метров технологического потока' называют пара м.е т р и ч н о -
стью потока.
Конструкционные и технологические параметры элементов ХТС
позволяют управлять процессом. Поэтому их называют также
управляющими параметрами.
Связи между аппаратами внутри ХТС называют внутрен-
ними связями, связи между аппаратами различных ХТС и
внешней средой — внешними связями.
От характеристик элементов ХТС и характера технологических
связей зависит качество функционирования химико-технологиче-
ской системы. Эффективность работы ХТС можно повысить: 1) пу-
тем изменения технологических связей между существующими
в системе технологическими операторами; 2) путем улучшения
функционирования основных элементов ХТС (за счет изменения
технологических параметров их работы или изменения типа аппа-
ратов); 3) введением в ХТС дополнительных операторов или об-
разованием новых внешних связей.
7.2. СПОСОБЫ ОТОБРАЖЕНИЯ СТРУКТУРЫ
ХИМИКО-ТЕХНОЛОГИЧЕСКИХ СИСТЕМ
Структуру ХТС, т. el все элементы и способ их соединения
между собой, чаще всего изображают с помощью структурных,
технологических или операторных схем.
153
Рис. 7.3. Структурная (а) и технологическая (б) схемы ХТС синтеза аммиака
под средним давлением:
1—колонна синтеза; 2 —паровой котел; 3~ холодильник; 4 — сепаратор; 5—турбо циркуляцион-
ный компрессор; 6—конденсационная колонна; 7 — испаритель.
Рис. 7.4. Операторная схема узла дистилляции I ступени ХТС производства кар-
бамида:
/—ректификационная колонна! подогреватель; 3—сепаратор.
На структурной схем.е все элементы ХТС графически
представлены в виде блоков, имеющих несколько входов и выхо-
дов материальных и тепловых потоков. Технологические связи
между блоками обозначены линиями со стрелками, указывающими
направление движения потоков. Структурная схема дает самое
общее представление о ХТС и практически не содержит количе-
ственной информации ни о потоках, ни об особенностях элемен-
тов ХТС.
Технологическая схема более информативна. На ней
все элементы ХТС представлены в виде условных стандартных изо-
бражений, позволяющих получить информацию о принципиальном
устройстве основных аппаратов и даже о соотношении размеров.
Аппараты связаны технологическими потоками, изображаемыми
в виде линий со стрелками, указывающими направления потоков.
Изображения дополняются краткой информацией о химическом
составе исходного сырья и продуктов реакции. В качестве при-
мера на рис. 7.3 показаны принципиальные структурная и техно-
логическая схемы ХТС синтеза аммиака.
Еще большую информацию несет операторная схема
ХТС. На такой схеме каждый аппарат изображается в виде не-
скольких типовых технологических операторов. Например, много-
полочный контактный аппарат с промежуточными теплообменни-
ками можно представить в виде совокупности нескольких опера-
торов химического превращения и теплообмена. Технологические
связи, как и в технологической и структурной схемах, изобра-
жаются линиями со стрелками направлений потоков. Операторная
схема ХТС позволяет более полно судить о физико-химической
154
сущности работы сложных агрегатов и о протекающих в них про-
цессах. На рис. 7.4 показан один из вариантов операторной схемы
узла дистилляции I ступени ХТС производства карбамида.
7.3. ОСНОВНЫЕ ТИПЫ СВЯЗЕЙ МЕЖДУ ЭЛЕМЕНТАМИ ХТС
Связи между аппаратами бывают различных типов: последо-
вательная, последовательно-обводная (байпасная), параллельная,
обратная (рециклическая), перекрестная. В существующих ХТС
характер технологических связей представляет собой сложную
комбинацию рассмотренных ниже типовых связей.
7.3.1. Последовательная связь
Последовательная связь характеризуется тем, что выходящий
из предшествующего элемента поток является входящим для по-
следующего элемента, и все технологические потоки проходят
через каждый элемент не более одного раза. Этот тип связи — наи-
более распространенный, и в самом общем случае его применяют,
когда степень превращения вещества в каждом предшествующем
аппарате (или операции) достаточно велика для эффективной
переработки полученных продуктов в каждом последующем аппа-
рате (операции).
Последовательную связь применяют также, когда необходимо
повысить степень превращения, избирательность, скорость про-
цесса за счет секционирования реакционных зон, создать опти-
мальный температурный режим, обеспечить промежуточный отвод
продуктов реакции.
Характерным примером последовательного соединения техно-
логических операторов химического превращения является каскад
реакторов смешения (см. рис. 6.30), обеспечивающий повышение
скорости процесса за счет увеличения движущей силы процесса
при последовательном секционировании реакционной зоны. Опе-
раторная схема каскада реакторов представлена на рис. 7.5. При-
мером гетерогенного процесса, проводимого в последовательно
соединенных аппаратах, является абсорбция нитрозных газов
в производстве разбавленной азотной кислоты (рис. 7.6). Степень
абсорбции оксидов азота в каждом абсорбере невелика, но после
прохождения шести последовательно установленных абсорберов из
газовой смеси отделяется более 90 % оксидов азота. Оставшиеся
оксиды поглощаются щелочью в других башнях, которые подклю-
чены также последовательно.
Аналогичный способ соединения аппаратов используют в про-
изводстве серной и соляной кислот, минеральных солей и удобре-
ний, при синтезе ряда органических продуктов.
При проведении обратимых экзотермических процессов для
приближения к оптимальному температурному режиму между
реакторами устанавливают промежуточные теплообменники. По-
163
1
3
%
Рис. 7.5. Операторная схема каскада реакторов.
Рис. 7.6. Абсорбция ннтрозных газов водой в производстве азотной кислоты по
схеме с последовательным соединением башен:
1 — абсорбционные башни; 2—кислотные холодильники.
a
4
Рис. 7.7. Операторная схема абсорбционного отделения ХТС производства серной
кислоты:
l—l-я ступень контактирования; 2—l-й абсорбер; 3—2-я ступень контактирования; 4—2-й аб-
сорбер.
j
следовательное соединение технологических операторов химиче-
ского превращения и теплообмена обеспечивает увеличение сте-
пени превращения, интенсивности протекания процесса (см. 1
разд. 6.10).
Еще больший эффект достигается при промежуточном отводе
продуктов реакции. Введение дополнительно оператора разделения
продуктов реакции в сочетании с оптимальным температурным ре-
жимом позволяет не только резко увеличить интенсивность работы
ХТС, но и достичь очень высоких степеней превращения. Приме-
нительно к ХТС производства серной кислоты последовательная
связь операторов химического превращения, теплообмена и раз-
деления показана на рис. 7.7,
Аналогичное соединение элементов используют в ряде случаев
для повышения избирательности при проведении сложных парал-
лельных или последовательных реакций.
7.3.2. Последовательно' обводная технологическая связь
Этот вид связи, часто называемый байпасом, характеризуется
тем, что часть технологического потока минует один или не-
сколько аппаратов по ходу технологической схемы. Байпас позво-
ляет эффективно управлять температурным и концентрационным
режимами работы аппаратов. Его используют, например, при адиа-
батическом проведении экзотермических химических превращений
совместно с последовательным соединением операторов химиче-
ского превращения (рис. 7.8,с). Благодаря байпасированию хо-
лодного потока сырья, высокая температура потока реагентов на
1Вв ,
Рис. 7.8. Последовательно-обводное соединение операторов:
а — регулирование температурного режима работы последовательно соединенных реакторов;
6 — V егулнрование температуры после теплообменника.
выходе из адиабатического реактора уменьшается, концентрация
реагирующего сырья на выходе из адиабатического реактора уве-
личивается. Байпас также можно использовать для регулирования
температуры на выходе из теплообменного узла (рис. 7.8,6).
7.3.3. Параллельная технологическая связь
Параллельную технологическую связь применяют для повыше-
ния производительности, увеличения ассортимента продуктов, по-
лучаемых на основе одного сырья, обеспечения повышенной надеж-
ности работы ХТС. Как правило, параллельную технологическую
связь используют для улучшения функционирования ХТС, состоя-
щей из последовательно установленных аппаратов, имеющих по
условиям масштабирования разную предельную мощность. В этом
случае мощность всей системы будет определяться элементом
с наименьшей мощностью. Для увеличения мощности ХТС уста-
навливают два или более одинаковых параллельно работающих
элемента, суммарная мощность которых равна мощности наиболее
производительного элемента (рис. 7.9, п).
Если на базе одного исходного вещества получают несколько
промежуточных продуктов, которые можно в дальнейшем исполь-
зовать для производства одного и того же целезого продукта, то
параллельно устанавливают несколько различных элементов, ра-
ботающих при разных режимах (рис. 7.9,6).
Если один из элементов ХТС обладает пониженной технологи-
ческой надежностью и его необходимо часто останавливать на
ремонт, то для обеспечения непрерывной работы системы устанав-
ливают параллельно несколько элементов.
Рис. 7.9. Параллельная технологическая связь:
а —прн повышении мощности ХТС; б—при переработке побочных продуктов.
Рис. 7.10. Обратная технологическая связь:
1» 3 — входной и выходной прямые потоки; 2—главный поток; <—обратный поток*.
167
7.3.4. Обратная (рециклическая) технологическая связь
Обратная технологическая связь характеризуется наличием
обратного технологического потока, связывающего выход какого-
либо последующего элемента с входом одного из предыдущих
элементов ХТС. Реакторный узел с рециклом показан на рис. 7.10.
Этот вид связи предусматривает многократное возвращение в один
и тот же элемент ХТС непрореагировавшей части одного или всех
реагентов. Если в ХТС имеется хотя бы одна обратная связь, то си-
стему называют замкнутой.
Применение обратной технологической связи позволяет более
полно использовать сырье при работе реакторов в условиях тер-
мических или термодинамических ограничений по степени превра-
щения. Примерами могут служить ХТС синтеза аммиака, мета-
нола и другие. Степень превращения азото-водородной смеси
в колонне синтеза аммиака за один проход через реакционную
зону не превышает 20%. Возврат на повторное контактирование
непрореагировавшей части смеси (рис. 7.11) обеспечивает общую
степень превращения более 99 %.
Другим примером является циркуляционная система произ-
водства серной кислоты с применением технического кислорода
(рис. 7.12). При степени превращения SO2 в контактном аппа-
рате 1 90—95 % степень превращения в контуре, включающем
контактный аппарат 1, охладитель газа 2 и абсорбер 3, дости-
гает 99 % •
Во многих случаях проведение процессов с большим выделе-
нием теплоты при высоких концентрациях реагентов затруднено
из-за сильного повышения температуры в реакционной зоне. Раз-
мещение же теплообменников в реакторах не всегда возможно
или малоэффективно. Применение обратной технологической
связи позволяет отводить теплоту из реактора 1 за счет нагрева
продуктов реакции и непрореагировавших компонентов, охлаж-
денных в выносном теплообменнике *2 (рис. 7.13), снизить удель-
ное выделение теплоты вследствие уменьшения концентра-
ций реагентов после смешения прямого входного и обратного
потоков.
Наличие обратного потока в циркуляционных ХТС позволяет
варьировать газовыми потоками не только с позиции массовых
расходов, но и менять их состав и соотношения концентраций. Для
простых реакций становится возможным избыток одного из ре-
агентов, необходимый для увеличения скорости процесса и дости-
жения наиболее полного выхода других реагентов. Так, в цирку-
Ма+ЗН3
Рис. 7.11. Циркуляционная схема синтеза ам-
миака:
I—компрессор; 2—колонна синтеза; 3—холодильник-
конденсатор аммиака*. 4—циркуляционный насос.
158
Кислота
Рис. 7.12 Обратная технологическая связь в циркуляционных системах производ-
ства серной кислоты:
1 — контактный аппарат; 2 — охладитель газа; 3 —абсорбер; 4—пиркуляционный инжектор.
Рис. 7.13. Применение рецикла для проведения экзотермических ХТП:
1— реактор; 2—теплообменник; 3—циркуляционный насос.
ляционной системе производства серной кислоты (см. рис. 7.12)’
реагенты (SO2 и О2) в прямом входном потоке находятся практи-
чески в стехиометрическом соотношении (избыток кислорода со-
ставляет 0,5—1,0%), а в главном потоке перед входом в контакт-
ный аппарат 1 избыток кислорода достигает 150—200 % От сте-
хиометрии при коэффициенте циркуляции 2—3. Это обеспечивает,
с одной стороны, интенсификацию процесса, а с другой стороны,
способствует достижению наиболее высоких степеней превраще-
ния оксида серы (IV) в контактном аппарате. Характерная зави-
симость избытка кислорода от коэффициента циркуляции пока-
зана на рис. 7.14.
При проведении сложных операций обратная технологическая
связь дает возможность управлять соотношением реагентов, доби-
ваясь высокой избирательности, и одновременно поддерживать
высокий выход целевых продуктов (см. разд. 4.2.2 и 6.4.2).
Рис. 7.14. Зависимость избытка кислорода (а) пе-
ред контактным аппаратом циркуляционной ХТС
производства серной кислоты от коэффициента
циркуляции (Ац).
169
7.3.5. Перекрестная технологическая связь
Этот вид технологической связи является вспомогательным
и встречается практически во всех сложных ХТС. Перекрести] ю
связь применяют главным образом для обеспечения эффективного
использования энергии (см. рис. 2.10). Так, введение в ХТС теп-
лообменников с перекрестной связью позволяет использовать теп-
лоту продуктов химической реакции для предварительного нагрева
исходной смеси реагентов, поступающих в реактор.
7.4. ПОНЯТИЕ О МАТЕМАТИЧЕСКОЙ МОДЕЛИ ХТС
Уравнения технологических связей записывают в следующем виде:
= (7.2)
Здесь Xf — i-я входная переменная k-ro элемента ХТС; Ylf — j-я выходная
переменная 1-го элемента ХТС.
Совокупность математических моделей элементов ХТС (7.1) и уравнений тех-
нологических связей между элементами (7.2) представляет математическую мо-
дель химико-технологической системы.
7.S. ЗАДАЧИ, РЕШАЕМЫЕ ПРИ ИССЛЕДОВАНИИ ХТС
Среди разносторонних задач, связанных с разработкой новых
и совершенствованием существующих ХТС, следует выделить за-
дачи синтеза, анализа и оптимизации.
На этапе синтеза четко формулируется цель задачи и усло-
вия ее достижения, разрабатываются конкурентноспособные ва-
рианты ХТС, подлежащие исследованию на следующем этапе.
На этапе анализа детально исследуются отобранные ва-
рианты ХТС и постепенно исключаются те их них, которые не от-
вечают в полной степени поставленным условиям.
Для получения наилучших показателей работы оставшихся
вариантов ХТС необходимо выполнить следующий этап — опти-
мизацию ХТС.
Задача оптимизации является комплексной. Она включает как
задачу оптимизации структуры ХТС, так и оптимизацию режимов
функционирования элементов. При оценке эффективности работы
ХТС используют различные экономические показатели. Следует
учитывать, что оптимизация работы только аппаратов без учета их
связей между собой может привести к тому, что вся ХТС будет
работать далеко не в оптимальном режиме.
7.6. ПРОЕКТИРОВАНИЕ ХТС
7.6.1. Основные этапы создания производств
и стадии проектирование
Знание основных закономерностей химической технологии осо-
бенно необходимо при разработке новых химико-технологических
систем. Создание новых производств протекает в несколько после-
160
довательных этапов и включает: научно-исследовательские, проект-
но-конструкторские работы, изготовление оборудования, строитель-
но-монтажные, пуско-наладочные работы, освоение проектных мощ-
ностей и достижение расчетных технико-экономических показателей.
Научно-исследовательские работы выполняют
с целью получения исходных данных для проектирования произ-
водства. Исходные данные должны включать всю информацию
по технологии получаемого продукта. В них содержатся рекомен-
дации: по химической и технологической схемам процесса, по ра-
циональным режимам проведения всех химико-технологических
процессов и их управлению, по типам и конструкциям основных
аппаратов.
Реализацию результатов научно-исследовательских работ
в промышленности осуществляют через проектно-конструкторские
разработки, являющиеся вторым этапом создания новых произ-
водств.
Проектно-конструкторские работы проводят с
целью выполнения проекта производства и разработки конструк-
ций основных и вспомогательных машин и аппаратов.
Проект любого химического производства — это совокупность
расчетно-пояснительных и графических материалов, необходимых
для создания промышленного объекта, обеспечивающего выпуск
продукта требуемого качества и в заданном количестве с наимень-
шей возможной себестоимостью и с обеспечением необходимых
требований по охране окружающей среды и труда обслуживаю-
щего персонала. В проекте обосновывается техническая возмож-
ность и экономическая целесообразность создания производства,
а также даются графические решения по строительству зданий,
сооружений, компоновке и монтажу основного и вспомогательного
оборудования.
Изготовление оборудования — производят машино-
строительные заводы. Все химическое оборудование делят на две
основные группы: нестандартизованное и стандартизованное, се-
рийно выпускаемое промышленностью. Нестандартизованное обо-
рудование изготавливают по чертежам проектного института, раз-
рабатывающего проект производства.
Строительные и монтажные работы предусматри-
вают строительство зданий и сооружений и осуществление мон-
тажа оборудования. Эти работы выполняют строительно-монтаж-
ные организации.
Пуско-наладочные работы предусматривают подго-
товку, опробование и вывод на устойчивые рабочие режимы всего
оборудования производства после завершения строительно-мон-
тажных работ. Пуск и наладка оборудования осуществляют спе-
циалисты управлений пуско-наладочных работ (УПНР).
Освоение проектных мощностей ссуществляют, как
правило, работники данного промышленного предприятия с при-
влечением специалистов научно-исследовательских, проектных ор-
ганизаций и УПНР.
6 Зак. 1869
161
Проектирование — основное звено, связывающее науку с про
изводством. Проектные работы выполняют в несколько стадий:
предпроектная разработка, разработка проекта, составление рабо-
чей документации и послепроектная стадия.
7.6.2. Предпроектная разработка
На стадии предпроектной разработки определяют технико-эко-
номическую целесообразность строительства нового или рекон-
струкции действующего производства, выбирают площадку для
строительства, проводят анализ исходных данных для проектиро-
вания и составляют задание на проектирование промышленного
объекта.
Эффективность проектных решений зависит от глубины тех-
нико-экономического обоснования (ТЭО), целесообразности наме-
чаемого создания ХТС.
ТЭО состоит из следующих основных разделов:
а) анализ потребности народного хозяйства в продукции, на-
мечаемой к выпуску, обеспеченности сырьем, топливом и энерго-
ресурсами;
б) выбор района строительства;
в) определение проектной мощности ХТС, обоснование ассор-
тимента продукции и требований к ее качеству.
Анализ потребности в продукции проводят на основе
перспективных планов развития отраслей промышленности по
экономическим районам. Из балансов производства и потребле-
ния данного вида химической продукции определяют недостаток
ее по состоянию на текущий или предшествующий год и на завер-
шающий год планируемого периода (не менее чем на 15 лет).
Анализ обеспеченности сырьем, топливно-
энергетическими ресурсами дает информацию о нали-
чии сырьевой базы и ее развитии, источниках обеспечения буду-
щей ХТС электроэнергией, топливом, водой и др.
Выбор района строительства проводят с учетом бли-
зости проектируемой ХТС к сырьевой базе и районам потребления
продукции, наличия топливно-энергетических ресурсов, транс-
портных коммуникаций, рабочих кадров, местных строительных
материалов, обеспеченности жильем, возможностей местных
строительных организаций и требований по охране окружающей
среды.
Проектную мощность ХТС определяют исходя из мак-
симального годового объема выпускаемой продукции при наибо-
лее полном использовании в течение года основного технологи-
ческого оборудования. При этом учитывают запасы сырья в районе
строительства, мощность энергетической базы, источников водо-
снабжения, требования по предельно допустимым выбросам вред-
ных веществ.
Ассортимент продукции и ее качество обусловлены
возможностями выпуска побочных продуктов, переработки отхо-4
162
дов производства, затратами на дополнительную очистку и кон-
центрирование основных и побочных ПрОД) ктов.
После выполнения ТЭО и определения района строительства
ХТС выбирают площадку для строительства. При этом
учитывают: топографические и геологические данные района
строительства; размеры площадки с учетом возможности расши-
рения предприятия, уровня залегания грунтовых вод и глубины
промерзания грунта; принадлежность земель и пригодность их
для сельского хозяйства; необходимость сноса строений; близость
площадок к городу; санитарно-гигиенические и технические требо-
вания и др.
Перед составлением задания на проектирование выполняют
анализ исходных данных для проектирования
Анализ исходных данных заключается в рассмотрении
всей имеющейся информации по технологии получаемого продукта
с точки зрения ее полноты и соответствия основным задачам, кото-
рые могут возникнуть при проектировании. При этом тщательно
изучаются регламенты (отчеты) научно-исследовательских органи-
заций, а также разделы монографий, статьи в периодической пе-
чати, патенты, на которые имеются ссылки в исходных данных
для проектирования. Особое внимание уделяют анализу качества
исходного сырья, на основе которого выполнены исследователь-
ские работы, и соответствию его источникам сырья для промыш-
ленных производств. Анализируют также: рекомендуемую прин-
ципиальную технологическую схему производства; основное тех-
нологическое оборудование; схему очистки выбросов; рекоменда-
ции по контролю и автоматизации процессов переработки сырья,
по технике безопасности и охране окружающей среды; результаты
проверки технологии на опытных или опытно-промышленных уста-
новках, а также расходные коэффициенты по сырью, пару.лоде,
топливу, -электроэнергии.
В результате анализа должны быть получены ответы на
основные вопросы, стоящие перед проектировщиком: можно ли по
рекомендованному методу организовать производство продукта
с заданным качеством, существует ли работоспособное оборудо-
вание для достижения заданной мощности производства; соответ-
ствует ли технология существующим санитарным нормам; будет
ли обеспечена приемлемая себестоимость продукта.
От полноты исходных данных и качества их анализа зависят
сроки освоения проектных мощностей, затра/Ы на строительство
ХТС, технико-экономические показатели ее работы.
После завершения анализа исходных данных и утверждения
ТЭО составляется задание на проектирование ХТС.
В задании на проектирование указывают: утвержден-
ное основание для создания ХТС или предприятия; район и пло-
щадку строительства; ассортимент продуктов и мощность произ-
водства; основные источники сырья, воды, топлива, электроэнер-
гии; условия по очистке выбросов; основные -технологические про-
цессы; оборудование и режим работы предприятия; требования по
••
163
разработке автоматизированных систем управления; сроки строи-
тельства и освоения; ориентировочный объем капитальных вло-
жений; основные технико-экономические показатели; последова-
тельность проектирования; наименование строительно-монтажной
организации.
После утверждения министерством задания на проектирование
приступают к разработке проекта.
7.6.3. Разработка проекта
На этой стадии выполняют проект со сводным расчетом стои-
мости (сокращенно «проект»), В состав проекта включены еле- <
дующие разделы: а) общая пояснительная записка с планом раз-
мещения существующих зданий, сооружений, коммуникаций, ин-
женерных сетей, а также проектируемой ХТС и- очистных соору-
жений; б) технологические решения с принципиальными схемами
ХТС, компоновки основного оборудования, автоматизации процес-
сов, электроснабжения, связи и сигнализации, распределительных
тепловых сетей и др.; в) строительные решения с принципиаль-
ными планами, разрезами зданий и сооружений, трасс внутрипло-
щадочных сетей; эскизных решений по антикоррозионной защите
строительных конструкций; г) организация строительства; д) смет-
ная документация.
В общей пояснительной записке приводят: исходные
данные для проектирования с указанием мощности ХТС, номен-
клатуры и качества продукции, источников и запасов сырья; све-
дения о потребности в топливе, воде, тепловой и электрической
энергии, трудовых ресурсах; характеристики основных принятых
технологических решений и сравнении их с лучшими отечествен-
ными и зарубежными аналогами; данные по экономике производ-
ства; краткие характеристики планировочных решений мероприя-
тий по гражданской обороне; основные решения по использованию
вторичных энергоресурсов и охране окружающей среды.
В разделе «Технологические решения» приводят: крат-
кую характеристику принятой технологии получаемого продукта;
предложения по механизации и автоматизации технологических 1
процессов; обоснование выбора оборудования, схем теплоэнерго- I
снабжения, доставки сырья, переработки отходов производства,
решений по охране, окружающей среды. В этом разделе приводят
топливно-энергетические и материальные балансы всех процессов, I
обосновывают численность производственного персонала, дают ре-
комендации по освоению проектных мощностей.
Особое внимание уделяют разработке технологической схемы
ХТС. Составляют возможные варианты, конкурентно-способных
схем. После синтеза ХТС проводят их анализ и выбирают наи-
более рациональную схему и оборудование, которые должны от- 1
вечать следующим основным требованиями: возможность надеж-
ного масштабирования по всему основному оборудованию при
переносе технологии с пилотных установок (на которых получены
164 I
исходные данные) к промышленным узлам, обеспечивающим тре-
буемую мощность; возможность соблюдения действующих сани-
тарных норм работы как внутри предприятия, так и с позиции
воздействия на окружающую среду при обеспечении минималь-
ной себестоимости продукта.
В разделе «Строительные решения» приводят: обосно-
вание основных архитектурно-строительных решений по зданиям,
сооружениям; перечень мероприятий по электро-, взрыве-, пожаро-
безопасности; решения по водоснабжению, канализации, отопле-
нию, вентиляции, по объектам гражданской обороны.
Раздел «Организация строительства» содержит ре-
комендации по последовательности строительства зданий, соору-
жений, установке основного оборудования. Этот раздел разраба-
тывают в соответствии с инструкцией Госстроя СССР.
Раздел «Сметная документация» содержит документа-
цию, необходимую для определения стоимости предприятия, зда-
ний, сооружений. Сметы, на строительство являются основными
документами, на базе которых осуществляется финансирование
строительства.
Чертежи к проекту, выполняемые на этой стадии проектирова-
ния, не являются рабочей документацией, на основании которой
осуществляют строительство и монтаж оборудования, сетей, ком-
муникаций и т. п. Их используют в качестве основы для составле-
ния рабочей документации на последующей стадии проектирова-
ния, а кроме того, они выполняют функции иллюстративного мате-
риала к принятым технологическим решениям.
7.6.4. Составление рабочей документации
Рабочую документацию составляют после утверждения про-
екта. Она содержит: рабочие чертежи, задания на разработку не-
стандартного оборудования, сметы, ведомости объемов строитель-
ных и монтажных работ, потребностей в материалах, спецификации
на оборудование.
Рабочие чертежи, на которых изображают элементы,
узлы, конструкции объекта, несут всю необходимую информацию
для его сооружения. По рабочим чертежам осуществляют: строи-
тельно-монтажные работы; установку оборудования; прокладку
сетей и устройств тепло-, газо-, электроснабжения, связи, сигнали-
зации, радиофикации; монтаж схем автоматизации технологиче-
ских процессов.
Задания на разработку нестандартного обору-
дования выдаются на основании технологических расчетов
и должны включать: 1) подробные эскизы разрабатываемых аппа-
ратов с указанием основных размеров и параметров работы наи-
более ответственных узлов; 2) характеристики и режимы прове-
дения процессов в разрабатываемых аппаратах; 3) физико-хими-
ческие свойства перерабатываемого сырья и получаемых продук-
тов, необходимые для выбора коррозионно-стойких материалов;
4) способ установки оборудования.
1бэ
В проектной организации конструкторский отдел разрабатывает
общую конструкцию аппарата и чертежи наиболее сложных и
принципиальных узлов. Подробные рабочие чертежи нестандарт’
ного оборудования разрабатывают на машиностро дельных заво-
дах с учетом имеющейся на них оснастки.
7.6.5. Послепроектная стадия
На этой стадии проектная организация осуществляет автор-
ский надзор за строительством. УПНР проводит пуск и наладку
оборудования, вывод его на рабочие режимы. Последующее
освоение проектной мощности и достижение расчетных технико-
экономических показателей введенной в эксплуатацию ХТС осу-
ществляют работники предприятия.
7.6.6. Методы проектирования
Применяемые технические методы проектирования можно объ-
единить в три группы: графические, плоскостного макетирования
и объемные макетные.
При графическом методе вычерчивают планы, разрезы -л
зданий, проекции аппаратов, установок, общие виды оборудования
и т. д. Основной недостаток метода — необходимость разработки
большого числа чертежей для каждого варианта технологической
схемы и .компоновки оборудования.
При плоскостном макетировании чертежи монтируют
из унифицированных заранее подготовленных типовых элементов
чертежей (ТЭЧ). Типовыми элементами могут быть самоприклеи-
вающиеся аппликации с изображениями оборудования, пластинки
с проекциями элементов конструкции и необходимыми надписями
(темплеты), монтируемые на магнитных досках. Полученные та-
ким способом чертежи из ТЭЧ фотографируют и размножают.
При разработке проектов ХТС со сложными технологическими
схемами и наличием большого числа трубопроводов используют
метод объемного макетного проектирования, преду-
сматривающий создание моделей ХТС путем сборки и компоновки
из готовых модельных элементов (примерный масштаб l:50).J
С готовой модели объекта делают фотоснимки, используемые
В качестве чертежей. Этот метод обеспечивает наиболее наглядное
представление о проектируемом объекте и упрощает поиск наи-
лучших компоновочных решений.
ГЛАВА 8
ПРИНЦИПЫ ЭКОЛОГИЧЕСКОЙ ТЕХНОЛОГИИ
8.1. ОСНОВНЫЕ НАПРАВЛЕНИЯ ОХРАНЫ
ОКРУЖАЮЩЕЙ СРЕДЫ ОТ ПРОМЫШЛЕННЫХ ВЫБРОСОВ
Окружающая среда включает атмосферу, источники воды, поч-
ву и земные недра.
Защита атмосферы имеет важное значение в решении вопросов
охраны окружающей среды. Большее количество выбросов в атмо-
1G6
сферу приходится на долю энергетических предприятий (тепловых
электростанций) и транспорта. Химические предприятия выбрасы-
вают меньше вредных веществ, но в большем ассортименте и бо-
лее токсичные.
Огромные количества отходящих газов энергетических, метал-
лургических и сернокислотных предприятий, содержащих диоксид
серы, приводят не только к коррозии металлических конструкций
и сооружений, но и к заболеванию людей, гибели животных и зе-
леных насаждений. Количество диоксида серы, выбрасываемого
в атмосферу, существенно превышает общий расход SO2 на про-
изводство серной кислоты.
Защита Мирового океана — одна Из глобальных экологиче-
ских проблем, волнующая общественность всех стран мира. В Ми-
ровом океане осуществляется основной процесс «дыхания» зем-
ного шара — фотосинтез, при котором усваивается большая часть
диоксида углерода атмосферы и вырабатывается больше поло-
вины ее кислорода.
Особую опасность представляет загрязнение источников воды
такими металлами, как ртуть, свинец, кадмий, медь, цинк и хром.
Например, ртуть в воде превращается в прочное соединение —
диметилртуть, которое, попадая в организм, накапливается там.
Значительные количества ртути выбрасывают целлюлозно-бумаж-
ные производства, использующие ее в качестве антисептиков;
электролитические предприятия при производстве хлора, щелочи
и др. Соединения меди, цинка, кадмия и хрома попадают в во-
доемы со сточными водами соответствующих металлургических
и химических предприятий, свинец — с дождевыми водами, содер-
жащими токсичные компоненты выхлопных газов двигателей внут-
реннего сгорания.
Защита почвы и земных недр имеет важное значение для сохра-
нения и воспроизводства флоры и фауны, полезных ископаемых.
Загрязнение почвы происходит сточными водами и твердыми отхо-
дами (шлаки, шламы) предприятий, бытовыми отходами, внесе-
нием избытка удобрений и ядохимикатов, атмосферными осад-
ками, загрязненными вредными выбросами. Все это губительно
действует на растительный и животный мир.
Охрана окружающей среды осуществляется государством в со-
ответствии с основными задачами социально-экономического раз-
вития. В нашей стране запрещено вводить в строй новые пред-
приятия, цеха, установки, не обеспеченные очистными устрой-
ствами для предотвращения загрязнения водного и воздушного
бассейнов. Ученые Советского Союза впервые разработали и утвер-
дили предельно допустимые концентрации (ПДК) вредных ве-
ществ в воздухе, воде и почве. Применительно к загрязнению воз-
духа ПДК определяется как концентрация, которая не оказывает
прямого или косвенного вредного и неприятного действия на чело-
века, не снижает его работоспособности, не влияет на его само-
чувствие и настроение. В СССР принят самый низкий уровень
ПДК из четырех, рекомендованных Всемирной организацией здра-
167
воохраиения при ООН в 1963 г. За выполнением соответствующих
норм и правил в нашей стране установлены различные виды Го-
сударственного надзора на всех стадиях: проектирования, строи-
тельства и эксплуатации производств
Каждое предприятие и объединение разрабатывает паспорт-до-
кумент, в котором содержатся объективные данные о технологии
и ее возможностях по выпуску продукции, в том числе и такие
основные показатели взаимодействий с окружающей средой, как:
использование территории; потребление воды, воздуха, электро-
энергии, тепловой энергии, топлива (угля, нефти, природного
газа — с указанием качества), сырья (всех видов); выбросы
в окружающую среду (с указанием состава, свойств, количества
и координат точки выбросов в атмосферу, на поверхность почвы
или на определенную глубину) и т. д. Наличие паспортных данных
по всем видам взаимодействий с окружающей средой поднимает
на более высокий уровень управление охраной среды во всех
звеньях (организация, планирование, контроль и прогноз измене-
ний состояния окружающей среды, правовая охрана).
В нашей стране разработано несколько основных направлений
защиты окружающей среды: 1) совершенствование технологиче-
ских процессов с целью уменьшения вредных выбросов; 2) приме-
нение методов очистки вредных выбросов и утилизации отходов
действующих производств; 3) создание безотходных производств,
основанных на замкнутых процессах и комплексном использова-
нии сырья.
Совершенствование технологических процес-
сов с целью уменьшения вредных выбросов — важ-
ное направление для решения экологической проблемы действую-
щих производств. Так, в производстве серной кислоты для дости- ;
жения максимальной степени превращения SO2 в SO3 стадию
окисления проводят в несколько ступеней (многостадийно). Таким
образом, уменьшается количество выбросов SO2 (после абсорбции
SO3 в серную кислоту). Такой метод широко используют в хими-
ческой технологии.
Уменьшить содержание диоксида серы и других токсич-
ных соединений в выхлопных газах позволит совершенствова-
ние методов предварительной очистки жидкого и твердого топ- 1
лива.
Другим примером может служить совершенствование техноло-
гии химических производств с целью экономии потребления воды
в промышленности. Замена водяного охлаждения воздушным
и применение замкнутого водооборота (оборотной воды)—самый
рациональный и прогрессивный путь охраны водоемов от загряз-
нения и истощения.'
Применение методов очистки вредных выбро- ]
сов и утилизации отходов действующих произ-|
водств — наиболее распространенный прием уменьшения вредных
выбросов в окружающую среду. Это направление основано на ис-
пользовании различных очистных сооружений. В настоящее время
168
в большинстве отраслей народного хозяйства имеются возможности
решения экологической проблемы таким путем.
Утилизация отходов действующих производств, т. е. перера-
ботка их в полезные продукты, имеет и экономическое значение:
сокращаются издержки на складирование отходов, которые подчас
составляют миллионы рублей; уменьшаются площади земли, зани-
маемые отвалами. Например, разрабатываются способы перера-
ботки фосфогипса (CaSO4-2H2O), накапливаемого в больших
количествах в производствах фосфорной кислоты и фосфорных
удобрений. В настоящее время предложен метод получения из него
сложного удобрения. В нашей стране разработано много способов
утилизации отходов различных предприятий. Для уменьшения
вредных выбросов применяют и другие методы обезвреживания —
отходы сжигают, подвергают термохимической и биологической
обработке.
Безотходные производства основаны на комплексном
использовании всех компонентов сырья, комбинировании пред-
приятий на базе использования отходов и создании замкнутых тех-
нологических процессов с целью повышения степени превращения
исходных реагентов. Эти производства не имеют никаких выбро-
сов в окружающую среду. В настоящее время по замкнутому
циклу работают производства аммиака, метилового и этилового
спирта и др.
8.2. ОЧИСТКА ГАЗООБРАЗНЫХ ПРОМЫШЛЕННЫХ ВЫБРОСОВ
Очистка газообразных промышленных выбросов остается пока
наиболее распространенным направлением в решении вопросо
охраны окружающей среды.
В газообразных промышленных выбросах могут содержаться
различные примеси: а) взвешенные, твердые и жидкие вещества
(аэрозоли); б) газо- или парообразные примеси. Аэрозоли пред-
ставляют собой дисперсные системы с газообразной (воздушной)
дисперсионной средой и твердой (пыль, дым) или жидкой (туман)
дисперсной фазой. Скорость оседания частиц аэрозоля очень мала,
и они могут неопределенно долгое время находиться во взвешен-
ном состоянии.
Пыль — это дисперсная малоустойчивая система, содержа-
щая больше крупных частиц, чем дымы и туманы. Неорганическая
пыль образуется при переработке неорганических веществ, напри-
мер металлов, минеральных солей, удобрений и др. Примером
пыли органического происхождения может служить угольная, тор-
фяная, древесная, сажа и др.
Дым — представляет собой дисперсную систему с малой ско-
ростью оседания твердых частиц под действием силы тяжести
Размер частиц много меньше, чем в пыли и тумане (<10,1 мкм)
Дымы образуются при сжигании топлива, в результате химических
реакций, например при окислении паров металлов в электрической
дуге и т. д.
169
Туманы содержат мельчайшие капельки жидкости, образую-
щиеся при конденсации паров или распылении жидкости. Так,
в производстве серной кислоты при охлаждении печного газа об-
разуется сернокислотный туман.
К наиболее распространенным примесям в промышленных
выбросах относят г азо- или парообразные (галогены и их произ-
водные, оксиды, альдегиды, кетоны, спирты, кислоты и др.).
Снижение концентрации вредных примесей в промышленных
выбросах методами очистки до значений ПДК, установленных
Минздравом СССР для воздуха населенных мест, часто дости-
гается методом их рассеивания через высокие трубы в верхних
слоях атмосферы.
Разнообразие вредных примесей в промышленных газовых вы-
бросах приводит к большому разнообразию методов очистки, при-
меняемых реакторов и химических реагентов. Выбор способа
очистки определяется физико-химическими свойствами примесей,
их агрегатным состоянием, дисперсностью, химическим составом
и др. Классификация методов очистки газов приведена ниже:
j,£i
Л
1
У
5
1
Загрязнения Характеристика загрязнений Метод очистки
Аэрозоли
неорганического происхождения Абразивная пыль, минераль- ные соли (фосфаты, арсенаты, мышьяк, ртуть), аэрозоли ме- таллов Механический сухой и мокрый, электростатиче- ский, коагуляция
органического происхождения Газо- или парооб- разные примеси Древесная, угольная, табачная и мучная пыль Механический сухой, ад- сорбционный, дожига- ние в печах
неорганического происхождения HF, HI, НС1, HBr, F2, CL, I,, SO2. SO3. NO, NO2, Р2Об Адсорбционный, хемо- сорбционный, электро- статический
органического происхождения Органические кислоты, альде- гиды, кетоны, углеводороды, бензол, толуол и т. д. Адсорбционный, катали- тический, дожигание в печах
комбинирован- ные Хладоны, амины, тиазол, пира- золы, пиридины, пирролы и т. д. Адсорбционный, катали- тический, дожигание в печах с последующей адсорбционной доочист- кой
1
8.2.1. Очистка .'азообразных выбросов
от взвешенных частиц (аэрозолей)
Наиболее широко распространены следующие способы очистки!
механическая и электростатическая.
Механическую очистку осуществляют сухим и мокрым
методами. К сухим методам относят: I) гравитационное осажде-
170 . '
<5
У
ние; 2) инерционное и центробежное пылеулавливание; 3) фильт-
рование. Чаще комбинируют несколько методов. Различают гру-
бую и тонкую очистку газов.
Гравитационное осаждение — осаждение взвешенных частиц
под действием силы тяжести при малой скорости потока и без
изменения его направления. Процесс производят в отстойных га-
зоходах и пылеосадительных камерах. Гравитационное осаждение
применяют для крупных частиц диаметром более 50—100 мкм,
степень очистки при этом 40—50 %, т. е. это лишь грубая очистка,
требующая дополнительной установки аппаратов для тонкой
очистки.
Инерционное осаждение основано на свойстве взвешенных ча-
стиц сохранять первоначальное направление движения при изме-
нении направления газового потока. Наиболее часто применяют
жалюзийные пылеуловители. Газы обеспыливаются, выходя через
щели. Степень очистки составляет 20—70%, т. е. производится
лишь грубая очистка. Кроме этого, есть еще недостаток — быст-
рое истирание и забивание щелей.
Центробежные методы очистки газов основаны на действии
центробежной силы, возникающей при закручивании газового по-
тока в аппарате или при вращении частей в самом аппарате.
Для этой цели применяют циклоны (батарейные циклоны). Они
просты по устройству, надежны в работе, отличаются высокой про-
изводительностью. Циклоны широко применяют для грубой очист-
ки газов от пыли с диаметром частиц более 30 мкм, степень очист-
ки 50—60%. После циклона необходима тонкая очистка газов от
пыли.
Фильтрование — прохождение очищаемого газа через фильт-
рующие пористые материалы. В качестве таких материалов при-
меняют хлопчатобумажные и шерстяные ткани, химические волок-
на, стекловолокно, керамику, металлокерамику, пористые перего-
родки и т. д. Очистка газов фильтрованием наиболее эффективна
при размере частиц примеси не менее 0,1 мкм.
Мокрую) очистку газов от аэрозолей осуществляют, промы-
вая газ жидкостью (чаще водой). Это распространенный метод
очистки газа от мелких частиц пыли, дыма, тумана. Для этой цели
используют различные аппараты: башни с насадкой (насадочные
скрубберы), пенные аппараты, скоростные прямоточные аппараты
(скрубберы Вентури) и др. Конструкции этих аппаратов анало-
гичны конструкциям реакторов, рассмотвенным в разд. 6.8.1.
Электростатическая очистка газов является универ-
сальной для многих аэрозолей, включая трудноулавливаемые ту-
маны кислот. Метод основан на ионизации аэрозоля при прохо-
ждении газа через электрическое поле высокого напряжения, соз-
даваемое коронирующими электродами. Осаждение частиц проис-
ходит на заземленных осадительных электродах. Электрофильтры
состоят из ряда заземленных пластин или труб, через которые
пропускают очищаемый газ. Между осадительными электродами
подвешены проволочные коронирующие электроды с напряжением
171
25—100 кВ (обычно отрицательным). При размере частиц менее
0,1 мкм степень очистки газа достигает 99,9%. Недостатком ме-
тода является значительный расход электроэнергии — 0,1—0,5 кВт
на 1000 м3 очищаемого воздуха — и большие капиталовложения на
содержание очистных установок.
8.2.2. Очистка газов от газо- или парообразных примесей
Наиболее часто применяют следующие методы очистки: 1) аб-
сорбцию жидкостями; 2) адсорбцию твердыми поглотителями;
3) каталитическую очистку; 4) термические методы.
Абсорбция жидкостями основана на избирательной рас-
творимости примесей в жидкости (физическая абсорбция) или на
избирательном извлечении примесей химическими реагентами
(хемосорбция).
Абсорбционную очистку широко применяют для извлечения
ценных веществ из отходящих газов, очистки реакционных газов
от каталитических ядов и санитарной очистки газов перед вы-
бросом в атмосферу. Этим методом производят очистку газов от
диоксидов серы, сероводорода и других сернистых соединений,
оксидов азота, паров кислот (НС1, HF, H2SO4), диоксида и оксида
углерода, различных органических соединений и др.
В качестве абсорбентов используют зоду, растворы аммиака,
едких и карбонатных щелочей, масла, суспензии гидроксида каль-
ция и др. Очистная аппаратура аналогична применяемой в методе
мокрой очистки газов от аэрозолей. Наиболее распространен на-
садочный скруббер, однако из-за малой скорости массообменных
процессов объемы аппаратов велики и установки громоздки. Осо-
бенно перспективны для очистки газов от аэрозолей и газообраз-
ных примесей пенные аппараты со стабилизатором пенного слоя;
они сравнительно просты по конструкции и работают в режиме
высокой турбулентности.
Абсорбционные методы характеризуются непрерывностью, эко-
номичностью и возможностью извлечения разнообразных ценных
примесей. Однако эти методы имеют ряд недостатков: схемы, как
правило, сложны, аппараты имеют большие размеры (особенно
скрубберы), очистка производится в несколько ступеней (для до-
стижения высокой степени извлечения).
Любой процесс абсорбционной очистки выхлопных газов целе-
сообразен только в случае его цикличности и безотходности.
Адсорбция твердыми поглотителями основана на
избирательном извлечении компонентов газовой смеси при помощи
адсорбентов — твердых поглотителей, обладающих высокой по-
ристостью. В качестве адсорбентов применяют активированный
уголь, силикагель, алюмогель, природные и синтетические цео-
литы (молекулярные сита). Промышленные адсорбенты должны
обладать высокой поглотительной способностью, избирательностью
действия (селективностью), термической устойчивостью, длитель-
ным сроком службы и легкой регенерируемостью.
572
Адсорбцию проводят в реакторах нескольких типов: полочного,
трубчатого и с движущимся или взвешенным слоем адсорбента
(см. разд. 6.9). Наиболее перспективным является последний тип
реактора, так как характеризуется высокими скоростями газового
потока, высокой производительностью и интенсивностью работы в
непрерывном режиме.
Адсорбционные методы имеют ряд общих достоинств: высокую
степень очистки газов от вредных примесей и сравнительно легкую
регенерацию этих примесей с превращением их в товарный про-
дукт или возвратом в производство, что позволяет осуществить
принцип безотходности. Недостатки этих методов —• периодичность
для большинства адсорбционных установок и соответственно ма-
лая их интенсивность. Непрерывный способ адсорбционной очистки
затруднен из-за отсутствия высокопрочных сорбентов.
Каталитическая очистка газов основана на реакциях
в присутствии твердых катализаторов, т. е. на закономерностях
гетерогенного катализа (см. разд. 5.5 и 6.9). В результате ката-
литических реакций токсичные примеси превращаются в безвред-
ные соединения. Этот метод очистки выхлопных газов перспекти-
вен ввиду высокой эффективности и возможности очищать боль-
шие объемы газов, содержащие малые концентрации примесей
[вплоть до 0,1—0,2 % (по объему)]. Установки для каталитиче-
ского разложения находят широкое применение, например, в про-
изводстве азотной кислоты для очистки газов от оксидов азота.
Термические методы очистки (сжигание отходов) —
один из наиболее распространенных и эффективных методов очист-
ки отходящих газов, содержащих большое количество вредных
примесей. Сущность метода заключается в разложении токсичных
веществ до безвредных. В тех случаях, когда образуются вредные
компоненты (диоксиды серы и азота, галогены и др.), необходима
вторичная обработка, включающая, например, дожигание. Про-
стейшим методом является факельное сжигание. При содержании
в отходящих газах большого количества горючих газов сжигание
осуществляют без затраты теплоты извне, при небольшом содер-
жании горючих примесей добавляют горючий газ и сжигают его
в потоке очищаемого газа. Теплоту сжигаемых газов утилизируют.
Метод сжигания экономически выгоден при достаточно высо-
кой концентрации горючих примесей в очищаемом газе, иначе уве-
личивается дополнительный расход топлива, стоимость которого
в настоящее время постоянно растет.
На практике широко применяют комбинирование перечислен-
ных выше методов очистки газов от газо-парообразных примесей.
Так, для грубой очистки используют абсорбционные методы, а за-
тем для тонкой доочистки — адсорбционные или каталитические.
8.3. ОЧИСТКА СТОЧНЫХ ПРОМЫШЛЕННЫХ ВОД
В промышленных сточных водах могут содержаться разнооб-
разные органические и неорганические примеси в твердом, жид-
ком и газообразном состоянии. Неорганические примеси (мине-
179
ральные кислоты, соли, гидроксиды и др.) при попадании в
водоемы изменяют свойства воды, влияют на живые организмы и
образуют нерастворимые осадки. Для органических примесей ха-
рактерны окислительные процессы, на которые расходуется рас-
творенный в воде кислород, что приводит к дефициту кислорода, "
необходимого для жизнедеятельности растений и живых орга-
низмов.
Загрязнение водоемов сточными'водами может нанести ущерб
многим отраслям народного хозяйства и создать дефицит чистой
питьевой воды. Недостаток питьевой воды может возникнуть также
из-за огромного потребления пресной воды промышленностью. Наи-
более радикальный путь охраны водоемов от истощения и загряз-
нения— это резкое уменьшение объема промышленных сточных
вод, вплоть до полной их ликвидаций на основе циркуляционных
процессов, работающих по замкнутому циклу, т. е. бессточных про-
цессов.
В настоящее время в промышленности стали широко приме-
нять оборотное и многократное использование сточных вод. Воз-
можны и другие направления уменьшения объема или исключения
сброса сточных вод — их полное испарение, спуск особо загряз-
ненных стоков в геологически изолированные пласты и др.
Применяемые в промышленности методы очистки сточных вод
определяются объемами стоков, количеством, дисперсностью и со-
ставом примесей. Ввиду многочисленности примесей и сложности
их состава, используют как правило, комбинирование методов
очистки.
Методы очистки сточных вод разделяют на механические, фи-
зико-химические, химические, биологические и термические.
Механические методы (процеживание, отстаивание,
осветление и фильтрация) применяют для очистки от крупнодис-
персных взвешенных частиц. С этой целью используют решетки,
отстойники, гидроциклоны, фильтры.
Физико-химические методы служат для очистки вод
от мелкодисперсных, коллоидных и растворенных веществ. К та-
ким методам относят: флотацию, -коагуляцию и флокуляцию, экс-
тракцию растворителями, дистилляцию и ректификацию, адсорб-
цию, обратный осмос и др.
Флотация в практике очистки сточных вод получает все боль-
шее распространение. Сущность метода та же, что и при обога-
щении твердого сырья (см. разд. 2.1.2). Концентрирование взве-
шенных частиц основано на использовании пузырьков газа для
увеличения подъемной силы, действующей на взвешенные частицы.
Газовые пузырьки прилипают к частицам и поднимают их на по-
верхность воды, где они и удаляются.
Коагуляцию и флокуляцию применяют для удаления мелко-
дисперсных и коллоидных примесей. Благодаря введению коагу-
лянтов и флокулянтов происходит слипание взвешенных частиц,
после чего укрупненные частицы отделяют отстаиванием или.
фильтрацией. Эти методы широко используют при очистке сточных
174
вод производств нефтехимии, синтетических смол, волокон, пласт-
масс, целлюлозы и в других случаях.
Экстракция с помощью растворителей избирательного действия
основана на поглощении загрязнений жидкими, реже, тверды-
ми, экстрагентами. Сточную воду смешивают с экстрагентом,
в котором загрязнения растворяются лучше, чем в воде, а сам
экстрагент не смешивается с водой. Отделяя экстрагент от воды,
а с ним и загрязняющее вещество, можно добиться значительной
степени очистки сточной воды. Экстрагент можно регенерировать,
а экстрагированное из него вещество использовать для нужд про-
изводства.
Дистилляция и ректификация основаны на удалении компо-
нентов примесей сточных вод, имеющих различную температуру
кипения.
Адсорбционные методы очистки применяют для удаления рас-
творенных органических соединений из сточных вод. Сорбен-
тами служат вещества с развитой поверхностью: активированный
уголь, опилки, зола, торф, глина, а также синтетические высоко-
пористые полимерные адсорбенты. В настоящее время наиболее
широко используют два основных режима адсорбционной обра-
ботки сточных вод: адсорбцию в неподвижном слое и адсорбцию
в движущемся слое сорбента. Сорбционные методы очистки яв-
ляются наиболее эффективным способом глубокой очистки сточных
вод. Эти методы будут одним из главных элементов систем глу-
бокой очистки сточных вод, призванных сыграть огромную роль
в создании безотходных производств.
Обратный осмос — основан на способности молекул воды
проникать через полупроницаемую мембрану, которая непрони-
цаема для более крупных молекул веществ, растворенных в воде.
При'этом внешнее давление должно превышать осмотическое дав-
ление раствора. В качестве материала для полупроницаемых
мембран в основном используют синтетические смолы в виде пле-
нок. Если фильтрованием можно удалить примесные частицы раз-
мером выше 5—10 нм, то обратным осмосом—частицы, размер
которых не превышает 0,1—0,5 нм.
Химические методы очистки сточных вод основаны на
проведении химических реакций, в результате которых образуются
новые вещества, легко удаляемые доступными методами. При хи-
мической очистке протекают реакции нейтрализации, окисления —
восстановления, конденсации, осаждения и др. Чаще проводят
реакции нейтрализации кислых стоков. Одновременно с нейтрали-
зацией происходит осаждение гидроксидов и карбонатов соответ-
ствующих металлов. Поэтому нейтрализация сопровождается от-
стаиванием, уплотнением и обезвоживанием полученных осадков.
Химические методы очистки характеризуются большим расходом
реагентов и громоздкой аппаратурой, особенно отстойной. Кроме
того, возникает проблема хранения и применения образующихся
осадков.
176
Из химических методов важное значение имеют хлорирование
и озонирование сточных вод. Их используют для доочистки и обез-
вреживания органических примесей, цианидов и дурно пахнущих
неорганических веществ.
Биологические методы очистки — наиболее распро-
страненные методы очистки промышленных и хозяйственно-быто-
вых сточных вод. При этом используют разные виды микроорга-
низмов для улавливания или переработки вредных примесей до
безвредных— воды, диоксида углерода, нитрат- и сульфат-ионов.
Существуют два основных метода биологической очистки — аэроб-
ный (при непрерывном притоке кислорода воздуха) и анаэробный
(в отсутствие кислорода). Из них наиболее распространен аэроб-
ный метод, позволяющий достигнуть максимальной скорости
очистки и максимальной эффективности обеззреживания приме-
сей. Анаэробный метод используют для предварительной очистки
примесей с большой концентрацией вредных веществ перед аэроб-
ной очисткой сточных вод.
Термическую очистку сточных вод применяют для
обезвреживания органических веществ. Сущность метода заклю-
чается в полном окислений (сжигании) органических веществ до
безвредных — Н2ОЦ СО2, N2 и зольного остатка. Недостатком ме- 1
тода является существенный расход топлива и большой объем
печей. Термические методы неэкономичны, особенно при больших
объемах стоков. Они целесообразны при содержании более 6 %
токсичных органических веществ, удаление которых невозможно
другими методами.
В реальных условиях схема очистки сточных вод состоит, как
правило, из нескольких ступеней, включающих различные методы •
очистки от разных веществ. После очистки сточные воды подвер-
гают контролю качества очистки от вредных примесей. Спуск в !
водоемы допускается при наличии примесей, не превышающих
установленные нормы.
Несмотря на значительные затраты, в настоящее время все 1
вновь сооружаемые предприятия снабжаются системами очистки I
сточных вод.
8.4. ПРЕДОТВРАЩЕНИЕ ТЕПЛОВОГО ЗАГРЯЗНЕНИЯ
Для уменьшения тепловых потерь в окружающую среду ши-
роко используют теплоизоляцию аппаратов и трубопроводов.
В химической технологии большое значение приобретает ути-
лизация вторичных энергоресурсов, образующихся при осуществ-
лении химико-технологических процессов, в первую очередь экзб-
термических. Рациональное использование вторичных энергоре- ]
сурсов'способструет максимальному уменьшению тепловых загряз-
нений окружающей среды и значительному снижению затрат на
топливо (см. разд. 2.3).
176
ГЛАВА 9
ПРОИЗВОДСТВО СЕРНОЙ кислоты
9.1. ОБЩИЕ СВЕДЕНИЯ
На практике под серной кислотой подразумевают любые смеси
оксида серы (VI) с водой. Безводную серную кислоту называют
моногидратом. Он хорошо смешивается с водой в любых со-
отношениях. При разбавлении серной кислоты водой выделяется
большое количество теплоты. Раствор SOa в 100 %-ной серной кис-
лоте называют олеумом.
Серная кислота является одним из крупнотоннажных продук-
тов химической технологии. В СССР ее производят около 26 млн. т
в год. Мировое производство составляет около 150 млн. т.
Серная кислота относится к числу сильных кислот и является
самой дешевой. Она реагирует почти со всеми металлами, всту-
пает в реакции обменного разложения. Почти половину всей вы-
рабатываемой кислоты используют для. производства минеральных
удобрений. Серную кислоту применяют также в нефтехимической,
металлургической отраслях промышленности, для производства
химических волокон, красителей, взрывчатых веществ и других
продуктов.
Промышленность выпускает несколько сортов серной кислоты,
различающихся концентрацией H2SO4 или свободного SO3, а так-
же содержанием примесей. Основное количество производится в
виде технической контактной кислоты (содержание H2SO4 92,5 %),
аккумуляторной кисдоты (H2SO4 — 92 4- 94 %) с пониженным со-
держанием примесей, олеума (SO3 —19-^24%), а также башен-
ной кислоты (H2SO4 — 75%).
9.2. СПОСОБЫ ПРОИЗВОДСТВА
Первой стадией производства серной кислоты по любому спо-
собу является получение оксида серы (IV) путем сжигания серу-
содержащего сырья:
S + О2 —> SO2 + q\ • (а) -
После очистки сернистого газа, т. е. смеси оксида серы(IV)
с азотом, кислородом и другими газами, оксид серы (IV) окисляют
до оксида серы (VI):
SOj -f- 0,5Ог < SO3 + <у2 (б)
Оксид серы (VI), соединяясь с водой, образует серную кислоту:
SO3 -|- Н2О —► H2SO4 -f- ^з (в)
По методу окисления оксида серы (IV) различают контактный
и нитрозный способы производства серной кислоты. В современных
177
производствах реакцию окисления SO2 проводят в присут-
ствии твердого катализатора, и способ производства называют
контактным. В СССР этим способом получают более 95 %
всей серной кислоты.
При нитрозном способе катализатором служат оксиды
азота. Окисление проводят в жидкой фазе с помощью нитрозы,
выполняющей функции передатчика кислорода. Процесс осуществ-
ляют в башнях с насадкой, и по это.му признаку нитрозный спо-
соб часто называют башенным.
9.3. СЫРЬЕ ДЛЯ ПРОИЗВОДСТВА СЕРНОЙ КИСЛОТЫ
В качестве сырья для производства серной кислоты исполь-
зуют элементарную серу, колчедан, отходящие газы печей цветной
металлургии, газы, содержащие сероводород. В СССР более 50 %
серной кислоты получают из серы, около 30 % — из колчедана.
Сера — используют природную серу после флотации серных
руд, а также серу, полученную из серусодержащих газов плавки
медного колчедана и сероводородных газов.
Колчедан (основной составной частью является FeS2)—
используют колчедан, получаемый в виде отходов при обогащении
руд цветных металлов (флотационный колчедан). Содержание
серы в таком колчедане составляет от 35 до 47 % в зависимости
от марки.
Отходящие газы — используют газы, образующиеся при
обжиге руд цветных металлов (содержание SO2 в отходящих газах
составляет 3—16%), при кислородной плавке медных и медно-
цинковых концентратов руд (SO2 — 70—DO %).
Сероводород — получают при очистке промышленных газов
(коксовых, генераторных, природных), а также газов нефтепере-
работки.
9.4. ПРОИЗВОДСТВО СЕРНИСТОГО ГАЗА
9.4.1. Сжигание серы
В настоящее время самые мощные предприятия по производ-
ству серной кислоты в качестве сырья используют серу. Сжигание
серы в промышленных условиях протекает в две стадии: а) испа-
рение с поверхности раздела жидкой серы и газовой фазы; б) го-
могенное окисление паров серы в газовой фазе. При высоких тем-
пературах (более 500°C) первая стадия является лимитирующей
и в промышленных условиях процесс, как правило, посмекает в
диффузионной области.
Интенсивное испарение серы начинается при температуре ее
кипения, которая при атмосферном давлении составляет 445°C.
Пары серы сгорают с образованием оксида серы(IV) по реак-
173
Сернистый
газ
Рис. 9.1. Схема форсуночной печи для сжигания серы:
I—форсунка; 2—корпус.
Рис. 9.2. Зависимость температуры в печи и молярной доли оксида серы(1У) в
сернистом газе от коэффициента Избытка воздуха а.
ции (а). При этом выделяется q\ = 3G2 кДж теплоты. Так как
сжигание жидкой серы в целом является гетерогенным процессом,
то для его интенсификации необходимо увеличивать поверхность
контакта фаз. С этой целью жидкую серу распыляют, пропуская
через форсунки. Чем меньше размеры частиц распыленной серы,
тем выше скорость процесса. В форсуночных печах (рис. 9.1),
применяемых для сжигания серы, развиваются температуры до
1400°C. Температура в печи зависит от избытка воздуха относи-
тельно стехиометрически необходимого для окисления серы до
SO2. Чем выше коэффициент избытка а, тем ниже температура
в печи и содержание SO2 в печном сернистом газе Характерная
зависимость показана на рис. 9.2. Повышение температуры выше
1400 °C недопустимо из-за разрушения футеровки и конструктив-
ных элементов печи. Содержание SO2 в газе при работе на воз-
душном дутье не превышает 15%. Интенсивность печей состав-
ляет 0,2—0,8 т/(м3-ч).
Так как по тепловому режиму форсуночные печи приближаются
к адиабатическим, то даже частичное обогащение воздуха техни-
ческим кислородом приводит к перегреву печей. Для производ-
ства высококонцентрированного сернистого газа, получаемого сжи-
ганием серы в техническом кислороде, используют двухступенча-
тые печи. На первой ступени серу испаряют, а на второй сжигают
пары серы в кипящем слое инертного материала с отводом избы-
точной теплоты теплообменными элементами, расположенными в
кипящем слое. Схема такой печи показана на рис. 9.3.
Рис. 9.3. Двухступенчатая печь для производства высоко-
концентрированного сернистого газа:
/ — барботажный испаритель серы; 2—теплообменные элементы;
3—кипящий слой инертного материала.
179
При анализе процесса горения серы и разработке математиче-
ской модели используют модель частицы с невзаимодействующим
ядром без учета образования зольной оболочки (см. разд. 5.7.2).
Сернистый газ, получаемый сжиганием серы, практически не
содержит пыль и другие примеси и может быть направлен для
окисления SO2 без предварительной очистки. Перед поступлением
в контактный аппарат газовый поток проходит через котел-утили-
затор, использующий теплоту газового потока для выработки пара.
9.4.2. Обжиг колчедана
Процесс обжига колчедана характеризуется суммарным урав-
нением:
4FeS2 + 11О2 —> 2Fe2O3 + 8SO3 + 3400 кДж
Фактически при обжиге протекает несколько реакций. Сначала
при нагревании до температуры выше 500°C происходит разло-
жение FeS2 с образованием сульфида железа и выделением паров
серы:
2FeS2 —>• 2FeS + S2 — 104 кДж
Сера интенсивно сгорает в газовой фазе по уравнению (а),
а сульфид железа окисляется
4FeO4-7O2 —> 2Fe2O3 + 4SO2
Наряду с FeS2 разлагаются и сульфиды других металлов, со-
держащихся в колчедане. Их оксиды совместно с оксидами железа
неразложившейся части FeS2 и другими примесями (например,
кварц, алюмосиликаты) образуют огарок. Содержание серы в огар-
ке'В зависимости от типа печей и режима обжига составляет от 0,3
до 3 % •
Скорость гетерогенного процесса обжига описывается уравне-
ниями, которые в общем виде можно записать в форме:
t/ = —-^- = КГудДС (9.1)
v dx 3
Здесь mSO2—масса полученного оксида серы(IV); v — объем реакционной
зоны; Гуд — удельная поверхность контакта фаз; ДС — движущая сила процесса;
К — коэффициент скорости процесса.
Конкретный вид уравнений скорости зависит от области про-
текания процесса. При низких температурах (менее 500 °C) обжиг
протекает в кинетической области, а при высоких (более 800 °C) —
в диффузионной На начальной стадии обжига процесс лимити-
руется скоростью диффузии паров серы с наружной поверхности
частиц колчедана в газовый поток, т. е. процесс протекает во
внешнедиффузионной области. По мере горения сульфидов на по-
верхности зерен образуется слой оксидов (огарок). Общая ско-
рость процесса определяется скоростью диффузии паров серы че-
рез слой огарка или скоростью диффузии кислорода внутрь ча-
стиц. Таким образом, процесс протекает во внутридиффузпонной
180
Рис. 9.4. Зависимость начальной скорости окисления колчедана от размера его ча-
стиц.
Рис. 9.5. Зависимость скорости окисления сульфидов железа в кипящем слое при
700 °C от молярной доли кислорода в дутье.
области Рассматривая процесс в целом, можно считать, что общая
скорость лимитируется скоростью диффузии газообразных компо-
нентов в порах оксидной оболочки. Процесс можно описать на
основе модели с невзаимодействующим ядром и пористой оболоч-
кой (см. разд. 5.6.2).
Так как внутридиффузионные торможения снижаются с умень-
шением толщины оксидной оболочки, то эффективным способом
интенсификации процесса обжига является уменьшение размера
обжигаемых частиц колчедана. При этом одновременно со сни-
жением диффузионных торможений (приводящем к росту К) уве-
личивается и внешняя поверхность частиц, что существенно ин-
тенсифицирует начальную стадию процесса (рис. 9.4). Для ис-
ключения внешнедиффузионных торможений линейная скорость
газового потока, обтекающего частицы колчедана, должна быть
не менее 0,1 м/с.
Для повышения коэффициента скорости К процесса эффектив-
но также увеличивать температуру. Однако при температуре выше
1000 °C материал в печи спекается, частицы оплавляются и ин-
тенсивность работы печей резко падает из-за возрастания диф-
фузионных торможений.
Для увеличения движущей силы процесса ДС целесообразно
повышать концентрацию кислорода в Зоне обжига (рис. 9.5).
В современных, сернокислотных системах для обжига колче-
дана применяют только печи с кипящим слоем (см. разд. 7.3.3 и
рис. 7.22). В печах КС процессы тепло- и массообмена, от кото-
рых сильно зависит интенсивность обжига, протекают с большой
скоростью.
Температуру в печи поддерживают в пределах 650—800°C.
Большое количество теплоты, выделяющееся в процессе обжига,
отводят с помощью охлаждающих элементов, расположенных не-
посредственно в кипящем слое. Это позволяет интенсифицировать
работу печей путем обогащения воздуха техническим кислородом.
При этом молярная доля оксида серы(IV) может быть увеличена
с 12—14 до 60—70 %.
Около 90 % образующегося огарка уносится потоком обжи
гового газа. Эта пыль выделяется сначала в котле-утилизаторе.
181
Рис. 9.6. Схема печного отделения производства серной кислоты?
1— нагнетатель; 2—печь КС; 3—котел-утилизатор; 4—циклон; 5—сухой электрофильтр.
в циклоне, а затем в электрофильтре. Крупные частицы огарка от-
водятся в бункер через отверстия в газораспределительной ре-
шетке. Схема установки для сжигания колчедана в кипящем слое
показана на рис. 9.6.
Остаточное содержание серы в огарке составляет 0,5—1,0%.
После выжигания серы и агломерации огарок используют для вы-
плавки чугуна.
В сернистом газе, полученном после обжига колчедана, содер-
жатся примеси, присутствие которых недопустимо из-за отравле-
ния катализатора при окислении оксида серы (IV). К этим при-
месям прежде всего относятся соединения мышьяка и фтора. Кро-
ме того, в газе содержатся ценные примеси — селен, теллур и др.
Поэтому обжиговый газ подвергают специальной очистке. Схема
очистительного отделения показана на рис. 9 7 Очистка coci оит
из следующих операций: 1) осаждение огарковой пыли в циклоне 1
(содержание пыли снижается с 300 до 10—20 г/м3); 2) очистка f;
газа в 4—5-секционном электрофильтре 2 до остаточного содержа-
ния пыли 0,05—0,1 г/м3; 3) двухступенчатая промывка газа 50 %-
и 15 %-ной серной кислотой в башнях 3 и 4, сопровождающаяся J
образованием мельчайших капелек серной кислоты (туман) и рас-
творением в них примесей; 4) улавливание тумана в электрофильт-
ре 5; 5) осушка газа в насадочной сушильной башне 6, орошаемой
93—95 % -ной серной кислотой.
После очистки и осушки сернистый газ поступает на окисление
в контактный аппарат.
Рис. 9.7. Схема очистительного (промывного) отделения:
1—цчклои; 2—сухой электрофи«1ьтр; 3 — первая промывная башня; 4 — вторая пром л злая
башня; 5—мокрый электрофильтр; 6 — насадочная башня; 7—гззэдувка; 8^холодильники
кислоты; 9 — сборники кислоты.
182
9.5. ОКИСЛЕНИЕ ОКСИДА СЕРЫ (IV]
Окисление оксида серы(IV) в оксид серы(VI) —основной про-
цесс в производстве серкой кислоты. Окисление проводят после
тщательной очистки газа от пыли, тумана серной кислоты, кон-
тактных ядов и осушки. Контактное окисление является типичным
примером гетерогенного экзотермического катализа. Оно протекает
по уравнению (б). Тепловой эффект q2 реакции зависит от тем-
пературы. В интервале 400—700 °C q2 (в Дж/моль) можно рас-
считать по формуле:
q2= 101 420 — 9.26Г (9.2 j
Равновесие реакции окисления SO2 в соответствии с~принци-
пом Ле Шателье сдвигается в сторону образования SO3 при пони-
жении температуры и повышении давления. Равновесная степень
превращения X* реагентов зависит также от соотношения SO2 и О2
в газе, которое, в свою очередь, зависит от вида обжигаемого
сырья и избытка кислорода. Характерные зависимости Xso2 от
температуры, давления и состава газовой смеси приведены на
рис. 9.8—9.10.
Константа равновесия реакции окисления SO2 выражается со-
отношением:
Хр = ^Оз/[^о2Ш°-6| О.З)
Зависимость К₽ от температуры, вычисленная по уравнению
Вант-Гоффа (3.53), показана на рис. 9.11.
Гомогенное некаталитическое окисление оксида серы(IV) про-
текает столь медленно, что в производственных масштабах его
осуществлять нецелесообразно. Энергия активации гомогенного
окисления Е т 300 кДж/моль. Поэтому процесс проводят в при-
сутствии катализаторов. В сернокислотной промышленности в
разное время применяли лишь три вида катализаторов, основу
которых составляли металлическая платина, оксиды железа и
оксид ванадия(V). Самыми активными катализаторами являются
платиновые (Е т 70 кДж/моль). Температура, при которой ката-
лизатор проявляет заметную активность (температура зажигания),
Рис. 9.8. Влияние температуры на равновесную степень окисления SO2 для газа,
содержащего 7 % SO2. 11 % О2, 82 % N2 (Р = 0,1 МПа).
Рис. 9.9. Зависимость равновесной степени превращения от давления для газа,
содержащего 7 % SOa, 11 % О2 и 82 % N2.
183
КР
Рис. 9.10. Зависимость равновесной степени превращения XSO2 от состава газо-
вой смеси, получаемой при обжиге колчедана (Г = 475 °C, Р — 0,1 МПа).
Рис. 9.11 Зависимость константы равновесия Кр от температуры (Р=0,1 МПа).
составляет 350 °C. Однако из-за высокой чувствительности к кон-
тактным ядам, в частности к мышьяковистым соединениям, эти
катализаторы уже более 40 лет не применяют.
Катализаторы на основе оксидов железа (их вводят в виде
колчеданного огарка) практически не отравляются, но их энергия
активации велика (Е « 150 кДж/моль), и катализатор'проявляет
достаточно высокую активность лишь при температуре выше
625 °C. При такой температуре равновесная степень превращения
не превышает 70%, и использование катализаторов на основе
оксидов железа возможно лишь для так называемого предвари-
тельного окисления сернистого газа со степенью превращения
50—60%.
Ванадиевые катализаторы содержат 5—10 % V2O5 и 5—10 %
КгО. Энергия активации в рабочем диапазоне температур 420—
530°C составляет в среднем около 90 кДж/моль. Температура
зажигания 390—410°С. В условиях работы контактная масса пред-
ставляет собой пористый носитель, внутренняя поверхность кото-
рого смочена пленкой раствора V2O5 в расплаве пиросульфата
калия. Носителями, как правило, служат высокопористые алюмоси-
ликаты. Ванадиевую контактную массу выпускают в виде цилинд-
рических гранул, таблеток, колец, шариков и др. Размеры гранул
катализатора имеют большое значение для процесса катализа.
Чтобы исключить внутридиффузионные торможения (см. разд. 5.3
и 5.5.2) при окислении промышленных газов с' содержанием SO2
7—11 % в диапазоне температур 500—550°С размеры гранул ка-
тализатора не должны превышать 1—1,5 мм. Однако при исполь-
зовании мелкозернистых катализаторов создается большое гидрав-
лическое сопротивление газовому потоку, проходящему через не-
подвижные (фильтрующие) слои катализатора. Это приводит к
неоправданно высоким энергозатратам. Поэтому при проведении
процессов в фильтрующих слоях применяют катализаторы с раз-
мерами гранул 5—10 мм. При таких размерах для начальных и
средних стадий окисления степень использования внутренней по-
.верхности гранул (см. разд. 5.5.2) не превышает 30—50%.
Кинетическое уравнение, широко применяемое технологами
при расчетах процесса окисления SO2 в SO3, выведено Г. К- Бо-
184
ресковым:
где
г > _ dX Робщ I — X Г___________X2______
U- dr-R /VSO2 1 - 0.2Х [Р Ро3шКр (1 - Л)2
(9.4)
(9.5)
Здесь X — степень превращения, доли единицы; т — время контаитирования,
с; k — константа скорости реакции, с-1 -Па-1; ^so2, ^о2— молярные доли SO2 и
О2 в исходной газовой смеси, %; Ро6ш — общее давление, Па; Кг—константа
равновесия, Па-0-5.
Из уравнений (9.4) и (9.5) следует, что скорость процесса
сильно зависит от константы скорости реакции k, увеличиваю-
щейся с ростом температуры в соответствии с уравнением Арре-
ниуса (4.11).
С увеличением температуры от 400 до 500 °C константа
скорости возрастает более чем в 30 раз. Однако при этом уменьша-
ется константа равновесия КР. Противоположное влияние темпе-
ратуры на k и Кр обусловливает экстремальный характер зависи-
мости U от Т. Из уравнения (9.4) следует, что скорость окисле-
ния SO2 тем выше, чем меньше достигаемая степень окисления.
Характерная серия кривых, выражающих зависимость скорости
реакции от температуры и степени превращения для газовой сме-
си, содержащей 7 % SO2 и 11 % О2, показана на рис. 9.12.
Линия АА, соединяющая максимумы, характеризует оптималь-
ный температурный режим проведения процесса. Линии ВВ и СС
ограничивают область допустимых колебаний температуры, в пре-
делах которой скорость реакции составляет не менее 90 % от мак-
симальных значений при оптимальных температурах.
Оптимальная температура зависит не только от X, но и от
состава газовой смеси (рис. 9.13).
Рис. 9.12. Зависимость относительной скорости окисления SO2 от температуры
при различных степенях превращения ASO2-
Рис. 9.13. Зависимость оптимальной температуры от степени превращения SO2
при различном составе газа:
1 — 7% SC2 и 6,0% О2: 2—7% SO2 и 14% О2.
185
Рис. 9.14. Диаграмма Х—Т для окисления
SO2 в пятислойном контактном аппарате с
промежуточным теплообменом.
В современных сернокислотных системах перерабатывают сер-
нистые газы с содержанием SO2 не более 12%. При такой кон-
центрации адиабатический разогрев контактной массы относитель-
но невелик, и окисление оксида серы (IV) можно проводить при
адиабатическом температурном режиме в контактных аппаратах
с фильтрующими слоями (см. рис. 6.52).
В соответствии ' с линией оптимальных температур (см.
рис. 9.13) процесс следует начинать с высокой температуры и по-
нижать ее по мере роста степени превращения. Адиабатический
температурный режйм,_однако, это выполнить не позволяет, так
как с увеличением степени превращения температура в слое воз-
растает. Для приближения температурного режима к оптималь-
ному газовую смесь после нагрева до определенной температуры
выводят из" слоя на охлаждение, а затем подают в следующий
слой катализатора и т. д. На практике газ нагревают до темпе-
ратуры, несколько превышающей температуру зажигания катали-
затора, и направляют в 1-й слой контактной массы. Так как
в аппаратах с фильтрующими слоями каждый слой работает в адиа-
батическом температурном режиме, то по мере окисления SO2 тем-
пература растет вследствие выделения теплоты. Процесс проводят
до тех пор, пока температура не превысит оптимальную, но при
этом не станет слишком близкой к равновесной. Обычно превы-
шение температуры над оптимальной выбирают с таким расчетом,
чтобы скорость реакции составляла не менее 70—80 % от макси-
мальной. Затем газовую смесь охлаждают в промежуточном теп-
лообменнике до такой температуры, чтобы процесс на следующей
полке шел с начальной скоростью, составляющей не менее 70—
80 % от максимальной. После второго слоя газ опять охлаждают
и подают на третий слой и т. д. На рис 9.14 изображена диа-
грамма ' X— Т, характеризующая протекание процесса в пятипо-
лочном контактном аппарате с фильтрующими слоями катали-
затора.
Конечная максимальная степень превращения X макс на выходе
из последнего слоя ограничивается равновесием, соответствующим
минимальной температуре, при которой катализатор сохраняет
активность. Для существующих катализаторов X макс в системах
без промежуточной адсорбции SOs составляет 98—98,4 %.
les
С повышением концентрации SO2 в исходном газе появляется
необходимость увеличения числа слоев катализатора. Это связано
с ростом адиабатического 'разогрева при увеличении концентра-
ции оксида серы (IV). Как рассмотрено в разд. 6.2.3, повышение
температуры при адиабатическом процессе выражается уравне-
нием:
+ (9.6)
Коэффициент адиабатического разогрева А применительно
к процессу окисления SO2 примерно прямо пропорционален его
начальной концентрации [см. уравнение (6 12)]. Если выражать
степень окисления SO2 в долях единицы, то значения X для раз-
личных молярных долей SO2 составят:
tfso? % 2 5 8 10 14 20 62,5
1 58 144 225 279 373 503 1234
При окислении высококонцентрированных газов с содержанием
SO2 более 15 % использование контактных аппаратов с фильтрую-
щими слоями нецелесообразно из-за большого числа полок и
опасности перегрева катализатора в условиях адиабатического
проведения процесса. Отводить же избыточную теплоту непосред-
ственно из слоев катализатора невозможно вследствие низкой теп-
лопроводности контактной массы и неизбежности затухания ката-
лизатора в области расположения теплообменных элементов.
В этом случае следует применять контактные аппараты с ки-
пящими слоями катализатора (см. разд. 6.9.2 и рис. 6.59). Высо-
кая теплопроводность кипящих слоев позволяет отводить избы-
точную теплоту реакции непосредственно из реакционной зоны.
При этом становится возможным окисление сернистых газов с мо-
лярной долей SO2 до 60 %. Независимость гидравлического сопро-
тивления от размера частиц катализатора и линейной скорости
газа обусловливает использование мелкозернистого катализатора
и проведение процесса'с высокой скоростью в кинетической обла-
сти. Высокая пылепропускная способность кипящего слоя дает
возможность перерабатывать запыленные газы без их предвари-
тельной тонкой очистки.
Возможность подавать газ с температурой ниже температуры
зажигания катализатора и изотермический температурный режим
позволяют достигать высоких степеней окисления при малом числе
полок (см. рис. 6.60 и 6.61).
Все катализаторы, применяемые для фильтрующих слоев,
практически непригодны для эксплуатации в условиях кипящих
слоев ввиду их быстрой истираемости. Для аппаратов с кипящими
слоями используют износоустойчивые ванадиевые катализаторы на
сферических алюмосиликатных носителях с размерами частиц
0,5—1,5 мм.
Из-за перемешивания газа и проскока его части в пузырях
максимальная степень превращения оксида серы(IV) в аппаратах
187
с кипящими слоями при отсутствии промежуточной абсорбции ок-
сида серы(VI) не превышает 97—97,5 %•
Для повышения степени окисления как при использовании ап-
паратов с фильтрующими слоями, так и аппаратов КС применяют
метод двойного контактирования. Сущность его состоит в том,
что окисление SO2 проводят в два этапа. Сначала SO2 окисляют
на 2—3-х полках до степени превращения X] = 90—95 % > затем
абсорбируют полученный SO3, а непрореагировавшую часть SO2
окисляют еще на 1—2-х полках до Х2 = 90—95 % с последующим
выделением SO3 во второй ступени абсорбции. Промежуточный
отвод продуктов реакции повышает движущую силу процесса,
увеличивает его скорость и достигаемую степень окисления. Об-
щую степень окисления ХОбщ можно рассчитать по формуле:
Хо6ш = Х, + (1 - X,) Х2 (9.7)
Общая степень превращения в системах с промежуточной аб-
сорбцией составляет 99—99,8 %.
9.6. АБСОРБЦИЯ ОКСИДА СЕРЫ (VI)
Абсорбция оксида серы (VI) является заключительной стадией
производства серной кислоты. Это типичный гетерогенный про-
цесс, протекающий в системе газ — жидкость. Абсорбция SO3
протекает в два этапа. На первом этапе оксид серы (VI) раство-
ряется в серной кислоте, на втором — взаимодействует с водой,
содержащейся в кислоте:
nSO8 + Н2О —> H2SO« + (n — 1)SO3
I
U
,3
л!
г.»
м
1
При п < 1 получают водный раствор серной кислоты, т. е.
разбавленную кислоту, при п = 1 образуется моногидрат
(100 %-ная серная кислота), при п > 1 — олеум.
Наибольшей способностью к поглощению газообразного оксида
cepbi(VI) обладает 98,3%-ная серная кислота. При содержании
H2SO4 ниже 98,3 % равновесная концентрация SO3 над серной
кислотой практически ровна нулю, а равновесное давление паров
воды велико. С поверхности серной кислоты происходит испаре-
ние молекул воды, которые препятствуют потоку SO3 к поверхно-
сти раздела фаз. При соединении воды и SO3 в газовой фазе по-
лучают пары серной кислоты по уравнению (в) с последующим
образованием сернокислотного тумана. Туман серной кислоты
трудно достаточно полно уловить, и значительная часть серной
кислоты уносится отходящими газами в атмосферу. При содержа-
нии серной кислоты более 98,3 % максимально возможная степень
абсорбции определяется равновесным давлением SO3. Поэтому
с повышением концентрации кислоты степень абсорбции умень-
шается.
Таким образом, независимо от температуры абсорбции опти-
мальное содержание серной кислоты равно 98,3 % и кинетику
188 t
Рис. 9.15. Зависимость степени абсорбции SO3 от молярной доли серной кислоты
в растворе и температуры.
Рис. 9.16. Зависимость степени абсорбции SO3 олеумом от температуры и моляр-
ной доли SOj в газе.
Рис. 9.17. Зависимость максимального содержания свободного оксида серы(У1)
в олеуме от температуры (молярная доля SO3 в газе 7 %).
абсорбции можно описать с помощью общего уравнения массо-
переноса:
77 = Ka6FAP (9.8)
Здесь Кае — коэффициент абсорбции; ДР — движущая сила процесса.
Коэффициент абсорбции Каб сильно зависит от линейной ско-
рости газа w и интенсивности перемешивания I в зоне контакта
фаз. С ростом w и I интенсифицируются процессы обновления по-
верхности контакта фаз (см. разд. 5.7.3). Поверхность контакта
фаз F зависит от размера элементов насадки в насадочных аппа-
ратах, размера капель в аппаратах распыливающего типа и др.
(см. разд. 6.8.1). Максимальные значения F достигаются при пен-
ном режиме взаимодействия жидкой и газовой фаз. В этом случае
обеспечиваются и благоприятные условия для интенсивного обнов-
ления поверхости контакта, а следовательно, и для роста Каб.
Движущая сила процесса ДР определяется разностью парци-
альных давлений SO3 у поверхности контакта фаз (оно равно
равновесному давлению) и в потоке газа, поступающего на аб-
сорбцию. С ростом температуры движущая сила падает и ско-
рость абсорбции, а следовательно, и степень абсорбции умень-
шаются. На рис. 9.15 показано характерное влияние содержания
H2SO4 и температуры на степень абсорбции SO3.
На практике абсорбцию проводят при температуре ниже 80°C.
При абсорбции оксида серы (VI) олеумом максимальная сте-
пень абсорбции и содержание свободного SO3 в получаемом про-
дукционном олеуме зависят от начальной концентрации SO3 в
газе и температуры. С ростом температуры увеличивается парци-
альное давление SO3 над жидкой фазой. Движущая сила и сте-
пень абсорбции при этом уменьшаются. Максимальное содержание
SO3 в олеуме определяется равновесным давлением SO3 над жид-
кой фазой. Зависимость степени абсорбции SO3 олеумом от
189
Рис. 9.18. Схема абсорбционного от-
деления:
/ — теплообменник для охлаждения газа;
2—олеумный абсорбер; 3—моногидратный
абсорбер; 4—кислотные холодильники;
5—сборники; 6 — фильтр.
температуры и содержания ок-
сида серы (VI) в газе показана
на рис. 9.16.
На рис. 9.17 представлены
данные о максимальном содер-
жании свободного SO3 в олеу-
ме, достигаемом при различ-
ных температурах.
Для поглощения SO3 в со-
временных сернокислотных си-
стемах используют два после-
довательно соединенных абсор-
бера насадочного типа (см.
разд. 6.8.1 и рис. 6.31). Схема абсорбционного отделения показана
на рис. 9.18. Первый абсорбер 2 орошается 17—20 %-ным олеумом,
второй 3— 98,3 %-ной серной кислотой. Реакция образования
H2SO4 сопровождается выделением большого количества теплоты.
Для отвода ее устанавливают кислотные холодильники 4. Степень
абсорбции при нормальном концентрационном и температурном
режимах, как правило, не ниже 99,9 %•
Перспективным направлением интенсификации процесса аб-
сорбции SO3 является применение пенных аппаратов, которые
обеспечивают не только развитую поверхность контакта фаз, но
и возможность отвода избыточной теплоты с помощью холодиль-
ных элементов, расположенных непосредственно в реакционной
зоне.
9.7. ХИМИКО-ТЕХНОЛОГИЧЕСКИЕ СИСТЕМЫ
ПРОИЗВОДСТВА СЕРНОЙ КИСЛОТЫ
Как отмечалось выше, основное количество серной кислоты
получают из серы и колчедана. Принципиальное различие техно-
логических схем производства кислоты из этих видов серусодер-
жащего сырья заключается в отсутствии промывного отделения
в системах, использующих серу. Поэтому в качестве примера
рассмотрим только современную сернокислотную систему, рабо-
тающую на колчедане (рис. 9.19).
Колчедан из бункера подают в печь КС 2 для обжига. Полу-
ченный сернистый газ содержит 13—14 % SO2 и при 800—850 °C
поступает в котел-утилизатор 3, где охлаждается- до 430—470 °C.
Избыточную теплоту расходуют для выработки пара. Далее газ
1W
Отходящие
газы
Рис. 9.19. Технологическая схема производства серной кислоты из колчедана по
методу двойного контактирования (ДК):
1 — воздуходувка; 2—печь КС; 3—котел-угилизатор; 4—циклон; 5—сухой электрофильтр;
б, 7—промывные башни; 8—мокрые электрофильтры; 9—сушильная башня; 10—брызгоуловн-
телн; /7—-отдувочная башня; 12—газодувка; 13—теплообменники; 14—контактный_аппарат;
15—олеумный абсорбер; 16, /7—моногндратные абсорберы; 18—холодильники; 19—насосы;
20 — сборники кислоты.
поступает для очистки от пыли в циклон 4 и сухой электро-
фильтр 5, а затем — в промывное отделение, где в башнях 6, 7
очищается от мышьяка и фтора. В промывных башнях при охлаж
дении газа содержащийся в нем оксид серы (VI), соединения
мышьяка, фтора, селена переходят в туманообразное состояние.
Для выделения тумана газ направляют в мокрые электрофиль-
тры 8. Поскольку промывные башни орошаются разбавленной
кислотой, то газ для осушки подают в сушильную насадочную
башню 9, орошаемую концентрированной серной кислотой. После
осушки и улавливания брызг в волокнистом фильтре 10 газ на-
гревают до необходимой температуры (390—420°C) и подают б
контактный аппарат 14, в котором, проходя через 4—5 полок с ка-
тализатором, оксид серы (IV) окисляется до оксида серы (VI).
Теплоту реакции окисления используют для нагрева газа, посту-
пающего из сушильной башни. Нагрев осуществляют в выносных
теплообменниках 13.
В системах небольшой мощности (до 500 т кислоты в сутки)
газовая смесь после окисления SO2 в контактном аппарате на
98—98,2 % поступает в абсорбционное отделение, после которого
непрореагировавшая часть оксида серы (IV) вместе с другими
компонентами выбрасывается в .атмосферу. При этом около 7 т
SO2 в сутки безвозвратно терялось, нанося одновременно окру-
жающей среде большой ущерб. В последние годы строят сернокис-
лотные системы с двойным контактированием и промежуточной
абсорбцией (системы ДК). Как отмечалось выше, в этих системах
после окисления оксида серы(IV) на двух-трех слоях катализа-
тора на 90—95 % газ направляют на первую (промежуточную)
191
абсорбцию оксида серы (VI) в олеумный 15 и моногидратные
абсорберы 16, 17. Затем газ снова нагревают и направляют
на окисление еще на один-два слоя катализатора контактного ап-
парата и далее на вторую стадию абсорбции в моногидратный
абсорбер 17. В результате отвода SO3 из газовой смеси на первой
стадии абсорбции изменяется состав газа, и равновесие реакции
сдвигается в сторону более высоких степеней окисления оксида
серы (IV).
В системах ДК обеспечивается степень окисления SO2 до
99,8%. При этом содержание SO2 в выхлопных газах снижается
по сравнению с системами с одинарным контактированием в
10 раз и составляет 0,02—0,03%. Увеличение скорости окисления
за счет повышения движущей силы процесса позволяет, не изме-
няя загрузки катализатора и размеров контактного аппарата,
окислять до высокой степени превращения газ с молярной долей
SO2 до 11—12%. Благодаря возрастанию содержания сернистого
газа мощность сернокислотных систем повышается на 20—25%.
Современные системы ДК имеют мощность 1000 т H2SO4 в сутки
при работе на колчедане и до 1500 т/сут при работе на сере. При
этом диаметр контактных аппаратов составляет 14—17 м, высо-
та— до 30 м, общая масса — до 1000 т.
9.8. ОСНОРНЫЕ НАПРАВЛЕНИЯ ИНТЕНСИФИКАЦИИ
СЕРНОКИСЛОТНЫХ ПРОИЗВОДСТВ
Наиболее эффективные направления развития производства
серной кислоты связаны с повышением концентрации оксида се-
ры (IV), проведением процессов под давлением, применением тех-
нического кислорода на стадии обжига и окисления SO2, исполь-
зованием высокоинтенсивных реакторов с кипящими слоями, но-
вых катализаторов, организацией производства по новым схемам,
в том числе с рециркуляцией газовой смеси. Между этими фак-
торами существует следующая причинно-следственная связь. По-
вышение крнцентрации SO2 пропорционально увеличивает произ-
водительность контактного и абсорбционного отделений при сни-
жении энергозатрат и потерь теплоты. Однако окисление высоко-
концентрированного газа возможно лишь в реакторах с кипящими
слоями катализатора, работающих при изотермическом темпера-
турном режиме. Пылепропускная способность кипящего слоя по-
зволяет резко упростить систему очистки газа, а его высокие
теплотехнические свойства обеспечивают наиболее полное исполь-
зование энергоресурсов производства. Получение же концентриро-
ванного газа возможно лишь при обогащении воздушного дутья
кислородом или полной замене воздуха техническим кислородом.
В последнем случае интенсивность основных аппаратов может
быть увеличена в 5—7 раз и появляется возможность замены мно-
гостадийных схем на одностадийную с циркуляцией непрореаги-
ровавшего за один проход газа, что резко уменьшает металлоем-
192
рис. 9.20. Принципиальная схема циркуляционного
контура сернокислотной системы:
/—печь сжигания серусо держащего сырья; 2—инженеры,
3—контактный аппарат с кипящим слоем катализатора;
4 — охладитель КС; 5—пенный абсорбер; 6—хвостовая
установка.
кость системы. Повышение рабочего дав-
ления увеличивает скорость всех про-
цессов, позволяет работать при высоких
температурах с обеспечением требуемой
конверсии и наиболее эффективного ис-
пользования теплоты. Вследствие умень-
шения объемов газа пропорционально
снижаются размеры всех основных аппаратов. Комплексное при-
менение всех этих факторов обеспечивает выполнение задачи соз-
дания высокоинтенсивного, практически, безотходного производства
большой единичной мощности.
Принципиальная схема энерготехнологической циркуляционной
системы, включающей однополочный контактный аппарат КС 3,
охладитель газа 4 с кипящим слоем инертного материала, устанав-
ливаемый с целью повышения эффективности использования энер-
горесурсов, абсорбер с пенными слоями 5, инжектор 2 для орга-
низации циркуляции газовой смеси, показана на рис. 9.20. Кон-
тактный аппарат 3 с теплообменной насадкой, расположенной в
кипящем слое, одновременно выполняет функции испарительной
секции ко^ла-утилизатора. Секции пароперегревателя размещены
в печи /. Функции подогревателя исходной воды выполняет теп-
лообменная насадка, расположенная в охладителе 4. При работе
под давлением 1—1,5 МПа для обеспечения мощности 2500 т кис-
лоты в сутки диаметр основных аппаратов может не превышать
3,5 м. Интенсивность работы оборудования в результате комплекс-
ного воздействия всех рассмотренных выше факторов возрастает
в 30—50 раз. Степень превращения оксида серы (IV) составляет
99,9 % без учета работы малогабаритной хвостовой установки 6.
Общая же степень превращения 99,98 %. Выбросы оксидов серы
снижаются до 0,04 кг на 1 т кислоты, что более чем в 50 раз
меньше, чем у существующих систем ДК.
глава ю
ПРОИЗВОДСТВО АММИАКА
10.1. ОБЩИЕ СВЕДЕНИЯ ОБ АММИАКЕ.
ЗНАЧЕНИЕ СОЕДИНЕНИЙ АЗОТА
Аммиак NH3 — бесцветный газ с резким запахом, действует
раздражающе на слизистую оболочку, токсичен. При температуре
—33,19°С кипит, а при —77,75°С затвердевает. Растворяется в
воде с образованием аммиачной воды. При обычной температуре
7 Зак. 1869
193
устойчив, а при 300 °C в присутствии катализатора диссоциирует
на водород и азот,. Аммиак реакционноспособен, в частности на
катализаторах окисляется до оксида азота. В смеси с воздухом
(15—28 % по объему) взрывоопасен.
Аммиак имеет важное значение в народном хозяйстве. В основ-
ном его используют для производства азотной кислоты и мине-
ральных удобрений. Жидкий аммиак сам является высококон-
центрированным азотным удобрением. Его также применяют в
производстве азотсодержащих солей, мочевины, соды, синильной
кислоты, взрывчатых веществ. Жидкий аммиак и его растворы ис-
пользуют в качестве хладоагентов. Производство аммиака отно-
сится к крупнотоннажным: оно развивается высокими темпами,
значительно опережающими средние темпы промышленности
в целом.
Азот — один из важных элементов (наряду с углеродом, во-
дородом, кислородом), без которых невозможно существование
живой и растительной клетки. Элементарный азот не усваивается
растениями. Пополнение запасов азота в почве в виде минераль-
ных азотных удобрений представляет исключительно важную за-
дачу, от решения которой во многом зависит благосостояние об-
щества. Источников связанного азота, имеющих промышленное
значение, в природе крайне мало. В Чили и Южной Африке име-
ются крупные запасы натриевой селитры, истощающиеся в резуль-
тате интенсивной добычи. Основным же неисчерпаемым источни-
ком азота служит воздух. Вследствие большой инертности азота
долгое время не удавалось найти способы соединения его с дру-
гими элементами. Инертность азота объясняется наличием проч-
ной тройной связи между атомами азота в молекуле (N=N).
10.2. СПОСОБЫ ПРОИЗВОДСТВА
Существует три метода получения связанного азота: дуговой,
цианамидный и аммиачный. По первым двум получают соединения
азота в виде NO и CaCN2. Методы отличаются большими затра-
тами энергии и вытеснены аммиачным методом, в основе кото-
рого лежит прямой синтез аммиака из азота и водорода
по реакции (при 500°C и 30 МПа):
3H2 + N2 2NH3 + q (1)-
Для проведения данной реакции предварительно получают азот
и водород с последующим приготовлением азотоводородной смеси.
10.3. СЫРЬЕ ДЛЯ СИНТЕЗА
Источником сырья для получения азота является ат-
мосферный воздух, в котором объемная доля азота составляет
78,05 %. Ресурсы воздуха практически неограниченны.
194
Сырье для производства водорода отличается боль-
шим разнообразием: природный и попутные газы, коксовый газ,
кокс, уголь и др. Основным видом сырья служат природный и по-
путные газы, на их долю в настоящее время приходится 92 %
всего производимого аммиака. В перспективе будет осуществ-
ляться переход на уголь.
10.3.1. Получение азота
Азот получают из воздуха двумя методами: ректификацией
сжиженного воздуха и конверсией метана в присутствии кисло-
рода воздуха.
Ректификация сжиженного воздуха основана на
различии температур кипения азота и кислорода, входящих в со-
став воздуха: азот кипит при —195,6°C, кислород — при —182,8 °C.
Разделение воздуха проводят в ректификационных колоннах.
Конверсия метана в присутствии кислорода
воздуха позволяет одновременно получать водород и азот, ко-
торые, смешиваясь образуют азотоводородную смесь, непосред-
ственно участвующие в синтезе аммиака. Этот метод является
экономически выгодным и находит все большее распространение.
10.3.2. Получение водорода
Водород может быть получен несколькими методами: 1) кон-
версией метана с водяным паром и воздухом (и кислородом);
2) крекингом метана; 3) электролизом воды; 4) разделением кок-
сового газа. В этом разделе рассмотрен метод двухступенчатой
каталитической конверсии метана природного газа с водяным па-
ром и кислородом (или воздухом), поскольку он является веду-
щим в промышленности. Метод представляет также наибольший
интерес с точки зрения обсуждения закономерностей протекания
химико-технологических процессов и аппаратурного оформления.
В основе конверсии метана лежат следующие реакции:
СН4 -|- Н2О СО -|- ЗН2 — 206,4 кДж (2)
СН4 + 0,5О2 СО + 2Н2 + 36,6 кДж (3
Источником метана служит природный газ, в котором 98 %
составляет метан, остальное — этан и пропан (в попутном нефтя-
ном газе метан присутствует несколько в меньшем количестве.
Примеси этана и пропана участвуют в реакциях аналогично ме-
тану. В качестве окислителей используют водяной пар и кислород.
Последний добавляют для компенсации теплоты, _поглощаемой
при конверсии метана.
Образующийся по реакциям (2) и (3) оксид углерода конвер-
тируется водяным паром:
СО + Н2О СО2 + Н2 + 41.0 кДж (4)
Все три реакции обратимы. Для каждой из них существует
определенное равновесное соотношение между концентрациями
7* 19а
веществ, которое при постоянной температуре остается неизмен-
ным и определяется константой равновесия:
v _ ₽сорна v рсорнг . PCOS₽H2
Л/>- 1 — 7 р ’ 2 —р Р5^’ Кр- 3 — р р
ГСН4ГН2О rCH/O2 rCOFH->O
Константа равновесия реакции (3) при температурах в инте-
ресующих нас пределах так велика, что практически реакция идет
вправо до конца. Реакции (2) и (3) идут с увеличением числа
молей, реакция (4)—без изменения числа молей. Суммарный про-
цесс конверсии метана — эндотермический.
На практике широко внедряется двухступенчатая каталитиче-
ская конверсия метана с применением в качестве окислителей во-
дяного пара и воздуха (вместо чистого кислорода). На первой
ступени конверсию проводят водяным паром в трубчатом реакторе
при 800 °C со степенью конверсии метана 90%. На второй сту-
пени конверсию остаточного метана осуществляют с воздухом
в шахтном реакторе при 1000 °C В конвертированном газе содер-
жится 0,3 % СН4. При одноступенчатой конверсии в качестве
окислителя применяют водяной пар и воздух, обогащенный кисло-
родом до 40—50%. Таким образом, в двухступенчатой конверсии
отпадает необходимость в сооружении дорогостоящей и энерго-
емкой установки для получения кислорода, что в значительной сте-
пени улучшает экономические показатели производства по сравне-
нию. с одноступенчатой каталитической и высо <отемпературной
конверсией (где большой расход энергии на создание высоких
температур 1350—1400°C). Кроме того, использование воздуха
в.качестве окислителя, позволяет получить конвертированный газ
с содержанием азота (поступающего с воздухом) в таком коли-
честве, которое необходимо для получения азотоводородной смеси
для синтеза аммиака, т. е. 75 % водорода 25 % азота.
Конвертированный газ наряду с азотом и водородом пример-
но содержит (%) следующие примеси: СО2 — 30, СО — 0,2 4-0,4,
СН4 — 0,5, Аг — 0,5, H2S — следы. Для синтеза аммиака необхо-
дима возможно более полная очистка азотоводородной смеси от
кислород- и серусодержащих соединений, являющихся каталити-
ческими ядами. Очистку осуществляют различными методами:
абсорбционными, адсорбционными, каталитическими и т. д.
Конверсия метана. Исходя из принципа Ле Шателье для до-
стижения максимального выхода водорода теоретически необхо-
димы следующие условия при ко- версии метана: понижение дав-
ления, повышение температуры и. избыток водяного пара по
сравнению со стехиометрическим количеством.
На практике конверсию проводят в основном при повышенном
давлении (2—3 МПа), несмотря на то, что содержание водорода
уменьшается с увеличением давления (равновесие смещается вле-
во). Процесс конверсии выгодно проводить при повышенном дав-
лении, так как в этом случае увеличивается скорость реакции
(в результате возрастания концентрации реагирующих веществ),
а кроме того, используется естественное давление природного
газа, с которым он подается на завод. Это влияет на уменьшение
объема аппаратов и трубопроводов .и сокращение капиталовло-
жений при строительстве завода. Помимо этого, снижается расход
электроэнергии на сжатие конвертирозанного газа перед следую-
щей стадией синтеза аммиака, проводимой при повышенном дав-
лении.
По температурному режиму различают два вида конверсии
метана: высокотемпературную при 1350—1400°С без катализатора
и каталитическую конверсию при 800—1000°С в присутствии ка-
тализатора. Для создания высоких температур и проведения эн-
дотермической реакции (2) необходимо подводить теплоту извне.
В целях экономии процесс осуществляют, сочетая конверсию ме-
тана с водяным паром и кислородом в соответствии с реакциями
(2) и (3), т. е. с использованием эндотермического и экзотерми-
ческого процессов.
Применение каталитической конверсии имеет преимущества
перед высокотемпературной. Катализатор ускоряет реакцию, сни-
жает температуру процесса и подавляет побочную реакцию СН4->-
->С4-2Н2, т. е. предотвращает образование технического угле-
рода и дополнительную стадию его очистки. В качестве катализа-
тора применяют никель, нанесенный на оксид алюминия или оксид
магния (катализатор ГИАП-3 для низкого давления, ГИАП-5 для
высокого давления). Процесс протекает во внутридиффузионной
области. Катализатор применяют в форме цилиндров диаме-
тром 8—12 мм и высотой 9—12 мм или в форме колец с на-
ружным диаметром от 8 до 20 мм и той же высоте. Это позво-
ляет уменьшить гидродинамическое сопротивление системы по
сравнению с тонкоизмедьченным катализатором и, таким об-
разом, ускорить протекание процесса во внутридиффузионной об-
ласти.
Для увеличения выхода водорода (смещения равновесия впра-
во) конверсию проводят с избытком водяного пара в соотношении
СН4: Н2О = 1:2 от стехиометрического количества.
При выборе условий для проведения конверсии метана важно
учитывать также состав конвертированного газа на выходе. Не-
обходимо, чтобы остаточное содержание метана в нем не превы-
шало 0,5 % (по объему), так-к-ак метан, постепенно накапливаясь
в агрегатах на стадии синтеза аммиака, затрудняет проведение
в них процесса. Содержание метана в конвертированном газе за-
висит от давления, температуры и соотношейия СН4: Н2О. Опти-
мальный режим для содержания метана не более 0,5 % соответ-
ствует температуре 800—1000 °C, давлению 0,1 МПа и соотноше-
нию СН4: Н2О =1:2.
Конверсию метана проводят в реакторах двух типов: трубча-
тых и шахтных.
Трубчатый реактор (трубчатая печь) (рис. 10.1)—аппарат,
в трубках которого помещают катализатор, а в межтрубное про-
странство подводят теплоту топочных газов (чаще при сжигании
197
Рис. 10.1. Трубчатая печь конверсии при-
родного газа:
1 — горелки; 2—газопадводящий коллектор;
3~подогреватель воздуха; 4 — испаритель паро -
вого котла; 5—пароперегреватель парового
котла; б—дымовая труба; 7—экономайзер;
S—труба с катализатором; 9 — камера сжига-
ния; 10— газоотводягцчй коллектор.
Рис. 10.2. Шахтный реактор:
/-термопары; 2—смеситель; 3—футеровка;
4 -слой огнеупоров; 5 —катализатор.
природного газа). По режиму движения реагентов это реактор
вытеснения, температурный режим — политермический.
Шахтный реактор (рис. 10.2) представляет собой аппарат ем-
костного типа, футерованный изнутри огнеупорным кирпичом,
снабженный водяной рубашкой, исключающей перегрев корпуса
в случае местных’ дефектов в футеровке. В нижнюю часть реак-
тора впрыскивают конденсат для снятия теплоты конвертирован-
ного газа и его увлажнения. По режиму движения реагентов —
реактор вытеснения, по температурному режиму — адиабатиче-
ский.
Конверсия оксида углерода(П). Конвертированный газ после
конверсии метана содержит около 20—40 % оксида углерода.
Проводя конверсию водяным паром по реакции (4) можно полу-
чить дополнительное количество водорода.
Процесс конверсии оксида углерода — гетерогенный, каталити-
ческий, обратимый я экзотермический. Он идет без изменения
числа молей. Поэтому увеличение давления не влияет на равно-
весную степень превращения оксида углерода, хотя скорость ре-
акции при этом возрастает. Равновесная степень превращения
оксида углерода растет с уменьшением температуры (реакция эк-
зотермическая), а фактическая степень превращения и скорость
имеет максимум (см. рис. 4.4 и 4.5).
На практике конверсию проводят при атмосферном или повы-
шенном давлении в зависимости от метода конверсии метана. Для
1
1
1
из
Рис. 10.3. Конвертор оксида углерода радиального
типа:
I — затвор; 2—катализатор.
достижения достаточной скорости процесса
применяют катализаторы и стремятся обес-
печить оптимальные температуры. Процесс
проводят в реакторе (конверторе) с ради-
альным ходом газа или с понижением тем-
пературы по полкам (согласно принципу
Ле Шателье и оптимизации температурного
режима) вследствие испарения впрыскивае-
мого в реактор конденсата.
На рис. 10.3 показан конвертор оксида
углерода радиального типа. Он представ-
ляет собой вертикальный цилиндрический
аппарат, заполненный катализатором. Парогазовая смесь подается
сверху вниз по центральной трубе и через отверстия по всей ее
высоте поступает на катализатор. Реакционный газ выходит из
аппарата через кольцевой зазор вдоль наружных стенок. При ра-
диальном потоке газа через катализатор сопротивление слоя ката-
лизатора меньше, чем при движении газа сверху вниз. Для пре-
дупреждения прохождения газа сверху вниз в верхней части аппа-
рата расположен слой катализатора, выполняющий роль затвора.
К конвертору применима модель реактора вытеснения, а по тем-
пературному режиму — адиабатического.
Для повышения степени превращения оксида углерода конвер-
сию проводят в две ступени. На первой ступени применяют высо-
котемпературный (450—500 °C) железохромовый катализатор,
обеспечивающий остаточное содержание оксида углерода в газо-
вой смеси порядка 2—4 %. После отвода теплоты В котле-утилиза-
торе вторую ступень конверсии проводят на низкотемпературном
(до 200—300°С) цинкхромоьомедном катализаторе, получая в от-
ходящем газе 0,2—0,4 % СО. Однако низкотемпературный ката-
лизатор очень чувствителен к серусодержащим соединениям,
в связи с чем требуется предварительная очистка газа
Поскольку процесс экзотермический, он осуществляется авто-
термично, т.е. без подвода теплоты извне, а избыток ее исполь-
зуют в котлах-утилизаторах для выработки пара.
10.4. ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ СИНТЕЗА АММИАКА
Синтез аммиака по реакции (1)—это обратимый гетерогенный
каталитический процесс, протекающий с выделением теплоты и
значительным уменьшением числа молей. По принципу ле Ша-
телье для увеличения выхода аммиака (смещения равновесия
вправо) необходимо повышение давления и понижение температуры.
199
Рис. 10.4. Зависимость молярной доли аммиака в равновесной газовой смеси Л%н,
от температуры Т для различных давлений.
Pi.c. 10.5. Зависимость молярной доли аммиака в газовой смеси от давления
(объемная скорость 30 000 ч_|, 500 СС, катапизатор — железный).
При повышении давления и постоянной температуре равновес-
ное содержание аммиака в смеси с азотом и водородом увели-
чивается (рис. 10.4). При 450 °C и 10 МПа в равновесной газовой
смеси содержится лишь около 16 % NH3, при 30 МПа равновесный
выход аммиака уже составляет 36%, а при 100 МПа — 70 %- Но
влияние давления на фактический выход аммиака характеризуется
затухающей кривой (рис. 10.5). Оптимальные давления опреде-
ляются не только выходом аммиака, но и затратами энергии на
сжатие газа, стоимостью изготовления оборудования.
Теоретически равновесный выход аммиака увеличивается
с уменьшением температуры, но при этом падает скорость реакции
(рис. 10.6). Синтез аммиака вследствие высокой прочности связи
атомов азота даже при высоких температурах (до 800°С) проте-
кает крайне медленно. В промышленных условиях для ускорения
процесса синтез аммиака проводят при 450—520 °C в присутствии
катализатора. Из существующих катализаторов наибольшую ак-
тивность проявляют контактные массы на основе пористого же-
леза с добавлением промоторов А12О3, К2О, CaO, M.gO и SiO2.
Но под действием высокой температуры и каталитических ядов
железный катализатор быстро теряет свою активность. Соедине-
ния серы отравляют' катализатор необратимо, кислород, водяной
пар и оксид углерода — обратимо. Поэтому для увеличения срока
службы катализатора азотоводородную смесь перед синтезом тща-
тельно очищают от каталитических ядов. Срок службы железного
катализатора составляет два года.
Синтеч аммиака протекает через ряд последовательных стадий:
1) диффузию азота и водорода из потока к поверхности зерен ка-
тализатора и внутри его пор; 2) активированную (химическую) ад-
200
Рис. 10.6. Зависимость молярной доли ам-
миака в газовой смеси от температуры при
различных объемных скоростях U азотово-
дороднон смеси (Р = 30 МПа).
сорбцию газов поверхностью катализатора; 3) химическое взаимо-
действие азота и водорода через промежуточные соединения их
с катализатором с образованием аммиака; 4) десорбцию аммиака
и диффузию его в газовый поток.
В результате многочисленных исследований установлено, что
самой медленной стадией является активированная диффузия га-
зов внутри' пор катализатора, т. е. весь процесс синтеза протекает
во внутридиффузионной области. Если скорость процесса лимити-
руется внутренней диффузией, то для ускорения всего процесса
в целом стремятся уменьшить размеры зерен катализатора или
развить их пористость (см. гл. 5).
В промышленных условиях оптимальным является стехиомет-
рическое соотношение N2 : Нг = 1 : 3. Катализатор применяют гра-
нулированный или дробленый с размером частиц 5—7 мм. Важ-
ная характеристика процесса — производительность П катализа-
тора, характеризующая съем (кг) аммиака с 1 м3 контактной
массы в течение 1 ч:
n = 0,77VNNH3
Здесь 0,77 — масса 1 м3 аммиака при нормальных условиях.
Формула. отражает прямо пропорциональную зависимость
между производительностью П катализатора, объемной ско-
ростью V газа и мольной долей аммиака Mnh3- С увеличением
объемной скорости уменьшается время пребывания газа в зоне
катализа и соответственно снижается содержание аммиака (см.
рис. 10.6).
Однако количество аммиака, получаемого с единицы объема
катализатора за единицу времени, растет за счет линейного уве-
личения количества газа, проходящего через катализатор. Соот-
ветственно возрастает и производительность установки. Содержа-
ние NH3 в интервале температур 450—490°C с увеличением
объемных скоростей 15000 до 120 000 ч-1 снижается с 25 до
15%. Одновременно растет количество непрореагировавших за
один цикл газов N2 и Н2. Это предопределяет необходимость
201
использования циркуляционных схем производства, позволяющих
непрореагировавшую азотоводородную смесь снова врзвращать на
синтез после выделения из нее образовавшегося аммиака.
Первые производства работали при У = 5000 4- 10000 ч~*.
В настоящее время большинство производств работает с V =
= 15 000 4-30000 ч-1.
Выбор оптимальной объемной скорости определяют исходя из
экономических соображений. Так,-с увеличением объемной ско-
рости более 40000 ч-1 значительно возрастает количество не-
прореагировавших газов, для возвращения которых необходимы
большие затраты электроэнергии на транспортировку, сжатие, на-
гревание и охлаждение газов. Кроме того, возрастают объемы тру-
бопроводов и аппаратов, нарушается также автотермичность про-
цесса, так как снижение количества прореагировавших газов ска-
жется на уменьшении выделяющейся при их взаимодействии
теплоты, которой будет недостаточно для поддержания необходи-
мой температуры в зоне катализа.
Важным условием для проведения синтеза аммиака является
также малое содержание инертных примесей в азотоводородной
смеси (Аг и СН4). Перед колонной синтеза инертных примесей не
должно быть более 7—10 %. Эти примеси, хотя и не снижают ак-
тивности катализатора, но, накапливаясь в системе, замедляют
процесс синтеза, поскольку понижают парциальное давление азота
и водорода.
10.5. РЕАКТОРЫ ДЛЯ СИНТЕЗА
В промышленных условиях синтез аммиака осуществляют в
аппаратах с фильтрующим слоем катализатора. Аппарат может
быть полочного или трубчатого типа (в виде колонны). Для обес-
печения надежной и безопасной работы колонны синтеза аммиака
предъявляют большие требования R стали, из которой она изго-
товлена. При этом учитывают агрессивные свойства ..водорода и
аммиака при высоких температурах. Особенно опасно обезугле-
роживание стали под действием водорода. Поэтому для снижения
влияния высокой температуры на корпус холодную азотоводород-
ную смесь подают по кольцевому зазору между корпусом колонны
и корпусом катализаторной коробки. Корпус колонны изготовляют
из высоколегированной стали — хромовоникелевомолибденовой
1Х18Н12М2Т.
На рис. 10.7 показана полочная колонна синтеза аммиака. Она
состоит из двух частей: в нижней части (диаметр до 2,4 м, высота
до 22 м) размещена катализаторная коробка с четырьмя слоями
катализатора, в верхней (диаметр до 1 м, высота до 6,5 м) теп-
лообменник. Азотоводородную смесь подают снизу вверх по коль-
цевому зазору, после чего она поступает в межтрубное простран-
ство теплообменника, нагреваясь в нем до 420—440°C. Для регу-
лирования температуры газовой смеси имеется байпас, через ко-
202
Рис. 10.7. Колонна синтеза аммиака:
1 — теплообменник; 2—кожух теплообменника; 3—корпус;
4—кожух каталнзаторной коробки; 5—3- и 4-й слои ката-
лизатора.
Газ
3
4
Газ
Байпасы
торый газ идет в зону катализатора, ми-
нуя теплообменник. Нагретый газ филь-
труется через четыре слоя катализатора,
затем по центральной трубе поднимается
вверх, поступает в трубки теплообмен-
ника, охлаждается в нем до 330 °C и вы-
ходит через верхний штуцер колонны
синтеза. Таким образом, в конструкции
колонны заложена возможность утилиза-
ции теплоты реакционных газов. Для ре-
гулирования температуры в слоях ка-
тализатора размещают теплоотводящие
трубки (в этой конструкции отсутствуют)
или в слой катализатора подводят хо-
лодную азотоводородную смесь через
байпас (байпасный газ).
Газовая смесь проходит через колон-
ну синтеза аммиака с фильтрующими
слоями катализатора в режиме вытеснения,
жим —• политермический.
В настоящее время почти все новые агрегаты синтеза аммиака
имеют мощность 1360 — 1500 т/сут.
Наиболее интенсивным реактором для синтеза аммиака может
быть аппарат кипящего слоя. Однако из-за отсутствия
устойчивого катализатора, необходимого для работы в
псевдоожижения при 450—520 °C, такого типа реактопы
мышленности пока не используют.
Температурный ре-
износо-
режиме
в про-
10.6. ХИМИКО-ТЕХНОЛОГИЧЕСКИЕ СИСТЕМЫ СИНТЕЗА АММИАКА
В зависимости от применяемого давления различают три типа
химико-технологических систем синтеза аммиака: 1) низкого дав-
ления (10—20 МПа); 2) среднего давления (25—36 МПа); 3) вы-
сокого давления (45—100 МПа). Наиболее распространены си-
стемы, работающие под средним давлением 30—32 МПа. Принци-
пиальная схема такой установки мощностью 1500 т/сут показана
на рис. 10.8.
Природный газ под давлением 4 М.Па после предварительной
очистки от серусодержащих примесей (на схеме не показано) сме-
шивается с паром в соотношении 1 :2, подогревается в теплооб-
меннике / отходящими дымовыми газами и поступает на 1-ю сту-
пень конверсии метана в трубчатую печь 2 с топкой 3, в которой
сжигается природный газ. Процесс конверсии метача протекает
203
Дымовые газы
Рис. 10.8. Принципиальная схема конверсии метана и синтеза аммиака под дав-
лением 30—32 МПа:
/“Теплообменник; 2 —трубчатая печь; 3—топка; 4— шахтный конвертор метана; 5, 6—котлы-
утилизаторы; 7 — конвертор СО 1-й ступени; 8—конвертор СО 2-й ступени; 9— турбокомпрессор
с газовой турбиной; 10—паровая турбина; 11— воздушный холодильник; 12—аммиачный холо-
дильник; 13—вторичный сепаратор; 14—первичный сепаратор; 15—«холодный* теплообменник;
16—воздушный холодильник; 17—«горячий* теплообменник; 18— водоподогреватель питатель-
ной воды; 19— колонна синтеза.
на никелевом катализаторе при 700—800 °C. Содержание остаточ-
ного метана в газе на выходе после 1-й ступени конверсии состав-
ляет 9—10%. Далее газ смешивается с воздухом и поступает на
2-ю ступень конверсии в шахтный конвертор 4, где происходит
конверсия остаточного метана кислородом воздуха при 900—
1000 °C. Конвертированный газ направляется в котел-утилизатор 5,
в котором теплота газа утилизируется для выработки пара, ис-
пользуемого далее в турбине 9 центробежного компрессора 8. Из
котла-утилизатора 5 газ поступает на двухступенчатую конверсию
оксида углерода. Конверсия оксида углерода осуществляется сна-
чала в конверторе 1-й ступени 7 на высокотемпературном железо-
хромовом катализаторе при 450—500 °C, затем в конверторе
2 й ступени 8 на низкотемпературном цннкхромовомедном ката-
лизаторе при 200—300 °C. Для утилизации теплоты конвертирован-
ного газа между 1- и 2-й ступенями конверсии устанавливают
котел-утилизатор 6. Газ после 2-й ступени конверсии подвергается
очистке от СО> и СО (на схеме не показано). Для сжатия азото-
водородной смеси до 30—32 МПа и циркуляции газа в агрегате
синтеза используют центробежный компрессор 9 с приводом от
паровой турбины 10. Свежая азотоводородная смесь смешивается
с циркуляционной смесью перед системой вторичной конденсации,
состоящей из воздушного холодильника 11, аммиачного холодиль-
ника 12 и сепаратора 13, проходит далее два теплообменника 15,17
и направляется в полочную колонну синтеза 19. Прореагировав-
ший газ, Содержащий около 20 % NH3, при 320—380 °C выходит
из колонны синтеза и проходит последовательно водоподогрева-
тель питательной воды 18, «горячий» теплообменник 17, воздуш-
ный холодильник 16 и «холодный» теплообменник 15, первичный
сепаратор 14 и поступает на циркуляционное колесо компрессора
для сжатия. Жидкий аммиак после первичного и вторичного се-
параторов направляется в хранилище аммиака.
204
10.7. ОСНОВНЫЕ НАПРАВЛЕНИЯ СОВЕРШЕНСТВОВАНИЯ
ПРОИЗВОДСТВА АММИАКА
Основные направления совершенствования производства ам-
миака определяются удовлетворением всевозрастающей потреб-
ности в минеральных удобрениях и нужд других отраслей народ-
ного хозяйства. По прогнозам до 2000 г. темпы роста производства
аммиака будут возрастать и опережать темпы развития дру-
гих крупнотоннажных производств. Основными направлениями
увеличения темпов роста *и совершенствования производства
аммиака являются: 1) кооперирование азотной промышленности
с промышленностью основного органического синтеза на базе ис-
пользования природного газа и газов нефтепереработки в каче-
стве сырья; 2) увеличение мощности отдельных технологических
ниток до 3000 т/сут NH3; 3) разработка и внедрение новых более
активных и устойчивых катализаторов; 4) применение колонн
синтеза с кипящим слоем катализатора; 5) наиболее полное ис-
пользование теплоты реакций для получения пара.
ГЛАВА П
ПРОИЗВОДСТВО АЗОТНОЙ кислоты
11.1. ОБЩИЕ СВЕДЕНИЯ
.Азотная кислота — один из важнейших продуктов хими-
ческой промышленности. По объему производства азотная кислота
находится на втором месте после серной кислоты.
Химически чистая азотная кислота (мол. масса 63) представ-
ляет собой бесцветную жидкость плотностью 1,52 г/см3 с сильным
едким запахом, замерзающую при 41 °C, кипящую при 86°С. На
воздухе концентрированная азотная кислота дымит, смешивается
с водой в любых соотношениях с выделением теплоты, образуя
гидраты (HNO3-H2O, HNO3-2H2O и др.). При упаривании раз-
бавленной кислоты содержание ее в растворе повышается до
68,4 % HNO3, соответствующей азеотропной смеси, кипящей при
121,9 °C.
Техническая азотная кислота вследствие содержания в ней
растворенного NO2 имеет желтоватый цвет.
Азотная кислота является сильным окислителем. Она легко
разрушает многие органические вещества, некоторые из них под
действием концентрированной кислоты способны воспламеняться.
Азотная кислота растворяет все металлы, кроме золота, платины,
титана, тантала, радия и иридия. Разбавленная азотная кислота
хорошо растворяет железо. Концентрированная кислота образует
на поверхности железа тонкий оксидный слой, нерастворимый в
концентрированной кислоте, защищающий металл от дальнейшего
205
разъедания. Это свойство железа пассивироваться используют
для защиты его от коррозии. Концентрированную азотную кис-
лоту (особенно с добавлением 10 % H2SO4) перевозят в стальных
цистернах.
Азотная кислота является сырьем для выработки многих про-
дуктов, применяемых в промышленности и сельском хозяйстве.
В нашей стране около 40 % вырабатываемой азотной кислоты
расходуют на производство азотсодержащих минеральных удоб-
рений, нитратных солей (нитратов натрия, калия и кальция).
Концентрированную азотную кислоту применяют в производстве
соединений ароматического ряда для синтеза красителей; в произ-
водстве взрывчатых веществ (нитроглицерина, продуктов нитро-
вания толуола), уротропина, диметиланилина, ксилола; в фарма-
цевтической промышленности; для получения нафталина, пла-
стических масс, нитролаков, кинопленки и других важнейших
продуктов.
Промышленность выпускает азотную кислоту двух видов: раз-
бавленную с содержанием 46—57 % HNOg и концентрированную,
содержащую 61—68% HNO3 и 98,9 % HNO3. В начале 70-х годов
в СССР разработана энерготехнологическая система производи-
тельностью 380 тыс. т/год.
11.2. СПОСОБ ПОЛУЧЕНИЯ
В современных производствах используют метод получения
азотной кислоты из оксидов азота, полученных окислением ам-
миака на платине.
Метод включает три стадии:
1) контактное окислёние аммиака до оксида азота(II)
4NH3 + 5О2 = 4NO + 6Н2О + 907,3 кДж
2) окисление оксида азота(II) до оксида азота(IV)
2NO + О2 2NO2 + 112 кДж
3) абсорбция оксида азота(IV) водой:
3NO2 + Н2О 2HNO3 + NO + 136 кДж
Выделяющийся при этом оксид азота(II) окисляется до оксида
азота(IV) и снова абсорбируется.
Данный метод производства азотной кислоты является безот-
ходным, в нем отсутствуют затраты на побочные дорогостоящие
продукты.
11.3. СЫРЬЕ ДЛЯ ПРОИЗВОДСТВА
Сырьем для производства азотной кислоты являются синтети-
ческий аммиак, воздух и вода. К чистоте сырья предъявляют
высокие требования. Аммиак, поступающий из цеха синтеза,
206
предварительно очищают от катализаторной пыли и масла, если
для транспортировки газа применяют поршневые компрессоры.
Эти примеси являются ядами для катализаторов на первой стадии
окисления аммиака до оксида азота(II).
Воду, необходимую для абсорбции диоксида азота и получе-
ния азотной кислоты, подвергают обессоливанию.
Воздух, применяемый для окисления аммиака, очищают от
пыли и газообразных примесей, попадающих в него от выбросов
из цехов, находящихся на территории завода. Эти примеси яв-
ляются каталитическими ядами на стадии окисления аммиака до
оксида азота (II).
11.4. ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ ПРОИЗВОДСТВА.
АППАРАТУРНОЕ ОФОРМЛЕНИЕ
11.4.1. Контактное окисление аммиака
В зависимости от условий окисления аммиака возможно проте-
кание следующих параллельных реакций:
4NH3 + 5О2 = 4NO + 6Н2О + 907,3 кДж
4NH3 + 4О2 = 2N2O + 6Н2О + 1104,9 кДж
4NH3 + ЗО2 = 2Na + 6Н2О + 1269,1 кДж
Для производства азотной кислоты необходимо возможно более
полное окисление аммиака до NO (N2O и N2 обладают инертными
свойствами). Для осуществления первой реакции и подавления
второй и третьей следует применять катализатор, обладающий
селективным (избирательным) действием. Окисление аммиака на
катализаторе — необратимый процесс, экзотермический, типично
гетерогенно-каталитический, протекающий последовательно в не-
сколько стадий: 1) диффузия реагирующих веществ к поверхности
катализатора; 2) адсорбция кислорода; 3) химическое взаимодей-
ствие молекул на поверхности катализатора; 4) десорбция про-
дуктов реакции с поверхности катализатора. Скорость окисления
аммиака определяется самой медленной стадией процесса—диф-
фузией аммиака к поверхности катализатора, т. е. процесс окисле-
ния аммиака протекает во внешнедиффузионной области.
Для ускорения процесса окисления аммиака и увеличения вы-
хода необходимы следующие условия: повышение температуры,
давления, концентрации реагирующих веществ, применение ката-
лизатора.
Повышение температуры — эффективное средство уве-
личения скорости процесса окисления NH3, однако при более 800—
850 °C выход NO снижается, а выход элементарного азота и дис-
социация NH3 возрастают (рис. 11.1). При атмосферном давлении
поддерживают температуру до 800 °C, при повышенном—до
900 °C.
207
Рис. 11.1. Зависимость выхода NO от температуры.
Рис. 11.2. Зависимость выхода NO от мольного отношения кислорода и аммиака.
Повышение давления ускоряет процесс окисления NH3
(в результате роста концентрации реагирующих веществ), увели-
чивает выход NO, но при этом возможно возрастание температуры
окисления аммиака. Увеличивается расход платинового катализа-
тора вследствие уноса его мельчайших частичек с газами, повыша-
ются требования к прочности аппаратуры..Все это приводит к удо-
рожанию аппаратов.
Концентрация реагирующих веществ NH3 и О2
влияет на скорость процесса окисления NH3 и выход NO. По
стехиометрическому уравнению для окисления аммиака необхо-
димо иметь в составе возДушно-аммиачной смеси 1,25 моль кис-
лорода на 1 моль NH3. Зависимость вйхода NO. от соотношений
кислорода и аммиака в исходной воздушно-аммиачной смеси для
платинопдного катализатора, работающего под атмосферным дав-
лением, приведена на рис. 11.2. На практике для увеличения вы-
хода NO и повышения скорости процесса соотношение О2I NH3
принимается равным 1,8—2,0, что соответствует содержанию ам-
миака в воздушно-аммиачной смеси 9,5—10,5 % (по объему).
Следует учитывать, что при обычной температуре смесь аммиака
с воздухом взрывоопасна в интервале 16—27 % NH3. При повы-
шении температуры границы взрывоопасности расширяются.
Катализаторы окисления аммиака, как указывалось
выше, должны обладать избирательным свойством, т. е. ускорять
только одну из трех возможных реакций. Наиболее селективным
и активным является платиновый катализатор, представляющий
собой сплав платины с палладием и родием. Чистая платина при
высоких температурах быстро разрушается. Примесь в платине
незначительного количества железа снижает активность катализа-
тора. Сплав платины с родием делает катализатор в процессе
окисления аммиака до NO активным и стойким к высоким тем-
пературам.
208
Рис. 11.3. Контактный аппарат:
1 — распределительная решетка; 2—корпус аппарата*
3 — платиновая сетка; 4 — слой неплатинового катализатора
5 — насадка; 6~ опорная решетка.
Наибольшее распространение получили следующие катализа-
торы окисления аммиака: при атмосферном давлении — Pt+
+ 4 % Pd + 3,5 % Rh; при повышенном давлении — Pt + 7,5 % Rh.
Для увеличения прочности платиновые катализаторы выпол-
няют в виде сеток из тонкой проволоки диаметром 0,06—0,09 мм,
имеющих 1024 отверстия в 1 см2. Сетки скрепляют в форме пакета
для обеспечения необходимого времени контактирования. При
работе под атмосферным давлением в аппарате устанавливают
пакет из 3—4 сеток, при. повышенном давлении — из 18 сеток.
При окислении NH3 под давлением 0,8 МПа производитель-
ность катализатора в 5 раз выше, чем при атмосферном давлении,
но и унос платины сильно возрастает. Если при атмосферном дав-
лении унос платины на 1 т азотной кислоты составляет 0,04—
0,06 г, то при повышенном он достигает 0,3—0,4 г. Часть платины
улавливается и регенерируется (10—15 %-ным раствором соляной
кислоты), но и при этом расходы ее составляют значительную
часть себестоимости азотной кислоты. Учитывая высокую стои-
мость и дефицитность материалов платиновой группы, проводят
исследования синтеза с участием неплатиновых катализаторов.
Для уменьшения потерь платины окисление аммиака предло-
жено проводить в две ступени. На первой ступени используют
платиновые сетки, на второй — гранулированный неплатиновый
катализатор (например, железохромовый). Применение двухсту-
пенчатого окисления дает возможность сократить загрузку пла-
тины в аппарат в три раза, уменьшить ее потери на 10—20 % при
сроке службы катализатора 3—5 лет.
На рис. 11.3 показано расположение катализаторов в контакт-
ном аппарате при двухступенчатом окислении.
При двухступенчатом контактировании (при повышенном дав-
лении) число платиновых сеток может быть снижено до 1—3.
Окисление NH3 проводят в контактном аппарате непрерывного
действия. Режим движёния реагентов — вытеснение. Температур-
ный режим — адиабатический. Газ в контактный аппарат подается
сверху. Катализаторные сетки во избежание провисания опираются
на металлический колосник.
209
11.4.2. Окисление оксида азота (II)
Реакция окисления оксида азота(II) до оксида азота(IV)—
самая медленная из всех реакций, которые протекают при пере-
работке нитрозных газоо:
2NO + O2 ч=± 2NO2 + 112,3 кДж
Окисление NO является обратимой реакцией, сопровождаю-
щейся выделением теплоты и уменьшением числа молей (газового
объема). В соответствии с принципом Ле Шателье для смещения
химического равновесия в правую сторону необходимо снижение
температуры и повышение давления.
До 100°C реакция практически полностью протекает^ сторону
образования NO2. При более высокой температуре равновесие
смещается в левую сторону, и при температуре выше 700 °C окис-
ления NO до NO2 практически не происходит. Следовательно,
горячие нитрозные газы, выходящие из контактного аппарата, не-
обходимо охладить до температуры ниже 100 °C.
Окисление NO в NO2 идет через образование промежуточного
продукта — димера (NO)2
2NO (NO)2 + <7
который затем окисляется до NO2:
(NO)2 + О2 ч * 2NO2 -|- q
Образование димера оксида азота — процесс обратимый, про-
текающий с выделением теплоты. С повышением температуры рав-
новесная концентрация димера в газовой смеси будет понижаться.
Скорость же дальнейшего окисления димера в NO2 зависит от
концентрации димера. Таким образом, с повышением темпе-
ратуры скорость окисления NO в NO2 уменьшается.
Степень окисления NO в значительной степени возрастает при
повышении давления (что эквивалентно повышению кон-
центрации), а одновременно увеличивается и скорость реакции. ч
На установках, работающих при повышенном давлении, NO прак-
тически полностью окисляется до NO2. При этом объем аппаратуры
в сотни раз меньше по сравнению с процессом окисления при
атмосферном давлении. Степень окисления при атмосферном дав-
лении составляет лишь 92—93%, что требует установки допол-
нительной громоздкой аппаратуры для очистки оставшихся нитроз-
ных газов.
Наряду с окислением NO в NO2 могут протекать следующие
реакции:
1. Оксид азота (IV) может ассоциировать с образованием ди-
мера:
2NO2 (NO2)2 + 57 кДж
Скорость этой реакции очень велика. С повышением давления
и понижением температуры равновесие смещается в правую сто-
рону.
210
2. Оксид азота (IV) может взаимодействовать с оксидом
азота (II):
NO2 + NO N2O3 + 40,2 кДж
Таким образом, в нитрозных газах, поступающих на абсорб-
цию, могут содержаться: NO2, N2O4, N2O3, NO, N2O, N2, пары
воды и др. Соотношение этих веществ изменяется в зависимости
от условий, однако основным компонентом является NO2.
Окисление NO и абсорбция NO2 водой осуществляются прак-
тически одновременно в одной и той же аппаратуре. Однако зако-
номерность этих процессов целесообразно рассмотреть раздельно.
11.4.3. Абсорбция оксида азота (IV) водой
Процесс абсорбции нитрозных газов водой осуществляется по
следующей схеме:
2NO2 + Н2О HNO3 + HNO2 +116 кДж
N2O4 + Н2О HNO3 + HNO2 + 59 кДж
Получающаяся азотистая кислота неустойчива и разлагается:
3HNO2 HNO3 + 2NO + Н2О — 76 кДж
Суммарное взаимодействие NO2 с водой можно представить
уравнением:
3NO2 + Н2О +=± 2HNO3 + NO + 136 кДж
NO, N2O и N2 практически нерастворимы в воде.
Поглощение оксида азота (IV) водой представляет собой типич-
ный хемосорбционный процесс в системе газ — жидкость.
Для смещения равновесия в сторону образования HNO3 необ-
ходимо снижать температуру и повышать давление. Процесс це-
лесообразно проводить при пониженной температуре
(20—30°C), но вследствие экзотермичности реакции абсорбция
в большинстве азотнокислотных производств идет в интервале тем-
ператур 50—70 °C. Большое количество кислоты, выделяющееся
в процессе (9-106 кДж на 1 т HNO3), расходуется на нагревание
орошающих растворов кислот. В связи с этим для создания опти-
мальных температур в зоне абсорбции орошающие кислоты охлаж-
дают в холодильниках.
Скорость абсорбции NO2 увеличивается с повышением
давления. В системах, работающих при атмосферном давлении,
абсорбируется только 92—93 % нитрозных газов от поступающего
количества оксидов. При этом получают азотную кислоту, содер-
жащую лишь около 50 % HNO3. При проведении абсорбции под
давлением 0,6—0,8 МПа абсорбируется 98—99 % исходных окси-
дов азота и образуется 58а—60%-ная кислота. Однако при повы-
шении давления возрастает растворимость NO2 в азотной кислоте.
Растворенный диоксид азота в абсорбции не участвует, поэтому в
системах, работающих при повышенном давлении, продукционную
211
азотную кислоту подвергают отбеливанию в специальной колонне,
где растворенный NO2 отдувают воздухом и снова направляют
на абсорбцию. -
Абсорбцию нитрозных газов под- давлением осуществляют
в барботажной колонне с тарелками колпачкового или ситчатого
типа по принципу противотока — снизу поступает газ, а сверху —
вода или конденсат. К колонне применима модель смешения по
жидкой фазе и вытеснения по газу. _
Процесс абсорбции при атмосферном давлении проводят в
6—8-ми абсорбционных башнях насадочного типа, заполненных
кислотоупорными кольцами Рашита, также по принципу противо-
тока. Режим движения газовой и жидкой фаз в башне соответ-
ствует вытеснению.
Явные преимущества имеют абсорбционные системы, работаю-
щие под повышенным давлением: высокая степень превращения
нитрозных газов (98—99%), небольшое число абсорбционных ап-
паратов (1—2 шт.), отсутствие громоздкой установки для очистки
нитрозных газов.
11.5. ХИМИКО-ТЕХНОЛОГИЧЕСКИЕ СИСТЕМЫ
ПРОИЗВОДСТВА КИСЛОТЫ
Производство разбавленной азотной кислоты из аммиака
можно осуществить по одной из трех систем: 1) при атмосферном
давлении; 2) при повышенном давлении, 3) при комбинировании
атмосферного и повышенного давлений. 3 основе всех ХТС лежит
схема с открытой цепью и последовательными технологическими
связями аппаратов.
Схема производства разбавленной азотной
кислоты при атмосферном давлении представлена на
рис. 11.4.
Атмосферный воздух поступает в систему через заборную
трубу, установленную вне территории завода. Для очистки от газо-
образных примесей и пыли воздух пропускают через пенный про-
мыватель / и картонный фильтр 2. Аммиак также очищают от
примесей в картонном фильтре 3.
Смешение воздуха с аммиаком до содержания 10—12 % NH3
и дальнейшая их транспортировка осуществляется аммиачко-воз-
душным компрессором 4. Далее газовая смесь проходит в кон-
тактный аппарат 5. Образующиеся при контактном окислении
нитрозные газы выходят с температурой около 800 °C. Утилизация
теплоты отходящих газов происходит в котле-утилизаторе 6 где
вырабатывается водяной пар. В результате температура газов
снижается до 250°C Затем газы охлаждаются водой до 30°C
в кожухотрубных теплообменниках 7 (на схеме показан один из
двух). При этом происходит частичная конденсация водяных паров
и в небольшой степени окисление NO до NO2, который, поглощаясь
конденсатом, дает разбавленную азотную кислоту (до 30 % HNO3).
212
Рис. 11,4. Принципиальная схема производства разбавленной азотной кислоты
при атмосферном давлении:
/ — пенный промиватель; 2, 3—картонные фильтры; 4—компрессор; 5—контактный аппарат:
6 — котел-утилнзатор; 7—кожухотрубный теплообменник; 8—газодувка; 9— абсорбционные
башни; 10— водяные холодильники; //-‘-циркуляционные насосы: /2—окислительная башня;
/J — башня с насадкой для щелочной абсорбции.
Далее нитрозные газы подаются газодувкой 8 в абсорбцион-
ные башни 9, заполненные насадкой из кислотоупорных колец.
Число башен кислотной абсорбции составляет от 6 до 8. Последняя
по ходу газа башня орошается водой; образующаяся слабая кис-
лота проходит последовательно противотоком газу все башни
и, наконец, поступает в первую башню, из которой выводится про-
дукционная 50 %-ная азотная кислота. Для снятия выделяющейся
теплоты при окислении NO и абсорбции NO2 башни снабжены
водяными холодильниками 10. Циркуляция кислоты в башнях осу-
ществляется насосами 11.
В абсорбционных башнях перерабатывается около 92 % окси-
дов азота. К оставшимся оксидам добавляют NO, выделяющийся
в процессе абсорбции NO2. Поэтому после абсорбционных башен
установлена окислительная башня 12, где NO частично окисляется
до NO2. Затем нитрозные газы поступают на щелочную абсорбцию
раствором с;оды в башни 13 (обычно две). В башнях щелочной
абсорбции поглощается NO2 и смесь NO2 ф- NO(N2O3), при этом
исключается выделение NO в газовую фазу, как это видно из
реакций:
NO + NO2 + Na2CO3 —-» 2NaNO2 + СО2
2NO2 + Na2CO:. —> NaNO2 + NaNO? + CO2
Образующиеся растворы нитрита и нитрата натрия выводят
для дальнейшей переработки NaNO2 в NaNO3. Нитрат натрия
используют в сельском хозяйстве в качестве азотного удобрения.
Установки, работающие при атмосферном давлении, имеют ряд
недостатков:
низкая степень превращения оксидов азота в азотную кис-
лоту — всего 92 %;
громоздкость производства из-за башен щелочной абсорбции
и соответственно большие капитальные затраты на их сооружение;
низкое содержание азотной кислоты — 50 %;
213
высокое содержание оксидов азота после щелочной абсорб-
ции— до 0,15 %.
Достоинством данного метода является небольшой унос пла-
тины и невысокий расход электроэнергии.
Схемы производства разбавленной азотной
кислоты, работающие при повышенном давлении,
имеют ряд достоинств по сравнению с установками, работающими
при атмосферном давлении:
1. Степень переработки оксидов 'азота в азотную кислоту по-
вышается до 98—99 %, а содержание полученной азотной кис-
лоты — до 60—62 % Отпадает необходимость в щелочной абсорб-
ции (схема значительно короче).
2. Содержание оксидов азота после каталитической очистки
низкое —0,01 % (по объему).
3. Объем абсорбционных колонн в десятки раз меньше, чем
башен с насадкой в системах, работающих под атмосферным дав-
лением.
4. Уменьшаются -капитальные затраты на постройку установки
и расход специальной стали (хромовоникелевой).
5. Упрощается обслуживание установки.
К недостаткам метода следует отнести увеличение потерь пла-
тины и расхода энергии с повышением давления.
Комбинированный способ производства раз-
бавленной азотной кислоты получил распространение
в последние годы. Контактное окисление аммиака проводят при
атмосферном давлении, а окисление NO и абсорбцию NO2 водой —
при повышенном давлении.
Схема производства по данному методу сочетает достоинства
систем высокого давления и систем атмосферного давления.
Концентрированную азотную кислоту производят
прямым синтезом концентрированного NO с кислородом и водой
под давлением. Помимо прямого синтеза применяют концентриро-
вание разбавленной HNO3 путем перегонки ее с концентрированной
серной кислотой, выполняющей роль водоотнимающего средства.
11.6. ОСНОВНЫЕ НАПРАВЛЕНИЯ
СОВЕРШЕНСТВОВАНИЯ ПРОИЗВОДСТВА
В связи с всевозрастающими потребностями сельского хозяй-
ства в минеральных удобрениях объем выработки азотной кис-
лоты растет быстрыми темпами. С 60-х годов действуют агрегаты
мощностью 120 тыс. т в год, с 1975 г.— агрегаты на 380 тыс. т/год.
Проектируются новые компактные системы, работающие при
повышенном давлении с мощностью одного агрегата 66 тыс. т
кислоты в год. Создание таких агрегатов сокращает капитало-
вложения.
В настоящее время проектируются системы на основе примене-
ния кислорода и концентрированных оксидов азота, что позволит
214
значительно сократить объемы аппаратов и трубопроводов и соот-
ветственно снизить капиталовложения.
Ведутся работы по замене платинового катализатора термо-
стойкими механически прочными неплатиновыми, а также по со-
кращению потерь платины путем ее эффективного улавливания.
ГЛАВА 12
ХИМИЧЕСКАЯ ПЕРЕРАБОТКА ТОПЛИВ
12.1. КЛАССИФИКАЦИЯ ТОПЛИВ
Топливом называют существующие в природе или искус-
ственные горючие органические вещества, являющиеся источником
тепловой энергии и сырьем для химической промышленности. Ниже
приведена классификация топлив.
Общая классификация топлив
Происхождение
Агрегатное состояние природное искусственное
Т вердое Древесина, бурый уголь, каменный уголь, антра- цит, горючие сланцы, торф Древесный уголь, кокс, полу- кокс
Жидкое Нефть Бензин, керосин, мазут и дру- гие продукты переработки неф- ти, метиловый спирт, каменно-
угольная смола
Г азообразное Природный и попутный газ Коксовый, доменный, водяной, генераторные газы, газы неф- тепереработки
Целесообразность практическогр использования определенных
видов топлив определяется прежде всего их запасами, доступ-
ностью, а также географическим положением месторождений.
Уголь, нефть и газ имеют повсеместно первостепенное значение
(см. разд. 2.3.1).
Не все добываемое топливо используют непосредственно в ка-
честве горючего. Значительную часть его предварительно подвер-
гают сложной обработке. Например, из нефти получают бензин
для авиационных и автомобильных двигателей, керосин для реак-
тивных самолетов, дизельное топливо для тепловозов. Кроме того,
в процессе переработки нефти получают газы, применяемые в ка-
честве сырья в химической промышленности и частично используе-
мые в виде топлива. Таким образом, из одного вида естественного
топлива-—сырой нефти — получают несколько видов жидкого и
газообразного топлива.
Природный и попутный газы — наиболее молодой энергетиче-
ский ресурс. Газообразное природное топливо очень эффективный
и наиболее простой заменитель нефти. Наметилась тенденция уве-
личения потоебления газа в качестве химического сырья. Из
215
природного газа получают синтез-газ, являющийся основой для
производства метанола. Почти весь водород, используемый для
синтеза аммиака, получают из природного газа. Сжатый и сжижен-
ный природный газ можно использовать как моторное топливо.
Развитие газовой промышленности и наличие больших запасов
дешевых углей в СССР позволило развернуть научно-исследова-
тельские и опытные работы по получению синтетических жидких
топлив (СЖТ).
В качестве дополнительных источников углеводородного
сырья будут использоваться природные битумы и горючие сланцы.
В состав природных битумов входит высоковязкая нефть, из ко-
торой можно получить около 40 % моторных топлив. Для получе-
ния качественного моторного топлива из горючих сланцев необхо-
дима дополнительная переработка с использованием процессов
очистки и гидрирования. Поэтому в настоящее время сланцепере-
работку ведут с целью получения химического -сырья, значитель-
ная часть которого производится из нефти. Это позволяет высво-
бодить дополнительные ресурсы нефти для производства мотор-
ного топлива.
Все более актуальным и важным становится изыскание путей
повышения эффективности использования и экономии топливно-
энергетических ресурсов, а также поиски новых видов энергоно-
сителей.
В последнее время бурно развиваются исследования по водо-
родной энергетике. Одним из основных сдерживающих факторов
в использовании водорода в качестве топлива в настоящее время
является его дороговизна. .Себестоимость водорода может быть
существенно снижена в результате использования энергии атом-
ных электростанций.
С экологической точки зрения водород является идеальным
энергоносителем, так как при его сгорании образуется только
вода, которая возвращается в цикл естественного кругооборота.
Теплота сгорания водорода почти в четыре раза выше, чем
углерода.
12.2. ПЕРЕРАБОТКА ТВЕРДОГО ТОПЛИВА
12.2.1. Виды, свойства, состав и методы переработки
твердого топлива
Твердое топливо в зависимости от его происхождения делят
на две большие группы: природное и искусственное. Искусствен-
ное топливо получают из природного путем его переработки.
Природным топливом являются древесина, торф/бурый и ка-
менный уголь, антрацит, сланец, искусственным — брикеты, пыле-
видное топливо (прошедшие значительную механическую обра-
ботку), кокс и полукокс (прошедшие тепловую обработку).
Горючая часть твердого топлива (горючая масса) состоит из
сложных веществ (соединений), в состав которых входят углерод,
216
водород, кислород, азот и сера, негорючая часть (балласт) — из
минеральных веществ. При пирогенети^еской переработке топлива
минеральные вещества входят в состав золы, при этом многие
соединения разлагаются с образованием оксидов.
•На свойства топлива и качество получаемых продуктов осо-
бенно сильно влияет сера. Она входит в состав твердого топлива
в виде сульфидов (сульфидная смола), сульфатов (сульфатная
сера) и органических соединений (органическая сера).
Органическая масса в основном определяет теплотворную спо-
собность твердого топлива. Энергетическая характеристика топли-
ва зависит от его состава и влажности.
Летучие вещества получаются при нагревании топлива без до-
ступа воздуха. Это газообразные и жидкие продукты, причем по-
следние находятся при высокой температуре в парообразном со-
стоянии. Вместе с летучими веществами удаляется и влага. Выход
летучих (потери массы топлива при нагревании его без доступа
воздуха при 850 °C в течение 7 мин за вычетом влаги) позволяет
решить вопрос о целесообразности химической переработки топ-
лива для получения жидких и газообразных продуктов.
Торф является продуктом разложения растительных остатков
в присутствии влаги без доступа воздуха. Для газификации при-
меняют достаточно разложившийся торф.
В естественных условиях торф обычно сильно увлажнен (85—
90%). Торф с малым содержанием влаги — Очень хорошее сырье
для получения высококалорийного горючего газа.
Бурый уголь — также продукт разложения веществ расти-
тельного происхождения под действием воды, воздуха при повы-
шенной температуре и давлении. По степени разложения он зани-
мает промежуточное место между торфом и каменным углем.
Вследствие значительного содержания влаги и золы бурые угли
не являются высококачественными топливами.
Каменный- уголь — разложение растительного вещества
в каменных углях прошло значительно дальше, чем в бурых, со-
деожание углерода в них больше, кислорода — меньше, влажность
каменного угля меньше, чем бурого. В СССР в ряде районов име-
ются богатые залежи угля.
Антрацит — разновидность каменных углей с’ содержанием,
не более 2 % водорода. Антрацит содержит около 95 % углерода
и дает малый выход летучих.
Горючие сланцы — представляют собой минеральную по-
роду, пропитанную горючими веществами, подобными нефтепро-
дуктам. Из сланцев можно получать нефтеобразные продукты.
При нагревании они часто распадаются на пластинки и растре-
скиваются. Высокое содержание золы и влаги в ряде случаев
затрудняет' использование сланцев для сжигания и газификации.
• Свойства и систАв топлив способны изменяться под воздей-
ствием высокой температуры и химических реагентов. Ниже рас-
смотрены основные методы переработки твердых топлив, при ко-
торых происходят изменения химических свойств топлива.
217
•Сухая перегонка топлива — нагревание топлива без до-
ступа воздуха. В результате протекают глубокие химические де-
структивные превращения компонентов топлива с получением га-
зообразных, жидких и твердых продуктов. В основном это реакции
расщепления молекул топлива, полимеризации, деалкилирования.
Суммарный тепловой эффект этих реакций эндотермический, и по-
этому для сухой перегонки твердого топлива необходим подвод
теплоты извне.
Такой переработке подвергают почти все виды твердого топ-
лива, за исключением бурого угля и антрацита. Состав и свойства
получаемых продуктов в значительной степени зависят от исход-
ного сырья и условий его перегонки. Принципиальная же схема
процесса во всех случаях одинакова. Его можно разделить на
следующие стадии:
1) до 100 °C — сушка;
2) от 100 °C до 350 °C — разложение менее стойких кислород-
содержащих органических соединений и отгонка легколетучих ве-
ществ;
3) до 500—550 °C — образование твердого остатка, смолы и
газа, так называемое полукоксование;
4) до 1000—1100°С — окончательное прокаливание твердого
остатка (коксование с выделением из кокса труднолетучих ве-
ществ). Летучие вещества, соприкасаясь с раскаленным коксом,
превращаются из соединений жирного ряда в "Циклические соеди-
нения — происходит ароматизация смолы.
Газификация — воздействие на горючую часть твердого
топлива кислооодом воздуха, водяным паром и диоксидом угле-
рода или их смесями при высокой температуре с целью получения
газообразного топлива — генераторного газа. Устройства, в кото-
рых получают генераторный газ (газогенераторы), представляют
собой шахту, куда сверху загружают топливо, а снизу подают
дутье. В результате газификации от топлива остаются остатки:
зола и несгоревшее топливо, которые лежат на колосниках и пе-
риодически или непрерывно удаляются.
Процесс газификации зависит от ряда факторов — темпера-
туры, состава дутьевой смеси, размера кусков топлива, способно-
сти его взаимодействовать с газами, спекаемости топлива, плавко-
сти золы, равномерности распределения газов по сечению и т. д.
В промышленных условиях работы газогенераторов (900—1000 °C,
1 X Ю5 Па) в основном получают синтез-газ (СО + Н2).
Гидрогенизация — процесс гидрирования и киекинга под
высоким давлением водорода в присутствии катализатора. В ре-
зультате этого процесса высокомолекулярные горючие вещества
превращаются в смесь насыщенных водородом низкомолекуляр-
ных соединений.
Исходя из природы перерабатываемого топлива и каче-
ства конечного продукта, подбирают соответствующие давление,
температуру, катализатор, концентрацию реагирующих ве-
ществ.
218
12.2.2. Коксование каменного угля
Основным методом переработки каменного угля является кок-
сование. В процессе коксования уголь нагревают без доступа воз-
духа до 900—1050 °C. При этом образуются летучие вещества и
твердый остаток — кокс.
Сырьем для коксования служат угли, способные хорошо спе-
каться. Они составляют около 20 % от общих запасов угля. В про-
мышленной практике используют смесь-шихту, содержащую не
только коксующиеся угли, но и угли других марок. Это позволяет
расширить сырьевую базу коксохимической промышленности.
Каменный уголь загружают в закрытые камеры — коксовые
печи. При постепенном нагревании компоненты угля претерпе-
вают глубокие физические и химические превращения: до 250°C
происходит испарение влаги, выделение оксида и диоксида угле-
рода, около 300 °C начинается выделение паров смолы, выше
350 °C уголь переходит в пластическое состояние, при 500—550 °C
наблюдается бурное разложение пластической массы с выделе-
нием газа и смолы и твердение ее с образованием полукокса. При
дальнейшем повышении температуры из полукокса выделяются
остатки летучих веществ, и он превращается в кокс. Летучие про-
дукты в зоне печи, нагретой до 1000 °C, превращаются в сложную
смесь паров с преобладанием ароматических соединений и газов,
содержащих в основном водород и метан.
Методом коксования получают следующие продукты: кокс,
коксовый газ, каменноугольную смолу, сырой бензол, надсмоль-
ную воду и соли аммония (большей частью сульфат аммония).
Главной целью коксования является получение металлургиче-
ского кокса, который должен обладать высокой прочностью, что-
бы не разрушаться под давлением слоя материалов, загружаемых
в доменную печь.
Кокс представляет собой твердый матово-черный пористый
продукт. Из 1 т сухой шихты получают 650—750 кг кокса. Основ-
ные показатели качества кокса — горючесть и реакционная спо-
собность. Первый показатель характеризует скорость горения
кокса, второй — скорость восстановления им диоксида углерода.
Качество кокса определяется также содержанием в нем серы,
золы, влаги и выходом летучих. Зола в коксе — это балласт, и
содержание ее равно примерно 10—11 %. Допустимое содержание
влаги в коксе — 5 %. Удаление летучих из кокса понижает его
теплотворную способность.
Состав и выход коксового газа определяютсяzглавным
образом температурой коксования. Из камеры, в которой прово-
дят коксование, выходит «прямой» коксовый газ, содержащий га-
зообразные продукты, пары каменноугольной смолы, сырого бен-
зола, воды, аммиака. После удаления из него смолы, бензола,
воды и аммиака получают «обратный» коксовый газ, который
используют в основном как промышленное топливо для обогрева
коксовых, сталеплавильных и других печей.
219
Каменноугольная смола — вязкая черно-бурая, со спе-
цифическим запахом жидкость, содержащая около 300 различных
веществ. Наиболее ценными компонентами смолы являются аро-
матические и гетероциклические соединения: бензол, толуол, кси-
лолы, фенол, крезол, нафталин, антрацен, карбазол, фенантрен,
пиридин, кумарон, хинолин и др. С повышением температуры
углубляется пиролиз углеводородов, что снижает выход смолы и
увеличивает выход газа.
В настоящее время из каменноугольной смолы выделяют около
двухсот продуктов различных наименований, куда входят смеси
и индивидуальные вещества, служащие сырьем для синтеза кра-
сителей, фармацевтических препаратов, пластических масс, хими-
ческих волокон и т. п.
Сырой бензол — это смесь, состоящая из сероуглерода,
бензола, толуола, ксилолов, кумарона и других веществ. Выход
его зависит от состава и свойств исходного угля и температурных
условий процесса. При ректификации сырого бензола получают
индивидуальные ароматические углеводороды (бензол, толуол,
ксилолы). Сырой бензол и смола, несмотря на развитие нефте-
химического синтеза, служат главнейшими источниками для полу-
чения ароматических углеводородов в химической промышлен-
ности.
Надсмольная вода — слабый водный раствор аммиака и
солей аммония с примесью фенола, пиридиновых оснований и не-
которых других продуктов. При ее переработке выделяют фенол
и аммиак, который совместно с аммиаком коксового газа исполь-
зуют для получения сульфата аммония и концентрированной ам-
миачной воды.
Коксование углей представляет собой высокотемпературный
химический процесс. Реакции протекают сначала только на по-
верхности твердой фазы. По мере повышения температуры обра-
зуются газо- и парообразные продукты, протекают сложные реак-
ции внутри твердой и газовой фаз, а также происходит взаимодей-
ствие между фазами. Повышение температуры — основной фактор,
определяющий протекание процесса коксования. Высокая темпе-
ратура необходима для нагрева шихты до температуры сухой пе-
регонки и проведения эндотермических реакций коксования. Од-
нако повышение температуры ограничивается рядом факторов,
среди которых следует указать на снижение выхода смолы и сы-
рого бензола, изменение состава продуктов коксования, нарушение
прочности огнеупорных материалов, используемых для кладки
коксовых печей.
Коксовые печи относят к печам косвенного нагрева — в них
теплота к коксуемому углю от греющих газов передается через
стенку. Коксовые печи представляют собой батарею, которая вклю-
чает до 70 работающих камер в виде длинных узких каналов
прямоугольного сечения, выложенных из огнеупорного кирпича.
Схема коксовой печи показана на рис. 12.1.
220
Рис. 12.1. Схема коксовой печи:
1—бункеры; 2 — стояк; 3—дверцы камеры; 4—коксовыталкиватель.
Шихта ссыпается порциями через загрузочные бункеры 1, ко-
торые закрываются после загрузки. Уголь выдерживают в печи
14—!Б ч, т. е. до тех пор, пока он не спечется в сплошную массу,
так называемый коксовый пирог, в котором к концу коксования
образуются трещины. Летучие продукты коксования покидают
камеру через стояк 2 и отводятся на. коксохимический завод. Кок-
совый пирог выгружается из печи коксовыталкивателем 4. При
этом дверцы 3 камеры открываются. Далее кокс охлаждают во-
дой (мокрое тушение) или инертными газами (сухое тушение).
Охлажденный кокс подают на сортировку по размерам кусков.
Поперечный разрез коксовой камеры показан на рис. 12.2.
Уголь в камере 1 нагревается через стенки дымовыми газами, ко-
торые проходят по обогревательным простенкам 2, находящимся
между камерами. Горячие дымовые газы образуются при сжига-
нии доменного, обратного коксового или, реже, генераторного га-
зов. Теплоту дымовых газов, выходящих из обогревательных про-
стенков, используют в регенераторах 3, для нагрева воздуха и
газообразного топлива, идущих на обогрев- коксовых
даря этому увеличивается тепловой к. п. д. печи.
Равномерное распределение греющих газов дости-
гается разделением обогревательных простенков
вертикальными перегородками на ряд каналов, на-
зываемых вертикалами.
Обычно толщина стенки, выполненной из дина-
сового кирпича, около 0,1 м. Ширина камеры около
400 мм, длина примерно 14 м, высота около 4 м.
Высота камеры определяется в основном условиями
равномерного ее обогрева. Поверхность теплопере-
дачи зависит от размеров камеры.
Коксовая камера представляет реактор периоди-
ческого действия, и потому температура Ту уголь-
ной загрузки изменяется во времени. В связи с
этим ДТ также изменяется во времени. По мере
печей. Благо-
Рис. 12.2. Попеоечный оазрез коксовой камеры:
1 — камера; 2—обогревательный простенок; 3— регенераторы.
221
Hnt Рис. 12-3. Разрез камеры с шихтой:
*— стенка камеры; 2—кокс; 3 — полукокс; 4—уголь в пластиче-
|! Н - s / 3 ском состоянии; 5—шихта.
______________________________________________
|ё И-5
; увеличения Ту уменьшается количество тепло-
1|к: = н ты, передаваемой в единицу времени, но посте-
КЕчВИ пенно повышается температура по сечению ка-
меры. Если рассматривать состояние материала
в камере в процессе коксования, то видно (рис. 12.3), что у стенок
находится слой образовавшегося кокса, далее по мере снижения
температуры от стенок к оси камеры располагается слой полу-
кокса, затем угля, находящегося в пластическом состоянии, и на-
конец в центре камеры неизмененная шихта. С течением времени
температура по сечению выравнивается, слои перемещаются к оси
камеры и постепенно угольная загрузка прококсовывается. Про-
должительность коксования составляет в среднем 10—15 часов.
В прямом коксовом газе содержатся: водород, метан, этилен
и другие углеводороды, а также смола, аммиак, сероводород,
циановодород и т. п. Газ выходит из коксовой печи при 700 °C.
Из него конденсируют смолу, воду, сырой бензол, улавливают
аммиак и сероводород. Затем подвергают разделению надсмоль-
ную воду, каменноугольную смолу и сырой бензол с получением
индивидуальных веществ или их смесей. Во всех процессах раз-
деления продуктов коксования основным фактором улучшения
технологического режима и увеличения скорости процесса служит
температура. Именно при понижении температуры увеличивается
движущая сила процесса при абсорбции, а при повышении темпе-
ратуры ускоряются процессы десорбции. Для снижения диффу-
зионного сопротивления на границе фаз и соответственного
увеличения коэффициента массопередачи применяют усиленное пе-
ремешивание фаз путем увеличения скоростей подачи газа и жид-
кости. Особенно хорошо сказывается этот прием при противотоке
газа и жидкости в башнях с насадкой. Для создания развитой
поверхности соприкосновения газа и жидкости при переработке
коксового газа применяют башни с различными видами насадок,-
барботажные аппараты, а также разбрызгивание жидкости в по-
токе газа.
Процесс разделения прямого коксового газа начинается в га-
зосборнике 1 (рис. 12.4), в который интенсивно впрыскивается хо-
лодная надсмольная вода. Газ охлаждается примерно до 80 °C,
благодаря чему из него частично конденсируется смола. Одновре-
менно в газосборнике из газа удаляются твердые частицы угля.
Для повышения степени конденсации смолы газ охлаждают до
20—30°C в трубчатых холодильниках 2. С понижением темпера-
туры газа увеличивается растворимость аммиака в конденсирую-
щейся воде и снижается равновесная упругость его над жидкой
фазой. Это приводит к увеличению движущей силы и степени
абсорбции. Смола и надсмольная вода из холодильников стекают
в сборник 3, где разделяются благодаря разной плотности. В хо-
222
Рис. 12.4. Схема переработки прямого коксового газа:
1 — газосборник; 2, 8—холодильники; 3—сборник; 4~электрофильтр; 5 —эксгаустер; 6— подо-
греватель; 7—сатуратор; 9—скрубберы.
лодилышках не удается полностью сконденсировать смолу, так
как она частично превращается в туман. Смоляной туман удаля-
ется из коксового газа электростатическим осаждением в электро-
фильтрах 4.
Для отсоса газа из печей и транспортирования его через аппа-
ратуру устанавливают эксгаустер 5 (турбогазодувка). Аммиак,
остающийся в газе после холодильников, улавливают в сатура-
торе 7, башенной серной кислотой, которая взаимодействует с ам-
миаком, давая кристаллы сульфата аммония. Вместе с аммиаком
в сатураторе улавливают пиридиновые основания с образованием
сульфата пиридиния. Сатуратор — аппарат барботажного типа.
За счет предварительного нагрева коксового газа паром в труб-
чатом подогревателе 6 и теплоты реакции температура в сатура-
торе поддерживается на уровне 60°C. Кристаллы (NH4)2SO4 вме-
сте с маточным раствором выводят из сатуратора, отделяют от
раствора на центрифугах и используют как азотное удобрение.
Коксовый газ, очищенный от аммиака, направляют на улав-
ливание сырого бензола. Наиболее распространенным методом
улавливания является абсорбция сырого бензола поглотительными
маслами при 20—25 °C в скрубберах 9. В качестве поглотителей
применяют каменноугольное (фракция каменноугольной смолы,
кипящая при 230—300 °C) или соляровое масло (фракция, кипя-
щая при 300—350°C). Газ, поступающий в бензольные скрубберы,
предварительно охлаждается водой в холодильниках 8 непосред-
ственного смешения. При этом из газа вымываются нафталин и
мельчайшие брызги серной кислоты, увлеченные из сатуратора.
Освобожденный от сырого бензола коксовый газ (обратный кок-
совый газ) в большинстве случаев очищается от сероводорода и
223
других серусодержащих соединений и поступает потребителю.
Раствор сырого бензола в поглотительном масле направляют
в дистилляционную колонну, где из него отгоняют сырой бензол,
а масло после охлаждения возвращают на орошение бензольных
скрубберов.
12.3. ПЕРЕРАБОТКА НЕФТИ И ГАЗООБРАЗНЫХ ТОПЛИВ
12.3.1. Состав нефти и нефтепродуктов
I Нефть состоит главным образом из парафиновых, нефтеновых
и ароматических углеводородов с большей или меньшей примесью
кислородсодержащих и сернистых соединений. В зависимости от
месторождения соотношение в нефти упомянутых углеводородов
значительно изменяется. Каждый класс углеводородов представ-
лен в нефти многочисленными гомологами и изомерами. Пара-
фины присутствуют в виде нормальных и разветвленных изомеров;
обычно преобладают углеводороды с нормальной цепью.
Нефтены, впервые открытые в нефти В. В. Марковниковым,
представляют собой производные циклопентана, циклогексана, а
также 'более сложных полициклических углеводородов с двумя,
тремя и большим числом колец, имеющих одну или несколько бо-
ковых цепей.
Ароматические углеводороды — бензол и его гомологи, а также
с конденсированными циклами — нафталин, антрацен, фенантрен
и их гомологи — находятся во всех нефтях, но содержание их
значительно меньше, чем углеводородов других классов.
Кроме углеводородов, из остальных соединений, входящих
«в состав нефти, наибольшая доля приходится на кислородсодержа-
щие соединения — смояисто-асфальтеновые вещества, нафтеновые
кислоты и фенолы. Смолисто-асфальтеновые вещества представ-
ляют собой многоядерные гетероциклические соединения со зна
чительной молекулярной массой, содержащие в качестве гетеро-
атомов кислород, серу и азот. Содержание фенолов в нефтях
незначительно (до 0,1 %). Нафтеновые кислоты представлены кар-
боновыми кислотами алициклического ряда. При большом содер-
жании смолистых веществ переработка нефти сильно осложняется.
Весьма вредной примесью нефти являются серусодержащг.е
соединения. Их присутствие отрицательно сказывается в процес-
сах переработки нефти, так как они вызывают коррозию аппара-
туры и влияют на качество получаемых нефтепродуктов. Сера при-
сутствует в нефти в виде сероводорода H2S, меркаптанов RSH,
сульфидов RSR', полисульфидов RSnR', тиофена и его гидропро-
изводных. В нефти встречается и элементарная сера.
Кроме того, нефть содержит минеральные примеси (NaCl,
MgCl2, СаС12, SiO2, А12О3) и воду.
Главные элементы, из которых состоят все компоненты неф-
ти,— углерод и водород, их содержание колеблется в узких пре-
224
'Л
делах: 84—87 % для углерода и 12—14 % для водорода. Наряду
с углеродом и водородом во всех нефтях присутствуют сера (до
8 %), кислород (до 2 %) и азот (до 1 %).
Большое значение для переработки и использования нефти
и нефтепродуктов имеют следующие свойства.
Молекулярная масса М нефти и нефтепродуктов — это
усредненное значение, которое зависит от состава и количестоен-
нЬго соотношения компонентов смеси. Для многих нефтей М =
= 250 300.
Фракционный состав содержащихся в нефти углеводо-
родов определяет качество нефти. Особенно ценны нефти с пре-
обладанием легкокипящих углеводородов (с низкими температу-
рой кипения и плотностью), так называемые легкие. Чем больше
высококипящих углеводородов содержит нефть, тем более тяже-
лой она считается. Ниже представлены температурные пределы
кипения нефтяных фракций:
Дистиллят Температура отбора, ос Фракции нефти Примерный выход от массы нефти, %
Бензин До 170 14,5
Лигроин 160—200 7,5
Керосин 200-300 18,0
Г азойль 300—350 5,0
Остаток—мазут — 55,0
Фракции мазута
Веретенный 230—250 10—12
Машинный 260—305 5
Легкий цилиндровый 315—325 3
Тяжелый цилиндровый 350—370 7
Остаток—гудрон 350-370 27—30
Примечания: Выход дан для грозненской нарафннистой нефти. Температура отбо-
ра фракций мазута — при остаточном давлении 0,008-“0.011 МПа.
Относительная плотность при 20°C (относительная
к плотности воды при 4 °C) р2° характеризует состав, качество
нефти и легкость ее' отделения от воды. Для большинства нефтей
р2о = 0,85-4-0,9.
Вязкость определяет расход энергии при транспортировке
нефти по трубопроводам. Зависит от температуры, резко умень-
шаясь при ее повышении.
Температура вспышки, воспламенения, самовос-
пламенения, пределы взрываемости определяют по-
жаровзрывоопасные свойства нефти и нефтепродуктов.
Теплотворная способность — количество теплоты, ко-
торое выделяется при полном сгорании 1 кг жидкого топлива.
Теплотворная способность основных видов жидкого топлива со-
ставляет 43—44 тыс. кДж/кг.
Сложность состава и разнообразие нефтей обусловливают воз-
можность получения из них большого числа различных продуктов.
^2 8 Зак. 1869 225
- Важнейшие нефтепродукты можно подразделить на следующие
группы: 1) горючие газы; 2) жидкое топливо; 3) смазочные мате-
риалы; 4) парафины и церезины; 5) битумы; 6) нефтяной кокс;
7) нефтехимическое сырье.
Области применения нефтепродуктов определяются их свой-
ствами, которые регламентируются соответствующими стандар-
тами и техническими услсьиями.
Горючие газы, состоящие в основном из пропана и бутана,
образуются в процессах вторичной переработки нефти и применя-
ются в качестве топлива для энергетических установок, комму-
нально-бытового потребления и сырья для промышленности орга-
нического синтеза. Газоснабжение городов осуществляется в га-
зовой фазе системой газопроводов и в сжиженном состоянии —
при помощи баллонов.
Жидкое топливо является главным продуктом перера-
ботки нефти. К этой основной группе нефтепродуктов относят:
бензины (карбюраторное топливо), реактивное, дизельное, газо-
турбинное, котельное и печное топливо.
Авиационные и автомобильные сорта бензина предназначены
для двигателей внутреннего сгорания. Важнейшей характеристи-
кой бензина как топлива является его стойкость к детонации.
Детонация — это взрыв горючей смеси паров бензина с воздухом
в цилиндре двигателя. Детонация приводит к преждевременному
износу двигателя и падению его мощности.
•Антидетонационные свойства, относящиеся к основным пока-
зателям качества бензинов, характеризуются октановым числом.
Чем оно больше, тем в большей степени может быть сжата горю-
чая смесь. Октановое число бензина находят путем сравнения
с различными смесями из гептана (октановое число равно 0) и
изооктана (октановое число равно 100). Оно равно объемной
доле (%) изооктана в смеси, которая детонирует как данный
бензин.
Для повышения октанового числа бензинов их смешивают ,
с продуктами, обладающими высоким октановым числом, или со
специальными веществами, называемыми антидетонаторами. В ка-
честве антидетонатора обычно применяют тетраэтилсвинец ( ГЭС)
РЬ(С2Нб)<, чрезвычайно токсичное вещество. ТЭС вводят в виде
смеси (этиловой жидкости) с галогенированными углеводородами,
антиокислителем и наполнителем. Галогенированные углеводороды
(чаще бромид этила и а-хлорнафталин) способствуют удалению
из двигателя образующихся оксидов свинца путем перевода и>
в летучие соединения.
• По аналогии с бензином свойства дизельного топлива харак- 1
теризуются цетановым числом. Для дизельного топлива важна (
способность самовоспламеняться при сжатии в цилиндре дви- Д
гателя. В качестве эталонного легко самовоспламеняющегося
углеводорода выбран цетан (гексадекан), углеводородом, обладаю- ।
щим нулевой способностью к самовоспламенению, служит а-ме-
тилнафталин. Объемная доля цетана в смеси с а-метилнафталигД
226
ном является цетановым числом дизельного топлива, воспламе-
няющегося в аналогичных условиях со стандартным образцом.
Смазочные материалы применяют для смазки всевоз-
можных машин и механизмов, при обработке металлов резанием,
для трансформаторов и масляных электрических выключателей,
для- герметизации различных систем, при обогащении руд. Важ-
нейшей характеристикой смазочных масел, наряду с температурой
вспышки и застывания, является их вязкость. Наибольшей вяз-
костью и температурой вспышки обладают масла, служащие для
смазки двигателей внутреннего сгорания и паровых машин.
Парафины и церезины — смеси высших парафиновых
углеводородов. Парафины содержат углеводороды нормального
строения с более чем 16-ю атомами углерода в молекуле. Цере-
зины являются смесями твердых углеводородов, главным образом
разветвленных.
Вырабатывают как жидкие, так и твердые парафины. Жидкие
парафины, получаемые карбамидной и адсорбционной депарафи-
низацией нефтяных фракций, являются сырьем для получения
белково-витаминных концентратов, синтетических жирных кислот
и поверхностно-активных веществ.
Твердые парафины и церезины широко используют в электро-
технической, бумажной, текстильной промышленности, для изго-
товления свечей.
Битумы получают из тяжелых нефтяных остатков путем их
окисления. Используют в дорожном строительстве, для гидро- и
электроизоляции, для изготовления кровельных материалов и по-
лиграфических красок.
Нефтяной кокс получают из остаточных продуктов кре-
кинга и пиролиза нефтяных фракций, применяют для изготовле-
ния анодов (для выплавки алюминия), графитированных электро-
дов (для получения стали, хлора, магния).
Нефтехимическое сырье — бензол, толуол, ксилолы,
нафталин и т. д. — применяют в промышленности органического
синтеза и в лакокрасочном производстве.
12.3.2. Методы переработки нефти
Начальной стадией переработки нефти является разделение ее
перегонкой на фракции, выкипающие в определенных темпера-
турных пределах. Прямая перегонка нефти относится к физиче-
скому методу переработки нефти, так как основана на раз-
нице в температурах кипения отдельных компонентов нефти,
входящих в ее фракции. Наиболее дефицитны легкокипящие
(бензиновые) фракции. Для увеличения их выхода высококипящие
фракции перегонки нефти подвергают химической переработке.
В основе химических методов переработки сырья лежат
глубокие химические деструктивные превращения, которые претер-
певают углеводороды, содержащиеся в нефтепродуктах, под влия-
нием температуры, давления, катализаторов. При этом различают
1/28* * 227
термические процессы, когда преобразование углеводородов про-
исходит под действием высоких температур, и термокаталитические,
протекающие при высоких температурах , в присутствии катали-
заторов.
Термические процессы включают: термический крекинг при по-
вышенном давлении различных нефтяных фракций перегонки
нефти с получением дополнительных количеств дегких нефтепро-
дуктов; коксование тяжелых нефтяных фракций при невысоком
давлении с целью получения кокса; пиролиз — высокотемператур-
ный крекинг при невысоком давлении различных нефтяных фрак-
ций с целью получения олефиновых углеводородов.
Основными термокаталитическимн процессами являются: ката-
литический крекинг различных нефтепродуктов — высокотемпера-
турный процесс в присутствии катализатора с целью получения
высококачественного бензина; каталитический риформинг различ-
ных нефтепродуктов — высокотемпературный процесс в присут-
ствии катализатора с целью получения высококачественного бен-
зина и ароматических углеводородов.
Наибольшее распространение среди'этих методов получили
различные виды крекинга. Эти процессы успешно применяют для
переработки самых тяжелых нефтяных остатков — мазута и гудро-
на— с целью получения дополнительных количеств светлых неф-
тепродуктов. Бензины термического и каталитического крекинга
обладают более высокой детонационной стойкостью, чем бензины
прямой гонки, благодаря тому, что в них имеются ароматические
углеводороды и углеводороды разветвленного строения. Октано-
вое число таких бензинов более 70. Газы термического и катали-
тического крекинга — смеси предельных и непредельных углево-
дородов. Крекинг-остаток используют как котельное топливо.
Наиболее жесткий из термических процессов переработки неф-
ти— пиролиз нефтяного сырья. Высокотемпературный режим про-
цесса при атмосферном давлении сырья в паровой фазе позволяет
получить пиролизный газ с большим содержанием олефинов: эти-
лена, пропилена, бутилена. Значение пиролиза нефтяного сырья
за последние годы возросло в результате увеличения потребности
в олефиновых углеводородах для промышленности органического
синтеза. Поэтому внимание отечественной науки привлечено к со-
зданию новых методов пиролиза, позволяющих перерабатывать
тяжелые нефтепродукты и сырую нефть. В настоящее время внед-
рены термоконтактный пиролиз, в котором используется твердый
теплоноситель (шамот, кокс, кварцевый песок), и гомогенный пи»
ролиз в токе водяного пара.
Важнейшей тенденцией в переработке нефти в настоящее время
является стремление возможно полнее использовать все составные
части нефти и максимально увеличить выход светлых нефте-
продуктов, в первую очередь бензина. С этой целью часть образую-
щихся тяжелых остатков (мазут, гудрон) подвергают коксова-
нию— процессу глубокого разложения путем нагревания до 50Э°С
при атмосферном давлении. При коксовании помимо беззольного
228 si
АД
8 Зак. 1869
Мегодм переработки нефти и нефтепродуктов
Г Газойль *"|
J”Рсаюивиое-топлнво j
Бензин ।
। Керосин
। Ароматические j
I углеводороды I
।----------------7
| Жидкий парафин }-*------1 Дизельное топливо j-
Бензол, толуол, "j
ксилолы |
Каталитический крекинг <
I Мазут Вакуум-эерегонка
I Гудрон ।
Дистиллятные j
смазочные масла I
{Твердый парафин [
1 (дпяхзштети- |
। ческнх жирных |
I кислот) I
Термический крекинг
। Газы нефтепе- j
I реработки |
| Авиационный и автомобиль-^
I ный бензин с высоким
1 октановым числом I
г-------------1—।
| Крекинг-остаток ь
Коксование
j Пд ХИМИЧССКуЮ ।
I переработку | | Топливо
j Высокооктановые j | На химическую
I компоненты I | “ '
'Продувка,
воздухом
Остаточные |
J :мазочш’е часла{
г
।--------------' ———у
I Нефтяной кокс для угольных электродов i
Очистка J । Битум I
- — I (для электро- и |
*1 тидроизояядии, |
1получения асфальта)!
U—н । , «--.и
нефтяного кокса, применяемого для изготовления угольных элек-
тродов, получают газ и жидкие нефтепродукты — бензин и га-
зойль — сырье для каталитического крекинга.
Взаимосвязь основных методов переработки нефти и нефте-
продуктов показана на приведенной схеме.
12.3.3. Перегонка нефти
' Сырая нефть не может быть направлена на переработку без
отделения попутного газа, являющегося ценным самостоятельным
продуктом, и вредных примесей.'Газ удаляют из нефти с помощью
сепарации и стабилизации. Метод сепарации заключается в сни-
жении давления нефти, в результате чего растворенные газы отде-
ляются от Нее. Однако даже после многоступенчатой сепарации
в нефти остается значительное количество углеводородов Ci—С,.
,'Поэтому нефть подвергают стабилизации на специальных установ-
ках, расположенных обычно недалеко от места ее добычи.
Установки стабилизации оборудованы трубчатыми печами для
подогрева нефти и ректификационной колонной для отделения
фракций углеводородов.. Смесь углеводородов Ci—С4 (головка ста-
билизации) разделяют на индивидуальные углеводородные фрак-
ции: этановую, пропановую, изобутановую, бутановую.
Вредными примесями нефти являются вода, присутствующая
в ней в виде крупных капель, минеральные соли (NaCl, MgCl2,
СаС12 и др.) и механические примеси (SiO2, А12О3). Воду и соли
удаляют на промыслах и на нефтеперерабатывающих заводах ме-
тодами обезвоживания и обессоливания, в основе которых лежит
разрушение нефтяных эмульсий: при обезвоживании — природной
нефтяной, при обессоливании — искусственной, полученной из оиез-
воженной нефти и волы.
3 промышленности для разрушения нефтяных эмульсии при-
меняют следующие процессы: механические — фильтрование, обра-
ботка ультразвуком; термические — подогрев, промывка горячей
водой с последующим отстаиванием нефти; электрические — обра-
ботка в электрическом поле переменного и постоянного тока; хи-
мические— обработка поверхностно-активными веществами (де-
эмульгаторами) .
Чаще всего для удаления воды и солей применяют способ, со-
четающий термохимические процессы с обработкой в электриче-
ском поле. При прохождении нефти между электродами, включен-
ными в цепь переменного тока высокого напряжения (30—40 кВ),
капельки эмульсии деформируются, сливаются, в крупные капли,
которые затем отделяются от нефти при ее отстаивании.
Схема установки для обезвоживания и обессоливания нефти
(ЭЛОУ) электрическим методом изображена на рис. 12.5.
Нефть забирается из резервуара 1 насосом 2, смешивается
с деэмульгатором и со щелочью или содой (если в нефти содер-
жатся неорганические кислоты), затем нагревается в паровом по-
догревателе 3 и поступает в электродегидратор 4. В этом аппарате
230
Рис. 12.Б. Схема установки для обезвоживания и обессоливания нефти электриче-
ским методом:
1—резервуар для сырья; 2 —васос; 3—паровой подогреватель» 4 — электро дегидратор; 5 — пла-
стинчатые электроды; 6 —водоотделитель; 7—хранилище обезвоженной и обессоленной нефти.
удаляется основная масса воды и солей. Внутри электродегидра-
тора находятся электроды б — металлические пластины, подвешен-
ные на фарфоровых изоляторах. Обессоленная нефть из электро-
дегидратора и вода направляются в водоотделитель 6 для допол-
нительного отстоя. Обработанная таким образом нефть поступает
на перегонку, вода — на очистку.
Эффективное обессоливание позволяет значительно уменьшить
коррозию технологического оборудования, предотвратить дезакти-
вацию катализаторов, улучшить качество топлив, нефтяного кокса,
битумов и других продуктов. Легкие нефти обессоливаются при
80—100 °C и давлении до 0,6 МПа, тяжелые — при 120—140 °C
и давлении 1,2—1,4 МПа.
Для нейтрализации кислых и сернистых примесей, также вызы-
вающих коррозию аппаратуры и снижающих качество нефтепро-
дуктов, в нефть добавляют растворы щелочи или соды.
После удаления примесей на нефтеперерабатывающих заводах
производят сортировку и смешение нефтей для получения равно-
мерного по составу сырья.
Перегонка — первая стадия переработки нефти. Разделение
перегонкой на фракции, отличающиеся по пределам выкипания,
основано на том, что при кипении смесей жидких веществ обра-
зуются пары. Эти пары содержат все компоненты смеси, но с пре-
обладанием легколетучих низкокипящих компонентов. Отгоняемые
пары конденсируются с получением фракции. Каждая последую-
щая фракция содержит более высококипящие компоненты, чем ра-
нее отогнанные. В установке, состоящей из кипятильника и конден-
сатора, нельзя четко разделить компоненты нефти на фракции.
Для достижения этого между кипятильником и конденсатором
устанавливают ректификационную колонну, в которой осуществ-
ляются многократные испарение и конденсация благодаря проти
воточному контактированию паров н жидкости.
Ректификационная колонна 1 (рис 12:6) представляет собой
вертикальный цилиндрический аппарат, заполненный насадкой илг
23)
Рис. 12.6. Ректификационный узел:
/ — ректификационная колонна: 2—конденсатор; 3—прием1
ная емкость для флегмы: 4—насос; 5—тарелки колонны;
6—кипятильник.
Рис. 12.7. Колпачковая тарелка:
1— тарелка; 2—паровые патрубки; 3—колпачки; 4—перс*,
.пивные патрубки. Сплошными стрелками показано дгн’ке
ине жидкости, пунктирными—движение паров
снабженный тарелками 5, расположенными одна на другой. В вер-
тикальную часть колонны подается орошающая жидкость (флег-
ма), которая стекает по насадке или перетекает с тарелки на
тарелку навстречу поднимающимся из кипятильника 6 парам. Оро-
шение колонны ведется легкокипящей жидкостью, которая посте-
пенно обогащается высококипящими компонентами, а поднимаю-
щийся вверх паровой поток — легкокипящими.
Наиболее распространены колпачковые тарельчатые ректифи-
кационные колонны. Колпачковые тарелки (рис. 12.7) имеют па-
ровые патрубки 2, перекрытые колпачками 3. Пары, выходя из-под
колпачка, барботируют через слой жидкости на тарелке 1. Жид-
кость (флегма) стекает с тарелки на тарелку по переливным пат-
рубкам 4. Чем больше в колонне тарелок, тем более четко удается
выделить компоненты исходной смеси.
• Перегонку нефти обычно ведут в две стадии. Сначала на уста-
новке, работающей под атмосферным давлением при 300—350 °C,
отгоняют более легкие нефтепродукты, а затем при остаточном
давлении 0,05—0,08 МПа и 400—420 °C — мазут, чтобы предотвра-
тить разложение отгоняемых масляных фракций при перегреве.
Данные перегонки представлены в разд. 12.3.1.
Установки, в которых последовательно соединены атмосферный
и вакуумный нефтеперегонные аппараты, называют атмосферно-
вакуумными трубчатками (АВТ). В них протекают следующие
процессы: предварительный нагрев сырья за счет отдачи теплоты
продуктами перегонки в теплообменнике, основной нагрев сырья
в трубчатых печах, отделение образовавшихся паров от жидкого
остатка и их рзктификация в колоннах, конденсация и охлаждение
продуктов перегонки в теплообменниках, которые служат подогре-
вателями сырья. Принципиальная схема перегонки нефти при атмо-
сферном давлении представлена на рис. 12.8.
Нефть насосом последовательно прокачивается через теплооб-
менники 4, где она, отнимая теплоту от полученных фракций, подо-
232
Рис. 12.8. Схема установки для перегонки
нефти:
/—трубчатая печь; 2—ректификационная колон-
на; 3, 5 — холодильники; 4— теплообменники.
Рис. 12.9. Схема трубчатой печи:
1 — форсунка; 2— каркас и огнеупорная кладка;
3—камера радиаиян; 4—радиантные трубы-
5—конвекционные трубы; 6 — пароперегрева;
тель; 7— камера конвекции; 8 —газоход; 9 —пе-
ревальная стенка; 10— факел.
4
3
2
7
в
Сырье~
5
Водяной
пар
Вода
Н? '
Сырье
гревается до 170—175 °C и далее поступает в трубчатую печь 1.
В печи нефть нагревается до 300—350 °C и давление ее снижается
почти до атмосферного. Под давлением до 1 МПа нефть поступает
в нижнюю часть ректификационной колонны 2, где происходит
испарение фракций и отделение их от жидкого остатка — мазута.
Пары фракций поднимаются вверх и ректифицируются, стекающая
вниз по колонне испарившаяся жидкость продувается острым во-
дяным паром для удаления легколетучих компонентов. Чем ниже
тарелка, тем более обогащена жидкость на тарелках высококипя-
щими компонентами смеси. Продукты перегонки охлаждаются
сначала в теплообменниках 4 поступающей туда нефтью, а затем
в холодильниках 5 водой. Пары бензина охлаждаются в холодиль-
нике 3, превращаются там в жидкость, после чего отводятся из
верхней части колонны. Жидкий бензин частично в виде флегмы
подается на орошение колонны.
• Принципиальная схема вакуумной перегонки мазута аналогична
рассмотренной схеме перегонки нефти. Мазут из ректификацион-
ной колонны направляют в трубчатую печь через серию тепло-
обменников, где он нагревается до 400—420 °C, и подают в ректи-
фикационную колонну, работающую под вакуумом (остаточное дав-
ление около 0,05—0,08 МПа). Из нижней части этой колонны
выводят гудрон, а по высоте отбирают масляные фракции. Благо-
даря достаточно глубокому вакууму и вводу в колонну перегретого
водяного пара температура в ее нижней части составляет только
380 °C, а в верхней части 220 °C.-Поэтому, а также вследствие
кратковременного пребывания масляных фракций в зоне нагрева,
обеспечивается высокое качество получаемых из них масел.
Важным агрегатом нефтеперегонной установки является труб-
чатая печь (рис. 12.9). Нефть нагревают в печи дымовыми газами.
Передача теплоты от газов к сырью, движущемуся по трубам,
происходит через стенки труб, а также путем радиации (излуче-
ния) или конвекцией.
233
12.3.4. Химические методы переработки нефти
Термические процессы. Переработка нефтяного сырья под дей-
ствием высоких температур значительно расширила возможности
использования нефти как химического сырья. При изучении терми-
ческих реакций углеводородов нефти прежде всего возникает воп-
рос о влиянии условий процесса на направление реакции и на
степень превращения исходного сырья при достижении равновесия
Главными факторами, влияющими на скорость и глубину превра-
щения углеводородов сырья, являются температура, давление, дли-
тельность нахождения в зоне реакции.
Для всех обратимых эндотермических реакций при увеличении
температуры сверх определенного предела равновесие реак-
ции смещается слева направо, т. е. в сторону образования продук-
тов реакции. Такие реакции можно назвать высокотемпературными, 'i
Для большинства реакций синтеза (гидрирование, алкилирование,
полимеризация), являющихся экзотермическими, наблюдается об-
ратная картина, поэтому их называют низкотемпературными. Тер-
мическое разложение углеводородов начинается при 380—400 °C.
С увеличением температуры скорость крекинга быстро растет. По-
вышение температуры крекинга при постоянном давлении приводит
к повышению содержания легких компонентов, к снижению выхода
тяжелых фракций и кокса, причем растет содержание в газе непре-
дельных углеводородов. Для практического осуществления терми-
ческих процессов требуется, чтобы они протекали с достаточной
скоростью и при этом достигалась высокая степень превращения и
избирательность. Для увеличения скорости реакции при жидко-
фазном термическом крекинге и коксовании нефтяного сырья тем-
пературу повышают до 470—550 °C, парофазный процесс веду" при
температуре более 550°С, пиролиз — при 700—900°C. Выход газа
в этих условиях заметно увеличивается, растет содержание в нем.
олефиновых углеводородов.
Для реакций, идущих с увеличением или уменьшением объема,
на состояние равновесия оказывает влияние не только температура,
но и давление. Повышение давления необходимо, например, ,
для реакций гидрирования, алкилирования, полимеризации, пони-
жение — для реакций распада, дегидрирования. Если стремятся
увеличить выход жидких продуктов, процесс проводят при повы-
шенном давлении и, наоборот, если желательно получить больше
газов, целесообразно понижать давление.
Следует учитывать, что при увеличении давления повышается
температура кипения реакционной смеси в зоне термического про-
цесса, а также уменьшается объем паров сырья и продуктов кре-
кинга. Последнее обстоятельство позволяет уменьшить объем
аппаратуры, увеличить производительность установки. Поэтому
термический крекинг проводят при давлении 2—5 МПа, коксова-
ние нефтяного сырья — при 1—2 МПа. Пиролиз осуществляют при
давлении близком к атмосферному, так как целью процесса яв-
ляется получение газообразных олефиновых углеводородов.
л
234
'Рис. 12.10. Зависимость выхода Ф бензи-
на от температуры Т и времени т пребыва-
ния сырья в реакторе.
Оптимальное время пребы-
вания продуктов в зоне реакции,
обеспечивающее высокую избира-
тельность процесса и выход - целе-
вых продуктов, зависит от темпера-
туры. Характер изменения выхода
бензина во времени и его зависи-
мость от температуры показаны на
рис. 12.10.
При увеличении времени пребывания т в зоне высоких темпера-
тур (при Р, Т — const) до оптимального значения возрастает сте-
пень разложения X тяжелых углеводородов и повышается выход Ф
бензина. При дальнейшем увеличении т легкие углеводороды рас-
щепляются с выделением газов и значение Ф падает. В процессах
крекинга степень превращения за один проход через аппарат под-
держивают на уровне 50—70 %. После отделения продуктов кре-
кинга оставшуюся часть нефти смешивают со свежим сырьем
и подают опять в реактор крекинга (крекинг с рециркуляцией).
Несмотря на сложность процессов превращения углеводородов
при термическом крекинге, можно установить некоторые законо-
мерности поведения отдельных групп углеводородов. Парафины
могут при высоких температурах подвергаться реакциям разрыва
цепей и дегидрирования:
^п^2п+2 CmH2m+2 + CmzH2m>
С„Н2„+2 < СпНгп + Н,
В результате в газах накапливается водород. Распад парафи-
нов может происходить по всем связям С — С. Место разрыва
цепи зависит от температуры и давления. Чем выше температура
и ниже давление, тем значительнее увеличивается выход газообраз-
ных углеводородов. Для углеводородов с числом углеродных ато-
мов более четырех при обычных температурах крекинга
470—550 °C распад происходит в центре углеродной цепи.
Для олефинов, полученных в результате разрыва цепей па-
рафинов, характерно большое разнообразие химических превраще-
ний, зависящее от температуры Т и времени пребывания т сырья
в зоне высоких температур. Чем больше Гит, тем глубже идет
распад длинных цепей олефинов:
t
CnHan > 2СтНаот
• CnH2(J >• CmH2m + Cm,H2m«
^n^2n * ^m^2m-2 4" ^m'^2m'+2
^n^2n ' * ^m^2m+2 + Qm’^2m' + ^•n"^2m"-2
t
СдНгп ---► СщНгт—2 + На
235
При температуре 600—700 °C можно осуществить химические
превращения низших олефинов С2 — С4. Повышение температуры
пиролиза до 900 сС и понижение давления способствуют также
протеканию реакций дегидрирования олефинов:
t
С«Н2П —> CnH2n—s + Н2
При этом в продуктах пиролиза накапливаются диолефины,
которые вступают в реакции полимеризации и конденсации с оле-
финами с образованием циклических углеводородов:
t
CnHJn—а —► n(CnH2n-a)
^л^2п-2 + ^п'^2п' * п
Циклоолефины легко дегидрируются до ароматических углево-
дородов:
Нафтены дегидрируются с образованием ароматических
углеводородов, например при дегидрировании циклогексана полу-
чают бензол:
Для них также характерны:
реакции деалкилирования или укорочение боковых парафино-
вых цепей
R
частичная или полная дециклизация полициклических нафтенов
после деалкилирования
распад моноциклических нафтенов на олефины
С2Н4 +
2C3ti6
СН4 +
С4Н8
СвНа
сгн4 + с4н8
236
Ароматические углеводороды наиболее термически
устойчивы. Поэтому они накапливаются в жидких продуктах кре-
кинга в больших количествах.
Ароматические углеводороды с длинными боковыми цепями
способны деалкилироваться:
+ К'
Затем происходит конденсация ароматических углеводородов
с выделением водорода:
Ароматические полициклические углеводороды составляют глав-
ную часть смолы пиролиза и образуют кокс при коксовании неф-
тяного сырья (если целью процесса не является получение нефтя-
ного кокса, то коксообразование весьма нежелательно).
Основные закономерности протекания термического крекинга
справедливы и для термокаталитических процессов.
Термокаталитические процессы. Увеличение выхода и улучше-
ние качества бензина могут быть достигнуты при крекинге нефтя-
ного сырья в присутствии катализаторов. Термокаталитические
превращения углеводородов нефти в настоящее время являются
основными, так как обеспечивают получение высококачественных
бензинов (с октановым числом 74—82), содержащих значительные
количества углеводородов разветвленного строения и ароматиче-
ских соединений. Преимущество каталитического крекинга заклю-
чается в том, что в результате общего ускорения процесса удается
снизить температуру и давление крекинга при селективном уско-
рении реакции образования ароматических, изопарафиновых и изо-
олефиновых углеводородов.
Каталитический крекинг проводят в паровой фазе, в присут-
ствии алюмосиликатного катализатора, при 490—540 °C, под давле-
нием не выше 0,2 МПа.
Алюмосиликатные катализаторы представляют собой твердые
высокопористые вещества с сильно развитой внутренней поверх-
ностью. Эти катализаторы делят на природные и синтетические.
В состав последних входят цеолиты, обладающие тонкопористой
структурой, что делает их прекрасными адсорбентами. Катализа-
торы на основе цеолитов (шариковые, микросферические) позво-
ляют получать большие выходы высококачественного бензина.
237
В СССР наиболее широко применяют цеолитсодержащие шарико-
вые катализаторы марок АШНЦ-3 и ЦЕОКАР-2,. микросферчче-
ские РСГ-2Ц.
Носителем активности поверхностного слоя катализатора, неза-
висимо от марки, является соединение (HAlSiO,)*. Катализатор
вступает в реакции водородного обмена согласно теории карбоний-
иона. Химические превращения происходят за счет иона водорода
катализатора:
(НА1S 1О4)Х —> - хН+ -I- Al S iO;
Протон катализатора взаимодействует с углеводородами на по-
верхности катализатора с образованием ионов карбония:
R—СН=СН2 + Н+ —> R—СН—CHS
Далее ионы взаимодействуют с нейтральными молекулами
с образованием новых активных центров и новых молекул:
R—СН—СН3 + СзН» —> СзН; + R—СН2—СН,
R—СН—СНз + C4Hio —> C4HJ +R—СН2—СНз
R—СН—СНз + С4Нз —> С4Н2 + R—СН=СН2
Развитие цепи происходит в результате разнообразных превра-
щений ионов карбония и водородного обмена с нейтральными мо-
лекулами. Затем ион карбония (катион) взаимодействует с анио-
ном алюмосиликата:
R—СН— СНз + AlSiO; —► R—CH=CH2 + HAlSiO4
Характерная особенность рассмотренного процесса — изомери-
зация парафиновых и олефиновых углеводородов путем перегруп-
пировки водорода в ионе карбония:
R—СН—СНа—СНз —> R—СН—СН2 —► R—С—СН3
£н3 СНз
Вероятно, лимитирующими стадиями термокаталитических про-
цессов являются стадии диффузии реагентов в порах катализатора
и диффузии продуктов реакции из внутренних областей пор к
внешней поверхности, т. е. процесс протекает во внутридиффу-
зионной области. Закономерности этих процессов рассмотрены в
разд. 5.3. Киветика каталитического крекинга, так же как и терми-
ческого, приближенно описывается уравнением первого порядка.
Склонность к превращениям у различных классов углеводоро-
дов при термокаталитическом и термическом крекинге различна.
Ниже приведены ряды основных классов углеводородов по убыва-
нию их склонности к химическим превращениям:
при термическом крекинге
Парафины > Олефины > Нафтены > Ароматические углеводороды
238
при термоката л итическом крекинге
Олефины > Ароматические углеводороды > Нафтены > Парафины
Тем не менее скорость распада парафинов при каталитическом
крекинге в десятки раз больше, а олефинов в тысячи раз больше,
чем при термическом.
Реакции деалкилирования, дегидрирования, распад кольца, ха-
рактерные для крекинга нафтенов, ускоряются в присутствии ката-
лизатора в 500—4000 раз. Дегидрирование и деалкилирование на-
фтенов протекают с перераспределением водорода. В результате
накапливаются ароматические и парафиновые углеводороды. На-
пример:
Изомеризация нафтенов и распад кольца с образованием изо-
олефинов также улучшают качество бензинов:
С—С— с=с— с—
Ароматические углеводороды с боковыми цепями распадаются
с отрывом колец от боковых цепей без расщепления самих колец.
Это приводит к накоплению бензола и олефинов. Бензол практиче-
ски в реакции не вступает и поэтому накапливается в крекинг-
бензине. Гомологи бензола легко изомеризуются с перераспределе-
нием метильных групп. Так, при крекинге лг-ксилол а получают
толуол, триметилбензол, л-ксилол:
239
Продукты кренима
на разделение
Рис. 12.11. Реактор каталитического крекинга
с кипящим слоем катализатора:
I- 7—решетки; 2—колодец с эжектором; 3, 4—катали-
заторопроводы; 5, В—циклоны; 6—контактный аппа-
рат; 9— регенератор.
Указанные выше процессы обусловь
ливают получение стабильных бензи-
нов благодаря малому содержанию в
них олефинов.
При протекании процессов крекин-
га на поверхности катализатора откла-
дывается кокс, который блокирует ак-
тивные центры катализатора и сни-
жает его активность. Поэтому в этих
процессах предусмотрена регенерация
катализатора.
Кратность циркуляции катализато-
ра определяет время его пребывания
в зоне реакции, а следовательно и ско-
рость крекинга. Кратностью цир-
куляции катализатора назы-
вают отношение количества регенери-
рованного катализатора, введенного
в аппарат, к количеству поступающего
вместе с ним сырья. Это отношение
регулируют увеличением скорости по-
дачи катализатора или уменьшением
количества поступающего в реактор
сырья. Кратности циркуляции катализатора обычно принимают в
пределах 4—6.
Основным аппаратом, в котором в настоящее время проводят
каталитический крекинг, является реактор с кипящим слоем ката-
лизатора (рис. 12. 11). В таком аппарате зона регенерации катали-
затора расположена над зоной катализа. Закоксованный катали-
затор из зоны катализа реактора 6 подается горячим воздухом
по катализаторопроводу 4 в зону регенерации 9. Туда же через рас-
пределительную решетку 1 поступает воздух, необходимый для
выжигания кокса. Образующиеся при этом дымовые газы осво-
бождаются в циклонах 5 и 8 от захваченных ими частиц катализа-
тора, которые по трубам поступают обратно в псевдоожиженный
слой. Регенерированный катализатор по катализаторопроводу 3 по-
падает обратно в зону катализа, куда через решетку 7 подаются
пары перерабатываемого сырья. Продукты крекинга проходят цик-
лон 5, где они освобождаются от частиц катализатора, и направ-
ляются на переработку. Отработанный катализатор в нижней части
аппарата обрабатывается паром для удаления с его поверхности
углеводородов.
240
В аппаратах с кипящим слоем алюмосиликатного катализатора
длительность пребывания катализатора в зоне реакции составляет
1,5 — 6 мин.
Основными эксплуатационными показателями реактора яв-
ляются температура и давление. Средняя температура в реакцион-
ной зоне определяется количеством введенных в аппарат сырья
и катализатора, их температурой и свойствами. Температурный
режим работы реактора при постоянном сырье и катализаторе ре-
гулируют изменением температуры предварительного нагрева
сырья и кратности циркуляции катализатора. При высокой крат-
ности циркуляции катализатора средняя температура в реакцион-
ной зоне возрастает и осуществляется более глубокий крекинг.
Однако это приводит к сильному абразивному износу оборудова-
ния.
При повышении давления увеличиваются выход бензина и со-
держание в нем парафиновых углеводородов, но снижается выход
углеводородов Ci — Сз, а также олефиновых и ароматических. Вы-
ход кокса не зависит от давления в условиях промышленного про-
цесса.
Наибольшее распространение в нашей стране получили установ-
ки каталитического крекинга с кипящим слоем микросферического
катализатора мощностью 600—1200 тыс. т/год. Построены укруп-
* ненные комбинированные установки предварительной гидроочистки
сырья, каталитического крекинга и газофракционирования мощ-
ностью 1500—2000 тыс. т/год.
Большое влияние оказывают катализаторы на условия катали-
тического риформинга. При риформинге происходят реакции изо-
меризации парафиновых углеводородов и изомеризация (одновре-
менно с дегидрированием) олефинов, а также гидрирование —
дегидрирование парафинов (с получением ароматических углево-
дородов) и олефинов (с получением парафинов). В процессе ис-
пользуют изомеризующий катализатор (платина, палладий, молиб-
ден) на кислотном носителе (оксид алюминия, алюмосиликат, цео-
лит), который ускоряет реакции гидрирования—дегидрирования.
Кислотная функция катализатора усиливается добавлением промо-
тора (фтора или хлора). Для углубления изомеризующей способ-
ности к катализаторам добавляют редкоземельные элементы (ре-
ний, иридий).
Реакции дегидрирования парафинов и нафтенов сопровож-
даются интенсивным поглощением теплоты. Это вынуждает вести
процесс с участием оксидных катализаторов при 540 °C, платнно-
рениевого катализатора — при 470—520 °C, платинового — при
480—530 °C.
Повышение давления препятствует быстрому отравлению ката-
лизатора, однако при этом снижается выход ароматических угле-
водородов и увеличивается скорость реакции изомеризации и деал-
килирования. Снижение давления смещает равновесие реакций
Дегидрирования и дегидроциклизации в сторону ароматических
углеводородов, т. е. увеличивает селективность каталитического
241
риформинга. Однако при этом резко возрастает скорость закоксо-
вывания катализатора.
При установлении технологических параметров надо учитывать
свойства применяемых катализаторов. Так, при использовании
медленно закоксовывающегося, хорошо регенерирующегося пла-
тино-рениевого катализатора процесс проводят при давлении
1,5—2,0 МПа.
В СССР эксплуатируются установки каталитического рифор-
минга для получения высокооктанового бензина мощностью 300—
1000 тыс. т/год, комбинированные установки каталитического ри-
форминга и выделения ароматических углеводородов мощностью
300 тыс. т/год.
12.3.5. Методы разделения газообразных топлив
К газообразному топливу относят природные и попутные газы,
газы нефтепереработки, коксовый и генераторный газы. Состав при-
родных и попутных газов очень разнообразен и зависит от условий
залегания.
Нефтяные газы в последние годы стали важнейшим видом
сырья для нефтехимического синтеза. Увеличение объема газа, до-
бываемого на промыслах и вырабатываемого в процессах перера-
ботки нефти, а также повышение требований к качеству моторных •
топлив н непрерывный рост потребностей на разнообразное угле-
водородное сырье для нефтехимической промышленности — все
это стимулирует совершенствование существующих и разработку
новых высокоэффективных процессов переработки углеводородных
газов.
Прогресс нефтехимической промышленности неразрывно свя-
зан с интенсификацией процессов газоразделения. Так, газообраз-
ные парафиновые (метан, этан, поопан, бутаны и пентаны), оле-
финовые (этилен, пропилен, бутилены и амилены) и диеновые (ди-
винил, изопрен) углеводороды являются сырьем для получения
различных продуктов, необходимых для народного хозяйства на-
шей страны. Это — спирты, кетоны, кислоты, альдегиды, оксиды,
пластические массы, синтетические каучуки, волокна, моющие
средства и т. д. В связи с этим в последние годы возникла необ-
ходимость создания процессов газоразделения, обеспечивающих
максимальное извлечение углеводородных компонентов.
Природные, попутные газы и газы нефтепереработки состоят
в основном из углеводородов и небольшого количества примесей.
Переработка газового сырья включает очистку газа от примесей,
удаление тяжелых углеводородов, осушку и разделение на фрак-
ции или индивидуальные компоненты. Разделение проводят ком-
бинированием различных методов: адсорбции, абсорбции, конден-
сации и ректификации. Ниже приведена краткая характеристика
отдельных методов переработки газов.
Абсорбция основана на поглощении газообразных веществ
жидким селективным абсорбентом (поглотительный раствор). Пе-
242
Рис. 12.12. Схема абсорб-
ционного метода разделения
природного газа:
1 — абсорбер; 2— холодильник;
3—подогреватель; 4—теплооб-
менник; 5 — десорбер; 6—насосы.
реход молекул газа в раствор характеризуется изменением объема
и уменьшением кинетической энергии молекул, что сопровождается
выделением теплоты абсорбции. Процесс идет эффективно при
противоточном движении газового и жидкого потоков и зависит
от поверхности их соприкосновения. Для увеличения поверхности
абсорбер заполняют насадкой или контактными устройствами —
тарелками.
Разделяемую газовую смесь пропускают через, абсорбционную
колонну, которая орошается растворителем. Низшие углеводороды
(С) — С2) не поглощаются и их отводят из верхней части колон-
ны. Более тяжелые углеводороды абсорбируются орошающей
жидкостью, выводятся из нижней части колонны и затем в десор-
бере выделяются из раствора. Растворители охлаждают и возвра-
щают на абсорбцию.
Для очистки и концентрирования углеводородных газов исполь-
зуют схемы с циркуляцией абсорбента, в которых чередуются про-
цессы абсорбции и десорбции (рис. 12.12). Газ пропускают снизу
вверх через абсорбер (скруббер) 1, заполненный насадкой или
оборудованный тарелками. Противотоком ему движется абсорбент.
После насыщения углеводородами абсорбент поступает в де-
сорбер 5, где из него при повышенной температуре удаляют угле-
водороды. Большей частью применяют тарельчатые десорберы.
Поскольку в процессе очистки газа абсорбент то нагревается, то
охлаждается, большое значение приобретает более полное исполь-
зование физической теплоты раствора. Горячий регенерированный
раствор, движущийся в теплообменнике 4 по одну сторону труб,
отдает теплоту раствору, направляемому на регенерацию. Окон-
чательный подогрев раствора до температуры десорбции и его
охлаждение, необходимое для лучшего поглощения, производятся
соответственно в подогревателе 3 и холодильнике 2. Циркуляция
раствора в системе осуществляется при помощи насосов 6.
Адсорбция — поглощение газов твердыми мелкопористымп
телами с последующей отгонкой и ректификацией поглощенных
углеводородов.
Различают физическую и химическую адсорбцию. При физи-
ческой адсорбции поглощающая способность поверхности объяс-
243
Отходящие
газы.
Рис. 12.13. Схема работы гиперсорбера:
1—зона охлаждения; 2—зона адсорбции; 3—зо-
на десорбции.
няется особым состоянием моле-
кул, находящихся на границе
раздела фаз адсорбент — адсорб-
тив. Внутри поглощаемого веще-
ства каждая молекула испыты-
вает одинаковое притяжение по
разным направлениям. Силы при-
тяжения могут удерживать на по-
верхности адсорбента один или
несколько слоев молекул погло-
щаемого вещества, причем моле-
кулы поглощаемого вещества и
адсорбент не взаимодействуют
между собой. Процесс протекает
быстро, с выделением небольшого
количества теплоты. Физическая
адсорбция не избирательна, на свойства адсорбента влияет только
состояние его поверхности. Скорость физической адсорбции мало
зависит от температуры, так как в основном она определяется ско-
ростью диффузии.
Химическая адсорбция (хемосорбция) сопровождается химиче-
ским взаимодействием поглощаемого вещества с адсорбентом, в
результате последний покрывается пленкой продуктов реакции.
Скорость процесса существенно зависит от температуры и харак-
теризуется значительной энергией активизации.
Химическую адсорбцию широко применяют для очистки, осуш-
ки газов и разделения углеводородных газовых смесей, а также
в процессах гетерогенного катализа. В качестве адсорбентов ис-
пользуют пористые вещества с развитой внутренней поверхностью:
активированный уголь, силикагель, активный оксид алюминия,
алюмосиликаты, цеолиты. В промышленности эксплуатируют уста-
новки по адсорбционному выделению на активированном угле:
пропана из природного газа, этилена из метано-водородных фрак-
ций и продуктов пиролиза метана. Наибольшее применение в про-
мышленности находит гиперсорбция — непрерывное разделение
газовых смесей избирательным поглощением отдельных компонен-
тов газа медленно движущимся слоем активированного угля.
Гиперсорбер — это вертикальный цилиндр (рис. 12.13) высотой
30 м и более, в котором непрерывно движется сверху вниз зерни-
стый активированный уголь. В верхней части аппарата уголь, про-
ходя вертикальные трубки, сушится и охлаждается (зона охлаж-
дения /). Ниже находится зона адсорбции 2. Поступающая в нее
газовая смесь движется противотоком углю, который адсорбирует
более тяжелые компоненты смеси. Непоглощенные легкие углево-
дороды отводятся из верхней части. В нижней части аппарата
244
(зона десорбции <?) уголь продувается паром и из него выделяются
поглощенные углеводороды.
Конденсация — метод, основанный на различной способно-
сти углеводородов к переходу из газообразного в жидкое состоя-
ние. Из многокомпонентной смеси в первую очередь конденси-
руются высококипящие углеводороды.
Конденсация многокомпонентных смесей может быть осуще-
ствлена подбором соответствующих температуры и давления. Про-
цесс проводят в сепараторах. Г аз сначала осушают для предотвра-
щения образования гидратов при низких температурах и освобож-
дают от примесей диоксида углерода и сероводорода, затем
сжимают и дросселируют в сенаратор. Здесь при снижении давле-
ния температура газа понижается и выделяется конденсат. Если газ
обогащен компонентами Cs и выше, его конденсацию ведут при
невысоком давлении и умеренных температурах. При небольшом
содержании высококипящих углеводородов смесь разделяют ме-
тодом низкотемпературной конденсации при 4,0—4,5 МПа и
—70°С и ниже. Газовый конденсат направляют далее на разде-
ление методом ректификации.
Ректификация — метод разделения, основанный на различ-
ных температурах кипения компонентов смеси. При переработке
нефтяного сырья используют низкотемпературную ректификацию
под давлением. Сначала сжатую газовую смесь охлаждают при-
мерно до —100 °C. При этом углеводороды С2 — Cs сжижаются,
а метан и водород остаются в газовой фазе. Сжиженные углеводо-
роды затем подвергают ректификации под давлением при низких
температурах, поддерживаемых путем испарения сжиженных газов
(аммиак, этилен и т. д.) или быстрого понижения давления газов
(дросселирование).
В промышленности для разделения близкокипящих компонен-
тов газов используют аппараты с большим числом решеток и вы-
сокой кратностью орошения {четкая ректификация). Ее проводят
с целью получения индивидуальных компонентов со степенью чис-
тоты 95 % и выше (до 99,99 %). В зависимости от температуры
и давления изменяется относительная летучесть компонентов
смеси: она уменьшается при повышении общего давления и тем-
пературы. Поэтому для лучшего разделения необходимо понижать
давление и температуру, но целесообразность этих мер зависит от
экономических показателей процесса. При глубоком вакууме и
низких температурах легко разделяются углеводороды Ci — С5.
Разделение углеводородов, имеющих небольшую разность
в температурах кипения (5—15°C), а также компонентов с оди-
наковыми температурами кипения и азеотропных смесей, у которых
коэффициент относительной летучести близок или равен единице,
не всегда экономично, а иногда и просто невозможно обеспечить
обычной ректификацией. Для увеличения разности в давлении на-
сыщенных паров разделяемых компонентов в них вводят третий ком-
понент (растворитель или разделяющий агент). Он обладает раз-
личной растворяющей способностью по отношению к разделяемым
245
компонентам, вследствие чего и изменяется их летучесть. В зави-
симости от летучести третьего компонента различают азеотропную
и экстрактивную ректификацию.
При азеотропной ректификации летучесть третьего компонента
сравнительно высока, и он выводится вместе с верхним потоком
из колонны. В экстрактивной ректификации используют малоле-
тучий третий компонент с высокой температурой кипения, который
селективно поглощает определенный компонент и выводится с ним
с низа колонны. Азеотропную ректификацию применяют для раз-
деления близкокипящих углеводородов различного строения. В ка-
честве разделяющего агента для ректификации, например, азео-
тропной смеси углеводородов С< можно применять диоксид серы,
который значительно изменяет температуры кипения компонентов.
Экстрактивную ректификацию широко применяют в нефтепе-
рерабатывающей промышленности, в том числе для разделения
, пропан-пропиленовой и бутан-бутиленовой фракций и для выде-
ления бутадиена. В обычных условиях давление насыщенных
паров компонентов бутан-бутиленовой фракции увеличивается в та-
кой последовательности: бутан < а-бутилен < изобутан. При вве-
дении в бутан-бутиленсвую фракцию фурфурола летучесть «-бу-
тилена (бутена-1) становится самой низкой, и он вместе с фур-
фуролом удаляется с нижней части колонны, а бутаны — с верхней.
Химическая переработка компонентов газов приобретает все
большее значение и является одним из основных направлений
дальнейшего развития химической промышленности. Себестои-
мость продуктов, получаемых из углеводородных газов, значи-
тельно ниже, чем из других видов сырья.
ГЛАВА 13
ОРГАНИЧЕСКИЙ СИНТЕЗ
13.1. ХАРАКТЕРИСТИКА СЫРЬЯ И ОСНОВНЫХ ПРОЦЕССОВ
Отечественная промышленность органического синтеза с каж-
дым годом увеличивает выпуск и ассортимент химических продук-
тов. Эти продукты находят широкое применение в различных
отраслях народного хозяйства. Среди них важнейшими являются:
мономеры и на их основе синтетические смолы, каучуки, пласт-
массы, волокна, клеи, красители, лакокрасочные материалы;
растворители, используемые в качестве реакционной среды во
многих технологических процессах, как экстрагенты для извлече-
ния из нефтепродуктов ценных веществ, а также для изготовле-
ния лаков;
синтетические моющие средства, применяемые в разнообраз-
ных отраслях промышленности и быту;
антидетонаторы и высокооктановые добавки, улучшающие ка-
чество моторных топлив;
246
фреоны для холодильных установок;
антифризы для охлаждения автомобильных и авиационных
двигателей;
высококипящие органические теплоносители для химической
и нефтехимической промышленности;
химические средства защиты растений для борьбы с сорня-
ками, болезнями и вредителями растений и животных.
До середины текущего столетия основным источником сырья
органического синтеза была смола коксования и полукоксования.
Широко использовалось сырье растительного и животного проис-
хождения. В последние десятилетия преобладающее значение при-
обрели жидкие углеводородные нефти, природный и попутный
газы, газы нефтепереработки. Наряду с этим применяют коксовый
и сланцевый газы, смолу коксования, сланцевую и древесную смо-
лу, торфяной деготь.
Главными исходными веществами для производства многочис-
ленных продуктов органического синтеза являются оксид угле-
рода, водород и различные углеводороды: парафины (от метана
до пентаноз), олефины, диолефины (бутадиен, изопрен), аце-
тиленовые и ароматические углеводороды (бензол, толуол, кси-
лол). Использование нефтяного сырья для получения разно-
образных продуктов иллюстрирует схема, приведенная в
разд. 12.312.
Кроме того, в больших количествах применяют неорганические
соединения (кислоты, щелочи, соду, хлор), а также катализаторы.
В качестве катализаторов употребляют газы (оксиды азота, хло-
роводород, аммиак), жидкости (кислоты, щелочи) и твердые
вещества (металлы, их соли, оксиды).
Для проведения технологических процессов необходимо выде-
лить индивидуальные углеводороды из смесей. Это достшается
при помощи методов: сжатие с охлаждением, абсорбционно-де-
сорбционный, адсорбционно-десорбционный, перегонка, ректифи-
кация.
Технология органических веществ базируется на реакциях
синтеза, но в производствах используют и реакции разложения. Ти-
пичные реакции органической химии: галогенирование, сульфиро-
вание, окисление и восстановление, гидрирование и дегидрирова-
ние, гидратация и дегидратация, нитрование, алкилирование, кон-
денсация, полимеризация, поликонденсация.
В процессах органического синтеза, как правило, протекает не
одна химическая реакция, а несколько параллельных и последо-
вательных реакций. В результате, кроме целевого прод)кта, по-
лучают еще побочные продукты и отходы производства. Соответ-
ственно числу реакций коэффициент скорости процесса К может
быть сложной функцией констант скоростей нескольких реакций:
К = f(ki, k2, кз, kt
Селективность процесса по целевому продукту определяется
соотношением скоростей целевой и побочных реакций. Поэтому
для интенсификации процессов органического синтеза применяют
247
селективные катализаторы, ускоряющие лишь основную реакцию.
Для ускорения реакции и увеличения равновесного выхода продук-
та полимеризацию, гидрирование и другие процессы, происходя-
щие с уменьшением объема, проводят при повышенных и высоких
давлениях. При повышенном давлении целесообразно проводить
и процессы абсорбции газов, а под вакуумом — процессы десорб-
ции и дегидрирования.
В газовых реакциях окисления, хлорирования, гидрирования
и других движущую силу ДС и скорость процесса U увеличивают,
варьируя температуру и давление и смещая тем самым равнове-
сие в сторону целевого продукта. При проведении процессов
сорбции это достигают повышением концентрации реагирую-
щих веществ или отводом готового продукта из зоны реакции.
Применение различных средств интенсификации производ-
ственных процессов нередко ограничивается стойкостью орга-
нических соединений, что особенно проявляется в высокотемпе-
ратурных процессах ввиду разложения исходных веществ и
продуктов.
Наряду с катализаторами для увеличения константы скорости
процесса в производстве органических продуктов используют ини-
циаторы, фотосинтез и радиационное облучение. Под действием
облучения можно проводить окисление парафиновых углеводоро-
дов, хлорирование бензола, полимеризацию этилена, получение
привитых полимеров, вулканизацию каучука и т. д. Образую-
щиеся продукты обладают более ценными свойствами, чем полу-
ченные обычным путем. Так, после облучения сульфохлорирован-
ный полиэтилен обладает повышенной теплостойкостью, а также
стойкостью к действию кислот, сильных окислителей и в том числе
к озону.
Для ускорения гетерогенных процессов, идущих во внешнедиф-
фузионной области, применяют усиленное перемешивание фаз для
замены молекулярной диффузии конвективной, что снижает диф-
фузионные сопротивления, препятствующие взаимодействию ком-
понентов. Возможность применения тех или иных способов интен-
сификации определяется и экономической эффективностью, в част-
ности сложностью аппаратурного оформления. Новые более
совершенные аппараты обеспечивают непрерывный процесс по всей
технологической цепочке при комплексной переработке сырья.
Современные заводы органического синтеза представляют собой
соединение различных технологических цехов, не только выраба-
тывающих определенный (основной) продукт, но и включающих
установки, тщательно улавливающие большинство побочных про-
дуктов, бывших ранее отходами.
Процессы органического синтеза влияют на технологические
схемы нефтеперерабатывающих заводов, вызывая глубокое пере-
плетение топливного и химического производства. В связи с тем,
что производства органического синтеза многочисленны и разно-
образны, ниже рассмотрены примеры типичных производств, имею-
щих большое народнохозяйственное значение.
248
$3.2. ПРОИЗВОДСТВО МЕТАНОЛА
Метанол представляет собой бесцветную жидкость с запахом,
подобным запаху этилового спирта. Он горюч, дает с воздухом
взрывоопасные смеси. Концентрационные пределы взрываемости
от 6 до 34,7 % (по объему). Представляет большую опасность
из-за своей высокой токсичности.
Основное количество метанола расходуют для производства
формальдегида. Он также является промежуточным продуктом
для получения сложных эфиров, таких, как диметилсульфат, диме-
тилтерефталат, метилметакрилат. Используют как метилирующий
агент при получении метиламинов, диметиланилина. В чистом виде
применяют в качестве растворителя и высокооктановой добавки
к моторному топливу.
Синтез метанола по физико-химическим условиям его прове-
дения и по технологическому оформлению аналогичен процессу
синтеза аммиака. Синтез-газ (смесь оксида углерода и водорода),
являющийся сырьем для получения метанола, получают из' при-
родного газа или генераторных газов конверсией. Кроме того,
метанол является одним из продуктов при окислении низших па-
рафинов в газовой фазе.
Образование метанола из синтез-газа протекает по обратимой
экзотермической реакции:
СО + 2Н2 СН3ОН + 9
Взаимодействие смеси тщательно очищенных газов происходит
при высоких давлении и температуре в присутствии катализато-
ров. Реакцию никогда не ведут до полного превращения, а приме-
няют непрерывную циркуляцию непрореагировавшей смеси в зоне
катализатора.
Равновесная степень превращения исходной газовой смеси
в метанол растет с увеличением давления и уменьшается с повы-
шением температуры. При низких температурах равновесие сме-
щено в сторону метанола, но реакция протекает слишком медленно.
Для увеличения скорости используют катализаторы, которые ста-
новятся активными только при 300—400 °C (цинк-хромовый ката-
лизатор), когда константа равновесия очень мала. Приходится по-
этому повышать давление, что способствует не только росту сте-
пени превращения, но и подавляет протекание побочных реакций.
Синтез метанола проводят в присутствии цинк-хромовых и мед-
ных катализаторов. Они максимально ускоряют образование мета-
нола и одновременно подавляют побочные реакции образования
метана, формальдегида, диметилового эфира и высших спиртов.
Цинк-хромовый катализатор состава 82пО-Сг2Оз-СгО3 мало чув-
ствителен к контактным ядам и высоким температурам и отрав-
ляется обратимо. Он легко регенерируется и имеет высокую изби-
рательность. Медный катализатор, активированный добавками
Cr2O3, ZnO, V2Os, более активен, чем цинк-хромовый, но менее
стоек к ядам и высоким температурам, имеет меньшую избира-
тельность.
24»
5^2, вО
5?
Ss
§1 t.te
К 0,82
4ООЮЧ~Т
30000
30
20-
10
0
А/* о/
’VCH3OHt%
20 30 40 50
Р,МПа
20000
100004~1
I
320 360 ->00
Температура^
Рис. 13.1. Зависимость объемной доли метанола в реакционной смеси от дав те-
ния и температуры.
Т, ®С: 1—200; 2—24f); 3—280; 4—300* 5—350; 6—400.-Кривая конденсации метанола.
Рис. 13.2. Зависимость производительности катализатора при давлении 30 МПа
от объемной скорости.
>0,25
CH3OH
Общая скорость реакции синтеза определяется адсорбцией во-
дорода на поверхности катализатора. По опытным данным полу-
чено кинетическое уравнение основной реакции:
dX Рц<,Рсп
U “ = kl ' ka р№
“т ^СНзОН гсо
Здесь kt и Аа — константы скорости прямой и обратной реакций; Р — давле-
ние соответствующего компонента смеси.
Зависимость объемной доли метанола в равновесной газовой
смеси от общего давления при равной температуре изображен;
на рис. 13.1. Так как реакция синтеза обратимая экзотермическая
и протекает с уменьшением объема, то для каждого давления
имеется своя оптимальная температура, при которой фактическая
степень превращения синтез-газа или концентрация метанола бу-
дут максимальны.
Синтез метанола на цинктхромовом катализаторе проводят при
370—400 °C и 25 МПа, а на медном — при 220—300 °C и 5—10 МПа
Процесс ведут при объемных скоростях 10 000—40000 ч-1, o6ecnei|
чивающих достаточно высокую производительность катализатора
(рис. 13.2). При дальнейшем возрастании объемной скорости (на
пример, до 100 000 ч-1) существенно повышается расход электро
энергии на циркуляцию газов из-за снижения степени превраще-
ния за один проход исходной смеси.
Устройство колонны синтеза метанола аналогично устройству
колонны синтеза аммиака. Колонна, в корпусе которой совмеще;..
катализаторная коробка, электроподогреватель и теплообменник;
лучше позволяет осуществлять теплообмен и тем самым обеспечи-
вает приближение к оптимальному температурному режиму.
ЯбО
S
Рис. 13.3. Схема колонны синтеза метанола:
1— катализаторная коробка: 2—корпус колонны: 3—электро-
подогреватель; 4—теплообменник.
На рис. 13.3 приведена схема полочной
колонны синтеза с фильтрующими слоями ка-
тализатора при адиабатическом режиме в
каждом слое. Синтез-газ вводится сверху меж-
ду катализаторной коробкой 1 и корпусом ко-
лонны 2, затем поступает в межтрубное про-
странство теплообменника 4, где подогрева-
ется за счет теплоты контактных газов, прохо-
дящих по трубкам теплообменника. Охлаж-
денные контактные газы выходят из нижней
части колонны. Во избежание перегрева ката-
лизатора предусмотрена подача холодного
газа. Внутри аппарата смонтирован электро-
подогреватель 3 для разогрева газа в пуско-
вой период. Внутренняя поверхность колонны
и теплообменника облицована красной медью
или выполнена из высоколегированной стали
для защиты от коррозии.
В настоящее время производство метанола
осуществляют в основном на низкотемпера-
'Основной поток
синтез-гава
Байпасный
поток
дурных медьсодержащих катализаторах в cawres—газа.
высокоэффективных агрегатах большой еди-
ничной мощности. Выход метанола составляет около 4 % за один
проход, общий — более 95 %.
Технологическая схема производства метанола на низкотемпе-
ратурном медном катализаторе представлена на рис. 13.4.
НепрореагцроВабший синтез-юз
Легкокипящие продукты
Продукты синтеза
метанола
Вода с органичес-
кими примесями
Товарный
метанол
Свежий
синтез-газ
Метанол-
сырец
Рис. 13.4. Технологическая схема синтеза метанола с использованием низкотемпе-
ратурного медного катализатора:
1—центробежный компрессор: 2—теплообменник; 3—реактор синтеза метанола; 4—холодиль-
ник; 5—сепаратор; е, 7—ректификационные колонны.
25k
Газ сжимается центробежным компрессором 1 до давления
5 МПа, нагревается в теплообменнике 2 отходящими газами до
250 °C и поступает в реактор синтеза 3. Синтез проводят при
220—300 °C. Регулирование температуры в реакторе осуществ-
ляют с помощью струи холодного газа, подаваемсго по всей вы-
соте реактора через распределители. Производительность реактора
500 т метанола в сутки. Продукты синтеза после теплообменника
охлаждаются в холодильнике 4. Сконденсированный метаиол со-
бирается в сепараторе 5, а непрореагировавшие газы смешиваются
со свежим синтез-газом и вновь направляются в реактор синтеза.
Метанол-сырец из сепаратора подается на ректификационную
колонну 6. В ее верхней части отгоняются легкокипящие примеси
(главным образом, диметиловый эфир и растворенные газы), а ку-
бовый остаток поступает на питание колонны 7, из которой отби-
рается товарный метанол. В качестве дистиллята колонны 7 от-
гоняется вода, а в виде кубового продукта отводится небольшое
количество смеси высших спиртов.
Метод синтеза с использованием низкотемпературного медного
катализатора позволяет получать метанол высокой степени чис-
тоты (99,85%).
Основные опасности процесса получения метанола нз синтез-
газа обусловлены: 1) высокой температурой процесса и возмож-
ностью значительного ее повышения в колонне синтеза вследствие
высокой экзотермнчности основной реакции; 2) высоким давле-
нием водорода, что сопряжено с возможностью его утечки н само-
воспламенения на воздухе.
Для обеспечения эффективного отвода теплоты и поддержания
необходимого температурного режима в кслонне синтеза между
слоями катализатора в пространство между полками устанавли-
вают охлаждающие змеевики. В отдельных случаях для отвода
теплоты реакции предусматривают принудительную циркуляцию
реакционной смеси через выносной холодильник.
Для поддержания необходимого температурного режима про-
цесса подбирают скорость парогазовой смеси в аппарате и высоту
слоя катализатора такими, чтобы не происходило чрезмерного
перегрева реагирующих веществ.
13.3. ПРОИЗВОДСТВО ФОРМАЛЬДЕГИДА
Простейший из алифатических альдегидов — формальдегид
<СН2О — представляет собой бесцветный газ с резким запахом,
вызывающий слезотечение, хорошо растворимый в воде и спирте,
температура кипения —19 °C.
Формальдегид применяют главным образом в виде водного рас-
твора — формалина, который содержит 37 % (по массе) формаль-
дегида. Как и исходный продукт, его используют в многочислен-
ных органических синтезах, в качестве дезинфицирующего и дез-
инсекционного средства. В больших количествах формальдегид
252
употребляют для производства полимеризационных и поликонден-
сационных полимеров, получения изопрена н бутадиена, синтети-
ческих смол для химических волокон, этиленгликоля, глицерина,
пентаэритрита, красителей, взрывчатых веществ, фармацевтиче-
ских препаратов и т. п.
В промышленности формальдегид получают двумя путями: не-
полным окислением метана и его гомологов и окислительным
дегидрированием метанола.
Окисление метана в формальдегид осуществляют в газовой
фазе при атмосферном давлении и температуре порядка 500 °C
в присутствии различных катализаторов: металлов, их оксидов
и солей. Окислителями чаще всего служат воздух и технический
кислород.
При неполном окислении метана сначала образуется метанол,
а из него формальдегид:
СН* + 0,5О2 —> СН3ОН + <7 (1>
СН3ОН + 0,бО2 —> СН2О + Н2О + q (2}
При атмосферном давлении основным продуктом является
формальдегид, при высоком давлении и большом избытке метана
в газовой смеси — метанол.
Для преимущественного протекания реакции (2) в качестве
катализаторов применяют соединения меди и серебра, а также
оксиды азота, задерживающие процесс окисления на стадии обра-
зования формальдегида. Дальнейшее окисление формальдегида
может быть частично предотвращено при быстром удалении про-
дуктов реакции из зоны высоких температур. Для этого метан
и кислород пропускают через реакционную зону с большой ско-
ростью. Способ экономи щеки невыгоден из-за малого выхода
формальдегида.
Основным промышленным способом получения формальдегида
в настоящее время является окислительное дегидриро-
вание метанола. Процесс протекает в две ступени:
2СН4ОН + О2 —> 2СН2О + 2Н2О + q (3)
СН3ОН ч=± СН2О + Н2-9 (4)
Около 90 % формальдегида образуется по реакции (3). Теоре-
тически выход формальдегида по реакции (4) возрастает с по-
вышением температуры, достигая 99 % при 700 °C. Однако при
этом возрастает скорость его распада:
НС ' СО -ь Н2 - q
\н
Лишь в присутствии кислорода, связывающего выделяющийся
водород, выход формальдегида по реакции (4) увеличивается.
Особенностью процесса является протекание его во внешне-
диффузионной области в режиме адиабатического разогрева ка-
тализатора до 600—700 °C. Серебро в качестве катализатора
26S-
Формалин
Рис. 13.5. Схема получения формальдегида окислением метанола:
/ — испаритель; 2—подогреватель; 3—контактный аппарат; 4—холодильник; 5—барботажный
холодильник; 6—абсорбер; 7—теплообменники; 8—ресивер; 9 —вакуум-компрессор; /0—водо-
отделитель.
окисления метанола используют в нескольких модификациях: губ-
чатое, в виде сеток, крупнокристаллическое, а также нанесенное
на крупнопористые носители. В промышленности наибольшее рас-
пространение получил серебряный катализатор на пемзе, отли-
чающийся высокой производительностью и при отсутствии в ме-
таноле вредных примесей (высшие спирты, кетоны, эфиры, непре-
дельные соединения, пентакарбонил железа) — большим сроком
службы. Температура процесса определяется составом спирто-воз-
душной смеси и возрастает с увеличением содержания кислорода.
Метанол с воздухом образует взрывоопасные смеси, пределы
взрываемости которых 5,5- -36%. Смесь, поступающая на контак-
тирование, содержит не менее 36—40 % метанола, что превышает
верхний предел взрываемости спирта. Времц пребывания продук-
тов в зоне реакции должно быть минимальным. Для увеличения
скорости прохождения паровоздушной смеси через зону реакции
реактор выполняют сверху и снизу в виде конуса. Так как форм-
альдегид склонен к процессам поликонденсации, необходимо
после реакции резко снизить температуру контактных газов. Ис-
ходный метанол содержит не менее 10—12 % (по массе) воды,
служащей для подавления побочных реакций окисления метанола
до СОг, окисления формальдегида, образования метана:
2СН3ОН + 30, —► 2СОа + 4Н2О
2СНаО + Оа —> 2НСООН
2СНаО + 2Оа —► 2СОа + 2НаО
СН3ОН-ЬНа —► СН4-ЬНаО
Схема получения формальдегида окислением метанола пред-
ставлена на рис. 13.5.
Очищенный от пыли воздух при 45 °C барботирует через мета-
нол в испарителе 1. При этом 70—90 % водного раствора мета-
254
ноля испаряется в токе воздуха с образованием спирто-воздушной
смеси. Она подогревается до ПО °C в подогревателе 2 и поступает
в контактный аппарат 3. Большое значение для нормального ве-
дения процесса имеет постоянство состава спирто-воздушной смеси,
так как его изменение может понизить выход формальдегида,
а уменьшение концентрации спирта в смеси создает опасность
взрыва. Это постоянство достигается автоматическим регулирова-
нием уровня спирта в испарителе, температуры воздуха и спирта,
а также давления в системе.
В контактном аппарате происходит неполное окисление мета-
нола и формальдегида. Чтобы затормозить дальнейшее превраще-
ние формальдегида, продукты реакции из зоны катализатора сразу
же отводят в холодильник 4, смонтированный как одно целое
с контактным аппаратом. Из холодильника 4 контактные газы,
охлажденные до 125 °C, поступают в холодильник 5, а далее в аб-
сорбер 6 колонного типа, орошаемый холодной водой, которая
поглощает формальдегид. Теплота абсорбции контактных газов
отводится в выносных холодильниках.
Из нижией части абсорбера вытекает формалин и около 10 %
СНзОН. Метанол стабилизирует формальдегид, предотвращая его
полимеризацию. Выход формальдегида составляет 82—84 %. Для
получения концентрированного раствора формальдегида формалин
подвергают ректификации. Освобожденные от формальдегида газы
(смесь N2, Нг, СН4, СОг, Ог) удаляются в атмосферу.
Вследствие побочных реакций суммарный тепловой эффект
процесса значительно в яше, чем тепловой эффект окисления ме-
танола Теплоту реакций используют для обогрева испарителя t
и подогревателя 2, т. е. для испарения спирта. Степень превраще-
ния метанола за один проход реакционной смеси через серебря-
ный катализатор не превышает 86 %, что объясняется невысокой
избирательностью катализатора.
Для процесса характерно низкое гидравлическое сопротивле-
ние слоя катализатора и незначительные затраты электроэнергии
при эксплуатации, малые габариты и высокая производительность
контактных аппаратов при небольших удельных капитальных за-
тратах. Недостатками процесса являются неполное превращение
метанола, невысокая избирательность по формальдегиду, чувстви-
тельность серебра к каталитическим ядам.
В настоящее время разработан промышленный процесс окисле-
ния метанола на оксидном железомолибденовом катализаторе.
Катализатор применяют в виде гоанул. Активным компонентом
является твердый раствор МоО3 в ге2(МоО4)3. Процесс протекает
без внешнедиффузионного торможения при 220—370 °C в отличие
от процесса окисления на серебряном катализаторе. Это увеличи-
вает степень превращения метанода за проход до 99,5 %.
Окисление метанола на оксидных катализаторах протекает по»
окислительно-восстановительному механизму:
СНзОН+ 2MoOs —> СН2О + Н2О + MosOj
Мо»О» + 0.5О2 —► 2МоОз
МЬ
Лимитирующая стадия как основной, так и побочной реак-
ции— взаимодействие метанола с поверхностным кислородом.
Образующаяся вода оказывает тормозящее действие на обе реак-
ции. Кроме того, метанол тормозит окисление формальдегида до
оксида углерода и воды.
Для поддержания активности катализатора реакцию необхо-
димо проводить в избытке кислорода, поскольку в противном слу-
чае под действием метанола и формальдегида катализатор быстро
восстановится. Из этого с учетом пределов взрываемости реак-
ционных смесей следует важнейшая особенность получения форм-
альдегида на оксидных катализаторах: реакцию проводят при со-
отношении реагентов и воздуха ниже нижнего предела взрываемо-
сти, а именно при содержании метанола в исходной смеси не
выше 7—8 %.
Образование формальдегида протекает по реакциям:
2СНзОН + О2 —> 2СН2О + 2Н2О + q
2СН2О + О2 —> 2СО + 2Н2О + q
Окисление формальдегида является единственной побочной
реакцией. Это позволяет исключить стадию очистки формальде-
гида от метанола. Реакция окисления метанола имеет порядок по
спирту несколько меньший 1, скорость ее не зависит от концентра-
ции СН2О и при мольном отношении кислород/метанол, большем 2,
не зависит также от концентрации кислорода. Окисление форм-
альдегида тормозится водой и метанолом, но эти реакции проте-
кают на разных участках поверхности катализатора. Это не вызы-
вает заметного внешнедиффузионного торможения основной реак-
ции получения формальдегида.
Технологическая схема производства формальдегида на желе-
зомолибденовом катализаторе включает следующие стадии:
1. Нагрев спирто-воздушной смеси в теплообменнике за счет
теплоты отходящих из реактора продуктов окисления.
2. Окисление метанола в реакторе.
3. Охлаждение продуктов окисления в теплообменнике путем
отдачи теплоты метанолу, воздуху и циркуляционной газовой
смеси.
4. Поглощение формальдегида водой в абсорбционной колонне
с образованием формалина и циркуляционной газовой смеси.
5. Окисление СО и следов формальдегида в дожигателе перед
выбросом в атмосферу (одна треть газов после абсорбции), воз-
вращение циркуляционной газовой смеси через теплообменник
в реактор.
Для проведения процесса разработан комбинированный реак-
тор. Он состоит из последовательно расположенных трубчатой
части и адиабатической секции. Применение такой конструкции
позволяет резко понизить гидравлическое сопротивление системы
и расход электроэнергии. Чистый сухой формальдегид стоек при
хранении, но в присутствии следов влаги начинается его полиме-
ризация с образованием полиоксиметилена. ,
256
Железомолибденовый катализатор мало чувствителен к каче-
ству метанола и к каталитическим ядам, однако его производи-
тельность существенно ниже, чем серебряного. Недостатками
процесса являются более высокие удельные капитальные затраты,
повышенный расход электроэнергии и более сложная технологиче-
ская схема, чем при производстве формальдегида на серебряном
катализаторе.
13.4. ПРОИЗВОДСТВО АЦЕТИЛЕНА
Ацетилен — бесцветный газ, обладающий в чистом виде сла-
бым эфирным запахом. Сжижается при —83,6 °C. В жидком виде
может сохраняться лишь при повышенном давлении. Ацетилен
растворим в воде: при 18°С и атмосферном давлении в 1 объеме
воды растворяется 1 объем ацетилена. Наличие тройной связи
в молекуле ацетилена обусловливает его высокую реакционную
способность: его широко применяют в многочисленных промышлен-
ных синтезах.
Ацетилен интересен с термодинамической точки зрения. В отли-
чие от других углеводородов реакции его образования из элемен-
тов и из метана являются сильно эндотермическими:
2С 4- Н2 < х С2Н2 — q~,
2СН« С2Н2+ЗН2- q
Ацетилен является исходным сырьем для многих производств.
Галогенпроизводные на его основе — хорошие растворители;
уксусный альдегид, получаемый из ацетилена по реакции Куче-
рова, перерабатывают в этиловый спирт и уксусную кислоту, а из
винилхлорида, также полученного при участии ацетилена, произ-
водят высокомолекулярное соединение — поливинилхлорид. При-
соединение к ацетилену циановодорода приводит к образованию
акрилонитрила, полимер которого идет на производство волокна
нитрон. Ацетилен используют также в производстве простых
и сложных эфиров, полимеры которых применяют в медицине,
лакокрасочной промышленности, в производстве пластмасс, в ав-
тогенной сварке и резке металлов либо в смеси с кислородом,
либо вместе с кислородом и водородом. Такие смеси при горении
развивают очень высокую температуру (до 2800 °C).
В настоящее время ацетилен получают двумя методами: из
карбида кальция (более половины производства) и пиролизом
низкомолекулярных газообразных алифатических углеводородов.
При производстве ацетилена из карбида каль-
ция процесс идет по реакциям:
СаС2 4- Н2О —> CaO 4- С2Н2 4“ q
СаО4-Н2О —► Са(ОН)24-<7
Суммарная реакция
СаС 2 4“ 2Н2О —► Са(ОН)2 4- С2Н2 4- q
267
Выход ацетилена из технического карбида кальция зависит от
чистоты исходного продукта и полноты его разложения. Литраж
технического карбида кальция (объем ацетилена, полученного при
полном разложении водой 1 кг карбида кальция) составляет от
230 — до 320 л/кг. Разложение карбида кальция осуществляют
в ацетиленовых генераторах мокрым и сухим способом.
При мокром способе дробленый карбид кальция равномерно
подают в генератор типа «карбид в воду», заполненный большим
количеством воды, что обеспечивает высокую безопасность работы.
Это очень важно, если учесть, что концентрационные пределы
воспламенения ацетилена 2,5—100 % (по объему), минимальная
энергия зажигания 0,01 мДж.
Выделяющаяся при реакции теплота отводится за счет нагре-
вания воды. Удаление гидроксида кальция в виде пульпы (содер-
жит до 70 % воды) осуществляется благодаря 10-кратному ее
количеству по отношению к м-ассе СаСг-
Недостатком прсцесса является высокий расход электроэнер-
гии (10—11 кВт-ч на 1 кг ацетилена) на удаление образующегося
шлама и циркуляцию воды, громоздкость оборудования, достоин-
ством — получение чистого ацетилена с малым содержанием при-
месей.
При сухом способе производства применяют генераторы, ра-
ботающие по принципу «вода на карбид», и воду добавляют только
для проведения реакций и отвода теплоты за счет испарения
избытка воды. Схема производства представлена на рис. 13.6.
Карбид кальция из бункеров 1 и 3 шнеком 4 подается на верх-
нюю полку генератора 5 и обильно смоченный водой перемещается
гребками по спирали сверху вниз через отверстия в полках.
Пройдя 11 полок, он практически полностью (на 98%) превра-
щается в С2Н2 и Са(ОН)г. Гидроксид кальция в виде сухого по-
рошка выходит из нижней части генератора. Ацетилен вместе
с водяными парами и известковой пылью при 100 °C поступает
в скруббер 7, орошаемый водой. Образовавшееся известковое мо-
локо из нижней части скруббера непрерывно выводится в отстой-
ник. Осветленную воду из отстойника через холодильник снова
направляют в скруббер.
Ацетилен, полученный из карбида кальция, имеет высокую чис-
тоту (99,5%) и содержит в качестве примесей NH3, РН3, H2S.
Если ацетилен идет на химическую переработку, его очищают,
промывая водным раствором гипохлорита с добавлением актив-
ного хлора или раствором дихромата натрия в разбавленной сер-
ной кислоте.
Выход ацетилена увеличивается при повышении температуры
и понижении давления.
Методы пиролиза низкомолекулярных предель-
ных углеводородов с целью получения ацетилена разли-
чаются способами подвода теплоты к реагирующей газовой смеси.
Высокие температуры, требуемые для разложения углеводородов,
достигаются тремя способами:
258
Газы крекинга
Рис. 13.6. Схема производства ацетилена из карбида кальция:
1— приемный бункер; 2—автоматический затвор; 3—буферный бункер; 4—шнек; 5—ацетиле-
новый генератор; 6—шнек для удаления извести; 7—скруббер; 8—отстойник; 9—холодильник.
Рнс. 13.7. Печь электрокрекинга:
1— катод; 2—реакционная камера; 3—заземленный анод; 4—пусковой электрод.
1. При помощи электрической дуги (электрокрекинг).
2. Прямым или регенеративным нагревом (собственно пи-
ролиз).
3. Сжиганием части сырья (окислительный пиролиз).
Общим условием достижения максимальных выходов ацетилена
является применение высоких температур при кратковременном
пребывании сырья в реакционной зоне, а также быстрое
охлаждение («закалка») продуктов реакции. Это предотвращает
протекание побочных реакций и разложение ацетилена на эле-
менты.
Электрокрекинг заключается в быстром пропускании углеводо-
родов через электрическую дугу, с помощью которой получают
высокую температуру в зоне реакции. Электрическая дуга соз-
дается постоянным током напряжением 7000—8000 В. Такая элек-
трическая печь (рис. 13.7) имеет мощность по метану 2800 м1 2 3/ч,
что дает производительность по ацетилену 15 т/сут. Производство
ацетилена электрокрекингом обходится дешевле, чем карбидным
методом.
Для осуществления собственно пиролиза необходима печь такой
конструкции, в которой газ в течение весьма малого времени мог
бы нагреваться до 1400—1500 °C. Этим условиям отвечает регене-
ративная печь периодического действия.
259
При окислительном пиролизе метан расщепляется за счет теп-
лоты, выделяющейся при сжигании части rasa с кислородом, по-
даваемым в реактор.
Газы, подвергаемые пиролизу, проходят через раскаленную
огнеупорную насадку, нагревающуюся при сжигании газообразного
топлива. Насадка выполнена из чистого оксида алюминия в виде
параллельно расположенных фасонных пластин, между которыми
для прохода газа образованы каналы диаметром 6 мм. Лучшие
результаты получены прн пиролизе пропана, так как для пиролиза
метана требуется более высокая температура, а максимальная
температура в печи не может быть выше 1250—1300 °C.
Процесс, протекающий в реакторе, можно выразить следую-
щей суммарной реакцией:
11СН« + 7О2 —► 2С2Н2 + 6СО+ 14Н2 + СО2 + 6Н2О + <7
Для получения максимального выхода ацетилена требуется:
Содержание кислорода в газе, % 98
Соотношение кислорода и метана (0,6-5-0,64): 1
Температура, °C
кислорода 400—600
процесса 1450— 1500
Время пребывания газа в зоне реак- 0,004—0,006
ции, с
Чтобы предотвратить обрыв пламени в реактор вводят не-
большое количество кислорода: это обеспечивает поджигание газа,
т. е. стабилизацию факела.
Для получения 1 т ацетилена окислительным пиролизом ме-
тана требуется 1000 м3 природного газа (600 м3 на процесс
и 400 м3 на подогрев) и 3600 м3 98%-ного кислорода, при этом
в качестве побочного продукта образуется 10300 м3 синтез-газа.
Расход электроэнергии составляет 1570 кВт-ч на 1 т ацетилена,
т. е. меньше, чем при карбидном методе и электрокрекинге. Окис-
лительный пиролиз метана является более экономичным, а потому
и более предпочтительным методом получения ацетилена. Однако
при наличии дешевой электроэнергии электрокрекинг выступает
как конкурентоспособный процесс.
13.S. ПРОИЗВОДСТВО УКСУСНОЙ кислоты
Безводная уксусная кислота замерзает при 16,6 °C, образуя
кристаллы в виде льда. Такую кислоту называют ледяной.
Уксусная кислота является важнейшим продуктом промышлен-
ности органического синтеза. Она широко применяется в пищевой
и текстильной промышленности и является ценным промежуточ-
ным продуктом в органическом синтезе. Из нее получают моно-
хлоруксусную кислоту, сложные эфиры, винилацетат, уксусный ан-
гидрид и ряд других ценных продуктов.
С давних пор уксусную кислоту получают сухой перегонкой
дерева и брожением этанола. Эти способы не смогли удовлетво-
260
рить растущую потребность в ней, и с начала XX века производят
синтетическую уксусную кислоту.
Синтетические методы производства уксусной кислоты можно
классифицировать следующим образом:
методы, основанные на окислении ацетальдегида
СНз—СНО + 0,5О2 —► СН3—СООН
жидкофазное окисление бутана
СНз—СН,—СНг—СН3 + 2,5О2 —*> 2СН3—СООН + Н»О
синтез из метанола и оксида углерода
СНзОН+СО —► СНз—СООН
Наиболее эффективны первые два метода. По имеющимся
данным себестоимость уксусной кислоты, полученной из бутана,
на 30 % ниже, чем из ацетальдегида. Тем не менее синтез уксус-
ной кислоты из ацетальдегида сохраняет свое значение, в особен-
ности при том варианте, когда совместно получают кислоту и ан-
гидрид.
Первым молекулярным продуктом окисления ацеталь-
дегида является надуксусная кислота
СНз—СНО + О2 —► СНз—СОООН
которая в присутствии солей металлов переменной валентности
(ацетаты кобальта и особенно марганца) становится нестабиль-
ной. Она окисляет находящийся в избытке ацетальдегид в уксус-
ную кислоту и сама тоже восстанавливается до кислоты:
СНз—СНО + СНз—СОООН —*• 2СНзСООН
На этом основан промышленный метод получения уксусной
кислоты из ацетальдегида.
Один из продуктов окисления ацетальдегида — уксусный ангид-
рид. Выход его может быть увеличен применением специальных
катализаторов, представляющих собой смесь металлов переменной
валентности (марганца и меди, кобальта и меди). Поскольку при
образовании уксусного ангидрита выделяется вода, происходит
его гидролиз с образованием кислоты:
2СН3—СНО + Оз —> (СНзСО)зО + Н»О —► 2СН3—СООН
Побочными продуктами являются: метилацетат, этилиденди-
ацетат, муравьиная кислота и диоксид углерода.
Выбор, количество катализатора и температура во многом
определяются необходимостью создания благоприятных соотно-
шений между скоростями всех стадий процесса. Так, применение
в качестве катализатора солей кобальта, меди, железа и снижение
температуры ведут к накоплению надуксусной кислоты. Это уве-
личивает взрывоопасность производства. Повышение температуры
ограничивается высокой летучестью ацетальдегида и развитием
побочных реакций, снижающих выход уксусной кислоты.
Производство состоит из трех стадий:
261
Рис. 13.8. Схема получения уксусной кис-
Отходящий газ
Ацетальдегид
ц катализатор
лоты окислением ацетальдегида:
/ — окислительная колонна; 2—конденсатор;
3—сепаратор.
1. Окисление ацетальдегида.
2. Выделение непрореагиро-
вавшего ацетальдегида из паро-
газовой смеси.
3. Выделение уксусной кисло-
ты из реакционной смеси.
В качестве катализатора ис-
пользуют 5 % раствор ацетата
марганца в циркулирующей ук-
сусной кислоте.
Реакцию проводят при 60—80 °C и давлении 2—3 МПа. Окис-
лителем служит технический кислород. Окислительная колонна 1
(рис. 13.8) выполнена из алюминия или нержавеющей стали
и снабжена полками, между которыми расположены змеевиковые
холодильники. Верхняя часть колонны расширена и играет роль
брызгоуловителя.
Выделяющаяся теплота отводится за счет циркуляции реак-
ционной смеси и охлаждения окислительной колонны. В верхнюю,
расширенную часть поступает азот для предупреждения образо-
вания взрывоопасных концентраций надуксусной кислоты в ко-
лонне. В нижнюю часть колонны вводят раствор ацетальдегида
и катализатор. Парогазовую смесь, содержащую уксусную кис-
лоту, отводят из нижней части брызгоуловителя.
Технический кислород поступает в 4—5 мест по высоте ко-
лонны через специальные распределительные трубки. Колонна
заполнена жидкостью примерно до верхней расширительной части.
При движении снизу вверх жидкость все более обогащается уксус-
ной кислотой, а концентрация в ней альдегида постепенно умень-
шается. Степень превращения альдегида достигает 98 %.
Непрореагировавший кислород захватывает пары ацетальде-
гида и уксусной кислоты. Эта парогазовая смесь из брызгоуло-
вителя окислительной колонны 1 поступает в конденсатор 2, где
конденсируются пары уксусной кислоты и значительная часть
ацетальдегида. Конденсат возвращается на окисление, разбавляя
ацетальдегид в нижней части окислительной колонны. Остаточ-
ный газ после сепаратора 3 промывают водой и сбрасывают в ат-
мосферу.
Реакционная масса из окислительной колонны поступает на
Ью ступень ректификации для отгонки легколетучих продуктов,
главным образом непрореагировавших ацетальдегида и метилаце-
тата. Их пары конденсируются: часть конденсата служит флегмой
ректификационной колонны, а остальное количество выводится из
системы. Кубовая жидкость, содержащая уксусную кислоту, по-
£62
ступает на 2-ю ступень ректификации, где уксусная кислота отго-
няется от тяжелых продуктов и ацетата марганца. Окончательная
очистка уксусной кислоты состоит в ее обработке перманганатом
калия с целью окисления примесей и завершающей ректификации.
В СССР разработан метод жидкофазного окисления
бутана с высоким выходом уксусной кислоты Окисление ведут
при 150—160°С и давлении около 5 МПа; выход кислоты дости-
гает 80 %.
Большой практический интерес представляет синтез уксус-
ной кислоты из метанола и оксида углерода. При
оптимальных условиях выход составляет до 99 %. Этот метод поз-
воляет базировать производство уксусной кислоты на метане
{метансинтез-газметанол->уксусная кислота).
Реакцию карбонилирования спиртов осуществляют в широком
интервале температур (80—300°C) и давлений (0,1—70 МПа)
в присутствии галогенводородных кислот. Особенно эффективны
катализаторы на основе родия, промотированного иодом. В их
присутствии синтез уксусной кислоты из метанола успешно проте-
кает при сравнительно низких давлениях (3 МПа и ниже), при-
чем достигается практически высокий выход уксусной кислоты
(99 %). Катализатор может быть использован многократно.
13.6. ПОЛУЧЕНИЕ ЭТАНОЛА
Из многочисленных реакций, в которые вступают олефины,
наибольшее практическое значение имеют процессы гидратации.
Таким способом получают этанол, изопропиловый спирт и др.
В настоящее время этанол по объему производства занимает пер-
вое место среди всех других органических продуктов.
С каждым годом спирт, получаемый из пищевого сырья, все
более и более заменяется синтетическим. 1 т этилена позволяет
сэкономить более 4 т зерна. Синтетический спирт из этилена в не-
сколько раз дешевле пищевого и требует меньших затрат*труда.
Этанол С2Н5ОН является жидкостью, кипящей при 78,3 °C.
Область взрываемости находится в пределах концентраций 3—20 %
(по объему). Относится к классу малоопасных токсичных ве-
ществ, обладает наркотическим действием на организм человека.
Этанол в больших количествах применяют в пищевой и меди-
цинской промышленности, он служит горючим в жидкостных ра-
кетах, антифризом, растворителем. Как Полупродукт используется
для получения сложных эфиров этилового спирта, хлороформа,
хлораля, диэтилового эфира, ацетальдегида и уксусной кислоты.
Наиболее крупный потребитель этанола — промышленность син-
тетического каучука. Значительную часть бутадиена-1,3 для син-
тетического каучука получают из этанола:
2С2Н5ОН —> СЧ2=СН—СН=СН2 + 2Н2О + Н2
263
Получение этанола гидратацией этилена осуществляют двумя
способами, сернокислотной и прямой гидратацией. Сернокис-
лотный способ, открытый А. М. Бутлеровым, освоен промыш-
ленностью только в 50-х годах XX века. Он основан на сернокис-
лотной гидратации этилена:
H2SO4
СН2=СНа + Н2О -----► С2Н5ОН
Взаимодействие между этиленом и серной кислотой состоит
из двух этапов: первый — физическое растворение этилена в сер-
ной кислоте и второй — гомогенное взаимодействие обоих компо-
нентов (Ж — Ж) с образованием алкилсульфатов по уравнениям:
CjIU + HjSO, C2H6OSO3H - <7
втилсульфат
C2HsOSO3H + С2Н4 (C,H5O)2SO2 - q
диэтилсульфат
Затем проводят гидролиз полученных эфиров по уравнениям:
C2H6OSO3H + Н2О С2Н6ОН + H2SO4
(C2H5O)2SO2 + 2Н2О 2С2НбОН + H2SO4
Кроме этих основных реакций идет образование диэтилового
эфира:
(C2H5O)2SO2 + С2НВОН С2Н5ОС2Нв + C2H6OSO3H
(C2H5O)2SO2 + Н2О С2Н5ОС2Н8 + H2SO4
Для уменьшения выхода диэтилсвого эфира гидролиз ведут
по возможности быстрее, сразу же отгоняя образующийся спирт
с тем, чтобы его концентрация в растворе была небольшой.
В процессе производства этанола сернокислотной гидратацией
этилена можно выделить следующие стадии (рис. 13.9):
1. Абсорбция этилена серной кислотой с образованием этил-
сульфатов в абсорбере /.
2. Гидролиз этилсульфата с образованием этанола и выделе-
нием серной кислоты в гидролизере 3.
3. Отделение этанола от серной кислоты и его очистка в отпар-
ной колонне 4.
4. Регенерация серной кислоты в нейтрализационной колонне 5.
С образованием этилсульфата скорость поглощения увеличи-
вается, так как этилен в нем растворяется лучше, чем в чистой
серной кислоте. Однако этилсульфат разбавляет серную кислоту,
в связи с чем общая скорость реакции снижается. Поэтому по
высоте абсорбционной колонны условия для поглощения этилена
неодинаковы.
Скорость абсорбции этилена зависит также от концентрации
серной кислоты, от давления, температуры и интенсивности пере-
мешивания. Практически серная кислота должна быть 97—
98 %-пая. Температура абсорбции 70—80°С. При повышений тем-
пературы скорость абсорбции увеличивается, но при этом усилн-
264
Рис. 13.9. Схема производства этанола сернокислотной гидратацией этилена;
1 — абсорбер; 2—теплообменник; 3—гидролизер; 4—отпарная колонна; 5—нейтрализационная
колонна.
вается полимеризация этилена и растет выход диэтилового эфира.
На практике поддерживают парциальное давление этилена при-
мерно 1,5 МПа, так как повышение давления сверх этого не ока-
зывает заметного влияния. В случае использования этан-этилено-
вой фракции с 50—60 % этилена давление составляет 2,5—
3,0 МПа. Длительность реакции 3 ч. При абсорбции этилена сер-
ной кислотой выделяется большое количество теплоты, которая
непрерывно отводится водяными холодильниками, расположен-
ными над каждой тарелкой таким образом, чтобы температура
в абсорбере не превышала 80 °C. Обычно применяют абсорберы
с 20 холодильниками.
В этиленовой фракции не должно содержаться пропилена и
ацетилена. Пропилен ухудшает качество спирта из-за образую-
щихся полимеров и изопропилового спирта. Ацетилен способствует
образованию взрывоопасного ацетиленида меди при соприкосно-
вении с медной футеровкой аппаратуры.
Сернокислотный раствор из абсорбера направляют в гидроли-
зер с керамической насадкой, где его разбавляют водой до содер-
жания H2SO4 50 %. Температуру в гидролизере поддерживают
100—110°С, давление-0,2—0,3 МПа. Время гидролиза 0,5 ч.
После гидролиза смесь направляют на промывку, нейтрализа-
цию гидроксидом натрия и конденсацию, а затем на ректифика-
цию. На установках сернокислотной гидратации получают
95,6 %-ный этанол. Это так называемый спирт-ректификат, кипя-
щий при 78,1 °C. Выход спирта достигает 97 %, выход диэтилового
эфира всего 1—2 %.
Отработанную 50%-ную серную кислоту отводят на очистку
от смолистых примесей и после концентрирования и добавки
олеума снова подают в абсорбер.
Недостатки метода сернокислотной гидратации этилена (высо-
кие капиталовложения при сооружении установок, необходимость
9 Зак. 1869
265
Рис. 13.10. Зависимость равновесной степени
конверсии этилена спирз от температуры и
давления.
применения кислотной аппаратуры,
большие энергозатраты на упарива-
ние кислоты) вызвали необходимость
разработки процесса прямой гидрата-
ции этилена. Трудность состояла в
том, что при этой реакции не удава-
лось достигнуть хорошего выхода эта-
нола. Прямая гидратация эти-
лена отличается от сернокислотной
тем, что реакция протекает в одну
стадию, без промежуточного выделе-
ния каких-либо соединений.
Присоединение воды к этилену является гетерогеннокаталити-
ческой экзотермической реакцией:
С2Н4-|-Н2О С2НеОН + <7
Как следует из уравнения реакции и термодинамйческих дан-
ных рис. 13.10, чтобы равновесие сдвинуть в сторону гидратации
этилена, необходимо понижение температуры и повышение дав-
ления, однако при ниже 280 °C скорость гидратации очень мала,
а применение давления более 8 МПа экономически не рентабель-
но. Наилучший катализатор, увеличивающий скорость гидрата-
ции,— фосфорная кислота на носителе. Важными показателями
при катализе являются скелет и поверхность носителя. Широко-
пористый носитель (силикагель, алюмосиликат) облегчает диф-
фузионные процессы. Катализ осуществляют свободной кислотой,
находящейся на поверхности носителя в жидком состоянии. Кон-
центрация кислоты — один из важных показателей активности
катализатора — зависит от парциального давления паров воды.
Если массовая доля кислоты в растворе ниже 83 %, выход спирта
резко уменьшается. Поэтому нельзя поддерживать большие мо-
лярные соотношения воды и этилена, как это вытекает из термо-
динамики процесса.
В результате обширных исследований и промышленных ис-
пытаний установлены основные условия взаимодействия газооб-
разного этилена и водяных паров:
Катализатор Фосфорная кислота на
носителе
Температура, °C 280—290
Давление, МПа 7—8
Молярное отношение Н2О : С2Н4 (0,6—0,7) : 1
Объемная доля С2Н4 в газе, % 80—85
Массовая доля Н3РО4 в растворе, %' Не ниже 83
Объемная скорость подачи этилена, ч-1 1800—2500
Перечисленные условия позволяют получать водно-спиртовой
раствор с массовой долей спирта 15 % при степени превращения
266
Рис. 13.11. Принципиальная схема установки для производства этанола каталити-
ческой гидратацией этилена в паровой фазе:
1 — трубчатый теплообменник; 2—печь; 3 — гидратор; 4—сборник; 5—холодильник; 6 — промы
ночная колонна с насадкой.
за один проход 4—5%. Низкая степень превращения делает не-
обходимой многократную циркуляцию этилена через зону ката-
лизатора. Благодаря рециркуляции непрореагчровавших продук-
тов выход этанола достигает 95 %.
Реакцию гидратации проводят в аппарате непрерывного дей-
ствия, который называют гидрататором. Он представляет собой
полую стальную колонну диаметром 1,6 м и высотой 10 м. Во из-
бежание коррозии под действием фосфорной кислоты корпус и
днище выкладывают листами красной меди. Катализатор насы-
пают в реактор слоем ня высоту 8,5 м. Смесь этилена и паров
воды проходит сверху через слой катализатора и выводится из
нижней части гидрататора. Ввиду малой степени превращения и
небольшой теплоты реакции гидрататор не нуждается в охлажде-
нии. По режиму работы гидрататор приближается к адиабатиче-
скому реактору идеального вытеснения.
Метод прямой гидратации этилена весьма перспективен, од-
нако дальнейшее его усовершенствование заключается в подборе
наиболее активных катализаторов и изыскании путей осуществле-
ния жидкофазного процесса, что позволит уменьшить температуру
процесса и увеличить степень превращения этилена. Метод позво-
ляет создавать установки большой производительности, осуще-
ствить водооборот технологической воды, избавляет от сернокис-
лотного хозяйства.
Сравнение технико-экономических показателей сернокислотной
и прямой гидратации показывает, что себестоимость 1 т спирта
при прямой гидратации на 20 % ниже. Недостатком прямой
9*
267
гидратации является частая замена катализатора и использование
более концентрированного этилена.
Технологическая схема прямой гидратации этилена представ-
лена на рис. 13.11.
Этилен и воду нагревают в теплообменнике I за счет теплоты
реакционной смеси, выходящей из гидрататора 3. В печи 2 с огне-
вым нагревом испаряют оставшееся количество воды и нагревают
смесь до 280—29С°С. Затем парогазовая смесь поступает в гидра-
татор 3. Выходящие из гидрататора.реакционные газы содержат
этилен, пары воды, этанол, диэтиловый эфир, фосфорную кислоту.
Во избежание коррозии последующей аппаратуры проводят ней-
трализацию фосфорной кислоты водным раствором щелочи (на
схеме не показано). После того как горячие реакционные газы
отдадут свою теплоту исходной смеси в теплообменнике 1, пэры
спирта и воды конденсируются в сборнике 4 и холодильнике 5.
Полная промывка газа идет в промывочной колонне 6 с насадкой.
Непрореагировавший этилен вновь возвращается после сжатия
и нагревания в гидрататор. Водный раствор спирта направляют
на ректификацию.
ГЛАВА 14
ТЕХНОЛОГИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
14.1. МЕТОДЫ ПОЛУЧЕНИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
По методу получения различают полимеризационные и поли-
конденсационные высокомолекулярные соединения.
Полимеризация — это процесс образования полимера пу-
тем соединения молекул мономера, не сопровождающийся выделе-
нием побочных продуктов. Поэтому макромолекулы полимера
имеют тот же элементарный состав, что и исходный мономер:
Р, Т, катализатор
n(CH2=CHR) ------------> (—СН2—CHR—)„
В реакции полимеризации могут вступать соединения, имею-
щие кратные связи. Простейшим примером таких реакций явля-
ется полимеризация моно- и диолефинов. При этом двойные связи
в молекулах «раскрываются» и за счет образующихся свободных
валентностей большое число молекул мономеров соединяется друг
с другом. При полимеризации циклических соединений свободные
валентности образуются в результате разрыва цикла.
Полимеризация может протекать по механизму ступенчатых
процессов (ступенчатая полимеризация) и цепных реакций (цеп-
ная полимеризация).
Ступенчатая полимеризация сопровождается переме-
щением атомов водорода и образованием промежуточных продук-
тов, являющихся устойчивыми соединениями. Например, при сту-
268
пенчатой полимеризации изобутилена миграция водорода проис-
ходит следующим образом:
СН3 СНз
СН2=£ + СН2=С —►
* I 1 I
I СНз { СНз
СНз СНз
} I
СНз— С—СН=С
Анз СНз
При ступенчатой полимеризации главными продуктами явля-
ются полимеры с низкой молекулярной массой.
Ступенчатую полимеризацию применяют для получения ком-
понентов моторных топлив, промежуточных продуктов для синтеза
моющих веществ, присадок к маслам.
Цепная полимеризация — это процесс получения высо-
комолекулярных соединений, при котором макромолекула обра-
зуется путем последовательного присоединения молекул мономеров
к растущей цепи. Сначала происходит активирование какой-либо
одной молекулы, вызывающей полимеризацию большого числа
других молекул, с которыми она сталкивается. В этом случае про-
межуточные продукты нестабильны. Цепная полимеризация явля-
ется наиболее распространенной разновидностью реакций полиме-
ризации.
При цепной реакции молекулярная масса образовавшегося
полимера очень высока, причем она достигается мгновенно.
Цепная полимеризация может протекать через промежуточное
образование свободных радикалов (радикальная) или через
промежуточное образование ионов (ионная). В зависимости от
заряда иона, различают катионную и анионную поли-
меризацию.
Общий вид цепной реакции можно представить следующим
образом:
Константа
скорости Скорость
Инициирование цепи:
М —> М' (радикал)
М —> М+ (ион) kg UB
М —> М- (ион)
Рост цепи:
М* + М —> ММ*
м+ + м —> мм+
м- + м —> мм-
Обрыв цепи
М* + ММ* —► ММ
м+ + ММ+ —> ММ ko
М~ + мм- —> мм
и0
Цепная полимеризация может быть вызвана облучением све-
том, лучами солнца, действием тока высокой частоты, нагрева-
нием, действием инициаторов и катализаторов.
269
Инициаторы, как и катализаторы, ускоряют реакции полиме-
ризации, но в отличие от катализаторов необратимо расходуются
в процессе. Инициаторы (органические пеооксиды, гидроперочси-
ды и азотсодержащие соединения) в ходе реакции распадаются
на реакционноспособные радикалы, которые входят в состав мо-
лекул полимера в виде конечных групп (цепная радикальная по-
лимеризация).
Предполагая, что константа скорости роста цепи kP одна и
та же для всех ступеней и не зависит, от радикала, общую ско-
рость расходования мономера, т. е. скорость полимеризации,
можно описать уравнением:
~=^и+(/р = ^ + ^[М]1М‘1 (14.1)
Здесь [М*]—концентрация всех полимерных радикалов; ГМ]— концентра-
ция молекул мономера.
Уравнение (14.1) можно представить в ионном виде, введя зна-
чение длины молекулярной цепи X, определяемой как среднее
число молекул мономера, расходуемых в каждом процессе иниции-
рования, т. е. X = UP/Un. Тогда
Un = - = £/р (1 + U„/Up) =VP (1 + 1/Х) (14.2)
Если степень полимеризации образующегося полимера велика,
значение 1/Х становится ничтожно малым и выражение (1+ 1/Х)
приближается к единице, т. е.
г/п«г/р (14.3)
Скорость роста цепи пропорциональна концентрации радика-
лов. Радикалы возникают при инициировании. Чем больше кон-
центрация инициатора, тем больше в единицу времени появляется
свободных радикалов, инициирующих процесс полимеризации.
В результате возрастает суммарная скорость полимеризации и
уменьшается средняя молекулярная масса образовавшихся макро-
молекул (рис. 14.1,а). Скорость полимеризации и молекулярная
масса полимера увеличиваются при повышении концентрации мо-
номера (рис. 14.1,6):
г/п = А[М]т, где/п>1 (14.4)
При ионной полимеризации (катионной или анионной), проте-
кающей в присутствии катализаторов, активными промежуточ-
ными продуктами служат ионы. Для ионной полимеризации ис-
пользуют кислые и основные катализаторы. Катализаторами ка-
тионной полимеризации являются сильные минеральные кислоты.
Чем сильнее кислота, тем легче образуется катион карбония:
СН2—С(СНа)2 + Н+ СНа—С(СН8),
270
Рис. 14.1. Зависимость скорости полимеризации Un и молекулярной массы М по-
лимера от концентрации инициатора [U] (а) и мономера [М] (6).
Такой катион присоединяется к другой молекуле с образова-
нием нового катиона:
снэ
+ . , (—(+)Й/СНз I + zCHg
сн3—С(СНз)2 + сн3=сс —>- снэ—с—сн2—с/
хснэ | чсн3
сн3
Ионная полимеризация осуществляется по цепному или сту-
пенчатому механизму. Реакцию полимеризации необходимо про-
водить таким образом, чтобы шло образование полимеров с регу-
лярным расположением замещающих групп во всех звеньях мак-
ромолекул относительно плоскости основной цепи (стеребре-
гулярные полимеры). Стереорегулярные полимеры получают
обычно по анионному механизму
Новые комплексные катализаторы, состоящие из металлорга-
нических соединений |например, А1(С2Н5)3] и из хлоридов метал-
лов (например, TiCl3, TiCl4), позволили получить стереорегуляр-
ные полимеры со строго линейной структурой. Такие полимеры
отличаются повышенной прочностью и плотностью и обладают
более высокой температурой плавления.
С повышением температуры увеличивается число элементар-
ных реакций полимеризации, при этом значительно, зозрастает
скорость образования активных центров. Рост их концентрации
увеличивает скорость реакции, рост цепи и особенно быстро ско-
рость обрыва цепи. В результате уменьшается средняя молекуляр-
ная масса полимера и средняя степень полимеризации. При повы-.
шенных температурах протекают побочные реакции между функ-
циональными группами мономера и полимера и образуются
сравнительно низкомолекулярные продукты полимеризации.
Высокое давление значительно ускоряет полимеризацию, и при
этом молекулярная масса полимера не уменьшается.
Таким образом, скорость процесса полимеризации и средняя
молекулярная масса получаемого полимера зависят от темпера-
туры, давления, активности катализатора или инициатора, кон-
центрации инициатора, характера среды.
271
В зависимости от характера среды многочисленные методы по-
лимеризации можно разделить на четыре группы: I) полимериза-
ция в массе (блочный метод); 2) полимеризация в растворе;
3) полимеризация в эмульсии; 4) полимеризация в суспензии.
. Полимеризацию в массе проводят при подаче мономера
в форму в жидкой или газовой фазе с катализатором или инициа-
тором (в отсутствие растворителей). Масса мономера при строго
регулируемой температуре превращается в полимер в виде блока,
трубок, листов, гранул. Этим методом' получают полистирол, по-
лимеры метакриловой кислоты, бутадиеновый каучук.
Полимеризацию в растворе проводят в таком раство-
рителе, в котором растворим мономер и полимер или растворим
только мономер, а полимер при его получении выпадает в осадок.
В первом случае растворителем служит готовый лак. Этот метод
широко применяют в лакокрасочной промышленности. Во втором
случае осадок полимера отделяют фильтрованием, промывают и
высушивают. Метод используют, например, для производства по-
лимеров винилацетилена в метаноле.
Полимеризацию в эмульсии применяют наиболее ши-
роко. Мономер с добавками распределяется в воде или водных
растворах солей в присутствии эмульгатора и образует эмульсию.
Скорость процесса больше, чем при полимеризации в массе, а
образовавшийся полимер имеет более высокую молекулярную
маСсу. Получив полимер с нужными свойствами, разрушают
эмульсию, добавляя кислоты или другие электролиты. Данным
способом получают поливинилхлорид, некоторые марки полисти-
рола и многочисленные сополимеры бутадиена, винилацетата,
акрилонитрила.
При п о л и м е р и з а ц и и в суспензии используют инициа-
торы, нерастворимые в воде, но растворимые в мономере. Полу-
чают крупные капли мономера. Образующийся полимер с высокой
молекулярной массой осаждается в виде частиц нерастворимого
в воде твердого вещества. Это намного облегчает отделение и
очистку образовавшегося полимера при сохранении большой ско-
рости полимеризации.
Поликонденсация — это процесс образования полимера
путем соединения молекул мономера, сопровождающийся выделе-
нием низкомолекулярного вещества (воды, спирта, аммиака, хлор-
водорода и др.). Поэтому элементарный состав полимера отли-
чается от состава исходного мономера.
В реакции поликонденсации могут вступать соединения, содер-
жащие в своем составе различные функциональные группы (—ОН,
—СООН, —NH2 и др.). Для образования полимера необходимо,
чтобы каждый мономер содержал не менее двух функциональных
групп.
Наличие низкомолекулярных соединений в реакционной смеси
обусловливает обратимость процесса. Процесс обратимой поли-
конденсации, как и обычная конденсация, характеризуется кон-
стантой равновесия Кр и константами скоростей прямой и обрат-
272
ной реакций. В момент равновесия скорость образования высоко-
молекулярного соединения равна скорости его деструкции. Зави-
симость предельной степени поликонденсации Хп от константы
равновесия К и концентрации С низкомолекулярного соединения
в реакционной смеси выражается уравнением:
Хп~л/К1С (14.5)
Следовательно, для получения полимера с большой молеку-
лярной массой необходимо более полное удаление низкомолеку-
лярного вещества из зоны реакции.
С возрастанием молекулярной массы полимера увеличивается
вязкость реакционной среды, уменьшается подвижность образо-
вавшихся макромолекул и одновременно затрудняется отвод по-
бочных продуктов. Это может привести к прекращению реакции
поликонденсации. Чтобы предотвратить затухание процесса, необ-
ходимо повышать температуру реакционной смеси, что снижает
вязкость среды и несколько увеличивает скорость диффузии низ-
комолекулярных соединений к поверхности раздела фаз. Но при
этом следует учитывать, что повышение температуры может вы-
звать деструкцию исходных веществ и промежуточных соединений.
Поэтому для достижения высокой скорости процесса стараются
сначала проводить поликонденсацию при повышенной темпера-
туре, понижая ее затем для получения продукта с большой моле-
кулярной массой.
На основании того, что на каждом этапе поликонденсации уча-
ствуют две функциональные группы, кинетическое уравнение реак-
ции имеет вид
--^- = КС2 или (14.6)
дт с
и после интегрирования:
1/С = Кт + const (14.7)
Так как концентрация реагирующих веществ С пропорцио-
нальна общему числу их молекул z за время т, уравнение (14.7)
примет вид:
1 /г = К'т + const (14.8)
Поскольку число молекул z через время т связано с начальным
числом молекул zq в реакционной смеси уравнением zo — zn, то
z-=z^/n. ТогДа уравнение (14.8) преобразуется следующим об-
разом:
n/z0 = Кт' + const (14.9)
Следовательно, стедень поликонденсации пропорциональна
времени, что подтверждается и практическими данными.
Реакция поликонденсации носит ступенчатый характер. Каж-
дый акт взаимодействия функциональных групп приводит к нара-
щиванию цепи на одно звено: сначала образуется димер, затем
273
тример, тетрамер и т. д. Лишь в конечной стадии получения из-
делий могут протекать реакции, в результате которых образуется
трехмерная структура.
Одновременно с ростом цепи полимера протекают побочные
реакции — деструкция полимера и сшивание макромолекул. Де-
струкцию можно уменьшить путем тщательного удаления низко-
молекулярного соединения из зоны реакции или проведением ре-
акции в атмосфере азота.
Основные факторы, влияющие на скорость и направление реак-
ции поликонденсации: число функциональных групп, температура,
давление, степень перемешивания, продолжительность, тип ката-
лизатора и его активность.
Чтобы сдвинуть реакцию в сторону образования полимера, сле-
дует повысить температуру или понизить давление. Это способ-
ствует уменьшению вязкости среды, что позволяет удалить низко-
молекулярные продукты.
Поликонденсацию проводят с эквивалентными количествами
исходных веществ. Катализаторами служат минеральные кислоты,
кислые соли, щелочные металлы, оксиды металлов и ароматиче-
ские сульфокислоты, щелочи.
В промышленности поликонденсацию осуществляют следую-
щими основными способами: в расплаве, в растворе, на границе
раздела фаз, в твердой фазе. Чаще всего проводят поликон-
денсацию в расплаве при 200—280°C. в среде инертного
газа. Заканчивают процесс в вакууме для более полного удаления
побочных продуктов.
Поликонденсация на границе раздела фаз проте-
кает при комнатной температуре, благодаря чему исключается
возможность деструкции полимера и, кроме того, отпадает необ-
ходимость поддержания эквимолекулярного соотношения исход-
ных веществ (оно устанавливается автоматически на границе раз-
дела фаз).
14.2. ПРОИЗВОДСТВО СИНТЕТИЧЕСКИХ СМОЛ И ПЛАСТМАСС
14.2.1. Полимеризационные смолы
и пластмассы на их основе
Из полимеризационных смол наиболее широко применяют по-
лиэтилен, полистирол, полимеры и сополимеры винилхлорида, по-
лиакрилаты, полипропилен, полиизобутилен, поливинилацетат.
Пластмассы на основе перечисленных смол термопластичны, вы-
пускаются без наполнителя, обладают хорошими диэлектриче-
скими свойствами, но у большинства из них низкая теплостой-
кость. .
Производство полиэтилена. Полиэтилен — один из наиболее
распространенных в настоящее время полимеров, так как на его
основе изготовляют пластмассы, из которых в свою очередь полу-
274
чают различные изделия путем литья под давлением, экструзией,
вакуумным формованием или выдуванием.
Полиэтилен представляет собой роговидный продукт белого
цвета. Устойчив к действию кислот, щелочей, растворов солей и
органических растворителей. Он разрушается только под дей-
ствием сильных окислителей — концентрированных H2SO4 и HNO3,
а также хромовой кислоты. При комнатной температуре полиэти-
лен нерастворим в известных растворителях, а при температуре
больше 70 °C растворяется в бензоле, ксилоле, хлоруглеводородах,
декалине, тетралине. При нагревании полиэтилен размягчается,
в этом состоянии легко изменить его форму, а при охлаждении
вновь затвердевает.
Основные методы его переработки в изделия — экструзия и
литье под давлением. Полиэтилен обладает рядом ценных техни-
ческих свойств,, обеспечивающих разнообразное применение его
в промышленности. Высокая влагостойкость, химическая стой-
кость, высокая прочность на разрыв, устойчивость к действию
микроорганизмов — все'это в сочетании с эластичностью, сохраняю-
щейся при понижении температуры до —60 °C, позволяет приме-
нять полиэтилен для изготовления труб, блоков, емкостей, в ка-
честве упаковочного материала, защитных покрытий для электро-
изоляции кабелей.
В настоящее время промышленное производство полиэтилена
осуществляют полимеризацией этилена. При этом возможно про-
текание процесса по трем вариантам:
1) при высоком давлении (120—250 МПа) в присутствии не-
больших количеств О2 в качестве инициатора;
2) при среднем давлении (3,5—7 МПа) в углеводородных рас-
творителях с оксидными металлическими катализаторами (не по-
лучил в СССР широкого применения).
3) при низком давлении (0,05—0,2 МПа) с использованием
комплексных металлорганических катализаторов.
Все способы имеют свои достоинства и недостатки. Целесооб-
разность получения полиэтилена тем или иным способом опреде-
ляется прежде всего предъявляемыми к нему требованиями. Так,
по сравнению с полиэтиленом высокого давления (ПЭВД) поли-
этилен низкого (ПЭНД) и среднего (ПЭСД) давления обладает
большой прочностью, более высокой плотностью и температурой
плавления. Однако ПЭВД имеет самые высокие показатели ди-
электрических свойств. Наличие примесей катализатора в ПЭНД
снижает его химическую стойкость. Наибольшее распространение
в промышленности получило производство ПЭВД.
Полимеризация этилена при высоком давлении
представляет собой цепную реакцию, протекающую по радикаль-
ному механизму с выделением большого количества теплоты:
пС2Н4 —> (—С2Н4—)„ + q
Инициатором полимеризации служит кислород (0,05—0,1 %)
или пероксиды. Степень конверсии этилена 8—15%, при более
275
высоком значении затрудняется отвод теплоты и поддержание
постоянной температуры процесса (180—240°C).
Макромолекула ПЭВД представляет собой длинную метилено-
вую цепь с небольшим числом метильных групп. С увеличением
молекулярной массы полимера механические свойства его улуч-
шаются.
Процесс сопровождается побочными .реакциями разложения и
окисления этилена:
СН2=СН2 —> СН4 + С + <?
СН2=СН2 —► 2С + 2Н2 — q
С2Н4 + О2 —> 2HCHO4-V
Реакция образования формальдегида является крайне нежела-
1ельной, так как он замедляет полимеризацию. Поэтому для нор-
мального хода процесса важно строгое регулирование темпера-
туры, что обеспечивается систематическим отводом теплоты.
Скорость процесса полимеризации и выход полимера зависят
от степени чистоты этилена [концентрация его в исходном газе
должна быть не менее 99,9 % (по объему)], количества инициа-
тора, температуры и давления.
Полимеризацию проводят в аппаратах с мешалкой или в труб-
чатых реакторах змеевикового типа. Первые более производитель-
ны, их конструкция предусматривает ввод этилена и инициатора
по зонам. Это позволяет применять более эффективный пероксид-
ный инициатор и обеспечивает его одинаковую концентрацию
в реакционном объеме, а следовательно, и получение более одно-
родного продукта. Трубчатые реакторы проще по конструкции и
в эксплуатации, но очень громоздки. Производительность реактора
с мешалкой составляет 6000—7000 т полиэтилена в год, трубча-
того реактора — 3000—4000 т/год.
В реактор с мешалкой можно подавать этилен при 30—40 °C,
так как температура вследствие перемешивания быстро выравни-
вается. В результате этого часть теплоты реакции расходуется на
нагрев сырья. Благодаря перемешиванию устраняются местные
перегревы и улучшаются условия отвода теплоты через стенку.
Реактор с быстроходной мешалкой для полимеризации этилена
представлен на рис. 14.2. Аппарат имеет длину 5—6 м, толщину
стенок 125—150 мм. В реакционном объеме происходит интенсив-
ное перемешивание этилена и продуктов реакции. Этилен под дав-
лением около 150 МПа при 50—70°C вместе с кислородом по-
дается в несколько точек по высоте реактора. В реакторе поддер-
живается температура около 220 °C. Степень конверсии этилена
за один цикл до 20%. В таком аппарате полимеризация этилена
идет полнее, чем в трубчатом, но остановка мешалки приводит
к полному нарушению процесса, вплоть до взрыва.
Трубчатый реактор представляет собой змеевик, в котором
диаметр труб по ходу полимеризации постоянно расширяется во
избежание забивки трубопрбвода. Диаметр трубок изменяется
в пределах от 17 до 50 мм.
276
Рнс. 14.2 Реактор с мешалкой для
полимеризации этилена под высо-
ким давлением:
I—мешалка; 2—рубашка.
Различают три зоны ре-
актора:
1 зона — подогрев реак-
ционной смеси до 180 °C для
возбуждения полимериза-
ции. Трубки снабжены ру-
башкой, в которой цирку-
лирует вода, нагретая до
200 °C.
2 зон'а — температура
смеси 220—280 °C, осущест-
вляется процесс полимери-
зации. Отвод теплоты осу-
ществляется путем циркуля-
ции в рубашках воды с тем-
пературой 100—125 °C.
3 зона — температура
.смеси до 200°C, процесс по-
лимеризации заканчивается.
Зона интенсивно охлажда-
ется водой.
За один цикл полимеризуется до 15 % этилена.
• Процесс полимеризации в массе (блочной) с использованием
трубчатого реактора проводят по следующей принципиальной тех-
нологической схеме (рис. 14.3). Этилен, содержащий 0,1 % кис-
лорода, сжимается компрессорами 1 и 2 до 150 МПа. Затем его
подают в реактор 3, охлаждаемый водой.
Из реактора продукты реакции через дроссельные вентили по-
ступают в сепаратор высокого давления 4, затем в сепараторы
низкого давления 5, 6. Непрореагировавший этилен направляется
снова в процесс. Из сепараторов 5, 6 жидкий полиэтилен посту-
пает на грануляцию, которая заключается в продавливании поли-
этилена через насадку с отверстиями. Калиброванный полиэтилен
охлаждают проточной водой и режут на гранулы. Поскольку кон-
версия этилена за один цикл невелика, доля газа рециркуляции
составляет около 90%. Суммарное превращение этилена в ре-
зультате неоднократной циркуляции достигает 95—97 %.
Полиэтилен среднего давления с высокой плотно-
стью получают полимеризацией этилена в растворителе (Р = 3,5 4-
4-7 МПа). Преимущества этого метода: дешевизна и нетокснч-
ность катализатора, возможность многократного его использова-
ния путем регенерации, простота технологического и аппаратур-
ного оформления процесса, меньшая взрыво- и пожароопасность.
ПЭСД имеет более высокие физико-механические свойства, чем
277
Рис. 14.3. Схема получения полиэтилена при высоком давлении:
1 — компрессор первого каскада; 2—компрессор второго каскада; 3—реактор; 4—6—сепара-
торы; 7—холодильник; 8—колонна сухой очистки возвратного этилена.
ПЭВД. Растворитель, в котором растворяется этилен и полиэти-
лен, способствует равномерному распределению катализатора и
отводу теплоты. В качестве растворителей используют бензин,
ксилол, циклогексан. Катализаторами служат оксиды металлов
переменной валентности, нанесенные на пористый алюмосиликат-
ный носитель, в котором оксиды кремния и алюминия находятся
в массовом отношении 90: 10. В промышленности в качестве ка-
тализатора используют оксид хрома.
На скорость полимеризации, выход и свойства полиэтилена
оказывают влияние активность катализатора, температура, дав-
ление. Активность катализатора зависит от пористости носителя
и температуры активации. С повышением активности катализатора
увеличивается скорость процесса полимеризации, но снижается
молекулярная масса полиэтилена. Рост температуры сначала уве-
личивает скорость полимеризации, а затем снижает вследствие
дезактивации катализатора. С увеличением давления возрастает
молекулярная масса полимера. Давление поддерживается на
уровне 7 МПа. Для создания постоянной температуры (135—
145 °C) необходим интенсивный отвод теплоты, поэтому в реакци-
онную смесь вводят дополнительное количество этилена и раство-
рителя, который, нагреваясь, уносит часть теплоты.
.Ионная полимеризация этилена при низком дав-
лении происходит в присутствии металлорганического катализа-
тора [TiCl4 и А1(С2Н6)з] в среде инертного растворителя (бензин,
гептан, гексан, циклогексан). Процесс проводят практически при
атмосферном давлении и 60—70 °C. Реакция сопровождается вы-
делением значительного количества теплоты. Полимеризация эти-
лена в этих условиях протекает по анионному механизму.
Молекулярная масса полимера зависит от соотношения три-
этилалюминия и тетрахлорида титана в катализаторе. Обычно мо-
лярное отношение компонентов равно 1 : 1, что обеспечивает полу-
чение полиэтилена с молекулярной массой 70 000—350 000.
278
Растворитель
Рис. 14.4. Схема получения полиэтилена низкого давления:
7, 2—емкости для компонентов катализатора; 5—смеситель для получения катализатор»;
4—реактор; 5—брызгоуловитель; 6—холодильник; 7—газодувка; 8 — аппарат для разложения
катализатора.
Реакцию проводят в вертикальном аппарате колонного типа и
в реакторе с перемешивающим устройством.
Особенность технологического процесса полимеризации заклю-
чается в том, что при его осуществлении необходимо полное ис-
ключение из системы воды, спирта, кислорода. Их присутствие
приводит к разложению катализатора. Поэтому полимер получают
в виде суспензии в атмосфере азота.
Отвод теплоты из реактора производят путем отдувки части
этилена с последующим его охлаждением и возвращением в ре-
актор.
Полученный полиэтилен очищают от катализатора. Катали-
затор разлагают спиртом (чаще пропиловым) при 50—70 °C, а по-
том бензином. Эта операция имеет большое значение для получе-
ния полиэтилена высокого качества.
На рис. 14.4 представлена принципиальная технологическая
схема получения полиэтилена низкого давления непрерывным спо-
собом.
Растворы компонентов катализатора из емкостей 1 и 2 посту-
пают в смеситель 3. Готовую суспензию непрерывно подают в ре-
актор полимеризатор 4, куда также вводят этилен и бензин. Па-
рогазовую смесь из верхней части реактора направляют в брызго-
уловитель 5 для отделения капель бензина от газа. Бензин
возвращает в рейктор. Парогазовая смесь охлаждается в холо-
дильнике 6 и направляется в нижнюю часть реактора.
Суспензия полимера поступает в аппарат 8, в котором проис-
ходит разложение катализатора смесью спирта и бензина. После
сушки подогретым азотом порошок полиэтилена направляют на
грануляцию.
Способ получения ПЭНД имеет ряд преимуществ по сравне-
нию со способом получения ПЭВД: высокая (100%-ная) степень
279
конверсии этилена за один цикл и отсутствие необходимости ап-
паратов высокого давления. Однако при получении ПЭВД не
требуется катализатора и растворителя.
ПЭВД обладает отличными электроизоляционными свойства-
ми, эластичностью (гибкость сохраняется даже при —60 °C) и
высокой химической стойкостью к различным агрессивным сре-
дам, поддается всем видам обработки, склеивается и сваривается.
ПЭНД отличается от ПЭВД более высокими плотностью и
прочностью, высокой температурой размягчения.
Производство полистирола. Выпускаемый промышленностью
полистирол — аморфный полимер с молекулярной массой 50000—
200 000. Обладает исключительной водостойкостью и высокими
диэлектрическими свойствами, позволяющими применять его
в качестве высокочастотного диэлектрика. При обычной темпе-
ратуре представляет собой твердый прозрачный материал, по-
хожий на стекло, пропускающий до 90 % лучей видимой частоты
спектра.
Кроме высоких физико-механических показателей, а также
превосходных изоляционных и оптических свойств одним из до-
стоинств полистирола является его высокая устойчивость к дей-
ствию агрессивных сред (концентрированных растворов щелочей
и всех кислот, за исключением азотной). Атмосферостойкость и
химическая стойкость полистирола объясняется насыщенностью
углеводородной цепи и присутствием фенильных групп. Полисти-
рол обладает удовлетворительной светостойкостью.
К недостаткам полистирола можно отнести: невысокую тепло-
стойкость, хрупкость, возникновение внутренних напряжений, при-
водящих к разрушению материала в изделиях,, склонность к ста-
рению.
Благодаря прочности и высокому коэффициенту преломления
полистирол применяют для изготовления оптических стекол, про-
зрачных моделей и галантереи. Из полистирола производят детали
радио- и электроаппаратуры, счетных машин, панели приборов,
плёнки для конденсаторов. Физиологическая безвредность поли-
стирола позволяет получать из него санитарно-гигиенические из-
делия, тару и упаковку для фармацевтических препаратов и пи-
щевых продуктов. Из ударопрочного полистирола изготовляют
камеры и дверцы холодильников, санитарно-техническое оборудо-
вание. Полистирольные лаки применяют для получения электро-
изоляционных и противокоррозионных покрытий
Полимеризация стирола может протекать по радикальному и
ионному механизму.
Радикальная полимеризация стирола происходит при
образовании свободных радикалов под действием высоких темпе-
ратур. При этом осуществляется термическое инициирование, за-
ключающееся в самоинициировании чистых мономеров без вве-
дения в реакционную среду специальных инициаторов. В этом
случае радикалы образуются вследствие разложения небольших
количеств пероксидных примесей, которые могут возникать при
280
взаимодействии мономера с кислородом воздуха:
О О о
II II II
С6Н5—С—О—О—С—С6Н6 —> 2C6HS—С—О' —> 2Свн; + 2СО2
Метод термического инициирования полимеризаиии требует
больших затрат энергии, а скорость полимеризации невелика. Ее
можно увеличить, повышая температуру, но при этом снижается
молекулярная масса образующегося полимера.
При химическом инициировании процесс радикальной полиме-
ризации протекает при более низких температурах, что позволяет
получить высококачественный полистирол. Наиболее распростра-
ненными инициаторами являются: пероксид водорода, пероксид
бензоила, персульфаты аммония и калия. Эти инициаторы исполь-
зуют в производстве полистирола эмульсионным методом.
При анионной полимеризации в качестве активного
центра выступает ион карбония — соединение с трехвалентным
углеродом, несущим отрицательный заряд, а сама растущая цепь
представляет собой ’ макроанион. Так, полимеризация стирола
в среде жидкого аммиака, катализируемая амидом натрия, про-
текает следующим образом:
NaNH2 Na* + NH2
CH2=CH + Na* + NH2"
I
CeH5
NH2CH2—CHNa*
I
CfiHB
Рост цепи:
NH2CH2—CHNa* + CH2=CH —> NH2—CH2—CH—CH2—CHNa* и т. д.
C6H6 i6H5 С6Н5 СВН5
т. е. молекула мономера внедряется между ионами ионной пары.
Обрыв цепи невозможен путем соединения растущих макро-
анионов из-за наличия у них одинакового заряда. Он чаще всего
происходит в результате реакций передачи цепи на растворитель
или мономер:
NH2r—СН2—CH—"I СН2—CHNa* + HNH2 —>
I I •
L свн5 Jn—। c6h5
—I NH2r—CH2—CH—1 CH2—CH2 + NaNH2
I I
L cbhb Jn c6hs
NH2r—CH2—CH—1 CH2— CHNa* + CH2=CH —>
I I ।
L cbhb Jn c0h6 ceH8
NH2r— СН2—CH— 1 CH=CH + CH.,—CHNa*
I I I
L cbhb j,n свнв СбН8
281
Стирол
Рис. 14.5. Схема блочной
полимеризации стирола с
полной конверсией:
1—реакторы периодического
действия; 2— обратный холо-
дильник; 3—полимеризацион-
ная колонна непрерыв-
ного действия; ~4 — экструдер;
5—ваина охлаждения; 6—гра-
нулятор.
Анионная полимеризация эффективна при пониженных темпе-
ратурах и тщательно освобожденных от воздуха и осушенных
растворителях основного характера.
Промышленное применение получили следующие методы по-
лимеризации стирола по радикальному механизму: блочный
(в массе) с полной конверсией, блочный (в массе) с неполной
конверсией, суспензионный, блочно-суспензионный и в меньших
масштабах эмульсионный.
Схема блочной полимеризации с полной конвер-
сией представлена на рис. 14.5.
Полимеризацию проводят в две стадии. Первая стадия — фор-
полимеризация — протекает в реакторах 1 периодического дей-
ствия, куда поступает стирол. Температуру в реакторе поддержи-
вают в пределах 60—80 °C. Алюминиевые реакторы снабжены ло-
пастными мешалками и обогревающими змеевиками.
Форполимеризация протекает при термическом инициировании
в атмосфере азота и длится около 30 ч.' Процесс проводят при
перемешивании, что обеспечивает равномерное распределение теп-
лоты в реакционной смеси. Вязкий раствор полимера (около
30%) в мономере из реакторов поступает в многосекционную ко-
лонну непрерывного действия 3. Она состоит из шести-восьми сек-
ций, в каждой из которых поддерживается определенный темпера-
турный режим — от 100°C в первой секции до 30 °C в последней.
Снизу колонна заканчивается конусом, в котором температура
достигает 235 °C. Для обеспечения непрерывной работы колонны
ее соединяют с двумя реакторами форполимеризации. Форполимер
медленно стекает по колонне в течение 30 ч, постепенно обога-
щаясь полимером. Пары мономера поднимаются вверх по колонне
и отводятся на охлаждение и конденсацию в обратный холодиль-
ник 2. Конденсат возвращается в. реакторы. Из конуса колонны
282
Рис 14.6. Схема блочной
полимеризации сгирола с
неполной конверсией:
/ — теплообменник; 2-4 — ре*
акторы; 5—вакуумная ка?
мера; 6 — экструдер грануля-
тор; 7—обратные холодиль-
ники.
расплавленный полистирол, полностью освобожденный от моно-
мера, непрерывной струей стекает на экструдер 4, который выдав-
ливает полимер в ванну охлаждения 5. Здесь полистирол охлаж-
дается, образуя прозрачную стекловидную массу, которая затем
измельчается в грануляторе 6.
Метод блочной полимеризации с неполной кон-
версией гораздо производительней. По данному методу поли-
меризацию доводят до степени конверсии 80—95 %, после чего
свободный стирол отгоняют и возвращают в производство. Из-
вестные преимущества имеет процесс блочной полимеризации
в присутствии 5—10 % растворителя, благодаря которому сни-
жается вязкость реакционной массы, улучшается теплообмен.
Схема процесса представлена на рис. 14.6. Стирол дозировоч-
ным насосом непрерывно подается через теплообменник 1 (80—
100 °C) в реакторы 2, 3 и 4. Полимеризация протекает по следую-
щему режиму:
1-й реактор 2-й реактор 3-й реактор
Температура, °C 120—140 160 180
Степень конверсии, %' 40—50 80 90
Время, д 2 2 2
Из последнего реактора полистирольный расплав и незаполи-
меризованный стирол перекачиваются в вакуумную камеру 5.
В камере отгоняется стирол и далее возвращается в цикл; рас-
плав полистирола поступает в экструдер 6 на грануляцию.
Суспензионная полимеризация стирола получила
широкое распространение. Проведение процесса в водной среде
облегчает регулирование технологического режима, что позволяет
получать полистирол различных марок в одном аппарате боль-
шого объема. Осуществление непрерывного процесса суспензион-
ной полимеризации затруднено недостаточной устойчивостью сус-
пензии и налипанием полимера на мешалку и стенки аппарата,
поэтому получение суспензионного полистирола производят перио-
дическим методом по схеме, представленной на рис. 14.7.
Подготовка сырья проводится в смесителях 1 и 2 и заклю-
чается в приготовлении водного раствора стабилизатора. Инициа-
283
Фильтрат
на очистку
Рис. 14 7. Схема получения по-
листирола суспензионным мето-
дом:
/, 2—смесители; 3—реактор; 4—сито;
5—промежуточная емкость; 6 — цен«
трифуга; 7—сушилка.
Рис. 14.8. Схема производства
вспенивающего полистирола
блочно-суспензионным методом:
1—реактор; 2—смеситель; 3— авто-
клав; 4—енто; 5 —промежуточная
емкость; б—центрифуга; 7—сушил-
ка-
торами полимеризации служат пероксиды (бензоила, лаурила)
или динитрил азобисизомасляной кислоты. В реактор 3 загружают
суспензию стабилизатора и мономерную смесь. Реактор снабжен
рубашкой и лопастной мешалкой. Полимеризацию стирола прово-
дят при 50—130 °C в течение 9—12 ч.
Полученная суспензия сливается через сито 4 в промежуточ-
ную емкость 5, где разбавляется водой для повышения текучести.
Отжим и промывка полимера происходит на центрифуге 6. Влаж-
ный полистирол передается в воздушную сушилку 7.
Основные недостатки суспензионной полимеризации — много-
стадийность и большое количество сточных вод.
Блочно-суспензионную полимеризацию стирола
применяют в основном для получения вспенивающегося полисти-
рола (рис. 14.8).
284
В реакторе 1 осуществляют блочную полимеризацию стирола
(80—85 °C) в присутствии инициатора — пероксида бензоила — до
степени конверсии 30—40 % • Затем форполимер сливают в авто-
клав 3 и перемешивают с раствором стабилизатора. В автоклав
также добавляют новую порцию раствора инициатора в стироле
и изопентан, служащий газообразователем при вспенивании поли-
стирола. Полученную суспензию нагревают до 90 °C и перемеши-
вают в течение 7—9 ч. Далее суспензия направляется через про-
межуточную емкость 5 на центрифугу 6 и сушилку 7.
При эмульсионной полимеризации в качестве эмуль-
гаторов используют мыла, некаль-натриевые соли дибутилнафта-
линсульфокислот и некоторые другие органические сульфосоеди-
нения. Инициаторами являются водорастворимые пероксиды (на-
пример, пероксид водорода), а также персульфаты аммония и
калия.
Эмульсионный метод в отличие от блочного не получил широ-
кого распространения из-за пониженных диэлектрических свойств
и ухудшения прозрачности полимера, загрязненного остатками
эмульгатора. Его используют в основном при получении АБС-пла-
стиков и вспенивающегося полистирола.
14.2.2. Пол икон денсационные смолы
и пластмассы на их основе
К поликонденсационным смолам относят фенолоформальдегид-
ные, полиэфирные, эпоксидные, полиамидные, кремнийорганиче-
ские смолы и полиуретаны. Изделия из пластмасс на основе этих
смол после отверждения могут эксплуатироваться длительное
время в более широком интервале температур и при повышении
температуры они меньше изменяют свои физико-механические
свойства, чем изделия из большинства полимеризационных смол.
Большая часть поликонденсационных смол термореактивна. Для
них характерна быстрая потеря текучести при повышенных тем-
пературах. Это затрудняет формование изделий из пластмасс на
их основе методом литья под давлением или экструзией. Для
этого используют метод прессования. В процессе прессования тер-
мореактивных материалов происходит не только формование из-
делий, но и протекают химические превращения сравнительно
низкомолекулярных полимеров в полимеры пространственной
структуры.
Фенолоформальдегидные полимеры широко используют для
получения термореактивных пластических масс. Эти материалы
отличаются высокой механической прочностью, малым водопогло-
щением, хорошими диэлектрическими свойствами, стойкостью
к действию химических веществ.
Их получают реакцией поликонденсации фенола и формальде-
гида в присутствии катализаторов (кислых и щелочных).
Термопластичные смолы, известные под названием но-
во л ачных, образуются при избытке фенола в исходной смеси
285
и применении кислых катализаторов. Реакцию образования ново-
лачных смесей — полимера линейного строения — можно предста-
вить в общем виде:
Выделяющаяся вода оказывает небольшое влияние на равно-
весие и скорость реакции. Основными факторами, определяющими
направление реакции, являются температура, время реакции, при-
рода и концентрация применяемого катализатора.
Скорость процесса увеличивается при повышении содержания
в исходной смеси катализатора и формальдегида. Таким образом,
получение фенолоформальдегидной смолы — это процесс гомоген-
ного катализа. Чем меньше молярное отношение фенола к фор-
мальдегиду; тем больше молекулярная масса полученной смолы.
Увеличение времени поликонденсации ведет к полному связыва-
нию фенола с формальдегидом и повышению молекулярной массы
конечных продуктов.
Термореактивные смолы, называемые резольными,
получают при избытке формальдегида в присутствии щелочного
катализатора. В этих условиях происходит связывание длинных
линейных цепей поперечными связями с образованием на послед-
ней стадии поликонденсации пространственных полимеров:
Молекулярная масса резольных смол колеблется от 700—1000.
Поликонденсация протекает ступенчато и ее можно остановить
на любой стадии процесса.
Отечественная промышленность выпускает новолачные и ре-
зольные смолы в сухом и жидком состоянии, а также в виде
эмульсии и лаков.
Производство новолачных и резольных смол непрерывным ме-
тодом (рис. 14.9) включает следующие стадии:
286
Фенол
Рис, 14.9. Схема производства новолачных и ре-
зольных смол непрерывным методом:
J —реактор-колонна смешения; 2, 3 —холодильники; 4—се-
паратор; 5—насос; €~сушильный аппарат; 7 — смолопри
емник; 8—охлаждающий барабан: 9 — транспортер.
Рис. 14.10. Реактор для непрерывной поликонден-
сации:
J — двигатель с редуктором; 2 — секция колонны; 3—ме-
шалка; 4 —сбщий вал; 5—патрубок.
1. Подготовка сырья (плавка фенола, подготовка формали-
на— на схеме не показаны).
2. Поликонденсация в реакторе 1.
3. Отделение образовавшейся смолы от надсмольной воды
в сепараторе 4.
4. Сушка смолы в сушильном аппарате 6, слив ее в смоло-
приемник 7.
5. Охлаждение смолы на охлаждающем барабане 8 водой с по-
следующей подачей ее на транспортер 9 для дальнейшей перера-
ботки.
6. Охлаждение непрореагировавших исходных веществ из реак-
тора / и смолоприемника 7 в холодильниках 2,3 с последующим
возвращением их в реактор 1.
Фенолоформальдегидные смолы используют в различном виде:
без наполнителей, как связующее для слоистых пластиков, в виде
287
клеев и лаков для покрытий по металлу и дереву, для изготовле-
ния поропластов, в виде прессовочных материалов.
Для непрерывной поликонденсации используют реакторы, со-
стоящие из секций полного смешения. Таким образом, аппарат
представляет собой колонну, состоящую из расположенных одна
над другой секций 2 (рис. 14.10). Мешалки 3 всех секций имеют
общий вал 4 и приводятся в движение от одного двигателя 1. Все
исходные вещества поступают в колонну при атмосферном дав-
лении и 95—98 °C.
ЛИТЕРАТУРА
1. Материалы XXVII съезда КПСС. М.: Политиздат, 1986.
2. Безденежных Л. А. Математические модели химических реакторов. Киев: Тех-
ника, 1970.
3. Закгейм А. Ю. Введение в моделирование химико-технологических процессов.
М.: Химия, 1979.
4. Карапетьянц М. X. Введение в теорию химических процессов. М.: Высшая
школа, 1975
5. Левеншпиль О. Инженерное оформление химических процессов. М.: Химия,
1969.
6. Технология катализаторов/Под ред. И. П. Мухленова. 2-е изд. — Л.: Химия,
1979.
7. Катализ в кипящем слое/Под ред И. П. Мухленова. — 2-е изд. — Л.; Химия,
1978.
8. Расчеты химико-технологических процессов/Под ред. И. П. Мухленова. — 2-е
изд. — Л.: Химия, 1982.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсорбер насадочного типа 190, 212
Абсорбция 172, 242, 243
Автоматизация 15
Адиабатический температурный режим
99, 102—104
Адсорбция 172, 173, 175, 243
Азот, получение
методы 195
сырье 194
Азотная кислота
получение 206
концентрированной 214
сырье 206
физико-химические основы 207—
212
химико-технологические системы
производства
комбинированная 214
на основе кислорода и концен-
трированных оксидов 214, 215
при атмосферном давлении 212,
213
при повышенном давлении 214
Активаторы 82
Активность катализатора 30, 81
Аммиак-
контактное окисление 206—209
синтез из азота и водорода 194,
199-202
сырье см. Азот, Водород
физико-химические основы 199—
202
Антидетонатор 226
Антрацит 215, 217
Атмосферная вода 22
Атмосферно-вакуумные трубчатки
(АВТ) 232
Ацетилен, получение
из карбамида кальция 257, 258
окислительным пиролизом 260
собственно пиролизом 259
электрокрекингом 259
Аэрозоль 169
Барботажная колонна 212
Байпас 156, 157, 202, 203
Безотходные производства 14, 169,193,
206
Бензин 215, 225, 226
Битумы 227
Бурый уголь 215, 217
Внешнедиффузионная область протека-
ния процесса см. Области протека-
ния процессов
Внешнедиффузионное торможение 73,
83, 84
Внутридиффузионная область протека-
ния процесса см. Области протека-
ния процессов
Внутридиффузионное торможение 76,
84, 85
Вода 21, 207
очистка 23—26
показатели качества 22, 23
Водоподготовка 23—26
Водород, получение
конверсия метана 195—198
высокотемпературная 197
двухступенчатая 196
каталитическая 197
конверсия оксида углерода(П) 198,
199
реактор >1 конверсии 197—199
сырье 195
Водяной газ 215
Воздух 17, 21, 26, 194, 207
Высокомолекулярные соединения
268 сл
Выход продукта
анализ влияния
времени 67, 235
давления 200, 235, 276, 278
температуры 53, 58, 59, 200, 208,
235, 276, 278
типа технологической связи 158,
159
равновесный 37, 45, 47
Газификация топлива 218
Газогенераторы 218
Генераторный газ 218
Генераторы ацетиленовые 258
Гетерогенный процесс 33
каталитический 80, 83—88
моделирование 88—95
области протекания 72—80, 83—88
определение 75, 77, 79
основные признаки 74, 77, 79
реакторы 124—150
факторы интенсификации 75—79
характеристика 71—73
Гидратор 267
Гидрогенизация 218
289
Гиперсорбер 244
Гиперсорбция 244
Гомогенный процесс 33, 49 сл
анализ влияния
времени 64—67
давления 59—61
катализатора 61, 63, 64
концентрации 50—53
температуры 53—59
расчет оптимальных параметров
концентрации 70
температуры 69
реакторы 122—124
характеристика 49, 50
Горючие газы 226
Горючие сланцы 215, 217
Гравитационное обогащение сырья 19
Гравитационное осаждение 171
Графический метод проектирования
166
Грохоты 19
Грохочение 19
Дегазация воды 25
Детонация 226
Диффузия
кнудсеновская 76—79
механизм 76
молекулярная 74, 76—78
Доменный газ 215
Древесина 215
Древесный уголь 215
Жесткость воды 22
Жидкое топливо 215, 226
Замкнутая система 158
Замкнутый объем см. Химико-техноло-
гический процесс в замкнутом объ-
еме
И збирательность
анализ влияния
времени 64—67
диффузионных торможений 83—
88
катализаторов 86
концентрации 52, 53
температуры 53, 58, 59
типа технологической связи 158,
159
катализатора 81, 207, 208
мгновенная 41
суммарная 40, 41
Изотермический температурный режим
99—102
Инерционное осаждение 171
Инициаторы полимеризации 270
стирола 281, 284, 285
этилена 275, 276
Интенсивность работы реакторов 121
290
Интенсификация. См. также Совершен-
ствование процесса
гетерогенных процессов 75—79
необратимых процессов 54
обжига колчедана 181
производства серной кислоты 192,
193
процесса сжигания серы 179
процессов органического синтеза
247, 248
работы аппаратов 13, 15
Каменноугольная смола 215, 219, 220
Каменный уголь 215, 217, 219
Катализ
гетерогенный 80, 83—88
гомогенный 61—64
механизм 62, 63
Катализатор(ы)
выбор 81
гетерогенных процессов 80—83, 86
гидратации этилена 266
гомогенных процессов 61—64
избирательность 81, 207, 208
ионной полимеризации 270, 271
конверсии
метана 197
оксида углерода(П) 199
кратность циркуляции 240
крекинга нефтяного сырья 237—
242
методы изготовления 83
нанесенные 83
окисления
аммиака 207—209
ацетальдегида 261, 262
метанола 253—257
оксида серы (IV) 183, 184, 187
органического синтеза 247
осажденные 83
отравление 81, 82, 92
плавленные 83
поликонденсации 274, 286
полимеризации
стирола 281
этилена 275, 278, 279
риформинга 241, 242
свойства 81, 82
синтеза
аммиака 200, 201, 204
метанола 249—251
уксусной кислоты из метанола
263
скелетные 83
смешанные 83
Керосин 215
Кинетическая область протекания про-
цессов см. Области протекания про-
цессов
Кинетическое уравнение 50
Коагуляция 23
Кокс 215, 219
Коксование 219—224
Коксовый газ 215, 219
схема переработки 222, 223
Колчедан
в качестве сырья 178
обжиг 90. 180—182
модель процесса 181
способы интенсификации 181
установка для сжигания 182
Конвекция 74
Конденсация 245 ,
Константы равновесия 45—49
Контактные аппараты
многоступенчатые 144—150
окисления аммиака 209
поверхностного контакта 136
полочные 137, 138, 145—147
радиальные 137
с внутренним теплообменом 148—
150
с движущимся катализатором 143,
144
с кипящими слоями катализатора
141 сл, 187
с промежуточным теплообменом
145—148
с фильтрующими слоями катали-
затора 135 сл, 145, 146, 186, 202
трубчатые 139, 140
шахтного типа 135
Контактные массы см. Катализгтооы
Контактные яды 82, 184, 200, 207
Кооперирование химических произ-
водств 14, 18, 205
Котлы-утилизаторы 31, 180, 199
Крекинг 228, 234-241
Критерии оптимизации 67, 68
Лимитирующая стадия 72, 73 сл
Линия оптимальных давлений 60
Линия оптимальных температур 56
Мазут 215
схема вакуумной перегонки 233
Материальный баланс 42
Метаиол
образование из сиитез-газа 249—
252
Метод (ы)
концентрирования 21, 53
очистки
абсорбция жидкостями 172
адсорбция твердыми поглотите-
лями 172, 173, 175
анаэробный 176
аэробный 176
биологические 176
вымораживание 24
гравитационное осаждение 171
дегазация 25, 26
Метод (ы)
очистки
дистилляция 24, 175
дожигание в печах 170
инерционное осаждение 171
ионообменные 24
каталитический 170, 173
классификация 170
коагуляция 23, 170, 174
механический 170, 174
мокрый 170, 171
обезз фаживание 26
о^ессол..зание 23, 24
обратный осмос 175
озонирование 176
осветление 174
отстаивание 23, 174
процеживание 174
ректификация 175
сухой 170
термический 24, 173, 176
умягчение 23, 24
фильтрация 23, 171, 174
флокуляция 174
флотация 174
хемосорбционный 170
химические 25, 175, 176
хлорирование 176
центробежный 171
экстракция 174
электрокоагуляция 24, 25
электростатический 170, 171
разделения 21, 53, 242—246
умягчения воды 24—26
Механизация 15 *
Моделирование процессов
взаимодействия между твердыми
фазами 95
в системе газ (жидкость) — жид-
кость 93, 94
в систе-.е газ (жидкость) — твер-
дое 89—93
математическое 89
физическое 88
Модель 88
квазигомогенная 92, 93
пленочная 94
потока с идеальным вытеснением
98-106, 114-119
потока с полным смешением 98
107-119
химико-технологической системы
160
частицы с невзаимодействующим
ядром 90, 91
ячеечная 120, 127
Молекулярная диффузия 74
Монтажные работ! ’61
Надсмольная вода 219, 220
Научно-исследовательские работы 161
291
Необратимые процессы (реакции) 34,
51, 54, 59, 64, 69, 102, 104, 105
Непрерывные процессы 15
Нефтехимическое сырье 227
Нефть (и) 215
важнейшие продукты 226, 227
качество 225
коксование тяжелых фракций 228,
234
крекинг 228. 237—241
легкие 225
обезвоживание 230, 231
обессоливание 230, 231
перегонка 227, 230, 232, 233
пиролиз 228, 234
риформинг 228, 241
свойства 225
состав 224
термическое разложение углеводо-
родов 234—238
термокаталитическое превращение
углеводородов 237—242
Нефтяной кокс 227, 230
Новолачные смолы см. Смолы термо-
пластичные
Носители 82
Обезвоживание нефти 230, 231
Обеззараживание воды 26
Обессоливание
воды 24, 25
нефти 230, 231
Обжиг 90, 180—182
Области протекания процессов. См.
1акже Гетерогенный процесс
внешнедиффузионная 72—76, 83—
88
внутридиффузионная 72, 76—79,
84—88
кинетическая 33, 72, 79, 80, 84,
86-88
переходная 73, 87
промежуточная см. переходная
Обогащение сырья 18—21
Обратимые процессы 34, 51, 52, 55—
58, 60, 64, 69, 102, 105
Обратная технологическая связь 158,
159
Объемное макетное проектирование 166
Окисляемость воды 23
Октановое число 226
Операторная схема ХТС 154
Оптимальные параметры, принципы
расчета 67—71
Оптимизирующие факторы 68, 69
Оригинал 88
Отравление катализатора 81, 82, 92
Отходы 14, 16, 17, 18
утилизация 168, 169
Охрана окружающей среды 14, 18,
166-176
Очистка. См. также Методы очистки
газообразных выбросов 169—173
обжигового газа 182
от аэрозолей 169—172
от газов 14, 23
от газообразных примесей 169,
170, 172, 173
от механических примесей 23
от парообразных примесей 169,
170, 172, 173
от' растворенных солей 23
промышленной воды 23—26
сточных вод 14, 174—176
Параллельная технологическая связь
157
Параллельные реакции 34, 52, 58, 61,
64—66, 85
Параллельные технологические линии 13
Параметричность потока 153
Параметры состояния технологических
потоков 153
Парафины 227, 235
Пенные аппараты 190
Перекрестная технологическая связь
160
Перемешивание 97
Периодические процессы 15
Печь(и)
барабанные вращающиеся 132
ванные 134
для сжигания серы 179
для электрокрекинга 259
камерные 133
коксовые 219—221
распылительные 130, 131
с кипящим слоем 131, 132
трубчатая 230, 233
туннельные 134
шахтные 129, 130
Пиролиз 228, 234
Питьевые воды 23
Плоскостное макетирование 166
Поверхностные воды 22
Подземные воды 22
Поликонденсация 272—274
Полимеризация
блочная см. Полимеризация в мас-
се
блочно-суспензионная 284, 285
в массе 272, 277, 282, 283
в растворе 272, 277
в суспензии (суспензионная) 272,
283, 284
в эмульсии (эмульсионная) 272,
281, 285
ионная 267—271, 278, 281
радикальная 269, 270, 280
стирола 280—285
ступенчатая 268
цепная 269
292
Полимеризация
этилена 275—280
Полистирол 280—285
Политермический температурный ре-
жим 99, 104—106
Полиэтилен
высокого давления 275—277, 280
низкого давления 275, 278—280
среднего давления 277, 278
Полукокс 215, 219
Полупродукты 16, 17
Полуфабрикаты см. Полупродукты
Попутный газ 215, 230. 242
Порядок реакции 50, 51
Последовательная технологическая
связь 155, 156, 212
Последовательно-обводная технологи-
ческая. связь 156, 157
Последовательные реакции 34, 53, 59,
61, 66, 67, 83—85
Предельно допустимые концентрации
(ПДК) 167
Природный газ 215, 242
Проектирование ХТС 160 сл
выполнение проекта 164, 165
графический метод 166
задание на проектирование 162
надзор за строительством 166
объемное макетное 166
плоскостное макетирование 166
рабочая документация 165, 166
Проектная мощность ХТС 161, 162
Проектно-конструкторские работы 161
Прозрачность воды 23
Производительность
аппаратов 13
катализатора 201, 250
реактора 121
Производство (а)
азотной кислоты 205 сл
аммиака 193 сл
ацетилена 257—260
безотходные 14, 169, 193, 206
метанола 249—252
пластмасс 274—288
полистирола 280—285
полиэтилена 274—280
сернистого газа 178—182
серной кислоты 177 сл
синтетических смол 274—288
уксусной кислоты 260—263
формальдегида 252—257
этанола 263—268
Промоторы см. Активаторы
Промышленные воды 23
Прочность катализатора 82
Пуско наладочные работы 161
Пыль 169. 170
Рабочая документация 165
Рабочие чертежи 165
Рассеивание см. Грохочение
Расходные коэффициенты 41, 42
Реактор (ы) 95. См. также Контактные
аппараты. Печи
адиабатические 121
барботажные 125
гетерогенно-каталитических про-
цессов 135—144
гомогенных процессов 122—124
идеального вытеснения 98, 121,
123, 124 с л
изотермические 121
камерные 123, 124
каскад 122, 123
каталитического крекинга 240, 241
колонные 123
многоступенчатые каталитические
144-150
некаталитических гетерогенных
процессов 124—134
непрерывнодействующие 120
непрерывной поликонденсации 287,
288
оксида углерода 199
основные показатели работы 121,
122
пенные 127
периодического действия 120
пленочные 125
полимеризации этилена 276, 277
политермические 121
полного смешения 98, 121, 124 сл
разбрызгивающего типа 126
с двухвалентными шнеками 128
синтеза аммиака 202, 203
с лопастной мешалкой 128
температурная устойчивость ISO-
152
трубчатые 123, 124, 127, 197, 198
шахтный 198
Реакция воды 23
Регенераторы 31
Резбльные смолы см. Смолы терморе-
активные
Ректификационная колонна 231, 232
Ректификация 245, 246
Рекуператоры 31
Риформинг 228, 241
Селективность см. Избирательность
Селективность катализатора см. Изби-
рательность катализатора
Сера
в качес’ве сырья 178
в составе топлива 217, 224
печи для сжигания 179
сжигание 178—180
интенсификация процесса 179
модели процесса 180
Серная кислота
моногидрат 177
293
Серная кислота
олеум 177
производство
интенсификация 192, 193
способы 177—190
сырье 178—182
технологическая система с двой-
ным контактированием (ДК)
188, 191, 192
энерготехнологическая циркуля-
ционная система 193
сорт 177
Сернистый газ 177
производство 178—180
Скорость процесса (реакции) 37—40
анализ влияния
времени 64—67
давления 59—61, 208, 210, 211,
234, 271, 276, 278
диффузионных торможений 84, 85
катализатора 63, 64, 86, 183,184,
207—209 249, 271
концентрации 50, 70. 208, 271
режима работы контактного ап-
парата 143—150
температурного режима по дли-
не реакционной зоны 101, 103,
106,107
температуры 53—59, 105, 185,
207, 210, 211, 234, 271, 273,
276, 278
типа технологической связи
155-158
числа ступеней 112—114
инвариантная 39
мгновенная 38
Смазочные материалы 227
Сметная документация 165
Смолы
пол иконденсац ионные 285—288
полимеризационные 274—285
термопластичные 285—288
термореактивные 286—288
Совершенствование процесса 205, 214,
215, 267
Солесодержание воды 22
Способ получения 9
Степень использования внутренней по-
верхности катализатора 84, 85
Степень межфазного перехода см. Сте-
пень превращения
Степень превращения 36, 50—52
анализ влияния
времени 64—67, 234, 273
давления 183, 210, 211, 234, 249,
266
концентрации 50, 51, Г88,189,273
неравномерности времени пребы-
вания реакции 97, 98
режима работы контактного ап-
парата 143—150
Степень превращения
анализ влияния
температурного режима по дли-
не реакционной зоны 101, 104,
107
температуры 55—58, 102, 105,
109, ПО, 183—190, 210, 211,
234, 249, 266
типа технологической связи .155,
- 156г 158, 159
числа ступеней 112—114
равновесная 45—49
Стереорегулярные полимеры 271
Структурная схема XIС 154
Сухая перегонка топлива 218
Сырой бензол 219, 220
Сырье
аммиак 206, 207
бензин 230
битумы 216
бурый уголь 17
вода 21—26, 207
воздух 26, 194, 207
газойль 230
горючее 17
горючие сланцы 216
древесина 17
древесная смола 247
животное 17, 18, 247
запасы 17
каменноугольная смола 220
каменный уголь 17
качество 30
классификация 16, 17
кокс 195
коксовый газ 195, 247
колчедан 178
комплексное использование 14, 18
концентрированное 18
минеральное 16, 17
надсмольная вода 220
нерудное 16, 17
нефтехимическое 227
нефть 17, 234, 247
нефтяные газы 242, 247
обогащение 18—21
отходящие газы 178
парафины 227
пищевое 17, 18
попутный газ 195, 247
природный газ 17, 195, 216, 247
растительное 17, 18, 247
рудное 16
сера 178
сероводород 178
сланцевая смола 247
сланцевый газ 247
сланцы 17
смола коксования 247
сырой бензол 220
техническое 17
394
Сырье
технологическое 16
торф 17, 217
торфян >й деготь 247
угли 195, 219
церезины 227
Температура зажигания катализатора
81
Тепловой баланс 42—44
Тепловые загрязнения 176
Термическое обогащение сырья 21
Термодинамический анализ 44
Термостойкость катализатора 81
Технологическая схема ХТС 154
Териологический режим 33
Технология 9
Топливный эквивалент 27
Топливо. См. также Сырье
антрацит 215, 217
бензин 215, 225, 226
бурый уголь 217
водород 216
водяной газ 215
газообразное 215, 242—246
газотурбинное 226
газы нефтепереработки 215
генераторный газ 215
горючие газы 226
горючие сланцы 215, 217
дизельное 226
доменный газ 215
древесина 215
древесный уголь 215
Жидкое 215, 226
искусственное 215, ?16
каменноугольная смола 215, 219,220
каменный уголь 215 217, 219
керосин 215, 225 ,
классификация 215
кокс 215, 219
коксовый газ 215, 219
котельное 226
мазут 215, 225, 233
метиловый спирт 215
методы переработки 217, 218
нефть 215, 224 сл
печное 226
полукокс 215, 219
попутный газ 215
природное 215, 216
природный газ 215
реактивное 226
синтетическое жидкое (QKT) 216
твердое 215, 216—224
торф 215, 217
Трегеры см. Носители
Туман 170
Уксусная кислота, получение
жидкофазным окислением бутана
263
Уксусная кислота, получение
окислением ацетальдегида 261, 262
синтезом из метанола и оксида уг-
лерода 263
Умягчение воды 24, 25
Уровни протекания процесса 34, 35, 50,
89
Фактор линейной скорости 97
Флотационные машины 21
Флотационные реагенты 21
Флотация 20
Формальдегид, получение
неполным окислением метана 253
окислением метанола на оксидных
катализаторах 255—257
окислительным дегидрированием
метанола 253—255
Хемосорбция 244
Химизация 11
Химико-технологическая система (ХТС)
152 сл
анализ данных для проектирова-
ния 162, 163
блочной полимеризации стирола
282, 283
модель 160
перегонки нефти 233
переработки прямого коксового га-
за 222, 223
площадка для строительства 163
получения
полистирола 284
полиэтилена 278, 279
*” ”3 уксусной кислоты 262
- формальдегида 254
производства
азотной кислоты 212—214
) ацетилена 259
новолачных смол 287
резольных смол 287
серной кислоты 190—192
этаиола 265, 267
разделения природного газа 243
район строительства 162
связи между элементами 1э2, 153,
155-160
синтеза
аммиака 203, 204
метанола 251
структура 153 "
схемы 154
технико-экономическое обоснова-
ние создания 162
циркуляционного контура 193
элементы 152
энерготехнологнческие 27
Химико-технологический процесс 32 сл
в 'замкнутом объеме 38, 50 сл,
71 сл, 95, 96
295
Хнмико-технологнческий процесс 32 сл
в потоке 95 сл
каталитический 61—64, 80—88
классификация 33, 34
константа равновесия 45—49
материальный баланс 42
основные показатели, характер из-
менения
в зависимости от типа потока
116-119
в кинетической области 50—71,
79
во внешнедиффузионной области
73-76
во внутридиффузионной области
76-79
по высоте контактного аппарата
145-150
по длине реакционной зоны 99—
НО, 115, 116
при секционировании 112—114
при теплообмене 110—112
тепловой баланс 42—44
уровни протекания 34, 35
Химическая технология 9
Целевая функция 69
Церезины 227
Цетановое число 226
Экзотермические процессы (реакции)
14, 26, 34, 55, 56, 102, 104—106, 109
Экологические проблемы см. Охрана
окружающей среды
Электрокрекинг 259
Электромагнитное обогащение сырья 20
Электростатическое обогащение сырья
20
Эндотермические процессы (реакции)
26, 34, 55, 57, 102, 109
Энергоресурсы 27 сл
вторичные (ВЭР) 30—32, 176
Энергосберегающие
производства 14
технология 29, 30
Энерготехнологические ХТС 27
Энерготехнологическое комбинирование
30
Энерготехнология 27
Этанол, получение
прямой гидратацией 266—268
сернокислотной гидратацией 264,
265
УЧЕБНИК
ВАЛЕРИЙ ЕЕГЕНЬЕРИЧ СОРОКО
СВЕТЛАНА ВАСИЛЬЕВНА ВЕЧНАЯ
НИНА НИКОЛАЕВНА ПОПОВА
Основы
химической
технологии
Редактор Л. Ф. Травина
Технический редактор Л. Ю. Линева
Корректор Л. С. Александрова
ИБ № 1816
г. Могилев
БИВЛИОТГКА
вх-*’ е.г.ясмюгвчес*ого
Инв. ----------------
Сдано в набор 26.12.85. Подписано в печать 22.05-86. М-33823. Формат бумаги 60X90/16. Бумага
тип. № 2. Гарн. литературная. Печать высокая. Усл. печ. л. 18.5. Усл. кр.-отт. 18,5. Уч.-изд. л. 22,0.
Тираж 17 800 экз. Заказ №1859 Цена 1 р. Изд. № 2724.
Ордена «Знак Почета» издательство «Химия», Ленинградское отделение, 191186, г. Ленинград,
Д-186, Невский пр., 28.
Отпечатано с матриц Ленинградской типографии № 2 головного предприятия ордена Тру-
дового Красного Знамени Ленинградского объединения «Техническая книга* им Гвгрнии
Соколовой Союзполиграфпрома при Государственном комитете СССР по делам издательств,
полиграфии и книжной торговли. 198052. г. Ленинград, Л-52, Измайловский проспект, 29
в Ленинградской типографии № 4 ордена Трудового Красного Знамени Ленинградского
объединения «Техническая книга» им. Евгении Соколовой Союзполиграфпрома при Госу-
дарственно^ м комитете СССР по делам издательств, полиграфии и книжной торговли.
191126, Ленинград, Социалистическая ул., 14.