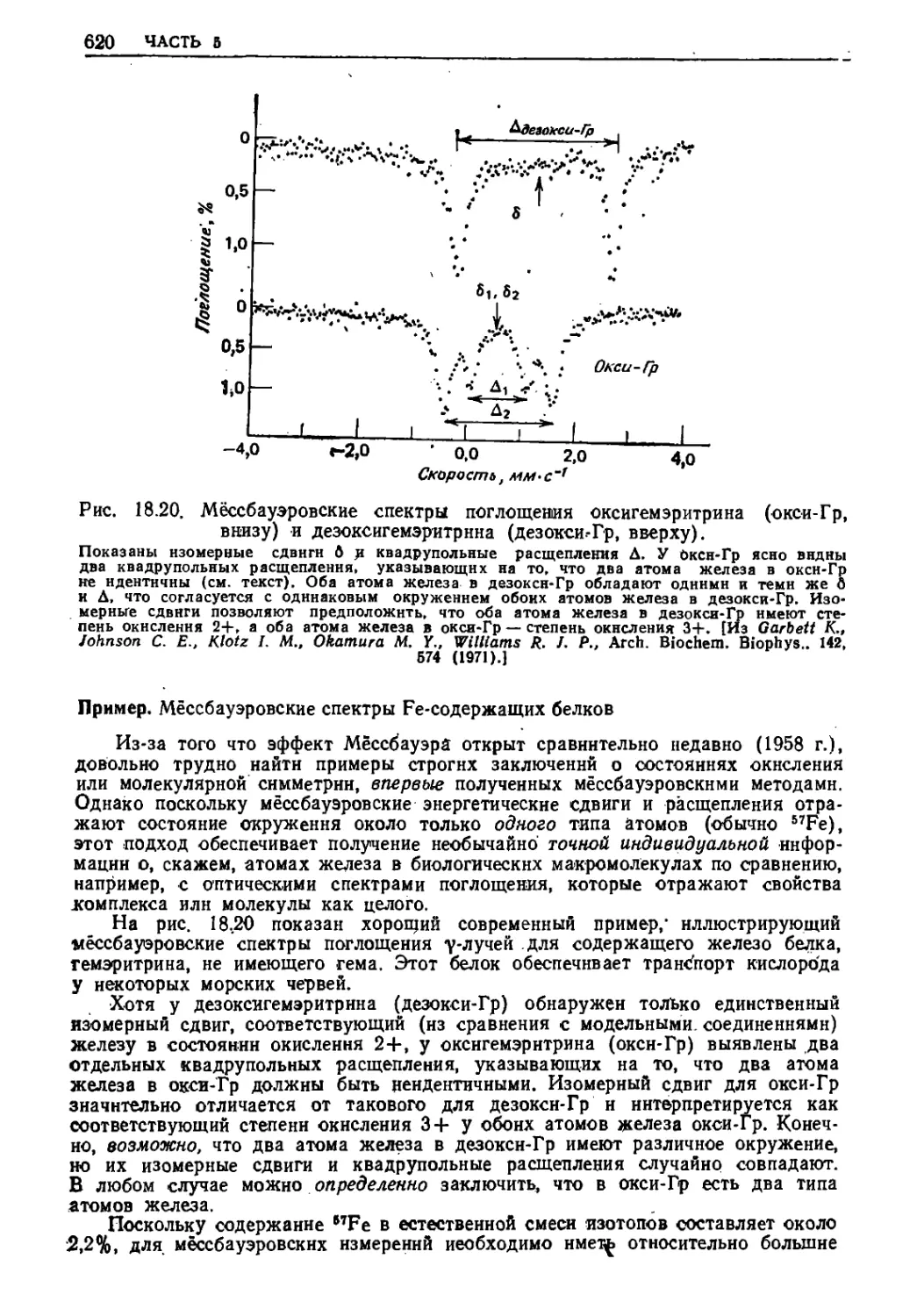
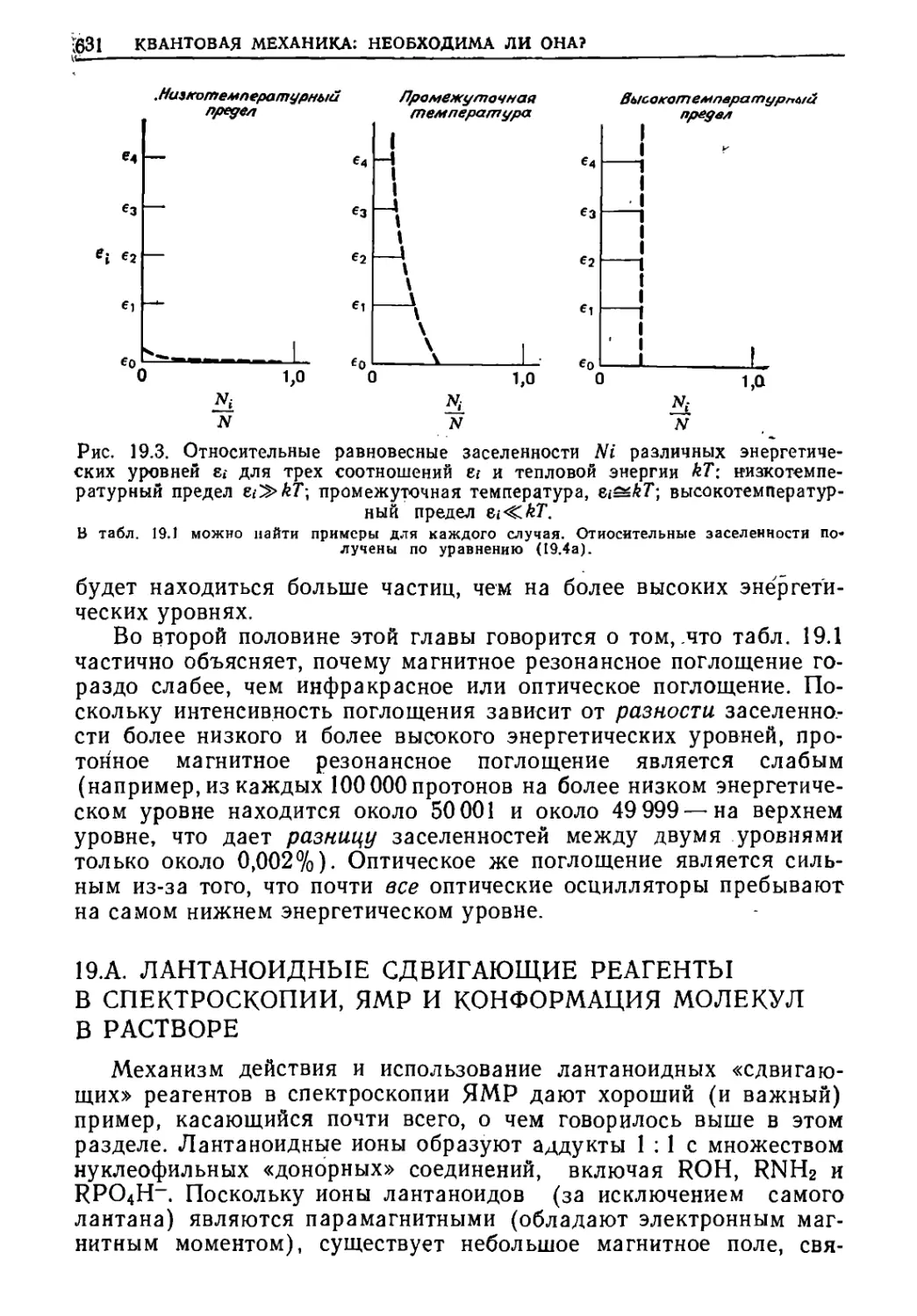
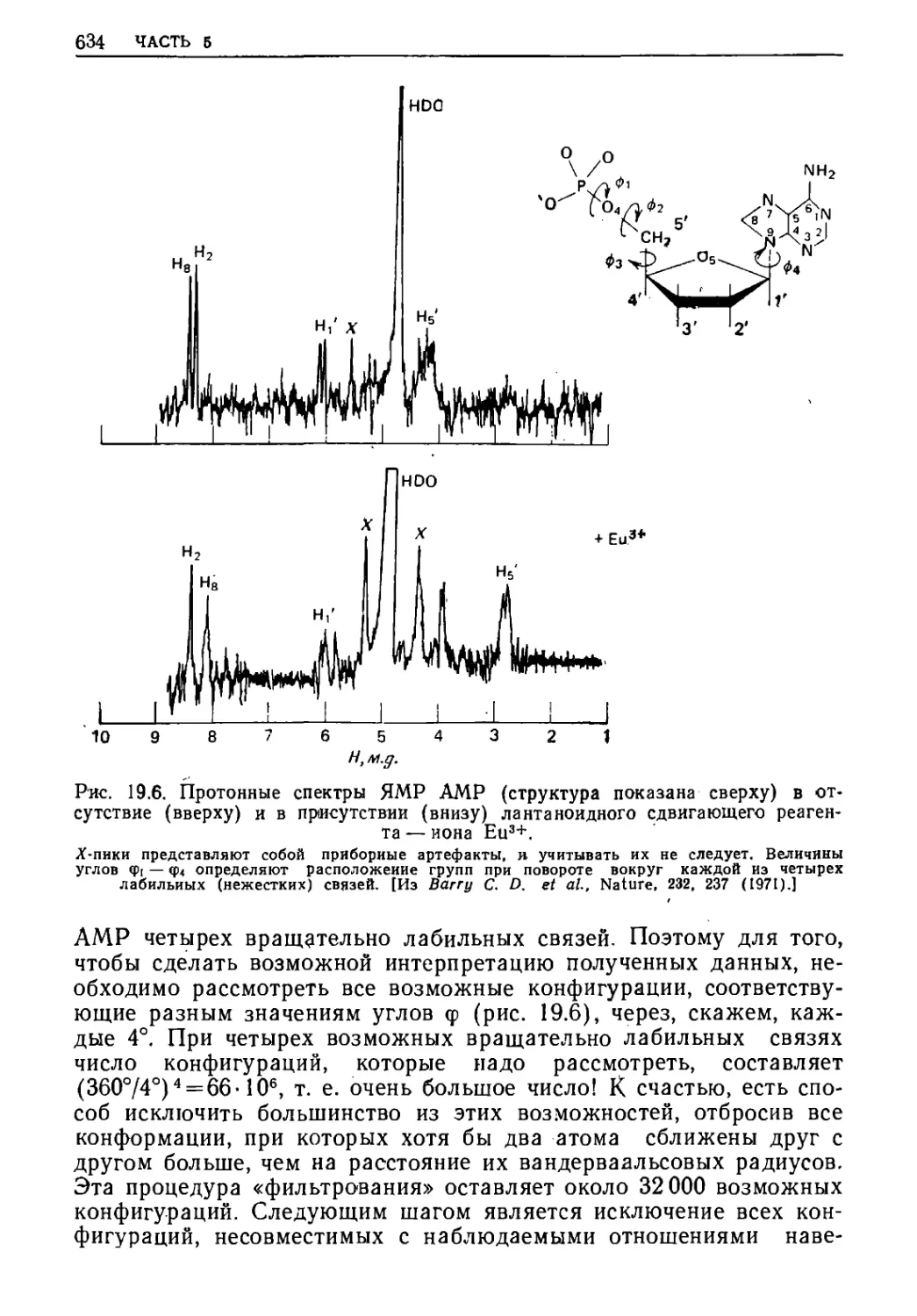
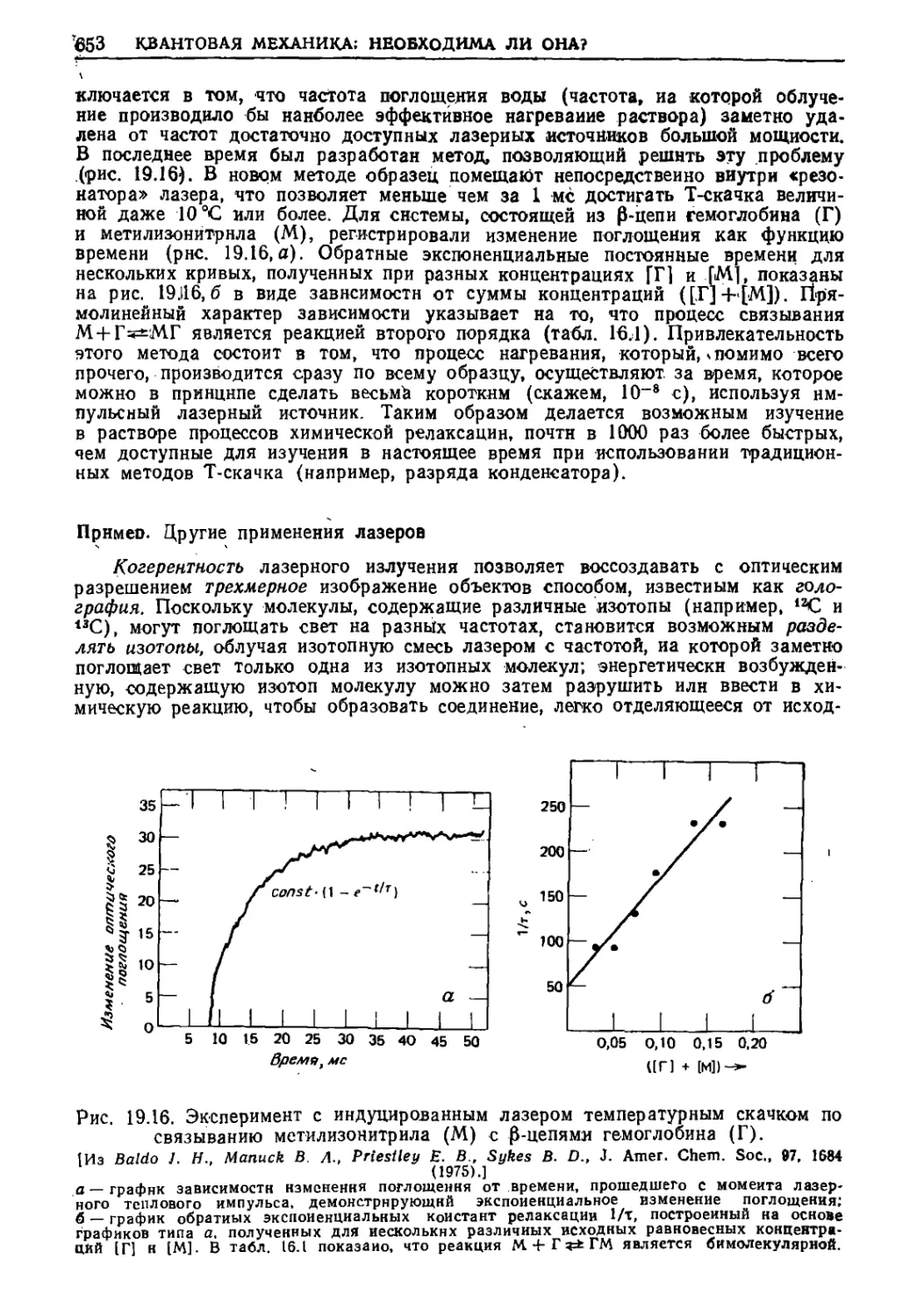
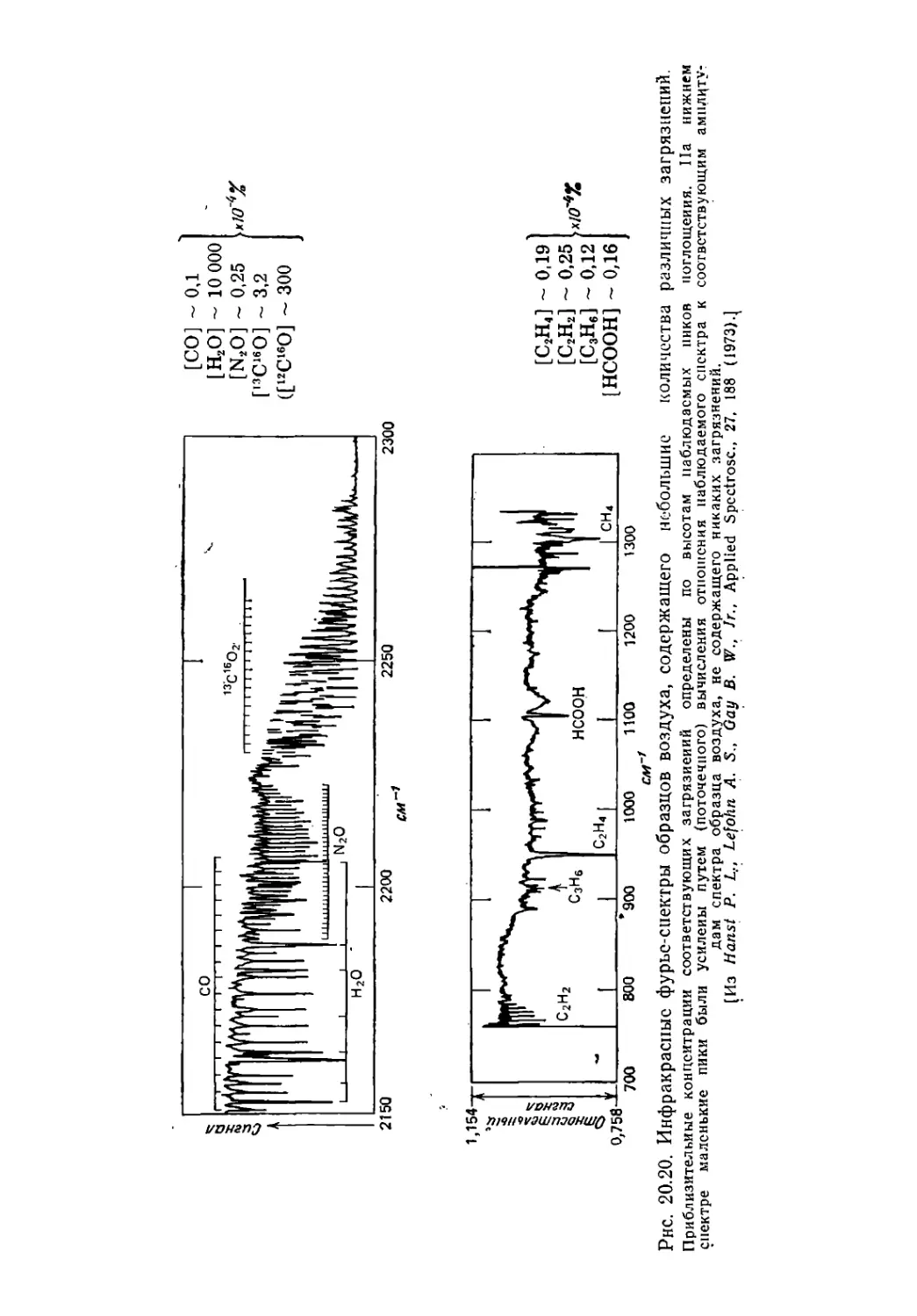
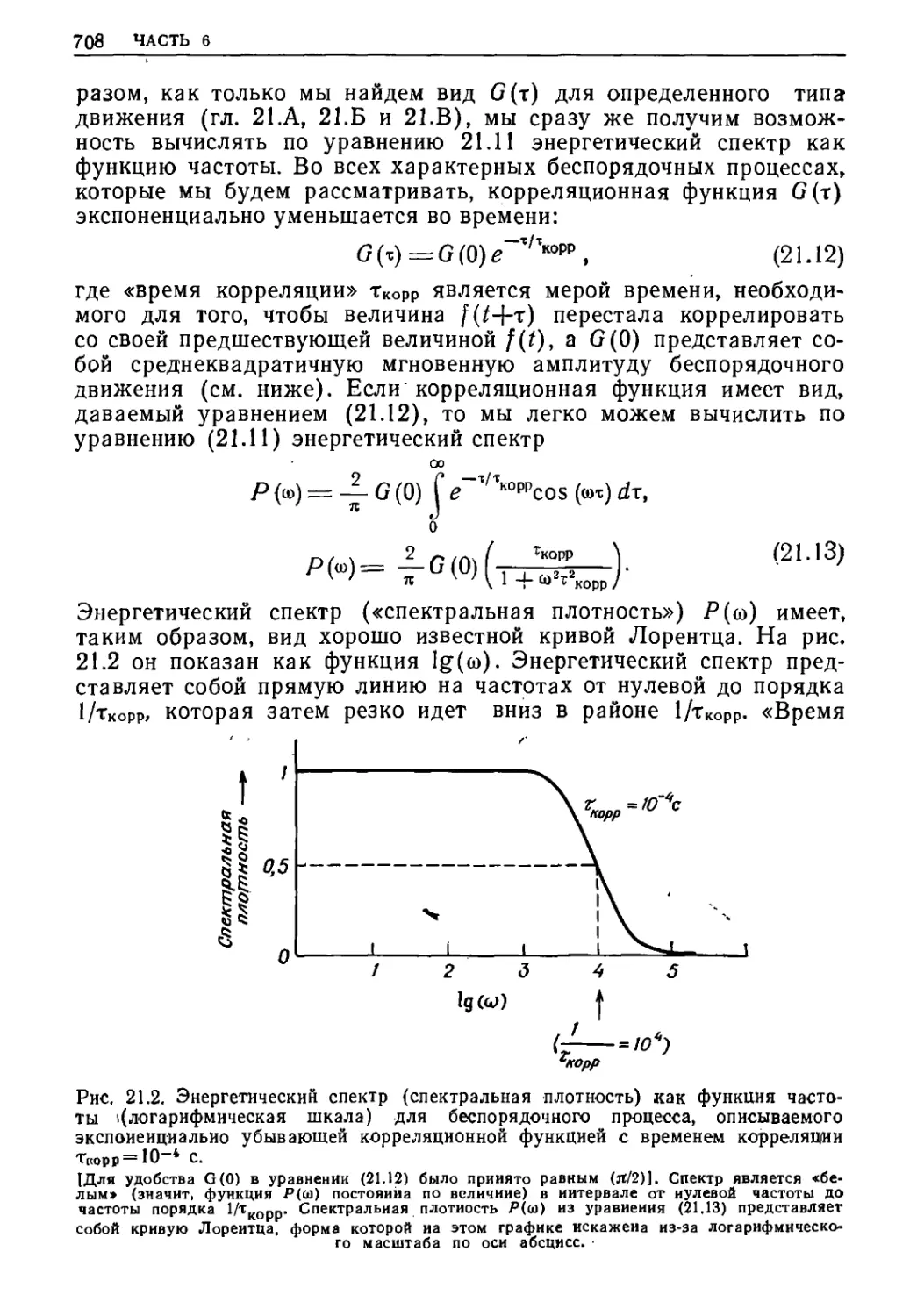
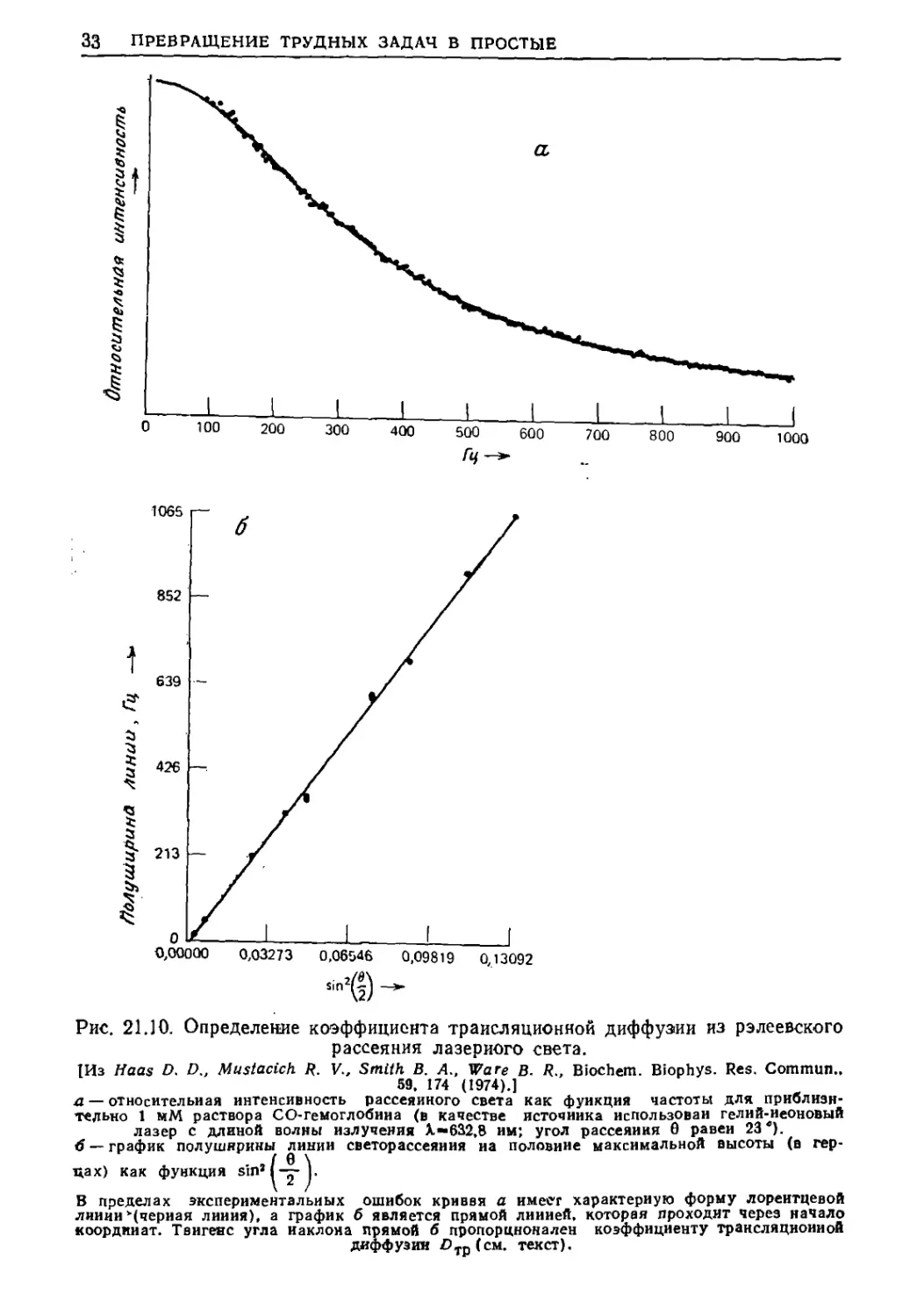
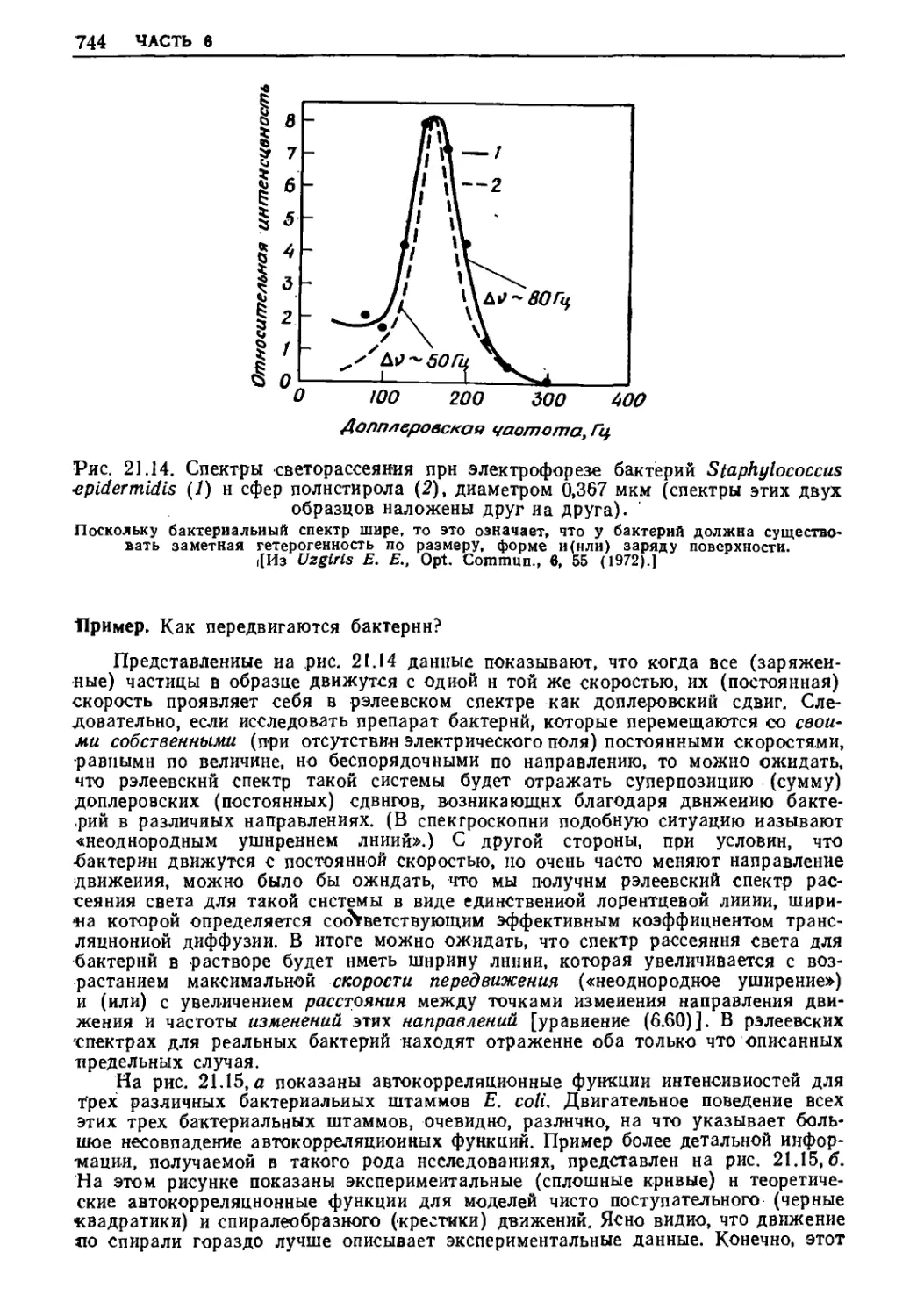
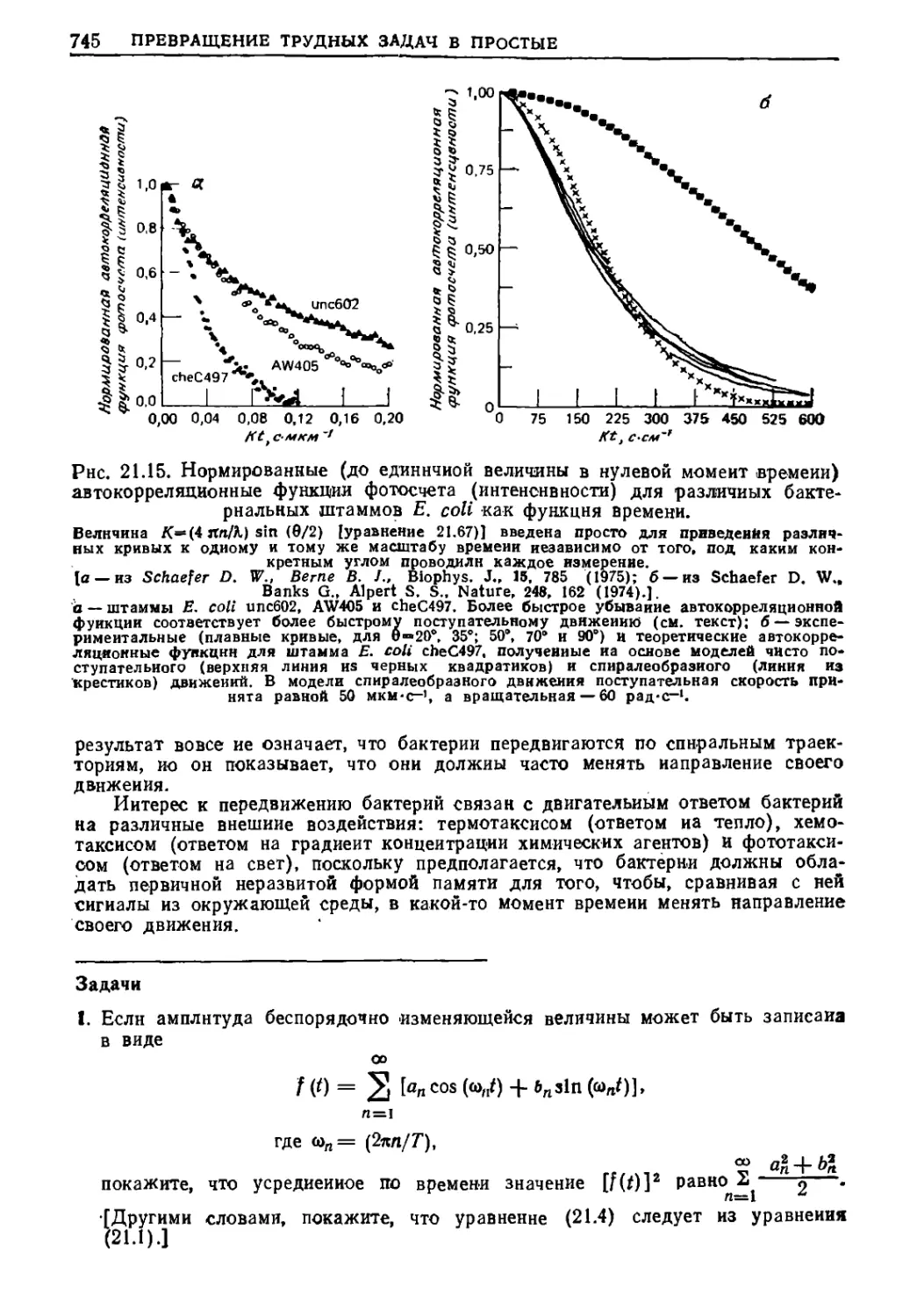
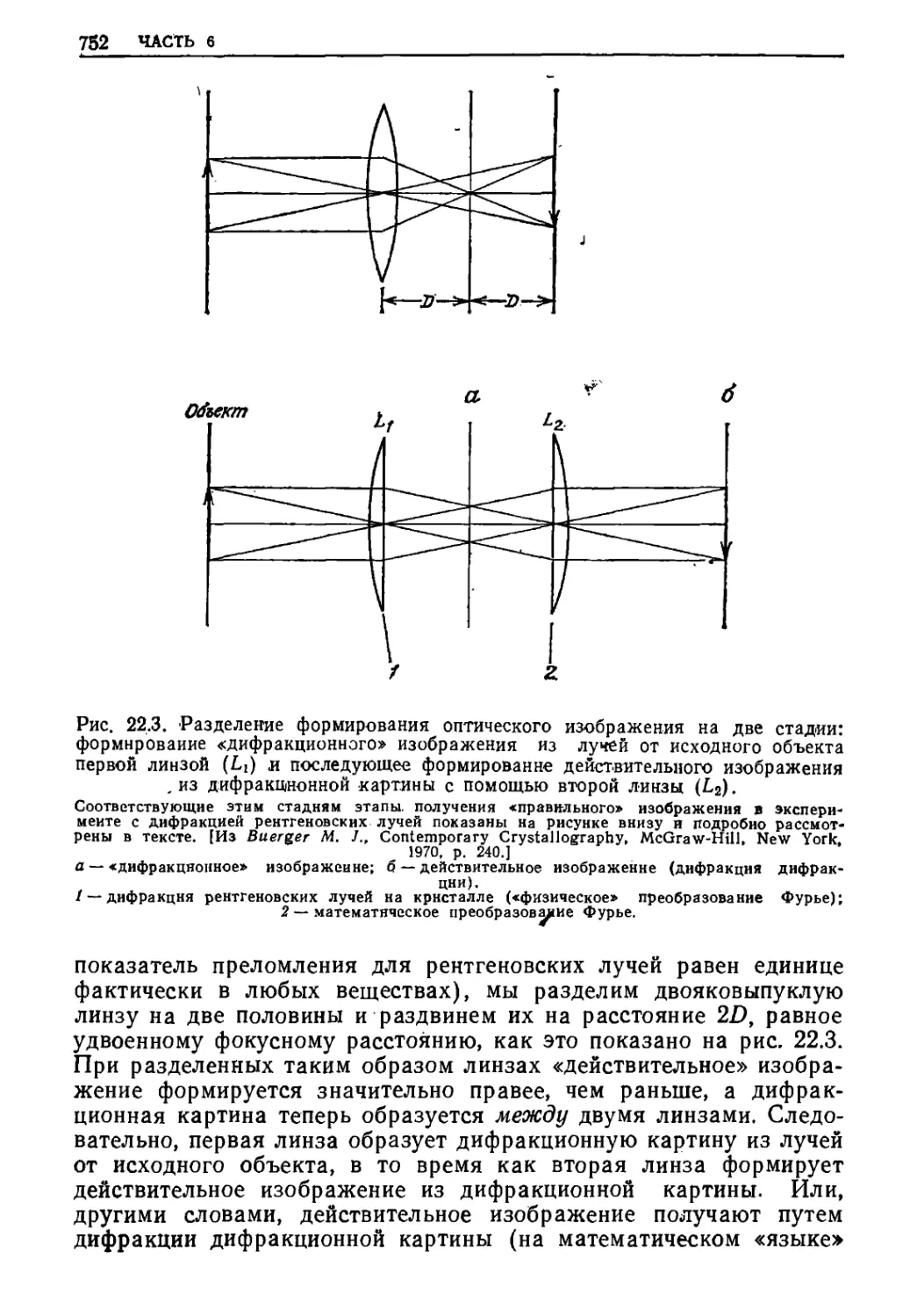
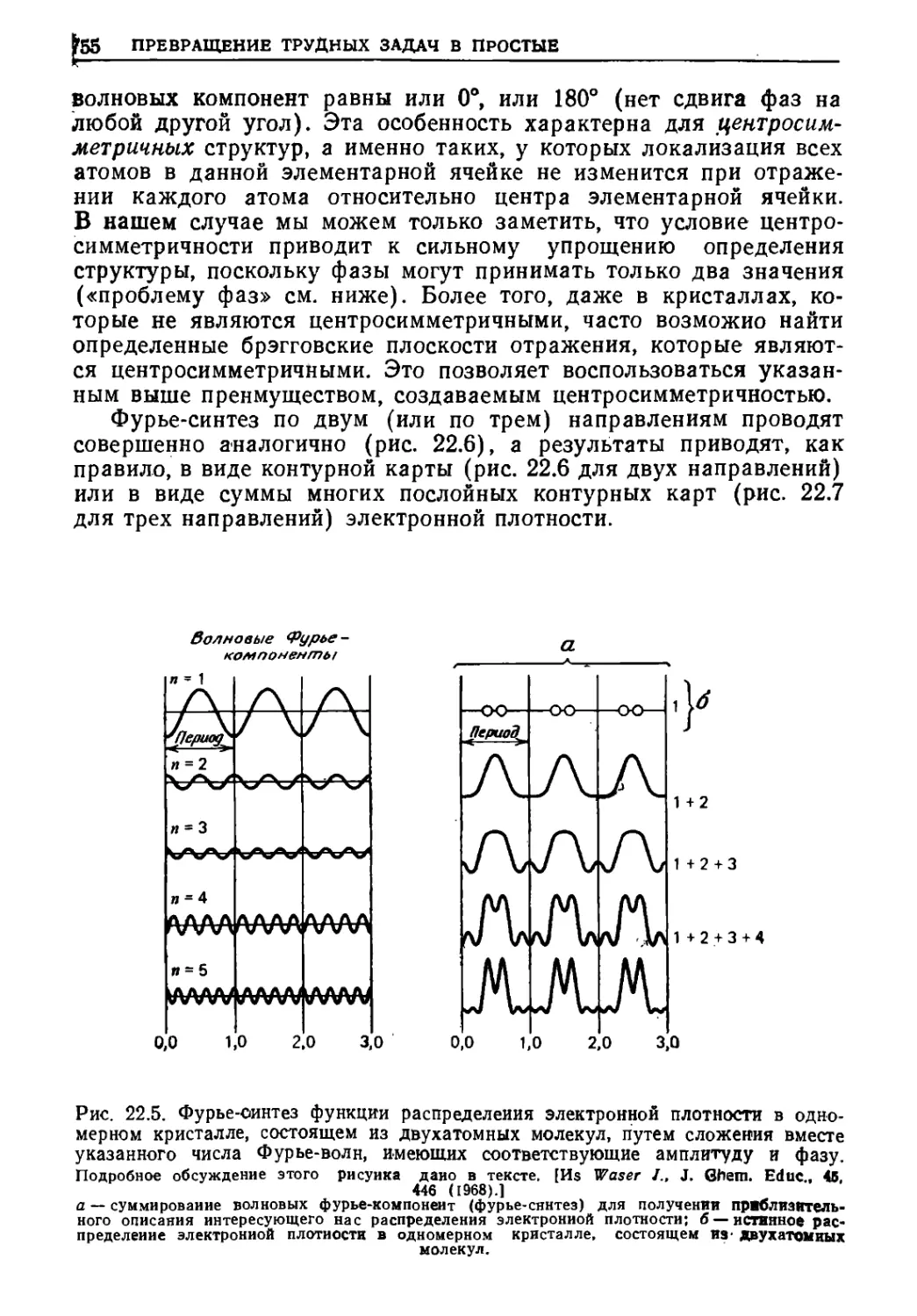
/











































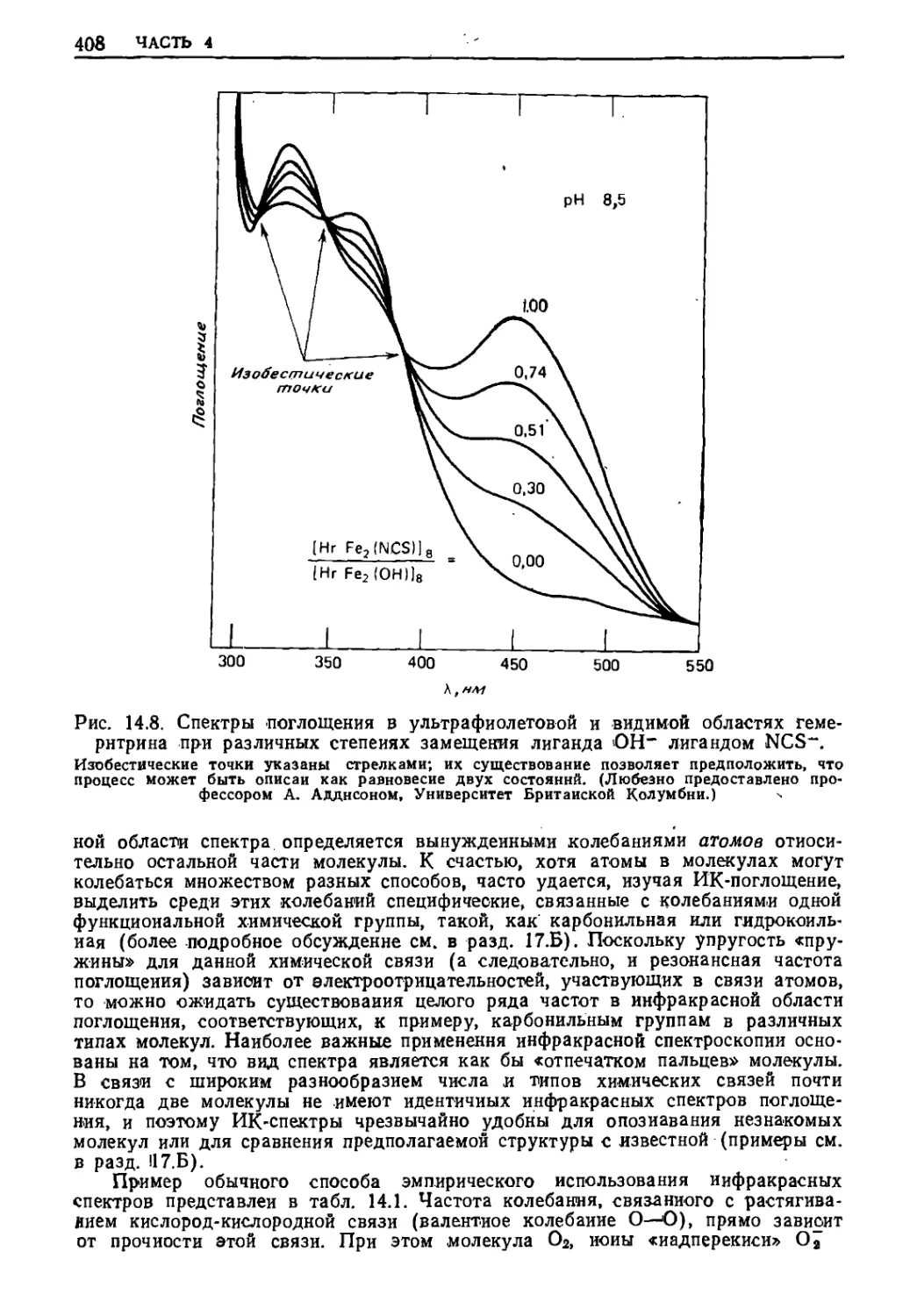
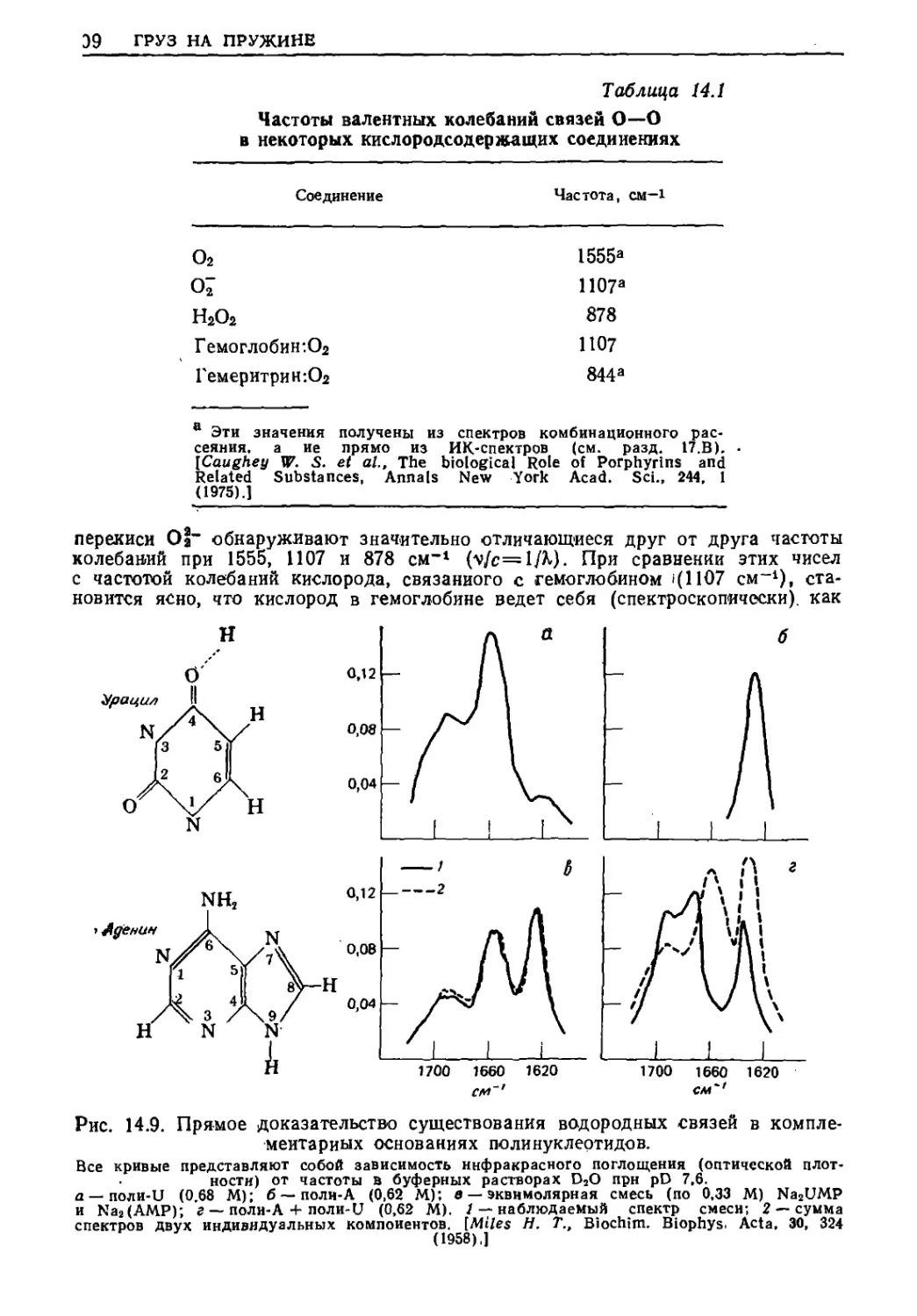



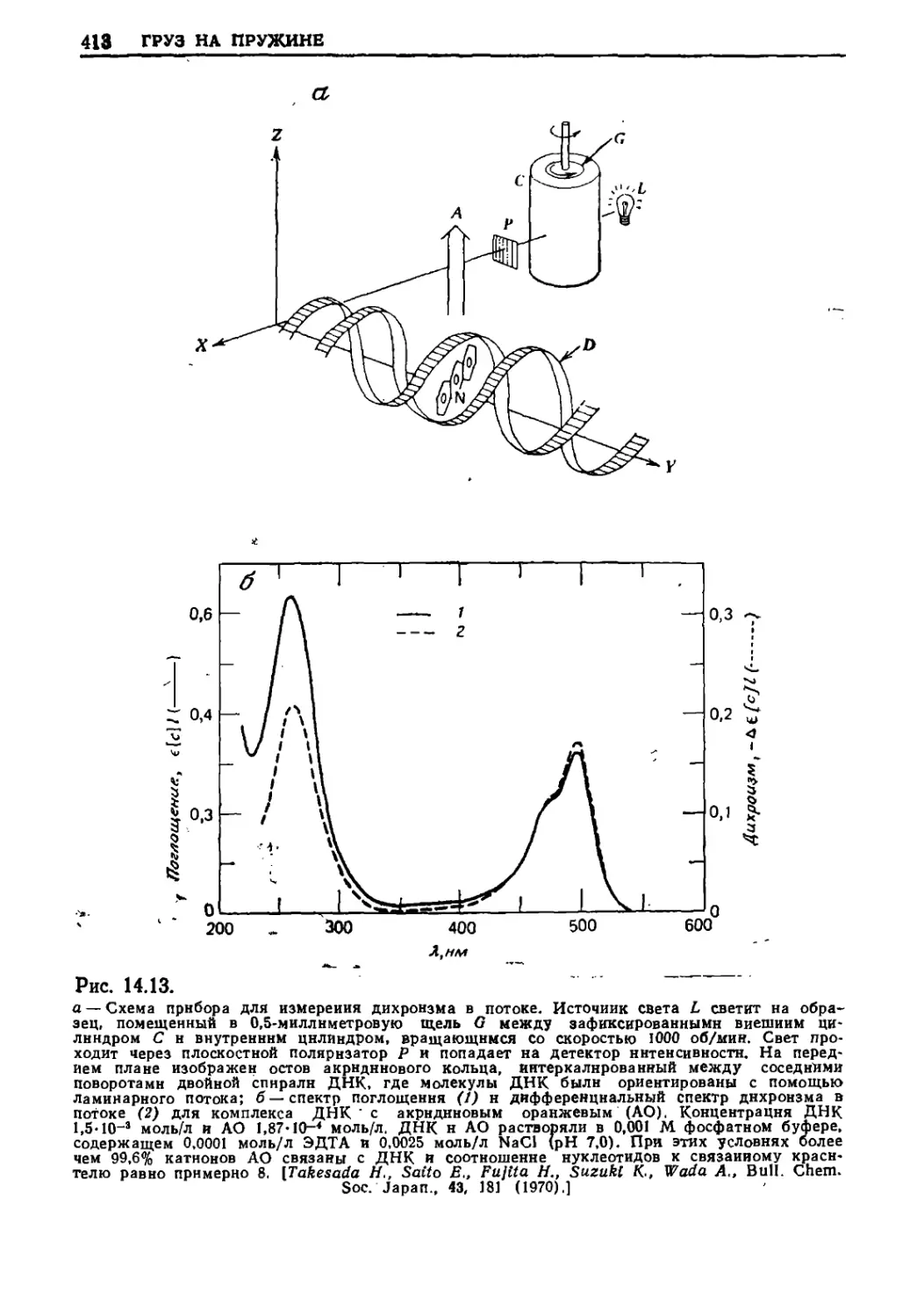
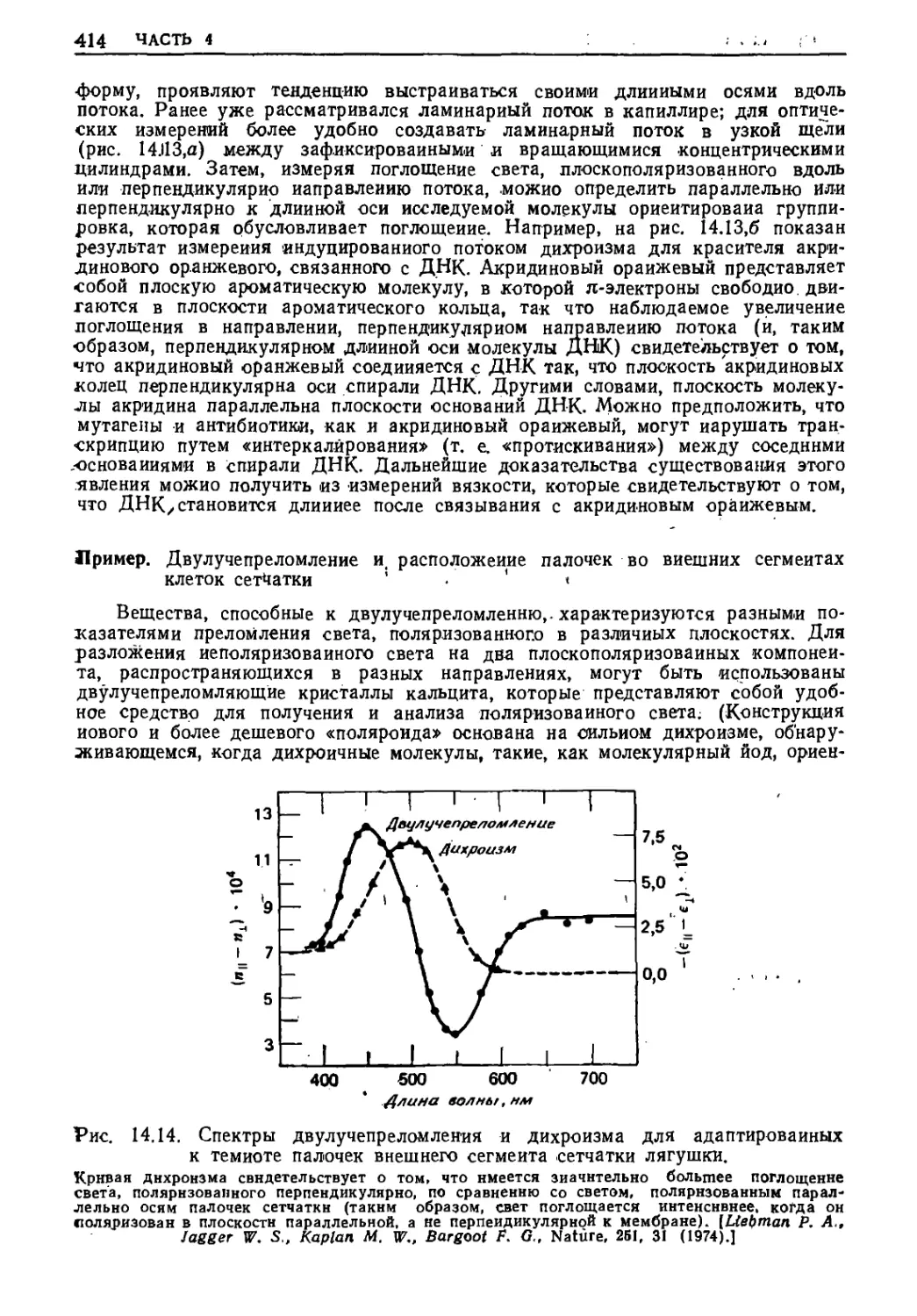

















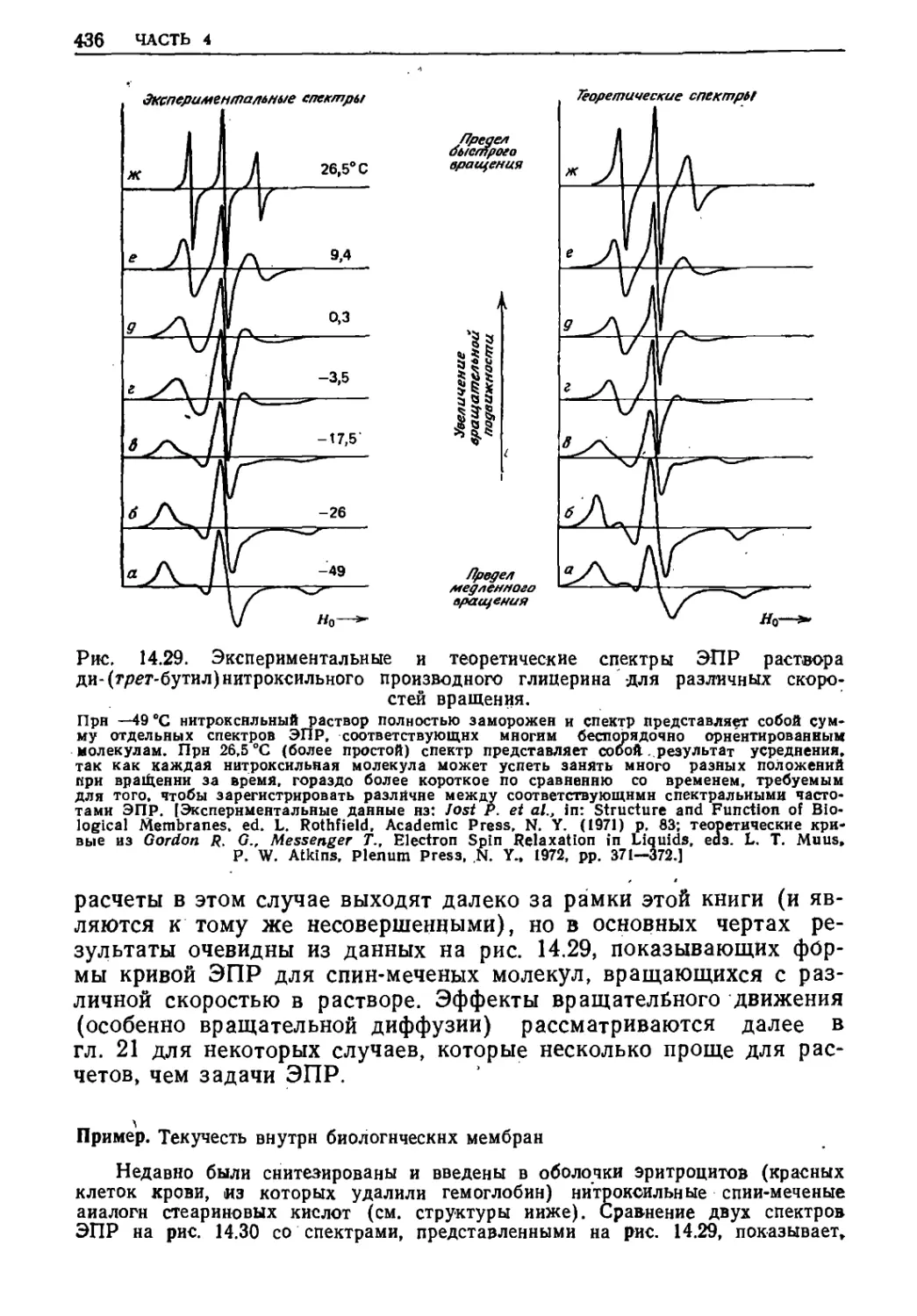
















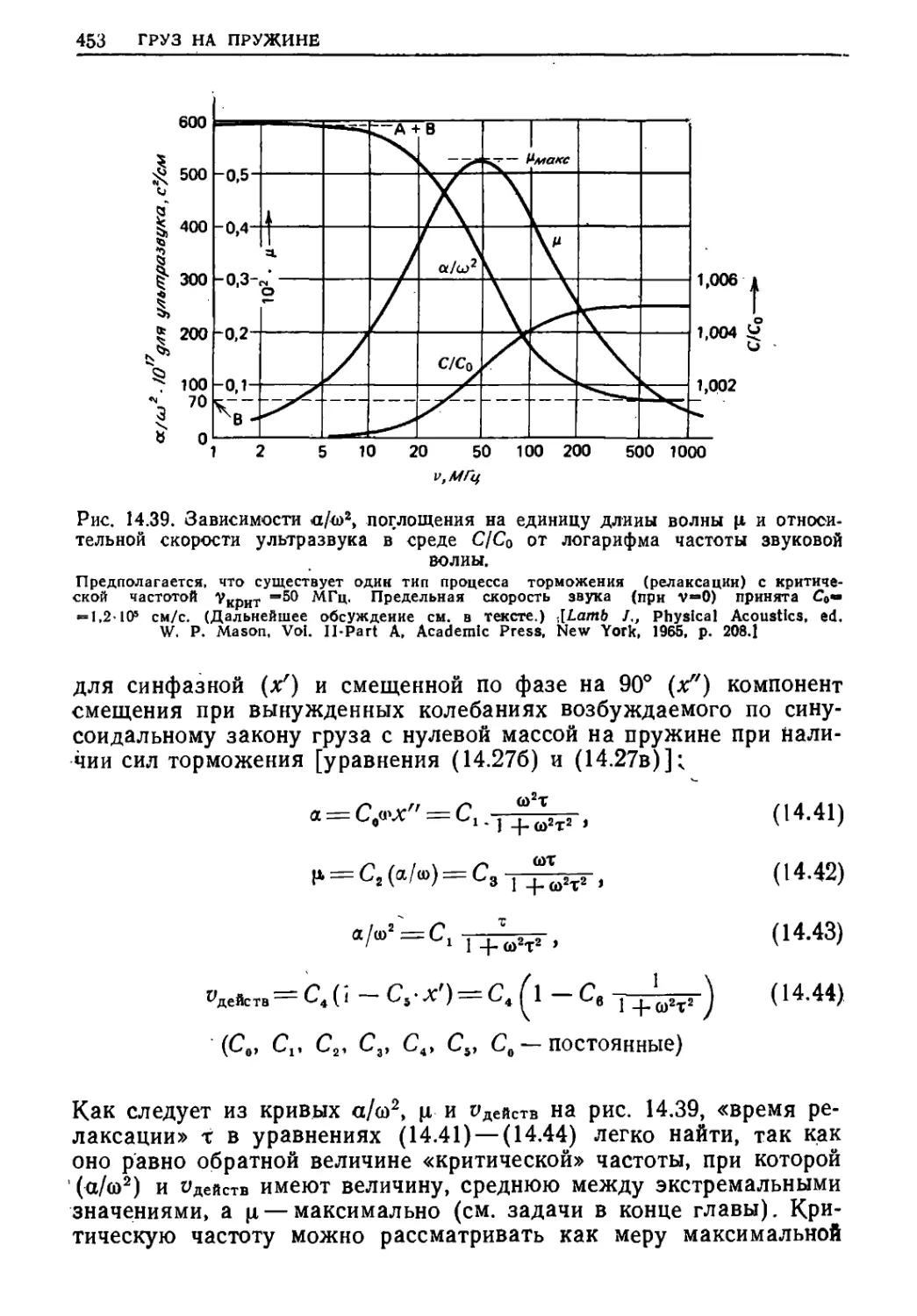



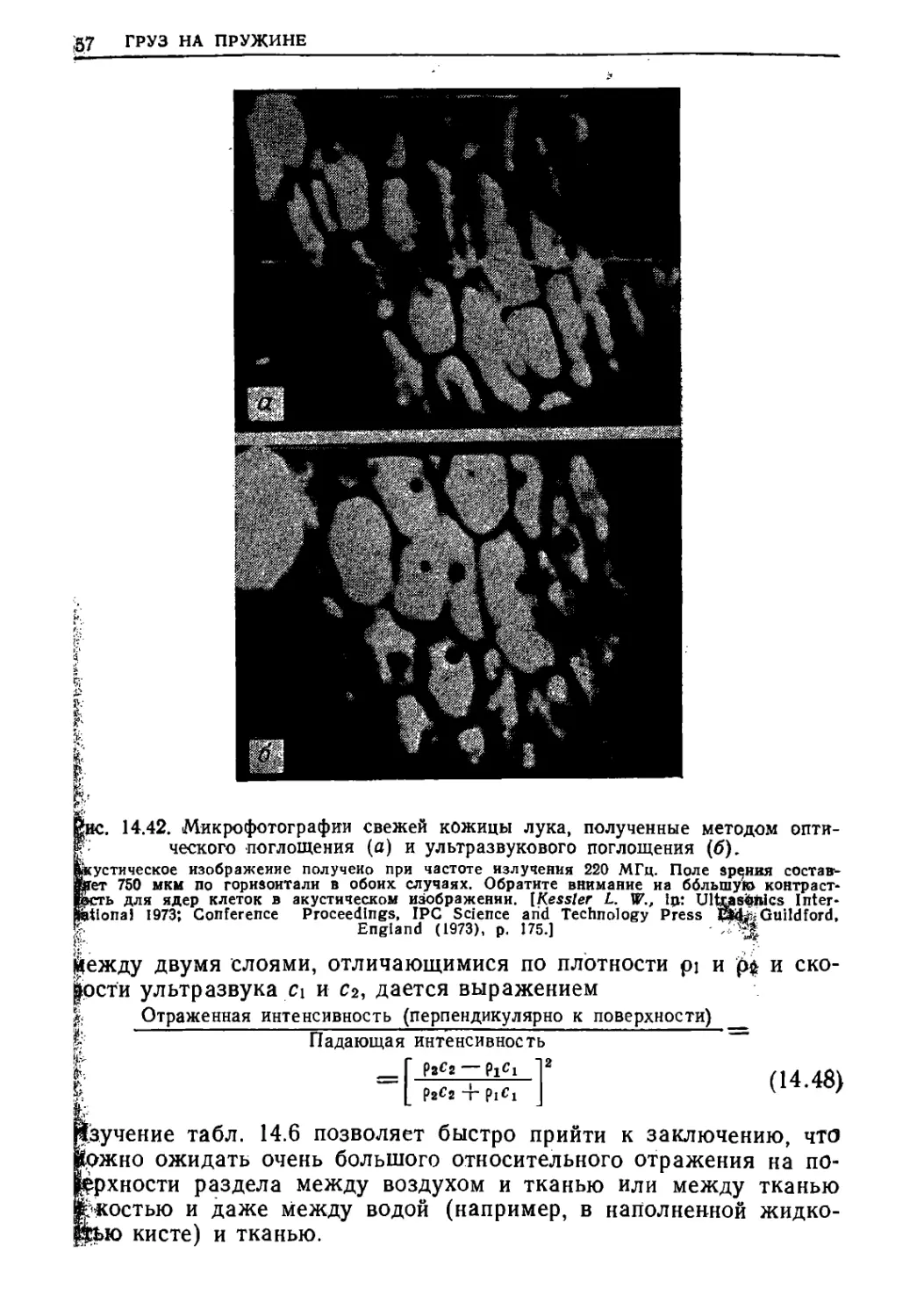
































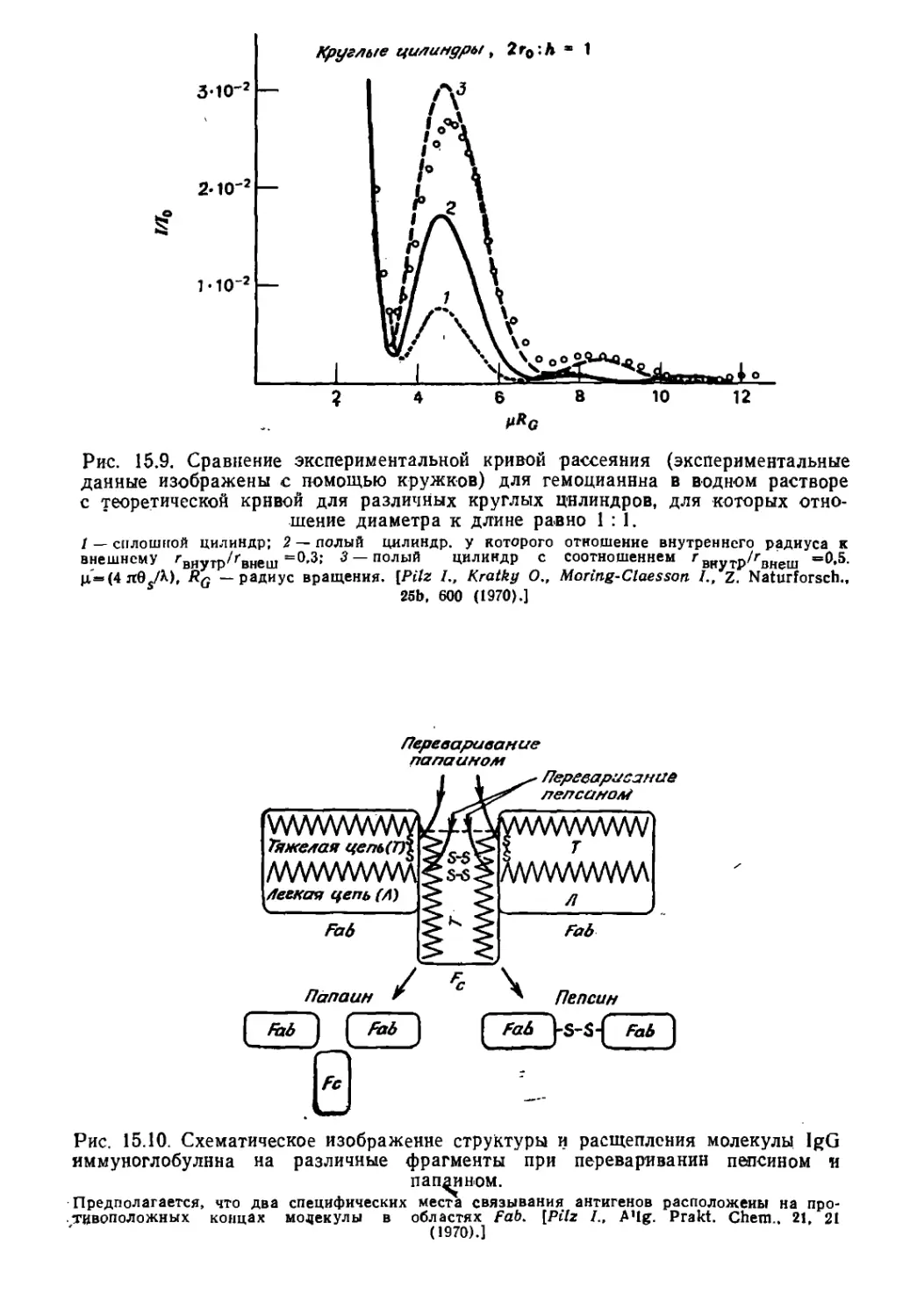






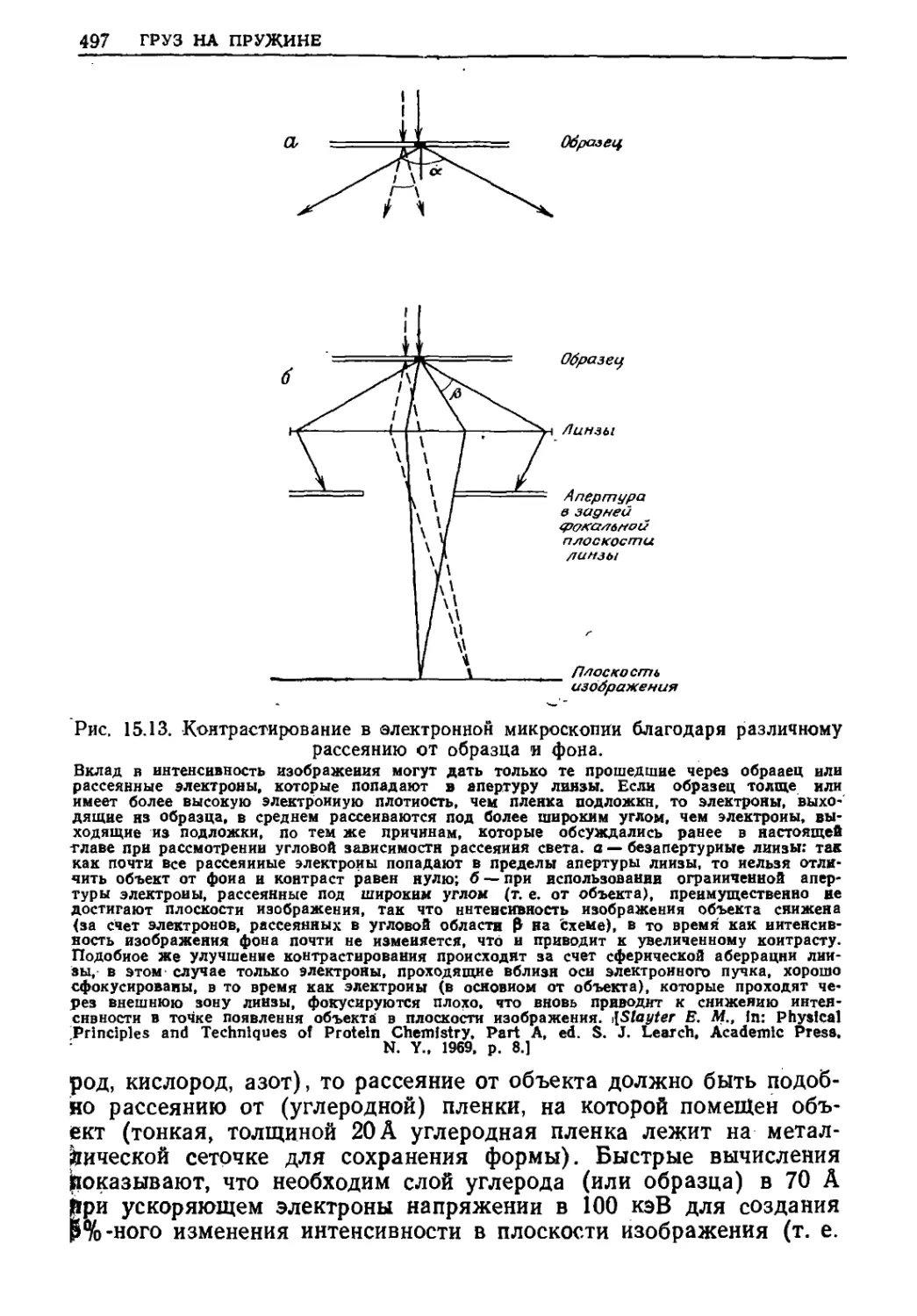
















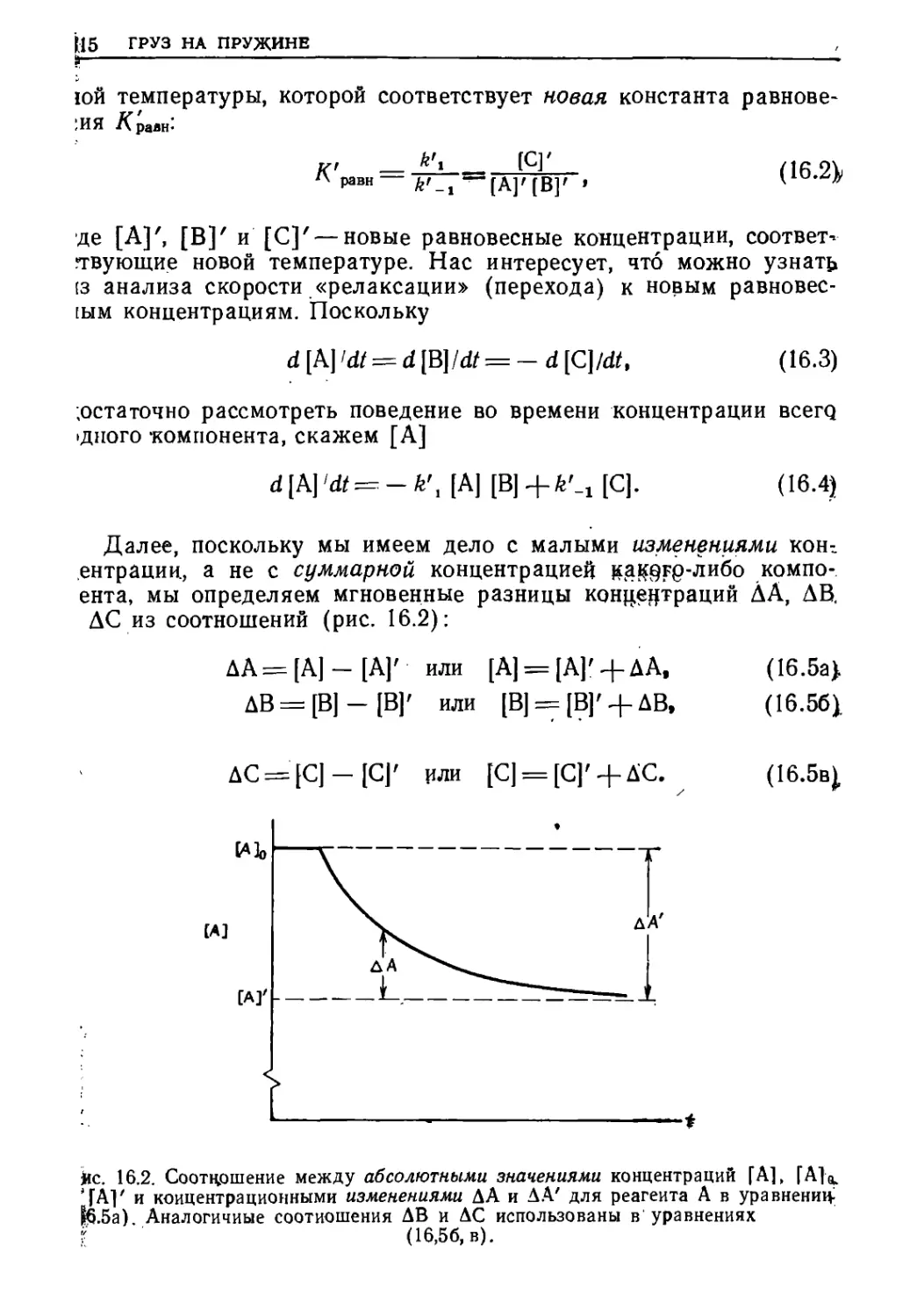

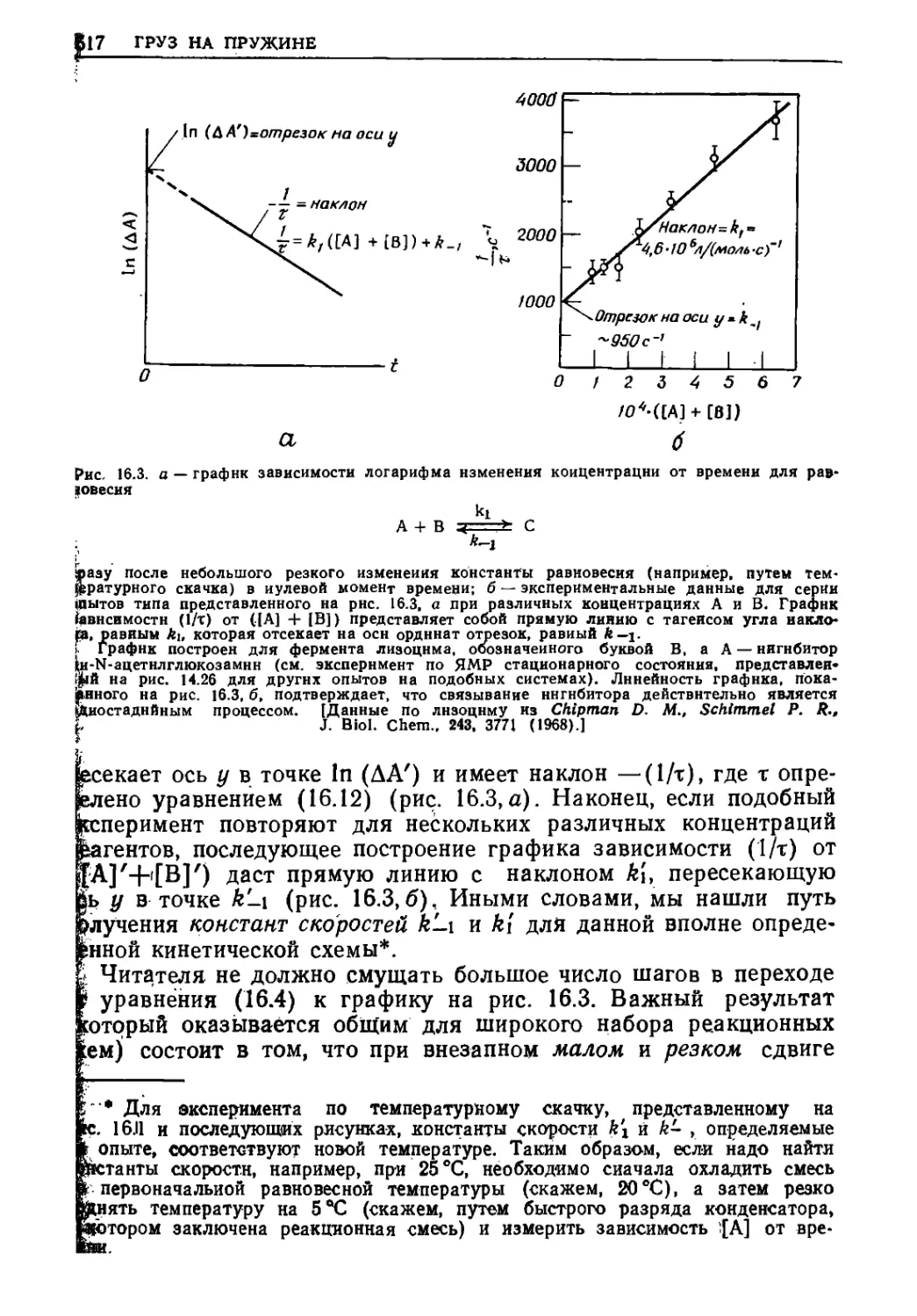






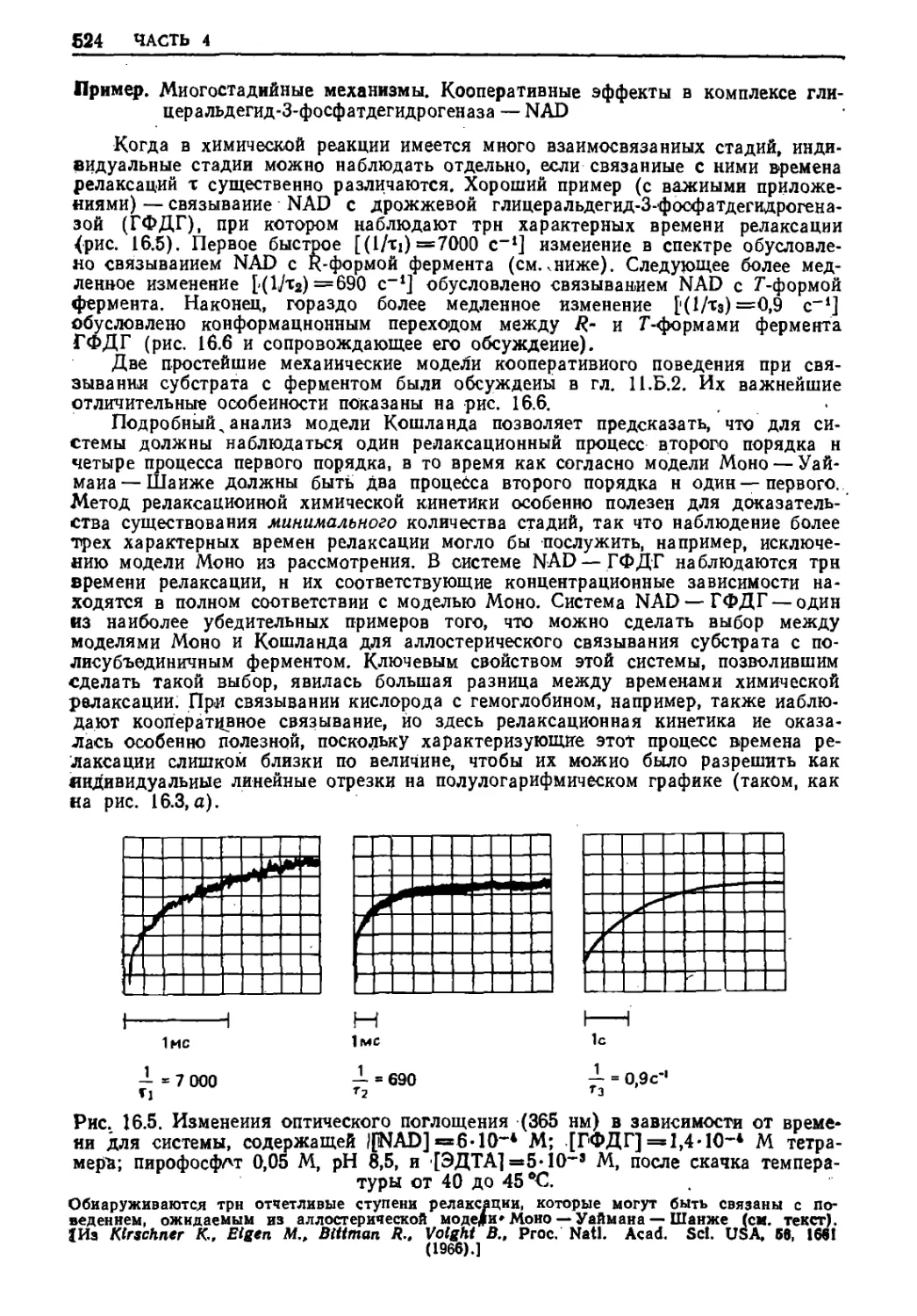

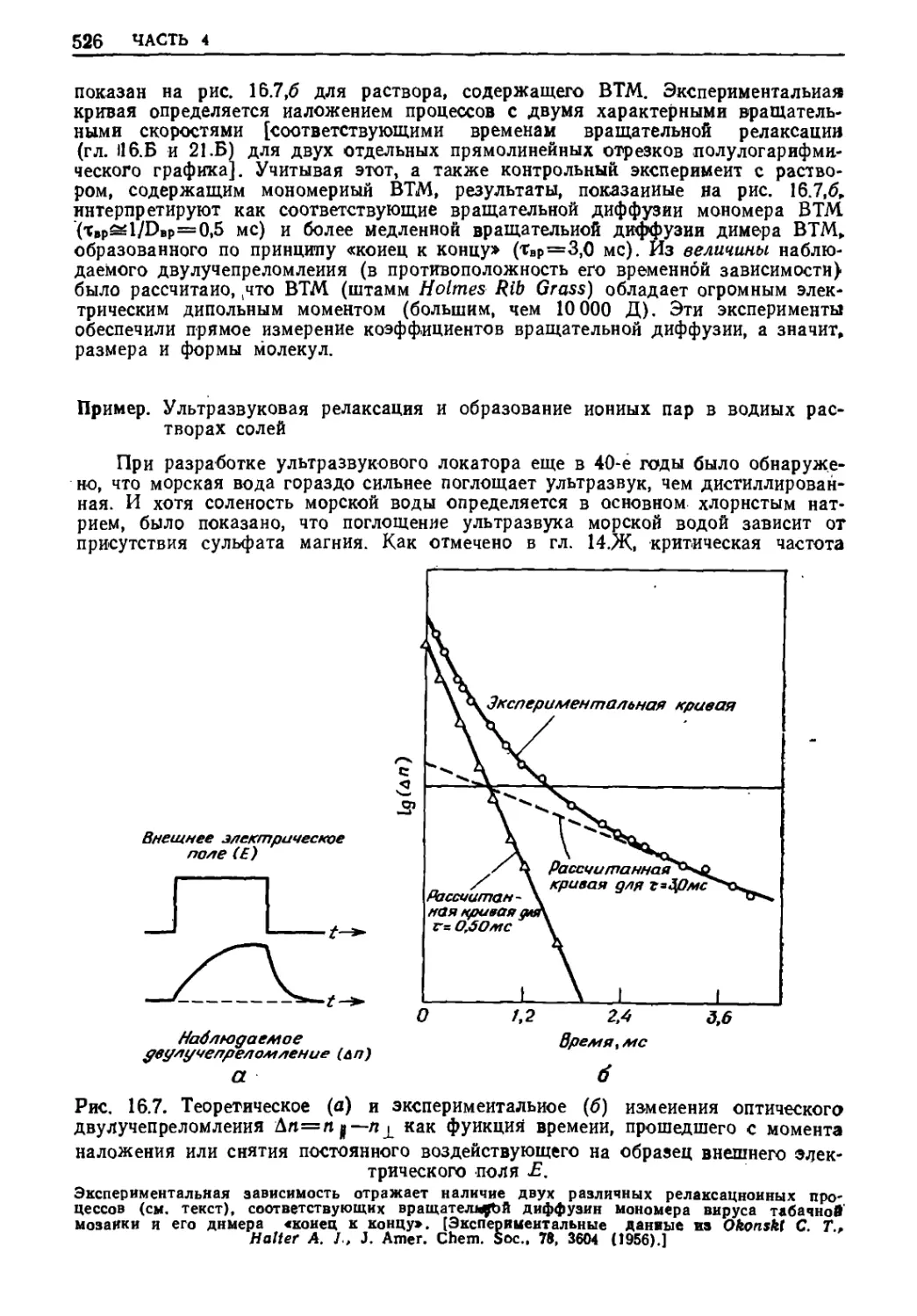


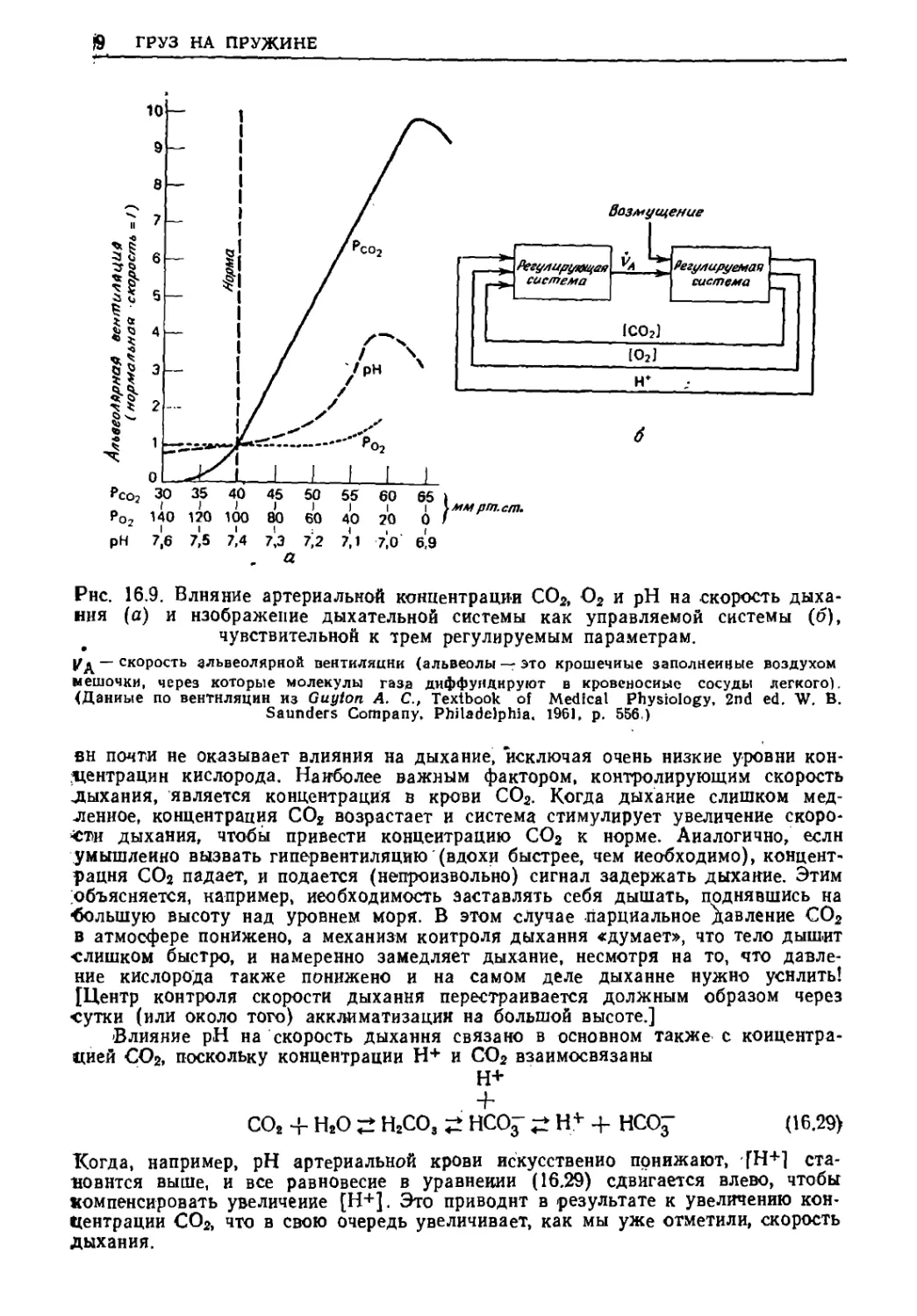




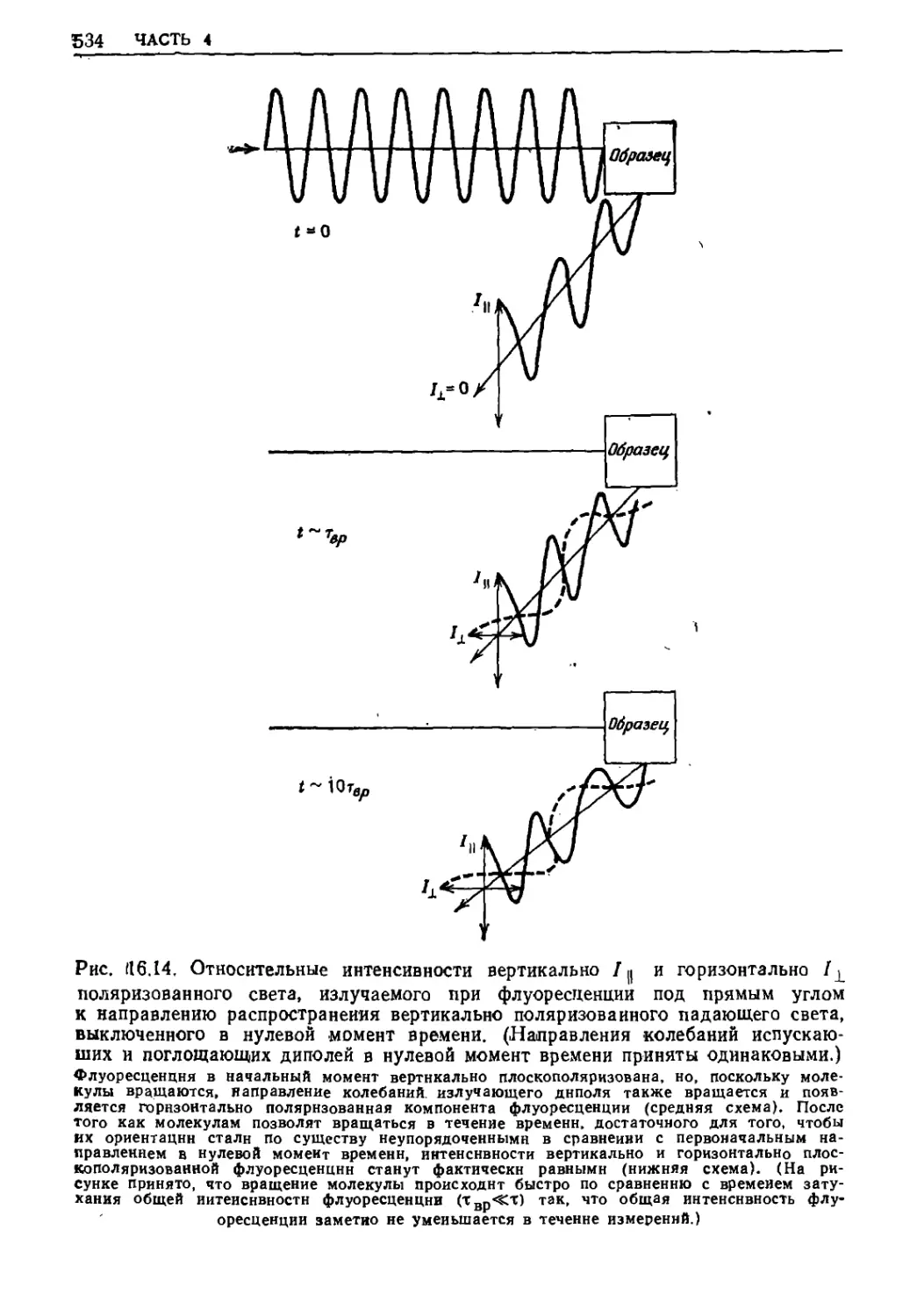



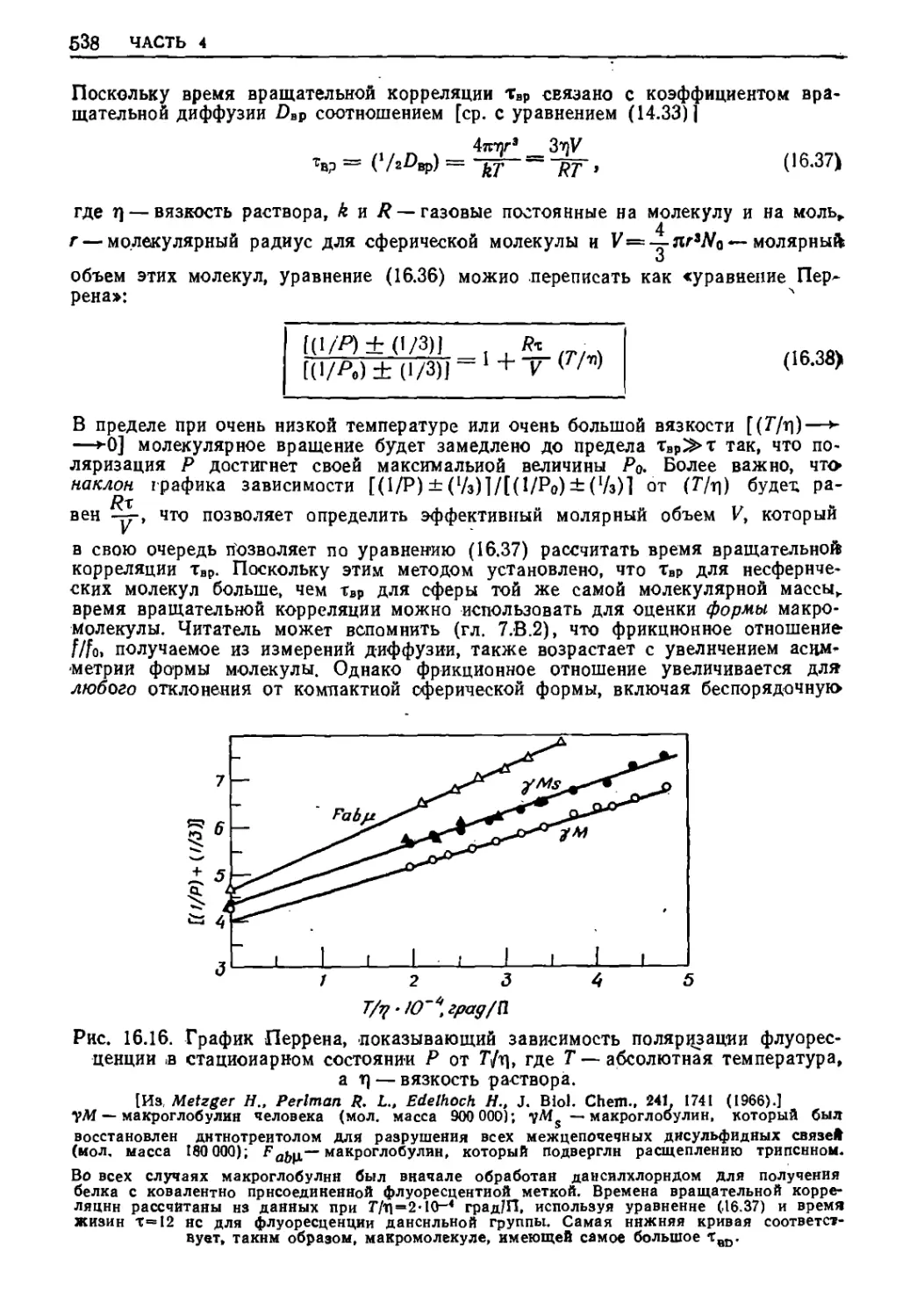



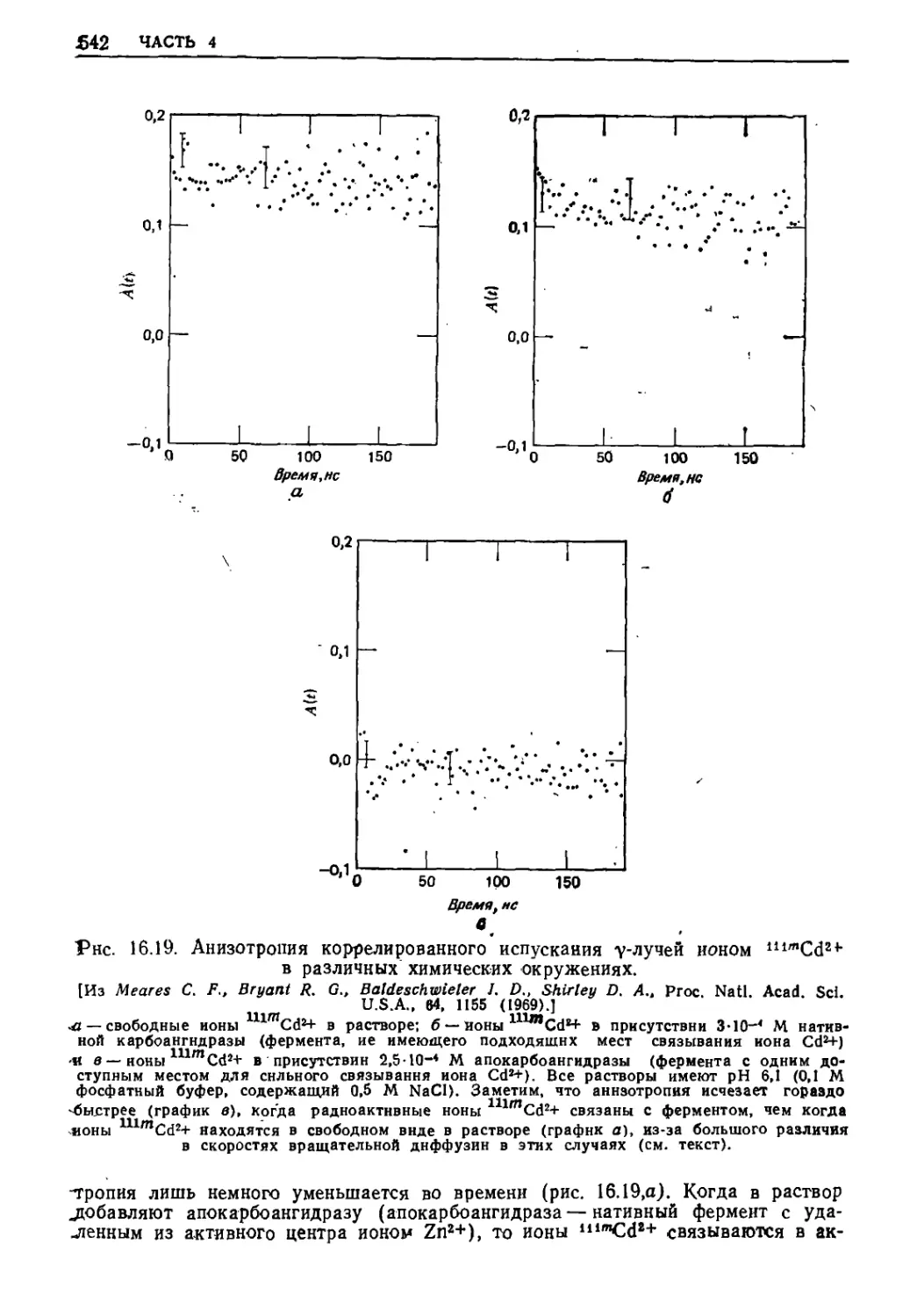


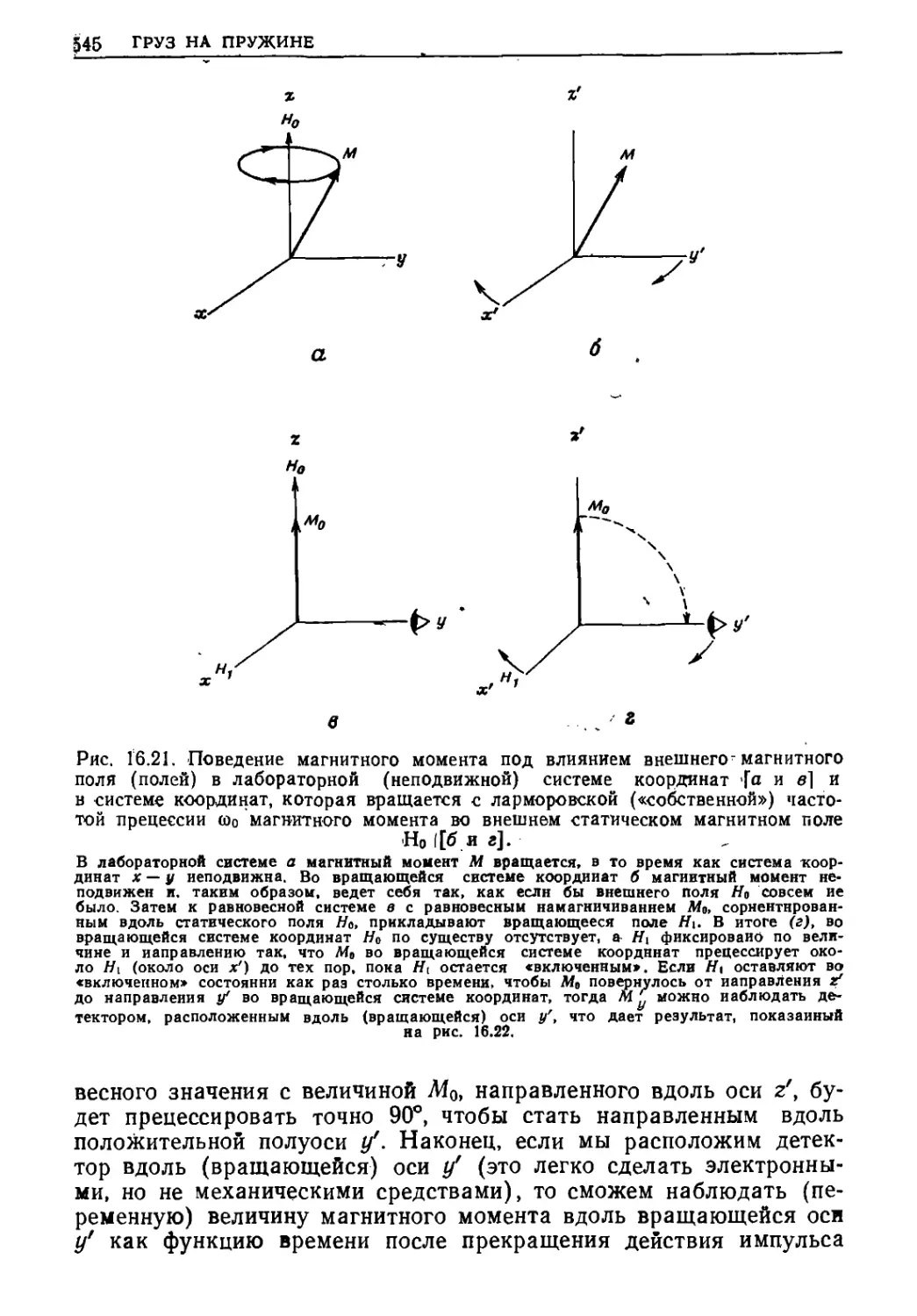




















































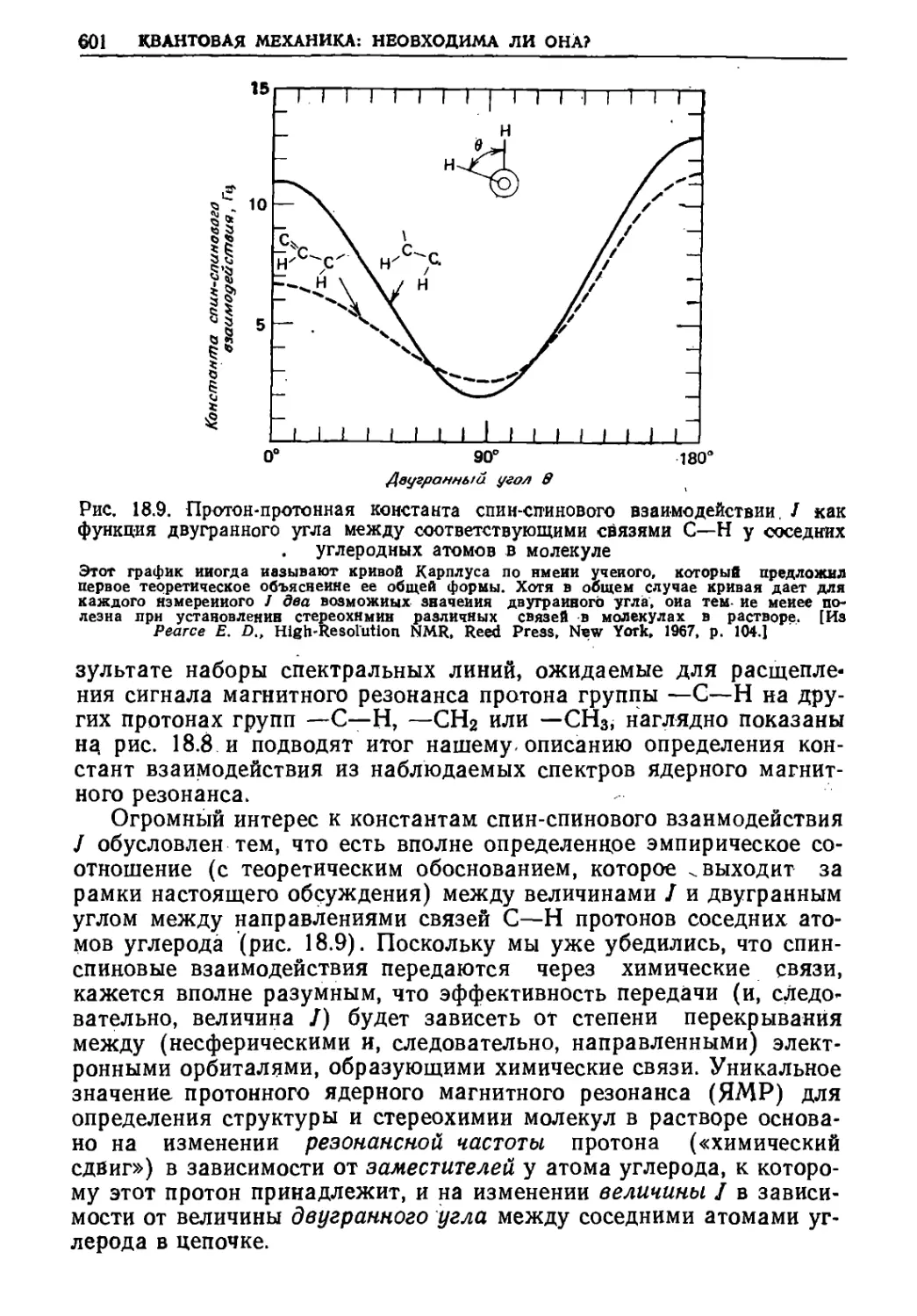
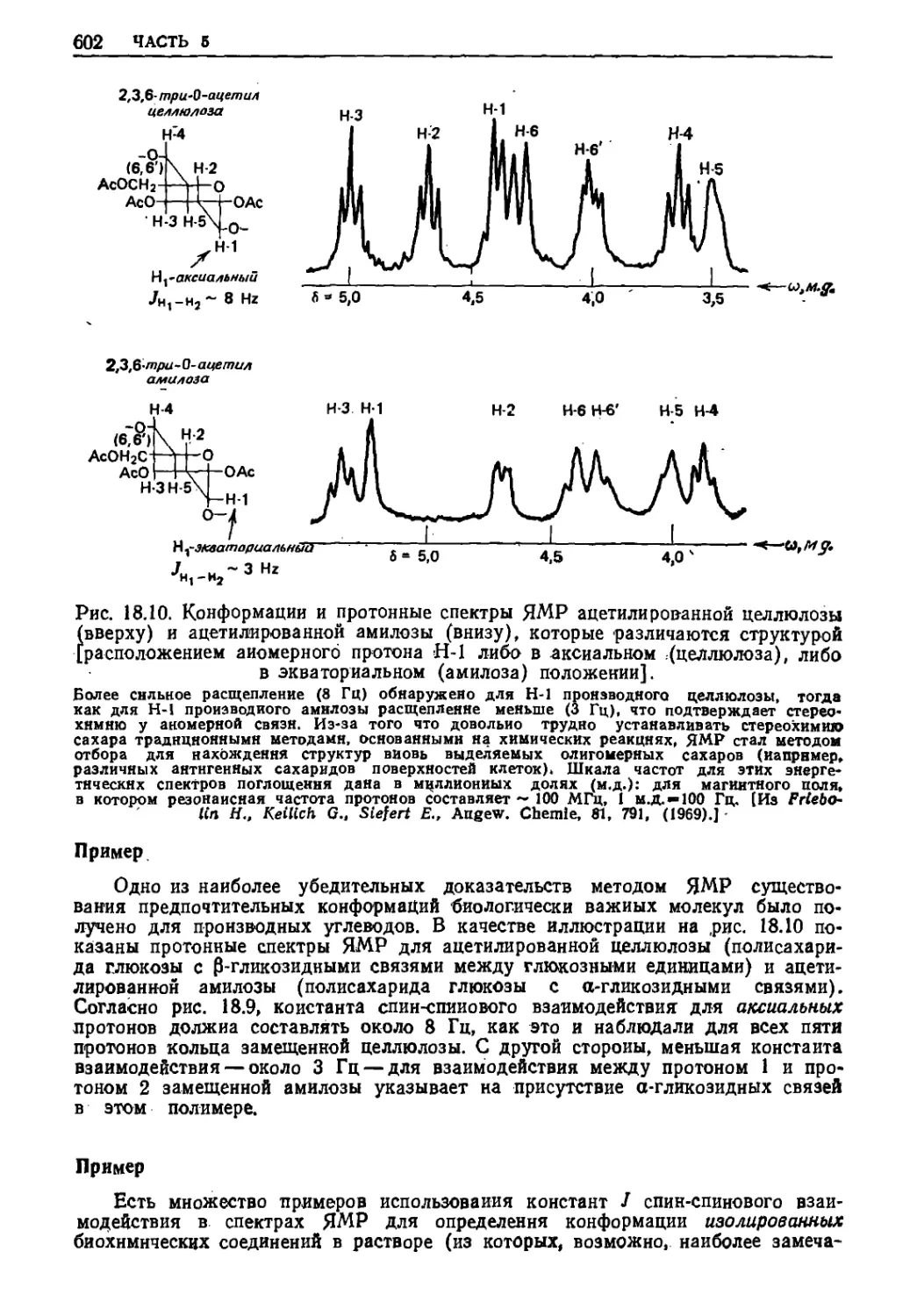






















































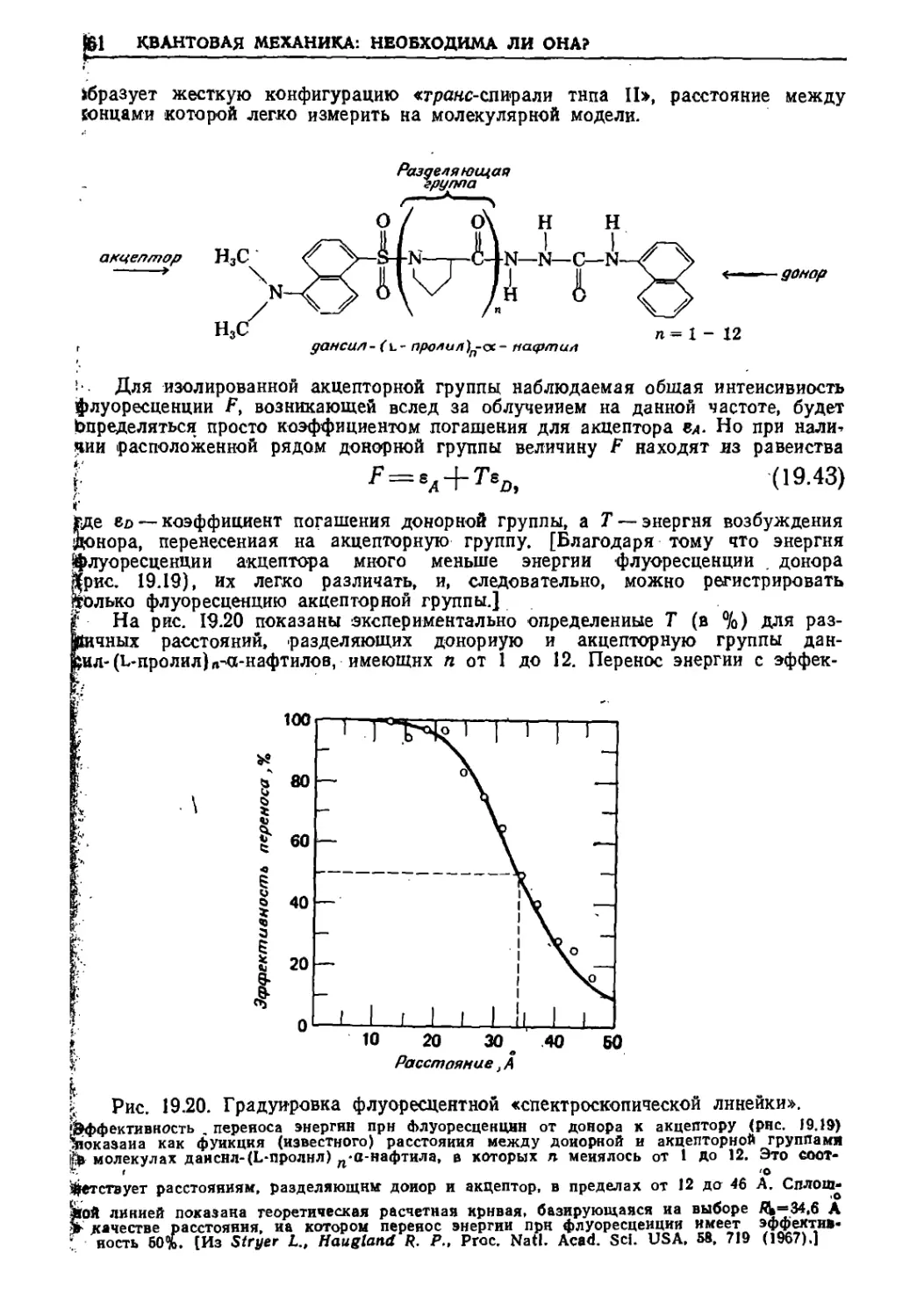




















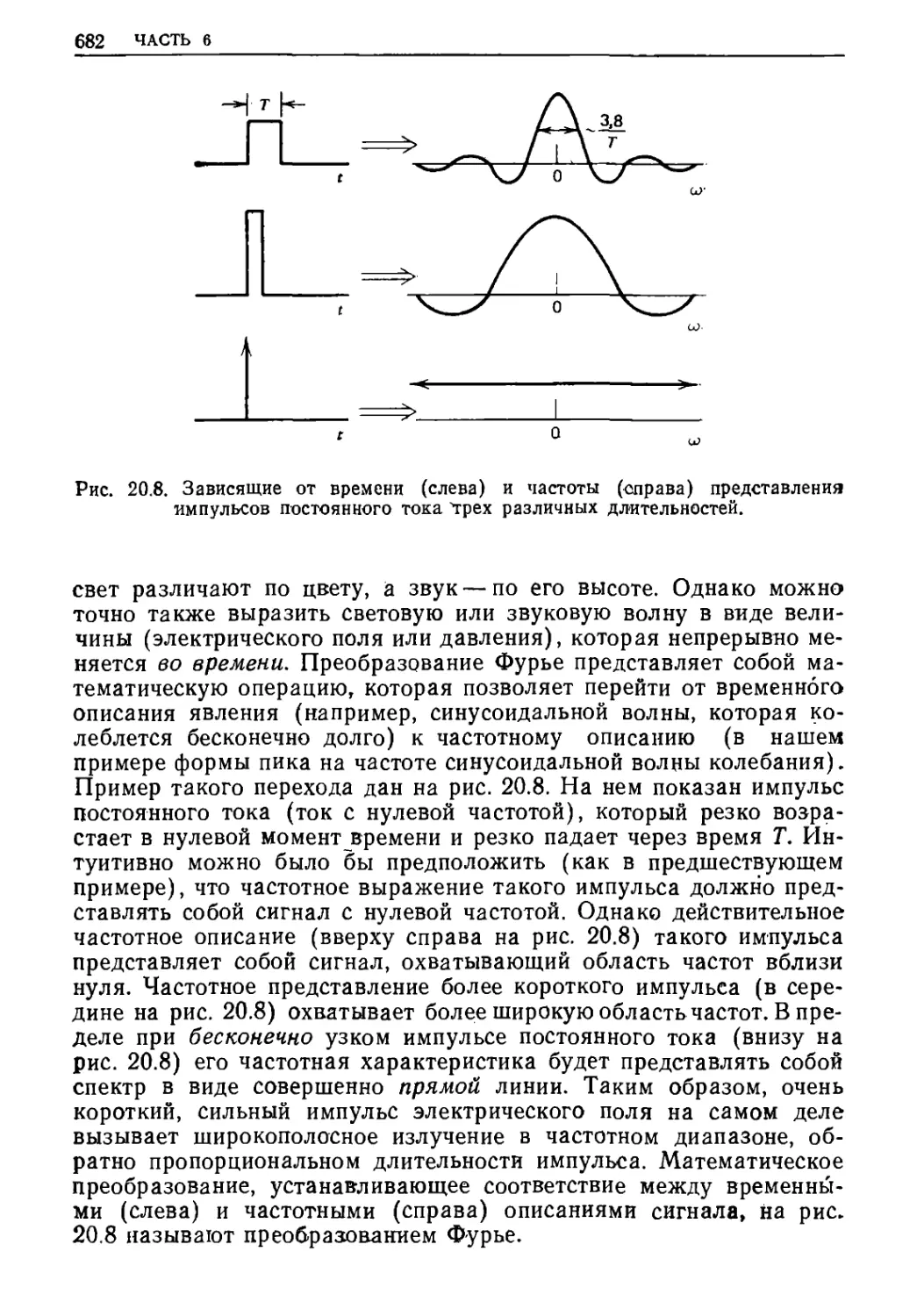





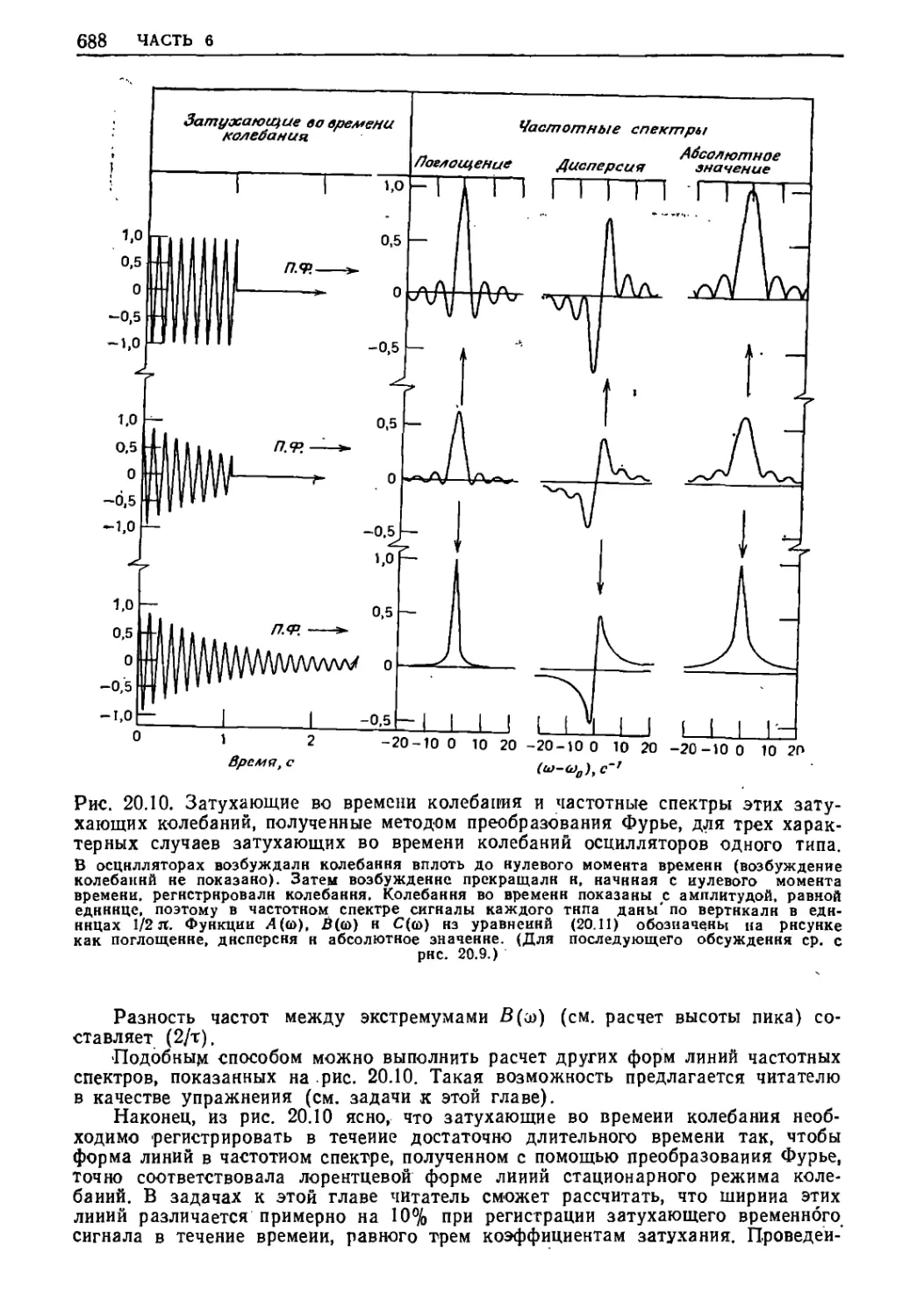




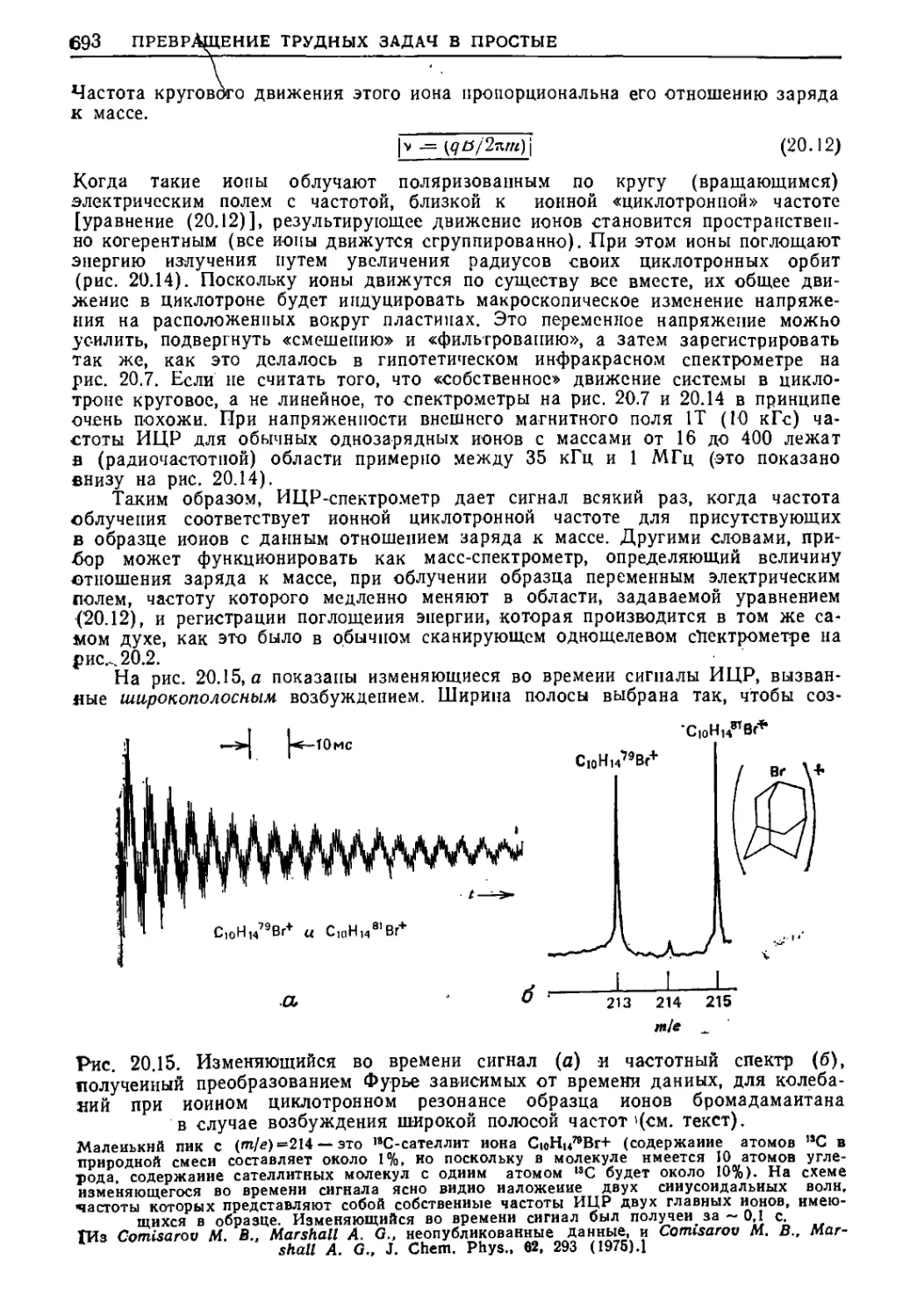

















































































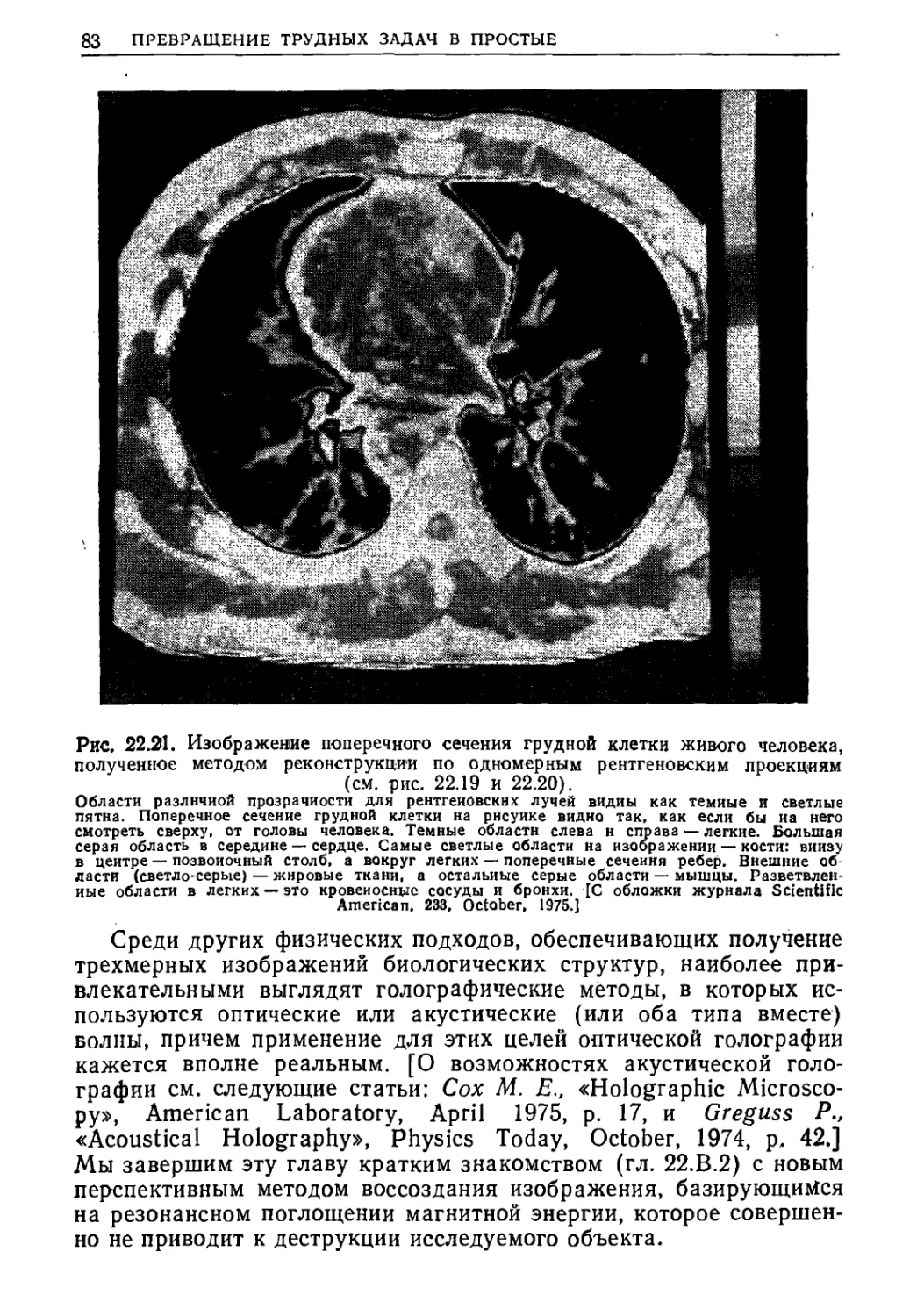


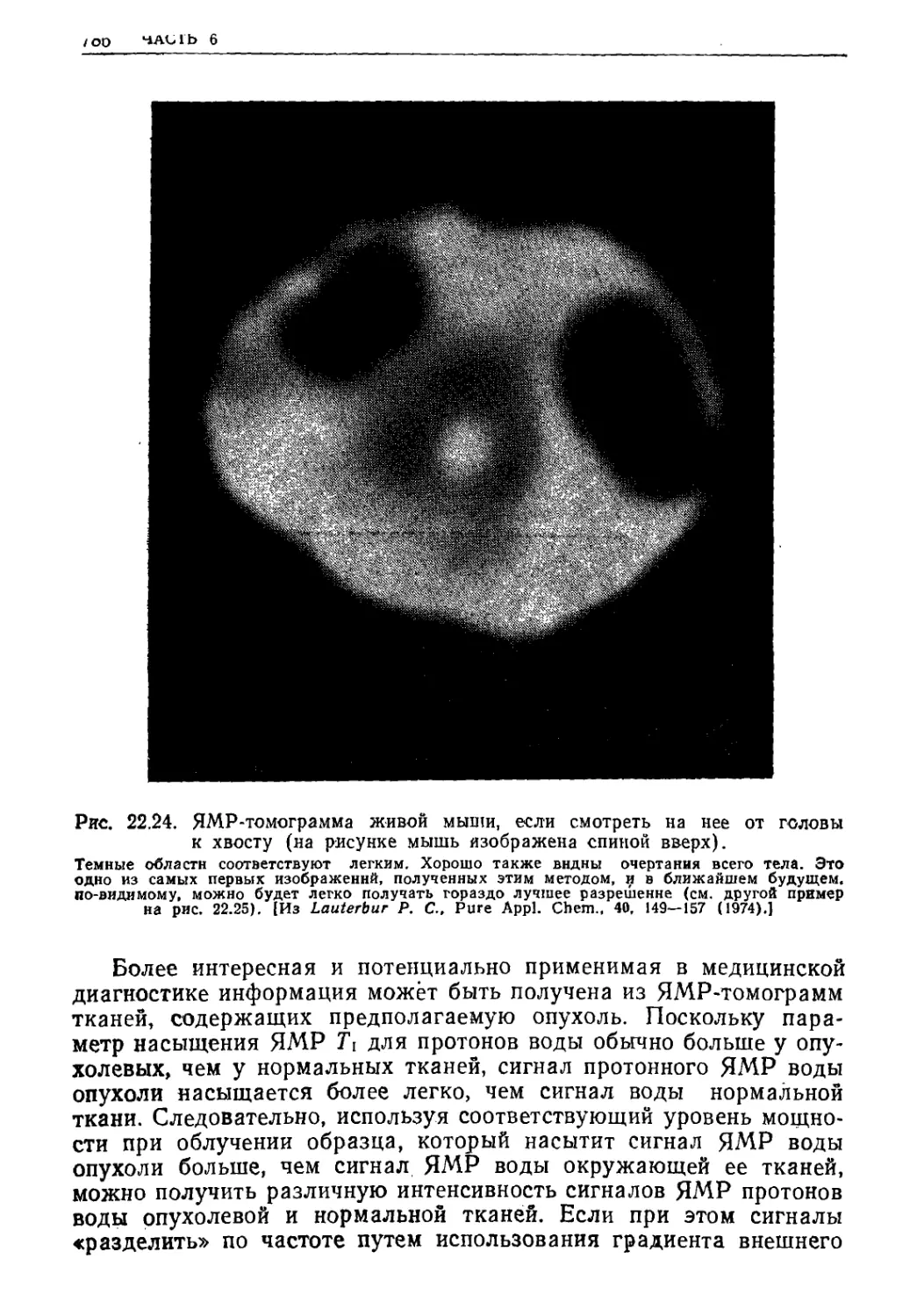



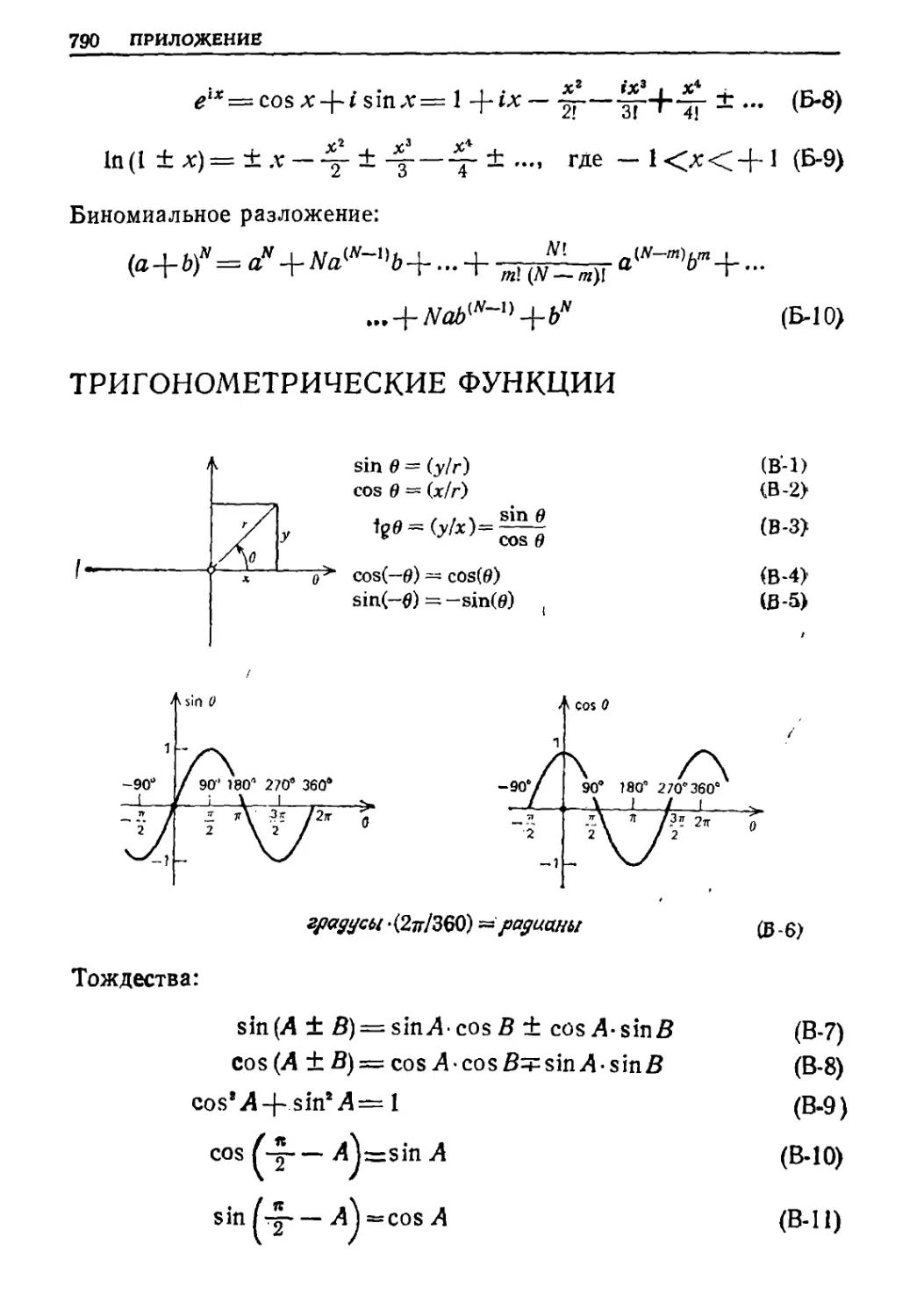


































Текст
Э. Маршелл
Биофизическая
химия
2
ИЗДАТЕЛЬСТВО
МИР»
BIOPHYSICAL CHEMISTRY
Principles, Techniques, and Application
ALAN G. MARSHALL
University of British Columbia
John Wiley & Sons
New York Santa Barbara Chichester
Brisbane Toronto
Э. МАРШЕЛЛ
БИОФИЗИЧЕСКАЯ
ХИМИЯ
ПРИНЦИПЫ, ТЕХНИКА И ПРИЛОЖЕНИЯ
В 2-Х ТОМАХ
2
Перевод с английского
канд. хим. наук В. Л. ДРУЦЫ
и канд. биол. наук Л. Я. ЛОМАКИНОЙ
под редакцией
д-ра хим. наук Б. В. ЛОКШИНА
ИЗДАТЕЛЬСТВО «МИР» МОСКВА 1981
УДК 578.088
Книга американского автора представляет собой курс физической
химии применительно к биологическим объектам. В русском
переводе выходит в двух томах.
Том 2 посвящеи новейшим методам исследования, используемым
в современной биологии, таким, как квантовая механика, все виды
спектроскопии, фурье-анализ, светорассеяние и т. д.
Предназначена для преподавателей и студентов биологических
вузов и научных работников биохимических специальностей.
Редакция литературы по химии
© 1978, by John Wiley and Sons, Inc. All
Rights Reserved.
Authorized translation from English
language edition published by John Wiley
and Sons, -Inc.
1805000000
20503-187 л „ *_
M 04Н0П-81 83'81 ® Перевод на русский язык, «Мир», 1981
|АСТЬ 4
РУЗ НА ПРУЖИНЕ
^ЛАВА 13
КОЛЕБАНИЯ ГРУЗА, ПОДВЕШЕННОГО
jfA ПРУЖИНЕ, ПОД ДЕЙСТВИЕМ
Возбуждающих и тормозящих сил —одна
р наиболее важных физических проблем
у Восприятие людьми окружающего мира происходит через вза-
модействия людей и окружающих предметов. Физика изучает
^оисхождение сил в природе и их взаимодействие. Так же как и
юбую другую величину, которая изменяется с расстоянием, функ-
рю, описывающую взаимодействия, можно в общем виде
разложить в степенной ряд, выбрав в качестве начала координат
продольную точку в пространстве
Обобщенная сила = т {d*xjd?) = А0 -{-А,х-f- Агхй +..., (13.1)
|е .дс — смещение от некоторой произвольной точки (например, от
ггра атома). Первое, на что следует обратить внимание, это то,
j даже такое относительно простое взаимодействие,.как куло-
^вское, между двумя заряженными частицами rii(d2x/dt2)=qq'/x2
Ькно представить в виде суммы бесконечного числа слагаемых
степенным рядом) аналогично уравнению (13.1). К счастью, мно-
§е из того, что уже известно об обычных взаимодействиях, можно
|нять, сделав два упрощения уравнения (13.1). \
Во-первых, путем выбора подходящей системы координат в
>странстве часто можно исключить постоянный член Л0. Напри-
|р, при рассмотрении электрофоретических опытов не
принимать во внимание сила тяжести (которая является постоянной),
-вторых, если взаимодействие настолько слабо, что
наблюдаете смещение х очень мало, можно пренебречь всеми членами
jtciuero порядка, ограничившись одним членом
Обобщенная сила =т {d2xfdf) = Агх. (13.2)
вконец, если теперь вместо постоянной А\ ввести новую
постоянно— k, то уравнение (13.2) сразу превратится в известное урав-
|ние для (упругой) силы, действующей на груз (с массой т),
решенный на пружине
Обобщенная сила == « {d*x/dt2) = — kx (13.3)
366 ЧАСТЬ 4
Например, даже зная, Что сила, связывающая электрон с атомом
или молекулой, является кулоновской, можно продолжать
рассматривать электроны в веществе так, как будто они связаны с
соответствующими атомами пружинками, при условии, что любые
силы, которые прилагаются, не сместят электроны далеко от
равновесного положения. В связи с тем, что электрические и магнитные
силы, возникающие под действием электромагнитных волн
(световое и рентгеновское излучения, радиоволны, микроволны,
инфракрасное излучение и т. д.) на атомах и молекулах, действительно
слабы, можно успешно описать многие молекулярные эффекты,
вызываемые электромагнитным излучением, используя тот же
математический аппарат, который описывает в механике ответную
реакцию обычного груза на пружине на действие приложенных
извне «раскачивающих» сил. Перед обсуждением движения
«раскачиваемой» пружины полезно восстановить в памяти термины,
служащие для описания волнового движения, поскольку
электромагнитное излучение можно рассматривать как обычные волны на
поверхности воды.
13.А. ТЕРМИНОЛОГИЯ ВОЛНОВОГО ДВИЖЕНИЯ
Следует рассмотреть два типа волн: продольные и поперечные.
Продольная волна (например, звуковая) может быть определена
как возмущение, при котором смещение происходит вдоль
направления распространения возмущения, а поперечная волна
(например, волна на воде или электромагнитная волна) — как
возмущение, при котором смещение происходит в направлении,
перпендикулярном направлению распространения возмущения. Таким
образом, при распространении звуковой волны молекулы воздуха
двигаются вперед и возвращаются на место, в то время как пробка,
плавающая на поверхности воды, двигается в вертикальном
направлении (вверх и вниз) при распространении волн на
поверхности воды. Оба типа волн показаны на рис. 13.1. *
Амплитудой волны называют максимальное смещение от
равновесного положения. Длина волны Я (для монохроматической
волны; см. ниже)—это расстояние (по направлению
распространения возмущения) между двумя последовательными точками с
максимальными амплитудами в определенный момент времени.
Скорость с монохроматической волны можно определить как
расстояние, которое определенный волновой гребень проходит в
единицу времени. Частота v монохроматической волны — это число,
показывающее сколько раз амплитуда волны достигает
максимального значения (например, в определенной точке пространства) в
единицу времени. Период Г —это время, необходимое для
завершения одного цикла колебания монохроматической волны.
Наконец, фаза (или фазовый угол) Ф волны в определенный момент
времени t — число радиан, которые накопились, начиная с произ-
ГРУЗ НА ПРУЖИНЕ
Г'родольная (звуковая) Щ
волна
I
\-т •*■ *)■
I4 *|
^'^Л;:?й^;%С-л--^ Направление
^^^т^Ш^^<Шй:^Ш Рас»РОстрс1ния
Колебания частиц
$р/*еречная
I- волна:
U
Ч) «~
и
11
с^ЛО
Л/SF
Амплитуда
Л
Т
Направление —
распространения
«I
5.
r-i
Амплитуда
\ЫЧ*
? (колебаний/с)
и) =2тг$(радиан/с)
<P=0o+<ot
\Поперечная (электромагнит-
I ная волна)
Направление
распространения'
13.1. Схематическое изображение продольной (звуковой), поперечной (на
поверхности воды) и поперечной (электромагнитной) воли.
4ждом случае предполагается, что волна монохроматическая (см. текст). Для простоты
>ажения электромагнитная волна взята плоскополярнзованной (см. текст). Все волны
устраняются слева направо. Параметры, связанные с движением волн, см. далее в
1 ходе дальнейшего обсуждения будет полезно возвращаться либо к изображению
5вого смещения в зависимости от пройденного расстояния' в данный момент времени
fox в центре слева), либо к другому, эквивалентному изображению волнового смеще-
! в зависимости от времени при заданном расстоянии >от нсточиика волны (в центре
справа).
|ьно взятого нулевого момента времени (каждое колебание со-
сит 2я радиан и, таким образом, циклическая или круговая
^ота w=2jtv радиан/с для волны с частотой v колебаний/с). Со-
ннения между данными выше определениями выражаются с
рщью следующих уравнений
>асстояние/с= (расстояние/цикл) (циклы/с)
(13.4)
с'цикл (13.5)
число радчан, накопившихся с нулевого момента (13.6)
времени
радиач/с (13.7)
368 ЧАСТЬ 4
Согласно принципу суперпозиции, при прохождении двух волн
через одну и ту же область пространства их амплитуды
складываются, например для двух звуковых волн различной частоты
(высота тона) или для двух световых волн различной частоты (цвет).
Волна называется монохроматической если все ее компоненты
имеют одинаковую частоту (и, следовательно, ту же длину волны).
Интенсивность волны характеризует энергию, проходящую через
единицу площади, перпендикулярной направлению
распространения волны. Интенсивность пропорциональна квадрату амплитуды
волны, что наглядно видно на примере волн на поверхности воды:
если амплитуда волны удваивается, то молекулы воды за данный
отрезок времени должны пройти в два раза больший путь и, таким
образом, будут иметь в среднем в два раза большую скорость.
Поскольку, кинетическая энергия пропорциональна квадрату
скорости, то интенсивность должна быть пропорциональна квадрату
амплитуды волны.
Что касается других терминов, то далее в настоящем разделе
основное внимание уделяется электромагнитным (поперечным)
волнам. Прежде всего отметим, что, как правило, взаимодействия
магнитной компоненты электромагнитного поля с веществом
значительно слабее, чем для электрической компоненты поля.
Поэтому, за исключением особых случаев, когда специально
рассматриваются магнитные эффекты (разд. 14.Г.4 и 16.Б.З), здесь магнитной
компонентой электромагнитного поля полностью пренебрегают.
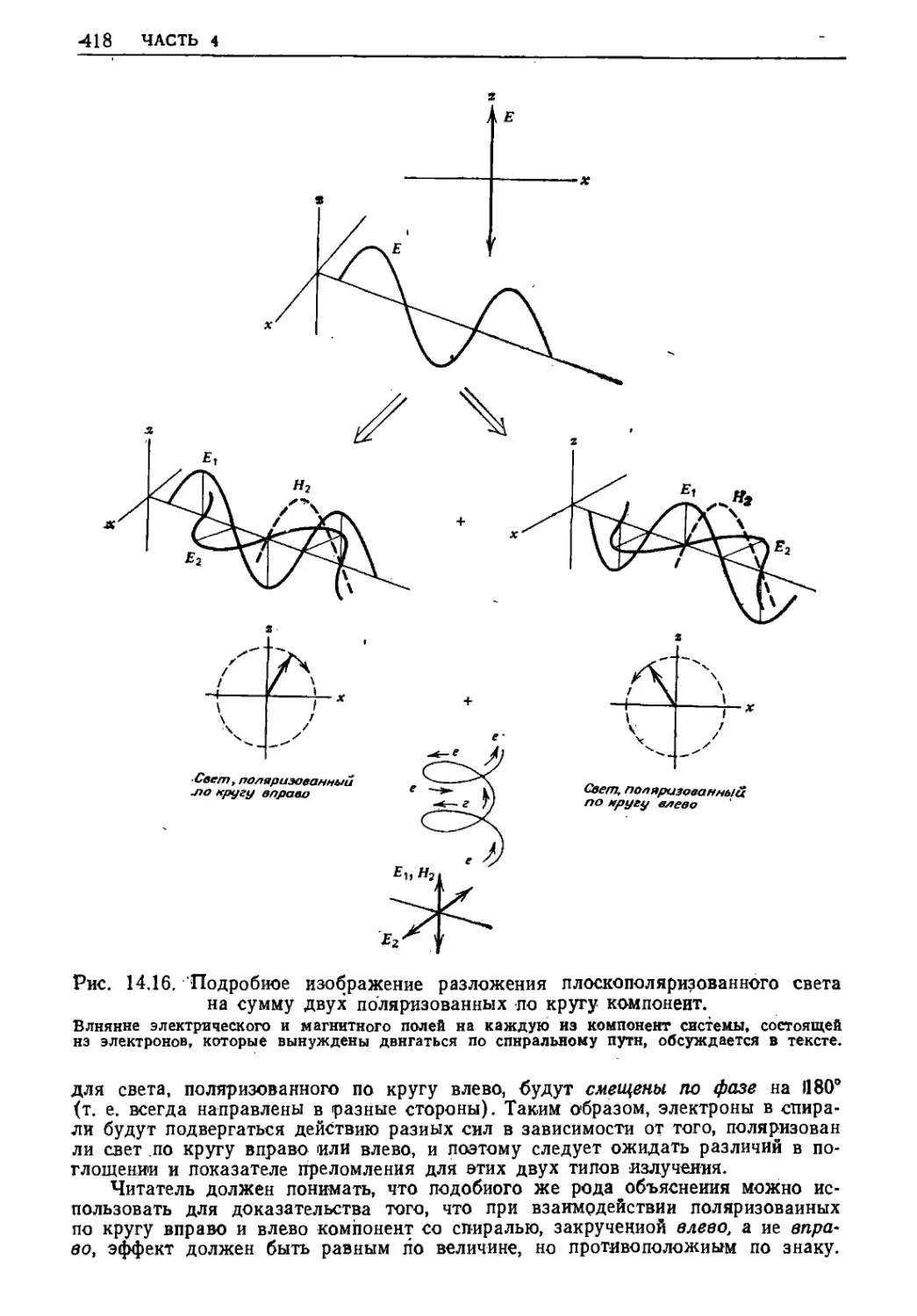
Электромагнитное излучение называется плоскополяризован-
ным, если направления векторов электрического поля в разных
точках вдоль направления его распространения в данный момент
времени лежат в одной плоскости. Плоскополяризованное
излучение не обязательно монохроматично, что очевидно из приведенной
ниже схемы.
Bug ago/id Плоскополяризованное
направления электромагнитное
распростране- > ■ Р^^^Л лоле^
/тия волна/
'Распросглра -
нение волны
Когда через одну и ту же точку пространства проходят много
компонент электромагнитной волны, то они могут суммироваться
самым различным образом, как показано на рис. 13.2. В том случае,
когда все компоненты имеют одинаковую частоту (длину волны)
и одинаковую фазу (т. е. все компоненты волны имеют нулевую
амплитуду в одной и той же точке пространства в данный момент
времени), сложение амплитуд называют когерентным; в
результате образуется синусоидальная волна с той же фазой, которую
имеют индивидуальные компоненты, но со значительно большей а м-
j[9 ГРУЗ НА ПРУЖИНЕ
Результирующая волна=
=Волн а Л/*7 + Волна N*2
Волна /Vs2
волна /Vs/
Когерентное сложение двух волн
одинаковой частоты и
амплитуды дает синусоидальную волну с
увеличенной амплитудой.
волна Л"I
Результирующая волна*
'Волна /Vs/ + Волна N±2
Некогерентное сложение двух волн
с одинаковой частотой, но с равной
фазой дает синусоидальную волну,
отличающуюся по амплитуде и
фазе от обеих компонент
-*/*Л2 = Л^,/+/у9^
н—lt"a'+"*> H
результирующая волна*
^ВолнаЛ/'/*Волна /Vх2
'Волна Л/г/
,волна№2
Некогервнтное сложение двусе волн
с разной частотой дает
периодическую несинусоидальмую волну
Рис. 13.2, Сложение двух поперечных волн.
Показана зависимость электрического поля электромагнитной волны от расстояния вдоль
% направления распространения волны в заданный момент времени.
|литудой. Если же компоненты волн не совпадают по фазе, то
суммирование является некогерентным и образующаяся волна может
ыть либо синусоидальной той же частоты, но другой амплитуды
фазы, либо периодической (но не синусоидальной) волной, в
зависимости от того,' имеют ли компоненты волн одинаковые или
различные частоты (рис. 13.2). Лазеры (разд. 19.Б.2)
представляет собой устройства, в которых компоненты монохроматических
)лн складываются когерентно, в результате чего образуется волна
■ амплитудой на несколько порядков больше, чем могут дать обык-
шенные некогерентные источники, например нити лампы нака-
|ивания. Можно показать (см. задачи в конце главы), что некоге-
Жтное сложение монохроматических волн всегда вызывает обра-
звание синусоидальной волны, но амплитуда этой синусоидаль-
волны будет всегда меньше, чем при когерентном сложении,
рфокое применение лазеров основано на их чрезвычайно высокой
Гтенсивности (разд. 19.Б.2).
370 ЧАСТЬ 4
В случае поляризованного по кругу (или по эллипсу) света
конец вектора электрического поля перемещается по кругу
(эллипсу), если смотреть на него вдоль направления распространения.
Несмотря на то, что поляризованный по кругу (по эллипсу) свет
не обязательно является монохроматическим, для иллюстрации
удобнее использовать монохроматическое излучение, как
показано на рис. 13.3.
Наконец, исходя из определения
(o = 2xv,
(13.7)
где © является круговой (циклической) частотой волны,
выражаемой в радианах в секунду, напряженность электрического поля
Г
**
Ллоскополяризованная
монохроматическая
электромагнитная волна
Циркулярно поляризованная
монохроматическая
злектро-магнитная eo/r/ta
Рис. 13.3. Общий вид (справа) и вид вдоль направления распространения волны
(слева) для разных типов, поляризованного электромагнитного (для простоты
монохроматического) излучения.
'1 ГРУЗ НА ПРУЖИНЕ
', образованного электромагнитной монохроматической волной,
южно выразить как функцию времени в данной (произвольной)
точке пространства
| Е = Е0 cos (urf) при у= 0 (13.8)
V
|где Е — амплитуда волны.
Поскольку для того, чтобы эта волна достигла точки,
расположенной на расстоянии у от заданного начала отсчета, требуется
л//с (секунд), где с — скорость распространения электромагнитного
^излучения, читателю нетрудно убедиться в том, что напряженность
'электрического поля в точке у описывается уравнением
* Е = Е0 cos [ш (/ — у/с)] (13.9)
^{обратите внимание, что Е имеет то же значение при 2=0 и у = 0,
£то и при t=y/c и у=у). Уравнения (13.8) и (13.9) показывают,
ррочему при математическом описании движения пружины почти
всегда используется круговая частота. Частота электромагнитной
^олны v появляется в уравнениях в виде 2лу = <в.
i- Введя таким образом необходимые термины, можно перейти к
Обсуждению колебаний груза, подвешенного на пружине, сначала
£ отсутствие (а затем и в присутствии) дополнительных
раскачивающих и тормозящих сил.
^Собственное» колебание груза на пружине
|; Уравнение, описывающее колебание груза с массой пг,
прикрепленного к пружине, имеющей упругость (константу упругости)
ft в отсутствие каких-либо раскачивающих или тормозящих сил,
^ножно написать в виде
f Упругая сила = — kx
Или
\ md'xjdt^kx^O. (13.3)
|1ожно удостовериться, что уравнение (13.10) является решением
'Этого уравнения движения. [Поскольку математические
преобразования для решения дифференциальных уравнений, подобных
уравнению (13.3), довольно стандартны, их решение приводится
]Прямо без описания способа его получения. Решения
используемых в этой главе уравнений можно найти в любом учебнике по
Дифференциальным уравнениям.] При максимальном
первоначальном смещении х=х0 во время /=0
X = JC„ COS (d)0t) ,
где
со0 = V (k/m) = «собственная частота» груза на пружине
(13.10)
(13.11)
372 ЧАСТЬ 4
Поскольку, отсутствует трение, вызывающее затухание, то груз
будет неопределенно долго колебаться с «собственной» частотой
vo=(u)0/2n) (циклов/с), а величина его смещения будет при этом
изменяться по синусоидальному закону.
Эксперименты для определения параметров движения груза,
подвешенного на пружине
Для того чтобы правильно сформулировать математическую
задачу, сначала необходимо кратко проанализировать типы
экспериментов, с помощью которых можно оценить параметры
движения подвешенного на пружине груза. Наиболее удачными
являются эксперименты двух типов: а) неожиданно толкнуть груз
(т. е. резко сместить его), а затем наблюдать за положением
пружины в -зависимости от времени или б) постоянно «раскачивать»
груз (с возбуждающей частотой, которая может отличаться от
«собственной» частоты самого груза на пружине); когда система
придет в состояние стационарного движения (вынужденные
колебания) измерить амплитуду движения в зависимости от возбуж-
дающей частоты. Эти два экспериментальных подхода
иллюстрируются схемой на рис. 13.4.
Примем, что электроны в веществе, ведут себя так, как если бы
они были связаны с помощью пружинок с соответствующими
атомами. Тогда действие переменного электрического поля
электромагнитной (например, световой) волны приводит к раскачиванию
электрона на удерживающей его пружине. Электрон, после того
как его привели в движение, будет испускать электромагнитное
излучение в разных направлениях, подобно тому как камертон
после удара испускает во всех направлениях звук. Оказывается,
что возможно определить местонахождение колеблющегося на *
пружине электрона путем измерения амплитуды (или, попросту,
интенсивности, которая пропорциональна квадрату амплитуды)
«рассеянного» излучения возбужденного электрона, в зависимости
от направления. Подобного рода эксперименты составляют основу
для определения расположения атомов в кристаллах с помощью
рентгеновского излучения и дифракции нейтронов. Добавим, что
собственная частота vo
v0 = (l/2ic) V(kfm) (колебаний/с) (13.12)
может быть определена а) путем подсчета числа колебаний в
секунду после неожиданного удара, или б) путем определения
приложенной извне частоты возбуждения, при которой достигается
максимальная амплитуда вынужденных колебаний (см. ниже)".
Поскольку эксперименты с «неожиданным ударом» (импульсом)
и с возбуждением вынужденных колебаний дают одинаковую
информацию, то не удивительно, что они связаны между собой мате-
ГРУЗ НА ПРУЖИНЕ
I
Груз на пружине под действием
возбуждающих и тормозящих
md'y/dt^fdy/dt+ty= F(t)
сил
w„
Sim
Возбудитель колебаний действует
постоянно
/
возбудитель колебаний внезапно
выключен
Стационарное состояние
(вынужденные колебания )
Преобразование
Фурье
ч
со = оза
S' Поглощение и дисперсия
К (лоренцов контур)
^и) далеко от со
Рассеяние
Ло«и)п
чо> » ь>0
Нестационарное состояние
(затухающие колебания")
Предел нулевой массы
(т=и); быстрая химическая
реакция; релаксационная
кинетика
Предел Рзлея
Предел Томсона
:с. 13.4. Взаимоотношения между различными опытами по рассеянию, спектро-
копии и затуханию, основанные на аналогии с движением возбуждаемого по
Гнусоидальиому закону (или неожиданно смещенного) груза иа пружине при
Р. наличии сил торможения.
ty3 имеет массу гп, подвешен на пружине с коэффициентом упругости k, погружен в сре-
с коэффициентом трения f и подвергается либо неожиданному смещению, либо дей-
гвню силы, изменяющейся по синусоиде в зависимости от времени F(t)~F<>cos{(ut),
Явления, связанные с вынужденными колебаниями, обсуждались в гл. 14 и 15, а явления,
связанные с затухающими колебаниями, — в гл. 16 и методы Фурье — в ч. 6.
Ь.
Шатически. Эта связь хорошо известна и описывается с помощью
преобразования Фурье (разд. 6), Схема на рис. 13.4 помогает по-
рпъ математическую связь между различными явлениями, описы-
1емыми на основе модели груза на пружине под действием воз-
гждающих и тормозящих сил.
Предположим, что пружина имеет возможность совершенно
|вободно двигаться, тогда дри условии, что частота возбуждения
равняется собственной частоте пружины vo (находится с ней в
резонансе»), можно ожидать, что пружина будет непрерывно
юглощать энергию и амплитуда ее колебаний будет непрерывно
возрастать (бесконечно). Однако движение любой реально
существующей пружины (включая и наш электрон, привязанный к мо-
|екуле) затормаживается силами трения, причем тормозящая си-
la пропорциональна скорости движения груза (электрона)
I Сила торможения = — f (dx(dt). (13.13)
коэффициент торможения / можно определить с помощью любого
|ё ранее описанных экспериментов: после неожиданного удара
374 ЧАСТЬ 4
уменьшение амплитуды движения груза (электрона) зависит от
величины f; также при непрерывном раскачивании пружины под
действием силы, изменяющейся по синусоидальному закону,
максимум смещения обратно пропорционален /, так как из всей
энергии, прилагаемой извне, при увеличении трения растет доля этой
энергии, расходуемой на нагревание, и уменьшается та ее часть,
которая идет на поддержание движения тела, подвешенного на
пружине.
В различных типах спектров, о которых пойдет речь далее,
собственные частоты различных пружин соответствуют частотам с
максимальным поглощением энергии — пикам. С коэффициентом
торможения данной пружины связана ширина спектральной
линии. Пространственное расположение пружинок (электронов) в
молекуле отражается на картине рассеяния излучения в
зависимости от направления, о чем было сказано выше. Практический
интерес к этим параметрам определяется тем, что собственные
частоты свидетельствуют об упругости рассматриваемой пружины и
связаны с цветом, прочностью химической связи, оптической
активностью, а также зависят от среды, окружающей пружину.
Ширину спектральных линий можно использовать для определения
скорости процессов, слишком быстрых для прямого измерения, таких,
как: столкновения, перемещения и вращения молекул и очень
быстрые химические реакции. Рассеянное излучение можно
использовать для определения пространственного расположения атомов
в молекуле (т. е. ее трехмерной структуры), когда молекулы
находятся в кристаллах, рибосомах или даже в растворе.
13.Б. ГРУЗ НА ПРУЖИНЕ ПОД ДЕЙСТВИЕМ
ВОЗБУЖДАЮЩИХ И ТОРМОЗЯЩИХ СИЛ:
ВЫНУЖДЕННЫЕ КОЛЕБАНИЯ
Кратко рассматриваемая в настоящем разделе
математическая обработка основана на схеме, изображенной на рис. 13.5.
Если на груз, подвешенный на пружине, действовать постоянной,
синусоидально изменяющейся во времени силой Focos(v)t), то ои
постепенно придет к состоянию незатухающих (вынужденных)
колебаний, частота которых будет равна частоте возбуждения. Эта
ситуация отлична от случая с пружиной, на которую силы извне
не действуют [уравнения (13.10) и (13.11)] и которая колеблется
с собственной частотой. При отсутствии трения (торможения)
пружина, на которую действует сила, колеблется точно в фазе с
частотой приложенной извне силы (рис. 13.5,6), но когда имеется
трение, то смещение пружины будет, как правило, отставать от
возбудителя (ср. рис. 13.5,6 и в). Удобно с точки зрения
математики (и полезно с физической точки зрения) «разложить»
смещение х в стационарном состоянии на две компоненты (частота
каждой из которых совпадает с частотой возбудителя). Эти компонен-
375 ГРУЗ НА ПРУЖИНЕ
а, шшж
t 6
Смещение при п
вынужденных
колебаниях ж
t г
ч «?
0,5
0
0,5
1- *-
ы0
= OJ
-(w0- о>)
Ы = Wq
Рис. 13.5. Диоперсия и поглощение на примере вынужденных колебаний груза,
подвешенного на пружине и находящегося под действием возбуждающих и
тормозящих сил.
Под действием синусоидально изменяющейся во времени возбуждающей силы (б) груз,
подвешенный иа пружине (а), при наличии тормозящих сил постепенно войдет в
стационарный режим вынужденных колебаний, имеющих ту же частоту, что н возбуждающая
частота, но отличающихся по фазе (в). Смещение можно разложить на компоненты, одна
из которых точно совпадает по фазе с колебаниями возбудителя (г, слева), а другая
смещена по фазе точно на 90° (г, справа). Зависимость от (возбуждающей) частоты
амплитуды синфазной компоненты называется спектром «дисперсии» (6\ слева), а для амплитуды
компоненты, смещенной по фазе на 90", называется спектром «поглощения». См. текст.
где приведены примеры по рассеянию и спектроскопии. Для механического аналога
<Оо=» (fe/m)1'a н (l/T)-f/2m.
ты либо точно находятся в фазе (амплитуда=х') или точно
смещены по фазе на 90° (амплитуда=х") относительно колебаний
возбудителя (рис. 13.5,г). Зависимости амплитуд компонент х' и х"
от частоты движущей силы показаны на рнс. 13.5, д. Эти
графические зависимости известны как кривые «дисперсии» и
«поглощения». Происхождение этих названий будет понятно из
последующего изложения. Чрезвычайное удобство этой механической
модели состоит в том, что она позволяет выразить в математической
376 ЧАСТЬ 4
форме xf и х", а когда такие выражения будут получены, то без
дополнительной математической обработки можно сопоставлять х'
и х" с разными типами спектроскопических сигналов, заменяя
лишь названия переменных. С этой целью приступим к
математическим выкладкам, необходимым для описания графических
зависимостей, изображенных на рис. 13.5.
Для груза с массой т, подвешенного на пружине с
коэффициентом упругости k и находящегося под действием переменной
возбуждающей силы F:
F = Fecos(to^),
(13.14)
а также силы торможения (трения) пропорциональной скорости
движения тела, уравнение движения записывается следующим
образом:
m&x/dt* = Обобщенная сила = — kx — fdx/dt + F0 cos (со*) (13.15)
где —kx — упругая сила пружины; —fdx/dt — сила торможения,
направленная в сторону, противоположную движению, и
F0cos((ot)—приложенная (движущая) сила. Читатель может
удостовериться в том, что уравнение (13.16) является решением
уравнения (13.15), причем уравнение (13.12) (Oo=~\/(k/m) использовано
для упрощения окончательного выражения
х =
где
х'
и
х" =
= х' cos (со*) + х" sin (cof),
= F0
= F0
т. (со20 — со2)
raa(cog — соа)а + /2со2
/со
тъ (со»0 — со2)8 + f2co2
общий случай
(13.16а)
(13.166)
(13.16в)
Уравнения (13.16а ■—13.16в) математически описывают
явления, которые изображены на рис. 13.5, т. е. показывают, что при
вынужденных колебаниях смещение х можно разложить на две
компоненты (компоненты смещения): jt'cos((o£) и *"sin.(©*),
которые либо точно совпадают по фазе с колебаниями возбудителя
[cos (©*)], либо смещены по фазе точно на 90° относительно этих
колебаний [поскольку cos(W — (я/2)) =sin(«>f)]. Компоненты х' и
х" представляют физический интерес и будут в дальнейшем
употребляться для обозначения синфазных (совпадающих по фазе) и
смещенных по фазе на 90° амплитуд смещения.
Замечание. Следующий короткий раздел не обязателен для понимания
вопросов, поднятых в настоящей главе. Однако ои очень полезен для читателя,
который надеется следить за литературой, посвященной этим проблемам, по*
377 ГРУЗ НА ПРУЖИНЕ
скольку в нем предложено простейшее математическое описание явлений
поглощения и дисперсии, особенно нужное при введении методов преобразования
Фурье в части 6.
До сих пор все формальные преобразования проводились с использованием
действительных (в противоположность комплексным) переменных, которые
применимы для описания явлений, рассматриваемых в этом разделе. Однако
гораздо удобнее, особенно при рассмотрении материала части 6, ввести обозначения
с использованием комплексных величин. Естественно возникает вопрос: каков
физический смысл мнимой части комплексных чисел? Ответ состоит в том, что
с результатами физических измерений будут связаны только действительные
величины или действительные части комплексных величин.
Попросту говоря, первая причина удобства использования комплексных
чисел состоит в том, что производная по времени от exffftof] является той же
самой функцией, только умиожеииой на константу ш ехр)[(<о/]. В
противоположность этому производная от sin(<of) является другой функцией со&(о>0;
подобным же образом производная от cos(cuf) тоже равна другой функции —sin(cof).
Таким образом, для любой задачи (такой, как задача о гармонических
колебаниях груза на пружине с возбуждением и торможением), .включающей
производные по времени от синусов и косинусов, проще употребить функцию ехр\[ьсо/]:
ехР [± tot] = cos («О ± t sin ((at).
(13.17)
;- Вторая причина удобства использования комплексных переменных состоит
:*в том, что они позволяют автоматически разделить две части задачи (в виде
^действительной и мнимой частей комплексного числа). Комплексные числа, таким
Цобразом, служат для тех же целей, для которых используются две
перпендикулярные оси координат в элементарной геометрии при разложении вектора
|в плоскости на две компоненты. Оба вышеописанных преимущества комплекс-
рой системы обозначения станут яснее прн разборе задачи о гармонических
^колебаниях (с возбуждением и торможением) с использованием комплексных
|чисел. Необходимо решить задачу
m {d2x/dt2) + f (dx/dt) + kx = F0 cos (со/),
(13.15)
где х — действительное число. Предположим, что к правой части уравнения
|(13.15) добавлено слагаемое (F0sin((u/); ищем решение для (комплексной)
величины х:
m (d2x/dt2) -f f (dx/dt) + kx = F0 exp [to*], (13.18)
|дс x — комплексное число. Понятно, что в уравнение (13.18) входит
комплексная переменная, ио после решения задачи интерес будет представлять
только действительная час*ть х. Теперь предположим, что для вынужденных
колебаний существует решение:
Комплексное х = у. exp [Ш], (13.19)
эгда (комплексную) амплитуду % легко найти путем подстановки уравнения
[1-3J19) в (13.18):
1 1
г~ F° {k — rato2) + ito
= F
0 m (to20 — to2) -f if (л '
Используя тождество
1
a — it
a + ib a2 -f b2 >
)жно переписать уравнение (13.20) в виде
m (to20 — to2) — if<a
X = fV
/я2 (to20 — <e2)2 4. /2co2
(13.20)
(13.21)
(13.22)
378 ЧАСТЬ 4
Найдем теперь действительную часть решения уравнения (13.19) для
комплексного х:
Re (комплексное х) = Re (x-exp [мог-] =
F0m (ш20 — to2) Fofco
= —ттг 2\г I <2^ч2 cos (<of) + ^.2 / г ,>2\2 i tzTZz sin (cof) = (13.23)
= x'cos(W) + x"sin((uO. (13.24)
Другими словами, действительная часть решения комплексного уравнения (13.18)
идентична с (действительным) решением (действительного) уравнения (13.15),
что и надо было показать.
Более того, теперь уже очевидно, что % можно выразить как
X = х' — ix"
(13.25)
т. е. х' и х" могут быть записаны в виде действительной и мнимой компонент
комплексной амплитуды в комплексном решении уравнения (13.18). На первый
взгляд может показаться, что х" связана с мнимым членом математического
выражения, однако если посмотреть на уравнение ,(13.23), то очевидно, что эта
величина соответствует (действительному) физическому решению, получаемому
из действительной части (%-exp[i<i>f]), так что х" в конечном счете является
амплитудой компоненты, смещенной по фазе на 90°. Комплексная амплитуда %,
таким образом, обнаруживает уже упомянутое удобное свойство — она
содержит раздельно х' и х в качестве действительной и мнимой частей комплексного
числа; более того, математические трудности перехода от уравнения (13.18) к
(13.20) значительно меньше, чем трудности перехода от уравнения (13^Л5) к
(13.16) (см. задачи в конце главы), что отвечает последнему из указанных
преимуществ использования комплексных величин.
Один из распространенных подходов к анализу сложных
формул [подобных уравнению (13.16)]" состоит в изучении их
поведения при различных физически значимых предельных значениях
переменных. Например, в предельном случае отсутствия
торможения (f—Ю) смещенная на 90° при фазе компонента х"
обращается в нуль
lim;c" = 0, (13.26)
так что колебания системы при этом совпадают по фазе с
возбуждающими колебаниями. Обобщим теперь этот результат для фи-,
зических ситуаций, когда опыты проводят при возбуждающей
частоте, либо очень близкой, либо очень далекой от собственной
частоты пружины.
13.Б.1. ПРЕДЕЛЫ РАССЕЯНИЯ: НЕЗНАЧИТЕЛЬНОЕ ТОРМОЖЕНИЕ
И/ИЛИ ВОЗБУЖДАЮЩАЯ ЧАСТОТА ДАЛЕКА ОТ РЕЗОНАНСА
^Обратите внимание, что для достижения предела в уравнении
V~BJ в общем виде требуется, чтобы второе слагаемое в Днаме-
, уравнения (13.16 в) было много меньше первого, а именно
}$1$!^*}Ь> -Т'вКвМ образом, если торможение незиавдтель-
'Щ,|;'Ца&)заяй если частота возбуждения зна-
379 ГРУЗ НА ПРУЖИНЕ
чительно отличается от собственной частоты колебания пружины
(разность j (©о—со) | велика), то компонента, смещенная по фазе
на 90°, пренебрежимо мала, а компонента, совпадающая по фазе,
достигает предельного значения:
Предел
рассеяния
Нт
f<
f«
т. (сй^о-
(0
Нт
<0 (Сй20 -
-Сй2)
-Сй2)
(*') — т (со20 — со2)
(х")=0
а>
(13.27а)
(13.276)
Для рассматриваемых случаев важно, что электромагнитное
излучение (например, видимый свет или рентгеновское излучение)
создает переменное электрическое поле, которое при
взаимодействии с принятой здесь моделью вещества, где электроны
подвешены на пружинках, будет возбуждать движение (заряженных)
электронов с частотой приложенного излучения. Уравнения
Максвелла позволяют предсказать, что такой движущийся электрон
будет испускать («рассеивать») новое излучение с амплитудой
электрического поля, пропорциональной второй производной от
координаты электрона:
Л2 р ц)8
£nacc°< -TJ7T \ХГ CO.S (шЛ|= ° к COS Ы)
расе ^2 l V п т ^(|)2() — w2) v /
ИЛИ
£расс «■ w((co0/(oV-l) C°S М-
(13.28)
В окончательном виде, принимая во внимание, что интенсивность
излучения пропорциональна квадрату амплитуды излучения (для
любых поперечных волн), получаем физически полезный результат
, const
/Расс [((•>„/(•>) 2 — I]2
(13.29)
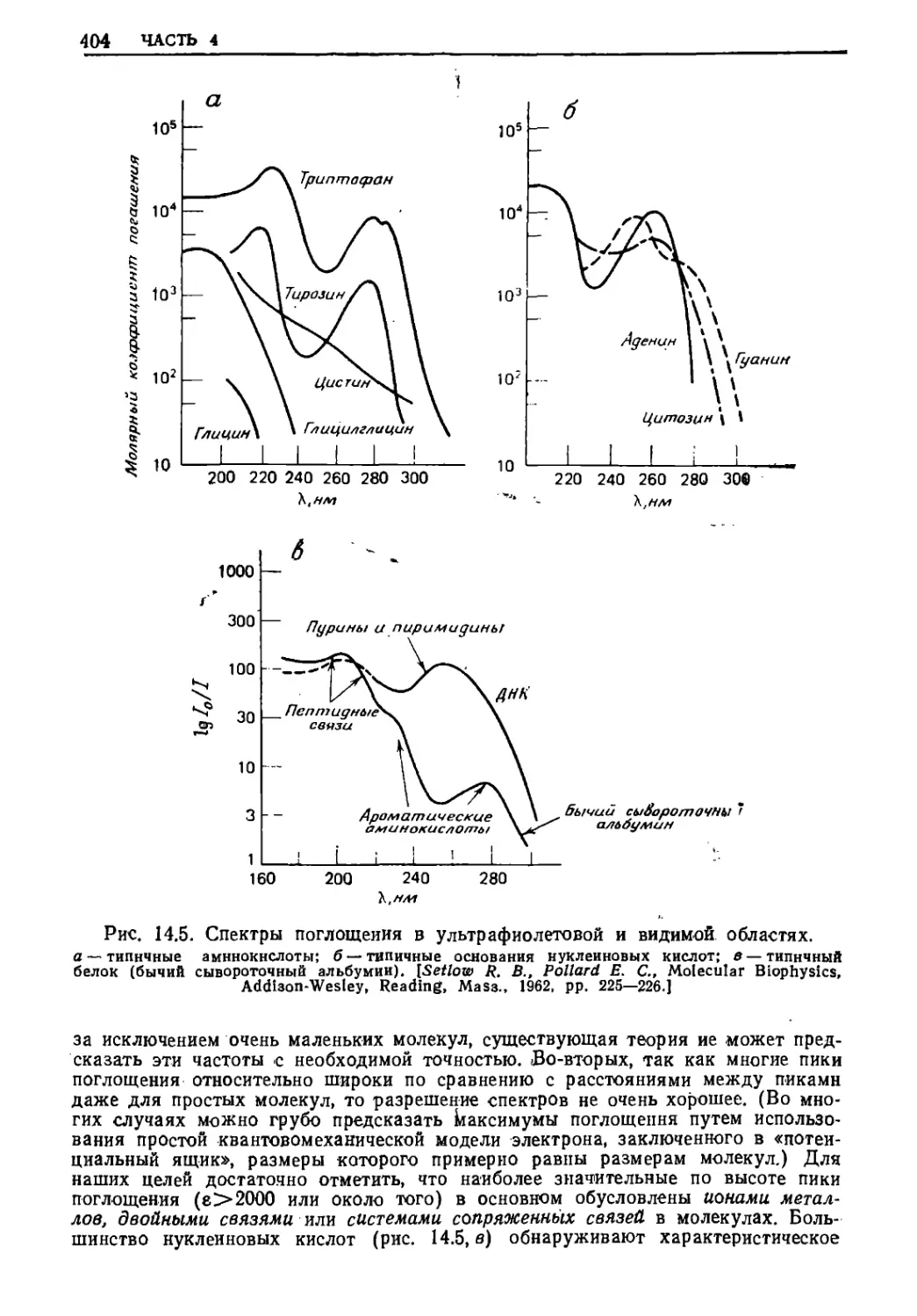
Почти в любом учебнике химии для студентов 1-го курса
показано, что собственная частота (частоты) электрона, связанного с.
водородным (или водородоподобным) атомом, появляется в
коротковолновой части оптического спектра (точнее, в ультрафиолетовой
области). Теперь полезно исследовать уравнение (13.29) в
предельных случаях, когда падающее на вещество излучение имеет
намного более низкую или более высокую частоту, чем частота
собственного движения электрона.
380 ЧАСТЬ 4
13.Б.1. а. ПРЕДЕЛ РЭЛЕЯ: ЧАСТОТА ВОЗБУЖДЕНИЯ МЕНЬШЕ,
ЧЕМ СОБСТВЕННАЯ ЧАСТОТА
В пределе, когда частота возбуждающего излучения
(например, видимый свет) значительно меньше, чем собственная частота
электрона, привязанного к пружинке (например, частота,
соответствующая переходам электронов в атоме или молекуле, которая
находится в ультрафиолетовой области), уравнение (13.29)
сводится к
[рэлеевское рассеяние (видимая (13.30)
область спектра)}
Из уравнения (13.30) следует хорошо известное явление, что
голубой свет рассеивается более сильно, чем красный свет (о)Голуб^>
^>'(0краси), так как рассеяние видимого света пропорционально
четвертой степени частоты возбуждающего излучения. В частности,
этим объясняется голубой цвет неба: свет, попадающий в глаз
наблюдателя со всех сторон, кроме прямого направления на
солнце, возникает от возбужденных колеблющихся электронов в
молекулах атмосферных газов. Более того, поскольку длина волны
видимого света совпадает по порядку величины с размерами очень
крупных макромолекул в растворе, таких, как молекулы
синтетических и биологических полимеров, то световые волны, рассеянные
различными частями такой гигантской молекулы, обнаруживают
интерференцию, при этом интенсивность света изменяется
(рис. 13.2) в степени, которая зависит от размера и формы
макромолекул. Таким образом, измерение интенсивности рассеяния
света может дать точную, хотя и не слишком детализированную
информацию (из-за сложности процесса сложения многих волн,
определяющего окончательную интенсивность рассеяния) о форме и
размерах макромолекул в растворе (разд. 15.А). Наконец,
поскольку частота света слегка изменяется, если рассеивающая молекула
движется по направлению к наблюдателю или от него в момент
рассеяния (эффект Допплера), свет, рассеянный молекулой (или
даже бактерией), при облучении монохроматическим источником
(например, лазером) будет содержать целый диапазон частот; и
ширина этого диапазона может быть использована для того,
чтобы вычислить среднюю скорость молекулы (или бактерии) и грубо
установить тип ее движения (эти вопросы будут обсуждаться в
гл. 21).
13.Б.1.6. ПРЕДЕЛ ТОМСОНА: ВОЗБУЖДАЮЩАЯ ЧАСТОТА БОЛЬШЕ,
ЧЕМ СОБСТВЕННАЯ
Другой простой предельный случай [уравнение (13.29)]
отвечает условию, когда частота возбуждения значительно больше,
чем у электронов, подвешенных на пружинках, например при стол-
Нт /расс = const- (со4/ад40)
(О < Щ
381 ГРУЗ НА ПРУЖИНЕ
кновении рентгеновских лучей или быстрых электронов с
веществом. (Следует помнить, что электрон имеет волновой характер
и длина волны может быть вычислена из его импульса согласна
соотношению де Бройля k = h/mv, где h — постоянная Планка.)
[рассеяние Томсона (рентгеновские (13.31)
лучи, быстрые электроны)]
Um /расс = const
СО><00
При очень высоких частотах, которые имеют рентгеновские лучи
или быстрые электроны, длина волны излучения мала по
сравнению с размерами типичной молекулы, что позволяет измерить
рассеяние от отдельных атомов или молекул. Поскольку не
существует линз для рентгеновских лучей, то нельзя построить
рентгеновский микроскоп, и для изучения молекулярной структуры с
помощью рентгеновского излучения необходимо обратиться к опытам
по дифракции (разд. 15.Б). Однако, поскольку электроны
являются заряженными частицами, их можно отклонять (фокусировать)
с помощью электрических и магнитных полей, что может
подтвердить каждый обладатель телевизора. Это дало возможность
сконструировать электронный микроскоп, изображение в котором
получается после столкновения быстрых электронов (рассматриваемых
здесь как электромагнитное излучение с очень короткой длиной
волны) с образцами (разд. 15.В). Уравнение (13.31) показывает,
что электронный микроскоп «не различает цветов», т. е. что
рассеяние не зависит от «цвета» (частоты) падающего излучения.
13.Б.2. ПРЕДЕЛ ЛОРЕНЦА: ВОЗБУЖДАЮЩАЯ ЧАСТОТА
БЛИЗКА К СОБСТВЕННОЙ
Рассмотрим другой крайний случай, когда частота
возбуждения очень близка к собственной, например находится рядом с
линией поглощения в спектре. Тогда общее выражение [уравнение
(13.16)] переходит в другой предельный случай. Во-первых, если
|ш0-ш|«К+со), (13.32)
то можно приближенно принять, что <о«аю. Тогда
(ш20 — со2)=(ш0 — со)(со0-1-оо) = 2ш0(ш0 — ш). (13.33)
Подставляя уравнение (13.33) в (13.166) и (13.16в), получаем
lim (*')= , .2Г?("*~?Г'>, . . (13.34а)
luli v ' 4/й2(о% (со0 — со)2 + /2ш20 ' v '
|соо —юЦ^ 1
too + <о
lim
Voo — со|
(Х") 4m!m! ((Л " tl\* -L *•«« ' (13.346)
4«»а>»0(а>0 — co)2 + f2co20
Щ + СО ^
382 ЧАСТЬ 4
Наконец, принимая во внимание, что коэффициент трения
(торможения) / имеет размерность масса-время-1, можно упростить
уравнение (13.34), введя характеристическое время релаксации т
•t = 2mff,
(13.35)
которое служит для получения окончательных функций
«дисперсии» и «поглощения» (Графическое представление членов,
стоящих в скобках в уравнении (13.36), дано на рис. 13.5, д).
Дисперсия = lim (х') = -^—~
coo + со ^ 1
Поглощение = lim (х*) =
No — И „ j
too + co
F.
2moia
Г ((о0 — со) т2 1
[l+((O0-O))2^J
(спектроскопия)
[l + K-«)42J
(13.36a)
(13.366)
Выражения, заключенные в квадратные скобки в уравнении
(13.36), часто называют лоренцовым контуром линии, и они
встречаются в разных видах спектроскопии. Компонента, смещённая по
фазе на 90°, известна как контур поглощения, поскольку
зависимость х" от частоты возбуждения часто может быть обусловлена
поглощением энергии при взаимодействии излучения с веществом
(гл. 14). Как следует из рис. 13.5, д, частота возбуждения, при
которой поглощение максимально, позволяет непосредственно
измерять собственную частоту образца. Это условие иногда называют
резонансом по аналогии с камертоном, для которого
максимальный переход энергии имеет место при его возбуждении
колебаниями, частота которых совпадает с его собственной частотой.
Подобным же образом полуширина полосы поглощения (т. е. ширина
полосы на половине ее максимальной высоты для зависимости
поглощения от частоты) позволяет прямо измерять время
релаксации 1/т, которое пропорционально коэффициенту трения системы,
и таким образом, предоставляет информацию относительно
окружающей среды, в которой совершает движение пружина.
На рис. 13.5, д показано, что те же самые данные (а именно ©о
и /) могут быть получены путем изучения синфазной компоненты
в зависимости от частоты возбуждения (сигнал дисперсии).
Термин «дисперсия» происходит от того, что зависимость (п—1) от
частоты падающего излучения имеет ту же форму, что и кривая
зависимости синфазной компоненты от частоты возбуждения, где
п — показатель преломления образца (т. е. параметр,
описывающий «дисперсию» белого света на составляющие его цвета с по-,
мощью призмы).
В этом проявляется взаимосвязь таких свойств вещества, как
поглощение энергии и показатель преломления (в чем можно убе-
383 ГРУЗ НА ПРУЖИНЕ
диться при более строгом рассмотрении; см. разд. 14.А).
Преимущества принятого здесь формального подхода теперь становятся
очевидными, что подтверждает табл. 13.1. Например, если
упругость пружинок, которые связывают электрон с молекулой,
различна в разных направлениях и если все молекулы одинаковым
образом ориентированы, то можно ожидать, что электроны будут
колебаться с разными амплитудами в зависимости от того, с какой
стороны их толкнуть (используя плоскополяризованное излучение
как средство возбуждения колебаний в определенном
направлении). Таким образом, при различных ориентациях плоскости
поляризации падающего на молекулу света следует ожидать разной
величины поглощения (дихроизм) и различных коэффициентов
преломления (двулучепреломление); примеры экспериментов
представлены в разд. 14.Б.
Вышеописанные примеры объясняют резонансные процессы, у
которых естественное движение моделируется электронами на
пружинках. Однако химические связи между различными атомами
в молекуле также можно представить в виде пружинок,
коэффициенты упругости которых позволяют измерить силу этих связей.
Частоты, соответствующие таким коэффициентам упругости
пружинок и массам ядер, лежат в инфракрасной области спектра.
Поглощение энергии и коэффициент преломления в инфракрасной
области дают, таким образом, информацию о прочности связей в
молекулах (разд. 14.А).
Рассматриваемое собственное движение не обязательно
должно быть линейным, оно может быть и круговым. Например,
свободные молекулы (в газовой фазе) вращаются с определенной
собственной частотой (квантовомеханические расчеты позволяют
выяснить, каковы эти частоты). Частота вращения зависит от
моментов инерции по разным осям молекулы. Моменты инерции в
свою очередь зависят от длины связей и углов между связями в
молекуле. Собственные частоты такого «чистовращательного»
спектра находятся в микроволновой области. Микроволновая
спектроскопия представляет собой очень точный метод измерения
длин связей и углов между связями в изолированных малых
молекулах, основанный на поглощении микроволновой энергии.
Принцип действия циклотрона основан на том, что ионы с
определенным соотношением заряда к массе вращаются в присутствии
статического магнитного поля с постоянной собственной частотой,
которая определяется отношением заряда иона к его массе. Для
однозарядных ионов при обычно используемых напряженностях
приложенного магнитного поля такие частоты попадают в
диапазон радиочастот. Таким образом, измерение поглощения
радиочастотной энергии в зависимости от частоты падающей радиации
будет давать спектр «ион-циклотронного резонанса» (ИЦР) с
пиками, соответствующими каждому из присутствующих ионов,
характеризующихся определенным отношением заряда к массе. Та-
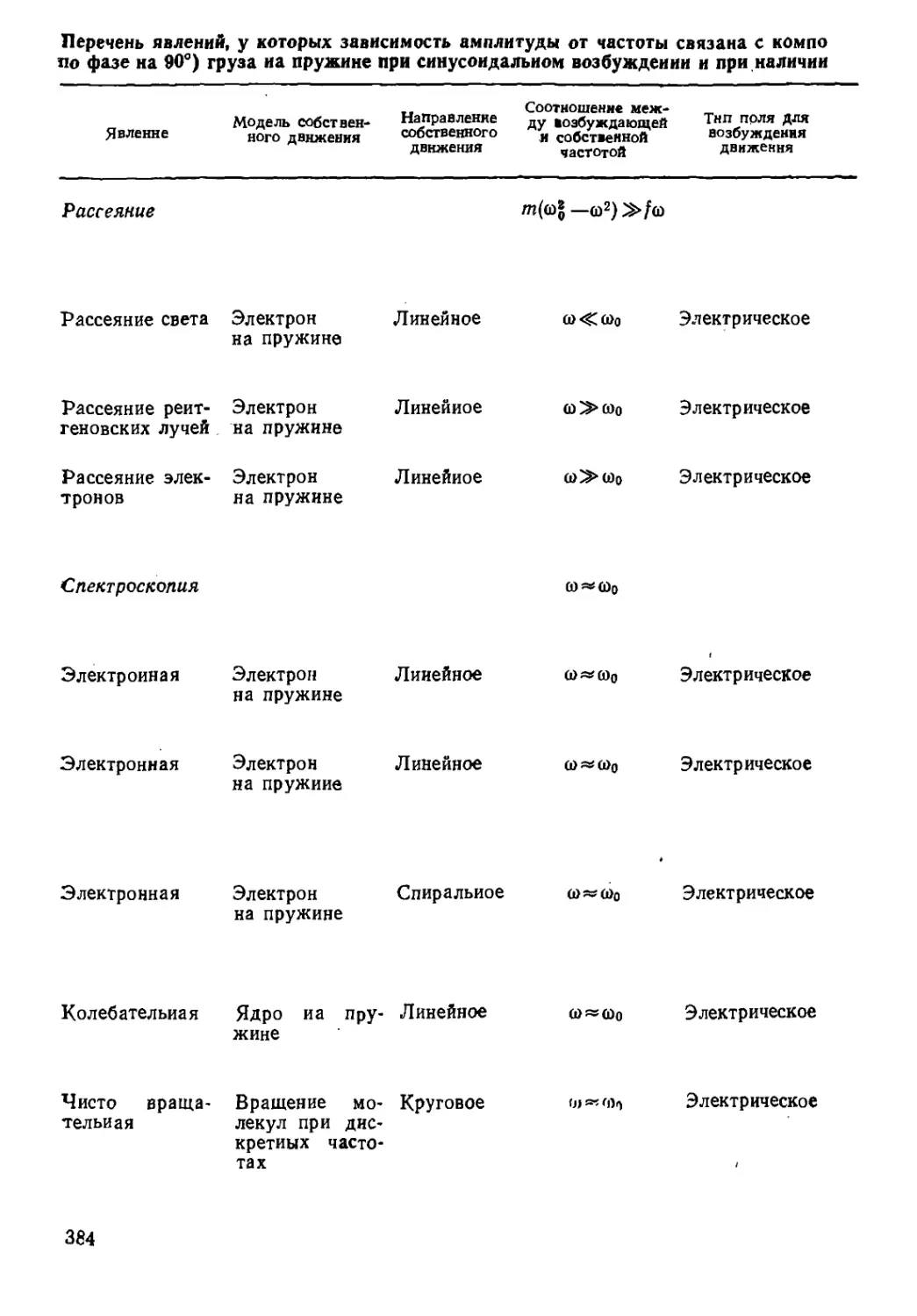
Перечень явлений, у которых зависимость амплитуды от частоты связана с компо
по фазе на 90°) груза иа пружине при синусоидальном возбуждении и при наличии
„ _ Соотношение меж- _
Модель собствен- Направление ду юзбуждающей Тнп поля Д™
собственного "'„ собственной возбуждения
движения частотой движения
Явление
ного движения
Рассеяние
т{(й\— со2)»/©
Рассеяние света Электрон
на пружине
Линейное
о><^о>о Электрическое
Рассеяние рейт- Электрон
геновских лучей на пружине
Линейное
со "> юо Электр ическое
Рассеяние элек- Электрон
тронов на пружине
Линейное
(о~>щ Электрическое
Спектроскопия
ю ««©о
Электронная Электрон Линейное
на пружине
0>«(0о
Электрическое
Электронная Электрон Линейное
на пружине
0>«0>о
Электрическое
Электронная Электрон Спиральное о>~о>о Электрическое
на пружине
Колебательная Ядро иа пру- Линейное
жине
0>Я5(|)0
Электрическое
Чисто
вращательная
Вращение мо- Круговое
лекул при
дискретных
частотах
0) я» (а^
Электрическое
384
Таблица 13.1
нентамн амплитуды смещения (находящейся в фазе или смещенной
торможения3
Диапазон Компонента, сме-
собственных Синфазная компонента щенная по фазе Получаемая информация
частот иа 90°
^рассОС
рассЧЛ. / ю
(■*)'-
Видимый свет,
Рентгеновские
лучи
Рентгеновские
лучи
^расс ОС (w/(i>o)2
("редел Рэлея)
£расс ие зависит от о>
(предел Томсона)
£"расс не зависит от ш
(предел Томсона)
, (Щ — <а)т2
* «■ 1 + К — Ю)2 Т*
хГ<Х
0 Положение центров
рассеяния и определение их
распределения по
картине интерференции или
фокусирования
0 Размер, форма и средняя
скорость очень больших
молекул (или бактерий)
в растворе
0 Трехмерная структура
кристаллических молекул
по картине дифракции
0 Изображение в
электронном микроскопе при
фокусировке прошедших
через образец или
рассеянных электронов
т Информация о силе пру-
1+(соо—<о)2та жины из а>о и об
окружающей пружину среде
из т.
Видимый свет,
ультрафиолетовое излучение
Видимый свет,
ультрафиолетовое излучение
Видимый свет,
ультрафиолетовое излучение
Дисперсия^п— 1;
» — показатель
преломления
Поглощеиие-^-е
8,1—8
(двулучепреломление) (дихроизм)
Ъ"\
Инфракрасное
излучение
Микроволновый
(оптическое
вращение)
Дисперсия=л — 1
Дисперсия
(круговой
дихроизм)
Поглощение
Поглощение
Данные о связях я-элек-
тронов в сопряженных
системах из
электронного спектра поглощения
Определение и
количественная оценка степени
линейного порядка в
молекулярных рядах,
используя дихроизм и
двулучепреломление
Определение и
количественная оценка «хи-
ральиости» органических
молекул и
неорганических комплексов
Сила химических связей
из силовых постоянных
k пружины (из
определений ©о)
Длины химических
связей и углы между ними
из значений ©о,
полученных при локализации
пиков в спектрах
поглощения
385
386 ЧАСТЬ 4
Явление
Модель
собственного движения
Направление
собственного
движения
Соотношение
между возбуждающей
и собственной
частотой
Тип поля для
возбуждения
движения
Ионный
циклотронный
резонанс
Ион в постоян- Круговое
иом магнитном
поле
W«0>o
Электрическое
Электронный
парамагнитный резонанс
Прецессия маг- Круговое
иитного
момента электрона
(0«й)0
Магнитное
Ядерный
магнитный
резонанс
Прецессия маг- Круговое
нитного ядра
(|)«й)о
Магнитное
Явления классифицируются в соответствии с несколькими указанными критериями и
f — коэффициент трения (торможения) груза на пружине; от —масса груза, подвешенного
возбуждения и торможения при коэффициенте упругости k; £pacc —амплитуда (в фазе)
нанса; т=2т//~ время «релаксации»; е — коэффициент погашения данного вида излучения.
ким образом, прибор функционирует как масс-спектрометр, и
имеет ряд уникальных преимуществ при изучении ион-молекулярных
реакций в газовой фазе (разд. 20.Б.2).
Наконец, поскольку фиксированный магнитный момент будет
прецессировать по круговому пути вокруг фиксированного
направления магнитного поля и поскольку можно «возбудить» такое
движение, используя колеблющуюся (или вращающуюся) компоненту
магнитного поля электромагнитного излучения, то график
зависимости поглощения энергии от частоты падающего излучения будет
давать спектр магнитного резонанса, по пикам которого можно
различать частицы с разными магнитными моментами. Если
частицы являются парамагнитными электронами или свободными
радикалами, то собственные частоты попадают в микроволновой
диапазон, а спектр электронного парамагнитного резонанса (ЭПР)
служит для идентификации различных химических веществ,
содержащих неспаренные электроны (парамагнитные ионы металлов или
свободные радикалы в растворе), и установления различий между
ними. Если частицы имеют парамагнитные ядра (например, ,Н, 2Н,
13С, 15N, 19F, 31P и т. д.), то собственные частоты находятся в
радиочастотном диапазоне, а спектр ядерного магнитного резонанса
387 груз на пружине
Продолжение табл. 13.1
Диапазон
собственных
частот
Радиочастотный
Микроволновый
Радиочастотный
Синфазная компонента
Дисперсия
'
Дисперсия
и-мода
(дисперсия)
Компонента,
смещенная по фазе
на 90°
Поглощение
Поглощение
и-мода
(поглощение)
Получаемая информация
Масс-спектры
ионизированных молекул из
локализации пиков в
спектрах поглощения
Идентификация иеспа-
ренных электронов и
различий между ними
в металлах или
свободных радикалах из
положения и формы спектров
поглощения
Идентификация ядер с
магнитным моментом
(например, 13С) и
различий между ними в
индивидуальной молекуле по
картине пиков спектров
поглощения
известными приложениями.
на пружине; <oo=(fe/m)V2 — собственная частота колебаний груза на пружине в отсутствие
рассеянного излучения от электрона на пружине с частотой возбуждения далекой от резо-
(ЯМР) может быть использован для наблюдения, например,
сигналов 13С, причем разные пики будут соответствовать разным
химически отличным атомам углерода в органической молекуле
(разд. 14.Г).
Широкая применимость модели «груз на пружине» видна из
того, что поглощение и скорость звуковых волн (разд. 14.Ж) или
электрические колебания (разд. 14.Д и 14.Е) также могут быть
описаны по аналогии с движением подвешенного на пружине
груза, масса которого равна нулю. Соответствующие уравнения
могут быть получены простым введением т=0 в левую часть
уравнения (13.20), что обсуждается в гл. 14.
Суммированные в табл. 13.1 данные показывают, что
зависимости, приведенные на рис. 13.5, выглядят почти одинаково для
большого набора процессов, несущих полезную для химии
информацию, и являются качественно понятными из одной простой
модели, если ввести ограничение, что наблюдаемая частота
(возбуждения) близка по значению к собственной частоте
рассматриваемого движения. Для другого экстремального случая из таблицы
видно, что синфазная компонента амплитуды смещения
возбужденной пружины, квадрат которой пропорционален интенсивности
.388 ЧАСТЬ 4
рассеянного излучения, может давать ценную информацию при
условии, что наблюдаемая частота значительно больше (рассеяние
Томпсона) или значительно меньше (рассеяние Рэлея), чем,
рассматриваемая собственная частота. Следует подчеркнуть, что для
лонимания этих примеров не требуются никакие квантовомехани-
ческие вычисления: классическая модель «груза на пружине»
достаточна для расчета описанных выше частотных зависимостей.
(Значение квантовой механики состоит в том, что с ее помощью
можно предсказать количество и значения «собственных» частот,
которые могут встречаться для данного типа собственного линей-
лого и кругового движения.)
ЧЗ.В. ГРУЗ НА ПРУЖИНЕ ПОД ДЕЙСТВИЕМ
ТОРМОЗЯЩИХ СИЛ: ЗАТУХАЮЩИЕ КОЛЕБАНИЯ
Как видно из рис. 13.4 и последующего обсуждения,
существует второй тип экспериментов, из которых можно получить
информацию относительно массы т, коэффициента упругости k и
коэффициента трения /, для тела, подвешенного на пружине и
находящегося под действием тормозящих сил. В этом типе экспериментов
после смещения тела из положения равновесия его отпускают
(нулевой момент времени) и наблюдают за его (затухающим)
колебанием в зависимости от времени. Задача определяется
уравнением, описывающим затухающее колебание груза, подвешенного на
пружине (в отсутствие возбуждающих сил):
mdH/dt2 + fdx/dt -f kx = 0 (13.37)
Читатель может в качестве упражнения (см. задачи в конце
главы) удостовериться в том, что уравнение (13.38) является
решением уравнения (13.37):
х = х0ехр[—tf*]cos'(Wt)t (13.38)
где
(1/х):=//2да (13.35)
и
W^=V^\-{\h)\ (13.39)
Этот результат становится особенно простым в обычном
физическом случае, когда
(1/х)<^ш0 (слабое торможение) (13.40)
для которого
Urn х = х0 ехр [—t/t] cos (mj)
i
т <(0°
(13.41)
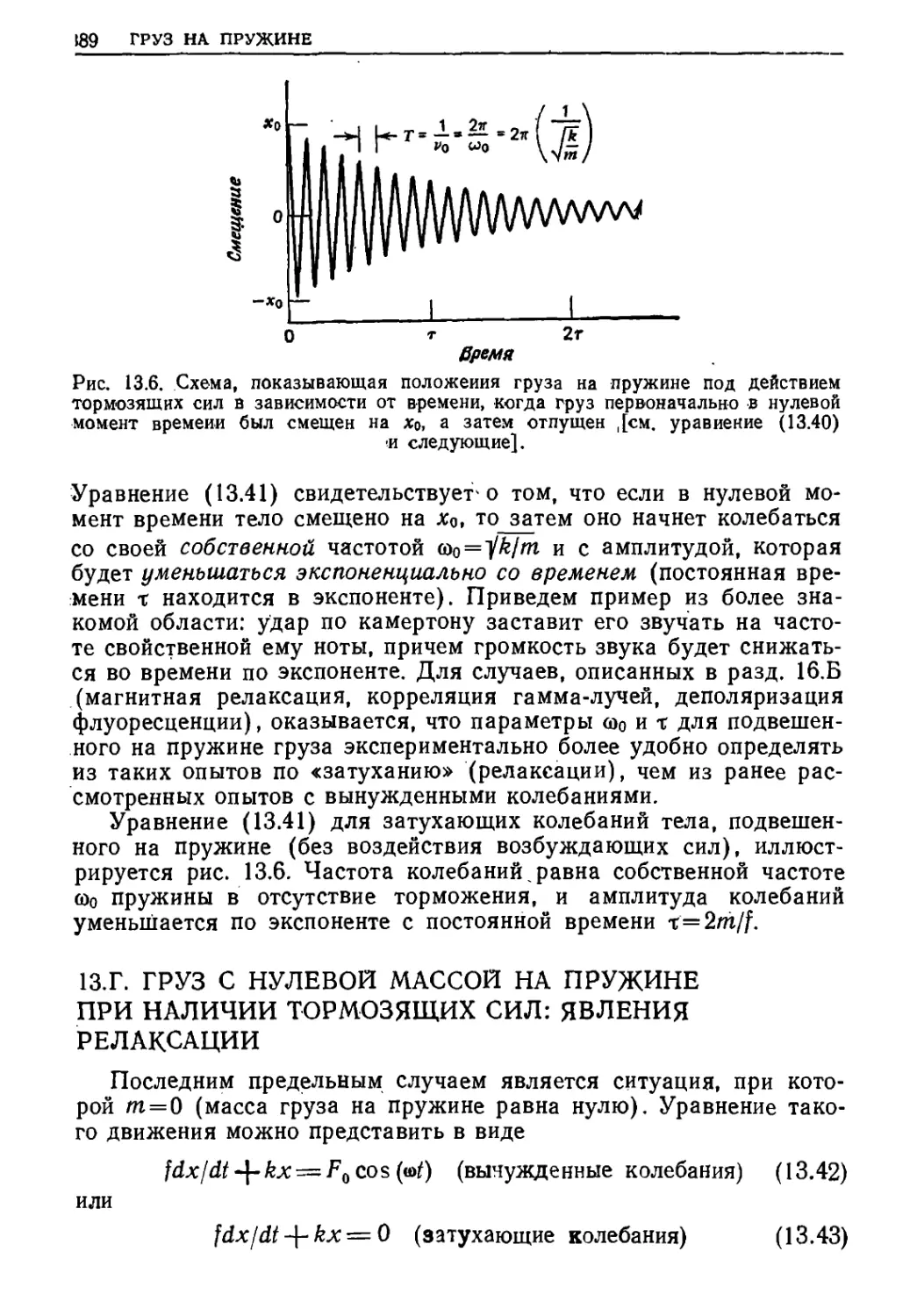
189 ГРУЗ НА ПРУЖИНЕ
О т IT
Время
Рис. 13.6. Схема, показывающая положения груза на пружине под действием
тормозящих сил в зависимости от времени, когда груз первоначально в нулевой
момент времени был смещен на дс0, а затем отпущен ,[см. уравнение (13.40)
■и следующие].
Уравнение (13.41) свидетельствует о том, что если в нулевой
момент времени тело смещено на jc0, to затем оно начнет колебаться
со своей собственной частотой щ=^Щт и с амплитудой, которая
будет уменьшаться экспоненциально со временем (постоянная
времени т находится в экспоненте). Приведем пример из более
знакомой области: удар по камертону заставит его звучать на
частоте свойственной ему ноты, причем громкость звука будет
снижаться во времени по экспоненте. Для случаев, описанных в разд. 16.Б
(магнитная релаксация, корреляция гамма-лучей, деполяризация
флуоресценции), оказывается, что параметры ©о и т для
подвешенного на пружине груза экспериментально более удобно определять
из таких опытов по «затуханию» (релаксации), чем из ранее
рассмотренных опытов с вынужденными колебаниями.
Уравнение (13.41) для затухающих колебаний тела,
подвешенного на пружине (без воздействия возбуждающих сил),
иллюстрируется рис. 13.6. Частота колебаний. равна собственной частоте
©о пружины в отсутствие торможения, и амплитуда колебаний
уменьшается по экспоненте с постоянной времени x=2ni/f.
13.Г. ГРУЗ С НУЛЕВОЙ МАССОЙ НА ПРУЖИНЕ
ПРИ НАЛИЧИИ ТОРМОЗЯЩИХ СИЛ: ЯВЛЕНИЯ
РЕЛАКСАЦИИ
Последним предельным случаем является ситуация, при
которой пг = 0 (масса груза на пружине равна нулю). Уравнение
такого движения можно представить в виде
fdxfdt -f- kx = F0 cos (Ы) (вынужденные колебания) (13.42)
или
fdx[dt-\-kx=0 (затухающие колебания)
(13.43)
390 ЧАСТЬ 4
Можно показать (см. задачи в конце главы), что решения
уравнений (13.42) и (13.43) имеют вид
х = х' cos (ш/) -]-х" sin (W),
где
и
или
x'-^F k
x"s=F.
0 k2 4- /2w2
A»
0 k* -f- f20)a
(13.44)
(вынужденные (13.45a)
колебания)
(13.466)
х==хпехр[—ktff\ (затухающие колебания) (13.46)
Таблица 13.2
Типичные диапазоны для констант скоростей различных химических
и физических процессов
Образование Образование
связи g связи
красите/16 -ДНК краситель -ДНК
I 1 И ■ 1
Переходы спираль-клубок для белков нук/ieuHosi/x кис/юта
Перераспределение зле/стронов в молекулах (флуоресценция ифссфсресиенция)
Фермент-субстрат (антитела-гаптен)
Скорость „соединения" Фермент-субстрат(антитело-гаятен)
( | Скорость „диссоциации1"1
Кислотно-основной катализ
ассоциация Кислот но-основной катализ, диссоциаиия
Ч
Перенос протонов, ассоциация
Гидратация и гидролиз*
Поверхностная диффузия в мембранах6
Перенос протонов, включающих диссоциацию Н и ОН"
Трансляционное gi/ффуз- - I I
ное перемещение молекул .
в растворе Перенос электронов *
Вращательная диффузия вращательная диффузия
молекул в растворе' в мембранах*
I 1 I 1
J I I I I I I I \ \ \ \ \ \ \
ю'г ю'° /ов ю6 ю" /о' I tcr*
Аонс/паннгб/ скоростей (логарифмическая шкала)
а Для процессов первого порядка константа скорости-(1/Время жизни).
6 Константа скорости равна константе скорости процесса второго порядка (исшМ-л.<*•■!),
в Константа скорости-(I/Время''вращательноЯ корреляции).
391 ГРУЗ НА ПРУЖИНЕ
Таблица 13.3
Типичные диапазоны скоростей, доступных для измерений
с помощью различных методов
Деполяризац ия
флуоресценции
Угловые корреляции
■ f-лучей
Электронный парамаг-
митнь/и резонанс .
Скачок электриче-
,ского поля |
Ультразвуковое поглощение,
1 .' Концентрационный скачок
Диэлектрическая (метод остановленного потока)
релаксация | ^
Ядерный магнитный резонанс
Спектроскопические методы
Скачок давления
Рассеяние, лазерного I I РУ?ртпаы
изличения - методы
| 2_— \ Температурный скачок |
■*>
« /л» /0е /0' /С* /С2 / Л>"2
Применения случая вынужденных колебаний груза с нулевой
[массой при наличии сил торможения рассмотрены в разд. 14.Д—
|4.3. Применения случая затухающих колебаний этой же модели
разобраны в разд. 16.А. Последние основаны на том, что если
система, находящаяся в химическом равновесии, подвергается вдруг
Небольшому воздействию, вызывающему сдвиг равновесия, то
достижение компонентами новых значений концентраций может быть
рписано с помощью уравнения (13.43). Константы скоростей
таких химических реакций часто связаны с экспоненциальной
постоянной времени затухающего колебания очень простыми
соотношениями [уравнение (13.46)]. Одно из основных преимуществ
релаксационного метода химической кинетики состоит в том, что он
позволяет определить константы скоростей реакций, которые
протекают слишком быстро для того, чтобы можно было их измерять
Ьутем смешивания реагентов и изучения изменений концентраций
Компонентов во времени. Из табл. 13.2 видно, что константы
скоростей большинства химических и физических процессов,
представляющих наибольший интерес для химиков или биохимиков,
слишком велики для прямого измерения. В табл. 13.3 указан диапазон
Скоростей таких процессов, которые для вынужденного колебания
могут быть определены по ширине линий, а для затухающего
колебания— по экспоненциальной константе затухания.
Таблица 13.4
а**ФоРмУлы Для смещения х груза (масса т) иа пружине (константа упругости k) при наличии торможения
(коэффициент трения f). Движение было начато или путем неравновесного смещения {х0ФО) в нулевой момент времени
(«затухающие» колебания), или путем постоянного приложения возбуждающей силы Focos(co0 («вынужденные» колебания)
Стационарное состояние
(вынужденные колебания)
°(*-mo>2)2 + /*V со =*
„V ^^Ф^У1^^''-
const
^л7 *J
m=o J
**оь1
k
x" = Fc
k* + f*a>*
f*
^Л
**%&&
Ke&
(k - mw1)1 + pшг <о
>=oJ
f*
'к* + Р<ог
F0 {щ - а>)т*
,2тш0 1 + (а>0 — а>¥тг
bh^lpacc* const
не зависит от <*>
Нестационарное состояние
(затухающие колебания)
т ^ О
x=Xo(e-w'>)cos[«x/a,5-(l)*]-I
1
- « too
*x0e-UMcoe{a>0t)
то = О
* = *„*-<*«/>
Для специального случая предела рассеяния (верхний правый угол) нитеисивиость рассеянного излучения от колебаний возбужденного
~гтПттнпнальна квадрату второй производной смещения по времени, что обсуждается в тексте. Выражения для соб-
" " " '"•'•"тм-пие тормозящих и возбуждающих сил (<йо) н для «времени релаксации»
|g3 груз на пружине
. В этой главе рассчитаны параметры вынужденных и
затухающих колебаний груза (с нулевой или конечной массой),
подвешенного на пружине, под действием тормозящих сил. Основные
формулы, необходимые для справок, собраны в табл. 13.4. Эти
вычисления составляют основу для обсуждения чрезвычайно широкого
Itpyra физических методов исследования. Эксперименты,
основанные на вынужденных колебаниях при частотах возбуждения,
близких к «собственным» частотам свободных колебаний груза на пру-
кине, рассмотрены в гл. 14. Эксперименты, основанные на
вынужденных колебаниях при частотах возбуждения, значительно
отливающихся от резонансных, обсуждены в гл. 15. Эксперименты,
связанные с затуханием колебаний, рассмотрены в гл. 16, а
специальный (но также очень важный) случай колебания двух или
уолее пружинок, соединенных вместе, разобран в гл. 17?
i
L
Задачи
р. Покажите, что сумма двух синусоидальных волн одинаковой частоты (но от-
f личающихся по фазе и амплитуде) дает результирующую синусоидальную
1 волну той же частоты, т. е. покажите, что существует ty, так что
A sin (9) + В sin (9 + Ф) = С sin (6 + Ф)
| [Эта формула является алгебраическим основанием среднего графика иа
f рис. 13.2. Наиболее важен специальный случай этой формулы ф = л/2, с по-
i мощью которого можно разбить произвольную синусоидальную волну иа си-
Г нусоидальную и косинусоидальную компоненты (рис. 13.5); такой подход
I лежит в основе изложения материала в гл. 14 и 15.]
J2-. Движение груза с массой тп, подвешенного на пружине (коэффициент упру-
\ гости k), в отсутствие возбуждающих и тормозящих сил описывается урав-
[■ нением (13.3) (где х — смещение)
m (d2x/dt2) +kx = 0. (13.3)
i
г
а. Покажите, что выражение x=Xocos(<aot) является (действительным)
решением уравнения (113.3) и найдите выражение для щ.
б. Покажите, что x=Xoexp[i<uot] является (комплексным) решением уравнения
(13.3) и вновь найдите выражение для <ао. Обратите внимание, что
действительная часть комплексного смещения совпадает с (действительным) решением
исходного (действительного) уравнения (13.3).
9. Для груза с массой т, подвешенного на пружине (коэффициент упругости k),
| под действием синусоидальной возбуждающей силы Focos(<oO и тормозящей
- силы (коэффициент трения f) уравнение движения при смещении запишется
следующим образом:
m (d2x/dt2) + f (dx/dt) + kx = F0cos (<ot) (13.15)
а. Покажите, что (действительное) решение уравнения (13.15) есть х=
=;t/cos(<i)tf)-fx"sin(<i^), и определите выражения для х' и х".
б. Исходя из комплексной формы уравнения (13.15):
m (d2x/dt2) + f (dx/dt) +kx=F0 exp [Ш] (13.16)
покажите, что (комплексное) решение уравнения (13.18) имеет вид
Я = ХехР Рш*]|
394 ЧАСТЬ 4
где
X = х' — ix"
и
Re [x exp (i<ot)[ = x' cos (<at) + л" sin (<at),
а х' и х" те же, что и в пункте а, т. е. покажите, что действительная часть
комплексного смещения совпадает с действительным решением исходного
действительного уравнения. Обратите внимание на большую простоту
алгебраических операций в случае использования комплексных величин. Эти
преобразования положены в основу дайной главы.
4. Подтвердите приведенные в тексте выражения для х' и х" '(дисперсия и
поглощение) в случае предела Лореица, когда частота возбуждения
близка к собственной частоте колебаний груза на пружине, т. е. получите
уравнение ;(13.36) «з (13.16). Эти два выражения описывают поведение
показателя преломления и поглощения энергии в зависимости от частоты внешнего
излучения при спектроскопических применениях, описанных в гл. 14.
5. Лоренцов контур линии (разд. 13.Б.2) может быть записан в виде
А И = 1 _j_(№ №)2t2 = контур поглощения
и
(со0 — со) г2
В (<о) = 1 , ,ш _(0ч2х2 = контур линии дисперсии
а. Постройте зависимость А{<о) от а л найдите высоту пика '(максимум
Л(й))) и ширину полосы на половине ее максимальной высоты.
б. Постройте зависимость В(<о) от <а, найдите экстремумы (максимальные и
минимальные значения) В(<а) и вычислите разность частот между двумя
экстремумами. Вышеприведенные вычисления включают в себя основные
свойства спектрального лоренцова контура (гл. 14).
в. В определенных спектроскопических экспериментах представляется
удобным определить первую производную от Л(<о) (по частоте возбуждения), что
делают, например, в спектроскопии электронного парамагнитного резонанса
(разд. 14. Г). Полученная кривая dA((a)Jd<a похожа иа дисперсионный контур
линии, построенный в пункте б. Вычислите положение, величины и разность
частот между экстремумами на кривой зависимости dA(<a)ld(o от со и
сравните с дисперсионным контуром (пункт б). •■
6. Движение груза с массой т, подвешенного на пружине (коэффициент
упругости k), под действием тормозящих сил (коэффициент трения f) в
отсутствие возбуждающих сил описывается уравнением (U3.3.7) (где х — смещение)
т ((Px/dt2) + / (dx/dt) + fee = 0. (13.37)
а. Покажите, что (действительное) решение уравнения (13.37) есть х=
=х0ехр[—tlx]cos(Wt), и найдите выражение для % и W, Затем найдите
предел, к которому' стремится решение при слабом затухании колебаний
,[;(1/т)«о0].
б. Покажите, что (комплексное) решение уравнения (13.37) есть х=
=х0ехр [ict], и найдите выражения для с. Покажите, что действительная
часть этого комплексного решения совпадает с (действительным) решением
исходного (действительного) уравнения.
(5 ГРУЗ НА ПРУЖИНЕ
. Движение при затухающих колебаниях (коэффициент трения f) груза с иуле-
: вой массой (т=0) на пружине (коэффициент упругости k) описывается
уравнением (13.42) и (13.43) (х— смещение):
/ (dx/dt) + kx = F° cos (<at) (синусоидальная возбуждающая сила) (13.42)
f (dx/dt)-\-kx = О (возбуждающая сила отсутствует) (13.43)
а. Покажите, что (действительное) решение уравнения (13.42) есть
х=х1'cos(<at)+x"sin(Ш), и найдите выражения для х' и х"'.
б. Покажите, что (действительное) решение уравнения (13.43) есть
х=х0ехр[—ktlf].
> в. Покажите, что (комплексное) решение комплексной формы уравнения
\ (13.42)
: f (dxfdt) + kx = F0 exp [Ш] (13.42a)
Ьть
top
x = jc" — tV
и x' и x" имеют тот же смысл, что и в пункте а. Вновь обратите внимание
иа простоту и быстроту алгебраических выкладок при использовании комп-
' лексной системы обозначения. Эти вычисления составляют основу для всех
связанных с затуханием (релаксационных) явлений, описанных в гл. 16.
Литература
1 Фейнман Р. П., Лейтон Р. Б., Сэндс М. Фейнмановскне лекции по физике. М.:
2 Мнр. Рассмотрение задачи о колебаниях груза на пружине можно найти почти
'[ в любом элементарном курсе физики, содержащем раздел «Механика». Однако
! наиболее четкое и общее изложение содержится в указанной книге.
ГЛАВА 14
ПОГЛОЩЕНИЕ И ДИСПЕРСИЯ:
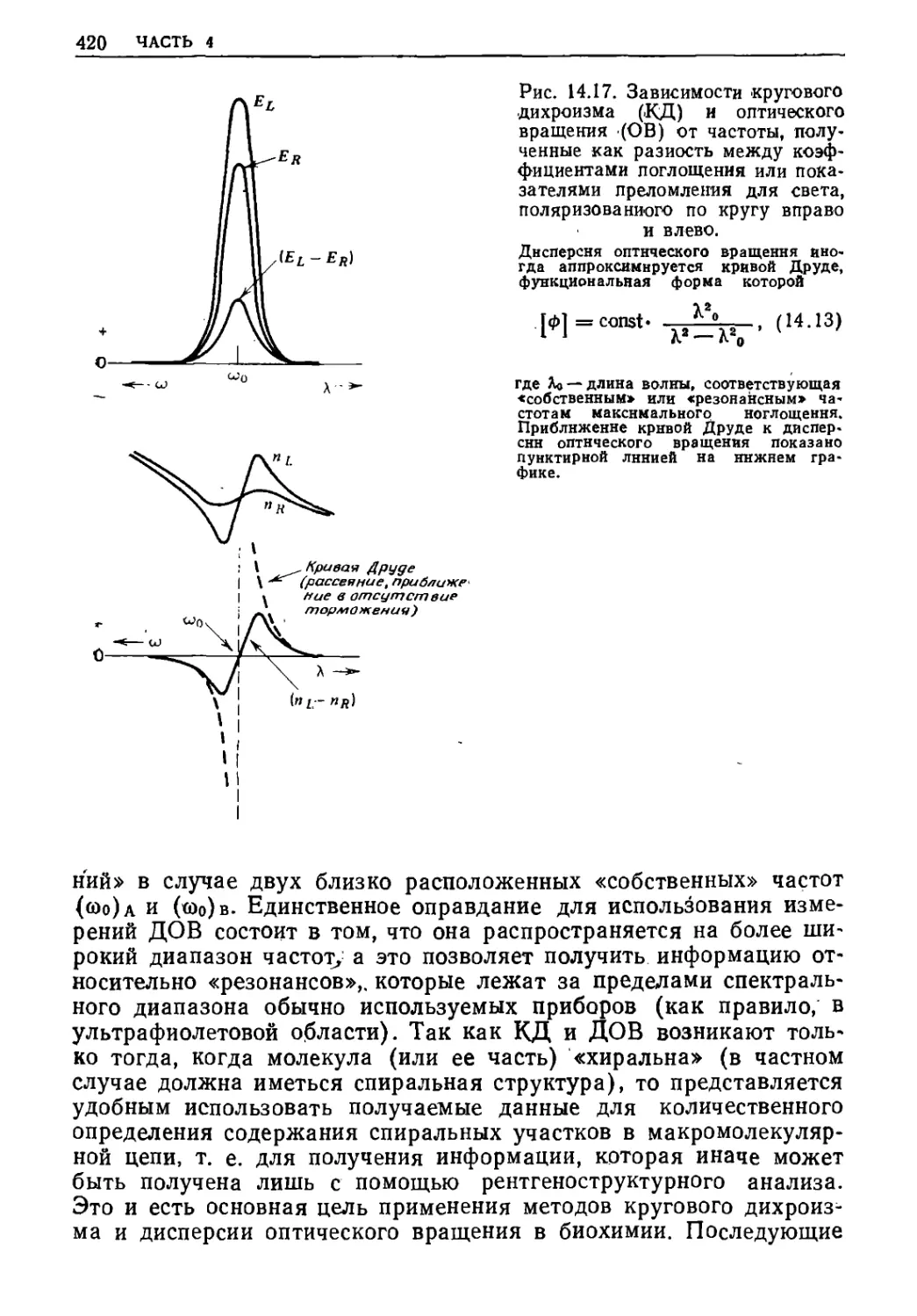
ВЫНУЖДЕННЫЕ КОЛЕБАНИЯ ГРУЗА НА
ПРУЖИНЕ ПОД ДЕЙСТВИЕМ ВОЗБУЖДАЮЩИХ
И ТОРМОЗЯЩИХ СИЛ
Из квантовой механики (см. часть 5) следует, что энергия
данной молекулы может изменяться только дискретно (в соответствии
с ее энергетическими уровнями), а не непрерывно. «Переход»
молекулы с низшего на высший энергетический «уровень» может
произойти, когда фотон с определенной энергией (Av = £„epxH —
—£нижн) сталкивается с молекулой, при условии что
удовлетворяются определенные «правила отбора» (см. часть 5). Поскольку при
таком переходе молекула поглощает энергию, можно
предсказывать появление пика при построении зависимости мощности
поглощений (т. е. энергии, поглощаемой в единицу времени) от частоты
фотонов v в том случае, когда энергия фотона приблизительно
равна энергии «разрешенного» перехода. Хотя совершенно верно,
что для определения частот и интенсивностей пиков в «спектрах»
(мощности поглощения) молекул необходимы
квантовомеханические расчеты, можно для объяснения формы любого из «пиков»
использовать методы классической механики, ранее примененные
для расчета смещения тела, подвешенного на пружине,
находящейся под действием возбуждающих и тормозящих сил в
стационарном состоянии. В частности, мы часто можем извлекать
необходимую информацию о молекулярном окружении из ширины
спектральной линии, поскольку ширина определяется конкретными
силами «трения», которые действуют между молекулой и ее
окружением.
Ранее было показано, что при вынужденных колебаниях
смещение груза, подвешенного на пружине, под действием
синусоидальной возбуждающей силы при наличии сил торможения можно
разложить на две компоненты хг и х", которые или точно
совпадают по фазе {х), или смещены по фазе на 90° {х") относительно
возбуждающей силы (см. рис. 13.5). В этой главе будет показано,
что «поглощение» х" можно определить, исходя из мощности
поглощения оптического излучения (в ультрафиолетовой, видимой,
инфракрасной областях), дихроизма, кругового дихроизма,
диэлектрических потерь, поглощения ультразвука и поглощения при
магнитном резонансе. «Дисперсию» хг можно найти из величины
(п—1), где п — показатель преломления, а также из данных по
ГРУЗ НА ПРУЖИНЕ
^лучепреломлению, оптическому вращению, диэлектрической
©ницаемости, дисперсии ультразвука и дисперсии при магнит-
It резонансе. Начинаем изложение с рассмотрения явлений по-
Ьщения света и показателя преломления света, от которых про-
ршли названия «поглощение» и «дисперсия» для х," и хг.
.А. ПОГЛОЩЕНИЕ И ПОКАЗАТЕЛЬ ПРЕЛОМЛЕНИЯ:
DHOBbI СПЕКТРОСКОПИИ И МИКРОСКОПИИ*
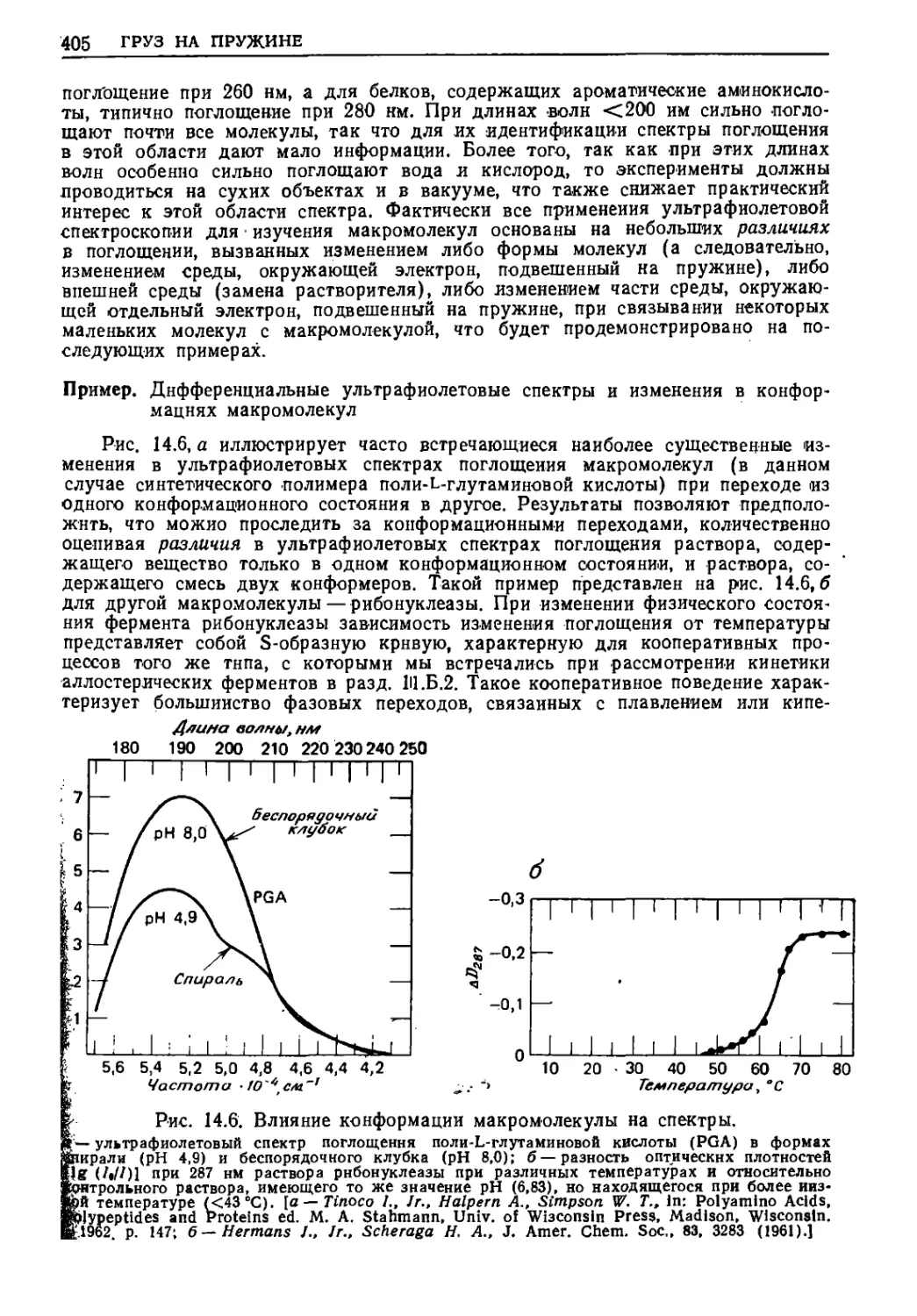
Предположим, что монохроматическое излучение направлено
^ез тонкую пластину толщиной Д*/(см. нижнюю схему). В этом
Эделе нас интересует амплитуда электрического поля прошедшей
аны на расстоянии у от пластины. Поскольку наблюдаемое в
|мент t электрическое поле на расстоянии у образовалось ранее
£ = Е0 cos(ujt)r>put/=0
(при ysy)
колебаниях электронов в пластине в момент [t—{у/с)], где
скорость света (в вакууме), то очевидно, что, если пластина
1к не влияет на падающее излучение, выражение для напря-
шости электрического поля на расстоянии у можно записать
Че
в точке
,)=£eCOs[<o(*--f)]=:
= Re [E,exp [to (*--£-)]]
(13.9)
(14.1)
1КО если скорость движения волны уменьшается при
прохожий пластины, то из обычного определения показателя
преломил п
Показатель преломления=п'--
Скорость света в вакууме
Скорость света в пластине
(14.2)
^Там, где это возможно, в этом разделе результаты представлены как
гвительиой, так и в комплексной системе счисления для ознакомления
|йя, и с интуитивно понятным («действительным») описанием, и с матема-
~ компактной '(«комплексной») формой записи.
398 ЧАСТЬ 4
следует, что после прохождения пластины волна уже будет
запаздывать на время t = (п'—1) (Ay)/ct так что выражение для
электрического поля в точке у теперь примет вид
£(.». „ = В. cos [. (, - fi^ilM-i.)]- (14.3a)
= Не[Ялехр[^((-<=1^Ш)_Х)]] =
== Re [exp [- ш (nr - 1) (Ду)/с] Ей ехр \fo ft - -jj-) 11 (14.36)
Влияние показателя преломления^ 1 *" •"" -*
Электрическое поле,
создаваемое волной,
если пластина
отсутствует
Уравнение (14.36) показывает, что с помощью комплексной
системы обозначения легко выделить член, описывающий поведение
волны при отсутствии преломления, и член, определяющий
уменьшение скорости света при прохождении через среду.
Во время прохождения волны через пластину кроме
уменьшения скорости [уравнения (14.3а) — (14.36)] будет наблюдаться
уменьшение dl интенсивности /. Для достаточно тонкой пластины
можно ожидать, что изменение интенсивности пропорционально
интенсивности падающего света, толщине пластины dy и
концентрации поглощающих молекул m (в молях на литр):
dl — — k/mdyt (14.4)
где k — константа пропорциональности, характерная для
данного вещества. Уравнение (14.4) в интегральной форме известно как
закон Б ера:
In (///0) = ~ km&.y
ЮЗ) __ 1П-(етД#),
(14.5)
.Пропускание» = ///0 = ю-^^.зоз) = 1(ГС«па*>,
_ Ю~Л =£-(2.3ОЗеД0ЛГ/Л'о)
где е называют молярным коэффициентом погашения (ранее его
называли молярным коэффициентом экстинкции), /0 —
интенсивность падающего на пластину света, А — поглощение или
оптическая плотность, N— число молекул в 1 см3 и No — число Авогад-
ро. Поскольку интенсивность электромагнитного излучения
пропорциональна квадрату амплитуды электрического поля
прошедшей через пластину волны и, кроме того, ^е~х= (e~x)lb =erW*)t то
ГРУЗ НА ПРУЖИНЕ
уравнение (14.3) можно теперь дополнить включением в него чле-
[а, описывающего поглощение энергии согласно уравнению (14.5):
|?(ВТ. у, = ехр [-2,3038^ (Ду)/2ЛГ#]£# cos L ft - (п' ~ 1) Ау -
■)]■
= Re [схр [-2,303вЛ/Дг//2Л/01 ехр [-ш {и' - \)Ьу1с\ X (14.6а)
Влияние поглощения
х£,ехр
Влияние показателя
преломления ^ 1,0
К(-^)
(14.66)
Электрическое поле,
создаваемое волной, если
пластина отсутствует
Рассмотрение первых двух множителей [уравнение (14.66)]
показывает, что значительная экономия при записи может быть
достигнута путем введения комплексного «показателя
преломления» п:
п = п' — in"
(14.7)
|огда уравнение (14.6) упрощается:
•ев точке у) = Re [ехр [-*» {и - 1) (АуЦс] Е0 ехр [да ^/ ?")]]• (14'8)
2,303cW
П ■=. „,„,— е.
2coW0
(14.9)
| уравнении (14.8) «мнимая» часть п" комплексного «показателя
|еломления» п прямо пропорциональна молярному коэффициен-
| погашения для поглощения энергии веществом [уравнение
14.9) ], в то время как «действительная» часть п' комплексного
жазателя преломления» является обычным показателем пре~
Ьмления из уравнения (14.2). Мы вновь убедились, что и п' и п"
|ляются «действительными» как в физическом, так и в
математиком смысле; мы просто экономим место при подстановке урав-
Иия (14.7) в (14.8).
I Вышеизложенное решение основано на допущении, что элек-
[агнитная волна замедляется и что ее энергия частично теряет-
при прохождении через тонкую пластину вещества. Если же
теперь предположим, что электроны в веществе ведут себя по-
>но пружинкам под действием возбуждающих и тормозящих сил
£и внесении 'в переменное электрическое поле падающей
электролитной волны, то легко понять, почему энергия поглощается
*еством: потому что энергия растрачивается на преодоление
1ия при возбуждении колебаний пружины. Значительно труд-
400 ЧАСТЬ 4
■Рис. 14.1. Взаимосвязь рассеяния вперед и преломления.
Волна / после прохождения через тонкую пластинку вещества сопровождается рассеянной
вперед волной 5, которая смещена относительно нее по фазе иа 90°. Эту рассеянную
вперед волну можно рассматривать как сумму всех элементарных волн W, рассеянных
частицами в пластинке. Результирующая волиа R образуется прн сложении падающей и
рассеянной вперед воли. Она слегка отстает по фазе, [van Holde К. Е., Physical Biochemistry,
Prentice-Hall. Englewood Cliffs. N. J., 1971, p. 187.]
нее понять, почему падающая волна замедляется, и ответ на этот
вопрос несколько выходит за рамки обсуждаемого материала. Для
этого требуется провести расчет напряженности электрического
поля, возникающего в месте нахождения отдаленного наблюдателя
в результате одновременного когерентного вынужденного
движения равномерно распределенных в нарисованной выше пластине
электронов на пружинках при наличии тормозящих сил.
Оказывается, что результирующее электрическое поле, образованное от всех
этих пружин (в предельном случае очень тонкой бесконечной
пластины) может быть записано очень просто в виде синусоидальной
волны, которая на 90° не совпадает по фазе с падающей волной.
Поэтому, учитывая доводы, подобные приведенным при
обсуждении рис. 13.2, в результате суммирования электрического поля этой
рассеянной волны и падающего (возбуждающего) электрического
поля также должна образовываться синусоидальная волна, фаза
которой в общем случае отличается от фазы падающей волны
(рис. 14.1). Общий эффект состоит в том, что электрическое поле
достигает наблюдателя, находящегося далеко справа на рис. 14.1,
несколько позже, чем поле падающей волны.
Здесь опущен математический аппарат, имеющий отношение
к предыдущим рассуждениям и рисунку, поскольку ои служит
только для нахождения константы пропорциональности в
уравнении (14.10):
л-1= -п \ , -г -о^-, n = n'-in", (14.10)
(<о20 — со2) -f- i[<a 2е0т » » v . /
где шо, «) и [ те же, что и в уравнении (13.16), q — заряд
электрона, N — число молекул в единице объема, ео — диэлектрическая
ГРУЗ НА ПРУЖИНЕ
»■
оницаемость и т — масса электрона. Строго говоря (как мы уз-
[аем из следующего раздела), даже единичный атом может иметь
июго дискретных «собственных» частот (йо, так что на практике
(ля оценки (п—1), согласно уравнению (14.10), необходимо в
Правую часть уравнения включить сумму многих членов,
соответствующих разным ©о.. Как следует из сопоставления уравнений
J14.10) и (13.20), зависимости показателя преломления пг и
молярного коэффициента погашения е (который пропорционален
t") от частоты подобны тем, которые изображены на рис. 13.5
Ьнизу) (см. также рис. 14.4). Происхождение терминов
«поглощение» и «дисперсия» теперь очевидны: п" — пропорционален моляр-
[ому коэффициенту погашения и п' — показателю преломления,
[зменение которого приводит к «дисперсии» (разложению) бело-
b света на цветные компоненты при прохождении через призму.
|6лыпая часть применений показателя преломления или оптиче-
|еой плотности основаны либо на изменениях пг и п" (или чаще е)
jF зависимости от (возбуждающей) частоты падающего света либо
Щ использовании пг или п" для измерения числа центров
расселся в единице объема N (или, чаще, концентрации поглощающих
|олекул).
1оказатель преломления и его применения
ример. Зависимость показателя преломления от концентрации: теневая оптика
При использовании центрифугирования в аналитических целях (см.
зд. 7.Б) желательно иметь возможность управлять процессом седиментации
исромолекул по мере их приближения к дну (внешнему краю) седиментацион-
й ячейки. При последующем рассмотрении рэлеевского рассеяния света будет
Казано, что при низких концентрациях показатель преломления раствора
меняется линейно с его концентрацией:
п =5r /z0 + (дп/д [с]) [с],
(14.11)
> п — показатель преломления раствора, По — показатель преломления раство-
геля и [с] — (молярная) концентрация растворенного вещества. [Поскольку
I
"
~=
Кювет
а
\\с\\п
1 1
\ \
\ 1
1 1
1
\с\ш
а о
йс. 14.2. Поведение фронта плоской волны при прохождении через раствор,
в котором имеется градиент концентраций.
скольку показатель преломления изменяется линейно с концентрацией раствора (а), то
dht выходящей волны имеет форму, показанную на рис. 6. .Стрелки укаэывают
направление нормалей (перпендикуляров) к фронту волны.
402 часть 4
фокусное
расстояние
\7
— о —
Л
1
Фокусное
расстояние
Кювета
Острие
ножа
Интенсивность
изображения
Рис. 14.3. Оптическая система теневого метода («шлирен»-метода) для
определения областей с высоким градиентом концентраций, основанного на линейной
зависимости показателя преломления от концентрации.
Линза Lj фокусирует изображение ячейки на экране, а острие ножа отсекает свет,
проходящий через область с высоким градиентом концентрации. [Setlow R. В., Pollard В. С.,
Molecular Biophysics, Addisgn-Wesley, Reading, Mass., 1962, p. 102.]
правая часть уравнения (14.11) представляет собой первые два члена
разложения п в ряд Тейлора относительно п0, то очевидно, что допущение будет
действительно при (п—«„«CI]. Другими словами, показатель преломления разбавленного
раствора изменяется линейно с изменением концентрации растворенного вещества..
На рис. 14.2 изображено поведение «плоской» электромагнитной волны (т. е. такой
волны, для которой напряженность электрического поля одинакова в любой точке
вдоль пунктирной вертикали) при прохождении через раствор, в котором (как.
и в седимеитационной ячейке) концентрация увеличивается при переходе of
верхней части раствора к нижней. Поскольку волны, которые проходят через
разбавленный раствор, затухают меньше, чем при прохождении через области
с большей концентрацией, то фронт прошедшей волны частично изгибается, как
показано на рис. 14.3.
■Оптическая схема теневого метода («шлирен»-метода), представленная на
рис. 14.3, позволяет простым способом получать теневое изображение зоны
раствора с •большим градиентом концентрации i(t. е. зоны, где концентрация
меняется от одной области к другой), и основана на явлениях, понять которые
помогает рис. 14.2. Получаемое изображение состоит из набора «пиков», подобных
концентрационным пикам прн исследованиях с помощью электрофореза или
хроматографии.
Пример. Зависимость показателя преломления от частоты: фазово-контрастный
микроскоп
Хотя электронный микроскоп (разд. 15.В) позволяет различать тонкие
детали, однако условия высокого вакуума, необходимые для его работы, делают
исследования большинства живых биологических объектов невозможным.
Оптический микроскоп можно использовать для изучения живых тканей, ио
контрастирование обычно основано на различиях в поглощении света объектом и
фоном. К сожалению, как мы увидим из следующего раздела, «собственные»
частоты электронов в веществе щ чаще всего лежат в ультрафиолетовой области,
так что неокрашенные препараты, как правило, дают изображение с плохим
контрастом, поскольку видимый свет (скажем, зеленый свет с длиной волны
около 5.10~5 см), так далек от «резонанса», что поглощение слишком слабо
(рис. 14.4). С другой стороны, показатель преломления (п'—1)^1 существенно
изменяется в гораздо более широком диапазоне частот (рис. 14.4) я, таким
образом, потенциально делает возможным достаточное усиление контраста при
работе с оптическим микроскопом в видимом свете по сравнению с обычным
поглощением.
ГРУЗ НА ПРУЖИНЕ
in'- 1)
Зеленый ^w^-"*""^
свет '
Ультрафиолетовый
свет
ЗРис. 14.4. Схема показывает возможность сильного отклонения показателя
лреломления от единицы, даже когда поглощение пренебрежимо мало для
видимой области (зеленый свет).
Это явление лежит в основе фаэово-коитрастной микроскопии и обусловливает ее
преимущество над обычной микроскопией поглощения (см. текст).
Например, в большей части видимой области спектра (скажем, зеленой
с длиной волны 5-Ю-5 см) бактерия практически не поглощает. Однако та же
•бактерия имеет показатель преломления 1,34, сравнимый с показателем
преломления окружающей среды (показатель преломления воды равен 11,33). Если
толщина бактерии ~10-4 см, то свет, проходящий через бактерию, будет зату-
<;хать сильнее, чем свет, проходящий через среду, и разность оптического пути
-между двумя лучами составит 10-4-(1,34—1,33) = 10-в см. Для
рассматриваемого зеленого света эта разность равна 10—3/(5-10-5) =0,02 длины волны. В фа-
Гзово-контрастном микроскопе свет, проходящий через образец, складывается
^с опорным лучом, путь которого установлен так, что имеется смещение по фазе
|на 180° (т. е. разность пути точно равна половине длины волны) между светом,
^проходящим через фон, и светом опорного луча. Свет, проходящий через фон,
1 таким образом, полностью гаснтся в результате деструктивной интерференции.
^Однако свет, проходящий через объект, например через рассматриваемую выше
бактерию, проходит, путь, длина которого не точно отличается на половину
длины волны от опорного луча и потому не полностью уничтожается опорным
;-лучом. Вследствие этого объекты при фазово-контрастном микроскопировании
.выглядят ярко освещенными на темном фоне. Преимуществом этого метода
■является отсутствие необходимости в окрашивании препаратов, что позволяет
снизить возможность артефактов изображения.
Рис. 14.4 позволяет объяснить, почему невозможно сконструировать
рентгеновские микроскопы. Поскольку показатель преломления мало отличается от
«единицы при очень высоких частотах (рентгеновские лучи) или очень низких
частотах по сравнению с «собственными» частотами электронов в молекулах,
расположенными в ультрафиолетовой области, то не существует в природе
вещества, которое могло бы быть использовано для изготовления линз,
обладающих способностью преломлять (изгибать) рентгеновские лучи, а без линз, нет
микроскопов. Вот почему взаимодействие рентгеновского излучения с веществом
'поддается изучению только косвенными методами, путем изучения рассеяния, а
не прямыми методами микроскопии (см. разд. 15.Б и 22.А).
Поглощение света и его применения: ультрафиолетовая
я видимая области спектра
Пример. Типичные спектры макромолекул в растворе в видимой и
ультрафиолетовой областях
На рис. 14.5 изображена зависимость поглощения от частоты для некоторых
веществ, представляющих биологический интерес. Точное теоретическое
определение значений резонансных частот ©о невозможно по двум причинам. Во-первых,
Рентгеновское
излучение
404 ЧАСТЬ 4
Триптофан
ю5
ю4
in3
10'
1П
-
—
- —
6
I
Аденин \ \\
1 \ \ Гуанин
\ \
Цитозин \ •
I I ! !
200 220 240 260 280 300
220 240 260 280 309
1000
300
100
30
10
6
Пурины а пиримидины
JHK
Пептидные
связи
Ароматические
аминокислоты
бычий сывороточны *
члббумин
160
200
240
\,н/и
280
Рис. 14.5. Спектры поглощения в ультрафиолетовой и видимой областях.
а — типичные аминокислоты; б — типичные основания нуклеиновых кислот; в — типичный
белок (бычий сывороточный альбумин). [Setlow R. В., Pollard E. С, Molecular Biophysics,
Addi3on-Wesley, Reading, MaS3., 1962, pp. 225—226.]
за исключением очень маленьких молекул, существующая теория ие может
предсказать эти частоты с необходимой точностью. Во-вторых, так как многие пики
поглощения относительно широки по сравнению с расстояниями между пиками
даже для простых молекул, то разрешение спектров не очень хорошее. (Во
многих случаях можно грубо предсказать максимумы поглощения путем
использования простой квантовомеханической модели электрона, заключенного в
«потенциальный ящик», размеры которого примерно равны размерам молекул.) Для
наших целей достаточно отметить, что наиболее значительные по высоте пики
поглощения (е>2000 или около того) в основном обусловлены ионами
металлов, двойными связями или системами сопряженньис связей в молекулах.
Большинство нуклеиновых кислот (рис. 14.5, а) обнаруживают характеристическое
405 груз на пружине
поглощение при 260 нм, а для белков, содержащих ароматические
аминокислоты, типично поглощение при 280 нм. При длинах волн <200 им сильно
поглощают почти все молекулы, так что для их идентификации спектры поглощения
в этой области дают мало информации. Более того, так как при этих длинах
волн особенно сильно поглощают вода и кислород, то эксперименты должны
лроводиться на сухих объектах и в вакууме, что также снижает практический
интерес к этой области спектра. Фактически все применения ультрафиолетовой
спектроскопии для ■ изучения макромолекул основаны на небольших различиях
в поглощении, вызванных изменением либо формы молекул (а следовательно,
изменением среды, окружающей электрон, подвешенный на пружине), либо
внешней среды (замена растворителя), либо изменением части среды,
окружающей отдельный электрон, подвешенный на пружине, при связывании некоторых
маленьких молекул с макромолекулой, что будет продемонстрировано на
последующих примерах.
Пример. Дифференциальные ультрафиолетовые спектры и изменения в конфор-
мацнях макромолекул
Рис. 14.6, а иллюстрирует часто встречающиеся наиболее существенные
изменения в ультрафиолетовых спектрах поглощения макромолекул (в данном
случае синтетического полимера поли-Ь-глутаминовой кислоты) при переходе из
одного конформационного состояния в другое. Результаты позволяют
предположить, что можно проследить за конформационными переходами, количественно
оценивая различия в ультрафиолетовых спектрах поглощения раствора,
содержащего вещество только в одном конформационном состоянии, и раствора,
содержащего смесь двух конформеров. Такой пример представлен на рис. 14.6,6"
для другой макромолекулы — рибонуклеазы. При изменении физического
состояния фермента рибонуклеазы зависимость изменения поглощения от температуры
представляет собой S-образную кривую, характерную для кооперативных
процессов того же типа, с которыми мы встречались при рассмотрении кинетики
аллостерических ферментов в разд. Ш.Б.2. Такое кооперативное поведение
характеризует большинство фазовых переходов, связанных с плавлением или кипе-
Длина волны, нм
180 190 200 210 220 230240 250
I I
—
—/
-4
i !
I I I I
/рН 8,оу
/рН 4,9\
Спираль
, \ ■■ ; i !
i | i | i | ч i
беспорядочные/
■^ клубок
yPGA
Д —
I I Mb^J
5,6 5,4 5,2 5,0 4,8 4,6 4,4 4,2
Частота
-0,1 —
10
20 ■ 30 40 50 60 70
Температура, °С
80
Рис. 14.6. Влияние конформации макромолекулы на спектры.
:— ультрафиолетовый спектр поглощения поли-Ь-глутаминовой кислоты (PGA) в формах
лиралн (рН 4,9) и беспорядочного клубка (рН 8,0); б—разность оптических плотностей
\g (/•//)! при 287 нм раствора рибонуклеазы при различных температурах и относительно
«трольного раствора, имеющего то же значение рН (6,83), но находящегося при более ииз-
й температуре (<43°С). [a— Tinoco /., Jr., Halpern A., Simpson W. Т., In: Polyamlno Acids,
•lypeptides and Proteins ed. M. A. Stahmann, Univ. of Wisconsin Press, Madison, Wisconsin.
1962 p. 147; 6—Hermans /., Jr., Scheraga H, A., J. Amer. Chem. Soc, 83, 3283 (1961).]
406 ЧАСТЬ 4
вием: если процесс уже начался, ои продолжается и скорость процесса
увеличивается по мере повышения температуры. Фазовые переходы уже разбирались
в разд. 2.А; предполагалось, что они обусловлены текучестью биологических
мембран (и, таким образом, скоростью транспорта через них).
Пример. Дифференциальные ультрафиолетовые спектры и фрагменты
ароматических аминокислот в белковых структурах-
Одно из самых удачных применений ультрафиолетовой спектроскопии
основано на том, что изменение растворителя [обычно замена воды иа 20%-ный (по
объему) раствор этиленгликоля] приводит к небольшому смещению (как пра-
Триптофан
>уч
J L
Тирозин _
/А
Де
J_-L
400
200
0
-200
Де
Де
240 260 280 300 320
X, нм
0,06
0,04
РЭ 0.02
0
-0.02
б
1 '
i
~\ 1
1 ' 1
Лизоцим
1 < 1 1
1
1 -
250
270 290
310
Рис. 14.7. Происхождение и образование дифференциальных ультрафиолетовых
спектров.
а — разность Де=8г— ei между нормальным (е^ и смещенным (е2) путем смены
растворителя ультрафиолетовыми спектрами (когда смещение мало, дифференциальные спектры
имеют приблизительно ту же форму, что и график наклона кривой обычного спектра в
зависимости от длины волны) {Donovan J. W. et al, J. Amer. Chem. Soc, 83, 2686 (1961)];
б — примеры дифференциальных спектров триптофана и тирозина-в присутствии н
отсутствии 20%-ного (по объему) этилеигликоля; так как замена растворителя вызывает
небольшое увеличение (а также смещение) е, то отрицательная часть дифференциального спектра
смещается вверх по оси ординат [Donovan J. W., Physical Principles and Techniques of
Protein Chemistry, ed. S- J. Leach, Academic Pre3S, New York. 1969, Part A, p. 1251; а —
дифференциальный спектр лизоцима в присутствии и в отсутствие гликолевого субстрата
хитина \Hayasht К. et al., J. Biochem. (Tokyo), 64, 381 (1963)].
407 ГРУЗ НА ПРУЖИНЕ
вило, в сторону более длинных волн) полос поглощения, связанных с
ароматическими аминокислотами. Из рис. 14.7,а видно, что, когда смещение невелико,
то дифференциальный спектр, полученный путем вычитания исходного спектра
из смещенного, напоминает кривую зависимости величины наклона исходной
кривой поглощения от длины волиы. Это можно увидеть, сопоставив
дифференциальные спектры триптофана и тирозина при смещении под действием
растворителя (рис. 14.7,5) « исходные спектры (рис, 14.5,а) тех же аминокислот
в том же диапазоне длин воли. Поскольку смещение полос поглощения должно
иметь место только для тех остатков ароматических аминокислот, которые
доступны для растворителя, то дифференциальные спектры при смене растворителя
позволяют определить число «экспонированных наружу» (в противоположность
«спрятанным внутри») ароматических аминокислот в белке, находящимся в
растворе. Одни из наиболее убедительных примеров получения информации такого
рода представлен на рис. 14.7.S, где изображен дифференциальный спектр лизо-
цима в присутствии и отсутствии субстрата. Ясно видно, что этот спектр
напоминает разность между спектром триптофана, смещенным под действием
растворителя, и спектром свободного триптофана (рис. 14.7,5). Путем сопоставления
дифференциальных спектров лизоцима с заменой растворителя в присутствии и
отсутствии субстрата было показано, что в присутствии субстрата триптофан
перестает быть доступным для растворителя. Несколько лет спустя изучение
=лизоцима в присутствии и в отсутствие субстрата с помощью рентгеноструктур-
яого анализа позволило обнаружить, что активный центр фермента имеет вид
;расщелнны, в которой триптофановый остаток (№ 62 на полипептидной цепи)
расположен на дне, и что этот триптофан закрывается при присоединении
^субстрата. Так как определить последовательность аминокислот белка гораздо
йроще, чем его третичную структуру, то понятно, насколько важно выяснить,
какие нз аминокислот находятся на поверхности, с тем чтобы ограничить число
возможных и отличающихся друг от друга способов складывания полипептид-
йых цепей для образования третичной структуры.
;
{Пример. Изобестическая точка в ультрафиолетовых спектрах: доказательство
существования химического равновесия между двумя состояниями
.
Для определения механизма любого биологического процесса необходимо
ыяснить число ступеней или стадий, через которые проходит реакция. Когда
еакции достаточно медленны, то информацию о них можно получить с по-
щыо различных модификаций метода «скачка», описанных в разд. 16.А.1.
лее простой и доступный метод основан на исследовании «изобестической»
очки в видимой и . ультрафиолетовой областях спектра. Предположим, что
'[1ехтр поглощения молекулы, находящейся в одном химическом состоянии,
ересекается со спектром поглощения той же молекулы в другом химическом
^стоянии (например, крайние кривые на рис. 14.8 для двух химически разли-
йющихся состояний дыхательного белка беспозвоночных гемеритрина). Нет
Иего необычного в том, что у двух различных объектов могут оказаться
^инаковые оптические плотности при одной или нескольких частотах. Предпо-
ожив, что обе формы молекулы находятся в динамическом равновесии, изу-
аем ее оптические спектры. Если все спектры продолжают пересекаться в од-
рй точке при определенной частоте, то такая точка называется изобестической
i. ее обнаружение свидетельствует о том, что молекула может существовать
только двух отличающихся друг от друга формах. Другими словами, хотя
рзможно, что обе формы одной молекулы могут иметь одно и то же поглоще-
!ие при определенных частотах, но маловероятно, чтобы одинаковое поглощение
|>и одной частоте имели три или более форм молекулы.
шмер. Поглощение в инфракрасной области спектра и химическая связь
Можно считать, что поглощение в ультрафиолетовой и видимой областях
стра возникает при действии внешней силы, вызывающей колебания электро-
относительно атомного или молекулярного остова. Поглощение в иифракрас-
408 ЧАСТЬ 4
300 350 400 450 500 550
Рис. 14.8. Спектры поглощения в ультрафиолетовой и видимой областях геме-
рнтрина при различных степенях замещения лиганда ОН" лигандом NCS~.
Изобестические точки указаны стрелками; их существование позволяет предположить, что
процесс может быть описай как равновесие двух состояний. (Любезно предоставлено
профессором А. Адднсоном, Университет Британской Колумбии.) -.
ной области спектра определяется вынужденными колебаниями атомов
относительно остальной части молекулы. К счастью, хотя атомы в молекулах могут
колебаться множеством разных способов, часто удается, изучая ИК-поглощение,
выделить среди этих колебаний специфические, связанные с колебаниями одной
функциональной химической группы, такой, как карбонильная или гидрокоиль-
иая (более подробное обсуждение см. в разд. 17.Б). Поскольку упругость
«пружины» для данной химической связи (а следовательно, и резонансная частота
поглощения) зависит от електроотрицательностей, участвующих в связи атомов,
то можно ожидать существования целого ряда частот в инфракрасной области
поглощения, соответствующих, к примеру, карбонильным группам в различных
типах молекул. Наиболее важные применения инфракрасной спектроскопии
основаны на том, что вид спектра является как бы «отпечатком пальцев» молекулы.
В связи с широким разнообразием числа и типов химических связей почти
никогда две молекулы не имеют идентичных инфракрасных спектров
поглощения, и поэтому ИК-спектры чрезвычайно удобны для опознавания незнакомых
молекул или для сравнения предполагаемой структуры с известной (примеры см.
в разд. И7.Б).
Пример обычного способа эмпирического использования инфракрасных
спектров представлен в табл. 14.1. Частота колебания, связанного с
растягиванием кислород-кислородной связи (валентное колебание О—О), прямо зависит
от прочности этой связи. При этом молекула 02, иоиы «иадперекиси» Оа
D9 ГРУЗ НА ПРУЖИНЕ
Таблица 14.1
Частоты валентных колебаний связей О—О
в некоторых кислородсодержащих соединениях
Соединение
Частота, см—1
о2
Н202
Гемоглобин :02
Гемеритрин-.Од
1555а
1107а
878
1107
844а
Эти значения получены из спектров комбинационного
рассеяния, а ие прямо из ИК-спектров (см. разд. 17.В).
[Caughey W. S. et al., The biological Role of Porphyrins and
Related Substances, Annals New York Acad. Sci., 244, 1
(1975).]
перекиси Ol~ обнаруживают значительно отличающиеся друг от друга частоты
колебаний при 1555, 1107 и 878 см-1 (\Jc=lJK). При сравнении этих чисел
с частотой колебаний кислорода, связанного с гемоглобином i(H07 см-1),
становится ясно, что кислород в гемоглобине ведет себя (спектроскопически), как
1700 1660 1620
см"
1700 1660 1620
см''
Рис. 14.9. Прямое доказательство существования водородных связей в
комплементарных основаниях полинуклертидов.
Все кривые представляют собой зависимость инфракрасного поглощения (оптической
плотности) от частоты в буферных растворах DjO прн pD 7,6.
а — поли-U (0.68 М); б — полн-А (0,62 М); в — эквнмолярная смесь (по 0,33 М) Na2UMP
и Na2(AMP); г — поли-А + поли-U (0,62 М). 1 — наблюдаемый спектр смеси; 2 — сумма
спектров двух индивидуальных компонентов. [Miles H. Т., Biochim. Biophys. Acta, 30, 324
(1958),]
410 ЧАСТЬ 4
ион иадперекиси 01. В противоположность этому кислород, связанный с
дыхательным- белком беспозвоночных — гемеритрином, имеет частоту колебаний при
844 см-1, которая ближе к частоте колебания пероксидного иона.
Пример. Инфракрасное поглощение и водородная связь
Поскольку образование водородной связи приводит к изменению прочности
химических связей, в которых участвует связанный с водородом атом, то можно
ожидать изменения частот колебаний этих связей, что проявляется в виде
смещения «пиков» инфракрасного поглощения, соответствующих этим колебаниям.
К наиболее важным водородным связям, встречающимся в природе, относятся
водородные связи, стабилизирующие конформацию двойной спирали нуклеиновых
кислот. Прямым доказательством существования водородных связей в двойной
спирали являются спектры, представленные на рнс. 14.9. Хотя-уридинмонофос-
фат (UMP) и аденозинмонофосфат (AMP) не соединяются попарно в растворе и,,
таким образом, не обнаруживают водородных связей, смесь поли-А и поли-U
имеет инфракрасный спектр, значительно отличающийся от суммы
индивидуальных спектров поглощения каждого из этих двух веществ. Отнесение полос в
инфракрасном спектре к определенным колебаниям карбонильных групп и колец
показало, что взаимодействие между цепями осуществляется скорее с помощью
группы С^О, чем через группу Сг=0 в основаниях урацила.
14.Б. ДИХРОИЗМ И ДВУЛУЧЕПРЕЛОМЛЕНИЕ:
ВЫЯВЛЕНИЕ ЛИНЕЙНОГО ПОРЯДКА В МОЛЕКУЛЯРНЫХ
СТРУКТУРАХ
В рассматриваемых ранее примерах с электроном,
подвешенным на пружине, предполагалось, что каждый электрон связан с
пружиной только одного типа. Однако интересный класс явлений
основан на более реальном случае, изображенном схематически на
рис. 14.10. Пусть каждый-электрон связан несколькими
пружинами, обладающими различными свойствами в зависимости от
направления. Например, если при освещении одной из этих систем
плоскополяризованным в плоскости yz излучением (рис. .14.11)
возбуждение электрона распространится по пружине,
направленной вдоль оси z, то при освещении светом, плоскополяризованным
в плоскости ху, возбужденный электрон будет растягивать
пружину вдоль оси х. Поскольку принято, что две перпендикулярные
пружины имеют разные упругости, то поглощение и показатель
преломления света, плоскополяризованного в двух
перпендикулярных направлениях, будут различаться при любой из заданных
частот. Вышеизложенное соображение справедливо только тогда,
когда большая часть подобных молекул каким-то образом
выстраивается так, что их «сильные» (или «слабые») пружины
направлены в одну и ту же сторону. Так, для молекул беспорядочно
ориентированных в растворе, количество сильных пружин,
направленных вдоль оси z и вдоль оси х, одинаково, и поэтому не будет
различий в поглощении и показателе преломления для света, плоско-
поляризованного в двух перпендикулярных направлениях
(рис. 14.10,fl). Дихроизм («два цвета») основан на различиях в
Ш ГРУЗ НА ПРУЖИНЕ
««
А
(чЛ
-OJ
<4>)и
(л)
(л)
(<*><л
("о)ц
ei
1щ,У
'о"-
■ со
(о)0\
-из
/±
(О)0)|
-(О
(ь>0к
Дихроизм = *-ц - ех
а
-О)
<Г„-£|
J\
(О
б
Рис. 14.10. Типы оптического поглощения света, плоскополяризоваиного в
любом из двух взаимноперпендикулярных направлений.
а — идентичное поглощение света, плоскополяризованного в любых направлениях
(изотропное поглощение)—дихроизм отсутствует; б — электроны, которым задано движение в
различных направлениях, связаны с пружинками, имеющими различную жесткость
[(Шо) ||=?ЧШо)j_ 1; в—электроны, которьТм задано движение в различных направлениях,
затормаживаются под действием разных тормозящих сил (f « =jbf j_); г — электроны, которым
задало движение в различных направлениях, имеют различные величины максимального
смещения. Анизотропные случаи (б, в, г или любые их взаимные комбинации) приводят
к дихроизму, отличному от нуля. Явления, изображенные на схемах б н в, можно понять
на основе классической механики, а явление, изображенное на схеме г, требует кванто-
вомеханнческого рассмотрения.
Рис. 14.11. Схема, объясняющая одну из причин дихроизма и двулучепрелом-
ления (рис. 14.10,5).
Предполагается, что электроны в оптически анизотропном веществе связаны пружинками,
обладающими разной упругостью в различных направлениях. Свет, плоскополяризоваиный
в направлении одной нз пружин, поглощается и замедляется по-иному, чем свет, плоско-
поляризованный в направлении пружины другого типа, (на рис. 14.10, б обратите внимание
На различия в поглощении для обеих плоскополяризоваиных компонент после прохождения
через вещество). Различие в поглощении называется дихроизмом, а связанное с ним
различие в показателе преломления — дву лучепреломлением.
412 ЧАСТЬ 4
Рис. 14.12. Дихроизм
инфракрасного поглощения для
ориентированной пленки поли-Ь-аланииа.
/ — электрический вектор плоскополя-
ризованного падающего света
перпендикулярен направлению ориентации
осей волокон; 2 — электрический
вектор параллелен направлению осей
волокон. а-Форма — а-спнральная
конфигурация, полипептида, 0-форма— 0-
растянутая структура складчатого
листа. [Elliot A, Proc. Roy. Soc. A226,
408 (1954).]
3200
3300
3400 см~'
поглощении (или коэффициенте погашения) для лучей света,
плоскополяризованных в двух взаимно перпендикулярных
направлениях и перпендикулярно по отношению к направлению падения
луча. Если поглощение при данной частоте различно в зависимости
от направления, то вещество будет менять окраску в зависимости
от направления поляризации плоскополяризованного света
(отсюда термин «дихроизм»). Двулучепреломление связано с различием
показателей преломления для лучей света, плоскополяризованных
в двух взаимно перпендикулярных направлениях. "Для применения
этих явлений необходимо создать частичный порядок в
расположении таких оптически анизотропных молекул. С их помощью
также можно определять уже существующий порядок. Способы
упорядочивания молекул включают в себя ориентацию с
использованием тонких пленок (в капиллярном потоке) при наложении
электрического поля, что иллюстрируется следующими примерами.
Пример. Дихроизм в инфракрасной области и конфигурация полипептидов
Данные, приведенные на рис. 14.12, показывают, что для синтетического
полипептида поли-Ь-аланина, свет, плоскополяризованиый в одном направлении
относительно оси ориентации волокон, поглощается иначе, чем свет,
поляризованный в плоскости, перпендикулярной первой. Особенно важно, что знак этих
различий меняется на обратный при переходе из а-спирали в |3-конфигурацию
складчатого листа (разъяснение этих терминов см. в части 6), поэтому
инфракрасный дихроизм может быть использован для идентификации этих двух
основных полипептидных конфигураций.
Пример. Дихроизм в потоке в ультрафиолетовой и видимой областях и иитер-
каляция красителя в ДНК
Если форма молекул отличается от сферической, то их ориентацию в
растворе можно упорядочить просто путем создания ламинарного потока (см.
разд. 7.В.1). При этом молекулы, имеющие удлиненную (палочкообразную)
413 ГРУЗ НА ПРУЖИНЕ
а
I
I
0,6
0,4
0,3
п
6
1 ] ■ 1 1 ]
/ \ 2
/ *s 1
/ ' м
г 1 \\
1 Ч
1 \ у
\\ у
1
л
1 V
1
—*—
—
1
0,3
— 0,2
— 0.1
<!
I
Ч
200
300
400
Л,нм
500
600
Рис. 14.13.
а —■ Схема прибора для измерения дихроизма в потоке. Источник света L светит на
образец, помещенный в 0,5-миллнметровую щель G между зафиксированными внешним
цилиндром С н внутренним цилиндром, вращающимся со скоростью Ю00 об/мин. Свет
проходит через плоскостной поляризатор Р и попадает на детектор интенсивности. На
переднем плане изображен остов акридинового кольца, йнтеркалнрованный между соседними
поворотами двойной спирали ДНК, где молекулы ДНК были ориентированы с помощью
ламинарного потока; б—спектр поглощения (1) н дифференциальный спектр дихроизма в
потоке (2) для комплекса ДНК " с акрнднновым оранжевым (АО). Концентрация ДНК
1,5-10—а моль/л и АО 1,87'I0-* моль/л. ДНК и АО растворяли в 0,001 М фосфатном буфере,
содержащем 0,0001 моль/л ЭДТА и 0,0025 моль/л NaCl {pH 7,0). При этих условиях более
чем 99,6% катионов АО связаны с ДНК и соотношение нуклеотидов к связанному
красителю равно примерно 8. [Takesada H., Saito Б., Fujita И., Suzuki К., Wada A., Bull. Chem.
Soc. Japan., 43, 18] (1970).]
414 ЧАСТЬ 4
форму, проявляют тенденцию выстраиваться своими длинными осями вдоль
потока. Ранее уже рассматривался ламинарный поток в капилляре; для
оптических измерений более удобно создавать ламинарный поток в узкой щели
(рис. 14ЛЗ,а) между зафиксированными и вращающимися концентрическими
цилиндрами. Затем, измеряя поглощение света, ллоскополяризованного вдоль
или перпендикулярно направлению потока, можно определить параллельно или
перпендикулярно к длинной оси исследуемой молекулы ориентирована
группировка, которая обусловливает поглощение. Например, на рис. 14.13,6 показан
результат измерения индуцированного потоком дихроизма для красителя
акридинового оранжевого, связанного с ДНК. Акридиновый оранжевый представляет
собой плоскую ароматическую молекулу, в которой я-электроны свободно,
двигаются в плоскости ароматического кольца, так что наблюдаемое увеличение
поглощения в направлении, перпендикулярном направлению потока (и, таким
образом, перпендикулярном длинной оси молекулы ДНК) свидетельствует о том,
что акридиновый оранжевый соединяется с ДНК так, что плоскость акридиновых
колец перпендикулярна оси спирали ДНК. Другими словами, плоскость
молекулы акридина параллельна плоскости оснований ДНК. Можно предположить, что
мутагены и антибиотики, как и акридиновый оранжевый, могут нарушать
транскрипцию путем «интеркалйрования» (т. е. «протискивания») между соседними
.основаниями в спирали ДНК. Дальнейшие доказательства существования этого
явления можно получить из измерений вязкости, которые свидетельствуют о том,
что ДНК/становится длиннее после связывания с акридиновым оранжевым.
Пример. Двулучепреломление и, расположение палочек во внешних сегментах
клеток сетчатки ' • ' «
Вещества, способные к двулучепреломленню,. характеризуются разными
показателями преломления света, поляризованного в различных плоскостях. Для
разложения иеполяризоваиного света на два плоскополяризоваиных
компонента, распространяющихся в разных направлениях, могут быть использованы
двулучепреломляющйе кристаллы кальцита, которые представляют собой
удобное средство для получения и анализа поляризованного света; (Конструкция
нового и более дешевого «поляроида» основана на сильном дихроизме,
обнаруживающемся, когда дихроичные молекулы, такие, как молекулярный йод, ориен-
13
и
о
а
l_ 7
jT
5
3
400 600 600 700
Алана волны, н/и
Рис. 14.14. Спектры двулучепреломления и дихроизма для адаптированных
к темноте палочек внешнего сегмента сетчатки лягушки.
Кривая днхронзма свидетельствует о том, что имеется значительно большее поглощение
света, поляризованного перпендикулярно, по сравнению со светом, поляризованным
параллельно осям палочек сетчатки (таким образом, свет поглощается интенсивнее, когда он
поляризован в плоскости параллельной, а не перпендикулярной к мембране). [Liebman P. А.,
/agger W. S., Kaplan M. W., Bargoot f. <?., Nature, 261, 31 (1974).]
1 \ ~
Двулучепреломление
Дихроизм
7.5„
о
—«
2.5" 1=
-4J
0,0 '
J I ! 1
415 ГРУЗ НА ПРУЖИНЕ
тируются в поливинильнои или других полимерных пленках.) Биологические-
липидиые двухслойные мембраны, в которых углеводородные цепи
преимущественно упорядочены перпендикулярно к плоскости мембраны, обладают хорошо-
выраженным дихроизмом (и, таким образом, двулучепреломлением, см,
рис. 14-14), когда много подобных мембран сложены в стопку друг иа друга,
как это имеет место в палочковых клетках внешнего сегмента сетчатки глаза.
Важно распознать, возникает ли двулучепреломлеиие в результате
упорядоченности молекул, которые сами по себе обладают двулучепреломлением
(«внутреннее» двулучепреломлеиие) или в результате необычного взаимного
расположения молекул '(типа сферических бусин на нитке), которые в отдельности
оптически изотропны («структурное» двулучепреломлеиие). В случае лалочек
внешнего сегмента знак структурного двулучепреломления, соответствующего стопке
многих слоев мембран, противоположен знаку наблюдаемого в
действительности общего двулучепреломления, что говорит о преобладании внутреннего
двулучепреломления, возникающего в результате оптической анизотропии самих
липидных молекул. Дихроизм и двулучепреломлеиие палочек внешних сегментов
иллюстрируются рис. 14.14. Недавно было сделано интересное наблюдение,
которое показало, что немедленно после облучения палочковых клеток сильным
светом наблюдаемое двулучепреломлеиие изменяется на 1%. Интересно, что
в палочках внешних сегментов на одну молекулу родопсина имеется — ШО фос^
фолипидных молекул; таким образом, это небольшое изменение
двулучепреломления может соответствовать переориентации одного фосфолипида при
поглощении света родопсином. В любом случае измерения двулучепреломления
являются, по всей видимости, чрезвычайно чувствительными к небольшим изменениям,
в расположении молекул.
14.В. КРУГОВОЙ ДИХРОИЗМ И ОПТИЧЕСКОЕ ВРАЩЕНИЕ:
ОПТИЧЕСКАЯ АКТИВНОСТЬ И «ХИРАЛЬНОСТЬ» МОЛЕКУЛ
В двух предыдущих разделах поглощение и показатель
преломления объясняются в предположении, что электроны в
веществе подвешены на пружинках и приводятся в движение силами,
возникающими при действии переменного электрического поля,
создаваемого падающим электромагнитным излучением. Дихроизм
и двулучепреломлеиие вытекают из различий в поглощении и
показателе преломления для света, плоскополяризованного в двух
взаимноперпендикулярных направлениях. В настоящем разделе
постараемся объяснить круговой дихроизм и дисперсию
оптического вращения («круговое двулучепреломлеиие»), используя ту
же терминологию и уже знакомые уравнения. Затем рассмотрим,,
каким образом естественная оптическая активность может быть
использована для установления спиральной структуры
макромолекул и как могла возникнуть существующая в настоящее время R
природе сильная асимметрия («хиральность») биологических
молекул.
Чтобы дать простое описание этого явления, удобно
представить плоскополяризованный свет как (векторную) сумму двух
вращающихся в противоположных направлениях компонент,
каждая из которых характеризуется постоянной скоростью (рис. 14.15).
При вхождении в «оптически активное» вещество эти две
поляризованные по кругу компоненты (см. рис. 13.3) могут поглощаться
в разной степени (круговой дихроизм) и могут замедляться в раз-
416 ЧАСТЬ 4
ной степени (оптическое вращение). При наблюдениях за светом,
выходящим из вещества, с помощью поляризатора, который
пропускает только свет, плоскополяризованный в одном
направлении, обнаруживается, что свет имеет максимальную интенсивность
яри повороте поляризатора на угол Ф относительно
первоначального направления. На первый взгляд может показаться, что
единственный эффект состоит в том, что плоскость плоскополяризован-
ного света поворачивается на угол * («оптическое вращение»)*.
Однако если затем измерить интенсивность выходящего света в
направлении, перпендикулярном направлению максимальной
интенсивности, то наблюдается, как правило, отличная от нуля
интенсивность, так что выходящий свет в действительности
эллиптически поляризован, поскольку одна из компонент
электрического поля в данном случае меньше другой. Так как различие в
показателе преломления (или поглощения) между лево- и правопо-
ляризованным по кругу излучением весьма мало, то
экспериментальные определения кругового дихроизма EL—Er основаны на
измерениях «эллиптичности» G, а кругового двулучепреломления
nL—nk) — на измерениях «оптического вращения» Ф, что
суммировано в уравнениях (14.11) и (14.12) (см. задачи в конце главы).
* Оптическое вращение обычно характеризуют одной из следующих величин:
^ = (я'лев — «'прав) яД (радиан/см)
п'лев И П'прав— (14. 11 а)
соответствующие показатели
«преломления для луча, поляризованного
по кругу влево и вправо, h — длина
волны
■5=(* (I) (радиан)
«=180 б/я (град)
/ (см)—длина пути
(14.116)
(Н.Пв)
Га] = 10-' a/d[C]0 (град-см2-г-') =
—Удельное вращение
[С]а (г/см3) —концентрация, (14. 11 г)
d (см)—длина оптического пути
1'(*1 = 10001аДГс1(град-см2-моль-1) =
=Молярное вращение
[с] (моль/л) —концентрация
(14. Пд)
Подобно же для эллиптичности [8] =
= 3298 (еЛев — епРав)=Моляриая
эллиптичность-10 (град-см2-моль_1) =
= 100t|>//[C]
еЛев и Бправ — молярные (14.12а)
коэффициенты погашения для лево-
и правополяризованного света, ч&> =
-1800
——-—, [С] (моль/л)—концентрация
(14. 126)
417 ГРУЗ НА ПРУЖИНЕ
х-
2
1
1
1
Плоскополяризованныи
луч можно разложить
на две противоположно
вращающиеся
компоненты ELuEK
Компоненты ££ а с~я при
рекомбинации дают
эллиптически
поляризованный свет
Компоненты £L и ER при
. происхождении через
оптически активное
l вещество замедляются
l и поглощаются в различ-
I ной степени-.
Рис. 14.15. Схематическое изображение, объясняющее поворот на угол Ф плоско-
поляризоваииого падающего света при прохождении через оптически активное
вещество (оптическое вращение) и также тот факт, что выходящий луч
становится слегка эллиптически поляризованным, что количественно характеризуют
эллиптичностью 6.
Качественное объяснение механизма кругового дихроизма
и оптического вращения для оптически активных молекул
На рис. 14.15 изображена полная феноменологическая картина, которая
разъясняет явление кругового дихроизма (КД) и оптического вращения,,
однако, пользуясь этим рисунком, нельзя понять, почему «правая» молекула
неодинаково взаимодействует со светом, поляризованным по кругу вправо и
влево. Качественное объяснение представлено на рис. 14.16.
На ряс 14.16 плоскополяризоваиное электрическое поле раскладывается на
две поляризованные по кругу компоненты (показаны на рисунке). Читатель
должен сам проследить, каким образом две схемы, изображенные внизу, прямо
получаются из соответствующих схем иад ними. Изображено также магнитное
поле #2, связанное с компонентой электрического поля Е2. Электрические поля
£i и Е2 заставляют электроны двигаться или вверх и вниз, или внутрь и наружу-
относительно плоскости рисунка. Магнитное поле Нъ вынуждает электроны
двигаться по круговому пути в плоскости, перпендикулярной основной оси спирали.
Однако поскольку электроны вынуждены двигаться по спиральному пути, то>
действие Я2 заставляет электроны двигаться вверх и вниз по спирали, что
аналогично действию £|. Поскольку сила, возникающая под действием #а,
определяется производной от #2 по времени я в связи с тем, что производная от
синусоидальной волны дает ту же самую функцию, смещенную на 90°,. то силы от Ё\
И #2 Для света, поляризоваииого по кругу вправо, будут действовать в фазе
(т. е. всегда в одном и том же направлении). В то же время силы от Ех и Нх
418 ЧАСТЬ 4
Рис. 14.16. Подробное изображение разложения плоскополяризованного света
на сумму двух поляризованных по кругу компонент.
Влияние электрического и магнитного полей на каждую из компонент системы, состоящей
нз электронов, которые вынуждены двигаться по спиральному пути, обсуждается в тексте.
для света, поляризованного по кругу влево, будут смещены по фазе на 1180°
(т. е. всегда направлены в разные стороны). Таким образом, электроны в
спирали будут подвергаться действию разных сил в зависимости от того, поляризован
ли свет по кругу вправо или влево, и поэтому следует ожидать различий в
поглощении и показателе преломления для этих двух типов излучения.
Читатель должен понимать, что подобного же рода объяснения можно
использовать для доказательства того, что при взаимодействии поляризованных
по кругу вправо и влево компонент со спиралью, закрученной влево, а не
вправо, эффект должен быть равным по величине, но противоположным по знаку.
419 груз на пружине
Внимательное рассмотрение показывает, что вышеупомянутые рассуждения
основаны на предположении о способности электрона двигаться одновременно
вверх-вниз и в стороны. Например, в случае плоской молекулы электроны
вынуждены двигаться в ее плоскости, так что индуцированные электрические и
магнитные дипольиые моменты находятся под прямым углом друг к другу и
вышеприведенные аргументы не применимы. И действительно, плоские молекулы
не проявляют оптической активности. Может показаться, что рассуждения
относительно спирали становятся несостоятельными при перевертывании спирали
«вверх ногами», но на самом деле правая спираль всегда отличается от левой
вне зависимости от угла зрения. Согласно общему химическому критерию, для
того чтобы молекула проявляла оптическую активность, достаточно, чтобы она
содержала «асимметрический» атом углерода, т. е. атом углерода с четырьмя
разными заместителями*.
Зависимость кругового дихроизма и оптического вращения
от частоты
Согласно рис. 14.15, КД возникает в результате различий в
двух типах поглощения, а оптическое вращение (ОВ) в результате
различий в двух типах показателей преломления. Поэтому можно
ожидать, что зависимость этих двух явлений от частоты будет
похожа на зависимость от частоты обычных поглощений и
показателя преломления, что и показано на рис. 14.17. Зависимость
оптического вращения от частоты называется дисперсией оптического
вращения (ДОВ). Скоро станет ясно (разд. 15.А), что
«приближение Друде» имеет такое же отношение к кривой ДОВ, какое
имеет приближение «рассеяния» или «отсутствия торможения» к
колебаниям электрона на пружине, совпадающим по фазе с
синусоидальной возбуждающей силой (см. разд. 13.Б.1). Ясно, что
уравнение Друде теряет смысл вблизи от резонанса, так как при
приближении к резонансным частотам начинает преобладать
компонента, смещенная по фазе на 90° (в этом случае КД). Подобно
поглощению и показателю преломления, КД и ДОВ являются
двумя проявлениями одного и того же свойства молекул и дают
одну и ту же информацию (т. е. резонансную частоту и константу
«затухания», связанную с шириной спектральной линии). ДОВ
была исторически раньше замечена и описана, но сравнение двух
самых нижних кривых на рис. 13.5 и 14.4 вновь свидетельствует
о том, что эффект поглощения в КД проявляется в более узком
диапазоне частот, чем «дисперсия» в ДОВ, так что КД дает более
четкие данные, на которые меньше влияет наложение «поглоще-
* Однако наличие асимметрического атома углерода не является
необходимым условием появления оптической активности. Оптическую активность
проявляет любая молекула, зеркальное изображение которой не совмещается с ее
первоначальным изображением. Необходимым условием для этого является
отсутствие у молекулы таких собственных элементов симметрии, как центр
инверсии и плоскость зеркального отражения. Такие молекулы, обладающие
«молекулярной асимметрией», называют хиральными. Примером могут служить
биологические макромолекулы, которые могут существовать в форме правой или
левой спирали. — Прим. ред.
420 ЧАСТЬ 4
Кривая Друде
(рассеяние,
приближение в отсутствие
торможения)
Рис. 14.17. Зависимости кругового
дихроизма ('КД) и оптического
вращения (ОВ) от частоты,
полученные как разность между
коэффициентами поглощения или
показателями преломления для света,
поляризованного по кругу вправо
и влево.
Дисперсия оптического вращения
иногда аппроксимируется кривой Друде,
функциональная форма которой
[Ф] =
= const*
1\
кг-г.г0
(14.13)
где Яо — длина волны, соответствующая
«собственным» или «резонансным»
частотам максимального ноглощення.
Приближение кривой Друде к
дисперсии оптического вращения показано
пунктирной линией на нижнем
графике.
ний» в случае двух близко расположенных «собственных» частот
(юо)а и (<оо)в. Единственное оправдание для использования
измерений ДОВ состоит в том, что она распространяется на более
широкий диапазон частот, а это позволяет получить информацию
относительно «резонансов»,. которые лежат за пределами
спектрального диапазона обычно используемых приборов (как правило, в
ультрафиолетовой области). Так как КД и ДОВ возникают
только тогда, когда молекула (или ее часть) «хиральна» (в частном
случае должна иметься спиральная структура), то представляется
удобным использовать получаемые данные для количественного
определения содержания спиральных участков в макромолекуляр-
ной цепи, т. е. для получения информации, которая иначе может
быть получена лишь с помощью рентгеноструктурного анализа.
Это и есть основная цель применения методов кругового
дихроизма и дисперсии оптического вращения в биохимии. Последующие
421 ГРУЗ НА ПРУЖИНЕ
примеры иллюстрируют наиболее типичные случаи используемого
полуэмпирического подхода*.
Пример. Использование измерений КД и ДОВ для определения количества
спиральных структур в макромолекулах
Как следует из данных, представленных на рис. 14.17, КД и ДОВ
наблюдаются при тех же частотах, что и обычное поглощение. Хотя природные
полипептиды и белки имеют центры асимметрии у каждого тетраэдрического
«-углеродного атома, этот атом и связи, в которых ои участвует, не обнаруживают
сильного поглощения в видимой и ультрафиолетовой областях спектра; поэтому
■о
«S
Q
5
У
i
S
Бесгюр ядочн ый
к/tydoK .
190
210 230
Л.нм
250
190 210 230
250
Рис. 14.18. Спектры КД (a) [Greenfield ЛГ., Fasman G. D„ Biochemistry, 8, 4108
11969)] и ДОВ (б) [Greenfield N., Davison В., Fasman G. D.t Biochemistry, 6,
1630 (1967)1.
Поли-Ь-лнзин в трех структурных формах: а-спнраль (рН 11), антнпараллельиая 8-структу-
ра (рН 11, нагревание при 50 °С в течение 12—25 мин, а затем охлаждение до 22 ^С) и
беспорядочный клубок (рН 5).
* Оптическая активность (отличные от нуля круговой дихроизм и дисперсия
Оптического вращения) может быть индуцирована во всех веществах, как «хи-
Ьальиых», так и «нехиральных», путем приложения статического сильного
магнитного поля. «Магнитный» круговой дихроизм—относительно новый метод
Ёз применении к биологическим проблемам, и известно относительно небольшое
Ршсло случаев, где спектры магнитного кругового дихроизма использовали для
выяснения функции макромолекул в зависимости от структуры. Многообещаю-
|цим является тот факт, что спектры магнитного кругового дихроизма для тиро-
1ина ,и триптофана различны, хотя обычно спектры оптического поглощения
Кля этих веществ близки (рис. 14.5а).
Таблица 14.2
Содержание различных структур для некоторых белков, определенное с помощью рентгеноструктурного анализа
и путем сравнения спектров КД и ДОВ нативного белка с теми же спектрами нескольких белков
в трех перечисленных конформациях (а-спираль, р-форма и беспорядочный клубок)
(ср. типичные контрольные спектры на рис. 14.19)а
Рентгеноструктурный анализ вдб ДОВв
беспоря- беспоря- беспоря-
о-спираль, Э-форма, дочный а-спираль, р-форма,, дочный а-спираль. р-форма, • дочный
% % клубок, % % клубок, % % клубок,
% % %
Миоглобин
Лнзоцим
Лактатдегидрогеназа
Папайя
Рибоиуклеаэа
Инсулин
Нуклеаза
Цитохром с
а-Хнмотрипснн
Химотрилсиногеи
А
• 77
29
29
21
19
22—45
24
11—39
9 *
6
0
16
20
5
38
12
15
0
23
55
51
74
43
66-43
61
89—61
77
29
31
21
18
31.
27
27
8
, 9
2
16
6
10
44
18
10
6
10
36
21
55
63
69
38
51
63
67
82
55
77
25
30
22
20
25
27
26
11
8
5
0
8
19
51
7
8
8
0
26
18
75
62
759
29
68
65
66
89
66
\
a [Chen Y. H., Yang J. Т., Martinez H. M., Biochemistry, 11, 4120 (1972).]
Основано иа контрольных значениях между 205 и 240 нм при интервалах 1 нм.
в Основано иа контрольных значениях между 225 и 240 нм при интервалах ! нм.
423 груз на пружине
может показаться, что КД и ДОВ для белков будут проявляться очень слабо.
Однако асимметричная природа а-углеродного атома частично связана с
близлежащими амидными группами, которые поглощают в видимой и
ультрафиолетовой областях, что приводит к значительным КД и ДОВ. Кроме того,
дополнительное усиление оптической активности^ может быть обусловлено вторичной
структурой белка (как а-спираль, так и (5-структура оптически активны,
поскольку они не совмещаются с их зеркальными изображениями). Итак,
оптическая активность обусловлена «точечной» асимметрией отдельных аминокислот,
а также асимметрией «формы» всего пептидного каркаса. На рис. 14.18
представлены спектры КД и ДОВ в дальней ультрафиолетовой области для трех
разных конформаций полипептида — поли-Ь-лизина: а-спираль, |}-антипараллель-
ная структура (см. часть 6) и беспорядочный клубок. Ясно видно, что спектры
КД и ДОВ чрезвычайно чувствительны к изменениям структуры полимера.
Вполне можно предположить, что спектры КД или ДОВ модельного
полипептида, представленные на рис. 14.18, можно использовать для количественного
определения содержания трех встречающихся в природе вторичных структур
(а-спираль, Р-структура и беспорядочный клубок) путем сравнения спектров на
рис. 14.18 со спектрами КД и ДОВ белков в растворе. Приведенные в
табл. 14.2 результаты показывают, что спектры КД, как правило, приводят
к более точным данным, чем ДОВ при сравнении со структурой, установленной
рентгеноструктурным анализом. Этого и следовало ожидать, учитывая, что
полосы в спектрах КД уже и лучше разрешены, чем в спектрах ДОВ.
Возникновение оптической активности в природе
в процессе эволюции
Каждого, кто серьезно задумывался об оптической активности природных
молекул, удивляет, каким образом могло возникнуть такое почти полное
превосходство (химических) изомеров определенной формы относительно других
(наиболее типичным примером служит очень существенное преобладание
L-аминокислот иад D-амииокислотами в большинстве живых организмов). Ответ на
этот вопрос очень сложен: традиционные объяснения основаны на том, что
оптическая активность возникла в результате преимущественного существования
света, поляризованного по кругу, скажем, влево, который пронизывал изначальную
атмосферу (поскольку было показано, что поляризованный по кругу свет
может избирательно разрушать молекулы определенного оптического антипода),
или на основании подобных же эффектов взаимодействия с природными
частицами с определенной круговой поляризацией (оптической активностью), такими,
как нейтрино, мюоны или (5-лучи (электроны, поляризованные по кругу влево).
Хотя нет никаких свидетельств о том, что солнечный свет, падающий на землю,
■был когда-либо в заметной степени поляризован по кругу, недавние
эксперименты показали, что (естественно возникающие при Р-распаде) электроны,
поляризованные по кругу влево, вызывают несколько большее разложение молекул
D-лейцина, чем L-лейцина. [В качестве контроля было показано, что электроны,
искусственно поляризованные по кругу вправо, вызывают слегка большее
разрушение ь-изомера.]
Однако для объяснения происхождения оптической активности совсем не
обязательно привлекать какие:либо внешние воздействия. Недавно были
проведены изящные эксперименты, которые показали возможность спонтанного
появления оптической активности в исходной «рацемической» смеси (т. е. смеси
равных количеств двух оптических антиподов), которая не обладала
первоначальной оптической активностью (рис. 14.19). Чтобы избежать загрязнения
природным оптически активным материалом, который может создать «зародыши»
для преимущественного образования определенного оптического шомера,
использовались молекулы, полученные синтетическим путем. Оптическая активность
кристаллов, полученных из исходной рацемической смеси, -иногда указывает на
значительное преобладание того или другого оптического изомера с
приближенно статистическим (гауссовым) распределением результатов. В дальнейших
424 ЧАСТЬ 4
Оптические R- и S- изоллеры
1,1'-бинафти/1а
30 (—
-240-192-144-96 -48 0 48 96
Удельное вращение осл
144 192 240
Рис. 14.19. Спонтанное расщепление рацемической смеси в кристаллах, которые
при последующем растворении обнаруживают заметную оптическую активность.
Исследовали 200 полиостью рацемических образцов, которые кристаллизовали из расплава
при 150 °С. Статистическая обработка полученных результатов дает плавную кривую
приблизительно гауссовой формы с максимумом при а.£)= + 0,14 (т. е. около нуля) и
стандартным отклонением 86,4. [Pincock R. E„ Perkins R. R., Ma A. S„ Wilson К. R., Science,
174, 1018 (1971).]
опытах, которые здесь ие обсуждаются, показано, что добавление малых
количеств оптически активного вещества может контролировать стереоспецифичность,
т. е. выбор «правого» или «левого», продукта реакции. Эти эксперименты
указывают на возможный механизм образования вещества с преимущественным
содержанием одного из оптических изомеров из ранее оптически неактивной
смеси, а также показывают, как присутствие оптически активной частицы может
привести к преимущественному отбору определенного оптического изомера из
многих других типов молекул.
Наконец, интересно отметить, что в другом недавно выполненном
исследовании обнаружено, что кристаллизация рацемического смешанного иатрий-ам-
монийного ь,о-тартрата приводит к значительному я систематическому
преобладанию кристаллов L-изомера в случае, когда опыты сопровождались излучением
радиоактивного 32Р. Таким образом, возможно, что оптическая активность
возникла спонтанно, хотя преобладание в природе L-фо-рм могло произойти и под
влиянием существующего в природе поляризованного по кругу излучения.
14.Г. ПОГЛОЩЕНИЕ И ДИСПЕРСИЯ ПРИ МАГНИТНОМ
РЕЗОНАНСЕ: ЯДЕРНЫЙ «МЯЧИК» НА «ВЕРЕВОЧКЕ»;
ИСПОЛЬЗОВАНИЕ СПИНОВЫХ МЕТОК ДЛЯ,
ОПРЕДЕЛЕНИЯ ПОДВИЖНОСТИ МОЛЕКУЛЯРНЫХ
СТРУКТУР; СЕЛЕКТИВНЫЙ рН-МЕТР
Свойства электронов и ядер описываются с помощью многих
статических параметров, к числу которых относятся масса, заряд,
четность (свойство, определяющее пространственную симметрию).
Электрон и некоторые ядра (такие, как 4Н, 2D, l3C, 31P, 23Na, 35C1
и др.) обладают дополнительным свойством, известным как
магнитный момент. Ранее уже рассматривалось линейное движение
заряженной частицы под действием электрической компоненты
электромагнитного излучения; теперь рассмотрим круговое
движение магнитной частицы при взаимодействии с магнитной
компонентой электромагнитного излучения. Природу магнитных взаимодей-
425 груз на пружине
ствий удобно представить себе, используя аналогию с движением
подвешенного на веревочке мячика, находящегося под постоянным
действием возбуждающей силы (рис. 14.20).
Как показано на рисунке, магнитный момент Мо, возникающий
при действии статического магнитного поля Но, описывается
уравнением
М. = ХоЯ0, , (14.14)
где %о — магнитная восприимчивость. При отсутствии трения
мячик на веревочке, или магнитный момент, будет прецессировать с
«собственной» частотой и>о, которая зависит от длины веревки
(или от Mq)\ следовательно, получается, что частота магнитной
прецессии о)0 пропорциональна Но:
». = ТЯ.. О4-15)
где у называется гиромагнитным отношением, и значительно
отличается у ядер разных изотопов*.
Важная особенность, которая следует из рисунка, состоит в
том, что магнитная система «линейна», т. е. амплитуда
наблюдаемого отклика (которая является х-компонентой прецессирующего
М0) пропорциональна амплитуде возбуждающей силы по крайней
Рис. 14.20. Иллюстрация магнитной прецессии на модели мячика на веревочке.
Если мячик иа веревочке (крайний слева) подвергается действию постоянной
тангенциальной силы (а силы трения пропорциональны скорости мячика), мячик движется по
установившемуся круговому пути под углом в к вертикали. Группа магнитных ядер или электро-
Н0В ""Cjч>иксиР°ванном магнитном поле проявляет магнитный" момент (т. е. ведет себя как
магнитный стержень), величина которого М0 пропорциональна напряженности
приложенного магнитного поля Я0 [уравнение (14.14)], как показано иа средней схеме. Магнитный
момент можно заставить двигаться по круговому пути подобно мячику иа веревочке при
приложении тангенциальной силы, как показано на схеме крайней справа. (Тангенциальная
сила образуется -путем приложения дополнительных небольших магнитных полей,
колеблющихся в плоскости х — у.) При малых тангенциальных силах обе системы прецессируют
с углом В, а для достаточно малых в амплитуда смещения по оси х (наблюдаемая
экспериментально) пропорциональна (sin в «* в) амплитуде приложенной тангенциальной силы.
Другими словами, система является линейной (отклик пропорционален возбуждающей
силе) и ведет себя как рассмотренный в гл. 13 груз иа пружине в присутствии
возбуждающих и тормозящих сил, однако «собственное» движение является круговым, а не прямее
линейным.
Уравнение (14.15) требует квантовомеханического решения (см. разд. 18.AJ.
426 ЧАСТЬ 4
мере при условиях, когда возбуждающая сила мала и прецессия
происходит при малых углах. Возвращаясь к ранее рассмотренной
модели груза на пружине под действием возбуждающих и
тормозящих сил, можно обнаружить, что в случае магнитной прецессии
имеют место те же особенности. Движение с собственной частотой
(оо при отсутствии трения неопределенно долго продолжается и
точно совпадает по фазе с движением возбудителя; при наличии
трення движение, вообще говоря, отстает от возбудителя и его
можно выразить с помощью компонент, которые вращаются точно
в фазе либо смещены на 90° по фазе (с отставанием на одну
четверть оборота) от движения возбудителя. Таким образом,
единственное существующее различие заключается в том, что
«собственное» движение является круговым, а не линейным, но это
совершенно неважно для рассмотрения. Опуская подробные
алгебраические преобразования, которые во всех случаях близки к
тем, которые выполнялись ранее, можно выразить изменение
магнитного момента Мх вдоль оси х под действием возбуждающего
магнитного поля с амплитудой Hi, вращающегося в том же
направлении, в котором происходит магнитная прецессия, в виде
- Мх— (x'cosarf + X"sinarf)(tfi). (14.16)
где
vf — Хо^О К — (О) Т\
*■ — 2 1 + К-со)2Г2
И
* — 2 1-f (со0-со)г722
и (1/Г2) можно рассматривать как величину трения, которое
стремится вернуть магнитный момент обратно к положению «покоя»
вдоль направления Я0 согласно модели мячика* на веревочке.
В уравнениях (14.17а) и (14.176) можно сразу распознать'лорен-
цеву частотную зависимость (рис. 13.5). х" называется модой
поглощения или v-модой сигнала магнитного резонанса и %' — модой
дисперсии или и~модой сигнала магнитного резонанса.
Резонансные («собственные») частоты различных магнитных ядер и
электрона представлены в табл. 14.3; пики ядерного магнитного
резонанса (ЯМР) лежат в радиочастотном диапазоне, в то время как
пики электронного парамагнитного резонанса (ЭПР)—в
микроволновом диапазоне (при лабораторных исследованиях
используются обычно магнитные поля Но с напряженностями в области
23,5 кГс).
Первая существенная особенность приведенных в табл. 14.3
данных заключается в том, что различия в частотах сигналов маг-
(14,17а)
(14.176)
%Z7 ГРУЗ НА ПРУЖИНЕ
Свойства различных магнитных изотопов
Таблица 14.3
Магнитные
изотопы
Частота магнитного
резонанса Vo (МГц)
для приложенного
магнитного поля в
23,5 кГс
Типичная
ширина линии
поглощения,
Гц
Естественное
содержание
магнитного
изотопа
Типичные диапазоны
значений Vo для
различных веществ,
измеренные в величинах
типичных ширин линий
Щ 100
2D 15,4
"С 25,1
"N 10,1
ifiF 94,1
asNa 26,5
Щ> 40,5
8*С1 9,8
1BBHg 17,9
Свободный 65 749,2
электрон
а Гидратированиый иои.
0,5
2
0,5
0,5
0,5
15а
0,5
15а
0,5
2-10е
0,9998
0,0002
0,01
0,004
1,00
1,00
1,00
0,75
0,17
—
-
2 000
20 000
40 000
40 000
10 (нитроксиды)
нитного резонанса для разных магнитных изотопов намного
превышают ширину сигнала поглощения; поскольку радиочастоты
^ можно легко измерить с точностью до 0,1 Гц, то достаточно
легко различить, к примеру, сигналы магнитного резонанса протонов
|от сигналов 13С. Еще более важным является то, что в любой
молекуле имеются свои малые естественные или индуцированные
^магнитные поля, и эти поля слегка различны для магнитных ядер,
^входящих в состав разных химических групп. Рассмотрим
действие внешнего магнитного поля на я-электроны ароматического
углеродного кольца. Приложенное магнитное поле будет заставлять
•электроны двигаться по кругу в плоскости кольца, создавая при
этом небольшое индуцированное магнитное поле, действующее в
^направлении, противоположном приложенному полю (рис. 14.21).
{Если проследить направление этого магнитного поля за пределами
.ароматического кольца, то становится ясно, что в месте
нахождения протона внешнее и индуцированное поля будут складываться,
протон будет находиться в несколько увеличенном магнитном поле
Г(и поэтому резонировать со слегка большей частотой vo), чем в
[отсутствие ароматического кольца. Несмотря на то что этот
эффект незначителен (всего лишь ~ 0,000007 от напряженности
приложенного магнитного поля Но), сигналы магнитного резонанса
для протонов настолько узки (ширина полосы составляет
~0,00000001 ширины резонансной полосы), что легко удается
отличить протоны, участвующие в разных химических связях или.
428 часть 4
Но Hq Но
Рис. 14.21. Приложение статического внешнего магнитного поля Я0 к
ароматическому углеродному кольцу приводит к циркуляции я-электронов, и этот
электронный ток вызывает появление небольшого направленного в
противоположную сторону (индуцированного) магнитного поля, которое добавляется к
приложенному магнитному полю в месте расположения протона за наружным краем
кольца, как показано справа на рисунке. Протон, таким образом, проявляет
большую частоту магнитного резонанса, чем в случае отсутствия ароматического
кольца.
расположенные вблизи различных химических функциональных
групп. На рис. 14.22 представлен спектр ядерного магнитного
резонанса 13С, на котором различные резонансные частоты
соответствуют различным, химически неэквивалентным атомам углерода
в органической молекуле. Таким образом, ядерный магнитный
резонанс представляет собой метод позволяющий с исключительной
селективностью получать сведения одновременно о разных частях
молекулы. (Ср. рис. 14.22, например, со сравнительно мало
выразительными спектрами в ультрафиолетовой, видимой или
инфракрасной областях, приведенных на рис. 14.5 или 14.9). Однако, как
мы выясним в разд. 19.Б.4, метод магнитного резонанса имеет и
слабую сторону, заключающуюся в его довольно низкой
чувствительности; концентрации образцов обычно должны превышать
10~3 моль/л, в то время как оптические методы часто позволяют
получать достоверную информацию при концентрациях
<10~7 моль/л. В тех случаях, когда количество вещества' (и/или
его растворимость) позволяют применить при исследованиях
метод ядерного магнитного резонанса, этот метод нельзя переоценить.
Гармонические колебания любой пружины, находящейся под
действием возбуждающих и тормозящих сил, характеризуются
достаточно полно с помощью ее резонансной частоты vo (получаемой
из значения возбуждающей частоты при вынужденных
колебаниях) и константы затухания (получаемой из ширины спектральной
линии). Преимущества высокой избирательности спектров ЯМР,
являющихся «отпечатками пальцев» молекул при определении
структуры небольших органических молекул, очевидны; примеры,
приведенные ниже, показывают, что это же свойство может быть
использовано для получения данных относительно отдельных
небольших частей биологических макромолекул.
429 ГРУЗ НА ПРУЖИНЕ
Холестерин
27 18
\~-J
Рис. 14.22. Спектр ядерного магнитного резонанса (ЯМР) углерода-13
/(поглощение в зависимости от частоты облучения) для холестерина. -
Получены и .идентифицированы резонансы от отдельных атомов углерода. Шкала частот
представлена'в миллионных долях резонансной частоты. [Reich H. J. et al., J. Атпег. Chem
Soc, 91, 7445 (1969).]
Пример. Селективный рН-метр: спектры протонного ЯМР рибонуклеазы
Имидазольное кольцо гистидина играет особую роль при функционировании
ферментов, участвующих в деградации белков и нуклеиновых кислот. С
помощью тех же доводов, которые приводятся при обсуждении рис. 14.21, можно
объяснить, почему сигналы протонного ЯМР для протонов кольца гнстидина
появляются при более высокой резонансной частоте (при фиксированной
напряженности магнитного поля) или, как показано на рис. 114.23, в области более
слабых магнитных полей (при фиксированной частоте излучения). Можно
определить все четыре гистидиновых фрагмента фермента рибонуклеазы (РНКаза)
в виде отдельных пиков поглощения, поскольку окружение каждого из четырех
гистидииов значительно отличается друг от друга.
Спектр протонного ЯМР РНКазы, таким образом, позволяет одновременно
и по отдельности определить окружение в четырех разных частях белковой
Иолекулы. Например, поскольку гистидин является единственной аминокислотой,
для которой боковая цепь может выступать в роли кислоты или основания в
430 ЧАСТЬ 4
- 1 1
HIS 119
HIS-12 \
~Г\ i
I I I
HIS-105
/ HIS-48 1
1 1 t
' 1 1 ■
1 РНКаза А
R pH# 3,52
'i Vi
4)
-9,0 -8,0 -7,0 -6,0
H0 , м.д. (относительно контрольного сигнала)
Рис. 14.23. Спектр ЯМР-1Н (Я0=58,75 кГс) для области ароматических
протонов рибонуклеазы А из поджелудочной железы быка.
Идентифицированные пики соответствуют протонам при С-2 гистидинов (HIS) в
положениях 12, 119, 105 и 48 аминокислотной последовательности. рН* относится к показанию
рН-метра дли раствора в D20 (см. текст). {Markley J. L., Accounts Chem. Res., 8, 70
(1975).]
физиологическом диапазоне рН, то можно использовать резонансную частоту
протонного ЯМР (положение пика поглощения) каждого из протонов при атоме
С-2 гистидииа в качестве индикатора при титровании белка во всем диапазоне
рН. Хотя при использовании обычного стеклянного электрода значения рН,
измеренные в D2P, по сравнению с теми же значениями в Н20 различаются при-
<~NH—CH—С—■
сн2 н
/'\'. -У3 +н*
Н
NH—СН—С
сн2 н
\
Г
V
кислота
/
N
з
?2
Н
основание
мерно на 0,4 единицы рН, величины p/d, измеренные при диссоциации дейтеро-
на гистидина в D^O и при диссоциации протона гистидина в Н20, различаются
примерно на столько же (см. задачи в конце раздела). Таким образом, данное
показание рН-метра в D2P соответствует примерно той же степени диссоциации
дейтеронов при атоме С-2 гистидина, что и для протонов при С-2 для того же
отсчета по рН-метру в Н20. Другими словами, буфер следует подобрать так,
чтобы рН-метр давал одинаковые показания в D2O и Н^О, если белок имеет
одинаковую степень диссоциации в обоих растворителях. На рис. 14.24
представлены результаты такого титрования. Очевидно, что для разных гистидинов
величины рКа различны. Еще более интересным является тот факт, что когда,
431 ГРУЗ НА ПРУЖИНЕ
например, His-12 модифицирован химически (что. подтверждается
аминокислотным анализом), то величина рКа для His-119 изменяется, в то время как
рКа Для двух других гистидинов остается без изменения; подобным же образом
при химической модификации His-119, изменяется только рКа для His-12. Эти
результаты позволяют предположить, что в интактном ферменте His-12 и
His-119 расположены близко в пространстве, несмотря иа то что эти два
остатка далеко отстоят друг от друга в аминокислотной последовательности.
Подобного рода информация представляет особую ценность, когда требуется
установить способ складывания полипептидной цепочки в нативном феРменте. Кроме
того, для His-105 р/Св=6,72:£0,02, что соответствует рКа небольшого
модельного трипептида. Эти данные позволяют предположить, что His-105
относительно доступен в растворе, в то время как His-112 и His-119 находятся в особом
внутреннем окружении, которое представляет собой активный центр фермента.
Подобные же эксперименты, проведенные с другими белками, показали, что
для двух гистидинов гемоглобина рКа гистидина могут изменяться от <4,0
(а-литическая протеаза) до 8,1. Химотрипсин и трипсин, например, имеют
значения рК« гистидина 4,5—5,5, т. е. меньше «нормальных» более чем на целую
единицу рН, что почти наверняка отражается на участии гистидина в катализе
при функционировании этих ферментов.
Пример. ЯМР и быстрый обмен: химический усилитель
Рассмотрим молекулу, которая может существовать в одном из двух
состояний (например, небольшая молекула, которая либо существует свободно
в растворе, либо связана с макромолекулой). Далее предположим, что
имеются значительные различия в окружении молекулы в этих, двух состояниях, так
что по крайней мере один из протонных сигналов ЯМР для этой молекулы
меняет свою резонансную частоту при переходе из одного состояния в другое:
Д=(ов—(Од, где (Оа и <1>в — частоты протонного резонанса для двух состояний
этого протона. Наконец, предположим, что молекула может быстро
перескакивать (туда и обратно) из одного состояния в другое. Если проследить за
протоном отдельно взятой молекулы, то можно заметить, что его ядерный
магнитный момент прецессирует с угловой частотой соа до тех пор, пока молекула
находится в состоянии А, а затем с частотой <лв, как только молекула перейдет
в состояние В, и т. д. Если выполняется условие, что «скорость обмена», или
Рис. 14.24. Кривые титрования i(c резонансной частотой протонов при С-2
Гистидинов в качестве индикатора) для различных гистидяиов РНКазы А в
растворе DaO.
*—HIS-105: 2— HIS-119; 3 — HIS-12; 4 — HIS-48. [Meadows D. H. et al., Proc. Natl. Acad.
f ' Scl. U.S.A.. 60, 766 (1968).]
432 ЧАСТЬ 4
а
I Нет обмена
А
Oil
Скорость обмена>>Д
I
^ср=Л^+Л<
Рис. 14.25. Схематическое
изображение спектров ЯМР для
молекулы, которая может
пребывать в двух различных
состояниях А и В.
а ~ между состояниями А и В нет
химического обмена;
б—химический обмен между А и В
происходит быстро по сравнению с
различиями в резонансных частотах
состояний А я В (предел «быстрого
обмена» в ЯМР): наблюдается
средний сигнал, положение
которого зависит от относительной доли
ядер в каждом нз двух состояний.
JA WA ' JBWB
скорость перескока, между двумя состояниями велика по сравнению с
разностью частот прецессирования в этих двух состояниях, то представляется,
разумным, что протонный магнитный момент прецессирует со средней частотой шср:
[Л] с _ [В\
[А] + [В] ' 'в— [А\ + [В\
'Л,
/а=
(14.18а)
(14.186)
где /а и /в показывают относительное содержание молекул в состояниях А и В
по сравнению с общим числом молекул. В итоге, если химический обмен между
состояниями А и В отсутствует, то будут наблюдаться два четких сигнала ЯМР;
при существовании быстрого химического обмена между двумя положениями
будет появляться только один сигнал ЯМР (рис. 14.25). Очевидно, что ЯМР
таким образом дает подход к изучению скоростей химического обмена.
Низкая чувствительность ЯМР уже упоминалась как одно из неудобств
использования этого -метода для изучения разбавленных растворов
макромолекул. Быстрый химический обмен позволяет ловко обойти эту трудность во
многих случаях. Напр-имер, предположим, что два состояния отличаются по частоте
на 100 Гц (вспомним, что частоты ЯМР для одного ядра могут варьировать
в интервале нескольких тысяч герц) н предположим, что одно из состояяий
(скажем, А) имеет в 100 раз более высокую концентрацию, чем другое (В).
Тогда, согласно уравнению (14.18), /а«1 и fB« [В]/[А]==0,01, т. е. можно
ожидать, что усредненный сигнал смещен иа ~1 Гц [уравнение (14.18)] от
сигнала А. Поскольку типичная ширина линии в спектре ЯМР составляет
s^0,5 Гц, то определить подобное смещение частоты нетрудно. Таким образом,
можно получить информацию о состоянии с низкой концентрацией (В),
наблюдая за небольшим изменением частоты ЯМР сильного сигнала от состояния А
с более высокой концентрацией. Так как сигналы протонного ЯМР можно
получить при концентрациях 10~3 моль/л; то вышеприведенные вычислении
показывают, что можно надеяться получить методом ЯМР данные о свойствах
макромолекул при более низких коицеитрациях порядка Ю-? моль/л, что уже, ближе
к реальным концентрациям как с препаративной, так н с физиологической точки
зрения. Одним из наиболее раиних и самых поразительных примеров
применения этого типа вычислений является изучение методом ЯМР связывания
небольших молекул 'ингибиторов сахарной природы с ферментом (лизоцимом),
который разрушает полисахаридиые клеточные стенки (рис. 14.26). Лизоцим —
фермент, разрушающий связи сахар—сахар у полисахаридов клеточных стенок,
433 груз на пружине
49 Гц -Ч
/■Эквимолекулярная
у смесь ас-АГ и fi-АГ
д.дГц
■*- *
Добавлен
лизоцим
\fi-АГ ot-АГ
'Рис. 14.26. Спектры протонного магнитного
резонанса для 0,05 М рацемического АГ
(вверху) и для 0,05 М рацемического АГ
в присутствии 0,003 М лизоцима (внизу).
Оба аномера сахара свяаываются с лизоцнмом
в пределе «быстрого обмена» ЯМР, и
наибольший сдвиг частоты наблюдается для а-аномера
(сигнал ЯМР во всех случаях принадлежит ме-
тнльному протону ацетамидной группы).
Благодаря быстрому обмену можно получить частоты
протонного магнитного резонанса как для сс-АГ,
так и для 0-АГ, связанных с лизоцимом, а
интенсивность сигнала соответствует концентрации
свободного сахара 0.05 моль/л, несмотря иа то
что концентрация фермента в десять раз меньше.
[Raftery M. A., Dahlquist F. W., Chan S. I.,
Parsons S. M., J. Biol. Chem.. 243. 4175 (1968).]
■Частота
.состоит в основном из а-связанных полимеров М-ацетил-О-глюкозамина (АГ),
■изображенного ниже. Для мономеров АГ оба свободных аномера обнаруживают
одинаковую частоту протонного магнитного резонанса (верхний спектр иа
рнс. 114.26). Однако при добавлении лизоцима к раствору происходит быстрый
химический обмен между оАГ « {J-АГ, с одной стороны, и ферментом —с
другой, н резонансные частоты обоих аномеров смещаются (но в разной степени)
прн связывании с ферментом (нижний спектр).
СН2ОН
с-
-о
н
уи
н
Н NH СО СН,
т
а-А Г • R=H Протон, сигнал
тл* АГ . Xi -OU "Некоторого
Ме-а- А/ . К — Urt, выдран для
наблюдения
сн2он
А—
1\ ОН
но\|
н
fi-АГ : R =
Me-fi-АГ : R =
\
-О
\ OR
\
Н /[
У н
NH CO CKj
t
H Протон, сигни л
-,-j. ЯМР которого
*~>*1з выдран для
наблюдения
Используя обычно применяемую прн изучении равновесий, при которых один
из компонентов находится в избытке, форму представления данных (рис. 14.27),
возможно определить константу связывания илн частоту ЯМР для связанных
состояний (относительно частоты ЯМР свободного сахара), откладывая ГПо
в зависимости от обратнбй величины наблюдаемого смещения частоты при
дайной концентрации для ряда экспериментов при постоянной концентрации
фермента [Е]0~и варьировании концентрации ингибитора [1]о (ом. задачи и конце
■Главы). Результаты экспериментов для двух пар ингибиторов представлены иа
рис 14.27.
434 ЧАСТЬ 4
Из спектров ЯМР, представленных на рис. 14.27, следует ряд выводов..
Во-первых, хотя а-АГ и р-АГ имеют близкие константы связывания ~с лизоци-
мом, частота а-аномера при связывании смещается гораздо сильнее (0,68 м. д.
против 0,51 м. д. для Р-аномера). Вполне возможно, что это смещение частоты
возникает из-за близости ацетамиднои метильнои группы к ароматическому
кольцу триптофана № 108 в аминокислотной последовательности лизоцима, так
что ацетамидиая метильная группа а-аномера должна лежать ближе к Try-108,
чем у Р-аномера. (Независимые эксперименты показала, что а-АГ и р-АГ
конкурируют за одно и то же место связывания.) В противоположность этому
смещения частоты при связывания для а-Ме-АГ л р-Ме-АГ одинаковы, несмотря
на то что их константы связывания различны. Из этих данных видно, что как
заместители, так и связывание в аномерных положениях влияют на взанмодей-
стине моносахаридов с лизоцимом, но механизмы этих влияний различны.
Все примеры, приведенные в этом разделе, касаются ядерного
магнитного резонанса, однако из табл. 14.3 следует, что неспарен-
ный электрон также имеет магнитный момент и, таким образом,
должен проявлять магнитный резонанс в приложенном
статическом магнитном поле при возбуждении излучением с частотой,
равной частоте прецессии электрона в микроволновом диапазоне. Как
увидим из гл. 19, интенсивность поглощения при электронном
магнитном резонансе [обычно называемом «электронным
парамагнитным резонансом» (ЭПР) или «электронным спиновым
резонансом» (ЭСР)] на несколько порядков выше, чем для ядерного
магнитного резонанса, поэтому ЭПР можно наблюдать без
затруднений при концентрациях <10~6 моль/л. Другое различие между
ЭПР и ЯМР состоит в способе записи спектров; так как спектры
ЭПР обычно получают при низкочастотной модуляции с малыми
амплитудами (см. разд. 17.В), то наблюдаемую экспериментально
линию поглощения регистрируют в виде первой производной (по
частоте) обычной лоренцовой линии поглощения, приведенной на
рис. 13.5, что показано на рис. 14.28.
0,2 0.4 0,6 0,8
1/Сдвигна6/,
1,0
0,1 0,2 0,3 0,4 0,5 0,6 0,7 0.8 0,9
1/Сдвигна6л
Рнс. 14.27. Зависимости концентрации ингибитора от обратной велячнны сдвига
частоты протонного ЯМР (Гц-1), полученные для связывания а-АГ и Р-АГ
с лизоцимом (слева) или связывания с а-Ме—АГ н Р-Ме—АГ с лизоцимом
(справа).
Пересечение с осью у дает константу диссоциации комплекса фермент — ингибитор, а
наклон пропорционален сдвигу частоты ЯМР для «связанной» формы в комплексе фермент —
ингибитор (см. задачи в конце главы); объяснения см. в тексте. [Данные для а-АГ и р-АГ
из: Dahlqutst F. W., Raftery M. A., Biochemistry, 7, 3269 (1968); данные для а-Ме—АГ и
Э-Ме— АГ из: Raftery M. A., Dahlquist F. W., et at., J. Biol. Chem., 243, 4175 (1968).]
435 груз на пружине
А- •
<^М/2 ~~
CJ-
Рис. 14.28. Лоренцов контур линии спектрального поглощения (слева) и первая
производная лоренцова контура линии (справа).
Спектры ЭПР обычно приводят в виде первой производной контура линии поглощения
(ср. с рис. 13.5).
Другая особенность спектров ЭПР состоит в том, что один не-
спаренный электрон может давать более чем один пик поглощения.
Это явление имеет простое квантовомеханическое объяснение
(разд. 18.А), для которого также имеется классическая аналогия
(разд. 17.А), и здесь более не рассматривается.
Большая часть применений ЭПР в биохимии развилась из
изучения группы веществ, называемых нитроксильными
радикалами RN-»-0, где точка над связью N—О свидетельствует о
присутствии неспаренного электрона, для которого можно наблюдать
поглощение ЭПР. Нитроксильный радикал имеет анизотропные
магнитные свойства, и поэтому энергия поглощения различна в
зависимости от того, ориентирована ли связь N—О параллельно или
Перпендикулярно к приложенному статическому магнитному полю
jf/o. Таким образом, можно определить направление и степень
литейного порядка в твердых веществах, содержащих нитроксиль-
|ше метки, путем наблюдения за разницей в поглощении ЭПР при
различных ориентациях образцов — примерно таким же образом,
|сак мы уже поступали при использовании в тех же целях
дихроизма и двулучепреломления.
£■ Поскольку форма линии ЭПР изменяется при вращении
молекулы, содержащей, скажем, связь N—О, то можно ожидать, что
форма линии ЭП1? будет чувствительной к скорости вращательной
Переориентации нитроксильномеченых («спин-меченых») молекул
в растворе. Исходя из ранее сделанного анализа влияния
быстрого обмена на спектры ЯМР, можно предположить, что при очень
^медленных скоростях вращательного движения (медленных по
уравнению с разностью частот спектральных пиков поглощения
рПР при разных углах между связью N—О и магнитным полем)
^пектр ЭПР должен представлять собой сумму всех индивидуаль-
ых спектров, соответствующих всем возможным молекулярным
жентациям; с другой стороны, для быстрых молекулярных
вращений можно ожидать какого-то усреднения всех возможных по-
>жёний пиков ЭПР и получение более простого спектра. Хотя
436 ЧАСТЬ 4
Жслеришнталбш/е спектры
Теоретические спектрбг
Преде
1ыетрс
дьГетрога
вращения
* 5 F
5j S Й
Р S §
а О ч
* 3-Й.
Лредел
медленного
вращения
Рис. 14.29. Экспериментальные и теоретические спектры ЭПР раствора
ди-(тргг-бутил)нитроксильного производного глицерина для различных
скоростей вращения.
Прн —49 "С нитрокснльный раствор полностью заморожен и спектр представляет собой
сумму отдельных спектров ЭПР, соответствующих многим беспорядочно ориентированным
молекулам. Прн 26,5 "С (более простой) спектр представляет собой результат усреднения,
так как каждая нитроксильная молекула может успеть занять много разных положений
при вращении за время, гораздо более короткое по сравнению со временем, требуемым
для того, чтобы зарегистрировать различие между соответствующими спектральными
частотами ЭПР. [Экспериментальные данные нз: lost P. et al., in: Structure and Function of
Biological Membranes, ed. L. Rothfield, Academic Press, N. Y. (1971) p. 83; теоретические
кривые из Gordon R. G., Messenger Т., Electron Spin Relaxation in Liquids, eds. L. T. Muus,
P. W. Atkins, Plenum Press, N. Y., 1972, pp. 371—372.]
расчеты в этом случае выходят далеко за рамки этой книги (и
являются к тому же несовершенными), но в основных чертах
результаты очевидны из данных на рис. 14.29, показывающих
формы кривой ЭПР для спин-меченых молекул, вращающихся с
различной скоростью в растворе. Эффекты вращательного движения
(особенно вращательной диффузии) рассматриваются далее в
гл. 21 для некоторых случаев, которые несколько проще для
расчетов, чем задачи ЭПР.
1
Пример. Текучесть внутри биологических мембран
Недавно были синтезированы и введены в оболочки эритроцитов (красных
клеток крови, из которых удалили гемоглобин) нитроксильные спии-меченые
аналоги стеариновых кислот (см. структуры ниже). Сравнение двух спектров
ЭПР на рис. 14.30 со спектрами, представленными на рис. 14.29, показывает»
ГРУЗ НА ПРУЖИНЕ
jjto когда нитроксильная метка ближе к полярной головной группе молекуляр-
)й цепи, то метка полностью иммобилизуется, но когда метка расположена
fJLa. другом (неполярном) конце цепи, то наблюдается большая степень
вращательной свободы. Серия таких экспериментов позволила сделать целый ряд
^интересных заключений относительно структуры и функции биологических
мембран. Во-первых, было показано, что в мембране существует относительно
Жесткий порядок от поверхности внутрь мембраны на расстоянии около 8
углеродных атомов цепи. Во-вторых, некоторые лекарства, такие, как обычные
Анестетики (обезболивающие препараты), вызывают увеличение текучести
внутри двухслойной мембраны, тогда как другие вещества, типа ДДТ и холестерина,
вызывают снижение текучести внутри мембраны. Пожалуй, наиболее интересным
квляется то, что ряд функций биологических мембран (например, транспорт
f-ЛЮкозы), как теперь показано, тесно связан с относительной текучестью
Мембраны. Мембраны можно рассматривать как многофазную систему (см.
разд. 2.А.2). При температуре фазового перехода в мембране от «твердой» фазы
[очень затрудненная вращательная прдвижность) к «твердо-жидкой» (когда
отдельные части мембран имеют большую степень вращательной свободы)
наблюдаются значительные изменения в степени трансмембранного транспорта
Раких веществ, ках глюкоза. Фазовый переход можно обнаружить в спектрах
}ПР спин-меченых фосфолипидных двухслойных везикул.
Ьример. Спиновые метки в ЭПР в качестве молекулярного зонда
Карбоангндраза представляет собой фермент, у которого активный центр
расположен в глубокой щели, на дне которой имеется один атом цинка.
Ароматический сульфонамндный ингибитор обладает способностью связываться с
активным центром этого фермента, непосредственно координируясь с атомом
шка. При сопоставлении спектров ЭПР ряда спин-меченых сульфонамидных
эоизводных" (рис. 14.31), которые характеризуются различным расстоянием d
|еста присоединения сульфонамида к ферменту от пирролидинового кольца спи-
" »вой метки, было обнаружено, что вращательное движение нитроксильной
)уппы можно использовать для определения размера активного центра. В пред-
эложенин, что активный центр карбоангидразы, выделенной из эритроцитов
келовека, имеет форму воронки, было найдено, что его глубина составляет 14 А.
О n-Vq \0Н
1—|-Ме
Me
10 Гс
!с. 14.30. Спектры ЭПР для двух спин-меченых производных стеариновой
кислоты, которые включены (по отдельности) в мембраны эритроцитов.
*ектр производного, меченного около карбоксильной группы, напоминает спектр сполн-
^исталла» («застеклованное состояние», «порошок»), характерный для высокоиммобилнзи-
Яанных нитроксильных групп (см. спектры при низких температурах на рис. 14.29). в то
&мя как спектр производного, меченного на другом конце углеводородной цепочки, напо-
«ает спектр интроксила с высокой степенью свободы вращения (ср. со спектрами при
Йкжих температурах на рис. 14.29). [Chignell С. F., in: Aldrichimlca Acta, 7, 1 (1974);
Aldrich Chemical Company, 940 W. St. Paul Ave., Milwaukee, Wisc.l
438 ЧАСТЬ 4
Рнс. 14.31. Схематическое изображение связывания сульфонамидной спиновой
•метки с активным центром фермента карбоангидразы.
По мере увеличения длины «молекулярного зонда» наблюдается увеличение вращательной
подвижности и интрокснльиая часть молекулы начинает выступать над поверхностью
фермента. [Erlich R. H., Starkweather D. К., Chignell С. F., Mol. Pharmacol., 9. 61 (1973).]
Пример. Использование спиновых меток в ЭПР для определения склонности
организма к морфину
Иммунная система млекопитающих с готовностью образует антитела против
макромолекулярных антигенов. Это свойство может быть использовано для
создания антитела против морфина, для чего морфин сначала связывают с
белком — бычьим сывороточным альбумином, который после этого вводят кроликам
(см. структуры, приведенные ниже). Затем синтезируют спин-мечеиое
производное морфина. При связывании спин-меченого морфина с антиморфиновым
антителом наблюдается высокая степень его иммобилизации, отражающаяся в
широкой асимметрической форме спектра ЭПР. При добавления морфина он
замещает спин-меченый морфин и спектр ЭПР быстро переходит в спектр с тремя
узкими симметричными линиями, характерный для раствора этого
низкомолекулярного соединения (верхняя кривая на рис. 14.29). Концентрацию свободного
спин-меченого морфина легко определить по высоте пика поглощения ЭПР в
области низких полей. Таким же способом можно определять концентрации
морфина в любой биологической пробе (например, в крови человека, привыкшего
к героину). Поскольку заменители морфина, такие, как метадон и пропоксифен
{а также наркотики других типов, такие, как барбитураты или амфетамины),
не связываются антнморфиновыми антителами, то метод чрезвычайно
избирателен. Благодаря простоте и дешевизне рн нашел широкое применение*.
N—СН,
R = Н(Морср'ин)
R = —СН2СО—Y£.Pi(Mogqpun,ce*3aH-
тг = рн рл ныи с бычьим
л ^Xla^M сывороточным
альбумином)
| JMe (Спин"
меченый
мор ери и)
Л,
* Более подробно см. Leate R. К., Ullman E. F., Goldstein А., Иеггеп-
berg L. A., Nature New Biol., 236, 93 (1972).
,439 груз на пружине
Пример. Фосфолипндный «переворот» в мембранных везикулах
Ярким примером успехов, достигнутых исключительно благодаря^ методу
ЭПР, служит описанное ниже использование фосфолипидной спиновой метки.
Корнберг и Мак-Конелл приготовили двухслойные везикулы, содержащие фос-
фатндилхолин и его спин-меченый аналог.
СНгОСОССН^мСНз
I „„Me Me
СН3(СН2)14СООСН СНз
сн
2ОРООСН2СН2—N
о-
Проводилось восстановление ннтроксильных групп спни-меченых фосфоля-
пндных молекул, расположенных во внешнем слое везикулы, с помощью
восстановителя (аскорбата), который не обладает способностью проникать в
везикулы. После восстановления происходило значительное уменьшение сигнала
ЭПР, так как восстановленные иитрокснльные группы уже более не являются
парамагнитными (не имеют неспаренного электрона). Строилась зависимость
интенсивности сигнала ЭПР от времени; обнаружилось, что при 30 °С сигнал
уменьшается на половину через 6,5 ч. Ряд контрольных опытов
продемонстрировал, что это снижение вызывается медленным «переворотом» (flip-flop) спин-
меченых фосфоляпидов из внутреннего слоя везикулы (где нитроксильная
группа все еще имеет неспаренный электрон, который, таким образом,
обусловливает сигнал ЭПР) во внешний слой (где нитроксильная группа быстро
восстанавливается аскорбатом из окружающего раствора). Один из возможных
механизмов подобного «переворота» показан на рис. 114.32 (справа), где изображено
Внутренняя
часть
Ш -
Внешняя
часть
внутренняя
часть
Рис. 14.32. Схематическое изображение двухслойной везикулы мембраны (слева),
образованной из спин-меченых (темные кружочки) и обычных (светлые
кружочки) фосфатидилхолинов.
Кружочки изображают полярные «головные группы», а извилистые линии соответствуют
неполярным углеводородным цепочкам. Добавление аскорбата снижает количество
спиновой метки во внешнем слое. Возможный механизм фосфолипндного переворота показан
справа, где внутренние н внешние слон фосфолнпидов одновременно меняют положение.
[Kornberg R. D., McConnell H. M., Biochemistry, 10, 1111 (1971).]
Одновременное движение одной фосфолипидной молекулы из внешнего во
внутренний слой, в то время как другая фосфолипидная молекула движется из
внутреннего слоя везикулы во внешний. Этот эксперимент представляет первое
прямое определение фосфолипндного «переворота». Небезынтересно отметить, что
скорость, с которой фосфолипидная молекула переворачивается туда и сюда
между внутренним и внешним слоем везикулы, достаточно велика, чтобы
объяснить транспорт хлора через подобные везикулы. Можно предположить, что
такой «переворот» объясняет механизм пассивного транспорта некоторых ионов
|Яерез биологические мембраны.
440 ЧАСТЬ 4
14.Д. МОДЕЛИРОВАНИЕ КОЛЕБАНИЙ ПРУЖИНЫ
С ПОМОЩЬЮ ЭЛЕКТРИЧЕСКИХ ЦЕПЕЙ: ЕМКОСТЬ,
СОПРОТИВЛЕНИЕ, ИНДУКТИВНОСТЬ, РЕЗОНАНС
И РЕЛАКСАЦИЯ
Исторически свойства электрических цепей очень скоро стали
понятными, поскольку было установлено, что с математической
точки зрения прохождение электрического тока Ьчень напоминает
поток воды. Электрическое напряжение является аналогом
механического давления. Накопление заряда на двух пластинках
конденсатора приводит к тому, что напряжение V пропорционально
заряду q, подобно тому как накопление воды за плотиной создает
гидростатическое давление, которое пропорционально количеству
воды за плотиной. Поскольку площадь поверхности и
конденсаторов и резервуаров может изменяться, то константа
пропорциональности 1/С (где С — емкость в случае электричества) представляет
собой свойство определенного конденсатора (резервуара):
ч
+
+
+
о
i +
I
I
1
i +
V = qlC
(14-19)
Точно так же, как поток воды, протекающий через трубу,
пропорционален приложенному гидростатическому давлению и
обратно пропорционален сопротивлению трубы, так.и электрический
ток I=dq/dt пропорционален приложенному напряжению и
обратно пропорционален сопротивлению цепи; это соотношение (закон
Ома) принято записывать в следующей форме:
-^-VW
V = RI = Ridqldt)
(14-20)
Математически уравнения (14.19) и (14.20) обнаруживают
близость к ранее рассмотренному уравнению (13.15), описывающему
движение груза на пружине при наличии тормозящих сил (при
условии, что вместо смещения пружины х стоит заряд q, вместо
dx/dt — dq/dt, вместо коэффициента трения F — сопротивление R,
а вместо постоянной упругости пружины k — обратная емкость
1/С). Как ни удивительно, но аналогия имеется еще и между
индуктивностью L и массой для механического движения (табл. 14.4).
Образно можно представить себе катушку индуктивности в виде
441 ГРУЗ НА ПРУЖИНЕ
Таблица 14.4
Сравнение свойств механических и электрических осцилляторов
Осно вные характеристики
Независимая переменная
Зависимая переменная
Инерция
Сопротивление
Жесткость
Резонансная частота
Постоянная времени для
возвращения к равновесию
поели удаления
возбуждающей силы (ЯС-цепь)
Постоянная времени для
LR- или 1Л?С-цепейа
Изменяющийся во времени
возбудитель
а См. разд. 14.Е.
Груз на пружине
Время t
Смещение х
Масса т
Коэффициент треиия f
Коэффициент
упругости к
cl>o = k/m
т-f/ft
-t = 2m/f
F = F0 cos {(ot)
Электрическая цепь
Время t
Заряд q
Индуктивность L
Сопротивление R
(емкость)-1, (L/C)
(o§ = l/LC
x = RC
т = 2 L/#
V = VQ cos {Ы)
f
спирали, которая «сопротивляется» изменениям тока с константой
пропорциональности L:
У = Ш1Ш) = Ш1ц1Мг)
(14-21)
Таким образом, уравнение, описывающее электрическую цепь,
содержащую все три элемента, при условии меняющегося в
зависимости от времени напряжения V(t) полностью аналогично
уравнению, описывающему движение груза на пружине в присутствии
тормозящих сил, при действии на него меняющейся во времени
силы F(t):
я
х
-VW-
W
L{d2qldt2) + R(dq/dt) + (l/CX? = V(t)
m(d2xldt*) + f(dxldt) + kx = F(t)
(14-22)
(13-15)
Поскольку для механических и электрических «движений»
дифференциальные уравнения имеют одинаковый вид, то следует
ожидать, что приложение возбуждающего напряжения, изменяюще-
442 ЧАСТЬ 4
гося по синусоидальному закону, вызовет колебания
электрического заряда, аналогичные колебаниям механической пружины. По
аналогии с уравнением (13.20) можно сразу же написать
решение для электрических колебаний:
9 = Г/I/O— «tl/l-L/ftm (14.23)
f(l/C) — o)2Z.] + //?o)
или
9 =
L (о>20 — а)2) + /7?о>
ш20 = (1/1С),
(14.24)
в котором комплексная система обозначения вводится для
компактности записи. Так же как и в случае механического движения,,
можно разделить смещение зарядов на компоненты, которые
изменяются либо в фазе (дисперсия), либо отличаются по фазе на
90° (поглощение) относи 1ельно возбуждающего синусоидального-
напряжения. Следовательно, электрические цепи могут служить-
удобной моделью всех явлений, рассмотренных в этой главе, а
также для недорогих' лекционных и лабораторных демонстраций,
связанных с контурами линии поглощения и дисперсии. . Далее
рассмотрение этой аналогии продолжается в разд. 16.А.2.
Предел нулевой массы
Особенно интересны предельные случаи уравнения (13.15) и
(14.22), когда масса (индуктивность) стремится к нулю.
Перепишем эти уравнения в следующей форме:
dx ' " Fo ' -' - f " (14.25)
dt
(14.26)
Тогда решения для вынужденных колебаний можно написать в
виде (см. табл. 13.4)
V//////////
f
I
Т
R
-лл/v-
V0 cos (<jr)
х--*
х' cos {(ot) + х" sin i(ot)
1
k 1 + <о2т2
xn = &
(ОТ
[_ (груз de3 массы поуве-
T l шеи к пружине)
k 1 + <о2т2
q = q' cos bit + q" sin (ot
* = СУ'ТТЪ?
я" = cv„
(ОТ
*■ T — RC (Rc-церочка без
индуктивности )
1 + (oh2J
(14-27а>
(14-27d);
(14-27о>
(14-28а>
(14-286)
(14-286)
443 ГРУЗ НА ПРУЖИНЕ
Уравнения (14.28) использованы в разд. 14.Е («Диэлектрическая
релаксация»),, а уравнение (44.27)—в разд. 14.Ж
(Ультразвуковая релаксация»).
Для уточнения следует сказать, что термин «релаксация» (он
постоянно используется в литературе) применяют для описания
опытов двух типов: 1) эксперименты, в которых изучаются
вынужденные синусоидальные колебания груза с нулевой массой,
подвешенного на пружине (например, диэлектрическая и
ультрафиолетовая релаксация) и 2) эксперименты, в которых
изучаются затухающие колебания груза, подвешенного на пружине, в
отсутствие возбуждающих сил при определенной массе груза
(например, ядерная магнитная релаксация, деполяризация
флуоресценции, гамма-лучевая корреляция) или для груза с нулевой
массой (например, релаксационная кинетика химических реакций).
Диэлектрическая и ультразвуковая релаксации рассматриваются
в последующих двух разделах этой главы, явления,
сопровождающие затухающие колебания, обсуждаются в гл. 16.
14.Е. ДИЭЛЕКТРИЧЕСКАЯ РЕЛАКСАЦИЯ:
ГРУЗ С НУЛЕВОЙ МАССОЙ НА ПРУЖИНЕ.
ВРАЩАТЕЛЬНАЯ ДИФФУЗИЯ МАКРОМОЛЕКУЛ
Начинаем с рассмотрения очень простой цепи, состоящей из
конденсатора, к которому приложено статическое напряжение.
Емкость пустого конденсатора (рис. 14.33, слева) обозначена Со.
а 6 6
_1 I 1_
+ + + + + + + + + + + + +
~I I I
с0 с
О Со
Рис. 114. 33. Схема распределения зарядов в конденсаторе, когда он пуст (а)
и когда он заполнен полярными молекулами (б и в).
В конденсаторах а и б-приложенное напряжение одинаково, в конденсаторе о оно выше.
При наложении разности потенциалов полярные молекулы стремятся «выстроиться» между
пластинками и на конденсаторе создается дополнительный заряд &q> который эффективно
нейтрализует часть заряда конденсатора (см. текст). Диэлектрическая проницаемость
определяется согласно уравнению (14.29).
444 ЧАСТЬ 4
Однако если пространство между пластинами конденсатора
заполнено полярными молекулами (т. е. имеющими электрический
дипольный момент), то приложенное к конденсатору напряжение
создает заряд на пластинах, а заряженные пластины образуют
электрическое поле, которое генерирует силу, стремящуюся
выстроить диполи в направлении, соединяющем обе пластины (рис.
14.33, в центре). Поскольку беспорядочное тепловое движение
заставляет молекулы беспорядочно «кувыркаться», то
упорядочивание, которое можно создать при приложении даже очень
сильного напряжения, далеко от совершенства (ср. рис. 14.33 в центре и
справа). Важно отметить следующее: в связи с тем, что полярные
молекулы между пластинами частично ориентируются вдоль поля,
часть заряда, созданного на конденсаторе, эффективно
нейтрализуется упорядоченными диполями. При этом, чтобы общий заряд,
остался таким же, как в случае пустого конденсатора, необходимо»
чтобы на пластинах конденсатора появился дополнительный заряд
(ср. заряд пластины на рис. 14.33 слева и в центре). Один из
способов описания этого явления состоит в том, что говорят об
увеличении эффективной емкости конденсатора, когда между его
пластинами помещают вещество с полярными молекулами. Это
увеличение емкости пропорционально диэлектрической
проницаемости*
*' = С/С„ (14.29)
где С и Со — емкость конденсатора в присутствии и в отсутствие
полярного вещества между пластинами.
Увеличенный заряд в заполненном конденсаторе (по
сравнению с пустым) зависит от квадрата дипольного момента
индивидуальных полярных молекул:
e'__l=W«L, (14.30)
где ро — электрический дипольный момент данной полярной
молекулы, N — число молекул в единице объема, k — постоянная Больц-
мана (отнесенная к 1 молекуле) и Т — абсолютная температура.
Таким образом, прежде всего, исходя из диэлектрической
проницаемости (т. е. отношения емкостей заполненного полярными
молекулами и пустого конденсатора), можно определить
электрический дипольный момент полярных молекул.
* Более точно, относительная диэлектрическая проницаемость Гуравнение
(14.29)] в системе СГСЭ является безразмерной величиной; она принимается
равной единице в вакууме. Абсолютное значение диэлектрической проницаемости
для вакуума 8,84-10~1* Ф/см. Подробности см. в любом учебнике по
электричеству и магнетизму.
U5 ГРУЗ НА ПРУЖИНЕ
л
г
с =
А
Рис. 14.34. Электрические цепи с конденсаторами.
Слева — емкость конденсатора меняется с частотой приложенного и изменяющегося во
времени напряжения; справа — емкость н сопротивление удовлетворяют соотношению CR=%
•(где т — постоянная времени, приблизительно равная среднему времени, требуемому, чтобы
'дипольная молекула в заполненном конденсаторе днффузно реориентировалась на угол
порядка одного радиана). Если приложено переменное электрическое поле (соответствующее
(приложенному переменному напряжению) £„ cos {(Ot), то общее электрическое поле в
заполненном конденсаторе можно выразить как гЕа [где е=е' cos ((at) + г" sin (at), уравнение
;;(14.31)]. е' н е" определяются путем измерения емкости С и сопротивления R цепн
(справа), поскольку диэлектрическая постоянная е'=С/С0, диэлектрические потерн e"=\/RCo(a
Йгде Со—емкость пустого конденсатора и со — (угловая) частота синусоидально изменяю-
I щегося приложенного напряжения].
\-
1 Уравнение (14.30) описывает диэлектрическую проницаемость
£ля приложенного статического напряжения. При приложении
синусоидально изменяющегося во времени напряжения V0cos(u)/)
диполи должны переориентироваться так, чтобы их положительно
Наряженные концы были направлены то к одной, то к другой
пластине конденсатора, поскольку знак приложенного к конденсатору
Напряжения меняется во времени. В этой новой ситуации имеется
два важных-момента. Во-первых, необходимо преодолеть
сопротивление трения при повороте мйлекул, так что альтернативное
математическое описание цепи, содержащей заполненный
конденсатор, к которому приложено напряжение, есть цепь, включающая
конденсатор с фиксированной емкостью и сопротивление
|рис. 14.34). Во-вторых, свойства цепи, содержащей конденсатор и
ропротивление, при приложении напряжения (см. предыдущий
|аздел этой главы), которое может быть описано через смещение
|арядов, движущихся в фазе и смещенных по фазе на 90°
[уравнение (14.28)], уже известны. При таком описании цепи,
«эквивалентной» реальному заполненному конденсатору под напряжением,
Достоянная времени x=RC характеризует среднее время, которое
Необходимо для того, чтобы данная полярная молекула изменила
|вое направление (путем вращательной диффузии) на угол
порядка одного радиана. Экспериментально синфазную и смещенную по
|>азе на 90° компоненты е, а именно г' (диэлектрическую
проницаемость) и е" (диэлектрические потери) в
е = е' COS (<ot) -f- e" sin (<ot)
(14.31)
[соответствующие q' и q" в уравнении (14.28а), согласно уравне-
1иям (14.28 6) и (14.28в) (см. задачи в конце главы)] можно оп-
)еделить путем измерения емкости и электропроводности
Ерис. 14.34).
446 ЧАСТЬ 4
Выражения для е' и е" в уравнении (14.31) обычно
записываются в следующей форме:
О 00
&Г ~ еоо + 1 4-й)2г2
,'г
(*t~«g»)<ar
1 + «>гт2
(14.32а)
(14.326)
где ео и е<» могут быть определены графически (см. рис. 14.35, где
показано поведение полярных макромолекул в водном растворе).
Вновь используя аргумент, основанный на классической
вращательной диффузии (см. разд. 21.Б), можно показать, что «время
релаксации», или постоянная времени x=RC для эквивалентной
цепи, приводящей к уравнению (14.32), дается формулой
(14.33)
где г] — вязкость раствора, а—радиус рассматриваемой полярной
молекулы, к — постоянная Больцмана и Т — абсолютная темпера-
Is
4J
4s
5 6 7 5
и>крит= О/Гмакро)
la о}
9 IO f II
О}крат = (,/Гнго)
Рис. 14.35. Зависимость диэлектрической проницаемости е' (вверху) и
диэлектрических потерь е" (внизу) от частоты (логарифмическая шкала).
Левые части каждой кривой построены с использованием уравнения (14.32) н соответствуют
экспериментам, проведенным иа макромолекулах, растворенных в воде; критическая
частота ©кпит, соответствующая точке перегиба зависимости е' от оо (и максимуму на
зависимости е" от ш), является обратной величиной диэлектрического времени релаксации т„ „„„
необходимого макромолекуле для вращательной переориентации на угол порядка одного
радиана. Критическая частота (не наблюдаемая) н время диэлектрической релаксации для
растворителя (воды) проявляются в области более высоких частот (так как молекулы воды
могут переориентироваться гораздо быстрее, чем макромолекулы), т. е. в правой части
кривых. Опыты обычно проводятся при частотах < ~ 200 Мгц.
447 ГРУЗ НА ПРУЖИНЕ
Рис. 14.36. Теоретические зависимости диэлектрической проницаемости е' от
частоты для удлиненных эллипсоидов с различным отношением ajb (см.
рис. 7.22).
Плавная кривая, полученная для сферы [(а/Ь — 1], начинает сраспадаться» на две сступень-
ки» по мере увеличения отношения а/Ь. Вид зависимости е' от частоты, таким образом,
может служить указанием на асимметрию формы макромолекул. [Oncley /. L., in:
Proteins, Amino Acids, and Peptide, ed. E. J. Conn, J. T. Edsall, Reinhold, New York, 1943,
p. 543.]
тура. Для воды т]=0,01 (в единицах СГСЭ). Считая, что а=2-
•10-8 см, получаем, согласно уравнению (14.33), что время
релаксации для реориентации молекул воды равно т«2,4«10~м с. На
кривой зависимости диэлектрической проницаемости е' от
частоты этому времени релаксации соответствует ступенька при
частоте, равной приблизительно v= (72ят)=6,6- Ю9 Гц; сопротивление
проводов очень велико для столь высоких частот, и поэтому они
лежат за пределами экспериментальных измерений. Так как
диапазон экспериментально определяемых частот обрывается, не
достигнув второй ступеньки на кривой е'—v, то экспериментально
наблюдаемая диэлектрическая проницаемость е' всегда будет
содержать значительный постоянный вклад еоо, что представлено в
явной форме в уравнении (14.32). Однако для такой
макромолекулы, как химотрипсин, имеющий молекулярный радиус ~20 А,
можно ожидать, что т = 2,4«10~8 с, что соответствует частоте
$,6 МГц. Эту область частот несложно наблюдать с помощью
электронных приборов, так что весь диапазон вблизи критической
частоты v=(V2Hx), показанной на рис. 14.35, поддается изучению.
; Время диэлектрической релаксации т удобно использовать для
измерения размеров молекул (уравнение (14.33); т легко
определяется экспериментально с помощью кривых, подобных
изображенным на рис. 14.35. Дальнейшая информация может быть
поручена из кривых типа представленных на рис. 14.36, где
показало теоретически рассчитанное изменение е' в зависимости от
частоты при удлинении макромолекул.
'Пример. Определение размера и формы макромолекул с помощью
диэлектрической релаксации
I В табл. 14.5 приведены значения времен диэлектрической релаксации и
Отношения длин осей молекул, определенные путем сопоставления эксперимен-
448 ЧАСТЬ 4
тальных зависимостей е' от частоты с кривыми типа представленных на
рис. 14.36.
Таблица 14.5
Времена диэлектрической релаксации и осевые отношения для различных белков,
полученные путем сравнения экспериментальных данных с зависимостями типа
показанных на рис. 14.36
Белок
Молекулярная
масса (.103)
т-108, с
alb
Яичный альбумин
Альбумин лошадиной
сыворотки
Лошадиный карбоксигемогло-
.бин
Псевдоглобулин лошадиной
сыворотки
Инсулин
Р-Лактоглобулин
Миоглобин
[Oncley I. Е-, Proteins, Amino Acids
New York. 1943. p. 543.]
45,0
70,0
67,0
142,0
40,0
40,0
17,0
and Peptides, E. I
18; 4,7
36; 7,5
8,4
250; 28
1,6
15; 5,1
2,9
[. Cohn. Г. Т. Edsall
5
6
1,6
9
4
(Eds.), Reinhold,
Пример. Зависимость Коул — Коула для миоглобина
Тщательное изучение рнс. 14.36 позволяет обнаружить, что трудно
определить небольшие отклонения от сферической формы [при (а/&)^5] путем
прямого сопоставления экспериментальных данных с теоретической зависимостью е'
от логарифма частоты. Быстрый расчет (см. задачи в конце главы) показывает,
что одним из свойств величин е" и е' является
[■' - (V.) (*2 +«Л1 + ■'"=К1/») («. - «J]'.
(14.34)
т. е. при условии, что система характеризуется единственным временем
релаксации т, зависимость ,е" от е' имеет вид окружности с радиусом ('/г) (во—е») и
центром на оси х в точке е"=»(72)(во+бо»). Если для системы имеется два или
более разных времен релаксации, кривая по-лрежнему в целом приближается
к окружности, однако центр этой окружности смещен ниже оси г". Построение
этой так называемой зависимости Коул — Коула* оказывается достаточно
чувствительным и удобным методом определения наличия нескольких времен
релаксации, что следует ожидать для молекул, имеющих несфернческую форму.
Пример использования этих кривых представлен на рис. 14.37 для диэлектрической
релаксации водного раствора миоглобина. Хорошее совпадение экспериментально
полученной зависимости е' от логарифма частоты с теоретическим уравнением
[уравнение (114.32)], содержащим одно время релаксации т, подтверждается
* Такой способ представления данных был впервые предложен в работе
Cole К. S., Cole R. И., J. Phys. Chem., 9, 341 (1941). — Прим. ред. -
149 ГРУЗ НА ПРУЖИНЕ
к
41
-о
Е
и
I
$ 88
СУ
§ 86
5"
. в:
5 84
I 82
ъ
S 80
з
78
100
Рис. 14.37. Диэлектрическая дисперсия (т. е. зависимость диэлектрической
проницаемости е' от частоты) для раствора миоглобина.
Кривая вычислена с использованием уравнения (14.32). В верхнем правом углу кривая
Коул —Коула (см. текст). [Takashima S., Physical Principles and Techniques of Protein
Chemistry. Part A, ed. S. J. Leach. Academic Press. New York. 1969. p. 313.J
кривой Коул — Коула, которая имеет вид полуокружности с центром иа оси х.
Этот график представляет возможно наиболее четкое свидетельство того, что
молекула миоглобина в растворе имеет форму, близкую к сферической.
Пример. Зависимость дисперсии от поглощения (в спектроскопии)
Уравнение (14,34) описывает особый случай (предел нулевой массы) общей
зависимости синфазной компоненты смещения от компоненты, сдвинутой по фазе
на 90°, при вынужденных колебаниях груза на пружине под действием
синусоидальной возбуждающей силы и при наличии торможения. В самом деле (см.
задачи в конце главы), кривая зависимости х* от х" [уравнение (13.36)] всегда
будет представлять собой полуокружность для любой спектральной линии,
имеющей лоренцов контур. Хотя такой способ представления данных спектральных
контуров был применен лишь недавно (ядерный магнитный резонанс), однако
возможно, что построение таких кривых удобно как метод «диагностики»
происхождения уширения линий в простых, спектрах. Например, уже отмечалось, что
когда контур возникает в результате суперпозиции многих лоренцовых контуров
с различной шириной линий, то экспериментальные данные, представленные
графически в виде зависимости дисперсии от поглощения, имеют вид кривой
смещенной ниже «контрольной» полуокружности, ожидаемой для одного лоренцова
контура; однако когда контур является результатом суперпозиций многих
лоренцовых контуров, отличающихся резонансной частотой (рис. 14.38), то
построенные зависимости дисперсии от поглощения имеют вид кривой, которая
смещена над «контрольной» полуокружностью.
кГц \МГц \0МГц УОйМГц
Частота ( \а-шкапа)
450 ЧАСТЬ 4
-5,0
-2,5 0,0 2,5
Частота со
0,25 0,5 0,75
Поглощение
1,0
Рис. 14.38. Зависимости нормированного поглощения от частоты (а) и
дисперсии от поглощения ;(б).
Пунктиром даны зависимости для одиночной линии с лоренцовым контуром
[уравнение (13.36)]. Сплошные лнннн представляют собой суперпозицию многих лниий с
лоренцовым контуром, резонансные частоты которых (т. е. положение пиков поглощения)
описываются гауссовым распределением (как для а, так н для б самые внешние кривые
соответствуют самому большому разбросу в частотах ©о). Частота выражена в 1/т j(rfle 1/т—
полуширина отдельной линии поглощения с лоренцовым контуром на середине высоты пнка
в максимуме). Объяснение значения таких кривых см. в тексте. (Mapшелл, неопублнкован-
'> ные результаты.)
14.Ж. УЛЬТРАЗВУКОВОЕ ПОГЛОЩЕНИЕ И ДИСПЕРСИЯ
СКОРОСТИ: ВНОВЬ ГРУЗ С НУЛЕВОЙ МАССОЙ
НА ПРУЖИНЕ,—НОВЫЙ МЕТОД МЕДИЦИНСКОЙ
ДИАГНОСТИКИ И ЛЕЧЕНИЯ
Как указано в разд. 13.А, звуковые волны образуют
продольные возмущения, состоящие из чередования сжатий и разрежений
среды вдоль направления движения волны. Известно*, что такая
система удовлетворяет волновому уравнению; эту аналогию
можно продолжить на такие явления, как зависимость поглощения
звуковой энергии от частоты, отражение и преломление
(рефракция) на границе раздела двух сред, в которых звук
распространяется с разной скоростью (разные показатели преломления для
звука), а также изменение скорости звуковой волны в
зависимости от частоты. В этом разделе сделана попытка раскрыть
механизмы этих явлений и их применение.
В противоположность электромагнитным волнам, которые
могут проникать через пространство даже в вакууме, звуковые
волны передаются посредством молекул. Для достижения сжатия и
разрежения, требуемых для распространения звуковой волны,
молекулы жидкости (наиболее интересный в связи с
рассматриваемыми вопросами случай) должны скользить друг относительно друга
(«сдвигаться»). Жидкость оказывает сопротивление подобному
* Фейнман Р. П., Лейтон Р. Б., Сендс М. Фейнмановские лекции по физике. —
М.: Мир. Т. 1.
Й51 ГРУЗ НА ПРУЖИНЕ
|
сдвигающему усилию в соответствии с величиной сдвига и в
соответствии с внезапностью возникновения этого изменения. Такая
ситуация хорошо описывается уже ставшей здесь обычной моделью
груза, подвешенного на пружине, под действием возбуждающих и
тормозящих сил. Сопротивление пружины растягиванию
соответствует величине смещения (т. е. константа упругости пружины
k=f=0)\ сопротивление вязкой среды, окружающей пружину,
зависит от скорости изменения длины пружины (т. е. коэффициент
трения f¥=b). Но точно так же, как пружина не останется в
вытянутом состоянии, если на одном ее конце не подвешен груз,
жидкость не будет постоянно противостоять сдвигающему давлению.
Поэтому математическое описание отклика жидкости на постоянно
приложенные возбуждающие силы звуковой волны сводится к
задаче о грузе с нулевой массой, подвешенном к пружине,
находящейся под действием возбуждающих и тормозящих сил
[уравнение (13.25)]. Для подобного случая ранее уже получено
выражение для величины смещения х при вынужденных колебаниях:
х = х'cos(wt)-\-x" ът(Ы), (14.27а)
где
*'=T"T+W (14.276)
*"=-tttW <14-27в>
сравнения (14.27 6) и (14.27 в) напоминают соответствующее вы-
ожение для линии с лоренцовым контуром [уравнение (13.36)],
юторый уже хорошо известен по предыдущим обсуждениям дис-
юрсии и поглощения ультрафиолетового, видимого, инфракрасно-
о, микроволнового и радиочастотного электромагнитного
излучений, с тем лишь отличием, что «резонанс» находится теперь при
гулевой возбуждающей частоте, а не там, где ранее были «собст-
енные» частоты пружины с определенной массой. Далее сделана
рпытка найти связь уравнения (14.27) с зависимостью скорости
поглощения звука от частоты в веществе.
Во-первых, звуковую волну можно рассматривать как периоди-
еское изменение давления (или плотности) вдоль направления
вижения. По аналогии с ранее проведенной обработкой оптиче-
кого поглощения и показателя преломления (разд. 14.А), можно
писать мгновенное значение давления в виде действительной
асти комплексного выражения
' Давление = Re (/>) = Re \P0exp limit ~ir)|]> 04.35)
;е t — время, у—расстояние от рассматриваемой точки до ис-
(чника звуковой волны, a v—(«фазовая») скорость (т. е. ско-
452 ЧАСТЬ 4
рость движения «гребня» волны) волны. С целью учета эффекта
поглощения энергии [ср. с ранее проведенным переходом от
уравнения (14.3) к (14.6) ] запишем v в виде комплексной величины
l/t> = (l/o«fcTB)-«(*/«>) (14.36)
где а — «амплитудный коэффициент поглощения». Подставляя
уравнение (14.36) в (14.35) и помня, что энергия на единицу
объема волны пропорциональна квадрату амплитуды (давления),
находим, что
Интенсивность = / = const • Р -Р*
или
/ = /0 ехр [—2ау] (14.37)
Уравнение (14.37) просто свидетельствует о том, что
интенсивность звуковой волны снижается экспоненциально (с
экспоненциальной постоянной 2а), по мере того как волна проходит через
вещество, так как энергия расходуется на то, чтобы заставить
молекулы смещаться одну за другой для достижения периодических
изменений плотности, которые создают волну.
Данные ультразвукового поглощения редко записывают
непосредственно с использованием значений а. Обычная запись
основана на «поглощении на единицу длины волны»:
/ = /0 ехр [—2ау] — /0 ехр [—2аА (у/Х)]
или
/ = /0ехр[-н.(г//Я)]; ц = 2аЛ, (14.38)
ц характеризует уменьшение энергии звуковой волны, когда
гребень волны проходит одну длину волны по направлению
распространения движения. Так как
Я = 2те?двйст?/ш
то получаем
Р = 4тодейства/ш (14.39)
Поскольку скорость звука идейств лишь слегка варьирует (обычно
на <1%; см. рис. 14.39) в широком диапазоне частот, так что для
И можно записать
ft ^= const-(а/ш). (14.40)
Ультразвуковое поглощение обычно выражается либо в виде
(а/ш2) или в виде (i=const- (а/ю) (рис. 14.39). В том же
приближении, которое приводит к уравнению (14.40), можно показать (см.
литературу), что скорость звука идейств и энергия поглощения на
единицу длины волны |i непосредственно связаны с выражениями
453 ГРУЗ НА ПРУЖИНЕ
2 5 10 20 50 100 200 500 1000
v, МГц
Рис. 14.39. Зависимости а/<о2, поглощения на единицу длины волны р, и
относительной скорости ультразвука в среде С/С0 от логарифма частоты звуковой
волны.
Предполагается, что существует один тип процесса торможения (релаксации) с
критической частотой 7криТ =50 МГц. Предельная скорость звука (при v«=0) принята С0—
= 1,2-105 см/с. (Дальнейшее обсуждение см. в тексте.) .[Lamb /., Physical Acoustics, ed.
W. P. Mason, Vol. II-Part A, Academic Press, New York, 1965, p. 208.1
для синфазной (x') и смещенной по фазе на 90° (х") компонент
смещения при вынужденных колебаниях возбуждаемого по
синусоидальному закону груза с нулевой массой на пружине при
наличии сил торможения [уравнения (14.276) и (14.27в)]^
а = £>*" = (?!
С02Т
сот
Ц = Сг(а/со) = С3 1+й>Ч2 ,
а/«)2=С1 -т—;—2~г.
удейств= Wl' L>i'-X ) = Ь4 ( 1 Ce j I Щ2Т2 J
(С0, Сх, С2, С3, С4> С5, С0 — постоянные)
(14.41)
(14.42)
(14.43)
(14.44)
Как следует из кривых а/оД ц и vдейств на рис. 14.39, «время
релаксации» х в уравнениях (14.41) — (14.44) легко найти, так как
оно равно обратной величине «критической» частоты, при которой
' (а/©2) и Удейств имеют величину, среднюю между экстремальными
значениями, а ц — максимально (см. задачи в конце главы).
Критическую частоту можно рассматривать как меру максимальной
454 ЧАСТЬ 4
скорости, с которой вещество может изменять плотность (или, что
равнозначно, свой объем).
Для многих химических реакций (таких, как переход спираль—
клубок, который в дальнейшем обсуждается) происходит
изменение объема при переходе от исходных к конечным продуктам
реакции. Таким образом, можно ожидать, что система
спираль—клубок должна поглощать энергию, по мере того как проходящие
звуковые волны сдвигают химическую реакцию попеременно в
противоположных направлениях в ответ на периодическое изменение
давления, вызываемые звуковой волной. Однако, когда частота
звуковой волны выше, чем константа скорости реакции,
химическая система не успевает «следовать» быстрым изменениям
давления, и потому в случае высокочастотных звуковых волн энергия
поглощается сравнительно слабо. Таким образом, можно
определить константу (константы) скорости для очень быстрых реакций
путем определения соответствующей критической частоты
(частот) по одной из кривых на рис. 14.39. Наконец (также как для
диэлектрической релаксации, рассматриваемой в предыдущем
разделе этой главы), критические частоты, соответствующие
изменениям плотности растворителя (для воды ~0,5-1012 с-1)» настолько
велики, что обычно их нельзя достичь в эксперименте; таким
образом, экспериментальные кривые, например зависимость а/а>2
от о), описывается суммой двух слагаемых [уравнение (14.43)],
где Т2<С1/й). Так что
О)2 0l
1 +G>2t21
+с,
2 1 + G>V
liffl
t2 <. 1/co
(£)-c.
1 + а>*х\
-f- С 2Тг
1+G>!
,2.2
В
(14.45)
(14.46)
Пример. Использование ультразвукового поглощения для определения констант
скоростей быстрых химических реакций: переход спираль — клубок для
поли-1-,-глутаминовой кислоты
На рис. 14.40,а представлена зависимость оптического поглощения от рН
для раствора поли-1*-глутаминовой кислоты (ГШК). По этой кривой можно
определить долю /н полимерных сегментов ПГК, которые имеют структуру
спирали (в противоположность беспорядочному клубку), что обозначено
пунктирной линией иа графике. Видно, что при рН 5,1 ~50% ПГК существует в виде
спирали и 50%—в виде беспорядочного клубка. Ультразвуковое поглощение
в зависимости от частоты для ПГК при рН 5,1 представлены на рис. 14.40,6;
кривая описывается уравнением (14.46) с единственным временем релаксации,
равным ~ 10_в с. Отложим детальное рассмотрение методов вычисления
констант скоростей химических реакций из времен релаксаций до разд. 16.А.1,
однако отметим, что найденная из настоящих данных константа скорости роста
спирали kf описывает процесс.
?f
hhc
или
chh
(14.47)
455 ГРУЗ НА ПРУЖИНЕ
1,14
1,10
1.06
J 1,02
0,98
0,94
0,90
—
—
А205>
4
1 1
ч g
А
/ \
•^«^
1 1
5 6
РН
а
1
—г
_w___
1
7
~~
_ 1
1,0
0,8
°'6/
0.4 Н
0,2
0
Рис.
150
£
t4 100
ъ
n 50
3
Э
0
14.40.
1 III II
—-4^ki
^Х
высокочастотная ^К4
экстраполяция ^v^ t L±
1" t""~I I "i **t
20 50 100 200 500 1000
Частота i>, кГц
6
Xa) — зависимость от рН оптического поглощения прн 205 вм (1) и доли спиральной
конфигурации (2) прн 37 °С, если содержание остатков ПГК составляет З.Э'Ю-* моль/л. (б) —
^типичная зависимость а/©* от частоты ультразвука прн 37 °С, рН 5,11 и 0,03 М NaCl, если
Содержание остатков ПГК составляет 2,7—10—* моль/л [расчет по уравнению (14.46)1.
\Barksdale A. D., Stuehr J. Е., J. Amer. Chem. Soc, 94, 3334 (1972).]
"где h — участок полимера в спиральной конформации (т. е. с водородными
связями), а с — участок в виде беспорядочного клубка.
b Существует сравнительно мало примеров четкого определения конформа-
^ционных изменений макромолекул в растворах ультразвуковым методом,
поскольку для большей части изученных макромолекул свой вклад в поглощение
[ультразвука дают также другие (обычно менее интересные) процессы. Сюда
зтносятся изменения в природе связывания молекул растворителя с
макромолекулой (следует запомнить, что любой процесс, протекающий с изменением
объема, оказывает влияние на ультразвуковое поглощение). Наиболее удачные
|ирнмеры использования ультразвукового излучения в биологии основаны иа облу-
;иии целого образца ткани. Для оценки этих методов необходимо иметь под1
?укой некоторые типичные данные об ультразвуковых свойствах различных тка-
1ей (табл. 14.6).
Ультразвуковые свойства некоторых обычных материалов
Таблица 14.6
Образец
Скорость
распространения ультразвука,
м/с
Коэффициент
ультразвукового поглощения
прн 1 МГц, а8/см
юздух
^ровь
<ир
1ыпщы (среднее значение)
1ода (дистиллированная)
Сости черепа
1гкие ткани человека (среднее зиа-
сие)
330
1570
1450
1590
1480
4080
1540
12
0,2
0,6
2,3
0,002
13
0,8
fslls P. N. Г., Ultrasonics In Clinical Diagnosis, Churchill Livingstone, Edinburgh, 1972,
456 члсть 4
Пример. Ультразвуковая микроскопия
Разрешающая способность любого микроскопа зависит от длины волны
излучения. Поскольку длина волны X, частота v и скорость волны с связаны
соотношением с=м>, ясно, что разрешение тем лучше, чем выше используемая
частота. Так как разные биологические ткани по-разному поглощают
ультразвук, то можно создать ультразвуковой микроскоп, в котором контрастирование
основано иа различиях в поглощении ультразвука разными частями объекта.
Источником ультразвукового излучения является «пьезоэлектрический» кристалл,
который просто механически изменяет свои размеры при воздействии
электрического потенциала. Так как ионные кристаллы состоят из отдельных зарядов, то
представляется логичным, что приложение внешнего электрического поля
должно приводить к деформации кристалла; последний выполняет роль
«преобразователя», превращающего электрические колебания в механические колебания,
которые затем проходят через образец (рис. 14.41).
Детектировать проходящие волны не просто, поскольку непрактично
создавать' многоканальную систему из крошечных пьезокристалличееких детекторов;
вместо этого обычно позволяют прошедшему через образец излучению (после
прохождения через микроскоп) ударяться в пластмассовое зеркало.
Деформацию зеркала под действием ультразвуковых воли можно наблюдать оптическими
методами с высоким пространственным разрешением. В наиболее типичных
биологических тканях длина звуковой волны при частоте 2 ГГц равна ~ 750О А,
что хорошо сопоставимо с длиной волны электромагнитного излучения в
оптическом диапазоне. Результаты исследования свежей кожуры лука с помощью
ультразвукового и оптического микроскопов сопоставлены на рис. 14.42.
Совершенно очевидно, что на изображении (внизу), полученном с помощью
ультразвука, ядра клеток видны значительно более четко (черные точки). Этот метод
относительно нов, и представляется разумным ожидать, что во многих случаях
его применение будет предпочтительнее, чем использование оптической
микроскопии, поскольку он не требует, чтобы объект исследования был окрашен.
Путем геометрических построений, таких же, как в случае
преломления и отражения электромагнитного излучения на
поверхности раздела двух сред, отличающихся по показателю
преломления (т. е. скорости света), можно показать, что относительная
интенсивность ультразвукового излучения, отраженного на границе
Рис. 14.4]. Взаимосвязь силы и распределения электрического заряда в
пьезоэлектрическом преобразователе.
На схеме изображен кварц (днокснд кремния). Стрелки показывают направление
приложенных сил, а результирующие поверхностные заряды указаны на электродах. Обратный
эффект приводит к деформации преобразователя в ответ иа приложенное напряжение.
J57 ГРУЗ НА ПРУЖИНЕ
|»с. 14.42. Микрофотографии свежей кожицы лука, полученные методом
оптического поглощения (а) и ультразвукового поглощения (б).
сустическое изображение получено при частоте излучения 220 МГц. Поле зрения состав^
рет 750 мкм по горизонтали в обоих случаях. Обратите внимание иа ббльшуго контраст-
нггь для ядер клеток в акустическом изображении. [Kessler L. W., Ijv: Ulteflsirtiics Inter-
pjtional 1973; Conference Proceedings, IPC Science arid Technology Press W&SiGuildford,
I England (1973), p. 175.] '^Ш
щежду двумя слоями, отличающимися по плотности pi и р% и
скорости ультразвука С\ и Сг, дается выражением
Отраженная интенсивность (перпендикулярно к поверхности)
Падающая интенсивность
__Г paCg — РхСх
[ ргСг + piCi
(14.48)
Изучение табл. 14.6 позволяет быстро прийти к заключению, что
иожно ожидать очень большого относительного отражения на
поверхности раздела между воздухом и тканью или между тканью
Ёмкостью и даже между водой (например, в наполненной жидко-
Цью кисте) и тканью.
Рис. 14.43. Диагностическая ультразвуковая томография.
а — киста в левой груди (правая грудь нормальная); сканирование проводилось при
погружении в воду на частоте 2 МГц; артефакты, наблюдающиеся по обе стороны изображения
(но больше слева), возникли из-за отражения от поверхности воды [Wells P. N, Т.,
Evans К. Т., Ultrasonics, 6, 220 (1969)]; б — две головки близнецов в матке иа 25-й неделе
беременности (поперечное сечение живота при 2,5 МГц). [Wells P. N., Ultrasonics in
Chemical Diagnosis. Churchill Livingstone, Edinburgh, 1972, p. 83.]
Груз на пружине
V
|}ример. Ультразвуковая томография
С Соиар (эхо-локация) у летучих мышей и дельфинов и ультразвуковая
томография основаны на том, что вперед посылается импульс ультразвука, а затем
регистрируется время (которое является мерой расстояния от источника до
объекта и обратно), затрачиваемое на то, чтобы ультразвуковое эхо, отраженное
Ьт объекта, вернулось к источнику ультразвука, который служит одновременно
а приемником и передатчиком. При достаточно высокой частоте
(соответствующей малым длинам волн) возможно различать относительно тонкие детали
|(дельфины издают звуки с частотами
|о 150 кГц и могут распознавать
объекты размером до 0,2 мм даже тогда,
|ЕОгда их глаза закрыты
специальными чашечками). Регистрация эха от
рдного импульса ультразвука,
позволяет получить одномерное изображе-
|&ие границ тканей, расположенных на
Вазных расстояниях от передатчика,
аписывая несколько подобных одно-
|ерных изображений для нескольких
юложений передатчика при его ли-
|ейном движении вдоль поверхности
•бъекта (см. разд. 22.В.1, где опяса-
io подобное устройство, основанное
\а рассеянии рентгеновских лучей),
; затем кодируя полученные сигналы
|ля передачи на развертывающее
устройство экрана телевизора, где ин-
нсивность отраженной волны преоб-
зуется в яркость телевизионного
бражения в зависимости от места
ражения звука, создается возмож-
€ть для получения изображений
перечного сечения биологического
ъекта. Слово «томография» проис-
ит от греческих корней, означаю-
х «резать» (поперек) и «записы-
ь» (регистрировать).
Ультразвуковая томография в
дицинской диагностике особенно
[обна для изучения мягких тканей
гкие, почки, мозг и др.), где изо-
жение при рентгенографическом
ении малоконтрастно. Вырази*
ршиый пример для оценки этого ме-
а представлен на рис. 14.43,а, где
азано изображение, полученное
сканировании тканей груди чело-
Ы. ГПоскольку произведение плот-
и на скорость распространения
а в воздухе и коже различаются
ительно, а в воде и коже лишь
ачительно [уравнение (14.48)1, то
ходимо погрузить объект в воду,
того, чтобы снизить отражение
гразвуковых воли сразу после
доения ими объекта.] Хотя клиии-
ая пальпация груди позволяет
Рис. 14.44. Дифференциальная
диагностика с использованием ультразвуковой
томографии.
а — нормальная грудь человека (поперечное
сечение); б —киста в груди; в —
фиброаденома (опухоль) груди. Все эхо-граммы
выполнены с использованием ультразвукового
излучения с частотой 5 МГц. [Wagai T in-
Ultrasonics in Medicine, ed. M. de Vlieger
D. N. White, V. R. McReady, Excepts Medica-
American Eisevier Pub. Company, Inc New
York, 1974, p. 188.]
460 ЧАСТЬ А
определять наличие заболевания относительно просто, но не так уж просто
решить, вызвано ли оно доброкачественной, либо злокачественной опухолью или
кистой. Поскольку все три рассматриваемых типа ткани различаются между собой
по плотности (скорости звука), а также отличаются от нормальной ткани груди,
то часто удается диагностировать тип поражения ткани путем изучения
двухмерного изображения поперечного сечеиия, полученного с помощью ультразвуковой
томографии (рис. 14.44).
Другой областью, где использование ультразвуковых изображений очень
полезно, является наблюдение за расположением и состоянием здоровья
эмбрионов в матке, а также обнаружение близнецов (рис. 14.43,6). Все это имеет
особое значение в частности ненадолго до родов, когда важно правильно определить
расположение младенца.
Пример. Разнообразные применения ультразвука в биологии
Если объект, рассматриваемый с помощью ультразвукового отражения,
движется навстречу или прочь От передатчика, то будет наблюдаться сдвиг
Доплера Av в измеренной частоте v возвращающейся волны:
Lv — \\\±{vjc)\, (.14.49)
где v и с — скорость объекта и испускаемой волиы соответственно. Эффект
Доплера использован для изучения тока крови и движения митрального
клапана сердца при диагиосцнровании его функции, В связи со значительным
поглощением энергии ультразвука-костью и фиброзной и нервной тканями по
сравнению с другими окружающими тканями ультразвуковые передатчики высокой
интенсивности были использованы в качестве хирургических ножей при тонких
операциях, в которых механические манипуляции с применением обычных
режущих инструментов затруднительны (например, при операциях на среднем ухе,
глазе, гипофизе). Более того, из-за относительно низкого поглощения
ультразвука кровеносными сосудами "ультразвуковые волны оказывают лишь
незначительное разрушающее действие на них, что в значительной степени сокращает
постоперационное заживление раиы и во многих случаях сокращает число
осложнений. Наиболее распространенное применение ультразвуковое излучение
нашло при мытье и стерилизации посуды и инструментов. Очищение происходит
в следствие «кавитации» (внезапного образования и разрушения «пузыреподоб-
йых» пустот), по мере того как молекулы растворителя взаимно растаскиваются
дру> от друга, а затем вновь сталкиваются вместе под действием «давления» от
ультразвуковых волн. Наибольший эффект достигается при низких частотах
(20—40 кГц), что достаточно для разрушения бактерий и для удаления твердых
веществ ^Поверхностей (особенно когда поверхности трудно доступны для
других спЩ^ббв очистки).
Задачи
1. Плоскополяризованная монохроматическая световая волиа двигается вдоль
оси у
[ ГРУЗ НА ПРУЖИНЕ
Предполагается, что эта волиа проходит через двулучепреломляющее
вещество, толщина которого такова, что компонента Ех отстает по фазе ровно на
90* ('А длины волны) от компоненты Ег. Покажите, что выходящий луч
поляризован по кругу. Далее предполагается, что Ех отстает по фазе на 180°
('/г длины волны). Покажите, что выходящий луч теперь плоскополяризоваи,
но в плоскости, обращенной на 90° от первоначальной плоскости поляризации.
Отсюда следует, что один из способов наблюдения двулучепреломлеиия
заключается в определении интенсивности света, прошедшего через
скрещенные поляризаторы (т. е. через два фильтра, каждый из которых пропускает
свет, поляризованный только в одном направлении, при условии, что одно
из направлений зафиксировано под углом 90° относительно другого) при
этом двулучепреЛомляющее вещество расположено между поляризаторами.
Если вещество ие обладает двулучепреломлением, то свет не будет проходить;
если вещество обладает двулучепреломлением, то для части плоскополярнзо-
ваниого света, прошедшего через первый поляризатор, при достижении
второго поляризатора плоскость поляризации окажется повернутой и некоторое
количество света пройдет через второй поляризатор.
Разберите следующий кажущийся парадокс. Свет не проходит через
скрещенные поляризаторы. Однако, если поместить между ними третий поляризатор,
пропускающий свет, плоскость поляризации которого повернута, скажем, на
30° относительно первого поляризатора, то некоторое количество света будет
теперь проходить через второй поляризатор. Создастся впечатление, будто свет
появился без использования источника. Дайте объяснение.
Круговое двулучепреломлеиие и круговой дихроизм обычно измеряют
экспериментально в виде «оптического вращения» и «эллиптичности». Смысл этих
терминов помогает понять диаграмма, изображающая зависимость
напряженности -электрического поля от времени для плоскополяризованного света после
того, как он прошел через оптически активную среду.
Ф — оптическое вращение,
6 — эллиптичность
Ь
jfc. Покажите, что эллиптичность записывается в виде
t
fl_. _, Г(ехр [-2,303/^/2] - ехр [-2,303/лев/2]) I
0 — tg L(exp [-2,303/пр/2] -(- ехр [-2,303/лвв/2]) J '
It Iяг* и /Пр — относительные интенсивности света, поляризованного по кругу
№раво или влево.
^Поскольку круговой дихроизм значительно меньше, чем поглощение, то
Цзкно раскрыть экспоненциалы (в затем и арктангенсы) в выше написанном
' >ажении, сохранив только два первых слагаемых. Приняв такое приближе-
jj& получите уравнение (14.12а), т. е. покажите, что круговой дихроизм про-
щонален эллиптичности.
462 ЧАСТЬ А
4. Чтобы избежать перекрывания исследуемых пнков поглощения очень
сильным пиком растворителя Н20, эксперименты по протонному магнитному
резонансу обычно проводят, используя в качестве растворителя D^O. Однако
такая замена сопровождается двумя осложнениями. Во-первых, показания
рН-метра различаются в D20 н Н^О примерно на 0,41 единицы рН
Р"покаэания в Н,0 == Р"в Н.О == % I" I»
НО
PHno_ в D.0 = PDb D,0 ~ <>,41 = - lg [D+] - 0,4 1.
Во-вторых, константы диссоциации дейтернрованных кислот в D20 меньше,
чем дли протонированных кислот в Н^О:
pKD=*\,№pKH + 0A2t
где pK=\g К и К—константа диссоциации кяслоты в D20 (Kd) или
Н20 (Кн).
а. Предположив, что раствор в НгО имеет рН 7,5, рассчитайте, какое
значение покажет рН-метр в растворе в D^O, если концентрация нона D+ в
растворе DaP будет та же, что н концентрация Н+ в растворе Н^О?
б. Ситуация, соответствующая полученному в пункте а результату, конечно,
является нежелательной. Обычно стремятся к тому, чтобы данный буфер нлн
слабая кислота имели ту же степень ионизации [A-]£(i[A-]-H[DA]) в D2O,
что и в Н20. Получите выражение для показания рН-метра в D^Q, которому
будет соответствовать одинаковая степень диссоциации в D20 н в НгО, т. е.
для слабой кислоты с р/Ся=6,5 ([A-]/[DA])bd2o=([A-]/[HA])b нао.
Найдите показание рН-метра в D20 при 50%-ной степени диссоциации [A~]/[DA] =
=(1,0. Основываясь на этих расчетах, определите, насколько важно вводить
поправку в значение рН раствора при переходе от Н20 к DjO?
5. В присутствии сильного статического магнитного поля Но в
направлении z группа магнитных ядер (или электронов) приобретает
макроскопический магнитный момент M<j вдоль осн z. Если приложить теперь
дополнительное переменное магнитное поле Hicos{(at)i—H\sin(a)t)j, вращающееся
с частотой со относительно осн г, то из геометрического построения можно
прямо предсказать, что должны существовать компоненты магнитного
момента и я v, которые вращаются относительно осн z либо точно в фазе, либо
не совпадают по фазе иа 90° с возбуждающим магнитным полем, согласно
уравнениям
du/dt = К — ш) v — (и/Т2)
и
dv/dt = - (<d, - ш) и - (v/T2) -f тЯХ.
где Т2 — постоянная, a #i— мала по величине. Решите этн уравнения для
стационарных условий и получите выражения для и и v (поглощение
энергии при магнитном резонансе пропорционально v).
в. Ядерный магнитный резонанс особенно удобен для вычисления скоростей
химических «обменов» нз стационарных спектров поглощения. Перепишем
уравнения предыдущей задачи (уравнения Блоха) в комплексной форме:
Af = «-j-/i> („комплексный" магнитный момент)
ш0 = и)0 — i/Tt („комплексная" частота)
dMjdT +1Я - <o)Af=iytf, M,
3 ГРУЗ НА ПРУЖИНЕ
Предположим, что ядро может находиться в одном из двух состояний с
разными резонансными частотами соа и сов. Если не существует перескоков
между состояниями А и В, то уравнения Блоха для Мк и Мв записываются
просто:
dMJdt + i (шА - ш) Af А = *Y# ,К
dMJdt + i (\ - ») AfB = tYtfi< "
Если теперь позволить ядру перескакивать туда и обратно между
состояниями А и В:
*АВ
A*=fcB,
*ВА
то доля ядер в этих двух состояниях равна
г ^ВА с ^АВ
'*""" *АВ+*ВА ' В^ *АВ + *ВА
я общий магнитный момент в направлении z распадется на компоненты в
соответствии с
/
Наконец, так как намагниченность, перенесенная из состояния А в
состояние В, пропорциональна числу ядер, которые переходят из состояния А в
состояние В, можно записать
dMjdt+1 (шА - ») м А + кквмх - кшмв = ifjHM,
dMJdt + i (ьв - ») Мв + kBPMB - kABMA = ifB уН,М0
Используйте последние два уравнения для вычисления общего магнитного
момента в стационарном состоянии М=Ма+А1в.
б. Упростите полученное выше (пункт а) выражение для М, предположив,
что оба типа состояний одинаково заселены (т. е. &лв=£вл=&), и
предположив, что «естественная» ширина линии для каждого из состояний прн
отсутствии обмена равна нулю: (1/7,2А) = (1/7,2В)=0.
я. Наконец, разложите упрощенное выражение для М на действительную и
^мнимую части (т. е. найдите и и t>, так что М = ы+ю) и покажите, что для
^относительно быстрых скоростей обменов &> (о>л—©в) энергия поглощения
^магнитного резонанса при стационарных условиях (пропорциональная v)
состоит из одного усредненного резонанса. Выполненные операции используются
здш нахождении констант скоростей химических реакций из спектров ЯМР
Щал. рис. 14.25—14.27).
в&бычио преобразования, сделанные в предыдущей задаче, применяются для
рыстрых процессов присоединении и диссоциации некоторых небольших
молекул I к макромолекуле Е. Если скорость химического обмена очень велика,
Цйектры ЯМР для данного протона в молекулах I будут содержать один
464 ЧАСТЬ 4
пик, резонансная частота которого соответствует средневзвешенной величине
резонансных частот coi н coei для свободной I и связанной EI частиц:
Е + I*==fcEI
к _[Е][1]
Al~~ [EI] »
Шнабл = /1Ш1+/Е1ШЕ1-
Покажите, что при [1]о>[Е]о (т. е. небольшие молекулы находятся в
большом избытке) зависимость '[1]0 от 1/6 имеет наклон, равный [Е]0 А, н отсекает
на оси у отрезок — /G, где
8 = шнабл— "г И Д —шы —шь
Эта зависимость, таким образом, позволяет просто графнческям путем
определить /G и Л (см. рнс. 14.26 и 14.27) из экспериментальных данных по
положению пика ЯМР. Подобный же способ представления данных может быть
использован для вычисления из данных оптического поглощения.
8. Исходя из комплексного варианта уравнения для #С-цепочки (груз с нулевой
массой на пружине)
х {dqjdt) -f- q = CV0 exp [Ш],
покажите, что для стационарного состояния (комплексное) решение имеет
вид (q'—iq")exfl[mt], где q', и q" — действительные величины. [Как обычно,
действительная часть этого решения
Re (?) = ?' cos (arf) -f ?" sin (arf)
совпадает с действительным решением действительного уравнения
-§- + 9^CV0cos(<ot)
9. а. Покажите, что для диэлектрической проницаемости е' и диэлектрических
потерь е" в уравнении (114.32) выполняется соотношение [уравнение (14.34)]
[•' - 7* (•. - • J'l + (е")2■= (V. (•. - • »))'■ (14.34)
б. Покажите, что зависимость е" от е' представляет окружность с радиусом
'/а (во—е») с центром при е'—Чз (во+е^) на оси е' (ось абсцисс) (см.
(рис. 14.37).
10. Покажите, что кривая зависимости лоренцова контура линии дисперсии
ют? г
l-t-coM от Ф°РМЫ лоренцова контура линии поглощения j _j_ ш2та также
будет окружностью с центром при т/2 на оси поглощения и с радиусом т/2 (см.
рис. 14.38).
11. Параметры ультразвуковой и диэлектрической релаксации обнаруживают за-
ГРУЗ НА ПРУЖИНЕ
висимость от частоты, которая может иметь вид одного из следующих трех
выражений:
Диэлектрический параметр Ультразвуковой
параметр
А= '
1 +со2г2 s' а/ш2= скорость звука
ш;
В= , ■ " о 2 е" U,
1 + <* *
О)2 С
С= ■ , п Электропроводность а
Изобретите способ вычисления времени релаксации t из зависимости А (или
! В или С) от lgx. [Обычно используются зависимости А или В от lgo>.]
Литература
;: Груз на пружине при наличии возбуждающих и тормозящих сил:
дисперсия и поглощение
\1. Фейнман Р. П., Лейтон Р. Б., Сэндс М. Фейнмановские лекции по физике.—
р М.: Мир.
?. Спектроскопия в видимой и ультрафиолетовой областях
|2. Donovan J. W., Physical principles and techniques of protein chemistry, vol. A,
Г: S. J. Leach (Ed.), Academic Press, New York, 1969, pp. 102—170.
Инфракрасная спектроскопия
Parker F. S., Applications of infrared spectroscopy in biochemistry, Biology and
Medicine, Plenum Press, New York, 1971.
Круговой дихроизм и оптическое вращение
Imahori К., Nicola N. A., Physical principles and techniques of protein chemistry,
art C, S. J. Leach (Ed.), Academic press, New York, 1973, pp. 368—445;
ears D. W., Beychok S., ibid., pp. 446—594.
Магнитный резонанс
James Т. L., Nuclear magnetic resonance in biochemistry, Academic press, New
York, 1975. .
Spin labeling: theory and applications, L. J. Berliner (Ed.), Academic press,
New York, 1976.
Электрические цепи как модели пружины
Фейнман Р. П., Лейтон Р. Б., Сэндс М. Фейнмановские лекции по физике, М.:
Мир.
Диэлектрическая релаксация
Takashima S., Physical principles and techniques of protein chemistry, part A,
jS.ed.'S. J. Leach, Academic Press, New York, 1969, pp. 291—334.
Поглощение и дисперсия ультразвука
Blandamer M. J., Introduction to chemical ultrasonics, Academic press, New
York, 1973.
|; Ultrasonics in clinical diagnosis, ed. P. N. T. Wells, Churchill Livingstone,
^Edinburgh and London, 1972.
Etgen M., de Maeyer L., Relaxation methods, Techniques of organic chemistry,
|; vol. VIII, S. L. Friess, E. S. Lewis, A. Weissberger (Eds.), Interscience, New
York, 1963, pp. 952—964.
ГЛАВА 15
РАССЕЯНИЕ: -
ВЫНУЖДЕННЫЕ КОЛЕБАНИЯ ГРУЗА
НА ПРУЖИНЕ В ОТСУТСТВИЕ СИЛ ТОРМОЖЕНИЯ
В гл. 13 рассматривалось смещение в стационарном состоянии
груза на пружине под действием изменяющейся по
синусоидальному закону возбуждающей силы при наличии сил торможения.
Было отмечено, что основное решение приобретало наиболее простую
форму в любом из двух предельных случаев: 1) когда
возбуждающая частота была близка к собственной частоте о)о груза на
пружине в отсутствие тормозящих сил и 2) когда возбуждающая
частота была настолько далека от резонанса, что торможением
можно пренебречь. Примеры явлений (спектроскопия,
диэлектрическая и ультразвуковая релаксации), которые могут быть описаны
лоренцовым контуром линии [уравнение (13.36)], для первого
предельного случая были обсуждены в гл. 14. В этой главе
рассматриваются явления рассеяния (рассеяние света, рентгеновских
лучей, электронов), которые могут быть описаны формулами (13.27)
для второго предельного случая. Хотя в гл. 14 для более
компактного проведения преобразований и представления результатов
часто использовалась комплексная система обозначений, все
рассматриваемые в настоящей главе явления можно описать
исключительно с помощью действительной системы обозначений.
Прежде всего (как и в гл. 14) предположим, что электроны в
веществе связаны пружинками с соответствующими атомами и что
они лишь немного смещаются от равновесного положения.
Поскольку электрон имеет заряд е, то под действием
электрического поля Е на него будет действовать (возбуждающая) сила еЕ.
Тогда под действием изменяющейся по синусоиде во времени
компоненты монохроматической электромагнитной волны
электрического поля (а именно световой волны одного цвета) электрон на
пружине будет смещаться в соответствии с уравнением
m (d*x/dt2) = — kx + еЕй cos (o>/) (15.1)
Обобщенная [сила = «Восстанавливающая* + Возбуждающая сила от сину-
сила пружины соидально меняющегося во
времени электрического поля
монохроматической
электромагнитной волны
£67
ГРУЗ НА ПРУЖИНЕ
где х — смещение электрона от положения равновесия, k —
коэффициент упругости пружины, с которой соединен электрон, т и
« — масса и заряд электрона. Предполагается, Что возбуждающая
^частота достаточно далека от «собственной» о)о=У&/т, так что
-можно пренебречь любыми силами торможения (см. разд. 13.Б.1).
Уже показано [уравнения (13.16а) и (13.27)], что стационарное
смещение х [уравнение (15.1)] полностью совпадает по фазе с
возбуждающим электрическим полем [т. е. x=x/cos((at)], тогда
£•* 1 л/'
х = — /~g _^ cos (0)0, <•>„ = у
т.
(сог0-о>4)
т.
(15.2)
:Далее необходимо предположить, что общий заряд атома, с
которым связан электрон, равен одному положительному
электронному заряду е и что этот положительный заряд распределен по та-
Цкой большой массе (по сравнению с массой электрона), что атом
южно считать неподвижным (это допущение известно как
приближение центрального поля еще из курса общей химии, где рассмат-
швается атом водорода). Другими словами, имеется
электрический дипольный момент (смещение заряда = елг), величина
которого синусоидально меняется с частотой возбуждающего
электрического поля. Но, согласно уравнениям Максвелла, осцил-
гирующий электрический диполь образует осциллирующее элект-
шческое поле, которое создает осциллирующее магнитное поле,
гаправленное под прямым углом. Эти два поля образуют электро-
гагнитную волну, которая распространяется от колеблющегося
возбужденного источника (электрона). Амплитуда электрического
1оля этой новой излучаемой, или рассеянной, волны пропорцио-
[альна второй производной диполя ех по времени:
^расс — ( Vе )
d2 {ex) sin 9
dt* r
(15.3)
\t с — скорость света, г — расстояние между колеблющимся
электроном и точкой пространства, в которой измеряется амплитуда
[ектрического поля, а sin в представляет собой проекцию ампли-
^ды рассеянной волны на линию, вдоль которой ведется наблюде-
ге (рис. 15.1). Подставим ех из уравнения (15.2) в (15.3):
sin 8 г
'расе
пит
ft)'"']
COS (arf),
(15.4)
468 ЧАСТЬ 4
Вертикально поляризованный
луч света
Экваториальная,
плоскость
Рнс. 15.1. Относительная амплитуда рассеяния вертикально (плоско)
поляризованного излучения, падающего вдоль у.
Возбуждаемый электрон расположен в начале координат. Длина радиус-вектора из начала
координат пропорциональна относительной амплитуде рассеяния. (Передняя половина
распределения удалена для большей- ясности схемы, она идентична нарисованной части и
точно к ней примыкает.) [Eggers D. F. et at., Physical Chemistry, John Wiley and Sons, New
York, 1964, p. 496.J
Наконец, так как интенсивность электромагнитного излучения
пропорциональна квадрату амплитуды электрического поля (разд.
13.А),то
'расе
Интенсивность вновь излучаемого (рассеянного) излучения
Интенсивность первоначального (возбуждающего) излучения
расе
Е4
возб
(15.5)
или
г ei у I slna 9 \
/расс='о [ mc2 J у Г2 )
интенсив- единицы геомет-
ность па- измере- рия
дающей ния
волны
_2
l(v)
зависимость
от частоты
Поляризованное в
вертикальной плоскости па- •
дающее излучение
(см. рис. 15.1)
(15.6)
|69 ГРУЗ НА ПРУЖИНЕ
Где принято во внимание, что величина возбуждающей силы
падающего излучения равна eEQcos(iit. Уравнение (15.6) лежит в
основе обсуждения явлений, связанных с рассеянием света, диф-
|фракцией и рассеянием рентгеновских лучей и нейтронов, а
также с электронной микроскопией, и потому заслуживает
тщательного изучения.
Г Как показано, уравнение (15.6) можно разложить на четыре
Множителя, каждый из которых имеет свое смысловое значение.
Так как /расс пропорциональна интенсивности падающего света
jfo, то система является, как говорят, «линейной». Это окажется;
удобным при сопоставлении рассматриваемых незатухающих
колебаний с затухающими колебаниями, которые будут обсуждаться
р разд. 16.Б. Множитель (е2/тс2)2 просто отвечает в данном
случае за использование правильных единиц измерения.
«Геометрический» множитель содержит знакомую обратноквадратичную
зависимость интенсивности электромагнитного излучения от
расстояния между наблюдателем и. источником излучения
(колеблющимся электроном). Рис. 15.1 позволяет оценить значение множителя
ип20. Поскольку электромагнитное излучение представляет собой
щоперечные волны, то движение должно происходить в
направлении, перпендикулярном направлению колебаний диполя,
создающего электрическое поле; но так как движение электрона,
возбужденного падающим излучением, плоскополяризовано в плоскости
|—z и не имеет компоненты в направлении х, то не может быть
Ьлучения, рассеянного (вновь излученного) в направлении
||[sin 0^0]. С другой стороны, излучение, рассеянное той же си-
ремой, имеет максимальную интенсивность в любом направлении
■•плоскости х—у [sin 90°= 1], так как все эти направления пер-
рндикулярны направлению колебаний электрического дипольного
источника рассеяния.
Ц- Ясно, что. если бы исходное излучение было выбрано плоскопо-
Цризованным в направлении х, то наблюдалась бы подобная же
юртина, за исключением того, что интенсивность рассеяния
изменилась бы теперь как cos2 Ф в экваториальной плоскости
1ис. 15.2).
щДля более общего и обычного случая, когда падающее излуче-
юе неполяризовано, можно считать, что оно состоит из двух
Шимно перпендикулярных линейнополяризованных компонент.
Интенсивность рассеянного излучения имеет тогда величину,
средино между значениями для каждой из перпендикулярных компо-
■рт. Например, в экваториальной плоскости (ху) угловая зави-
гаюсть интенсивности рассеяния равна или sin290°= 1, или cos2*
ря каждой из двух индивидуальных компонент и интенсивность
JM неполяризованного излучения (сумма двух компонент) в эк-
■|?рриальной плоскости равна (\-\-cos2 Ф)/2. В случае
произвольно угла Qs между линией наблюдения и направлением распро-
470 ЧАСТЬ 4
Полюс
г
Горизонтально ^ N
поляризованный
луч света
Экваториальная
плоскость
Рис. 15.2. Относительная амплитуда рассеяния от электрона, находящегося
в начале координат и возбужденного с помощью горизонтально (плоско)
поляризованного падающего в направлении у излучения.
Длина радиус-вектора из начала координат пропорциональна относительной амплитуде
рассеяния. (Передняя половина распределения удалена; см. подпись к рнс. i5.i.) [Eggers D. F.
et at.. Physical Chemistry, John Wiley and Sons, New York, 1964, p. 496.]
странения исходного падающего луча (рис. 15.3) общий угловой
множитель равен (l+cos2<9s/2)
Неполяризованное падаю- 'расе
щее излучение (см.
рис. 15.3)
= 1.
тс
"2 / 1 -Ь cos2 Qs \
ыч
(15.7)
Угловая зависимость интенсивности рассеяния, согласно
уравнениям (15.6) и (15.7), свидетельствует о двух главных
качественных свойствах. Во-первых, свет, рассеянный под прямым углом к
направлению распространения падающего света, всегда плоско-
поляризован, даже если падающий свет кеполяризован (читатель
может отметить, что видимая яркость ясного неба при
рассмотрении через поляризационные солнечные очки изменяется при
повороте очков). Во-вторых, процесс рассеяния можно
экспериментально изучать двумя способами: либо наблюдая снижение
интенсивности вдоль направления распространения падающего
излучения (как делают при измерении «мутности» для рассеянного
света; см. ниже), либо путем наблюдения за светом, излученным
471 ГРУЗ НА ПРУЖИНЕ
(вновь) из центра рассеяния под разными углами к направлению
распространения падающего света (как поступают в различных
экспериментах по дифракции рентгеновских лучей или нейтронов
или при работе с электронным микроскопом).
Зависимость рассеянного излучения от частоты, последний
множитель в уравнениях (15.6) и (15.7), достаточно легко
определяется для двух простых предельных случаев [уравнения (13.30) и
(13.31)]:
'расе
'расе
ное N [ е2
1ение J J J mc*
Рассеяние Рэлея (<о<^<о,)
неполяризован
падающее излучение
Рассеяние Томсона (со^><о0)
неполяризованное
падающее излучение
(
(
= и
,2. 12
ГнС*
I"
1 + cos2 Bs
l -h cos2 es
] /со
(i 5.8)
(15.9)
Так как собственная частота электронов, «на пружинке» в
веществе соответствует ультрафиолетовой области спектра, то рэлеев-
ское рассеяние можно ожидать для видимого света и излучения
с более низкой частотой, в то время как томсоновское рассеяние
Ьжидается для рентгеновского и более высокочастотного
излучений, связанных с волновыми свойствами частиц типа нейтронов и
^лектронов. Отметим далее, что если рентгеновские лучи и элект-
/?0/1ЮС
■
\
• Неполяризованныи
Ь свет
Ел А
Ш" i^^J "^^Д^ыДЮаиВд
1
г
- Рассеяние
^ поляризовано
горизонталь но
Штт^
Экваториальное
рассеяние
ЛО/гяризовано
вертикально
Выход неполяризо-
ванного, но
ослабленного луча
Экваториал ь нал
плоскость
_, 15.3. Относительная амплитуда рассеяния от электрона, находящегося
*ачале координат и возбужденного с помощью неполяризованного падающего
в направлении у излучения.
Aggers D. F. ei at.. Physical Chemistry, John Wiley and Sons, New York, 1964, p. 497.1
472 ЧАСТЬ 4
Рис. 115.4. Схематическое изображение суммирования амплитуд рассеянных
электрических полей от излучения одновременно рассеянного двумя (/ я 2)
электронами, разделенными в пространстве.
Предполагается, что возбуждающее излучение когерентно (т. е. оба электрона возбуждены
с одной и той же фазой), с тем чтобы прямо показать различия в фазах Д ф, которые
возникают для двух воли рассеянных по направлению к одной и той же точке наблюдения.
На верхней схеме показан большой угол рассеяния 9$1 а иа нижней — малый угол рае-
сеяния. (См. объяснения в тексте.)
роны рассеиваются независимо от их энергии (частоты), т. е. все
объекты выглядят «серыми» в рентгеновских лучах и в
электронном микроскопе, то видимый свет рассеивается пропорционально
четвертой степени его частоты. Таким образом, синий свет
рассеивается гораздо более интенсивно, чем красный, что объясняет
синий цвет неба, если смотреть на него под значительным углом от
направления на солнце, и красный цвет заходящего солнца, если
смотреть под углом, близким к направлению на Солнце
(поскольку синий свет предпочтительно устраняется в направлении
падения солнечного света). (Облачность — это другой вопрос, который
обсуждается позже.)
Для оценки излучения, рассеянного от макроскопического
образца, необходимо рассмотреть суммарное рассеяние от двух
разных электронов, разделенных в пространстве (рис. 15.4). Для
облегчения иллюстрации использован когерентный источник, так
что первоначальное возбуждение обоих электронов происходит в
фазе. Каждый возбужденный электрон рассеивает независимо, и
амплитуды двух рассеянных волн суммируются, как показано на
рисунке для случаев наблюдения рассеяния под большим
(верхняя схема) или под малым углом (нижняя схема). Из рисунка
качественно ясно, что когда угол рассеяния велик (и/или когда
длина волны возбуждающего излучения мала по сравнению с рас-
473 ГРУЗ НА ПРУЖИНЕ
стоянием между двумя электронами), то две рассеянные волны
в той или иной степени гасят друг друга в результате
интерференции. Напротив, при малом угле рассеяния (и/или когда длина
волны возбуждающего излучения велика по сравнению с
расстоянием между двумя электронами) происходит когерентное
суммирование амплитуд двух рассеянных волн. Таким образом, можно
представить себе, что суммарная интенсивность рассеянного
излучения в зависимости от положения наблюдателя может меняться
от нуля до величины, в четыре раза превышающей интенсивность
рассеяния от любого из одиночных электронов.
Из рис. 15.4 непосредственно следует ряд новых выводов. Во-
первых, если два электрона находятся очень близко друг к другу
(например, два электрона в одном атоме) по сравнению с длиной
волны возбуждающего излучения (Дф^СЯ), то суммирование
рассеянных электрических полей происходит когерентно независимо
от угла рассеяния:
£расс (суммарное) = NE^^ (от любого из возбужденных электронов)
(15.10)
и интенсивность суммарного рассеяния пропорциональна квадрату
числа центров рассеяния N2:
/расе (суммарное) = N'-I^^ (от любого из возбужденных электронов)
(15.11)
Уравнение (15.11) позволяет понять, почему тяжелые атомы
(которые можно рассматривать как центры рассеяния с многими
электронами, расположенными близко друг к другу) рассеивают
видимый свет, рентгеновские лучи и электроны так интенсивно,
что их часто используют в качестве «меток» в экспериментах по
рентгеновской дифракции и электронной микроскопии. С другой
стороны, если расстояние между двумя возбужденными
электронами меняется беспорядочно (например, когда два возбужденных
электрона связаны с двумя разными молекулами газа или
жидкости), то суммарная амплитуда рассеяния в данной точке
пропорциональна квадратному корню из числа таких электронов (это
вытекает из тех же соображений, которые были использованы при
вычислении беспорядочного движения). Поскольку интенсивность
изменяется пропорционально квадрату амплитуды, то получаем
^расс (суммарное) = Л//расс (от любого из возбужденных электронов)
(15.12)
Уравнение (15.11) приложимо к рентгеновской дифракции и
рассеянию электронов от твердых образцов, в то время как
уравнение (15.12) применимо для многих видов светорассеяния и
рентгеновского рассеяния под малыми углами.
474 ЧАСТЬ 4
15.А. МАЛЫЕ ОБЪЕКТЫ, БОЛЬШИЕ ВОЛНЫ, ЛИНЗЫ
НЕ ПОМОГАЮТ: РЭЛЕЕВСКОЕ РАССЕЯНИЕ СВЕТА.
МУТНОСТЬ, РАДИУС ВРАЩЕНИЯ, СООТНОШЕНИЕ ЦИММА:
РАЗМЕР, ФОРМА И МОЛЕКУЛЯРНАЯ МАССА
МАКРОМОЛЕКУЛ В РАСТВОРЕ
Зависимость рассеяния света от молекулярной массы
макромолекулы
В этом разделе рассматривается рассеяние видимого света
растворами макромолекул (т. е. то дополнительное рассеяние,
которое появляется помимо слабого рассеяния света чистым
растворителем). Для того, чтобы показать, почему растворы молекул,
имеющих большую молекулярную массу рассеивают свет
значительно более интенсивно, чем растворы, содержащие молекулы с
небольшой молекулярной массой, необходимо придать задаче
новую форму путем замены (как часто делают) некоторых
переменных.
Удобно выразить величину электрического дипольного
момента ех, возникающего при приложении электрического поля Е,
через молекулярную поляризуемость а, которую можно
рассматривать как меру силы, необходимой для того чтобы оторвать
электрон от его ядра
ex = <zE^aE0 cos (Ы), (15.13)
где Е — возбуждающее электрическое поле, образованное
падающим светом. Подставляя ех в уравнение (15.3) получим
амплитуду рассеянного от возбужденного электрона стационарного
электрического поля
/ вертикально \
Urn EV(X I поляризованный I = — (<»2/c*) я^о (s*n ^lr) cos (ait). (15.14)
^ ' \пз дающий свет /
Используя те же преобразования, которые привели от уравнения
(15.4) к (15.8), получим интенсивность рассеяния для одного
возбужденного электрона /расс для неполяризованного падающего
света
(«поляризованный N = £]д,ГJ+^|
<"><<!>„ \ падающий свет J Lc J l *r j
Продолжение расчетов имеет целью выразить /раСс и а через
подходящие и измеряемые экспериментально величины. Для- этого
необходимо учесть еще две зависимости Максвелла, приняв ре-
475 ГРУЗ НА ПРУЖИНЕ
зультат без доказательства [уравнения (15.3) и
(15.16)—единственные приведенные без объяснения]:
е' = п* = 1 -{- 4гс#а (для газов) (15.16)
где е' — диэлектрическая проницаемость, п — показатель
преломления и N — число рассеивающих центров в единице объема. Таким
образом, если бы существовал только один возбужденный
электрон на молекулу, то
п* i
л= „ (для молекулы газа) (15.17а)
или аналогично
л==—4гсдг°- (для молекулы в растворе) (15.176)
где «о — показатель преломления чистого растворителя и п —
показатель преломления раствора. Уравнение (15.176) можно
переписать в виде
*=(n+n;lkZ')M' <15-l8>
где
N (частиц/объем)^ М (г/объем) ^ Састяц/моль) (15Л9)
v ' . ' М (г/моль) v '
Для разбавленного раствора
п +лв ^ 2па (15.20)
(п—tio)/[c]=xif (приращение показателя преломления, т. е.
изменение показателя преломления при изменении концентрации
растворенного вещества) (15.21)
Комбинируя уравнения (15.18) -*■ (15.21):
«=^г с5-22'
и подставляя в уравнение (15.15), получаем
СГ"^л$- '+7'к 1^<на одау частац*> <15-23>
Путем умножения интенсивности рассеяния (одной частицей)
на число частиц в единице объема [уравнение (15.19)] получим
интенсивность рассеяния для единицы объема раствора:
^ешляриз^ а^_ 1 + cos2 9, _п£У[с] М единицы объема)
расе ° с4 r 8n*Nb
(15.24)
476 ЧАСТЬ 4
Наконец, из уравнения (13,4) Xv=c и уравнения (13.7) <o = 2rtv
имеем
ю = 2тс/Я,
(15.25)
и после подстановки о) в уравнение (15.24) получаем результат в
желаемой форме:
'расе
иеполяризованный
падающий свет
2п>п*0У 1 + cos2 Bs
= '• ПС?^ 7* W М
(для
единицы объема)
(15.26)
Таким образом, рассеяние света прямо пропорционально
концентрации раствора и молекулярной массе растворенного вещества.
В качестве небольшого добавления следует заметить, что
уравнение (15.26) выведено при допущении (мысленном), что в
молекуле имеется лишь один возбужденный электрон. Если же имеется /
возбужденных электронов на молекулу, то амплитуды рассеяния
от всех этих электронов когерентно суммируются [уравнение
(15.11)] и интенсивность рассеяния пропорциональна /2. Как
правило, последнюю величину избегают вводить, а включают / в
поляризуемость а, так как постоянные, входящие во второй
сомножитель уравнения (15.26), обычно определяются
экспериментально в виде одного параметра К:
К^
2п*п\ф2
(15.27)
Поскольку относительные интенсивности измерять проще, чем
абсолютные, то уравнение (15.26) часто выражают в терминах
рэлеевского отношения (интенсивностёй) R(QS)
■расе
/?(М =
(неполярлзованный
падающий свет
1+С05гб/ = /С^М
(15.28)
Уравнение (15.28) показывает, что отношение интенсивностёй
при рассеянии света можно использовать для определения
молекулярной массы макромолекул в растворе. Если раствор
содержит смесь макромолекул с различными молекулярными массами,
то общее рассеяние [как следует из рэлеевского отношения
#(6s)] представляет собой сумму интенсивностёй рассеяния от
молекул с определенной молекулярной массой Mi с учетом соответ-
$77 ГРУЗ НА ПРУЖИНЕ
ствующих концентраций [c]i (масса/объем) частиц с данной
молекулярной массой:
К2И«]1Л*/
Я (W = У, R (U = S * Mi М<= -^~ [с] = /С [с] Л*„, (15.29а)
где
2[е]/А1, 2^/А«»|
Л*« = -^ =^ (15.296)
и
2М = И. (15«29в>
где N— число молей компонента JW/.
Если принять заведомо неверное допущение, что раствор —
гетерогенная система и содержит только один тип молекул с
концентрацией, [с] (масса/объем) со (средней) молекулярной массой
Mw> то Mw связана с молекулярными массами индивидуальных
молекул Mi соотношением (15.296). Уравнение (15.296) показывает,
почему необходимо содержать растворы для измерения рассеяния
света в совершенной чистоте, не допуская попадания частиц пыли,
так как даже очень небольшая концентрация частиц пыли с
высокой «молекулярной» массой значительно отразиться на
определении средней молекулярной массы Mw в растворе макромолеку-
.лярного вещества (см. задачи в конце главы).
Один из наиболее простых экспериментальных подходов к
изучению рассеяния света заключается в измерении снижения
интенсивности падающего луча по направлению его падения. Хотя та-
'кие измерения и напоминают измерение поглощения света,
природа этих явлений совершенно различна. При поглощении света
излучение входит в образец, но часть его никогда не выходит; при
рассеянии света все падающее излучение либо целиком проходит,
либо рассеивается в различных направлениях, но поскольку этот
(рассеянный) свет в основном имеет направления, отличные от
первоначального, то наблюдается кажущееся уменьшение
интенсивности в направлении падения света при его прохождении через
образец. Из уравнения (15.28) следует основной вывод о том, что
интенсивность рассеяния пропорциональна интенсивности
падающего света; поэтому можно считать, что бесконечно малая потеря
интенсивности в «прямом» направлении —dl (знак минус свиде-
478 ЧАСТЬ 4
тельствует о потере интенсивности) пропорциональна бесконечно
малой толщине образца (раствора) dyt а также интенсивности
света, входящего в бесконечно малую область, / и мутности х
[аналогична «коэффициенту поглощения» в спектроскопии; ср. с
уравнением (14.4) и следующими], представляющей собой
интенсивность рассеяния для. образца с единичной толщиной и при
.интенсивности падающего света, равной единице
Падающий
сбст *" *•
— dl=hdy. (15.30)
Проинтегрируем уравнение (15.30), с тем чтобы перейти к образцу
конечной толщины
/ = /.ехр[-^1, (15.31)
где / — интенсивность в направлении падения света с
интенсивностью /о после прохождения через образец толщиной у и мутностью
х. Читателю предлагается в качестве упражнения доказать (см.
задачи в конце главы), что в этом простом опыте помутнение
связано прямой пропорциональной зависимостью с рэлеевским
отношением
, = !|?Д(90о), (15.32)
так что измерение мутности [т. е. кажущегося «поглощения»; см.
уравнение (14.5)] делает доступным прямое измерение
молекулярной массы макромолекул в растворе. В дальнейшем будет
показано, что измерение интенсивности рассеяния в зависимости от
угла рассеяния относительно направления падения 95 позволяет
получить дополнительную информацию о форме и размерах
макромолекул в растворе.
Зависимость рассеяния света от размера и формы макромолекул
На рис. 15.5 показано, что амплитуды электрических полей
двух световых волн, рассеянных от двух возбужденных
электронов одной макромолекулы, для отдаленного наблюдателя в
основном не совпадаютчпо фазе по двум причинам. Во-первых, так как
два электрона часто расположены на разных расстояниях от
источника излучения, то их возбужденные колебания имеют
неодинаковую фазу. Во-вторых, так как два электрона обычно находятся
на разных расстояниях от наблюдателя, то появится
дополнительная разность по фазе между двумя,рассеянными волнами, вос-
(I-d/h
Прошедший
-^- (^рассеянный
вперед) свет
*h-
179 ГРУЗ НА ПРУЖИНЕ
Когерентное Молекулярная
излучение частица
I&-*. \^,А^>^^~^~^,'\^^У^^А^Г?~*'1оасс(0') Фазовое смещение от-
'л■ ' ■ ' » ' v ' » ^V^L-iil-*^-/.\j » Patx сутствует про нулевом
г В- 45* Уеле рассеивания
= /55" ^
С <"~ "U Рассеянные волны
Рассеянные .—) ) «лл совпадают по tpaee
волны не сое- ' { \^--\
падают по^^' О *"*\
1ра«, №> fa" m'}
Рис. 15.5. Фазовые взаимоотношения между падающими и рассеянными
световыми волнами от двух возбужденных электронов, расположенных в разных
частях одной макромолекулы.
Для большей наглядности выявления смещении предполагается, что падающие световые
волны когерентны. Для указанных углов расположения (между направлением падения и
линией, соединяющей электроны) н углов наблюдения 6S волны рассеяния отличаются по
фазовым углам на 0° при 9S™45° и на 180* при 6s»*135e. Дли упрощенного формального
решения см. уравнение (15.33) и следующие.
принимаемыми наблюдателем под заданным углом рассеяния ds
к направлению падающего света.
Для света, рассеянного в направлении к наблюдателю,
который расположен под произвольно выбранным углом рассеяния 9*,
электрическое поле £ от двух разделенных в пространстве
возбужденных электронов одной макромолекулы (рис. 15.5) можно
записать так:
£ = £,cos(orf+*i) + £,cos(arf4-*I), (15.33)
где *! и Ф2— соответствующие разности фаз (для света от
электронов № 1 и № 2) между возбуждающими колебаниями и
колебаниями в точке наблюдения, причем оба электрона возбуждены
с одной и той же первоначальной амплитудой Es. Используя
тригонометрическую формулу
cos A + cos B = 2cos [(А - В)/2] cos [(А +Д)/2], (15.34)
получаем
Да=2Ядсо8((Ф1-Ф,)/2)со8((2^+Ф1-И,)/2). (15 35)
амплитуда временная зависимость
Уравнение (15.35) свидетельствует о том, что результирующая
амплитуда обеих рассеянных волн варьирует от нуля до удвоенного
значения амплитуды Es для любого из источников рассеянных
волн. При (Фх—Фг)=0 между двумя рассеянными волнами нет
различий по фазе в месте расположения наблюдателя и
амплитуда равна 2Я5, но при Ф4 — Ф2=180°, обе рассеянные волны
гасятся полностью, что приводит к тому, что результирующая
амплитуда равна нулю. Результирующая интенсивность рассеяния, таким
480 ЧАСТЬ 4
образом, варьирует от нуля до 4IS, где Is есть интенсивность
рассеянной волны от каждого электрона. Этот вывод совпадает с
результатом прежнего качественного рассуждения.
Уравнение (15.35) описывает результирующее электрическое
поле от двух возбужденных и определенным образом
расположенных электронов. У макромолекул в растворе векторы,
направленные от одного электрона к другому, ориентированы во всех
направлениях, так что необходимо «усреднить» уравнение (15.35), введя
условие, что два электрона расположены в любом месте (на
противоположных сторонах) бесконечно малой сферической ячейки.
Чтобы завершить расчеты, надо найти выражение для разности
пути А, которая ответственна за разность фаз 2лА/Я волн в месте
расположения наблюдателя, для каждой возможной ориентации
межэлектронных векторов; эта задача решается путем прямого, но
скучного применения чистой тригонометрии и здесь опускается
(см. [Tanford, с. 298—302]). Результат наиболее удобно написать
в следующей форме:
/ для двух возбужденных электронов, расположенных в \
расс \любом месте на тонкой сферической оболочке с радиусом гц)
/для двух возбужденных электронов, расположенных близко \
Амплитуда £раСС
^ \ друг к другу по сравнению с А /
=——^ (15.3ба)
где
p,=:(4ic/a)sin(8s/2) (15.366)
и Гц — расстояние между двумя электронами. Для реально
существующей макромолекулы, у которой много возбужденных
электронов, необходимо было бы суммировать амплитуды рассеянных
электрических полей от всех возможных пар возбужденных
электронов. Например, для сферической макромолекулы, если принять,
что центры рассеяния равномерно распределены по объему сферы,
требуется лишь суммировать рассеяние сферическими ячейками
[уравнение (15.36)] от радиуса, равного нулю, до радиуса сферы:
Амплитуда £расс (от макромолекулярной твердой сферы диаметра D)
/ рэлеевское рассеяние от того же числа \
Амплитуда
£расс I возбужденных электронов, расположенных I
V близко друг к другу по сравнению с А /
D/2
fxr
= —j- [sina — a cos a]; a = (\iDj2) (15.37)
1
d/2 a
I 4nr%dr
6
(поскольку sin(tir)/V—+ 1 при г—>0)
181 груз на пружине
ш
Рис. 15.6. Функции рассеяния Р(0«) для макромолекул различной формы.
По оси абсцисс нанесены либо (цО/2) [где ц,-»(4 rt/A,)-sin (6s/2) и D — диаметр сферы,
длина палочки нли диаметр диска], либо У\1ЮЩ, где D* — среднеквадратичное расстояние
между концами беспорядочного клубка. Таким образом, по оси абсцисс представлены
комбинации углов рассеяния и размеров молекул: для макромолекул данного размера и
формы P(0S) снижается по мере увеличения угла рассеяния; подобным же образом для
данного угла рассеяния Я(6$) снижается по мере увеличения размера макромолекулы. Для
о
макромолекул с размером <50 А (верхняя пунктирная линия) фактор рассеяния равен
единице при любом угле рассеяния при использовании видимого падающего света
(см. текст). [Sadron С, Pouyet J., Ргос. 4th Intern. Congr. Biochem.. Vol. 9, Vienna, 1958
{ (Pergamon Press, Oxford), p. 52.]
Наконец, так как интенсивность изменяется пропорционально
квадрату амплитуды, то принято вводить фактор «внутренней
интерференции» P(fis), равный отношению интенсивности рассеянных
|волн, когда внутренняя (внутримолекулярная) интерференция
Принимается во внимание, к интенсивности, вычисленной без уче-
jra внутренней интерференции
P(h) =
'расе (внутренняя интерференция учтена)
/расе (внутренней интерференцией пренебрегли)
(15.38)
С учетом уравнения (15.28) рэлеевское отношение R($s) мож-
10 написать в виде
(15.39)
R(Bs) = K[c]MP(Bs)
Функции рассеяния Р(9$) для макромолекул различной формы
изображены на рис. 15.6 и даны в табл. 15.1. Из табл. 15.1 видно,
ято функции, описывающие P{Qs), весьма разнообразны в
зависимости от формы макромолекулы.
f Желательно было бы выразить фактор рассеяния более просто
|ерез какие-либо параметры формы, которые можно было бы лег-
jfco вычислить для каждой определенной формы молекулы.
Рассмотрим функцию рассеяния P(QS) для твердой сферы при углах,
близких к Gs=0. Тогда в уравнении (15.37) a<Cl и можно аппрок-
Функция рассеяния Р(в*) для различных макромолекулярных структур
Таблица 15.1
Структура
р(Ч
Размеры
Радиус вращения Rq
Сфера
г/о, чч / , v» M^ D — диаметр сферы
[(3/а3) (sin а — а cos а)2; а = -*тр м к «г к
(3/20)l/* D
Тонкая палочка
1 Г sin* _, / sin a \*
3-J—*-(—): —
Ц,Р
2
D — длина палочки
D//2
Беспорядочный клубок 2 ц,2 <D2) <Р2> — среднеквадратичное
0а 1ехР ( л) + а— ч; а— fj расстояние конец—конец
УЧ^Тб"
Тонкий диск
3-[l-(l/a)/i{2o)l;o = (fiZ)/2)
D — диаметр диска
/i — функция Бесселя первого
порядка
(D/2 /2)
3 ГРУЗ НА ПРУЖИНЕ
мировать sin а и cos а с помощью первых нескольких членов их.
зложения в степенной ряд (см. приложение)
а8 I а8 I
cosa=l--2|-+ir+---,
к что Р(д$) для сферы переходит в
Р(is) = (1 - £)*«! - ^ = 1.*£! sin'(0./2) »» «ФЧ*
(15.40а)
(15.406)
с диаметром D
(15.41)
феделим «радиус вращения» Rg'-
(15.42)
в rtii — молекулярная масса, расположенная на расстоянии г4- от
нтра массы макромолекулы. Тогда можно определить радиус
ащения сферы:
R\ (сферы) =
JJ*/2>r»4«r"dr (г»/5)|рт
$lQDM4nr4r
г3/* IS
^5r = (3D»/20), (15.43)
g. D — диаметр сферы, а масса, расположенная между г ц
фг от центра сферы, пропорциональна 4nr2dr.
гПодставим уравнение (14.43) в (15.41):
к--
/>(В,) **: 1 —g-^-
где ц = (4яД) sin (6s/2)
(15.44)
Ьнность уравнения (15.44) состоит в том, что, будучи выведен-
|й для сферической макромолекулы, оно действительно для мак-
молекул любой формы. Поскольку возможно прямое вычисление
1'для определенной формы молекул (см. задачи в конце главы),
^уравнение (15.44) дает непосредственный метод для определен
я размера макромолекулы условной формы путем измерения
реевского отношения [уравнение (15.39)] в пределе низких уг-
I рассеяния 6S или для макромолекул малых размеров с таки-
|Д что \iD^\ [где \i определяется уравнением (15.366)].
|*еньшей чувствительностью (обратите внимание на сходство
484 ЧАСТЬ 4
кривых на рис. 15.6 для молекул разной формы) можно
установить форму макромолекулы условного размера.,Быстрое
вычисление, основанное на уравнениях (15.366) и (15.44), показывает, что
если радиус вращения макромолекулы в водном растворе больше
7бо длины волны видимого света (/?g>80 А), то функция
рассеяния Р(в«) равна единице при любом угле рассеяния. Так как
Rg = 80 А, что соответствует молекулярной массе 400 000 для
сферического белка, то становится ясно, что измерение отношения ин-
тенсивностей рассеянного видимого света может служить для
определения размеров и форм только самых больших естественных
и синтетических макромолекул. В следующем разделе этой главы
увидим, что применение гораздо более коротких волн падающего
излучения (рентгеновских лучей) делает рентгеновское рассеяние
под малыми углами значительно более пригодным для
определения формы макромолекул типичных белков и ферментов.
Зависимость рассеяния света от концентрации
растворенного вещества: кривые Цимма
Уравнение (15.28), отражающее зависимость интенсивности рэ-
леевского рассеяния от массы макромолекул и их концентрации,
получено из уравнения (15.12), согласно которому общая
интенсивность рассеяния равна интенсивности рассеяния от одного
центра, умноженной на число центров рассеяния. Уравнение (15.12) в
свою очередь основано на допущении, что угловая ориентация и
расстояние между двумя центрами рассеяния (в данном случае
между двумя различными макромолекулами в растворе)
беспорядочно меняется. Однако ясно, что в реальных растворах межмо^
лекулярные расстояния и ориентация не могут беспорядочно
меняться: во-первых, сами макромолекулы занимают определенный
объем, так что самое малое возможное расстояние между двумя
макромолекулярными центрами не может быть меньше, чем
диаметр макромолекул, и,. во-вторых, существующие силы
притяжения и отталкивания между макромолекулой и молекулами
растворителя или между молекулами растворителя стремятся
упорядочить межмолекулярные расстояния и изменения углов. Эти
корреляции в относительном положении молекул растворенного
вещества описываются введением в уравнение (15.28) дополнительного
члена
1
Ж + 2В W
где В— молекулярная константа взаимодействия, известная как
второй вириальный коэффициент (разд 2.Б). Объединяя уравнение
(15.45), которое описывает.неидеальную природу любого
реального раствора, с уравнением (15.39), которое учитывает частичное
л(о,)=/си
(15.45)
185 ГРУЗ НА ПРУЖИНЕ
Начальный наклон (предел* нулевого
угла рассеяния 0S) пропорционален
второму вириальному коэффициенту б
6,0
Отрезок, отсекав
Яый на оси, odpam-
(о пропорционален 3,6
молекулярной массе О
cs/,75-/0 г/мл
135'
1'2
sin2(%/2)+/00[c] Начальный наклон
(предел нулевой
концентрации) пропорционален Rr
ис. 15.7. Зависимость Цимма {см. уравнения (15.44) н ((15.46)] для большого
синтетического полимера в разбавленном солевом растворе.
Молекулярная масса (нз, пересечения с осью ординат) равна 260 000. \Ehrlich О., Doty P.,
J. Araer. Chem. Soc, 76. 3764 (1954).]
ашение в результате интерференции световых волн, рассеянных
Г различных частей одной и той же молекулы, получаем резуль-
1Т, позволяющий получить информацию о трех свойствах макро-
Ьлекул в растворе:
К [с] 1 /_|_ \
■Я(в,)~я(е,) {m+2BWj
(15.46)
I;
|-первых, так как P(Q$)—И при д$—Я), уравнение (15.46)
порывает, что массу макромолекулы М можно определить путем
|страполяции рэлеевского отношения R(ds) к предельным усло-
|ям нулевой концентрации и нулевому углу рассеяния. Во-вторых,
i экспериментально полученных рэлеевских отношений при раз-
|чных углах рассеяния (но при экстраполяции к нулевым кон-
рнтрациям макромолекул) можно определить Р(9«) и получить
^формацию относительно размера и формы макромолекул* что
ike обсуждалось (рис. 15.6 и табл. 15.1). Наконец, определив
йлеевские отношения при различных концентрациях (но в этот
jb они все экстраполированы к нулевому углу рассеяния Gs),
$Жно найти константу взаимодействия В, которая дает инфор-
&цию относительно ассоциации макромолекул друг с другом и с
р'створителем (см. разд. 2.Б). Графическое представление
результатов (предложенное Циммом) для проведения такого анализа в
Цгчае большого синтетического полимера показано на рис. 15.7.
486 ЧАСТЬ 4
15.Б. КРУПНЫЕ ОБЪЕКТЫ, НЕБОЛЬШИЕ ВОЛНЫ, ЛИНЗЫ
ОТСУТСТВУЮТ: РАССЕЯНИЕ РЕНТГЕНОВСКИХ ЛУЧЕЙ;
ДЕТАЛЬНОЕ ОПРЕДЕЛЕНИЕ ФОРМЫ МОЛЕКУЛ
Как указывалось в предыдущем разделе, рассеяние в видимой
области дает возможность для определения молекулярных масс
типичных биологических макромолекул, но размер и форма
молекулы могут быть определены только для очень больших
макромолекул (Mw^l 000 000), для которых размеры частиц (скажем,
сферических с диаметром 100 А) составляют ^Vso длины волны
используемого облучения (например, 4000 А). Среди таких
крупных макромолекул трудно найти молекулы с Мда=106, которые
являются к тому же и монодисперсными в растворе (т. е. все
молекулы имеют одинаковую молекулярную массу); более того,
молекулы столь большого диаметра как 100 А могут быть видны
непосредственно в электронном микроскопе (см. следующий раздел)»
Следовательно, не остается другого выхода, как искать
информацию о форме макромолекул из данных по рассеянию более
коротковолновое излучение. Однако, как уже выяснили в разд. 13.Б,
эксперименты с рассеянием теряют смысл, когда частота
облучения приближается к собственным частотам электрона на
пружине. Таким образом, поскольку белки и нуклеотиды начинают
поглощать при длинах волн ^3000 А (и поскольку даже вода и
воздух начинают поглощать ^2000 А), то следует искать
подходящее для поставленных выше целей облучение в области еще более
высоких частот. Но здесь отсутствуют источники излучения вплоть
до области рентгеновских^ лучей (А,= 1,54 А для обычной Ка
линии меди).
Хотя формальное рассмотрение рассеяния в предыдущей
главе теоретически пригодно и для рассеяния рентгеновских лучей,
можно отметить ряд различий, имеющих практическое значение.
Во-первых, подробное рассмотрение рис. 15.6 [и уравнение
(15.44)] позволяет понять, что даже для относительно небольших
Макромолекул (например, молекулы лизоцима—небольшого
фермента с Afw=14100) интенсивность рассеяния рентгеновских
лучей падает практически до нуля при углах рассеяния всего в
несколько градусов от направления падения. Для более крупных
макромолекул рассеяние по существу исчезает при углах
рассеяния, больших доли градуса. Таким образом, эксперименты по
измерению интенсивности рассеяния должны проводиться при очень
малых углах рассеяния. К счастью, рентгеновские лучи можно кол-
лимировать до 0,001 рад, что делает возможным измерения
рассеяния под малыми углами. Во-вторых, снижение длины волны
облучения в 2000 раз при переходе от видимой области к
рентгеновским лучам делает возможным, определение размеров и форм
большинства ферментов, белков и нуклеиновых кислот; более
того, поскольку частицы пыли чрезвычайно велики по сравнению с
ГРУЗ НА ПРУЖИНЕ
шой волны рентгеновских лучей, то при таких маленьких уг-
рассеяние от пылевых частиц снижается до нуля и пыль
влияет на измерения при низкоугловом рентгеновском рассея-
|и, что упрощает приготовление растворов.
|;Что касается математической обработки данных, то при перехо-
|; к очень маленьким углам рассеяния можно сделать дополни-
1ьные упрощения (по сравнению с рассеянием света):
sin(6s/2)^6s/2, б,<1 радиан
|к что уравнение (15.44) преобразуется
j _16яа#а<?(Э5)/2)а
t:
РФ)
ЗА,3
-, для es < 1
(15.47)
(15.48)
I поисках более прямого экспериментального метода определения
I Гуньер принял во внимание, что поскольку
= 1-
16*2#20(е5/2)2 128п»/?*0(9,/2)*
"1 о>* г
ЗА2
9К*
^р[-(1б«>/г"0(в,/2)«/зя«]=
I (15.49)
^поскольку первые два слагаемых уравнения (15.49) те же, что
|в (15.48) (а в случае сферической формы молекулы и третье
к Таблица 15.2
I Рентгеновское рассеяние под малыми углами и рассеяние света8.
[ Определение молекулярных масс н радиусов вращения
;; для типичных макромолекул
Вещество
Состояние
Rr;, A
Молекулярная Масса
50ЦИМ
£имотрипсин
Йактоальбумин А
йоглобин
103ИН
ЯК
ipyc карликовой кусти-
Ьсти
Раствор
Кристалл
Раствор
Кристалл
Раствор
Раствор
Кристалл
Раствор
Раствор
Раствор
)ус табачной мозаики Раствор
14,3Ч±0,3
13,86
18,0
16,06
21,6±0,4
16,3±0,5
15,5±0,5б
468*
1170а
120
924*
14 100
24 500
36600 (мономер)
17 000
493 000
4 000 000
10 600 000
39000 000
Щд для кристаллов определены методом реитгеноструктурного анализа (см. разд. 22.А).
Ь
488 ЧАСТЬ 4
слагаемое в них совпадает), то можно получить радиус вращения
Rg, определив тангенс угла наклона зависимости /расс//о от (95/2)2
(кривая Гуньера), равный —16n2i?c /ЗА2. В табл. 15.2
представлены некоторые молекулярные массы и радиусы вращения
макромолекул в растворе, определенные с помощью рассеяния света или
рентгеновских лучей под малыми углами. Как следует из таблицы,
на основании данных по рассеянию можно определить
молекулярные массы и размеры молекул. Следует отметить, что если
кристаллическая структура и структура в растворе для миоглобина
остается одинаковой в пределах ошибки эксперимента, то для
\* 277,2 А
39,3 А
Вычисленные радиусы вращения Rg для различных тетрамерных
моделей
Модель
Rn. A
Сфера
а
б
в
г
д
Экспериментальные значения
27,7
81',0
47,1
48,7
35,4
40,5
34,4±0,4
Рис. 15.8. Различные модели сборки а-лактоглобулина А — тетрамера,
состоящего из четырех моиомерных единиц, которые в свою очередь образованы
связыванием двух сферических частей.
Сравнение вычисленных и экспериментальных значений радиусов вращения Rq,
полученных при рентгеновском рассеянии под малЫмн углами, позволяет предположить, что
наиболее вероятна модель сборки г. [Wttz /., Tltnasneff S., Luzzati V., J. Amer. Chem. Soc., 86,
168 (1964)0
ГРУЗ НА ПРУЖИНЕ
ЩЙПетур в кристалле^ и растворе фермента а-химотрипсина имеют-
н1$начительные различия (которые еще более значительны, если
Енюльзовать данные рентгеноструктурного анализа для обработке
■рультатов рентгеновского рассеяния по зависимости Гуньера).
шло показано, что осевое соотношение для химотрипсина изме^
рется от 1,3 до 2,0 при переходе от кристалла к раствору — пред-
рложительно потому, что молекула фермента частично развора-
ивается и приобретает асимметричную форму. На примерах пока-
■но, как на основании данных рентгеновского рассеяния под ма-
шми углами можно сделать важные качественные выводы
^структуре макромолекул в растворах.
Ёример. Сборка белковых субъеднниц: а-лактоальбумин А
г а-Лактоальбумин А — это белок, состоящий из субъединиц с молекулярной
рассой 36000. Этн субъединицы соединяются, образуя тетрамеры, имеющие ра-
j^yc вращения /?<з = 34,4±0,4 А. Из известного радиуса вращения субъединиц
$\МШ = 36 600 (см. табл. 15.2) н зная, что эти субъединицы могут в свою
очередь быть (разделены на две сферические единицы с молекулярной массой
£•"18 000 каждая, можно оценить вероятность нескольких возможных способов
}Яборки этих субъеднннц для образования тетрамерной единой молекулы
|рис. 15.8). Можно сразу же легко исключить возможность образования
различных вытянутых структур и прнйтн к прямому выводу о том, что восемь сфер
Щ молекулярной массой 18000) скорее всего группируются в кубическую
структуру. Этот пример показывает, как определение всего одного параметра (в
данном случае Ra) может значительно сузить число возможных макромолекуляр-
Ых структур.
г'
Пример. Обнаружение? пустот в макромолекулах: гемоцианин
i
Как обнаружено при—исследовании рентгеновского рассеяния, гемоциаиин,
^езгемный медьсодержащий голубой дыхательный фермент, обнаруженный
в улитках, имеет молекулярную массу 9-Ю6 и радиус вращения Яс=1б4 А.
Сравнение экспериментальной зависимости интенсивности рассеяния от угла
рассеяния с теоретическими кривыми для молекул, имеющих разнообразные
формы (см. следующий пример), показывает, что гемоцианин ведет себя в
растворе как молекула цилиндрической формы с высотой, примерно равной
диаметру. При этом сразу же возникает затруднение, так как из радиуса вращения и
ртношення диаметра" к высоте можно вычислить объем соответствующего
Сплошного цилиндра, равный 3,66» 10 А3, но это значение вдвое больше объема,
Полученного с помощью других способов, согласно которым объем равен
1,74- 10т А3. Это противоречие можно разрешить, только предположив, что в
середине цилиндра имеются пустоты. Как видно из рис. 15.9, наиболее точно
экспериментальные данные по интенсивности рассеяния в зависимости от угла
рассеяния соответствуют цилиндру с высотой 360 А, внутренним радиусом
гвнутр=74 А, а внешним радиусом г„нешн=164 А.
Пример. Более детальное определение формы макромолекул с помощью
рентгеновского рассеяния под малыми углами: IgG-нммуноглобулины
Благодаря доступности получения чрезвычайно гомогенных
иммуноглобулинов от пациентов с множественной миеломой, в 1969 г. установлены
аминокислотная последовательность и положение дисульфидных связей в 1вО-иммуногло-
булиновой молекуле (рис. 15.10).
С помощью переваривания папаином удалось определить размеры
индивидуальных единиц Fab по их радиусам вращения и форме кривой Гуньера. По-
s
,-2
3W
2-io-2|—
1-10-
Круглые цилиндры, 2r0:h * 1
Рис. 15.9. Сравнение экспериментальной кривой рассеяния (экспериментальные
данные изображены с помощью кружков) для гемодианнна в водном растворе
с теоретической кривой для различных круглых цилиндров, для которых
отношение диаметра к длине равно 1:1.
/ — сплошной цилиндр; 2 — полый цилиндр, у которого отношение внутреннего радиуса к
внешнему ''внутр^внеш =0'3: 3~ полый цилиндр с соотношением ''внутр^внеш =0»5-
р,'=(4я6.Л), Rq —радиус вращения. [Pilz /., Kratky О., Moring-Claesson /., Z. Naturforsch.,
25b, 600 (1970).]
Переваривание
папаино/и
Лереварусзнае
/телсаноМ
Тяжелая цепьОУ
ЛАМЛЛМЛЛЛЛ!
Легкая цепь (Л)
Fab
Папаин Г ~ л Пепсин
[ ГЬЬ ) ( Fab j [ Fab }s~S-[ Fab )
В
Рис. 15.10. Схематическое изображение структуры и расщепления молекулы IgG
иммуноглобулина на различные фрагменты при переваривании пепсином и
папсином.
Предполагается, что два специфических места связывания антигенов расположены на
противоположных концах молекулы в областях Fab. [Pilz I., A'lg. Prakt. Chera.. 21, 21
(1970).]
491 ГРУЗ НА ПРУЖИНЕ
следующие эксперименты по рентгеновскому рассеянию димеров F(ab')2,
полученных при пепсиновом переваривании, позволили получить радиус вращения
#о=53 А, который хорошо соответствует ранее предложенной структуре
«шарнирного» соединения между двумя фрагментами Fab
Расщепленный
пепсином
фрагмент F (аЬ')2
RG-55A
J Л6 = 5в,5А
', Яе(зксп.) * 53 А
Рис. 15.11. Сравнение
вычисленных
зависимостей «поперечных
сечений» lgirbacc-26sl
от квадрата угла
рассеяния 6| для
разнообразных
предполагаемых форм молекулы
IgG
иммуноглобулина.
Вертикальная шкала для
различных кривых
смещена, чтобы совместить
все кривые иа
небольшом рисунке для
наглядности. Пунктиром поха-
заны эллипсоидные
эквиваленты отдельных
фрагментов. Кривые Я и /0
построены по
экспериментальный данный.
(Кривая рассеяния 4
рассчитана для форм
молекулы, полученных путем
изменения угла между
эллипсоидами от 0 до
90°). [Ptlz /., Pachwetn 0„
Kraiky О., fferbst M.,
Haager О. Gall W. E„
Edelmann 0. М.,
Biochemistry, 9, 211 (1970).]
lg(I-20s)
-492 часть 4
Наконец, ряд возможных вариантов структуры для полностью интактной
молекулы IgG-иммуноглобулнна был затем применен для построения теоретических
Зависимостей lg(/pftoo) от (8s)a, используя известные размеры фрагментов Fab
и Fc. Полученные кривые были сопоставлены с опытными данными по
рассеянию (рис. 15.11). Наилучшее совпадение дают структуры, у которых концы
различных фрагментов находят друг на друга, как показано на рисунке для
гипотетической структуры 9. Интересно отметить, что последующий рентгено-
структурный анализ дал структуру, которая подтвердила структуру в растворе,
«олученную нз измерений рассеяния. Этим упомянутая работа выгодно
отливается от многих спекулятивных биофизических исследований, в которых
полученные методом рёнтгеноструктурного анализа структуры различных молекул
в кристалле подтверждались уже после того, как кристаллические структуры
были известны.
15.В. БОЛЬШИЕ ОБЪЕКТЫ, НЕБОЛЬШИЕ ВОЛНЫ, ЛИНЗЫ
МОЖНО ИСПОЛЬЗОВАТЬ: РАССЕЯНИЕ ЭЛЕКТРОНОВ
И РАЗЛИЧНЫЕ ЭЛЕКТРОННЫЕ МИКРОСКОПЫ
В предыдущих двух разделах разбирались основы проведения
экспериментов по изучению размеров и форм непрерывно
движущихся в растворе молекул; при этом обнаружилось, что можно
получать достаточно детальную информацию, когда длина волны
облучения имеет тот же порядок, что и размер частицы. Далее
следует выяснить, каковы же наилучшие способы изучения твердых
объектов при использовании тех же видов облучения. Для
твердых кристаллов, имеющих почти совершенный регулярный порядок
построения молекул, можно дать довольно простое краткое
объяснение природы рассеяния излучения (обычно рентгеновского) с
длиной волны того же порядка, что и расстояние между
соседними молекулами в упорядоченной структуре (см. разд. 22.А). Здесь
же будет рассмотрена более часто встречающаяся ситуация, когда
хотят знать строение некристаллических объектов, таких, как
биологическая клетка, органелла или макромолекула. Требуемые для
этого математические расчеты основаны на тех же принципах
сложения амплитуд рассеянных электрических полей от всех
возбужденных электронов объекта. Однако Детальный анализ уводит
далеко в сторону — в область, изучаемую геометрической .оптикой.
В настоящей дискуссии следует удовлетвориться принципиальным
результатом из геометрической оптики, который относится к
микроскопам, а именно тем, что разрешающая спосрбдаегб.-микроско:
ла (определяемая как минимальное расстояние d между двумя
точкаш! на объекте, которые могут быть четко видны у дается
выражением
где Л — длина волны использованного излучения, п — показатель
преломления среды между объектом и линзой, И а — угол между
осью инструмента и самым внешним лучом, который выходит из
объекта и еще проходит через линзы объектива микроскопа.
(93 ГРУЗ НА ПРУЖИНЕ
Уравнение (15.50) показывает, что, как и при изучении молекул
в растворе, самая тонкая деталь, поддающаяся выявлению в
твердом объекте, имеет размеры порядка длины волны используемого
облучения. Становится ясным, почему так часто между покровным
стеклом и линзой объектива светового микроскопа помещают
масло; это связано с тем, что масло вызывает увеличение как п, так
И а и.таким образом увеличивает разрешающую способность. Из
уравнения (15.50) видно, что разрешающая способность
пропорциональна длине волны, следовательно, самая высокая
разрешающая способность для светового микроскопа может быть
получена при использовании ультрафиолетового излучения {Я—2000 А).
При попытках увеличить разрешающую способность путем
использования излучения с более короткой длиной волны возникают
следующие трудности: во-первых, в области длин волн <2000 А вода
и воздух сильно поглощают; во-вторых, реально существующие
источники еще более коротковолнового излучения испускают
рентгеновские лучи с длиной волны примерно 1 А или гамма-лучи с
длиной волны во много раз короче, чем 1 А. Поскольку не
существует линз, способных сфокусировать рентгеновские или гамма-
лучи, то и нельзя построить микроскопа, работающего на этих
видах излучения. Читателю следует вспомнить, что основная
причина, по которой линзы применимы при оптических длинах волн,
заключается в том, что можно найти вещество, показатель
преломления "которого заметно больше единицы. Как показано в
разд. 14.А, показатель преломления >1,0 прямо связан с
поглощением излучения. Поскольку рентгеновские и гамма-лучи почти
не поглощаются при прохождении через самые мелкие объекты,
то нет вещества, из -которого можно было бы сделать линзу, а
следовательно, нельзя создать и микроскопа для этих лучей.
В ч. 5 говорилось,, что электромагнитное излучение проявляет
свойства частиц (квантов); это свойство наиболее заметно в
спектроскопии. Де Бройль (в 1927 г.) предположил, что частицы
могут проявлять и волновые свойства:
Х — Н/ти, (15.51)
где X — длина волны, соответствующая частице с массой т,
движущейся со скоростью v, h — постоянная Планка, равная 6,62Х
Х10~~27 эрг-с. Уравнение (15.51) позволяет предположить, что
излучение с очень малой длиной волны должно быть связано с
быстро движущимися частицами малой массы. Следовательно, в
качестве «источника света» для микроскопа, построенного на
основании уравнения (15.51), можно логически выбрать электроны, так
жак они заряжены и поэтому могут быть разогнаны до очень
больших скоростей (и соответствующая им длина волны очень мала);
шш этом движение электронов легко контролировать наложением
ййектрических и магнитных полей, что позволяет создать «линзы».
щ качестве упражнения читатель может показать, что эффектив-
494 ЧАСТЬ 4
Лампа
—(Конденсорная
линза
Образец
Н Линза
объектива
Промежуточно в
изображение
I Проекционная
линза
I Электронная '
J пушка
\\ Конденсорная
LJI "инза
Образец
1
/7инза
обгектиоа
Промежуто чное
изображение
[ ^ Промежуточное
I [ проекционные
С-1 /IUH3tl
л
3
Промежуто чное
изображение
Конечные
проекиионные
линзы
Фотопластинка
Глаз (фокусирует W/////M//////M
X /^ окончательное
\ / изображение на
V сетчатке)
Световой, микроскоп
Электрон Hit й микроскоп
Рис. 15Л2. Сравнительная схема, оптических частей светового микроскопа (а)
н просвечивающего электронного, микроскопа (б).
Каждый микроскоп состоит из источника (лампа, электронная пушка), коиденсориой линзы,
лииэ изображения (здесь показана двухлинзовая система для светового микроскопа и трех-
лннэовая система для электронного микроскопа) н детектора (глаз, фотографическая
пластинка). Несмотря иа то что устройство двух микроскопов похоже,, принципы их работы
совершенно различны (табл. 15.3). [Slayier Е. M., in: Physical Principles and Techniques of
Protein Chemistry, Part A., ed. S. J. Leach, Academic Press, New York, 1969, p. 5.]
ная длина волны X электрона,
(вольт), дается выражением
ускоренного напряжением V
(15.52)
Так как электронные микроскопы обычно работают при
напряжениях от~40 кэВ до ~3 МэВ, то соответствующая длина волны
излучения меняется от ~в,06 А до ^0,01 А. Устройства светового
и электронного микроскопа показаны схематически на рис. 15.12.
К сожалению, если" микроскопы, использующие видимую
область светового спектра, могут достигать разрешающей способно-
495 груз НА пружинь:
сти достаточно близкой к теоретической [уравнение (15.50)], то
электронный микроскоп обычно обнаруживает реальное
«разрешение» (минимальное расстояние в пространстве, при котором
изображения двух точек объекта четко различимы) для реальных
объектов (в противоположность разрешающей способности для
гипотетических объектов) в ~1000 раз хуже, чем присущая
электрону длина волны (табл. 15.3). Во-первых, теоретически
невозможно создать отклоняющую цилиндрически симметричную элек-
Таблцца 15.3
Сравнение параметров светового и электронного микроскопа
(см. объяснения в тексте)
Световой микроскоп
Свойство
Электронный микроскоп
2000—6000 А
O.filX
п sin a
2000 А
Длина волны А,
использованного излучения
Теоретическое
ограничение разрешающей
способности
Может быть исправлена Сферическая аберрация
с помощью
рассеивающих линз
Неодинаковое
поглощение различными частями
объекта; различные
длины пути (и потому
разные фазы) для
света, проходящего через
разные части объекта
Использование
красителей для усиления
поглощения объектом по
сравнению с фоном
(позитивные красители) нли фо-
■ном по отношению к
объекту (негативные
красители) для микроскопа
Поглощения; метод
темного поля для фазово-
жонтрастного микроскопа
Относительная
фокуса прн
увеличении
глубина
заданном
Пронсхожденне
контраста при изображении
Методы усиления
контрастности изображения
Электроны с энергией
40 кэВ—3 МэВ,
соответствующие А,=0,06—0,01 А
(0,61 Va)«2A(cM.
сферическая аберрация)
Ограничения связаны с
постоянством ускоряющего
напряжения и тока в
отклоняющих линзах (примерно
одна часть на 105), таким
образом, предельное
разрешение 2 ^ А
500 (улучшение путем
сужения апертуры по
сравнению со световым
микроскопом)
Различные интенсивности
рассеяния для электронов,
падающих на различные
части объекта
Использование позитивных
(или негативных)
красителей для увеличения
рассеяния электронов от объекта
по сравнению с фоном (или
наоборот); оттененне для
выявления рельефа
поверхности; нанесение металла
для выявления поверхности;
сферические аберрации линз
или снижение апертуры для
улучшения контрастности;
метод темного поля
496 ЧАСТЬ 4
тронную линзу, так что не существует способа для коррекции
сферической аберрации. Поскольку сферические аберрации наиболее
заметны для лучей, наиболее отдаленных от оптической оси, то это
(жесткое) ограничение требует уменьшения апертуры электронной
линзы до очень малого значения а [уравнение (15.50)], что
ограничивает практическое разрешение значениями 2 А даже для
электронов с длиной волны 0,02 А.
Поскольку «цвет» движущегося электрона (т. е.
соответствующая ему длина волны) зависит от ускоряющего напряжения
[уравнение (15.52)] и поскольку хроматическая аберрация
(фокусирование разных цветов в различных местах) в электронном
микроскопе не может быть исправлена использованием линз, то
единственный способ избавиться от хроматической аберрации
состоит в монохроматизации источника. Точность, в пределах
которой можно регулировать обычное ускоряющее напряжение 100 кэВ,
также ограничивает практическое разрешение электронного
микроскопа значением ^2 А. Одно из преимуществ ограниченной
апертуры, продиктованной необходимостью избегать сферической
аберрации, состоит в том, что глубина резкости (т. е. толщина
объекта, в пределах которой можно реализовать четкую
фокусировку) для электронного микроскопа значительно больше, чем для
светового микроскопа. Электронный микроскоп, следовательно,
значительно более приспособлен для изучения поверхностей (в
противоположность тонким срезам), чем световой микроскоп при
тех же увеличениях, (см. ниже о «сканирующем» электронном
микроскопе).
На этой стадии может показаться, что даже при ограничениях,
налагаемых сферической и хроматической аберрациями,
практическое разрешение электронного микроскопа должно быть
достаточным для того, чтобы различить отдельные химические связи и
атомы в молекулах. Причина, из-за которой электронные
микроскопы редко выявляют детали ^50 А, кроется в плохом
контрастировании изображения. Контраст изображения определяется как
разность яркости объекта и яркости фона:
•/. контрастам _W^2^_. Ю0. (15.53)
••фон
В световом микроскопе часто возможно (особенно при
использовании красителей) достичь 100%-ного контраста (белый фон,
черный объект), так как поглощение света объектом по сравнению
с фоном может значительно различаться. Однако в электронном
микроскопе контраст возникает, когда рассеянные электроны не
попадают на линзу (см. рис. 15.13). Трудность заключается в том,
что поскольку относительная интенсивность электронного
рассеяния прямо зависит от электронной плотности около атомов
объекта и так как молекулы,, представляющие биологический интерес,
состоят в основном из атомов с низким атомным номером (угле-
497 ГРУЗ НА ПРУЖИНЕ
Образец
Образец
\ Линзы
Апертура
в задней
плоскости
линзы
,^______ . Плоскость
' " ' изображения
Рис. 15.13. Контрастирование в электронной микроскопии благодаря различному
рассеянию от образца и фона.
Вклад в интенсивность изображения могут дать только те прошедшие через обрааец или
рассеянные электроны, которые попадают в апертуру линзы. Если образец толще или
имеет более высокую электронную плотность, чем пленка подложки, то электроны,
выходящие нз образца, в среднем рассеиваются под более широким углом, чем электроны,
выходящие из подложки, по теи же причинам, которые обсуждались ранее в настоящей
главе при рассмотрении угловой зависимости рассеяния света, а — безапертуриые линзы: так
как почти все рассеянные электроны попадают в пределы апертуры лиизы, то нельзя
отличить объект от фона и контраст равен нулю; б — при использовании ограниченной
апертуры электроны, рассеянные под широким углом (т. е. от объекта), преимущественно не
достигают плоскости изображения, так что интенсивность изображения объекта снижена
{за счет электронов, рассеянных в угловой области р" на схеме), в то время как
интенсивность изображения фона почти не изменяется, что и приводит к увеличенному контрасту.
Подобное же улучшение контрастирования происходят за счет сферической аберрации
линзы, в этом случае только электроны, проходящие вблизи оси электронного пучка, хорошо
сфокусированы, в то время как электроны (в основном от объекта), которые проходят
через внешнюю зону линзы, фокусируются плохо, что вновь приводит к снижению
интенсивности в точке появления объекта в плоскости изображения. \Slayter Б. М„ In: Physical
Principles and Techniques of Protein Chemistry, Part A, ed. S. J. Learch, Academic Press.
N. Y., 1969. p. 8.]
род, кислород, азот), то рассеяние от объекта должно быть
подобно рассеянию от (углеродной) пленки, на которой помещен
объект (тонкая, толщиной 20 А углеродная пленка лежит на
металлической сеточке для сохранения формы). Быстрые вычисления
Доказывают, что необходим слой углерода (или образца) в 70 А
)&ри ускоряющем электроны напряжении в 100 кэВ для создания
(5%-ного изменения интенсивности в плоскости изображения (т. е.
498 ЧАСТЬ 4
5%-ного контраста). Проблему контрастирования в электронной
микроскопии можно сравнить с трудностями, которые могли бы
встретиться, если при рассматривании объектов под микроскопом
пользоваться темными предметными стеклами.
До изучения полученных электронных микрофотографий
важно правильно оценить за-висимость между относительными
размерами объектов и соответствующими им массами (или
молекулярными массами). Для сферических объектов с молекулярной
массой М
Af = V3w3pW0, (15.54)
где г — радиус молекулы, р(г/см3)—ее плотность и N — число
Авогадро. Другими словами, видимый диаметр D макромолекулы
на электронной микрофотографии варьирует пропорционально
кубическому корню из величины молекулярной массы:
£ = (6Л?/*р#0)1/з (см), (15.55)
так что две макромолекулы должны различаться по молекулярной
массе в восемь раз, если их видимые размеры на
микрофотографии отличаются в два раза. Таким образом, электронные
микрофотографии плохо пригодны для определения гомогенности
макромолекул по молекулярной массе, так как макромолекулы,
отличающиеся даже в два раза по молекулярной массе, трудно различить.
Исходя из уравнения (15.55), легко показать, что
сферическая макромолекула с молекулярной массой ЛТ=100000 и с
плотностью 1,3 г/см3 (типичная плотность обезвоженного белка)
должна иметь диаметр, равный ~60 А.
Укажем на дополнительные экспериментальные особенности
электронной микроскопии: 1) электростатические и магнитные
линзы (подобно контролю «фокуса» и «сведения» цветов в цветном
телевизоре) позволяют непрерывно изменять фокусное расстояние,
так что увеличение можно непрерывно контролировать; 2) так как
электроны рассеиваются воздухом, то при работе давление.вдоль
оптического пути должно быть ^10_3 мм рт. ст.; 3) «предметное
стекло» оптического микроскопа заменено тонкой углеродной
пленкой, которая лежит на медной сеточке, и 4) выявление
изображения производится на флуоресцирующем экране (как в
телевидении) или с помощью фотографической пленки.
Пример. Позитивное окрашивание — визуализация синтеза РНК
Многие этапы репликации и транскрипции нуклеиновых кислот н синтеза
белка недавно были продемонстрированы на фотографиях, полученных с
положительно окрашенных препаратов (т. е.^Д1репаратов, на которых объекты были
окрашены атомами тяжелых тиеталлов сильнее, чем фон). На поразительной
картине, представленной на рис. 15.14, можно различить экстрахромосомный ядрыш-
499 ГРУЗ НА ПРУЖИНЕ
Направление
синтеза
Рис. 15.14. Увеличение (Х48 000) позволяет наблюдать, каким образом
молекулы РНК-полимеразы активно участвуют в транскрипции ДНК © молекулы-
предшественникн рибосомальной РНК.
Молекулы полимеразы видны как плотные гранулы, прикрепленные к ДНК геиа (длинная
тонкая центральная нить). Каждая молекула полимеразы иа схеме движется слева направо
«о мере прохождения транскрипции. Синтезированные молекулы РНК выглядят как
отдельные нити структуры, напоминающей бахрому, вырастающую из гена. .[Miller О. L., The
[Visualization of Genes in Action, Copyright © March 1973 by Scientific American, Inc. All
t rights reserved.]
500 ЧАСТЬ 4
ковый ген. На микрофотографии видно, как с помощью 80—100 молекул РНК-
полнмеразы одновременно транскрибируется несколько генов с образованием
нитей рнбоиуклеопротеидных молекул-предшественниц, из которых в
дальнейшем образуется рибосомальная РНК. Последовательные стадии синтеза четко
видны в виде растущего ряда молекул продукта. Идентификация различных
компонентов, таких, как ДНК, РНК и белок, основана на использовании красителей,
которые способны избирательно окрашивать эти молекулы.
Пример. Позитивное окрашивание—биологические мембраны
На рнс. 15.15 представлена одна из наиболее четких, электронных
микрофотографий поперечного среза плазматической мембраны клетки ворсинок
кишечника, так называемого ресничатого эпителия. Поверхностные белкн каждой мем-
Внутренняя побсрхпооть мембраш
Т \^^rjf\~^ белки
(20 А)
Внешняя поверхность мемра/л/
|Рис. 15.15. «Трехслойная» структура клеточной мембраны.
Белки внутренней и внешней поверхвости каждой мембраны положительно окрашиваются
солями тяжелых металлов. Электронная микрофотография с увеличением в 230 000 раз.
Фотография сделана со среза кишечника кошки в плоскости, параллельной основной
внутренней поверхности (из которой торчат множественные небольшие полые отростки клетки,
называемые микроворсииками). [Микрофотографии из Pawcett D. W„ An Atlas of Pine
Stirncture: The Cell, W. B. Saunders Co., Philad^tphia, 1966, p. 342: схема из Jain M. K... The
Blmolecular Lipid Membrane: A System, Van Nostrand Reinhold Co.. New York, 1972, p. 11.J
601 ГРУЗ НА ПРУЖИНЕ
браны окрашены ураннлацетатом и цитратом свинца (так как присоединение
тяжелых металлов увеличивает рассеяние). Эта структура понадобится в
дальнейшем при обсуждении движения белков в мембранах (следующая глава).
Пример. Негативное окрашивание — сборка субъединнц фермента.
Некоторые особенно впечатляющие электронные микрофотографии
индивидуальных макромолекул были получены прн использовании негативного
окрашивания (илн, точнее, негативного контрастирования), при котором краситель,
содержащий тяжелые металлы, избирательно связывается с пленкой фона,
оставляя молекулы объекта неокрашенными. Так как почти все электроны,
проходящие через объект, затем фокусируются в плоскости изображения, то на
фотопленке самым темным будет место, куда.падает изображение от объекта, и
потому при отпечатке это место будет наиболее светлым на фотографии.
Сборка ферментов, имеющих близкие к сферическим субъединицы,
представлена на рис. 15.16. Объяснение предполагаемой полимерной структуры
становится более убедительным при рассмотрении молекулы под несколькими углам
зрения.
Рис. 15.16. Электронные микрофотографии и объясняющие нх модели строения
ферментов пируватдегидрогеназы (а и б) н дигндролипоилтрансацетилазы (в иг),
в —показаны отдельные изображения (Х700 000); б — тетраэдрнческие модели, снятые в
соответствующих ориентациях; ей г — показаны различные ориентации трамсацетилазы
Г (Х350 000) и модели в соответствующих ориентациях.
Ъ02 ЧАСТЬ 4
Рис. 15.17. Негативно контрастированиые электронные микрофотографии
(верхние три фотографии) и гипотетическая модель сборки субъединиц фермента;
у нативного фермента два тримера расположены один над другим как бы
затеняя один другой и поддерживаются вместе с помощью трех регуляторных
субъединиц.
-Фото слева Х400 000, остальные данные X360 000. [Микрофотографии взяты из:
Richards К. Е., Williams R. С, Biochemistry, 11, 3393 (1972); модель из: Cohlberg J. A., et aL,
ibid, 3407.]
Сборка фермента, состоящего из несферических субъединиц, показана на
рис. 15.17. Это фермент — аспартаттранскарбамилаза (АТКаза). Отростки регу-
ляториой субъеднннцы четко различимы на электронной микрофотографии при
рассмотрении сверху (при негативном контрастировании фосфовольфраматом),
а «сэидвичевая» структура ясно видна при рассмотрении сбоку, Дальнейшие
подтверждения представлены на микрофотографии каталитической субъединицы,
имеющей треугольную форму (без выступающих отростков). Организация
субъеднииц, показанных иа рис. 15.17, недавно была подтверждена € помощью
рентгеноструктурного анализа иа препарате кристаллического фермента.
Применение метода оттенения и замораживания-скалывания
в электронной микроскопии
Один из старых методов электронной микроскопии, а именно
метод оттенения, недавно- вновь Получил широкое применение в
связи с разработкой метода замораживания-скалывания при изу-
503 ГРУЗ НА ПРУЖИНЕ
чении внутреннего строения биологических мембран. Основная
идея метода оттенения связана со способностью атомов тяжелых
металлов сильно рассеивать электроны. Атомы тяжелых
металлов напыляют на объект под углом к нему, при этом металл
накапливается и образует слой толщиной ~20 А, как показано на
рис. 15.18. Когда покрытый металлом объект затем изучают
сверху в просвечивающем электронном микроскопе, любой выступ на
поверхности становится видимым вследствие дополнительного
накопления металла на объекте, а также вследствие отсутствия
металла на непокрытых («затененных») областях, лежащих рядом с
объектом. При этом контраст значительно увеличивается и
позволяет определять рельеф поверхности высотой в ~20 А, что часто
позволяет увидеть молекулы белков обычных размеров (с
молекулярной массой > 50 000).
Несмотря на то что при изучении макромолекул в электронной
микроскопии метод оттенения в значительной степени был
вытеснен методами окрашивания, он продолжает оставаться очень
удобным методом в сочетании с методом замораживания-скалывания.
Техника замораживания-скалывания основана наследующем
явлении: если биологический объект быстро заморозить, а затем
сломать с помощью резкого удара, то линии излома твердого образца
а
Г////Ъгъ
&
'/У/У/У/У:
Рис. 15.18. Схема метода оттенения.
а — траектории атомов тяжелых металлов, напыляемых на объект для оттенения; б
—накопление металла после завершения оттеисния; в — относительная электронная плотность
образца при рассмотрении под прямым углом к а н б. [Slayter E. M.> in: Physical Principles-
and Techniques of Protein Chemistry, Part A, ed. S. J. Leach, Academic Press, New York,
1969, p. 23.}
504 Ч^Г.ТЬ 4
Рис. 15.19. Схематическое изображение результатов замораживания-скалываиия
клеточной мембраны.
На поверхности вндны выступы и дыры на местах, где определенные молекулы белка были
сорваны с одного из расщепленных слоев. [McNutt N. S., Weinstein R. S., Progr. Biophys.
& Mol. Biol.,, 26, 57 (1973).]
будут следовать пути наименьшего сопротивления. Так как силы,
скрепляющие внешние и внутренние половинки биологических
мембран, явно слабее, чем силы, связывающие молекулы
(«кристаллического») растворителя или сил, которые связывают
растворитель с поверхностью мембраны, то трещина часто возникает
внутри мембраны и при этом открывается ее внутренняя поверх-
506 ЧАСТЬ 4
ность, как показано на рис. 15.19. С помощью метода оттенения
такой замороженной и сколотой поверхности можно увидеть
молекулы белков, которые лежат на внутренней поверхности
мембраны. Таким способом удалось увидеть, как упакованы белки
мембраны в местах соединения клеток. Например, между двумя
клетками печени они образуют типичную плотноупакованную
гексагональную организацию, которая, по всей вероятности,
участвует в ускорении транспорта молекул от одной клетки к другой.
Пример. Изучение топографии родопсина в синтетических двухслойных
мембранах с помощью электронной микроскопии методом
замораживания-скалывания
Предполагается, что когда свет попадает на клетки-палочкн сетчатки, то
его восприятие происходит вследствие действия света на белок — родопсин,
который находится во множестве уплощенных дисков, расположенных друг над
другом и образующих наружный сегмент клетки-палочки. Так как диски
структурно не связаны с наружной плазматической мембраной клетки-палочки, от
которой посылается действующий сигнал к зрительному нерву и затем и мозгу, то
предполагается, что при попадании света из дисков высвобождается какой-то
химический медиатор (вероятно, Са2+), который затем переносится к наружной
мембране клеткН-палочки и инициирует нервный импульс. Некоторые из
наиболее прямых молекулярных свидетельств о ранних этапах вышеописанной схемы
были получены при изучении расположения родопсииовых молекул в
синтетических фосфолипидных двухслойных мембранах методом заморажнвания:скалыва-
ния (рис. 15.20).
Темнопольная и сканирующая (растровая) электронная
микроскопия
На рис. 15.21 представлены две схемы усиления контраста при
электронной, микроскопии в результате смещения оптической
системы детектирования в сторону от оси электронного луча, с по-
Детектор сканирующего
электронного микроскопа
Электроннь/й
прошедший vepee
образец
Пучок параллельно
падающих
электронов
Темнопольный
детектор
Рис. 115.21. Схема детектирования рассеянных назад электронов (сканирующий
электронный микроскоп) или электронов, рассеянных через пленку (темиополь-
ный детектор), как метод для улучшения иентрастиости изображения путем
значительного снижения иитеисивиости фона.
750 ГРУЗ НА ПРУЖИНЕ
Рнс. 15.22. Темноиольные электронные микрофотографии (а — в) небольшой
гетероциклической серусодержащей молекулы меченой ацетатом ртути.
о
Ртуть и сера хорошо видны (отрезок равен 10 А); б — микрофотография одного образца,
в — изображение, полученное путем наложения и усреднения данных с 64 отдельных*
микрофотографий; так как «шум» от фона является беспорядочным, то его интенсивность свшжа-
ется по сравнению с изображением объекта при усреднении. Атом серы (иа рис. в) является
■до сих пор самым маленьким атомом, для которого было получено электроиномикрографи-
,ческое изображение. [OUensmyer F. P., Canadian Research & Development, в, 37 (1973).]
мощью которого ведут наблюдение. Если наблюдают за
электродами, рассеянными в обратном направлении, то этот метод
называется сканирующей или растровой электронной микроскопией.
Измерения обычно проводятся путем сканирования тонкого пучка
электронов вдоль поверхности и посылки соответствующих
сигналов в телевизионную камеру, чей растр (сканирование строки за
строкой на телевизионном экране) контролируется положением
^сканируемого электронного луча, образуя непрерывное
телевизионное изображение образца. В методе «темного поля» оптический
детектор расположен как в обычном просвечивающем электроном
микроскопе (рис. 15.13), но, поскольку детектор находится
достаточно далеко от оси, то падающий луч и большая часть
электродов, рассеянных от фона, проходит мимо него и контраст
усиливается благодаря значительному снижению интенсивности фона.
^Пример. Темиопольная электронная микроскопия при сверхвысоком разрешении
В связи с увеличением контраста с помощью метода темного поля
становиться возможным приблизиться к пределу разрешения электронной оптики,
составляющему ~2 А. На .рис. 15.22, в приведена поразительная картина, на которой
.удается увидеть отдельные атомы в небольшом молекулярном кольце.
'Пример. Сканирующая электронная микроскопия (СЭМ): получение
изображения отдельных бактериофагов
Разрешающая способность сканирующих электронных микроскопов (СЭМ)
недавно была усовершенствована до такой степени, что позволяет увидеть при
высоком разрешении изображения отдельных бактериофагов ЗС (стафиллококко-
вый фаг) и Т4 (фаг кишечной палочкн), как полазано на рис. 15.23. На этой и
других микрофотографиях можно увидеть «головку», «хвост», «базальную пластии-
>ку», «воротничок» и небольшие дополнительные «отростки», отходящие от
«банальной пластинки». Если рассматривать бактериофаг Т4 со стороны хвоста, то
508 ЧАСТЬ 4
ясно выявляется наличие симметрии базальиои пластинки с осью шестого
порядка. (См. разд. 21.В.1 об изучении сборки бактериофага Т4.)
Рис. 15.23. Микрофотография интактного Т4
фага кишечной палочки, полученная на
сканирующем электронном микроскопе.
Видны головка, воротничок (белые стрелка), хвост
и базальная пластинка, которые могут быть
нитевидными. (Более подробно о деталях структуры
см. разд. 21.В.1.) \Broers A. N., Panes В. /., Qenna*
го J. F.. Jr.. Science. 189, 637 (1975).]
Задачи
1. Для данного растворителя, если известны прирост показателя преломления,
длина волны света и угол рассеяния, наблюдаемое рэлеевское отношение (для
интенсивностей) имеет вид
я (6,)=*'им,
где с — концентрация (масса/объем) растворенного вещества, М —
молекулярная масса вещества и К' — константа. Используйте это уравнение для
доказательства того, что значение М, определенное для смеси растворенных веществ,
имеющих разную молекулярную массу, будет обозначать средневесовую
молекулярную массу Mw
Mw=-t- = i . (15.296)
2 [с,] J^'
Замечание. Рассмотрите раствор, содержащий произвольные концентрации
трех веществ, имеющих различную молекулярную'массу, ср. с уравнением
(2.41) и следующими.
2. Поскольку молекулярные массы, полученные методом рассеяния света,
представляют собой средиевесовые молекулярные массы (см. предыдущую
задачу), то, если требуется получить точные значения молекулярных масс,
необходимо в эксперименте исключить присутствие даже следовых количеств
примесей веществ с высокой молекулярной массой.
а. Покажите, что если образец р-аствора фермента содержит 5% примеси —
тримера, то вычисляемая (средиевесовая) молекулярная масса будет
завышена на 10% по сравнению с действительной молекулярной массой мономера.
509 ГРУЗ НА ПРУЖИНЕ
Эти расчеты показывают, каким образом рассеяние света может быть
использовано для изучения олигомеризации макромолекул.
б. Найдите молекулярную массу, определяемую методом рассеяния светаг
для образца с молекулярной массой 34 000, содержащего 1% примеси
вещества с молекулярной массой 1 000 000.
3. В тексте показано, что интенсивность рассеяния света от электронов,
равномерно распределенных по твердой сфере с диаметром D, будет снижена в
число раз, определяемое множителем, который дается левой частью указанного-
ниже уравнения, по сравнению с интенсивностью, которую можно было бы
наблюдать при условии, что все электроны расположены в центре сферы.
Покажите, что этот множитель равен величине, представленной в правой
части нижеследущего уравнения:
acosa); a~pD[2t н- = -?-siti(6J2),
4. Путем (Интегрирования (суммирования) интенсивности .рассеяния света
[уравнение (15.26)] по всем углам
а. покажите, что
■к 2п
VA Sin ^dbsdO =^/pace 90°
8^=0 Ф=0
б. Затем, рассмотрев образец, разбавленный настолько, что ехр[—т]«1— т,
где т — «мутность» [см. уравнение (15.31)],покажите, что
v = Мутность = (16гс/3) R (90°),
где R (90°) — рэлеевское отношение [уравнение (15.28)] для угла рассеяния
в 90°.
5. Вычислите кажущийся диаметр каждой из следующих макромолекул (пред-
положите,- что каждая макромолекула представляет собой твердую сферу/
с плотностью в 1,3 г/см3):
а. М=25 000;
б. М=75 000;
в. М=500 000;.
г. ЛГ=10 000 000.
в. Вирус табачной мозаики является длинной палочковидной макромолекулой,,
у которой экспериментально установленный (путем измерений светорассеяния)
радиус вращения равен 924 А. Выведите зависимость между длиной этой
вытянутой макромолекулы и ее радиусом вращения [используя уравнения
(15.42) и (15.43)]. Затем вычислите длину частицы вируса табачной мозаики
из экспериментального .радиуса вращения и сравните с длиной в 3200 А, по-
лучевдой методом электронной микроскопии.
7. Оцените размеры определенных макромолекул на основании различных
предположений об их форме. Для макромолекулы с молекулярной массой 500 000
а. вычислите радиус молекулы, предполагая, что молекула является твердой1
сферой с однородной плотностью.
б. Вычислите радиус вращения (т. е. видимый размер молекулы из
определений методом светорассеяния), при условиях, что молекула имеет форму
беспорядочиого клубка (предположите, что длина звена равна 4,6 А, молеку-
D/2
Sill ({АГ)
ЦГ
Аш2йг
D/9
= (sina-
1 шЧг
»общ
расе
«НО ЧАСТЬ 4
лярная масса звена равна 50), длинной палочки диаметром 25 А, длинной
палочки диаметром 15 А, длиной палочки диаметром 10 А.
в. Имеет ли значение в этих вычислениях тот факт, что длинная палочка
может иметь квадратное или круглое поперечное сечеиие?
•S. Если кофермеит NAD связывается с дрожжевой глицеральдегид-3-фосфатде-
гидрогеназой (GPDH), то наблюдается изменение в объеме фермента. Это
изменение можно определить количественно путем измерения рентгеповского
рассеяния под малыми углами. Для различных связанных фракций NAD были
получены следующие результаты [Durchschlag Н., Puchwein G., Kratsky О.,
Schuste I., Kirschner К., FEBS Lett., 4, 75 (1969)]:
Фракция NAD,
связанная с GPDH
€ (апофермент)
0,23
0,46
0,72
0,99 (холофермент)
Объем, (А*)
2,64- 10s
2,58-105
2,53 -105
2,49-105
2,45- Ю5
Степень уменьшения
объема, %
0
100
Последовательный аллостерический механизм Кошланда (см. рис. 11.12)
позволяет предположить линейную зависимость между связыванием NAD и
степенью уменьшения объема, в то время как согласно модели, Моно
предполагается нелинейная зависимость. Нанесите иа график приведенные выше
данные с тем, чтобы показать, следует ли аллостерический фермент механизму
Кошланда или нет.
D. Из соотношений X^h/mv (Де Бройля) и eV=mvz/2, где V — ускоряющее
напряжение, е—заряд электрона, m — его масса, h— постоянная Планка,
v—скорость электрона и X'—длина волны, соответствующая движущемуся
электрону, докажите, что
Я = (12,3/}/У(А).
Найдите длину волны в ангстремах, соответствующую движению электрона,
ускоренного с помощью напряжения в 15 000, 40 000 и 1000 000 В.
Литература
Рассеяние света
1. Eggers D. F., Gregory N. W„ Halsey G. D. Jr., Rabinovitch B. S., Physical
chemistry, John Wiley and Sons, New York, 1973, chapter J 5.
Рассеяние рентгеновских лучей под малыми углами
2. Pilz I., Physical principles and techniques of protein chemistry, part A, ed.
S. J. Leach, Academic Press, New York, 1973, pp. 141—245.
Электронная микроскопия
3. Slayter E. M., Physical principles and techniques of protein chemistry, part A,
ed. S. J. Leach, Academic Press, New York, 1969, pp. 2—J58.
-4. Reed L. J., Cox D. L, The enzymes, vol. 1, 3rd ed., ed. P. D. Boyer, Academic
Press, New York, 1970, pp. 213—240.
ГЛАВА 16
НЕСТАЦИОНАРНЫЕ ПРОЦЕССЫ.
ПЕРВОНАЧАЛЬНЫЙ ОТКЛИК
ВНЕЗАПНО СМЕЩЕННОГО ГРУЗА НА ПРУЖИНЕ
Мы уже приводили многочисленные примеры разнообразных
сведений, получаемых из опытов в стационарном состоянии. В них
наблюдают компоненты отклика системы, находящиеся в фазе-
и (или) смещенные по фазе на 90° по отношению к малой,
синусоидальной, постоянной по амплитуде действующей силе. В этих
экспериментах амплитуда отклика всегда была пропорциональна
амплитуде движущей силы (т. е. система была линейна). Отклик
всегда, изменялся во времени по синусоидальному закону с той же
самой частотой, что и воздействие. В этом разделе мы получим эту
же информацию («собственную» частоту колебаний и
коэффициент затухания) из экспериментов другого типа, когда груз на:
пружине внезапно смещают (например, резким ударом) и отклик
пружины регистрируют в отсутствие какой-либо внешней
движущей силы. При этих условиях мы находим, что пружина
колеблется с частотой, близкой к ее собственной частоте <оо с
амплитудой, которая уменьшается во времени экспоненциально.
Постоянную времени экспоненциального затухания называют «временем
релаксации» (поскольку система «релаксирует» обратно к своему
равновесному состоянию), и она обратно пропорциональна
коэффициенту трения (затухания) f.
Разумно спросить, зачем нужно проводить эти новые
«импульсные» эксперименты, если та же самая информация может быть
получена при использовании методов стационарного состояния, на
анализ которых мы потратили так много времени в двух
предыдущих главах. Ответ обычно заключается в самом эксперименте,
поскольку приборы, необходимые для проведения одного типа
опытов, могут быть более доступны, чем для другого. Иногда
интерпретация данных оказывается более простой или более общей,.
Как это имеет место в случае химической релаксации по
сравнению с кинетикой в стационарном состоянии. Кроме того,
«нестационарный» эксперимент может оказаться более удобным; так,.
f гл. 20 мы узнаем, что этим методом данный спектр можно
полупить почти в 10 000 раз быстрее, чем при использовании
аппаратуры для стационарного состояния.
I 5II
512 часть 4
Выведенный из равновесия груз на пружине.
Отсутствие внешней движущей силы
Все примеры этой главы могут быть сведены к решению
задачи о грузе на пружине, когда груз был смещен из положения
равновесия, а затем отпущен при отсутствии в дальнейшем
воздействия внешней возбуждающей силы:
т (cfxjdt*) + / {dxjdt) -f kx *= 0. (13.37)
16.A. ГРУЗ С НУЛЕВОЙ МАССОЙ ПРИ НАЛИЧИИ
СИЛ ТОРМОЖЕНИЯ
СКОРОСТЬ ВОЗВРАЩЕНИЯ К РАВНОВЕСИЮ
Важным частным случаем является (опять) предел нулевой
:массы, когда основное дифференциальное уравнение (13.37)
упрощается до
f{dxldt) + kx = 0, (13.43)
решением которого, как легко проверить, является
* = л:вехр [-*/%]; % = (fjk). (13.46)
Мы начнем эту главу с Применений, основанных на более простом
-случае предела «нулевой массы» (главным образом, для релакса-
дионной кинетики быстрых химических реакций), а затем
перейдем к применениям решения более общего уравнения (13.37)
(оптическая и магнитная деполяризации). [Общее решение
уравнения (13.37) точно такое же, как и решение уравнения (13.43), за
исключением того, что пружина колеблется с частотой, близкой
к ее собственной частоте <ао=Ук/т, в то время как груз экспонен-
диально релаксирует обратно к своему равновесному положению
(гл. 16.Б).]
16.А.1. РЕЛАКСАЦИОННАЯ КИНЕТИКА БЫСТРЫХ
ХИМИЧЕСКИХ РЕАКЦИИ
Медленные химические реакции можно, очевидно, наблюдать,
просто смешивая реагенты и контролируя концентрацию реагента
(реагентов) или продукта (продуктов) во время протекания
реакции. Однако, как видно из задачи 16.1 и табл. 13.2, большинство
лредставляющих интерес химических реакций протекает со
скоростями во много раз большими, чем время, обычно необходимое
(скажем, несколько секунд), чтобы смешать реагенты. Мы уже
видели в части 3, что методы стационарного состояния
обеспечивают нас мощным инструментом для изучения быстрых реакций,
включающих образование промежуточных продуктов, таких, как
•фермент-субстратные комплексы/ К Сожалению (гл. 10.Б.2), фер-
&13 ГРУЗ НА ПРУЖИНЕ
■•)
ментативная кинетика стационарного состояния по Михаэлису—
Ментен, например, совершенно не дает сведений о разнообразных
промежуточных продуктах, которые различаются или химически
(например, различной степенью протонирования, различным
химическим связыванием), или физически [различной конфигурацией:
фермента и (или) субстрата]. Более того, в части 3 было
показано, что для математического описания даже простых
стационарных схем взаимосвязанных химических реакций могут
потребоваться слишком сложные алгебраические преобразования.
Основными преимуществами наблюдения за химическими реакциями
лосле быстрого выведения системы из состояния равновесия по>
сравнению с методами стационарного состояния являются: 1)
возможность изучать скорости быстрых химических реакций с
константами скоростей первого порядка вплоть до 106 с-1; 2)
возможность обнаружить промежуточные соединения между реагентом и:
^продуктом, что резко снижает круг вероятных механизмов, и
!3) простая экспоненциальная зависимость от времени
алгебраического описания возвращения концентраций любых компонентов-
{смеси к равновесным для случая быстрого и небольшого
выведения системы из состояния равновесия, что видно из уравнения
113.46) и строго выведено для общего случая ниже.
IХимический релаксационный эксперимент
Методика проведения опыта в любом импульсном эксперимент
е по химической релаксации всегда одна и та же: а) позволяют*
истеме прийти в состояние равновесия; б) равновесие системы
езко нарушают (нарушают быстро по сравнению со скоростями
юбых интересующих экспериментатора химических реакций), на
езначительно (так, чтобы любые наведенные изменения
концентраций были малы по сравнению с общими концентрациями каких--
[ибо компонентов системы); в) определяют постоянную
(постойное) времени экспоненциального спада [время (времена) «релак-
&ции»], характеризующую приближение концентрации какого-ни-
удь компонента (скажем, одного из реагентов) к новой равно-
есной концентрации; г) повторяют предыдущие операции для-
ескольких концентраций каждого компонента и из концентраци-
нных зависимостей каждого «времени релаксации» выводят меха-
изм химической реакции в системе.
г Поскольку в общем случае на констаыы химического
равновесия влияют давление, температура, электрическое поле и кон-
ентрация (изменение концентрации одного компонента приведет
[изменениям концентраций всех остальных компонентов в данном
равновесии), то можно представить себе получение необходимого-
йлого и быстрого изменения в равновесии посредством короткого,
игльного импульса давления («Р-скачок»), температуры («Т-ска-
1ро>), электрического поля («Е-скачок») или концентрации («ос-
£14 ЧАСТЬ 4
\
I
{
S
Температура
"Рис. 16.1. Зависимость концентрации реагентов А и В и продукта С от времени,
-прошедшего с момента внезапного, малого изменения в константе равновесия
для системы А+В^С.
Разрыв на оси ординат сделай для демонстрации того, что новые равновесные
концентрации отличаются от первоначальных равновесных концентраций на величину, малую по
.сравнению с абсолютными величинами [А]0, [В]0 и [С]0, Константа равновесия внезапно
изменилась до новой величины в момент времени t=0.
•тановленная струя» или «концентрационный скачок»). Перед тем
как перейти к описанию этих экспериментов, желательно
выработать единообразный алгебраический подход, чтобы связать «время
релаксации» с константой скорости и концентрациями в
определенной химической кинетической схеме. Схема, выбранная для
подробного анализа, представляет собой простейший случай, который
включает все элементы, вовлекаемые в более сложные схемы. Она
дает возможность (в отличие от наших задач для стационарного
состояния, требовавших отдельного решения для каждого
изменения в схеме реакций) предсказать без добавочных расчетов
ожидаемые результаты для множества других кинетических схем.
Простейший общий пример химической релаксации: А+В=<=*С
Сначала рассмотрим систему химических реакций в состоянии
равновесия: '
к-г
_ *, _ [С].
Аравн— k_x [А]0[В]0
(16.1)
в которой первоначальные равновесные концентрации такие, как
показано на рис. 16.1. Теперь допустим, что в этой системе
произвели малое, внезапное температурное изменение до повой постоян-
И 5 ГРУЗ НА ПРУЖИНЕ
юй температуры, которой соответствует новая константа равнове-
:ия К
де [А]', [В]' и [С]' — новые равновесные концентрации,
соответствующие новой температуре. Нас интересует, что можно узнать
[з анализа скорости «релаксации» (перехода) к новым равновес-
[ым концентрациям. Поскольку
d [k]idt = d \B]ldt = - d [C]/dt, (16.3)
;остаточно рассмотреть поведение во времени концентрации Bcerq
•дного -компонента, скажем [А]
d\kydt = -k\ [А] [В]+^_! [С]. (16.4)
Далее, поскольку мы имеем дело с малыми изменениями кон-
.ентрацин, а не с суммарной концентрацией ^ак^рр-либо компо-
ента, мы определяем мгновенные разницы концентраций АА, ДВ.
ДС из соотношений (рис. 16.2):
ДА=[А]-[А]' или [А] = [А]' + ДА, (16.5а>
ДВ = [В] - [В]' или [В] = [В]' + АВ, (16.56).
ДС = [С] —[С]' или [С] = [С]' + Д'С. (16.5b).
[А]о
[А]
[А]'
iic. 16.2. Соотношение между абсолютными значениями концентраций [А], \Pi\x
TAT и концентрационными изменениями ЛА и АА' для реагента А в уравнение
|б.5а) Аналогичные соотношения ЛВ и АС использованы в уравнениях
I (16,56, в).
£16 ЧАСТЬ 4
Подставляя значения [А], [В] и [С] из уравнений (16.5) в
уравнение (16.4), получим
дГ([А]' + АА) _d[k]' .rfAA
dt dt + dt
-(поскольку [А]' постоянна во времени)
- =^=-Л'.([А]' + ДА)([В]' + ДВ) + Л'_1(И' + ДС). (16.6)
Кроме того, поскольку одна молекула С получается при реакции
каждой молекулы А с молекулой В, то
ДА = ДВ = -ДС (16.7)
и уравнение (16.6) после упрощения даст
^-=--Л'1([А]' + ДА)([В]' + ДА) + Л'.1([С]'-ДА) =
= -*'ЛА]' [BJ''+^.ЛСГ -ДА^аВГ + ^' + Д^ + Л'.,). (16.8)
Наконец, поскольку скорости прямой и обратной реакций при
равновесии должны быть равны
скорость прямой реакции=скорость обратной реакции
Л', [А]'[В]':=*'_, [С]' (16.9)
и поскольку мы приняли, что изменение концентрации мало по
сравнению с абсолютной величиной самой концентрации
ДА<[А]\ [В]', (16.10)
то, подставляя уравнения (16.9) и (1£.10) в уравнение (16.8),
получим
^■^-ДА(А\ [8]' + *',^]'+*'-,) = -;ДА/« (16.11)
где
|(1/т) .-= к\ [В]' + k\ [A]' + k'-t, г —время химической «релаксации» (16.12)
Уравнение (16.11)—разновидность уравнения —dy/dx=—ky,
которое было решено в гл. 9
ln(AA)/==ln(AA),=0-4-
(16.13)
Уравнение (16.13) показывает, что график зависимости In (ДА) от
времени, прошедшего с момента небольшого, быстрого изменения
.константы равновесия, должен быть прямой линией, которая пе-
17 ГРУЗ НА ПРУЖИНЕ
In (&А')=отрезок на оси и
— -наклон
4000
3000
£=*,([А]+[В])+*_, 'Ч 200°
/000
'Нак/юн= к,"
\6-10 6л/(мо/1б -с)'1
- Отрезок на оси y*k.
~950с-1
а
/23456
/0Ч[А] + [8])
6
Рис. 16.3. а — график зависимости логарифма изменения концентрации от времени для
равновесия
А + В 4 > С
. *-1
»азу после небольшого резкого изменения константы равновесия (например, путем тем-
йгратурного скачка) в нулевой момент времени; б — экспериментальные данные для серии
щытов типа представленного на рнс. 16.3. а при различных концентрациях А и В. График
{авнсимостн (1/т) от ([А] + [В]) представляет собой прямую линию с тагеисом угла
наклона, равным k\, которая отсекает на осн ординат отрезок, равный k—\.
Г График построен для фермента лизоцнма, обозначенного буквой В. а А — ингибитор
bi-N-ацетнлглюкозамнн (см. эксперимент по ЯМР стационарного состояния, представлен»
шй на рис. 14.26 для других опытов на подобных системах). Линейность графика,
покаянного на рис. 16.3, б, подтверждает, что связывание ингибитора действительно является
шиостаднйным процессом. [Данные по лнзоцнму из Chipman D. M., Schimmel P. R.,
|? J. Biol. Chem., 243. 3771 (1968).]
ьсекает ось у в точке In (ДА') и имеет наклон —(1/т), где т опре-
лено уравнением (16.12) (рис. 16.3,а). Наконец, если подобный
гперимент повторяют для нескольких различных концентраций
,-агентов, последующее построение графика зависимости (1/т) от
|Д]'_|_([В]') даст прямую линию с наклоном k[, пересекающую
|ь у в точке k'-\ (рис. 16.3,6), Иными словами, мы нашли путь
^лучения констант скоростей k'-\ и k[ для данной вполне опреде-
1нной кинетической схемы*.
| Читателя не должно смущать большое число шагов в переходе
\ уравнения (16.4) к графику на рис. 16.3. Важный результат
который оказывается общим для широкого набора реакционных
|ем) состоит в том, что при внезапном малом и резком сдвиге
"• Для эксперимента по температурному скачку, ^ представленному на
... 16.11 и последующих рисунках, константы скорости k\ и k- , определяемые
Ь опыте, соответствуют новой температуре. Таким образом, если надо найти
гстанты скорости, например, при 25 °С, необходимо сначала охладить смесь
первоначальной равновесной температуры (скажем, 20 °С), а затем резко
«ять температуру на 5°С (скажем, путем быстрого разряда конденсатора,
этором заключена реакционная смесь) и измерить зависимость :[А] от вре-
518 ЧАСТЬ 4
равновесия концентрации всех участвующих в равновесии
соединений изменяются до своих новых равновесных величин
экспоненциально и постоянная времени для экспоненциальной кривой
непосредственно связана с константами скоростей для данного
механизма реакции.
Интерпретация времени релаксации т для химической реакций
Предшествующий метод представления данных, приведший в
результате к понятию времени химической релаксации т, которое
в свою очередь дает способ определения констант скоростей для
определенных простых кинетических схем, может показаться
отвлеченным расчетом, особенно из-за того, что включает некоторые
неясности и приближения. Однако это не так, и результат
содержит все необходимое для предсказания поведения химических
систем в аналогичных экспериментах для разнообразных
возможных кинетических схем. Чтобы убедиться в этом, необходимо
вернуться назад к кинетике простейших химических реакций первого
и второго порядков.
Время релаксации реакции первого порядка. В начале нашего
обсуждения кинетики стационарных состояний в гл. 9 мы нашли,
что увеличение или уменьшение концентрации компонента А,
подчиняющееся уравнению для скорости реакции первого порядка
d[A]ldt = ±k[A] (9.1)
А-*
Или
* А
можно было охарактеризовывать временем релаксации первого
порядка Та, так что
[А] = [А]0ехр[±г/тА], (9.9)
где
... ч £ I константа скорости первого порядка увели- ,q m
! чения или уменьшения количества компонента А
В случае уменьшения концентрации в реакциях первого
порядка та можно рассматривать как время, необходимое для того,
чтобы концентрация компонента А уменьшилась в е раз по
сравнению со своей первоначальной величиной [А]0.
Время релаксации реакции второго порядка. Для химической
реакции второго порядка, такой, как
k ./
A -f A—*■ продукты, (16.14)
19 ГРУЗ НА ПРУЖИНЕ
рротекание реакции во времени больше не является
экспоненциальным, поскольку
d[A]/dt = -k[AY, или d[A]/{AY^-kdt. (16.15)
Это уравнение легко интегрируется, давая
(1/[А]) = (1/ГА].) + &, (16.16)
[ описываемый этим уравнением процесс, конечно, не является
(Кспоненциальным. Однако если бы мы не знали, что реакция име-
ta второй порядок, и описали бы ее как процесс первого порядка
d[Al/£ft=-(*[A])[A] = -(l/xA)[A]l (16.17)
о в результате мы определили бы время релаксации псевдопер-
юго порядка тд в виде
(1/тА) = £[А]. (16.18)
В обычной кинетике, где изменения концентраций велики и,
озможно, даже сравнимы с первоначальными концентрациями
«агентов, ошибочность этих определений [уравнения (16.17) и
16.18)] быстро бы выявилась при построении графика изменений
о времени определенной экспериментально концентрации како-
6-либо компонента [см. уравнение (16.16)]. Несмотря на это, за-
;ечательной особенностью импульсного кинетического эксперимен-
й, описанного ранее в этом разделе, является то, что малость
наблюдаемых изменений концентраций с достаточной степенью точ-
рсти позволяет применять уравнения (16.17) и (16.18) в описании
Установления равновесия химической реакции второго порядка,
ия более общего случая
A-f В-^продукты (16.19)
!меем
d[A]/dt=-k[B\ [А] = -(1/т ) [А], (16.20)
(1/хА) = k [В]
нелогично из скорости исчезновения В находим
(l/tB) = A[Aj
(16.21)
(16.22)
езюмируя уравнения (16.14) — (16.22), заключаем, что общее об-
|тное время релаксации (1/т) для (экспоненциального) восста-
|вления концентраций всех химических компонентов для
скороди химических процессов первого порядка будет определяться
сожителем k, а для реакции второго порядка (А+В) —множите-
520 ЧАСТЬ 4
лем k[В], где k[В] следовало бы называть обратным временем
релаксации для (экспоненциального) изменения компонента А:
1п(Д/), = 1п(Д/)'— (1/х)/,
в котором (1/х) =2(1/Ti)'
I
де (1 At) — * (Д^ каждой реакции
первого порядка компонента i)
и 0А/) = ^ [/] (для каждой реакции
второго порядка компонента i
с компонентом /)
В уравнении (16.23) суммирование проводят по всем компонентам,
участвующим в равновесии.
Например, мы уже показали, что концентрации и А, и В, и С
ki
в ряде реакций А+В^С будут изменяться во времени до новых
k-i
равновесных величин после небольшого быстрого изменения
константы равновесия, подчиняясь экспоненциальному закону с общей
постоянной времени t
0Л) = МА1 + МВ1 + Л-,. (16.12)
-Теперь ясно, что величины &i[A], &i[B] и k-\ представляют собой
вклады в общее обратное время релаксации (1/т) за счет
обратного времени релаксации (второго порядка) для исчезновения В,
(1/тв) =&i[A], обратного времени релаксации (второго порядка)
для исчезновения А, (1/ta)=&i[B], и обратного времени
релаксации (первого порядка) для исчезновения С, (l/tc)=£-i. Другими
словами, мы теперь умеем предсказывать вид выражения общего
«времени химической релаксации» для многих типов простых
равновесий, показанных в табл. 16.1.
Очевидно, можно полагать, что для равновесия, включающего
два процесса первого порядка (первая реакция в табл. 16.1),
общее обратное время релаксации будет суммой двух отдельных
обратных времен релаксации первого порядка для исчезновения
А или В. В другом примере — равновесии А+В^±:С, в котором кон-
*-i
центрацию В поддерживают постоянной (например, используя
буфер, табл. 16.1), в общее обратное время релаксации будут вносить
вклады отдельные обратные времена релаксации для
исчезновения A, (1/ta)=£i[B] и С, (1/tc)=£-i, но неВ, поскольку его
концентрация постоянна и «время релаксации» для изменения
концентрации В фактически бесконечно, так что вклад от (1/тв)
равен нулю. В табл. 16.1 приведен также ряд других полезных
результатов вычислений, а общий ход расчетов должен теперь быть
понятен по крайней мере для простых равновесий.
(16.13)
(16,23)
(16.24а)
(16.246)
Таблица 16.Т
Соотношение между временем химической релаксации т, константами скоростей)
и концентрациями для различных кинетических схем*
Схема реакций Обратное время химической релаксации (1/т)
Л 7-=» В (1/Т) = *! + £_!
А + В :?=>= С (1/т) = К [А] + К [В] + А-!
А + В z^ C + D (l/T) = ft1[A] + A1[B] + *_I[C]+A_1 |D]
*-i
f
A + C *^fc B + C (1/т) = МС] + *-i[C]
(С — катализатор)
iA+B ^C (1/т) = МВ] + *Ч
5([B] остается
постоянной)
A + B + C^D О/т) - MA] [В] + MA] 1С]+МВ][С| + *_*
\- Ь~Х
|A + A^fe A, (1/t) = 4MA] + *-i
v.
|^+B + B ^ AB2 (1/t) = 4MA][B] + MB]2 + A-i
, j^Tr^E+A+B^ZZ^: (1/т) = (*a + I^AT+tlBf)<lEA] + lB]) +
Ht*-i Mr
feA+B =^EB + A +(^-з+Л[А]*Д[В])([ЕВ]+[А]>
^реакция обмена) К W+ < Wl - *k Ю + *x [ВД + A-, + *a + *-,
LID *i [(1/т1)Х(1/т2)] = ^ ([A] + [B]) (A, + ft_8) + *_!*_,
&
A_! (Ti — время релаксации для (экспоненциальиого>
k перехода либо fA], либо [В| к новому равновесному
± АВ <■ ' > С значению; та— время релаксации для перехода ГАВ"|
ft_? к новому равновесному значению)
622 часть 4
Продолж. табл. 16.1
Схема реакций Обратное время химической релаксации (1/т)
fei *a Имеются три времени химической релаксации, самое
с + 5 ч >- Xj ч ■ * медленное из котооых тя получают по уоавнению
*—1 *—2
медленное из которых тз получают по уравнению
=t Х2 ч=ь Е + Р
(механизм Михаэли- Aj
са — Ментен с двумя (W- ^ [S] -f *-1^-а IE] + *-i*-s [p] + *-!*-» +
еКеТияТм°и)НЫМИ C°" + WE] + M-, [E] [[EJ + [S] + [PJ]
, *z2
+ . , Kb [S] + k^k-, [P] + Мз [El + *-Л
■+■ А-^-з [EJ + W [E] [[E] + [S] + [PJ]
* Вторая схема подробно разобрана в тексте; остающиеся четыре верхние схемы дают
выражения для (1/т). которые могут быть выведены из примера в тексте без
дополнительных разъяснений. Схема вторая снизу используется в задаче к этой главе н демонстрирует
общий подход к рассмотрению схемы произвольной сложности. Необходимо отметить, что
see констзиты скоростей для любых механизмов, приведенных выше, ыожно определить
экспериментально из графика зависимости (1/т) от концентраций различных участвующих
в реакциях компонентов (примеры см. в тексте).
Пример. Константы скорости для равновесий фермент — субстрат
Мы видели в гл. 10, что стационарная ферментативная кинетика дает в
общем случае константу скорости появления продукта и константу
псевдодиссоциации /Са, которая является мерой прочности связывания субстрата с
ферментом. Изучение быстрой химической релаксации после резкого небольшого
импульса, который нарушает существующее равновесие, дает подход к
определению индивидуальных констант скоростей взаимодействия фермент — субстрат
вместо неразделимой комбинации констант скоростей Кк*={к-\ + к2)1к\ в
экспериментах со стационарным состоянием. Диапазон констант скоростей для
связывания субстрата с ферментом и диссоциации этого комплекса показан
в табл. 13.2. Для физиологического (т. е. «природного») субстрата константа
скорости второго порядка для процесса связывания обычно находится в
диапазоне от 107 до 108 М-1-с-1, т. е. немного ниже вначения, ожидаемого для
контролируемого диффузией связывания реагентов (см. задачи к этой главе).
Однако, когда используют искусственные субстраты, лишь незначительно
отличающиеся по способности к связыванию илн по конфигурации от физиологических
субстратов, константа скорости их «посадки» при связывании может быть
значительно меньше, что подтверждает критическое значение «подгонки» между
субстратом и активным центром фермента для процесса связывания.
Константа скорости «отхода» для .реакции диссоциации первого порядка
фермент-субстратного комплекса является мерой прочности связи субстрата с ферментом.
Наконец, из анализа зависимости константы скорости «посадки» в процессе
связывания от рН можно получить информацию о значениях рКа, способных
к ионизации групп фермента, которые определяют эффективность процесса
связывания. Интересно, что эти значения рд0 могут отличаться от наблюдаемых
для тех же самых аминокислот в малых полипептндиых цепях, что указывает
иа определенный способ укладки полипептидных цепей в третичной структуре,
при котором два или более заряженных звена в цепи помещаются рядом.
Многочисленные примеры всех этих эффектов интересующийся читатель может
найти в литературе к этой главе.
523 груз на пружине
Пример. Химическая релаксация как индикатор конформационного изменения
в макромолекуле
В экспериментах по химической релаксации изменения концентраций обычно-
обнаруживают по изменению оптического поглощения, рН или проводимости
раствора. Однако изменение оптического поглощения может быт*, результатом»
конформационного изменения вблизи поглощающей химической группы. Это дает
возможность изучать механизмы, включающие стадии, проходящие без
изменений в ковалентном связывании, такие, как изменение конформацни фермента
после связывания с субстратом:
E + S^X,^X2. (16.25)
Существует хорошо определяемая постоянная времени химической релаксации,
связанная со второй стадией уравнения (16.25), численное значение которой
имеет порядок от — НО— * до 10~* с для ряда изученных конформационных
изменений в ферментах. Убедительным примером такого процесса является время
релаксации для конформационной изомеризации рибонуклеазы. Константы
скоростей конформационного изменения зависят от рН в соответствии с механизмом
Е + H+t;EH^T (EH)', (16.26)
*-i
в котором EH и (EH)' представляют собой различные конформации фермента*
Время релаксации для этой схемы (где реакции первой стадии считают горазда
более быстрыми, чем второй) определяют по уравнению
(l/*)==*-i+*i(l+pprj)- (16.27)
На рис. 16.4 показаны экспериментальная и теоретическая рН-зависимости (1/т)».
и совпадение их просто превосходное. Вместе с другими независимыми
исследованиями эти результаты привели к тому, что был предложен механизм^ в
котором конформационное изменение связывают с открытием и закрытием желобка
в структуре фермента, соединенного с активным центром для связывания
субстрата.
2,8
2.4
*. 2,0
'о
►Г 1,6
0,8
5,5 6,0 6,5 7,0 7,5
РН
Рис. 16.4. Зависимость обратного времени химической релаксации 1/т от рН\
характеризующая конформациониую изомеризацию фермента рибонуклеазь»
при 25 °С.
Сплошная линия рассчитана согласно теоретической модели [уравнение (16.26)] при нс-
нользования уравнения (16.27) с ft,-2468 с-1. *—1—780 c-i и рКа=6,1. [Из French Т. С, Нат~
mes О. О., J. Amer, Chem. Soc, 87, 4669 (1965).]
J I I I I I I L
£24 ЧАСТЬ 4
Пример. Многостадийные механизмы. Кооперативные эффекты в комплексе гли-
церальдегид-3-фосфатдегидрогеназа — NAD
Когда в химической реакции имеется много взаимосвязанных стадий»
индивидуальные стадии можно наблюдать отдельно, если связанные с ними времена
релаксаций х существенно различаются. Хороший пример (с важными
приложениями) — связывание NAD с дрожжевой глицеральдегид-3-фосфатдегидрогена-
зой (ГФДГ), при котором наблюдают трн характерных времени релаксации
{рис. 16.5). Первое быстрое [(1/ti) =7000 с-1] изменение в спектре
обусловлено связыванием NAD с R-формой фермента (см.чниже). Следующее более
медленное изменение ['(1Ут2)=690 с-1] обусловлено связыванием NAD с Г-формой
фермента. Наконец, гораздо более медленное изменение ['(1/тз)=0,9 с-1]
обусловлено конформацнонным переходом между Я- и Г-формами фермента
ГФДГ (рис. 16.6 и сопровождающее его обсуждение).
Две простейшие механические модели кооперативного поведения при
связывании субстрата с ферментом были обсуждены в гл. 11.Б.2. Их важнейшие
отличительные особенности показаны на рис 16.6.
Подробный,анализ модели Кошланда позволяет предсказать, что для
системы должны наблюдаться один релаксационный процесс второго порядка н
четыре процесса первого порядка, в то время как согласно модели Моно— Уай-
маиа — Шаиже должны быть два процесса второго порядка н один — первого.
Метод релаксационной химической кинетики особенно полезен для
доказательства существования минимального количества стадий, так что наблюдение более
трех характерных времен релаксации могло бы послужить, например,
исключению модели Моно из рассмотрения. В системе NAD — ГФДГ наблюдаются трн
времени релаксации, н их соответствующие концентрационные зависимости
находятся в полном соответствии с моделью Моно. Система NAD — ГФДГ — один
из наиболее убедительных примеров того, что можно сделать выбор между
моделями Моно и Кошланда для аллостерического связывания субстрата с по-
лисубъединичным ферментом. Ключевым свойством этой системы, позволившим
сделать такой выбор, явилась большая разница между временами химической
релаксации. При связывании кислорода с гемоглобином, например, также
наблюдают кооперативное связывание, йо здесь релаксационная кинетика ие
оказалась особенно полезной, поскольку характеризующие этот процесс времена
релаксации слишком близки по величине, чтобы их можно было разрешить как
индивидуальные линейные отрезки на полулогарифмическом графике (таком, как
на рис. 16.3,а).
1мс
Н
1мс
г
I—I
1с
- =7000
*1
— = 690
- = 0,9 с-'
Рис. 16.5. Изменения оптического поглощения (365 нм) в зависимости от
времени для системы, содержащей |[NAD]*=6-10-* М; [ГФДГ] =1,4-10-* М тетра-
меръ; пирофосфлт 0,05 М, рН 8,5, и [ЭДТА1=5-10-' М, после скачка
температуры от 40 до 45 •С.
Обнаруживаются трн отчетливые ступени релаксации, которые могут быть связаны с
поведением, ожидаемым из аллостерической модеДи» Моно — Уаймана — Шанже fcH. текст).
1Из Ktrschner К, Eigen Af., Bittman R.. Volght В., Ргос. Natl. Acad. Scl. USA. 66, 1681
(1966).]
ГРУЗ НА ПРУЖИНЕ
ЙШкШ 88 88
ffihsffi 88
tfKffl\8R
х
-J
L L
Nl_J—I
\
L
-ll
аш\
\<а
ffffi>\
4L +
3L +
2L +
L +
'
L
L
L
L
■^ (L
+ 4L
+ 3L
+ 2L
+ L
1
Ъ
а
в
ffl*
Щ£> *. CD®
Г.'
Рис. 16.6. Набор (гл. Ц1. Б. 2) аллостерических моделей для кооперативного
j:. связывания субстрата с тетрамером фермента.
ЙМодели Моно — Уаймеиа — Шанже (б) и Кошланда (в) рассматриваются как
специальные предельные упрощения обобщенной модели (а), в которых лиганд (L) связывается
| субъединицами фермента, существующими в двух конформациях, обозначенных на рн-
■ сунке кружками и квадратами.
Г.:'
|1ример. Электрическое двулучепреломление и дихроизм. Вирус табачной мозаи-
J ки (ВТМ)
г
I: В гл. 14.Е бьшо показано, что «коэффициент трения», связанный с откликом
Совокупности электрических диполькых моментов на действие внешнего
стационарного переменного электрического поля, отражает скорость вращательной
переориентации диполей в растворе. Когда эта переориентация происходит
посредством многочисленных маленьких произвольных по величине угла ступеней,
процесс можно рассматривать как произвольное вращательное движение, которому
^соответствует коэффициент вращательной диффузии DBp, изменяющийся обратно
Молекулярному объему (крупные молекулы вращаются медленнее). В этом
£азделе мы просто добавим, что очень похожую информацию можно получить,
оздействуя на раствор, содержащий электрические диполи, резким малым
импульсом электрического поля. На самом деле, даже нет необходимости в том,
|йтобы дипольиые моменты существовали постоянно — они могут возникнуть в
результате «поляризации» заряда на молекуле при включении внешнего
электрического поля. Затем фиксируют степень ориентации молекул как функцию
времени так же, как мы фиксировали изменение концентрации в зависимости от
Времени, прошедшего с момента сдвинувшего равновесие импульса, в примерах
Но химической релаксации. В эксперименте с электрическим полем,
генерирующим стационарное синусоидальное возмущение (гл. 14.Е), измеряют
электрический отклик (емкость, проводимость). В нестационарном эксперименте с
возмущением, создаваемом быстрым импульсом электрического поля, легче
регистрировать оптическое двулучепреломление или дихроизм после наложения или
снятия импульса электрического поля, как показано на рис. 16.7,а.
Экспериментальный пример уменьшения оптического двулучепреломлеиия в зависимости от
Рремеии, прошедшего после снятия внешнего постоянного электрического поля,
526 ЧАСТЬ 4
показан на рис. 16.7,6 для раствора, содержащего ВТМ. Экспериментальная
кривая определяется наложением процессов с двумя характерными
вращательными скоростями [соответствующими временам вращательной релаксации
(гл. И6.Б и 21.Б) для двух отдельных прямолинейных отрезков
полулогарифмического графика]. Учитывая этот, а также контрольный эксперимент с
раствором, содержащим мономериый ВТМ, результаты, показанные на рис. 16.7,6,
интерпретируют как соответствующие вращательной диффузии мономера ВТМ
"(tbpse1/Dbp=0,5 мс) и более медленной вращательной диффузии димера ВТМ,
образованного по принципу «конец к концу» (твр=3,0 мс). Из величины
наблюдаемого двулучепреломлеиия (в противоположность его временной зависимости)
было рассчитано, ,что ВТМ (штамм Holmes Rib Grass) обладает огромным
электрическим дипольным моментом (большим, чем 10 000 Д). Эти эксперименты
обеспечили прямое измерение коэффициентов вращательной диффузии, а значит,
размера и формы молекул.
Пример. Ультразвуковая релаксация и образование иониых пар в водных
растворах солей
При разработке ультразвукового локатора еще в 40-е годы было
обнаружено, что морская вода гораздо сильнее поглощает ультразвук, чем
дистиллированная. И хотя соленость морской воды определяется в основном хлористым
натрием, было показано, что поглощение ультразвука морской водой зависит от
присутствия сульфата магния. Как отмечено в гл. 14.Ж, критическая частота
Экспериментальная кривая
Внешнее электрическое
поле (£)
Наблюдаемое
двулучелреломление (an)
а
f,2 2.4
Время, мс
Рис. 16.7. Теоретическое (а) и эксперимеитальиое (б) изменения оптического
двулучепреломлеиия An=rt|— п ^ как функция времени, прошедшего с момента
наложения или снятия постоянного воздействующего на обрааец внешнего
электрического поля Е.
Экспериментальная зависимость отражает наличие двух различных релаксационных
процессов (см. текст), соответствующих вращателырЬй диффузии мономера вируса табачной
мозаики и его днмера «конец к концу». [Экспериментальные данные из Okonskt С. Т.,
Halter А. /., J. Amer. Chem. See. 78, 3604 (1956).]
ГРУЗ НА ПРУЖИНЕ
В
II
Сближение /
Вытеснение^, Знедренце
I
ш
Вытеснение
IV
Рис. 16.8. Поглощение ультразвука в зависимости от частоты (а) для
типичного электролита 2:2, такого, как MgS04l в воде и предполагаемый механизм-
{б), который объясняет число и концентрационную зависимость трех
характерных времен- ультразвуковых релаксаций, соответствующих трем ступенькам
в частотной зависимости поглощения ультразвука.
[Основано на работе: Eigen M., Z. Phys. Chem. Frankfurt. 1, 176 (1954).]
Состояние I, свободные ноны; состояние II. сольватно разделенные ионные пары;
состояние III, нонные пары с объединенными сольватными оболочками; состояние IV,
контактные ионные пары.
<йКр при частотной зависимости поглощения ультразвука позволяет определить
время релаксации хкр= (1/о)кр), характеризующее процесс, в котором поглощение
ультразвука делается возможным благодаря изменению объема. Если этот
процесс является химической реакцией, сопровождающейся общим изменением
•©бъема, тогда время ультразвуковой релаксации можно интерпретировать на
основании времени химической релаксации для интересующего нас механизма
реакции. На рис. 16.8,а показана схематическая диаграмма поглощения
ультразвука в зависимости от частоты для раствора, содержащего электролит 2:2,.
такой, как сульфат магния. На графике видны три отдельные ступени,
соответствующие по крайней мере трем отдельным химическим равновесиям.
Общепринятый механизм, которому соответствуют эти ступени, был выведен Эйгеном
«з концентрационных зависимостей трех времен релаксации:
А+В
D
(16.28)
я показан более подробно на ряс. 16.8,6. Одним из наиболее интересных
выводов из таких экспериментов является представление о существовании в самой
воде относительно упорядоченных или неупорядоченных областей, находящихся
■между собой в равновесии. Это равновесие устанавливается слишком быстро
(т=10-12 с), чтобы следить за ним методом частотной зависимости поглощения
-ультразвука. Однако смещение равновесия может быть замедлено добавлением
* воде глицерина с последующей экстраполяцией результатов назад к нулевой
-концентрации глицерина. Добавление хорошо известных денатурирующих белок
.агентов, таких, как мочевина и хлоргидрат гуанидииа, приводит к «раэрушеник»
528 часть 4
«упорядоченной» структуры воды. Таким образом, может оказаться, что
основным эффектом действия этих денатурирующих агентов является изменение
структуры растворителя, который в свою очередь изменяет структуру
макромолекул. В настоящее время проводятся многочисленные эксперименты по
изучению этого интересного явления.
16.А.2. КИБЕРНЕТИКА. МОДЕЛИ «ЧЕРНОГО ЯЩИКА»
В ФИЗИОЛОГИИ
В гл. 12 была использована близкая аналогия между фермент-
субстратной моделью и моделью лекарство-рецептор для переноса
метода математической обработки по способу Михаэлиса — Мен-
тен на физиологические проблемы. В этом разделе мы покажем,
как те же самые математические операции, которые используются
для описания изменений химической концентрации в реакционном
сосуде, можно применять для анализа изменений концентрации
С02 при дыхании in vivo. Математическое описание
физиологических процессов изучает «теория контроля». В основе этой теории
лежит предположение о том, что все физиологические процессы
можно моделировать «черным ящиком» и описывать отношения
между тем, что поступает в «черный ящик» и что из него выходит
с помощью соответствующих дифференциальных уравнений.
Поскольку дифференциальные уравнения в физиологическом случае
почти всегда слишком трудны для точного решения, стало модным
строить «эквивалентные электронные цепи» (гл. 14.Д), такие, как
мы строили для диэлектрической релаксации (гл. 14.Е). В этих
цепях подводимое и снимаемое напряжения соотносят с
физиологическими входными и выходными данными (концентрациями
метаболитов, давлением крови и другими) с помощью различных
сопротивлений и конденсаторов, которые в свою очередь ставят
в соответствие со свойствами интересующего нас
физиологического «ящика». Модель оценивают по тому, соответствуют ли ее
выходные данные физиологическим выходным данным в широком
интервале предписанных входных данных. Модели для ряда
физиологических систем, включая модели для дыхательной (см.
ниже) и сердечно-сосудистой систем, рефлекса сокращения мышцы,
регуляции содержания глюкозы в крови, регуляции температуры
тела и многочисленных функций нервной системы, дают довольно
хорошее приближение.к экспериментальным результатам.
Пример. Модели «черного ящика» для дыхательной функции
Для понимания моделей дыхательной функции сначала необходимо
познакомиться с природой биологического контроля скорости дыхания. Действия трех
наиболее важных факторов, влияющих на скорость дыхания («вентиляции»)
человека, показаны на рис. 16.9а. На первый взгляд кажется, что, поскольку
снабжение кислородом крови и затем тканей является основной функцией
дыхания, скорость дыхания должна контролироваться концентрацией кислорода
в крови. Вопреки этому из рис. 16.9 а видно, что концентрация кислорода в кро-
|9 ГРУЗ НА ПРУЖИНЕ
Рнс. 16.9. Влияние артериальной концентрация С02, 02 и рН на скорость
дыхания (а) и изображение дыхательной системы как управляемой системы (б),
чувствительной к трем регулируемым параметрам.
Уд — скорость альвеолярной вентиляции (альвеолы — это крошечные заполненные воздухом
мешочки, через которые молекулы газа диффундируют в кровеносные сосуды легкого).
(Данные по вентиляции из Gugton А. С, Textbook of Medical Physiology, 2nd ed. W. B.
Saunders Company, Philadelphia. 1961, p. 556)
вн почти не оказывает влияния на дыхание, исключая очень низкие уровни
концентрации кислорода. Наиболее важным фактором, контролирующим скорость
дыхания, является концентрация в крови С02. Когда дыхание слишком
медленное, концентрация С02 возрастает и система стимулирует увеличение
скорости дыхания, чтобы привести концентрацию С02 к норме. Аналогично, если
умышленно вызвать гипервентиляцию (вдохи быстрее, чем необходимо),
концентрация СОг падает, и подается (непроизвольно) сигнал задержать дыхание. Этим
объясняется, например, необходимость заставлять себя дышать, поднявшись на
•большую высоту над уровнем моря. В этом случае парциальное Давление С02
в атмосфере понижено, а механизм контроля дыхания «думает», что тело дышит
«лишком быстро, и намеренно замедляет дыхание, несмотря на то, что
давление кислорода также понижено и на самом деле дыхание нужно усилить!
[Центр контроля скорости дыхання перестраивается должным образом через
<утки (или около того) акклиматизации на большой высоте.]
Влияние рН на скорость дыхання связано в основном также с
концентрацией СОь поскольку концентрации Н+ и С02 взаимосвязаны
Н+
+
С02 + Н20 £ Н2С03 ^ НСОг £ Н* + НСО:
(16.29)
Когда, например, рН артериальной крови искусственно понижают, ГН+1
становится выше, и все равновесие в уравнении (16.29) сдвигается влево, чтобы
компенсировать увеличение [Н+]. Это приводит в результате к увеличению
концентрации С02, что в свою очередь увеличивает, как мы уже отметили, скорость
дыхания.
530 ЧАСТЬ 4
Три фактора: [COJ, [Н+] и [Оа],— которые контролируют скорость
дыхания, могут быть использованы для построения модели системы контроля
дыхания, простейший вариант которой представлен на рис. 16.9,6. УпрощаяN модель,
еще больше, мы пренебрежем эффектами [Н+] и [OJ и рассмотрим только»
систему обмена СОа, который переходит из легких в артерии, потом в ткаии,
потом в вены и потом обратно в легкие, как это показано на рис 116.10. Эту
систему (рис. 16Л0) можно описать пятью уравнениями (два дифференциальных
и три алгебраических) с пятью неизвестными: [С02]л (концентрация С02 в
легких); [C02]tic (концентрация С03 в тканях); q\ (скорость удаления СОя из
легких с выдыхаемым воздухом); 9а (скорость, ухода СОя из легких с артериальной
кровью); q% (скорость поступления С02 в легкие с венозной кровью). Например,
d [С08]тк
dt
Цг — Я» + (скорость образования метаболического СОа)
(объем тканевого резервуара С02)
Второе уравнение:
d [СОа]л ^ ^СО,+ Ь - ft— Q*
dt (объем легочного резервуара)
(16.30а)
(16.306)
в котором ^со2 — содержание С02 во вдыхаемом воздухе. Наконец, есть еще
три равновесных соотношения (16.30 в, г, д)
[СО,]л= (ft/Уд).
(1б.30в)
Уравнение (16.30в) выражает равенство концентраций СОг в альвеолах и
выдыхаемом воздухе.
[^/(производительность сердца)] = Р (наклон) [С02]л + пересечение с осью
ординат. (16.30 г)
Уравнение (16.30г) описывает равновесие между артериальной и альвеолярной
концентрациями С02 посредством вымышленного линейного графика для
поглощения СОя как функции внешнего барометрического давления Р (это уравнение
Скорость MemqSojtU'
ческой выработки СОг
Чз
Венозная
Легочный
резервуар
Тканевый
резервуар
Ссог]тк
Артериальная L
9г
Рис. 16.10. Схематическое изображение обмена С02 между легкими, кровыо>
н тканями.
Термины разъяснены в тексте. |[Из Grodins F. S., firay J. S., Schroeder К. R., Nortns A. L.„
Jones R. W., J. Appl. Physiol., 7, 283 (1954).]
ГРУЗ НА ПРУЖИНЕ
Л,
■ЛЛАЛг
eAt)
e2U)
to
t[t)
-ллл/v
=^c,
Рис. 16.11. Электронная схема-аналог изолированной регулируемой системы (см.
|>ис. 16.10) как части дыхательной системы, в которой регулирование основано
; на изменении концентрации С02.
Поведение этой схемы будет точно соответствовать поведению модели, описываемой
уравнениями (,16.30).
Обозначения: R\ — Vд— скорость альвеолярной вентиляции; Rt — производительность
сердца; Ci — объем легочного резервуара; Cj — объем тканевого резервуара; {— скорость
метаболической выработки СОа; ei(0 = [COa] л— концентрация COi в легких; <г3(0-[СО31 тк —
концентрация СО» в тканях и E[t)=F*&. (г) — скачкообразно изменяющаяся функция
воздействующего извне резкого изменения уровня содержания С02 во вдыхаемом воздухе.
[Из Grodins F. S., Control theory and Biological Systems, Columbia University Press, New
York, 1963, p. 131.]
■описывает наклон графика и отрезок, отсекаемый прямой на оси ординат) и,
наконец, третье уравнение
[С02]тк = [^/(производительность сердца)] (16.30д)
«ыражает равенство концентраций С02 в тканях и венах.
Выбрав [С02]л и [С02]тк в качестве величин, измеряемых на выходе
системы, а ^со8 в качестве небольшого резкого изменения, приложенного к системе
(внезапное увеличение концентрации С02 во вдыхаемом воздухе), а также
считая все остальные величины постоянными и независящими от времени, можно
объединить пять уравнений (16.30) в дифференциальное уравнение с одной
переменной, изменяющейся под действием «внешней движущей силы» ^со„ . Хотя
в этом частном случае можно решить уравнение при условии, что возмущающее
воздействие Fcbs мало, проще сконструировать эквивалентную электронную
-аналогичную цепь (рис. 16.11), компоненты которой можно отождествить с
различными переменными наших уравнений и отклик ее можно, следовательно,
«оитролировать. Это даст тот же самый результат, что и при использовании
уравнений, ио с тем преимуществом, что нам совершенно не надо будет решать
уравнения!
Доказательство точности соответствия опытным данным результатов,
полуденных для модели несколько более сложной, чем на рис. 16.11, представлено
на рис. 16.12. Так как различные величины: нормальная концентрация 002,
производительность сердца, скорость метаболизма и т. д., — установлены заранее,
•единственным регулируемым параметром является объем тканевого резервуара,
который дает наилучшее соответствие теории и опыта" при значении 30 л.
Математические модели предназначены, конечно, для того, чтобы получить
совпадение с экспериментальными данными прн использовании наименьшего числа
возможных параметров. Так, если биологическая система обладает каким-либо
избытком в своих механизмах контроля, этих сверхконтрольных контуров не
окажется в математической модели. По этой причине ие всегда возможно скоррели-
•ровать отдельные «черные ящики» в физиологических моделях с
функционированием определенных органов. Более того, очевидно, что очень неестественно
изолировать данную систему, например такую, как дыхательную, от остального
тела. Так, концентрация [Н+] в крови контролируется (со значительно более
медленной зависимостью от времени) посредством преимущественного выделе-
532 ЧАСТЬ 4
о
•£
Время, мин
Рис. 16.12. Экспериментальное (черные точки) и теоретическое (сплошная
кривая) изменения вентиляции как отклик на внезапное увеличение (слева) и
последующее внезапное уменьшение (справа) уровня содержания С03 во
вдыхаемом воздухе на 5%.
Вентиляция приведена относительно средней величины н нормирована по уровню
стационарного, состояния. '[Из Grodlns F. S., Gray /. S„ Schroeder К. R-, Norlns A. L., Jones R. W.f
J. Appl. Physiol., 7, 283 (1954).]
ния ионов NH4 почками. Учитывая это и другие ограничения, кажется еще более
замечательным то, что такие сложные физиологические функции можно
описывать достаточно точно с помощью относительно простых электрических цепей,
базируясь только на физиологических данных о поступлениях и выделениях.
16.Б. ЗАТУХАЮЩИЕ КОЛЕБАНИЯ ГРУЗА
С КОНЕЧНОЙ МАССОЙ НА ПРУЖИНЕ.
ЗВУЧАНИЕ КОЛОКОЛА
16.Б.1. ДЕПОЛЯРИЗАЦИЯ ФЛУОРЕСЦЕНЦИИ.
НАПРАВЛЕННОЕ ЗОНДИРОВАНИЕ УЧАСТКОВ МАКРОМОЛЕКУЛ
В гл. 13.В рассматривалась задача о (нестационарном)
поведении груза с конечной массой на пружине, первоначально
выведенного из равновесия и отпущенного без приложения в
дальнейшем движущей силы. В противоположность эксперименту со
стационарным, состоянием,, в котором пружина в конце концов
приспосабливается к колебаниям с частотой возбуждающего
воздействия, груз, выведенный из равновесия в нестационарных
условиях, колеблется с частотой, близкой к его собственной частоте
колебаний на данной пружине (как камертон, по которому резко
нанесен удар, колеблется со своей собственной частотой). В
эксперименте по нестационарной деполяризации флуоресценции (рис. 16.13,
16.14) на образец направляют поляризованный падающий свет
(обычно ультрафиолетовый), близкий по частоте к резонансной
частоте поглощения. В противоположность обычному
эксперименту по светорассеянию излучение, ожидаемое от данного диполя-
поглотителя, не появляется немедленно. Часть энергии данного
диполя очень быстро теряется, и последующее излучение, таким
образом, имеет более низкую (чем у падающего света) частоту, так
«-«■"•■
i i i i i
j_j
5 10 15 20 25 300 5 10 15
ГРУЗ НА ПРУЖИНЕ
lk часть энергии была потеряна до того, как излучение вышло
[з молекулы. Времена, связанные с каждым из этих процессов,
жазаны на рис. 16.13. Первоначальное поглощение падающего
^вета происходит приблизительно за Ю-15 с. Затем необходимо
жоло Ю-12 с для перераспределения внутренней энергии молеку-
[ы, чтобы вывести диполь, все еще колеблющийся с пониженной
|астотой, из-под (уменьшенного) влияния первоначального
внешнего возбуждающего электрического поля. Если падающий свет
[теперь убрать, излучение останется, распространяясь от
низкочастотного диполя, в течение времени 0,1-г-200-10-9 с в зависимости
t>T типа вовлеченных в процесс молекул.
Предположим для простоты, что для молекулы, находящейся в
покое, направление, вдоль которого колеблется приведенный в
движение «электрон на пружине», одно и то же для первоначально
возбуждаемого «поглощающего» диполя и второго возбужденного
«испускающего» диполя. Теперь предположим, что вертикально
поляризованный падающий свет направлен на неподвижные
молекулы и что после этого мы измеряем интенсивность
флуоресценции, которая излучается после выключения падающего света, как
для вертикально поляризованной его части с интенсивностью /ц>
так и для горизонтально поляризованной части с интенсивностью
/± (рис. 16.14). Поскольку направление колебаний испускающих
и поглощающих диполей совпадает, можно рассчитывать на то, что
вся флуоресценция, возникающая вдоль направления,
перпендикулярного направлению распространения падающего света,
должна быть полностью плоскополяризована в вертикальном
направлении (ср. с рис. 15.1, где тот же самый довод использовался ранее
S
/
Возбужденное состояние
возбужденное
синглетное
состояние
■ Основное состояние
Рис. 16.13. Диаграмма поглощения и замедленного испускания света
флуоресцирующей молекулой.
Основное и возбужденное состояние — квантовомеханические понятия, которые обсуждены
в часть 5. Частота колебания второго «излучающего» диполя меньше частоты
возбуждаемого «поглощающего» Диполя. Направление, вдоль которого происходит колебание «элек*
трона на пружине», также может быть различным для излучающего н поглощающего
диполей, даже если молекула не вращается в течение всего времени между поглощением и
испусканием (см. текст).
/ — поглощение падающего света (10-1S с); 2 — безызлучательный переход части
поглощенной энергии в энергию внутренних движений молекулы (Ю-11 с); 3 — флуоресценция —
вторичное излучение света с частотой более низкой, чем частота падающего света (время
жизни 10-» с).
534 ЧАСТЬ 4
t ~10т,
Рис, (16.14, Относительные интенсивности вертикально /ц и горизонтально /^
поляризованного света, излучаемого при флуоресценции под прямым углом
к направлению распространения вертикально поляризованного падающего света,
выключенного в нулевой момент времени, (Направления колебаний
испускающих и поглощающих диполей в нулевой момент времени приняты одинаковыми.)
Флуоресценция в начальный момент вертикально плоскополяризована, но, поскольку
молекулы вращаются, направление колебаний, излучающего диполя также вращается и
появляется горизонтально поляризованная компонента флуоресценции (средняя схема). После
того как молекулам позволят вращаться в течение времени, достаточного для того, чтобы
их ориентации стали по существу неупорядоченными в сравнении с первоначальным
направлением в нулевой момент времени, интенсивности вертикально и горизонтально плос-
кополяризованной флуоресценции станут фактически равными (нижняя схема). (На
рисунке принято, что вращение молекулы происходит быстро по сравнению с временем
затухания общей иитеиснвностн флуоресценции (твр«:т) так, что общая интенсивность
флуоресценции заметно не уменьшается в течение измерений.)
ГРУЗ НА ПРУЖИНЕ
1ля рассеяния света). Далее предположим, что мы позволили мо-
гекуле вращаться какое-то время (около Ю-9 с) между погло-
[ением и испусканием. Ясно, что интенсивность света, испускае-
юго вдоль направления, перпендикулярного падающему лучу, бу-
?дет иметь некоторую компоненту, плоскополяризованную в
горизонтальном направлении. Наконец, если молекуле позволить
произвольно вращаться в течение очень долгого времени, направление
•колебаний испускающего диполя станет полностью
неупорядоченным. При этом можно ожидать, что интенсивности флуоресценции
при наблюдениях как в вертикальной, так и в горизонтальной
плоскостях будут равными. Определим анизотропию
флуоресценции, наблюдаемую вдоль направления, показанного на рис. 16.14,
как
/n(0-/j_(0
анизотропия флуоресценции = A (t) j—/м1Г~о/—/77
(16.31)
Тогда общая интенсивность флуоресценции [/ц(0+2/1 (t)] будет
уменьшаться экспоненциально во времени с постоянной т:
•с = время жизни возбужденного синглетного состояния, (16.32)
а анизотропия A (t) будет экспоненциально уменьшаться с
постоянной времени твр. Последняя представляет собой время,
необходимое для того, чтобы молекула диффузно (посредством
маленьких угловых скачков) повернулась на угол порядка одного
радиана (гл. 21.Б). Итак,
A(t)=A0exp[-thJ. (16.33)
Пример. Нестационарная анизотропия флуоресценции как метод исследования
подвижности белка в мембране
Молекула 5-диметиламино-1-нафтилсульфохлорида (дансилхлорида)
:\N/
НаСч уСН3
дансилхлорид
S02CI
становится способной к интенсивной флуоресценции при (ковалентном)
связывании с белком и имеет время жизни возбужденного сииглетного состояния около
И2 не. Поскольку времена «вращательной корреляции» тВр для белков имеют тот
же порядок величины, флуоресценция от дансил-мечеиого белка будет
«деполяризоваться» (A(t) будет заметно уменьшаться) перед тем, как общая
флуоресценция исчезнет. Временную зависимость A(t) в уравнении (16.33) можно,
таким образом, использовать для определения времени вращательной корреляции
данснл-меченого белка.
Б36 ЧАСТЬ 4
а
ю5
1 .10" - -
•в
I
* з
=1
ю2 —
101
1 Л-»— Импуллс
\ \ падающего
I \ света
1 \ \ (/„</> +2/,(/)J
IH^-hWK \.
^ 1и<«+214|*> Ж"
1 1 1 1 1 1
«
■ Импульс
падающего
света
U,M+2lxU)]
О 20 40 60 80 100 120 140
Время, не
— flflR '
О 20 40 60 80 100 120 140
Время, не
Рис. 16.15. Графики различных комбинаций иитенсивиостей вертикально [/ц (t)]
и горизонтально i[fj_(/)] поляризованной флуоресценции после действия очень
короткого (около 5-Ю-9 с) импульса падающего света.
'[Из Wahl P., Kasai M., Changeux J. P., Eur. J. Biochem.. 18. 332 (1971).]
a — флуоресценция даисил-меченых белков во фрагментах биологических мембран из
электрического органа морского угря Etectrophorus electricus; б — флуоресценция свободных
данснл-меченых мембранных белков в растворе. Как в а, так и в б уменьшение
анизотропии A(t) показывает наличие компонентов с быстрым вращательным движением (т„_«3 не),
вр
соответствующим локальной гибкости в "месте присоединения метки к белку, и с
медленным вращательным движением |[т® >700 не в а н т^ =45 не в б],соответствующим враща-
вр вр
тельной диффузии молекулы белка как целого. Очень медленное вращение белка в а
показывает, что этот дансил-меченый соединенный с мембраной белок очень сильно
иммобилизован в мембране даже несмотря на то, что сама мембрана (рнс. Н.ЗО) может быть
высокотекучей в своей внутренней части.
На рис. 16.15,а показаны графики общей интенсивности флуоресценции
V О (0+2/±(0]> разности иитенсивиостей флуоресценции между вертикально и
горизонтально поляризованным испущенным светом [/11(0—»fi(0] и
анизотропии Л(0, как функций времени, прошедшего с момента прекращения действия
импульса падающего света, для дансил-меченых белоксодержащих фрагментов
при возбуждении биологических мембран. Оказывается, что анизотропия
уменьшается в соответствии с двумя значениями характеристического твр, одно нз
которых гораздо больше другого (что соответствует гораздо более медленному
вращательному движению):
(16.34)
(16.34а)
в
Л (0 = tA ехР Г- 'Ail + h ехР b '/V =
^Твр] "Г IB "t"1 "вр *=" "вр1
= fA exp [- t/*) -f fB при xf > £,
где Tgp и Tfp— времена вращательной корреляции для двух типов
вращательного движения, а §а и /в представляют собой соотвественно относительные доли
наблюдаемой анизотропии, свойственные этим движениям. Видно, что одно из
движений гораздо медленнее другого и что анизотропия выравнивается до
постоянного значения, как это и следует из уравнения (16.34а). Представляется
; 537 ГРУЗ НА ПРУЖИНЕ
наиболее вероятным, что два типа движения соответствуют локальному
быстрому вращению благодаря гибкости в месте прикрепления даисильной метки к
белку (твр=3'Ю-в с) н значительно более медленному вращательному
движению дансил-меченого белка как целого (т^,>700-10-9 с). Когда дансил-меченые
белки извлекают из мембраны экстракцией тритоновым детергентом
(рис. 16.15,6), анизотропия снова указывает на два типа вращательного
движения, но медленное движение белка становится теперь достаточно быстрым,
чтобы вызвать заметное уменьшение в анизотропии (т^,=45'10-9 с и
соответствует вращательной диффузии белка с молекулярной массой около 75000). Эти
результаты представляют особый интерес при сравнении с данными метода
спиновой метки в спектрах электронного парамагнитного резонанса (ЭПР)
(рис. 14.30). Результаты ЭПР показывают, что большинство самих мембран
являются «выоокотекучими» (обладают заметной вращательной подвижностью),
в то время как результаты по флуоресценции показывают, что мембранные
белки оказываются очень сильно . связанными (иммобилизованными) в мембране.
Разрешить это кажущееся противоречие можно благодаря недавно выполненным
экспериментам по ЭПР со спин-мечеными фосфолипидами. В этих опытах
Гриффит показал, что липиды в непосредственной близости от белка оказываются
значительно более сильно связанными, чем в отдалении от него. Поэтому
современную модель -биологической мембраны можно представить себе в виде
массивных медленно вращающихся белковых частиц, плавающих в относительно жидкой
внутренней среде мембраны.
Пример. Поляризация флуоресценции в стационарном режиме. График Перрена
В последнее время были проведены современные эксперименты по
поляризации флуоресценции с измерениями нестационарной анизотропии после
импульсного возбуждения. Однако существует более старый метод, в котором
измеряют наблюдаемую флуоресценцию в стационарном режиме при непрерывном
облучении образца падающим, светом. Определяя «поляризацию» как аналог
анизотропии при стационарном режиме, можно записать
/,, -11
поляризация = Р = j—, ,
(16.35)
где / и и / , обозначают теперь величины в стационарном состоянии'(не
зависящие от времени) для интенсивности флуоресценции, поляризованных
вертикально и горизонтально. Ясно, что поляризация Р будет максимальной (Ро),
когда вращательное движение очень медленно в сравнении со средним временем
жизни, которое отдельная молекула проводит в синглетном возбужденном
состоянии, приводящем к флуоресценции (ко времени заметного поворота
молекулы испускание флуоресценции уже должно произойти с сохранением
поляризации). С другой стороны, если молекула вращается быстро ло сравнению с
временем жизни флуоресценции (твР<?), то наблюдаемая в стационарном,режиме
флуоресценция должна быть полностью деполяризована (за время, пока
происходит испускание флуоресценции, направления колебаний излучающих диполей
становятся неупорядоченными). Таким образом, качественное поведение
системы, предсказываемое уравнением (16.35) (выведено Перреиом в 1926 г. и
распространено иа анизотропную вращательную диффузию несферических молекул
Вебером в 1952 г.), должно быть понятно (вывод этого уравнения здесь не
приводится):
[(1/Я)±(1/3)] .,„, . v MfiOfi4
[(1/л)±(|/з)]^1+3(^)- (,6-36)
638 ЧАСТЬ 4
Поскольку время вращательной корреляции тар связано с коэффициентом
вращательной диффузии DBp соотношением [ср. с уравнением (14.33)]
Ч? = (Vi^ep) =
4тгг)гэ _ 3^V
RT '
(16.37)
где г)— вязкость раствора, k и R — газовые постоянные на молекулу и на моль,.
А
г — молекулярный радиус для сферической молекулы и V=—-JirWo*— молярный*
О
объем этих молекул, уравнение (16.36) можно переписать как «уравнение Пер^
рена»: N
((l//>)±(l/3)j ft, ■
[(1/Л)±(1/3)]~1+ V V/v>
(16.38)
В пределе при очень низкой температуре или очень большой вязкости [(Т/г\)—»-
—»-0] молекулярное вращение будет замедлено до предела Твр^т так, что
поляризация Р достигнет своей максимальной величины Р0. Более важно, что»
наклон графика зависимости [(1/Р) ±(1/з)1/[(1/Ро)±(7з)1 от (Т/ц) будет; ра-
вен -^-, что позволяет определить эффективный молярный объем V, который
в свою очередь позволяет по уравнению (16.37) рассчитать время вращательной
корреляции тВр. Поскольку этим методом установлено, что твр для несфернче-
ских молекул больше, чем тВр для сферы той же самой молекулярной массы,,
время вращательной корреляции можно использовать для оценки формы макро-
Молекулы. Читатель может вспомнить (гл. 7.В.2), что фрикционное отношение-
f/fo, получаемое из измерений диффузии, также возрастает с увеличением
асимметрии формы молекулы. Однако фрикционное отношение увеличивается для
любого отклонения от компактной сферической формы, включая беспорядочную»
7/7 - Ю'^град/й
Рис. 16.16. График Перрена, показывающий зависимость поляризации
флуоресценции <в стационарном состоянии Р от TJx\, где Т — абсолютная температура,
а т) — вязкость раствора.
[Из, Metzger H., Perlman R. L., Edelhoch H., J. BioJ. Chem.. 241 1741 (1966).]
УМ — макроглобулин человека (мол. масса 9О0 00О); уМ — макроглобулин, который был
восстановлен днтнотреитолом для разрушения всех межцепочечных днсульфидяых связей
(иол, масса 180 000); Ffl£„—макроглобулин, который подвергли расщеплению трипсином.
Во всех случаях макроглобулия был вначале обработан даисилхлорндом для получения
белка с ковалентно присоединенной флуоресцентной меткой. Времена вращательной
корреляции рассчитаны из данных при 7*/tl=2-10—4 град/П, используя уравнение (,16.37) и время
жизин т=12 не для флуоресценции данснльной группы. Самая нижняя кривая
соответствует, таким образом, макромолекуле, имеющей самое большое TeD.
19 ГРУЗ НА ПРУЖИНЕ
«спнрализацию, тогда как время вращательной корреляции увеличится только
^тогда, когда молекула асимметрична, но еще остается жесткой. Таким образом,
£с помощью измерения т8р можно различать не только сферические и несфериче-
!-ские, но также жесткие и гибкие молекулы.
■ На рис. 16.16 показан график Перреиа для макроглобулина человека (мол.
масса 900 000) и также его субъединиц, полученных разрывом межцепочечиых
дисульфидных связей (мол. масса 180 000), и для фрагментов, полученных
трипсиновым гидролизом (мол. масса 50000). Максимальную поляризацию Ро
определяли прямым измерением величин /1| и Jl Для белков, растворенных
в 90%-ном глицерине при 8°С [(Т/ц) -Ю-4 = 0,0051, как показано
экспериментальными точками (крайними слева) на рис. 16.16. Ро также определяли
экстраполяцией данных, полученных для разных белков в воде при различных
температурах (экспериментальные точки справа на рис. 16.16). Время вращательной
корреляции вычисляли По уравнению (16.37), используя величины Р,
полученные для белков в воде при комнатной температуре (самые левые
экспериментальные точки в правой части рис. 16.16). Времена вращательной корреляции были
следующими: мол. масса 900 000, тВр=80 не; мол. масса 180 000, твр = 69 не;
мол. масса 50000, твр=58 не. Другими словами, хотя трипсин разрывает на-
тивиый макроглобулин иа фрагменты почти в 20 раз более мелкие, наблюдается
только 27,5%-ное уменьшение во времени вращательной корреляции (а не
100-[900000/50 000]1'3=260%-ное уменьшение, которое мы могли ожидать, если
бы оба типа молекул были жесткими). Аналогичное рассуждение справедливо
и для субъединицы с мол. массой 1180000. Из этих значений твр следует, что на-
тнвный макроглобулин с мол. массой 900 000 представляет собой очень гибкую
молекулу.
16.Б.2. КОРРЕЛЯЦИЯ ИСПУСКАНИЯ у-ЛУЧЕИ ПО НАПРАВЛЕНИЯМ:
ТОТ ЖЕ ЭКСПЕРИМЕНТ ДЛЯ НЕПРОЗРАЧНОЙ СРЕДЫ
Эксперимент по корреляции испускания 7_лУчей по
направлениям по своему принципу и аппаратуре аналогичен эксперименту
по нестационарной флуоресценции с импульсным возбуждением.
В обоих случаях первичная электромагнитная волна используется
для «поляризации» (определенного «ориентирования») образца.
Последующая эмиссия второй волны происходит преимущественно
в определенном направлении до тех пор, пока молекулы образца
не получили достаточно времени для поворота, после которого
первоначальная ориентация нарушается и испускание становится
изотропным (равным по интенсивности во всех направлениях).
Главные характерные особенности экспериментов с 7_лУчами с0_
стоят в том, что а) «ориентирующая» и «наблюдаемая»
электромагнитные волны исходят от образца (тогда как при
флуоресценции первичная плоскополяризованная волна поступает извне) и
6) у-лучи исходят от колеблющихся «на пружинах» ядер, а не
электронов и энергии волн находятся, следовательно, в ином
частотном диапазоне по сравнению с флуоресценцией (7-лучи, а не
видимый свет).
Принцип и схема эксперимента по корреляции испускания
7-лучей по направлениям показаны на рис. 16.17. Подходящие
ядра (например, 62Zn и inCd, рис. 16.18) спонтанно испускают два
7-луча один за другим (так же как флуоресцирующие молекулы
спонтанно испускают один световой луч из возбужденного синг-
Б40 ЧАСТЬ 4
Ъ
\
I
Счетчик tff
Ъ
Счетчик /г
Счетчик fz
-J.
Рис. 16.17. Причина возникновения анизотропии в направлении испускания
Y-лучей от первоначально ориентированных ядер магнитных диполей.
Хотя сначала направления магнитных днполей беспорядочны (вверху слева), ограничение
регистрации лучами в единственном направлении отбирает ядра, которые должны быть
сориентированы согласно этому направлению испускания (в середине слева). v-Лучи,
-испущенные позже из этих сориентированных ядер (лучи 7г), будут наблюдать
преимущественно под углом 180°, а не 90° к направлению между образцом и счетчиком лучей Vi (справа).
Поскольку мечеииые радиоизотопом молекулы вращаются, ориентация ядерных магнитных
диполей нарушается, и наблюдаемая анизотропия в испускании лучей 7г [уравнение (16.39)|
понижается. В большинстве случаев энергии лучей Vi и 7г отличаются достаточно^ чтобы
их легко было различить регистрирующими устройствами (рис. 16.18).
летного состояния). Если испускающие (магнитные) диполи
ориентированы беспорядочно (как в случае набора сигналов от
беспорядочно ориентированных шСс1-меченых макромолекул в
растворе), тогда оба ?-луча будут испускаться с одной и той же
интенсивностью во всех направлениях. Однако если мы регистрируем
первые лучи (71), которые испущены только в определенном
направлении, то они должны исходить от ядер, магнитные диполи
которых были сориентированы в соответствии с этим
направлением, как показано в середине левой части рис. 16.17. Наконец,
если мы теперь наблюдаем луч 72, который испущен тем же
самым ядром, то наиболее вероятно, что он тоже будет направлен
определенным образом (скажем, под углом 180° по отношению к
направлению лучей 71), а не как-то по-другому (скажем, под углом
90° по отношению к направлению лучей у\). Модель испускания
7-лучей для ориентированных излучающих ядер аналогична
модели для светорассеяния (рис. 15.1). Пусть направления
зафиксированы так, чтобы направлению 0° соответствовала регистрация
только лучей 7ь а в направлениях 90 и 180° регистрировались
только лучи 72. Если появление луча 72 регистрируют только
после (но не раньше) испускания луча у\, тогда, естественно, есть
уверенность в том, что зарегистрированные лучи 71 и 72 пришли от
одного- и того же ядра. В этом случае нужно ожидать наличия
анизотропии A(t) между лучами, испускаемыми под углами 90 и
,^41 ГРУЗ НА ПРУЖИНЕ
180° по отношению к направлению на детектор лучей yi- Если при
этом молекулы способны в промежуток времени между
испусканием сориентированного луча 71 и наблюдением луча 72
повернуться, тогда анизотропия будет частично утрачиваться, т. е. нужно
ожидать уменьшения A(t) во времени. Как и в случае
деполяризации флуоресценции, уменьшение A(t) во времени во многих
случаях экспоненциально.
A(f\ [(Ya-счет под ушюм 180°)—fo-счет под углом 90°)] ПКЗДЪ
Л^~~ (Ya-счет под углом 90°) U°-°yJ
Наконец, существенное различие между деполяризацией
флуоресценции и деполяризацией 7_лУчей состоит в интерпретации
экспериментальных результатов. При затухании флуоресценции более
быстрое молекулярное вращение приводит к более быстрому
исчезновению анизотропии флуоресценции; в исследованиях с 7"ЛУ"
чами более быстрое молекулярное вращение обычно ведет к
более медленному экспоненциальному исчезновению анизотропии.
Объяснение такого поведения несложно и будет дано в гл. 21.БГ
где этот эффект обсужден вместе с рядом других аналогичных
явлений. Теперь можно рассмотреть некоторые экспериментальные
результаты.
Пример. Корреляции испускания У"лУЧеи по направлениям для 1ИС(1-меченоЙ1
карбоангидразы
Существует изотоп кадмия lllmCd (лг обозначает, что данный изотоп мета-
стабилен), дающий подходящий каскад Y-лучей (рис. 16.18) со временем
полураспада промежуточного продукта 85 не, т. е. достаточно большим, чтобы
молекулярная вращательная диффузия белков могла заметно повлиять на
анизотропию испускания -у-лучей до того, как интенсивность излучения Y2 слишком
сильно уменьшится (сравните с аналогичным рассуждением для времени жнзнк
флуоресценции 12 не для дансильной флуоресцентной метки). Когда ионь*
nim,c(j2+ находятся в свободном виде в растворе, вращательная диффузия
сольватированных ионов радиоактивного металла происходит быстро и анизо-
111т
21'. = 150кэВ
I-»-
§. ?г=247кэВ
Cd
■'■%■*■«*<
■ Стабильный изотоп
Рис. 16.18. Каскад 7"лУчеи Для последовательного испускания двух у-лу'че*
ядрами метастабильиого изотопа lllmCd.
Энергии луяей Vi и Vj не зависят от химического окружения ядра кадмия, в то время как
анизотропия интенсивности в зависимости от направления испускания Vj-лучей с энергией
247 кэВ чувствительна к молекулярному вращательному движению (см. текст).
£42 ЧАСТЬ 4
50 100 150
Время,не
а
V,i
0,1
0.0
П 1
I I l
I
]'•'' .*•! •'" ■• •'.-. • •••
• • • • •• VL «~ • * •
■ •« • • . • • ••
— • • «
•
• • • .
•
J
~~ *"~
I" I , f
50
100
Время, не
150
0 50 100 150
Время, не
«.
"Рнс. 16.19. Анизотропия коррелированного испускания \-лучей ионом mmCd2f
в различных химических окружениях.
[Из Meares С. F., Bryant R. С, Baldeschwieler I. D., Shirley D. A.t Proc. Natl. Acad. Sci.
U.S.A., 64, 1155 (1969).]
-й-свободные ионы limCd2+ в растворе; б —ионы1 Cd*+ в присутствии 3-10—"* М натив-
ной карбоангндразы (фермента, ие имеющего подходящих мест связывания нона CdH)
•и в —ноны lllmCds+ в присутствии 2,510-* М апокарбоангидразы (фермента с одним
доступным местом для сильного связывания иона Cd*+). Все растворы имеют рН 6,1 (0,1 М
фосфатный буфер, содержащий 0,5 М NaCI). Заметим, что анизотропия исчезает гораздо
^ьцстрее (график в), когда радиоактивные ноны imCd2+ связаны с ферментом, чем когда
ионы 1UmCd2+ находятся в свободном виде в растворе (график а), из-за большого различия
в скоростях вращательной диффузии в этих случаях (см. текст).
тропия лишь немного уменьшается во времени (рис. 16.19,а). Когда в раствор
.добавляют апокарбоангидразу (апокарбоангидраза—нативный фермент с
удаленным из активного центра ионом Zn2+), то ионы "lmCd8+ связываются в ак-
43 ГРУЗ НА ПРУЖИНЕ
тивном центре в положении, обычно занимаемом ионами Zn2+. Поскольку вра-
щательная диффузия 11ШС(1-меченого белка теперь гораздо меньше,
анизотропия испускания у-лУчей уменьшается во времени гораздо более быстро-
.(рис. 16.19, в). Для доказательства того, что изменение, внесенное ферментом,
вызвано не просто увеличением вязкости раствора (которое также привело бы
к понижению вращательной диффузии ионов in"»Cd2+ и, таким образом, к более
быстрому исчезанию анизотропии испускания у-лучей), был сделан контрольный
эксперимент (рис. 16.19,6), в котором раствор содержал ионы lllmCd2+ и на-
тивную карбоангидразу (карбоангидразу, уже содержащую в активном центре
ион Zn2+, что предотвращает какое-либо связывание иона Cd2+). Контрольный
эксперимент показывает, что увеличение вязкости раствора действительно влияет
на анизотропию, но в значительно меньшей степени, чем эффект связывания
иона Cd2+ в активном центре фермента.
Поскольку графики анизотропия — время, подобные показанным на рис. 16.19,.
строят для получения единственного числа — времени вращательной
корреляции
'вр=(1/6Аф). (16.40Г
где DBp — коэффициент вращательной диффузии, может показаться излишним
показ кривых целиком для определения одной только цифры. Существует
другой, более удобный для анализа способ представления данных. Он показан на
рис. 16.20, где по вертикальной оси отложены величины, пропорциональные
площади под графиком A(t), а по горизонтальной оси — время, соотнесенное
со временем жизни для затухания излучения Y2-
Gi2 (оо) = (const) Г A (t) ехр (— tf%) dt. (16.41>
о
0,24
0,20
1 0,16
£ 0,12
0,08
J.04
0
\\
— 0 = 90° -*-~Т
— 0 = 30°-^
— в = о°-*"'
I I I
L ^0 = 60°
Л I-*
у 0=0°
/ ^0 = 30°
/ /V д = 60°
/ 11 f в = 90°
Mi 1
-12
-10
ЬЬ
-8
ер
-6
-4
Рис, 16.20, Графики зависимостей интегральной анизотропии от времени
вращательной корреляции Твр (логарифмическая шкала) для ядра lllmCd (О),
прикрепленного под углом 9 к главной оси симметрии вытянутого молекулярного»
эллипсоида (рис. 7.22).
Еще более широкая вариация в положении графиков была найдена, когда зависимость
G»(°°) от т строили для ядер 111OTCd, связанны^ со сферическими молекулами, но у них:
была возможность вращаться с разными скоростями в месте прикрепления, [Из
Marshall A. G„ Werbelow L. С, Meares С. P., J. Chem. Phys., 57, 364 (1972),]
* При экспериментах с улУчамя и магнитным резонансом (гл. 21,Б)
соотношение между tap и DBp задается уравнением (16,40). Для экспериментов по»
диэлектрической релаксации и деполяризации флуоресценции DBp имеет тот же
самый смысл, ио твр может быть пропорционально D~£ с коэффициентом или?
7з, или 1/е в зависимости от определения или (в «диэлектрическом» случае) or
природы наведенного дипольного момента.
544 ЧАСТЬ 4
Значение графика иа рис. 16.20 заключается в том, что если просто проводить
измерение A{t), интегрированное за время, длительное по сравнению с т, то
можно затем определять время вращательной корреляции твр из графика
зависимости 0^2(0°) от твр. Кроме того, как видно из дополнительных кривых на
рис. 16.20, форма кривой зависимости {^(оо) от твр (где тВр является лишь
мерой размера молекулы) может сильно зависеть от угла, под которым метка
прикреплена относительно общей молекулярной оси симметрии. Аналогичные
кривые получены для прикрепленных меток с различающимися степенями
внутренней гибкости в .месте прикрепления. Читателю нужно помнить, что
спиновые метки в ЭПР и эксперименты по поляризации флуоресценции также
обеспечивают доступ к информации о молекулярной форме и гибкости. Уникальная
•особенность экспериментов с применением Y-лучей состоит в том, что оии дают
ту же самую информацию при очень низких концентрациях (вплоть до Ю-12 М)
и в непрозрачных средах (что особенно важно при измерениях in vivo).
Корреляции испускания Y-лучей по направлениям уже применяли иа практике для
изучения связывания ионов т1п3+ белками сыворотки крови живых мышей, и
кажется, что этот очень новый метод будет обладать необыкновенными
преимуществами для таких систем.
16.Б.З. МАГНИТНАЯ РЕЛАКСАЦИЯ.
МОЛЕКУЛЯРНО-СПЕКТРОСКОПИЧЕСКАЯ «ЛИНЕЙКА»
Понятие движения магнитного момента М во внешнем
статическом магнитном поле Но было введено в гл. 14.Г при
рассмотрении в стационарном состоянии магнитного отклика на
переменное возбуждающее магнитное поле. Магнитный момент прецесси-
рует около приложенного статического поля с частотой прецессии
. % = Y^o- (14.15)
При обсуждении нестационарного магнитного отклика на
импульс магнитного поля особенно удобно перейти к системе
координат, которая сама вращается со скоростью ах> так, что в новой
«вращающейся системе координат» магнитный момент становится
«неподвижным (рис. 16.21,а, б). Поскольку магнитный момент
теперь фиксирован по направлению (т. е. он не прецессирует), то как
бы отсутствует и приложенное статическое поле Но (которое,
конечно, на самом деле присутствует). Это и есть то упрощение, к
•которому мы стремились. Если теперь представить себе второе
малое наложенное магнитное поле Hi, которое вращается с той же
«самой скоростью и в том же самом смысле, что и прецессирующий
момент, в лабораторной системе координат (рис. 16.21, в), то при
переходе к вращающейся системе координат Hi станет малым
статическим полем (рис. 16.21,г). Теперь, поскольку поле Н0 по
своему действию равно нулю, а пояе Н\ по своему действию —
статическое поле во вращающейся системе координат, магнитный момент
Mq во вращающейся системе координат будет просто прецессиро-
ъатъ около Н\ (в плоскости tf—z') с частотой
«I = Y^,. (16.42)
Таким образом, при использовании магнитного поля Н\ в течение
времени 7(90°) = (1,57/©I) сек намагничивание, начиная от равно-
545 груз на пружине
г'
а
Z
Но
к"'
—!>»•
W,
\,
н
Ма
*" \
£>У'
/
Рис. 16.21. Поведение магнитного момента под влиянием внешнего7 магнитного
поля (полей) в лабораторной (неподвижной) системе координат |а и el и
в системе координат, которая вращается с ларморовской («собственной»)
частотой прецессии ш0 магнитного момента во внешнем статическом магнитном поле
■Но [[б и г].
В лабораторной системе а магнитный момент М вращается, в то время как система
координат х — у неподвижна. Во вращающейся системе координат б магнитный момент
неподвижен и. таким образом, ведет себя так, как если бы внешнего поля Нв совсем ие
было. Затем к равновесной системе в с равновесным намагничиванием Afo,
сориентированным вдоль статического поля Но, прикладывают вращающееся поле Н\. В итоге (г), во
вращающейся системе координат Но по существу отсутствует, & Н\ фиксировано по
величине и направлению так, что Мь во вращающейся системе координат прецессирует
около Н\ (около оси х') до тех пор, пока Hi остается «включенным». Если Н\ оставляют во
«включенном» состоянии как раз столько времени, чтобы AfD повернулось от направления z?
до направления у1 во вращающейся системе координат, тогда М{\ можно наблюдать
детектором, расположенным вдоль (вращающейся) оси у', что дает результат, показанный
на рис. 16.22.
весного значения с величиной М0, направленного вдоль оси z\
будет прецессировать точно 90°, чтобы стать направленным вдоль
положительной полуоси у'. Наконец, если мы расположим
детектор вдоль (вращающейся) оси t/ (это легко сделать
электронными, но не механическими средствами), то сможем наблюдать
(переменную) величину магнитного момента вдоль вращающейся оси
у' как функцию времени после прекращения действия импульса
546 ЧАСТЬ 4
-Ч К-
90°-ный импульс
к-
90°-ный импульс
Рис. 16.22. Поведение t/'- и 2'-компоиент намагничивания после действия
-сЭ^-ного импульса», в котором малое вращающееся поле Н\ использовано для
поворота равновесной намагниченности М0 с направления г' до направления у'
во вращающейся системе координат (рис. 16.2)1).
"После того как намагниченность повернули до нового (фиксированного) направления вдоль
вращающейся оси у' и сняли поле Н{, намагниченность М' уменьшается до нуля экспонен-
■циально с постоянной времени Т2, в то время как намагниченность Mz возвращается
обратно к своему равновесному значению экспоненциально с постоянной времени Т\. Rnn
небольших молекул ТУ и Г» обычно равны, тогда как для макромолекул Т\ может быть
много больше Г3. Связь между этими «временами магнитной релаксации» и молекулярным
вращением обсуждена в тексте.
Н\ (рис. 16.22). Единственная цель применения импульса
#i — повернуть М0 в плоскость х'—у'. Теперь мы обсудим
результат этого эксперимента и его приложения.
Вслед за наложением и снятием вращающегося поля Hi,
которое используют для создания системы с намагниченностью Мо,
направленной вдаль (вращающейся) оси у' в нулевой момент
времени, намагниченность вдоль оси у' уменьшается до нуля, в то
время как намагниченность вдоль оси г' восстанавливается до
своего равновесного значения согласно экспоненциальным
соотношениям
M'y=M0exp(-t/Ts)
Af',«JIUl-exp[-*/7\J)
(16.43а)
(16.436)
Поскольку (гл. 13) 72 в нестационарном эксперименте связано с
шириной полосы поглощения в эксперименте со стационарным
состоянием До уравнением (задача 13.5а)
(1/Г,) = (Асе/2) =«Av
(16.44)
и поскольку мы уже показали, что ширина полосы магнитного
резонанса в стационарном состоянии пропорциональна времени
вращательной корреляции твр= (1/6 DBp), ясно, что Ti прямо связано
со скоростью молекулярного вращательного движения: более
длительное Т2 соответствует более быстрому вращению молекулы в
растворе.
547 ГРУЗ НА ПРУЖИНЕ
Пример. Количественная оценка текучести внутренней части мембраны, исходя
из величин Т\ для углерода-13
Наиболее простое определение Т2 основано на прямой регистрации
экспоненциально уменьшающейся до нуля величины М'у во вращающейся системе
координат. Тх можно определить аналогично, выжидая в течение различного
времени после первоначального «90°-ного импульса» (чтобы позволить М'г достигнуть
некоторой частичной величины своего конечного равновесного значения Мо)
и применяя затем другой «90°-ный импульс», чтобы повернуть сигнал М'г в
направлении вращающейся оси у' для его регистрации. Из значений М'г,
соответствующих различным временам выжидания, воссоздают по точкам кривую
восстановления М'г и из нее определяют Т\. Практически для малых молекул
более легко проводить и интерпретировать измерения Т\, чем измерения 7V
Для молекул с молекулярной массой менее чем ~10О00 в протонном
магнитном резонансе Т\ = Т2, и, таким образом, Г] несет в себе ту же самую
информацию о молекулярном движений, которая была бы получена из измерений
ширины полосы в стационарном режиме. Преимущество нестационарного
эксперимента в том, что его можно проводить в 1000—10000 раз быстрее, чем
соответствующий эксперимент со стационарным состоянием (гл. 20), и это резко
повышает его практическое значение.
э.» СН3 CHj
1.8 сн2 сн»
1,1 СН2 СН, •
о,б (CH2)io (CHJio
с: СН2 СН2
«,. сн2 сн2
с=о с=о
о о
о,. Н2С : С— СН, о..
о-р=о
Н 6 рис< ]6.23. Значения Т\ из данных по ЯМР-13С для
различных выделенных сигналов индивидуальных
атомов углерода в фосфолипидной молекуле.
Растворы дипальмитоиллецитина были обработаны
ультразвуком для получения пузырьков с двуслойной"
структурой, аналогичной той, которую имеют природные-
/Н2 м биологические мембраны. Значения 7*1 постепенно
возрастают при продвижении от атомов углерода,
расположи женных вблизи поверхности, к атомам, наиболее углуб-
i 2 ленным во внутреннюю часть двойного слоя (см. текст),
I [Из Lee Л. G., Birdsall N. J. M., Metcalfe J. С, Chem.
hN(CH3), o,7 Brit., 9, 116 (1973).]
i
c.
c.
Мы уже видели, что часто возможно разрешить сигналы ядерного
магнитного резонанса для индивидуальных атомов углерода в малой молекуле
(мол. масса ^! 1000) (рис. 14.22), Поэтому возможно также определять и
индивидуальные значения 7, для каждого из этих атомов углерода в спектре ЯМР
на ядрах углерода-13. Ряд значений Т\ для индивидуальных сигналов различных
атомов углерода фосфолипида в двухслойной структуре типа мембраны
показан на рис. 16.23. Очевидно, что имеет место 30-кратное увеличение Т\ (и
соответственное увеличение во вращательной гибкости), которое развивается от
648 ЧАСТЬ 4
лолярной «головной» группы глицеринового остова до коицевой метальной
труппы, локализованной во внутренней части двойного слоя. Эти измерения
позволили получить некоторые количественные данные о сильно возрастающей
текучести во внутренней части фосфолипидного двойного слоя по сравнению
с относительно ограниченным вращательным движением рядом с внешней
поверхностью модельной мембраны. Недавно эти результаты были подтверждены
аналогичными экспериментами с использованием ЯМР на ядрах дейтерия в дей-
терированиых молекулах фосфолипида. Дальнейшее распространение метода на
липиды смешанного состава, которые характерны для биологических мембран,
должно оказаться чрезвычайно интересным для установления склонности (или
•ее отсутствия) липидов определенного типа к агрегации друг с другом в
природных мембранах.
Пример. Времена релаксации для протонных спектров ЯМР и молекулярно-
спектроскопическая «линейка»
Мы уже обсуждали преимущество «химического усилителя» для случая
быстрого- химического обмена в связи с измерением в стационарном режиме
резонансной частоты, методом ЯМР. Доводы тогда были основаны на идее о том,
что магнитное ядро может «прыгать» туда и обратно между двумя химически
различными местами (состояниями) с различными частотами магнитного
резонанса. Если частота скачков велика по сравнению с различием в резонансных
частотах между двумя состояниями, будет наблюдаться единственный сигнал
с промежуточной частотой резонанса. Она является «средневзвешенной» между
частотами для обоих состояний согласно тому, насколько велико относительное
время, которое ядро провело в каждом состоянии. Аналогичное рассуждение
можно применить к влиянию быстрого обмена на скорость экспоненциального
уменьшения М'ц или экспоненциального\роста М'г вслед за «90°-ным импульсом».
Сигнал My будет уменьшаться с постоянной времени Т2, соответствующей
первому состоянию, до тех лор, пока ядро там остается. Затем, при «перепрыгивании»
ядра в другое состояние сигнал М'у будет уменьшаться оо скоростью,
соответствующей Гг другого состояния и т. д. Пока частота скачков k велика по
сравнению с различием во «временах релаксации» в обоих состояниях
ВД1 (1/^2 ) —1(1 /7?) I («быстрый7обмен»), (16.45)
наблюдаемое уменьшение М'в будет следовать единому (среднему)
экспоненциальному уменьшению
Af'„=Afeexp[— (f/Tf)CpI, (16.46a)
где
(1/Ъ)ер =• fA №)4 + !в (1/ГОв. (16.466)
Аналогично
(I/7,i)cp = f<4(l/7'.)A + fB(V7,1)fif (16.46b)
где [а и \в снова представляют собой относительные доли ядер в состояниях
А н В. Как и в случае приложений быстрого обмена к резонансной частоте
ЯМР в гл. 14.Г, феиомеи обмена может быть использован для измерения
«времен релаксации» Tib и Т2В макромолекул в разбавленных растворах путем
наблюдения малых изменений в средних «временах релаксации» Т\ и Т2 для
небольших молекул (имеющих гораздо более высокую концентрацию и, значит,
дающих более сильный сигнал ЯМР), которые могут обратимо связываться
с макромолекулами. Примем (как это имеет место в нижеследующем примере),
что (1/7,8)в>-(1/Г8)л. Благодаря этому становится возможным измерить время
649 грУЗ НА ПРУЖИНЕ
Рис. 16.24. Конфигурация молекулы NADH, связанной с дрожжевой
алкогольдегидрогеназой, которая была спин-мечена парамагнитным производным иитроксила,
ковалентно присоединенным к цистеину-43 полипептидной цепи фермента.
[Данные ЯМР из Sloan D. L., Mlldvan A. S., Biochemistry, 13, 1711 (1974): данные рентгено-
структурного анализа из Branden С. I., Ektund H„ Nordstrom В., Boiwe Т., Soderlund G.,
Zeppezauer E., Ohlsson I., Akeson A., Proc. Natl. Acad. Scf. U.S.A., 70, 2439 (1973).]
Слева: расстояния от нитроксила до соответствующих протонов NADH, определенные из
значений Тх протонов, при использовании уравнения (16.47). Справа: сравнение конформа*
цин NADH в спин-меченой дрожжевой алкогольдегидрогеназе (из ЯМР-данных по Т\)
с конформацией" ADP-рнбозы, связанной с алкогольдегидрогеназой печени (н'з данных рент»
геноструктурного анализа).
релаксации «связанного состояния» для макромолекул, концентрация которых
гораздо меньше, чем та, при которой их можно было бы непосредственно
исследовать.
«Метрическая» информация, получаемая из Т\ и Т2, вытекает из
соотношений типа
(1/7*,). = const (1/r8), (16.47)
где г — расстояние между интересующим иас магнитным диполем (т. е. ядром)
и магнитным диполем (гораздо более сильным) ближайшего неспареиного
электрона. Из-за того что можно разрешить много отдельных резонаисов ядер,
скажем, для различных протонов в одной и той же молекуле, стало возможным
измерить расстояния между каждым из этих протонов и неспареиным электроном,
локализованным, например, у определенного аминокислотного остатка фермента.
На рис. 16.24 показаны результаты серии таких определений, исходя из
«связанных» значений Т{ и Тг для малой молекулы NADH при ее связывании с
ферментом алкогольдегидрогеназой. Эти измерения стали возможны благодаря
быстрому обмену свободных и связанных молекул NADH. При этом концентрация
фермента была более чем в 10 раз меньше, чем у молекул NADH, по которым вели
наблюдение. Вследствие зависимости от г в шестой степени, согласно уравнению
(16.47), возможно очень точно определить расстояния даже из значений Тх
низкой точности. Поразительно сходство конформаций молекул NADH, связанных
с алкогольдегидрогеназой (в растворе), и похожих молекул ADP-рибозы
(данные рентгеноструктуриого анализа кристаллического фермента), связанных с де-
гидрогеиазой другого происхождения. На рис. 16.24 представлено
доказательство сходства структуры фермента в кристалле и растворе. Это пример точной
геометрической информации, получаемой из данных ЯМР на основе значений
7"i я Т2.
550 ЧАСТЬ 4
Задачи
1. В разд. 16.A.I1 рассмотрена связь между понятиями времени релаксации
химической частицы т и константой скорости k изменения концентрации этой
частицы в реакции первого порядка. Среднее время релаксации всех молекул
реагента А, присутствующих в нулевой момент времени, можно выразить как
среднее из всех возможных времен релаксации молекул А, каждое из
которых вносит свой вклад, пропорциональный числу молекул с этим
индивидуальным временем релаксации. Для простого процесса уменьшения
концентрации А, описываемого уравнением для реакции первого порядка, среднюю
величину вычисляют по сравнению
о
Г — td[A]
|AJf=0
т= щ:—> <а>
где [А] о—концентрация А в нулевой момент времени. Нужно подставить-
концентрацию А в момент времени t, [A(/)]
[A(t)]=A0exP[-kt] (б)
в скорость исчезновения [А]
— d[A)=k[A(t)]dt '(в)
и затем уравнение (в) в уравнение (а) для того, чтобы показать, что»
т=(1/£), согласно уравнению (9.10).
Замечание. I х ехр [— ах] dx = [ 1 /а2].
2- Вывести уравнение Смолуховского, которое предсказывает константу
скорости для контролируемых диффузией быстрых химических реакций. Из
основного закона диффузии [см. уравнение (6.51)]
поток = — 4ws/> (dn/dr), (a>
где «поток» представляет собой число частиц, диффундирующих к
рассматриваемой частице и сталкивающихся с ней, D — коэффициент диффузия,
л —число частиц в 1 мл, а г — радиус сферической поверхности вокруг частицы.
Интегрируя, находим число частиц, локализованных на произвольном
расстоянии г от поверхности данной частицы (откуда другие частицы непрерывно
удаляют посредством химической реакции) ,
п= п0— (поток) /4nrD. (б)
В контролируемой диффузией реакции, когда концентрация частиц на
реакционной поверхности п равна нулю, а яо — концентрация частиц в массе
растворителя (на большом удалении от данной частицы),
поток = BnDron0, (в)
где расстояние наибольшего сближения между двумя сферическими
частицами радиуса г0 составляет г=2г0. Далее, поскольку центральная частица не
находится в покое, но также непрерывно диффундирует, число столкновений
оказывается в два раза большим, чем предсказывает уравнение (в)
поток = 16яОг0пв. (г)
(gl ГРУЗ НА ПРУЖИНЕ
Скорость исчезновения первоначальных частиц, таким образом, будет
dn9/dt = —l6nDr9n\. (д)
Сравнивая уравнение (д) с уравнением для химической реакции второго
порядка
d[C\/dt^ — k[C\K (e)
в котором [С] — молярная концентрация, покажите, что
k=\6-lO-3nDr9N0, (ж)
где No — число Авогадро. Наконец, из уравнения Стокса для коэффициента
диффузии большой сферической молекулы
D = RT/&trnN„ (7.13, 7.55)
4\де ti — вязкость раствора, равная — 0,00^01 при 30 СС, покажите, что
^в.Ю-'ЯТуЗчз^З^.Ю'М-^с-1 (для воды при 30 *С)
(з)
Этот удивительный результат товорит о том, что в контролируемых диффузией
реакциях скорость химической реакции не зависит от размера молекулы!
Хотя более крупные молекулы и двигаются путем диффузионного переноса
медленнее, площадь их поперечного сечения больше, и эти два эффекта
компенсируют друг друга так, что скорость реакции не зависит от размера
частиц.
I. Покажите, как два различных времени релаксации получаются из
кинетической схемы, включающей два равновесия. После этого расчета должно
казаться естественным то, что схема с N равновесными состояниями будет в общем
случае обладать N отдельными временами химической релаксации (которые
могут достаточно отличаться или не отличаться настолько, чтдбы быть
определимыми при выделении N отдельных линейных отрезков на графике
зависимости логарифма изменения концентрации от времени для конкретных
экспериментальных данных). Начнем с рассмотрения схемы
E+S^X.^X,, (a)
*.i *.■
которая могла бы соответствовать связыванию субстрата с ферментом с
последующей перестройкой фермент-субстратного комплекса, как обсуждалось
в примере с ГФДГ (рис. 16.5 и 16.6). Определим разность концентраций AS,
ЛХ[ и ДХ2 между мгновенными концентрациями ([S], [JE], ГХф \Xt\j
и конечными (новыми) равновесными концентрациями ([S]', [Е] , [Xij,
[Х2]') как
AS = [S]-[S]' = [E]-[E1\
AX.^IX.J-IX,]', (б)
АХ, = [Х,1-[Х,]'.
а. Покажите, что скорость изменения AS описывается уравнением
d (AS)/rf* = - k\ (AS + [E]') (AS + [S]') + ft'-. (AX, + [X,]'). (в)
<>. Предполагая, что AS<[S]', покажите, что уравнение (в) приводится
к виду
d(AS)M = -AS(fc'J([E]'-b[S]'))+AX1(b/-,). (r)
552 ЧАСТЬ 4
Аналогично покажите, что
d (АХ,)/Л-= AS (k\ ([E]' + [S]')) - АХ, (*'_, + *',) + AX2(fc'_2) (д)
и
d (АХ,)/Л = АХ, (*',) — АХг (/г'2) (е)
Уравнения (г), (д) и (е) представляют собой систему из трех
дифференциальных уравнений с тремя неизвестными: AS, AX! и АХг. Однако из уравнения
сохранения вещества
AS + АХ, + АХг = 0 (ж)
получается, что
d (AS)/dt + d (ДХ,)/Л + d (АХ,)/Л =0 (з)
и в действительности есть только система из двух независимых уравнений
с двумя неизвестными. Стандартными методами (здесь они ие описаны)
оставшиеся два дифференциальных уравнения могут быть сведены к системе из Двух
алгебраических уравнений, решениями которой являются корни определителя:
(«и +~ J (<*!!■+ — 1 — Оцвц. (н)
в. Определитель можно легко решить, получив два значения (1/г), где
an A'i([SJ' + [Ej'),
al2 = £'_,,
afl=ft'1([S]' + [E]')-A/-«, (к)
а22 = — (*'-! +А'.4-А'-,).
г. Убедитесь, что оба корня (1/т) и уравнения '(и) удовлетворяют свойствам,
перечисленным для этого примера в табл. 116.1. Для более сложных примеров
с многочисленными состояниями равновесия проблема по-прежнему сводится
к решению определителя типа, показанного в уравнении (и), за исключением
того, что в общем случае существует N решений для системы с N
уравнениями.
Таким образом, вы решили примеры наиболее типичных и общих задач
релаксационной химической кинетики, выведя уравнения (16.12), (16.13) и
данное выше уравнение (и).
Литература
Нестационарная химическая кинетика быстрых реакций
1. Havsteen В. Н., In Physical Prindples and Techniques of Protein Chemistry,
Part A, ed. S. J. Leach, Academic Press, New York (1969), p. 245—290.
2. Hammes G. G„ Wu C.-W. «Kinetics if Allosteric Enzymes». Ann. Rev. Biophys.
Bioeng., 3, 1—34 (1974).
Деполяризация флуоресценции
3. Rigler R., Ehrenberg M., «Fluorescence Relaxation Spectroscopy in the Analysis
of Macromolecular Structure and Motion», Quart. Rev. Biophys., 9, 1—20
(1976).
4. Chen R. F., Edelhoch H., Steiner R. F„ In Physical Principles and Techniques of
Protein Chemistry, Part A, ed. S. J. Leach, Academic Press, New York (1969),
p. 171—244.
ГРУЗ НА ПРУЖИНЕ
т. Pesce A. J., Rosen C.-G., Pasby T. L. Fluorescence Spectroscopy: An Introduction
Г for Biology and Medicine, Marcel Dekker, New York (1971).
Магнитная релаксация
6. Sykes В. D., Scott M. D. «Nuclear Magnetic Resonance Studies of the Dynamic
Aspects of Molecular Structure and Interaction in Biological Systems», Ann. Rev.
Biophys. Bioeng., 1, 27—50 (1972).
7. James T. L. Nuclear Magnetic Resonance in Biochemistry, Academic Press, New
York (1975), Chaptes 2, 6 and 8.
Модели «черного ящика» в физиологии
в. Grodins F. S. Control Theory and Biological Systems, Columbia University Press,
New York (1963).
ГЛАВА 17
СВЯЗАННЫЕ ПРУЖИНЫ
В гл. 14—16 мы рассматривали определенные электронные,
ядерные и атомные движения раздельно, считая каждое из них
независимым от всех остальных. Однако можно извлечь много
дополнительной информации, изучая взаимодействия (обычно
слабые) между различными типами молекулярных «пружин»,
включая информацию о прочности химических связей, их длинах,
углах между связями, а также химическом окружении определенных
функциональных групп. В этой главе мы показываем, что такие
«взаимодействия» проявляют себя, как правило, в виде
«расщепления» единственной собственной частоты исходной пружины на две
частоты с двумя максимумами поглощения в стационарном
состоянии. Сила взаимодействия оказывается связанной с
величиной расщепления и поэтому легко определяется из
спектроскопических экспериментов в стационарном режиме. Мы обсудим
далее результат последовательного соединения нескольких пружин
(например, химических связей), дающих составное движение,
которое может быть разбито на различные «нормальные» колебания,
становящиеся новыми собственными частотами (с
соответствующими спектральными «пиками» поглощения) для системы.
Наконец, будет показано, что особый тип взаимодействия между
вынужденными колебаниями электронов (т. е. рассеянием света) и
колебаниями атомов, которым принадлежат эти электроны,
известный как эффект комбинационного рассеяния, или раман-эффект,
ведет себя подобно обычной «амплитудной» модуляции, давая
«центральную полосу», соответствующую исходному вынужденному
колебанию электронов, плюс «боковые полосы», расположенные по
обеим сторонам центральной полосы иа шкале частот. Применения
этих явлений показаны и подробнее обсуждены в гл. 18.А, где
описана обработка экспериментальных данных для определения
«констант взаимодействия» из спектров поглощения.
17.А. КОНСТАНТЫ ВЗАИМОДЕЙСТВИЯ
И СПЕКТРАЛЬНЫЕ РАСЩЕПЛЕНИЯ
Рассмотрим систему, показанную на рис. 17.1, состоящую из
двух одинаковых масс т, подвешенных на двух идентичных
пружинах (с силовой постоянной £о), в которой обе пружины к тому
ГРУЗ НА ПРУЖИНЕ
Рис. 17.1. Схематическое изображение двух равных масс, подвешенных на двух
идентичных пружинах \(k0) и соединенных третьей пружиной \(k).
Координата расположения левой массы — у, правой массы — х. Как следует из уравнений,
на каждую массу действует сила, пропорциональная (с коэффициентом пропорционально»
стн ко) смещению ее собственной пружины, плюс сила, пропорциональная разнице
смещений (у — *) двух масс из-за наличия соединяющей их пружины (с коэффициентом
пропорциональности- к), (х и у принимают за нуль, когда массы занимают свое равновесное
положение.)
же связаны между собой третьей пружиной (с силовой постоянной
k). Вначале рассмотрим обобщенную силу m{d2yfdt2),
действующую на левую массу:
m(d2yjdt*) = — kbu — ky-\-kx — — k0y — k{y — x). (17.1)
Другими словами, сила [левая сторона уравнения (17.1)],
действующая на левую массу, является суммой возвращающей силы
ее собственной пружины —koy и добавочной силы, которая зависит
от того, где находится другая масса в соответствии с различием
в смещении двух масс относительно друг друга (например, если
обе массы сдвигаются вправо на одно и то же расстояние (у—х) =
= 0, соединяющая пружина не растягивается совсем, и сила,
возникающая из-за наличия этой пружины, равна нулю).
Аналогичное уравнение можно записать для обобщенной силы,
действующей на правую массу на рис. 17.1
т (d2x(dt2) = — k0x — k (x — у).
(17.2)
Далее попытаемся найти «собственную» частоту (частоты) этой
системы, делая простейшее - возможное предположение, что при
данном «собственном» колебании («нормальное колебание») обе
массы колеблются с одинаковой частотой (мы вернемся к
разъяснению смысла этого предположения в следующем разделе).
Другими словами, мы предполагаем, что смешения хну имеют одну
и ту же частоту колебаний
х = А cos (Ы),
y=Bcos\(tot)
(17.3а)
(17.36)
в нашей обычной записи, где частота ю одинакова в обоих
уравнениях, но амплитуды А и В движений двух масс могут быть раз-
556 ЧАСТЬ 4
личны. Для определения ю подставим в уравнения (17.1) и (17.2)
выражения для х и у из уравнений (17.3а) и (17.36) и получим
I т т \ т
И
В
J m ml m
А
т т-
Перемножая уравнения (17.4а) и (17.46), находим
(17,4а)
(17.46)
L« _ *•_—L\aB = (—)
(17.5)
Отбрасывая простейшее решение (Л = В=0), мы можем сократить
множитель АВ в обеих частях уравнения (17.5) и получить
решения:
со
=Vv
т
» = K[(V») + (2A/«)J .
две частоты
нормальных колебаний
(17.6а)
(17.66)
Конечный результат [уравнения (17.6а) и (17.66)] описания
движения двух связанных пружин таков, что массы могут теперь
проявлять две собственные частоты колебаний, которые
различаются (по частоте) на величи-ну, прямо связанную с силой пружины
соединяющей движения двух изолированных пружин. Качественная
картина результата этого вычисления, базирующегося целиком на
классической механике, сохраняется и при кваитовомеханическом
описании энергетических уровней и спектральных расщеплений,
являющихся результатом взаимодействия, например, магнитной
прецессии одного ядра с магнитной прецессией другого.
В гл. 18.А мы покажем, как величину такого «скалярного
взаимодействия» в магнитном резонансе легко определяют из
спектральных расщеплений отдельных компонент мультиплетной
структуры резонансов для единственного магнитного ядра и как
получающаяся «константа взаимодействия» обеспечивает прямое
измерение двугранного угла (угла, на который заместители на одном
конце углерод-углеродной связи повернуты относительно
заместителей иа другом ее конце).
17.Б. НЕПОСРЕДСТВЕННО СОЕДИНЕННЫЕ ПРУЖИНЫ.
НОРМАЛЬНЫЕ КОЛЕБАНИЯ
Особенно наглядно описывает внутримолекулярные движения
модель, в которой каждый атом молекулы соединен пружиной с
другими атомами так, что сила пружины, обозначающей данную
ГРУЗ НА ПРУЖИНЕ
©■*-
©*-
Рис. 17.2. Направления смещений колеблющихся атомов в каждом из шести
нормальных колебаний иона СО|"
[Из Cotton F. A., Chemical Applications' of Group, Theory, Wiley-Intersdence, New York,
1963, R. 247.1
химическую связь, характеризует прочность этой связи (сила, при
приложении которой связь разорвалась бы). Существуют два пути
описания движений всех атомов в молекуле. Трудный путь —
следить за изменением положений (т. е. за координатами х, у и z
каждого атома) всех N атомов в молекуле, требующий 3N
переменных величин для описания всех изменений в положениях
атомов. Более легкий путь — следить за поступательным
перемещением молекулы как целого, когда все атомы двигаются вместе в
одном направлении. Таким образом, если мы отделим
поступательные перемещения молекулы как целого (в любом из трех
независимых направлений) от остающихся движений, будет
необходимо только (3N—3) переменных. Аналогично после отделения
трех независимых вращательных движений молекулы как целого
мы останемся только с {SN—6) переменными, связанными с
колебательными движениями атомов относительно друг друга
(изменения длин связей и углов между связями — «валентные» и
«деформационные» колебания). Можно описать все эти валентные и
деформационные движения как набор «собственных»
колебательных частот, называемых «нормальными колебаниями», идя тем же
самым путем, что и при отыскании собственных колебательных
частот двух масс в предыдущем примере со связанными
пружинами. При любом нормальном колебании все атомы в молекуле ко-
558 часть 4
л
V
|<
/
V
1/"'
\
*
/1
' IN
(СН валентное)
Л
н*
АС
\ валентн
=2950см
/
И
1/Н^
\
Л
/•
'СН2деаюрмаццоцное)
*= 1460 см4
/1
"I
ое)
-/
/
1/
\
'
(СН2 крутильное)
»-7270см-'
л
/1
Н2 маятниковое)
> = 800см->
'У/
\
/^ (CHzeeepHot
0
Рис. 17.3.. Направления движения атомов для каждого из различных
нормальных колебаний изолированной метнленовой группы,
Частоты нормальных колебании, указанные здесь же, почти ие зависят от строения
остальной части молекулы и, таким образом, могут помочь идентифицировать эту химическую
группу по наличию пиков спектрального поглощения с этими частотами. '[Из Barrow G. М.,
Introduction to Molecular Spectroscopy, McGraw-Hill, New York, 1962, p. 200.]
леблются с одинаковой собственной частотой, но, вообще говоря,
в разных направлениях (см. ниже).
На рис, 17.2 показаны [3(4)—6] =6 типов нормальных
колебаний иона СО§~. Важными особенностями этих колебаний
являются, во-первых, то, что для каждого из нормальных колебаний
любое данное внутримолекулярное движение — безразлично какой
сложности — можно выразить как сумму движений с
«собственными» частотами, равными частотам «нормальных колебаний» с
соответствующими амплитудами, и, во-вторых, то, что реакция
молекулы в стационарном состоянии иа непрерывное синусоидальное
внешнее воздействие будет состоять из пиков поглощения (и
дисперсии) (рис. 13.5) с центрами при каждой из частот
«собственных» или «нормальных колебаний» (см, примеры в гл. 20.Б.З и
14.А).
Наиболее полезное свойство нормальных колебаний
становится очевидным, когда мы рассматриваем колебания молекул,
содержащих большое число атомов. В этих случаях часто возможно
рассматривать определенные химические функциональные группы
(такие, как карбонильная или Метиленовая) так, как если бы они
были связаны с очень массивным молекулярным остовом. Чита-
ГРУЗ НА ПРУЖИНЕ
Ель может оценить обоснованность этой точки зрения, решив за-
ючу о связанных пружинах (рис. 17.1) при условии, что одна из
расе гораздо больше другой (см. задачу к этой главе). В
результате получается, что существуют нормальные колебания, которые
реперь можно связать с данной химической группой почти
независимо от ее окружения в молекуле. Колебания, связанные с
изолированной метиленовой группой, показаны иа рис. 17.3. Существует
несколько «собственных» частот, достаточно отделенных друг от
круга, чтобы их можно было наблюдать по отдельности методом
инфракрасной спектроскопии (стационарное состояние) (см.
|л.. 20). Это формальная основа для «дактилоскопического»
применения инфракрасной спектроскопии при идентификации
химических функциональных групп в молекулах: оказалось, что частота
Электрического поля (электромагнитной волны), необходимая для
|ого, чтобы возбуждать движение положительно заряженных ядер
относительно их равновесных положений на соответствующих
межатомных «пружинах», лежит в инфракрасной частотной области.
:-
17.В. АМПЛИТУДНАЯ МОДУЛЯЦИЯ.
СПЕКТРОСКОПИЯ КОМБИНАЦИОННОГО РАССЕЯНИЯ
I Существует особый тип рассеяния света, в котором иа
вынужденное колебание «электронов на пружинах» оказывают
воздействие происходящие одновременно с ним движения при нормальных
колебаниях (со значительно меньшей частотой) атомов, к
которым эти электроны прикреплены. «Взаимодействие» этих ■ двух
нвижеиий не является, однако, прямым, как это было в примерах,
данных в гл. 17.А и 17.Б, и требует другого толкования (рис. 17.4).
| При нашем первом рассмотрении светорассеяния оказалось
удобным определить «поляризуемость» как коэффициент линейной
пропорциональности между величиной возбуждающего
электрического поля E=Eq cos («0 и наведенным дипольным моментом ех.
ех^аЕ = аЕь cos (arf)/ (15.13)
^Исходя из этого уравнения, мы затем показали, что излучение,
испускаемое наведенным диполем, происходит лишь на одной
частоте о, одинаковой с возбуждающей частотой. Однако после
обсуждения явления дихроизма стало ясно, что свойства молекулы,
[рюобще говоря, могут быть различны, если на молекулу смотреть
§вдоль разных направлений. В частности, мы можем ожидать, что
(наведенный дипольный момент ех для колебания вдоль
направления х может быть отличным от наведенного момента еу для
движения вдоль направления у или ег для направления z. Мы, таким
^образом, ожидаем, что соответствующие поляризуемости
(которые определяют амплитуду движения возбуждаемого электрона
|вдоль соответствующей оси) цх, щ и az могут быть различны для
560 ЧАСТЬ 4
1
i
"cf
Рэлеевская
Ьиния
1лМЫд
Рэлеевское
рассеяние
Частота
I
<*>,
и)
—^—CHMiL^O *%%ж
Стоксова
линия
Рэлеевская
линия
■ионное
рассеяние
Антистоксава
линия
(л),
to
Частота
Рис. 17.4. Схематическое изображение возникновения эффекта комбинационного
рассеяния.
«Собственная» частота возбуждаемого электрона на неподвижном атоме, как правило,
лежит в ультрафиолетовой области спектра и приводит к рассеянию света с максимальной
интенсивностью при собственной частоте о. как показано на верхней диаграмме справа.
Колебание (с частотой ©i) атома, которому принадлежит электрон, действует как
синусоидальная модуляция амплитуды колебания возбуждаемого электрона, и эта амплитудная
модуляция обнаруживает себя в появлении двух новых компонент (комбинационного)
рассеяния, расположенных при более высоких (антистоксовы линнн) или более низких
частотах (стоксовы линии) и отстоящих от основной частоты иа ± ©i соответственно.
Поскольку Ю1<£Сю, иа левых диаграммах «пружина» колебания атома показана гораздо более
слабой, чем «пружина» колебания электрона.
несферических молекул. Бол^е того, поскольку атомы молекулы
непрерывно колеблются и меняют при этом распределение
электронов в молекуле, мы ожидаем, что поляризуемость изменится, как
только атомы сдвинутся со своих равновесных положений. Если
изменение поляризуемости с расстоянием невелико, можно
приблизительно выразить а как функцию расстояния, ограничившись
только первыми двумя членами разложения ее в ряд Тейлора
вблизи положений равновесия атомов грав (см. приложение):
« = ««,.+ [ iiftr^ ]r=rjr-r>"> +
(17.7)
где г— (неравновесное) положение колеблющихся атомов, а
(г—Грав) представляет собой отклонение от положения
равновесия. Но поскольку атомы колеблются с частотой, скажем, о)ь
отклонение от равновесия (г—грав) также колеблется с частотой о>ь
r-rtn = Acos{*lt)t (17.8)
где А— амплитуда атомного колебания. Подставляя уравнение
(17.8) в уравнение (17.7) и затем уравнение (17.7) в уравнение
(15.13), получим новое уравнение
ех = аг=Грав Е0 cos К) + Е,А [dafd (г - грав)]г=грав cos К) cos(a> Д
(17 9)
ГРУЗ НА ПРУЖИНЕ
■ророе можно переписать, используя тригонометрическое
тожество для произведения косинусов (см. приложение):
I cos(a)cos(6)=Va[cos(a — b)-\-cos(a-\-b)], (17.10)
Шолучив желаемый результат:
I в* = *г=ГраЕ* C0S И + И/2) Ео [*Ф {Г ~ '■рав)]г=грав [COS (ш - «J f +
: + cos (ю+ ©,)*]. (17.11)
^посредственное влияние колебаний молекулы состоит в том, что
[место стационарного рэлеевского рассеяния света с излучением
[а одной частоте о излучение происходит на трех частотах и со-
:тоит из «центральной полосы» обычного рэлеевского рассеяния©
[ двух новых дополнительных полос с частотами сь
локализованных на расстоянии в одну частоту атомных колебаний по обе
стороны от рэлеевской центральной полосы. Эти новые
компоненты рассеяния называют компонентами комбинационного рассеяния
или рамановскими компонентами, и теперь ясно, что они получа-
ртся просто в результате синусоидальной «модуляции»
(изменения) амплитуды а рэлеевского отклика на возбуждение. Ситуация
Ьдесь формально идентична использованию голоса или музыки
Для модуляции амплитуды радиочастотной волны-«носителя» в
системе АМ-радиопередатчик — АМ-приемник, когда принимают
Модулированные сигналы и восстанавливают первоначальный
«звуковой» сигнал, использовавшийся для их получения, с помощью
Электронного процесса, который по существу преобразует
уравнение (17.9) в уравнение (17.11).
\ Читатель может вспомнить, что наличие нормальных
колебаний атомов в молекулах можно непосредственно обнаруживать,
изучая поглощение в инфракрасной области, (гл. 14.А) легче, чем
этим косвенным путем, измеряя рассеянное излучение в видимой
-и ультрафиолетовой областях. Логично поинтересоваться, зачем
нужно проводить оба типа экспериментов. На это есть две
главные причины. Первая — в то время как рэлеевское рассеяние
наблюдают благодаря наличию у выведенного из равновесия
«электрона на пружине» электрического дипольного момента
[уравнение (15.13)], уравнение (17.11) показывает, что появление «пиков,»
комбинационного рассеяния требует изменения в поляризуемости,
когда атомы колеблются с частотой «нормального колебания»
©1 (т. е. da/d(r—грав)¥=0). Более того, квантовомеханические
«правила отбора» (гл. 19.Б.4) предсказывают, что поглощение (в
инфракрасной области) на данной частоте нормального колебания
может происходить только в том случае, когда в результате
колебания возникают изменения дипольного момента. Так,
колебание vi для иона СОз" (рис. 17.2) «неактивно» (т. е. ненаблюдае-
мо) в инфракрасном поглощении, поскольку при этом колебании
562 ЧАСТЬ 4
дипольный момент не изменяется. (По той же самой причине
частоты «валентных колебаний» 02 в табл. 14.1 определены
методом комбинационного рассеяния в видимой и ультрафиолетовой
областях, а не путем непосредственного измерения поглощения в
инфракрасной области.) Различные нормальные колебания
стараются наблюдать с помощью обоих методов (комбинационного
рассеяния и инфракрасного поглощения), поскольку для полного
описания молекулярных колебаний желательно применение обоих
типов измерения. Вторая причина заключается в том, что
непосредственное получение инфракрасных спектров молекул в водных
растворах затруднено из-за очень сильного инфракрасного
поглощения водой, тогда как при снятии, спектров комбинационного
рассеяния эта проблема несущественна. Фактически (см. нижесле-
! п ' I • I i I ' i I i I i I Ч
600 800 1000 1200 1400 1600
Рис. 17.5. Спектры комбинационного рассеяния фермента рибонукдеазы А
в растворе (а) и в шоликрметаллической форме (б).
Различия в этих спектрах интерпретированы (см. текст) следующим образом: коиформацня
основной полипептидной цепн в общих чертах сохраняется при переходе от кристалла к
раствору, в то время как днеульфидные связи и окружение тирозина меняются. 1Из
Уи. N.. /о В. Н., J. Am. Chem. SoC, 95. БОЗЗ (1973).J
53 ГРУЗ НА ПРУЖИНЕ
|ующий пример) спектроскопия комбинационного рассеяния
является одним из немногих методов, одинаково пригодных для
детального исследования молекул как в жидкой, так и в твердой фа-
sax.
х-
^Пример. Применение спектров комбинационного рассеяния для сравнения
р- структуры белка н кристаллах и растворе
{ На рис. 17.5 показаны спектры комбинационного рассеянвя, полученные для
'фермента рибоиуклеазы А в растворе (верхний спектр) и в поликристалличе-
кжом (порошок) состоянии (нижний спектр). Их главная особенность состоит
;"в том, что колебания полипептидного «остова», помеченные как область
НЕамид III», показывают очень хорошее соответствие как по положениям, так и
:ЛО ширине пиков между кристаллической и растворенной формами. Это
позволяет предположить идентичность конформаций остова RNa3U А в обеих фазах,
;Это чрезвычайно важное заключение потому, что, хотя определения структуры
^макромолекул методом рентгеноструктуриого анализа и могут быть очень
точными, всегда остается вопрос об обоснованности сравнения структуры
кристаллической молекулы с ее структурой в растворе: Аналогичные выводы не следует,
однако, обобщать: существует несколько белков, структуры которых в
кристаллах и в растворе различаются. Например, небольшое отличие области
колебаний «амид III» в спектрах комбинационного рассеяния обнаружено у
растворенной и кристаллической форм другого фермента—карбоксипептидазы А.
[-Возвращаясь к спектрам на рис. 17.5, заметим, что в области 500—700 см-1
^имеются и различия между растворенной и кристаллической формами. Они
связаны с изменениями в способах расположения связей S—S и С—S, а также
р колебаниями кольца тирозинового остатка. Это позволяет предположить, что,
Зсотя конфигурация остова и сохраняется, при переходе от кристаллической
Эфазы к раствору появляются изменения в геометрии дисульфидных связей и
в окружении «спрятанных» тирозиновых остатков. Подобные измерения
становятся все более популярными по мере появления лазерных источников света,
^которые обеспечиваю»- весьма высокую интенсивность возбуждения,
необходимую для усиления (слабых) сигналов комбинационного рассеяния.
Задачи ,
1. а. Для системы двух неравных масс, подвешенных на одинаковых пружинах
(с силовой постоянной k0) и связанных третьей пружиной (с силовой
постоянной k), рассчитайте возможные частоты «собственных» («нормальных»)
колебаний.
б. Покажите, что в пределе, когда взаимодействие становится бесконечно
слабым (k—-н0), результатом являются два независимых движения с
«собственными» частотами двух изолированных пружии.
в. Более интересно показать, что в пределе, когда одна из масс (скажем,
т2) гораздо больше другой, движение легкой массы происходит с частотой,
определяемой k0, k и m,. Другими словами, этот расчет показывает, почему
во многих случаях хорошим приближением является рассмотрение
функциональной группы молекулы (например, карбонильной группы) как
изолированного «груза иа пружине», связанного с каркасом бесконечной массы.-
I
В
V к — Ь г— k S
/'/:—^S&$u—0—-Ши—["71—кЩи—Ь,
Ь,
564 ЧАСТЬ 4
Литература'
Спектроскопия комбинационного рассеяния
1. Moore В. Chemical and Biochemical Applications of Lasers, Academic Press, New
York (1974).
2. Spiro T. G., Gaber B. P. «Laser Raman Scattering as a Probe of Protein
Structure», Ann. Rev. Biochem., 46, 553 (1977).
3. Frushour B. G., Koenig J. L. Advances in Infrared and Raman Spectroscopy, ed.
R. J. H. Clark, Hester R. E., Haydon & Son, New York (1975), vol. 1, pp. 35—
97.
ЧАСТЬ 5
КВАНТОВАЯ МЕХАНИКА:
КОГДА ОНА НА САМОМ ДЕЛЕ
НЕОБХОДИМА?
«Было время, когда газеты говорили, что только
двенадцать человек понимают теорию относительности. Я не верю,
что такое время было. Конечно, сначала ее понимал только
один человек, единственный, кто мог написать о ней статью.
Но после того, как статью прочли, многие люди так или
иначе поняли теорию относительности, и их, несомненно, было
больше двенадцати... С другой стороны, я думаю, что, могу
без опасений сказать, что никто не понимает квантовую меха-
НИКУ>:>- Ричард Фейнман.
В части 4 мы нашли, что многие специальные свойства,
обычно вводимые с помощью квантовой механики, можно понять
исходя из простых классических моделей, основанных на
рассмотрении груза на пружине. Однако классические модели
неудовлетворительны в целом ряде областей, важных для биофизических
приложений (как и в других областях, представляющих интерес,
например, для физиков). Так, в классической модели электрона на
пружине электрон имеет только одну собственную частоту, в то
время как реальный электрон в простом атоме водорода имеет
бесконечное 'число «собственных» частот. Квантовая механика
дает средства для предсказания числа и величины этих частот
(гл. 18). Поскольку установлено, что частицы могут обладать
только определенными, дискретными (в противоположность
непрерывным), «квантованными» энергиями, немедленно возникает
статистическая задача о числе частиц, обладающих каждой
определенной энергией, учитывая, что общее число частиц и суммарная
энергия этого набора частиц постоянны. Получающееся в
результате" распределение Больцмана (гл. 19) позволяет предсказать
множество молекулярных свойств, например средний электрический
дипольный момент, который нельзя получить из классической
механики, и приводит естественным путем к пониманию лазеров я
других неравновесных процессов. Наконец, воздействие внешних
или уже существующих в самом образце флуктуирующих
электрических или магнитных полей на переходы между энергетическими
уровнями (соотносящимися с интенсивностями спектрального
поглощения и связанными с экспериментами по хромофорам и
«меткам») логически вытекает из рассмотрения действий маленьких
резких (синусоидальных или беспорядочных) возмущений на
статические энергетические уровни, полученные в гл. 18.
ГЛАВА 18
ОБОБЩЕННАЯ ГЕОМЕТРИЯ:
СУЩЕСТВОВАНИЕ И ПОЛОЖЕНИЕ
СПЕКТРАЛЬНЫХ «ЛИНИЙ» ПОГЛОЩЕНИЯ
ЭНЕРГИИ. КВАНТОВОМЕХАНИЧЕСКИЙ
ГРУЗ НА ПРУЖИНЕ
Квантовую механику наиболее часто вводят, используя язык
исчислений. Обычный метод — предложить подходящее
дифференциальное уравнение (гамильтониан) и затем показать, что его
решение возможно только для определенных дискретных
(«квантованных», часто целочисленных) значений некоторого параметра.
Многие читатели этой книги, возможно, уже решали этим путем
задачи о «частице в ящике» и о «центральной силе» (т. е. о
водородном атоме) в начальных курсах химии или физики. Сложность
этого подхода помимо значительных математических трудностей
становится сразу очевидной из табл. 18.1, которая показывает, что
основное содержание квантовой механики излагается с
применением операторов, а не функций. Узнав совсем немного об
операторах, мы сможем решить квантовомеханическую задачу о грузе на
пружине, вообще не используя дифференциальных уравнений.
Затем, пользуясь полным соответствием свойств линейных
операторов и матриц, мы используем матрицы для решения простых и
важных «спиновых» задач, которые не имеют классических
аналогий, но имеют много биофизических приложений.
Как показано ниже, квантовая механика базируется на двух
постулатах: 1) «состояние» частицы (из которого мы можем
предсказать вероятность получения данного результате при
экспериментальном измерении энергии, координаты, импульса и т. д.)
описывается некоей функцией; 2) все физические «наблюдаемые
величины»* (энергия, координата, импульс и т. д.) представлены
операторами. Оперировать этими категориями станет легче, если
понять, что их можно представлять себе как обобщение обычной
геометрии (табл. 18.2).
- Основная идея табл. 18.2 состоит в том, что полезно
вообразить себе'«пространство», состоящее из функций (а ие векторов),
в котором любая индивидуальная функция д|> может быть
выражена через ее компоненты вдоль неких базисных функциональных
«осей» от ф1 до фп так же, как обычный вектор можно выразить
* В литературе физические наблюдаемые величины часто называют просто
«наблюдаемыми». — Прим. ред.
Таблица 18.2
Наглядное сравнение свойств обычных векторов в трехмерном пространстве и свойств
функций в n-мерном функциональном «пространстве»»
Обычная геометрия
Обобщенная геометрия (квантовая механика)
Любой вектор можно выразить его компонентами РАЗЛОЖЕНИЕ НА
вдоль трех осей через векторы единичной длины КОМПОНЕНТЫ Гурав-
2 нення (18.1), (18.2)]
i — вектор единичной
длины вдоль оси х
j — вектор единичной
длины вдоль осн у
к — вектор единичной
—У длины вдоль оси z
Любую функцию г|> можно выразить ее
компонентами единичной «длины» по направлениям
«базисных» функций цц (см. ниже) {ty может быть
комплексной — см. ниже)
■ф = fli<Pi + а2ф2 + .. a„tp„
*=2«iW£
Ой
г = at\ + a2j + a3k = f<h\— «вектор-столбец»
х-, у- и «-компонентами г являются alt a2 и а3
| г |2 = г г = (а&аз) /аЛ = а\ + а| + а\
(9
ДЛИНА
[уравнения (18.3) —
(18.5)]
<tjj, ф> = (aj, а\.. .а*п) /^Ч = J] fyi =
*=i
Длиной вектора г является \г\ = у г-г
Для единичного вектора г-г = 1
ЕДИНИЧНАЯ ДЛИНА
= К12+ 1«а|2+---|а«1а
«Нормой» функции является У"^»^)
Для «нормированной» функции (ф, "ф> = 1
Продолжение табл. 18.2
Обычная геометрия
Обобщенная геометрия (квантовая механича)
Замена вектора г на вектор s
Tr = s
ПРЕОБРАЗОВАНИЯ
(
Тгг
ти
7«
Тгг
Т
1 23
Гэа
^13
т»
Тзз.
[уравнения
(18.8)]
(18.6)-
Ь2 = Т^ -j- 7,з2а2+ Тм<Н
ba — Тзгвг + Ttofh + ^за0*
3
bt = 2 TVai
_J является линейным преобразованием
Тц — элемент (t-й строки и /-го столбца)
ставления Т в виде матрицы
Замена функции 1]з на
Тф=Ф
~?Тгг Т12... Т1П\/аЛ
1т21 т22... г*\|в1
\Тпг Тпг... Тпп/ \ап/
Ьх = Т1хах + Т12а2 -f- • •
функцию Ф
l-ffl
п
Jj TfPs
У-1
предал = Тщ^г + ^WV • • + Tnnfln
Т является линейным оператором
Тц— элемент представления Т в виде матрицы
Изменение направления вектора г, с сохранением
той же самой его длины (т. е. поворот вектора г)
Ur = s,
так что |s| =|£/г| = )г|
УНИТАРНОЕ
ПРЕОБРАЗОВАНИЕ
Замена функции i|) на функцию Ф с сохранением
нормы функции
U-ф = Ф
Если <Ф,ф> = <[/+, U^> = <+. +>.
то-1/является «унитарным» оператором
572 ЧАСТЬ Б
через его компоненты вдоль осей х, у и. г. Операции, используемые
в квантовомеханических расчетах (например, нормирование,
проверка на ортогональность, действие операторов на волновые
функции, решение задач на собственные значения), можно понять по
аналогии с различными хорошо известными геометрическими
понятиями, такими, как длина, угол, перпендикулярность, поворот
и изменение длины. Поскольку геометрия проще алгебры
(сравните рисунок эллипса с формулой для эллипса!), большинство
квантовомеханических расчетов проводят, используя векторную и
матричную запись, показанную в табл. 18.2, а не решая
соответствующие дифференциальные уравнения (в действительности
дифференциальное уравнение даже не всегда может быть составлено —
см. «спиновые» задачи ниже). Правила обращения с векторами
и матрицами проиллюстрированы примерами в табл. 18.2.
Операции с матрицами
л-Мерный вектор можно представить себе в виде колонки,
цифры которой обозначают величины компонент вектора («проекций»)
в направлении каждого из определенных «базисных векторов» или
«базисных функций». При записи уравнения (18.1) читатель
должен помнить, что эти компоненты будут различны, если мы
выберем различные базисные наборы [так, в обычной геометрии
единичный вектор вдоль оси х нужно записывать как (1,0,0) в базисе
(i, j, k), но следует записывать как (0, 1, 0) в базисе (j, i, k)].
В квантовой механике обычная задача состоит в том, чтобы
сначала найти собственные функции [уравнения (18.14) и
последующие] для определенного оператора (гамильтониан — см. табл. 18.3)
на основе некоторого удобного набора базисных функций и затем
выразить любое данное состояние (функцию) реальной частицы
(частиц) через его компоненты вдоль каждой из собственных
функций, образующих (новый) набор базисных функций.
Ф = «I?! + а*Ъ + » • + ЯП?Л> ( 1 8. 1)
П
Ф=2 aib> 08.1а)
где понятно, что это компоненты
для определенного набора базисных (18.2)
функций <р„ <Pi. ••••> 9п-
Иногда говорят, что уравнение (18.1) выражает tp в виде
«линейной комбинации» базисных функций ср*.
Для получения скалярного произведения векторов мы сначала
построим вектор-строку, состоящую из величин, комплексно
сопряженных компонентам левого вектора скалярного произведения
Перечень физических наблюдаемых величин и соответствующих им квантовомеханических операторов-
Физическая наблюдаемая величина
Квантовомеханяческий оператор
х — компонента расстояния вдоль оси х
Рх= компонента импульса вдоль осн х — mvx
г — векторное расстояние
р — векторный импульс
t — время
(р%/2т) + V(x) — гамильтониан (т. е. кинетическая плюс
потенциальная энергии) для одной частицы в одном измерении
(p-p/2m) + V(x, у, г) — гамильтониан для одной частицы в
трехмерном пространстве
L = г-р— классический («орбитальный») угловой момент
х (г. е. умножение на х)
Рх= —iHdfdx)
г = х\ -f-Ч\ + zk (т. е* г — вектор, компоненты которого —
операторы)
р = (-/ft) (d/dx)i + (—/ft) (д/dt/)} + (-/ft) (д/дг)Ь
t (т. еф умножение на t)
р%/2т) + V{x) = — (ft8/2m) (dW) + V(x), где ft = ft/2 л, aft —
постоянная Планка ft = 1,055 Ю-34 Джс
(pp/2m) + V(x, y. z) = -(ky2m)V2+ V(x, y, z),
где V2 -= дг/дх* -f дуду2 -+- d*/dz*
L=rp = Lx\ + Lyj + Lzk,
где Lx = (- /ft) \y{d/dz) - z{d)dy)\,
Ly = {-/ft) Wfdx)-х(д/дг)\,
Lz=(~Л) [Ф/ду)-У(д/дх)},
откуда видно (см. текст), что
[LXt Ly\ = LxLy — LyLx = in,Lz,
[Ly, Lz\ ■= LyLz — LzLy — inLx,
[Lz> Lx] = LZLX — LXLZ = ihLy,
где компоненты / удовлетворяют соотношениям
Vy, Iz) = lylz-lzIy = iMx,
[/*. Ix) = hh - hh = Щ
a r, p, L и I — «векторные операторы:». Это значит, что с каждым из них надо обращаться как с обычным трехмерным вектором,
компоненты которого являются операторами. Для объяснения наблюдаемых измерений необходимо постулировать дополнительный (чисто кван-
товомеханический) угловой момент I в добавление к угловому моменту L, полученному по аналогии с классической механикой. Другими
словами, реальные частицы обладают угловым моментом J = L + I. Значение операторов, перечисленных в этой таблице, в том, что результат
любого единственного измерения любой из наблюдаемых величин должен быть собственным значением соответствующего оператора 1см.
физическую интерпретацию № 1 в тексте).
I — квантовомеханяческий «спиновый»
которого нет классического аналога
угловой момент, для
574 часть s
(т. е. получающихся заменой pzhiq на pz^iq), затем умножим
первый элемент вектор-строки на первый элемент вектора-столбца,
соответствующего правой функции в скалярном произведении,
затем умножим второй элемент вектор-строки на второй элемент
вектор-столбца и т. д. и, наконец, сложим все получившиеся числа
вместе, как показано в уравнениях (18.3) — (18.5а). Все это
выглядит сложно, но на самом деле представляет собой весьма
быстрый процесс, если его записать не словами, а в виде уравнений
Ч>4 а- V <Р=1 ? I (18-3)
<Ь?> = «а\...а*п)\ : |= (18.4)
К
Можно представить преобразование одного вектора в другой
вектор (также известное как «операция» по замене одной функции
другой функцией) как умножение матрицы, соответствующей
этому преобразованию, на вектор-столбец таким образом, чтобы
получить конечный вектор-столбец. Правило для этой матричной
«операции» простое: мы получим, скажем, второй элемент нового
вектора, взяв сумму произведений после поэлементного умножения
второй строки матрицы на исходный вектор-столбец, как показано
в следующих уравнениях:
Тф — <р (Т — «оператор») , (18.6)
Т» Т1а ... Tin \ уах\ ГК
121 12е ••• * tn I / ^а |_»[ ^«
(18.7)
' м * пл '•• * пп &п *п
"го
Ьх = Tuat -f Tisfl2 4-... И- Tlnar
b*^T21a1 + Tnas + ... + T2nan, (18.8)
К — 7\* А + ТпгОг + • •• + Tnr&n
п
или более компактно 6, =2 ^цаг (1^.8а)
175 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
|.. В квантовой механике часто желательно применить два
последовательных преобразования, скажем преобразование В, за
которым следует преобразование А (этот процесс можно, например,
■использовать для нахождения результата измерения физической
переменной, соответствующей В, за которым следует измерение
физической переменной, соответствующей А, см. ниже)..
Определим произведение двух операторов АВ как
Щ = А(ВЦ).
(18.9)
Другими словами, мы действуем на функцию г|э оператором В и
затем воздействуем на результирующий вектор-столбец, используя
оператор А. В этом случае мы можем действовать на вектор г|э по-
другому, используя единственный оператор С, в котором каждый
элемент матрицы С (скажем, C2i) получен умножением (второй)
строки матрицы А поэлементно на (первый) столбец матрицы В.
АВ = С (18.10)
•"И **12 •" ^\П
Л2, Л22 . . Л2П
Г Г Г
с
г г
^21 ^2 2 '"
*п . (18.11)
'пп
А А А / \В В I \С С
Заметим, что в общем случае АВФВА (см. ниже).
Для данного определенного набора базисных функций фЬ Ф2,..,*
фп можно непосредственно выразить произвольную функцию ф
через ее компоненты а\, йч, ..., ап путем проектирования д|> на
каждый вектор (функцию) базисного набора. Например, вторую
компоненту д|) можно найти из проекции
<?1, ф> = (0100 ... 0)
а,
а»
—а2.
(18.12)
Аналогично данный элемент произвольной матрицы
преобразования можно найти, действуя матрицей на базисный вектор
единичной длины и затем вычисляя скалярное произведение
результирующего вектора на другой единичный базисный вектор, как
показано ниже:
(18.13)
<?i. Ap»> = (ooi) /л. А* 4.
I \l Аг -"г»
\Atl Ап A
Наконец, когда выбран базис, найдены компоненты ф и
элементы всех нужных операторов, квантовомеханической задачей,
0\ (001)/Д,
1 1= |Д8|=ЛМ.
576 ЧАСТЬ 5
представляющей для нас интерес, является нахождение тех
отдельных, особых «собственных» функций, для которых действие на
них данной матрицы приводит к получению снова той же самой
функции, отличающейся от исходной только на постоянный
действительный числовой множитель.
Если Я'ф=Л'ф, а А,— действительное число, тогда г|) является
собственной функцией, а X — собственным значением, связанным с
этой собственной функцией- (18.14)
#,, Н12 ... Hlft\ /ai \ /ai
ИИ Н \ I а2 \ / а2 .
21 "22 "• л,я ' I > = А . • (18.15)
"«1 **П2 •'• "пп аП Л«
Тщательный анализ уравнения (18.15) показывает, что задача
на собственное значение теперь просто сводится к решению
набора из п уравнений с п неизвестными (отсылаем читателя к почти
любому элементарному учебнику линейной или матричной
алгебры). Тогда п собственных значений Я-ь ta, — > ^«» которые
удовлетворяют уравнению (18.15), получают как корни полинома из
определителя матрицы
"|| " ^ "12 *•• "1П
det | Я21 Н22-Х...Н2П | = Q (18.16)
Ч/7Л1 Пп2 ... Лпп А,
в то время как собственные функции -фь ярг, ..., tyn, соответствующие
собственным значениям щ, пъ ..., пп, находят (см., например,
задачи к этой главе) как
Ф| = апЪ + ЯпЪ + •■ • + амУп>
(18.17)
где
л// = <ср/,ф£>- (18Л8)
Огромное преимущество матричного подхода к квантовой
механике состоит в том, что он дает возможность получать имеющие
важнейшее физическое значение величины, а именно собственные
значения из данного базисного набора и уравнений (18.13) и
(18.16) даже не зная собственных функций ifr, тр2> •••. tyn [уравне-
РЁ>77 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
it
ние (18.17)]. В противоположность этому подход с применением
дифференциальных уравнений в квантовомеханических задачах
требует, чтобы сначала были получены собственные функции, а
уже потом они были использованы для получения собственных
значений. Поскольку собственные функции могут быть алгебраически
сложны (даже в такой простой задаче, как груз на .пружине) или
даже не могут быть выражены «классическим языком» (как,
например, собственные функции «спиновых» операторов), матричный
подход в общем случае короче, проще и часто является
единственно возможным.
Мы окончим этот вводный раздел, показав соответствие между
физическими величинами (и физическими измерениями) и
обобщенной геометрией функций, которую мы ввели и
классифицировали.
Физическая реальность и операции с матрицами.
Постулаты квантовой механики
Здесь мы не будем подробно останавливаться ни на
обосновании постулатов (в любом случае подразумевается, что постулаты
принимают на веру, а не понимают, причем «веру» можно
впоследствии проверить сравнением с экспериментом), ни на философских
аспектах теории, базирующейся частично на идее о том, что
поведение вселенной управляется вероятностью (и, следовательно,
неопределенностью в любой данный момент), а вовсе не
определенностью, основанной на предшествующем поведении системы. Мы
просто установим соответствие между реальными физическими
измерениями и связанными с ними квантовомеханическими
матричными операциями, которые можно использовать для предсказания
результатов этих измерений. Затем мы перейдем к решению двух
небольших подсобных задач, для которых решение существует:
квантовомеханического груза на пружине и энергетических
спектров для некоторых простых «спиновых» задач.
Физическая интерпретация М .1: возможные результаты
измерения физической наблюдаемой величины. Существует
определенный оператор, соответствующий каждой физической наблюдаемой
величине (см. табл. 18.3). Результат любого единственного
измерения этой наблюдаемой величины должен быть одним из
собственных значений соответствующего оператора. Поскольку эти
собственные значения действительны (в математическом смысле) для
реальных физических измерений, связанные с ними операторы
являются «эрмитовыми». Это означает, что матрица,
соответствующая такому оператору, обладает следующим свойством: если
переставить в ней строки и столбцы, то элементы полученной матрицы
будут отличаться от первоначальных в исходной матрице только
комплексным сопряжением [это свойство, очевидно, уменьшает
число подлежащих вычислению элементов матрицы вдвое, посколь-
578 ЧАСТЬ 5
ку можно получить остальную половину, воспользовавшись
уравнением (18.19)]:
'#п #12 • • • Я1л\ Н имеет действительные собственные значения
^2i "22 • • • "an
Jinx Нпг . . .Нпп/(т. е. Я является„эрмитовыма)
'Я,, Я12 ... Ят\
Я*18 Ни ... Hin \ Я//==/*/< (18,i9)
t/* Н* Я I если Н — эрмитов оператор
Ktl in ** ЪП • ' • 1ЛПП/
Физическая интерпретация № 2: вероятность получения
определенного результата при измерении физической наблюдаемой
величины. Частицу (ее природу и состояние) описывают функцией W.
Для измерения данной наблюдаемой величины W можно выразить
через ее компоненты сь Сг, ..., сп вдоль базисного набора
собственных функций оператора, соответствующего этой наблюдаемой
величине*. Преимущество такого выражения
*-*«*.+*.*. + .. .+с^„ (18.20)
где % — собственная функция желаемого оператора, заключается
в том, что мы можем теперь предсказать вероятность получения
i-ro собственного значения данного оператора при единичном
измерении для этой частицы. Она дается уравнением
|<ЧГ, ■ф/>Г=1с1|*. (18.21)
в котором предполагается, что собственные функции были
«нормированы» по «длине»:
<Ф/,ф|>=Ь (18-22)
Уравнение (18.21) дает повод для возникновения, термина
«волновая функция», обозначающего квантовомеханическую функцию
«состояния», так как \а\2 является аналогом интенсивности волны
(гл. 13), а С{ можно рассматривать как «амплитуду вероятности»,
квадрат которой дает вероятность найти частицу в состоянии i|x
при любом отдельном измерении.
* Одно из свойств собственных функций данного оператора состоит в том,
что каждая собственная функция «ортогональна» ((«перпендикулярна») другим
собственным функциям с другими собственными значениями (т. е, (i|)/, *ф/ > =
=0). По этой причине собственные функции образуют удобный набор «взаимно
перпендикулярных осей», вдоль которых и раскладывают произвольную
функцию («функцию состояния»).
}Q КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
Физическая интерпретация М 3: средний результат из многих,
измерений физической наблюдаемой величины. Теперь, зная
вероятность получения любого отдельного собственного значения а*
Вператора А, мы в состоянии вычислить среднее значение многих
(измерений наблюдаемой величины А:
среднее значение многих __ ^ ш АЧ>*~> —
измерений А ^ ' ^ ~~
(18.23)
= <с,ф1+с2ф2 + --- + сЖ. Л(с,ф, + с2ф2 + ... + с„фл)> =
.=< с,ф, + саф2 + ... + сяфп, А?,ф, + Ас£2 + ... + Асп<\>п > =
=< с$г + с»ф2 + • • • + C«V aicAi + «Лф1 + ■ • • + апсп$п >=
= < <Vk» Л,С,ф, > + < С2ф2э Я2С2ф2 > + ... + <С„ф„, Я„С„ф„ >,
но
1
<«К.Ф<>=1 H<t„ t/> = 0*.
так что
<ЧР, ЛФ> = |с1|2«1 + [с2|2д2 + .. . + |с„|2ди
(18.22>
(18.24>
Уравнение (18.23) есть лишь сокращенный способ записи
уравнения (18.24). Уравнение (18.24) следует сравнить с примером
;бросания игральных костей, приведшем к уравнению (6.38),
которое дает неквантовомеханический пример аналогичного типа
расчета среднего значения результата наблюдения, способного
принимать только определенные дискретные (в противоположность
непрерывному спектру) значения. Другими словами, если |с<|2
является вероятностью получения результата а\, тогда уравнение
(18.24)—способ для вычисления среднего значения многих
измерений А. Таким образом, интерпретация № 3 является прямыми
следствием уравнения (18.21) интерпретации № 2.
Интерпретации № 1, 2 и 3 предписывают, как выяснить
возможные результаты измерения данной наблюдаемой величины
(интерпретация № 1), вероятности получения любого
индивидуального из этих возможных результатов (интерпретация № 2) и
среднего результата многих таких измерений (интерпретация № 3).
Все эти предписания справедливы для частицы, которая «остается*
неподвижной», так что результат последующего измерения будет
тем же самым, что и предыдущего (точнее, вероятность получения
данного результата постоянна во времени). Поскольку реальные-
частицы непрерывно меняют свое положение и другие свойства,
следующей задачей является предсказание изменения результатов*
* См. предыдущую сноску.
580 ЧАСТЬ Б
измерений во времени. Самое простое — сформулировать эту
задачу, основываясь на временной зависимости функции «состояния»
ЧЛ В результате этого мы сможем использовать интерпретации
№ 1, 2 и 3 для нахождения результатов измерений в более поздние
моменты времени.
^Физическая интерпретация М 4: зависимость результатов
измерений физической наблюдаемой величины от времени. «Уравнение
движения» (уравнение, которое предсказывает зависимость от
времени) для функции состояния У записывают для единственной
частицы как
ШдФ (t)/dt = 3VQ>{t) | = — ^ VzO (t) -+- V (x, y, z,t) Ф (t) (уравнение
' Шрёдингера) (18.25)
Наиболее часто мы будем иметь дело с «консервативными»
системами, в которых потенциальная энергия V не зависит (явно) от
времени. В этом случае возможно представить решение в виде
произведения двух функций
Ф^¥<р, (18.26)
где ф зависит только от времени, a W от времени не зависима.
Подставив уравнение (18.26) в уравнение (18.25) и разделив
на ^¥<p, получим
^(-£W+Vv)=m-L%. (18.27)
Поскольку левая сторона уравнения (18.27) является только
функцией координат, а правая сторона — только функцией времени, обе
стороны должны быть равны одной и той же постоянной величине,
которая не является функцией ни координат, ни времени.
Обозначив эту константу через Е, уравнение (18.27) можно переписать в
виде двух отдельных уравнений:
\Ж"¥^Е^ (18.28)
£—т*- <18-29>
Первое уравнение (18.28) называют независимым от времени
уравнением Шрёдингера. Второе уравнение (18.29) легко
интегрируется, давая
<р(0 = ехр [-(///*)#] (18.30)
или
Ф«)=е-<11Н)В<ф№
(18.31)
Л КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
Таким образом, уравнение (18.31) дает нам желаемый способ для
Получения функции состояния Ф(0. в момент времени t, если дана
функция состояния в нулевой момент времени Ф(0).
Физическая интерпретация № 5: последняя и наименее ясная
Интуитивно интерпретация, касающаяся точности одновременных
Измерений двух наблюдаемых величин. Мы уже установили, что
измерение любой единственной наблюдаемой величины будет давать,
^ообще говоря, ряд (собственных) значений со средним
результатом
[ среднее значение Л = <% Л¥> (18.23)
й среднеквадратичной «шириной» ДЛ, определяемой формулой
[Ср. с уравнением (6.44)]
l)2 = <(m-<m»a> = <(m2-2m<m> + <m2»> =
= <m2>-2<m><m> + <m>2=:
= 02>-<m>2
(6.44)
(среднеквадратичное отклоне-
v ' ' ние в измеренных значениях Л)
| (18.32)
как показано схематически на рис. 18.1.
Для результатов измерения любой конкретной наблюдаемой
ветчины будет существовать распределение, аналогичное показанно-
гу на рис. 18.1. В частности, с наблюдаемой величиной В будут
вязаны ее средняя величина <С¥, BW> и неточность ДВ. Теперь
[опустим (а часто так и бывает), что мы в состоянии выполнять
езависимые одновременные измерения как В, так и Л и что нет
акой-то особой взаимосвязи между точностями этих двух измере-
ий. Однако неожиданным,4но неизбежным принципом квантовой
Механики является то, что сам акт измерения меняет состояние ча-
гицы так, что иногда получают различные результаты измерения
до измерения Л и после измерения Л. Математически это
обнаруживается при последовательном проведении операции В, а по-
м Л по сравнению с действием сначала А, а затем В (А к В —
ераторы, соответствующие отдельным физическим наблюдаемым
личинам — координате и импульсу).
Измерим сначала импульс (линейный), а затем координату:
xpj(х) = х[pxf (х)] = х |- ih2M] =1 hxV (x). (18.33)
|еперь измерим сначала координату, а затем импульс:
7xxf (х) = рх \xf (х)] = - in Щр- = - Ш (х) - ihxf (х). (18.34)
682 ЧАСТЬ 5
а, аг а3 а4 Qs^a6 a7 а8 а9 аю а,, аг
{ip,Atfr>*s средний результат многих измерении'А
Рис. 18.1. Схематическое изображение относительно числа измерений
оператора А, дающего определенный результат т, как функции аи
Среднее значение н ширина этого распределения значений <ь указаны на рисунке.
Предсказываемый результат измерения можно, таким образом, выразить в виде
предсказываемое значение A"<1V, ХЧ?^=±\А.
Запишем разность между результатами этих двух операций:
xpj (х) -\pxxf (х)щ== (хрх - рхх) f (х) = ihf (х) (18.35)'
или более кратко
(18.36)
хрх- Рх* =
[х, px] — (h
Скобки в уравнении (18.36) называют «коммутатором» и
говорят, что «коммутатор х и рх есть ik*. Оставляем читателю в
качестве упражнения (см. задачи к этой главе) возможность проверить
коммутаторы операторов «орбитальных» угловых моментов в
табл. 18.3.
Из уравнения (18.36) можно показать, что, когда два
оператора не коммутируют, существует взаимозависимость между
точностями, с которыми две соответствующие переменные могут быть
измерены:
bxkpx^>h. (18.37)
Уравнение (18.37) называют «принципом неопределенности
Гейзенберга для координаты и импульса». Существуют
аналогичные принципы неопределенности для других пар некоммутирую-
щих (т. е. [АВ]Ф0) операторов, например принцип неопределенно-
КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
т «время — энергия», обсуждение которого приведено в гл. 20.Б,
юкольку его можно понять в рамках классической механики.
Интерпретации и терминология, данные ранее в этой главе,
беспечивают нас основными орудиями, с которыми можно успеш:
;0 анализировать два важных типа задач, требующиеся для ре-
гения квантовой механики (гл. 18.А).
•.
1озникновение и положение спектральных линий.
(вантовомеханический груз на пружине
I Обычным исходным пунктом любого квантовомеханического
расчета является вычисление общей энергии системы, исходя из
Свойств молекул или частиц (например, линейной и вращательной
;коростей, размера и жесткости «сосуда», в котором они
находятся). Поскольку общая энергия обычно принимает только вполне
>пределенные дискретные (собственные) значения, мы можем
определить разности между различными значениями энергии («энер-
•етическими уровнями»), измеряя поглощение при облучении
системы электромагнитным излучением с частотой v,
соответствующей разнице между энергетическими уровнями Et и Еу.
hv^E.-E}.. (18.38)
|Тогда, подгоняя наблюдаемую картину энергетических
промежутков к картине энергетических уровней, предсказанной нами на
основе расчета собственных значений энергии, можно вычислить
интересующий нас молекулярный параметр (скажем, прочность
отдельной химической связи). Для того чтобы продемонстрировать
&ид общего подхода, мы рассчитаем собственные значения энергии
квантовомеханического груза на пружине, используя язык,
введенный ранее в этой главе.
[ Классическая кинетическая энергия частицы массой пг,
движущейся со скоростью v вдоль оси х, дается уравнением
кинетическая энергия (классическая) = (то8/2)=-?^-==у^-.
: (18.39)
На основе соответствий, Определенных в табл. 18.3, квантовомеха-
нический оператор для кинетической энергии должен иметь
следующий вид:
кинетическая энергия (квантовая)=(p*J2m) = — (/t2/2m) (d2/^)-
(18.40)
Потенциальная энергия V(x) для частицы, удерживаемой
пружиной с силовой постоянной k, может быть выведена из уравнения
584 ЧАСТЬ 5
.силы для пружины:
сила = — kx = —
_ dV (s)
dx
или
V (х) = ('/,) to' + const = (V.) kx\
(18.41)
(18.42)
в котором константа интегрирования принята равной нулю при
х=0^ Оператор энергии (т. е. гамильтониан) можно теперь
записать как сумму кинетической и потенциальной энергий:
Я? = р*х/2т + kx2/2
(18.43)
На этом этапе вычисления можно значительно упростить,
выразив общую энергию в единицах h(k/m)1/2. Линейный импульс Р
будет выражен теперь в единицах hxl% (mk)1!*, а линейная
координата X — в единицах h1/2 (mk)~y*t как показано уравнениями
(18.44), (18.45) и (18.46):
(18.44)
Ж = (1/2) (Р2+Х2)
где
и
P = trlh(mk)-iupx^~ih1,'(mk)-iud!dx=-1 Х- (18.45)
X = trl,>(mk)lux.
(18.46)
Читатель может сам удостовериться в том, что Р к X
удовлетворяют соотношению
PX — XP= — t
(18.47)
Уравнение для определения собственных значений
энергетических величин [уравнения (18.28) и (18.44)] примет вид
(Р" + АГ1)Т = 2£ЧГ.
(18.48)
Теперь мы прибегнем к хитрости, основанной на уравнении
(18.47):
(P±iX)(P + iX)± l = P2±i(XP-PX) + X2± 1 =
= Pa±i(i) + X*± 1=P* + X*
Из уравнения (18.49) мы можем вывести, что
(Р ± iX) (Я2 + А*2) = (Р+ iX) VP ± IX) (Р + iX) ± I] =
— {Рц. iX) (P± iX) (Р + iX) ± (P :р IX) —
= \{Р + iX) (P\± iX) ±\]{P + iX) =
= [P2 + i (XP - РХ) + Х2{±: 1] (Р + IX)
(18.49)
КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
(Р + IX) (Р2 +'Х*) = (Р2 + Х2±2)(Рц: iX). (18.50V
гперь исследуем уравнение (18.50) для случая (P-\-iX):
(P + iX)(P2 + X2)W = (P2 + X2-2){P + iX)V. (18.51)
[о с учетом уравнения (18.48) можно переписать левую сторону
&ак
2Е{Р + {Х)Ч = {Р2 + Х2){Р-\-1Х)Ч-2{Р + 1Х)Ч
или как
; (Я' + АГ1)[(Р + хА,)*1 = 2(£+1)[(Р+.^Т1. (18.52)
Иными словами, если W есть собственная функция (Р2-\-Х2) с
собственным значением (энергией) Е, тогда и (P-\-iX)W является
собственной функцией (Р2-\-Х2) с собственным значением
(энергией) £+1. (P+iX) иногда называют «повышающим оператором»,
поскольку он приводит к повышению энергии системы на одну
единицу. Мы можем построить энергетическую «лестницу»
возможных квантовых энергий, применяя оператор (P-\-iX)n, который
будет увеличивать энергию на п единиц, и распространять эту
лестницу вверх без ограничений (рис. 18.2).
И*) = "U*2
^^П-ЦрУ"''1
*-£)> Н<~л
„3 \ 1/4
#,^)'VA
№
*о=0 «-««^««-^
о—
Рис. 18.2. Собственные значения энергии Еп и соответствующие собственные
функции энергии ifn для квантовомеханической массы (т) на пружине с
силовой постоянной k:
Для удобства собственные значения энергии выражены в единицах *(ft/m) '2. Оии
образуют «лестницу», начинающуюся от значения £о™'/а н имеющую равноудаленные,
отличающиеся на одну единицу «ступеньки», которые поднимаются вверх до бесконечности.
586 ЧАСТЬ Б
Аналогично можно показать, что «понижающий оператор»
(Р—IX) приводит к уменьшению энергии на одну единицу. Таким
образом, мы можем начать с любой «ступеньки» энергетической
«лестницы» и передвигаться по ней или вверх, или вниз на одну
энергетическую единицу («ступеньку») за раз:
(Я2-f Хг) \(Р ± IX)Ч]=2(Е±\)[(Р ± IX)Щ. (18.53)
Сначала может показаться, что и вниз лестницу также можно
продолжать неограниченно. Однако мы уже постулировали, что
собственные значения квантовомеханических операторов, которые
соответствуют физическим наблюдаемым величинам (таким, как х
я рх), являются математически действительными {в
противоположность комплексным или мнимым), а квадрат любого
действительного числа должен быть больше нуля. Поэтому разрешенные
собственные значения для {Р2-\-Х2) должны быть больше нуля и
лестницу надо остановить или на нулевой энергии, или выше (рис.
18.2).
После установления интервалов между «ступеньками» нам
необходимо только определить абсолютное значение энергии любого
одного «уровня», чтобы узнать энергии всех возможных
собственных значений гамильтониана (Р2-\-Х2). Поскольку действие
«понижающего» оператора на собственную функцию Vo с
наименьшим возможным собственным значением должно быть равно нулю
(так.как мы не можем продолжать лестницу в область
отрицательных энергий), то
(Я-ОДУв = (1//)^+х]^0-0. (18.54)
Легко проверить, что решением этого уравнения является
выражение
j^F0 = c6nst-exp[— Хг/2]\ („основное" состояние „волновой* функ-
ции). (18.55)
Кроме того,
('/,) (Р* + Xй) W0 = E.V. = (1 /2) Т. [в единицах h (k/m)4*]
или
£0=(l/2)ft(jfe//7Z),/» в общепринятых единицах.
Хотя необязательно знать собственные функции энергий для
поиска соответствующих им собственных значений энергии, мы
можем теперь получить их для гармонического осциллятора,
используя «повышающий» оператор столько раз, сколька нужно:
ЧЬ^(P + lXf% = const-(^L-xJlF,. (18.56)
КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
Несколько первых собственных функций подробно показано на
tec. 18.2.
F Для наших целей важны следующие особенности квантовомеха-
|шческого описания груза на пружине: 1) существует бесконечное
Число «собственных» частот \ц=*(Е$—E{)/h, около, которых мы
ржидаем найти сильное поглощение при действии
электромагнитного поля (например, инфракрасного облучения) в отличие от
единственной «собственной» частоты для груза на пружине в
классической механике*; 2) используя ненулевое коммутационное
соотношение между х и рх [уравнение (18.47)], мы способны
построить «повышающие» и «понижающие» операторы, которые
изменяют энергию системы на одну единицу вверх или вниз, давая в
результате «лестницу» равноотстоящих энергетических «ступенек»,
и 3) мы имеем возможность выводить энергетические интервалы
без точного знания какой-либо из собственных функций. Хотя
многие колебательные «собственные» частоты, предсказываемые в
квантовом описании, действительно можно наблюдать для малых
молекул в газовой фазе, обычно невозможно разрешить многие
частоты для каждой из индивидуальных «пружин» в большой
молекуле в жидкой фазе. По этой причине в отношении
качественного описания спектроскопии в ультрафиолетовой, видимой и
инфракрасных областях квантовая механика дает немногим больше,
чем наше классическое описание (гл. 13). Однако истинная
важность предшествующего упражнения заключается в том, что оно
совершенно аналогично квантовомеханическому описанию
«спинового» углового момента, для которого нет классического аналога
и для которого (в частности) мы не можем написать точной
собственной функции. Из-за особенности (3), указанной выше, нам
снова удастся вывести величины энергетических «уровней» даже
без- точного знания собственных функций, и это упражнение
покажется несколько менее непривычным после рассмотрения примера
груза на пружине, для которого имеется классический аналог*
В противоположность квантовомеханическому гармоническому
осциллятору квантовомеханическое описание «спина» приносит
непосредственную пользу в объяснении наблюдаемого магнитного
резонансного поглощения на ядрах, для которого возможно разре-
* В нашем дальнейшем обсуждении «правил отбора» (гл. 19.Б.4) мы
обнаружим, что разрешены только определенные из бесконечного числа возможных
переходов, а именно те из них, для которых (/—f) = + I («поглощение») или
(у—£)=—1 («испускание»). Таким образом, для простого квантомеханического>
груза на пружине в отсутствие сил торможения мы нашли бы поглощение
энергии только на одной частоте v=h(k!m) '2. Для реальных молекул
потенциальная энергия включает ненулевые члены, пропорциональные х3, х4 и т. д.,
наличие которых меняет интервалы между соседними энергетическими уровнями так,
что (£/—£/) перестает быть независимой от i и /. Таким образом, у реальных
молекул даже при наличии «правила отбора» (;—0==Н существует
бесконечное число различных частот поглощения для электромагнитного излучения,
действующего на имеющий заряд квантовомеханический груз на пружине.
B8 ЧАСТЬ 5
шение множества новых «собственных» частот, предсказываемых
на основе квантового расчета.
18.А. СПИНОВЫЕ ЗАДАЧИ.
ПРОСТЕЙШИЕ КВАНТОВОМЕХАНИЧЕСКИЕ РАСЧЕТЫ
Энергия, связанная с взаимодействием магнитного момента ц
*со статическим магнитным полем Н0, дается выражением —|хЯо
или только ц2Н0, если //& взято вдоль отрицательного направления
оси г (так что мы можем забыть о знаке минус в формуле).
Поскольку классические магнитные моменты возникают при
вращении зарядов, кажется разумным, что магнитный момент должен
быть пропорционален (константу пропорциональности называют
«гиромагнитным отношением» у) классическому угловому моменту
вращающейся заряженной частицы L:
H = 1ll> L = Lxi + Ly} + Lzk. (18.57)
Позже мы предположим, что неклассический угловой момент (см.
ниже) отражает то же самое свойство частиц, которое проявляет
неклассический «спиновый» угловой момент I
l^Y/1; I = /J + /„J + 4k (18.58)
Энергия, связанная с взаимодействием магнитного момента со
статическим магнитным полем, может, таким образом, быть
выражена в форме
га мильтониан = Ж0 = yLff0Lz (18.59)
для частицы, обладающей компонентой углового момента Lz вдоль
оси г.
Из уравнения (18.59) ясно, что для того, чтобы предсказать
набор энергетических уровней, ожидаемых в экспериментах по
магнитному резонансу, необходимо найти собственные значения для
оператора Lz. Учитывая наше более раннее успешное решение
задачи о грузе на пружине, при создании подхода к решению этой
задачи мы начнем с проверки коммутаторов для различных пар
операторов угловых моментов, свойства которых находят по
аналогии с классической механикой (см. табл. 18.3 и задачи к этой
главе):
(18.60а)
(18.606)
(18.60в)
[Lx,
[Ly
[L,.
Ly] = Hz .
Lz] = (Lx
Lx] = iLy
угловой момент в единицах %
|89 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
Если теперь определить новые операторы L+ и L- как
L.=Lx-tLy
(18.61а)
(18.62а)
тогда уравнения (18.606), (18.60в) и (18.61а) можно объединить,
получив
LZL+ — L+Lz = LZLX 4- iLzLy — LXLZ — iLyLz=
— (LzLx — LxLz) — i (LyLz — LzLy) =
= iLz> Lx] - i [Ly, Lz\ ■=
- ILB - i (i) LX = LX + iLy, (18.63)
LizLi^ L,^L,Z = .L+.
Преобразуя уравнение (18.63), получим
LZL+~L+ = (LZ-\)L+=L+LZ. (18.63a)
Применяя обе части уравнения (18.63а) к 0рп, которая является
собственной функцией Lz со связанным с ним собственным
значением (tin, мы делаем заключение, что
(Ia-1) L+% = L+ (Lz%) = цге (L+%)
или
Lx(L+^ = {j^+l){LM (18.6 4)
Другими словами, если Црп есть собственная функция Lz с
собственным значением (хп, тогда (L-H|)n) также является собственной
функцией Lz с собственным значением (|хп+1) и действие
«повышающего» оператора L+ состояло в увеличении собственного
значения Lz на одну единицу.
Как можно предвидеть из накопленного нами опыта, L_
окажется «понижающим» оператором, который уменьшает собственное
значение Lz на одну единицу. Таким образом, мы снова ухитрились
найти «лестницу» собственных значений Lz, которая пока кажется
простирающейся неограниченно вверх и вниз (рис. 18.3). Можно
показать (см. задачи к этой главе), что существуют верхний
предел (Хмакс и нижний предел Цмин для длины «лестницы» на рис. 18.3
и что
И-макс = — И-мин* (18.65)
Теперь, если «ступеньки лестницы» отстоят друг от друга на одну
единицу и середина «лестницы» соответствует нулю [уравнение
(18.65)] независимо от того, имеется там «ступенька» или нет,
ясно, что собственные значения 2-компоненты углового момента
могут принимать только целые или пдлуцелые значения (рис. 18.4).
590 часть 5
*
LI ф fi + 2
L+ф ■— Д + 1
ф fJL
L- ф д — 1
Ы ф д - 2
Рис. 18.3. Собственные функции (слева) и соответствующие собственные
значения (справа) для z-компоненты углового момента L2 после применения
«повышающего» или «понижающего» операторов L+ и L-.
Поскольку энергия взаимодействия магнитного момента с фиксированным магнитным полем
пропорциональна L , диаграмма энергетических уровней для этой системы будет также
представлять собой «лестницу» равноудаленных (собственных) значений энергии.
Отсюда можно показать (см. задачи к этой главе), что величина
ц для 2-компоненты «орбитального» углового момента Lz должна
принимать только целые значения.
Внимательное исследование уравнений (18.60)—(18.69)
показывает, что единственными свойствами углового момента,
необходимыми для построения лестницы собственных значений, были
коммутационные соотношения [уравнение (18.60)]. Реальные
частицы часто ведут себя так, как если бы они обладали
дополнительным угловым моментом (сверх того, который выведен по
аналогии с классической механикой), называемым «спиновым»
угловым моментом I. Связанный с ним квантовомеханический оператор
удовлетворяет, следовательно, тем же самым коммутационным
соотношениям
I = Ixj-}-I j-(-/rk (вектор, компонентами которого являются
операторы) (18.66)
где
[/„ /,]=*/, , (18.67а)
Vf 1ж] = Мх угловой момент в единицах h (18.676)
[/,. h\=U9 (18.67b)
Из этих коммутаторов [уравнения (18.67)] мы сразу можем
заключить, что собственные значения z-компоненты «спинового»
углового момента могут быть только целыми или полуцелыми
величинами, умноженными на h. Оказывается, что существуют частицы
обоих типов, как это отмечено в неполном перечне на рис. 18.4,
а также некоторые частицы (такие, как ядра углерода-12),
которые вообще не обладают «спином». Спин ядра является
существенным ядерным свойством, таким же, какими являются масса, заряд,
четность и т.- д. Он один и тот же для всех идентичных ядер [все
ядра, например, углерода-13 имеют максимальное собственное
/. = 3
+ 3
+ 2
• + 1
О
■-1
-- 2
L=l
Z.-0
+ 1
О
- 1
1 2
42
2
2
3
2
/ = 1
+ 1
О
1
'"5
1
2
/=0
о
■-3
f.Qp4uma/ib
р- ор(*италь
$-орбита ль
-3Na
«Си
8,Вг
-н
,4N
•Н.,3С
1»р 31 р
п,Сё.,!*Нв.е
•»С,,60
з»3,40Са
Рис. 18.4. Схематическое изображение собственных значений оператора г-компо-
ненты углового момента, соответствующих установленному максимуму г-компо-
ненты «орбитального» (L) и «спинового» (/) угловых моментов, показанных
вверху каждой «лестницы».
Согласно уравнению (18.59), магнитная энергия данной частицы пропорциональна
(собственному) значению z-компоненты углового момента для этой частицы и силе
приложенного внешнего статического магнитного поля. Таким образом, в отсутствие внешнего
магнитного поля магнитная энергия для каждой из возможных собственных функций
z-компоиенты углового момента для данной частицы будет одна н та же. При наложении
внешнего статического магнитного поля диаграмма (магнитных) энергетических уровней
для любой из указанных частиц будет такой, как показано на рисунке, где масштаб
энергии для данной частицы определяется величиной V в уравнении (18.59). На рисунке
показаны хорошо известные «орбиталн» водородного атома (см. L=»3, 1, 0 на рисунке),
которые соответствуют собственным значениям z-компоненты орбитального углового
момента, и в дополнение к ним — «спнновые> собственные значения z-компоненты углового
момента, соответствующие некоторым интересным ядерный изотопам. Актуальные задачи
для ядер с «половинным спином» разобраны в тексте этого раздела.
592 ЧАСТЬ 5
значение /z=/t( V2)]. но z-компонента «спина» может принимать
любое из значений, показанных на рис. 18.4. Простейшие и
наиболее широко распространенные применения для «спиновых»
операторов связаны с исследованиями магнитного резонанса ядер со
спином 7г, главным образом 'Н и 13С. В оставшейся части этого
раздела и будут рассмотрены частицы со спином 7г.
В заключение сделаем важное номенклатурное замечание. L и
I являются векторами (компоненты которых — операторы), но на
практике обычно говорят, что частица имеет орбитальный угловой
момент L или «спин» / (L и / являются целыми или полуцелыми).
Под этим подразумевают, что L или / есть максимальные
собственные значения Lz или 1г (в единицах Н).
Одно ядро со спином 7г в статическом магнитном поле
Как видно на рис. 18.4, единственными возможными
собственными значениями 2-компоненты «спинового» углового момента для
частицы со спином 7г являются +72 и —7г, относящиеся к
соответствующим собственным функциям /z, которые мы обозначим как
а и р:
/2Р = - V.P
(18.68а)
(18.686)
<*.
<*,
а> =
i
= <h
■-<h
Р> =
а> =
1
■О
Для простоты предположим, что аир нормированы до
единицы по «длине». Кроме того, можно показать, что собственные
функции, соответствующие двум различным собственным
значениям эрмитова оператора (такого, как /z), являются
ортогональными («перпендикулярными») (см. литературу к этой главе).
Другими словами,
(18.69а)
(18.696)
Мы можем теперь вычислить все матричные элементы оператора
a%>o=yH0Iz [уравнение (18.59)], необходимые для определения
собственных значений энергии из уравнения (18.16) для единственной
частицы со спином 7г в статическом магнитном поле Я0,
используя базисный набор
<р,=а, (18.70а)
<р8 = р, (18.706)
ур. (18.69а)
а> = (уЯ0)/2 (18.71 я)
= Y#oO> 7га>=
Т".
ИЗ КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
i
1
&
X
*ъ
а
1Но
2 t
-
6
— ос
-fi
6
♦*
-*
А
t .
b>0=<ftiH0
ш —^-
Рис. 18.5. Схематическое изображение энергетических уровней я
предсказываемый спектр поглощения для системы изолированных частиц со спином .(!/а)
в статическом магнитном поле Я0.
Форма линии поглощения обсуждена в гл. 13; интенсивность поглощения обсуждена в
гл. 19.Б.4; здесь- же нас интересует только положение спектральной линии.
а — собственные значения энергии (величина Iг приведена в единицах ft); б — собственные
функции энергии; в — собственные значения z-компоненты углового момента 1г.
или в матричной записи
= Т*.0 ^(К^О^ТЯ. (1 0)(£) = *£, (18.716)
ур.(18.696)
= ТЯ.<«» -7.Р> = -нг<АР> = 0, (18.71b)
или в матричной записи
= ТН.О °)(ol,y[i) = YW.(l 0)(_01/^ = ТЯ.(0) = (0). (18.71Г)
Аналогично
Жг1 = О,
^22 О
(18.71д)
(18.71е)
Энергетические собственные значения находим теперь, решая
определитель уравнения (18.16)
!#•
—А О
О -Л&-Я
=-0, (18.72)
594 ЧАСТЬ 5
корнями которого являются просто
Я, = ^ (18.73а)
1,= -^. (18.736)
Собственные значения и собственные функции [уравнение (18.17)]
энергии для этого случая показаны на рис. 18,5. Видно, что есть
два энергетических уровня и интервалы между этими уровнями
пропорциональны силе приложенного магнитного поля Но.
Два химически различных изолированных ядра со спинами 7г
В случае исследования образца, содержащего два типа ядер»
со спинами у каждого по 72» но с различными гиромагнитными
отношениями у\чьЧ2 интуитивно кажется очевидным, что, пока эти
два типа ядер не взаимодействуют друг с другом, спектр
поглощения будет состоять из двух отдельных пиков, локализованных при
<й\=у\Шъ и (u2=Y2^o, как в примере на рис. 18.5. (Другими
словами, мы предполагаем найти резонансные частоты в результате
наложения двух типов «камертонов», как показано на рис. 18.6.)
Для упрощения преобразований в этой и последующих задачах
полезно ввести гамильтониан {общей кинетической и потенциальной
энергий), представляющий собой сумму {индивидуальных)
гамильтонианов, которые мы могли бы записать для каждого типа ядер
в отдельности:
оь == Olx -j- ггГ8»
Ж = -{,Н0/г1-}-угН01гл {1г выражен в единицах h) (18.74)
где оператор IZl действует только на функции для частиц с Y=Yi»
a Iz% — только на частицы с y=v2- Поскольку для предсказания
7i^o t 7? Wo
ДЕ, = 7т Н0
<Х\а7
72 Н0 7i #0
Д£2 = У 2 Но
О —
\
/J,0f2
УуНр У^Нр ,.
мв2
$ 7'Я° 72Wo_L
72 Но
J
Д£, =7>^о
■<*,(Ь
Cl>-
wj = 7iftW0 w2 = У2^Н0
■hh
2 2
Рис. 18.6. Схематическое изображение энергетических уровней я
предсказываемый спектр поглощения для набора из двух типов изолированных (не
взаимодействующих) ядер со спином Va в статическом магнитном поле Я0.
Эти два типа ядер различаются величинами V и, таким образом, собственными значениям»
энергий, а следовательно, и промежутками между энергетическими уровнями, которые
определяют нх частоты поглощения.
КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
результатов любого данного измерения мы должны точно устано-
Ьить состояния обоих типов ядер, полезно определить базисный на-
>ор фг в виде «произведений» собственных функций /Zl для ядер
St vi и собственных функций /Za для ядер с Y2. как показано ниже:
?. = «,«.= • (18'75а)
cp2 = aIp2= I (18.756)
<Р.=РА= " • (18.75в)
<P< = PA=H " • (18-75Г)
На основе того, что мы уже знаем об операторах /z и собственных
функциях
Л «А = VaP.. ; VА = - VaP,»
'*M = ~ 7A«i. 'Z2P A = 7tPA>
W=- 7.PA. '„PA=- '/.PA.
(18.76)
мы можем сразу построить матрицы для операторов /Zl> /z2 и #? в
этом ф-базисе:
'V. о о о \ /у, о о ,о
/.-1° V, оо о -■/. о о
Zl ' 0 0 — V. О I *' 1.0 0 V. 0 '
оо о -v V о о о —v.-
696 ЧАСТЬ f
det
п О / Yi^o
О
(-
2
=0
Н/
5 — У'Я« |У»Я»
-л, 2 ~i" 2 »
л?~~ 2 2 »
Я8=~^+^, (18.78)
Л4 ■ О - О •
Энергетические уровни и спектр, соответствующие уравнениям
(18.78), показаны на рис. 18.6.
Два химически различных взаимосвязанных ядра со спинами '/г-
Спектр типа «АХ»
Если два химически различных* ядра, таких, как два протона,
входят в состав одной и той же молекулы, их поведение перестает
быть взаимно независимым. Результат взаимодействия между
двумя ядрами можно описать классическим способом по аналогии с
моделью для связанных пружин в гл. 17.А так, что резонансная
частота, относящаяся к данному протону, «расщепится» на две или
несколько резонансных частот из-за «взаимодействия» его с другим
протоном. Квантовая механика необходима для расчета силы взаи-
* Под «химически различными» здесь подразумеваются два ядра, у
которых y не идентичны: это случай ядер двух различных элементов «ли изотопов
(1аС и *Н или "С1 и 8'С1) или случай ядер одного и того же изотопа,
участвующих в образовании разных химических связей (*Н в группе СНа и 'Н в
группе СН8).
)7 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
юдействия (и, таким образом, величины расщепления). В класси-
1еской механике нет двух таких частиц, которые могли бы в одно
|и тоже время занимать одно и то же пространство. Однако кван-
втовомеханический электрон (см. любое элементарное описание
|частицы в потенциальной яме или водородного атома) имеет
конечную вероятность быть найденным около самого ядра и, таким
|образом, влиять неклассическим образом (известным как
«контактное» взаимодействие Ферми) на ядерный магнитный момент.
•Поскольку электрон может быть найден через некоторое время в
!точке, сильно удаленной от ядра, это «контактное» воздействие
.распространяется вдоль любых близлежащих химических связей и
передается вдоль любого внутримолекулярного пути с повышенной
электронной плотностью (через, химические связи, но не через
«пустое» пространство). В конечном итоге рассматриваемый электрон
(электроны) достигает второго магнитного ядра, которое (как в
нашем случае) может быть химически отличным от первого ядра.
Тогда из-за аналогичного «контактного» воздействия второе ядро
будет возмущено в результате взаимодействия, которое
первоначально возникло из-за наличия первого магнитного ядра. В первом
приближении (см. задачи к этой главе) это «контактное» явление
может быть описано очень простым путем через «скалярное»
взаимодействие между двумя ядерными спинами
#=tW«1+tA/,1+'V«, <7*в единш*ах «, (18-79)
где величина / (которая имеет размерность с-1) является мерой
силы ядерного «скалярного» или «спин-спинового» взаимодействия.
Теперь мы рассчитаем энергетические уровни и разности между
энергетическими уровнями («частоты переходов») для
гамильтониана в уравнении (18.79) и затем продемонстрируем чрезвычайно
большую ценность использования экспериментально определяемой
константы взаимодействия /.
Матричное представление произведения операторов hJZi
получают сразу же из правила умножения матриц [уравнение
(18.11)] и уже установленной формы матриц для индивидуальных
операторов hx и h2 [уравнение (18.77)]:
Г ~
'V, о о о\ /•/• о о '0\ /ги о оо
j j Л о v. о op -v. о ° L( ° ~V* о о
* * loo -v. о II о о 7, о I I о о -74 о
о о о -l/J\o о о -7а/ \о о о v^
(18.80)
Б98 часть б
Подставляя выражение (18.80) в левую часть уравнения (18.78),
мы решим его для собственных значений энергии гамильтониана из
уравнения (18.79):
/С
det
^+^+ о ° о \
+1—*
с
2 2
Ч->)
(-^+
Yi#o Тг^о
f
2 2
+*-0 1
=0
Л1 о Г о Т 4 »
3 Yi^o _Ya^o /-
Ла— 2 2 4 »
Л|~ 2 г 2 ~~ 4 »
2 — Yi^o Yg^o j '
Гамильтониан [уравнение (18.79)], который приводит к
спектру типа АХ на рис. 18.7, является хорошим приближением, когда
разница «собственных частот» двух ядер гораздо больше
величины воздействия между ними (см. задачи к этой главе):
«>. -в>»^ О8-81)
Поскольку резонансная частота ш пропорциональна силе
приложенного внешнего магнитного поля Но, разность частот в
уравнении (18.81) можно сделать больше, просто увеличив силу
магнитного поля, так что реальные эксперименты при использовании
КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
7i Wo J 72#о . J
2 4 /
Д£"з-+1
-7iWo ^ 7г"о J / „ Д£"г-+1
a,tt2
5 \ 7i Wo 72'/о У
5«
§• \2 2 4
Д£"4-+з '
J5
«102
-7iW0 72 «o
Д£4->
+ ^ ^~/M2
-H J K- -H/K-
<*>2 = 72^0
■Рис. 18.7. Схематическое изображение энергетических уровней и
предсказываемых спектров поглощения для магнитного резонанса двух химически
различных взаимодействующих ядер со спином 7а в статическом магнитном поле #о.
Константу спии-спннового взаимодействия / легко измеряют как расщепление каждого из
г резонаисных сигналов. Как и на рис. 18.6, переход АЕ ^-*1 «запрещен» (гл. 19.Б.4).
|рчень сильных полей (50 кГс и более) часто могут приближаться к
Ситуации, изображенной на рис. 18.7.
Имеется возможность расширить описание, ведущее к картине
[на рис. 18.7; на случаи, включающие взаимодействие более чем
двух ядер, без дополнительного привлечения каких-либо
математических операций, а используя лишь качественную аргументацию,
рти рассуждения основаны на том, что данное ядро со спином 7г
может иметь магнитный момент, Направление которого либо
совпадает (f), либо направлено в противоположную сторону (|) по
отношению к направлению внешнего магнитного поля
(соответствуя двум состояниям аире различными энергиями, как показано
на рис. 18.5). Если это первое ядро «скалярно"взаимодействует» со
вторым ядром, то второе ядро будет объектом действия внешнего
поля, которое чуть-чуть больше или чуть-чуть меньше (на величину
//2yh)t чем Я0, в соответствии с тем, был ли первый спин
направлен «вверх» или «вниз». Если теперь есть два спина первого типа,
то будет иметься четыре возможные взаимные ориентации
направлений их спинов, но две из этих ориентации продуцируют один и
тот же магнитный момент и, следовательно, оказывают одно и то
же воздействие на ядро второго типа (рис. 18.8). Получаемые в ре-
600 ЧАСТЬ б
®
1
{j.
н
с—с
/и)
и. ^
с—с
н'
н
с—с
р
Н'1
н
к
/
н
ч
Н—С ~ч
Н чО
Г1
1 1
■О) >■
iJU
1:2:1
to-
1:3 : 3 : 1
Н\ хО
О) ^-
7
^/-СНз
Общая площадь •= 1
.{j-
Общая площадь- 3
Рис. .18.8. Качественный метод для предсказания характера расщепления в
спектрах ЯМР из-за взаимодействия между двумя химически различными типами
протонов в одно.й и той же молекуле.
Каждый из спектров справа представляет собой спектр ЯМР, возникший благодаря
поглощению обведенного кружком протона слева. Не обведенный кружкой протон действует
магнитное поле, которое является суммой приложенного внешнего магнитного поля
(рисунок вверху) плюс или минус иебольшое поле в соответствия с тем, направлен ли спии
взаимодействующего с ним ядра (ядер) «вверх» нли «вниз». Например, поскольку общий
магнитный момент двух антнпараллельных спинов одинаков для обеих возможных
конфигураций t 1 и \ \ , то обе они будут создавать одно я то же магнитное поле на
обведенных кружком ядрах я сигнал магнитного резонанса, соответствующий этим
конфигурациям, будет вдвое больше по интенсивности, чем сигналы для отдельных
конфигураций t t H 1 1 (рисунок .в середине). На нижнем чертеже показан протонный спектр ЯМР
молекулы ацеталъдегнда. Резонансные сигналы для метальных протонов (относительная
интенсивность равна 3 для трех идентичных протонов) расщеплены иа две линнн в
соответствии с двумя возможными орнентациями спина альдегидного протона. В то же время
резонансный сигнал альдегидного протона (относительная интенсивность равна 1 для одного
протона) расщеплен на четыре линии в согласии с четырьмя возможными суммарными
магнитными моментами, связанными с восьмью возможными конфигурациями спилов трех
метильных протонов (рис. 18.7). Этот рисунок содержит достаточно информации, чтобы
предсказать протонный спектр ЯМР любой молекулы (пря достаточно большой Н») я
получить резонансные частоты для каждого невозмущеиного протона и константы спин-
спинового взаимодействия между всеми типами протонов в молакуле.
601 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
I I I I I I'll | I М П I ! I
* Г| 1 I i i 1 i I I I м i i I I I
0° 90° 180°
Двугранный угол 8
Рис. 18.9. Протон-протонная константа спин-спинового взаимодействии. / как
функция двугранного угла между соответствующими связями С—Н у соседних
углеродных атомов в молекуле
Этот график иногда называют кривой Карплуса по нмеии ученого, который предложил
первое теоретическое объяснение ее общей формы. Хотя в общем случае кривая дает для
каждого измеренного / два возможных значения двугранного угла, оиа тем- не менее
полезна при установлении стереохимии различных связей в молекулах в растворе. [Из
Реагсе Е. £>., High-Resolution NMR, Reed Press, New York, 1967, p. 104.1
зультате наборы спектральных линий, ожидаемые для
расщепления сигнала магнитного резонанса протона группы —G—Н на
других протонах групп —С—Н, —СН2 или — СН3, наглядно показаны
на рис. 18.8 и подводят итог нашему-описанию определения
констант взаимодействия из наблюдаемых спектров ядерного
магнитного резонанса.
Огромный интерес к константам спин-спинового взаимодействия
/ обусловлен тем, что есть вполне определенное эмпирическое
соотношение (с теоретическим обоснованием, которое ^выходит за
рамки настоящего обсуждения) между величинами / и двугранным
углом между направлениями связей С—Н протонов соседних
атомов углерода (рис. 18.9). Поскольку мы уже убедились, что спин-
спиновые взаимодействия передаются через химические связи,
кажется вполне разумным, что эффективность передачи (и,
следовательно, величина /) будет зависеть от степени перекрывания
между (несферическими и, следовательно, направленными)
электронными орбиталями, образующими химические связи. Уникальное
значение протонного ядерного магнитного резонанса (ЯМР) для
определения структуры и стереохимии молекул в растворе
основано на изменении резонансной частоты протона («химический
едбиг») в зависимости от заместителей у атома углерода, к
которому этот протон принадлежит, и на изменении величины I в
зависимости от величины двугранного угла между соседними атомами
углерода в цепочке.
602 ЧАСТЬ Б
2,3,6: mpU'0-ацетил
целлюлоза
Н:4
-о-„
(6,6')Г
АсОСН2-
АсО
ОАс
ь>,м.0.
2,3,8-три-О- ацетил
амилоза
ОАс
Н-3 Н-1
Н-6Н-6' Н-5 Н-4
Н ^жваториальнЗй~
^-и2~ЗНг
6= 5,0
-6», А/у.
Рис. 18.10. Конформации и протонные спектры ЯМР ацетилиров-анной целлюлозы
(вверху) и ацетилированной амилозы (внизу), которые различаются структурой
[расположением аиомерного протона Н-1 либо в аксиальном (целлюлоза), либо
в экваториальном (амилоза) положении].
Более сильное расщепление (8 Гц) обнаружено для Н-1 производного целлюлозы, тогда
как для Н-1 производного амилозы расщепление меньше (3 Гц), что подтверждает
стереохимию у акомерной связи. Из-за того что довольно трудно устанавливать стереохимию
сахара традиционными методами, основанными на химических реакциях, ЯМР стал методом
отбора для нахождения структур вновь выделяемых олигоыерных Сахаров (например,
различных антигенных сахаридов поверхностей клеток). Шкала частот для этих
энергетических спектров поглощения дана в миллионных долях (м.д.): для магнитного поля,
в котором резонансная частота протонов составляет ~ 100 МГц, 1 М.Д. — 100 Гц. [Из Frlebo-
Un И., Keilich G., Siefert £., Angew. Chemie, 81, 791, (1969).] -
Пример.
Одно из наиболее убедительных доказательств методом ЯМР
существования предпочтительных информации биологически важных молекул было
получено для производных углеводов. В качестве иллюстрации на ,рис. 18.10
показаны протонные спектры ЯМР для ацетилированной целлюлозы
(полисахарида глюкозы с {3-гликозидными связями между глкжозными единицами) и
ацетилированной амилозы (полисахарида глюкозы с а-гликозидными связями).
Согласно рис. 18.9, константа спин-спииового взаимодействия для аксиальных
лротонов должна составлять около 8 Гц, как это и наблюдали для всех пяти
протонов кольца замещенной целлюлозы. С другой стороны, меньшая константа
взаимодействия — около 3 Гц — для взаимодействия между протоном 1 и
протоном 2 замещенной амилозы указывает на присутствие а-гликозидных связей
в этом полимере.
Пример
Есть множество примеров использования констант / спин-спинового
взаимодействия в спектрах ЯМР для определения конформации изолированных
биохимических соединений в растворе (из которых, возможно, наиболее замеча-
i&03 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
тельным является доказательство того, что восстановленная форма пиридииди-
нуклеотида NADH существует в свернутой конформащш, основанное на
химической неэквивалентности двух (геминальных) протонов у С-4 дигидропиридиново-
го кольца). Недавно был опубликован важный пример доказательства конфор-
мационного изменения субстрата при связывании его с ферментом (рис. 18.11).
Как мы видели на рис. 18.10, а-замещенная глюкоза в своей обычной кон-
формации «кресла» имеет константу взаимодействия для аномерного протона
/~3 Гц. Однако если конформацню «кресла» исказить до «полукресла»
(рис. 118.11), то двугранный угол (который определяет /) между протоном 1 я
протоном 2 кольца молекулы сахара уменьшится, а / возрастет. Сайке с
сотрудниками непосредственно наблюдали за поведением тетрамерного олигосахарида
(АГ—АМКЬ ПРИ связывании с ферментом лизоцимом (который по своей
природе предназначен разрывать (АГ—АМК)л-полимеры в клеточной стенке).
Поскольку (АГ—АМКЬ является субстратом ,и быстро распадается на более
мелкие олигомеры, которые больше ие связываются так хорошо с лизоцимом (и
у которых в любом случае больше не искривлена форма в связанном состои-
йии), более высокое значение /, которое рассчитывали обнаружить сразу после
связывания субстрата с ферментом, должно было быстро возвращаться назад
Кресло"
сн2он
он
в
CH3CONHjiC3
он
„Полукресло"
н4
>^£н^3^он
Лизоцим
0 12 3 4 5 6
Время, мин
Рис. 18.11. Искажение молекулы субстрата при связывании с ферментом.
Неискаженное (вверху слева, конформацня «кресле») н искаженное (вверху справа, кон-
формация «полукресло>) кольцо 2-ацетамндо-2-дезокси-0-глюкопнранозы в тетрамере
(N-ацетилглюкозамии — N-ацетилмурамовая кислота )2 или (АГ — АМКЬ — субстрате
фермента лнзоцима. Величины /-взаимодействия в спектре ЯМР между протонами 1 и 2
восстанавливающего конца субстрата (верхняя кривая) и восстанавливающей способности смеси
субстрата и фермента (нижняя кривая) измерены в зависимости от времени. Обнаружено
очевидное увеличение / в случае образования комплекса фермент — субстрат, а
экстраполяция к нулевому моменту времени (моменту смешнваиия субстрата и фермента)
подтверждает, что концевое сахарное кольцо изменяет свою конформацню с «кресла> (в ~ 6(г)
на <полукресло> (в ~ 0°) при присоединении к лизоциму. Этот результат показывает, что
основу механизма действия лизоцнма составляет деформация молекулы субстрата для
облегчения разрыва связи по месту соединения Сахаров. |[Из Patt S. L., Dolphin D., Sy-
kes В. D., Ann. New York Acad. Scf., 222, 2ii (1973).]
604 ЧАСТЬ S
к величине 3 Гц, как только произойдет реакция. Таким образом, необходимо
было регистрировать константу спин-спинового взаимодействия как функцию
времени и затем экстраполировать ее обратно к начальному моменту
смешивания реагентов, чтобы рассчитать значение / для фермент-субстратного
комплекса. Экстраполяция стала возможнрй благодаря независимому слежению за
ходом реакции путем измерения восстанавливающей способности (при
осуществлении реакции каждый раз возникает новый сахарный конец-восстановитель) саха-
ридов как функции времени, прошедшего после смешивания субстрата и
фермента. Этот эксперимент важен по двум причинам. Первая заключается в том, что
он дает наиболее убедительное доказательство изменения конформации
субстрата при связывании его с ферментом. Вторая — в том, что, как оказалось,
изменение конформации в растворе, наблюдаемое методом ЯМР, является точно
таким же, какое, наблюдали в кристаллах методом реитгеиоструктурного
анализа. Отсюда весьма вероятно, что и другие механистические заключения,
базирующиеся иа реитгеноструктурных исследованиях лизоцима, будут применимы
к действительному поведению его в растворе.
18.Б. ТЕОРИЯ МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ
И АКТИВНОСТЬ ЛЕКАРСТВ
Предполагается, что читатель уже сталкивался с электронными
волновыми функциями («атомными орбиталями») водородного
атома. На рис. 18.12 показаны соответствующие им электронные
плотности и энергии электронов. Хотя эти функции получены для куло-
новского потенциала пары электрон — протон, они дают хорошее
описание поведения многих электронов в более крупных атомах
при условии, что большинство пар электронов располагаются
каждая на какой-нибудь одной «атомной орбитали». Возможно,
читатель уже знаком (например, из курса органической химии) и с
описанием «гибридных орбиталей». В этом случае из водородоподоб-
ных орбиталей формируют определенные сочетания атомных
орбиталей данного атома (sp3, sp2, sp). В любом случае можно
ожидать, что если сложные атомы можно описать комбинациями
атомных орбиталей водородного атома, то молекулы могут быть
описаны комбинациями атомных орбиталей составляющих их
атомов. Эта гипотеза лежит в основе теории молекулярных орбита-
лей, которая в свою очередь составляет основу теории химических
связей и молекулярных свойств. Примеры этого раздела
базируются на несколько упрощенной теории молекулярных орбиталей Хюк-
келя. Соответствующие расчеты являются относительно простыми,
но качественное соответствие их эксперименту, как ни странно,
хорошее.
Как оказалось, теория молекулярных орбиталей имеет очень
большое значение при описании молекул, в которых есть кратные
(двойные и тройные) связи между атомами углерода, и особенно
для молекул, содержащих «сопряженные» связи (чередующиеся
простые и двойные связи —С = С—С=С—С = ). Двойные связи
атомов углерода могут быть описаны исходя непредставления о
двух типах связей, как это показана на рис. 18.13.
605 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
р -орбита/16
р- орбита ль
6s
Ър
р -орбиталь
dj^-opSuma/ib
с?
5s
4j
3s
2s
Ь
Ар
Zp
2р
5d
Id
3d
*f
dx2 -z 2 ~°р6итадь
Лг2-орбита/1ь
Рис. 18.12. Волновые функции (слева) и соответствующие энергетические уровни
(справа) водородного атома.
Энергетические уровни изображают порядок, в котором водородоподобные орбитали должны-
заполняться при последовательной добавлении электронов в сложных атомах. Формы
орбнталей определяются поверхностью постоянное электронной плотности (так же как-
топографический коитур определяется линией постоянной высоты). .[Схемы орбнталейс
приведены из Kier L. В., Molecular Orbital Theory In Drug Research, Academic Press, New
York, 1971, p. 2Г; энергетических уровней — нз Davis J. С, Jr., Advanced Physical
Chemistry, Ronald Press, New York. 1965, p. 242,]
с-Связь образуется перекрыванием одной р-орбитали от
каждого из двух атомов углерода таким образом, что главные оси р-
орбиталей располагаются вдоль оси о-связи. Второй тип связиг
я-связь, образуется перекрыванием одной р-орбитали от каждого-
из двух атомов углерода таким образом, что главные оси р-орби-
талей располагаются перпендикулярно оси я-связи. (На
математическом языке можно было бы сказать, что а- и я-орбитали
молекулы «ортогональны».) В нашем упрощенном варианте теории
молекулярных орбиталей игнорируются все атомные орбитали, за
606 ЧАСТЬ 6
*-ecsx=>-
Рис. 18.13. Молекулярно-орбитальное описание углерод^углеродной двойной
связи.
При образовании ст-связи реализуется перекрывание двух р-орбнталей, главные оси
которых расположены вдоль направления связи. В it-связи реализуется перекрывание между
двумя р-орбнталямн, главные оси которых перпендикулярны направлению связи.
Упрощенная теория молекулярных орбиталеА ограничивается рассмотрением только я-свяэей
между атомами углерода, соединенными кратными связями.
исключением тех, которые вовлечены в образование связей я-типа.
Подобным образом выводят свойства элементов периодической
таблицы, учитывая только «валентные» орбитали и, таким
образом, рассматривая сложныеатомы по аналогии с атомом водорода.
В случае сопряженной цепи углеродных атомов ожидается, что
каждый атом углерода даст по одному р-электрону для связывания
по п-типу, так что можно записать молекулярную волновую
функцию в виде взвешенной суммы («линейной комбинации») атомных
2р-орбиталей каждого из углеродных атомов в цепи:
ty = Wi~hW* + • • - + Сп9п* (18.82)
где фг — волновая функция атомной 2/?-орбитали для i-ro
углеродного атома,, а д|> — молекулярная орбиталь. Обычно каждая из
возможных i)5 «нормирована» до единичной «нормы» (единичной
«длины»):
kl' + Kr + .-. + kl'^l. (18.83).
Нам (как обычно) интересно в первую очередь выявить
энергетические уровни системы, которые получают из определителя,
даваемого гамильтонианом Э6\
Щ~Е$, (18.28)
г МО ■ 1 Мб МО \
Ob\1 л «^12 ' ' * "*■ \П \
I МО МО 1 МО V
det I **«« Жгг — 1 . . . Жгп 1 = 0 (18.84)
мр ме 5 /
'п\ uvm • * • ""пч
Уровни энергии
Как читатель может вспомнить, решение определителя (пХ^п)
включает решение полинома п-й степени. Поскольку нас обычно
интересуют молекулы с более чем п=2 атомами^!), было бы же-
607 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
лательно сначала отыскать какое-нибудь упрощение для
уравнения (18.84). К счастью, важное упрощение следует сразу после
выяснения физического смысла различных матричных элементов
5#ij. Например, мы уже знаем, что средняя энергия системы в
состоянии ip будет < ij), 3#i|) > [уравнение (18.2)], так что логично
интерпретировать матричный элемент Жц\
#н = <?,. Жъ> (18.85)
как кулоновскую энергию, которая удерживает электрон на
атомной 2р-орбитали *-го углеродного атома в молекуле. Поскольку
я-электроны сопряженной молекулы поступают от идентичных
центров (атомы углерода), кажется вполне разумным, что куло-
новская энергия связи а должна быть приблизительно одной и той
же для каждого из углеродных атомов сопряженной цепи:
^fjj = ^?22 = • • • = &€пп ~ а \ 1о«оЬ)
По аналогичной логике матричный элемент Ж^ может быть
идентифицирован как энергия взаимодействия между атомной 2р-орби=
талью t-го и атомной 2р=орбиталью /-го углеродных атомов цепи.
Снова мы можем ожидать, что взаимодействие между, скажем, 2-м
и 3-м атомами углерода будет таким же, как и взаимодействие
между 7-м и 8-м углеродными атомами. Эту единую величину,
обозначенную как р, иногда (по недоразумению) называют энергией
«резонанса»
«#?,2 — &£iz = • • • =&€(п-\)п='- Р (18.87)
Наконец, поскольку электронная плотность для данной атомной
орбитали по существу простирается не более чем на длину одной
связи от данного атома, можно с уверенностью заключить, что,
когда атомы углерода разделены более чем одной связью, энергия
взаимодействия между двумя 2р-электрона ми в цепн углеродных
атомов будет равна нулю:
3@ц=0, кроме случая, когда t и /отличаются на ± 1 (18.88)
Пример. Этилен НгС=СН2
В молекуле этилена с двумя атомами углерода есть одна двойная связь и
по одному 2р-электрону, пригодному для образования я-связи, у каждого из
диух атомов углерода. Таким образом, можно записать вид молекулярной
орбиталн:
*■■=<?,¥, + *,¥, (18.89)
€08 ЧАСТЬ 5
« энергетические уровни, получаемые при решении определителя уравнения
(18.90) с использованием упрощений [уравнения (18.86)—(18.88)1
I в —Л
=0. (18.90)
Хотя мы могли бы решить определитель уравнения (18.90) прямо, полезно
ввести прием, который будет важен в последующих (более сложных) случаях,
а именно: разделим каждый член на р* и введем
■*»
X
-(^г).. <18-91>
так что уравнение (18.90) примет вид
\Х 1
|1 X
■я будет иметь решения
=0 = А"*—1 (18.92)
Х=± 1, ,7 . (18.93)
«ли просто
*.=1
Я, = а4-р, (18.94а)
Я2 = а-р. (18.946)
Чтобы найти соответствующие волновые функции ф молекулярных орбиталей
(МО), мы просто запишем уравнение собственных значений для энергии,
используя уравнения (18.89) и (18.93), и решим его для коэффициентов с\ и с*.
ЯМ>,'=Л.,*,. (18.95)
(VX: )-"■+»(; )•
или
(", + Р<Л Л« + МЧ (18.96а)
Уравнение (18.96а) показывает, что два вектора равны. Но я в обычном
трехмерном пространстве, и в пространстве функций два вектора равны тогда и
только тогда, когда равны их компоненты вдоль каждого базисного вектора
(в нашем случае вдоль базисных векторов <pi и
ас, + К= ас* + ?ci (18.97а)
v
Я
рс, -J- <xca = ос2 -|~ р£а. (18.976)
Решением этих уравнений является, очевидно,
С^Сг. (18.98)
Аналогично можно показать, что для другого собственного значения энергии
Ж$=)4$ другая молекулярная орбиталь будет иметь компоненты
сх=* — сг. (18.99)
)9 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
<t-fi . ^ = _.^_^)
V 2 ft (связывающая) цгг (ацтисайЗывающааУ
Этилен
о» Q О
• •
*+ув.
f-J—*/-3f'#,+*j ^ 0 С)
|' Рис. 18.14. Схематическое изображение энергетических уровней (слева) н jcoh-
|туров электронной плотности (справа) двух я-электронных молекулярных орби-
I талей этилена.
I Стрелками на картине энергетических уровней показаны два электрона (с противоположны-
^ мн спннамн), занимающие самую ннэкоэнергетическую МО. (Обсуждение см. в тексте.)
; Наконец, поскольку г|э нормирована до единичной «длины»,, мы должны иметь.
с.
и (18.100а>
(18.1006)
Из уравнений (18,100) мы можем теперь записать две молекулярные орбитали.
для этилена как
*i = 7f (*«+?.) (18.101а)
и
ф2 = ^(?1-92). (18.1016)
Собственные значения энергии и собственные функции для я-электронов
этилена показаны на рис. 18.14. Поскольку а и Р являются отрицательными
числами (по отношению к энергии я-электрона, который находится на бесконечном
удалении от молекулы), два электрона на самой нижней занятой молекулярной
орбитали «удерживаются» в пределах «связи» только энергией притяжения.
Карта для низкоэнергетической МО показывает, что электронная плотность
локализована главным образом в пространстве между двумя углеродными
атомами. Это хорошо согласуется с нашей обычной концепцией химической
связи. С другой стороны, в случае более высокоэнергетической МО расчет
электронной плотности показывает, что электрон на этой «антисвязывающей»
молекулярной орбитали (если бы электрон действительно был на этом
энергетическом- уровне) не может быть локализован в области пространства между угле*
родными атомами и не может, следовательно, обеспечить химическую связь.
(На молекулярных орбиталях электроны располагаются парами так же, как иа
более привычных нам водородоподобных атомных орбиталях.)
Этот пример показывает, как молекулярные орбитали, из которых мы можем
определить электронную плотность вокруг каждого из атомов углерода, дают
энергию связывания и, кроме того, предсказывают, что я-электроны чаще всего
будут находиться в области между двумя соединенными химической связью
атомами углерода, как это и ожидается интуитивно. Отсюда расчеты по мето-
610 ЧАСТЬ 5
ду МО оправданы вовсе не потому, что они правильно предсказывают
электронные энергии или электронные плотности в реальных молекулах (хотя именно это
чаще всего имеется в виду), но потому, что некоторые рассчитываемые методом
МО свойства могут хорошо объяснять определенные биологические функции
молекул. После нескольких дополнительных примеров проведения конкретных
расчетов читатель будет в состоянии выполнить самостоятельно широкий круг
расчетов по методу МО. В качестве примера будут приведены также типичные
корреляции полученных методом МО параметров с действием лекарств.
Пример. Бутадиен СН2=СН—СН=СНа
Идя точно таким же путем, как и в случае этилена, выразим молекулярную
орбиталь (молекулярные орбитали) в виде линейных комбинаций атомных орби-
талей (обычно это называют расчетом МО в приближении ЛКАО):
Ф = ci?i + с*Ъ +■ сз?э + с4?4
(18.102)
и, используя уравнения (18.86) — (18.88), сразу запишем определитель, из
которого вычисляют энергии
а—I р 0 0
Р « —X Р 0
О р а—X р
0 0 р а—I
= 0.
а—А,
Разделяв обе части на р и учитывая, что по определению Я= —q-
(18.91)], получим
А" 1 0 0
1 А" 0 0
0 1 X 1
0 0 1 X
= 0.
(18.103)
[уравнение
(18.104)
Уравнение (18.104) решают, раскрывая определитель по элементам, например,
верхней строки (см. любой справочник по определителям или линейной
алгебре):
= *
X
1
0
0
1
X
1
0
0
1
X
1
0
0
1
X
X 1 0
1 X 1
0 1 X
— 1
1 1 0
0 X 1
0 1 X
+ 0
1 X 0
0 1 1
0 0 X
—0
1 X 1
0 1 X
0 0 1
=х
X 1 0
1 X 1
0 1 X
1 1 0
0 X 1
0 1 X
— X*
X 1
1 X
— X
1 1
0 X
— 1
X 1
1 X
= Xй (X* — 1)— Х(Х — 0) — (X* — 1) =
X* — ЗХ* + 1 = 0; X ='± 1,62; ± 0,62;
Х = о±1,62р; о±0,62р, /
(18.105)
[ВЦ КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
а- 1,620-
04 - 0,3701 - 0,6002 + О.6О0з - 0.37^4
t
а - 0,620 ■
Фз * O,6O0i - 0,37^2 - О,370з +
о,'бО04 СИЗ
е>о
^ а+0,620 +—4 ^2-О,6О01+О,370г-О.370з-О,6О04 ^э (2)<i>£~\
О§>©0
«+1,620-
Т
■VI = 0,3701 + О,6О02 + О,6О0з + 0,3704
Рис. 18.15. Схематическое изображение энергетических уровней (слева),
собственных функций энергии (в середине) и контуров электронных плотностей
(справа) четырех я-электронных молекулярных орбнталей бутадиена.
Стрелками обозначена занятость двух самых нижних МО четырьмя я-электроиами.
Кате и в случае этилена; волновые функции МО бутадиена находят, применяя
оператор Гамильтона к энергии МО Х\, затем к энергии МО Х2 и т. д.
Например: «?«ь. =>.*. (18.106)
= («+1,620) | „_ |, (18.107)
ас,+.?с2 = (а+ 1,62р)с,
рсх4-*с.4-Рс, = (о4-1.62р)с.
рс,+ «<?. +К = (*+!.620) с,
рса«с4=(о+1,62р)с4
и |cI|4-|c,|«+|ce|, + U4|a=l
(18.108)
;
Оставляем читателю в качестве упражнения вычислить из уравнений (18.108)
четыре неизвестных с и с2, с% и с4. Остающиеся три молекулярные орбитали
получают аналогично. Результаты вычислений показаны на рис. 18.15.
Пример. Бензол
Оставляем читателю (см. задачи к этой главе) показать, что определитель,
нз которого получают собственные значения энергии для я-электронов бензола,
имеет вид Л 1 0 0 0 1 I
1*1 0 0 0
0 1 X 1. 0 О
0 0 1 X \ 0
0 0 0 1 Л" 1
1 0 0 0 1 Л"
= о = х» —:6Х* + ^*2 - 4.
(18.109)
612 ЧАСТЬ Б
e_2(I — ♦•-fciH
02 + 03 - 04 + *5 - 06^
а_0 *в = (1/>А2)(20, - ф2 - 03+,2tf4 - 05 - 0„)
| *4 =(-у)'02 - 03 + 05 - Фб)
| „ . (18-Ш)
I1 1111 ^3=(-)(02+*3-05 -0б)'
^ а + Д -4—i— -f—Jr /—
IT If ^2= (1/V12M20, + 0? -03 - 204 -0s + 06)
a + 20 -j J- ^, =(^=)<01 + 02 + 03 + 04 + 05 + 06)
Рис. 18.16. Схематическое изображение энергетических уровней |(слева) и
собственных функций энергии (справа) шести я-электронных молекулярных орбита-
лей бензола.
Стрелками обозначена занятость трех самых нижних МО шестью я-электронами.
Заметим, что молекулярные орбиталн ф2 и ф3 имеют одинаковые энергии (то же можно сказать
« про 1|э* и 1|э5). В таком случае говорят, что энергетические уровни для ч|>2 и ф» являются
-«вырожденными». Несмотря на то что эти две МО имеют одинаковые энергии, иа каждой
из них все же может находиться по два электрона.
Решениями его являются значения Х=2, 1, 1, —1, —1, — 2, (18.110)
дающие в результате энергетические уровни и молекулярные орбнтали,
показанные на рис. 18.16.
Пример. Пиридин
Если в систему химических связей молекулы я-электроны вносят не только
атомы углерода, но и другие атомы, то нельзя больше считать, что « (нли В)
одинаковы для всех участвующих в сопряжения атомов. Поправка, форма
которой позволяет сохранить простоту анализа МО, вводится, исходя из
предположения, что значения а и В для любого нового атома могут быть получены из
значений для атома углерода в соответствии с линейными соотношениями
«(гетероатом) = а(углерод) +Л(гетероатом)-8(углерод— углерод) (18.112)
^(углерод — гетероатом) = k (углерод — гетероатом) • В (углерод — углерод) (18.113)
Относительно согласованные между собой значения h и k были определены
эмпирически для ряда гетероатомов (см. значения Л и А в табл. 18.4). Величины
а табл. 18.4 не являются единственными: существуют и другие
самосогласованные наборы [наибольшего внимания заслуживает набор, данный Пульманом
(Pullman В., Quantum Biochemistry, Wiley-Interscience^ N. Y., 1963)], которые
иногда лучше подходят для определенных серий гомологичных соединений.
613 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
= 0
(18.114)
,Используя значения h и k для N в табл. 18.4, можно сразу записать
необходимый определитель для расчета собственных значений энергии пиридина
s виде
* + 0,5 10 0 0 1
1 X 1 0 0 0
0 1 X 1 0 0
О 0 1 X 1 О
0 0 0 1 X 1
1 0 0 0 1 X
и получить в результате энергетические уровни н волновые функции для
занятых уровней, приведенные на рнс. 18.17.
Таблица 18.4
Параметры МО для упрощенного описания гетероатомов
(неуглеродиых атомов) [уравнения (18.112) и (18.113)]а
N = 0,5
N=1,5
6 = 1,0
6 = 2,0
N® = 2,0
СНЭ = 2,0
С— N = 1,0
С —N = 0,8
С—6 = 1,0
С— О = 0,8
С — N©= 1,0
С —СНЭ = 0,7
а Из книги Стрейтвизер Э. Теория молекулярных орбит для
химиков-органиков. — М.: Мир, 1965. --'"
си - 1,93д
I»
а-р.
-0,840
«с*з= -0,502 -0.503 + 0,505 +О,506
I ■♦'ifaK
«% а+ 1,16(3-|—F- ^2= -0.570, -0,19^2+0,3503+0.6004+0,3505-0,19^6
а+ 2,10 -4—Х- ф, =0,5201 +0,4202 + 0.3603 + 0,3404 + О,3б05 + О,4206
(18-115)
Рис. И8.17. Схематическое изображение энергетических уровней (слева) и
собственных функций энергии занятых молекулярных орбнталей (справа) для шести
я-электронов пиридина.
Заметим, что вырожденность энергетических уровней, наблюдавшаяся для бензола, исчезает
при введении в кольцо гетероатома.
614 ЧАСТЬ 5
Применение: корреляция галлюциногенной активности с энергией молекулярной
орбитали
Образование связи одной молекулы с другой иногда объясняют процессом,,
известным как «перенос заряда», при котором электрон, первоначально
локализованный на «донорной» молекуле, теперь частично становится связанным с
другой «акцепторной» молекулой, D+A^DA. (Взаимодействия с переносом заряда
составляют, например, основу ксерографии.) Отсюда, вообще говоря, можно
было бы ожидать, что идеальная донорная молекула должна иметь относительно
высоколежащую «высшую занятую молекулярную орбиталь» (ВЗМО). В этом
случае необходимо затратить сравнительно мало энергии для удаления части
или всех электронов с этой орбитали для переноса к акцепторной молекуле.
Из многих примеров, в которых донорные свойства в комплексах с переносом
заряда были соотнесены с энергетическими уровнями ВЗМО, рассчитанными
согласно теории МО, возможно, наиболее интересным для биологов является
пример, показанный в табл. 18.5. Таблица демонстрирует явное соответствие между
энергией ВЗМО для я-электронов наркотика и его галлюциногенной
активностью, определяемой как минимальная действующая доза (гл. 12.Б) "по
сравнению с мескалнном.
Таблица 18.5
Соотношение энергий высших занятых молекулярных орбиталей (ВЗМО)
и биологической (галлюциногенной) активности (отношение эффективной дозы
мескалина к эффективной дозе исследуемого наркотика) для нескольких
известных галлюциногенов3 (структуры ТМА и ТМА-2 изображены внизу)
Соединение Биологическая активность ^ВЗМО
ЛСД 3700 0,2180
Псилосин 31 0,4603
6-Оксидиэтилтриптамин 25 0,4700
ТМА-2 17 0,4810
ТМА 2,2 0,5357
Мескалин 1 0,5357
ОСН3
СН30—(С^)— CH2—CH-NH2 ТМА-2
/— I
СН3
сн3о
СН30—(О)—СН2—СН—NH2 ТМА
I сн„
сн3о "3
в Из Snyder S. H., Merril С. R., Ргос. Natl. Acad. Sci. USA. 54, 258 (1965).
Несмотря на поразительное (в данном конкретной случае) соответствие
между энергией ВЗМО и активностью наркотика, важно отметить, что
корреляции фактически не раскрывают причины и механизм действия наркотика.
Например, есть сотнн молекул с энергией ВЗМО, столь же малой или еще меньшей,
чем у ЛСД, но которые ие обладают галлюциногенным действием. Другими
$15 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
i. ; —
«
словами, теория МО дает корреляции, которые могут говорить о каком-то
аспекте возможного механизма процесса, но эта теория не может быть основой
длн предсказания большей нли меньшей активности новых лекарств по
сравнению с известными.
Электронная плотность на различных атомах, получаемая
на основе теории молекулярных орбиталей
Мы уже рассчитали энергетические уровни и соответствующие
им собственные функции энергии (МО) для я-электронов в
нескольких сопряженных органических молекулах. Из выражения
каждой МО для данной молекулы как линейной комбинации
атомных орбиталей мы можем сразу определить вероятность
нахождения я-электрона у любого отдельного атома. Она определяется как
квадрат коэффициента, который описывает проекцию МО на эту
отдельную атомную орбиталь. Например, в бутадиене есть по два
я-электрона на каждой из занятых молекулярных орбиталей.
Таким образом, электронная плотность, например, на атоме № 1,
дается выражением
электронная плотность на атоме № 1 — qt = 2 (0,37)2 -|- 2 (0,60)г — 1,
(18.116а)
где (0,37)2 считают вероятностью нахождения я-электрона на
МО \j)i, локализованной у атома № 1, а (0,60)2 — вероятностью
нахождения я-электрона на МО г|)2, локализованной у атома № 1.
Аналогично читатель может проверить, что
^ = 2(0,37)г + 2(-0,60)г=1, (18.1166)
а также что
д = 0,= 1.. (18.116в)
Более интересен пример с пиридином (рис. 18.17), для которого
читатель должен сам проверить, что из МО на рис. Л8.17
электронные плотности на различных атомах кольца получаются
такими, как указано ниже.
0,950
Вооруженный параметрами из табл. 18.4, читатель может в
принципе вычислить, энергетические уровни, собственные функции
энергии (МО) и затем электронные плотности на каждом атоме для
очень большого числа сопряженных органических молекул. Далее
мы опять сосредоточим наше внимание на корреляции электронной
616 ЧАСТЬ Б
плотности с биологической функцией, а не на сравнении
рассчитанных и экспериментальных электронных плотностей реальных
молекул.
Пример. Электронная плотность н канцерогенность ароматических полнцнкличе-
скнх углеводородов
Можно надеяться, что какие-то ключи к реагентам, вызывающим рак, нлн
к механизму инициации рака могут быть найдены путем выявления решающих
особенностей молекул с известным (очень часто высоким) канцерогенным
(вызывающим рак) потенциалом. Для производных антрацена, показанных в табл. 18.6,
Таблица 18.6
Соотношение между общей электронной плотностью в /С-областн
и канцерогенной активностью некоторых производных антрацена.
Электронную плотность рассчитывали с помощью теории МО почти так же,
как и в случае пиридина [уравнение (18.117)]а
Соединение
Электронная
плотность
Канцерогенная
активность
Антрацен (СЖ(ЭК))
1,259
1,2-Бензантрацен
5-Метил-1,2-бензантрацен
Ю-Метил-1,2-бензантрацен
5,9,10-Трйметнл-1,2-бензантрацей
1,283
1,296
1,306
1,332
+
++
+++
++++
а.Из Doorenbos N. J., «Physical Properties and Biological Activit)^, in A. Burger, ed..
Medicinal Chemistry, 2nd ed., Interscience, N. Y.. 1960, pp. 46—71.
имеется четкая корреляция между электронной плотностью (вычисленной на
основе теории МО) в «it-области» ароматического кольца н канцерогенной
активностью. Более сложные корреляции, включающие больше соединений,
наводят на мысль, что канцерогенную активность, по-виднмому, можно уменьшить,
Повышая электронную плотность во второй «L-областн». Эти результаты,
кажется, позволяют предположить, что канцерогенная активность находится в
определенной связи с образованием ковалентной связи в -/(-области, в то время
как образование ковалентной связи в i-областн могло бы быть альтернативной
«антиканцерогенной» реакцией. Эта область является в настоящее время
объектом активного изучения.
L-область
1,2-бензантрацсм
5 lA°j \*/К-сАчасть
617 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
18.В. БИОЛОГИЧЕСКОЕ ЖЕЛЕЗО.
МЕССБАУЭРОВСКАЯ СПЕКТРОСКОПИЯ* •
В предшествующих "разделах мы рассмотрели энергетические
уровни, которые возникают в результате действия силы,
удерживающей электроны в молекулах, и силы, воздействующей на
магнитный момент в статическом магнитном поле. Поскольку атомные
ядра сами по себе составлены из набора протонов и нейтронов,
можно было бы ожидать, что силы взаимодействия между этими
нуклонами также могут приводить к набору энергетических
уровней, между которыми можно было бы надеяться индуцировать
переходы, воздействуя на ядро излучением подходящей частоты.
Хотя такие ядерные энергетические уровни и существуют на самом
деле и хотя поглощение и испускание излучения с частотами,
соответствующими разностям ядерных энергетических уровней,
действительно наблюдают, существующие сейчас теоретические расчеты
не могут удовлетворительно объяснить расположение
энергетических уровней ядер. Проблема состоит в том, что поскольку все
нуклоны по существу имеют равные массы, то рассмотрение
отдельного нуклона таким образом, как если бы его поведение
управлялось взаимодействием с усредненным силовым полем всех
остальных нуклонов, не является хорошим приближением. [Такое
приближение успешно описывает поведение электронов в атомах
потому, что ядро гораздо более массивно, чем электрон
(электроны), и, следовательно, подход является хорошим приближением.
Можно считать, что сила, действующая на электрон, возникает
благодаря наличию «центрального силового.поля» (т. е. благодаря
действию заряда ядра, локализованного как точечный источник в
его середине). Это приводит к квантовомеханическому описанию
водородного атома, которое можно найти в большинстве учебников
химии.] Мёссбауэровская спектроскопия просто описывает
эксперимент, при котором фиксируют поглощение электромагнитного
излучения в области 7_излУчения (около 10 кэВ) подходящими
ядрами как функцию частоты падающего излучения, Поглощение
наблюдают на частотах, соответствующих разностям ядерных
энергетических уровней ядра мишени. Поскольку эти уровни
отражают состояние очень близкого к ядру окружения (электронное
состояние окисления, симметрию окружения, природу лигандов),
мёссбауэровские методы обеспечивают весьма специфичное
зондирование только одного типа атомов (обычно 57Fe) в сложной
молекуле.
Хотя теория реально не предсказывает абсолютные величины
ядерных энергетических уровней с хорошей общностью, можно
понять (по крайней мере качественно) природу небольших относи-
* Мёссбауэровские спектры иногда называют также спектрами ядерного
гамма-резонанса (ЯГР). — Прим. ред.
618 ЧАСТЬ Б
» .
!
Рнс. 18.18. Схематическое изображение двух относительных ориентации
несферического ядра в градиенте электрического поля.
Энергии взаимодействия между несфернчески распределенным зарядом ядра н градиентом
электрического поля различны для двух взаимных"ориентации.
тельных различий ядерных энергетических уровней для данного
ядра в разных отличающихся друг от друга окружениях.
Например, если распределение ядерного заряда не является сферическим
и если ядро находится в градиенте электрического поля
(возникшего, например, в результате образования химической связи),
тогда для ядра будут существовать различные энергии в зависимости
от его ориентации по отношению к ближайшим соседям (рис.
18.18).
Аналогично энергии взаимодействия заряда q с электрическим
полем Е, которая имеет вид q-E-x, где х — расстояние, на котором
находится q, энергия взаимодействия несферически
распределенного заряда с градиентом электрического поля dEjdz может быть
записана в виде q-x-z- {dEjdz). С помощью матричных методов, не
используемых в этой книге, можно показать, что собственные
значения энергии (энергетические уровни), возникающие из этого
потенциального члена в ядерном гамильтониане, дают «квадруполь-
ное расщепление», проиллюстрированное на рис. 18.19 для случая
S7Fe (наиболее общего для мёссбауэровской спектроскопии). Если,
скажем, атом железа находится в сферически симметричном
окружении, то градиент электрического поля на таком атоме железа
равен нулю, и никакого квадрупольного расщепления не будет.
Для искаженных распределений электронов расщепление будет
вполне определенным. Таким образом, квадрупольное
расщепление отражает симметрию распределения электронов вокруг мёссба-
уэровского ядра. Вместе с тем, поскольку ядра взаимодействуют с
собственными электронами, будут иметь место небольшие
изменения в ядерной энергии («изомерный сдвиг» по терминологии
мёссбауэровской спектроскопии), соответствующие изменениям в числе
и распределении электронов вокруг этого ядра. Таким образом,
положение (энергия) мёссбауэровского пика поглощения дает
информацию о химических связях и состоянии окисления
исследуемого атома. v
619 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
Последнее дополнительное замечание о мёссбауэровских
спектрах касается характерной шкалы частот спектра, несущих
информацию. В наиболее хорошо знакомых нам типах спектроскопии
(поглощение в ультрафиолетовой, видимой и инфракрасной
областях) источником излучения служит просто нагретое «черное
тело», которое излучает в относительно широкой области спектра.
Для спектроскопии ядерных энергетических уровней в мёссбауэ-
ровском эксперименте источником спектрального излучения
являются возбужденные ядра (скажем, 57Fe), получающиеся в
результате распада с выделением энергии из достаточно долгоживущих
высоковозбужденных ядер (57Со с периодом полураспада 270 сут
в случае 57Ре-мёссбауэровской спектроскопии). Этот источник
излучения является предельно монохроматическим, и для того, чтобы
изменить частоту источника, его физически двигают в направлении
поглотителя или от него так, что частота излучения,
«принимаемая» поглотителем, сдвигается благодаря эффекту Доплера
соответственно в область более высоких или более низких частот (так
же, как звук гудка поезда меняется по высоте тона от более
высокого до более низкого в зависимости от того, приближается ли
поезд к наблюдателю или удаляется от него). Поскольку сигналы
мёссбауэровского поглощения лежат в пределах относительно
маленького частотного интервала, возможно перекрыть необходимую
спектральную область, двигая источник к поглотителю или от него
со скоростью всего несколько мм/с. Таким образом (см.
последующий пример), положение пиков мёссбауэровского поглощения и
величины их расщепления приводят в единицах мм/с, где скорость
1 мм/с соответствует изменению энергии приблизительно на
5-Ю-8 эВ.
г А
1 а 5 • ю'8зд
т
Д£ = 14,4 кэВ
6
Рис. 18.19. Схематическое изображение уровней энергии ядра для "Fe.
Из-за небольшого (около 5-10—8 эВ) квадрупольного расщепления возбужденного ядерного
состояния излучение может поглощаться ядрами s7Fe прн двух отдельных энергиях, обе из
которых очень близки к 14,4 кэВ. («Собственная» ширина линии поглощения составляет
около 5-10—" эВ, н поэтому квадрупольное расщепление по сравнению с ней почти в десять
раз больше. Это приводит к получению двух хорошо разрешенных пиков поглощения — см.
примеры н обсуждение собственной ширины линии в гл. 19.Б и 20.)
а — квадрупольное расщепление возбужденного несфернческого состояния ядра; б — основ--
ное сферическое состояние ядра.
1
620 ЧАСТЬ 5
0,5
«1
1
*)
зг
Q
1
1,0
0
0,5
1*0
, ljt Ьдеаокси-Гр ,
А ' ■
6ь 6г
\
J*..
-4,0
J L
r-2,0
lr-l I'
.-^л^ч^-
Окси-Гр
0,0 2,0
Скорость, мм-с"1
4,0
Рис. 18.20. Мёссбауэровские спектры поглощения оксигемэритрина (окси-Гр,
внизу) и дезоксигемэритрнна (дезокси-Гр, вверху).
Показаны изомерные сдвнгн ft и квадрупольные расщепления Д. У оксн-Гр ясно видны
два квадрупольных расщепления, указывающих на то, что два атома железа в оксн-Гр
не идентичны (см. текст). Оба атома железа в дезоксн-Гр обладают одними и теми же в
и Д, что согласуется с одинаковым окружением обоих атомов железа в дезокси-Гр.
Изомерные сдвиги позволяют предположить, что оба атома железа в дезокси-Гр имеют
степень окисления 2+, а оба атома железа в окси-Гр — степень окисления 3+. [Из Garbett К.,
Johnson С. Е., Klotz I. M., Okamura М. У., Williams R. J. P., Arch. Biochem. Biophys.. 142,
Б74 (1971).)
Пример. Мёссбауэровские спектры Fe-содержащих белков
Из-за того что эффект Мёссбауэра открыт сравнительно недавно (1958 г.),
довольно трудно найти примеры строгих заключений о состояниях окисления
или молекулярной симметрии, впервые полученных мёссбауэровскнми методами.
Однако поскольку мёссбауэровские энергетические сдвиги и расщепления
отражают состояние окружения около только одного типа атомов (обычно 57Fe),
этот подход обеспечивает получение необычайно точной индивидуальной
информации о, скажем, атомах железа в биологических макромолекулах по сравнению,
например, с оптическими спектрами поглощения, которые отражают свойства
комплекса илн молекулы как целого.
На рис. 18,20 показан хороший современный пример," иллюстрирующий
мёссбауэровские спектры поглощения у-лучей для содержащего железо белка,
гемэритрина, не имеющего тема. Этот белок обеспечивает транспорт кислорода
у некоторых морских червей.
Хотя у дезоксигемэритрнна (дезокси-Гр) обнаружен только единственный
изомерный сдвиг, соответствующий (из сравнения с модельными, соединениями)
железу в состоянии окисления 2+, у оксигемэритрина (оксн-Гр) выявлены два
отдельных квадрупольных расщепления, указывающих на то, что два атома
железа в окси-Гр должны быть нендентичными. Изомерный сдвиг для окси-Гр
значительно отличается от такового для дезоксн-Гр н интерпретируется как
соответствующий степени окисления 3+ у обоих атомов железа окси-Гр.
Конечно, возможно, что два атома железа в дезоксн-Гр имеют различное окружение,
но их изомерные сдвиги и квадрупольные расщепления случайно совпадают.
В любом случае можно определенно заключить, что в окси-Гр есть два типа
атомов железа.
Поскольку содержание 67Fe в естественной смеси изотопов составляет около
2,2%, для мёссбауэровскнх измерений необходимо нмет^ относительно большие
КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
эличества образца (около 100 мг содержащего железо белка). Кроме того,
5разец- должен быть нлн кристаллическим, или очень вязким. В противном
[рлучае ширина линии поглощения становится слишком большой по сравнению
небольшими энергетическими сдвигами, которые хотят наблюдать. [Этот
эффект связан с импульсом «отдачи», возникающим при поглощении ядром
-луча. В твердом теле отдачу принимает на себя кристаллическая решетка как
{елое, а не только один атом нлн молекула. Прн этом энергия отдачи
(расширяющая диапазон частот, на которых происходит поглощение) становится нсче-
$ающе маленькой, и поэтому наблюдаемая линия поглощения будет очень
сой.] Развитие этого метода требует совершенствования теоретического описа-
шя поведения электронов в комплексах металлов.
Задачи
■I. а. Покажите, что следующие три базисных вектора образуют ортонормальный
1' набор (т. е. что они взаимно «перпендикулярны» н каждый имеет единичную
«длину») ,[табл. 18.2 н уравнения (18.3) — (18.5а)]:
б. Покажите, что следующие трн базисных вектора образуют другой
ортонормальный набор. Этот пример соответствует конструированию «орбнталей»
рх, рг и ру из комбинаций" первоначальных p+h р0 и p-i функций,
полученных решением угловой части гамильтониана водородного атома (см. любой
элементарный учебник химии):
Ж. Исходя из определения компонент Lx, Ly н Lz квантовомеханнческого
оператора углового момента, полученного на основе принципа соответствия с клас-
;" снческой механикой (табл. 18.3), проверьте «коммутационные» соотношения
\LX> Ly] = LxLy — LyLx = ifiLg,
\Ly, Lz\ = LyLz — LzLy= ihLx
[Lz, Lx\ = LZLX LXLZ = ifiLy.
3. Для частицы со спином '/а (рнс. 18.4) существуют два возможных
собственных значения (±ft/2) оператора для z-компоненты «спинового» углового
момента 1г. Таким образом, если собственные функции 1г, соответствующие
этим собственным значениям, обозначить как единичные векторы-столбцы (п| и
/0\ л W'
J11, то матричное изображение оператора должно принять вид
, _ /А/2 0 \
622 ЧАСТЬ Б
так что
/'"= ■r(JJ)(o)="5"(o)""5"'
Исходя из свойств «повышающего» и «понижающего» операторов /+ н /_,
а именно:
/_Р = 0,
можно сразу построить матричное представление для /+ и /- '[уравнение
(18.13)]:
А
и-
/0 IV /0 0\
= [о о)' 7" =(i oj'
нз которых вы должны затем сконструировать матрицы для операторов 1Х
и 1У:
а. Используя правило умножения матриц [уравнение (18.11)], покажите, что
матрицы, изображающие 1%, 1у и U для частицы со спином '/г, удовлетворяют
установленным коммутационным соотношениям для компонент углового
момента:
[Ixly] = Мг\ [Iylx] = Шх) [IzIx] = Oily.
б. Аналогично может быть показано, что матрицы, изображающие операторы
1х, 1У и /2 для частицы со «спнном», равным единице, имеют вид
(1 0 0\ /0 1 0\ /0 —I ON
о о —1 / r z \ о 1 о/ r M о 1 о
Покажите, что этн три матрицы удовлетворяют коммутационным правилам
для компонент оператора вектора углового момента.
4. а. Используя обычные соотношения для перехода от декартовых к угловым
координатам (х=г sin 9 cos<p, y=r sin 9 sinq) и z=r cOs9), покажите, что
оператор Lz, полученный в декартовых координатах из классической механики
(табл. 18.3), может быть выражен в угловых координатах гораздо более
просто:
Lz = - i% [* (д/ду) - у (д/дх)] = - ih (d/df)
б. Теперь решите уравнение собственных значений для jU в угловых
координатах
Lz$ = п%Ф,
где п принимает целые или полуцелые значения, чтобы получалось выражение
для собственной функции Ф. Далее покажите, что если собственная функ-
|3 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
ция Ф является однозначной (т. е. Ф(0)=Ф(2л;)), то собственные значения
L* ограничены только целыми числами, умноженными на h. (Заметьте, что
для «спинового» углового момента собственные значения /2 могут быть илн
целыми, или полуцелымн числами, умноженными на h.)
а. Поскольку большинство представляющих интерес органических молекул
содержат более чем два типа протонов в пределах четырех химических
связей один от другого, желательно расширить описание двух спинов гл. 18.А
до случая, включающего трн типа взаимодействующих магнитных ядер,
каждое из которых имеет спин 72. Начиная с основного гамильтониана «первого
порядка»
* = -{jPJ.a + iMn + Te*V,c + Т V* +
+т'Л+т'Л; где ('• с">
и базисного набора произведений собственных функций hK, /zB н hc Для
трех ядер А, В и С, показанного ниже
ф2 = аладрс, ф6 = Рл?даС)
Фз = *Ah*C, *7 = алЫс,
h = haB«c, ** = hh$c,
вычислите собственные значения энергии (их существует восемь) для этой
системы ядер при наличии внешнего магнитного поля Н0.
б. Когда различия в резонансных частотах между ядрами А, В и С велики
■по сравнению с константами взаимодействия /ав, /вс и /ас, единственными
разрешенными переходами между энергетическими уровнями будут такие, при
которых только одни нз трех спинов меняет собственное значение оператора
/г. Используя это «правило отбора» (гл. 19.Б.4), найдите энергии для 12
разрешенных переходов. Далее принимая, что |/ав| > |/вс|>|/ас|, покажите,
что энергетический спектр поглощения для этой системы можно анализировать
графически, полагая, что каждый ядерный резонанс «расщеплен»
взаимодействием с одним нз двух других ядер и затем снова «расщеплен»
взаимодействием с оставшимся ядром, как схематически показано ниже. Несмотря на
то, что получение такой схемы расчетным путем может быть весьма
утомительным, вы теперь в состоянии анализировать любой протонный (нлн
углеродный, или фторный, или фосфорный) спектр ЯМР как спектр первого
порядка, решая обратную задачу, т. е. исходя из схемы, показанной ниже, чтобы
получить резонансные частоты («химические сдвиги») и константы спин-
стшнового взаимодействия (/) для спектра ЯМР, не прибегая к каким-либо
квантовомеханичсским расчетам!
в
р-1Ли»1-*1 Р",Л|в1~П 1 Н7вс1
Ua6 UkI иЛС1
624 ЧАСТЬ 5
в. Решите систему уравнений .[уравнения (18Л08)], чтобы получить собственные
функции молекулярных орбнталей для молекулы бутадиена.
7. а. Покажите, что определитель, из которого получают собственные значения
энергии для я-электронов бензола, имеет вид
X I О О О I
1X10 0 0
0 1X10 0
0 0 1X10
0 0 0 1 XI
10 0 0 1 X
= 0 = Х« — 6Х4 + 9Х2 — 4.
6. Покажите, что решениями этого определителя являются Х=2, 1, 1, —1,
—I,—2, и используйте эту информацию, чтобы получить энергетические
уровни и молекулярные орбнталн бензола.
8. а. Исходя из собственных значений эиергнн для я-электронов пиридина
(рнс. 18.17), покажите, что молекулярные орбнталн, соответствующие трем
самым низким собственным значениям энергии, являются такими, какие даны
уравнениями (18.115).
б. Используйте этн трн собственные функции для вычисления электронных
плотностей на каждом из шести атомов кольца пиридина (ответ показан
ниже).
0,95
Ъ,\9
Литература
Спиновые задачи (спектры магнитного резонанса)
1. Carrington A., McLachlan A. D., Introduction to Magnetic Resonance, Harper &
Row, New York, 1967.
2. Becker E. D. High Resolution NMR: Theory and Chemical Applications,
Academic Press, New York (1969).
3. Попл Дж., Шнейдер В., Бернстейн Г. Спектры ядерного магнитного
резонанса высокого разрешения.—М.: ИЛ, 1962. Особенно ценна гл. 6, где приведены
хорошо систематизированные расчеты спектров ЯМР.
Теория молекулярных орбиталей
4l Kier L. В., Molecular Orbital Theory in Drug Research, Academic Press, New
York, 1971.
Основы квантовой механики
5. Pauling L., Wilson E. В., Jr., Introduction to Quantum Mechanics, McGraw-Hill,
New York, 1935. До снх пор остается превосходной работой.
6. Davis J. С, Jr., Advanced Physical Chemistry, Ronald Press, New York, 1965.
1ЯАВА 19
росание монеты: «орел или решка»?
Цаспределение болыдмана
V.
и
? .
«-
t-
К-
!t
/ ■.
t
i
В предшествующей главе на примерах груза на пружине,
группы магнитных ядер со спином 7г и я-электронов в сопряженных
рганических системах мы показали, что энергия частицы
квантована (может принимать только определенные дискретные
значения). В этих типичных примерах мы до сих пор рассматривали
поведение только одной частицы или молекулы. Пытаясь понять
поведение большого числа таких частиц (химия, в конце концов, име-
X дело с количествами в 1020 и более частиц), мы сразу же
сталкиваемся со следующим вопросом: в данной обычной ситуации
ри фиксированном общем числе частиц N и фиксированной общей
нергии Е для всех частиц как можно предсказать относительное
йсло частиц Ni, которое описывается собственной функцией энер-
ии i|)i с собственным значением ei? Другими словами, как решить,
колько частиц имеют определенную энергию? [Для простоты мы
(редположим, что существует только одна собственная" функция
U» связанная с данным собственным значением энергии ei, т. е. что
;и один из энергетических уровней не вырожден (рис. 18.16).}
2^ =±=# (фиксировано), (19.1)
i
2 #*«, — £ (фиксировано). (19.2)
Сколько частиц Nt имеет данную энергию е< при
условии постоянства общего числа частиц N и
общей энергии £?
Чтобы ответить на этот вопрос, необходимо вернуться
(вкратце) к ситуации задачи о бросании монеты (о мешках и шарах или
^случайном перемещении) в гл. 6. Там мы рассматривали задачу
■ подбрасывании монеты N раз и определении вероятности того,
625
!
е5
е 4
N5
N3
626 ЧАСТЬ Б
Доли общего числа бросков с выпавшими
„ орлами "
Рис. 19.1. «Пикообразование» для относительной^вероятности получения данной
доли «орлов» при подбрасывании монеты N раз.
Обратите внимание на то, что при увеличении числа N становится все более вероятным
получить в отдельном эксперименте результат ближе к ожидаемому (среднему). [Взято с
изменениями нз Nash L. К., Elements of Statistical Thermodynamics, Addison-Wesley.
Reading, Mass., 1968, p. 11.]
что т раз из N бросков выпадает «орел». Было найдено, что
среднее число выпадения «орлов»^ равно N/2, но среднеквадратичное
отклонение изменяется как "|W. Графическое представление этой
задачи дано на рис. 19.1, где показаны относительные вероятности
получения данной доли бросков с выпавшими «орлами» как
функции относительной доли бросков, в которых «орел» действительно
выпал. В этот раз мы нормировали вероятность так, чтобы
наиболее вероятный результат (половина всех бросков — «орлы») имел
вероятность, равную единице. На рис. 19.1 показано, что по мере
того, как число бросков увеличивается, становится все более
вероятным то, что любой отдельный эксперимент дает результат,
близкий к среднему результату, так же как относительная точность
эксперимента по счету радиоактивности (гл. 8.А) увеличивается
при возрастании числа наблюдаемых отсчетов.
Согласно рис. 19.1, при очень большом числе бросков мы
получим наиболее вероятный (N/2 «орлов») результат, и можно с очень
высоким уровнем надежности не принимать в расчет результаты,
отличные от наиболее вероятного.
Похожая ситуация складывается и в задаче распределения
частиц по энергетическим уровням. Например, на рис. 19.2
проиллюстрирован простой (относительно!) случай трех гармонических
осцилляторов (грузы на пружинах) с установленной общей
энергией, состоящей из трех энергетических «квантов» по (7г) {К) (£/m)Va
КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
|аждый (гл. 18). В конфигурации № 1 один осциллятор имеет
Энергию 63 = 3 единицы, а два других — энергию ео = 0 единиц. Есть
&ри пути выбора одной из трех частиц с энергией ез, два пути
выбора двух частиц с энергией ео и единственная возможность для
Остающегося осциллятора иметь энергию е0. Однако безразлично,
£ каком порядке брать последние два осциллятора, и поэтому есть
^только 3!/2!= (3-2-1)/(2-1) =3 действительно различающихся
комбинаций, а вовсе не 31 = 6 комбинаций, которые были бы
возможны, если бы мы могли отличать осцилляторы один от другого.
Аналогично другие две конфигурации могут быть получены шестью
или одной различными комбинациями, как показано на рисунке.
В этом примере, поскольку число частиц ограничено величиной
N = 3, а общая энергия частиц £ = 3 единицы, наиболее вероятной
является конфигурация № 2 (поскольку существует больше
различных способов образования именно этой, а не любой из двух
Других возможных конфигураций). Как и в только что
рассмотренном примере с бросанием монеты, существует ярко выраженное
кпикообразование» в распределении вероятности около наиболее
^вероятной конфигурации, когда число частиц возрастает до очень
большой величины (скажем, до 1020 молекул). Таким образом, при
Конфигурация №} Конфигурация М-2 Конфигурация Л/-3
е3 = 3 *
еге2-
е- * 1 ■
е0 = 0—*—*-
XXX
У ч у-
3—* —*— х—
_^ч_
О—**— * х- —им— —и —м *— —*— *_ч ц- •
яЬе abc abc abc abc abc abc abc abc abc'
3-2*1
Э!/2!= 2§1 "3 3! = 3-2-1=6 31/3! -1
1Рис. 19.2. Конфигурации .(верхние схемы) и число комбинаций, приводящих
(к этим конфигурациям (нижние схемы), для системы из трех (а, Ь, с)
идентичных гармонических осцилляторов (N=3) с фиксированной общей энергией
Е=3 единицы.
^Конфигурация просто показывает, сколько частиц занимают каждый энергетический
уровень. В атом примере конфигурация № 2 является наиболее вероятной, поскольку
возможностей ее образования больше, чем любой другой конфигурации. Когда число частиц ста-
i ловится очень большим, наиболее вероятная конфигурация приобретает подавляюще
большую значимость по сравнению со всеми другими вариантами, и ее называют
распределением Больцмана [см. уравнения (19.3)].
628 ЧАСТЬ Б
рассмотрении большого числа молекул свойства системы можно
определять с предельно большой точностью, исходя из наиболее
вероятного распределения частиц по энергетическим уровням.
Можно показать (см. литературу к этой главе), что наиболее
вероятное распределение общего числа частиц N по различным
энергетическим уровням еь ег, ... при условии, что общая энергия Е
постоянна [уравнения (19.1) и (19.2)], имеет свойства
NJN, = ехр [- ptJ/exp [- реу] = ехр [- р (t, -t,)], (19.3a)
так что
NifN= exPbM t (19,3б)
t
где Nt и Nj представляют собой число частиц на энергетических
уровнях. Bi и Bj, а р — константа. Сравнив классические и квантово-
механические свойства идеального газа в закрытом сосуде (см.
задачи к этой главе), мы обнаружим, что константу р можно
отождествить с (1/kT). (Этот статистический вывод на практике иногда
используют в качестве определения температуры, в чем мы скоро
убедимся при описании эксперимента с лазером,) Уравнения (19.3)
выражают в этом случае «распределение Больцмана»:
Ni/Nj= ехр [— (ег — tf)/kT], к — постоянная
Больцмана
Ni/N — ехр [—ti/kT] I 2 ехр [—tt/kT] = Г—абсолютная
/ ' температура
q — функция
= (1/<7)ехр [—ei/kT) распределения
Основываясь на распределении Больцмана, температуру
считают «низкой», когда большинство частиц имеет энергию гораздо
большую, чем тепловая энергия (z%^>kT), а.«высокой»
температуре соответствуют частицы с энергией гораздо меньшей, чем
тепловая энергия (Ei<g.kT). При «низкой» температуре большинство
частиц будет находиться на самом низком энергетическом уровне
(уровнях), в то время как при «высокой» температуре
большинство энергетических уровней будет заселено равномерно, (рис. 19.3).
Из примеров с энергетическими уровнями, которые мы до сих пор
рассматривали, условие «высокой» температуры имеет место в экс^
периментах по магнитному резонансу, тогда как условие «низкой»
температуры осуществляется для экспериментов в инфракрасной,
видимой и ультрафиолетовой областях, (табл. 19.1). В любом слу.-
чае, когда система находится в тепловом равновесии с
окружающей средой (в рассматриваемых нами случаях — почти всегда),
как видно из рис. 19.3, на более низких энергетических уровнях
(jy.taj
(19.46)
(19.4в)
Таблица ЙС.
Относительные заселенности двух соседних энергетических уровней
для различных типов природных осцилляторов [уравнение (19.4а)]
Природа
осциллятора
* / \ Относительные заселенности ^х/Л^о самого низкого и следующего.
Типичная собственная Разность энергии (ei—во). более высокого энергетических уровней для разных температур
частота осциллятора связанная с частотой, где ^
(еа—e0)=/iv. эрг ' : i
комнатная <295 К) N2 (жидк.) (77 К) Не (жидк.) (4,2 К)
2000 А = 1,5-10» ГЦ
10-"
Ю-1о«^о
Ю-ш^о
\0-1Ш^о
1500 см-1 = 4,5-10» Гц
310-
13
0,00066
7-10-MsO
lQ-238^0
Продолжение табл. 19.1
Природа
осциллятора
Типичная собственная
частота осциллятора
Относительные заселенности N\INq самого низкого и следующего.
Разность энергии (ei—ео). более высокого энергетических уровней для разных температур
связанная с частотой, где
(ei—eo)=f»v, эрг
комнатная (295 К) Na (жидк.) (77 К) Не (жидк.) (4.2 К)
65,8 ГГц (электронный
парамагнитный ■ резонанс
в магнитном поле
23,5 кГс)
4,4-10-и
0,989
0,960
0,471
ядра
100 МГц (протонный
магнитный резонанс в
магнитном поле 23,5 кГс)
6,6.10-»
0,99998
0,99994
0,9989
1631 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
С2 —
ei
ео
.Низкотемпературный
преде/1
Л.
1,0
ез
*2
61
*о
Промежуточная
температура
I
1,0
ез
С2
ei
во
высокотемпвратурпй/И
предел
2±
N
К
1,Q
i
N;
(
N
Рис. 19.3. Относительные равновесные заселенности Ni различных
энергетических уровней 8,- для трех соотношений а и тепловой энергии kT; кизкотемпе-
ратурный предел ei^>kT; промежуточная температура, e&kT;
высокотемпературный предел Zi<.kT.
В табл. 19.) можно найти примеры для каждого случая. Относительные заселенности
получены по уравнению (19.4а).
будет находиться больше частиц, чем на более высоких
энергетических уровнях.
Во второй половине этой главы говорится о том, что табл. 19.1
частично объясняет, почему магнитное резонансное поглощение
гораздо слабее, чем инфракрасное или оптическое поглощение.
Поскольку интенсивность поглощения зависит от разности
заселенности более низкого и более высокого энергетических уровней,
протонное магнитное резонансное поглощение является слабым
(например, из каждых 100 000 протонов на более низком
энергетическом уровне находится около 50001 и около 49 999 —на верхнем
уровне, что дает разницу заселенностей между двумя уровнями
только около 0,002%)- Оптическое же поглощение является
сильным из-за того, что почти все оптические осцилляторы пребывают
на самом нижнем энергетическом уровне.
19.А. ЛАНТАНОИДНЫЕ СДВИГАЮЩИЕ РЕАГЕНТЫ
В СПЕКТРОСКОПИИ, ЯМР И КОНФОРМАЦИЯ МОЛЕКУЛ
В РАСТВОРЕ
Механизм действия и использование лантаноидных
«сдвигающих» реагентов в спектроскопии ЯМР дают хороший (и важный)
пример, касающийся почти всего, о чем говорилось выше в этом
разделе. Лантаноидные ионы образуют аддукты 1:1с множеством
нуклеофильных «донорных» соединений, включая ROH, RNH2 и
RP04H~. Поскольку ионы лантаноидов (за исключением самого
лантана) являются парамагнитными (обладают электронным
магнитным моментом), существует небольшое магнитное поле, свя-
632 ЧАСТЬ 5
за иное с магнитным моментом лантаноида. Это магнитное поле
будет складываться (или вычитаться) с магнитным полем,
действующим на каждое из ядер, расположенных поблизости от
лантаноидного иона, в соответствии с тем, будет электронный спин
направлен «вверх» или «вниз» (ср. с рис. 18.8). Однако направление
электронного «спина» не постоянно, и, как мы увидим в гл. 19.Б,
направление спина быстро меняется «вверх-вниз». Таким образом,
небольшое магнитное поле, связанное с этим направлением
электронного спина, также быстро меняет знак, и общее эффективное
магнитное поле (приложенное магнитное поле плюс или минус
небольшое магнитное поле, обусловленное электронным спином
лантаноида) у данного близлежащего протона усреднится до единого
эффективного магнитного поля, величину которого определяет
относительное число электронов в двух возможных электронных
«спиновых» состояниях лантаноида. Наконец, поскольку в
состоянии с более низкой энергией находится чуть-чуть больше
электронов, чем в состоянии с более высокой энергией, средний электрон-
Направление
спина
электрона
Т
e%*Ho/2kT
Рис. 19.4. Схема возникновения индуцируемых лантаноидом «сдвигов» линий
ядерного магнитного резонанса.
Данный протон в окрестности иона лантаноида «вндит» одно из двух возможных магнитных
полей (/ или 2) в соответствии с тем, направлен электронный спин лантоиоида «вверх»
«ли «вниз». Поскольку направление электронного спина быстро меняется «вверх-вина»,
магнитное поле вблизи протона усредняется до единственного эффективного значения (5).
Кроме того, поскольку нижний энергетический уровень занимает несколько большее число
электронов, чем верхний (табл. 19.1), среднее магнитное поле будет слегка сдвинуто по
отношению к величине приложенного внешнего поля Но (см. задача 14.6).
/ — суммарное магнитное поле при электронном спине |; 2 —суммарное магнитное поле
яри электронном спине f. 3 —быстрое изменение магнитного поля нз-за изменения
направления спина электрона; 4 — лантаноидный сдвнг ДЯ; б — среднее эффективное магнитное
поле; 6 — относительные заселенности энергетических уровней электронного спина.
Z КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
Лантаноид q
Рис. 19.5. Параметры гиб для комплекса лантаноидного иона с монофосфатом
[" нуклеотида.
'(
[Взято с изменениями из Barry С. D. et at., Nature, 232, 237 (1971).]
йый магнитный момент будет отличен.от нуля и эффективное
среднее магнитное поле на близлежащем протонном ядре будет
слегка сдвинуто по величине относительно внешнего магнитного поля
по (отсюда название «сдвигающий», или «шифт-реагент») (рис.
|9.4).
I Причина, по которой все сказанное выше оказалось весьма
полезным практически, состоит в том, что величина индуцированного
|сдвига» связана с геометрией комплекса между лантаноидом и
^рлекулой-донрром уравнением (19.5). Поскольку длины связей
^нтаноид — донор хорошо известны, для определения
конфигурации дрнорной молекулы могут быть использованы относительные
индуцированные сдвиги для различных ядер в той же самой до-
)рной молекуле, как показано в примере на рис. 19.5.
Afl/fl0:= (const) З^6-1 (19.5)
а.е г — расстояние между ионом лантаноида и интересующим нас
дром в донорной молекуле, а 0 —угол между осью связи ланта-
ид — донор и вектором лантаноид — исследуемое ядро (см. ниже
имер с нуклеотидом). Поскольку константа имеет одно и то же
ЦКачение для различных протонов в одной и той же донорной моле-
Руле, отношения наведенных сдвигов для различных протонов в
|олекуле будут отражать различия в 0 и г двух соответствующих
|ротонов. Сравнивая наблюдаемые отношения наведенных сдвигов
| отношениями, рассчитанными для различных возможных
молекулярных конформаций (см. следующий ниже пример), можно оп-
(рделять преимущественную конформацию молекулы в растворе.
| На рис. 19.6 показаны протонные спектры ЯМР аденозинмоно-
юсфата (AMP) в отсутствие (вверху) и присутствии (внизу)
Ьна Еи3+ в качестве сдвигающего реагента. Ясно видно, что сиг-
&лы ЯМР разных протонов сдвинуты по-разному (например,
^ижайший к иону лантаноида 5'-протон сдвинут больше всего).
I. Очевидной проблемой при попытке сравнить наблюдаемые и
несчитанные отношения сдвигов является наличие в молекуле
634 ЧАСТЬ Б
Рис. 19.6. Протонные спектры ЯМР AMP (структура показана сверху) в
отсутствие (вверху) и в присутствии (внизу) лантаноидного сдвигающего
реагента — иона Еи3+.
Я-пнки представляют собой приборные артефакты, и учитывать их не следует. Величины
углов Фг — ф4 определяют расположение групп при повороте вокруг каждой из четырех
лабильных (нежестких) связей. [Из Barry С. D. et ah, Nature, 232, 237 (1971).]
AMP четырех вращательно лабильных связей. Поэтому для того,
чтобы сделать возможной интерпретацию полученных данных,
необходимо рассмотреть все возможные конфигурации,
соответствующие разным значениям углов ф (рис. 19.6), через, скажем,
каждые 4°. При четырех возможных вращательно лабильных связях
число конфигураций, которые надо рассмотреть, составляет
(360°/4°)4 = 66-106, т. е. очень большое число! К счастью, есть
способ исключить большинство из этих возможностей, отбросив все
конформации, при которых хотя бы два атома сближены друг с
другом больше, чем на расстояние их вандерваальсовых радиусов.
Эта процедура «фильтрования» оставляет около 32 000 возможных
конфигураций. Следующим шагом является исключение всех
конфигураций, несовместимых с наблюдаемыми отношениями наве-
135 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
Идейных лантаноидом сдвигов для различных сдвинутых протонов,
|ito уменьшает число возможных конфигураций до 195. Наконец,
|после сравнения наблюдаемой и рассчитанной ширины линий
прогонного ЯМР в присутствии Gd3+ (как в примере с NAD:NADH,
•рис. 16.24) остается только 12 очень близких возможных
конфигураций. Усреднение этих 12 слегка отличных конформаций дает
Структуру (рис. 19.7), очень близкую к той, которая была найдена
[у кристаллической формы AMP при использовании рентгенострук-
турного анализа (гл. 22). Некоторое представление о значимости
этого метода можно получить из того факта, что публикация этого
примера в журнале «Nature» была одной из наиболее часто
цитируемых научных статей в мире через год после ее опубликования.
"С тех пор метод был усовершенствован (изменились некоторые из
первоначальных выводов рис. 19.7), но тем не менее развитие
этого подхода должно быть полезно при установлении структуры в
растворе широкого круга биологически важных метаболитов,
гормонов и лекарств.
19.Б. ПЕРЕХОДЫ МЕЖДУ ЭНЕРГЕТИЧЕСКИМИ
УРОВНЯМИ
19.Б.1. ОБЩИЕ ФОРМУЛЫ
В гл. 18 был дан способ определения квантовомеханических
ролновых функций и энергетических уровней некоторых отдельных
Независимых от времени гамильтонианов (некоторых отдельных
Г
AMP
(кристаллы)
AMP
(раствор)
ряс. 19.7. Стереоизображение конфигурации молекулы AMP в кристаллах,
Определенной методом рентгеноструктурного анализа (вверху), и в растворе,
частично определенной при использовании лантаноидных сдвигающих реагентов
(внизу).
гГИэ Barry С. D„ North А. С. Т., Glasel J. A., Williams R. J. P., Xavier A. V., Nature, 232,
236—245 П97П.1
636 ЧАСТЬ Б
типов потенциальной энергии). Однако не было найдено способа
для того, чтобы установить, как данная частица может изменить
свое состояние на другое состояние с отличной энергией (перейти
с одного энергетического уровня на другой). На самом же деле,
поскольку для независимого от времени гамильтониана в
отсутствие электрического и магнитного полей мы исходим в нулевой
момент времени из системы в данном собственном энергетическом со-
стбянии Ф„, то уравнение (18.31) предопределяет, что состояние
ip(f) в любой более поздний момент времени примет вид
ехр[-(ЗДедФя,
где Еп — энергия в собственном состоянии Фп- Вероятность найти
систему в первоначальном состоянии Фп через время t
представляет собой просто квадрат проекции волновой функции
упц)=;е-{11П)Еп'фп (!9.б)
в момент времени t на Фп = ^п(0):
I < % (0. *« (0) > I" = К е~"Ь)Еп'фп, Ф„ > |- =
= е[-{ИЬ)Еп'^МЕгЛ\<фпГФпу\\
|Ш0. <Ы0)>|'=!, (19.7)
где предполагается, что первоначальная собственная функция Ф„
нормирована. Уравнение (19.7) подтверждает, что, если система
находится на каком-либо данном энергетическом уровне в
нулевой момент времени, она все еще будет оставаться там в течение
неопределенного времени t. [Можно было бы легко обобщить
уравнение (19.7), чтобы показать, что даже если система в исходном
состоянии описывается некоторой линейной комбинацией
(взвешенное усреднение) собственных энергетических состояний, то
вероятность нахождения этой системы в любом энергетическом
состоянии через время / будет точно такой же, как и в нулевой
момент времени.]
На рис. 19.8 схематически показаны механизмы, по которым
осуществляются переходы. Все механизмы каким-либо образом
связаны с наличием возмущающего, изменяющегося во времени
электрического или магнитного полей на заряженных частицах, из
которых состоит вещество образца. Индуцированное поглощение,
индуцированное испускание и спонтанное (самопроизвольное)
испускание соответствуют захвату (индуцированное поглощение) или
высвобождению (индуцированное или спонтанное испускание)
фотона, энергия которого связана сротношеиием
Вт — Ек — fl\
h — постоянная Планка = по я\
= 6,62-10" эрг-с 1У j
637 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
и
W
W
л
Щи.
' ''
Wf
•П
'ч
Рис. 19.8. Схемы различных механизмов переходов между энергетическими
собственными состояниями системы с двумя состояниями.
Wm _, п и Wn _,m представляют собой константы скоростей для нспускання и поглощения
(фотона), индуцированных переменным электрическим или магнитным Полем, частота
которого v задается уравнением (19.8): Ет—ЕпшAv. Wqh — константа скорости для «спон»
тайного» нспускання. Wj и W i — константы скоростей для безызлучательного поглощения
или испускания энергии Ет—Еп. вызванного взаимодействием системы с другими
частицами. Показано (см. текст), что Wm^,n -W„_»m, на Wj/Wj «exp(—{Em — En)lkT\
[уравнения (19.29) н (19.33)1. Спонтанное излучение существенно главным образом иа оптических
частотах, где оно предопределяет максимальную скорость флуоресценции. В этом разделе
рассмотрены скорости индуцированных процессов поглощения и испускания. Скорости
безызлучательиых процессов поглощения и нспускання наиболее легка вычисляются для
спектроскопии на радиочастотах (гл. 19.Б.З н 21.Б). Nn и Nm обозначают число Частиц
иа энергетических уровнях Еп н Ет (см. текст).
с разностью энергий между двумя энергетическими уровнями
системы. Остающиеся безызлучательные поглощение и испускание
соответствуют захвату или высвобождению количества энергии
\Ет—Еп\, когда поглощенная или испущенная энергия
соответственно отдается или поглощается другими молекулами так, что в
процессе не участвуют фотоны.
Поскольку механизм индуцированных процессов поглощения и
испускания по существу один и тот же для всех форм
спектроскопии, в этом разделе мы рассмотрим его в первую очередь. Мы
кратко покажем, что прочие механизмы переходов обеспечивают
возможность установления равновесного распределения Больцма-
на для заселенностей энергетических уровней. Вероятность
спонтанного излучения можно рассчитать [2]. Заметим только, что она
пропорциональна \Ет—Еп\г. Таким образом, оказывается, что
спонтанное излучение пренебрежимо мало для низкоэнергетической
спектроскопии (т. е-. ЯМР). Более того, скорости безызлучательных
переходов в этом диапазоне энергий часто можно рассчитать (гл.
21.Б), и поэтому в этой главе мы рассмотрим радиочастотные не-
реходы несколько позже. В оптической области
(ультрафиолетовая, видимая, инфракрасная) рассчитывать скорости
безызлучательных переходов предельно трудно, и мы ограничим наше
обсуждение оптических переходов качественным анализом лазеров в
следующем разделе этой главы.
Мы начнём с индуцированных переходов, которые получаются в
результате введения изменяющейся во времени энергии [такой,
как изменяющаяся энергия взаимодействия между (переменным)
638 ЧАСТЬ Б
электрическим полем монохроматического электромагнитного
излучения и (постоянным) электрическим дипольным моментом
молекулы]. Обозначив зависимую от времени энергию как
возмущение Ж'((), добавленное к независимым от времени кинетической и
потенциальной энергиям Ж°((), можно записать зависимую от
времени волновую функцию дЯя системы [уравнение Шредингера
(18.25)] в виде
ih
dV(t)
dt
={&e*+w (t))V(t)
(19.9)
В общем виде уравнение (19.9) может быть решено при
условии, что возмущение Ж'(t) либо быстрое, либо малое. Если Ж' (t)
мало, то простейшими являются случаи, когда возмущающее
воздействие постоянно (исключая моменты его «включения» и
«выключения») и когда оно либо периодически, либо беспорядочно
меняется во времени. В этом разделе мы рассмотрим действие малого
периодического возмущения энергии, получающегося в результате
воздействия электромагнитного излучения на квантовомеханичес-
кую систему. Случай беспорядочно изменяющегося воздействия
обсужден в гл. 21 вместе с шумовыми явлениями. При условии что
возмущающее воздействие Ж'(t) мало, возможно выразить
волновую функцию в более поздний момент времени t через
энергетические собственные функции невозмущенного гамильтониана Ж0
w (0 =ct (t) «мо + сг (о ф, (/) + ...+ сп (t) ф„ (О
согласно уравнению
ччо
00
= SCn {t) *«{t)
я=1
00
= Зся(0^-(//А)£«.гФя
п=1
медленно быстро изме-*
изменяю- няюшаяся
щаяся временная
временная зависимость,
завнси- обусловлен-
мость, ная невозму-
обуслов- щенным га-
ленная мильтониа-
№(t) ном 96*
(19.10)
В уравнении (19.10) задача упрощена выделением быстро
изменяющейся временной зависимости (i|)n(0 =el~l{ ft)£V]G>n),
обусловленной невозмущенным гамильтонианом [уравнение (19.6)], в
отдельный сомножитель от гораздо более медленно изменяющегося
во времени сомножителя, обусловленного гораздо меньшим Ж'{1).
639 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
Подставляя выражение для ^(t) из уравнения (19.10) в
уравнение (19.9), получим
ih ^рм-ы+ihYfn со^-Фгг (о=Е сп (о ww)+2 сп№'«т).
п п п п
(19.11)
Но, поскольку уравнение Шредингера [уравнение (18.25)] для
волновой функции i|>n(0 невозмущенного гамильтониана Жй имеет
вид
два средних члена в уравнении (19.11) сокращаются, давая
йЕ^ФЛО^Х'ЛОапОФЛО. (19.13)
п п
По причине, которая вскоре станет ясной, мы теперь возьмем
проекции векторов, выраженных каждой стороной уравнения (19.13),
на невозмущенную базисную функцию г|>т(0
< ф™ (th ^ Е %г- <мо > =ih Е ^г- < *» (0. ф» (о->=
(19.14а)
где мы используем свойство ортогональности собственных
функций Ж [ср. с уравнениями (18:69)]
и <<ыо. 2мо#'(о<ыо> = 2*я(0<<мо. #'(0<ыо>=
-S^(0^««(0 (19.146)
л
(m-я строка и /г-й столбец матрицы представления оператора Ж it)
в базисном наборе г|>п(0-)
Из уравнения (19.14а) видно, что действие этой процедуры
состояло в выборе временной зависимости только одной из амплитуд
волновой функции dcm(t)ldt. Приравнивая затем уравнения
(19.14а) и (19.146), получим желаемый общий результат
^—A-Emo*w<o
(19.15)
Уравнение (19.15) образует систему из п дифференциальных
уравнений (по одному для каждого выбранного т), каждое из которых
640 ЧАСТЬ Б
описывает поведение во времени, амплитуды для одной из
первоначальных (невозмущенных) собственных функций энергии. Зная
данный набор начальных условий (например, ci(0) = l, а все
другие d(0)=0 в нулевой момент времени^т: е. система
первоначально находится на самом низком энергетическом уровне) и зная
конкретное, вполне определенное зависимое от времени возмущающее
воздействие Ж'{1), мы можем, решая уравнение (19.15) для_
получения каждого из cm(t), найти относительные заселенности
Cm{t)cm{t) каждого из энергетических уровней в момент времени t,
как это показано в имеющих принципиальное значение
последующих вычислениях.
Индуцированное испускание и поглощение.
Переходы, индуцированные переменным электрическим полем
(электромагнитным излучением)
В гл. 14.А рассматривалось поглощение электромагнитного
излучения с точки зрения классической механики; согласно которой
вещество состоит из электронов на пружинах, а излучение
представляет собой переменное электрическое поле. В этом разделе
будут использованы выведенные ранее выражения амплитуд
энергетических собственных функций, чтобы вычислить «вероятность
перехода» для поглощения (или испускания) электромагнитного
излучения, падающего на квантовомеханическую систему. Этот
расчет обеспечивает основу для понимания механизма действия
лазера, а также явлений «насыщения», которые могут быть
использованы для определения «времен релаксации» для
спектральных энергетических уровней. Затем будут описаны биологические
применения таких явлений.
Для простоты записи предположим, что система первоначально,
т. е. в нулевой момент времени, находилась в состоянии с самой
низкой энергией («основном» состоянии) е вероятностью, равной
единице (рис. 19.9). Затем образец помещают в электромагнитное
поле, создавая зависящее от времени возмущение энергии 3@'(t).
Теперь цель состоит в том, чтобы рассчитать вероятность
нахождения системы через какое-то время t на любом другом
энергетическом уровне. . '
Ограничиваясь рассмотрением плоскополяризованного
монохроматического электромагнитного излучения с частотой со и
амплитудой электрического поля 2Ео и пренебрегая (как в гл. 14.А)
магнитной компонентой излучения, можно написать выражение
для электрической компоненты излучения
£x = 2£ecos(arf) (19.16а)
или, что более удобно, в комплексной форме
Ех = Е,[ехр(Ш) + ехр(—Ш)\. (19.166)
641 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
а
I
с*(0)сп(0) = 0
. „ Действие J€'(t)
i venue времени
с3*(0)с3(0) = 0
с2*(0)с2(0) = 0
с,*(0)с,(0) = /
s me-
t
^s f
Энергия —
*п
•
»
•
*3
€,
c*(t)c„(t)«l
c?(t)c3(t)«i
C2*lt)Cz(t)<<1
Рис. 19.9. Схема энергетических уровней и относительных заселениостей этих
уровней перед (слева) и после (справа) наложения электромагнитного поля,
действующего непрерывно в течение интервала времени t
Система полностью находится в своем основном состоянии в нулевой момент времени
[при /=0 ci(0)-l, av все остальные Cj„(0)-0j. Возмущающее действие электромагнитного
поля достаточно мало, так что заселенность основного состояния остается по существу
постоянной в течение времени t. Мы вычисляем амплитуды ст(0 каждой из остальных
энергетических собственных функций, чтобы получить вероятность c^iO)eM(t) вайтя систему
на т-и энергетическом уровне через время г.
Относительные заселенности энергетических уровней (а) в нулевой момент времени и (б)
через время / после начала действия электромагнитного излучения.
Энергию взаимодействия этого излучения с электрическим диполь-
ным моментом р,х некоторой молекулы или атома можно записать
так:
энергия взаимодействия=(электрическое поле)X(электрический
дипольный момент)
Я'(') = *,-14с.
или
Я' (0 = ^ [ехр (№) + ехр (— fcrf)], (19.17)
где цх= (заряд) • (х), а х—расстояние, разделяющее два заряда
диполя.
Компонента электрического
поля монохроматического
плоскополяризоеанного
электромагнитного излучений
Электрический
дипольный момент
атома или молекулы
0
После получения подробного выражения для изменяющейся во
времени возмущающей энергии &$'{$) остается только подставить
642 часть б
уравнение (19.17) в уравнение (19.15) и решить получившееся
уравнение, используя начальные условия
са (0) = 1, сст(0) = 0 при/rt^l. (19.18)
Подставляя, получим
^ЭГ^жХ *»<0<Ф»('). Е#х(ем + е~М)}ят=* (19.19а)
п
но
и
Ъп«) = е {1П)Ет*Фт (19.20а)
%У) = е~Ш)Вп'фп [из уравнения (18.31)]. (19.206)
Подставляя уравнения (19.20) в уравнение (19.196) if за способом
преобразования отсылаем к уравнению (18.24)], получаем
*-Ш=|.[,"•'*"* "^-•""<ФЯ> ,чФ^+
+ «'-'"'+,'/ft,M<W,<M. (19.2!)
Обозначая тп-к матричный элемент оператора \ix в базисном
наборе Фт как
(|xj_ —(фт, }л,хфп) — (электрический) дипольный момент перехода
(19.22)
х гт
(сейчас у нас нет необходимости останавливаться, чтобы оценить
его величину), можно упростить уравнение (19.21) до следующего
вида:
dCm{t)__E, (f/ft) <Bm-Bn+l») t , («7ft) (Em-En-hv)t
~t Jh\Px)mn[e ~Te V (Jy.^o)
Уравнение (19.23) дает мгновенную скорость изменения по
амплитуде собственной функции cm{t) для m-го энергетического уровня.
Для того чтобы определить суммарное изменение, в cm(t) после
того, как 2%>'(t) было приложено в течение времени t, необходимо
643 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
проинтегрировать уравнение (19.23) от нуля до t в соответствии с
рис. 19.9, что и приводит к желаемому результату:
£m-'£„ + Av + Em-En-hv j
Индуцированное испускание* Индуцированное поглоще-
излучения при ние* излучения при ftv -*
ftv-> — (Ет — Еп), где £«>£„ -> (Ет~Еп), где Ет>Еп
(19.24)
У уравнения (19.24) есть две очень важные особенности.
Во-первых, оно показывает, что скорость переходов [а отсюда и величина
cm(t)] между m-м и /г-м энергетическими уровнями будет заметной,
только когда один из знаменателей в уравнении (19.24) станет
малым (числители меняются в пределах от 0 до 2), так что
именно знаменатели оказывают наибольшее влияние на cm(t)*.
Знаменатели же становятся малыми, либо когда энергия излучения очень
близка к энергии, инициирующей переход частицы с уровня Еп на
более высокий энергетический уровень Ет (поглощение), либо
когда энергия электромагнитного излучения очень близка к той,
которая высвобождается при переходе частицы вниз по энергии из
состояния Ет в состояние Еп (испускание), как указано в
объяснениях к уравнению (19.24). Процесс поглощения, несомненно,
хорошо известен большинству читателей, тогда как индуцированное
испускание (вынужденная эмиссия) — явление новое. Другими
словами, когда на нижнем энергетическом уровне Еп находится
больше частиц, чем на верхнем уровне Ет, амплитуда внешнего
излучения будет уменьшаться вследствие поглощения. Но когда
станет больше частиц не на нижнем уровне Еп, а на верхнем
уровне Ет, амплитуда внешнего излучения будет, наоборот,
увеличиваться при прохождении его через образец! На индуцированном
испускании излучения и основаны мазеры и лазеры (см.
следующий раздел). Вторая главная особенность уравнения (19.24)
состоит в том, что коэффициенты первого и второго членов одинаковы.
Это просто указывает на равную вероятность (см. следующий раз-
* Из уравнения (19.24) может показаться, что, когда частота
«возмущающего» излучения будет действовать точно «в резонанс», т. е. h\=\Em—Е„\,
амплитуда пг-й функции базисного набора cm{t) станет бесконечной. Этот
феномен аналогичен классической модели движения груза на пружине, амплитуда
которого также увеличивается неограниченно, когда незадемпфированную
пружину раскачивают точно в резонанс. Как и для классической пружины, эта
аномалия исчезает, когда мы вводим ненулевой демпферный член (в нашем случае
спонтанное испускание или безызлучательный переход). При этом всегда будет
существовать конечная (т. е. не бесконечно малая) вероятность найти
«возбуждаемый» электрон на каком-нибудь одном из энергетических уровней,
соответствующем данной базисной функции состояния невозмущенного гамильтониана
(энергетическом уровне при отсутствии каких-либо воздействующих полей).
644 ЧАСТЬ 5
дел) вынужденных переходов «вверх» и «вниз» (это свойство
существенно также при расчете лазеров). Наконец, для того чтобы
переход осуществлялся с определенной конечной вероятностью
[т. е. был «разрешен» (cm(t)ФО)], совершенно необходимо, чтобы
матричный элемент оператора электронного дипольного момента
был отличен от нуля (ц*т ^0). Таким образом, чтобы определить,
какие из многих возможных переходов на самом деле
«разрешены», всегда необходимо исследовать цх (гл. 19.Б.4). Часто выбор
легко сделать на основе простых «правил отбора» (см. ниже).
19.Б.2. ИНВЕРСИЯ ЗАСЕЛЕННОСТИ. ЛАЗЕРЫ И ИХ ПРИЛОЖЕНИЯ
Лазеры и мазеры [название составлено из начальных букв слов
фразы Light (или Microwave) Amplified Stimulated Emission of
Radiation] основаны на испускании излучения, индуцированного
(стимулированного) внешним электромагнитным излучением на
частоте, соответствующей разности энергий между двумя участвующими
в процессе энергетическими уровнями [первый член в уравнении
(19.24)]. Чтобы понять этот процесс, мы сначала должны найти
относительные заселенности каждого из энергетических уровней
после облучения системы. Эти относительные заселенности мы
сразу получаем из уравнения (19.24):
С\ W Cm (t) =* £\ (рж)тш {- (£ДД,-.М» -'
(19.25а)
где переход от уравнения (19.25а) к (19.256) осуществлен путем
преобразований с использованием хорошо известных
тригонометрических тождеств (см. задачи к этой главе) и при рассмотрении
только второго члена уравнения (19.24) (процесс поглощения).
Поскольку поглощение, очевидно, происходит во всем диапазоне
частот воздействующего, излучения v для получения общего
поглощения, соответствующего переходам от Еп к Ещ, необходимо
суммировать поглощение по всем частотам падающего излучения:
sin
ffi{Em-En-hv)t
]o\{t)om[t)dv^E\{K)mn^ {Em-En-H,y V О9'26)
где мы предполагаем, что источник внешнего излучения является
однородным («белым» — дающим излучение, амплитуда которого
не зависит от частоты) для очень узкого частотного диапазона, в
$45 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
^ _
пределах которого подынтегральная функция заметно больше ну-
!ля. Наконец, поскольку можно пренебречь поглощением на
частотах, сильно отличающихся о-т hv=Em—Еп, мы можем
распространить пределы интегрирования до ±оо и затем использовать
определенный интеграл
00
f^djc = « (19.27)
—00
для упрощения уравнения (19.26) до вида
I
*„«№№>= ±Е>,(\>Х) Н, (19.28)
тп
Значение уравнения (19.28) заключается в том, что оно
показывает пропорциональность времени для вероятности перехода из
состояния Еп в состояние Ет (т. е. поглощение фотона); поэтому для
данной молекулы при данной амплитуде внешнего
воздействующего излучения вероятность перехода для индуцированного
поглощения за единицу времени Wn—».m является величиной постоянной.
Преобразуя подобным образом первый член уравнения (19.24)
{индуцированное испускание), можно показать, что вероятность
индуцированного испускания за единицу времени Wm—►» задается
той же самой константой:
w =w
=W. (19.29)
Этот принципиальный результат [уравнение (19.29)] позволяет
поставить вопрос: почему мы обычно наблюдаем (индуцированное)
поглощение, а не (индуцированное) испускание при облучении
образца? Теперь можно ответить на этот вопрос.
Рассмотрим два энергетических уровня, о которых идет речь
(Еп и Em, показанные на рис. 19.8).-Относительные заселенности
этих уровней (число частиц на каждом уровне) составляют Nn и
Nm. Тогда число вынужденных переходов «вверх» будет Wn—+mNn
в секунду, а число вынужденных переходов «вниз» Wm—>nNm в.
секунду, и, таким образом, скорость изменения заселенностей Nn
и Nm двух энергетических уровней описывают выражения
dNnldt = Wm^nNm-Wn^mNn = W(Nm - Nn), (19.30a)
dNJdt^Wn^mNn-Wm^nNm = W(Nn-Nm), (19.306)
в которых при получении второго равенства.в каждом случае
использовано уравнение (19.29). Возьмем разность между
уравнениями (19.30а) и (19.306)
dJ^_dJ^ = d{Nn-Nm) =„W{Nn_Nm)t (I9#31)
646 ЧАСТЬ 5
9
Интегрируя уравнение (19.31) по времени, в течение которого
внешнее излучение воздействовало на систему (от t=0 до t = t),
обнаружим, что различие в заселенности (Nn—./Vm) между двумя
затронутыми уровнями описывается уравнением
№ - #«),=, = (*„ - AU-*
—'2Wt
(19.32)
В гл. 19.А уже говорилось о том, что для системы,,
находящейся в тепловом равновесии, выполняется соотношение
N
= е
п Тепловое
равновесие
(Em-Er)lkT
(19.4а)
Следовательно, поскольку большинство изучаемых систем
первоначально находится в тепловом равновесии (#п >#?»), уравнение
(19.32) предсказывает, что облучение вызовет больше переходов
«вверх», чем «вниз». Это создает результирующий эффект
понижения амплитуды внешнего излучения при прохождении его сквозь
образец (рис. 19.10). Таким образом, уравнение (19.32)
описывает в основном поглощение излучения веществом: даже несмотря на
то, что вероятность (константа скорости) "квантовомеханических
переходов «вверх» и «вниз» одна и та же, имеется больше частиц
*-т
"♦*]
а
£п-
"п>"т
"п<"т
6
Путь внутри
образца
Путь
внутри
образца
Рис. 19.110. Графическое представление выводов из уравнения (19.32).
Когда (как в условиях теплового равновесия) на нижнем энергетическом уровне находится
больше частиц, чем на верхнем, внешнее электромагнитное излучение вызывает переходы
в основном «вверх» и амплитуда проходящего через образец излучения со временем
уменьшается (А). Однако, когда (в неравновесных первоначальных условиях) больше частиц
находится на верхнем, а не на нижнем энергетическом уровне, внешнее излучение
вызывает переходы в основном «вниз» и амплитуда излучения по мере его прохождения через
образец в результате возрастает со временем (Б). Уравнение (19.32) показывает, что в
обоих случаях внешнее излучение действует так, чтобы уравнять заселенности двух
энергетических уровней. (В обоих случаях частота внешнего излучения соответствует разности
энергий между двумя уровнями.)
Л — вынужденное поглощение; Б — вынужденное испускание.
Суммарное действие электромагнитного излучения состоит в стимулировании переходов (а)
«вверх» (поглощение) и (б) «вниз» (испускание); в — электрическое поле внешнего
излучения.
i47 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
|в более низком энергетическом состоянии и, значит,
непосредственно реализуется больше переходов «вверх», чем «вниз». В более
^общем случае уравнение (19.32) предсказывает, что
индуцированное поглощение или испускание будет продолжаться до тех пор,
пока заселенности двух энергетических уровней не станут равны-
\ми независимо от того, какой из двух уровней с самого начала был
более заселен. Если бы (пока неизвестным нам способом) мы
смогли сделать так, чтобы в нулевой момент времени верхний
уровень был более заселен, чем нижний, то внешнее облучение
стимулировало бы переходы в основном «вниз» {испускание излучения
образцом) до тех пор, пока снова заселенности обоих уровней не
выравнялись бы (рис. 19.10). В этом случае амплитуда внешнего
излучения действительно возрастала бы при прохождении его
сквозь образец. Создание лазеров и мазеров основано просто на
оригинальных приемах для достижения такого дисбаланса
первоначальных заселенностей между двумя энергетическими уровнями,
чтобы в более высокоэнергетическом состоянии находилось
больше частиц. Тогда последующее облучение системы на частоте,
равной разности энергий между состояниями, приводит к
вынужденному испусканию, а отсюда и к усилению (увеличению) амплитуды
внешнего излучения.
Общее устройство лазеров
На рис. 19.11 показана типичная схема энергетических уровней,
на которой основано большинство лазеров. Поскольку степень
усиления лазером (т. е. амплитуда индуцированного испускания),
очевидно,, будет иаиболылей, когда заселенность нижнего уровня
энергии для перехода (Еп) будет в начальный момент близка к
нулю [уравнение (19.32)], желательно выбрать уровень Еп так,
чтобы он на несколько величин kT превышал энергию основного
состояния [уравнение (19.4а)] и чтобы при этом Nn<^Nm.
Работа лазера начинается с использования (вынужденного)
поглощения для увеличения заселенности («накачки») верхнего
состояния Ет или непосредственно облучением на частоте (Ет—
—EQ)]h, или облучением на частоте (Ер—E0)fh, за которым
следует некий спонтанный переход вниз до желаемого уровня Ет. Тогда
при действии излучения на частоте (Ет—En)/h будет происходить
вынужденное испускание, так как это излучение начнет
выравнивать заселенность уровней Ет и Еп.
Накачивающее облучение может оставаться непрерывным в
течение того времени, пока происходит желаемое вынужденное
испускание (лазеры непрерывного действия, используемые главным
образом в качестве лазерного «ножа» или как источник света в
спектроскопии высокого разрешения). В другом варианте сначала
используют процесс накачки для заполнения верхнего уровня Ет,
а затем этот уровень очень быстро опорожняют, применяя частоту,
648 ЧАСТЬ Б
I
1
А/рШО
Nm~0
Частота излучения
лазера
i> = (Em-En)/h
Nn = 0
N0*1
Рис. 19.11. Энергетические уровни (слева) и начальные относительные
заселенности (справа) системы, пригодной для использования в качестве лазера.
Вначале облучение проводят на частотах, приводящих к поглощению с заселением
состояний Ет или Ер (в последнем случае далее следует спонтанный переход на уровень Ет).
Таким путем возможно заселить уровень Е.т в гораздо большей степени, чем уровень £„.
Затем, используя электромагнитное поле с частотой. (Ет— En)/h вызывают на этой
частоте вынужденное испускание. При обеспечении быстрого перехода частиц, достигших
уровня Еп, на основной нижний уровень (волнистая линия внизу) можно поддерживать весь
процесс непрерывным.
Интенсивность вынужденного испускания будет максимальна, когда ЛГП » 0. Для этого
необходимо, чтобы Еп— Ей » kT.
стимулирующую излучение («импульсные» лазеры, используемые
для изучения очень быстрых реакций с участием
электронно-возбужденных молекул).
Наконец, поскольку необходимо иметь как можно большую
длину пути света внутри рабочего вещества лазера, чтобы получить
наибольший «выигрыш» в усилении вынужденного излучения
(рис. 19.10,5), но длина камеры для помещения этого вещества
ограничена, большинство лазеров сконструировано с отражателями
на обоих концах камеры. При такой схеме излучаемый свет много
раз проходит через вещество туда и обратно перед тем, как выйти
наружу сквозь (слегка прозрачное) зеркало на одном из концов
камеры. Чтобы отраженный свет не интерферировал с другими
световыми волнами в трубке и не ослаблялся из-за этого, расстояние
между зеркалами на двух концах рабочей камеры тщательно
выставляют на определенное число длин волн вынужденного
излучения, поэтому интенсивность лазера возрастает когерентно даже
при наличии зеркал. В этом случае источник накачки может
просто представлять собой лампу-вспышку, навитую вокруг
цилиндрической камеры с рабочим веществом (рис. 19.12). Применения
лазеров базируются главным образом на одном из следующих
свойств лазерных лучей: 1) высокой интенсивности, 2)
когерентности, 3) высокой монохроматичности (очень небольшом «разбросе»
частот в выходящем пучке) и 4) малой длительности излучения
?Й49 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
а
L
1<У\ЛЛЛР
А ! !
i i
i i
Л/2
I
б
Исходящий ^
^> когерентный
лазерный луч
L = п(Х/2)
Рис. 19.12. Схематическое изображение лазера.
Вначале от лампы-вспышки, навитой вокруг образца, в область между зеркалами (в
«резонатор» лазера) подводят энергию («накачка»), заселяя уровень Ет (рис. 19.11). Когда
подводят свет с частотой (Ет—En)lh (обычно все начинается со спонтанного испускания
с уровня Ет на уровень Еп), он стимулирует последующие переходы на этой частоте, и.
если длина резонатора лазера выбрана правильно (см. рисунок), фаза каждой из этих
воли сохраняется при отражении от любого конца. В этом случае амплитуда вынужденного
испускания когерентно возрастает с каждым отражением. Сделав одно, из зеркал слегка
прозрачным, можно выпускать часть когерентнбго излучения, которое и образует
исходящий лазерный луч.
а — источник излучения иакачки; б—полиостью отражающее зеркало; в — частично отра-
- жающее зеркало.
(в случае импульсных лазеров). Поскольку «перестраивание»
(изменение частоты исходящего излучения) лазеров становится все
более легким делом, их все чаще выбирают в качестве источников
излучения для спектроскопии в оптической области благодаря их
высокой избирательности (поскольку рассматривается каждый pai
только совершенно ничтожная по ширине полоса частот спектра)
и высокой интенсивности (при исследовании слабых сигналов).
Медицинские применения лазеров базируются в основном на большом
'^количестве тепла, выделяемого на небольшой регулируемой
площади, при использовании сфокусированного лазерного пучка. Лазеры
"можно использовать, чтобы создать короткий тепловой импульс в
Химической реакционной смеси для проведения эксперимента по
Температурному скачку (гл. 16.А.1), благодаря поглощению
лазерного света растворителем. Очень короткие импульсы (Ю-12 с или
|менее) используют для инициации и изучения сверхбыстрых реак-
гций с малыми молекулами — эти эксперименты ведут к лучшему
.'пониманию первичных событий, которые происходят сразу же п<ь
еле первоначального поглощения излучения веществом, таких,
например, как первичные реакции при фотосинтезе.
Пример. Бескровная хирургия. Лазерный скальпель
Поглощение любого излучения приводит в результате к генерированию
некоторого количества энергии в виде тепла, которое рассеивается от
возбужденных молекул в окружающее пространство. Когда излучение исходит от высоко-
650 ЧАСТЬ Б
. . . f
Рис. 19.13. Бескровный надрез кожи крысы При использовании непрерывного
С02-лазера.
Мощность сфокусированного пучка прн резке составляет около 2500 Вт/см21 (Из American
Optical по публикации Goldman L., Rockwell R. I., Jr., Lasers in Medicine, Gordon and
. Breach, New York, 1971.)
интенсивного сфокусированного лазерного источника, количество выделяющегося
тепла столь велико, что ткань может практически испариться вблизи точки, на
которую сфокусирован луч. Использование такого пучка в хирургии в качестве
скальпеля имеет несколько преимуществ. Главное из них — глубину резаиия
йожно легко регулировать, используя хорошо сфокусированный пучок, и
поэтому во время тонких операций легче разрезать ткань слой за слоем. Более того,
поскольку кровь при нагревании коагулирует, любые мелкие кровеносные
сосуды, перерезаемые пучком, будут сразу же накрепко запечатаны коагуляцией,
сопровождающей разрезание (впечатляющий пример приведен на рис. 19.13).
Наиболее известный н важный клинический вклад лазера состоит в лечении
отслоений сетчатки — состояния, при котором сетчатка глаза (слой, содержащий
светочувствительные клетки) отделена от клеток слоя сосудистой оболочки,
располагающейся сразу за сетчаткой на задней стенке глазного яблока. При
использовании всего лишь одного лекарства для расширения зрачка операция
заключается просто в фокусировке интенсивного лазерного пучка на место
«тслоення: вызванная лазером коагуляция накрепко присоединяет сетчатку
лазад к сосудистому слою, предотвращая полное отслоение сетчатки на
большой площади.
Лазеры должны в принципе оказаться полезными при вырезании опухолей
-или для 'избирательного разрушения любой ткани, цвет которой достаточно
отличен от цвета ее окружения. Лазеры уже применяли для осветления
красно-коричневых родинок и татуировок, а также для лечения меланомы кожи.
-Пример. Микрохирургия
Благодаря небольшому диаметру сфокусированного лазерного пучка
<(50 мкм) появилась возможность облегчить предельно тонкие операции,
особенно в тех случаях, когда объект операции труднодоступен (например, гипо-
651 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
физ). С помощью лазера можно также избирательно разрушать определенные
клеточные оргаиеллы (хромосомы, митохондрии, ядрышко), оставляя другие
неловрежденными. Это открывает новую широкую область физиологических-
исследоваиий, образцом которых служит пример на рис. 19.14.
Пример. Лазерное светорассеяние
Мы еще будем обсуждать эту тему в гл. 21 .В. Сейчас отметим только, что
интенсивность светорассеяния под данным углом (гл. 15.А) зависит от размера
объекта, на котором это рассеяние происходит. Когда используют видимый свет,
длины его волн оказываются достаточно большими для того, чтобы, измеряя
светорассеяние, различать клетки крови по размеру. В подходящей проточной
системе возможно найти распределение клеточной популяции по размерам
клеток. Легко представить себе варианты этого метода, в которых клетки
окрашены светорассеивающим красителем, специфичным, скажем, к ДНК, так что
клетки можно изучать и даже разделять в соответствии с размером, формой и
содержимым. Поскольку ненормальные клетки часто имеют необычную форму
и(нли) размер, метод кажется многообещающим для предварительного
исследования при диагностике болезни. Метод, например, применим для распознавания
полного набора различных типов клеток (от нормальных до предраковых
аномальных и раковых) среди клеток из женских половых путей.
Пример. Лазерная спектроскопия комбинационного рассеяния
Внутримолекулярные колебания обеспечивают амплитудную модуляцию
движения электронов молекулы, возбуждаемых оптическим излучением. Это
делает возможным получение лежащих в инфракрасной области спектра частот
Рис. 19.14. Эффекты
микрооблучения аргоновым лазером
различных мест клетки
(диаметр пораженной области
около 1 мкм).
Зависимость эффектов облучения
от места облучения позволяет
установить связь ассоциированных с
ядрышком частей хромосом с
особенностями клеточного деления,
[Из Ohnukl Y., Berns M. W.,
Rounds D. E., Olson R. S„ Exptl.
Cell. Res.. 71. 132 (1972).]
а и б —клеточное деление
подавлено; в — клетка разделилась, но
выделение ядрышка подавлено; г и
д—клетка разделилась, ядрышки
нормальные.
652 ЧАСТЬ 5
Рис. 19.15. Лазерные спектры комбинационного рассеяния ДНК тимуса теленка
в 2,5%-ном водном растворе при рН 7,2 (вверху) и дрожжевой тРНК
в 2,5%-ном водном растворе при рН 7,0 (внизу).
Отмечены пики, приписываемые определенным нуклеотидам. (Взято с изменениями иа
Peticolas W. L., in Advances in Raman Spectroscopy, Vol. 1. ed. J. P. Mathleu, Heydon arid
Son, Ltd., London, 1973, p. 286.)
Обратите внимание иа детальность этого спектра по сравнению со спектрами поглощения
в видимой области на рис. 14.16,6).
колебаний молекулы при наблюдении боковых полос в спектре рассеяния
образца, который был облучен светом в оптическом диапазоне (rjL17JB). Поскольку
взаимодействие двух типов движения (вынужденного движения электрона я
колебательного движения ядер) слабо, амплитуда боковых полос мала и
необходим очень мощный оптический источник света. По этой причине в
экспериментах по спектроскопии комбинационного рассеяния, которые проводят в
последнее время, в качестве оптического источника все чаще используют лазер.
Поскольку интенсивное излучение лазера сконцентрировано в частотном
диапазоне шириной в 10е раз меньше, чем у обычного лампового 'источника, то при
использовании лазерного источника мощность, непосредственно идущая на
возбуждение комбинационного рассеяния, возрастает по величине на много
порядков. По этой причине спектры комбинационного рассеяния (имеются в виду
боковые полосы) также гораздо* более интенсивны. На рис. 19.15 показаны
спектры комбинациоиного рассеяния типичных ДНК и RHK. Видно, что отличить
«сигналы четырех оснований один от другого очень легко (сравните с худшим
разрешением прн обычном поглощении в видимой области, например, на
рис. 14.5,б). Фиксируя изменение относительных интенсивностей сигналов от
•различных оснований как функцию возмущающих воздействий, можно сделать
заключение о структуре молекул в растворе.
;Пример. Кинетика химической релаксации при индуцированном лазером
температурном скачке
Лазеры являются очень удобным инструментом для создания внезапного
■температурного изменения («Т-скачок»), с которого начинаются эксперименты
що химической релаксации (гл. 16.А.1). Единственная проблема при этом за-
?653 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
ключается в том, что частота поглощения воды (частота, иа которой
облучение производило бы наиболее эффективное нагревание раствора) заметно
удалена от частот достаточно доступных лазерных источников большой мощности.
В последнее время был разработан метод, позволяющий решить эту проблему
(рис. 19.16). В новом методе образец помещают непосредственно внутри
«резонатора» лазера, что позволяет меньше чем за 1 мс достигать Т-скачка
величиной даже 10 °С или более. Для системы, состоящей из Р-цепи гемоглобина (Г)
и метилизонитрнла (М), регистрировали изменение поглощения как функцию
времени (рнс. 19.16,а). Обратные экспоненциальные постоянные времени для
нескольких кривых, полученных при разных концентрациях ГП и [Ml, показаны
на рис. 19.116,6 в виде зависимости от суммы концентраций ([Г]-Ь[М]).
Прямолинейный характер зависимости указывает на то, что процесс связывания
М + Гз^МГ является реакцией второго порядка (табл. 16.1). Привлекательность
этого метода состоит в том, что процесс нагревания, который, v помимо всего
прочего, производится сразу по всему образцу, осуществляют, за время, которое
можно в принципе сделать весьма коротким (скажем, Ю-8 с), используя
импульсный лазерный источник. Таким образом делается возможным изучение
в растворе процессов химической релаксации, почти в 1000 раз более быстрых,
чем доступные для изучения в настоящее время при использовании
традиционных методов Т-скачка (например, разряда конденсатора).
Прнмео. Другие применения лазеров
Когерентность лазерного излучения позволяет воссоздавать с оптическим
разрешением трехмерное изображение объектов способом, известным как
голография. Поскольку молекулы, содержащие различные изотопы (например, 12С и
i3C), могут поглощать свет на разных частотах, становится возможным
разделять изотопы, облучая изотопную смесь лазером с частотой, иа которой заметно
поглощает свет только одна из изотопных молекул; энергетически
возбужденную, содержащую изотоп молекулу можно затем разрушить илн ввести в
химическую реакцию, чтобы образовать соединение, легко отделяющееся от исход-
35
а 30
*
$ 25
II20
*| 15
4)Q
i| ю
*» R
*
■а
* о
-1 1
—
—
1 \\
-
1 ! 1
/const- (1 -
1 1 1
1 1 !
-е-*'т)
1 ! 1
I 1_
^^г^^^^Г. ■
:
—
а —
1 1
250
200
„ 150
100
50
1 1 I I ■
•/•
—■ / —
л/
ГЛ
б
1111
5 10 15 20 25 30 35 40 45 50
Время, мс
О,05 О,10 0.15 0.20
1[Г1 + [М])->-
Рис. 19.16. Эксперимент с индуцированным лазером температурным скачком по
связыванию метилизонитрнла (М) с 0-цепями гемоглобина (Г).
1Из Baldo J. H., Manuck В. Л., Priestley Е. В., Sykes В. D., J. Amer. Chem. Soc, 97, 1684
(1975).]
а—график зависимости изменения поглощения от времени, прошедшего с момента
лазерного теплового импульса, демонстрирующий экспоненциальное изменение поглощения;
б — график обратных экспоненциальных констант релаксации 1/т, построенный на основе
графиков типа а, полученных для нескольких различных исходных равновесных
концентраций [Г] н [М]. В табл. 16.1 показано, что реакция М. + Г*±ГМ является бимолекулярной.
654 ЧАСТЬ Б
ных молекул с другими изотопами. Был выполнен целый ряд таких разделений
изотопов. Однако пока еще ни одно из ннх не является достаточно
эффективным, чтобы ааменить существующие технологии. Наконеи, предполагают, что
с помощью лазеров можно будет осуществлять управляемый термоядерный
синтез, и есть надежда, что лазерный термоядерный синтез станет реальностью через
одно-два десятилетия.
л
19.Б.З. ЯВЛЕНИЯ НАСЫЩЕНИЯ
И СПЕКТРАЛЬНЫЕ ВРЕМЕНА РЕЛАКСАЦИИ
В предшествующем разделе мы рассчитали воздействие
монохроматического излучения на относительные заселенности уровней
системы из изолированных частиц, характеризующихся двумя
энергетическими уровнями Ет и Еп- Было выяснено, что главным
результатом этого расчета является уравнение (19.32), которое
показывает, что внешнее облучение на частоте (Em—En)/h ведет к
экспоненциальному уменьшению до нуля любого различия
первоначальных заселенностей (Nn—Nm) в течение всего времени
действия внешнего излучения. Однако для реальной системы,
находящейся в тепловом равновесии с окружающей средой, найдено, что
в отсутствие воздействия внешнего излучения две заселенности не
являются совершенно одинаковыми, но подчиняются соотношению
Больцмана [уравнение (19.4а)]. Мы, следовательно, пришли к
заключению, что должно существовать «взаимодействие», или
связь, между системой и ее окружением, которое делает переходы
«вниз» более вероятными, чем переходы «вверх», в соответствии
с фактором Больцмана, который описывает две заселенности в
состоянии теплового равновесия:
-~==^=е п m (без воздействия внешнего излучения)
(19.33)
Уравнение (19.33) по своему смыслу идентично обычному
соотношению между константой равновесия и константами скоростей
образования компонентов при обыкновенном химическом равновесии
А1В> т=& <|9-34>
в котором W] и W± символизируют «константы скоростей» для
переходов «вверх» и «вниз» соответственно (рис. 19.8).
При отсутствии внешнего облучения мы можем записать
уравнения для скорости изменения заселенностей энергетических
уровней как dNnldt и dNmldt, где константы скоростей W \ и W ± для
(безызлучательных) переходов между уровнями отражают
наличие некоторого типа взаимодействия между системой и окружаю-
655 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
щей средой (это взаимодействие более детально обсуждено в гл.
21.Аи21.Б):
или
dNmldt = -W^m + W^n
(19.35а)
(19.356)
d(N»dtN")=(Nm + Nn)(Wi-Wi)~(Nn--Nm)(Wi+W^ (19.36)
Удобно переписать уравнение (19.36) в виде
d (Nn - Nm)/dt =
{Nn — Nm)Pam — (Nn ~ Nm)
7\
(19.37)
где
w\+Xr\ J "(19.37a)
(l/7\) = (H^+^t)l
(19.38)
Например, если исходить из положения, в котором Afn=vVm
в.нулевой момент времени (первоначально оба уровня заселены
одинаково), уравнение (19.37) легко решается и дает выражение
(Nn - Nm) = (Nn - tf JpaBH (1 - e~tlTl)) (19.39)
из которого ясно, что (Nn—МттОравн представляет собой разность
равновесных заселенностеи двух уровней. Уравнение (19.39)
описывает, например, экспоненциальное увеличение магнитного
момента системы ядер со спином Уг в нулевой момент времени
внезапно помещенной в постоянное магнитное поле (рис. 19.17).
(^гГ^т'равн
Nn-Nm
\
; Рис. 19.il7. График зависимости различия заселенностеи от времени для
системы с двумя уровнями и различием первоначальных заселенностеи, равным
нулю в нулевой момент времени.
'. .Через бесконечное время (при наступлении теплового равновесия) различие заселенностеи
•Достигает своего значения для теплового равновесия (Nп— ЛГт)равн [уравнение (19.37а)].
656 ЧАСТЬ Б
Теперь мы можем объединить уравнения (19.31) и (19.37),
чтобы выяснить поведение системы с двумя уровнями, на которую
действуют как внешнее излучение на.частоте (Ет—En)/h, так и
взаимодействие между системой и ее окружением,
характеризующееся параметром Т\ уравнения (19.38):
«№-*Ц = _2Г (ЛГ„ -ЛЦ+ У--"-Ь-+ <""-*"> (,9.40)
Решение этого уравнения в условиях стационарного состояния
(разность заселенностей наблюдают после того, как внешнее
электромагнитное поле действовало в течение времени, достаточно
большого по сравнению с Т\) представляет собой выражение
(М —N ) — ^П~~ ^)равн
vvn '*т)стгц —
i + ттл
(19.41)
Уравнение (19.41) приводит к нескольким важным
заключениям и приложениям. Первое: оно объясняет явление «насыщения» в
спектроскопии. Если исходить из вероятности индуцированного
излучением перехода в изолированной системе [уравнение (19.32)],
то стационарного состояния быть не может, и разность
заселенностей (а отсюда и наблюдаемый спектроскопический сигнал
поглощения) экспоненциально уменьшается до нуля во время
воздействия излучения. Однако, поскольку для низкоэнергетического
уровня существует противоположная тенденция — стать относительно
более заселенным, чем верхний уровень, из-за взаимодействия
системы с окружающей средой — уравнение (19.41) предсказывает,
что со временем возникает стационарная ситуация, в которой
эффект выравнивания заселенностей благодаря действию внешнего
облучения совершенно сводится на нет тенденцией к возрастанию
различия заселенностей до величины, определяемой тепловым
равновесием при взаимодействии с окружением. До тех пор, пока
2WT]<^]> для этого стационарного состояния соблюдается
соотношение Nn—Nm2*(Nn—Л^т)Равн, и внешнее облучение заметно не
меняет равновесных заселенностей энергетических уровней.
Однако, поскольку скорость (индуцированного) поглощения энергии
[ордина'та в типичном (энергетическом) спектре «поглощения»]
dE/dt задается уравнением
%= {Nn - Nm) (Em - En) W = (Nn - Nm)pam (Em - En) ( rq^rf;)
(19.42)'
[основанным на уравнении (19.31)] и поскольку W
пропорциональна квадрату амплитуды электрического (или магнитного)
поля внешнего излучения [уравнение (19.28)], ясно, что сигнал
энергетического спектра поглощения будет увеличиваться с увеличе-
(557 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
1 ■—-
нием интенсивности внешнего облучения до тех пор, пока 2WTi<g.\.
Однако если внешнее облучение становится достаточно
интенсивным, чтобы 2WT\^l, то поглощение энергии выходит на плато, и
при более высоких уровнях облучения поглощение энергии
перестает повышаться, хотя интенсивность излучения все более
возрастает. Эту последнюю ситуацию называют насыщением: поглоще1
ние энергии определяется (при этих условиях) не силой
облучающего поля, а скоростью, с которой система может вновь заселять
свой нижний энергетический уровень благодаря взаимодействию
с окружающей средой.
Одно из прямых следствий уравнения (19.42) состоит в том, что
оно дает простой способ для определения константы Т\
взаимодействия система -^окружение. График зависимости скорости
поглощения энергии dE/dt от квадрата силы приложенного
переменного поля будет при высоких напряженностях поля выходить на
.плато с-величиной (Nn—^m)paw(Em—Еп)(2Ть из которой можно
определить Т\.
Когда параметр Т\ определен, его величину можно соотнести с
информацией или о движении, или о расстоянии (гл. f6.B.3 и 21.Б).
С другой стороны, возможно изучать различие между разными
областями макроскопического объекта при условии, что значения 7\
различны в этих двух областях (см. пример «ЯМР-томографии» в
гл. 22.В.2). Наконец, можно фиксировать скорости различных
химических или физических процессов, когда интересующая нас
скорость того же порядка, что и 1/7"ь как это показано в следующем
ниже примере с цитохромом с.
Перенос насыщения как мера скоростей реакций
Предположим, что молекула может существовать в двух
равновероятных формах, которые различаются интервалами энергий
между двумя энергетическими уровнями Ет- и Еп, только что
обсуждавшимися в гл. 19.Б.З, и что молекула может изомеризоваться
из одной формы в другую, как схематически изображено ниже.
Предположим далее, что находящуюся в тепловом равновесии
смесь обеих форм молекулы мы облучаем на частоте, которая
насыщает переходы между Efn и Ев так, что эти два уровня
становятся равнозаселенНыми.
ЯЪрма А Форма В
<-т
-п
658 ЧАСТЬ б
Поскольку молекулы формы В (насыщенные) непрерывно
превращаются опять в форму А, энергетические уровни формы А будут
иметь тенденцию стать более равномерно заселенными в
соответствии с тем, насколько скорость обмена А+*В велика по сравнению
со скоростью теплового уравновешивания заселенностей (т. е.
l/Tf). Таким образом, скорость обмена между формами Лив
можно определять, регистрируя разность заселенностей между
двумя энергетическими уровнями формы А сразу после облучения
формы В до такого уровня, при котором достигается насыщение
переходов между энергетическими уровнями формы В, если только
k\ и k-i имеют тот же порядок величины, что и \/Tf. Постановку
подобного эксперимента можно без труда представить себе из
следующего примера.
Пример. Электронный обмен между восстановленной и окисленной формами ци-
тохрома с
На рис. 19.18,а показаны части спектра ядерного магнитного резонанса
окисленного цн^рхрома с. Пик а относят к протонам метальной группы метио-
ннна, пики от bi до bi, — к метильным группам лорфиринового кольца, а лики
Ci и сг — к метильным группам боковой цепи порфирина. Спектр
восстановленной формы аналогичен описанному, за исключением того, что для диамагнитной
восстановленной формы (окисленная форма парамагнитна) пики метильных
протонов теперь наблюдают при несколько иных частотах из-за отсутствия
наведенных парамагнитностыо сдвигов, которые обсуждались в гл. 19.А. Например, пик
протонов метальной группы метионина сдвигается с 23,4 м, д. в окисленном
цитохроме с до 3,3 м. д. в восстановленной его форме, что соответствует случаю»
Рис. 19.18.
а — спектр протонного ядерного магнитного резонанса окисленного цнтохрома с в водном
растворе (обозначения пиков см. в тексте), б—часть протонного спектра ЯМР раствора
цитохрома с, содержащего приблизительно равные концентрации окисленной и
восстановленной форм. Интенсивность пнка протонов метнльной группы метионина в восстановленной
форме заметно уменьшается сразу после насыщающего облучения метильиой группы
метионина в окисленной форме. Это указывает на то, что скорость взаимного превращения
окисленной и восстановленной форм составляет около 0,25 с (это есть Tt для
восстановленной формы —см. текст). [Из Gupta R. К., Redfield A, G., Science, 169, 1204 (1970).]
/ — сигнал протонов метнльной группы метионина окисленного цитохрома с; 2 — сигнал
протонов метнльной группы метнонииа восстановленного цитохрома с; 3 — без облучения
окисленной формы; 4 — сразу после насыщающего облучения окисленной формы.
659 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
показанному на рис. 19.18. Когда присутствуют и окисленная, и восстановленная
формы, спектр смеси представляет собой просто наложение индивидуальных
спектров двух форм, поскольку обмен между формами является медленным по
сравнению с различием в резонансных частотах между этими двумя формами
(подробное рассмотрение этого положения дано в гл. 14.Г и задаче 14.6).
Если теперь провести облучение высокой мощности на частоте резонанса
протонов метильной группы метионина в окисленной форме (23,4 м. д.) и затем
следить (рис. 19.18,6) за той же самой метильной группой в восстановленной
форме (3,3 м. д.), то можно обнаружить, что сигнал протонов метильной
группы метионина в восстановленной форме имеет пониженную интенсивность
благодаря частичному выравниванию заселенностей энергетических уровней
протонов метильной группы метионина в восстановленной форме из-за обмена
с (насыщенными и поэтому одинаково заселенными) энергетическими уровнями
протонов метиониновой метильной группы окисленной формы. Теперь, если бы
метионнновая метильная группа восстановленной формы имела Ti гораздо
меньшее, чем время протекания процесса обмена (окисленная форма)
^^((восстановленная форма), то интенсивность сигнала протонов метиониновой метильной
группы восстановленной формы была бы неизменной при облучении окисленной
формы. С другой стороны, если бы скорость обмена была очень большой по
сравнению с 1/Т] для восстановленной формы, интенсивность сигнала протонов
■метильной группы метионина окисленной формы стремилась бы к нулю при
насыщении окисленной формы. Поскольку наблюдаемый эффект, показанный иа
рнс. 119.18,6, промежуточный между этими двумя предельными случаями, ясно,
что время жизни для электронного перехода между окисленной и
восстановленной формами цнтохрома с того же порядка, что и Т\ для метильной группы
метионина в восстановленной форме, и составляет около 0,25 с.
В настоящее время-делаются попытки проводить эксперименты этого типа
на зрительных пигментах и пигментах фотосинтеза, которые имеют гораздо
более короткие параметры насыщения 7\ переходов между оптическими
(электронными) .энергетическими уровнями, чтобы обнаружить и количественно
описать очень быстрые изменения, которые следуют за поглощением фотона такими
молекулами. Эти изменения происходят за время меньше 10~9 с.
Использование переноса энергии при флуоресценции
в качестве «спектроскопической линейки»
При магнитном резонансе осуществление связи между данными
ядерным спином и спинами соседних ядер (^-процесс)
приписывают, как правило, диполь-дипольному взаимодействию между
магнитным диполем данного ядра и дипольными моментами
окружающих ядер. Как будет показано в гл. 21.Б, в этом случае Т\
меняется пропорционально шестой степени расстояния между двумя
взаимодействующими ядерными диполями (см., например, гл. 16.Б.З).
Аналогичное взаимодействие между двумя электрическими
диполями электронно-возбужденных групп (в противоположность
магнитным, как в примере с ядрами), как было недавно показано,
является чувствительным инструментом для измерения расстояния
между двумя флуоресцирующими фрагментами одной и той же
молекулы. Так, если в макромолекулу ввести две флуоресцентные
метки, то, основываясь на результатах эксперимента по
флуоресценции, можно в принципе измерить расстояние между метками в
макромолекуле. Схема происходящих при этом процессов
показана на рис. 19.19.
660 ЧАСТЬ Б
Донор
Акцептор
I
Рис. 19.19. Энергетическая диаграмма основных процессов, вовлеченных в
перенос энергии при флуоресценции.
Акцепторная область молекулы может достичь возбужденного состояния либо путем
прямого вынужденного поглощения внешнего излучения (прямая стрелка вверх справа), либо
через перенос энергии от аналогичным образом возбужденной, расположенной поблизости
донорной области той же самой молекулы (стрелка в). После возбуждения происходит
быстрая безыэлучательная (излучения не происходит) перестройка акцептора (е) так, что
молекула переходит на самый нижний энергетический уровень возбужденного состояния,
с которого и происходит спонтанное (самопроизвольное) испускание (д). Так как процесс
переноса энергии очень сильно зависит от расстояния между донорным и акцепторным
концами молекулы, то это расстояние может быть определено путем сравнения
флуоресценции акцепторной части в присутствии и в отсутствие близлежащей донорной группы
(см. текст).
а — поглощение донором (вынужденное); б — флуоресценция донора (спонтанная);
в—перенос энергии (безызлучательный); г —флуоресценция акцептора после переноса энергии
от донора (спонтанная); д — флуоресценция после поглощения акцептором (спонтанная);
е — быстрый безызлучательный переход вниз до самого нижнего энергетического уроаня
возбужденного состояния.
Сама по себе флуоресценция является примером спонтанного
излучения (излучения в отсутствие какого-либо воздействия
внешних переменных полей). Хотя подробное описание этого явления
немного выходит за рамки настоящего обсуждения, нам все же
необходимо знать, что скорость спонтанного излучения меняется
пропорционально кубу разности энергий участвующих в процессе
уровней- По указанной причине такой процесс, очевидно, гораздо
более важен на оптических частотах, а не на частотах микроволн
или радиочастотах (более того, спонтанное излучение при
магнитном резонансе слабее приблизительно в 105 раз вследствие того,
что магнитные взаимодействия во столько раз слабее электричес\
ких). Рис. 19.19 представляет собой основу для
экспериментального применения переноса энергии при флуоресценции в качестве
индикатора расстояний в молекулах в растворе.
Пример. Градуировка «флуоресцентной линейки»
Несмотря на то что зависимость процесса переноса энергии от расстояния
довольно хорошо установлена, необходимо калибровать наблюдаемую
интенсивность флуоресценции как функцию известного расстояния между доиорными и
акцепторными частями модельного негибкого соединения, чтобы установить
константу пропорциональности. Такое соединение, показанное ниже, содержит до-
норную и акцепторную группы, разделенные известным числом L-пролиновых
звеньев. Это соединение было выбрано из-за того, что поли-Ь-пролиновая цепь
КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
образует жесткую конфигурацию «гране-спирали типа II», расстояние между
концами которой легко измерить на молекулярной модели.
Разделяющая
группа
акцептор
»
н3с
\ >=
о / о\ н
•S-J-N—I—c4n—N— С—N
• донор
их
дансил- (\.~ пролил)п-<х- rfatpma/i
п=1- 12
!•. Для изолированной акцепторной группы наблюдаемая общая интенсивность
флуоресценции F, возникающей вслед за облучением на данной частоте, будет
(определяться просто коэффициентом погашения для акцептора ел- Но при
наличии расположенной рядом донорной группы величину F находят лз равенства
Р = *л + Т*п
(19.43)
г
г
Где во — коэффициент погашения донорной группы, а Т — энергия возбуждения
донора, перенесенная на акцепторную группу. [Благодаря тому что энергия
флуоресценции акцептора много меньше энергии флуоресценции . донора
жрис. 19.19), их легко различать, и, следовательно, можно регистрировать
|!сйлько флуоресценцию акцепторной группы.]
На рис. 19.20 показаны экспериментально определенные Г (в %) для раз-
^дчных расстояний, .разделяющих донориую и акцепторную группы
данаид- (Ь-пролил)л-а-нафтилов, имеющих л от 1 до 12. Перенос энергии с эффек-
100
It
10 20 30
Расстояние, А
Рис. 19.20. Градуировка флуоресцентной «спектроскопической лннейки».
'Ь*фективн0сть переноса энергии прн (Ьлуоресценцин от донора к акцептору (рнс. 19.19)
доказана как функция (известного) расстояния между донорной и акцепторной группами
р молекулах даиснл-(Ь-пролнл) „-а-нафтнла, в которых л менялось от 1 До 12. d-ro
соответствует расстояниям, разделяющим доиор и акцептор, в пределах от 12 до 46 А.
Сплошной линией показана теоретическая расчетная кривая, базирующаяся на выборе Ji-JJJA
£ качестве расстояния, иа котором перенос энергии прн Флуоресценция имеет »ФФехпш-
' ность 60%. [Из Stryer L., Haugland R. P., Proc. Natl. Acad. Scl. USA. 58, 719 (1967).]
662 ЧАСТЬ Б
тивностью 50% имеет место при расстоянии между донором и акцептором,
равном 34,6 А, а экспериментальные данные могут быть описаны теоретическим вы-
ражением вида
Т— (R*/r)* П9 44Ъ
*
в котором г — расстояние, разделяющее донор и акцептор, a Ro — разделяющее
расстояние, прн котором эффективность переноса энергии равна 50% [в наймем
случае Г=0,5 при #0=34,бА].
Прекрасное соответствие между экспериментально определенной
эффективностью переноса энергии при флуоресценции и теоретической зависимостью от
шестой степени расстояния, разделяющего донор и акцептор, указывает на то, что
этот тип измерений можно использовать при определении расстояния между
двумя метками (одна метка—донор, а другая — акцептор) макромолекулы
в растворе, когда расстояние составляет приблизительно 25—40 А. При
использовании разных пар донор—акцептор с различными величинами /?о было бы
возможно конструировать и другие «линейки» для измерения более коротких или
более длинных расстояний; так что эту методику можно было бы использовать
для определения расстояний в интервале от 10 до 60 А, который включает в себя
размеры большинства наиболее крупных белков.
19.Б.4. ИНТЕНСИВНОСТИ ЛИНИИ В СПЕКТРАХ ПОГЛОЩЕНИЯ
Правила отбора и относительные интенсивности линий.
Согласно ранее выведенному нами уравнению (19.24), переход (точнее—
электрический дипольный переход) между энергетическим
состоянием Еп и энергетическим состоянием Ет. будет иметь место,
когда на систему действуют переменным электромагнитным полем с
соответствующей частотой (Ет—En)/h, при условии что матричный
элемент оператора электрического дипольного момента [уравнение
(19.22)] между волновыми функциями tyn и i|)m не является, «*/лг-
вым. Это и есть правило отбора для переходов между
энергетическими уровнями. Поскольку поглощение энергии квантовомехани-
ческой системой пропорционально вероятности индуцированных
переходов [уравнение (19.42)]—при условии, чтб мощность
облучения поддерживают ниже порога * насыщения (2WTi<Cl),—
и так как вероятность перехода пропорциональна квадрату
матричного элемента оператора дипольного момента между i|)n и tym
[уравнение (19.28)], правило отбора предсказывает наличие (или
отсутствие) и относительные интенсивности возможных переходов
между различными, энергетическими уровнями, получающимися в
результате квантовомеханических расчетов. Легче всего
продемонстрировать правила отбора для относительно простых схем
энергетических уровней, возникающих в результате явлений
магнитного резонанса, в нижеследующих примерах. Главная особенность
этих расчетов состоит в том, что для оценки матричных элементов
рассматриваемого дипольного оператора необходимо определять
вид волновых функций tyi, соответствующих вычисленным энерге-
|бЗ КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
Этическим уровням £*. Пока мы имели дело только с
энергетическими уровнями, не было необходимости записывать связанные с
ними волновые функции. Однако последние необходимо знать,
чтобы определить, какие переходы «разрешены» (переходы, для
которых | \ixmn 12Ф0) и каковы их относительные интенсивности.
Используя модель ядра «мячик на веревочке», мы в гл. 14.Г
Показали, что для передачи энергии классическому магнитному
моменту, который прецессирует вокруг статического внешнего
магнитного поля, необходимо приложить силу в направлении,
перпендикулярном как оси вращения, так и радиусу прецессии. (Другими
словами, лучший способ ускорить движение мячика на веревочке—
это подтолкнуть его в направлении, параллельном земле.)
Используя квантовомеханический язык, можно было бы сказать, что
энергия возбуждения обеспечивается приложенным внешним
переменным* магнитным полем Hxcos((ut), взаимодействующим с лс-ком-
^онентой магнитного дипольного момента частицы (частиц) \ix =
&=hlx, где Ix — ^-компонента оператора квантовомеханического уг-
дового момента («спина»)
• * возбуждающая энергия =^ННХ1Х cos (cor). (19.45)
Но уравнение (19.45) описывает взаимодействие, формально
идентичное уже разобранному [уравнение (19.17) и последующие] при
действии переменного электрического поля на электрический ди-
рольный момент системы частиц, за исключением того, что
вероятность перехода для магнитного случая теперь пропорциональная
квадрату матричного элемента Ux^l2, а не матричного элемента
Il^mJ2, Мы теперь в состоянии оценить спектральные
интенсивности поглощения для некоторых простых примеров спиновых си-
|тем.
i-. '
f-
У
пример. Одни тнп ядер со спином 7г в статическом магнитном поле
?,:
rj. На рис. 18.5 показаны энергетические уровни и волновые функции для
одного типа магнитных ядер со спином 7а в статическом магнитном поле Н0>
направленном вдоль оси г. Существует только один переход, участвующий в
поглощении, а именно переход из состояния Р в состояние а. Нам, следовательно,
Необходимо просто оценить матричный элемент (скалярное произведение,
проекцию)
* Можно использовать обычный математический прием: считать это
переменное поле состоящим из двух вращающихся в противоположных направле-
рлях полей равной величины. Поскольку только одно из двух вращающихся
Полей будет вращаться так же, как и прецессирующий момент, можно показать,
<что другое вращающееся поле можно не принимать во внимание.
664 ЧАСТЬ в
чтобы определить, «разрешен» ли этот переход. Такие расчеты резко
облегчаются, если выразить оператор /* через «повышающий» и «понижающий» операторы
[уравнения (18.61а) и (18.616)], обсужденные в гл. 18.А:
(19.46)
Тогда, использовав простые свойства повышающего и понижающего операторов
(см. задача 18.3):
/+Р=а, (19.47а)
/+а = 0. (19.4Гб)
/_Р = 0, (19.47в)
/_а = р (19.47г)
и учтя теперь хорошо нам знакомые свойства ортогональности
(перпендикулярности) базисных функций !а и 0:
<а, р> = <р, а> = 0,
<а, а>=<р, р>=Г
(19.48а)
(19.486)
мы немедленно получим желаемый матричный элемент:
<а, /х р>=<а, (/+|7-)р> = 4- «*' Л-Р> + <а. /-Р» =
= -9-«*. *>+'<*• О»
<а, /,р> = -f (l+0) = -f =£ О
2 ♦
i
этот-
пере-
ход
решен"
(19.49)
Пример. Два типа химически различных изолированных ядер со спином 7г
Более интересный случай включает в себя схему энергетических уровней,
показанную на рис. 18.6 для двух невзаимодействующих ядер с различной
резонансной частотой. В этом случае имеется шесть возможных переходов между
четырьмя энергетическими уровнями. Индивидуальный расчет вероятности
перехода для каждой пары уровней проводят, как описано выше, принимая, что
в этом процессе участвуют две частицы таким образом, что общая ж-комионеита
углового момента является суммой ^-компонент IXl и /*2 для двух частиц в
отдельности:
'< = '„ + '*,
2
/*"=
(/+ + /-).
(19.50а)
(19.506)
665 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
Так как четыре волновые функции представляют собой ахщ, aiP2, Pia2 и р,р2, то
вероятность перехода, например, между состояниями ai,a2 И piP2
пропорциональна
|<a,a„ /,№>Г = |<«А, (/л + /»,)№>|!^=
1
«a,a2, /+lp1p2>H-<a1a2, /_ip,pi> +
+<aia2, /+2№>+<a1a2, /-2PiP2»2= -j- ««.a,, a,p2> +
-|-<a1a2, 0> + <a|a2, p,a2> + <aia2, 0»2,
|<а1а2,/жр1р2>|2=^(0 + 0 + 0 + 0)2 =■ °- (19.51)
t
этот
переход
запрещен
(не
разрешен)
При получении последней строки [уравнение (119.51)] были использованы
следующие соотношения ортогональности, базирующиеся на взаимной
«перпендикулярности» четырех собственных функций (см. примечание к стр. 578).
<*Л. *А> = <«А. &««> = <>= О А. РА> (19.52)"
Аналогично вероятности остающихся переходов пропорциональны [с той же
самой константой пропорциональности (уравнение (19.24)] величинам:
l<a,a2, /xa.p2>|2=-4-»
1<*A, W2>|2 = 4 , : WMi _ ^ =1:1:1:1
<(W /,PiP»>P=-4\
4
1<«А. /А«8>Г = т (19.53)
Заметим, что в этом описании «первого порядка» из шести возможных
переходов только четыре разрешены: запрещены «двухквантовый» переход Р1Р2—*■ «lCfc,
а также переход с^Ря—>-Pi«2. Остающиеся разрешенные переходы и
соответствующий им спектр поглощения показаны на ряс. 18.6. В спектре (рис. 18.6)
есть две линии поглощения равной интенсивности, поскольку переходы как
^PiBA-atffl' так и ^Pi02-»aiafl происходят на одной и той же частоте
(разности энергий) и аналогично №Plpfl _ pltt2 н ^aiPfl -► aiafl также происходят на
одной и той же частоте. Оставляем читателю в качестве упражнения (см.
задачи к этой главе) показать, что спектр типа «АХ», получающийся в результате
:'всех возможных переходов для гамильтониана уравнения ,(18.79), состоит из
^четырех пиков равной интенсивности, как показано на рис. 118.7.
666 ЧАСТЬ 5
Основываясь на этих примерах, читатель теперь в состоянии вычислить
относительные интенсивности всех переходов* включающих энергетические
уровни для любого числа ядер со спином '/г. Эти расчеты охватывают очень
большой класс задач по определению положения линий и их ннтенсивностей для
протонных и |3С-спектров ядерного магнитного резонанса (а также спектров
электронного парамагнитного резонанса). Как правило, существуют доступные
программы для компьютеров, которые подгоняют наблюдаемый спектр ЯМР
к вычисленному для системы, состоящей нз установленного числа ядер со
спином У2, взаимодействие между которыми задается выражениями типа /;//;«//«•
Такие программы оказались весьма эффективными при вычислении из
спектральных данных интересующих нас положений спектральных линий и констант спнн-
спинового взаимодействия с целью использования их для установления
конфигураций молекул в растворе (гл. 18.А). Некоторые общие информативные случаи
можно найти в задачах к этой главе.
Задачи *
1. Получите отношения заселенности первого «возбужденного» состояния к
заселенности «основного» состояния Ni/Nq, данные в табл. 19.1, из разностей
энергий для разных типов спектроскопии, приведенных в таблице, для всех
трех указанных там температур (295, 77 и 4,2 К).
2. Предположим, что действие парамагнитного атома лантаноида, связанного
с какой-то органической молекулой, состоит в расщеплении сигнала
магнитного резонанса каждого протона (или углерода-13) этой молекулы на два
резонанса, отстоящих друг от друга по частоте на величину, равную
/C(3cos29—1)/г3, где г — расстояние от атома лантаноида до интересующего
нас протона (или 4SC), 9 — угол между направлением лантаноид — протон
(лантаноид — углерод) и осью симметрии комплекса, а К~- константа,
характерная для данного лантаноидного атома. Предположим далее, что быстрые
переходы между двумя электронными спиновыми состояниями (благодаря
очень короткому электронному Т\* — гл. 19.Б.З) вызывают «химический» обмен
между двумя резонансными частотами протонов (углеродов-13). Используйте
эту информацию для того, чтобы показать, что этот «обмен» приводит к
превращению двух протонных (углеродных) пиков в единый пик, который
смещен относительно «несмещенной» первоначальной частоты на величину,
пропорциональную (3 cos2б—1)/г', при условии что I/Tia>/C(3cos26—l)/rs. Вы
можете считать, что разница в энергиих между двумя электронными
спиновыми состояниями удовлетворяет «высокотемпературному пределу»
*-<*""■> Sl-A./W.
3. Перейдите от комплексной к действительной записи выражения, выведенного
в тексте для относительной заселенности т-го энергетического уровня после
наложения переменного электрического поля, т. е. получите уравнение
(19.256) из (19.25а).
4. Покажите, что четыре из шести возможных переходов с поглощением энергии
для спектра ЯМР типа «АХ» иа рис. 18.7 ([уравнение (18.79) для
гамильтониана] разрешены и имеют равную вероятность. Поскольку каждый из
разрешенных переходов происходит с характерной частотой, покажите, что
предсказываемый энергетический спектр поглощения состоит из четырех
линий равной интенсивности, расположенных так, как показано на рис. 18.7.
5. Для частиц со спином xk ([уравнения (18.61) и последующие] и со спином,
равным 1 (задача 18.3), мы показали, что операторы I+=IX+Uy и
I-=IX—ily приводят соответственно к повышению или понижению
собственного значения /«. Считая, что этот результат имеет силу и для частиц с
произвольным спииом (рис. 18.4), покажите, что индуцированные магнитные
резонансные переходы могут происходить только между соседними «ступень-
567 КВАНТОВАЯ МЕХАНИКА: НЕОБХОДИМА ЛИ ОНА?
камн» на «лестнице» собственных значений /«. Аналогичный расчет
показывает, что переходы между энергетическими уровнями гармонического
осциллятора (рис. 18.2) также могут происходить только между соседними
ступеньками энергетической «лестницы». Эти два правила отбора иногда
формулируют как Ат=±1 (илн Av=±l), где гп (или v) обозначает просто целые
(нли полуцелые) числа, которые характеризуют определенные «ступеньки» на
соответствующей «лестнице».
6. Восемь собственных функций гамильтониана первого порядка дли трех
взаимодействующих ядер со спином V2 имеют вид
ф4 = р1а2а3, ф, = р,р1р1.
Используя правило отбора <tym, lxtyn>¥:0 для разрешенных переходов,
покажите, что из возможных 28 вынужденных поглощательных переходов
разрешены 12, и выведите их относительные интенсивности (вид спектра см. в
задаче 18.56).
Замечание:
Литература
Распределение Больцмана
1. Nash L. К, Elements of Statistical Thermodynamics, Addison-Wesley, Reading,
Mass., 1968.
2. Davis J. C, Jr., Advanced Physical Chemistry, Ronald Press, New York, 1965.
Переходи между энергетическими уровнями
3. Davis J. C, Jr., Advanced Physical Chemistry, Ronald Press, New York, 1965.
4. Pauling L., Wilson E. В., Jr., Introduction to Quantum Mechanics, McGraw-Hill,
New York, 1935.
Лазеры
5. Siegman A. E., An Introduction to Lasers and Masers. McGraw-Hill, New York,
1971.
6. Goldman L., Rockwell R. J., Jr., Lasers in Medicine, Gordon and Breach, New
York, 1971.
7. Berns H. W., Biological Microirradiation, Prentice-Hall, Englewood Cliffs, N. J.,
1974.
Правила отбора
8. Feenberg E., Pake G. E., Notes on the Quantum Theory of Angular Momentum,
Stanford University Press, Stanford, Calif., 1959.
9. Попл Дж., Шнейдер В., Бернстейн Г. Спектры ядерного магнитного
резонанса высокого разрешения. —М.: ИЛ, 1962, гл. 6.
10. Chang R., Basic Principles of Spectroscopy, McGraw-Hill, New York, 1971.
ЧАСТЬ 6
ПРЕВРАЩЕНИЕ ТРУДНЫХ
ЗАДАЧ В ПРОСТЫЕ.
ПРЕОБРАЗОВАНИЯ
И НАГЛЯДНЫЕ ПРИМЕРЫ
ИХ ПРИМЕНЕНИЙ
В этом разделе мы в основном будем иметь дело с
приложениями методов, основанных на преобразовании Фурье. В
математике преобразование Фурье рассматривается как «взвешенная»
сумма (или интеграл) дискретного (или непрерывного) набора
экспериментальных точек. В физике необходимость в
преобразованиях Фурье, как и следовало ожидать, появляется * при решении
разного рода задач. Например, обычная линза работает как
преобразователь Фурье, и поскольку не существует линз, пригодных
для использования в области рентгеновских лучей, для
превращения картин дифракций рентгеновских лучей в трехмерные
изображения изучаемых объектов используют математическое
преобразование, которое обсуждается в гл. 22.А. Человеческое ухо или глаз
также работают как преобразователи Фурье, анализируя
взвешенные суммы когерентных звуковых или электромагнитных волн по
соответствующим высотам и цветам (частотам). Аналогичное
математическое преобразование сложных смесей спектральных
колебаний можно использовать для анализа частотных компонент
спектра смеси. Эти спектроскопические фурье-м.етоды (гл. 20),
появившиеся в 1965 г., дали мощный импульс многочисленным
исследованиям. Математическая статья, сделавшая эти методы
общедоступными, является и сейчас наиболее часто цитируемой работой
во всей, математической литературе. Более того, эти
преобразования можно использовать для того, чтобы выбрать из беспорядочно
изменяющихся электрического или магнитного полей
определенную частотную компоненту (компоненты), которая относится к
собственной частоте электронного или ядерного движений. С их
помощью можно затем объяснить наблюдаемую ширину линий
при явлениях рассеяния или резонанса (спектроскопии) (гл. 21).
Наконец, необходимо отметить, что важнейшие особенности
преобразований и их приложений можно понять, не прибегая к
пространным математическим выкладкам (отметьте, например, что в
гл. 20 практически нет уравнений). Поэтому при ознакомлении с
этим предметом мы будем чаще прибегать не к алгебраическим
выражениям, а к наглядным картинкам.
ГЛАВА 20 •
ГРУЗЫ НА ВЕСАХ:
ВЗВЕШИВАНИЕ В СПЕКТРОСКОПИИ.
МНОГОКАНАЛЬНЫЕ И МУЛЬТИПЛЕКСНЫЕ
МЕТОДЫ
Чтобы понять, почему методы Хадамарда и Фурье оказались
незаменимыми в спектроскопии, необходимо сначала понять,
почему невыгодно наблюдать спектр через узкое окошко путем
сканирования, как это делается в обычных методах. Как ни странно,
этот же самый недостаток присущ и способу, которым мы
взвешиваем предметы на обычных двухчашечных весах. Поэтому мы
начнем с поисков лучших путей использования весов, но эти
усовершенствования более или менее прямо применимы и в
спектроскопии.
Грузы на весах
Рассмотрим задачу определения веса трех неизвестных
предметов при использовании весов. В обычном случае мы решили бы
эту задачу путем поочередного взвешивания предметов, кладя их,
скажем, на левую чашку весов и нагружая правую подходящими
гирями с известным весом, как это показано на рис. 20.1.
Очевидное преимущество этой процедуры состоит в том, что каждое
измерение позволяет определять вес исследуемого предмета
(никакой обработки полученных результатов не требуется). Однако (см.
ниже) невыгодность такого подхода в том, что каждый
исследуемый предмет взвешивают только по одному разу.
При любых экспериментальных измерениях,
характеризующихся определенным уровнем неточности (случайный шум),
желательно повторять измерение много раз для получения более точного
'результата. Как показано в гл. 8.Б.4, сигнал (в нашем случае вес
данного предмета) будет накапливаться с ростом числа
взвешиваний N. Но если «шум» имеет случайный (беспорядочно
изменяющийся) характер, то его величину можно считать беспорядочно
блуждающей около нуля (средний уровень шума), а среднее
абсолютное отклонение от нуля после N ступеней случайного
блуждания (более точно — среднеквадратичное отклонение)
оказывается пропорциональным (N)l/i, Таким образом действительная мера
Прочности повторяемого измерения (а именно — отношение сигнал/
|цум) пропорциональна (N/yN), т. е. ~]/N.
670 ЧАСТЬ 6
ODD
1 2 3
idDdD
Рис. 20.1. Схематическое изображение двухчашечных весов, набора стандартных
гнрь (справа) и трех предметов неизвестной массы (слева).
Измерение
№ 1
№ 2
№ 3
Число взвешиваний каждого
неизвестного
груза
Неизвестный груз
№ 1
1
0
0
1
№ 2 № 3
0 0
1 0
0 1
1 1
Возвращаясь к задаче со взвешиванием, предположим, что
теперь мы кладем по два неизвестных предмета на левую чашку
весов и измеряем веса трех линейно независимых комбинаций
неизвестных грузов, взвешивая их по два одновременно:
Измерение
Неизвестный груз
№ 1 № 2 № 3
№ 1 110
№ 2 10 1
№ 3 0 11
Число взвешиваний каж- 2 2 2
дого груза
На этот раз три неизвестных веса связаны с результатами трех
взвешиваний тремя линейными алгебраическими уравнениями. Эти
уравнения можно затем решить, пЪлучив желаемые значения
весов неизвестных грузов. Однако, поскольку каждый груз был
взвешен дважды, точность (отношение сигнал/шум) для каждого
рассчитанного неизвестного веса будет теперь лучше в (2/УЗ) раз, чем
в первоначальном обычном эксперименте (т. е. сигнал больше в
2 раза, тогда как шум больше только в УЗ раз, поскольку
проводилось три взвешивания). Более того, поскольку требуется то же
самое общее число взвешиваний (три), суммарное время, необхо-
'671 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
димое для проведения нового эксперимента, также остается тем же
самым. Для произвольного числа N неизвестных грузов, из
которых приблизительно половина одновременно кладется при
взвешивании на весы, общее улучшение отношения сигнал/шум для
принятой нами схемы взвешивания составляет (N}2) ftN=~УN12 по
сравнению с экспериментом, который включает в себя
проведение не более чем N обычных взвешиваний предметов по одному*.
Хотя очевидно, что спектроскопический эксперимент отличен от
взвешивания (см. ниже), приведенная схема проведения опытов
с кодированием и декодированием результатов все еще
применяется и составляет основу спектроскопии с преобразованием Хада-
марда (гл. 20.А).
Продолжая логически предшествующую аргументацию, можно
думать, что если положить все три неизвестных груза с двух
сторон на весы в различных комбинациях и добавить к ним гири
известного веса для уравновешивания любых получающихся
комбинаций неизвестных грузов, то можно получить еще одну систему
взвешивания:
Измерение Неизвестные грузы
№ 1
№ 2
№ 3
левая чашка
№ I, № 2
№ 1, № 3
№ 2, № 3
правая чашка
№ 3
№ 2
№ 1
|; Число взвешиваний каждого
I" неизвестного груза=3
ё
\ * Строго говоря, точность расчета веса предмета возрастает только тогда,.
|^огда средняя ошибка при отдельном взвешивании зависит только от
взвешивания и не зависит от величины измеряемого веса неизвестного предмета
(сигнала). Это общее условие называют «шумом детектора», и его отличают от
«шума источника», при котором среднеквадратичный шум пропорционален
квадратному корню нз величины сигнала. Когда имеется «шум источника», приведенная*
йыше схема взвешивания не будет улучшать отношение сигнал/шум для
рассчитываемых весов по сравнению с теми, которые получены прн более простом
способе взвешивания по одному грузу за раз. Примерами спектроскопии, в
которых имеется только «шум детектору», являются (низкоэнергетические):
инфракрасная, микроволновая и спектроскопия ядерного магнитного резонанса, а
жакже циклотронные эксперименты по резонансу ионов. Примеры, в которых
имеется «шум источника», включают (высокоэнергетические): оптическую (в
видимой я ультрафиолетовой областях) спектроскопию и спектроскопию
заряженных частиц (фотоэлектронная, рентгеноэлектронная и спектроскопия
электронного удара). Шум становится «шумом источника», когда энергия фотона (или
^астицы) становится настолько большой по сравнению с тепловой энергией.:
£kT), что возможно подсчитывать отдельные частицы, как в примере с
«импульсным шумом» гл. 8.Б.1. Таким образом, можно ожидать, что «трюки» с
проведением эксперимента будут наиболее полезны для относительно кизкоэнергетиче-
|ких видов спектроскопии (это обсуждается ниже).
<672 ЧАСТЬ в
В этом случае снова можно получить три интересующих нас
индивидуальных значения весов неизвестных грузов решением трех
взаимосвязанных линейных алгебраических уравнений. Однако,
поскольку каждый груз теперь был взвешен трижды, отношение
сигнал/шум для каждого рассчитанного неизвестного веса будет
улучшено в 3/УЗ = УЗ раз по сравнению с обычным взвешиванием
ло одному грузу за раз. Для произвольного числа неизвестных N
следует, что общее улучшение отношения сигнал/шум равно yw.
Это улучшение также применяется в (экспериментально отличных
от вышеописанных) спектроскопических экспериментах с
преобразованием Фурье, разобранных в гл. 20.Б.
Методы Хадамарда и Фурье дают возможность улучшить
точность, (отношение сигнал/шум) в «весовых» экспериментах
соответственно в yN/2 или yyv раз и в то же время требуют того же
самого общего времени для измерений, как и обычный метод
поочередного «взвешивания» для каждой неизвестной величины. "Это
улучшение известно как «выигрыш» Фёлжета*. Все, что нам
теперь остается,— это показать, что спектроскопические измерения,
выполняемые обычным способом, также неэффективны, как и
используемые обычным способом двухчашечные весы, и затем
исследовать имеющиеся в этой ситуации способы для использования
потенциального выигрыша Фелжета.
Прямые многоканальные спектрометры
На рис. 20.2 показана очень упрощенная схема устройства
спектрометра. Диспергирующим элементом может быть, например,
призма или дифракционная решетка (в инфракрасной или
оптической областях), а щелью — фильтр, пропускающий только
определенную полосу в области низких частот (микроволн или
радиочастот в случае ядерного магнитного резонанса). Ширину щели
выбирают достаточно узкой, чтобы после объединения отсчетов
детектора для ряда отдельных положений щели (рис 20.2) дрстичь
достаточно полного разрешения (достаточное число,
экспериментальных точек на линии поглощения) для выявления интересных
особенностей спектра. Наиболее важная особенность такого
спектрометра состоит в том, что для получения с его помощью
спектра поглощения необходима процедура, формально идентичная
методу индивидуального (по одному за раз) определения весов
{спектральных интенсивностей) для N различных неизвестных
объектов (мест расположения «щели» на спектре). Таким образом,
было бы желательно увеличить ширину щели до полной ширины
необходимого-спектрального окна, используя N одноканальных де-
* И опять выигрыш Фелжета может быть реализован только тогда, когда
шум является «шумом детектора», и его нельзя использовать в случае «шума
источника» (см. предшествующую сноску).
673 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
600
Положение щели
1000
Рис. 20.2.
Вверху: схематическое изображение одиощелевого сканирующего спектрометра для
излучения поглощения. Одиощелевой сканирующий спектрометр для изучения испускания
неимеет только широкополосного источника. Внизу: запись показаний детектора для ряда
различных последовательных положений щели.
/ — источник; 2 — образец; 3 — диспергирующий элемент; 4 — окно; 5 — щель; 6 —
широкополосный детектор.
текторов, как показано на рис. 20.3. Поскольку в этом случае
фиксируются сигналы от всех положений щели сразу, а не один за
другим по очереди, «многоканальный» спектрометр на рис. 20.3,
дает возможность реализовать преимущество Фелжета, улучшив
отношение сигнал/шум в УЛГ раз по сравнению со спектром,
полученным за то же самое время сканированием только одной щелью
за один раз как на рис. 20.2. С другой стороны, можно было бы
получить спектр, имеющий то же самое отношение сигнал/шум за 1/Л/"
времени, необходимого, чтобы просканировать N отдельных
положений щели одно за другим.
Учитывая простоту схемы многоканального спектрометра (рис,
20.3), логично поинтересоваться возможностью его реального соз-
Z > N кана/юь
;Рис. 20.3. Схематическое изображение детекторной части гшямого
многоканального спектрометра, составленного из множества отдельных детекторов.
/ — диспергирующий элемент: 2 — окно; 3 — многоканальный детектор.
^674 ЧАСТЬ 6
дания. При этом всегда необходимо иметь возможность разрешить
детали спектра, близкие по ширине к ширине линии поглощения.
•Следовательно, минимальное число каналов, которое
потребовалось бы для многоканального спектрометра; определяется просто
шириной интересующего нас спектрального диапазона, поделенной
на ширину одной спектральной линии. Получающееся в результате
такой операции число каналов для различных видов
спектроскопии показано в табл. 20.1. »
Таблица 20.1
Минимальное число каналов, необходимое для различных типов
прямых многоканальных спектрометров
Энергия для Типичный Ширина одной Минимальное
Тип спектроскопии резонансного спектральный лннин ^ каИаЛОва
поглощения, Гц диапазон, Гц число каналов
.Мёссбауэровская 6-10й» 108 107 10
Рентгеноэлектронная 3,5- 10й 1017 1014 1 000
Фотоэлектронная 5I015 31016 101* 3 000
Электронная 1*6-10» 1,2-1015 10"» 1250 000
Колебательная 2-10" 1,6-10м 3-10» 50*000
Вращательная 4'10й 3-Ю10 10* 300 000
ЯМР-13С 8I07 2-10* 0,5 40 000
Ионный циклотронный
резонанс 2-10* 2 10е 102 20 000
Число каналов получали делением типичного спектрального диапазона на ширину одной
линии.
Из табл. 20.1 может показаться, что для электронной
спектроскопии (спектроскопии в видимой и ультрафиолетовой областях
спектра) постройка прямого многоканального спектрометра
наименее вероятна, но фактически, создание многоканального
детектора ультрафиолетового излучения легко осуществляется на
практике с помощью фотографических методов. Разрешения
мелкозернистой фотографической пластинки вполне достаточно
для обеспечения необходимого громадного числа каналов. При
этом рассматриваемый спектр можно без особых усилий растянуть
до необходимой длины (несколько метров).
В рентгеноэлектронной* и фотоэлектронной спектроскопии
электроны выбивают из атомов или молекул с помощью
рентгеновских или ультрафиолетовых лучей. Испускаемые электроны
обладают поступательной энергией (и, таким образом, линейной
скоростью), которая зависит от энергетического состояния
электрона в первоначальном атоме или молекуле. Измеряя энергии на-
* В иностранной литературе часто используют для этого вида спектроскопии
■название ESCA (Electron Spectroscopy for Chemical Analysis — электронная
«спектроскопия для химического анализа). — Прим. ред.
675 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
блюдаемых выбитых электронов, можно определять энергии
электронных уровней в исходной молекуле. Выбитые же электроны
можно диспергировать в пространстве в соответствии с их
скоростями путем пропускания пучка этих электронов между двумя
параллельными заряженными пластинами. Это позволяет
использовать схему, показанную на рис. 20.3. Такая многоканальная схема
регистрации электронов стала осуществимой только недавно
после создания «видиконового» детектора, в котором поступающие
электроны ударяются о флуоресцентный экран на поверхности
телевизионной камеры. Поскольку электроны с различными
скоростями можно разделить так, чтобы они попадали в разные области
экрана, их поступление будет фиксироваться независимо
различными элементами телевизионной камеры. Таким образом в
настоящее время реально создание многоканального спектрометра (рис.
20.3) для рентгено- и фотоэлектронной спектроскопии, поскольку
для них необходимо лишь небольшое число каналов-детекторов
(табл. 20.1).
Менее заманчиво использование прямых многоканальных
приборов для низкоэнергетических видов спектроскопии,
перечисленных в табл. 20.1. Например, для микроволновой (электронного
парамагнитного резонанса или «чисто вращательной») спектроскопии
не существует подходящего широкополосного источника излучения.
Можно было бы использовать в качестве источника излучения
«черное тело», такое, как используется для генерации излучения
с более высокими энергиями [ксеноновый или водородный разряд
для ультрафиолетовой области, нагретая вольфрамовая проволока
для видимой области, глобар для ближней (к видимой)
инфракрасной и инфракрасной областей, пары ртути для дальней
инфракрасной области]. Однако из-за того, что черное тело работает
только при достаточно высокой температуре, получаемый с его
помощью поток излучения оказывается слишком мал для
использования в качестве источника излучения для микроволновой области
л областей спектра с более низкими энергиями. С другой стороны,,
можно было бы в качестве «широкополосного» источника
излучения применить набор отдельных («узкополосных») микроволновых
рпередатчиков. Однако число необходимых для работы каналов,
йтабл. 20.1) показывает, что цена такого прибора была бы
слишком высокой. Что касается многоканальных детекторов, то надо-
заметить, что фотографические пленки нельзя использовать при
й;лине волны более ~ 12000 А. Поэтому фотографические методы
неприменимы в длинноволновой спектроскопии. Для инфракрасной
спектроскопии имеются подходящие широкополосные источники,
ро при этом необходимо было бы растягивать спектр
приблизительно до 100 метров, чтобы разрешить нужные детали спектра с
ромощью существующих индивидуальных детекторов
(термоэлементов) с размерами около 1 мм. Однако цена такого
многоканального инфракрасного спектрометра опять оказывается слишком
676 ЧАСТЬ 6
высокой. Наконец, для
радиочастотной спектроскопии
(ядерный магнитный
резонанс, ионный циклотронный
резонанс) также имеются
широкополосные источники,
но стоимость конструкции
из десятков тысяч
отдельных узкополосных
детекторов-фильтров (см. ниже)
снова слишком высока.
Таким образом, для
инфракрасных,
микроволновых и радиочастотных
спектрометров прямой подход
с использованием
многоканальных приборов
неосуществим или по
геометрическим, или по экономическим
соображениям. Теперь мы рассмотрим два новых и весьма
полезных непрямых подхода: спектроскопию с преобразованием Ха-
дамарда и спектроскопию с преобразованием Фурье.
Рис. 20.4. Диспергирующий элемент (/),
окно (2), маска (3) и (один
широкополосный) детектор спектрометра Хадамар-
да (4).
Открыто около половины возможных позиций
щелн.
20.А. СПЕКТРОМЕТРЫ С КОДИРОВАНИЕМ
И ДЕКОДИРОВАНИЕМ («МУЛЬТИПЛЕКСНЫЕ»),
ИСПОЛЬЗУЮЩИЕ ПРЕОБРАЗОВАНИЕ ХАДАМАРДА
На рис. 20.4 показана схема прибора, которая позволяет
использовать метод Хадамарда: между «окном», вырезающим
желаемый участок спектра, и специальным (недорогим)
широкополосным детектором (рис. 20.2) помещена маска. Эта маска
сконструирована так, что наименьшее отверстие в ней равно ширине
(узкой) щели условного спектрометра (рис. 20.2) и при этом
остается открытой приблизительно половина всех возможных
местоположений щели. Последовательность открытых и закрытых
участков образует определенный беспорядочный узор (см. ниже).
Пусть спектр передаваемых интенсивностей (точки на нижней
диаграмме рис. 20.2) представлен спектральными элементами Х\,
%2, ..., xN (то есть х\ представляет собой интенсивность,.,
попадающую на детектор при i-м положении щели). При наличии маски
(рис. 20.4) детектор регистрирует общий сигнал у,
представляющий собой сумму всех желаемых спектральных элементов
(интенсивностей), помноженных на фактор щ, равный нулю или единице
в зависимости от того, закрыто ли соответствующее положение
щели или открыто:
y=atxt-\-atXi-\-.,.-\-aNXit, где а{ = 0 или 1 (20.1)
677 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Около половины а{ равно единице, а другая половина равна нулю.
Другими словами, детектор регистрирует одну наблюдаемую
величину, выраженную через N неизвестных величин (х\, х%, ...,xN)
посредством определенного «кода» [коэффициенты а\, ач, ..., fljv
в уравнении (20.1)]. Эта,ситуация совершенно аналогична случаю
одновременного взвешивания на весах половины неизвестных
грузов, который уже был разобран выше. Затем, чтобы получить
желаемый спектр неизвестных иптенсивностей (х\, х2, ..., xN),
удаляют первую маску и вводят на ее место вторую снова с
беспорядочным (но отличным от предыдущего) расположением открытых и
закрытых щелей (открыто вновь около половины щелей). Эту
новую маску выбирают так, чтобы расположение в ней щелей было
линейно независимо от первой маски. Такой путь позволяет
получить N наблюдаемых величин (общих интенсивностей излучения
У\> Уъ •■•> Уя, проходящих через N различных масок), выраженных
через N неизвестных (желаемых элементов интенсивностей спектра
Х\, х2, ..., xN) посредством «кода», в котором все коэффициенты
представляют собой нули или единицы, причем в любом уравнении
около половины коэффициентов равно нулю:
г yt =L(2iiX1 -J- й12Х2 -\- ... -\~Q]f{Xfr
уй — агххх + а2гх2 + • • • + a2NxN (20.2)
yN ==■ Q>Hvxx -\- атхг -\-... -\- Q>NNXN.
В соответствии с табл. 20.1 очевидно, что сконструировать,
скажем, 50 000 масок для получения таким путем спектральных
интенсивностей в инфракрасной области на практике невозможно. Более
того, даже крупнейшие из имеющихся вычислительных машин не
в состоянии надежно решать систему со столь большим чиблом
линейных алгебраических уравнений. Подход Хадамарда основан на
таком чрезвычайно удачном выборе узора щели, что обе эти
проблемы могут быть решены одновременно.
Особенно удобный способ получения N линейно независимых
расположений щелей для случая N=3 показан на рис. 20.5.
Вместо N отдельных масок достаточно сделать только одну
подвижную маску, состоящую из (2N—/) потенциально открытых
положений щели. Тогда при продвижении маски поперек
(неподвижного) спектрального окна в первое положение, показанное на
рисунке, мы создадим первое расположение щелей (вверху справа на
рис. 20.5). Сдвигая маску во второе (третье) положение, мы
создадим второе (третье) расположение щелей. Таким образом,
просто механически сдвигая единственную маску с (2АГ—1)
«отверстиями» поперек окна на одну ширину щели за раз, можно пере-
Положение подвижной
маски
Диспергирующее
устройство
л/*1 Ф? лАт Фиксированное
/v i г* с /у о спектральное —•
окно (А/-8)
А
Фиксированный __
широкопо/госный
детектор
„Код"маски =* ПОП- МИМ
y,=xt+x2 + 0
в Лв*х+0 +х3
Уз-0 + х2+х3
Х\ Х2 Х3 ,*,
Уг -> 1 1 0~
у,~+ 1 О 1
Уз->0 1 1J
.„Код" я
Хадамардл
2*1= У1+У2-У3
f 2хг = Уу-Уг+Уз
2*з = ~У1-1-у2+Уз '
Рис. 20.5. Схема работы 3-канального спектрометра Хадамарда.
Мы можем получить три желаемых закодированных методом Хадамарда комбинации общих
иитеисивностей в детекторе, располагая маску с пятью щелями ,1(2 N — 1)«=51 тремя
подходящими способами относительно фиксированного спектрального окна. Простота
вычислений по этой схеме показана на рисунке внизу слева. Экспериментальные интенсивности
(У\> Уг. Уз) представляет собой «взвешенные» суммы отдельных искомых иитенсивиостей
(*i. Хг, *3). «Весовые» коэффициенты равны либо нулю, либо единице (а, справа).
Отдельные искомые интенсивности легко получить из наблюдаемых иитенсивиостей путем
простого сложения или вычитания (б) с использованием оригинального «кода» Хадамарда,
в котором 0 заменен на — I (см. текст): а—наблюдаемые экспериментально общие
интенсивности света, прошедшего через окно с маской; б—интенсивности света для каждого
фиксированного положения щели в окне, рассчитанные из данных для всех трех
положений маски.
ходить от одного расположения щелей к другому. С точки зрения
вычислений эти специфические расположения щелей делают
возможным получение желаемых элементов спектральных
иитенсивиостей (xh хч., ..., xN) просто путем комбинирования полученных
экспериментальных общих иитенсивиостей (уь Уч, ..., Un),
прошедших через маску, согласно «коду», полученному замещением 0 на
—1 в первоначальном «коде», которым выражены у\ через х{ (рис.
20.5). Другими словами, мы можем получить х4 из
экспериментальных величин у\ не прибегая, как в обычном случае, к решению
системы из N уравнений с N неизвестными. По причинам как
вычислительным, TajK и механическим подход Хадамарда (рис. 20.4 и
20.5) дает в УМ/2 раз лучшее отношение сигнал/шум по сравнению
с обычными спектрометрами (рис. 20.2), сканирующими спектр
при единовременно открытой только одной щели. Это происходит
потому, что в спектрометре Хадамарда в течение каждого измере-
679 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
ния открыты около половины из N возможных щелей, а не только
лишь одна. Причем оба этих типа экспериментов требуют одного
и того же времени для получения N измерений отдельных интен-
сивностей (вычисления столь просты, что могут быть выполнены
небольшим, предназначенным для этой цели компьютером за
время, незначительное по сравнению с требуемым для получения
экспериментальных данных). С другой стороны, с помощью
спектрометра Хадамарда можно получать то же самое отношение
сигнал/шум за время в N/4 раз меньше, чем потребовалось бы
обычному прибору с одной щелью.
Внимательный читатель может быть заметил, что три
расположения щелей, показанные на рис. 20.5, отличаются друг от друга
«циклической перестановкой». Она состоит в том, что каждое
последующее расположение щелей получается из предыдущего
перестановкой первого выбранного (открытого или закрытого) состояния
щели с левой стороны данного ряда в крайнее правое
положение, чтобы получить следующий ряд. Эта характерная
особенность является общей для всех экспериментов Хадамарда.
Оказывается всегда, когда N= (2П—1), возможно составить N линейно
независимых расположений щелей, которые обладают этим
свойством «циклической перестановки» (см. задачи к этой главе).
Описанный выше метод Хадамарда еще очень молод. Поскольку до
сих пор его использовали главным образом в инфракрасной
спектроскопии, экспериментальные подробности этого метода будут
рассмотрены ниже.
Некогерентные и когерентные спектрометры
Для того чтобы перейти к методам Фурье, важно понять, что
рассмотренные к настоящему моменту спектрометры (рис. 20.2—
20.5) могут работать с некогерентными источниками излучения
(различные компоненты излучения источника не согласованы по
фазе). Для таких спектрометров с некогерентными источниками
методы кодирования Хадамарда создают возможность
эффективного увеличения ширины щели, не жертвуя при этом разрешением
(рис. 20.6). Однако использование сканирующих спектрометров с
«когерентным» источником имеет свои важные преимущества (см.
ниже). Когерентный источник делает возможным другой тип схемы
кодирования-декодирования (Фурье) для увеличения
спектрального окна при сохранении разрешения (рис. 20.6). Наконец, еще
одна причина того, что не только метод Хадамарда (некогерентный
источник), но и метод Фурье (когерентный источник) можно
применять в инфракрасной спектроскопии — это возможность
использования интерферометра Майкельсона (прибор, применяемый в
инфракрасных спектрометрах с преобразованием Фурье). В
настоящем контексте его можно считать средством для эффективного
преобразования некогерентного излучения в когерентное (гл. 20.Б).
680 ЧАСТЬ в
Спектроскопии
Некогерентная Когерентная
Рис. 20.6. Классификация видов
спектроскопии по источникам излучения
и ширине полосы регистрации
сигнала.
Источник когерентного излучения и когерентный детектор в
спектрометре обеспечивают два важных практических
преимущества. Во-первых, поскольку с помощью электроники частоту
излучения когерентного источника легко измерять с весьма высокой
точностью, положения линий в спектре можно определить очень
точно, просто измеряя частоту источника при (медленном)
сканировании через спектральное окно. Во-вторых, когерентное
излучение создает возможность применения системы электронных
фильтров, которая обеспечивает сколь угодно высокое разрешение
спектрометра. Таким образом, сводя уширение спектральных линий за
счет прибора к минимуму, можно сделать так, чтобы форма
наблюдаемой спектральной линии была близка к . характерному
спектру образца, а не отражала приборные артефакты.
На рис. 20.7 показана схема устройства спектрометра с
когерентным источником на примере гипотетического инфракрасного
спектрометра с лазерным источником излучения для
использования в колебательной спектроскопии. Излучение источника
представляет собой плоскополяризованное электрическое поле,
величина которого синусоидально меняется во времени. При
взаимодействии с электрическим диполем полярной молекулы электрическое
поле будет заставлять его колебаться с частотой излучения.
Амплитуда колебания этого диполя будет наибольшей, когда частота
воздействующего электрического поля совпадет с собственной
частотой колебания диполя (поле действует «в резонанс»—
гл. 13.Б.2). Если источник дает когерентное излучение, то все
диполи в данной области пространства будут колебаться вместе,
создавая в образце макроскопический переменный электрический
диполь. В этом случае макроскопический переменный диполь
индуцирует переменный заряд на параллельных пластинах конденсатора,
в который помещен образец (рис. 20.7), и, таким образом,
соответствующее переменное напряжение. Это индуцированное
напряжение можно затем усилить и (это наиболее важная ступень)
умножить (в «смесителе») на переменный сигнал от источника, а
произведение с помощью электроники разложить на сумму и разность
частот двух синусоидальных волн так же, как произведение двух
Узко- Широко- Узко- Широко-
/толосная полосная полосная по-лоСная
Преобразование Преобразование
Хадамарда Фурье
681 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
1
А г
t—
л:
—*-
V
с
*) i
Рис. 20.7. Гипотетический инфракрасный спектрометр с лазерным источником
излучения.
/ — монохроматический перестраиваемый когерентный источник; 2 — смеситель; 3 — фильтр;
4 — самописец.
синусоидальных волн можно было бы алгебраически (с помощью
тригонометрических тождеств) разложить на синусоидальные
волны с «суммарной» и «разностной» частотами. Низкочастотный
фильтр задерживает «суммарную» (более высокую) частоту и
пропускает только «разностную» (более низкую) частоту, которая
затем и регистрируется. ОписанньРй выше процесс смешения с
высокой эффективностью выделяет маленькую область спектральных
частот, центр которой сосредоточен на частоте источника и ширина
которой определяется шириной полосы пропускания
низкочастотного фильтра.
На более привычном языке сказанное выше означает, что в этом
типе спектрометров положение щели определяется с помощью
частоты (измеряемой очень точно) излучения источника и ширина
щели определяется шириной полосы пропускания электронного
низкочастотного фильтра, которая, следовательно, может быть
сделана произвольно широкой или узкой без каких-либо
механических регулировок геометрии спектрометра. Спектрометры, в
которых макроскопическое изменение какого-то физического свойства
образца индуцируется излучением когерентного источника и затем
регистрируется электронной системой способом, описанным выше
(например, рис. 20.7), уже давно применяются в радиочастотной
спектроскопии ядерного магнитного резонанса (ЯМР) и
спектроскопии ионного циклотронного резонанса (ИЦР), а недавно были
введены в практику микроволновой и инфракрасной
спектроскопии.
20.Б. ФУРЬЕ-СПЕКТРОСКОПИЯ
На первый взгляд кажется, что методы с преобразованиями
Фурье чужды нашему пониманию, поскольку мы привыкли
полагаться на глаза и уши, которые анализируют окружающий нас мир
по частотам:
682 ЧАСТЬ 6
u>
Рис. 20.8. Зависящие от времени (слева) и частоты (оправа) представления
импульсов постоянного тока ^грех различных длительностей.
свет различают по цвету, а звук—по его высоте. Однако можно
точно также выразить световую или звуковую волну в виде
величины (электрического поля или давления), которая непрерывно
меняется во времени. Преобразование Фурье представляет собой
математическую операцию, которая позволяет перейти от временного
описания явления (например, синусоидальной волны, которая
колеблется бесконечно долго) к частотному описанию (в нашем
примере формы пика на частоте синусоидальной волны колебания).
Пример такого перехода дан на рис. 20.8. На нем показан импульс
постоянного тока (ток с нулевой частотой), который резко
возрастает в нулевой момент времени и резко падает через время Т.
Интуитивно можно было бы предположить (как в предшествующем
примере), что частотное выражение такого импульса должно
представлять собой сигнал с нулевой частотой. Однако действительное
частотное описание (вверху справа на рис. 20.8) такого импульса
представляет собой сигнал, охватывающий область частот вблизи
нуля. Частотное представление более короткого импульса (в
середине на рис. 20.8) охватывает более широкую область частот. В
пределе при бесконечно узком импульсе постоянного тока (внизу на
рис. 20.8) его частотная характеристика будет представлять собой
спектр в виде совершенно прямой линии. Таким образом, очень
короткий, сильный импульс электрического поля на самом деле
вызывает широкополосное излучение в частотном диапазоне,
обратно пропорциональном длительности импульса. Математическое
преобразование, устанавливающее соответствие между
временными (слева) и частотными (справа) описаниями сигнала, на рис.
20.8 называют преобразованием Фурье.
ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
&• Схемы рис. 20.8 наводят на мысль, что широкополосное
частотное возбуждение для многоканальных или мультиплексных
спектрометров можно генерировать, используя достаточно короткий
импульс электромагнитного излучения. [Если импульс представляет
робой по форме волны не постоянный, а переменный ток, то схемы
на рис. 20.8 все еще будут оставаться в силе, за исключением того,
что теперь «центр» описывающих импульс частот будет
расположен на частоте переменного тока, а не на нулевой частоте — см.
Верхний чертеж на рис. 20.9.]
Из табл. 20.1 видно, что, например, для спектроскопии ЯМР
Необходима полоса возбуждения шириной около 10 кГц. Рис. 20.9
(верхний чертеж) показывает, что такое возбуждение можно
обеспечить просто путем применения радиочастотного импульса,
длительность которого имеет порядок 10~5 с. В качестве другого
примера можно привести спектроскопию электронного удара, в
основе которой лежит быстрое пропускание электрона вблизи
исследуемой молекулы. Этот пролетающий электрон продуцирует
Ьчень короткий, резкий импульс электрического поля в районе
молекулы и, таким образом, действует как источник очень
широкополосного почти равномерного облучения. В этом случае ширина
полосы частот вполне достаточна для возбуждения того же самого
типа переходов, которые традиционно изучаются фото- и рентгено-
электронной спектроскопией. Имея описанный выше метод для
Широкополосного возбуждения, мы теперь рассмотрим
широкополосную регистрацию резонансных колебаний образца с различны-
ми частотами.
При облучении одиночного осциллятора на его резонансной
частоте амплитуда колебаний будет возрастать (гл. 13.Б). Если та-
jkoe облучение прекратить, то колебание осциллятора будет про-
y(t) = cosjtot);0<r < Т
yit) = О ; t < OuMJt > т
yit)=e-
y{t) = 0
-Ш
,t>0
;t<0
y(t) = <H'/r)cos (u>t); 0<t<T
y{t) = 0 ;.t< OmuI > T
о
Рис. 20.9. Частотные представления (справа) трех типов спектрометрических
сигналов, зависящих от времени (слева).
684 ЧАСТЬ 6
должаться с амплитудой, уменьшающейся по времени (обычно
экспоненциально — гл. 13.В). Этот процесс показан на рис. 20.9
(слева) для граничных условий трех характерных типов. Так, если
вынужденные колебания осциллятора заметно не уменьшаются за
время наблюдения Т (верхний чертеж рис. 20.9), соответствующее
частотное представление имеет форму функции, имеющую
сходство с распределением амплитуды интенсивности света при
дифракции (Фраунгофера) от щели. С другой стороны, если колебание
наблюдают в течение времени, равного нескольким временам
релаксации его затухания (средний чертеж на рис. 20.9), спектр
частотного представления приближается по форме к хорошо
известной кривой Лорентца, характерной для многих типов
спектроскопии. Наконец, нижний чертеж на рис. 20.9 иллюстрирует
промежуточный случай, когда время наблюдения Г (время получения
экспериментальных данных) имеет тот же порядок, что и время
релаксации т процесса затухания колебаний. Необратимое затухание
колебаний может происходить вследствие «спонтанного
испускания» («излучательное затухание» — гл. 19.Б.1) или такого
взаимодействия образца (ядро, ион, молекула) с его окружением,
которое прерывает непрерывные регулярные колебания образца.
Взаимодействие с окружением может осуществляться за счет
столкновений между нейтральными частицами (микроволны, свет
в инфракрасной, видимой и ультрафиолетовой областях),
столкновений ион — молекула (ионный циклотронный резонанс),
вращательной диффузии (ЯМР, ЭПР) или расхода возбужденных частиц
на химическую реакцию.
Теперь можно легко понять преимущество (Фелжета) многока-
нальности при подходе Фурье. Предположим, что временную
зависимость для набора вынужденных колебаний различных
осцилляторов y(t) измеряют в N равноотстоящих точках в течение
времени наблюдения Т. Каждую из этих отобранных точек
зависимости от времени y(tn), где л = от 1 до N, представляют затем в виде
суммы всех дискретных, зависящих от частоты спектральных
амплитуд х((йп) согласно показанному ниже «коду»:
У &) = Яп* К) + 0i2* W + •• • + alNx (<oN)
У (tt) = я2г* К) + Д22* К) + • •. Н- я»гг К) (20-3)
V {tN) = amx (ш,) + aN2x (ш2) -f... -l aNNx {<oN)f
где
апт=ехр[2ъШп/Т] - (20.4)
или
агм — ехр [2nmn/N].
(20.5)
685 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Уравнения (20.3) следует сравнить с уравнениями (20.2):
поскольку опять имеется N независимых, отобранных, зависящих от
времени экспериментальных точек [y(ti), y(t2), ...*/(ЗД] и каждая
выражена через все N дискретных зависимых от частоты амплитуд
[х(ац), лг(со2)> ..., Jc(oajv)], то снова имеется возможность
«декодировать» наблюдаемые данные, чтобы получить желаемый спектр,
путем решения системы линейных алгебраических уравнений
(20.3). К счастью, процедуру декодирования (известную как
«дискретное» преобразование Фурье) можно выполнить очень легко с
помощью предназначенного для этой цели компьютера, используя
знаменитый алгоритм, полученный в 1965 г. Кули и Тыоки. Более
того, в противоположность методу Хадамарда, в котором в течение
любого данного наблюдения регистрируется половина возможного
спектра [половина апт в любом отдельном ряду системы
уравнений (20.2) равна нулю], в эксперименте Фурье величина каждого
аПт в уравнениях (20.3) равна единице
| апт | = | exp \2wrm]N\ \ = 1 (20.6)
Таким образом, эксперимент Фурье проводят так, что все
возможные «щели» открыты.
Учитывая соображения, использовавшиеся ранее в примере с
двухчашеч-ными весами, теперь становится ясным, что
регистрация зависящих от времени вынужденных колебаний с
последующим преобразованием Фурье для получения колебаний, зависящих
от частоты, дает возможность получить частотный спектр,
имеющий в УЫ раз большее отношение сигнал/шум за тот же самый
общий период наблюдения, или спектр, имеющий то же самое
отношение сигнал/шум, но за время в N раз меньшее, чем требуется
обычному спектрометру, который сканирует спектр непрерывно с
помощью одной щели, при использовании узкополосного
детектора*.
Последнее замечание относительно частотных спектров,
получаемых с помощью фурье-преобразований, зависящих от времени
вынужденных колебаний, касается сравнения формы их линий с
соответствующими линиями, получаемыми на обычном
спектрометре с медленной разверткой. При том общем условии, что
вынужденные колебания системы линейны (амплитуда вынужденных
колебаний пропорциональна амплитуде возбуждающего облучения;
см. гл. 19.Б.З), обычные спектры при медленной развертке и спект-
* В частном случае инфракрасного Фурье-спектрометра, основанного на
интерферометре Майкельсона, спектр получают дискретным преобразованием Фурье
(пространственной) интерферограммы. Поскольку половина интенсивности
сигналов спектра теряется на полупроницаемом зеркале интерферометра,
инфракрасный спектр с преобразованием Фурье обеспечивает только половину
полного, (в N раз) экономящего время выигрыша Фелжета (гл. 20 н рис. 20.17а).
686 ЧАСТЬ 6
ры после преобразования Фурье идентичны при достаточно
большом времени наблюдения (средний чертеж на рис. 20.9).
Пример. Форма спектральной линии после преобразования Фурье
В этом примере мы получим форму спектральной линии, показанную па
среднем чертеже рис. 20.9 (два других случая читателю предлагается разобрать
самостоятельно в задачах к этой главе). Рассмотрим, следовательно, зависящее
от времени колебание, которое представляет собой экспоненциально
затухающую синусоидальную волну, наблюдаемую во времени неопределенно долго
f(f)=*-(*/*>casK,f) 0<*<co. (20.7)
Можно определить формы трех отдельных линий в частотном спектре А(ш),
В (со) и С(и>), полученные из зависимого от времени колебания:
00
А{и>)
И
f(t) cos{wt)dt,
—ОО
00
5Н=4" jf (0s^ (©/)л,
— 00
С (<о) = VА2 (ш) + В2 (ш) ,
(20.8а)
(20.86)
(20.8в)
Л (со) называют «косинусоидальным преобразованием Фурье» f(t),
B(u>)—«синусоидальным преобразованием Фурье» /(/), а С(ш)—«величиной» или
«абсолютным значением» частотного представления f(t).. [С(со)2 называют
«энергетическим спектром» f(t)]. Используя определенные интегралы (таблица интегралов)
00
je"a/ cos {bt)dt = [aj{a2-\-b2)\
и
00
^e-ats'm{bt)dt = lbi{a2-{-b%
(20.9а)
(20.96)
а также тригонометрические тождества
cos A cosB=-2- [cos (A — В) + cos {А-{-В)] (20.10а)
и
smAcosB = ^-[s'm{A--B) + sm(A-{-B)\ (20.106)
легко получить А (со) и В (со). Для этого надо подставить уравнение (20.7)
в уравнения (20.8) и затем, используя уравнения (20.10) для упрощения инте-
687 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
гралов, привести их к виду, данному в уравнениях (20.9) (см. задачи к этой
главе):
4°)=-£-
2л
(т) +((0-й)о)2 ("Г )2 + («+«<,)2J
5(0))=-'-
(<■> — Q>o) («> + "o)
"Г/ 1 N2
.-j +(©-4,)» (—) + (co + co0)2jS
_J_/ (CO-CO,),* ^ -
Из уравнений (20.11) сразу же можно получить С((о)
СИ- j- Л, * ) (20. И в)
Поскольку нас интересует, как правило, частота колебаний образца вблизи его
резонансной частоты (со^со0), то можно с полным основанием пренебречь
вторым членом в выражениях для Л (со) и В(со), как это и показано в уравнениях
(20.11а) и (20.116).
Форма линий для Л(со), £(со) и С(со) в частотном спектре показана на
рис. 20.10 (нижняя диаграмма). Здесь читатель должен обратить внимание на
то, что Л (со) и В(со) по своей форме представляют собой «поглощение» и
«дисперсию», которые мы первоначально вывели для стационарных колебаний при
непрерывном возбуждении (гл. 13.Б, рис. 13.5, <Э). Таким образом, мы доказали
один пример эквивалентности преобразования Фурье затухающих колебаний при
импульсном возбуждении и стационарных колебаний при непрерывном
возбуждении.
Как показано ниже, свойства «лорентцевых» форм линий уравнения (20.11)
могут быть легко вычислены.
Высота пика. Экстремумы (максимум и минимум) находят сразу, изучая
наклон каждой кривой, и в точке (точках), где он равен нулю, вычисляют
величину функции.
Л (со), очевидно, имеет максимум при со=со0, где высота пика равна (1/2л)т.
С(<о) также имеет максимум при со=(0о, где также имеет высоту пика
(1/2я)т.
1
Легко можно найти, что В (со) достигает экстремумов при (со—со0) = ±—,
где его величина равна
±(1/2я)ф.
Ширина пика. Определяется простейшим образом, как ширина пика на
половине максимальной высоты (для Л (со) и С(со) или как разность частот между
экстремумами для В(со). Приняв Л (со) (или С(со)) равной половине ее
максимальной высоты и решив уравнения для со, легко получить, что:
пик Л (со) имеет ширину (2/т) на половине его максимальной высоты;
пик С(ю) имеет ширину (2УЗ/т) на половине его максимальной высоты.
688 часть б
Затухающие во времени
колебания
Частотные спектры
\ 2
Время, с
-20-10 0 10 20 -20-10 0 10 20 -20-10 0 10 2П
Рис. 20.10. Затухающие во времени колебания и частотные спектры этих
затухающих колебаний, полученные методом преобразования Фурье, для трех
характерных случаев затухающих во времени колебаний осцилляторов одного типа.
В осцилляторах возбуждали колебания вплоть до нулевого момента времени (возбуждение
колебаний не показано). Затем возбуждение прекращали и, начиная с нулевого момента
времени, регистрировали колебания. Колебания во времени показаны с амплитудой, равной
единице, поэтому в частотном спектре сигналы каждого типа даны' по вертикали в
единицах 1/2 я. Функции А(а>), В(а>) и С(ю) нз уравнеинй (20.11) обозначены на рисунке
как поглощение, дисперсия и абсолютное значение. (Для последующего обсуждения ср. с
рис. 20.9.)
Разность частот между экстремумами В(&) (см. расчет высоты пика)
составляет (2/т),
Подобным способом можно выполнить расчет других форм линий частотных
спектров, показанных на рис. 20.10. Такая возможность предлагается читателю
в качестве упражнения (см. задачи к этой главе).
Наконец, из рис. 20.10 ясно, что затухающие во времени колебания
необходимо регистрировать в течение достаточно длительного времени так, чтобы
форма линий в частотном спектре, полученном с помощью преобразования Фурье,
точно соответствовала лорентцевой форме линий стационарного режима
колебаний. В задачах к этой главе читатель сможет рассчитать, что ширина этих
линий различается примерно на 10% при регистрации затухающего временного
сигнала в течение времени, равного трем коэффициентам затухания. Проведен-
689 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
иые расчеты дают относительно полное описание форм линий спектра,
полученного с помощью преобразования Фурье, и мы можем теперь -рассмотреть
некоторые экспериментальные спектры.
Пример. Ядерный магнитный резонанс с преобразованием Фурье (ЯМР-ПФ)
Методы Фурье оказали, вероятно, большее влияние на спектроскопию ЯМР,
чем на любой другой вид спектроскопии. ЯМР-эксперименты основаны на
возбуждении, представляющем собой короткий импульс радиочастотного
переменного магнитного поля с частотами, близким^ к «собственным» ларморовским
частотам прецессии ядер в образце. После возбуждения проводят регистрацию
сигналов, используя схемы со смесителем и фильтром того же типа, что и в
гипотетическом инфракрасном спектрометре, показанном на рис. 20.7 (отличие
заключается лишь в том, что ядерная прецессия — это движение круговое, г не
линейное).
Важно, чтобы короткий импульс радиочастотного магнитного поля с
частотами, близкими к ларморовским частотам интересующих иас ядер (скажем, ядер
углерода-13), действовал иа образец в течение времени, достаточного для того,
чтобы вызвать поворот равновесного магнитного момента ядер углерода-13 на
Серин, pD = 7
Рис. 20.11. Зависимые от
частоты (а) и времени {б и в)
сигналы спектра ЯМР-13С для
аминокислоты серина (0,5 М,
15%-ное обогащение по угле-
роду-13, нейтральный pD).
[Взято с изменениями из Ног-
sley W., Sternlicht H., Cohen J. S.,
J. Amer. Chem. Soc, 92, 680 (1970).]
a — обычный спектр ЯМР-,3С серн-
на, снятый с медленной разверткой
по частоте прн непрерывном
возбуждении, б н в — зависимые от
времени сигналы, полученные
возбуждением образца
широкополосным импульсом излучения при
использовании смесителя, эталонной
частоты и низкочастотного фильтра,
выбранного так, чтобы выделить
частоты области, обозначенной А
[для зависимого от времеин сигнала
на чертеже (б)] или Б [для
зависимого от времени сигнала на
чертеже (в)] (см, текст).
Преобразование Фурье, например,
зависимого от времени сигнала (в), дало
бы частотный спектр области,
помеченной Б на верхней части
рисунка. Видно, что два
различающихся пика поглощения ЯМР-13С
регистрируются одновременно (в).
В то же время прн использовании
обычной регистрации в
стационарном режиме было бы необходимо
сканировать эти пики поочередно.
-Н"К
H*h
а
Серинt pD = 7
690 ЧАСТЬ 6
а
*'^^^P'^rtй^%ч^
ЮООс
Юс
CHCh
jK^
-1000 Гц-
Рис. 20.12. Временное преимущество применения преобразования Фурье при
регистрации спектра в сравнении с традиционным методом ЯМР.
[Из Hill #., Freeman R., Introduction to Fourier Transform NMR, Varian Associates
Analytical Instrument Division, Palo Alto, Calif., 1970.]
a — протонный спектр ЯМР холестернлацетата (0,0037 М) с медленной разверткой прн
непрерывном возбуждении; б—протонный спектр ЯМР-ПФ, полученный за 1/100 времени,
потребовавшегося для получения обычного спектра (а) н имеющий то же самое отношение
сигнал/шум. С другой стороны, можно получить спектр ЯМР-ПФ с гораздо лучшим
отношением снгнал/шум путем накопления множества зависимых от времени сигналов (каждый
из которых несет информацию о всем спектре) за то же самое время, которое было бы
необходимо для получения единственного традиционного спектра (рис. 8.5).
90° в плоскость х'—у', где расположен детектор (рис. 16.21). Если
возбуждающий импульс достаточно короткий, его частотное представление будет
соответствовать широкополосному источнику возбуждения с равномерным по существу
распределением мощности в интересующей нас спектральной области (рис, 20.9).
После прекращения этого импульса ядерные магнитные моменты прецессируют
с соответствующими им ларморовскими частотами. Используя схему со
смесителем, низкочастотным фильтром и детектором согласно рис. 20.7, регистрируют
сигналы только в нужной Полосе частот и накапливают их в небольшом
компьютере. Сигналы, изменяющиеся во времени, с помощью преобразования Фурье
превращают в сигналы частотного спектра ЯМР в интересующем нас диапазоне
частот, как показано на рис. 20.11.
На рис. 20.11а дан обычный (при непрерывном возбуждении) спектр
поглощения ЯМР-13С серииа при нейтральном pD. На рис. 20.11,6 показан
изменяющийся во времени сигнал ЯМР, полученный при импульсном возбуждении и
последующей регистрации с применением системы смеситель — фильтр при
использовании низкочастотного фильтра с верхним пределом по частоте около
100 Гц. Поскольку частота эталонного сигнала (рис. 20.7) отлична от
интересующей нас частоты ЯМР-13С примерно на 14 Гц, изменяющийся во времени
сигнал представляет собой затухающее колебание с частотой примерно 14 Гц.
Аналогично, на рис. 20.111, е показан изменяющийся во времени сигнал,
получающийся в результате действия возбуждающего импульса с частотой в
окрестности резонансных частот двух других атомов 1ЧС с последующей регистрацией
с помощью системы смеситель — фильтр. При этом рассматриваемая частота
была выбрана на ~ 10 Гц меньше резонанса а и па ~ 68 Гц меньше резонанса £,
так что оба сигнала были пропущены фильтром. В этом случае зависимый от
691 ПРЕЙРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
времени сигнал представляет собой наложение двух колебаний с приблизительно
равной амплитудой. Преобразование Фурье зависимого от времени сигнала на
рис. 20.11,6 дает один пик поглощения, соответствующий области, помеченной
на рис. 20.11, а как А. В то же время преобразование Фурье затухающего сиг-
нала на рис. 20.11, в дает спектр с двумя пиками поглощения в области,
помеченной на рис. 20.111, а как Б.
Рис. 20.11, в иллюстрирует преимущество многоканальной фурье-спектроско-
пии: два пика поглощения с различными частотами регистрируются
одновременно, а не лоочередно, как это имеет место в обычном спектрометре с медленным
сканированием. Еще более наглядный пример показан на рис. 20.12, в котором
методом преобразования Фурье изменяющегося во времени сигнала (рис. 20.12, б)
необходимый спектр был получен в 100 раз быстрее по сравнению с обычным
медленным сканированием непрерывно возбуждаемой системы с поочередной
регистрацией пиков (рис. 20.12,а). С другой стороны, в спектрах ЯМР-ПФ есть
возможность получать гораздо лучшее отношение сигнал/шум, чем в
традиционных спектрах ЯМР. Этого можно достичь, накапливая для получения одного
спектра ЯМР-ПФ много зависимых от времени сигналов, каждый из которых
по существу эквивалентен одному обычному спектру ЯМР (см. рис. 8.5).
Прекрасная селективность, обеспечиваемая ЯМР-13С (рис. 20.113), была
известна химикам уже на протяжении 20 лет, ио спектры ЯМР-ПС почти любых
(за исключением самых маленьких) молекул были практически недоступны до
тех пор, пока в 1965 г. не были введены в практику методы Фурье. В настоя-
^с=
№
М(СО)„
К\\\\Ч
R2CO
■КЗ
RCHO
■ Q
Алкены
У/////У////////////Л
apu/i С
Y////////A
арцл
С-О
У//////Л
арил
C-N -CsN
Y//A UUU
—с=с—
Алканы
L(CH2)„J
-с-о
□
и-<-3
-C-N
RCOOH
С-Р
У7\
-С-Р
RCOOR
6
в
КЗ
cs2
200
С6Н6 ССЦ CHC!3 CH,OH C6H1?
I J I I I
100
H-
CH4
0 Af.ff.
TMC
Рис. 20.13. Диапазоны частот поглощения ЯМР-13С в нейтральных органических
соединениях.
Для обозначения гибридизации атомов углерода использованы символы: а для sp3, б для
sp1 и $ для sp; карбонильные углеродные атомы различают как насыщенные г и сопряжен-
иые д Подобная разница в частотах поглощений для различных типов связей атомов
углерода наблюдается при поглощении в инфракрасной области, но в водных растворах
инфракрасное поглощение большинства соединений невозможно наблюдать вследствие очень
сильного поглощения в инфракрасной области спектра самой водой (см. примеры по
инфракрасной спектроскопии с преобразованием фУРье>-
Шз Stothers J. В., Carbon-13 NMR Spectroscopy, Academic Press, New York, 1972, p. 13.]
Тетраметилсилаи (TMC) — это соединение, используемое в качестве эталона для получения
частоты от которой отсчитывают остальные «химические сдвиги> атомов углерода-13.
692 ЧАСТЬ 6
шее время в повседневной лабораторной работе используют сотни спектрометров
ЯМР-13С-ПФ. Применение спектров ЯМР-ПФ для биохимических исследований
продемонстрировано в гл. 14.Г.
Пример. Масс-спектрометрии ионного циклотронного резонанса с
преобразованием Фурье (ИЦР'ПФ)
ЯМР и масс-спектроскопия являются, вероятно, двумя ндиболее полезными
для химиков аналитическими спектроскопическими методами. Мы не можем
здесь- подробно обсудить работу обычных масс-спектрометров. Скажем только,
что в основе их работы лежит прямая или непрямая ионизация нейтральных
газообразных молекул потоком ускоренных электронов. Затем эти ионы
отклоняют в соответствии с характерным для них отношением заряда к массе
[используя электрически заряженные пластины и (или) магнитные поля так, как
это делают для контроля «сведения» или «фокуса» в цветной телевизионной
трубке], после чего они проходят через маленькую щель в детектор ионов.
В этом случае получающиеся ионы с различным отношением заряда к массе
можно регистрировать по очереди, изменяя электрическое или магнитное
отклоняющее поле (или оба поля) по существу так же, как это делается в фотонном
однощелевом спектрометре на рис. 20.2. [Существует тип масс-спектрометра,
в котором ионы с различным отношением заряда к массе одновременно
разделяются в пространстве и регистрируются на фотопленке, как в многоканальном
детекторе (рис. 20.3). Однако из-за того, что перед тем, как увидеть спектр,
надо обработать фотопленку, в этом приборе почти не удается использовать
обсужденную ранее экономию времени, которую дает выигрыш Фелжета.]
Теперь следует рассмотреть возможность использования в масс-спектрометрах
мультиплексных схем регистрации. Отметим, что метод Хадамарда здесь
неприменим, поскольку шум в масс-спектрометрах — это «шум источника» (см.
сноску в разд. «Грузы на весах») и поэтому преимущество Фелжета теряется.
Однако существует один тип масс-спектрометра, в котором возможно
осуществить когерентное широкополосное возбуждение и регистрацию, — спектрометр
«ионного циклотронного резонанса» (ИЦР), позволяющий использовать методы
Фурье почти точно так же, как их используют в спектроскопии ЯМР.
На рис. 20.14 показана принципиальная схема ИЦР-спектрометра.
Движущийся в магнитном поле ион вынуждают перемещаться по круговой траектории.
г&
XXX V
В ххх
XXX
&
Самописец
v=
2/тт
Детектор
со смеситем
и фильтром
т
1 L
400 100 60 АО
L
(
20Q
до
_L
20
±
400
600
600
16
>
кГц
\
1Т
Рис. 20.14. Схематическое изображение масс-спектрометра ионного
циклотронного резонанса (ИЦР). Работа его описана в тексте.
693 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Частота кругового движения этого иона пропорциональна его отношению заряда
к массе.
|у ■= (gfl/2n"i»j] (20.I2)
Когда такие ионы облучают поляризованным по кругу (вращающимся)
электрическим полем с частотой, близкой к ионной «циклотронной» частоте
[уравнение (20.12)], результирующее движение ионов становится
пространственно когерентным (все ионы движутся сгруппированно). При этом ионы поглощают
энергию излучения путем увеличения радиусов своих циклотронных орбит
(рис. 20.14). Поскольку ионы движутся по существу все вместе, их общее
движение в циклотроне будет индуцировать макроскопическое изменение
напряжения на расположенных вокруг пластинах. Это переменное напряжение можно
усилить, подвергнуть «смешению» и «фильтрованию», а затем зарегистрировать
так же, как это делалось в гипотетическом инфракрасном спектрометре на
рис. 20.7. Если не считать того, что «собственное» движение системы в
циклотроне круговое, а не линейное, то спектрометры на рис. 20.7 и 20.14 в принципе
очень похожи. При напряженности внешнего магнитного поля IT (10 кГс)
частоты ИЦР для обычных однозарядных ионов с массами от 16 до 400 лежат
в (радиочастотной) области примерно между 35 кГц и 1 МГц (это показано
внизу на рис. 20.14).
Таким образом, ИЦР-спектрометр дает сигнал всякий раз, когда частота
облучения соответствует ионной циклотронной частоте для присутствующих
в образце иоиов с данным отношением заряда к массе. Другими словами,
прибор может функционировать как масс-спектрометр, определяющий величину
отношения заряда к массе, при облучении образца переменным электрическим
полем, частоту которого медленно меняют в области, задаваемой уравнением
{20.12), и регистрации поглощения энергии, которая производится в том же
самом духе, как это было в обычном сканирующем однощелевом спектрометре на
рис... 20.2.
На рис. 20.15, а показаны изменяющиеся во времени сигналы ИЦР,
вызванные широкополосным возбуждением. Ширина полосы выбрана так, чтобы соз-
-4 Н
C,0Hi4,9Bf+
С10Н,479Вг+ и СшНи8,Вг+
а
б -
213
214 215
т/е _
Рис. 20.15. Изменяющийся во времени сигнал (а) и частотный спектр (б),
полученный преобразованием Фурье зависимых от времени данных, для
колебаний при ионном циклотронном резонансе образца ионов бромадамаитана
в случае возбуждения широкой полосой частотней, текст).
Маленький пик с (т/е) =214 —это ,8С-сателлит иона Ci«Hu7*Br+ (содержание атомов "С в
природной смеси составляет около 1%, ио поскольку в молекуле имеется 10 атомов
углерода, содержание сателлитных молекул с одним атомом 13С будет около 10%). На схеме
изменяющегося во времени сигнала ясно видно наложение двух синусоидальных волн,
частоты которых представляют собой собственные частоты ИЦР двух главных ионов,
имеющихся в образце. Изменяющийся во времени сигнал был получен за ~ 0,1 с.
ГИз Comisarov М. В., Marshall A. G., неопубликованные данные, и Conusarov М. В.,
Marshall A. G., J. Chem. Phys.. 62, 293 (1975U
a
200
250
m
в
242
300 350
N4J и' Yi
о- "VV**
400;
о=р
i
о
л1 1
■O-CHjOpN^
NH^ но
I I I I I I I I I I 1 г I I I I I I \ I I I I I I I I I f Г г I I I I I I I I I I I I I I I м I
200 250 300 350 400 450 550 500 606 "650
т
е
Рис. 20.16. Обычные масс-спектры ИЦР морфина (о) и нуклеотида (б).
Давление паров образца в этих экспериментах было около I0-7 торр (очень иизкое па
обычным масс-спектрометрическнм меркам).
[Из Mclver R. Т., Jr., Ledford Е. В., Jr., Miller J. S., Anal. Chem., 47, 692 (1975).J
-»■
2
i
Путь№]
— >
/
/
/
/
.i
3
/
—^' —
Путь N-Z
f
^= 5
'. S
Рис. 20.117a. Схема интерферометра Майкельсоиа.
Делитель пучка (полупрозрачное зеркало) разбивает поток исходящего из образца света
на даа потока равной амплитуды. Эти потоки отражаются от двух зеркал, возвращаются
обратно складываются и направляются в детектор тем же делителем пучка. (В делителе
пучка при этом теряется половииа интсненвиости отраженных от зеркал потоков света.)
Таким образом этот прибор обеспечивает возможность сложения двух потоков света от
образца которые прошли на своем пути к детектору разные по длине пути.
/ — источник; 2 — образец; 3 — делитель пучка; 4 — фиксированное зеркало;
5—подвижное зеркало; 6 — детектор.
695 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
'AUIE
Путь ti-t
Путь N-2
•Суммарный
поток
Путь Л/?/
Путь Л/*2
Суммарный
поток
Низкая частота
Высокая частота
Путь А/'/
Путь А/"2
fiymbtfil
■^Ч/Х/Х
-\
Путь а/'2
У
Суммарный
поток
Суммарный
поток
Рис. 20.176. Схематическое изображение амплитуд совмещенных в детекторе
(рис. 20.17а) низкочастотного (слева) и высокочастотного (справа) потоков,
которые па своем пути к детектору прошли равные (а) и неравные (б)
расстояния.
Если длины путей равны, потоки с частотами обоих типов сложатся когерентно, дав в
результате максимальную амплитуду в детекторе (а). Когда же длины путей немного
отличны, низкочастотные потоки все еще будут складываться в одной и той же фазе, давая
<уммариую волну большей амплитуды. В отлнчие от них высокочастотные потоки будут
складываться почти в разных фазах {здесь показан сдвиг фаз иа 180°), давая
результирующий суммарный поток с сильно уменьшенной амплитудой. По мере увеличения разности
•фаз (при перемещении подвижного зеркала) даже относительно низкочастотные
компоненты потоков будут складываться в сильно различных фазах. Таким образом, амплитуда ин-
терферограммы (рис. 20.18) будет, вообще говоря, уменьшаться от максимального значения
(для равных путей) до все меньших и меньших значений по мере увеличения разницы в
длинах путей между двумя потоками. Подобным образом амплитуда зависящего от
времени сигнала ЯМР или ИЦР в общем уменьшается со временем, когда спектр состоит из
сигналов со многими отличающимися друг от друга частотами.
дать условия ионных циклотронных резоиансов для ионов с (т/е) в области
от 213 до 215. Колебание, показанное на рис. 20,15, а,—это результат
наложения двух сигналов колебаний ИЦР легкого иона (т/е=213) с большей
частотой ИЦР и тяжелого иона (т/е=215), имеющего меньшую частоту ИЦР. Обе
696 ЧАСТЬ 6
f
|V»'|^*<
•^V>»<4A'AA/V
Разность
длин ^
путей
Преобразование Турье
*-»
4000 см
Ш 000 см'
Рис. 20.18. Инфракрасная ннтерферограмма (а) (сигнал, идущий в детектор
на рис. 20.17а как функция различия в длинах путей между двумя
разделенными потоками) и инфракрасный спектр (б) планеты Юпитер.
Спектр был получен путем преобразования Фурье данных интерферограммы. Поскольку
этот спектр получен через атмосферу Землн, пики поглощения в нем обусловлены главным
образом газами земной атмосферы, а не являются характерными пиками излучения
источника (см. последующие примеры).
[Из Pellgett Р., /. dc Physique, Colloque С2, Tome 2», p. C2 — 165 (1967).l
частоты ИЦР были подвергнуты процедурам смешения и фильтрования,
которые сильно понижают обе частоты ИЦР до величин, удобных для работы.
Преобразование Фурье изменяющегося во времени сигнала на рис. 20.15, а дает
частотный (масс-)спектр на рис. 20.15,6. В качестве примера был выбран спектр
бромадамантана, поскольку оба изотопа брома имеют приблизительно равное
содержание в природной смесн и, таким образом, дают лики приблизительна
равной интенсивности.
Хотя методы Фурье используются в масс-спектроскопии совсем недавна
(первая опубликованная статья относится к 1974 г.), они уже обещают
уменьшить время, необходимое для получения масс-спектра ИЦР, почти в 10 000 раз
(ср. с 1000-кратной экономией времени за счет выигрыша Фелжета при
использовании методов Фурье в ЯМР). Кроме того, разрешение по массам, получаемое
с помощью методов Фурье в ИЦР, £гочти в 100 раз лучше, чем при
традиционном методе регистрации ИЦР (методы Фурье фактически ие улучшают разре-
шеиие в спектрах ЯМР). Из рис. 20.16 видно, почему эти улучшения должны
представлять интерес даже если не принимать в расчет обычную сферу
использования масс-спектроскопии ИЦР как средства изучения ионно-молекулярных
реакций в газовой фазе (области, не имеющей прямого отношения к биохимии).
Масс-спектры морфина и нуклеотида, показанные на рис. 20.16 (они получены
на обычном масс-спектрометре ИЦР), демонстрируют, по-видимому, главное
потенциальное преимущество использования этого типа масс-спектрометра,
а именно возможность работать при очень низких давлениях паров образца,
что позволяет исследовать практически нелетучие вещества (пептиды, иуклеоти-
ды, углеводы и другие). До сих пор это достоинство ие могли реально
использовать, поскольку у обычных спектрометров ИЦР было слишком плохое
разрешение по массе (обратите внимание, что иа рис. 20.16 ширина одного масс-пика
порядка нескольких единиц массы) и при работе иа них требовалось слишком
много времени (вплоть до часа) для получения спектра. Можно ожидать, что
697 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
при более быстром получении спектров и лучшем разрешении по массе на
спектрометрах ИЦР-ПФ будет возможно поднять аналитические возможности
ИЦР при масс-спектроскопическом изучении природных объектов.
Пример. Инфракрасная фурье-спектроскопия
Использование методов Фурье в инфракрасной спектроскопии (поглощение
излучения на частотах, характерных для собственных колебаний «пружин»,
которые посредством химических связей соединяют атомы в молекуле) по
существу предполагает обработку данных, аналогичную описанной выше, но с
помощью другой аппаратуры. Инфракрасный спектр можно разложить, используя
прибор, известный как интерферометр Майкельсона (для более подробного
ознакомления с этим прибором, схематически показанным на рис, 20.17,а,
можно обратиться к любому элементарному учебнику физики). Этот
интерферометр представляет собой средство для сложения амплитуд двух пучков света,
которые, покинув образец, проходят различные по длине пути. Если потоки
проходят один и тот же путь (равные расстояния между делителем пучка и
фиксированным или подвижным зеркалами на рис. 20.17 а), то два потока
после сложения дадут максимальную амплитуду, поскольку электромагнитные
волиы любой частоты складываются в одной и той же «фазе». Однако если
расстояние между делителем потока и фиксированным зеркалом отличается от
расстояния между делителем потока и подвижным зеркалом, то амплитуда
суммарного света будет зависеть от частоты волны (рис. 20.17 6). По мере того как
пути потоков света становятся1 все более различными, в суммарном потоке все
больше и больше частотных компонент оказываются не в фазе. В результате
получается «интерферограмма» суммарной интенсивности как функции
положения подвижного зеркала (рис. 20.18). Как и в предыдущих примерах с ЯМР и
ИЦР, интерферограмма представляет собой сумму амплитуд всех частотных
компонент излучения от образца, так что имеет место выигрыш Фелжета.
Различные частотные компоненты оказываются не в фазе по мере увеличения
разницы путей, точно так же, как изменяющиеся во времени сигналы ЯМР и ИЦР
оказываются не в фазе по мере увеличения времени. Единственное отличие
состоит в том, что интерферограмма является «двусторонней» (она симметрична
относительно положения зеркала, которое дает нулевую разность путей), в то
время как явления ЯМР и ИЦР регистрируют только после нулевого момента
времени и онн, таким образом, являются «односторонними». Таким образом,
преобразование Фурье инфракрасной интерферограммы должно (и это
действительно так) давать инфракрасный спектр образца.
Инфракрасные спектры планет и звезд
На рис. 20.18 показан инфракрасный фурье-спектр планеты Юпитер по
наблюдениям с Земли (таким образом, в спектре преобладает инфракрасное
поглощение земной атмосферы, а не спектр испускания самой планеты). Как показано
на рис. 20.19, больше информации можно получить, изучая инфракрасный спектр
п низкочастотном диапазоне, где атмосферные Н20 и С02 не поглощают
большей части спектра излучения планеты. Спектр на рис. 20.19 был скорректирован
с учетом (небольшого) поглощения атмосферой, что стало возможным благодаря
недавно полученным данным по инфракрасному поглощению, собранным на
большой высоте, практически за пределами атмосферы. Пики на спектре
рис. 20.19 возникли главным образом благодаря переходам между
возбужденными состояниями молекул аммиака в атмосфере Юпитера. Из таких
инфракрасных спектров можно извлечь только ограниченную информацию о
составе и температуре атмосферы (о заселенности энергетических уровней как
функции температуры см. гл; 19). Более подробную информацию можно
получить о изотопном составе различных элементов, поскольку колебательные
частоты, скажем, 1ZC190 и 13С1вО немного отличаются из-за различия масс на концах
соответствующих «пружин» (химических связей). Отношения 1ZC/13C для иекото-
<698 часть б
860
880
900
920
см
940
960
980
Тле. 20.19. Фурье-спектр испускания планеты Юпитер в длинноволновой
инфракрасной области с поправками на атмосферное поглощение.
•Наиболее характерные линии поглощения обусловлены присутствием в атмосфере Юпитера
.аммиака. Этот инфракрасный Фурье-спектр был получен за 3 ч. Для получения
аналогичного спектра с помощью обычного однощелсвого сканирующего спектрометра
потребовалось бы 500 ч.
[Из Ridgeway S. Т., Capps R. W., Rev. Sci. Instrum., 45, 676 (1974).]
■рых характерных типов звезд даны в табл. 20.2. Эти результаты используют
для подтверждения теорий о путях эволюции звезд.
Таблица W.2
Соотношение изотопов углерода в некоторых типах звезда
Тип
Пример
12С/13С
.Мира Кита
Сверхгиганты спектрального класса М
Углеродные звезды
% Лебедя
а Ориона
о) Ориона
30
5
12-15
Определено но инфракрасным спектрам на основании сравнения интененвностей
линий 12С'Ю и 13С160 в спектрах звезд, наблюдаемых с Земли (с поправкой на атмосферное
поглощение). [Из данных Johnson H. L., Mendez M. E„ Astronom. J., 75, 785 (1970); в этой
■.-статье представлены инфракрасные спектры 32 звезд.]
Пример. Обнаружение методом инфракрасной фурье-споктроскопии небольших
атмосферных загрязнений
Поскольку химически различные малые молекулы имеют различные
эффективные массы (атомы) на концах своих межатомных «пружин» (химических
связей), они характеризуются различными «собственными» колебательными
частотами. Электромагнитным излучением можно возбудить колебания на этих
частотах (они лежат в инфракрасной области электромагнитного спектра).
Однако для того, чтобы наилучшим образом различать малые молекулы одну от
другой, желательно записывать спектр с очень большим .разрешением (табл. 20.1).
3 этом могут помочь фурье-методы, поскольку они обеспечивают одновременную
работу большого числа «каналов», что дает высокое разрешение, ие уменьшая
высокой чувствительности (отношение сигнал/шум), необходимой для изучения
таких разбавленных объектов, как атмосферные загрязнения. На рис. 20.20
показано, как с помощью инфракрасных спектров можно идентифицировать
атмосферные загрязнения и оценить их количество (приблизительные концентрации
загрязнений показаны на рис. 20.20 справа). Очень важно то, что детектор
инфракрасного излучения может быть установлен в месте, удаленном от
источника загрязнений (скажем, дымовой трубы)., что облегчает контроль особенно
а ночные часы.
2150
"■c1<=o2.
I I I ! I I II ! 1 I I П I 1 1 Г
I 1 I
2200
CM
~1
2250
2300
[CO] ~ 0,1
LH20] ~ 10 000
[N.,0] ~ 0,25
[,3C160] ~ 3,2
([12C16OJ ~ 300
>x!0
-4i
CM'
[C2H<] ~ 0,19
[C2H2] ~ 0,25
[C3H6] ~ 0,12
[HCOOH] ~ 0Д6
\*ior*Z
Рнс. 20.20. Инфракрасные фурье-спектры образцов воздуха, содержащего небольшие количества различных загрязнений.
Приблизительные концентрации соответствующих загрязнений определены по высотам наблюдаемых пиков поглощения. Па нижнем
спектре маленькие пики были усилены путем (поточечного) вычисления отношения наблюдаемого спектра к соответствующим
амплитудам спектра образца воздуха, не содержащего никаких загрязнений.
(Из Hanst P. L., Lefohn A. S., Gay В. W., Jr., Applied Spcctrosc, 27, 188 (1973).J
700 часть -6
а
I
Рис. 20.21. Инфракрасные
фурье-спектры поглощения (а)
водного раствора гемоглобина,
(б) чистой воды и (в)
разностный спектр, полученный путем
поточечного вычитания
спектра б из верхнего спектра а для
устранения очень сильного
поглощения растворителя.
Обратите внимание иа то, что на
разностном спектре видно гораздо
больше деталей, чем на верхнем
спектре водного раствора
гемоглобина.
[Из Koenig J. L,. Applied Spectrosc.,
29, 293 (1975).]
Пример. Разностные инфракрасные спектры с преобразованием Фурье.
Гемоглобин
В обычном эксперименте по оптическому поглощению поглощение
растворителя устраняют, взяв разность между поглощением раствора и поглощением
растворителя. Однако в тех случаях, когда поглощение растворителя очень
велико (как при поглощении инфракрасного излучения водой), возникает
необходимость определить разность между двумя очень маленькими числами
(интенсивность света, прошедшего через растворитель). В этом случае наличие
помех со стороны небольших количеств рассеянного света в обычном однощеле-
вом приборе делает сравнение по существу бесполезным. Интерферометр
гораздо менее чувствителен к наличию рассеянного света и, как видно из 'рис. 20.21,
позволяет получить инфракрасные спектры молекул, растворенных в воде.
Т^а рисунке хорошо видно, что на инфракрасном спектре гемоглобина в водном
-растворе, полученном при использовании преобразования Фурье, нельзя
разглядеть характерных деталей. Однако после вычитания из него спектра растворителя
получают спектр, в котором ясно различимы полосы колебаний амидных связей
■с частотами 1657 и 1547 см-1. В настоящее время появилась возможность
делать заключения о вторичной структуре белков на основе детального изучения
чувствительных к конформации частот инфракрасного поглощения белка в
водном растворе.
Задачи
1. Маски Хадамарда можно создавать для случаев, когда число потенциально
открытых щелей равно (2я—1), где п — целое число. На рис. 20.5 показано,
как схема работает при я=2 (система имеет 3 щели). Приняв, что одна из
необходимых линейных комбинаций позиций щелей для случая п = 7 является
циклической перестановкой расположения 1 ООН 0 11, покажите, что семь
1800 1400 1000
Vac тот а, см4
600
=701 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Р ! —
циклических перестановок этого расположения являются линейно
независимыми, и найдите способ (как слева внизу на рис. 20.5) получения желаемой
интенсивности, прошедшей через данную щель, из измерений общей
интенсивности, прошедшей через все щели при их семи расположениях.
2. Рассчитайте синусоидальное и косинусоидальное преобразования Фурье
изменяющейся во времени функции
f(t) = cos (a>J)f при 0<^<Г и /(0 = 0, при /<0 или t>T
и скомбинируйте их, чтобы получить амплитудный спектр этого
изменяющегося во времени сигнала. Если воспользоваться уравнениями 20.8, получившиеся
Л(о)), В (со) и С((о) должны дать верхний ряд кривых на рис. 20.10. Эти
кривые описывают (помимо всего прочего) спектр короткого импульса
незатухающих во времени колебаний, дифракцию от щели, рассеяние от точечного
источника.
3. Рассчитайте синусоидальное и косинусоидальное преобразования Фурье
изменяющейся во времени функции
f(t) = exp(—t/*)cos(aiJ), при 0</<оо и /(0 = 0, при /<0
н скомбинируйте их, чтобы получить амплитудный спектр этого
изменяющегося во времени сигнала. Получившиеся в результате функции Л (со), В (со) и
С((о) представляют собой кривые, показанные в нижнем ряду на рис. 20.10.
Эти кривые описывают формы множества спектральных линий при
магнитном резонансе, рассеянии света, флуоресценции и других типах
спектроскопии.
4. Рассчитайте синусоидальное н косинусоидальное преобразования Фурье
изменяющейся во времени функции
f(0 = exp(—//*) cos К/), при 0<г<Г и /(0 = 0,
при ^<0 или />Г
и скомбинируйте их так, чтобы получить амплитудный спектр этого
изменяющегося во времени сигнала. Получившиеся функции Л (со), В (со) и С (а)
представляют собой кривые, покаранные в среднем ряду на рис. 20.10, когда
х=Т. (Расчеты в задачах 2, 3 и 4 даны в порядке возрастания
алгебраической трудности.) Эти кривые описывают формы линий, наблюдаемых в фурье-
спектрах магнитного резонанса и ионного циклотронного резонанса.
Полученные в этой задаче выражения представляют собой общие формулы, из
которых, используя соответствующие пределы (1/т—Ю или Т—>-оо), можно
вывести выражения, полученные ранее в задачах 2 и 3.
5. Для каждого из трех спектров, полученных путем преобразований Фурье
в задачах 2, 3 и 4 рассчитайте полную ширину на половине высоты
{для A ((d) и С((й)] и расстояние между экстремумами [для В (а)]. Далее
следует показать (это видно из рис. 20.10), что представление линии
поглощения в виде Л (со) всегда обеспечивает самый узкий сигнал и, таким
образом, предпочтительнее, когда надо получать спектры с высоким
разрешением.
6. Задача 5 показывает также, что при данной постоянной затухания т
минимальная ширина линии (скажем, для Л (со)) получается при условии, что
время сбора экспериментальных данных Т приближается к бесконечности.
Покажите, насколько большим надо выбирать Г, чтобы получить ширину линии в
пределах, скажем 10 или 1% от минимально возможной предельной ширины
сигнала, поскольку иа практике мы можем зафиксировать только столько
данных, на сколько хватит нашего терпения или объема памяти компьютера!
Для этого рассчитайте полную ширину линии на половине максимальной
высоты для Л (со) из задачи 4, используя следующие значения Т: a) T=»t,
702 часть б
б) Т=2х, в) Г=3т, г) Т—>-оо. Затем используйте эти четыре точки для
построения графика зависимости ширины линии от времени измерения 7",
чтобы ответить на вопрос, поставленный выше.
7. В реальном спектроскопическом эксперименте мы имеем дело с
преобразованием Фурье конечного числа (дискретных) точек из зависимых от времени
данных (выборок), полученных измерением непрерывных по форме волн,,
описываемых уравнениями из задач 2, 3 и 4, через одинаковые промежутки
времени. Общий принцип при определении частоты выборок состоит,
очевидно, в том, что необходимо измерять волну по крайней мере дважды в
промежутке, равном периоду ее колебания. Это нужно для того, чтобы
правильно оценить частоту волны (то есть для того, чтобы знать, что имеет мест»
колебание, мы за время измерения должны суметь определить, что волна
идет «вверх» и «вниз»!). Другими словами, мы должны делать измерения:
с частотой по крайней мере не менее чем
1 ^ v.waKC>
где Vm3kc — самая высокая частота в интересующем нас спектре. Кроме того,,
число точек, которые мы можем накопить, определяется емкостью памяти
имеющегося в наличии компьютера. Таким образом, наибольшее возможное
время накопления данных (и, следовательно, наилучшее возможное
разрешение или наименьшая ширина линий; см. задачу 6) ограничено числом jV:
FT = N.
Используя эти уравнения, получите выражение для ширины спектральной
линии в простейшем случае, когда ширина линии определяется исключительно
временем накопления (задачи 2 и 5). Используйте эту формулу для
вычисления наименьшей возможной ширины спектральной линии спектра ЯМР,
максимальная частота которого составляет 10 000, 1000 или 100 Гц, при
использовании компьютера с емкостью памяти в 4096 слов для накопления данных по
зависящему от времени спектру.
Литература
Спектроскопия с преобразованиями Хадамарда и Фурье
1. Marshall A. G., Comisarow M. В., Multichannel Methods in Spectroscopy, in:
Transformations in Chemistry, ed. P. R. Griffiths, Plenum, New York, 1978.
Магнитный резонанс с преобразованием Фурье
2. Hill H., Freeman R., Introduction^ Fourier Transform NMR, Varian Associates,
Analytical Instrument Division, Palo Alto, Calif., 1970.
3. Фаррар Т., Беккер Э. Импульсная и фурье-спектроскопия ЯМР. — М.: Мир,
1973.
Ионный циклотронный резонанс с преобразованием Фурье
4. Comisarow М. В., Marshall A. G., J. Chem. Phys., 64, ПО (1976) и ссылки к
ней.
Инфракрасная фурье-спектроскопия
5. Koenig J. L., Applied Spectroscopy, 29, 293 (1975). Общий обзор.
6. Hunten D. M. «Fourier Spectroscopy of Planets», Science, 162, 313 (1968).
ГЛАВА 21
ФУРЬЕ-АНАЛИЗ БЕСПОРЯДОЧНОГО ДВИЖЕНИЯ.
АВТОКОРРЕЛЯЦИЯ И СПЕКТРАЛЬНАЯ
ПЛОТНОСТЬ
К настоящему моменту читатель должен был твердо усвоить,
что пружину ["классическую (гл. 13) или квантовомеханическую
(гл. 18)] можно заставить колебаться под действием выводящего
«е из равновесия возмущения, которое содержит частотные
компоненты, близкие к «собственной» частоте колебаний этой пружины.
В ,гл. 13 и 18 в качестве такого возмущающего воздействия было
рассмотрено когерентное переменное электрическое поле,
возбуждавшее колебания пружин в любой данной области пространства
таким образом, что они осуществлялись в фазе. [Импульс
электрического (или магнитного) поля, служивший в качестве
широкополосного источника частот в спектроскопии ЯМР-ПФ (гл. 20.Б),
также был когерентным, хотя это и не оговаривалось специально].
В этом разделе мы хотим установить частотные компоненты
некогерентных источников излучения или молекулярных движений. Да-
jiee станет очевидным, что беспорядочно изменяющееся во времени
электрическое (или магнитное) поле может иметь частотные ком-
лоненты, которые также индуцируют переходы между
энергетическими уровнями (в терминах квантовой механики) или колебания
-«электронов на пружинах» с частотами, близкими к их
«собственным» частотам (в терминах классической механики) даже в
отсутствие внешнего облучения, что объясняет «Г]»-процесс, который
-«отвечает» за «естественную ширину» линий спектра,
рассмотренных в гл. 14 и 19. Вначале мы дадим общие формулы для
вычисления частотных компонент любых изменяющихся во времени
беспорядочных процессов и затем покажем, каким образом
определенные типы беспорядочного движения (например, химический обмен,
вращательная диффузия, трансляционная диффузия) связаны с
уширеиием спектральных линий в магнитном резонансе,
деполяризацией флуоресценции и экспериментами по квазиупругому рэлеев-
■скому рассеянию света.
Беспорядочным или стохастическим процессом называют такой
процесс, в котором величина f(t) мгновенного изменения
координаты или направления движения молекулы или амплитуда
электрического поля не зависит от времени каким-то определенным
образом. Например, три изменяющиеся со временем траектории на
704 часть б
Молекула
A/'/ fit) 0
л"!2 fu) О
А/*3
Рис. 21.1. Гипотетическая запись координаты как функции времени для трех
молекул, участвующих в одномерной трансляционной диффузии.
У разных молекул поведение разное. Кроме того, частотные компоненты движения каждой
молекулы меняются во времени (иногда молекулы колеблются медленно, иногда — быстро).
Следовательно, осмысленный частотный анализ такого движения должен включать в себя
как усреднение по времени, так и усреднение по различным частицам («усреднение по
ансамблю»).
рис. 21.1 могут представлять собой мгновенные координаты
каждой из трех различных молекул при одномерном беспорядочном
движении как функции времени (гл. 6.А). В этом примере мы
обнаруживаем, что среднее отклонение молекулы от начального ^по-
ложения равно нулю. Однако можно вычислять
среднеквадратичную координату, которая прямо связана-со скоростью
трансляционной диффузии молекулы. Предположим, что теперь мы
регистрируем произвольную среднюю амплитуду f(t) в течение точно
установленного интервала времени Т. Оказывается, что можно
выразить f(t) через бесконечный набор синусоидальных колебаний,
имеющих амплитуды ап и Ьп при соответствующих частотах ©„;
как указано в уравнении (21.1) [ср. с уравнением (13.16а)]:
(21.1а)
/(0
где
«я
00
•-
=2 (а«cos (<о
п=\
= (2wi/7-)
4) + Ьп
sin
КО).
(21.16)
Как и в случаях описания амплитуды вынужденного (гл. 13.Б)
или свободного (гл. 13.В и 20.Б) колебаниями груза на пружине,
мы разбиваем процесс иа компоненты, которые меняются во
времени по косинусоидальному или по синусоидальному законам, но
с одним-принципиальным отличием. В предшествующих примерах
движение или колебание было когерентным. Это означало, что все
705 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
частицы (или электромагнитные волны) в одной области
пространства колебались в одной и той же фазе. Поэтому можно было
предсказывать поведение любой выбранной группы («ансамбля»),
зная только поведение какой-то одной частицы (или волны).
Теперь, однако, величина беспорядочного сигнала изменяется не
только во времени, но и от одной частицы к другой (рис. 21.1) так,
ччто наблюдаемый экспериментальный результат базируется на
некотором среднем (по времени и по частицам) поведении
отдельных частиц (волн). Если попытаться теперь провести простое
усреднение по времени функции f(t) в уравнениях (21.1) в течение
периода наблюдения от нуля до времени Т, то средняя по времени
величина < /(7) > будет стремиться к нулю из-за того, что в
течение этого интервала времени средние значения cos[(2лл/Т)/] и
&in[-(2nn/T)t] будут стремиться к нулю:
Г Cos /-^Л dt = f sin №f\ dt = 0 (21.2)
о о
Величины ап и bn в настоящем анализе представляют собой
точечные амплитуды косинусоидальных и синусоидальных колебаний
наборов равноудаленных дискретных частот [уравнения (21.1)] и
являются дискретными вариантами соответствующих Л (со) и 5 (со),
которые мы вывели в уравнениях (20.8) для описания непрерывной
зависимости амплитуды от частоты. Новая характерная
особенность их состоит в том, что, хотя ап и Ьп для любой отдельной
частицы в любой отдельный момент времени не являются нулевыми,
усредненные по времени величины ап и Ьп равны нулю, поэтому
мы не можем более получать ап и Ьп непосредственно из
наблюдения усредненного поведения какого-либо ансамбля частиц.
Однако если проводить эксперимент так, что измеряется
квадрат f(t), как, например, при измерении интенсивности (но не
амплитуды) излучения, то в этом случае существует возможность
определения усредненных по времени частотных компонент
суммарного спектра (см. задачи к этой главе) по уравнению
jW(-^)df = jsm°(-^)* = -i-, (21-3)
о о
так что
оо
<[/(0r>=J](^4^)-const (21.4)
для любой отдельной частицы. Наконец, когда мы возьмем еще
одно усреднение по различным частицам (или излучениям, испу-
706 ЧАСТЬ 6
щенпым различными частицами), то получим так называемый
«энергетический спектр» беспорядочного движения Р{ы):
^K)-(^SS), (2 J.5)
так что
<[/№>= S я К). (21.6)
где черта над соответствующим выражением обозначает
усреднение по частицам*. Таким образом, P((on) представляет собой
усредненную по времени и по частицам частотную компоненту (на
частоте <ап) квадрата амплитуды рассматриваемого нами
беспорядочного движения. {Название «энергетический спектр» возникло
из примера по прохождению электрического тока (f(t)) через
единичное сопротивление, в котором мгновенное рассеяние энергии
пропорционально [f(t)]2.} В наших примерах Р(ып) чаще всего
будет представлять собой энергетический спектр (или спектр ин-
тенсивностей) излучения, поскольку интенсивность излучения
пропорциональна квадрату амплитуды электрического (или
магнитного) поля (гл. 13.А). P((on) для беспорядочного некогерентного
излучения соответствует [С(а>)]2 для монохроматического
когерентного излучения, описываемого уравнением (20.8в). Ясно, что
для некогерентных источников невозможно определить задаваемые
уравнениями (20.8а) и (20.86) величины Л (to) и В (со), и поэтому
нам приходится иметь дело исключительно с интенсивностью, но
не с амплитудой излучения.
Из схем, представленных на рис. 21.1, становится ясным, что
мы не можем заранее установить величину, которую /(7) примет
для определенной частицы в определенный момент времени. Тем
не менее если известна, например, величина коэффициента
диффузии в случае трансляционной диффузии беспорядочного процесса,
то мы можем сказать' что /(7) имеет данное значение в
определенный момент времени для данной частицы, и через небольшой
промежуток времени она будет иметь близкую величину (величина
в момент времени t-\-x будет «коррелировать» с величиной в
момент времени /)• Однако через достаточно большой промежуток
времени уже невозможно предсказать величину f(t-\-%),
основываясь на ее первоначальной величине f(t), так как при достаточно
большом т f(t-\-x) «не коррелирует» с /(т), Поскольку величина
временного интервала, через который /(/-f-т) перестает
коррелировать с /(т), является мерой скорости, характеризующей
беспорядочный процесс (например, коэффициент диффузии в случае транс-
* Здесь и далее в этом обсуждении мы обозначаем величину, усредненную
по времени, угловыми скобками вида < >, а усредненную по частицам —
чертой сверху.
'07 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
чяционной диффузии), можно вывести частотные компоненты
беспорядочного процесса, изучая «корреляционную функцию» G(t):
G(') = <f(0f(' + ')>.
(21.7)
в которой угловые скобки обозначают усреднение по времени, а
черта сверху — усреднение по всем частицам, участвующим в
беспорядочном процессе. Замечательное свойство корреляционной
функции проявляется, если выразить f(t) и /(/-f-т) через их
частотные компоненты, как это сделано в уравнениях (21.1):
GW = <f(0f(* + ')> =
7/00 \ . оо
— j I J^ancosa>nt-\-bnsma>at UV ат cos шт (t + х) 4-
+ &OTsina>m(^+x)\ctf-
(21.8)
00
=
см.
задачи к
этой
главе
£(a2"V2")c°w =
л=1 уравне
ния
(21.5)
00
«=1
(21.9)
Заменяя суммирование в уравнении (21.9) интегрированием,
получим выражение
00
<f(t)f{t-\-*)>^ G (х) = f P (о>) COS «v«R (21.10)
которое чаще записывают в следующей форме*:
Р(со)
= 2/.
с»
0
G(t)
cos
((a:)dz
(21.11)
Уравнение (21.11) описывает способ нахождения относительного
количества энергии на любой данной частоте, получающейся в
результате беспорядочного движения частицы или изменения
электромагнитной волны с корреляционной функцией G(t). Таким об-
* Взаимосвязь между уравнениями (21.10) и (21.Ill) является свойством
преобразований Фурье; математическое доказательство возможности такого
перехода см. в литературе к этой главе в любой ссылке на фурье-методы.
708 ЧАСТЬ б
разом, как только мы найдем вид G(t) для определенного типа
движения (гл. 21.А, 21.Б и 21.В), мы сразу же получим
возможность вычислять по уравнению 21.11 энергетический спектр как
функцию частоты. Во всех характерных беспорядочных процессах,
которые мы будем рассматривать, корреляционная функция G(r)
экспоненциально уменьшается во времени:
G(t)=G(0)£~t/TkoPP, (21.12)
где «время корреляции» Ткорр является мерой времени,
необходимого для того, чтобы величина f(t-\-r) перестала коррелировать
со своей предшествующей величиной f(t), a G(0) представляет
собой среднеквадратичную мгновенную амплитуду беспорядочного
движения (см. ниже). Если корреляционная функция имеет вид,
даваемый уравнением (21.12), то мы легко можем вычислить по
уравнению (21.11) энергетический спектр
00
P(i») = |g(0) Г£~t/Tk°ppcos ((ox)dx,
/>(«,)= —G(0)
п
'■корр
1 Ч-со2т
2,2
корр
(21.13)
Энергетический спектр («спектральная плотность») Р(о>) имеет,
таким образом, вид хорошо известной кривой Лорентца. На рис.
21.2 он показан как функция lg((o). Энергетический спектр
представляет собой прямую линию на частотах от нулевой до порядка
1/ткорр, которая затем резко идет вниз в районе 1/тКорР. «Время
г = ю~*с
Рис. 21.2. Энергетический спектр (спектральная плотность) как функция
частоты ^логарифмическая шкала) для беспорядочного процесса, описываемого
экспоненциально убывающей корреляционной функцией с временем корреляции
T(tOpp = 10-4 с.
1Для удобства G(0) в уравнении (21.12) было принято равным (Л/2)]. Спектр является
«белым» (значит, функция Р(ш) постоянна по величине) в интервале от нулевой частоты до
частоты порядка 1/тк0пр. Спектральная плотность Р(ш) из уравнения (21.13) представляет
собой кривую Лорентца, форма которой на этом графике искажена из-за
логарифмического масштаба по оси абсцисс.
709 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
корреляции» Ткорр является, таким образом, мерой наименьшего
времени, за которое может существенно измениться
среднеквадратичная амплитуда, характеризующая положение или направление
частицы (или электромагнитной волны).
На рис. 21.2 видно, что такой беспорядочный процесс является
эффективным широкополосным источником частот вплоть до
приблизительно (1/ткорр) с-1. Таким образом, если беспорядочное
движение связано с изменением амплитуды электрического (или
магнитного) поля, то в указанном выше диапазоне частот оно может
выступать в качестве широкополосното источника
электромагнитного излучения. Если окажется, что «собственная» частота
интересующего нас образца меньше, чем приблизительно (1/тКОрр) с-1*
такой беспорядочный процесс заставит систему колебаться с ее
собственной частотой (в терминах классической механики) так,
чтобы вызвать переходы между соответствующими
энергетическими уровнями (в терминах квантовой механики) даже в отсутствие
источника внешнего облучения. Поскольку эти переходы
происходят со скоростью, которая определяется «временем релаксации»
Гь описывающим различия заселенностей между двумя
участвующими в переходе энергетическими уровнями (гл. 19.Б.З), можно-
объяснить теперь «естественную ширину линии» (2/Ti) для
нескольких типов линий поглощения в спектре через
микроскопическое движение частиц в образце. Остается только показать, как
ширина линии в спектре образца (связанная со скоростью
перехода между двумя энергетическими уровнями) соотносится со
временем корреляции для беспорядочного движения частиц и затем"
(см. последующие разделы) как это время корреляции
определяется тем или иным конкретным типом беспорядочного движения.
Для данного беспорядочного процесса существует хорошо опре-'
деляемая среднеквадратичная величина мгновенной амплитуды
движения
|f(0|2 = const, (21.14)
но 1/(0 I2 B уравнении (21.14) как раз и является величиной
корреляционной функции G(t) при т = 0. Возвращаясь к уравнениям
(21.7) и (21.10), мы, таким образом, прямо придем к важному
заключению, что
00
j7WIa = G (0) |= j" />(«) <fo (21.15)
6
во /площадь под кривой графика зависимости N
ГЯ(о))^0)=( функции спектральной плотности от ча- |=COnst (21.16)
J V стоты J
и не зависит от времени корреляции ткорр для этого процесса.
Уравнение (21.16) графически представлено на рис. 21.3, где даны
графики зависимости спектральной плотности от частоты (в логариф-
71$ ЧАСТЬ б
PtUi)
Ряс. 21.3. Зависимость спектральной плотности Р{<й) от частоты
(логарифмическая шкала) для беспорядочных процессов, характеризуемых тремя
различными временами корреляции.
Исследуется величина (см. текст) спектральной плотности на фиксированной частоте Шо
^-собственных колебаний определенного осциллятора в образце. И для большого (самая
верхняя кривая), и для маленького (самая нижняя кривая) времени корреляции tKopp
-спектральная плотность на частоте Шо мала. Спектральная плотность на частоте О)о
максимальна, когда (1/ткорр) ~ со0. Эти кривые объясняют показанную на рис. 21.4 зависимость
константы скорости индуцированных переходов 1/7"] от времени корреляции.
.'мическом масштабе) для трех различных времен корреляции
данного беспорядочного процесса. Поскольку площадь под каждой
;кривой должна быть одной и той же [уравнение (21.16)], в то
жремя как частоты «отключения» (1/тКОрр) меняются от графика к
трафику, высота каждой кривой также должна меняться так, как
.показано на рисунке. Если мы теперь будем считать, что в данном
беспорядочном процессе участвует флуктуирующее электрическое
(или магнитное) поле, то эти графики будут показывать
относительные энергетические спектры излучения, продуцируемого таким
■процессом. Наконец, если мы рассмотрим энергию излучения на
•определенной частоте щ, выбранной в качестве собственной
частоты движения определенного типа молекул, то константа скорости
для индуцированных (безызлучательных) переходов (1/Ti) будет
пропорциональна количеству энергии, переданному образцу (гл.
19.Б.1). Например, когда время корреляции очень велико
''[(1/ткорр)<С<оо], образцу передается слишком мало энергии,
переходы индуцируются относительно редко и (1/7\) мало. Когда
?(1/тКорр) ~сйо, наблюдается максимальная спектральная плотность
при ©о и скорость индуцирования переходов также максимальна.
Наконец, когда время корреляции очень мало [(1/ткорр)^>©о],
-спектральная плотность при ©0 снова мала и (l/7i) также мало.
Из следствий уравнения (21.16), показанных на рис. 21.3, ясно, что
константа скорости для индуцирования переходов (в отсутствие
внешнего облучения) (1/7\) должна зависеть от времени
корреляции, как это показано на рис. 21.4. Эта зависимость приводит к
максимальному уширению спектральных линий (Т\ —
минимально), когда беспорядочное движение имеет обратное время
корреляции того же порядка, что и собственная частота колебаний
образца [(1/ткорр) ~юо]. Таким образом, рис. 21.4 дает объяснение
711 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
естественной ширине линий спектра Д©= (2jT\) на основе
характеристического времени корреляции для изменения
среднеквадратичной амплитуды беспорядочно изменяющегося локального
электромагнитного поля. В нескольких последующих разделах мы
покажем, как рассчитывать корреляционные функции (и, таким
образом, времена корреляции, а значит, и ширину спектральных
линий) для некоторых определенных типов беспорядочного движения:
молекул (химический обмен, вращательная и трансляционная
диффузия), которые приводят к появлению соответствующих
беспорядочно флуктуирующих электромагнитных полей. Кроме того,
можно использовать полученные результаты в обратном порядке для7
того, чтобы определять характеристические константы скоростей
(например, константу скорости химической реакции, коэффициенты
вращательной или трансляционной диффузии) из измеряемой
ширины линий в спектрах.
21.А. БЕСПОРЯДОЧНЫЕ ПЕРЕСКОКИ
МЕЖДУ ДВУМЯ СОСТОЯНИЯМИ
С РАЗЛИЧНЫМИ «СОБСТВЕННЫМИ» ЧАСТОТАМИ
Для ясного понимания описания беспорядочного шума
необходимо (возможно более, чем в любом другом случае, с которым мье
до сих пор сталкивались) в первую очередь разобрать несколько-
характерных примеров. В нашем первом примере рассмотрены
эффекты, возникающие при химическом «обмене» (беспорядочных
скачках), скажем, магнитного ядра между двумя состояниями с
-г,
Рис. 21.4. График зависимости вероятности перехода \\Т\ (логарифмическаяг
шкала) от времени корреляции тКорр (логарифмическая шкала) для
определенного беспорядочного процесса.
Как видно из рисунка, вероятность перехода максимальна, когда (I/*'корр) ~ *>•
(объяснение этого явления см. на рис. 21.3 и в тексте). Эти переходы осуществляются и пр»
отсутствии внешнего облучения, что, следовательно, и предопределяет «естественнук»-
шнрину рассматриваемых спектральных линий (см. текст).
712 ЧАСТЬ в
различными напряженностями магнитного поля. Если эти два
состояния соответствуют, например, положениям протона в двух
разных молекулах, то для беспорядочного процесса обмена
существует вполне определенная (усредненная по времени) константа
скорости k. Задача, следовательно, состоит в том, чтобы найти
выражение корреляционной функции для напряженности магнитного
поля путем усреднения для частиц по всем возможным (в нашем
случае по двум) исходным состояниям в нулевой момент времени
и всем возможным «конечным» состояниям через время т. Из
корреляционной функции магнитного поля мы можем получить
энергетический спектр магнитного излучения [уравнение (21.11)] и,
таким образом, рассчитать вероятность переходов между двумя
энергетическими состояниями протона в фиксированном внешнем
магнитном поле.
При расчете среднего значения любой функции (в- нашем
случае магнитного поля) мы, как это было показано в гл. 6.А,
составляем сумму членов типа (величина функции) X (вероятность
получения этой величины). Следовательно, в первую очередь нам
необходимо вычислить требуемые вероятности нахождения ядра в
любом из двух исходных состояний и нахождения ядра в каждом из
двух конечных состояний через время т при условии, что в нулевой
момент времени ядро находилось в данном исходном состоянии.
Обозначим теперь через РА и Рв вероятности того, что протон
находится в состоянии А или В. Из химического равновесия между
двумя состояниями
А ^В (21.17)
k
мы можем сразу записать уравнения для скорости изменения Ра
и Рв во времени
dPA
dPB
-^-= — kPB -j- kPA
Поскольку мы рассматриваем простой случай, когда скорости
прямого и обратного перемещений равны, вероятности нахождения
протона в положении А или в положении В в первоначальный
момент будут равны
РА(0) = Рв(0) = -^. (21.19)
Далее, уравнения 21.18 легко решаются (гл. 10.А.З), давая
вероятности того, что протон локализован в установленном конечном
состоянии через время т при условии, что он находился в данном
исходном состоянии в нулевой момент времени [Р (конечное поло-
(21.18)
713 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
жение через время т; исходное положение — нулевой момент
времени)]
Р{А, *; Л, 0) = ±(l + e-2fn), (21.20а)
—Vtx-,
Р(В, t; Д 0) = -И1-е-*"),
—2kx.
Я (Л, х; В, 0) = -f (1-<Г""),
—2feT%
Р(В, t; В, 0) = -^-(\+е-™).
(21.206)
(21.20в)
(21.20г)
Теперь можно составить корреляционную функцию для
флуктуации магнитного поля из-за химического обмена между двумя
состояниями с различным магнитным полем, используя
вероятности, даваемые уравнениями (21.19) и (21.20), для вычисления
усредненных по частицам значений произведений магнитного поля в
исходном положении в нулевой момент времени ЯНСх и магнитного
поля в конечном положении через время т, Нкон:
G (Т) = <И (0) //(*)>=* 2 S (Рисх"нсх) [Р ("кон. *; "нсх. 0) ИК0Н]
исход-
конечные по- ные по-
ложе- ложе-
ния ния
(21.21)
Если ради удобства обозначить магнитные поля НА в положении
А и Нв в положении В как
Яд = + Я, (21.22а)
Яв=-Л, (21.226)
то корреляционную функцию, задаваемую уравнением (21.21),
можно переписать в виде
+ ?№&„ «;
+ /УУ>(ЯД, ,;
ЯА, 0)ЯВ +
Яв, 0)ЯА +
Яв, 0)ЯВ =
1 , <> , « . ~2fet4 , 1
-2ftv
X(i-«-a,)+~(->t!) <•-*'") +
—2ftx4
+ 7-(A)i(l+«-",)=AV
, —2ftx
(21.23)
714 ЧАСТЬ &
-г/ г
<// (0) Я (г)> = Л2*? ' К°РР = G (t),
где 1Дкорр = 2*
(21.24а)
(21.246)
Из корреляционной функции, описываемой уравнением 21.24а, и
уравнения 21.11
-г/т
Я (Ш) ;= 2/11 j fc«tf K°PP COS (<Ot) rfx
можно быстро получить спектральную плотность Р(ах)
/»--=— л
скорр
я 1 -[- ш2^
2,2
= 2А
корр '■корр
(21.25)
Пример. Беспорядочные флуктуации магнитного поля и времена релаксации
в ЯМР
Рассмотрим набор ядер со спином '/г, на которые действует сильное
однородное магнитное поле, направленное (как обычно) вдоль оси г, и небольшое
магнитное поле, значение которого меняется (со временем корреляции тКОрр)
по величине от +h до —h вдоль каждого из трех ортогональных направлений
(х, у и z). Используя методы, очень похожие на те, которые мы только что
рассмотрели, можно показать, что ядерное магнитное «время релаксации» Т\
определяется преобразованием Фурье корреляционной функции для флуктуации
магнитного поля иа частоте <Оо=тН0:
корр
корр
(21.26)
поскольку вероятность переходов пропорциональна (I/^i) ^уравнение (19.38)].
При магнитном резонансе существует и другой характеристический
коэффициент затухания Г» который описывает (экспоненциальное) уменьшение со
временем магнитного момента, выведенного в плоскость х— у [уравнение (16.43а)].
Можно показать (используя методы теории зависимых от времени
возмущений, представленной в гл. 19.Б), что Т2 связано со спектральной плотностью
для того же самого беспорядочного процесса при частоте <оо н нулевой частоте:
(l/r2)=^-f[P(0)+P(<ob)},
<№)
= fh* Г
корр
+
1 + «
скорр I
<• et коррJ
(21.27)
Уравнение (21.26) показывает, что спектральное «время релаксации» Т\ в ЯМР
определяется спектральной плотностью на резонансной частоте щ. Другими
словами, беспорядочно меняющееся магнитное поле воздействует на ядра
пропорционально (среднеквадратичной) амплитуде частотной компоненты
магнитного поля на «собственной» частоте ядерной магнитной прецессии. С другой
стороны, 1/Гз определяется спектральной плотностью и на частоте ©о, и на нуле-
715 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
вой частоте. Это объясняется тем, что намагниченность вдоль оси у может
уменьшаться двумя способами: благодаря возвращению Мг обратно к своему
значению при тепловом равновесии (для этого необходимы переходы между
двумя энергетическими уровнями и, таким образом, этот процесс определяется
спектральной плотностью на соответствующей этим переходам частоте шо) и
вследствие уменьшения Му в результате того, что различные ядра могут пре-
цессировать с немного различными скоростями так, что суммарное значение
Му уменьшается во времени по мере взаимной «расфазировки» ядер в процессе
их прецессии. Поскольку энергия магнитного момента не изменяется при
изменении его направления в плоскости х—у, можно считать, что собственная частота
для этой потери фазовой когерентности равна нулю и, таким образом, Г2
■частично определяется и спектральной плотностью на нулевой частоте.
Уравнения (21.26) и (21:27) имеют вид, который является общим при
описании воздействия разнообразных неупорядоченных процессбв на спектральное
«время релаксации» в ЯМР (и других типах резонансов) и ширину линий в
спектрах. Важной особенностью этих выражений является в первую очередь то,
что в пределе для очень высоких скоростей флуктуации (для очень маленьких
времен корреляции «oTkopp'CI) они принимают вид
т~= I -2T2A2:KO(V; совгкорр<1 [(21.28)
Однако, поскольку спектральная плотность на нулевой частоте продолжает
возрастать, в то время как спектральная плотность на частоте ш0 незначительна
для больших времен корреляции ((ОоЯКарр>1), как это наглядно показано иа
рис. 21.3, \jT2 при увеличении тКОрР продолжает расти, тогда как МТ\ в этой
области уменьшается (рис. 2.1.5).
Особенно полезным приложением предшествующего анализа является
использование его для определения скоростей химического обмена на основе
измерений методом ЯМР значений 7\ и Т2 для случая «быстрого обмена»
Ю/Тобм)»!/?^ (или 1/7\).]. [Мы уже разбирали пример определения скоростей
'г
или ■
in —
h
A Jp ZKapp
Рис. 21.5. Зависимости скоростей релаксации ЯМР \/Т\ и 1/Т2 (логарифмическая
шкала) от времени корреляции (логарифмическая тикала) для процесса, в
котором магнитное поле беспорядочно изменяется между значениями ^ +h и —h
в каждом из трех взаимно перпендикулярных направлений.
В интервале «предельного сужения» (в левой части рисунка) ©оТкарр « 1, a Tt = Ti. Для
больших времен корреляции с увеличением ткорр Г, уменьшается, в то время как Г*
увеличивается Следовательно, Т2 более чувствительно к медленным 'флуктуациям, в то время
как и Г] и Т2 одинаково чувствительны к быстрым изменениям. Практические приложения
измерений Г, н Г2 в спектрах ЯМР основаны именно «а этом фундаментальном свойстве.
716 ЧАСТЬ 6
химического обмена из времен релаксации, полученных методом ЯМР для случая
смедленного обмена» (гл. 119).]
Наиболее важным аспектом настоящего обсуждения является то, что
использованная аргументация совсем не зависит от природы флуктуирующего
магнитного поля. В следующем разделе мы разберем флуктуации магнитного поля,
которые возникают в результате вращения молекулы, и сделаем заключения,
формально очень похожие на те, которые получены в уравнениях (21.26)—=■
(21.28) и на рис. 21.5, за исключением того, что теперь время корреляции будет
служить мерой времени, необходимого для того, чтобы молекула изменила свой
угол ориентации в пространстве приблизительно иа один радиан. Это даст
возможность вывести скорость вращения молекулы в растворе из измерений Tt
и Т2 в спектрах ЯМР.
-21.Б. БЕСПОРЯДОЧНОЕ ВРАЩАТЕЛЬНОЕ ДВИЖЕНИЕ.
ОПРЕДЕЛЕНИЕ КОЭФФИЦИЕНТА ВРАЩАТЕЛЬНОЙ
ДИФФУЗИИ МЕТОДАМИ ДИЭЛЕКТРИЧЕСКОЙ
РЕЛАКСАЦИИ, МАГНИТНОГО РЕЗОНАНСА И
ДЕПОЛЯРИЗАЦИИ ФЛУОРЕСЦЕНЦИИ
Предположим, что некоторое свойство молекулы (энергия, ди-
польный момент и т. д.) меняется при изменении ориентации
молекулы. Тогда беспорядочные флуктуации по углу между
некоторой вполне определенной осью молекулы и фиксированными
лабораторными осями будут приводить к флуктуациям, скажем,
дипольного момента, которые могут иметь частотные компоненты
на «собственных» частотах для интересующего нас образца. Таким
образом, беспорядочное вращательное движение может служить в
качестве широкополосного источника излучения на частотах вплоть
до самого верхнего частотного предела, определяемого
максимальной скоростью вращения молекулы. Мы рассмотрим эти
положения сначала для случая вращательной диффузии вокруг только
одной оси (сравните с одномерной трансляционной диффузией, гл.
6) и затем распространим наши выводы на трехмерную
вращательную диффузию реальных молекул в растворе. '
Одномерная вращательная диффузия
Как и в предыдущем примере с беспорядочными перескоками
между двумя разными состояниями, мы начнем с вычисления для
некоторого молекулярного свойства корреляционной функции /(ф),
которая изменяется с углом ф по отношению к одной
лабораторной оси. Это вычисление включает в себя усреднение по всём
первоначальным ориентациям и всем конечным ориентациям точно
так же, как раньше мы проводили усреднение по всем исходным и
конечным состояниям. Поскольку число возможных ориентации
бесконечно (ориентация меняется непрерывно в отличие от
конечного числа дискретных состояний в предыдущем примере), вместо
717 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
суммы ориентации необходимо взять интеграл по всем углам [ср.
с уравнением (21.21)]
2и 2и
<f(0)f(x)>= Jf(«p)/>(<P,t;<Po.O)d«pJ f(?0)P(?0)d?0, (21.29)
в котором Р(ф0, 0)^ф0 является вероятностью нахождения
молекулы в нулевой момент времени, повернутой на угол между ф0 и фо+
Н-^Фо, а Р(ф, т; фо, 0) —вероятностью ориентации молекулы между
углами ф и ф+^ф через время т при условии, что в нулевой момент
времени она была сориентирована между углами ф0 и фо+^фо-
Поскольку в обычном случае мы имеем равную вероятность
найти молекулу сориентированной в исходный момент в любом
направлении, Р(фо) не будет зависеть от фо и поэтому
<Ро=0
или просто
=*>(«?.) J <*Ро = 2*Р(«Р.) (21.306)
о
Все, что нам теперь остается,— это найти Р(ф, т; фо, 0) точно
таким же путем, каким мы вычисляли Р(НК0И,х; Нисх,0) в
предшествующем примере, т. е. из уравнения, которое описывает
изменение во времени интересующего нас параметра (в этом случае
угла ориентации молекулы ф). Основываясь на опыте и по
аналогии с уравнением движения для трансляционной диффузии
[уравнение (6.55)]
J^MdL^D-jjLpfat), (21.32)
мы можем ожидать, что вращательная диффузия (а именно
переориентация как результат многочисленных крошечных угловых
перемещений по часовой стрелке или против) описывается
аналогичным уравнением
дР{<?, 0 д* п
(21.33)
в котором DBP называют коэффициентом вращательной диффузии.
Читатель может легко проверить, что решением уравнения (21.33)
718 ЧАСТЬ в
является выражение*
Р(Ь * «Р.. 0) = 2 е-1п«>е1п*е-пЮ«>< . (21.34)
га=0
Теперь мы в состоянии рассмотреть некоторые конкретные
зависимые от угла молекулярные свойства /(<р). Например,
электрический дипольный момент, направленный под углом ф к направлению
некоторой фиксированной лабораторной оси, имеет вдоль этой
лабораторной оси компоненту, которая меняется с углом по закону
cos(<p)**. Так как молекула (и связанный с ней электрический
дипольный момент) вращается, компонента электрического поля
вдоль лабораторной оси изменяется так, что беспорядочные
повороты молекулы приводят к излучению вдоль фиксированной
лабораторной оси с частотами, которые можно предсказывать с
помощью спектральной плотности, рассчитываемой из
корреляционной функции для беспорядочного вращательного движения.
Сами расчеты гораздо проще, чем их описание
2и 2те оо
<ро=0 <р=0 га=0
со 2те 2и
Но поскольку
п=0 <р=0 9о=0
2я
J
А (га-1) ю
gl <я-"*rf(p= о при л^ 1 (2.1.36а)
«р=0
и
2те 2и
Г el'(B"I)?dp = Jdp=^2ic при пр=\, (21.366)
* Так же как и в гл. 13 и 19, мы используем в уравнениях (21.34) и
последующих комплексную запись. Аналогичный результат можно было бы
получить, используя вместо экспонент косинусы, однако комплексная запись
позволяет сильно упростить алгебраические преобразования. Поскольку конечный
результат должен быть всегда действительным, нам необходимо только снова
определить корреляционную функцию [уравнение (21.7)], как < /(x)*f(0) ) =
= G(x) и энергетический спектр, как P((u)=lltiKyZt:DG(x)e~i<i(tcl%.
** Или в ехр(мр) в комплексной записи.
*** Мы выразили корреляционную функцию через среднеквадратичные
амплитуды беспорядочно изменяющейся переменной так, что уравнение (21.35) и
последующие будут справедливы для любой беспорядочно измениющеися угловой
функции, мгновенная (комплексная) амплитуда которой пропорциональна
*хр(«р>.
719 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Рис. 21.6. Определение сферических
координат г, 0 и ф.
Компонента электрического диполя вдоль
произвольной оси изменяется как
функция только одного угла, в то время как
параметры магнитного резонанса и
флуоресценции меняются как сферические
гармонические функции обоих углов 9 и ф.
уравнение 21.35 упрощается до выражения с единственным членом
<П')*П0)> — e~D*>\
<(f(0))2>
t
одномерный
случай
(21.37)
Уравнение (21.37) имеет точно такой же функциональный вид,
что и уравнение (21.24) для беспорядочных перемещений между
двумя состояниями. Таким образом, все выводы и рассуждения,
касающиеся уравнения (21.24), применимы и к воздействию
вращательной диффузии (вокруг одной оси) на спектральную плотность
как функцию частоты.
В других рассматриваемых нами примерах (магнитной
релаксации, деполяризации флуоресценции) интересующий нас
молекулярный параметр меняется в зависимости от угла несколько более
сложным путем, а именно как функция двух пространственных
углов Э и ф (ф и 0 определены на рис. 21.6), в соответствии с
функциональным видом «сферических гармонических функций»
[уравнения (21.38)]. Заметим, что изменение дипольного момента по
направлению можно считать сферической гармонической функцией
первого «ранга» (/=1 в табл. 21.1). Магнитные же и
флуоресцентные параметры меняются, оказывается, как сферические
гармонические функции второго «ранга» (1=2 в табл. 21.1).
Трехмерная вращательная диффузия
Поскольку очевидно, что реальные молекулы в растворе
вращаются вокруг более чем одной оси, мы теперь подошли к
рассмотрению трехмерной вращательной диффузии, такой, при которой
720 ЧАСТЬ 6
Таблица 21.1
Функциональный вид некоторых сферических гармонических функций Yim (О, <р)
для / = 0, 1 и2>
4 = 0
1=1
/ = 2
л 2л
п
9=0 ф=0
УооО, Ф) = V4&
т = 0
т= 1
т = 0
m = — 1 Ki-^U, Ф) = VVtftsin 8 е-'»
m = 2 У22(8, ф) = /15/^Tsin2 9 е2/ *
« = 1 К21(8, ф) = —VlT/^T sin 8 cos 8 е/ф
т = 0 Г20(6, ф) = /5717л(3 cos2 6 — 1)
т = — 1 У2_1(8, ф) - У1»^ sin 9 cos 6 е~'ф
m = —2 К2_2(8, ф) = У1Ъ1г2п sin2 6 е-2'*
/п=(/), (/-1), (/-2), . . . , 0, -1, -2,
. , -(/-1), -/
У;т(8, ф) У,,т, (9, ф) sin е^ЭйГф = 0 при 1Ф1' и (или) тфт! (21.38а)
= 1 при /=/' и т—'т' (21.386)
Зависящие от угла параметры для диэлектрической релаксации меняются как Ую(б, ф),
в то время как те же параметры для магнитной и флуоресцентной релаксации меняются
как У ^(9. ф). В этом разделе мы вычисляем спектральную плотность, полученную из
корреляционной функции для подходящей сферической гармонической функции У;т(6, ф),
используя диффузионную модель для вращательного движения рассматриваемой молекулы.
(Читатель должен узнать в этих функциях «угловую» часть квантовомеханнческого
решения волновых функций водородного атома, даваемых во всех вводных курсах по квантовой
механике.) Некоторые свойства функций У\т (в, Ф) также даны в таблице.
молекула может менять свою ориентацию благодаря
многочисленным маленьким скачкообразным изменениям углов относительно
каждой из трех взаимно перпендикулярных осей. (В гл. 6 мы
разобрали трехмерную трансляционную диффузию,
осуществляющуюся благодаря маленьким линейным скачкам вдоль каждого из
трех взаимно перпендикулярных направлений.) Снова используя
соображения, которые привели уравнение (6.55) для
трансляционной диффузии к виду
«PJb£_»iA=D„VV(A-.». г, <) =
=°44гг+тр+тЬ) Р{х' у-г- "■ (2|-39)
721 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
мы получим желаемое уравнение для вращательной диффузии в
трех измерениях
dP^^'/)=DBPVaP(e, ь t), (21.40)
в котором P(Q,(p,t)smQdQdq> представляет собой вероятность
ориентации молекулы в направлении, располагающемся между
углами 8 и 0+^6 и ф и ф+^Ф в момент времени /; DBp — коэффициент
вращательной диффузии (быстрой диффузии соответствует
большой DBP). Оператор V2 в уравнении (21.40) выражен в полярных
координатах (рис. 21.6 при г=1).
Как и в одномерном случае, мы предполагаем, что все исходные
направления ориентации молекулы равновероятны, так что
Р(6о, фо, 0) не зависит от 0 и ф:
я 2я
Г Г P(eo,<p0)sineodeod<p0= l (таким образом, исходное направле-
e0ioPo=o ние ориентации обязательно
находится в пределах углов, указанных
на двух интегралах),
(21.41)
(21.42)
о о
Я(80,<ро,0)=1/4*
Р (8,
У-
*;
ео.?о.0)
оо оо
= 22 к*"»
;=0 т=0
(во
.?о) У»Я1
(9.*)*
-и/+0 V
Наконец, можно показать, что решение уравнения (21.40) для
вращательной диффузии можно записать в виде
(21.43)
где Р(0,ф, t\ 0о, Фо, 0) есть вероятность того, что молекула будет
сориентирована в направлении, близком к 0 и ф в момент времени
/ при условии, что в нулевой момент времени она была
сориентирована в направлении около 0О и <р0. Используя уравнения (21.38),
(21.42) и (21.43), читатель может теперь показать сам, что
корреляционная функция для любой из сферических гармонических
угловых функций будет иметь вид
<У/«(е, ?, *)у/и(е0,?0,о)>
ЧШЗЗЛ^о, %)У1т{Ъ, 9)е '
-'< + »> Vx
О <f 0о f о / т
ХУ*Ш(Ъ, 9)У1т(Ъ„ <?.)-(smMM?Hsmb&A?J. (21.44).
722 ЧАСТЬ 6
<Yim* (в**) Yim(b0,b,Q)>^clme l (/+0 V
(21.45a)
вр
где
= c/eiexpH/xJ (21.456)
■—*■ 1/2 Z)Bp для диэлектршеской релаксации
-вр — / (/+1ШВ„ |
—т* 1/6 DBp для ЯМР, ЭПР и деполяризации флуоресценции
(21.46)
а с/т является функцией / и т, но не зависит от времени.
Алгебраические преобразования, приводящие к уравнению
(21.45) для трехмерной вращательной диффузии, являются
значительно более сложными, чем при получении уравнения (21:24) для
скачков между двумя состояниями или уравнения (21.37) для
одномерной вращательной диффузии. Тем не менее уравнение
(21.45) снова показывает, что корреляционная функция
экспоненциально уменьшается во времени, а это приводит к тому, что
спектральные плотности вновь описываются простыми
зависимостями, как показано на рис. 21.2.
Наконец, есть возможность соотнести коэффициент
вращательной диффузии с вращательным коэффициентом трения
[стационарной скоростью вращения при единичной вращающей силе
(крутящем моменте)]. Это соотношение описывается уравнением по
существу таким же, как и соотношение [уравнение (7.13)]
коэффициента трансляционной диффузии и линейного коэффициента
трения (стационарной линейной скорости при единичной линейной
силе):
/тр
Аф^Г". (21-47)
•', ;• /вр
Далее, аналогично тому, как мы определяли зависимость
линейного коэффициента трения для сферической макромолекулы от
размера молекулы и вязкости раствора по уравнению
fтр (сфера) = 6ят)#, (7.55)
где R — радиус сферы, а г\ — вязкость раствора, мы получим
похожую зависимость вращательного коэффициента трения от
молекулярного размера и вязкости раствора для сферических
макромолекул:
/ (сфера) = 8mjfl\ (21.48)
723 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Объединяя уравнения (21.47) и (21.48), можно получить
желаемое соотношение между коэффициентом вращательной диффузии
DBp (его можно получать из различных уже рассматривавшихся
нами экспериментов) и размером R сферической макромолекулы:
(для сферических макромолекул) (21.49)
Пример. Диэлектрическая релаксация
В гл. 14.Е мы обсудили эксперименты (диэлектрические потери,
диэлектрическую проницаемость), отражающие скорость, с которой молекула с
постоянным электрическим дипольным моментом может изменять свою ориентацию
(вращаться) в электрическом поле. Поскольку компонента электрического диполыюге
момента вдоль фиксированного лабораторного направления (направления,
перпендикулярного двум параллельным пластинам конденсатора на рис. 14.33)
изменяется в зависимости от угла по закону.cos(6), где 9 — угол между
вектором диполыюго момента и вектором нормали к пластинам, ширину линий
стационарного спектра получают из спектральной плотности, рассчитанной из
корреляционной функции /(6)=cos(6) [см. уравнения (21.45) для случая 1=1,
m=0]:
<cos (9, т) cos (9„0)> = с10е u+ bp '
где
(21.50а)
(21.506)
(21.51)
Таким образом, при исследовании диэлектрической релаксации характерная
ширина линии (половина ширины линии на половине максимальной высоты). для
диэлектрической проницаемости как функции частоты переменного внешнего
электрического поля (рис. 14.35) задается уравнением
"^вр
вр"
kT
4nrft31
.(21.52)
которое выведено в гл. 14.Е [уравнение (14.33)]. Умозрительно картину этого
явления можно представить себе так: внешнее электрическое поле непрерывно
стремится выстроить диполи вдоль своих силовых линий, ио, с другой стороны,,
беспорядочное вращательное движение (вращательная диффузия) самих
молекул непрерывно расстраивает их упорядоченное расположение. Наблюдаемая
в стационарном режиме диэлектрическая проницаемость по данной частоте
является мерой этого процесса упорядочения. Однако, поскольку из
преобразования Фурье уравнения (21.506) нам теперь известен энергетический спектр для
процесса беспорядочной вращательной диффузии, то, следовательно, мы можем
дать объяснение диэлектрической проницаемости в стационарном состоянии как
функции частоты внешнего электрического поля с фиксированной амплитудой
колебаний [если амплитуда (и связанная с ией тенденция к упорядочению)
действующего внешнего поля постоянна, то любое изменение диэлектрической
проницаемости должно быть результатом изменений энергетического спектра
беспорядочного вращательного движения]. Поскольку энергетический спектр [кор-
724 ЧАСТЬ 6
реляционная функция из уравнения (21.506)] должен, согласно уравнению
(21.13), иметь вид хорошо известной кривой Лорентца, мы можем теперь дать
объяснение функциональным зависимостям от частоты диэлектрических потерь
и диэлектрической проницаемости, которые ранее обсуждены в гл. 14 [уравнение
014.32)].
Информация, которую получают при измерении диэлектрической
релаксации, и приложения для коэффициента вращательной диффузии уже
обсуждались ранее (гл. 14.Е) и примеры читатель может найти в соответствующих
разделах.
Пример. Магнитная релаксация
Существует маленькое «локальное» магнитное поле, связанное с любым
ядерным (или электронным) магнитным моментом. Его напряженность
изменяется по закону (Л/г3), где г — расстояние между магнитным моментом и точкой,
в которой измеряют это локальное поле. В молекуле, обладающей двумя
расположенными рядом магнитными ядрами, каждое магнитное ядро создает
«биполярное» магнитное поле на другом ядре. Если теперь такие молекулы
поместить в большое внешнее статическое магнитное поле, то результирующее
магнитное поле на данном ядре будет представлять собой векторную сумму
большого внешнего магнитного поля и малого поля, направление которого
постоянно меняется вследствие вращения молекулы в растворе. Среднеквадратичное
усреднение этого малого флуктуирующего поля будет, таким образом, меняться
по закону (1//*), где г теперь является расстоянием между двумя ядрами.
Энергетический спектр флуктуирующего поля будет определяться спектральными
плотностями, которые получают с помощью преобразования Фурье
корреляционной функции подходящих сферических гармонических функций Y2m (6, ф),
описывающих зависимость взаимодействия от ориентации молекулы. Поскольку
система из двух частиц со спинами п/2) [рис. 18.6] описывается фактически тремя
различными энергиями, различия между которыми (энергии «переходов»)
соответствуют «собственным» частотам (о0, 2(о0 и 0, где (Oo=V^^o, можно было бы
ожидать, что скорости релаксации ЯМР (1/Т]) и (1/Г2) должны быть прямо
связаны со спектральными плотностями на этих частотах. Для корректного
расчета необходимо привести полную квантовомеханическую обработку зависимого
от времени возмущающего воздействия. Однако конечный результат расчета
вполне соответствует приведенным выше умозрительным рассуждениям и
качественно аналогичен (как и следовало ожидать) скоростям релаксации,
полученным ранее [уравнения (21.16) и (21.27)] для похожего случая обмена между
двумя состояниями:
77= "g- U{B) К) + /(С) (2со0)], *ч
h= Т [4 /И> W + Т-/(В> ("о) + 4 '(С) (*■*>],
где
оо
/(»)э j <f(8,f,T)*f(e0,?o.O)> e-tax(h (21.54)
—00
и
fW (e,rt =^- j/"^ У20 (9, ?). <21.55a)
вследствие
взаимодей- (21.53а)
ствия двух
магнитных ,01 «я
диполей для 1А1Л#>)
частиц со
1
СПИНОМ ~7Г
JT25 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
г
• ,R. y2h2 -|/"8тг
/(В) (8,Т) = ^г- |/ l5- У,., (8, <?), (21.ЕБ6)
/(C)(M=-V"K Т5"У»-*<9- V). (21.5Бв)
В этих уравнениях мы снова для краткости использовали комплексную запись.
Уравнения (21.5.3) и (21.45) можно использовать либо для вычисления
межъядерного расстояния г {если из уравнений (21.46) и (21.49) известно
вращательное время корреляции твр], либо для вычисления вращательного
времени корреляции (если известно межъядерное расстояние). В гл. 16 (рнс. 16.24)
мы приводили пример определения полной конфигурации кофактора в активном
центре фермента, который был основан на измерениях ядерных магнитных
«времен релаксации» Т} и Т2 и известной величине молекулы фермента [из нее по
уравнениям (21.4.6) и (21.49) можно вычислить вращательное время
корреляции]. В этом примере расстояния протон--электрон были получены из
уравнений, аналогичных уравнениям (21.53).
Проведенное недавно исследование связывания различных регуляторов
с ферментом аспартаттранскарбамилазой дает пример практического
использования параметра хвр для случая, когда известно межъядерное расстояние.
[Структура (рис. 15.17) и функция (рис. 11.9, 11.6 и 16.6) этого фермента уже были
описаны ранее.] Этот фермент обратимо ингибируется продуктом его
метаболизма цитидинтрифосфатом ЦТФ, образующимся в результате многоступенчатой
реакции. Измерение Т\ и Т2 для протонов различных молекул, которые
связываются с местом «посадки» обратимого ингибитора, показало, что протоны 5-ме-
тилцитидина имели вращательное время корреляции фактически точно такое же,
как и протоны белка как целого (эта молекула «жестко» связывается в месте
.посадки обратимого ингибитора). В то же время протоны тимидина имели
вращательное время корреляции но величине почти на порядок меньше (более
быстрая вращательная диффузия), чем протоны белка как целого (т. е. в том
же самом месте тимидин связывается относительно свободно). Поскольку
5-метилцитидин более близок по структуре к природному ингибитору ЦТФ, чем
тимидин, эти результаты позволяют предположить, что точное соответствие
обратимого ингибитора месту его связывания существенно для аллостерйческой
,функции молекулы. Более того, мы уже отмечали, что этот интересный фермент
можно разделить и получить отдельно «каталитическую » и «регуляторную»
субъединицы. Ферментативный катализ в этом случае осуществлялся
практически с той же эффективностью, что и целым ферментом, только одной
каталитической субъединицей. В рассматриваемом ЯМР-исследовании авторы сравнили
также связывание двух ингибиторов с изолированной регуляторяой субъединицей
и с целым ферментом. Было показано, что 5-метилцитидин снова связывается
с изолированной регуляторяой субъединицей жестко, а тимидин — свободно, что
полностью совпадает с результатами, полученными на целом ферменте. Таким
образом, оказалось, что у этого фермента можно полностью разделить при
изучении каталитическую и регуляторную функции, даже несмотря на то, что при
связывании с целым ферментом субстрат и регулятор взаимно влияют на
связывание друг друга.
Когда на промежуточных стадиях в химической реакции участвует
макромолекула, можно извлечь много информации о механизме из кинетических
исследований (разд. 3 и гл. 16.А.1). Если же химической реакции не происходит,
большую часть информации того же типа можно получить, следя за другими
параметрами (таким, например, как твр из данного выше примера). Другой
пример информации, получаемой с помощью вращательного времени корреляции
{в свою очередь выведенного из времен магнитной релаксации), уже приведен
«а рис. 16.23, где изменение Т\ для различных атомов углерода в фосфолипид-
ной молекуле интерпретировалось как изменение степеней вращательной
свободы для .различных частей цепи жирной кислоты как составной части мембраны.
726 ЧАСТЬ «
По существу те же самые аргументы были использованы для объяснения
примеров по электронному (в отличие от ядерного) парамагнитному резонансу,
которые также были разобраны ранее (гл. 14.Г).
Пример. Деполяризация флуоресценции
Эксперимент по изучению деполяризации флуоресценции был описан в гл. 16
(рис. 16.14). Образец облучали вертикально поляризованным светом; этот свет
поглощается и затем, после отключения внешнего источника света,
возбужденный образец испускал другой, «флуоресцентный» свет в течение интервала
времени длительностью до нескольких наносекунд. Если в период времени между
поглощением и флуоресцентным испусканием молекула вращается, то будет
наблюдаться уменьшение «поляризации» (т. е. отношения интенсивиостей
вертикально и горизонтально поляризованных флуоресцентных потоков)
флуоресцентного испускания. Таким образом, поляризация флуоресцентного света
уменьшается за время, прошедшее после поглощения, со скоростью, которая
характеризует скорость вращательной переориентации молекулы флуоресцирующего
вещества.
Интенсивность флуоресценции, поляризованной в разных плоскостях,
обусловлена сложным усреднением по различным ориентациям молекул, как
вовремя поглощения, так и позже в момент испускания. Компонента внешнего-
электрического поля вдоль направления «поглощающего диполя» (то есть вдоль
оси «электрона на пружине» для процесса поглощения) будет меняться по
закону cos(6C-»n)> где 6с->п —это угол между осью поглощающего диполя и;
осью падающего плоскополяризованного света. Однако интенсивность этой
компоненты зависит от квадрата амплитуды электрического поля и, следовательно,,
меняется по закону cos2(oc_>n). Диалогично можно показать, что интенсивность
флуоресценции, плоскополяризованной в двух взаимно перпендикулярных (в
лабораторной системе координат) направлениях, меняется по законам со52(6с_+ф)
и 81пг(0с_^ф)-со82(фс_+ф), где 6С^.Ф и Фс->.ф— это углы, описывающие
ориентацию испускающего диполя по отношению к лабораторной системе координат.
Таким образом, даже несмотря на то, что компоненты амплитуды действующего
электрического поля меняются как У\оФ, ф), наблюдаемая анизотропия
флуоресценции
меняется так же, как интенсивность излучения, и в результате- зависимость А\х)
от углов описывается сферической гармонической функцией Y2m(Q, Ф), точно
такой же, как в примере с магнитным резонансом. Анизотропия флуоресценции
уменьшается (по интенсивности) таким образом, как и среднеквадратичная
степень упорядоченности испускающих диполей в возбужденной молекуле флуо--
ресцирующего вещества. Она, следовательно, пропорциональна корреляционной
"функции распределения по углам ориентации испускающих диполей:
А(х) ос е~™^ = <Гг/Ч (21.56)
■■■л
где
z r—- ' . Г для магнитного резонанса или [ (21.57)
вр 60вр [деполяризации флуоресценции J . "
Таким образом, измерения деполяризации флуоресценции являются способом
определения коэффициента вращательной диффузии (так же как к эксперииен-
ы.
ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
ты по магнитному резонансу или диэлектрической релаксации). Особенность
этих экспериментов состоит в том, что в них, как и в экспериментах по
магнитному резонансу, определяемое экспериментально вращательное время
корреляции связано с коэффициентом вращательной диффузии соотношением т„р=
= 1/6/)Вр, тогда как в случае диэлектрической релаксации оно меняется как
.1/2 Dnp). Это отличие возникает из-за того, что рассматриваемые времена
корреляции для двух типов корреляционных функций получают из
экспериментальных параметров, которые меняются как сферические гармонические функции
второго и первого «ранга».
Оказалось, что к разным макромолекулам можно ковалентно присоединять
.целый ряд флуоресцентных меток. Поскольку вращательные времена
корреляции, рассчитанные по уменьшению деполяризации флуоресценции после
импульса возбуждения внешним поляризованным светом, очень близки по величине
к рассчитанным на основании простой сферической модели макромолекулы
в непрерывной среде [по уравнениям (21.49) и (21.57)]
ЯР'
4Я7)£3
(21.57)
Таблица 21.2
Вращательные времена корреляции твр для белков, определенные
из экспериментально измеренных скоростей затухания деполяризации
флуоресценции или рассчитанные на основании теоретической
фрикционной модели сферического белка в непрерывной средеа.
Белок
Апомиоглобин
ji-Лактоглобулин
Трипсин
Химотрипсин
Карбоангидраза
Р-Лактоглобулин
Апопероксидаза
(мономер)
(димер)
Сывороточный альбумин
Молекулярная
масса
17 000
18 400
25 000
25 000
30 000
35 000
40 000
66 000
(из
деполяризации
флуоресценции)",
не
8,3
8,5
12,9
15,1
11,2
20,3
25,2
41,7
твр
<нз уравнения
21.58Д
НС
4,4
4,7
6,4
6,6
8,0
9,7
10,5
17,4
т(эксп)
вр
т(рассч)
вр
1,9
1,8
2,0
2,3
1,4
2,1
2,4
2,4
а Экспериментально определенные значения г» всего в~2 раза отличаются от
теоретических величии и, таким образом, отражают вращательное диффузионное движение
молекулы белка как целого. [Взято с изменениями из Yguerabtde /., in Methods in Enzymology,
Vol. 26, part С ed. C.H.W. Hirs and S.N. Timasheff, Academic Press, N.Y.. 1972, p. 528.]
<> Все экспериментальные твр приведены к значениям, которые они должны были бы иметь
в воде при 25*С.
* Рассчитанные тВр получены для жестких иегидратированных сфер с той же молекуляр-
яой массой, что и у белка. Парциальный удельный объем белка принят равным 0,73 см'/г
<уравиенне 21.58).
728 часть б
что видно из данных табл. 21.2, то можно заключить, что в большинстве
случаев флуоресцентные метки жестко прикреплены к макромолекулярному остову
и могут использоваться для прямого измерения «вращательной гибкости» самой
макромолекулы.
Так как вращательные времена корреляции определяют из экспериментов
различных типов, надежность определения их истинных значений возрастает.
Например, выполненное недавно методом ЯМР определение твр для
сывороточного альбумина дало значение 4» Ю-8 с, которое прекрасно согласуется со
значением, полученным из эксперимента по деполяризации флуоресценции.
Вращательное время корреляции твр определяют как постоянную времени
экспоненциального уменьшения корреляционной функции для вращательной
диффузии. Однако Твр можно также считать характеристическим временем, которое
необходимо для (диффузионного) поворота выбранной макромолекулы на угол
порядка одного радиана (см. задачи к этой главе).
Выполненные недавно исследования по деполяризации флуоресценции
антитела, к которому был присоединен флуоресцирующий гаптен, явились одним
из наиболее убедительных доказательств гибкости молекулы иммуноглобулина.
Для этих экспериментов были выращены антитела, специфичные к дансильиому
гаптену (строение дансильной группы см. гл. 16.Б.1). Затем образовывали
комплексы дансильной молекулы со специфичным к ней антителом и со
связывающими гаптен фрагментами этого антитела, которые изображены ниже:
Интактное - Чра****™ Фрагмент
антитело
На каждом чертеже предполагаемая область связывания гаптена обозначена
черным. Далее изучали анизотропию флуоресценции связанной дансильной
молекулы как функцию времени, прошедшего после импульса внешнего плоскополя-
ризованного света. На рис. 21.7 показано поведение анизотропии для
различных меченых фрагментов антитела.^Тангенс угла наклона каждой кривой
является мерой коэффициента вращательной диффузии для связанной с дайной
молекулой дансильной метки. Для самого маленького фрагмента (Fab)
обнаружена быстрая вращательная диффузия с твр=33 не, и экспериментальные
данные для него хорошо соответствуют теоретической кривой, рассчитанной для
вращательной диффузии сферической макромолекулы Fab. Однако для
дансильной метки, связанной с макромолекулами F(ab')2 и IgG, уменьшение
анизотропии флуоресценции ие является экспоненциальным процессом. Тем не менее эти
экспериментальные данные можно описать как сумму двух экспоненциальных
процессов: бытрого первоначального уменьшения благодаря (по-видимому)
внутренней гибкости макромолекулы в области «шарнира», который соединяет два
фрагмента Fab (тВр=33 не), и более медленного уменьшения, соответствующего
вращению интактного антитела как целого (самая верхняя кривая на рис. 21.7
с ТвР=168 не). Эти эксперименты дают, таким образом, убедительное
доказательство того, что существует внутренняя гибкость сегментов в молекуле
антитела. Возможно, что такое гибкое соединение имеет биологическое значение,
облегчая образование комплекса аитигеи-—антитело-.-
729 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
и,«
0,3
53
S 0,2
5Г
>
«
■5
1 °п
1
*
1
|^0,05
'о
5
*
"* 0,03
1
_
~"
1
1
h ^Ьк^^У
1
I
^ч^^р
V3
Fab\n
а
1
1 1
*
_
IgG
^g> _
^*^^^Л.«
?(аЬ'>2 о^^°
за
\о
°^ч
nrfO О
\а_
1 • 1
20
40 60
t,HC -
ВО
100
Рис. 21.7. Зависимость от времени анизотропии флуоресценции A (t)
(логарифмическая шкала) дансил-Ь-лизинового гаптена, связанного с антидансильным
антителом IgG или с указанными иа рисунке фрагментами этого антитела.
Сплошными лнинями показаны полученные методом наименьших квадратов аппроксимации
для убывания по одной экспоненте (кривая для фрагмента Fab) илн по закону суммы двух
экспонент (кривые для IgG и фрагмента F(ab')2). Интерпретация этих результатов на
основе подвижности сегментов в молекуле IgG даиа в тексте.
[Из Yguerabidc J., Epstein Я. F., Siryer L„ J. Mol. Biol., 51, 573 (1970).]
21.B. БЕСПОРЯДОЧНОЕ ПОСТУПАТЕЛЬНОЕ ДВИЖЕНИЕ.
ТРАНСЛЯЦИОННАЯ ДИФФУЗИЯ
21.В. 1. РАССЕЯНИЕ СВЕТА. КОЭФФИЦИЕНТЫ ТРАНСЛЯЦИОННОЙ
ДИФФУЗИИ ДЛЯ МАКРОМОЛЕКУЛ (ИЛИ БАКТЕРИЙ) В РАСТВОРЕ
В гл. 15.А мы уже подробно обсуждали стационарную
интенсивность света, рассеянного под некоторым углом 9 по отношению
к падающему потоку. Однако если исходить из соображений,
наглядно представленных на рис. 21.8, то может показаться, что
наблюдаемая интенсивность света, рассеянного однородным
раствором, должна быть нулевой. Теперь мы в состоянии разрешить этот
парадокс, предположив, что в любой выбранный момент времени
плотность растворителя в любых двух маленьких областях (рис.
21.8,6) не идентична. Поэтому взаимная компенсация амплитуд
рассеянного света, показанная на рисунке, не является полной, так
как некоторое количество света все же рассеивается в направлении
наблюдателя. Аналогично, в растворе, содержащем растворенные
макромолекулы, любое дополнительное (к тому, которое дает
растворитель) светорассеяние должно быть обусловлено флуктуация-
ми концентрации растворенного вещества. Поскольку единствен-
730 ЧАСТЬ 6
Рис. 21.8. Рассеяние длинноволнового излучения (света) совершенным
бесцветным кристаллом (а) .и чистой жидкостью (б).
1Из van Holde К. Е., Physical Biochemistry, Prentice-Hall, Englewood Cliffs, N. J., 1971,
p. 186.]
л — если кристалл достаточно велик, а интервалы между частицами достаточно малы, то
все частицы в кристалле могут быть разделены на пары таким образом, что для каждого
рассеивающего элемента i найдется такой другой рассеивающий элемент /, что амплитуды
рассеянного от них света будут полностью взаимно гасить друг друга в выбранной точке
«аблюдения Р. Поскольку этот аргумент справедлив для любого выбранного
рассеивающего элемента t, общая интенсивность света, рассеянного под любым углом, будет равна
нулю.
•б — разобьем чистую жидкость на множество маленьких кубиков так, чтобы каждый из
«их содержал лишь небольшое число молекул, а размеры самих кубиков были малы по
.сравнению с длиной волны падающего излучения. Будем считать, что каждый такой кубнк
является рассеивающим элементом. Предположим, что такие рассеивающие элементы i
-и / [как и в (а)] расположены так, что рассеянные от иих волны снова попадают в точку
яаблюдеиия Р в противоположных фазах. Теперь, если бы ( и / содержали равное число
молекул (т. е. i и / давали бы равные амплитуды рассеянного света), пришедшие от иих
я точку Р волны полностью гасились бы. Однако, поскольку флуктуации плотности
растворителя (а также концентрации растворенного вещества) препятствуют полному взаимному
гашению рассеянных волн, некоторое количество света все же рассеивается.
ной причиной изменений концентраций в данном случае может
■быть только перемещение (поступательное) растворенных молекул
•с одного места на другое и так как интенсивность наблюдаемого
светорассеяния зависит от величины этих изменений, оказывается,
■что из измерений интенсивности светорассеяния раствором можно
получать информацию о скорости трансляционной диффузии
макромолекул.
731 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Падающий
веет
2
Рис. 21.9. Схематическое изображение направлений потоков падающего и
рассеянного света для раствора, концентрация которого в данной точке
пространства изменяется во времени синусоидально (см. текст).
[Из Ford N. С. Jr., Chemica Scripta, 2, 193 (1972).]
1 — направление распространения синусоидально меняющейся во времени
«концентрационной волны; 2—направление распространения наблюдаемого рассеянного света.
Интуитивно можно ожидать, что длина волны рассеянного
света должна быть связана с расстоянием между концентрационными
максимумами и минимумами, которые образуются в различных
областях раствора в данный момент времени. Отсюда кажется
логичным анализировать все концентрационные флуктуации исходя
из бесконечного числа компонент, когда каждая компонента
рассматривается как «концентрационная волна» с соответствующей
амплитудой и длиной волны. Такой подход формально очень похож
на рассмотренный нами ранее (гл. 20.Б и 21.А) анализ
произвольного когерентного или беспорядочного сигнала путем разбивки его
на синусоидальные компоненты с набором различных частот.
Следовательно, если теперь рассмотреть (рис. 21.9) малое
синусоидальное изменение концентрации растворенного вещества (и,
таким образом, показателя преломления раствора), то обнаружится,
что при рассеянии света выполняется соотношение
Л = т2-даг <21-59>
где {kin)—это длина волны падающего света внутри раствора
с показателем преломления п, 8 — угол рассеяния, а Л — длина
волны (рис. 21.9) синусоидальной функции (волны) концентрации
растворенного вещества, являющегося причиной рассеяния. Это
соотношение весьма похоже на хорошо известное уравнение
Брэгга (гл. 22.А) для дифракции рентгеновских лучей на атомной
решетке (кристалле), которая происходит фактически Но той же
самой причине, что и рассеяние света-.
732 ЧАСТЬ 6
Очевидно, что концентрация растворенного вещества в любом
реальном жидком растворе меняется во времени не синусоидально.
Напротив, она скорее меняется беспорядочно (благодаря
броуновскому движению) в соответствии с явлением трансляционной
диффузии, рассмотренным в гл. 6. Однако мы уже знаем, что можно
анализировать любой беспорядочно изменяющийся во времени
процесс исходя из его среднеквадратичных компонент на различных
частотах. Таким образом, для описания результата, получаемого
на любой отдельной длине волны полного спектра частот
флуктуации, нам не потребуется ничего, кроме уравнения (21.59).
Для определенности рассмотрим компоненту флуктуации
концентрации, соответствующую длине волны Л, которую обозначим
как бс(Л,/). Если использовать систему координат, в которой
направление у совпадает с направлением распространения
«концентрационной волны», показанной на рис. 21.9, то можно описать
флуктуацию концентрации уравнением для одномерной
(трансляционной) диффузии
t№=z>Tp|L6c(j/, t) (2Ш)
Решение уравнения (21.60) удобно выразить в виде произведения
сомножителей, зависящего от времени и зависящего от
координаты:
Ьс[у, t) = bc{\,t)sm№\ |(21:61)
Подставляя уравнение (21.61) в уравнение (21.60) и решая его
относительно части, зависящей от времени, получим
бс(Л, t) = bc(\, 0)бГ^(2л/А,г' (21.62)
Теперь мы можем составить корреляционную функцию для
концентрационных флуктуации:
Gm(*) = <*c(K i)tc(A, 0)> = <[ИЛ, 0)]2>бГ^(2л/А)'' (21.63)
ИЛИ \
C?(x)=G(0)<T*,ZV ; К =^sin 4- (21.63а)
Наконец, считая, что изменения показателя преломления
раствора пропорциональны изменениям концентрации растворенного
вещества, и вспомнив, что интенсивность рассеянного света
зависит от квадрата показателя преломления [уравнение (15.26)],
можно заключить, что корреляционная функция для флуктуации
интенсивности рассеянного света должна иметь вид
I Якоиц W I2 = (const) exp [-2tf2DT- x]
Таким образом, наблюдаемая интенсивность рассеянного света
(рис. 21.10) имеет частотную зависимость, определяемую путем
33 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
I
It
■в
4
а.
100
200
300
400
500
Гц
600
700
800
900
1000
1065 г—
«?
t3
а;
I
639 —
0,00000 0,03273 0,06546 0,09819 0,13092
«-©
Рис. 21.10. Определение коэффициента трансляционной диффузии из рэлеевского
рассеяния лазерного света.
{Из Haas D. D., Mustacich R. V., Smith В. A., Ware В. R., Biochem. Biophys. Res. Commun..
59. 174 (1974).]
a — относительная интенсивность рассеянного света как функция частоты для
приблизительно 1 мМ раствора СО-гемоглобииа (в качестве источника использован гелий-иеоновый
лазер с длиной волны излучения Х«632,8 им; угол рассеяния в равен 23*).
б — график полуширины линии светорассеяния иа половине максимальной высоты (в
герцах) как функция sin* 1—1.
В пределах экспериментальных ошибок кривей а имеет характерную форму лореитцевой
линии "(черная линия), а график б является прямой линией, которая проходит через начало
координат. Твигеяс угла наклона прямой б пропорционален коэффициенту трансляционной
диффузия £тр(см. текст).
734 ЧАСТЬ «
преобразования Фурье этой корреляционной функции. Такое
преобразование Дает зависимость в форме очень хорошо нам знакомой
кривой Лорентца, ширина «линии» которой пропорциональна
коэффициенту трансляционной диффузии DTp, Что же касается
эксперимента, то следует сказать, что ранние измерения ширины
линии интенсивности рэлеевского рассеяния были выполнены путем
поточечного определения всего контура спектральной линии. В
противоположность им более современные измерения базируются на
непосредственном расчете корреляционной функции из
регистрации меняющейся во времени интенсивности рассеяния. Из
последующих примеров станет понятно, что этот новый подход к
методу определения коэффициентов трансляционной диффузии развит
для получения высокоточных результатов. Там же будут даны и
некоторые другие приложения этого метода.
Прежде чем перейти к примерам, необходимо сделать
последнее практическое замечание. На характерных углах рассеяния
ожидаемая расчетная ширина спектральной линии (на половине
максимальной высоты), обусловленная флуктуациями
концентрации, которые в свою очередь возникают благодаря трансляционной
диффузии макромолекул растворенного вещества, составляет
величину порядка 1000 Гц для макромолекулы с молекулярной
массой 50000. Поскольку сам падающий (видимый) свет имеет
частоту около 5'1014 Гц, прямое определение формы спектральной
линии рассеянного света потребовало бы разрешающей силы,
большей чем 1014/103=10п, что почти в 10 000 раз больше, чем у
лучших существующих решеток или интерферометров. Поэтому
обычно измеряют «биение» интересующего нас сигнала с излучением
какого-либо другого когерентного источника (два потока
складывают вместе). Если обозначить частоты двух сигналов (потоки от
образца и упомянутого источника), как on и юг, то получающееся в
результате электрическое поле будет представлять собой простую
сумму его компонент
Е = Ех cos (mj) + £2 cos КО- ' (21 *64)
Поскольку фотоумножитель измеряет интенсивность (а не
амплитуду) электрического поля, регистрируемая интенсивность будет
пропорциональна квадрату амплитуды электрического поля,
описываемой уравнением (21.64)
/регос | Я |в =s | £, cos ш,^ + £"t cos <ot/ j*. (21.65)
Раскрывая скобки в уравнении (21.65) и используя
тригонометрическое тождество для выражения coswi/'COsg^, получим
/регос Е\ cos2 К/) + Е\ cos2 Ы) + ^ [cos (ш, - <о2) t +
-j-COS(a),-j-(D2)/].
(21.66)
!85 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Однако анод фотоумножителя не реагирует на частоты, большие
аем 1010 Гц, и, значит, сигналы с частотами ац, (Ог и {coi-f-сог) в
сравнении (21.66) будут регистрироваться усредненными по
времени. Но так как усредненное по времени значение cos2(cui/) =
= (72), a cos[(<ui-f-co2)f] =0, анодный ток детектора будет состоять
зз постоянной компоненты и переменной компоненты, которая
колеблется с частотой «биения» или разностной частотой (coi—юг)*.
Поскольку разностная частота близка по порядку величины к
ширине спектральной линии рассеянного света (несколько кГц) и
поскольку такие частоты можно измерять очень точно (в пределах
«собственной» ширины линии испускания самого оптического
лазерного источника), имеется возможность изучать интенсивность
рассеянного света на частотах, предельно близких к частоте
падающего (лазерного) света. Для точного измерения ширины линии
в 1000 Гц необходимо, чтобы монохроматичность потока
падающего света была лучше, чем 100 Гц. Поэтому такие эксперименты
стали возможны только после создания практически
монохроматических (лазерных) источников света.
Рример. Коэффициенты трансляционной диффузии и молекулярные массы
Как уже было показано [уравнения (21.63) и последующие], можно
ожидать, что спектр интенсивностей рэлеевского светорассеяния на растворенных
макромолекулах будет иметь вид кривой Лорентца с шириной на половине
максимальной высоты, пропорциональной KZDTV, где К определяется соотношением
К = (4я/!/Х) sin (8/2), (21.67)
в котором я— это показатель преломления раствора, "к — длина волны
падающего света, а 8 — угол между падающим и рассеянным потоками света. На
рис. 21.10, а показан характерный спектр СО-гемоглобина для рассеяния под
углом 0 = 23°. Если построить график зависимости полуширины таких спектров
на половине максимальной высоты от sin2!-?,-), то получится прямая линия
(рис. 21.10,6), из которой можно вычислить коэффициент трансляционной
диффузии. В рассматриваемом случае он оказался равным D2q, а> = 6,9±0,ЗХ
ХЮ^7 см2с-1. Точность этого вычисления, очевидно, достаточно высока и,
судя по прекрасному совпадению экспериментальных точек с соответствующими
кривыми на рис. 21.10, а и 211.10,6, составляет в большинстве случаев величину
порядка 1%- В табл. 21.3 даны коэффициенты трансляционной диффузии для
некоторых объектов, полученные описанным выше способом. Определение
методом светорассеяния имеет следующие достоинства: а) необходимы низкие
концентрации растворенных макромолекул (обычно около 1 мг/мл), и поэтому
можно проводить точные измерения, используя относительно небольшие образцы;
б) нет необходимости точно контролировать температуру, поскольку, если
использовать подходящую оптическую ячейку, макроскопическая конвекция ие
влияет иа спектр; в) измерение можно проводить очень быстро (на все
операции после приготовления образца обычно затрачивают около 1 ч).
В гл. 7 было отмечено, что эксперименты по определению скорости
седиментации обеспечивают подкупающе быстрое определение молекулярной массы
* Этот процесс приводит к тому же самому конечному результату, что и
система смеситель — фильтр на рис. 20.7 (хотя механизмы ях действия различны).
736 ЧАСТЬ 6
Таблица 21.3
Коэффициенты трансляционной диффузии D1p некоторых природных
и синтетических макромолекул9
Образец
D , 10-' см» с-1
по данный рассеяния по гидродинамический
света данным
Лизоцим
Овальбумин
Сывороточный альбумин крупного
рогатого скота
Вирус табачной мозаики
ДНК (тимус теленка)
( г = 440 ± 40 А
г = 630 ± 30 А
г = 1830 ± 30 А
Сферы полнстн-
рольного
латекса
П,5±0,3
7,1±0,2
б,7±0,1
0,40±0,02
0,2±0,1
0,59±0,02
0,368±0,006
0,134±0,004
11,6
8,3
(данных для
сравнения нет)
0,3
0,13
0,56±0,06°
0,38 ±0,026
0,134±0,002б
л Dubin S. В., Lunacek 1. Н.ш Benedek G. В., Рг0с. Natl. Acad. Sci. USA, 57, 116*
(1967).
c Рассчитано по известному размеру частиц нз уравнений (7.13) и (7.55).
при условии, что коэффициент диффузии растворенного вещества известен. Таким
образом, объединение констант седиментации, полученных путем измерения
скоростей седиментации, и коэффициентов диффузии, полученных методом
светорассеяния, дает в рукн исследователя новый удобный способ определения:
молекулярных масс макромолекул в растворе. Он особенно удобен для наиболее
Таблица 21.4
Коэффициенты трансляционной диффузии н молекулярные массы вирусов*
Вирус
D20, w> IO-'cm'c-i
5 л . I0-I3C
20, W*
вируса'
смзг—l
Молекулярная масса,
10-6
RI7
Q
BSV
РМ2
Т7
1,53± 0,026
1,42±0,01
1,25±0,01
0,65±0,01
0,64±0,01
ч 79±1
88±2
133±2
294±3
487±5
0,689±0,012
0,668± 0,009
0,706± 0,002
0,771±0,007
0,639±0,006
4,02±0,17в
4,55±.0,16'
8,81±0,17
47,9±1,7
50,9±1,1
а Из Camerini-Otero R. D., Pusey P. N., Koppel D. E., Schaefer D. W., Franklin /?. M.,
Biochemistry, 13, 960 (1974).
Получено из автокорреляционной функции для рэлеевского рассеяния света [D
вычислено из тангенса угла наклона графика зависимости логарифма автокорреляционной функции
для наблюдаемой интенсивности рассеяния от времени (см. уравнения (21.63) и
последующие).!
ь Рассчитано по уравнению (7.31). В этом расчете использовали коэффициент
седиментации, полученный при измерении скорости седиментации.
Г37 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
фупных макромолекул (с молекулярной массой больше 1000 000), когда
обычные методы определения молекулярной массы неприменимы совсем или неточны.
Определенные недавно молекулярные массы нескольких различных вирусов в
[растворе даны в табл. 21.4. Из этих данных (гл. 7.Б и 7.В), принимая, что
|молекулы имеют сферическую форму, можно рассчитать степень гидратации,
^которая в большинстве случаев составляет около 1 ' см3 Н20 иа грамм сухого
^вируса.
i
Пример. Определение констант скоростей макромолекуляриых реакций из
данных по рэлеевскому рассеянию света
Так как ширина линии рэлеевского рассеяния света зависит от
коэффициента трансляционной диффузии макромолекулы, можно надеяться, что измерения
ширины этой линии окажутся полезными прн изучении любых реакций,
связанных со значительным изменением коэффициента диффузии. Логично, что такие
реакции должны быть среди процессов, включающих образование комплекса
двумя большими молекулами с молекулярной массой, существенно большей, чем
у каждой из реагирующих макромолекул. Замечательный пример реакций
подобного типа дает нам недавно выполненный эксперимент с фрагментами
бактериофага.
Для того чтобы частица бактериофага T4D могла инфицировать бактерию
(прикрепиться к поверхности бактерии и впрыснуть в нее свою нуклеиновую
кислоту для воспроизводства новых частиц бактериофага), она должна быть
полиостью собрана из трех основных компонентов, показанных ниже. Поскольку
частично реконструированные, ие содержащие фибрилл отростка частицы не
являются инфекционными, необходимо рассмотреть как можно более подробно
•процесс сборки для выяснения причины иифекционности бактериофага.
Традиционные кинетиче-ские исследования процесса сборки основаны на биоиспытаннях
продукта на инфекцнонность и, таким образом, отражают скорость суммарного
процесса. Однако, поскольку фибриллы отростка имеют гораздо меньшую
молекулярную массу, чем головка или отросток, метод светорассеяния
малочувствителен к присутствию фибрилл отростка. Следовательно, данные по
светорассеянию будут отражать главным образом степень протекания показанной ниже
на схеме реакции I. Сопоставление констант скоростей реакций для стадии I
'определены методом светорассеяния) и для стадий I и II (получены на основе
биоиспытаний конечного продукта) должно было показать, какая из этих
стадий является критической (скорость определяющей) для суммарного процесса.
В первую очередь необходимо определить коэффициенты диффузии фрагментов,
которые вносят наибольший вклад в наблюдаемое светорассеяние, а именно
головок и частиц, не содержащих фибрилл. Этн коэффициенты были получены
I Головка
Отросток-
*ч^
т
Частица без
фибрилл
ошростка
^
Неинфекционная
частица
738 ЧАСТЬ 6
Tj Частица без
11 <pudptt/i/i
отростка
6 фибрилл
отростка
Фаговая'
частица
Полностью
реконструированная
инфекционная фаговая частица
аналогично данным в табл. 21.4 н оказались равными 3,60- 10_s и
3,14-Ю-8 см2 с-1, что соответствовало молекулярным массам 1,76- 10е и
1,95 - 10е. Зная, что автокорреляционная функция интенсивности
светорассеяния сферическими частицами одного типа может быть представлена в виде
(21.68)
можно показать, что автокорреляционная функция Для раствора, содержащего
смесь двух типов сферических макромолекул, будет иметь внд
1(0)
ИЛ*
~K*Dtx
+ А2е
-К*0»г,2
(21.69)
где /(0)есть величина /(т) в нулевой момент времени, a D[ и
D2—коэффициенты диффузии сферических макромолекул двух типов в растворе. Константы
в уравнении (21.69)—это средневесовые концентрационные доли компонентов
№ 1 и № 2 в данный момент времени:
Л =
с,М,
CiMs
/в моменте
с1М1 + сгМ21 2 с1М1-\-с2М2 [времени tj'
Отметим, что данные по автокорреляции можно получить приблизительно за
Ю-3 с (что соответствует ширине спектральной линии рэлеевского
светорассеяния в несколько сотен герц), а для исходных концентраций головок и отростков
как реагентов интересующая нас реакция имеет «время релаксации» порядка
1000 с. Таким образом, имеется возможность измерять автокорреляционную
функцию интенсивности на отдельных стадиях реакция. Преобразуем
уравнение (21.69):
[/ (t)//(0)jx/2_ exp [—K2D2z] = Л,(ехр [—K2D,z] — ехр [—**ОД). (21.70)
Строя график зависимости левой части уравнения (21.70) от величины в
круглых скобках с правой стороны этого уравнения, можно регистрировать
средневесовые концентрации реагирующих частиц в течение всего времени протекания
ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
а
IOOO 2000 3000
Время реакции, с
400ё
V
*= 1,28 •1бтЬЛ'л.сл
при Г*30'С
: I I I 1—1
о /о го зо 4о so во
Время реакции, мин
Рис. 21.11. Определение константы скорости реакции второго порядка для
сборен частиц бактериофага T4D из головки и отростка путем построения графика
швиснмостн (1/[/"|) от времени, где Г — это концентрация головок в момент
времени i.
| [Из Aksiyote-Benbasat J., Bloomfield V. A., J. Mol. Biol., 95, 335 (1975.)]
fa — концентрации получены по данным измерения автокорреляционной функции
рассеянного свет* (только реакция I — см. текст.) О —для получения концентрации полиостью
^собранного фага использованы данные биоиспытаиий иа иифекциоииость (реакция I и II).
реакции. Далее обычным путем (гл. 16.А.1), на основе зависимости концентрации,
«кажем, головок \\Г] от времени реакции t, можно определить, как это показано
■на рис. 21.Ш,а, константу скорости реакции (второго порядка) k из уравнения
0/[Л*)-(1/[Л.) = «.-
(21.71)
Сравнительные данные для скорости суммарной реакции (полученные путем
определения концентрацнн конечного продукта методом биоиспытаний) показаны
на рис. 21.11,6. Учитывая поправку на небольшое отличие в температурах
между обоими экспериментами, можно констатировать, что в пределах
экспериментальной ошибки обе скорости одинаковы. Это говорит о том, что реакция I
(реакция головок и отростков, ведущая к образованию частиц, не содержащих
фибрилл) является скорость лимитирующей стадией во всем нашем
сверхупрощенном механизме. Более того, поскольку рассчитанная константа скорости
&=1.107 М-1 с-1 почти в 500 раз меньше той, которую можно было бы
ожидать для лимитируемой диффузией реакции, становится ясно, что для
осуществления реакции частицы не только должны подойти друг к другу, но н принять
правильную относительно друг друга ориентацию, с тем чтобы нужная часть
головкн прореагировала с правильным концом отростка. Отметим, что головка
имеет форму, близкую к удлиненному икосаэдру, а отростки представляют собой
740 ЧАСТЬ 6
вытянутые стерженьки. Если предположить, что реакционноспособна только
одна грань икосаэдра (всего 12 граней) и активен только один из концов
отростка, то вероятность того, что обе частицы будут должным образом
сориентированы при столкновении, будет только 1/24. При требовании, например,
коллинеарности обеих молекул для успешного столкновения этот фактор еще более
уменьшается. Приведенный пример дает первое убедительное определение
константы скорости реакции макромолекул, выполненное методом измерения
ширины линии спектра рэлеевского светорассеяния. Логично предположить, что уже
в ближайшие годы такой подход будут широко использовать при изучении
ассоциации белков, нуклеиновых кислот и других полимеров в растворе.
Пример. Измерение коэффициентов вращательной диффузии макромолекул в
растворе по деполяризации светорассеяния
В предыдущих примерах проводилось измерение общей интенсивности
рассеянного света как функции угла., рассеяния. Однако очень легко использовать
в подобных экспериментах источник плоскополяризоваиного падающего света и,
ориентируя должным образом поляризатор, регистрировать вертикально или
горизонтально поляризованный рассеянный свет по точно такой же схеме, как
в экспериментах по измерению поляризации флуоресценции (регистрируя или
вертикально, или горизонтально плоскополяризованный свет, рассеянный под углом
90° к направлению падающего потока) (рис. 16.14). При этих условиях можно
ожидать, что ширина линии спектра интенсивностей рассеянного света будет
зависеть как от вращательной (как в случае флуоресценции), так и от
трансляционной диффузии. Единственное существенное отличие этого эксперимента от
ранее описанных состоит в том, что рассеянный свет вновь испускается из тех
же самых возбужденных состояний, которые были заселены при поглощении
падающего света, в то время как при флуоресценции испускание света
происходит из состояний с более низкой энергией, чем у состояний, первоначально
заселенных при поглощении падающего света. Как и в случае флуоресценции,
вращательный ориеитационный фактор для рассеянного света выражается
сферической гармонической функцией второго ранга и поскольку трансляционное
перемещение и вращение можно считать независимыми (они не коррелируют),
их корреляционные функции просто перемножаются. Таким способом получают
спектральную плотность для интенсивности (плоскополярнзованного) света,
рассеянного раствором с концентрацией макромолекул [с]:
(интенсивность горизонтально поляризованного \ , .
рассеянного света при возбуждении вертикаль- ) [интенсивность деполяри-\_
но поляризованным светом / V зованного света у —
(/C2DTp+6Dep)
=, /деп ос [с] (а „ - а±)3 (ц_ц§)1 + ^.^ + fo^.' (21.72)
где а || и <Jj_ представляют собой \оляризуемости молекулы в двух взаимно
перпендикулярных направлениях. Величина (ац—a L) является мерой оптической
«анизотропии» молекулы (мерой того, насколько рассеяние в различных
направлениях неодинаково для разных ориентации молекулы). Если молекула
оптически «изотропна» [(а||—а1)=0], то рассеяние не зависит от ориентации
молекулы и, следовательно, вращение молекулы на него не влияет. Возвращаясь
к уравнению (21.72), можно заметить, что величина члена /С2/)тР уменьшается
(при очень малых углах рассеяния 0) по закону sin2(0/2) и, следовательно,
пренебрежимо мала при измерениях под достаточно маленькими углами рассеяния:
6/>вр
Пто/деп ее [с] (.,, - *±У ((0_(0о)2+(6/)вр)2 (21.73)
Уравнение (21.73) дает, таким образом, простой способ для определения
коэффициента воашательной диффузии D»P из измерения интенсивности деполяризо-
541 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
-100 О ЮО
] Частота, Mfy
Рис. 21.12. Спектр интенсивности рэлеевского рассеяния деполяризованного
света от образца связывающего кальций белка мышцы.
' Плавная кривая — это наилучшая аппроксимация экспериментальных точек одной лорент-
цевой линией. Коэффициент вращательной диффузии для молекулы белка был вычислен из
ширины этой кривой на половине ее максимальной высоты- (см. текст).
(Из Bauer D. R., Opella S. J., Nelson D. J., Pecora R., J. Amer. Chem. Soc, 97, 2580
(1975).]
ванного рассеянного света. Спектр /деп состоит нз единственной лорентцевой ли-
нни, ширина которой непосредственно связана с коэффициентом вращательной,
диффузии /)Вр. Теперь возникает вопрос: как переход к пределу в уравнении
(21.73) зависит от размера макромолекулы? Мы уже знаем, что для
макромолекулы сферической формы выполняются соотношения-
И
а значит
DT0=AT/6iciirl
D„ = kTI8vw;
^вр 1 /для сферической макромолекулы
DTp = г2
радиуса г
У
(21.74)
(21.49)
(21.75).
Длина волны видимого света Я. имеет порядок Ю-5 см, и, значит, параметр.
Л?=((2я/Л)2 [где Л задана уравнением (21.59)] меняется в этом случае между
нулем и 1010 для рассеяния под углами от нуля до 180°. Таким образом, для;
малых молекул с г=10~а см 1/л2=1016 и в уравнении (21.72) можно
пренебрегать членом K2DTp во всех случаях (в сравнении с членом 6 DBP). Однако в
случае более крупных агрегатов молекул, таких, как вирус табачной мозаики
с эффективным радиусом около 3-10-5 см П/г2= (1/9) •10101, член K2DTP будег
пренебрежимо мал по сравнению с 6Dep только для очень малых углов
рассеяния [скажем, 8<(1/10) радиана, что дает Двр//С2£)тр=1001. Таким образом,
для всех макромолекул 1(за исключением самых крупных .макромолекул или
агрегатов макромолекул с молекулярной массой, 106 и более) уравнение (21.73))
удовлетворительно описывает форму лнннн спектра интенсивностей
деполяризованного рассеянного света, и оно может быть использовано для определения
коэффициента вращательной диффузии макромолекул в растворе.
На рис. 21.12 показан экспериментальный пример спектра
деполяризованного рэлеевского рассеянного света для связывающего кальций белка мышцы
(почти сферического маленького белка с молекулярной массой 12 000).
Экспериментальные точки очень хорошо ложатся на лорентцев контур, линии. Вычисленное
742 ЧАСТЬ 6
яз ее ширины вращательное .время корреляции оказалось равным тВр='12±1 не.
Из этого результата по уравнению (21.58) было подсчитано, что раднус
молекулы составляет ~ 22 А. Вычисленный раднус почти на 5 А больше среднего
кристаллографического радиуса «сухого» белка. Это позволяет предположить, что
в растворе с белком прочно связано значительное количество воды. В
противоположность некоторым, другим методам определения коэффициента
вращательной диффузии, которые мы обсуждали (деполяризации флуоресценции и введению
парамагнитной спиновой метки), в экспериментах по светорассеянию нет
необходимости, прикреплять к макромолекуле хромофорную или какую-либо другую
метку. Это преимущество позволяет надеяться, что описанный здесь подход
станет основным методом определения коэффициентов вращательной диффузии
макромолекул в растворе.
21.В.2. СВЕТОРАССЕЯНИЕ ПРИ ЭЛЕКТРОФОРЕЗЕ.
ЭЛЕКТРОФОРЕЗ БЕЗ ГРАНИЦ
Заряженная макромолекула в электрическом поле Е будет
двигаться с постоянной средней скоростью vE, задаваемой
уравнением (7.3) (это явление обсуждалось в гл. 7.А):
х>Б = \*.Е, (21.76)
где (х — электрофоретическая подвижность. Если мы теперь хотим
наблюдать свет, рассеянный этими молекулами, в любом
направлении, кроме перпендикулярного к их электрофоретическому
движению, то можно ожидать, что частота рассеянного света будет
сдвинута за счет эффекта Доплера на величину,
пропорциональную скорости движения молекулы vE. Если учесть эту особенность
при описании формы линии рэлеевского спектра, то общая
интенсивность света, рассеянного под определенным углом, будет иметь
вид
/С2/>Тр
/-(const)[с]l{u-»j±KVsr + ivw , (21.77)
где [с] —концентрация растворенного вещества -(прочие
обозначения имеют свой обычный смысл). Поскольку ширина рэлеевской
линии меняется как К2, ясн\ что самые узкие линии (и, таким
образом, наилучшее разрешение рэлеевских линий для молекул с
различной электрофоретической подвижностью) будут
наблюдаться под малыми углами рассеяния.
Пример. Быстрый электрофорез плазмы кровя человека
На рис. 21.13 показан рэлеевский спектр образца плазмы крови человека
в сильном (350 В/см) электрическом поле. Наиболее быстро движущийся komj
.понент (альбумин) дает наибольший доплеровский сдвиг и образует первый
(самый правый) пик в спектре. Остающиеся пики (еще ие охарактеризованные)
«соответствуют различным глобулинам (ср. с данными рис. 7.4 и в табл. 7.1 для
юбычкого электрофореза). Интересная особенность (и, вероятно, достоинство)
j'743 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
&
Альбумин
180
300 420
Частота , Гц —*-
540
Рис. 21.13. Спектр светорассеяния при электрофорезе плазмы крови человека
при рН 8,4 и ионной силе 0,006 (угол рассеяния 7°30', напряженность
электрического поля 350 В/см).
[Из Ware В. R., Adv. Colloid and Interface Science, 4, 1 (1974).]
,этого эксперимента по светорассеянию при электрофорезе состоит в том, что>
относительная интенсивность светорассеяния для любых объектов
пропорциональна их весовым концентрациям, умноженным на их молекулярные массы. Таким'
образом, наиболее массивные объекты будут гораздо лучше вндны при
сравнимых весовых концентрациях, чем ннзкомолекулярные объекты. Это, например,
объясняет большую относительную величину пнков глобулинов на рис. 21.13 по>
сравнению с пнком имеющего гораздо большую концентрацию альбумина
(табл. 7.1). Еще одно достоинство этого метода заключается в том, что для!
■разделения не используют гелн, и поэтому можно достичь разрешения электро-
форетнческих пиков очень больших молекул (макромолекулы с молекулярной,
массой заметно большей, чем ~ 100 000, не могут свободно проходить сквозь,
полнакрнламндные гелн при электрофорезе). В недавних экспериментах было'
продемонстрировано движение целых бактерий при электрофорезе (рис. 21.114).
Эти исследования открыли новую область для экспериментов по определению4
заряда и формы поверхности бактерий на количественной основе. Наконец,
следует отметить, что проведенные эксперименты выполнены в растворах при:
очень низних концентрациях солей (данные, представленные на рис. 21.14,
получены при использовании в качестве растворителя дистиллированной воды)-
в противоположность традиционным электрофоретнческим экспериментам, в
которых необходимо использовать высокие солевые концентрации, чтобы
обеспечить хорошую проводимость раствора. Таким образом, метод светорассеяния прн
электрофорезе хорошо моделирует условия окружения макромолекул in vivo.
Электрофоретические эксперименты со светорассеянием можно выполнять за
несколько минут (ср. с несколькими часами, необходимыми для проведения
традиционного электрофореза), что обусловливает возможность их клинического'
применения.
744 ЧАСТЬ 6
I8
И
1
2 -
Lyf-вОГц
/00
200
300
400
Допплеровская чаотота, Гц
Рис. 21.14. Спектры светорассеяния прн электрофорезе бактерий Staphylococcus
■epidermidis (J) н сфер полистирола (2), диаметром 0,367 мкм (спектры этих двух
образцов наложены друг иа друга).
Поскольку бактериальный спектр шире, то это означает, что у бактерий должна
существовать заметная гетерогенность по размеру, форме и(нли) заряду поверхности.
,[Из Uzgiris Е. Е., Opt. Comirmn., в, 55 (1972).]
Пример. Как передвигаются бактернн?
Представленные иа рис. 21.14 данные показывают, что когда все
(заряженные) частицы в образце движутся с одной н той же скоростью, их (постоянная)
скорость проявляет себя в рэлеевском спектре как доплеровский сдвиг.
Следовательно, если исследовать препарат бактерий, которые перемещаются со
своими собственными (при отсутствии электрического поля) постоянными скоростями,
•равными по величине, но беспорядочными по направлению, то можно ожидать,
что рэлеевскнй спектр такой системы будет отражать суперпозицию (сумму)
доплеровских (постоянных) сдвигов, возникающих благодаря движению бакте-
,рий в различных направлениях. (В спектроскопии подобную ситуацию называют
«неоднородным ушнреннем линий».) С другой стороны, при условии, что
^бактерии движутся с постоянной скоростью, но очень часто меняют направление
движения, можно было бы ожидать, что мы получим рэлеевскнй спектр
рассеяния света для такой системы в виде единственной лорентцевой линии,
ширина которой определяется соответствующим эффективным коэффициентом
трансляционной диффузии. В итоге можно ожидать, что спектр рассеяния света для
бактерий в растворе будет иметь ширину линии, которая увеличивается с
возрастанием максимальной скорости передвижения («неоднородное уширение»)
и (или) с увеличением расстояния между точками изменения направления
движения и частоты изменений этих направлений [уравнение (6.60)]. В рэлеевских
спектрах для реальных бактерий находят отражение оба только что описанных
предельных случая.
На рис. 21.15, а показаны автокорреляционные функции интенсивиостей для
трех различных бактериальных штаммов Е. colL Двигательное поведение всех
этих трех бактериальных штаммов, очевидно, различно, на что указывает
большое несовпадение автокорреляционных функций. Пример более детальной
информации, получаемой в такого рода исследованиях, представлен на рис. 21.15,6.
На этом рисунке показаны экспериментальные (сплошные кривые) н
теоретические автокорреляционные функции для моделей чисто поступательного (черные
чсвадратики) и спиралеобразного (крестики) движений. Ясно видно, что движение
яо спирали гораздо лучше описывает экспериментальные данные. Конечно, этот
745 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
0,00 0,04
0,08 0,12 0,16
Ht,C-A)KM''
0,20
600
Рнс. 21.15. Нормированные (до единичной величины в нулевой момент времени)
автокорреляционные функции фотосчета (интенсивности) для различных
бактериальных штаммов Е. coli как функция времени.
Величина К— (4 Яп/к) sin (8/2) [уравнение 21.67)] введена просто для приведения
различных кривых к одному и тому же масштабу времени независимо от того, под каким
конкретным углом проводили каждое измерение,
[а —из Schaefer D. W., Berne В. 1., Biophys. J.. 15. 785 (1975); б —из Schaefer D. W„
Banks G., Alpert S. S.. Nature. 248. 162 (1974).].
a — штаммы E. coli unc602, AW405 и cheC497. Более быстрое убывание автокорреляционной
функции соответствует более быстрому поступательному движению (см. текст); б—
экспериментальные (плавные кривые, для 6->20°, 35°; 50", 70" и 90°) и теоретические
автокорреляционные функции для штамма Е. coli cheC497, полученные иа основе моделей чисто
поступательного (верхняя линия hs черных квадратиков) и спиралеобразного (линия из
крестиков) двнжеиий. В модели спиралеобразного движения поступательная скорость
принята равной 50 мкм-с-1, а вращательная — 60 рад-с-1.
результат вовсе не означает, что бактерии передвигаются по спиральным
траекториям, ио он показывает, что они должны часто менять направление своего
движения.
Интерес к передвижению бактерий связан с двигательным ответом бактерий
на различные внешние воздействия: термотаксисом (ответом иа тепло),
хемотаксисом (ответом на градиент концентрации химических агентов) И
фототаксисом (ответом на свет), поскольку предполагается, что бактерии должны
обладать первичной неразвитой формой памяти для того, чтобы, сравнивая с пей
сигналы из окружающей среды, в какой-то момент времени менять направление
своего движения.
Задачи
I. Если амплитуда беспорядочно изменяющейся величины может быть записана
в виде
оо
f(t)= 2 К cos КО + 6„з1п («„*)].
где <ол= ряп/Т),
покажите, что усредненное по времени значение [/(f)]2 равно 2 « •
п=.\ *
[Другими словами, покажите, что уравнение (21.4) следует из уравнения
(21.1)J
746 ЧАСТЬ 6
2. Для той же, что и в задаче 1, /■(/) покажите, что корреляционная функция
для нее имеет вид
С(*) = </(0/(<-Ь*)>
у 2 )cos{<ant),
где угловые скобки указывают на усреднение по времени от нулевого
момента до Т. [Другими словами, покажите, что корреляционная функция — это
преобразование Фурье энергетического спектра (т. е. получите уравнение
(21.9) из уравнения (21.11).]
со
8. Рассчитайте энергетический спектр Р {a) =.{2fn) J G{x)dx и площадь под
о
графиком зависимости Я(ш) от частоты (между нулем и бесконечной
частотой) для корреляционной функции вида
(?(*) = С? (0)ехр(-т/ткорр),
в которой G(0)—это среднеквадратичная амплитуда беспорядочно
изменяющейся величины. В этой задаче необходимо показать, что площадь под
энергетическим спектром не зависит от тКОрр. Это важно для понимания рис. 21.3
•и 21.4.
4. В главе 21.А мы вывели корреляционную функцию и спектральную плотность
для процесса химического обмена между двумя состояниями, при котором обе
константы скоростей были равны [уравнение (21.17)]. Теперь вычислите
корреляционную функцию и энергетический спектр :[виовь пройдите все стадии
от уравнения (21.18) до уравнений (21.24) и (21.25)] для двухпозиционного
обмена, при котором равновесные заселенности обоих состояний не
одинаковы:
кг
Замечание. О заселенности двух состояний в зависимости от времени cmi
уравнения (10.10) и i(HO.ll). Алгебраические преобразования можно
упростить, вводя новые переменные: >
НА = -^^ h и Нв = -4£-i (-A).
Б. Рассчитайте корреляционную функцию и спектральную плотность для трех-
позициониого обмена между тремя кинетически эквивалентными состояниям»
Л, В и С, в которых величина магнитного поля равна +А, 0 и —А:
к
^47 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
| Замечание. При [А] =|[Л]о, а [В]—[С]=0 (начальные условия) концентра-
I ции во всех трех состояниях будут:
г w=mj. (4—г *-•*').
f [ci=№ ^4-" 4" *"■*')•
| ([Эти расчеты очень похожи иа те, которые приводят к корреляционной функ-
f ции и энергетическому спектру для вращения трех протонов метильной труп-*
\. пы около оси, связывающей эту метильную группу с остальной молекулой,
| Результат этого расчета важен для интерпретации формы и гибкости изучае-
I. мой молекулы на основе измерения «времен релаксации» в спектрах ЯМР.]
& В тексте было показано, что вращательная диффузия приводит для сфериче-
\ ских гармонических угловых функций к корреляционной функции вида
[ <f(e,?)*f(ei,?.)> = exp[-V'.J.
Ь в которой Твр называют «вращательным временем корреляции». Для выясие-
■(■ ния физического смысла тВр рассчитайте средний угол, на который молекула
повернется за время твр, когда физический параметр, который мы регистриру-
г ем, меняется по закону сферической гармонической функции первого или
- второго «ранга».
Замечание. Считайте '9о=0 н рассмотрите только случай, когда т=0.
р. Рассчитайте вращательное время корреляции (уравнение 21.58) для
макромолекул с молекулярной массой 25000; 40 000 и 66000, приняв, что вязкость
воды равна 0,01 пуаз (в единицах СГС), температура 25 °С, а парциальный
[ удельный объем каждого белка составляет приблизительно 0,73 см3/г.
Полученные результаты должны быть близки к значениям, указанным в
таблице 21.2.
Литература
Преобразование Фурье
■1. Champeney D. С, Fourier Transforms and Their Physical Applications,
Academic Press, London, 1973.
12. Bracewell R., The Fourier Transform and Its Applications, McGraw-Hill, New
York, 1965.
Спектральные плотности и корреляционные функции
3. Slichter С. P., Principles of Magnetic Resonance, Harper & Row, N. Y. (1963),
Chap. 5. (Кваитовомеханический подход ие используется; основное
содержание— классическое.) В приложении С (две страницы) дан вывод
корреляционной функции для химического обмена между двумя состояниями.
Корреляционные функции для беспорядочных вращательного
и поступательного движений
4. Abragam A., The Principles of Nuclear Magnetism, Clarendon Press, Oxford,
1961, pp. 298—302.
748 часть б
Деполяризация флуоресценции и вращательное время корреляции
Б. Yguerabide С, In: Methods in Enzymology, 26C, 528 (1972).
Лазерное светорассеяние и диффузия макромолекул
6. Ford N. С, Jr., «Biochemical Applications of Laser Rayleigh Scattering», Chemica
Scripta, 2, 193—206 (1972). Трансляционная диффузия.
7. Pecora R., «Light Scattering Spectra and Dynamic Properties of Macromolecu-
lar Solutions», Disc. Faraday Soc, 49, 222—228 (1970). Вращательная днффу-
зия.
Рассеяние света при электрофорезе
«. Ware В. /?., Adv. Colloid and Interface Science, 4, 1—44 (1974).
ЛАВА 22
^КОНСТРУКЦИЯ ОБЪЕКТОВ ПО
130БРАЖЕНИЯМ
'' В предыдущих главах мы уже описывали некоторые изображе-
|:ия, получаемые на основе взаимодействия волн (ультразвуковых
ли электромагнитных) и вещества. В этом разделе будет рассмот-
ен способ получения информации о структуре объекта с разреше-
ием около 1 А при использовании электромагнитного излучения
очень короткой длиной волны («рентгеновские» лучи, длина
вольт около 1А), рассеянного упорядоченными ансамблями (кристал-
ами) макромолекул. Процесс рассеяния падающих лучей упоря*
оченной системой рассеивающих центров называют «дифракци-
й». Качественный анализ экспериментов по дифракции
рентгеновских лучей будет относительно прост, если мы объединим вместе
рее то, что уже знаем о рассеянии (гл. 15) и фурье-преобразовани-
j*x (гл. 20 и 21).
i
|22.А. РЕНТГЕНОВСКАЯ КРИСТАЛЛОГРАФИЯ.
ЬПРЕДЕЛЕНИЕ УГЛЕРОДНОГО ОСТОВА
МАКРОМОЛЕКУЛЫ
На рис. 22.1 показан простейший чертеж, который объясняет
возникновение дифракционных картин рентгеновских лучей.
Условия дифракции были определены в 1912 г. Брэггом-сыном, когда
он был еще аспирантом.
Если на атомы падает поток рентгеновских лучей, то излучение
(с той же самой длиной волны) рассеивается во всех
направлениях в соответствии с закономерностями, .выясненными нами в
гл. 15. Однако в кристаллах, когда атомы равноудалены друг от
друга, существуют особые углы падения, для которых рассеяние
рентгеновских лучей осуществляется «в фазе» целыми плоскостями
атомов так, что общая интенсивность рассеяния становится
максимальной. На рис. 22.1 показано, что при определенных условиях
волны рентгеновских лучей, рассеянных двумя атомами,
расположенными в соседних плоскостях, будут «в фазе» [так что
амплитуды их складываются и дают «пятно» интенсивности на картине
рассеянного излучения (дифракции)]. Необходимо лишь,, чтобы
750 Часть б
Рлс. 22.1. Модель Брэгга для отражения рентгеновских лучей соседними
плоскостями атомов в кристалле.
Волны, рассеянные двумя атомами (см. рисунок), складываются. Амплитуда получившейся
в результате волны максимальна в том случае, если обе рассеянные волны имеют
одинаковые фазы. Это получается всегда, когда разница в длинах путей обоих волн [ОА + ОВ =
= 2d-sin9] равна длине волны рентгеновских лучей, умноженной на целое число пК
[уравнение (22.1)].
различия в длинах пути, пройденного двумя волнами, были
кратны длине волны рентгеновского излучения X:
2dsmQ = nX, (22.1)
где d—расстояние между двумя атомными слоями, а п — целое
число. Исходя из этого условия оказалось возможным определять
расстояния между атомами в кристалле, меняя углы падения
рентгеновских лучей на образец и определяя углы, при которых
интенсивность рассеяния максимальна (образуются дифракционные
«пятна»). Более того, поскольку уже было показано (гл. 15.Б),
что интенсивность рассеяния данным атомом пропорциональна его
атомному номеру, оказывается, что потоки рассеянного света от
слоев атомов в кристалле, различающихся химически (т. е.
имеющих разные атомные номера), отличаются по относительной
интенсивности. Таким образом, принцип решения задачи о получении
структуры молекулы в кристалле из межатомных расстояний,
которые определяют по картине дифракции рентгеновских лучей,
вполне ясен. Однако на практике решение этой -задачи стало
относительно легко выполнимым только после того, как Брэгг-отец
предложил приблизительно в 1915 г. способ объединения данных
по дифракции рентгеновских лучей с теорией Фурье.
22.А.1. ДИФРАКЦИОННАЯ КАРТИНА.
ЛИНЗА КАК ПРИБОР ДЛЯ ПРЕОБРАЗОВАНИЯ ФУРЬЕ
Уравнение (22.1), выведенное Брэггом-сыном, показывает, что
сумма амплитуд рентгеновских лучей, рассеянных различными
атомными слоями в кристалле, образует «дифракционную картину»
(картину интенсивностей). Пики интенсивности на этой
дифракционной картине расположены так, что отражают расстояния
между пиками электронной плотности (а именно положениями атомов)
в исследуемом объекте. Чем ближе друг к другу расположены ато-
ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Фокальная Плоскость
плоскость изображения
Объект
Дифракцион-
Действительное изображе- мое
изображение ние
Рис. 22.2. Изображения, формирующиеся за оптической двояковыпуклой линзой.
(Действительное изображение (самое дальнее справа) образуется при пересечении
различных лучей, идущих от одной и той же части изучаемого объекта (каждая точка объекта
>дает одиу точку изображения). «Дифракционное» изображение расположено в фокальной
'плоскости. Оио образуется при пересечении лучей, идущих в одном и том же направлении
-от различных частей изучаемого объекта. Эти два типа изображений связаны
преобразованием Фурье (см. текст). D — фокусное расстояние линзы. [Из Buerger M. 1., Contemporary
Crystallography, McGraw-Hill, New York, 1970, p. 230.1
мы в исследуемом кристалле, тем дальше друг от друга
расположены пики интенсивности на дифракционной картине (чтобы
удовлетворялось уравнение (22.1), наименьшему d должен
соответствовать наибольший угол 8). Аналогичное соотношение хорошо
известно читателю из предыдущего обсуждения зависящих от
времени и от частоты сигналов спектра (гл. 20.Б). Мы тогда
отмечали, что сжатие одной области (более короткий по времени сигнал)
'соответствует расширению другой области (более широкий по
частоте сигнал). Фактически математическая связь между исходным
изучаемым объектом и его дифракционной картиной представляет
собой то же самое преобразование Фурье, с которым мы
сталкивались ранее в примерах по определению формы линий спектра.
Читатель наверняка уже знает о получении оптических
изображений из школьного курса физики. Поэтому для того, чтобы
понять как получают рентгеновское изображение объекта из
рентгеновской дифракционной картины (рентгенограммы), надо
вспомнить этапы получения оптического изображения, На рис. 22.2
представлены два типа изображений, формирующихся за
двояковыпуклой линзой: обычное изображение, образованное в плоскости
«изображения» и отличное от него «дифракционное» изображение
в другой особой «фокальной» плоскости. С помощью
соответствующего геометрического анализа (см. ссылку на литературу к
рис. 22.2) можно показать, что эти два типа изображений
являются такой же парой для преобразования Фурье, как зависящие от
времени и частоты сигналы, которые мы анализировали ранее.
Чтобы понять соответствующий процесс воссоздания
изображения по рентгенограмме (в котором нет никаких линз, поскольку
752 ЧАСТЬ 6
Объект
Рис. 22.3. Разделение формирования оптического изображения на две стадии:
формирование «дифракционного» изображения из лучей от исходного объекта
первой линзой (L2) я последующее формирование действительного изображения
, из дифракционной картины с помощью второй линзы (L2).
Соответствующие этим стадиям этапы, получения «правильного» изображения в
эксперименте с дифракцией рентгеновских лучей показаны на рисунке внизу и подробно
рассмотрены в тексте. [Из Buerger M. Л, Contemporary Crystallography, McGraw-Hill, New York.
1970, p. 240.]
a — «дифракционное» изображение; б — действительное изображение (дифракция
дифракции).
/ — дифракция рентгеновских лучей на кристалле («физическое» преобразование Фурье);
2 — математическое преобразование Фурье.
показатель преломления для рентгеновских лучей равен единице
фактически в любых веществах), мы разделим двояковыпуклую
линзу на две половины и раздвинем их на расстояние 2D, равное
удвоенному фокусному расстоянию, как это показано на рис. 22.3.
При разделенных таким образом линзах «действительное»
изображение формируется значительно правее, чем раньше, а
дифракционная картина теперь образуется между двумя линзами.
Следовательно, первая линза образует дифракционную картину из лучей
от исходного объекта, в то время как вторая линза формирует
действительное изображение из дифракционной картины. Или,
другими словами, действительное изображение получают путем
дифракции дифракционной картины (на математическом «языке»
превращение трудных задач в простые
южно было бы сказать, что преобразование Фурье преобразова-
гия Фурье функции даст снова исходную функцию). В любом слу-
гае рис. 22.3 помогает понять суть проблем при рентгеноструктур-
j&oM анализе. Дело в том, что при дифракции рентгеновских лучей
[ристалл действует как первая линза на рис. 22.3. Однако,
поскольку для рентгеновских лучей не существует линз (гл. 15.Б),
которые могли бы выполнять функцию второй линзы в оптическом
'^налоге на рис. 22.3, следующим этапом при восстановлении
действительного изображения в эксперименте с рентгеновскими лучами
^является превращение дифракционной картины (данных по интен-
|ивности) в реальную картину распределения электронной
плотности молекул в кристалле с помощью математического
преобразования Фурье. Теперь уже известно, что для выполнения
преобразования Фурье какой-либо функции необходимо знание и амплитуды,
М фазы функции в каждой точке (см. следующие ниже
графические примеры). Однако детекторы рентгеновских лучей дают ин-
юрмацию только об интенсивности, и поэтому не существует
метода непосредственного экспериментального определения фаз рент-
ченовских волн в различных точках дифракционной картины. Мы
Дернемся к «проблеме фаз» позже. Будем считать, что фазы можно
жаким-то образом определить, и сосредоточимся на том, как
реконструировать исходное изображение по дифракционной картине.
I..
&2.А.2. ФУРЬЕ-СИНТЕЗ (ГРАФИЧЕСКИЙ)
;: В гл. 20.Б мы рассматривали фурье-анализ, согласно которому
[определенного вида волна (зависящая от времени)
анализировалась для получения ее периодических (зависящих от частоты)
амплитудных компонент и фаз («спектра» сигнала). В рентгенострук-
<гурном анализе нас интересует обратная задача — определить
истинное изображение по сумме волновых компонент, полученных из
дифракционной картины рентгеновских лучей (фурье-синтез).
\В настоящий момент нам необходимо ясно представлять себе, что
из-за того, что сам по себе кристалл состоит из повторяющихся
рядов атомов, узор интенсивности на дифракционной картине также
будет периодичен в пространстве. Начнем, следовательно, с
рассмотрения того, как можно синтезировать произвольную
периодическую функцию (рис. 22.4), зная амплитуды и фазы
«составляющих ее» синусоидальных волн, каждая из которых имеет период
колебания, отличающийся в целое число раз от периода колебания
интересующей нас произвольной периодической функции.
Рентгеновские лучи рассеиваются (подвергаются дифракции)
реальными кристаллами в соответствии с электронной плотностью
на отдельных атомах. На рис. 22.5 представлен фурье-синтез
электронной плотности в одномерном кристалле, состоящем из простых
(двухатомных) молекул, на основе амплитуд и фаз волновых
компонент, получаемых (см. ниже) из картины дифракции рентгенов-
754 часть б
и = О
в-1
и = 2
—>
я = 3
• Период-
II = 5
■ -^/iOw^/wwvw
Определенная периодическая
синтезируемая функция
f{x) U-/7ey*«y->4 1
Суммирование волновых
<Рурь е - компонент
Сумма
Рис. 22.4. Фурье-синтез данной периодической функции (вверху справа) из
суммы волн, периоды колебаний которых в п раз (где п=0, 1, 2...) меньше
периода исходной функции.
[Из Waser /., J. Chem. Educ, 45, 446 (1968).]
ских лучей. На этом рисунке можно проследить ряд типичных
особенностей определения структуры с помощью рентгеновских лучей
(рентгеноструктурного анализа). Первая: необходимо
относительно немного волновых компонент Фурье (вплоть до я = 2), чтобы
установить приблизительное расположение молекул в
кристаллической решетке. Вторая: добавление все большего' и большего
числа фурье-компонент (соответствующее вовлечению в рассмотрение
все большего и большего числа «пятен интенсивности» на
экспериментальном узоре дифракции рентгеновских лучей) приводит в
результате все к лучшему, и лучшему разрешению. Так, увеличение
числа волн до п — Ь дает достаточно точное определение
локализации отдельных, атомов в кристалле. (На математическом «языке»
это означает, что прогрессия Фурье быстро сходится к желаемому
результату—функции распределения электронной плотности.)
Третья: можно пренебречь нулевой (п = 0) фурье-компонентой,
поскольку нас интересуют только положения атомов (пики
электронной плотности), но не абсолютная, величина (постоянная)
базисной линии, определяемая нулевой компонентой. Наконец, можно
отметить, что в рассматриваемом особом случае фазы каждой из
55 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
волновых компонент равны или 0°, или 180° (нет сдвига фаз на
любой другой угол). Эта особенность характерна для центросим-
метричных структур, а именно таких, у которых локализация всех
атомов в данной элементарной ячейке не изменится при
отражении каждого атома относительно центра элементарной ячейки.
В нашем случае мы можем только заметить, что условие центро-
симметричности приводит к сильному упрощению определения
структуры, поскольку фазы могут принимать только два значения
(«проблему фаз» см. ниже). Более того, даже в кристаллах,
которые не являются центросимметричными, часто возможно найти
определенные брэгговские плоскости отражения, которые
являются центросимметричными. Это позволяет воспользоваться
указанным выше преимуществом, создаваемым центросимметричностью.
Фурье-синтез по двум (или по трем) направлениям проводят
совершенно аналогично (рис. 22.6), а результаты приводят, как
правило, в виде контурной карты (рис. 22.6 для двух направлений)
или в виде суммы многих послойных контурных карт (рис. 22.7
для трех направлений) электронной плотности.
волновые Фурье -
компоненты
■У
1 + 2
1+2 + 3
1+2+3+4
0,0 1,0 2.0 3,0 0,0 1,0 2,0 3,0
Рис. 22.5. Фурье-синтез функции распределения электронной плотности в
одномерном кристалле, состоящем из двухатомных молекул, путем сложения вместе
указанного числа Фурье-волн, имеющих соответствующие амплитуду и фазу.
Подробное обсуждение этого рисунка дано в тексте. [Ms Waser /., J. Ghent. Educ. 46
446 (1968).]
a — суммирование волновых фурье-компонеит (фурье-сянтез) для получении
приблизительного описания интересующего нас распределения электронной плотности; б— истинное
распределение электронной плотности в одномерном кристалле, состоящем из- двухатомных
молекул.
я = 1
'flepuog^
я = 2
» = 3
Ч/Ч/ЧУ 4/\/V Sf\/\t
и = 4
|(\ЛЛЛ|^ЛЛЛАЛЛ^
я = 5
WWVAAA/VWWM
I—ОО
ПериоВ
-оо-
-оо—
AAA
JXSXSX
N VW VW V>
AAA
756 ЧАСТЬ 6
' <Х
CL
^э
Qu
VI
^э
м
о.
о.
^э
Q.
CL
^0
■ и
0,0 1,0 2,0
а
Рис. 22.6. Фурье-синтез распределения электронной плотности в двумерном
кристалле, состоящем из трехатомных молекул. (Разъяснения см. ниже).
[Из Waser J., J. Chem. Educ, 45, 446 (1968).]
a — модель двумерного кристалла. Показаны четыре элементарные ячейки этого
«кристалла», каждая из которых содержит одну линейную трехатомную молекулу АВ2. На
чертежах 6, в. г к д дано представление распределения электронной плотности в этом
«кристалле» путем наложения волы.
g—четыре самые большие волны. Слева показаны четыре волны с самыми большиии
амплитудами, которые необходимы для описания распределения электронной плотности в
«кристалле» на чертеже а. а их сумма дана справа. Получающееся в результате сложения
расположение «горбов» уже показыаает приблизительное очертание молекул. Пары делых
чисел (1,0), (0,1), (1.1) и (2,2) слева характеризуют различные волны. Первая цифра
равна числу гребней или впаднн на протяжении повторяемого интервала ,вдоль оси х, вторая
цифра — то же самое вдоль оси и.
!57 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
«—следующие по размеру две волны. Добавление еще двух волн к уже имеющимся
показанным слева волнам улучшает определение контуров молекул. Однако детали структуры
Молекул все еще не видны. Причина состоит в том, что. за исключением самой
низкочастотной, все до сих пор использовавшиеся волны были ориентированы более или менее
вдоль наметившихся «горбов», которые отражают положение молекул.
г — еще три волны. Следующие по размеру три волны сориентированы поперечно
направлению расположения получившихся «горбов». Суммирование нх с предшестаоааашнми
волнами позволяет получить на месте каждого «горба» трн отдельных пика, которые
отражают наличие определенных атомоа а молекуле. Черточками над вторыми цифрами
(см. пары цифр в круглых скобках на этом чертеже слева) обозначен знак минус. Это
должно означать, что гребень волны, который пересекает ось х в области ее
положительных значений, одновременно должен пересекать ось у в области ее отрицательных
значений и наоборот, если гребень волны пересекает ось х а отрицательной области, то off
должен отсечь отрезок в положительной области иа оси у. [Например, волну (2,3) можи»
было бы обозначить и как (2.3). Аналогично, на чертеже б к в ориентация воли такова,
что данный гребень одновременно должен пересекать либо полуоси + х и + у, либо
полуоси — х в —у. Этн аолны можно было бы, следовательно, обозначнть_и парами
отрицательных, а не положительных чисел. Например, символы (2,1) и (2,1) обозначают одну
н ту же волну.]
758 ЧАСТЬ в
2,0
1,0
t
У
0,0 1,0 2,0
д — контурная карта для описания нашего кристалла. Воавышеииые области и пики на
чертеже г (справа) показаны здесь линиями (контурами), соединяющими точки с одинаковой
амплитудой суммарной волны (в нашем случае амплитуда — синоним электронной
плотности). Это обычный прием, которым пользуются для демонстрации полученных
результатов в работах по реитгеноструктурному анализу. Приращение электронной плотности
постоянно при переходе от одной контурной линии к другой.
Рис. 22.7. На чертеже справа видно, что гемовая группа (она показана так,
что ее плоскость перпендикулярна плоскости чертежа) соединена со спиральной
областью полипептидной цепи миоглобина через гистидиновый остаток. Это
удалось доказать с помощью трехмерного фурье-синтеза с разрешением в 2 А,
показанного слева.
[Из Kendrew J. С. Dickerson R. Е., Strandberg В. Е., Hart R. G., Davies D. R., Phillips D. C.e
ins Shore V. C, 185. 422 (I960).]
?59 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
В2.А.З. ОПРЕДЕЛЕНИЕ ПОЛОЖЕНИЯ АТОМОВ
р ЭЛЕМЕНТАРНОЙ ЯЧЕЙКЕ. СИНТЕЗ ПАТТЕРСОНА
\ В предыдущем разделе обсуждалось, как можно получить кар-
йу электронной плотности молекулы, исходя из суммы подходящих
рмплитуд и фаз компонент картины дифракции рентгеновских лу-
|*ей на кристалле. Принципиальная схема установки для
получения дифракционных картин показана на рис. 22.8, а. На рис.
Р2.8, б приведен соответствующий «узор», полученный для
определенным образом ориентированного кристалла лизоцима.
Размеры элементарной ячейки* определяют по расстояниям между
различными соседними пятнами интенсивности на дифракционной
картине. Вращая кристалл вокруг разных осей, определяют раз-
Рис. 22.8.
й — Принципиальная схема получения дифракционной картины.
На кристалл направляют коллимнрованный поток монохроматических рентгеновских лучей.
В результате получают множество рассеянных потоков под углами, зависящими от
ориентации кристалла [уравнение (22.1)]. Для получения всех возможных «рефлексов» кристалл
необходимо вращать вокруг разных осей. Дифракционные картины обычно регистрируют
фотографическими методами. В центре пленки вырезают отверстие, чтобы предотвратить
засвечивание ее самим падающим потоком рентгеновских лучей. [Из Випп С. W., Crystals,
Their Role In Nature and Science, Academic Press, New York, 1965.J
б — Картина дифракции рентгеновских лучей при их прохождения вдоль
(определенной) оси 4-го порядка тетрагонального кристалла белка лизоцима
(выделен из белка куриного яйца).
Ясно видно влияние осн симметрии 4-го порядка иа дифракционную картину. Расстояния
между следующими один за другим пятнами связаны с расстояниями между
последовательными плоскостями Брэгга (рис. 22.1), тогда как интенсивности различных пятеи
определяются положениями отдельных атомов в элементарной ячейке (см. текст). [Из Fra-
ser R. D. В., MacRae R. P., «X-Ray Methods», In Physical Principles and Techniques of Pro»
tein Chemistry, Vol. A, ed. Leach S. J., Academic Press. New-York, 1969, p. 68.]
* Элементарная ячейка — это наименьшая повторяющаяся трехмерная
структурная единица кристалла. Она может содержать одну или большее число
молекул.
760 ЧАСТЬ в
Г
ФА
<Г -
%вс
'<4
лв |;
%АС
Рис. 22.9. Изображения распределения электронной плотности в простой трех-
атомиой системе.
Слева: распределение электронной плотности, полученное путем преобразования Фурье-
функции s | F | е*Ф, которая описывает амплитуду и фазу рассеянной волны в каждой
точке на дифракционной картине, удовлетворяющей уравнению (22.1) [фурье-сннтез].
Справа: функция Паттерсона, полученная путем преобразования Фурье функции | F \\
где | F | — амплитуда рассеянной волны. Интенсивности различных пятен на
дифракционной картине рассеяния рентгеновских лучей кристаллом пропорциональны (каждая) функция
5 | F Р для выбранной точки дифракционной картины. Таким образом, функции | F |*
легко определить из экспериментально полученных внтенсивностей рассеяния
(дифракционной картины) [синтез Паттерсона]. Относительные амплитуды функций, описывающих
карту распределения электронной плотности, которая получена методом Фурье,
пропорциональны относительной рассеивающей способности соответствующего атома (т. е. 2 «
Zg или Zq, где 2 — это атомный номер, или, точнее, число электронов в атоме).
Относительные амплитуды функций, описывающих карту распределения электронной плотности,
полученную методом Паттерсона, пропорциональны произведениям Zj^Zg, Z^Zq нли
ZJ>~ • Расположение точек на карте Фурье прямо соответствует расположению атомов в
элементарной ячейке кристалла. Точки же на карте Паттерсона получаются путем
поочередного «помещения» каждого атома в центр (на рисунке справа это точка О) и
определения положений остальных атомов элементарной ячейки относительно данного
«центрального» атома. Если число рассматриваемых атомов невелико (как, например, в нашем
случае), то из карты Паттерсона легко вывести истинные положения атомов в элементарной
ячейке кристалла (на рисунке слева), которые могли бы дать полученное расположение-
пятен Паттерсона (на рисунке справа) [см. задачу к этой главе]. Этот принцип лежит в
основе метода изоморфного замещения для определения структуры белков (см. текст),
поскольку положение тяжелого атома (с большим 2) легко определить по большой
интенсивности (амплитуде) соответствующего ему пятна на карте Паттерсона.
меры элементарной ячейки по всем трем направлениям.
Положение каждого атома внутри элементарной ячейки (т. е. структуру
молекулы) получают путем анализа относительных интенсивностей
различных пятен. Если амплитуда и фаза рассеянных
рентгеновских лучей имеют в одном из пятен на дифракционной картине
величину ys|/r|ei(P (где s — «масштабйый» фактор, см. рис. 22.9),
то измерение интенсивности даст информацию только о функции
^s\F\e1^-is\F\e-^=s\F\2. Таким образом, информация о фазе
оказывается потерянной. Это, очевидно, лишает нас возможности
провести фурье-синтез. Однако, как было показано Паттерсоном,
часто есть возможность извлечь некоторую информацию о
положениях атомов в элементарной ячейке, производя преобразование
Фурье не функции l/7!^, а функции \F\2. Эту новую функцию
называют функцией Паттерсона. Результат подобных
преобразований проиллюстрирован графически на рис. 22.9. Функция
Паттерсона простым образом связана с отыскиваемой нами картой
электронной плотности. Поэтому в случае элементарных ячеек, со-
761 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
держащих всего несколько атомов, обычно можно догадаться об
относительных положениях атомов в элементарной ячейке,
разглядывая карту Паттерсона (см. задачу к этой главе). Далее,
используя преобразование Фурье для полученных методом Паттерсона
электронных плотностей, определяют дифракционные амплитуды
\F\, которые можно сравнивать с экспериментальными
дифракционными интенсивностями, поскольку интенсивность
пропорциональна квадрату амплитуды. Систематически варьируя положения
атомов в предполагаемой теоретической структуре, можно получить
минимальное различие между наблюдаемыми и вычисленными
функциями \F\2. Это позволяет получить теоретическую структуру,
которая наилучшим образом объясняет наблюдаемую картину
дифракции. Хотя этот метод «проб и ошибок» вполне пригоден для
малых молекул, совершенно очевидно, что он быстро теряет свое
значение с ростом величины молекулы, поскольку число точек на.
карте Паттерсона увеличивается как п2 [с п пиками в начале
координат и остающимися (я2—п) пиками в других точках], где п—
число атомов в элементарной ячейке. При исследовании очень
больших молекул необходим, следовательно, другой подход,
рассмотренный в следующем разделе.
22.А.4. ОПРЕДЕЛЕНИЕ СТРУКТУРЫ БОЛЬШИХ МОЛЕКУЛ.
РЕШЕНИЯ ПРОБЛЕМЫ ФАЗ
Очень сильная зависимость амплитуды функции Паттерсона от
атомного номера (рис. 22.9) наводит на мысль, что если большая
органическая молекула (содержащая главным образом
относительно легкие атомы с Z=\, 6, 7 и 8) каким-либо образом кова-
лентно помечена тяжелым атомом М (имеющим, скажем, z,m— 25
или более), то паттерсоновские пики, связанные с расстоянием
М—М в кристалле, должны быть гораздо сильнее, чем
паттерсоновские пнки, связанные с расстояниями М—С или С—С [в нашем
примерев (25)2/(25) • (6) =4,2 раз или (25)2/(6)2 = 17,4 раза].
Этот вывод приводит в результате к двум важнейшим методам
определения структур больших (мол. масса ^1000) и очень
больших (мол. масса >1000) органических молекул.
Метод тяжелого атома (мол. масса около 1000). Для
последующих рассуждений удобно представить дифракционную волну в
месте данного пятна дифракционного узора в виде комплексного
числа, модулем которого является дифракционная амплитуда, а
фазой— фаза дифракционной волны. В этом случае мы сможем
изображать наши рассуждения графически, используя диаграммы
Аргана (рис. 22.10). Тогда интересующий нас фазовый угол
становится просто углом между комплексным дифракционным
«вектором» и осью действительных чисел на диаграмме Аргана.
Если органическая молекула содержит тяжелый атом и если
число входящих в нее легких атомов еще достаточно малб, то
762 часть б
F = IЛ в'*'= „структурный фактор" (22- 2а)
•в
F = \F\ cos4> + i\F\sin<p (22-26)
Дейстштель- Мнимая
ная часть част*
Рис. 22.10. Представление комплексного числа в соответствии с его
действительной (ось х) и мнимой (ось у) частями. Интересующая нас комплексная
величина— дифракционная волна (с амплитудой \F\ и фазой <р) в месте данного
пятна на картине дифракции рентгеновских лучей.
Когда в элементарной ячейке кристалла находится несколько атомов, результирующая
функция F определяется по амплитуде и фазе векторной суммой дифракционных воли от
всех атомов в данной ячейке.
вклад тяжелого атома FM в общий «структурный фактор» (рассеян
ние) F (дифракционную волну в месте данного пятна
дифракционной картины) будет достаточно велик по сравнению с общим
рассеянием на всех легких атомах Fji В этом случае есть возможность
отобрать пики М—М среди пиков М—С или С—С на карте
функции Паттерсона, рассчитанной путем преобразования Фурье
функций \F\2, в свою очередь полученных из наблюдаемой картины
дифракционных интенсивностей. Если использовать соображения,
представленные на рис. 22.9, то после этого, как правило, можно
локализовать положение (положения) тяжелого атома в
элементарной ячейке. Затем, применяя (обратное) преобразование Фурье
^полученной функции электронной плотности для тяжелого атома,
можно вычислить амплитуду и фазу рентгеновских лучей,
рассеянных тяжелым атомом, для каждого пятна на дифракционной
картине. В дальнейших рассуждениях используют следующее
соображение: если |Fjfj»|/\f|* то фаза ф дифракционной волны F=
= Fm-\-Fji (ее вектор показан в диаграмме Аргана на рис, 22.11)
молекулы как целого будет почти такой же, как и фаза волны
самого тяжелого атома фдг. Следовательно, в первом приближении
можно считать фазу волны в месте каждого дифракционного пятна
равной величине фазы, рассчитанной исходя из (известного)
положения тяжелого атома. Тогда после преобразования Фурье
величин |F|eiq>, где функцию \F\ получают из наблюдаемой картины
пятен дифракционных интенсивностей, а фаза ф рассчитана из фаз
для тяжелых атомов, мы получим карту приблизительного
распределения электронной плотности всех атомов молекулы.
Поскольку теперь на карте электронной плотности должны стать
видимыми по крайней мере некоторые легкие атомы, мы можем
использовать их приблизительные положения для вычисления вклада
части легких атомов в функцию Fji* Далее, вновь обратившись к
рис. 22.11, мы можем рассчитать лучшее приближение по фазе для
Ось действительных
чисел
763 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Ъ
Рис. 22.11. Графическая иллюстрация принципов определения фазы в методе
тяжелого атома.
Пока соблюдается соотношение | Fj^ | » | Fjj |, фааа фд^ будет достаточно хорошо
описывать необходимую нам фазу q> структурного фактора F для всей молекулы. [Фазу фд| по-
лучают путем преобразования Фурье положений тяжелого атома, которые в свою очередь
определяют по карте Паттерсоиа- см. теквт>.]
функции F всей молекулы путем включения теперь уже оцененного-
нами значения функции Fл вместе с функцией FM в суммарный
вектор функции г. Начертив новую карту электронной плотности с
помощью преобразования Фурье этого нового набора функций F
с уточненными фазами, можно теперь определить положения
большего числа легких атомов. Уточняя расчеты до тех пор, пока не
будут определены координаты всех легких атомов, можно
определить полную структуру исследуемой молекулы.
Наиболее сложной структурой, которую удалось расшифровать
этим методом, была структура витамина Bi2, полученная Ходжкин>
с сотрудниками. Молекула витамина Bi2 содержит около ста
атомов (не считая атомов водорода), один из которых — атом
кобальта с атомным номером 27 — выполнял функцию «тяжелого атома».
Описанный метод имеет, очевидно, верхний предел по
молекулярной массе. Когда исследуемая молекула слишком велика или
тяжелый атом недостаточно «тяжел», или и то и другое вместе, мы
не можем уже воспользоваться соотношением \FM\ ~> |^л|,
которое необходимо, чтобы фаза, определенная для самого тяжелого
атома, была хорошим приближением для фазы функции F
молекулы как целого.
Метод изоморфного замещения (мол. масса более 1000). Для-
более крупных молекул (таких, как, например, белки) описанный
выше метод непригоден по двум причинам. Во-первых, уже нельзя
больше легко определять положение тяжелого атома на карте-
Паттерсона, рассчитанной на основе наблюдаемых дифракционных
интенсивностей. Суммарное рассеяние на всех легких атомах не
является больше пренебрежимо малым по сравнению с рассеянием
на тяжелом атоме (несмотря на то что рассеяние на любом
отдельном легком атоме гораздо слабее, чем на тяжелом атоме,
легких атомов присутствует так много, что общее суммарное
рассеяние на них может быть большим, чем рассеяние на единственном:
764 ЧАСТЬ 6
тяжелом атоме. Во-вторых, поскольку, как только что выяснено,
больше уже не выполняется условие \FM\ > \Рл\, графическое
построение на рис. 22.11 показывает, что и соотношение ф=<рм уже
не является хорошим приближением. Требуется, следовательно,
дальнейшее развитие этого подхода.
Предположим, что рентгеноструктурный анализ белка проводят
до и после введения в его структуру (предпочтительно)
единственного тяжелого атома. Причем такое введение проводят
«изоморфно», т. е. так, чтобы не изменить ни конформацию самого белка,
ни размеры элементарной ячейки («изоморфный» — имеющий ту
же самую форму). Целью этих рентгеноструктурных экспериментов
является, во-первых, определить положение тяжелого атома, и, во-
вторых, использовать полученную информацию для определения
положения легких атомов. Структурные факторы F (для каждого
-«пятна») Для соединения белка с тяжелым атомом можно
выразить (как н раньше) в виде суммы вкладов от самого белка Fb и
ютдельно от тяжелого атома Fm-
F~ рм + РБ. (22*3)
Единственное отличие подходов на основе уравнения (22.3) и рис.
22.11 состоит в том, что функция Fm не является большей по
сравнению с функцией Fb, и поэтому теперь уже нелегко выбрать
положения пятен М—М на карте Паттерсона, построенной прямо на
основе величин \F\2, Однако если мы вычтем структурный фактор
для белка из структурного фактора для соединения белка с
тяжелым атомом, получив разность \F—Fb\, to появится возможность
устанавливать местоположение тяжелых атомов на основе
функции Паттерсона, составленной из этих разностей структурных
факторов \F-Fb |2. Поскольку мы не знаем фазы ни для функции F,
ни для функции Fb , мы не сможем реально вычислить
необходимую разность (F—Fb ). Однако можно сделать почти то же самое,
вычисляя разность (\F\ — \Fb |)2 и составляя из этих величин
функции Паттерсона для определения положения тяжелого атома.
Этот прием позволяет решить первую половину проблемы,
обрисованной выше.
Так как теперь положения тяжелых атомов в их соединении с
•белком установлены, мы можем выполнить преобразование Фурье
этих положений и вычислить вклад тяжелых атомов (как по
амплитуде, так и по фазе) в структурный фактор F соединения
тяжелого атома с белком (рис. 22.12). Как видно прн рассмотрении
этого рисунка, амплитуды F, Fu и Fb определены и известно, что
вектор F должен быть суммой FM н F в, еще известны фазы
(направления на диаграмме Аргана) для FM- Рассмотрим в первую
очередь белковый структурный фактор. Известно, что вектор Fb
должен оканчиваться в точке О (см. рис. 22.12) и иметь
неопределенную еще фазу (принимающую значение от 0 до 360°). Таким
образом, возможные ориентации вектора Fb \ можно изобразить
765 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Рис. 22.12. Определение фазы, при использовании одного (чертеж слева) или
двух (чертеж справа) изоморфных тяжелоатомных производных изучаемого
белка.
Прн использовании только одного соединения белка с тяжелым атомом получают два
значения, которые может принимать фаза (на левом чертеже юин обозначены как X и К,
а на правом — как X я Z). Если же использовать два соединения белка с тяжелыми
атомами, то фазу можно определить полностью (разъяснения см. в тексте). |[Из Wilson H. R.,
Diffraction of X-rays by Proteins, Nucleic Acids, and Viruses, Edward Arnold Publishers,
London, 1966, p. 58.1
в виде окружности радиуса \Fe | с центром в точке. О.
Аналогично, ориентации структурного фактора F соединения тяжелого
атома с белком можно изобразить в виде окружности радиуса \F\
с центром в точке А (см. рис. 22.12). Наконец, поскольку сложе*
ние векторов F и Fe должно дать вектор FM> можно выбирать
только такие фазы на диаграмме Аргана, в которых две
окружности пересекаются. Таким образом, количество возможных фаз для
соединения тяжелого атома с белком (а значит, и для самого
белка) удается снизить до двух возможных значений. Повторив те же
самые эксперименты и графические построения для второго
соединения белка с тяжелым атомом, можно сделать выбор и между
двумя оставшимися возможными фазами для функции Fe и,
следовательно, продолжить выяснение структуры, как описано ранее
(используя эти фазы, построить фурье-карту электронной
плотности, затем по положению некоторых легких атомов получить
уточненные фазы для' различных функций Fe , вновь получить
уточненную карту электронной плотности и так далее до тех пор, пока
не будут найдены положения всех атомов). Из приведенных здесь
рассуждений должно быть понятно, почему необходимо
использовать по крайней мере два различных изоморфных (для обоих
соединений функции Fe должны быть одинаковы) кристаллических
соединения макромолекулы с тяжелыми атомами при
установлении ее структуры в кристаллах (см. последующие примеры).
Пример. Миоглобин
Миоглобии (как чнтатель, несомненно, уже знает) яиляется мономериой
единицей, из которых построен .(тетрамерный) белок гемоглобин. Этот белок
с мол. массой 17 000 имеет в своем состаие 153 аминокислоты или околв
766 ЧАСТЬ 6
1260 атомов (не считая атомов водорода). '[Как в ври рентгеноструктурном
анализе большинства других молекул, рассеяние атомами водорода многлобнна
столь слабо по сравнению с рассеянием атомами углерода, азота или кислорода
(2 = 1 в отлнчне от Z=6, 7 нли 8), что нет возможности определить положения-
атомов водорода на карте электронной плотности.] Элементарная ячейка в
кристалле многлобина содержит две молекулы белка, упакованных в виде
моноклинной структуры с размерами ячейки 64,6X31,3X34,8 А. Для определения
структуры многлобина Кендрю с соавторами применил метод изоморфного
замещения с использованием трех типов производных белка с тяжелыми
атомами в различных комбинациях, чтобы получить шесть типов, кристаллов. Для
каждого типа кристаллов экспериментально измеряли интенсивности около
400 «рефлексов». На этом этапе было получено разрешение около 6 А. Его было
достаточно для установления взаимного расположения сильно скрученных поли-
пептндных цепей, которые, как оказалось, образуют довольно компактную
белковую структуру. Дальнейшее исследование с привлечением данных по
9600 «рефлексам» позволило улучшить разрешение до приблизительно 2 А,
которого вполне достаточно для идентификации и разрешения всех
индивидуальных аминокислотных звеньев и расположения их боковых цепей. Примеры
использования соединений белка с тяжелыми атомами при установлении фаз
структурных факторов, необходимых для построения фурье-карты электронной
плотности многлобина, даны на рис. 22.13. Из этого рисунка (особенно из
части Ь) видно, почему в данном случае необходимо использовать более чет два
а
&
Рис. 22.13. Определение фазы структурных факторов, соответствующих грем
различным «рефлексам» (а, б, а в) для кристаллов многлобина и его соединений
с тяжелыми атомами.
На каждом чертеже жирной окружностью показана амплитуда рефлекса для иемоднфн-
цнрованного белка. Остальными окружностями показано то же для следующих
производных: 1 — миоглобнн + ПХМБС (л-хлормеркуробензолсульфат); 2 — миоглобин + HgAm2 (дн-
амнд ртути): 3 — многлобнн + AuGlj [хлорид золота(Ш)]; 4 — многлобнн + ПХМБС +
+ HgAm2; 5 — миоглобин + ПХМБС + AuCl».
Маленькими стрелками, направленными к центру, обозначены векторы для тяжелых
атомов. Жирными линиями на каждом чертеже показаны выбранные в конечном итоге
фазовые углы. [Из Bodo G., Dlntzis H. M., Kendrew J. С, Wyckoff Я. ТР., Ргос. Roy. Soc. (A).
253. 91 (I9S9).]
Таблица 22.1
Хронология определений кристаллических структур белков
методом рентгеноструктуриого анализа
Год
1960
1965
1967
1968
1969
1970
1971
1972
Белок
Миоглобин
Лизоцим
Карбоксипептидаза А
а-Химотрипсин
Рибонуклеаза А
Рибонуклеаза S
Оксигемоглобин
Папаин
у-Химотрипсин
Гемоглобин
Инсулин
Рубредоксин
Субтилизин BPN'
Химотрипсиноген
Эластаза
Дезоксигемоглобин
Дезоксигемоглобин
Лактатдегндрогеназа
Ингибитор трипсина
Карбоангидраза С
Цитохром h
Феррицитохром с
Ферроцитохром с
Флаводоксин
Гемоглобин
Гемоглобин
Связывающий железо
белок
Миоген
Нуклеаза
Субтилнзин Novo
Трипсин
Конканавалин А
Конканавалин А
Ферроцитохром с
Источник белка
Кашалот
Белок куриного яйца
Крупный рогатый скот
Крупный рогатый скот
Крупный рогатый скот
Крупный рогатый скот
Лошадь
Папайя
Крупный рогатый скот
Хирономус
Свинья
Clostridium pasteurlanum
Bacillus amyloliquefaciens
Крупный рогатый скот
Свинья
Лошадь
Человек
Акула
Поджелудочная железа
крупного рогатого скота
Человек
Печень теленка
Лошадь
Тунец
Clostridium pasteurianum
Glycera dibranchiata
Минога
Хроматиум
Карп
Staphylococcus aureus
Bacillus subtllis
Крупный рогатый скот
Канавалня мечевидная
Канавалия мечевидная
Пеламида
Молекулярная
масса
17 800
14 600
34 600
25 000
13 700
13 700
66 500
23.000
25 000
17 000
5 800
6 000
27 500
25 000
25900
66 500
66 500
140 000
6 500
30 000
11000
12 500
12 500
16000
18200
18 000
9 650
11000
16 800
27 500
25 000
108 000
108 000
12 500
Разрешение,
о
Л
2,0;
2,0
2,8;
2,0
2,0
3,5;
2,8;
2,8
2,7
2,8;
2,8;
3,0;
2,5;
2,5
3,5
2,8
3,5
2,8;
2,5;
2,0
2,8;
2,8
2,5;
3,25
2,5
2,0
2,25
2,0;
2,0;
2,8
2,7
2,0
2,4
2,3
1,4
2,0
2,0
2,5
2,5
1,5
1,5
2,0
2,0
1,5
2,0
2,0
; 1,9
; 2,о
1,85
1,5
Продолжение табл. 22.1
Год
1972
1973
1974
Белок
Ферредоксин
Флаводокснн
Инсулин
Мелатдегндрогеназа
Рнбонуклеаза А
Термолнзнн
Алкогольдеги др оге-
наза
Белок Бенс-Джонса,
Meg
а-Хнмотрнпснн
Ферроцитохром с
Цнтохром сз
ЦнТОХрОМ С550
Фрагмент Fab
Глнцеральдегид-3-
фосфатдегидрогена-
за
Лактатдегидрогена-
за, тройной комплекс
Трипсин —
панкреатический ингибитор
трипсина (комплекс)
Аденилаткнназа
Белок Бенс-Джонса,
REI
Фрагмент Fab, Mc
PC 603
Флаводокснн
окисленный
Инсулин
Лизоцнм
Лизоцнм
Фосфоглицераткнна-
за
Фос фогл ицер аткин а-
за
Фосфоглицератмута-
за
Преальбумнн
Источник белка
Peptococcus aerogenes
Desulfovibrio vulgaris
Свинья
Свинья
Крупный рогатый скот
Bacillus
thermoproteolytics
Печень лошади
Человек
Крупный рогатый скот
Тунец
Rhodospirillum rubrum
Micrococcus denitrificans
Человек
Омар
Акула
Крупный рогатый скот
Свинья
Человек
Мышь
Clostridium MP
Свинья
Человек
Бактериофаг Т4
Мышца лошади
Дрожжи
Дрожжи
Человек
Молекулярная
масса
6 000
16 000
5 800
72 000
13 700
34 600
80 000
23 000
25 000
12 020
12 480
J14 000
55 000
143 000
140 000
30 000
22 000
11000
55 000
16 000
5 800
14 500
18 700
Г8000
45'* 0
110 700
54 000
Разрешение,
о
А
2,5; 2,0
2,5
3,1
3,0; 2,5
2,5
2,3
2,9; 2,4
3,5; 2,3
2,8
2,45; 2,0
2,0
4,0; 2,5
2,8; 2,0
3,0; 2,9i
3,0
2,8;:i,9-
•3,0
2,8; 2,0
3,0
1.9
1.8
2.5
2,5
з.о
3.5
з.>
2,&
769 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Продолжение табл. 22.1
Год
1974
1975
Белок
Роданеза
Трипсин — ингибитор
трипсина из сои
(комплекс)
Белок бактериохлоро-
филла
Фрагмент Бенс-Джон-
са, Аи
Фрагмент Бенс-Джон-
са, Rhe
Карбоангидраза В
Карбоксипептидаза В
Глюкагон
Гоксокиназа
Лизоцим, триклинный
Пепсин
Фосфолипаза Аг
Протеаза SGPB
Пероксид-дисмутаза
Тиоредоксин S2
Триозофосфатизоме-
раза
Трипсин
{Из Matthews В. W.. «X-Ray Cry
Chem., 27, 493-523 (1976).]
Источник белка
Печень крупного
рогатого скота
Свинья, бобы сои
Chlorobium limicola
Человек
Человек
Человек
Крупный рогатый скот
Свинья
Дрожжи
Белок куриного яйца
Свинья
Свинья
Streptomyces griseus
Крупный рогатый скот
Escherichia coli
Мышца цыпленка
Молекулярная
Масса
32 000
43 500
127 000
11000
12 500
30 000
34 000
3 600
51000
14 600
35 000
14 500
18400
16 000
11700
52 000
Крупный рогатый скот 25 000
stallographic Studies of Proteins», in Ann
Разрешение,
о
А
3,9; 3,0
2,6
2,8
2,5
3,0
2,2
2,8
3,0
2,7
1,5
2,7
3,0
2,8
3,0
2,8
2,5
1,8
. Rev. Рпуз.
соединения белка с тяжелым атомом," чтобы получить точную информацию
о фазе. Изображение самой трехмерной структуры молекулы миоглобнна
сейчас можно найти почти во всех новейших учебниках бнохимнн вместе с
обсуждением укладкн многлобнновых субъединиц в гемоглобине и его функции по
связыванию кислорода.
Б табл. 22.1 объединены некоторые (вплоть до 1975 г.) данные по белкам,
трехмерные структуры которых известны с атомным разрешением (около 2А).
В настоящее время возросшая скорость проведения рентгеноструктурного
анализа объясняется главным образом уменьшением стоимости (а также увеличением
объема памяти и быстродействия) вычислительных машин, с помощью которых
обсчитывают дифракционные картины.
Кроме того, чрезвычайно мощные потоки рентгеновских лучей, создаваемые
с помощью синхротронов, делают сейчас возможным получение данных на очень
маленьких образцах.
770 ЧАСТЬ 6
Пример. Нуклеотидсвязывающне ферменты лактатдегидрогеназа (ЛДГаза) и
глнцеральдегид-3-фосфатдегндрогеназа (ГФДГаза)
Взаимосвязь структуры и функции фермента прекрасно иллюстрируется
проведенными недавно структурными исследованиями (с использованием метода
рентгеноструктурного анализа) двух нуклеотндсвнзываюшнх ферментов ЛДГазы
и ГФДГазы. Оба фермента являются дегндрогеназамн гликолиза, которые
связывают молекулу 'NAD+; оба фермента представляют собой тетрамеры,
построенные из четырех химически идентичных субъединнц, причем каждая субъеднннца
имеет один активный центр. На рис. 22Л4 схематически показана
конфигурация NAD+, связанного с ГФДГазой. Хотя очевидно, что можно было бы начать
объяснять механизмы каталитических реакций и на основе детальной
информации, даваемой этой схемой, но на рис. 22.15 схематически показаны особенности
структуры, которые имеют более общее значение. Россман и сотрудники
установили, что структуры областей связывания NAD+ обоих ферментов очень
похожи (аналогичное заключение, как оказалось, можно сделать н относительно
некоторых других нуклеотцдсвязывающих ферментов, структура которых была
определена совсем недавно). Более того, интересно отметить, что у
некооперативного фермента ЛДГазы активные центры в значительной мере отделены один
от другого в тетрамере ЛДГазы (рис. 22.15). В противоположность этому
у кооперативного фермента ГФДГазы активные центры .располагаются по-со-
седству с центрами связывания молекул кофермента NAD+ и в
непосредственной близости друг от друга в тетрамере фермента. Таким образом, оказывается,
что для ГФДГазы (как и для более раннего и поэтому лучше известного
примера с гемоглобином, изученного Перутцем и сотрудниками) кооперативное
связывание лигандов может быть прямо объяснено на основе контактов субъедн-
Рис. 22.14. Схематическое изображение места связывания NAD+ в ферменте
глицеральдегид-3-фосфатдегидрогеназе \[ГФДГазе].
Положения аминокислот в полипептидной цепи указаны маленькими цифрами около соот»
ветствующих остатков. [Из Buehner M.. Ford G. С., Moras D., Olsen К. W., Rossmann M. О.»
J. Mol. Biol., 90. 25 (1974).]
771 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Рис. 22.15. Схематическое сравнение ассоциации субъединиц в ГФДГазе (слева)
и ЛДГазе (справа).
В некооперативной ЛДГазе активные центры далеко отстоят друг от друга. Напротив,
в кооперативной ГФДГазе активные центры находятся рядом Друг с другом. ГФДГаза
оказалась первым многосубъединичиым ферментом, структура которого была определена
методом рентгеноструктурного анализа. :[Иэ Buehner M., Ford G. С., Moras D., Olsen К. W.,
kossmann М. С, J. Mol. Biol., 90, 2S (1974).]
ниц, обнаруженных методом рентгеноструктурного анализа.
Приведенные выше данные о том, что трехмерная структура таких
ферментов в их нуклеотидсвязывающих областях одинакова, позволяют предположить,
что структура в этой области должна была быть «заморожена» на очень ранней
стадии эволюционного развития нз-за необходимости обеспечить подходящий
по форме «карман» для связывания нуклеотида. Однако аминокислотная
последовательность фермента не была «заморожена» в столь давние времена, и к
настоящему времени аминокислотные последовательности этих ферментов стали
различаться, хотя форма связывающего «кармана» сохранилась по существу
одинаковой. Сравнивая аминокислотные последовательности в таких
«замороженных» областях различных ферментов и выявляя, какие из аминокислотных
остатков изменились, генетики смогли бы определить, на какой эволюционной
стадии и в каком порядке определенные ферменты перешли в другие типы,
а следовательно, н выявить прямую эволюционную связь ферментов друг с
другом. Такой анализ для других белков уже был проделай. Он позволил охватить
период в .1,5 миллиарда лет, но применение его к нуклеотндсвязывающим
ферментам обещает увеличить эту цифру, возможно, вдвое.
Пример. Нуклеиновые кислоты J
Наиболее знаменитым (и вполне заслуженно) примером макромолекулярной
структуры, определенной методом рентгеноструктурного анализа, без сомнения
является структура двойной спирали ДНК, открытая Уотсоном и Криком. К
несчастью, анализ этой структуры можно проводить только на основе знания
преобразования Фурье спиральной кривой, а результат этого анализа
математически довольно сложен нз-за применения соответствующих функций Бесселя.
Такой расчет был впервые проведен Криком и сразу же привел к правильному
определению структуры ДНК как спнралн. Аналогичная структура была
предложена Полингом для полипептндных цепей в различных белках (например,
около 77% полипептидных цепей миоглобина находится в конформацни а-спи-
ралн). Мы поэтому не будем здесь давать количественное описание дифракции
от спирали, а интересующемуся этим вопросом читателю можно рекомендовать
монографию Вильсона i[3].
772 часть б
Дрожжевая тРНК
Ч>ЕН
5-конец-* pG
С
G
G
А
U
U
10 U
G А \ А
hU С UCm2G
hU ....
G
G G A G С
20
,G A
m,G
-3-0 H акцепторном конца
.70
ТУС-лелмя
\
./
60
G А С АС
m'A
m5C U G U G «j,
с 11 >-50 T
Дополнительная
ftcm/m.
Ш-петля
петля
Рнс. 22.16.
a — иуклеотидная последовательность дрожжевой фенилаланнновой тРНК, показанной в
конфигурации «клеверного листа». Нуклеотндные звенья пронумерованы начиная с 5'-конца.
б — фотография проволочной модели Кендрю дрожжевой фенилаланнновой тРНК, построен-
Вой на основе карты электронной плотности с разрешением в 3 А. Модель выполнена в>
масштабе 2 см/А. Акцепторный ССА-конец расположен вверху справа, hU-петля и TYC-пет*
ля — внизу, а антнкодоновая петля — вверху слева. На фотографии хорошо видна L-образ*
вая форма молекулы тРНК. [Из Suddath F. L., QuCgley G. J., McPherson A., Sneden D.,
Kim J. J., Kim S. H„ Rich A., Nature, 248; 20 (1974).]
773 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Одним нз наиболее интересных выполненных в последнее время рентгено-
структурных исследований явилось определение структуры транспортной
рибонуклеиновой кислоты (тРНК). С помощью таких тРНК иа рибосоме при
взаимодействии с информационной (или матричной) РНК наращиваются
полипептидные цепи вновь синтезируемых белков (подробнее с этим процессом читатель».
может познакомиться в любом современном учебнике биохимии). На*
рис. 22.16, а показана нуклеотидная последовательность одной из тРНК,
которая отвечает за транспорт аминокислоты фенилаланииа в клетках дрожжей.
Нуклеотнды на этом рисунке обозначены своими обычными символами (отметьте-
наличие в тРНК множества модифицированных нуклеотидов в дополнение
к обычным A, G, С и U). Обнаружено, что существуют «стержнеобразные»
области, в которых реализуются структуры двойной спирали, удерживающие
различные части цепи вместе. Это приводит в итоге к L-образной конфигурации-
молекулы тРНК. Отметим также, что антикодоновая петля (функционально
важная часть молекулы) является единственным местом, где рибозные и фосфатные-
группы находятся как бы внутри структуры, в то время как основания «смот-.
рят» наружу. Такая конфигурация вполне соответствует предполагаемому
механизму функционирования тРНК. Проволочная модель трехмерной структурьг
дрожжевой феннлаланнновой тРНК показана иа рис. 22.16,6. Расчет этой-
структуры основан на данных 4902 отдельных «рефлексов», что позволило»
достичь на картах электронной плотности пространственного разрешения в 3 А.
22.Б. ДИФРАКЦИЯ НЕЙТРОНОВ.
ОПРЕДЕЛЕНИЕ ПОЛОЖЕНИЙ АТОМОВ ВОДОРОДА.
ВОДОРОДНЫЕ СВЯЗИ
Степень спектрального разрешения, которое обеспечивается r
экспериментах по рассеянию (светорассеяние, дифракция
рентгеновских лучей, электронная микроскопия), определяется
эффективной длиной волны используемого излучения. Мы уже отмечали, что
видимый свет может обеспечить получение деталей
пространственного изображения размером порядка ~5000 А, в то время как
рентгеновские лучи или ускоренные электроны — порядка 1 А
или даже менее. Важнейшая особенность этих форм излучения в-
том, что природа процесса рассеяния во всех случаях одна—это
вызываемые переменным электрическим полем излучения
вынужденные движения (колебания) связанных электронов исследуемого
образца. Поэтому если сравнить разные атомы образца, то
окажется, что рассеяние быстро возрастает с увеличением атомного
номера (с увеличением числа электронов в рассеивающем атоме).
Эта характерная особенность лежит в основе уже рассмотренных
нами методов рентгеноструктурного анализа с использованием
тяжелых атомов и окрашивания в электронной микроскопии. Однако
эта же самая особенность делает определение положения
водородных атомов в больших молекулах очень трудным,- поскольку эти
атомы вносят слишком маленький вклад в наблюдаемое рассеяние.
С другой стороны, движущиеся нейтроны также имеют
характерную длину волны порядка 1 А (см. табл. 22.2). Следовательно,
можно ожидать, что они также могут обеспечить атомное
разрешение, если провести эксперимент, в котором макромолекулярный
кристалл будет рассеивать (дифрагировать) поток нейтронов, Не*
Таблица 22.2
Сравнение методов рентгеновской и нейтронной дифрактометрии
Свойство
Рентгеновские лучн
Нейтроны
А. Длина волны
0,71 А (Мо—К-источник)
1,54 А (Си—К-нсточннк)
2. Источник
излучения
3. Некогерентный
фон
4. Проблема фаз
Электронами с энергией
50 кэВ бомбардируют
мишень из Си нлн Мо,
выбивая внутренние электроны.
Рентгеновские лучи
испускаются, когда другие
электроны перескакивают вниз,
заполняя вакантные
орбита л и
Низкий по сравнению с
«нейтронными»
экспериментами
Разрешима, при
использовании метода тяжелого
атома, дающего гораздо более
сильное рассеяние
5. Энергия облучения
(определяет
радиационное
повреждение образца)
«6. Радиационный
поток от источника
7. Фаза излучения,
рассеянного
атомом
Высокая (8 кэВ), что на
5 порядков по величине
больше, чем для тепловых
нейтронов
1010 квантов-см~2-с-1
На 180° не в фазе с
возбуждающим излучением
(гл. 13). Все атомы
проявляются как положительные
области на карте
электронной плотности
От 0,5 до 5 А («тепловые»
нейтроны — это нейтроны,
кинетическая энергия
которых такая же, как у
молекул газа с температурой,
близкой к комнатной)
Высокоэнергетические
нейтроны после ускорителя
частиц пропускают сквозь
«замедлитель» (скажем,
D20 при комнатной
температуре), который замедляет
их до скоростей,
соответствующих длине волны,
указанной выше
Высокий вследствие
некогерентного рассеяния на
атомах водорода (можно
сильно понизить заменой про-
тия на дейтерий)
Трудно разрешимая
проблема, поскольку рассеяние
почти на всех атомах
практически одинаково.
Необходимо использовать прямой
подход (метод проб и
ошибок) при определении фаз
структурных факторов из
экспериментальной
дифракционной картины
Радиационное повреждение
образца незначительно, что
позволяет использовать
гораздо более крупные
образцы
10е нейтронов-см^-с-1, так
что необходимы гораздо
более крупные кристаллы из-
за небольшой интенсивности
источника радиации
Обычно на 180° не в фазе с
воздействием. Исключением
является водород (протнй),
для которого наблюдается
рассеяние точно ~ в фазе с
воздействием. По этой
причине на карте атомной
плотности появляются
отрицательные области
775 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
поскольку нейтроны взаимодействуют с ядрами атомов, а не с
электронами, то интенсивность рассеянного потока по существу
будет мало зависеть от атомного номера (см. табл. 22.3). Таким
образом, атомы водорода (особенно если заменить протий на
дейтерий) будут теперь гораздо более заметны на картине рассеяния и,,
следовательно, могут быть разрешены на окончательной карте
атомов, получаемой с помощью преобразования Фурье дифракцион-^
ной картины.
Существуют значительные отличия в ограничениях,
накладываемых на метод рассеяния нейтронов по сравнению с методом
дифракции рентгеновских лучей, хотя основной эксперимент и способы
получения конечных данных в этих методах очень похожи (см.
табл. 22.2). Из-за того, что величина интенсивности рассеяния
нейтронов почти одинакова для всех атомов, не существует реального-
аналога методу тяжелого атома, столь успешно используемому в-
рентгеноструктурном анализе (см., однако, пример с дейтерирова^
нием). Выполнение эксперимента по нейтронному рассеянию также
более трудоемко из-за 1) необходимости иметь в качестве источ-
Таблица 22S
Сравнение факторов атомного рассеяния для нейтронов и рентгеновских лучей
(в обоих случаях при нулевом угле рассеяния)»
Атом Рентгеновские лучн (электроны) Нейтроны, ферми"
Н 1 —0,37
D 1 +0,67
С 6 +0,66
N 7 +0,94
О 8 +0,58
Р 15 +0,51
S 16 +0,28
а Основано на данных из работы Engleman D. M.t Moore P. В., in: Ann. Rev. Blophys»
& Bioenglneering, 4. 219 (1975).
Ь Ферми=10-12 см. Для нейтронного пучка с потоком N нейтронов на единицу площади в
единицу времени число нейтронов, рассеянных в стерадиан (единица углового пространства}-
за единицу времени, будет определяться произведением N на квадрат числа в
«нейтронной» колонке таблицы.
ника излучения для получения частиц с высокой энергией
ускоритель (!), 2) большего фонового рассеяния (делающего более
трудной локализацию и измерение интенсивностей дифракционных
пятен) и 3) (что наиболее важно) необходимости работать с очень
большими кристаллами вследствие слабых нейтронных потоков от
источника (они почти в 10s раз меньше потоков от
соответствующих источников рентгеновского излучения). Тем не менее,, посколь-
776 ЧАСТЬ 6
-ху относительный вклад данного атома в общую интенсивность
-рассеянного потока меняется как
парциальная интенсивность — Я/Р//2ЛЛ» (22.4)
тде щ— нисло атомов с одним и тем же атомным номером в
кристалле, a fi — атомные факторы рассеяния, данные в табл. 22.3,
очевидно, что водородные атомы будут давать гораздо больший
■вклад в рассеяние нейтронов, чем в дифракцию рентгеновских
лучей. Например, для аминокислоты аланина (химическая формула
^СзНтМОг) парциальная интенсивность рассеяния атомами
водорода составляет только 0,02 для дифракции рентгеновских лучей, но
уже 0,25 для дифракции нейтронов.
Вследствие трудности решения фазовой проблемы методом
дифракции нейтронов исследованы пока только относительно
небольшие молекулы (отдельные аминокислоты, например). Однако в
настоящее время делаются попытки определять методом дифракции
нейтронов положения водородных атомов в макромолекулах тогда,
когда с помощью рентгеноструктурного анализа положения других
атомов уже определены. Повышенный интерес к экспериментам
по нейтронному рассеянию обусловлен тем, что они дают
возможность определять положения атомов водорода, что в свою очередь
важно для выяснения природы и степени образования водородных
связей в макромолекулярных структурах. Широкие исследования
индивидуальных аминокислот показали, что многие из водородных
связей N—НО изогнуты [т. е. угол между связями может быть
значительно (на 30° и более) отличен от 180°]. Интересно, что
такие изогнутые связи, которые обычно считают невыгодными по
энергетическим соображениям, как оказалось, вносят
значительный вклад в стабилизацию определенных молекулярных конформа-
ций.
Пример. Водородные связи между комплементарными нуклеотидами. 9-метил-
аденин и 1-метилтимин
Структура кристаллов из «модифицированных» пар адении-тимин была
недавно исследована методом дифракции нейтронов. На карте распределения
ядерной плотности (рис. 22.17) ясно видиы положения атомов углерода колец
пурина и пиримидина, а также остальные атомы С, Н, N и О. Интересная
особенность этой карты состоит в том, что хотя два комплементарных основания и
удерживаются вместе двумя водородными связями (N—Н'-О и N—H"-N), но
хонформация их отлична от постулированной Уотсоном и Криком, согласно
которой в образовании рассматриваемой пары должен участвовать не атом N 7
аденина, а атом N 1. Однако образование канонических («по Уотсону—Крику»)
пар также было обнаружено с помощью рентгеноструктурного анализа высокого
разрешения в кристаллическом динуклеозидфосфате ApU, имеющем
«нормальные» основания.
Большой интерес к водородным связям в нуклеиновых кислотах отчасти
связан с гипотезой Лёдина, согласно которой два протона, участвующие в обра-
777 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
л -К ■ •
Рис. 22.17. Часть карты плотности нейтронного рассеяния для кристалла пар
9-метиладении-1-метилтимин.
Отрицательная плотность (водородные атомы) обозначена штриховыми контурными
линиями, а положительная плотность (атомы С, N и О) — сплошными контурными линиями.
Отметим хорошее разрешение положений всех атомов. [Из Koetzle T. F., in: Spectroscopy
in Biology and Chemistry: Neutron, X-Ray, Laser, Academic Press, New York, 1974, p. 177.]
зовании пар оснований, могут «перепрыгивать» от одного основания к другому
одновременно. Если бы такой процесс действительно происходил во время
репликации ДНК, комплементарность оснований была бы нарушена и в
результате могла бы синтезироваться новая ДНК с другой последовательностью
оснований (произошла бы мутация). В этой связи интересно заметить, что
результаты, полученные методом дифракции нейтронов для пары оснований,
показанных на рис. 22.17, говорят о том, что протоны прочно фиксированы на своих
местах и ие проявляют никакой тенденции к «туииелированию» через
промежуточный энергетический барьер, чтобы достичь комплементарного основания.
778 ЧАСТЬ б
Таблица 22.4
Среднее нейтронное рассеяние на единицу объема некоторых обычных компонентов
[содержащих протий или(и) дейтерий] биологических объектов*
Химическое соединение
Величина рассеяния
нейтронов, ферма
Н20
D20
Детергент ДДАО6, протонированный
Детергент ДДАО6, дейтерированный
Углеводороды, протонированные
Углеводороды, дейтерированные
«Головки» липидов, протонированные
«Головки» липидов, дейтерированные
Белки, протонированные
Белки, дейтерированные на 20%
Белки, дейтерированные иа 100%
Нуклеиновые .кислоты, протонированные
Нуклеиновые кислоты, дейтерированные на 100%
—0,6
6,3
—0,2
8,0
—0,3
7,0
1,7
2,6
4,0
5,0
9,0
4,2
7,2
а Из Schoenborn В. P., «Neutron Scattering and Biological Structures», Chem. and Eng.
News (Jan.. 24, 1977), pp. 31—41.
6 ДДАО — додецилднметиламиноксид.
Пример. Дейтерирование и подбор контрастных пар
Как видно из табл. 22.4, существует очень большое различие между
рассеянием, даваемым Н20 и DgO как растворителями. Фактически, поскольку
контрастность изображения (аналогичная ситуация наблюдается, например, для
рассеяния электронов; гл. 15.В) основана иа разнице наблюдаемых иитенсивно-
стей, рассеянных различными участками образца, мы можем сделать
«невидимыми» любые «нормальные» (содержащие только атомы протия) липиды, белки,
нуклеиновые кислоты или их комплексы. При наблюдении иейтроиного
рассеяния это достигается путем подбора такого соотношения H2O/D2O в растворителе,
которое дает ту же самую интенсивность рассеяния, что и интересующие нас
«нормальные» молекулы. Этот прием называют подбором контрастных пар. Если
в таком эксперименте в смесь ввести липид (или белок, или нуклеиновую
кислоту), содержащий вместо протия дейтерий, то за его поведением можно
следить отдельно на общем «сером» фойе рассеяния растворителем и содержащими
протий макромолекулами. ^Некоторые данные по нейтронному рассеянию дейте-
рированными и «нормальными» липидами, белками или нуклеиновыми кислотами
можно найти в табл. 22.4.]
Одним из наиболее замечательных приложений метода подбора контрастных
пар явилось «картирование» относительных положений различных белков в
рибосомах. Рибосома — это сложный комплекс из трех рибонуклеиновых кислот
и около 54 различных белков. Можно разложить этот комплекс на
составляющие компоненты, заменить в нем определенные белки их аналогами,
содержащими вместо протия дейтерий, и затем вновь полиостью реконструировать
«правильные» рибосомы. Поскольку только меченные дейтерием белки будут хорошо
видны на контрастно подобранной картине рассеяния, можно определить рас-
|79 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
стояния между этими мечеными белками. Повторяя эту процедуру по очереди
*ля различных пар белков (табл. 22.5), можно установить полную четвертичную
структуру расположения белков в рибосоме просто на основе данных о белок —
"влковых расстояниях. В настоящее время исследования в этой области успешно
>азвиваются и в ближайшие несколько лет можно ожидать получения интересных
очень важных результатов.
|Ь.В. РЕКОНСТРУКЦИЯ ИЗОБРАЖЕНИЯ ПО ПРОЕКЦИЯМ
22.В.1. РЕНТГЕНОВСКАЯ ТОМОГРАФИЯ
В гл. 14.Ж мы упоминали о методе, названном «ультразвуковая
«томография», в котором изображение исследуемого объекта ре-
онструировалось по одномерным эхо-проекциям от ультразвуко-
ых волн, направляемых на объект с разных сторон. В принципе
хема реконструкции изображения всегда одна и та же независи-
о от того, основана ли она на поглощении или рассеянии волн
'{ультразвуковых — гл. 14.Ж; рентгеновских — гл. 22.В.1; магнит-
ых радиочастотных — гл. 22.В.2). В данном разделе мы будем
^рассматривать только изображения макроскопических объектов
Г(объектов, размеры которых гораздо больше длин зондирующих
их волн). Один из самых простых методов реконструкции
изображений— метод «суммирования» (используемый также и для полу-
Таблица 22.5
Определение расстояний между различными парами
рибосомных белков, выполненное методом
рассеяния нейтронов иа контрастно
подобранных рибосомах, реконструированных
из нормальных и дейтерированных белков*
Пара белкое
S2 —S5
S3 —S4
S3 — S5
S3 —S7
S3 —S8
S4 —S7
S4 —S8
S5 —S6
S5 —S8
а Из Schoenborn
gical Structures»,
pp. 31—41.
В.
Ch
P.,
em.
о
Расстояние между белками, А
«Neutron
and Eng.
Scatte
News
105
61
57
115
78
95
63
111
35
ring and Biolo-
(Jan.. 24. 1977).
780 часть в
Исходная
-'картинка
Реконструированное
изображение
Рис. 22.18. Метод суммирования.
Метод является довольно грубым способом восстановления изображения по серии проекций.
Здесь показаны три проекции простой двумерной картинки, содержащей только одну
точку. Каждая проекция дает одномерное распределение плотности (илн аатемиення) поперек
картинки, если ее рассматривать под определенным углом. В случае нашей конкретной
рассматриваемой картинки проекции со всех направлений будут одинаковы. Исходную
картинку можно восстановить по этим проекциям: плотность в каждой точке на
восстанавливаемом изображении оценивают, складывая плотности всех «лучей», проходящих
через эту точку, При реконструкции одной точки изображение похоже иа «звездочку» или
на спицы колеса. Эта «звездочка» и является «функцией поточечной развертки» в методе
реконструкции изображения. Указанные выше процедуры в общих чертах описывают суть
метода суммирования. [Из «Image Reconstruction from Projections» by Richard Gordon, Ga-
hor T. Hermau and Steven A. Johnson. Copyright October 1975 by Scientific American, Inc.
All rights Reserved.]
чения ЯМР-томографических изображений; гл. 22.В.2), показан на
рис. 22.18. Согласно этому методу, изображение реконструируют
путем вычерчивания на рисунке линий соответствующей плотности
под углами, отвечающими направлениям наблюдения такого
распределения плотности при исследовании исходного объекта.
Очевидный недостаток этой схемы в плохой контрастности
изображения, так как, хотя плотность изображения в местах расположения
объекта и является максимальной, но и вокруг изображения
объекта она имеет небольшую, но вполне определенную величину (а
не равна нулю, как нам хотелось бы). Один из нескольких
способов преодоления этого недостатка показан на рис. 22.19.
Согласно этому методу, с помощью вычислительной машины строят
модель объекта, которая дает при рассмотрении с данного
направления такое же распределение плотности, какое наблюдается при
изучении поглощения или рассеяния направляемого на объект
излучения. Далее этот процесс повторяют, для всех возможных
«рассматриваемых» направлений, постоянно корректируя форму
модели до тех пор, пока не будет достигнуто наилучшее совпадение
между наблюдаемыми и рассчитанными интенсивностями на
проекциях. В промышленных приборах для рентгеновской томографии
p
1 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
изображение поперечного сечения части тела человека (обычно
j-оловы, грудной клетки или живота) получают, собирая данные в
160 равноудаленных точках при данном угле наблюдения (в 160
точках по абсциссам на проекциях, показанных на рис. 20.20) и
цля 180 равноудаленных углов (на один градус по
полуокружности) вокруг тела. В итоге регистрируют 160-180=28800 значений
интенсивностей лучей. После обработки этих данных на
вычислительной машине по схеме, показанной на рис. 22.19, получают изо-
5ражение, аналогичное представленному на рис. 22.21 (детали
этого изображения разъяснены в подписи к этому рисунку).
Источник
рентгеновских
лучей
Детектор
■Рис. 22.19. Метод алгебраической реконструкции '(MAP).
Метод разработан для того, чтобы преодолеть неточность метода суммирования. MAP
осуществляют с помощью вычислительной машины, в которой изображение хранится в виде
двумерных наборов чисел, причем каждое число представляет собой рентгеновскую
плотность в одном «кусочке», т. е. небольшом элементе изображения (квадратике).
Одномерная проекция картинки хранится в памяти вычислительной машины в виде перечня цифр,
каждая из которых представляет собой «лучевую сумму», т. е. общую рентгеновскую
плотность вдоль направления луча для узкой полоски картины, вырезанной под
определенным углом. MAP представляет собой итерационный метод. Вр соответствии с этим методом
сначала строят двумерную картинку путем сложения одномерных проекций. Затем для
реконструированного изображения вычисляют новые «лучевые суммы» для всех точек
одномерной проекции, которые сопоставляют с «лучевыми суммами» реального объекта,
хранящимися в памяти вычислительной машины. После этого вычисляют разности, которые
распределяются в виде поправок к значениям плотности по всем элементам изображения,
пересекаемым лучом. При этом «лучевая сумма» приводится в соответствии с измеренной
на опыте. Указанные выше операции повторяют для всех лучей во всех проекциях до тех
пор пока не будет реконструировано изображение, дающее хорошее соответствие со всеми
измеренными проекциями. (Из «Image Reconstruction from Projections by Gordon R
Herman Т., Johnson S. A., Copyright October 1975 by Scientific American, Inc. АП rights
reserved.]
782 ЧАСТЬ 6
о№
ование чере*
Сканирование А/в60
Источник
рентгеновских
лучей
Голова
Емкость с водой.
760 позиций
За
/сканирование
Детектор
/1инейное
перемещение
Рис. 22.20. Сканирующее устройство, позволяющее определить интенсивности
прошедших через исследуемый объект рентгеновских лучей в 160 точках иа
каждой проекции.
За пять с половиной минут можно получить такие проекции со 180 направлений
облучения головы пациента, которые сдвинуты друг относительно друга но углу на один градус.
[Из «Image Reconstruction from Projections» by Gordon R., Herman Т., Johnson S. A.
Copyright October 1975 by Scientific American, Inc. All rights reserved.]
Ценность описанного метода очевидна из того факта, что сотни
таких приборов уже проданы ведущим медицинским учреждениям
несмотря на их высокую стоимость. В будущем предполагается
использовать описанный метод для изучения работающего сердца.
Предварительные эксперименты в этой области (в которых
изучали изолированное сердце собаки, вращая его для получения
необходимых проекций, пока оно еще билось) проиллюстрированы на
рис. 22.22. На приведенных фотографиях ясно видны процессы
расширения и сокращения. Однако для того, чтобы накопить
необходимое число проекций сердца живого человека, потребовалось
бы использовать несколько независимых источников и детекторов
рентгеновских лучей (поскольку ни человека, ни систему
рентгеновских источников и детекторов невозможно вращать достаточно
быстро по сравнению с длительностью цикла биения сердца). Это
заставляет предположить, что даже если этот метод когда-нибудь
и будет доведен до практического использования, то стоимость его
окажется слишком высокой.
83 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
Рис. 22.21. Изображение поперечного сечения грудной клетки живого человека,
полученное методом реконструкции по одномерным рентгеновским проекциям
(см. рис. 22.19 и 22.20).
Области различной прозрачности для рентгеновских лучей видны как темные и светлые
пятна. Поперечное сечение грудной клетки на рнсуике видно так, как если бы на него
смотреть сверху, от головы человека. Темные области слева н справа — легкие. Большая
серая область в середине — сердце. Самые светлые области на изображении — кости: внизу
в центре—позвоночный столб, а вокруг легких — поперечные сечеиня ребер. Внешние
области (светло-серые) — жировые ткани, а остальные серые области — мышцы.
Разветвленные области в легких — это кровеносные сосуды и бронхи. [С обложки журнала Scientific
American, 233, October, 1975.]
Среди других физических подходов, обеспечивающих получение
трехмерных изображений биологических структур, наиболее
привлекательными выглядят голографические методы, в которых
используются оптические или акустические (или оба типа вместе)
волны, причем применение для этих целей оптической голографии
кажется вполне реальным. [О возможностях акустической
голографии см. следующие статьи: Сох М. Е., «Holographic
Microscopy», American Laboratory, April 1975, p. 17, и Greguss P.,
«Acoustical Holography», Physics Today, October, 1974, p, 42.]
Мы завершим эту главу кратким знакомством (гл. 22.В.2) с новым
перспективным методом воссоздания изображения, базирующимся
на резонансном поглощении магнитной энергии, которое
совершенно не приводит к деструкции исследуемого объекта.
784 ЧАСТЬ 6
Рис. 22.22. Фотография поперечного сечения изолированного бьющегося левого
желудочка сердца, сделанные с интервалами в 1/15 с в течение одного цикла
сердечного сокращения.
Частота сокращений сердца — около 120 биений в минуту. Все поперечные сечеиия — это
одно и то же анатомическое сечение посередине между основанием и верхушкой желудочка.
Изображения демонстрируют сжатие левой камеры желудочка (светлая область в центре.)
и последующее расширение камеры во время расслабления сердца. [Из «Image Reconatru-
ctlon from Projections» by Gordon R., Herman Т., Johnson S. A., Scientific American,
October. 1975.]
22.B.2. МАГНИТНАЯ РЕЗОНАНСНАЯ ТОМОГРАФИЯ*
РЕКОНСТРУКЦИЯ ИЗОБРАЖЕНИИ ДЛЯ МЕДИЦИНСКИХ ЦЕЛЕЙ
Как известно, основной составной частью биологических
тканей является вода. По этой причине основным сигналом
протонного магнитного резонанса от ткани будет единственный сигнал
поглощения молекулами воды. [Поскольку оба протона в воде
идентичны, индивидуальные сигналы поглощения каждым
протоном расположены на одной и той же частоте, давая в результате
единый сигнал.] Далее, поскольку частота ядерного магнитного
резонанса пропорциональна напряженности действующего
внешнего магнитного поля, путем создания в ткани градиента магнитного
поля можно получить спектр ЯМР, в котором интенсивность
сигнала на данной частоте будет характеризовать относительное
количество воды в той части ткани, которая находится в области
определенного значения магнитного поля.
Рассмотрим первый простой случай, показанный на рис. 22.23,а.
Здесь изображен образец, представляющий собой два отдельных
близко расположенных и наполненных водой капилляра. Если
величина действующего на образец внешнего статического
магнитного поля линейно меняется вдоль линии, соединяющей оба ка-
• В литературе можно встретить .и другие названия этого метода: «эойгма-
тография» и «ЯМР-интраскония». — Прим. ред.
85 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
.илляра, то частота протонного ЯМР для воды в левом капилляре
!удет меньше, чем в правом, и протонный спектр ЯМР для этого
•бразда будет иметь вид, показанный на средней из кривых в ииж-
:ей части рисунка. С другой стороны, если создан градиент маг-
итного поля, действующего перпендикулярно линии, соединяющей
;ва капилляра, то магнитное поле в обоих капиллярах будет одним
; тем же и сигналы протонного ЯМР в обоих капиллярах будут
сходиться на одной и той же частоте (кривая справа на нижней
асти рис. 22.23,а). Другими словами, мы получили разные
проекции объекта путем использования градиентов магнитного поля,
;ействующего в разных направлениях. Комбинируя проекции,
порченные для четырех направлений градиента, указанных на рис.
!2.23, а, можно воссоздать исходные поперечные сечения капилля-
юв (рис. 22.23,6).
Таким образом, ЯМР-томография обеспечивает возможность
лучения различных частей макроскопического объекта на основе
[азличий амплитуды сигнала ядерного магнитного резонанса в
юзличных частях образца. На рис. 22.24 показано полученное
тим методом изображение живой мыши, если смотреть на нее от
оловы к хвосту. Внутри контура тела отчетливо видны полости
(егких.
Рис. 22.23.
— связь между трехмерным объектом (вверху), представляющем собой два наполненных
одой капилляра, его двумерным сечением поперек осн у (в середине) н четырьмя одномер-
ыми проекциями в плоскости х — г (остальные внешние чертежи). Одномерные проекции
оответствуют спектрам протонного ЯМР (зависимости ЯМР-логлощения от частоты облу-
ення) для четырех направлений градиента магнитного поля, которые иа рисунке покаааны
сплошными линиями.
— изображение поперечного сечеиия исходного объекта, реконструированное по четырем
го проекциям, показанным в части а. Изображения, получаемые таким способом, называют
MP-томограммами илн зойгматограммами, от греческого слова zeugma, означающего «то,
то объединяют» (поле радиочастотного облучения и магнитное поле). [Иа Lauterbur P. С.,
Nature. 242. 190 (1973).]
/OD ЧАС1Ь 6
Рис. 22.24. ЯМР-томограмма живой мыши, если смотреть на нее от головы
к хвосту (на рисунке мышь изображена спиной вверх).
Темные области соответствуют легким. Хорошо также видны очертания всего тела. Это
одно из самых первых изображений, полученных этим методом, и в ближайшем будущем.
по-видимому, можно будет легко получать гораздо лучшее разрешение (см. другой пример
на рис. 22.25). [Из Lauterbur P. С. Pure Appl. Chem., 40, 149—157 (1974).]
Более интересная и потенциально применимая в медицинской
диагностике информация может быть получена из ЯМР-томограмм
тканей, содержащих предполагаемую опухоль. Поскольку
параметр насыщения ЯМР Т\ для протонов воды обычно больше у
опухолевых, чем у нормальных тканей, сигнал протонного ЯМР воды
опухоли насыщается более легко, чем сигнал воды нормальной
ткани. Следовательно, используя соответствующий уровень
мощности при облучении образца, который насытит сигнал ЯМР воды
опухоли больше, чем сигнал ЯМР воды окружающей ее тканей,
можно получить различную интенсивность сигналов ЯМР протонов
воды опухолевой и нормальной тканей. Если при этом сигналы
«разделить» по частоте путем использования градиента внешнего
'87 ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ
>ис. 22.25. Серия трансаксиальных ЯМР-томограмм живой мьщш линии
3H/HEJ, в которую была в нулевой день трансплантирована опухоль СЗНВА
(самая левая картинка в верхнем ряду),
(стальные картинки на рисунке (последовательно слева направо сначала в верхнем, а затем
; нижнем ряду) получены спустя 6, 15, 18, 24, 27, 30 и 38 дней. Различия в яркости на
артннах обусловлены различиями в магнитных «временах релаксации» протонов Тг (спра-
а на темном фоне дан «ключ», с помощью которого можно оценить 7\ в различных
метах ЯМР-томограмм). Поскольку 7"i гораздо больше у опухолевых клеток, то их легко
бнаруживать в виде затемненных областей на ЯМР-томографическом изображении.
Фотография любезно предоставлена профессором Паулем Лаутербуром, химический
факультет Нью-Йоркского государственного университета в Стони Брук.]
4агнитн6г6 поля, то можно будет установить положение и разме-
>ы опухоли (рис. 22.25). На этом рисунке представлены ЯМР-то-
юграммы мыши, в которую была имплантирована опухолевая
гкань. На реконструированных изображениях четко видны
участки с опухолевой тканью.
Этот метод воссоздания изображений с помощью ЯМР
является весьма новым. Для определения положения тканей разных ти-
юв его можно использовать почти во всех тех случаях, когда
применяют рентгеновскую или ультразвуковую томографию. Важным
[достоинством описываемого метода является то, что
радиочастотное электромагнитное облучение не вызывает каких-либо
известных радиационных повреждений исследуемого образца (в отличие
от рентгеновских лучей). Кроме того, радиочастотные сигналы легко
проходят сквозь костные ткани и воздух (в противоположность
ультразвуковым волнам, которые от костей отражаются), и
поэтому этим методом можно было бы легко получать изображения
органов внутри грудной клетки или в полостях черепа, то есть в
областях, относительно недоступных при получении ультразвуковых
Изображений.
788 ЧАСТЬ 6
Задача
1. Используя правила, данные в подписи к рнс. 22.9, составьте ожидаемую
двумерную карту функции Паттерсона для плоской молекулы, показанной ниже.
[Можно принять, что атомы углерода имеют sp2 гибридизацию, т. е. что все
углы у атомов углерода равны по 120°.] Обязательно выпишите отдельно
относительные интенсивности каждого пятна.
Литература
Рентгеноструктурный анализ и кристаллография
1. Waser J., J. Chem. Educ., 45, 446 (1968). Фурье-сиитез.
2. Buerger M. J., Contemporary Crystallography, McCraw-Hill, New York, 1970,
Chap. 12. Хорошее изложение проблемы фаз и оптических аналогов дифракции
рентгеновских лучей.
3. Wilson H. R., Diffraction of X-Rays by Proteins, Nucleic Acids and Viruses,
Edward Arnold Publishers; London, 1966. Рассмотрена дифракция от спирали и
приведено очень наглядное краткое математическое описание восстановления
структуры изучаемого объекта по экспериментальным результатам.
4. Matthews В. W., «X-Ray Crystallographic Studies of Proteins», Ann. Rev. Phys.
Chem., 27, 493—523 (1976). Новейший обзор по структурам белков в
кристаллическом состоянии.
Дифракция нейтронов
б. Schoenborn В. P. «Neutron Scattering and Biological Structures», Chem. and
Eng. News (Jan., 24, 1977). pp. 31^—41. Современный, без технических деталей
обзор биологических приложений.
6, Chen $i-H., Yip S., eds., Spectroscopy in Biology and Chemistry: Neutron, X-Ray,
Laser, Academic Press, New York, 1974. Хороший подбор литературы. Обратите
особое внимание на гл. 5, р. 177.
Реконструкция изображения по проекциям: рентгеновская томография
7. Gordon R„ Herman G. Т., Johnson S. A., Scientific American, 233, 56—68 (1975).
Превосходный популярный обзор.
Реконструкция изображения по проекциям: Я MP-томография
8. Lauterbur P. С, «Magnetic Resonance Zeugmatography», Pure Appl. Chem., 40,
149—157 (1974).
ПРИЛОЖЕНИЕ
ЛОГАРИФМЫ
Определение: если у=ах, тогда x=logay
\ogex =^ In х
Свойства:
log (а • Ь) = log a-{-l°g &
log (#/&)= loga — log 6
log («к") ^ я log л;
Переход от одного основания к другому:
пусть z=ax=bv так, что *=logaz и #=log&2
теперь возьмем loga от ах и Ьу
logaax = logaby
далее, применяя свойство (А-5), получим:
xlogaa=x=ylogab
(А-1)
(А-2)
(А-3)
(А-4)
(А-5)
I0ga2 = (l0ga6)(l0g62) (А-6)
Например: logi0*=logi0e-log*=0,434 loge*
РАЗЛОЖЕНИЯ В РЯДЫ И ПРИБЛИЖЕНИЯ
Разложение в ряд Тейлора (приближение f(x) в окрестностям
точки х=а):
f(x)=f(a)+f' (а) (х-а) + ± f" (а) (х-а)'+...+-L f<»> (а) (х - а)" (Б-1)
Факториалы:
N[^N{N - 1) (N ~ 2) ...32.1 (Б-2)
О! ^ 1 (Б-3)
In (N\) = N In (N) - N для больших tf (формула Стирлийга) (Б-4)
Представление простых функций степенными рядами:
/,±*__1_l_r1*2 + *Lj-*l+ 1 (±Х)П « е*
х8
вовл=.1-^+^.-^-±... (Б-6)
790 ПРИЛОЖЕНИЕ
хг ix3 i x4
eix = cos x + i s'mx = 1 -\-ix~- -^—"зГ"1""4Г -*- "• (^)
Щ\±х)=±х-^г±-^—~±.^ где-1<л:<+1 (Б-9)
Биномиальное разложение:
... + Nabl"-l)+bN (Б-10)
ТРИГОНОМЕТРИЧЕСКИЕ ФУНКЦИИ
sin в = (у/г)
cos в = (х/г)
sin в
cos 0
COS(-e) =: COS(0)
sin(-0) =■ -sin(0)
(B'-l)
(B-2)
(B-3>
(B-4)
Ш-5)
>f sin 0
-90"
-?
л cos 0
90' 180" 270" 360*
-90'
2k
-1
90° ?80" 270" 360°
P? 2тг
Тождества:
градусы -(2я/360) ^радианы
sin (Л ± B) = sini4cosB ± cosi4-sin5
cos (Л ±#) = cosj4-cos£=Fsin,A-sin5
cosM-f-sinM— 1
cos (-JJ— A)=sin A
sin [ r^— Л J =cos A
Ф-6)
(B-7)
(B-8)
(B-9)
(B-10)
(В-П)
; 791 ПРИЛОЖЕНИЕ
Для перехода от декартовых к полярным координатам (см. рис.
21.6):
х = г sin б cos <jp (B-12)
*/=rsin0sin<p (B-13)
z=rcos6 (B-14)
ПРОИЗВОДНЫЕ
Определение: -^f{x)=f'{x);^f(x)=f"{x); и так далее (Г-1)
Производные простых функций (в уравнениях с Г-2 по Г-6 а=
=const) :
^rxa^=axia'1) (Г-2)
jLeax = aeax (Г-3)
£l*ax=± (Г-4)
■т— sin ах = а соs ал (Г-5)
-з— cos ax = — а sin ад: (Г-6)
Производная суммы: ^[ф) + ф)]=-^«(*)+-^(х) (Г-7)
произведения: "h^u'u^=sV('w) + u(^e) ^Г'^
частного: -^1 — I = -* ^ '— (Г-9)
Основные правила:
^[f(a(*). c(4 w(x),...)] = (dfidu) (du/dx)+
+ 0f/A>) (do/Лс) + 0//А») (da;/dJc) +... (Г-11)
ИНТЕГРАЛЫ
Основные свойства:
$f(x)dx=-\f(x)dx (Д-1)
792 ПРИЛОЖЕНИЕ
ь ь ъ
j \и(х) + v(x)\dx=lu(x)dyfr$v(x)dx (Д-2)
а а а
Ъ Ь
f cf {x)dx = c С / (х) dx, где с = const (Д-3)
а а
\fa)dx = ^f{x)dx+^{x)dx (Д-4)
а а
Чётные и нечётные функции:
/ (х) — четна я, если / (—х) = / (х) (Д-5)
(примеры: я2, cos x, е-х2)
. f(*) — нечётная, если /(—х) =—/(*) (Д-6)
(примеры: х, sin x)
Пусть £— чётная функция, а О — нечетная функция. Тогда:
£•£ — четная (Д -7а)
£.0 — нечетная (Д-76)
0-0^ четная (Д-7в)
и интегралы четной и нечетной функций имеют свойства:
• а
J Е (х) dx = 2 J £(*)** (Д-8)
—а О
fO(*)d*: = 0 (Д-9)
—а
Замена переменных:
Пусть дан интеграл \%ba](x)dx. Требуется заменить переменную
х на переменную и = и(х) и интегрировать по и от значения и —
— и(а) до значения и — и(Ь). Для этого выражают х как функцию
х—х(и) и затем используют соотношение:
х=Ь и=и ф)
J f(*)dr = j f\x(u)\^da (Д-10)
лг=а usu (a)
793 ПРИЛОЖЕНИЕ
Интегрирование по частям:
ь ь
\ u(x)v'(x)dx=u(x)v(x)\ba- ^v(x)u'{x)dx (Д-1Г)
а а
Неопределенные интегралы некоторых функций:
\xndx=T^J' при/г^-Ь (Д-Г2)
Г^-=1п|д:|, придг^О (Д-13>
f smaxdx = -^~- (Д-14>
f cos axdx = s-^ (Д-15>
jV*<br = ?^ (Д-16>
Г . 4 j cos (a-f>)x cos (a — d) x г , t2 / rr i т\
j sinaA:cos^^ = ^ТЦ 2(a-4 * ири a=^b (Д"17>
(Для интегрирования sinajt-sinfo или cosaX'Cosbx применяют
тригонометрические тождества:
cosAcosB^1U[cos(A+B)-{-cos(A-B)]
и
sin A sinfi = xJt [cos (Л - Я) - cos (Л+В)]'
и затем используют интеграл Д-15.)
J в» Sinfcc sinatrf* = ^Ub-e)sm(.-c)^+acos(t>-c)x] _
еах [{b + с) sin {b + с) x -f a cos (ft -f c) x] ,n 1ftv
2[a2 + (& + c)2] 1Д-1<5>
j g" cos 6* cos aato = '""»-<> ЗД~ fll+« "»C>-«)«1+
In (fr-f-c) x-f-g<
2 [a2+[*+*]*]
i i"*[(ft + c)sin(fr-f-c)x-f-gcos(ft-f-c)x] m 1QV
794 ПРИЛОЖЕНИЕ
I
e°*sinbxcoscxdx = *a4***(ь-с) х-(ь-c)cos{t
2[a2 + (fr — c)2]
e™ \a sin {b + c) x — (b + c) cos (b + c) x]
c)x]
i
2[a2 + (fr + c)*]
f In axdx = л: In ax — *
J
,0*
Ar^ajcdjc = ^-(ajc— 1)
+
(Д-20)
(Д-21)
(Д-22)
Определенные интегралы некоторых функций:
оо
1
.\e-axdx = ~
а
(Д-23)
1С 1С
i sin2/«jrdA: = I cos*mxdx
(Д-24)
\е-"*йх=У*
2а
о
тэо
f *£ xtdx = ~2-
(Д-25)
(Д-26)
ТЭО
Гл *'<£*: =
Vn
О
ТЭО
О
оо
1
a
0"a*cos&xd* =
a2 + Ь*
в ~ах sin b xdx =
а2 + Ъ*
СД-27)
СД-28)
(Д-29)
796 ПРИЛОЖЕНИЕ i
Величины некоторых единиц в системе СИ
1 дюйм
1 ангстрем (1 А)
1 фунт
1 торр (мм рт. ст.)
1 атм
1 эрг
1 кал (термохимическая)
1 Б.Т.Е.
1 кВт-ч
1 эВ
1 Гц
1 см-1
1 К
1 кг
1 D (дебай)
0,0254_м
Ю-10 м
0,45359 кг
13332 Н-м-2
101325 Н-М-2
10-7 дж
4,184 Дж
1055,1 Дж
3,6-10е Дж
1,6022-10-19 Дж
6,6262-Ю-34 Дж
1,9865-Ю-2» Дж
1,3807-НН3 Дж
8,9876-10" Дж
3,3356-Ю-30 А-м-с
У
я-3,1416
е=2,7183
ln*/lg*=2,3026
Температура замерзания воды (1 атм) =0°С=273,16К
Приставки для обозначения кратных единиц в системе СИ
Мнвжитель
10-1
10-2
10-з
10-е
Ю-9 .
ю-1*
Ю-15
Ю-»»
Приставка
Деци
саити
МИЛЛИ
микро
наио
пико
фемто
атто
Обозначение
Д
с
м
мк
н
п
ф
а
Множитель
10
102
10»
10«
10»
1012
Пристает
дека
гекто
кило
мега
гига
тера
Обозначение
да
г
к
М
Г
т
1
Таблица символов, порядковых номеров и атомных масс химических элементов
Элемент
Символ
Актиннй
Алюминий
Америций
Сурьма
Аргон
Мышьяк
Астат
Барий
Берклий
Бериллий
Висмут
Бор
Бром
Кадмий
Кальций
Калифорний
Углерод
Церий
Цезий
Хлор
Хром
Кобальт
Медь
Кюрий
Диспрозий
Эйнштейний
Эрбий
Европий
фермий
Фтор
Франций
Гадолиний
Галлий
Германий
Золото
Гафний
Ганий*
Ас
А1
Am
Sb
Аг
As
At
Ва
Bk
Be
Bi
В
Br
Cd
Ca
Cf
С
Се
Cs
CI
Cr
Co
Cu
Cm
Dy
Es
Er
Eu
Fm
F
Fr
Gd
Ga
Ge
Au
Hf
Ha
Порядковый
номер
89
13
95
51
18
33
85
56
97
4
83
5
35
48
20
98
6
58
55
17
24
27
29
96
66
99
68
63
100
9
87
64
31
32
79
72
105
Атомная Масса
(227)а
26,98154
(243)а
121,75
39,948
74,9216
(210)а
137,34
(247)а
9,01218
208,9804
10,81
79,904
112,40
40,08
(251)*
12,011
140,12
132,9054
35,453
51,996
58,9332
63,546
(247)*
162,50
(254)а
167,26
151,96
(253)*
18,99840
(223)*
157,25
69,72
72,59
196,9665
178,49
(260)*
Продолжение табл.
Элемент
Символ
Гелий
Гольмий
Водород
Иидий
Иод
Иридий
Железо
Криптой
Курчатовий6
Лаитан
Лоуренсий
Свинец
Литий
Лютеций
Магний
Марганец
Менделевий
Ртуть
Молибден
Неодим
Неон
Нептуний
Никель
Ниобий
Азот
Нобелий
Осмий
Кислород
Палладий
Фосфор
Платина
Плутоний
Полоний
Калий
Празеодим
Прометий
Протактиний
Не
Но
Н
In
I
Ir
Fe
Кг
Ku
La
Lr
Pb
Li
Lu
Mg
Mn
Md
Hg
Mo
Nd
Ne
Np
Ni
Nb
N
No
Os
0
Pd
P
Pt
Pu
Po
К
Pr
Pm
Pa
Порядковый
номер
2
67
1
49
53
77
26
36
104
57
103
82
3
71
12
25
101
80
42
60
10'
93
28
41
7
102
76
8
46
15
78
94
84
19
59
61
£1
Атомная масса
4,00260
164,9304
1,0079
114,82
126,9045
192,22
55,847
83,80
(260)а
138,9055
(257)*
207,2
6,941
174,97
24,305
54,9380
(2Е6)а
200,59
95,94
144,24
го,^
237,0482s
58,71
92,9064
14,0067
(254)а
190,2
15,9994
106,4
30,97376
195,09
(242)а
(210)*
39,098
140,9077
(147)а
231,0359*
Продолжение табл.
Элемент Символ номер Атомная масса
Радий
Радон
Рений
Родий
Рубидий
Рутений
Самарий
■Скандий
Селен
Кремний
Серебро
Натрий
Стронций
Сера
Тантал
Технеций
Теллур
Тербий
Таллий
Торий
Тулий
Олово
Титан
Вольфрам
Уран
Ванадий
Ксенон
Иттербий
Иттрий
Цинк
Цирконий
Ra
Rn
Re
Rh
Rb
Ru
Sm
Sc
Se
Si
Ag
Na
Sr
S
Та
Тс"
Те
Tb
Tl
Th
Tm
Sn
Ti
W
и
V
Xe
Yb
Y
Zn
Zr
88
86
75
45
37
44
62
21
34
14
47
11
38
16
73
43
52
65
81
90
69
50
22
74
92
23
54
70
39
30
40
226,0254B
(222)a
186,2
102,9055
85,4678
101,07
150,4
44,9559
78,96
28,086
107,868
22,98977
87,62
32,06
180,9479
98,9062B
127,60
158,9254
204,37
232,0381B
168,9342
118,69
47,90
183,85
238,029
50,9414
131,30
173,04
88,9059
65,38
91,22
Атомная масса наиболее стабильного или наиболее изученного изотопа.
Условное наименование элемента.
Атомная масса наиболее широкораспространеиного долгоживущего изотопа.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Аберрация
сферическая 495 и ел.
хроматическая 496
Автокорреляция см. Корреляционная
функция
Агароза 248
Аденин 409
Адеиозиндифосфат 101 и ел., 186, 333
в колебательных реакциях при
гликолизе 336—338
Аденозинмонофосфат 186
в колебательных реакциях при
гликолизе 336—338
протонные спектры ЯМР 633—635
рентгеноструктуриый анализ 635
Аденозинтрифосфат 333
в колебательных реакциях при
гликолизе 336—338
свободная энергия гидролиза 101—103
Адиабатический процесс 12
Адреналин 350, 352
Акридиновый оранжевый, интеркаляция
в ДНК 412—414
Активаторы 320—334
Активация ферментов см. Аллостерия
изменением рН 318—321
ионами металлов 332—334
субстратом 320—325
эффекторами 327—329
Активированный комплекс 112—117
Активный центр 292
Активность
ионов 66—68
коэффициент 63
оптическая 417—419, 421—424
растворенного вещества 65, 66
растворителя 65, 66
средняя 67 и ел.
Алкоголь, фармакокииетика 343—346
Алкогольдегидрогеназа 196
дрожжей 547
печени 547, 768
Аллостерия 321—329, 340, 524 и ел.
Амберлит 250
Аминокислоты
анализ 251—253
изоэлектрическая точка 96
кривые титрования, рКа 90 и ел.
последовательность в белке 291
хроматография 244, 253
Амплитуда волны 366
связь с интенсивностью 368, 468
Амплитудная модуляция 559—563
Амфолиты 180
Анизотропия
испускания -у-лучейг 539—543
оптического поглощения и показателя
преломления 410—415
светорассеяния 740—742
флуоресценции 530, 535, 537, 726—729
Анод 135, 179, 215 и ел., 221
Антитела (иммуноглобулины)
гибкость молекулы 489—492, 728 и ел.
иммуиодиффузия 161—164
иммуноэлектрофорез 180 и ел.
рентгеиоструктурный анализ
фрагмента 768
специфичность 161—163
Аргана диаграмма 761 и ел.
Аррениуса уравнение 109
Арчибальда метод 195
Аспарагиновая кислота 271
Аспартаттранскарбамилаза (аспартат-
карбамилтрансфераза), аллостери-
ческие активация и иигибирование
322, 329, 725
Аспирин, комплексообразование с
сывороточным альбумином 56 и ел.
Асимметрический атом углерода 417—
419
Аффинная хроматография 255—258
1^-Ацетил-о-глюкозамин 433 и ел., 602—
604
N-Ацетилмурамовая кислота 602—604
Ацетилхолинэстераза
ннгибирование физостигмииом 306
очистка 256, 258
центрифугирование в градиенте
плотности 196
Бактерии
кинетика роста 261—264
определение подвижности по
светорассеянию 744 и ел. '
фазовое состояние мембраны 40
электрофорез 743 и ел.
Бактериофаги
сборка 737—740
структура 507 и ел.
Барбитурат 356 и ел.
Барьер энергетический 112—117
Белки
Бен с-Джонса 178, 768 и ел.
вращательные времена корреляции 727
денатурация см. Денатурация
конформация см. Конформация
мембранные 502—506
методы разделения 257
молекулярная масса см. Молекуляр-
мая масса
растворимость 68—73
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 801
ренатурация 78
реитгеноструктурный анализ 767—769
структура первичная 291 и ел.
— вторичная 421—423
— третичная 76—78
— четвертичная 184 и ел., 488 и ел.,
501 и ел., 770 и ел., см.
Субъединицы
Бенези—Хильдебранда кривые 83, 86
Бензол, молекулярные орбитали 611
и ел.
Вера закон 398
Биологические часы 334—340
Блоха уравнение 462
Больцмана постоянная 795
— распределение см. Распределение
Больцмана
Броуновское движение 159—161
Брэгга уравнение 731, 749 и ел.
Бутадиен, молекулярные орбитали 610
и ел.
Буферная емкость 87—91
Бьеррума кривые титрования 82—88, 91,
431
Вант-Гоффа уравнение 103—105, 116—
118
Вектор, свойства 568—571, 573
Вероятность
аксиомы 141
амплитуда 578
нормировка 144, 147, 568, 592, 606,
608 и ел., 611
ВЗМО (высшая занятая молекулярная
орбиталь) 614 и ел.
Вирус
гидродинамические свойства 736
полиомиелита 248
табачной мозаики 166, 487, 526, 736
Витамин В12, 220, 763
Витамин С, полярография 221
Вода
в тканях 784
обессоливание 255
структура 527 и ел.
теплота образования 24
ультразвуковые свойства 455
фазовая диаграмма 34 и ел.
Водородные связи
в белках 412
— нуклеиновых кислотах 106—108,
409 и ел., 772, 776
Возраст объектов, определение
радиоизотопным методом по
отношению содержания изотопов
калия и аргона 266—268
по радиоактивному 14С 264—266
химическим анализом по степени
рацемизации L-амииокислот 268—272
Волновая функция 578—581, 584—588,
605
Волновые свойства 366—371
Время
вращательное см. Корреляция
отрезвления 345
релаксации см. Релаксация
Вычисление
интегралов 791—794
производных 791
Вязкость
измерение 200 и ел.
и поток жидкости в капилляре 200—
203
истиннаи 210
определение формы макромолекул
203—212
приведенная 207, 212
удельная 207
Газожидкостная хроматография 241—
245
Газы
идеальные 14—16, 44, 47, 170—172
реальные 50 и ел.
Гамильтониан 573, 584, 588, 597, 606
и ел., 623
Гаптен 729
Гейгера счетчик 230 и ел.
Гейзенберга принцип неопределенности
582
Гель ацетата целлюлозы 177
Гель-электрофорез 175—179
Гемоглобин
гидродинамические свойства 166,
208—211
кооперативное связывание кислорода
94, 323, 524
разделение компонентов изоэлектри-
ческим фокусированием 180
разностный ИК-спектр 700
реитгеноструктурный анализ 765—769
форма молекулы в растворе 211
Гемоцианин 489
Гемэритрин
изобестическая точка в УФ-спектрах
407 и ел.
мёссбауэровский спектр 620 и ел.
связывание кислорода 409
Гепатит, диагностика 177, 289 н ел.
Гиббса—Дюгема уравнение 65
Гидролиз
адеиозинтрифосфата 101—103, 333
пептидных связей 290—293
фосфорнлированных соединений 103
Гидрофобиость
боковых цепей аминокислот 74—76
определение 73 и ел.
при мнцеллообразовании 78—80
802 ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Гиперболическая зависимость,
преобразование в линейную 82, 86, 88,
286—288, 312—314
Гиромагнитное отношение 588
Гистамии, влияние на кровяное
давление 350 и ел.
Гистидаза, рН-зависнмость
каталитической активности 318 и ел.
Гистидин 429—431
Гликолиз, колебательные реакции 336—
338
Глицеральдегид-3-фосфатдегидрогеиаза
аллостерическая активация 524 и ел.
ассоциация субъединиц 770 и ел.
реитгеноструктурный анализ 768
Глицерин, спектры ЭПР дн-(грег-бу-
тнл)нитроксильного производного
436
Y-Глобулин
в плазме крови 176
обнаружение 127
светорассеяние при электрофорезе 742
и ел.
форма молекулы 211
электрофорез 176 н ел., 181, 183 и ел.
Глюкоза 11, 334
Гофмейстера ряды 71—73
Градиент
концентрации 156, 164—166, 189, 401
и ел.
напряжения (электрического поля)
183
плотности 195—198
Граница раздела
перемещение при изотахофорезе 185—
187
электрофорезе 172 и ел.
Гуанидингидрохлорид 155, 179, 248
Гуньера кривая 487 и ел.
Даисил-меченые белки, флуоресценция
537—539, 660—662, 728 и ел.
Дансилхлорид 535
Дауэкс 250
Двулучепреломлеиие 410—415
электрическое 525 и ел.
Де Бройля длина волиы 493
Дебая—Хюккеля уравнение 69
Дегидрогеназы см. Алкогольдегидроге-
наза; Глицеральдегид-3-фосфатде-
гидрогеназа; Лактатдегидрогеназа
Дезоксирибонуклеииовая кислота
визуализация транскрипции 498—500
интеркаляция красителя 412—414
коэффициент вязкости 212
— диффузии 166, 736
радиус вращения 487
реитгеноструктурный анализ 771
репликация 197 и ел.
спектр комбинационного рассеяния 651
и ел.
Дейтернроваиие 778 и ел.
Денатурация
гуанидиигидрохлоридом 155, 179, 248,
527
детектирование 455
додецнлеульфатом натрия 155^ 178
и ел., 250
кислотно-основна1я 455
мочевиной 155, 527
температурная 115 и ел., 118, 405
и ел.
Деполяризация
Y-лучей 539—544
светорассеяния 740—742
Флуоресценции 532—537, 541
Детергент 155, 178 и ел., 250
Диализ 55 и ел.
равновесный 56 и ел.
Дигидролипоилтрансацетилаза 501
Диксона зависимость 314 и ел.
Днполь-дипольное взаимодействие 548
и ел., 662 и ел.
Дипольиый момент
магнитный 424 и ел., 588, 663—666
электрический 467, 641—644
Диск-электрофорез 182—185
Дисперсия
и поглощение 375, 449 и ел., 686—689
оптического вращения 419—423
типы 384—387
Дисульфидные связи
в иммуноглобулине 489—492
восстановление 179
и электрохимическое определение
белков 127
при вулканизации резины 154
Дифракция
нейтронов 773—779
рентгеновских лучей см.
Реитгеноструктурный анализ
Диффузия
вращательная 716—729
— коэффициент см. Коэффициент
вращательной диффузии
— одномерная 716—719
— трехмерная 719—723
иммуио- 161—164
— двухмерная 163 и ел.
— радиальная 161—163
радиальная 163 и ел., 167 и ел.
трансляционная 164—166, 214—216
— двухмерная 156, 166 и ел.
. — коэффициент см. Коэффициент
трансляционной диффузии
— трехмерная 167 и ел.
уравнение 158
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 803
Фика закон 156
Дихроизм
в потоке 412—414
н электрическое двулучепреломление
525 и ел.
круговой 415—424
Диэлектрическая проницаемость 444—
449
Дозы логарифм, связь с откликом био-
снетемы 349 и ел., 354
Доннана равновесие 51—55
Доплера эффект 460, 619, 742
Друде уравнение 420
Емкость 440—446
ED50 450
Желатин 211
Железо биологическое 617—621
Желтуха, диагностика 289 и ел.
Загрязнения
атмосферные 698
радиоактивные 272—276
Заселенность энергетических уравнений
инверсия 644—647
равновесная 628—631, 654
Зеии, молекулярная масса и форма
молекулы 211
Зойгматография 784—787
Излучение
когерентное 369, 647—649, 679—681
некогереитное 369, 679 и ел.
электромагнитное см.
Электромагнитное излучение
Изобестнческая точка 407 и ел.
Изотопное разбавление 231—234
Изоэлектрическая точка 96
Изоэлектрическое фокусирование 180
Ильковича уравнение 216 и ел.
Имидазол 429—431 -
Иммуноглобулин G 181, 489—492, 728
и ел., 768
Ингибирование ферментов 301—317
аллостерическое 321—329
бесконкурентное 303
графический анализ 312—314
избытком субстрата 330—332
изменением рН 318—321
конкурентное 304—308, 314
— антагонизма к гистамину
димедролом 350 и ел.
— ацетилхолинэстеразы физостигми-
ном 306 и ел.
— механизм 304—308
— противоопухолевыми препаратами
315—317
— психотропными препаратами 352—
354
неконкурентное 308—310, 314
предельные случаи 309
продуктом реакции 293 и ел.
смешанное 310—315
Индуктивность 440 и ел.
Инсулин
конформация 422
молекулярная масса и форма
молекулы 211
рентгеноструктурный анализ 768
Интерференция 368 и ел., 697
внутренняя 478—484
и теневая оптика 401 и ел.
Иод
накопление в щитовидной железе 233,
276
связывание с белками 233
Ионная пара 526—528
Ионная подвижность 171 н ел., 178 и ел.
Иоииая сила 69
Ионный радиус 71
Кавитация 460
Кадмий, корреляция испускания у-лучей
539—544
Калориметрия 20, 23
дифференциальная сканирующая 105—
108
Карбоангидраза
рентгеноструктурный анализ 767, 769
число превращения 292
Карбоксипептидаза 291 и ел.
рентгеноструктурный анализ 767, 769
число превращения 292
Карбонат-ион, нормальные колебания
557
Карта
плотности нейтронного рассеяния 777
потенциальной энергии 113
электронной плотности 755—758
Каталаза 220, 292 и ел.
гидродинамические свойства 178, 208,
210
седиментация в градиенте плотности
196
число превращения 292
Катехол-О-метилтрансфераза 353
Каучук натуральный 153 и ел.
Квадрупольное расщепление 618
Кибернетика 528—532
Кинетика
двусубстратных ферментативных
процессов 296—299
804 ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
иекатализируемых реакций
второго порядка 518 н ел.,
737—740
одностадийных прямых одно-
компонентных 280
последовательных 282 и ел.,
343—346
прямых и обратных 281 и ел.
с одним промежуточным
соединением 283 и ел.
таблица времен химической
релаксации 521
стационарная 284—289, 304
— ферментативных реакций 295 и ел.
Кинетические процессы первого порядка
261—277
время релаксации 263, 518—520
— полупревращения 263
— радиоактивный распад 264—268
рацемизация аминокислот в организме
268—272
рост бактерий 262—265
химические реакции 111 и ел.
Кислород
в гемэритриие 409
кооперативное связывание с
гемоглобином 94, 409
образование при диспропорциониро-
вании перекиси водорода 220
при дыхании 528—532
Клетки
деление
— влияние лазерного облучения 651
— кинетика 261—264
ионный баланс 124
палочковые сетчатки глаз 414 и ел.
Клубок беспорядочный 422
переход в а-спираль 405 и ел., 455
радиус вращения 482
среднеквадратичное расстояние
конец—конец 152—155, 482
Коллаген, гидродинамические свойства
166, 208, 210
Комбинационное рассеяние 559—563, 651
и ел.
Комплекс с переносом заряда 614
Компоненты системы 33
Конденсатор 440
Константа
ассоциации 57, 59, 80—95
диссоциации, метод определения
косвенный 309, 310—314
прямой 56 и ел. 80—95
ионизации см. Буферная емкость;
Кривые титрования
Михаэлиса 285
равновесия 60—63
— концентрационная зависимость 63—
73
— определение 56 и ел., 80—95, 129—
133
— связь с гидрофобиостью 73—76
свободной энергией 64
— температурная зависимость 103—
105
спин-спинового взаимодействия
— — определение по спектрам ЯМР
599 и ел., 623
связь с двугранным углом 601—
604
Контраст изображения 402 и ел., 496—
502 ,
Конформация
белков, определение по вязкости 209
и ел.
— КД и ДОВ 421—423
— — — коэффициенту диффузии 208
радиусу вращения 482—492
— теоретический расчет 76—78
нуклеиновых кислот 197 и сл„ 211
и сл„ 635, 701—703
определение по константам
спин-спинового взаимодействия 602—604
химическим сдвигам ЯМР,
индуцированным лантаноидом 631—
635
Кооперативный процесс
переход спираль—клубок 405 и ел.,
455
— фазовый 33—41
Корреляционная функция
угловая 719—722
флуктуации концентрации 729—732
— между состояниями 711—716
— по направлениям 716—723
Корреляция
вращательное время 448, 727
и вязкость 446, 538, 543, 722
и ел., 727, 740 и сЛ.
связь с коэффициентом
вращательной днффузин 538, 543, 722
и ел., 726 и ел., 740 и ел.
испускания у-лучеп 539—544
Коул—Коула зависимость 448—465
Коэффициент
активности 63
— иоиов 66—68
— определение 64—66
диффузии
— вращательной
определение по деполяризации
светорассеяния 740—742
диэлектрической
релаксации 443—449
магнитной релаксации
724—726
флуоресценции 532—539,
726-729
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 805
— электрическому двулуче-
преломлению 525 и ел.
\. — трансляционной 156, 164—166
' в одномерном пространстве 159
; двухмерном пространстве
159, 163, 167 и ел.
[ трехмерном пространстве
г' 159, 167 и ел.
' и вязкость 191
скорость седиментации 190
— — определение по светорассеянию
735—737
связь с коэффициентом трения
172
— формой и размером
макромолекул 208, 736 и ел.
температурная зависимость 191
— молярного погашения 398
седиментации 190
треиия 203—208, 726
— вращательный 722
— при электрофорезе 169—172
— трансляционный 170—172, 203—
208, 722
Кривые титрования 82—88, 91, 431
Кристалл
рассеяние света 730
элементарная ячейка 759
Кристаллография см. Дифракция
нейтронов; Рентгеноструктуриый анализ
Кровь
определение объема в организме 232
1 ультразвуковые свойства 455
Круговой дихроизм см. Дихроизм
Кулоиовскне взаимодействия 169—172,
365 и ел., 466
энергия 607
Пайнуивера—Берка зависимость 288,
298, 306, 309, 313—315
Лазер
в хирургии 649—651
индукция температурного скачка 652
и ел.
применение 653 и ел.
принцип действия-и-общее устройство
644—649
Лазерное светорассеяние 651, 729—745
П актатдегидрогеназ а
конформация 422
определение в сыворотке крови 289
и ел.
рентгеноструктуриый анализ 767
х-Лактоглобулин А, сборка субъединиц
488 и ел.
5-Лактоглобулин, гидродинамические
свойства и форма молекулы 208—
211
Лантаноидные сдвигающие реагенты *-
ЯМР 631—635
Лейкемия, химиотерапия 315—317
Лекарственные препараты
антибиотики 308
взаимодействие с рецептором 347—352"
кинетика введения и вывода 343—346*
полярографическое определение 220—
222
противоопухолевые 315—317
с замедленным действием 253
сульфамидные 307 и ел.
теория действия 347—357
— занятости 347—350
Ле Шателье принцип 105
Лизоцим
взаимодействие с субстратом 432—434J.
602—604
дифференциальный УФ-спектр 406
коэффициент диффузии 736
конформация 422
молекулярная масса 194
последовательность аминокислот 292
радиус вращения 487
рентгеноструктуриый анализ 759,.
767—769
Линдемана модель 111 и ел.
Линза 492—498, 750—753
Линия
спектральная 375 и ел., 683 и ел., 688,;.
701
— форма 687, 701
— Фраунгофера 684, 688, 700 и ел.
лоренцов контур 383—388
ЛКАО-МО (линейная комбинация
атомных орбиталей для получения
молекулярных орбиталей) см.
Молекулярные орбитали; Хюккеля теория-
молекулярных орбиталей
Логарифм, определение и свойства 789*
Лотка схема, 334 и ел.
ЛСД 350, 614
Y-Лучи, корреляция испускания по на~
правлениям 539—544
Майкельсона интерферометр 697
Макромолекулы
гетерогенность по свойствам 257
методы разделения 257
определение формы, гибкости и
размера 437—439, 447—449, 489—492,.
535—537, 538 и ел., 543, 547 и сл.„
723—729
растворимость 68—73
связывание лигандов 80—95
кооперативное 91—95
— — независимое 85—91
равновесное 312 и ел.
— растворителя 206—210
806 ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Масс-спектрометр 692—697
Масс-спектрометрия ионного
циклотронного резонанса 692—697
Математическое ожидание переменной
величины
дискретной 149—152
непрерывной 149 и ел.
Матрица 570—577
Мембранные везикулы
подвижность белков 536
текучесть 35—41, 436 и ел.
фосфолипидный переворот 439
Мембраны
биологические, активный перенос 39
и ел.
— двуслойные
подвижность фосфолипидов 547
и ел.
структура 41, 500 и ел.
— электронная микроскопия 502—
506
— латеральное разделение фаз 39
— текучесть 35—41, 436 и ел.
— фазовые переходы 35—41
ионообменные 137 и ел., 235
полупроницаемые
— диализ 55—57
■ равновесный 56 и ел.
.Мениск 189, 193
Метатрексат, противоопухолевая
активность 317
'9-Метиладенин, дифракция нейтронов
777
.Метнлеиовая группа
нормальные колебания 558
расщепление сигнала ЯМР 599—601
1-Метилтимин, дифракция нейтронов 777
Метильиая группа, расщепление сигнала
ЯМР 599—601
-Механизм двусубстратиых реакций
296—299
Механический сдвиг 198
Микроскоп
ультразвуковой 456 и ел.
фазово-контрастиый 402 и ел.
электронный 492—498
Миозин, гидродинамические свойства
178 и ел., 208—210, 487
Миоглобин 765—769
конформация 422
радиус вращения 487
реитгеноструктурный анализ 767
Михаэлиса константа 285
Михаэлиса—Ментен модель 284—289,
293—299
Мицеллообразование, критическая
концентрация 79
Мицеллы 78^В0
Многоканальная спектроскопия 669—Щ
Мода
дисперсии 426
поглощения 426
Модель дыхательной функции 528—
Молекулярная масса, виды 49 и ел. |
Мольная доля 42, 61, 64
Моляльная концентрация 47, 61, 63 ^
Момент i
магнитный 424—426, 462—464 *
угловой орбитальный 573, 588—590;?
— спиновый 573, 590—592 %
Моно—Уаймана—Шанже модель л
аллостерической активации и инги
рования 327—329
кооперативного связывания 524 и ;г
Моноаминооксндаза 352—354 \"|
Моча, белки 178 |
Мочевина 155, 527 щ
Мультиплексная спектроскопия %
Фурье-преобразование; Хадамард
преобразование
Муравьиная кислота, теплота pea
образования в клетках 23 и ел,
Мутность 478, 509
Надперекиси 408
Напряжение 440—442
Насыщение
в спектроскопии 654—657
перенос 657—659
фермента 278
Нейромедиатор 351—354
Нейтронная дифрактометрия,
фаз 774
Нернста уравнение 123, 128
Норадреналин 350, 352—354
Нормальное распределение
159, 167, 227 и ел.
Нормальные колебания 554—559, 563
Нормировка 144, 147, 568, 592, 606,
и ел., 611
Нуклеиновые кислоты
конформация 197 и ел., 211 и ел., 6
771—773
спаривание оснований 106—108,
и ел., 772 ■-*,
хроматография 252—254 |
Нулевой порядок процесса 344—346 ''■
NAD ' j
в колебательных реакциях при глик|
лизе 336—338
связывание с глицеральдегид-3-ф1
фатдегидрогеиазой 524 и ел., 7{
и ел.
NADH
в колебательных реакциях при гли:
лизе 336—338
%
проблей
147, \\
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 8&Г
связывание с алкогольдегидрогеназой
547, 549
Обратимый процесс 11—14, 17 и ел.
Обратная зависимость 82—88, 351
Объем
исключенный 153 и ел.
парциальный моляльный 27 н ел.
— удельный 189 н ел., 192, 208 н ел.,
736
Овальбумин
коэффициент диффузии 166, 740
молекулярная масса и форма
молекулы 211, 247
энергия активации денатурации 118
Окружающая среда Н
Ома закон 440
Оператор
гамильтониан 573, 584, 588, 597, 606
и ел., 623
коммутатор 573, 582—584, 589 н ел.
повышающий 585, 589, 621 н ел.
понижающий 586, 589, 621 н ел.
собственные значения 572—578, 583—
592, 606—615, 624
соответствие матрице 570 н ел.
спиновый 588—592
среднее значение 579—582
эрмитов 571—577 н ел.
Оптическая плотность 398
Оптические изомеры 417—419
возникновение в природе 423 и ел.
Оптическое вращение 415—417
молярное 417
Оптическое поглощение 396—410
Орбиталь
атомная, линейная комбинация 604—
616
молекулярная антневязывающая 609
— высшая занятая 614 и ел.
— н электронная плотность 615 и ел.
— связывающая 609
— теория Хюккеля 604—607
Ортогональность функций 569
Осадки радиоактивные 272—276
Осевое отношение 204, 205, 208, 210, 448
Осмотическое давление 42—51
влияние заряженных частиц 54 и ел.
определение сред нечисловой
молекулярной массы 48—50
Оствальда внекознметр 202 н ел.
Отклик типа «все илн ничего» 354—357
Палочковые клетки сетчатки глаз
двулучепреломленне и дихроизм 414
н ел.
родопсин 506
Папанн
конформация 422
расщепление иммуноглобулина G490
рентгеноструктурный анализ 767
Парамагнитные ядра 427
Парамагнитный сдвиг ЯМР,
индуцированный лантаноидом 631—635
Паттерсона функция 759—769
Пепсин
расщепление иммуноглобулина G
489—492
рентгеноструктурный анализ 769
специфичность 290 и ел.
электрофоретическая подвижность 178*
Пептидная связь
гидролиз протеазами .290 н ел.
КД н ДОВ 421—423
спектр поглощения 404
Перегонка фракционная 243
фазовая диаграмма 57
Передача нервного импульса 352 н ел.
Перенос
активный 40, 123 н ел.
заряда 614
пассивный см. Диффузия трансляцион1-
ная
электрона 130, 220, 390, 614
энергии 659—662
Переход между уровнями энергии
безызлучательный 637, 654—659
дипольный момент 642
запрещенный 587, 635—643
индуцированный 636—647, 655—657,.
662—667, см. Лазер; Правила
отбора
разрешенный дипольный магнитный*
662—666
электрический 635—642
спонтанный 636, 649
Переходное состояние 112—117
Перрена график 537—539
Печень, ферментативная диагностика
заболеваний 289 и ел.
Пинг-понг, механизм двусубстратных
ферментативных реакций 296—299
Пиридин
молекулярные орбиталн 612 и ел.
электронная плотность 615
Пируватдегидрогеназа 501
Плавление
транспортной РНК 107 и ел.
фосфолипидов в биомем^ранах 35—41
Поглощение 398, 687
и дисперсия 375
оптическое см. Оптическое поглощение-
спектр 375, 450, 686 и ел.
типы 384 н ел.
УФ-света 403—405
Позитрон, аняигнляция 233
808 ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Показатель преломления 397—403, 475
Лолнакриламид 175
Полиизопрен 153
Полн-Ьглутаминовая кислота 405, 454
и ел.
Поли-ь-лизин 421, 423
Поли-Ь-пролин 660 и ел.
Полимеры
вязкость 209—212
исключенный объем 153 и ел.
«информация 152—155
Полннуклеотиды 409
Полисахариды 602—604
Полный дифференциал 9, 14, 19
Поляризуемость 474 и ел., 559 н ел.
Полярные координаты 719, 791
Постулаты квантовой механики 577—583
Потенциал
мембранный 124
.окнелнтельно-восстановнтельный 129—
137
— стандартный 131
— электродный 134
химический 27—33
электрохимический 121—137
Потерн диэлектрические 443—449
— ультразвуковые 450—455
Поток
диффузионный 156—158, 179
капиллярный 200—202
— измерение вязкости 202 и ел.
ньютоновский ламинарный 202
остановленный 391, 512—523
турбулентный 202
электрофоретический 174
Правила отбора 643, 662 н ел.
Правило фаз 33—35
Предел
Лорентца 381—388, 392
рассеяния
— Рэлея 380, 392, 471
— Томсона 380 и ел., 392
Предэкспоненциальиый множитель 109
н ел.
Приближения математические 789 и ел».
Пробит 355—357
Прогестерон 239
Произведение растворимости 69
Протеазы см. Карбокснпептидаза;
Папайи; Пепсин; Трипсин; Химотрнпсин
Протои
константа скорости переноса 390
магнитный момент 424—427, 462—464
Прямой метод 81 и ел.
W
и денатурация 454 и ел.
измерение в D20 462
Н20 124 и ел.
и регуляция скоростей
ферментативных реакций 318—321
титрование 82—87, 91, 431; см.
Буферная емкость
Работа
виды 12
и тепло 11—14
максимальная 19
против центробежной силы 194
Равновесие
кислотио-основное 82 и ел.
критерий 18, 32
механическое 31 и ел.
седимеитацнонное 192—195
тепловое 31 и ел.
химическое 31 и ел., 60—68
Радиоактивные изотопы
измерение радиоактивности 225—234
ошибки 225—231
изучение механизмов ферментативных
реакций 299
метод изотопного разбавления 231 —
234
— радиоактивных индикаторов 231—
234
период полураспада 264—268, 272—
276
распад 264—268
Радиус вращения 482 и ел., 487—492,
509
Рак
диагностика 222
радионзотопная локализация 233
химиотерапия 315—317
ЯМР-томографня 786—788
Распределение
биноминальное 144, 227 и ел., 790
Больцмана 625—631, 646, 654—657
Гаусса 147, 151, 159, 227 и ел.
Пуассона 225—228
— в теории электрических шумоЕ
234—239
хроматографии 241—245
— график вероятностей 240 и ел
— при измерении радиоактивное^
228—234
Рассеяние 378 и ел., 466—473
зависимость концентрационная 484 i
ел.
— угловая 469 и ел., 478—484
— частотная 468, 471
предел Рэлея 373, 380, 474—485
— Томсона 373, 380 н ел.
рентгеновских лучей 486—492
рэлеевское см. Рэлеевское светорас
сеяние света 474—485
типы 384 и ел.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 809
электронов см. Электронная
микроскопия
Раствор
гипотонический 47
идеальный 42, 60—63
изотонический 47
реальный 63—73
Рауля закон 42
Рацемическая смесь 423
Редуктаза фолевой кислоты 317
Релаксация 389—393, 443
время 263, 518—522
— связь с константой скорости
реакции 263
диэлектрическая 443—449, 723 и ел.
магнитная 544—549, 724—726
скорость 521—525, 551 и ел.
— в ЯМР 544—549, 724—726
— и анизотропия испускания у-лучей
539-544
деполяризация флуоресценции
532—539, 726—729
электрическое , двулучепреломле-
ние 525 и ел.
спии-решеточная, время Т&
определение 655
■ и спектральная плотность
709-711, 714—716, 724—726
экспериментальные примеры
544—549, 656 и ел., 758—761
спин-спиновая, время Tit определение
655
н спектральная плотность
714—716, 724—726
ультразвуковая 450—455, 526—528
химическая 512—520
Рентгеновское рассеяние под малыми
углами 486—492
Рентгеноструктурный анализ
белков (хронология) 767—769
дифракционная картина 750—753
закон Брэгга 749 и ел.
метод изоморфного замещения 763—
765
— тяжелого атома 761—763
миоглобина 765—769
нуклеиновых кислот 771—773
проблема фаз 759—769
синтез Паттерсона 759—761
Рибонуклеаза
молекулярная масса 194, 208
размер молекулы 208, 210
рентгеноструктурный анализ 767
спектр протонного ЯМР 429—431
число превращения 292
Рибонуклеиновая кислота
рнбосомальная 778 и ел.
транспортная
— ионообменная хроматография 252
— плавление 107 и ел.
— рентгеноструктурный анализ 772
и ел.
— спектр комбинационного рассеяния
651 и ел.
Рибосома 778 и ел.
Родопсин
в палочковых клетках сетчатки глаз
415
топография в двуслойных мембранах
505 и ел.
Рэлеевское светорассеяние
зависимость от концентрации 484 и ел.
молекулярной массы 474—478,
508
формы и размера макромолекул
478—484
кривые Цима 484 и ел.
мутность 478, 509
определение радиуса вращения 483—
509
Сахароза 194, 196 и ел.
Сведберг 191
Свет см. Электромагнитное излучение
Свойства интенсивные 30
— экстенсивные 30
Седиментация
в градиенте плотности 295—198
изокинетнческом 197
изопикническом 197
коэффициент 190
метод Арчибальда 195
равновесие 192—195
скорость 188—192
Сердце
рентгеновская томография 783 и ел.
. ферментативная диагностика
заболеваний 289
Сефадекс 245, 250
Силы 365 и ел.
Кулона см. Кулоновские
взаимодействия
магнитные 424 и ел., 544 и ел., 588,
592 594
центробежные 188—190, 194
Система, определение И
Скачок
давления 391, 513
концентрационный 391, 513—522
температурный 391, 513
электрического поля 391, 513
Скорость
волны 366
релаксации см. Релаксация
седиментации см. Седиментация
теплового движения 170 и ел.
угловая 188
810 - ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
ультразвука 450—455, 457—460
ферментативной реакции 286—290
начальная 284, 293 н ел.
Скэтчарда кривые 84 и ел,, 313, 315
Случайное блуждание
одномерное 147—151
трехмерное 152
Смола ионообменная 250
Снотворные препараты 354—357
Собственные значения 572—578, 583—
592, 606—615, 624
Собственные функции 572—578, 585, 615
и ел., 624
Солевой мостик 122, 128
Сопротивление 440 и ел.
Состояние
возбужденное 533, 585, 647—649. См.
Уровни энергии
основное 533, 585, 548
переходное 113, 116 и ел.
стандартное 20 и ел.
— ионов водорода 99
— растворителя 44
термодинамическое 14—17
Спектральная плотность 708—711, 714—
716
Спектроскопия
вращательная 674
ионного циклотронного резонанса
692—697
колебательная 23, 410, 412, 674, 697—
700
комбинационного рассеяния см.
Комбинационное рассеяние
мёссбауэровская 617—621
многоканальная 669—676
радиочастотная 672, 681
разрешение .672, 680
рентгеноэлектронная 674
типы 384—387, 674
флуоресцентная 537—539
фотоэлектронная 674
электронная 403—410/412—415, 421—
423
электронного парамагнитного
резонанса 434—439
электронного удара 683
ядерного магнитного резонанса см.
Ядерный магнитный резонанс
Спин
задачи 588—604
квантовые числа 591 и ел.
угловой момент 588 и ел.
электрона 586
ядра 590 и ел.
Среднее значение
переменной дискретной 149 и ел.
— непрерывной 149 и ел.
— случайной 703—707
результатов измерений физической
величины 579 н ел.
Среднеквадратичное расстояние
между концами полимера в
статистическом клубке 152—155
при случайном блуждании 151 и ел.
Стационарная область ферментативной
реакции 285
Степени свободы 34
Степень гидратации 206, 208—210, 736
Стирлинга приближение 145, 166, 789
Структурный фактор 759—769
Стокса закон 205, 722
Субстрат
аллостерическая активация фермента
323—327
взаимодействие с ферментом 602—604
ингибнрование фермента 330—332
модель Михаэлиса—Ментен 278, 284*-
289
меченый 299
Субъединицы 184 и ел.
аспартаттранскарбамилазы
каталитические 321—323, 329, 502, 725
— регуляторные 327—329, 502, 725
Сыворотка кровн
белки 176, 724 и ел.
— гель-электрофорез 175—177
— диск-электрофорез 183 и ел., 742 и
ел,
— иммуноэлектрофорез 180 и ел.
электрофорез 176, 742 и ел.
концентрация аспартатаминотрансфе-
разы 289
объем 232
Сывороточный альбумин
гидродинамические свойства 208—211»
247, 736
комплексообразованне с аспирином 56
и ел.
определение 127
Сфера
коэффициент трения вращательный
722
трансляционный 205, 722
Тейлора ряд 789
применение 146, 158, 174, 227, 483, 487
Теоретическая тарелка 241—245
Теория
действия см.. Лекарственные
препараты
занятости см. Лекарственные
препараты
переходного состояния 112—117
скорости 350 н ел.
столкновений 109—112 550 и ел.
Теплоемкость 105—108
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 811
Теплота
и работа 12—14
образования 21
фазового перехода 105—108
химической реакции 19—25
Термодинамика, законы
первый 13
второй 13—19
Термотаксис 745
Термохимия 19—26
Тизелиуса аппарат 172 н ел.
Тимиднлатсиитетаза, ннгибирование 316
и ел.
Тирозин 404, 406 н ел.
Томография
груди (жеищнны) 458—460, 783
магнитная резонансная 784—787
определение воды в тканях 784
рентгеновская 779—784
ультразвуковая 458—460
L. -Треониндезаминаза, аллостерическое
ннгибирование 327—329
Тригонометрические функции 790
Трипсин
рентгеноструктурный анализ 767—769
специфичность действия 290 и ел.
Трипсина ингибитор
панкреатический 76—78, 767 и ел.
соевый 247, 769
Триптофан 404—407
Тройная точка 35
Тропомиозин, гидродинамические
свойства 166, 208—210
Тяжелого атома метод 473
в рентгеноструктуриом' анализе 761—
769
— электронной микроскопии 498—508
Углеводы, определение конформацин па
данным ЯМР 602^604
Углекислый газ, влияние на
дыхательную функцию 528—532
Угловой момент
орбитальный 573, 588—590'
спиновый 573, 590—592
Удельное вращение 417
Ультразвуковая микроскопия 456 и ел.
Ультрафильтрация см. Дналнз
Урацил 409
Уреаза, самоторможение субстратом 330
Усреднение сигнала 238—240, 670—679,
685 и ел.
Ухтерлони метод 163 и ел.
Фазовая диаграмма 35, 36, 38
Фазовый переход 34—41
Фазовый угол 366, 375 и ел., 754 и ел.
Фармакокинетика
введение и вывод лекарственных
препаратов 343—346
дифференцированный отклик, теория
занятости 347—350
скорости 350—354
логарифм дозы и отклик, взаимосвязь
349 и ел., 354
отклик «все-или-ничего» 354—357
последовательные процессы 343—346
пробит 355—357
Фелжета выигрыш 672
Фенобарбитал 356 н ел.
Ферментативный катализ
модель Михаэлиса—Ментен 284—289
при наличии двух субстратов 295—299
нескольких промежуточных
соединений 295 и ел.
обратной реакции 293 и ел.
эффективность 291
-Ферментная диагностика 289
Ферменты
ннгибирование см. Ннгибирование
катализ см. Ферментативный катализ
определение в сыворотке крови 289
и ел.
регуляция действия см. Активация
ферментов; Аллостерня; Ингнбиро-
рование
Фибриноген
в плазме кровн 176
гидродинамические свойства 166,
208—211
Физические постоянные, таблица 795
Физостигмин 306
Фика закон 156
Флуктуации ч
заселенности состояний 711—714
концентрационные 729—735
напряжения 236 и ел.
по направлениям 716—729
при измерении радиоактивности 225—
231
спектральная плотность 708—711,
714—716
фликкер-шум 237 и ел.
электрического тока 234—236
энергетический спектр 703—708
Флуоресценция
анизотропия 532—535, 726—729
перенос энергии 659—662
поляризация 537—539
Форма и размер макромолекул
определение по вязкости 207—212
гель-хроматографии 245—250
деполяризации флуоресценции
535—539, 726—729
диэлектрической релаксации
446-^449, 723 :
812 ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
коэффициенту диффузии 205—
207, 208, 722 и ел.
магнитной релаксации 547—549,
724—726
радиусу вращения 483—492
рэлеевскому светорассеянию
733, 735—742
седиментации 188—198
электрофорезу 178 и ел.
Фосфатаза щелочная 289 н ел.
Фосфолипиды 36—41,.439
Фототаксис 745
5-Фторурацил 252, 317
Фурье-аналнз беспорядочных процессов
см. Корреляционная функция;
Флуктуации; Энергетический спектр
в инфракрасной спектроскопии 697—
700
— спектроскопии ионного
циклотронного резонанса 692—697
дифракционного изображения 751—
753
и спектроскопия ядерного магнитного
резонанса 689—692
относительных интенсивностей 759—
769
Фурье-синтез 753—758
фрагмент Fab 489—492, 728 н ел., 768
фрагмент Fc 490
Хадамарда метод
маска 677 и ел., 700
преобразование 671 и ел., 676—679
Хемотаксис 745
Хендерсона—Хассельбалха уравнение 82
Хилла зависимость 93—95
Химические реакции
кинетические схемы 261—263, 281—
283, 521 и ел.
колебательные 334—340
i обратимость, полярографический
критерий 218 н ел.
порядка первого 261—263
— второго 518 и ел., 737—740
— псевдопервого 519
последовательные 282 и ел.
релаксационная кинетика 512—526
самопроизвольные 97—103
температурная зависимость константы
ранновесия 103—105
скорости 108—117
теплота 19—25
цепные свободно-радикальные 112
эндотермические 20
Химические связи
двойные 605 н ел.
прочность 21—23, 557
сопряженные 604
угол между ними 601—604
энергия 21—23, 101—103
энтальпия 21—23
Химический обмен
быстрый 431—434
корреляционная функция 711—716
определение константы скорости по
переносу насыщения 657—659
уравнение 462 и ел.
• Химический сдвиг
индуцированный лантаноидом 631—
635
таблица 691
Химотрипсин
конформация 422
механизм действия 291
радиус вращения 487
рентгеноструктурный анализ 767 и ел.
специфичность 290
число превращения 292
Хнмотрипсиноген
гидродинамические свойства 166, 178,
194, 247
конформация 422
молекулярная масса 194
рентгеноструктурный анализ 767
Хиральность 415, 419
Холестерин 429
Хроматография 241—258
аффинная 255—258
газожидкостная 241—245
гель-проникающая 245—250
ионообменная 250—255
общие принципы 241—245
разрешение 260
Хюккеля теория молекулярных орбита-
лей 604—616
Цвнттер-нон 96
Целлобиознлгндролаза 191
Целлюлоза 602
Центросимметричные структуры,
определение фазового состава 753—755
Центрифугирование см. Седиментация
Циклическая перестановка 678 и ел.
Циклотрон 692—697
Цимма кривые 484 и ел.
Цинк
замещение на кадмий в карбоангидра-
зе 541—544
полярографическое определение 219
Цирроз 289
Цитохром с
восстановление 130 и ел., 658 и ел.
конформация 422
рентгеноструктурный анализ 768
Частота
возбуждающая 375, 466
круговая 367, 370 и ел.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 813
магнитного резонанса 427
магнитной прецессии 425
перехода через энергетический барьер
резонансная 441
собственная 374, 466
Число превращения 291 н ел.
Ширина полосы
пропускания частот 235
спектральная 374, 396, 672—674, 679—
683
Шлирен-метод 401 н ел.
Шредингера уравнение 580, 638 и ел.
Шум
детектора 671
источника 671
отношение к сигналу 239 н ел., 670,
685
устранение
— многоканальными мультиплексными
методами 672—686
— модуляцией 238
— усреднением сигнала 238—240
электрический
— импульсный 234—236, 671
— сопротивления (Джонсона,
Найквиста, тепловой) 236 н ел.
— фликкер 237 и ел.
энергетический спектр 703—711
Щитовидная железа, диагностика
гиперфункции 233
Эди зависимость 313, 315
Эйнштейна уравнение вязкости 207
Электрическая цепь
закон Ома 440
модель системы регуляции дыхания
531
сравнение с грузом на пружине 440
и ел.
элементы 440 и ел.
Электрическое поле
градиент 618
компонента электромагнитного
излучения' 367 и ел.
наведение двулучепреломлення 525
и ел.
— дипольного момента 442, 467, 474,
559
ориентация молекул 443—449
перемещение заряженных молекул см.
Электрофорез
Электрод
ионоселективный 124—126
капельный ртутный 216—218
поляризация 213
стандартный водородный 129 и ел.
стеклянный, измерение рН 124 и ел.
Электромагнитное излучение
дисперсия см. Дисперсия
когерентное и некогерентное 368 и ел.,
648, 679 и ел.
плоскополярнзованное 368—370, 415—
424, 460 и ел., 466—469, 525 и ел.,
532—539, 726—729, 740—742
поглощение см. Поглощение
поляризация по кругу и по эллипсу
370, 415—424, 460 и ел.
рассеяние 373, 378—380, 384—387. См.
Рассеяние
характеристики 366—368
Электронная микроскопия 492—508
глубина резкости 496
контраст 496—502
метод замораживания — скалывания
. 502—506
— оттеиения 502—506
окрашивание 498—502
разрешение 492—495
сканирующая 506—508
сравнение с оптической 495
темнопольная 506 и ел.
Электронная плотность 615 и ел., 755—
758
Электронный парамагнитный резонанс
434—439
Электрофорез 169—187. См.
Гель-электрофорез; Диск-электрофорез
изотахофорез 185—187
изоэлектрическая фокусировка 180
иммуио- 180 и ел.
и светорассеяние 742—745
разрешение 182
с подвижной границей 172 и ел.
Электрофоретическая подвижность 171
и ел.
связь с молекулярной массой 178
Электрохимический элемент
концентрационный 121—127
топливный 127—137
Эллипсоид вытянутый и сплющенный
204—206
Эллиптичность 417, 461
Энергетический спектр 686, 703—708
Энергия
активации 109, 115, 119
кинетическая 170, 203, 368, 573, 583
перенос 659—662
потенциальная 573, 580, 583—588
— поверхность 113
свободная Гельмгольца 16—19
— Гиббса 16, 19, 30—33
и самопроизвольность химиче-
814 ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
ских процессов 97—105
определение 16 и ел.
парциальная моляльная 30 и ел.
связь с константой равновесия
64, 97, 100
стандартного состояния 99, 101
и ел.
уровни 533, 539—541, 585, 590, 593,
594, 599, 605, 609, 611 н ел., 619,
637, 646 и ел.
— вырожденные 612
Энтальпия
денатурации химотрипсиногена 118
критерий равновесия 18
определение 16 и ел.
стандартная 20 и ел.
фазового перехода 105—108
фермент-субстратного взаимодействия
116 и ел.
Энтропия
и степень неупорядоченности 14—16
как функция состояния 13 и ел., 17
критерий равновесия 18
Эритроциты, время жизни 234
Этанол, фармакокинетика 343—346
Этилен, молекулярные орбитали 607—
610
Эффект
всаливания 68—70
высаливания 70—73
Эффектор аллостернческий 327—329
Ядерный магнитный резонанс
константа спнн-спииового
взаимодействия 599 н ел., 601—604, 623
связь с двугранным углом
601—604
поглощение и дисперсия 424—429
правила отбора 662—667
преобразование Фурье 689—692
расчет спектров (типа А, АА, АХ,
АМХ) 592—601, 623
релаксация см. Релаксация
свойства магнитных изотопов 427
13С 427, 429, 547 и ел., 691
— время релаксации, в фосфолнпндах
547
— преобразование Фурье 689—692
— спектр холестерина 429
— химические сдвиги 691
Ячейка элементарная 759—761 „
СОДЕРЖАНИЕ
Том 1
Предисловие редакторов перевода ... . 5
Предисловие . . ■ 7
ЧАСТЬ I. ТЕРМОДИНАМИКА В БИОЛОГИИ . . 9
Глава 1. Работа, тепло и энергия П
Литература .... . 26
Глава 2."Химический потенциал ... . ; . . 27
2.А. Фазы и фазовые переходы 33
2.А.1. Сколько существует фаз? (Правило фаз Гиббса) 33
2.А.2. Фазовые переходы в биологических мембранах . . 35
2.Б. Полупроницаемые мембраны и нейтральные молекулы в
растворе: осмотическое давление 42
2.В. Полупроницаемые мембраны н заряженные молекулы в
растворе: равновесие Доннана . 51
Литература . .59
Глава 3. Химические реакции и константы равновесия . .60
З.А. Константы равновесия .... 60
З.А.1. Активность: термодинамическая концентрация . . 63
З.Б. Растворимость макромолекул 68
З.Б.1. Всаливание и высаливание 68
З.Б.2. Гидрофобность: нековалентная ассоциация пеполяр-
кых молекул 73
З.В. Связывание небольших молекул или ионов с
макромолекулами . 80
Литература 96
Глава 4. Самопроизвольное протекание химических реакций:
температурная зависимость констант равновесия и скоростей реакций . • 97
4.А. Критерии самопроизвольности химической реакции ... 98
4.Б. Изменения констант равновесия с температурой . . . 103
4.В. Температурная зависимость индивидуальных констант
скоростей реакций . . . . 108
4.8.1. Теория столкновений и константы скорости реакций 109
4.8.2. Теория переходного состояния н скорости
химических реакций ..... 112
Литература ... . . 119
Глава 5. Электрохимический потенциал . 121
5.4. Концентрационные элементы . , 121
5.Б. Топливные элементы 127
5.Б.1. Аналитические применения: AGrx, AHrx, А&* и Ка
из электрохимических измерений 129
5.Б.2. Препаративные применения: электроосаждениё и
устройство аккумуляторов , 133
Литература 139
816 СОДЕРЖАНИЕ i
ЧАСТЬ 2. УСПЕХИ И НЕУДАЧИ 140
Глава 6. Задача о случайных блужданиях . 140
6.А. Равные шансы: перемещение на расстояние . . . 147
6.Б. Трансляционная днффузия ь ... 156
Литература . . . . .168
Глава 7. Вынужденное передвижение ., . . . . , 169
7.А. Электрофорез 169
7.Б. Седиментация 187
7.Б.1. Скорость седиментации , 188
7.Б.2. Седиментационное равновесие 192
7.Б.З. Седиментация в градиенте плотности . . . . 195
73. Вязкость . . . 198
7.8.1. Поток жидкости в капилляре 200
7.8.2. Вязкость, коэффициент трення .и форма
макромолекул в растворе , 203
7.Г. Вынужденное передвижение. Полярография . . . . 212
Литература 224
Глава 8. Низкая вероятность: распределение Пуассона . ... 225
8.А. Измерение радиоактивности 225
8.Б. Электрический шум 234
8.Б.1. Импульсный шум i 234
8.Б.2. Шум сопротивления (шум Джонсона, шум Найкви-
ста, тепловой шум) 236
8.Б.З. (1//>Шум (Фликкер-шум) 237
8.Б.4. Устранение шума: усреднение сигнала . 238
8.В. Помогло ли лечение пациентам? 240
8.Г. Хроматография 241
Литература . . . . 260
ЧАСТЬ 3. РОСТ И РАСПАД . . . . . . 261
Глава 9. Кинетические процессы первого порядка . . . 261
Литература . . . . 277
Глава НО. Катализаторы . 278
10.А. Кинетика (стационарная) Михаэлиса—'Ментен .... 278
10.А.1. Необходимость гипотезы о стационарности . . . 278
10.А.2. Одностадийная однокомпонентная прямая реакция 280
10.А.З. Прямая и обратная реакции 281
10.А.4. Последовательные реакции 282
'10.А.5. Некатализируемая реакция с одним
промежуточным соединением 283
10.А.6. Каталитическая реакция с одним промежуточным
соединением 284
10.Б. Снятие ограничений Михаэлиса — Ментен относительно
ферментативных реакций 293
10.Б.1. Существование обратной реакции ... . 293
10.Б.2. Реакции с числом промежуточных соединений
больше одного 295
817 СОДЕРЖАНИЕ I
10.Б.З. Два субстрата: почему односубсхратная схема
вообще удовлетворяет экспериментальным данным? 296
Литература 300
Глава 11. Регуляция скоростей ферментативных реакций 30L
ПА. Лекарственные вещества, яды и гормоны: типы инги-
бирования и активации ферментов . . ЗОГ
II1.А. 1. Конкурентное ингибирование . '. . . . . 304
11.А.2. Неконкурентное ингибирование 308
П.А.З. Смешанное ингибирование . 310
11.Б. Другие типы регуляции ферментов 31&
П.Б.1. рН-Регуляция скоростей ферментативных реакций 318-
11.Б.2. Почему у ферментов есть субъединицы? ... 32]
11.Б.З. Самоторможение субстратом 330'
11.Б.4. Активация ионами металлов 332'
111.В. Биологические часы? Причуда природы или явление,
обусловленное химическими процессами? 334-
Литература . . 34t
Глава 12. Фармакокинетика: кинетика химических реакций с
переименованными переменными ... ^. ....... . 34&
12.А. Продолжительность действия лекарственных веществ:
введение и вывод лекарственных препаратов 343-
12.Б. Теория действия лекарственных препаратов . 347
12.Б.1. Дифференцированный отклик: теория занятости и
теория скорости 347
12.Б.2. Отклик типа «все-или-ничего»: сон и смерть . . 354
Литература . . . .... 368
Том 2
ЧАСТЬ 4. ГРУЗ НА ПРУЖИНЕ . . . . 365
Глава 13. Колебания груза, подвешенного на пружине, под действием
возбуждающих н тормозящих сил — одна из наиболее важных
физических проблем . 365
13.А. Терминология волнового движения 366
13.Б. Груз па пружине под действием возбуждающих и
тормозящих сил: вынужденные колебания 374
13.Б.1. Пределы рассеяния: незначительное торможение
и/или возбуждающая частота далека от резонанса 378
13.Б.2. Предел Лоренца:- возбуждающая частота близка
к собственной . 381
13.В. Груз на пружине под действием тормозящих сил:
затухающие колебания 388
13.Г. Груз с нулевой массой на пружине при наличии
тормозящих сил: явления релаксации 389
Литература . . . . ... 395
Глава 14. Поглощение и дисперсия: вынужденные колебания груза на
пружине под действием возбуждающих и тормозящих сил . . .396
Ц4.А. Поглощение и показатель преломления: основы
спектроскопии и микроскопии 397
€18 СОДЕРЖАНИЕ
14.Б. Дихроизм и двулучепреломлеиие: выявление линейного
порядка в молекулярных структурах 410
14.В. Круговой дихроизм и оптическое вращение: оптическая
активность и «хиральность» молекул .• 415
14.Г. Поглощение и дисперсия при магнитном резонансе:
ядерный «мячик» на «веревочке»; использование спиновых
меток для определения подвижности молекулярных
структур; селективный рН/метр 424
14.Д. Моделирование колебаний пружины с помощью
электронных цепей: емкость, сопротивление, индуктивность,
резонанс и релаксация 440
14.Е. Диэлектрическая релаксация: груз с нулевой массой на
пружине. Вращательная диффузия макромолекул . . . 443
14.Ж. Ультразвуковое поглощение и дисперсия скорости: вновь
груз с нулевой массой, иа пружине, — новый метод
медицинской диагностики и лечения 450
Литература .... ...» 465
Глава 15. Рассеяние: вынужденные колебания груза на пружине в
отсутствие сил торможения- ..-.'.« . ^ . . . 466
15.А. Малые объекты, большие волны, линзы не помогают: ре-
леевское рассеяние света; мутность, радиус вращения,
соотношение Цимма: размер, форма и молекулярная масса
макромолекул в растворе
15.Б. Крупные объекты, небольшие волны, линзы отсутствуют:
рассеяние рентгеновских лучей; детальное определение
формы молекул 486
15.В. Большие объекты, небольшие волны, линзы можно
использовать: рассеяние электронов и различные электронные
микроскопы 492
Литература . .510
Глава 16. Нестационарные процессы. Первоначальный отклик внезапно
смещенного груза на пружине 511
16.А. Груз с нулевой массой при наличии сил торможения.
Скорость возвращения к равновесию 512
16.А.1. Релаксационная кинетика быстрых химических
реакций 512
16.А.2. Кибернетика. Модели «черного ящика» в
физиологии 528
16.Б. Затухающие, колебания груза с конечной массой на
пружине. Звучание колокола 532
16.БЛ. Деполяризация флуоресценции. Направленное
зондирование участков макромолекул 532
16.Б.2. Корреляция испускания у-лучей по направлениям:
тот же эксперимент для непрозрачной среды . . 539
16.Б.З. Магнитная релаксация. Молекулярно-спектроскопи-
ческая «линейка» .... * 544
.Литература 552
Глава 17. Связанные пружины 554
17.А. Константы взаимодействия и спектральные расщепления 554
17.Б. Непосредственно соединенные пружины. Нормальные
колебания 556
819 СОДЕРЖАНИЕ
17.В. Амплитудная модуляция. Спектроскопия комбинационного
рассеяния 659
Литература . 664
ЧАСТЬ б. КВАНТОВАЯ МЕХАНИКА: КОГДА ОНА НА
САМОМ ДЕЛЕ НЕОБХОДИМА? 565
Глава 18. Обобщенная геометрия: существование и положение
спектральных «линий» поглощения энергии. Квантовомеханический груз
иа пружиие .....»»,.„ 566
18.А. Спиновые задачи. Простейшие квантовомеханические
расчеты 588
18.Б. Теория молекулярных орбиталей и активность лекарств 604
18.В. Биологическое железо. Мёссбауэровская спектроскопия 617
Литература . 624
Глава 19. Бросание монеты: «орел или решка»? распределение Больцмана 625
19.А. Лантаноидные сдвигающие реагенты в спектроскопии
ЯМР и конформация молекул в растворе. ...... . 631
19.Б. Переходы между энергетическими уровнями .... 635
19.БЛ. Общие формулы 635
19.Б.2. Инверсия заселенности. Лазеры и их приложения 644
19.Б.З. Явления насыщения и спектральные времена
релаксации 654
19.Б.4. Интенсивности линий в спектрах-поглощения . . 662
Литература ..... 667
ЧАСТЬ 6. ПРЕВРАЩЕНИЕ ТРУДНЫХ ЗАДАЧ В ПРОСТЫЕ.
ПРЕОБРАЗОВАНИЯ И НАГЛЯДНЫЕ ПРИМЕРЫ ИХ
ПРИМЕНЕНИИ 668
Глава 20. Грузы на весах: взвешивание в спектроскопии. Многоканальные
и мультиплексные методы 669
20.А. Спектрометры с кодированием и декодированием
(«мультиплексные»), использующие преобразование Хадамарда 676
20.Б. Фурье-спектроскопия 681
Литература ч 702
Глава 21. Фурье-анализ беспорядочного движения. Автокорреляция и
спектральная плотность ~ . 703
21 Л. Беспорядочные перескоки между двумя состояниями с
различными «собственными» частотами . ... 711
21.Б. Беспорядочное вращательное движение. Определение
коэффициента вращательной диффузии методами
диэлектрической релаксации, магнитного резонанса и деполяризации
флуоресценции . . 716
21.В. Беспорядочное поступательное движение. Трансляционная
диффузия • 729
21.В. 1. Рассеяние света. Коэффициенты трансляционной
диффузии для макромолекул (или бактерий) в рас-
! творе 729
21.В.2. Светорассеяние при электрофорезе. Электрофорез
без границ 742
,;. Литература 747
820 СОДЕРЖАНИЕ J
Глава 22. Реконструкция объектов по изображениям 749
22.А. Рентгеновская кристаллография. Определение углеродного
остова макромолекулы 749
' 22.А.1. Дифракционная картина. Линза как прибор для
преобразования Фурье 760
22.А.2. Фурье-синтез (графический) 753
22.А.З. Определение положения атомов в элементарной
ячейке. Синтез Паттерсона 759
22.А.4. Определение структуры больших молекул.
Решения проблемы фаз 761
22.Б. Дифракция нейтронов. Определение положений атомов
водорода. Водородные связи 773
22.В. Реконструкция изображения по проекциям 779
22.В.1.Рентгеновская томография 779
22.В.2. Магнитная резонансная томография.
Реконструкция изображений для медицинских целей . . . 784
Литература - . , 788
Приложение ♦ 789
Логарифмы 789
Разложения в ряды и приближения .... , 789
Тригонометрические функции . . , 790
Производные 791
Интегралы 791
Предметный указатель . * 800
Элаи Маршелл
БИОФИЗИЧЕСКАЯ ХИМИЯ
Научные редакторы Т. И. Почкаева,
Т. Т. Орловская
Мл. научн, ред. И. И. Землячева
Художник В. А. Медников
'Художественный редактор М. Н. Кузьмина
Технический редактор 3. И. Резник
Корректор А. Ф. Рыбальченко
ИБ № 2767
Сдано в набор 25.06.80.
Подписано к печати 23.01.81.
Формат 60X90/16.
Бумага типографская № 2.
Гарнитура латинская. Печать высокая.
Объем 14,50 бум. л. Усл. печ. л. 29.
Уч.-изд. л. 32,19. Изд. № 3/1253.
Тираж 8000 экз. Зак. 709. Цена 3 р. 50 к.
ИЗДАТЕЛЬСТВО «МИР»
Москва, 1-й Рижский пер., 2.
Московская типография № 11 Союэполиграфпрома
при Государственном комитете СССР по делам
издательств, полиграфии и книжной торговли.
Москва, 113105. Нагатинская ул., д. I.