/




































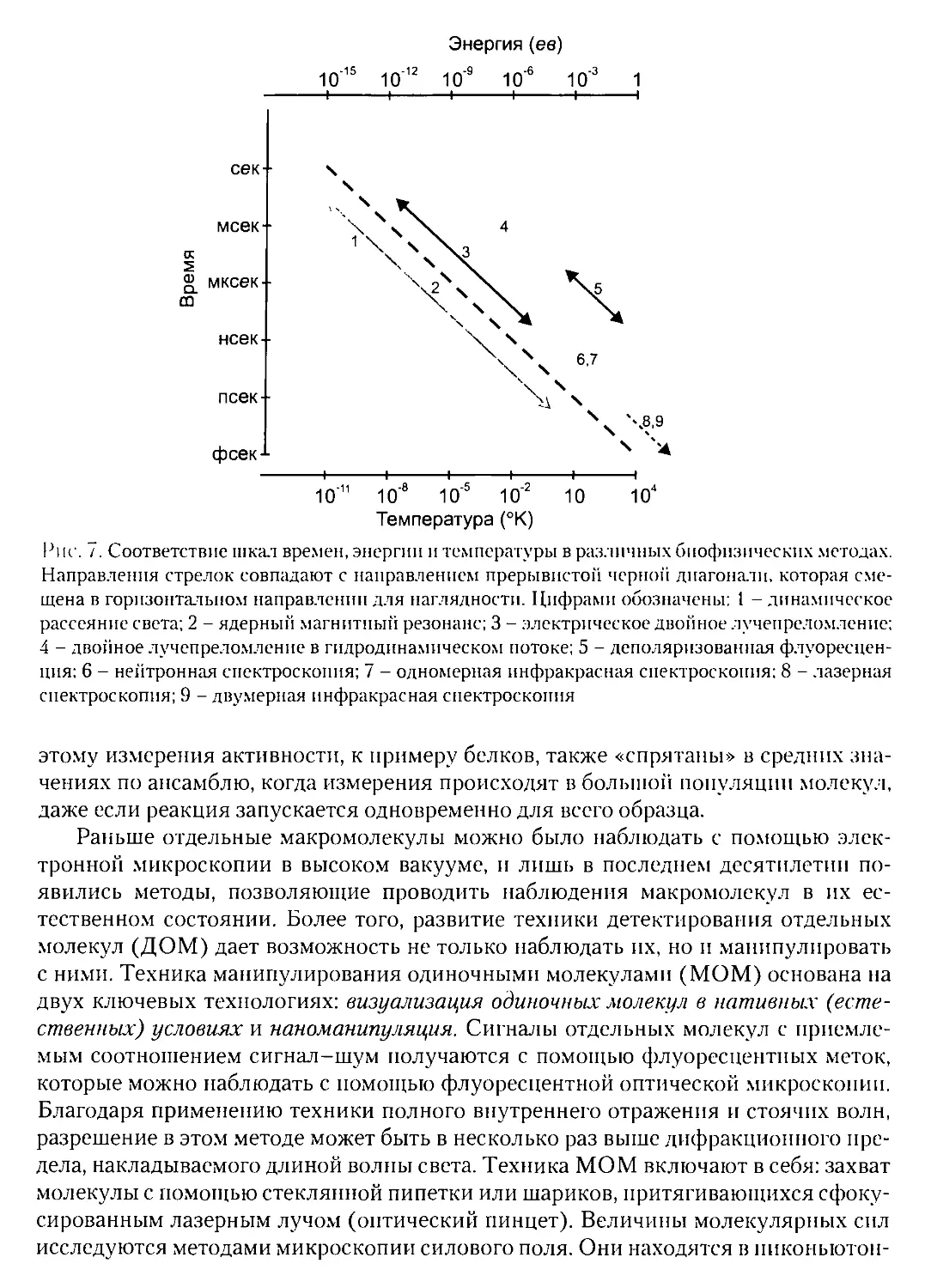





































































































































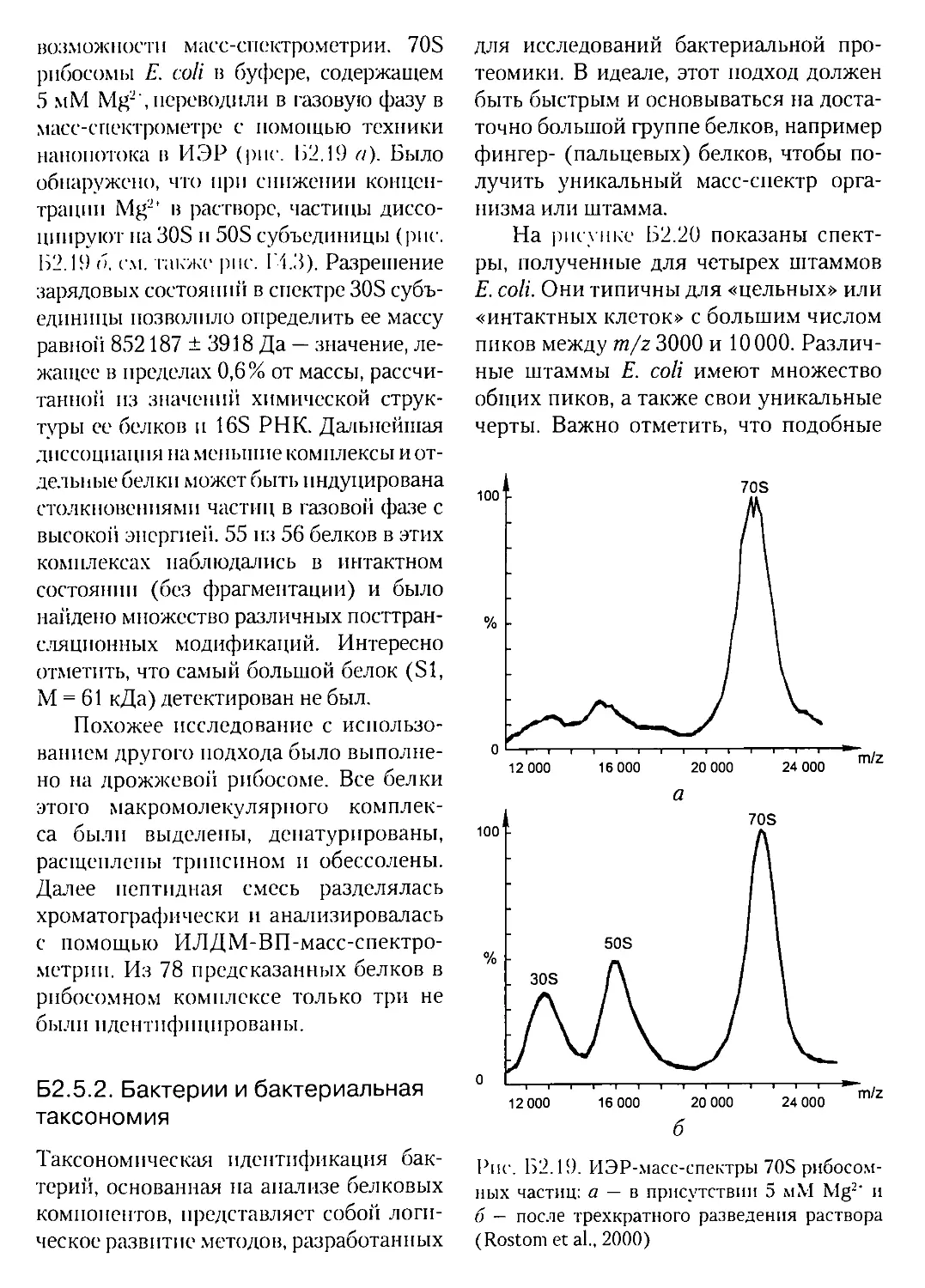


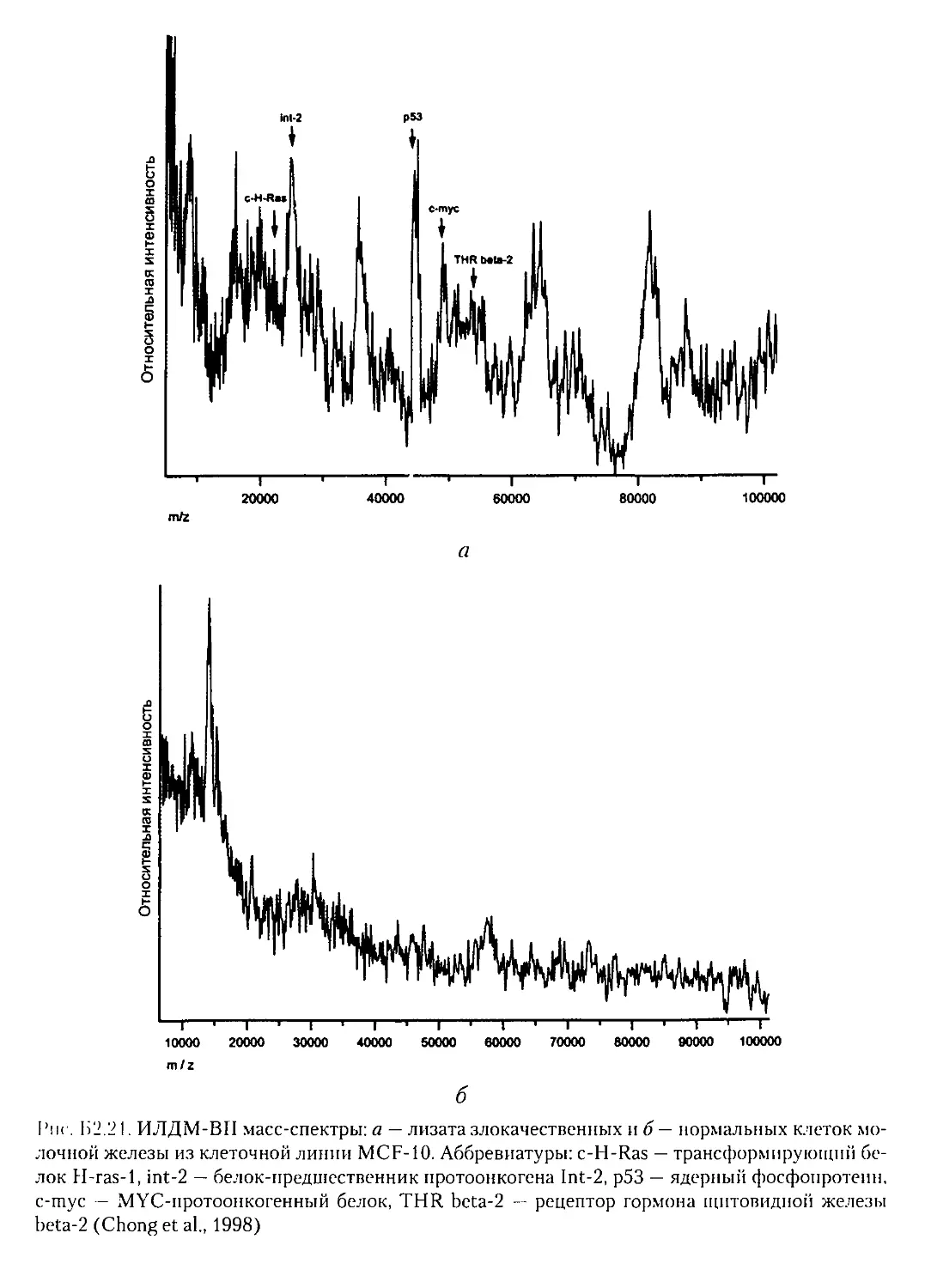










































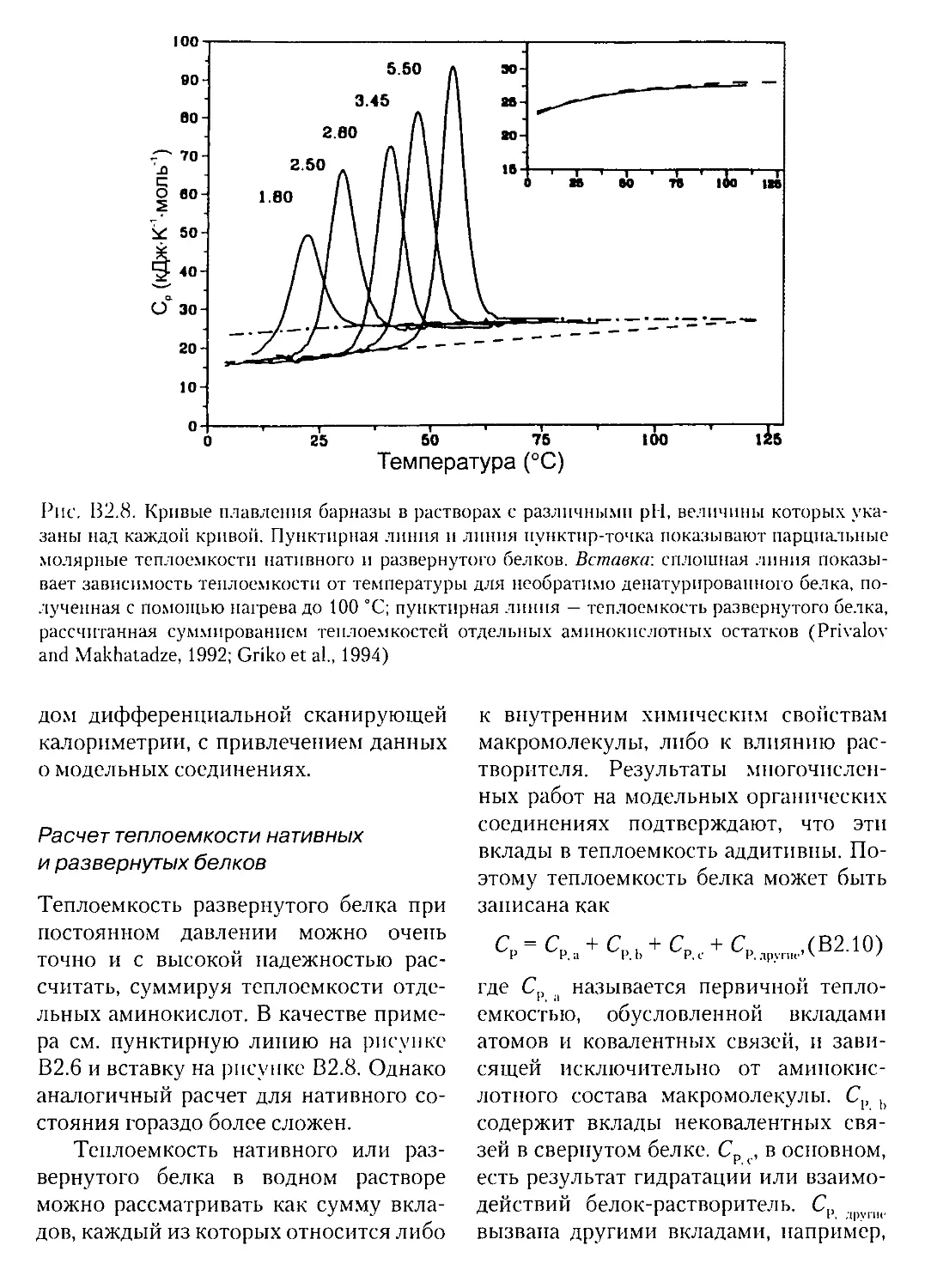















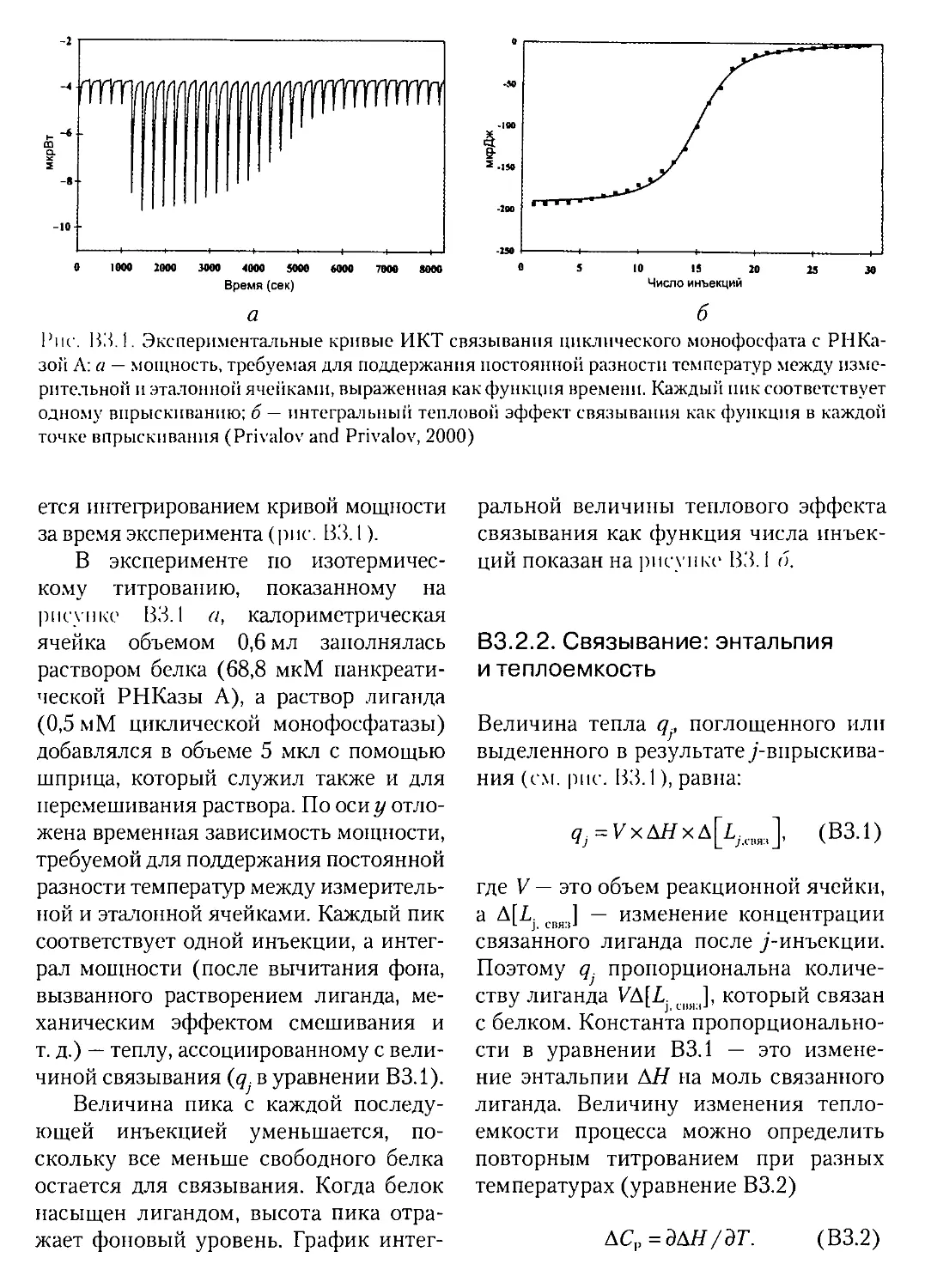







































































































































































































































































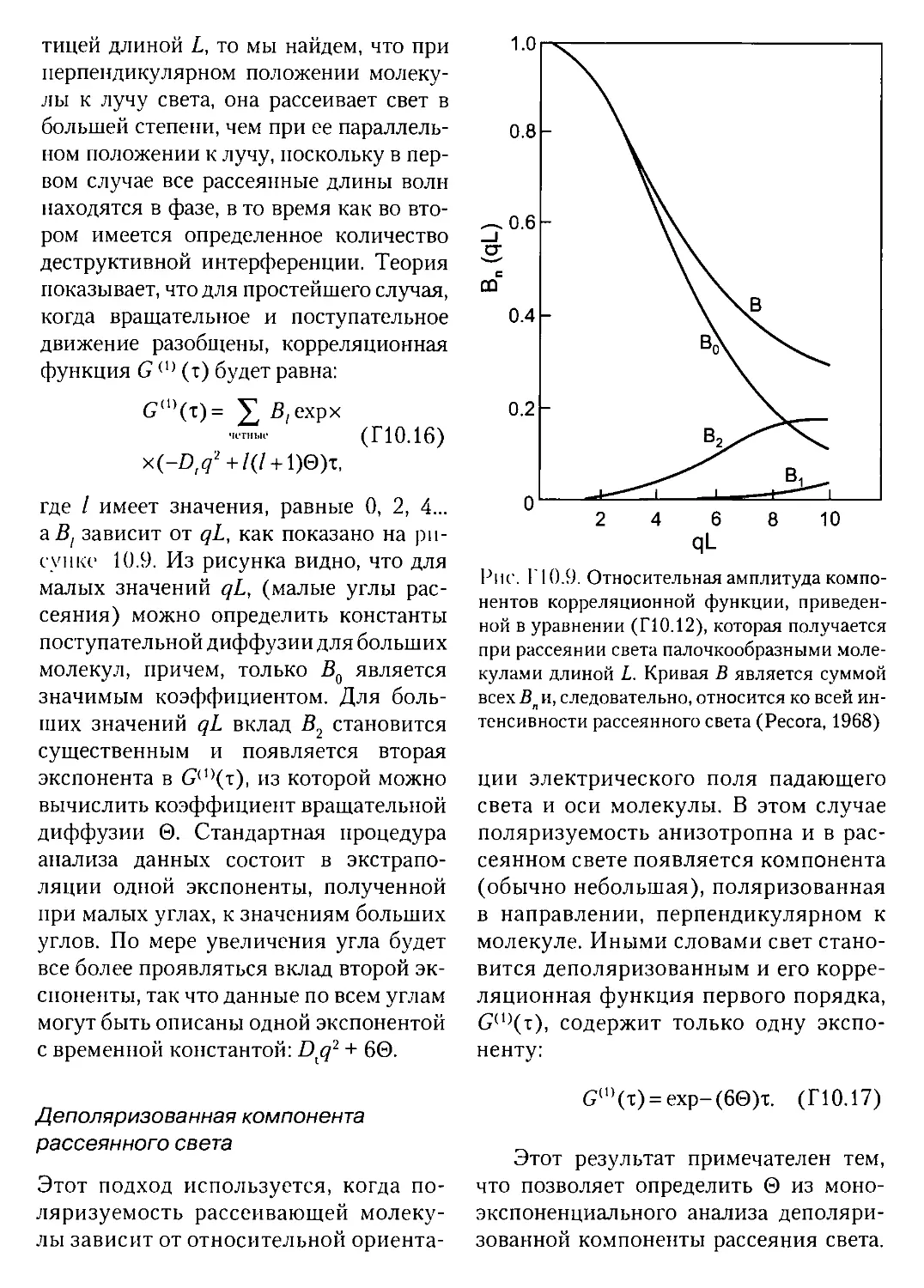



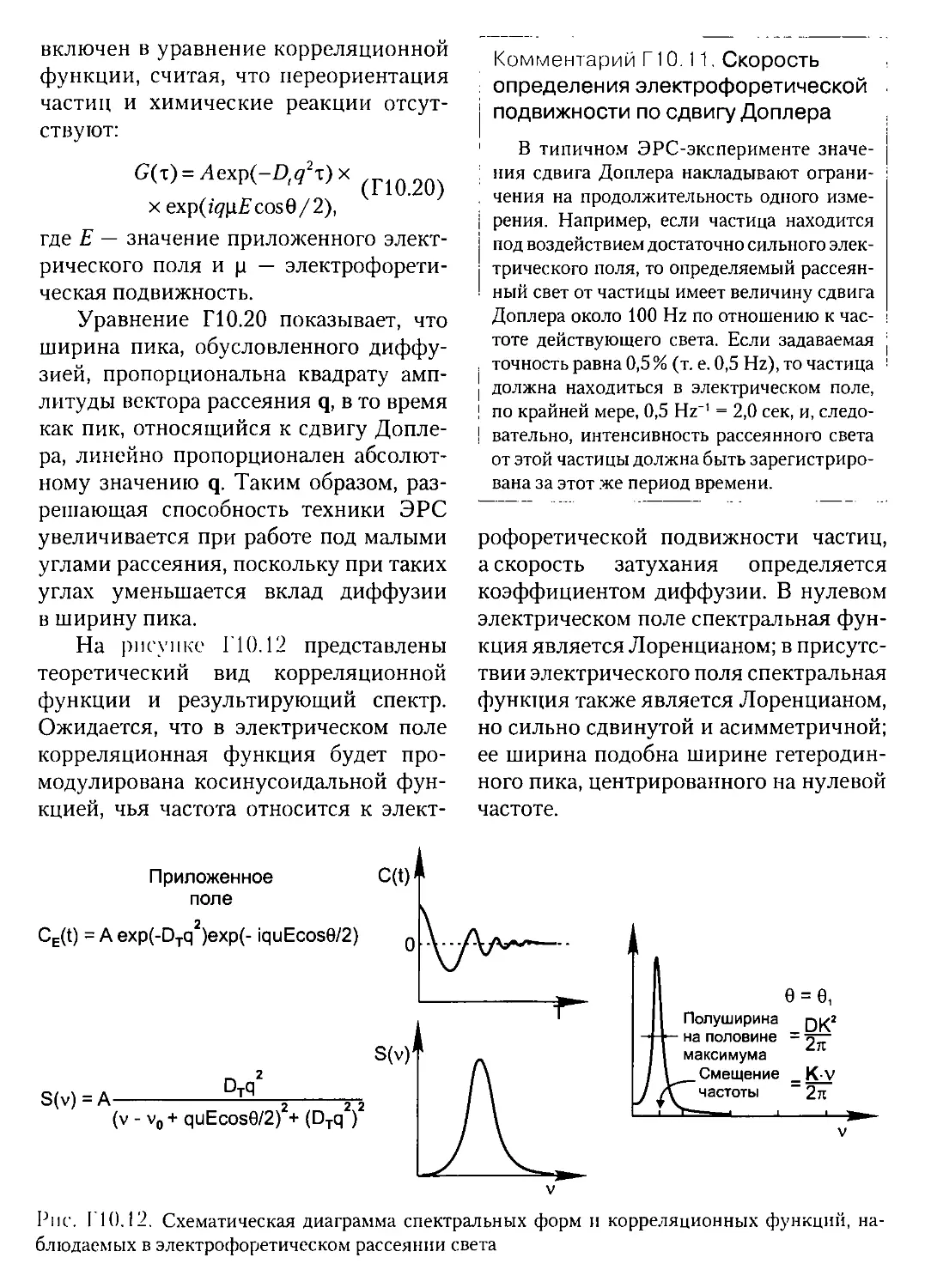






















Текст
И. Сердюк
Н. Заккаи
Дж. Заккаи
Методы
в молекулярной
биофизике
Структура. Функция. Динамика
Учебное пособие
Том I
СОДЕРЖАНИЕ
Предисловие .....................
ПРЕДИСЛОВИЕ РЕДАКТОРА РУССКОГО ПЕРЕВОДА
ОБ АВТОРАХ.......................... ............
ПРЕДИСЛОВИЕ ОТ Д. М. ЗИГЕЛЬМАНА
ПРЕДИСЛОВИЕ ОТ ПЬЕРА ЖОЛИО........
ОТ АВТОРОВ...............
Введение. Молекулярная биология в начале XXI века:
от ансамбля — к одиночным молекулам................24
КРАТКАЯ ИСТОРИЯ И ПЕРСПЕКТИВЫ......................24
ЯЗЫКИ И ИНСТРУМЕНТАЛЬНЫЕ СРЕДСТВА..................28
ШКАЛЫ ДЛИН И ВРЕМЕНИ В БИОЛОГИИ....................29
СТРУКТУРНО-ФУНКЦИОНАЛЬНЫЕ ГИПОТЕЗЫ.................31
КОМПЛЕМЕНТАРНОСТЬ ФИЗИЧЕСКИХ МЕТОДОВ
ИССЛЕДОВАНИЯ.......................................32
Термодинамика...................................32
Гидродинамика.............................-.....34
Рассеяние излучения.............................Зо
Спектроскопия...................................36
Детектирование одиночных молекул................$7
ЧАСТЬ А. БИОЛОГИЧЕСКИЕ МАКРОМОЛЕКУЛЫ
И ФИЗИЧЕСКИЕ ИНСТРУМЕНТЫ 41
Глава А1. Макромолекулы и их окружение 43
А1.1. ИСТОРИЧЕСКИЙ ОБЗОР...........................43
А1.2. РАСТВОРЫ МАКРОМОЛЕКУЛ........................44
А1.2.1. Концентрация.............................
А1.2.2. Парциальный объем...... *............ 4
A 1.2.3. Коллигативные свойства...........................46
А 1.2.4. Химический потенциал и активность................46
А 1.2.5. Температура......................................47
А 1.2.6. Осмотическое давление............................48
А1.2.7. Вириальные коэффициенты...........................50
А1.3. МАКРОМОЛЕКУЛЫ, ВОДА И СОЛИ.............................50
А1.3.1. Ионная сила и теория Дебая-Хюккеля................51
А1.3.2. Полиэлектролиты и эффект Доннана..................52
А 1.3.3. Взаимодействия между макромолекулами и растворителем.53
А 1.3.4. Вода, соль и гидрофобный эффект......................54
А1.4. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ............................... 58
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ............................61
Глава А2. Макромолекулы как физические частицы...................62
А2.1. ИСТОРИЧЕСКИЙ ОБЗОР..............................................62
А2.2. БИОЛОГИЧЕСКИЕ МОЛЕКУЛЫ И ПОТОК ГЕНЕТИЧЕСКОЙ
ИНФОРМАЦИИ............................................................64
А2.3. БЕЛКИ67
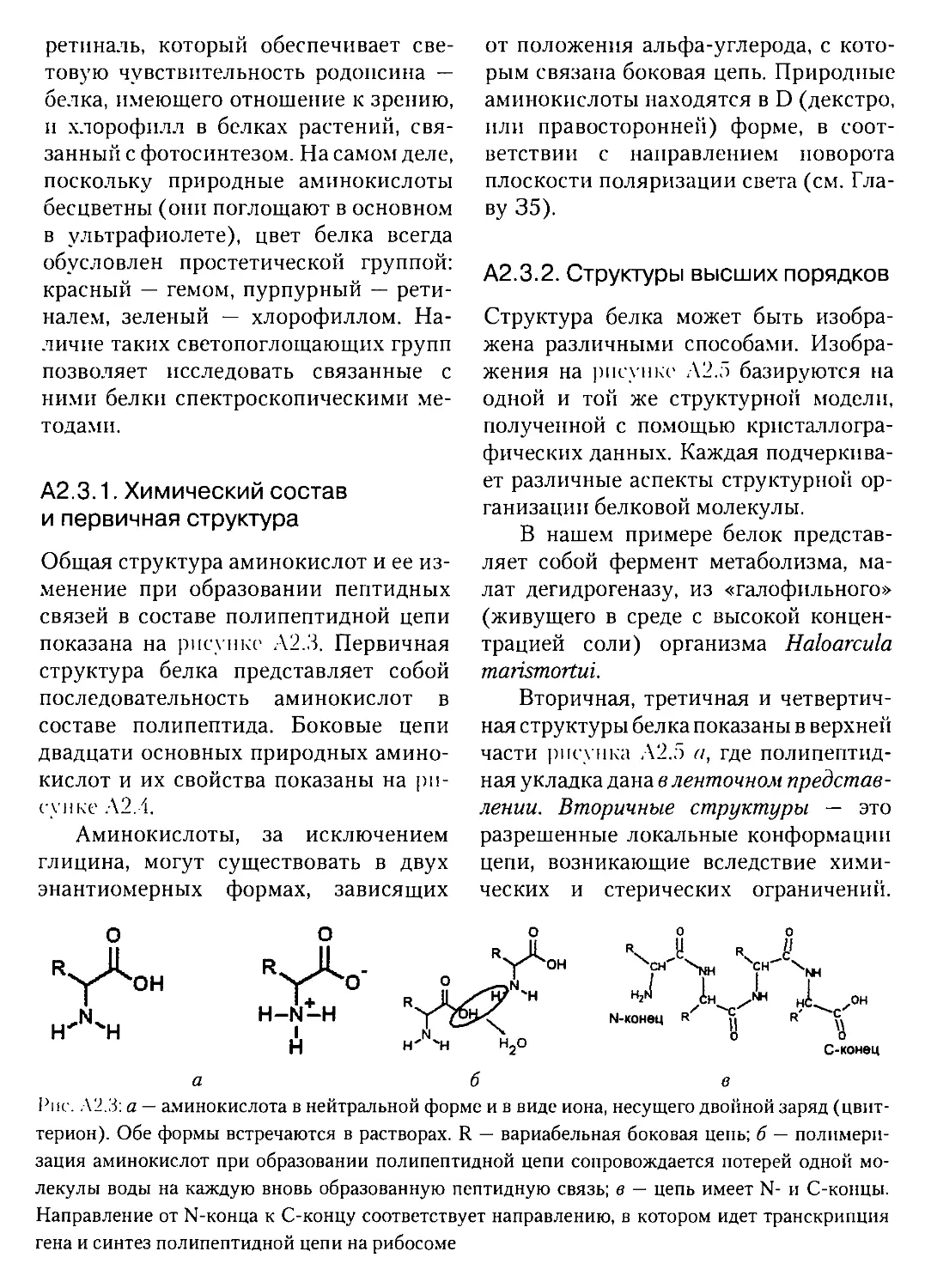
А2.3.1. Химический состав и первичная структура....................®8
А2.3.2. Структуры высших порядков..................................68
А2.4. НУКЛЕИНОВЫЕ КИСЛОТЫ.............................................74
А2.4.1. Химический состав и первичная структура нуклеиновых кислот.75
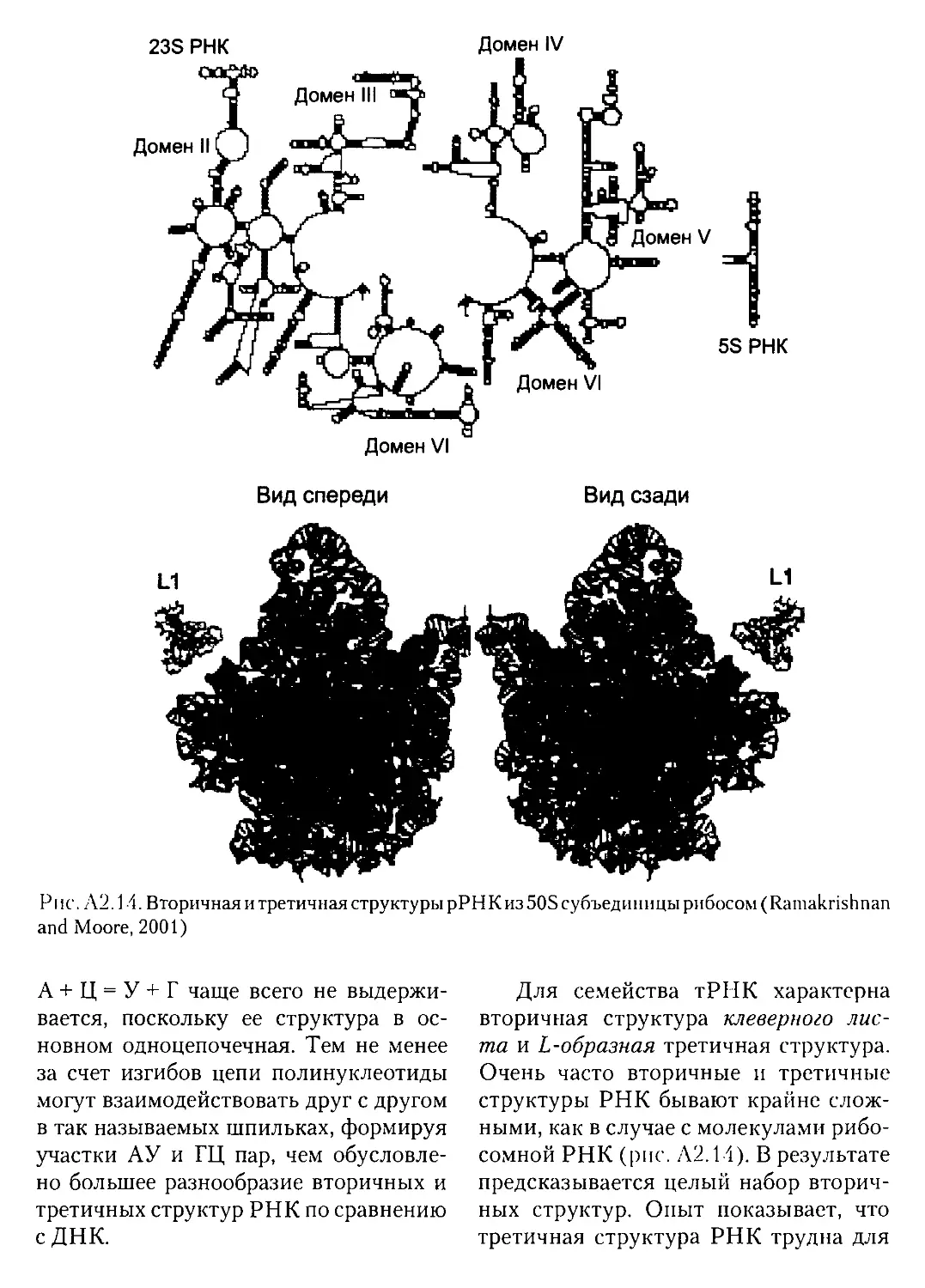
А2.4.2. Структуры высших порядков..................................76
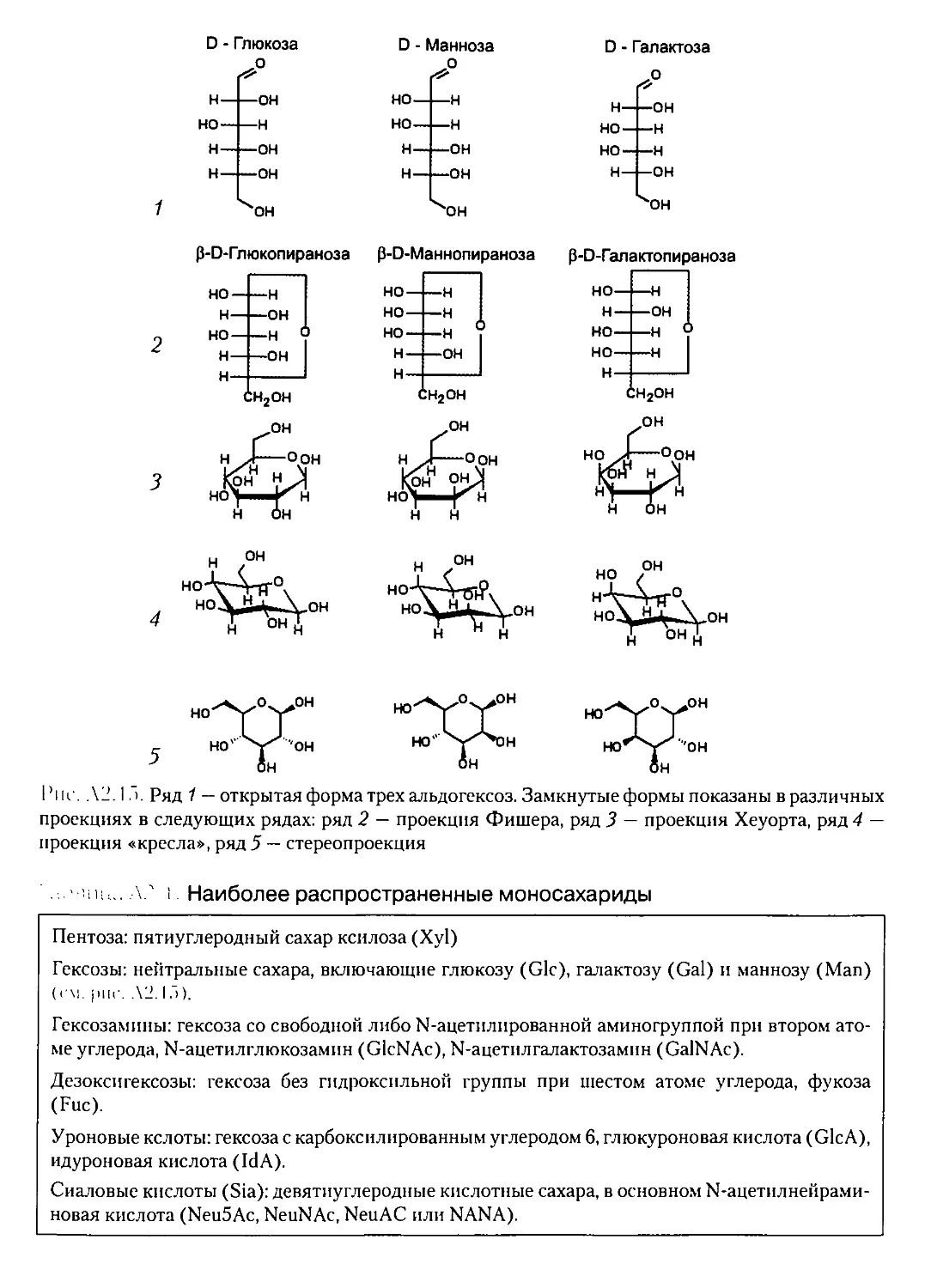
А2.5. УГЛЕВОДЫ........................................................79
А2.5.1. Химический состав и первичная структура углеводов..........80
А2.5.2. Структуры высших порядков..................................82
А2.6. ЛИПИДЫ..........................................................83
А2.6.1. Химический состав липидов..................................83
А2.6.2. Структуры высших порядков..................................84
А2.6.3. Липиды и мембранные белки..................................86
А2.7. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ..........................................86
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ.....................................89
Глава АЗ. Понимание макромолекулярной структуры.90
А3.1. ИСТОРИЧЕСКИЙ ОБЗОР.....................................90
А3.2. ОСНОВНЫЕ ФИЗИЧЕСКИЕ
И МАТЕМАТИЧЕСКИЕ ИНСТРУМЕНТЫ................................92
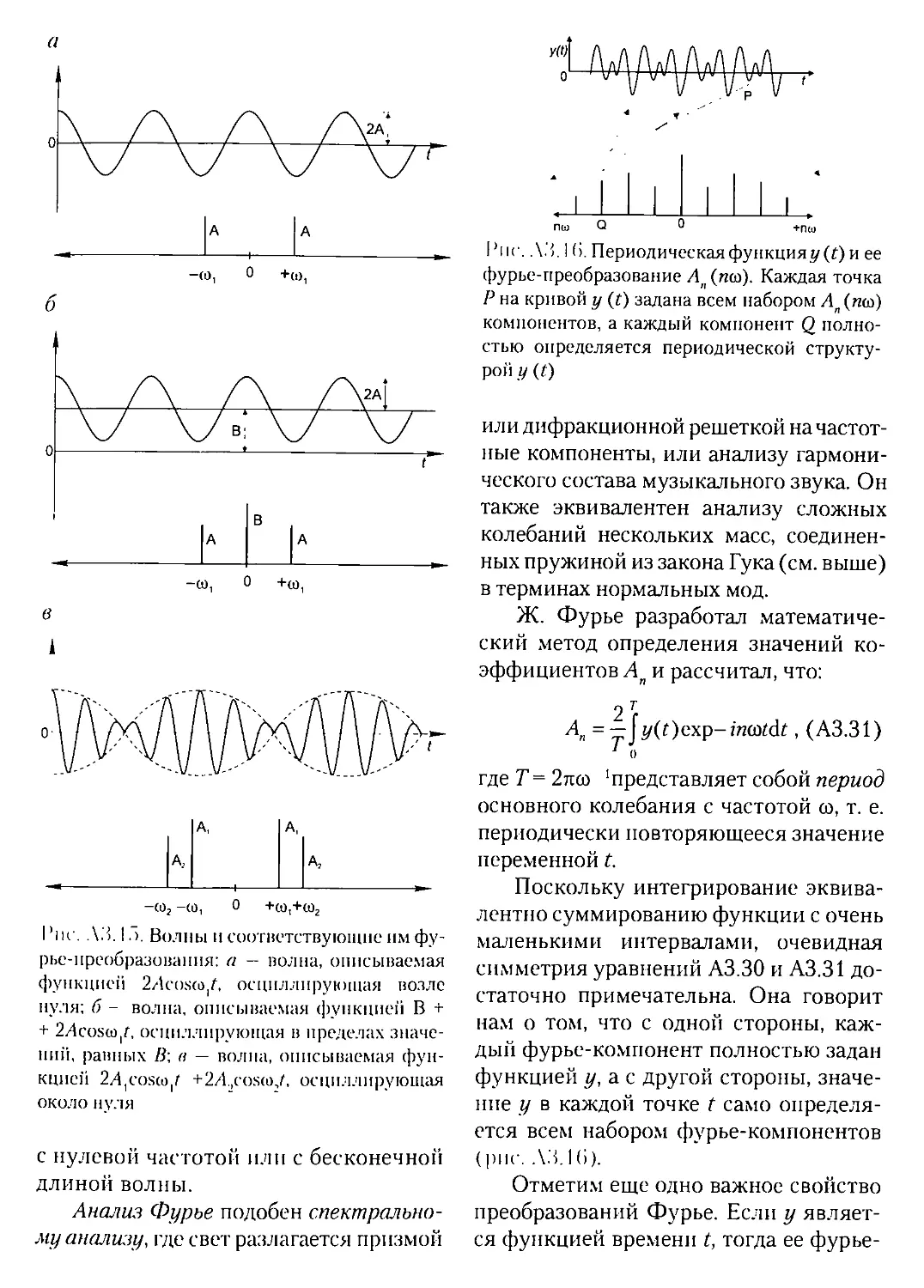
АЗ.2.1. Волны............................................92
АЗ.2.2. Простые гармонические колебания.................100
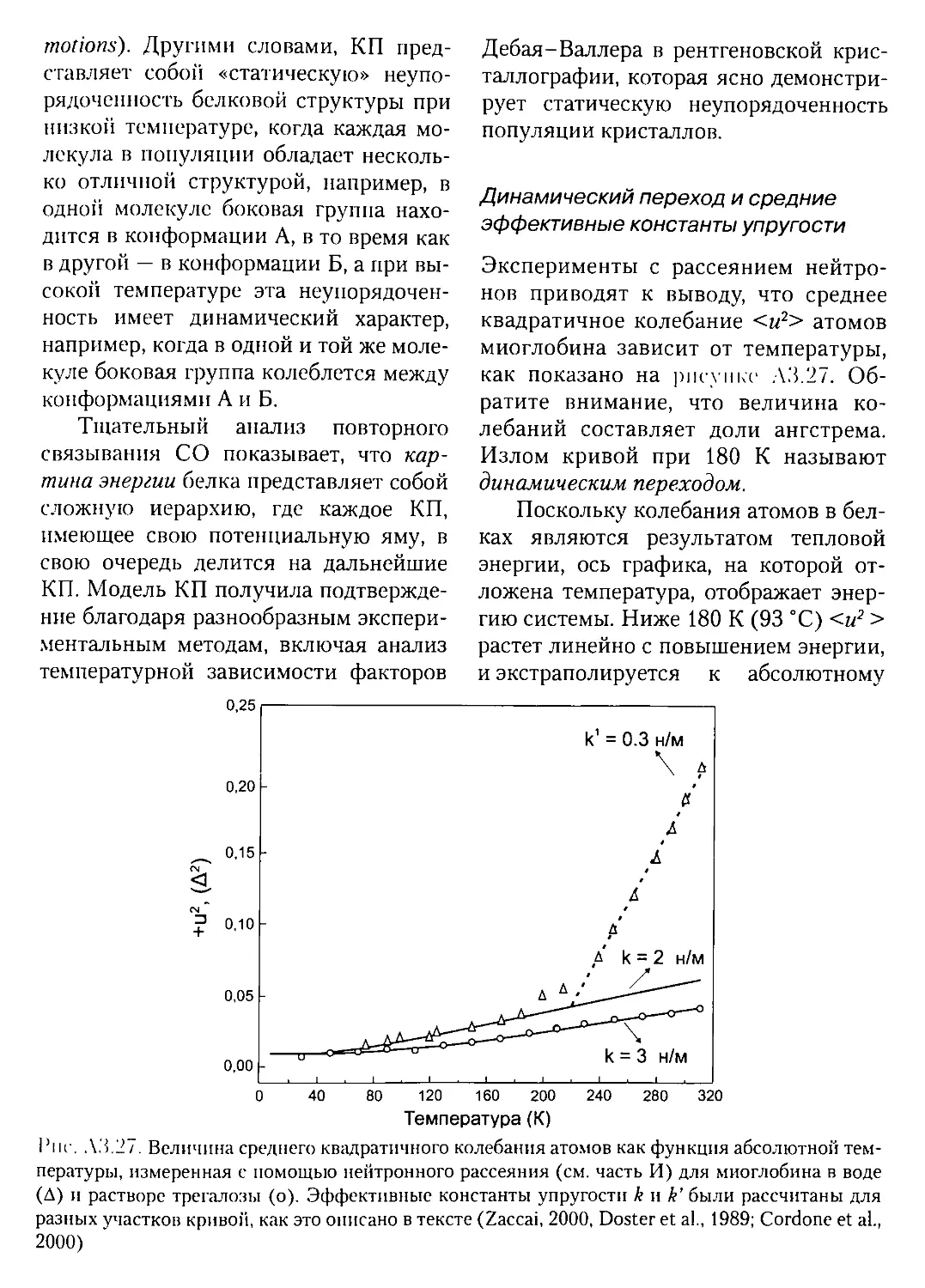
А3.2.3. Анализ Фурье....................................104
АЗ.2.4. Квантовая механика................................Ш
АЗ.З. ДИНАМИКА И СТРУКТУРА, КИНЕТИКА,
КИНЕМАТИКА, РЕЛАКСАЦИЯ.....................................116
А3.3.1. Силы, стабилизирующие макромолекулы...................117
А3.3.2. Шкала длины и времени в молекулярной динамике....... 118
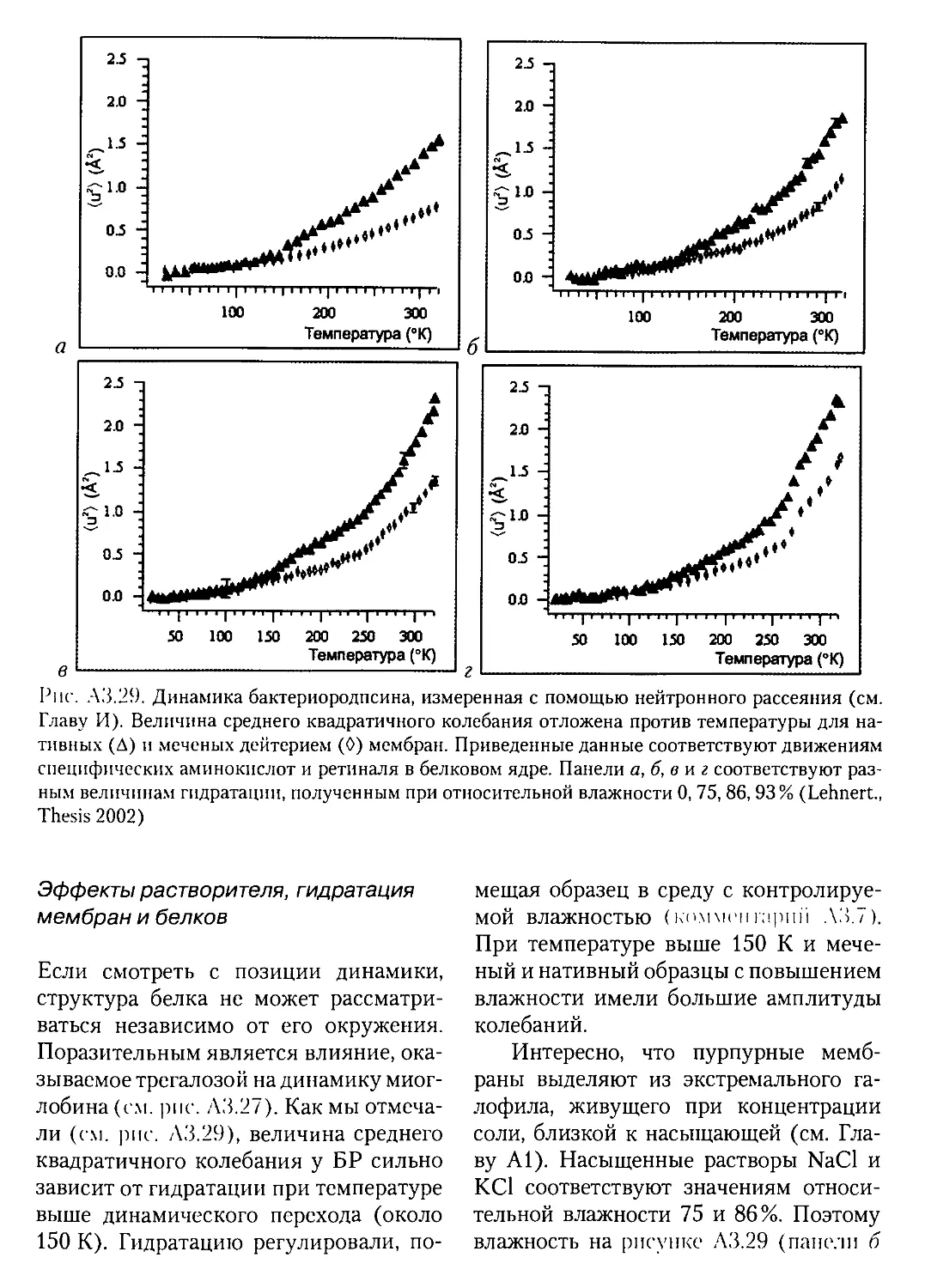
АЗ.З.З. Физическая модель динамики белков.....................119
А3.4. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ.................................... 131
ЧАСТЬ Б. МАСС-СПЕКТРОМЕТРИЯ 133
Глава Б1. Масса и заряд...........................................135
Б1.1. ИСТОРИЧЕСКИЙ ОБЗОР..........................................135
Б1.2. ВВЕДЕНИЕ В БИОЛОГИЧЕСКИЕ ПРОБЛЕМЫ...........................137
Б1.3. ИОНЫ В ЭЛЕКТРИЧЕСКОМ И МАГНИТНЫХ ПОЛЯХ......................138
Б1.4. МЕТОДЫ ИОНИЗАЦИИ............................................138
Б 1.4.1. От ионов в растворе к ионам в газовой фазе...........138
Б 1.4.2. Электронная ионизация................................139
Б 1.4.3. Ионизация полем......................................140
Б 1.4.4. Бомбардировка быстрыми атомами.......................140
Б1.4.5. Плазменная десорбция..................................141
Б 1.4.6. Ионизация лазерной десорбцией при содействии матрицы.141
Б 1.4.7. Ионизация электрораспылением.........................143
Б1.5. ИНСТРУМЕНТЫ И ПЕРЕДОВЫЕ ТЕХНОЛОГИИ..........................145
Б 1.5.1. Масс-спектрометры с одиночной и двойной фокусировкой.145
Б 1.5.2. Квадрупольный масс-фильтр............................148
Б 1.5.3. Квадрупольная ионная ловушка.........................148
Б 1.5.4. Масс-спектрометр с использованием
ионно-циклотронного резонанса.................................148
Б 1.5.5. Время-пролетная масс-спектрометрия...................151
Б 1.5.6. Масс-спектрометрия с фурье-преобразованием...........152
Б 1.5.7. Тандемная масс-спектрометрия.........................183
ПРИЛОЖЕНИЕ 1. РАЗРЕШЕНИЕ И ТОЧНОСТЬ
ОПРЕДЕЛЕНИЯ МАССЫ.................................................I54
Разрешение определения массы..................................184
Точность измерения массы......................................155
Моноизотопная масса...........................................155
Измеренная масса...............................................
Средняя масса.................................................157
Б1.6. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ......................................158
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ.................................158
Глава Б2. Структурно-функциональные
исследования.....................................................160
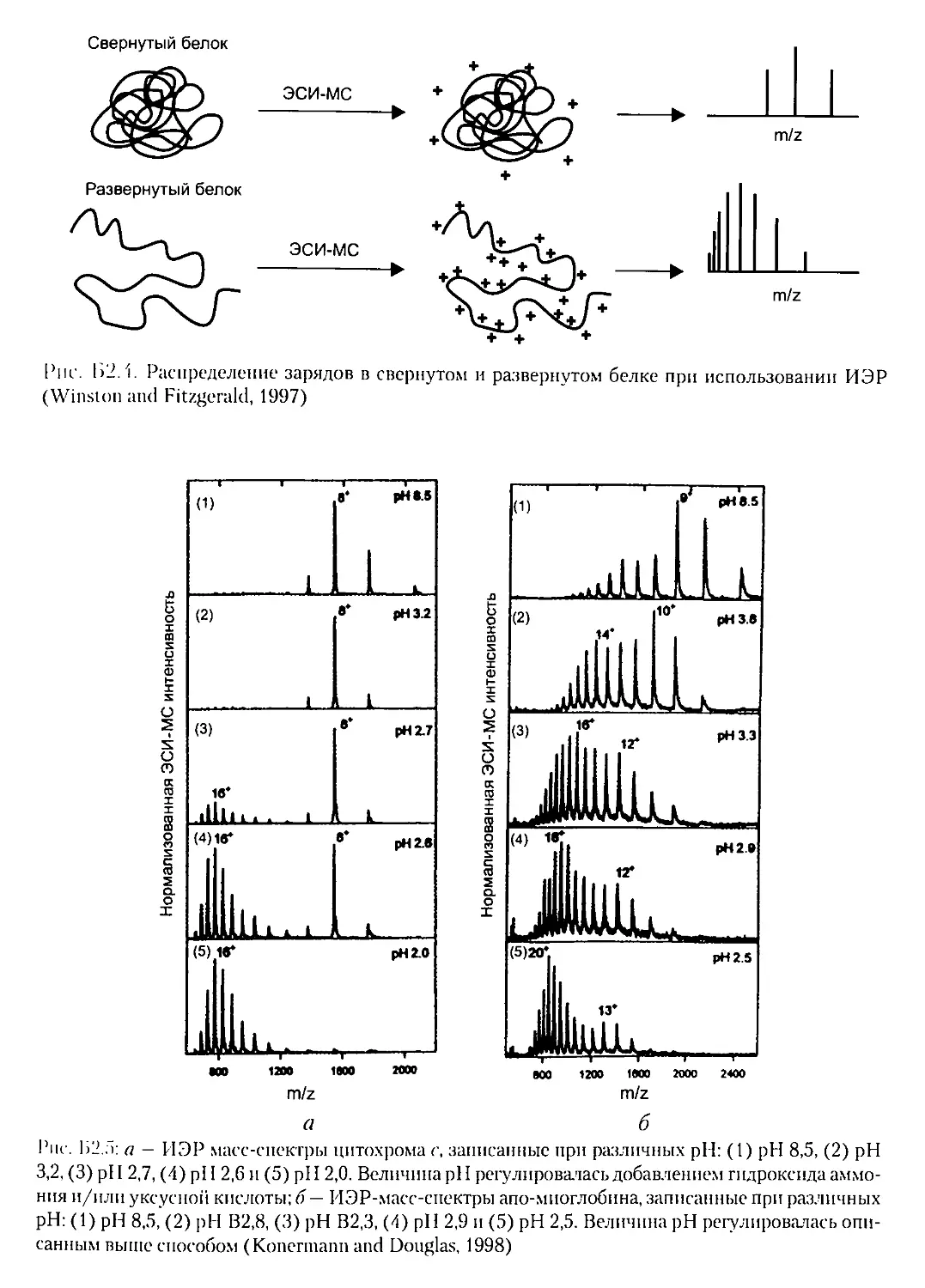
Б2.1. СТРУКТУРА И ФУНКЦИЯ БЕЛКОВ.................................160
Б2.1.1. Определение массы....................................160
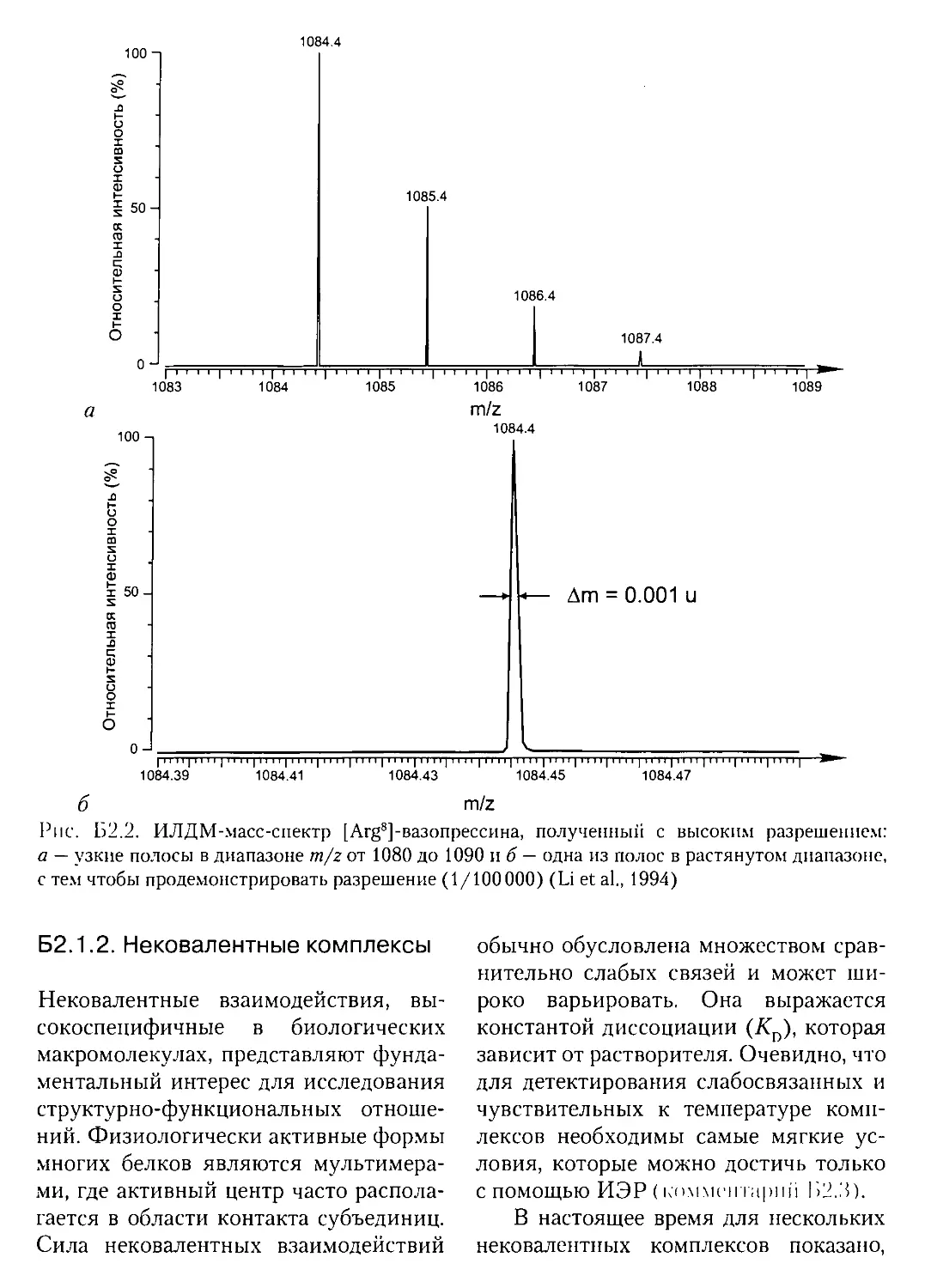
Б2.1.2. Нековалентные комплексы..............................162
Б2.1.3. Сворачивание и динамика белков.................163
Б2.1.4. Секвенирование белков..........................169
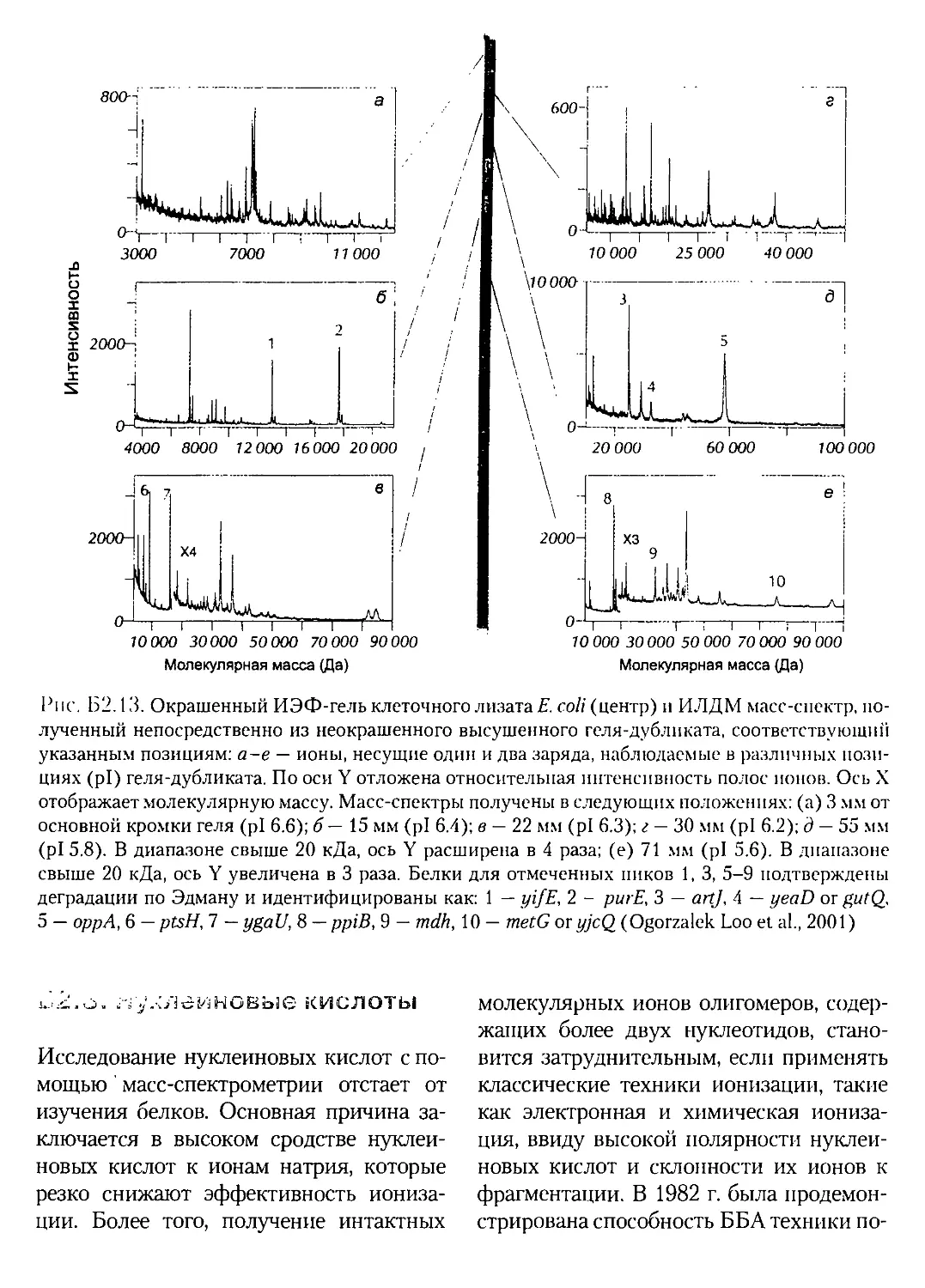
Б2.1.5. Идентификация белков в двумерном электрофорезе.171
Б2.2. ФУНКЦИОНАЛЬНАЯ ПРОТЕОМИКА...........................173
Б2.2.1. Роль масс-спектрометрии........................174
Б2.3. НУКЛЕИНОВЫЕ КИСЛОТЫ.................................176
Б2.3.1. Анализ смеси олигонуклеотидов..................177
Б2.3.2. Нековалентные комплексы........................177
Б2.3.3. Большие и очень большие нуклеиновые кислоты....177
Б2.3.4. Секвенирование ДНК.............................179
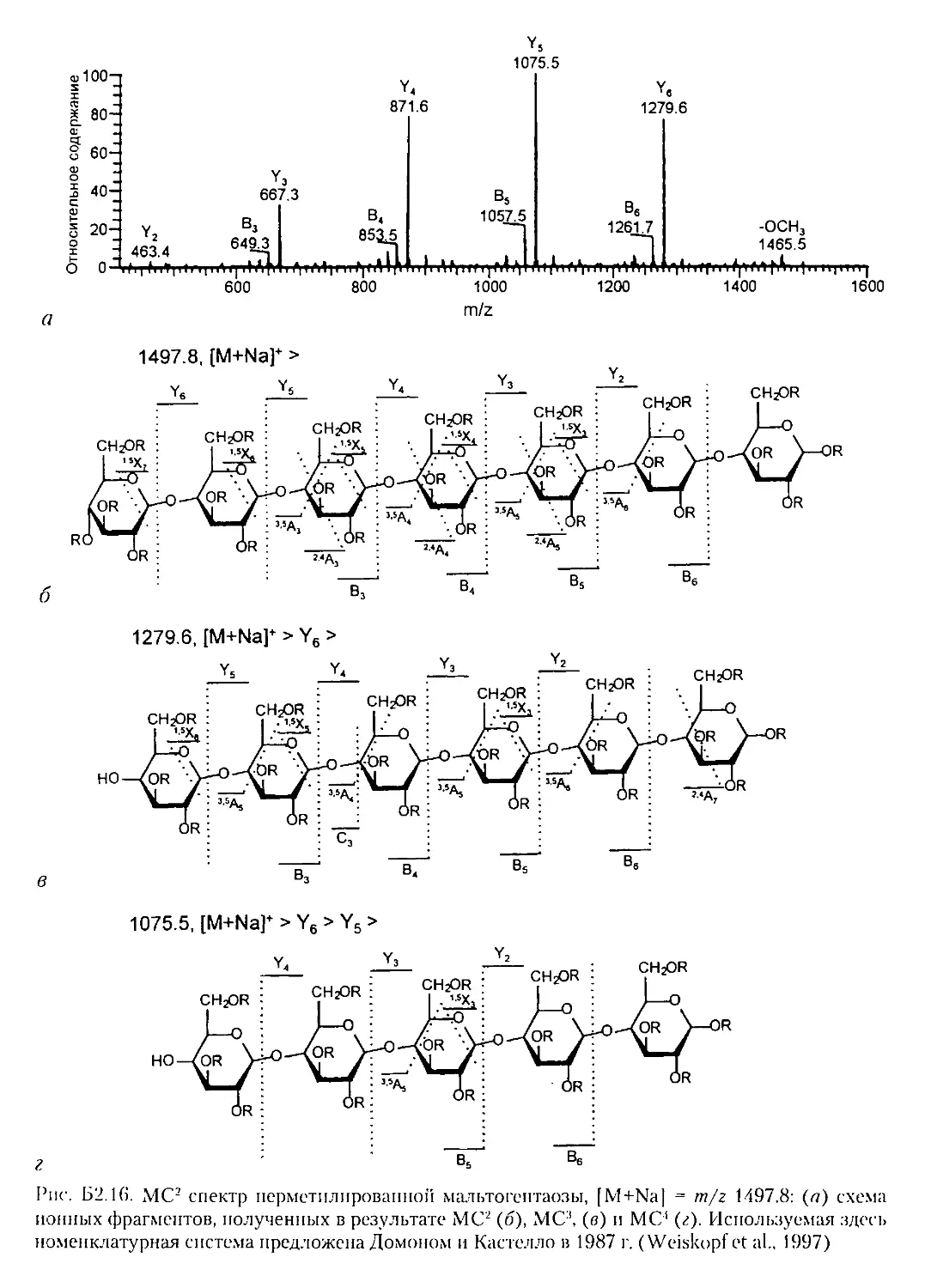
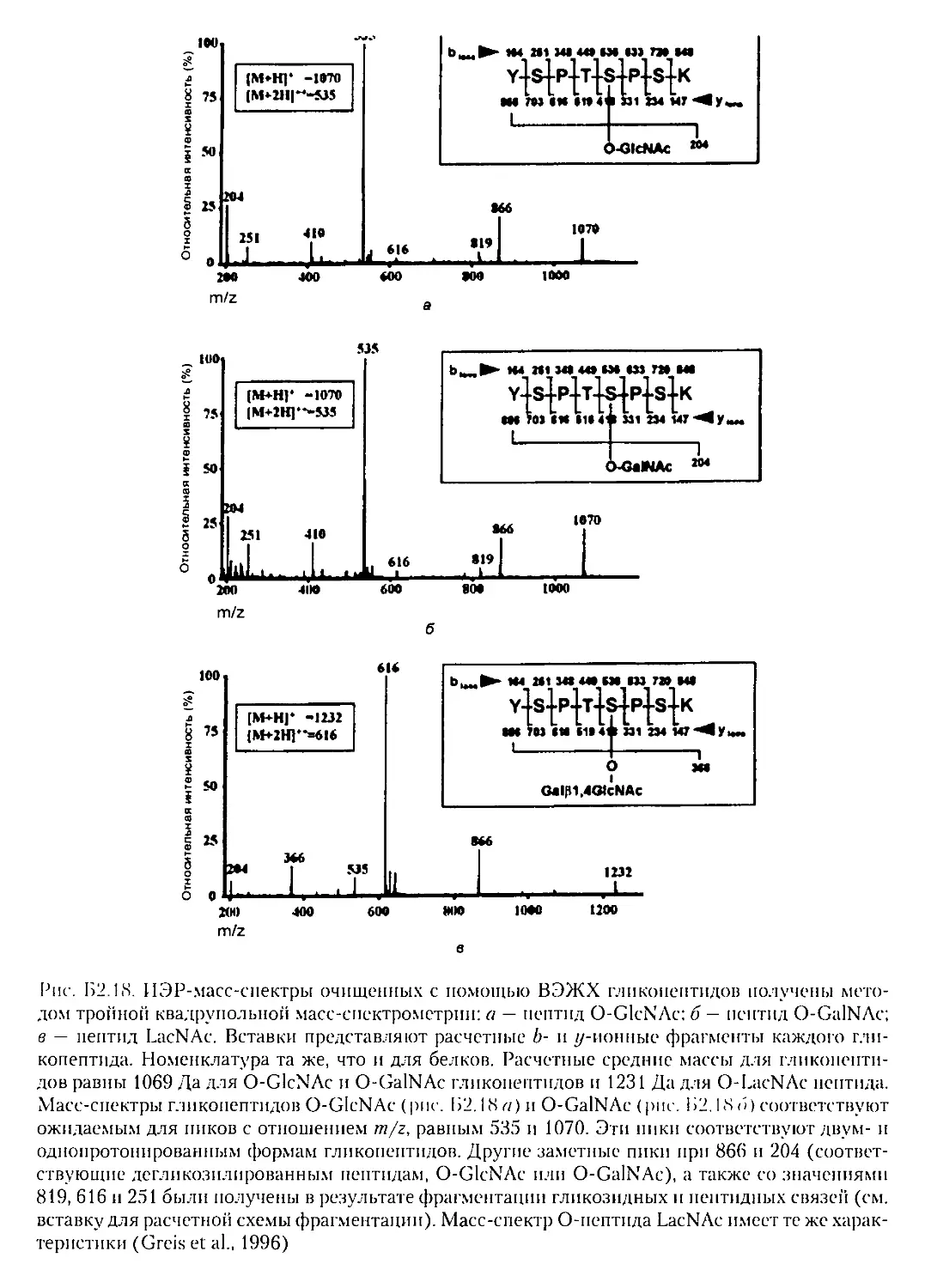
Б2.4. УГЛЕВОДЫ............................................180
Б2.4.1. Олигосахариды..................................181
Б2.4.2. Гликопептиды...................................181
Б2.5. СУБКЛЕТОЧНЫЕ КОМПЛЕКСЫ И ОРГАНЕЛЛЫ..................183
Б2.5.1. Рибосомы, рибосомные субъединицы и рибосомные белки.
Б2.5.2. Бактерии и бактериальная таксономия............|8^
Б2.6. МАСС-СПЕКТРОМЕТРИЯ В МЕДИЦИНЕ....................... ‘ 186
Б2.6.1. Клиническая онкология..........................
Б2.7. МАСС- СПЕКТРОМЕТРИЧЕСКАЯ ВИЗУАЛИЗАЦИЯ
ИЗОБРАЖЕНИЙ.....................................................
Б2.7.1. Клетка...............................................
Б2.7.2. Ткань......................................... 18д
ПРИЛОЖЕНИЕ 1. ВЫЧИСЛЕНИЕ МОЛЕКУЛЯРНОЙ
МАССЫ БЕЛКА ИЗ ЕГО МАСС-СПЕКТРА.......................... 190
ПРИЛОЖЕНИЕ 2. ВЫЧИСЛЕНИЕ НУКЛЕОТИДНОЙ
ПОСЛЕДОВАТЕЛЬНОСТИ....................................... 192
Б2.8. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ..............................’ ’' 193
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ.........................’ 193
ЧАСТЬ В. ТЕРМОДИНАМИКА 195
Глава В1. Термодинамическая стабильность
и взаимодействия макромолекул...................................
В1.1. ИСТОРИЧЕСКИЙ ОБЗОР........................................
В1.2. ЗАКОНЫ ТЕРМОДИНАМИКИ..................................
В 1.2.1. Основные определения и нулевой закон термодинамики.200
В1.2.2. Первый закон и энергия.........................202
Bl.2.3. Второй закон и энтропия.............................203
В 1.2.4. Третий закон и абсолютный нуль......................
В1.3. ОСНОВНЫЕ ПОНЯТИЯ И УРАВНЕНИЯ..............................
В 1.3.1. Свободная энергия и родственные понятия.............
В1.3.2. Изучение связывания..................................
В1.3.3. Термодинамика и связывание...........................
В1.3.4. Термодинамика переноса неполярных групп в воду.215
Bl.3.4, Термодинамика и активация.
В1.4. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ
Глава В2. Дифференциальная сканирующая
калориметрия....................
- .216
217
.219
В2.1 ИСТОРИЧЕСКИЙ ОБЗОР............
В2.2 ОСНОВЫ ТЕОРИИ..................
В2.3 . ЭКСПЕРИМЕНТАЛЬНЫЕ ПОДХОДЫ ’ ’
В2.3 .1. Инструменты......................
В23.2. Чувствительность приборов и точность измерения теплоемкости
В233. Требования к образцам................
В2.4 . ТЕПЛОЕМКОСТЬ БЕЛКОВ..........,,,,
В2.4.1. Зависимость теплоемкости от температуры..................
В2.4.2. Анализ кривой теплоемкости с помощью функции распределения ....
В2.43. Кооперативность плавления......................_
В2.4.4. Конформационные переходы, при которых калориметрическая
энтальпия и энтальпия Вант-Гоффа равны между собой.......
В2.4.5. Конформационные переходы, при которых калориметрическая
энтальпия и энтальпия Вант-Гоффа не равны между собой....
................220
................220
................220
аав g'sSS g S gtgtSSSSSgg S SSSSSSS
В2.4.6. Интермедиаты в процессе сворачивания..........................
В2.4.8. Сложные мультидоменные белки............................
В2.4.9. Влияние растворителя на кривые плавления белков.........
В2.4.10. Расчет теплоемкости из структурных данных.............
В2.4.11. Силы, стабилизирующие нативную структуру белка......
В2.4.12. Холодовая денатурация...................
В25. НУКЛЕИНОВЫЕ КИСЛОТЫ И ЛИПИДЫ...................
В2.6. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ........................
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ...................
ГЛАВА ВЗ. Изотермическая калориметрия
титрования................................................
В3.1. ИСТОРИЧЕСКИЙ ОБЗОР..................................
В3.2. ЭКСПЕРИМЕНТАЛЬНЫЕ ПОДХОДЫ
И ОСНОВНЫЕ УРАВНЕНИЯ
ВЗ.2.1. Проведение измерений и требования к образцам..
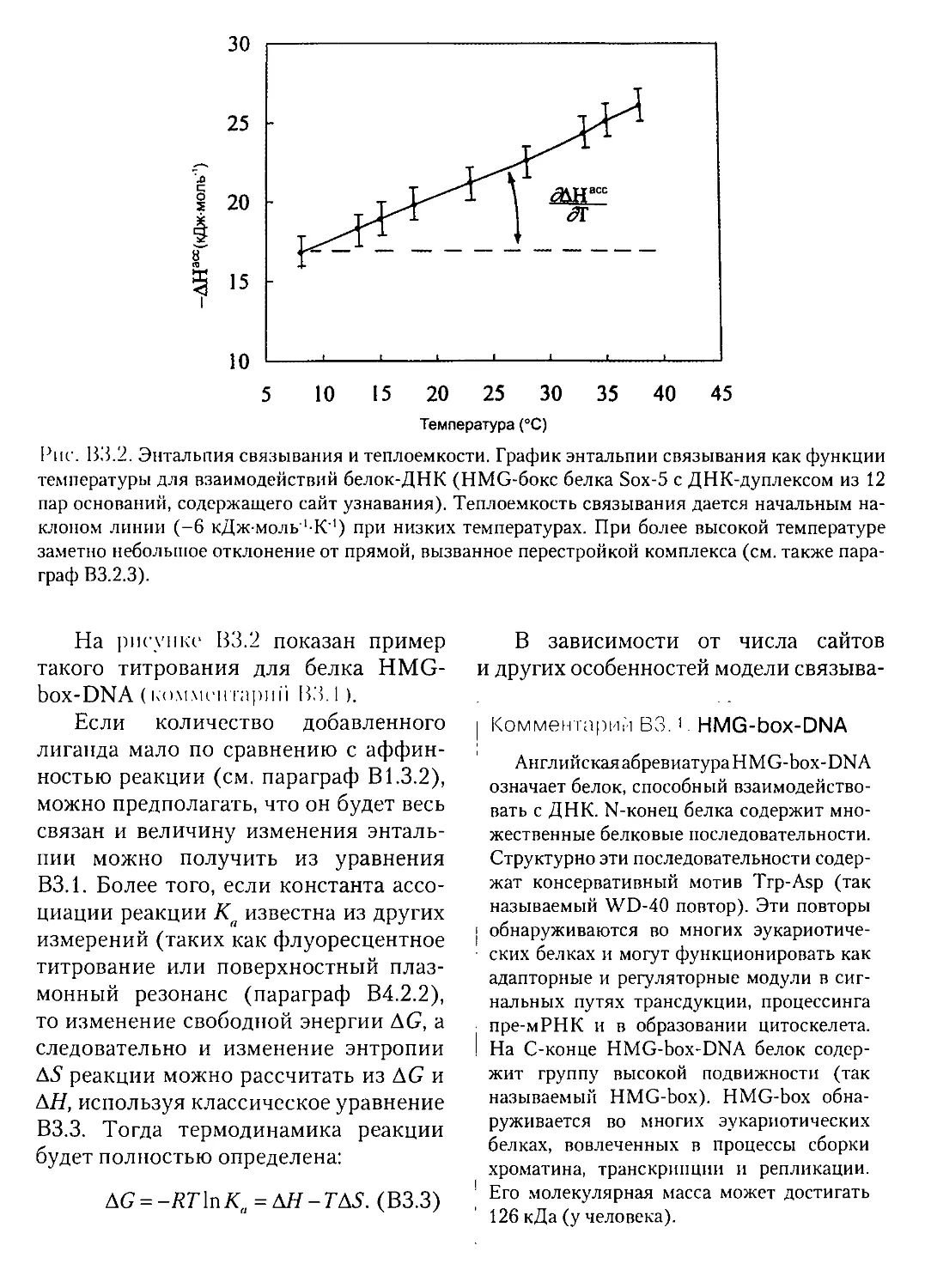
ВЗ.2.2. Связывание: энтальпия и теплоемкость. - - • —
В3.23. Константы сродства (аффинности)..........
ВЗ.2.4. Изотермическая калориметрия титрования и дифференциальная
сканирующая калориметрия.......•....•.................
ВЗЗ. ПРИЛОЖЕНИЯ МЕТОДА...................................
В33.1. Конструирование лигандов .............. -.....
В3.4. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ......
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ
258
259
Глава В4. Биосенсоры и поверхностный
плазмонный резонанс...............................................260
В4.1. ИСТОРИЧЕСКИЙ ОБЗОР И ВВЕДЕНИЕ В БИОЛОГИЧЕСКУЮ
ПРОБЛЕМАТИКУ..........................................................260
В4.2. ИЗМЕРЕНИЕ ПОВЕРХНОСТНОГО СВЯЗЫВАНИЯ.........................261
В4.2.1. Биосенсор как инструмент исследования взаимодействий между
молекулами.....................................................261
В4.2.2. Биосенсор на основе поверхностного плазмонного резонананса.262
Б4.2.3. Биосенсоры на основе интерферометров...................265
В4.2.4. Лиганды и поверхность..................................265
В4.3. ВЗАИМОДЕЙСТВИЕ МЕЖДУ МОЛЕКУЛОЙ И ПОВЕРХНОСТЬЮ.. .266
В4.3.1. Термодинамика взаимодействий с поверхностью............266
В4.3.2. Измерение константы равновесия.........................267
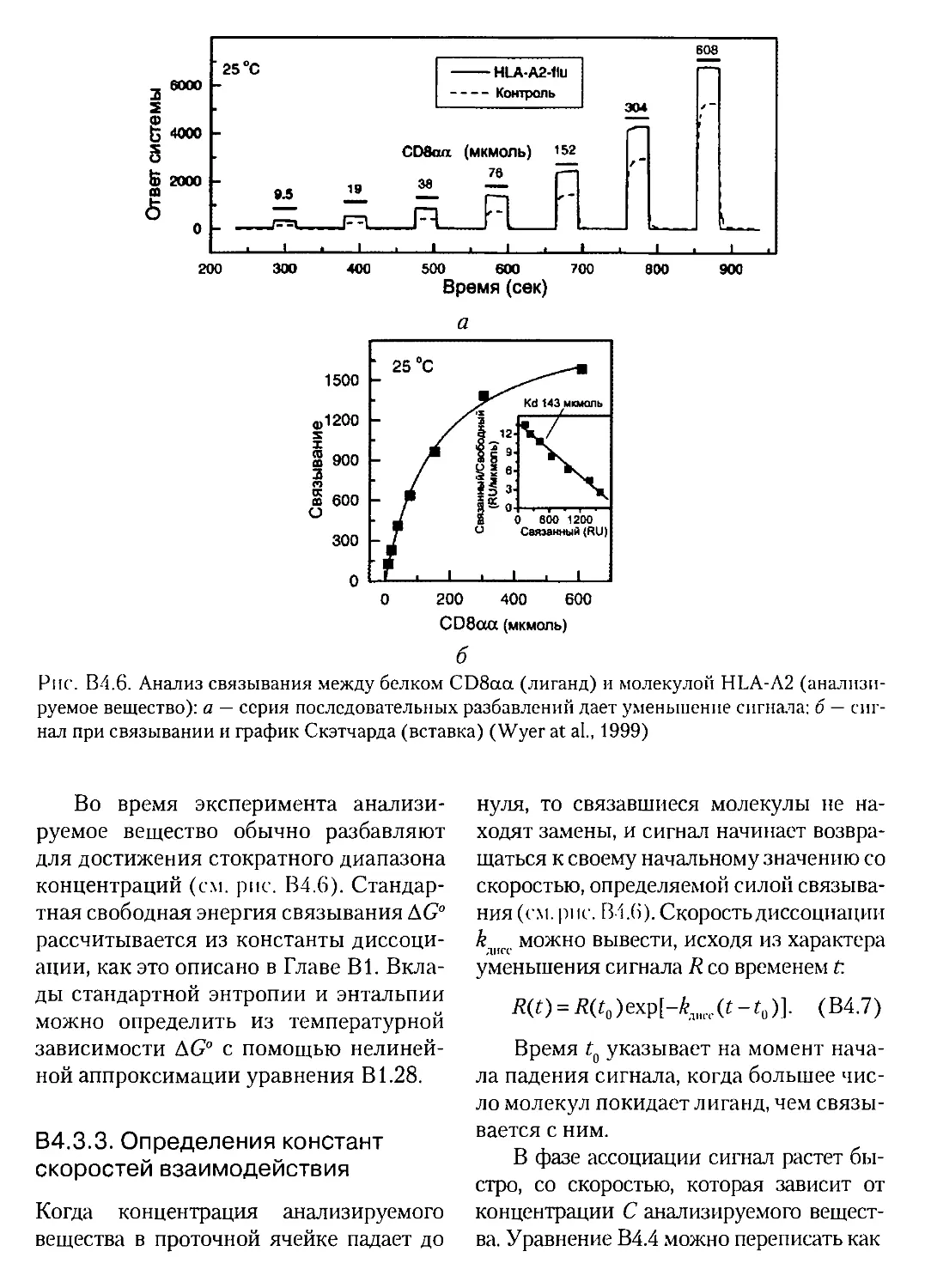
В4.3.3. Определения констант скоростей взаимодействия..........268
В4.4. ЭКСПЕРИМЕНТАЛЬНЫЙ АНАЛИЗ....................................269
В4.4.1. Диапазон анализируемых веществ.........................269
В4.4.2. Экспериментальный контроль и «подводные камни».........269
В.4.4.3 . Межклеточные взаимодействия..........................270
В4.4.4. Поверхностный плазмонный резонанс и масс-спектрометрия.271
В4.5. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ......................................272
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ.................................272
ЧАСТЬ Г. ГИДРОДИНАМИКА 273
Глава Г1. Биологические макромолекулы
как гидродинамические частицы.....................................275
Г1.1. ИСТОРИЧЕСКИЙ ОБЗОР И ВВЕДЕНИЕ В БИОЛОГИЧЕСКИЕ
ПРОБЛЕМЫ 275
Г1.2. ГИДРОДИНАМИКА ПРИ НИЗКИХ ЧИСЛАХ РЕЙНОЛДСА...................279
Г1.2.1. Число Рейнолдса.......................................279
Г1.2.2. Движение при низких значениях числа Рейнолдса.........279
Г1.3. ГИДРАТАЦИЯ..................................................280
Г1.4. ОПРЕДЕЛЕНИЕ ФРИКЦИОННЫХ СВОЙСТВ ЧАСТИЦ......................283
Г1.4.1. Граничные условия: «полное прилипание» — «свободное скольжение» 283
Г1.4.2. Гидродинамические эксперименты........................284
Г1.4.3. Гидродинамические параметры...........................284
Г1.4.4. Гидродинамически эквивалентные тела...................288
Г1.5. ВЫЧИСЛЕНИЕ ФРИКЦИОННЫХ СВОЙСТВ ЧАСТИЦ.......................289
Г 1.5.1. Частицы «правильной» округлой формы..................289
Г1.5.2. Частицы «правильной» граненой формы...................289
Содержание
9
П.5.3. Частицы произвольной сложной формы......................290
П.5.4. Частицы с известной трехмерной структурой ..............291
Г1.6. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ......................................291
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ.................................292
Глава Г2. Фундаментальная теория..................................294
Г2.1. ИСТОРИЧЕСКИЙ ОБЗОР.....................................294
Г2.2. ПОСТУПАТЕЛЬНОЕ ТРЕНИЕ..................................298
Г2.2.1. Частицы правильной формы.........................298
Г2.2.2. Жесткие частицы произвольной формы...............899
Г2.2.3. Коэффициент поступательного трения
и электростатическая емкость..............................
Г22.4. Частицы с известной структурой.................. XTZ
Г2.2.5. Жесткие частицы с сегментальной подвижностью......
Г2.2.6. Экспериментальные методы исследования коэффициента
поступательного трения...............................
Г23. ВРАЩАТЕЛЬНОЕ ТРЕНИЕ.................................
Г2.3.1. Вращательное движение в одном измерении......
Г2.3.2. Вращательное движение в трех измерениях..... -.
Г2.3.3. Вращательное движение и время релаксации.....
Г2.3.4. Жесткие частицы регулярной формы.............
Г2.35. Частицы произвольной формы....................
Г2.3.6. Экспериментальные методы для измерения коэффициентов
Г2.4. ВЯЗКОСТЬ....................................................
Г2.4.1. Вязкость как локальные потери энергии.................
Г2.4.2. Относительная, удельная и характеристическая вязкость.
Г2.4.3. Частицы правильной формы..............................
Г2.4.4. Частицы произвольной формы............................
Г25. ОТ ПРИБЛИЖЕНИЯ «ГИДРОДИНАМИЧЕСКИ ЭКВИВАЛЕНТНАЯ
СФЕРА» К ПРИБЛИЖЕНИЮ «ЦЕЛОЕ ТЕЛО».................................
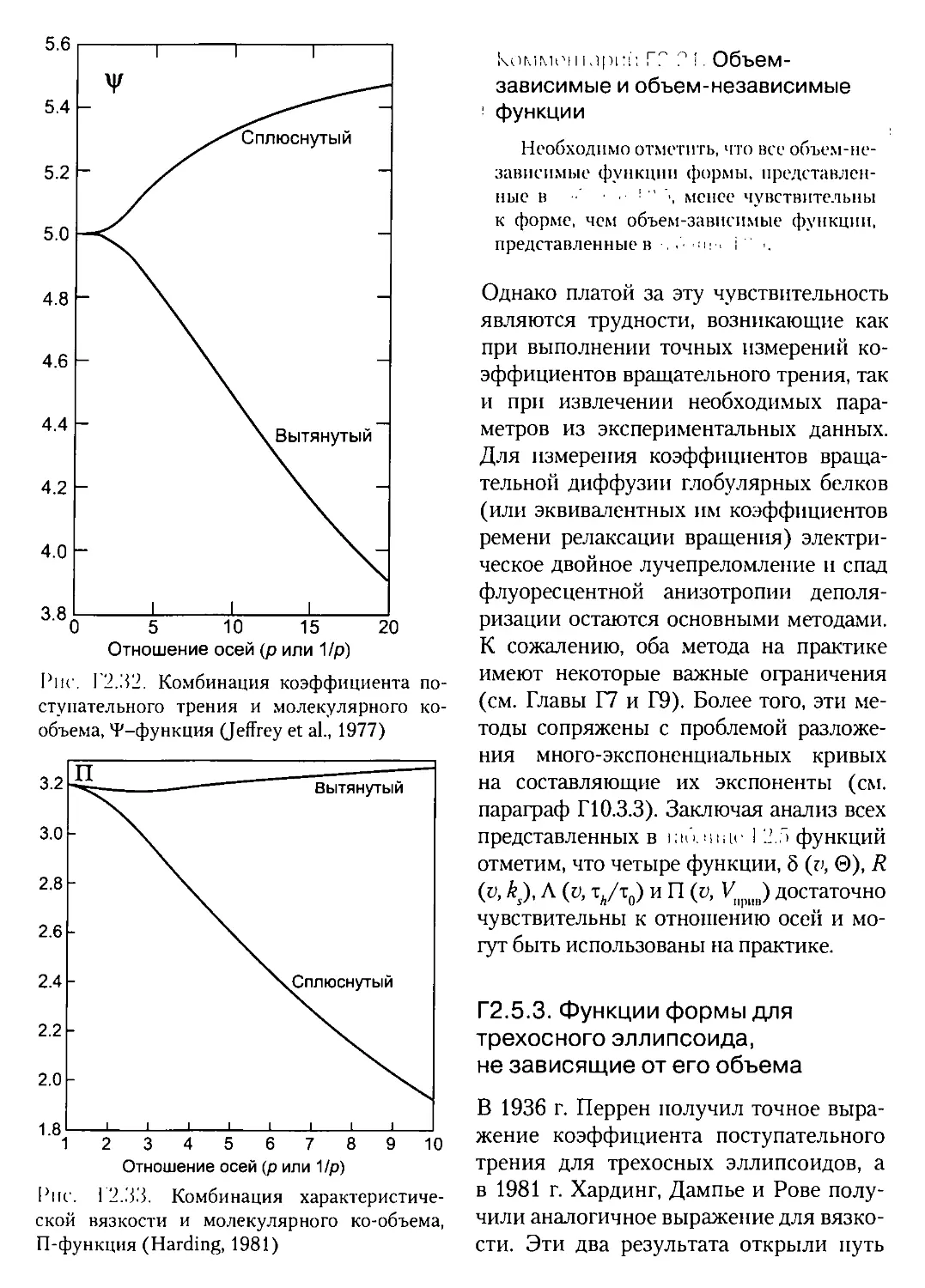
Г2Л.1. Функции формы, зависящие от объема для двухосного эллипсоида- -
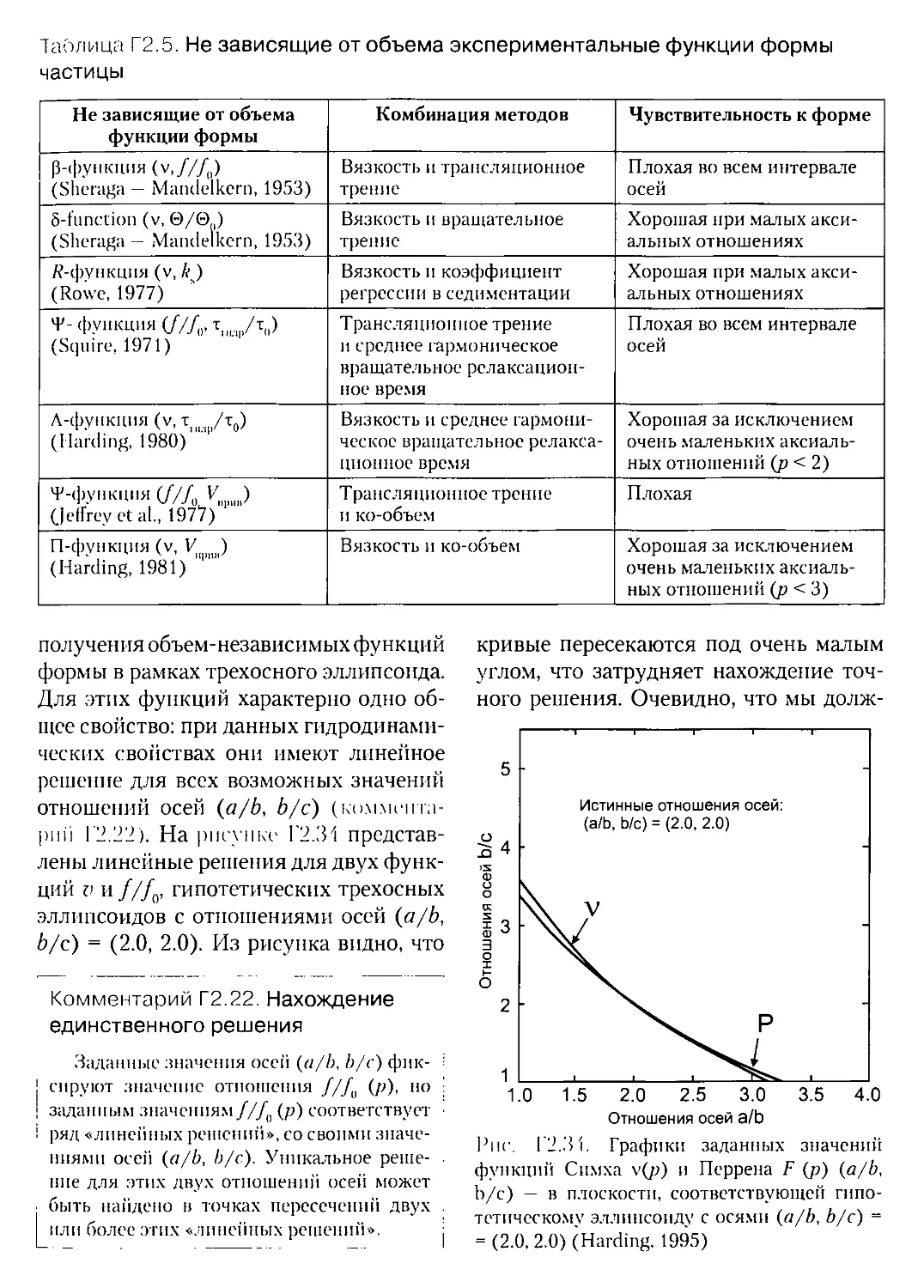
Г2Л.З. Функции формы для трехосного эллипсоида,
не зависящие от его объема.........................................
Г2.5.4. Приближение «целого тела» и моделирование тела «бусинками».
Г2.6. ГОМОЛОГИЧНЫЕ РЯДЫ................................................
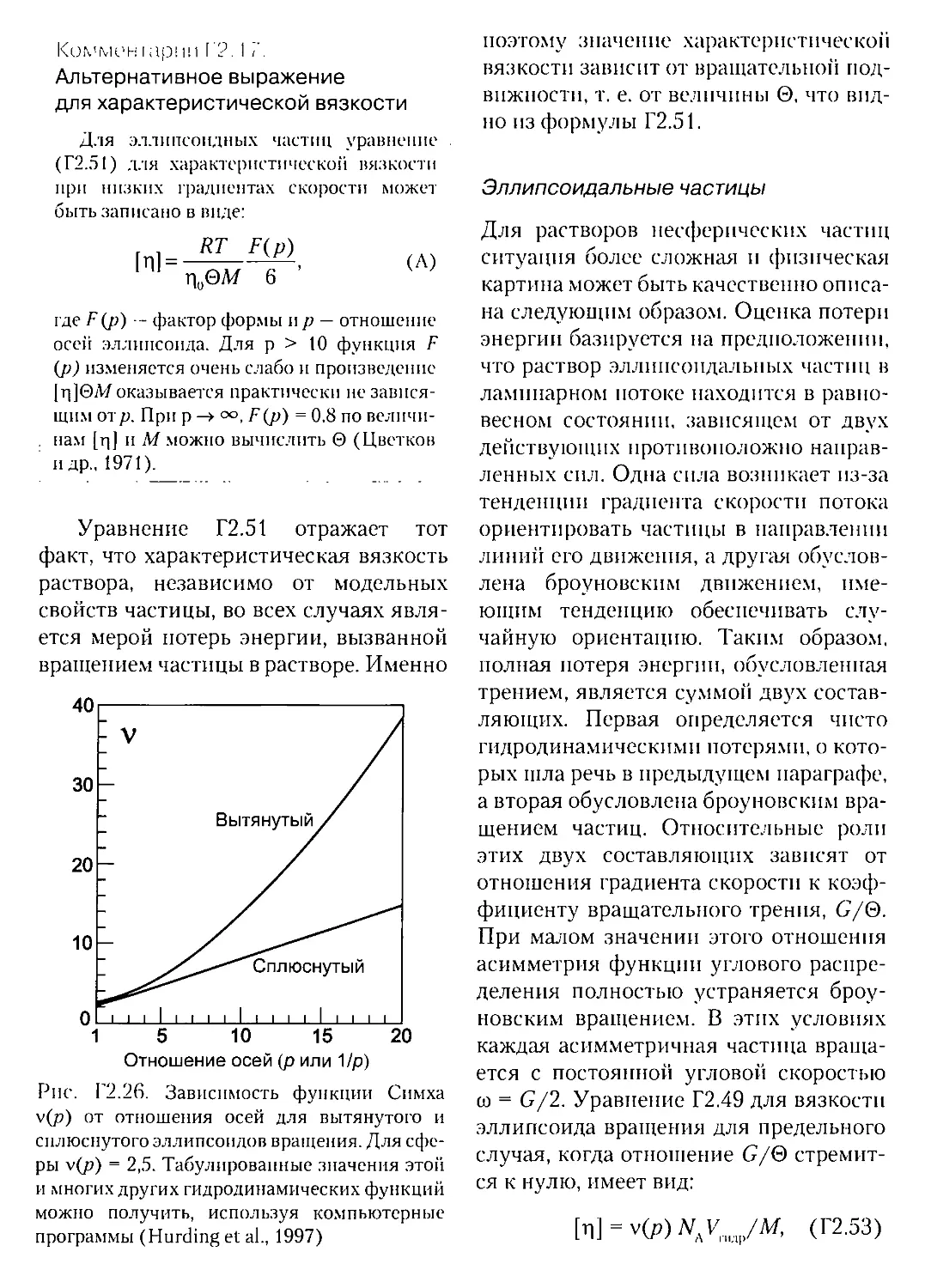
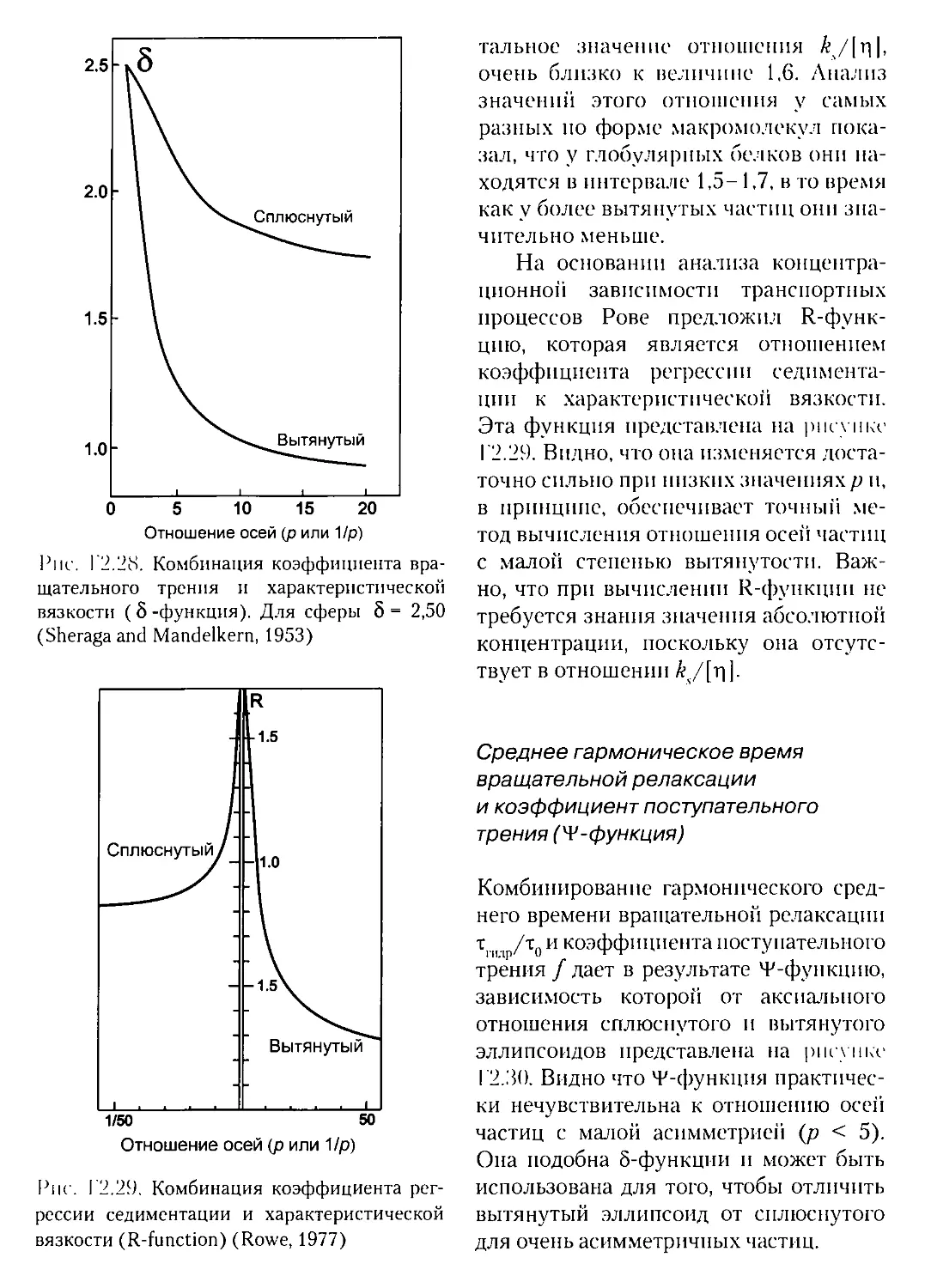
Г2.6.1. Характеристическая вязкость................................
314
316
316
317
318
319
331
338
341
341
343
ПРИЛОЖЕНИЕ 1.0 КОРРЕКТНОСТИ УРАВНЕНИЯ
КИРКВУДА-РАЙЗМАНА...............................344
Г2.7. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ....................345
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ...............347
Глава ГЗ. Макромолекулярная диффузия
ГЗ-1- ИСТОРИЧЕСКИЙ ОБЗОР
ГЗ 2. КОЭФФИЦИЕНТ ПОСТУПАТЕЛЬНОЙ ДИФФУЗИИ........................350
ГЗ з. МИКРОСКОПИЧЕСКАЯ ТЕОРИЯ ДИФФУЗИИ...........................352
Г3.4. МАКРОСКОПИЧЕСКАЯ ТЕОРИЯ ДИФФУЗИИ...........................353
ГЗ.4.1. Первое уравнение Фика............................... 353
ГЗ.4.2. Второе уравнение Фика.................................354
Г3.4.3. Нестационарные решения уравнения Фика.................354
ГЗ.4.4. Стационарные решения уравнения Фика...................355
Г3.5. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
КОЭФФИЦИЕНТОВ ДИФФУЗИИ...........................................356
ГЗ.5.1. Метод расширяющейся границы...........................356
ГЗ.5.2. Метод восстановления флуоресценции после выцветания красителя .. 357
Г3.5.3. Метод привязки золотых кластеров («нановидная микроскопия»).358
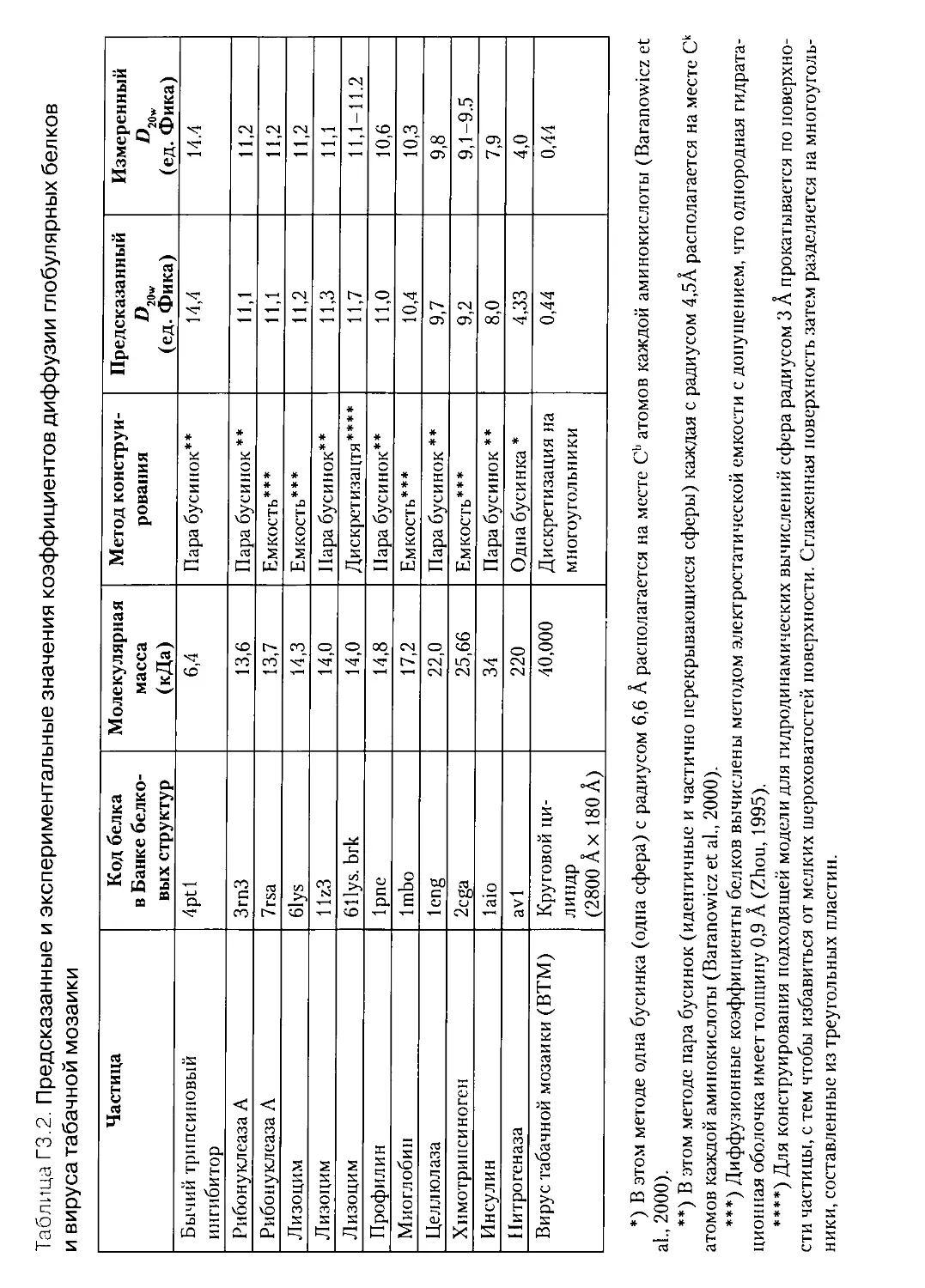
Г3.6. ПРЕДСКАЗАНИЕ КОЭФФИЦИЕНТОВ ДИФФУЗИИ
ГЛОБУЛЯРНЫХ БЕЛКОВ...............................................359
Г3.7. ТРАНСЛЯЦИОННОЕ ТРЕНИЕ И ДИФФУЗИОННЫЕ
КОЭФФИЦИЕНТЫ.....................................................361
ГЗ.7.1. Уравнение Эйнштейна—Смолуховского.....................361
ГЗ.7.2. Диффузионные коэффициенты биологических макромолекул..362
Г3.7.3. Зависимость коэффициентов диффузии глобулярных белков от
молекулярной массы............................................363
ГЗ.7.4. Зависимость коэффициентов диффузии ДНК от молекулярной массы 363
ГЗ.7.5. Предел применения закона Стокса: «малые», «средние» и «большие»
молекулы......................................................364
Г3.8. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ.....................................367
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ................................369
Глава Г4. Аналитическое ультрацентрифугирование..................371
Г4.1. ИСТОРИЧЕСКИЙ ОБЗОР.........................................371
Г4.2. ОБОРУДОВАНИЕ И ИННОВАЦИОННАЯ ТЕХНИКА.......................374
Г4.2.1. Роторы и ячейки.......................................375
Г4.2.2. Системы оптического детектирования...................376
Г4.2.3. Накопление и обработка данных........................378
Г4.3. УРАВНЕНИЕ ЛАММА............................................378
Г4.4. РЕШЕНИЕ УРАВНЕНИЯ ЛАММА ДЛЯ РАЗЛИЧНЫХ
ГРАНИЧНЫХ УСЛОВИЙ................................................380
Г4.4.1. Точные решения.......................................380
Г4.4.2. Аналитические решения................................381
Г4.4.3. Численные решения....................................384
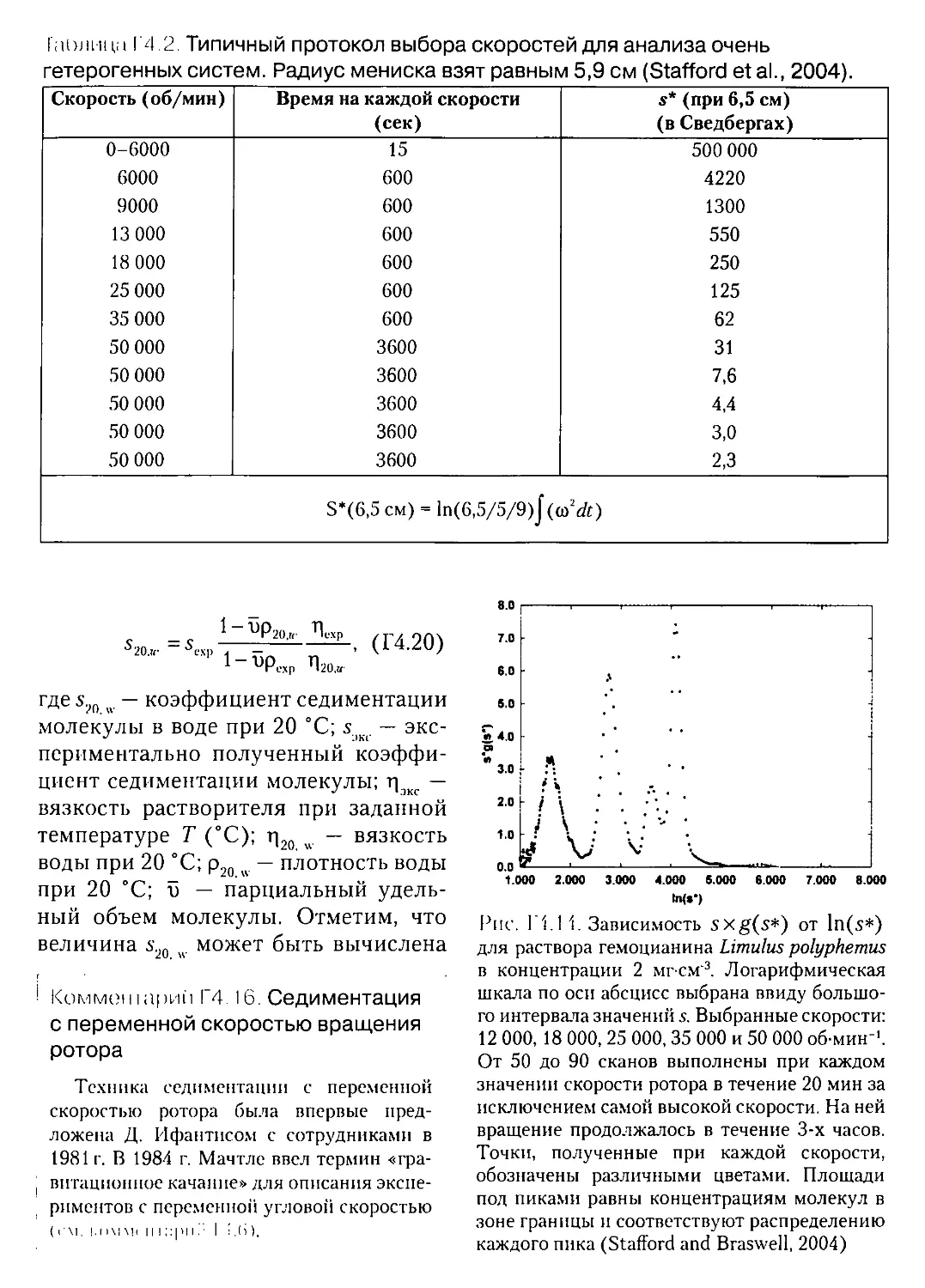
Г4.5. СКОРОСТНАЯ СЕДИМЕНТАЦИЯ..........................................384
Г4.5.1. Макромолекулы в сильном гравитационном поле..........384
Г4.5.2. Определение седиментационных и диффузионных коэффициентов из
данных скоростной седиментации...............................387
Г4.5.3. Очень гетерогенные системы...........................390
Г4.5.4. Коэффициенты седиментации биологических макромолекул.392
14.5.5. Дифференциальная седиментация
Г4 6 ВЫЧИСЛЕНИЕ МОЛЕКУЛЯРНОЙ МАССЫ ПО ядuut,
СЕДИМЕНТАЦИИ И ДИФФУЗИИ ‘ ПОДАННЫМ
Г4.7. РАВНОВЕСНАЯ СЕДИМЕНТАЦИЯ ................
Г4.7.1. Молекулярная масса... ..............
Г4.7.2. Константы связывания. ..............
Г4Л ПАРЦИАЛЬНЫЙ УДЕЛЬНЫЙ ОБЪЕМ ................
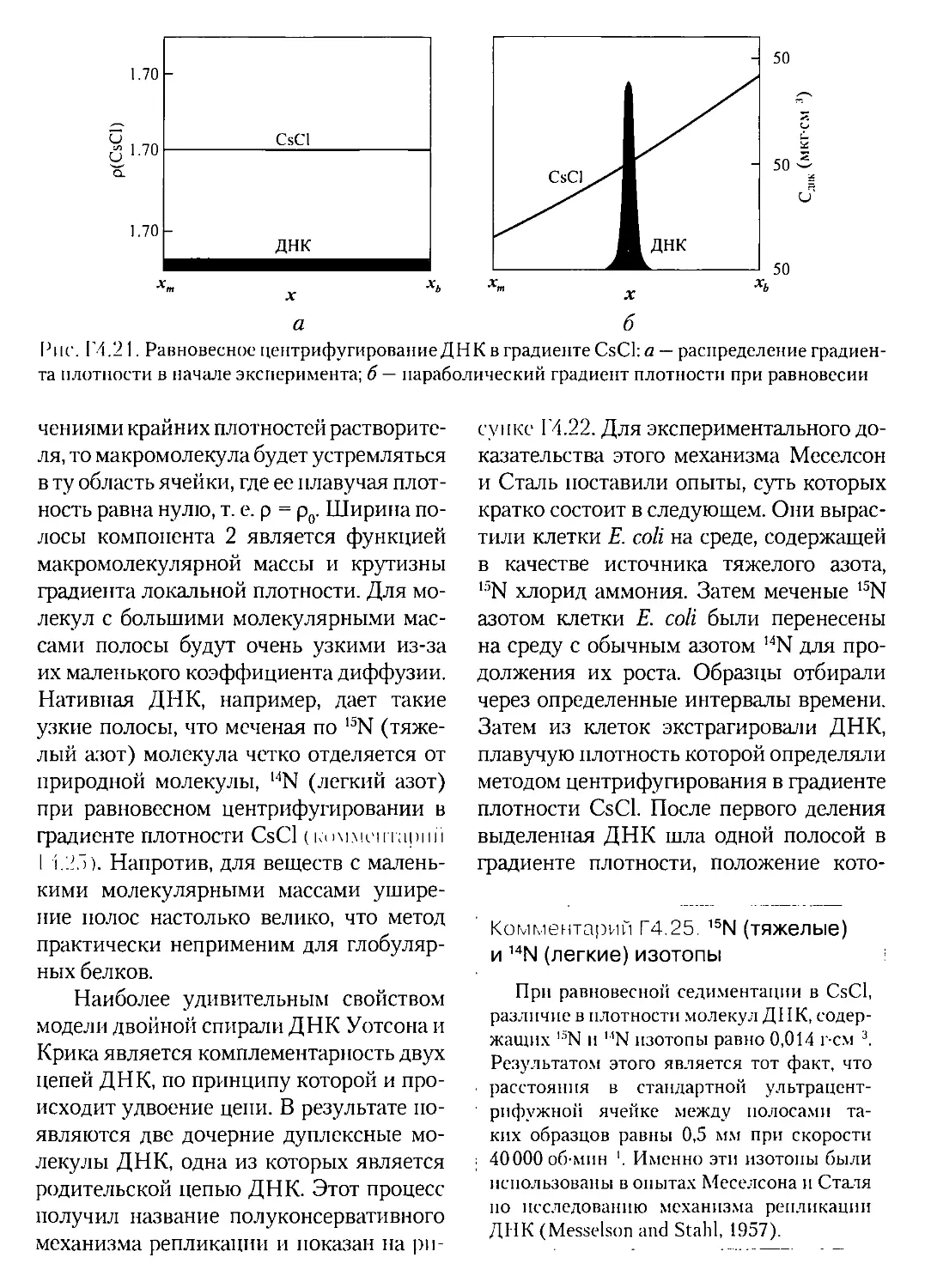
Г4.9. СЕДИМЕНТАЦИЯ В ГРАДИЕНТЕ ПЛОТНОСТИ
.........394
..........397
..........398
..........398
.........400
.........401
.........404
Г4.9.1. Метод скоростного зонального центрифугирования.....404
Г4.9.2. Метод равновесной седиментация в градиенте плотности.406
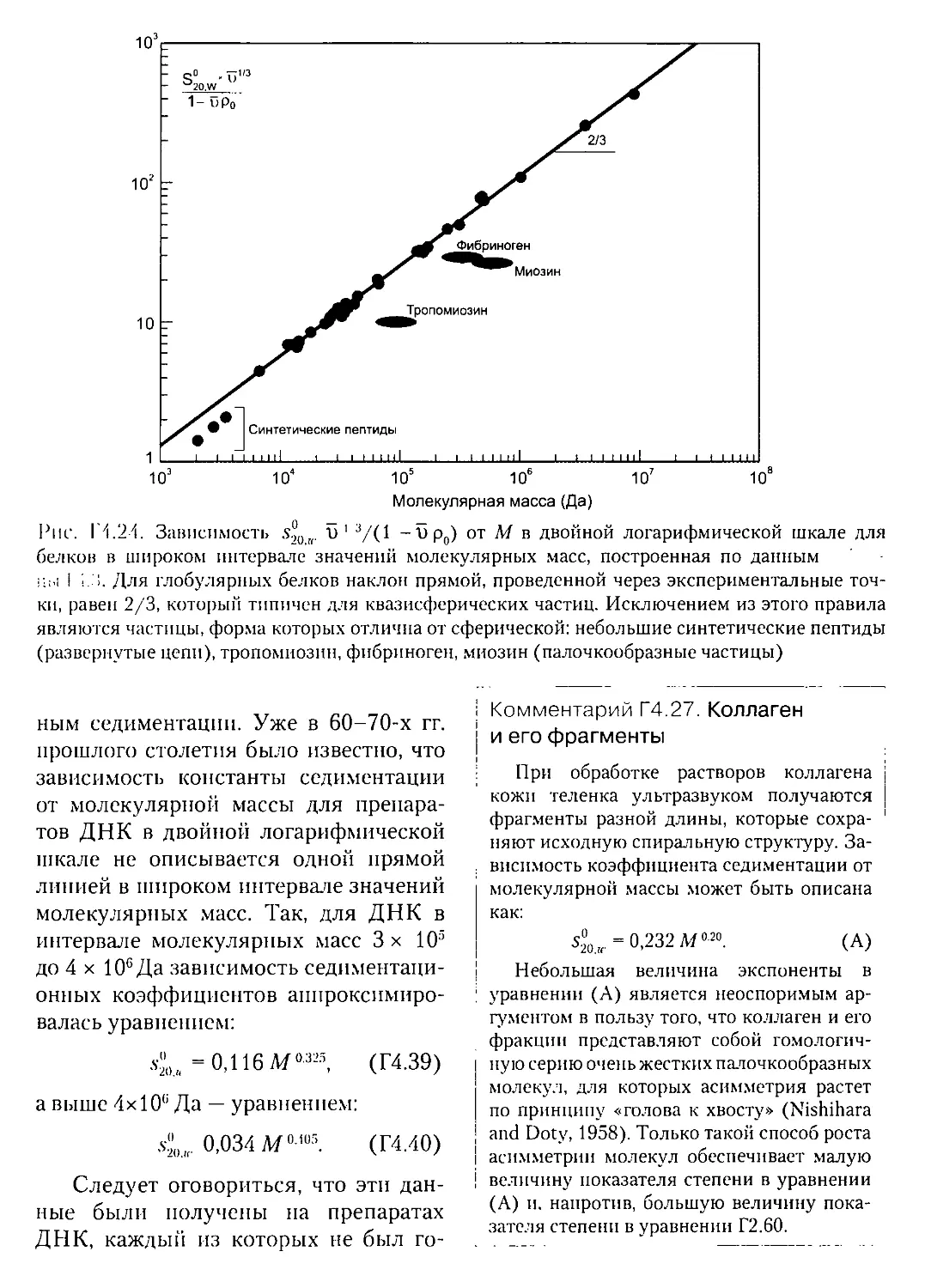
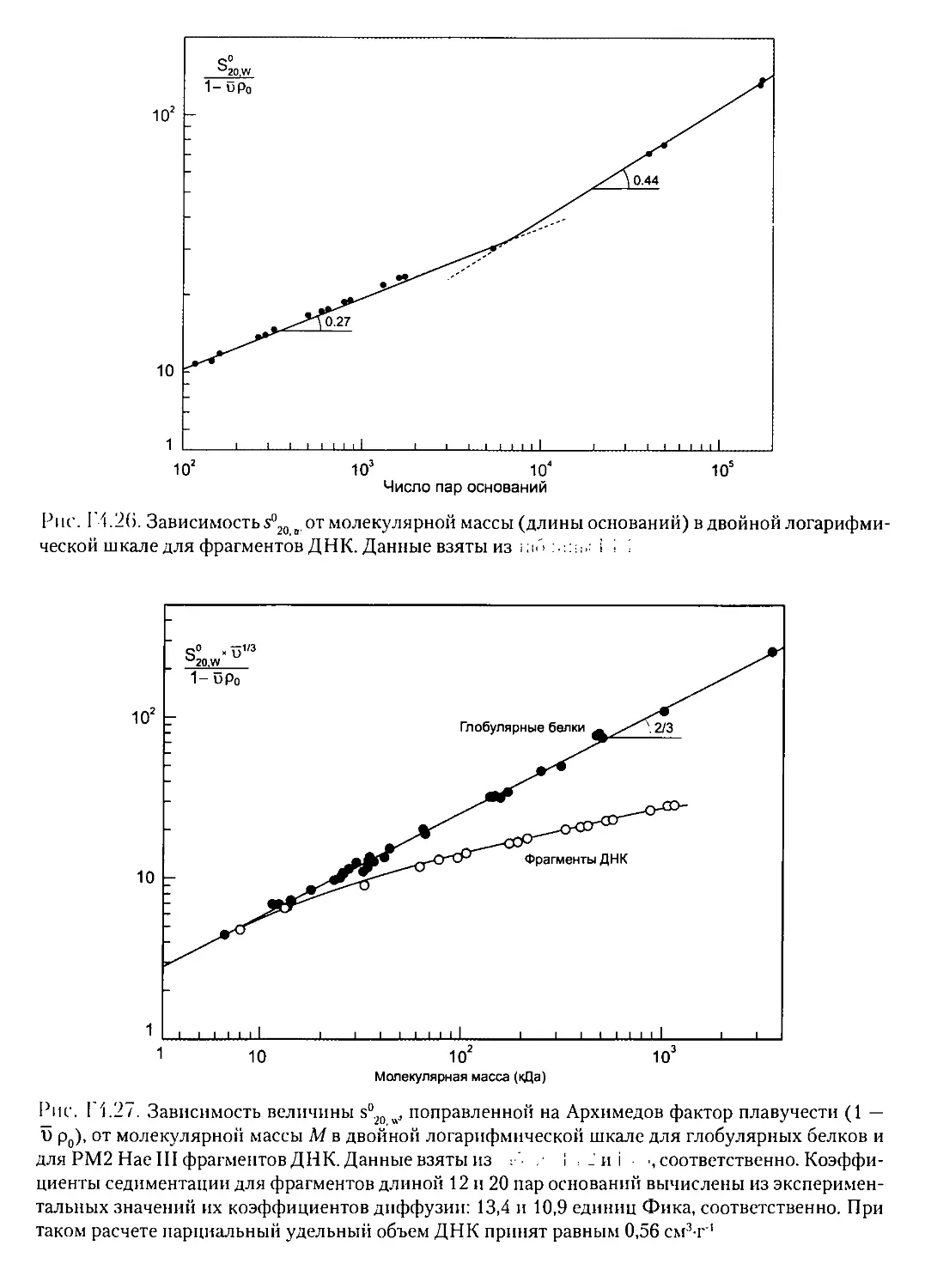
Г4.Ю. ФОРМА МОЛЕКУЛ ПО ДАННЫМ СКОРОСТНОЙ
СЕДИМЕНТАЦИИ..............................................408
Г4.10.1. Гомологичный ряд квазисферических частиц: глобулярные
белки в воде...........................................408
Г4.10.2. Гомологичный ряд клубкобразных молекул: белки в 6М
гуанидингидрохлориде или в 8М мочевине................. 410
Г4.10.3. От коротких жестких палочек к гибким клубкам: конформация ДНК 410
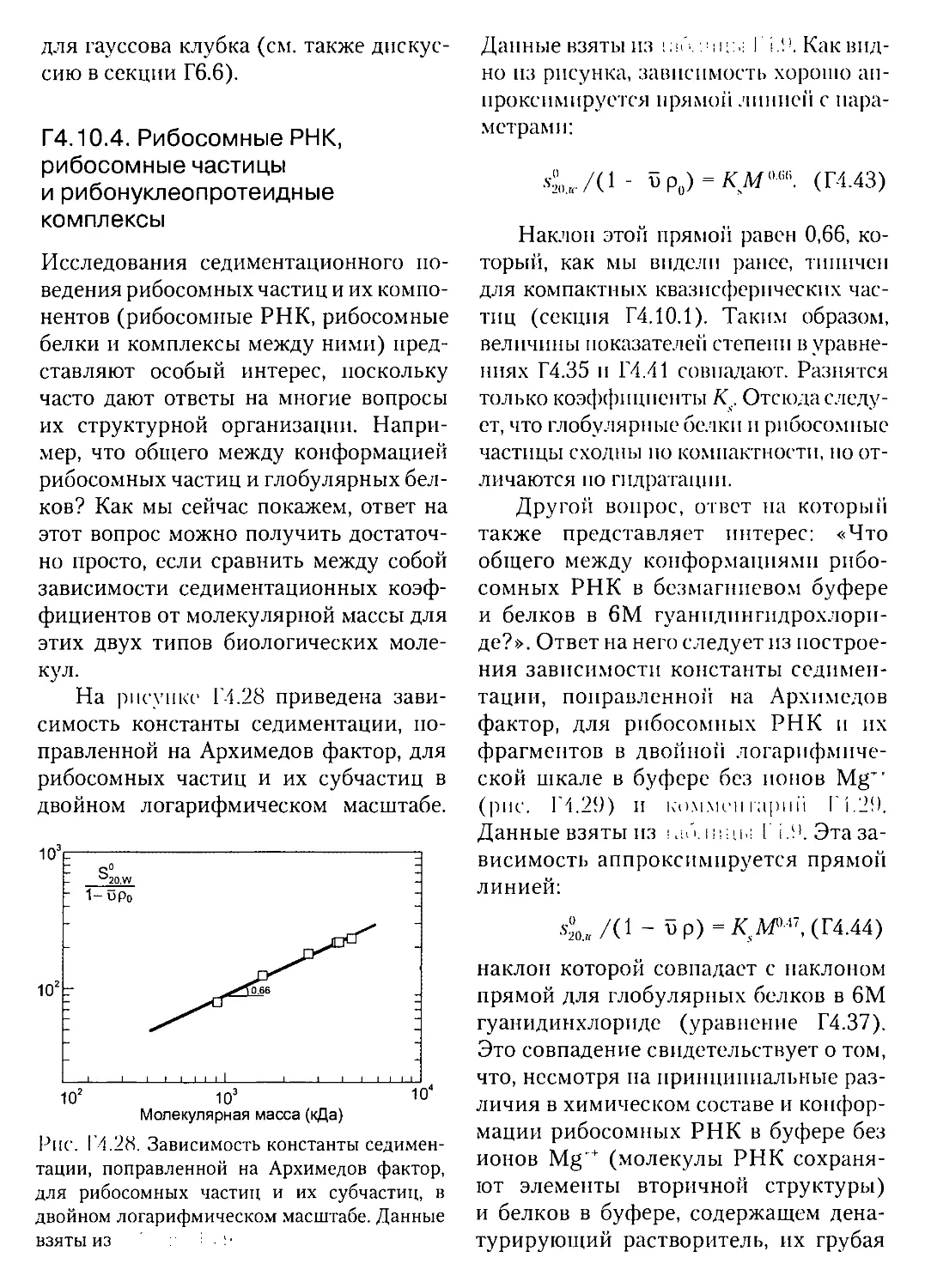
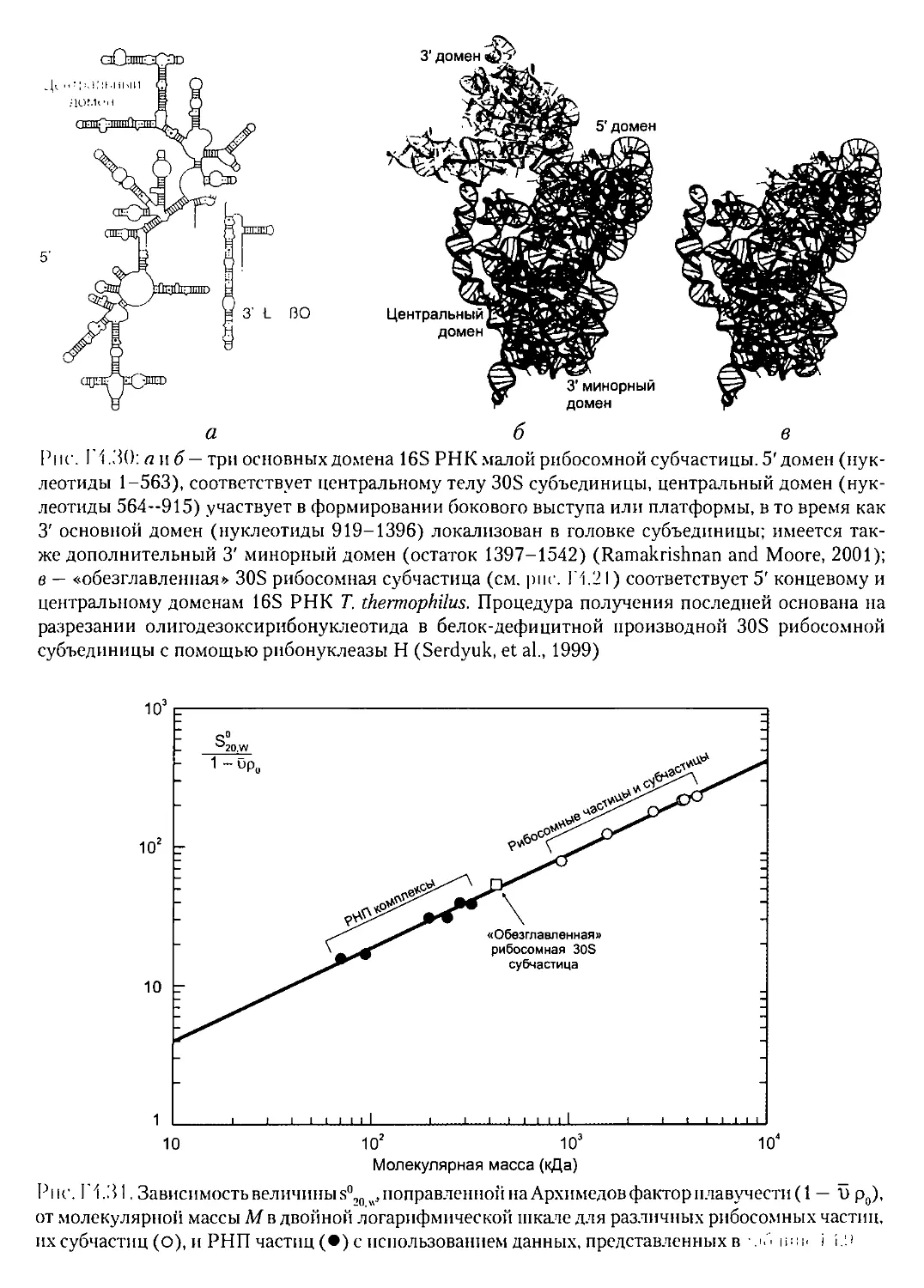
Г4.10.4. Рибосомные РНК, рибосомные частицы и рибонуклеопротеидные
комплексы................................................414
Г4.11. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ................................418
ЛИТЕРАТУРА ДЛЯ ПОСЛЕДУЮЩЕГО ЧТЕНИЯ...........................419
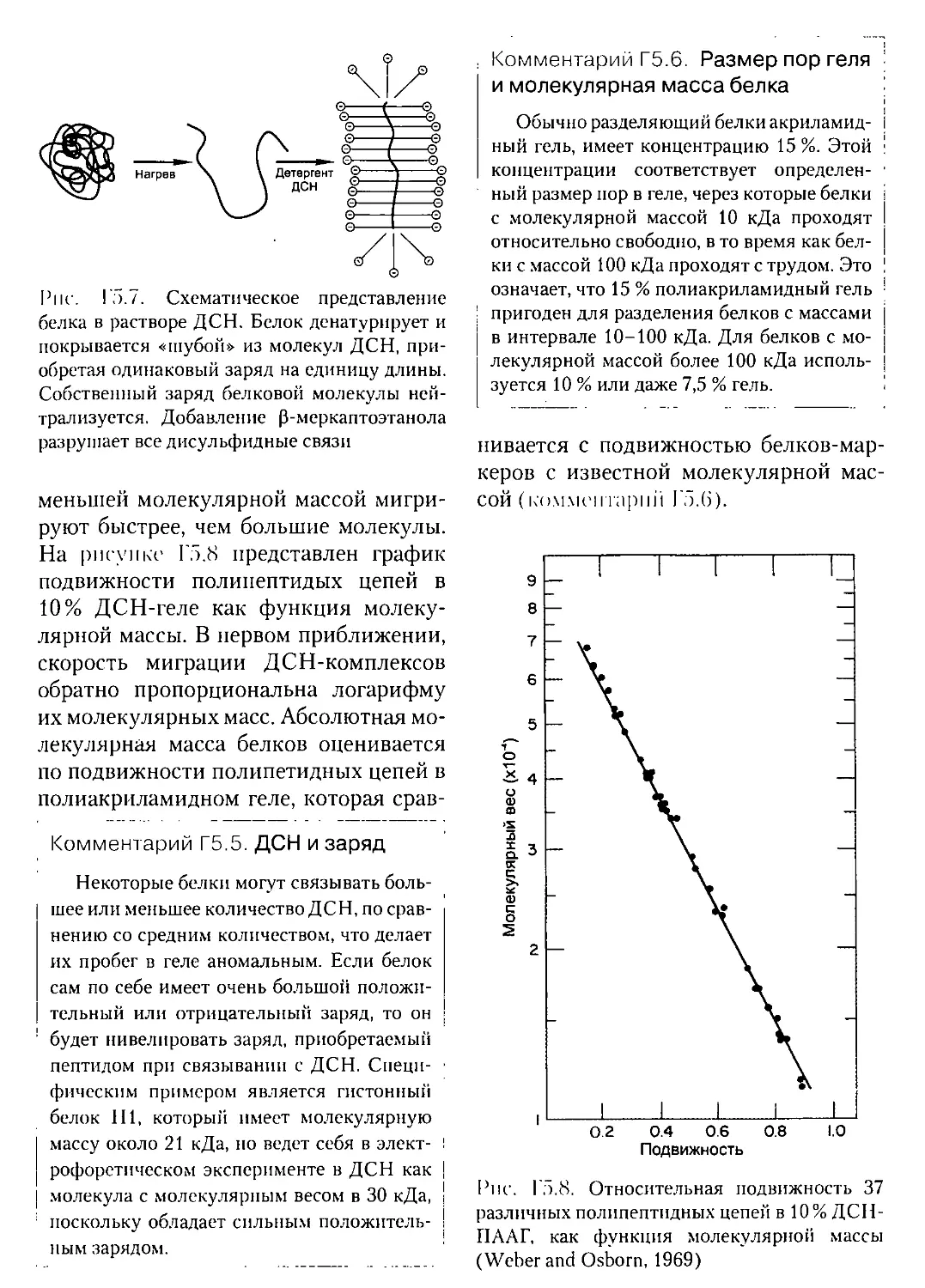
Глава Г5. Электрофорез
Г5.1. ИСТОРИЧЕСКИЙ ОБЗОР.......................422
Г5.2. ВВЕДЕНИЕ В БИОЛОГИЧЕСКИЕ ПРОБЛЕМЫ........424
Г5.3. ЭЛЕКТРОФОРЕТИЧЕСКИЕ ЭКСПЕРИМЕНТЫ.........425
Г5.4. МАКРОМОЛЕКУЛЫ В ЭЛЕКТРИЧЕСКОМ ПОЛЕ......
Г5.4.1. Заряд молекулы и ее электрофоретическая подвижность.423
Г5.4.2. Моделирование электрофоретической подвижности........427
Г5.5. ЭЛЕКТРОФОРЕТИЧЕСКИЕ МЕТОДЫ В ОТСУТСТВИЕ
ПОДДЕРЖИВАЮЩЕЙ СРЕДЫ: СВОБОДНЫЙ ЭЛЕКТРОФОРЕЗ...................428
Г5.5.1. Метод движущейся границы.............................429
Г5.5.2. Метод стационарного электрофореза....................430
Г5.6. ЭЛЕКТРОФОРЕТИЧЕСКИЕ МЕТОДЫ В ПРИСУТСТВИИ
ПОДДЕРЖИВАЮЩЕЙ СРЕДЫ: ЗОНАЛЬНЫЙ ЭЛЕКТРОФОРЕЗ...................431
Г5.6.1. Разделение по массе: ДСН — электрофорез..............432
Г5.6.2. Разделение по заряду: изоэлектрическое фокусирование.434
Г5.6.3. Одновременное разделение по заряду и массе:
двумерный гель-электрофорез...............................435
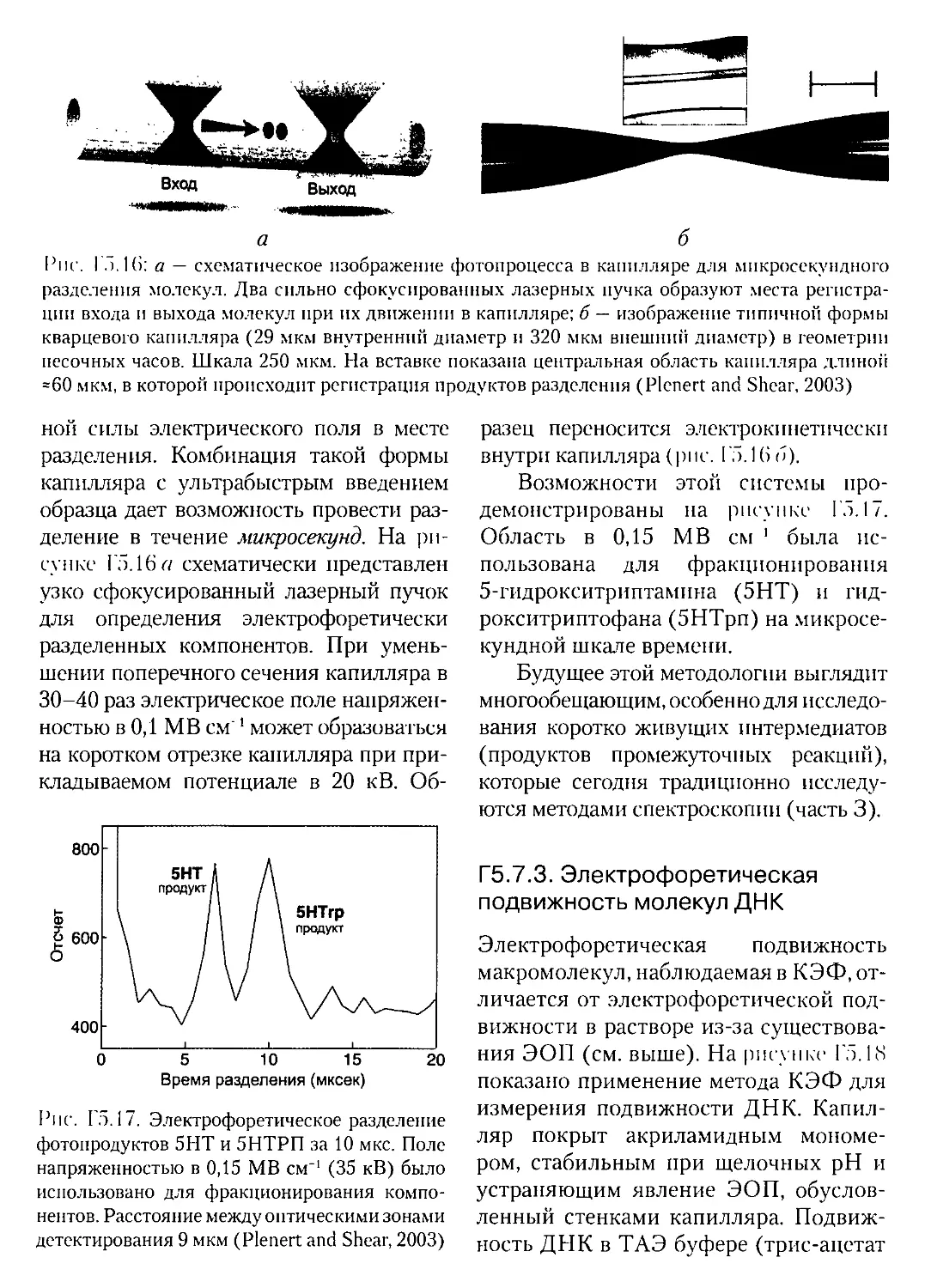
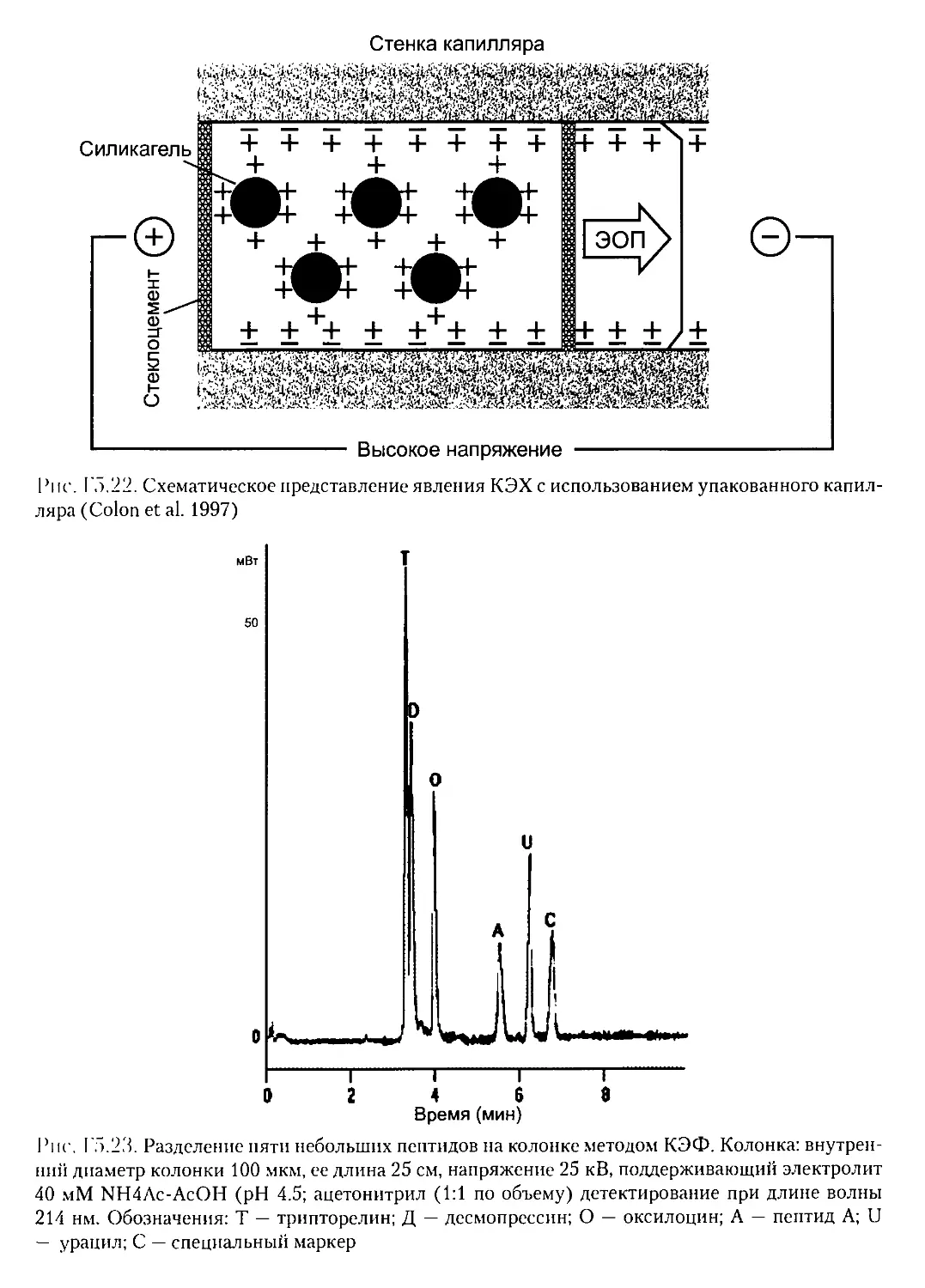
Г5.7. КАПИЛЛЯРНЫЙ ЭЛЕКТРОФОРЕЗ................................436
Г5.7.1. Одномерный капиллярныйэлектрофорез................436
Г5.7.2. Сверхбыстрый капиллярный электрофорез.............439
Г5.7.3. Электрофоретическая подвижность молекул ДНК.......440
Г5.7.4. Двумерный капиллярный электрофорез.................441
Г5.7.5, Разделение хиральимх молекул ....................... 4H
Г5Л КАПИЛЛЯРНАЯ ЭЛЕКТРОХРОМАТОГРАФИЯ........................... 444
ГМ ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ.......................................446
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ.............................. 417
Глава Г(>. Ориентация макромолекул
электрическим нолем................................... 44$
Гв.1. ИСТОРИЧЕСКИЙ ОБЗОР И ВВЕДЕНИЕ
В БИОЛОГИЧЕСКИЕ ПРОБЛЕМЫ...............................44$
Г6.2. МАКРОМОЛЕКУЛЫ В ЭЛЕКТРИЧЕСКОМ ПОЛЕ........................451
Гб,2.1. Дипольный момент биологических макромолекул..........451
Г6.2.2. Электрическое двойное лучепреломление и дихроизм.....452
Гб.З. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ИЗМЕРЕНИЙ ДВОЙНОГО
ЛУЧЕПРЕЛОМЛЕНИЯ........................................453
Г63.1. Стационарное двойное лучепреломление..................454
Г6.3.2. Процесс спада двойного лучепреломления...............454
Г6.33. Процесс нарастания двойного лучепреломления...........455
Г634. Двойное лучепреломление в реверсивных полях............456
Г6.4. ИЗМЕРЕНИЯ ЭЛЕКТРИЧЕСКОГО ДВОЙНОГО
ЛУЧЕПРЕЛОМЛЕНИЯ........................................456
Г6Л.1. Оптические схемы......................................456
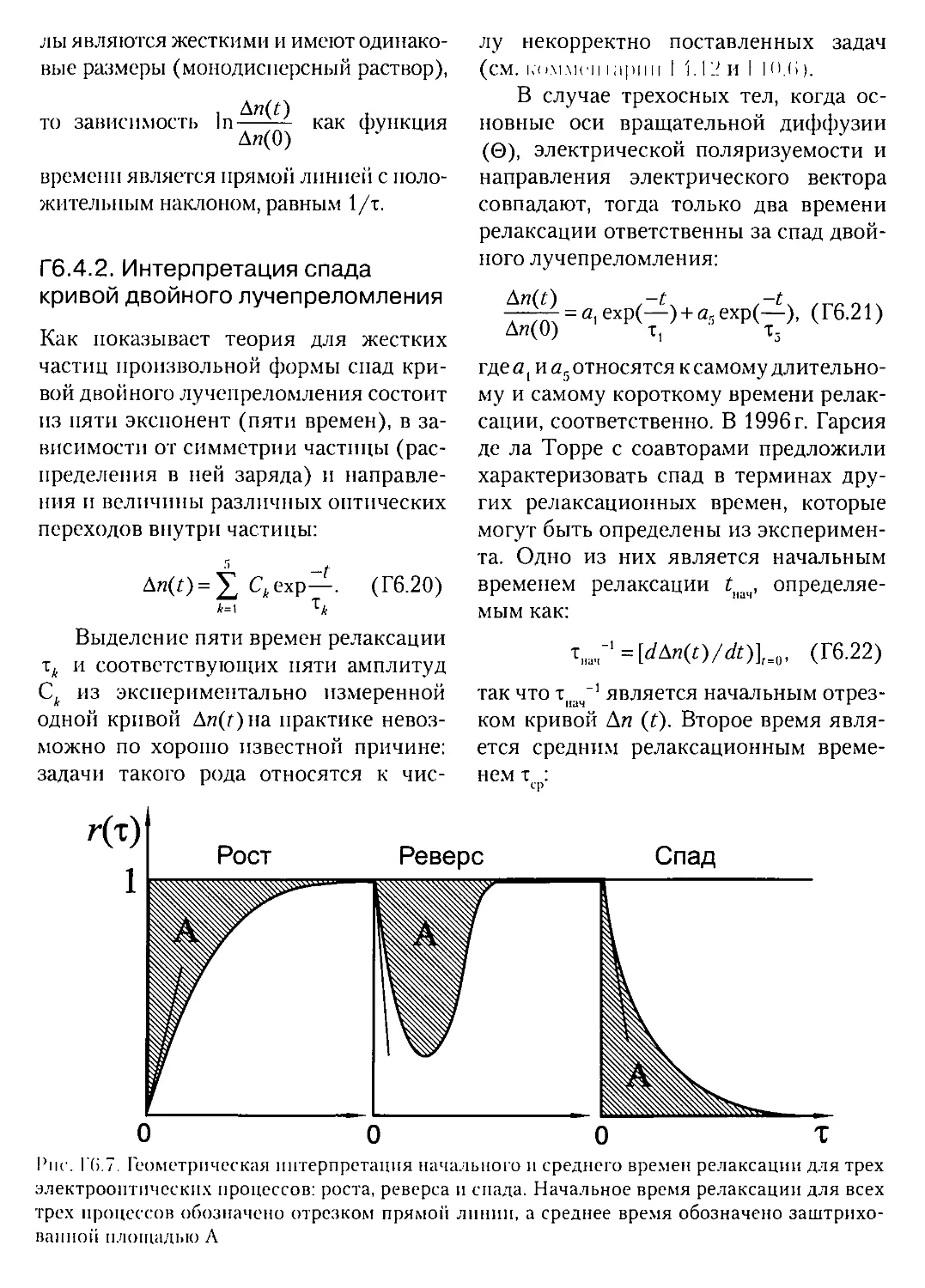
Г64.2. Интерпретация спада кривой двойного
лучепреломления.....................................459
Г6.5. ГЛОБАЛЬНАЯ СТРУКТУРА И ГИБКОСТЬ БЕЛКОВ...........460
Г6.5.1, Исследование внутренней структуры «моторного» домена.460
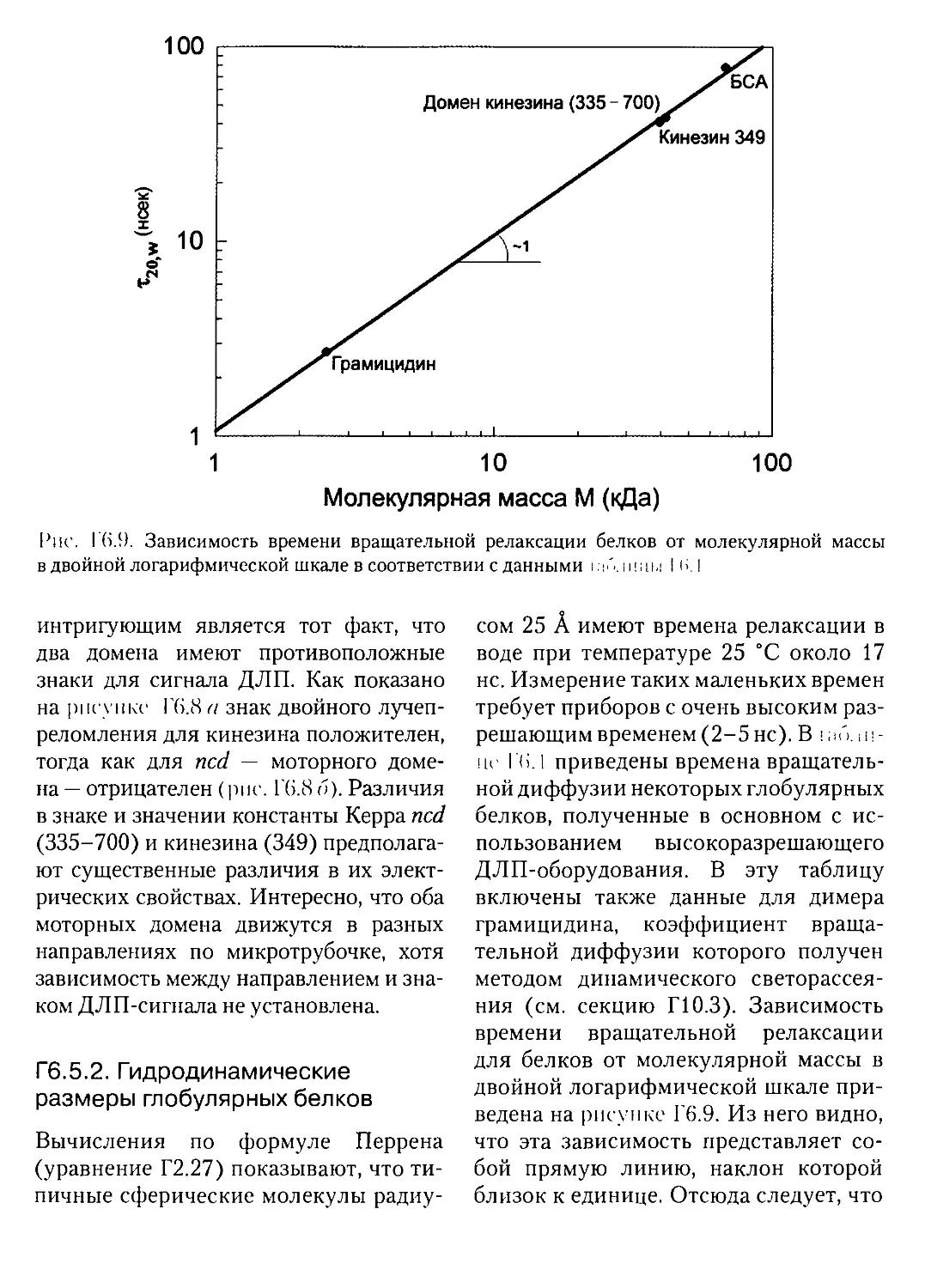
Г6.5.2. Гидродинамические размеры глобулярных белков.........461
Г6.6. ГЛОБАЛЬНАЯ СТРУКТУРА И ГИБКОСТЬ
НУКЛЕИНОВЫХ КИСЛОТ.....................................462
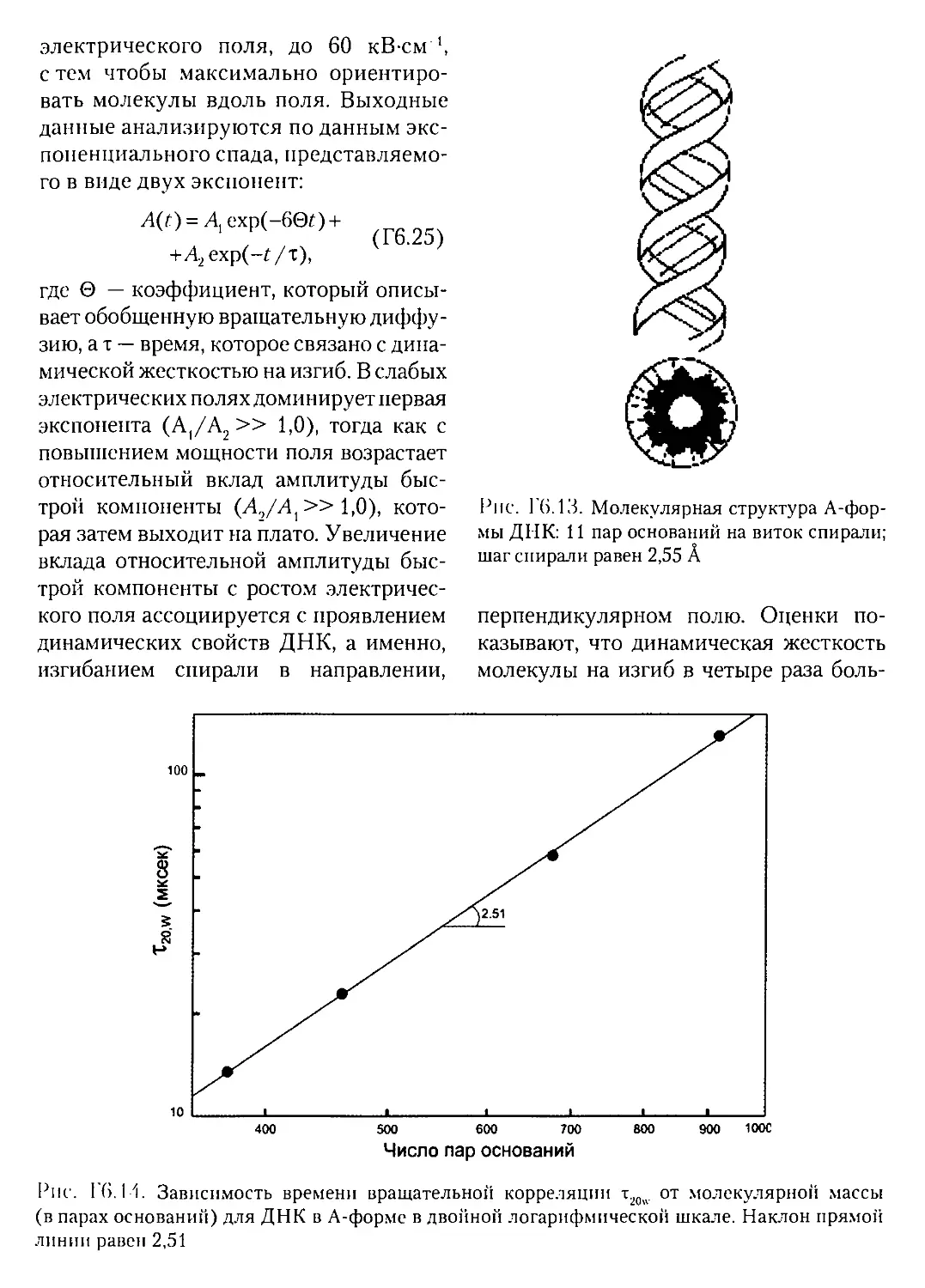
Г6.6.1. Гибкость молекулы ДНК в В- и А-форме.........462
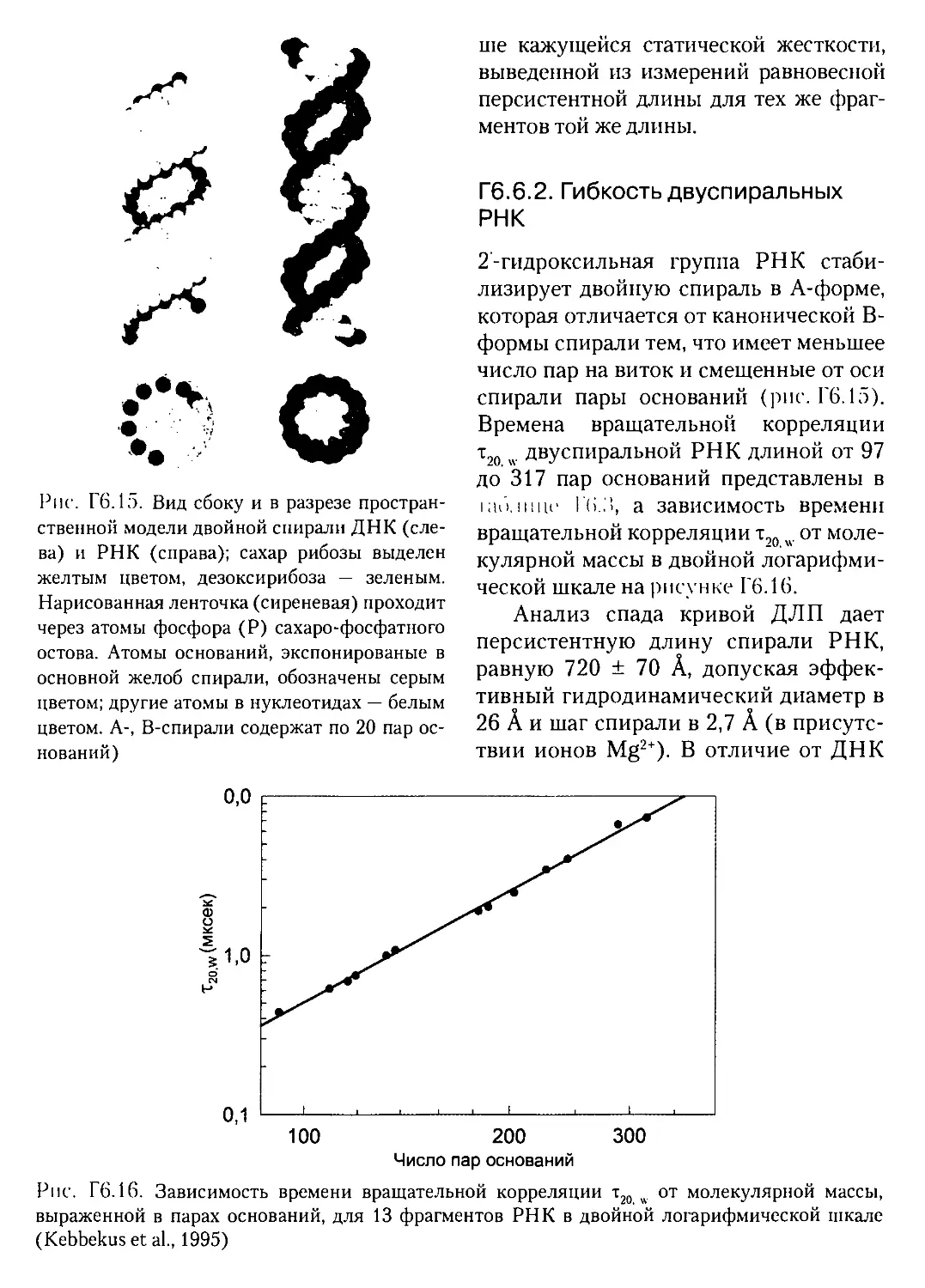
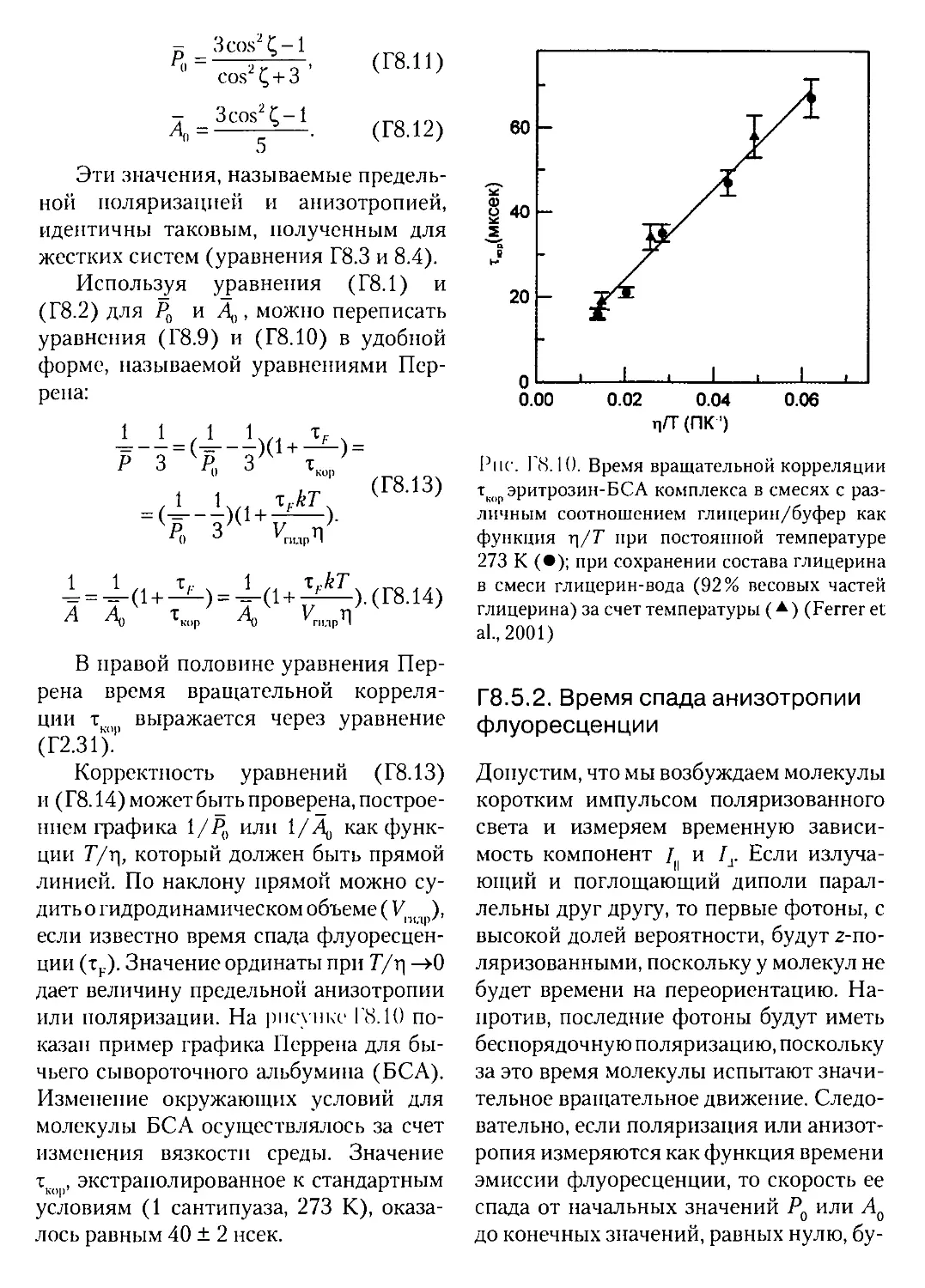
Г6.6.2. Гибкость двуспиральных РНК..................468
Гб.бЗ. Конформации неспиральных элементов в РНК..............469
Гб.7. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ...........................470
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ......................472
Глава 17. Ориентация макромолекул
гидродинамическим нолем................................473
ПЛ. ИСТОРИЧЕСКИЙ ОБЗОР..........................................473
П.2, ВВЕДЕНИЕ В БИОЛОГИЧЕСКИЕ ПРОБЛЕМЫ..........................474
ПЛ. СТАЦИОНАРНОЕ ДИНАМИЧЕСКОЕ ДВОЙНОЕ
ЛУЧЕПРЕЛОМЛЕНИЕ........................................474
П.ЗЛ. Угол ориентации и коэффициент вращательного трения.....476
ПЛ. СПАД ДИХРОИЗМА В ПОТОКЕ................................... 478
ПЛ. ОСЦИЛЛЯТОРНОЕ ДВОЙНОЕ ЛУЧЕПРЕЛОМЛЕНИЕ В ПОТОКЕ.. .479
[7.6. ОРИЕНТАЦИИ МАКРОМОЛЕКУЛ В ПОТОКЕ
КАК ДИНАМИЧЕСКОЕ ЯВЛЕНИЕ 480
Г7.7- ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ.. / ... .482
ЛИТЕРАТУРА ДЛЯ ПОСЛЕДУЮЩЕГО ЧТЕНИЯ. ..................482
Глава Г8. Деполяризованная флуоресценция........
Г8.1. ИСТОРИЧЕСКИЙ ОБЗОР...........................................
Г8.2. ВВЕДЕНИЕ В БИОЛОГИЧЕСКИЕ ПРОБЛЕМЫ............................
Г8.3. ТЕОРИЯ ДЕПОЛЯРИЗОВАННОЙ ФЛУОРЕСЦЕНЦИИ........................
Г8.3.1. Флуоресценция как физическое явление....................
Г8.3.2. Время жизни флуорофоров и время вращательной корреляции.
Г8.3.3. Стационарная деполяризованная флуоресценция.............
Г8.3.4. Деполяризованная флуоресценция, разрешенная во времени..
Г8.4. ЭКСПЕРИМЕНТАЛЬНАЯ ТЕХНИКА....................................
Г8.5. ДЕПОЛЯРИЗОВАННАЯ ФЛУОРЕСЦЕНЦИЯ
И БРОУНОВСКОЕ ДВИЖЕНИЕ...............................
Г8.5.1. Стационарная или статическая поляризация..
Г8.5.2. Время спада анизотропии флуоресценции.....
484
485
486
486
488
490
491
492
494
494
495
Г8.5.3. Вращательные корреляционные времена
глобулярных белков................................
Г8.6. ДЕПОЛЯРИЗОВАННАЯ ФЛУОРЕСЦЕНЦИЯ И МОЛЕКУЛЯРНЫЕ
ВЗАИМОДЕЙСТВИЯ................................
Г8.7. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ..................
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ.............
..496
..501
..502
..503
Глава Г9. Вязкость
Г9.1. ИСТОРИЧЕСКИЙ ОБЗОР.................................
Г9.2. ВВЕДЕНИЕ В БИОЛОГИЧЕСКИЕ ПРОБЛЕМЫ..................
Г9.3. ИЗМЕРЕНИЕ ВЯЗКОСТИ.................................
Г9.3.1. Капиллярные вискозиметры.....................
Г9.3.2. Ротационные вискозиметры.....................
Г9.3.3. Дифференциальный балансный вискозиметр.......
Г9.3.4. Вязкоупругая релаксация......................
Г9.3.5. Концентрационная зависимость характеристической вязкости • • •
Г9.4. ВЫЧИСЛЕНИЕ ХАРАКТЕРИСТИЧЕСКОЙ ВЯЗКОСТИ
БЕЛКОВ ИЗ ИХ АТОМНЫХ КООРДИНАТ....................
Г9.5. ОПРЕДЕЛЕНИЕ КОНФОРМАЦИИ МАКРОМОЛЕКУЛ ИЗ
ХАРАКТЕРИСТИЧЕСКОЙ ВЯЗКОСТИ...............................
Г9.5.1. Синтетические полипептиды....................
Г9.5.2. Белки в глобулярной конформации..............
Г9.5.3. Белки в клубкообразной конформации...........
Г9.5.4. Природно-неструктурированные белки...........
Г9.5.5. ДНК..........................................
..505
....505
....506
....506
....506
....508
....510
...511
...512
...513
...513
...515
...515
...516
...517
Г9.6. КОНФОРМАЦИОННЫЕ ИЗМЕНЕНИЯ В БЕЛКАХ
И НУКЛЕИНОВЫХ КИСЛОТАХ.....................
Г9.7. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ...............
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ...........
Глава ПО. Динамическое рассеяние света.......
Г10.1. ИСТОРИЧЕСКИЙ ОБЗОР............................
Г10.2. ВВЕДЕНИЕ В БИОЛОГИЧЕСКИЕ ПРОБЛЕМЫ.............
ПО З. ДИНАМИЧЕСКОЕ РАССЕЯНИЕ СВЕТА
КАК СПЕКТРОСКОПИЯ ОЧЕНЬ ВЫСОКОГО РАЗРЕШЕНИЯ...............52б
ПО.3.1. Флуктуации и временные корреляционные функции. 52g
ПО.3.2. Измерение динамической составляющей рассеянного света..........531
П 0.3.3. Коэффициенты диффузии, вычисляемые из динамической
составляющей рассеяния света...........................533
П0.4. ДИНАМИЧЕСКОЕ РАССЕЯНИЕ СВЕТА
В УСЛОВИЯХ ГАУССОВОЙ СТАТИСТИКИ...........................................535
Г10.4.1. Частицы, размеры которых малы по сравнению с длиной волны света... .536
Г10.4.2. Жесткие частицы, размеры которых сравнимы с длиной волны света 536
ПО.4.3. Гибкие частицы, размеры которых сравнимы с длиной волны света..539
ПОЛЛ. Электрофоретическое рассеяние света..............540
Г10.5. ДИНАМИЧЕСКОЕ РАССЕЯНИЕ СВЕТА
В УСЛОВИЯХ НЕГАУССОВОЙ СТАТИСТИКИ.........................543
Г10.5.1. Рассеяние малым числом частиц.................543
Г10.5.2. Кросс-корреляция (метод двух детекторов)......545
И 0.5.3. Рассеяние одной частицей......................545
П0.6. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ..............................546
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ.........................548
Глава Г11. Флуоресцентная корреляционная
спектроскопия................................550
П 1.1. ИСТОРИЧЕСКИЙ ОБЗОР................................550
Г11.2. ВВЕДЕНИЕ В БИОЛОГИЧЕСКИЕ ПРОБЛЕМЫ.................551
Г11.3. ТЕОРИЯ И ПРАКТИКА МЕТОДА..........................552
Г11.3.1. Трехмерная и двумерная диффузия..............553
Г11.3.2. Различные виды движений......................553
Г11.3.3. Динамический интервал времен.................554
Г11.3.4. Исследование многокомпонентных систем........^55
Г11.4. ДВУХЦВЕТНАЯ ФЛУОРЕСЦЕНТНАЯ
КРОСС-КОРРЕЛЯЦИОННАЯ СПЕКТРОСКОПИЯ...............
Г11.5. ПЕРЕЧЕНЬ КЛЮЧЕВЫХ ИДЕЙ....................
ЛИТЕРАТУРА ДЛЯ ДАЛЬНЕЙШЕГО ЧТЕНИЯ................
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ.............................
АВТОРСКИЙ УКАЗАТЕЛЬ..............................
556
558
559
560
564
ПРЕДИСЛОВИЕ
Эту книгу авторы посвящают женам
Ольге, Бринде и Мисси, которые
помогли осуществить задуманное
предисловие редактора русского перевода
Предлагаемая российскому читателю книга является переводом с английского кни-
ги И. Н. Сердюка (I. N. Serdyuk) Джузеппе Заккаи (J. Zaccai), Натана Заккаи (N. Zac-
cai), «Methods in Molecular Biophysics. Structure, Function, Dynamics», вышедшей в
издательстве Cambridge University Press (Кембридж, Англия) в 2007 году.
Основная задача, которую ставили перед собой авторы, состояла в том, чтобы
«собрать под одной крышей» физические методы исследования структуры биоло-
гических макромолекул, ставшие уже классическими, а также появившиеся относи-
тельно недавно. К первым мы отнесли методы гидродинамики и оптики, малоугловое
рассеяние рентгеновских лучей и нейтронов, рентгеноструктурный и нейтроност-
руктурный анализ, ядерный магнитный резонанс и электронную микроскопию. Для
более полного охвата в книгу включены термодинамические методы (сканирующая
и титрующая микрокалориметрия), которые обычно отсутствуют в классических
учебниках по структуре макромолекул. При описании методов авторы исходили из
принципа их равной значимости, не отдавая предпочтения ни одному из них.
Излагая фундаментальные основы физических методов, мы старались выбрать
наиболее яркие результаты, полученные с их помощью. Так, в книге описаны впе-
чатляющие успехи в исследовании структуры и функции биологических макро-
молекул с применением масс-спектрометрии, новые возможности капиллярного
электрофореза в геометрии «песочных часов», возрождение метода аналитичес-
кого ультрацентрифугирования, реконструкция одиночных молекул с помощью
электронной микроскопии, оптическая активность с применением синхротронных
источников, методы, основанные на явлениях поверхностного плазменного и маг-
нитного резонанса в магнитном поле Земли, преодоление классического светового
дифракционного барьера, составляющего, как известно, половину длины волны
падающего излучения. Не остались без внимания и фантастические достижения
рентгено-структурного анализа и ядерного магнитного резонанса, обеспечиваю-
щие сегодня основной поток структурных данных атомарного разрешения для бел-
ков, нуклеиновых кислот и комплексов между ними. Набирает силу нейтронно-
структурный анализ, обладающий уникальными возможностями по локализации
атомов водорода в молекулах.
Особое внимание в книге уделено физическим методам, которые фактически
открыли новую страницу в исследовании структуры биологических макромоле-
кул. К ним мы отнесли, в первую очередь, методы детектирования и манипули-
рования одиночными молекулами с применением оптических и магнитных лову-
шек (твизеров). На их основе совершен принципиальный переход от исследования
макромолекул в больших объемах (доли миллилитра) к исследованию макромо-
лекул в предельно малых объемах (доли аттолитра), что дает возможность пе-
рейти от описания свойств макромолекул в ансамбле к описанию их свойств на
одиночном уровне. Не сбылось предсказание великого Шредингера о том, что мы
никогда не сможем работать с одиночными электронами, атомами и молекулами.
Сегодня с помощью конфокальной микроскопии и микроскопии ближнего поля мож-
но детектировать флуоресцентно меченые одиночные молекулы, с помощью мик-
роскопии силового поля визуализовать одиночные молекулы, с помощью оптиче-
ских и магнитных ловушек растягивать, расстегивать, скручивать, раскручивать
биологические макромолекулы. В результате применения этих методов появилась
уникальная возможность визуализации и описания работы линейных и роторных
биологических моторов в терминах сил (пиконьютоны) и расстояний (нанометры),
сопровождающих каждую фазу их рабочего цикла. Все это привело к появлению
нового термина «сила» для описания механики биологических молекул. В повсе-
дневную практику вошли такие величины как ammo- и зептомоль, составляющие
10’18 и 10'21 моля, соответственно, которые ранее на практике не использовались.
Физика — быстро развивающаяся наука. Следствием такого развития является
появление двумерной инфракрасной спектроскопии, которая может рассматри-
ваться как аналог двумерного ядерного магнитного резонанса в оптической облас-
ти, и флуоресцентной корреляционной спектроскопии, которая является разви-
тием метода динамического рассеяния света для случая негауссовой статистики
(малое число частиц).
Излагая новые методы, авторы сохранили фундаментальные результаты, которые
получены около 50 лет назад и вошли в «золотой» фонд молекулярной биологии. Сре-
ди них: открытие П. Доти с сотр. (1956) переходов «спираль-клубок» в полипептидах,
обнаружение практически полного разворачивания белков в 6М гуанидингидрохло-
риде Ч. Тенфордом с сотр. (1968), доказательство непрерывности рибосомных РНК
А. Спириным (1963), получение X. Блумом и X. Шуллери (1955) спектра этанола на
частоте 60 МГц, открывшее метод ЯМ Р для химиков. И, конечно же, разработка М. Пе-
рутцем и Дж. Кендрю (1957 г.) метода рентгеновской кристаллографии, с помощью
которого структуры первых белков с атомарным разрешением были расшифрованы,
получение Р. Франклин и Р. Гослингом (1953) рентгеновских структур волокон ДНК,
приведшее к открытию Дж. Уотсоном и Ф. Криком (1953) двойной спирали ДНК. И,
наконец, подтверждение полуконсервативного механизма редупликации ДНК в блес-
тящих экспериментах М. Месселсона и Ф. Сталя, (1958) методом аналитического цен-
трифугирования в градиенте плотности.
В книге мы попытались донести до читателя тесную взаимосвязь всех физичес-
ких методов и их комплементарность. Например, понятие корреляционных враща-
тельных времен, лежащее в основе ряда гидродинамических методов, принципиально
важно для понимания метода ядерного магнитного резонанса, а понятие поступа-
тельной диффузии необходимо для понимания метода флуоресцентной корреляци-
онной спектроскопии. Анализ возможностей методов, имеющихся сегодня в нашем
распоряжении, свидетельствует о том, что не существует одного единственного ме-
тода, обеспечивающего всю необходимую информацию о макромолекулах и их вза-
имодействиях н только их совокупность дает видение системы в пространстве и во
времени. Именно комплементарность является основополагающей идеей, оправды-
вающей существование на практике всех физических методов.
Поскольку физики и биологи часто «разговаривают» на разных языках, книга
снабжена комментариями, предназначенными как для биологов, так и для физи-
ков. Главная их задача служить подсказками для тех и других.
Каждая глава, посвященная тому или иному методу, начинается с истории его
возникновения, вклада отдельных групп ученых в становление и развитие того или
иного физического метода. Заканчивается глава изложением ключевых идей, в ко-
торых сформулированы основные достоинства и ограничения каждого метода.
В русское издание включены дополнительно ссылки на некоторые русские ис-
точники, с тем чтобы сделать пользование литературой более доступным для на-
шего читателя.
Издание книги на русском языке в двух томах привело к некоторому ее пере-
форматированию, на что мы обращаем внимание читателя, знакомого с однотом-
ным английским вариантом книги.
Авторы выражают искреннюю благодарность Генеральному директору Компа-
нии «Базовый Элемент» О. В. Дерипаске и Генеральному директору Благотвори-
тельного фонда «Вольное Дело» Т. Д. Румянцевой за финансовую помощь в созда-
нии макета книги.
Моя личная, искренняя признательность коллеге по лаборатории Г. А. Енину за
оформление рисунков и иллюстраций, вклад которого в книгу трудно переоценить.
Особая признательность А. Ю. Хмельницкому, О. П. Сердюк, И. И. Проскуря-
кову и Ю. Н. Шарпинской за перевод книги, без большого труда которых не могло
бы состояться ее издание книги на русском языке.
Я благодарен Издательству «Книжный Дом “Университет”» и его ведущему ре-
дактору М. С. Климкин за творческое отношение к делу.
Отдельная глубокая благодарность моим коллегам, взявшим на себя труд про-
честь отдельные главы русского издания книги: В. В Филимонову, В. Д. Василь-
еву, О. М. Селивановой (Институт белка РАН), В. В. Волкову (Институт крис-
таллографии РАН), А. М. Балагурову (Лаборатория нейтронной физики ОИЯИ),
С. Н. Рязанцеву (Калифорнийский университет, США). Особое признание
Т. А. Хмельницкой за редактирование некоторых глав книги.
Написание книги такого объема с неизбежностью ведет к некоторой доле ошибок
и неточностей (надеемся не очень большой), за которые мы заранее приносим извине-
ния нашим читателям. Мы с благодарностью примем любые замечания и пожелания.
Профессор И. Н. Сердюк,
Пущино, 1 июня 2008 г.
Об авторах
Наше знание биологических макромолекул и их взаимодействий базируется на
применении физических методов в широком диапазоне -- от классической тер-
модинамики до недавно разработанных техник детектирования и манипуляции
одиночными молекулами. Описанные в этой книге методы включают в себя масс-
спектрометрию, гидродинамику, микроскопию, дифракцию и кристаллографию,
электронную микроскопию, молекулярную динамику и ядерный магнитный резо-
нанс. Каждый из них имеет свои преимущества и ограничения.
Предлагаемый учебник, объединивший эти подходы, дает описание и приме-
ры использования ключевых физических методов в современной биологии. Это
бесценный ресурс для студентов и аспирантов, изучающих молекулярную био-
физику в научных и медицинских высших учебных заведениях, а также для ис-
следователей, которым требуется знание основ методик, лежащих за пределами
их специальности. В соответствии с междисциплинарным подходом, которому
следовали авторы книги, в ней присутствуют короткие отступления (коммента-
рии), объясняющие физические аспекты биологам и биологические аспекты фи-
зикам.
Игорь Сердюк родился в Одессе (Украина) и обучался физике в Одесском госу-
дарственном университете. В 1969 г., после окончания аспирантуры на факультете
физики полимеров в Ленинградском институте высокомолекулярных соединений,
он защитил кандидатскую диссертацию. Затем, будучи научным сотрудником Ин-
ститута белка Академии наук СССР в Пущино, занимался исследованием физи-
ки белков. Здесь же впоследствии он возглавил свою исследовательскую группу
по физике нуклеопротеидов и получил степень доктора физ.-мат. наук в 1981 г.
С тех нор он возглавляет лабораторию физики нуклеопротеидов в Институте бел-
ка в Пущино. Степень профессора Игорь Сердюк получил в Московском госу-
дарственном университете в 1981 г., а 1985 — награжден Государственной премией
СССР в области науки и техники. На протяжении последних 25 лет он читает курс
лекций по применению физических методов в структурной биологии на биологи-
ческих факультетах Московского и Путинского государственных университетов.
Известен своими работами в области структурной нейтронографии биологических
макромолекул.
Натан Заккаи получил начальное образование во Франции, затем поступил
в Эдинбургский университет и в 1997 г. окончил его со степенью бакалавра фи-
зики. В 2001 г. ему присуждена степень доктора философии в области биохимии
в Оксфордском университете. Потом он занимался проблемами иммунологии,
вирусологии и структурной биологии, специализируясь на рентгеноструктурном
анализе. Его первая работа касалась структурных исследований рецепторов кле-
точной поверхности в иммунной системе. По программе структурной геномики в
Оксфорде Натан Заккаи занимался изучением вирусов герпеса и гриппа. В насто-
ящее время он является научным сотрудником университета в Кембридже и рабо-
тает над созданием вакцины против человеческого папиломавируса.
Джозеф (Джузеппе) Заккаи родился в Александрии (Египет), учился в английс-
кой школе сначала там, а затем в Риме, после чего поступил в Эдинбургский уни-
верситет, где получил сначала степень бакалавра (1968), а затем доктора философии
(1972) в области физики. Работа в национальной лаборатории Брукхэвэна в Нью-
Йорке пробудила у него интерес к биофизическим исследованиям биологических
мембран и разработке методов нейтронного рассеяния в биологии. Он продолжил
эту работу и после возвращения в Европу — сначала в Институте Лауэ-Ланжевена
в Гренобле, ведущем институте по нейтронному рассеянию. С 1992 г. он возглав-
ляет лабораторию молекулярной биофизики Института структурной биологии
(Гренобль) Национального центра научных исследований Франции. В настоящее
время научные интересы профессора Заккаи связаны с исследованиями в области
экзобиологии, а также динамики белков, их структуры и стабильности в организ-
мах, живущих в экстремальных условиях — при высокой концентрации солей, вы-
сокой температуре и давлении. Джозеф Заккаи имеет большой опыт преподавания
биологии физикам и физики — биологам. Он первым ввел эти курсы в программу
для студентов и аспирантов в университете Гренобля в 1980-х гг.
Предисловие от Д. М. Энгельмана
С первого взгляда может показаться, что эта книга — очередной обзор методов биофи-
зики, и, как обычно полагают, область этой науки определяется простым их набором.
Однако тщательно разработанное содержание учебника демонстрирует нам более глу-
бокое представление о предмете обсуждения. Методы, описанные здесь, определяют
то, что мы знаем о мире биологических молекул, а биофизика предстает областью зна-
ний, которая объединяет фрагменты информации, с тем чтобы наполнить содержани-
ем наше толкование биологии в терминах макромолекулярного пространства и вре-
мени: структуры, взаимодействия и динамики. Две взаимосвязанные идеи, которые
используют биофизики, — это структурно-функциональная гипотеза и эволюция.
Структурно-функциональная гипотеза обсуждается в прозрачно-понятном введе-
нии: идея заключается в том, что каждая закодированная в геноме молекула обладает
функцией, которую можно понять, рассматривая химическую структуру макромоле-
кул, изучая их взаимодействия и динамику. Эволюция лежит в основе этого подхода,
поскольку функция является фундаментом естественного отбора. Таким образом, био-
физические методы исследования дают нам представление - в пределах той информа-
ции, которую они способны предоставить, — о функции и эволюции. Особая надежда,
как награда за достигнутые успехи, заключается в том, что понимание каких-то част-
ных случаев приведет к основополагающим идеям — например, спаривание оснований
в нуклеиновых кислотах, связывание кислорода гемом или самосборка липидов.
Эта книга учит методам и создает надежный интеллектуальный каркас для си-
стематизации наших знаний, что позволяет ей быть путеводителем в данной обла-
сти. Предыдущие работы — Кона и Эдселла, Тэлфорда, Эдселла и Ваймана, а также
Кантора и Шиммела — выполняли эту исключительно важную роль в прошлом.
Но время не стоит на месте, и технологии, описанные в их классических трудах, не-
сколько утратили свою актуальность в процессах обучения и в практике. Поэтому
можно только приветствовать ясно написанный, вдумчивый и современный текст,
который сослужит хорошую службу как на специальных курсах, так и в качестве
справочника. Авторы описали каждый метод, исходя из фундаментальных при-
нципов и предпосылок, успешно охватив впечатляющий диапазон исследований.
И за это они будут вознаграждены вниманием аудитории, которая так жаждет но-
вых фундаментальных текстов по молекулярной биофизике.
Йельский университет,
Нъю-хейвен, США
Предисловие от Пьера Жолио
Как написали во введении авторы книги, идеальный биофизический метод мог бы
предоставить возможность наблюдать структуры биологических молекул и их ди-
намику на атомном уровне в физиологичной для них среде, т. е. in vivo. Конечно, та-
кого метода не существует, и, возможно, никогда не будет существовать из-за бес-
численных технических ограничений, поэтому для характеристики структурных
и функциональных свойств биологических макромолекул необходимо применять
целый арсенал комплементарных методов. Мы должны, однако, заметить, что на
практике многие высокопродуктивные группы молекулярных биофизиков скон-
центрированы на какой-либо одной технике, из которой они стараются «выжать»
все, что возможно. Эти исследователи разрабатывают, в сущности, методологиче-
ский подход, с помощью которого стараются охарактеризовать как можно большее
число биологических молекул. Примером такого подхода может служить высоко-
производительная кристаллография или структурная геномика. Тут цель одна —
дать точную информацию для базы трехмерных белковых структур, аналогично
базам данных по первичным структурам геномных последовательностей, которые
широко используются всеми биологами.
Другой подход в решении трудной биологической задачи — мультидисцип-
линарный, и ему в молекулярной биофизике принадлежит ведущая роль. Здесь
цель иная — как можно более точно определить функциональные, структурные
и динамические свойства молекул, участвующих в физиологических процессах, а
также их взаимодействия. Именно такой подход поддерживает эта книга, которая
дает важный и исчерпывающий обзор современных методов исследования, а так-
же обсуждает преимущества и недостатки каждого. Обычно первым шагом бывает
изучение каждой молекулы в очищенном виде. Для большинства биофизических
исследований требуются образцы, состоящие из большого числа идентичных мо-
лекул (в 1 мг белка с молекулярным весом 60 000 содержится 1016 молекул), что
позволяет достигнуть необходимой чувствительности измерений при миними-
зации повреждений, нанесенных самим экспериментом (например, излучением).
Следовательно, эти молекулы изучают в условиях, которые отличаются от естест-
венных. Следующий шаг — наблюдение за ассоциацией молекул и в особенности
за надмолекулярными структурами, которые, как сейчас считают, присутствуют в
клетке. Там, где невозможно организовать эти комплексы в упорядоченные двух- и
трехмерные структуры, их можно наблюдать только с низким пространственным
разрешением.
Эта книга содержит также главы, посвященные новым многообещающим направ-
лениям, например детектированию одиночных молекул и манипуляциям с ними.
Последний шаг, который все еще трудно сделать, связан с исследованием молекул и
комплексов in vivo. В этом контексте нужны не только новые технические подходы,
но и новые способы мышления, даже если в результате будет разработано всего не-
сколько биофизических методов, способных давать информацию о молекулах в их
клеточном окружении. Применяя функционально-структурный подход в дополне-
ние к традиционному структурно-функциональному, ученые получают возможность
изучить, какие структурно-организационные модели совместимы с функциональ-
ными свойствами ансамбля молекул. Например, благодаря термодинамическому и
кинетическому анализу реакций переноса электрона в фотосинтетическом аппарате
теперь известно, что мобильные носители, составляющие фотосинтетическую цепь
переноса электрона, объединены в домены. Эти домены могут быть как небольшими
мембранными компартментами, изолированными друг от друга, так и суперкомплек-
сами, сформированными в результате ассоциации нескольких больших мембранных
белков, которые захватывают мобильные носители. Во многих случаях мембраны,
также как и цитозоль, выглядят высоко компартментализованными системами. Оп-
ределение надмолекулярной организации в этих компартментах, несомненно, будет
одной из главных задач современной биофизики.
Институт биологической и физической химии,
Франция
От авторов
Андре Гииье, чьи фундаментальные исследования внесли большой вклад в разви-
тие методов, основанных на дифракции рентгеновских лучей и которые составля-
ют основу современной структурной молекулярной биологии, умер в Париже в
начале июля 2000 г. — всего лишь через несколько недель после того, как в прессе
было объявлено о завершении проекта по секвенированию человеческого генома.
Вскоре ушли из жизни Макс Перутц, Френсис Крик и Дэвид Блоу, самый млад-
ший из первых исследователей в области кристаллографии белка. С их именами
было связано рождение молекулярной биологии и с их уходом совпало открытие
новой эпохи, которую называют постгеиомным секвенированием. В ней физиче-
ские методы исследования играют все более важную роль в понимании биологи-
ческой функции на молекулярном и клеточном уровне.
Классические учебники по молекулярной биофизике, опубликованные в пре-
дыдущих десятилетиях, во многом устарели, но не только вследствие значительно-
го развития методов, вызванного, например, приходом синхротронных источников
для рентгеновской кристаллографии или мощных сверхпроводящих магнитов для
ЯМР. Родились совершенно новые методы, такие как масс-спектрометрия, мани-
пуляции с одиночными молекулами и их детектирование, флуоресцентная корел-
ляционная спектроскопия. В соответствующих главах мы описали, как класси-
ческие, так и самые «продвинутые» техники, основанные на масс-спектрометрии,
термодинамике, гидродинамике, спектроскопии, микроскопии, радиационном рас-
сеянии, электронной микроскопии, молекулярной динамике и ЯМР. Однако быс-
трый прогресс в этой области, который происходил также и в течение тех несколь-
ких лет, которые понадобились нам для написания и подготовки книги, а также
желание сохранить книгу в разумном объеме означает, что некоторые методы были
опущены по этим причинам, либо потому, что некоторые из них устарели.
Ключевое слово в молекулярной биофизике — комплементарность. Для нашей
области метафорой может служить индийская история о шести слепых и слоне.
Каждый слепой прикасался к различной части животного и на основании своих
ощущений делал вывод о сто природе. «Большая змея», — сказал человек, который!
прикоснулся к хоботу. Бивни казались копьями, бок — стеной, хвост — кисточкой,
уши — опахалами, а ноги — стволами деревьев. Мы могли бы добавить еще и седь-
мого, очень близорукого человека, который видел бы целого слона как туманное
серое облако, чтобы проиллюстрировать дифракционные методы. На самом деле,
повторяем, идеального метода в молекулярной биофизике не существует. Мы не
только не можем следить за положением атомов в молекулах in vivo, но и не спо-
собны увидеть их движение, и конформационные изменения, которые происходят
в процессе химических реакций, связанных с их биологической функцией, — при-
чем на любой временной шкале. Ни одна экспериментальная техника не способна
дать такую информацию. Каждая обладает ограниченным полем зрения, со своими
четкими участками, выступающими из глубокой тени. Физические методы XXI
в. должны справиться со множеством сложных биологических проблем, решение
которых будет зависеть от их способности переносить структурную и функцио-
нальную информацию из масштабов одной клетки на клеточный уровень, а затем
и на весь организм. Величие и невероятная сложность задачи не заставят ученых
отступиться, вызов времени будет принят.
Мы благодарны профессору Дону Энгельману из Йельского университета
(США) и профессору Пьеру Жолио из Института биологической и физической
химии (Франция), которые согласились написать предисловия к нашей книге.
Выдающиеся ученые и педагоги, они одновременно участники биофизических ис-
следований и наблюдатели развития современной биологии. Не можем пи сказать
слова благодарности коллегам-эксиертам за критичное обсуждение различных
методов: Мартине Блэкледжу и членам лаборатории ЯМР, Кристине Эбель, Дику
Уэйду, Хью Лорта-Жакобу, Патрисии Амара, сотрудникам лаборатории масс-спек-
трометрии, всему Институту структурной биологии (Гренобль, Франция), Регине
Виллумайт, Национального центра нейтронных исследований GKSS (Гисстхахт,
Германия), Виктору Аксенову из Объединенного института ядерных исследова-
ний (Россия), Лесли Грин, Кристине Редфилд, Гиллому Стюарт-Джонсу, Ивон-
не Джонс и Дэвиду Стюарту из Оксфордского университета (Англия), Джоната-
ну Рупрехтанду и Ричарду Хендерсону из Лаборатории молекулярной биологии
(Англия), Симону Хэнслип и Роберту Фалконеру из Кембриджского университе-
та (Англия), Владимиру Филимонову из Института белка РАН.
Мы признательны также Бринде Мутузаме, которой принадлежит идея фрон-
тисписа, Ольге Сердюк, которая на протяжении всех лет активно участвовала в
поиске литературных источников, их техническом оформлении, Миссии Заккаи,
помогавшей в редактировании отдельных мест текста. Л также признательны со-
трудникам Института белка РАН: Александру Тимченко, Маргарите Шелестовой,
Маргарите Ивановой, Татьяне Кувшинкиной и Альбине Овчинниковой за техни-
ческую помощь.
Выражаем также глубочайшую признательность за финансовую поддержку
фонду Радульфа Обертюра (Бавинкель, Германия), Институту структурной био-
логии и Институту Лауэ-Ланжевена (Гренобль, Франция), Лаборатории нейтрон-
ной физики ОИЯИ (Дубна, Россия), Институту белка РАН (Пущино, Россия), Пу-
тинскому центру научных исследований в лице Вячеслава Корнилова (Пущино,
Россия), компании «Кирилл Сердюк» (Украина). Наконец, без теплых слов благо-
дарности мы не вправе оставить наших коллег, друзей, членов своих семей, а также
команду Издательства Кембриджского университета — всех тех, кто поддерживал
пае своим терпением, пониманием и одобрением нашего скромного труда.
Введение
МОЛЕКУЛЯРНАЯ БИОЛОГИЯ
В НАЧАЛЕ XXI В.: ОТ АНСАМБЛЯ -
К ОДИНОЧНЫМ МОЛЕКУЛАМ
Краткая история и перспективы
Рождение молекулярной биологии связано с созданием модели двойной спира-
ли ДНК, обеспечившей элегантное объяснение механизма сохранения и передачи
генетической информации (рис. 1). Модель Уотсона и Крика, а также параллель-
ные дифракционные исследования М. Вилкинса, А. Стокса и X. Вилсона, а также
Р. Франклина и Р. Гослинга (М. Wilkins, A. Stokes & Н. Wilson, and R. Franklin &
R. Gosling), опубликованные в серии статей в апреле 1953 г. в Nature, явились три-
умфом использования физических методов в биологии. Модель Уотсона и Крика
только частично базировалась на дифракционных диаграммах рентгено-структур-
ного анализа волокон ДНК. Спиральные молекулы с постоянным шагом и диамет-
ром не были однозначным подтверждением точности предлагаемой модели. Гени-
альной догадкой в этом открытии было предположение об одинаковых размерах
пар оснований А-Т и G-С, что предопределило постоянный диаметр и шаг в звень-
ях цепи двойной спирали.
ДНК
Рис. 1. а — химическая формула одиночной цени ДНК; б — схематическое изображение двух
спаренных цепей ДНК в виде двух лент, состоящих из остатков фосфорной кислоты и сахаров.
Горизонтальные черточки, обозначают пары оснований нуклеотидов, удерживающих цепи вме
сте. Вертикальная линия обозначает ось молекулы ДНК; в - химическая формула спаренных
цепей ДНК. Водородные связи обозначены линиями, состоящими из точек; г — дифракция нити
В-формы ДНК в рентгеновских лучах. Рисунки скопированы из оригинальных работ Уотсона
и Крика (1953), Франклина и Гослинга (1953)
С точки зрения «дифракционной физики» разнообразные спиральные моде-
ли согласуются с дифракционными диаграммами волокон ДНК. Одной из таких
альтернативных моделей была модель «спина к спине» («side-by-side model»), со-
стоящая из двух спаренных одиночных спиралей ДНК. Несмотря па то, что такая
модель впоследствии оказалась неверной, она ясно показала, что для дальнейше-
го становления молекулярной биологии очень важно уметь определять структуру
биологических молекул с более высоким пространственным разрешением, чем это
возможно из дифракционных методов на волокнах.
Для определения расстояний между атомами в кристаллах наиболее подходя-
щим является метод рентгено-структурного анализа, разрешающая способность
которого около 1А (0,1 нм). Однако при этом основной проблемой остается полу-
чение высококачественных кристаллов, что существенно для точного определения
положения всех атомов в макромолекуле и решения фазовых проблем.
Первые кристаллы белков были получены уже в 1930-е гг. Однако только
к 1957 г. Макс Перутц и Джон Кендрю (Max Perutz и John Kendrew) нашли путь к
решению фазовых проблем в кристаллографии, используя производные с тяжелы-
ми атомами. Это позволило получить детальную трехмерную структуру миоглоби-
на. Проблемы, связанные с кристаллизацией белков (всего несколько исследовате-
лей в мире умели это делать), и тяжелый труд кристаллографических исследований
сам по себе (это была докомпьютерная эра и все вычисления выполнялись руками
аспирантов) обрекали белковую кристаллографию на получение информации о
трехмерной структуре очень немногих биологических макромолекул. Тем не ме-
нее «структурщики» продолжали развивать и улучшать методы, хотя полностью и
не обеспечивающие желаемого разрешения на атомном уровне, но имеющие свои
преимущества при изучении макромолекулярных структур. Эти методы, увязыва-
ющие между собой термодинамику и структуру, уже играли важнейшую роль в
столетии, предшествующем созданию модели двойной спирали. Открытие биоло-
гических макромолекул само по себе тесно переплеталось с приложением физи-
ческих концепций и методов к биологии (молекулярной биофизике).
Использование физики для решения инструментальных проблем в биологии
определенно происходило раньше, чем появился сам термин «биофизика». В бри-
танской энциклопедии предполагается, что изучение биолюминесцении Атанаси-
усом Киршером (Athanasius Kircher) в XVII в. может считаться одним из первых
биофизических исследований. Им было показано, что экстракт, полученный из
светлячков, не может быть использован для домашнего освещения.
Установление связи между биологией и тем, что впоследствии станет извес-
тно как электричество, принадлежит физикам. Исаак Ньютон (Isaac Newton)
в заключительном параграфе своих Principia (1687), отмечает, что «...все чувства
есть результат возбуждения, при котором каждая частица животного организма
движется по команде воли, что проявляется в вибрации духа, и его распростране-
нии вдоль твердых нервных волокон от внешних органов чувств к мозгу и от мозга
к мускулам. Однако это те вещи, которые не могут быть объяснены несколькими
словами, или обоснованы надежными экспериментами, которые необходимы для
точного определения и демонстрации законов, по которым этот электрический
и эластичный дух действует».
Столетием позже Луиджи Гальвани и Алессандро Вольта (Luigi Galvani,
Alessandro Volta) провели эксперименты на лапках лягушки, что привело к изоб-
ретению электрической батарейки. Эти эксперименты стали фундаментом для
становления электрофизиологии как науки, хотя опыты по возбуждению с приме-
нением электрических батареек стали возможны только в XIX в., и были впослед-
ствии продолжены известным ученым Эмилем Генрихом Дюбуа-Реймондом
(Emil Heinrich Du Bois-Reymond), исследовавшим животное электричество. Дру-
гая ветвь биофизики XIX столетия, связанная с исследованиями диффузии и ос-
мотического давления в растворе и, частично совпадающая с физической химией,
имеет более непосредственное отношение к открытию и изучению биологических
макромолекул. Публикация первой статьи в Zeitschrift fur Physikalische Chemie
(1887), о реакциях в растворе, была обусловлена тем, что биологические процессы
внутри живущей клетки, в основном, происходят в водной среде.
Тепловое движение частиц в растворе (броуновское движение) было открыто
Робертом Броуном (Robert Brown) в 1827 г. Аббе Ноллет (Abbe Nollet), про-
фессор экспериментальной физики, впервые описал осмотическое давление в на-
чале XIX столетия, используя в эксперименте мембраны животных для разделения
спирта и воды. Дальнейшее изучение и обозначение названия этого феномена при-
надлежит доктору медицины и физиологу Рене Дютроше (Rene J. Н. Dutrochet)
(1828), который осознал важность осмотического давления в живых системах и
предположил, что основные биологические процессы могут быть объяснены в тер-
минах физики и химии. Теория осмотического давления была развита Джоном
Вант-Гоффом (1880) (J. Van’t Hoff).
Георг Габриель Стокс (George Gabriel Stokes) в середине XIX столетия ус-
тановил важнейший закон движения частиц в вязкой среде. Закон диффузии в
градиенте концентрации был описан Адольфом Фиком (Adolf Fick) в 1856 г., по
аналогии с законами, действующими для описания потоков тепла. Во второй по-
ловине XIX столетия были также сделаны открытия двойного лучепреломления в
потоке Джеймсом Клерком Максвеллом (James Clerk Maxwell) и электрического
двойного лучепреломления в растворе Джоном Керром (John Kerr). Оба эти фе-
номена проявляются, как правило, только при достаточно большой асимметрии
растворенных частиц.
Большинство макромолекул хотя и являются большими молекулами, но они
намного меньше, чем длина световой волны. Они не могут быть обнаружены пря-
мым наблюдением в микроскопе, хотя при его помощи можно разглядеть клетки в
биологических тканях и структуры внутри клетки, такие как хромосомы (в пере-
воде с греческого — «окрашенные тела»). Постепенно накопленные данные, полу-
ченные Эмилем Фишером (Emil Fischer) (1882) из его экспериментов на раство-
рах, показали, что биохимическая активность белков, на самом деле, обусловлена
обособленными макромолекулами. В 1908 г. Жан Перрен (Jean Perrin) приложил
теорию Альберта Эйнштейна (Albert Einstein) (1905) к броуновскому движению
для вычисления числа Авогадро. Теория макромолекул, основанная на концепции
Германа Штаудингера (Hermann Staudinger) (1920), позволила Вернеру Куну
(Werner Kuhn) в 1930 г. утвердить представления о макромолекулах как о дискрет-
ных частицах. Открытие рентгеновских лучей Вильгельмом Конрадом Рентгеном
(Wilhelm Conrad Rontgen) (1895) и их приложение к атомистической кристалло-
графии в 1910-х гг. в исследованиях Петера Эволда, Макса фон Лауэ, X. Уильяма
и В. Брэгга (Peter Ewald, Max von Laue, H. William и W. Laurence Bragg) зало-
жили основы для работ по атомной организации структуры макромолекул.
Теодор Сведберг (Theodor Svedberg) (1925) впервые определил молярную
массу белковой макромолекулы, используя созданный им метод аналитического
центрифугирования. В эти годы теория атомного строения вещества становится
признанным фактом. Методы дифракции рентгеновских лучей и кристаллогра-
фии, атомная спектроскопия быстро прогрессировали. Неизвестные ранее инс-
трументальные методы, применение которых стало возможно благодаря новому
пониманию взаимодействия радиации и вещества, тщательно оттачивались, с тем
чтобы исследовать биологические структуры на молекулярном и атомном уровнях.
Физики, вдохновленные примером Макса Дельбрюка (Max Delbriick), который
выбрал в 40-х гг. прошлого века для изучения генетики бактериофаги (бактери-
альные вирусы) как наиболее простые модели, и книгой Эрвина Шрёдингера «Что
такое жизнь?» (1940), в которой поднимался вопрос о том, возможно ли обойтись
известными законами физики при описании биологических процессов, принялись
за решение биологических проблем самым активным образом.
К концу XX в. в биофизике доминировали два метода исследования, рентге-
но-структурный анализ и ЯМР, которые играли ключевую роль в определении
трехмерной структуры биологических макромолекул с высоким пространственным
разрешением. Но, даже если представить, что все белковые структуры в различных
геномах были бы определены, все равно оставались бы невыясненными ключевые
вопросы: что происходит со структурой и какова динамика каждой макромолекулы
при соприкосновении живой клетки с окружающей средой; как меняется структура
при проявлении биологической активности; как макромолекулы взаимодействуют
друг с другом в пространстве и во времени? Эти вопросы могут быть разрешены
только при комбинированном и адекватном использовании практически всех био-
физических методов. Масс-спектрометрия может определять макромолекулярную
массу с изумляющей точностью. Высокочувствительный метод микрокалориметри-
ческого сканирования и титрования может быть использован для определения тер-
модинамики разворачивания макромолекулы, ее стабильности и, при объединении
с биосенсорной техникой, для исследования взаимодействий при связывании белка
с лигандом (партнером). Наблюдается возрождение метода аналитического центри-
фугирования с приходом новых высокоточных и автоматизированных инструментов.
Его комбинация с методами малоуглового рентгеновского и нейтронного рассеяния
обеспечивает новый виток исследования структуры макромолекул, их взаимодей-
ствия в растворе и роли гидратации. Кинетика быстротекущих процессов методом
оптической спектроскопии исследуется в шкале фемтосекунд. Световой микроскоп
в комбинации с флуоресцентными зондами позволяет локализовать одиночные мо-
лекулы внутри клетки. Методом сканирующей микроскопии силового поля мож-
но определить профиль поверхности макромолекулы и его изменения во времени.
Электронная микроскопия с ее разрешением, близким к атомарному, скорее всего
явится мостом, соединяющим исследования на молекулярном и клеточном уровнях.
Нейтронная спектроскопия обеспечивает информацией о динамике функциониро-
вания белков внутри живой клетки. Метод кругового дихроизма с использованием
синхротронного излучения может быть использован в исследованиях электронных
переходов в полипептидном остове.
Вплоть до конца 70-х гг. прошлого века биофизика и биохимия имела дело только
с большими молекулярными ансамблями, для которых законы термодинамики легко
применимы. Так, 100 микролитров раствора гемоглобина с концентрацией 1 мг см 1
содержат 1018 молекул белка, атипичный белковый кристалл содержит порядка 1015
макромолекул. В природном окружении биологические молекулы действуют в виде
десятков копий, что вызывает необходимость в развитии новых методов, позволя-
ющих исследовать молекулы на одиночном уровне. Для детектирования и мани-
пуляции с одиночными молекулами разработаны методы сверхчувствительной
флуоресцентной спектроскопии. Применение механических зондов в микроскопии
силового поля позволяет растягивать или скручивать одну молекулу и придавать ей
новую конформацию. При переносе энергии в опытах по флуоресценции может быть
измерено расстояние между донором и акцептором внутри единичной молекулы, in
vitro или в живой клетке. Оптическая микроскопия ближнего поля применяется для
идентификации и исследования динамики единичных молекул в конденсированной
фазе. История развития каждого биофизического метода, упомянутого выше, будет
более детально представлена в соответствующих главах книги.
Языки и инструментальные средства
По-гречески физика — «physike» имеет происхождение от «physikos» — природное.
Физика является наукой, наблюдающей и описывающей природу. Во времена,
когда один из авторов (Джо Заккаи) был студентом Эдинбургского университе-
та, физика изучалась на кафедре натурфилософии. Слово «философия» — «лю-
бовь к мудрости» — буквально передает суть научного процесса, когда мудрость
«наблюдателя» приводит его к познанию нового. Этому способствует использо-
вание «наблюдателем» в эксперименте специального оборудования и языка для
описания полученных им результатов. Современная наука охватывает так много
различных областей познания, что невозможно овладеть знанием всех используе-
мых инструментальных средств и языков. Для студентов-биофизиков не представ-
ляет трудность язык общения с «чистыми» физиками или «чистыми» биологами
из-за десятка изучаемых ими в университетах междисциплинарных курсов. Сле-
дует отметить, что каждой дисциплине соответствует богатство и глубина, выража-
ющиеся в своем собственном специфическом языке и развивающие свой собствен-
ный набор инструментальных методов исследования. Ясно, что физики и биологи
«разговаривают» на разных языках, однако важно понимать, что и внутри каждой
дисциплины также имеются различные языки. Эти языки влияют на развитие ин-
струментальных методов, которые в свою очередь вносят вклад в описание концеп-
ций языков. Биофизики владеют языками физиков и биологов и могут адекватно
«переводить» с одного на другой. Иногда это трудная и непосильная задача даже
для очень хорошего «переводчика», поскольку каждый язык имеет свою собствен-
ную специфику и неповторимость.
Молекулярная биофизика изучает в большинстве случаев структуру, динамику
и взаимодействие макромолекул. Что такое биологические макромолекулы? Их био-
логическая активность описывается на языке биохимии и молекулярной спектроско-
пии, они были открыты с использованием гидродинамических и термодинамических
свойств, они визуализируются по их способности рассеивать рентгеновские лучи.
Таким образом, физические частицы вырисовываются как картины с разноцветным
орнаментом. Каждому языку соответствует набор инструментальных средств, обо-
рудования и методов экспериментального наблюдения. Успехи в изучении и пони-
мании биологических макромолекул несомненно базируются на развитии методов
исследования. Физические методы исследования, приводящие к правильным и точ-
ным результатам, требуют параллельного применения биохимических методов (часто
имеющих основу физических методов, например, электрофорез или хроматография)
для обеспечения эксперимента со «значимыми» образцами. Слово «значимый» явля-
ется ключевым в предыдущем предложении. Оно отражает значимость и уважитель-
ное отношение к исследованиям в области биологии (с греческого bios — «жизнь», а
logos — «слово», «причина»), т. е. биофизика ставит своей целью привести нас к более
полному пониманию процессов жизни. Молекулярную биофизику следует отличать от
биологической физики, которая имеет дело со свойствами биологического материала,
с целью создания наноустройств, например, на основе ДНК.
Шкалы длин и времени в биологии
Биологические явления происходят в широком диапазоне шкал длин и времени — от
ангстрем для расстояний между атомами до размера Земли в масштабе экосистемы, от
фемтосекунд, за которые происходят перестройки в электронных оболочках атомов
в первичных реакциях фотосинтеаза до 109лет эволюции. Инструменты для наблю-
дений за процессами развивались и адаптировались к различным шкалам времени
и длин. Клетка представляет собой центральный, отправной пункт в биологических
Рис. 2. «Реалистическая» картина бактерии
Escherichia coli, представленная на основе экс-
периментальных данных. Жгутик бактерии,
представляющий двойную мембрану, и ассо-
циированные с ней белки и гликопротеины
представлены на рисунке зеленым цветом; ри-
босомы и другие белки и нуклеиновые кисло-
ты, являющиеся цитоплазматическими компо-
нентами, обозначены фиолетовым и голубым;
набор полипептидных цепей — белым; ДНК и
ассоциированные с ней белки окрашены в жел-
тый и оранжевый цвета. Размер бактерии на
рисунке равен около 1 мкм, что соответствует
толщине двойного слоя мембраны около 10 нм
(http://www. scripps. edu/mb/goodsell/)
исследованиях (рис. 2). Имея размеры от
1 до 10 микрон, клетки могут быть рас-
смотрены в световом микроскопе. То же
и со скоростями клеточных процессов,
протекание которых лежит в интервале
времени от секунд до минут, что дает воз-
можность достаточно легко их измерять.
Если бы мы смогли взглянуть внутрь
эукариотической клетки через ее плаз-
матическую мембрану, то увидели бы и
другие мембранные структуры, разде-
ляющие различные компартменты, та-
кие как ядра и митохондрии, большие
макромолекулярные ансамбли, такие
как хроматин, рибосомы, шапероны пли
мультибелковые комплексы. Рассмат-
ривая все меньшие и меньшие структу-
ры, мы обнаружили бы молекулы РНК
и белков, затем пептиды и другие малые
молекулы, молекулы воды и ионы, и, на-
конец, атомы, составляющие их (рис. .3).
Наименьшие длины и наиболее
короткие промежутки времени более
трудны для изучения и требуют исполь-
зования более сложного оборудования
и методов исследования.
Фемтосекунды (10-15 сек) — наибо-
лее короткое время, представляющее
интерес для молекулярной биологии, со-
ответствующее преобразованиям элект-
рона в светочувствительной молекуле
ретиналя при поглощении им фотона.
Этот интервал времени может быть из-
мерен методом лазерной спектроскопии
(за одну фсек свет покрывает расстояние
3 х 10’7м, или 300 нм, около половины
длины волны видимого света). Тепло-
вые колебания находятся в пикосекунд-
ном интервале (10“12 сек), сворачивание
ДНК в микросекундном, скорости эн-
зиматического катализа равны порядка
1000 реакций в секунду, синтез белка
происходит за десятые доли секунды и т. д. Наиболее протяженное время, представ-
ляющее интерес для молекулярной биологии, — геологическое время, соответству-
10“
ю4
ю2
1.0
® 102
КУ4
106
108
Земля как экосистема
Кит
Человек
Длина ДНК в геноме человека
Нематода
Растительная клетка
Животная клетка
Бактериальная клетка
Вирус
Рибосома
Белок
Атом
Рис. 3. Шкалы длин в биологии
Ю’6 . Молекулярная эволюция
Синтез белка
Катализ ферментов
Разворачивание ДНК
10'9
10'12
Тепловое движение макромолекул
Колебания связей
, п-15
10 Электронные перестройки при фотосинтезе
Рис. 1. Шкалы времен в биологии
ющее тысяче миллионов лет молекулярной эволюции (рис. 1).
..j ктуэно-функциональные гипотезы
Эта книга описывает применение классических и новых физических методов для
наблюдения за биологическими структурами, динамикой и взаимодействием на
молекулярном уровне. Интенсивные исследования в течение 50 последних лет
подчеркнули фундаментальную значимость биологической активности на этом
уровне. Структурно-функциональные гипотезы являются фундаментом молеку-
лярной биологии. Одна из них сводится к утверждению, что, если какой-либо бе-
лок в данный момент существует в организме, то это связано с выполнением им
определенной биологической функции и его структура была отобрана эволюци-
ей. Открытие и исследование нуклеиновых кислот и белков как макромолекул с
четко определенными структурами позволили дать беспрецедентное понимание
таких процессов, как хранение и передача генетической информации, регуляция
экспрессии генов, ферментный катализ, иммунный ответ или передача сигнала.
В параллель стало очевидной возможность управлять биологическими процес-
сами, воздействуя на макромолекулярные структуры, и, в связи с этим, мощные
инструментальные средства стали развиваться не только для дальнейшего фунда-
ментального научного понимания процессов, но и для приложения этих знаний
к биотехнологии или разработки лекарств в фармакологии.
Понятие «структура» должно рассматриваться в широком смысле. Трехмер-
ная структура белка не абсолютно жесткая. Она адаптируется к своему лиганду
(партнеру) в соответствии с гипотезами «конфигурационной адаптивности» или
«индуцированной подгонки». Многие белки в клетке не обладают уникальной тре-
тичной структурой в изолированном состоянии, хотя и имеют при этом четкую
функцию при физиологических условиях. Такие белки получили название белков
с природной или внутренней неупорядоченностью. Доля неупорядоченных облас-
тей в таких белках может быть разной, начиная от последовательности в несколь-
ко аминокислот и заканчивая полностью неупорядоченной последовательностью
длиной в десятки, а иногда и в сотни аминокислот. Главное отличие этих белков
от структурированных (глобулярных) белков состоит в том, что они не имеют уни-
кальной третичной структуры в изолированном виде, а приобретают ее после вза-
имодействия с лигандами. Их конформация в комплексе определяется партнером
по взаимодействию, а не только с собственной аминокислотной последовательнос-
тью, как это характерно для структурированных (глобулярных) белков. Белки с
внутренней неупорядоченностью, как правило, полифункциональны, т. е. кроме
основной функции имеют дополнительные функции. Они получили название бел-
ков «по совместительству».
Большинство событий, измеряемых на шкале длин в ангстремах и шкале вре-
мен в пикосекундах, имеют глубокие последствия для жизни клетки, организма,
всех путей, определяющих взаимоотношения между организмами и их окруже-
нием во времени и пространстве. Недавнее появление высокопроизводительного
оборудования для определения последовательностей целых геномов и их анализа
(биоинформатика), для идентификации всех белков, присутствующих в клетке
(функциональная протеомика) и их ответа на внешние условия (динамика бел-
ков), для определения структуры белков (структурная протеомика) открыло но-
вую эру в молекулярной биологии, чьи революционные последствия еще пред-
стоит оценить.
Биологические макромолекулы, представляемые на рисунках, предстают как
статические структуры. Более правильно определять их как структуры, «усред-
ненные по ансамблю и по времени». Атомы в макромолекулярной структуре под-
держиваются в своих усредненных позициях балансом различных сил. При воз-
действии тепловой энергии атомы могут отклоняться от этих позиций. Динамика
с греческого «Suvapia» обозначает прочность как эквивалент понятию «сила». Это
общепринятое понятие в биофизике. Однако для разделения понятий «структу-
ра» и «динамика» первое соизмеряют со шкалой длины (например, конфигурация,
усредненная по времени), а второе со шкалой времени (например, энергия и флук-
туация). Разделение на два отдельных понятия обосновывается тем фактом, что
методы для исследования структуры и динамики обычно совсем разные и специа-
лизированные. Современные эксперименты, однако, часто направлены на исследо-
вание усредненной структуры и на ее изменений во времени.
Комплементарность физических методов исследования
О существовании макромолекул мы знаем только благодаря методам, с помощью ко-
торых их можно наблюдать. Однако не существует единого метода, обеспечивающе-
го всю необходимую информацию о макромолекулах и их взаимодействии. Каждый
метод дает свое видение системы в пространстве и во времени: методы — комплемен-
тарны. Биологические макромолекулы становятся активными структурами толь-
ко в подходящем растворителе. Стабилизирующие их силы слабы (порядка кТ, где
к — константа Больцмана, а Т— абсолютная температура) и возникают они частично
при взаимодействии с растворителем. Исследования биологических макромолекул,
следовательно, не могут проводиться без изучения окружающих их водных раство-
ров. Макромолекулы исследуются обычно в разбавленных или концентрированных
растворах, в липидном окружении мембран или в кристаллах. Молекулы белков и
нуклеиновых кислот в кристаллах окружены значительным количеством молекул
воды. Исходя из выбранного экспериментального метода исследования мы будем
рассматривать биологические макромолекулы в растворе как «физические частицы»
(масс-спектрометрия, детектирование отдельных молекул), «термодинамические
частицы» (измерение осмотического давления, калориметрия), «гидродинамические
частицы» (вискозиметрия, диффузия, седиментация) или «частицы, рассеивающие
излучения» (спектроскопия, дифракция, и микроскопия).
Достигаемое пространственное разрешение, перечень используемых при этом
приборов и требуемая для их применения масса образца для некоторых биофизи-
ческих методов приведены на рисунке 5.
Термодинамика
Из классической термодинамики следует, что многие свойства растворов, такие как
температура кипения или замерзания, а также осмотическое давление зависят от
Шкала длин
ю'6 ю’7 ю’8 ю'9 1О10 (м)
—I-------1----1----1---1-------
10 000 1000 100 10 1 (А)
I МУРР, '
МУРН I
ЯМР I
, Н-крист
Р-крист|
эм !
! дом ' ’
Рис. 5. Количество биологического материа-
ла, необходимое для определения его струк-
туры при использовании в эксперименте раз-
личных физических методов и оборудования
с определенной разрешающей способностью.
Сокращения: г — граммы; N — число молекул
(при допущении, что их молекулярный вес
равен около 100 000); PC — рассеяние света;
ГД — гидродинамика; МУРР, МУРН — ма-
лоугловое рассеяние рентгеновских лучей и
нейтронов, соответственно; ЯМР — ядерный
магнитный резонанс в растворе; Н-крист —
нейтронная кристаллография; Р-крист — рент-
геновская кристаллография; ЭМ — электрон-
ная микроскопия; ДОМ — детектирование
одиночных молекул
(Масса, г.) (Число молекул)
концентрации растворенного вещества. При постоянной массовой концентрации
эти термодинамические параметры чутко реагируют на изменения его молекуляр-
ной массы. Поэтому, например, массы и взаимодействия макромолекул впервые
были определены с помощью измерения осмотического давления.
Само сворачивание макромолекул и стабилизация биологически активных
структур строго следуют термодинамическим правилам, в которых свойства
растворителя играют определяющую роль. Тонкие калориметрические изме-
рения теплоемкости как функции температуры очень четко показывают, что
стабилизационная свободная энергия достигает максимума при температуре,
близкой к физиологической, а стабильность свернутой частицы падает как при
понижении, так и при повышении температуры. Существует следующая интер-
претация этого явления. Поведение цепи, окруженной растворителем, гораздо
сложнее, чем в вакууме. Энтальпия может вырастать, падать или даже не ме-
няться после сворачивания, потому что связи способны образовываться с оди-
наковым успехом как внутри макромолекулы, так и между отдельными цепями
и компонентами раствора. То же самое справедливо и для энтропии: потеря кон-
фигурационной свободы белковой цепи после сворачивания может с избытком
компенсироваться потерей степеней свободы у молекул растворителя вокруг
несвернутой цепи, к примеру, через обеспечение доступа молекулам воды не-
полярных групп. Находясь в растворе, молекула воды способна образовывать
водородные связи с соседними молекулами во всех направлениях. Неполярные
группы не обладают такой способностью, поэтому вблизи них молекулы воды
частично теряют возможность образовывать водородные связи, что сопровож-
дается снижением энтропии.
В белковом растворе теплоемкость почти целиком определяется водой и лишь
в незначительной степени — макромолекулами. Для проведения экспериментов
с такими растворами потребовалось создание прецизионных калориметров. Ран-
нис же калориметрические исследования оиологическпх макромолекул оыли
сконцентрированы на сравнительно больших эффектах, таких как переходы
под действием температуры, что, тем не менее, все же позволило понять основы
энергетики белкового сворачивания. В настоящее время в этой области достиг-
нут значительный прогресс. Созданы не только чувствительнейшие нанокалори-
метры, но и программы анализа для обработки термодинамической информации
и соотнесения ее со структурными данными. Энергетика внутримолекулярных
конформационных изменений, процесса формирования комплексов и взаимо-
действий с соседними молекулами белков и нуклеиновых кислот теперь может
быть детально исследована. Однако мы должны всегда помнить, что калоримет-
рия (как и все методы, основанные на применении термодинамических законов)
пригодна для измерения среднего по ансамблю очень большого числа частиц
(обычно порядка 1013), даже если результаты описываются в терминах поведения
одной частицы.
Гидродинамика
Первые намеки на существование биологических макромолекул как отдельных
частиц появились из наблюдений их гидродинамического поведения. Язык макро-
молекулярной гидродинамики — это язык динамики жидкостей в особых условиях
низкого значения числа Рейнолдса. Число Рейнолдса представляет собой безраз-
мерный параметр, выражающий относительную величину действия сил инерции
и вязкости на тело, движущееся сквозь жидкость. Тела с одинаковым числом Рей-
нолдса демонстрируют одинаковое гидродинамическое поведение. Поэтому воз-
можно, к примеру, определить поведение крыла самолета из опытов, проведенных
в гидродинамической трубе с помощью уменьшенной модели. Числа Рейнолдса
кита и маленькой рыбки равны 109 и 105 соответственно.
Для биологических макромолекул и их комплексов в водных растворах -- от
небольших белков до больших вирусных частиц и даже бактерий — они очень
малы. Например, для бактерии, плывущей со скоростью 10 ’ см сек ‘, число Рей-
нолдса составляет 10-3. В таких условиях силы инерции пренебрежительно малы
и движение частицы в жидкой среде определяется исключительно силами, дей-
ствующими на нее в данный момент. Роль такой силы выполняют силы броунов-
ского движения. Диффузия частиц в жидкой среде под воздействием градиента
концентрации или электрического поля, а также процесс осаждения в ноле уль-
трацентрифуги под действием гравитационной силы могут быть описаны с помо-
щью сравнительно простых уравнений в терминах молекулярной массы частиц,
их коэффициентов трения, зависящих от формы и размеров. Разрешение опреде-
ляет, насколько детально описывается структура частицы. Гидродинамические
методы описывают биологические макромолекулы с очень низким разрешением,
например, в виде двух- или трехосного эллипсоида, но при этом они также очень
чувствительны к гибкости частицы и к взаимодействиям частиц друг с другом.
Современная гидродинамика содержит ряд новых экспериментальных методов.
В дополнение к классическим подходам, таким как аналитическое ультрацентри-
фугирование — для определения коэффициента седиментации и динамическое
рассеяние света, для измерения коэффициента диффузии, в настоящее время раз-
работан ряд новых: свободный электрофорез — для измерения явлений переноса
в растворе иод действием электрического поля, метод восстановления красителя
после фотовыцветания — для наблюдения за подвижностью отдельных молекул
в живых клетках, метод деполяризованной флуоресценции и электрического
двойного лучепреломления — для определения коэффициентов вращательной
диффузии, а также флуоресцентная корреляционная спектроскопия — для изме-
рения макромолекулярной динамики ансамбля, состоящего из небольшого числа
частиц.
Рассеяние излучения
Мы видим окружающий мир благодаря тому, что он рассеивает свет, который по-
падает нам в глаза и анализируется мозгом. В экспериментах с дифракцией волны
излучения, рассеянные различными объектами, интерферируют, создавая разли-
чимый рисунок, с помощью которого может быть установлено относительное рас-
положение (или структура) этих объектов.
Интерференционная картина возникает тогда, когда длина волны излучения
совпадает с расстоянием между объектами или меньше его. В некоторых случаях
волны, формирующие такой рисунок, можно собрать с помощью линзы, чтобы по-
лучить непосредственный образ объекта. Длина атомных связей близка к одному
ангстрему (10~10м или 0,1 нм), поэтому для исследования структуры макромолекул
с помощью дифракционных экспериментов на практике используют три типа из-
лучения: рентгеновские лучи с длиной волны около 1 А, электроны с длиной волны
примерно 0,01 А и нейтроны с длиной волны от 0,5 до 10 А. Рассеяние видимо-
го света с длинами волн в районе 400-800 нм позволяет получать информацию о
больших макромолекулярных ансамблях и их динамике. Рентгеновские же лучи,
поскольку с их помощью возможны исследования структуры на атомном уровне,
дали возможность заложить тот фундамент, на котором структурная биология
была построена и развивается и сегодня.
Исследования биологических мембран, волокон, макромолекул и их комплек-
сов в кристаллах и растворах с помощью нейтронной дифракции стали возможны
в 70-х гг. прошлого столетия с разработкой методов, максимально использующих
особые свойства нейтронов.
Вслед за ограничениями, накладываемыми техникой окрашивания, была раз-
работана криоэлектронная микроскопия, позволяющая наблюдать субклеточные
и макромолекулярные структуры с высоким разрешением.
Свет, рентгеновские лучи и нейтроны слабо рассеиваются материей, поэто-
му требуются образцы, содержащие большое количество частиц, чтобы получить
приемлемое соотношение сигнал-шум. Такие эксперименты дают усредненные
по ансамблю данные о структуре. В последнем десятилетии XX в. создание высо-
коинтенсивных синхротронных источников произвело революцию в макромоле-
кулярной кристаллографии, значительно повысив скорость определения молеку-
лярных структур. Были разработаны эффективные методы модификации белков,
кристаллизации, сбора данных и анализа. Чрезвычайно высокая скорость в самом
процессе сбора данных позволила изучать кинетические интермедиаты ферментов
с помощью кристаллографии, разрешенной во времени. Параллельно развивалась
электронная микроскопия с применением электронных пушек с полевой эмиссией,
которая позволила исследовать одиночные молекулы и восстанавливать их трех-
мерную структуру. Значительно повысить скорость сбора данных в нейтронной
дифракции обещают мигающие источники для нейтронного рассеяния.
Спектроскопия
В спектроскопии излучение обменивается частью своей энергии с образцом через
эффекты поглощения или излучения вследствие внутренней или глобальной ди-
намики частицы, что отражается в изменении длины волны (частоты или цвета)
исходящего луча по отношению к падающему. Поскольку поглощение зависит
от расположения атома в структуре, определенные типы спектроскопических эк-
спериментов могут также использоваться для изучения самой структуры. Раз-
решение такого чувствительного спектроскопического метода, как ядерный маг-
нитный резонанс, приближается к атомному. Частота поглощенного излучения
может быть измерена как функция времени с точностью выше, чем одна мил-
лионная. Точная природа сигнала зависит от химического окружения ядра, что
позволяет получать структурную информацию. В технике построения изображе-
ния с помощью магнитного резонанса (томография) миллиметровое разрешение
достигается с помощью излучения метровой длины волны, когда исследуемое
тело помещается в градиент магнитного поля, и излучение на ядре фокусирует-
ся в определенном химическом окружении. В этих условиях резонансное погло-
щение соответствует определенной величине поля и, как следствие этого, дает
точное положение исследуемого вещества. Что касается дифракции, где длина
волны соответствует требуемому разрешению, то его энергия для спектроскопии
выбирается таким образом, чтобы разность, возникающая из-за возбуждения или
поглощения в образце, могла быть измерена без задержки. Поэтому излучение
различной длины волны используется в основном для дифракционных и спект-
роскопических экспериментов.
Когерентная спектроскопия, где применяется излучение с одной и той же
фазой, обеспечила беспрецедентные возможности для изучения динамики и из-
менения структур во времени. Когда когерентные лазеры стали доступны, метод
«спинового эха» в приложении к ЯМР и нейтронной спектроскопии был допол-
нен методом «фотонного эха». Двумерная спектроскопия, сначала разработанная
для ЯМР, позволяет теперь измерять сопряжение в системе колебательных мод.
Недавно данный метод был применен в инфракрасном диапазоне для определе-
ния структуры небольших молекул. Наиболее захватывающим аспектом двумер-
ной инфракрасной спектроскопии можно назвать ее чувствительность к структуре
и временное разрешение вплоть до фемтосекунды.
Если взять в качестве примера электромагнитное излучение, то для атомной
дифракции наиболее пригодна длина волн рентгеновских лучей, тогда как для
изучения внутримолекулярных колебаний требуются длины волн инфракрасного
диапазона (рис. 6).
Электромагнитное излучение
А.(м) IO'15 IO’12 IO’9 10-6 IO’3 1 103
v (сек ’) 1024 1 021
X
X
0J
106
Е(эв) Ю9 106 103 1 IO’3 IO’6 10-9
Е/Л(°К) 1013 1О10 107 104 10 ю-2 ю-5
Нейтроны
Z.(m) 1O’io-1O’9
т(мсек’) 4000-400
Е(эв) ЗхЮ’МхЮ-4
ЕЛН°К)
400-4
Рис. 6. Длина волны, энергия и частота электромагнитного и нейтронного излучения. Шка-
лы на рисунке представляют приблизительные порядки значений. Точные значения констант
получены из равенств: vX = с, где v и X — частота и длина волны электромагнитного излуче-
ния, соответственно, и с — скорость света (3 х 108м-сек-1); Е = hv (где Е — энергия и h — кон-
станта Планка (6,626 = 10~34 Дж-сек = 4,136 х Ю 15 эВ сек); температурный эквивалент энергии,
1 эВ/к= 11604,5 К, где k — константа Больцмана. В случае нейтронного излучения, Х= h/mu (где
и мсек 1 — скорость нейтрона), и Е = Угти2, где т — масса нейтрона (1,6726 х Ю“27кг)
В ЯМР спектроскопии падающее электромагнитное излучение находится
в радиочастотном диапазоне, что соответствует метровой длине волн. Надо заме-
тить, что нейтронное излучение с длиной волны примерно 1 А (что соответствует
межатомным расстояниям и амплитудам колебаний) обладает энергией примерно
1 мэВ (что соответствует энергиям атомных колебаний), поэтому дифракционные
и спектроскопические эксперименты могут проводиться одновременно, давая ин-
формацию об атомных амплитудах и частотах движения макромолекул. Молеку-
лярная шкала времени, соответствующие энергии и температуры для разных био-
физических методов, показаны на рисунке 7.
Детектирование одиночных молекул
До 90-х гг. прошлого века биохимические и биофизические исследования биоло-
гических макромолекул проводились с очень большим количеством частиц, тогда
как в условиях in vivo макромолекулы функционируют как одиночные частицы в
динамическом гетерогенном окружении. Структура, динамика и взаимодействия
наблюдались и измерялись как средние по ансамблю, и такое положение дел в ос-
новном сохраняется до сих пор. Более того, энзиматические, сигнальные реакции,
а также реакции связывания носят преимущественно стохастический характер, по-
сек
мсек--
к
2
мксек-
m
нсек
псек--
фсек
Энергия (ев)
10‘15 10'12 10'9 10’6 10’3 1
-и------1------1-------1------1------1
-и------1-----1------1-----1------1
1011 10 е 10'5 10'2 10 104
Температура (°К)
Рис. 7. Соответствие шкал времен, энергии и температуры в различных биофизических методах.
Направления стрелок совпадают с направлением прерывистой черной диагонали, которая сме-
щена в горизонтальном направлении для наглядности. Цифрами обозначены: 1 - динамическое
рассеяние света; 2 - ядерный магнитный резонанс; 3 - электрическое двойное лучепреломление:
4 - двойное лучепреломление в гидродинамическом потоке; 5 - деполяризованная флуоресцен-
ция; 6 - нейтронная спектроскопия; 7 - одномерная инфракрасная спектроскопия; 8 - лазерная
спектроскопия; 9 - двумерная инфракрасная спектроскопия
этому измерения активности, к примеру белков, также «спрятаны» в средних зна-
чениях по ансамблю, когда измерения происходят в большой популяции молекул,
даже если реакция запускается одновременно для всего образца.
Раньше отдельные макромолекулы можно было наблюдать с помощью элек-
тронной микроскопии в высоком вакууме, и лишь в последнем десятилетии по-
явились методы, позволяющие проводить наблюдения макромолекул в их ес-
тественном состоянии. Более того, развитие техники детектирования отдельных
молекул (ДОМ) дает возможность не только наблюдать их, но и манипулировать
с ними. Техника манипулирования одиночными молекулами (МОМ) основана на
двух ключевых технологиях: визуализация одиночных молекул в нативных (есте-
ственных) условиях и наноманипуляция. Сигналы отдельных молекул с приемле-
мым соотношением сигнал-шум получаются с помощью флуоресцентных меток,
которые можно наблюдать с помощью флуоресцентной оптической микроскопии.
Благодаря применению техники полного внутреннего отражения и стоячих волн,
разрешение в этом методе может быть в несколько раз выше дифракционного пре-
дела, накладываемого длиной волны света. Техника МОМ включают в себя: захват
молекулы с помощью стеклянной пипетки или шариков, притягивающихся сфоку-
сированным лазерным лучом (оптический пинцет). Величины молекулярных сил
исследуются методами микроскопии силового поля. Они находятся в пиконыотон-
Энтропийная сила
Типичной энергией для макромо-
лекул является их тепловая энергия:
К,,'Г “ 4 х 10 21 Дж, Поскольку шкала длин
для биологических макромолекул порядка
1 нм, шкала сил находится в пределах ии-
коныотонов (10 12 И). Следовательно, энт-
ропийная сила может быть вычислена как
к Т/(1 нм), что равно 4 нН при 300 К.
ном диапазоне, что сравнимо с тепловы-
ми силами, стабилизирующими актив-
ную макромолекулярную структуру.
Выдающийся ученый Эрвин Шре-
дингер писал в 1952 г., что мы никогда
не сможем проводить эксперименты с
одним электроном, одним атомом или
одной молекулой. Однако изобретение
Г. Биннингом и X. Рорером сканиру-
ющей туннельной микроскопии в 1980 г.
радикально изменило взгляд ученых
на этот предмет. Стали возможны эксперименты по измерению пиконьютонных
сил, структурирующих макромолекулы i':i !э:<1ь; ). В этих экспериментах
используется один или два оптических пинцета (твизера), позволяющих рас-
тягивать биологические макромолекулы и измерять прикладываемую при этом
силу (рис. <S п).
В инструменте, называемом магнитным пинцетом (рис. 8 б), один конец моле-
кулы прикрепляется к стеклянной пипетке, в то время как другой — к магнитному
шарику, на который действует постоянное магнитное поле. Затем измеряется рас-
тяжение или вращение молекулы как функции приложенной силы. В микроско-
пии силового поля один конец молекулы прикрепляется к поверхности, а другой —
к кронштейну (консоли). Когда поверхность сдвигается, отклонение кронштейна
(консоли) отслеживается с помощью отраженного лазерного луча (рис. 8 «)
Эти эксперименты позволяют получить новый структурный параметр для оди-
ночной молекулы: силу ( ибл. : >. Верхний предел измерения силы в микромани-
пуляциоиных экспериментах — это прочность ковалентной связи (в пределе эВ/А
или около 1000-2000 nil). Предел наименьшей измеряемой силы устанавливается
силой Ланжевена (около 1 фН), которая ответственна за броуновское движение
сенсора (размер порядка 1 мкм).
Стоит заметить, что диапазон сил в < ш'. । ш I покрывает всего лишь три поряд-
ка величины. До того как стала доступна техника работы с единичными молекула-
ми, информация о стабильности белковых молекул могла быть получена только с
помощью измерения потери структуры в денатурирующих условиях (используя
температуру, химические агенты и pH), откуда рассчитывалась свободная энергия
сворачивания для молекул. Свободная энергия, однако, не снабжает нас прямой
информацией о механической стабильности. Для определения последней важ-
но знать изменение полной энергии как функции пространственных координат.
С целью определения силы, необходимой для разворачивания одиночной молеку-
лы, было исследовано несколько белков. Эксперименты показали очень большой
разброс данной величины (достигая множителя порядка десяти) для различных
белковых доменов, чья температура плавления очень схожа. Эти результаты де-
монстрируют, что механическая стабильность белковой молекулы не коррелирует
напрямую с ее термодинамической стабильностью. Можно ожидать, что анализ
механических свойств макромолекул заложит фундамент новой области исследо-
ваний — механохимической биохимии.
Таблица I. Шкала сил для биологических макромолекул
Разрыв ковалентных связей 1000-2000 нН
Деформация сахарного кольца 700 п Н
Разрыв двойной спирали ДНК 400- 580 нН
Сила связи между авидином и биотином 140-180 нН
Структурный переход в двуспиральной ДНК при растяжении 60-80 nil
Структурный переход двусппральной ДНК при торзионном скручивании -20 нН
Разрушение индивидуальных нуклеосом 20-40пН
Разворачивание тройной спирали спектрина 25-35 пН
Сила, необходимая для остановки мотора РНК-полимеразы 14—27 нН
Структурный переход в рибозимной шпильке РНК при растяжении -14 нН
Разделение комплементарных цепей ДНК (Т = 20 °C, 150 мМ NaCl)
(зависит от конкретной нуклеотидной последовательности) 10-15 нН
Сила, необходимая для остановки мотора миозина 3-6 нН
Сила, генерируемая при полимеризации белка в растущих микротрубочках 3-4 нН
Объективы
Рис. 8. Схематическое изображение трех основных способов, используемых при манипуляции
с одиночными биологическими молекулами: а — оптический пинцет (tweezer); б — магнитный
пинцет и в — атомный силовой микроскоп
Часть А
БИОЛОГИЧЕСКИЕ
МАКРОМОЛЕКУЛЫ
И ФИЗИЧЕСКИЕ ИНСТРУМЕНТЫ
Глава А1
МАКРОМОЛЕКУЛЫ И ИХ ОКРУЖЕНИЕ
A'i .1. Исторический обзор
Исследования биологических макро-
молекул тесно переплетаются с исто-
рией физической химии, которая фор-
мально возникла в 1887 г., когда вышел
журнал Zeitschrift fitr Physikalische Che-
mie, основанный Жакобом Вант-Гоф-
фом (Jacobus Van’t Hoff) и Вильгель-
мом Оствальдом (Wilhelm Ostwald).
Интересно, что первые выпуски были
посвящены реакциям в растворах, пос-
кольку биологические процессы внут-
ри живой клетки протекают в водном
окружении.
Исследования в XIX в. свойств рас-
творов привели нас к познанию свойств
биологических макромолекул, о чем мы
писали во Введении. Стоит также упо-
мянуть Франсуа-Мари Рауля (Francois-
Marie Raoult) (1886), химика из Гре-
нобля, который сформулировал закон
понижения (повышения) температуры
замерзания (кипения) раствора при уве-
личении концентрации растворенных в
нем молекул, что легло в основу разви-
тия метода определения молекулярно-
го веса растворенных веществ, а также
Ганса Гофмайстера (Hans Hofmeister)
(1895), доктора медицины и физиолога,
который, изучая эффекты мочегонных и
слабительных солей, классифицировал
их в соответствии с влиянием на рас-
творимость белков в водных растворах.
Позже серии Гофмайстера были при-
няты в качестве порядка ранжирования
ионов по эффективности «высаливания»
или осаждения. Гилберт Ньютон Льюис
(Gilbert Newton Lewis) ввел понятия ак-
тивности в 1908 г. и ионной силы вместе
с Мерлом Ренделом (Merle Randall) в
1921 г. В1911 г. Фредерик Джордж Дон-
нан (Frederick George Donnan) опубли-
ковал статью о мембранном потенциале,
возникающем во время диализа элект-
ролитов. Питер Дебай (Peter Debye)
и Эрих Хюккель (Erich Huckel) (1923)
предложили теорию растворов электро-
литов. Наконец, в последние десятилетия
новые методы, такие как динамическое
рассеяние света и нейтронное рассеяние
под малыми углами, разработанные для
изучения полимеров — в основном поли-
электролитов, — внесли весомый вклад в
наше понимание свойств биологических
макромолекул в растворе.
В настоящее время растет интерес
к поведению белков в неводных рас-
творах и даже в вакууме, в основном
для выяснения вопроса: необходима ли
вода для жизни или нет? Однако белки
как биологически активные частицы
возникли в присутствии воды, и прак-
тически не могут рассматриваться от-
дельно от своего водного окружения.
Можно вспомнить, что даже кристаллы
биологических макромолекул содержат
ощутимое количество воды, и поэтому
должны рассматриваться как организо-
ванные макромолекулярные растворы.
А1.2. Растворы макромолекул
Раствором является гомогенная на мо-
лекулярном уровне смесь двух или бо-
лее компонентов. Компонент, присут-
ствующий в наибольшем количестве,
называется растворителем, а осталь-
ные — растворенными веществами. Мы
будем иметь дело преимущественно с
водными растворами макромолекул, где
растворитель — вода, а растворенные
вещества — макромолекулы и другие,
меньшие ио размеру молекулы, такие
как простые соли.
А1.2.1. Концентрация
Концентрация растворенного вещест-
ва может быть выражена несколькими
Комментарий А1.1. Единицы
молекулярной массы ।
Молярная масса выражается в граммах
на моль или, в системе единиц СИ, кг на
моль. '
Относительная молекулярная масса,
: или молекулярный вес — безразмерная ве-
личина, определяемая как отношение мае- ;
сы молекулы к 1/12 массы изотопа углеро- |
! да 12С. Молярная масса |2С очень близка к
12 г/моль. Поэтому молярная масса (в кг ।
на моль) может быть преобразована в .мо- ।
лекулярный вес делением на ПУ3 г на моль i
; (тот же результат получим, умножив на
1000 и отбросив единицы измерения).
Биохимики пользуются молекулярной
। массой, выраженной в дальтонах — Да.
Один Да = одной атомной единице массы =
= 1/12 массы одного атома |2С (см. также
| Главу Б1). |
способами. Весовая, или массовая кон-
центрация, — это масса растворенного
вещества на единицу веса растворителя
(или на 100 весовых единиц раствори-
теля, если она выражена в процентах).
Обычной единицей молекулярной мас-
сы в биохимии является дальтон (к.л:
мен inpiiii ,\ 1.1 У
Молярность — это количество мо-
лей растворенного вещества на литр
раствора. Выражение концентрации в
молях на литр имеет то преимущество,
что оно более уместно при описании
коллигативных свойств раствора (т. е.
свойств, зависящих только от количе-
ства частиц растворенного вещества,
а не от его массы и свойств. См. также
параграф Л1.2.3).
Моляльность — это количество
молей растворенного вещества на ки-
лограмм растворителя. Преимущест-
во выражения концентрации в таких
единицах заключается в том, что мо-
ляльность получается взвешиванием
растворенного вещества и раствори-
теля, тогда как молярность зависит от
объема раствора. Массы инвариантны,
в то время как объем раствора является
функцией температуры и давления.
Понятия молярной доли и объемной
доли сходны с понятием весовой доли,
но при этом равны количеству молей
или объему растворенного вещества на
количество молей или полный объем
раствора. Стандартные способы изме-
рения концентраций растворов белков
и нуклеиновых кислот обсуждаются
в коммен r.ipini Л 1.2.
А1.2.2. Парциальный объем
Парциальный объем растворенного ве-
щества равен изменению объема раст-
вора после добавления растворенного
! Коммсн । арий А1.2. Измерение концентраций белков и нуклеиновых кислот
Концентрацию белка или нуклеиновой кислоты нельзя определить простым взвешивани-
ем материала, находящегося в растворителе. Порошки белков и нуклеиновых кислот, полу-
ченные в результате лиофилизации или осаждения, всегда содержат какое-то количество гид- >
ротированной воды и ионов соли, необходимых для поддержания их активной конформации.
Более того, для многих экспериментальных методов достаточно крайне малой концентрации
белка или нуклеиновой кислоты. Однако точное взвешивание нескольких микрограммов, а
тем более еще меньшего количества материала сопряжено с относительно большой погреш-
ностью. На практике концентрации белков определяют с помощью колориметрического ана-
лиза (окрашиванием по Брэдфорду), в котором индикатор химически взаимодействует с по-
липептидом, или с помощью спектрофотометрии, где количество растворенного материала
пропорционально поглощению света при определенной длине волны. Поглощение при 280
нм особенно чувствительно к присутствию аминокислотных остатков триптофана, тирозина
и цистеина и для большинства белков оно составляет величину порядка одной оптической
единицы поглощения для раствора с концентрацией 1 мг-мл’1 и длиной оптического пути в
1,0 см. Величина поглощения зависит от количества таких остатков в белке. Нуклеиновые
кислоты демонстрируют сильное поглощение при 260 нм (одна оптическая единица погло-
щения соответствует примерно 40 мг-мл ’). Поглощение при этой длине волны используется
для определения концентрации ДНК и РНК в растворе. Колориметрические и спектрофо-
тометрические измерения дают относительные значения по сравнению с калибровочным
образцом. Когда требуются значения концентрации для конкретной макромолекулы, напри-
мер, для интерпретации данных малоуглового рассеяния (Глава Е2) или вязкости (Глава Г9),
колориметрические и спектрофотометрические данные должны быть калиброваны с учетом '
аминокислотного или нуклеотидного состава образца.
вещества. Парциальный объем — это
не просто объем, занимаемый добав-
ленным веществом, поскольку его
присутствие может также приводить к
изменению объема растворителя. Пар-
циальные объемы заряженных молекул
в водных растворах служат интересной
иллюстрацией эффектов растворите-
ля. Молекулу воды можно представить
в виде небольшого электрического ди-
поля. Причем в жидкой фазе вода пред-
ставляет собой малоупорядоченную
структуру, удерживаемую водород-
ными связями. В присутствии ионов
молекулы воды группируются вокруг
зарядов, занимая при этом меньший
объем. Такой эффект носит название
электрострикции. Поэтому парциаль-
ный объем растворенных заряженных
ионов может быть отрицательным, как
в случае ионов Na* и Mg2+. Для этих
ионов уменьшение объема вследствие
электрострикции больше того объема,
который они реально занимают в рас-
творе. Парциальный объем иона К+
слегка положителен, поскольку элек-
трострикция недостаточно компенси-
рует занимаемый ионом объем. Следу-
ет заметить, что величину изменения
объема раствора при добавлении не-
скольких солей нельзя предсказать из
соответствующих величин для каждой
соли.
Парциальный удельный объем рас-
творенного вещества равен изменению
объема раствора при добавлении од-
ного грамма вещества. Парциальный
моляльиый объем растворенного веще-
ства равен изменению объема раствора
при добавлении одного моля вещества.
Парциальные объемы ионов приведе-
ны в коммсн 1 ;ipi111 .\ 1.3, а парциальные
удельные объемы биологических мак-
ромолекул обсуждаются в секции Г4.8.
с.:с:л,м.,j л 1 .Л Парциальные моляльные объемы ионов
ион
Na‘ в воде
Na* в морской воде (-0,725 моляльный NaCl)
К* в воде
К* в морской воде (-0,725 моляльный NaCl)
Mg* * в воде
Mg’* в морской воде (-0,725 моляльный NaCl)
Cl* в воде
С1* в морской воде (-0,725 моляльный NaCl)
Из Millero Е J. (1969).
парциальный моляльный
объем (мл. моль*1)
- 5,7
-4,4
4,5
5,9
-30,1
-27,0
22,3
23,3
А1.2.3. Коллигативные свойства
Коллигативные свойства растворов (от
латинского ligare — связывать) — это
свойства, зависящие только от количе-
ства молекул растворенного вещества
в объеме, а не от их массы или приро-
ды. Открытие этих свойств сыграло су-
щественную роль на заре физической
химии, потому что позволило точно оп-
ределить молекулярный вес, который,
в свою очередь, послужил доказатель-
ством самого существования атомов и
молекул. Закон Рауля утверждает, что
в идеальном растворе при постоянной
температуре парциальное давление
компонента в жидкой смеси пропорцио-
нально его мольной доли. Коллигатив-
ными свойствами, описывающимися
законом Рауля и применимыми к раз-
бавленным растворам нелетучих моле-
кул, являются повышение температуры
кипения и понижение температуры за-
мерзания раствора. Разница значений
температуры идеального раствора и
чистого растворителя в каждом случае
пропорциональна количеству молекул
растворенного вещества. Этот закон
может быть применим и к неидеальным
растворам, если ввести понятия хими-
ческого потенциала и активности.
А1.2.4. Химический потенциал
и активность
Рассмотрим сосуд, показанный на ри-
сунке Al.l, — с перегородкой, разделя-
ющей растворы с двумя различными
молярными концентрациями: и Сн.
Если мы проделаем отверстие в пе-
регородке, возникнет поток молекул
растворителя из области с высокой в
область с низкой концентрацией. Он
похож на поток воды, текущей вниз в
градиенте потенциала тяготения. Та-
кой потенциал можно связать с кон-
центрацией растворенного вещества.
Химический потенциал, р, растворен-
ного вещества равен выигрышу сво-
бодной энергии после добавления в
раствор одного моля вещества (см. так-
же Главу В1):
• ® ф • • • •
________________ЦСА > цСв______________
Рис. А 1.1. Химический потенциал (см. текст)
И = до/
ц /дс
Разница свободной энергии двух
растворов с концентрациями С( и С
(в молях на литр раствора) в термодн-
нам и ко вы ражается:
Ga -G„ =AG=-RT In (Al.l)
Сц
где R — газовая константа, T— абсолют-
ная температура. Выражение получено
в результате интегрирования уравне-
ния Больцмана, которое описывает рас-
пределение молекул идеального газа
согласно величинам свободных энергий
при постоянной температуре:
— = ехр (-^7=^4 (А1.2)
Рв I RT )
где рА, — величины давления при по-
стоянном объеме (пропорциональные
молярной концентрации), связанные
с состояниями свободных энергий Gx
и GB. После простого преобразования
уравнение А 1.2 становится:
ДС = ц(Сд-Сд ) = -/??’In—. (А1.3)
Ся
Выражение этой зависимости в тер-
минах разности свободной энергии или
химического потенциала, а не в абсо-
лютных величинах, позволяет обойти
необходимость определения стандарт-
ной свободной энергии или химическо-
го потенциала (к примеру, связанных с
идеальным раствором при данной кон-
центрации, температуре и давлении).
Такие уравнения применимы к идеаль-
ным растворам, т. е. в которых молеку-
лы растворенного вещества ведут себя
как точечные частицы и не взаимодейс-
твуют ни друг с другом, ни с сосудом.
В 1908 г. Г. Льюис ввел понятие ак-
тивности, чтобы объяснить отклонения
в поведении растворов от идеального.
Переписав выражение А1.3 в терминах
активности, получим:
ДС =-7?Т In —. (А1.4)
ав
Активность растворенного вещест-
ва А дается выражением
«я=Ул С.,,
где у., — коэффициент активности,
равный единице для идеального раство-
ра. Коэффициенты активности получа-
ются экспериментально, например, из
наблюдений за отклонением от закона
Рауля.
Уравнения для идеальных раство-
ров на практике могут быть использова-
ны для реальных растворов при замене
в них концентрации на активность.
А1.2.5. Температура
Повышение температуры кипения и по-
нижение температуры замерзания ра-
створов, обусловленные присутствием
растворенного вещества, на самом деле,
дают мало полезной информации при
изучении свойств таких биологичес-
ких макромолекул, как белки и нукле-
иновые кислоты. Во-первых, потому,
что мольная доля таких молекул даже
в самом концентрированном раство-
ре чрезвычайно низка (комментарий
А I. i). Во-вторых, биологические мак-
ромолекулы, как правило, нестабиль-
ны в чистой воде, поэтому данный
эффект будет обусловлен в основном
буферным веществом и ионами. И на-
конец, белки и нуклеиновые кислоты
обычно нестабильны при темпера-
турах кипения и замерзания воды. В
процессе эволюции они приобрели
способность приобретать стабильную
и активную конформацию в весьма
if .с ; i л >, ii1 '': 1 Значения мольной доли и молярности обычных растворов
биологических макромолекул
Мольная доля макромолекул с массой 30 кДа при концентрации 300 гл 1 в водном растворе
составляет 1/4000; .молярность этого раствора равна ЮмМ. Такая концентрация считается
высокой для большинства биофизических экспериментов и схожа с концентрацией белков
в цитоплазме. Мольная доля макромолекул, растворе, при концентрации 3 мг-мл 1 является
обычной концентрацией для многих биофизических экспериментов и равна 1/400 000; мо-
лярность раствора составляет 100 мкМ.
ограниченных условиях, касающих-
ся состояния растворителя, а также в
узком диапазоне таких термодинами-
ческих параметров, как температура
и давление (см. Часть В). Интересно
отметить, однако, что существуют ор-
ганизмы, называемые экстремофила-
ми (любители крайностей), которые
адаптировались к различным экстре-
мальным условиям окружающей сре-
ды, включая температуру ( iu’.vmcp i ;i-
;1111 i \ 1.5 ).
A1.2.6. Осмотическое давление
Осмотическое давление является кол-
лигативным свойством, которым обла-
’/ А / 5. Экстремофилы
Микроорганизмы адаптировались к различным экстремальным условиям окружающей
среды, включая очень высокие и очень низкие значения pH (алкалофилы и ацидофилы), вы-
сокие концентрации солей (галофилы, см. Секцию А1.3), высокое давление в глубоких оке-
анских впадинах (барофилы), температуры близкие к 0 °C в полярных или ледниковых водах
(психрофилы, от греческого психро - прохладный; приставка крио, что значит холод, обычно
применяется для температур ниже нуля), температуры около 70 °C в термальных источни-
ках, показанных на картинке (термофилы), и даже температуру ИЗ °C (максимальную для
известных сегодня живых организмов) для Methanococcus jannaschi, которая обитает в глубо-
ководных морских гидротермальных источниках (гипертермофилы). Структура белков экс-
тремофильных организмов чрезвычайна схожа со структурой белков у мезофилов (живущих
при температуре около 37 °C). Сдвиг их оптимума стабильности и активности в зависимости
от условий окружающей среды обусловлен аминокислотными заменами, которые изменяют
внутренние стабилизирующие силы (см. Главу В2)
дают как небольшие молекулы раство-
ренного вещества, так и макромолекулы
(рис. А1.2).
Важность данного свойства для
биологических процессов была оце-
нена с самого момента его открытия.
Осмос, названный так от греческого
осмос, — импульс, представляет собой
явление, возникающее, когда два рас-
твора разной концентрации отделены
полупроницаемой мембраной, про-
пускающей молекулы растворителя
(воды), но не пропускающей молеку-
лы растворенного вещества. Осмоти-
ческое давление представляет собой
гидростатическое давление раствори-
теля, которое образуется на мембране
со стороны более концентрированного
раствора в результате того, что вода
движется в область с более высокой
концентрацией из области с более низ-
кой, с тем чтобы уравновесить свой
химический потенциал. Давление воз-
никает со стороны более высокой кон-
центрации, поскольку объемы отсеков
постоянны. Осмотическое давление 11
дано в выражении
(А1.5)
где R — газовая константа, Т— абсолют-
ная температура, a N — количество мо-
лей растворенного вещества в объеме V.
Массовая концентрация С и молярная
масса М растворенного вещества связа-
Случай, когда только один отсек со-
суда содержит растворенное вещество
(концентрация Св равна нулю), позво-
ляет точно измерить молярную массу,
что широко используется при исследо-
вании полимеров.
Надо заметить, что очевидное ма-
тематическое сходство уравнения А1.5
с уравнением идеального газа, выра-
женного в молярных единицах, не име-
ет под собой физического основания.
Как мы указывали выше, осмотическое
давление П представляет собой гидро-
статическое давление растворителя, а
не молекул растворенного вещества,
которые, подобно молекулам газа, «тол-
кают» мембрану со стороны высокой
концентрации.
Когда в растворе присутствует бо-
лее одного вида молекул растворенно-
го вещества, уравнение А1.5 преобра-
зуется в:
ПК = ХЛГ7?7; А1.6)
}
где суммируются все недиффундирую-
щие растворенные вещества, для которых
мембрана непроницаема. Реальное осмо-
тическое давление зависит и от свойств
мембраны по отношению к различным
растворенным веществам. Например, что-
бы подсчитать осмотическое давление,
действующее на мембрану, отсекающую
молекулы с молекулярной массой свыше
нысЛГи Vотношением — = —.
V М
Разница осмотических давлений
в двух отсеках, показанных на рисун-
ке А 1.2, равна pgh, где р — плотность
растворителя, ag — ускорение свобод-
ного падения:
(П,-ПЙ) = ^--^7?Г (А1.5а)
М
с. > с.
Рис. А 1.2. К определению осмотического дав-
ления (см.текст)
10 кДа, мы используем уравнение А1.5,
но учитываем только молекулы с молеку-
лярной массой, превышающей 10 кДа.
А1.2.7. Вириальные коэффициенты
Уравнения А1.5 и А1.6 предполагают,
что мы имеем дело с идеальными раст-
ворами, в которых частицы растворен-
ного вещества нс взаимодействуют друг
с другом. Их можно считать хорошим
приближением, когда растворы сильно
разбавлены. Хотя межмолекулярные
взаимодействия можно описать с по-
мощью химических потенциалов, более
широкое распространение при исследо-
вании макромолекулярных растворов
получило использование коэффициен-
тов активности, основанных на вири-
альных коэффициентах (от латинского
vires — силы). Теперь мы перепишем
уравнение А1.5 для неидеального рас-
твора, осмотическое давление которо-
го выражается с помощью разложения
концентрации в степенной ряд:
П 1
— = — + А,С + А.,С1+... (А 1.7)
CRT М 2 3
где Ау А3 — второй, третий и после-
дующие вириальные коэффициенты.
Они определяются экспериментально
и дают ценную информацию о взаи-
модействиях частиц. Коэффициенты
могут быть положительными, что го-
ворит об отталкивании между части-
цами, либо отрицательными, указыва-
ющими на их притяжение.
А 1.3. Макромолекулы,
вода и соли
Растворы солей оказывают значительное
влияние на стабильность конформации
макромолекул. Белки экстремальных га-
лофилов ( K.eiMi ч .ер;.и \; .ii) в процессе
эволюции выработали особый адапта-
ционный механизм, позволяющий им
оставаться стабильными, растворимыми
и активными даже при высоких концен-
трациях соли, свойственным цитоплаз-
ме этих организмов. Связывание ионов
макромолекулами приводит к специфи-
ческим солевым эффектам, зависящим
от конкретного типа солей и макромо-
лекул. Например, для образования ста-
бильной конформации тРНК в растворе
необходимо присутствие ионов магния,
а также калия или натрия — в неболь-
ших концентрациях. При 0,IM NaCl и
1мМ MgCl., противоионы Na и Mg"
нейтрализуют отталкивание между фос-
фатными группами основной цени нук-
леиновых кислот, тем самым позволяя
им принять компактную конформацию.
Данный эффект не является целиком
электростатическим и включает также
стерические взаимодействия. Если в
качестве соли используется NCCH.^Cl
(хлорид тетрамстиламмония), тогда
противоион N(CH3)j не дает тРНК свер-
нуться правильно, но всей вероятности
из-за того, что ион тетрамстиламмония
занимает слишком большой объем.
Специфическое ионное связывание
играет ключевую роль в механизмах
регуляции активности и стабилизации
некоторых белков, таких как калмо-
дулин, амилазы или парвальбумины,
которые связывают Са". Неспецифи-
ческими называют солевые эффекты,
схожие для всех макромолекул, хотя
они и могут сильно зависеть от приро-
ды ионов. Сюда относятся, например,
эффекты, связанные с ионной силой
при низких концентрациях солей. Соли
также способны оказывать влияние на
макромолекулы, изменяя структуру
воды, что становится очевидным при
их высоких концен трациях.
Экстремальными галофилами являются археи и бактерии (см. Главу Л2), способные жить
исключительно в местах с высокой концентрацией соли, таких как соленые озера, наподобие
Мертвого моря, Великого Солевого озера и Розового озера в Сенегале (показано на картинке),
а также естественные и искусственные солончаки. Эти организмы содержат каротеноиды, бла-
годаря которым среда их обитания приобретает розовый цвет. Кроме того, каротеноиды вклю-
чаются в пищевую цепь, в результате чего в розовый цвет окрашиваются такие животные орга-
низмы как фламинго и лосось. Экстремальные галофилы отличаются от умеренных галофилов
и галотолерантных организмов тем, что для компенсации высокого осмотического давления
окружающей среды, вызванного концентрацией NaCl, близкой к насыщающей, они вынуждены
использовать высокие цитоплазматические концентрации КС1. Таким образом, биохимичес-
кие реакции протекают в условиях, которые пагубно сказываются на стабильности и раствори-
мости белков. Однако белки галофилов способны адаптироваться к экстремальным условиям
среды. Вместо того чтобы «защищать», как вполне можно было бы ожидать, свою структуру от
соли, — к примеру, окружая себя прочно связанной водной оболочкой, — они, наоборот, вклю-
чают в нее большое количество ионов соли и молекул воды, которые стабилизируют их струк-
туру и поддерживают растворимость.
В биохимии для осаждения и крис-
таллизации белков часто используют
сульфат аммония. Ионы сульфата умень-
шают растворимость неполярных (гидро-
фобных) групп, влияя на структуру воды.
Так, на заре биохимии белки делили на
глобулины пли альбумины в соответс-
твии с концентрацией сульфата аммония,
необходимой для их высаживания.
А1.3.1. Ионная сила
и теория Дебая-Хюккеля
Открытие диссоциации сильных элек-
тролитов на отдельные ионы в водном
растворе оказало огромное влияние
на последующее развитие физической
химии. Из-за электростатического вза-
имодействия между зарядами раство-
ры ионов далеки от идеальных, даже
когда они сильно разведены. Понятие
активности (см. выше) было введено
для сильных электролитов. Для учета
влияния ионов различных валентнос-
тей Льюис и Ренделл предложили счи-
тать, что средняя активность полностью
диссоциировавшего электролита в раз-
бавленном растворе зависит только от
ионной силы раствора г.
<4Хсл;. <а1.8)
где С, zj — молярная концентрация н за-
ряд иона j соответственно. Значение /,
рассчитанное для 1 мМ раствора NaCl,
составляет 1мМ. Значение i для 1мМ
раствора MgCl., — 2,5 мМ.
Конечно, ионная сила может быть
рассчитана для растворов любой концен-
трации, и, к сожалению, некоторые иссле-
дователи пишут о ионной силе растворов
с концентрацией 1 М и даже выше. Мы
говорим «к сожалению», потому что не-
льзя забывать о том, что понятие ионной
силы применимо только к сильно разбав-
ленным растворам, для которых позво-
лительно пренебречь «природой» ионов
и их взаимодействием с водой, чтобы
рассматривать их как точечные заряды.
Комментарий А1.7. Уравнение
Пуассона
Линейное дифференциальное уравне- ।
ние в частных производных первого поряд- '•
ка, названное в честь физика XIX в. Симо-
на-Дени Пуассона, возникает при решении i
проблем электростатики. Закон электро-
статики Гаусса утверждает, что поток элек-
трического вектора Е через поверхность ра- |
вен 4л величины заряда, окруженного этой
поверхностью. Это может быть записано .
в следующем виде: I
ЭЕ ЭЕ ЭЕ.
—- + —- +—-
Эх ду dz
4 ла,
(А)
где Е. — компоненты Е и а — плотность
заряда в (х, у, z).
Теперь Е можно записать в виде про-
изводной потенциала и, взятой со зна-
ком минус:
Ех
ди „ ди ди
—',Е =----;Е =-----.
Эх у ду 2 dz
(Б) |
Уравнение Пуассона получается подста-
новкой выражения (Б) в (А). Оно связыва- I
ет потенциал с плотностью заряда: I
д2и д2и д2и . .
гт+тт+тт+4ла=0-
Эх ду dz
(В) ।
Строго говоря, расчет ионной силы дол-
жен ограничиваться концентрациями
в миллимолярном диапазоне.
Теория Дебая—Хюккеля была раз-
вита для понимания поведения разбав-
ленных растворов электролитов. Она
базируется на уравнении Пуассона
(коммепrapin'! A 1.7), наиболее общей
форме закона Кулона из электростати-
ки и статистической механике и служит
для расчета электростатического потен-
циала в определенном месте раствора в
терминах концентрации и распределе-
ния зарядов ионов, а также диэлектри-
ческой константы растворителя. Тео-
рия устанавливает соотношение между
ионной силой и коэффициентом актив-
ности раствора электролита при низких
значениях ионной силы, т. е. когда это
понятие действительно уместно. Она
предсказывает уменьшение коэффици-
ента активности с повышением ионной
силы, что находит экспериментальное
подтверждение. При высоких значени-
ях ионной силы (когда данное понятие
уже ошибочно) коэффициент актив-
ности растет и в некоторых случаях мо-
жет достигать значений, превышающих
единицу. При разработке подхода, с ис-
пользованием теории, похожей на тео-
рию Дебая—Хюккеля, и включающего
в себя область высоких концентраций,
должны учитываться специфические
свойства ионов, такие как взаимодей-
ствие растворенного вещества с водой
(сольватация).
А1.3.2. Полиэлектролиты и эффект
Доннана
Полиэлектролиты представляют собой
макромолекулярные ионы. К примеру,
ДНК в виде высушенного порошка яв-
ляется нейтральной солью, поскольку
отрицательно заряженные фосфатные
группы молекулы нейтрализованы по-
ложительными противоионами,напри-
мер Na*. Противоионы и макромолеку-
лы диссоциируют в растворе, образуя
отрицательно заряженные полиионы
(или макроионы) и «свободные» ионы
Na*. Распределение противоионов в
электростатическом поле макроионов
может быть рассчитано при низкой
ионной силе с помощью теории Де-
бая—Хюккеля.
Рассмотрим эксперимент с диа-
лизом на рисунке А 1.3. Здесь показан
разбавленный раствор NaCl в сосуде,
разделенном на два отсека мембраной,
которая полностью проницаема для
воды и ионов Na* и СГ. тРНК длиной
70 нуклеотидов в виде натриевой соли
находится в левом отсеке (красный
эллипсоид). В этих условиях макро-
молекула диссоциирует на полиион
тРНК с 70 отрицательными зарядами
и на 70 ионов Na* (отмечены красным).
Полиионы не проходят через мембра-
ну. Раствор содержит три электроней-
тральных компонента: 1 — вода; 2 —
макромолекула в виде натриевой соли
тРНК; 3 — NaCl. Область отрицатель-
ных зарядов в компоненте 2 ограниче-
на левым отсеком, и для сохранения
электронейтральности с обеих сторон
мембраны в этом отсеке должно быть
больше положительных ионов (Na'),
чем несвязанных отрицательных (СР).
Поскольку на одну макромолекулу
Рис. А 1.3. Эффект Доннана (ем. текст)
приходится 70 положительных проти-
воионов, то в результате будет проис-
ходить отток компонента 3 из левого
отсека в правый. Это явление называет-
ся эффектом Доннана. Распределение
ионов по обе стороны от мембраны, не-
обходимое для достижения равновесия
химического потенциала между левой
и правой частями сосуда, называется
распределением Доннана.
А1.3.3. Взаимодействия
между макромолекулами
и растворителем
Давайте снова рассмотрим два раство-
ра, разделенные диализной мембраной
(рис. А 1.1). Слева находится раствор,
состоящий из трех компонентов: воды
(1), макромолекулы (2) слишком боль-
шой, чтобы проникать через диализ-
ную мембрану и небольших молекул
растворенного вещества (3), например,
соли, которые свободно проникают
через мембрану. Раствор в правой час-
ти сосуда не содержит макромолекул.
Вода и диффундирующие молекулы
растворенного вещества перемещают-
ся между отсеками, уравновешивая
химические потенциалы ц1 и ц3. Мак-
ромолекула будет специфически вза-
|’пс. А 1.1. Растворитель, состоящий из ком-
понентов 1 (вода) и 3 (соль), показан свет-
ло-голубым. Макромолекула (компонент 2)
изображена красным эллипсоидом, а ее соль-
ватационная оболочка, состоящая из связан-
ной воды и соли, — темно-синим
имодействоватьс водой и небольшими
молекулами растворенного вещества
в основном через гидратацию, связы-
вание ионов и/или аффект Доннана.
Возникает вопрос, как все эти взаи-
модействия влияют на распределение
компонентов (1) и (3) с обеих сторон
мембраны?
Уравнение А1.9 определяет увеличе-
ние плотности раствора с левой сторо-
ны мембраны из-за присутствия макро-
молекул с концентрацией С,
' др
Р'~Р
с.,
(А1.9)
Ц|.М3
где р' и р — плотности растворов в ле-
вой и правой частях, соответственно.
Такое увеличение плотности явля-
ется свойством всего объема раствора
в целом и может быть точно измерено
с помощью денситометра путем взве-
шивания данного объема раствора при
постоянной температуре. Присутствие
макромолекул приводит к изменению
свойств окружающего их раствори-
теля. Например, если макромолекула
связывает соль и воду в соотношении,
отличном от такового в растворителе,
тогда результатом будет отток или при-
ток воды или соли в данный диализный
отсек — для компенсации и поддержки
химического потенциала диффунди-
рующих компонентов по обе стороны
мембраны. Приращение массовой плот-
ности выражает увеличение массовой
плотности на единицу концентрации
макромолекул. Оно учитывает не толь-
ко присутствие самой макромолекулы,
но и ее взаимодействие со способными
к диффузии компонентами растворите-
ля. Эта величина может быть выражена
следующим образом:
(cp/<?CJp = (1 + 4,) - р0( О, + 4, и ,). (А1.10)
где индекс ц условно обозначает кон-
станты р, и ц,, а — параметр взаимо-
действия в граммах воды на грамм мак-
ромолекул, й х — парциальный удельный
объем (в мл на г) компонента .г.
Параметр представляет воду, не
«связанную» с макромолекулами, а за-
полняющую диализный объем, с тем что-
бы скомпенсировать изменение в составе
растворителя, вызванное связыванием
(или, наоборот, отталкиванием) воды и
небольших молекул растворенного ве-
щества с макромолекулой. Изменение
плотности представляет собой разность
в массах между объемом одного грамма
макромолекул и граммов воды с од-
ной стороны и тем же объемом основной
массы растворителя с другой.
Уравнение, похожее на уравне-
ние А1.10, может быть получено в
терминах величины ф(, выраженной
в граммах молекул растворенного ве-
щества на грамм макромолекул; и
^связаны соотношением = -ффгс.,,
где — моляльность растворителя
в граммах компонента (3) на грамм
воды. Параметры предпочтительного
взаимодействия и ф, являются тер-
модинамическим выражением взаи-
модействий макромолекулы с раство-
рителем в данных условиях.
А1.3.4. Вода, соль
и гидрофобный эффект
При очень низких концентрациях солей
(без учета специфических эффектов,
связанных со свойствами различных
ионов) понятие ионной силы вполне
обосновано. Однако при высоких кон-
центрациях это не так. Структура воды
в жидкой фазе и ее специфические взаи-
модействия с растворенным веществом
играют ключевую роль при описании
поведения макромолекул в растворе.
Несмотря па многочисленные по-
пытки молекулярного моделирова-
ния для объяснения обширных тер-
модинамических данных в отношении
воды п растворов, структура воды на
молекулярном уровне остается до кон-
ца непонятой. Мы даем лишь качест-
венное описание ее характерных черт,
подразумевая, что оно основано на тща-
тельно измеренных эксперименталь-
ных термодинамических величинах.
Жидкая вода представляет собой край-
не динамичную систему направленных
водородных связей.
Каждая молекула может прини-
мать участие в образовании четырех
таких связей, и мы можем рассмат-
ривать ее как маленькое «тело» (атом
кислорода) с двумя «руками» (атомы
водорода), вытянутыми под фикси-
рованным углом по отношению друг
к другу (рис. . \ I ,э). Водородная связь
молекул воды носит электрическую
природу. Это прямо следует из того,
что температуры кипения легкой и
тяжелой воды близки, тогда как мас-
сы ядер отличаются в два раза. Заря-
ды на полярных атомах появляются в
результате того, что электроотрица-
тельные атомы кислорода оттягивают
электронные облака от соседних водо-
родных атомов. В результате на первых
появляется отрицательный заряд, а на
последних — небольшие положитель-
ные заряды. Электронная оболочка
атома водорода очень мягкая, и она
легко деформируется при сближении
с атомом кислорода. При таком сбли-
жении и появляется водородная связь.
Напомним, что энергия водородной
связи составляет около 5 ккал/моль
( KOMMcii I арий ,\ I).
Молекулы воды в жидкой фазе
постоянно меняют положение в про-
странстве, образуя водородные связи с
разными партнерами, подобно челове-
Комментарии A 1.8. Энергия
водородной связи
Эта оценка получается из сравнения
экспериментальных теплот испарения ве-
ществ, состоящих из одинаковых атомов,
из которых одно может образовывать водо-
родные связи, а другое нет. Классический
пример —диметиловый эфир (И ,С-О-СН().
теплота испарения которого -5 ккал/моль
и этиловый спирт (CH ,-С11,-011). тепло-
та испарения которого в два раза больше,
око.чо 10 ккал/моль. Причина различия
проста - молекула спирта может образо-
вывать водородную связь (одну), тогда
как диметиловый эфир — ни одной. По-
этому цена одной водородной связи —
5 ккал/моль.
а б и г
I’.n . \ IСтруктуры воды: а -одиночная молекула воды, большой темный атом кислород,
два светлых атома а томы водорода: б — направленные водородные связи вокруг одной молеку-
лы, образуют тетраэдр; и двумерная модель упорядоченной структуры воды или льда: / дву-
мерная модель неупорядоченной структуры воды. Рисунки любезно предоставлены профессо-
ром Джоном Финни (John Finney)
Pm. A 1.6. Трехмерная модель воды. Данная
структура высоко динамична благодаря водо-
родным связям, которые постоянно образу-
ются, разрушаются и восстанавливаются уже
в других положениях. В макрообъеме воды
каждая молекула обладает конфигурационной
свободой образования таких связей во всех на-
правлениях, которая не сохраняется в присут-
ствии растворенного вещества. Рисунок любез-
но предоставлен профессором Джоном Финни
(John Finney)
ку, стоящему в центре толпы: каждое
мгновение он соприкасается с людь-
ми вокруг себя, тогда как они сами
вынуждены ощущать чужие сопри-
косновения (рис. А 1.6). В термодина-
мических терминах образование водо-
родной связи приводит к уменьшению
энтальпии (отрицательной АН), в то
время как конфигурационная свобо-
да образования связей с различны-
ми партнерами вызывает повышение
энтропии (положительной А5). Оба
эффекта способствуют уменьшению
свободной энергии (АС = АН — TAS) и
поэтому являются термодинамически
выгодными (см. Часть В).
Что произойдет со структурой
воды в присутствии растворенного
вещества? Его молекула изменит ди-
намику молекул воды, находящихся с
ним в контакте. Если вещество поляр-
но (т. е. несет заряд), либо нейтрально,
но способно образовывать водородные
связи, оно может упорядочивать окру-
жающие ее молекулы воды. Мы видели
в разделе, посвященном парциальному
объему, как небольшой заряженный
ион (Li* или Na*) «тянет» на себя моле-
кулы воды, вызывая электрострикцию
(см. параграф А 1.2.2). Если вещество
неполярно (т. е. не обладает свойства-
ми иона и не способно образовывать
водородные связи), оно все равно бу-
дет приводить к изменению структуры
воды, поскольку находящиеся вблизи
него молекулы будут иметь меньше
возможностей образовывать водород-
ные связи. Молекулы теряют конфи-
гурационную свободу образования та-
ких связей во всех направлениях, и их
энтропия падает. По этой же причине
неполярные вещества плохо растворя-
ются в воде. Более того при температу-
ре, близкой к комнатной, они обладают
аномальной кривой растворимости,
т. е. их растворимость падает с повы-
шением температуры (рис. А1.7). И
тогда термодинамику системы опреде-
ляют эффекты, связанные с влиянием
растворенного вещества на энтропию
растворителя (уменьшение конформа-
ционной свободы или числа возмож-
ностей образования водородных мос-
тиков).
Явление, называемое гидрофоб-
ным эффектом, обусловлено низкой
растворимостью неполярных веществ
в воде. Вследствие того, что контакт
между растворенным веществом и
водой не выгоден с точки зрения
энтропии (см. Часть В), неполяр-
ные вещества склонны к агрегации
для минимизации поверхности, вза-
имодействующей с окружающими
молекулами воды и приводящей к
возникновению гидрофобной (бук-
вально — «боящейся воды») силы,
соединяющей! их вместе.
Соли изменяют структуру окружа-
ющей нх воды в соответствии со спе-
цифическими ионными свойствами, в
особенности такими, как заряд и объем.
При высоких концентрациях солей та-
кое воздействие испытывает большая
часть молекул воды ( ко.ммсн lapini ,\ I.!).
Л 1.10), что, в свою очередь, влияет на
растворимость других растворенных ве-
ществ. Ионы, еще сильнее понижающие
растворимость неполярных веществ в
воде, называются высаливающими. До-
бавление таких солей приводит к выса-
ливанию неполярных веществ, т. е. к их
осаждению. Поверхность биологичес-
ких макромолекул (белков и нуклеи-
новых кислот) гетерогснна и содержит
различные заряженные и неполярные
участки, что обусловливает их сложные
взаимодействия при гидратации, свора-
чивании и растворении (см. также Часть
Т/°С
Рис. А 1.7. Растворимость неполярного ве-
щества в воде на примере бензола. X означает
растворимость, выраженную через мольную
долю. Свободная энергия переноса Гиббса
представлена как RT In X. (Franks F„ Gent M.,
Johnson H.H., 1963)
В). Сериями Гофмайстера называют
классификацию ионов согласно их спо-
собности стабилизировать нативную
Комментарий А1.9. Моляльность солей и воды
Килограмм воды в жидкой фазе содержит около 55 молей данного компонента. Таким об-
разом, 5-моляльный раствор NaCl состоит из 10 молей ионов соли и 55 молей воды. Учитывая,
что в среднем каждый ион окружен шестью молекулами воды, становится ясно, что такой
раствор не содержит молекул воды свободных от контактов с ионами.
Комментарий А1.10. Хаотропы и космотропы
Структура воды в жидкой фазе представляет собой высокодинамичное образование на-
правленных водородных связей (см. выше). В соответствии с воздействием на нее ионы де-
лят на космотроны и хаотропы. Космотропы — это «организаторы» водной структуры. Сюда
относятся в основном небольшие ионы с высокой плотностью заряда (например, Na*, Li’, F ). ,
Хаотропы — это большие ионы - «разрушители» водной структуры. К ним относятся Rb’,
Br, C.s’ и I. Ионы К' и С1 с трудом вписываются в эту категорию. Существует сложная взаимо-
связь между способностью ионов к упорядочиванию водной структуры и их высаливающи-
ми и всаливающими свойствами. Вспомним, что серии Гофмайстера имеют феноменологиче-
ский характер. Космотропные свойства ионов коррелирует с их высаливающими эффектами.
В случае же с катионами, чей сложный характер связывания со свернутыми и развернутыми
макромолекулами смазывает картину, взаимосвязь высаливающих и всаливающих явлений
прослеживается не столь отчетливо.
Тионнца ALL Серии Гофмайстера
Серии Гофмайстера для катионов и анионов. Среди катионов высаливающие свойства наи-
более выражены у фосфата, тогда как хлорид в этом отношен ни нейтрален, а тиоцианид вса-
ливает. Что касается анионов, то аммоний, калий и натрий нейтральны, а кальций и магний
более всаливаюшие, чем литий.
катионы:
фосфат > сульфат > ацетат > хлорид < бромид < хлорат < тноцианнд
анионы:
аммоний, калий, натрий < литий < кальций, магний
Комментарий А1.11. Буферные
растворы
Все эксперименты с белками и нуклеи-
новыми кислотами проводятся в буфер-
ных растворах, подчеркивая тот факт, что
макромолекулы поддерживают нативную
конформацию только в определенном ок-
ружении. Буфер представляет собой набор
химических веществ, которые способны
поддерживать pH в относительно узком
диапазоне. Другим компонентом буферно-
го раствора может служить определенная
соль в соответствующей концентрации.
Иногда добавляется восстанавливающий
агент для защиты SH-групп (дитиотрейтол,
или меркантоэтанол).
структуру и снижать растворимость
белков в водных растворах ( г; > A. i. A I . I ).
Эти серии были установлены эк-
спериментально и верны для многих
самых разнообразных процессов, от
растворимости белков до переходов
«спираль-клубок» некоторых поли-
меров и стабилизации свернутой или
развернутой формы макромолекул.
Явление всаливания заключается в
увеличении растворимости макромо-
лекул при добавлении соли. Сильные
всаливаюшие ионы в высоких концен-
трациях дестабилизируют свернутую
структуру, предположительно из-за
того, что развернутая цепь предостав-
ляет больше возможностей для связы-
вания. Порядок ионов в сериях Гоф-
майстера не является строгим и может
варьировать для разных белков. Тем
не менее классификация анионов как
стабилизирующих (высаливающих) и
дестабилизирующих (всалпвающих)
ионов в общем верна (см. ком.\н д
piiii \ !. I 0 ).
Эффекты влияния солей на биологи-
ческие макромолекулы широко исполь-
зуются в биохимии и биофизике, как
важнейшая часть набора методов фрак-
ционирования, солюбилизации, кри-
сталлизации и т.д. Но ключевую роль
растворителя в поддержании целостно-
сти активных биологических макромо-
лекул нельзя преувеличить. В биохимии
очень важно работать в четко охарак-
теризованном, буферном растворителе
(а >мм<ч 11;11м । н ALII).
А1.4. Перечень ключевых идей
Биологические молекулы как биологически активные частицы не могут рас-
сматриваться отдельно от своего водного окружения.
* Концентрация растворенного вещества в растворе может быть выражена
как масса на объем (весовая концентрация) или как число молекул на объем
(молярность или моляльность).
* Коллигативные свойства растворов зависят от количества молекул
растворенного вещества.
* Парциальный объем растворенного вещества равен изменению объема раствора
после добавления вещества.
* Закон Рауля утверждает, что в идеальном растворе при постоянной температуре
парциальное давление компонента в жидкой смеси пропорционально его
мольной доли.
* Следствием закона Рауля является понижение температуры замерзания
(или повышение температуры кипения) раствора по сравнению с чистым
растворителем, пропорциональное молярной концентрации растворенного
вещества.
• Химический потенциал растворенного вещества представляет собой свободную
энергию, создаваемую при его добавлении в раствор.
® Понятие активности учитывает отклонение поведения растворов от идеальных;
активность растворенного вещества равна концентрации, помноженной на
коэффициент активности, который для идеального раствора равен единице.
* Законы, выраженные в терминах концентрации и полученные для идеальных
растворов, остаются действительными и в неидеальных условиях, если
концентрация заменяется на активность.
* В неидеальных растворах осмотическое давление может быть выражено
разложением концентрации в степенной ряд, коэффициенты разложения
которого называются вириальными коэффициентами.
• Биологические макромолекулы активны и имеют нативную конформацию в узком
диапазоне условпй, касающихся свойств растворителя, температуры и давления.
° Экстремофилы — это организмы, живущие в экстремальных условиях,
связанных со свойствами растворителя, с температурой или давлением.
• Осмотическое давление представляет собой гидростатическое давление,
оказываемое на полупроницаемую мембрану со стороны более высокой
концентрации вещества.
Осмотическое давление является коллигативным свойством; при комнатной
температуре оно пропорционально разности молярных концентраций раство-
ров с обеих сторон мембраны.
Соли оказывают значительное воздействие на стабильность и растворимость
макромолекул в растворах, в соответствии с их концентрацией и химической
природой; влияние солей обусловлено их взаимодействием с водой или пря-
мым связыванием с макромолекулами.
Понятие ионной силы верно только при очень низких (миллимолярных) кон-
центрациях солей, когда учитываются только концентрация и валентность ио-
нов, но не их конкретная природа.
Теория Дебая-Хюккеля позволяет рассчитать электростатический потенциал
в любом месте раствора электролита в терминах концентрации и распределе-
ния зарядов ионов и диэлектрических констант; она применима для раство-
ров с низкой концентрацией ионов, когда понятие ионной силы еще остается
верным.
Структура воды в жидкой фазе и ее взаимодействия с небольшими молекулами
растворенного вещества играют ключевую роль в определении поведения мак-
ромолекул в растворе.
Вода в жидкой фазе представляет собой высокодинамичную систему направ-
ленных водородных связей.
Гидрофобный эффект обусловлен низкой растворимостью неполярных групп
в воде ввиду невозможности образования ими водородных связей; он являет-
ся результатом действия гидрофобной силы, соединяющей вместе неполярные
группы в водном растворе.
Растворенные соли влияют на гидрофобный эффект в соответствии со своей
природой и концентрацией.
Космотропные ионы, «созидатели» водной структуры, представляют собой не-
большие ионы с высокой плотностью заряда, повышающие растворимость не-
полярных групп в воде; хаотропные ионы, «разрушители» водной структуры,
усиливают гидрофобный эффект.
Сериями Гофмайстера называют феноменологическую классификацию ио-
нов в соответствии с их способностью стабилизировать нативную структуру
и осаждать белки и нуклеиновые кислоты в растворах.
Литература для дальнейшего чтения
Von Hippel Р., Schleich Г. The effects of neutral salts on the structure and confor-
mational stability of macromolecules in solution./In: S.N. Timasheff, G. D. Fasman ed.
Structure of Biological Macromolecules. N. Y.: Marcel Dekker Inc., 1969. P. 417-575.
Эта глава из книги, несмотря на то, что ей уже около 40 лет, до сих пор является
прекрасным источником информации об эффектах воздействия солей на макро-
молекулы.
Финкельштейн А.В. Физика белка. М.: Изд-во «Книжный дом «Университет»,
2007.
Глава А2
МАКРОМОЛЕКУЛЫ
КАК ФИЗИЧЕСКИЕ ЧАСТИЦЫ
'ЧЭСХ/lVs обзор
Р. Бойль (R. Boyle) подверг сом-
нению химическую теорию, принятую в
его времена, и утверждал, что настоящей
задачей химии является определение
состава веществ.
А.-Л. Лавуазье (A.-L. Lavoisier)
пришел к верному пониманию процес-
са окисления. Он продемонстрировал
количественное сходство между хими-
ческим окислением и дыханием у жи-
вотных. Лавуазье считается отцом со-
временной химии.
,Щ-Л
Дж. Пристли (J. Priestley), И.Ин-
генхауз (J. Ingenhousz) и Ж.Сенебье
(J. Senebier) установили, что фотосин-
тез представляет собой процесс, обрат-
ный дыханию.
Несмотря на сильное противодейс-
твие виталистов, считавших, что транс-
формация веществ в живых организмах
не подчиняется законам химии и фи-
зики, а является результатом действия
живительной силы, развитие органи-
ческой химии привело к рождению био-
химии. В 1828 г. Ф. Вёлер (F. Wohler)
произвел первый лабораторный синтез
органической молекулы — мочевины.
В 1840 г. И. Ф. Либих (J.V. Liebig)
заложил прочный фундамент для раз-
вития органической химии и описал
наиболее значимые природные хими-
ческие циклы. В 1869 г. из клеток гноя
было выделено вещество, названное
нуклеиновой кислотой. Благодаря эк-
спериментам по трансформации Pneu-
mococcus О. Авери (О. Avery) в 1944 г.
представил веские доказательства того,
что нуклеиновая кислота является хра-
нителем генетической информации.
В 1860-х г.г. Л. Пастер, считающий-
ся отцом бактериологии, доказал, что
микроорганизмы вызывают брожение,
гниение, а также инфекционные за-
болевания, и разработал химические
методы их изучения. В 1877 г. пасте-
ровские ферменты были переимено-
ваны в энзимы (от греческого эн «в»
и зим «дрожжи»), В 1897 г. Е. Бюхнер
(Е. Buchner) обнаружил, что фермен-
тация может происходить в препа-
рате дрожжей, лишенных клеточной
стенки. Наконец, в 1882 г. Е. Фиш
(Е. Fisch) показал, что белки пред-
ставляют собой очень большие моле-
кулы, состоящие из аминокислот. Он
открыл также феномен стереоизоме-
рии углеводов.
1926
Дж. Б. Самнер (J.B. Sumner) вы-
явил, что уреаза — фермент, впервые
закристаллизованный в чистом виде, —
является белком.
I 9 К)
Ф.А. Липман (F.A. Lipmann) пред-
положил, что аденозин трифосфат
(/\ТФ), впервые выделенный из мышеч-
ной ткани в 1929 г., представляет собой
молекулу, выполняющую роль перено-
счика энергии во многих типах клеток.
1953
Дж. Д. Уотсон и Ф. Крик (J. D. Wat-
son и Е Crick) опубликовали структуру
двойной спирали ДНК, полученную бла-
годаря «химической интуиции» и данным
Р. Франклин и М. Уилкинса (R. Franklin
и М. Wilkins) по дифракции на волокнах.
Работы этих ученых были удостоены Но-
белевских премий.
195.)
Ф. Сэнгер (F. Sanger) определил
аминокислотную последовательность
инсулина, доказав тем самым, что белки
представляют собой линейные амино-
кислотные полимеры. Это достижение
было отмечено Нобелевской премией.
Его дальнейшие исследования приве-
ли к разработке эффективного метода
определения нуклеотидных последо-
вательностей ДНК н РНК. За эту рабо-
ту Сэнгер получил вторую Нобелевс-
кую премию, разделив ее с П. Бергом и
В. Гилбертом (Р. Berg и W. Gilbert).
I 960-е
М. Перутц и Дж. Кендрью (М. Ре-
rutz и J.Kendrew) получили моле-
кулярные структуры гемоглобина и
миоглобина методом рентгеновской
кристаллографии, доказав тем са-
мым, что белки имеют вполне оп-
ределенную строго упорядоченную
пространственную структуру, за что
были награждены Нобелевской пре-
мией.
1970-е
К. Вуз (С. Woese), основываясь на
анализе нуклеотидной последователь-
ности рибосомной РНК (рис. А2.1),
разделил все живые организмы на три
домена: Бактерии, Археи и Эукариоты.
Правильность этой классификации
была подтверждена и уточнена, когда
стали доступны данные о последова-
тельностях многих белков.
1980-е
С. Альтман (S. Altman) выделил
рибонуклеазу Р — фермент, состоящий
из белка и РНК-компонентов, — по-
ложив начало целому ряду открытий
других типов каталитических РНК.
Он был награжден Нобелевской пре-
мией. В 1986 г. В. Гилберт (W. Gilbert)
ввел в обиход понятие РНКового мира,
обозначающее гипотетическую стадию
в эволюции жизни, за существование
которой уже высказывались такие уче-
ные, как К. Вуз, Ф.Крик и Л. Е. Ор-
гель (С. Woese, F. Crick и L. Е. Orgel).
В этом мире РНК сочетала роль храни-
теля информации с каталитическими
свойствами, необходимыми для само-
репликации. Гипотеза существования
РНКого мира способствовала возрож-
дению интереса к изучению происхож-
дения жизни.
Впечатляющее развитие молеку-
лярной биологии сопровождалось не
менее впечатляющим прогрессом в
методах определения структуры био-
Археи
Эукариоты
Рис. А2.1. Филогенетическое дерево, построенное на основе анализа последовательности ри-
босомальной РНК, отображает три Надцарства. Результаты дальнейших исследований позво-
ляют предположить, что его корни находятся где-то между доменами Бактерий и Архей, про-
кариотическими одноклеточными организмами, чей генетический материал располагается не в
ядре. Археи были названы так потому, что они считаются ближайшими родственниками первых
живых организмов. Они имеют как свои специфические характеристики, так и те, которые они
разделяют с Бактериями и Эукариотами. Большинство известных организмов экстремофилов
(см. Главу А1) относятся к Археям. Некоторые из них, как, например, анаэробные метаногены,
найденные в кишечнике коров и болотах, распространены очень широко
логических макромолекул и изучения
их взаимодействий. Эти методы как
раз и составляют содержание данной
книги и поэтому не будут обсуждаться
в этой главе. Здесь мы опишем струк-
турные компоненты и организацию
биологических макромолекул.
А2.2. Биологические
молекулы и поток
генетической информации
Ген представлен четко определенной
нуклеотидной последовательностью в
клеточной ДНК. В ДНК существуют
четыре разновидности нуклеотидов.
Генетический код соотносит шестьде-
сят четыре возможных нуклеотидных
триплета {кодона) с двадцатью ами-
нокислотами, найденными в белках.
Одной и той же аминокислоте могут
соответствовать несколько триплетов,
что обеспечивает белкам стабильность
в отношении мутаций на уровне ДНК.
Существуют также кодоны, соответ-
ствующие сигналам «стоп», которые от-
мечают конец цепи. В белковом синтезе
генетическая информация сначала пе-
реписывается в нуклеотидную последо-
вательность матричной РНК в процессе
транскрипции, а затем уже переводится
в аминокислотную последовательность
белка на рибосоме (трансляция), в со-
ответствии с центральной догмой моле-
кулярной биологии:
ДНК о- ДНК <-> РНК -> белок.
Первая стрелка обозначает репли-
кацию материнской ДНК с образова-
нием идентичных дочерних молекул,
вторая — транскрипцию ДНК, в резуль-
тате чего образуется РНК, и обратную
транскрипцию РНК -> ДНК, обнару-
женную у ретровирусов, а последняя
стрелка обозначает трансляцию.
Схема показывает поток информа-
ции, а не последовательность эволюции
макромолекул, поскольку конечный
продукт этой схемы — белок — необхо-
дим на каждом этапе. Все эти процессы
катализируются и регулируются бел-
ковыми транскрипционными факто-
рами, белковыми ферментами, такими
как полимеразы, белковыми трансля-
ционными факторами и рибосомами,
которые сами состоят из белков и РНК.
Конечным результатом этого процесса
является белок, имеющий аминокис-
лотную последовательность (первич-
ную структуру); он один в один соот-
ветствует гену, его кодирующему.
Перед тем как приобрести биоло-
гическую активность, белок должен
претерпеть процесс сворачивания в
результате межцепочечных взаимо-
действий и взаимодействий с раство-
рителем. Его вторичная структура
организована локальными конфор-
мациями, такими как а-спирали или
В-слои, скрепленными внутри- и меж-
цепочечными водородными связями.
Третичная структура формируется
за счет элементов вторичной струк-
туры, образующих компактную трех-
мерную укладку, тогда как четвертич-
ная структура получается в результате
объединения в комплекс разных бел-
ков или копий одной и той же белко-
вой молекулы.
Часто при обсуждении структуры
ДНК используется терминология из об-
ласти полимеров. Объясняется это тем,
что ДНК состоит из линейной цепочки
повторяющихся структурных единиц,
нуклеотидов. Каждый нуклеотид обра-
зован фосфатрибозной группой, свя-
занной с одним из четырех азотистых
оснований: аденин (А), гуанин (Г), ци-
тозин (Ц) и тимин (Т). ДНК однако
отличается от обычных синтетических
полимеров тем, что она способна фор-
мировать специфические структуры
в соответствии со своим окружением
и взаимодействиями с белками, непо-
средственно связанными с ее биологи-
ческой функцией.
Зачастую удобно определять фи-
зический объект, описывая, чем оп не
является. Белок, вероятно, даже в боль-
шей степени, чем ДНК, не является
классическим полимером (рис. А2.2).
Данное утверждение может показать-
ся противоречивым, поскольку белок
часто описывают как полимер из ами-
нокислотных остатков. Между тем его
биологическая активность наделяет
белковую молекулу специфическими
свойствами, отличными от тех, которы-
ми обладают классические полимеры.
Их изучение привело к формулировке
законов науки о полимерах. Отметим,
что химики уже занимаются синтезом
полимеров со свойствами, специфич-
ными для белков.
Укажем несколько свойств, кото-
рые отличают белки от классических
полимеров.
Во-первых, различие в составе и мо-
лекулярной массе. Обычная полимер-
ная цепь состоит из небольшого числа
различных элементов, повторяющихся
много раз. Типичный образец состоит
из цепей различной длины, поэтому
молекулярная масса определяется как
среднее статистическое значение рас-
пределения цепей по длинам (см. при-
мер в параграфе Г.4.71). Белковая цепь
Белок
Обычные гомополимеры
Длина цепи (молекулярная масса)
N . С - _______
- , Статистическое распределение
Определено с точностью лучше, м н
чем один аминокислотный остаток
Состав
Двадцать аминокислот
А А А ААААААА
▼▼▼▼▼▼▼▼▼▼
Повторяющиеся блоки
Цепь в растворе
Уникальная трехмерная
структура при
физиологических
условиях
Разупорядоченная
структура при
денатурирующих
условиях
Рис. А2.2. Белок не является классическим полимером
имеет совершенно конкретные начало
(N-конец) и конец (С-конец), ее первич-
ная структура четко обозначена (напри-
мер, все 1013 молекулярных цепей в од-
ной десятой миллиграмма белка имеют
идентичную массу с точностью, ограни-
ченной только естественной изотопной
вариабельностью). Эти свойства, экспе-
риментально подтвержденные с помо-
щью масс-спектрометрии (см. пример в
параграфе Б2.2.1), следуют из структу-
ры гена, кодирующего белок.
Во-вторых, говоря о конформации
белка в растворе, стоит обратить внима-
ние на то, что классическая полимерная
цепь принимает в растворе конформа-
цию, которая зависит от растворите-
ля и, опять-таки, может быть описана
как среднее значение в распределении
различных конформаций. Напротив,
водорастворимые и мембранные белко-
вые цепи сворачиваются в присутствии
растворителя, в котором они активны,
и формируют строго определенные
компактные структуры. Эти структуры
в основном идентичны для всех моле-
кул в чистом препарате белка. Кристал-
лографические и ЯМР исследования
белков (образцов, также содержащих
около 1015 молекул) позволяют разре-
шать структуры, где позиции атомов
можно определить с точностью лучше
1 ангстрема.
В физиологических условиях бел-
ковая цепь принимает определенную
третичную и четвертичную структуру,
которая разворачивается в денатури-
рующих условиях (при повышенной
температуре или, например, в раство-
рах, содержащих мочевину или гуани-
дингидрохлорид). Денатурированный
белок принимает конформацию, по-
хожую на таковую гомополимеров
(гауссов клубок). Однако неоргани-
зованные белковые структуры не обя-
зательно должны быть биологически
неактивны. Доступные в настоящее
время полные геномные последова-
тельности указывают на то, что боль-
шая часть генов кодирует длинные
аминокислотные последовательности,
и они остаются несвернутыми в фи-
зиологических условиях (см. примеры
в параграфе Г9.5.4). Это утверждение
особенно справедливо для эукариоти-
ческих организмов.
Кроме ДНК, РНК и белков, клет-
ка содержит еще два больших класса
молекул — углеводы и липиды. Они
играют важнейшую структурную и
функциональные роли. Углеводы и ли-
пиды не кодируются непосредственно
в геноме, а являются субстратами и
продуктами белковых ферментов и
синтезируются в сложных метаболи-
ческих цепях.
А2.3. Белки
Все молекулы живых организмов пред-
ставляют собой в основном либо белки,
либо продукты деятельности белков. За
исключением рибозимов, образующих
небольшой класс РНК-молекул, все
ферменты (биохимические катализато-
ры метаболических реакций) являются
белками. Некоторые гормоны (моле-
кулы, осуществляющие регуляторные
функции через взаимодействие с бел-
ковыми рецепторами), сами белки, в
то время как другие — лишь белковые
составляющие, такие как одиночные
аминокислоты или небольшие пепти-
ды. Два больших класса гормонов пред-
ставлены стероидами и аминами.
Белки могут выполнять также
и транспортные функции (например,
гемоглобин — переносчик кислорода в
красных клетках крови). Они состав-
ляют основу мышечной и соединитель-
ной ткани, поддерживающей структуру
тела животных, а также кожи и волос.
Белки являются компонентами удиви-
тельно продуманных механизмов рас-
познавания и сигнальных путей, как,
например, в иммунной системе, а также
в разнообразных клеточных ответах на
внешние раздражители или стимулы,
которые могут быть как химическими,
так и физическими, как в случае с фо-
тонами в зрительном процессе или в
фотосинтезе. Из белков состоят мемб-
ранные насосы и каналы, устанавлива-
ющие электрохимические градиенты,
необходимые для биоэнергетики клет-
ки. Они играют важную роль в переда-
че нервных импульсов, но этим список
биологических функций, выполняе-
мых белками, не исчерпывается.
Как мы указывали, в состав бел-
ков входят в основном 20 разных ами-
нокислот, объединенных в линейную
цепь с помощью пептидных связей. Их
биологическая активность — результат
организации более высокого уровня,
которую формирует цепь в физиоло-
гических условиях. Белок определяет-
ся как объект, обладающий биологиче-
ской активностью, тогда как понятие
полипептид указывает на его химиче-
ский состав. С белком может быть
связана простетическая неаминокис-
лотная группа, необходимая для его
активности. В качестве примера можно
привести кислород-связывающие гемо-
вые группы гемоглобина, миоглобина
и некоторых белков дыхательной цепи,
ретпналь, который обеспечивает све-
товую чувствительность родопсина —
белка, имеющего отношение к зрению,
и хлорофилл в белках растений, свя-
занный с фотосинтезом. На самом деле,
поскольку природные аминокислоты
бесцветны (они поглощают в основном
в ультрафиолете), цвет белка всегда
обусловлен простетической группой:
красный — гемом, пурпурный — рети-
налем, зеленый — хлорофиллом. На-
личие таких светопоглощающих групп
позволяет исследовать связанные с
ними белки спектроскопическими ме-
тодами.
А2.3.1. Химический состав
и первичная структура
Общая структура аминокислот и ее из-
менение при образовании пептидных
связей в составе полипептидной цепи
показана на рисунке А2.3. Первичная
структура белка представляет собой
последовательность аминокислот в
составе полипептида. Боковые цепи
двадцати основных природных амино-
кислот и их свойства показаны на ри-
сунке А2.4.
Аминокислоты, за исключением
глицина, могут существовать в двух
энантиомерных формах, зависящих
от положения альфа-углерода, с кото-
рым связана боковая цепь. Природные
аминокислоты находятся в D (декстро,
или правосторонней) форме, в соот-
ветствии с направлением поворота
плоскости поляризации света (см. Гла-
ву 35).
А2.3.2. Структуры высших порядков
Структура белка может быть изобра-
жена различными способами. Изобра-
жения на рисунке А2.5 базируются на
одной и той же структурной модели,
полученной с помощью кристаллогра-
фических данных. Каждая подчеркива-
ет различные аспекты структурной ор-
ганизации белковой молекулы.
В нашем примере белок представ-
ляет собой фермент метаболизма, ма-
лат дегидрогеназу, из «галофильного»
(живущего в среде с высокой концен-
трацией соли) организма Haloarcula
marismortui.
Вторичная, третичная и четвертич-
ная структуры белка показаны в верхней
части рисунка А2.5 а, где полипептид-
ная укладка дана в ленточном представ-
лении. Вторичные структуры — это
разрешенные локальные конформации
цепи, возникающие вследствие хими-
ческих и стерических ограничений.
С-конец
Рис. А2.3: а — аминокислота в нейтральной форме и в виде иона, несущего двойной заряд (цвит-
терион). Обе формы встречаются в растворах. R — вариабельная боковая цепь; б — полимери-
зация аминокислот при образовании полипептидной цепи сопровождается потерей одной мо-
лекулы воды на каждую вновь образованную пептидную связь; в — цепь имеет N- и С-концы.
Направление от N-конца к С-концу соответствует направлению, в котором идет транскрипция
гена и синтез полипептидной цепи на рибосоме
Аланин Ala А Аргинин Arg R Аспарагин Asn N
Аспарагиновая
кислота Asp D
Цистеин Cys C
Глутамин Gin Q
h2n
Гистидин His H
Глицин Gly G
Лизин Lys К
Изолейцин He I
Пролин Pro P
Метионин Met M Фенилаланин Phe F
Треонин Thr T
Триптофан Trp W
Тирозин Туг Y
Валин Vai V
Рис. A2.1. Двадцать основных природных аминокислот. Под каждой указан соответствующий
трех- и однобуквенный коды. Основная цепь показана темно-красным, неполярные боковые
цепи — черным, нейтральные полярные — зеленым, положительно заряженные (основные) — си-
ним, отрицательно заряженные (кислые) — красным. Гистидин изображен в заряженной форме
(значение pH ниже 6,0). Рядом с написанием каждой аминокислоты дано ее трех- и однобуквен-
ное обозначение в английской транскрипции
а б
Рис. А2.5. Разные способы изображения одной и той же белковой! структуры используются для
того, чтобы подчеркнуть ее различные особенности: а — верх: ленточная схема показывает ор-
ганизацию элементов вторичной структуры (закрученные ленты обозначают а-спирали, стрел-
ки — p-листы) в третичную структуру, каждой из четырех субъединиц, которые все вместе обра-
зуют тетрамерную четвертичную структуру; низ: шарики и палочки — «лупа» атомных размеров,
увеличивающая область контакта димеров друг с другом, с тем чтобы показать сложную картину
организации солевых мостиков между ионами в этом месте белковой структуры; б — атомное
представление — наполнение пространства белка атомами', отрицательно заряженные атомы по-
верхности белка изображены красным, положительно заряженные атомы — синим
Альфа-спирали и бета-слои (см. ниже)
являются наиболее распространенны-
ми белковыми структурами, они пока-
заны, соответственно, как ленточные
спирали и стрелки. Представленный бе-
лок — тетрамер. Его третичная струк-
тура представляет собой трехмерную
конформацию субъединицы, а четвер-
тичная структура этого белка образу-
ется за счет объединения субъединиц
в тетрамер.
Увеличенный фрагмент белко-
вой структуры, детально иллюстриру-
ющий взаимосвязи различных хими-
ческих групп, показан в представлении
шарики-палочки в нижней части ри-
сунка А2.5 а. Подобные иллюстра-
ции часто используются, например,
для демонстрации взаимодействий в
активном центре фермента. В нашем
случае картина состоит из множест-
ва солевых мостиков (ионных связей
между заряженными аминокислота-
ми) и ионных связей с солями раство-
рителя, от которых зависит стабиль-
ность белка в присутствии высокой
концентрации соли.
В атомном представлении на ри-
сунке А2.56 вокруг каждого атома
нарисована ван-дер-ваальсовая сфе-
Рис. A2.(i. Плоские пептидные группы поли пептидной цепи
ра — для более наглядного отображе-
ния белковой поверхности. Отрица-
тельно заряженные атомы показаны
красным, а положительно заряжен-
ные — синим цветом. Поверхность
данного белка имеет суммарный от-
рицательный заряд, что, по-видимому,
характерно для белков тех организ-
мов, которые адаптированы к высокой
концентрации соли.
Вторичная структура
Атомы в основной полипептидной цепи
(N — Са — С — N -...) не могут нахо-
диться на прямой линии или свободно
вращаться, что обусловлено свойства-
ми связи между ними. В особенности
это касается атомов пептидной связи в
ОН
группе, которые лежат в одной плос-
кости, что ограничивает гибкость цепи
(рис. А2.6).
Конформация цепи может быть
задана значениями углов ф и у, меж-
ду плоскостью пептидной связи и Са
атомами с каждой из сторон. Угол ф
соответствует вращению NH-группы
относительно С атома и отсчитыва-
ется против часовой стрелки, если
смотреть с позиции альфа-углерода.
А угол \|/ соответствует вращению СО-
группы также относительно Сц атома
и отсчитывается по часовой стрелке,
если смотреть с той же позиции. По-
стоянные значения ф и ц/ углов прида-
ют цепи регулярную спиралеобразную
конформацию. Картой Рамачандрана
называют двумерное отображение пар
углов ф—\|/ и соответствующих им вто-
ричных структур (см. секцию Е3.5).
Некоторые из этих структур, например
а-спираль, стабилизируются за счет
виутрицепочечных водородных связей
и относятся к энергетически выгодным
(рис. А2.7). «Прямая» цепь с череду-
ющимися по обе стороны боковыми
группами аминокислот называется
Р-слоем. p-листы состоят из р-слоев,
скрепленных вместе водородными свя-
зями. Они могут быть параллельными
Рис. А2.7. а-спираль. Карбоксильная группа
j основной цепи связана водородной связью
с NH-группой аминокислоты j + 4
ПО ПР n
Put-. A2.8. Параллельный (верх) и антипарал-
лельный (низ) р-листы
и антипараллельными (рис. А2.8). В то
же время а-спирали и p-слои являют-
ся наиболее распространенными вто-
ричными структурами, если судить по
содержимому банков данных белковых
структур, находящихся в Интернете
( ।м м и 11та р11 ii А2.1 ).
Вторичные структуры асиммет-
ричны не только вследствие направ-
ленности основной цепи, но также
из-за асимметричного расположения
Комментарий А2.1. Аллостерия
Термином «аллостерия» обозначают
вызванное эффектором изменение струк-
туры, способствующее определенной ак-
тивности, например, связыванию субстрата
с ферментом. В качестве примера возьмем ।
гемоглобин, белок — переносчик кислорода :
в крови. Гемоглобин является тетрамером,
с которым связаны четыре гемовые груп-
пы. Субъединица может принимать либо 1
релаксированную (R), либо напряженную
(Т) конформацию. Кислород действует как I
аллостерический эффектор, одновременно ।
являясь и субстратом. Когда он связывает-
ся с одной из субъединиц, это вынуждает
остальные субъединицы принять R-koh-
формацию, которая имеет высокое срод- :
ство к кислороду.
цепи боковой группы аминокислот
относительно альфа-углерода (за ис-
ключением глицина). Следует напом-
нить, что природные аминокислоты
имеют D- (правостороннюю) энан-
тиомерную конфигурацию, поэтому
в правосторонней альфа-спиральной
конфигурации боковые группы будут
направлены наружу, а боковые цепи
бета-слоя направлены наружу пооче-
редно вверх и вниз. Таким образом,
бета-слой, в котором чередуются по-
лярные и неполярные аминокислоты,
будет иметь полярную и неполярную
стороны.
Третичная структура
Третичная структура белка является
результатом слабых (иековалентных,
за исключением дисульфидных свя-
зей между двумя цистеинами) взаи-
модействий между аминокислотами в
полипептидной цепи. Она лежит в ос-
новании структурно функциональной
гипотезы. Интересно отметить, что ее
общие черты в пределах одного бел-
кового семейства могут быть высоко
консервативны, даже если первичные
структуры этих молекул сильно варьи-
руют. Большинство белков структурно
схожи с другими белками. Изучение
этих общих черт важно не только для
понимания и установления эволюци-
онного и функционального родства, но
также для структурного анализа нук-
леотидных последовательностей, полу-
чаемых в геномных проектах.
Третичные структуры белковых
субъединиц можно разделить на одно-
доменные и мультидоменные. Напри-
мер, дегидрогеназы — ферменты мета-
болизма имеют общий динуклеотидный
кофакторный домен, названный в честь
его открывателя укладкой Россмана.
СН5
б
Рис. А2.9: <7 — ленточная модель димера дифтерийного токсина (DT), демонстрирующая трех-
доменную структуру (С, Т, R). Один мономер показан серым, другой — белым. Данный димер
является иллюстрацией феномена доменного обмена (domain swopping), где мономеры обмени-
ваются доменами R. Каждый домен связан с определенной функцией токсина. Домен бета R (по
третичной структуре похожий на домены иммуноглобулина) отвечает за распознавание рецеп-
тора на первой стадии проникновения токсина в клетку. В процессе эндоцитоза DT переносится
в эндосому. Домен альфа Т (по третичной структуре похожий на трансмембранные белки) по-
зволяет токсину проникать через мембрану эндосомы для высвобождения каталитического до-
мена С в цитоплазму после расщепления нолипептидной цепи между С и Т. Альфа-бета домен С
представляет собой фермент, катализирующий летальную для клетки реакцию. Интересно, что
активный центр домена С скрыт за доменом R, поэтому целостный белок не активен. Подобным
образом и димер во время проникновения в клетку менее активен, поскольку домены R закрыва-
ют друг друга; б — ленточная модель «открытого» мономера (Bennet et al., 1994)
Белковая субъединица дифтерийного
токсина поразительно четко делится на
три домена, каждый из которых имеет
характерную организацию, связанную
с его функцией (рис. А2.9).
Доменные структуры классифици-
руют но группам или семействам, в со-
ответствии с анализом белковых струк-
тур по базам данных. Они доступны на
веб-страницах банков белковых струк-
тур. САТН классификация белковых
структур представляет собой иерархи-
ческую программу доменной класси-
фикации, состоящей из пяти уровней:
класса (уровень С), архитектуры (уро-
вень А), топологии семейства укладки
(уровень Т), надсемейства гомологов
(уровень Н) и семейства аминокислот-
ных последовательностей (уровень S).
Последние два уровня предназначены
для белков, имеющих общего предка, и
основаны на сравнении последователь-
ностей и структур.
Класс определяется в соответствии
с составом вторичной структуры и ук-
ладкой внутри домена. Насчитывается
четыре больших класса: а) преимуще-
ственно альфа, куда входят домены, чья
вторичная структура в основном пред-
ставлена альфа-спиралями; б) преиму-
щественно бета, который включает до-
мены, организованные бета-листами; в)
альфа-бета, состоящий из чередующих-
ся альфа и бета вторичных структур; г)
этот класс состоит из доменов с низким
содержанием вторичных структур. Ар-
хитектура описывает общую форму
домена, определяемую ориентацией
элементов вторичной структуры. Топо-
логия зависит как от общей формы, так
и от связи вторичных структур между
собой (см. параграф. 35.7.4).
SCOP (структурная классификация
белков) представляет еще одну класси-
фикацию доменных организаций, осно-
ванную на сравнении всех известных
структур. Уровнями иерархии SCOP
являются виды, белки, семейства, ук-
ладки и классы.
Четвертичная структура
Организация белковых субъединиц на
уровне четвертичной структуры была
отобрана эволюционно ввиду ее функ-
циональных преимуществ, начиная от
простой стабилизации — как в случае с
олигомеризацией ферментов из гипер-
термофильных организмов, сохраня-
ющих стабильность при температурах,
близких к 100 °C (см. Главу А1) — и
тонкой регуляции активности — как
в случае с аллостерией (от греческо-
го — «другая структура» (см. i.hmmcii-
rapnii Л 1.1 ), — до тщательно согла-
сованных сложнейших реакционных
механизмов — как в случае с большими
структурами в клетке, такими как ри-
босомы, протеосомы, термосомы (см.
Главу Ж2) и шаперонные комплексы.
Большие организованные структуры,
состоящие из различных белковых
молекул, подобные тем, что найдены
в мышцах и микротрубочках, тоже мо-
гут рассматриваться как четвертичные
структуры.
А2.4. Нуклеинозые кислоты
Нуклеиновые кислоты подразделяют-
ся на два основных класса — дезокси-
рибонуклеиновую кислоту (ДНК) и
рибонуклеиновую (РНК). Как и бел-
ки, имеющие полипептидную первич-
ную структуру, нуклеиновые кислоты
представляют собой полинуклеоти-
ды, неразветвленные полимеры субъ-
единиц определенного химического
типа — нуклеотидов. ДНК — это хра-
нилище генетической информации, и
в клеточных хромосомах она связана
с белками.
Структура ДНК преимущественно
двухцепочечная и имеет спиральную ор-
ганизацию. РНК обладает целым рядом
биологических функций (до сих пор от-
крываются новые) и еще большим чис-
лом структурных организаций. Обычно
она состоит из одной цепочки, которая,
изгибаясь и взаимодействуя сама с со-
бой, образует замысловатые вторичные
структуры. Матричная РНК (мРНК)
считывается с ДНК и переносит генети-
ческую информацию дальше — на рибо-
сому. Транспортная РНК (тРНК) пред-
ставляет собой семейство небольших
молекул РНК, состоящих примерно из
семидесяти нуклеотидов. Каждый тип
тРНК переносит одну специфическую
аминокислоту на рибосому, действуя
как адаптер между мРНК и растущей
полипептидиой цепью. Сами рибосомы
состоят из больших и маленьких моле-
кул РНК, называемых рибосомными
РНК(рРНК).
С тех пор как был выделен фермент
РНКаза П, состоящий из РНК и белка,
было найдено множество каталитичес-
ких молекул РНК. Сюда относятся так
называемые рибозимы и часть рРНК,
играющая важную каталитическую
роль в трансляции, а также siPHK, не-
большая молекула РНК, вовлеченная
в процесс молчания генов через РНК-
интерференцию с мРНК. РНК-интер-
ференция стала крайне популярной
темой современных исследований в
связи с ее возможным применением
в терапии рака и вирусных инфекций.
Удивительной молекулой является
тмРНК, так как сочетает в себе функ-
ции тРНК и мРНК.
Молекулы малой ядрышковой РНК
локализованы в ядрышках эукариоти-
ческих клеток и участвуют в биогенезе
рРНК, определяя сайты модификации
нуклеотидов.
Геном некоторых вирусов состоит
не из ДНК, а из двуспиральной РНК.
Такие вирусы называются ретровиру-
сами. Примером может служить вирус
иммунодеффицита человека (ВИЧ),
который имеет механизм рстротранс-
крипции вирусной РНК, приводящий
к образованию ДНК, способной встра-
ваться в генетический материал инфи-
цированной клетки.
А2.4.1. Химический состав
и первичная структура
нуклеиновых кислот
Как и аминокислоты, нуклеотиды со-
стоят из постоянных и вариабельных
частей. Постоянная часть формирует
основную цепь полинуклеотида и со-
стоит из нуклеозидов, которые обра-
зованы двумя ковалентно связанными
группами: дезоксирибозной (в ДНК)
или рибозной (в РНК) и фосфатной
группой. Вариабельная часть представ-
лена азотистым основанием, связанным
с сахарным остовом. В природной ДНК
встречается в основном четыре разно-
видности азотистых оснований: аденин,
тимин, гуанин и цитозин. Такой же на-
бор оснований характерен и для РНК,
за исключением тимина, который заме-
нен на урацил.
В определенных молекулах могут
встречаться химически модифициро-
ванные формы оснований (пример — ме-
тилированная ДНК или более сложные
модификации в транспортной РНК).
Как правило, химическое модифициро-
вание имеет важное биологическое зна-
чение для взаимодействий нуклеиновых
кислот с другими молекулами. Вспом-
ним, что клетки разных типов одного
организма содержат один и тот же набор
молекул ДНК. Разница в природе этих
клеток определяется тем, что в каждом
их типе транскрибируются свои гены.
Считается, что метилирование ДНК, т. е.
добавление метильной группы к опреде-
ленному остатку цитозина в цепи ДНК,
участвует в ингибировании транскрип-
ции в клетках позвоночных. Причем с
каждым типом клеток связана своя схе-
ма метилирования. Нуклеотиды, а также
элонгация полинуклеотидной цепи по-
казаны на рисунке А2.10.
А2.4.2. Структуры высших
порядков
ДНК
ДНК в основном двухцепочечное образо-
вание, и ее вторичная и третичная струк-
туры являются результатом спаривания
оснований. По Уотсону-Крику класси-
ческими парами являются А-Т, G-С, от-
куда возникает правило А + С = Т + G,
которому подчиняется состав ДНК.
Важное стереохимическое свойство, ко-
торое в свое время способствовало от-
ДНК
РНК
3' конец
Рис. Л2.10: а — химические структуры нуклеотидов. Синим показана постоянная часть, состоя-
щая из дезоксирибозы (в ДНК) и рибозы (в РНК) и фосфотной группы, связанной с 5’-углеродом
сахара (углероды молекулы сахара нумеруются по часовой стрелке - Г, 2’ и т.д., начиная справа
от атома кислорода в кольце). Четыре природных азотистых основания представлены черным.
Соединение, состоящее из азотистого основания и сахара, называется нуклеозидом; 6 — элонга-
ция цепи идет через потерю молекулы воды и присоединение кислорода при З'-атоме углерода к
фосфатной группе следующего нуклеотида в цепи. Полинуклеотидная цепь имеет 5' и 3’ концы.
Ген читается в направлении 5’ — 3’, которое также является направлением роста цепи во время
репликации и транскрипции
крытию двойной спирали, заключается
в том, что размер пары А-Т, соединенной
двумя водородными связями, такой же,
как у пары G-С с ее тремя водородными
связями. Поэтому обе пары могут обра-
зовывать одну регулярную двуспираль-
ную структуру (рис. А2.11).
Существуют белки, способные спе-
цифически узнавать нуклеотидную
последовательность ДНК за счет нали-
чия у спирали большой и малой кана-
вок (рис. А2.12).
В зависимости от нуклеотидного
состава и гидратации ДНК может при-
нимать форму различных двуспираль-
ных третичных структур (рис. А2.1.3).
Существуют и искусственные двуспи-
ральные структуры ДНК, получаемые
растяжением В-формы ДНК (S-ДНК)
или скручиванием (Р-ДНК) (см. сек-
цию Д5.3).
РНК
Геном некоторых РНК вирусов пред-
ставлен двухцепочечными молекула-
ми. Однако в случае с РНК правило
Pili’. A2.1 1. Уотсон-Криковские пары осно-
ваний, А-Т и Г-Ц. Обратите внимание на оди-
наковый размер этих структур, делающий воз-
можным формирование регулярной двойной
спирали
Рис. А2.12. Рисунок двойной спирали ДНК,
показывающий большую и малую канавки,
благодаря которым основания (серый цвет)
доступны для взаимодействий с другими мо-
лекулами
А-ДНК
Правая
11 пар оснований
на виток
Шаг = 28,2 А
В-ДНК
Правая
10 пар оснований
на виток
Шаг = 34 А
Z-ДНК
Левая
12 пар оснований
на витоко
Шаг = 43 А
Рис. А2.13. Различные структуры ДНК и соответствующие им дифракционные картины. За-
крученная вправо А-ДНК переходит в В-ДНК при повышении влажности с 75 до 92 %; Z-ДНК
представляет собой закрученную влево спираль, наблюдаемую у поли-GC последовательностей
при высоких концентрациях соли (Fuller et al., 2004)
23S РНК
Домен IV
Вид спереди
Вид сзади
Рис. Л2.14. Вторичная и третичная структуры рРНКиз 50S субъединицы рибосом (Ramakrishnan
and Moore, 2001)
А + Ц = У + Г чаще всего не выдержи-
вается, поскольку ее структура в ос-
новном одноцепочечная. Тем не менее
за счет изгибов цепи полинуклеотиды
могут взаимодействовать друг с другом
в так называемых шпильках, формируя
участки АУ и ГЦ пар, чем обусловле-
но большее разнообразие вторичных и
третичных структур РНК по сравнению
с ДНК.
Для семейства тРНК характерна
вторичная структура клеверного лис-
та и L-образная третичная структура.
Очень часто вторичные и третичные
структуры РНК бывают крайне слож-
ными, как в случае с молекулами рибо-
сомной РНК (рис. А2.14). В результате
предсказывается целый набор вторич-
ных структур. Опыт показывает, что
третичная структура РНК трудна для
изучения из-за своей относительной
нестабильности.
А2.5. Углеводы
Общую формулу семейства молекул
углеводов можно выразить формулой
Сх(Н.,О)х с некоторыми более или менее
значимыми модификациями. Углеводы
живых организмов служат структурны-
ми компонентами, источником энергии и
углерода, сигнальными молекулами, ме-
диаторами межклеточных взаимодейс-
твий и взаимодействий между разными
организмами. Сахариды (от латинского
saccharum, ведущего начало от гречес-
кого «сахарон», что значит «сахар») яв-
ляются основными элементами крайне
сложных молекул углеводов, изучаемых
гликобиологией (термин, принятый в
1988 г. для обозначения области знаний,
которая сочетает традиционную биохи-
мию углеводов с современным понима-
нием роли сложных сахаров в клеточной
и молекулярной биологии).
Биологические углеводороды де-
лятся на моносахариды (единичные са-
хара, пример — глюкоза, от греческого
glycys — «сладкий» и -оза — принятое в
номенклатуре родовое окончание моно-
сахаридов), дисахариды (две ковален-
тно связанные сахарные субъединицы,
пример — сахароза), олигосахариды
(несколько ковалентно связанных саха-
ров) и полисахариды («множество» ко-
валентно связанных сахаров). Отметим,
что черта, разделяющая олиго и полиса-
хариды, довольно размыта.
Целлюлоза, представляющая собой
большой линейный полимер, состоя-
щий из мономеров глюкозы, является
основным структурным компонентом
растений и наиболее распространенным
природным полисахаридом. Гликоген
(от «сладкий» и корня gennaein, «про-
изводить» или «рождать») — сложный
разветвленный полисахарид из глюко-
зы, запасающийся в клетках печени и
мышц в качестве источника энергии. Из
смеси линейных и разветвленных поли-
сахаридов состоят гранулы крахмала
(от старого английского слова stercan,
что значит «затвердевать»), выступаю-
щие в роли основного запасаемого ис-
точника энергии у растений.
Во всех типах клеток и у многих
биологических макромолекул встре-
чается множество олигосахаридных
цепей, называемых гликанами, которые
могут существовать и по отдельности.
Большинство гликанов связаны с экс-
кретируемыми макромолекулами, а
также с белками и липидами, находя-
щимися на внешней стороне клетки,
окруженной особой богатой углевода-
ми оболочкой, называемой гликокалик-
сом (от греческого kalyx — покрытие, а
не от латинского calix — чашка). Глика-
ны участвуют в механизмах узнавания
клетка-клетка, клетка-макромолекула,
и во взаимодействиях между разны-
ми организмами, например между па-
разитом и хозяином. Эти механизмы
интенсивно исследуются в связи с уча-
стием гликанов в заболеваниях — через
иммунный ответ или разрастание опу-
холи, к примеру. Гликаны иммуноген-
ны, поэтому специфически узнаются и
связываются молекулами антител.
Полипептиды и полинуклеоти-
ды — это линейные цепи, каждая из
которых содержит один тип связи
между составляющими их аминокис-
лотами или нуклеотидами. Гликаны,
с другой стороны, являются резуль-
татом огромного числа связей между
сахарами, которые могут создавать
бесконечно сложные разветвленные
полимерные структуры. Между дву-
мя молекулами моносахаридов, обра-
зующими полисахарид, существуют
два типа связей, называемых а и р.
Легко представить себе, насколько
возрастают возможности образования
различных структур с увеличением
числа мономеров. К счастью, встре-
чающиеся в природе гликаны содер-
жат сравнительно небольшое число
разновидностей сахаров, поэтому на-
бор комбинаций структур ограничен.
Если бы это было не так, то структур-
ные исследования гликанов, которые
и без того чрезвычайно затруднены,
были бы почти невозможны.
А2.5.1. Химический состав
и первичная структура углеводов
Химические структуры моносахаридов
впервые описаны в конце XIX в. Е. Фи-
шером, который также обнаружил их
стереоизомерию. К примеру глюкоза,
фруктоза и галактоза (вспомним, что
суффикс -оза характерен для номенк-
латуры углеводородов; например, мо-
лекулы, имеющие пять и шесть атомов
углерода, называются пентозами и
гексозами) являются стереоизомерами.
Они имеют одинаковый химический со-
став (С6Н12О6) и отличаются структур-
ной организацией атомов, которая обу-
словливает их специфические свойства
и характеристики. Вообще, биологиче-
ские системы очень чувствительны к
стереоизомерии (мы уже отмечали, что
все природные аминокислоты нахо-
дятся в D энантиомерной форме), по-
скольку узнавание молекул в активном
центре фермента зависит, например, от
точного расположения атомов.
Моносахариды делятся на два клас-
са — альдозы и кетозы, в зависимости
от того, содержат они альдегидную (-
СН=О) или кетогруппу (>С=О). Далее
они подразделяются на подклассы в со-
ответствии с числом атомов углерода:
альдотриозы (3 углерода), альдотетро-
зы (4), кстотриозы, кетотетрозы и т.д.
Открытые и замкнутые (циклические)
формы глюкозы, маннозы и галактозы
показаны на рисунке А2.15.
Проекционные формулы Фишера
и Хеуорта точно отражают ориентацию
групп, но являются обманчивыми в том
смысле, что на самом деле кольцо не
плоское, а может принимать различные
конформации типа «кресло», как пока-
зано на рисунке.
Наиболее распространенные мо-
носахариды, найденные в гликанах
высших организмов, приведены в i.io-
nine \2.1. Эти молекулы преоблада-
ют у высших животных, несколько
других типов найдены в организмах
низших животных, растениях и мик-
роорганизмах. Химические модифика-
ции гидроксильных, карбоксильных и
аминогрупп еще больше увеличивают
структурное разнообразие и обогаща-
ют биологическую функциональность
полисахаридов.
В растворе большинство молекул са-
харов формируют пяти- или шестичлен-
ные кольцевые структуры, не имеющие
свободных альдегидных или кетогрупп.
Замыкание в кольцо способствует даль-
нейшему образованию центра асиммет-
рии у исходного карбонильного угле-
рода, называемого аномерным, за счет
чего D-глюкоза, например, может суще-
ствовать в двух формах — а и р. Очень
часто гликаны находят связанными с
неуглеводным компонентом, обычно с
белком или липидом. Такие соединения
носят название гликоконьюгатов и оп-
ределяются согласно типу связи между
элементами. Белок или липид, образу-
ющий гликоконьюгат, называют глико-
зилированным.
D - Глюкоза
D - Галактоза
н----он
но----н
н----он
н----он
чон
P-D-Глюкопираноза p-D-Маннопираноза p-D-Галактопираноза
1’нс. ,\2.1.). Ряд 1 — открытая форма трех альдогексоз. Замкнутые формы показаны в различных
проекциях в следующих рядах: ряд 2 — проекция Фишера, ряд 3 — проекция Хеуорта, ряд 4 —
проекция «кресла», ряд 5 — стереопроекция
' иц... । Наиболее распространенные моносахариды
Пентоза: пятиуглеродный сахар ксилоза (Xyl)
Гексозы: нейтральные сахара, включающие глюкозу (Glc), галактозу (Gal) и маннозу (Man)
(см. рис. ,\2,|.->).
Гексозамины: гексоза со свободной либо N-ацетилированной аминогруппой при втором ато-
ме углерода, N-ацетилглюкозамин (GlcNAc), N-ацетилгалактозамин (GalNAc).
Дезоксигексозы: гексоза без гидроксильной группы при шестом атоме углерода, фукоза
(Fuc).
Уроновые кслоты: гексоза с карбоксилированным углеродом 6, глюкуроновая кислота (GlcA),
идуроновая кислота (IdА).
Сиаловые кислоты (Sia): девятиуглеродные кислотные сахара, в основном N-ацетилнейрами-
новая кислота (Neu5Ac, NeuNAc, NeuAC или NANA).
Микрогетерогенность, обусловлен-
ная тем, что один и тот же сайт гли-
козилирования может содержать раз-
личные гликаны, делает исследование
гликозилирования белков особенно
трудным.
Определенные первичные структу-
ры углеводородов помещаются в ком-
пьютерные базы данных. Ветвление
молекул приводит к существенному
усложнению их первичных структур, и
для их анализа было разработано спе-
циальное программное обеспечение.
А2.5.2. Структуры высших
порядков
Целлюлоза представляет собой гомопо-
лимер, образованный из глюкозы, орга-
низованной в структуру, которая была
определена методами электронной мик-
роскопии и дифракции (рис. А2.16).
Гомо- и гетерополисахариды, связан-
ные с белками или липидами, также
образуют конструкции, выполняющие
структурную роль в соединительной
ткани или клеточных стенках.
Структуры белковых гликоконью-
гатов, полученные с высоким разреше-
нием и показывающие природу связи
и трехмерную атомную организацию,
определяются с помощью рентгенов-
ской кристаллографии и ЯМР. Более
предпочтительным является исполь-
зование техники ЯМР в растворе, по-
скольку олигосахариды, в отличие от
моно- и дисахаридов, представляющих
собой сравнительно жесткие молекулы,
проявляют некоторую гибкость, что за-
трудняет их кристаллизацию.
Рис. А2.16. Структура целлюлозы и схема водородных связей. Атомы углерода, кислорода, во-
дорода и дейтерия представлены черным, красным, белым и зеленым цветами, соответственно.
Водородные связи показаны пунктирной линией (Nishiyama et al., 2002)
Инфицирование организма бактерия-
ми, паразитами и вирусами часто сопро-
вождается специфическим связыванием
микробных белков с гликанами поверх-
ности клеток хозяина. Так, холерный
токсин — это белок, который связывается
с клеточным олигосахаридом. Структу-
ра связанного с сахаром белка холерного
токсина, определенная с высоким раз-
решением при помощи рентгеновской
кристаллографии, является хорошей ил-
люстрацией взаимодействий между ато-
мами сахара и белка (рис. А2.17).
в клетке. Соответственно, биохимия
липидов достаточно богата: липидные
молекулы служат источниками энер-
гии, участвуют в сложных процессах,
таких как формирование кровяных
сгустков, тромбов, без них невозмож-
но нормальное функционирование не-
рвной системы. Что касается структуры
и динамики макромолекул, то здесь нас
интересует прежде всего структурная
роль липидов в биологических мембра-
нах, а также их взаимодействие с мемб-
раносвязанными белками.
А2.6. Липиды
Если давать липидам широкое опреде-
ление, то это класс молекул, чьи свой-
ства обусловлены водонерастворимым
компонентом. Они составляют весь-
ма разнообразную группу структур,
выполняющих множество функций
А2.6.1. Химический состав липидов
В упрощенном представлении основные
липидные компоненты биологических
мембран состоят из водорастворимой
заряженной или полярной головки и во-
доиерастворимых неполярных углево-
дородных цепей. Обобщенные структу-
ры фосфоглицерида или фосфолипида
а б
Рис. А2.17. Сравнение рецептор-связывающих сайтов семейств холерного СТ и тифозного SHT
токсина. Связанные с белком молекулы сахара изображены в виде представления шарик-па-
лочка: а — СТ пентамер показывает 5 связывающих участков в ганглиозидных олигосахаридах;
б — SHT пентамер показывает 15 связывающих участков в ганглиозидах (три на мономер, обоз-
наченными I, II и III (Fan et al., 2000)
a б в
Рис. A2.IS. Фосфатидилэтаноламин (а),
фосфатидилхолин (б) и кардиолипин (в) (см.
текст)
даны на рисунке А2.18. Цепи образо-
ваны жирными кислотами (окрашено
черным), которые могут быть насыщен-
ными (содержащими только единичные
связи углерод-углерод) или ненасы-
щенными (содержащими одну двойную
связь или более). Они находятся в
сложной эфирной связи с группой гли-
церина (окрашено красным). Головные
группы показаны синим.
В биологических мембранах при-
сутствует большое количество различ-
ных липидов. К примеру, в дополнение
к фосфатидилэтаноламину и фосфати-
дилхолину мембраны организмов вы-
сших животных содержат значительное
количество сфинголипидов, а также хо-
лестерин (рис. А2.19). Принадлежащий
Рис. А2.19. Холестерин
к стероидам холестерол встраивается
между цепями жирных кислот, изменяя
текучесть мембранной структуры. Не-
полярные цепи липидов не обязательно
должны быть производными жирных
кислот. Например, углеводородные цепи
в мембранах архей — это производные
изопрена, которые похожи на фитольные
цепи хлорофилла растений, содержащие
чередующиеся двойные и одинарные свя-
зи углерод-углерод, а также метильные
группы по всей длине цепи. Другое хими-
ческое отличие липидов мембран архей
от липидов мембран эукариот и бактерий
заключается в том, что их цепи соедине-
ны с глицерольной группой эфирной, а не
сложноэфирной связью.
Интересно, что некоторые археи
содержат «двойные» молекулы липи-
дов, с двумя головками с обеих сторон
углеводородных цепей. Эти глицерол-
диацил- глицерол тетраэфиры впер-
вые были обнаружены в гипертермо-
фильных организмах, в которых, как
полагают, они способствуют повыше-
нию стабильности мембран при очень
высоких температурах. Совсем недав-
но подобные структуры были обнару-
жены в археях, живущих в довольно
прохладных условиях — ниже 20 °C
(см. Главу А1).
А2.6.2. Структуры высших порядков
Молекулы липидов в водных раство-
рах склонны к спонтанному объедине-
нию для изоляции своих неполярных
гидрофобных компонентов от термо-
динамически невыгодных контактов с
молекулами воды. Как следствие этого
существует целый ряд структурных фаз
в смеси липид-вода, которые зависят
от липидного состава, соотношения
компонентов, температуры и давления
(рис. А2.20).
Жидкая изотропная упаковка - FI
Рис. Л2.2О. Структурные фазы схематически показаны слева, а справа — диаграммы фаз сме-
сей моноолсин-вода. Моноолеин это липид, имеющий одну углеводородную цепь, который не
встречается в природных мембранах, но широко используется при кристаллизации мембранных
белков. Жидкая ламеллярная двуслойная фаза (L) соответствует состоянию большинства при-
родных мембран (Caffrey, 2000)
В биологических мембранах наибо-
лее вероятны ламеллярные двуслой-
ные образования в жидкой фазе (La) с
неупорядоченными углеводородными
цепями, играющие роль эффективного,
имеющего плоскую структуру, прони-
цаемого барьера, который, тем не менее,
позволяет мембраносвязанным белкам
сохранять функциональную гибкость
и способность диффундировать в мем-
бранной плоскости. В естественных
мембранах образованию La фазы спо-
собствуют: наличие в смеси липидов
ненасыщенных углеводородных цепей,
воды, а также высокая температура.
Интересно, что, когда организм подвер-
гается воздействию низкой температу-
ры, структура его липидных мембран
адаптируется к ней для поддержания
жидкой двуслойной структуры. У ги-
пертермофильных архей «двуслойную»
структуру образует обнаруженный в
этих организмах монослой липида с
двумя полярными головками. Предпо-
лагается (но не доказано), что для не-
которых природных мембран возможно
существование кубической фазы (см.
рис. .\2.20).
А2.6.3. Липиды и мембранные
белки
Мембранные белки тесно связаны
с липидами. Они гидрофобии, что за-
трудняет их исследование обычными
биохимическими методами. По этой
причине исследования мембранных
белков отстают от исследований во-
дорастворимых белков. Развитие ме-
тодов. в которых применяются детер-
генты. позволило выделить активные
мембранные белки и даже закристал-
лизовать некоторые из них. Получение
первой структуры .мембранного белка
с высоким разрешением при помощи
рентгеноструктурного анализа крис-
таллов, содержащих детергент, отме-
чено Нобелевской премией. В 1988 г.
ее получили М. Хармут (совместно с
11. Дайзенхофером и Р. Хубером) за оп-
ределение трехмерной структуры фо-
тосинтетического реакционного цен-
тра. Однако и по сей день получение
кристаллов мембранных белков пред-
ставляет собой сложнейшую задачу.
Диаграмма фаз, изображенных на ри-
сунке А2.20 была изучена в контексте
биофизики мембранных белков. Было
обнаружено, что моноолеин в кубичес-
кой фазе способствует кристаллизации
мембранных белков, необходимой для
получения структур с высоким разре-
шением.
А2.7. Перечень ключевых идей
• Все живые организмы согласно анализу их геномных последовательностей де-
лятся на три Домена (Царства): Эубактерии, Археи и Эукариоты.
* Поток генетической информации в организмах идет от ДНК через РНК к бел-
ку; эта схема является результатом длительной эволюции, поскольку на каждом
ее этапе — от репликации ДНК, транскрипции РНК и обратной транскрипции
до трансляции — требуется участие конечного продукта-белка, выполняющего
сложно регулируемые каталитические функции.
• Несмотря на то что биологические макромолекулы представляют собой по-
следовательность структурных единиц, в процессе эволюции они «научились»
выполнять специфические функции и приобрели свойства, отличные от тех,
которыми обладают классические полимеры.
э Белки состоят из особым образом свернутых полипептидов, которые построе-
ны из аминокислотных остатков, и могут включать простетические группы,
имеющие специфические свойства (как, например, гем, связывающий кисло-
род).
* Цвет белков (например, красный цвет гемоглобина или зеленый хлорофилл-
связывающих белков) обусловлен простетической группой, поскольку амино-
кислоты поглощают в УФ-диапазоне.
В природных бедках встречается двадцать разных аминокислот с различными
химическими характеристиками. Аминокислоты могут быть кислыми и основ-
ными, полярными и неполярными, алифатическими и ароматическими.
° Первичная структура — это последовательность аминокислот в макромоле-
куле; вторичная структура представляет собой упорядоченную локальную
конформацию цепи, возникающую вследствие химических и стерических огра-
ничений; третичная структура является трехмерной организацией макромо-
лекулярной цепи, заданная координатами ее атомов; четвертичную структуру
образуют несколько одинаковых или разных цепей, организованных в макро-
молекулярный комплекс.
- Вторичные структуры белков могут быть представлены па карте Рамачандра-
на в терминах углов вращения пептидной плоскости в цепи вокруг так называ-
емого альфа-углерода, с которым связана боковая группа аминокислот.
° Основными вторичными структурами, найденными в белках, являются аль-
фа-спирали и бета-листы.
3 Третичная структура возникает в результате слабых, нековалентных (за ис-
ключением дислульфидной связи между двумя цистеинами) взаимодействий
между аминокислотами в полипептидной цепи.
с Белковые домены делят на группы или семейства но уровням, в соответствии
с их архитектурой, топологией, гомологией и аминокислотной последователь-
ностью.
13 Организация белков в четвертичную структуру — результат эволюционного
отбора вследствие функциональных преимуществ в отношении стабильности
аллостерического контроля и координации сложных реакционных механизмов
большими молекулярными клеточными комплексами, такими как рибосомы,
протеосомы, термосомы и шапероны.
° ДНК является хранилищем генетической информации, ее структура преиму-
щественно двухцепочечная и имеет спиральную организацию, конформация
которой зависит от окружения.
* РНК выполняет множество биологических функций и образует целый ряд
структур.
° Молекула РНК чаще всего одноцепочечная структура, тем не менее, она может
формировать сложные вторичные структуры — за счет поворотов и внутрице-
почечных взаимодействий между основаниями.
• Рибозимы — это молекулы РНК, обладающие каталитической активностью,
чье открытие в какой-то степени подтверждает гипотезу о существовании
РНКового мира — примитивных форм жизни, использующих РНК для хране-
ния генетической информации и катализа биологических реакций.
• ДНК представляет собой полинуклеотидную цепь, состоящую в основном из
четырех нуклеотидов: аденина, тимина, гуанина и цитозина; в РНК тимин за-
менен урацилом.
• Основания могут иметь химические модификации, связанные с биологической
активностью, такие как метилирование в ДНК или другие более сложные мо-
дификации, найденные в тРНК.
• Углеводы живых организмов служат структурными компонентами, источни-
ком энергии и углерода, сигнальными молекулами и медиаторами межклеточ-
ных взаимодействий, а также взаимодействий между разными организмами.
• Полисахариды могут представлять собой линейные или более сложные развет-
вленные цепи одинаковых или разных ковалентно связанных моносахаридных
субъединиц.
° Все типы клеток и многие биологические макромолекулы образуют гликоко-
ньюгаты через гликозилирование — связывание сложных олигосахаридных це-
пей, называемых гликанами.
* Гликаны иммуногенны, узнаются и специфически связываются молекулами
антител.
• Моносахариды могут существовать в различных структурных формах, называ-
емых стереоизомерами.
* Инфицирование организма паразитами, бактериями и вирусами сопровожда-
ется специфическим связыванием между микробными белками и гликанами
поверхности клеток хозяина.
0 Липиды — основные составляющие биологических мембран, обеспечивающие
их пассивную проницаемость.
° Основные липидные компоненты биологических мембран можно схематиче-
ски представить состоящими из водорастворимой заряженной или полярной
головки и нерастворимой неполярной углеводородной цепи.
3 Молекулы липидов в водном растворе склонны спонтанно объединяться для
того, чтобы изолировать свои неполярные гидрофобные группы от термодина-
мически невыгодных контактов с молекулами воды.
• Воднолипидные смеси, в зависимости от липидного состава, соотношения воды
с молекулами липидов, а также от температуры и давления, могут демонстри-
ровать ряд специфических структурных фаз.
• Для естественных биологических мембран особенно характерна так называе-
мая Ьафаза, в которой углеводородные цепи липидов находятся в текучем, жид-
кокристаллическом состоянии. Так называемые кубические фазы, полученные
в искусственных условиях, оказались чрезвычайно полезными для кристалли-
зации мембранных белков.
Литература для дальнейшего чтения
Нуклеиновые кислоты
Woese С. (1967). The Genetic Code. New York, Harper and Row.
Crick F.H. C. (1968). The origin of the genetic code, J. Mol. Biol., 38, 367-379.
Orgel L. E. (1968). Evolution of the genetic apparatus, J. Mol. Biol., 38, 381-393.
Gilbert IE. (1986). The RNA World, Nature, 319, 618.
Hirao I. and Ellington A. D. (1995). Re-creating the RNA World, Curr. Biol., 5, 1017-
1022.
Dykxhoom D. M. and Novina C. D. (2003). Killing the messenger: short RNAs that
silence gene expression. Nat. Rev. Mol. Cell Biol., 4,457-467.
Белки
Dickerson R. and Geiss I. (1969). The Structure and Action of Proteins. Menlo Park:
Benjamin Cummings.
Branden C. and ToozeJ. (1999). Introduction to Protein Structure. New York: Garland
Science.
Углеводы
Varki A., Cummings R. et al. (eds) (1999). Essentials of Glycobiology. New York:
CSHL Press.
Langan P, Nishiyama Y. and Chanzy H. (1999). A revised structure and hydrogen
bonding system in cellulose II from a neutron fibre diffraction analysis./. Am. Chem Soc.,
121, 9940-9946.
Merrit E. A., Sarfaty S. et al. (1994). Crystal structure fo cholera toxin B-pentamer
bound to receptor GM1 pentasaccharide. Protein Sci. 3, 166-175.
Липиды
Caffrey M. (2000). A lipid’s eye view of membrane protein crystallization in meso-
phases. Curr. Opin. Struct. Biol., 10, 486-497.
Schouten S., Hopmans E. C., Pancost R. D. and DamsteJ.S. S. (2000). Widespread oc-
currence of structurally diverse tetraether membrane lipids: Evidence for the ubiquitous
presence of low-temperature relatives of hyperthermophiles. Proc. Natl. Acad. Sci. USA,
97, 14421-14426.
Глава АЗ
ПОНИМАНИЕ МАКРОМОЛЕКУЛЯРНОЙ
СТРУКТУРЫ
А3.1. Исторический обзор
Ричард Фейнман, один из выдающих-
ся физиков прошлого столетия, считал
Пифагора (с 500 до н. э.) первоот-
крывателем численных соотношений в
природе. Открытие великого древнего
мыслителя, которое привело его к осно-
ванию философско-религиозной шко-
лы с мистической верой в силу чисел,
заключалось в том, что одновременное
звучание двух одинаковых струн раз-
личной длины приятнее для слуха, если
длины этих струн относятся друг к дру-
гу как небольшие целые числа. Сейчас
мы говорим, что отношение 1:2 соот-
ветствует октаве, 2:3 — квинте и т. д. Все
эти интервалы считаются гармонически
звучащими аккордами.
Согласно Фейнману это открытие
имеет три составляющие: основано на
экспериментальном наблюдении, мате-
матика используется как метод для по-
нимания природы явления, и оно име-
ет эстетическое начало («приятность»
звука). Фейнман писал, что если бы
Пифагор был более всего впечатлен си-
лой экспериментального наблюдения,
физика как наука началась бы гораздо
раньше.
С помощью эксперимента мы узна-
ем о существовании макромолекул и
пытаемся понять их с биофизической
точки зрения, при этом мы используем
математический аппарат не только для
подготовки эксперимента и анализа его
результатов, по и для описания самого
объекта исследования. Вполне веро-
ятно, что основы эстетики до сих пор
остаются такими же загадочными, как
и во времена Пифагора. Несомненно,
однако, что «красота» двойной спирали
ДНК и то, каким элегантным образом
она дала объяснение хранению и переда-
че генетической информация, послужи-
ли основным источником вдохновения
в развитии современной молекулярной
биологии. Точно также эстетичны и
красочные иллюстрации структурных
моделей белков. Конечно, они играют
важную роль в понимании и принятии
этих моделей биологами, которых мог-
ла бы оттолкнуть чисто математическая
интерпретация тех же данных, пусть
и более точная.
Эксперименты с биологическими
макромолекулами трудны как для по-
становки, так и для осмысления их ре-
зультатов. Материал образца хрупок,
нестабилен и поэтому хороший био-
физический опыт должен покоиться на
прочном биохимическом фундаменте.
Биологические макромолекулы гораздо
меньше длины волны света и их нельзя
«увидеть». Мы получаем информацию
об их структуре и динамике, облучая
образец электромагнитным или другим
излучением и анализируя то, что полу-
чается на выходе. Образец поглощает и
рассеивает излучение. Разность энергий
падающего и выходящего излучения
анализируется с помощью спектроско-
пии, тогда как в дифракционных экс-
периментах, где нет разности энергий,
анализируется зависимость интенсив-
ности рассеянного излучения от угла
рассеяния. Интерпретация таких экс-
периментов целиком и полностью осно-
вана на физике и математике волн и их
взаимодействий, будь то классическое
описание в терминах интерференции
рассеянных волн, применяемое в крис-
таллографии, или квантово-химическое
описание, необходимое для электромаг-
нитной спектроскопии. Работа Жозефа
Фурье (Joseph Fourier) (1768-1830)
по волновому описанию процессов теп-
лопередачи привела к появлению за-
мечательного набора математических
инструментов для описания взаимо-
действий излучения с вещес твом.
Один из плодотворных подходов
к исследованию структуры макромоле-
кул заключается в поиске сил стабили-
зирующих ее структуру, т. е. таких, кото-
рые поддерживают атомы в положениях,
соответствующих правильно свернутой
активной конформации молекулы. Если
мы добьемся этого понимания, то смо-
жем узнать, за счет чего атомы в ее струк-
туре двигаются, сохраняя свои средние
положения. Выражаясь языком физики,
наша цель состоит в достижении доста-
точно ясного представления о том, что
собой представляет силовое поле, или
какова функция потенциальной энергии
вокруг каждого атома в структуре моле-
кулы. Это позволило бы моделировать
ее молекулярную динамику.
В 1975 г. Дж. А Мак Кэммон,
В.Р.Гелин и М.Карплюс(5. A. McCam-
mon, В. R. Gelin и М. Karplus) впер-
вые опубликовали свои результаты мо-
делирования молекулярной динамики
(МД) движения атомов биологичес-
кой макромолекулы, небольшого бел-
ка, трипсинового ингибитора из под-
желудочной железы теленка (BPTI),
структура которого к тому времени
была определена с высокой точнос-
тью с помощью рентгеноструктурного
анализа. Несмотря на недостаточную
вычислительную мощность тех лет
(моделирование было ограничено пе-
риодом времени менее 10 пикосекунд)
эти исследования вместе с первыми эк-
спериментами К. Линдерстрёма-Лан-
га (К. Linderstrom-Lang) (1955) и его
сотрудников по водородному обмену
подтвердили представление о белках
как динамичных структурах, чьи внут-
римолекулярные движения играют
важную роль в их биологической ак-
тивности.
В последующие десятилетия после
публикации модели трипсинового ин-
гибитора аналогичные расчеты были
сделаны для более крупных белков и
для более длительных периодов, по мере
того как становились доступны новые
структуры с высоким разрешением и все
более мощные компьютеры. Параллель-
но развивались экспериментальные под-
ходы, предоставляющие информацию о
макромолекулярных энергиях и внут-
римолекулярных движениях, необхо-
димых для обоснования таких моделей.
Сюда можно отнести микрокалоримет-
рию (Часть В), анализ температурных
факторов в кристаллографии (Часть Е),
ЯМР (Часть К), неупругое нейтронное
рассеяние (Часть И), инфракрасную
спектроскопию с фурье-преобразовани-
ем (Часть 3) и различные методы изме-
рения быстрой кинетики с использова-
нием лазерных импульсов для запуска
реакций. Были установлены синерге-
тические отношения между расчетами
молекулярной динамики и ЯМР и рен-
тгеновской кристаллографией, с исполь-
зованием минимизации энергии при
уточнении макромолекулярной струк-
туры. Динамические переходы в белках
были открыты благодаря Мессбауэровс-
кой спектроскопии (Ф. Парак (F. Parak)
с сотр.) и неупругому нейтронному рас-
сеянию (В. Достер (W. Doster) с сотр.).
X. Фраунфельдер (Н. Frauenfelder)
опубликовал модель конформационно-
го подсостояния (КП) белков, послу-
жившей физической основой для пони-
мания макромолекулярной структуры и
динамики.
Начиная с 2000 г. моделирование
молекулярной динамики применяется
на надмолекулярном и даже клеточном
уровне. Экспериментальные методы
постоянно совершенствуются и возни-
кают новые, такие как кристаллогра-
фия в реальном времени и кинетичес-
кая криокристаллография, в которой
промежуточные структуры каталити-
ческого ферментного цикла заморажи-
ваются и исследуются по отдельности.
АЗ.2. Основные физические
и математические
инструменты
Практически все экспериментальные
методы анализа биологических мак-
ромолекул (за исключением калори-
метрии и методов классической фи-
зической химии растворов, таких как
определение осмотического давления,
вязкости и т. д.) полностью или частич-
но основаны на наблюдении их взаимо-
действия с излучением. Это наиболее
очевидно для тех из них, которые ос-
нованы на дифракции и спектроскопии
Рис. А3.1. Образец представляет со-
бой «черный ящик», в который попада-
ет излучение с известной длиной волны:
а — дифракционный эксперимент: интен-
сивность рассеянного излучения измеряется
как функция угла; б — спектроскопия: ана-
лизируется энергия выходящего излучения
(кристаллография и ЯМР), но также
справедливо и для гидродинамических
методов, таких как аналитическое уль-
трацентрифугирование или динамиче-
ское рассеяние света, где присутствие
макромолекул в растворе детектирует-
ся благодаря их поглощению или рас-
сеянию света. В большинстве случаев
образец представляет собой «черный
ящик», через который мы пропускаем
излучение с известными свойствами
и анализируем то, что получается на
выходе (рис. А3.1). Для исследования
взаимодействий между излучением и
веществом нам потребуется некоторое
знание математических средств описа-
ния волн, а также квантовой механики,
где рассматриваются корпускулярные
свойства излучения и волновые свой-
ства движущихся частиц.
АЗ.2.1. Волны
Синусы и косинусы
Свойство периодичности волн проще
всего описать с помощью функций сину-
са или косинуса (рис. АЗ.2). Кроме того,
волна может быть охарактеризована еще
несколькими параметрами. Ее амплиту-
да А является мерой максимального от-
клонения функции от среднего значения;
интенсивность волны равна квадрату ее
амплитуды. Длина волны X представляет
собой расстояние, на котором соверша-
ется одно полное колебание; ее частота
f равна числу полных колебаний, совер-
шаемых в единицу времени. Таким обра-
зом, скорость распространения волны и
(расстояние, покрываемое точкой цикла
за единицу времени) равна ее частоте,
помноженной на длину волны
u=fk (А3.1)
Значения функций sin X и cos X из-
меняются с периодом, равным 2л (см.
рис. АЗ.2).
Рассмотрим сначала аргумент Xдля
случая стоячей или распространяющей-
ся волны, наблюдаемой в определенный
момент времени. Выражая X в терми-
нах длины волны как kx = (2л/Х)х, где
х — расстояние, можно описать волну
с помощью выражения
Y=coskx
или (А3.2)
Y - sinfcc,
где для простоты амплитуда принята за
единицу.
Волновой вектор к, с абсолютным
значением k = (2л/Х), указывает на-
правление распространения волны.
Теперь рассмотрим случай, ког-
да параметр X описывает время. Здесь
волновые колебания уже будут иметь
временную зависимость и совершать-
ся в определенной точке пространства.
Учитывая, что период колебаний равен
2л, мы можем записать Х= a>t = 2nft, где
Рис. АЗ.2. Волны, описываемые следующими функциями: а — K=XsinX; б — K=XcosX; в —
Y = (A/j2) (cosX-sinX)^- y = (4/2)(cosX-V3sinX);d- Y = (Д/2)(V3cosX-sinX)
t — время. И опять, принимая амплиту-
ду равной единице, опишем волну как:
Y = cos cot
(АЗ.З)
или
Y = sin соЛ
Величина со = 2л/ называется угло-
вой частотой.
Разность фаз 5 показывает на-
сколько две волны с одной частотой
и длиной не совпадают друг с другом
(см. рис. АЗ.З). Например, если одна из
них описывается функцией coscot, тог-
да другая — функцией cos(cot + 5). Вол-
ны с разными фазами представляются
функциями cos(cot + стД cos(cot + ст,),
cos(cot + ст3) и т. д. Стоит заметить, что
функции синуса и косинуса соотносят-
ся друг с другом через разность фаз 5 =
л/2 (см. рис. АЗ.2 а, б).
Вспомним, что в тригонометрии
cos(cot + 3,) =
(А3.4)
= coso( coscot - sin5, sincot.
Поскольку cos 5j и sin 5, константы,
то мы можем записать:
cos(cot + 5.) =
, А (А3.5)
= а, coscot + »! sincot.
Другими словами, волна с данной
частотой и фазой может быть представ-
лена суммой косинуса и синуса, помно-
женных на соответствующие коэффи-
циенты а(и bv
Волна, описываемая функцией
(cosX — sinX) отстает по фазе на ве-
личину 5 = л/4 от волны, описывае-
мой функцией cosX (см. рис. АЗ.2 б,
в). Это можно также рассчитать из
уравнения АЗ.4: sin5 = cos5 =1/V2
для 5 = л/4 (45°). Точно также, вол-
ны, различающиеся по фазе от cosX на
л/3 и л/6, описываются функциями
(cosX-V3sinX и (>/3cosX-sinX) (см.
рис. АЗ.2 г, д).
Таким образом, сумму волн одина-
ковой длины и частоты, но с разными
амплитудами и фазами можно выразить
математически как сумму функций си-
нуса и косинуса с соответствующими
коэффициентами. На рисунке АЗ.З по-
казаны два крайних случая: в результа-
те сложения двух волн с разностью фаз
гт, где п — ноль или любое целое чет-
ное число, возникает конструктивная
интерференция, сложение двух волн с
разностью фаз п’к, где п’ — целое нечет-
ное число, приводит к деструктивной
интерференции.
Интерференция волн различной
частоты порождает более сложную
картину. Рассмотрим две волны с оди-
наковыми амплитудами и разными уг-
ловыми частотами СО] и со,. Применяя
математическое правило косинусов, их
сумму можно представить следующим
образом:
cosco,t + cosco,t =
‘ (А3.6)
= 2 cos/(со, + co,)tx
XCOsX(Cl), -CO2)t.
Сумма воли с разными частотами
и амплитудами А и А2 описывается вы-
ражением
A, cosco,t + A., cos со, t =
(АЗ 7)
= COSTCO, + со2 )t[A, cosX х
х(со1 — co2)t + А2cos/,(cot -co2)t].
Сумма таких волн, но с одинаковы-
ми амплитудами, показана на рисун-
ке АЗ.4. В очень хорошем приближе-
нии можно считать среднюю частоту
результирующей волны, равной /2(соt +
+ со2), а амплитуду, осциллирующую с
более низкой частотой, равной ,/2(со1 -
- со2). Говорят, что итоговая волна мо-
дулируется колебаниями с более низ-
кой частотой.
Феномен биений возникает, когда
значения со( и со2 достаточно близки
6
Рис. АЗ.З: а — конструктивная интерференция двух волн, находящихся в фазе (или с разностью
фаз 2л, 4л, 6л и т. д.)
At cosX + A2cos(X + n7t) = (A1 + A2)cosX,
где n равно нулю или целому четному числу; б — деструктивная интерференция двух волн, име-
ющих разные фазы (разность фаз п’п, где п’ — нечетное целое число)
Л, cosX + A, cos(X + n'it) = (Al- А.2 )cosX
i
г
Рис. АЗ/i. Две волны (а) и (б) с небольшой
разностью частот и их сумма (в). Измене-
ние интенсивности (квадрата амплитуды)
результирующей волны на относительной
шкале (г)
(—со), например, когда мы слышим зву-
чание двух струн с немного разным на-
тяжением во время настройки гитары.
Частота звучания ноты равна -со, но
ее интенсивность будет пульсировать
с гораздо меньшей частотой (со, - со2),
которая в два раза больше частоты
осцилляции амплитуды, поскольку
интенсивность равна квадрату ампли-
туды (см. рис. А3.1). Феномен биений
можно наблюдать в экспериментах с
динамическим рассеянием света, где
волны света с несколько различными
частотами интерферируют после рас-
сеяния молекулами, движущимися с
разными скоростями (эффект Допле-
ра, см. параграф П 0.4.4).
Скорость волны, описанная в урав-
нении А3.1, соответствует скорости рас-
пространения точки постоянной фазы
(например, гребня волны) и называется
фазовой скоростью волны. Используя
определения волнового вектора и уг-
ловой частоты, мы можем переписать
уравнение А3.1 как:
u = a/k, (А3.8)
где и фазовая скорость.
Когда мы наблюдаем суперпози-
цию волн с различными значениями со
и k, бегущих в одном направлении, как
на рисунке АЗ. 1, то групповая скорость
w результирующей волны равна ско-
рости распространения модуляции.
Можно показать, что:
_ dco
•' dF
Соотношение между значениями со
и k называют дисперсионным соотноше-
нием. Понятно, что фазовые и группо-
вые скорости равны в случае волн, у ко-
торых со пропорционально k.
Для того, чтобы рассчитать группо-
вую скорость результирующей волны
на рисунке АЗ.!, мы прежде всего долж-
ны учесть в уравнении А3.7 колебание
в продольном направлении .г:
A, cos(co/ - k, х) + А2 cos х
x(co2£-&,x) = cosX х
х [(со, +со2 )£-(&, + &>)х] х
x{A,cosX[(®, -co2)t-
- (k} - k2 )x] + A2 cosX x
x [(co,-co2)£-(£,-&2)x]}.
Члены выражения, обозначающие
время и расстояние, имеют разные зна-
ки потому, что гребень волны, находя-
щийся в определенной точке, в преды-
дущий временной период был позади
нее на расстоянии одной длины волны.
(А3.10)
Модуляция описывается функцией ко-
синуса в фигурных скобках в правой
части уравнения АЗ. 10, а ее скорость
равна иМ1>;1 = (со, — со2)/(Хг, -k2). Если час-
тоты и волновые векторы двух волн от-
личаются незначительно, тогда им рав-
на групповой скорости, описываемой
уравнением А3.9.
Комплексные экспоненты
Комплексные числа были изобретены
для решения некоторых уравнений, та-
ких как, например х' = -1, но благодаря
своим уникальным свойствам они стали
мощным математическим инструмен-
том, позволяющим решать множество
физических задач.
Комплексное число а имеет вид:
x + iy,
где i равно квадратному корню из -1;
х называют «действительной» частью
комплексного числа, а у — «мнимой».
Конечно, с математической точки зре-
ния ничего мнимого в этой части нет!
Суммы и произведения комплексных
чисел сами являются комплексными:
(х + п/)+(х '+iy') =
= (x + x') + i(y + y'),
(x + iy)(x'+iy') =
= (хх- уу') + i(xy'+ х'у).
а* = x-iy (A3.ll)
а а* = х? + у2.
Число а* называют комплексно со-
пряженным с а числом.
Комплексное число а можно предста-
вить на плоскости через координаты х и
у как радиальную линию длиной А, на-
зываемую амплитудой, и образующую с
осью х угол ф, называемый фазой. В этом
Рис. Комплексное число а = х + iy, вы-
раженное геометрически на плоскости ху. Оно
может быть записано также как а = Аехргф, где
А называют амплитудой, а ф фазой.
.V -= Acos ф
у = Asin ф
A = V(.v’ +у2).
случае ось х является «действительной»,
а ось у — «мнимой» (рис. А3.5).
Такой график называют диаграм-
мой Аргана. Из рисунка видно, что А =
= т[(х^ + у2), и комплексное число может
быть записано как а = А (созф + zsin ф).
Из математики известно, что комплекс-
ную экспоненту можно выразить как:
е’* = созф +/зтф, (А3.12)
поэтому а = Аехрг ф.
Фейнман называл формулу АЗ. 12
«самой замечательной в математике, на-
шей жемчужиной»! Формула связывает
алгебру (комплексные числа) и геомет-
рию (синусы и косинусы, определяе-
мые отношением сторон треугольника).
Удивительным является то, насколько
ее свойства позволяют упростить со-
ответствующие математические опера-
ции. Основное из этих свойств:
e^,+o;)=e.o,eio2_ (А3.13)
Умножая экспоненту на комплексно
сопряженную с ней, мы получим:
е-ое-й = е0 = 1
Таким образом, комплексное число
может быть записано в виде действи-
тельной и мнимой частей какх- iy, или
Ком moi ; i ?.:н и: ЛЗ.1. Сложение комплексных экспонент
Уравнение АЗ.7 может быть записано простым образом через комплексные экспоненты:
(А)
At ехр/ф, + А., схр/ф, =
1 1
= A, exp i—(ф, + ф, + ф, - ф,) + A, exp z—(ф, + ф, - ф, + ф,) =
= ехр/^ф, +ф,)][А, ехр/|(ф,-ф,) + А,ехр-Д(ф,-ф,) .
Действительная часть выражения (А) соответствует уравнению АЗ.7, полученному ра-
нее с использованием правил косинуса и синуса. Однако вывод последнего намного более
сложен.
в терминах амплитуд и фазовых углов
в комплексной экспоненте как Аехр/ф.
Обратите внимание, что квадрат амп-
литуды комплексной экспоненты по-
лучается не возведением экспоненты в
квадрат, а умножением ее на комплекс-
но сопряженную:
аа* = Аехр/фАехр-/ф = А2. (АЗ. 14)
Поскольку с математической точки
зрения с экспонентами работать проще,
чем с суммами синусов и косинусов,
описание волн через комплексные эк-
споненты обладает большим преиму-
ществом (коммеп lapilli .1 ).
Волна в пространстве с амплитудой
А и волновым вектором с абсолютной
величиной Q записывается как AexpiQx.
Волна, осциллирующая во времени
с амплитудой А и угловой частотой со
записывается как Aexp-icot.
Волну, осциллирующую во време-
ни и пространстве, можно выразить как
Аехр/ (fer-cot).
Опять мы видим, что член выраже-
ния, зависящий от времени, имеет отри-
цательный знак, потому что гребень вол-
ны, находящийся в определенной точке, в
предыдущий период времени отставал от
нее на расстояние одной длины волны.
Две волны с амплитудами А, и А2
и фазами ф1 и ф2 могут быть представле-
ны на диаграмме Аргана как а, = А]ехр/ф|
и а,, = А,ехр/ ф, очень просто рассчитать.
Их сумма Аехр/ф может быть записана
алгебраически или геометрически, если
рассматривать й, и а., как векторы на
плоскости ху (рис. АЗ.б).
Аехр/ф = А, ехр/ф, + А2ехр/ф2. (АЗ.15)
Рис. АЗ.6: а — две волны, представленные
в виде комплексных чисел а, и а2 на диаграмме
Аргана и б — их сумма а, выраженная как век-
торная сумма а, и а2. Обратите внимание, что
а, а, и а2 обозначают комплексные числа, а не
длины отрезков, которые обозначены как А1 и
А2. Фазы ф, и ф, соответствуют векторам а, и а2,
соответственно разность фаз равна 5 = ф, — ф.
Рис. АЗ.7. Распространение плоскополяризованной электромагнитной волны. Электрическое
поле Е осциллирует вдоль оси у. Магнитное поле Н осциллирует вдоль оси х. Волна распростра-
няется вдоль оси z
Приравнивая действительные ча-
сти, ПОЛуЧИМ АсОЗф = А) СО5ф| + А2 созф2,
а приравнял мнимые части Азтф =
= Дзтф, + А,зтф2.
Свойства результирующей волны
выводятся с помощью анализа, описан-
ного в i.uMMrii inpiiii АЗ. I.
Теперь запишем две волны в терми-
нах разности их фаз 5 = ф, — ф, и найдем
амплитуду результирующей волны:
а = а{ +а2 = А, ехргф, +
+А.г ехр/(ф, +8),
аа* = [А, ехргф, + А,ехр/х
х (ф, + 8) | [ A, exp- гф, +
+А..ехр-г(ф. +8) =
1 1 ' (аз.16)
= ехргф, ехр- /фДА, +
+А2 ехр /5] [ А, + А2 ехр- /8] =
= А(2 + А2 + А]А2(ехргЗ +
+ехр-г'8) =
= А2 + А2 + 2 А, А2 cos 8.
Из формулы АЗ. 16 видно, что амп-
литуда результирующей волны равна
7А2 + А2 +2A,A2cos3.
В качестве еще одной иллюстра-
ции преимущества использования ком-
плексных экспонент отметим, что по-
следняя строчка формулы АЗ. 16 — это
просто правило косинусов, применен-
ное к треугольнику со сторонами а , й2
и а (см. рис. АЗ.6).
Поляризация
Электрические и магнитные поля элект-
ромагнитной волны осциллируют вдоль
оси, перпендикулярной направлению
распространения волны (рис. АЗ.7).
Изображенная на рисунке волна на-
зывается линейно- или плоскополяризо-
ванной, поскольку оба поля колеблются
вдоль прямых линий (оси х и у). В общем
случае электрическое или магнитное поле
излучения описывается осциллирующим
вектором, который может иметь любое
направление, при условии, что оно пер-
пендикулярно направлению распростра-
нения излучения. Другими словами, если
волна распространяется вдоль оси z, век-
тор электрического поля Е может иметь
компоненты £.и Еу вдоль осей х и у.
Волна будет линейно- или плоско-
поляризована, когда £., ^осциллируют
в фазе. Примеры различных значений
амплитуды каждого компонента поля
показаны на рисунке АЗ.З.
Когда Е. и Еу осциллируют с раз-
ностью фаз 3, такая волна называется
эллиптически поляризованой. Частным
случаем является круговая поляризация,
когда 3 = +л/2 или —тг/2. Конец вектора
I'i:c. \3.,Y Линейно поляризованная волна: компоненты электрического поля осциллируют
в фазе вдоль осей .v и у с разными амплитудами: а - Е = 1,£ = 0:6 — Е = 1, £.-'/.,: в— £ = 1,£ = 1:
г- Е =0.Е = 1:8-£ = !,£ = -!;?-£ = -!,£ = 1 “
у < у I ул
а
Рис. А РР. Волна, имеющая линейную, эллиптическую и круговую поляризацию. Считается, что
£ и Еи имеют одинаковые амплитуды и осциллируют с фазами ют и ют + 5: а — 3 = 0; б — 5 = л/4;
в — 3 = тг/2; г — 8 = Зтт/4; д — 8 = л; е — 8 = 5л/4; ж — 8 = Зтг/2; з — 8 = 7 л/4. В примерах а и д волна
поляризована линейно; в случаях в и ж — по кругу. Во всех остальных случаях волна поляризо-
вана эллиптически
электрического поля описывает либо
эллипс, либо круг (рис. АЗ.9). Исполь-
зуя функцию косинуса или комплек-
сную экспоненту и принимая значение
амплитуды, равное единице, получим:
Е = созсоГ или 1,
•V
Е = cos(cof + 5) или expz'8.
Конец вектора электрического поля
поворачивается по часовой стрелке,
если фазовый угол 0 < 5 < л и против
часовой стрелки, если л < 5 < 2л. Волна
будет линейно- или плоскополяризо-
ванной, если 5 =0 или л.
АЗ.2.2. Простые гармонические
колебания
Простой гармонический осциллятор
Раскачивающийся маятник или масса
на пружине, двигающаяся периодиче-
ски вверх и вниз, — это механические
системы, являющиеся хорошими при-
мерами простых гармонических ко-
лебаний. Рассмотрим движение тела
Рис. АЗ. 10. Диаграмма потенциальной энер-
гии простого гармонического колебания (гар-
монический осциллятор)
с массой т, прикрепленное к пружине
(рис. ЛЗ.10).
Тело испытывает простые гармони-
ческие колебания в одном измерении,
описываемые уравнением движения:
F = = (А3.17)
dr
где F — сила, приложенная к грузу,
вызывающая растяжение пружины,
//-отклонение пружины от положе-
ния равновесия и k — константа, ко-
торая зависит от жесткости пружины.
Отрицательный знак указывает, что
F — возвращающая сила. Эта сила про-
порцианальна смещению и действует в
противоположном направлении, стре-
мясь вернуть груз в первоначальное
положение. Уравнение АЗ. 17 являет-
ся выражением закона Гука, утвержда-
ющего, что для эластичного тела растя-
жение (деформация) пропорционально
нагрузке. Решением уравнения (АЗ.17)
будет выражение, описывающее пери-
одическое движение:
у = A cos со „у, (АЗ. 18)
где соm — собственная колебательная
угловая частота движения груза и А —
максимальное отклонение от положе-
ния равновесия — амплитуда движе-
ния. Разумеется, мы можем записать
то же выражение через комплексные
экспоненты.
Чтобы найти соотношение между
со„, и константой k мы должны получить
вторую производную у по времени (ус-
корение) в уравнении АЗ.18 и подста-
вить ее в уравнение движения (АЗ. 17).
Тогда это соотношение будет выглядеть
как:
(0,„ = Jk/m. (АЗ. 19)
Мы произвольно приняли значение
потенциальной энергии £системы, рав-
ной нулю, когда масса находится в по-
ложении равновесия. В то время, когда
пружина сжимается или растягивается,
Е увеличивается на величину, равную
работе, необходимой для перемещения
массы:
dE=-Fdy. (А3.20)
Комбинируя это уравнение с урав-
нением АЗ. 17 и интегрируя, мы получим
следующее отношение для потенциаль-
ной энергии осциллятора как функции
смещения массы:
E=Viky\ (А3.21)
Полученное уравнение имеет вид
параболы (см. рис. АЗ. 10). Потенци-
альная энергия максимальна, когда
пружина сжата или растянута до зна-
чения, равного амплитуде А, и она
уменьшается до нуля в положении
равновесия.
Полная энергия системы равна сум-
ме ее потенциальной и кинетической
энергий. Обратите внимание, что в от-
личие от потенциальной, кинетическая
энергия достигает максимума, когда
масса проходит положение равновесия
и обращается в ноль при у = +А и у =
= -А, где груз останавливается, меняя
направление движения. Полная энергия
осциллятора £ю ]|1 все время колеблется
между потенциальной и кинетической
энергиями и постоянна во всех фазах
движения:
(АЗ.22)
Для описания поведения системы,
состоящей из двух масс т{ и соеди-
ненных пружиной в отсутствии грави-
тации (рис. Л3.1 I ), необходимо вместо
т подставить приведенную массу т{.,:
Рис. АЗ.1 1. Две массы, соединенные пружи-
ной с константой упругости k
Колебательная угловая частота сис-
темы теперь дается выражением:
I k lk(m, +т.,)
®я =-----= J—-------- -(А3.24)
у т12 у т,/т?2
Нормальные моды в одном измерении
Периодические движения масс, соеди-
ненных пружинами, или простые гар-
монические потенциалы, как тот, что
представлен на рисунке ЛЗ. 10, следует
рассматривать как суперпозицию ос-
новных колебаний, называемых нор-
мальными модами.
Анализ нормальных мод линейной
цепи атомов при определенных гранич-
ных условиях показан на рисунке ЛЗ. 12.
Согласно этим условиям, концевые ато-
мы не движутся, подобно концам ги-
тарной струны. Также как и у звучащей
гитарной струны основные моды соот-
ветствуют набору волн в линии атомов,
концы которой неподвижны.
Продольные или поперечные вол-
нообразные смещения атома s даются
выражением:
us = sinsQa, (АЗ.25)
где Q — волновой вектор (мы не обо-
значили его как вектор, поскольку наш
пример ограничен одним измерением;
длина волны смещения атомов равна
А = 2n/Q). Первое граничное условие
(zzs = 0 для 5 = 0) автоматически удов-
летворяется в уравнении АЗ.25. Второе
граничное условие (ws = 0 для s = N) ус-
танавливает предел значениям Q, рав-
ный:
Q = —(А3.26)
* Na Na Na Na ’
Стоит заметить, что при максималь-
ном значении Q (Q = л/а) решением
уравнения АЗ.26 является = 0 для
всех атомов. Таким образом, существу-
ет N - 1 нормальных мод для линии ато-
мов при данных граничных условиях.
Каждая нормальная мода определяется
своим вектором Q и соответствует стоя-
чей волне, описываемой уравнением:
us = zz(0)exp(-zcoQZ)sinsQa, (АЗ.27)
где и (0) — начальная амплитуда, coQ —
угловая частота моды и t — время.
Вспомним, что математическое от-
ношение между coQ и Q называется дис-
персионным соотношением. Для нашего
примера дисперсионное соотношение
выражается формулой:
Na , „ л _ л
со0 =— 4; О — <Q<—,
2 2л а * а (А3.28)
coQ = 0 в любом другом случае.
Нормальные моды в трех измерениях
Уравнение простого гармонического ко-
лебания массы т в одном измерении да-
ется выражением АЗ. 17. Применяя наш
анализ к системе N гармонически свя-
занных масс, движущихся в трех изме-
Зафиксировано
Зафиксировано
s = 0 1 2
s = п
Рис. АЗ. 12. Линейная цепь из ^+l атомов. Граничным условием является неподвижность ато-
мов при $ = 0 и $ = — смещение атома, а — расстояние между атомами
рениях, мы заменим т и k на две 3Nх 3N
матрицы эффективных масс и констант
упругости между всеми парами масс, а
у — на вектор, имеющий 3N координат.
По аналогии с уравнением АЗ.18 реше-
ние для трехмерного случая составля-
ет набор 3N периодических функций.
Однако шесть из них соответствуют
поступательным и вращательным сте-
пеням свободы всего тела целиком,
когда массы не смещаются относитель-
но друг друга, поэтому система будет
иметь 3N - 6 нормальных мод. Можно
показать, что если массы составлены
в линию (как в нашем примере) число
нормальных мод равно 3N - 5.
Для двухатомной молекулы число
3N соответствует шести степеням сво-
боды (рис. АЗ. 13), из которых три соот-
ветствуют поступательным движениям
(вдоль осей .г, у, и z), две - вращатель-
ным модам (вокруг осей у и z), и только
одна — колебательной моде (растяже-
ние связи между атомами).
В гармоническом приближении ко-
лебания атомов в макромолекуле явля-
О-----О
t г
О-----О
х-трансляция
у-трансляция
z-трансляция
у-вращение
z-вращение
О--------о > растяжение
’1
Рис. АЗ. 13. Внутренние степени свободы
двухатомной молекулы. Ось z перпендикуляр-
на плоскости страницы. Поскольку атомы рас-
сматриваются как точечные, вращение вокруг
оси .г отсутствует
ются результатом суперпозиции нор-
мальных мод. Совокупные движения
атомов, рассчитанные с использованием
гармонических потенциалов, для двух
низкочастотных мод небольшого бел-
ка — трипсинового ингибитора подже-
а б
Рис. АЗ. 11. Коллективные смещения атомов для двух низкочастотных нормальных мод, рассчи-
танные для трипсинового ингибитора. Частоты мод равны 3,56 нсек 1 (а) и 0, 21 псск 1 (б) (Go et
al., 1983)
дудочной железы теленка показаны на
рисхпке \3.| i. Обратите внимание на
относительно большие смещения неко-
торых атомов. В каждой моде все атомы
движутся с одинаковой фазой!, т. е. они
достигают максимума и минимума и
проходят через точки равновесия одно-
временно.
АЗ.2.3. Анализ Фурье
Периодические функции, ряды Фурье
и преобразования Фурье
Выше мы продемонстрировали, что
периодическая функция может быть
представлена суммой синусов и коси-
нусов. Па самом деле, согласно теореме,
сформулированной Фурье, любая пе-
риодическая функция может быть пред-
ставлена суммой синусов и косинусов,
известной как ряд Фурье (уравнение
А3.29):
у(/) = «0 + а, cosco/+ /;, sin со/ +
+ а2 cos2co/ + b2 sin2соГ +
+ а3 cos3co/ + b3 sin3co/ + (АЗ.29)
+... = ^(а„ cos«cot + bn sin «со/).
п=()
Синусы и косинусы описывают вол-
ны с возрастающими частотами, крат-
ными целым числам «со с базовой часто-
той со, и амплитудами ап и b . Мы также
можем записать ряд Фурье, используя
комплексные экспоненты:
y(t) = У, A„expz«cor, (АЗ.ЗО)
П = -оо
где А — амплитуда волны с частотой
«со, при этом « может принимать все це-
лочисленные значения — отрицатель-
ные и положительные, поскольку это
необходимо для тождественности меж-
ду уравнениями А3.29 и АЗ.ЗО. В качес-
тве иллюстрации соотношения между
рядами в виде комплексных экспонент
и функций синуса и косинуса рассмот-
рим простую косинусоидальную волну:
y(t) = czcosco/ = У («„ cos «со/ + Л„ sin «со/).
«=()
Очевидно, что в этом случае а=а,
а все остальные значения а и b равны
нулю. Выражение, переписанное в тер-
минах комплексных экспонент:
//(/) = «cos со/= У А,(ехр«/со/,
м--~
имеет два члена при « = ±1, поэтому си-
нусы, составляющие мнимую часть, со-
кращаются:
//(/) = ncosco/ = А_, exp(-zco/) +
+A1exp(+zco/) = 2Acoscoz
где = А, = А, и а = 2А. Другой способ
описания разложения функции у (/) в
ряд заключается в рассмотрении каждо-
го компонента волны, имеющего свою
амплитуду А и соответствующую час-
тоту «со, по отдельности. Мы получим
набор слагаемых, называемых фурье-
компонентами, амиплитуда каждого из
которых называется коэффициентом
Фурье, а функция А/;(«со) — фурье-пре-
образованием у (/).
К примеру, фурье-преобразование
волны, описываемой формулой Acosco^,
содержит компоненты с амплитудой А
при «со = ±сог Волна, полученная в ре-
зультате сложения двух функций ко-
синуса, как в уравнении АЗ.7, имеет
фурье-компоненты с «со = ±со15 ±со,, ам-
плитудами А! и А2 (рис. АЗ.15).
Из рисунка АЗ. 15 и видно, что
результатом фурье-преобразования
постоянной функции y(t) = В явля-
ется одиночная линия в точке начала
координат («со = 0), т. е. постоянный
уровень можно описать как волну
-<о2 -<0, 0 +w,+«>2
I’nc. A.'J. I.>. Волны и соответствующие им фу-
рье-преобразования: а — волна, описываемая
функцией 2zlcos(o/, осциллирующая возле
нуля; 6 - волна, описываемая функцией В +
+ 2Aco.sw|r, осциллирующая в пределах значе-
ний, равных В; в — волна, описываемая фун-
кцией 2A|cos(o|Z +2 A,cosw/, осциллирующая
около нуля
с нулевой частотой или с бесконечной
длиной волны.
Анализ Фурье подобен спектрально-
му анализу, где свет разлагается призмой
WVWH
• 111111111'
пш Q 0 +пш
Рис. Л.Ч. 1 В. Периодическаяфункцияу(^и ее
фурье-преобразование Ап(и<о). Каждая точка
Р на кривой у (t) задана всем набором ЛДлсо)
компонентов, а каждый компонент Q полно-
стью определяется периодической структу-
рой у (t)
или дифракционной решеткой на частот-
ные компоненты, или анализу гармони-
ческого состава музыкального звука. Он
также эквивалентен анализу сложных
колебаний нескольких масс, соединен-
ных пружиной из закона Гука (см. выше)
в терминах нормальных мод.
Ж. Фурье разработал математиче-
ский метод определения значений ко-
эффициентов Апи рассчитал, что:
2 Т
АП = —jy(r)exp-mcozdf, (А3.31)
о
где Т= 2лсо ‘представляет собой период
основного колебания с частотой со, т. е.
периодически повторяющееся значение
переменной t.
Поскольку интегрирование эквива-
лентно суммированию функции с очень
маленькими интервалами, очевидная
симметрия уравнений АЗ.30 и А3.31 до-
статочно примечательна. Она говорит
нам о том, что с одной стороны, каж-
дый фурье-компонент полностью задан
функцией у, а с другой стороны, значе-
ние у в каждой точке t само определя-
ется всем набором фурье-компонентов
(рис. АЗ.16).
Отметим еще одно важное свойство
преобразований Фурье. Если у являет-
ся функцией времени t, тогда ее фурье-
преобразование — функция частоты,
т. е. параметра пропорционального 1 /Г.
Анализ Фурье находит широкое
применение в различных областях фи-
зики. Для волны, распространяющейся
в пространстве, ее фурье-преобразова-
нием будет функция волнового вектора
с абсолютной величиной Q, пропорцио-
нальной l/.v. Уравнение, аналогичное
АЗ.ЗО записывается как:
z/(x) = X A expwQ.r. (А3.32)
п=-«
Вследствие этого взаимного соотно-
шения пространство, выраженное через
значения л* иногда называют действи-
тельным пространством, а пространст-
во величины Q, в котором описывается
фурье-преобразование, называют про-
странством волновых векторов или об-
ратнее пространством.
Рис. АЗ. 17. Дельта-функция Дирака. Она бес-
конечно узка — ее «полуширина» равна 1 /А
(А->°°) и бесконечно высока — высота А—>°°,
а интеграл по всем ее значениям равен 1
Дельта-функция Дирака
Линии, обозначающие коэффициенты
Фурье на рисунке АЗ. 15, имеют опре-
деленную высоту, пропорциональную
их величине. Хотя такое изображение
довольно наглядно и приятно для глаза,
математически оно неверно. На самом
деле, каждая линия соответствует бес-
конечно тонкой кривой, которая, тем не
менее, имеет площадь, равную значению
фурье-коэффициента. Математическая
функция, называемая дельта-функци-
ей Дирака, описывает такую кривую
единичной площади. Дельта-функция
Дирака в позиции .v = .г' обозначается
как З(л-л'). Функция обладает следу-
ющими свойствами:
если .г = л’, тогда 8(.г -,г') = °°;
если л" * х’, тогда 8(.т - л ’) = 0;
площадь, ограниченная кривой, равна
единице, поэтому j8(.т-x')dx = 1.
Поскольку ширина функции крайне
мала, не обязательно определять интеграл
на всем промежутке от до +°°, доста-
точно взять небольшой участок л' вокруг
л'’(рис. АЗ. 17). Можно показать, что:
8(л-л')= JexpiQ(x-x')dQ, (АЗ.ЗЗ)
где Q — абсолютная величина волново-
го вектора, описанная выше. Нетрудно
заметить поразительное сходство между
уравнениями А3.32 и АЗ.ЗЗ. Кажется, что
уравнение АЗ.ЗЗ тоже является рядом
Фурье. Однако на этот раз члены ряда
отделяют не целые значения прираще-
ния п, а очень маленькие интервалы dQ.
Интеграл Фурье и непрерывное
фурье-преобразование
Уравнение АЗ.ЗЗ описывает интеграл
Фурье. Оно показывает, что анализ
Фурье не ограничен функциями перио-
дического характера. Вспомним, что
любая периодическая функция может
быть представлена суммой волн с часто-
тами, значения которых увеличиваются
на целый множитель. Теперь, обобщая
это утверждение, можно сказать, что
любую функцию у (х) можно предста-
вить в виде интеграла Фурье в терми-
нах суммы волн с постоянно уменьша-
ющимися длинами (или с постоянно
увеличивающимися частотами (если х
заменить на г)):
,г/(-г) = Г F(Q~)expiQxdQ,
1 ... (А3.34)
y(t) = — | Т(а>)ехр/(о^(о,
где 1/2тг — множитель, возникший
вследствие замены суммирования ин-
тегрированием.
В соответствующих фурье-преобра-
зованиях вместо коэффициентов Ап те-
перь стоят непрерывные функции F(Q)
(или Т(со)):
f (Q) = f z/(r)exp(-/Qr)a'r,
J (А3.35)
Р (“) = J г/(Г)ехр(-/®Г )dt.
Уравнение АЗ.35 называют обрат-
ным фурье-преобразованием F (Q).
Фурье-преобразования некоторых не-
периодических функций показаны на
рисунке* АЗ. 18.
Колоколообразные функции Гаус-
са и Лоренца очень часто встречаются в
молекулярной биофизике. Статистиче-
ское распределение данных, получен-
ных на большом числе молекул (ансам-
бле), описывается гауссовой кривой.
Почти все явления, имеющие времен-
ную зависимость, могут быть описаны
как экспоненциальное затухание во
времени: динамическое рассеяние света
(Глава ПО) и квазиупругое нейтронное
рассеяние (Глава И2), поступательная
диффузия (Глава ГЗ), деполяризован-
ная флуоресценция (Глава Г8) и элек-
трическое двойное лучепреломление
(Глава Гб).
Фурье-преобразование экспоненци-
альной функции представляет собой кри-
вую Лоренца, поэтому явления, имеющие
временную зависимость, могут быть опи-
саны с помощью Лоренциана в терминах
частот или энергий.
Фурье-преобразование в трех изме-
рениях записывается с использованием
векторов:
f(Q) = J*/(r)exp(-zQ r)dr. (А3.36)
Свертка (конволюция)
В решении различных задач теории
дифракции находит широкое примене-
ние операция свертывания функций.
Сверткой или конволюцией называется
результат распределения одной функ-
ции /,(х) по закону, задаваемому другой
функцией/2(х). Проще всего понять эту
операцию, рассмотрев сначала некото-
рые свойства 8-функции. Интеграл от
произведения какой-либо функции с
8-функцией «вырезает» значение этой
функции при х = 0. Если 8-функция
задана не в точке х = 0, а в точке х = х’,
то такой интеграл «вырезает» значение
функции /(х) при х = х’. Если в таком
интеграле рассматривать х’ как перемен-
ную величину, то он осуществляет «пе-
ревод» /(х) в пространство аргументах’.
Рассмотрим функции, представлен-
ные на рисунке АЗ. 19. Функция С (х) на
рисунке АЗ. 19 « математически связана
с функциями А (х) и В (х), показан-
ными на рисунках АЗ. 19 а и АЗ. 19 б, с
помощью операции свертки. С (х) есть
продукт свертки А (х) и В (х).
Прямое пространство
Обратное пространство
о
a f(x) = К ехр(-х2)
F(Q)= exp(-—l-^Q2)
4лК
1
f(x) = 1 для |х| < а
f(x) = 0 для |х| > а
sinQa
Qa
1
о
f(x)= ехр(-а|х|)
f(x) = 1 для |x| < R
г f(x) = 0 для |х| > R
<D(QR) =
3(sinQR - QRcosQR)
(QR)3
Рис. АЗ. 18. Некоторые непериодические функции и их фурье-преобразования (ФП):
а — ФП гауссиана шириной 1 /К само является гауссианом шириной К;
б — ФП щели шириной 2а есть осциллирующая функция с интенсивным центральным максиму-
мом Q, шириной 2л/а и вторичными быстро затухающими максимумами;
в — ФП экспоненты является колоколообразная кривая, называемая функцией Лоренца;
г — ФП сферы радиуса R представляет собой функцию, обладающую сферической симметрией
и называемую функцией Бесселя, которая имеет интенсивный центральный максимум Q шири-
ной 2n,/R и слабые осцилляции с обеих сторон от него
Математически свертка двух одно-
мерных функций / (х) и g (х) записы-
вается как:
/(x)®g(x) = j/(w)g(x-w)t/w, (А3.37)
где ® — значок свертки. Операция свер-
тки показана на рисунке АЗ.ЗО для двух
простых функций с центрами в точках х
= 0 и х’. На рисунке АЗ.20 « вы видите
один из членов интеграла при х = х’ и и
= и’. Функция g (х — и} имеет конечное
значение, поскольку она была смещена
и теперь ее центр находится в точке и’\
однако мы видим, что в этой точке f (и’)
равна нулю, поэтому данный член интег-
рала тоже будет равен нулю. Понятно,
что он будет отличаться от нуля, только
если обе функции g (х — и} и f (и) от-
личны от нуля. Такая ситуация может
возникнуть лишь в том случае, если зна-
в
Рис. АЗ. 19. Функция (в) является результа-
том свертки функции (а) и набора дельта-функ-
ций (б).
чение х близко к х’, иначе g будет равна
нулю, и если и принимает значения при
которых g перекрывается с/ (и), т. е. и”
близка к нулю (рис. АЗ.20 г). В результа-
те операции свертки мы имеем функцию
в которой каждая точка — это точка фун-
в г
х’ х
Рис. Л3.20. Иллюстрация операции свертки. На рисунке а я б представлены две функции,
чьи продукты свертки необходимо получить. Рисунки в и г демонстрируют члены интеграла
в уравнении АЗ.37 для х = х’, и = и’ и и”. Рисунок д показывает, что операцию свертки можно
рассматривать как умножение каждой точки функции g на функцию/. Рисунок е отображает
результат свертки
Kming, помноженной на/(рис, АЗ.20 с).
Если центр/ (.г) находится не в нуле, а в
.г = д ”, центр свертки разместится в точ-
ке Л-’ + х”. Можно показать, что / (.v)®g
(.v)=g(.r)®/(.v).
Процедура свертки играет очень
важную роль в физических измерени-
ях. Рассмотрим спектроскопический
эксперимент с образцом, поглощающим
в очень узком диапазоне длин волн. Из-
мерительный инструмент всегда сам об-
ладает чувствительностью к определен-
ным длинам волн, которая может быть
шире полосы поглощения исследуе-
мого образца. В результате измерений
мы получим продукт свертки функции
формы полосы поглощения и функции
разрешения инструмента.
Продукт свертки в трех измерениях
записывается в векторной форме:
/(r)®g(r)= j/(u)g(r-u//u.(A3.38)
Фурье-преобразование продукта
свертки приводит к чрезвычайно важ-
ному результату, для простоты пока-
занному в одном измерении:
F7'[/(.v)®g(.v)| =
= F7'[/(-v)]x/T[g(.v)|, (A3’39)
где FT обозначает операцию фурье-пре-
образования. Уравнение А3.39 говорит
о том, что для выполнения фурье-ире-
образования продукта свертки двух
функций мы просто умножаем друг на
друга независимо полученные фурье-
преобразования каждой из них. Ис-
пользуя понятия действительного про-
странства и пространства волновых
векторов, мы можем сказать, что свер-
тка в реальном пространстве приводит
к перемножению в пространстве волно-
вых векторов и наоборот.
Проиллюстрируем результат при-
менения уравнения А3.39 примером из
кристаллографии (рис. А3.21). Кристал-
лическая решетка описывается набором
дельта-функций Дирака. Помещение од-
ной из функций в узловую точку решетки
можно рассматривать как продукт сверт-
ки решетки и функции (левая часть ри-
сунка). В правой части рисунка показаны
соответствующие фурье-преобразования.
Фурье-преобразование i ювторяющейся
Рис. А3.21. Левая панель: свертка функции (красная кривая) и решетки, представленной на-
бором дельта-функций. Правая панель: фурье-преобразование повторяющейся функции равно-
значно фурье-преобразованию красной кривой, умноженной па фурье-преобразование решетки
функции получается умножением фу-
рьс-преобразования функции (широкая
красная колоколообразная кривая) на
фурье-преобразование решетки (набор
дельта-функций, разделенных при nQp
где п — целое число, a Q,; = lit/d, где d по-
казывает повторение решетки в данном
одномерном примере). Конечное преоб-
разование обозначено набором красных
линий (дельта-функций с уменьшаю-
щимися амплитудами) в нижней правой
части рисунка.
АЗ.2.4. Квантовая механика
Квантово-механические понятия необ-
ходимы для описания взаимодействий
.между излучением и материей на атом-
ном уровне. Развитие квантовой меха-
ники в значительной степени подстеги-
валось необходимостью интерпретации
данных спектроскопических наблюде-
ний, в особенности дискретных линий
в атомных спектрах, и она необходима
для описания многих методов, которые
приводятся в настоящем издании. Од-
нако здесь мы дадим лишь обзор ключе-
вых понятий квантовой механики.
Константа Планка, квант энергии
и фотоны
В квантовой механике энергия может
передаваться в течение определенно-
го времени от одной системы к другой
только дискретными порциями, крат-
ными некой минимальной величине.
Эта величина — константа Планка
(// = 6,626* 10 31 Дж-сек) может рассмат-
риваться как неделимый пакет энергии,
умноженный на время. В квантовой ме-
ханике волна с частотой v, осциллиру-
ющая соответственно v раз в секунду,
несет порцию энергии hv, которая на-
зывается квантом. Энергия волны опи-
сывается двумя компонентами. Первый
компонент — энергия кванта, полностью
определяемая ее частотой. Второй же
зависит от числа квантов, передаваемых
в единицу времени, и пропорционален
интенсивности волны.
Квант волны электромагнитного излу-
чения называется фотоном. Энергия
кванта видимого света зависит от его
цвета. Квант красного света обладает
меньшей энергией по сравнению с кван-
том синего цвета, однако более интен-
сивный красный луч все же может нести
больше энергии. Связанная с фотоном
длина волны обратно пропорциональна
ее частоте — X = c/v, где с — скорость
света. Таким образом, энергия кванта
электромагнитного излучения полно-
стью определяется частотой (/zv) или
длиной волны (hc/V).
Корпускулярно-волновой дуализм
При анализе излучения его удобно
представлять в виде потока частиц
(корпускул) или волны. Поскольку все
события с участием излучения включа-
ют временные периоды и расстояния
гораздо меньшие тех, что мы способны
осознать с помощью повседневного
опыта, вопрос «Так из чего же состоит
луч света — волн или частиц?» следует
считать бессмысленным. Достаточно
сказать, что электромагнитное излуче-
ние можно описать двумя способами.
А поскольку оба описания должны быть
самосогласованными, существуют пра-
вила их связывающие. Так, импульс
и энергия, выраженные в терминах
массы и скорости частицы, соотносят-
ся через уравнение, содержащее кон-
станту Планка с волновым вектором
и частотой волны. Эти соотношения
даны в : т > Л :. для трех случаев:
потока нейтронов (частиц с массой тп,
Частицы и волны * (добавить синусоиду)
Частицы ••••••••• Волны
Интенсив- ность Поток (число частиц в единицу времени) Квадрат ампли туды (/V)
Момент Нейтроны: р = mnU I к’йтроны: р = йк = (й / Х)и
т и Электроны р = , — — А1-"/ V /с Электроны: р = йк = (й/X)u
Фотоны = р = йк = (й/Х)и Фотоны: р = hk = (Й/Х)и
Энергия Нейтроны: р, 1 2^2/ 2 Е = -ти ~-р /ти 2 л 2 * п Нейтроны: г- , . Е = hv = ha> = —г,— = 2Гт„ 2т„
Электроны: 1 2 , mu2 Е= Р = г-^-х 2 Д1-«/, V / с~ Электроны: г- , . Е - hv = ha>- —г.— = 2E'mt. 2mt,
Фотоны: Е = hv = Йсо — hc/E Фотоны: E = hv = ЙСО = hc/X
*) Уравнения, описывающие соотношения интенсивности, импульса и энергии с волновой
и корпускулярной точек зрения для нейтронов («классических» частиц), электронов («реляти-
вистских» частиц) и фотонов (частиц с нулевой массой покоя). Уравнение Де Бройля соотносит
длину волны X с импульсом р; и — единичный вектор в направлении распространения волны; к —
волновой вектор (й=2л/Х); h с черточкой h - константа Планка, деленная на 2л; тп и т — массы
нейтрона и электрона, соответственно. Остальные символы сохраняют свои обычные значения
летящих «медленно» со скоростью и),
потока электронов (частиц с массой т ,
движущихся со скоростью, близкой к
световой), и потока фотонов (частиц
с массой покоя, равной нулю и движу-
щихся со скорость света).
Соотношение Де Бройля утвержда-
ет, что длина волны, связанная с дви-
жущейся частицей, обратно пропор-
циональна ее импульсу, с константой
пропорциональности, равной константе
Планка — \=h/p. Таким образом, быст-
рые частицы ассоциируются с короткой
длиной волны и наоборот. Отметим, что
из-за своей большой массы нейтрон об-
ладает уникальными свойствами для ис-
следования материи. Если мы выразим
кинетическую энергию нейтрона через
температуру, нейтроны при комнатной
температуре (так называемые тепло-
вые нейтроны) будут иметь длину вол-
ны между 1 и 2 ангстремами, холодные
нейтроны (около ЮК) будут обладать
длиной волны близкой к 10 ангстремам,
тогда как для горячих нейтронов длина
волны составит доли ангстрема.
Принцип неопределенности
ГЭйзенберга
Для того чтобы определить положе-
ние, импульс или энергию частицы или
волны мы так или иначе на нее воздей-
ствуем. Это вмешательство во время
измерения не может быть сколь угодно
малым, и его минимальное значение ог-
раничено константой Планка, которой
на атомном уровне нельзя пренебречь.
Следствием этого является принцип
неопределенности Гейзенберга. Выра-
женный в двух неравенствах, он соотно-
сит координаты иространство-импульс
и энергия-время:
ДрДг > h,
&ЕЫ > h,
где .г — пространственная координата.
Принцип неопределенности утвержда-
ет, что для того чтобы увеличить точ-
ность определения положения частицы
на атомном уровне (т. е. сделать Аг как
можно меньше) мы должны пожертво-
вать знанием ее импульса и наоборот.
Другими словами, можно надеяться на
то, что мы узнаем либо скорость движе-
ния частицы, либо ее положение, но мы
не можем рассчитывать на получение
всей информации одновременно.
Иллюстрацией принципа неопре-
деленности является волновое описа-
ние материи. Из раздела по анализу
Фурье мы помним, что непериодиче-
ская функция может быть представ-
лена суммой волн с различными
длинами. Таким образом, частицу, ло-
кализованную в пространстве, можно
представить суммой волн. Поскольку
длина этих волн лежит в некотором
диапазоне, мы можем определить ее
импульс лишь с большой степенью не-
определенности. Теперь рассмотрим
частицу, чей импульс известен точно.
В этом случае она становится волной
совершенно определенной длины, на-
пример, одиночной синусоидальной
волной. Но такая волна простирается
в пространстве до бесконечности, по-
этому мы не можем определить где эта
частица находится.
Аналогичные аргументы справедли-
вы и для измерения пары энергия-время.
Принцип неопределенности также
можно проиллюстрировать на приме-
ре условий, определяющих разреше-
ние в кристаллографии (см. Главу ЕЗ и
рис. АЗ.22). Из взаимного соотношения
между структурой кристалла и волнами,
которые он рассеивает (в действитель-
ности представленными ее фурье-пре-
образованием), следует, что наимень-
шее расстояние d , которое можно
разрешить кристаллографически, соот-
носится с максимальным наблюдаемым
значением величины вектора рассеяния
О через выражение:
j 2л
- (А3.41)
*Смакс
где
О = к — к
а*макс макс О’
где к0 и к — волновые векторы пада-
ющего и дифрагированного лучей.
Из ' 2 । видно, что импульс вол-
ны выражается через ее волновой вектор,
поэтому Q можно рассматривать как
величину, пропорциональную разно-
сти импульсов двух лучей — падающего
и дифрагированного. Пространственное
жение атомов в кристалле, если знаем, ка-
ким образом они рассеивают излучение. Луч,
описываемый волной с вектором к0, падает на
кристалл (обозначенный прямоугольником),
и расстояние между атомными плоскостями
измеряется посредством определения интен-
сивности рассеянных волн как функции вол-
нового вектора kr Вектор рассеяния Q нахо-
дится как разность к, и к0
разрешение dvitn можно рассматривать
как минимальное расстояниеЛг, которое
определяется экспериментально:
2Л.£2м.,к."
2л
= • (А3.42)
*4маке
поэтому
Д/;Дт = h.
Подходы Шредингера и Дирака
Следствием принципа неопределенно-
сти является то, что результаты кван-
тово-механических расчетов (напри-
мер, решение уравнений движения
системы атомов) выражаются в виде
вероятностен, а не точных величин.
Поэтому, если различные вероятно-
стные траектории рассчитаны для по-
тока частиц, падающих на экран через
небольшую щель, то профиль вероят-
ности будет соответствовать экспери-
ментально наблюдаемым дифракци-
онным полосам. В волновой механике
Шредингера плотность вероятности
для потока частиц до и после попада-
ния в щель изображается в виде квад-
рата амплитуды волновой функции.
В подходе Дирака описываются веро-
ятности различных конечных состо-
яний, при данном начальном состоя-
нии, выраженных в виде дискретных
элементов математической матрицы.
Энергетические уровни
Фундаментальная концепция кванто-
вой механики заключается в том, что
энергия систем, таких как атомное ядро
или электроны квантована и разбита на
уровни, которые заполнены в соответ-
ствии с определенными статистически-
ми законами. В основе всех спектроско-
пических экспериментов (см. Часть 3)
лежит гот факт. что переход на более
высокие уровни может происходить за
счет поглощения излучения, а испус-
кание излучения происходит при пере-
ходе системы на более низкий уровень.
Квант энергии hv, поглощенный или из-
лученный, соответствует в точности
разности уровней энергии.
Энергетические уровни электрона
в атоме характеризуются орбитальны-
ми и спиновыми квантовыми состояния-
ми или числами. Пользуясь классиче-
ской аналогией, можно сказать, что они
могут быть описаны как дискретные
энергетические состояния электрона,
вращающегося вокруг ядра и своей оси
одновременно. Эта аналогия, однако,
не должна уводить нас слишком дале-
ко, поскольку самой сутью квантовой
механики является невозможность ее
описания в классической манере. За-
полнение электронных уровней опреде-
ляется строгими правилами. Например,
уровень с определенным орбитальным
и спиновым квантовыми числами мож-
но заполнить только одним электроном.
Состояние с наименьшей энергией на-
зывается основным состоянием, а состоя-
ния с большими квантовыми числами
называются первым, вторым и т. д. воз-
бужденными состояниями.
Атомы, связанные друг с другом
в молекулах, колеблются и вращаются
подобно шарикам, соединенным пру-
жинками. Такую систему мы рассмат-
ривали выше в разделе, посвященному
простым гармоническим колебаниям.
В квантовой механике эти колебания и
вращения также будут иметь дискрет-
ные уровни энергии с соответствующи-
ми квантовыми числами. «Пружинки»
атомных связей образованы общими
электронными парами, чьи энергети-
ческие уровни сами находятся под влия-
пнем колебательной и вращательной
анергии молекулы (рис. АЗ.23). Пив-
шее (основное) п первое возбужден-
ное электронные состояния молекулы
изображаются в виде потенциальной
ямы относительно конформационных
координат, где электроны колеблются
н вращаются вместе со всей молекулой.
В отличие от классического случая, в
квантовой механике только дискретные
уровни могут быть заняты в данной по-
тенциальной яме. Обратите внимание
на большое пространство между элек-
тронными уровнями по сравнению с
колебательными н вращательными пе-
реходами, н на разницу между колеба-
тельными энергетическими уровнями,
которая больше промежутка между вра-
щательными уровнями.
Атомные колебания в молекуле мож-
но в первом приближении рассматри-
вать как простые гармонические колеба-
ния. Выше мы уже получили функцию
потенциальной энергии классического
простого гармонического осциллятора.
Квантовая механическая потенциальная
энергия такого осциллятора, состоящего
из двух связанных атомов с приведенной
массой иц.,, имеет вид:
Е = р/ + - I Асо,
I 2J
где (Л3.43)
Здесь и — колебательное квантовое
число, которое может принимать только
положительные целые значения, вклю-
чая ноль, h — это константа Планка, де-
ленная на 2л, k — силовая константа, а
ти|9 — приведенная масса (сравните с
уравнением Л3.24). В отличие от клас-
сической механики, где колеблющаяся
система может принимать любые зна-
ние. АЗ.23. Диаграмма потенциальной энер-
гии низшего (,$’) и первого возбужденного (5Д
электронных состояний в молекуле, г, v, и е —
вращательные, колебательные и электронные
переходы
чения потенциальной энергии, кванто-
вые системы обладают вполне опреде-
ленными дискретными энергиями (см.
рис. АЗ.23). Переходы между энерге-
тическими уровнями могут осущест-
вляться через поглощение излучения,
при условии, что его энергия в точности
равна разности энергий АД'двух кванто-
вых состояний.
Однако описание потенциальной
энергии электронных колебаний связи
между двумя ат омами с помощью гармо-
нической модели не является полным.
Например, когда два атома сближаются,
кулоновское отталкивание между двумя
ядрами действует в том же направлении,
что и восстанавливающая сила связи, по-
этому можно ожидать, что потенциаль-
ная энергия будет расти быстрее, чем это
предсказано с помощью гармонического
приближения. При этом кривые потен-
циальной энергии принимают негармо-
ническую форму, показанную па рисун-
ках АЗ.23 и АЗ.21. Эти кривые в разной
степени отклоняются от гармонических,
<-------Межатомное расстояние г — -►
Рис. Л3.21. Диаграмма потенциальной энергии. Кривая 1 — гармонический осциллятор. Кривая
2 — негармонический осциллятор. Обратите внимание: гармоническая и негармоническая кри-
вые почти совпадают при низкой потенциальной энергии. Энергия диссоциации — это энергия,
необходимая для разрыва «пружины» между атомами
в зависимости от природы атомов и об-
разованной ими связи.
АЗ.З. Динамика и структура,
кинетика, кинематика,
релаксация
Понятие динамики часто используется
при описании временных зависимостей
(или движения), в отличие от струк-
туры, которая описывает статичную
или, точнее, усредненную по времени
картину. Греческий корень этого сло-
ва Suvapiq — сила. Динамика является
разделом физики, где рассматривается
движение объектов и сил, которые при-
водят их в движение.
Динамика в свою очередь делится
на кинетику (от греческого kineein, дви-
гать), которая занимается установлени-
ем соотношений между движущимися
объектами, их массами и силами на них
действующими, и кинематику, где рас-
сматривается только движение объек-
та, без учета сил. Кинематика и слово
кино имеют один и тот же греческий
корень — kinema, движение.
В химии кинетика относится к изу-
чению скоростей химических реакций.
Релаксация означает возврат сис-
темы из возмущенного состояния в
равновесное. Простейший процесс ре-
лаксации может быть описан как экспо-
ненциальное затухание:
Л(О = Л(0)ехр(-Г/т). (А3.44)
Параметр А (?) отражает отклоне-
ние свойств системы от равновесных в
момент времени ?. Он равен А в момент
времени 0 и затухает до нуля при бес-
конечном значении времени. Время ре-
лаксации т — это время, при котором ве-
личина А затухает в 1/е раз (примерно
в 1/2,7 раза) от первоначального значе-
ния А (см. Главы Гб и Г8).
В этой главе мы обсудим понима-
ние структуры биологических макро-
молекул в терминах действующих сил.
Тем самым мы сконцентрируемся на
внутренней (атомной) динамике пра-
вильно свернутой макромолекулы, а не
на динамике движущейся жесткой час-
тицы. Последняя обсуждается в Час-
ти Г, посвященной гидродинамике.
А3.3.1. Силы, стабилизирующие
макромолекулы
В настоящее время известны силы,
удерживающие атомы на своих местах
в биологически активных макромо-
лекулах (см. Главу В). Они являются
результатом ван-дер-ваальсовых взаи-
модействий, водородных связей, элек-
тростатических и гидрофобных взаи-
модействий, а также S-S связей между
остатками цистеина (рис. АЗ.25).
Стабилизационная энергия макро-
молекулярной структуры — это раз-
ность свободных энергий свернутой
и развернутой форм молекулы. Кова-
лентные связи, крайне стабильные в
отношении температурных условий,
обычно не разрушаются при развора-
чивании макромолекулы, поэтому они
(за исключением S-S связей) не вносят
вклад в стабилизационную энергию
третичной и четвертичной структу-
ры макромолекулы. Биологические
макромолекулы являются достаточно
«мягкими», поскольку энергии стаби-
лизирующих сил у них слабы — в том
смысле, что они составляют величину,
сравнимую с тепловой энергией при
физиологической температуре. Такая
мягкость не кажется удивительной,
поскольку стабилизационные силы
сильно зависят от окружения, несмот-
ря на то, что кристаллографические
и калориметрические исследования
показывают, что белки образуют ком-
пактные структуры, составляющие
ядро с плотноупакованными атомами
и учитывая, что электростатическое
(кулоновское) взаимодействие между
двумя зарядами достаточно сильное
и действует на относительно больших
расстояниях, a S-S связи являются ко-
валентными.
Электростатические взаимодейст-
вия в биологических макромолекулах
Водородные связи в а-спиралях и р-плоскостях
Соленые мостики
между АСП и АРГ
остатками в
контактируемых
субъединицах
Связанные ионы
растворителя и
молекулы воды
Гидрофобные и ван-дер-ваальсовы
взаимодействия в белковом ядре
Рис. АЗ.25. Взаимодействия, стабилизирующие свернутую форму тетрамера малат дегидро-
геназы из галофильной бактерии археи Halobacterium marismortui. Белок не содержит остатков
цистеина. Его физиологическое окружение представляет собой среду, содержащую КС1 в кон-
центрации, близкой к насыщающей. Такая высокая концентрация соли необходима для того,
чтобы белок оставался правильно свернутым и стабильным. Слабые электростатические взаи-
модействия приводят к специфическому связыванию ионов растворителя, которые участвуют в
стабилизации белка. Ионы хлора представлены в виде шариков между субъединицами (Richard
et al., 2000)
экран провал ы o.i а годаря д 11 эле к гр 11 -
ческим свойствам воды и противо-
ионам растворителя. S-S связи легко
разрушаются в восстанавливающей
среде, образуя S11 группы. Водородные
связи различной прочности образу-
ются как между донорными и акцеп-
торными группами самой макромоле-
кулы. так и между макромолекулой и
ионами растворителя. Гидрофобные
взаимодействия по определению за-
висят от окружения, поскольку они
связаны с низкой растворимостью оп-
ределенных химических групп в воде
(см. Часть В). Даже ван-дер-ваальсовы
взаимодействия, которые проявляются
при плотной упаковке атомов, зависят
от окружения, поскольку некоторые из
атомов упакованы плотнее, чем другие.
Они. как считается, являются важной
движущей силой сворачивания белков
(см. Часть В).
.Моделирование молекулярной ди-
намики отражает наше теоретическое
знание о макромолекулах как о физи-
ческих частицах. Несмотря на то что
известны все разновидности макромо-
лекулярных стабилизационных сил,
наше представление о соответству-
ющих соотношениях далеко не полно,
в основном из-за сложности эффек-
тов, связанных с окружением. Поэто-
му моделирование должно иметь под
собой прочное экспериментальное ос-
нование.
АЗ.3.2. Шкала длины и времени
в молекулярной динамике
Амплитуды атомных колебаний в мак-
ромолекулах при комнатной темпера-
туре (-300 К) находятся в диапазоне
от 0,01 А до 5 А для временных перио-
дов 10 103 секунд (от фемтосекун-
ды для электронных переходов до при-
мерно 20 мин при сворачивании белка
или его денатурации) < гм i \.-У).
Для различных временных периодов
и расстояний существуют своп био-
физические методы (см. Введение).
Лазерная оптическая спектроскопия
в наше время может достигать суб-
фемтосекуидного разрешения при
измерении кинетики продолжительно-
стью несколько минут, что составля-
ет диапазон в 18 порядков величины
временной шкалы. ЯМР чувствителен
к динамике от пикосекунды и более.
Нейтронное рассеяние способно раз-
решать времена в районе от 10 9 до
10 12 сек и чувствительно к расстояни-
ям от 1 до 5 А.
Таблиц.. АЗ.2. Времена и расстояния в молекулярной динамике
Тип Расстояние (А) Время (сек)
Электронные переходы 0,01 10 13
Колебания связанных атомов 0,01-0.1 10 " 10 13
Движения боковых цепей 1 10 12- 10 9
Вращения боковых групп в белковых структурах 5 10 ‘ -1
Движения доменов в белках 1 5 10 9 1
Сворачивание белка, образование комплексов, конфор- мационные изменения 1-5 10 ,;-103
АЗ.3.3. Физическая модель
динамики белков
Наше понимание бе.чковоП динамики
основано на результатах эксперимен-
тов с использованием разнообразных
биофизических методов, в особеннос-
ти оптической спектроскопии, раз-
решенной во времени, нейтронной и
мессбауэровской спектроскопии, ЯМР
и кристаллографии, которые будут об-
суждаться в основном в Главе К.
Белок представляет собой плотно
упакованный объект, в котором атомы
занимают некие усредненные места,
соответствующие нативной структуре.
В результате действия на атомы зна-
чительных удерживающих сил моле-
кулярная динамика белка напоминает
динамику аморфного твердого тела.
Поэтому только в первом приближе-
нии белок можно рассматривать как эла-
стичную сплошную среду, где движения
боковых цепей аминокислот и доменов
подчиняются закону Гука (коммент
Г.: \ >.? ).
Конформационные подсостояния
на картине энергии и специфические
движения в белках
Модель конформационных подсостоя-
ний (КП) была предложена для белков
после экспериментов со связыванием
монооксида углерода с гемовой груп-
пой миоглобина ( i. <' М м 1 : | . 11 > I i,Л.'...’ ;.
Анализ кинетики повторного связыва-
ния указывает на гетерогенность белко-
вой популяции при низкой температуре
(ниже 200 К). При более высоких темпе-
ратурах лиганд преодолевает несколько
энергетических барьеров па пути к пов-
торному связыванию, и характер этого
процесса позволяет предположить, что
белок колеблется между несколько раз-
Коммеи;арий АЗ.2. Закон Гука
Закон Гука гласит: растяжение (дефор-
мация) упругого тела пропорционально
приложенной силе. Этот закон справедлив
в случае сравнительно небольших растяже-
ний.
личающимися конформациями. Схема-
тическая картина энергии модели КП
показана на рисунке АЗ.26.
При температуре ниже 200 К белок
остается в состоянии с низким значени-
ем минимума энергии — в одном из КП
(нижняя панель на рис. АЗ.26).
Разница в структурах между двумя
КП может быть очень небольшой, на-
пример, в положении одной боковой
группы аминокислотной цепи. С по-
вышением температуры наступает мо-
мент, когда макромолекула уже при-
обрела достаточно тепловой энергии,
чтобы преодолеть барьер, отделяющий
одно КП от другого. Соответствующие
движения белковой молекулы были на-
званы специфическими (protein specific
G
б кк
1’иг. .\3.26. Картина одномерного профи-
ля потенциальной свободной :>нергнн белка
для набора ((/) и одного из нодсостояннн (б).
Конформационная координата обозначена КК
(I;raiienl'ekleret al.. 1988)
Комментарий АЗ.З. Флэш-фотолиз миоглобина при низкой температуре
Миоглобин представляет собой небольшой связывающий кислород белок с молеку-
лярной массой около 17 кДа, найденный в мышцах. Это один из первых белков наряду
с гемоглобином, родственным белком — переносчиком кислорода красных кровяных те-
лец. чьи структуры были разрешены с помощью рентгеновской кристаллографии. За эти
исследования Макс Перутц и Джон Кендрью получили Нобелевскую премию. Кристал-
лографические исследования гемоглобина и миоглобина пролили свет на функционально
важные структурные различия между несвязанным (дезокси) состоянием и различными
состояниями, в которых белок связан с лигандом. Миоглобин содержит простетпческую
гемовую группу с атомом железа, отвечающую за связывание молекулы кислорода. Срод-
ство миоглобина к монооксиду углерода выше, чем к кислороду, и в этом кроется причи-
на токсичности монооксида, поскольку он блокирует кислород, связывающий сайт. Его
способность к связыванию СО сыграла важную роль в изучении белка. Оно оказывает
влияние на спектр поглощения белка в видимой области. В экспериментах с флэш-фо-
толизом наблюдают кинетику повторного связывания после того, как связь между СО и
гемом разрушена лазерным импульсом. При низких температурах (ниже 200 К) СО не по-
кидает белок. В результате флэш-фотолиза СО переходи т из позиции Л, где он был связан,
в позицию В в гемовом кармане белка.
Вопреки ожиданиям, навеянным простой моделью релаксации (уравнение Л3.34), было
обнаружено, что кинетика повторного связывания не соответствует кривой, описываемой од-
ной экспонентой, а представляет собой сумму быстрого и медленного процессов. Это можно
объяснить одним из следующих способов: либо белок составляет гомогенную популяцию с
несколькими В-сайтами в каждом, например, «быстрый» и «медленный» сайты повторного
связывания либо белок составляет гетерогенную популяцию, где разные молекулы имеют
разные сайты. На рисунке справа внизу «быстрые» и «медленные» сайты изображены буква-
ми f их соответственно
Выбор одной из моделей был осуществлен благодаря экспериментам с импульсами разной
длины. Ожидалось, что для моделей гомогенного связывания (два сайта на белок в гомоген-
ной популяции) и гетерогенного связывания (один сайт на белок в гетерогенной популяции),
поведение миоглобина будет различаться. В результате экспериментов с импульсами разной
длины было установлено, что последняя модель является верной. Таким образом, популяция
белка при низких температурах состоит из молекул с несколько отличной структурой, отра-
жающей различные конформационные подсостояния (КП). При более высоких температурах
повторное связывание СО описывается одной экспонентой, когда гомогенная белковая попу-
ляция колеблется между различными КП (Frauenfelderet al., 1988).
morions). Другими словами, КП пред-
ставляет собой «статическую» неупо-
рядоченность белковой структуры при
низкой температуре, когда каждая мо-
лекула в популяции обладает несколь-
ко отличной структурой, например, в
одной молекуле боковая группа нахо-
дится в конформации А, в то время как
в другой — в конформации Б, а при вы-
сокой температуре эта неупорядочен-
ность имеет динамический характер,
например, когда в одной и той же моле-
куле боковая группа колеблется между
конформациями А и Б.
Тщательный анализ повторного
связывания СО показывает, что кар-
тина энергии белка представляет собой
сложную иерархию, где каждое КП,
имеющее свою потенциальную яму, в
свою очередь делится на дальнейшие
КП. Модель КП получила подтвержде-
ние благодаря разнообразным экспери-
ментальным методам, включая анализ
температурной зависимости факторов
Дебая-Валлера в рентгеновской крис-
таллографии, которая ясно демонстри-
рует статическую неупорядоченность
популяции кристаллов.
Динамический переход и средние
эффективные константы упругости
Эксперименты с рассеянием нейтро-
нов приводят к выводу, что среднее
квадратичное колебание <и2> атомов
миоглобина зависит от температуры,
как показано на рисунке АЗ.27. Об-
ратите внимание, что величина ко-
лебаний составляет доли ангстрема.
Излом кривой при 180 К называют
динамическим переходом.
Поскольку колебания атомов в бел-
ках являются результатом тепловой
энергии, ось графика, на которой от-
ложена температура, отображает энер-
гию системы. Ниже 180 К (93 °C) <и2 >
растет линейно с повышением энергии,
и экстраполируется к абсолютному
Рис. АЗ.27. Величина среднего квадратичного колебания атомов как функция абсолютной тем-
пературы, измеренная с помощью нейтронного рассеяния (см. часть И) для миоглобина в воде
(Д) и растворе трегалозы (о). Эффективные константы упругости k и k’ были рассчитаны для
разных участков кривой, как это описано в тексте (Zaccai, 2000, Doster et al., 1989; Cordone et al.,
2000)
Комментарий АЗ.4. Точка нулевой
энергии
В простом гармоническом колебании
величина среднего квадратичного отклоне-
ний линейно зависит от температуры и экс-
траполируется до значения, равного нулю
при температуре абсолютного нуля. Одна-
ко из-за квантовых эффектов ожидается,
что такая зависимость на практике не будет
иметь места в области абсолютного нуля.
Это следствие принципа неопределенности
Гейзенберга (см. параграф АЗ.2.4), посколь-
ку любые измерения при абсолютном нуле
приведут к возмущению системы, и темпе-
ратура равняться нулю уже не будет.
нулю ( коммун ripiiij Л.!, i). Линейную
зависимость от температуры можно
объяснить с помощью модели, в которой
атомы движутся в простых гармониче-
ских потенциалах, для которых возвра-
щающая сила пропорциональна смеще-
нию относительно среднего положения.
Энергия гармонических осцилляторов
пропорциональна среднеквадратично-
му отклонению с константой <k>, рав-
ной средней константе упругости пли
константе пружины (параграф АЗЛ.2).
Следовательно, <k> можно рассчитать
из обратной величины наклона кривой
зависимости <и’> от Т (см. рис. АЗ.27).
Среднюю константу упругости, подде-
рживающую атомы в структуре миогло-
бина можно рассчитать, если перевести
Т в энергию, умножив ее на константу
Больцмана. Эта константа упругости
оказывается равной 2 Н м ’, что соот-
ветствует таковой для достаточно же-
сткой, но эластичной ленты ( пшчс: i,
pi।и ,\3./>).
Существует природный дисахарид,
называемый трегалозой, который син-
тезируется некоторыми организмами,
живущими в пустыне, помогая им пе-
реносить засуху. Трегалоза покрывает
и защищает клеточные структуры этих
организмов пока их метаболизм замед-
лен в ожидании лучших, более влаж-
ных времен. Миоглобин, заключенный
в оболочку из трегалозы (кривая на
рис. АЗ.27, обозначенная кружками)
Комментарий. АЗ.о. Константы упругости эластично связанных атомов
Эффективная константа упругости, равная примерно 2 Нм ', позволяет удержива ть атомы
в структуре миоглобина при низкой температуре. Эластичная пружина с такой констан той
I упругости растянется на один сантиметр под действием веса в 2 грамма. Сила, прилагаемая
таким весом, составляет 0,002 х 10 Н
имеет большую жесткость, чем белок
в воде, его <k> равна 3 Н м '.
Чтобы избежать применения тер-
мина «жесткост ь», который часто упот-
ребляется при качественном описа-
нии, среднюю эффективную константу
упругости, полученную из данных по
температурной зависимости среднего
квадратичного колебаний атомов, иног-
да называют эластичностью структуры.
Отсутствие динамического перехода в
присутствии трегалозы ведет к предпо-
ложению, что защитный эффект этого
сахара обусловлен «замораживанием»
макромолекулы в определенном энер-
гетическом состоянии вплоть до доста-
точно высоких температур.
Рассуждая в терминах модели КП,
можно сказать, что атомы белка, на-
ходясь ниже динамического перехода,
осциллируют в гармонической потен-
циальной яме. Тогда динамический пе-
реход на картине потенциальной энер-
гии соответствует колебаниям атомов
между различными потенциальными
ямами КП. Данные на рисунке АЗ.27
получены в результате использования
модели двух потенциальных ям (двух
КП) для всех атомов белка, окруженных
водой (рис. АЗ.28). Такая модель явля-
ется слишком большим упрощением,
поскольку различные атомы обладают
разной потенциальной энергией. Тем не
менее данный подход является хорошей
иллюстрацией связи между динамиче-
ским переходом и КП моделью. Даже
несмотря на то, что на участке кривой
выше динамического перехода движе-
ния атомов не являются простыми гар-
моническими колебаниями, вполне за-
конно применение полугармонического
приближения для расчета эффективной
феноменологической константы упру-
гости из обратной величины наклоны
кривой зависимости <и2> от Т. Это зна-
чение для миоглобина равно 0,3 Н-м-1,
что в десять раз меньше, чем для белка
при низкой температуре.
Динамические переходы наблю-
даются для многих других белков, и,
по-видимому, являются общей чертой
их молекулярной динамики. Значения
амплитуд среднего квадратичного ко-
лебаний и эффективных констант уп-
Рис. АЗ.28: а — кривые свободной потенциальной энергии G атомов белка. На практике такой
потенциал означает, что боковые цепи колеблются между двумя разными локальными энерге-
тическими минимумами; б — эффективную константу упругости можно рассчитать с помощью
кназигармонического приближения по формуле <k ' > -AG/2(P
Комментарий АЗ.6. Пурпурная мембрана и бактериородопсин
Пурпурная мембрана (ИМ) экстремального галофила Halobacterium salinarurn (см. Главу
А1) образована специфическими липидами на высокоорганизованном двумерном каркасе.
Наличие такого каркаса сделало возможным кристаллографические исследования естест-
венных мембран с высоким разрешением с помощью электронной микроскопии (см. Главу
Ж2). Белок ПМ назван бактериородопсином (БР), поскольку он связывает молекулу хро-
мофора-ретиналя, который придает мембране пурпурный цвет. До этого открытия ретиналь
был найден только в зрительном белке животных. У Н. salinarum БР выполняет функцию
активируемого светом протонного насоса, откачивающего один протон из клетки против гра-
диента его потенциала на каждый поглощенный ретиналем фотон. Связанный с активностью
протонного насоса миллисекундный фотоцикл, который сопровождается изменением цвета,
позволяет исследовать структурно функциональные соотношения в ПМ спектроскопически-
ми методами (см. Главу 32).
ругости скорее всего специфичны для
каждого белка и связаны с его стабиль-
ностью и активностью.
Эксперименты по криокристал-
лографии (см. Главу ЕЗ) показали, что
рибонуклеаза А не способна связы-
вать субстрат при температурах ниже
динамического перехода. Если же про-
вести связывание с субстратом при
более высокой температуре, а затем
белок охладить до низкой температу-
ры, то субстрат остается связанным.
Динамическая гибкость белка позво-
ляет отбирать состояния с различной
энергией, которые по всей вероят-
ности необходимы для ферментной
активности. Доказательства корреля-
ции между динамическим переходом
и активностью были получены для
мембранного белка бактериородопси-
на (БР), функционирующего как уп-
равляемый светом протонный насос
(цом.мен lapnii .\.3.(S. рис. АЗ.29).
Динамические переходы в белках
напоминают стеклование аморфных
материалов, но эти два процесса не-
льзя считать полностью аналогичны-
ми. Белки — это сложные, гетероген-
ные структуры, чья динамика является
результатом эволюции и служит для
выполнения специфических функ-
ций. На рисунке АЗ.29 показано, что
для бактериородопсина величины
средних квадратичных колебаний и
эффективные константы упругости
отличны для разных аминокислотных
групп. Активное ядро протонного на-
соса располагается вокруг ретиналя,
который остается связанным с ос-
татком лизина через так называемое
шиффовое основание, и погружается
в мембрану на глубину примерно в
половину ее толщины. Во время фо-
тоцикла БР (CM. \. !.!>>,
протон шиффового основания меняет
ориентацию: если раньше он был до-
ступен с внеклеточной стороны мемб-
раны, то теперь — с внутриклеточной.
Таким образом, ретиналь выполняет
роль насосного клапана, обеспечивая
одностороннюю передачу протона.
Эксперименты показали, что актив-
ное ядро белка имеет более низкие
значения среднего квадратичного ко-
лебания и более высокую эффектив-
ную константу упругости, чем белок в
среднем (рис. АЗ.ЗО). Предполагается,
что такая жесткость ядра необходима
для регуляции функции клапана рети-
наль-связывающего сайта.
Рис. АЗ.29. Динамика бактериородпсина, измеренная с помощью нейтронного рассеяния (см.
Главу И). Величина среднего квадратичного колебания отложена против температуры для на-
тивных (Д) и меченых дейтерием (0) мембран. Приведенные данные соответствуют движениям
специфических аминокислот и ретиналя в белковом ядре. Панели а, б, в и г соответствуют раз-
ным величинам гидратации, полученным при относительной влажности 0, 75, 86, 93% (Lehnert.,
Thesis 2002)
Эффекты растворителя, гидратация
мембран и белков
Если смотреть с позиции динамики,
структура белка не может рассматри-
ваться независимо от его окружения.
Поразительным является влияние, ока-
зываемое трегалозой на динамику миог-
лобина (см. рис. АЗ.27). Как мы отмеча-
ли (см. рис. АЗ.29), величина среднего
квадратичного колебания у БР сильно
зависит от гидратации при температуре
выше динамического перехода (около
150 К). Гидратацию регулировали, по-
мещая образец в среду с контролируе-
мой влажностью ( коммсп riipnii АЗ.7).
При температуре выше 150 К и мече-
ный и нативный образцы с повышением
влажности имели большие амплитуды
колебаний.
Интересно, что пурпурные мемб-
раны выделяют из экстремального га-
лофила, живущего при концентрации
соли, близкой к насыщающей (см. Гла-
ву А1). Насыщенные растворы NaCl и
КС1 соответствуют значениям относи-
тельной влажности 75 и 86%. Поэтому
влажность на рисунке А3.29 (панели б
н+
Рис. АЗ.30. Схематическое представление пурпурной мембраны. Молекула БР отображена в ви-
де ленточной диаграммы, а липиды показаны синим. Две липидные молекулы и меченые группы
представлены в виде шариков и палочек (см. рис. АЗ.5). Превращая энергию света в химическую
энергию, БР откачивает из клетки один протон на каждый поглощенный фотон. Ядро и внеклеточ-
ная часть БР (внутри красного эллипса), которые действуют подобно насосному клапану, облада-
ют большей жесткостью (менее эластичны), чем вся мембрана в целом (Zaccai, 2000)
ин) очень близка к физиологическим ус-
ловиям для пурпурных мембран. Образ-
цы последних в экспериментах по ней-
тронному рассеянию состояли из стопок
чередующихся мембран и водных слоев
(рис. А3.31 а). Фотоцикл протонного
насоса пурпурных мембран, исследо-
ванный на таких образцах, ингибирует-
ся при значениях влажности менее 75%,
при которых в мембранных стопках при-
сутствует всего лишь один водный слой.
По-видимому, такой уровень гидратации
Кв; АЗ. I Соли и относительная влажность
Парциальное давление водяного пара над насыщенным раствором соли является постоян-
ной величиной при данной температуре и давлении. Таким образом, в экспериментальных ус-
ловиях относительную влажность (ОВ) можно контролировать, помещая образец в закрытый
термостатируемый сосуд в присутствии подходящего насыщенного раствора соли. К приме-
ру, при 25 °C и нормальном давлении относительная влажность над насыщенным LiCl равна
15%; для NaCl — 75 %, для КС1 — 86%, для KNO.,— 93%. Преимущество использования этих
солей состоит в том, что соответствующие значения ОВ не слишком чувствительны к неболь-
шим температурным отклонениям. Значения относительной влажности 0 % («сухое») и 100 %
(«влажное») поддерживать гораздо труднее. «Сухая» атмосфера может быть получена при
помощи силикагеля, но еще более сухих условий можно достичь, создавая вакуум над двуоки-
сью фосфора Р2О5.100 % ОВ в принципе может быть получена над чистой водой, однако даже
небольшие температурные градиенты в стенках сосуда приведут к конденсации и колебанию
относительной влажности.
минимален для обеспечения активности
белка в мембране. Аналогичный вывод
получен и для лизоцима и других раство-
римых ферментов. Сегодня общепринято,
что для активности глобулярного белка
достаточно одного гидратационного слоя
средней толщиной 1,2 Л, что соответству-
ет величине гидратации 0,3 грамма воды
на грамм белка (см. Главу П). Экспери-
ментально показано, что плотность этого
слоя воды на 10% больше плотности воды
в основном объеме (рис. АЗ.ЗI б).
Видеоизображение промежуточных
структур
Кинетическая кристаллография деталь-
но обсуждаемая в Главе ЕЗ более всего
похожа на видеоизображение макромо-
лекул. Этому методу больше подходит
название кинематической кристалло-
графии. Кристаллография макромолекул
дает красивые изображения структур с
атомной детализацией, и вполне логич-
ным было бы использовать этот метод
для получения изображений структуры
как функции времени. Очень высокая
интенс!।вность рентгеновского 1влече-
ния в широком волновом диапазоне, по-
лучаемого в синхротроне и возможность
добиться импульсного излучения с про-
должительность несколько пикосекунд
сделали кристаллофафию, разрешенную
во времени вполне достижимой целью.
Однако кристалл содержит пример-
но 1013 молекул, а информация о поло-
жении атомов является результатом ус-
реднения не только по времени, но и по
всему набору молекул в кристалле. По-
этому, чтобы наблюдать структурные
изменения во времени, все молекулы
должны колебаться в фазе, т. е. синхрон-
но. Синхронность реакции может быть
достигнута благодаря использованию
очень короткого лазерного импульса,
б
Рис. А.3.31: а -- образцы пурпурных мемб-
ран для нейтронного рассеяния состоят из
упорядоченных стопок мембран и водных
слоев с периодичностью чередования равной
d. Толщина мембраны d постоянна и рав-
на примерно 50 А; толщина водного слоя ds
изменяется вместе с относительной влажно-
стью от одного водного слоя (толщина око-
ло 3 А) при 75% относительной! влажности
до трех слоев при 93%; б - схематическое
изображение водной оболочки (голубая) вок-
руг молекулы лизоцима (красная). Толщина
оболочки соответствует примерно одному
молекулярному слою, его плотность прибли-
зительно на 10% выше плотности воды в ос-
новном объеме (Weik et al., 2004); Svergun et
al.. 2001; см также Главу E2)
который запускает процесс фотоактива-
ции в светочувствительных молекулах
(например гем в гемоглобине и миогло-
бине, см. коммеп гэрпг АЗ.З. или рети-
наль в родопсине и бактериородопсине,
см. коммси uipiiii АЗ.б). Если молекула
не имеет природного хромофора, то ис-
пользуют внешний (подсаженный) хро-
мофор. Связывание его блокирующей
химической группыссубстратомспособ-
ствует эффективному экранированию
последнего и заметно снижает сродство
к ферменту и препятствует проведению
реакции. Если блокирующая группа об-
ладает светочувствительной связью, то
диффундирующий в кристалл хромо-
фор может быть освобожден с помощью
лазерного импульса подходящей длины
волны, что приводит к одновременному
и быстрому запуску реакции.
Кристаллография в реальном време-
ни позволяет получать видеоизображе-
ние работающих светочувствительных
белков с наносекундиым разрешением.
Однако по различным техническим при-
чинам, большинство кристаллографиче-
ских исследований ферментных реакций
с использованием внешних хромофоров,
опубликованных к середине 2004 г., огра-
ничивались секундным и даже минутным
разрешением, и только одно было сдела-
но с миллисекундным разрешением.
Более обещающий подход к полу-
чению видеоизображений белков ос-
нован на замораживании структурных
интермедиатов при криотемпературах.
Этот подход оказался очень плодотвор-
ным для изучения природных светочув-
ствительных белков и в исследованиях
с использованием внешних хромофо-
ров. Достигаемое временное разреше-
ние обусловлено выбором температуры.
С помощью кристаллографии в реаль-
ном времени при комнатной темпера-
туре или криокристаллографии различ-
ные авторы исследовали структурные
интермедиаты, когда связанный с мио-
глобином монооксид углерода (СО)
диссоциирует и отделяется от белко-
вой молекулы. Результаты этих работ
запечатлены на видеоролике. Внима-
тельный просмотр этих кадров дает
хорошее представление о некоторых
общих свойствах белковой динамики,
связанных с биологической функци-
ей. Первый кадр фильма соответствует
СО-миоглобину, последний кадр отра-
жает структуру несвязанного миогло-
бина (см. ко.ммсн'1 ариii А.З..З).
Рентгеновская кристаллография
дает информацию о распределении элек-
тронной плотности в структуре. Струк-
турные изменения в реальном време-
ни выглядят как пики положительной
электронной плотности, указывающие
на присутствие новых атомов в этом по-
ложении, или, наоборот, отрицательной,
которая говорит о том, что атомы были
удалены из этой позиции. Короткий ла-
зерный импульс запускает диссоциацию
СО. Самые первые изменения — пе-
ремещение атома железа и изменение
кривизны плоскости молекулы гема
происходят слишком быстро, чтобы их
можно было увидеть в видеоролике с
таким временным разрешением. Затем
следуют изменения в структуре крис-
талла, происходящие в интервале меж-
ду 1 нсек и 1 мсек. На этой временной
шкале колебания в структуре белка
открывают путь для диффузии моле-
кулы СО, которая выходит наружу из
связывающего кармана, делая останов-
ки в нескольких специфических «сты-
ковочных» местах, где она взаимодейс-
твует с боковыми группами некоторых
аминокислот. Были найдены четыре
потенциальных стыковочных места,
которые способны связывать ксенон.
Они обозначены как Хе1, Хе2, ХеЗ, Хе4
(рис. АЗ.32 а).
Попутно структура белка меняется
в сторону конформации несвязанного
белка. Эти изменения затрагивают боко-
вую цепь Leu89, способствующую свя-
зыванию СО в сайте Хе1. Кадры, зафик-
сировавшие изменения интегральной
положительной электронной плотности
в Хе1 связывающем сайте и интеграль-
ной отрицательной плотности Leu89, по-
казаны на рисунке АЗ.32 а. На рисунке
АЗ.32 б показаны пики, соответствую-
щие разностной электронной плотности
при 362 нсек.
1.5
a
Рис. АЗ.32: а — временные изменения интегральной положительной электронной плотности в Хе1
связывающем сайте и интегральной отрицательной электронной плотности в Leu89. Для сравне-
ния показаны такие же изменения для трех других Хе связывающих сайтов — Хе2, ХеЗ и Хе4. Они
указывают на то, что на данной временной шкале сайты не заняты СО; б — карта разностной элек-
тронной плотности в области сайта Хе1 при 362 нсек. Положительная плотность в Хе1 сайте обо-
значена символом X, в то время как положительные и отрицательные плотности, указывающие на
движения боковой цепи Leu89, обозначены как L1 и L2 и соответствуют двум конформационным
подсостояниям. Структуры СО- и дезоксимиоглобина представлены в виде красных и синих ша-
риков и папочек. Положительная и отрицательная разности электронной плотности изображены
синей и красной решеточной конструкцией. Гем с атомом железа (Fe) в его центре и положение
связанного СО можно увидеть в верхней части структуры (Srajer et al., 2001)
Вспомним, что структура, получен-
ная в кристаллографических экспери-
ментах, соответствует структуре, прини-
маемой значительным числом молекул в
кристалле. Поэтому кадры видеоролика
отображают специфические интермедиа-
ты (или энергетически выгодные состоя-
ния КП) в процессе релаксации между
структурами СО- и дезоксигемоглобина.
В кристаллографических роликах нельзя
увидеть непрерывные изменения в поло-
жениях атомов между двумя интермедиа-
тами, что указывает на то, что отдельные
молекулы в кристалле пересекают потен-
циальный барьер разными путями. Такая
картина хорошо вписывается в гипотезу
о том, что большое число колебательных
движений на наносекундной временной
шкале приводит к гораздо более медлен-
ным конформационным изменениям
между интермедиатами и образованием
коридора для выхода молекул СО.
На рисунке АЗ.33 можно видеть
другой тип кристаллографических ви-
деороликов. Он отображает временные
изменения, протекающие под действи-
ем освещения двух участков структуры
фермента, называемого ацетил холинэ-
стеразой, который участвует в перено-
се нервного импульса. Боковые цепи
аминокислот в начальной структуре
показаны в виде шариков и палочек, а
наблюдаемая электронная плотность
изображена синей решеткой.
Рис. ЛЗ.ЗЗ. Видеоролик радиационного повреждения боковой цепи глютаминовой кислоты
(верхняя панель) и связи цистенн-цистснн (нижняя панель) в ацетилхолинэстсразе при 100 К
(Weik et al., 2000)
Рис. АЗ.34. Схематическая диаграмма, иллюстрирующая характерные черты физической моде-
ли белковой динамики
В верхней части показано последова-
тельное декарбоксилирование боковой
цени глютаминовой кислоты, вследствие
радиационного повреждения, а в нижней
отображено разрушение S-S связи меж-
ду двумя цистеинами. Все данные были
получены при 100 К. При 150 К повреж-
дение затрагивает еще большее количе-
ство остатков, включая расположенные
в активном центре, что свидетельствует
о том, что боковые цепи способны при-
нимать различные конформации при бо-
лее высокой температуре.
Основные характеристики совре-
менной физической модели белковой
динамики приведены на рисунке .\3.3i.
АЗ.4. Перечень ключевых идей
* Молекулярная биофизика является экспериментальной наукой, в которой
даже теоретические подходы, такие как моделирование молекулярной динами-
ки .должны основываться на наблюдении.
* Практически все эксперименты в молекулярной биофизике, за исключением
калориметрии и классических методов физической химии растворов, таких как
определение осмотического давления, вязкости и т. д„ целиком основаны на
наблюдении за взаимодействием излучения с макромолекулами.
* К необходимым математическим инструментам относятся те из них, которые
имеют дело с общими свойствами волн, комплексными экспонентами, простым
гармоническим колебанием, нормальными модами и анализом Фурье.
° Эксперименты в молекулярной биофизике проводятся в пространстве изме-
рения, которое соотносится с действительным пространством через матема ти-
ческое преобразование; например, пространство волновых векторов в кристал-
лографии соотносится с действительным пространством через преобразования
Фурье.
• Понятия квантовой механики необходимы для описания взаимодействий меж-
ду материей и излучением на атомном уровне.
• Динамика — раздел механики, который имеет дело с движением объекта и сила-
ми, приводящими его в движение. Динамика делится на кинетику, которая за-
нимается соотношениями между движущимися объектами, их массами и дейс-
твующими на них силами, и кинематику, в которой исследуют исключительно
движение объектов без учета действующих на них сил.
• В химии термин кинетика относится к изучению скоростей химических ре-
акций.
° Релаксация означает возвращение возмущенной системы в состояние равновесия.
Свернутые нативные структуры биологических макромолекул поддерживают-
ся водородными связями, солевыми мостиками или экранированными элек-
тростатическими взаимодействиями, гидрофобными и ван-дер-ваальсовыми
взаимодействиями.
Амплитуды движений атомов в макромолекулах при температуре окружающей
среды лежат в диапазоне от 0,01 А до 5 А для временных периодов от 10 15 до
103 секунд (от одной фемтосекунды для электронных переходов до примерно
двадцати минут для сворачивания белка или для денатурации).
Наше понимание белковой динамики базируется на результатах спектроскопи-
ческих и кристаллографических исследований структур и их интермедиатов,
усредненных по времени.
При низкой температуре, ниже 200 К, белок находится в одном относитель-
но мало отличающихся по свойствам конформационных подсостояний (КП):
выше 200 К белок флуктуирует между несколькими разными КП. Переход от
низкотемпературного колебательного режима, где белок заморожен в одном из
КП, к высокотемпературному режиму, где белок флуктуирует между различ-
ными КП, называется динамическим переходом.
Ниже динамического перехода средняя амплитуда атомных флуктуаций, изме-
ренная нейтронным рассеянием, увеличивается линейно с температурой, как и
ожидается в случае простого гармонического осциллятора; средняя константа
упругости может быть вычислена из зависимости обратной величины <и2 > от
температуры.
Выше динамического перехода эффективную константу упругости можно рас-
считать, используя квазигармоническое приближение. Ниже динамического
перехода константы упругости белков составляют величину порядка несколь-
ких Н м1; выше динамического перехода эти константы порядка 0,1 Н-м '.
Белковая динамика чувствительна к окружающему растворителю; измеряемые
амплитуды флуктуаций и константы упругости свидетельствуют об увеличе-
нии гибкости молекулы при повышении влажности среды.
Для сохранения биологической активности белковой молекулы необходим по
крайней мере один окружающий ее водный слой; заключенный в трегалозную
оболочку белок сохраняет большую жесткость при высоких температурах, что
защищает его от разворачивания.
Часть Б
МАСС-СПЕКТРОМЕТРИЯ
Глава Б1
МАССА И ЗАРЯД
Б1.1. Исторический обзор
l 897
Дж. Дж. Томсон (J. J. Thomson)
первым произвел измерения и рассчитал
отношение массы к заряду для [элемен-
тарных частиц — «корпускул», которые
позднее стали известны как электроны.
Это событие с полным правом можно
считать рождением масс-спектромет-
рии.
1918 1919
А. Демпстер и Ф. Астон (A. Demp-
ster и F. Aston) разработали первые
масс-спектрографы. В качестве детектора
ис!Ю.ш>зовачасьфото графическая пласти-
на. Прибор применяли для определения
относительного содержания изотопов.
1951
В. Паули и X. Штайнведель
(W. Pauli and Н. Steinwedel) опи-
сали созданный ими квадрупольный
масс-спектрометр. Четыре стержня, к
которым прикладывали постоянное и
переменное радиочастотное напряже-
ния, служили разделителями масс, где
с постоянной амплитудой осциллиро-
вали только ионы, чья масса укладыва-
лась в определенный диапазон. Затем
эти ионы собирались анализатором.
1959
К. Биеман (К. Biemann) применил
электронно-ионизационную масс-спек-
трометрию для анализа пептидов. Позд-
нее было показано, что для определения
аминокислотной последовательности
пептиды должны быть фрагментированы
перед анализом.
1968-1970
М. Доле (М. Dole) был первым, кому
удалось перевести синтетические и при-
родные полимеры в газовую фазу при
атмосферном давлении. Эксперимент
проводился путем распыления раствора
образца из маленькой трубки в сильном
электрическом поле в присутствии пото-
ка горячего азота, который способство-
вал десольватации. Первые же опыты
показали, что лизоцим способен приоб-
ретать множественный заряд.
1974
Д. Торгерсон (D. Torgerson) ввел
в научную практику плазменную десорб-
ционную масс-спектрометрию, когда для
десорбции больших молекул использу-
ются осколки от деления ядра 23-'Cf. Это
была первая методика, показавшая воз-
можность получения молекулярных ио-
нов белка в газовой фазе из твердого об-
разца с помощью элементарных частиц.
1 97 !
Б. Мамырин (В. Mamyrin) сделал
наиболее значительный вклад в разви-
тие время-пролетной (ВП) масс-спек-
трометрии. Он сконструировал так
называемый рефлектрон, ранее предло-
женный С. Алихановым (S. Alikhanov)
в 1957 г. Прибор позволил значительно
улучшить разрешающую способность
ВП масс-спектрометрии.
197S
Н. Комисаров и А. Маршалл
(N. Comisarow и A. Marshall) примени-
ли фурье-преобразование к спектромет-
рии с ионно-циклотронным резонансом
и построили первый инструмент подоб-
ного типа с фурье-преобразованием.
С тех пор интерес к этой технике рос
экспоненциально, так же как и число
таких инструментов.
1981
М. Барбер (М. Barber) открыл бом-
бардировку быстрыми атомами (ББ А): но-
вый источник ионов для масс-спектромет-
рии. Масс-спектр нефрагментированного
пептида из одиннадцати остатков — Мет-
Лиз-брадикинина (Met-Lys-bradykinin)
(М = 1318) был получен бомбардировкой
маленькой капли глицерина, содержащей
несколько микрограммов пептида, с по-
мощью пучка атомов аргона с энергией
в несколько кэВ. Эта техника совершила
революцию в масс-спектрометрии и от-
крыла ее для биологии.
1984
Р. Виллубай (R. Willoughby) и,
независимо от него, М. Александров
(М. Aleksandrov) предложили объеди-
нить жидкостную хроматографию и
масс-спектрометрию для анализа мате-
риалов с большими молекулярным ве-
сами в жидкой фазе.
1988
Дж. Фенн (J. Fenn) и, независимо от
него, М. Ямашита (М. Yamashita) осу-
ществили перевод биологических макро-
молекул в газовую фазу при атмосферном
давлении. Они предложили новый тип
ионизации, названной ионизацией элек-
трораспылением (ИЭР), для получения
нативных биологических молекулярных
ионов при помощи распыления сильно
разбавленного раствора через кончик
иглы сквозь электростатическое поле
с градиентом в несколько киловольт.
1988
М. Карас (М. Karas) и Ф. Хиллен-
камп (F. Hillencamp) и, независимо от
них, К. Танака (К. Tanaka) разработали
новую технику ионизации, названную
методом ионизации лазерной десорб-
цией в матрице (ИЛДМ). Было пока-
зано, что белки с молекулярной массой
вплоть до 60 кДа могут быть ионизованы
при облучении лазерным лучом, если их
предварительно поместить в УФ-погло-
щающую матрицу. Масс-спектрометрия
с использованием техники ИЛДМ (МС-
ИЛДМ) обладает высоким разрешением,
точностью измерения,способностью за-
хватывать ионы, что позволяет получать
информацию не только о молекулярной
массе, по также и о структуре различных
пептидов и олигонуклеотидов. В 2002 г.
К. Танака получил Нобелевскую пре-
мию по химии за вклад в масс-спектро-
метрию.
1992-1999
Молекулярнаяспецифичностьи чув-
ствительность МС-ИЛДМ породила
новую технологию прямого картирова-
ния и получения изображений распре-
деления биологических макромолекул
в клетках и тканях. Благодаря сканиро-
ванию образца ионным лучом и сбору
масс-спектра по точкам, откуда десорби-
рованы ионы, удалось «увидеть» распре-
деление молекул по массе на клеточной
поверхности или в образце ткани.
2000 и по настоящее время
Масс-спектрометрия стала важ-
нейшим аналитическим инструментом
в науке о жизни. Методы мягкой иони-
зации, такие как ББА, ИЭР и ИЛДМ,
превратили в рутинную еще недавно
недоступную процедуру по измерению
масс белков и нуклеиновых кислот с
высокой точностью и разрешением.
Масс-спектрометрия как один из са-
мых мощных экспериментальных ме-
тодов дает возможность для прямого
наблюдения биологических комплек-
сов в газовой фазе, их сборки и разбор-
ки в реальном времени. В последние
годы данные по масс-спектрометрии
все чаще совмещаются с таковыми по
изотопному и аффинному мечению и
данными геномной информации. Со-
вершенно очевидно, что фаза быстро-
го роста в развитии биоаналитической
масс-спектрометрии еще не достиг-
ла своего пика. Несомненно, в следу-
ющем десятилетии масс-спектромет-
рия будет развиваться невероятными
темпами и из области структурной
Комментарий Б1.1. Термин J
«масс-спектроскопия»
Нам хотелось бы предостеречь читателя 1
против использования термина «масс-спек-
| троскопия». Он неверный, поскольку не
| имеет отношения к настоящим спектро- ;
[ скопическим методам, описанным в Частях ,
3 и К. Масс-спектр в значительной степени |
зависит от стабильности ионов, полученных :
и собранных во время эксперимента. Ста- :
билыгость, в свою очередь, зависит от усло-
вий опыта, поэтому предсказать масс-спектр
вещества практически невозможно.
биологии шагнет в область медицины
и терапии.
Б1.2. Введение
в биологические проблемы
В течение последних двух десятиле-
тий масс-спектрометрия (коммсша-
pniilil.l) стала важнейшим аналити-
ческим инструментом в структурной
биологии. Этому способствовало раз-
витие методов получения интактных,
высокомолекулярных ионов различных
биологических макромолекул в газовой
фазе. Некоторые техники ионизации,
такие как бомбардировка быстрыми
атомами (ББА), ионизация лазерной
десорбцией в матрице (ИЛДМ), и элек-
трораспыление (ИЭР) произвели рево-
люцию в масс-спектрометрии и откры-
ли ее для биологии. Были разработаны
новые методы характеристики белков,
основанные на спектрометрии с ионно-
циклотронным резонансом фурье-пре-
образования (МС-ИЦР-ФП), имеющие
предел детектирования 30 зептомолей
(30 х 10'21 молей) для белков с молеку-
лярной массой от 8 до 20 кДа. Исполь-
зуя эту технику, можно выделить отде-
льные ионы — от полиэтиленгликоля до
ДНК с массой свыше 108 Да (ком мен и-
рии 1> 1.2 и Б1.3).
Разрешение и точность определения
массы с помощью масс-спектрометрии
обсуждаются в Приложении Б1.
Комментарий Б1.2. Абсолютная
и относительная массы
Масс-спектрометр не измеряет абсолют-
ную массу М, поэтому инструмент должен
быть калиброван с помощью стандартных
веществ, чьи значения М известны с боль-
I шой точностью. Чаще всего используется
• углеродная шкала. Масса |2С = 12,000000.
Комментарий Б 1.3. Молекулярная
масса и молекулярный вес
Когда Л/ используется для обозначения
относительной молекулярной массы, может
возникнуть некоторая путаница. Л/ являет-
ся относительной безразмерной! величиной.
Однако по абсолютной величине Л/ равна
Л/, которая имеет размерность, - в частно-
сти, для биологических макромолекул она
выражается в дальтонах. Надо заметить, что
молекулярный вес, который есть сила, а не
масса, в этом случае термин неправильный.
В настоящее время масс-спектромет-
рия стала основным методом в исследова-
ниях структуры биологических макромо-
лекул: в определении молекулярных масс
белков и нуклеиновых кислот с высокой
точностью и в широком диапазоне, в пеп-
тидном и нуклеотидном секвенировании,
при идентификации посттрансляционных
модификаций белков, в исследованиях из-
менений! белковой структуры, фолдинга
(сворачивания) и динамики, в идентифи-
кации субпикомольных количеств белков
в двумерном электрофорезе, при иденти-
фикации изотопного мечения.
В последнее время масс-спектромет-
рия позволила охарактеризовать полно-
стью функциональные биологические
субклеточные комплексы и органеллы,
а также интактные бактерии. Были от-
крыты большие перспективы для при-
ложений этого метода в протеомике,
таксономии бактерий и медицине.
Б1.3. Ионы в электрическом
и магнитных полях
Ускоряющийся ион, покидая источник,
приобретает кинетическую энергию
г, „ ти! ..
F = zuB =----, (Б1.1)
г
гдег - это заряд иона, V' — ускоряющая
разность потенциалов, т масса иона,
а и — его скорость. В однородном маг-
нитном поле В, перпендикулярном его
траектории, ион испытывает действие
силы Лоренца F ( immmcii r.nniii 1 > I. i),
которая перпендикулярна как В, так и и
(рис. Б 1.1 а). Результирующая траекто-
рия иона в магнитном поле представляет
собой окружность с радиусом г, посколь-
ку сила Лоренца просто уравновешивает
центробежную силу:
г- v "1ц1 /1'1 оч
Е = zv =-------. (bl.2)
КИП \СК 2 X • /
Величина отношения массы к заря-
ду m/zдается выражением:
Из уравнения Б 1.3 следует, что са-
мые легкие ионы имеют траекторию с
наименьшим радиусом, который уве-
личивается в ростом массы иона и на-
пряженности электрического поля
(рис. Ы. 1 б).
Б1.4. Методы ионизации
Б1.4.1. От ионов в растворе
к ионам в газовой фазе
Переход ионов из газовой фазы в раствор
процесс естественный. В присутствии
молекул растворителя, таких как Н.,О,
। Комментарий Б1.4. Сила Лоренца
Сила Лоренца (F) - это сила, испытыва-
емая анионом с зарядом (г), движущимся в
: электромагнитном ноле. F = z(E + и*. В), '
где Е — напряженность электрического
поля, а ихВ — векторное произведение
напряженности магнитного поля В и ско-
рости иона и.
I ’; i('. I > 1 1: <i сила Лоренца, действующая на заряженную частицу в магнитном поле; б — заря-
женные час тины в электрическом и магнитном нолях. В обоих случаях магнитное поле направ-
лено перпендикулярно плоскости рисунка
«голые» (дегидратированные) ионы
в газовой фазе, например Na', спонтан-
но образуют молекулярные кластеры
ион-растворитель — Na‘(H.,O)n. Если
давление паров растворителя несколько
больше давления насыщенного пара, эти
кластеры вырастут до размеров неболь-
ших капель.
Перенос ионов из раствора в газовую
фазу представляет собой процесс десоль-
ватации, требующий затрат энергии (эн-
дотермический процесс), и поэтому не
происходит спонтанно. Например, для
переноса ионов Na' из водной фазы в га-
зовую требуется большая свободная энер-
гия - около 98 ккал/моль. В большинстве
методов аналитической масс-спектро-
метрии для переноса в газовую фазу тре-
буется очень большая энергия, которая
обеспечивается за счет каскадов высоко
энергетических столкновений или силь-
но локализованного нагрева.
Ионы можно получать тремя раз-
ными способами. Во-первых, отрывая
электрон от молекулы с образованием
положительно заряженного катиона,
который может быть ускорен в расту-
щем градиенте отрицательного поля,
либо в понижающимся градиенте по-
ложительного поля. Во-вторых, добав-
ляя электрон, получая при этом анион.
В этом случае ускоряющие поля прямо
противоположны тем, что используют-
ся для катионов. В-третьих, удаляя или
добавляя протон. В этом случае масса
результирующего иона будет отличать-
ся на 1 от исходного нейтрального иона.
Ниже мы опишем наиболее часто ис-
пользуемые пути для получения ионов
в масс-спектрометрах.
Б1.4.2, Электронная ионизация
Электронная ионизация (ЭИ) — наи-
более часто используемая техника в
масс-спектрометрии. Она является отно-
сительно простой иллюстрацией общего
принципа ионизации при электронной
бомбардировке. Энергия электронов, по-
Комментарий Б 1.5. Электронвольт
(эВ) и джоуль (Дж)
Электронвольт (эВ) — это единица энер-
гии, равная кинетической энергии электро-
на, приобретенной им вследствие прило-
i женной разности потенциалов в 1 В:
| 1 эВ = 1,6 х 10 19 Дж.
। Джоуль (Дж) — стандартная единица
; энергии в системе СИ. 1 Дж = 1 ньютон на
: метр.
лученных в результате нагревания нити,
находящейся в источнике ионов, обычно
составляет 70 эВ ( кчммсн rii’iiii 1>1.5).
Под воздействием таких электронов га-
зообразная молекула может терять или
приобретать один электрон. Возможные
последствия этого описаны ниже.
Ковалентные связи образуются спа-
риванием электронов. При образовании
катиона в результате ионизации проис-
ходит потеря электрона с образованием
связи с одним неспаренным электро-
ном. Тогда:
М (нейтральная) + е —> М'т + 2е ,
где М"+ — это положительно заряженный
молекулярный ион. В случае захвата элек-
трона образуется анион через добавление
неспаренного электрона и поэтому:
М (нейтральная) + е-—> М”_,
где М*" обозначает отрицательно заря-
женный молекулярный ион. В услови-
ях электронной бомбардировки такие
ионы относительно нестабильны и об-
разуют серии дочерних ионов, которые
и регистрируются в виде спектра.
Б1.4.3. Ионизация полем
Для ионизации полем (ИП) образец
должен находиться в газовой фазе. Мо-
лекулы подвергаются действию элект-
рического поля высокой напряжен-
ности порядка 107—108 B-см-1. Такая
напряженность достигается использо-
ванием металлической (Pt, W) иглы
с радиусом 100-1000 нм, к которой
прилагается напряжение около 5 кВ.
В этих условиях электроны внешней
оболочки испытывают действие сил,
достаточных для образования молеку-
лярных катионов.
Б1.4.4. Бомбардировка быстрыми
атомами
В масс-спектрометрии с использовани-
ем бомбардировки быстрыми атомами
(МС-ББА) ионизация происходит при
бомбардировке поверхности образца
пучком атомов Аг или Хе, разогнан-
ных до энергии в несколько кэВ. В этом
случае первичные частицы способны
вызвать каскад столкновений в неболь-
шом объеме.
Образец помещается в жидкую мат-
рицу (обычно глицерин) для поддержа-
ния относительно постоянной концент-
рации молекул в поверхностном слое, за
счет того, что на смену испарившимся
молекулам приходят молекулы из ос-
новного объема. Эта процедура позво-
ляет получать стабильный масс-спектр
в течение продолжительного времени
(около одного часа).
В ББА-ионизации капли образца
бомбардируются атомами Аг или Хе, об-
ладающими кинетической энергией от
8-10 кэВ. При такой энергии изначаль-
но заряженные молекулы образца будут
десорбированы, тогда как формирование
протонированных молекулярных ионов
(М+Н)+ при получении положитель-
ных ионов и (М-Н) — при получении
отрицательных ионов происходит вслед-
ствие перехода молекул в газовую фазу
и химических процессов в растворе.
Основной недостаток техники ББА
(очень высокая концентрация органичес-
кой жидкой матрицы) преодолевается за
счет использования проточной ББА, ког-
да раствор образца постоянно подается к
месту бомбардировки с небольшой скоро-
стью до 10 мкл-мин '. В этом случае тре-
буется меньше органической матрицы, в
результате чего отношение сигнал-шум
возрастает. На рисунке Б1.2 показаны ос-
новные элементы техники ББА.
о
о
Рис. Б 1.2. Механизм десорбции в процессе ББА-ионизации. А' — отрицательно заряженный
ион. С' — положительно заряженный ион. Капля бомбардируется атомами Хе или Аг, облада-
ющими большой энергией (Caprioli and Suter, 1995)
Самым большим преимуществом ме-
тода ББА является его простота. Спект-
ры легко интерпретируются. В настоящее
время ББА-МС широко используется
для анализа компонентов с молекуляр-
ной массой ниже 5 кДа.
Б1.4.5. Плазменная десорбция
В масс-спектрометрии с плазменной де-
сорбцией образец наносится на металли-
ческую поверхность и бомбардируется
осколками ядер радиоактивного изото-
па. В этом контексте термин «плазма»
означает атомные ядра, лишенные элек-
тронов. В результате спонтанного распа-
да ядер 252Cf возникают две атомные час-
тицы с большой кинетической энергией
(80 МэВ) U4Cs и |08Тс, которые летят в
противоположных направлениях. Такая
энергия позволяет частицам проникать
сквозь тонкую металлическую фольгу и
ионизировать биологический материал,
нанесенный с обратной стороны.
Деление ядра — это дискретное со-
бытие, происходящее с частотой (1-
5) х 103 сек ’, результатом которого
является поток ионов, пульсирующий
с той же частотой. По этой причине
в методе целесообразно использовать
время-пролетный масс-спектрометр
(параграф Б 1.5.5). Измеряя время по-
лета вторичных ионов и зная их энер-
гию и траектории, можно преобразо-
вать время-пролетный спектр иона в
масс-спектр. На рисунке Б 1.3 показа-
ны основные характеристики техники
МС-ПД с время-пролетным масс-ана-
лизатором.
Метод МС-ПД обладает хорошей
чу вствител ьностью для пептидов и отно-
сительно небольших белков (7-20 кДа).
Для определения молекулярной массы
этим методом обычно требуется около
10 пМ материала. Разрешение при этом
составляет около 1000.
Б1.4.6. Ионизация лазерной
десорбцией при содействии
матрицы
При ионизации лазерной десорбци-
ей лазерное излучение очень большой
плотности фокусируется на маленьком
участке, что приводит к крайне высо-
кой скорости нагрева. В результате
формируется локализованный лазер-
Детектор
Фрагменты деления
образца
Ускоряющая
решетка
десорбированных
Рис. IS 1.3. Схема метода МС-ПД с источником 25-Cf и время-иролстным масс-анализатором
(Caprioli and Suter, 1995)
ный «факел» испарившихся молекул
из адсорбированного материала или из
самого твердого субстрата. Непосредс-
твенная ионизация интактных биоло-
гических молекул без использования
матрицы ограничена молекулярными
массами около 1 кДа. Для преодоления
этого ограничения используется иони-
зация лазерной десорбцией в матрице
(ИЛДМ).
ИЛДМ отличается от прямой лазер-
ной десорбции тем, что здесь использу-
ется специальный материал, смешан-
ный с образцом, который называют
матрицей. С этой точки зрения тают
техника ионизации похожа на технику
а б
Рис. Ы.1. Схема механизма ИЛДМ с использованием лазера: а - поглощение излучения мат-
рицей; б — диссоциация матрицы, переход в фазу сверхсжатого газа и перенос заряда на молеку-
лы образца; в — увеличение объема матрицы со сверхзвуковой скоростью с вовлечением молекул
образца в расширяющийся «факел» и переносом заряда на молекулу
ББА, которая также использует жид-
кую матрицу для мягкой ионизации.
На практике ИЛДМ обеспечивает еще
более мягкую ионизацию, чем ББА, что
позволяет анализировать большие мо-
лекулы с массой вплоть до 1000 кДа при
минимальной фрагментации.
Детальные механизмы превращения
энергии, десорбции образца и ионизации
до сих пор полностью неизвестны. Общее
представление механизма дано на рисун-
ке Б 1.1. Энергия лазерного луча погло-
щается хромофором в матрице, которая
очень быстро переходит в газовую фазу,
унося с собой молекулы образца. Иони-
зация происходит за счет переноса про-
тона от возбужденных молекул матрицы
к молекулам образца, скорее всего еще в
твердой фазе, а также через соударения
молекул в расширяющемся «факеле».
Матрица - ключевой компонент
техники ИЛДМ. Она действует как по-
глотитель энергии, увеличивая время
жизни образца. Анализируемый матери-
ал смешивается с избытком матричного
материала, который поглощает лазерное
излучение. В качестве такого материала
обычно используют ароматические ве-
щества, содержащие функциональные
карбоксильные группы. Ароматические
кольца матричного материала играют
роль хромофора, поглощая лазерное из-
лучение и способствуя переходу его мо-
лекул и молекул образца в газообразное
состояние. Матрица по только повышает
выход ионов образца, но п предотвраща-
ет их излишнюю фрагментацию.
Для десорбции биологического ма-
териала более всего подходят два типа
лазеров: инфракрасный, который спо-
собен возбуждать колебательные моды,
и ультрафиолетовый лазер, возбужда-
ющий электронные моды в ароматиче-
ских молекулах. Используют импульсы
длительностью 100 нсек и меньше для
обоих диапазонов волн, поскольку бо-
лее длительное воздействие излучения
сопровождается нагревом, способным
привести к пиролитическому разруше-
нию биологических молекул.
Поскольку большинство лазерных
источников импульсные, с ИЛДМ чаще
всего используют время-пролетные
масс-спектрометры с ионно-циклотрон-
ным резонансом и фурье-преобразо-
ванием (ИЦР-ФП-МС) (раздел Б1.6).
В настоящее время точность измерения
массы в благоприятных условиях состав-
ляет ±0,01% (±1 Да при молекулярной
массе 10 кДа). Если позволяют условия,
возможно разрешение индивидуальных
пиков изотопов углерода (секция Б2.1).
Техника ИЛДМ находится в процессе
развития и постоянно улучшается.
Б1.4.7. Ионизация
электрораспылением
Ионизация электрораспылением (ИЭР)
дает возможность получать интактные
ионы из молекул образца, находящихся
в растворе при атмосферном давлении
( коммсн lapnii Б1.(>). Ионы образуются
при подаче напряжения в 1-5 кВ к рас-
твору образца, выходящего через капил-
ляр с низкой скоростью (1-20 нл-мин ').
Большой электрический потенциал,
приложенный между концом капил-
ляра и противоположным электродом,
расположенным на небольшом расстоя-
нии, вызывает превращение жидкости,
выходящей через кончик капиллярной
трубочки, в аэрозоль, состоящий из за-
ряженных капель (рис. Б 1.5). Раствор
испаряется по мере того, как капли пе-
ремещаются из области ионизации с
атмосферным давлением в вакуумную
камеру, где располагается масс-анализа-
тор. Испарению способствует встречный
ток осушающего инертного газа либо
Ионизация распылен- ного потока Распад на мелкие капли Распад на ионы в камере соударений Анализ массы
о © о°
@ О °о ©
о ° °(») ° ©° ©°®°©°
r~W ©С) ° ®о°о©о°© ° ®
° о °© ®о®°©®
Атмосферное давление
1е'7 - 1е’’° мбар
Рис. Ь 1.5. Схематическое изображение траекторий ионов от иглы электрораспылителя до де-
тектора масс-спектрометра. Раствор белка, обычно 1-2 мкл с концентрацией 5 мкМ, помещается
в очень тонкий капилляр с диаметром примерно 10 мкм. К позолоченной игле прикладывает-
ся напряжение в несколько кВ, вызывая электрораспыление маленьких капель. Положитель-
но заряженные капли электростатически притягиваются, десольватируются и фокусируются
в масс-спектрометре для детектирования (Rostom, 1999)
нагрев трубки, через которую капли из
источника ионов попадают в вакуумную
камеру анализатора. Заряд полученных
ионов определяется полярностью при-
ложенного к капилляру напряжения.
Привлекательным свойством мето-
дики ИЭР является образование моле-
кулярных ионов, несущих множествен-
ный заряд, при условии если данные
молекулы могут принимать более од-
ного заряда (см. комментарий 1>1.(S).
Это свойство позволяет детектировать
ионы с большой массой в низком диа-
пазоне величин m/z. Сдвиг шкалы де-
лает возможным детектирование тя-
желых ионов с высоким разрешением,
поскольку разрешение обратно пропор-
ционально отношению m/z (см. пример
в разделе Б2.7). Точность определения
молекулярной массы с помощью техни-
ки ИЭР и масс-спектрометрии с фурье-
преобразованием достигает примерно
Комментарий Б1.6. Число присоединяемых протонов !
Как правило, максимальное число протонов, которое может присоединить пептид или белок 1
в условиях ИЭР, хорошо коррелирует с количеством основных аминокислот (Apr, Лиз, Гис)
плюс N-концевая аминогруппа, если, конечно, она не ацилирована. Однако важным фактором
в этом процессе является еще и доступность этих протон-связывающих участков. Поэтому I
распределение зарядов зависит от pH, температуры и присутствия денатурирующих агентов 1
в растворе. Такое распределение можно использовать для исследования конформационных |
изменений в белке. Например, наиболее распространенный ион бычьего цитохрома с несет 10 ,
положительных зарядов во время электрораспыления при pH 5,2 и 16 зарядов при pH 2,6. По-
хожий эффект наблюдается при восстановлении дисульфидных связей. Максимум распределе-
ния зарядов ионов лизоцима куриного яйца с четырьмя дисульфидными связями находится в
области 12 положительных зарядов (12+), но после восстановления при помощи дитиотрейтола
(АТТ) этот максимум смещается в область 15* (см. комментарий в секции Б2.9).
0,001 0,005%. В благоприятных усло-
виях можно разрешать индивидуальные
ники изотопов углерода (раздел Б1.8).
Наконец, надо отметить, что ИЭР —
один из самых мягких методов иониза-
ции, доступных сегодня, поскольку он
не приводит к молекулярной фрагмен-
тации. Техника ИЭР также как и техни-
ка ИЛДМ активно развивается.
О £ л О . !/ ? Те С й *7 LV'bxs' С □
Первый масс-спектрометр был пост-
роен в 1913 г., когда Дж. Дж. Томпсон
предложил использовать постоянные
магнитные и электрические поля для
разделения двух изотопов благородно-
го газа Ne, используя различие в пове-
дении заряженных частиц с разным им-
пульсом и энергией в электромагнитном
поле. В любом масс-спектрометре ионы
сначала переводят в газовую фазу, затем
разгоняют до определенной скорости
в электромагнитном поле, разделяют в
масс-анализаторе, а затем детектируют
и получают масс-спектр. Поэтому все
масс-спектрометры состоят из пяти ос-
новных частей: впрыскивателя образца,
ионизационной камеры, масс-анализа-
тора, детектора ионов и блока обработ-
ки данных (рис. 1> 1 .(>).
Система введения образца состо-
ит из устройства, осуществляющего
впрыск раствора, зондов для инъек-
ции малолетучих жидкостей и различ-
ных хроматографических приспособ-
лений. Ионы, образованные разными
способами в ионизационной камере
(раздел Б 1.5), анализируются в соот-
ветствии с отношением масса-заряд
в масс-анализаторе.
В настоящее время используют-
ся пять типов масс-анализаторов: маг-
Рис. !>!.(>. Схема основных блоков масс-спек-
трометра
нитный сектор, квадрупольный масс-
фильтр, квадрупольная ионная ловушка,
время-пролетные и ионно-циклотрон-
ные резонансные устройства. Детектиро-
вание ионов после масс-анализа может
осуществляться с помощью деструктив-
ных и недеструктивных методик (см.
ниже). Современные масс-спектромет-
ры полностью управляются компьюте-
рами, а для обработки и интерпретации
данных используются специальные про-
граммы.
Б1.5.1. Масс-спектрометры
с одиночной и двойной
фокусировкой
В инструментах с одним фокусиро-
вочным сектором ионы с массой т,
зарядом z и определенной кинетичес-
кой энергией помещаются в магнитное
поле В. Как следует из уравнения Б 1.3,
при заданных значениях В, V и г ионы с
различными массами будут двигаться по
траекториям с различными радиусами г.
Смеси ионов с большим диапазоном
масс будет соответствовать траектории
с разными радиусами. Поэтому опреде-
ление масс-спектра такой смеси потре-
бовало бы большого числа детекторов.
На практике проблему можно решить,
если заставить все ионы следовать по
траекториям с одинаковым радиусом.
Этого можно добиться двумя способами.
В электромагнитном анализаторе (ЭМА)
используются электромагниты, позволя-
ющие изменять магнитное поле В таким
Магнит
б
Рис. 151.7. Общая схема масс-спектрометра с одной фокусировкой, основанного на принципе
электромагнитного анализатора (а) и электростатического анализатора (б) (Caprioli and Suter,
1995)
образом, чтобы ионы с разными массами,
но с одинаковой скоростью, следовали по
заданному радиусу (рис. 151.7 г/). В элек-
тростатическом анализаторе (ЭСА) ус-
коряющее напряжение V изменяют так,
чтобы разгонять ионы с разными масса-
ми до различных конечных скоростей и
(рис. 151.9). Отметим, что масс-спектро-
метр с одиночной фокусировкой облада-
ет ограниченным массовым диапазоном
и низким разрешением.
В масс-спектрометрах с двойной фо-
кусировкой используется комбинация
ЭСА и ЭМА (рис. 151.8). В таком при-
боре обе траектории линия А и линия Б
пересекаются в одной точке. Инстру-
менты с двойной фокусировкой облада-
ют намного большим разрешением.
Б1.5.2. Квадрупольный
масс-фильтр
Разделение масс в квадрупольном масс-
фильтре основано на достижении ста-
бильной траектории для ионов с опре-
деленными значениями m/z в быстро
меняющемся электрическом поле. Иде-
альный квадрупольный масс-фильтр со-
стоит из четырех цилиндрических стерж-
ней круглого сечения, как показано на
рисунке Б 1.9. К одной паре диагонально
расположенных стержней прилагается
Рис. Ы ,<S. Общая схема масс-спектрометра с двойной фокусировкой (Caprioli and Suter, 1995)
постоянное отрицательное напряжение
(-г/с) и переменное радиочастотное на-
пряжение (</). К другой паре прикла-
дывается постоянное напряжением с
противоположной! полярностью (+(/с) и
переменное //-напряжение с разностью
фаз в 180”. Фильтрация масс происходит
при изменении постоянного и перемен-
ного напряжений, при сохранении по-
стоянства отношения de к rf. При данных
характеристиках поля только траектории
ионов с определенными массами будут
Электронная
линза
Верх и низ
концевых
электродов
цоколя
усилителю
а б
Риг. I > I.!): а схема квадрунольного масс-фильтра. В идеальной ситуации используются стерж-
ни с гиперболическим сечением. Для сканирования значений m/z от 1 до 500, постоянное напря-
жения варьируют между 0 и 300 В, а переменное — от 0 до 1500 В. Частота переменного поля rf
находится в мегагерцовом диапазоне; б - продольное сечение квадруиолыюй ионной ловушки.
Ионы образуются благодаря радиальной инжекции электронов через отверстия в кольцевом
электроде или с помощью аксиальной инжекции через наконечник. При данных значениях m/z
ноны удерживаются на постоянных орбитах, если правильно подобраны амплитуды и частоты
сигналов, приложенных между наконечником и кольцом (Gordon, 2000)
1’ис. Б 1.10. Схематическое изображение ИЭЛДМ-масс-спектрометра с ионной ловушкой
стабильными, они и попадут в детектор.
Ионы с нестабильными траекториями не
пройдут сквозь стержни, поэтому такое
устройство и называется фильтром.
Одно из преимуществ квадруполь-
ного масс-фильтра заключается в низ-
ком напряжении, прилагаемом к источ-
нику ионов — 5-20 В по сравнению с
несколькими кВ в ЭСА-приборах. Низ-
кое напряжение к тому же делает бо-
лее простым соединение инструмента
с жидкостным хроматографом.
Б1.5.3. Квадрупольная ионная
ловушка
Квадрупольная ловушка первоначаль-
но была разработана физиками, которые
были заинтересованы в увеличении вре-
мени наблюдения в спектроскопических
исследованиях элементарных частиц.
Она основана на том же принципе, что
и квадрупольный масс-фильтр, за тем
исключением, что квадрупольное поле
здесь генерируется в трехмерном уст-
ройстве, состоящем из кольцевого элек-
трода и двух электродов-наконечников,
как показано на рисунке Ы.9 б. Кольце-
вой электрод представляет собой одно-
полостный гиперболоид. Такой электрод
похож на тор, за исключением того, что
сечение кольца представляет собой не
окружность, а гиперболоид. Ионы, об-
разованные в ловушке или во внешнем
источнике, остаются в ней же. По мере
повышении напряжения //-траектории
ионов с различными значениями m/z
постепенно становятся нестабильными.
Прошедшие через ловушку ионы детек-
тируются электронным умножителем.
В этом состоит отличие принципа дейс-
твия ионной ловушки от квадрупольного
фильтра, где детектируются ионы со ста-
бильными траекториями. Схематическое
изображение масс-спектрометра с ион-
ной ловушкой и ИЭЛДМ при атмосфер-
ном давлении показано на рисунке Б 1.10.
Б1.5.4. Масс-спектрометр
с использованием ионно-
циклотронного резонанса
Масс-спектрометрия с использованием
ионно-циклотронного резонанса (МС-
ИЦР) основана на захвате ионов и этим
отличается от техники, в которой для
разделения масс используется пропу-
скание ионов ( K(>MM('iri;ipiiii Б 1.7).
В И ЦР ионы, захваченные магнитным
и электрическим полем, детектируются
в том случае, когда частота приложен-
ного //-поля приходит в резонанс с час-
тотой циклотрона < ।,<।м\:-I ।;।:л।i। M.S).
Резонансная частота со прямо пропорци-
ональна напряженности магнитного поля
(обычно от 3 до 7 Тесла) и обратно про-
порциональна отношению m/z ионов:
со. = —. (Б1.4)
т
Ионы, однажды образованные
в ячейке анализатора МС-ИЦР, вы-
нуждены двигаться по круговым ор-
битам с радиусом г равным:
г = —. Б15)
zB
Их подвижность ограничена плос-
костью, перпендикулярной магнитно-
му полю (плоскость ху) (рис. 1S1.11).
Захват ионов вдоль оси z достигается
приложением электростатического по-
тенциала к двум пластинам на концах
ячейки. Захваченные ионы могут нахо-
s.;'’ о Р.1 .7. Принцип
работы ионного циклотрона
В 1932 г. Е. Лоренс (Е. Lawrence) и С. Ли-
вингстон (S. Livingstone) продемонстриро-
вали. что траектория заряженной частицы,
движущейся перпендикулярно однородно-
му магнитному полю, ограничена круговой
орбитой, где угловая частота ее движения не
зависит от радиуса орбиты частицы и выра-
жается уравнением циклотрона Б1.4. Лоренс
открыл, что частица в циклотроне может
двигаться по большей орбите, если прило-
жить поперечное переменное электрическое
поле, чья частота равна циклотронной час-
тоте частицы. Важность открытия заключа-
лась в том, что оказалось возможным сооб-
щить частице очень большую кинетическую
энергию, используя довольно умеренную
напряженность электрического поля. За счет
переменного напряжения в 1 кВ после 1000
циклов частица можетбыть разогнана до ки-
нетической энергии в 1 МэВ.
диться в ячейке несколько часов, при
условии поддержания высокого вакуума
(10 8-10 9 Торр) для уменьшения чис-
ла дестабилизирующих столкновений
между ионами и остаточными нейтраль-
ными молекулами. Захваченные ионы,
имеющие определенные значения m/z,
• Коымонiсфин Б 1.8. Циклотронные
частоты ионов
Из уравнения Б 1.4 следует, что ионы
с разными значениями т/г будут иметь
уникальные циклотронные частоты. При
напряженности магнитного поля в 6 Т ион
с отношением т/г = 36 имеет циклотронную
частоту 2.6 MHz, тогда как ион с т/г = 3600
будет обладать циклотронной частотой
26 kHz. Уравнение Б1.5 также показывает,
что линейное увеличение магнитного поля
приводит к линейному увеличению циклот-
ронной частоты ионов, что делает ионы с
большими массами более легкими для детек-
тирования в низкочастотном (кГц) диапазо-
не. Еще одним преимуществом повышения
напряженности магнитного поля, является
увеличение разрешающей способности при-
бора, а также расширение верхнего предела
измеряемых масс. Надо отметить, что урав-
нение Б 1.4 не учитывает присутствие элек-
трического поля, образуемого двумя захва-
тывающими пластинами, и поэтому может
считаться приближенным.
Единицы напряженности магнитного
поля и давления
Тесла(Т)
Стандартная единица плотности потока
магнитного поля в системе СИ
1 Т = 1 кг-сек2 х А 1
Торр
Единица давления. Один Торр равен
давлению, необходимому для поддержа-
ния столбика ртути с высотой в 1 мм при
температуре 0 °C и стандартной величине
тяготения.
1 Торр равен 133.322 Паскалей
Паскаль (Па)
Стандартная единица давления в систе-
ме СИ. 1 Па = 1 кг • .м 1 сек 2.
I’iic. 15f.ll. Ячейка ИЦР-ФП-масс-спектрометра. Показаны захватывающие, передающие
и принимающие пластины, также указано направление магнитного поля В (Buchanan and
Hettich, 1993)
Ки.-.к днrapiii। t> 1.9. Сходства
и различия между ЯМР и ИЦР
Ядсрная магнитная резонансная спек-
троскопия (ЯМР) это техника, которая
позволяет напрямую измерять расщепле-
ние Зеемана вырожденных энергетических
уровней ядра, обладающего магнитным мо-
ментом (см. Главу К1). В ЯМР резонансная
частота ядра дается выражением
со = уВ(1-о), (Л)
где со — резонансная частота в рад • сек ',
а у — гиромагнитное отношение, характер-
ное для конкретного ядра,В— приложенное
магнитное поле, а — параметр химического
сдвига, который характеризует химическое
окружение ядра.
Заметьте, что в уравнении А, как и в ура-
внении Б1.4, угловая частота пропорцио-
нальна приложенному продольному маг-
нитному полю В. Однако ширина линий в
ЯМРи ИЦРсовершенноразная. Например,
для ‘Н ЯМР в жидкой фазе при магнитном
поле в 2,3 Т все ‘Н ЯМР частоты уклады- ;
ваются в очень узкий диапазон 100 MHz ±
5 kHz. Тогда как при 2.3 Т диапазон масс
15-1500 Да покрывает область ИЦР-час-
тот в 20 kHz — 2 MHz для ионов, несущих i
один заряд. ;
обладают одинаковой циклотронной
частотой, но располагаются в ячейке
случайным образом, поэтому их движе-
ние не приводит к образованию сигнала
на принимающих пластинах МС-ИЦР
ячейки. Для детектирования циклотрон-
ного движения к МС-ИЦР ячейке при-
кладывается возбуждающий импульс с
тем, чтобы собрать ионы в когерентно
движущийся пакет. Результатом коге-
рентного движения ионов является об-
разование на принимающих пластинах
сигнала, имеющего временную зависи-
мость.
Масс-спектрометрия ионно-цикло-
тронного резонанса с фурье-преобра-
зованием (ИЦР-ФП-МС) — резуль-
тат дальнейшего развития техники
ИЦР. Сигналы, регистрируемые как
функция времени, оцифровываются и
подвергаются фурье-преобразованию,
в результате чего получается сигнал
в частотной области, который потом
преобразуют в масс-спектр (параграф
Б1.6.6). Преимуществами ИЦР-ФП-
МС являются: более высокое разреше-
ние, широкий диапазон одновременно
детектируемых значений m/z, возмож-
ность изучения ион-молекулярных
реакций при низком давлении. В
> : о; 1 проводится параллель
между ИЦР и ЯМР.
Б1.5.5. Время-пролетная
масс-спектрометрия
Анализ масс во время-пролетном масс-
спектрометре основан на том, что ионы
с разными значениями m/z обладают
одинаковой энергией, но разными ско-
ростями, приобретенными в результате
ускорения. Отсюда следует, что время
прохождения ионов через время-про-
летную базу будет различным для час-
тиц с разной массой: легкие ионы до-
летят до детектора быстрее тяжелых.
Из уравнения Б 1.1 мы получаем выра-
жения для скорости и иона с массой т
и зарядом z:
и для времени I, необходимого для прео-
доления базового расстояния L
,=Н:)L <Б17)
Уравнение Б1.7 говорит нам о том,
что при ускоряющем напряжении 20 кВ
и L равным 1 м, ион с одним зарядом и
массой 1кДа будет иметь скорость око-
ло 6 х 10'1 м-сек ', а время, необходимое
для прохождения через пролетную базу,
составит 1,4 х 10 5 сек.
Очевидно, что для время-пролет-
ного масс-анализатора наиболее под-
ходящими техниками получения ио-
нов будут те из них, которые работают
в импульсном режиме: использование
осколков деления ядра 252Cf и лазерный
импульс подходящей частоты. При вве-
дении ионов из постоянных источников
(ПЭ, ЭР, ББА и т. д.) необходимо им-
пульсное отражение пучка ионов или
экстракция импульсов из источника.
Метод по времени пролета облада-
ет рядом преимуществ по сравнению с
технологиями, использующими после-
довательное сканирование спектра, вви-
ду «неограниченного» диапазона масс,
высокого пропускания (детектирует-
ся большинство ионов, попадающих в
анализатор), высокой скорости (весь
спектр масс детектируется одновремен-
но в течение нескольких микросекунд)
и потенциально большего коэффициен-
та заполнения (процент детектируемых
ионов). Между тем большим недостат-
ком метода является его низкое раз-
решение. Из уравнения Б 1.7 следует,
что величина m/z пропорциональна t~,
откуда получаем выражение для разре-
шения: R = = -1— . Для стандартных
Ат 2
линейных ВП-инструментов оно со-
ставляет не более 1000.
Значительного улучшения разре-
шения при ВП-.методе можно добиться,
используя электростатическое зеркало
или «рефлектрон» и ортогональный
время-пролетный масс-спектрометр (о-
ВП-МС).
В основе работы рефлектронного
время-пролетного масс-спектрометра
лежит тот факт, что ионы с большой
энергией проникают глубже в отража-
ющее электрическое поле, находясь там
дольше низкоэнергетических ионов.
Поскольку при этом они вынуждены
преодолевать большее расстояние, ионы
с высокой энергией достигнут детек-
тора в то же самое время, что и ионы с
низкой энергией. Применение рефлек-
трона позволяет увеличить разрешение
ВП-МС до 6000. ‘
Радиочастотное
возбуждение
WWlzz
а
t
Выходной
сигнал
1
Рис. I > I. I 2: а общая схема движения возбужденных ионов в ПЦР-ФП ячейке; б - подученный
временной сигнал в результате фурье-преобразования трансформируется в частотный сигнал, из
которого получают масс-спектр
Характерной чертой о-ВП-МС яв-
ляется параллельное расположение де-
тектора к оси ионного пучка. Под дейс-
твием короткого электростатического
импульса (10-100 нсек), направленно-
го строго перпендикулярно исходному
потоку, ионы будет испытывать ортого-
нальное ускорение. Ионы отклоняются
под углом 90" и попадают на детектор,
работающий в режиме времени проле-
та. Такая геометрия спектрометра имеет
ряд преимуществ, главное из которых
состоит в возможности использования
небольшой начальной скорости ион-
ного пучка и как следствие этого — ис-
пользование метода электрораспыления
для получения исходных ионов. Все это
приводит к повышению разрешающей
способности и высокой линейности
спектрометра. Разрешение таких ин-
струментов составляет примерно 4000.
Б1.5.6. Масс-спектрометрия
с фурье-преобразованием
В большинстве масс-спектрометров
ионы детектируются благодаря элект-
рическому току, который они вызывают
при столкновении с поверхностью уст-
ройства, такого как электронный умно-
житель (деструктивный метод). Хотя
такой подход широко применяется и
обладает высокой чувствительностью,
его недостатком является разрушение
детектируемых ионов. Другими слова-
ми, сигнал иона может быть измерен
только один раз. Метод детектирования
в масс-спектроскопии с фурье-преобра-
зованием работает по другому принци-
пу: сильное магнитное поле захваты-
вает ионы внутри ячейки анализатора,
а электрические сигналы, образуемые
за счет их циклотронного движения,
детектируются парой электродов, под-
соединенных к усилителю с большим
внутренним сопротивлением (импедан-
сом) (рис. Б1.12).
Данный метод детектирования по-
зволяет «чувствовать» число ионов, не
удаляя их из ячейки анализатора и не
уничтожая их (недеструктивный ме-
тод). Сигнал от одного и того же иона
может быть измерен несколько раз. Та-
кое детектирование подобно таковому
в методеЯМР ((м. ьо.ммсн ;i;>ini HI.!1 и
Главу Б1.9). МС-ФП даст возможность
детектировать несколько тысяч ионов
для получения полезного сигнала, ко-
торый содержит полную информацию
о частотах и относительном содержа-
нии всех ионов, захваченных в ячейке.
Поскольку частота может быть измере-
на с высокой точностью, то массу иона
можно определить с точностью до од-
ной миллиардной или еще точнее. Надо
отметить, что разрешение в ИЦР-МС с
фурье-нреобразованием зависит от мас-
сы, поэтому сверхвысокое разрешение
достигается при ее низких значениях
(параграф Б2.1.1).
I ’нс I > 1.13. Принцип тандемной масс-спектрометрии. MCI и МС2 — первый и второй масс-спек-
трометры. КС — камера столкновений. Смесь пяти пептидов сканируется для получения спект-
ров пяти (М + Н)' ионов, обозначенных на рисунке как (Р -Р5). После сканирования только один
отобранный ион (Р ) проникает в камеру столкновений. Фрагменты полученные в ре-
зультате деструкции иона предшественника Р( (часть молекул которого сохраняет целостность),
затем анализируются с помощью МС2 для записи спектра ионного продукта (Biemann, 1992)
Чувствительность ИЦР-ФП-МС
настолько высока, что данный метод с
успехом применяется для исследова-
ния одиночных макроионов, несущих
множественный заряд.
Б1.5.7. Тандемная
масс-спектрометрия
Для получения структурной информа-
ции с помощью масс-спектрометрии
молекула должна претерпеть фрагмен-
тацию одной или нескольких связей с
образованием ионов, значения m/z ко-
торых может быть соотнесено со струк-
турой. Вспомним, что методы «мягкой»
ионизации, такие как ББА, ИЭЛДМ и
ИЭР, производят одиночные молеку-
лярные ионы, энергии которых недоста-
точно для фрагментации. Однако фраг-
ментация становится достижимой при
использовании специальной камеры
столкновений в тандемной масс-спект-
рометрии (МС-МС).
Самым типичным экспериментом
в МС-МС является полное сканирова-
ние ионных продуктов. Ионы с опре-
деленным значением m/z отбираются
в первом масс-спектрометре (MCI,
рис. Б1.13). Затем они попадают в каме-
ру столкновений (КС), обычно напол-
ненную гелием, аргоном или ксеноном.
Ионы активируются в результате стол-
кновений и фрагментируются. Ионные
продукты фрагментации анализируют-
ся вторым масс-спектрометром (МС2),
который рассчитан на сканирование оп-
ределенного диапазона масс. Посколь-
ку для записи всего спектра требуется
всего лишь 1-2 мин, можно настроить
MCI на регистрацию следующего иона
предшественника и получить его спектр
столкновений и т. д.
Существуют два типа таких инстру-
ментов. Первый представляет собой
два масс-спектрометра, работающих в
тандеме. Часто используются комбина-
ции двух квадруполей для масс-анали-
за или двух магнитных анализаторов,
либо комбинации из одного магнит-
ного и одного квадрупольного спект-
рометра. С этой точки зрения связка
магнитного и электрического секторов
может рассматриваться как тандемная
масс-спектрометрия (МС с двойной
фокусировкой).
Инструменты второго типа состо-
ят из анализатора, который способен
хранить ионы: масс-спектрометры
с ионно-циклотронным резонансом и
квадрупольной ионной ловушкой. Эти
устройства позволяют отбирать опре-
деленные ионы, выбрасывая из ловуш-
ки все остальные. Далее происходит
возбуждение отобранных ионов и их
фрагментация в течение определенно-
го времени, после чего фрагменты ио-
нов можно наблюдать в масс-спектре.
Этот процесс может быть повторен,
чтобы проанализировать фрагменты
фрагментов.
Альтернативный подход заключа-
ется в использовании тройной квад-
рунольной схемы (рис. Б 1.11 и па-
раграф Б 1.5.2), которая, несмотря на
низкую чувствительность и узкий
диапазон масс, является гораздо более
дешевой. Первый квадруполь QI ис-
пользуется в качестве масс-спектро-
метра, отобранный пик вводится в ка-
меру столкновений (КС), а продукты
деструкции анализируются во втором
квадруполе QIT.
В заключение упомянем «гибрид-
ные» инструменты, называемые так по-
тому, что в них сочетаются магнитные
сектора, квадруполи и время-пролет -
ные инструменты в линейной и ортого-
нальной проекциях.
ПРИЛОЖЕНИЕ 1. Разрешение
и точность определения
массы
Разрешение определения массы
Способность к разделению масс опре-
деляется разрешающей способностью
масс-спектрометра. Разрешение R здесь
определяется как R = т/\т, где Ат —
разность масс двух соседних пиков т и
т + Ат с равной интенсивностью и пе-
рекрыванием сигналов в 10%. Разреше-
ние, равное 100000, позволяет отличить
ион с массой 100000 Да от иона с мас-
сой 100001 Да, т. е. с точностью одной
стотысячной.
s +vdc + V^os cot
Рис. Б1.14. Схема тройной квадрупольной системы. QI и QII — первая и вторая квадрупольные
системы. Третий квадруполь q используется в качестве камеры столкновений (коллизионная
ячейка). S — источник; D — детектор; rf — радиочастота. Такая конфигурация имеет название
Q(qQ2 (Gordon, 2000)
Поскольку пики в масс-спектрах
имеют вполне определенную ширину и
форму, необходимо определить степень
перекрывания двух соседних пиков для
оценки разрешения. Существуют два
общепринятых способа определения
перекрывания, и необходимо знать,
какой из них используется, когда ссы-
лаются на разрешение в реальном
масс-спектре (рис. Л). Первый — это
так называемый критерий 10%-ного
перекрывания, когда каждый из двух
смежных пиков вносит 5%-ный вклад
в область перекрывания. Второй — это
критерий «полуширины на половине
максимума». Разрешение в этом слу-
чае равно массе пика в Да, деленной на
ширину в Да, измеренную на половине
высоты пика.
Полезно запомнить, что величина
разрешения, определенная с помощью
критерия полуширины на половине
максимума, примерно в два раза боль-
ше величины, полученной при исполь-
зовании критерия 10%-ного перекры-
вания. Разрешение, равное 1000 при
использовании критерия полушири-
ны на половине максимума, примерно
равно 2000. Надо отметить, что обна-
ружение изотопов в протонированном
спектре возможно лишь при очень вы-
соком разрешении с использованием
ИЦР-ФП масс-спектрометров (пара-
граф Б 1.6.6).
Точность измерения массы
Точность измерения массы определя-
ется как разность между измеренной и
расчетной массами иона. Она выража-
ется в процентах от измеренной мас-
сы (например, молекулярная масса =
= 10000 ± 0,01 %) или в долях на мил-
лион (например, молекулярная мас-
са = 10000 ± 100 миллионных долей).
По мере увеличения массы абсолютная
ошибка в процентах или в миллионных
долях также будет пропорционально
увеличиваться.
Обычно для точного измерения
массы не требуется максимального раз-
решения, при условии, что наблюдае-
мый сигнал при данной массе связан
только с одним типом молекул. Основ-
ным фактором, ограничивающим эту
точность для больших биологических
макромолекул, является перекрывание
пиков. При использовании ИЛДМ по-
лучаются следующие пики: [М + Н]*,
[М + Na]* и [М + матрица]*. Однако
в подходящих условиях, когда ионы об-
разца полностью отделены от фоновых
ионов, измерения, сравнимые по точ-
ности, могут быть сделаны и при более
низком разрешении
Моноизотопная масса
В большинстве химических элемен-
тов присутствуют природные изотопы,
каждый из которых обладает уникаль-
ной массой и природным уровнем обо-
гащения. Моноизотопная масса — это
масса наилегчайшего стабильного
изотопа элемента. Например, сущест-
Рис. А. Два определения разрешающей спо-
собности в масс-спектрометрии (подробности
см. в тексте) (Carrand Burlingame, 1996)
вуют два основных изотопа углерода:
|2С и 13С, с массами 12 000 000 (моно-
изотопная) и 13003355 и природным
содержанием каждого 98,9% и 1,1%,
соответственно. Аналогично есть два
природных изотопа азота — 14N и 15N, с
массами 14 003074 (моноизотоппая) и
15 000 109 и их соотношением 99,6% и
0,4 %, соответственно. Моноизотопный
пик означает, что все атомы углерода
в молекуле являются атомами 12С, все
атомы азота — 14N, все атомы кислоро-
да — ,бО и т. д. Моноизотоппая масса
молекулы, следовательно, получается
сложением моноизотопных масс всех
ее элементов.
Измеренная масса
Когда измерения проводятся набольших
статистических ансамблях молекул, то
они, как правило, содержат не только на-
илегчайшие изотопы элементов, по так-
же один или более атомов тяжелых изо-
топов. Вклад пиков тяжелых изотопов в
молекулярный ионный кластер зависит
от суммы значений содержание-вес для
каждого элемента. Теоретическая веро-
ятность появления кластеров этих изо-
топов может быть точно вычислена с по-
мощью следующего выражения:
{а + Ь}"'. (Л)
Разрешение = 25 000
Максимальная масса - 2530,91
Средняя масса = 2531,67
Моноизотопная масса
2524 2525 2526 2527 2528 2529 2530 2531 2532 2533 2534
4-------
2535 2536 2537
2536 2539
2524 2525 2528 2527 2526 2529 2530 2531 2532 2533 2534 2536 2536 2537 2536 2539
Рис. !>. Кластер молекулярных ионов дляокислеинойр-цепиинсулина(формулаС,)7Н1-|К25О4(.8)),
показанный с различным разрешением. Асимметрия кластера становится менее очевидной по
мере уменьшения разрешения, при этом масса наибольшего пика и средняя масса становятся
практически идентичными (Carr and Burlingame, 1996)
где а — это процентное естественное со-
держание легкого изотопа, b — процент-
ное естественное содержание тяжелого
изотопа, а т — число атомов элемента
в молекуле. Вычисления показывают,
что для небольших молекул, таких как
«-бутан (С4Н|()), существует некоторая
вероятность (-4%), что одна молекула
природного «-бутана будет содержать
атом 13С. Вероятность нахождения двух
или трех таких атомов в одной молеку-
ле пренебрежительно мала. Для био-
логических макромолекул, состоящих
из нескольких сотен атомов углерода
и азота, распределение изотопов стано-
вится крайне сложным, однако может
быть рассчитано с помощью коммерче-
ских программ.
Средняя масса
Средняя химическая масса элемен-
та — это просто сумма масс всех его ста-
бильных изотопов с учетом содержания
(например, 98,9% для 12С и 1,1% для
13С дадут среднюю массу 12,011 для уг-
лерода). Таким образом, средняя масса
молекулы равна сумме средних химиче-
ских масс ее элементов.
Соотношения моноизотопной, сред-
ней массы и массы наибольшего пика
показаны на рисунках В и В для белков
с массами 2500 и 25000. Важным след-
ствием вклада пиков тяжелого изотопа
является то, что для пептидов с массой
больше чем 2000 Да, пик, соответству-
ющий моноизотопной массе, более не
1’п< . 15. Молекулярный ионный кластер белка вируса и.ммунодиффицита человека HIV-p24
(формула С ,,1JH|W).,N.|||.S1.1), показанный с различным разрешением. Положение моноизотопной
массы обозначено стрелкой (Carrand Burlingame, 1996)
является преобладающим в изотопном
кластере (см. рис. 15).
С повышением молекулярной мас-
сы самый большой пик сдвигается
по отношению к пику моноизотоп-
ной массы. При значениях масс выше
8000 Да вклад моноизотонной массы в
спектр становится пренебрежительно
малым (см. рис. В). Использование
той или иной массы для характеристи-
ки образца зависит от массы вещества
и разрешающей силы масс-спектро-
метра. Отметим, что при очень высо-
ком разрешении становятся видимы-
ми сателлитные пики (см. параграф
Б2.2.1).
61.6. Перечень ключевых идей
° Масс-спектрометр не измеряет абсолютную массу. Инструмент должен быть
калиброван с помощью стандартных веществ, чьи значения массы известны
с большой точностью.
* Техника ионизации с электрораспылением (ИЭР) позволяет получать интакт-
ные ионы из образца в растворе при атмосферном давлении путем распыления
очень разбавленного раствора из тонкой иглы в градиенте электростатического
поля с напряжением в несколько кВ.
- Уникальным свойством процесса ИЭР является образование молекул с мно-
жественным зарядом. ИЭР — наиболее мягкий метод ионизации, не приводя-
щий к фрагментации молекул.
J Техника ионизации лазерной десорбцией в матрице (ИЯД М) дает возможность
получать интактные ионы из образца, смешанного со специальным материалом
матрицы, который поглощает лазерное излучение.
Литература для дальнейшего чтения
Исторический обзор
Griffiths I. W. J. J. Thomson — the centenary of his discovery of the electron and his
invention of mass spectrometry. Rapid Commun. Mass Spectr, 1997,11, 2-16.
Comisarow M. B. and Marshall A. G. The early development of Fourier transform ion
cyclotron resonance (FT-ICR) spectroscopy. J. Mass Spectr., 1996, 31, 581-585.
Методы ионизации
Smith D. R., LooJ. A., Loo R. R. 0., Busman M. and Udseth H. R. Principles and practice
of electrospray ionization — mass spectrometry for large polypeptides and proteins. Mass
Spectr. Rev., 1991.10, 359-451.
Muddiman D. C., Gusev A. I. and Hercules D. M. Application of secondary ion and
matrix-assisted laser desorption-ionization time-of-flight mass spectrometry for the
quantitative analysis of biological molecules. Mass Spectr. Rev., 1995.14, 383-429.
Gordon D. В. Mass spectrometric techniques. In Principles and Techniques of
Practical Biochemistry, Ch. 11., ed. K. Wilson and J. Walker Cambridge University
Press, 2000.
Инструменты и передовые технологии
Caprioli R. M. and Suter M. J.-F. Mass Spectrometry, Ch.4. In Introduction to
Biophysical Methods for Protein and Nucleic Research, Ed. Academic Press.
Amster I. J. Fourier transform mass spectrometry. J. Mass Spectr., 1996. 31,
1325-1337.
Hofmann E. Tandem mass spectrometry: a primer. J. Mass Spectr., 1996, 31,
129-137.
Dienes T., Pastor J. S., Schurch S., Scott J. R., Yao J., Cui S. and Wilkins C. L. Fourier
transform mass spectrometry — advancing years (1992 — mid.1996). Mass Spectr. Rev.,
1996,15, 163-211.
GuilhausM., Mlynski V. and Selbi D. Perfect timing: time-of-flight mass spectrometry.
Rapid Commun. Mass Spectr., 1997,11, 951-962.
Belov M. E., Gorshkov M. V., Udeseth H. R., Anderson G. A. and Smith R. D. Zeptomole-
sensititivity electrospray ionization — Fourier transform ion cyclotron resonance mass
spectrometry proteins. Anal. Chem., 2000. 72, 2271-2279.
Guilhaus M., Selby D. and Mlynski. Orthogonal acceleration time-of-flight mass
spectrometry. Mass Spectrometry Reviews, 2000,19, 65-107.
ГЛАВА Б2
СТРУКТУРНО-ФУНКЦИОНАЛЬНЫЕ
ИССЛЕДОВАНИЯ
52.1. Структура и функция
бэлкое
Ионизация электрораспылением (ИЭР)
и ионизация лазерной десорбцией в
матрице (ИЛДМ) находят все большее
применение в масс-спектрометрии бел-
ков. Эти два метода ионизации широко
используются для изучения процесса
сворачивания белков, картирования
их функций, описания нековалентных
белковых комплексов и многих других
приложений (комментарий Б2.1).
Б2.1.1. Определение массы
Благодаря методам ИЭР и ИЛДМ ана-
лиз масс-белков превратился в рутин-
ную процедуру. По ряду причин пеп-
тиды и белки особенно подходят для
этих методов ионизации, которые были
впервые разработаны и продемонс-
трированы на молекулах именно этого
типа. На рисунке Б2.1 приведен участок
масс-спектра цитохрома с и миоглобина
лошади, полученных с помощью ИЭР-
ФП-масс-спектрометрии. Концентра-
ция образца составляла 0,4 нМ. Общее
i I,-.' ,:i Б2 I Масс-спектрометрия и рентгеновская кристаллография
До проведения рентгеноструктурных исследований полезно снять масс-спектр образ-
ца. В самом деле, определение структуры белка с атомным разрешением требует значи-
тельных затрат (см. Главу ЕЗ). Масс-спектрометрия позволяет осуществить быструю
проверку того, насколько точно была определена первичная структура белка, а также оп-
ределить чистоту препарата с высоким разрешением. Такая информация особенно полез-
на для белков, полученных с помощью рекомбинантных техник, где существует большое
число источников ошибок, включая непредвиденные мутации, модификации, термина-
цию и протеолитическую деградацию. Простое измерение массы сберегает время и яв-
ляется настолько информативным, что имеет смысл получать спектр масс почти каждого
белка перед применением рентгеновской кристаллографии. Это особенно важно для тех
из них, которые содержат особые аминокислотные остатки, например, селен-метионин,
для рентгеновской кристаллографии, 13С и/или l5N для ЯМР и -^для нейтронной кри-
сталлографии и малоуглового нейтронного рассеяния. В последнем случае доля вклю-
чения дейтерия в биологические макромолекулы, как правило, определяется с помощью
масс-спектрометрии.
m/z
[М + 17Н]17+
а б
1’нс. 112.1. Участки масс-спектра цитохрома с (а) и миоглобина лошади (б), полученных с помо-
щью ИЭР-ИЦР-ФП-масс-спектрометрии (Belov et al. 2000)
число исследованных молекул равня-
лось 135 зептомолей, что составляет
примерно 80000 молекул t
рии I >”.2 I.
На рисунке 1)2.2 приведен ИЛДМ-
масс-спектр [Arg8]-Ba30iipecciiHa с вы-
соким разрешением. Основной пик при
m/z =1084,446 — это «моноизотопный»
пик (см. Приложение Б1) интактного
протонированного пептида. Три боль-
ших пика, каждый из которых отлича-
ется от соседнего на 1 Да, соответствуют
природным редким изотопам углерода,
присутствующим в молекуле в коли-
честве одного пли нескольких, среди
которых наибольший вклад вносит 13С
с содержанием 1,108%.
Пример вычисления молекулярной
массы белка из его масс-спектра дан
в Приложении 1 этой главы.
koi.-j-.ieiiiapia: Б2.2 От наномолей
к зептомолям
Напоминаем доли молей веществ, ко-
торые используются в сегодняшней масс-
спектроскопии
10 9 нано п
10 12 пико р
10 15 фемто f
Ю is атто а
10’21 зспто z.
1084.4
100-1
1085.4
1086.4
1087.4
1083 1084 1085 1086 1087 1088 1089
Рис. Б2.2. ИЛДМ-масс-спектр [Аг§8]-вазопрессина, полученный с высоким разрешением:
а — узкие полосы в диапазоне m/z от 1080 до 1090 и б — одна из полос в растянутом диапазоне,
с тем чтобы продемонстрировать разрешение (1/100000) (Li et al., 1994)
Б2.1.2. Нековалентные комплексы
Нековалентные взаимодействия, вы-
сокоспецифичные в биологических
макромолекулах, представляют фунда-
ментальный интерес для исследования
структурно-функциональных отноше-
ний. Физиологически активные формы
многих белков являются мультимера-
ми, где активный центр часто распола-
гается в области контакта субъединиц.
Сила нековалентных взаимодействий
обычно обусловлена множеством срав-
нительно слабых связей и может ши-
роко варьировать. Она выражается
константой диссоциации которая
зависит от растворителя. Очевидно, что
для детектирования слабосвязанных и
чувствительных к температуре комп-
лексов необходимы самые мягкие ус-
ловия, которые можно достичь только
с помощью ИЭР ( комментарии Б2.3).
В настоящее время для нескольких
нековалентных комплексов показано,
что они остаются интактными в ИЭР-
МС экспериментах. К ним относятся ме-
таллы, гемовые группы и пептиды, свя-
занные с белком, а также мультимерные
белковые комплексы, олигонуклеотиды,
комплексы фермент-субстрат, рецептор-
лиганд н большие комплексы РНК- бе-
лок.
Одной из наиболее впечатляющих
иллюстраций ИЭР-МС стало наблю-
дение очень прочных комплексов меж-
ду белками и другими молекулами.
ПЭР-спектр человеческого цитоплаз-
матического рецептора (FKBP) де-
монстрирует наличие (М + 7Н)7' иона
при m/z =1688.7 (рис. Б2..3 а). После до-
бавления лекарства — иммуносупрессо-
ра (М = 804 Да) появляется новый пик
при m/z = 1803.1, соответствующий ком-
плексу FKBP-FK506 (1:1) с зарядом 7*
(рис. Б2..3 б). Тот же эффект наблюдает-
ся с рапамицином (М = 913 Да). В этом
случае появляется возможность оце-
нить отношение их констант связыва-
ния из относительной величины пиков
при добавлении в смесь рапамицина в
соотношении 1:1 (рис. Б2.3 в). Кроме
того, в обоих случаях детектировались
значительные количества свободного
FKBP в масс-спектрах. Используя эту
методологию, удалось наблюдать за
гидролизом гекса-М-ацетилглюкозами-
на лизоцимом куриного яйца.
Б2.1.3. Сворачивание и динамика
белков
Скорость, точность и высокая чувст-
вительность ИЭР и ИЛДМ исполь-
зовались в разработках различных
подходов к изучению процесса свора-
чивания белков, их динамики и функ-
ционального картирования, основан-
ных на масс-спектрометрии. Во многих
случаях масс-спектрометрия позволяла
। Комментарий Б2.3. Детектирование
, слабосвязанных комплексов
. В большинстве случаев свойства раство-
| ра, необходимые для поддержания интак-
' тных комплексов, не являются оптималь-
ными для ИЭР. Поэтому для достижения
, максимальной чувствительности при ана-
| лизе полипептидов берут раствор с pl 1 2-4
। для положительной и pH 8-10 для отри-
цательной ионизации. Многие белковые
. комплексы денатурируют в растворе со
, значениями pH, лежащими вне диапазона
, pH 6-8. То есть совершенно очевидно, что
растворы белков для ИЭР-МС должны
поддерживаться в условиях, близких к фи-
энологическим с нейтральным pH и при
соответствующий температуре, для того
чтобы белки оставались в состоянии, близ-
: ком к нативному. Однако для ИЭР-МС с
; растворами белка, имеющими нейтраль-
ный pH, требуются масс-спектрометры с
увеличенным диапазоном m/z по сравне-
: нию с инструментами для работы в тради-
: ционных условиях.
До сих пор не совсем понятно, какие
из известных слабосвязанных комплек-
' сов, существующих в растворе, можно
наблюдать с помощью ИЭР-МС и какая
минимальная связывающая сила в этих
комплексах достаточна для этого. Литера-
турные данные дают право полагать, что
ИЭР-МС наблюдения таких слабосвязан-
ных систем в некоторой степени отражает
природу взаимодействий в конденсиро-
ванной фазе. Однако результаты ИЭР-МС
экспериментов показывают, что каждая
биомолекулярная система обладает свои-
ми характерными экспериментальными
чертами, и опыт, полученный при иссле- i
довании одного белкового комплекса, не
обязательно будет полезен при исследова-
| НИИ свойств другого.
получать данные о функциональных
свойствах белка в дополнение к тем,
что были получены с помощью тради-
ционных техник, таких как круговой
дихроизм, ядерный магнитный резо-
нанс и флуоресцентная спектроскопия.
a
в
Рис. 152.3: а — масс-спектр, полученный
с помощью ионизации электрораспылением
белка, связывающегося с человеческим ци-
топлазматическим рецептором (FKBP) цик-
лоспорина при pH 7,5. Молекулярная масса
FKBP равна И 812 Да. Этот массив ионов,
несущих множественный заряд, простирает-
ся от (Л/ + 6Н) 6' до (Л/ + 8Н) “’для FKBP;
б — ионизация распылением FKBP с веще-
ством FK506 (М = 804 Да); в — конкурентное
связывание FKBP с FK506 и рапамицином
(RM) (Ganem et al., 1991)
Кроме того, относительная быстрота
и легкость, с которыми ИЭР и ИЛДМ
могут быть использованы для получе-
ния очень точной информации о моле-
кулярной массе очень ограниченного
количества исследуемого материала,
сделали возможным применение этих
методов там, где не так-то просто до-
быть необходимые сведения с помощью
других методов.
Свернутые и развернутые состояния
В начале 1990-х гг. исследователи при-
шли к заключению, что свернутые и
развернутые белки проявляют разнос
распределение зарядов в ИЭР-спектрах.
В спектрах нативных белков, получен-
ных элсктрораспыленнием из раство-
ров, это распределение обычно наблю-
дается в достаточно узком диапазоне
и с низким суммарным зарядом, тогда
как белки, находившиеся до ионизации
в денатурирующих условиях, обладают
более широким распределением заряда
в спектре (рис. Б2.4).
Считается, что разница в распреде-
лении зарядов связана с доступностью
способных к ионизации групп. Вполне
возможно, что в развернутом состоянии
основные аминокислоты (остатки Apr,
Лиз, Гис) легче аккумулируют заряд
(см. рис. 152.4).
Свернутое состояние белка в рас-
творе можно наблюдать через распре-
деление зарядов во время ИЭР. Так, в
случае с разворачиванием цитохрома с
(М = 12 360 Да) при низком значении
pH наблюдаемые изменения отчет-
ливо указывают на высокую коопера-
тивность этого процесса (рис. 152.5 а).
И наоборот, разворачивание апо-миог-
лобина (М = 16951,5 Да), миоглобина,
потерявшего группу гема, сопровожда-
ется постепенным сдвигом максимума
Свернутый белок
Рис. 1>2.1. Распределение зарядов в свернутом и развернутом белке при использовании ИЭР
(Winston and Fitzgerald, 1997)
Рис. B2..5: a - ИЭР масс-спектры цитохрома с, записанные при различных pH: (1) pH 8,5, (2) pH
3,2, (3) pH 2,7, (4) pH 2,6 и (5) pH 2,0. Величина pH регулировалась добавлением гидроксида аммо-
ния и/или уксусной кислоты; б — ИЭР-масс-спектры апо-миоглобина, записанные при различных
pH: (1) pH 8,5, (2) pH В2,8, (3) pH В2,3, (4) pH 2,9 и (5) pH 2,5. Величина pH регулировалась опи-
санным выше способом (Копеппапп and Douglas, 1998)
hMb11
0.07 в
hMb20
hMb11
аМЬ20
15.1 в
О
hMb20
аМЬ20
ll pill
600 800 1000 1200 1400 1600 1800 2000
m/z
Рис. 152.(5. Иллюстрация ИЭР-МС разрешен-
ной во времени денатурации миоглобина в
кислых условиях. ЬМЫ1 — свернутый комп-
лекс, ЬМЬ20 — частично развернутый интерме-
диат гем-миоглобина, АтЬ20 — развернутый!
миоглобин. Затухание интенсивности пика,
принадлежащего интермедиату, происходило
примерно за 0,4 сек, что хорошо коррелирует
со временем жизни, определенным в растворе
(Konerman and Douglas, 1998)
наблюдаемого распределения зарядов
(рис. Б2.5 б). Эти результаты свиде-
тельствуют, что ИЭР-МС можно рас-
сматривать как общий метод для оцен-
ки кооперативности переходов белка
в свернутое состояние.
Интересно, что с помощью ИЭР-МС
удалось наблюдать процесс, напомина-
ющий сворачивание и разворачивание
цитохрома с в вакууме, что заставляет
поднять вопрос о роли воды в сворачи-
вании белков.
Интермедиаты процесса
сворачивания белка
Тот факт, что распределение зарядов
зависит от укладки белка, дал иссле-
дователям основание предложить но-
вый подход для изучения процесса
сворачивания, использующий ИЭР-
МС с разрешением во времени. Метод
применялся для наблюдения процес-
сов сворачивания цитохрома с и ми-
оглобина с временным разрешением
0,1 сек. Конформационные интерме-
диаты между свернутым и разверну-
тым состояниями в случае цитохрома
с обнаружены не были. Напротив, экс-
перимент с миоглобином показал при-
сутствие интермедиатов во время дена-
турации в кислых условиях (рис. 152.(5).
Результаты этих опытов дают только
качественную информацию. Извлече-
ние количественной информации из
ИЭР-масс-спектров стало возможным
после разработки процедуры деконво-
люции распределения зарядов. Ниже
мы кратко ее опишем.
ИЭР-масс-спектр любого белка
можно представить линейной комбина-
цией различных распределений зарядов,
называемых «базисными функциями»,
которые могут быть аппроксимирова-
ны с помощью гауссова распределения.
Изменения интенсивности выражают-
ся через средневзвешенные коэффици-
енты, которые учитывают относитель-
ный вклад в суммарное распределение
зарядов. Таким образом, наблюдаемый
ИЭР-масс-спектр можно рассматривать
как сумму вкладов каждой из белковых
конформаций (конформеров). Рису-
нок 152.7 отображает ИЭР-масс-спект-
ры холо-миоглобина (/гМЬ) и апо-миог-
Рис. 152.7. ИЭР-масс-спектры положительных ионов холо-миоглобина (ЛМЬ) (а-д)
и апо-миоглобина (<?МЬ) (е-к), полученные при pH 7,4 (а, е), 4,5 (б, ж), 4,0 (в, .?), 3,5 (г,
и) и 2,5 (d, к). Пики интактного иона ЛМЬ обозначены сплошными квадратиками (Dobo and
Kaltashov, 2001)
лобина (аМЬ) в большом диапазоне pH
2,5-8,6.
При pH 4,5 и выше спектры /гМЬ
проявляют распределение зарядов в
очень узком диапазоне. Спектр содер-
жит только два пика зарядовых состоя-
ний: +8 и +9. Понижение pH раствора
до значения 4 приводит к масштабным
конформационым изменениям, заклю-
чающимся в появлении сильно прото-
нированных (с низким значением m/z)
ионов белка и частичной диссоциации
гемовой группы (рис. 152.7 «). Даль-
нейшее понижение pH до 2,5 приводит
к повышению среднего заряда ионов
белка и исчезновению ионов комплекса
гемм-белок.
В отличие от /?МЬ, спектр «Mb име-
ет мультимодальный характер даже
при нейтральных pH (рис. 152.8 е).
В дополнение к ионам с зарядами +9
и +8 в спектре заметно наличие пиков
других ионов с зарядами от +10 до +23
(рис. 152.7).
Число заряженных состояний (п) Число заряженных состояний (п)
Рис. Ь2.К. График кривых аппроксимации распределения зарядов положительно заряженных
ионов в ИЭР- масс-спектре ЛМЬ (a-в) и «Mb (г-е), полученных при pH 7,4 (а, <?), 4,0 (б, д) и 2,5
(в, е). Экспериментальные точки показаны квадратиками ( для интактных ионов ЛМЬ и □ для
ионов аМЬ). Гауссовы кривые представляют средневзвешенные базисные функции, использо-
ванные для анализа кривой (зачернены для ЛМЬ). Толстыми сплошными линиями обозначены
суммы средневзвешенных базисных функций (Dobo and Kaltashov, 2001)
При pH 2,5 спектр аМЬ неотличим
от /гМЬ, что полностью согласуется с
ожиданиями, основанными на том, что
любые взаимодействия между гемовой
группой и дестабилизированным кис-
лотой белком будут минимальны.
Результаты аппроксимации некото-
рых распределений зарядов с исполь-
зованием базисных функций показаны
на рисунке Б2.8. Только одна базисная
функция использовалась для аппрокси-
мации спектра /гМЬ при pH 4,5 и выше,
тогда как для анализа данных, полу-
ченных в диапазоне pH от 4,5 до 3,5,
необходимы три такие функции. Нако-
нец, чтобы проанализировать данные,
полученные при низких значениях pH
для /гМЬ и аМЬ, придется добавить чет-
вертую базисную функцию. Эти четыре
основные функции приписывают че-
тырем различным конформационным
состояниям белка (в порядке его разво-
рачивания): нативному (N), так называ-
емому «pH 4-интермедиату» (/), растя-
нутой конформации (£) и развернутому
состоянию (U). Все характерные черты
спектра ИЭР, интерпретируемые как
линейная комбинация ионных вкладов
N, I, Е и U, полностью согласуются с
картиной разворачивания в кислых ус-
ловиях, полученной с помощью многих
других экспериментальных методов.
Эти эксперименты являются пре-
красной! иллюстрацией возможностей
11ЭР-масс-сиектрометрии для наблюде-
ния за интермедиатами сворачивания.
Б2.1.4. Секвенирование белков
Отсутствие фрагментации белков при
мягкой ионизации очень полезно для
точного определения молекулярной
массы, но не дает структурной инфор-
мации, что является ее большим недо-
статком. Сначала эту проблему решали
с помощью техники ББА, которая, к
сожалению, применима для исследова-
ния относительно коротких пептидов и
требует большого количества образца
(2-50 нМ).
Наиболее подходящей техникой
для секвенирования белков оказалась
тандемная масс-спектрометрия (па-
раграф Б2.6.7). С помощью МС-МС
можно определять не только все 20
основных аминокислот, но и неизвест-
ные модифицированные аминокислоты
в соответствии с их массой. МС-МС
характеризуется высокой скоростью,
чувствительностью и позволяет напря-
мую анализировать смеси пептидов.
Был разработан ряд масс-спектро-
метрических подходов для секвени-
рования больших пептидов и белков.
К ним относятся: тандемные МС под-
h'or.o и : ।к и 1 1 Деградация по
Эдману
В 1950 г. П. Эдман предложил химиче-
ский метод для последовательного от-
щепления аминокислотных остатков от
N-копца полипептида или белка. Серия
химических реакций, известных как де-
градация по Эдману, до сих пор остается
наиболее эффективной техникой секвени-
рования полипептидов. Для ее примене-
ния необходимо наличие свободной амино-
группы naN-конце. Пептиды, чья концевая
аминогруппа блокирована (например фор-
матирована, или ацилирована), должны
быть расщеплены энзиматически, либо хи-
мически. Деградация по Эдману проводит-
ся в автоматическом анализаторе. Сегодня
для определения аминокислотной последо-
вательности требуется совсем немного бел-
ка — от 10 до 100 нг.
ходы, комбинируемые с энзиматиче-
ской и химической деградацией для об-
разования олигопептидов (<3 кДа) на
первом этапе и с масс-спектрометрией
полученного продукта на втором или
использование ИЭР-ФП-МС в каче-
стве первого этапа деградации в комби-
нации классической деградации по Эд-
ману ( !,и\|\|( (I С тандемной
масс-спектроскопией.
Вычисление аминокислотной
последовательности белка
Для получения структурной информа-
ции с помощью масс-спектрометрии
изучаемая молекула должна претер-
петь фрагментацию одной или более
связей с образованием ионов. Получен-
ные в результате этого пептиды обла-
дают следующими характеристиками:
их линейный скелет образован только
повторяющимися связями а-амино-
кислота-амид, и замещающие группы
каждого а-углерода представляют со-
Таблица Б2.1. Массы аминокислотных остатков [-NH-CHR-CO-]
Аминокислота Буквенный код Масса (Да)
трех ОДНО
Глицин Gly (5 57
Аланин Ala A 71
Серин Ser S 87
Пролин Pro P 97
Валин Vai V 99
Треонин Thr T 101
Цистеин Cys c 103
Изолейцин He I ИЗ
Лейцин Leu L 113
Аспарагин Ash N 114
Аспарагиновая кислота Asp D 115
Глютамин Gin Q 128
Лизин Lys к 128
Глютаминовая кислота Glu E 129
Метионин Met M 131
Гистидин His H 137
Фенилаланин Phe F 147
Аргинин Arg R 156
Тирозин Tyr Y 163
Триптофан Trp W 186
бой 20 естественных аминокислот. При
использовании методов «мягкой» ио-
низации образуются одно- или поли-
протонированные молекулярные ионы,
у которых недостаточно энергии для
фрагментации. Однако фрагментация
достижима, если воспользоваться спе-
циальной камерой столкновений в тан-
демной масс-спектрометрии (параграф
Б 1.5.7) или прибегнуть к бомбардиров-
ке образца быстрыми атомами Аг или
Ne (параграф Б1.4.4).
Несмотря на то что фрагментация
может происходить по-разному, суще-
ствуют зоны предпочтительного рас-
щепления пептидной связи. Единовре-
менная потеря одного аминокислотного
остатка сопровождается образованием
пиков в спектре, которые отличаются
друг от друга на массу одной аминокис-
лоты минус Н,,О (так называемая масса
остатка — табл. Г>2.1). Это различие в
массе каждых последующих пиков яв-
ляется точным отражением первичной
структуры пептида.
Фрагментация идеального пепти-
да представлена на рисунке Б2.9. Рас-
щепление связей в точках, указанных
на рисунке, приводит к образованию
фрагментов с массами ап, Ьп\\ сп, если за-
ряд остается на N-концевом фрагменте.
Исключение составляет ион сп, к массе
которого нужно добавить два Дальтона,
поскольку он сохраняет исходный про-
тон и присоединяет еще один с другой
стороны пептидной связи.
+2Н
+2Н
х3 Уз z3 x2 У2 Z2 X, y, Zj
+2H ”
*3
h2n-ch
I2
--NH--CH--C--NH--CH--C--NH--CH-CO2H
ai th
+2H
Cl
+2H
a>2 bi
a3 b3
+2H
c3
Puc. 1)2.9. Фрагментация идеально протонированного пептида (Biemann, 1992)
Ноны .v(, уп и гп образуются, когда
заряд остается на С-конце с двумя ато-
мами водорода, добавленными к иону
уп, аналогично случаю с ионом сп. Имея
одну последовательность ионных фраг-
ментов, мы можем рассчитать амино-
кислотную последовательность пептида,
за исключением лейцина и изолейцина,
а также лизина и глутамина, которые не-
льзя отличить таким образом. Обычно
получается не одна серия ионов, а набор
перекрывающихся N- и С-терминальных
ионов, они и используются для опреде-
ления полной последовательности.
Процесс фрагментации протониро-
ванных пептидов в настоящее время на-
столько понятен и воспроизводим, что
был выработан набор следующих эмпи-
рических правил.
1) Пик с наибольшей массой
в ББА-спектре представляет (М + Н)'.
2) Разность масс (А) между после-
довательными ионами отражает массу
аминокислотного остатка (ао).
3) Сумма всех аминокислотных
остатков должна давать полную моле-
кулярную массу.
Ниже показано приложение этих
правил в случае с ББА-МС пептида со
значениями m/z, равными 128; 185; 299;
396 и 528.
Самый большой пик в ББА-спектре
имеет т/г = 528, отсюда М = 527. С по-
мощью набора т/г данных можно по-
лучить пептидную последовательность
следующим образом:
т/г 128 185 299 396 527
А 57 114 97 131
ао Gly Asn Pro Met.
Ион с наименьшей массой т/г = 128
является лизином (Lys), отсюда после-
довательность пептида с N-конца будет
следующая:
Met-Pro-Asn-Gly-Lys.
Совокупная масса аминокислотных
остатков равна 527 Да, что соответствует
масс-спектрометрическому значению М.
Б2.1.5. Идентификация белков
в двумерном электрофорезе
Двумерный гель-электрофорез (2-ГЭФ)
дает возможность разделять белки в со-
ответствии с их химической структурой.
В 1970-х гг. идентификация белков в геле
проводилась на основе коэлектрофореза
известных очищенных белков или при
помощи иммунного окрашивания (свя-
зывания белков на электрофореграм-
ме с антителами и последующее спе-
цифическое окрашивание комплекса).
Позднее 2-ГЭФ был усовершенствован
Изолированный
пептид
Его фрагменты
141.10 5 а2 [А,Р ]
169.102 Ь2 [А,Р ]
175.112 у, R
240.137 Ьэ [А,Р] А
288.205 у2 IR
935.471 М,
База данных
141.10 5
169.102
175.112
240.137
288.205
935.471
Рис. 152.10. Принцип «концевого секвенирования». Очищенные пептиды фрагментируются.
Массы легких ионных фрагментов, таких как у(, у2, b и Ь3, плюс точно определенная масса ин-
тактного пептида, используются для «концевого секвенирования». Положение фрагментов и мо-
лекулярная масса позволяют идентифицировать пептид в базе данных (Nielsen et al., 2002)
до такого уровня, что позволяло разре-
шать до 10000 белковых пятен на одном
геле благодаря окрашиванию серебром
и увеличению размера геля (см. Главу
Г5). Разделенные таким способом пятна
идентифицировались при помощью «мо-
лекулярной дактилоскопии» на уровне
фемтомолей посредством ИЛДМ-ВП-
масс-спектрометрии или ИЭР-тандем-
ной масс-спектрометрии.
Высокая чувствительность и про-
пускная способность системы, ос-
нованной на ортогональной ИЛДМ
тандемной масс-спектрометрии (па-
раграф Б1.5.7), обеспечивает автома-
тическое распознавание фрагментов,
соответствующих N- и С-концевым
аминокислотным остаткам. Импульс-
ный характер работы инструментов
позволяет улучшить разрешение в об-
ласти низких значений масс пример-
но на порядок. Эта область в спектре
очень легко интерпретируется, когда
наблюдаемый легкий фрагмент состо-
ит из всего несколько аминокислот.
Как правило, можно определить сум-
му масс второго N-концевого иона (Ь.,
ион), третьего N-концевого попа (bt
ион) и двух С-концевых фрагментов
пептида (у и у2). Учитывая точность
определения массы в нижнем диа-
пазоне, достаточно концевого секве-
нирования одного или двух трипти-
ческих пептидов для идентификации
белка в образце геля из области, сла-
бо окрашенной серебром (несколько
нанограммов белка на полосу). Ри-
сунок 152.10 иллюстрирует принцип
«концевого секвенирования».
Рисунок 152.1 1 позволяет судить об
анализе окрашенных серебром и выре-
занных из двумерного геля фрагментов
с помощью ортогональной время-про-
летной МС. Изучение масс-спектра пят-
kDa
125
pl 5
в
Рис. 1>2.I 1. Анализ окрашенного серебром фрагмента, вырезанного из двумерного геля,
с помощью о-ИЛДМ; а — увеличенная область геля. Вырезанное пятно отмечено стрелкой;
б — масс-спектр пептидов, полученных из пятна; в — тандемный масс-спектр области низких
значений масс наиболее интенсивного иона-предшественника с т/г =1298,762. Расчетные трип-
тические массы пептидов этого белка отмечены звездочкой (Nielsen et al., 2002)
на из геля (см. рис. Б2.11 о) показывает,
что полученная пептидная карта не поз-
воляет найти соответствие в базе бел-
ковых данных. Поэтому был выделен
ион-предшественник tm/z = =1298,762
и получен его тандемный масс-спектр
(см. рис. Б2.1 1 н). Комбинация N- и С-
концевого секвенирования позволила
однозначно идентифицировать пептид
как VHLVGIDIFTGK (Val-His-Len-
Val-Gly-Ile-Asp-Leu-Phe-Thr-Gly-Lys),
а белок — как 5А эукариотический фак-
тор инициации трансляции.
Б2.2. Функциональная
протеомика
В отличие от генома, протеом крайне
динамичен и сильно зависит не только
Клетка или образец ткани
Рис. 152.12. Две стратегии в анализе протеома образцов клеток и тканей: слева — двумерный
гель. Белки разделяются и визуализируются с помощью гель-электрофореза. Индивидуальные
белки выделяются из геля, фрагментируются трипсином и анализируются масс-спектрометри-
чески; справа — жидкостная хроматография. Вся белковая смесь обрабатывается трипсином,
разделяется с помощью жидкостной хроматографии и анализируется масс-спектрометрически
(Godovach-Zimmermann and Brown, 2001)
от типа клетки, но и от ее состояния.
Протеом определяется как совокупное
множество клеточных белков, представ-
ляющих экспрессию генома с посттран-
сляционными модификациями. В эру
постгеномного секвенирования одной
из принципиальных задач науки о жиз-
ни является понимание живых систем
на геномном и протеомном уровнях.
Б2.2.1. Роль масс-спектрометрии
В отличие от геномики, где вклад
масс-спектрометрии был достаточно
ограничен, ее роль в изучении протеома
является поистине ключевой. Обычно
используют две основные стратегии для
высокопроизводительного анализа про-
теома (рис. 152.12). В первой (на рисун-
ке слева) двумерный гель-электрофорез
служит для разделения и количествен-
ного анализа белков. Идентификация
пятен в геле чаще всего производится
через их вырезание, фрагментирование
белка трипсином, экстракцию пепти-
дов и анализ фрагментов с помощью
ИЛДМ-ВП и/или ИЭР-масс-спектро-
метрии.
Во второй стратегии (на рисунке
справа) фрагменты из белковой смеси,
обработанной трипсином, разделяются
путем жидкостной хроматографии, пос-
ле чего пептиды напрямую попадают
в масс-спектрометр.
Недостаток использования дву-
мерных rejieii заключается в том, что
данный метод предполагает денату-
рацию белка с сопутствующим раз-
рушением белок-белковых взаимодей-
ствий. Более тщательное исследование
белков, обычно идентифицируемых
в протеомных исследованиях, пока-
зывает, что комбинацию двумерного
геля и масс-спектрометрии нельзя в
действительности назвать всеобъем-
лющей техникой. Некоторые классы
белков — например, очень кислые или
основные, а также чересчур большие
или слишком маленькие, отсутствуют
в геле, а белки с низким относитель-
ным содержанием в клетке вообще не
детектируются.
Новая стратегия визуализации и ана-
лиза протеома с помощью масс-спектро-
метрии была разработана в начале этого
века. Она основана на масс-спектромет-
рическом анализе поверхностей гелей,
полученных в результате изоэлектри-
ческого фокусирования, и называется
«виртуальным двумерным гелем». Та-
кой подход, когда вместо разделения
белков в геле по размеру, используется
масс-спектрометрия, обладает преиму-
ществом в разрешении и точности по
сравнению с классическим двумерным
гель-ЭФ, и, кроме того, он может быть
легко автоматизирован.
Классический двумерный элек-
трофорез в полиакриламидном геле
(ПААГ) включает в себя изоэлектри-
ческое фокусирование (ИЭФ) в первом
направлении и разделение в соответ-
ствии с размером молекул в присут-
ствии додецил сульфата натрия во
втором направлении (Глава Г5). Ана-
логичным образом в методе скани-
рования поверхности геля, основан-
ного на масс-снектрометрии, белки
десорбируются непосредственно из
ИЭФ-геля. При этом аналитические
данные представляются в виде кар-
тинки, похожей на ту, что получается
в результате окрашивания двумерно-
го геля. Рисунок Б2.13 демонстриру-
ет детектирование белков с помощью
сканирования поверхности ИЭФ-геля
клеточного лизата Е. coli и ИЛДМ-
масс-спектр, соответствующий ука-
занным позициям.
В соответствии с особенностью дву-
мерного геля, когда при каждом зна-
чении pH наблюдаются белки во всем
массовом диапазоне, в масс-спектре
на рисунке Б2.13 б можно увидеть 33
белка. Белки с маленькой массой, ко-
торые необходимы для экспрессии не-
больших открытых рамок считывания
или процессинга, очень часто теряются
при разгонке во втором направлении в
классическом двумерном геле, но легко
детектируются с помощью масс-спект-
рометрического сканирования поверх-
ности ИЭФ-геля. Из рисунка Б2.13 <)
и с, где разрешаются белки с массами
82.1 и 84.6 кДа, а также восемь белков
от 30 до 50 кДа, становится совершенно
очевидным превосходство данного ме-
тода в разрешении и точности по срав-
нению с СДС-ПААГ. Метод позволяет
анализировать белки с массой свыше
100 кДа, а также маленькие полипеп-
тиды (<8 кДа), которые очень часто
теряются при разгонке во втором на-
правлении в СДС геле, если не созда-
ны специальные электрофоретические
условия.
Технологии сочетания двумерного
геля и масс-спектрометрии в настоя-
щее время стали движущей силой про-
теомики и, скорее всего, останутся ею
в ближайшем будущем. Однако, ког-
да-нибудь новые подходы, основанные
на применении антител и белковых чи-
пов, смогут бросить им вызов.
10000 30000 50000 70000 90000 10 ООО 30 000 50 000 70 000 90 000
Молекулярная масса (Да)
Молекулярная масса (Да)
Рис. 52.13. Окрашенный ИЭФ-гель клеточного лизата Е. coli (центр) и ИЛДМ масс-спектр, по-
лученный непосредственно из неокрашенного высушенного геля-дубликата, соответствующий
указанным позициям: а-е — ионы, несущие один и два заряда, наблюдаемые в различных пози-
циях (pl) геля-дубликата. По оси Y отложена относительная интенсивность полос ионов. Ось X
отображает молекулярную массу. Масс-спектры получены в следующих положениях: (а) 3 мм от
основной кромки геля (р! 6.6); 6—15 мм (р! 6.4); в — 22 мм (р! 6.3); г — 30 мм (р! 6.2); д — 55 мм
(р! 5.8). В диапазоне свыше 20 кДа, ось Y расширена в 4 раза; (е) 71 мм (р! 5.6). В диапазоне
свыше 20 кДа, ось Y увеличена в 3 раза. Белки для отмеченных пиков 1, 3, 5-9 подтверждены
деградации по Эдману и идентифицированы как: 1 — yifE, 2 - purE, 3 — artj, 4 — yeaD or gutQ.
5 — oppA, 6 — ptsEI, 7 — ygall, 8 — ppiB, 9 — mdh, 10 — metG or yjcQ (Ogorzalek Loo et al., 2001)
кислоты
Исследование нуклеиновых кислот с по-
мощью ’ масс-спектрометрии отстает от
изучения белков. Основная причина за-
ключается в высоком сродстве нуклеи-
новых кислот к ионам натрия, которые
резко снижают эффективность иониза-
ции. Более того, получение интактных
молекулярных ионов олигомеров, содер-
жащих более двух нуклеотидов, стано-
вится затруднительным, если применять
классические техники ионизации, такие
как электронная и химическая иониза-
ция, ввиду высокой полярности нуклеи-
новых кислот и склонности их ионов к
фрагментации. В 1982 г. была продемон-
стрирована способность ББА техники по-
лучать интактные ноны нуклеотидов, на
основе которой вскоре было предложено
ультраскорос пюе секвсч111 рова!iне оли го-
нуклеотндов. Внедрение ИЭР и ИЛДМ
значительно расширило диапазон масс
в анализе нуклеиновых кислот. Затем по-
следовал заметный прогресс в секвениро-
вании олигонуклеотидов, анализе смесей,
исследовании нековалентных комплексов
и манипуляции с микрообразцами.
Б2.3.1. Анализ смеси
олигонуклеотидов
Казалось, что комбинация жидкостной
хроматографии и техники ионизации
электрораспыл ей нем в масс-спектро-
метрии должна быть очень информа-
тивной в анализе нуклеотидных смесей.
Однако дальнейшее развитие показало
ограниченность такого подхода вви-
ду несовместимых требований двух
методов. Анионно-обменная и обрат-
но-фазная хроматография использу-
ют солесодержащую подвижную фазу
для эффективного разделения, которая
ухудшает качество масс-спектра. Реше-
ние последней проблемы было найде-
но добавлением специальной добавки
(гексафлуро-2-пропанол) в подвижную
фазу. Потенциал нового метода был
продемонстрирован на синтетическом
гомополимере тимидина длиной до 75
оснований, для фрагментов плазм одной
ДНК pBR322 и фосфотиоэфирах анти-
смысловых олигонуклеотидов. Оказа-
лось, что техника наиболее подходит
для характеристики ДНК зондов, ПЦР
праймеров, причем для тестирования
достаточно менее 10 нМ материала.
Б2.3.2. Нековалентные комплексы
Благодаря тому, что ИЭР достаточно
мягкий способ ионизации, становится
возможным переводить олигонуклеоти-
ды и их комплексы с белками в газовую
фазу без нарушения структуры. Очень
точное измерение массы ДНК-белко-
вых комплексов дает возможность оп-
ределить идентичность и стехиометрию
(эквимолярность) составных компонен-
тов. Так например, известны результаты
ряда измерений, проведенных набелок-
нуклеотидных комплексах, в которых
использовались два белка: белок гена
V (молекулярная масса 9688 Да) и бы-
чий сывороточный альбумин (молеку-
лярная масса 66497 Да). Как оказалось,
результаты таких экспериментов силь-
но зависят от условий их проведения,
поэтому вопрос о том, отражают ли эти
измерения реальную структуру макро-
молекулярных комплексов в растворах,
остается открытым.
Б2.3.3. Большие и очень большие
нуклеиновые кислоты
Попытки анализа больших нуклеино-
вых кислот с помощью ИЛДМ-МС с
лазерным излучением в ультрафиоле-
товой области имели весьма умерен-
ный успех. Верхний предел измеряе-
мых масс находился в районе 90 кДа
для ДНК и 150 кДа для РНК, и был
ограничен в основном фрагментацией
ионов.
Оказалось, что использование инфра-
красного лазера и глицериновой матри-
цы представляет собой наиболее мягкую
комбинацию для интактной десорбции и
ионизации нуклеиновых кислот в боль-
шом диапазоне от небольших олиго-
нуклеотидов до макромолекул длиной
в 2000 нуклеотидов. Так, для синтетичес-
кой ДНК длиной в 21 нуклеотид (моле-
кулярная масса 6398 Да) разрешение со-
ставило 1200, что сходно с разрешением
для белков, имеющих сравнимую массу.
Для детектирования очень боль-
ших молекул ДЫК наиболее подходя-
щим является метод ИЦР-ФП, когда за
один проход можно провес ти более 450
повторных измерений одного и того же
иона. Это позволило значительно повы-
несущпх более 250 зарядов, полученный
с низким разрешением - около 25.
На первый взгляд, для интактной
десорбции больших нуклеиновых кис-
лот ИЭР более подходит, чем ИЛДМ.
Но точность отнесения отдельных ни-
сить отношение «сигнал — шум» и точ-
кой к определенным значениям массы в
ность измерения массы и заряда очень
этом случае очень невелика и неопреде-
Масса (МДА)
g 80,0 -
ф
S
I
га
§.60,0-
CD
о 40,0-
g 20,0-
О 1
0,0 -1—।——.—।—>—।—.—11 ।—।—.——।—.—।—.—।.—।—।—।—।—।—।—г»
2000,0 2500,0 3000,0 3500,0 4000,0 4500,0
m/z
б
Рис. Б2.14: а - гистограмма средней массы, полученная для образца ДНК. Основной ник ври
2.88 МДа соответствует ионам натриевой соли бактериальной плазмиды pBR322, а гораздо мень-
ший пик при 5,85 Мда — ионам димера pBR322 (Benner, 1997); б — ИЦР-ФП-спектр иона с т/
г =2883, полученный для ДНК Т4 бактериофага. Расчетная молекулярная масса примерно равна
90,9 (±9,1) 10° Да (Smith et al., 1996)
тированные с помощью ИЭР-МС име-
ли массу около 40 кДа.
Комбинирование ИЭР и ИЦР-ФП
в масс-спектрометрии дает возмож-
ность анализа отдельных макроио-
нов, несущихе множественный заряд.
Рисунок 152.14 б показывает ИЦР-
ФП-спектр, полученный для ДНК бак-
тсрпофагаТ4 (ожидаемая молекулярная
масса примерно 111,5МДа). Отметим,
что сигнал индивидуальных ионов этой
ДНК регистрировался иод давлением
<1,0 х 10 9 Торр через 1,5 часа после
первоначального возбуждения.
Б2.3.4. Секвенирование ДНК
Для секвенирования ДНК необходима
техника ионизации, способная полу-
чать ионы, содержащие несколько ты-
сяч оснований, с достаточным разреше-
нием и точностью для идентификации
пиков, соответствующей разнице в одно
основание или еще выше. С методоло-
гической точки зрения, масс-спектро-
метрические подходы подразделяются
на две группы, в соответствии с полу-
чением информации о нуклеотидной по-
следовательности непосредственно из
фрагментов в газовой фазе, либо путем
измерений массы продуктов расщепле-
ния цепи, образованных в конденсиро-
ванной фазе (масс-аиализ продуктов
реакции секвенирования по Сэнгеру
(л!\ц'ц । арнн I >2..”> ।! | /’.(>).
Масс-спектроскопическое ступен-
чатое секвенирование включает по-
следовательное ферментативное отщеп-
ление нуклеотидов от цепи с сопут-
ствующей регистрацией изменений ее
массы. В этом процессе предъявляют-
ся более строгие требования к разре-
шению и точности измерения массы,
чем метод Сэнгера. Для того чтобы от-
личить Л от Т с помощью ступенчатого
секвенирования, необходимо уловить
разницу в 9 Да, тогда как для секвени-
рования но Сэнгеру достаточно найти
массовую разность в -300 Да (один
нуклеотид) для определения нуклео-
тидной последовательности.
В ]методах фрагментации в газо-
вой фазе весь процесс секвенирования
проводится в масс-спектрометре через
идентификацию ионных фрагментов,
полученных в результате диссоциации,
вызванной столкновениями. Потенци-
ально он является более быстрым по
сравнению с методами, основанными не
на масс-спектрометрических методах.
Коммеи।;ipiпi Б2.5. Секвенирование по Сэнгеру
Существует несколько распространенных не масс-спектрометрических методов секве-
нирования ДЫК, принятых в молекулярной биологии. Для крупномасштабного экспери-
мента предпочтительным является метод Сэнгера. ДНК, комплементарная интересующей
цепи, синтезируется с помощью праймера, меченого с З’-конца каждого нуклеотида. Затем
реакционная смесь разделяется на четыре аликвоты, куда добавляются 2’,3’-дидезоксинук-
леозид трифосфаты (ddNTP). Когда трифосфат включается в растущую цепь вместо своего
дезокси аналога, отсутствие гидроксильной группы с З’-конца молекулы приводит к тер-
минации цепи. Каждый нуклеотид метится радиоактивной или флуоресцентной меткой.
Идентификация производится разделением различных фрагментов с использованием элек-
. трофореза в полиакриламидном геле. Визуализация полос достигается либо радиоактив-
ным подсчетом в случае с радиоактивно мечеными праймерами или детектированием его
флуоресценции, если используется соответствующая метка. Для секвенирования по Сэнге-
; ру обычно требуется 1,0 пМоль ДНК, а длина определяемых фрагментов может достигать
1000 нуклеотидов пли более.
Комментарий Б2.6. Проект «Геном человека»
В этом проекте ДНК-секвенирование производилось путем электрофоретического раз-
деления фрагментов, полученных методом Сэнгера. Четыре смеси реакционных продуктов
наносились на четыре дорожки и разделялись в соответствии с их электрофоретической
подвижностью - свойст вом, которое зависит в основном от длины фрагмента ДНК (см. Гла-
ву Г5). В зависимости от длины т еля, разделение может занимать до нескольких часов. Время
ожидания можно компенсировать, проводя несколько анализов параллельно. Автоматиче-
ские секвенаторы, использующие одновременно 96 дорожек, работали днями и ночами — в
результате все 3 миллиарда оснований были идентифицированы напрямую из геля. В начале
проекта «Геном человека» .масс-спектрометристы полагали, что они внесут основной вклад
в секвенирование ДНК. Но когда проект был завершен, все 3 миллиарда оснований были
определены напрямую из теля без использования масс-спектрометрии. Ключевым преиму-
ществом секвенирования с помощью электрофореза стала возможность анализа ДНК длиной
около 1200 нуклеотидов. Такие длины недоступны для метода масс-спектрометрии, который
ограничен 100 основаниями. МС более всего подходит для быстрого и точного определения
массы коротких фрагментов ДНК. Тем не менее многие масс-спектрометристы продолжают и
сегодня считать, что масс-спектрометрическое секвенирование ДНК обладает солидным по-
тенциалом и в будущем все-таки заменит электрофоретический метод.
Однако каждый подход имеет свои
преимущества и недостатки. Так, с эк-
спериментальной точки зрения, прямое
секвенирование путем фрагментации в
газовой фазе является более простым,
но зато оно ограничено цепями длиной
-80 нуклеотидов. Это следствие того,
что разрешение ИЛДМ быстро падает
с ростом молекулярной массы и очень
трудно разделить два нуклеотида, кото-
рые отличаются лишь на одно основа-
ние, если размер молекулы ДНК превы-
шает 80 оснований. Непрямые методы
секвенирования требуют для приме-
нения масс-спектрометрии достаточ-
но сложной обработки образца, но при
этом способны определять длину цепи
-100 нуклеотидов. Правда, в некоторых
случаях качество секвенирования ог-
раничено отсутствием специфических
сайтов энзиматического расщепления.
Б2.4. Углеводы
Биологические углеводы составля-
ют разнообразную группу полимеров,
выполняющих множество функций
в клеточных процессах — от запасания
энергии (гликоген) или структурной
функции (целлюлоза, хитин) до пере-
дачи сигналов, адгезии и модификации
белков. Они могут быть простыми мо-
носахаридами и мегадальтонными го-
мополимсрамн, строго структурирован-
ными сигнальными олигосахаридами и
сложной смесью углеводно-модифици-
рованных белков и липидов.
Основная сложность в структур-
ной идентификации углеводных оли-
гомеров состоит в большом числе изо-
меров, возникающих как вследствие
вариаций в позиции образования связи
между остатками мономера, так и вви-
ду возможности образования множест-
венных связей с одним остатком (вет-
вление). Например, для структурной
идентификации прямой олигомерной
цепи требуется знание последователь-
ности и типов гликозильной связи,
т. е. места образования связи и аномер-
ная конфигурация (параграф А2.5.1),
а также последовательность мономер-
ных остатков.
Многие биологические углеводы,
интересные со структурной точки зре-
ния, являются конъюгатами с белками,
липидами или теми и другими вместе —
гликопротеины, гликолипиды или гли-
колипопротеины и т. д. Л конъюгация
представляет дополнительное измере-
ние в структурной изомерии в соответ-
ствии со специфическим положением
углеводавгликоконыогате. Комбинация
конъюгации и изомерической структур-
ной сложности препятствовала разви-
тию стандартных методик секвенирова-
ния углеводов, подобных тем, что давно
уже существовали для полипептидов и
нуклеиновых кислот.
По сравнению с современными ме-
тодами секвенирования белков и нук-
леиновых кислот, методы анализа
гликанов более разнообразны, но все
еще находятся в начальной фазе раз-
вития. Ниже мы приведем два приме-
ра исследований углеводов с помощью
масс-спектрометрии.
Б2.4.1. Олигосахариды
По сравнению с пептидами, нативные
олигосахариды трудно поддаются иони-
зации. В литературе описаны различных
стратегии ионизации, включая пермети-
лпрование. Перметилированные олиго-
сахариды претерпевают специфическую
перекрестную фрагментацию, которая
позволяет определить типы связей,
встречающиеся в кольцевой структуре
углевода. В результате расщепления
гликозидной связи образуются фраг-
менты, которые легко отличить друг от
друга благодаря разнице в массе, обус-
ловленной метилированием.
Линейные олигосахариды и смеси
олигосахаридов (рис B2.I.5), присо-
единенные к N-концу белка, успеш-
но исследовались с помощью ИЭР-
масс-спектрометрии с квадрупольной
ионной ловушкой.
Тандемная масс-спектрометрия с по-
рядком свыше двух (параграф Б1.5.7)
предлагает несколько путей для улучше-
ния МС-спектров (рис. 62.16). Актива-
ция перметилированных молекулярных
ионов олигосахаридов через столкно-
вения в тандемном масс-спектрометре,
показанная на примере мальтогептаозы
(см. рис. 62.16), приводит к образованию
большого количества фрагментов после
расщепления гликозидной связи. На ри-
сунке показан состав и последователь-
ность продуктов перекрестно-кольцевого
расщепления, указывающие на специфи-
ческие связи внутри олигосахарида. Пу-
тем захвата и дальнейшей диссоциации
этих фрагментов в МСХ можно подтвер-
дить наличие данных продуктов и их от-
носительное содержание.
Метилирование позволяет также
установить характер ветвления данного
сахара. Поскольку оно происходит толь-
ко в незанятых сайтах образованной
связи, степень метилирования связана
обратной зависимостью с количеством
ветвлений данного моносахарида.
Б2.4.2. Гликопептиды
Множество ядерных и цитоплазматиче-
ских белков модифицируются путем
присоединения одиночного остатка
М-ацет11 л гл юкозам ина к п i дроке 11 лу
боковой цепи серина или треонина (О-
GlcNAc). Считают, что эта иосттранс-
GIC01 — 4GIC01— 4GIC01— 4GIC01— 4GIC01— 4GIC01— 4Glc
Рис. 62.15. Структура мальтогептаозы. Примеры сокращений названий моносахаридов даны
в ;iA i. \'.Т i
Ys
1075.5
1497.8, [M+Na]+ >
1279.6, [M+Naf >Y6>
B4 B:
1075.5, [M+Naf > Y6 > Y5 >
Рис. 52.16. MC2 спектр перметилировапной мальтогентаозы, [M+Na] = m/z 1497.8: (а) схема
ионных фрагментов, полученных в результате МС2 (б), МС3, (о) и МС1 (г). Используемая здесь
номенклатурная система предложена Домоном и Кастелло в 1987 г. (Weiskopf et al., 1997)
К< Г. . 1-| 1-2. Л ЩеЛОЧНЭЯ
0-элиминация
Щелочная р-элиминация - один из наи-
более распространенных методов для от-
щепления гликанов от белков и пептидов,
соединенных О-связыо. В результате гли-
козилированные остатки серина и треони-
на превращаются в аланин и 2-амнномас-
ляную кислоту.
ляционная модификация выполняет
регуляторную функцию, наподобие фос-
форилирования, поскольку она быстро
реагирует в ответ на внешний клеточный
стимул и связана с клеточным циклом.
Современный метод картирования
сайтов, который включает мечение га-
лактозилтрансферазой, образование
гликопептидов через протеолиз, очистку
с помощью высокоэффективной жид-
костной хроматографии в несколько
циклов, а также секвенирование в газо-
вой фазе и методом Эдмана, очень тру-
доемок и требует около 10 пикомолей
чистого меченого гликопептида.
Для избирательного детектирования
гликопептидов и идентификации точных
сайтов гликозилирования на пикомоль-
ном уровне разработан чувствительный
ИЭР-МС метод. Были получены синте-
тические гликопептиды (рис. Б2.17), ко-
торые использовались для демонстрации
того, что O-GlcNAc-модифицированные
пептиды можно быстро идентифициро-
вать в сложных смесях с помощью ском-
бинированной с ВЭЖХ-ИЭР-масс-спек-
трометрией благодаря частичной потере
связанного О-гликана (М = 204) при уме-
ренной напряженности электрического
ноля. Полученные масс-спектры пред-
ставлены на рис. 152.18, После удаления
O-GlcNAc с помощью щелочной 0-эли-
минации (ы ।хаз । i3i>i H i I >2.7) сайт глико-
зилирования был напрямую идентифи-
цирован путем диссоциации, вызванной
Туг - Ser - Pro - Thr - Ser - Pro - Ser - Lys
a
О
Туг - Ser - Pro - Thr - Ser - Pro - Ser - Lys
6
Tyr - Ser - Pro - Thr - Ser - Pro - Ser - Lys
в
Рис. 152.17. Структуры трех синтетических
гликопептидов: а — пептид O-GlcNAc; б — пеп-
тид O-GalNAc; е -- пептид LacNAc (Greis et al.,
1996)
столкновениями в коллизионной камере
(параграф Б2.6.7) гликопептида.
Превращение гликозилсерина в
2-аминопропионовую кислоту (см.
структуры на рис. Б2.17) через 0-эли-
минацию одновременно уменьшает
массу гликопептида на 222 единицы
молекулярной .массы. Такой результат
доказывает, что описанный метод спо-
собен стать мощным инструментом для
определения сайтов O-GlcNAc моди-
фикации белков, численность которых
в клетке невысока — например, транс-
крипционных факторов и онкогенов.
в Б« Суб КЛвТСИ' j Н bi‘3
комплексы и органеллы
Б2.5.1. Рибосомы, рибосомные
субъединицы и рибосомные белки
Исследование рибосомы является хо-
рошим примером, демонстрирующим
б
m/z
в
Рис. Б2.18. ИЭР-.масс-спектры очищенных с помощью ВЭЖХ гликопептидов получены мето-
дом тройной квадрупольной масс-спектрометрии: а — пептид O-GlcNAc: б — пептид O-GalNAc;
в — пептид LacNAc. Вставки представляют расчетные Ь- и //-ионные фрагменты каждого гли-
копептида. Номенклатура та же, что и для белков. Расчетные средние массы для гликопепти-
дов равны 1069 Да для O-GlcNAc и O-GalNAc гликопептидов и 1231 Да для О-LacNAc пептида.
Масс-спектры гликопептидов O-GlcNAc (рис. 1)2. 18 а) и O-GalNAc (рис. I>2.18 б) соответствуют
ожидаемым для пиков с отношением т/z, равным 535 и 1070. Эти пики соответствуют двум- и
однопротонированным формам гликопептидов. Другие заметные пики при 866 и 204 (соответ-
ствующие дегликозилированным пептидам, O-GlcNAc или O-GalNAc), а также со значениями
819, 616 и 251 были получены в результате фрагментации гликозидных и пептидных связей (см.
вставку для расчетной схемы фрагментации). Масс-спектр О-пептида LacNAc имеет те же харак-
теристики (Greis et al., 1996)
возможности масс-спектрометрии. 70S
рибосомы Е. соН в буфере, содержащем
5 мМ Mg-’, переводили в газовую фазу в
масс-спектрометре с помощью техники
нанопотока в ИЭР (рис. Б2.19 а). Было
обнаружено, что при снижении концен-
трации Mg2' в растворе, частицы диссо-
циируют на 30S н 50S субъединицы (рис.
Б2.19 б. см. также1 рис. П.З). Разрешение
зарядовых состоянии в спектре 30S субъ-
единицы позволило определить ее массу
равной 852187 ± 3918 Да — значение, ле-
жащее в пределах 0,6% от массы, рассчи-
танной из значений химической струк-
туры ее белков и 16S РНК. Дальнейшая
диссоциация на меньшие комплексы и от-
дельные белки может быть индуцирована
столкновениями частиц в газовой фазе с
высокой энергией. 55 из 56 белков в этих
комплексах наблюдались в интактном
состоянии (без фрагментации) и было
найдено множество различных посттран-
сляционных модификаций. Интересно
отметить, что самый большой белок (S1,
М = 61 кДа) детектирован не был.
Похожее исследование с использо-
ванием другого подхода было выполне-
но на дрожжевой рибосоме. Все белки
этого макромолекулярного комплек-
са были выделены, денатурированы,
расщеплены трипсином и обессолены.
Далее пептидная смесь разделялась
хроматографически и анализировалась
с помощью ИЛДМ-ВП-масс-спектро-
метрии. Из 78 предсказанных белков в
рибосомном комплексе только три не
были идентифицированы.
Б2.5.2. Бактерии и бактериальная
таксономия
Таксономическая идентификация бак-
терий, основанная на анализе белковых
компонентов, представляет собой логи-
ческое развитие методов, разработанных
для исследований бактериальной про-
теомики. В идеале, этот подход должен
быть быстрым и основываться на доста-
точно большой группе белков, например
фингер- (пальцевых) белков, чтобы по-
лучить уникальный масс-спектр орга-
низма или штамма.
На рисунке Б2.20 показаны спект-
ры, полученные для четырех штаммов
Е. соИ. Они типичны для «цельных» или
«интактных клеток» с большим числом
пиков между m/z 3000 и 10000. Различ-
ные штаммы Е. соН имеют множество
общих пиков, а также свои уникальные
черты. Важно отметить, что подобные
12 000 16 000 20 000 24 000
а
12 000 16 000 20 000 24 000
б
Рис. 152.19. ИЭР-масс-спектры 70S рибосом-
ных частиц: а — в присутствии 5 мМ Mg2’ и
б — после трехкратного разведения раствора
(Rostom et al., 2000)
a
4000 5000 6000 7000 8000 9000
Масса (ДА)
Рис. Б2.20. ИЛДМ-ВП-масс-спектры кле-
точных суспензий четырех штаммов Е. coir,
а - BLR; 6 - XL1 Ыне; в - RZ1032 и г - CSH23,
показывающие типичный «клеточный» про-
филь в интервале m/z от 3000 до 10000 (Lay,
2001)
спектры зависят от фазы роста клеток.
Бактерии очень быстро реагируют на
изменения в окружающей среде, по-
этому производство стрессовых белков,
когда клетки хранятся, подвергаются
манипуляциям или культивируются в
течение различных периодов времени,
приводит к тому, что образцы далеко
не идентичны. Это нужно учитывать,
когда ИЛДМ-ВП-МС применяется для
идентификации бактерий.
Б2.6 Масс-спектрометрич
в медицине
Ряд проблем клинической химии, свя-
занных с анализом белков и нуклеино-
вых кислот, могут решаться с помощью
масс-спектрометрии. Этот метод уже
заменяет электрофорез при анализе про-
дуктов во время рутинных молекулярно-
биологических процедур, таких как опре-
деление размеров амплифицированной с
ПОМОЩЬЮ ПЦРДНК( la >\| MCI! I; I ’ Ч ! 11 |>2.Ч I,
секвенирование коротких олигонук-
леотидов, детектирование генетических
изменений и мутаций и многие другие.
Разработаны технологические платфор-
мы для полного сравнительного анализа
ДНК, основанные на производстве корот-
ких диагностических продуктов и точном
измерении их массы с помощью ИЛДМ-
ВП. Они представляют собой первые
шаги в предклинических и клинических
испытаниях лекарств, которые движутся
в направлении от подходов, основанных
на статистике к нестатистическим под-
ходам, позволяющим получать профиль
ответа на уровне индивидуального паци-
ента для каждого лекарства-кандидата.
Комментарий Б2.8. Полимеразная цепная реакция
Полимеразная цепная реакция (ИЦР) используется для амплификации фрагментов ДНК
из сложной смеси исходного материала, который обычно называют ДНК-матрицей. Метод
требует частичного знания последовательности ДИК, находящейся у концов амплифици-
руемого фрагмента. На основе этой информации химически синтезируют два олигонуклео-
тидных праймера, каждый из которых комплементарен небольшому участку на З’-концс
ДНК-матрицы — по одному олигонуклеотиду на каждую цепочку ДНК. ПЦР состоит из трех
этанов. На первом двухцепочная ДНК-матрица подвергается денатурации. На втором проис-
ходит гибридизация двух олигонуклеотидных праймеров со своими комплементарными кон-
цевыми последовательностями. На третьем этапе идет синтез ДНК с помощью полимеразы.
Другим потенциальным приложе-
нием масс-спектрометрии может стать
ранняя диагностика некоторых разно-
видностей рака, который возникает
вследствие постепенного накопления
генетических изменений в одной-
единственной клетке. Были иденти-
фицированы несколько мутаций,свя-
занных с раком, такие как точечные
мутации в K-ias и р53 генах. Зачастую
образцы раковой ткани содержат все-
го несколько мутированных клеток,
окруженных большим количеством
здоровых Поэтому прикладывают-
ся большие усилия для разработки
масс-снектрометрпческого метода де-
тектирования злокачественных кле-
ток на ранней стадии.
Б2.6.1. Клиническая онкология
ПЛДМ-ВП-масс-снектрометрия дает
возможность быс трого детектирования
и наблюдения отклонений в экспрес-
сии онкобелков ( гиммсн rapiiii I ).
Она использовалась, например, для
исследования белкового профиля че-
ловеческих клеток по мере превраще-
ния нормального эпителия молочной
железы в полностью злокачественную
ткань. Рисунки 152.21 а и б показывают
ИЛДМ-ВП-масс-спектры лизатов зло-
качественных и нормальных клеток,
соответственно. В масс-спектре лиза-
та раковых клеток молочной железы
доминируют несколько белковых пи-
ков, специфически связанных с канце-
рогенезом (рис. Б2.21 а). Напротив, в
масс-спектре лизата нормальных кле-
ток лишь небольшое число белковых
пиков выбивается из общего фона (см.
рис. Б2.21).
Б2.7. Масс-
спектрсметрическая
визуализация изображений
Несмотря на то, что обычными метода-
ми создания изображений биологиче-
ских образцов служат трансмиссионная
и растровая электронные микроскопии,
они. как правило, дают только морфо-
логическую информацию (см. Часть
Ж). В настоящее время молекулярная
специфичность и чувствительность
ИЛДМ-МС используется для разработ-
ки новой технологии прямого картиро-
вания и визуализации биомакромоле-
кул в отдельной клетке или в срезах
тканей.
Б2.7.1. Клетка
Биологические образцы, полученные
в результате замораживания-оттаива-
ния, представляют собой наилучший
материал для их молекулярного опи-
сания с помощью визуализацнонной
ИЛДМ-ВП-масс-снсктрометрии. По-
тенциал метода был продемонстриро-
ван на клетках Paramecium, которые
Коммон трин Б2.9. Онкогены и онкобелки
Онкогены представляют собой мутированные формы нормальных генов, которые участву-
ют в регуляции клеточной пролиферации и/или дифференциации. Онкобелки, кодируемые
онкогенами и генами опухолевой супрессии, выполняют ключевую роль в контроле нормаль-
ного и злокачественного роста через регуляцию различных сложных путей сигнальной тран-
сдукции. Потеря кон троля зачас тую оборачивается некон тролируемой клеточной пролифера-
цией. Возможность ИЛДМ-ВП масс-спектрометрии детектировать отклонения в экспрессии
онкогенов и следить за ними была продемонстрирована Чонгом и сотрудниками в 1998 г.
6
I’m . 1)2.21. ИЛДМ-ВП масс-спектры: a — лизата злокачественных и б — нормальных клеток мо-
лочной железы из клеточной линии MCF-10. Аббревиатуры: c-H-Ras — трансформирующий! бе-
лок H-ras-1, int-2 — белок-предшественник протоонкогена Int-2, р53 — ядерный фосфопротеин,
с-шус — iMYC-иротоонкогенный белок, THR beta-2 — рецептор гормона щитовидной железы
beta-2 (Chong et al., 1998)
прошли процедуру размораживание/
оттаивание и были помещены в ВП-
ионный масс-спектрометр. Однокле-
точные организмы являются идеальным
объектом, поскольку их большой раз-
мер (-180-310 мкм в длину) позволяет
точно расположить индивидуальную
клетку прямо на пути пучка первичных
ионов. Были получены изображения
распределений молекул на клеточном
срезе с помощью субмикронного пучка
ионов. Также удалось получить изобра-
жения распределений диметилсульфок-
сида и кокаина, добавленных к клеткам
Paramecium.
Б2.7.2. Ткань
Существует несколько способов приго-
товления образцов ткани для визуализа-
ции распределения молекул с помощью
масс-спектрометрии. Типичная проце-
дура включает в себя нанесение заморо-
женного среза ткани на пластину из не-
ржавеющей стали, покрытой матричным
раствором, который затем высушивают и
вводят в вакуумный вход масс-спектро-
метра. Молекулярные изображения по-
лучаются сканированием по поверхнос-
ти образца лазерным лучом диаметра -25
мкм (рис. Б2.22). При частоте лазерных
импульсов 20 Гц на сканирование одной
точки уходит примерно 2,5 сек, включая
сбор данных и передачу их в компьютер,
а также обработку информации и изме-
нение положения образца. Типичный
набор данных состоит из 1000-30000
пятен, в соответствии с требуемым раз-
решением. Изображение формируется
из значений интенсивностей ионов, де-
Рис. Б2.22. Схематическое изображение методики, разработанной для анализа ткани с помо-
щью ИЛДМ-МС. Замороженный срез помещается на металлическую пластинку, покрывается
УФ-ноглощающей матрицей и устанавливается в масс-спектрометр. Импульсный УФ-лазер
десорбирует и ионизирует молекулы ткани, а ВП-анализатор определяет значения m/z. После
сканирования ткани и измерения интенсивностей пиков на тысячах участках получают спект-
рометрические изображения распределений молекулярных масс по срезу ткани (Stoeckli et al.,
2001)
Рис. 152.23. Масс-спектрометрические изображения среза мозга мыши: а оптическое изобра-
жение среза, нанесенного на позолоченную пластину: б — область с т/г -8528 коры головного
мозга и гиппокампа: в — область с т/г 6716 черной субстанции и медиального коленчатого
тела: г — т/г 2564 — зона среднего мозга (Stoeckli et al., 2001)
сорбированных из каждого пятна в диа-
пазоне от 500 Да до 80 кДа.
Рисунок Б2.23 демонстрирует МС-
изображение среза мозга мыши. Хотя
сигналы многих белков расположены
по всей поверхности, некоторые из
них являются высокоспецифичными
для данной области мозга. К примеру,
белок с m/z = 8528 (см. рис. 152.23 б)
присутствовал в коре головного моз-
га и гиппокампе; белок с m/z = 6716
локализовался в черной субстанции
и медиальном коленчатом теле; пеп-
тид с m/z = 2564 находился в среднем
мозге. Идентификацию белков можно
проводить через экстракцию, фракци-
онирование с помощью жидкостной
ЖХВД хроматографии, протеолиза,
масс-спектрометрического фракцио-
нирования одного или более фрагмен-
тов и поиска в базе данных по белкам.
Потенциал новой технологии был про-
демонстрирован на примере иденти-
фикации специфических опухолевых
маркеров в пролиферирующей ткани.
Приложение 1. Вычисление
молекулярной массы белка
из его масс-спектра
Масс-спектр — это график зависимости
интенсивности сигнала от отношения
масса-заряд. Пик с наибольшей интен-
сивностью называется базовым пиком.
Как правило, спектр нормируется по вы-
соте базового пика, чтобы получить зна-
чения относительных интенсивностей.
Молекулярную массу белка лег-
ко рассчитать из масс-спектра белка
(рис. А) при помощи математических
формул, приведенных ниже. При этом
нужно помнить, что значения z всегда
целые. Подразумевается, что ионы пред-
ставляют собой продукт взаимодействпя
нейтральной молекулы и протонов.
Молекулярная масса нейтральной
молекулы М может быть найдена из
значений регистрируемых масс ///, и т.,
(что эквивалентно значениям m/z) и
числа добавленных зарядов или прото-
нов /7,11/7,:
M = n.,(m,-Y), (Л)
/77,-1
где /7, = /?., + 1 и п., - —1.
tn., - mt
Можно рассчитать /?,, и, следователь-
но, М, если брать ники регистрируемых
масс попарно.
Применяя формулу (А) для расче-
та молекулярной массы цитохрома с с
использованием двух соседних пиков с
777 , = 952,3 и //?., = 1031,3, получим для п2
следующее значение:
- /77.,-7/7, 1031,3-952,3
или Z = 12.
Это означает, что 12 положитель-
ных зарядов ассоциированы с относи-
тельной массой, равной 1031,3.
Молекулярная масса в этом случае
рассчитывается как М- п2(т2 -1) и
равна 12 363,6.
Рассматривая следующие два пика
с относительными массами /п, = 884,3 и
т,2 = 952,3, мы получим:
883,3
2 7/72 -777, 952,3-884,3
или Z = 13, т. е. 13 положительных за-
рядов ассоциированы с относитель-
ной массой 952,3. Рассчитанная из
этих пиков молекулярная масса равна
12366,9.
Продолжая процедуру для других
пар пиков, получаем:
из двух пиков с относительными
массами //?, = 825,5 и т2 = 884,3, молеку-
лярную массу 12 366,2;
Рис. А. Масс-спектр цитохрома с (16 951,5 Да), несущего множественный заряд от ^12 до +18,
полученный с низким разрешением при помощи ионизации электрораспылением (Gordon, 2000)
330 619 923 1212 1541 1854 2158 - Н 2407
Рис. В. Схематическая структура октамерного олигодезоксирибонуклеотида d (A-C-T-C-G-A-T-
G), с отмеченными точками разрыва связей, в результате которых образуются основные ионные
фрагменты. Пунктиром отмечены основные фрагменты с указанием массы: сверху — фрагменты,
соответствующие ионам с 5’-фосфатными концами (5’ - Р), снизу — ионы с З’-фосфатными кон-
цами (3' — Р) (Grotjahn et al., 1982)
из двух пиков с относительными
массами т{ = 773,9 и т2 = 825,5, молеку-
лярную массу 12 367,5;
из двух пиков с относительными
массами = 825,5 и т2 = 884,3, молеку-
лярную массу 12 366,4.
Отсюда мы можем заключить, что
наблюдаемая средняя молекулярная
масса, рассчитанная из пяти пиков, рав-
на 12 366,1. Теоретическая масса ци-
тохрома с равна 12366.
Приложение 2.
Вычисление нуклеотидной
последовательности
На рисунке Б схематически представле-
на структура олигодезокси-рибонуклео-
тида d (A-C-T-C-G-A-T-G), на которой
отмечены связи, расщепление которых
приводит к образованию основных ион-
ных фрагментов.
Ниже показан пример определения
последовательности олигодезоксири-
бонуклеотида с помощью ББА-МС из
следующей серии данных:
m/z (5’ - Р): 2407, 2174, 1885, 1581,
1292, 963, 650, 346;
m/z (3’ - Р): 330, 619, 923, 1212,
1541, 1854,2158, 2407.
Правила состоят в следующем:
1. Разность масс (А) в значениях m/z
между последующими ионами представ-
ляет собой массу нуклеотидного остатка.
Следовательно, набор m/z данных может
быть преобразован в олигонуклеотидную
последовательность следующим образом:
(5’ - Р) А 233* 289 304 289 329 313
304;
m/z 2407 2174 1885 1581 1292 963
650 346*;
нуклеотиды ACTCGATG.
Первый нуклеотид — А. Молеку-
лярная масса = 233 + 96 (РО(Н) — 17
(ОН) + 1 (Н) = 313. Последний нуклео-
тид — G. Молекулярная масса = 346-17
(ОН) = 329.
Получаем последовательность
d (A-C-T-C-G-A-T-G).
Читатель может проверить после-
довательность нуклеотидов, применяя
те же правила для ряда значений m/z
для (3’ — Р) конца.
2. Наибольший пик в ББА-спектре
m/z = 2407, который представляет (М —
Н)\ Отсюда М= 2408 + 96 (РО4Н) — 34
(2ОН) = 2470.
Сумма нуклеотидов дает 2470 Да,
что соответствует масс-спектрометри-
ческому значению М.
Б2.8. Перечень ключевых идей
• Самые разнообразные биологические молекулы - от пептидов и олигонуклео-
тидов до субклеточных комплексов и органелл - могут быть переведены в газо-
вую фазу без фрагментации техникой ионизации с электрораспылением (ИЭР)
или техникой ионизации лазерной десорбцией в матрице (ИЛДМ).
• Масс-спектрометрия с использованием метода ИЭР наиболее подходит для
получения информации о молекулярной массе, структуре различных пептидов
и олигонуклеотидов.
• Масс-спектрометрия с использованием метода ИЛДМ благодаря своей ско-
рости, точности и чувствительности широко используется для исследования
функции, структуры и динамики белков; она позволяет проводить прямое кар-
тирование и визуализацию распределения биологических макромолекул в оди-
ночной клетке или тканях млекопитающих.
• Объединение двух технологий — двумерного геля электрофореза и тандемной
масс-спектрометрии — стало основой современной протеомики на субпико-
мольном уровне.
• Особенно большие перспективы открываются для использования масс-спект-
рометрии в бактериальной таксономии и медицине.
• В следующем десятилетии ожидается развитие масс-спектрометрии гигантски-
ми темпами и ее широкое применение в разных областях науки — от структур-
ной биологии до медицины.
Литература для дальнейшего чтения
Структура и функция белков
Chait В. Т. Mass spectrometry — useful tool for protein X-ray crystallographer and
NMR spectroscopist. Structure, 1994, 2, 465-467.
Winston R. L. and Fitzgerald M. C. Mass spectrometry as a readout of protein structure
and function. Mass Spectr. Rev., 1997.16, 165-179.
Rostom A. A. and Robinson С. V. Disassembly of protein complexes in the gas phase.
Curr. Opin. Struct. Biol., 1999. 9. 135-141.
MirankerA.D. Protein complexes and analysis of their assembly by mass spectrometry.
Curr. Opin. Struct. Biol., 2000. 10, 601-606.
DoboA. and Kaltashov I. A. Detection of multiple protein conformational ensembles
in solution via deconvolution of charge-state distribution in ИЭР MC. Anal. Chem.,
2001.73, 4763 477B2.
Nielsen M. L„ Bennet K. L., Larsen B„ Monniate M. and Mann M. Peptide end sequencing
by orthogonal ИЛДМ tandem mass spectroscopy. J. Proteome Res., 2002.1, 63-71.
Link A. J., EngJ., Schieltz D. M„ Carmack E., Mize G.J., Morris D. R., Garvic В. M.,
Yates, J. R. III. Direct analysis of protein complexes using mass spectrometry. Nature
Biotechnol., 1999.17, 676-682.
Функциональная протеомика
Jungblur P. and Thiecle, B. Protein identification from 2-I'3gels by ИЛДМ mass
spectrometry. Mass Spectr. Rev., 1997. 16, 145 -162.
Godovach-ZimmermannJ. and Brown L. R. Perspectives for mass spectrometry and
functional proteomics. Mass Spectr. Rev., 2001. 20, 1-57.
Ogorzalek Loo R. R., CavalcoliJ. D„ Van Bogelen R. A., Mitchell C„ LooJ. A., Moldover B.
and Andrews P. C. Virtual 2-D gel electrophorllDPs: visualization and analysis of the E.
coli proteome by mass spectrometry. Anal. Chem., 2001. 73, 4063-4070.
Нуклеиновые кислоты
NordhojJ E„ Kirpekar F. and Roepstorff P. Mass spectrometry of nucleic acids. Mass
Spectr. Rev., 1996.15,67-138.
Murray К. K. DNA sequencing by mass spectrometry. J. Mass Spectr., 1996. 31,
1203-1215.
Berkenkamp S., Kirpekar F. and Hillenkamp F. Infrared ИЛДМ mass spectrometry of
large nucleic acids. Science, 1998. 281, 260 -262.
Crain P. F. and MccloskayJ.A. Application of mass spectrometry to the characterization
of oligonucleotides and nucleic acids. Curr. Opin. Biothechnologv, 1998. 9, 25-34.
Углеводы
Karlsson K. A. Animal glycosphingolipids as membrane attachment sites for bacteria.
Annu. Rev, Biochem., 1989. 58, 309-350.
Solouki T., Reinhold В. B., Costello С. E., O’malley M., Guan Sh. and
Marshall A. G. Electrospray ionizatio and matrix-assisted laser desorption/ionisation
Fourier transform ion cyclotron resonance mass spectrometry of permethylated
oligosaccarides. Anal. Chem., 1998. 70, 857-864.
Субклеточные комплексы и органеллы
Rostom A., Fucini Р., Benjamin D., Juenmann R., Nierhaus K„ Haiti E, Dobson C.
and Robinson C. Detection and selective dissociation of intact ribosomes in a mass
spectrometer. Proc. Natl. Acad. Sci. USA, 2000. 97, 5185-5190.
Lay J. O. ИЛДМ-TOF mass spectrometry of bacteria. Mass Spectr. Rev., 2001. 20,
172-194.
Масс- спектрометрия в медицине
Guo В. Mass spectrometry in DNA analysis. Anal. Chem., 1999. 71, 333-337R.
Масс-спектрометрическая визуализация
Pacholski M. £. and WinogradN. Imaging with mass spectrometry. Chem. Rev., 1999.
99, 2977-3005.
Часть В.
ТЕРМОДИНАМИКА
Глава В1
ТЕРМОДИНАМИЧЕСКАЯ СТАБИЛЬНОСТЬ
И ВЗАИМОДЕЙСТВИЯ МАКРОМОЛЕКУЛ
В 1.1. Исторический обзор
Термин термодинамика происходит
от греческого therme (тепло) и dynamis
(сила). Эта наука берет свое начало в
XIX в., когда были поставлены первые
эксперименты по исследованию соотно-
шения между теплотой и работой, опре-
делены понятия температуры и энергии,
а также четко сформулированы ее первые
и вторые законы. В последующие годы
термодинамика обобщила понятие рабо-
ты, включив в нее кроме механической
работы все остальные формы энергии,
такие как электрическую, магнитную, хи-
мическую и энергию излучения. Приме-
нимость термодинамики была установле-
на для всех типов систем — от идеального
газа до термоядерной плазмы, включая
магнитные системы, системы «жид-
кость-пар», растворы макромолекул,
химические реакции и т. д. В этой главе
мы будем иметь дело в основном с прило-
жениями классической и статистической
равновесной термодинамики для иссле-
дования биологических макромолекул,
их растворов и взаимодействий.
Начало XVII в.
Галилео Галилей (Galileo Galilei)
и его последователи сконструировали
первые термометры, позволяющие де-
лать воспроизводимые измерения тем-
пературы. Основателем калориметрии
считается Джозеф Блэк (Joseph Black)
(1728-1799), который сделал первый
калориметр. Его измерения привели к
созданию калорической теории, или тео-
рии калорической жидкости, обладаю-
щей весом и связанной с температурой.
1780
Антуан-Лоран Лавуазье (Antoine-
Laurent Lavoisier) впервые применил
калориметрические измерения в биоло-
гии. Он изучал энергетический обмен у
животных (хотя понятие энергии тогда
еще не было сформулировано), помещая
морскую свинку в ледяной калориметр,
разработанный вместе с П. С. Лапла-
сом (Р. S. Laplace). Выделение тепла
измерялось с помощью оценки скоро-
сти таяния льда. Тепловой баланс был
рассчитан из разности между теплом,
выделенным экскрементами, и теплом,
поступившим с пищей.
1807
Томас Янг (Thomas Young) ввел
в обиход понятие энергии (от гречес-
кого еп «в» и ergon «работа»), однако
в то время четкого различия между
силой и энергией еще не было. Сади
Карно (Sadi Carnot), военный ин-
женер, ввел в науку концепцию цик-
ла тепловой машины, которая была
опубликована в его .мемуарах «Sur la
puissance motrice du feu» («Размыш-
ления о движущей силе огня») (1824).
Своп результаты он интерпретировал
с точки зрения все той же калориче-
ской теории.
1842
Юлиус Роберт фон Майер (Julius
Robert von Mayer) доктор медицины
заявил о равнозначности тепла п меха-
нической энергии. В 1845 г. он опубли-
ковал численное значение эквивалента
теплоты.
1845
Джеймс Прескотт Джоуль (James
Prescott Joule) — пивовар и ученый-
любитель из .Манчестера, предъявил
убедительные экспериментальные до-
казательства того, что теплоту можно
перевести в работу и обратно.
1849
Лорд Кельвин (Вильям Томсон)
(Lord Kelvin (William Thomson))
указал на несоответствие результа-
тов Джоуля калорической теории.
Проблема была решена Рудольфом
Юлиусом Клаузиусом (Rudolf Julius
Clausius) (1850), который установил
первый и второй законы термодина-
мики. Его работа означала рождения
новой науки — термодинамики. Клау-
зиус определил свойства энтропии
(от греческого еп (в) и tropos (превра-
щать)), которая использовалась в зна-
чении трансформации, т. е. энтропия
имела трансформационное содержа-
ние. Понятие энергии было определе-
но Кельвином. Кроме того, он пред-
ложил температурную шкалу, позже
названную его именем.
1865 1875
Джеймс Клерк Максвелл (James
Clerk Maxwell) определи;! математи-
ческие соотношения между термоди-
нам и чес ki 1 м и параме трам 11, ко торые
теперь носят его имя. Дж. Уиллард
Гиббс (J. Willard Gibbs) (1873, 1874)
опубликовал три выдающиеся работы,
в которых математическое описание ус-
ловий равновесия было настолько эле-
гантным, что они стали основой совре-
менной классической термодинамики
и физической химии.
1918
Вальтер Нернст (Walther Nemst)
установил третий закон термодинами-
ки, тем самым придав ей законченную
форму. Статистическая термодинами-
ка, основанная на соотношении между
теплом и движением атомов и молекул,
развивалась параллельно с классиче-
ской термодинамикой, в основном бла-
годаря работам Германа Людвига Фер-
динанда фон Гельмгольца, Клаузиуса,
Максвелла, Людвига Больцмана и Гиб-
бса (Hermann Ludwig Ferdinand von
Hemholtz, Clausius, Maxwell, Ludwig
Boltzmann and Gibbs).
1930-e
Калориметрия была изобретена в
XX в. для изучения физической хи-
мии растворов. Джон Т. Эдсалл (John
Т. Edsall) впервые измерил термоди-
намические параметры растворов ами-
нокислот и других органических со-
единений в воде. Кристиан Анфинсен
(Christian Anfinsen) в 50-х гг. XX в.
осуществил эксперименты с рибонукле-
азой и выдвинул «термодинамическую
гипотезу». Согласно ей, вся инфор-
мация, необходимая для правильного
сворачивания белка в свою активную
в физиологических условиях структу-
ру, содержится в его аминокислотной
последовательности. За свое откры-
тие Анфинсен совместно с С. Муром и
У. Стайном в 1972 г. был удостоен Но-
белевской премии по химии.
1970-е
11ервый высокочувс твительный кало-
риметр для исследования белковых рас-
творов был построен в 70-х it. прошлого
века Петром Приваловым (Р. Privalov)
и его сотрудниками. В последующие
десятилетия дифференциальная ска-
нирующая калориметрия, — а позже и
калориметрия изотермического титро-
вания применялась для исследования
процесса сворачивания белка и взаимо-
действий в процессе .межмолекулярного
связывания. Точность измерений тер-
модинамических параметров возросла с
изобретением еще более чувствительных
калориметров — микрокалориметров, а с
наступлением 2000 г. и нанокалоримет-
ров (приставки микро и нано говорят о
степени точности измерения тепла в этих
приборах). Фундаментальные работы
по калориметрии белков были выполне-
ны Петром Приваловым, Джулианом
Стюртевантом, Ернесто Фрейре (Julian
Sturtevant, Ernesto Freire) и их сотруд-
никами.
В 1990-е
Появились чувствительные био-
сенсоры для измерения констант свя-
зывания молекул с иммобилизованны-
ми лигандами в растворе, получившие
впоследствии широкое распростране-
ние во многих биохимических лабора-
ториях.
2000 и по пасюящее время
Открываются большие перспективы
для современных термодинамических
исследований в биофизике, основанные
на комбинации структурной информа-
ции, полученной с высоким пространс-
твенным разрешением (см. Часть Е) и
методов молекулярной биологии. На-
пример, стабильность белка можно из-
менять путем направленного введения
мутаций в область функционального
центра и последующего изучения кон-
формационных переходов при сворачи-
вании и разворачивании молекулы. По-
этому для термодинамики, так же как и
для многих других биофизических мето-
дов, структура с высоким разрешением
является скорее отправной точкой! для
исследований, чем конечной целью.
Неравновесная термодинамика ле-
жит вне поля зрения данной книги, но она
должна быть упомянута в нашем кратком
историческом обзоре, благодаря важно-
сти ее применения во многих областях,
включая экспериментальную физику,
химию реакций, биологию, экологию и
даже социологию. Необратимая термо-
динамика неравновесных процессов ин-
тенсивно развивается с конца 1950-х гг.,
в основном, в работах Ильи Пригожина
(I. Prigogine) и его сотрудников. Он ввел
термин диссипативные структуры для
определения открытых систем, которые
могут поддерживаться в состояниях, да-
леких от равновесных, благодаря прито-
ку энергии и вещества извне. Интересно,
что впечатляющие примеры самооргани-
зации в биологии, такие как клеточное
деление, дифференциация и морфогенез,
можно смоделировать в терминах дисси-
пативных структур.
В заключение заметим, что термо-
динамика не рассматривает величины и
свойства энергетических тепловых ба-
рьеров, которые должны быть преодо-
лены, прежде чем реакция начнет про-
текать самопроизвольно. Скорости и
механизмы реакций являются привиле-
гией специального раздела физической
Комментарии В1.1. Термодинамика '
и математика
Пытаться объяснять термодинамику i
без использования математики - значит
вводить читателя в заблуждение. Триумф
и полезность термодинамики объясняется \
тем, что она сформулирована в несколь-
ких законах, имеющих вид математических
уравнений, которые устанавливают точные 1
отношения между измеряемыми величи-
нами. Дифференциальные уравнения в час-
тных производных, которые сами следуют ;
определенным математическим правилам,
являются простым и элегантным способом
выразить изменения экспериментального 1
параметра, например, давления газа, когда
меняется другой, например, температура,
в то время как третий параметр остается .
неизменным, например, объем. Уравнения
говорят нам не только о том, что давление
вырастет, как мы и ожидали интуитивно, но
и о точной величине этого изменения для
данного температурного сдвига. Иаш при-
мер — просто иллюстрация, однако похожие
предсказания можно сделать в гораздо более !
сложных ситуациях, таких как химические
реакции или процесс сворачивания белка.
химии, называемого кинетикой, кото-
рая в этой книге не рассматривается.
В1.2. Законы термодинамики
Несмотря на свою сложность, язык клас-
сической и статистической термодина-
мики является чрезвычайно полезным
(на самом деле без него просто нельзя
обойтись). Он необходим для пони-
мания экспериментальных данных об
энергетике определенных систем — от
больших ансамблей частиц до одиноч-
ных молекул. Работая над этой главой,
авторы не пытались дать ничего, кроме
краткого описания принципов, лежащих
в основе этого языка. Мы приветствуем
его тщательное изучение, благо на дан-
ную тематику написано множество хоро-
ших книг, поскольку термодинамика иг-
рает фундаментальную роль в развитии
и приложении биофизических методов.
Классическая термодинамика пред-
ставляет собой феноменологическую
науку, занятую точным описанием опре-
деленных систем, например, растворов
макромолекул. Статистическая же тер-
модинамика старается объяснить про-
исходящее в терминах движения частиц
системы, например, молекул раствора и
растворенного вещества.
В данной главе дан лишь беглый об-
зор законов классической термодинами-
ки. Мы обсудим наиболее важные пара-
метры и математические соотношения
между ними, необходимые прежде всего
для понимания экспериментов по кало-
риметрии в исследовании связывания,
сворачивания/разворачивания и взаимо-
действия биологических макромолекул
(комменrapnii BI.I). Представленные
здесь основные принципы также пригод-
ны и полезны для изучения макромоле-
кул в растворе (Часть А), для гидроди-
намических экспериментов (Часть Г),
экспериментов но рассеянию (Часть Е),
опытов по спектроскопии (Часть 3) и
изучения одиночных молекул (Часть Д).
Мы ограничим описание равновесной
термодинамикой, которая обеспечивает
соответствующую базу для исследований
биологических макромолекул и их взаи-
модействий в основном in vitro.
В1.2.1. Основные определения
и нулевой закон термодинамики
Адиабатические и изотермические
системы, а также понятие равновесия
Мы рассматриваем систему как материю,
отделенную от внешнего мира стенками, с
которыми она не взаимодействует хими-
чески. Адиабатическая (от греческого «не
позволяет проходить») стенка полностью
изолирует систему от внешнего воздейс-
твия. Неадиабатические стенки называ-
ют диатермическими (от греческого dia
«сквозь» и theme «тепло»). Две системы,
отделенные такой перегородкой, являют-
ся изотермическими. Условие, в котором
находится система, называется ее состо-
янием. Свойства конкретного состояния
можно определить из эксперименталь-
ных измерений. Два состояния одинако-
вы, когда все измеренные значения, ха-
рактеризующие их свойства, одинаковы.
Система, заключенная в адиабатический
сосуд, со временем достигает состояния,
в котором ее свойства перестают менять-
ся. Тогда говорят, что система пришла в
равновесие. Обратите внимание, что рав-
новесие обозначает статичность условий
(их неизменность) только в отношении
макроскопических свойств системы. Газ
или раствор, находящиеся в равновесии,
представляли бы собой высокодинамич-
ные системы, если бы мы наблюдали бро-
уновское движение молекул или частиц
растворенного вещества. Именно о таких
системах идет речь в Главе ПО, посвящен-
ной динамическому рассеянию света.
Нулевой закон термодинамики
Нулевой закон термодинамики был
установлен после первого и второго.
В основном он определяет понятие тем-
пературы. Поскольку он важен для по-
нимания других законов, его называют
нулевым. Он утверждает, что если два
тела А и Б находятся в равновесии с
третьим телом В, тогда А и Б находятся
в равновесии друг с другом.
Следствием данного закона является
существование некоторой эмпирической
функции состояния системы — темпера-
туры, которая принимает одинаковые
Комментарий В1.2. Температура
; То, что мы сегодня называем темпера-
’ турой, по сути дела есть мера средней ки-
I нетической энергии молекул некоего тела.
Вполне ясные статистические понятия тем-
пературы и тепла были осознаны далеко не
сразу. Существовавшие ранее представле-
| ния о температуре было основано на опре-
! деленных свойствах тела, находящихся в
: равновесии. Тепло рассматривалась как не-
видимая калорическая жидкость — тепло-
род, которая перетекает от более нагретого
тела к холодному, находящемуся в контак-
i те с первым, и это приводит к изменению их
i температуры. Нулевой закон термодинами-
' ки может быть сформулирован через поня-
тие температуры и теплового равновесия:
«Две системы, находящиеся в тепловом
равновесии с третьей, находятся в тепло-
вом равновесии друг с другом».
значения для различных систем в равно-
весии (комментарий 151.2).
Функции состояния и сопряженные
переменные
Наблюдаемый параметр, значение ко-
торого зависит только от состояния
системы, а не от способа, которым оно
достигнуто, называется функцией со-
стояния. Температура является такой
функцией. Внутренняя энергия, энт-
ропия, давление и объем представляют
собой другие примеры функций состоя-
ния. Параметр, который зависит от раз-
мера системы, называется экстенсивной
переменной, в то время как параметр,
зависящий от условий, но не от размера
системы, называется интенсивной пе-
ременной. Объем, внутренняя энергия
и энтропия — все это экстенсивные пе-
ременные. Давление, температура или
значение pH раствора — интенсивные
переменные.
• Комментарий В 1.3. Обозначения :
для термодинамических функций
В этой главе мы будем использовать
следующие обозначения, необходимые для ,
записи уравнений, в которых фигурируют
теплота и другие формы энергии. Приведен-
ные ниже символы отражают следующие
термодинамические функции: U- внутрен-
няя энергия, Н — энтальпия, 5 -- энтропия,
G — свободная энергия Гиббса, F - свобод-
ная энергия Гельмгольца, VI7 работа, Q —
теплота. Функции U, Н. G. IVи Q имеют раз- * 1
мерность энергии и чаще всего выражаются
в калориях. Функция 5 выражается в кало-
риях или в джоулях, деленных на градусы. ,
К сожалению, в мировой литературе не на- ;
блюдается однозначности в этих символах,
за исключением энтальпии и энтропии.
Комментарий В 1.4. Определение
теплоты
Уравнение В 1.1 определяет теплоту сис-
темы. Для системы, окруженной адиабати-
ческими стенками, изменение внутренней
энергии дается выражением АС/ = АН7, по-
этому значение величины Q — эго мера того,
насколько стенки неадиабатны и тепло сио- ’
собно поступать в систему снаружи. Можно
; показать, что Q обладает свойствами, кото-
! рые мы обычно ассоциируем с теплотой:
— добавление тепла к системе изменяет |
ее состояние:
— тепло передается от одной системы
к другой благодаря проводимости, конвек-
ции или излучению: |
— в калориметрическом эксперименте I
I с адиабатной системой тепло сохраняется.
В уравнениях термодинамики, кото-
рые описывают отношения между различ-
ными функциями состояния, его элементы
(члены) выражаются в единицах энергии
(комментарий BI.3). Когда такой член вы-
ражения содержит два параметра, эта пара
состоит из так называемых сопряженных
переменных. Давление и объем, а также
энтропия и температура представляют со-
бой сопряженные переменные (в качестве
примера см. уравнение В1.5).
В1.2.2. Первый закон и энергия
Первый закон термодинамики форму-
лировали по-разному. По существу, он
утверждает, что «количество работы,
необходимое для изменения состояния
системы от начального до конечного,
зависит исключительно от самого про-
изведенного изменения, а не от способа,
которым проделана работа, и не от про-
межуточных состояний, через которые
система могла пройти».
Первый закон определяет внутрен-
нюю энергию (U) как свойство началь-
ного и конечного состояний системы,
чье значение изменяется под действием
произведенной над ней работы.
Сохранение энергии
Сохранение энергии, безразлично ка-
кой — механической, химической,
тепловой или любой другой, является
следствием первого закона термодина-
мики. Интересно, однако, что сам этот
закон нельзя получить из утверждения
о сохранении энергии.
В соответствии с первым законом,
изменение внутренней энергии систе-
мы АС/дается уравнением:
AC/ = AQ + AIK, (В1.1)
где 1У- количество механической рабо-
ты (или химической, или любой другой,
не имеющей форму тепла), произведен-
ной над системой, a Q — добавленное к
системе тепло ( коммеи гарпй В I. i).
Химический потенциал
Химический потенциал растворенного
вещества ц — это энергия, необходимая
для того, чтобы добавить одну молекулу
(или один моль молекул, в зависимости
от определения термина) в раствор. Мы
можем разложи ть работу И7 в уравне-
нии В 1.1 на два вклада — химический
потенциал и механическую работу
Д[/=Д(2 + цА« -PAV, (В1.2)
где Д// — ото разность в количестве мо-
лекул растворенного вещества между
начальным и конечным состояниями
(в зависимости от используемой шка-
лы, данная величина может быть выра-
жена и в молях). Механическая работа
представлена в терминах давления Р и
изменения объема ДИ Химический по-
тенциал - это интенсивная переменная.
Сопряженная с ней концентрация — эк-
стенсивная переменная. Для изменений
при постоянном давлении член, содер-
жащий химический потенциал, связан с
функцией, называемой свободной энер-
гией Гиббса (параграф В1.3.1).
В1.2.3. Второй закон и энтропия
Прямым следствием понятия равнове-
сия является то, что многие типы изме-
нений, которые удовлетворяют первому
закону, на практике произойти просто не
могут. Наблюдаемый факт — движение
изолированной системы к равновесию,
например, стремление молекул раство-
ренного вещества равномерно заполнить
сосуд после удаления стенки, ограничи-
вавшей их в отсеке, исключает обратный
путь (в нашем примере движение моле-
кул обратно в отсек сосуда), если даже
он и не нарушает первый закон.
Второй закон
Мы можем выразить направленность оп-
ределенных термодинамических измене-
ний в терминах субъективных понятий —
горячее и холоднее. Существует четкая
тенденция перетекания тепла от более го-
рячего тела к более холодному. Формули-
ровка второго закона, сделанная Клаузиу-
сом, включает следующее определение:
«Невозможно создать машину, которая,
работая циклично, способна произвести
другой эффект кроме переноса тепла от
горячего тела к холодному».
Обратимость
Благодаря своим экспериментам Кар-
но продемонстрировал, что кратчайший
путь к превращению теплоты в работу
заключается в использовании машины, в
которой каждый процесс обратим. Обмен
тепла в таком идеальном устройстве про-
исходил бы между телами с почти одина-
ковой температурой (рис. Bl. I). Если мы
слегка охладим одно тело, тепло потечет
к нему от другого; если же мы немного
нагреем его, тепло потечет в обратную
сторону. Этот процесс обратим. Точно
таким же обратимым образом мы можем
произвести работу над объемом газа, изо-
I’ ч . BLI. Обратимый перенос тепла между
двумя телами
Рис. В 1.2. Работа, совершаемая над объемом
газа в обратимой манере
термическим со своим окружением при
температуре Т, очень медленно двигая
поршень в отсутствие трения в одном или
другом направлении, заставляя тепло
Q покидать газ или попадать в него, в то
время как его температура оставалась бы
очень близкой к Т(рис. В 1.2).
растет со временем, пока не достигнет
максимума в состоянии равновесия
Д5>0. (В1.3)
Равенство (нулевое изменение энт-
ропии) соблюдается в пределах обрати-
мых изменений, во время которых систе-
ма считается находящейся очень близко
к равновесию.
Изучая следствия второго закона,
было найдено, что энтропия соотно-
сится с членом, содержащим теплоту
в уравнении В1.1 как:
Д5 = Д(2/Г (В1.4)
Уравнение В1.4 определяет единицы,
в которых выражается энтропия, в тер-
минах энергии и температуры. Теперь
мы можем переписать уравнение В 1.2:
ДС/ = T&S + цД« — РДV. (В1.5)
Энтропия
Клаузиус выразил тенденцию изолиро-
ванной системы, которая не обменива-
ется энергией или веществом со своим
окружением, к достижению равнове-
сия через определение функциональ-
ного состояния — энтропии S, которая
' Комментарий В1.5. Энтропия
и вероятность i
| Связь между энтропией и вероятностью
I легко просматривается исходя из нашего
житейского опыта о наиболее вероятных
событиях. Связь между энтропией и ве-
роятностью описывается уравнением 5 = I
= А lnp+В, где р — вероятность события, А и '
В — постоянные. Логарифмическая форма
। этого уравнения следует из того, что для ,
| получения вероятности конечного события ,
| вероятности надо перемножить, а энтропии i
1 отдельных событий надо сложить. '
Энтропия и беспорядок
Наш пример, в котором молекулы двига-
лись так, чтобы заполнить весь доступ-
ный объем по мере достижения системой
равновесия, есть отражение простого
факта — равновесное состояние — это
максимально неупорядоченное состоя-
ние. Ее компоненты «пробуют» все
возможные из доступных им конфигу-
раций. Больцман в своей трактовке ста-
тистической термодинамики выразил
эту мысль количественно и соотнес энт-
ропию S с числом всех доступных систе-
ме конфигураций р через уравнение:
S = kB\np, (Bl.6)
где kB — универсальная константа Боль-
цмана ( KI >.М М('11 гарнп В I .."> и В 1 .(>).
В отличие от внутренней энергии (U)
и энтальпии (Н) энтропия имеет размер-
ность энергии, деленной на температуру,
так как она представляют собой тепловую
энергию, рассеянную при определенной
температуре, или сумму энергетических
потерь, приходящихся на градус в данном
температурном интервале. Поскольку
S имеет размерность энергии, деленной
на температуру, для получения энергии,
надо умножить ее на Т, и, следовательно,
TS имеет размерность энергии.
Уравнение В1.6, объединенное
с уравнением второго закона термодина-
мики В 1.3, показывает, что при равнове-
сии число доступных системе конфигу-
раций максимально. При рассмотрении
уравнений В 1.4 и В 1.6 мы приходим к
выводу, что TS — это «распределенная»
тепловая энергия системы, необходимая
для того, чтобы перебрать все конфигу-
рации р при температуре Т, выраженной
в единицах Кельвина. Ниже, в парагра-
фе В 1.3.1, мы увидим, что эта энергия не
является «свободной энергией», т. е. с ее
помощью нельзя произвести работу.
Второй закон термодинамики — за-
кон статистический, он соблюдается
только тогда, когда мы рассматриваем
большое число событий. Когда в ка-
ком-то макрообъеме содержится не-
сколько молекул газа, то вполне вероя-
тно, что в какой-то момент времени
они соберутся у одной стенки. Однако,
если в объеме очень много молекул, то
их распределение будет случайным и
практически однородным. Если рас-
сматриваемый объем сильно умень-
шить, то в какой-то данный момент вре-
мени в двух маленьких объемах будет
содержаться разное число молекул (см.
Главы ПО и Г11).
В1.2.4. Третий закон
и абсолютный нуль
Тепловая теорема Нернста, известная
также как третий закон термодинами-
Комментарий В 1.6. Константа
Больцмана и определение
температурной шкалы
| Константа kB имеет ту же размерность,
что и энтропия: kB = 1,3806503 * 10-23Дж-К1.
На молекулярном уровне энергия, связан-
ная с одной степенью свободы и темпера-
: турой Т (в единицах Кельвина), равна (Уг)
kgT. Число Авогадро (число частиц на один
! моль) = 6,02214199 х 1023моль’. Это чис-
ло, умноженное на константу Больцмана,
равно газовой константе R, R = R =
' = 8,34624 Дж-К'моль1. Заметим, что раз-
: личие между k и R только в одном: k упот-
ребляют, когда говорят об одной частице, а
R — о моле частиц.
ки, утверждает, что «энтропия любого
тела равна нулю при нулевой темпера-
туре на абсолютной шкале».
Классическая формулировка не со-
держит утверждения о нулевой энтро-
пии (или температуры) — там опреде-
ляется только разность этих величин
между двумя состояниями. Третий за-
кон дает нам нулевую точку на абсо-
лютной шкале энтропии. Поскольку
было показано, что не существует про-
цесса, результатом которого была бы
нулевая энтропия, альтернативная фор-
мулировка третьего закона звучит так:
«Не существует такой конечной серии
процессов, с помощью которой можно
достичь абсолютного нуля». Другими
словами, когда температура стремится
к нулю, величина изменения энтропии
в любом обратимом процессе тоже стре-
мится к нулю.
Абсолютная температура
Третий закон определяет абсолютный
нуль на абсолютной шкале. В ней абсо-
лютная температура выражается в еди-
ницах Кельвина и растет с шагом в один
градус, который определяется как одна
Комментарии В1.7. Закон
Шарля-Гей-Люссака и шкала
Кельвина
вытекает из закона
В современном виде
процессе охлаждения
давлении на каждый
Шкала Кельвина
Шарля-Гей-Люссака,
этот закон гласит: в
газа при постоянном
градус Цельсия его объем Vo уменьшается
на 1/273,15 того объема, который он зани-
мает при О °C. Математически это выража-
t
ется уравнением VT = 1(, + х 1-( (при
постоянном давлении). Это уравнение пока-
зывает, что при понижении температуры до
-273,15 °C газ должен вообще не занимать
никакого объема. Ясно, что этот закон не мо-
жет выполняться точно. В качестве нижне-
го предела температурной шкмы, которая
получила название абсолютной темпера-
турной шкалы, была принята температура
-273,15'С, ставшая в ней нулем. Позднее
эту шкалу стали называть шкалой Кельви-
на, в честь лорда Кельвина, обосновавшего
ее важное теоретическое значение в термо-
динамике.
сотая разности температур замерзания
и кипения воды при атмосферном дав-
лении ( комментарии В1.7).
Третий закон выполняет важную
роль в физической химии. Устанав-
ливая нулевую точку энтропии и аб-
солютную температурную шкалу, он
позволяет рассчитать константы рав-
новесия из тепловых свойств реагентов
(параграф В1.3.2) и калориметричес-
кой полной внутренней энергии (параг-
раф В1.3.3).
В1.3. Основные понятия
и уравнения
Фундаментом классической термодина-
мики является набор простых с матема-
тической точки зрения уравнений, ко-
торые устанавливают то, как различные
переменные соотносятся друг с другом.
Эти уравнения можно записать и в тер-
минах переменных с помощью правил
дифференциальной геометрии. К при-
меру, уравнения Максвелла соотносят
друг с другом изменения давления Р,
объема V, энтропии 5 и температуры Т
жидкой системы. Здесь мы сосредото-
чимся на нескольких концепциях из хи-
мического приложения классической и
статистической термодинамики, кото-
рые помогут нам понять биологические
макромолекулы и их взаимодействия.
В1.3.1. Свободная энергия
и родственные понятия
Энтальпия
Энтальпия системы И определяется как
H=U + PV. (В1.7)
Термин «энтальпия» в переводе с
греческого означает «теплый внутри».
При постоянном давлении — условии,
характерном для растворов биологиче-
ских молекул, энтальпия системы при
температуре Т отражает полную энер-
гию, необходимую для нагрева системы
от абсолютного нуля до Т.
Уравнение В1.7 есть не что иное, как
просто полезное определение, характе-
ризующее Н как функцию состояния.
Поскольку абсолютные значения U и II
неизвестны, то это уравнение лучше ис-
пользовать на языке приращений:
АН = АС/+ A(PV). (Bl.7а)
Подобно АС/, величина А//, зависит
только от начального и конечного состоя-
ний. Уравнение В1.7 имеет характерную
особенность. Поскольку при постоян-
ной температуре объем идеального газа
изменяется обратно пропорционально
давлению, все члены PV равны друг дру-
гу и Л(РУ) = 0. Следовательно, в данном
случае энтальпия и внутренняя энергия
изменяются одинаково. Если газ расши-
ряется или сжимается против внешнего
давления, то АЯ = АС + РАЕ. В этом вы-
ражении — РАЕ — это работа системы,
расширяющейся при постоянном дав-
лении. Поэтому, если РАЕ = VE, то АЯ =
= Q, поскольку АС= Q + IE. Поэтому для
этого случая АЯ = Q}, где Qp означает ко-
личество тепла, поглощенного при пос-
тоянном давлении. Соответствующей
величиной для процесса, протекающего
при постоянном объеме, будет Qv
Изменение энтальпии АЯ во время
химической! реакции — это тепло, по-
глощенное или освобожденное при раз-
рушении и образовании связей. В ка-
лориметрии АЯ — экспериментально
измеряемая величина.
Свободная энергия Гиббса
Свободная энергия Гиббса G определя-
ется как:
G = U + РЕ - TS = Я - TS. (В 1.8)
G представляет собой ту часть энер-
гии системы, которая не используется
различными энтропийными конфигура-
циями. Отсюда следует, что, поскольку
энергия Гиббса не вовлечена в процесс
теплового переноса, она может быть
трансформирована в полезную работу,
отсюда и ее название — свободная энер-
гия ( К(>м.\к'11 г;Iр 11п В1.8).
Понятие свободной энергии Гиббса
привлекательно потому, что дает прак-
тический критерий самопроизволь-
ности данного процесса. Приращение
свободной энергии при постоянной тем-
пературе равно АС = АЯ — 7А5. Иными
словами, как уменьшение энтальпии,
так и увеличение энтропии способ-
Коммежарий В1.8. Свободная
: энергия Гиббса и свободная
энергия Гельмгольца
К понятию свободной энергии пришли :
независимо друг от друга Гиббс и Гель- ;
мгольц. Свободная энергия по Гельмгольцу
есть /•'= U — TS. На практике измеряется не
изменение внутренней энергии U изучае-
мого вещества, а измененное его энтальпии
; U + PV. Для большинства биологических
; макромолекул при атмосферном давле-
нии величину PV можно игнорировать, по-
скольку она намного меньше, чем тепловая
энергия тела. Поэтому представляется не-
существенным различие между свободной
энергией Гиббса и свободной энергией по
Гельмгольцу. И то и другое целесообразно
именовать просто свободной энергией.
ствуют уменьшению свободной энер-
гии, от чего зависит самопроизвольное
протекание реакции. Отрицательный
или положительный знак АС позволяет
судить, может ли реакция происходить
самопроизвольно или нет. Существуют
три общих правила для систем, функ-
ционирующих при постоянном давле-
нии и постоянной температуре: если
AG <0, то реакция может протекать са-
мопроизвольно, если AG >0, то реакция
не может протекать самопроизвольно,
если AG =0, то система находится в рав-
новесии.
Химическая реакция включает в себя
создание и разрушение различных типов
связей между атомами. В этом смысле
сворачивание биологических макромо-
лекул и их взаимодействия с лигандом
во время связывания являются хими-
ческими реакциями (рис. В 1.3). Энер-
гия ковалентных связей составляет
величину порядка 10’&вТ, ван-дер-ва-
альсовые, водородные и электроста-
тические связи в водных растворах, а
также так называемые гидратационные
и гидрофобные связи являются слабы-
б — Белок + Лиганд о Белок * Лиганд
Рис. В 1.3. Разворачивание белков (а) и их взаимодействие с лигандами во время связыва-
ния (б)
ми, поскольку их энергия составляет
величину порядка kBT при температуре
Т, близкой к комнатной — 300 К. Сколь
ни мала энергия этих связей в макро-
молекуле, она оказывается достаточ-
ной для того, чтобы противостоять
нарушению структуры, которое могут
вызывать столкновение с другими мо-
лекулами при комнатной температуре.
Однако по мере повышения темпера-
туры возрастающая сила молекуляр-
ных столкновений и возрастающая
энергия внутримолекулярных колеба-
ний вызывает разрыв и водородных и
неполярных связей, что служит при-
чиной тепловой денатурации биологи-
ческих макромолекул. Разворачивание
(рис. В 1.3 а) и взаимодействие с ли-
гандом (рис. В 1.3 б) могут включать в
себя формирование и разрушение как
нескольких ковалентных, так и мно-
жественных слабых связей. Обсужде-
ние относительных вкладов ван дер
ваальсовых сил, водородных связей,
гидратационных и гидрофобных вза-
имодействий в структуру белка и его
разворачивание будет обсуждаться
в Главе В2. При этом, как мы увидим
ниже, важнейшую роль играет взаимо-
действие с растворителем.
Обратимую мономолекулярную хи-
мическую реакцию можно записать как:
А # В. (В1.9)
При постоянной температуре, когда
система находится в изотермическом
контакте с большим объемом с посто-
янной температурой, как, например, в
случае с молекулами в разбавленном
растворе, свободная энергия, высво-
бождаемая в реакции, дается выраже-
нием
AG = АН -TAS. (В1.10)
Второй закон утверждает, что сис-
тема будет изменяться, с тем чтобы
максимизировать свою энтропию. Ана-
логично система, которая обменивается
энергией со своим окружением, тоже
изменяется, чтобы максимизировать
свою свободную энергию. Поэтому
реакция В 1.9 будет идти в том направ-
лении, для которого AG < 0. Когда два
состояния системы находятся в равно-
весии, разность их свободных энергий
равна нулю (AG = 0).
Картина свободной энергии
и распределение Больцмана
Когда системе доступны несколько
состояний с различными уровнями
энергией, они заселяются в разной сте-
пени в зависимости от температуры
(рис. В1.1).
Вероятность того, что состояние
со свободной энергией G. при темпе-
ратуре Т будет заселено, можно пред-
ставить Больцмановским распределе-
нием:
С=ехр - G}/RT, (Bl.11а)
так что
G.= -/?TlnC.. (Bl.116)
Отсюда следует, что вероятность
состояния с энергией 2RTбудет отно-
ситься к таковой с энергией RT, как
е~2.
Конформация
Рис. В1.4. Диаграмма свободной энергии сис-
темы как функции ее конформации. В нашем
примере показаны два состояния, разделен-
ные разностью свободных энергий ДС. Избы-
ток свободной энергии, или энергетический
барьер Е*, называют энергией активации двух
состояний
Функция распределения
(статистическая сумма)
Функция распределения Q определя-
ется как сумма статистических вкла-
дов всех доступных системе состоя-
ний:
Q = XC;=ZeXP(-AG;//?7')- <В112)
;=0 ;=0
Определяя состояние j = 0 как репер-
ное, по отношению к которому термо-
динамические переменные выражаются
как избыточные величины, уравнение
В 1.12 можно переписать:
Q-l + Xexp(-AG;//?r). (Bl.12а)
7=1
Заселенность j состояния Р. дается
соотношением статистического вкла-
да этого состояния к сумме статистиче-
ских вкладов всех состояний, доступ-
ных системе:
P^expf-AGyPTj/Q. (Bl.126)
Заметим, что^Д = 1. a P( = \/Q.
В общем случае, средние экспери-
ментальные значения свойства а сис те-
мы выражаются как
И = (В1.12в)
,-о
где а. — значение а, относящееся к со-
стоянию/.
Уравнение В1.12 (в) является осно-
вой для теоретических или модельных
расчетов величины <а>, проводимых
для сравнения с экспериментальными
наблюдениями.
Средняя свободная энергия системы
<ДС> = <G - G’( > записывается как:
(AG) = X/JA=-7?7'lnQ- (Bl-12г)
7 = 1
Средняя избыточная энтальпия вы-
ражается как:
,-1 (В1.12д)
= /?7'2(ainQ/ar),
а средняя избыточная энтропия дается
выражением
(Д5) = УДД5, =
Х (В1.12е)
= RT(d\nQ/dT)+R\nQ.
Уравнения В 1.12 (г-е) применимы
к любому свойству системы.
Химический потенциал
Из обсуждения уравнения В 1.2 мы
помним, что химический потенциал р
растворенного вещества — это энергия,
требуемая для добавления одной мо-
лекулы в раствор. На самом деле такая
энергия является свободной энергией,
поэтому химический потенциал может
быть приравнен к изменению свобод-
ной энергии в результате добавления
одного моля растворенного вещества
в раствор. Химический потенциал или
изменение свободной энергии в резуль-
тате растворения вещества до конечной
концентрации /V. (в молях на объем) да-
ется формулой
^р;=дС; = -7?ПпУ,. (В1.12ж)
Заметьте, что сравнивая уравнения
Bl.ll (б) и В1.12 (ж), мы приходим
к выводу об эквивалентности N и С\
Иными словами, раствор с концентра-
цией N. представляет собой состояние
С системы.
1
Для раствора, содержащего несколь-
ко различных веществ:
AG = -7?7’^lnA\ =
= -РТ\пК^.,К3...= (В 1.13)
= -7?7'1пПУ,.
Свободная энергия связана с хими-
ческим потенциалом ц простым образом:
р = G/N.
Если в этой формуле У означает чис-
ло молекул, то химический потенциал
относится к одной молекуле. Если же
Nозначает число молей, то химический
потенциал относится к молю молекул.
Биологическое стандартное
состояние
Поскольку мы имеем дело с разностью
потенциалов или уровней энергий, по-
лезно определить стандартное состоя-
ние, по отношению к которым они из-
меряются. Если не определено иначе,
температура Т принимается равной
298,15 К (25 °C). Для газов принято
парциальное давление в 1 атмосферу.
Концентрация вещества в водных рас-
творах равна 1М. Если в реакции участ-
вуют протоны, биологическое стандарт-
ное состояние имеет pH 7.
Химические реакции
Запишем реакцию В 1.9 в виде
/1| + /Е + + ••• ч- ВI +...
где А — реагенты, а В — продукты. Ис-
пользуя уравнение В 1.13, рассчитаем
изменение свободной энергии реакции,
идущей слева направо, в терминах кон-
центраций реагентов и продуктов по
формуле:
да = -ять пв- яги гы =
= -ЯПпх(ПВ/ПЛ)= (В1.14)
= -Я7’1пЯ,
(гдеЯ= [BJ [В,] [BJ.../HJ [Л,] [А,]...).
Как следует из нашего определения,
в стандартных условиях К = 1. Стандарт-
ная свободная энергия ДС" определяется
как изменение свободной энергии, когда
реакция начинается со значения К = 1 и
приходит к равновесию при К = КР.иш, где
нет различий в свободной энергии меж-
ду продуктами и реагентами:
\G" = -RT\nKv (В 1.15)
Теперь запишем изменение свобод-
ной энергии для реакции, начинающей-
ся с любого значения К
лс = лС"-(-«о„Ю= (В116а)
= ДС" + ЯТ1пЛ'
или
дс = -ят1пл;./к. (В1.166)
Обратите внимание, что ДС" равно
нулю только если Я = 1 (т. е. когда
произведение концентраций В^Ву.. =
= Л, Л,ЛГ..).
Если К > 1, Д(7 отрицательна и реак-
ция протекает спонтанно в стандартных
условиях. Если К < 1, ДС положительна
и реакция требует затрат энергии для
протекания в тех же условиях.
Анализ Вант-Гэффа
Уравнение В1.15 является основой ана-
лиза Вант-Гоффадля систем с двумя со-
стояниями (А <-> В). Здесь стандартная
свободная энергия и, соответственно,
энтальпия и энтропия рассчитываются,
исходя из полученных значений равно-
весных концентраций двух состояний.
Напомним, что стандартная свобод-
ная энергия, энтальпия и энтропия свя-
заны уравнением ВЕЮ:
ДС"=ДЯ"-7’Д5".
Комбинируя его с уравнением В 1.14,
получим:
1пКр.1|Ш = -ДЕТ/RT + Д5"/R, откуда
и найдем уравнение Вант-Гоффа:
АЯ„.1.1„Мш = /?7’2Э1пК1)./дТ. (В1.17)
В1.3.2. Изучение связывания
Константы связывания и сродства
(афинности)
Если молекула А связывается с молеку-
лой В, формируя комплекс АВ, термоди-
намическая равновесная константа дис-
социации К] определяется в терминах
равновесных концентраций отдельных
компонентов — [А], [В] н [АВ] и равна:
|ЛВ] '
(В1.18)
Здесь мы следовали условиям, при-
нятым для экспериментов по изучению
связывания, и записали равновесную
константу как константу диссоциации.
Глядя на уравнение В1.14 и учитывая,
что Kf = //К , приходим к выводу, что
константа ассоциации К )н — это мера
«силы» связывания или сродства взаи-
модействия.
Константа К, соотносится с хими-
ческой константой скорости ассоциа-
ции А и В, k , и константой скорости
диссоциации комплекса АВ, k , через
уравнение В1.19 ( мппкт inpiiii В I.9):
К, = k /k . (Bl.19)
Связывание со многими сайтами
Если молекула А имеет более одного
сайта для связывания с молекулой В, то
существует несколько условий связыва-
ния для В. Говорят, что оно кооператив-
но, если заполнение одного сайта способ-
ствует связыванию с другим. Если же,
наоборот, происходит снижение аффин-
ности другого сайта, то связывание анти-
кооперативно. Все эти процессы можно
идентифицировать и проанализировать
Комментарий В 1.9. Константы
равновесия и скорости
Рассмотрим одноступенчатую реакцию
связывания: ;
А+В<->АВ.
Скорость прямой реакции, которая пре-
вращает А и В в АВ, равна
[А] [В].
Скорость реакции, превращающей АВ
обратно в А и В, равна
*,,JAB].
Равновесие достигается, когда эти две
скорости равны и
^[А][В] = ^,сс[АВ].
Перестановка даст нам
W^=[A][B]/[AB] = K,.
экспериментально. В том случае, когда
сайты идентичны и независимы, констан-
та диссоциации для каждого из них оди-
накова и не зависит от того, занят другой
сайт или нет. Если молекула А имеет п
идентичных и независимых сайтов для
лиганда В, то среднее число лигандов п ,
действительно связанных с молекулой А,
дается выражением
>7 = -^-,
К,/+[В]
(В1.20)
где [В] — это равновесная концентра-
ция свободного лиганда. Используя
подход Скэтчарда перепишем это урав-
нение как
п п п
[В] Kd к,-
(В1.21)
Среднее число связанных лигандов
п, также как значение концентрации
свободного лиганда [В], получают из
экспериментов по связыванию в равно-
весном состоянии. Откладывая на гра-
фике как функцию от п, получим
1
прямую линию с наклоном —— , пере-
секающую ось х в точке п, которая дает
число сайтов. Пример графика Скэт-
чарда можно найти в Главе В4.
Свободная энергия связывания
Зная Kd, нетрудно вычислить энергети-
ку взаимодействия между лигандом и
рецептором. Разность свободной энер-
гии Гиббса между связанным и несвя-
занным состояниями можно приписать
различным химическим и физическим
факторам. Вспомним уравнение ВЕЮ
AG = АГ/ — TAS, а также то, что реакция
протекает спонтанно в направлении
убывания свободной энергии, т. е. ког-
да AG < 0. Энтальпия АН отражает из-
менения свободной энергии системы,
сопровождающие внутримолекуляр-
ные взаимодействия при связывании.
Энтропия Л5 дает описание степеней
свободы компонентов, участвующих в
реакции связывания. Произойдет ли
между ними взаимодействие — зави-
сит от AG, а не от каждого члена по от-
дельности. Поэтому при связывании
не исключены значительные отри-
цательные (невыгодные) изменения
энтропии, если они компенсируются
соответствующими изменениями эн-
тальпии, которые, в свою очередь, мо-
гут отражать формирование выгодных
взаимодействий, таких как солевые
мостики и водородные связи. Посколь-
ку для подавляющего большинства
биологических макромолекул реакции
происходят в водной среде, то очевид-
но, вода с ее уникальными свойствами
(см. Главу А2) должна вносить фунда-
ментальный вклад в такие взаимодей-
ствия (параграф В 1.3.4 и секция В2).
Расчет стандартных изменений
энтальпии и энтропии после
связывания
Вспомним, что для стандартной свобод-
ной энергии реакции уравнение имеет
вид:
AG'' = -,mnK|K.
а в терминах Kd
AG" = RT\nKd. (В1.22)
Переписав уравнение Bl.10, мы по-
лучим:
АН" AS"
InK, (В1.23)
'' RT R
Таким образом, график зависимости
In Kd от 1 /Т(график Аррениуса) даст пря-
мую линию с наклоном, пропорциональ-
ным изменению стандартной энтальпии
после связывания. Сопутствующее изме-
нение стандартной энтропии можно оп-
ределить из уравнений В1.10 и В1.22.
Авидность
Когда процесс связывания обусловлен
другими менеезначительными события-
ми, происходящими в месте контакта
двух молекул, совокупная сила связы-
вания называется авидностью и, также
как аффинность, она измеряется через
константу равновесия. То есть авид-
ность получается в результате учета
аффинностей для различных сайтов, и
она больше суммы всех аффинностей
(।з|\1М( Нтарш| В 1.10).
Важным следствием взаимосвязи
между стандартной AG и авидностью
Комментарий В1.10. Авидность
। Стандартная свободная энергия связы-
. вания ряда слабых независимых сайтов да-
ется выражением:
AG = AGn = AG, + AG. + AG3...
п-1
II
AG = Y(-RT-\nK^= :
И=1 .
= -RT InKdi - RT-InKd2 -
-RT\nKd3...
где Kd — равновесная константа диссоциа-
ции связывания /.
Совокупная Kd называется авидностью
реакции. Она получается произведением
(ио не суммированием) отдельных аффин-
ностей:
Kl = Kll^Kd.,xKd.ix...
является то, что даже неоолыпое изме-
нение в одном из сайтов этой цепочки
способно привести к очень большим из-
менениям в совокупной константе рав-
новесия.
В1.3.3. Термодинамика
и связывание
Полный набор термодинамических урав-
нений, описывающих систему, можно по-
лучить из функциональной зависимости
энтальпии от температуры, которую, в
принципе, могут дать калориметричес-
кие измерения.
Сканирующая калориметрия изме-
ряет тепловой обмен в системе при из-
менении температуры. Температура (Т)
и энтальпия (//) - это сопряженные эк-
стенсивные и интенсивные переменные
(параграф В1.2.1). Когда изменение в
системе, например, сворачивание белка
пли связывание лиганда, происходит в
результате изменения температуры, то
соответствующим образом изменяется и
энтальпия. Тепло поглощается (ДН > О'),
если реакция вызвана повышением тем-
пературы, или выделяется (ДН< 0), если
она результат понижения температуры.
Теплоемкость
В калориметрии макромолекулярных
растворов измеряется величина, пред-
ставляющая собой теплоемкость при
постоянном давлении С
С„=(ЭЯ/Э7')р. (В1.24)
Функции, соответствующие энталь-
пии и энтропии, Н (Т) и 5 (Т) получают
интегрированием С по температурно-
му диапазону:
Н = H(Tn) + J СР (T)dT. (В1.25 а)
Г
5 = 5(T) + J(C,,(T)/Ty/7’. (В 1.25 б)
Мы с вамп видели в параграфе В 1.3.1,
что стандартная температура Tt в физике
равна абсолютному нулю, поэтому при
постоянном давлении энтальпия сис-
темы при температуре 7' отражает пол-
ную энергию, которая необходима для
нагрева системы от абсолютного нуля
до Т. Однако в биофизике обычно стан-
дартную температуру Г принимают рав-
ной комнатной температуре (298,15 К).
В случае протекания реакции, чле-
ны И и 5 в этих уравнениях заменяют
на ДН и А 5, которые соответствуют
изменениям во время реакции. Изме-
нение теплоемкости в результате ре-
акции при температуре Г дается выра-
жением
АСГ = АС,,.,. + J d^Cr’%T\(lT, (В 1.26)
где подстрочные индексы указывают
температуру и давление.
Влияние температуры на связывание
Из указанных выше уравнений и выра-
жения В 1.15, которое соотносит конс-
танту диссоциации с изменением стан-
дартной свободной энергии, получим
для последней уравнение:
ag; = ан; +тдд; +
+7'дс;
(В1.27)
бДС
Если в данном температурном диа-
пазоне теплоемкость постоянна, выра-
жение упрощается до выражения:
AG"r = a//" + т x да; + т x дс; x
(Bl.28)
которое является формой уравнения
Вант-Гоффа. АН‘‘ , А5" и АС" оцени-
вают с помощью нелинейной аппрок-
симации AG'/-, измеренной при разных
температурах.
В1.3.4. Термодинамика переноса
неполярных групп в воду
Рассмотрим характерные термодина-
мические эффекты переноса в воду
простых алифатических соединений
на примере типичной неполярной мо-
лекулы бутана. Для молекулы бутана
величина AG для такого переноса рав-
на +6 ккал на моль при 25 °C. Как мы
видели ранее, положительная величи-
на АС означает низкую растворимость
бутана в воде. Однако из этого не сле-
дует; что низкая растворимость бута-
на в воде связана с неблагоприятными
изменениями энтальпии. При 25 "С
величина АН невелика (около 0,8 ккал
на моль), что должно благоприятс-
твовать переходу в воду. Однако пе-
ренос сопровождается очень боль-
шим уменьшением энтропии (Д5 =
= 22 кал-град '-моль '), которое пере-
вешивает с лихвой небольшое отрица-
тельное значение АН, что, в конечном
счете, приводит к положительному AG
и не дает бугану растворяться в воде.
Напоминаем, что энтропия входит в
свободную энергию со знаком минус
(уравнение В1.10).
Причина сильного падения энтро-
пии проста: молекула бутана экрани-
рует своей неполярной поверхностью
часть водного пространства и частич-
но «замораживает» примыкающие
к ней водородные связи. Это и прпво-
Рпс. В1.5. Зависимость свободной энергии
AG и ее составляющих АП и AS от темпера-
туры для переноса пентана в воду. Величины
даны в ккал в расчете на моль переносимых
молекул пентана (Privalov and Gill, 1988)
дит к сильному энтропийному эффекту
( комментарий В 1.1 1 ).
Гидрофобный эффект сильно зави-
сит от температуры. На рисунке В1.5 по-
казана термодинамика переноса другой
неполярной молекулы, пентана в широ-
ком интервале температур. Как видно,
AG растет с ростом температуры, до-
стигает своего максимума в точке, где
AS = 0. Особенно сильно температура
Комментарий В 1.1 1. Гидрофобность
и доступная поверхность
Анализ характерных термодинами-
ческих параметров переноса неполярных
групп в воду показывает, что AG растет с
размером неполярной группы. Под разме-
ром неполярной группы следует понимать
доступную для воды поверхность полярных
групп. Та же закономерность имеет место
для аминокислотных остатков, если толь-
ко считать только ту доступную для воды
поверхность, которая создается истинными
iieiюлярпым11 атомам и.
Конформационная координата
Рис. В 1 .(>. Профиль свободной энергии ре-
акции или конформационного изменения.
Е* — это энергия активации или энтальпия
активации, в зависимости от используемой те-
ории (см.текст)
леть. Конформационное пли другое из-
менение зависит значения равновесно-
го состояния AG, которое должно быть
меньше нуля, однако скорость проте-
кания реакции обусловлена высотой
энергетического барьера (рис. В 1.6).
Наблюдаемые при различных темпе-
ратурах временные константы реак-
ции или изменения являются мерами
соответствующих термодинамических
параметров.
Зависимость константы скорости k
от температуры дается уравнением Ар-
рениуса:
k = А exp
-£*)
RT )'
(В1.31)
сказывается на величине и знаке компо-
нент свободной энергии. Если при низ-
кой температуре гидрофобный эффект
создается энтропией, то при повыше-
нии температуры все большую роль на-
чинает играть энергия разрушающихся
водородных связей. При температуре
свыше 140 °C, когда большинство свя-
зей разрушено, гидрофобный эффект
выходит на плато.
Температурная зависимость, пред-
ставленная на рисунке В1.5, играет
важную роль в понимании гидрофоб-
ного эффекта в глобулярных белках,
поскольку перепое неполярной груп-
пы в воду моделирует разворачивание
белковой глобулы, при котором часть
гидрофобных аминокислотных остат-
ков оказывается в водном окружении
(см. секцию В2.4).
В1.3.4. Термодинамика и активация
Термодинамический путь реакции
(конформационное изменения в моле-
куле) часто включает энергетический
барьер, который необходимо преодо-
где А — константа. В статистической
термодинамике А включает в себя ве-
роятность изменения (например, вероя-
тность столкновения двух партнеров
по взаимодействию), а Е* является
энергией активации. В теории переход-
ного состояния А зависит от энтропии
активации, а Е* от энтальпии актива-
ции. Величина А определяется кванто-
во-механической моделью переходного
состояния Эйринга как:
Ъ Т f АС *)
(В1-32)
где kl} — константа Больцмана, h — кон-
станта Планка, а Д5* — энтропия акти-
вации. Комбинируя уравнения В 1.31 и
В1.32 и подставляя энтальпию актива-
ции Е* = АН*, получим
I (k
In —
I Г
Д5* АН*
Т +—-ТгГ'<в1М)
Энтальпию и энтропию активации
можно получить из наклона и точки пе-
ресечения линии In (k/T), отложенной
против 1/Т (график Эйринга).
В1.4. Перечень ключевых идей
® Классическая термодинамика — это феноменологическая наука, которая зани-
мается описанием систем в определенных условиях; растворы макромолекул
можно считать термодинамическими системами.
• Адиабатическая система идеально изолирована от внешних воздействий; изо-
термическая система находится в тесном тепловом контакте со своим окруже-
нием.
• Две системы пребывают в одинаковых состояниях, когда все измеренные вели-
чины, характеризующие их свойства, одинаковы.
• Заключенная в адиабатический сосуд система со временем достигнет состоя-
ния, в котором ее свойства больше не будут меняться; тогда говорят, что систе-
ма находится в равновесии.
* Нулевой закон термодинамики утверждает, что если два тела А и В по отде-
льности находятся в равновесии с третьим телом В, тогда А и В тоже находятся
в равновесии друг с другом.
• Первый закон термодинамики гласит, что величина работы, требуемой для
изменения системы от начального до конечного состояний, зависит исключи-
тельно от производимого изменения, а не от промежуточных состояний, через
которые система может пройти.
• Согласно второму закону, невозможно создать машину, которая, работая цик-
лически, произведет другой эффект кроме передачи тепла от горячего тела
к холодному.
• Третий закон термодинамики гласит, что энтропия любого тела равна нулю
при нулевой температуре на абсолютной шкале.
• Третий закон устанавливает нулевую точку для энтропии и абсолютной тем-
пературной шкалы (шкалы Кельвина) и позволяет рассчитать константы рав-
новесия, исходя из тепловых свойств реагентов и полной внутренней энергии,
определяемых калориметрически.
• Энтропия системы — это функция состояния, которая растет со временем, пока
не достигнет максимума в состоянии равновесия; энтропия напрямую зависит
от числа различных доступных системе конфигураций, т. е. является мерой бес-
порядка в системе.
Энтальпия системы при температуре Т отражает полную энергию, необходи-
мую для того, чтобы нагреть тело от абсолютного нуля до Т; изменение энталь-
пии во время химической реакции обусловлено поглощенным или выделенным
теплом при разрыве или образовании связей.
Свободная энергия Гиббса — это часть энергии системы, которая не расходует-
ся на заселение различных энтропийных конфигураций; если система обмени-
вается энергией со своим окружением, то она будет изменяться для того, чтобы
максимизировать свою свободную энергию.
Когда два состояния системы находятся в равновесии, разность их свободных
энергий равна нулю.
Все термодинамические переменные системы можно записать в терминах фун-
кции распределения, которая определяется как сумма статистических вкладов
всех доступных системе состояний.
Химический потенциал растворенного вещества — это свободная энергия, не-
обходимая для добавления одного моля или одной молекулы этого вещества
в раствор.
Стандартное биологическое состояние определяется при температуре 298,15 К
(25 °C), парциальном давлении газов в 1 атмосферу, концентрации вещества
в водном растворе 1 М и pH 7.
Константа равновесия химической реакции равна математическому произведе-
нию концентраций продуктов реакции, деленному на произведение концентра-
ций реагентов, когда свободные энергии тех и других равны.
Уравнение Вант-Гоффа соотносит изменение энтальпии с константой равнове-
сия химической реакции.
Константа равновесия реакции ассоциации является мерой силы связывания
или аффинностью взаимодействия.
Связывание называют кооперативным, если заполнение одного сайта способ-
ствует связыванию с другим; если же, наоборот, происходит снижение аффин-
ности другого сайта, то оно антикооперативно.
В том случае, когда сайты идентичны и независимы, константа диссоциации для
каждого из них одинакова и не зависит от того, занят другой сайт или нет.
График Скэтчарда соотносит число сайтов связывания с константой диссоциа-
ции и концентрацией свободных лигандов.
Термодинамические равновесные параметры можно получить из графика Ар-
рениуса, который соотносит константы диссоциации с температурой.
• Когда процесс связывания обусловлен взаимосвязью других менее значитель-
ных событий, происходящих в месте контакта двух молекул, совокупная сила
связывания называется авидностью, которая также как аффинность измеряет-
ся через константу равновесия.
• Калориметрия растворов макромолекул основана на измерении теплоемкости
при постоянном давлении.
• В калориметрии соответствующие функции энтальпии и энтропии получают
интегрированием теплоемкости, деленной на температуру, по всему темпера-
турному диапазону эксперимента.
• Скорость реакции или конформационного изменения зависит от высоты энер-
гетического барьера активации на пути между состояниями с большей и мень-
шей свободной энергией; термодинамические параметры активации можно
получить из графика Эйринга, который соотносит константы скорости с тем-
пературой.
Литература для дальнейшего чтения
Feynman R. Р., Leighton R. В., Sands М. The Feynman lectures on physic,
Volume 1. Addison Wesley, Reading Massachusetts, 1963.
Pace N. C., Dwek R. A., Ratcliffe R. G., Wormaid M. R. Principles and problems in
physical chemistry for biochemists. Oxford University Press. 3r<1 Edition. Oxford, 2001.
Литература, добавленная редактором русского перевода
Финкельштейн А. В. и Птицын О. Б. Физика белка: курс лекций. М.: КДУ,
2007.
Глава В2
ДИФФЕРЕНЦИАЛЬНАЯ СКАНИРУЮЩАЯ
КАЛОРИМЕТРИЯ
В2.1. Исторический обзор
Исследования белков с помощью диф-
ференциальной сканирующей калори-
метрии берут свое начало в середине
1960-х гг., когда инструменты, облада-
ющие достаточной чувствительностью,
были разработаны независимо друг от
друга Петром Приваловым и Стенли
Гиллом (Peter. Privalov, and Stanly
Gill). К этому времени в биофизике на-
копилось множество проблем, которые
нельзя было решить без измерения не-
больших тепловых эффектов в относи-
тельно разбавленных растворах. В пос-
ледующие десятилетия с развитием все
более чувствительных калориметров
стали возможны эксперименты по оп-
ределению термодинамических пере-
менных с все более высокой точнос-
тью. Появились микрокалориметры,
а с наступлением нового тысячелетия
сделаны заявки на нанокалориметры
(приставки микро и нано указывают па
точность, с которой можно измерять
тепловые эффекты этими приборами).
В2.2. Основы теории
Полное количественное термодина-
мическое описание макромолекулы
в растворе в данном температурном
диапазоне можно получить из изме-
рения зависимости ее парциальной
удельной теплоемкости при посто-
янном давлении Ср от температуры
методом дифференциальной сканиру-
ющей калориметрии. Из Главы В1 мы
помним, что
А// = |дСР(Г)й'Г, (В2.1а)
A5 = JAC^r) dT, (В2.16)
где ДСр — это изменение теплоемкости
вследствие определенного макромо-
лекулярного процесса (например, раз-
ворачивания белка), а ДН и Д5 — со-
ответствующие изменения энтальпии
и энтропии.
В2.3. Экспериментальные
подходы
В2.3.1. Инструменты
Общие требования к дифференциаль-
ному сканирующему калориметру для
исследования биологических макромо-
лекул можно сформулировать следу-
ющим образом: высокая чувствитель-
ность и точность, работа в диапазоне
отрицательных и положительных тем-
ператур, возможность работы в режиме
как повышения, так и понижения тем-
пературы.
В настоящее время существуют
коммерчески доступные инструменты
различных производителей. Согласно
их характеристикам, на одно измерение
требуется около 50-500 мкг материала,
температурный диапазон составляет
от -10 °C до 130 °C, чувствительность
приближается к 10 мкДж • градус'1. Из-
мерения при температурах более 100 °C
требуют повышенного давления. Все
калориметры оснащены компьютерным
управлением.
Обратимость процессов плавления
должна контролироваться сканирова-
нием вниз и вверх по температурной
шкале. Вследствие возможных вто-
ричных эффектов, таких как агрега-
ция или химическая деградация при
высокой температуре, необходимо
соблюдать определенную предосто-
рожность.
Калориметрические измерения
в водных растворах вплоть до -10 °C
(в переохлажденных растворах) менее
распространены. Однако они имеют
особый интерес, так как позволяют от-
слеживать некоторые процессы,такие
как холодовая денатурация, полное
сворачивание а-спирали или одноце-
почечного олигонуклеотида. Измере-
ния свыше 100 °C сегодня рутинны,
поскольку разработаны специальные
ячейки для работы под давлением.
Измерения при высокой температуре,
вплоть до 130 °C (выше этой отметки
происходит быстрая химическая де-
градация биологических макромоле-
кул), важны для исследования белков
и комплексов гипертермофильных
организмов и мутантов, которые оста-
ются стабильными и активными при
температуре свыше 100 °C.
В2.3.2. Чувствительность
приборов и точность измерения
теплоемкости
На практике в калориметре измеряет-
ся тепловая мощность, которую следует
подать в одну из ячеек калориметра, с
тем чтобы разность температур между
ячейками калориметра поддержива-
лась равной нулю. Следует различать
чувствительность и точность, последняя
определяется, в основном, воспроизводи-
мостью базовой (нулевой) линии прибо-
ра. Последняя особенно важна при иссле-
довании небольших белков.
Переменная Ср (Т) набп, определяе-
мая методом дифференциальной ска-
нирующей калориметрии, — это тем-
пературная зависимость разности
теплоемкости между раствором мак-
ромолекул и тем же объемом чистого
растворителя при постоянном давле-
нии. Если теплоемкость растворителя
выше теплоемкости раствора того же
объема, то разность будет отрицатель-
ной. Она соотносится с парциальной
удельной теплоемкостью макромоле-
кулы как:
СЛТ\ай1 -
= СР(Т)^т(Т)^~ (В2.2)
где С (Т) мо1 — это парциальная удельная
теплоемкость, т. е. теплоемкость на еди-
ницу массы растворенных макромоле-
кул при температуре Т, т (Д\ип — масса
макромолекул в измерительной ячейке
при температуре Т, С (Т) я — парци-
альная удельная теплоемкость раство-
рителя при температуре Т, а Ат (Г) —
это масса растворителя, вытесненного
объемом макромолекул. Массу вытес-
ненного растворителя можно подсчи-
тать исходя из парциальных удельных
объемов макромолекулы и растворите-
ля й (7’) и й (Г)
4 7 мол. 4 7 расти
Am (Г)
' 7 расгн
= ™(Пй (Пмо.,/й (П1Ж1„. (В2.3)
Комбинируя уравнения В2.2 и В2.3
и подставляя типичные значения этих
параметров, мы увидим, что наблюда-
Комментарий В2.1. Вклады
парциальной теплоемкости
В экспериментах по дифференциалыюй ।
сканирующей микрокалориметрии бел- I
ковых растворов требуется очень высокая
чувствительность и точность, поскольку
наблюдаемый сигнал очень мал. Рассчи- !
таем с помощью уравнения В2.2 величи- .
ну избыточной теплоемкости для белка и '
растворителя в водном растворе объемом
1 см3, содержащего 1 мг макромолекул.
Парциальная удельная теплоемкость бел- !
ка взята равной 2 Дж-К'т1, а парциаль- ।
ный удельный объем белка — 0,73 см:,-г’.
Парциальная удельная теплоемкость воды
равна 4,2 Дж-К'т'1 (1 калория на градус на |
грамм), а ее парциальный удельный объем ।
равен 1 с.м’-г1. I
Ср(Т)^т(Т)^2^10^ДЖ-К-'
СР(Т)^^т(Т)^ =
= 4,2 х 103х 0,73 Дж-К’1 |
СР(Т) , =-1 х 10 3Дж-К '. !
Теплоемкость, измеренная для 1 см3
раствора, будет равна -4 Дж-К '-г1. Сле-
довательно, разность теплоемкостей,
обусловленная присутствием макромо-
лекул, — это -10 3 от теплоемкости раство-
рителя. Для измерений с точностью в 0,1 % :
чувствительность инструмента должна 1
быть порядка 10 А
емая разность теплоемкости раствора
п растворителя очень мала (коммента-
рий В2.11.
В2.3.3. Требования к образцам
Образцы растворов макромолекул для
калориметрических измерений должны
быть чистыми, гомогенными и содер-
жать не более 5% посторонних приме-
сей. Абсолютная концентрация должна
быть известна с точностью до несколь-
ких процентов, макромолекулы долж-
ны оставаться растворимыми во всем
температурном диапазоне эксперимен-
та. Раствор должен быть тщательно
диализован против растворителя, а для
снижения риска агрегации — достаточ-
но разбавленным.
На первых этапах развития метода
дифференциалыюй сканирующей кало-
риметрии, когда разрабатывались фун-
даментальные основы термодинамики
процессов разворачиванпя/сворачива-
ния белков, измерительные ячейки име-
ли объем порядка одного см3, стандартная
концентрация макромолекул составляла
1 мг-см’3, что требовало 1 мг белка. С тех
пор характеристики инструментов за-
метно выросли за счет аппаратных и
программных новшеств и теперь в благо-
приятных случаях возможны измерения
образца белка в количестве 50-200 мкг.
В2.4. Теплоемкость белков
В2.4.1. Зависимость теплоемкости
от температуры
Типичная кривая зависимости тепло-
емкости от температуры, полученная
для небольшого белка с помощью диф-
ференциалыюй сканирующей калори-
метрии, показана на рисунке В2.1.
Нативному состоянию чаще всего
приписывается статус эталонного, по
отношению к которому рассчитывают-
ся все термодинамические параметры
системы (см. Главу В1), поэтому столь
важный параметр как ДСр может быть
определен непосредственно из кривой
на рисунке В2.1. Вклады в ДСр можно
разделить на две составляющие:
<ДСР(Г)> =
' (В2.4)
= 8СР(Г)1Г+8СР(7’)Ь|.
Первая относится собственно к из-
быточной теплоемкости системы, тог-
да как вторая описывает сдвиг базовой
линии (сигмоидальная кривая меж-
ду двумя пунктирными линиями на
рис. 132.1).
В2.4.2. Анализ кривой
теплоемкости с помощью функции
распределения
Согласно аналитическому подходу,
описанному в Главе В1, усредненное
значение термодинамической пере-
менной <а> выражается в виде сум-
мы вкладов различных состояний си-
стемы, которые, в свою очередь, можно
представить в терминах функции рас-
пределения (уравнения Bl.ll е, Bl.ll б
в главе В1):
.V
<а> = £/>,,
7=0
/>=exp(-AG,//?r)/Q.
где Q — функция распределения, Р —
величина относительной заселеннос-
ти, а а. — термодинамическая перемен-
ная, связанная с состоянием j. Если
состояниеу=0 (как правило нативное)
принять за эталонное состояние и
суммирование производить от у = 1 до
N, то <а> = <Д//> будет представлять
Температура (°C)
Рис. 132.1. Молярная теплоемкость белка Ср
как функция температуры Ср ц и Ср ц — моляр-
ные удельные теплоемкости развернутого и
нативного состояний. ЛС — это изменение
р
теплоемкости, связанное с переходом. Если
при температуре выше основного перехода
существует множество состояний, то избыточ-
ная теплоемкость выражается как среднее по
всем этим состояниям <ДСр>
собой избыточную энтальпию, связан-
ную с заселением других энергетичес-
ких уровней, отличных от нативного.
Надо отметить, что, строго говоря, все
величины, представленные на рисун-
ке В2.1 и в уравнении В2.4 следует
заключить в скобки <> (усреднить),
поскольку может существовать целый
ансамбль нативных и денатурирован-
ных состояний для данного белка.
По определению теплоемкости мож-
но записать
< ДС’Р > - < Ср Ср0 > -
= (д<АН> /ЭТ)Р.
По аналогии с уравнением В2.4 раз-
ложим правую часть уравнения В2.5 и
выразим его члены как суммы различ-
ных состояний:
<дср> = £дя,(Эр,./Э7’)р +
7=1 (В2.6)
+Х^дс(!, = 5С7+5с;1.
7=1
Первый член называется избыточ-
ным теплопоглощением и относится к
энергии, которую нужно затратить, что-
бы изменить распределение молекул по
состояниям, а второй член называется
собственной теплоемкостью и отражает
распределение молекул по состояниям
с разными теплоемкостями.
Анализ кривой с помощью функции
распределения является общепринятым
и применяется к любой (чаще всего мо-
номерной) системе в равновесии, незави-
симо от механизмов перехода и от числа
промежуточных состояний. Под моно-
мерной системой понимается система,
в которой число молекул в растворе не
меняется в ходе разворачивания.
В2.4.3. Кооперативность плавления
Денатурация небольших белков явля-
ется кооперативным переходом с одно-
временным достаточно резким «5-об-
разным» изменением практически всех
характеристик молекулы (см. например
рисунок Г9.11). «5-образность» экспе-
риментальных кривых доказывает, что
соответствующие характеристики мо-
лекулы меняются от тех, которые ха-
рактерны для нативного белка, до тех,
Комментарий В2.2.
Денатурирующие агенты
Денатурировать белок можно разными i
способами: добавлением в раствор денатури-
рующих агентов типа гуанидингидлохлори-
да или мочевины, нарушающих водородные
связи в белке или додецилсульфата натрия,
гидрофобные хвосты которых проникают в ;
молекулу белка и разрушают его ядро. При i
этом молекулы белков принимают специфи-
ческие конформации, обсуждаемые в соот-
ветствующих главах этой книги (параграфы
Г4.10.5, Г5.6.1, Г9.5.3). Кроме того, сильное ,
изменение pH также может вызывать кис- ;
лую или щелочную денатурацию белка.
которые характерны для денатуриро-
ванного белка, а узость кривых свиде-
тельствует о том, что она охватывает
сразу много аминокислотных остатков
в белке ( коммеи lapini В2.2). Однако в
отличие от истинного фазового пере-
хода, наблюдающегося, например, при
плавлении кристаллов, «5-образность»
перехода имеет не нулевую, а конечную
ширину. Последнее означает, что этот
переход охватывает не всю макроско-
пическую систему, а только ее часть.
Главный вопрос, который здесь возни-
кает, прост: «Какую часть молекулы за-
трагивает такой переход?» и можно ли
получить ответ на него из данных ска-
нирующей микрокалориметрии.
В2.4.4. Конформационные
переходы, при которых
калориметрическая энтальпия
и энтальпия Вант-Гоффа равны
между собой
Первые исследования небольших
однодоменных белков с помощью
дифференциальной сканирующей
калориметрии установили, что раз-
ворачивание их структуры под воз-
действием температуры можно удов-
летворительно описать в рамках
модели кооперативного перехода
между двумя состояниями. Это озна-
чает, что заселенности всех промежу-
точных состояний крайне малы. Такой
переход получил название «все или
ничего».
Моделирование процесса пере-
хода основано на сравнении кало-
риметрической энтальпии перехода,
полученной интегрированием кривой
плавления, с величиной, вычислен-
ной согласно уравнению Вант-Гоф-
фа (см.Главу В1). Если оба значения
совпадают, то считается, что переход
происходит только между двумя со-
стояниями, а вклад интермедиатов
пренебрежительно мал.
Экспериментальная калориметри-
ческая энтальпия перехода дается вы-
ражением
(В2.7)
где ДСр п 1ксн (Г) — это величина пика
перехода над экспериментальной базо-
вой линией ДСр Ь1 СХ,(Г) (см. уравнение
В2.4 и рис. В2.1).
Уравнение Вант-Гоффа соотносит
энтальпию и константу равновесия
между двумя состояниями (коммента-
рий 152.3):
Д/У, = RT2d\nKeq/ЭТ. (В2.8)
Из уравнения видно, энтальпия
перехода равна наклону кривой зави-
симости In К от обратной температуры
1/RT.
Мы помним из Главы В1, что для
одностадийного перехода А <-> В кон-
станта равновесия при температуре Т
выражается как:
( *
где [В(Т)] и [А(Т)] — равновесные кон-
центрации состояний. Таким образом,
константу равновесия К(Г) можно из-
мерить в любом эксперименте, кото-
рый дает отношение [В(Т)]/[А(Т)] как
функцию температуры. Например, К(Т)
можно рассчитать из спектров круго-
вого дихроизма, если состояния Л и В
значительно различаются, как в случае
с разворачиванием белков.
Предполагая существование только
двух состояний, можно получить отно-
шение значений их заселенностей не-
посредственно из формы кривой плав-
ления.
. Комментарий В2.3. Константы !
i равновесия и термодинамические
переменные
Мы помним из раздела В1.3.1, что
&G = -RT\nK.
Комбинируя это выражение
с фундаментальными соотношением >
ДС = Д#-ГД5, ’
мы получим I
1пК = -ДЯ/RT + AS/R.
I Из него получается уравнение Вант- 1
Гоффа:
; Д//уН =ЯГ2Э1пК/ЭГ.
! Величина АН вычисляется из наклона
кривой зависимости In К от обратной тем-
пературы 1/RT.
На рисунке В2.2 а показана избы-
точная кривая теплоемкости белка бар-
назы, а на рисунке В2.2 б ее интеграл
<Д//(Т)>. Как видно из рисунка В2.2 б
калориметрическое значение теплово-
го эффекта находится из амплитуды
функции <Д7/ (Т)>. С другой стороны
значение Вант-Гоффовой энтальпии
определяется из производной в точке
Гт но формуле:
ДЯ „ =4/?Г2<АСр(^)> (В28 а)
<АН(Тт)>
где <ДСр(Г)> — значение теплоемкости
при температуре перехода Г, а ДНт —
полное тепло, поглощенное в этой точке.
Следует отметить, что Вант-Гоффо-
ва энтальпия может быть определена из
производной температурной зависимо-
сти любой функции состояния (напри-
мер, вязкости или оптической плотности)
в предположении об одностадийности
перехода. Однако только калориметрия
позволяет одновременно определить оба
значения энтальпии и, в случае их ра-
венства, сделать заключение о справед-
ливости модели двух состояний.
Рис. В2.2. I Нахождение энтальпии Вант-Гоффа из формы кривой плавления: а — темпера турная
зависимость теплоемкости барназы при pl 1-=4.5 (Martinez et al.. 1994); б интеграл функции
<ДС > = С,(1~) — С ,('Г). Тт (середина перехода) соответствует условию .\G'(7’n) = О
Более современный подход заклю-
чается в подгонке экспериментальной
кривой теплоемкости по формулам, запи-
санным для модели двух состояний с по-
мощью минимизационных алгоритмов.
В2.4.5. Конформационные
переходы, при которых
калориметрическая энтальпия и
энтальпия Вант-Гоффа не равны
между собой
Для мономолекулярной одностадийной
реакции кривая плавления, определя-
емая с помощью дифференциальной
сканирующей калориметрии, отражает
действительное изменение тепла, свя-
занное с конформационным переходом
при денатурации моля макромолекулы,
а расчетная энтальпия Вант-Гоффа соот-
ветствует количеству тепла, связанному
с плавлением одного моля кооператив-
ной единицы в макромолекуле. Поэто-
му, когда кооперативный домен меньше
молекулы, как в случае с мультидомен-
ными белками, то энтальпия Вант-Гоф-
фа меньше калориметрической. Если же
энтальпия Вант-Гоффа больше калори-
метрической, то это указывает на то, что
в случае обратимости кооперативный
домен больше молекулы, т. е. является
доказательством присутствия межмо-
лекулярных взаимодействий, таких как
олигомеризация (рис. В2.3).
Во всех случаях, когда эти энталь-
пии не равны, переход идет через про-
межуточные состояния
В2.4.6. Интермедиаты в процессе
сворачивания
Мультидоменные белки
Уравнения функции распределения, по-
лученные в секции В 1.3, используются
для расчета модельных функций тепло-
емкости переходов в тех случаях, когда
заселено более двух состояний. Анализ
экспериментальных кривых с помощью
таких функций является эффективным
подходом для детального анализа тер-
модинамических данных дифференци-
альной сканирующей калориметрии.
Точность сегодняшних калориметров
такова, что позволяет наблюдать и ин-
терпретировать достаточно малые раз-
личия между модельной и эксперимен-
тальной кривыми (рис. В2.4).
Рис. В2.3. Калориметрическая ;>нталы1ия и энтальпия Вант-Гоффа для однодоменных, двухдо-
менных макромолекул и их димеров
Лизоцим куриного яйца является
двухдоменным белком, и как свидетель-
ствуют данные, представленные на ри-
сунке В2.4, температурная денатурация
обоих его доменов происходит не совсем
кооперативно. В родственном белке — ли-
зоциме лошади, независимость процесса
разворачивания обоих доменов выраже-
на еще больше (см. рис. В2.8).
Существуют ли заселенные
интермедиаты сворачивания
в однодоменных белках?
В настоящее время считается, что про-
цесс сворачивания небольших однодо-
менных белков должен проходить по
крайней мере через одно переходное
состояние, энергия которого выше, чем
у нативного и денатурированного со-
стояний. В качестве такого интерме-
диата была предложена расплавленная
глобула, которая, как считают некоторые
исследователи, может наблюдаться и в
определенных равновесных условиях.
Она может образоваться в результате
быстрого сворачивания цепи в компакт-
ную жидкообразную структуру, которая
затем переходит в нативное состояние.
Существование такого состояния
позволило бы разрешить парадокс Ле-
впнталя ( коммсп lapnii 112. i), значи-
телыюсужая поиск конфигурационного
пространства. Однакосвойства расплав-
ленной глобулы и само ее существова-
ние ДОВОЛЬНО спорны (kii.\:\liinapiin
1)2.5). Различные авторы определяли
по-разному ее динамику и структуру.
Так П. Привалов, анализируя калори-
метрические данные в поиске интерме-
диатов процесса сворачивания, сделал
вывод, что эти промежуточные состо-
яния представляют собой либо непра-
вильно свернутые формы, образующи-
еся в неблагоприятных условиях, либо
частично развернутые структуры, ко-
торые сохранили правильно свернутые
участки молекулы. Эти участки обла-
дают всеми характерными чертами ко-
оперативно сворачивающихся доменов.
Комментарий В2.4. Парадокс
Левинталя
Скорость сворачивания полинептидной
цепи в уникальную трехмерную белко-
вую структуру, определяемую аминокис-
лотной последовательностью, исключает
вероятность того, что этот процесс имеет
стохастическую природу, где молекула пе-
ребирает огромное число всевозможных
конформаций. Это утверждение известно
как парадокс Левинталя.
25
Комментарий 2.5. Расплавленная
глобула
Свойства расплавленной глобулы до-
статочно противоречивы. Так, в ней су-
ществует развитая вторичная структура,
практически в том же количестве, как и в
нативном белке. Она слегка мопсе компакт-
на - ее гидродинамический объем всего на
30 % больше, чем объем в нативном состоя-
нии. Зато в ней почти полностью отсутству-
ет упорядоченность боковых групп, явля-
ющаяся характерной для нативного белка,
а довольно высокая скорость водородного
обмена в расплавленной глобуле свиде-
тельствует о том, что некоторое количество
молекул растворителя могут свободно про-
никать вглубь нее. Эти противоречия, в из-
вестной степени, примиряются предполо-
жением о неоднородности расплавленной
глобулы, а именно, введением представ-
ления о более или менее нативоподобного
ядра и опушенной оболочки. Но даже этой
простой модели противоречат некоторые
эксперименты по круговому дихроизму и
ядерному магнитному резонансу (Фин-
кельштейн, 2007).
Свободная энергия промежуточного
состояния выше нативного состояния,
или денатурированного, или их обоих,
поэтому его заселенность всегда мала.
В2.4.7. Влияние мутаций в белках
на кривые плавления
Замена одного или двух аминокислот-
ных остатков в белке может приводить
к значительным различиям в экспе-
риментальных кривых плавления (см.
рис. В2.4). Так и хочется приписать эн-
тальпийный или энтропийный вклад
замен аминокислотных остатков изме-
нению белковой стабильности. Однако
делать это нужно с величайшей осторож-
ностью, поскольку изменение стабиль-
ности может с таким же успехом быть
результатом изменения кооперативное-
Рпс. В2.4. Экспериментальные и модельные
кривые плавления лизоцима куриного яйца.
Кривые, полученные экспериментально при
pH 2,5, показаны сплошными линиями. Пунк-
тирные линии — результат компьютерного мо-
делирования: вверху переход с двумя состоя-
ниями, внизу переход с тремя состояниями
(Privalov et al., 1995)
тн процесса разворачивания. Такое из-
менение стабильности может указывать
на то, что аминокислотная замена суще-
ственно влияет на междоменные взаимо-
действия внутри белка. Обратите внима-
ние на то, как замена серина на аланин в
лизоциме Т4 не только дестабилизирует
белок (он разворачивается при более
низкой температуре, чем белок дикого
типа), ио и увеличивает полуширину
перехода, тем самым указывая на сниже-
ние кооперативности (рис. В2.5).
В2.4.8. Сложные мультидоменные
белки
Кривую зависимости теплоемкости от
температуры для белков со сложной
структурой можно разделить на со-
ставляющие кривые, которые соответ-
ствуют переходам между различными
состояниями. Такое разделение дела-
ется без всяких априорных посылок,
используя специальные компьютерные
алгоритмы. Надежность данного анали-
за (некоторыс авторы называют его «де-
конволюцией», но это не деконволюция
в строгом смысле слова, а аппроксима-
ция) зависит от точности определения
экспериментальной кривой плавления
и, следовательно, от микрокалоримет-
ра. В идеале каждый! компонент кри-
вой! соответствует переходу между
двумя состояниями со своими термо-
динамическими параметрами. Компо-
ненты кривой плавления могут быть
соотнесены со структурными единица-
ми при обработке модельной инфор-
мации о белке или с помощью исследо-
вания его фрагментов. Информацию о
структурной связи между фрагментами
можно получить из данных по взаимо-
зависимости температуры и энтальпии
отдельных компонентов кривой при из-
менении условий, например pH. Этот
метод с успехом применялся для муль-
тидоменных белков, комплексов белок-
белок и белок-нуклеиновая кислота,
а также для фибриллярных белковых
структур, таких как мышечные белки и
коллагены. Калориметрический анализ
процесса плавления миозина в качестве
примера показан на рисунке В2.6.
Как видно из этого рисунка, слож-
ность кривой плавления нарастаете уве-
личением длины фрагмента от LF-3 до
LMM. Все кривые могут быть разложе-
ны на сумму пиков, каждый из которых
описывается переходом между двумя
состояниями. Затем каждому пику при-
писывается отдельная кооперативная
единица в структуре путем приблизи-
тельной! оценки размера кооперативного
Рис. 132.5. Кривые плавления лизоцима фага
Т4. Сплошной линией обозначены данные,
полученные при pH 2,8; 3,0; 3,5; 3.7. Кружками
показаны данные для мутанта белка S44A при
pH 4,0 (Carra et al., 1996)
блока из энтальпии перехода, принимая
значение удельной энтальпии плавле-
ния постоянной но всей длине молеку-
лы. Наименее стабильный кооператив-
ный блок (компонент 1) лежит близко к
середине молекулы. Примечательно, что
блок, соответствующий компоненту 4,
отделен от других блоков эластичными
областями (легко расщепляемыми про-
теазами и заштрихованными на рисунке
В2.6), которые не принимают участие в
процессе плавления. Для более точной
структурной интерпретации результа-
тов деконволюции весьма желательно
использовать дополнительные, напри-
мер оптические, данные.
В2.4.9. Влияние растворителя
на кривые плавления белков
Растворитель играет важную роль
в стабилизации конформационных (на-
тивное и свернутое) состояний белка
(см. Главу В1), а также в определении
природы перехода между ними. Взаи-
Миозин
Рис. В2.6. Калориметрический анализ миозина.
Верхняя панель: схематический вид молекулы миозина. Заштрихованные места подвержены
полному протеолизу. ЛММ и ТММ (легкий и тяжелый меромиозин) являются основными
фрагментами после обработки трипсином или пепсином. Фрагмент ЛММ далее расщепляется
папаином, как это показано на рисунке. Там же можно наблюдать фрагменты дальнейшего рас-
щепления ЛММ с помощью трипсина. Пронумерованные фигурные скобки обозначают обна-
руженные с помощью калориметрического анализа кооперативные блоки, которые показаны на
нижней панели.
Нижняя панель: кривые плавления фрагментов миозина в 25 шМ калий фосфатном буфере, pH
6.5 и 0,5 М КО:
а — фрагмент ЛММ после трипсинового расщепления;
6 — фрагмент ЛММ после пепсинового расщепления;
в — фрагмент LF-3;
г — нефрагментрированная молекула миозина ТР.
Пики переходов между двумя состояниями пронумерованы согласно возрастанию стабильности
блоков и приписаны различным элементам структуры, как показано на верхней панели
(Privalov, 1982)
модсйствия белок-растворитель могут
быть специфическими, как в случае
со связыванием ионов с отдельными
участками, или более общими, которые,
в свою очередь, влияют на гидратацию
полярных и неполярных групп белка.
Стабилизация структуры через
связывание низкомолекулярных
лигандов
Лизоцим лошади состоит из двух доме-
нов, которые разворачиваются квази-
независимо и дают два пика на кривел')
плавления. Стабильность одного из до-
менов изменяется благодаря связыва-
нию ионов кальция, поэтому положение
соответствующего пика (первого), его
высота и ширина зависят от концентра-
ции СаС1.,в растворителе (рис. В2.7).
Влияние pH
Температура разворачивания барназы
(Г) возрастает от 20 °C при pH 1.8 до
55 "С при pH 5,5 (рис. В2.8).
Вспомним, что знание функции
<ДСр (Т)> обеспечивает нам полную
характеристику термодинамики пере-
хода. Однако чистое нативное и развер-
нутое состояния обычно реализуются в
узких температурных диапазонах, по-
этому температурные зависимости С п
и С u можно оценить только экстрапо-
ляцией или с помощью модельных рас-
четов (в качестве примера см. пунктир-
ные и пунктирно-точечные линии на
рис. В2.7 н В2.8). Протоны также явля-
ются лигандами, и они сильно влияют
на стабильность белка (Г), если в пос-
леднем есть группы с аномальным зна-
чением рК. Вариации pH, сдвигая пик
тенлопоглощения, позволяют получить
семейство кривых, как это показано на
рисунке В2.8, и таким образом опреде-
50 □
Ю -1............-г.......................
260 300 340 ЗВО
Температура (°К)
Рис. В2.7. Кривые плавления лизоцима
лошади при pH 4,5 в присутствии различ-
ных концентраций СаС1.,: кривая 1) 0,00 .мМ,
2) 0,10 мМ, 3) 0,75 мМ, 4) 1,50 мМ. Пунктир-
ная линия функция теплоемкости от темпе-
ратуры, рассчитанная для несвернутого пеп-
тида, а линия пунктир-точка — для нативного
состояния, полученная линейной экстраполя-
цией с использованием величины наклона, ха-
рактерного для нативных глобулярных белков
(см. параграф В2.4.8) (Griko et al., 1995)
лить температурные зависимости тер-
модинамических параметров перехода.
В2.4.10. Расчет теплоемкости
из структурных данных
Теплоемкость макромолекул обуслов-
лена в основном внутренними колеба-
тельными и вращательными модами, а
также взаимодействием с растворите-
лем. Надежные предсказания величин
свободной энергии являются обяза-
тельными в подходах к дизайну белков
и лигандов. Невозможно, однако, пред-
сказать теплоемкость сложной систе-
мы путем умозаключений (ab initio).
Поэтому были разработаны методы
для расчета теплоемкостей белков в
различных состояниях, основанные на
кривых плавления, полученных мето-
100
Рис. В2.8. Кривые плавления барназы в растворах с различными pH, величины которых ука-
заны над каждой кривой. Пунктирная линия и линия пунктир-точка показывают парциальные
молярные теплоемкости нативного и развернутого белков. Вставка: сплошная линия показы-
вает зависимость теплоемкости от температуры для необратимо денатурированного белка, по-
лученная с помощью нагрева до 100 °C; пунктирная линия — теплоемкость развернутого белка,
рассчитанная суммированием теплоемкостей отдельных аминокислотных остатков (Privalov
and Makhatadze, 1992; Griko et al., 1994)
дом дифференциальной сканирующей
калориметрии, с привлечением данных
о модельных соединениях.
Расчет теплоемкости нативных
и развернутых белков
Теплоемкость развернутого белка при
постоянном давлении можно очень
точно и с высокой надежностью рас-
считать, суммируя теплоемкости отде-
льных аминокислот. В качестве приме-
ра см. пунктирную линию на рисунке
В2.6 и вставку на рисунке В2.8. Однако
аналогичный расчет для нативного со-
стояния гораздо более сложен.
Теплоемкость нативного или раз-
вернутого белка в водном растворе
можно рассматривать как сумму вкла-
дов, каждый из которых относится либо
к внутренним химическим свойствам
макромолекулы, либо к влиянию рас-
творителя. Результаты многочислен-
ных работ на модельных органических
соединениях подтверждают, что эти
вклады в теплоемкость аддитивны. По-
этому теплоемкость белка может быть
записана как
С=СР +СР,+СР +С„ ,(В2.10)
Р Р. а Р. b Р, с Р.другие*7
где Ср ч называется первичной тепло-
емкостью, обусловленной вкладами
атомов и ковалентных связей, и зави-
сящей исключительно от аминокис-
лотного состава макромолекулы. Ср ь
содержит вклады нековалентных свя-
зей в свернутом белке. Ср с, в основном,
есть результат гидратации или взаимо-
действий белок-растворитель. Ср (.
вызвана другими вкладами, например,
протонированными состояниями ами-
нокислотных групп с ионизуемыми бо-
ковыми цепями, такими как гистидин,
аспарагиновая кислота, глютаминовая
кислота, аргинин и лизин. Обычно по-
следний вклад мал по сравнению с дру-
гими вкладами в уравнение В2.10, и мы
будем пренебрегать им в последующем
обсуждении.
Энергия полипептидной цепи
и первичная теплоемкость
Первичная теплоемкость Ср ч — это теп-
лоемкость безводного несвернутого по-
липептида. Ее можно точно рассчитать
суммированием вкладов отдельных
аминокислот (табл. B2.I) и процесса
образования пептидной связи (коммен-
lapiiit В2.6).
Внутренние белковые взаимодействия
и теплоемкость безводного
свернутого белка
Как следует из уравнения В2.10, тепло-
емкость безводного свернутого белка
равна Ср + Ср ь. Теплоемкости различ-
ных растворимых белков, отнесенные
к единице массы, оказываются доволь-
но близкими: 1,17 кДж-кг-1 для тепло-
емкости, рассчитанной из первичной
последовательности; 1,21 кДж-кг"1 для
экспериментального значения безвод-
Комментарий В2.6. Теплоемкость ;
образования пептидной связи
' Средний вклад процесса образования
пептидной связи в теплоемкость после ос-
вобождения молекулы воды может быть
рассчитан из разности между эксперимен-
тальными теплоемкостями дипептидов и
составляющих его отдельных аминокислот
• в безводном состоянии. Эта величина теи-
1_лоемкости равна 39,2 ±0,74 Дж • К 1 моль1. J
него состояния при 25 °C; 1,46 кДж-кг-1
для свернутого состояния в растворе
и 1,96 кДж-кг-1 для развернутого состо-
яния в растворе. Обратите внимание,
что измеренные теплоемкости безвод-
ных белков очень близки к расчетным
значениям первичной теплоемкости.
Это говорит о том, что основная часть
абсолютной теплоемкости обусловлена
первичной последовательностью. В слу-
чае с безводным белком Ср а составляет
97,5% от экспериментальной теплоем-
кости (см. также коммеп гарий В2.6).
Заметим, что теплоемкость безвод-
ных белков растет линейно с темпе-
ратурой — с наклоном, равным 4,10 х
х 10"3Дж'К"1 г-1, который практически
постоянен для всех исследованных бел-
ков.
Теплоемкость нативного белка
в растворе
В соответствии с уравнением В2.10 теп-
лоемкость нативного белка в растворе
равна Сп + С„, + С.. . Это значение
выше такового для безводных белков
(табл. В2.2), поскольку вклад гидра-
тации Ср с, в основном, имеет положи-
тельную величину. При 25 °C вклад
гидратации составляет около 15 % от со-
вокупной теплоемкости нативного бел-
ка в растворе. Гидратация также отвеча-
ет за более выраженную температурную
зависимость, наблюдаемую для этой
теплоемкости (6,80 х 10 3Дж-К"1-г |) по
сравнению с безводным свернутым со-
стоянием (4,1 х Ю~3Дж-К-1 • г ’).
Вклад гидратации варьирует в за-
висимости от состава поверхности мак-
ромолекулы и ее взаимодействия с рас-
творителем, которое в первую очередь
определяется доступностью полярных
и неполярных групп для растворителя.
Вклад гидратации каждой группы, ус-
Таблица В2.1. Экспериментальные
значения теплоемкости аминокислот
Аминокислота Г * (Дж-К’-моль1)
Ала 122,7
Apr 234.4
Аси 160,9
Аси 155,8
Цис 163,0
Гли 184,9
Гл го 175,7
Гл и 99,5
Гис 216,2
Иле 189.0
Лей 201,7
Лиз 205,4
Мет 290,6
Феи 203,7
Сер 136,1
Тре 147,8
Три 239,0
Тир 217,1
Вал 169,5
*) Значения для безводных аминокислот
при 25 °C взяты из Hutchens (1970).
Hutchens JO in «Handbook of Chemistry
and Selected Data for Molecular Biology» (ed.
Sober H. A., Cleveland OH, Chemical Rubber
Co., 1970).
Теплоемкости аминокислотных остатков
в растворе приведены в Makhatadze G. I, and
Privalov Р (1990); Privalov Р. L. and Makhatadze
(1990); Privalov P. L. and Makhatadze G. I.,
«Physical Properties of Polymers Handbook»
(E. Mark, ed.) AIP Press, Woodbury, New York,
1996
тановленный на основе исследований
модельных соединений, пропорциона-
лен площади ее поверхности, которая
доступна для растворителя. Теплоем-
кость гидратации полярной группы
имеет отрицательное значение, тогда
как в случае с неполярной группой она
положительна. Таким образом, более
высокое значение теплоемкости натив-
ных белков в растворе по сравнению
с безводным свернутым состоянием
объясняется тем, что в среднем 55 %
поверхности растворимых белков со-
стоят из неполярных групп.
Теплоемкость развернутого пептида
в растворе
Теплоемкость развернутого пептида в
растворе равна Ср ч + Ср с ш, где вклад
гидратации Ср . н представляет собой
сумму соответствующих значений для
отдельных аминокислот в белковой
последовательности, а также для N и С
концевых групп. Парциальные тепло-
емкости аминокислотных остатков в
растворе очень точно измерены и хоро-
шо известны в температурном диапазо-
не от 5 до 125 °C (см. гиб.:. Г>2.1).
Теплоемкость развернутого состоя-
ния в растворе примерно на 0.4 Дж-Кг 1
больше ее теплоемкости в нативном со-
стоянии ( габ. I. В2.2 и 1,( >м х',< 11 i:i|>nr IT. /).
Преобладающий вклад в теплоемкость
вносит гидратация, в результате которой
возникают контакты неполярных групп
с растворителем. Однако в отличие от
нативного состояния в растворе, зави-
симость от температуры теплоемкости
развернутого состояния в растворе име-
ет нелинейный характер. В диапазоне
0-100 °C она хорошо аппроксимируется
с помощью полинома второго порядка.
Глобальная аппроксимация
теплоемкости с помощью одной
математической функции
Для зависимости теплоемкости белков
в разных состояниях от температуры
ТаЬлица 132.2. Параметры,
наилучшим образом описывающие
термодинамическую базу данных по
теплоемкости белков
Пара- метр Значение(единицы)
с ГС !> <1 а., "л /л, К 1,18 (Дж-К’1 г ') 41,0 х Ю '(Дж-К --г ') 36,5 х К) "(Дж-К 1 моль 1 А"2) 27,0 х К) '(Дж-К 1 моль 1 А 2) 1,89 (Дж-Дж 1 моль’1 А’2) 11,0 х К) '(Дж-К 2-моль 'А 2) -17,6 х Ю 5(Дж- К '-моль 'А 2) -1,11 (Дж-К 1 моль 1 А 2) 12,0 х Ю '(Дж-К 2-моль 1 А 2) 18,1 х К) -’(Дж-К ' моль 1 А 2)
(Gomez el al.. 1995)
была получена одна математическая
функция, основанная на уравнении
В2.9. Исключая последний член из
уравнения В2.9, получим
СР= G-.a + СР.Ь + Qc- (В2-П)
Выражая каждую из теплоемкостей
с правой стороны в виде набора некоторо-
го числа параметров: г, «',р, q,a,h и струк-
турных свойств белка: молекулярной
массы М, полной площади поверхности
молекулы, недоступной для раствори-
теля Л||(..1(к| и площадей неполярных и
полярных групп, доступных растворите-
лю — Л.1|ю и Л||() |) следующие равенства:
С,, а = |г + ?е (Т- 25) ] М, (В2.12)
Ср.ь= 17> + <7 (т~ 25) 1 Л (В2.13)
С„ = а(Г)А +Ь(Т)Л .(В2.14)
г. с х ' апо 1>|р v ' iiixbip v 7
где
а (Г) = at +а., (Т — 25) +
+ 6/ДГ-25)2,
(В2.14а)
b(T) = h1 +b2(J-25) +
+ b.j(T—25)2.
Эти уравнения были сопоставлены
со всей доступной термодинамичес-
кой базой данных. Лучшие значения,
найденные для этих параметров, даны
в iаб.шцг В2.2.
Полученный набор параметров по-
зволяет в большинстве случаев хорошо
предсказывать не только измеренную
теплоемкость данного состояния белка,
но и ее зависимость от температуры.
Хотя средние значения параметров,
представленные в ia'>.ищи В2.2, явля-
ются хорошим приближением, нужно
признать, что в действительности гидра-
Комментарий В2.7. Значения
теплоемкости
Средняя экспериментальная теплоем-
кость безводных свернутых белков, выра-
. женная в единицах на грамм, составляет
; 1,22 Дж-К 1 г ', а их средняя теплоемкость,
рассчитанная из первичной структуры,
равна 1,19 Дж-К 1 г* о м. 1Г..’.'Л. Соот-
ветственно, вклад нековалентных взаимо-
j действий составляет всего лить около 0.03
Дж-К 1 г '.
Теплоемкости нативного и развернуто-
го состояний в растворе больше значений,
I соответствующих безводным состояниям.
Из-за положительного вклада теплоемко-
стей неполярных групп значения варьиру-
1 ют в зависимости от состава поверхности ;
для нативного состояния или аминокис-
лотного состава для развернутого состоя-
, ния. Разность теплоемкостей развернутого
и нативного состояний в растворе лежит в
диапазоне 0,4-0,7 Дж-К ' г ’.
Расчеты теплоемкости для макромоле-
! кул, основанные на знании состава, .значе-
ний гидратации и структурных параметров,
таких как доступные для растворителя по-
верхности различных групп, производились
I в основном группами Фрейре (Freire, 1995)
' и Привалова (Privalov and Privalov, 2000).
тационный вклад в теплоемкость зави-
сит от конкретной химической группы.
Поэтому несколько лучшую аппрокси-
мацию можно получить, если разделить
неполярные группы на алифатические
и ароматические, так как последние, на
самом деле, имеют слегка полярный ха-
рактер. Соответствующие средние пара-
метры при 25 °C равны 2,14 Дж-К' ‘-моль
‘•А-2 для алифатических групп и 1,55
Дж-К_,-моль '-А'2 для ароматических,
поэтому параметр а, (который дан выше
как среднее для обеих групп) можно раз-
делить на два вклада.
Разворачивание белка сопровождается
изменением теплоемкости
Разность теплоемкости между развер-
нутым и нативным состояниями белка в
растворе обусловлена в основном разли-
чиями в гидратационной теплоемкости:
ДС„
Р, разв-натив
= ДСР. +ДС„ , (В2.15)
Р. Ь. разв-натив Р, с, разн-нашв’ ' 7
где ДСЦ и обозначает такую раз-
ность.
Вклад Д Ср с в суммарную Д Ср при 25 °C
составляет более 90%. В среднем вклад
гидратации неполярных групп уменьша-
ется наполовину с повышением темпе-
ратуры — от 1,89 Дж-К 1-моль-1 А-2 при
25 °СдоО,96Дж-К“1-моль"1-А“2при 100 °C,
тогда как гидратация полярных групп
уменьшается с 1,11 Дж-К_|-моль_1-А“2 при
25 °C почти до нуля при 100 °C (табл.
В2.3 и уравнение В2.14). Значение ДСр ь
отрицательно, поскольку нековалентные
взаимодействия разрушаются при пере-
ходе от нативного состояния к разверну-
тому. Оно меньше теплоемкости, обус-
ловленной гидратацией, и увеличивается
с температурой до среднего значения -
0,25 Дж-К-'-А’2 на моль недоступной по-
верхности при 100 °C. Результирующее
влияние, оказываемое гидратацией и
нековалентными взаимодействиями на
температурную зависимость ДСр при раз-
ворачивании исчезает по мере приближе-
ния температуры к значению 120 °C (см.
рис. В2.8).
Достоверность расчетов
Из-за важности предсказания свобод-
ной энергии для дизайна белков и лиган-
дов чрезвычайно полезными являются
расчеты теплоемкости, основанные на
знании детальной структуры белка. До-
статочно точно их можно произвести
для белка, полностью развернутого в
растворе (когда все аминокислоты до-
ступны для растворителя), основыва-
ясь на знании его последовательности
и табличных значениях теплоемкости
отдельных аминокислот. Между тем
теплоемкость нативного белка в рас-
творе нельзя рассчитать из его последо-
вательности, поскольку она зависит от
поверхности макромолекулы и других
свойств белка. Однако, когда структура
нативного белка известна, описанный
выше параметризированный подход мо-
жет служить хорошим приближением
(см. рис. В2.8), если белок принадлежит
к тому же структурному семейству, что
и белки в базе данных, используемые
для получения этих параметров. Такие
расчеты, однако, необходимо восприни-
мать с осторожностью, поскольку тепло-
емкости индивидуальных химических
групп могут существенным образом от-
личаться от значений, полученных для
белков в базе данных. Ожидается, что
разность в теплоемкости, обусловлен-
ная динамикой цепи, между нативным
и развернутым состояниями должна
быть мала, поэтому вычисленные зна-
чения разности теплоемкости пос-
ле разворачивания более надежны по
сравнению с расчетами для нативного
состояния, поскольку последние в ос-
новном обусловлены гидратационным
вкладом ( i.ommi'ii Hipin'i l>2.<S). Вклад в
теплоемкость гидратации данной груп-
пы пропорционален ее площади, кото-
рая доступна для растворителя. В раз-
вернутом состоянии подразумевается,
что группа доступна растворителю пол-
ностью, тогда как в нативном белке ее
доступная площадь может быть точно
определена из структурных координат.
В2.4.11. Силы, стабилизирующие
нативную структуру белка
Силы, стабилизирующие нативную
структуру белка, возникают из-за кон-
куренции между различными взаимо-
действиями, такими как белок-белок и
белок-растворитель. Они поддерживают
свернутую структуру в данном темпера-
турном интервале. Калориметрические
исследования внесли фундаменталь-
ный вклад в наше понимание этих сил.
Как мы уже видели выше, АСр между
свернутым и развернутым состояниями
обусловлена в основном гидратационны-
ми взаимодействиями. На рисунке В2.9
приведена зависимость этой величины
от температуры для небольших однодо-
менных белков, а также ее стремление к
маленьким значениям при высоких тем-
пературах. Первый возникающий воп-
рос: «Какие силы определяют процесс
сворачивания белка?»
Сворачивание цепи в вакууме
Прямой анализ термодинамики сво-
рачивания белка в вакууме (или в ус-
ловиях, когда влиянием растворите-
ля можно пренебречь) представлен на
рисунке. В2.9. Цепь поддерживается в
Комментарий В2.8. Соотношения I
между АН, AS, и ЛСР j
Вспомним, что члены энтальпии и энт-
ропии рассчитываются с помощью измере-
ний теплоемкости в соответствии с уравне- i
ниями В2.1 ।
АН = |дСр(7’)47’, (В2.1а)
AS = dT. (В2.16)
Из уравнений следует, что ДСр может
быть найдена из наклона кривых зависи-
мостей АН и Д5 от температуры. i
[’нс. В2.9. Расчетные (линии) и измеренные
теплоемкости развернутого (верхняя кривая
на каждой панели) и нативного состояний
(нижняя кривая) различных растворимых
белков. Вычисления произведены на основе
параметров, приведенных в 2...Точ-
ки кривой для нативного состояния выше
50 °C были экстраполированы. В случае с раз-
вернутым миоглобином квадратиками обозна-
чены расчетные значения, взятые из Privalov
and Makhatadze (1990) и из Freire (1995)
своей свернутой конфигурации благо-
даря внутренним силам, таким как во-
дородные связи, ван-дер-ваальсовы или
электростатические взаимодействия.
Их разрушение ведет к независимому от
температуры росту внутренней энергии,
поэтому при разворачивании величина
АН положительна и постоянна при лю-
бой температуре. Что касается энтропии,
то очевидно, что развернутое состояние
обладает большим числом степеней сво-
боды, чем свернутое, поэтому А5 в про-
цессе разворачивания также будет по-
ложительна. Следовательно, изменение
свободной энергии (АС = АН - 7А5) во
время этого процесса будет уменьшаться
с повышением температуры и пересека-
Рис. 132.10. Термодинамика процесса свора-
чивания белка в вакууме: а — схематическое
представление равновесия; б — изменения
энтальпии (Д/f), энтропии (Д5) и свободной
энергии (AG) в процессе разворачивания. Тт —
температура плавления цепи
ст нуль при Т , как показано на рисунке
В2.10. Свернутое состояние поддержи-
вается при температуре выше Т , развер-
нутое, наоборот, — ниже этой величины,
поэтому Гп можно рассматривать как
температуру плавления цепи.
Влияние растворителя на процесс
сворачивания белка
С помощью тщательных калориметри-
ческих измерений было установлено,
что простая картина сворачивания, по-
казанная на рисунке В2.10, не примени-
ма к белкам в естественном окружении.
С помощью анализа калориметрических
кривых теплоемкости было выяснено,
что температурная зависимость AZ/, A.S,
а, следовательно, и AG для данного про-
цесса имеет сложный характер. Это сви-
детельствует о том, что влиянием рас-
творителя на сворачивание белка нельзя
пренебречь.
Общие кривые термодинамиче-
ских функций, полученные из калори-
метрических данных для миоглобина
(рис. B2.ll), довольно типичны для
небольших глобулярных белков, хотя
они могут быть и сдвинуты по величине
теплоемкости или температуре в соот-
ветствии с природой каждого белка (см.
комментарий В2.7).
Нативноесостояниесуществуетв до-
статочно узком температурном диапа-
зоне благодаря положительной свобод-
ной энергии разворачивания, которая
достигает максимума при температуре,
близкой к комнатной, и надает при ее
повышении или понижении. Интерес-
но, что максимальная стабилизирующая
свободная энергия неожиданно мала.
К примеру, ее значение 50 кДж -моль-1
соответствует выигрышу энтальпии в
результате разрушения двух или трех
водородных связей в интерьере белка
( !;н'>. i. I12.3), тогда как внутрибелковые
взаимодействия и взаимодействия меж-
ду белком и растворителем характери-
зуются энергией в тысячи кДжмоль 1
(см. рис. В2.10 и нюл. В2.3). Для срав-
нения, энергия ковалентной связи со-
ставляет около 300 кДж -моль '. За ис-
ключением S-S связей, которые могут
образовываться между боковыми цепя-
ми цистеина в невосстанавливающих
условиях, ковалентные связи не прини-
мают участие в стабилизации нативной
укладки белка. Связи, вовлеченные в
эту стабилизацию, являются слабыми,
обладают энергиями, соответствующи-
ми нескольким kT.
Гдцрофобный эффект
Стабилизацию нативного состояния
белка можно описывать в терминах рас-
творимости. Белок сворачивается, что-
бы «спрятать» как можно больше «не-
растворимых» неполярных групп цепи
от контакта с растворителем. С этой
точки зрения, тот факт, что свободная
энергия разворачивания имеет макси-
мум около 30 °C, есть следствие ано-
мальной температурной зависимости
растворимости неполярных компонен-
тов (см. рис. А1.7).
Растворимость неполярных ком-
понентов в воде минимальна при тем-
пературе, близкой к комнатной, что
соответствует максимуму свободной
Рис. В2.1 1. Температурная зависимость тер-
модинамических функций в процессе разво-
рачивания миоглобина. Разности энтальпии,
энтропии и свободной энергии, сопровож-
дающие разворачивание, были рассчитаны,
исходя из характера зависимости теплоем-
кости от температуры. Пунктирные линии
соответствуют функциям, которые были бы
получены в отсутствии растворителя, поэтому
8AG представляет собой результат взаимо-
действий белка с растворителем (Privalov and
Khechinashvili, 1974)
Табшп ia В2.3. Энергии сил стабилизирующих белок
Связь Энергия (кДж -моль ')
Электростатичес кая -20
Водородная -4-25
Гидрофобная -4
Ван-дер-ваальсова -0,5
Рис. В2.12. Разворачивание белка в раство-
рителе. Внутренние белковые контакты раз-
рушаются при разворачивании и заменяются
на контакты с растворителем. Изменения эн-
тальпии и энтропии при разворачивании мо-
гут быть положительными и отрицательными.
Выигрыш в конфигурационной энтропии цепи
может компенсироваться потерей конфигура-
ционной свободы молекулами растворителя
энергии переноса. При такой темпера-
туре доминирует большое отрицатель-
ное изменение энтропии, которое, как
считают, обусловлено упорядочивани-
ем молекул воды вокруг неполярных
групп (см. главу В1). Структура жид-
кой воды характеризуется большой эн-
тропией благодаря конфигурационной
свободе образования водородных свя-
зей у каждой ее молекулы с ближайши-
ми соседями во всех направлениях (см.
Главу А2). Но эта свобода уменьшается,
когда молекула находится рядом с не-
полярной группой, которая не дает ей
возможности образования водородных
связей. Данное явление называется гид-
рофобным эффектом.
Система будет меняться таким
образом, чтобы исключить термоди-
намически невыгодный контакт непо-
лярных групп с водой. Гидрофобный
эффект собирает вместе неполярные
молекулы в водном растворе для ми-
нимизации поверхности их контакта с
водой. Если спрятать СН, СН2 или СН3
группу от контакта с ней, произойдет
потеря около 4 кДж-моль-1 свободной
энергии (см. табл. В2.3). Хотя в гид-
рофобном эффекте доминирует отри-
цательное изменение энтропии при
гидратации неполярных групп, нельзя
пренебрегать и вкладом энтальпии от
ван-дер-ваальсовой упаковки непо-
лярных молекул в ядре. Считается, что
гидрофобный эффект является основ-
ной движущей силой в стабилизации
белка. Тщательный анализ калори-
метрических данных, приведенных на
рисунке В2.12, позволил дать количес-
твенную оценку его роли в стабилиза-
ции нативной укладки белка.
Энтальпия и энтропия стабилизации
нативного состояния белка:
гидратация неполярных и полярных
групп и компактность их упаковки
Из кривых, изображенных на рисун-
ке В2.10, мы видим, что при температуре,
близкой к комнатной, соответствующие
изменения энтальпии и энтропии вслед-
ствие взаимодействий между белком и
растворителем, в значительной степени
компенсируют изменения энтальпии,
обусловленные динамикой внутренних
связей белка, и энтропии, связанными
с конфигурационной свободой цепи.
При разворачивании молекулы при
максимальном значении ДС, Д5 равно
нулю, поэтому нативный белок стаби-
лизируется за счет небольшого вклада
энтальпии. При более низких темпера-
турах Д5 отрицательно и стабилизация
обусловлена энтропией, тогда как при
более высоких температурах домини-
рует энтальпия. Обратите внимание на
то, что на рисунке В2.10 Дб” — свобод-
ная энергия стабилизации в отсутствие
взаимодействий с растворителем всюду
выше ДС и что их разность стремится к
нулю с повышением температуры. Сле-
довательно, гидрофобный эффект и вза-
имодействия с растворителем делают
нативное состояние менее стабильным
относительно состояния в вакууме.
Дальнейший анализ еще более под-
черкивает роль гидратации полярных
групп, которой нельзя пренебречь.
Из параграфа В2.4.8 мы знаем, что в от-
личие от положительного вклада гид-
ратации неполярных групп, гидратация
полярных групп вносит отрицательный
вклад в ДСр который исчезает при очень
высокой температуре. Вспомним что ДСр
определяется из наклона кривой зави-
симости энтропии от температуры. На-
личие полярных и неполярных групп в
зоне контакта с водой приводит к упоря-
дочиванию ее молекул, хотя их действие
прямо противоположно: неполярные
группы упорядочиваются потому, что не
могут образовывать водородные связи,
а полярные, наоборот, дают молекулам
воды такую возможность. При темпе-
ратуре, приближающейся к комнатной,
энтропии гидратации обеих групп отри-
цательны и близки по значению. Но энт-
ропия гидратации полярных групп име-
ет отрицательный наклон и понижается
еще сильнее с повышением температу-
ры, тогда как та же величина для непо-
лярных групп обладает положительным
наклоном и приближается к нулю выше
100 °C. Необходимо подчеркнуть, что
«общие» значения, принятые для тепло-
емкостей гидратации тех и других групп
для расчетов в параграфе В2.4.8, явля-
ются, в лучшем случае, лишь хорошими
приближениями. На самом деле они за-
висят от природы конкретной группы,
и, как мы с вами уже видели в случае с
семейством неполярных боковых цепей,
значение и температурная зависимость
теплоемкости гидратации существенно
отличаются для алифатических и арома-
тических групп.
Анализ калориметрических дан-
ных после учета влияния гидратации
показал, что плотная ван-дер-вааль-
сова упаковка внутри белка играет су-
щественную роль в стабилизации его
нативной укладки. С точки зрения
гидрофобного эффекта, мы должны
ожидать, что как можно большее чис-
ло неполярных групп будет группиро-
ваться вместе, прячась таким образом
от воды. Однако гидрофобный эффект
в основном является результатом нена-
правленного влияния энтропии, а оно
не может служить основой механиз-
ма специфических взаимодействий.
В случае с находящимися в воде угле-
водородами агрегация неполярных мо-
лекул приводит к формированию жид-
кой фазы. С внутренней частью белка
этого не происходит. Она, как показали
исследования тысяч структур белковых
кристаллов, скорее похожа на компакт-
ное аморфное тело или апериодический
кристам. Отдельные ван-дер-ваальсо-
вы взаимодействия крайне слабы (,м.
i;i6.I. B2..S). Однако сумма их энергий,
как результат специфической упаковки
всех групп внутри белка, довольно зна-
чительна и вполне способна быть опре-
деляющей движущей силой, стоящей за
стабильностью нативной укладки.
Для описания энергетики процесса
стабилизации белка и более полной ин-
терпретации калориметрических дан-
ных требуется детальный анализ струк-
туры поверхности макромолекулы и
плотности ее внутренней упаковки.
Значительные усилия были посвящены
развитию методов для такого анализа
белков, структуры которых были полу-
чены с помощью кристаллографии пли
ЯМР ( коммгр га он ।' В2.Р ).
В2.4.12. Холодовая денатурация
С приближением к низкотемператур-
ному концу кривой Лб доминирует эн-
Ко:.и ко।’;;р!о• !'2 9.
Термодинамика и структура белка
Пространственно заполненная модель
белковой структуры, полученной с помо-
щью рентгеновской кристаллографии. Об-
ратите внимание на компактную упаковку
атомов. Красные и синие шарики на поверх-
ности белка отображают отрицательно и
положительно заряженные боковые цепи
аминокислот. Укажем основные процессы,
обусловливающие разность в стабильности
между нативным и развернутым состоя-
ниями. Первое — гидратация групп на по-
верхности. Она пропорциональна площади
каждой группы, доступной растворителю,
которую можно рассчитать из структуры
в нативном состоянии, а в развернутом
состоянии ее принимают равной всей тео-
ретически доступной площади цепи. Вто-
рое — детальная упаковка внутренней час-
ти белка. Ее можно проанализировать, зная
трехмерную структуру.
тропия сольватации неполярных групп.
Это ведет к дестабилизации нативной
укладки белка по мере снижения темпе-
ратуры, которая была названа холодовой
денатурацией ( ш н rapnii IЭ. I о).
Причина холодовой денатурации
состоит в том, что гидрофобные взаи-
модействия, компактизирующие белок,
сильно растут с повышением температу-
ры и, следовательно, так же сильно пада-
ют с ее понижением. В результате теплота
плавления белка сильно падает с пони-
жением температуры. Ее уменьшение на-
столько велико, что может изменить знак
при низких температурах. На рисунке
В2.13 а показано изменение удельной (на
грамм белка) разности энергий нативно-
го и денатурированного состояний белка
с температурой для пяти белков. Из ри-
сунка видно, что при температурах выше
комнатной более упорядоченная струк-
тура (нативный белок) имеет более низ-
кую энергию, чем менее упорядоченная
(денатурированный белок), но при низ-
ких температурах (ниже 20 °C) энергия
упорядоченной структуры может быть
выше, чем денатурированной. Этот эф-
фект особенно выражен для миоглобина,
обратимая холодовая денатурация кото-
рого показана на рисунке B2.I3 Г>. После
холодовой денатурации (нижняя кривая)
наблюдается ренатурация белка (верхняя
кривая), которая имеет два пика. Левый
пик относится к собственно к ренатура-
ции, а правый пик - к обычной тепловой
денатурации.
Полная холодовая денатурация бел-
ка не наблюдается в обычных физиоло-
гических растворителях, поскольку она
может происходить лишь при темпера-
туре ниже нуля. Однако она отмечалась
также в некоторых денатурирующих
'<() ' В2.10. Дестабилизация
белковых комплексов при низкой
температуре
Механизмы холодовой денатурации
действительны и в случае с белковыми
комплексами. Хорошо известно, что неко-
торые из них диссоциируют, если раствор
поместить в холодную комнату.
Е^АТФаза, растворимая часть связан-
ной с мембранной F Е0-АТФазы, является
примером такой чувствительной к холоду
структуры. Можно заключить, что гидро-
фобные силы доминируют над белок-белко-
выми взаимодействиями внутри комплекса.
Рис. 132.13: а — зависимость удельной разности энергий нативного и денатурированного состоя-
ния для пяти белков: 1 — РНКаза; 2 — лизоцим; 3 — а-химотриисин; 4 — цитохром с; 5 — миогло-
бин (Griko et al., 1974); б — денатурация апомиоглобина при понижении температуры (нижняя
кривая). Верхняя кривая показывает обратимость процесса. Стрелки показывают направление
изменения температуры. Концентрация белка 9,5 мг-см’3 (Griko et al., 1988)
растворителях, таких как концентри-
рованный раствор гуанидин гидрохло-
рида, который сдвигает термодинами-
ческие кривые в сторону более высоких
температур.
В2.5. Нуклеиновые кислоты
и липиды
Те аргументы и аналитические под-
ходы в дифференциальной сканиру-
ющей калориметрии, которые были
описаны выше для анализа стабилиза-
ции укладки белков, полностью при-
менимы к другим макромолекулам,
например, таким как нуклеиновые
кислоты (рис. 132.11), а также к взаи-
модействиям, участвующим в обра-
зовании комплексов макромолекул и
между макромолекулами и меньшими
по размеру лигандами.
Изучение структуры олигонуклео-
тидов привело к пониманию энергети-
ки образования двойной спирали ДНК.
Остаточная структура олигонунуклео-
тидов оявляется, когда температура
снижается до 80 °C. Это диффузный
процесс с достаточно небольшой эн-
тальпией образования, который закан-
чивается при температуре ниже 0 °C,
требуя отрицательных температур для
наблюдения.
Рис. 132.14. Зависимость теплоемкости от
температуры для одноцепочечного дезокси-
олигонуклеотида 5'-GCGAACAATCGG-3' с
концентрацией 1,88 мг-мл'. Полная энталь-
пия разворачивания в диапазоне от -10 °C до
80 °C равна 77 кДж моль'1 (Privalov & Privalov,
2000)
Дифференциальная сканирующая
калориметрия является также важным
инструментом для изучения образования
мембран в смеси воды с липидами. При
этом получают сложные фазовые диа-
граммы как функции температуры и соот-
ношения вода/липид. Энергетика таких
систем исследуется посредством измере-
ния температурной зависимости тепло-
емкости для каждой фазы и связанных с
переходами пиков. Ссылки на обзоры по
данной тематике даны в разделе В2.7.
В2.6. Перечень ключевых
® Полное количественное термодинамическое описание макромолекулы в рас-
творе в заданном температурном интервале можно получить из измерения за-
висимости ее парциальной удельной теплоемкости от температуры при посто-
янном давлении с помощью дифференциальной сканирующей калориметрии.
• Характерными чертами современного дифференциального сканирующего ка-
лориметра являются: использование менее 50-200 мкг материала на одно из-
мерение, возможность использования температурного диапазона от -10°С до
130 °C, чувствительность около 10 мкДж-градус-1 и возможность сканирования
вверх и вниз по температурной шкале для контроля обратимости процесса.
• Нативному состоянию макромолекулы чаще всего приписывают статус эта-
лонного и по отношению к нему определяют все термодинамические парамет-
ры системы.
* Анализ кривой теплоемкости с помощью функции распределения применим
к любым мономерным (когда число присутствующих в растворе частиц не ме-
няется после сворачивания) системам в равновесии, независимо от механизма
перехода и промежуточных состояний.
• Калориметрическая энтальпия и энтальпия Вант-Гоффа равны, когда вся мо-
лекула представляет собой один кооперативный домен, и переход происходит
по принципу «все или ничего»; в остальных случаях, когда эти две энтальпии
не равны, переход происходит через промежуточные состояния.
’ Когда кооперативный домен меньше молекулы, как в случае с мультидомен-
ными белками, энтальпия Вант-Гоффа меньше калориметрической; если же
кооперативный домен больше молекулы, то энтальпия Вант-Гоффа больше ка-
лориметрической, что служит доказательством межмолекулярных взаимодейс-
твий, таких как олигомеризация.
3 Не существует калориметрических доказательств того, что расплавленная гло-
була является интермедиатом при разворачивании небольших однодоменных
белков.
Различие в стабильности между нативным белком и его мутантом может быть
следствием как изменения вклада энтальпии и энтропии при замене одних ами-
нокислотных остатков на другие, так и следствием изменения кооперативности
разворачивания.Кривую зависимости теплоемкости от температуры для белков
со сложной структурой можно разбить при помощи компьютерных программ на
несколько кривых, соответствующих переходам различных состояний системы.
Данные по дифференциальной сканирующей калориметрии белка и модель-
ных соединений дают возможность рассчитать теплоемкость белка в различ-
ных состояниях.
Теплоемкость безводного развернутого полипептида называется первичной
теплоемкостью и может быть точно рассчитана суммированием вкладов отде-
льных аминокислот с учетом формирования пептидных связей; таким же обра-
зом можно рассчитать теплоемкость свернутых безводных белков.
Теплоемкость развернутого пептида в растворе складывается из первичной
теплоемкости и гидратационных вкладов тех же аминокислот с учетом N и С
концевых групп.
Поскольку гидратационный вклад в теплоемкость положителен, то теплоем-
кость нативного белка в растворе выше, чем у безводного свернутого белка.
Вклад гидратации зависит от взаимодействия поверхности белка с растворите-
лем и, особенно, от величины площади доступной поверхности полярных и не-
полярных групп.
Теплоемкость нативного состояния белка в растворе зависит линейно от тем-
пературы, теплоемкость же развернутого состояния в растворе хорошо аппрок-
симируется полиномом второго порядка в диапазоне температур 0-100 °C.
Теплоемкость нативного белка в растворе зависит от свойств поверхности мак-
ромолекулы и особенностей свернутой структуры, поэтому ее нельзя рассчи-
тать непосредственно из его аминокислотной последовательности; для этих
целей разработан специальный параметризированный подход.
Для описания энергетики стабилизации белка и более полного использования
калориметрических данных необходим детальный структурный анализ поверх-
ности макромолекулы и ее интерьера.
Гидрофобный эффект в водном растворе приводит к объединению неполярных
групп в гидрофобное ядро для минимизации поверхности их контакта с водой.
Энтропия сольватации неполярных групп вызывает дестабилизацию нативной
укладки белка по мере понижения температуры. Этот процесс называют холо-
довой денатурацией.
Литература для дальнейшего чтения
Tristram-Nagle S., Nagle J. F. Lipid bilayers: thermodynamics, structure, fluctuations,
and interactions. Chem. Phys. Lipids. 2004.127,3-14.
Weber P. C., Salemme F. R. Applications of calorimetric methods to drug discovery and
the study of protein of protein interactions. Curr. Opin. Struct. Biol. 2003.13,115-121.
Privalov G. P. and Privalov P. L. Problems and prospects in microcalorimetry of
biological macromolecules in «Energetics of biological macromolecules», Methods in
Enzymology 31-62, 2000.
Privalov P. L. Scanning microcalorimeters for studying macromolecules, Pure &
Appl. Chem., 1980. 52, 479-497.
MartinezJ. C., el HarrousM., Filimonov V. V.,MateoP.L. &Fersht, A. R. A calorimetric
study of the thermal stability of barnase and its interaction with 3’GMP. Biochemistry,
1994.33,3919-3926.
Финкельштейн А. В. и Птицын О. Б. Физика белка: курс лекций. М.: КДУ,
2007.
ГЛАВА ВЗ
ИЗОТЕРМИЧЕСКАЯ КАЛОРИМЕТРИЯ
ТИТРОВАНИЯ
-г ?/ обзор
, y/S i
Р. Б. Спокан и С. Дж. Гилл
(R. В. Spokane and S. J. Gill) описали
первый микрокалориметр для измере-
ния теплоты титрования реагентов на
уровне наномолей.
i TS9
Т. Вайсман, С. Уиллистон, Дж.
Ф. Брандте и Л. Н. Лин (Т. Wiseman,
S. Williston, J. F. Brandts and L. N. Lin)
опубликовали работу под названием
«Быстрое измерение констант и теплот
связывания с использованием нового
калориметра титрования».
!!>90
Е. Фрейре (Е. Freire) с коллегами
ввели термин «изотермическая кало-
риметрия титрования» (ИКТ). В 1990—
2000 гг. был опубликован ряд важных
обзоров ио основам метода ИКТ, ре-
зультатов его применения и перспектив
дальнейшего развития.
2000 н по насгоящее время
Уникальность метода заключается
в том, что он дает возможность опре-
делять не только величину изменения
энтальпии после связывания, ио так-
же, если позволяют условия экспери-
мента, значения аффинности связыва-
ния и величины изменения энтальпии.
Поскольку эти параметры полностью
определяют энергетику процесса свя-
зывания, изотермическая калоримет-
рия титрования играет все более важ-
ную роль в детальных исследованиях
белок-лигандных взаимодействий и в
подходах по конструированию новых
лекарств.
В3.2. Экспериментальные
подходы и основные
уравнения
ВЗ.2.1. Проведение измерений
и требования к образцам
Реакционная ячейка в приборе для изо-
термической калориметрии титрования
имеет объем около 1 мл и содержит один
из реагентов. Небольшой объем другого
реагента (1-10 мкл) впрыскивают в ячей-
ку и перемешивают. Калориметр измеря-
ет мощность (в мкДж-сек'1 или в мик-
роваттах), требуемую для поддержания
постоянной разности температур между
реакционной и эталонной ячейками. Теп-
ло, поглощенное пли выделенное в ре-
зультате химической реакции, определи-
Время (сек)
а б
Рис. В3.1. Экспериментальные кривые ИКТ связывания циклического монофосфата с РНКа-
зой А: а — мощность, требуемая для поддержания постоянной разности температур между изме-
рительной и эталонной ячейками, выраженная как функция времени. Каждый пик соответствует
одному впрыскиванию; б — интегральный тепловой эффект связывания как функция в каждой
точке впрыскивания (Privalov and Privalov, 2000)
ется интегрированием кривой мощности
за время эксперимента (рис. ВЗ. 1).
В эксперименте по изотермичес-
кому титрованию, показанному на
рисунке ВЗ. 1 а, калориметрическая
ячейка объемом 0,6 мл заполнялась
раствором белка (68,8 мкМ панкреати-
ческой РНКазы А), а раствор лиганда
(0,5 мМ циклической монофосфатазы)
добавлялся в объеме 5 мкл с помощью
шприца, который служил также и для
перемешивания раствора. По оси у отло-
жена временная зависимость мощности,
требуемой для поддержания постоянной
разности температур между измеритель-
ной и эталонной ячейками. Каждый пик
соответствует одной инъекции, а интег-
рал мощности (после вычитания фона,
вызванного растворением лиганда, ме-
ханическим эффектом смешивания и
т. д.) — теплу, ассоциированному с вели-
чиной связывания (д в уравнении В3.1).
Величина пика с каждой последу-
ющей инъекцией уменьшается, по-
скольку все меньше свободного белка
остается для связывания. Когда белок
насыщен лигандом, высота пика отра-
жает фоновый уровень. График интег-
ральной величины теплового эффекта
связывания как функция числа инъек-
ций показан на рисунке ВЗ. 1 б.
ВЗ.2.2. Связывание: энтальпия
и теплоемкость
Величина тепла q? поглощенного или
выделенного в результате у-впрыскива-
ния (см. рис. ВЗ. 1), равна:
= V х АН хА (В3.1)
где V — это объем реакционной ячейки,
а А[Н спя|] — изменение концентрации
связанного лиганда после у-инъекции.
Поэтому q. пропорциональна количе-
ству лиганда КА[£. который связан
с белком. Константа пропорционально-
сти в уравнении В3.1 — это измене-
ние энтальпии АН на моль связанного
лиганда. Величину изменения тепло-
емкости процесса можно определить
повторным титрованием при разных
температурах (уравнение В3.2)
ДСР=ЭДН/ЭГ. (В3.2)
Температура (°C)
Рис. В3.2. Энтальпия связывания и теплоемкости. График энтальпии связывания как функции
температуры для взаимодействий белок-ДНК (HMG-бокс белка Sox-5 с ДНК-дуплексом из 12
пар оснований, содержащего сайт узнавания). Теплоемкость связывания дается начальным на-
клоном линии (-6 кДж моль ’ К1) при низких температурах. При более высокой температуре
заметно небольшое отклонение от прямой, вызванное перестройкой комплекса (см. также пара-
граф В3.2.3).
На рисунке В3.2 показан пример
такого титрования для белка HMG-
box-DNA (коммептприп B3.I ).
Если количество добавленного
лиганда мало по сравнению с аффин-
ностью реакции (см. параграф В 1.3.2),
можно предполагать, что он будет весь
связан и величину изменения энталь-
пии можно получить из уравнения
В3.1. Более того, если константа ассо-
циации реакции Кп известна из других
измерений (таких как флуоресцентное
титрование или поверхностный плаз-
монный резонанс (параграф В4.2.2),
то изменение свободной энергии AG, а
следовательно и изменение энтропии
А5 реакции можно рассчитать из AG и
АН, используя классическое уравнение
ВЗ.З. Тогда термодинамика реакции
будет полностью определена:
AG = -7?TlnH„ =АН-ТА5. (ВЗ.З)
В зависимости от числа сайтов
и других особенностей модели связыва-
। Комментарий ВЗ. i. HMG-box-DNA
Английскаяабревиатура HMG-box-DNA
означает белок, способный взаимодейство-
вать с ДНК. N-конец белка содержит мно-
жественные белковые последовательности.
Структурно эти последовательности содер-
жат консервативный мотив Trp-Asp (так
называемый WD-40 повтор). Эти повторы
। обнаруживаются во многих эукариотиче-
ских белках и могут функционировать как
адапторные и регуляторные модули в сиг-
нальных путях трансдукции, процессинга
пре-мРНК и в образовании цитоскелета.
I На С-конце HMG-box-DNA белок содер-
жит группу высокой подвижности (так
называемый HMG-box). HMG-box обна-
руживается во многих эукариотических
белках, вовлеченных в процессы сборки
хроматина, транскрипции и репликации.
1 Его молекулярная масса может достигать
126 кДа (у человека).
Рис. ВЗ.З. И КТ-кривые для различных констант аффинности. Здесь приведены три искусствен-
ные кривые ИКТ для иллюстрации эффектов возрастающей аффинности связывания. Для моде-
лирования были использованы следующие параметры:
[Р] = 0,05 мМ; (Z.] = 0,6 мМ; А// = -42 кДж моль К и с показаны на рисунке, где с = |Р|.
Аффинность связывания устанавливается только в случае, изображенном на первой панели
(с <1000), где величина ников последовательно уменьшается. Если же аффинность слишком ве-
лика для экспериментальных условий (с > 1000), тогда можно оценить только ее минимальное
значение. Напомним, что микрокалории в секунду можно перевести в микроватты умножением
на 4,2 (Leavitt and Freire, 2001)
ния, математическое выражение, кото-
рое связывает константы аффинности с
уравнением В3.1, может быть достаточ-
но сложным.
qj = V(\H)PM.mx
(В3.5)
В3.2.3. Константы сродства
(аффинности)
В самом простом случае, когда белок Р
имеет один сайт для лиганда L, можно
записать:
P + L—^PL,
к -- [Р£]
° №]’ (В3.4)
Pof„n =[?] + [?£].
£о6,„=[£] + [?£],
где Рабщ и — совокупные концент-
рации белка и лиганда, а квадратные
скобки обозначают молярные концен-
трации. Комбинируя уравнения В3.1
и В3.4, получим:
где [£]; — это концентрация свободного
лиганда после /титрования.
Данный анализ также применим
для реакций с числом сайтов более
одного с одинаковыми или разными
константами связывания. В этом слу-
чае уравнение В3.5 принимает более
сложный характер и для его решения
требуются дополнительные данные,
поскольку количество параметров ста-
новится велико.
В тех случаях, когда энтальпия ре-
акции недостаточно большая для того,
чтобы ее можно было измерить во всем
диапазоне концентраций лиганда, сама
константа может быть определена с по-
мощью серии титрования (рис. ВЗ.З).
Для соотнесения константы связыва-
ния с условиями эксперимента вводят
дополнительный параметр с, равный:
с = К[Р]. (В3.6)
Значение с должно быть <1000 для
К* реакции, чтобы эту константу можно
было определить с помощью калори-
метрии титрования. На практике дан-
ное соотношение устанавливает верх-
ний предел в 10к— 109 М 1 для константы
связывания (см. рис. ВЗ.З).
Реакции с высокими константами
связывания
Когда значения с превышают 1000, кри-
вые калориметрического титрования те-
ряют свою характерную форму и больше
не содержат информацию о константах
связывания (см. рис. ВЗ.З). Это становит-
ся существенным ограничением в приме-
нении метода ИКТ для конструирования
лекарств, поскольку «лидирующие ком-
поненты» должны иметь наиомолярные
или даже более высокие значения аффин-
ности (см. об этом ниже). Тем не менее в
рамках метода изотермической калори-
метрии титрования разработан протокол,
позволяющий определять высокие конс-
танты связывания с помощью конкурен-
ции лигандов. Для этого производят три
титрования раствора белка. В первом оп-
ределяют термодинамические параметры
связывания для слабого лиганда, во вто-
ром для сильного, а в третьем, вытесняю-
щем титровании, белок, заранее связан-
ный с более слабым лигандом, титруется
лигандом с высокой аффинностью. Ри-
сунок В3.4 демонстрирует применение
данного метода на примере ингибиторов
О 20 40 60 80 100
Рис. ВЗ. 1. Вытесняющее титрование для определения высоких констант аффинности связывания
(см. текст). Эксперименты по исследованию взаимодействия ингибиторов (Acaet, KNI-764) с HIV-1
протеазой проводили при 25 °C в ЮмМ ацетате, pH 5,0,2% диметилсульфоксида (Velazquez-Campoy
et al., 2001)
HIV-1 протеазы, которая исследуется с
целью создания лекарств в борьбе против
СПИДа.
Энтальпия связывания при взаимо-
действии ингибитора с HI V-1 протеазой
может быть точно измерена с помощью
ИКТ (см. рис. В3.4, верхняя правая па-
нель, ось у здесь в мкал-М-1) в противо-
положность константе ассоциации ин-
гибитора Ка, которая слишком велика,
чтобы ее можно было определить дан-
ным методом. Кажущуюся константу
связывания К (см. рис. В3.4, нижняя
панель) определяют в присутствии сла-
бого ингибитора (вытесняющее титро-
вание) ацетил-пепстатина с константой
связывания Ка Дем. рис. В3.4, верхняя
левая панель). Выбор слабого конку-
рентного ингибитора с энтальпией свя-
зывания, имеющей противоположный
знак, позволяет увеличить чувствитель-
ность эксперимента. Ка может быть оп-
ределена из Ккаж с помощью уравнения:
К =— Ка
1+ка,Лх]”
где [X] — концентрация слабого ин-
гибитора, которую можно довести до
необходимого К . Величины коне-
каж
тант аффинности оказались равными:
К = 2,4 х Ю6М-‘ и К = 3,1 х 10'0М-'.
Конструирование (дизайн)
лекарственных препаратов
Знание трехмерных структур фермен-
тов, участвующих в метаболических ре-
акциях при заболевании, помогает со-
зданию высокоаффинных ингибиторов,
которые могут стать потенциальными
фармацевтическими агентами. Чтобы
лекарство было пригодным для лече-
ния, его аффинность должна лежать в
наномольном диапазоне (константы
диссоциации порядка 10“9 М). Если
описывать кратко, то конструирование
лекарственных препаратов включает в
себя несколько шагов. После того как
выбран целевой белок, ведется комби-
наторный поиск из сотен тысяч аген-
тов, которые могут взаимодействовать
с его активным сайтом. Многие из них
представляют собой природные моле-
кулы, получаемые из растений. Иногда
структуры таких ингибиторов настолько
сложны, что их трудно вообразить. При-
мером такой структуры может служить
таксол (паклитаксел), антимитотичес-
кий препарат, получаемый из коры тиса.
Перспективные комплексы белок-
ингибитор характеризуют различными
методами, включая метод ИКТ, который
дает информацию об энергетике связыва-
ния. Затем идет химическое моделирова-
ние для нахождения структурных вари-
антов, способных повысить аффинность
ингибитора к белку. Такие модифициро-
ванные ингибиторы синтезируются и ис-
пытываются, затем процесс повторяется
до тех пор, пока не будет найден удовлет-
ворительный фармацевтический агент.
ВЗ.2.4. Изотермическая
калориметрия титрования
и дифференциальная
сканирующая калориметрия
Температурную зависимость энтальпии
связывания можно интерпретировать
как теплоемкость этого процесса только
в том случае, когда изменение темпера-
туры не вызывает перестройку компо-
нентов реакции. Если же теплоемкость
изменяется с температурой, как в реак-
ции, изображенной на рис. В.3.2, то это
означает, что изменения компонентов
под действием температуры могут иметь
место. В этом случае целесообразно ис-
пользовать сочетание изотермической
калориметрии титрования и дифферен-
циальной сканирующей калориметрии,
которое позволяет исследовать тепло-
емкость каждого компонента реакции в
подходящем температурном диапазоне.
Здесь мы вернемся к примеру с HMG
box-DNA (гм. м aim ci! ।; 11 >1111 I >.'i. I), темпе-
ратурная зависимость энтальпии связы-
вания которого была показана на pi icvi 1 ке
В3.2. Функции молярной парциальной
теплоемкости компонентов реакции,
определенные посредством дифферен-
циальной сканирующей калориметрии,
представлены на рисунке 133.5.
Очевидно, что интерпретация изме-
нения теплоемкости в процессе связы-
вания существенно затруднена развора-
чиванием белка (точечная линия на рис.
13,3.5) и частичной диссоциацией комп-
лекса (сплошная линия) в исследуемом
температурном диапазоне. Реакции взаи-
модействия HMG с ДНК были выраже-
ны как набор равновесных уравнений
наподобие уравнения В3.4, а все термо-
динамические параметры системы были
оценены с помощью аппроксимации на-
блюдаемых функций теплоемкости ком-
плекса и его свободных компонентов.
Анализ показал, что в изменениях
комплекса, вызванных температурой,
присутствуют две стадии (сплошная
линия на рис. В3.5). На первой стадии
происходит постепенное увеличение
теплоемкости, отражающее накопление
тепловой энергии. На второй наблю-
дается диссоциация комплекса с сопут-
ствующим разворачиванием одного из
компонентов — белка.
ВЗ.З. ПРИЛОЖЕНИЯ МЕТОДА
В3.3.1. Конструирование лигандов
Как видно из уравнения ВЗ.З, одну
и ту же константу связывания можно
Рис. 133.5. Парциальные молярные тепло-
емкости компонентов взаимодействия HMG
box DNA. Линия пунктир-точка обозначает
дуплекс ДНК из 12 пар оснований, точечная
линия — белок Sox-5 HMG, а сплошная — их
комплекс (Privalov and Privalov, 2000)
получить путем различной комбина-
ций вкладов энтропии и энтальпии.
Энтальпия связывания отражает кон-
куренцию между взаимодействиями
белок-лиганд при образовании связи
между ними (например, ван-дер-вааль-
совой и водородной) с одной стороны,
и взаимодействиями белок-раствори-
тель и лиганд-растворитель с другой.
Энтропия процесса отражает в основ-
ном динамику энтропии сольватации,
например, через экранирование гидро-
фобных групп от контакта с раствори-
телем и изменения в конформационных
энтропиях белка и лиганда.
Оптимизация вкладов энтальпии
и энтропии
Оптимизация аффинности связывания
включает различные тактические под-
ходы, в зависимости от того, достигает-
ся ли она за счет изменения вкладов эн-
тальпии или энтропии. Сверхвысокая
аффинность требует благоприятной эн-
тальпии и энтропии связывания. Высо-
кой энтропии связывания способствует
Когшзснi<ipia B3.2. Возбуждение
ВИЧ-инфекции, цитокины
и хемокины
ВИЧ (вирус иммунодефицита человека)
является причиной заболевания под на-
званием СПИД (синдром приобретенного
иммунодефицита). Инфицирование начи-
нается со специфического взаимодействия
между внешним гликопротеином оболочки
вируса (gpl20), мембранным клеточным
рецептором (CD4) и облигатными рецепто-
рами хемокинов (CCR5 или CXCR4). Свя-
зывание вирусного белка с CD4 запускает
конформационные изменения в оболочке
вируса, которые приводят к его узнаванию
рецепторами хемокинов и впоследствии —
к слиянию ВИЧ с клеточной мембраной
и его проникновению в клетку.
Хемокины — разновидность цито-
кинов, противовоспалительных белков,
ответственных за хемотаксис клеток
иммунной системы. Они являются необ-
ходимыми компонентами фагоцитарной
реакции; предполагается их участие в
элиминации опухолевых клеток, повреж- >
денных при цитотоксической терапии. |
В 1996-1997 гг. опубликованы результа-
ты комплексных исследований, которые
вскрыли уникальную роль в развитии .
инфекции вирусом иммунодефицита че- ,
ловека рецепторов молекулярных аттрак-
тантов — хемокинов. Выяснилось, что для
инфицирования клеток первичными штам-
мами ВИЧ, т. е. штаммами, выделенными
непосредственно от больного и не адапти- ;
рованными к размножению в культурах пе- 1
ревиваемых лимфоцитов, необходимо при-
сутствие на поверхности клеток не только
известных ранее рецепторов CD4, но и ре-
цепторов для хемокинов — CCR-5 (CKR-5). i
Только при наличии этих рецепторов в ин- |
тактном состоянии возможно заражение
чувствительных клеток первичными мак-
рофаготропными вариантами ВИЧ.
дизайн конформационно ограниченных
(напряженных) лигандов, имеющих вы-
сокую степень гидрофобности. Именно
такими свойствами обладают ингиби-
торы протеазы IIIV-1 (см. рис. 133.1),
которые одобрены для клинического
использования.
Жесткость лигандов обеспечивает
их высокую специфичность в отноше-
нии белка-цели. Однако эта специфич-
ность делает лиганд малоэффективным
в отношении родственных или мутиро-
ванных целевых белков, что может при-
водить к лекарственной устойчивости.
Иногда желательно, чтобы лиганд об-
ладал определенной гибкостью. Тогда
более низкая аффинность, обусловлен-
ная снижением энтропийного фактора
связывания, должна быть скомпенси-
рована другими энергетически выгод-
ными взаимодействиями. Стоит также
отметить, что гидрофобность лиганда
будет влиять и на его растворимость.
Поэтому выбор определенного соотно-
шения между энтропией и энтальпией
определяет не только аффинность свя-
зывания, но и более общие свойства са-
мого лиганда.
Соотношение между энергией
связывания и структурой
Знание энтальпии связывания несом-
ненно оказывает большую помощь для
дизайна лигандов. К сожалению, ее ин-
терпретация далеко не однозначна, по-
скольку самые разные процессы вносят
почти равноценные вклады в энталь-
пию связывания. Структурное иссле-
дование комплексов белков с низкомо-
лекулярными лигандами показало, что
сайты связывания в свободных белках
характеризуются низкой структурной
стабильностью. Их стабилизация после
присоединения лиганда отражается в
термодинамике процесса как конфор-
мационный энтальпийный вклад свя-
зывания. Несмотря на присущие этой
проблеме трудности, сегодня предпри-
нимаются попытки параметризовать
энтальпию связывания в терминах
структурных характеристик, как это
было сделано в сканирующей калори-
метрии (см. параграф В2.4.9).
Изотермическая калориметрия
титрования в комбинации с другими
биофизическими методами
В качестве примера комплексного под-
хода в изучении белок-белковых вза-
имодействий опишем исследование
энергетики реакции связывания комп-
лекса гликопротеина внешней оболоч-
ки вируса иммунодефицита человека
(gpl20) и мембранного клеточного
рецептора CD4 ( к<>x:xn-:i 1 ир.э: II.';.2).
Здесь использовались различные фи-
зические методы, включая масс-спек-
трометрию и аналитическое ультра-
центрифугирование для определения
масс белков и комплексов в растворе,
изотермическую калориметрию титро-
вания для характеристики энергетики
процесса и аффинностей связывания,
поверхностный плазмонный резонанс
для характеристики кинетики связы-
вания и круговой дихроизм (Глава Е6)
для идентификации структурных пере-
строек в ходе связывания.
Термодинамика взаимодействия
при связывании определялась с помо-
щью изотермической калориметрии
титрования. На рисунке В3.6 пред-
ставлены графики тепловой мощнос-
ти и интегральной энтальпии титрова-
ния белком CD4 зрелого а и исходного
б gpl20. Растворимые формы двух
белков были получены путем экспрес-
сии в клетках яичников китайского
хомячка и клеточной линии дрозофи-
лы. Белок gpl20 был получен в двух
формах: как полноразмерный глико-
зилированный белок и как исходный
gp 120, с некоторыми аминокислотны-
ми делециями и без сахарных групп.
Молекулярные массы CD4 и исход-
ного gpl20 по отдельности и в комп-
лексе между ними оказались равными
45 кДа, 38 кДа и 83 кДа, соответствен-
но. Линии аппроксимации показаны
для значений энтальпий связывания,
равных 63 и 62 ккал на моль белка
CD4 для полноразмерного и исход-
ного белка gpl2O. Равновесные конс-
танты диссоциации KD для двух бел-
ков оказались равны 5 нМ и 190 нМ
соответственно. Рисунок В3.6 д отоб-
ражает температурную зависимость
энтальпии связывания CD4 для пол-
норазмерного (пустые кружки) и ко-
рового (закрашенные кружки) gpl20.
Наклоны линий аппроксимации дают
значения теплоемкостей связывания,
равные 1,2 и 1,8 ккал-моль'-К’1.
В приведены термо-
динамические параметры, рассчитан-
ные с помощью данных, показанных
на рисунке В3.6. Стоит отметить, что
. . Термодинамические параметры взаимодействия CD4
с белком др 120 при 37°С
^~~~~~\Параметр Молекула~~~\^_ AG° (ккал-моль-1) дя° (ккал-моль-1) -T&S° (ккал-моль-1) ДС° (ккал-моль-1) (нМ)
полноразмерный gpl20 -11,8 ±0,3 -63 ±3 51,2 ± 3 -1,2 ±0,2 5±3
коровый gpl20 -9,5 ±0,1 -62 ±3 52,5 ± 3 -1,8 ±0,4 190 ±30
Рис. В3.6. Изотермическая калориметрия титрования gpl20 белком CD4; а) графики тепловой
мощности (вверху) и интегральной энтальпии (внизу) титрования полноразмерного гликолизи-
рованного белка gpl 20 белком CD4; б) то же для исходного белка; в) температурная зависимость
энтальпии связывания для белков двух типов (Myszka et al., 2000); г) рентгеновская кристалло-
графическая структура комплекса gpl20 (красный) с CD4 (желтый) при разрешении 2,5 А. В
этой проекции видно, что остаток Phe 43 белка CD4 находится в «кармане» белка gpl20. Пло-
щадь контакта между двумя белками составляет около 800 А2 (Kwong et all., 1998)
эти параметры для полноразмерного и
корового gpl20 в основном схожи, что
указывает на общий механизм связы-
вания. Большая разность между зна-
чениями констант диссоциации (5 нМ
и 190 нМ) соответствует разности сво-
бодной энергии связывания, равной
всего лишь 2 ккал-моль-1, что меньше
энергии образования одной водород-
ной связи.
Термодинамика процесса связыва-
ния для комплекса CD4-gpl20 качест-
венно отличается от других комплексов
белок-белок, для которых характерны
небольшие положительные значения
АН или -T&S. Однако она похожа на
термодинамику взаимодействий между
антителом и антигеном, а также меж-
ду рецептором Т-клеток и пептидом
главного комплекса гистосовместимо-
сти (МНС). Благоприятная энтальпия
связывания CD4 с gpl2O поразительно
велика (&Ни ~ -63 ккал-моль"1), тогда
энтропийный вклад данного процес-
са (~TASo ~ 52 ккал-моль *) хотя тоже
очень большой, но неблагоприятный.
Таким образом, образование комплекса
CD4 — gpl20 включает большое число
взаимодействий при связывании, та-
ких как водородные связи и ван-дер-
ваальсовы взаимодействия, а также су-
щественную потерю степеней свободы.
Как показывают кристаллографические
данные, такое уменьшение энтропии
объясняется значительными конфор-
мационными перестройками белков,
а не захватом молекул воды. Большие
значения энтальпийного и энтропий-
ного вкладов в связывание уравнове-
шивают друг друга и дают в результате
низкие значения констант диссоциации
и свободной энергии, похожие на со-
ответствующие показатели для других
комплексов.
Теплоемкости связывания в дан-
ном случае (см. i;i5. с IJ.'J. i ) значитель-
но выше значений, обычно наблюдае-
мых для комплексов белок-белок (от
-0,2 до -0,7 ккал-моль-1 К). Исходя из
структурной интерпретации измене-
ний теплоемкости, обсуждаемой в раз-
деле ВЗ.З, можно предположить, что
после образования комплекса CD4 —
gpl20 обширная неполярная площадь
Время (сек)
т----1----1----1 I I
О 50 100 150 200 250
Время (сек)
а б
Рис. В.3.7. Кинетики связывания CD4 с gpl20, полученные методом поверхностного плазмон-
ного резонанса: (а) нормированные сигналы связывания CD4 (1мкМ) с полноразмерным gpl20
(черная кривая), гликозилированным коровым белком (красная кривая), пегликозилированным
коровым белком gp 120 (синяя кривая) и [^модифицированным сенсором (серая кривая); (б) ки-
нетика связывания корового gpl20 с поверхностью белка CD4 при различных концентрациях —
от 975 нМ (верхняя черная кривая) до 36 нМ (вторая нижняя черная кривая). Также показана
базовая линия для концентрации 0 нМ. Данные аппроксимированы (красные линии) с помощью
модели взаимодействия с одним сайтом (Myszka et al., 2000)
Комментарий ВЗ.З. Структурное
моделирование термодинамических '
параметров
Сполар Р. С. л Рекорд М. 'Г, независимо
от подхода Фреире и его сотрудников, соот- :
несли термодинамические параметры с ко-
личеством остатков, которые подвергаются
реструктуризации во время связывания, на
основе анализа структуры, полученной с I
высоким пространственным разрешением
(Spolarand Record, 1994).
поверхности белков становится недо-
ступной для растворителя.
В другом подходе, основанном на
моделировании (комментарий 1)3.3),
теплоемкость и энтропия связывания
интерпретировали в терминах числа
остатков, которые подвергаются рест-
руктурированию во время связывания.
Применение данного подхода для комп-
лекса CD4 — gp 120 дало значение числа
таких остатков, близкое к 100. Это одно
из самых больших значений для взаи-
модействий белок-белок.
Кинетика связывания CD4 — gp 120
была проанализирована также методом
поверхностного плазмонного резонан-
са (см. рис. В3.7 и Главу В4). Получен-
ные константы скорости ассоциации
для полноразмерного и корового бел-
ка gpl20 оказались малыми и близки-
ми по значениям: 6,72 х Ю4 М-1'сек-1 и
6,27 х Ю4 М-1сек-1 соответственно, что
характерно для больших структурных
перестроек во время связывания.
Различные значения равновес-
ных констант диссоциации (СМ. i.lu.i.
113.1 ) согласуются с данными, полу-
ченными с помощью изотермической
калориметрии титрования. Они воз-
никают в результате различных конс-
тант скоростей диссоциации, возмож-
но, из-за потери термодинамически
выгодных контактов в коровом белке
gpl20. В то же время влияние глико-
зилирования на кинетику связывания
оказалось минимальным.
Гибкость белка gp 120, а также маски-
рование его рецептора и сайтов связы-
вания хемокинов могут способствовать
тому, что свободные мономеры этого
белка из вирусной оболочки будут вы-
зывать появление в основном антител,
которые неспособны к его нейтрализа-
ции, и, следовательно, плохо стимули-
руют образование нейтрализующих ан-
тител. Такая стратегия-ловушка может
помогать вирусу избегать гуморального
иммунного ответа.
Комбинация результатов гидродина-
мических, термодинамических, спектро-
скопических и кристаллографических ис-
следований комплекса CD4 — gpl20 дает
нам захватывающую картину его роли в
биологии ВИЧ-инфекции. Как пишут
авторы исследования, которое мы цити-
ровали в разделах В3.5-3.7: «Конфор-
мационная гибкость gpl20 служит при-
влекательной гипотезой для понимания
иммунной невосприимчивости вируса и
механизма его проникновения в клетку».
В3.4. Перечень ключевых идей
• В изотермической калориметрии титрования мощность, необходимая для подде-
ржания постоянной разности температур между реакционной ячейкой и эталон-
ной, измеряется калориметром; тепло, поглощенное или выделенное в результате
химической реакции, находится интегрированием кривой мощности по времени.
Изменение теплоемкости связывания в процессе реакции можно определить пов-
торным титрованием при разных температурах, в тех случаях, когда энтальпия
реакции слишком велика для измерения в диапазоне концентрации лиганда.
• Перспективные комплексы компонент-белок для конструирования лекар-
ственных препаратов можно охарактеризовать с помощью изотермической ка-
лориметрии титрования, которая дает информацию об энергетике связывания.
• Одинаковую константу связывания можно получить различными комбинация-
ми вкладов энтропии и энтальпии.
• Энтальпия связывания отражает конкуренцию между взаимодействиями,
обусловленными образованием связи белок-лиганд с одной стороны и взаимо-
действиями белок-растворитель и лиганд-растворитель с другой.
• Энтропия связывания прежде всего говорит об изменениях в энтропии сольва-
тации и конформационных энтропиях белка и лиганда.
• Для получения как можно более полного описания энергетики и структурных
изменений во время реакции связывания целесообразно комбинировать изотер-
мическую калориметрию титрования с другими биофизическими методами.
• В реакциях, где теплоемкость компонентов реакции может меняться с темпе-
ратурой, целесообразно использовать изотермическую калориметрию титрова-
ния совместно с дифференциальной сканирующей калориметрией.
Литература для дальнейшего чтения
Spokane R. В and Gill S.J. Titration microcalorimeter using nanomolar quantities of
reactants. Rev. Sci. Instrum., 1981. 52, 1728-1733.
Seelig J. Thermodynamics of lipid-peptide interactions. Biochim. Biophys. Acta,
2004.1666, 40-50.
Velazquez-Campoy A., Leavitt S. A., Freire E. Characterisation of protein-protein
interactions by isothermal titration calorimetry. Methods Mol. Biol., 2004. 261, 35-54.
Глава В4
БИОСЕНСОРЫ И ПОВЕРХНОСТНЫЙ
ПЛАЗМОННЫЙ РЕЗОНАНС
В4.1. Исторический обзор
и введение в биологическую
проблематику
1801
Томас Янг (Thomas Young) открыл,
что что при помещении светового пучка
двух щелей на экране наблюдается ин-
терференционные полосы. Это явление
лежит в основе современных интерферо-
метров для измерения взаимодействия
лиганд-рецепторов.
1902
Р. В. Вуд (R. W. Wood) впервые
наблюдал явление поверхностного
плазмонного резонанса (ППР). Позже
это явление легло в основу простого и
чувствительного метода, который по-
зволил регистрировать малые измене-
ния коэффициента преломления, про-
исходящие вблизи поверхности тонкой
металлической пленки.
1968
Е. Кречманн и X. Ц. Рэтзер
(Е. Kretschmann и Н. Z Raether) пред-
ложили схему сенсора, основанного на
принципе поверхностного плазмонного
резонанса с оптической системой, со-
держащей призму, соединенную с реак-
ционной ячейкой.
J982 f9;S3
Б. Лидберг (В. Liedberg) с сотруд-
никами выяснили потенциал явления
поверхностного плазмонного резонан-
са для исследований связывания мак-
ромолекул. Они обратили внимание
на то, что изменение коэффициента
преломления вблизи поверхности тон-
кой металлической пленки зависит от
свойств молекул, локализованных на
поверхности металла и впервые детек-
тировали связывание индивидуальных
специфических иммуноглобулинов с
антителами, пришитыми к поверхности
золотой пленки.
P.hSO 1990-е
В эти годы было создано несколько
коммерческих биосенсоров, основан-
ных либо на явлении интерферомет-
рии, либо на явлении поверхностного
плазмонного резонанса для простого и
быстрого, а главное, не требующего ме-
чения анализа биологических макро-
молекул, таких как белки, ДНК и дру-
гие молекулы.
! 993
С. Дэвис (S. Davies) с коллегами
продемонстрировали биосенсор, ос-
нованный на явлении поверхностного
плазмонного резонанса для анализа
слабых взаимодействий, на примере
связывания рецепторов клеточной по-
верхности.
.1 по inc । оящее прело!
Были разработаны различные оп-
тические схемы биосенсоров с высо-
кой чувствительностью и избиратель-
ностью для одновременного анализа
взаимодействий между тысячами мо-
лекул, для исследования внутримоле-
кулярных взаимодействий с разным
уровнем аффинности в самых раз-
личных температурных и химических
условиях. Использование биосенсо-
ров делает их особенно удобными для
быстрого перебора предполагаемых
лигандов. За последние несколько лет
эти инструменты стали важнейшими
в изучении протеома. Используемые
в комбинации с другими биофизичес-
кими приборами, такими как кало-
риметр изотермического титрования,
аналитическая ультрацентрифуга и
масс-спектрометр, они позволяют про-
водить очень точный термодинами-
ческий анализ процесса связывания, а
также характеризовать и идентифици-
ровать партнеров. К преимуществам
использования бисенсоров по сравне-
нию с другими методами относятся:
использование небольшого количест-
ва материала (в нанограммовом диа-
пазоне), возможность исследования
разнообразных систем (от молекул с
массой от 200 Да до одноклеточных
организмов), отсутствие мечения и
низкие требования к чистоте препара-
та (можно исследовать связывающие
свойства даже клеточного экстракта).
Их несомненным достоинством яв-
ляется также возможность изучения
кинетика связывания в реальном вре-
мени.
3 .. й. И sм е ре н и е
поверхностного связывания
В4.2.1. Биосенсор как инструмент
исследования взаимодействий
между молекулами
Для исследования взаимодействий меж-
ду биологическими макромолекулами
разработаны различные типы биосенсо-
ров. Биосенсор как инструмент вклю-
чает в себя проточную кювету, присо-
единенную к оптическому устройству.
Буфер и анализируемое вещество вхо-
дят в кювету и покидают ее, а об их
биохимическом взаимодействии су-
дят по изменению коэффициента пре-
ломления, которое происходит из-за
изменения концентрации вещества
в кювете.
Для измерения показателя прелом-
ления в кювете используются два спо-
соба. Первый основан на поверхностном
плазмонном резонансе, а другой — на
высокочувствительной разностной ин-
терферометрии. Биохимические пара-
метры связывания, такие как равновес-
ная константа диссоциации, константа
скорости реакции (см. Главу В1) мож-
но рассчитать напрямую из изменений
коэффициента преломления раствора,
зависящих от времени.
Эксперименты по изучению повер-
хностного связывания обычно включа-
ют иммобилизацию первой молекулы
(лиганд) на поверхности и наблюде-
ние за ее взаимодействием со второй
молекулой в растворе (анализируемое
вещество). Преимущество такой схе-
мы заключается в том, что она служит
моделью многих биологических про-
цессов узнавания, таких как, например,
взаимодействия между клеточными ре-
цепторами и растворимыми сигнальны-
ми белками.
В4.2.2. Биосенсор на основе
поверхностного плазмонного
резонананса
Биосенсор, работающий с использова-
нием поверхностного плазмонного резо-
нанса, завоевал большую популярность
прежде всего благодаря простоте и вы-
сокой чувствительности. Сначала рас-
смотрим само явление поверхностного
плазмонного резонанса.
Явление поверхностного плазмонного
резонанса
Прежде всего вспомним хорошо изве-
стное явление в оптике — явление
полного внутреннего отражения. Оно
происходит на границе раздела между
двумя прозрачными средами с разны-
ми коэффициентами преломления и
детально описано в параграфе ДЗ.З.З.
Когда свет падает на поверхность разде-
ла таких сред под углом, который пре-
вышает некий критический угол, почти
весь свет полностью отражается. Одна-
ко некоторое количество света все же
проникает в среду с более низким коэф-
фициентом преломления в виде элект-
рического поля, называемого стоячей
волной.
Если поверхность раздела двух сред
покрыть тонким слоем металла, то стоя-
чая волна, поляризованная в плоскости
падения и отражения лучей (р-поляри-
зованный свет, коммсп rapnii 11 1.1), про-
никнет в слой металла (рис. В4.1). При
определенном угле падения такой свет
возбуждает делокализованные элект-
роны поверхности металла, названные
плазмонами, что приводит к сильному
увеличению амплитуды стоячей волны
(отсюда возникает резонанс). В это,м
угле интенсивность отраженного све-
та резко падает в результате передачи
энергии плазмонам. Такой угол паде-
ния луча называют углом резонанса.
Область
резонанса
Рис. 134.1. Стоячая волна, поляризованная в плоскости падения и отражения (р-полярпзация),
проникает в слой металла и возбуждает электроны в поверхностном слое металла. Поляризован-
ный свет падает под углом 0р к поверхности золотой пленки. кюв -- это волновой вектор поверх-
ностного плазмона, а к — волновой вектор падающего луча. Компоненты кх и kv — параллельны
и перпендикулярны поверхности, соответственно
Условия для возбуждения поверх-
ностных плазмонов на границе между
металлом и раствором выполняются
при совпадении проекции волнового
вектора падающего света в направле-
нии границы раздела (£р) и волнового
вектора (&„,„) осцилляции поверхност-
ного плазмона:
*,=*„„„ (В4.1)
где К. = xsin0>
Ip X F
_ I -чстдлл ^ячейка (В4 2)
1ЮН С V £ 4- £
L V мег:1Л.ч еячгйка
sin ер = р"™" хе,1"‘йка,
У ^металл ^ячейка
где со — угловая частота падающей вол-
ны, с — скорость света, ас — зависящая
от длины волны комплексная диэлектри-
ческая проницаемость. Угол падающего
света и, соответственно, отраженного све-
та 0р, который удовлетворяет условиям
поверхностного плазмонного резонанса,
зависит, таким образом, от коэффициен-
та преломления материала с неосвещен-
ной стороны металла (проточная ячейка
в нашем случае). Любые локальные изме-
нения показателя преломления приводят
к изменению интенсивности света, попа-
дающего на детектор.
На рисунке В4.2 схематически по-
казан биосенсор, работающий на основе
принципа поверхностного плазмонного
резонанса. Две среды в биосенсоре обра-
зованы стеклянной призмой и проточной
кюветой, в которой проходит биохими-
ческая реакция. Их разделяет тонкая ме-
таллическая пленка. В диапазоне длин
волн видимого света условия резонанса
выполняются всего для несколько метал-
лов, из которых наиболее подходящими
являются золото и серебро ( ком мен lapnii
В 1.2). Сигнал, измеренный в резонан-
Комментарий В4.1. s- и p-волны в •
явлениях отражения и преломления !
'п
I При изучении закономерностей поляри- ;
зации света в результате отражения и пре-
ломления естественного света последний
удобно рассматривать как совокупность
одинаковых по интенсивности линейно по- :
I ляризованных волн двух типов: s- и р-волн
(рис. 1 а и б). Согласно представлениям •
классической электронной теории обра- i
зование отраженной волны обусловлено :
вторичными волнами, которые излучают
молекулы — осцилляторы отражающей
свет среды. Волне 5-типа соответствуют ос-
цилляторы (колеблющиеся электрические
; диполи), оси которых перпендикулярны к
| плоскости падения. Эти осцилляторы по-
казаны на рисунке а зелеными стрелками.
Волне p-типа соответствуют осцилляторы,
оси которых лежат в плоскости падения
и перпендикулярны преломленному лучу.
! Рис 1: а — р-поляризованный свет: осцил- '
ляторы электрического поля (синие стрел- |
ки) лежат параллельно плоскости, образо- |
ванной падающим и отраженным лучами i
(красные стрелки); б — 5-поляризованный '
свет: осцилляторы электрического поля
(зеленые стрелки) лежат перпендикуляр-
но плоскости, образованной падающим и
отраженным лучами, поэтому параллельны
. поверхности раздела сред.
сных единицах (РЕ), пропорционален
количеству белка, связавшегося у повер-
хности (обычно 1000 РЕ = 1 нг связанно-
го белка на квадратный миллиметр). Ре-
зультаты обычно представляются в виде
сенсограммы, которая отражает измене-
KoMMeiiianiin. B-I.L’.
Распространение поверхностных
плазмонов
Плазмоны, распространяющиеся по по-
верхности металла, постепенно затухают
из-за потерь, вызванных поглощением. Ве-
личина затухания зависит от диэлектри-
ческой проницаемости металла при данной
частоте осцилляции плазмона. Золото и
серебро являются самыми предпочтитель-
ными кандидатами на роль металлических
пленок в диапазоне видимого света. Хотя се-
ребряная пленка дает более четкий сигнал,
на практике она не используется ввиду ее
химической нестабильности. Поэтому для
биосенсоров лучший выбор — золото.
ния резонансного сигнала как функции
времени (см. рис. В1.2 в).
В заключение отметим одно инте-
ресное явление, связанное с явлением
поверхностного плазмонного резонанса.
На его основе созданы светящиеся час-
тицы фантастической яркости, во много
тысяч раз превосходящую таковую всех
известных флуорофоров. Так, сфериче-
ские частицы диаметром 100 нм имеют
яркость, эквивалентную яркости 100000
флуоресцентным молекулам. Вдвойне
интересно, что форма частиц оказывает
сильное влияние на спектральные ха-
рактеристики частиц (рис. В4.3).
Рис. В4.2: а — общий принцип детектирования биомолекулярных взаимодействий с помощью
поверхностного плазмонного резонанса. Увеличившаяся концентрация образца на поверхности
сенсорного чипа вызывает рост коэффициента преломления, который изменяет резонансный угол;
б — такое изменение регистрируется положением детектора для минимума интенсивности отра-
женного луча (из позиции I в позицию II); в — изменение резонансного сигнала от времени.
а
Рис. В4.3: а — сферические частицы излучают преимущественно голубое свечение (415—490 нм),
пентатонные частицы светят зеленым цветом (480-600 нм, а треугольные частицы излучают
красное излучение (620-720 нм); б — зависимость интенсивности светящихся частиц от длины
волны (Shultz, 2003)
Б4.2.3. Биосенсоры на основе
интерферометров
В интерферометре с двойным ходом
лучей лазерный луч движется по двум
параллельным путям — А и Б, один из
которых — А — содержит образец (рис.
В4.4). Напомним, что скорость света в
среде с коэффициентом преломления п
дается выражением
с = -7Ё, (В4.3)
п
где со — это угловая частота волны света,
а е — диэлектрическая проницаемость
среды. Поскольку образец имеет свой
коэффициент преломления, он изменя-
ет скорость света, приводя к появлению
разности фаз лучей А и Б.
Две волны интерферируют, давая
классическую интерференционную кар-
тину Янга. Любые изменения показа-
теля преломления будут влиять на ско-
рость распространения лазерного луча и
вызывать сдвиги в интерференционной
картине. Величину сдвига калибруют
по известным значениям коэффициента
преломления. Оптическая конфигура-
ция биосенсора, основанная на интерфе-
рометрическом принципе, показана на
рисунке В4.4.
В таком биосенсоре образец пред-
ставляет собой проточную ячейку,
в которой происходят молекулярные
реакции. Рецепторы прикреплены к
клеточной поверхности, а анализируе-
мые вещества обтекают их. В отличие
от биосенсора, основанного на прин-
ципе поверхностного плазмонного ре-
зонанса, такой эксперимент чувстви-
телен к изменениям коэффициента
преломления по всему объему ячейки.
В4.2.4 . Лиганды и поверхность
В биосенсорах лигандами, присоеди-
ненными к поверхности проточной
ячейки, служат в основном белки, но
могут использоваться и другие разно-
образные молекулы. Их длительное и
высокостабильное связывание с по-
верхностью позволяет проводить эф-
фективное измерение и контроль ко-
личества связанного анализируемого
вещества.
Белки могут быть присоединены
к поверхности благодаря своим гидро-
фобным участкам или ковалентно при-
шиты через свои амино-, карбоксиль-
ные или сульфгидрильные группы.
При таком связывании лиганды, ве-
роятнее всего, принимают случайную
ориентацию на поверхности, и их мес-
то связывания не всегда будет доступ-
но для анализируемого вещества. Это
оказывает влияние на максимальную
Рис. В 1.4. Биосенсорная интерферометрия (см. текст)
емкость связывания, но не на измеря-
емые константы скорости и диссоциа-
ции (см. ниже).
Для пришивания лиганда к поверх-
ности часто используют тэги (от анг-
лийского tag — ярлык, конец). При пра-
вильной подборке они могут придать
белку положение, которое является для
него естественным на поверхности кле-
точной мембраны. Специально разра-
ботаны элементы биосенсоров (чипы),
способные связывать биотин, глута-
тион S-трансферазу и полигистидино-
вые тэги. Разработаны также и более
близкие к природным системы, включа-
ющие лиганды, погруженные в липид-
ный слой и обладающие латеральной
подвижностью.
Биосенсорный чип можно исполь-
зовать много раз подряд, поскольку
после каждого эксперимента лиганд-
ную поверхность регенерируют, чтобы
тщательно удалить оставшееся анали-
зируемое вещество.
В4.3. Взаимодействие между
молекулой и поверхностью
В4.3.1. Термодинамика
взаимодействий с поверхностью
Вспомним из главы В1, что термодина-
мическая равновесная константа диссо-
циации для связывания молекул А и В
дается выражением
к _И][В]_
d МВ1 kvm
В случае биосенсоров под Л.11ч. пони-
мают скорость связывания анализиру-
емого вещества с поверхностью, а под
А — скорость, с которой оно уходит
оттуда. Типичный диапазон значений
этих констант: Кл = 10’7-10’9Л/, km. =
= 103-108М~‘сек-1 и k =10°-10’6сек1.
ЛИСС
Типичная сенсограмма показана
на рисунке 134.5. Биосенсор калиб-
рован таким образом, что сигнал R
пропорционален концентрации свя-
Рис. В4.5. Сенсограмма представляет собой график зависимости сигнала от времени. Увеличе-
ние числа резонансных единиц по сравнению с начальной базовой линией отражает связывание
молекул. Плато показывает стационарную фазу взаимодействия, когда ассоциация уравновеше-
на диссоциацией молекул комплекса. Дальнейшее уменьшение сигнала указывает на удаление
анализируемого вещества из ячейки
данного анализируемого вещества.
Сигнал будет меняться со временем в
соответствии с концентрацией моле-
кул в растворе и с тем, как быстро мо-
лекула ассоциирует и диссоциирует
с рецептором, который пришит к по-
верхности проточной ячейки.
В4.3.2. Измерение константы
равновесия
Равновесная константа диссоциации Kd
определяется из равновесной величины
сигнала. Стандартное уравнение для
изменения сигнала со временем, при
стехиометрии взаимодействии 1:1 дает-
ся выражением
dR(t)/dt = k С (t) х
а" (В4.4)
х [R -R(t)]-k R(t)
для концентрации анализируемого ве-
щества С. /? — это максимальная ве-
личина сигнала. Равновесный сигнал
R достигается, когда dR (t)/dt =0.
Таким образом, предыдущее уравнение
можно переписать как
R =(!/(£ /k + C\yCR . (В4.5а)
равн х / \ Диес/ асе 7 7 макс ' 7
Или, используя константу равнове-
сия Кл= kvm_/k.wc,
R = CR (Д/(К.+ С)). (В4.56)
что подобно изотерме Ленгмюра (ком-
ментарий В 1.3). Равновесная констан-
та диссоциации Кл рассчитывается из
кривой зависимости равновесного сиг-
нала в точке на плато от концентрации
анализируемого вещества. Стандарт-
ный метод оценки Kt[ состоит в изме-
рении равновесного сигала R пн для
различных концентраций лиганда Ск.
Преобразовав предыдущее уравнение
в линейную форму, получим:
(VCk)/U(ck) =
= (-Ж)Я;... (Ск)+ (В4.6)
+ (1/7С)Я .
х ' d7 макс
Анализ Скэтчарда основан на опре-
делении сигнала связывания в равно-
весных условиях и соответствующих
концентраций анализируемого веще-
ства (см. Главу В1). Прямая R /Cv
отложенная против R , имеет наклон,
равный -1 /Ка, и пересекает ось х в точ-
ке RmKC/Kd (пример графика приведен
на рис. В4.6). Надо отметить, что урав-
нение В4.6 позволяет рассчитать Kd и
R из значений равновесных сигна-
лов при различных концентрациях,
не достигая насыщения лигандов по-
верхности анализируемым веществом.
На самом деле, при низкой аффинно-
сти взаимодействий, когда Kd лежит в
диапазоне--0,1-500 мМ, насыщение
поверхности сенсора достигается очень
редко.
Комментарий В4.3. Изотерма
Ленгмюра
Изначально изотерма Ленгмюра была
предложена для описания состояния рав-
новесия газа, находящегося в контакте
с твердой средой:
Г=Г^*С/(а + С) (Л),
где Г11кс — максимальная величина адсорб-
ции газа, а а — отношение скоростей десор-
бции и адсорбции (аналогично отношению
распада и образования комплекса), С —
равновесная концентрация, остающаяся в
растворе. Соотношение (А) называют изо-
термой, поскольку оно справедливо только
при постоянной температуре. Уравнение
(А) носит название адсорбционной изотер-
мы Ленгмюра и характеризует состояние
термодинамического равновесия при ра-
венстве скоростей адсорбции и десорбции
на адсорбенте.
CD8aa (мкмоль)
б
Рис. В4.6. Анализ связывания между белком CD8aa (лиганд) и молекулой HLA-A2 (анализи-
руемое вещество): а — серия последовательных разбавлений дает уменьшение сигнала: б — сиг-
нал при связывании и график Скэтчарда (вставка) (Wyer at al., 1999)
Во время эксперимента анализи-
руемое вещество обычно разбавляют
для достижения стократного диапазона
концентраций (см. рис. В4.6). Стандар-
тная свободная энергия связывания ДС°
рассчитывается из константы диссоци-
ации, как это описано в Главе В1. Вкла-
ды стандартной энтропии и энтальпии
можно определить из температурной
зависимости ДС° с помощью нелиней-
ной аппроксимации уравнения В 1.28.
В4.3.3. Определения констант
скоростей взаимодействия
Когда концентрация анализируемого
вещества в проточной ячейке падает до
нуля, то связавшиеся молекулы не на-
ходят замены, и сигнал начинает возвра-
щаться к своему начальному значению со
скоростью, определяемой силой связыва-
ния (см. рис. В4.6). Скорость диссоциации
k можно вывести, исходя из характера
уменьшения сигнала R со временем t.
R(t) = R(t0)exp[-kvm. (t -10)]. (B4.7)
Время t указывает на момент нача-
ла падения сигнала, когда большее чис-
ло молекул покидает лиганд, чем связы-
вается с ним.
В фазе ассоциации сигнал растет бы-
стро, со скоростью, которая зависит от
концентрации С анализируемого вещест-
ва. Уравнение В4.4 можно переписать как
dR(t) _ , \
dt U-c 4 + (B48)
x7?(O + ^cG7?MaKc.
а константу скорости k определить
из графика линейной регрессии dR
(?)/dt от R (t) в фазе ассоциации. Од-
нако быстрые скорости процесса де-
лают такую оценку неточной, поэтому
&асс лучше устанавливать с помощью
выражения
^асс Хадисе-
Процессы активации
Зависимость скорости реакции от
температуры дает информацию о тер-
модинамических параметрах актива-
ции. Вспомним уравнение В1.31 (Гла-
ва В1):
В эксперименте по изучению свя-
зывания константа скорости k в урав-
нении равна k ,с для диссоциации и
С“&асс для реакции ассоциации, где
С° — это стандартная концентрация
(1 М). Тогда значения энтальпии и
активации могут быть вычислены из
угла наклона кривой зависимости In
(k/T) от 1/Т на графике Эйринга, а
энтропию в точке ее пересечения (см.
параграф В 1.3.4).
84.4. Экс пер у. ментальный
анализ
В4.4.1. Диапазон анализируемых
веществ
В первых экспериментах биосенсоры
использовали преимущественно для
анализа достаточно больших молекул
(>5 кДа), поскольку локальная ве-
личина коэффициента преломления
зависит от массы. Биосенсоры могли
идентифицировать штаммы Е. соН в
соответствии с типом связываемых
лигандов и измерять аффинность свя-
зывания между вирусными частицами.
Чувствительность современных био-
сенсоров позволяет определять вещес-
тва с массой до 200 Да. Еще меньшие
молекулы можно охарактеризовать с
помощью конкурентного ингибирова-
ния (см. параграф В3.2.3).
В4.4.2. Экспериментальный
контроль и «подводные камни»
На практике применение биосенсоров
сопряжено с необходимостью особен-
но тщательного контроля за ходом
эксперимента. В частности неспеци-
фичное связывание определяют с по-
мощью клеток, не содержащих лиганд.
При построении экспериментальных
кривых наподобие графика Скэтчар-
да предполагают, что максимальная
теплоемкость связывания рецептора с
лигандом постоянна во времени. Од-
нако лиганд может менять свою теп-
лоемкость в результате денатурации
или деградации, поскольку обычно
эксперименты проводят при комнат-
ной температуре. Для оценки степени
деградации лиганда опыт должен быть
повторен при схожей концентрации
анализируемого вещества.
Слишком высокие концентрации
лиганда или анализируемого вещества
способны вызывать стерические помехи
и препятствовать связыванию молекул
из-за их избытка у поверхности проточ-
ной ячейки. Вследствие этого диффузия
вещества из основного объема раствора
к поверхности может быть затруднена,
и оценка скоростей реакции будет лож-
ной. Такие связанные с массопередачей
эффекты можно идентифицировать с
помощью варьирования скорости про-
тока вещества через ячейку. Очень вы-
сокая скорость связывания приводит к
конкуренции лиганда за ограниченное
число молекул анализируемого веще-
ства и повторному связыванию, а вслед-
ствие этого и к занижению константы
диссоциации.
Измерение связывания с высокой
аффинностью компонентов может
быть затруднено из-за низкой скоро-
сти диссоциации. Например, веществу,
константа диссоциации & которого
равна 10 6 сек ', потребуется около трех
часов (104 сек) на диссоциацию только
1 % его молекул. Для подтверждения
результатов наблюденных величины
аффинности, крайне желательно поме-
нять местами лиганд и анализируемое
вещество.
Заметим, что биосенсоры не подхо-
дят для исследования некоторых типов
молекулярных взаимодействий. На-
пример, когда присоединение лиган-
да к поверхности меняет его связыва-
ющие свойства, или когда связывание
прекращается из-за значительных кон-
формационных изменений лиганда.
В.4.4.3. Межклеточные
взаимодействия
Биосенсоры успешно применяются
в исследованиях межклеточных взаи-
модействий, поскольку иммобилизация
лиганд, которые в естественных усло-
виях связаны с мембраной, в проточной
ячейке в определенной степени воспро-
изводит ситуацию in vivo.
Узнавание антигена Т-клетками,
представляющее собой ключевой ме-
ханизм контроля адаптивного иммун-
ного ответа, интенсивно изучалось с
помощью биосенсоров. Рецептор Т-
клеток узнает антигены, которые не-
сут молекулы главного комплекса гис-
тосовместимости (МНС). Антигеном
обычно является пептид, полученный
в результате клеточного белкового
синтеза. Т-клетки всегда «проверяют»
наличие необычных пептидных анти-
генов, поскольку это указывает на ка-
кие-то отклонения от нормы. Напри-
мер, они распознают и уничтожают
клетки, имеющие вирусный антиген.
Взаимодействие между комплексом
МНС-пептида и рецептором Т-клеток
определяет судьбу клетки, несущей
МНС. Измеренная аффинность при
этом обычно мала (ТС, -0,1—500 мкМ)
вследствие медленной ассоциации и
быстрой диссоциации. Большое зна-
чение К . (-0,01-5 сек1), соответ-
ствующее периоду полураспада 70-0,1
сек, указывает на то, что сформирован-
Рис. 134.7. Сравнение усредненных термоди-
намических параметров тридцати взаимодейс-
твий белок-белок и взаимодействия рецепто-
ра Т-клеток с пептидом МНС (Willcox et al.,
1999)
Рис. IH.iS. Связывание саквинавира и индинавира протеазой HIV-1: а — масс-спектр, получен-
ный методом ИЛДМ-ФП, показывающий элюированные с поверхности саквинавир (670 Да) и
индинавир (613 Да); б — смоделированная с помощью данных масс-спектрометрии сенсограмма
диссоциации саквинавира и индинавира с поверхности чипа. Вертикальная линия обозначает
время, при котором соотношение между двумя ингибиторами должно быть 0.4, что соответс-
твует отношению концентраций между двумя молекулами в масс-спектре. Пунктирная линия
соответствует смеси саквинавира с индинавиром, линия с длинным пунктиром — саквинавиру,
линия точка-пунктир — индинавиру (Mattei et al., 2004)
ный комплекс рецептор Т-клеток-пеп-
тид-МНС более стабилен, чем другие
комплексы, участвующие в межкле-
точном узнавании. Измерения связы-
вания при различных температурах с
помощью биосенсора, основанного на
явлении поверхностного плазмонного
резонанса, показали, что оно характе-
ризуется необычно выгодными изме-
нениями энтальпии и крайне невыгод-
ными — энтропии (рис. В 1.7).
Исследование ряда других комп-
лексов МНС-пептида с рецептором Т-
клеток обнаружило корреляцию меж-
ду соотношением аффинность/время
полураспада и функциональным эф-
фектом. Полагают, что взаимодейс-
твия по схеме «рецептор Т-клеток-пеп-
тид-МНС» определяют длительность
связывания. Более продолжительное
взаимодействие дает возможность
другим молекулам Т-клеток собрать-
ся в месте контакта двух клеток и
инициировать гибель той, которая не-
сет антиген.
В4.4.4. Поверхностный плазмонный
резонанс и масс-спектрометрия
Сочетание биосенсора с масс-спектро-
метром дает возможность проводить
одновременно анализ связывания ана-
лизируемого вещества, его кинетику и
идентификацию молекул. Биосенсор
позволяет выделить партнеров по свя-
зыванию для рецептора из клеточного
лизата. Интересующие исследователей
молекулы вылавливают из общего экс-
тракта благодаря тому, что они удержи-
ваются лигандом в проточной ячейке.
Затем анализируемое вещество элюи-
руют и идентифицируют с помощью
масс-спектрометрии.
В качестве примера на рисунке В4.8
показаны результаты конкуренции меж-
ду двумя лекарствами (саквинавир и
индинавир) за поверхность сенсора, со-
держащую иммобилизованную HIV-1
протеазу. Масс-спектрометрический ана-
лиз позволил установить вклад каждого
ингибитора в процесс диссоциации.
iwuO4SBb!X идей
' Явление поверхностного плазмонного резонанса возникает, когда монохромати-
ческий р-поляризованный свет отражается от тонкой металлической пленки, на-
ходящейся на границе двух сред с различными коэффициентами преломления.
0 Эксперимент по связыванию с помощью биосенсора включает иммобилизацию
молекулы на поверхности (лиганд) и наблюдение ее взаимодействия со второй
молекулой в растворе (анализируемое вещество).
° Об изменении локальной концентрации макромолекул в биосенсоре судят по
изменению коэффициента преломления.
° Изменение коэффициента преломления в биосенсоре можно измерить с помо-
щью поверхностного плазмонного резонанса или двухлучевой интерферомет-
рии.
° Кривая, отражающая процесс связывания во времени, называется сенсограммой.
4 Для точного измерения термодинамических параметров используют серию
последовательных разведений анализируемого вещества в стократном диапа-
зоне концентраций.
Скорость диссоциации k рассчитывают из кривой, показывающей затухание
сигнала связывания, а скорость ассоциации вещества с лигандом kMc из фазы
ассоциации, используя различные концентрации анализируемого вещества.
( k А
° График Эйринга — зависимость Ini — I от \/Т, где k — это kvm. или O’kan. — по-
зволяет определить значения энтропии и энтальпии реакции активации.
° Диапазон анализируемых веществ в биосенсоре варьирует от молекул с массой
в несколько сотен дальтон до целых клеток.
с Комбинация биосенсора с масс-спектрометром позволяет совместить термоди-
намический анализ связывания с идентификацией анализируемого вещества.
° Применение биосенсоров требует тщательного экспериментального контроля
специфичности связывания.
j 'ут-ература для дальнейшего чтения
Mullet W. М., Lai Е. О. С. and Yeung J. М. Surface Plasmon Resonance-Based
Immunoassays, Methods, 2000. 22, 77-91.
Schultz D. A. Plasmon resonant particles for biological detection, Current opinion in
biothechology, 2003.14, 13-22.
Barnes W. L., Dereux A., Ebbesen, Surface Plasmon Subwavelength Optics, Nature,
2003. 424, 824-830.
Часть Г
ГИДРОДИНАМИКА
Глава Г1
БИОЛОГИЧЕСКИЕ МАКРОМОЛЕКУЛЫ
КАК ГИДРОДИНАМИЧЕСКИЕ ЧАСТИЦЫ
Г1.1. ИСТОРИЧЕСКИЙ
ОБЗОР И ВВЕДЕНИЕ
В БИОЛОГИЧЕСКИЕ
ПРОБЛЕМЫ
В традиционном представлении гид-
родинамика изучает поведение тел в
жидкостях, и в частности, какие силы
действуют на тела при их движении
в среде с определенной вязкостью и
плотностью. Многие выдающиеся уче-
ные-физики, такие как Исаак Ньютон,
Джеймс Клерк Максвелл, лорд Рэлей
(Дж. У. Струтт) и Альберт Эйнштейн
(Isaac Newton, James Clerk Maxwell,
Lord Rayleigh (J. W. Strutt) and Albert
Einstein) начинали свои карьеры ис-
следованиями в области гидродинами-
ки и внесли фундаментальный вклад в
науку. Следует отметить, что хотя эти
открытия сделаны давно, однако они до
сих пор сохраняют свое фундаменталь-
ное значение и продолжают стимулиро-
вать развитие этой области знаний.
1731
Гидродинамика как наука была
впервые описана в классической книге
Дениэла Бернулли (Daniel Bernoulli)
«Гидродинамика», которая содержала
изложение законов о давлении и вязко-
сти в несжимаемых жидкостях, а также
их многочисленные следствия. В сле-
дующем веке фундаментальный вклад
в эту область внес сэр Гораций Лэмб
(Sir Horace Lamb), опубликовавший
в 1879 г. другую классическую книгу,
также названную «Гидродинамика».
1821
Ботаник Роберт Броун (Robert
Brown) описал беспорядочное тепловое
движение маленьких растительных час-
тиц, суспендированных в воде, явление,
которое позже получило название броу-
новского движения. В 90-х гг. следующе-
го столетия метод усовершенствованной
видеомикроскопии позволил непосред-
ственно наблюдать броуновское движе-
ние макромолекул в мембране, меченых
коллоидным золотом.
1855
Адольф Е. Фик (Adolf Е. Fick)
опубликовал феноменологическое опи-
сание поступательной диффузии и вы-
вел фундаментальные законы транс-
порта частиц в растворах.
18 К)
Дж. Л. М. Пуазель (J. L. М. Poise-
uille) создал теорию движения жид-
кости в капилляре. На базе этой тео-
рии Вильхельм Оствальд (Wilhelm
Ostwald) изобрел вискозиметр и ввел
его в физическую и химическую прак-
тику. Позднее прибор был назван его
именем.
1856
Сэр Георг Стокс (Sir George
Stokes) показал, что коэффициент пос-
тупательного трения частицы зависит
от се линейных размеров. Описание
гидродинамического поведения частиц
в терминах радиуса эквивалентной сфе-
ры (радиус Стокса) вошло в физико-
химическую практику. В 1880 г. Стокс
проанализировал также вращательное
трение для частиц сферической формы
и пришел к выводу о связи коэффици-
ента вращательного трения сферы с ее
объемом. В 1930-х гг. Франсуа Перрен
(Frangois Perrin) расширил примене-
ние формулы Стокса на случай трехос-
ного эллипсоида вращения.
1856
Джеймс Клерк Максвелл (James
Clerk Maxwell) обнаружил, что неко-
торые жидкости в гидродинамическом
потоке становятся двулучепреломля-
ющими. В 1960-1970-х гг. Вик-
тор Цветков и Роже Серф (Victor
Tsvetkov и Roger Cerf) разработа-
ли детальный метод для измерения
двойного лучепреломления в потоке
макромолекулярных растворов. Эф-
фективность метода была подтверж-
дена в исследованиях эластичности
и оптических свойств синтетических
полимеров и биологических макро-
молекул, не имеющих фиксированной
жесткой структуры.
1887
Осборн Рейнолдс (Osborne Rey-
nolds) обратил внимание на то, что от-
ношение сил инерции и вязкости (позже
названное числом Рейнолдса) является
ключевым для понимания особеннос-
тей движения частиц в вязкой среде.
В 1970-х гг. Ховард Берг и Эдвард Пар-
селл (Howard Berg and Edward Purcell)
рассчитали это число в случае движения
различных объектов (от молекул до жи-
вых организмов) в разных средах. Было
найдено, что при движении частиц в рас-
творителе с молекулярными размерами
10-10 000 А число Рейнолдса очень мало.
Это означает, что биологические макро-
молекулы «живут» в мире без инерции.
В таком мире решающее влияние на их
поведение оказывает броуновское дви-
жение и вязкость среды.
188!
Альберт Видеманн (Albert Wiede-
mann) предложил термин «люминесцен-
ция», с тем чтобы подчеркнуть различие
между тепловым равновесием и неравно-
весным испусканием излучения. Среди
неравновесных процессов он исследовал
эмиссию света молекулами при комнат-
ной температуре, вызываемую падающей
радиацией. В 1945 г. Сергей Вавилов
(Serguei Vavilov) предложил способ оп-
ределения гидродинамического объема
частиц из экспериментальных данных по
поляризованной флуоресценции.
1896
Джон Керр (John Kerr) обнаружил,
что некоторые растворы становятся дву-
лучепреломляющими под действием
электрического поля. После этого откры-
тия измерение электрического двойного
лучепреломления стало способом полу-
чения информации о природе диполь-
дипольных взаимодействий и гибкости
макромолекул, обладающих как посто-
янным, так и индуцированным диполь-
ным моментом. Введение импульсного
режима измерения в практику элект-
рического двойного лучепреломления
и развитие основ теории для макромо-
лекул позволило Генри Бенуа (Henri
Benoit) в 1950 г. изучать биологические
молекулы в растворе методом электри-
ческого двойного лучепреломления.
1 905
Альберт Эйнштейн (Albert Einstein)
разработал теорию броуновского дви-
жения. Он количественно охарактери-
зовал движение молекул в раствори-
телях и газах. Годом позже он получил
уравнение, связывающее коэффициент
диффузии макромолекул в раство-
ре с коэффициентом поступательного
трения, а также показал, что удельная
вязкость суспензии жестких сфер про-
порциональна их объемной доле и не
зависит от их радиуса.
!!)'!()
Роберт Симха (Robert Simha) полу-
чил уравнение для вязкости растворов эл-
липсоидов вращения, а в 1981 г. Стефан
Хардинг и Артур Рове (Stephen Harding
and Arthur Rowe) получили уравнение
для вязкости трехосного эллипсоида.
1920
Теодор Сведберг (Theodor Sved-
berg) изобрел высокоскоростную
центрифугу, открывшую эпоху анали-
тического и препаративного ультрацен-
трифугирования. Он предложил метод
совместного использования коэффици-
ентов седиментации и диффузии для
вычисления молекулярных масс частиц.
В1929 г. Отто Ламм (Otto Lamm) вывел
основное уравнение, описывающее пове-
дение движущейся границы в поле уль-
трацентрифуги, которое, как оказалось
впоследствии, не имеет аналитического
решения. Появление в 90-х гг. прошлого
века нового поколения полностью ав-
томатизированных аналитических уль-
трацентрифуг фирмы Beckman (Optima
XL), а также развитие численных реше-
ний уравнения Ламма способствовали
возрождению метода аналитического
ул ьтрацентрифу ги ро ван ия.
1937
Альберт Тизелиус (Albert Tiselius)
использовал различия в зарядах макро-
молекул для их разделения на фракции
методом электрофореза, что привело
к его широкому применению в биохи-
мии. В 1950 г. Оливер Смитис (Oliver
Smithies) показал, что электрофорети-
ческое разделение белков по принципу
молекулярного сита обладает гораздо
большей разрешающей способностью,
чем электрофорез в свободном растворе.
1975
Патрик О’Фаррелл (Patrick O’Far-
rell) развил метод двумерного электро-
фореза, который привел к революции
в биохимии белков и настоящее время
интенсивно используется в протеомике.
Впоследствии на смену электрофорезу в
геле пришел капиллярный электрофо-
рез.
i 9G2
Бруно Зимм (Bruno Zimm) предло-
жил оригинальную модель ротационно-
го вискозиметра, с помощью которого
можно было проводить измерения при
очень низких градиентах скорости. Та-
кой прибор оказался незаменимым для
измерения вязкости асимметричных
структур, таких как ДНК, фибрилляр-
ные белки и палочкообразные вирусы.
1964
Герман 3. Каммингс (Herman
Z. Cummings), исходя из теории Ро-
берта Пекоры (Robert Pekora), опуб-
Комментарий Г1.1. Единицы силы
и вязкости в гидродинамике
Поскольку гидродинамика является нау-
кой, развиваемой на протяжении десяти-
летий, вычисления в ней традиционно де-
лаются в сгс единицах. Дина есть единица
силы в этой системе. Одна дина есть сила,
которую надо приложить к частице массой
один грамм, для того чтобы она получила
ускорение один см-сек -
дина = Г'С.м-сек 2. !
Все жидкости сопротивляются изме-
нению их формы. Это свойство жидкости
называется вязкостью. Единица вязкос-
ти есть пуаз. Один пуаз определяется как 1
тангенциальная сила на единицу площади
(дин-см -’), которая требуется, чтобы жид-
кость приобрела скорость 1 см еею' между
двумя параллельными плоскостями, нахо-
дящимися на расстоянии 1 см
1 пуаз = 1 дина-сек-см 1 = 1 г - смсек '.
ликовал первую экспериментальную
статью, в которой показал, что коэффи-
циент диффузии частиц латекса в рас-
творе может быть получен из спектров
динамического рассеяния света при
использовании лазера в качестве источ-
ника излучения. Эта работа подтверди-
ла блестящее теоретическое предсказа-
ние Леонида Мандельштама (Leonid
Mandelstamm), сделанное в 1923г., от-
носительно модуляции интенсивности
рассеяния света броуновским движе-
нием частиц. С этого момента метод
динамического рассеяния света стал
общепризнанным методом быстрого и
точного определения коэффициентов
диффузии макромолекул.
1967 1985
За эти годы были развиты новые тео-
ретические подходы для вычисления
гидродинамических свойств биологи-
ческих макромолекул (Виктор Блюм-
филд, Гарсиа де ла Торре, Стюарт
Аллисон) (Victor Bloomfield, Garcia
de la Torre, Stuart Allison). Было пока-
зано, что гидродинамические свойства
макромолекул могут быть вычислены
при заполнении всего объема частицы
набором сфер (шариков) одинаковых
или разных радиусов. В другом подхо-
де поверхность частицы моделируется
набором равных маленьких сфер (метод
бусинок) или набором маленьких тре-
угольных пластинок (метод чешуек).
Альтернативный подход был предложен
Стефаном Хардингом и Артуром Рове
(Stephen Harding and Arthur Rowe),
выдвинувшими концепцию описания
биологической макромолекулы в рам-
ках сфероида (трехосного эллипсоида)
и обосновавших принципиальную воз-
можность экспериментального опреде-
ления параметров такого эллипсоида из
гидродинамических данных.
1993
Джозеф Хаббард и Джек Дуглас
(Joseph Hubbard and Jack Douglas) об-
ратили внимание на существование ма-
тематического подобия между уравнени-
ями гидродинамики и электростатики.
Это открыло новые возможности вычис-
лений гидродинамических параметров
частиц разной формы на основе формул
электростатики, расчеты по которым
оказались гораздо менее трудоемкими
(Хуан-Ксианг Жу (Huan-Xiang Zhou)).
1990 2000
Впечатляющий прогресс в расшиф-
ровке структур белков и нуклеиновых
кислот с высоким пространственным
разрешением методами рентгенострук-
турного анализа и ЯМР стимулировал
развитие новых подходов для вычисле-
ния гидродинамических параметров био-
логических макромолекул. Было пока-
зано, что фрикционные свойства белков,
структура которых известна с атомарным
разрешением, могут быть вычислены с
точностью около 1-3% при условии кор-
ректного учета вклада гидратации.
2000 и по настоящее время
Современная гидродинамика нахо-
дится в стадии расцвета, являясь наукой
с четкими подходами для определения
размеров, формы, эластичности и ди-
намики биологических макромолекул.
Опа включает много новых эксперимен-
тальных физических методов, как, на-
пример, восстановление флуоресценции
красителя после фотовыцветания, раз-
решенная во времени деполяризованная
флуоресценция, флуоресцентная корре-
ляционная спектроскопия. Однако, не-
смотря на все эти достижения, мы всегда
должны помнить, что гидродинамика от-
носится к методам, дающим структуру
биологических макромолекул с низким
пространственным разрешением (ком-
мепlapnii I'I.I ). Эта структура описы-
вается всего несколькими параметрами:
одним — в случае сферы, двумя — в слу-
чае эллипсоида вращения и тремя — в
случае трехосного эллипсоида. Для оп-
ределения последних требуется комби-
нация нескольких (трех или более) гид-
родинамических методов.
Г1.2. Гидродинамика
при низких числах рейнолдса
При конструирования обоснованных
физических моделей тел, движущихся в
жидкой среде, включающих биологиче-
ские частицы, необходимо пользоваться
многочисленными упрощениями. В дан-
ной секции делаются два основных допу-
щения. Первое допущение состоит в том,
что ноток является ламинарным, н вто-
рое, что он достаточно «медленный», с
тем чтобы инерционными эффектами
можно было пренебречь. Эти допуще-
ния оправданы, поскольку интересую-
щие нас биологические системы состоят
из очень малых частиц и, хотя частицы и
движутся быстро по отношению к стенке
сосуда, как, например, в методах виско-
зиметрии и гидродинамического двой-
ного лучепреломления, они движутся
медленно по отношению к окружающей
их жидкости.
Г1.2.1. Число Рейнолдса
Рассмотрим объект, движущийся с ка-
кой-либо скоростью в растворителе с
определенной плотностью и вязкостью.
Для описания относительного вклада
эффектов инерции и вязкости более
чем сто лет назад Рейнолдсом был пред-
ложен безразмерный параметр R:
Число Рейнолдса -
плотность ж идкости
=------------------х
вязкость
скорость ..
х--------х (Г1.1)
* размер частицы _ _
Х 1 =
_ pxuxl
П
Как видно из формулы Г1.1, этот
параметр будет мал для частиц малых
размеров и, следовательно, при их дви-
жении должны доминировать силы вяз-
кости. Напротив, для частиц больших
размеров будут доминировать силы
инерции ( к<>мди:11 apini 1'1.2).
Г1.2.2. Движение при низких
значениях числа Рейнолдса
Вычислим число Рейнолдса для вируса
диаметром 500 А (5 х 10 6 см), переме-
щающегося в воде со скоростью порядка
Коммешарий Г1.2. Движение
различных частиц в водной среде
Бактерия
Э. Парсел был первым, кто провел ,
расчеты чисел Рейнолдса для бактерий
(Purcell Е. М„ 1977). Пусть бактерия
имеет диаметр 1x10’* см и плывет со ско-
ростью около 1x10 3 смсек Принимая |
р= 1 гем-3 и г|0 = 10 2тсм 'сек получаем для >
этого случая число Рейнолдса равное 10
т. е. очень маленькое. Отсюда мы заключа-
ем, что бактерия живет в мире, в котором
инерционные силы не действуют.
Рыбка
Те же вычисления для рыбки, имеющей
размеры / =10 см и движущейся в воде со i
скоростью около 100 см сек-1, дают число, I
равное 105. Такой случай в гидродинамике
называется гидродинамикой при высоких
числах Рейнолдса. Отсюда мы заключаем,
что рыбка живет в среде, где вклад инерци-
онных сил весьма существенен.
Кит
Для кита, имеющего размеры 10 м
(1000 см), движущегося со скоростью
3,6 км час-1 (100 см сек-1) в воде, число Рей-
нолдса равно 108. Поэтому плавание кита оп-
ределяется очень большими инерционными
силами.
Комментарий Г1.3. Определение
движения при очень низких числах ।
Рейнолдса I
Наилучшее определение движения при
низких числах Рейнолдса принадлежит ।
Э. Парселу: «То, что вы делаете в данный .
момент полностью определяется силами '
действующими на вас в этот момент, но
никак не силами, действующими в про-
; шлом» (Purcell Е. М., 1977).
10-3см-сек-1. Учитывая, что р = 1 г-см 3,
а Т| = 10-2 г-см 1 сек-1 мы получим чис-
ло Рейнолдса равным 5 х 10-7, т. е. это
число для вируса пренебрежимо мало.
Маленькое число означает, что моле-
кула вируса должна остановиться не-
медленно при исчезновении действия
силы. Однако поскольку частица виру-
са постоянно испытывает соударения с
другими молекулами растворителя, то в
действительности она никогда не оста-
навливается.
Вычисления показывают, что для
крупных биологических образований,
например, бактерий в воде, значение
числа Рейнолдса также мало (ем. ким-
\ieii lapiiii Г1.2 ). Таким образом, все
биологические макромолекулы от не-
больших белков до больших бактерий
живут в «мире без инерции», где доми-
нируют силы вязкости. Напротив, для
больших биологических объектов это
число велико и для таких объектов го-
раздо более важными становятся силы
инерции (см. коммен i apnii Г1
Г 1.3. ГИДРАТАЦИЯ
В гидродинамических экспериментах
биологические макромолекулы переме-
щаются с определенным количеством
связанной воды. Такую гидратирован-
ную частицу можно представить со-
стоящей из ядра (собственно материал
частицы) и оболочки (связанная вода).
На рисунке Г1.1 видно, что гидратация
может рассматриваться как увеличение
размера или объема ядра. Объем части-
цы в гидратированном состоянии, V ,
больше, чем объем «сухой» частицы, У vs,
который можно рассчитать из молеку-
лярной массы белка, М, и его парциаль-
ного удельного объема частицы, о.
Vm=Mv/NA. (Г1.2)
Гидратация белка, 8 (г/г), выража-
ется через отношение массы связанной
воды к массе сухого белка
g _ грамм (воды) (П 3)
грамм (белка)
Если р — это плотность растворите-
ля, тогда мы имеем:
8 = (V.1U/Vy!i-I)pu (Г1.4)
Возможны две трактовки значения
величины 8. Первая трактует эту вели-
чину как изоморфное расширение всего
белка (рис. Г 1.2). Она берет начало из
классического представления о глобу-
лярных белках как эллипсоидальных
частицах с небольшой степенью вытяну-
тости. Для частицы произвольной фор-
мы изоморфное расширение предпола-
гает, что линейные размеры, I, частицы
увеличиваются на константу, h, равную
h = utJu^ (п-5)
так что
= V1;1/V,.x. (П.6)
Из этого следует, что константа h
связана с параметром 8 как:
Л = (1+ 8/й р)1'3. (Г1.7)
Комментарий 1 1.4. Однородное
расширение для компактных
и вытянутых частиц
Принимая значения р =1 см’т1, Й =
= 0,73 гем 3 и 6 = 0,3 г-гтипичные для
глобулярных белков, получаем величину
для h = 0,12, которая соответствует расши-
рению, равному 12% в линейных размерах и
41 % в объеме. Для очень вытянутых частиц
с размерами 20 А в диаметре и 200 А в длину
такая величина гидратации ведет к увеличе-
нию диаметра частицы на 2 А, а в длину на
40 А. Очевидно, что для вытянутых частиц
однородное расширение ведет к аномальной
гидродинамической модели частицы.
Изоморфное расширение примени-
мо для компактных частиц, но не годит-
ся для очень удлиненных или палочко-
образных частиц ( ком мен । арии 1 1.1).
Вторая трактовка значения 8 бази-
руется на предположении, что негид-
ратированное ядро молекулы покры-
то (одето) водной оболочкой, которая
Рис. 1 1.2. Иллюстрация трактовки значения
6 как единообразное расширение для глобу-
лярных белков. При такой трактовке части-
ца увеличивает свои размеры (расширяется)
в разной степени (Garcia de LaTorre. 2001)
Рис. I 1.1. Схематическое представление же-
стких гидратированных частиц. Гидратация
влияет на общую форму белка, сглаживая не-
которые структурные детали, такие как углуб-
ления или полости. Темным цветом изобра-
жен сухой объем частицы. Серым - связанная
с ним вода (Garcia de La Torre. 2001)
Put'. Г1.3. Иллюстрация к интерпретации
значения 5 как толщины единообразной гид-
ратированной оболочки; — усредненное зна-
чение толщины гидратированной оболочки
(Garciade La Torre, 2001)
имеет постоянную толщину d, измеряе-
мую в перпендикулярном направлении
но отношению к поверхности белка
(рис. П.З). Гидратированная оболочка
рассматривается как свойство, прису-
щее всем белкам (Глава ГЗ).
Прежние интерпретации данных
по гидратации белков привели к пред-
Комментарий Г1.5. Вычисление I
величины гидратации
। Вычисления гидратации из гидродина-
мических свойств белков весьма чувства- '
тельны к ошибкам измерений, поскольку i
; величина гидратации вычисляется как «ма- I
। ленькая разность двух больших величин»
между гидратированным и негидратиро-
ванным (сухим) объемом частицы (уравне-
ние Г.1.4). Как было отмечено (Garsia de la I
| Torre, 2001), основной источник ошибок в |
величинах гидратации кроется в ошибках в
определении гидродинамических парамет-
ров. Большинство литературных данных
для белков базируется на опытах почти 50- ,
летней давности и очевидно, что для точ-
ной оценки гидратации требуются более
точные гидродинамические данные. ,
ставлснпю о том, что величина гидра-
тации может варьировать от белка к
белку. Эти результаты были получены
при моделировании белков в виде сфер
или эллипсоидов вращения и базирова-
лись в основном на экспериментальных
данных по диффузии и характеристиче-
ской вязкости.
Использование моделей белков,
структура которых была получена
из данных по кристаллографии пли
ЯМР, привело к величине гидрата-
ции от 0,3- 0,4 грамма воды на грамм
белка, что соответствует менее од-
ному гидратационному слою воды в
гидратационной оболочке. Развитие
такого рода вычислений (Глава ГЗ)
позволило сопоставить их с экспери-
ментальными данными таких методов
как ЯМР, инфракрасная спектроско-
пия, калориметрия, малоугловое рен-
тгеновское и нейтронное рассеяние.
В результате такого сопоставления
унифицированная картина гидрата-
ции для белков представляется следу-
ющим образом. Белок содержит опре-
деленное количество связанной воды
в среднем (0,3 г-г ’). Такое количество
связанной воды соответствует толщи-
не его гидратационной оболочки око-
ло 1,2 А. Локальная плотность воды
в оболочке приблизительно на 10%
выше, чем плотность воды в раство-
ре. По современным представлениям,
если в гидродинамическом экспери-
менте вычисленное значение величи-
ны гидратации сильно отличается от
величины 0,3 гг ’, то это, скорее все-
го, является показателем того, что
используемая гидродинамическая мо-
дель ошибочна ( комментарии Г1.5).
Молекулы РНК и ДНК благодаря
наличию суммарного отрицательного
заряда содержат гораздо больше ассо-
циированой воды, что приводит к зна-
чению гидратации около 0,6 ± 0,2 г-г
Для гликозилированных белков и кар-
богидратов значение гидратации со-
ставляет 0,5 г-г
Г1.4. Определение
фрикционных свойств частиц
Г1.4.1. Граничные условия: «полное
прилипание» — «свободное
скольжение»
В классической теории гидродинамики
взаимодействие частицы с растворите-
лем обычно рассматривается при двух
граничных условиях: «полное прилипа-
ние» и «свободное скольжение» (ком-
ментарий I I.6). В случае «свободного
скольжения» молекулы растворителя
не взаимодействуют с поверхностью
частицы, и частица перемещается или
вращается в нем независимо от моле-
кул растворителя (рис. Г1.1 ц). Другой
крайний случай представлен «полным
прилипанием», при котором первый
слой растворителя примыкает к повер-
хности частицы и перемещается (вра-
щается) вместе с ней (рис. 11.1 б).
Коммешприй Г1.6. Математическое
определение условий «свободного
скольжения» и «полного прилипания»
Строгая математическая трактовка усло-
вий «свободного скольжения» и «полного
прилипания» представляет собой сложную
математическую задачу. Читатель, интере-
сующийся этой проблемой, отсылается к
специальной литературе (Hu and Zwanzig,
1974).
Важно отметить, что значения кон-
стант в уравнениях, связывающих из-
меренные величины с вычисляемыми
гидродинамическими параметрами,
сильно зависят от выбора граничных
условий (см. ниже, уравнения Г1.1, Г1.2,
Г1.4-Г1.6).
В литературе можно найти утверж-
дения, что для молекул белков с мо-
лекулярной массой 2000 Да и менее в
большей степени приемлемы граничные
условия свободного скольжения, тогда
как для молекул белков с молекулярной
массой 5000 Да и больше более обос-
нованы условия полного прилипания.
Этот вопрос носит принципиальный ха-
рактер, поскольку связан с нижней гра-
ницей применения гидродинамических
Рис. I 1.1. Гидродинамическое представление условий «свободного скольжения» (<т) и «прили-
пания» (б): (о час то та вращения.
уравнений и будет обсужден подробно
в параграфе ГЗ.7.5).
Г1.4.2. Гидродинамические
эксперименты
Эксперименты в гидродинамике можно
разделить на четыре группы. В первую
группу входят эксперименты, в которых
измеряется стационарная скорость дви-
жения частиц под действием некоторой
силы. Если движущей силой является
градиент концентрации макромолекул,
то такое явление называется трансляци-
онной диффузией. Когда действующая
сила имеет гравитационную природу,
то такое явление называется седимента-
цией. Если на частицу действуют силы
электрической природы, то явление на-
зывается электрофорезом.
Вторая группа включает экспери-
менты, в которых вращение частиц осу-
ществляется под действием на них пары
сил. Если градиент скорости в раствори-
теле играет роль ориентирующей силы,
то такое явление известно под названи-
ем эффекта Максвелла или двулучепре-
ломления в гидродинамическом потоке.
Если ориентирующая сила имеет элект-
рическую природу, то явление называет-
ся эффектом Керра или двулучепрелом-
лением в электрическом поле.
Третья группа представлена экс-
периментами, измеряющими потерю
энергии, обусловленную трением моле-
кулы в растворе. Это явление называет-
ся вязкостью.
К четвертой группе мы относим
явления, в которых на частицу не
действуют никакие внешние силы и
их перемещение и вращение проис-
ходят только под влиянием теплового
воздействия, связанного с броуновским
движением. Поведение частиц в экс-
периментах по флуоресценции и ди-
намическому рассеянию света имеет
такую природу.
В .Hi iiiih I I I собраны гидродина-
мические методы, используемые в на-
стоящее время (первая колонка), а также
представлены параметры, которые могут
быть вычислены на их основе (третья ко-
лонка). Измеряемые величины представ-
лены во второй колонке. В первой колон-
ке в скобках приведены также главы, в
которых каждый метод описан в деталях.
Следует заметить, что несмотря на разно-
образие представленных методов, в ко-
нечном счете, только три параметра могут
быть определены из гидродинамических
опытов: коэффициенты поступательного
и вращательного трения, а также характе-
ристическая вязкость.
В ;,in. ।::;I.' I I I приведен также ме-
тод, который мы называем методом
гидродинамического моделирования.
В этом методе определяют фактиче-
ский коэффициент трения частицы,
наблюдая за движением ее макроско-
пической модели в растворителе с высо-
кой вязкостью, с тем чтобы обеспечить
измерения при маленьких числах Рей-
нолдса. Следует отметить, что в таких
экспериментах коэффициент трения
модели определяют прямым способом,
без каких-либо промежуточных уравне-
ний или вычислений. Краткое описание
метода и некоторые результаты приве-
дены в Главе Г2.
Г1.4.3. Гидродинамические
параметры
Гидродинамические методы, представ-
ленные в т ;ьб. 1111 к ГI. I, позволяют вычис-
лить коэффициенты поступательного и
вращательного трения, а также харак-
теристическую вязкость биологических
макромолекул. Эти параметры в свою
очередь зависят от вязкости растворите-
I, it ни и j.u I l. Современные гидродинамические методы, используемые
в структурной биологии
Экспериментальный метод Измеряемые величины Вычисляемые параметры
Трансляционная диффузия (Глава ГЗ) Коэффициент трансляционной диффузии Коэффициент поступательного трения
Скоростная седиментации (Глава Г4) Коэффициент седиментации Коэффициент поступательного трения
Электрофоретическая подвижность (Глава Г5) Электрофоретическая подвижность Коэффициент поступательного трения
Флуоресцентная корреляционная спектроскопия (Глава П 1) Время диффузии Коэффициент поступательного трения
Восстановление свечения красителя после выцветания (Глава ГЗ) Время диффузии Коэффициент поступательного трения
Прямые гидродинамические модельные эксперименты (Глава Г2) Прямое определение коэффициента поступательного трения
Ориентация молекул в электрическом поле (эф- фект Керра) и в гидродинамическом потоке (эффект Максвелла) (Главы Гб и Г7, соответственно) Угол ориентации и величина двойного лучепреломления Коэффициент вращательного трения
Деполяризованная флуоресценция (Глава Г8) Коэффициент деполяризации Коэффициент вращательного трения
Динамическое рассеяние света (Глава ПО) Корреляционная функция и флуктуации числа частиц Коэффициенты поступательного и/или вращательного трения
Вязкость (Глава Г9) Удельная вязкость Характеристическая вязкость
ля, а также от структуры частиц (размеры
и форма) и взаимодействия между ними.
Коэффициент поступательного трения
Поступательное трение лежит в основе
явлений диффузии, скоростной седи-
ментации и электрофоретической под-
вижности. В каждом явлении на части-
цу действует сила F, которая заставляет
ее ускоряться. Перемещение частицы
массой т, обусловленное этой силой,
описывается в механике вторым зако-
ном Ньютона:
F=mdu/dt, (Г1.8)
Рис. Г 1.5. Линии, изображающие поток во-
круг сферы. Сфера движется слева направо
с постоянной скоростью и в потоке вязкой
жидкости
Комментарии Г1./. Размерности
гидродинамических величин
В системе сгс размерность коэффици-
ента поступательного трения — г сек Раз-
мерность коэффициента вращательного
трения — сек1. Удельная вязкость — вели- I
чина безразмерная.
. Комментарий Г 1.8. Время,
необходимое для достижения
молекулой постоянной скорости
Вычисления на основе уравнения Г1.10
показывают, что время, которое необхо-
димо для достижения равновесия очень
мало. Так для белка из 300 аминокислот- ;
вых остатков с молярной массой около 1
33000 г моль1 величина константы посту-
пательного трения равна 5 х 10 "гсек1, а т
равна 5 х Ю 20 г. Конечная скорость для него .
достигается за время не более 10 12 сек (1
пикосек). Эти времена очень малы и близки
к временам тепловых вибраций молекул.
где т — масса частицы, и — скорость,
t — время.
В вязком растворе движение части-
цы тормозится растворителем. Моле-
кула будет ускоряться до тех пор, пока
приложенная сила не уравновесится си-
лой вязкого сопротивления среды. Сила
трения, F пропорциональна скоро-
сти частицы, и направлена в сторону,
противоположную приложенной силе
(рис. П.5). Коэффициент поступатель-
ного трения, /, определяется как конс-
танта пропорциональности в уравнении:
/• =-Ju. (Г1.9)
Знак минус означает направлен-
ность силы в сторону, противополож-
ную направлению скорости ( ком.меп ги-
ри й Г1.7).
Когда эти две силы становятся рав-
ными по величине, частица будет двига-
ется с постоянной скоростью и согласно
уравнению
w=F//, (Г1.10)
где F — значение силы, когда F = -F ия.
Уравнение Г1.10 соотносит эту кон-
станту с коэффициентом трения и сум-
марной величиной двух, действующих
в противоположном направлении, сил.
Это уравнение является основой для
экспериментального определения ко-
эффициентов поступательного трения
(см. i.ii'i.i. 11.1 ). Из уравнения Г 1.9 сле-
дует, что чем большим трением обла-
дает частица, тем большую силу требу-
ется приложить к частице, с тем чтобы
она двигалась с постоянной скоростью.
Если отношение силы к коэффициенту
трения F/f будет большим, то необхо-
димая устойчивая скорость будет очень
велика и недостижима в эксперименте.
Однако, если отношение F/f будет ма-
лым, то конечная скорость достигается
почти мгновенно после приложения
силы ( комментарий I I .8).
В своей классической работе Стокс
установил связь между коэффициентом
трения сферы и ее радиусом R() для двух
крайних случаев взаимодействия с моле-
кулами растворителя. В случае «полного
прилипания» уравнение имеет вид:
/ = 6лПо/?о. (Г1.11)
Тогда как это уравнение для случая «сво-
бодного скольжения» выглядит как:
f = 4тг По Ro. (Г1.12)
Как видно из этих уравнений изме-
нение граничных условий не меняет их
физическую сущность — пропорциональ-
ность коэффициента трения сферы ее ра-
диусу, а лишь меняет коэффициент.
Коэффициент вращательного трения
Вращательное трение лежит в основе
эффекта Керра, эффекта Максвелла
и деполяризованной флуоресценции.
По аналогии с поступательным дви-
жением мы можем определить коэф-
фициент вращательного трения. Если
постоянный вращающий момент F щ
приложен к частице, находящейся в
вязкой среде, то частица будет иметь
постоянную угловую скорость, со, через
короткий интервал времени (рис. Г 1.6).
Коэффициент вращательного трения, 0,
определяется как константа пропорцио-
нальности в уравнении
F =0со. (Г1.13)
пращ х '
Уравнение Г1.13 аналогично урав-
нению Г1.4. Уравнение ПЛЗ лежит в
основе определения коэффициентов
вращательного трения методами, пред-
ставленными в 1.Ю. nine Г:. I.
Согласно Стоксу для сферической
частицы в случае «полного прилипания»
связь между коэффициентом вращатель-
ного трения и ее объемом V имеет вид:
0 = 8 лт|0 V. (Г1.14)
Отметим, что для сферической час-
тицы в случае «свободного скольже-
Рис. I 1.6. Линии, изображающие поток во-
круг сферы. Сфера вращается вокруг оси, пер-
пендикулярной к плоскости страницы с посто-
янной угловой скоростью Сй
Рис. Г1. /. Линии потока вокруг сферы, дви-
жущейся в жидкости
ния» коэффициент вращательного тре-
ния равен нулю.
Характеристическая вязкость
При измерении вязкости определя-
ются потери локальной энергии на
трение, которые происходят, когда
большие частицы движутся в раство-
рителе, состоящем из маленьких мо-
лекул (рис. Г 1.7). Обозначим вязкость
растворителя в отсутствие частиц как
Гемоэритрин
а
Арабинозо -
связывающий
белок
б
Рис. П.8: (а) Гидродинамическим эквива-
лентом белка гемоэритрина является сфера;
(б) гидродинамическим эквивалентом белка,
связывающего арабинозу, является эллипсоид
вращения с отношением осей 2:1
г|0, а в их присутствии с определенной
концентрацией как гр Если раствор
содержит монодисперсную суспен-
зию сферических частиц, то уравне-
ние Эйнштейна для удельной вязкости
(П — т1о)/т1о’ в слУчае «полного прили-
пания», имеет вид:
(П-По)/П0 = 2,5 0., (Г1.15)
где Q — объемная доля сферических
частиц в растворе.
Для случая «свободного скольже-
ния» удельная вязкость также пропор-
циональна объемной доле сферических
частиц в растворе
(П - По)/По = 1 (П.16)
Важно отметить, что значение ра-
диуса частицы не входит в уравнения
Г1.15, Г1.16. Последнее ведет к важным
физическим следствиям, обсуждаемым
в параграфе Г9.5.2.
Г1.4.4. Гидродинамически
эквивалентные тела
Обычно уравнения Г1.11, Г1.14 и
Г1.15 используются при работе с от-
носительно большими биологиче-
скими макромолекулами (более, чем
несколько кДа, см. дискуссию в Главе
ГЗ). Они позволяют выразить измеря-
емые значения коэффициентов тре-
ния и характеристической вязкости
через радиус гидродинамически экви-
валентной сферы /?(), обычно называ-
емый радиусом Стокса. Понятие гид-
родинамически эквивалентных сферы
может быть распространено и на дру-
гие гидродинамически эквивалентные
формы (рис. П.8).
Необходимо ясно себе представ-
лять, что описание частиц на основе
гидродинамических эквивалентов не
учитывает их конкретную форму, а
только одинаковые гидродинамические
свойства. Несмотря на то, что такое при-
ближение является достаточно грубым,
однако оно во многих случаях оказыва-
ется очень близким к поведению частиц
в реальных экспериментах.
В случае жестких сфер только один
параметр (объем сферы или ее радиус)
необходим для полного описания их
гидродинамических свойств. Для опи-
сания эллипсоида вращения (форма
мяча в регби) необходимо определить
два параметра — объем и соотношение
осей. Для того, чтобы описать вращение
эллипсоида в трех плоскостях (осях),
требуются три параметра — его объем
и отношение осей в двух измерениях.
(Глава Г2).
Методы, представленные в габ.ni-
ne Г1 отличаются по их относительной
Рис. Г1.9. Сфера (а) и два эллипсоида вращения (б и в) с равными объемами. Вытянутый эл-
липсоид (б) имеет сигарообразную форму, образуемую при вращении эллипса вокруг его корот-
кой полуоси Ь. Сплюснутый эллипсоид (в) имеет дискообразную форму волчка, образуемую при
вращении эллипса вокруг его длинной полуоси а. Для каждого вида эллипсоида отношение осей
(р) определяется как отношение а/b — длинной и короткой полуосей. Ближний правый октант
каждого эллипсоида вырезан, чтобы продемонстрировать длинную (а) и короткую (Ь) оси
чувствительности к этим параметрам.
Так, все методы, базирующиеся на
поступательном трении, чувствитель-
ны к линейным размерам частицы в
первой степени, тогда как методы, ба-
зирующиеся на вращательном трении,
чувствительны к линейным размерам в
кубе, или, иными словами, к объему час-
тицы.
Г1.5. ВЫЧИСЛЕНИЕ
ФРИКЦИОННЫХ СВОЙСТВ
ЧАСТИЦ
Современная гидродинамика позволяет
вычислять коэффициенты трения био-
логических макромолекул различной
формы. Процедура вычисления зависит
от конкретной формы макромолекулы.
Г1.5.1. Частицы «правильной»
округлой формы
Многие биологические макромолеку-
лы, как, например, глобулярные бел-
ки или сферические вирусы, имеют
округлую форму. В этом случае они
хорошо аппроксимируются гидроди-
намически эквивалентной сферой или
гидродинамически эквивалентным
двухосным или трехосным эллипсо-
идом (рис. П.9). Гидродинамические
свойства таких частиц могут быть вы-
числены аналитически (Глава Г2).
Г1.5.2. Частицы «правильной»
граненой формы
Другие биологические макромолекулы
в первом приближении могут модели-
роваться как палочки или цилиндры.
Такими молекулами являются малень-
кие жесткие мономеры, олигомеры
ДНК, а-спиральные полипептиды, па-
лочкообразные белки и вирусы. Труд-
ность всех гидродинамических теорий,
Рис. I I. К). Частицы «ломаной» формы име-
ют резко очерченные края. К ним относятся —
куб (а) и круговой цилиндр (б). Их фрикци-
онные свойства могут быть вычислены только
приблизительно
a
б
Рис. Г1.1 1. Примеры частиц произвольной формы: тРНК (<•/) и иммуноглобулин (б). 11х фрик-
ционные свойства могут быть вычислены только приблизительно
в которых предпринимается попытка
описать фрикционные свойства таких
молекул, возникает из-за эффекта кон-
цов палочкообразной молекулы. Опи-
сание трения куба, который имеет фор-
Рнс. I 1.12. Структура молекулы лизоцима
с разрешением на атомном уровне. Фрикци-
онные свойства молекул с таким разрешением
могут быть предсказаны с точностью около не-
скольких процентов
му с четкими гранями и углами. — одна
из трудноразрешимых проблем гидро-
динамики (рис. Г1.10). По мере перехо-
да от куба к параллелепипеду эффект
концов становится все менее заметным.
Аналогичный эффект имеет место и для
кругового цилиндра.
Г1.5.3. Частицы произвольной
сложной формы
Детальное описание частиц произволь-
ной формы требует определения гораз-
до большего числа параметров, чем мы
можем получить из гидродинамичес-
ких измерений. Концепция гидродина-
мически эквивалентных тел в данном
случае работает плохо (рис. Г1.1 I).
В настоящее время разработано не-
сколько подходов для вычисления
фрикционных свойств таких частиц.
В первом подходе все пространство
частицы заполняется сферами одинако-
вого или разного диаметра, с тем чтобы
максимально правдоподобно описать
ее форму. Во втором подходе только ее
поверхность заполняется маленькими
сферами (метод бусинок) или малень-
кими панельными элементами (метод
чешуек). При определенных условиях
такие подходы позволяют вычислять
свойства трения частиц произвольной
формы с хорошей точностью (Глава Г2).
Г1.5.4. Частицы с известной
трехмерной структурой
Впечатляющий прогресс в расшиф-
ровке структур белков и нуклеиновых
кислот с высоким пространственным
разрешением методами рентгенострук-
турного анализа (Глава ЕЗ) и ЯМР
(Глава КЗ) стимулировал развитие но-
вых подходов для вычисления гидроди-
намических параметров таких молекул.
На рисунке 1'1,12 в качестве примера
приведена структура лизоцима в ато-
марном разрешении. Для таких струк-
тур предложены различные алгоритмы
вычисления их гидродинамических
свойств, а именно: атомы в С -положе-
нии заменяются сферами соответству-
ющего диаметра, поверхностные атомы
моделируются маленькими сферами
или треугольными панельными элемен-
тами (Глава Г2). Сравнение расчетов с
экспериментом показало их хорошее
совпадение: с точностью около 1 % для
поступательного трения (Глава ГЗ) и
около 2-3% для характеристической
вязкости (секция Г2.4). Специальные
подходы предложены для расчетов мо-
лекул ДНК (Глава Г2).
Г1.6. Перечень ключевых идей
• Безразмерный параметр, число Рейнолдса, характеризует соотношение сил
инерции и вязкости, действующих на частицу в вязкой среде.
° Движение биологической частицы с молекулярными размерами 10-10000 А
описывается гидродинамикой при «низких» числах Рейнолдса, когда силы вяз-
кости преобладают над силами инерции.
• Коэффициенты в уравнениях, описывающих зависимость гидродинамических
параметров от вязкости растворителя и размеров частиц, зависят от граничных
условий, два из которых называются «свободным скольжением» и «полным
прилипанием».
* В методах, основанных на поступательном трении, измеряется стационарная
скорость движения частицы под действием сил разной природы: гравитацион-
ной, в случае скоростного ультрацентрифугирования, градиента осмотического
давления, в случае поступательной диффузии и электрического поля, в случае
электрофореза.
* В эффекте Максвелла (двойное лучепреломление в потоке) или эффекте Керра
(электрическое двойное лучепреломление) измеряют скорость вращения час-
тицы иод действием пары сил.
• В экспериментах по вязкости определяют локальные потери энергии на тре-
ние, обусловленные движением частиц в вязкой среде.
В экспериментах по флуоресценции и динамическому рассеянию света измеря-
ют движение и вращение частиц только под действием собственно броуновско-
го движения (без воздействия внешних сил).
• Результаты всех гидродинамических методов могут быть описаны в терминах
только трех гидродинамических параметров — коэффициентов поступательно-
го и вращательного трения, а также характеристической вязкости.
• Гидродинамические свойства частиц «правильной» округлой формы (сферы,
двух- и трехосные эллипсоиды) могут быть вычислены аналитически.
• Гидродинамические свойства частиц «правильной» граненой формы (кубы,
круговые цилиндры) могут быть рассчитаны только приблизительно из-за кон-
цевых эффектов.
• Гидродинамические свойства частиц произвольной формы могут быть вычис-
лены методами бусинок или чешуек с хорошей точностью.
• Гидродинамические свойства глобулярных белков с известной атомной струк-
турой могут быть вычислены различными способами с точностью около 1-3%,
при условии корректного учета толщины гидратационной оболочки (около 1 А)
для всех белков.
Литература для дальнейшего чтения
Гидродинамика при низких числах Рейнолдса
Purcell Е. М. Life at low Reynolds number. Am. J. Phys., 1977. 45, 3-11.
Гидратация
Kuntz I. D. and Kauzmann W. Hydration of proteins and polypeptides. Adv. Prot.
Chem., 1974. 28, 239-345.
Finny J. L. Overview lecture. Hydration processes in biological and macromolecular
systems. Faraday Discuss. Chem. Soc., 1996.103, 1-395.
Wutrich K., Billeter M., Guntert P., Luginbuhl P., Reik R. and Wider G. NMR studies
of the hydration of biological macromolecules. Faraday Discuss. Chem. Soc., 1996.103,
245-253.
De la Torre G. Hydration from hydrodynamics. General consideration and applications
to bead modelling to globular proteins. Biophys. Chem., 2001. 93, 159-170.
Zhou H.-X. A unified picture of protein hydration: prediction of hydrodynamic
properties from known structures. Biophys. Chem., 2001. 93, 171-179.
Perkins S.J. X-ray and neutron scattering analyses of hydration shells: a molecular
interpretation based on sequence predictions and modelling fits. Biophys. Chem., 2001.
93, 129-139.
Engelsen S. В., Monteiro C., Herve de Penhoat C. and Perez S. The dilute aqueous
solvation of carbohydrates as inferred from molecular dynamics simulations and NMR
spectroscopy. Biophys. Chem., 2001.93, 103-127.
Экспериментальное определение фрикционных свойств частиц
HappelJ. and BrennerH. Low Reynolds number hydrodynamics. 2nd edn. Noordhoff
Int., Groningen, Leyden. 1973.
Harding S. E. The intrinsic viscosity of biological macromolecules. Progress in
measurement, interpretation and application to structures in dilute solution. Prog.
Biophys. Mol. Biol., 1998. 68, 207-262.
Hu C.-M. and Zwanzig R. Rotational friction coefficients for spheroids with the
slipping boundary conditions. J. Chem. Phys., 1974. 60, 4354-4357.
Расчет фрикционных свойств частиц
Brune D. and Kim, S. Predicting protein diffusion coefficients. Proc. Natl. Acad. Sci.
USA, 1994. 90, 3835-3839.
Zhou H-X. Calculation of translational friction and intrinsic viscosity. II. Application
to globular proteins. Biophys J., 1995. 69, 2298-2303.
De la Torre G., Huertas M. L. and Carrasco B. Calculation of hydrodynamic properties
of globular proteins from their atomic-level structure. Biophys J., 2000. 78, 719-730.
Allison S. A. Boundary element modelling of biomolecular transport. Biophys. Chem.,
2001.93,197-213.
ГЛАВА Г2
ФУНДАМЕНТАЛЬНАЯ ТЕОРИЯ
Г2.1. ИСТОРИЧЕСКИЙ ОБЗОР
! S 15
Г. Стокс (G. Stokes) показал, что
поступательное трение сферы пропор-
ционально ее радиусу и вязкости ок-
ружающего ее растворителя. В 1856 г.
он нашел, что ее вращательное трение
при малых угловых скоростях может
быть охарактеризован единственным
параметром, который пропорциона-
лен кубу линейных размеров, т. е. ее
объему.
1<S!)3
Д. Эдвардс (D. Edwardes) рассчи-
тал два коэффициента трения эллипсо-
ида вращения: один для вращения вок-
руг малой оси, а другой для вращения
вокруг большой оси. В 1906 А. Эйн-
штейн (A. Einstein) показал, что вра-
щение сферы в приближении Стокса
можно охарактеризовать одной кон-
стантой, которая имеет размерность
времени. В 1928 г. Р. Ганс (R. Gans)
использовал коэффициенты трения
для эллипсоида вращения, рассчитан-
ные Эдвардсом, с тем чтобы вычислить
отношение релаксационных времен эл-
липсоида ко времени релаксации сфе-
ры равного объема.
1936
Ф. Франсуа Перрен (F. Froncois
Perrin) получил уравнения, которые со-
держали три коэффициента вращатель-
ного трения и три времени релаксации
как функции осей трехосного эллипсои-
да. Эти уравнения не могли быть выра-
жены в значениях элементарных функ-
ций. В 1960 г. Л. Д. Фавро (L. D. Favro)
показал, что коэффициент диффузии,
относящийся к вращательному движе-
нию частицы как целого, включает пять
времен релаксации; в случае, когда два
коэффициента диффузии равны, чис-
ло времен релаксации уменьшается до
трех. В 1977 г. Е. Смолл и И. Айзенберг
(Е. Small and I. Isenberg) решили уравне-
ния Перрена для вращательной диффу-
зии обобщенного эллипсоида, используя
методы численного интегрирования. Ко-
эффициенты вращательной диффузии,
вращательные времена релаксации и пять
экспоненциальных вкладов в флуорес-
центную анизотропию были табулирова-
ны как функции отношения осей эллип-
соида. В 1981 г. П. Хагерман и Б. Зимм
(Р. J. Hagerman and В. Н. Zimm) разра-
ботали подход для анализа вращательной
диффузии червеобразных цепей методом
Монте Карло. Была установлена количе-
ственная взаимосвязь между продольным
(наибольшим) вращательным временем
релаксации линейного полимера и его
эластичностью (персистентной длиной).
! 1.4*6
А. Эйнштейн впервые исследовал
вязкость суспензии жестких сфериче-
ских частиц, которые по своим размерам
были намного больше размеров молекул
растворителя. Он показал, что удельная
вязкость раствора пропорциональна объ-
емной доле раствора, занимаемой части-
цами, и не зависит от диаметра сферы.
В 1945-1951 it. В. Кун и Г. Кун,
Дж. Кирквуд и П. Ауэр (W. Kun and
Н. Kun, J. J. Kirkwood and P. L. Auer)
завершили теорию характеристической
вязкости для длинных палочек и эллип-
соидов.
В 1967 г. В. А. Блумфилд, В. Даль-
тон и К. Ван Холде (V. A. Bloomfield,
W. Dalton and К. Е. Van Holde) разра-
ботали метод «оболочек» или «бусинок»,
который с теоретической точки зрения
должен был стать идеальным решением
для строгих вычислений вязкости макро-
молекул. Однако его применение на прак-
тике было сильно затруднено тем, что
требовалось использование очень боль-
шого числа бусинок для построения точ-
ных моделей структур и, соответственно,
очень длительного времени вычислений.
В 1970-1977 гг. несколько незави-
симых групп К. Тсуда, Дж. МакКам-
мон и Дж. Дейтч, X. Накаджима и
И. Вада, Гарсиа де ла Торре и В. Блум-
филд (К. Tsuda, J. A. McCammon and
J. М. Deutch, Н. Nakajima and Y. Wada,
J. Garcia de la Torre and V. A. Bloomfield)
развили общую теорию вычисления
характеристической вязкости жестких
макромолекул произвольной формы.
Во всех этих исследованиях форма мак-
ромолекулы задавалась расположением
в ее пространстве относительно малого
количества сферических бусинок.
В 1981 г. С Хардинг, М. Демпье и
А. Рове (S. Harding, М. Dampier and
A. Rove) решили задачу вычисления
вязкости для трехосного эллипсоида.
< 927
С. Озеен (С. Oseen) рассчитал гид-
родинамический тензор трения, опи-
сывающий гидродинамическое взаи-
модействие между фиксированными в
пространстве источниками трения.
В 1932 г. Дж. Бюргерс (J. Burgers)
применил тензор О.зеена к объектам раз-
личной геометрии. В 1953 г. Дж. Кирквуд
(J. Kirkwood) дат гидродинамическое
описание цепных макромолекул, состо-
ящих из ансамбля равновеликих сфер,
и предложил простую формулу, базиру-
ющуюся на оригинальном тензоре Озеена,
для вычисления усредненно-ориентиро-
ванного поступательного трения частиц.
В 1963 г. Дж. Хирст (J. Е. Hearst)
применил формализм Кирквуда для опи-
сания вращательного трения гибких и
полужестких макромолекул, что открыло
путь к исследованию ДНК различными
гидродинамическими методами.
В 1967 г. В. Блумфилд (V. Bloom-
field) предложил способ вычисления
гидродинамических свойств частиц про-
извольной формы, получивший название
метода оболочек. При этом поверхность
частицы описывалась большим количе-
ством маленьких сфер или бусинок.
В конце 1960-х гг. Дж. Ротне и
С. Прагер (J. Rotne and S. Prager) и, не-
зависимо, X. Иамакава (Н. Yamakawa)
предложили новый улучшенный под-
ход для изучения гидродинамических
взаимодействий.
1932
X. Штаудингер (Н. Staudinger) пред-
ложил использовать характеристиче-
скую вязкость для определения молеку-
ляриых масс полимеров. Он обнаружил,
что зависимость характеристической
вязкости [г|] от молекулярной массы М
для гомологичной серии полимеров мо-
жет быть выражена простой! формулой
типа [г|] = Ма. Как позже было выясне-
но Г. Марком, Р. Хаувинком, В. Ку-
ном и X. Куном (Н. Mark, R. Houwink,
W. Kuhn and H. Kuhn) константа а не-
посредственно связана с молекулярной
конформацией макромолекул в растворе.
1934
Ф. Перрен (Е Perrin) аналитиче-
ски решил проблему гидродинамичес-
кого движения твердого эллипсоида
вращения. Он получил уравнения,
которые давали три коэффициента
поступательного трения как функции
размеров трехосного эллипсоида. Эти
уравнения включали набор эллипти-
ческих интегралов и не могли быть
выражены в виде элементарных фун-
кций.
В 1977 г. Е. Смолл и И. Айзенберг
(Е. Small and I. Isenberg) решили урав-
нение Перрена методом численного
интегрирования. В начале 1980-х гг.
С. Хардинг и А. Рове (S. Harding and
A. Rowe) ввели в практику трехосный
гидродинамический эллипсоид как бо-
лее реалистическую модель по срав-
нению с двухосным эллипсоидом для
описания структуры биологических
макромолекул.
1936
А. Петерлин (A. Peterlin) ввел по-
нятие коэффициента вращательной
диффузии для описания интенсивности
броуновского движения. Он вычислил
функцию распределения для ориентации
главных осей эллипсоида при заданном
значении градиента скорости, принимая
во внимание энергию диссипации, обус-
ловленную только вращением частиц в
гидродинамическом поле.
В 1940 г. Р. Симха (R. Simha) рас-
ширил терию Петерлина, принимая во
внимание полную энергию диссипации
при броуновском движении. Он полу-
чил уравнение для вязкости раствора
эллипсоидов вращения для предель-
ного случая, когда частицы имеют слу-
чайную ориентацию, и следовательно
вращаются с постоянной угловой ско-
ростью. Позже результаты, полученные
Симха, были строго воспроизведены
Н. Сайто (N. Saito) для различных гра-
ничных условий.
1941
Дж. Онсли (J. L Oncley) сделал пер-
вую попытку решить проблему вычисле-
ния степени гидратации для белков. Он
показал, что всегда существует выбор
между вытянутым и сплюснутым эллип-
соидом, каждый из которых имеет свое
соотношение осей, даже тогда, когда пред-
полагается разумная степень гидратации.
В 1953 г. X. Шерага и Л. Мандель-
керн (Н. A. Sheraga and L. Mandelkem),
пытаясь преодолеть неоднозначность
при решении проблемы объем-отноше-
ние осей, предложили объединить коэф-
фициенты трения и характеристической
вязкости. Однако эта первая объем-неза-
висимая функция оказалась мало чувс-
твительной к отношению осей и могла
быть использована только для того, что-
бы отличить вытянутый эллипсоид от
сплюснутого при очень большой степе-
ни асимметрии последних. В дальней-
шем была предложена целая серия ком-
бинаций различных гидродинамических
параметров: коэффициентов регрессии
седиментации и характеристической
вязкости (М. Валес и К. Ван Хольде,
1954) (М. Wales and К. Е. Van Holde,
1954), коэффициента поступательного
трения и среднего гармоничного времени
вращательной релаксации (П. Сквайер,
1970) (Р. Squire), регрессии седимен-
тации и характеристической вязкости
(А. Рове (A. Rowe, 1977), коэффициен-
та трения и операционалистически оп-
ределяемого молекулярного ко-объема
(П. Джефри с сотр., 1977) (Р. Jeffrey
with colleagues); гармонического значе-
ния времени вращательной релаксации
и характеристической вязкости, харак-
теристической вязкости и молекуляр-
ного ко-объема (С. Хардинг, 1980-81);
(S. Е. Harding). Некоторые из этих
комбинаций привели к функциям, с по-
мощью которых можно было различить
вытянутые и сплюснутые эллипсоиды
даже при относительно небольших сте-
пенях вытянутости.
1960
С. Броерсма (S. Broersma) был
первым, кто исследовал фрикционные
свойства круглого цилиндра. Из полу-
ченной им формулы следовало, что по-
перечный (наименьший) коэффициент
вращательной диффузии обратно про-
порционален кубу длины. Позже однако
было обнаружено, что его формула кор-
ректна только для длинного цилиндра.
В 1965 г. X. Бреннер (Н. Brenner)
внес существенный вклад в гидроди-
намику жестких частиц. Он получил
аналитические решения характерис-
тической вязкости для эллипсоидов,
длинных цилиндров и гантелей. Он
также показал, что поступательное и
вращательное движение жестких мак-
ромолекул, погруженных в вязкую
жидкость, сопряжено, если частица
имеет сложную форму.
1975
Г. Ионгрен и А. Акривос (G. Youn-
gren and A. Acrivos) разработали ме-
тод граничных элементов и применили
его для описания поверхности частиц.
В 1992-1995 г. Д. Бруне, С. Ким и
Н.-Х. Чжоу (D. Brune, S. Kim and
Н.-Х. Zhou) использовали эту тех-
нику для вычисления фрикционных
свойств белков, а в 1996 г. С. Аллисон
(S. Allison) — для моделирования сво-
бодного электрофореза в растворе для
коротких фрагментов ДНК.
1978
Д. Теллер (D. Teller) впервые опуб-
ликовал метод вычисления коэффи-
циентов трения белков по их атомным
координатам. Р. Венейбл и П. Пастор
(R. Venable and R. Pastor) вычисли-
ли коэффициенты поступательного
трения белков, используя детальную
картину распределения аминокислот в
белках, а Гарсиа де ла Торре (J. Garsia
de la Torre) произвел аналогичные вы-
числения для нуклеиновых кислот.
В 1995-1999 гг. ряд авторов предложи-
ли улучшенные алгоритмы для конс-
труирования гидродинамических мо-
делей биологических макромолекул на
основе их атомных координат. Специ-
альные подходы были разработаны для
моделирования фрикционных свойств
коротких фрагментов и замкнутых
кольцевых ДНК.
1982
С. Хардинг и А. Рове (S. Harding
and A. Rowe) предложили новый метод
моделирования биологических макро-
молекул, основанный на использовании
трехосного эллипсоида. Параметры этого
эллипсоида находили из графического
пересечения линий трех гидродинами-
ческих функций, включающих значения
характеристической вязкости, седимен-
тации и временного спада электричес-
кого двойного лучепреломления. Годом
позже эти же авторы предложили аль-
тернативный метод, е использованием
характеристической вязкости, седимен-
тации и гармонического значения време-
ни релаксации. В 1997 г. они же и Гарсиа
де ла Торре ввели новую концепцию для
описания биологических макромолекул
в растворе с использованием универсаль-
ных функций формы. В настоящее вре-
мя для пользователей доступны различ-
ные компьютерные программы ELLIPS
(Хардинг с corp.) и SOLPRO (Гарсиа де
ла Торре с сотр.) для оценки трехмерных
размеров биологических макромолекул,
исключающие вычисления значений гид-
ратации и объемов.
19‘):;
Дж. Дуглас и Дж. Хаббард (J. Doug-
las and J. Hubbard) обнаружили взаимо-
связь между гидродинамикой и электро-
статикой, что открыло новые возможнос-
ти для гидродинамических вычислений.
На этой основе в 1995 г. Н.-Х. Чжоу
(Н.-Х. Zhou) вычислил коэффициенты
трения для различных белков. Он также
предложил алгоритм для вычисления ха-
рактеристической вязкости частиц произ-
вольной формы и глобулярных белков.
200!) и по настоящее время
Гидродинамические свойства био-
логических макромолекул могут быть
вычислены с хорошей точностью при
моделировании их поверхности боль-
шим числом маленьких бусинок или ма-
леньких треугольных элементов (чешу-
ек). Предложены автоматизированные
методы такого моделирования на основе
кристаллографических данных для нук-
леиновых кислот и белков. Трехмерные
модели биологических макромолекул
без предварительных вычислений значе-
ний их гидратации и объемов могут быть
рассчитаны из комбинации различных
гидродинамических параметров.
5 2.Z. ! iov /Ts'j.Al
Г2.2.1. Частицы правильной формы
Сфера
В простейшем случае сила трения в жид-
кости возникает из-за притяжения между
ее молекулами. Движение твердого объ-
екта в жидкости сопровождается переме-
щением молекул раствори теля (рис. Г2.1).
Молекулы растворителя, находящиеся
вблизи движущейся частицы, испытыва-
ют большее возмущение, чем молекулы,
находящиеся вдали от нее. На больших
расстояниях это возмущение падает до
нуля. Для вычисления силы трения мы
должны вычислить силу, необходимую
для сохранения скорости возмущенных
молекул растворителя. Эта сила связа-
нно. Г2.1. Линии потока вокруг сферы ра-
диусом Rtl, движущейся слева направо через
несжимаемую вязкую жидкость с постоянной
скоростью иг Число на линях потока в каждой!
точке при 0 =- -90" выражено в единицах
Даже на самых дальних линиях потока, пока-
занных на рисунке, соответствующих расстоя-
нию, равному диаметру сферы, поток дви-
жется на 30'% быстрее, чем движется сфера.
Беспорядочное движение молекул раствори-
теля, обусловленное броуновским движением,
не принимается во внимание
на со свойством жидкости, называемым
вязкостью. При движении сферических
частиц нам необходимо найти уравнение,
которое связывает коэффициент посту-
пательного трения частицы / и вязкость
жидкости г|0. Получение этого уравне-
ния связано с большими трудностями,
поскольку необходимо точно вычислить
как движение сферы деформирует гради-
ент скорости. В соответствии с законом
Стокса поступательное трение сферы
пропорционально ее радиусу, Ra, и вяз-
кости растворителя, г|0, через который
движется частица:
/() = 6лП(Дг (Г2.1)
Следует отметить, что уравнение
Стокса справедливо при трех допу-
щениях. Первое, коэффициент (6л) в
уравнении (Г2.1) зависит от граничных
условий и [5 данном случае соответству-
ет условию полного прилипания (см.
параграф Г1.4.1). Второе, с математи-
ческой точки зрения подход Стокса
корректен, когда растворитель можно
считать неструктурированной средой.
Очевидно, что последнее верно, если
размеры сфер гораздо больше разме-
ров молекул растворителя. Считается
общепринятым, что движение белков с
молекулярной массой, превышающей
5000 Да (так называемые «большие»
молекулы), можно рассматривать как
движение сквозь бесструктурную среду.
Верность такого утверждения — какие
частицы в этом смысле можно считать
«большими», а какие «малыми» — об-
суждается в Главе ГЗ. Третье, уравне-
ние Г2.1 было получено с допущением,
что взаимодействие между самими час-
тицами отсутствует. Па практике это
условие реализуется экстраполяцией
к сильно разбавленным растворам.
Поскольку силы трения напрямую
связаны с площадью поверхности изу-
чаемой частицы, а сфера имеет из всех
геометрических объектов наименьшую
поверхность при равном объеме, то
можно прийти к выводу, что трение для
сферических частиц будет наименьшим
по сравнению с таковым для несфери-
ческих частиц такого же объема.
Эллипсоид вращения
Влияние асимметрии частиц на их гид-
родинамические свойства может быть
строго учтено, если они моделируют-
ся эллипсоидами вращения (сплюс-
нутыми или вытянутыми). Из общих
соображений следует, что сила трения
асимметричных частиц зависит от их
ориентации по отношению к направле-
нию движения потока. Так, вытянутые
частицы имеют наименьшее трение, ког-
да они ориентированы по направлению
движения потока, и наибольшее, когда
они ориентированы перпендикулярно
потоку. В усредненном случае имеем:
/0 = б7П10Д0Д(р). (Г2.2)
Теоретически вычисленные значе-
ния функции Перрена F (р) для вытяну-
тых и сплюснутых эллипсоидов враще-
ния представлены на рисунке Г2.2. Три
важных заключения можно сделать
исходя из вида этой функции. Первое,
при увеличении аксиального отношения
численные значения функции Перрена
увеличиваются относительно медленно.
Например, функция Перрена изменяется
всего на 4%, если форма частицы меня-
ется от сферической к эллипсоидальной
с аксиальным отношением 2. В то же са-
мое время для такого эллипсоида функ-
ция, описывающая вращательное трение
и характеристическую вязкость, изменя-
ется приблизительно на 12% (см. ниже).
Второе, при том же аксиальном отноше-
нии, коэффициент поступательного тре-
5
Рис. I 2.2. Функция Перрена F(p) для сплюс-
нутых и вытянутых эллипсоидов вращения.
N. В. В настоящее время нет необходимости
использования таблиц для функции Перре-
на. Необходимые гидродинамические функ-
ции для частиц разной формы читатель мо-
жет найти из базы компьютерных программ
(Harding et al., 1977)
ния для вытянутого эллипсоида всегда
больше, чем для сплюснутого. Третье,
для асимметричной частицы коэффици-
ент поступательного трения зависит от
двух параметров: ее объема, выраженно-
го через радиус эквивалентной сферы, и
отношения осей. Следовательно, вычис-
ленный коэффициент поступательного
трения частицы может быть всегда ин-
терпретирован в терминах сферической
частицы соответствующего объема или
в терминах асимметричной частицы
меньшего объема ( kommcii iapin'i Г2.1).
Круговой цилиндр
Разнообразные биологических макро-
молекулы, включающие как короткие
фрагменты ДНК, так и палочкообраз-
ные вирусы, подобные вирусу табачной
мозаики, существуют в виде длинных
вытянутых молекул, имеющих посто-
янную толщину. Для описания таких
макромолекул наиболее подходящей
моделью является круговой цилиндр с
Коммепlapnii Г2.1. Трехосный
гидродинамический эллипсоид
В начале 1980-х гг. Хардинг и Рове пред-
ложили использовать трехосные гидроди-
намические эллипсоиды как модели струк-
туры биологических макромолекул. Эти
модели гораздо более правдоподобно опи-
। сывают глобулярные белки, чем модели на
основе двухосных эллипсоидов. Более под-
робное описание поступательного трения
, для трехосных эллипсоидов можно найти
; в литературе (Harding and Rowe, 1982).
плоскими концами. Для цилиндра дли-
ной L, радиусом г и отношением L/r = р
усредненный по всем ориентациям ко-
эффициент трения fравен:
/=6ш]0£/(21пр + у), (Г2.3)
где у является функцией р, зависящей
от краевых эффектов.
Для относительно короткого ци-
линдра:
у(р) = 1,65 - 4 (1 /1пр — 0,4З)2 -
(Г2.4)
- 8 (1/1пр — 0,30)2.
Уравнение (Г2.4) справедливо толь-
ко в том случае, когда р^°°. Исследо-
вания последних лет показали, что пре-
дельное значение для у равно:
у = 0,386, когда р-^°°, (Г2.5)
а в интервале 2 < р < 20 хорошим прибли-
жением для у(р) является выражение:
у(р) = 0,312 +0,561/р + 0,100/р2. (Г2.6)
Г2.2.2. Жесткие частицы
произвольной формы
Рисунок Г2.3 показывает структурные
модели различных биологических макро-
молекул, полученные методом рентгено-
структурного анализа. Из него видно, что
с помощью сфер и эллипсоидов вращения
можно описать форму многих биологи-
ческих макромолекул, а с помощью моде-
ли трехосного эллипсоида можно хорошо
передать реальную форму большинства
глобулярных белков. Однако многие био-
логические молекулы, например, тРНК,
иммуноглобулины, фосфоглицератмута-
за, форма которых далека от сферической
или эллипсоидальной (см. рис. 1’2.3), тре-
буют более сложных моделей для их опи-
сания и, следовательно, для вычисления
их гидродинамических свойств.
В течение последних трех десятиле-
тий был предложен ряд методов для опи-
сания гидродинамического поведения
частиц сложной формы. В методе «буси-
нок», который берет свое начало из пио-
нерской работы Кирквуда и Райзмана,
частица заполняется сферами (бусинка-
ми) равного или неравного диаметра.
В модельном подходе «оболочек»,
который впервые был предложен в
работах Блумфилда, поверхность час-
тицы заполняется малыми равнове-
ликими сферами. Формально модель-
ные подходы «бусинок» и «оболочек»
тесно связаны фундаментальной гид-
родинамической единицей — сферой.
В модельном подходе «граничных эле-
ментов», разработанном Йонгреном и
Акривосом, поверхность частиц моде-
лируется набором панельных элемен-
тов. В этом случае фундаментальной
гидродинамической единицей является
пластинка, обычно треугольной формы
(чешуйка). Рассмотрим каждый из трех
упомянутых методов по отдельности.
Моделирование целой частицы
набором сфер (метод «бусинок»)
В общих чертах модель «бусинок»
представляет собой частицу, объем ко-
торой заполнен сферическими элемен-
тами (бусинками), каждая из которых
обладает своим, индивидуальным, в
соответствии с законом Стокса, коэф-
фициентом трения. Первая ключевая
проблема при конструировании такой
модели состоит в том, как учитывать
гидродинамическое взаимодействие
между различными сферическими эле-
ментами. Для этого имеются два под-
хода: строгий, тензорный метод, и при-
ближенный метод двойных сумм.
Вторая ключевая проблема состо-
ит в построении модели таким образом,
чтобы ее размер и форма были наиболее
близки к таковым у реальной частицы.
При таком построении всегда возникает
конфликтная проблема: получение вы-
сокого разрешения требует увеличения
числа сфер, описывающих модель, что
в свою очередь сильно увеличивает рас-
четное компьютерное время (коммента-
рии 1'2.2). Ниже мы кратко опишем воз-
можности для решения этих проблем.
Начнем с первой. Решая ее, Дж. Кир-
квуд предложил простую модель для
описания гидродинамических свойств
гибких молекул: он моделировал линей-
ные и клубкообразные полимеры набо-
ром одинаковых сфер, соединенных в
цепочку. Все бусинки в модели имели
идентичные радиусы и, следовательно,
одинаковые фрикционные свойства.
Гидродинамическое взаимодействие
между бусинками учитывалось по ме-
тоду Озеена-Бюргерса.
Рисунок Г2.4 схематически по-
казывает гидродинамическое взаи-
модействие между двумя сферами
макромолекулы, фиксированными
в пространстве. В соответствии с под-
ходом Озеена-Бюргерса скорость рас-
творителя ir в положении сферы i есть
сумма невозмущенной скорости и и
вклада возмущенной скорости, кото-
рый возникает при гидродинамиче-
белок
Рис. 1'2.3. Формы различных биологических молекул, полученные методом рентгеноструктурного
ция каждой молекулы выбрана таким образом, чтобы подчеркнуть своеобразие ее формы (Goodsell
анализа (увеличение 4 х 10G). 11ри таком увеличении каждый атом имеет диаметр 1-2 мм. Ориента-
and Olson, 1993)
коммоймпии. Г2.2. Классический i
и строгий расчет модели «бусинок»
При строгом расчете моделей «бусинок»
должно быть использовано тензорное урав-
нение, поскольку скорость пертурбаций не
всегда параллельна силам, их вызывающим.
В этом случае силы трения получаются в
результате решения системы ЗЛГ линейных
уравнений. Соответственно, необходимое 1
для вычисления компьютерное время про-
порционально У’, и быстро увеличивается
с ростом У. Это противоречие возникает
из-за необходимости использовать боль-
шое количество «бусинок», для получения
структуры объекта с высоким разрешением.
В классической теории Кирквуда Райзма-
на приближения, сделанные при трактовке
гидродинамических взаимодействий, при- i
водят к простым уравнениям, в которых
гидродинамические свойства вычисляются
из двойной суммы обратных расстояний.
Это требует операций порядка У2, так что
компьютерное время для вычислений при
большом значении N намного меньше, чем
при строгом расчете. :
ском взаимодействии с другими сфе-
рами. Возмущение сферы i зависят от
сил трения F, производимых всеми
другими сферами j (в нашем конкрет-
ном случае это вторая сфера) через
гидродинамическое взаимодействие
тензоров:
и( = и-Т.хЕ. (Г2.7)
Тензор гидродинамического взаи-
модействия Т по Озесну-Бюргерсу
Т = (1/8лг|п г.) (I + г..г/г.2), (Г2.8)
где I является единичным тензором,
а г. — вектор расстояния между взаимо-
действующими сферами i и j.
Выражение коэффициента поступа-
тельного трения частиц, состоящих из
У-сфер, каждая из которых обладает ко-
эффициентом трения ф имеет простой
вид ( коммен lapilli Г2.3):
(Г2.9)
6тгг)Л“£
Рис. Г2.1. Схематическое изображение гид-
родинамического взаимодействия между дву-
мя фиксированными в пространстве сфера-
ми, каждая из которых является источником
трения. Две сферы движутся со скоростями
и и и. г^есть расстояние между сферами. При
движении через жидкость каждая сфера изме-
няет скорости потока вблизи себя (Cantor and
Schimmel, 1980)
Уравнение Г2.9 может быть просто
интерпретировано с физической точ-
ки зрения. При отсутствии гидроди-
намического взаимодействия между
сферами коэффициент трения части-
цы является суммой N коэффициен-
Комментарий Г2.3.
Взаимодействие между бусинками
по Озеену-Бюргерсу
, Основными характерными чертами гид-
: родинамического взаимодействия между
бусинками по методу Озеена-Бюргерса
являются: (1) каждая бусинка являет-
! ся источником трения; (2) только парное
1 взаимодействие между этими источниками
принимается во внимание. В результате в ,
уравнении Г2.9 всегда содержится двойная '
| сумма расстояний между частицами.
тов трения сфер, Уф в то время как
в присутствии гидродинамического
взаимодействия между сферами коэф-
фициент трения частицы меньше, чем
Уф Это происходит потому, что каж-
дая сфера, в среднем, является объек-
том взаимодействия с растворителем,
и, следовательно, имеет меньшее тре-
ние. В этом случае гидродинамическое
взаимодействие учитывается дважды,
что и отражено в формуле Г2.9 в виде
двойной суммы.
Простота уравнения Г2.9 очень
привлекательна. Она позволяет произ-
водить необходимые вычисления для
малого числа сферических субъединиц
вручную. Следует однако помнить, что
простота уравнения Г2.9 очень иллю-
зорна ( С /. I ! . >I j. 1.1 ,',. 11, ! ).
В дальнейшем тензор гидродинами-
ческого взаимодействия Озеепа-Бюр-
герса был модифицирован, с целью ус-
транения двух основных недостатков
подхода Кирквуда: 1) источники трения
считались точечными, несмотря на то
что сферы имели конечные размеры,
2) гидродинамические взаимодействия
учитывались как поправка первого по-
рядка для невозмущенных сил. В моди-
фицированном варианте в случае сфер
равного размера с радиусом Ra тензор
гидродинамического взаимодействия Т.
имеет вид:
т,
1
(Г2.10)
Тензор (уравнение Г2.10) называ-
ется по имени их создателей тензором
Ротне-Прагера-Ямакавы, или модифи-
цированным тензором Озеена-Бюргер-
са. Имеется также вариант уравнения
Г2.10 для случая сфер разных радиусов,
ст. и ст,
' j
v ЗтЩф
i(CT-+gi!)p/ у/
I3
Заметим, что уравнение Г2.11 дей-
ствительно, если расстояние между
частицами больше, чем сумма радиусов
г > ст. + ст.,
и 1 j
Сейчас мы подведем итоги обсужде-
ния проблемы учета гидродинамическо-
го взаимодействия между различными
сферическими элементами при пост-
роении модели, концентрируя внима-
ние только на поступательном трении.
Вращательное трение и вязкость будут
обсуждаться позже.
Когда частица произвольной фор-
мы движется со скоростью и через
жидкость, сила трения, производимая
потоком, зависит от и, и выражается
через тензор поступательного трения
ф как:
F = Eiu. (Г2.12)
Трансляционная динамика трения
частиц, описываемая при помощи тензо-
ра поступательной! диффузии £ф равна:
D, = kT/Et, (Г2.13)
где k — это константа Больцмана, а Т —
температура по Кельвину.
Уравнение Г2.13 является строгим
в отсутствие эффекта сопряжения пос-
тупательного движения и вращения
Л.: I Е и D( являются
симметричными тензорами, компонен-
ты которых .зависят от ориентации час-
тицы ПО отношению К ОСЯМ ГЮММ.'!
>!|||2 )). След матрицы D является
однако инвариантным, так что коэффи-
циент поступательной диффузии может
быть определен как:
D = l/3tr(D,). (Г2.14)
Поэтому ориентационно-усреднен-
ное сопротивление поступательному
движению может быть выражено через
скалярную величину — коэффициент
поступательного трения, /, который
связан с коэффициентом диффузии
уравнением Эйнштейна.
Как мы указывали выше, кроме
проблемы учета гидродинамического
взаимодействия при моделировании
имеется вторая ключевая проблема,
Комментарий Г2.4. Частицы
произвольной формы
Для частиц произвольной формы гидро-
динамическое сопротивление выражается
6 х 6 тензором трения Подобно этому броу-
новская диффузия выражается 6x6 диф-
фузионной матрицей D. которая связана с Е
зависимостью Эйнштейна D kT=.'. И Е, и
D могут быть разделены на 3 х 3 блока, каж-
дый из которых соответствует поступатель-
ному движению, вращению и сопряжению
движение—вращение (Navarro et al., 1995).
Комментарий Г2.5. Тензор
поступательной диффузии
Отметим, что тензор поступательной диф-
фузии строго поддается интерпретации толь-
ко тогда, когда соотнесен со специфической
точкой, так называемым центром диффузии.
Последний совпадает с центром симметрии
для центросимметричных частиц. Во всех
других случаях теоретическое обоснование
гидродинамических вычислений может быть
найдено в специальной литературе (см., на-
пример, Navarro et al., 1995).
связанная с построением самой мо-
дели из сферических элементов. Оче-
видно, что при ее решении мы должны
одновременно стремиться к решению
двух задач. Первая связана с объемом
частицы, который должен быть макси-
мально близким к реальному. Вторая
связана с формой частицы, которая
должна быть передана с максималь-
ным правдоподобием. Схема такого
моделирования для гипотетической
двумерной частицы представлена на
рисунке Г2..5. Очевидно, что модель,
показанная на рисунке 1'2.5 б, более
точно передает реальные объем и фор-
му частицы, чем модель, показанная на
рисунке 12..5 а, однако ее расчет пот-
ребует намного большего компьютер-
ного времени. В заключение заметим,
что при моделировании удлиненных
структур, таких как эллипсоид или па-
лочка, важными критериями является
совпадение длины и объема у частицы
и модели.
Моделирование поверхности частицы
набором малых равных сфер (метод
«оболочек»)
У компактной твердой частицы гидро-
динамическое трение фактически про-
исходит на ее поверхности. Это спра-
ведливо для многих биологических
макромолекул и, в первую очередь, для
глобулярных белков, поскольку внут-
ренняя часть белка недоступна раство-
рителю. Даже если в молекуле есть норы,
пропускающие растворитель, жидкость
захватывается, движется вместе е пей,
становясь частью гидродинамической
частицы.
В методе «оболочек» макромолеку-
ла окружена оболочкой, состоящей из
У маленьких сфер радиусом /‘(рис. Г2..5
в и /). При этой процедуре большое чис-
а б в г
Рис. I 2..>. Схематическое представление двумерной частицы: а — модель бусинок, составленная из
неравных по размеру сфер; б модель бусинок, составленная из равновеликих сфер; в — идеальная
оболочечная модель; г — приближенная оболочечная модель (Carrasco and Garcia de la Torre, 1999)
ло сфер или бусинок расположены на
поверхности и плотно упакованы. Пос-
тупательное трение ансамбля бусинок
вычисляется при использовании моди-
фицированного тензора Озеен-Бюр-
герса. Процедура расчета повторяется
несколько раз с использованием все
меныпих размеров бусинок. В конечном
итоге, трение вычисляется экстраполя-
цией к предельно большому количеству
бусинок бесконечно малого размера.
Процедура моделирования относи-
тельно проста для тел, имеющих ось вра-
щения, подобную таковой эллипсоидов
или цилиндров, для которых бусинки
могут быть расположены параллельно
замкнутой кривой, которая определя-
ется плоскостью, перпендикулярной к
оси симметрии. Таким образом, методом
наложения бусинок на кольца с различ-
ными радиусами мы можем построить
гладкую оболочечную модель эллипсои-
да. Пример такого построения представ-
лен на рисунке Г2.6.
Рис. I 2.6. Оболочечная модель для эллипсои-
да вращения с отношением осей р = 2 (Carrasco
and Garcia de la Torre, 1999)
Моделирование поверхности частиц
набором малых панельных элементов
(метод «панелей» или «чешуек»)
В методе «панелей» поверхность мо-
лекулы описывается как N взаимосвя-
занных малых трехгранных панелей.
a 6
I’не. I 2.7: a — сфера диаметром в 20 А моделируется 256 треугольными пластинками: б — за-
крытый цилиндр, моделирующий ДНК с 20 нарами оснований, представлен 96 треугольными
пластинками (Allison, 2001)
На рисунке Г2.7 а и б, соответственно,
представлены примеры сфер и закры-
тых с концов цилиндров. Как и в пре-
дыдущем случае, процедура расчета
повторяется несколько раз с использо-
ванием все меньших размеров панелей.
В конечном итоге трение вычисляется
экстраполяцией к предельно большому
количеству панелей бесконечно мало-
Рис. 1 2.8. Пример дискретизации вируса та-
бачной мозаики. Поверхность вируса с прибли-
зительно цилиндрической формой поделена
на п приз.м, число которых в процессе расчета
увеличивалось. Уже при п = 36 результаты ком-
пьютерных вычислений коэффициента диффу-
зии для вируса табачной мозаики находились в
хорошем соответствии с его эксперименталь-
ными значениями (Brune and Kim, 1993)
го размера. Метод «панельных элемен-
тов» одинаково хорошо работает как
в условиях полного прилипания, так
и свободного скольжения (параграф
Г1.4.1).
Для белков, поверхности которых
нерегулярны, процедура дискретиза-
ции нетак проста и требует применения
специальных программ для расчета ко-
эффициентов трения исходя из коор-
динат атомов (см. ниже). Для больших
макромолекул не обязательно знать
точное положение каждого атома, с тем
чтобы дискретизировать поверхность.
Например, вирус табачной мозаики яв-
ляется достаточно большой частицей
(3000 А х 180А) и по этой причине его
мелкие нерегулярности поверхности
не влияют на его гидродинамические
свойства. Поэтому этот вирус может
моделироваться цилиндром, поделен-
ным на относительно небольшое коли-
чество панелей (рис. 1'2.8).
Г2.2.3. Коэффициент
поступательного трения
и электростатическая емкость
Традиционно коэффициент трения час-
тиц с произвольной формой вычисли-
ется методами, описанными в предыду-
щей секции. Эти методы требуют особой
тщательности и большого времени для
получения адекватных результатов.
Электростатические вычисления бо-
лее простые и имеют длинную историю
и богатые традиции.
Электростатическая емкость мно-
гочисленных геометрических форм,
включая камень преткновения гидроди-
намики — куб, хорошо известна ( -
i:i|4:i’, I Взаимосвязь между гид-
родинамикой и электростатикой была
установлена в 1993 г. и сразу же были
предложены простые и точные методы
вычисления поступательного гидродина-
мического трения. Коэффициент посту-
пательного тренияJ частицы соотносится
с ее электростатической емкостью с как:
/=6пцос. (Г2.15)
Это отношение точно выполняется
для сферы и двух- и трехосного эллип-
соидов ( \• I. ;, I.: i': I. ' ~
Уравнение (Г2.15) открывает прос-
той способ вычисления гидродинами-
ческого трения частицы произвольной
формы через электростатическую ем-
кость с. Важно подчеркнуть, что для тел
произвольной формы обычно намного
проще вычислить его емкость с, чем
компоненты тензора трения. Точность
таких вычислений весьма велика, она
составляет 1 % для случаев, когда ана-
литическое решение известно.
Г2.2.4. Частицы с известной
структурой
Моделирование белков
по координатам атомов
Доступность баз данных, содержащих
атомные координаты большого числа
Электростатическая емкость
Электрический заряд q генерирует на
теле электростатический потенциал U, про-
порциональный q:
U = q/c. (А)
Константа пропорциональности в этом
уравнении обратно пропорциональна с, где
с есть электростатическая емкость тела, с
имеет размерность длины и равна заряду,
требуемому для поддержания единицы
электростатического потенциала тела по
отношению к бесконечности, или иначе,
с является эквивалентом электростатиче-
ской емкости частицы в единицах, равных
радиусу сферы.
' : г 2 , О сходстве
уравнений, описывающих
свойства сферы в кинетических,
электростатических и
гидродинамических явлениях
Интересным является тот факт, что ма-
тематические уравнения, описывающие
свойства сфер в электростатических и гид-
родинамических явлениях сходны. В самом
деле уравнение, описывающее скорость час-
тиц в диффузионно-контролируемых реак-
циях с константой диффузии D, движущих-
ся по направлению к абсорбирующей сфере
(Глава ГЗ), приведено ниже:
k=inDRu. (Л)
В электростатике емкость с электропро-
водящей сферы равна:
с = Яп. (В)
Коэффициент поступательного трения
/ сферы в растворителе с вязкостью г|0,
при граничных условиях «полного прили-
пания» в соответствии с законом Стокса
(уравнение Г2.1) равен:
/ = блц,,/?,,. (В)
Как видно, все три величины пропорци-
ональны радиусу сферы.
белков, позволяет конструировать гид-
родинамические модели на основе этих
координат. Моделирование каждого
атома белков приводит к получению
слишком большого количества цент-
ров трения и трудновыполнимым вы-
числениям. С целью уменьшения коли-
чества этих центров было предложено
несколько подходов: моделирование
белков набором бусинок (известный
как метод кубиков) и моделирование
поверхности белков набором панель-
ных элементов.
Моделирование белков сферами
(бусинками)
Программа «от А к Б» — «от атомных
координат к белку» разработана для
конструирования бусинками сплош-
ных моделей с различным пространс-
твенным разрешением из атомных
Рис. 12.9. Пример конструирования белка
альдолазы на базе программы «от А к Б» - «от
атомных координат к белку» с разным про-
странственным разрешением (5-30 A) (Byron,
1997)
координат. Принцип работы програм-
мы «от Л к Б» состоит в следующем.
Пространство, занимаемое молекулой,
делится на маленькие кубы. В каждом
из них содержится бусинка, размер и
положение которой определяется по-
ложением всех атомов в этом кубе. Та-
кой подход позволяет точно регулиро-
вать размер куба, который и определяет
пространственное разрешение модели.
Реальная форма макромолекулы дик-
тует необходимый уровень разрешения.
Для более округлых глобулярных час-
тиц используются модели с большими
размерами бусинок. На рисунке Г2.9
приведен пример моделирования для
альдолазы. Модели, построенные по
атомным координатам на основе про-
граммы «от Л к В», имеют регулируемое
разрешение от 5 до 30 А. Как видно из
этого рисунка, разрешение от 10 до 20 А
является приемлемым для построения
модели, имеющей достаточное сходс-
тво с оригинальной кристаллической
структурой. При меньшем разрешении
важные структурные детали могут быть
утрачены.
Моделирование поверхности белков
панельными элементами — метод
«чешуек»
При моделировании поверхности мо-
лекул методом «чешуек» также ис-
пользуются атомные координаты.
Рассмотрим такое моделирование на
примере молекулы лизоцима. Повер-
хность этой молекулы при высоком
пространственном разрешении демон-
стрирует большое число малых впадин
и выступов (рис. 1’2.10 а). На уровне
континуумной гидродинамики (раз-
меры объекта намного больше разме-
ров молекул растворителя) наличие
небольших участков нерегулярности
на поверхности белка слабо сказыва-
ется на конечном результате, однако
значительно увеличивает время вы-
числений. Для получения поверхности
белка, которую можно использовать
для гидродинамических вычислений
методом панельных элементов, мелкие
нерегулярные участки сглаживаются
обкаткой пробной сферы радиусом 3 А
(см. рис. Г2.10 б). Затем поверхность
белка делится на граничные элементы
(см. трехгранные поверхности на рис.
1'2.10 и). Коэффициент диффузии мо-
жет быть впоследствии вычислен при
использовании техники граничных
элементов, онncaiiпой ранее.
Моделирование структуры ДНК
Большинство динамических свойств
длинных молекул ДНК определяется
ее контурной длиной и гибкостью, в
то время как толщина и, еще в боль-
шой степени, детали ее поверхност-
ной структуры не являются важными.
Напротив, для динамических свойств
коротких фрагментов ДНК структура
се поверхности становится определя-
ющей. Для моделирования гидроди-
намических свойств ДНК использу-
ются два разных подхода. В первом из
них акцент делается на ДНК молекуле
как на спирали, параметры которой
могут быть заданы геометрией распо-
ложения бусинок, тогда как во втором
координаты атомов молекулы ДНК
используются для локализации тех же
бусинок.
Моделирование ДНК как спиральной
структуры методом «бусинок»
При моделировании ДНК методом «бу-
синок» ДНК моделируется как двойная
спираль, в которой каждый нуклео-
тид представлен отдельной бусинкой
.ipi'i; 12.--). Радиус СПИрЭЛИ
рассматривается как подгоночный па-
раметр. На рисунке Г2.1 1 показаны две
двойные спирали, в которых на один
оборот приходится 15 бусинок, ради-
ус спирали и ее шаг в обеих моделях
одинаковы (Д = 10 А, Р = 34 А). Первая
спираль симметрична (ф =180°), с дву-
мя равными бороздками, в то время как
другая (ф = 120°) имеет две бороздки
разной толщины. При ф = 120° модель
описывает В-форму ДНК.
Рис. I 2.10. Поверхность молекул лизоцима: a - при атомном разрешении: б после обкатки
поверхности пробным шаром радиусом 3 А: « — после дискретизации поверхности на составные
треугольные элементы (Brune and Kim, 1993)
I’iic. I 2.1 1. .Аппроксимация двойной спирали
моделью «бусинок», в которой каждый нук-
леотид представлен одной бусинкой. Модель
слева приближенно описывает В-форму ДНК
(Garcia de la Torre et al., 1994)
Моделирование ДНК методом
«бусинок» по атомным координатам
При моделировании ДНК методом «бу-
синок» по атомным координатам бусинки
располагаются в геометрических центрах
групп атомов (от 10 до 30 атомов), с тем
чтобы обеспечить сходство с реальной
формой поверхности молекулы. Схема-
тический вид фрагмента ДНК из 20 нук-
леотидов, описывающий ее бусинковую
модель представлен на рисунке Г2.12 с/.
Наилучшее согласие с эксперименталь-
ными данными обнаружено при радиусе
бусинок 7,3 А. В двойной бусинковой мо-
дели (рис. Г2.12 б), в которой нуклеотиды
разделены на две группы атомов (одна
содержит основания, другая — сахара и
фосфатные остатки) наилучшее согласие
с экспериментальными данными дости-
гается при радиусе бусинок 5,6 А. Ис-
пользование второй сферы целесообраз-
но только при моделировании коротких
фрагментов ДНК.
Моделирование ДНК по атомным
координатам методом «пластинок»
Пример разбиения гидродинамиче-
ской поверхности фрагмента ДНК, со-
' ' < . ' Геометрия ДНК
Геометрия одиночной спирали может
быть описана следующим набором пара-
метров: .1, радиус спирали; Р, ее шаг; //, чис-
ло ви тков (не обязательно целочисленное).
Если ось спирали направлена вдоль осп z,
тогда параметрические уравнения спирали
будут иметь вид:
Л'—Л cos (г + ф).
Д sin (г + ф). (1'2.19)
Z=Pt/2n.
где значение фазового угла ф может быть
произвольным, а I есть параметр, который
находи тся в пределах от I °- 0 до t -2mir Две
сплетенные нити двойной спирали могут
бы ть описаны с помощью итого же уравне-
ния, если задать разные значения (разового
угла для каждой нити. В спирали ДНК бу-
синки радиусом /?(| помещаются вдоль ли-
нии контура спирали и располагаются так.
чтобы бусинки соприкасались. Два подхода
при моделировании структуры ДНК опи-
саны в тексте.
стоящего из 20 пар оснований, на 352
треугольные пластинки приведен на
рисунке Г2.13 г/. Применение этого под-
хода не ограничивается только такими
относительно малыми фрагментами.
На рисунке 1'2.1.3 б показана торзионно
напряженная суперскрученная коль-
цевая ДНК, состоящая из 375 пар ос-
нований с 4 зацеплениями. Молекула
сплетается в закольцованный кабель с
радиусом 10 А, покрытый 640 треуголь-
ными пластинками. Контурная длина
такой ДНК составляет 1275 А.
Г2.2.5. Жесткие частицы
с сегментальной подвижностью
При гидродинамическом описании
клубкообразных молекул необходимо
принимать во внимание как раство-
ритель внутри клубка, движущий-
а б
Рис. I 2.1 2: а — одиночная бусинковая модель фрагмента ДНК длиной 20 нуклеотидов. Атомы
представлены шариками, а связи между ними — палочками; б — эти же атомы встроены в двой-
ную бусинковую модель (Banachowicz et al., 2000)
ся вместе с ним, так и растворитель,
свободно протекающий через клубок.
Кроме того, необходимо также учи-
тывать статистическое распределение
элементов трения внутри клубка. Вы-
числения гидродинамических свойств
таких молекул, базирующиеся на
теории Кирквуда-Райземапа, мето-
дах Монте-Карло и молекулярной
динамики, представляют специаль-
ную ветвь гидродинамики и здесь не
рассматриваются. Однако в биологии
существуют промежуточные структу-
ры, когда молекула может рассматри-
ваться как жесткая структура с опре-
деленной подвижностью ее сегментов.
К ним относятся иммуноглобулины, и
некоторые глобулярные белки,напри-
мер калмодулин. Структурная под-
вижность таких молекул напрямую
связана с их функцией, поэтому изу-
чение их гидродинамических свойств
представляет большой интерес.
Наиболее простой и широко изу-
ченной моделью, имитирующей сегмен-
тальную подвижность, является жест-
кая палочка, состоящая из двух ветвей,
соединенных шарниром под углом а
(рис. 1'2.11). Вычисления, основанные
на простом усреднении всех возможных
конформаций такой модели, показыва-
ют, что коэффициент поступательного
трения такой палочки изменяется все-
го на 3%, по сравнению с таковым для
жесткой палочки такого же размера и
мало зависит от места положения шар-
нира. Поскольку экспериментальная
ошибка при использовании этих мето-
дов в лучшем случае равна 1-2 %, то это
означает, что экспериментальное изуче-
а
Рис. Г2.13: а пластинчатая (чешуйчатая) модель фрагмента ДНК из 20 пар оснований ДНК
(pd (A) 21) pd (Т) ,,(|) в виде 352 плоских треугольных пластинок: б — чешуйчатая модель кольце-
вой ДНК, состоящая из 375 пар оснований с 4 зацеплениями. Модель состоит из 640 взаимосвя-
занных чешуек (Allison, 2001)
Рис. 12.14. Палочка, сломанная под углом а,
моделируется набором бусинок
ступателыюй диффузии изогнутой па-
лочки, вычисленный из динамических
экспериментов, например, динамиче-
ского рассеяния свеча, должен зависеть
от времени. Это означает, что автокор-
реляционная функция, описывающая
временную зависимость флуктуаций
рассеянного света от ансамбля таких
частиц, должна состоять, ио крайней
мере, их двух экспонент (Глава ПО).
Подобные временные эффектчя обнару-
живаются и в явлениях электрического
двойного лучепреломления. Читатель
может найти обсуждение эксперимен-
тальных исследований с применением
этих методов в Главе Гб.
Модель червеобразной цени
(рис. Г2.15, см. также рис. Гб. 1 1), в ко-
торой гибкость распределена ио всей
длине цепи, представляет особый инте-
рес с точки зрения изучения гидродина-
мики, поскольку она хороню описывает
поступательное н вращательное трение
молекул ДНК. Сравнение этих моделей
с моделями изогнутых палочек показы-
вает, что их равновесные усредненные
конформации очень близки, ио их ди-
намические свойства, выраженные че-
рез спектр времен релаксации, заметно
отличаются. Динамические свойства
молекул ДНК будут обсуждаться в па-
раграфе Г6.7.1.
Рис. 12.15. Плавно изгибающаяся полужест-
кая цепь может быть уподоблена червяку. От-
сюда название — червеобразная цепь
ние равновесной подвижности сегмен-
тов у частиц с такой структурой метода-
ми седиментации или поступательной
диффузии вряд ли имеет смысл. Одна-
ко гибкость влияет гораздо сильнее на
динамические свойства частицы. Рас-
четы показывают, что коэффициент но-
Г2.2.6. Экспериментальные методы
исследования коэффициента
поступательного трения
В i:i'7 ни|. I 2.1 приведены экспери-
ментальные методы, используемые в
настоящее время для определения ко-
нфе}) и ш ченгов п осту 11 ател ы iог<) трен ня,
/, биологических макромолекул (пер-
вая колонка), их измеренные величины
(вторая колонка), и области измеряе-
мых значений (третья колонка). Четвер-
Liojiuua Г2.1. Современные методы, используемые для определения
коэффициента поступательного трения, f, биологических макромолекул
Экспериментальный метод Измеряемое значение Область измеряемых значений Область применения
Расширя ющаяся граница (Глава ГЗ) Коэффициент постунательной диффузии D 5 х 10 5 - 5 х Ю 12 (см2сек ') Макромолекулы различной формы и размеров
Скоростная седиментация (Глава Г4) Коэффициент седиментации (в единицах Сведберга) 0,5-200000 (10 сек) Макромолекулы различной формы и размеров
Электрофоретиче- ская подвижность (Глава 1’5) Электрофоретиче- ская/подвижность р 1 х Ю 1 - 5 х Ю-’ (cm2V ’сек') В основном молекулы ДНК
Флуоресцентная корреляционная спектроскопия (Глава ГН) Коэффициент поступательной диффузии D 5 х Ю ’ - 5 х Ю-12 (см2-сек ) Флуоресцентно меченые макромо- лекулы различной формы и размеров
Восстановление свечения красителя после фотовыцве- тания (Глава ГЗ) Коэффициент поступательной диффузии D 5 х Ю 5 - 5 х Ю-'2 (см2-сек ') Флуоресцентно меченые макромо- лекулы различной формы и размеров
Метод ядерного магнитного резонанса (Глава КЗ) Коэффициент поступательной диффузии D 5 х Ю’5 - 5 х Ю 7 (см2-сек_|) Белки, ДНК
Гидродинамическое моделирование (Глав Г2) П ря мое о и редел сч и ie коэффициента поступательного трения/ Макромолекулы ква- зисферической формы
| Комментарий 1'2,9. Прямое определение коэффициентов поступательного
| трения гидродинамическим моделированием
I
Стандартный аппарат состон г из стеклянного цилиндра (с внутренним диаметром прибли-
зительно 10 см и высотой 100 см), погруженного в термостат. Два круглых маркировочных зна-
ка приблизительно на расстоянии 7-10 см от каждого конца цилиндра показывают расстояние,
которое должна пройти частица за расчетное время. Жесткие молекулы конструируются из
шаров различного материала. Диаметр шаров колеблется от 0,25 см до 1 см. Частица из многих
I сфер конструируется склеиванием подобранных шаров. Стационарные скорости для несфери-
ческих частиц (цилиндры, димеры и тетрамеры молекул) определяются в двух ориентациях
в жидкостях различной вязкости с числом Рейнолдса в интервале значении 0,0001-0,01.
Для каждой мультисферной частицы интересующее нас значение трения будет равно отно-
шению скорости одной сферы к скорости многосферной частицы. На отношение не должны
, влиять условия эксперимен та, особенно краевые эффекты и размеры частицы. Такой простой
। эксперимен т на макроскопической модели позволяет определить свойства поступательного
| трения микроскопической частицы произвольной формы. Теоретически полученные значе-
ния совпадают с экспериментальными при минимальной ошибке (0,1 %) во всех случаях, ког-
да известен аналитический результат.
тая колонка включает ооласть размеров
и формы .макромолекул, в которых при-
менение каждого метода наиболее це-
лесообразно. Главы, где каждый метод
описывается в деталях и обсуждается
область его применения, даны в скобках
в первой колонке.
Особое место в i;u'>. пни - I 2.1 занима-
ет гидродинамическое моделирование,
в котором поступательное трение жес-
тких микроскопических частиц опре-
деляется наблюдением скоростей осаж-
дения их макроскопических моделей в
очень вязких растворителях при низких
числах Рейнолдса (Глава П). Описание
типичного эксперимента для ансамблей
жестких сфер дано в ю'Х'мел .,..41; i
Г2.3, ВРАЩАТЕЛЬНОЙ
ТРЕНИЕ
Г2.3.1. Вращательное движение
в одном измерении
Броуновское движение включает в себя
не только поступательное движение мо-
лекул, но также и их вращательное дви-
жение. Классическое утверждение, со-
Рис. Г2.16. Одномерное вращение. Все палоч-
ки лежат в ху плоскости. Вращение происхо-
дит вокруг z-оси, которая перпендикулярна
плоскости ху. Вращательное движение палоч-
ки может быть описано одним параметром,
углом 6
держащееся во всех учеониках, гласит:
«Броуновское движение частиц само но
себе нельзя наблюдать, за исключением
частиц настолько больших, что они ста-
новятся видимыми в оптический мик-
роскоп, однако о его присутствии мож-
но судить по физическим свойствам
раствора». Сегодня его можно считать
устаревшим, поскольку впечатляющий!
прогресс в течение последних 10 лет
позволяет визуализовать единичные
молекулы в растворе и на поверхности
(см. Главу Д4).
Мы начинаем этот параграф с об-
суждения вращательной диффузии в
одном измерении, поскольку это позво-
лит провести нам аналогию с явлением
поступательной диффузии.
Вращательное движение частицы
может быть описано как движение в од-
ном измерении, если движение каждой
точки частицы есть простое вращение
вокруг одной оси, которая проходит че-
рез центр тяжести частицы (рис. Г2.16).
Движение такого тина может быть опи-
сано одним углом 0 между соответству-
ющими осями частицы (в нашем случае
это линия, проходящая через длинную
ось палочки) с любой другой подхо-
дящей осью в пространстве (в пашем
случае это горизонтальная линия). Из-
менение 0 во времени может быть вы-
ражено в терминах угловой скорости
w = dQ/dt.
Теперь введем переменную величи-
ну р(0), таким образом, что число час тиц
в 1 см3 раствора, обладающих ориента-
цией между углами 0 and 0 + dQ будет
равно р(0) dQ. Физический смысл этой
величины очень прост. В равновесном
состоянии все значения угла 0 будут
равновероятны и величина р(0) будет
постоянной. В неравновесном состоя-
нии под влиянием вращающего момента
некоторые ориентации становятся пред-
почтительными и значение р(0) стано-
вится зависимым отО. Если вращающий
момент, производящий ориентацию мо-
лекулы, удалить, то система постепенно
теряет преимущественную ориентацию
и, в конце концов, р(0) снова становит-
ся постоянной. Этот процесс называется
в ран цггел ы I ой д иффуз ней.
Трактовка одномерной вращатель-
ной диффузии подобна трактовке одно-
мерной поступательной диффузии (Гла-
ва ГЗ). Если мы определим J (0) dl как
суммарное число молекул в 1 см3, которое
за время dt проходит через угол ориента-
ции 0 в направлении положительного 0,
то феноменологический закон, аналогич-
ный первому закону Фика (уравнение
Г3.7), може т быть записан в виде:
J(O) = -0[^p(0)/j0](. (Г2.16)
В этом уравнении 0 называется вра-
щательным коэффициентом диффузии.
Второй закон Фика может быть опи-
сан аналогично уравнению (Г3.9):
т/Р(0)М = 0с/2р(0)/с/О2. (Г2.17)
Продолжая аналогию с поступа-
тельным движением, мы можем опре-
делить коэффициент вращательного
трения ф Вращательный момент Fв щ,
действующий на любую частицу в ху
плоскости (см. рис. 1'2.1 (>) в направле-
нии положительного угла 0, приведет к
ее угловому ускорению. В вязкой среде
этому ускорению противостоит трение
между частицей и молекулами раство-
рителя. Сила, обусловленная этим тре-
нием, F |||п|, пропорциональна угловой
скорости со. Константа пропорциональ-
ности определяется как коэффициент
вращательного трения ф
F =Ссо. (Г2.18)
В равновесном состоянии эти две
противоположные но направлению
силы равны по величине, ускорение при
этом становится равным нулю, и макро-
молекулы вращаются с постоянной уг-
ловой скоростью, равной:
со = F/ф (Г2.19)
Это уравнение эквивалентно урав-
нению Г1.10. Оно является основой для
определения коэффициента вращатель-
ного трения во многих эксперименталь-
ных методах, представленных в ia6.ni-
m I (>. I. Связь между коэффициентами
вращательного трения и коэффициен-
том вращательной диффузии подобна
таковой в уравнении (Г3.24):
0 = kT/ф (Г2.20)
Г2.3.2. Вращательное движение
в трех измерениях
Вращение молекулы в пространстве
должно в общем случае состоять не
из одного, а трех независимых видов
вращения, рассмотренных в предыду-
щем разделе. Кроме того, эти три вида
не могут быть описаны с помощью
обычной прямоугольной системы ко-
ординат, а требуют специальной сис-
темы координат, в качестве которой
выбираются углы Эйлера. Необходи-
мое рассмотрение сделано в большин-
стве учебников по механике и здесь не
описывается. Скажем, что для опреде-
ления ориентации молекулы в общем
случае требуются три параметра и что
вращательное трепне для трехосного
эллипсоида описывается тремя коэф-
фициентами вращательного трения
относительно каждой из его принци-
пиальных осей. Экспериментально
определяемые коэффициенты враща-
тельной диффузии и коэффициенты
вращательного трения даются следу-
ющими соотношениями:
0 = (0, + ©, + ©.,)/3, (Г2.21а)
или
0 = кт (1/;, + 1/Q, +1/У/3.(Г2.21б)
Для двухосного эллипсоида враще-
ния два параметра являются идентич-
ными
(0, = 03) и 0 = (0, + 202)/3,(Г2.22в)
или
0 = kT(l/;, + 2/Q/3. (Г2.226)
Г2.3.3. Вращательное движение
и время релаксации
Часто описание вращательного трения
удобнее рассматривать при помощи
представления о времени релаксации,
которое мы сейчас определим. Предпо-
ложим, что у нас есть набор макромоле-
кул в растворе и некоторое выделенное
направление, жестко связанное с моле-
кулой. Поскольку молекулы распреде-
лены в растворе хаотически, то все воз-
можные направления равновероятны
(рис. Г2.17 а). Допустим, что за какое-то
короткое время мы сможем сориенти-
ровать макромолекулы в заданном на-
правлении, используя, например, силу
электрического поля (рис. Г2.17 б).
После выключения поля и. следова-
тельно, исчезновения действия ориен-
тирующей силы через короткое время
наступит частичное нарушение ориен-
тации как показано па рисунке Г2.17 в.
Для того, чтобы охарактеризовать ста-
дии бив мы введем угол, 0, который бу-
дет характеризовать связь между исход-
ной ориентацией молекулы в положении
б и новым направлением в положении в.
Среднее значение косинуса этого угла
<cos0> может служить мерой ориента-
ции молекулы; <cos0> равен единице,
когда все молекулы полностью ориенти-
рованы и стремится к нулю, когда ориен-
тация беспорядочная (рис. Г2.17 /). Вре-
мя, необходимое для перехода системы
от состояния б к состоянию г называется
временем релаксации системы. Итак,
время релаксации, т, которое определя-
ется уравнением
<cos0>t/<cos0> (|=е ’ (Г2.23)
является временем, необходимым для
того, чтобы значение <cos0> уменьши-
лось до 1/е от его начальной величины.
а б в г
Рис. 1 2.17. Различные состояния макромолекул в растворе: а все молекулы расположены
случайным образом; б - все молекулы ориентированы вертикально; в — некоторые молекулы
частично разориентированы; г - полная (случайная) разориентания. Примечание, интервал вре-
мени между состояниями бив — короткий, временной интервал между в и г — длинный по отно-
шению к вращательным свойствам макромолекул
Как указано в главах, посвященных
различным экспериментальным мето-
дам, таким как электрическое двойное
лучепреломление (Глава Гб), деполя-
ризованная флуоресценция (Глава Г8)
наблюдаемые эффекты всегда описыва-
ются м ул ьт и э кс 11 о 11 е и ци ал ы । ы м и фун-
кциями, содержащими до пяти времен
релаксации < о< х о;. 11;i р: i; i ! 2.111 >. Одна-
ко выделение пяти времен релаксации
и соответствующих им пяти амплитуд
из экспериментально измеренного вре-
мени спада невозможно. Причина это-
го лежит в том, что с математической
точки зрения разложение такого рода
относится к некорректно поставлен-
ным или плохо обусловленным задачам
(Глава Г10). Упрощение этой проблемы
приводит к двум временам: к «среднему
времени релаксации», которое опреде-
ляется как взвешенное арифметическое
среднее из пяти времен релаксации и
к «начальному времени релаксации»,
которое определяется как взвешенное
гармоническое среднее всех релаксаци-
онных времен (другие времена релакса-
ции обсуждаются в параграфе Г6.4.2).
Г2.3.4. Жесткие частицы
регулярной формы
Вращение твердого объекта в вязкой
жидкости сопровождается также враще-
нием молекул растворителя относитель-
но друг друга. Молекулы растворителя,
находящиеся вблизи вращающейся
частицы, оказываются возмущенными
в наибольшей степени. Это возмущение
исчезает при удалении молекул раство-
рителя от частицы. На рисунке Г2.18 а
линии потока обозначены прерывис-
тыми линиями вокруг сферы радиусом
Ro, вращающейся с постоянной угловой!
скоростью со. Линии потока в данном
случае предс тавляют собой окружности
Комментарий Г2.10. Форма тела
и времена релаксации
Вращение трехосного эллипсоида мо-
жет быть описано тремя временами ре-
лаксации, в то время как вращение так
называемой «полной жесткой частицы
произвольной формы» — пятью време-
нами релаксации. Из этих пяти времен
релаксации три очень близки и в экспери-
менте проявляются как одно. Поэтому в
большинстве случаев может наблюдаться
только три времени. Поскольку к тому же
два из них взаимозависимы, на практике
наблюдаются только два времени релакса-
ции (Small and Isenberg, 1977).
(рис. Г2.18 о). У поверхности сферы мо-
лекулы растворителя движутся вместе
со сферой, в соответствии с граничными
условиями полного прилипания. На ри-
сунке Г2.18 а показаны также линии по-
тока (непрерывные линии) вокруг сфе-
ры, когда она движется в потоке слева
направо в плоскости страницы. Видно,
что возмущение линий потока при пос-
тупательном движении уменьшается
относительно более медленно, чем воз-
мущение при вращательном движении.
Сфера
Напоминаем (параграф Г1.4.3), что вра-
щательный коэффициент трения С, сфе-
ры радиусом 7?0 в граничных условиях
полного прилипания равен:
;0=8rni()/?()3. (Г2.24)
или
;о = 6ЛоУ. (Г2.25)
С учетом закона Стокса враща-
тельный диффузионный коэффици-
ент 0 и вращательное время релакса-
ции т(| равны:
Q. = kT/^0R^ = kT/^V, (Г2.26)
Рис. Г2. IS. Сравнение вращательного и по-
ступательного потоков. На рисунке показан
профиль вычисленных скоростей потока, вы-
зываемый сферическими макромолекулами,
подчиняющимися приграничным условиям
полного прилипания. Прерывистыми линиями
представлены линии потока для сферы, вра-
щающейся вокруг нормали оси в плоскости
страницы. Сплошными линиями обозначены
линии потока для сферы, движущейся сле-
ва направо в плоскости страницы. В каждом
случае лабораторная система координат была
выбрана таким образом, чтобы сфера была ста-
ционарной. Показан только один квадрант, на-
ходящийся в поле наблюдения. Радиус сферы
равен Ло, а расстояние выражается в единицах
Rq. Вверху показано поперечное сечение пото-
ков, совпадающее с центром сферы; рисунки б
и в показывают профиль скоростей (линии по-
тока) для случая чистого вращения и поступа-
тельного движения, соответственно (Kuntz and
Kauzman, 1974)
ИЛ _ Ц„У
3/?7' kT ’
(Г2.27)
Сравнение формул Г2.30 н Г2.31
показывает, что вращательное трение
гораздо более чувствительно к размеру
частицы, чем поступательное. Причина
этого лежит в том, что величина про-
порциональна кубу линейных размеров
молекулы, тогда как величина / зависит
от линейных размеров в первой степени
(см. уравнения ПО. 11, Г10.12).
Вращательные коэффициенты диф-
фузии некоторых биологических молекул
(от грамицидина до ДНК из бактериофа-
га Т4) представлены в .... Ин-
тервал их значений составляет восемь
порядков, что во много раз превосходит
таковой для коэффициентов поступа-
тельной диффузии или коэффициен-
тов седиментации (см. и :. ).
Причина этого с физической точки зре-
ния проста: поступательное трение про-
порционально линейным размерам час-
тицы, а вращательное — кубу линейных
размеров (объему).
Эллипсоид вращения
Для эллипсоида вращения с осями
a,b = c существует не один, а два коэф-
фициента трения: один Д — для враще-
ния вокруг полуоси а, а второй С* — для
. Константы вращательной диффузии некоторых макромолекул
в воде
Макромолекулы 0 (сек1)
Грамицидин(димер) Лизоцим Кинезин (349) Фрагмент ДНК (104 п. о.) Т7 бактериофаг Вирус табачной мозаики ДНК бактериофага Т7 ДНК бактериофага Т4 60000000 16700000 5000000 172000 5290 330 5,2 0,41
вращения вокруг полуоси Ь. На практи-
ке очень трудно измерить величины С,а
и C,h независимо. Поэтому обычно вы-
числяют среднее значение коэффици-
ента вращательного трения С, и среднее
значение коэффициента вращательной
диффузии 0 по уравнениям:
<; = 6т|0Г/У(р), (Г2.28)
® = <Г2-29
где функция Перрена, которая
зависит от отношения осей эллипсои-
да вращения. Теоретическое значение
\/J(p) представлено в виде графика на
рисунке Г2.1!). Сравнение зависимостей
вращательного трения, представленных
на рисунке 1'2.19, с таковыми для посту-
пательного трения (см. рис. Г2.2) ясно
показывает, что вращательное трение
гораздо более чувствительно к форме
молекулы, чем поступательное трение
( кчммрп rapnii I 2.1 1 ).
Для вытянутого эллипсоида враще-
ния с полуосями а и b {а > 5й), вращение
вокруг одной b оси хорошо описывается
уравнением:
Q _3kT\(2\n(2a / Ь)-У]
~16тгт]0а3
из которого видно, что малая ось b по-
является под знаком логарифма. Отсю-
да следует, что ее вклад в величину 0
относительно мал и, следовательно, кон-
станту вращательной диффузии следу-
ет использовать для вычисления длин-
ной, а не короткой оси эллипсоида.
Круговой цилиндр
Напоминаем, что круговой цилиндр от-
носится к телам, для которых константа
вращательного трения (или диффузии)
не может быть представлена в анали-
тическом виде. Причина этого — на-
(Г2.30)
личие краевых эффектов, которые мы
обсуждали ранее (секция Г1.5). Поэто-
му константа вращательной диффузии
круговых цилиндров (палочек) обычно
выражается как:
ЧАТ
0 =----^(In^/^-y), (Г2.31)
7СТ]о£
где г|0 — вязкость растворителя; kT —
тепловая энергия; L — длина палочки;
d — ее диаметр и у — фактор трения, ко-
торый зависит от способа учета краевых
эффектов.
Как и в случае эллипсоида враще-
ния диаметр палочки, d, появляется в
уравнении (Г2.31) только под знаком
логарифма и поэтому константу враща-
тельной диффузии следует использо-
вать в первую очередь для вычисления
длины кругового цилиндра.
Все существующие сегодня теории
согласуются с функциональной формой
уравнения (Г2.31) и отличаются только
фактором трения у и его зависимостью от
параметра/? = L/d(г<>м.\н!гг:цшi"i Г2.1 2 ).
Для очень длинных цилиндров
(р = L/d > 30) может быть использова-
ние. 12.19. Зависимость усредненного по
ориентациям коэффициента вращательно-
го трения С, от отношения осей вытянутого и
сплюснутого эллипсоидов. Эта гидродинами-
ческая функции вычислена с помощью ком-
пьютерной программы (1 larding et al., 1997)
Комментарий Г2.1 1. Коэффициенты
вращательного трения и граничные
условия
Коэффициенты вращательного трения
более чувствительны к изменениям гранич-
ных условий. Так. при переходе от условий
скольжения к условиям прилипания пос-
тупательное трение изменяется только в
1.5 раза (см. уравнения Г1.11, Г.12). Однако
при таком же переходе при вращательном
движении имеет место другая ситуация, о
которой мы упоминали для случая сферы
(уравнение Г1.14). Для вытянутого эллип-
соида в условиях скольжения нет сопротив-
ления при вращении его вокруг длинной
оси, а для сплюснутого — вокруг короткой.
Вычисления вращательных свойств макро-
молекул различной формы при разных гра-
ничных условиях можно найти в литерату-
ре (Allison, 1999).
; Комментарий Г2.12. Теоретическая
| трактовка вращательного трения
правильного кругового цилиндра
: Имеется, по крайней мере, пять теоре-
i тическпх подходов, относящихся к расчету
коэффициента вращательной диффузии пра-
, вильногокругового цилиндрасопределенной
длиной и диаметром. Для длинных цилинд-
ров большинство из них дают практически
одинаковые результаты, но для коротких ци-
линдров каждая теория дает свои результаты
(Elias and Eden, 1981; Lu et al., 2002).
на усовершенствованная формула Бро-
ерсма:
( 1 Y
7 = 0,757-7 -----0,27 . (Г2.32)
( In 2 р )
Для относительно коротких цилин-
дров (2<д<30) наиболее приемлемой
является формула Тирадо Гарсиа де ла
Торре:
0,917 0,05 zrQQQ.
у = 0,667-----+ —5-. (Г2.33)
Р Р
В заключение следует упомянуть, что
имеются две формулы, описывающие
длинные (в рамках червеобразной моде-
ли) и короткие (в рамках модели слабо
скрученной палочки) молекулы ДНК.
Однако эти формулы слишком громоз-
дки, чтобы воспроизводить их здесь.
Применение уравнений Г2.32—Г2.34 для
исследования молекул ДНК в интервале
пар оснований длиной от 8 до 120 мил-
лионов будет обсуждено в Главе Г7.
Короткие цилиндры
Понимание гидродинамических свойств
коротких цилиндров представляет
особый интерес, поскольку такие ци-
линдры хорошо моделируют короткие
олигонуклеотиды ДНК. Для коротких
цилиндров (р - 2) существует практи-
ческий способ определения размеров
цилиндра, основанный на комбинации
поступательного и вращательного ко-
эффициентов трения Dt и 0. Объеди-
няя уравнения Г2.3 и Г2.31, определим
функцию р(/?), как:
'°'
kT 0
1пр + ф
(lnp + 7)* 1
(Г2.34)
Функция р(р), приведена на ри-
сунке Г2.20. из которого видно, что
она обладает достаточно высокой чув-
ствительностью к асимметрии части-
цы, особенно в области малых асим-
метрий р. Таким образом, уравнение
Г2.34 открывает возможность прямо-
го определения асимметрии короткой
палочки, если коэффициенты посту-
пательного и вращательного трения
определяются экспериментально, на-
пример методом динамического рас-
сеяния света (Глава ПО).
Г2.3.5. Частицы произвольной
формы
Техника моделирования, используемая
для вычисления вращательного тре-
ния частиц произвольной формы с ис-
пользованием набора бусинок, такая же
как и для вычисления поступательного
трения (параграфы Г2.3.1 —Г2.3.3). Од-
нако, для того, чтобы вычислить коэф-
фициент вращательного трения части-
цы произвольной формы, необходимо
знать истинную ось вращения. На этой
оси находится центр трения. Для неко-
торых частиц, таких как сферы, палоч-
ки или кольца, центр трения совпадает
как с геометрическим центром, так и с
центром масс. Для структур с меньшей
симметрией, таких как бактериофаги,
это обычно ие так. Очевидно, что моле-
кула вращается таким образом, чтобы
при этом растрачивать минимум энер-
гии на трение с растворителем. Таким
образом, подлинной осью вращения
является та, которая дает минимальное
значение для величины С, или макси-
мальное значение для величины D.
Другое важное различие между вра-
щательной и поступательной диффузи-
ей связано с вкладом в трение дополни-
тельных масс. Так, добавление массы
в центр частицы мало сказывается на
ее вращательных свойствах, тогда как
ее добавление на периферию частицы
оказывает гораздо большее воздействие
(ком Mi'll repi । ii Г2.13).
Г2.3.6. Экспериментальные методы
для измерения коэффициентов
вращательного трения
Экспериментальные методы для изме-
рения коэффициентов вращательного
трения подразделяются на две группы.
К первой группе относятся эксперимен-
Рпс. Г2.20. График функции р(р) отр. Сплош-
ная линия построена по уравнению (Г2.38);
прерывистая линия — то же самое, но при на-
ложении относительной ошибки, равной ±1 %
в D и ±3% в 0 (Garcia de la Torre and Martinez,
1984)
ты, в которых определяется скорость
вращения частицы под действием пары
сил. Если градиент скорости в раствори-
теле играет роль ориентирующей силы,
то явление называется двойным луче-
преломлением в потоке, или эффектом
Максвелла. Если сила имеет электриче-
скую природу, явление называется элек-
Комментарий Г2.13. Метод
граничных элементов для
вычисления вращательного трения
I Альтернативный подход вычисления
' вращательных свойств частиц базируется
на методе граничных элементов. В настоя-
щее время алгоритм для вычисления кон-
станты вращательной диффузии является
доступным. В случае, когда их вращатель-
ные свойства известны, точность алгоритма
i весьма высока и составляет несколько про-
, центов (Allison, 1999).
Методы определения коэффициентов вращательного трения
Эксперимен- тальный метод Измеряемый параметр Интервал измеряемых коэффициентов диффузии 0 в сек'1 Интервал измеряемых релаксационных времен т в сек Область применимости метода
Электрическое двойное луче- преломление (Глава Гб) Релаксаци- онное время, т 1,7 х 106-0,3 100 нсек — 500 м сек Большие асиммет- ричные молекулы, такие как ДНК и РНК
Двойное лу- чепреломление в потоке (Глава Г7) Угол ориен- тации, X 5 х 104,7 3 реек — 100 мсек Большие асимме- тричные молекулы
Деполяризо- ванная флюо- ресценция и фосфоресцен- ция (Глава Г8) Релаксаци- онное время, т 50 х Ю6-1,7х Ю6 3-100 нсек Малые сфериче- ские молекулы
Динамическое рассеяние света (Глава ПО) Корреля- ционная функция или коэффици- ент деполя- ризации 50 х 10G—0,17 1 нсек — 1 сек Малые квазисфе- рические моле- кулы или большие и очень асиммет- ричные молекулы
Ядерный магнитный резонанс (Глава КЗ) Релаксаци- онное время, т 50 х 10е-1,7хЮ6 3-100 нсек Маленькие ква- зисферические молекулы
трическим двойным лучепреломлением
или эффектом Керра. Ко второй группе
относятся явления, в которых внешние
силы не воздействуют на частицу и ее
вращение связано только с броуновским
движением. Поведение частицы при
флуоресценции и динамическом рассея-
нии света относится к этому типу.
В iao 12.3 приведены пять ме-
тодов для определения коэффициентов
вращательного трения. В ней перечис-
лены экспериментальные методы (пер-
вая колонка), измеряемые параметры
(вторая колонка), доступные интерва-
лы экспериментальных значений вра-
щательных коэффициентов диффузии
(третья колонка), времена релаксации
(четвертая колонка) и области приме-
нимости каждого метода. В таблице
также указаны главы, в которых деталь-
но описаны экспериментальные методы
и их применение.
Следует заметить, что исследова-
тели, работающие в разных областях,
часто используют различные опреде-
ления времени релаксации. Так, при
описании экспериментальных данных,
полученных с помощью эффекта Кер-
ра или эффекта Максвелла, предпо-
читают пользоваться вращательным
релаксационным временем, обознача-
емым как р, тогда как при описании
аналогичных данных ЯМР и деполя-
ризованной флюоресценции обычно
пользуются вращательным корреляци-
онным временем, обозначаемым как ф .
Эти времена связаны между собой как
Ф =
- . Их связь с вращательными диф-
фузионными коэффициентами имеет
1
вид: Ф = ЕН
b/J
р = —. В этой книге т оз-
н 2D
начает вращательное реалаксациоппое
время, а т. означает вращательное кор-
реляционное время.
Г2.4.1. Вязкость как локальные
потери энергии
: л.н.ь. -о 'Щ >.:i: I 2. i4. Ламинарный
и турбулентный потоки
Когда жидкость, наблюдаемая в экс-
перименте, течет медленно между двумя
параллельными поверхностями, то такое
течение называется ламинарным. В этом
случае т)0 не зависит от G и такой поток по
своим вязкостным свойствам называется
ньютоновым. При достаточно высокой ско-
рости свойства потока начинают изменять-
ся. Такой поток называется турбулентным.
В этом случае т|0 начинает зависеть от G и
такой поток называется неньютоновым.
В цилиндрических трубах переход от ла-
минарного потока к турбулентному про-
исходит, когда число Рейнолдса достигает
2000. Поток жидкости в узких капиллярах
или протекающий между двумя медленно
вращающимися цилиндрами, расположен-
ными близко друг к другу', является лами-
нарным потоком при любых скоростях, не-
обходимых для измерения вязкости.
Внутреннее трение или вязкость жид-
кости проявляется тогда, когда она
находится в состоянии потока с от-
личным от нуля градиентом скорости.
Самым простым примером являет-
ся ламинарный поток с постоянным
градиентом скорости G = dujdy под
прямым углом по отношению к пото-
ку (рис. Г2.21 и м>г\и г I С I
Скорость жидкости и находится из
выражения:
Рис. I 2.21. Скорость и градиент распределе-
ния скорости в ламинарном потоке в беско-
нечно малом зазоре между цилиндрами
и = и,.= Gy,
Uy ~ U: ~ б-
(Г2.35)
Вязкость жидкости является мерой
внутреннего трения, которое опреде-
ляет значение тангенциальной силы F,
необходимой для поддержания гради-
ента скорости G между плоскостями
жидкости. Чем большим внутренним
трением обладает жидкость, тем боль-
шую силу надо приложить к ней с тем,
чтобы поддерживать в потоке заданный
градиент скорости G. Эта связь выража-
ется формулой Ньютона:
F = f>=no^ = noG. (Г2.36)
ay
Константа пропорциональности
г|0 называется коэффициентом вяз-
кости или просто вязкостью жидкости.
Ее размерность — дина сек'1-см’2 или
эрг-см"3-сек *. Таким образом, вязкость
может быть определена как потери энер-
Рис. Г2.22. Ламинарный ноток (объяснения
в тексте)
гни на единицу объема за единицу времени
в потоке с единичным градиентом скоро-
сти. Для поддержания градиента скоро-
сти G в жидкости должна быть затрачена
энергия. Чтобы вычислить ее, рассмотрим
два слоя — 1 и 2, разделенных третьим,
толщиной dy над участком поверхности
ху и площадью 1 см2 (рис. 12.22). За время
t смещение слоя 2 относительно слоя 1 в
х-направлении будет равно:
dx = t(du./dy) - tgdy.
Таким образом, в слое толщиной dy
и площадью 1 см2 проделанная работа
будет равна: dA = Fdx = F Gtdy для объе-
ма жидкости, равного dy-см1. При этом
количество проделанной работы для
преодолении трения на единицу объема
равно: А = FGt. Подставляя Гиз уравне-
ния (Г2.36), получаем выражение для
проделанной работы за единицу време-
ни на единицу объема, обусловленной
направленным потоком:
£ = ^ = ПоС2. (Г2.37)
dt
Это выражение является централь-
ньъм в обсуждении вязкости макромо-
лекул.
Г2.4.2. Относительная, удельная
и характеристическая вязкость
Вязкость чистого растворителя обо-
значается как т]0. Добавление макромо-
лекул будет увеличива ть вязкость до но-
вой величины г|. В результате вязкость
раствора макромолекул всегда больше,
чем чистого растворителя (коммента-
рий Г2.15). Отношение вязкости раство-
ра к вязкости чистого растворителя на-
зывается относительной вязкостью
Л„„, = Л/По- (1’2.38)
Значение, на которое повышается
вязкость, называется удельной вязкос-
тью и выражается:
= П/1%-1 =П11П1 - 1- (Г2.39)
При достаточно низкой концентра-
ции, С, г| , пропорционально С. мы мо-
жем определить характеристическую
вязкость | г| | как значение, на которое уве-
личивается вязкость раствора при внесе-
нии одного грамма макромолекул (ком-
мен гарий Г2.1(>). Абсолютная величина
характеристической вязкости зависит от
того, в каких единицах выражена кон-
центрация растворенного вещества. Так,
если концентрация выражена в мг-мл ',
то характеристическая вязкость глобу-
лярного белка [г|] будет равна 3 мл-мг '.
Если же концентрация выражена в %
I . !
; Комментарий Г2.15. Отрицательное I
значение характеристической j
j вязкости
На практике вязкость раствора может
быть ниже вязкости чистого растворителя.
Явление отрицательной вязкости было от- ।
крыто давно на примере простых жидких ।
; смесей, таких как бензол в этаноле, изобу-
тилен в бензоле. Это явление возникает в
результате специфического взаимодейс-
твия между рас творяемым веществом и рас- ,
творителем, при котором жидкая структура j
растворителей некоторых видов нарушает-
ся вблизи растворяемой молекулы. Такой
результат не может обсуждаться в рамках
классической гидродинамики.
(мгдецилитр' ’), то аналогичное значение
вязкости будет равно 0,03 децилитр.-мг'1.
Общепринятое выражение для характе-
ристической вязкости обычно имеет вид:
lnJ = ]inl21z2k (Г2.40)
П(1С
С^О,
отражающий факт необходимости эк-
страполяции получаемых величин к ну-
левой концентрации растворенных мак-
ромолекул.
Г2.4.3. Частицы правильной формы
Сфера
В ламинарном потоке, определяемом
уравнением (Г2.35), сферические час-
тицы движутся в слоях жидкости с раз-
личными скоростями. Поэтому силы
трения со стороны растворителя, обте-
кающего частицу, приводят ее не толь-
ко в поступательное, но и также во вра-
щательное движение (рис. Г2.23).
Когда сумма всех вращательных мо-
ментов, действующих на частицу, равна
нулю, то угловая скорость сферических
частиц будет постоянной:
w = |g. (Г2.41)
На рисунке Г2.23 изображен не-
возмущенный ламинарный поток до
введения в него частиц в системе ко-
ординат, начало которых совмещено с
центром сферической частицы и кото-
рые движутся поступательно вместе с
нею в потоке. Нетрудно видеть, что в
выбранной системе линейные скорости
в точках a, b, с п d поверхности частицы
направлены по касательным к ней и по
абсолютной величине равны и0 = a>R0 =
/1 х GRW где 7?(| — радиус сферической
частицы. Скорости невозмущенного
Комментарий Г2.16. Термин
«характеристическая вязкость»
Термин «характеристическая вязкость»
был предложен Кремером в 1938 г. Строго
говоря, использование термина «вязкость»
для т]шн, r|vi и [т|] является некорректным,
поскольку эти величины являются просто
числами и не имеют размерности вязкости.
В 1957 г. Международный союз чистой
и прикладной химии предложил переиме-
новать характеристическую вязкость [т|]
в «предельное число вязкости». Последнее
четко отражает размерность термина. Од-
нако предложенный термин так и не стал
общепринятым.
потока и в системе этих координат раз-
личны в разных точках. Так, в точках с
и d скорость и = 0, в то время как в точ-
ках а и b абсолютное значение скорости
равно GR0 (т. е. вдвое больше скорости
частицы), а направление скорости сов-
падает со скоростью поверхности час-
тицы. Таким образом, в точках а, Ь, с,
и d скорость невозмущенной жидкости
1’пс. 1'2.23. Сферические частицы в ламинар-
ном потоке (Цветков, 1989)
отличается от скорости соприкасаю-
щихся участков частицы на величину
/2 х С/?(). В других точках поверхности
частицы относительная скорость невоз-
мущенного обтекающего растворителя
отличается от значения У> х GR0, но так-
же пропорциональна GR0.
В присутствии частицы распреде-
ление истинной скорости несколько
отличается от таковой!, приведенной на
рисунке Г2.21, поскольку частица воз-
мущает поток растворителя. Измене-
ние скорости точек среды на величину
Уг х б7?опри переходе от частицы к рас-
творителю совершается на ее границе
не скачкообразно (как в точках а, Ь, с,
и d рис. Г2.23), а непрерывным образом в
некотором слое жидкости, окружающем
частицу. На самой же границе раздела
скорость частицы совпадает со скоро-
стью прилегающего возмущенного рас-
творителя. Однако для оценки потерь па
трение это не имеет принципиального
Рис. 1'2.21. Направления скоростей невозму-
щенного растворителя по отношению к поверх-
ности сферической частицы в ламинарном
потоке. Линии потока показывают значение
относительной скорости и-и0 в ху плоскости
(Цветков, 1989)
значения. На рисунке 1 2.21 показан не-
возмущенный поток растворителя в ху
плоскости по отношению к поверхности
частицы. Из диаграммы видно, что сумма
вращающих моментов, обусловленная
силами вязкости, равна нулю. Однако
результатом проделанной этими силами
работы является дополнительная поте-
ря энергии и, следовательно, увеличение
вязкости раствора.
Качественная оценка рассматри-
ваемого эффекта показывает, что ско-
рость растворителя ио отношению к
поверхности сферы всегда пропорцио-
нальна произведению G/?o, что экви-
валентно вращению частицы в рас-
творителе с относительной угловой
скоростью,равной:
co()=aG. (Г2.42)
Уравнение Г2.42 имеет универсаль-
ный характер. Для частиц различной
формы коэффициент пропорциональ-
ности а имеет различные значения, но
пропорциональность между со0 и G всег-
да сохраняется.
На рисунке Г2.25 показано истин-
ное распределение скоростей для чисто
вращательного движения и движения
в потоке. Здесь ясно прослеживается
основное различие между ними: изме-
нение профиля скоростей в потоке про-
исходит более быстро, чем при чистом
вращении. Заметим, что в потоке мо-
лекулы растягиваются при движении
вдоль одного направления и сжима-
ются при движении вдоль другого на-
правления. Этот эффект очень выражен
для гибких молекул и будет рассмотрен
в секции Г7.6.
Для вращения твердых частиц, фор-
ма которых отлична от сферической, в
вязком растворителе работа, проделан-
ная в единицу времени по преодолению
трения, равна:
W = w0Q, (Г2.43)
где Q — вращательный момент, связан-
ный с коэффициентом вращательного
трения частицы, Q в соответствии с за-
висимостью:
Q = ^%- (Г2.44)
Если 1 см3 раствора содержит Динас-
тии, тогда потеря энергии, ДЕ, в единицу
времени, обусловленная вращательным
трением, будет равна W(V0. Используя
уравнения (Г2.42—Г2.44), находим:
ДЕ =a2G2 (Г2.45)
где ДЕ обозначает разность между поте-
рей энергии, обусловленной трением в
растворе и в растворителе. Таким обра-
зом, из уравнения (Г2.45) следует:
ЛЕ = рС2-ПцС2, (Г2.46)
где р является вязкостью раствора. При-
равнивая уравнения (Г2.41) и (Г2.42),
получим значение для удельной вязко-
сти раствора:
= (Г2.47)
По По
Уравнение Г2.47 носит общий ха-
рактер и для частиц разной формы от-
личается только значением коэффици-
ента а2. Для сферических частиц 6a2 =
2,5 и с учетом уравнений Г2.47 и урав-
нения Стокса 0=6 np(lV получаем
nvi = 2,5W0, (Г2.48)
Уравнение Г2.48 известно как урав-
нение Эйнштейна. Напоминаем, что
Л70 =CNК/М, где С — это концентрация
раствора в граммах на кубический сан-
тиметр, N — это число Авогадро, а М —
молекулярная масса частицы. Из урав-
нения Г2.48 следует, что:
[П| = Нт
П.
= 2,5WV„, (Г2.49)
Рис. Г2.25. Сравнение чисто вращательного
потока и потока в градиенте скорости. Профиль
скорости жидкости был вычислен для сфери-
ческих макромолекул, находящихся в пригра-
ничном состоянии прилипания. Прерывистые
линии представляют собой линии потока для
сфер, вращающихся вокруг оси перпендику-
лярной к плоскости страницы. Сплошные ли-
нии представляют линии градиента скорости
потока. Радиус сферы равен Ro, а расстояние
выражено в единицах Ro. Возмущенный поток,
обусловленный присутствием сфер, является
функцией скорости компонентов потока вдоль
трех осей. Эта формула слишком громоздка и
не может быть воспроизведена здесь. Читате-
ли, интересующиеся математическими дета-
лями, отсылаются к специальной литературе
(Kuntz and Kauzmann, 1974)
где [р] — это характеристическая вяз-
кость раствора макромолекул, а V —
гидродинамический объем частицы.
Для жестких несферических частиц
из уравнения Г2.47 следует:
a2N С
[П1 = —f (Г2.50)
По Л/
Используя уравнение Г2.20, мы
имеем:
DT
1п] = а2-7^7- (Г2.51)
П()0М
Комментарии 1'2. I 7.
Альтернативное выражение
для характеристической вязкости
Для эллипсоидных частиц уравнение
(Г2.51) для характеристической вязкости
при низких градиентах скорости может
быть записано в виде:
[nl =
RT F(p)
т\овМ 6
(А)
где F (р) --- фактор формы и р — отношение
осей эллипсоида. Для р > 10 функция F
(р) изменяется очень слабо и произведение
[ц]0Моказывается практически независя-
щим от р. При р —> оо, F(р) = 0.8 по величи-
нам [г|] и М можно вычислить 0 (Цветков
и др„ 1971).
Уравнение Г2.51 отражает тот
факт, что характеристическая вязкость
раствора, независимо от модельных
свойств частицы, во всех случаях явля-
ется мерой потерь энергии, вызванной
вращением частицы в растворе. Именно
Отношение осей (р или 1/р)
Рис. 1'2.26. Зависимость функции Симха
v(p) от отношения осей для вытянутого и
сплюснутого эллипсоидов вращения. Для сфе-
ры v(p) = 2,5. Табулированные значения этой
и многих других гидродинамических функций
можно получить, используя компьютерные
программы (Hurtling et al., 1997)
поэтому значение характеристической
вязкости зависит от вращательной под-
вижности, т. е. от величины 0, что вид-
но из формулы Г2.51.
Эллипсоидальные частицы
Для растворов несферических частиц
ситуация более сложная и физическая
картина может быть качественно описа-
на следующим образом. Оценка потери
энергии базируется на предположении,
что раствор эллипсоидальных частиц в
ламинарном потоке находится в равно-
весном состоянии, зависящем от двух
действующих противоположно направ-
ленных сил. Одна сила возникает из-за
тенденции градиента скорости потока
ориентировать частицы в направлении
линий его движения, а другая обуслов-
лена броуновским движением, име-
ющим тенденцию обеспечивать слу-
чайную ориентацию. Таким образом,
полная потеря энергии, обусловленная
трением, является суммой двух состав-
ляющих. Первая определяется чисто
гидродинамическими потерями, о кото-
рых шла речь в предыдущем параграфе,
а вторая обусловлена броуновским вра-
щением частиц. Относительные роли
этих двух составляющих зависят от
отношения градиента скорости к коэф-
фициенту вращательного трения, G/Q.
При малом значении этого отношения
асимметрия функции углового распре-
деления полностью устраняется броу-
новским вращением. В этих условиях
каждая асимметричная частица враща-
ется с постоянной угловой скоростью
to = G/2. Уравнение Г2.49 для вязкости
эллипсоида вращения для предельного
случая, когда отношение G/Q стремит-
ся к нулю, имеет вид:
[n] = v(p)^Vi;ip/M, (Г2.53)
где Ал — число Авогадро, М — молеку-
лярная масса и V — гидродинамиче-
ский объем макромолекулы (коммеп i а-
рий Г2.17).
Функция ч(р), называемая функци-
ей Симха для вытянутых и сплюснутых
эллипсоидов вращения, представлена на
рисунке Г2.26. Из рисунка видно, что эта
функция гораздо более чувствительна
к форме частицы, чем функция Перре-
на (см. рис. Г2.2). Для отношения осей
больше 15, функция Симха для вытяну-
тых эллипсоидов может быть представ-
лена асимптотическим уравнением:
(Г2'53)
15 1п(2</ / /;)
Из уравнения Г2.53 следует, что для
вычисления формы макромолекулы в
рамках эллипсоида вращения нет необ-
ходимости знать молекулярную массу
макромолекулы.
Палочкообразные частицы
В 1951 г. Кирквуд и Ауэр вычислили
характеристическую вязкость палоч-
кообразной частицы как функцию от-
ношения осей. В этой модели палочка
рассматривалась как набор линейно
расположенных сфер, состоящих из п
звеньев диаметром d и длиной L=nd.
Асимптотическая форма уравнения для
характеристической вязкости имеет
вид:
। | =---7tN,L-d--- (Г2.54)
1 1 2250Л/„1п(£/с/)
где Ах — число Авогадро и М — молеку-
лярная масса мономера.
Это уравнение идентично уравне-
нию Симха для вытянутых эллипсои-
дов с очень большой длиной полуоси
L/2. Если выразить Ми как npb~L/3n и
вспомнить, что для больших значений
a/b In (2a/b) = 1п2 +ln (a/b) = In (a/b),
то выражение для характеристической
вязкости принимает вид:
24 г/
<Г2-55>
9000р In р
Это уравнение можно использовать
для определения отношения осей палоч-
кообразных частиц, а если молекуляр-
ная масса частицы известна, то можно
вычислить и ее длину. Характеристи-
ческая вязкость молекул другой формы
обсуждается в ком мет арии Г2.1
Г2.4.4. Частицы произвольной
формы
Возможности для вычисления значений
характеристической вязкости для частиц
произвольной формы более ограничены,
чем при вычислении коэффициентов
вращательной диффузии и, особенно,
коэффициентов поступательной диффу-
зии. Во-первых, это связано с необходи-
мостью экстраполировать величину [ц]
к нулевому градиенту скорости и усред-
нять ее по всем ориентациям частицы.
Во-вторых, для несферических молекул
величина [г|] зависит от выбора центра
частицы, называемого в данном случае
«центром вязкости», который может от-
личаться от «центров» поступательной
или вращательной диффузии для частиц
произвольной формы.
При вычислении характеристи-
ческой вязкости частиц произвольной
формы используются три подхода для
моделирования, два из которых (мо-
делирование с помощью бусинок и
треугольных пластинок) аналогичны
таковым, используемым в вычислении
поступательной или вращательной
диффузии ( ком.мен inpiiii 1'2. i !>). В пер-
вом из них жесткая молекула моделиру-
ется Асферическими элементами соот-
Характеристическая вязкость
частиц специфической формы
Трехосные эллипсоиды
Значение вязкости трехосного эллипсо-
ида равно v = |г||/1-' = Y («, Ь, с), где [г|| —
характеристическая вязкость, выраженная
в см1 на грамм, V — удельный объем в см3
на грамм и У (а, Ь, с) — эллиптические ин-
тегралы, которые могут быть вычислены
численным интегрированием. При данных
значениях величин (а/Ь, Ь/с) они одно-
значно фиксируют значение функции v,
но при заданном значении функции v, она
будет иметь линейное решение по отно-
шению к значениям двух осей (а/b и Ь/с).
К сожалению, пересечение этих двух линий
имеет место только при больших значени-
ях отношений а/Ь и Ь/с, а не при их малых
значениях. Последнее представляло бы наи-
больший практический интерес (Harding
and Rowe, 1982).
Сфероидно-цилиндрические молекулы
В литературе имеются данные ио харак-
теристической вязкости цилиндров, концы
которых аппроксимированы разными фор-
мами (полусфера, полуэллипсоид). Эти дан-
ные выявляют влияние краевых эффектов
на значение характеристической вязкости.
Как и следовало ожидать, сильные краевые
эффекты наиболее выражены для относи-
тельно коротких цилиндров. Примечательно,
что эти эффекты не так сильно сказываются
на значении коэффициента поступательной
диффузии (Yoshizaki and Yamakawa, 1980).
Молекулы в виде гантелей
Известны значения характеристической
вязкости для гантелевидных частиц, кото-
рые можно представить в виде двух оди-
наковых сфер, находящихся на различном
расстоянии друг от друга (Wakia, 1971).
ветствующего радиуса. Размер и форма
модели должны быть как можно ближе
к таковым реальной макромолекулы
(см. рис. Г2.5 а). Гидродинамическое
взаимодействие между сферами учи-
тывается классическими гидродинами-
ческими методами. К сожалению, все
теории характеристической вязкости
. ' Использование
моделей оболочек при вычислении
характеристической вязкости
Подход, основанный на модели оболо-
чек, теоретически должен быть идеаль-
ным решением для вычисления вязкости
макромолекул. В этой процедуре большое
число бусинок располагается на повер-
хности объекта как можно ближе друг к
другу (см. рис. Г2.7). Поэтому вычисле-
ние характеристической вязкости при
экстраполяции к нулевому размеру бу-
синок должно давать самое точное значе-
ние. К сожалению, такая процедура тре-
бует так много расчетного времени, что на
практике не используется.
дают хорошие результаты только для
случая сильно асимметричных частиц
и приводят к относительно большим по-
грешностям в случае одной сферы или
нескольких сфер.
Второе приближение основано на
моделировании поверхности частицы
набором треугольных пластинок. Ха-
рактеристическая вязкость такой мо-
дели вычисляется для ряда структур
с экстраполяцией числа пластинок к
бесконечности. Основное преимуще-
ство такого подхода состоит в том, что
он применим для вычислений харак-
теристической вязкости при разных
граничных условиях, а также при дейс-
твии различных сил, действующих на
окружающую частицу жидкость. Точ-
ность таких вычислений достаточно
велика. Так для эллипсоидов враще-
ния, где аналитическое решение из-
вестно, точность составляет около 1 %.
Для палочкообразных частиц удовлет-
ворительные результаты получены в
интервалер от 2,04 до 17,0, что соответ-
ствует фрагментам ДНК длиной от
12 пар оснований (р = 2,04) до 100 пар
оснований (р = 17,0).
: - j i'.’.’X «пониманию
формулы Г2.56
В формуле Г2.56 С измеряется в едини-
цах численной плотности, а характеристи-
ческая вязкость — в единицах объема.
Третий подход базируется на кор-
реляции между характеристической
вязкостью [т|] молекулы и ее поляри-
зуемостью а. Основываясь на прямом
сравнении [т|] и а в случае эллипсоидов
и гантелей, Дуглас и Хаббард предложи-
ли следующее выражение для сферы:
[r)|=liin2^L2k = 0,79а. (Г2.56)
G-о С;,Ц0
Ориентационное усреднение тензо-
ра Озеена дает несколько другое отно-
шение:
[г)] = 3/4а, (Г2.57)
отличающееся на 5% от предыдущего
( ?. . ..:'рс1; Г). Оба соотношения
однако отличаются от точного значения,
ожидаемого для сферы. Для нее поля-
ризуемость равна а = 3Vp. Тогда харак-
теристическая вязкость, предсказанная
уравнением (Г2.57), равна [ц J = 9/4 х у
которое отличается от точного резуль-
тата Эйнштейна [г|] = 5/2 V на 10%.
Поэтому Дуглас и Хаббард предложили
добавить константу в правую часть урав-
нения Г2.57, с тем чтобы иметь возмож-
ность его использовать для глобулярных
белков. Это приводит к следующей эм-
пирической зависимости между харак-
теристической вязкостью частицы [т]],
ее поляризуемостью а и объемом V:
[т)1 = 3/4 а + </4 V. (Г2.58)
Применение этого соотношения к
телам, имеющим форму эллипсоидов,
цилиндров, гантелей дает значения ве-
личин [г|] с точностью 3%.
5, Г»Т При б Л 3’7
/г Г-1/: ГГ . у , IX .Z
7 Т '.У/.
Г2.5.1. Функции формы, зависящие
от объема для двухосного
эллипсоида
Для двухосного эллипсоида вращения
каждая гидродинамическая функция
зависит от его объема и отношения осей
(уравнения Г2.2, Г2.26 и Г2.53). В
суммированы все эксперимен-
тальные гидродинамические функции,
каждая из которых нормирована на сфе-
ру, с тем чтобы высветить только вклад
отношения осей. Эти функции называ-
ются «функциями формы, зависящими
от объема». Такое название они полу-
чили, с тем чтобы отличать их от «фун-
кций формы, не зависящих от объема»,
когда различные гидродинамические
функции комбинируются между собой
путем исключения входящего в них гид-
родинамического объема. Различные
комбинации таких функций рассматри-
ваются в следующем параграфе. Главная
их цель — найти такую комбинацию гид-
родинамических функций, которая по-
зволила бы решить классическую про-
блему «объем — отношение осей» для
двухосного эллипсоида вращения.
Г2.5.2. Функции формы,
не зависящие от объема
для двухосного эллипсоида
Первая попытка решить проблему «объ-
ем — отношение осей» для двухосного
эллипсоида были предприняты Онслп в
1941г. Он вычислил значения отноше-
ния осей и гидратации для эллипсоида
вращения при различных значениях ко-
Г2.-i Экспериментально измеряемые функции формы, зависящие
от объема
Функция формы Экспериментальный параметр Экспериментальный метод
1. Отношение коэффициента поступательного трения части- цы к таковому для сферической частицы равного объема ///’, (для сферы ///0 = 1) (Главы ГЗ и Г4) / = А/(1-йр„) 4 л А |_31'„,.1р_ Г “I1 •* f _ kT 4п А Седиментация Диффузия
2. Отношение коэффициента вращательного трения частицы к таковому для сферической частицы равного объема 0,/0о (для сферы 0,/0„ = 1) (Главы Г7, Г8 и ПО) 00 kT Динамическое рассе- яние света. Эффекты Максвелла и Керра. Деполяризованная флуоресценция
3. Фактор формы характеристи- ческой вязкости v (для сферы v = 2,5) (Глава Г9) .. . [nbw i АГ, V;li;lp Вязкость
4. Приведенный исключенный объем V (ко-объем)*) (для сферы Ррнв = 8) v 1 X,,- д;, V;,,.,,,' где — исключенный макромо- лекулярный объем, а Л, - второй вириальный коэффициент Концентрационная зависимость осмо- тического давления, статического рассея- ния света и равновес- ной седиментации
5. Отношение гармонического среднего релаксационного вре- мени к таковому для сфериче- ской частицы равного объема т0 (для сферы ih/x0 = 1) (Главы Гб и КЗ) ч, _ 3 _kT z 1 Ч> (Ч>А„) + (2т0А,) Зц„ X.,,,’ Равновесная депо- ляризованная флюо- ресценция. Ядерный магнитный резонанс
6. Отношение корреляционно- го времени частицы к таковому для сферической частицы рав- ного объема т0 (для сферы т./т0 =1) (Глава Гб) _L Зт1„ Т' 1/.нлр i = 1-5 для общего тела; i = а, b для эллипсоида вращения Временной спад деполяризованной флуоресценции. Электрическое двой- ное лучепреломление
7. Коэффициент регрессии, k зависимости константы се- диментации от концентрации (Глава Г4) где sc и 50 — коэффициенты седи- ментации при конечной и экстра- полированной к нулевой! концент- рации, соответственно Концентрационная зависимость коэф- фициента седимен- тации
*) Исключенный объем представляет собой сумму гидродинамического объема, под которым
всегда понимается объем, занимаемый одной частицей в растворе, и дополнительного объема,
который является следствием взаимодействия между молекулами. Чем более асимметрична
частица, тем больше ее взаимодействие с другими молекулами и тем больше ее исключенный
объем, иногда называемый ко-объемом. Молярный ко-объем для системы макромолекул может
быть получен из термодинамического второго вириального коэффициента А,, после устранения
электростатических взаимодействий
эффициентов трения и вязкости и пред-
ставил данные этих исследований в виде
очень наглядных контурных карт. Этот
результат четко показал, что всегда су-
ществует проблема выбора между моде-
лью вытянутого и сплюснутого эллипсои-
да, каждый из которых будет иметь свое
отношение осей даже в том случае, когда
выбрана приемлемая степень гидратации.
В дальнейшем неоднократно предприни-
мались попытки комбинации различных
гидродинамических параметров с целью
найти такую, которая позволяла бы от-
личить вытянутый эллипсоид от сплюс-
нутого не только при больших степенях
вытянутости, но и при малых. Ниже мы
очень кратко опишем такие комбинации.
для коэффициента поступательного
трения/(уравнение Г2.2) и характери-
стической вязкости (уравнение Г2.53).
Эта попытка оказалась не совсем
удачной, поскольку результирующая
P-функция оказалась практически не-
чувствительной к отношению осей,
особенно для сплюснутого эллипсои-
да (рис. Г2.27). Однако это свойство
функции позволило авторам рекомен-
довать ее использовать (зафиксировав
ее значение) для определения молеку-
лярной массы макромолекул со слабой
степенью вытянутости. Долгое время
такая комбинация использовалась для
определения молекулярной массы гло-
булярных белков.
Коэффициент поступательного трения
и характеристическая вязкость
(р -функция)
В 1953 г. Шерага и Манделькерн пред-
ложили исключить объем из уравнений
Отношение осей (р или 1/р)
1’ие. Г2.27. Комбинация коэффициента пос-
тупательного трения и характеристической
вязкости (0-функипя). Для сферы величина
0 = 2,12 (Sheraga and Mandelkern, 1953)
Коэффициент вращательного трения
и характеристическая вязкость
(8-функция)
Комбинирование коэффициента враща-
тельного трения (уравнение Г2.24а) и ха-
рактеристической вязкости (уравнение
Г2.45), предложенная этими же авторами,
дало в результате функцию, которая ока-
залась несколько более чувствительной
к отношению осей особенно при малых
значениях последних (рис. Г2.28). Од-
нако практического использования эта
функция так и не получила, поскольку
к тому времени измерение коэффициен-
тов вращательной диффузии для белков
было нелегкой задачей.
Коэффициент регрессии
седиментации и характеристическая
вязкость (R-функция)
В 1954 г. Уолес и Ван Холде предложи-
ли совместно использовать коэффи-
циент регрессии седиментации kr (па-
раграф Г4.5.4) и характеристическую
вязкость. Они нашли, что эксперимен-
Рис. 1'2.28. Комбинация коэффициента вра-
щательного трения и характеристической
вязкости ( 8 -функция). Для сферы 8 = 2,50
(Sheraga and Mandelkern, 1953)
Рис. Г2.29. Комбинация коэффициента рег-
рессии седиментации и характеристической
вязкости (R-function) (Rowe, 1977)
тальное значение отношения &/|г]|,
очень близко к величине 1,6. Анализ
значений этого отношения у самых
разных но форме макромолекул пока-
зал, что у глобулярных белков они на-
ходятся в интервале 1,5-1,7, в то время
как у более вытянутых частиц они зна-
чительно меньше.
На основании анализа концентра-
ционной зависимости транспортных
процессов Рове предложил R-функ-
цию, которая является отношением
коэффициента регрессии седимента-
ции к характеристической вязкости.
Эта функция представлена на рисунке
Г2.29. Видно, что она изменяется доста-
точно сильно при низких значениях р и,
в принципе, обеспечивает точный ме-
тод вычисления отношения осей частиц
с малой степенью вытянутости. Важ-
но, что при вычислении R-функции не
требуется знания значения абсолютной
концентрации, поскольку она отсутс-
твует в отношении &./[г)].
Среднее гармоническое время
вращательной релаксации
и коэффициент поступательного
трения (^-функция)
Комбинирование гармонического сред-
него времени вращательной релаксации
т /т0и коэффициента поступательного
трения f дает в результате ^-функцию,
зависимость которой от аксиального
отношения сплюснутого и вытянутого
эллипсоидов представлена на рисунке
Г2.30. Видно что ^-функция практичес-
ки нечувствительна к отношению осей
частиц с малой асимметрией (р < 5).
Она подобна 8-функции и может быть
использована для того, чтобы отличить
вытянутый эллипсоид от сплюснутого
для очень асимметричных частиц.
Характеристическая вязкость
и среднее гармоническое время
вращательной релаксации
(Х-функция)
Комбинирование характеристической
вязкости и среднего гармонического
времени вращательной релаксации, дает
Л-функцию, которая представлена на ри-
сунке Г2.31. Эта функция несколько более
чувствительна к отношению осей, чем 'Р-
функция. Основная проблема в ее приме-
нении на практике, особенно для частиц с
небольшой асимметрией (р < 3), связана с
трудностью определения величины т с
высокой точностью (см. Главу Гб).
Коэффициент поступательного трения
и молекулярный ко-объем
(v -функция)
Теоретическая зависимость Т-функции,
основанной на комбинации коэффици-
ента поступательного трения и молеку-
лярного ко-объема, от отношения осей у
вытянутого и сплюснутого эллипсоидов
представлена на рисунке Г2.32. Из ри-
сунка видно, что функции Т может быть
использована для обнаружения разли-
чий между моделями двух видов эллип-
соидов при большем отношении осей
аппроксимирующего эллипсоида.
Характеристическая вязкость
и молекулярный ко-объем (П-функция)
Комбинирование ко-объема и характе-
ристической вязкости дает в результате
функцию, представленную на рисун-
ке Г2.33. Сравнение П- и 'Р-функции
показывает, что первая должна быть бо-
лее чувствительна для определения от-
ношения осей вытянутого эллипсоида.
В таблице Г2.3 суммированы экспе-
риментально измеряемые объем-незави-
Рис. Г2.30. Комбинация коэффициента пос-
тупательного трения и среднего гармони-
ческого времени вращательной релаксации
(*Р -функция) (Squire, 1970)
симые функции формы для двухосного
эллипсоида вращения. Из нее видно, что
явления, основанные на вращательном
трении, более чувствительны к форме час-
тицы, чем таковые, основанные на посту-
пательном трении (комментарий Г2.21).
Рис. Г2.31. Комбинация характеристической
вязкости и среднего гармонического времени
вращательной релаксации (Harding, 1980)
Рис. Г2..32. Комбинация коэффициента по-
ступательного трения и молекулярного ко-
объема, Ч'-функция (Jeffrey et al., 1977)
Рис. 12.33. Комбинация характеристиче-
ской вязкости и молекулярного ко-объема,
П-функция (Harding, 1981)
Ki>мми111.1 pi:i: Г? Pi. Объем-
зависимые и объем-независимые
! функции
Необходимо отметить, что все объем-не-
зависимые функции формы, представлен-
ные в . । , менее чувствительны
к форме, чем объем-зависимые функции,
представленные в .. и: . i •.
Однако платой за эту чувствительность
являются трудности, возникающие как
при выполнении точных измерений ко-
эффициентов вращательного трения, так
и при извлечении необходимых пара-
метров из экспериментальных данных.
Для измерения коэффициентов враща-
тельной диффузии глобулярных белков
(или эквивалентных им коэффициентов
ремени релаксации вращения) электри-
ческое двойное лучепреломление и спад
флуоресцентной анизотропии деполя-
ризации остаются основными методами.
К сожалению, оба метода на практике
имеют некоторые важные ограничения
(см. Главы Г7 и Г9). Более того, эти ме-
тоды сопряжены с проблемой разложе-
ния много-экспоненциальных кривых
на составляющие их экспоненты (см.
параграф ПО.З.З). Заключая анализ всех
представленных в инмиас I 2.5 функций
отметим, что четыре функции, 5 (v, 0), R
(v, ks), Л (v, xh/xQ) и П (v, Vpim) достаточно
чувствительны к отношению осей и мо-
гут быть использованы на практике.
Г2.5.3. Функции формы для
трехосного эллипсоида,
не зависящие от его объема
В 1936 г. Перрен получил точное выра-
жение коэффициента поступательного
трения для трехосных эллипсоидов, а
в 1981 г. Хардинг, Дампье и Рове полу-
чили аналогичное выражение для вязко-
сти. Эти два результата открыли путь
TciCviHLia Г2.5. He зависящие от объема экспериментальные функции формы
частицы
Не зависящие от объема функции формы Комбинация методов Чувствительность к форме
0-функция (v,///„) (Sheraga — Mandelkern, 1953) Вязкость и трансляционное трение Плохая во всем интервале осей
5-function (v, 0/0.,) (Sheraga — Mandelkern, 1953) Вязкость и вращательное трение Хорошая при малых акси- альных отношениях
/{-функция (v, k) (Rowe, 1977) Вязкость и коэффициент регрессии в седиментации Хорошая при малых акси- альных отношениях
У- Функция (f/f„, т,плр/т„) (Squire, 1971) Трансляционное трение и среднее гармоническое вращательное релаксацион- ное время Плохая во всем интервале осей
Л-функция (v, т , /т0) (Harding, 1980) Вязкость и среднее гармони- ческое вращательное релакса- ционное время Хорошая за исключением очень маленьких аксиаль- ных отношений (р < 2)
Ч'-функция (///,, V ) (Jeffrey et al., 1977) Трансляционное трение и ко-объем Плохая
П-фуикция (v, V|[|m„) (Harding, 1981) Вязкость и ко-объем Хорошая за исключением очень маленьких аксиаль- ных отношений (р < 3)
получения объем-независимых функций
формы в рамках трехосного эллипсоида.
Для этих функций характерно одно об-
щее свойство: при данных гидродинами-
ческих свойствах они имеют линейное
решение для всех возможных значений
отношений осей {а/Ь, Ь/с) (коммента-
рий 1'2.22). На рисунке 1'2.31 представ-
лены линейные решения для двух функ-
ций v и ///0, гипотетических трехосных
эллипсоидов с отношениями осей {а/Ь,
Ь/с) = (2.0, 2.0). Из рисунка видно, что
Комментарий Г2.22. Нахождение
единственного решения
Заданные значения осей {а/h. Ь/с) фик-
I сируют значение отношения ///„ (р), ио
I заданным значениям///() (р) соответствует
! ряд «линейных решений», со своими значе-
ниями осей (а/b, Ь/с). Уникальное реше-
ние для этих двух отношений осей может
быть найдено в точках пересечений двух
или более этих «линейных решений».
кривые пересекаются под очень малым
углом, что затрудняет нахождение точ-
ного решения. Очевидно, что мы долж-
Рис. 1'2.31. Графики заданных значений
функций Симха v(p) и Перрена F (р) (а/Ь,
Ь/с) — в плоскости, соответствующей гипо-
тетическому эллипсоиду с осями (а/Ь, Ь/с) =
= (2.0, 2.0) (Harding. 1995)
Димеры неирофизина
а б
Рис. Г2.35. Графики функций R и Л (а/Ь, b/с) — в плоскости для мономеров неирофизина
(а) и его димеров (б). Мономеру соответствует вытянутая модель, в которой отношения осей
приблизительно равны (а/Ь, b/с) = (4.0, 1.0), в то время как у димера, являющегося более ком-
пактной частицей, отношение (а/Ь, b/с) приблизительно равно (2.8, 2.5). Это указывает на то,
что образование димера происходит скорее всего по принципу «спина к спине», а не по типу
«хвост к хвосту» (Harding, 1995)
ны найти такие две объем-независимые
функции формы, пересечение которых
является как можно более ортогональ-
ным. Наилучший результат был найден
для комбинации двух функций (R и Л),
включающих вязкость, коэффициент
регрессии седиментации и среднее гар-
моническое время релаксации вращения.
На рисунке Г2.35 показана зависимость
функций R и Л (а/Ь, b/с) для мономе-
ров нейрофизина (а) и его димеров (б).
Такая комбинация линейных решений
была использована для нахождения наи-
более вероятного типа димеризации это-
го белка.
В заключение отметим следующее.
Проблема, поставленная еще Онсли —
сделать выбор между удлиненным и
сплюснутым эллипсоидом при низ-
ком значении отношения осей, харак-
терном для большинства глобулярных
белков, — на сегодняшний день все
еще далека от своего решения в силу
специфики гидродинамических урав-
нений и существующей на сегодня
точности измерений.
Г2.5.4. Приближение «целого тела»
и моделирование тела «бусинками»
Из изложенного выше следует, что в
сегодняшней гидродинамике имеются
два основных подхода для определения
структуры биологических макромоле-
кул. Один из них, моделирование струк-
туры с помощью бусинок с последу ющим
вычислением ее гидродинамических
параметров — характеристической вяз-
кости, поступательной и вращательной
диффузии. Затем эти экспериментально
определенные параметры сравниваются
с таковыми, теоретически рассчитанны-
ми для модели. Впоследствии модель со-
вершенствуется, с тем чтобы ее реальные
параметры совпали с рассчитанными.
Недостатком такого подхода является
то, что обнаруженным параметрам мо-
жет соответствовать не одна, а несколь-
ко моделей. На практике предлагаемая
модель используется в качестве старто-
вой модели с целью определения в даль-
нейшем ее структуры с более высоким
пространственным разрешением.
Второй подход базируется на вы-
числении структуры в соответствии
с известными гидродинамическими
свойствами. В этом подходе, получив-
шем название метода «целого тела»,
жесткая частица представляется в
виде трехосного эллипсоида с тре-
мя различающимися по длине полу-
осями. Уникальная структура может
быть предсказана из комбинации трех
соответствующих эксперименталь-
ных измерений, чтобы исключить
проблему, связанную с определением
гидродинамического объема (молеку-
лярная гидратация) и подобрать пару
отношений осей, соответствующих
трехосному эллипсоиду. Основным
препятствием при таком подходе яв-
ляется недостаточная чувствитель-
ность объем-независимых функций
при малых значениях отношений осей
и недостаточная точность экспери-
ментальных измерений.
Несмотря на значительные успехи
в развитии гидродинамических мето-
дов, особенно в области моделирования
структур биологических макромолекул,
их основным недостатком всегда будет
низкая разрешающая способность. По-
скольку гидродинамические методы не
требуют особых затрат и относятся к
«неразрушающим» методам, то они мо-
гут быть использованы для получения
моделей низкого разрешения, предва-
ряющих применение методов высокого
разрешения, таких как рентгеновская
кристаллография и ЯМР.
Г2.6. Гомологичные ряды
На рисунке 1'2.36 приведены три типич-
ных гомологичных ряда биологических
макромолекул. Первый ряд — сфери-
ческие частицы, сохраняющие свою
форму с ростом молекулярной массы,
второй — палочкообразные частицы с
длинной осью, увеличивающейся про-
порционально молекулярной массе, в то
время как длина поперечной оси сохра-
няется постоянной, и третий пример —
гауссовы клубки, асимметрия которых
постепенно повышается с увеличением
молекулярной массы.
Зависимость коэффициента по-
ступательного трения от молекулярной
массы М в гомологичных рядах обычно
выражается уравнением Куна-Марка-
Хаувинка (сокращенно КМХ):
Рис. 1 2.36. Гомологичные ряды макромолекул различной формы: а сферы; о - палочкообразные
молекулы, присоединяющиеся друг к другу по типу «голова-хвост»; в — модели гауссовых клубков
/=к^х. (Г2.59)
где k и X — константы. Как будет пока-
зано ниже, величина степенного множи-
теля Xзависит от формы макромолекул
в гомологичных рядах.
В соответствии с законом Стокса для
ряда сферических частиц их константа
Комментарий Г2.23. Острогой
интерпретации показателя степени
1/з в гомологичных рядах ।
При работе с гомологичными рядами
сферических частиц удобно представить
коэффициент трения в форме / = 6пт]0 .
(ЗУЛ/4п)''3. Отсюда следует, что коэффи-
циент трения для сферических частиц со
сходными величинами гидратации и парци-
ального удельного объема пропорционален ;
М1/3. Однако обратное неверно. Величина
показателя степени 1/3 для гомологичных ।
серий частиц не означает, что изучаемые мак-
ромолекулы действительно имеют сфериче-
скую форму. Так, например, в гомологичном
ряду эллипсоидов с фиксированным значе-
нием отношения осей показатель X также •
равен 1 /3. Строго говоря, показатель степени
1/3 означает только одно: частицы в гомоло-
гичном ряду сохраняют свою форму.
поступательного трения пропорциональ-
на молекулярной массе в степени 1/3
(ком.мен lapiu'i 12.23). Для гомологичного
ряда палочкообразных частиц величина
степенного множителя X в уравнении
Г2.59 равна приблизительно 0,85.
Для гомологичных рядов гауссовых
клубков в «идеальном» растворителе
показатель X равен 0,5, а в «хорошем»
растворителе он увеличивается до 0,8. По-
скольку коэффициент поступательного
трения может быть определен с помощью
экспериментально измеренных констант
седиментации и диффузии, то уравнение
(Г2.59) можно использовать для предска-
зания их зависимости от молекулярной
массы для различных гомологичных ря-
дов. Примеры использования уравнения
Г2.59 для определения формы различных
биологических макромолекул будут при-
ведены в Главах ГЗ и Г4.
По аналогии с поступательным тре-
нием зависимость коэффициента вра-
щательного трения от молекулярной
массы М в гомологичных рядах может
быть представлена в виде степенной за-
висимости типа:
e = keMy, (Г2.60)
где k и Y — константы, как и в случае
поступательного трения, зависящие от
формы макромолекулы. В соответс-
твии с формулой Перрена (уравне-
ние Г2.2) для ряда сферических частиц
их константа вращательного трения
пропорциональна молекулярной массе
в степени, равной 1. Для гомологич-
ного ряда непроницаемых гауссовых
клубков в «идеальном» растворителе
Y = 1,5. Для гомологичных рядов бес-
конечно тонких (L » d) палочкооб-
разных частиц Y = 3,0. Таким образом,
как и в случае поступательного трения,
чем быстрее растет асимметрия с рос-
том молекулярной массы, тем больше
величина показателя степени в уравне-
нии Г2.60.
Коэффициент вращательного тре-
ния частицы может быть определен из
экспериментально полученных коэф-
фициентов вращательной диффузии
или вращательных времен релаксации.
Поэтому уравнение Г2.60 можно исполь-
зовать для предсказания зависимости
этих коэффициентов от молекулярной
массы для различных гомологичных
рядов. Примеры определения формы
различных биологических макромоле-
кул с помощью уравнения Г2.60 приве-
дены в Главах Гб и Г8.
Г2.6.1. Характеристическая
вязкость
Зависимость характеристической вяз-
кости от молекулярной массы в гомо-
логичном ряду также может быть пред-
ставлена уравнением типа:
[т|]= |п| Л/а, (Г2.61)
где k и а — константы. Из уравнения
Г2.56 следует, что жесткие эллипсоиды
или палочкообразные частицы, напри-
мер такие, которые имеют постоянное
значение V/Мв гомологичном ряду, бу-
дут иметь зависимость [г|] от М, подоб-
ную зависимости v (р) от М (уравнение
Г2.52). Например, если масса и, следова-
тельно, размеры частиц увеличиваются
так, что их форма остается постоянной,
то v (р) = константа, [г|] = константа
и, следовательно, показатель степени
а в уравнении (Г2.61) равен нулю, не-
зависимо от формы частиц. С другой
стороны, для палочкообразных частиц
(цилиндров), длинная ось L которых
увеличивается пропорционально М, в
то время как диаметр d остается пос-
тоянным, величина асимметрии р уве-
личивается пропорционально М и, со-
ответственно, зависимость [г| j = f (М)
совпадает с зависимостью v = f (р).
Для таких молекул, при больших р, за-
висимость, приведенная в уравнении
Г2.56, совпадает с зависимостью урав-
нения Г2.51.
Для гомологичных рядов вытяну-
тых эллипсоидов, которые имеют оди-
наковую короткую ось, уравнение Г2.55
может быть аппроксимировано уравне-
нием:
v = 0,233 р 169820 < р < 100
или (Г2.62)
v = 0,2071732 2 0 < р < 300.
Таким образом, v должно увеличи-
ваться с ростом молекулярной массы
или длины частицы в степени 1,7.
Если, однако, при увеличении мас-
сы частиц увеличивается их диаметр
b при постоянной длине L, тогда а. бу-
дет < 0. Таким образом, как и в первых
двух случаях, чем быстрее растет асим-
метрия с ростом молекулярной массы,
тем больше величина показателя степе-
ни в уравнении Г2.61. Примеры опреде-
ления формы различных биологических
макромолекул с помощью уравнения
Г2.61 будут даны в Главе Г9.
Приложение 1. О корректности
уравнения Кирквуда-
Райзмана
В основу уравнения Г2.9 положено пред-
ставление о сферах как источниках трения
при гидродинамическом взаимодействии.
Это действительно для случаев, когда раз-
меры этих сфер значительно меньше, чем
расстояния между ними. Для большин-
ства изучаемых биологических структур
это условие не выполняется. Оно не вы-
полняется также и для четырех бусинок,
в классическом примере в уравнении
Г2.9, где размер каждой из них прибли-
зительно равен расстоянию между ними.
Возможно несколько простых вариантов
упаковок: линейная, тетраэдральная и
квадратно-плоская. Ниже представлены
вычисления для первых двух.
В этом случае набор расстояний между
сферами есть 27?О, 47?0, 67?0. Расчет по
формуле Г2.9 дает
f =___________4х6тгг|оДо_________
1+ (_^+Х+А.)-
4 х блгЛо/?^ v 272ц 47?o 67^
Поскольку fn = 6лг|07?0, то
Результаты компьютерных вычис-
лений поступательного трения для ли-
нейных тетрамеров при использовании
оболочечной модели (см. ниже) с экс-
траполяцией к бесконечно малому диа-
метру бусинки (при этом число буси-
нок на поверхности частицы стремится
к бесконечно большому числу) показы-
вают, что вычисления по формуле Кир-
квуда дают ошибку около 6 %.
Тетраэдрический тетрамер (тетраэдр)
2R.
В этом случае все расстояния равны 27?О.
Расчет по формуле Г2.9 дает:
4х6лт|()/?и
~п бтпцЛ ( 12 ’
4Х6ТЛ1Л 2V
А = 6пт1(А
/= 4
= 1,6
4 2/?н
4
4
Результаты компьютерных вычис-
лений поступательного трения для
тетраэдра при использовании оболо-
чечной модели с экстраполяцией к
бесконечно малому диаметру бусинки
(при этом число бусинок стремится
к бесконечно большому числу) пока-
зывают, что вычисления по формуле
Кирквуда дают еще большую ошибку,
около 12%. Эти результаты показыва-
ют, что вычисления по формуле Кирк-
вуда являются наименее точными при
малых расстояниях между сферами и
тесном контакте частиц (наиболее плот-
ная упаковка — это тетраэдрическая
упаковка). При увеличении расстояния
между сферами можно ожидать улуч-
шения точности при применении фор-
мулы Кирквуда.
В заключение заметим, что уверен-
ное суждение о типе упаковки частицы
может быть сделано в двух случаях: в
случае наиболее компактной тетра-
эдрической упаковки, когда коэффи-
циент трения наименьший, и в случае
наиболее вытянутой линейной упа-
ковки, когда коэффициент трения наи-
больший. Во всех остальных случаях,
включая плоско-квадратичную, сделать
однозначный вывод о типе упаковки не
представляется возможным.
Обратите внимание на то, что в этих
вычислениях полученные числа явля-
ются отношением трения частиц к тре-
нию сфер равного объема. Следователь-
но, ///0 следует разделить на константу
41/3 = 1,5873.
Г2.7. Перечень ключевых идей
• Коэффициент поступательного трения сферы пропорционален ее радиусу
и вязкости растворителя, в котором она движется.
• Коэффициент поступательного трения двухосного эллипсоида вращения зави-
сит от его объема и отношения осей.
• Поступательное трение частиц с «ломаной» поверхностью (различные типы
круговых цилиндров, кубы...) может быть вычислено только приблизительно.
• Для моделирования гидродинамического трения жестких частиц произвольной
формы применяются три различных, ио родственных подхода: в методе «буси-
нок» целая частица аппроксимируется с помощью «бусинок» (сфер) одного
или разных размеров; в модельном подходе «оболочек» поверхность частицы
аппроксимируется набором малых равновеликих сфер; в модельном подходе
«граничных элементов» поверхность частицы моделируется набором элемен-
тов из малых треугольных панелей (чешуек).
• Связь между гидродинамикой и электростатикой обеспечивает простой и точ-
ный способ вычисления коэффициента поступательного трения жестких час-
тиц произвольной формы.
• Коэффициент поступательного трения слабо чувствителен к сегментальной
подвижности; для ломаной палочки (две жесткие палочки, связанные шарни-
ром) он изменяется всего на 3 % от такового для жесткой палочки того же раз-
мера и слабо зависит от положения шарнира.
Прямое вычисление коэффициента поступательного трения жестких мик-
роскопических объектов можно определить из движения их макроскопиче-
ских моделей в растворителе с высокой вязкостью при низких числах Рей-
нолдса.
Вращение сферы в приближении Стокса может быть охарактеризовано одной
константой, которая имеет размерность времени.
Коэффициент вращательного трения двухосного эллипсоида вращения зави-
сит от его объема и отношения осей.
Поступательное трение коротких частиц «ломаной» формы (цилиндры, кубы
и т. п.) может быть вычислено только приблизительно.
Вращательное трение гораздо более чувствительно к размерам частицы, чем
поступательное трение.
Для длинных вытянутых частиц из константы вращательной диффузии можно
вычислить длинную ось молекулы.
Вязкость растворов биологических макромолекул, т|, всегда больше вязкости чис-
того растворителя, т|0. Доля увеличения вязкости называется удельной вязкостью
и обозначается как г] = Т|/г|0—1 = т|от11 -1, где г|отп есть относительная вязкость.
Характеристическая вязкость [г] ] определяется как увеличение вязкости рас-
твора, обусловленное добавлением 1 грамма макромолекул; она может быть
получена экспериментально при экстраполяции г) /С к нулевой концентра-
ции и имеет размерность — кубический сантиметр на грамм или децилитр на
грамм.
Удельная вязкость раствора сфер пропорциональна объемной доле частиц и не
зависит от абсолютного их размера.
Характеристическая вязкость двухосного эллипсоида вращения зависит от его
объема, отношения осей и описывается уравнением Симха.
Характеристическая вязкость белков может быть вычислена с хорошей точно-
стью, исходя из их атомных координат при корректном учете гидратации.
Связь между гидродинамикой и электростатикой обеспечивает простой метод
вычисления характеристической вязкости частиц правильной и неправильной
формы.
Характеристическая вязкость белков и коротких фрагментов ДНК может быть
наиболее просто вычислена с использованием моделей «бусинок» или «чешуек».
• Гидродинамические параметры макромолекул (характеристическая вязкость,
коэффициенты поступательной и вращательной диффузии) зависят от их фор-
мы и гидродинамического объема (включая гидратацию); отсюда следует, что
определить форму или объем измерением одного гидродинамического пара-
метра невозможно.
• Комбинированием разных гидродинамических параметров можно получить
семь (р, 5, R , Т, Л, и П) объем-независимых функций формы, каждая из
которых имеет различную чувствительность к отношению осей аппроксимиру-
ющих их эллипсоидов.
• Все объем-независимые функции формы менее чувствительны к форме, чем их
объем-зависимые предшественники.
• Для трехосного эллипсоида наилучшая комбинация двух объем-независимых
функций формы (R и Л) приводит к наилучшим результатам при вычислении
всех его осей.
• Для гомологичного ряда сферических частиц коэффициент поступательного
трения пропорционален их молекулярной массе в степени 1/3, а для палочко-
образных частиц — в степени 0,85.
• Для гомологичного ряда сферических частиц коэффициент вращательного
трения пропорционален их молекулярной массе в степени, равной 1, а для тон-
ких палочкообразных частиц — в степени равной 3. Аналогичная степень для
гауссовых клубков равна 1,5.
• Для гомологичного ряда сферических частиц характеристическая вязкость не
зависит от их молекулярной массы, тогда как для гомологичного разряда па-
лочкообразных частиц она зависит от молекулярной массы в степени, равной
1,7-1,8.
• Несмотря на значительные успехи в развитии теории гидродинамических ме-
тодов, они остаются методами низкого пространственного разрешения.
Литература для дальнейшего чтения
Частицы правильной формы
Harding S. Е. On the hydrodynamic analysis of macromolecular conformation.
Biophys. Chem., 1995. 55, 69-93
Garcia de la Tone J., Carrasco B. and HardingS. E. SOLPRO: theory and computer
program for the prediction of SOLution PROperties of rigid macromolecules and
bioparticles. Eur Biophys J., 1997. 25, 361-372.
Garcia de la Torre J. and Bloomfield V. A. Hydrodynamic properties of complex, rigid,
biological macromolecules: theory and applications, Quart. Rev. Biophys., 1981. 14,
81-13Г2.
Garcia de la Tom J. Hydrodynamic properties of macromolecular assemblies. In
Dynamic Properties of Biomolecular Assemblies/eds. S. E. Harding and A. J. Rowe,
1988. p. 3-31. Nottingham: The Royal Society.
Частицы произвольной формы
Swanson E., TellerD. C. and de Haen C. The low Reynolds number translation friction
of ellipsoids, cylinders, dumbbells, and hollow spherical caps. Numerical testing of the
validity of the modified Oseen tensor in computing the friction of objects modelled as
beads on a shell. J. Chem. Phys., 1978. 68, 5097-5102.
Carrasco B. and Garcia de la Tom J. Hydrodynamic properties of rigid particles:
comparison of different modelling and computational procedures. Biophys. J. 1999. 75,
3044-3057.
Allison S. A. Boundary element modelling of biomolecular transport. Bioph. Chem.,
2001.93, 197-213.
Трансляционное трение и электростатическая емкость
HubbardJ. В. and DouglasJ. F. Hydrodynamic friction of arbitrarily shaped Brownian
particles. Phys. Rev., 1993. 47, 2983-2986.
Zhou H.-X. Calculation of translational friction and intrinsic viscosity. I. General
formulation for arbitrarily shaped particles. Biophys. J., 1995. 69, 2286-2297.
Частицы с известной трехмерной структурой
Venable R. M. and Pastor R. IE. Frictional models for stochastic simulations of
proteins. Biopolymers, 988. 27, 1001-1014.
Garcia de la Tom J., Huertas M. L. and Carrasco B. Calculation of hydrodynamic
properties of globular proteins from their atomic level structure. Biophys. J., 2000. 78,
719-730. Rigid particles with segmental mobility.
Garcia de la Tom J. Hydrodynamics of segmentally flexible macromolecules. Eur.
Biophys. J., 1994. 23, 307-322.
Определение формы молекул из трансляционного трения: гомологичные
ряды
Tsvetkov V. N. Rigid-chain polymers. Hydrodynamic and Optical Properties in
Solution. New York: Consultants Bureau. 1989.
HearstJ. E. Rotary diffusion constants of stiff-chain macromolecules. J. Chem. Phys.,
1963.38, 1062-1065.
Garcia de la Tom, J. and Bloomfield, V. A. Hydrodynamic properties of complex,
rigid, biological macromolecules: theory and applications. Quart. Rev. Biophys., 1981.
14,81-139.
Hagerman P.J. and Zimm B. Monte Carlo approach to the analysis of wormlike
chains. Biopolymers, 1981.20, 1481-1502.
Harding, S. E. On the hydrodynamic analysis of macromolecular conformation.
Biophys. Chem., 1995. 55, 69-93.
Garcia de la Tom J. Hydrodynamics of segmentally flexible macromolecules. Eur.
Biophys. J., 1994. 23, 307-322.
Allison S. Low Reynolds number transport properties of axisymmetric particles
employing stick and slip boundary conditions. Macromolecules, 1999. 32, 5304-5312.
Глава ГЗ
МАКРОМОЛЕКУЛЯРНАЯ ДИФФУЗИЯ
Г3.1. Исторический обзор
1850
Т. Грэхэм (Т. Graham) обнаружил,
что яичный альбумин диффундирует в
водных растворах гораздо медленнее,
чем такие распространенные вещества,
как соль или сахар. В 1861 г. он применил
диализ для разделения смеси медленно
и быстро диффундирующих растворен-
ных веществ и количественно измерил
диффузию у многих соединений. На ос-
нове полученных данных он классифи-
цировал вещества как коллоиды или
кристаллоиды.
1855
А. Фик (A. Fick), в уравнениях, из-
вестных как его первый и второй законы,
ввел понятие коэффициента диффузии
D для описания потока растворенных ве-
ществ вдоль их градиента концентрации.
Дж. Стефан (J. Stefan) в 1879 г. и Л. Бо-
льцман (L. Boltzmann) в 1894 г. предста-
вили второй закон в интегральной форме,
тем самым открыв путь для эксперимен-
тального определения коэффициентов
диффузии различными методами.
1905 1906
А. Эйнштейн, В. Сазерленд и
М. ван Смолуховский (A. Einstein,
W. Sutherland and М. von Smoluchow-
ski) обнаружили, что решения уравне-
ния Фика для стационарных условий
аналогичны решениям для потоков
тепла вдоль температурного градиен-
та и установили взаимосвязь между
коэффициентами диффузии частиц и
их трением в броуновском движении.
В 1905 г. Сазерленд использовал зна-
чение коэффициента диффузии, вы-
численное Дж. Стефаном (J. Stefan)
по данным, собранным Т. Грэхэмом, и
получил значение молекулярной массы
для яичного альбумина, равное 33 кДа
(истинное значение — около 45 кДа).
1926
Л. Мандельштам (L. Mandelshtam)
пришел к пониманию, что коэффициент
поступательной диффузии макромоле-
кул может быть определен из спектров
рассеяния света. Однако отсутствие
пространственной когерентности и не
монохроматическая природа обычных
источников света предопределили не-
возможность проведения таких экспе-
риментов вплоть до 1964 г., пока Г. Кам-
мингс, Н. Кнабль и Й. Йе (Н. Cummins,
N. Knable and Y. Yeh) не использовали
лазер в методе оптического смешения,
с тем чтобы определить спектральный
состав света, рассеянного раствором
латексных сфер. В 1967 г. С. Дубин,
Дж. Луначек и Г. Бенедек (S. Dubin,
J. Lunacek and G. Benedek) измерили
коэффициенты поступательной диффу-
зии бычьего сывороточного альбумина,
лизоцима, вируса табачной мозаики
и ДНК.
1927
Т. Сведберг (Т. Svedberg) показал,
что молекулярная масса белка может
быть вычислен, исходя из его констан-
ты седиментации,парциального удель-
ного объема и коэффициента диф-
фузии. Это стимулировало развитие
новых усовершенствованных методов
и экспериментального оборудования
для исследования процесса диффузии
как такового.
1938 1960
За эти годы были разработаны раз-
личные экспериментальные методы для
измерения коэффициентов диффузии
малых и больших биологических мак-
ромолекул (метод шкалы Ламма, метод
шлирена Филпота и Свенссона, интер-
ферометрические методы Жамена, Гуи,
Рэлея и Лебедева).
1972-197/1
Д. Магде, Л. Элсон и В. В. Вебб
(D. Magde, L. Elson and W. W. Webb)
математически обосновали процессы
в методе флуоресцентной корреля-
ционной спектроскопии, подчеркнув
его широкие возможности для из-
мерения коэффициентов диффузии.
В 1990 г. Р. Риглер (R. Rigler) с со-
авторами впервые осуществили де-
тектирование одиночных молекул,
объединив метод флуоресцентной
корреляционной спектроскопии с ме-
тодом конфокальной флуоресцентной
микроскопии.
Середина 1970-х
Для измерения коэффициентов
диффузии флуоресцентно- меченых мо-
лекул был развит метод восстановления
флуоресценции красителя после фо-
товыцветания, который впоследствии
оказался идеальным инструментом
для определения двумерной латераль-
ной подвижности молекул в мембранах
и в индивидуальных живых клетках.
2000 и по настоящее время
Наряду с экспериментальными ме-
тодами, перечисленными выше, со-
временная гидродинамика позволяет
предсказать с высокой точностью ко-
эффициенты диффузии биологических
макромолекул с известной структурой.
Г3.2. Коэффициент
поступательной диффузии
Коэффициент поступательной диф-
фузии, Dho(i, может быть определен
различными способами. Исторически
сложилось так, что первое определе-
ние коэффициентов диффузии было
проведено методом расширяющейся
границы. В этом методе наблюдают
за изменением концентрации моле-
кул в макроскопическом объеме под
действием градиента осмотического
давления (рис. Г.3.1). Такой коэффи-
циент диффузии получил название
D . Позже появились другие спосо-
бы определения коэффициентов по-
ступательной диффузии. Один из них
получил название коэффициент сле-
довой диффузии, D.ici. Он вычисляет-
ся из наблюдения за одной частицей в
микроскопическом объеме (рис. Г.3.2).
Экспериментально D определяется
методами флуоресцентной корреля-
ционной спектроскопии (Глава Г11),
ние. 1 3.2. Диффузия как хаотическое движе-
ние одной частицы
Рис. Г3.1. Диффузия как макроскопическое
изменение концентрации вещества
нановидной микроскопии (пара-
граф ГЗ.5.2) и флуктуации числа час-
тиц (параграф Г.10.5.1). Другой коэф-
фициент диффузии получил название
коэффициента взаимной диффузии
£>В.)11М1) поскольку описывает взаимное
движение частиц в микроскопическом
объеме (риг. ГЗ.З). Экспериментально
£>В;)11М|1 определяется методом динами-
ческого рассеяния света в условиях
Гауссовой статистики (большое число
частиц) (параграф ПО.3.3).
В том случае, когда частицы не
взаимодействуют, все три типа измере-
Рис. I 3.3. Диффузия как совместное движе-
ние частиц в ансамбле
ний дают один и тот же коэффициент
поступательной диффузии, который
исторически получил название D ,
с тем чтобы отличать его от коэффици-
ента вращательной диффузии, £>вращ.
В lai'i.iime ГЗ.I представлены раз-
личные гидродинамические методы,
Таблица 13.1. Методы, используемые для определения коэффициентов
диффузии биологических макромолекул
Способ наблюдения Метод Диффузионный коэффициент
Наблюдение среднего микроско- пического движения частиц в ансамбле. Прямое наблюдение диффузи- онного движения одиночной флуоресцентной частицы. Стохастическое появление и исчезновение частиц в малом объеме. Стохастическое появление и ис- чезновение частиц в очень малом объеме Восстановление флюоресценции красителя после выцветания (Секция Г3.5). Конфокальная флуоресцентная микроскопия (Секция Д4.6.1). Флуктуация числа частиц в методе динамического рассеяния света (Секция Г10.5). Флуоресцентная корреляционная спектроскопия (Секция Г.11.3) Следовый D след Следовый D след Следовый D след Следовый D след
Взаимное движение частиц в ан- самбле Динамическое рассеяние света (Секция Г. 10.3) Взаимный D В. ШИМ
Макроскопическое изменение концентрации частиц Метод расширяющейся границы (Секция Г3.5) Трансляцион- ный D пост
используемые для определения коэф-
фициентов поступательной диффузии.
В ней приведены обсужденные выше
подходы, экспериментальные мето-
Комментарий ГЗ. 1.
, Среднеквадратичная скорость I
i частиц при броуновском движении
При абсолютной температуре Т сред-
няя кинетическая энергия частицы, при- |
холящаяся на одну степень свободы, рав-
на kT/2, (где k — константа Больцмана).
Частица с массой m и скоростью и имеет
кинетическую энергию ти2/2. В среднем
<ти2/2> = kT/2, где < > обозначает усред-
1 нение по времени или по ансамблю. Из это-
го выражения мы можем вычислить сред- ,
, неквадратичную скорость, равную: |
<u2> = kT/m (А) '
и корень квадратный из ее значения
<ui2>'/2 = (kT/m)i2. (Б) |
Комментарий ГЗ.2. Диффузия ;
i и скорость I
Из уравнения (А) комментария Г3.1
можно вычислить скорость, которую имеет
i в данный момент маленькая частица. При- j
ведем два примера.
Сахароза в вакууме
У сахарозы молекулярная масса рав-
на 342 Да. Это масса одного моля или
6,02хЮ23 молекул; масса одной моле-
кулы равна т = 5,7х10~23г. Значение kT
при комнатной температуре, 293 К, рав- .
но 4,04 х К)-'4 гсм2сек2. Следовательно, |
<ur2>1/2 = 8,ЗхЮ3см-сек-1. При отсутствии ;
столкновений молекулы сахарозы будут
пересекать большое помещение типа пла-
вательного бассейна приблизительно за
1 сек. В соответствии с уравнением (Б)
комментария Г3.1, скорость частицы об-
ратно пропорциональна корню квадратно-
му из ее молекулярной массы.
Биологические макромолекулы
Для белков с молекулярной массой 2,Ох 104
Да скорость равна 1,09 х 103 см сек'1. Для Д НК
с молекулярной массой 9хЮ8 Да скорость >
равна 33 см-сек *.
ды и типы коэффициентов диффузии.
Приведены также главы и секции, где
можно найти детальное описание ме-
тодов и их применение. Как мы уже
отмечали, для невзаимодействующих
частиц три коэффициента, Dc , £>ih.iiiw
и D|U)l.T, эквивалентны друг другу.
ГЗ.З. Микроскопическая
теория диффузии
Диффузия макромолекул в растворе
может быть представлена как явление,
связанное с броуновским движением.
Тепловая энергия молекул в растворе
обеспечивает их постоянное хаотиче-
ское движение (комментарий 1'3.1).
Как было показано Эйнштейном
в случае одномерной диффузии средне-
квадратичное смещение <г-’> пропор-
ционально времени Г:
<^> = 2£>/ (Г3.1)
и
<^>./2 = (2£у) I (Г3.2)
Уравнения (Г3.1) и (Г3.2) показы-
вают, что из величины среднеквадра-
тичного смещения <х2> можно опре-
делить коэффициент диффузии и
наоборот. Будем считать, что движения
в трех направлениях — х, у и z являются
независимыми. Если <х2> = 2D/, тогда
<г/2> = 2D2t, <z?> = 2D3t. Квадрат рассто-
яния от начала координат до точки (х,
у) в двумерном случае равен г1 = х2+у2,
и,следовательно:
<г?> = 4£)Г, (ГЗ.З)
где D есть среднее значение коэффици-
ентов диффузии D{ и D2 для двумерного
беспорядочного движения.
Для трехмерного случая г2 = х2 +
+ у2 + z2, и среднеквадратичное смеще-
ние равно:
<^> = 6Dt, (Г3.4)
где D есть среднее значение (Dv+ D2 +
+ £>3) коэффициентов диффузии для
трехмерного беспорядочного движения.
Опять же, знание величины <г2> доста-
точно для определения коэффициента
диффузии D и наоборот.
Примеры вычислений скорости
диффузии и зависимости диффузии
от времени даны в к<>.ммсн ириих 1'3.2
и Г.3.3.
Г3.4. Макроскопическая
теория диффузии
ГЗ.4.1. Первое уравнение Фика
Первое уравнения Фика гласит, что
суммарный поток Jx всегда пропор-
ционален первой степени градиента
концентрации растворенного вещества
Рис. Г.3.1. Поток молекул справа налево
обусловлен градиентом концентрации. Поток
возникает из-за того, что число частиц слева
меньше числа частиц справа
dC/dx, где D — константа пропорцио-
нальности:
Jx = -D [dC/dx], (Г3.5)
Если частицы распределены рав-
номерно, то наклон кривой градиента
концентрации равен нулю, т. е. dC/dx =
= 0 и система находится в равновесии.
Если наклон является постоянным,
т. е. dC/dx = константа, Jxтакже постоя-
Комменшрий ГЗ.З. Диффузия и время
Из уравнения (Г3.2) можно вычислить постоянную скорость диффузионного движения
малой частицы. Рассмотрим несколько примеров таких частиц, диффундирующих в водной
! среде. I
Мочевина 1
Коэффициент диффузии мочевины в воде равен 118 см2 сек’’ ( нюл 1.3.3). Частица с таким 1
коэффициентом диффузии диффундирует па расстояние .г = 10 4 см за время t =.v!/2D = 5x10"*
i сек, или около 0,5 мсек. На расстоянием = 1 см она диффундирует за время 5х105сек, или около
| 14 часов. Очевидно, что диффузионное перемещение частицы на большое расстояние занимает I
гораздо большое время. i
Глобулярный белок
Коэффициенты диффузии D порядка 10 6см2-сек_1 соответствуют небольшим глобулярным
। белкам ( i;h> I. ГЗ.З). Такие белки диффундируют на расстоянием = 1 х Ю 1 см за время Г= 5
I х Ю :’сек, или около 5 мсек. Для диффузии на расстоянием = 1 см это время составит 5 х Ю5 .
сек, или около 138 часов.
Вирус табачной мозаики
Вирус табачной мозаики имеет коэффициент диффузии, равной 0,44 х Ю 7с.м2 сек1 и диф-
| фундирует очень медленно. Так, расстоянием = 0,5 см он будет проходить за время t =106 сек,
। или около 700 часов. Этот факт объясняет, почему метод расширяющейся границы, в котором
минимально необходимое для измерения диффузии расстояние равно приблизительно не-
скольким миллиметрам, никогда не используется для измерения коэффициентов диффузии
, таких больших молекул.
.v л'+5
Рис. I 3.3. Поток через поверхность тонкого
бокса, имеющего размеры от.гдол-г 6. Площадь
каждой поверхности равна A (Berg, 1983)
С = 0
Рис. Г3.6. Диффузия молекул из объема жид-
кости, изначально содержащего частицы в кон-
центрации С(1 (внизу слева) в объем жидкости,
изначально не содержащей частиц (вверху
слева). Распределение молекул по прошест-
вии определенного времени (справа)
нен. Случай, когда С является линейной
функцией х, показан на рис. 1’3.4.
его справа. Число частиц на единицу
объема в боксе, следовательно, увели-
чивается на:
1/т [С (t+T)-C (т) ] =
= - 1Л|./1(.г+5)-/>(.г)]Дт/Л5 =
= - 1/5 I./, (x+8)-Jr(x) ].
При условии 5^0 и т->0 ото означа-
ет, что:
dC/dt = -dJx/dx (Г3.6)
или, если использовать первый закон
Фика (уравнение Г3.5), то:
dC/dt = D d-’C/dx’. (Г3.7)
Из уравнения Г3.7 следует, что ско-
рость изменения концентрации за еди-
ницу времени, dC/dt, пропорциональ-
на второй производной концентрации
раствора, d’C/dx2, где D есть константа
пропорциональности. Для диффузии в
трех измерениях концентрация изменя-
ется во времени как:
dC/dt = D V2C. (Г3.8)
где V2 — трехмерный Лапласиан, d^dv1 +
+ d1 dy2 + d^dz1.
Для сферически симметричного
случая поток будет радиальным:
J=-DdC/dr, (Г3.9)
и
dC/dt = D(\/г1) d (t3 dC/dr)/dr (Г3.10)
ГЗ.4.2. Второе уравнение Фика
Второе уравнение Фика следует из
первого при том условии, что общее
число частиц в системе постоянно,
т. е. они ие возникают и не разрушают-
ся. На рисунке Г3..5 рассматривается
бокс объемом А. За период времени т,
Jr(x)xAT частиц будут входить в объем
А слева, a Jx (х+5) х Ат будут покидать
Г3.4.3. Нестационарные решения
уравнения Фика
Предположим, что диффузия проис-
ходит из объема жидкости изначально
содержащего частицы в концентрации
Со (внизу слева) в объем жидкости из-
начально не содержащий частиц (рис.
ГЗ.б). В этом случае начальные условия
имеют вид: С = Со для х > 0, С = 0 для .г
Рис, I .3.7: а — концентрация как функция расстояния за время t = tn, t = t.vt = t.,. 11ача.тьная кон-
центрация частиц в период времени ф равна С’,. Через бесконечное время концентрация вырав-
нивается н становится равной С„/2; б — производная ио концентрации как функция расстояния
за время Г = Г(|, t = tt, t = t., для такого же процесса, что и в а
< 0 и уравнение (Г3.8) будет иметь ре-
шение:
С Г 2
C(x,t) = — 1—гтх
2 L Р
,r/2(Dr)' 2
х j ехр(-г/2)огг/
О
(Г3.11)
Интеграл в уравнении (Г3.11) из-
вестен как интеграл вероятности. Он
является функцией х/2 (Dt)' 2 и изме-
няется от 0 до !/2. Уравнение (Г3.11)
в графическом виде представлено на
рис. Г3.7 а. Взяв производные от С (х,
t) по х или t, мы получим:
dC/dx = CQ/(/vtDt)' 2 х
х exp (-.v2/4£V). (Г3.12)
Графическое изображение процес-
сов, описываемых уравнением Г3.12,
приведено на рисунке Г.3.7 б. Уравнение
показывает, что максимальное значение
dC/dx достигается при ,т = 0, и будет рав-
но С0/(4л£>?)1/2.
ГЗ.4.4. Стационарные решения
уравнения Фика
Для стационарного случая, когда
dC/dt = 0, уравнение (Г3.8) может быть
записано как:
V2C-0, (Г3.13)
которое в случае сферической симмет-
рии уравнение имеет вид:
l/rd(^dC/dr)dr=O. (Г3.14)
Особый интерес представляет слу-
чай, связанный с процессом диффузии
на адсорбирующей сфере. Предполо-
жим, что такая сфера имеет радиус /?0 и
находится в бесконечном объеме, как по-
казано на рисунке Г.3.8. Каждая частица,
достигающая поверхности сферы, захва-
тывается так, что концентрация частиц
на поверхности сферы при г = R{} равна
0. При г= оо эта концентрация равна Си.
В таких граничных условиях уравнение
(Г3.13) имеет следующее решение:
C(>) = C0O-Rtt/r). (Г3.15)
Рис. Г.3.8. Адсорбирующая сфера радиусом Ro,
находящаяся в среде бесконечного объема, содер-
жащего частицы с исходной концентрацией Со.
Прерывистые линии обозначают линии потока
Тогда поток частиц согласно урав-
нению Г3.9 равен:
JT(r)=DC0R0/r>. (Г3.16)
Если мы определим диффузионный
поток I как I = 4п г2Jr (?), тогда
1 = 4пГ)7?0С0. (Г3.17)
В уравнении Г3.17 концентрация
Со выражена как число частиц в кубиче-
ском сантиметре, а диффузионный по-
ток I — как количество частиц в секунду.
Как видно из уравнения Г3.17 этот поток
I пропорционален не величине поверх-
ности сферы, а ее радиусу. Это происте-
кает из того факта, что когда радиус сфе-
ры Ro увеличивается, то ее поверхность
растет как RQ2, но при этом градиент кон-
центрации, которому пропорционален
поток, уменьшается как 1 /RQ.
Важно заметить, что уравнение
(Г3.13) аналогично уравнению Лапласа
для электростатического потенциала в
незаряженном пространстве. Это оз-
начает, что диффузионный поток, на-
правленный в сторону изолированного
поглотителя какой-либо формы и раз-
мера, может быть описан как:
I=4nZ)cC0, (Г3.18)
где с -- электроемкость изолированно-
го проводника той же формы и размера,
которая в системе сгс выражается в сан-
тиметрах (сxi. кох!\нч।:.11>i।!i 13./ ). Подо-
бие уравнений Г3.17 и ГЗ. 18 очевидно.
Уравнение Г3.18 имеет большое зна-
чение на практике, поскольку электроем-
кость тел различной формы либо извес-
тна, либо легко может быть вычислена.
Последнее обстоятельство позволяет за-
менить трудоемкие гидродинамические
вычисления частиц разной формы более
легкими вычислениями электростатичес-
ких аналогов. Примеры таких вычисле-
ний были даны ранее в параграфе Г2.2.3.
Г3.5. Экспериментальные
методы определения
коэффициентов диффузии
ГЗ.5.1. Метод расширяющейся
границы
В методе расширяющейся границы пер-
воначально устанавливается резкая гра-
ница между раствором определенной
концентрации и чистым растворителем.
Концентрация, С, измеряется как фун-
кция расстояния, х, которое пройдено
от границы за данное время (уравне-
ние ГЗ. И). Значение коэффициента
диффузии D за каждый промежуток
времени t вычисляется из распределе-
ния С (.г).
Альтернативный подход базируется
на уравнении Г3.12, которое описывает
зависимость dC/dx от х (см. рис. Г3.7 б).
В этом случае максимальная высота
кривой, равна &С0/(4п£)Г)1/2, а пло-
щадь, А, под кривой равна kC. Тогда ко-
эффициент диффузии вычисляется по
простой формуле:
(A/Я 1аи.)2 = 4пЯГ. (Г3.19)
Run icoTPuMP Восстановление
Рис. Г3.9. Восстановление флуоресценции красителя после фотовыцветания. Флуоресциру-
ющие молекулы в малом объеме (белый кружок) выцветают под действием кратковременной
лазерной вспышки высокой интенсивности. Восстановление флуоресценции, обусловленное
диффузией частиц, не подвергшихся выцветанию, измеряется как функция времени (Bastiaens
and Pepperkok, 2000)
ГЗ.5.2. Метод восстановления
флуоресценции после выцветания
красителя
В методе, называемом восстановлением
флуоресценции после выцветания кра-
сителя (ВКПВ), наблюдение ведется за
частицами, мечеными флуоресцентным
красителем.
Метод прост как концептуально,
так и практически. Малый (по срав-
нению с общим) избранный объем, со-
держащий подвижные флуоресцент-
ные молекулы, подвергается действию
короткого мощного светового импуль-
са, с тем чтобы обеспечить разруше-
ние флуорофора, присоединенного к
молекуле (рис. Г3.9). После этого в
объем натекают флуоресцирующие
молекулы из оставшегося объема, не
подвергшегося действию лазерного
импульса.
В результате последующего обмена
между выцветшими и не выцветшими
флуоресцирующими популяциями мо-
лекул в объеме происходит восстанов-
ление уровня свечения объема прак-
тически до исходного. Построение
графика зависимости восстановления
относительной интенсивности флу-
оресценции как функции времени по-
зволяет вычислить коэффициент диф-
фузии меченых молекул (рис. ГЗ.Юг/).
Важно отметить, форма кривой ВКПВ
может быть также использована для
анализа характера подвижности моле-
кул в исследуемом объеме, поскольку
существует четко выраженное разли-
чие в ходе кривых для хаотического и
направленного движения (рис. Г3.10 б).
Если разные частицы мечены разны-
ми красителями, то их коэффициенты
диффузии могут быть получены отде-
льно наблюдением за каждым из них.
Метод восстановления флуорес-
ценции после выцветания красителя
наиболее широко используется для ис-
следований латеральной диффузии на
плоскости, соответствующей обшир-
ным областям цитоплазматических
мембран, реконструированных мемб-
ран и тонкослойных растворов. Этим
методом было показано, что различные
поверхностные белки и гликопротеиды
могут перемещаться латерально внут-
ри мембраны. Были проведены прямые
экспериментальные измерения коэф-
фициентов диффузии различных мемб-
ранных белков in situ. Полученные зна-
чения находились в пределах от 5 х 10-9
см2-сек 1 для родопсина в фоторецеп-
торных мембранах до 1 х 10 12 см2 сек~*
I ’нс. 1.3.1 (I: a - идеализированный график зависимости интенсивности флуоресценции /от вре-
мени, демонстрирующий количественную сторону В КП В метода. Начальная интенсивность вы-
деленного объема / измеряется до действия световой подсветки небольшой интенсивности. За-
тем объем подвергается действию короткого мощного импульса длительностью (г — г0). Начиная
с времени 10 рост интенсивности восстановленной! флюоресценции измеряется до тех пор, пока
она не выйдет иа плато и не достигнет значения / . Кажущийся коэффициент диффузии £>ku.
может быть определен разными методами, включающими в себя как метод определения того вре-
мени, при котором интенсивность достигает половины значения интенсивности /ф, так и метод
полной подгонки кривой (сплошная линия) (Bastiaens and Pepperkok, 2000); б — теоретические
кривые восстановления флуоресценции /к (/) для трех разных случаев: чистая диффузия, на-
правленный поток и суперпозиция первых двух (Koppel, 1979)
для флуоресцентно меченых белков на
поверхности эритроцитов человека. Ве-
личины 1 х 10 " см2-сек 1 были обнару-
жены для флуоресцентно меченого кои-
кавалина А рецепторного комплекса иа
мембране из культивированных миоб-
ластов крысы.
Г3.5.3. Метод привязки золотых
кластеров(«нановидная
микроскопия») В
В 1988г. М. Шитс (М. Sheets) разра-
ботал метод определения коэффици-
ентов диффузии биологических мак-
ромолекул с использованием малых
коллоидальных частиц золота, присо-
единенных к исследуемой молекуле.
За траекторией таких молекул можно
следить при помощи светового микро-
скопа. Метод получил название «нано-
видной микроскопии» (микроскопии
с разрешением на уровне нанометра).
Годом позже он применил этот метод
для исследования подвижности неко-
торых белков на поверхности живой
клетки (лектин, конканавалин А), на-
блюдая их с помощью за движением
частиц коллоидного золота диаметром
40 нм (рис. ГЗ. 1 1).
В заключение отмстим, что коэф-
фициенты диффузии макромолекул,
определяемые методами восстановле-
ния флуоресценции после выцветания
красителя и нановидной микроскопии,
строго говоря не имеют прямого отно-
шения к размерам и форме меченой мо-
лекулы. Они чаще всего характеризуют
Рис. I .'J. I I. Слева. Визуализация следов диффузионного движения частицы при направленном дви-
жении (а) и при беспорядочном движении частицы (б). Виет ре. Зависимость среднее-квадратичного
отклонения частицы от времени для диффузионного направленного движения и для беспорядочно-
го движения, построенная поданным (а) и (б). Справа. Теоретическая зависимость средне-квадра-
тичного отклонения частицы от времени дня трех случаев: свободная диффузия плюс направленное
движение (а), чисто свободная диффузия (б) и диффузия в ограниченном пространстве (в)
подвижность молекул в специальных
условиях, например при очень высоких
концентраций окружающих молекулу
других белков как в случае первого ме-
тода или привязки тяжелой! частицы,
как в случае второго метода.
Г3.6. Предсказание
коэффициентов диффузии
глобулярных белков
Гидродинамическое поведение белков
определяется различными фактора-
ми: их размером, неровностью повер-
хности, поверхностной гидратацией
заряженных и полярных групп. Су-
ществуют две стратегии предсказания
коэффициентов диффузии для белков,
трехмерная структура которых извес-
тна с высоким пространственным раз-
решением из кристаллографических
или ЯМР данных. Первая включает
детальный учет констант трения всех
атомов белка, доступных действию
растворителя. При таких вычислени-
ях необходимо учитывать не только
атомы молекулы белка, но и молекулы
воды, ассоциированные с ней. Во вто-
рой стратегии вода, ассоциированная
с белком, представлена в виде водной
оболочки с толщиной, одинаковой для
всех белков (секция Г1.3).
В настоящее время разработаны ав-
томатизированные методы обращения
координат атомов в дискретные цент-
ры трения, в роли которых выступают
либо бусинки, либо треугольные плас-
тинки. Детали такого моделирования
представлены в Главе Г2. Сравнение
предсказанных таким моделированием
констант диффузии с эксперименталь-
но измеренными для различных белков
представлено в пюпще Г.1.2. Из нее
видно, что имеющиеся сегодня алго-
ритмы, позволяют предсказывать ко-
эффициенты диффузии с точностью
до 1-2%. Особо примечательным яв-
ляется тот факт, что «окружение» всех
глобулярных белков тонкой оболочкой
воды(1 ± 0,1 А) с одинаковой толщиной
также приводит к хорошему согласию
между предсказанными и эксперимен-
тальными данными. Плотность воды в
окружающей оболочке для диффузии
не имеет значения.
Таблица Г3.2. Предсказанные и экспериментальные значения коэффициентов диффузии глобулярных белков
и вируса табачной мозаики
Частица Код белка в Банке белко- вых структур Молекулярная масса (кДа) Метод конструи- рования Предсказанный ^20w (ед. Фика) Измеренный Д20» (ед. Фика)
Бычий трипсиновый ингибитор 4 pt 1 6,4 Пара бусинок** 14,4 14.4
Рибонуклеаза А ЗгпЗ 13,6 Пара бусинок ** 11,1 11,2
Рибонуклеаза Л 7rsa 13,7 Емкость*** 11,1 11,2
Лизоцим 61ys 14,3 Емкость*** 11,2 11,2
Лизоцим llz3 14,0 Пара бусинок** 11,3 11,1
Лизоцим 611ys. brk 14,0 Дискретизацтя**** 11,7 11,1-11.2
Профилин Ipne 14,8 Пара бусинок** 11,0 10,6
Миоглобин Imbo 17,2 Емкость*** 10,4 10,3
Целлюлаза leng 22,0 Пара бусинок ** 9,7 9,8
Химотрипсиноген 2cga 25,66 Емкость*** 9,2 9,1-9.5
Инсулин laio 34 Пара бусинок ** 8,0 7,9
Нитрогеназа avl 220 Одна бусинка * 4,33 4,0
Вирус табачной мозаики (ВТМ) Круговой ци- линдр (2800 Ax 180 A) 40,000 Дискретизация на многоугольники 0,44 0,44
*) В этом методе одна бусинка (одна сфера) с радиусом 6,6 А располагается на месте Сь атомов каждой аминокислоты (Baranowicz et
al., 2000).
**) В этом методе пара бусинок (идентичные и частично перекрывающиеся сферы) каждая с радиусом 4,5Л располагается на месте С?
атомов каждой аминокислоты (Baranowicz et al., 2000).
***) Диффузионные коэффициенты белков вычислены методом электростатической емкости с допущением, что однородная гидрата-
ционная оболочка имеет толщину 0,9 A (Zhou, 1995).
****) Для конструирования подходящей модели для гидродинамических вычислений сфера радиусом 3 А прокатывается по поверхно-
сти частицы, с тем чтобы избавиться от мелких шероховатостей поверхности. Сглаженная поверхность затем разделяется на многоуголь-
ники, составленные из треугольных пластин.
Г3.7. Трансляционное
трение и диффузионные
коэффициенты
ГЗ.7.1. Уравнение
Эйнштейна—Смолуховского
Кинетическая теория диффузии, пред-
ставленная выше, позволяет вычислять
значения коэффициентов диффузии
частиц путем определения скорости, с
которой они движутся под действием
внешней силы. На практике эта скорость
пренебрежимо мала, по сравнению с сред-
неквадратичной скоростью, приведенной
в уравнении (А) комментария Г3.1. Это
значит, что частицы диффундируют быс-
трее, чем при отсутствии поля, но с малым
направленным смещением вдоль оси, как
показано на рисунке ГЗ. 12.
Сейчас мы выведем уравнение, свя-
зывающее коэффициент диффузии час-
тицы D с ее коэффициентом трения, /,
используя подход Берга, названный им
«беспорядочным блужданием со сме-
щением». Рассмотрим частицу массой
т в положении х, на которую действует
внешняя сила, приложенная в направ-
лении +х. В соответствии с определе-
нием, коэффициент поступательного
трения является коэффициентом про-
порциональности в уравнении:
Fx=fux, (Г3.20)
где F является силой, действующей в
направлении х, а их есть среднее значе-
ние скорости в этом направлении. С дру-
гой стороны, в соответствии со вторым
законом Ньютона, сила заставляет час-
тицу двигаться слева направо с ускоре-
нием а = Fx/m. В соответствии с подхо-
дом беспорядочного блуждания частица
делает один скачок за время т вправо при
начальной скорости +их или влево, с на-
чальной скоростью — их.
т
О ““^Fx
х+5 х х+5+
Рис. 13.12. Частица массой т, находящаяся
под действием внешней приложенной силы
Fx и испытывающая одномерное блуждание
вдоль оси х
Частицы, начинающие свое движе-
ние из положения х с начальной ско-
ростью +их, пройдут за время т рассто-
яние, равное 8+= их + а х1 /1, в то время
как частицы, начинающие свое движе-
ние из положения х, с начальной скоро-
стью -иг, за время т пройдут расстояние
8 = -их + а т2/2. Поскольку шаги вправо
и шаги влево равновероятны, то среднее
смещение за время т будет равно а т2/2,
а средняя скорость частицы при движе-
нии слева направо будет:
их = 1/2 а т = 1/2 F т/т. (Г3.21)
В модели беспорядочного блуж-
дания f = 2т/х Умножая числитель
и знаменатель уравнения Г3.21 в виде
мг=7//на(8/т2) и вспоминая, что их = 8/т
и D = 82/2 мы получаем, что/ = mux/D.
Но согласно уравнению (А) коммен-
тария Г3.1 тих = kT; поэтому / = kT/D,
или
D = kT/f. (Г3.22)
Этот результат известен как уравне-
ние Эйнштейна-Смолуховского
менripiiii ГЗ. i) иявляется принципиаль-
но важным для гидродинамики. Важно
подчеркнуть, что уравнение не зависит
от каких-либо допущений, сделанных
относительно структуры частицы или
деталей ее движения. Частица всегда
Комменшрий I 3,4. К выводу
уравнения Г3.22
Как указывается в книге Берга, вы-
вод уравнения (Г3.22) не имеет строго-
го физического обоснования. Конечно,
в действительности частицы не делают
шаги с фиксированными интервалами и
с фиксированной скоростью. Существу-
ет перераспределение интервалов шагов,
направлений их движения, и скоростей в
силу обмена энергии между молекулами.
Однако конечный результат оказывается
тот же самый. Это происходит потому,
что в результате приложения внешних
сил частицы ускоряются, но поскольку
прилагаемые силы малы (см. параграф
Г1.2.2), то при обмене молекулами энер-
гией это ускорение нивелируется. Это и
есть реальное движение молекул при низ-
ких числах Рейнолдса, когда силы инер-
ции малы (Berg, 1983)
• Комментарий Г3.5.
Концентрационная зависимость
, коэффициентов диффузии
Концентрационная зависимость коэф-
фициентов диффузии может быть охарак-
। теризована коэффициентом k в уравне-
' НИИ I
, DC = DO(1-^Q. (А)
Для низких концентраций растворенных |
веществ коэффициент k,. является суммой
двух вкладов — «исключенного объема» и
эффективного гидродинамического тре- !
ния и является аналогом коэффициента
i k , который описывает концентрационную
зависимость коэффициента седиментации .
j (laiMMi'ii । арий 1 1. H'S). И хотя в величины I
kD и ^вносятся оба вклада, однако их сум-
марный результат разный, поскольку в яв- ।
j лении седиментации они входят с одинако- I
выми знаками, а в явлении диффузии -- с
разными. Поэтому константа k обычно
; мала по сравнению с константой k. I
i __________________________ _
движется через жидкость со скоростью
пропорциональной силе, приложенной
извне. В случае диффузии константа
пропорциональности равна D/kT. В слу-
чае седиментации эта константа равна
s/m (1 — ц р0) (Глава Г4). В случае элек-
трофореза эта константа равна p/Q где
р — электрофоретическая подвижность
частицы, a Q — ее заряд (Глава Г5).
Уравнение Эйнштейна—Смолухов-
ского показывает, что измерение ко-
эффициента диффузии D позволяет
вычислить коэффициент трения ко-
торый дает нам информацию о размере
и форме молекулы.
ГЗ.7.2. Диффузионные
коэффициенты биологических
макромолекул
Коэффициенты диффузии макромоле-
кул зависят от вязкости растворителя и
температуры, при которой выполнены
измерения. Считается общепринятым
приводить измеренные коэффициенты
диффузии к стандартным условиям, в
качестве которых выбирается темпера-
тура 20 °C и вязкость чистой воды при
20°, nw:
Э20.„ = »1(293/П(т1;м(..Т/П„ 2(1). (Г3.23)
В этом уравнении Э.,(| w является кон-
стантой диффузии в чистой воде при
20 °C, Т — абсолютная температура,
r|w,,() — вязкость воды при температу-
ре 20"С, а г] т — вязкость реального
раствора при измеренной температуре
Т. Следует отметить, что уравнение Г3.23
не имеет какой-либо теоретической ос-
новы и базируется в основном на фено-
менологических соображениях. Следует
отметить, что величина D,o w может быть
вычислена даже в том случае, если био-
логическая макромолекула не может су-
ществовать в чистой воде при 20 °C.
Коэффициенты диффузии биоло-
гических макромолекул обычно вычис-
ляются при конечной концентрации и
должны быть экстраполированы к нуле-
вой концентрации (коммен lapnii I З.э).
Г3.7.3. Зависимость
коэффициентов диффузии
глобулярных белков
от молекулярной массы
Для сферических частиц комбинация
уравнения Эйнштейна—Смолуховского
(Г3.2) и уравнения Стокса (Г2.1) откры-
вает возможность вычисления радиуса
сферы Ro, исходя из экспериментально
измеренных коэффициентов диффузии
D и вязкости растворителя р0:
R{. = ^Г/6пт|0Э. (Г3.24)
Из О и т|() можно вычислить Ro,
и если плотность частицы р известна,
то ее масса будет равна:
тп = 4/3 и /?03р, (Г3.25)
а молекулярная масса М:
М = 4/3 п Д/р Ул. (Г3.26)
Сравнение уравнений Г3.24 и Г3.26
показывает, что коэффициент диффузии
обратно пропорционален корню кубиче-
скому из молекулярной массы сфериче-
ских частиц (см. также секцию Г2.6):
D-M-'l (Г3.27)
На рисунке ГЗ. 13 показана зависи-
мость экспериментальных значений
коэффициентов диффузии от молеку-
лярной массы, построенная по данным
н|б.шц|,| ГЗ..З, для глобулярных белков,
начиная с тетрадекапептида (соматоста-
тин) и кончая большими белками (оли-
гомерный белок глутаматдегидрогеназа
из печени быка) в двойной логарифми-
ческой шкале.
Рис. ГЗ. 13. Зависимость экспериментальных
значений коэффициентов диффузии от мо-
лекулярной массы для глобулярных белков в
двойном логарифмическом масштабе, постро-
енная поданным : !') Illi:!.: I '). !
Наклон прямой, проведенной через
экспериментальные точки, равен 0,336,
что хорошо согласуется с наклоном 1 /3,
предсказываемым уравнением Г3.27 для
сферических частиц.
ГЗ.7.4. Зависимость
коэффициентов диффузии ДНК
от молекулярной массы
На рисунке ГЗ. 14 показана зависимость
коэффициентов диффузии от молеку-
лярной массы ДНК в В-форме, построен-
ная поданным mu. iimi.i 1 Г 1. Для моле-
кул ДНК размером от 70 пар оснований
(молекулярная масса -46 кДа) до 6000
пар оснований (молекулярная масса
-4 МДа) эта зависимость описывается
одной прямой линией:
£) = 276М°72. (Г3.28)
Значение показателя степенной зави-
симости (0,72) близко к теоретическому
значению, ожидаемому для гомологич-
ного ряда палочкообразных частиц ко-
нечной толщины (см. секции Г2.6, Г4.10.3
и Г6.7.1). Интересным является также
то, что гидродинамические свойства
Таблица ГЗ.З. Коэффициенты диффузии и молекулярные массы белков*)
Белок Молекулярная масса (Да) Коэффициент диффузии (в ед. Фика)
1. Стоматостатин (тетрапептид) 1,636 24,5
2. Грамицидин(димер) 2,500 18,4
3. Бычий трипсиновый ингибитор 6,400 14,4
4. Липаза 6,669 14,5
5. Рибонуклеаза А (бычья панкреотическая) 12,640 13,1
6. Рибонуклеаза А 13,690 11,2
7. Цитохром с 13,370 11,4
8. Лизоцим 13,930 11,2
9. Лизоцим 14,320 11,2
10. Профилин 14,800 10,6
11. Миоглобин 17,190 10,3
12. Целлюлаза 22,000 9,8
13. Химотрипсиноген А 23,240 9,5
14. Химотрипсиноген А 25,660 9,0
15. Инсулин 34,400 7,9
16. Карбоксипептидаза В 34,280 8,2
17. а-Лактоглобулин 35,000 7,2
18. Овальбумин 45,000 7,8
19. Сывороточный альбумин 68,600 6,4
20. Гемоглобин 68,000 6,9
21. Цитратсинтетаза 97,938 5,8
22. Лактатдегидрогеназа 133,000 5,1
23. Глицератофосфат дегидрогеназа 136,800 5,0
24. Альдолаза 156,000 4,8
25. Нитрогеназа 220,000 4,5
26. Каталаза 250,000 4,1
27. Апоферритин 467,000 3,6
28. Уреаза 482,700 3,5
29. Глютаматдегидрогеназа 1 015 000 2,5
w) Диффузионные коэффициенты взяты из разных источников: 1) Zipper and Durchshlag
(2000); 2) Banachowicz et al. (2000); 3) Zhou (2001); 4) Hellweg et al. (1997); 5) Smith (1970);
6) Garcia de la Torre (2001); 7) Byron (1997); 8) Brune and Kim (1993)
коротких фрагментов ДНК (12-20 пар
оснований) сходны с таковыми для
глобулярных белков соответствующей
молекулярной массы (см. рис. Г3.14).
В то же время эти свойства очень отли-
чаются от таковых у молекул большого
молекулярного веса. Этот факт отражает
принципиальные различия между двумя
гомологичными рядами. В ряду глобу-
лярных белков асимметрия с увеличени-
ем молекулярной массы остается посто-
янной, в то время как в ряду фрагментов
ДНК асимметрия растет с ее увеличени-
ем. Читатель может обнаружить это же
явление в секции Г4.10.3, в которой об-
суждаются седиментационные свойства
молекул ДНК.
ГЗ.7.5. Предел применения закона
Стокса: «малые», «средние»
и «большие» молекулы
На рисунке ГЗ. 15 показана зависимость
коэффициентов диффузии от молекуляр-
Таблица Г3.4. Диффузионные коэффициенты гомогенных молекул ДНК
в В-форме
Частица Молекулярная масса (Да) Диффузионный коэффициент (ед. Фика)
8 н. о. 5304 15,26
12 п.о. 7956 13,41
20 п. о. 13260 10,86
89 н. о. 59007 4,27
104 п. о. 68952 3,88
124 п. о. 82212 3,41
2311 п.о. 1532193 0,46
ДНК - pUC19 1829880 0,35
ДНК-pDSl 2538637 0,29
ДНК (М„./М„< 1.15 3730000 0,223
Данные взяты из: Eimer and Ресога (1991); Tirado et al., (1984); Sorlie et al. (1988); Seils and
Dorfimiller (1991); Jolly and Eisenberg (1976)
ной массы для разнообразных веществ,
начиная от молекул газов до больших мак-
ромолекул в двойной логарифмической
шкале на основе данных, представленных
в таблицах 1.3.3. 13.1 и ГЗ..”). Как видно
из рисунка, зависимость подразделяет-
ся на три различные области. Область I
описывает зависимость коэффициентов
диффузии глобулярных белков от их мо-
лекулярной массы в широких пределах
(параграф Г3.7.3). Напоминаем, что на-
клон прямой в этой области равен 0,336.
Область II представляет аналогич-
ную зависимость для аминокислот и
сахаров в интервале значений молеку-
лярных масс от глицина (М = 75 Да) до
рафинозы (М = 504 Да). Наклон этой
прямой равен 0,47. И наконец, область III
(2Да — 100 Da) описывает движение га-
зов и малых молекул в воде: от молеку-
лы водорода Н2 (М = 2 Да) до глицерина
(М = 92 Да). В этом случае зависимость
не является монотонной функцией
М. Проведенная прямая в этой области
имеет наклон, равный 0,42.
Рис. ГЗ. 14 Зависимость коэффициентов диф-
фузии от молекулярной массы для глобуляр-
ных белков (•) и для молекул ДНК в В — фор-
ме (О) в двойном логарифмическом масштабе.
Для последних интервал молекулярных масс
простирается от 8 пар оснований до 6000 пар.
Данные взяты из пи ' зи. Г’...'! и I 3. i, принимая
парциальные удельные объемы ДНК и бел-
ка равными 0,56 см’ г1 и 0,73 см3 г ', соответ-
ственно
Три различных области I—III на
рисунке Г3.15 соответствуют трем
различным типам диффузионного
движения в жидкостях. Область I со-
ответствует движению «больших» мо-
Молекулярная масса (Da)
Рис. I 3.1э. Зависимость коэффициентов диффузии от молекулярной массы М для различных
веществ в двойной логарифмической шкале. Данные взяты из i.iO.nm II и i.;.H
лекул в растворителе в режиме «пото-
ка», когда последний рассматривается
как континуум. Для молекул таких
размеров их движение описывается
при низких числах Рейнолдса (см.
примеры в к< >.м Xi * • i 11; i ] > 11; । \ ГЗ.З и ГЗ. 1).
В этом случае движение молекул под-
чиняется закону Стокса с граничны-
ми условиями полного прилипания
(уравнение П.11). Отклонение от
линейности начинается в районе 1000
Да. Это означает, что закон Стокса
применим к молекулам с молекуляр-
ной массой 1000 Да и более (см. рис.
Г3.15). Такие молекулы мы отнесем к
категории «больших» молекул.
Таблица Г3.5. Диффузионные коэффициенты аминокислот и сахаров в водных
растворах
Молекула Молекулярная масса (Да) Коэффициент диффузии (в ед. Фика)
Глицин 75 94,3
Аланин 89 81,0
Пролин 97 78,1
Пентаэритриол 136 67,8
Фенилаланин 147 62,7
Глюкоза 180 59,8
Маннитол 182 58
Сахароза Рафиноза 324 46,3
504 38,6
Краситель Алеха 600 30
Данные взяты из: 1) Longsworth, 1953 и пересчитаны иа стандартные условия; 2) Nir and Stein,
1970; 3) Kelly et al., 1942
Таблица 1'3.6. Коэффициенты диффузии некоторых газов и малых молекул
Молекула Молекулярная масса (Да) Коэффициент диффузии „ (ед. Фика)
11, 2 510
Не 4 540
11,0 18 220
Ne 20 250
к. 28 190
О, 32 201
Лг 40 190
СО, 44 177
С1„ 71 122
Мочевина 60 118
Глицерин 92 83
Данные взяты из Nir и Stein (1971)
Область III относится к «решеточ-
ной» моде движения «малых» молекул
в жидкости. В этом случае молекулы
движутся в среде, состоящей из частиц,
сходных с ними по размеру (случай
самодиффузии). В таком растворите-
ле молекулы перемещаются способом
«прыгни и жди». Следовательно, моле-
кулы «не ощущают» растворитель как
непрерывную среду. Значение коэф-
фициента диффузии, вычисленное из
уравнения Эйнштейна—Смолуховско-
го, будет гораздо больше, чем значение,
предсказанное законом Стокса. Такие
молекулы мы будем называть «малы-
ми» молекулами. Их молекулярная
масса не более 100 Да.
Область II — это переходная область
между двумя модами диффузии в жид-
Г3.8. Перечень ключевых идей
костях. Здесь молекулы передвигаются
через растворитель по смешанному ме-
ханизму. Область II постепенно перехо-
дит в область III с переломом в районе
1000 Да (см. рис. Г3.15). Молекулы с
массой в интервале значений от 100 до
1000 Да мы определим, как молекулы
«среднего» размера.
Для молекул «среднего» разме-
ра в условиях «полного скольжения»
уравнение Г3.24 является наиболее
приемлемыми, однако на практике оно
не используется.
Из изложенного строго следует, что
истинные гидродинамические размеры
частицы могут быть определены только
для молекул «большого» размера, что
соответствует молекулярным массам
1000 и более Дальтон.
• Коэффициент диффузии, определяемый из стохастического движения одной
частицы, называется коэффициентом следовой диффузии D.ie|.
• Коэффициент диффузии, определяемый из взаимного движения частиц в ан-
самбле, называется коэффициентом взаимной диффузии Dnm .
" Коэффициент диффузии, определяемый по макроскопическим изменениям кон-
центрации молекул, называется коэффициентом поступательной диффузии D .
° Для невзаимодействующих между собой броуновских частиц все три коэффи-
циента диффузии, D ,D и £) являются идентичными.
J Согласно первому уравнению Фика общий поток макромолекул пропорциона-
лен наклону градиента концентрации с константой пропорциональности D.
0 Согласно второму уравнению Фика скорость изменения концентрации во вре-
мени пропорциональна градиенту концентрации с константой пропорциональ-
ности D.
° Решение уравнения Фика в стационарных условиях аналогично решению
уравнения Лапласа для электростатического потенциала в незаряженном про-
странстве; отсюда следует, что диффузионные свойства макромолекул могут
быть вычислены с использованием методов электростатики.
° Метод расширяющейся границы позволяет определить коэффициенты посту-
пательной диффузии биологических макромолекул.
* Метод восстановления флуоресценции красителя после выцветания наиболее
подходит для исследований латеральной диффузии в цитоплазматических и
реконструированных мембранах, а так же в тонкослойных растворах и в цито-
плазме.
• Коэффициенты диффузии макромолекул обычно приводятся к «стандартным
условиям», в качестве которых используется чистая вода при 20 °C.
• Коэффициенты диффузии глобулярных белков могут быть вычислены из их
атомных координат с точностью 1-2% при правильном учете гидратации.
* Зависимость коэффициентов диффузии глобулярных белков от молекулярной
массы в двойной логарифмической шкале является прямой линией с наклоном
0,336, что совпадает с теоретическим значением, равным 1 /3 для сферических
частиц.
9 Молекулы с молекулярной массой >1000 Да в гидродинамических экспери-
ментах могут рассматриваться как «большие»; такие молекулы движутся в рас-
творителе, который для них является континуумом.
° Молекулы с молекулярной массой < 100 Да называются «малыми» молекула-
ми; такие молекулы двигаются способом «прыгни и жди» и растворитель для
них не является континуумом.
• Молекулы с молекулярной массой от 100 до 1000 Да называются «средними»
молекулами; они двигаются в растворе по механизму смешанного типа «движе-
ние» — «прыжок-ожидание».
• Уравнение Эйнштейна—Смолуховского—Стокса применимо к биологическим
молекулам, имеющим молекулярную массу >1000 Да.
Литература для дальнейшего чтения
Коэффициенты диффузии
Costing L. J. Measurement and interpretation of diffusion coefficient of proteins.
Adv. Prot. Chem., 1956.11, 429-554.
Микроскопическая теория диффузии
Einstein A. Investigations on the Theory of the Brownian Movement, ed. R. Furth,
transl. hy A. D. Cowper. New York: Dover Publication, Inc. 1956.
BergH. C. Random Walks in Biology. Princeton: Princeton University Press, 1983.
Макроскопическая теория диффузии
BergH. C. Random Walks in Biology. Princeton: Princeton University Press. 1983.
Экспериментальные методы определения коэффициентов диффузии
Costing L.J. Measurement and interpretation of diffusion coefficient of proteins.
Adv. Prot. Chem., 1956.11,429-554.
Axelrod D„ Koppel D. E., Schlessinger J., Elson, E. and Webb IT. IT. Mobility
measurement by analysis of fluorescence photobleaching recovery kinetics. Biophvs. J.,
1976.16,1055-1069.
Kucik D. F., Elson E. and Sheetz M. P. Forward transport of glycoproteins on leading
lamellipoda in locomoting cells. Nature, 1989. 340, 315-316.
Lee G. M., Ishihara A. and Jacobson K. A. Direct observation of Brownian motion of
lipids in a membrane. Proc. Natl. Acad. Sci. USA, 1991. 88, 6274-6278.
Концентрационная зависимость коэффициента диффузии
Rowe A.J. The concentration dependence of transport processes: A general description
applicable to the sedimentation, translation diffusion, and viscosity coefficients of
macromolecular solutes. Biopolymers, 1977.16, 2595-2611.
Harding S. E. and Johnson P. The concentration dependence of macromolecular
parameters. Biochem. J., 1985. 231, 543-547.
Предсказание коэффициентов диффузии для белков и сравнение с экспери-
ментом
Teller D., Swanson Е. and Наеп С. The translation friction coefficients of proteins.
Meth. Enzymol., 1979.61, 103-124.
Venable R. M. and Pastor R. IT. Frictional models for stochastic simulations of
proteins. Biopolymers, 1988. 27, 1001-1014.
Garcia De la Tone J. Hydration from hydrodynamics. General consideration and
applications to bead modelling to globular proteins. Biophys. Chem., 2001. 93, 159—
170.
Zhou H.-X. A unified picture of protein hydration: prediction of hydrodynamic
properties from known structures. Biophys. Chem., 2001. 93, 171-179.
Коэффициенты диффузии и молекулярная масса
Gosling L. J. Measurement and interpretation of diffusion coefficient of proteins.
Adv. Prot. Chem., 1956.11, 429-554.
Einstein A. Investigations on the theory of the Brownian movement, ed. R. Furth,
transl. by A. D. Cowper. New York: Dover Publication, Inc., 1956.
ГЛАВА Г4
АНАЛИТИЧЕСКОЕ
УЛЬТРАЦЕНТРИФУГИРОВАНИЕ
Г4.1. Исторический обзор
НИЗ
А. Думанский (A. Dumansky) пред-
ложил использовать аналитическое уль-
трацентрифугирование (АУЦ) для опре-
деления размеров коллоидальных частиц.
1923
Т. Сведберг и Дж. Б. Николс
(Т. Svedberg and J. В. Nichols) скон-
струировали первую центрифугу с оп-
тической системой для наблюдения за
частицами в поле центрифуги, приво-
димую в действие сжатым воздухом. Го-
дом позже они наблюдали уменьшение
поглощения во время центрифугирова-
ния раствора гемоглобина.
1926
Т. Сведберг провел первые из-
мерения молекулярных масс белков
(гемоглобин, овальбумин) методом
Комментарий Г4.1. Сведберг
и Нобелевская премия
Интересно отметить, что Теодор Свед- •
берг получил Нобелевскую премию за его !
работу ио коллоидальным системам, а не
за изобретение аналитической центрифуги
и становление и развитие метода аналити-
ческого центрифугирования.
равновесного центрифугирования, а в
1927 г. определил молекулярную массу
гемоглобина сочетанием методов седи-
ментации и диффузии. Эти пионерские
работы привели его к пророческому вы-
воду, что белки являются макромолеку-
лами, состоящими из большого числа
атомов, соединенных ковалентными
связями (комментарий ГГ I ).
1929
О. Ламм (О. Lamm) вывел основ-
ное уравнение, описывающее поведение
движущейся границы в поле центрифу-
гирования. Точное решение уравнения
представляло собой серию интегралов,
которые можно было вычислить толь-
ко при помощи численного интегриро-
вания. Позже уравнение Ламма было
решено аналитически для специаль-
ных граничных условий (X. Факсен,
В. Арчибалд и X. Фуджита) (Н. Faxen,
W. J. Archibald and Н. Fujita).
1930
Шлиреновская оптическая систе-
ма была изобретена Дж. Филпотом
и X. Свенсоном (J. St. L. Philpot and
Н. Svenson), и независимо Л. Г. Лонг-
свортом (L. G. Longsworth). Она по-
зволила получить зависимость гради-
ента концентрации (или более точно,
градиента показателя преломления),
как функцию расстояния в ячейке цент-
рифуги. С помощью этой системы были
поставлены первые эксперименты по
седиментации белков для вычисления
их коэффициентов трения в терминах
формы и гидратации. В этом десяти-
летии важным достижением в области
исследования структуры молекул было
предсказание формы и размера вируса
табачной мозаики из седиментацион-
ных измерений до того, как эта палоч-
кообразная частица была визуализова-
на в электронном микроскопе.
1940-е
Для проведения экспериментов
в лаборатории была сконструирована
и стала коммерчески доступной анали-
тическая центрифуга Spinco, модель Е,
приводимая в действие электрическим
мотором. В 1942 г. В. Дж. Арчибальд
(W. J. Archibald) предложил новые под-
ходы для определения молекулярной
массы макромолекул по данным АУЦ.
В 1943 г. Е. Кон и Дж. Эдсалл (Е. J. Cohn
and J. Т. Edsall) показали, что парци-
альный удельный объем белков может
быть успешно вычислен из парциаль-
ных удельных объемов, составляющих
их аминокислотных остатков. Первый
учебник, посвященный аналитическо-
му центрифугированию, который на-
зывался «Ультрацентрифуга» Т. Свед-
берга и К. Педерсена (Т. Svedberg and
К. О. Pedersen), появился в 1942 г. Эта
монография стала Библией аналитиче-
ского ультрацентрифугирования.
1950-е
Эта декада отмечена как начало ши-
рокого применения метода седимен-
тации. Много блестящих достижений
в молекулярной биологии связано с
использованием метода АУЦ. М. Ме-
сельсон и Ф. Сталь (М. Meselson and
F. W. Stahl), используя метод центри-
фугирования в градиенте плотности и
изотопные метки, доказали полуконсер-
вативный механизм удвоения ДНК, а
эксперименты А. Тиссиера и Дж. Уот-
сона (A. Tissier and J. D. Watson,
1958), а также Ф. Чао и X. Шахмана
(F.-C. Chao and H. K. Schachman, 1959)
привели к открытию рибосом. Второй
учебник по ультрацентрифугированию
«Ультрацентрифугирование в биохи-
мии» X. Шахмана был опубликован в
1959 г. и стал на многие годы настольной
книгой для многих исследователей.
19(>0-е
Развиваются первые сканирующие
фотоэлектрические абсорбционные оп-
тические системы. В этот период ультра-
центрифуга стала широко использовать-
ся для изучения белков, рибосом, ДНК и
вирусов. Существенный вклад в теорию
и практику аналитического ультрацен-
трифугирования внесли Дж. Уильямс,
К. ВанХольде, X. Шахман, Д. Ифантис,
X. Фуджита (J. W. Williams, К. Е. van
Holde, Н. К. Schachman, D. A. Yphantis
and Н. Fujita).
1970-е
Эти годы были золотыми для метода
аналитического ультрацентрифугирова-
ния. К началу 1980-х гг. во всем мире име-
лось около 2000 аналитических ультра-
центрифуг. Включение монохроматора
в абсорбционную оптику позволяло ис-
следовать очень разбавленные растворы.
Рэлеевская интерференционная оптика
позволяла получать высокоточные ре-
зультаты для непоглощающих растворен-
ных веществ и использовалась в основном
для определения молекулярной массы
методом равновесного центрифугирова-
ния. В 1971г. введение дифференциалы
Коммун 1арии Г4.2. «Второе
рождение аналитического
ультрацентрифугирования»
X. Шахман в статье, названной «Второе
рождение аналитического ультрацентри-
фугирования», писал:
«...Но к 80-м гг., несмотря на неистовую
активность, связанную с конструированием
новых аналитических ячеек, разработкой
новых оптических систем, развитию новых
подходов к интерпретации эксперименталь-
ных данных применение метода ультрацент-
рифугирования для решения разнообразных
и множественных проблем биологии, резко
сократилось и метод стал терять свою по-
! пулярность. Что случилось? Получили ли
! развитие более надежные и универсальные
методы, которые могли быть использованы
вместо метода ультрацентрифугирования?
Или у исследователей, занимающихся хи-
мией белка и молекулярной биологией в
80-е гг. возникли вопросы, на которые не мог
ответить метод АУЦ? Молекулярные био-
логи в начале 80-х, которые интересовались
молекулярными взаимодействиями между
различными белками или между белками и
нуклеиновыми кислотами, стали удовлет-
воряться ответом “да” или “нет” и перестали .
i интересоваться константами равновесия и \
изменениями свободной энергии. Для них
использование фильтров, колонок с сефадек-
сом, полиакриламидных гелей обеспечивали
желаемую информацию. Сейчас, однако, с
; появившейся возможностью синтезировать
; сотни белков методом сайт-направленного
! мутагенеза, вновь появилась необходимость
в прецизионной технике для физико-хими-
ческих исследований» (Schachman, 1989).
кого метода седиментации открыло путь
для обнаружения небольших различий в
коэффициентах седиментации (М. Кир-
шнер и X. Шахман) (М. W. Kirschner
and Н. К. Schachman). В 1978 г. были
развиты новые методы анализа данных,
которые позволяли исключать вклад
диффузии в скоростную седиментацию,
и давали в результате интегральное рас-
пределение коэффициентов седимента-
ции (К. Ван Хольде и В. Вайшейт) (К.
Е. van Holde and W. О. Weischet). Тре-
тий фундаментальный учебник по уль-
трацентрифугированию «Основы анализа
ультрацентрифугирования» X. Фуджита
(Н. Fujita) появился в 1975 г.
1980-е
В начале 1980-х гг. метод седимен-
тации начал терять популярность среди
биохимиков. Для этого было две при-
чины: первая — развитие методов гель-
электрофореза и гель-хроматографии,
для которых требовалось малое коли-
чество исследуемого материала; вторая
состояла в том, что хотя процесс ультра-
центрифугирования и фиксировался
на компьютере, возможности быстрого
обсчета данных были весьма ограни-
чены. Кроме этого, обработка данных
была весьма трудоемким процессом,
поскольку сводилась к обработке фото-
графий образца (KoMMiMi iapnii Г 1.2).
1990-е
Ситуация драматически изменилась
с появлением нового поколения автома-
тизированных ультрацентрифуг фирмы
Beckman XL. Она содержит две оптиче-
ские системы детектирования: уль-
трафиолетовую (УФ) абсорбционную
систему, которая дает возможность ис-
следовать белки и нуклеиновые кислоты
при очень низкой концентрации (вплоть
до 2-3 мкг-см ') и рэлеевскую интерфе-
ренционную систему, с помощью которой
можно исследовать мало поглощающие
или вовсе непоглощающие макромолеку-
лы. Шлиреновская оптическая система,
по всей видимости, ушла в прошлое, по-
скольку зависимость инкремента показа-
теля преломления от расстояния в ячейке
ультрацентрифуги может быть получена
путем компьютерного дпфференцнрова-
ния интегральных кривых зависимости
показателя преломления от расстояния.
Стали доступны новые программы об-
работки данных, обеспечивающие чис-
ленные решения уравнения Ламма для
сложных (многокомпонентных) систем.
С 2000 но настоящее время
В настоящее время метод АУЦ на-
ходится в состоянии возрождения. Он
может применяться для определения мо-
лекулярных масс молекул от сотен до де-
сятков миллионов Дальтон. Метод АУЦ
является признанным методом для точ-
ного определения чистоты, массы, фор-
мы, самоассоциации и других свойств
молекул в растворе. На сегодняшний
день все это опять определяет востребо-
ванность метода в лабораториях.
Г4.2. Оборудование
и инновационная техника
Ниже мы кратко опишем основные ха-
рактерныечерты современной аналитиче-
ской центрифуги, обращая основное вни-
мание на центрифугу фирмы Beckman,
Optima XL( ко.ммеп iupiiii Г1.3).
Введение в практику в 1990-х гг.
этой центрифуги существенно увеличи-
ло точность измерений и объем данных,
которые могут быть получены в экспе-
рименте. Это стало возможным благода-
ря наличию в ней микропроцессорного
контроля за экспериментальными па-
раметрами и оптики высокого качества.
Кроме оптики поглощения она обору-
дована рэлеевской интерференционной
системой, которая позволяет выполнять
измерения растворов макромолекул
с низким оптическим поглощением.
Основными частями центрифуги яв-
ляются: ротор, находящийся в охлаждае-
мом вакуумном пространстве ее корпуса,
Коммоп।арий Г4.3. Техника
препаративного и аналитического
центрифугирования
Техника центрифугирования в молеку-
лярной биологии представлена двумя ос-
новными типами — препаративным и ана-
литическим. Процедура препаративного
центрифугирования состоит из стадий
разделения, выделения и очистки биоло-
гического материала (клеток, органелл,
нуклеиновых кислот и т. и.) и его после-
дующего исследования биохимическими
и физическими методами.
Коммешарпи Г4.4. Сила
и ускорение при центрифугировании
Гравитационное поле, образующееся
1 при центрифугировании (g). равно квадра-
ту угловой скорости ротора (со в радианах в
I сек) и радиальному расстоянию (г, в санти-
I метрах) частицы от оси вращения, и выра-
I жается уравнением:
I g = со2/'. (А)
Поскольку один оборот ротора равен 2п
радиан, то его угловая скорость в радианах
в секунду может быть выражена в об мин 1
и общее уравнение, выражающее скорость
вращения ротора, будет равно:
со =2п(обмин’)/60. (Б)
Тогда гравитационное поле g, выражен-
ное в об/мин, равняется:
g = 4п2 (об мин ') 2-//3600. (В)
Значение g обычно выражается в еди-
ницах гравитационного поля Земли (g =
= 981 см2сек•'), т. е. отношение веса части-
цы в иоле центрифуги к весу этой же час-
тицы в иоле с искусственной гравитацией
и является относительным центрифужным
полем, g*, или множителем при g.
g- = 4п2 (об мин”1)2 //3600 х 981 (Г)
или в сокращенном виде:
I g =(1,118 х 10’5) (об мин 'Уг.
: При скорости 60000 об мин 1 g* - 300000.
Рис. Г-1.1, Типичный ротор аналитической центрифуги и двухсекторная ячейка для образца
оптическая система для наблюдения за
распределением концентрации в образ-
це во время центрифугирования, а также
система накопления и обработки данных.
Рис. Г1.2. Два типа ячеек, используемых
в методе ЛУЦ: а — двухсекторная ячейка;
б — ячейка, для формирования границы непо-
средственно в ультрацентрифуге. В последней
имеются два маленьких канала, соединяющих
между собой компартменты ячейки. Движе-
ние раствора или растворителя осуществля-
ется через нижний канал и происходит только
при вращении ячейки с определенной скоро-
стью (обычно 6-8 тысяч оборотов в минуту).
Верхний канал служит для возврата потока
воздуха
Г4.2.1. Роторы и ячейки
Аналитические роторы должны выдер-
живать сильные гравитационные поля.
При 60000 об/мин типичный ультра-
центрифужный ротор создает поле в
ячейке, которое в 300000 раз превыша-
ет гравитационное поле Земли (кюм-
мс!i inpiiii Г i. ।). В этих условиях масса
в 1 г имеет вес 300 кг.
Схематическое изображение ротора
и ячейки для седиментации приведено
на рисунке Г1.1. Металлический ротор
содержит несколько отверстий, куда
вставляются кюветы с образцом. Самый
простой тип ротора состоит из двух яче-
ек: аналитической и контрольной, кото-
рые изображены на рисунке Г1.1. Мате-
риалом для верхних и нижних окошек
ячейки служит оптический кварц или
синтетический сапфир. Объем ячеек
составляет от 0,2 до 1,0 см3. Секторная
форма ячейки принципиально важна для
экспериментов по скоростной седимента-
ции, поскольку осаждаемые частицы дви-
гаются вдоль радиальных линий. В ячей-
ке с параллельными боковыми стенками
осаждаемые молекулы на периферии бу-
дут соприкасаться со стенками и вызы-
вать конвекцию. В седиментационной
практике обычно используется двухсек-
торная ячейка, которая позволяет учиты-
вать поглощение отдельных компонен-
тов в растворителе (рис. Г4.2 <-/). Кроме
нее иногда используется двухсекторная
ячейка, позволяющая формировать гра-
ницу между раствором и растворителем
непосредственно в кювете (рис. Г4.2 6)
при вращении ротора с умеренно медлен-
ной скоростью. Эти ячейки удобны для
получения четкой границы при опреде-
лении коэффициента диффузии белков
методом расширяющейся границы (па-
раграф ГЗ.5.1), или при исследованиях
малых молекул, для которых скорость
седиментации недостаточна, чтобы обра-
зовалась четко движущаяся граница.
Г4.2.2. Системы оптического
детектирования
В зависимости от типа используемой оп-
тической системы различают два спосо-
ба для определения седиментационных
свойств молекул: наблюдение за изме-
нением концентрации вещества или за
изменением его градиента вдоль ячейки.
Для этих целей используются три типа
оптических систем. Шлиреновская оп-
тическая система выявляет движение
границы в виде зависимости градиен-
та показателя преломления от радиуса,
рэлеевская — в виде зависимости пока-
зателя преломления от радиуса, абсор-
бционная оптическая система — в виде
зависимости поглощения от радиуса.
Шлиреновская оптическая система,
доминировавшая над другими в тече-
ние 70 лет, ушла в историю (коммента-
рий Г4.5). Многие блестящие работы,
включая открытие рибосом, были сде-
ланы при использовании этой оптики
(рис. Г4.3). Ультрацентрифуга Beckman
Комментарий Г4.5. Шлиреновская
оптическая система
Изобретение этой простой системы
Филпотом и Свенсоном было главным до-
। стижением в разработке метода аналити-
ческого центрифугирования, без которого
i этот метод, возможно, никогда не стал бы
таким полезным. Система иногда имену-
ется шлиреновской оптической системой :
(от немецкого слова schlieren, означающего |
i «полоса или щель»). Если на пути пучка !
I света есть вещество с градиентом показа-
теля преломления, которое как призма от-
клоняет проходящие через него лучи света,
то в этом месте изображение горизонталь-
i ной щели сместится и через наклонную
| щель пройдет выше или ниже оптической
। оси, получив при этом смещение вправо
или влево на расстояние, определяемое
углом наклона щели. Чем резче граница и .
чем больше наклон щели, тем более острый !
j максимум получается на градиентной кри-
I вой. Высота максимума пропорциональна ;
i величине dn/dr и тангенсу угла наклона
щели. Таким образом, шлиреновская опти-
ка обеспечивает измерение градиента пока-
зателя преломления, dn/dr, как функцию
i радиального расстояния, г, непосредствен-
j но в кювете. Заметим, что если бы не было
I наклонной щели, то изображение кюветы
не зависело бы от наличия градиента.
XL содержит только две оптические де-
текторные системы: оптику для погло-
щения в ультрафиолетовой (УФ) облас-
ти и рэлеевскую интерференционную
оптику. Шлиреновский профиль седи-
ментации (зависимость градиента пока-
зателя преломления от радиуса ячейки)
может быть получен прямым диффе-
ренцированием зависимости показателя
преломления, либо оптической плотно-
сти от радиуса (комм<чггариi! Г1.6).
Абсорбционные оптические систе-
мы основаны на пропорциональности,
существующей между концентрацией
растворенного вещества и его оптиче-
ской плотностью. Большинство био-
Рис. Г1.3. Профили седиментации (а) 70S ри-
босом Е. соИ и (6) 30S и 50S рибосомных суб-
частиц, полученные с помощью шлиреновской
оптической системы
логических макромолекул поглощают
свет в ближней УФ области. Нуклеино-
вые кислоты имеют пик поглощения в
области спектра 258-260 нм, в то время
как белки характеризуются поглоще-
нием около 280 нм (Глава 31). На ри-
сунке Г4.4 показан типичный профиль
седиментации, измеренный с помощью
абсорбционной оптической системы.
Интерференционная оптическая
система базируется на рэлеевском
интерферометре (комментарий Г 1.7).
Система может быть использована
для макромолекул, которые слабо по-
глощают в УФ и в видимой областях.
На рисунке Г4.5 а показан ход лучей
от образца, полученный с помощью
этой оптики, а на рисунке Г4.5 б — за-
висимость показателя преломления
как функция радиуса, вычисленная с
помощью программного обеспечения
ультрацентрифуги Optima XL.
Значительно более высокую чув-
ствительность можно ожидать при
использовании флуоресцентного де-
тектора, так как необходимая в этом
случае концентрация вещества на не-
сколько порядков ниже, чем при ис-
пользовании УФ — абсорбционной
оптики. Более того, молекулы с раз-
личными флуорофорами могут быть
зарегистрированы селективно, даже
если они присутствуют как минор-
ные компоненты в смеси соединений.
Профиль седиментации бычьего сы-
вороточного альбумина (БСА), изме-
ренный для двух его концентраций
флуоресцентной системой детектиро-
вания, приведен на рисунке Г4.6. На-
именьшая концентрация (6,5 нг-мл"1)
на 4 порядка ниже чем та, которую
можно зарегистрировать с помощью
УФ-абсорбционной оптики для этого
белка (коммгп iарпii Г i.S).
Рис. 1'1.1. Типичный профиль седиментации, полученный с помощью абсорбционной оптиче-
ской системы ультрацентрифуги Beckman XL
Комментарий Г4.6. Седиментация в поле переменной гравитации
В 1972 г. детектор, измеряющий светорассеяние от образца, был встроен в ультрацентри-
фугу для наблюдений за изменениями концентрации полимерных молекул в ячейке (Scholtan
and Lange, 1972). В дальнейшем этот метод был модифицирован с целью получения распреде-
ления полимерных молекул по размерам с более высоким разрешением (Machtle, 1999). Ха-
рактерной чертой модификации метода является седиментация в поле переменной гравитации
за счет постепенного экспоненциального увеличения скорости ротора от 0 до 40 000 об мин 1 в
течение 1 часа. Эта инновация позволила изучать дисперсионные свойства полимеров в ши-
роком интервале размеров (от 10 до 3000 нм) без их фракционирования (см. параграф Г4.5.3
для дальнейшей дискуссии).
Комментарий Г4./. Интерферометр Рэлея
Интерференция света в интерферометре Рэлея происходит при расщеплении луча коге-
' рентного света (на практике от одного источника) на два, каждый из которых проходит оба
сектора двухсекторной ячейки. После прохождения ячейки два луча смешиваются, образуя
интерференционную картину. Если оба сектора содержат идентичные растворы, интерферен-
ционная картина представляет собой прямые полосы. Однако, когда один сектор содержит
растворитель, а другой — раствор макромолекул, полосы искривляются пропорционально
разности концентраций между образцом и растворителем. Образуется интерференционная
картина, каждая интерференционная полоса которой является кривой зависимости показате-
: ля преломления от расстояния.
Комментарий Г4.8. Прототип флуоресцентного детектора для аналитической
। ультрацентрифуги Beckman XL-I
Прототип флуоресцентного детектора для аналитической ультрацентрифуги Beckman
XL-I недавно был испытан в опытах по скоростной и равновесной седиментации. Его чув-
ствительность такова, что он способен определить флуоресцентную метку в концентрации
меньшей, чем 300 пМ. Радиальное разрешение детектора сравнимо с таковым для абсорбци-
онной системы (MacGregor et al., 2004).
Г4.2.3. Накопление и обработка
данных
С введением аналитической ультра-
центрифуги Optima XL получение
экспериментальных данных и их об-
работка стали полностью автоматизи-
рованными. Компьютерные програм-
мы сегодняшнего дня преобразуют
экспериментальные данные в графи-
ческие изображения для получения
интегральных распределений компо-
нентов. Высокое качество исходных
седиментограмм позволяет получить
дополнительную информацию о коэф-
фициентах диффузии и парциальных
концентрациях компонентов и, в неко-
торых случаях, определить константу
ассоциации ( коммсн mpiiii I i.D).
Г4.3. Уравнение Ламма
Уравнение Ламма описывает транс-
портные процессы в центрифуге. Оно
может быть получено при введении
параметра смещения в уравнение диф-
фузии Фика. Как было отмечено в па-
Комментарий Г4.9. Домашняя
' страница для аналитического :
ультрацентрифугирования в сети '
, Internet
| В настоящее время имеются три домаш-
них страницы на веб-сайте, где в режиме
on-line можно найти полезную ииформа- I
цию об инновациях и экспертные оценки
в области ультрацентрифугирования.
Домашняя страница Beckman home page
(http://www.beckman.com) кроме этого со- i
। держит информацию о коммерческой про- j
| дукции фирмы, характеристики инстру-
| ментов XL-А и XL-I, а также представляет
основные научные статьи.
Сайт RASMB (reversible association
in structural and molecular biology) был i
создан как сервер электронной почты,
позволяющий осуществлять связь меж-
ду научными группами (http://www.eri.
harvard.edu).
Во Demeler's XL-А and RASMB/Analytical
Ultracentrifugation page является пшерсвя-
зью через RASMB (http://)ioc02.uthsca.edu).
Сайт имеет программное обеспечение,
j доступное для пользователей RASMB, и
1 дополнительно предоставляет адреса элек-
тронной почты для коммуникации, сотруд- |
ничества, сервисной информации.
Рис. Г1.5. Типичный профиль седиментации,
измеренный интерференционной оптикой:
а — интерференционная картина получена при
помощи интерферометра Рэлея. Различие в
показателе преломления (Ан) для двух точек
ячейки может быть вычислено исходя из числа
полос, N, пересекающих эти точки зависимости
N = а(Ди)/Х, где а — это толщина ячейки, а л —
длина волны используемого света. Поскольку
показатель преломления пропорционален кон-
центрации вещества, то число полос может быть
использовано для определения концентрации
как функции радиуса в ячейке; б — графическое
представление интерференционных полос как
функции радиуса, полученное при помощи про-
граммного обеспечения XL-I центрифуги
а б в
Рис. 1’1.6. Два седиментационных профиля бычьего сывороточного альбумина (БСА), получен-
ные с помощью флуоресцентной детектирующей системы. БСА был помечен флуоресцирующей
меткой FITC (Флуоресцеин-изотиоцианат): а — наибольшая концентрация, 65 нгмл’1; б — на-
именьшая концентрация 6,5 нгмл в — направление осей в опытах с флуоресцирующей опти-
кой. Флуоресцирующая детектирующая система разработана для аналитической центрифуги
Spinco модель Е и инсталлирована на место стандартной шлиреновской оптики (Schmidt and
Reisner, 1992)
раграфе ГЗ.4.1, поток пропорционален
градиенту концентрации е конетантой
пропорциональное™, равной коэффи-
циенту диффузии, D (уравнение Г3.5).
Если предположить, что все частицы в
ячейке движутся в направлении +х, со
скоростью и, тогда поток в точке х дол-
жен увеличиваться на значение иС (х),
где С(х) — локальная концентрация.
Тогда первое уравнение Фика прини-
мает вид:
Jx = -D[dC/dx\ + wC(x). (Г4.1)
Помня, что и = svbJx, мы получаем:
Jx = -D[dC/dx\ + sco2xC(x). (Г4.2)
Первый и второй члены с правой
стороны уравнения Г4.2 соответствуют
переносу вещества в процессе диффу-
зии и седиментации, соответственно.
Уравнение (Г4.2) было получе-
но для идеальной бесконечно длин-
ной ячейки без стенок и не является
строго корректным для описания про-
цессов седиментации — диффузии
в реально существующих условиях
эксперимента. Поперечное сечение
секторной ячейки пропорционально
г, поэтому уравнение непрерывности
будет иметь вид
(Г4.3)
( dt J г ( dr )
Объединяя уравнения (Г4.2) и
(Г4.3), мы получаем следующее уравне-
ние, известное как уравнение Ламма:
(02r2sC-Dr<
(Г4.4)
•t.
Уравнение Ламма описывает диф-
фузию со смещением в секторной ячей-
ке аналитического центрифугирования
в реально существующих эксперимен-
тальных условиях.
Г4.4. Решение уравнения
Ламма для различных
граничных условий
Г4.4.1. Точные решения
Основное решение уравнения Г4.4 пред-
ставляет собой бесконечную серию интег-
ралов и не существует в аналитическом
виде. Его решения могут быть получены
только методами численного интегриро-
вания. Однако точное решение уравне-
ния Г4.4 возможно при двух следующих
условиях: «отсутствие диффузии» или
«отсутствие седиментации».
Отсутствие диффузии
В отсутствие диффузии уравнение Лам-
ма для растворов гомогенных макромо-
лекул имеет следующее точное решение:
С2 (х, f) = 0, если хм < х < х, (Г4.5)
С2(х, t) = Соехр(~25со2?) если х <х<хл,
где Со — исходная концентрация, а х = хм
ехр(5С1А). В графическом виде уравне-
ние представлено на рисунке Г4.7. Кон-
центрация молекул изменяется резко от
нуля до значения, которое не зависит от
х в любой момент времени. Ступенчатая
функция определяет границу. Область
х> х называется плато. Уровень плато
понижается со временем из-за разбав-
ления раствора в секторной ячейке (см.
рис. Г4.7). Положение ступенчатой фун-
кции изменяется во времени как следс-
твие седиментации вещества.
Отсутствие седиментации
При таком ограничении уравнение
Ламма Г4.4 имеет простой вид:
Рис. I'4.7. Графическое представление урав-
нения Г4.5. В отсутствие диффузии форми-
руется четкая граница, которая переметается
вдоль ячейки в течение времени
(dC/dt) r = -D^cFC/dr2)', (Г4.6)
а уравнение для градиента концентра-
ции:
(dC/dr)t =
= -Со/2 (nDt)'A exp (-^/4L>0- (Г4.7)
Решение этих уравнений обсуждалось
в секции Г3.2. Коэффициент диффузии
может быть определен при измерении
стандартного отклонения гауссовой фун-
кции по отношению к функции границы,
которая равна (2Dt)'A. На практике кри-
вая градиента концентрации измеряется
как функция времени, в соответствии с
процедурой, описанной в секции Г3.4.3
для чистой диффузии (см. рис. Г3.7 а и (7).
Комментарий Г4.10. Аналитические
решения уравнения Ламма в
различных граничных условиях
Решение Факсена: центрифужная ячей- .
ка имеет бесконечные размеры, диффу-
зия мала (D/<B?s:r «1) и рассматривается I
только начальное время седиментации
((rfst «1) (Faxen, 1929).
Решение Арчибальда: s и D являются
постоянными (Archibald, 1938).
Решение Фуджита: расширенное реше-
ние Факсена для случая, когда D посто-
янно, а х зависит от концентрации как х =
= х(| (1 - kC) (Fujita, 1956)
Использование этого подхода к оп-
ределению коэффициентов диффузии
макромолекул в ультрацентрифуге
обосновано, если значение выражения,
описывающего поток седиментации
со^хС, существенно меньше потока
для диффузии D\dC/dx\. На практи-
ке эти условия выполняются для не-
больших глобулярных белков с кон-
стантой седиментации около 25 при
низкой скорости вращения ротора
(3000-6000 об-мин-1) в ячейке с пред-
варительным образованием границы.
Зависимое от времени распределение
границы измеряется после наслаива-
ния (или подслаивания) буфера на
раствор макромолекул.
Г4.4.2. Аналитические решения
Аналитические решения уравнения
Ламма при специфических граничных
условиях известны с 1930г. (коммен-
тарий Г!. 10). В настоящее время со-
временные компьютерные програм-
мы позволяют решать это уравнение
в условиях, более приближенных к
эксперименту. Для этой цели предло-
жены два метода, каждый из которых
дает возможность одновременного
определения коэффициентов седи-
ментации и диффузии и, как следс-
твие, молекулярной массы. Выбор
метода зависит от соотношения вкла-
да диффузии и седиментации в дви-
жущуюся границу.
Первый метод назван по имени ав-
торов (Ван Хольде и Вайшета) и бази-
руется на том факте, что седиментация
пропорциональна времени, в то время
как диффузия пропорциональна кор-
ню квадратному из времени (сравните
уравнения Г4.18 и Г3.12). Отсюда сле-
дует, что экстраполяция к бесконечно-
му времени должна исключать вклад
диффузии в форму границы. Метод
состоит из следующих основных ша-
гов.
На первом шаге расстояние между
базовой линией и плато для каждого
скана подразделяется на N (обычно не-
сколько десятков) горизонтальных се-
чений N и кажущиеся коэффициенты
седиментации, s*, вычисляются как:
5* = In ((r/rJ/oA), (Г4.8)
где г, гм — радиусы сечения и мениска,
соответственно. На втором шаге зна-
чения s* откладываются как функция
корня квадратного из времени для
каждого скана. Пересечение с г/-осью,
которое соответствует бесконечному
времени, дает величину 5, поправлен-
ную на диффузию. Для гомогенных
образцов все линии должны сойтись
к одной точке — одному значению
константы седиментации 5. Для него-
могенного образца линии не сходятся
в одной точке и среднее значение при
пересечении с (/-осью используется
для вычисления средне-весового зна-
чения коэффициента седиментации,
.s'w. Затем результаты представляются
в виде графика доли общего седимен-
тирующего материала как функции
5. Такой график характеризует качест-
во образца. На рисунке Г1.8 показаны
седиментационные профили (встав-
ки а, г) и анализ методом Ван Холь-
де-Вайшета для гомогенных (вставки
а, б) и гетерогенных (вставки г—е) об-
разцов биологических макромолекул
( коммеп Iapiiii Г i. 1 1 ).
Второй метод по имени автора ио-
сит название метода Стаффорда и
иногда называется методом временных
производных. В нем распределение ко-
эффициента седиментации вычисляет-
ся из производной по времени профиля
скорости седиментации. Кажущийся
коэффициент седиментации (т. е. не
Радиус (см)
г, (-0 5»
Время
Константа седиментации S
Радиус (см)
ВРемя Константа седиментации S
г
е
Рис. I 1.8. Анализ скорости границы седиментации по методу Ван Хольде-Вайшета: а-в — дан-
ные и анализ для белка капсиды II бактериофага Т7 MLD; г-е — данные и анализ для того же
препарата, но в котором к белку добавлено некоторое количество субгено.мной ДНК. Скорость
вращения 10000 об мин-1 (Hansen et al., 1994)
Коммунi;i|)lih Г4.1 I. Метод
Ван Хольде-Вайшета
При практическом применении метод
требует ярко выраженной области плато и
симуляции данных, которая должна быть
использована для демонстрации достовер-
ности полученных значений. Метод Ван
Хольде-Вайшета использован Демелером
в программном обеспечении ULTRASCAN
(http://www.camna.uthscsa. edu//), ко-
торое существует в трех коммерческих
версиях в зависимости от операционной
системы.
поправленный на диффузию), g*(s)f мо-
жет быть вычислен из (dc/dt\согласно
уравнению:
«'W,-W*>„„,p(l/C0)x
- (ш¥/|п <г„/г) (г/г <Г4 '1|
где 5 — коэффициент седиментации;
со — угловая скорость ротора; CQ — ис-
ходная концентрация; г и гм — теку-
щий радиус и радиус мениска, соответ-
ственно. В этом методе константы
седиментации получаются следующим
Метод временных
производных
0.35
0.30
К 0.25
ф
ю
m 0.20
О
g 0.15
ГО
А 0.10
< ад 0 05
V
0.00
-0.05
g(s*) анализ тАЬ +партнер
3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.С
з*(ед. Сведберга)
s (ед. Сведберга)
Рис. I 1.9. Иллюстрация метода Стаффорда (а—в см. текст). Типичный результат представлен
в г в виде функции g (s*) для моноклональных антител (mAb) и их комплекса с белком. Моле-
кулярная масса для inAb определена методом комбинации констант 5 и D и оказалось равной
144 кДа с ошибкой в 1,4%. Масса этого же белка, определенная с помощью масс спектромет-
рии равна 146 кДА. Для комплекса mAb с белком молекулярная масса в 245 кДа была вычис-
лена из комбинации констант s and D, с ошибкой 3,8%. Масса того же белка, определенная с
помощью метода масс-спектрометрии, равна 237 кДа. Высокая точность метода АУЦ связана с
тем, что были проанализированы все данные из 40 сканов, каждый из которых содержал около
1000 точек (Hensley, 1996). Отметим, что улучшенная коммерческая версия метода Стаффорда
(DCDTPLUS) доступна на сайте Philo (http://www. jphilo. mailway. com)
способом (рис. Г1.9<7). Близко рас-
положенные две седиментационные
кривые вычитаются друг из друга,
давая дифференцированную кривую
(отсюда название «метод производ-
ных») как показано на рисунке Г1.9 6.
Каждый седиментационный профиль
будет иметь гауссову форму, похожую
на таковую, получаемую при использо-
вании шлиреновской оптики (dC/dr)
(см. рис. Г1.3). Затем кривые нормали-
зуются на время и накладываются друг
на друга. На последнем этапе они нор-
мализуются на концентрацию. Резуль-
тирующий пик имеет гауссову форму,
положение и ширина которого связаны
с константой седиментации и диффу-
зии, соответственно (рис. Г4.9 «)•
Г4.4.3. Численные решения
Использование численных решений
уравнения Ламма для прямого анализа
данных аналитического ультрацентри-
фугирования известно уже около 30 лет.
Однако относительно недавно, в основ-
ном благодаря возросшей мощности
компьютеров, они стали использовать-
ся для анализа данных аналитического
скоростного ультрацентрифугирова-
ния. Главное преимущество численных
решений состоит в том, что с их помо-
щью можно учесть практически все
граничные условия, накладываемые ко-
нечной длиной центрифужной ячейки
и эффектами диффузии.
Г4.5. Скоростная
седиментация
В аналитическом ультрацентрифу-
гировании имеются два комплемен-
тарных метода с различными экс-
периментальными протоколами, но
с использованием одного и того же
оборудования: скоростная седимен-
тация и равновесная седиментация
(гибл. 11.1). В первом из них исполь-
зуется высокая скорость ротора и
длинные ячейки, с тем чтобы получить
максимальное разрешение, обеспечить
информацию о размере и форме моле-
кул, а также их взаимодействиях. При
равновесной седиментации, напротив,
используется низкая скорость ротора
и короткие ячейки, с тем чтобы свести
к минимуму время, необходимое для
установления равновесия и обеспечить
информацию о молекулярной массе,
константах ассоциации и стехиомет-
рии. На рисунке Г1.10 приведены ти-
пичные данные экспериментов по ско-
ростной седиментации (часть а} и по
равновесной седиментации (часть б}.
Г4.5.1. Макромолекулы в сильном
гравитационном поле
Стандартным способом определения
массы частицы является сравнение ее
относительного ускорения под дейс-
твием силы известной величины с уско-
рением частицы-стандарта с известной
массой. Наиболее удобно использовать
силы на макроскопической шкале, т. е.
под действием силы тяжести Земли.
Однако действие этой силы на биоло-
гические макромолекулы настолько
мало, что перевешивается силами, воз-
никающими от соударений между мо-
лекулами в результате их хаотического
движения.
Простые вычисления показывают,
что на молекулярном уровне тепловая
энергия намного превосходит грави-
тационную потенциальную энергию и,
следовательно, направленное движение
молекул в гравитационном поле Земли
невозможно (комментарий Г1.12). Од-
Г;пни и Li I •'!. 1. Два типа экспериментов в аналитической ультрацентрифуге
Обсуждаемая характеристика Скоростная седиментация Равновесная седиментация
Угловая скорость Большая (как правило); выбирается согласно седи- ментационным свойствам частицы Небольшая (как правило); выбирается согласно седи- ментационным свойствам частицы
Тип анализа Концентрация как функция времени и расстояния в тече- ние нескольких часов Концентрация как функция расстояния после достиже- ния равновесия (24 часа и более)
Тип измерения Наблюдение за движением границы Наблюдение за распределе- нием концентрации молекул в ячейке
Вычисляемые параметры Размеры и конформация частиц Молекулярная масса иполидисперсность
ним из способов обойти эту трудность
является повышение эффективной гра-
витационной потенциальной энергии
до значений больше, чем kT, путем пе-
ренесения частиц в ячейку, вращающу-
юся с высокой скоростью.
Рассмотрим частицу массой т и
плотностью р0, находящуюся в сектор-
ной ячейке, которая находится в роторе,
вращающемся вокруг z — оси, с угловой
скоростью со, и движущуюся через рас-
творитель С ПЛОТНОСТЬЮ р0 (KoMMcii i;,
рен I i : На молекулу действует три
силы: гравитационная F, выталкива-
ющая, Fb, и сила сопротивления среды, Fd.
(рис. Г1.11). Гравитационная сила в уль-
трацентрифуге пропорциональна массе
частицы т и ее линейному ускорению
со2г
F = nwFr, (Г4.10)
а
б
Рис. I 1.10: а — вид границы в экспериментах по скоростной седиментации. На рисунке воспро-
изведены восемь следующих друг за другом седиментационных профилей. Образец — олигонук-
леосомы в низкосолевом буфере. Скорость вращения 20 000 об/мин. Направление седиментации
слева направо. Сканирования проводились через 8 минут (самое первое сканирование обозначе-
но как Г,; последнее зарегистрированное сканирование как г ). Базовая линия регистрируется по
поглощению растворителя. Центрифужная кювета состоит из двух секторов; б — данные равно-
весной седиментации этого же образца, полученные после центрифугирования при 1600 об мин 1
в течение 72 часов. Радиальное положение границы раздела фаз воздух — растворитель в секторе
отмечено как «мениск образца» (Hansen et al., 1994)
Комментарий Г4.12. Сравнение гравитационной потенциальной и тепловой
энергий
Вычислим разницу в значении гравитационной потенциальной энергии между двумя точ-
ками, отделенными друг от друга вертикально расстоянием h для молекулы с молекулярной
массой М.
Пример!.Л/= 10;’ДаиЛ = 1см.ТогдаМ = 10'7(6.03.1026) = 1.6-10 22 кг(при использовании
числа молекул в кг-молях). Ускорение, обусловленное гравитацией Земли, равно 10 м сек 2, а
разница потенциальной энергии, £р, будет равна: £ = mgh =1.6-10 22-10-10 2 = 1.6-1 О’2:1 Джоуля
(J). Тепловая энергия -kT (где Т — абсолютная температура) и при комнатной температуре
она будет равна: kT = 1.38-10 2:,J/°K 293°К = 400-10’2:у. Отсюда следует, что гравитационная
потенциальная энергия меньше тепловой энергии в 250 раз, и, следовательно, эксперимен-
тальное определение седиментации такой частицы, обусловленное гравитацией поля земли,
невозможно.
Пример 2. М = 109 Да и h = 100 см. Аналогичные вычисления для такой молекулярной
массы и такого расстояния показывают, что в этом случае гравитационная потенциальная
энергия больше тепловой энергии в 400 раз и, следовательно, экспериментальное определе-
ние константы седиментации такой частицы при использовании естественной гравитации,
в принципе, возможно.
где г — расстояние от оси вращения.
Под влиянием этой силы частица будет
двигаться в окружающей ее среде по
направлению от оси вращения. Вытал-
кивающая сила равна по величине весу
Комментарий Г4.13. Механический
и термодинамический подходы
Исследования процессов седиментации в
этой главе обсуждается в терминах простых
механических моделей. Такая постановка i
вопроса сопряжена с некоторыми неод-
нозначностями при интерпретации, которые
можно избежать только при строгом термо- ,
динамическом подходе. ;
жидкости, вытесняемой частицей ( кюм
мен гариii 1'1.1 i)
Fh = — m° ы2г. (Г4.11)
Результирующая суммарная «гра-
витационная» сила равна:
F + F, = (m - т°) со2?’ =
С , (Г4.12)
= 7ИСО-Г(1 - р0/р).
Выражая величину плотности час-
тицы, р, ее обратной величиной, пар-
циальным удельным объемом, б (см.
секцию Г4.8) мы получаем:
Гравитационная сила: Fc = m<o2r
Выталкивающая сила: Fb = -m0<o2r
Сила трения: Fd = - fu
<о — угловая скорость
г —расстояние от центра вращения
и — скорость молекул
Рис. I 4.11. Три силы, действующие на частицу в ячейке центрифуги (см. текст)
I
I Комментарий Г4.14. Архимедова
сила: макроскопический и
микроскопический подходы |
• Принцип Архимеда гласит: «на тело,
| плавающее в жидкости, действует сила, рав-
ная весу жидкости в объеме, вытесненном
телом». Хорошо известно, что закон был
сформулирован для макроскопических тел,
находящихся в жидкости. В этой связи воз- I
I пикает вопрос: может ли микроскопическое |
! тело (в пашем случае молекула, помещенная
в жидкость) считаться физическим телом,
помещенным в эту же жидкость? Имеется
строгое определение архимедовых сил, лейс- ]
твующих на индивидуальные молекулы: «на !
! тело, помещенное в жидкость, действует вы- !
талкивающая сила, равная весу жидкости в ;
таком объеме, который становится недоступ- ;
ным его молекулам». Под недоступным объ- ।
емом в данном контексте понимается исклю- |
' ченный объем или ко-объем (см. параграф
Г2.5.1) (Luboshitz and Podgoretskii, 1991).
F + Fb = m<syr (1 - в p0). (Г4.13)
И, наконец, третья сила, сила тре-
ния, действующая на частицу, зависит
от ее скорости, и, и коэффициента тре-
ния f Fd= fи (уравнение Г1.9).
Частица достигнет постоянной скоро-
сти, когда суммарная сила, действующая
на частицу, станет равной нулю, т. е.
F.+ Fb+F(| = 0, (Г4.14)
mu>2v (1 — и р0) = fu. (Г4.15)
Умножая уравнение Г4.15 на число
Авогадро и помещая экспериментально
измеряемые параметры в левую часть
уравнения, а молекулярные параметры
частицы в правую, мы получаем:
№ и/агг = М (1 — и р0)/А\/. (Г4.16)
Отношение измеренной скорости
частицы к ее центрифужному ускорению
называется константой седиментации —
5. Ее размерность — секунда. Константы
седиментации большинства биологиче-
ских частиц очень малы, и для удобства
за единицу константы седиментации
принимается 1 х Ю 13 сек. Такая едини-
ца называется 1 сведберг (1S) в память
фундаментального вклада Тео Сведберга
в теорию и практику метода аналитиче-
ского ультрацентрифугирования.
Г4.5.2. Определение
седиментационных и диффузионных
коэффициентов из данных
скоростной седиментации
Простые системы: метод средней точки
В стандартном эксперименте по скоро-
стной седиментации ячейка центрифу-
ги вначале содержит раствор, концен-
трация которого постоянна по всей ее
длине. В процессе центрифугирования
частицы движутся под действием гра-
витационной силы, мениск осветляется
и формируется движущаяся граница
(рис. П.12). За скорость движения гра-
ницы и стандартно принимается из-
с/г
менение радиальной координаты —-£-
at
в средней точке. Значение скорости и
drcP 2
можно представить как и - -±- = а> rs,
где со — угловая скорость; 5 — константа
седиментации.
После интегрирования получаем
ln-b£2 = 0y5(f_f ). (Г4.17)
гг г
Построение зависимости In—1----
<р(^о)
от (t-tQ) позволяет вычислить кон-
станту седиментации макромолекул.
Заметим, что этот метод применим
только для «идеальных» растворов, т. е.
для растворов, содержащих одинаковые
Рис. Г1.12. Гипотетическая граница седиментации, показывающая мениск, эквивалентный по-
ложению границы, плато и базовую линию. Традиционное рутинное определение константы
седиментации базируется на графическом представлении логарифма радиального положения
средней точки границы седиментации от времени
и невзаимодействующие макромолеку-
лы. В случае смеси, содержащей два типа
макромолекул, которые имеют большие
молекулярные массы (100 кДа и более)
и достаточно сильно различаются меж-
ду собой по молекулярной массе, можно
вычислить две константы седиментации,
характеризующие каждый тип молекул
по отдельности. Однако в общем случае,
когда молекулярно-весовое распределе-
ние заранее неизвестно, применение ме-
тода «средней точки» может приводить
к большим ошибкам и в современной
практике аналитического центрифуги-
рования не используется. Кроме того, в
качестве обязательного условия метод
требует наличия на седиментограмме
четкого плато и базовой линии, что за-
трудняет его применение к белкам с мо-
лекулярной массой меньше 30 кДа.
Заметим, что для грубой характе-
ристики полидисперсных систем наи-
более подходящим является метод
вторых моментов, дающий седимента-
ционный коэффициент весового усред-
нения ( М ' I.ID.'I! Г i п.
Сложные системы: комплексный
анализ седиментационной границы
Анализ данных скоростной седимен-
тации сложных систем требует более
трудоемких подходов для анализа пара-
метров по сравнению с методом средней
точки. В случае очень больших частиц,
для которых разделение достигается в
течение времени эксперимента, анализ
с высоким разрешением может быть вы-
полнен рутинным методом средней точ-
ки. Рисунок Г1.3 является хорошим при-
мером такого случая, когда константы
седиментации двух рибосомных субчас-
тиц (50S и 30S) легко вычисляются из
седиментационного профиля в их смеси.
Однако для малых частиц диффузион-
ное расширение создает большие труд-
ности в решении задачи о распределе-
ния компонентов, делая невозможным
: Метод вторых
моментов
Метод вторых моментов является очень
удобным методом для точной оценки рас-
' пределения коэффициентов седиментации
! в случае слабо асимметричной популяции
в смеси. Его использование позволяет вы-
числить положение гипотетической грани-
цы. По скорости перемещения положения
этой границы можно найти средневесовой
коэффициент седиментации. Метод второ-
го момента наиболее удобен в применении
к данным, полученным из кривых dC/dx от
.г (шлиреновская оптическая система), по
которым вычисляется положение эквива-
: лентной границы (второй момент кривой
градиента показателя преломления):
Однако, когда данные получены из кри-
вых С отх (интерференционная оптика или
оптика поглощения), то удобнее вычислять
эквивалентное положение границы по сле-
дующему уравнению:
j г2 de
<г2>=^----. (Б)
j de
разделение компонентов, отличающихся
по константе седиментации в таком же
отношении, как и для рибосомных час-
тиц. Методы Стаффорда или Ван Холь-
де—Вайшета, описанные ранее в этой
главе (параграф Г4.4.2), являются очень
удобными для работы в этих режимах.
Хотя каждый из них имеет свои досто-
инства и недостатки, однако главная их
особенность очевидна: они являются
безмодельными, т. е. не требуют ника-
кого априорного знания о параметрах
седиментирующих компонентов.
Однако, если допустить, что имеются
заранее известные сведения о параметрах
компонентов в смеси (такой подход мы
будем называть модельным), то такой
анализ может обеспечить гораздо более
высокую точность в распределении, хотя
математическая обработка при этом го-
раздо более сложная, чем в методах Стаф-
форда или Ван Хольде-Вайшета.
Суть проблемы коротко сводится
к следующему. В отсутствие взаимо-
действия между макромолекулами экс-
периментально наблюдаемый профиль
непрерывного распределения масс может
быть описан суперпозицией профилей с
(М), молекулярная масса которого рас-
полагается между М и М + dM. Если L
(М, г, £) обозначает седиментационный
профиль монодисперсного вещества с
массой М, находящегося на расстоянии г
в момент времени t, то задача сводится к
решению уравнения Фредгольма первого
рода в условиях реального эксперимента:
s(r,t) = |с(М)£(М,г,ОЖ(Г4.18)
где 5 (г, t) обозначает экспериментально
наблюдаемый сигнал, получаемый с не-
которой ошибкой. Заметим, что задачи
такого рода часто встречаются в ряде
областей физики. Типичным примером
является метод динамического рассея-
ния света (Глава ПО).
В случае скоростного ультрацент-
рифугирования разбавленных раство-
ров ядро L (М, г, t) уравнения Г4.18 есть
решение уравнения Ламма, которое те-
перь имеет вид:
(Г4.19)
где s(M) и D(M) есть седиментационные
и диффузионные коэффициенты части-
цы, соответственно. Они обасильнозави-
сят от молекулярной массы (параграфы
Г3.7.3 и Г4.10.1) и связаны между собой
уравнением Сведберга (смотри ниже).
Программа SEDFITTL Шука (Р. Schuck)
для анализа частиц, имеющих непрерыв-
ное распределения по размерам, доступ-
на на сайте http://www. analyticalultrace
ntrifugation. com.
В программе SEDF1T проблема ус-
тойчивости достигается наложением
физически разумного ограничения на
решение — положительностью получа-
емых величин. Кроме того, в уравнение
Г4.18 добавляется стабилизирующий
член, называемый регуляризационным.
Проблема однозначности требует вве-
дением априорного знания парциально-
го удельного объема б и гидродинами-
ческой формы частицы, вычисляемой
как отношение ее реального коэффи-
циента трения к минимальному J//
Для сферической белковой частицы
с общепринятой величиной гидрата-
ции (0,3 грамма воды на грамм белка)
это отношение равно 1,2. Для большин-
ства глобулярных белков это отноше-
ние равно 1,25-1,35, свидетельствуя об
их малой асимметрии. Поскольку для
белков парциальный удельный объем
можно достаточно точно рассчитать из
аминокислотной последовател ьности
(см. секцию Г4.8), то именно отноше-
ние /// чаще всего используется как
подгоночный параметр. Заметим, что
отношение///м1 не тождественно отно-
шению ///0, которое отражает геометри-
ческий эффект отличия формы части-
цы от сферы. Для сферической частицы
///,-1,0.
На рисунке 14.13 приведено не-
сколько примеров, описывающих при-
менение программы SEDF1T для иссле-
дования изолированных белков и их
смесей. Из него видно, что с помощью
программы SEDFIT можно разрешать
дискретные компоненты смеси, отли-
чающиеся по молекулярной массе в два
раза и больше. В том случае, если это
отношение меньше двух (смесь оваль-
бумина и бычьего сывороточного аль-
бумина, отношение молекулярных масс
этих двух белков равно 1,55), программа
SEDF1T не разделяет компоненты смеси,
кроме визуализации димеров БСА, как и
в предыдущих случаях (рис. Г!. 13, ниж-
няя правая четверть). Основный пик яв-
ляется сложным, его параметры близки к
средним значениям для двух компонен-
тов смеси. Следует заметить, что во всех
экспериментах, проведенных на смесях,
исследованные белки имеют близкие ве-
личины парциальных удельных объемов
и не взаимодействуют друг с другом.
Г4.5.3. Очень гетерогенные
системы
В стандартных экспериментах по ско-
ростной седиментации используется
только одна заранее выбранная ско-
рость ротора. Это сразу ограничивает
область исследуемых молекул по кон-
стантам седиментации. Так, при скоро-
сти вращения ротора 50000 об мин 1 и
обычном значении ускорения ротора,
400 об-сек ’, частица, которая пройдет
путь, равный половине длины ячейки,
будет иметь константу седиментации
280S. Тем самым выбор одной угловой
скорости сильно ограничивает интер-
вал доступных наблюдению констант
седиментации. Этот интервал может
быть существенно расширен варьирова-
нием скорости ротора — от относитель-
но маленькой скорости, позволяющей
вести наблюдение за самыми большими
частицами, и до самой большой скоро-
Рис. П.1,3. Примеры применения программы SEDFIT для исследования изолированных белков
и их смесей.
Условные обозначения к рисунку Г4.13
Верхняя левая четверть, а — седиментограмма бычьего сывороточного альбумина. Молеку-
лярная масса, рассчитанная из первичной структуры, составляет 66,43 кДа. Концентрация
С = 1,5 мг-смЛ V = 0,733 см' г ', угловая скорость вращения ротора со = 40 000 обмин темпе-
ратура Т= 20 °C. Сканирование: 20 сканов через 240 сек, оптическая плотность по измерению в
ячейке АУЦ равна 1,0 о. е. б. в — восстановленные распределения в координатах с (х) отх и с (М)
от Л/, соответственно. Верхняя правая четверть. Седиментограмма смеси из двух белков, состо-
ящей из бычьего сывороточного альбумина и бычьей карбоангидразы. Концентрация БСЛ равна
2,0 мг-см *, карбоангидразы — 0,7 мгсм Угловая скорость вращения ротора со = 50 000 об мин ',
температура Т= 20 °C. Сканирование: 30 сканов через 240 сек. оптическая плотность по измере-
нию в ячейке ЛУЦ равна 1,3 о. с. б, в - восстановленные распределения в координатах с (х) отх и
с (М) от М, соответственно. Нижняя левая четверть. Седиментограмма смеси из трех белков, со-
стоящей из бычьего сывороточного альбумина, карбоангидразы и апоцитохрома. Концентрация
БСА равна 2,4 мг-см карбоангидразы — 1,1 мгсм ', апоцитохрома — 1.4 мг-см \ Угловая ско-
рость вращения ротора со = 50.000 об мин1, температура Т= 20 °C. Сканирование: 45 сканов через
480 сек, оптическая плотность по измерению в ячейке АУЦ равна 1,5 о. е. б, в — восстановленные
распределения в координатах с (х) от х и с (Л7) от М, соответственно. Нижняя правая четверть.
Седиментограмма смеси двух белков, состоящей из бычьего сывороточного альбумина (БСА) и
яичного овальбумина. Буфер: 50 мМ Трис IIC1, 100 мМ NaCl (pl 1 7,6). Концентрация БСА равна
2,8 мг-см •'*, овальбумина — 2,4 мгсм Угловая скорость вращения ротора со = 50 000 об/мин,
температура Т= 20 °C. Сканирование: 21 скан через 480 сек, оптическая плотность по измерению
в ячейке АУЦ равна 1,8 о. е. б, в — восстановленные распределения в координатах с (х) от х и с
(АГ) от М, соответственно. Прерывистой линией указано положение молекулярных параметров
для индивидуальных белков (Сердюк и Евсеева, 2006)
сти — для выявления самых маленьких
частиц. Данные, полученные для каж-
дой скорости, объединяются в одну не-
прерывную функцию распределения.
Протокол такого эксперимента, поз-
воляющего наблюдать частицы в ин-
тервале значений s от 200 000S до 2,0S,
приведен в nio. nine I 1.2. При каждом
разгоне на установление скорости ухо-
дит до 600 сек. На конечной скорости
50000 об-мин-1- центрифугирование
продолжается до тех пор, пока наимень-
шие частицы не достигнут середины
пути до дна кюветы. На рисунке Г4.14
показано применение этого протокола
к раствору гемоцианина. В соответс-
твии с методом Стаффорда (параграф
Г4.2.2) построены графики значений
xxg(x*) от 1п(х*). Как видно из этого
рисунка, в образце гемоцианина выяв-
ляются четыре компонента со значени-
ями констант седиментации 5, 16, 40
и 60S, соответствующими мономерам,
гексамерам, 24-мерам и 36-мерам. Этот
результат показывает, что при исполь-
зовании вышеописанного протокола,
можно исследовать очень гетерогенные
системы ( ки\|\|,-п । щи in I 1.1(1).
Г4.5.4. Коэффициенты
седиментации биологических
макромолекул
Коэффициенты седиментации биоло-
гических макромолекул обычно изме-
ряют в буферных растворах, вязкость
и плотность которых может отличать-
ся от таковых у воды. Коэффициенты
седиментации могут измеряться также
при различных значениях температу-
ры. Поэтому принято приводить реаль-
ные данные к «стандартным условиям»,
в качестве которых выбирается чистая
вода при 20 °C:
Гнолица 1’4.2. Типичный протокол выбора скоростей для анализа очень
гетерогенных систем. Радиус мениска взят равным 5,9 см (Stafford et al., 2004).
Скорость (об/мин) Время на каждой скорости (сек) s* (при 6,5 см) (в Сведбергах)
0-6000 15 500 000
6000 600 4220
9000 600 1300
13 000 600 550
18 000 600 250
25 000 600 125
35 000 600 62
50 000 3600 31
50 000 3600 7,6
50 000 3600 4,4
50 000 3600 3,0
50 000 3600 2,3
S*(6,5 см) = ln(6,5/5/9)J(<o2rft)
= J1" (Г4-2°)
1 ^Рехр 1Ъо.г<
где s2n w — коэффициент седиментации
молекулы в воде при 20 °C; s — экс-
периментально полученный коэффи-
циент седиментации молекулы; т]экс —
вязкость растворителя при заданной
температуре Т (°C); ц20 w — вязкость
воды при 20 °C; р20 п. — плотность воды
при 20 °C; й — парциальный удель-
ный объем молекулы. Отметим, что
величина может быть вычислена
20. w
Коммен 1 л|)и( 1 Г4.16. Седиментация
с переменной скоростью вращения
ротора
Техника седиментации с переменной
скоростью ротора была впервые пред-
ложена Д. Ифантисом с сотрудниками в
1981 г. В 1984 г. Мачтле ввел термин «гра-
витационное качание» для описания экспе-
риментов с переменной угловой скоростью
(i xi. 1.11\1 хк 11 I:: | и ।. • I
8.0
7.0
6.0
6.0
w 4.0
О)
V
3.0
2.0
1.0
0.0
1.000 2.000 3.000 4.000 6.000 6.000 7.000 8.000
ln(»’)
Рис. П.11. Зависимость sxg(s*) от ln(s*)
для раствора гемоцианина Limulus polyphemus
в концентрации 2 мгсм’3. Логарифмическая
шкала по оси абсцисс выбрана ввиду большо-
го интервала значений 5. Выбранные скорости:
12 000, 18 000, 25 000,35 000 и 50 000 об-мин’1.
От 50 до 90 сканов выполнены при каждом
значении скорости ротора в течение 20 мин за
исключением самой высокой скорости. На ней
вращение продолжалось в течение 3-х часов.
Точки, полученные при каждой скорости,
обозначены различными цветами. Площади
под пиками равны концентрациям молекул в
зоне границы и соответствуют распределению
каждого пика (Stafford and Braswell, 2004)
даже в случае, когда их седиментаци-
онные характеристики не могут быть
измерены в воде при 20 °C. В заклю-
чение заметим, что коэффициенты
седиментации биологических макро-
молекул обычно получаются при ка-
кой-либо конечной концентрации и
поэтому должны быть экстраполиро-
ваны к нулевой концентрации (ком
мен । ;i[ 1;Iн 14.17).
Коэффициенты седиментации био-
логических макромолекул охватыва-
ют очень широкую область значений.
Для малых пептидов, глобулярных бел-
ков они лежат в интервале значений от
0,45 до 100S; для рибосомных PHK-ot4S
до 30S; для рибосом и полисом — от 20S
Комментарий Г4.17.
Концентрационная зависимость
коэффициента седиментации
Считается общепринятым, что концент-
рационная зависимость коэффициента се-
диментации 5 выражается формулами:
5 = 5°(1 + ^С)-', (А)
5 = 5° (1 + k "С ), (Б)
где s° — величина при бесконечном разве-
дении, a k' и k" — эмпирические константы,
выраженные в см3 г '. Обычно их значения
положительны, т. е. х понижается при по-
вышении концентрации. Эти уравнения
эквивалентны при малых концентрациях
макромолекул, но расходятся при больших
; концентрациях. Уравнение (А) лучше ис- |
пользовать для компактных частиц, а урав- j
нение (Б) — для синтетических разверну- ।
тых полимеров и высоко-асимметричных ;
частиц. В некоторых случаях наблюдается
увеличение s с возрастанием С. Это харак-
терно для слабо взаимодействующих мак-
ромолекул. Эмпирические константы k’ и
> k” зависят от формы молекул. Это означает,
что они могут использоваться как гидроди-
намические параметры для вычисления ,
i молекулярной асимметрии частиц (см. па- |
I раграф Г2.5.2). |
до 100S; для вирусов — от 40S до 1000S;
для лизосом — от 4000S; для мембран от
100000S; для митохондрий — от 20000S
до 70000S; и для ядер -- от 4 000 000S
до 40000000S. Обычно, чем больше
частица, тем большим значением х она
обладает. Очень большие молекулы мо-
гут осаждаться в гравитационном поле
Земли (см. >мм(л। г;।р11н Г i. I.’>). В i;:0-
uii.i I i. ’i представлены значения кон-
стант седиментации, х,,0 w, парциальных
удельных объемов, й и молекулярных
масс М некоторых представителей на-
иболее типичных биологических макро-
молекул — начиная от самых маленьких
синтетических пептидов (пептида I, мо-
лекулярная масса 2 кДа) и заканчивая
большими олигомерными белковыми
комплексами (гемоцианин, молекуляр-
ная масса около 9 МДа). В i ш р ! i.i
представлены аналогичные данные для
нативных ДНК и ее фрагментов. Об-
суждение данных, представленных в
этих таблицах, будет дано позже (сек-
ция Г4.10).
Г4.5.5. Дифференциальная
седиментация
Неопределенность, обусловленная ско-
ростью и температурой ротора, являет-
ся причиной ошибок при измерении ко-
эффициентов седиментации. Их можно
избежать, если поместить образцы в
одну и ту же двухсекторную ячейку в
одном и том же роторе. В первых экс-
периментах была использована шлире-
новская оптика и двухсекторная ячейка.
Одна из ячеек была оснащена устрой-
ством, обеспечивающим возможность
получения двух отдельных шлиренов-
ских пиков на одном снимке. В даль-
нейшем для этой цели использовалась
интерференционная оптика. Каждое
отделение в двухсекторной ячейки за-
ТсЮлица Г4.3. Молекулярные массы М, константы седиментации s°Ori:, парциаль-
ные удельные объемы ъ , пептидов и белков в водных растворителях. Красным
цветом отмечены белки, не имеющие в растворе глобулярной конформации
Пептиды и белки’* (данные взяты из различных лите- ратурных источников) Молекуляр- ная масса (Да) Константа седиментации S« (ед. Сведберга) Парциальный удельный объем Ъ (см3-г‘)
Синтетический пептид I 2049 0,37 0,706
Синтетический пептид II 2777 0,45 0,715
Синтетический пептид III 3505 0,52 0,720
Липаза 6669 1,14 0,7137
Инсулин (димер) И 466 1,60 0,744
Цитохром С 12 400 1,80 0,707
Рибонуклеаза Л 13 683 1,78 0,696
Лизоцим 14 305 1,91 0,702
а-Лактальбумин 14 180 1,92 0,704
Миоглобин 17 836 1,98 0,741
Папаин 23 350 2,42 0,723
а-Хи.мотринсин 25 000 2,40 0,736
Химотрипсиноген Л 25 767 2,58 0,736
Эластаза 25 900 2,60 0,73
Субтилизин 27 537 2,77 0,731
Карбоангидраза 30 000 3,0 0,735
Рибофлавин связывающий белок 32 500 2,76 0,720
Карбоксипептидаза 34 472 3,2 0,725
Пепсин 34 160 2,88 0,725
р-лактоглобулин, (димер) 36 730 2,87 0,751
Домен кинезина (конструкт) К366 41 404 3,25 0,733
Яичный альбумин 44 000 3,6 0,74
Бычий сывороточный альбумин 66 300 4,50 0,735
Гемоглобин 64 500 4,60 0,749
Аптакс (защитный антиген) 85 000 5,01 0,762
Тропомиозин 93 000 2.(1 0,71
Лактатдегпдрогеназа 138 320 7,54 0,741
Р-лактоглобулип А. (октамер) 146 940 7,38 0,751
Апо-гл и нерал ифосфатдеп щрогеназа 142 870 7,60 0,737
Альдолаза 156 000 7,40 0,742
Малатсиитетаза 170 000 8.25 0,735
Каталаза 248 000 11,3 0,730
Глютаматдегидрогеназа 312 000 11,4 0,749
Продолжение таблицы Г4.3
Пептиды и белки‘> (данные взяты из различных лите- ратурных источников) Молекуляр- ная масса (Да) Константа седиментации 4»- (ед. Сведберга) Парциальный удельный объем й (см3-г_|)
Фибриноген 3.30 000 7.6 0,706
Апоферритин 467 000 17,6 0,750
Уреаза 482 700 18,6 0,742
Миозин .570 000 6.13 0,728
Глутаматдегидрогеназа 1 015 000 26,6 0,73
Гемоглобин улитки 3 500 000 58,9 0,747
Гемоцианин 8 950 000 105,8 0,72
* данные взяты из различных литературных источников.
Таблица Г4.4. Коэффициенты седиментации ДНК в форме двойной спирали
Образец Длина молекулы (в парах оснований) Коэффициент седиментации S°Ob. (ед. Сведберга)
Фрагменты, полученные 12 1,86“
разрезанием фага ДНК РМ2 20 2,54“
рестрикционной нуклеазой 50 3,51
Нае III (Kovacic et al., 1972) 94 4,58
117 4,92
145 5,20
160 5,41
263 6,24
288 6,37
322 6,70
498 7,62
592 7,89
642 8,03
794 8,59
854 8,72
1310 9,99
1606 10,67
1735 10,78
Нативная ДНК: 5386 14,3
0Х 174RF 39 937 32,0
XDNA 48 502 34,4
Т4 DNA 168 903 62,1
Т2 DNA 172 000 64,5
Образец Длина молекулы (в парах оснований) Коэффициент седиментации 520,г (еД- Сведберга)
ДНК, тщательно фракциони- 3681’ 7,2
рованная гсль-хроматогра- 2891’ 6,3
фней; 172ь 5,7
отношение М /М 1361’ 5,4
не более 1,1 133ь 5,4
1111' 4,7
68ь 3,92
а) седиментационные коэффициенты фрагментов длиной 12 и 20 пар оснований вычислены
из их диффузионных коэффициентов (гм. i.u'i । 13. i > согласно уравнению Сведберга (уравне-
ние Г.4.22);
б) отношение Mw/Mn, равное 1,1, означает весьма узкое распределение по молекулярной мас-
се I ч I "I V, i.:;i;ill i I ’ )
полняется двумя растворами. Если два
раствора идентичны по седиментиру-
ющим свойствам, то интерференци-
онное изображение будет состоять из
серии прямых полос, параллельных
друг другу. Если граница одного рас-
твора движется более медленно, чем
другого, разница в интерференционных
картинах даст результирующую карти-
ну, сходную с таковой, наблюдаемой с
помощью шлиреновской оптической
системы. Заметим, что такой подход
аналогичен дифференциальной спект-
роскопии (Главы 31 и 32).
Очень малые изменения в значе-
ниях коэффициентов седиментации
могут быть обнаружены методом диф-
ференциальной седиментации. Метод
был оттестирован на примере вируса
карликовой кустистости томата. Раз-
личия в величинах констант седимен-
тации имитировались добавлением в
буфер определенного количества тя-
желой воды. Результаты показали, что
как чувствительность, так и точность
метода дифференциальной седимента-
ции равны 5% даже для значений As/s
ниже, чем 0,002.
Г4.6. Вычисление
молекулярной массы
по данным седиментации
и диффузии
Заменяя в уравнении Г4.15 коэффици-
ент трения частицы на ее коэффициент
диффузии мы получаем для отношения
s/D следующее выражение:
(Г4.21)
D RT 7
Его можно переписать и получить
первое уравнение Сведберга, которое
позволяет вычислить молекулярную
массу комбинацией седиментации
и диффузии (к< >м м<• ।: ।;11)1111 I 1.1S и
I !.!')).
5 RT
D (1-йр0)’
(Г4.22)
Заметим, что при выводе уравнения
Г4.22 предполагается, что поступатель-
ное трение одинаково проявляет себя
в явлениях диффузии и седиментации.
Иными словами величины /(1 и /s яв-
ляются идентичными. Такое предпо-
ложение является строго корректным
Комментарии Г4.18. Молярная масса
Использование единиц системы СИ в уравнении Сведберга (Г4.22) приводит к получению I
значения М, обозначаемого, как молярная масса, в единицах кг-моль'. Мы напоминаем, что
(см. Главу Б1) относительная молекулярная масса или относительный молекулярный вес яв-
ляются безразмерными относительными величинами, определяемыми как отношение массы
молекулы к 1/12 изотопа массы углерода |2С (очень близкое к значению 1 г-моль '). I
Молярная масса (кг-моль1) может быть переведена в молекулярный вес делением на 10 3 :
г-моль 1 (что эквивалентно умножению на 1000 и устранению размерности). Биохимики про-
должают использовать молекулярную массу, выражая ее в дальтонах (Да) или в единицах атом-
ной массы (1 дальтон = 1 единице атомной массы, которая является эквивалентом массы ;
одного атома углерода |2С). ;
Комментарий Г4.19. Вычисление молекулярной массы
Для примера вычислим молекулярную массу макромолекулы, которая имеет парциальный
удельный Б = 0,74 см3/г и движется в воде при температуре 20 °C; ее коэффициент седимен-
тации s° равен 14.2S, а коэффициент диффузии равен D",,v ” 5,82 х Ю 7 см2-сек '.
В соответствии с уравнением Г4.20 имеем:
5°ои, RT 14,2х10"|3(8,31х107)293
М = —г---------=----------=---------------- zz / к Да.
Г>°ои, 1-йр0 5,82х10"7(1-0,74х0,998)
только с теоретической точки зрения.
В реальности, fd и /. измеряются в раз-
ных экспериментальных условиях: fd
под действием слабых или близких к
нулю термодинамических сил, а /. под
действием сильных гравитационных
полей. Такие поля могут деформиро-
вать эластичную молекулу или ориен-
тировать асимметричную молекулу.
Экстраполяция величины 5 к нулевой
угловой скорости позволяет устранить
этот эффект. Полученное таким путем
/. должно быть идентично fd.
Г4.7. Равновесная
седиментация
Г4.7.1. Молекулярная масса
Если центрифугировать долгое время
раствор, содержащий макромолекулы
разной молекулярной массы, то через
некоторое время самые большие мо-
лекулы осядут на дно центрифужной
кюветы, а макромолекулы средней или
малой молекулярной массы распреде-
лятся недалеко от дна кюветы и их по-
ложение не будет меняться во време-
ни, поскольку установится равновесие
между гравитационными и диффузион-
ными силами.
В экспериментах по равновесной се-
диментации объем изначально однород-
ного раствора макромолекул центрифу-
гируется при низкой угловой скорости
по сравнению со скоростью, которая не-
обходима для проведения эксперимента
по скоростной седиментации (коммеп-
шрпй Г 1.20). Начавшемуся процессу
седиментации противостоит процесс
диффузии. После определенного перио-
да времени достигается равновесие, в
котором концентрация макромолекул
повышается экспоненциально по на-
правлению к дну кюветы, по тому же
принципу, как и концентрация газов
распределяется в атмосфере под дей-
ствием гравитационного поля Земли.
Поэтому молекулярная масса осажда-
ющихся молекул может быть вычисле-
на по распределению их концентрации
на разных длинах ячейки.
Уравнение, описывающее распре-
деление концентрации молекул при
равновесной седиментации, получается
приравниванием к нулю суммарного
потока в уравнении Г4.13, поскольку
при равновесии не происходит измене-
ния концентрации во времени:
к = о
и,следовательно:
D (dC/dr), — со2гС$ = 0.
Перегруппировывая и подставляя
D из уравнения Сведберга (уравнение
Г4.22), получаем:
dln(C) _ 1 dC со2с _
d(r /2) г dr D (Г424)
_ М(1 - йр)со2
“ RT ‘
Распределение макромолекул по
концентрации между точками мениска
а и точкой г подчиняется экспоненци-
альному закону:
С (г) =С (а) ехр х
х {ш2М (i-й р)х (Г4.25)
х - a2)/2RT}.
Зависимость In [С (г)\ от г'/2 пред-
ставлена на рисунке Г1.15г/ для мо-
нодисперсного раствора, а на рисунке
Г4.15 б—для полидисперсного раствора.
Комментарий Г4.20. Время
достижения равновесия ।
Время, необходимое для достижения
равновесия, пропорционально квадрату ,
длины столба раствора в радиальном на-
правлении. Если раствор в столбе длиной
6 мм достигнет равновесия через 72 часа, то
тот же раствор в 3 мм столбе — за 24 часа.
В первом случае угол наклона прямой
линии пропорционален молекулярной
массе. В полидисперсных растворах, со-
держащих макромолекулы различной
молекулярной массы, каждая из них
будет распределяться в соответствии
с уравнением Г4.25. Макромолекулы с
наибольшей молекулярной массой кон-
центрируются у дна ячейки, а с малой
молекулярной массой — у поверхности,
и поэтому кривая зависимости 1п[ С(г)]
от i2/2 имеет изгиб направленный
вверх (см. рис. Г4.156). Тангенциаль-
ный наклон в каждой точке кривой дает
среднее значение молекулярной массы
в соответствующей точке ячейки ( ком-
ментарий Г i.2! ).
В уравнение Г4.25 не входит форма
молекулы. В этом особенность метода
седиментационного равновесия. Фор-
ма макромолекулы действует только
на скорость, с которой устанавлива-
ется равновесие, а не на конечный ре-
зультат.
Уравнение Г4.25 может быть исполь-
зовано для оценки скорости вращения ро-
тора, необходимого для установления рав-
новесия. Со стандартными оптическими
системами точное измерение может быть
выполнено, когда концентрация пони-
жается в два раза на 1 мм длины образца
а. Если молекулярнаямассачастицы равна
50000Да,а = 6см,и о =0,75см3-г ’,тогда
со = ЮООрадиан-сек 1 или 10 400 об-мин'1.
На рисунке Г4.16 приведены оптималь-
ные скорости для достижения равновесия,
2.0
Рис. I I. 1.5: а — кривая зависимости логарифма равновесной концентрации от квадрата радиуса
для монодисперсных растворов макромолекул. Наклон кривой пропорционален молекулярной
массе макромолекулы; б — кривая логарифма равновесной концентрации против квадрата радиу-
са для раствора полидисперсных макромолекул
если молекулярная масса или коэффици-
ент седиментации образца приблизитель-
но известен.
Г4.7.2. Константы связывания
Измерение концентрационной зависи-
мости среднего значения молекуляр-
ной массы является очень полезным
при описании ассоциативных явлений,
например, для определения количест-
ва мономеров, димеров и олигомеров.
Для системы гетерогенных ассоциаций
вида А + В о А*В равновесное седи-
ментационное распределение описы-
вается тремя экспонентами, одна для
каждого сорта ассоциации (экспоненты
в сжатой форме обозначены как ст):
Приближенный коэффициент седиментации (ед.Сведберга)
I I I I I I I I
Рис. 1’1.16. Рекомендуемая оптимальная скорость, при которой достигается равновесие, для мо-
лекул с известными молекулярной массой или константой седиментации
С(г) = Сл(г)сл +
в(Фв + Cie(r)o/1B
(Г4.26)
Предэкспоненциальные множители
для А*В-ассоциации могут быть пред-
ставлены в терминах концентраций А, В, и
константы равновесной диссоциации, КЛВ:
C(r) = C.1(r)G,1 +
П < Ч саСв (Г4.27)
+ ^в(Г)°В+ „ &ЛВ’
&АВ
а из закона действующих масс следует:
к С(г)лС(г)в
ЛВ С(г)АВ
"С(г)Лв = С(Г^(Г)в- (Г4.28)
Равновесные константы диссоциа-
ции могут быть прямо определены из
равновесных седиментационных дан-
ных, согласно уравнению Г4.28.
В качестве примера на рисунке 1'1.17
показаны данные и анализ равновесных
констант для двух ассоциирующих бел-
ков, образующих обратимо ассоцииро-
ванную систему типа мономер-димер-
тетрамер. Величины, определенные для
Kt,2 и K2i оказались равными 8,3 х 10-6М
и 2,0 х Ю"6М, соответственно. На ри-
сунке Г4.17 6 показана аппроксимация
данных суммой трех экспонент, а на ри-
сунке Г4.17 а показана разность между
экспериментальными и расчетными дан-
ными. Зависимость вычисленных отно-
сительных концентраций мономеров,
димеров и тетрамеров представлена на
рисунке Г1.17 б как функция общей кон-
центрации мономеров. Отметим, что в
экспериментах по равновесной седимен-
тации можно исследовать макромолеку-
лы в широком диапазоне концентраций
(10 3- 10"8М).
Во втором примере на рисунке 1’4.18
показана зависимость среднего молеку-
Комментарий Г4.21. Среднее
значение молекулярной массы
Вычисление среднего значения молеку-
лярной массы для полидисперсных раство-
ров базируется на разном способе подсче-
та суммарной массы разных компонентов.
Если вычисление основано на численном >
: усреднении, то такой тип среднего значе-
ния известен как средне-численная моле-
кулярная масса, Мп\
Ч = 27^/2™, = Епт, (А)
. где п. есть численная доля молекул с моле-
I кулярной массой т,
i Средне-весовая молекулярная масса Mw
определяется как
Ч =ENim\/EN.mrEwrni, (Б) '
где wi есть массовая доля молекул с массой т, !
; Например, для смеси из двух молекул с мае- I
' сами 10000 Да и 100000 Да М = 55000 Да, а
4= 91818 Да. Очевидно, что отношение
Ч /Ч является мерой полидисперсности
системы. Если это отношение меньше 1,1,
то это означает, что молекулы в смеси име-
ют близкие молекулярные массы.
лярного веса Мп, для карбоангидразы от
концентрации последней, в интервале
значений, перекрывающих три порядка.
Сплошная линия описывает равновесие
тример-гексамер, а прерывистая — рав-
новесие димер-тример. Очевидно, что
данные исключают вариант равновесия
димер-тетрамер. Данные коррелируют
с данными кристаллографии, показыва-
ющими димеризацию двух тримеров в
гексамер путем ассоциации N-концов
шести мономеров в шести цепочечную
структуру с гидрофобной центральной
частью.
Г4.8. Парциальный удельный
объем
Определение молекулярной массы мак-
ромолекул методом равновесной седи-
Рие. 1’1.17: a — данные равновесной седиментации для мутанта (легкая вариабельная цепь им-
муноглобулина) и домена REI (белок Бенс-Джонса), анализируемые в терминах мономер-димер-
тетрамер модели (см. текст); б — утолщенная часть кривой показывает область концентраций, при
которых был выполнен анализ. Утонченная часть кривой получена экстраполяцией (Hensley, 1996)
Рис. Г). 18. Концентрационная зависимость
молекулярного веса для карбоангидразы из
Methanosarcina thermophila. Сплошная линия
описывает равновесие тример-гексамер (Belhke
and Ristau, 1997)
ментации (секция Г4.6) или сочетани-
е*м методов седиментации и диффузии
(секция Г4.7) требует точных знаний
об их парциальном удельном объеме б.
Для простых двухкомпонентных систем,
содержащих один тип растворенного
вещества в одном типе растворителя (в
нашем случае буфер), о есть термоди-
намическая величина, определяемая как
увеличение объема раствора при добав-
лении одного грамма сухого вещества
(см. Главу П).
Стандартная экспериментальная
процедура определения величины пар-
циального удельного объема б обычно
базируется на серии точных измерений
плотности раствора, содержащего рас-
творенные вещества различной весовой
концентрации, zet
р = р0+Д’ (1-рои), (Г4.29)
где р и р0 есть плотности раствора
и растворителя, соответственно. Зна-
чения и получаются из зависимости р
от Z£). Для разбавленного раствора мак-
ромолекул и не зависит от концентра-
ции макромолекул, а зависимость р от
w представляет собой прямую линию.
Парциальный удельный объем, б,
обычно выражается в см3-г *. Анализ
уравнения Г4.29 показывает, что для
получения значение о с приемлемой
точностью необходима очень высокая
точность измерений плотности. Так,
для получения точности в 1 % в вели-
чине и, что соответствует точности
3-4 % в определении молекулярной
массы, плотности растворов должны
быть измерены с точностью более, чем
0,000001 часть при концентрации рас-
творенного вещества 1мг-мл Такую
точность очень трудно получить с по-
мощью стандартной техники, которая
включает пикнометры, магнитные де-
нситометры, электровесы с маятни-
ками, вибрационные денситометры
и аналитическое ультрацентрифуги-
рование в растворителях переменной
плотности. К тому же такие измерения
очень трудоемки.
Еще в 1950 г. Кон и Эдсалл выска-
зали предположение о том, что пар-
циальный удельный объем белков и
карбогидратов может быть вычислен с
необходимой точностью по их амино-
кислотному или моносахаридному со-
ставу, соответственно, при допущении
аддитивности объемов их остатков
й ^i^W./E.W, (Г4.30)
где IV и о . есть весовые доли и парци-
альные удельные объемы, соответствен-
но, I остатков. Объемы остатков приве-
дены в ia6.nine Г 1.5 для аминокислот,
а । ао. ипц- П.бдля карбогидратов.
За прошедшие годы было предпри-
нятомногопопытокулучшить изначаль-
ный подход Кона и Эдсалла, например,
учитывать разницу в объемах остатков в
зависимости от их расположения внут-
ри или снаружи молекулы. Однако все
они не привели к улучшению конечного
результата и на сегодня стало общепри-
нятым считать, что именно метод Кона
Таблица Г4.5. Парциальные удельные объемы при 25 °C, о
и молекулярные массы М аминокислотных остатков белков*
Аминокислота М(Да) О (см3-г‘)
Глицин 57 0,64
Аланин 71 0,74
Серин 87 0,63
Треонин 101 0,70
Валин 99 0,86
Лейцин ИЗ 0,90
Изолейцин ИЗ 0,90
Пролин 97 0,76
Метионин 131 0,75
Фенилаланин 147 0,77
Цистеин 103 0,61
Триптофан 186 0,74
Тирозин 163 0,71
Гистидин 137 0,67
Аргинин 156 0,70
Лизни 128 0,82
Аспарагиновая кислота 115 0,60
Глутаминовая кислота 129 0,66
Глутамин 128 0,67
Аспарагин 115 0,60
* Данные взяты из Cohn and Edsall (1965)
Таблица Г4.6. Парциальные удельные объемы при 25 °C, п и молекулярные
массы, М, сахаров*
Карбогидрат М и (см3-г*)
Фруктоза 180 0,614
Фукоза 164 0,671
Галактоза 180 0,622
Глюкоза 180 0,622
Гексоза 180 0,613
Сахароза (0.05 М) 342 0,613
Сахароза (0.15 М) 342 0,616
Сахароза (1 М) 342 0,620
Рафиноза 486 0,608
* Данные взяты из Chervenka (1969)
Таблице! Г4. /. Приблизительные значения парциальных удельных объемов v ,
для основных типов биологических макромолекул
Макромолекулы 1) (см3-г1)
Белки 0,73 (0,70-0,75)
Карбогидраты 0,61 (0,59-0,65)
РНК 0,54 (0,47-0,55)
ДНК 0,57 (0,55-0,59)
и Эдселла дает результаты, наиболее
близкие к экспериментальным значе-
ниям (>ммеи । npiiii Г !.:>?.). Типичные
значения величин 6 для основных ти-
пов биологических макромолекул пере-
числены в гиб.nine I 1.7.
Г4.9. Седиментация
в градиенте плотности
Метод седиментации в градиенте плот-
ности в исследовании нуклеиновых
кислот и белков имеет длительную
историю и сегодня находит широкое
применение в различных областях — от
определения состава генома до харак-
теристики взаимодействий белок-де-
тергент, белки-нуклеиновые кисло-
ты, белки-липиды. Ниже мы опишем
классический подход, базирующийся
на аналитическом скоростном зональ-
ном центрифугировании и равновесной
седиментации в градиенте плотности
( к< )М м('! Г1;11 в I i I I ‘ 1 .'.!).
Г4.9.1. Метод скоростного
зонального центрифугирования
Образец раствора, содержащий смесь час-
тиц, наслаивается на поверхность вещест-
KoMM(?Hiapi-in Г-4.22. Предсказание
величины парциального удельного
объема
Парциальный удельный объем в насто-
ящее время вычисляется по химическо-
му составу белков с помощью программы
, SEDNTEP (Harpaz et al., 1994).
ва, создающего градиент повышающейся
плотности (рис. Г4.19<7). При центри-
фугировании частицы проходят через
градиент, образуя зоны в соответствии
с их скоростью седиментации. Центри-
фугирование прекращается прежде чем
частицы, разделенные на зоны, осядут
на дно ячейки (рис. 1'4.196). Максималь-
ная плотность градиента выбирается так,
чтобы она не превышала ту плотность,
при которой частица находит свою зону.
Плотность и вязкость градиента среды, а
также скорость центрифугирования вли-
яют на разделение. Растворы сахарозы и
глицерина наиболее часто используются
для создания градиента при скоростном
зональном центрифугировании. Линей-
ный градиент 5-20 % сахарозы является
традиционным выбором для определе-
ния коэффициентов седиментации бел-
ков и нуклеопротеидов. Методом скоро-
стного зонального центрифугирования
можно разделить смесь молекул по их
: Комментарии Г4.23. Динамический
градиент плотности
Недавно было предложено еще одно по-
нятие градиента плотности, получившее
название динамического градиента плот-
ности для осаждающихся растворенных
веществ. Оно основано на численном ре-
шении уравнения Ламма с конкретными
пространственными и временными вариа- ।
циями плотности и вязкости локального J
растворителя. Его описание выходит за i
рамки этой книги, и читатель отсылается
к соответствующей литературе (Schuck, :
2004).
коэффициентам седиментации, которые
определяются их размером, формой и
плавучей плотностью, а также опреде-
лить относительную молекулярную мас-
су (iji.MMcnГ1.21). Глобулярные
белки с большей молекулярной массой и
большей плотностью осаждаются ближе
к дну пробирки, а таковые с меньшими
размерами и плотностью остаются ближе
Рис. Г4.19. Скоростное зональное разделение смеси частиц в градиенте плотности: а — образец
содержит смесь частиц различного размера, формы и плотности, который наслаивается на по-
верхность вещества, создающего градиент. Плотность увеличивается по направлению к дну про-
бирки; б — в результате частицы разделяются в зависимости от их размера, формы и плавучей
плотности. Поделенные на фракции молекулы отбираются из различных зон градиента
Комментарий Г4.24. Получение
коэффициентов седиментации
методом скоростного зонального
центрифугирования
Коэффициенты седиментации, полу-
чаемые методом зонального центрифуги-
рования в градиенте сахарозы, могут быть
ошибочными для белков с обратимой са-
моассоциацией или взаимодействующих
с другими макромолекулами. Источником :
ошибки является молекулярный эффект
уплотнения, обусловленный высокой кон- .
; центрацией сахарозы, из-за чего сдвигается •
равновесие в пользу ассоциированных мак-
ромолекул. В этом случае рекомендуется
использовать классический метод АУЦ для
точного определения коэффициентов седи-
ментации (см. секцию Г.4.5).
к поверхности пробирки. Для иллюст-
рации метода на рисунке Г4.20 показано
разделение 22S рибонуклеопротеидных
(РНП) частиц методом центрифугирова-
ния в градиенте сахарозы. Пик I соответс-
твует 22S РНП. Анализ этих частиц дан
ниже в параграфе Г4.10.4.
Г4.9.2. Метод равновесной
седиментация в градиенте
плотности
Большие силы, которые генерируются
при центрифугировании, могут быть ис-
пользованы для создания естественного
градиента плотности, образованного не-
большими растворенными молекулами.
Такой градиент может быть достаточно
просто реализован на практике при ис-
пользовании солей растворов тяжелых
металлов, таких как цезий хлор (CsCl)
или рубидий хлор (RbCl). В поле тако-
го градиента исследуемые макромоле-
кулы будут распределяться по соответ-
ствующим правилам. Рассмотрим
молекулярный раствор, состоящий из
трех компонентов: молекулы раствори-
теля (компонент 1), исследуемые макро-
молекулы (компонент 2), и низкомоле-
кулярные растворенные вещества (CsCl
и RbCl) (компонент 3).
Из уравнения Г4.23 следует, что
низкомолекулярные растворенные ве-
щества будут распределяться в ячей-
ке таким же образом, что и большие
молекулы. Для частиц с малой моле-
кулярной массой (компонент 3), мы
можем удержать только первые члены
разложения экспоненты в ряд в урав-
нении Г4.23 и тогда оно примет вид:
^Ц = 1 + Л/3(1-ир0)йГ х
Г~ - (Г
X——---.
2RT
(Г4.31)
Уравнение Г4.29 показывает, что
в ячейке распределение концентрации
компонента 3 подчиняется параболичес-
кому закону. Поскольку плотность рас-
творителя является линейной функцией
концентрации компонента 3, то при рав-
новесии достигается градиент плотности,
который в виде параболы представлен на
рисунке Г).21. Если плотность, р, компо-
нента 2 располагается между двумя зна-
2 5 ;
00..............................
0 4 s
Фракционное число
Рис. Г4.20. Выделение 22S рибонуклеопроте-
идных частиц методом центрифугирования в
градиенте сахарозы 5-20%, при 50000 об мин ’,
в течение 3 час, при температуре 20 °C, в роторе
SW 55 (Ulitin et al., 1997)
50
Рис. Г4.21. Равновесное центрифугирование ДНК в градиенте CsCl: а — распределение градиен-
та плотности в начале эксперимента; б — параболический градиент плотности при равновесии
чениями крайних плотностей растворите-
ля, то макромолекула будет устремляться
в ту область ячейки, где ее плавучая плот-
ность равна нулю, т. е. р = р0. Ширина по-
лосы компонента 2 является функцией
макромолекулярной массы и крутизны
градиента локальной плотности. Для мо-
лекул с большими молекулярными мас-
сами полосы будут очень узкими из-за
их маленького коэффициента диффузии.
Нативная ДНК, например, дает такие
узкие полосы, что меченая по 15N (тяже-
лый азот) молекула четко отделяется от
природной молекулы, MN (легкий азот)
при равновесном центрифугировании в
градиенте плотности CsCl (киммсп lapini
I 1.25). Напротив, для веществ с малень-
кими молекулярными массами ушире-
ние полос настолько велико, что метод
практически неприменим для глобуляр-
ных белков.
Наиболее удивительным свойством
модели двойной спирали ДНК Уотсона и
Крика является комплементарность двух
цепей ДНК, по принципу которой и про-
исходит удвоение цепи. В результате по-
являются две дочерние дуплексные мо-
лекулы ДНК, одна из которых является
родительской цепью ДНК. Этот процесс
получил название полуконсервативного
механизма репликации и показан на ри-
сунке Г4.22. Для экспериментального до-
казательства этого механизма Меселсон
и Сталь поставили опыты, суть которых
кратко состоит в следующем. Они вырас-
тили клетки Е. соН на среде, содержащей
в качестве источника тяжелого азота,
15N хлорид аммония. Затем меченые 15N
азотом клетки Е. соН были перенесены
на среду с обычным азотом 14N для про-
должения их роста. Образцы отбирали
через определенные интервалы времени.
Затем из клеток экстрагировали ДНК,
плавучую плотность которой определяли
методом центрифугирования в градиенте
плотности CsCl. После первого деления
выделенная ДНК шла одной полосой в
градиенте плотности, положение кото-
Комментарий Г4.25. 15N (тяжелые)
и 14N (легкие) изотопы
При равновесной седиментации в CsCl,
различие в плотности молекул ДНК, содер-
жащих l5N и "N изотопы равно 0,014 г-см 3.
Результатом этого является тот факт, что
расстояния в стандартной ультрацент-
рифужной ячейке между полосами та-
ких образцов равны 0,5 мм при скорости
: 40000 об-мин '. Именно эти изотопы были
использованы в опытах Меселсона и Сталя
по исследованию механизма репликации
ДИК (Messelson and Stahl, 1957).
рой было между зонами ДНК с тяжелым
и обычным азотом (рис. Г-1.23 «). После
двух делений на ”N среде выделенная
ДНК давала две полосы, одну с плотнос-
тью, равной легкой ДНК, другую с проме-
жуточной плотностью, равной гибридной
ДНК, наблюдаемой после одного деле-
ния (рис. Г1.23/). После трех делений
в UN среде ДНК давала еще две полосы,
подобные тем, которые наблюдались пос-
ле двух делений (рис. Г4.23()). Именно
такой результат — наличие полосы с про-
межуточной плотностью после первого и
последующих делений, и ожидался при
полуконсервативном механизме деления
ДНК.
Г4.10. Форма молекул
по данным скоростной
седиментации
Уравнение Г4.21 соотносит коэффици-
ент седиментации, 5, с такими парамет-
рами молекулы, как М, D и й . Как мы
указывали ранее, коэффициент седи-
ментации зависит от объема и формы
частицы. Он обычно выражается в по-
нятиях радиуса эквивалентной сферы
(радиус Стокса), но в равной степени
может соответствовать различным
комбинациям «объем-форма». Напри-
мер, удлиненная частица малого объ-
ема может иметь тот же радиус Стокса,
что и сферическая частица, но больше-
го объема (параграф Г2.2.1). Отсюда
следует, что прямое определение фор-
мы молекул по величине коэффициен-
та седиментации невозможно.
Как мы уже указывали ранее, моле-
кулы одинаковой формы, но различной
молекулярной массы, образуют гомоло-
гичные ряды (см. секцию Г2.6). В этом
случае соотношение между s и М следу-
ет из уравнения Г2.64:
Рис. Г-4.22. Полу консервативный механизм
репликации ДНК. Каждый вновь образован-
ный дуплекс на первой стадии деления (F )
содержит одну родительскую цепь. 11а второй
стадии (F2) генерация состоит из двух гиб-
ридных ДНК и двух полностью новых ДНК.
Вновь реплицированная цепь обозначена се-
рым цветом, родительская — черным
s = KsMa, (Г4.32)
где величина экспоненты, а, зависит от
формы молекул в гомологичном ряду
(секция Г2.6). Практическая польза
анализа экспоненты, а, для различных
гомологичных серий проиллюстриро-
вана ниже (ком мен гарпй Г i,2(i).
Г4.10.1. Гомологичный ряд
квазисферических частиц:
глобулярные белки в воде
Как следует из изложенного ранее (па-
раграф Г2.2.1), коэффициент трения,/(),
сферы радиусом, Ro, в растворителе вяз-
костью, г|0, выражается законом Стокса:
/0 = 6nr]0 Ro (уравнение Г2.1). Радиус
сферы связан с ее объемом, Го, как:
4/ЗпЯ03 = Va=Mv /Ny (Г4.33)
Рис. Г4.23 Схематическое представление результатов экспериментов Меселсона и Сталя:
а — контроль: смесь тяжелой (15N) и легкой (14N) ДНК; б — исходное деление начинается с ро-
дительской тяжелой ДНК; в — ДНК после первого деления на l4N среде; г — ДНК после двух
делений па 14N; д — ДНК после трех делений на 14N среде
Поэтому:
5* = 12,0 х 10~3M2''3. (Г4.36)
/0 = 6пг]0(ЗМи/4тт Д/)13. (Г4.34)
Подставляя этот результат в урав-
нение (Г4.16), мы получаем после пере-
группировки:
*’= 4,г й1/3/(1 - up) =
(Г4.35)
= М2/3/6лг]()У12/3(3/4тт)1/3.
Таким образом, теоретическая за-
висимость s’ от М в двойной логариф-
мической шкале для ряда сферических
молекул должна быть прямой линией с
наклоном 2/3. Когда s"Olr выражается
в единицах Сведберга, а все другие па-
раметры в системе СГС, то уравнение
(Г4.33) принимает вид:
Экспериментальная зависимость s’ от
М в двойном логарифмическом масштабе
приведена на pi icvhkc 1'4.24 для глобуляр-
ных белков в широком диапазоне моле-
кулярных масс по данным, приведенным
в пи'стце Г i.3. Она хорошо аппроксими-
руется прямой линией с параметрами:
s* = 9,1х10-3 М065, (Г4.37)
которые указывают на то, что реальные
глобулярные белки действительно об-
разуют гомологичный ряд.
Сравнение уравнений Г4.36 и Г4.37
показывает, что хотя экспоненты в этих
уравнениях совпадают, коэффициенты у
них различны. Уравнение Г2.1 предска-
зывает, что малые отклонения от совер-
шенной сферической формы и существо-
вание гидратационной оболочки меняют
соотношение между радиусом Стокса и
парциальным удельным объемом без из-
менения величины показателя степени.
Из этого факта мы приходим к заключе-
нию, что белки по форме очень близки к
сфере и гидратированы приблизительно
в одинаковой степени.
В ;;iC. 1И!1л I i .'i включены три белка,
(тропомиозин, фибриноген, миозин),
которые не подчиняются уравнению
Г4.37. Они отмечены в таблице крас-
ным цветом. Их коэффициенты седи-
ментации меньше, чем таковые для
глобулярных белков такой же молеку-
лярной массы (см. рис. Г4.24). Имеются
две предпосылки для такого поведения:
развернутость молекулы или удлинен-
ная (палочкообразная) форма. Особый
интерес представляет молекула кол-
лагена, структура которого и его фраг-
ментов, полученная ультразвуковой
обработкой, обсуждается в ьоммеи i;i-
рпи Г 1.127.
Г4.10.2. Гомологичный ряд
клубкобразных молекул: белки
в 6М гуанидингидрохлориде или
в 8М мочевине
Большинство белков диссоциируют на
отдельные пептидные цепи и развора-
чиваются в 6М гуанидингидрохлориде
или в 8М мочевине, содержащих 0,1 М
меркапоэтанола. На рисунке Г4.25 по-
казана зависимость s°2Q !i,/(1 — й р0) от
п, для белков в 6М гуанидингидрохло-
риде в двойной логарифмической шка-
ле (и — число аминокислотных остатков
в полипептидной цепи, а й — кажу-
щийся парциальный удельный объем).
Эта зависимость представляет собой
хорошую прямую линию, которая соот-
ветствует уравнению:
Комкп'ч;;ip:n। Г4.26. Уравнения
Куна-Марка-Хаувинка (КМХ)
Как мы обсуждали в секции Г2.6, для
гомологичных серий частиц нерпы следу-
ющие уравнения:
[41= (Л)
s=K'M“, (Б)
D = K„M (В)
Взаимосвязь между константами а, а и
Ь, выражается как:
b + а = 1, (Г)
Ь = (1+а)/3. (Д)
Таким образом, для гомологичных се-
рий должны выполняться следующие соот-
ношения.
Для жестких палочкообразных частиц:
а = 1,7; а = 0,15; b = 0,85. (Е)
Для жестких сферических частиц:
а = 0; а = 2/3;Л= 1/3. (Ж)
Для гауссовых клубков в «идеальных»
растворителях:
а = 0,5; а = 0,5; b = 0,5. (3)
Для гауссовых клубков в «хороших»
растворителях:
а = 0,8; а = 0,4; b = 0,6. (И)
$°/(1 - й р0) = 0,286 и0-47. (Г4.38)
Величина показателя степени а
в уравнении Г4.37 близка к таковой,
свойственной клубкообразным поли-
мерам в растворителях, близких к «иде-
альным» (см. уравнение И м >ммi: i
I' i /'() И 1,1 I i i; 11) i I ii I i./s ).
Г4.10.3. От коротких жестких
палочек к гибким клубкам:
конформация ДНК
Исследование молекул ДНК является
хорошим примером того, как получить
сведения о форме молекулы по дан-
Молекулярная масса (Да)
Рис. I'4.24. Зависимость 5®0г(, й 1 3/(1 -Йр0) от М в двойной логарифмической шкале для
белков в широком интервале значений молекулярных масс, построенная по данным
;ы I Для глобулярных белков наклон прямой, проведенной через экспериментальные точ-
ки, равен 2/3, который типичен для квазисферических частиц. Исключением из этого правила
являются частицы, форма которых отлична от сферической: небольшие синтетические пептиды
(развернутые цепи), тропомиозин, фибриноген, миозин (палочкообразные частицы)
ным седиментации. Уже в 60-70-х гг.
прошлого столетия было известно, что
зависимость константы седиментации
от молекулярной массы для препара-
тов ДНК в двойной логарифмической
шкале не описывается одной прямой
линией в широком интервале значений
молекулярных масс. Так, для ДНК в
интервале молекулярных масс Зх 10э
до 4 х Ю6 Да зависимость седиментаци-
онных коэффициентов аппроксимиро-
валась уравнением:
=0,116 М °-325, (Г4.39)
а выше 4x10° Да — уравнением:
0,034 М0105. (Г4.40)
Следует оговориться, что эти дан-
ные были получены на препаратах
ДНК, каждый из которых не был го-
i Комментарий Г4.27. Коллаген
| и его фрагменты
При обработке растворов коллагена
кожи теленка ультразвуком получаются
фрагменты разной длины, которые сохра-
няют исходную спиральную структуру. За-
. висимость коэффициента седиментации от
молекулярной массы может быть описана
как:
S°20„. = 0,232 Л/ 0 20. (А)
| Небольшая величина экспоненты в
! уравнении (А) является неоспоримым ар-
гументом в пользу того, что коллаген и его
фракции представляют собой гомологич-
ную серию очень жестких палочкообразных
молекул, для которых асимметрия растет
по принципу «голова к хвосту» (Nishihara
and Doty, 1958). Только такой способ роста
асимметрии молекул обеспечивает малую
i величину показателя степени в уравнении
(А) и. напротив, большую величину пока-
зателя степени в уравнении Г2.60.
Log n (число остатков в цепи)
Рис. Г4.25. Зависимость s20o, /(1 - йр0)отп,
для белков в 6 М GuHCl в двойной логариф-
мической шкале (п — число аминокислотных
остатков в полипептидной цепи, а и кск есть
кажущийся парциальный удельный объем)
(Tanford et al., 1967)
Комментарии 1'4.28. «Идеальные»,
«хорошие» и «плохие» растворители
Термин «идеальный» или «тета» раство-
ритель в полимерной химии означает ра-
1 створитель, в котором второй вириальный
коэффициент равен нулю (см. секцию
А1.2). Иными словами, в таком раство-
рителе контакты полимер-полимер будут
тождественны контактам «полимер-рас-
творитель». Термин «хороший» раство-
ритель означает растворитель, в котором
второй вириальный коэффициент положи-
тельный. В таком растворителе контакты
«полимер-растворитель» будут предпоч-
тительны контактам «полимер-полимер».
В этом случае молекула набухает и ее
। размеры будут всегда больше размеров
в идеальном растворителе. И, наконец,
в «плохом» растворителе контакты «по- ,
лимер-полимер» предпочтительнее кон-
тактов «полимер-растворитель». В таком 1
растворителе молекула поджимается и при
. определенных условиях может выпасть
в осадок (Цветков и др. 1975).
могенен. Большинство препаратов
были получены ультразвуковой де-
струкцией исходных нативных ДНК.
В настоящее время имеется возмож-
ность получать гомогенные фрагменты
двуспиральных ДНК и вычислять их
молекулярную массу из нуклеотидной
последовательностям, что дает воз-
можность получения более детальных
данных о зависимости коэффициентов
седиментации от молекулярной мас-
сы (рис. 1'1.26). По новым данным для
ДНК в интервале молекулярных масс
от 3 х 105 до 4 х 106 Да, зависимость се-
диментационных коэффициентов ап-
проксимируется уравнением:
4„. = 0,116 М °273, (Г4.41)
а выше 4 х 106 Да — уравнением:
=0,034 М° ио. (Г4.42)
Сравнение величин вновь получен-
ных степенных показателей (уравнения
Г4.42) и Г4.41) с ранее полученными
(уравнения Г4.39 и Г4.40) обнаруживает,
что новые данные лучше выявляют ха-
рактер как клубкообразного (увеличение
а от 0,405 до 0,440), так и палочкообраз-
ного состояния {а уменьшается от 0,325
до 0,273).
Интересно отметить, что седимен-
тационные свойства коротких фрагмен-
тов ДНК (12-20 пар оснований) близ-
ки к таковым для глобулярных белков
соответствующей молекулярной массы
(рис. Г4.27). В то же самое время эти
свойства очень отличаются для боль-
ших молекулярных масс. Этот факт
отражает принципиальные различия
между этими двумя гомологичными
рядами. В ряду глобулярных белков
асимметрия остается постоянной при
повышении молекулярной массы, а в
ряду ДНК асимметрия наиболее быст-
ро растет в ряду относительно коротких
фрагментов. Темп этого роста замедля-
ется по мере увеличения молекулярной
массы и для очень больших молекуляр-
ных масс темп роста асимметрии при-
ближается к таковому, характерному
Рис. I 4.26. Зависимость5°20 в от молекулярной массы (длины оснований) в двойной логарифми-
ческой шкале для фрагментов ДНК. Данные взяты из ню . i ; \
Рис. 1 4.27. Зависимость величины s020 и_, поправленной на Архимедов фактор плавучести (1 —
V р0), от молекулярной массы М в двойной логарифмической шкале для глобулярных белков и
для РМ2 Нае III фрагментов ДНК. Данные взяты из / i , и i , соответственно. Коэффи-
циенты седиментации для фрагментов длиной 12 и 20 пар оснований вычислены из эксперимен-
тальных значений их коэффициентов диффузии: 13,4 и 10,9 единиц Фика, соответственно. При
таком расчете парциальный удельный объем ДНК принят равным 0,56 см3 г1
для гауссова клубка (см. также дискус-
сию в секции Г6.6).
Г4.10.4. Рибосомные РНК,
рибосомные частицы
и рибонуклеопротеидные
комплексы
Исследования седиментационного по-
ведения рибосомных частиц и их компо-
нентов (рибосомные РНК, рибосомные
белки и комплексы между ними) пред-
ставляют особый интерес, поскольку
часто дают ответы на многие вопросы
их структурной организации. Напри-
мер, что общего между конформацией
рибосомных частиц и глобулярных бел-
ков? Как мы сейчас покажем, ответ на
этот вопрос можно получить достаточ-
но просто, если сравнить между собой
зависимости седиментационных коэф-
фициентов от молекулярной массы для
этих двух типов биологических моле-
кул.
На рисунке 1'4.28 приведена зави-
симость константы седиментации, по-
правленной на Архимедов фактор, для
рибосомных частиц и их субчастиц в
двойном логарифмическом масштабе.
Рис. I 4.28. Зависимость константы седимен-
тации, поправленной на Архимедов фактор,
для рибосомных частиц и их субчастиц, в
двойном логарифмическом масштабе. Данные
взяты из :
Данные взяты из i; л: i::Г i.9. Как вид-
но из рисунка, зависимость хорошо ап-
проксимируется прямой линией с пара-
метрами:
•С./(1 - йр0) = КМ"№. (Г4.43)
Наклон этой прямой равен 0,66, ко-
торый, как мы видели ранее, типичен
для компактных квазисфернческнх час-
тиц (секция Г4.10.1). Таким образом,
величины показателей степени в уравне-
ниях Г4.35 и Г4.41 совпадают. Разнятся
только коэффициенты К. Отсюда следу-
ет, что глобулярные белки и рибосомные
частицы сходны по компактности, но от-
личаются по гидратации.
Другой вопрос, ответ на который
также представляет интерес: «Что
общего между конформациями рибо-
сомных РНК в безмагнпевом буфере
и белков в 6М гуанидингидрохлори-
де?». Ответ на него следует из построе-
ния зависимости константы седимен-
тации, поправленной на Архимедов
фактор, для рибосомных РНК и их
фрагментов в двойной логарифмиче-
ской шкале в буфере без ионов Mg”
(рис. 1'4.29) и комментарии Г!.29.
Данные взяты из । ай. i и:iы Г i.9. Эта за-
висимость аппроксимируется прямой
линией:
•4л А1 - й р) = К М017, (Г4.44)
наклон которой совпадает с наклоном
прямой для глобулярных белков в 6М
гуанидинхлориде (уравнение Г4.37).
Это совпадение свидетельствует о том,
что, несмотря на принципиальные раз-
личия в химическом составе и конфор-
мации рибосомных РНК в буфере без
ионов Mg + (молекулы РНК сохраня-
ют элементы вторичной структуры)
и белков в буфере, содержащем дена-
турирующий растворитель, их грубая
100
С°
^20,W
1- иро
50
О
\,0.47
10
5 L
10
ю2
103
Молекулярная масса (кДа)
Рис. I 1.29. Зависимость величины х°,0 а, поправленной на Архимедов фактор плавучести
(1 — V рп), от молекулярной массы М в двойном логарифмическом масштабе для различных ри-
босомных РНК (•) и их фрагментов (°) по данным i. A. cci:,; I 2\ Парциальный удельный объем
РНК и ее фрагментов принят равным Й =0,537 см3-г‘
конформация описывается как развер-
нутое состояние, напоминающее гаус-
сов клубок.
В заключение заметим, что сравне-
ние характеристик седиментации РНП-
Комменшрии 94.29. фрагменты 16S
! рибосомной РНК
Фрагменты 16S рибосомной РНК из
Т. thermophilus, представленные 3' доменом
(нуклеотиды 890-1515), 5' доменом (нук-
I леотиды 1-539) и центральным доменом
(нуклеотиды 547-895), были получены
методом транскрипции in vitro. Инкуба-
; цией этих фрагментов с 30S рибосомными
! белками Т. thermophilus получены специфи-
ческие рибонуклеопротеидные комплексы
(Agalarov et al., 1999).
комплексов, моделирующих три основ-
ных домена, с таковыми для природных
рибосомных частиц показывает, что
комплексы имеют ту же компактность,
что природные частицы (рис. Г4.30).
Это следует из того факта, что констан-
ты седиментации для всех РНП-комп-
лексов лежат на продолжении прямой,
полученной для рибосомных частиц и
их субчастиц (рис. Г4.31). Этот резуль-
тат позволяет предположить, что каж-
дый из трех доменов 30S субъединицы
T.thermophilus способен к самоорганиза-
ции in vitro, независимо друг от друга, а
22S РНП-комплекс может рассматри-
ваться как компактное «обезглавлен-
ное» производное 30S рибосомной час-
тицы.
'аб• л id I 4.8. Константы седиментации, молекулярные массы и парциальные
удельные объемы рибосомных частиц, их субчастиц и РНП комплексов*
Частица Молеку- лярная масса (Да) Коэффици- ент седимен- тации, (ед. Свед- берга) Парци- альный удельный объем, Б (в см’-г1) Ссылка
Т4 РНП комплекс Е. coli 70000 5,8 0,63 1
ТЗ РНП комплекс Е. coli 93000 6,1 0,64 1
Центральный домен 30S Т. th. (547-895) 195000 12,0 0,61 2
5’-домен Т. th. (1-539) 242000 13,0 0,58 3
3’ домен Т. th. (890-1373) 280000 15,4 0,61 4
«Безголовая» 30S субчастица Т. th 427000 22S 0,59 6
30S субчастица Е. coli 910000 30,8 0,612 5
50S субчастица Е. coli 1550000 50,0 0,597 5
70S частица Е. coli 2650000 70,0 0,610 5
80S частица дрожжей 3700000 80,0 0,630 5
80 S частица Artemia salina 3800000 81,0 0,630 5
80S частица Euglena gracilis 4400000 86,0 0,630 5
‘Данные взяты из: 1) Agalarov and Williamson (2000); 2) Agalarov et al. (1999); 3) Agalarov et al.
(1998); 4) Selivanova et al. (неопубликованные данные); 5) Ulitin et al. (1997); 6) Serdyuk et al. (1999)
Таблица Г4.9. Рибосомные РНК и их фрагменты в безмагниевом буфере,
содержащем 100 мМ КО
Частица Число аминокислот- ных остатков в цепи Коэффициент седиментации s!^. (ед. Сведберга)
5S RNA хлорпластов 100 4,5
5S RNA Е. coli 120 5,0
5S RNA крысы 160 5,8
З’-фрагмент (1000-1542) 16S РНК Е. 542 9,0
coli
5’-фрагмент (1-530) 16S РНК Е. coli 530 9,5
З’-фрагмент (535-1542) 16S РНК Е. coli 1007 12,0
5’-фрагмент (1-996) 16S РНК Е. coli 996 12,0
16S РНК Е. coli 1542 16,0
16S РНК хлорпластов 1490 16,0
16S РНК Т. thermophilus 1515 16,0
18S РНК дрожжей 1789 18,0
23S РНК Е. coli 2904 23,0
28S РНК крысы 4750 28,0
РНК вируса табачной мозаики 6363 31,0
Данные по рибосомным РНК взяты из Spirin A. S in «Ribosomes» Kluwer academic/Plenum
Publishing, USA, 1999. Данные по фрагментам РНК получены И. Н. Сердюком (неопублико-
вано).
a
Рис. I 1.30: а и б — три основных домена 16S РНК малой рибосомной субчастицы. 5'домен (нук-
леотиды 1-563), соответствует центральному телу 30S субъединицы, центральный домен (нук-
леотиды 564—915) участвует в формировании бокового выступа или платформы, в то время как
3' основной домен (нуклеотиды 919-1396) локализован в головке субъединицы; имеется так-
же дополнительный 3' минорный домен (остаток 1397-1542) (Ramakrishnan and Moore, 2001);
в — «обезглавленная» 30S рибосомная субчастица (см. рис. П.21) соответствует 5' концевому и
центральному доменам 16S РНК Т. thermophilus. Процедура получения последней основана на
разрезании олигодезоксирибонуклеотида в белок-дефицитной производной 30S рибосомной
субъединицы с помощью рибонуклеазы Н (Serdyuk, et al., 1999)
Рис. Г1.31. Зависимость величины s°,(|w, поправленной на Архимедов фактор плавучести (1 — U р0),
от молекулярной массы М в двойной логарифмической шкале для различных рибосомных частиц,
их субчастиц (О), и РНП частиц (•) с использованием данных, представленных в А. i i
Г4.11. Перечень ключевых идей
• Внедрение в практику в 1990-х гг. новой ультрацентрифуги Beckman Instru-
ments, Optima XL, открыло новую эпоху в аналитическом ультрацентрифуги-
ровании (ЛУЦ).
• В АУЦ используются два комплементарных метода: скоростная седиментация
и равновесная седиментация.
• Метод скоростной седиментации обеспечивает информацию о размере, фор-
ме макромолекул и взаимодействии между ними; высокая скорость вращения
ротора и длинные центрифужные ячейки используются для улучшения раз-
решения.
• Равновесная седиментация обеспечивает информацию о молекулярной массе,
константах ассоциации и стехиометрии; низкая скорость вращения ротора и
короткие центрифужные ячейки используются для уменьшения времени до-
стижения равновесия.
• Отношение измеренной скорости частицы к ее ускорению в ультрацентрифуге
называется коэффициентом седиментации, х. За единицу константы седимен-
тации s принимается единица Сведберга = 1013 секунды, обозначаемая как 1S.
• Поведение макромолекул в ячейке аналитической ультрацентрифуги описыва-
ется уравнением Ламма, которое не имеет аналитического решения.
• Метод «средней точки», долгое время использовавшийся в качестве основного
способа вычисления констант седиментации, в современной практике АУЦ не
используется.
• Применение современных компьютерных программ дает возможность реше-
ния уравнения Ламма в различных граничных условиях методами численного
интегрирования; они позволяют существенно улучшить разрешающую способ-
ность АУЦ при исследовании белков и их смесей.
• Метод Ван Холде-Вайшета базируется на том, что скорость седиментации про-
порциональна времени, тогда как скорость диффузии - корню квадратному из
времени; поэтому экстраполяция к бесконечному времени позволяет исклю-
чить вклад диффузии в форму седиментационной границы.
• В методе Стаффорда распределение коэффициентов седиментации вычисляет-
ся из производной по времени профиля седиментационной кривой; этот метод,
как и метод Ван Холде-Вайшета, дает возможность одновременного определе-
ния коэффициентов диффузии и седиментации и, следовательно, молекуляр-
ных масс исследуемых макромолекул.
Парциальный удельный объем белков и карбогидратов можно вычислить из их
химического состава по методу Кона и Эдселла.
• Молекулярная масса макромолекулы может быть определена из уравнения
Сведберга из ее коэффициентов седиментации и диффузии или методом рав-
новесной седиментации.
• Равновесное центрифугирование в градиенте плотности является мощным тех-
ническим приемом для разделения макромолекул ио плавучей плотности.
• Метод дифференциальной седиментации позволяет уловить очень небольшие
различия в коэффициентах седиментации.
• Глобулярные белки в водном растворе образуют гомологичный ряд квазисфе-
рических частиц с показателем степени в уравнении КМХ, равным 2/3; такой
же ряд образуют в буфере с ионами Mg рибосомные частицы и их субчастицы,
«обезглавленная» 30S субчастица и РНП-комплексы, моделирующие три ос-
новных домена 30S субчастицы.
• Белки в 6М гуанидингидрохлориде образуют гомологичный ряд рыхлых клуб-
ков с показателем степени в уравнении КМХ, равным 0,47; такой же ряд обра-
зуют рибосомные РНК и их фрагменты в буфере без ионов Mg.
• Молекула коллагена и продукты его фрагментации ультразвуком образуют го-
мологичный ряд жестких палочкообразных частиц; показатель степени в урав-
нении КМХ для такого ряда равен 0,20.
• Зависимость 5 от М в двойном логарифмическом масштабе для молекул ДНК
в широком интервале молекулярных масс не описывается одной прямой лини-
ей, что свидетельствует об изменении ее структуры.
• Для ДНК в интервале молекулярных масс 1,3 х 104 < М <3,5 х Ю6 Да пока-
затель степени в уравнении КМХ равен 0,273; такая величина характерна для
относительно жестких, палочкообразных структур.
• Для ДНК в интервале больших молекулярных масс (>3,5 х Ю6 Да) показатель
степени в уравнении КМХ равен 0,440; такая величина характерна для гибких
структур, близких к гауссовому клубку.
Литература для последующего чтения
Исторический обзор
Schachman Н. К. Analytical ultracentrifugation reborn. Nature, 1989. 341, 259-260.
Schachman H. К. Is there a future for the ultracentrifuge? In Analytical
Ultracentrifugation in Biochemistry and Polymer Science/eds. S. E. Harding, A. J. Rowe
and J. C. Horton. Cambridge: Royal Society of Chemistry. 1992.
Инструменты и инновационная техника
Schachman Н. К. Ultracentrifugation in Biochemistry. New York: Academic Press,
1959.
Ralston G. Introduction to Analytical Ultracentrifugation. Fullerton, CA: Beckman
instruments. 1993.
GeibelerR. The Optima XL-А: A new analytical ultracentrifuge with a novel precision
absorption optical system. In Analytical Ultracentrifugation in Biochemistry and
Polymer Science, eds. S. E. Harding, A. J. Rowe and J. C. Horton. Cambridge: Royal
Society of Chemistry, 1992.
Van Holde К. E., Johnson IF. C. and Ho S. P. Principles of Physical Biochemistry.
New Jersey: Prentice Hall. 1998.
Скоростная седиментация
Van Holde К. E. Sedimentation analysis of proteins. In The Proteins, third edition.,
eds. H. Neurath and R. I. Hill, Volume VI, pp. 225-291. New York: Academic Press.
1975.
Perkins S.J. Protein volumes and hydration effects: the calculation of partial
specific volumes, neutron scattering matchpoints and 280 nm absorption coefficients
for proteins and glycoproteins from amino acid sequences. Eur. J. Biochem., 1986. 157,
169-180.
HardingS. E., Rowe A. J. and Horton J. C. (eds.) Analytical Ultracentrifugation in
Biochemistry and Polymer Science. Cambridge: The Royal Society of Chemistry. 1992.
Stafford, IT. F, III. Boundary analysis in sedimentation velocity experiments.
Methods Enzymol., 1994. 240, 478-501.
HardingS. E. Determination of macromolecular homogeneity, shape, and interactions
using sedimentation velocity analytical centrifugation. In Methods in Molecular
Biology, eds. C. Jones, B. Mulloy and S. Thomas A. H., volume 22. Microscopy, Optical
Spectroscopy, and Macroscopic Techniques. Totowa, NJ: Humana Press. 1994.
Hansen J. C, Lebowitz J. and Demeter B. Analytical utracentrifugation of complex
macromolecular systems. Biochemistry, 1994. 33, 13155-13163.
Hensley P. Defining the structure and stability of macromolecular assemblies in
solution: the re-emergence of analytical ultracentrifugation as a practical tool. Structure,
1996. 4, 367-373.
Laue T. M. and Stafford IT. F., III. Modern application of analytical ultracentrifugation.
Annu. Rev. Biophys. Biomol. Struct., 1999. 28, 75-100.
Carruthers L. M., Schirf V. R., DemellerB. and Hansen J. C. Sedimentation velocity
analysis of macromolecular assemblies. Methods Enzymol., 2000. 321, 67.
Schuck P. Size distribution analysis of macromolecules by sedimentation velocity
ultracentrifugation and Lamm equation modelling. Biophys. J., 2000. 78, 1606-1619.
Равновесная седиментация
Cantor C. and Schimmel P. Biophysical Chemistry. Part II. Technique for the Study
of Biological Structure and Function. San Francisco, CA: W. H. Freeman and company.
1980.
OberfelderR. W., ConslerT. G. and Lee,J. C. Measurement of changes of hydrodynamic
properties by sedimentation. Meth. Enzymol., 1985.117, 27-40.
Schuster T. M. and Laue T. M. Modern Analytical Ultracentrifugation. Boston, MA:
Birkhauser. 1994.
Laue T. M. Sedimentation equilibrium as thermodynamic tool. In Methods in
Enzimology, eds. G. K. Ackers and M. L. Jonson V, pp. 427-452. New York: Academic
Press. 1995.
Глава Г5
ЭЛЕКТРОФОРЕЗ
Г5.1. Исторический обзор
1920-е
М. Смолуховский и Е. Хюккель
(М. Smoluchowski and Е. Hiickel) пер-
выми разработали теорию электрофореза
для тонких и толстых бислоев. Д. С. Ген-
ри (D. С. Henry) расширил теорию элек-
трофореза сферических полиионов для
слоев произвольной толщины. В моде-
ли Генри распределение зарядов внут-
ри сферы предполагалось сферически
симметричным, а релаксацией ионов
пренебрегали. В 1950-х гг. ряд иссле-
дователей — Дж. Т. Овербиик, Ф. Бут,
П. Ч. Виерсма (J. Th. G. Oberbeek, F. Bo-
oth, P. H. Wiersma) изучили влияние
эффекта релаксации ионов на электро-
форетическую подвижность сфериче-
ских частиц, содержащих центросиммет-
ричное распределение зарядов.
1930
А. Тизелиус (A. Tiselius) предло-
жил новую технику для изучения элек-
трофоретических свойств белков. Он
показал, что резкая электрофоретиче-
ская зона передвигающихся ионизо-
ванных молекул может быть получена
с использованием U-образной трубки,
а границы движущихся белков могут
быть визуализованы в ультрафиоле-
товом свете. Тизелиус наблюдал при
электрофорезе несколько зон, соответ-
ствующих сывороточным альбуминам,
а-, р-, и у-глобулинам.
1940-е
Т. Виеланд и Е. Фишер (Т. Wieland
and Е. Fischer) предложили использо-
вать бумажные фильтры в качестве но-
сителей при электрофорезе. Простота
оборудования, компактность, относи-
тельная дешевизна и небольшое, менее
1 мг, количество белка, необходимого
для анализа, сделали электрофорез на
бумаге очень популярным методом ис-
следования.
1950-е
О. Смитис (О. Smithies) получил
хорошее разрешение белковых зон в
целлюлозных гелях. Использование
электрофореза на основе молекуляр-
ных сит давало гораздо большее раз-
решение, чем в свободном растворе.
В это десятилетие были предложе-
ны такие носители для электрофо-
реза, как ацетатцеллюлоза и агароза.
В 1959 г. С. Раймонд и Л. Вайнтрауб
(S. Raymond and L. Weintraub) доло-
жили о первых результатах, получен-
ных методом электрофореза в полиак-
риламидном геле (ПААГ).
1960-е
А. Л Шапиро, Е. Винуела и
Дж. В. Майзел (A. L. Shapiro, Е. Vinuela
and J. V. Maizel) обнаружили взаимо-
связь между электрофоретической под-
вижностью белков и их молекулярной
массой при использовании гомогенного
ПААГ в присутствии додецил сульфата
натрия (ДСН). Электрофорез в ПААГ в
присутствии ДСН начинает широко ис-
пользоваться для определения чистоты
и молекулярных масс белков. Возмож-
ность разделения нуклеиновых кислот
и нуклеотидов была продемонстриро-
вана на примере ДИК и даже хромосом
при использовании метода импульсно-
го электрофореза в агарозном геле.
1975
П. О’Фаррелл и Дж. Клозе
(Р. O’Farrell and J. Klose) развили метод
двумерного электрофореза. При движе-
нии в одном направлении белки дели-
лись по заряду, а во втором — по массе.
Уже в первых экспериментах удалось
разделить таким образом 1100 белков из
Е. coli. В это время молекулярные массы
белков в геле определяли, сравнивая их с
известными массами очищенных белков.
В 1977 г. Н. Г. Андерсен и Н. Л. Андер-
сен (N. G. Andersen and N. L. Andersen)
начали автоматизировать метод двумер-
ного электрофореза и разработали раз-
личные коммерческие аппараты. Методы
идентификации белков были существен-
но улучшены, благодаря применению
мембран, позволяющих определять ами-
нокислотную последовательность белков
cN-конца. Нижний количественный пре-
дел определения белков достиг 1 пмоль.
Благодаря увеличению размеров геля и
применению способа окрашивания бел-
ковых зон серебром удалось увеличить
разрешающую способность двумерного
электрофореза до 10000 пятен.
1981
Дж. В. Йоргенсен и К. Д. Лукас
(J. W. Jorgensen and К. D. Lukas) про-
вели электрофоретическое разделение
аминокислот в капилляре с высокой эф-
фективностью, которая ранее могла быть
достигнута только в газовой фазе. При
разделении белков в капилляре элект-
рическое поле приблизительно в 50 раз
больше, чем в традиционном пластинча-
том геле, что привело к разделению бел-
ков в течение нескольких минут. На ос-
нове капиллярного электрофореза были
созданы полностью автоматизирован-
ные, высокочувствительные инструмен-
ты, способные анализировать до одного
нанолитра сложного образца. Появились
фирменные приборы, объединяющие
методы разделения и секвенирования
белков и пептидов и хорошо сочетавши-
еся с оборудованием для масс-спектро-
метрического анализа.
1990
Принципиальный шаг в развитии ка-
пиллярного электрофореза был сделан
в группе Б. Л. Каргера (В. L. Karger),
создавшей линейный полиакриламид-
ный матрикс, который мог быть исполь-
зован для разделения ДНК в капилляре
с высокой разрешающей способностью.
В этом же году X. Свердлов и Р. Гестеланд
(Н. Sverdlow and R. Gesteland) описали
способ еще более быстрого разделения
ДНК в капилляре из плавленого силика-
геля с внутренним диаметром 75 мкм и
с большей эффективностью разделения,
чем в простом линейном пластинчатом
геле. В1993 г. X. Камбара и С. Такахаши
(Н. Kambara and S. Takahashi) сконс-
труировали устройство, объединившее
капилляр, сканирующий детектор и кон-
фокальный микроскоп. Усовершенство-
вание электрофоретического разделения
и визуализации нуклеиновых кислот
и нуклеотидов сделало возможным быс-
трое и эффективное картирование генов,
что способствовало ускорению решения
исторической программы «Геном челове-
ка».
2000 и по настоящее время
Электрофоретические методы яв-
ляются наиболее широко использу-
емыми при аналитическом и препа-
ративном разделении макромолекул.
Они внесли бесценный вклад в науки
о жизни — биохимию, молекулярную
биологию, генетику, генную инжене-
рию и медицину.
Г5.2. Введение
в биологические проблемы
Сутью электрофореза является переме-
щение заряженных частиц, растворен-
ных или суспендированных в электроли-
те. В 1791г. М. Фарадей (М. Faraday)
сформулировал два фундаментальных
закона электролиза, что дало название
указанному выше явлению. Фарадей
ввел такие понятия как электролит,
электролиз, анод и катод (для положи-
тельных и отрицательных электродов),
и анион и катион (для ионов, мигриру-
ющих к соответствующим электродам).
В 1897 г. Ф. Кольрауш (F. Kollraush)
сформулировал первую теорию, объяс-
няющую поведение передвигающихся
частиц в электрическом поле.
В 1926 г. Сведберг предложил свое-
му ученику Тизелиусу тему исследо-
ваний, связанную с использованием
электрофореза. При электрофорезе и
ультрацентрифугировании можно было
наблюдать перемещение зон белков и ре-
гистрировать их, однако ни один из этих
методов не обладал достаточным разре-
шением для полного разделения смеси
молекул. Уже через четыре года Тизели-
ус представил свою докторскую работу о
перемещении электрофоретической гра-
ницы белков, которая положила начало
использования этого метода в биологии.
После работы Тизелиуса развитие
метода электрофореза пошло по двум
направлениям. В первом, названном сво-
бодным электрофорезом, для разделения
использовались среды без геля. До не-
давнего времени этот метод широко не
применялся как структурный метод, по-
скольку сложные взаимодействия меж-
ду заряженными молекулами, раствори-
телем, подвижными солями и внешним
электрическим полем очень затрудняли
интерпретацию полученных данных.
Другое направление объединяло раз-
личные электрофоретические методы
в поддерживающей среде из различных
носителей. Двумерный электрофорез в
полиакриламидном геле обеспечивает
самое высокое разрешение в белковом
анализе и, следовательно, является ос-
новным методом в «протеомике». Метод
капиллярного электрофореза (КЭФ) по-
лучает все большее применение как ана-
литический метод в различных областях
исследований. Привлекательной чертой
этого метода является короткое время
анализа, высокая разрешающая способ-
ность, возможность работы в реальном
режиме времени и автоматическое вве-
дение образца в минимальном (мкл) объ-
еме. Метод капиллярного электрофореза
позволил приблизить окончание работ по
проекту «Геном человека» на 3-5 лет.
В 2003 г. М. Пленерт и Дж. Б. Шир
(М. L. Plenert and J. В. Shear) исполь-
зовали для электрофореза капилляр не-
обычной геометрии, в форме песочных
часов, для увеличения напряженности
электрического поля в локальной обла-
сти разделения белков и совместили его
с оптической приставкой для быстрого
введения образца. Все это позволило
делить образцы в течение нескольких
микросекунд и тем самым открыло путь
для изучения короткоживущих интер-
медиатов.
Появился также новый метод для
разделения макромолекул, капиллярная
электрохроматография (КЭХ), который
является гибридом методов высокоэф-
фективной жидкостной хроматографии
(ВЭЖХ) и капиллярного фореза. Метод
КЭХ сочетает лучшие свойства каждого
из них. Как в ВЭЖХ он делит незаря-
женные частицы, как в КЭ он обеспе-
чивает высокоэффективное разделение
заряженных частиц без использования
системы насосов высокого давления.
В этой главе мы сначала обсудим ме-
тод свободного электрофореза в раство-
ре. Недавние инновации в этом методе
вселяют надежду на его использование
для разделения относительно больших
и сильно заряженных биологических
макромолекул. Затем мы вернемся к
обсуждению различных типов оборудо-
вания для электрофоретического раз-
деления макромолекул, уделив особое
внимание капиллярному электрофоре-
зу и их применению для решения раз-
личных аналитических проблем.
Г5.3. Электрофоретические
эксперименты
Имеются три способа получения ин-
формации о структуре частиц, находя-
щихся в электрическом поле ( iao. 1. э. i ).
Первый базируется на измерении их
подвижности в постоянном электриче-
ском поле. Абсолютное значение элек-
трофоретической подвижности может
быть измерено по перемещению элект-
рофоретической границы, т. е. методом
свободного электрофореза. Этот метод
подобен методу скоростной седимента-
ции (секция Г4.5).
Второй базируется на электрофо-
ретическом прохождении тонкого слоя
или зоны макромолекулярного раствора
через среду. Среда действует по принци-
пу молекулярного сита и обеспечивает
стабильность против конвекции. Про-
цесс подобен таковому при делении ме-
тодом зонального центрифугирования
в градиенте сахарозы (секция Г4.9).
Третий базируется на измерении
ориентации, которая возникает, когда
молекулы, имеющие асимметричное
распределение заряда, помещаются в
переменное электрическое поле. Вра-
щающий момент стремится ориенти-
ровать молекулу по направлению поля.
Ориентация частиц под влиянием элек-
трического поля называется эффектом
Керра (Глава Гб). Этот процесс подобен
двойному лучепреломлению в потоке,
когда ориентирующей силой является
гидродинамическое поле (Глава Г7).
В инь 15.1 представлены ме-
тоды электрофореза, используемые в
настоящее время в научных исследова-
ниях (колонка 1). В ней приведены так-
же условия эксперимента (колонка 2),
вычисляемые параметры (колонка 3) и
аналогичные явления, встречающиеся
в гидродинамике (четвертая колонка).
Г5.4. Макромолекулы
в электрическом поле
Г5.4.1. Заряд молекулы и ее
электрофоретическая подвижность
Заряд — фундаментальное свойство мо-
лекул, которое тесно связано с их струк-
турой, растворимостью, стабильностью
и с характером взаимодействия с други-
ми молекулами.
Таблица Г 5.1. Современные методы электрофореза
Экспериментальный метод Условия эксперимента Вычисляемые пара- метры Аналогия с гидро- динамическими методами
Подвижная элек- трофоретическая граница Свободный электрофорез Электрофоретиче- ская подвижность Скоростная седиментация
Одномерный капиллярный электрофорез Свободный электрофорез Электрофоретиче- ская подвижность Скоростная седиментация
Равновесный электрофорез Аналитический электрофорез в про- странстве, ограни- ченный мембраной Коэффициент диффузии, заряд Диффузия с использованием ультрацентрифуж- ной ячейки, форми- рующей границу
Изоэлектрофокуси- ровка в геле Градиент pH И з озле ктр и ч ес кая точка, в которой под- вижность молекулы равна нулю Равновесное центри- фугирование в градиенте плотности
Зональный одномерный электрофорез в геле Додецил сульфат натрий (ДСН) Разделение в соот- ветствии с фрикци- онными свойствами (массой) Равновесное центрифугирование в градиенте плотности
Зональный двумерный электрофорез в геле Градиент pH (первое направле- ние) + ДСН (второе направле- ние) Разделение в соот- ветствии с зарядом (первое направле- ние) и фрикцион- ными свойствами (масса (второе направление)) Равновесное центрифугирование в градиенте плотности
Наиболее прямой способ оценки за-
ряда макромолекулы — получение отве-
та на прилагаемое электрическое поле.
Если частица имеет заряд Q, и находит-
ся в электрическом поле Е, то в резуль-
тате на нее будет действовать сила F:
F = QE. (Г5.1)
Комментарий Г5.1. Единица '
электрофоретической подвижности 1
Электрофоретическая подвижность риме- ,
ет размерность в системе СГС см2 сек ‘ В ,
Через короткое время после при-
ложения электрического поля частица
приобретает постоянную скорость и, с
которой она движется по направлению
к одному из электродов. При этой ско-
рости, силы трения равны и направле-
ны против приложенной силы:
u-QE/f, (Г5.2)
где f — коэффициент поступательного
трения макромолекулы.
Транспорт заряженной частицы под
действием электрического поля назы-
б
РI iv. ГЗ. 1. Макромолекулы в электрическом поле: а — при отсутствии электрического поля. Ма-
ленькие ионы растворителя образуют атмосферу вокруг макромолекул, в которой ионы имеют
заряд, противоположный заряду молекулы; б — в присутствии электрического ноля. Ионная ат-
мосфера искажается нолем и движением макромолекул (Cantor and Shimmel, 1980)
вается электрофоретической подвижно-
стью (комментарии ГЗ. I). Электрофо-
ретическая подвижность р может быть
определена как скорость на единицу
электрического ноля:
р = и/Е (Г5.3)
и, следовательно,
<2 = М/. (Г5.4)
Такое простое описание электрофо-
ретической подвижности справедливо
только для идеального случая и не соот-
ветствует реальной действительности.
В самом деле трудности возникают уже
при введении понятия суммарный заряд.
В растворах всегда имеются проти-
воионы. Часть из них тесно ассоцииро-
вана с макромолекулами, другие — нет
(рис. Г5.1). Чтобы ослабить взаимо-
действие между разноименно заряжен-
ными ионами, в раствор добавляется
некоторое количество поддерживающе-
го электролита.
Поскольку электролит образует атмо-
сферу вокруг макромолекулы, уравнение
Г5.2 может быть представлено в форме:
w = Q.llM,£//’ <Г5-5)
где Q. является эффективным заря-
дом (макроионы плюс противоионы)
«одетой» макромолекулы.
Несмотря на принципиальную важ-
ность понятия заряда, его измерение
сопряжено с большими трудностями.
К настоящему времени отсутствуют
экспериментальные методы, которые
определяют эффективный заряд, неза-
висимо от других свойств макроионов.
Недавние инновации в методах стацио-
нарного и капиллярного электрофо-
резов показали возможность их при-
менения для исследования молекул,
подобных ДНК (см. ниже параграфы
Г5.5.2 и Г5.7.2).
Г5.4.2. Моделирование
электрофоретической
подвижности
Для электрофоретической подвиж-
ности, р, наиболее часто используется
уравнение Хюккеля
р = Ze/f, (Г5.6)
где Z — число зарядов; е — заряд элект-
рона;/— коэффициент трения заряжен-
ной макромолекулы. Как и в случае ко-
эффициента седиментации, р является
отношением скорости движения части-
цы к силе прилагаемого поля (сравните
с уравнением Г4.16). Если частицы име-
ют сферическую форму, то, применяя
закон Стокса, можно записать:
Комментарий Г5.2. Влияние заряда
на константы седиментации
и диффузии
Эффекты заряда влияют также и на
константы седиментации, диффузии и на
j удельную вязкость, даже тогда, когда не
присутствует внешнее электрическое поле.
Понять это можно на примере фрагмента :
। осаждающейся высокозаряженной полии-
онной ДНК. Текущий поток растворителя
искривляет ионную атмосферу, в резуль- ,
| тате чего возникает дополнительное вязко-
; стное торможение, так называемое «элект-
ролитное трение». Электролитное трение
увеличивает константы седиментации и
। диффузии. Подобно этому ион релаксации '
повышает характеристическую вязкость
суспензии растворенных веществ жестких
। полиионов, помещенных в градиентный ;
поток поля, и это явление называется «пер- ।
вичным электровязкостным эффектом».
: С более полным описанием этих явлений
' читатель может ознакомиться в специаль-
ной литературе (Schmitz, 1993). Эффект ;
заряда на седиментацию и диффузию будет
| меньше или равен нулю для слабо заря-
женных полиионов в высоко солевых рас- :
творах, но проявление эффекта становится |
более ощутимым при повышении заряда
полииона или при понижении концентра-
ции соли.
д = Ze/6nr|0/?0, (Г5.7)
где Ro — радиус частицы, а т|0 — вязкость
растворителя.
Уравнение Г5.7 может быть обоб-
щено, если принять во внимание гид-
ратацию и отклонение от сферической
формы, как в случае седиментации и
диффузии. Однако для заряженных
частиц, двигающихся с постоянной
скоростью в электрическом поле, необ-
ходимо принимать во внимание иска-
жение ионной атмосферы от ее равно-
весного состояния(комментарии Г.5.2).
Этот эффект «ионной релаксации»
будет важен для слабо заряженных
макромолекул. Описание транспорта
заряженной частицы по сути является
гораздо более трудной проблемой, чем
незаряженной и до недавнего време-
ни классическая теория применялась
только к наиболее простым модельным
структурам.
Модели, используемые в классиче-
ской теории электрофореза, подобны
гидродинамическим, описанным в Гла-
ве Г2. В них принимается во внимание
распределение заряда и ионная атмос-
фера. Развитие новых методов модели-
рования в гидродинамике сложных сис-
тем на основе электростатики (параграф
Г.2.2.3) позволило пересмотреть теорию
свободного электрофореза. Проблема
полиионных частиц произвольной фор-
мы и распределения заряда была решена
введением в практику техники гранич-
ных элементов, описанной в параграфе
Г2.2.1. В ней приведен пример разбиения
гидродинамической поверхности фраг-
мента ДНК из 20 пар оснований на 352
треугольные чешуйки (см. рис. Г2.15 а).
Кроме того, в этой модели каждой че-
шуйке можно приписывать разный за-
ряд, что иллюстрируется на этом же ри-
сунке каркасной моделью треугольников
с числом элементов, равным от 5 до 9 над
одной из чешуек. Тем самым создается
возможность вариации напряженности
поля в каждой чешуйке при сохранении
общего числа чешуек.
Г5.5. Электрофоретические
методы в отсутствие
поддерживающей среды:
свободный электрофорез
Различные методы электрофореза по-
ка широко не используются в качест-
ве зонда структуры. Причина проста:
сложные взаимодействия между ио-
нами, растворителями, солями, гелем
(если последний присутствует) и вне-
шнее электрическое поле усложняют
интерпретацию электрофоретической
подвижности. Однако при отсутствии
геля, в методе электрофореза в раство-
ре, который мы в дальнейшем будем
называть свободным электрофорезом
(СЭФ), интерпретация данных может
быть более однозначной. Развитие ана-
литического электрофореза в мембран-
ной геометрии (см. параграф Г5.5.2) и
капиллярного электрофореза (секция
Г5.7) показало, что метод СЭФ может
эффективно применяться для исследо-
ваний относительно больших и сильно
заряженных биологических макромо-
лекул. СЭФ также является эффектив-
ным методом для разделения макромо-
лекул (см. к< 1.ММСП ।арий I).
Г5.5.1. Метод движущейся границы
В электрофоретическом эксперимен-
те на частицу действуют четыре силы
(рис. 1'5.2). Первая это электростати-
ческая сила/7! = QE.
Вторая — гидродинамическая сила
трения F2 = где /п(кт — коэффици-
ент поступательного трения, а и ско-
рость частицы.
Третья сила F3 возникает в резуль-
тате тенденции макроионов быть окру-
женными противоионами, в результате
чего поток противоионов движется в на-
правлении, противоположном их движе-
нию. По этой причине макроионы всегда
движутся против потока так, что их ско-
рость по отношению к их непосредствен-
ному окружению будет выше, чем тако-
вая в лабораторной системе координат.
И наконец, четвертая сила, Z7, возни-
кает при искажении ионной атмосферы
(эффект асимметрии поля). Эта сила
обусловлена дипольным моментом,
Коммешарий Г5.3. Свободный
; электрофорез в потоке
В так называемом свободном электрофоре-
зе в потоке, переносящий вещество буфер-
ный раствор заключен между двумя верти-
кальными пластинами, находящимися на
| близком расстоянии друг от друга. Элек-
трическое поле прилагается перпендику-
лярно переносящему потоку, в то время
как раствор образца вводится непрерывно
в переносящий буфер в виде узкой полосы.
Компоненты образца впоследствии разде-
ляются в электрическом поле в поперечном
направлении в соответствии с их элект-
i рофоретической подвижностью, и затем
। отбираются на выходе. Этот метод очень
| хорош для работы с белками и клетками,
поскольку в нем не используются органи-
ческие растворители, высокие концентра-
ции солей или поддерживающие среды.
который возникает при перераспреде-
лении противоионов под воздействием
внешнего электрического поля.
В результате общий заряд Q макромо-
лекулы можно определить из ее подвиж-
ности под действием всех четырех сил:
F2 + F3+F4 = F!=/M=Q/£. (Г5.8)
Тогда подвижность равна: ц = и/Е =
= Q/fxW гДеАм> - эффективный коэф-
фициент трения, производимый силами
F2>F3MF4-
Электрофоретический метод движу-
щейся границы наиболее часто применя-
ется для определения общего заряда на
ДНК олигонуклеотидах. Скорость пере-
Рис. Г5.2. Четыре силы, действующие на мо-
лекулу в опыте по электрофоретической под-
вижности (описание сил см. в тексте)
Рис. ГЗ.З. Профиль интенсивности в ячейке,
показывающей границу олигомера DNA (pd
(А) ,0 pd (Т) 20, С = 250 мкг-см3 в 100 мМ КО,
20 мМ Tris, pH 8,0, движущуюся слева напра-
во. Положение мембран, М, обозначено с каж-
дой стороны ячейки. Данные приведены для
профилей, полученных через 20, 80 и 150 сек
после приложения поля (£ = 3,0 В см ') (Laue
et al., 1996)
из мембран. Диффузия образует поток
макроионов в направлении, противопо-
ложном направлению электрического
поля. При наступлении стационарного
состояния эти два потока выравнива-
ются в каждой точке, и при этом уста-
навливается градиент концентрации.
Таким образом, в стационарном элект-
рофорезе потоки, также как и силы, сба-
лансированы (рис. Гэ/i). В любой точке
ячейки ноток, обусловленный электро-
форезом,/^^, равен Сх и', где С — это
концентрация макрононов в точке .г, а и'
их скорость. ПотокJ является эффек-
тивным потоком, складывающимся под
действием всех сил F{, F.,, F3 и Fv
еГГ
мещения границы фиксируется аппарату-
рой, приспособленной для измерения ана-
литического электрофореза и диффузии в
реальном режиме времени. Рисунок Г5.3
показывает профиль интенсивности в
ячейке при движении границы ДНК оли-
гомеров (pd(A)20-pd(T)20. Изданных, пред-
ставленных на этом рисунке вычисленная
электрофоретическая подвижность р для
(pd (А)20 pd (Т)20 оказалась равной 2,9910"
4 см2 - В-1 • сек"1 при 20 °C.
Г5.5.2. Метод стационарного
электрофореза
В противоположность свободному элек-
трофорезу, когда граница перемещается
свободно, в стационарном электрофо-
резе макроионы захватываются неболь-
шими камерами, поверхность и дно
которых закрыты полупроницаемыми
мембранами. Электрическое поле при-
лагается вдоль полости камеры, так что
макроионы скапливаются против одной
Рис. Гэ.!. Стационарные измерения, в ко-
торых потоки/1(|1()1и J,, сбалансированы
Поскольку
Г.|+/"'-2Е (Г5.9)
<Г5ЛО>
мы имеем:
ЛИ,-<2ВД.И.)С- (C5.lt)
В соответствии с первым законом
Фика (уравнение Г3.7) градиент кон-
центрации создает поток, обусловлен-
ный диффузией и равныйJ = -DdC/dx.
В стационарном состоянии J + Jn = 0,
отсюда:
{QE/f^)C = DdC/dx. (Г5.12)
Решением уравнения Г5.12 будет:
С = Со ехр [ст(х - х0)1, (Г5.13)
где Со — концентрация в точке х0 и эк-
спонента ст = QE/f ..D включает
' J :и|и|| эфф
Рис. Г.5.6. Определение коэффициента диф-
фузии для олигомеров ДПК (pd (А) 20 pd(T)20
из спада градиента, показанного на рисунке
Г.5.5. Наклон линии In (Л2/1— А1/3) как функция
времени пропорционален п2£>£'2, где А2;)и А
есть значения оптической плотности при уров-
нях последней, равной 2/3 и 1 /3, соответственно
(Laue et al, 1996)
Рис. Г.5..5. Зависимость оптического погло-
щения Л2(.(| от расстояния х для олигомеров
ДНК (pd (А) 2() pd (Т) 20 в буфере, содержащем
100 мМ КС1, 20 мМ Трис-НС1, pH 8,0, после
выключения поля. Начальный стационарный
градиент (пунктирная линия) был сформиро-
ван приложением поля силой 0,12 Всм 1 в те-
чение более 24 часов (Laue et al., 1996)
Z). , — коэффициент диффузии при
наличии электрического поля. Теперь
Предполагая, что уравне-
ние/^^ = kT/D,^ применимо для этого
случая, получаем:
Q = (Г5.14)
Уравнение Г5.14 обеспечивает прос-
той путь определения эффективного за-
ряда молекулы непосредственно из экс-
периментальных измерений величин ст,
Е, и Т, в противоположность измерени-
ям подвижности, которые требуют зна-
ния или -Рф],-
Стационарный концентрационный
градиент препарата олигомеров ДНК
(pd (А) 20 pd (Т) 20 показан на рисун-
ке Г.5.5. Коэффициент диффузии в от-
сутствие электрического поля можно
получить из значения релаксации равно-
весного концентрационного градиента
(рис. Г.5.б). Вычисленное из этих данных
значение D оказалось равным 9,6 х Ю-7
см2 сек~’, что находится в хорошем со-
гласии с таковым (10,86 х 10"7см2-сек ’),
полученным методом динамического
рассеяния (см. i;iAi. ГЗ. 1). Если предпо-
ложить, что уравнение Эйнштейна fD =
kT действительно также и для «одетых»
макроионов, находящихся в электри-
ческом поле, то методом стационарного
электрофореза можно оценить эффек-
тивный электрический заряд макро-
молекулы. Значение Q полученное из
уравнения Г5.14, дает отношение эффек-
тивного заряда к формальному заряду
Q/40 (для двойной спирали ДНК дли-
ной 20 пар оснований), очень близкое к
предсказанному (0,12), в присутствии
одновалентных солей без учета поправ-
ки на конечную длину макроиона.
Г5.6. Электрофоретические
методы в присутствии
поддерживающей среды:
зональный электрофорез
Первая работа Тизелиуса по электрофо-
резу была выполнена в свободном рас-
творе. Однако вскоре выяснилось, что
при использовании этого метода возни-
кает много проблем, которые могут быть
устранены только при стабилизации сре-
ды. Роль такого стабилизатора выпол-
няла пористая механическая подложка,
смоченная буфером для электрофореза.
Подложка снижала конвекционые по-
токи и диффузию, так что разделенные
компоненты предстали в виде четких
Комментарии Г5.4. Гели
и додецилсульфат натрия 1
Агарозные гели
Агароза — линейный природный по-
лисахарид, выделенный из содержащих
агар морских водорослей. Физические и
химические свойства агарозы, такие как
температура застывания, пористость, твер-
дость могут варьироваться. Агарозный гель
формируется помещением сухой агарозы в
буфер с последующим нагреванием. После
охлаждения до комнатной температуры на
плоской поверхности образуется прозрач-
ный студенистый гель. Агарозный гель
используется для электрофореза белков
и нуклеиновых кислот.
Полиакриламидные гели (ПААГ)
Сетчатый ПААГ получают путем по-
лимеризации акриламидных мономеров в I
присутствиии небольшого количества со-
полимера (бисакриламида). Бисакрилами-
ды являются бифункциональными агента- ।
ми, которые ковалентно сшивают соседние :
линейные акриламидные цепи. Мономеры
полимеризуются по принципу «голова-
хвост» в длинные цепи и бисакриламидные I
молекулы (сшивка) встраиваются изредка I
в растущую цепь, в результате чего образу-
ется место для разветвления цепи. Акри-
ламидные гели можно полимеризовать в ,
любой концентрации. Этим, а также коли- !
чеством добавленной сшивки, варьируется
размер пор в геле.
Додецилсульфат натрия (SDS или ,
ДСН)
ДСН (СН3-(СН2) 10-CH2OSO3-Na+) яв-
ляется анионным детергентом.
зон. Ранее используемыми подложками
были фильтровальная бумага и ацетат-
целлюлоза. В течение многих лет они ус-
пешно использовались для разделения
малых молекул, таких как индивиду-
альные аминокислоты, пептиды и кар-
богидраты. Однако введение в качестве
подложек различных гелей, таких как
агароза или полиакриламид, привело к
существенному усовершенствованию
электрофореза как метода для анализа
макромолекул ( коммсп lapiiii Гб. i).
Г5.6.1. Разделение по массе:
ДСН — электрофорез
В методе, называемом ДСН-электро-
форезом, изучаемые белки перед нане-
сением на гель прогреваются в буфере
с низкой ионной силой, содержащем в
разбавленном растворе до 2 % додецил
сульфата натрия (ДСН). Эта процеду-
ра разрушает нативную структуру бел-
ка с распадом элементов вторичной,
третичной и четвертичной структуры.
Добавление специального агента, р-
меркаптоэтанола, приводит к разрыву
дисульфидных связей. Такая обработ-
ка разворачивает белковые молекулы
и образует структуры в форме палочек
с включением отрицательно заряжен-
ных молекул ДСН полипептидную
цепь (рис. Гб.7). В среднем, одна мо-
лекула ДСН связывается с двумя ами-
нокислотными остатками. Природный
заряд нативной молекулы белка, сле-
довательно, подавляется отрицательно
заряженной молекулой ДСН, поэтому
в присутствии ДСН все белки имеют
сходную структуру, отрицательный
заряд и отличаются только по моле-
кулярной массе (комментарий Гб.5).
Разворачиваемые додецилсульфатом
натрия пептиды движутся через гель
по определенной схеме: молекулы с паи-
Рис. Г5.7. Схематическое представление
белка в растворе ДСН. Белок денатурирует и
покрывается «шубой» из молекул ДСН, при-
обретая одинаковый заряд на единицу длины.
Собственный заряд белковой молекулы ней-
трализуется. Добавление р-меркаптоэтанола
разрушает все дисульфидные связи
меныпей молекулярной массой мигри-
руют быстрее, чем большие молекулы.
На рисунке Г.5.8 представлен график
подвижности полипептидых цепей в
10% ДСН-геле как функция молеку-
лярной массы. В нервом приближении,
скорость миграции ДСН-комплексов
обратно пропорциональна логарифму
их молекулярных масс. Абсолютная мо-
лекулярная масса белков оценивается
по подвижности полипетидиых цепей в
полиакриламидном геле, которая срав-
Комментарий Г5.5. ДСН и заряд
Некоторые белки могут связывать боль-
шее или меньшее количество ДСН, по срав-
нению со средним количеством, что делает
их пробег в геле аномальным. Если белок
сам по себе имеет очень большой положи-
тельный или отрицательный заряд, то он
! будет нивелировать заряд, приобретаемый
пептидом при связывании с ДСН. Специ- 1
фнческим примером является гистонный
белок Ш, который имеет молекулярную
массу около 21 кДа, но ведет себя в элект- I
рофоретическом эксперименте в ДСН как |
молекула с молекулярным весом в 30 кДа, ।
: поскольку обладает сильным положитель- !
ным зарядом.
Комментарий Г5.6. Размер пор геля '
и молекулярная масса белка
Обычно разделяющий белки акриламид- I
ный гель, имеет концентрацию 15 %. Этой ;
концентрации соответствует определен- '
ный размер пор в геле, через которые белки
с молекулярной массой 10 кДа проходят
относительно свободно, в то время как бел-
ки с массой 100 кДа проходят с трудом. Это !
означает, что 15 % полиакриламидный гель
пригоден для разделения белков с массами
в интервале 10-100 кДа. Для белков с мо-
лекулярной массой более 100 кДа исполь-
зуется 10 % или даже 7,5 % гель. :
нивается с подвижностью белков-мар-
керов с известной молекулярной мас-
сой (комментарий Г.5.б).
Рис. Г.5.8. Относительная подвижность 37
различных полипептидных цепей в 10% ДСН-
ПААГ, как функция молекулярной массы
(Weber and Osborn, 1969)
Г5.6.2. Разделение по заряду:
изоэлектрическое фокусирование
Изоэлектрическое фокусирование (И ЭФ)
является электрофоретическим мето-
дом для разделения амфотерных мо-
лекул в градиенте pH. Общий заряд
амфотерной молекулы определяется
величиной pH ее локального окруже-
ния (коммеиlapnii Гэ.7). Белки явля-
ются наиболее известными примерами
амфотерных молекул, несущих положи-
тельный, отрицательный или нулевой
общий электрический заряд, в зависи-
мости от pH их окружения. Для каж-
дого белка имеется свое значение pH,
называемое pl, при котором он не несет
заряда. Следовательно, pl является ха-
рактеристическим свойством белка.
Комментарий Г5.7. Амфотерные
молекулы i
. Амфотерной называется молекула, ко- '
I торая способна нести положительный, от-
рицательный или нулевой заряд, в зависи-
мости от pH окружения i
В градиенте pH белок будет переме-
щаться электрическим полем, пока не
достигнет изоэлектрической точки (рис.
Гэ.9). Его общий заряд в этой точке бу-
дет равен нулю и электрическое поле
больше на него не будет действовать.
Ситуация сходна с таковой в экспери-
ментах по равновесной седиментации
в градиенте плотности, когда частица
движется к точке, в которой ее плот-
ность становится равной плотности
растворителя и где происходит ее оста-
новка (см. 1,14.1. I .">. I и секцию Г4.9).
Метод ИЭФ является идеальным
для разделения белков в соответствии
с их зарядом. Он обладает высокой раз-
решающей способностью, что позволяет
разделять белки с разницей в изоэлек-
трической точке в 0,01 pH. Наиболее
широко используемой системой в этом
методе является горизонтально распо-
ложенный на стекле гель. Разделение
достигается приложением разности по-
тенциалов к гелю, содержащему pH гра-
диент. Стабильный линейный градиент
pH — ключ к успеху в экспериментах по
ИЭФ. Создание такого градиента сопро-
Суммарный (+2) (0) (-2)
заряд
Рис. Гэ.!). Изоэлектрическое фокусирование. В градиент pH, создаваемый молекулами амфо-
литов под действием электрического поля, помещается молекула белка. Градиент изменяется от
кислого (pH 3) на аноде, до щелочного (pH 10) на катоде. Гипотетический белок общим зарядом
+2, 0 и -2 показан в трех положениях в градиенте pH. В изоэлектрической точке pl, общий (сум-
марный) заряд белка равен нулю, что приводит к его остановке в электрическом поле, т. е. фоку-
сированию (Garfin, 1995)
вождается введением в гель соединений,
известных как амфолиты (комментарий
17>.8). Поскольку белки при изоэлект-
рической фокусировке перемещаются
свободно в соответствии с их зарядом
в электрическом ноле, то, как правило,
используют низкую концентрацию геля,
чтобы избежать эффекта сита (см. ком-
ментарий Г5.(>). Рисунок Г5.10 демонс-
трирует высокую степень разрешения,
достигаемую в методе ИЭФ на примере
рибосомного белка S1.
Г5.6.3. Одновременное
разделение по заряду и массе:
двумерный гель-электрофорез
Двумерный электрофорез объединяет
два метода: изоэлектрофокусировку с
ДСН-электрофорезом в полиакрила-
мидном геле. Этот объединенный метод
называется кратко двумерным электро-
форезом и в настоящее время является
наиболее используемым аналитическим
методом для разделения белков. Уже
первые эксперименты по разделению
белков показали колоссальные возмож-
ности метода. Так, 1100 белков из Е. coli
были разделены в одном опыте и было
предсказано, что до 5000 белков могут
быть разделены методом двумерного
электрофореза. Несмотря на эти выда-
ющиеся возможности и широкое при-
менение для решения различных задач,
почти 20 лет прошло, пока метод дву-
мерного электрофореза стал составной
частью протеомики (см. секцию Б2.2).
Способность метода разделять од-
новременно большое число белков, за-
тем идентифицировать индивидуальные
белки, как химическим анализом их
концевых аминокислотных последо-
вательностей, так и с помощью поиска
этих последовательностей в компьютер-
ных базах данных, привела к быстрому
70S 30S 70S 30S 50S
Е coh Е coh Т th Т th Т th М
— 45
Рис. 15.10. Анализ белков рибосом и рибо-
сомных субъединиц из Е. coli и Т. thermophilus
методом ДС11-электрофореза в полиакрила-
мидном геле. М — молекулярная масса марке-
ров в кДа. Отчетливо видно присутствие белка
S1 из Е. coli в 70S рибосоме и в 30S субъедини-
це Т. thermophilus и его отсутствие в 50S субъ-
единице. Содержание белка S1 в препаратах
рибосом из Т. thermophilus обычно ниже, чем
содержание того же белка в стандартных пре-
паратах рибосом Е. coli (Shiryaev et al., 2002)
г-------------- -----------------------.
Комментарий Г5.8. Амфолиты i
| Амфолиты являются сложной смесью
| синтетических полиамино-поликарбоксиль-
1 пых кислот. Величина pH комплектов ком-
мерческих амфолитов может варьировать
от 3 до 10 или иметь более узкий интервал
। значений, например, от 5 до 6 единиц pH.
I Область pl I выбирается таким образом, что-
бы значение р! образца находилось внутри ;
выбранного интервала значений pl I.
пополнению наших знаний о белковых
компонентах клетки. Более того, сейчас
с помощью этого метода можно следить
за экспрессией практически всех белков
в клетке и сравнивать информацию, иду-
щую от «нормальных» и «аномальных»
клеток (см. примеры в параграфе Б2.6.1).
Нарисунке Г5.11 представлена типичная
картина двумерного гель-электрофореза
иа примере разделения белков из 30S
рибосомных частиц.
a
б
Pm-. 1’5.1 I. Электрофоретическое разделение белков 30S рибосомной частицы из Е. соН (а) из
Т. thermophilus (б). Электрофорез в первом направлении выполняется при кислом значении pH
(5.5). Во втором направлении используется 15% ДСН-ПАЛГ. Сравнение результатов электро-
форетического разделения белков 30S субчастицы из Е. coli u Т. thermophilus в этой системе пока-
зало, что пятно в положении, обозначенном (►) в правой половине рисунка, соответствует пятну
белка S1 из Е. соН в левой половине рисунка (Shiryaev et al., 2002)
Г5.7. Капиллярный
электрофорез
Несмотря на существенный прогресс
в методологии, техническая реализация
электрофореза в гелях является трудо-
емким процессом, занимает много вре-
мени и практически недоступна автома-
тизации. Капиллярный электрофорез
(КЭФ) выступает в настоящее время
как альтернатива электрофорезу в геле
со всеми преимуществами современ-
ных автоматизированных технологий.
КЭФ-технология может быть адапти-
рована и к двумерному электрофорезу.
Г5.7.1. Одномерный
капиллярныйэлектрофорез
Как предполагает название, при про-
ведении КЭФ образец перемещается в
очень узких капиллярах, внутренний
диаметр которых обычно составляет от
20 до 100 мкм. Капилляры из плавле-
ного кварца погружаются в резервуар
с электролитом и электродами, на кото-
рые подается напряжение. Электроды,
сделанные из такого инертного материа-
ла как платина, помещаются в электро-
литный резервуар. Небольшой объем
образца вводится в капилляр с одного
конца. На рисунке Г 5.12 представлена
схема типичного прибора для капил-
лярного электрофореза.
Разделенные молекулы регистриру-
ются детектором, находящимся на конце
капилляра, противоположном тому, куда
вводится образец. Термин «электрофо-
реграмма» представляет собой графи-
ческое изображение ответа детектора.
Теория капиллярного электрофоре-
за базируется на двух уравнениях. Пер-
вое определяет электрофоретическую
подвижность, д, как:
\i = L2/Vt, (Г5.15)
где t — время, необходимое для перене-
сения образца к детектору (в секундах);
Рис. ГЗ. 12. Схематическое изображение аппаратуры для капиллярного электрофореза
L — длина капилляра (в сантиметрах)
и V — прилагаемое напряжение. Второе
уравнение определяет эффективность
разделения в понятиях общего числа
теоретических тарелок, N, как:
N=pV/2£>, (Г5.16)
где D — коэффициент диффузии моле-
кулы (комментарии I 3.!)).
Из уравнения Г5.15 следует, что для
получения наилучшего разрешения не-
обходимо прикладывать наибольшее
напряжение к капилляру минимальной
длины. Однако имеются практические
ограничения для такого подхода. Когда
длина капилляра уменьшается, количе-
ство тепла, которое должно рассеяться,
увеличивается благодаря уменьшению
электропроводности капилляра. Высо-
кое напряжение вызывает больший ток, и
как следствие этого, большее нагревание.
На практике необходим компромисс при
выборе величины напряжения и длины
капилляра. Возможен вариант компро-
мисса - это напряжение 20-50 кВ при
длине капилляра 40-100 см. При этом
разделение занимает обычно несколько
минут. Процедура введения образца по-
казана на рисунке ГЗ. 13. Использование
плавленого кварца в качестве материа-
ла для капилляра позволяет выполнить
определение разделенных компонентов
прямо в капилляре. Часть полиамидного
покрытия удаляется с конца капилляра,
который расположен в освещаемой час-
ти оптического детектора. Количество
вводимого образца (обычно под давле-
нием) измеряется в нанолитрах. Ско-
рость прохождения через капилляр не-
больших объемов образца мала, около
1 мкл в минуту, что дает возможность
сочетать оборудование КЭФ с ионизиру-
Комментарпй Г5.9. Эффективность
разделения
Общее число тарелок или пластин, N, —
термин, используемый в хроматографии, —
является мерой эффективности разделе-
ния. Главным отрицательным эффектом,
снижающим разделение в капиллярном
электрофорезе, является молекулярная
диффузия растворенного вещества внутри
капилляра. Диффузия уменьшается с рос-
том молекулярной массы. Следовательно,
теоретически возможно подсчитать число
У для многих белков и нуклеотидов в ме-
тоде КЭФ исходя из их коэффициентов
диффузии. Для больших полинуклеотидов
число N может достигать 10 млн.
ющпми источниками масс-спектрометра.
Важным усовершенствованием метода
КЭФ является использование лазерных
источников для возбуждения лазер-ин-
1’ис. 13.1.3. Три способа введения образца
в капилляр, погруженный в сосуд с раствором.
В первом (верхний рисунок) раствор вводится
в капилляр под внешним давлением. Во вто-
ром (средний рисунок) это давление задается
разницей гидростатических давлений в высо-
тах столбов раствора и буфера (метод сифона).
В третьем способе введения (нижний рисунок)
капилляр погружается в раствор образца с по-
следующей подачей напряжения. Если обра-
зец ионизирован и выбрана соответствующая
полярность напряжения, то ионы мигрируют в
капилляр. Этот способ введения образца назы-
вается электрокинетическим
Комментарий Г5.10. Уровень
детектирования в КЭФ
При введении образца объемом около
нанолитра, реальное количество детектиру-
емого вещества, которое можно определить
методом КЭФ исчисляется в аттомолях
(10-18 моля) или зептомолях (10 21 моля).
Напоминаем, что объем в 1 зептомоль — это
около 600 определяемых молекул, т. е. ана-
лизируется именно число молекул.
дуцируемой флуоресценции непосред-
ственно в капилляре для определения.
Этот метод, включающий также окра-
шивание молекул, обеспечивает опреде-
ление концентрации молекул от 10 11 М
и ниже (комменгарпй ГЗ.10). Помимо
высокой чувствительности, флуоресцен-
тный анализ обладает высокой селектив-
ностью при введении различных меток в
детектируемую молекулу. Анализ ДНК
с использованием меченых нуклеотидов
в настоящее время является рутинной
практикой.
В отличие от гель-электрофореза
важной составляющей в капиллярном
электрофорезе является величина элек-
троосмотического потока (ЭОП). Этот
поток перемещается вдоль капилляра,
обычно по направлению к детектору
(рис. Го.И). Он значительно сокраща-
ет время анализа и может преодолеть
тенденцию ионов передвигаться к со-
ответствующему его заряду электроду
(комментарий 1'3.11). Поток эффек-
тивно прокачивает растворенные ионы
вдоль капилляра и может рассматри-
ваться как «электронасос». Для устра-
нения ЭОП капилляр внутри покры-
вается нейтральными гидрофильными
полимерами. Такое покрытие стенок
повышает вязкость слоев вблизи стен-
ки капилляра, устраняя явление ЭОП.
Рисунок ГЗ. 13 демонстрирует при-
мер высокоразрешающей способности
метода КЭФ. Разделение смеси бел-
ков выполнялось в открытой камере в
капилляре со стенками, предотвраща-
ющими адсорбцию образца. Разде-
ление было быстрым, а пики узкими.
Если адсорбция образца на стенке ка-
пилляра минимальна, эффективность
разделения достигает 106 теоретичес-
ких тарелок на метр (мера ширины
пика, см. уравнение Г5.16 и коммен-
тарий Г3.‘)). Этот показатель прибли-
Рис. ГЗ. 14. Схематическое изображение явле-
ния электроосмотического потока. ЭОН отно-
сится к потокам жидкости в разделительной
камере и возникает при взаимодействии заря-
женных групп со стенками камеры
зителыю в 10-100 раз лучше, чем в
жидкостной хроматографии высокого
давления.
Г5.7.2. Сверхбыстрый капиллярный
электрофорез
Как следует из уравнений Г5.15 и Г5.16
для достижения наиболее быстрого раз
Комментарий Г5.11. Поток ионов
и молекул воды вдоль капилляра ;
Стенки капилляра, состоящие из плав-
леного кварца, содержат силанолы, которые
ионизируются при контакте с раствором
электролита с высоким pH. При диссоциа- :
ции образуется отрицательный заряд на
стенке капилляра, который нейтрализуется j
ионами .металла. При подаче напряжения .
ионы металла и ассоциированные с ними
молекулы воды перемещаются ио направ- ;
ленто к катоду. Это движение ионов с ас- •
социированными молекулами воды, созда-
ст поток раствора к детектору.
деления молекул в методе КЭФ необхо-
димо максимально увеличить подаваемое
напряжение на капилляр минимальной
длины. Однако этому препятствует ряд
обстоятельств, обсужденных в предыду-
щем параграфе и в первую очередь со-
путствующее выделение тепла. Решение
этой проблемы достигается выбором не-
обычной геометрии капилляра — в форме
песочных часов для повышения локал ь-
Рпс. Г5. 15. Разделение белков при pH 4,5, методом КЭФ в открытом капилляре. Капилляре па-
раметрами: 50 см х 75 мкм покрыт линейным полиакриламидом. Напряженность электрического
поля — 530 V-см ’, лизоцим: 2 — цитохром, с; 3 — миоглобин: 4 трипсиноген; 5 — а-химотрип-
синоген A (Karger et al., 1995)
Рис. 1.5. l(i: a — схематическое изображение фотопроцесса в капилляре для микросекундного
разделения молекул. Два сильно сфокусированных лазерных пучка образуют места регистра-
ции входа и выхода молекул при их движении в капилляре; б — изображение типичной формы
кварцевого капилляра (29 мкм внутренний диаметр и 320 мкм внешний диаметр) в геометрии
песочных часов. Шкала 250 мкм. На вставке показана центральная область капилляра длиной
=60 мкм, в которой происходит регистрация продуктов разделения (Plenert and Shear, 2003)
ной силы электрического поля в месте
разделения. Комбинация такой формы
капилляра с ультрабыстрым введением
образца дает возможность провести раз-
деление в течение микросекунд. На ри-
сунке 1'5.16(7 схематически представлен
узко сфокусированный лазерный пучок
для определения электрофоретически
разделенных компонентов. При умень-
шении поперечного сечения капилляра в
30-40 раз электрическое поле напряжен-
ностью в 0,1 МВ см"1 может образоваться
на коротком отрезке капилляра при при-
кладываемом потенциале в 20 кВ. Об-
800
ф
5 600
о
400
0 5 10 15 20
Время разделения (мксек)
Рис. 15.17. Электрофоретическое разделение
фотопродуктов 5НТ и 5НТРП за 10 мкс. Поле
напряженностью в 0,15 МВ см-1 (35 кВ) было
использовано для фракционирования компо-
нентов. Расстояние между оптическими зонами
детектирования 9 мкм (Plenert and Shear, 2003)
разец переносится электрокинетически
внутри капилляра (рис. 1'5.16 б).
Возможности этой системы про-
демонстрированы на рисунке 1'5.17.
Область в 0,15 МВ см 1 была ис-
пользована для фракционирования
5-гидрокситриптамина (5НТ) и гид-
рокситриптофана (5НТрп) на микросе-
кундной шкале времени.
Будущее этой методологии выглядит
многообещающим, особенно для исследо-
вания коротко живущих интермедиатов
(продуктов промежуточных реакций),
которые сегодня традиционно исследу-
ются методами спектроскопии (часть 3).
Г5.7.3. Электрофоретическая
подвижность молекул ДНК
Электрофоретическая подвижность
макромолекул, наблюдаемая в КЭФ, от-
личается от электрофоретической под-
вижности в растворе из-за существова-
ния ЭОП (см. выше). На рисунке 1'5.16
показано применение метода КЭФ для
измерения подвижности ДНК. Капил-
ляр покрыт акриламидным мономе-
ром, стабильным при щелочных pH и
устраняющим явление ЭОП, обуслов-
ленный стенками капилляра. Подвиж-
ность ДНК в ТАЭ буфере (трис-ацетат
Рис. Г.5.1S. Зависимость электрофоретической подвижности от логарифма молекулярной массы
для фрагментов монодисперсной ДНК в буфере, содержащем трис-ацетат ЭДТА (Stellwagen et al.,
1997)
в присутствии ЭДТА) оказалась равной
(3,75 ± 0,04) х 10"4 х см2-В“’-сек~1 при
25 °C и не зависела от концентрации
ДНК и напряженности электрического
поля. Как видно из рисунка, подвиж-
ность ДНК не зависит от ее молекуляр-
ной массы в интервале от 48,5 килопар
оснований до 400 пар оснований, одна-
ко монотонно уменьшается при пони-
жении молекулярной массы. Причина
такого уменьшения пока не ясна.
Г5.7.4. Двумерный капиллярный
электрофорез
Напомним, что в стандартном двумерном
электрофорезе деление, как в первом, так
и во втором направлениях, выполняется в
полосе геля. К настоящему моменту раз-
работаны приборы, являющиеся прото-
типом двумерного гель-электрофореза в
формате капилляра. Они позволяют про-
вести разделение макромолекул в первом
направлении в одномерном капилляре,
физически разобщенном с капиллярами,
делящими макромолекулы во втором на-
правлении (рис. Г5.19). После окончания
первого разделения в горизонтальном
направлении, капилляр для деления в
первом направлении физически соеди-
няется с капиллярами для разделения во
втором направлении представляющем
собой набор параллельных, вертикально
расположенных капилляров. Использо-
вание полидиметилсилоксана для созда-
ния системы таких двумерных текучих
микросред позволило создать реально ра-
ботающий прототип прибора для разде-
ления в капилляре в одном направлении
и обеспечения пути миграции белков к
ортогонально выстроенным капиллярам
во втором направлении.
Г5.7.5. Разделение хиральных
молекул
Многие из широко используемых се-
годня лекарств содержат, по крайней
a
в
Рис. Г5.19. Традиционный гелевый и капиллярный электрофорез в двумерном формате. В тра-
диционной! системе полоска геля (а) используется для разделения в первом направлении, а
двумерная полоса геля (6), называемая иногда слаб-гелем, осуществляет разделение во втором
направлении. В капиллярном формате (в) полоска геля (а) заменяется капилляром (г), а слаб-
гсль заменяется несколькими параллельно расположенными капиллярами (д). На диаграмме
представлено наиболее простое решение для объединения капилляра, используемого для пер-
вого разделения с упорядоченными в пространстве капиллярами, используемыми для второго
разделения. Эффективность такой системы была продемонстрирована на примере смеси белков,
содержащих карбонангидразу и БСА, меченых флуоресцеином, а также яичный альбумин, мече-
ный красителем Texas-Red (Chen et al., 2002)
Комментарий Г5.12. Энантиомеры и рацематы
Изомерия химических соединений — явление, заключающееся в существовании веществ,
одинаковых по составу и молекулярной массе, но различающихся по строению или располо-
жению атомов в пространстве и вследствие этого по физическим и химическим свойствам.
Такие вещества называются изомерами. Различают два основных вида изомерии: структур-
ную и пространственную. Пространственная изомерия подразделяется на два вида: геометри-
ческую и оптическую. Оптическая изомерия свойственна молекулам органических веществ, !
не имеющим плоскости симметрии. Такие асимметричные молекулы обладают двумя фор- ।
мами оптической активности (см. Главу 35). Одна форма вращает плоскость поляризации |
влево (/ — форма), а другая — вправо (d — форма). Смесь равных количеств двух форм (двух ।
антиподов или энантиомеров) ведет себя как индивидуальное химическое соединение, ли- ।
шейное оптической активности. Такое вещество называется рацемическим соединением или i
рацематом.
а б в
Рис. Г5.20: а — химическая структура р-циклодекстрина (Ward,1994); б - его .модель; в — схема
взаимодействия между двумя лекарственными энантиомерами (Ad+ и А1+) и циклодекстрином
при капиллярной хроматографии (с сайта www.ceandcec.com)
мере, один хиральный центр, и около
80% отмечены как рацематы (коммен-
тарий 1'5.12), Как результат, каждый из
них может содержать примеси в виде
изомеров, не дающих терапевтического
эффекта, и потенциально вызывать от-
рицательные побочные эффекты. Так,
наиболее хорошо описанный кардиоло-
гический препарат, являющийся р-ад-
реноблокатором, является рацематным
пропанолом. D-энантиомер пропанола
является в 100 раз более сильным по
воздействию и слабо метаболизируе-
мым, чем L-энантиомер.
На сегодня основным подходом,
позволяющим разделять хоральные
молекулы, является добавление цик-
лодекстринов в разделяющий элек-
тролит и последующее проведение
капиллярного электрофореза (коммен-
тарий Г5. 13). Циклодекстрин является
идеальным хиральным селектором в
капиллярном электрофорезе благодаря
его способности формировать комплек-
сы по типу «хозяин-гость» со многими
соединениями.
Механизм хиралыюго узнавания
растворенных веществ циклодекстри-
ном, как в связанном, так и в свободном
состоянии, хорошо описан Армстрон-
гом с соавторам в 1985 г. и впоследствии
подтвержден рентгеноструктурным
анализом (рис. 1'5.20 с/ и б). Оба энан-
Комментарий Г5.13.
; Циклодекстрины
Циклодекстрины относятся к макроцик-
лическим соединениям углеводной при-
роды, получаемым путем воздействия на
1 крахмал некоторых ферментов микробного
происхождения и способные образовывать
комплексы включения типа «гость-хозя-
ин». Циклодекстрины представляют по-
лимергомогический ряд общей формулой
(С611|0О_), а также его производные. Общее
для них — наличие характерного цикло-
: декстриронового макроцикла, структурной
единицей которого является а-£)-глюко.за
। в пиранозной форме, имеющая конформа-
I цию кресла. Структурные формулы цик-
лодекстринов трех первых членов этого ,
гомологического ряда, представляющих
собой фрагменты цепей крахмала, свер-
' нутые в компактные кольца, показаны на
рисунке 1'5.20 г/ и б. Наибольший практи-
ческий интерес представляют три первых
возможных гомолога с п = 6, 7, 8, которые
обладают фиксированной и соответственно
осевой симметрией. Циклодекстрины при-
нято обозначать в направлении увеличения
длины кольца: а, р, у и т. д. Молекула таких
циклодекстринов представляют собой усе-
I ценный конус с относительно гидрофобной
| внутренней частью и относительно гидро-
I филыюй внешней.
тиомера циклодекстрина, продвигаясь
вдоль капилляра, будут взаимодейс-
твовать с молекулами в разной степе-
ни (рис. Г5.20«). Следовательно, более
Время (мин)
Рис. Г5.21. Разделение и разрешение пяти
дансил-ВЬ-аминокислот с использованием у-
циклодекстрина и ДСН. Капилляр из плавле-
ного кварца имел размер 57 см х 75мкм (Ward,
1994)
сильно взаимодействующий энантио-
мер подойдет к детектору быстрее, чем
слабовзаимодействующий. Стандарт-
но p-циклодекстрин и его химические
производные, такие как диметилиро-
ванные или гидроксипропилированные
циклодекстрины, добавляются к элект-
ролиту в концентрации от 1 до 100 мМ.
На рисунке Г5.21 показано разделение и
энантиоморфное разрешение пяти дан-
сил-ОЬ-аминокислот с использованием
у-циклодекстрина и ДСН.
Г5.8. Капиллярная
электрохроматография
Успехи в капиллярной жидкостной
хроматографии и в капиллярном элек-
трофорезе ускорили развитие новой
методики, получившей название капил-
лярная электрохроматография (КЭХ).
В обычной жидкостной хроматографии
элюент подается под высоким давле-
нием механическим насосом. Как мы
обсуждали ранее, подвижная фаза, од-
нако, может перемещаться и под дейс-
твием электрического насоса, приводи-
мого в действие силами электроосмоса
(параграф Г5.7.1). Жидкостная хрома-
тография, базирующаяся на эффекте
электроосмоса, называется электрохро-
матографией.
По существу, капиллярная элект-
рохроматография является гибридом
жидкостной хроматографии и капил-
лярного фореза и может проводиться в
открытой емкости, а также в капилляре
из традиционных материалов. Как и в
капиллярном электрофорезе, электри-
ческое поле прилагается поперек разде-
лительного капилляра, генерируя поток
растворителя, вызываемого электроос-
мосом. В капиллярной электрохрома-
тографии электроосмос используется
как «помпа», перемещающая раство-
ритель через разделительную колонку,
тогда как в жидкостной хроматографии
поток движется под давлением.
Следует отметить два процесса,
связанные с разделением в КЭХ. Один
включает удерживание растворенных
веществ, основанное на разнице взаи-
модействий между подвижной и стацио-
нарной фазами, как в ВЭЖХ. Другой
включает дифференциальную мигра-
цию, которая характеризуется электро-
форетической скоростью растворенных
веществ, как в КЭХ.
Стационарная фаза может быть об-
разована на основе шариков кварца,
упакованных внутри капилляра, или
фиксированного на стенках открытой
камеры (рис. 1'5.22). Подвижная фаза
обычно состоит из электролита, содер-
Стенка капилляра
Высокое напряжение
Рис. Г,).22. Схематическое представление явления КЭХ с использованием упакованного капил-
ляра (Colon et al. 1997)
Время (мин)
Рис. 1’5.23. Разделение пяти небольших пептидов на колонке методом КЭФ. Колонка: внутрен-
ний диаметр колонки 100 мкм, ее длина 25 см, напряжение 25 кВ, поддерживающий электролит
40 мМ NH4Ac-AcOH (pH 4.5; ацетонитрил (1:1 по объему) детектирование при длине волны
214 нм. Обозначения: Т — трнпторелин; Д — десмопрессин; О — оксилоцин; А — пептид A; U
— урацил; С — специальный маркер
жащего водно-органический раствор,
переносимый через капилляр элек-
тросмотическим потоком, который
генерируется в интерфазе «твердое
тело-жидкость» из кварцевого материа-
ла. Кварцевая поверхность заряжена
отрицательно благодаря депротони-
рованию поверхностных силанольных
групп. Следовательно, раствор в интер-
фазе несет общий положительный за-
ряд (вызываемый ионами в растворе), с
тем чтобы нейтрализовать постоянный
отрицательный заряд на кварцевой по-
верхности и образовать двойной элект-
рический слой.
Положительно заряженный раст-
вор вблизи поверхности проникает
внутрь подвижной диффузной об-
ласти двойного слоя. Под влиянием
электрического поля сольватирован-
ные катионы движутся к отрицатель-
ному электроду, неся за собой моле-
кулы растворителя. Так генерируется
ноток вблизи поверхности как у квар-
цевых частиц, так и у стенок капилля-
ра. На макромолекулы, обладающие
зарядом, может также действовать
электрическое ноле, приводящее к
дифференциальной миграции как при
электрофорезе. Поэтому заряженные
и незаряженные частицы могут быть
разделены в соответствии с их диффе-
ренциальной миграцией через колонку
и электрофоретической подвижностью
растворенных частиц. В качестве при-
мера на рисунке Гэ.23 показано разде-
ление пяти небольших пептидов на ка-
пиллярной колонке методом КЭХ.
Г5.9. Перечень ключевых идей
° Движение заряженных частиц в электрическом поле называется электрофо-
резом; оно происходит под действием кулоновских сил, действующих между
частицей и нолем.
* Стационарная электрофоретическая скорость заряженных частиц на единицу
электрического поля называется электрофоретической подвижностью-, в систе-
ме СИ она имеет размерность м2- сек"1- В"1.
• Абсолютное значение электрофоретической подвижности в растворе может
быть измерено методом движущейся электрофоретической границы или сво-
бодным электрофорезом; процесс подобен процессу высокоскоростной седи-
ментации.
“ Эффективный заряд частицы может быть вычислен из измерений ее электро-
форетической подвижности и коэффициента диффузии при использовании
техники электрофореза в мембранной геометрии (стационарный электрофо-
рез).
• Молекулярные массы белков могут быть оценены из относительной подвижно-
сти их полипептидных цепей в ДСН-электрофорезе при сравнении с подвиж-
ностью белков-маркеров, молекулярная масса которых известна.
• Метод изоэлектрической фокусировки является методом разделения белков
по их заряду; метод имеет высокое разрешение при разделении белков с раз-
личными значениями изоэлектрических точек (pl), различающихся не менее,
чем на 0,01 единицы pH.
* Комбинация разделения по заряду и разделения по массе приводит к возник-
новению одного из самых эффективных аналитических методов — двумерного
электрофореза в геле, пригодного для разделения белков.
* Одномерный капиллярный электрофорез обладает большой скоростью разде-
ления и высокой разрешающей способностью; разделение требует небольших
объемов образца (0,1-10 нанолитров), в противоположность слаб-электрофо-
резу, для которого необходим образец в объеме микролитров.
* Использование геометрии песочных часов в методе капиллярного электрофо-
реза позволяет достичь микросекундного временного разрешения, что откры-
вает новые возможности в области исследования короткоживущих молекул.
• Двумерный капиллярный электрофорез может рассматриваться как альтерна-
тивный двумерному электрофорезу в геле со всеми преимуществами современ-
ных автоматизированных технологий.
• Циклодекстрины являются идеальными хиральными селекторами в капилляр-
ном электрофорезе благодаря их способности образовывать комплексы «хозя-
ин-гость» с широким кругом соединений.
* Метод капиллярной хроматографии сочетает принципы методов высокоэффек-
тивной жидкостной хроматографии и капиллярного электрофореза; он не нуж-
дается в механическом насосе и имеет ряд преимуществ по сравнению с традици-
онной техникой, используемой для разделения заряженных макромолекул.
Литература для дальнейшего чтения
Исторический обзор и введение в биологические проблемы
Vesterberg О. Л short history of electrophoretic methods. Electrophoresis, 1993.14,
1243-1249.
Cantor C. and Schimmel P. Biophysical Chemistry. Part II. Technique for the Study
of biological Structure and Function. San Francisco. A. W. H. Freeman and Co., 1980.
Электрофоретические эксперименты
Van Holde К. E.,Johnson W. C. and Ho S. P. Principles of Physical Biochemistry. New
Jersey: Prentice Hall, 1998.
Skoog D. A., Holler F. J. and Neiman T. A. Principles of Instrumental Analysis, fifth
edition Philadelphia: Sounders College Publishing, 1998.
Walker J. M. Electrophoretic techniques. In Principles and Techniques of Practical
Biochemistry Ch. 12., eds. K. Wilson and J. Walker Cambridge: Cambridge University
Press, 2000.
Garfin D. E. Electrophoretic methods. In: Introduction to Biophysical Methods for
Protein and Nucleic Acid Research, eds. J. A. Glaser and M. P. Deutscher. San Diego:
Academic Press, 1995.
Макроионы в электрическом поле
Manning G. S. The molecular theory polyelectrolyte solutions with applications to
the electrostatic properties of polynucleotides. Q. Rev. Biophys, 1978.11, 179-246.
Schmitz K. S. Macroions in Solution and Colloidal Suspension. New York: VCH
Publishers, 1993.
Allison S. A. Boundary element modelling of biomolecular transport. Biophys. Chem.
2001.93,197-213.
Метод свободного электрофореза
Laue T. M., Ridgeway Т. М., Wool J. О., and Shepard, H. К. Insights into a new
analytical electrophoresis apparatus. J. Pharm. Sci., 1996. 85, 1331-1335.
Stellwagen N. C., Gelfi C. and Righetti P. G. The free solution mobility of DNA.
Biopolymers, 1997. 42, 687-703.
Метод зонального электрофореза
Weber К. and Osborn M. The reliability of molecular weight determinations by
dodecyl sulfate-polyacrylamide gel electrophoresis J. Biol. Chem., 1969. 244, 4406-
4412.
O’Farrell P. High-resolution two-dimensional electrophoresis of protein. J. Biol.
Chem., 1975.250,4007-4021.
CelisJ. E. and Gromov P. 2D protein electrophoresis: can it be perfected? Curr. Opin.
Biotech. 1999.10,16-21.
Капиллярный электрофорез
Grossman P. D. Free solution capillary electrophoresis. In Capillary Electrophoresis:
Theory and Practice, eds. P. D. Grossman and J. C. Colburn. San Diego, CA: Academic
Press., 1992.
Karger B. L., Chu Y.-H. and Foret F. Capillary electrophoresis of proteins and nucleic
acids. Annu. Rev. Biophys Biomol. Struct., 1995. 24, 579-610.
Zubritsky E. How analytical chemists saved the human genome project. Anal. Chem.,
2002. 74, 23A-26A.
Chen X., Wu H., Mao C. and Whitesides G. M. A prototype two-dimensional capillary
electrophoresis system fabricated in poly (dimethylsiloxane). Anal. Chem., 2002. 74,
1772-1778.
Plenert M. L. and Shear J. B. Microsecond electrophoresis. Proc. Natl. Acad. Sci.
USA, 2003.100, 3853-3857.
Капиллярная электрохроматография
Colon L. A., Guo Y. and Fermier A. Capillary electrochromatography. Anal. Chem.
News, 1997. 69, 461A-467A.
Unger K. K„ Huber M. Walhagen K., Hennessy T. P. and Hearn, M. T. IT. A cri-
ticalappraisal of capillary electrochromatography. Anal. Chem., 2002. 74, 200A-207A.
Глава Гб
ОРИЕНТАЦИЯ МАКРОМОЛЕКУЛ
ЭЛЕКТРИЧЕСКИМ ПОЛЕМ
Г6.1. Исторический обзор
и введение в биологические
проблемы
1875 1880
Дж. Керр (J. Kerr) был первым, кто
наблюдал эффект электрического двой-
ного лучепреломления (ДЛП) при ори-
ентации молекул электрическим полем.
Он показал, что наблюдаемая величина
двойного лучепреломления пропорцио-
нальна квадрату амплитуды приложен-
ного поля. Позднее это явление было
названо его именем.
1880 1920
В эти годы для измерения ДЛП ис-
пользовался постоянный ток высоко-
го напряжения. Это очень затрудняло
его применение для исследования вод-
ных растворов из-за их проводимос-
ти. В 1940-1950 гг. несколько групп
исследователей (У. Кайе и Р. Дева-
ни, Н. А. Толстой и П. П. Феофилов,
К. О’Конски и Б. Зимм, и Г. Бенуа,
(W. Kaye and R. Devaney, N. A. Tolstoy
and P. P. Feofilov, С. T. O’Konski and
В. H. Zimm and H. Benoit) независимо
друг от друга провели первые измере-
ния электрического двойного лучепре-
ломления в коллоидальных системах
с использованием короткого прямо-
угольного электрического импульса.
Это существенно уменьшило эффекты
нагревания и поляризации, которые
были наиболее уязвимыми местами в
исследованиях биологических макро-
молекул в водных растворах.
1910
Л. Ланжевен (L. Langevin) разрабо-
тал первую теорию электрического
двойного лучепреломления, которая
в 1918г. была обобщена М. Борном
(М. Вот). Было показано, что ориен-
тация оптически анизотропных моле-
кул может быть вызвана ее постоянным
дипольным моментом или электри-
ческой анизотропией, или обоими сра-
зу. В 1931г. А. Петерлин и Г. Стюарт
(A. Peterlin и Н. A. Stuart) распростра-
нили теорию на случай суспензии жес-
тких частиц.
1951
Г. Бенуа (Н. Benoit) развил теорию
переходных явлений в электрических по-
лях. Он решил уравнение вращательной
диффузии, связанное со скоростью изме-
нения ориентации аксиально симметрич-
ных жестких макромолекул при появле-
нии и исчезновении электрического поля.
Он показал, что коэффициент враща-
тельной диффузии может быть получен
из кривой спада (затухания) ориентации
при выключении поля, а параметры элек-
трической ориентации - из кривой на-
растания ориентации при его включении.
Впоследствии он применил эту теорию в
исследованиях водных растворов вируса
табачной мозаики и ДНК.
1959
O’KoHCKH(O’Konski) предложил ме-
тод раздельного определения дипольно-
го момента, анизотропии электрической
поляризуемости и фактора оптической
анизотропии из амплитуды двойного
лучепреломления для аксиально сим-
метричных жестких макромолекул. Этот
метод был применен к вирусу табачной
мозаики, синтетическим полипептидам
и коллагену. В 1963 г. И. Тиноко (млад-
ший) (I. Tinoco Jr.) распространил тео-
рию двойного лучепреломления на дру-
гие молекулярные модели и применил ее
для интерпретации данных по фибрино-
гену. В 1973 г. была опубликована книга
Е. Фредерик и К. Хусьера (Е. Fredericq
and С. Houssier’s) «Электрический
дихроизм и электрическое двойное луче-
преломление» — одна из лучших книг,
описывающих явление Керра и его при-
менение.
1979
В. А. Вегенер (W. A. Wegener) ре-
шил задачу спада кривой двойного лу-
чепреломления для жестких тел произ-
вольной формы. Он показал, что в этом
случае временная зависимость затуха-
ния ДЛП является экспоненциальной
функцией, состоящей из пяти компонен-
тов. В1981 г. Б. Зимм и П. Дж. Хагерман
(В. Zimm and Р. J. Hagerman) предста-
вили различные варианты зависимости
между коэффициентом вращательной
диффузии, вычисленной из спада оп-
тической анизотропии после короткого
импульса электрического поля и гибко-
стью (персистентной длиной) корот-
ких червеобразных цепей. В 1990-х гг.
П. Дж. Хагерман (Р. J. Hagerman) и
его группа внесли существенный вклад
в теорию и применение электрического
ДЛП для исследования макромолеку-
лярной структуры ДНК.
1990 2000
За эти годы в практику ДЛП были
внедрены два новых подхода. Во-пер-
вых, время ответа детектирующих сис-
тем было существенно сокращено (до
1 наносекунды), что позволило исследо-
вать электрооптические и гидродинами-
ческие свойства белков и нуклеиновых
кислот, имеющих небольшие размеры.
Во-вторых, керамика, не проводящая
электричество, но обладающая превос-
ходной тепловой проводимостью (нит-
рид алюминия) была введена в практику
электрического ДЛП. Это позволило про-
водить измерения в растворах с большей
концентрацией солей без значительного
при этом выделения джоулевого тепла во
время электрического импульса и, следо-
вательно, применять его при более высо-
ких значениях амплитуды и частоты. Все
эти инновации привели к значительной
стабилизации сигнала, более точному
разрешению экспоненциальных компо-
нентов из кривых ДЛП и открыли новые
пути для исследования конформации и
гибкости РНК и ДНК.
2000 и по настоящее время
На сегодня метод электрического
двойного лучепреломления является
одним из наиболее точных методов оп-
ределения коэффициентов вращатель-
ного трения многих биологических мак-
ромолекул и, в первую очередь, ДНК и
РНК. Стоит лишь сожалеть, что полный
прибор для измерения электрического
двойного лучепреломления коммерчески
недоступен и всякий, кто планирует за-
няться исследованиями в этой области,
должен сам сконструировать необходи-
мый оптический и электрический трак-
ты. Рекомендации по конструированию
аппаратуры читатель может найти в ли-
тературе (Hagerman, 2000).
Гб.2. Макромолекулы
в электрическом поле
Гб.2.1. Дипольный момент
биологических макромолекул
Наиболее важным понятием для даль-
нейшего рассмотрения является элек-
трический диполь, который состоит из
двух зарядов противоположного знака
(+q и -q), разделенных расстоянием L
(рис. Г6.1). Различные типы заряжен-
ных частиц, такие как ионизированные
группы или группы с асимметричным
распределением заряда, могут рассмат-
риваться как диполи. В этом случае
говорят, что молекула обладает посто-
янным дипольным моментом (коммен-
тарий Г(>. I ). Дипольный момент//, зада-
ется определенным значением зарядов
и расстоянием между ними:
р = 9£. (Г6.1)
Дипольным моментом обладают
только асимметричные молекулы. На-
пример, молекула Н2О изогнута и имеет
значение дипольного момента 1,85 де-
бая (D). Молекула СО., симметрична
и не имеет дипольного момента.
Когда молекула, не имеющая ди-
польного момента, переносится в
электрическое поле, то оно вызывает
смещение отрицательно заряженного
электронного облака от положитель-
но заряженного ядра, создавая таким
Рис. 16.1. Диполь в электрическом поле.
В электрическом поле диполь не перемеща-
ется, поскольку его суммарный заряд равен
нулю. Однако он обладает вращающим момен-
том, который имеет тенденцию ориентировать
молекулу в направлении приложенного поля
i Комментарий Гб. 1. Дипольный
| момент
! Значение дипольного момента, р, вы-
ражается в системе СГС — универсальной
системе, обычно используемой в этой об-
ласти физики. Дипольный момент выража-
I ется в единицах дебая, D (ID = 10-18 элект-
1 ростатических единиц заряда, умноженных
на сантиметр). Фактор перехода в систему
СИ 1D = 3,33564 х Ю'30 кулон-метров.
Комментарий Гб.2. Поляризуемость
Поляризуемость — это скалярная ве-
: личина для изотропных веществ, но она
становится тензорной для анизотропных
веществ. Размерность поляризуемости см3 '
в системе СГС.
образом дипольный момент. В этом
случае говорят, что молекула облада-
ет наведенным дипольным моментом.
Он существует до тех пор, пока присут-
ствует электрическое поле.
Взаимодействие молекул с внешним
полем зависит от их поляризуемости
или способности нейтральных молекул
становиться диполем во внешнем элек-
трическом поле (к'оммсн uipiii! Г(>,2).
Значение дипольного момента, р, инду-
цируемого внешним полем, пропорцио-
нально интенсивности этого поля. Кон-
станта пропорциональности называется
поляризуемостью:
р = аЕ. (Г6.2)
Очень сложные молекулы могут
быть представлены одним дипольным
моментом, представляющим сумму мо-
ментов индивидуальных диполей внут-
ри молекулы. Синтетические полипеп-
тиды в а-спиральной конформации
имеют очень большой дипольный мо-
мент, поскольку все маленькие диполь-
ные моменты аминокислотных остат-
ков вдоль цепи складываются вместе, в
то время как антипараллельная двойная
спираль ДНК имеет небольшой посто-
янный момент, но может приобретать
большой наведенный дипольный мо-
мент.
Гб.2.2. Электрическое двойное
лучепреломление и дихроизм
Некоторые кристаллические структуры
разлагают падающий свет на два пер-
пендикулярных поляризованных луча,
которые распространяются в среде с
различными скоростями. Это свойство
называется двойным лучепреломлением
или двойной рефракцией. Двойное лу-
чепреломление среды определяется как
разница показателей преломления этих
двух лучей:
Ди = пх-пу (Г6.3)
и связана с оптическим замедлением 5
(также называемым фазовым сдвигом)
уравнением:
Аи
5 = 2тг/у, (Г6.4)
где X — длина волны падающего луча
в вакууме, а I — толщина образца.
Когда два ортогональных коле-
бания, проходящих через образец, по-
глощают по-разному, то говорят, что
образец проявляет дихроизм, который
определяется как разность в поглоще-
нии двух лучей:
АА=Ах-Аи. (Г6.5)
Эти два явления лежат в основе опти-
ческой анизотропии. Они обычно называ-
ются линейным двойным лучепреломле-
нием или линейным дихроизмом, с тем
чтобы отличать их от кругового двойного
лучепреломления и кругового дихроизма,
которые характеризуют оптическую ак-
тивность (Глава 35). Подобно рефракции
и абсорбции, двойное лучепреломление
(если присутствует) может наблюдаться
при любой длине волны, в то время как
дихроизм (если присутствует) проявля-
ется только в полосе поглощения.
Оптическая анизотропия может ин-
дуцироваться в изотропных растворах
внешними силами, такими как гидроди-
намические (двойное лучепреломление
в потоке), электрические (электриче-
ское двойное лучепреломление) и маг-
нитными полями (магнитное двойное
лучепреломление). Эти силы создают
ориентацию частиц в растворе: в таких
случаях направление х и у ортогональ-
ных поляризаций выбирается, соот-
ветственно, параллельно и перпендику-
лярно по отношению к ориентирующей
силе, и двойное лучепреломление и
дихроизм определяются уравнениями:
Дп = Пц-и± (Г6.6)
и
М=лн-л± (Г6.7)
соответственно.
В зависимости от относительных
значений ц. (Аг|) и и± (А±) двойное лу-
чепреломление (дихроизм) может быть
положительным или отрицательным.
В принципе электрическое двойное лу-
чепреломление и электрический дих-
роизм дают одинаковую информацию
относительно макромолекулярной струк-
туры: идентичный набор релаксационных
времен, одинаковые знаки и амплитуды
двойного лучепреломления, тождествен-
ную информацию относительно ориента-
ции хромофорных групп внутри молеку-
лы. Однако на практике измерение ДЛП
дает более точную информацию относи-
тельно набора релаксационных времен,
следовательно является более приемле-
мым для исследования формы макромо-
лекул, в то время как дихроизм является
более точным способом определения ам-
плитуд и является более удобным в ис-
следованиях, связанных с определением
внутренней структуры.
В этой главе мы будем рассмат-
ривать только метод двойного луче-
преломления. Мы не будем обсуждать
магнитное двойное лучепреломление,
которое является результатом появле-
ния магнитной и оптической анизотро-
пии частиц в сильном магнитном поле.
Интересующиеся читатели могут обра-
титься к специальной литературе.
Г6.3. Теоретические основы
измерений двойного
лучепреломления
Два основных импульса используют
в измерениях ДЛП (рис. Гб.2). Первый
прямоугольный электрический импульс
(рис. Гб.2<7). После приложения импуль-
са величина ДЛ П изменяется во времени,
как показано на рисунке Гб.2 б. С помо-
щью этого импульса могут быть исследо-
ваны процессы, связанные с нарастанием
и спадом двойного лучепреломления, а
также с величиной амплитуды равновес-
ного состояния. Второй прямоугольный
импульс имеет сложную форму: после
достижения величины постоянного сиг-
нала двойного лучепреломления поле
быстро меняет знак на обратный, как
показано на рисунке Гб.2 в. После такого
импульса величина двойного лучепре-
ломления изменяется (рис. 1'6.2/). Эта
форма импульса наиболее полезна при
исследованиях механизма электриче-
ской ориентации.
Е
б
в
Рис. Гб.2: а — прямоугольный электрический импульс £ как функция времени; б — изменение ве-
личины сигнала двойного лучепреломления Ди в зависимости от времени, показывающее нарас-
тание сигнала и достижение стационарного значения Дп0; в — инверсионный электрический им-
пульс. Амплитуда сигнала изменяет свой знак на обратный в середине импульса; г — переходной!
сигнал двойного лучепреломления в ответ на инверсию поля
Ниже мы кратко просуммируем ос-
новные теоретические результаты, полу-
ченные для растворов жестких частиц.
Для упрощения изложения мы пред-
положим, что макромолекулы имеют
общую ось симметрии их электричес-
ких, оптических и гидродинамических
свойств и постоянный дипольный мо-
мент вдоль этой оси. Мы также будем
считать, что раствор разбавлен настоль-
ко, что взаимодействие между макромо-
лекулами отсутствует.
Г6.3.1. Стационарное двойное
лучепреломление
Зависимость между оптической ани-
зотропией и величиной электрического
двойного лучепреломления раствора
макромолекул выражается следующим
соотношением:
/тт* о\
Ди =----(g,-g2)®, (Г6.8)
п
где Q — это объемная доля растворен-
ных макромолекул, п — показатель пре-
ломления раствора в отсутствие электри-
ческого поля. Двойное лучепреломление
пропорционально произведению фак-
тора оптической анизотропии, g^-g2, на
функцию распределения ориентации,
Ф. При долговременном действии элек-
трического поля функция распределе-
ния ориентации, Ф, принимает вид:
Ф = |(3(со520}-1), (Г6.9)
где 0 — угол между осью симметрии
макромолекулы и направлением поля, а
(...) означает усреднение по ансамблю.
Когда эффект ориентации, обуслов-
ленный электрическим полем, уравно-
вешивается эффектом дезориентации,
обусловленным броуновским движени-
ем, то:
2тг£2. ч 1
Д« =-----(g( -£'2)77х
п Ю (Г6.10)
2
kT ’ ’
k'lT-
где а, и а,, — избыточная электрическая
поляризуемость вдоль оси симметрии и
продольных осей, соответственно. Та-
ким образом, двойное лучепреломление
пропорционально квадрату силы поля в
области малых полей. Этот закон назы-
вается законом Керра. Уравнение Г6.10
демонстрирует, что в ДЛП-эксперимен-
тах молекулы имеют тенденцию ориен-
тироваться в поле через взаимодействие
индуцированных (af - а.,), и/или посто-
янных, ц, диполей с приложенным элек-
трическим полем.
Константа пропорциональности К на-
зывается константой! Керра. Она равна:
" a a (Г6.11)
ц a. - a.
% + kT
Как видно из уравнения Г6.11 кон-
станта Керра является произведени-
ем двух вкладов, один из которых оп-
тической природы (g( - g.,), а второй
электрической природы (фЧ ~-Т 2 +
+ (at - a2)-k 1 Г1). Поэтому определить ин-
дивидуальный вклад каждого в констан ту
Керра невозможно (1юм.\н'п i.ipin'i
Гб.3.2. Процесс спада двойного
лучепреломления
При выключении электрического поля
будет происходить дезориентация мак-
ромолекул под действием броуновского
движения, что приводит к спаду двой-
ного лучепреломления. В этом случае из
кривой спада можно прямо вычислить
время релаксации молекулы т, посколь-
ку оно описывается формулой:
An = Дл„ехр(-/ / т), (Гб.12)
где Ли — значение величины двойного
лучепреломления в любое время t после
начала спада, а Az?(| — начальная величи-
на двойного лучепреломления в момент
времени t = 0.
Вспомним, что время релаксации
т связано с константой вращательной
диффузии ©(параграф Г2.3.3). Таким
образом, анализ кривой спада двойного
лучепреломления обеспечивает самый
прямой и удобный способ определе-
ния константы вращательной диффу-
зии, по сравнению с такими методами,
как двойное лучепреломление в потоке
(Глава Г.7) пли деполяризация флуо-
ресценции (Глава Г8). Связь константы
вращательной диффузии 0 с размера-
ми и формой макромолекулы была об-
суждена нами ранее в параграфе Г2.3.4.
Гб.3.3. Процесс нарастания
двойного лучепреломления
При низких значениях величины ноля
зависимость двойного лучепреломле-
ния Дп от времени t описывается более
сложным выражением:
Дд
Дд„
ЗА
2(Л + 1)
ехр(-20Г) +
Л-2
2(Л+1)
ехр(-60Г),
(Г6.13)
где Дд() является значением А/? при t —>°°
и эквивалентно постоянному значению,
приведенному в уравнении (Гб. 10). Па-
раметр/! является отношением вклада по-
стоянного диполя к вкладу индуцирован-
ного диполя н для стационарного случая
при низкой величине поля имеет вид:
А =-----------. (Г6.14)
(а, -а2)&Г
. Комментарий Гб.3. Эффект «формы» '
в двойном лучепреломлении
Если молекулы имеют средний показатель
преломления, отличающийся от такового у
растворителя, то раствор будет проявлять
эффект «формы» в двойном лучепреломле-
нии. Наилучший способ вычленить вклад
«формы» в двойном лучепреломлении — это
измерить двойное лучепреломление в раство-
рителях с различным показателем преломле-
ния. Этот эффект исчезнет, когда показатель
преломления растворителя станет равным
таковому у растворенных молекул. Растворы
глицерина и сахарозы в воде используют-
ся как добавки для увеличения показателя ,
преломления среды и, следовательно, для ,
изменения вклада формы. Эти же растворы !
используются дтя контрастирования при
рассеянии рентгеновских лучей (см. секцию
Е2.4). Если молекулы оптически анизотроп-
ны, то раствор будет проявлять также и внут-
реннее двойное лучепреломление, величина
которого будет определяться внутренней
структурой частицы.
Когда ориентация полностью обу-
словлена анизотропией электрической
поляризуемости (т. е. А = 0), уравнение
(Г6.13) сокращается до
— = 1-ехр(-60г). (Гб. 15)
Д«о
В этом случае нарастание и спад
кривых симметричны. С другой сто-
роны, когда ориентация обусловлена
постоянным дипольным моментом
(т. е. А = °°), уравнение (Гб. 13) транс-
формируется как:
Дц . 3 , ,
---= 1 —ехр(-20г)+
Д"° 2 (Гб. 16)
+|ехр(-60г).
В этом случае нарастание идет го-
раздо медленнее, чем спад, и параметр/!
Pin . 16.3. Нормализованные кривые нараста-
ния и спада Дл/Длов зависимости от времени
(метод площадей)
может быть вычислен из формы кривой
нарастания.
Другой метод определения парамет-
ра А есть метод площадей (рис. Гб.З).
Если площадь 5, — площадь под кривой
нарастания, а 5., — площадь под кривой
спада, то эти площади при низких полях
связаны отношением:
5, _ 4Л +1
S2 " А + 1 ’
(Г6.17)
Отметим, что отношение St/S2 рав-
но 1 для Л = 0 и равно 4 для Л=°°.
Рис. I (i.l. Схема эксперимента по электрическо-
му ДЛП. При воздействии электрического поля
молекулы в ячейке частично ориентируются и
раствор проявляет двойное лучепреломление.
При использовании стандартной оптической
схемы электрическое поле направлено вдоль х-
оси, которая находится под углом 45° по отноше-
нию к поляризатору и анализатору. Последние
расположены под углом 90° относительно друг
друга (Hagerman, 2000). Для детального озна-
комления с различными оптическими схемами,
используемыми в электрическом ДДП, чита-
тель отсылается к соответствующей литературе
(Fredericq and Houssier, 1973)
Гб.3.4. Двойное лучепреломление
в реверсивных полях
Уравнение для случая реверсии в поле
низкой мощности (рис. Гб.2 в), эквива-
лентно уравнению Бенуа для процесса,
описывающего ход двойного лучепре-
ломления в нарастающем поле. В этом
случае уравнение Гб. 13 имеет вид:
Ап . ЗА . z .
---= 1-----[ехр(-60г) -
Ди0 Д + 1 (Г6.18)
-ехр(-20Г)].
Наиболее характерной чертой кри-
вой реверсии является наличие мини-
мума, который достигается за время ?чн|1
с глубиной tZMi||)(pnc. Гб.2/). В соответ-
ствии с теорией при использовании по-
лей низкой мощности ?м1п| и б/мин не зави-
сят от мощности поля и, более того, (ми||
не зависит от типа диполя.
Гб.4. Измерения
электрического двойного
лучепреломления
Гб.4.1. Оптические схемы
Основная схема эксперимента по ДЛП
приведена на рисунке Гб. 1. Свет из лазер-
ного источника, распространяющийся
вдоль z-оси, проходит последовательно
поляризатор, ячейку Керра и анализа-
тор. Луч поляризуется поляризатором,
который располагается под углом 45°
по отношению к электрическому полю,
и затем попадает на ячейку. Когда рас-
твор после приложения импульса ста-
новится двулучепреломляющим, свет,
выходящий из раствора, оказывается
эллиптически поляризованным. Затем
свет проходит через анализатор, кото-
рый расположен под углом 90° по отно-
шению к поляризатору, и после этого
попадает на детектор. В эксперименте
используются две оптические схемы: без
и с пластинкой в четверть длины волны
( КО.ШК'П ГЭ|)иГ| I .(). i ).
В простейшем оптическом при-
боре без пластинки в четверть длины
волны изменения интенсивности све-
та, под действием электрического поля
Комментарий Гб.4. Двойное лучепреломление в анизотропных кристаллах,
пластинка в !4 длины волны
а б
, Рис. а. Явление двойного лучепреломления, которое состоит в том, что луч света, падающий
i на поверхность кристалла, раздваивается в нем на два преломленных луча — обыкновенный
о и необыкновенный е. Угол преломления г зависит от того, как ориентирована поверхность
пластинки по отношению к оптической оси кристалла.
Рис. б. Параллельный пучок света, прошедшего через поляризатор (П), падает нормально
I на поверхность плоскопараллельной пластинки, вырезанной из одноосного кристалла парал-
лельно его оптической оси MN.
В оптически анизотропных кристаллах (а их в природе большинство) наблюдается явление
двойного лучепреломления, которое состоит в том, что луч света, падающий на поверхность
кристалла, раздваивается в нем на два преломленных луча (рис. а). В одноосном кристалле
. один из лучей, образующихся при двойном лучепреломлении, подчиняется обычным законам
преломления света и поэтому его называют обыкновенным лучом и обозначают буквой о. Вто-
рой луч обозначают буквой е, так как он не лежит в плоскости падения. Если параллельный
пучок света, прошедшего через поляризатор, падает нормально на поверхность плоскопарал-
: дельной пластинки, вырезанной из одноосного кристалла параллельно его оптической оси MV,
то на входе в кристаллическую пластинку электрические векторы о и е волн колеблются в од-
ной фазе (рис. б). В пластинке о и е волны распространяются с разными скоростями, поэтому на
выходе из пластинки толщиной d волны будут колебаться со сдвигом по фазе. В зависимости от
толщины пластинки возможно несколько частных случаев. Один из них соответствует сдвигу7
фаз п/2. Такая пластинка называется пластинкой в четверть длины волны.
отношению к оси поляризатора Р. Приклады-
ваемое поле направлено под углом 45° к оси
А и оси Р
Рис. Г6.6. Кривая спада двойного лучепре-
ломления для фрагмента ДНК размером 98
пар оснований в полулогарифмическом мас-
штабе аппроксимируется одной экспонентой
в области свыше двух порядков спада интен-
сивности сигнала. Условия проведения экс-
перимента: 2,0 мМ Трис-НС1-буфер, pH 8,0,
4 °C; длительность импульса 2 мкс, амплиту-
да импульса 5 кВ сек-1. Вставка: та же кривая,
построенная в линейной шкале по оси ординат
(Hagerman, 1985)
Д/6, регистрируемые детектором, можно
выразить уравнением Д/6 =/osiir(3/2),
где 3 — оптическое замедление, обуслов-
ленное электрическим полем, и /0 — ин-
тенсивность света, который попадает на
детектор, если поляризатор и анализа-
тор параллельны, а 8 равняется нулю.
Для 8«1 мы получаем приближенную
формулу Д76 = /082 /4. Эта схема облада-
ет тем недостатком, что ее чувствитель-
ность мала при небольших значениях 8
и знак 8 остается неизвестным.
В другом варианте оптическое уст-
ройство включает пластинку в четверть
длины волны, которая расположена
между ячейкой Керра и анализатором
(см. к<>ммс11 i:!|ii।н Гб. ।). Анализатор
может вращаться на угол а от кресто-
образного положения, как показано на
рис. Гб.5. Эллиптически поляризован-
ный свет, исходящий из двулучепре-
ломляющего раствора, превращается в
линейно поляризованный свет, после
прохождения через пластинку в чет-
верть длины волны, угол поляризации
которой зависит от величины опти-
ческого замедления 8, обусловленного
электрическим полем. В этой оптиче-
ской схеме зависимость между Д/6 и 8
для 8 «1 описывается уравнением:
Д/6 = /osin2a(8/2). (Г6.19)
Следовательно, Д/6 пропорциональ-
на 8, а не 82. В этом случае чувствитель-
ность выше, чем без использования
пластинки в четверть длины волны.
Более того, световой сигнал усилива-
ется при положительном значении 8 и
уменьшается при его отрицательном
значении. Это позволяет определить
знак двойного лучепреломления.
Кривая спада двойного лучепрелом-
ления обычно представляется в полуло-
гарифмической шкале, как показано на
рисунке Гб.б. Если исследуемые молеку-
лы являются жесткими и имеют одинако-
вые размеры (моиодисперсный раствор),
то зависимость In—1— как функция
Дп(0)
времени является прямой линией с поло-
жительным наклоном, равным 1/т.
Гб.4.2. Интерпретация спада
кривой двойного лучепреломления
Как показывает теория для жестких
частиц произвольной формы спад кри-
вой двойного лучепреломления состоит
из пяти экспонент (пяти времен), в за-
висимости от симметрии частицы (рас-
пределения в ней заряда) и направле-
ния и величины различных оптических
переходов внутри частицы:
5 -t
Д?г(/) = Qexp—. (Г6.20)
*=i Т
Выделение пяти времен релаксации
тк и соответствующих пяти амплитуд
Ск из экспериментально измеренной
одной кривой Дп(г)на практике невоз-
можно по хороню известной причине:
задачи такого рода относятся к чис-
лу некорректно поставленных задач
(см. коммсн । при и i 1.1 2 и I 1 ().(;).
В случае трехосных тел, когда ос-
новные оси вращательной диффузии
(0), электрической поляризуемости и
направления электрического вектора
совпадают, тогда только два времени
релаксации ответственны за спад двой-
ного лучепреломления:
Дп(Г) -г -Г
—— = а,ехр(—) + а,ехр(—), (Г6.21)
Дп(0) т, т3
где а( и аГ) относятся к самому длительно-
му и самому короткому времени релак-
сации, соответственно. В 1996 г. Гарсия
де ла Торре с соавторами предложили
характеризовать спад в терминах дру-
гих релаксационных времен, которые
могут быть определены из эксперимен-
та. Одно из них является начальным
временем релаксации Гнач, определяе-
мым как:
тна,г‘ = [dbn(t)/dt)\._0, (Г6.22)
так что т|1ач_1 является начальным отрез-
ком кривой Да (Г). Второе время явля-
ется средним релаксационным време-
нем т :
ср
Рис. Г(>.7. Геометрическая интерпретация начального и среднего времен релаксации для трех
электрооптических процессов: роста, реверса и спада. Начальное время релаксации для всех
трех процессов обозначено отрезком прямой линии, а среднее время обозначено заштрихо-
ванной площадью А
Trp=fAn'm (Г6.23)
О
где Ди '(О = Дп(Г) характеризует область
спада, а и '(£) = 1 — Ди (Г) характеризует
область возрастания и область ревер-
сивного процесса, так что Ди'(0) = 1 и
An'(°°) = 0 во всех случаях. Следует
заметить, что значение т — это, факти-
чески, площадь под кривой спада Ди (Г).
Геометрический смысл тнач и тср иллюст-
рируется на рисунке Гб.7.
Для сферы радиусом Ro, все ^вре-
мена релаксации (включая т||1ч и т )
сводятся к одному значению, т0, которое
равно:
т"=бЬ- (гб-24>
Гб.5. Глобальная структура
и гибкость белков
Из уравнения Гб. 11 следует, что мето-
дом электрического ДЛП можно оп-
ределить постоянные и наведенные
дипольные моменты, электрическую
поляризуемость и оптическую анизот-
ропию макромолекул. Ниже мы приве-
дем несколько примеров применения
метода ДЛП в исследованиях белков,
а именно домена белка кинезина, отно-
сящегося к классу линейных моторов
(см. секцию Д6.3).
Г6.5.1. Исследование внутренней
структуры «моторного» домена
«Моторные» домены, а именно ncd до-
мен дрозофилы и домен кинезина чело-
века, осуществляют транспорт везикул и
органелл вдоль микротрубочек (см. сек-
цию Д6.3). Укороченная тяжелая цепь
кинезина человека, так называемый ки-
незин 349, имеет молекулярную массу
39200 Да и содержит 349 аминокислот-
ных остатков; тяжелая цепь моторного
домена Drosophila ncd (ncd (335-700))
имеет молекулярную массу 41308 Да и
содержит 366 аминокислотных остат-
ков. Хотя молекулярные массы двух
моторных доменов отличаются всего
на 5% и имеют высоко гомологичные
последовательности, они проявляют
различные оптические, электрические
и гидродинамические свойства. Немно-
го меньший по длине домен кинезина
(349) обладает значительно более про-
должительным вращательным временем
релаксации (к тому же зависит от мощ-
ности поля), чем ncd домен. Наиболее
Время (нсек) Время (нсек)
а б
Рис. Гб.8. Сравнение сигнала электрического ДЛП раствора: а — кинезина (349) и б — ncd (335-
700) в присутствии АДФ и Na2HPO4 после приложения импульса в течение 520 наносекунд. Кон-
центрация каждого белка 1,5 мг-мл1. Измерения проводились при 4 °C (Eden et al., 1995)
Рис. I 6.9. Зависимость времени вращательной релаксации белков от молекулярной массы
в двойной логарифмической шкале в соответствии с данными iucu.i I В. I
интригующим является тот факт, что
два домена имеют противоположные
знаки для сигнала ДЛП. Как показано
на рисунке Г6.8 а знак двойного лучеп-
реломления для кинезина положителен,
тогда как для ncd — моторного доме-
на — отрицателен (рис. Г6.8б). Различия
в знаке и значении константы Керра ncd
(335-700) и кинезина (349) предполага-
ют существенные различия в их элект-
рических свойствах. Интересно, что оба
моторных домена движутся в разных
направлениях по микротрубочке, хотя
зависимость между направлением и зна-
ком ДЛП-сигиала не установлена.
Гб.5.2. Гидродинамические
размеры глобулярных белков
Вычисления по формуле Перрена
(уравнение Г2.27) показывают, что ти-
пичные сферические молекулы радиу-
сом 25 А имеют времена релаксации в
воде при температуре 25 °C около 17
нс. Измерение таких маленьких времен
требует приборов с очень высоким раз-
решающим временем (2-5 нс). В
пс Гб. I приведены времена вращатель-
ной диффузии некоторых глобулярных
белков, полученные в основном с ис-
пользованием высокоразрешающего
ДЛП-оборудования. В эту таблицу
включены также данные для димера
грамицидина, коэффициент враща-
тельной диффузии которого получен
методом динамического светорассея-
ния (см. секцию Г10.3). Зависимость
времени вращательной релаксации
для белков от молекулярной массы в
двойной логарифмической шкале при-
ведена на рисунке Гб.9. Из него видно,
что эта зависимость представляет со-
бой прямую линию, наклон которой
близок к единице. Отсюда следует, что
г : Времена релаксации некоторых глобулярных белков
Белок Молекулярная масса (кДа) Вращательное время релаксации Т.,()11. (нсек)
Грамицидин(димер) 2,5 2,7
Кинезин (349) 39,2 41
ncd(335-700) 41,3 43
БСА 68,0 78
Комментарии Гб.5. Персистентная
длина цепи как мера ее гибкости
Расстояние между концами (h2} черве-
образной цепи может быть вычислено по
контурной длине LK, если известна персис-
тентная длина, Р.
{h2) = 2PLr(l-P/Lr)+ (А)
+P/Lr[exp(-L,./p)]
и в пределах больших значений Lk (клуб-
кообразная конформация) получаем урав-
нение: !
({h2}/Lc)L^=2P. (Б)
Для малых значений LKуравнение (А)
приводится к виду для LJP <1:
(A2) = L’[l-l/3(Lr/P)+
+ 1/12(Г,./Р)2].
При L «Р из уравнения (В) следу-
ет, что {h2^ = L2r, т. е. расстояние между
концами равно контурной длине. Это ха- ,
рактерно для палочкообразных моделей. I
Если Ьк увеличивается, (h2^<Cc проявля- I
ется эффект кривизны. При очень высо-
ких значениях LK действительно соотно-
шение (h2^L2 = 2Р/Lk, обозначающее, что
отклонение значения от такового для |
палочкообразных молекул определяется
отношением персистентной длины к кон-
турной длине. I
время вращательной релаксации про-
порционально объему, занимаемому
молекулой белка (см. секции Г2.3 и па-
раграф Г8.5.3).
Гб.6. Глобальная структура
и гибкость нуклеиновых
кислот
Ранние исследования конформацион-
ных свойств нуклеиновых кислот про-
ведены с относительно длинными,
фракционированными по размеру, моле-
кулами. Неоднозначность данных о дли-
не ДНК ограничивала точность оценки
гибкости по этим измерениям. Исполь-
зование метода электрического ДЛП
является более приемлемым для описа-
ния глобальной структуры и гибкости
нуклеиновых кислот по двум причинам.
Во-первых, недавние инновации в этом
методе позволили получать высокоста-
бильные сигналы и, как следствие этого,
более высокое временное разрешение эк-
споненциальных компонент. Во-вторых,
как мы уже упоминали, молекулы ДНК
и РНК можно легко получать фактиче-
ски любой последовательности и длины
с точностью до одного основания.
Г6.6.1. Гибкость молекулы ДНК
в В- и А-форме
Нативная ДНК в В-формс обладает от-
рицательным дихроизмом в видимой и
в ультрафиолетовой области спектра в
полосе ее поглощения (X = 259 нм) (Гла-
ва 31). Такие же эффекты — отрица-
тельное ДЛП и дихроизм — имеют мес-
то, когда молекула ДНК ориентируется
гидродинамическим полем (Глава Г7).
Эти факты привели к двум заключениям
относительно поведения ДН К в электри-
ческом поле: макромолекула ориентиру-
ется длинной осью в направлении поля
и плоскость оснований пар нуклеотидов
и составляют приблизительно правиль-
ные углы по отношению к длине молеку-
лы (рис. Гб. 10). Однако величины при-
веденных значений дихроизма, которые
измеряются и затем экстраполируются
к бесконечному значению напряжения
поля (полная ориентация), оказались
очень низкими. Основная причина этого
состоит в гибкости цепи ДНК.
Гибкость цепи ДНК удобно харак-
теризовать персистентной длиной. Это
понятие в 1948 г. было введено Кратки
и [Городом в их пионерской работе, в
которой они предложили модель черве-
образной цепи (см. рис. Г2.17). Модель
характеризуется двумя параметрами: ее
полной контурной длиной Lk и перси-
стентной длиной Р. Последняя опреде-
ляет, как далеко гибкая макромолекула
простирается вдоль цепи (рис. Гб. 11).
Равновесная персистентная длина
Р измерялась на протяжении многих
лет различными методами, которые
чувствительны к равновесным свой-
ствам макромолекул, включая стати-
ческое рассеяние света, ДЛП в потоке,
седиментацию, характеристическую
вязкость. Проблемы с интерпретацией
некоторых из этих данных и получение
противоречивых результатов, особенно
связанных с зависимостью Рот концент-
рации соли, неоднократно обсуждались
в литературе. Однако, несмотря на эти
проблемы, сегодня найден консенсус
относительно ее длины: равновесная
персистентная длина ДНК в В-форме
в буфере с умеренной концентрацией
соли принята равной около 500 А (ь< >м-
мен ।apiIii Гб..-)).
Рис. 1 б. 1 1. Персистентная длина Р опреде-
ляется как проекция вектора г, соединяющего
концы цепи в направлении первой связи при
бесконечной длине цепи. Если длина связи /
становится все меньше и меньше, цепь может
превратиться в нить с плавными изгибами.
Эта абстрактная модель называется моделью
Кратки-Порода
Существующая сегодня техника по-
лучения фрагментов ДНК практически
любой фиксированной длины позволя-
ет обойти проблему полидисперсности
и получить достоверные равновесные
характеристики молекул ДНК в ог-
ромном интервале молекулярных масс.
Молекулярная масса таких фрагментов
вычисляется из длины цепи и не требу-
ет применения физических методов для
ее определения.
Б ' представлены вре-
мена вращательной корреляции t,Ow
В-формы ДНК, начиная с короткого
фрагмента ДНК (8 пар нуклеотидов)
и заканчивая нативной ДНК плодовой
мушки Drosophila Melanogaster (около
120 миллионов пар нуклеотидов). Ко-
эффициенты вращательной диффу-
зии коротких фрагментов синтетичес-
кой ДНК (8, 12, 20 пар нуклеотидов)
измерены методом деполяризованной
компоненты в динамическом рассея-
нии света (Глава ПО). Фрагменты
ДНК в средней области значений мо-
лекулярных масс (64-5243 нар нук-
10° 101 ю2 ю3 ю4 ю5 ю6 ю7 ю8 ю9
Число пар оснований
Рис. Гб. 12. Зависимость времени вращательной релаксации от молекулярной массы ДНК (выра-
женной в парах оснований) в двойной логарифмической шкале для широкого интервала молеку-
лярных масс, начиная от коротких фрагментов (8 пар оснований) и заканчивая нативной! ДНК из
плодовой мушки Drosophila Melanogaster {около 120 миллионов пар оснований). Все измерения
выполнены в 1-2 мМ NaCl, pH 6-7. Времена релаксации приведены к вязкости воды при 20 °C.
Данные о препаратах ДНК, имеющих неопределенность молекулярной массе, в данную зависи-
мость не включены. Кривая спада ДЛП для каждого из фрагментов, имеющих длину более 200
пар оснований, содержит не одну, а две компоненты, быструю и медленную. Здесь во внимание
принята только медленная компонента. Вставка. Демонстрация вклада концевых эффектов во
вращательное трение для олигонуклеотидов ДНК.
кюлица Гб.2. Времена релаксации молекул ДНК: от олигонуклеотида
(8 пар оснований) до нативной из Drosophila melongaster
(119 710 000 пар оснований)
Число пар оснований Время релаксации Источник ДНК°)
8 3,2 нсек S
12 6,4 нсек S
Тридекамер (13) 1,9 нсек S
20 16,2 нсек S
64 294 нсек R
80 485 нсек R
89 703 нсек R
104 0,97 мксек R
124 1,34 мксек R
145 2,18 мксек R
188 3,44 мксек R
192 3,8 мксек R
213 4,8 мксек R
213 5,01 мксек R
213 5,01 мксек R
234 5,50 мксек R
234 6,2 мксек R
267 8,0 мксек R
267 7,15 мксек R
267 7,15 мксек R
410 20,6 мксек R
434 22,0 мксек R
520 33,8 мксек R
587 42,0 мксек R
587 42,5 мксек R
659 57,0 мксек R
782 90,0 мксек R
910 110 мксек R
1314 280 мксек R
4361 1,28 мсек R
5243 2,20 мсек R
39937 32 мсек Т7
168903 450 мсек Т4
168903 520 мсек Т4
4266814 40 сек В. subtilus.
4639221 65 сек Е. coli
119710000 6012 сек Drosophila melongaster
“) Источник ДНК: S — синтетические фрагменты; R — фрагменты, полученные с помощью
рестриктаз; Т7, Т4 — ДНК, выделенные из Т7 и Т4 бактериофагов, соответственно
леотидов) получены с помощью рест-
риктаз и их корелляционные времена
измерены методом электрического
ДЛП. Времена вращательной корре-
ляции нативных ДНК, выделенных из
различных источников, вычислены из
данных по упруговязкой релаксации
(см. Главу Г9).
На рисунке 1’6.12 показана зави-
симость вращательных корреляци-
онных времен т,0 w от молекулярной
массы ДНК в двойной логарифми-
ческой шкале, на которой отражают-
ся две характерные черты поведения
ДНК в растворе. Первая — для ДНК
размером 20-200 пар оснований. Эта
зависимость описывается прямой ли-
нией! с наклоном, равным 2,47, кото-
рый типичен для гомологичной серии
палочкообразных частиц конечного
диаметра (секция Г2.6). Для случая
очень тонкой палочки (d/L «1) на-
клон прямой равен 3 (см. уравнение
Г2.60). Вторая состоит в том, что в
районе 180-200 пар оснований начи-
нается отклонение в поведении ДНК
от палочкообразного. Область значе-
ний 200-8000 пар оснований являет-
ся переходной областью от палочки к
гауссовому клубку. В области 8-4700
килопар оснований зависимость вре-
мени вращательной корреляции моле-
кулы ДНК от ее молекулярной массы
представляется прямой линией с на-
клоном 1,47, который является типич-
ным для объемных гауссовых клубков
(секция Г2.6).
В заключение остановимся на корот-
ких фрагментах ДНК. Очевиден суще-
ственный вклад концевых эффектов во
вращательное трение для олигонуклео-
тидов (8 и 12 пар оснований) (рис. Гб. 12.
вставка). Шпилько-образная молекула,
состоящая из 13 пар оснований (триде-
камер), вращается быстрее, чем ДНК-
дуплекс, имеющий 8 пар оснований,
благодаря его компактной форме, близ-
кой к сферической (см. Гб.2). Эти
данные демонстрируют торжество тео-
рии описания гидродинамического по-
ведения коротких круговых цилиндров,
для которых электрическое двойное
лучепреломление выявляет заметный
вклад концевых эффектов во враща-
тельное трение.
Рисунок Гб. 13 демонстрирует мо-
лекулярную структуру ДНК в А-фор-
ме. Она имеет 11 остатков иа виток;
ее диаметр на несколько ангстрем
больше, чем диаметр ДНК в В-форме.
К сожалению, в литературе экспери-
ментальные данные по исследованию
ДНК в Л-формс методом электриче-
ского ДДII ограничены всего несколь-
кими фрагментами, поэтому на рис.
Гб. И показана зависимость времени
вращательной корреляции т.,0 w от мо-
лекулярной массы (в нарах основа-
ний) только для четырех фрагментов
ДНК длиной 373, 460, 676 и 917 пар
нуклеотидов. И хотя угол наклона
прямой линии (2,51) для ДНК в А-
форме близок к таковому для ДНК в
В-форме (2,47), что, как мы указывали,
типично для палочкообразных частиц,
однако имеется одно существенное
различие. В то время как зависимость
времени релаксации от молекуляр-
ной массы для ДНК в В-форме начи-
нает отклоняться от прямой линии в
районе 180-200 пар нуклеотидов (см.
рис. Гб. 12), аналогичная зависимость
для ДНК в A-форме продолжает ос-
таваться прямой линией вплоть до
последней измеренной точки (917 пар
нуклеотидов) (см. рис. Гб. 13). Таким
образом, данные по электрическому
ДЛП подтверждают, что ДНК в А-
форме более жесткая, чем ДНК в В-
форме. Действительно, персистентная
длина ДНК в A-форме равна около
1500 А, что в три раза больше, чем та-
ковая для ДНК в В-форме.
Обсужденные в предыдущих па-
раграфах экспериментальные данные
относятся к равновесной или стати-
ческой гибкости ДНК. Ни одно из этих
измерений не является динамическим
в том смысле, что оно выявляет зави-
симость гибкости ДНК от времени.
Динамическая гибкость может быть
получена из начального спада кривой
в экспериментах по электрическому
ДЛП. В таких экспериментах фраг-
менты ДНК подвергаются действию
короткого импульса очень сильного
электрического поля, до 60 кВ-см
с тем чтобы максимально ориентиро-
вать молекулы вдоль поля. Выходные
данные анализируются по данным экс-
поненциального спада, представляемо-
го в виде двух экспонент:
Л(Г) = Л,ехр(-60Г)+ /ГС9с;ч
(Г6.25)
+Агехр(-£/т),
где О — коэффициент, который описы-
вает обобщенную вращательную диффу-
зию, ат — время, которое связано с дина-
мической жесткостью на изгиб. В слабых
электрических полях доминирует первая
экспонента (А,/А2» 1,0), тогда как с
повышением мощности поля возрастает
относительный вклад амплитуды быс-
трой компоненты (А2/А>> 1,0), кото-
рая затем выходит на плато. Увеличение
вклада относительной амплитуды быс-
трой компоненты с ростом электричес-
кого поля ассоциируется с проявлением
динамических свойств ДНК, а именно,
изгибанием спирали в направлении,
Рис. Гб.13. Молекулярная структура А-фор-
мы ДНК: 11 пар оснований на виток спирали;
шаг спирали равен 2,55 А
перпендикулярном полю. Оценки по-
казывают, что динамическая жесткость
молекулы на изгиб в четыре раза боль-
Рис. 16.11. Зависимость времени вращательной корреляции т2(ь. от молекулярной массы
(в парах оснований) для ДНК в A-форме в двойной логарифмической шкале. Наклон прямой
линии равен 2,51
Рис. Гб. 15. Вид сбоку и в разрезе простран-
ственной модели двойной спирали ДНК (сле-
ва) и РНК (справа); сахар рибозы выделен
желтым цветом, дезоксирибоза — зеленым.
Нарисованная ленточка (сиреневая) проходит
через атомы фосфора (Р) сахаро-фосфатного
остова. Атомы оснований, экспонированью в
основной желоб спирали, обозначены серым
цветом; другие атомы в нуклеотидах — белым
цветом. А-, В-спирали содержат по 20 пар ос-
нований)
ше кажущейся статической жесткости,
выведенной из измерений равновесной
персистентной длины для тех же фраг-
ментов той же длины.
Гб.6.2. Гибкость двуспиральных
РНК
2'-гидроксильная группа РНК стаби-
лизирует двойную спираль в А-форме,
которая отличается от канонической В-
формы спирали тем, что имеет меньшее
число пар на виток и смещенные от оси
спирали пары оснований (рис. Гб. 15).
Времена вращательной корреляции
т20 w двуспиральной РНК длиной от 97
до 317 пар оснований представлены в
i;i(>.пик- !(>..!, а зависимость времени
вращательной корреляции т,0 w от моле-
кулярной массы в двойной логарифми-
ческой шкале на рисунке Гб. 16.
Анализ спада кривой ДЛП дает
персистентную длину спирали РНК,
равную 720 ± 70 А, допуская эффек-
тивный гидродинамический диаметр в
26 А и шаг спирали в 2,7 А (в присутс-
твии ионов Mg2+). В отличие от ДНК
Рис. Гб. 16. Зависимость времени вращательной корреляции т20 w от молекулярной массы,
выраженной в парах оснований, для 13 фрагментов РНК в двойной логарифмической шкале
(Kebbekus et al., 1995)
r.r jjiiiii.i Гб.'!. Времена релаксации
молекул РНК
Число пар оснований Время релаксации оснований т20 w (в мксек)
92 0,44
109 0,62
116 0,69
119 0,75
132 1,00
136 1,08
180 1,90
186 2,02
203 2,48
226 3,46
243 4,03
288 6,63
317 7,31
Данные взяты из Kebbekus et al. (1995)
в В-форме, зависимость времени вра-
щательной корреляции от молекуляр-
ной массы в логарифмической шкале
для РНК в A-форме может быть опи-
сана прямой линией для всего интер-
вала значений молекулярных масс с
наклоном прямой, равным 2,47, кото-
рый типичен для гомологичной серии
палочко-образных частиц конечного
диаметра (секция Г2.6). Из сказанно-
го выше можно сделать заключение о
том, что РНК в A-форме является бо-
лее жесткой молекулой, чем ДНК в В-
форме, но менее жесткой, чем ДНК в
А-форме.
Гб.6.3. Конформации неспиральных
элементов в РНК
Время спада ДЛП изучаемых молекул
обычно интерпретируется в понятиях
гидродинамических моделей, пара-
метры которых подбираются таким
образом, чтобы расчетные времена
вращательной диффузии совпали с
экспериментальными. Такая интер-
претация всегда содержит долю неоп-
ределенности, поскольку несколько
моделей с разными параметрами мо-
гут соответствовать одному измерен-
ному времени вращательной диффу-
зии (см. секцию Г2.3). В начале 2000-х
годов для исследования конформа-
ционных свойств однонитевой моле-
кулы РНК в растворе методом ДЛП
был предложен новый подход, назван-
ный т-отношением. На рисунке Гб. 17
показан принцип этого подхода. При
изучении роли неспиральных элемен-
тов (например, выступов) конструи-
руются полностью дуплексные моле-
кулы и гетеродуплексы, содержащие
выступ (рис. Гб. 17 а). Оба конструкта
должны совпадать по длине ( комлии-
iiipiiii I (>.(>). Затем из профиля спада
ДЛП для двух конструктов определя-
ются времена релаксации, отношение
Комментарий Гб.6. Выбор длины
РНК в методе «т-отношение»
Выбор длины спирали РНК зависит от
нескольких основных факторов. Во-пер-
вых, спираль должна быть достаточно ко-
роткой, чтобы можно было четко выявить
структуру центрального изгиба и аппрок-
симировать спад кривой одной экспонен-
той. Таким условиям соответствует РНК,
по своей длине приблизительно равная
одной персистентной длине (200-250 пар
оснований). Во-вторых, потенциальные
места изгибов должны располагаться в та-
ком месте спирали, чтобы обеспечить за-
метные различия в их конформационных
свойствах. Анализ показывает, что длина
одного плеча спирали должна быть не ме-
нее 70 пар оснований при суммарной длине
полного дуплекса в 250-300 пар оснований
(Hagerman, 2000).
Рис. Гб. 17: а — демонстрация основного принципа в исследовании конформационных свойств
ДНК и РНК методом т-отношения. Конечное время спада определяется для гетеродуплекса, со-
держащего выступ, (т ), и контрольного дуплекса, не содержащего выступ (ты„„). Каждый экс-
периментальный профиль для гетеродуплекса состоит из двух экспоненциальных компонент —
медленной амс1л и быстрой а,ы ; б — экспериментальное значение отношения двух времен спада
т-отношение s т /тк(Шт) сравнивается с теоретической зависимостью этого же отношения от угла
между плечами модели (Hagerman, 2000)
которых дает количественную оценку
угла между изгибами (рис. Гб. 17 б).
Применение этого подхода для ха-
рактеристики неспиральных элементов
(например, выступов, внутренних пе-
тель, и т. п.), обычно присутствующих
в РНК, показало, что выступы, содер-
жащие как адениновые, так и урацило-
вые остатки, монотонно увеличивают
углы изгибов от 10-15° для единичных
остатков в присутствии ионов Mg2+ до
80-90° для шести остатков в отсутствии
этих же ионов. При добавлении ионов
Mg2', вставка шести урацилов, U6, вызы-
вает примерно двухкратное уменьшение
угла изгиба, в то время как углы для шес-
ти аденинов, А6, остаются неизменными.
Это наблюдение представляет элементар-
ный пример структурной перестройки в
РНК, специфичность которой связана
как с последовательностью оснований,
так и с валентностью катионов.
Гб.7. Перечень ключевых идей
* Если молекула асимметрична, то она может рассматриваться как диполь, и тог-
да говорят, что она обладает постоянным дипольным моментом.
Если молекула не имеет постоянного дипольного момента, но может приоб-
ретать его в электрическом поле, тогда говорят, что она обладает наведенным
дипольным моментом', последний существует, пока существует электрическое
поле.
Величина наведенного дипольного момента пропорциональна интенсивности
поля; константа пропорциональности называется поляризуемостью.
Благодаря своей дипольной природе, биологические макромолекулы в раство-
ре ориентируются под действием электрического поля, проявляя эффект двой-
ного лучепреломления.
Закон Керра гласит, что величина двойного лучепреломления пропорциональ-
на квадрату интенсивности электрического поля для случая малых интенсив-
ностей полей; константа пропорциональности называется константой Керра.
Оба метода, ДЛП и дихроизм, дают одну и ту же информацию о структуре мак-
ромолекул; однако на практике метод ДЛП в большей степени подходит для
исследования формы макромолекул.
Скорость спада кривой двойного лучепреломления после прекращения электри-
ческого поля обеспечивает прямое измерение времени релаксации макромолекул.
Для жестких тел произвольной формы временная зависимость ДЛП описы-
вается мультиэкспоненциалыюй функцией, которая может содержать до пяти
компонент; на практике из кривой можно извлечь не более двух экспонент.
Время релаксации сферической макромолекулы пропорционально ее объему.
Для жесткой палочкообразной частицы вре,мя релаксации пропорционально ее
длине; вычисление длины не требует точного знания молекулярной массы.
Статическая персистентная длина ДНК в В-форме в буфере с повышенной
концентрацией соли равна 500 А; аналогичное значение для ДНК в А-форме
равно 1500 А.
Молекулы ДНК, имеющие молекулярную массу от 8 пар оснований (олигонук-
леотиды) до 120 миллионов пар оснований {Drosophila melanogaster), образуют
уникальный гомологичный ряд, время вращательной корреляции в котором на-
ходится в пределах от нескольких наносекунд до тысяч секунд (12 порядков).
В области небольших значений молекулярных масс (8-200 пар оснований)
поведение ДНК подобно поведению жесткой палочки, а в области больших
молекулярных масс (более 8 килопар оснований) подобно поведению гибкого
гауссова клубка.
Литература для дальнейшего чтения
Исторический обзор и введение в биологическую проблему
Fredericq Е. and Houssier С. Electric Dichroism and Electric Birefringence. Oxford:
Clarendon Press, 1973.
Hagerman P. J. Sometimes a great motion: application of transient electric
birefringence to study of macromolecular structure. Curr. Opin. Struct. Biol., 6, 643-
649, 1996.
Макромолекулы в электрическом поле
Belmonte Р. A., Martinez С. L. and Garcia de La TorreJ. Time course of the orientation
and birefringence of axially symmetric macromolecules upon reversal of an electric field
of arbitrary strength. J. Phys. Chem., 1991. 95, 5661-5664.
Основы теории
Charney E. Electric linear dichroism and birefringence of biological polyelectrolites.
Quart. Rev. Biophys., 1988. 21, 1-60.
Экспериментальные измерения электрического ДЛП
Hagerman Р. J. Application of transient electric birefringence to the study of
biopolymer structure, Methods in Enzymol, 1985.117, 199-215.
Глобальная структура и гибкость белков
Kooijman М., Bloemendal М., Van Amerongen Н., Traub Р. and Van Grondel-
le R. Characterization of multiple oligomeric vimentin intermediate filament units by
transient electric birefringence measurements. J. Mol. Biol., 1994. 236, 1241-1249.
Глобальная структура и гибкость нуклеиновых кислот
Klotz L. С. and Zimm В. Н. Size of DNA determined by viscoelastic measurements:
results on bacteriophage, Bacilus subtilus and Escherichia coli, 1972. 72, 779-800.
Hagerman P.J. and Zimm В. H. Monte Carlo approach to the analysis of the rotational
diffusion of wormlike chains. Biopolymers, 1981.20, 1481-1502.
Diekmann S., Hillen W., Morgeneyer Wells R. D. and Porschke D. Orientation
relaxation of DNA restriction fragments and the internal mobility of the double helix,
Biophys. Chem., 1982.15, 263-270.
Pecora R. DNA: a model compound for solution studies of macromolecules, Science,
1991.251,893-898.
Kebbekus P., DraperD. E. and Hagerman P.J. Persistence length of RNA. Biochemistry,
1995. 34, 4354-4357.
Hagerman P.J. Transient electric birefringence for determining global conformations
of nonhelix elements and protein-induced bends in RNA. Meth. Enzymol., 2000. 317,
440-453.
Lu Y., Weers B., Stellwagen N. C. DNA persistence length revisited. Biopolymers,
2001-2002.61,261-275.
Hagerman P. J. Investigation of the flexibility of DNA using transient electric
birefringence. Biopolymers, 1981. 20, 1503-1535.
Stellwagen N. C. Electric birefringence of kilobase-sized DNA molecules. Biophys.
Chem., 1996. 58, 117-124.
Глава Г7
ОРИЕНТАЦИЯ МАКРОМОЛЕКУЛ
ГИДРОДИНАМИЧЕСКИМ ПОЛЕМ
Г7.1. Исторический обзор
1870
Дж. Клерк Максвелл (J. Clerk
Maxwell) впервые описал эффект
двойного лучепреломления (ДЛП) в
потоке, используя в качестве объек-
та исследования канадский бальзам.
ДЛП возникало при помещении ка-
надского бальзама между двумя кон-
центрическими цилиндрами, один из
которых вращался, а второй был за-
фиксирован.
1932
П. Бедер (Р. Boeder) предста-
вил первую теории ДЛП в потоке. Он
сформулировал основную проблему
теории — нахождение связи между уг-
лом ориентации и величиной двойного
лучепреломления с размерами молекул
коллоидальных частиц. Между 1938
и 1941 г., А. Петерлин и Г. А. Стюарт
(A. Peterlrin and Н. A. Stuart), а так-
же О. Снельман и Е. Бьйорншталь
(О. Snellman and Y. Bjomstahl) ввели
в рассмотрение важный для описания
процессов ДЛП в потоке универсаль-
ный параметр — отношение градиента
скорости к коэффициенту вращатель-
ной диффузии.
1950-е
Г. А. Шерага, Дж. Т. Эдсалл и
Дж. О. Гадд (Н. A. Sheraga, J. Т. Edsall
and J. О. Gadd) дали количественную
оценку угла ориентации и величины
двойного лучепреломления как функ-
ции отношения градиента скорости к
коэффициенту вращательной диффузии
в широком интервале значений градиен-
та скорости. Это открыло возможность
вычисления молекулярных размеров эл-
липсоидальных частиц методом ДЛП.
Конец 19(>0-х
Г. Б. Тарстон (G. В. Thurston) пред-
ложил способ измерения двойного лу-
чепреломления в гидродинамическом ос-
цилляторном поле. В то же самое время
Н. Дэвидсон (N. Davidson) разработал
метод определения времен релаксации из
измерений спада дихроизма в потоке.
1970-е
Р. Е. Харрингтон и В. Н. Цветков
(R. Е. Harrington and V. N. Tsvetkov)
разработали приборы и оборудование дтя
измерения величин ДЛП и углов ори-
ентации в растворах высокомолекуляр-
ных полимеров при низких градиентах
скорости. Это позволили изучать сильно
разбавленные растворы ДНК. В. Кун,
Г. Кун Р. Серф, В. Цветков (W. Kun,
H. Kun, R. Serf, V. N. Tsvetkov) внесли
существенный вклад в теорию и практику
ДЛП-деформируемых макромолекул, и, в
первую очередь, линейных полимеров.
1980 2000
В начале 1980-х метод ДЛП в потоке
начал терять популярность среди биофи-
зиков. Причин для этого было две. Первая
была связана с появлением метода элек-
трического двойного лучепреломления,
который требовал гораздо меньших ко-
личеств исследуемого материала. Вторая
связана с принципиальными ограниче-
ниями самого метода ДЛП, поскольку с
его помощью можно исследовать только
вытянутые частицы, такие как палочко-
образные вирусы, молекулы ДНК и РНК.
Сегодня, однако, интерес к ДЛП в потоке
вновь возрос в связи с открывшимися воз-
можностями исследования конформаци-
онной динамики индивидуальных боль-
ших молекул ДНК в реальном режиме
времени. За такой динамикой можно вести
непосредственное наблюдение с использо-
ванием флуоресцентного микроскопа.
Г7.2. Введение
в биологические проблемы
Двойное лучепреломление наблюдается
в жидкости, содержащей асимметрич-
ные частицы, когда в жидкости устанав-
ливается градиент скорости. Градиент
скорости проще всего достигается, когда
жидкость помещается между двумя кон-
центрическими цилиндрами при враще-
нии одного и фиксированном положении
другого. Если приложить внешнее поле
к изначально изотропному раствору, то
он становится двулучепреломляющим.
В случае гидродинамического поля двой-
ное лучепреломление образуется в ре-
зультате действия градиента скорости,
которому подвергается система. Если
растворенные макромолекулы являют-
ся геометрически, и, следовательно, оп-
тически асимметричными, то двойное
лучепреломление устанавливается как
результат равновесия между эффектами
ориентации, происходящими благодаря
приложенному нолю, и беспорядочной
разориентации под действием броунов-
ского движения. Однако даже для сфери-
ческих частиц, которые в отсутствие поля
оптически изотропны, оптическая ани-
зотропия может возникать, как результат
деформации молекулы вследствие при-
ложенного градиента скорости. Следова-
тельно. ДЛП в потоке является методом
получения информации о геометриче-
ских, оптических свойствах и гибкости
биологических макромолекул в растворе.
К сожалению, область применения ме-
тода ДЛП при исследовании биологиче-
ских макромолекул относительно узка,
несмотря на его очевидные потенциаль-
ные возможности. Так, его практически
невозможно применять к глобулярным
белкам, поскольку скорость их враще-
ния очень велика, что делает ориентацию
этих молекул гидродинамическим полем
очень затруднительной. Наиболее ин-
тересные результаты получены для рас-
творов больших гибких макромолекул,
особенно для ДНК и РНК, причем в по-
следнее время особое внимание уделяет-
ся динамическому поведению единичных
флуоресцентно меченых молекул ДНК,
за которыми можно непосредственно на-
блюдать в флуоресцентном микроскопе.
Г7.3. Стационарное
динамическое двойное
лучепреломление
Стационарная ориентация в потоке
наиболее легко достигается при поме-
щении раствора, содержащего суспен-
дированные асимметричные частицы, в
зазор между двумя концентрическими
цилиндрами, один из которых вращает-
ся, а другой зафиксирован (рис. 17.1).
Когда оба цилиндра находятся в состо-
янии покоя, как показано на рис. 17.1 а,
частицы и имеют произвольную ори-
ентацию. Когда один из цилиндров на-
чинает вращаться, в жидкости создает-
ся ламинарный поток и, как следствие
этого, градиент скорости. В результате
этого частицы ориентируются, как схе-
матически показано на рис. 17.1 б. Поле
между цилиндрами можно наблюдать
при прохождении света, направленного
параллельно оси цилиндра, через скре-
щенные две призмы, представляющие
собой поляризатор и анализатор. Допус-
тим, что у пас есть набор молекул, ори-
ентированных таким образом, что их ось
создает угол 0 с осью у (рис. 17.2). Па-
дающий на молекулы поляризованный
свет с электрическим вектором, колеб-
лющимся в направлении //-оси, расщеп-
ляется на компоненты Е, и Ег посколь-
ку показатель преломления в этих двух
направлениях различен. В этом случае
мы говорим, что ориентированные мо-
Рис. 17.1. Частицы в жидкости располагают-
ся между двумя концентрическими цилиндра-
ми: а — схематическое представление частиц
в виде коротких отрезков, показывающих их
оптические оси в состоянии покоя цилиндров;
б — ориентация частиц, вызываемая вращени-
ем внешнего цилиндра
лекулы обладают свойством двулучеп-
реломления. Если обозначить действу-
ющее электрическое поле как £о, то тогда
Е, = £о cos0 и Е± = £ sin 0. Свет, который
прошел через ориентированные молеку-
лы, теперь должен пройти через анали-
затор, который располагается таким об-
разом, чтобы пропускать все колебания,
параллельные г-оси. Если 0 равно нулю
или 90°, то падающий поляризованный
свет не будет расщепляться на компо-
ненты. В этом случае падающее на ана-
лизатор поле будет параллельно оси у,
Рис. 17.2. Относительная ориентация поляризатора, молекулярных осей и анализатора. Поля-
ризованный свет, падающий на молекулу, может быть разложен на компоненты Е and Е±.
Частицы, ориентированные
под углом х к потоку
Рис. Г7.3. Схематическая иллюстрация пото-
ка, когда все частицы имеют один и тот же угол
X по отношению к линиям потока. На диаграм-
ме показано пересечение изоклинов, которые
показывают четыре положения, где оптиче-
ские оси частиц являются строго параллель-
ными к плоскости поляризатора и анализато-
ра (Tenford, 1965)
дается визуально при помощи оптиче-
ского компенсаторного оборудования
(см. выше). К сожалению, приемлемый
уровень точности не может быть достиг-
нут с помощью такого оборудования,
если величина лучепреломления мень-
ше, чем 10"8. Наилучшпм способом ре-
шения проблемы чувствительности из-
мерений ДЛП является использование
фотоэлектрических детекторов. С их по-
мощью можно определять ДЛП макро-
молекул с очень высокой молекулярной
массой и низкой концентрацией, с гра-
диентом скорости меньше чем 0,01 сек-1,
когда эффект Максвелла мал. Как и в
случае метода Керра коммерческие при-
боры, предназначенные для изучения
макромолекул методом Максвелла, на
сегодняшний день отсутствуют.
и свет не будет проходить. Для всех дру-
гих углов ориентации часть света будет
пропускаться анализатором. В результа-
те действия внешнего поля получаются
четыре темные области, в форме креста,
с перемежающимися светлыми областя-
ми (рис. Г7.3). В темных областях плос-
кость колебаний света, передаваемая как
поляризатором, так и анализатором, па-
раллельна оптической оси раствора.
Одним из ограничений метода ДЛП
при исследовании гидродинамических
свойств и оптического поведения мак-
ромолекул в растворе является его
невысокая чувствительность при из-
мерении очень низких значений вели-
чина ДЛП и углов экстинкции около
45° (смотри ниже), когда распределение
макромолекул по ориентации относи-
тельно широко.
В стандартном методе для измере-
ний ДЛП макромолекул используется
оборудование, в котором поток раствора
рассматривается через скрещенные по-
ляризаторы, а лучепреломление наблю-
Г7.3.1. Угол ориентации
и коэффициент вращательного
трения
В эксперименте по ДЛП жидкий слой
просматривается в направлении обра-
зующей цилиндра. При этом ламинар-
ный поток, возникающий в зазоре, мож-
но считать однородным с постоянным
градиентом скорости в направлении ра-
диуса (рис. Г7.4). Под действием напря-
жения сдвига в потоке жидкость ста-
новится оптически анизотропной. При
этом для светового пучка, нормального
направлениям градиента скорости G и
скорости потока и, т. е. параллельного
оси цилиндра, свойства анизотропного
слоя оказываются подобными оптичес-
ким свойствам пластинки кристалла,
главное сечение которого образует не-
который угол х с направлением потока
и. Этот угол называется углом ориента-
ции. Угол х — одна из двух основных
величин, измеряемых на опыте. Второй
величиной является разность двух глав-
ных показателей преломления Ди = пх -
п2, жидкости, из которых первый (пх)
соответствует лучу, электрический век-
тор которого параллелен главному се-
чению анизотропного слоя, второй (и2)
перпендикулярен к главному сечению
(оси) слоя. Величина Ди характеризу-
ет, в основном, оптические свойства
частиц, тогда как угол у имеет непосред-
ственное отношение к гидродинамиче-
ским свойствам частиц. В этой главе мы
сосредоточимся на угле у, поскольку,
как будет показано ниже, он связан не-
посредственно с коэффициентом вра-
щательного трения частицы.
Рассмотрим в качестве простого при-
мера случай вращения жестких палоч-
кообразных частиц, ограничившись для
простоты двумерным пространством
(рис. 17.5). Такая частица оказывается
под действием двух сил, одна из кото-
рых ориентирующей природы (градиент
скорости), а вторая — дезориентирую-
щей природы (броуновское движение).
Гидродинамические силы вращают час-
тицу по часовой стрелке, и очевидно,
что вращающий момент будет макси-
мальным при угле 0 = 90° по отношению
к линиям потока, и минимальным при
0 = 0°. Молекулы будут двигаться более
быстро в районе первого угла, и более
медленно в районе второго угла, созда-
вая преимущественную ориентацию
частиц по углам. Этой ориентации про-
тиводействует броуновское движение,
которое способствует случайному рас-
пределению частиц. Равновесная ориен-
тация достигается при балансе этих двух
сил. Она характеризуется параметром
а = G/Q, где G — градиент скорости, а
0 — коэффициент вращательной диффу-
зии. Когда а « 1, наиболее вероятный
угол ориентации равен 45°, при котором
гидродинамические и броуновские силы
равны по величине и противоположны
Рис. I 7/1. Поле градиента скорости в жидком
слое, заключенном между стенками непод-
вижного (/) и вращающегося (2) цилиндров.
u-скорость потока, G — направление градиен-
та скорости, х — угол ориентации
Рис. Г7.5. Силы, действующие на жесткую
палочкообразную частицу в градиенте скоро-
сти. Тангенциальная компонента градиента
скорости показана, как силы броуновского
движения показаны как---->
по знаку. Наименее вероятный угол —
135°, в котором силы складываются (см.
рис. Г7.5). В другом случае, когда а» 1,
гидродинамические силы существенно
подавляют броуновское движение, и все
оси располагаются вблизи угла ориента-
ции, равного нулю. Наименее вероятный
угол будет при 90°.
Зависимость угла экстинкции у от
а для различных значений отношения
осей р эллипсоида вращения показана на
1’iic. Г7.(>. Угол ориентации» % как функция
параметра а для различных значений отноше-
ния осей, р, эллипсоида вращения (Sheraga et
al., 1951)
Комментарии Г7.1. «Внутреннее»
двойное лучепреломление
и двойное лучепреломление «формы»
Большинство макромолекул имеет пока-
затель преломления п, который отличается
от такового обычных водных растворов. .
Поэтому, когда биологические макромо-
лекулы в растворе ориентируются в гради-
енте скорости, раствор становится двулуче-
преломляющим. Иными словами, он имеет
различные показатели преломления, па-
раллельно и перпендикулярно линиям по-
тока. Это явление называется двойным лу-
чепреломлением «формы», и возникает оно '
в растворах несферических макромолекул, 1
показатель преломления которых отлича- I
ется от такового у растворителя. Однако, I
даже если показатель преломления частиц i
равен таковому у растворителя, двойное
лучепреломление может возникать, ког-
да молекулы сами по себе анизотропны и ,
имеют различные показатели преломле-
ния, параллельные и перпендикулярные к '
их длинной оси. Это явление называется 1
«внутренним» двойным лучепреломлени- |
ем. Читатель может найти количественные |
характеристики эффектов внутреннего и
формы двойного лучепреломления в учеб-
никах (см. например, Цветков и др., 1971).
рисунке Г7.6. Из рисунка видно, что для
жестких частиц % приближается к 45°,
когда ориентирующая сила, G, стремит-
ся к нулю. Важно также то, что наклон
прямой при малых значениях параметра
а не зависит от формы частицы.
При малых значениях а угол экс-
тинкции вычисляется как:
y = 45°-—- + 4-) +••• (Г7.1)
л 120 \®у
где k — коэффициент, зависящий от фор-
мы макромолекул. Из уравнения Г7.1
следует, что зависимость между % и G
должна быть линейной для малых значе-
ний G, с углом наклона, равным -1/120.
Таким образом, коэффициент враща-
тельной диффузии может быть опреде-
лен непосредственно из измерений при
низких градиентах скорости. Однако
именно в этом случае проявляется один
из главных недостатков метода — низкая
чувствительность при измерениях, близ-
ких к 45° и очень низких градиентов.
Г7.4. Спад дихроизма
в потоке
Если молекулы обладают внутренней
оптической анизотропией (коммсн ia-
рпй Г7.1), то ориентацию таких моле-
кул можно рассматривать как дихроизм
в потоке. Если поток внезапно остано-
вить, то время релаксации, т. е. время, за
которое система возвращается к своему
равновесному распределению, может
быть определено. В этих условиях пове-
дение макромолекул описывается как:
^ = exp(-60t). (Г7.2)
где Д/1(0) — есть поглощение в нулевой
момент времени; ДЛ(7) — есть его изме-
нения во времени. Типичный ход кри-
вой для этого процесса представлен на
рисунке 1'7.7. Отметим, что уравнение
Г7.2 аналогично уравнению Гб. 12.
Г7.5. Осцилляторное двойное
лучепреломление в потоке
Осцилляторное ДЛП в потоке наблю-
дается, когда круговой поляризованный
свет пропускается через узкий гидроди-
намический зазор между осциллиру-
ющей плоской поверхностью и жестко
закрепленной поверхностью. Затем
определяется величина двойного луче-
преломления и его фаза, по отношению
к градиенту скорости в гидродинами-
ческом зазоре.
Зависимость осцилляторного двой-
ного лучепреломления от частоты для
двух образцов ДНК с молекулярными
массами 6 х 103 и 5 х 106 Да приведена
на рисунке Г7.8 а и б. Изданных рисунка
видно, что теоретическая кривая релак-
сации для жесткой частицы хорошо опи-
Рис. 1 7.7. График спада сигнала дихроизма
для молекул ДНК с различной молекулярной
массой в полулогарифмических координатах
(Callis and Davidson, 1969)
сывает поведение молекулы ДНК с мо-
лекулярной массой М = 6 х 105 Да (-900
пар оснований), тогда как для молекулы
Рис. 1 7.8: а — зависимость осцилляторного двойного лучепреломления от частоты приложен-
ного поля для молекул ДНК (М = 6 х 105 Да) в 89% растворе глицерина. Штриховые линии — тео-
ретические кривые релаксации жестких частиц. Частотная шкала, f, для измеряемой частоты,
шкала— эквивалент ожидаемой частоты для тех же самых молекул ДНК в водном растворе:
б — то же для ДНК (М = 5 х 10R Да) в водном растворе (Wilkinson and Thurston. 1976)
Рис. I 7.9. Растяжение и сжатие гибкой мо-
лекулы при вращении в потоке с градиентом
скорости
ДНК с молекулярной массой 5 х 10° Да
(-7500 нар оснований) уже заметно про-
является ее гибкость (см. параграфы
ГЗ.7.4, Г4.10.3, Г6.7.1 и Г9.5.5).
Г7.6. Ориентации
макромолекул в потоке
как динамическое явление
Двойное лучепреломление в потоке яв-
ляется динамическимявлением. Как вид-
но из рисунка Г7.9 макромолекулярная
цепь будет вытягиваться, когда ее концы
находятся в первом и третьем квадран-
тах, и сжиматься, когда она находится во
втором и четвертом квадрантах.
। г Ртутная
IL лампа
Объектив
Регистрирующее
устройство
Линза
в
Рис. I 7.10: а — сдвиг потока как суперпозиция чисто растягивающего потока и чисто враща-
тельного потока; б — схематическое представление гибкой макромолекулы, растягивающейся в
потоке. При растяжении молекулы и ее вращении могут возникнуть различные динамические
сценарии; в — схема измерительного прибора. Однородный градиент скорости создается в гидро-
динамическом зазоре -50 мкм между двумя параллельными пластинами при перемещении одной
стеклянной пластины относительно другой. Флуоресцентно меченые изолированные молекулы
Х-ДНК (48 502 пар оснований), представленные на рисунке в виде темных точек, визуализуются
в микроскопе с помощью видеокамеры (Smith et al., 1999).
На практике такая деформация
может быть наблюдена в иоле сдвига.
Простейшим случаем является сдвиг
между двумя параллельными пласти-
нами, одна из которых перемещается
параллельно другой. Этот сдвиг потока
можно представить как суперпозицию
чисто вращательного и растягивающе-
го потоков (рис. Г7.10 «). В растягива-
ющем потоке можно ожидать деформа-
цию гибкой молекулы, в то время как
в чисто вращательном потоке наблюда-
ется только вращение и деформации не
происходит. Когда вклады компонент,
обеспечивающих вращение, и растя-
жение близки, молекула находится в
нестабильном конформационном со-
стоянии. Конформация молекулы бу-
дет изменяться динамически вслед за
изменением гидродинамического поля
(рис. Г7.10 (5).
На рисунке Г7.11 приведены при-
меры, изображающие поведение
трех индивидуальных изолирован-
ных молекул флуоресцентно меченой
ДНК-бактериофага лямбда (Л.-ДНК)
в стационарном градиенте скорости
с числом Вайсенберга IV. = 19
vt и Lipiiii 1'7.2). Первая молекула ве-
дет себя следующим образом: будучи
клубком, она переходит в растянутое
состояние, возвращается в состояние
клубка и затем вытягивается опять
(рис. Г7.1 1 а). На второй молекуле ви-
ден сгусток, который передвигается
вдоль контура частично растянутой
молекулы (рис. Г7.1 I б). И, наконец,
на рисунке 1'7.11 « показана молеку-
ла, которая вначале имеет Y-образную
форму, затем принимает форму вытя-
нутого каната, и в конце концов опять
принимает Y-образную, но переверну-
тую форму.
Рис. 17.11. Изображение одиночной цени
X — ДНК бактериофага, меченой флуорес-
центным красителем в вязком растворе са-
хара. Молекула Х-ДНК имеет контурную
длину L, равную около 22 мкм и содержит
около 440 персистентных длин. Каждый ряд
представляет собой серию развернутых во
времени изображений слева направо. Вер-
тикальная полоса слева вверху равна 5 мкм
(Smith et al., 1999)
KoMMeHiapiui Г/ 2. Число
Вайсенберга
Число Вайсенберга W. = G/т является
безразмерным параметром, характеризу-
ющим отношение величины приложенного
градиента скорости, G, к скорости релак-
сации макромолекул т. Чем больше число
Щ, тем быстрее флуктуирует молекула по
отношению к ее времени релаксации и до-
I стигает большего растяжения.
Причина столь разного поведения
трех одинаковых молекул не очень ясна.
Предполагается, что такое поведение
отражает «индивидуализм» сложных
биологических макромолекул.
Г7.7. Перечень ключевых идей
• Ориентация макромолекул в гидродинамическом потоке называется двойным
лучепреломлением в потоке или эффектом Максвелла.
• Макромолекула в потоке оказывается под действием двух сил, одна из которых
ориентирующей природы (градиент скорости), а вторая — дезориентирующей
природы (броуновское движение); при равенстве этих сил возникает равновес-
ная ориентация частиц в потоке.
• Равновесная ориентация описывается параметром а = G/0, где G — градиент
скорости, а 0 — коэффициент вращательной диффузии макромолекул.
• Измеряемый на опыте угол ориентации частицы, /, непосредственно связан
с ее коэффициентом вращательного трения.
• Вращательный коэффициент диффузии твердых асимметричных частиц может
быть определен непосредственно в эксперименте при низких значениях гради-
ента скорости или при исследовании релаксации двойного лучепреломления
после резкой остановки потока.
• Вращательные скорости глобулярных белков настолько велики, что их ориен-
тация в потоке практически невозможна.
• Конформационная динамика флуоресцентно меченых индивидуальных боль-
ших молекул ДНК может наблюдаться в реальном масштабе времени непо-
средственно в видеофлуоресцентном микроскопе.
Литература для последующего чтения
Исторический обзор и введение в биологические проблемы
De Gennes Р. G. Molecular individualism, Science, 1997. 276, 1999-1992. Steady-
state flow birefringence.
Sheraga H. A., Edsall J. T. and Gadd J. O. Double refraction of flow: numerical
evaluation of extinction angle and birefringence as a function of velocity gradient.
J. Chem. Phys. 1951.19,1101-1108.
Цветков В. H., Эскин В. Е. и Френкель С. Я. Структура макромолекул в раство-
рах. М.: Наука, 1964.
Harrington R. Е. Measurement of flow birefringence at very low velocity gradients,
Biopolymers, 1970. 9, 141-157.
Спад дихроизма в потоке
Callis Р. R. and Davidson N. Hydrodynamic relaxation times of DNA from decay of
flow dichroism measurements. Biopolymers, 1969. 8, 379-390.
Caimey К. L. and Harrington R. E. Flow birefringence of T7 Phage DNA: dependence
on salt concentration. Biopolymers, 1982. 21,923-924.
Осцилляторное ДЛП
SchragJ. L., GuessJ. F. and Thurston G. B. Shear-wave interference observed by optical
birefringence induced in a viscoelastic liquid. J. Appl. Phys., 1965. 36, 1996-2000.
Wilkinson S. R. and Thurston G. B. The optical birefringence of DNA solutions induced
by oscillatory electric and hydrodynamic fields. Biopolymers, 1976.15, 1555-1572.
Ориентация макромолекул в потоке как динамическое явление
Smith D. Е., Babcock Н. Р. and Chu S. Single-polymer dynamics in steady shear flow,
Science, 1999. 283, 1724-1727.
Глава Г8
ДЕПОЛЯРИЗОВАННАЯ ФЛУОРЕСЦЕНЦИЯ
Г8.1. Исторический обзор
1926
Е. Гавиола (Е. Gaviola) создал пер-
вый прибор для измерения флуоресцен-
ции, который назвал флуориметром.
1933 1935
А. Яблонски (A. Jublonski) дал объ-
яснение длительному послесвечению, на-
блюдаемому в молекуле при некоторых
условиях. Он постулировал, что помимо
обычного синглетного возбужденного
уровня молекулы имеют некоторый мета-
стабильный, т. е. долгоживущий уровень,
лежащий несколько ниже первого возбуж-
денного. Физическая природа этого явле-
ния, иногда неправомерно называемого
фосфоресценцией, была дана А. Н. Тере-
ниным (A. N. Terenin) в 1943 г.
1934
Ф. Перрен (F. Perrin) установил
зависимость между деполяризованной
флуоресценцией и броуновским враща-
тельным движением для сферических мо-
лекул. В 1936г. он распространил эту за-
висимость на асимметричные молекулы.
1952
Г. Вебер (G. Weber) впервые при-
менил теорию Перрена к биологиче-
ским макромолекулам. Он также вы-
полнил ряд измерений поляризации
флуоресценции в стационарных усло-
виях при постоянном освещении для
определения значения среднего гармо-
нического времени вращательной ре-
лаксации. Классический обзор Вебера
о применении поляризованной флуо-
ресценции в исследовании макромоле-
кул, и белков в особенности, появился
в 1953 г.
1961
А. Яблонски предложил исследо-
вать процесс вращательной релаксации
путем измерения временной зависи-
мости спада поляризованной флуорес-
ценции как функцию времени после
действия короткого импульса. Он об-
ратил внимание на то, что полученная
таким способом анизотропия может
быть более точно интерпретирована,
чем ее усредненное значение, наблюда-
емое при постоянном освещении.
1966
П. Валь (Р. Wahl) и, двумя годами
позже, Л. Страйер (L. Stryer), провели
первые экспериментальные измерения
времени спада поляризованной флуо-
ресценции меченых глобулярных бел-
ков.
1969
1. Tao (T. Tao) установил взаи-
мосвязь между зависимой от времени
анизотропией поляризованной флуо-
ресценции и броуновской вращатель-
ной диффузией в макромолекулах.
Он показал, что для тел произвольной
формы временная зависимость спада
анизотропии описывается пятью экс-
понентами. В 1972 г. аналогичные ре-
зультаты были получены двумя други-
ми группами ученых (Г. Г. Белфорд,
Р. Л. Белфорд и Г. Вебер, Т. Дж. Чу-
анг, К. В. Эйзенталь) (G. G. Belford,
R. L. Belford and G. Weber, T. J. Chu-
ang, К. B. Eisenthal).
С средина 1990-х
Несколько групп доложили о синтезе
и спектральных свойствах долгоживущих
металл-лигандных флуоресцирующих
комплексов (Б. А. де Граф и Дж. Н. Дема;
группа Дж. Р. Лаковича (В. A. De Graff
and J. N. Demas, J. R. Lacowicz’s group).
С помощью этих очень устойчивых к вы-
цветанию красителей с микросекундным
временем жизни можно измерять време-
на вращательной корреляции до 2 мксек,
что открыло путь к детектированию вра-
щательного движения больших биологи-
ческих макромолекул.
1980 1995
Дж. Р. Лакович и его группа
(J. R. Lakowicz’s group) внесли суще-
ственный вклад в теорию и практику
деполяризованной флуоресцентной
спектроскопии. В 1987 г. вышел в свет
фундаментальный учебник Дж. Р. Ла-
ковича «Основы флуоресцентной спек-
троскопии».
2000 и no iiaciomnee время
Деполяризованная флуоресцентная
спектроскопия является адекватным
методом для исследования вращатель-
ного движения макромолекул в водных
растворах в наносекундном и микросе-
кундном интервале времен. Она откры-
ла новые возможности для измерения
вращения во внутримолекулярных до-
менах, латеральной диффузии в мемб-
ранах, больших белков и нуклеопроте-
идных комплексов.
Г8.2. Введение
в биологические проблемы
Теоретические основы флуоресцент-
ной спектроскопии были заложены в
первой половине XX в. такими учены-
ми-первооткрывателями, как Е. Гавио-
ла, Жан и Франсуа Перрены (отец и
сын), П. Принсгейм, С. Вавилов, Е. Фёр-
стер. В 1961 г. А. Яблонски предложил
измерять спад поляризованной флуо-
ресценции как функцию времени для
исследования процессов вращатель-
ной релаксации. Однако только с нача-
ла 1980-х стали доступными приборы
для измерения спада флуоресценции
благодаря широкому распространению
лазеров и достижениям в электронике.
Теперь исследователи могли делать
выбор между использованием времен-
ных анализаторов в режиме счета от-
дельных фотонов и многочастотным
фазовым флуориметром, и быть при
этом уверенными в получении одних и
тех же результатов.
Значительный экспериментальный
и теоретический вклад в флуоресцент-
ную спектроскопию, разрешенную во
времени, внесли Дж. Лакович, Д. Мил-
лар, Р. Шерри и их научные группы.
Были предложены два способа кон-
кретной реализации экспериментов.
Первый способ основывался на из-
мерении спада общей интенсивности
флуоресценции вслед за импульсным
возбуждением; такие измерения мог-
ли быть использованы, как сигнал
о каких-либо событиях (изменения
конформации белков, связывание с
лигандом, взаимодействие белков и
нуклеиновых кислот и т. д.). Второй
способ основывался на анализе вели-
чин параметров флуоресценции для
понимания деталей структуры и ди-
намики флуорофоров и их окружения.
Однако наше неполное знание деталей
фотофизики большинства природных
флуорофоров, таких как триптофан и
тирозин, явились причиной трудно-
стей в интерпретации данных по флуо-
ресценции в исследованиях структу-
ры и динамики белков.
За последние годы большое распро-
странение получил метод флуоресцен-
ции, разрешенной во времени, кото-
рый включает в себя измерения спада
анизотропии поляризации, с тем чтобы
характеризовать вращение молеку-
лы целиком или ее отдельных частей.
Если ранее большинство измерений
проводилось на наносекундной шкале
времени, то сегодня можно определять
динамику процессов на микросекунд-
ной шкале. Использование этой шкалы
стало возможным благодаря примене-
нию новых металл-лигандных комп-
лексов, которые имеют время жизни от
10 нсек до 10 мксек.
В этой главе мы сфокусируем наше
внимание на использовании методов
временного спада анизотропии флуо-
ресценции, которые обеспечивают полу-
чение гидродинамических данных, опи-
сывающих вращательную диффузию
биологических макромолекул. В секции
Д3.5 мы обсудим передачу энергии меж-
ду двумя флуорофорами (эффект Фёр-
стера).
Г8.3. Теория
деполяризованной
флуоресценции
Ниже мы кратко обсудим теоретиче-
ские основы флуоресцентной спект-
роскопии с целью понимания того, как
коэффициенты вращательного трения
можно определить из измерений депо-
ляризованной флуоресценции.
Г8.3.1. Флуоресценция
как физическое явление
При возбуждении электрон перехо-
дит на орбитали с высокой энергией.
В большинстве случаев эта энергия за-
тем переходит в тепло. Такой процесс
называется неизлучающим переносом
энергии. Неизлучающий перенос энер-
гии проиллюстрирован на рисунке
Г8.1 а. У некоторых молекул электрон
в возбужденном состоянии может воз-
вращаться на более низкий или исход-
ный энергетический уровень. Благодаря
различным электронным конфигураци-
ям возбужденного и основного состоя-
ний, электрон может легко переходить
на более низкие энергетические уровни
вплоть до перехода на самое нижнее
основное состояние с потерей энергии
на каждом этапе перехода. Такой про-
цесс называется излучающим перено-
сом энергии и схематически показан на
рисунке Г8.1 б. Новое поглощение фо-
тона приведет к переходу электрона в
возбужденное состояние и переносу не-
которого количества энергии в раство-
ритель в виде тепла. Однако наиболее
низкий колебательный уровень возбуж-
денного состояния все равно оказывает-
ся гораздо выше колебательного уровня
основного состояния, при котором энер-
гия полностью высвобождается, если не
произойдет новое поглощение фотона.
Поглощение и эмиссия света при излу-
чающем энергию переходе показана на
диаграмме (рис. Г8.2).
Акт поглощения света — очень
быстрый процесс, занимающий около
10-15 сек. После поглощения света ро-
Рис. Г8.2. Диаграмма поглощения и эмиссии света (диаграмма Яблонского). Основное первое
и второе электронные состояния обозначаются как 50,5, и 5,, соответственно. На каждом из этих
электронных энергетических уровней существует ряд энергетических колебательных подуров-
ней, обозначаемых как 0, 1, 2. и т. д. (Lakowicz, 1999). В русской литературе фосфоресценцию
принято называть метастабильиым состоянием
исходит ряд процессов. Флуорофоры
обычно возбуждаются до высокого ко-
лебательного уровня, соответствующе-
Г8.1. Синглетные
и триплетные состояния
Принцип запрета Паули означает: два
электрона в атоме не могут иметь один и
тот же набор из четырех квантовых чисел.
Это ограничение означает, что не более
двух электронов могут занимать одну ор-
биталь, поэтому эти два электрона долж-
ны иметь спины разных знаков, т. е. быть
спарены. Поскольку спины спарены, то
большинство молекул не проявляет маг-
нитных свойств и является диамагнети-
ками — т. е. они не притягиваются и не
отталкиваются статическим магнитным
полем. Электронное состояние молекул,
при котором все электронные спины спа-
рены, называется синглетным состоянием,
и на этом уровне не происходит диссоци-
ации энергии электрона. Когда один из
пары электронов возбуждается до более :
высокого энергетического уровня, возни-
кает синглетное и триплетное состояния.
В возбужденном синглетном состоянии
спин этого электрона еще остается спарен-
ным с электроном в основном состоянии.
В триплетном состоянии спины двух элек-
тронов становятся не спаренными и явля-
ются, таким образом, параллельными.
Основное
синглетное
состояние
Возбужденное
синглетное
состояние
Возбужденное
триплетное
состояние
Рис. 1'8.3. Три различных состояния электро-
на. Обратите внимание на то, что возбужден-
ное триплетное состояние является менее вы-
годным энергетически, чем соответствующее
возбужденное синглетное состояние
го синглетному состоянию S.,. Обычно
молекулы в конденсированной фазе
быстро релаксируют до низшего син-
глетного колебательного уровня
( коммсн rapiiii rs.i). Этот процесс на-
зывается внутренней конверсией и про-
исходит обычно за 10’12сек. Поскольку
время жизни флуоресценции равно
около 10“8 сек, внутренняя конверсия
завершается обычно до начала эмис-
сии. Поэтому эмиссия флуоресценции
обычно происходит из равновесного
термодинамического состояния.
Молекулы из 5, состояния могут
переходить также в первое триплетное
состояние Т, называемого метастабиль-
ным. Метастабильность уровня означает,
что вероятность перехода 5, —> Тна мно-
го порядков меньше, чем вероятность
перехода 5, —> 5,. Эмиссия из состояния
Т,, часто называемая фосфоресценци-
ей, обычно характеризуется сильным
сдвигом максимума поглощения в длин-
новолновую область (с более низкой
энергией). Переход в 1\ называется
внутрисистемным переходом. Переход
из состояния 1\ в основное состояние
является запрещенным, в соответствии с
правилами квантовой механики, и, в ре-
зультате, константа скорости для такой
эмиссии на несколько порядков ниже,
чем при флуоресценции. Различные мо-
лекулярные состояния показаны на ри-
сунке Г8.3, где стрелками представлены
направления спинов.
Г8.3.2. Время жизни флуорофоров
и время вращательной корреляции
Интервал времени между возбуждени-
ем и эмиссией для большинства малых
молекул в водных растворах равен при-
близительно 10“8 сек, тогда как время их
вращательной релаксации гораздо коро-
че. Поэтому можно ожидать, что за время
Рис. I 8.4. Анизотропия флуоресценции и молекулярное связывание. Свободный лиганд, мече-
ный флуоресцеин-иодобным флуорофором (слева), вращается в субнаносекундной шкале време-
ни (со временем вращательной корелляции т ) и излучает деполяризованную флуоресценцию.
Тот же лиганд, связанный с большой макромолекулой (справа), вращается гораздо медленнее
и, следовательно, сохраняет анизотропию (Sportsman, 2003)
эмиссии молекулы будут ориентированы
случайным образом и флуоресценция бу-
дет неполяризованной. Однако большие
молекулы, такие как белки, под действием
броуновского движения будут вращаться
гораздо медленнее и, следовательно, флу-
оресценция будет частично поляризован-
ной (рис. 1'8.4).
На практике очень немногие белки
обладают природными флуорофорами.
Для большинства белков надо полу-
чать флуоресцирующие производные,
вводя в них флуоресцентные группы,
время жизни которых обычно равно
нескольким наносекундам. В г tv. ;
lapin: IS.2 приведены примеры не-
которых типичных внутренних (при-
родных) флуоресцирующих молекул,
их синтетические аналоги и внешние
флуоресцентные зонды.
Комментарий Г8.2. Флуоресцентные метки
Флуоресцентные метки подразделяются на три категории: внутренние метки, их аналоги
и внешние метки. Внутренние метки — это природные молекулы, проявляющие значитель-
ную флуоресценцию, которую можно использовать на практике. В эту группу образцов вхо-
дят ароматические аминокислоты, в том числе триптофан и тирозин, восстановленный ни-
котипадеииннуклеотид (11АД-Н), и восстановленный флавинадениндинуклеотид (ФАД-Н),
некоторые порфирины, модифицированные нуклеиновые кислоты (такие как Y-основания
в некоторых тРНК) и хлорофиллы.
Аналоги внутренних меток являются их модификациями и проявляют уникальные спек-
трофотометрические свойства. Их примером являются 5-гидрокситриптофан и 7-азатрип-
тофан.
Внешние метки называются так потому, что могут быть введены в молекулу-мишень с об-
разованием комплексов с ковалентными или нековалентными связями. Тысячи меток до-
ступны в настоящее время. Примером нековалентно связанного флуорофора является диме-
тиламино-нафталии сульфонил-хлорид (дансил-хлорид), 8-анилино-1 -нафталин сульфонат,
1,6-дифенпл-1,3,5-гексатриен, этидиум-бромид. изотиоцианат, эфир сукцинимида, иодане-
тамид, малеимид. Эти и другие соединения являются мишенями для многих биологических
макромолекул.
Г8.3.3. Стационарная
деполяризованная флуоресценция
Вращательные свойства флуоресциру-
ющих молекул могут быть определены
методом Перрена, суть которого состоит
в следующем. Предположим, что на рас-
Рис. Г8.5. Экспериментальная схема для из-
мерения деполяризации флуоресценции света.
Углы ф и 0 определяют ориентацию вектора у
по отношению к фиксированным осям. Поля-
ризация не зависит от угла ф и изменяется с 0,
становясь равной нулю для излучения, направ-
ленного вдоль z-направления (Weber, 1953)
Рис. Г8.6. К определению анизотропии А:
неполяризованный свет, направленный вдоль
оси х, разлагается на у- и z-поляризованные
компоненты. Общая эмиссия находится сум-
мированием эмиссии, направленной вдоль
трех декартовых осей
твор падает плоско поляризованный свет
(рис. Г8.5) вдоль .v-направления с векто-
ром поляризации, направленным вдоль
г-оси. Измеряются две компоненты из-
лучаемого света: 1 — параллельная ком-
понента, поляризованная вдоль z-ocn,
и - перпендикулярная компонента,
поляризованная вдоль.v-оси; р — диполь
флуоресцентной молекулы. Анизотро-
пия или поляризация, обозначаемые как
А и Р, соответственно, вычисляются как
разница между значениями компонент,
деленная на их сумму:
р = (Г8.2)
+ Al
Знаменатель в уравнении Г8.1 явля-
ется суммарной интенсивностью света,
которая наблюдалась бы при использо-
вании неполяризованного света. Если
обратиться к рисунку Г8.6,то можно ви-
деть, что / и /± распространяются как
вдоль х-оси, так и вдоль (/-оси, но удво-
енная интенсивность 2 ^распространя-
ется вдоль z-оси. Общая интенсивность
/пропорциональна сумме всех трех ин-
тенсивностей, распространяющихся во
взаимно перпендикулярных направле-
ниях, т. е. 1 = 1ц + 2/±.
Чтобы вычислить поляризацию
флуоресценции, мы должны вычислить
вероятность возбуждения молекул с
конкретной ориентацией, и затем под-
считать вероятность, с которой! эти мо-
лекулы будут излучать свет, поляризо-
ванный в определенных направлениях.
Начнем с жестких и изотропных моле-
кул. Молекулы должны быть достаточ-
но большие, чтобы не было заметного
вращения молекул в шкале времен флу-
оресценции (<100 нсек). Вычисления
показывают, что если испускающие и
поглощающие диполи параллельны друг
другу (т. о. относятся к одному и тому же
электронному переходу), тогда = 3/5 и
IL = 1/5. Для полностью жестких мо-
лекул Р = 1/2 и А = 2/5, как это следует
из вычислений, основанных на уравне-
ниях Г8.1 и Г8.2. Когда испускающий
диполь молекулы будет перпендикуляр-
ным поглощающему диполю, то Р = 1/2
и А = 2/5. В общем случае, поляризация
Ро и анизотропия Ло для жестких молекул
будут равны:
p_3eos4-l
0 cos2£+3
и
(Г8.4)
где — есть угол между поглощающим
и излучающим диполями.
Противоположной является ситуа-
ция, когда диполь вращается достаточно
быстро, чтобы его ориентация стала ха-
отичной за время жизни возбужденного
состояния. В этом случае, за время, когда
происходит эмиссия, все сведения о про-
исхождении фотопроцессов утрачивают-
ся. = /± и поляризация и анизотропия
становятся равными нулю. Большинство
биологических макромолекул не попа-
дает в разряд этих двух крайних случа-
ев; время их вращательного движения
не пренебрежимо мало по сравнению со
шкалой времени флуоресценции и они
не вращаются так быстро, чтобы достичь
6eciюрядочной ориентации.
Г8.3.4. Деполяризованная
флуоресценция, разрешенная
во времени
Два типа экспериментов могут быть
выполнены с использованием флуо-
। Комментарий Г8.3. Флуоресцентная
анизотропия как рациометрическая
величина
Флуоресцентная анизотропия является
рациометрическим измерением, т. е. она не
зависит от концентрации белка, поскольку
определяется как отношение двух компо-
; нент, каждая из которых, сама по себе, про-
| порциональна концентрации белка.
।___________________________________
ресцентной техники, разрешенной во
времени. Первый включает измерение
времени спада эмиссии, следующее за
коротким импульсом возбуждения.
Такой тип экспериментов использу-
ется для определения времени жизни
флуорофора, которое отражает среднее
время, в течение которого молекула
остается в возбужденном синглетном
состоянии. Время жизни флуоресцен-
ции строго зависит от молекулярных
свойств окружения флуорофора и мо-
жет изменяться от нескольких пикосе-
кунд до десятков наносекунд.
Другой тип экспериментов по флуо-
ресценции, разрешенной во времени,
базируется на измерении спада анизот-
ропии флуоресценции. Спад анизотро-
пии отражает переориентацию эмиссии
диполя во время возбужденного состоя-
ния и дает информацию о локальном
сдвиге флуорофора, сегментальном пе-
ремещении и вращательной диффузии
макромолекул ( коммсп lapnii IS.3).
Предположим, что флуоресцентная
метка жестко прикреплена к макромо-
лекуле (см. рис. 1’8.1). Очевидно, что на-
блюдаемая поляризация является некой
промежуточной величиной между вели-
чиной, вычисленной из уравнения Г8.3 и
нулем. Чтобы вычислить это значение,
мы должны проанализировать относи-
тельную скорость эмиссии и вращатель-
ного движения макромолекулы. Изме-
рения поляризации являются функцией
как времени жизни флуорофора, так
и времени релаксации белков. Эта связь
выражается уравнением Перрена:
<ге-5>
В этом выражении Ло является ста-
ционарной анизотропией, а — ани-
зотропия флуорофора в отсутствие вра-
щательного движения. Из уравнения
Г8.5 видно, что если время жизни флуо-
рофора г,, много больше, чем время вра-
щательной корреляции, т , то анизот-
ропия Ло равна нулю. Если же время
жизни намного короче, чем корреляци-
онное время, то анизотропия равна
Отсюда следует весьма важный вывод:
анизотропия Ло будет чувствительна
к скорости вращательной диффузии
только в том случае, если т сравнимо
со временем жизни флуорофора тг
Г8.4. Экспериментальная
техника
Два наиболее популярных метода ис-
пользуются сегодня, чтобы зафиксиро-
вать флуоресценцию, разрешенную во
времени, — это методы гармонического
и импульсного ответа. В методе гар-
монического ответа параметры флуо-
ресценции во временном разрешении
анализируются через ответ эмиссии па
синусоидально модулированное воз-
буждение. Модуляторы света и обработ-
ка сигнала, необходимые для этих изме-
рений!, могут быть объединены в виде
стандартного спектрофлуорометра.
Метод импульсного ответа включает
прямое наблюдение за временем спада
эмиссии, которое следует за коротким
возбуждающим импульсом. Импуль-
сы возбуждения обычно получают от
лазера с синхронизованными модами
или титан-сапфировых лазеров, кото-
рые производят пикосекундные или
субпикосекундные импульсы и являют-
ся легко настраиваемыми в видимой и
УФ-областях спектра. Профиль спада
эмиссии регистрируется схемами, рабо-
тающими по принципу быстрого счета
одиночных фотонов. Использование та-
кого оборудования позволяет регистри-
ровать профили спада флуоресценции
с высокой степенью точности и времен-
ным разрешением порядка пикосекунд
и менее. Измерение флуоресцентной
Рис. Г8.7. Схема измерений анизотропии флуоресценции методом L-формата: верти-
кальное и горизонтальное возбуждение
Рис. 1 '8.8. Схема измерений анизотропии флуоресценции методом Т-формата: вертикальное
и горизонтальное возбуждение
анизотропии технически является еще
более простым. Обычно используются
два метода. Эти методы обозначаются
как L- и Т-форматы. В методе L-фор-
мата используется один канал эмиссии
(рис. Г8.7). Излучаемая образцом флуо-
ресценция измеряется под прямым уг-
лом по отношению к лучу возбуждения.
Для быстрых измерений анизотропии
флуоресценции обычно применяются
два фотоумножителя в Т-конфигурации
для одновременного измерения излу-
чаемой флуоресценции при двух углах,
соответствующих поляризации, парал-
лельной и перпендикулярной по отно-
шению к исходящему лучу (рис. Г8.8).
На практике, фактор коррекции,
К, должен вводиться в уравнения Г8.1
и Г8.2, чтобы учесть различия в чув-
ствительности систем детектирования
в двух поляризующих направлениях
( и>м\нч। Hipnii IS. i).
В упрощенной системе детектиро-
вания используется только один фото-
умножитель без поляризатора. В этом
случае поляризация возбужденного
света изменяется от вертикального (V)
до горизонтального (Н) состояния с по-
мощью фотоэластического модулятора
( м>мм( и’арий Из рисунке Г8.9 а
видно, что при вертикально поляризо-
ванном световом возбуждении детек-
тируется сумма интенсивностей излу-
чения IVH и 1п,, определяющая рх- и
^-компоненты, соответственно. Отсю-
да следует:
Комментарий Г8.4. фактор
коррекции, К
В реальных измерениях фактор коррек-
ции, К, вводится в уравнения Г8.2 и Г8.3:
7,,-КД
А = -4-----,
/р+ 2К7±
р _ Н;
I.. + KI
(А)
(В)
Выравнивание оптических систем осу-
ществляется с помощью исследования
рассеяния разбавленной суспензии не
флуоресцирующих соединений (например,
гликогена или коллоидального кремния в
воде). Рассеянный свет от таких суспензий
на 100% поляризован, и, следовательно,
измеренная анизотропия должна быть рав-
на 1,00 (Л = 1,00). Выравнивание оптиче-
ской системы можно считать законченным,
если измеренное значение/! равно 0,98 или
больше.
Комментарии I 8.5. Фотоэластичный
модулятор
В основе устройства, называемого фото- j
эластичным модулятором (ФЭМ) лежит ;
эффект фотоэластичности. Если образец
из прозрачного, материала сильно сжать '
или растянуть, то он становится двулучен-
реломляющим, т. е. скорость прохождения
света через материал будет разной для све-
та с разной линейной поляризацией. ФЭМ ,
может быть использован в каждом из двух i
обсуждаемых методов: для получения за-
. данного состояния поляризации падающе-
го луча света, или для определения состоя-
ния поляризации прошедшего светового
луча.
4 = (Г8.6)
Ситуация для горизонтально поля-
ризованного возбуждения отражена на
рисунке Г8.9 б. В этом случае сигнал,
определяемый для горизонтально поля-
ризованного возбуждения, равен:
Лн=2х/,.н (Г8.7)
и анизотропия вычисляется из /ун и /vv
из результирующего выражения:
Этот метод не требует фактора кор-
рекции G (см. коммои шр:ill IS. i).
Г8.5. Деполяризованная
флуоресценция
и броуновское движение
Г8.5.1. Стационарная
или статическая поляризация
При применении постоянного освеще-
ния измеряется стационарная или ста-
тическая поляризация. В этом случае
выражение для среднего значения ста-
тической поляризации и анизотропии
будет следующим:
Р =-----------------------, (Г8.9)
l + 10(l + Tf/TK„p)(3cos4-l)''
3cos2£-l
5(l + Tf/TKop)’
Если измерения проводятся при вы-
соком значении г|/7’, при котором моле-
кулы не способны вращаться в шкале
времени жизни возбужденного состоя-
ния, тогда т,./т.1)г = 0, и поляризация
и анизотропия будут равны:
Детектор
а б
Рис. I 8.9. Схема эксперимента по измерению анизотропии флуоресценции: а — вертикальная
поляризация возбуждения; детектирование без поляризатора дает величину 7 + 7VV; б — гори-
зонтальная поляризация возбуждения; детектирование дает величину 7Hi| +THV Последняя равно
2 XIVH , если предположить, что образец ориентирован хаотически (Canet et al., 2001)
p 3cos2i;-i
° cos4 + 3 ’
-л 3cos4-l
A> =---P—
(Г8.11)
(Г8.12)
Эти значения, называемые предель-
ной поляризацией и анизотропией,
идентичны таковым, полученным для
жестких систем (уравнения Г8.3 и 8.4).
Используя уравнения (Г8.1) и
(Г8.2) для Ро и Д), можно переписать
уравнения (Г8.9) и (Г8.10) в удобной
форме, называемой уравнениями Пер-
рена:
Х-- = (^--)(1 + —) =
Р 3 3 Т-Р 0.0
11 xkT (Г8-13)
Ро з УГ1ирЛ
Рис. Г8.10. Время вращательной корреляции
ткпрэритрозин-БСА комплекса в смесях с раз-
личным соотношением глицерин/буфер как
функция г\/Т при постоянной температуре
273 К (•); при сохранении состава глицерина
в смеси глицерин-вода (92% весовых частей
глицерина) за счет температуры (А) (Ferrer et
al., 2001)
£
А
Х(1+ТЧ=Х(1+^)'(Г8Л4)
кор А) гидр Ч
В правой половине уравнения Пер-
рена время вращательной корреля-
ции тк1)) выражается через уравнение
(Г2.31)’1’
Корректность уравнений (Г8.13)
и (Г8.14) может быть проверена, построе-
нием графика 1/Д или 1/Д) как функ-
ции Т/г\, который должен быть прямой
линией. По наклону прямой можно су-
дить о гидродинамическом объеме (V ),
если известно время спада флуоресцен-
ции (тн). Значение ординаты при Т/г| ->0
дает величину предельной анизотропии
или поляризации. На рисунке Г8.10 по-
казан пример графика Перрена для бы-
чьего сывороточного альбумина (БСА).
Изменение окружающих условий для
молекулы БСА осуществлялось за счет
изменения вязкости среды. Значение
т , экстраполированное к стандартным
условиям (1 сантипуаза, 273 К), оказа-
лось равным 40 ± 2 нсек.
Г8.5.2. Время спада анизотропии
флуоресценции
Допустим, что мы возбуждаем молекулы
коротким импульсом поляризованного
света и измеряем временную зависи-
мость компонент [ и 1А. Если излуча-
ющий и поглощающий диполи парал-
лельны друг другу, то первые фотоны, с
высокой долей вероятности, будут z-no-
ляризованными, поскольку у молекул не
будет времени на переориентацию. На-
против, последние фотоны будут иметь
беспорядочную поляризацию, поскольку
за это время молекулы испытают значи-
тельное вращательное движение. Следо-
вательно, если поляризация или анизот-
ропия измеряются как функция времени
эмиссии флуоресценции, то скорость ее
спада от начальных значений Ро или Ао
до конечных значений, равных нулю, бу-
дет определяться скоростью вращатель-
ного движения молекул.
Как мы видели в параграфе Г2.3.4,
вращательное поведение сферы может
быть описано одним параметром, вра-
щательным корреляционным временем
т Если это время определить как:
1 ^К.ирПо
к,,р 60 kT '
(Г8.15)
тогда
Л(г) = (2/5К' ^Зсо^ -1. (Г8.16)
Таким образом, спад анизотропии
флуоресценции сферической молеку-
лы описывается одной экспонентой.
Для жестких эллипсоидов результат
получается более сложным, и спад А
(Г) описывается суммой трех экспонент:
Л (О t
---= а1 ехр-----+
Д° Т1к',р (Г8.17)
t t
+а, ехр------+ а3 ехр-----,
*^2кор Тзкор
где предэкспоненциальные множите-
ли су зависят только от относительной
ориентации моментов электронных пе-
реходов во флуоресцентной метке. Если
белок помечен меткой беспорядочно, то
at = a2 = ос, = 2/5.
Времена вращательной корреляции
т связаны с коэффициентами враща-
тельной диффузии эллипсоида Dpi DL
как:
т. = (4D,+ 2
1кор ' 1 / ’
т9 = (Д + 5 О. ) ',
2кор v I' 1 / ’
т., = (6D
Зкор ' -L 7
Зависимость коэффициентов диф-
фузии от формы молекулы, температу-
ры и вязкости растворителя может быть
выражена в компактной форме уравне-
нием Стокса-Эйнштейна-Дебая:
kT
Ц=-----г;--- (М|.1). (Г8.18)
где Vnt — гидродинамический объем
и г](1 - вязкость растворителя. Фактор
Перрена g. зависит только от аксиаль-
ного отношения эллипсоида р = а/Ь
(где а •- ось симметрии) и равен:
g
.1
2(р2 -1)
Зр(р-5)’
(Г8.19)
2(/>s -1)
3p(2p2-l)S-p’
(Г8.20)
5..=(//-1)-' 21п[р + (/г’-1)-’ 2,(Г8.21)
М =(1-/>2) 1 -’arctgx
х|р-'(1-/) ' 2.
(Г8.22)
На практике, однако, точность дан-
ных такова, что кривую спада редко
удастся описать больше, чем двумя эк-
спонентами.
Для жестких вытянутых эллипсои-
дов с аксиальным отношением <5 и не-
которых сплюснутых эллипсоидов вид
функции А (?) является очень близким
к простой экспоненциальной функции.
В этих условиях наблюдаемый спад ап-
проксимируется функцией со средним
корреляционным временем, равным:
\пр = ЕаЛк.,рр- (Г8.23)
1
Среднее значение т отличается от
гармонического значения, которое по-
лучается из наклона прямой А (?) функ-
ции при t = 0 (параграф Г2.3.3).
Г8.5.3. Вращательные
корреляционные времена
глобулярных белков
На рисунке Г8.11 приведен пример одно-
го из самых ранних исследований спада
анизотропии для флуоресцентно мече-
Рис. I 8.1 1: а — затухание флуоресценции антранилоила-8ег193-а-химотрипсина для индиви-
дуальных поляризованных компонентов; б — анизотропия в логарифмической шкале после пре-
кращения действия импульса возбуждения (Stryer, 1968)
ного а-химотрипсина. Измеренное вре-
мя вращательной корреляции состави-
ло около 15 нсек. Измерение константы
спада позволяет вычислить гидродина-
мический объем V , если известна вяз-
кость раствора. Простое вычисление по-
казывает, что тк<) может быть вычислено
из уравнения (Ы.15):
СР = ^хГ2сек, (Г8.24)
т. е. если белок имеет приблизительно сфе-
рическую форму, то времени корреляции
ткор, равной 1 нсек, соответствует белок с
молекулярной массой 2500 Да с уровнем
гидратации 0,3-0,4 г-г 1 (Глава П). Сред-
ние значения времен корреляции ткор
Рис. ГН. 12. Зависимость времен вращательной корреляции для тридцати одного белка от мо-
лекулярной массы в двойном логарифмическом масштабе. Штрих-пунктирная линия имеет на-
клон, равный 1 в соответствии с уравнением Г8.24
Комментарий Г8.6. Вращательное
релаксационное время, трел i
и вращательное корреляционное
время, ткор
Отметим, что исследователи, применя-
ющиетот или иной метод, часто используют
два времени для описания вращательного
движения: вращательное релаксационное
время, т и вращательное корреляционное
время, тм11 Они связаны между собой сле-
дующим образом тк,„, = т|(..,/3, TKIV = (fiD) '. ,
В простейшем случае жесткой сферы, для i
которой А (?) затухает ио экспоненциаль-
ному закону:
/1(Г) = Ле’1Ю'=Л«" (А)
где 0 — коэффициент вращательный
диффузии, а Т есть вращательное кор- >
реляционное время.
Молекулярная масса (Да)
Рис. 18.13. Зависимость затухания анизот-
ропии от молекулярной массы для флуоро-
форов с разным временем излучения от 4 до
2700 нсек. Для каждой из кривой жирными
линиями выделены участки, определяющие
интервал молекулярных масс белков, у кото-
рых реально могут измерены их времена ре-
лаксации при данном времени жизни флуоро-
фора. Для этих расчетов было использовано
уравнение Г8,5. Объем V белка вычислен
1 I нлр
из его молекулярной массы при допущении,
что уровень гидратации равен 20% (Lakovicz
et al., 2000)
белков, определяемые с использованием
метода деполяризованной флуоресцен-
ции, представлены в m >лнцг 1 8. К Таб-
лица содержит также значения времен
корреляции для белков, полученные ме-
тодом ЯМР (секция КЗ.З) и методом де-
поляризованного динамического рассея-
ния света (Глава ПО).
Зависимость времен вращательной
корреляции для тридцати одного белка от
молекулярной массы изображена графи-
чески в двойной логарифмической шка-
ле на рисунке Г8.12 (коммеп lapnii Г8.(>).
Прерывистая прямая линия соответству-
ет данным, предсказываемым уравнением
(Г8.24). Из рисунка Г8.12 видно, что все
белки (за исключением карбоангидразы)
имеют времена вращательной корреля-
ции несколько большие, чем соответству-
ющие значения для сферических частиц.
Последнее не удивительно, поскольку
молекулы вытянутой формы вращаются
более медленно. Наклон прямой линии,
проведенной через экспериментальные
точки (сплошная линия),свидетельствует
о том, что в пределах экспериментальных
ошибок вращательное корреляционное
время глобулярных белков пропорцио-
нально их массе в соответствии с уравне-
нием Г8.24.
Большинство данных, представлен-
ных на рисунке ГК. 12, было получено в
наносекунд!юм интервале времени. Такие
условия эксперимента налагают очень
серьезные ограничения на исследуемые
биологические макромолекулы в отноше-
нии их молекулярных масс. Из уравнения
(Г8.7) следует, что макромолекулы с моле-
кулярными массами в области 10-50 кДа
имеют времена релаксации в интервале
4-20 нсек, т. е. в области, которая соот-
ветствует синглетному времени жизни
большинства органических красителей.
На рисунке Г8.13 показана теоре-
тическая зависимость анизотропии
Таблица 18.1. Времена вращательной корреляции ткор (20 °C) для белков,
определенные различными методами
Белок Молеку- лярная масса (кДа) Время вращательной корреляции ткпр (20 °C) (нсек) Метод Ссылки
Грамицидин 2,5 2,8 ДРС е)
Фрагмент белка Zn-нальцев ДНК 2,93 2,4 ЯМР а)
Трипсиновый ингибитор 6,16 4,4 ЯМР а)
Эглии С 8,15 6,2 ЯМР а)
Калбиидин- D9k apo 8,43 5,1 ЯМР а)
Убиквитин 8,54 5,4 ЯМР а)
Цитохром Ь. 9,61 6,1 ЯМР а)
Вастар с/40/82А 10,14 7,4 ЯМР а)
Тгр-реирессор 11,89 23,1 ЯМР а)
Рибонуклеаза 13,68 8,3 ЯМР а)
Азурин 13,95 4,8 нсек-ДПФ б)
Лизоцим 14,32 8,3 ЯМР а)
Лизоцим 14,30 10 ДРС в)
Стафилококковая иуклеаза 15,51 13,3 ЯМР а)
5. aureus нуклеаза В 20,00 9,9 ТРП-ДПФ з)
Интерлейкин-ip 17,4 12,4 ЯМР а)
р-лактоглобулин 18,40 9,4 нсек-ДПФ г)
Фактор лейкемии 19,1 14,9 ЯМР а)
р2Г" 21,00 16 нсек-ДПФ д)
H1V-1 протеаза 21,58 13,2 ЯМР а)
Трипсин 25,00 14,3 нсек-ДПФ г)
Химотрипсин 25,00 Г8,7 нсек-ДПФ г)
Сави паза 26,70 12,4 ЯМР а)
Карбонаигидраза 30,0 12,4 нсек-ДПФ г)
р-лактоглобулин (димер) 36,00 22,5 нсек-ДПФ г)
Апопероксидаза 40,00 28,0 неек-ДПФ г)
FU) фрагмент иммуноглобулина G 47,50 33 нсек-ДПФ г)
Бычий сывороточный альбумин 66,50 40 мксек-ДПФ ж)
р-лактоглобулин 18,40 8,6 мксек-ДПФ 3)
Яичный альбумин 43,5 30,2 мксек-ДПФ 3)
Сахароза-изомальтаза 221,0 112 мксек-ДПФ з)
Сокращения: (ДРС) - динамическое рассеяние света; (ЯМР) — ядерный магнитный резо-
нанс: (неек-ДПФ) - деполяризованная флуоресценция в наносекуидном диапазоне; (мксек-
ДПФ) — деполяризованная флуоресценция в микросекундном диапазоне; (ТРП-ДПФ) — депо-
ляризованная флуоресценция триптофанового остатка.
Данные взяты из:
a) Garcia de la Torre (2001);
6) Kroes et al. (1998);
в) Dubin et al. (1971);
r) Yguerabide et al. (1970); ж) Ferrer et al. (2001):
д) 1 lazlett et al. (1993): з) Cherry and Schneider (1976)
e) Michielsen et al. (1981):
Рис. Г8. 14. Спад анизотропии флуоресцен-
ции комплекса эритрозина с БСА и рассчитан-
ная на ее основе моноэкспоненциальная фун-
кция со временем вращательной корреляции
т = 16 ±1,5 мксек (92% глицерина по весу в
буфере; температура 18 °C. (Ferrer et al., 2001).
Эритрозин — это 2'4'5',7'- тетраиод-флуорес-
цеин
от молекулярной массы, построен-
ная в соответствии с уравнением Г8.5
для флуорофоров с разным временем
жизни. Из рисунка видно, что флуо-
ресценция белка с молекулярной мас-
сой 100 кДа, меченого флуорофором с
временем жизни 4 нсек, будет полно-
стью поляризованной. Отсюда следу-
ет, что флуораторы, имеющие время
жизни в наносекундном диапазоне, не
могут использоваться для измерения
вращательной диффузии белков с мо-
лекулярной массой больше 40-50 кДа.
Интервал молекулярных масс суще-
ственно расширяется при увеличе-
нии времени жизни флуорофора. Так,
для флуорофора со временем жизни
400 нсек оказывается возможным изу-
чение белков с молекулярной массой
до 10е Да.
В настоящее время можно изучать
флуоресценцию в микросекундной
шкале времени, используя флуорофо-
ры на основе комплексов металл-ли-
ганд со временем жизни до 10 мксек и
более, которое соответствует времени
жизни триплетного состояния. На ри-
сунке Г.8.14 показано затухание ани-
зотропии флуоресценции комплекса
эритрозин-БСА в микросекундной
шкале времени в буфере, содержащем
глицерин.
Результаты этих измерений свиде-
тельствуют об отсутствии независимо-
го вращения больших сегментов белков
Рис. Г8.15: а — вытянутый эллипсоид (140 X 40А), используемый как стандартная модель для
конформации человеческого и бычьего сывороточных альбуминов, представленная во всех учеб-
никах; б — структура человеческого сывороточного альбумина (ЧСА) в том же масштабе (Банк
белковых структур; файл lbm°; в — оболочечная модель БСА в виде трехгранника с размера-
ми 80 х 80 х 89 х 30 А; г — модель ЧСА, построенная по данным рентгеноструктурного анализа
(Ferrer et al., 2001)
в растворе во временном интервале от
наносекунд до миллисекунд. Вычис-
ленное вращательное время корреля-
ции т для БСА (стандартные условия:
1 сантипуаза, 20 °C) = 40 ± 2 исек (см.
также рис. 1'8.10).
Анализ значений времени враща-
тельной корреляции (40 ± 2 нсек) и ха-
рактеристической вязкости (4,1 см3-г-1)
позволяет сделать выбор между сердце-
образной структурой Ч С А, наблюдаемой
в кристаллическом состоянии, и хрис-
томатийной гидродинамической моде-
лью БСА, — сигарообразный эллипсоид
с размерами 140x40 А (рис. Г8.15 а и б).
Расчет коэффициента вращательной
диффузии и характеристической вяз-
кости БСА с помощью метода оболо-
чек (параграф Г2.2.2) показал, что обе
модели (одна — треугольная призма,
с размерами 84x84x84x31,5 А и вто-
рая — кристаллографическая модель)
(рис. Г8.15 « и /) дают близкие резуль-
таты, совпадающие с эксперименталь-
ными значениями. Однако эти значения
сильно отличны от таковых, получен-
ных для сигарообразной модели. От-
сюда следует, что модель удлиненного
эллипсоида, используемая десятилети-
ями в качестве канонической модели
сывороточного альбумина, скорее все-
го, не верна.
Г8.6. Деполяризованная
флуоресценция
и молекулярные
взаимодействия
Деполяризованная флуоресценция мо-
жет быть использована для изучения
молекулярных взаимодействий, вклю-
чая белок-белковые, ДНК-белковые, а
также связывания антигенов с антите-
лами. Деполяризованная флуоресцен-
ция является уникальным методом
для анализа молекулярного связыва-
ния, поскольку позволяет измерить
отношение меченых связанных форм
к меченым свободным. Большинство
таких измерений может быть прове-
дено в шкале реального времени, что
позволяет исследовать кинетику реак-
ций ассоциации и диссоциации. Ниже
мы приведем примеры применения
деполяризованной флуоресценции в
исследованиях белок-белковых взаи-
модействий на молекулярном уровне.
Исследования процессов связыва-
ния методом поляризации флуоресцен-
ции включает титрование небольшого
количества флуоресцентно меченого
белка другим белком. При увеличении
фракции белка, связанного с мечены-
ми атомами, поляризация возрастает.
Константы равновесного связывания
вычисляются по результирующей кри-
вой связывания (т. е. поляризации как
функции концентрации немеченых бел-
ков). На рисунке Г8.16 показано влия-
0.07
0.06-
S
S
о 0.05-
н
| 0.04-
га
о 0.03-
z 0.02-
о
£ 0.01-
0-1-----1------1-----1------1-----1------1-
0 0.5 1 1.5 2 2.5 3
Log гА2 или гА2 концентрации (нМ)
Рис. Г8.16. Влияние активации тромбина на
ассоциацию А- и В-субъединиц фактора XIII
тромбина. Аликвоты гА'., или тромбин-акти-
вированный гА'., были добавлены к раствору
В2. Горизонтальная линия соответствует 50 °о
изменению уровня анизотропии (Checovich
и др. 1995)
Комментарий Г8. /. Фактор XIII коагуляции крови
Фактор XIII коагуляции кропи, или фибрин - стабилизирующий фактор, является плаз-
матическим .зимогеном, ответственным за стабилизацию (усиление) процесса свертывания
крови и обеспечение ее устойчивости к лизису. Фактор XIII действует через промотор, объ-
единяя две различные субъединицы АВ в А.,В., тетрамер. Только А-субъедииица модифици-
руется тромбином через N-концевую активацию пептидов (АП). Гидролитически расщеплен-
ная субъединица обозначается А.
Рис. Г8.17. Время спада флуоресцеин-мечено-
го казеина при его переваривании трипсином.
Реакция начинается после добавления 5 пи-
комолей казеина, меченого флуоресцеином
к смеси: 200 иг трипсина (нижняя кривая);
100 иг трипсина (средняя кривая); без трипси-
на (верхняя кривая). По оси ординат значения
поляризация приведены в относительных еди-
ницах (Checovich et al., 1995)
ние активации тромбина на ассоциацию
А и В субъединиц фактора XIII (ком-
ментарии 1'8.7). Расщепление тромбина
(переход из состояния гА2 в гА',), при-
водит к драматическому понижению
аффинности г А., к меченому В., (294 нМ
для гА против 19 нМ для гА.,) объяс-
няя таким образом диссоциацию комп-
лекса А.,В.,.
Другой пример относится к дей-
ствию деградирующих энзимов, та-
ких как протеазы, ДНК-азы, РНК-азы.
Действие таких энзимов проявляется
обычно в изменении молекулярных
размеров, что, следовательно, может
быть зафиксировано методом деполя-
ризованной флуоресценции. На ри-
сунке Г8.17 показано затухание трип-
синовой активности с использованием
меченого флуоресцеином казеина. Поч-
ти любой субстрат может быть флуо-
ресцентно помечен и использован для
измерения разных энзимов. Стандарт-
ная кривая для тестирования действия
субстрата на казеин является линейной
по крайней мере в интервале 100-крат-
ного изменения концентрации, а метод
в 10 раз более чувствительным, чем у
широко распространенного метода пре-
ципитации.
Г8.7. Перечень ключевых идей
• Люминесценция является результатом излучения молекулой света, находя-
щейся в электронно-возбужденном состоянии.
• Эмиссия из синглетного состояния в основное называется флуоресценцией’, вре-
менной интервал между возбуждением и эмиссией обычно равен 10 8 сек.
• Эмиссия из триплетного состояния в основное называется фосфоресценцией',
при фосфоресценции интервал между возбуждением и эмиссией может состав-
лять от микросекунд до секунд.
• На практике немногие белки флуоресцируют; большинство белков и практи-
чески все нуклеиновые кислоты могут быть переведены во флуоресцирующие
производные введением специальной метки.
• Флуоресцирующие метки подразделяются на три категории: внутренние метки
(ароматические ipynnw в белках), их химические аналоги (например, 5-гидрокси
триптофан) и многочисленные внешние метки для белков и нуклеиновых кислот.
• Время вращательной корреляции т может быть измерено по спаду поляриза-
ции флуоресценции только в том случае, если вращательное корреляционное
время макромолекулы сравнимо со временем жизни флуорофора.
• Время вращательной корреляции глобулярных белков с молекулярной массой
в районе 10-50 кДа может быть измерено при использовании метки, излуча-
ющей в течение 4 -20 нсек.
• Время вращательной корреляции биологических макромолекул с молекуляр-
ной массой в районе 0,5-5 МДа может быть измерено при использовании мет-
ки, излучающей в течение микросекунд.
• Спад анизотропии флуоресценции сферических молекул описывается одной
экспонентой.
• Белок, имеющий приблизительно сферическую форму, с молекулярной массой
25 Да имеет время корреляции т около 10 нсек.
• Для жестких несферических молекул, экспериментальное время вращательной
корреляции всегда больше, чем для сферы; вытянутые молекулы вращаются
медленнее.
• Спад анизотропии флуоресценции жестких тел произвольной формы описыва-
ется многоэкспоненциальиой функцией (до 5 экспонент); однако на практике
экспериментальные данные могут быть аппроксимированы не более, чем двумя
экспонентами.
Литература для дальнейшего чтения
Исторический обзор и введение в биологические проблемы
Tao Т. Time dependent fluorescence depolarization and Brownian rotational diffusion
coefficients of macromolecules. Biopolymers, 1969. 8, 609-632.
Prendergast F. G. Time-resolved fluorescence techniques: methods and application in
biology. Curr. Opin. Struct. Biol., 1991. 1, 1054-1059.
Checovich IV.J., Bolger, R. E., and Burke T. Fluorescence polarization — a new tool for
cell and molecular biology. Nature, 1995. 375, 254-256.
Теория деполяризации флуоресценции
Weber G. Rotational Brownian motion and polarization of the fluorescence of
solutions. Adv. Protein Chem., 1953. 8, 415-459.
Small E. IF. and Isenberg, I. Hydrodynamic properties of a rigid molecule; rotational
and linear diffusion and fluorescence anisotropy. Biopolymers, 1977.16, 1907-1928.
Обоорудование
Cherry R.J. and Schneider G. A spectroscopic technique for measuring slow rotational
diffusion of macromolecules. 2: Determination of rotational correlation times of proteins
in solution. Biochemistry, 1976. 24, 3657-3661.
LakowiczJ. R., Gryczynski I. et al. Microsecond dynamics of biological macromolecules,
Meth. Enzymol., 2000. 323,473-509.
Sportsman J. R. Fluorescence anisotropy in pharmacologic screening. Meth. Enzymol.,
2003. 361, 505-529.
LakowiczJ. R., Gryczynski Let al. Microsecond dynamics of biological macromolecules,
Meth. Enzymol., 2000. 323, 473-509.
KroesS.J., Canters G. W., Giardi G., Van HoekA. and Visser A. J. IF. G. Time-resolved
fluorescence study of azurin variants: conformational heterogenity and tryptophan
mobility. Biophys. J., 1998. 75, 2441-2450.
Деполяризованная флуоресценция и броуновское движение
Lakovicz J. R. (ed.). Principles of Fluorescence Spectroscopy, second edition. New
York: Kluwer Academic/Plenum Publ., 1999.
Chevy R.J. and Schneider G. A spectroscopic technique for measuring slow rotational
diffusion of macromolecules. 2: Determination of rotational correlation times of protein
in solution. Biochemistry, 1976.15, 3657-3661.
Fever M. L., Duchowicz R., Cavasco B., Garcia de la Tove J. and Acuna A. U. The
conformation of serum albumin in solution: a combined phosphorescence depolarization-
hydrodynamic modeling study. BiophysicalJ., 2001. 80, 2422-2430.
Деполяризованная флуоресценция и молекулярные взаимодействия
Millar D. Р. Time-resolved fluorescence methods for analysis of DNA — protein
interactions. Meth. Enzymol., 2000. 323, 442-459.
Hazlett T. L., Moore K.J. M., Lowe P. N„ Jameson D. M. andEcclestonJ. F. Solution of
p21ras proteins bound with fluorescent nucleotides: a time-resolved fluorescence study.
Biochemistry, 1993. 32, 13575-13583.
Canet D., Doering K., Dobson С. M. and Dupont Y. High-sensitivity fluorescence
anisotropy detection of protein-folding events: Application to a-Lactalbumin. Biophys
J., 2001. 80, 1996-2003.
Глава Г9
ВЯЗКОСТЬ
Г9.1. Исторический обзор
(846
Дж. Л. Пуазейль (J. L. Poiseuille)
разработал теорию потока жидкости
в капилляре. На основе этой теории
В. Оствальд (W. Ostwald) (1933) и
Л. Уббелоде (L. Ubbelohde) (1936)
изобрели вискозиметры и ввели их в
практику физико-химических исследо-
ваний. Позднее приборы были названы
их именами.
1906
А. Эйнштейн (A. Einstein) первым
исследовал вязкость суспензии жестких
частиц, имеющих сферическую форму
и большой размер по отношению к раз-
мерам молекул растворителя. Он пока-
зал, что характеристическая вязкость
раствора пропорциональна объемной
доле, занятой частицами, и не зависит
от размера сферы.
1932
Г. Штудингер (Н. Staudinger)
предложил использовать характерис-
тическую вязкость для определения
молекулярной массы полимеров. Он
обнаружил, что зависимость характе-
ристической вязкость [т|] от молеку-
лярной массы М может быть выраже-
на простой формулой типа [т|] = М“.
Позднее из работ Г. Марка, Р. Ха-
увинка, В. Куна и Г. Куна (Н. Mark,
R. Houwink, W. Kuhn and H. Kuhn)
стало ясно, что константа а связана с
конформацией молекул в растворе.
1962
Б. Г. Зимм и Д. М. Крозерз
(В. Н. Zimm and D. М. Crothers) пред-
ложили оригинальную конструкцию для
ротационного вискозиметра, который
мог функционировать при низких гради-
ентах вязкости и который оказался очень
удобен для измерений асимметричных
структур, таких как ДНК, фибриллярные
белки и палочкообразные вирусы.
1985
М. А. Хани (М. A. Haney) с колле-
гами разработал «дифференциальный
вискозиметр», с помощью которого
непосредственно можно измерять от-
носительную вязкость. Преимущество
такого вискозиметра перед традицион-
ным стеклянным капиллярным виско-
зиметром состояло в увеличении его
чувствительности, точности, скорости
и оперативности, что позволило изме-
рять вязкость растворов с низкой кон-
центрацией образца (-1 мг-см 3для гло-
булярных белков).
2000 и но настоящее время
Характеристическая вязкость очень
чувствительна к конформации макро-
молекул и ее гибкости. Она может быть
измерена легко и с большой точностью
при использовании различных типов
вискозиметров. Однако к настоящему
времени метод вискозиметрии не очень
востребован, поскольку требует относи-
тельно большого количества образца.
Г9.2. Введение
в биологические проблемы
Характеристическая вязкость — один из
наиболее «старых» молекулярных па-
раметров. Так, Эйнштейн исследовал
характеристическую вязкость суспен-
зии сферических частиц еще в 1906 г.
Позднее стало ясно, что характеристи-
ческая вязкость очень чувствительна к
конформации биологических макромо-
лекул и может быть измерена с большой
точностью. Классический обзор Янга
(Yang), посвященный способам ее из-
мерения и практического применения, в
частности к белкам, был опубликован в
1961 г. В 1997 г. Хардинг (Harding) опуб-
ликовал работу, описывающую прогресс
в измерениях характеристической вяз-
кости и ее интерпретации в терминах
конформации, гидратации и гибкости
биологических макромолекул в растворе.
Традиционно вязкость растворов
биологических макромолекул измеряет-
ся двумя способами: временем прохож-
дения жидкости через капилляр опреде-
ленной длины и толщины или скоростью
вращения двух цилиндров друг относи-
тельно друга. Хотя измерение вязкости
обоими способами было полуавтомати-
зировано или автоматизировано, однако
основные конструкции вискозиметров
оставались прежними со времени опуб-
ликования фундаментальных статей
Янга и Зимма в 1960 г. Поэтому ни один
из этих способов не привлекает внима-
ния исследователей в области биохимии
белка, из-за большого количества мате-
риала, необходимого для проведения
эксперимента — обычно, 1 см1 образца
с концентрацией более 5-6 мг-см 3.
Для развития метода очень важным
было появление прибора, основанного
на другом принципе измерения, и на-
званного «дифференциальным» вис-
козиметром. На этом приборе можно
проводить измерения при более низких
концентрациях (около 1 мг-см 3) и в
малых объемах, что сразу повлекло его
широкое применение в исследованиях
биохимии белка. Однако, даже несмот-
ря на эти инновации, метод вязкости
до сих пор не имеет той популярности
в исследованиях структуры биологи-
ческих макромолекул, которую он имел
несколько десятилетий назад.
Г9.3. Измерение вязкости
Г9.3.1. Капиллярные вискозиметры
Капиллярный вискозиметр или вис-
козиметр Оствальда, наиболее широко
применяемый в исследованиях конфор-
мации биологических макромолекул яв-
ляется простым стеклянным прибором,
помещенным в термостатированные ус-
ловия (рис. Г9.1 с/). Он состоит из двух
сообщающихся отростков и функцио-
нирует следующим образом. Раствор
вводится в отросток 1 до тех пор, пока
уровень жидкости не достигнет положе-
ния риски С. Раствор всасывается через
отверстие 2, пока уровень жидкости чуть
превысит риску А. Затем измеряется
время t, за которое жидкость пройдет
расстояние между рисками А и В. По-
скольку скорость истечения жидкости
.зависит от разности высот столбов в двух
отростках вискозиметра, то разбавлять
раствор белка простым дополнением
растворителя непосредственно в отрос-
ток 1 вискозиметра не представляется
возможным. Для проведения измерений
при различных концентрациях целесо-
образно использовать вискозиметры,
называемые вискозиметрами Уббелоде
(рис. 1'9.1 б).
Вискозиметр Уббелоде функциони-
рует следующим образом. Раствор вво-
дится в отросток 1 до тех пор, пока жид-
кость не достигнет уровня С. Отверстие 3
закрывается и всасывание идет через от-
верстие 2, пока жидкость не превысит от-
метку Л. Затем отверстие 3 открывается и
определяется время Г, за которое жидкость
пройдет расстояние между рисками А и
В. Теперь скорость истечения жидкости
из объема Е не зависит от разности высот
столбов в отростках вискозиметра 1 и 2,
поэтому разбавлять раствор белка можно
простым дополнением растворителя не-
посредственно в отросток 1. Иногда вис-
козиметр Уббелоде называется вискози-
метром с «висячим» уровнем, поскольку
жидкость в процессе измерения течет по
стенкам нижней части отростка 2. Спра-
ведливости ради следует заметить, что в
случае отсутствия вискозиметра Уббе-
лоде, разбавление можно проводить и в
вискозиметре Оствальда, который в этом
случае требует достаточно трудоемкой
предварительной калибровки на разные
объемы жидкости.
Если вискозиметр Оствальда имеет
капилляр радиусом г и длиной L, через
который проходит объем жидкости Уза
время t, то вязкость q и среднее значе-
ние градиента скорости G в таком вис-
козиметре описываются уравнениями:
q = nhgpr't/8L V7, ( Г9.1 а)
Рис. Г9.1. Капиллярные вискозиметры: Ост-
вальда (а) и Уббелоде (б). Объяснение в тексте
Комментарий Г9.1. Кинематическая
. вязкость
: Относительная вязкость, без поправки
! на плотность, известна как «кинематиче-
ская» (в противоположность «динамиче-
ской») и равна цот]| = (t/t0). Полученные
из нее параметры: т)' , [q]' соответствуют
кинематическим величинам. В разумном
приближении при концентрации белка
< 1МГ-СМ’3 [Т|]’- [Т)].
G=8V73n?;ir, (Г9.16)
в которых h — среднее значение высоты
столба жидкости: g — сила тяжести, а
р — плотность жидкости. В точном вы-
числении констант h, г и L нет необхо-
димости, поскольку измерения норма-
лизуются на растворитель:
П.,,,, = Gp РЛо Ро)- <Г9-2)
где г и /() являются временем истече-
ния раствора концентрации С и рас-
творителя. соответственно, а р и р0
являются соответствующими плот-
ностями раствора и растворителя
( коммсн । арий Г9.1 ).
Вращающийся
внешний
цилиндр
Рис. Г9.2. Вискозиметр Куэтта: внешний ци-
линдр вращается, внутренний! неподвижен
Исследуемая
Термостатирующая рубашка
____Циркулирующая
------жидкость
Мениск
Ротор
Статор----
привода
1 см
Рис. Г9.3. Вискозиметр Зимма-Крозерса (см.
текст)
Г9.3.2. Ротационные вискозиметры
Вязкость раствора зависит от ориента-
ции в нем частиц. При больших гради-
ентах скорости, когда частицы ориен-
тированы параллельно линиям потока,
можно ожидать меньшего прироста вяз-
кости, чем при беспорядочной ориента-
ции частиц. Другими словами, частицы,
ориентированные параллельно в пото-
ке, демонстрируют неньютоновское по-
ведение, даже если растворитель нью-
тоновский.
Компактные недеформирующие-
ся молекулы (например, глобулярные
белки) всегда проявляют ньютонов-
ское поведение, и по этой причине нет
необходимости измерять их вязкость
при различных градиентах. Однако для
очень асимметричных молекул (таких
как коллаген), или очень деформиру-
ющихся (таких как ДНК), необходимы
измерения вязкости при разных гради-
ентах скорости с обязательной экстра-
поляцией данных к нулевому градиен-
ту скорости. Для этих целей наиболее
подходящими являются ротационные
вискозиметры.
Один из них, вискозиметр Куэтта,
предназначен для измерений при низ-
ких градиентах скорости. Он состоит
из двух концентрических цилиндров,
разделенных небольшим зазором, кото-
рый заполняется раствором (рис. Г9.2).
Один цилиндр фиксирован, другой
вращается. Очевидное преимущест-
во ротационных вискозиметров перед
капиллярными заключается в постоян-
стве градиента скорости в зазоре меж-
ду статором и ротором. Однако регис-
трация на приборах с вращающимися
цилиндрами значительно сложнее, чем
в капиллярных вискозиметрах.
Другой коаксиальный вискозиметр
Зимма-Крозерса удобен для работы
с водными растворами и не содержит
сложных механических конструкций
(рис. Г9.3). Он отличается принципи-
ально от вискозиметра Куэтта тем, что
в нем фиксировано напряжение сдвига
и измеряется его градиент, тогда как в
вискозиметре Куэтта фиксирован гра-
диент сдвига, а измеряется его напря-
женис ( м>мм‘и i;i|>nii Г!'.'!). Раствор
располагается между стационарным
внешним цилиндром и вращающим-
ся внутренним цилиндром, который
поддерживается в жидкости на плаву.
Уровень жидкости (мениск) совпадает
с верхним краем ротора. При таком за-
полнении благодаря поверхностному
натяжению обеспечивается автомати-
ческое центрирование ротора. Внут-
ренний цилиндр содержит стальной
стержень. Снаружи внешнего цилин-
дра находится вращающийся магнит,
который заставляет стержень и, сле-
довательно, внутренний цилиндр, вра-
щаться с постоянной угловой скоро-
стью to. Градиент скорости меняется
при изменении силы приложенного
магнитного поля. На этом вискозимет-
ре, как и на капиллярном, измеряется
относительная вязкость, определяемая
из отношения числа оборотов ротора
в течение какого-то фиксированно-
го времени в чистом растворителе и в
исследуемом растворе. Соответству-
ющие значения вязкости каких-либо
двух растворов могут быть вычислены
просто из равенства rjl/ri2 =
Как мы указывали, очень асим-
метричные молекулы (низкомоле-
кулярная ДНК; коллаген) или силь-
но деформирующиеся молекулы
(высокомолекулярная ДНК) имеют
тенденцию ориентироваться или де-
формироваться при высоком градиен-
те скорости. При этом в зависимости
[г]] от G для жестких асимметричных
частиц существенную роль играет на-
правленная ориентация их осей вдоль
потока. Действительно, при прибли-
жении продольной оси палочкообраз-
ной частицы к направлению потока
скорость, с которой окружающаяся
жидкость обтекает частицу, умень-
шается, что приводит к уменьшению
Коммон юрии Г9.2. Градиент сдвига
и напряжение сдвига
Мы определили вязкость ц (параграф
Г2.4.1) как силу F, которая необходима для
поддержания в жидкости градиента du/dx,
направленного по нормали к этой силе:
Г= т]Л (du/dx), (А)
где А — площадь поверхности, образован-
ной линиями тока. Сдвиговые усилия в
жидкости, развивающиеся внутри стан-
дартного капиллярного вискозиметра при
поддержании градиента скорости в потоке,
весьма значительны. Напряжение сдвига 5
определяют как силу, действующую на еди-
ницу площади в жидкости и возникающую
в результате течения последней. Скорость
сдвига — это градиент скорости, направлен-
ный по нормали к этой силе. Соотношение,
связывающее напряжение сдвига и ско-
рость сдвига: i
5= F/A = n(du/dx). (В)
В стандартном капиллярном вискози-
метре для водных растворов S составляет
-10 дин-см2. Гораздо меньшие значения S
можно получить в ротационных вискози-
метрах. Так в вискозиметре Зимма-Кро-
зерса можно проводить измерения при
значениях напряжения сдвига, не превы-
шающих 0,0006 дин-см 2.
потерь на трение и, следовательно,
и вязкости раствора. Таким образом,
уравнение Г2.40 должно быть перепи-
сано следующим образом:
[т|] = lim (т| - т|0)/т|0 С, (Г9.3)
С->0
G^0.
На рисунке Г9.4 приведены приме-
ры зависимости вязкости для ДНК из
бактерифага Т2 от величины напряже-
ния сдвига I ' ). Заметим,
что в случае использования больших
значений скорости сдвига может проис-
ходить даже разрыв молекул ДНК.
Рис. 19.1. Зависимость удельной вязкости от напряжения сдвига для ДНК из бактериофа-
га Т2, выраженная как доля соответствующего значения при пулевой величине напряжения
(Л )/(т] ) 0сдвига. Показана кривая для двух концентраций ДНК. Результаты измерений при
очень малых градиентах сдвига демонстрируются на вставке вверху. Видно, что вязкость прак-
тически перестает зависеть от напряжений сдвига при его значениях, меньших чем 0,001 дин см -.
Для исследованных препаратов ДНК найденная экспериментальная величина характеристиче-
ской вязкости оказалась равной 31 600 см'г 1 (Crother and Zimm, 1965)
Г9.3.3. Дифференциальный
балансный вискозиметр
В жидкостном дифференциальном ба-
лансном вискозиметре используется
тот же принцип, что и в хорошо изве-
стном электрическом мостике Уинсто-
на (рис. Г9.5 а). Дифференциальным
он называется так, поскольку в нем
напрямую измеряется относительная
вязкость, а балансным — поскольку
прикладываемое к одному из плечей
давление уравновешивает таковое, воз-
никающее из-за разности свойств рас-
твора и растворителя.
Прибор имеет высокую чувстви-
тельность, измерения выполняются
при низких концентрациях глобуляр-
ного белка (-1 мгмл-1). Когда рас-
творитель находится во всех четырех
капиллярах R(, R2, R3 и R4, то диффе-
ренциальное давление через мостик
равно нулю. Ситуация подобна отсут-
ствию тока в мостике Уинстона, ког-
да все четыре плеча в мостике имеют
одинаковые сопротивления. Когда же
раствор наполняет капилляры R , R., и
R3, то растворитель, поступающий из
накопительного резервуара, остается
в капилляре Rt. Различия в вязкости
между растворителем в R( и раствором
в R3 вызывают неравномерное распре-
деление давления ДР в мостике, кото-
рое связано с удельной вязкостью рас-
твора отношением:
= 4ДР/(Р-2ДР). (Г9.4)
Этот вискозиметр имеет высокую
скорость измерения. Кроме того, допол-
нительное количество раствора может
вводиться постепенно через проточную
кювету. Он также может быть совмещен
Рис. I 9.5: а — принцип работы дифференциального вискозиметра. Когда во всех четырех ка-
пиллярах вискозиметра Л,-/?, находится растворитель, дифференциальное давление через мост
равно нулю. Когда раствор молекул из колонки наполняет капилляры R2 и R3, в то время как
растворитель, поступающий из задерживающего (накопительного) резервуара (Резервуар В),
остается в капилляре Rf, то различие в вязкости раствора и растворителя вызывает неравномер-
ное распределение давления в мосте, которое пропорционально вязкости раствора образца. При
известном значении концентрации можно вычислить удельную вязкость раствора (Dutta et al.,
1991); б — принцип работы мостика Уинстона. В роли Rt~Rt выступают четыре омических сопро-
тивления, величина одного из которых (R}) подбирается таким, чтобы уравнять сопротивление
искомого (Я2). При равенстве этих сопротивлений ток в мостике будет равным нулю
в оперативном режиме с детектором
концентраций (базирующемся на изме-
рении показателя преломления или на
регистрации поглощения в ультрафио-
лете) для преобразования относитель-
ной вязкости в удельную вязкость.
Г9.3.4. Вязкоупругая релаксация
Если к ротору в ротационном виско-
зиметре, где находится раствор ДНК,
приложить внешний вращающий мо-
мент, то конфигурация цепей ДНК в
среднем изменится, перейдя от конфор-
мации статистического клубка к более
вытянутой. Энергия, которая затрачи-
вается на этот переход, обеспечивается
вращающим моментом, создаваемым
внешними силами и передается через
градиент скорости в жидкости. Внут-
ренняя энергия молекул ДНК при этом
увеличивается вследствие изменения
конфигурации цепей. Если убрать вне-
шний момент, то ротор не остановится
до тех пор, пока не произойдет релакса-
ция молекул ДНК обратно в состояние
статистического клубка. Те силы, кото-
рые возникли под действием исходно-
го вращающего момента, вызвавшего
вытягивание клубка, создают теперь в
роторе некоторый момент все то время,
пока происходит релаксация цепей. Ки-
нетика перехода статистического клуб-
ка, образованного цепями однородного
полимера из возмущенного состояния в
конечное невозмущенное равновесное
Рис. Г9.(>. Зависимость относительной вяз-
кости Г| для пяти глобулярных белков от
их концентрации С: 1 — гемоглобин челове-
ка; 2 — яичный альбумин; 3 — бычий сыворо-
точный альбумин; 4 — человеческий сыворо-
точный альбумин и 5 — у-глобулин человека
(Monkos and Turczynski, 1991)
состояние, представляет собой слож-
ный процесс. При низких градиентах
сдвига, при которых наблюдается не-
большая деформация цепи в непроте-
каемых гауссовых клубках, наибольшее
время релаксации т цепи равно:
т, = 0,423 (Г9.5)
RT
где [г| | — характеристическая вязкость
молекулы и г|0 — вязкость растворите-
ля. Для больших молекулярных масс
ДНК времена релаксации очень велики
' С'.'. । ;/>. г | 0.2 ).
Г9.3.5. Концентрационная
зависимость характеристической
вязкости
Вязкость частиц в растворе является
сложной функцией концентрации, по-
скольку при высоких значениях концен-
траций искажения скорости одной час-
тицы могут подействовать на скорости
соседних частиц. Ожидаемая вязкость
раствора выражается как степенной ряд
по отношению к вязкости чистого рас-
творителя:
П = По(1=^С + ^ +...). (Г9.5)
При таком разложении коэффи-
циент выражает вклад в суммарную
вязкость индивидуальных молекул,
k., — парный эффект взаимодействия
двух молекул, и т. д. На рисунке Г9.6
приведен график относительной вяз-
кости как функция концентрации
для различных глобулярных белков.
Из рисунка видно, что этот эффект об-
наруживается при концентрациях бел-
ка более чем 150 мг см 3.
Для разбавленных растворов спра-
ведливо уравнение Хаггинса:
Пуд/с = [П] + Х [р]2с, (Г9.6)
где г] — константа Хаггинса, которая
связана с конформацией макромолекул
в растворе.
Альтернативным уравнением для
концентрационной зависимости явля-
ется:
Ф,Л = [п](1+М’ (Г9-7>
где
vm,- <г9-8)
Концентрационная зависимость
k измеряется в тех же единицах
(мл • г1), что и эквивалентный пара-
метр скоростной седиментации, k, из
уравнения (А) комментария Г4.18, и
поступательной диффузии, kl} из урав-
нения (А) комментария Г3.6. Значе-
ния k и k связаны как:
Ч s
к V
(Г9.9)
где Vs/v — фактор набухания макро-
молекулы в растворе. Фактор набуха-
ния V /о, который есть не что иное как
отношение гидродинамического объема
к сухому объему частицы может быть
найден из отношения k /k. Заметим,
что эта процедура не требует знания
конформации частиц.
Г9.4. Вычисление
характеристической вязкости
белков из их атомных
координат
Несмотря на эмпирический характер
уравнения Г2.58, вычисления характе-
ристической вязкости через поляри-
зуемость дают хорошие практические
результаты для глобулярных белков.
B i;i6.nmc I S). 1 сравниваются значения
характеристической вязкости, пред-
сказанной по атомным координатам, с
экспериментально полученными. Эти
данные находятся в хорошем согласии,
если принять, что толщина гидратиро-
ванной оболочки (универсальной для
всех глобулярных белков) равна 0,9 А
(см. также i;i6. i. Г.3.2 для коэффициен-
тов диффузии).
Г9.5. Определение
конформации макромолекул
из характеристической
вязкости
Г9.5.1. Синтетические
полипептиды
Синтетические полипептиды являются
идеальной моделью для иллюстрации
высокой чувствительности вязкости к
конформационному состоянию макро-
молекулы. Еще в 1959 г. Доти с сотруд-
никами обнаружили, что синтетический
полипептид поли-у-бензилглутамат мо-
жет существовать в виде а-спирали По-
линга-Кори в растворителе со слабыми
водород-водородными связями.
В растворителях с сильными водо-
род-водородными взаимодействиями
внутримолекулярные водородные свя-
зи спиралей не так стабильны и кон-
формация полипептида соответствует
конформации беспорядочного клубка.
На рисунке Г9.7 представлена зависи-
мость характеристической вязкости
от молекулярной массы в двойной
логарифмической шкале для поли-у-
бензил-Ь-глутамата в двух типах рас-
творителей. В дихлоруксусной кисло-
те (открытые кружки) полипептиды
Таблица 19.1. Сравнение экспериментальных значений характеристической
вязкости с предсказанными по атомным координатам *)
Белок Код белка ЛГ(кДа) 19 ]„ред (СМЗ • Г‘ ) hUep. (CM3T-‘)
РНКаза А 7rsa 13,7 3,26 3,30
Лизоцим 61ys 14,3 2,99 2,98-3,00
Миоглобин 1 in bo 17,2 3,14 3,15
Химотрипсиноген 2cg 25,66 2,93 2,5-3,13
*) Характеристическая вязкость белков вычислена через электростатическую емкость. Толщина
гидратированной оболочки в этих вычислениях принята равной 0,9 A (Zhou, 1995)
Рис. 19.7. Зависимость характеристической
вязкости от молекулярной массы для поли-у-
бензил-Б-глутамата. Открытые кружки отно-
сятся к конформации беспорядочного клубка,
заполненные кружки к а-спиральной конфор-
мации (Doty et al., 1956)
находятся в форме гауссового клубка.
В этом растворителе [г|] = 2,78 10’3Л/°-87
и величина экспоненты соответствует
ожидаемой для гауссовых клубков в
«хороших» растворителях. В раство-
рителях, таких как диметилформамид
или хлороформ (закрытые кружки),
формируется структура, близкая к па-
лочкообразной. В этих растворителях
[г|] = 2,78 10"3Л/‘7. Показатель степени
1,7 полностью согласуется с теорети-
ческим значением в формуле Симха
для палочкообразных молекул (урав-
нение Г2.52).
Интересно, что две прямые на ри-
сунке Г9.7 пересекаются и характерис-
тическая вязкость в точке пересече-
ния (М равно около 70 кДа) остается
неизменной, несмотря на то, что клу-
бок трансформируется в спираль и на-
оборот. Справа от точки пересечения
прямых переход полипептида от спи-
ральной конформации к клубкообраз-
ной сопровождается уменьшением ха-
рактеристической вязкости, тогда как
такой же переход слева от точки пере-
Таблица Г9.2. Характеристическая вязкость некоторых белков и вирусов
в водных растворах’1
Белок М (Да) [г|] (см3 • г ‘)
Рибонуклеаза 13683 3,3
Лизоцим 14211 2,7
Миоглобин 17836 3,15
Химотрипсиноген 25660 2,5-3,15
Р-лактоглобулин 36800 3,4
БСА 66296 3,7
Гемоглобин 68000 3,6
Каталаза 250000 3,9
Вирус карликовой кустистости томата 10700000 3,4
Тропомиозин 70000 52
Фибриноген 330000 27
Коллаген 345000 1150
Миозин 490000 217
Вирус табачной мозаики 40000000 37
’* Значения [ц] для других белков читатель может найти в литературе (Harding, 1997)
сечения сопровождается увеличением
характеристической вязкости.
Г9.5.2. Белки в глобулярной
конформации
Экспериментальные значения характе-
ристической вязкости некоторых бел-
ков представлены в ।; н'1.11!; к Г9.2.Изнее
видно, что в соответствии с значениями
характеристической вязкости белки
можно разделить на две группы. Одна
группа характеризуется значениями [г]]
между 3,0 и 4,0 см3т ’. Это глобулярные
белки, форма которых близка к сфери-
ческой. Очевидно, что определение мо-
лекулярной массы глобулярных белков
по их характеристической вязкости не-
возможно.
Заметим, что сферический вирус
карликовой кустистости томата с моле-
кулярной массой 10,7 МДа имеет то же
значение характеристической вязкости,
что и рибонуклеаза с молекулярной мас-
сой 13,7 кДа. Это факт объясняется тем,
что объемная доля сферических частиц,
а не их радиус, определяет значение ха-
рактеристической вязкости (уравнение
Г2.48).
Вторая группа белков, в отличие от
первой, имеет гораздо большие значе-
ния [Г|].
В эту группу входят асимметричные
белки, форма которых далека от сфери-
ческой. Характеристическая вязкость
таких белков в первую очередь зависит
от их асимметрии.
Так, характеристическая вязкость
коллагена (палочкообразная молекула
с аксиальным отношением 100) имеет в
30 раз большее значение величины ха-
рактеристической вязкости, чем у виру-
са табачной мозаики (палочкообразная
молекула с аксиальным отношением,
равным 18).
log п (п - число остатков)
Рис. 19.8. Зависимость характеристической
вязкости от длины полипептидной цепи в
двойном логарифмическом масштабе в 6М гу-
нидингидрохлориде при 25 °C. Окисление ди-
сульфидных связей предотвращается добав-
лением р-меркаптоэтанолоа (0,15-0,20 мМ)
(Tanford, 1968)
Г9.5.3. Белки в клубкообразной
конформации
Полипептидная цепь, имеющая конфор-
мацию беспорядочного клубка (лишен-
ная вторичной и третичной структуры),
занимает гораздо больший объем, по
сравнению с тем, который она занима-
ет, находясь в конформации глобулы, и,
как следствие, в этом состоянии имеет
более высокое значение характеристи-
ческой вязкости. Данные по вязкости
[г|] денатурированных белков приведе-
ны в । 111! ic ГЗ.З.
На рисунке Г9.8 приведен график
зависимости [г|]М0 от п в двойной ло-
гарифмической шкале для 16 белков в
гуанидингидрохлориде, где Ма и п яв-
ляются значениями массы и числа ами-
нокислотных остатков белка, соответ-
ственно. На этом же рисунке показано
уравнение прямой линии, проведенной
через экспериментальные точки мето-
дом наименьших квадратов. Из него
Таблица Г9.3. Белки в конформации беспорядочного клубка в 6 М
гуанидингидрохлориде в присутствии 0,15мМ 0-меркаптоэтанола
Белок М (Да) мо (Да) п [П1 (см3-г~‘)
I 1нсулин 5 700 ИЗ 26 6.1
Рибонуклеаза 13 683 110 124 16,6
Лизоцим 14 211 111 129 17.1
Гемоглобин 15 500 108 144 18,9
Миоглобин 17 200 112 153 20,9
Р - л а кто гл обул ин 18 400 ИЗ 162 22,8
Химотрипсиноген 25 660 105 245 26,8
Фосфорибозил-трансфераза 35 000 109 320 31,9
Глицеральдегид-З-фосфат-дегидрогеназа 36 300 110 331 34,5
Тропомиозин 38 000 114 333 33,0
Альдолаза 40 000 107 376 35,5
Пепсиноген 40 000 108 370 31,5
БСА 66.296 114 605 114 605 52,2
Парамиозин 100 000 114 880 65,6
Тироглобулин 165 000 121 1364 82
Миозин 200 000 115 1739 92,6
видно, что величина экспоненты (0,666)
соответствует величине, ожидаемой для
клубков в «хорошем» растворителе.
Г9.5.4. Природно-
неструктурированные белки
До недавнего времени наши представ-
ления о структуре белков исходили из
того, что изначально свернутая уни-
кальным образом белковая третичная
структура необходима для их функ-
ционирования. Однако за последние
годы было найдено, что многие белки в
клетке не обладают такой структурой в
изолированном состоянии, хотя и име-
ют при этом четкую функцию при фи-
зиологических условиях. Такие белки
получили название белков с природной
или внутренней неупорядоченностью.
Доля неупорядоченных областей в та-
ких белках может быть разной, начиная
от последовательности из нескольких
аминокислот и заканчивая полностью
неупорядоченной последовательно-
стью длиной в десятки, а иногда и в
сотни аминокислот. Главное отличие
этих белков от структурированных
(глобулярных) белков состоит в том,
что они не имеют уникальной третич-
ной структуры в изолированном виде,
а приобретают ее после взаимодей-
ствии с партнерами. Их конформация в
комплексе определяется партнером по
взаимодействию, а не только собствен-
ной аминокислотной последовательно-
стью, как это характерно для структу-
рированных (глобулярных) белков(см
также секцию КЗ.5).
Убедительные доказательства су-
ществования таких белков были полу-
чены с помощью измерения вязкости
их растворов. В предыдущем парагра-
фе было показано, что все белки неза-
висимо от их исходной конформации
в растворе, будучи переведенными
а б
Рис. Г9.9: а — зависимость удельной вязкости от концентрации для рекомбинантного
фибронектин связывающего домена (rFNBD-A) в физиологическом буфере (О) и 6 М
гуанидингидрохлориде (•); б — то же для другого домена (rFNBD-B) (House-Pompeo
et al., 1996)
в универсальный растворитель (8М
мочевина или 6М гуанидингидро-
хлорид плюс 0,1 М меркаптоэтанол),
приобретают конформацию, близкую
к таковой гауссового клубка. Пото-
му сравнение величины характерис-
тической вязкости белка в нативных
физиологических условиях и в уни-
версальном растворителе дает прямой
ответ на вопрос о степени его разу-
порядоченности в растворе. Рисунок
1'9.9 показывает зависимость удель-
ной вязкости от концентрации для
двух белков в физиологическом буфе-
ре (О) и 6 М гуанидингидрохлориде
(•). Практически полное совпадение
прямых в двух растворителях строго
доказывает, что рассматриваемые бел-
ки могут быть отнесены к классу при-
родно неструктурированных белков.
В заключение заметим, что данные,
представленные на рисунке Г9.9, стро-
го говоря не означают, что рассмат-
риваемые белки не содержат некото-
рую долю флуктуирующей вторичной
структуры.
Г9.5.5. ДНК
В габлице 1’9.1 представлены значе-
ния вязкости пяти препаратов высо-
комолекулярных ДНК, выделенных
из фагов. Измерения проводились
при очень малых напряжениях сдви-
га с использованием вискозиметра
Зимма-Крозерса. На рисунке 1'9.10
показана зависимость характеристи-
ческой вязкости от молекулярной
массы в двойной логарифмической
шкале, построенная по данным этой
Таблица Г9.4. Характеристическая вязкость препаратов ДНК, выделенных
из некоторых бактериофагов
Источник М (МДа) [т|] (см3 • г ’)
Т2 115 31600
X 32 14000
1/4 Т2 27,2 11500
Т7 26,35 И 100
Т7 (узкая фракция) 3,7 2590
Рис. Г9.10. Зависимость характеристической вязкости |г|] от молекулярной массы ЛИК, выде-
ленной из бактериофагов, в двойной логарифмической шкале. Наклон прямой равен 0,73, что
соответствует таковому, ожидаемому для конформации набухшего гауссового клубка
Комментарий Г9.3. Взаимосвязь
, между экспонентами а и а для
молекул ДНК
Мы напоминаем, что для гомологичного
ряда молекул в конформации гауссового
клубка в «хорошем» растворителе справед-
ливы следующие уравнения КМХ (см. так-
же комментарий Г4.27):
[П] = Х[П|М°“, (А)
s = KA/04. (Б)
Экспериментальные значения экспонент
для ДНК в области молекулярных масс 4-
115 МДа очень близки к ожидаемым. Так, а
= 0,73 (см. рис. 1'9.9), а = 0,44 (см. рис. Г4.28).
Интересно, что в пределах эксперименталь-
ной ошибки зависимость между а и а описы-
вается уравнением (Е) в 1,<>мм('!1 lapiiii 11.27.
Это еще раз подтверждает плодотворность
i использования уравнений КМХ для анализа
^конформации биологических макромолекул.
таблицы. Из рисунка видно, что ДНК
с молекулярными массами в интервале
4-115 МДа описывается прямой с на-
клоном а = 0,73, который типичен для
гауссового клубка в «хорошем» раство-
рителе (комментарий Г9.3).
Г9.6. Конформационные
изменения в белках
и нуклеиновых кислотах
При денатурации глобулярных белков
их характеристическая вязкость увели-
чивается очень заметно. Масштаб этих
изменений иллюстрируется на рисун-
ке 1'9.11 а для белка (рибонуклеаза Н).
На рисунке Г9.11 б показано развора-
чивание нуклеиновой кислоты (рибо-
сомная 16S РНК).
Г9.7. Перечень ключевых идей
Поток жидкости, медленно движущийся вдоль капилляра или между двумя
параллельными плоскостями, называется ламинарным; в этом случае вязкость
а б
Рис. Г9.1 1: а — зависимость характеристической вязкости [ц] от температуры (температурная
денатурация белка) рибонуклеазы Н (pH 2) (Van Holde, 1985); б — зависимость удельной вяз-
кости [r|vJ от температуры (разворачивание 16S РНК) (Spirin, 1963)
жидкости т]0 не зависит от градиента скорости, и такая жидкость называется
ньютоновой.
* При достаточно высокой скорости исследуемый поток жидкости возмущается;
в этом случае вязкость жидкости т)0 зависит от градиента скорости; такая жид-
кость называется неньютоновой.
• Характеристическая вязкость [р] определяется как увеличение вязкости при
добавлении в него 1 г макромолекул; она имеет размерность см3 • г1.
• Очень асимметричные или деформирующиеся молекулы имеют тенденцию
к ориентации или деформации в градиенте скорости потока; для таких молекул
характеристическая вязкость должна экстраполироваться не только к нулевой
концентрации, но и к нулевому градиенту скорости.
• Удельная вязкость г] раствора сферических частиц пропорциональна их объ-
емной доле в растворе; г| не зависит от диаметра сферы.
• Характеристическая вязкость белков может быть предсказана с хорошей точ-
ностью по их атомным координатам при корректном учете вклада гидратации.
• Глобулярные белки и сферические вирусы образуют гомологичный ряд, пока-
затель степени в уравнении КМХ для такого ряда равен нулю. Определить мо-
лекулярную массу сферической частицы из измерений ее характеристической
вязкости невозможно.
• Различные белки в 6М гуанидингидрохлориде или 8М мочевине образуют го-
мологичный ряд, показатель степени которого в уравнении КМХ равен 0,67.
Такой показатель степени типичен для белков в конформации гауссова клубка
в «хорошем» растворителе.
® Характеристическая вязкость для гомологичных рядов палочкообразных час-
тиц пропорциональна их молекулярной массе в степени 1,7-1.8.
® Молекулы ДНК с большой молекулярной массой (М > 3 х 106Да) образуют
гомологичный ряд гауссовых клубков; показатель степени в уравнении КМХ
для такого ряда равен 0,73.
Литература для дальнейшего чтения
Исторический обзор и введение в биологические проблемы
Harding S. Е. The intrinsic viscosity of biological macromolecules. Progress in
measurement, interpretation and application to structure in dilute solution. Prog.
Biophys. Mol. Biol. 1997. 68, 207-262.
Сравнение теории и эксперимента
Zhou Н.-Х. Calculation of translational friction and intrinsic Viscosity. I. General
formulation for arbitrary shaped particles. BiophysicalJ., 1995. 69, 2286-2297.
ZhouH.-X. Calculation of translational friction and intrinsic Viscosity. II. Application
to globular proteins. BiophysicalJ., 1995. 69, 2298-2303.
Brenner H. Rheology of a dilute suspension of axisymmetric Brownian particles. Int.
I. Multiphase Flow, 1974.1, 195-341.
Allison S. A. Low Reynolds number transport properties of axisymmetric particles
employing stick and slip boundary conditions. Macromolecules, 1999. 32, 5304-5312.
Измерение вязкости
Cantor C. and Schimmel, P. Biophysical Chemistry. Part II Technique for the study of
Biological Structure and Function. San Francisco: W. H. Freeman and Co. 1980.
Dutta R. K., Hammons K., Willibey B. and Haney M. A. Analysis of protein denaturation
by high-performance continuous differential viscometry./ Cromatog. 1991.536, 113-121.
Конформация макромолекул, определяемая по их характеристической вяз-
кости
Цветков В. И, Эскин В. Е. и Френкель С. Я. Структура макромолекул в раство-
рах. М.: Наука, 1964.
Tanford С., Kawahara К. and Lapanije S. Proteins as a random coils. Intrinsic viscosity
and sedimentation coefficients in concentrated guanidine hydrochloride./ Amer. Chem.
Soc., 1967.89, 729-736.
RoweA.J. The concentration dependence of transport processes: a general description
applicable to the sedimentation, translation diffusion, and viscosity coefficients of
macromolecular solutes. Biopolymers, 1977. 16, 2595-2611.
Crothers D. M. and Zimm В. H. Viscosity and sedimentation of the DNA from
bacteriophages T2 and T7 and the relation to molecular weight./. Mol. Biol., 1965. 12,
527-536.
Doty P., Bradbury J. II. and Holtzer A. M. Polypeptides. IV. The molecular weight,
configruation and association of poly-y-glutamate in various solvents./. Am. Chem. Soc.,
1956. 78,947-954.
Природно-неструктурированные белки
Сердюк И. Н. Структурированные белки и белки с внутренней неупорядочен-
ностью. Молекулярная биология, 1997. 41, 1-17.
Конформационные изменения в белках и нуклеиновых кислотах
Yangi J. Т. The viscosity of macromolecules in relation to molecular conformation.
Adv. Protein. Chem., 1961.16, 323-400.
Tanford C. Protein denaturation. Adv. Protein Chem., 1968. 23, 121-282.
Van Holde K. Physical Biochemistry. Englewood Cliffs, NJ: Prentice-Hall., 1985.
Глава Г10
ДИНАМИЧЕСКОЕ РАССЕЯНИЕ СВЕТА
Г1О.1. Исторический обзор
1869
Дж. Тиндаль (J. Tyndall) выполнил
первые эксперименты по рассеянию све-
та аэрозолями. Он объяснил голубой цвет
неба присутствием пыли в атмосфере.
1871
Лорд Рэлей (Lord Rayleigh) создал
первую теорию рассеяния света ансамб-
лем невзаимодействующих частиц, име-
ющих малые размеры по сравнению с
длиной волны света. По Рэлею, среда рас-
сеивает свет благодаря колебаниям моле-
кул около положения равновесия. Он вы-
вел формулу, которая объясняла голубой
цвет неба преимущественным рассеяни-
ем молекулами атмосферы синих лучей
по сравнению с красными.
1906
Л. Мандельштам (L. Mandelshtam)
вновь поднял вопрос о природе рассея-
ния света. Он обратил внимание на
то, что аргументы Рэлея не полностью
объясняют феномен рассеяния света.
По Мандельштаму, рассеяние света бу-
дет происходить только тогда, когда в
объеме порядка А,3, где А, — длина волны
рассеянного света, в каждый данный мо-
мент времени будет содержаться разное
число молекул. Как следовало из рассуж-
дений Мандельштама, коэффициенты
поступательной н вращательной диффу-
зии макромолекул могут быть получены
из анализа спектров рассеяния света.
1924
Л. Мандельштам с коллегами раз-
вил теорию и практические основы ком-
бинационного рассеяния света, которое
позже в англоязычной литературе полу-
чило название рамановского рассеяния.
Он еще раз подтвердил, что коэффици-
ент поступательной диффузии макромо-
лекул может быть получен из спектров
рассеяния света. Однако отсутствие ко-
герентности и монохроматичности у тра-
диционных источников света, использу-
емых в то время, не давало возможности
провести такие эксперименты.
1914
Л. Бриллюен (L. Brillouin) предска-
зал дублет в спектре рассеянного света,
обусловленный рассеянием тепловых
звуковых волн в твердых телах. Этот
дублет позже был назван его именем.
В 1932 г. Е. Гросс (Е. Gross) провел се-
рию экспериментов по светорассеянию
жидкостями. Он обнаружил не только
дублет Бриллюена, но также и несме-
щенный центральный пик. В 1933 г.
Л. Ландау и Г. Плачек (L. Landau and
G. Placzek) дали теоретическое объяс-
нение присутствия центрального пика
с термодинамической точки зрения и
вычислили соотношение интегральной
интенсивности центральной линии и
таковой для дублета.
ИИ 7
Л. Горелик (L. Gorelik) выдвинул
идею спектроскопии оптического сме-
шения. Он назвал э тот метод — методом
демодуляционного анализа рассеянного
света. В 1955 г. А. Форрестер, Р. Гуд-
мунсен и П. Джонсон (A. Forrester,
R. Gudmunsenand Р. Johnson) реализо-
вали эту идею на практике. Они назва-
ли ее спектроскопией оптических само-
биений в противоположность обычной
спектроскопии. Однако в долазерную
эпоху отношение «сигнал- шум» в этих
опытах было плохим, и метод пе полу-
чил тогда широкого применения.
1964
Р. Пекора (R. Ресога) показал, что
частотный профиль рассеянного света
уширяется в результате поступательной
диффузии макромолекул. Он создал тео-
рию, согласно которой из полуширины
центрального пика на половине его вы-
соты можно вычислить коэффициент
поступательной диффузии. В том же
году Г. Каммингс, Н. Кнабль и Й. Йе
(Н. Cam mi ns, N. Knable and Y. Yeh)
использовали метод оптического смеше-
ния в опытах по рассеянию света на сус-
пензии сферического полистирольного
латекса. Они применили гелий-неоно-
вый (He-Ne) лазер в качестве источни-
ка света и технику, которая позже была
названа «гетеродинной» техникой оп-
тических биений. В 1967г. С. Б. Дубин,
Дж. Луначек и Г. Б. Бенедек (В. Dubin,
S. J. Lunacek and G. В. Benedek) опуб-
ликовали первые данные ио измерению
полуширин линий Рэлеевского рассея-
ния для ряда биологических макромо-
лекул. Эти пионерские теоретические и
экспериментальные работы обогатили
и разнообразили эту область исследова-
ний. В 1976г. Б. Дж. Берне и Р. Пекора
(В. J. Вете and R. Ресога) опублико-
вали свою знаменитую книгу «Динами-
ческое рассеяние света в приложении к
химии, биологии и физике».
1972
Б.Р. Вар и В. Г. Флайгар(В.В. Ware
and W. Н. Flygare) опубликовали ре-
зультаты исследований бычьего сыво-
роточного альбумина методом динами-
ческого рассеяния света в присутствии
статического электрического поля. Они
показали, что при приложении такого
поля молекулы начинают направлен-
но двигаться со скоростью, которая
пропорциональна электрофоретичес-
кой подвижности, сохраняя при этом
беспорядочное броуновское движении
заряженных частиц. Этот метод был на-
зван электрофоретическим рассеянием
света.
1976
С. Б. Дубин, Н. А Кларк
и Г. Б. Бенедек (S. В. Dubin and
N. A. Clark, G. В. Benedek) измерили
коэффициент вращательной диффузии
лизоцима, используя сферический ин-
терферометр Фабри-Перо. Комбини-
руя полученные данные с данными по
коэффициенту поступательной диффу-
зии, они пришли к выводу, что гидроди-
намические размеры молекулы лизоци-
ма в растворе очень близки к таковым
для лизоцима в кристалле. В 1991 г. Пе-
кора весьма успешно применил этот
подход к исследованию коротких оли-
гонуклеотидов ДНК.
1982
3. Кам и Р. Риглер (Z. Кат
and R. Rigler) использовали кросс-кор-
реляцию для выделения коэффициента
поступательной диффузии из других
динамических компонентов лазерного
рассеяния света. Они показали, что вра-
щательная диффузия асимметричных
частиц, конформационная подвижность
беспорядочных клубков, динамика про-
цессов ассоциации/диссоциации могут
быть определены методом кросс-корре-
ляции рассеяния света при измерении
интенсивности флуктуаций при раз-
личных углах рассеяния.
1997
Р. Бар-Зив (R. Bar-Ziv) с коллега-
ми предложили метод локализованного
динамического рассеяния света для ис-
следования динамических свойств оди-
ночных молекул в нанометровой шка-
ле. Позже этот метод был применен для
измерения констант биомолекулярных
сил и зондирования вязкостно-эластич-
ных свойств сложных сред.
2000 и но настоящее время
Метод динамического рассеяния
света используется для быстрого (не-
сколько минут) и точного (1-2%) из-
мерения в растворе коэффициентов по-
ступательной диффузии макромолекул
практически любых размеров. Для моле-
кул, размеры которых сравнимы с дли-
ной волны света, можно определять
также и коэффициенты вращательной
диффузии. Для малых глобулярных
белков и олигонуклеотидов, коэффи-
циенты вращательной диффузии кото-
рых очень велики, можно использовать
стандартную спектроскопию очень вы-
сокого разрешения. Метод динамиче-
ского рассеяния света незаменим также
для измерения скоростей направленно-
го движения макромолекул в линейных
и турбулентных потоках.
Г10.2. Введение
в биологические проблемы
В середине 1960-х гг. в молекулярной
биологии начал интенсивно использо-
ваться новый физический метод, полу-
чивший разные названия, но который
вслед за Р. Пекорой мы будем называть
методом динамического рассеяния све-
та (ДРС). Его успешное развитие в
последующие 15 лет показало, что ме-
тод добавил к классическому методу
некогерентного статического рассея-
ния света новую физическую размер-
ность — время. За эти годы были про-
ведены интенсивные исследования по
анализу динамики различных переход-
ных процессов, которые вносят вклад
во флуктуацию интенсивности рассе-
янного света. В процессе эксперимен-
тов было обнаружено влияние диффу-
зии на уширение спектров рассеянного
света и влияние направленного дви-
жения на сдвиг Допплера для широко-
го круга частиц различных размеров,
формы и гибкости. На сегодняшний
день метод ДРС — наиболее подходя-
щий инструмент для точных измере-
ний коэффициентов поступательной
диффузии макромолекул в растворе.
В последнее десятилетие появились
новые направления в методе ДРС, ко-
торые в основном связаны с переходом
от исследований свойств макромолекул
в растворе в больших объемах к иссле-
дованиям их индивидуальных свойств
в очень малых объемах. Появилась но-
вая техника, названная флуоресцент-
ной корреляционной спектроскопией
(ФКС), которая соединяет методы све-
товой и флуоресцентной микроскопии.
В этой главе мы опишем два типа экспе-
риментов по динамике рассеяния света.
В первом — интенсивность флуктуиру-
ет вследствие изменения ориентации и
положений молекул в пространстве объ-
ема порядка -V. Такие эксперименты
мы будем называть экспериментами по
ДРС в условиях гауссовой статистики
(большое число молекул). Во втором —
флуктуация интенсивности обуслов-
лена флуктуациями числа молекул в
микроскопическом объеме. Этот тип
экспериментов мы будем называть эк-
спериментами по динамическому рас-
сеянию света в условиях негауссовой
статистики (малое число молекул).
Метод флуоресцентной корреляци-
онной спектроскопии может рассматри-
ваться как дальнейшее развитие метода
динамического рассеяния света. Однако
между ними существует принципиальное
различие. Динамическое рассеяние света
по своей природе - когерентное явление,
тогда как флуоресцентная корреляци-
онная спектроскопия - некогерентное
явление. Как следствие этого, первое на-
блюдается в ансамбле молекул, тогда как
второе- только при небольшом числе
молекул. В идеале, метод флуоресцентной
корреляционной спектроскопии — это ме-
тод для исследования одиночных молекул
в микрообъемах. Его описание Когерент-
ного явлениябудет дано в Главе П1.
В настоящее время имеется доста-
точно много названий техники для
измерения спектрального состава рас-
сеянного света. Каждое из них подчер-
кивает ту или иную сторону метода,
концентрируясь или на способе детек-
тирования сигнала или на специфике
регистрируемых событий. Приведем
некоторые из них.
1. Динамическое рассеяние света:
это название является основополага-
ющим. Оно включает в себя все методы
исследования рассеяния света, которые
обеспечивают информацию о динамике
молекул. Противоположное ему — ста-
тическое рассеяние света или любого
другого типа излучения — дает информа-
цию о массе и геометрии рассеивающих
центров в молекуле (см. секцию Е2.2).
2. Квазиупругое рассеяние света:
это название обращает наше внимание
на то, что в процессе рассеяния возни-
кает новый спектр длин волн, у кото-
рого центральная частота такая же, как
у падающей световой волны, но ампли-
туда и фаза рассеянной волны модули-
рованы по частоте.
3. Фотонная корреляционная спек-
троскопия: это название фокусирует
наше внимание на том, что в этой экспе-
риментальной технике одиночные фото-
ны используются для подсчета автокор-
реляционной функции.
4. Оптическая спектроскопия сме-
шения: этот термин используется, чтобы
подчеркнуть очень высокую разреша-
ющую способность техники при опреде-
лении динамических процессов, которые
описывают подвижность макромолекул в
растворе. Имеется также и много других
названий: спектроскопия флуктуации
интенсивности рассеянного света, рэлеев-
ское уширение и т. д. Мы будем рассмат-
ривать все эти названия как синонимы.
В комментарии I I0.1 будет дана
краткая информация о взаимодействии
света с веществом, которая необходима
для дальнейшего обсуждения.
В заключение укажем, что иногда
употребляемое в литературе названиеля-
зерное светорассеяние никак не отражает
особенности рассматриваемого метода,
и мы его не рекомендуем для использо-
вания. Лазер как источник с успехом ис-
пользуется в экспериментах по анализу
статической (усредненной по времени)
составляющей рассеянного света.
Комментарии. ПО. 1. Свет и вещество
Свет является невозмещаемой субстанцией, которая может быт ь использована для получения
информации о структуре и динамике молекул. Уравнения Максвелла являются основой для опи-
сания всех электромагнитных явлений. Эти уравнения идентифицируют свет как поперечную
электромагнитную волну, которая колеблется как в пространстве, гак и во времени, т. е. направле-
ние колебания является перпендикулярным направлению распространения луча (см. рис. АЗ.7).
Способ взаимодействия света с материей зависит от электронной структуры вещества,
т. е. от его квантово-механических свойств. Если энергия, присущая фотону (/zv, где Л —кон-
станта Планка и v -- частота света), равна разнице между двумя энергетическим состояниями
в системе, тогда фотон может быть абсорбирован системой. Поскольку электрическое поле
совершает колебания, свет тоже искажает распределение зарядов в системе, изменяя их. так
что излучение принимает форму рассеянного света. Если обмен энергией между фотонами и
системой не происходит, тогда частота рассеянного света, v v, равна v(1, и процесс относится
к упругому рассеянию света. Если обмен между фотонами и системой происходит, тогда у|н
отличается от v0, и процесс относится к неупругому рассеянию света.
Ответ системы на внешнее электрическое поле называется поляризацией системы. Величи-
на поляризации зависит от амплитуды прилагаемого электрического поля и способности рас-
пределяемого заряда «деформироваться» внешними силами. Способность заряда деформи-
роваться носит название поляризуемости. Рассеяние света является результатом флуктуаций
поляризуемости среды. Если свет поляризован в направлении, перпендикулярном к плоскос-
ти, определяемой источником, рассеивающей кюветой и детектором, тогда процесс относится
к поляризованному рассеянию света и обозначается как lvv Деполяризованное рассеяние све-
та, проявляется в его горизонтальной компоненте и обозначается как 1V|). Деполяризованный
свет можно представить себе как комбинацию различных поляризованных лучей с беспоря-
дочной ориентацией вокруг оси нх распространения (см также главу 35).
Г10.3. Динамическое
рассеяние света
как спектроскопия
очень высокого разрешения
В соответствии с фундаментальными за-
конами статистической физики система
из многих частиц никогда не проявляет
полного однообразия. Описывающие
систему термодинамические параметры
постоянно флуктуируют относительно
своих наиболее вероятных значений.
Поскольку эти флуктуации происходят
очень медленно, по сравнению с часто-
той световой волны, то для регистрации
таких флуктуаций требуется спектро-
метры с очень высокой разрешающей
способностью. Требуемое разрешение
столь велико, что традиционная опти-
ческая спектроскопия не может решать
такого рода задачи и приходится ис-
пользовать технику спектроскопии оп-
тического смешения.
I Комментарий Г10.2. Разрешающая
I способность метода ДРС
' На характерных углах рассеяния (-90")
ожидаемая расчетная ширина спектраль-
; ной липни (на половине максимальной!
| высоты), обусловленная флуктуациями
концентрации, которые в свою очередь
возникают благодаря трансляционной :
, диффузии макромолекул растворенио- i
I го вещества, составляет величину около ,
[ 1000 Гц для белка с молекулярной мас-
' сой 50 кДа. Поскольку сам падающий
(видимый) свет имеет частоту около
। ,5x10” Гц, то прямое определение формы |
| спектральной линии рассеянного света
требует разрешающей силы, больше чем '
j 10”/10:!= 10".
Лазерный луч, проходящий в среде
рассеяния, может рассматриваться как
«несущая волна», колеблющаяся с опти-
ческой частотой 5 х 10й Гц (рис. 1'10.1 а).
Процесс рассеяния создает новую волну
рассеяния, основная частота которой сов-
падает с таковой для падающей волны, но
амплитуда и фаза рассеянной волны мо-
дулируются синхронно флуктуациями
среды. Такой модулированный сигнал
показан на рисунке ПО. 1 б). Его ушире-
ние для большинства биологических мак-
ромолекул очень мало (см. ниже). Столь
небольшое уширение вызывает необхо-
димость использования лазера в качес-
тве источника света. Очевидно, что для
получения информации, содержащейся в
модуляционном сигнале, его необходимо
демодулировать. Необходимость такой
демодуляции предопределила одно из
первоначальных названий метода — де-
модуляционный анализ рассеянного
света. Очевидно, что при использовании
обычного немонохроматического источ-
ника ширина его линии будет намного
больше полезного сигнала и демодуляция
последнего станет практически нерешае-
мой задачей (к< >м хн-ит api i ii Г10.2).
Демодуляция быстрых процес-
сов, протекающих в шкале времен
10-11-101() сек. осуществляется спект-
рометрами с дифракционной решеткой;
для исследования процессов, протека-
| Комментарий ПО.3. Оптическое 1
। и радиочастотное детектирование
I И лазер, и спектрометры оптического
: смешения можно рассматривать как рас-
ширение диапазона устройств и методов,
общепринятых в радиочастотных линиях ,
связи. Для того, чтобы понять, как можно
разрешать узкие спектральные линии в 1
оптическом диапазоне, мы должны веном- i
1 нить, что сходные проблемы возникают в
; радиочастотном диапазоне. В последнем
| случае решение проблемы сводится к при-
i менению «гомодинированных» и «гетеро-
. динированных» приемников. В этих уст- |
ройствах основная частота несущей волны
сдвигается в область более низких частот
с помощью нелинейного смещения. Затем
на этой низкой частоте спектр анализиру- i
ется с помощью узкополосных фильтров. I
Таким образом, тогда как оптический спек- ;
трометр фильтрует на оптической частоте, i
в радиочастотных устройствах фильтра- I
ция осуществляется только после того,
как несущая частота с помощью смесителя
сдвинута до подходящей низкой частоты. ’
' Поэтому спектроскопию оптического сме- '
! шения можно рассматривать как прило-
। жение радиочастотной техники к области •
оптической частоты, где смешивающим :
элементом является фотоумножитель. !
ющих в шкале времени 10 10-10-7 сек,
используются интерферометр Фабри-
Перо. (коммен гарпй Г10.1) Такие прибо-
ры действуют как оптические фильтры
и, следовательно, не могут быть исполь-
Рнс. Г10.1: а — сигнал одномодового лазера до рассеяния с частотой <о0; б — лазерный сигнал
после рассеяния на флуктуациях среды располагается на несущей частоте со0; в — де.модулиро-
ванный сигнал располагается в радиочастотном диапазоне со — со0
зованы для разрешения спектральных
компонентов уже, чем -10 МГц (мень-
ше чем 10'7сек).
Динамические процессы, описыва-
ющие подвижность биологических мак-
ромолекул в растворе, относятся к мед-
ленным процессам, имеющим ширину
спектральной линии уже, чем 10 Гц, что
на шесть порядков меньше, регистриру-
емых оптически, поэтому возникает не-
обходимость в технике с очень высоким
разрешением. Такая техника называется
спектроскопией оптического смешения
( коммун Kipnii Г10.З).
Г10.3.1. Флуктуации и временные
корреляционные функции
Для того, чтобы получить информацию
о динамике флуктуирующих систем, мы
должны выбрать один из двух различных,
но близких по физическому смыслу под-
ходов. Мы можем анализировать флук-
туирующие системы на основе частотных
компонентов с разными амплитудами,
получая спектр частот флуктуаций со
свойствами А, <Д/12 (v)>, где v — это час-
тота. Однако имеется другой, более точ-
ный метод, в котором измеряется непос-
редственно временная корреляционная
функция флуктуаций <А (Г), A (t + т)>,
где т — корреляционное время, характе-
ризующее флуктуации в системе.
В этой главе мы будем обсуждать
только эксперименты с использованием
техники временных корреляционных
функций, поскольку она обеспечива-
ет более высокую точность по сравне-
нию с техникой спектрального анализа.
Для визуализации результатов мы будем
иногда пользоваться спектром частот.
Корреляционные и кросс-
корреляционые функции
Флуктуирующий сигнал A (t), изоб-
раженный на рисунке Г10.2 а, облада-
ет следующими свойствами: свойство
А для времени t всегда отлично от свойс-
тва А для времени г+Дд Когда время
Рис. I 10.2: а — изображение некого свойства A (t), флуктуирующего во времени относительно
своего среднего <А>, равного нулю. Временная ось разбивается на ряд дискретных интервалов
ДЦ б —корреляционная функция <Л (0) А (т)>, описывающая флуктуационный процесс, изоб-
раженный в а. Начальное значение функции равно <А2>. Для времен значительно больших, чем
корреляционное время процесса т v корреляционная функция равна <А>2
т мало по сравнению с типичным вре-
менем флуктуаций переменной A (t),
A (t + т) будет близко к величине A (t).
Когда величина т увеличивается, откло-
нение A (t + т) от A (t) становится более
вероятным. Таким образом, можно ска-
зать, что величина A (t + т) коррелирует
с A (t) когда т мало, но эта корреляция
теряется, когда т становится большим
по сравнению с характерным периодом
флуктуаций. Мерой такой корреляции
является автокорреляционная функция,
определяемая как:
<A(0)A(t)> =
= Нт|р(г)А(Г + т)«/г. (Г10Л)
Если ось времени t разбить, как это
сделано на рисунке 10.2 а на дискрет-
ные интервалы At, так что t = j kt, т = п
kt, Т =Nkt, a t+ т = (j + n)kt, = kt, то ин-
тегрирование в уравнении Г10.1 можно
заменить суммированием:
<А>=Пт1Ул., (Г10.2а)
< Л(0)Л(г) limit л,.л,.+п, (ГЮ.26)
где Л. есть значение свойства в начале
у-го интервала. Уравнение ГЮ.26 при-
ближается к уравнению ПОЛ, по мере
того как At->0.
Мы ввели дискретизацию в описа-
ние флуктуационного процесса, с тем
чтобы продемонстрировать, как корре-
ляционная функция меняется со вре-
менем. Из рисунка ГК).2 а видно, что
некоторые члены в суммах уравнений
Г10.2а и ГЮ.26 отрицательны, напри-
мер А3 и А4. Следовательно, в этих сум-
мах часть вкладов будет положительна,
а часть отрицательна. С другой сторо-
ны величина <Л(0) Л(0)> всегда поло-
жительна, так как Л^> 0, и ожидается
большой. Таким образом, мы можем
ожидать, что корреляционная функция
будет постоянной, равной начально-
му значению для всего отрезка времен
или уменьшаться от максимального
значения <Л2> до минимального <Л>2
(рис. ПО.2 б).
Отметим, что в большинстве прак-
тических применений корреляционная
функция падает моноэкспоненциально:
<Л(0)Л(т)> = <Л>2 +
( л2 л 2} t (ГЮ.З)
+{<Л >-<Л > }ехр----,
Чор
где т есть корреляционное или релакса-
ционное время флуктуирующего сигнала.
Корреляционная функция двух
свойств А и В описывается кросс-корре-
ляционными функциями:
<А(0)В(т)> =
1г, , (ГЮ.4а)
= lim~ jA(t)B(t + i)dt
и
<В(0)А(т)> =
1 г , (Г10.46)
= lim-jB(t)A(t + T)</t?
0
Корреляторы
В лабораторных условиях превра-
щение флуктуаций сигнала в корре-
ляционную функцию выполняется
с помощью прибора, называемого
коррелятором (рис. ГЮ.З). Флукту-
ирующий сигнал в цифровом виде
передается по двум каналам: накопи-
тельному и общему каналу коррелято-
ра. Накопитель считает импульсы за
определенный промежуток времени,
kt, длительность которого задается
тактовыми кристаллическими часа-
ми. В конце этого промежутка содер-
жимое переносится в первый регистр
Рис. I 10.3. Схема коррелятора для анализа
флуктуирующих процессов (см. текст)
Рис. I 10.4. Типичная геометрия рассеяния
света. Ео — амплитуда падающего поля; к0 и
к] — падающий и рассеивающий волновые
векторы; q = к0 - к, — волновой вектор рас-
сеивания, определяемый углом рассеяния 0 и
длиной волны падающего света X; п — показа-
тель преломления среды
коррелятора. Затем коррелятор начи-
нает считать импульсы для второго
идентичного временного интервала.
Этот процесс продолжается в течение
всего эксперимента, так что регистры
коррелятора содержат информацию
о флуктуирующем сигнале за проме-
жуток времени nxAt, где п — число
регистров коррелятора, a At называ-
ется временем выборки или временем
сэмплирования, которое выбирается,
как правило, в интервале времени от
2 нсек до 1сек. Время выборки должно
соответствовать типичным корреля-
ционным временам системы. Очевид-
но, что для быстрых процессов время
выборки берется малым, а для медлен-
ных процессов — большим.
Корреляционные функции поля
и интенсивности
Для единичной частицы наведенный
диполь р, который является источни-
ком рассеяния, определяется падающим
полем Е и поляризуемостью частицы а:
р = а Е. (Г10.5)
Свет, излучаемый всеми индивиду-
ально индуцируемыми диполями, соби-
рается на детекторе, где результирующее
электрическое поле зависит от относи-
тельной позиции и ориентации всех час-
тиц в рассеивающем объеме. Типичная
геометрическая картина рассеяния све-
та представлена на рисунке П0.4. Кор-
реляционная функция рассеивающего
электрического поля, G (1>(т), называе-
мая корреляционной функцией первого
порядка, определяется, как:
G <'>(т) = <Е (Г) Е (t +т)>. (Г10.6)
На практике в силу квадратичной
природы детектирования измеряется
корреляционная функция флуктуаций
интенсивности. Для интенсивности I, ко-
торая пропорциональна квадрату вектора
электрического ноля <Е2>, нормализо-
ванная корреляционная функция, G(2)(t),
называемая корреляционной функцией
второго порядка, вычисляется как:
G (2)(т) = <1 (t) I (t + т)>. (Г10.7)
Для систем, содержащих большое
количество независимых источников
рассеяния света (гауссово приближе-
ние), связь между G(1,(t) и G(2)(t) зада-
ется соотношением Зигерта:
G(2>(t) = 1 + | G (т) |2. (Г10.8)
Корреляционные функции и частотный
спектр флуктуаций
Как было упомянуто выше, частотный
спектр I (v) и корреляционная функция
G (т) полностью эквивалентны при опи-
сании динамического поведения равно-
весных систем, поскольку они связаны
фурье-преобразовапием:
/(v) = (27t)-1 jG(r)exp х
х (-2mvt)dT, (Г10.9)
где v — частота в Гц. Так, если I (v) име-
ет форму Лорентца на нулевой частоте,
тогда G (т) имеет форму спадающей эк-
споненциальной функции с константой
времени, равной обратной величине по-
луширины линии Лорентца. Эти про-
стые функции верны для описания всех
элементарных стохастических процес-
сов и применимы для интерпретации
повеления большинства систем, кото-
рые будут обсуждаться в дальнейшем.
Простые примеры связи между кор-
реляционной функцией и частотным
спектром приведены на рисунке ПО.5.
Г10.3.2. Измерение динамической
составляющей рассеянного света
На рисунке Г10.6 приведены несколько
схем, иллюстрирующих три различных
подхода, используемых в эксперимен-
тах по динамическому рассеянию света:
(а) метол фильтра, (б) метод гомодини-
рования и (в) метод гетеродинирования.
Ниже мы кратко опишем каждый из них.
Техника фильтра
Классическая техника фильтра использу-
ется для демодуляции быстрыхпроцессов
(10_||-10"7сек). В интервале 10_1|-10_9сек
применяется дифракционная решетка,
для процессов в шкале времени 10~10-10 7
сек — интерферометр Фабри-Перо (ком-
ментарий Г10.1). Последний может быть
использован для измерения подвижно-
сти небольших глобулярных белков, пос-
кольку их вращательные времена лежат
в интервале 10 7-10“6 сек (см. гш'л. [’2.2).
Однако большинство биологических
макромолекул имеет корреляционные
времена, лежащие в интервале от 10"6 до 1
сек (см. ту же табл.) и, следовательно, тех-
ника фильтрации для них не применима.
Техника гомодинирования
и гетеродинирования
В методе динамического рассеяния
света используются две техники — го-
модинирования и гетеродинирования,
v = (2лт)1
а
5 - функция
при v = 0
Рис. I 10.5. Корреляционные функции и час-
тотный спектр для трех простейших моделей
флуктуационных процессов: синусоидальная
функция (л): постоянная (б); простейший сто-
хастический процесс (в)
Интерферометр Фотоумножитель Регистратор
Рассеянный।---------1 j 1 j——п
свет 1 1 1 1 1 1
Фотоумножитель Автокоррелятор Регистратор
Рассеянный i—— .
6 ---------4ZZ1--------------[=1-------------4=1
•AS»,
Xjb Фотоумножитель Автокоррелятор Регистратор
в ----------------HZZZJ------------4ZZ1-------------4ZZI
Рис. Г10.6. Схемы различных видов техники эксперимента: фильтр (л); гомодин (б) и гетеро-
дин (в)
каждая из которых имеет свои досто-
инства и недостатки. В большинстве
приложений, обсуждаемых ниже, ис-
пользуется гомодинная техника (тех-
ника самобиений), когда только рас-
сеянный свет коррелирует сам с собой
(рис. ПО.4 б). Поскольку любой фо-
топриемник не реагирует на частоты,
большие чем 1010 Гц, то он будет регис-
трировать только частоты колебаний,
обусловленные диффузией макромо-
лекул в растворе.
В другом экспериментальном вари-
анте используется техника оптического
гетеродинирования, при котором на фо-
токатоде смешиваются поле рассеянного
света и поле гетеродина (опорного пуч-
ка) (рис. Г10.4 в). В качестве последне-
го может служить часть первоначально
падающего лазерного луча. Смешение
Комментарий Г10.4. Интерферометр Фабри-Перо
Интерферометр Фабри-Перо состоит из двух плоскостей зеркального диэлектрика, выстро-
енных параллельно. Внутренние поверхности зеркал обладают высокой отражающей способ-
ностью, близкой к 99%. В экспериментах по рассеянию, свет обычно проходит через нормаль
интерферометра к зеркалам, затем отражается обратно и попадает в камеру интерферометра.
Волна, имеющая длину X, сохранится внутри интерферометра, если целое число половин длины
волны будет соответствовать размеру камеры, т. е. т (X/2) = d, где т — целое число nd — рас-
стояние между зеркалами. При таком соотношении между т, X и d будет иметь место максимум
интенсивности передаваемого света. Если d = 0,1 см и X = 5000 А, тогда т = 2 xlO3. Различия
между последующими длинами волн X, которые могут проходить через интерферометр, для
данного значения d, ДХ составляют около 2,5 А. Соответствующий частотный интервал равен
Av - 3 х 1011 Гц. Таким образом, в соответствии с выбранным d, интерферометр выбирает только
одну частотную компоненту (длину волны) из спектра рассеяния света. Для получения особо
высокого разрешения используется сферическое зеркало вместо плоского, как описано выше.
Сферические зеркала повышают светособирающую способность и дают возможность легко ска-
нировать спектр.
частот опорного и рассеивающего пуч-
ков приводит к двум важным следстви-
ям. Во-первых, максимум спектра сдви-
гается до значения разностной частоты
v0 — v’, которая теперь является легко де-
тектируемой радиочастотой. Во-вторых,
получаемая таким образом корреляци-
онная функция является корреляцион-
ной функцией поля, которая несет в себе
информацию не только об амплитуде,
но и о фазе сигнала. Недостаток техни-
ки гетеродинирования состоит в необ-
ходимости тщательного согласования
волновых фронтов рассеянного света и
опорного пучка, что само по себе являет-
ся нетривиальной задачей.
Фотонная корреляционная
спектроскопия
Техника фотонной корреляционной
спектроскопии используется для регис-
трации медленных процессов, чьи ха-
рактерные времена лежат в интервале
от 10 6 до 1 сек. В этой технике фильтр
не используется, и свет попадает непо-
средственно на фотодетектор. Аппара-
тура, необходимая для получения спек-
тров ДРС, хотя и не является сложной,
но требует выполнения некоторых ус-
ловий для получения оптимального со-
отношения сигнал-шум и сохранения
когерентности сбора рассеянного света
( ко.ммеп ।арий 1 11)..-)).
Схема типичной эксперименталь-
ной установки показана на рисун-
ке Г10.7. Линза £] фокусирует лазерный
луч в центре рассеивающей ячейки.
Апертурная линза!., и фотоумножитель
располагаются на плече гониометра,
вращающегося вокруг оси, проходящей
через центр ячейки. Положение плеча
определяет угол рассеяния 20. Располо-
жение элементов на плече отрегулиро-
вано таким образом, чтобы изображение
Коммен гарий Г10.5. Условия для
когерентного сбора света при
динамическом рассеянии света
При построении спектрометра по иссле-
дованию динамического рассеяния света
одним из принципиальных вопросов явля-
ется вопрос о когерентности собираемого
рассеянного света при заданном разреше-
нии прибора, поскольку он определяет пре-
дельную чувствительность метода. Сфор-
мулированные на сегодня критерии такого
сбора сводятся к нескольким простым пра-
вилам для угла сбора и размера фотокато-
да оптического спектрометра. На практике
эти критерии сводятся к выбору размеров
двух точечных диафрагм при заданном
расстоянии между рассеивающим объемом
рассеивающего объема фокусировалось
на точечной диафрагме. Сигнал рассе-
яния анализируется с помощью корре-
лятора электронной обработки сигнала.
Рисунок ПО.8 показывает типичный
ход корреляционной функции G(2)(t),
которая всегда состоит из двух частей:
зависящей и не зависящей от времени.
Не зависящая от времени часть, под-
ставка под корреляционной функцией,
определяется шумами прибора, стати-
ческим рэлеевским рассеянием и про-
порциональна числу частиц в рассеива-
ющем объеме.
Г10.3.3. Коэффициенты диффузии,
вычисляемые из динамической
составляющей рассеяния света
Как мы указывали ранее (Глава ГЗ).
существует много различных экспери-
ментальных методов для определения
коэффициентов диффузии макромоле-
кул, одним из которых и является метод
ДРС. Коэффициент диффузии, опреде-
ляемый этим методом, носит название
коэффициента взаимной диффузии.
Регистратор Коррелятор
Рис. I I 0.7. Схема спектрометра по корреляции фотонов, используемого в режиме самобиений.
Оптимальный сигнал получается тогда, когда точечная диафрагма имеет тот же радиус, что и
лазерный луч. Типичное значение расстояния, /. , между рассеивающим объемом и щелью рав-
но около 1м, размер щели, d, перед ФЭУ, равен около 0,1-0,2 мм. Для сохранения когерентно-
сти сбора точечная диафрагма перед ФЭУ должна иметь как можно меньший размер. Однако
ее очень сильное уменьшение приводит к падению регистрируемой интенсивности
D . Этим подчеркивается, что такой
коэффициент определяется из рассея-
ния в ансамбле частиц, взаимное движе-
ние которых приводит к биению между
гармониками излучения, исходящего из
разных частиц. Поэтому наиболее точ-
ные результаты по определению Р
получаются для разбавленных и полу-
разбавленных растворов макромолекул
(высокое отношение сигнал/шум). При
т (отн. ед.)
Рис. ПО.8. Типичный вид корреляционной
функции в эксперименте по динамическому
рассеянию света
уменьшении числа молекул в растворе
точность определения £>втаим падает и, в
пределе одной частицы, его определе-
ние становится принципиально невоз-
можным (см. ниже).
При высоких концентрациях рас-
творенных молекул можно ожидать из-
менений в корреляционной функции,
отражающих два различных эффек-
та. Во-первых, изменения константы
диффузии, обусловленные термоди-
намическими и гидродинамическими
причинами (см. комментарий Г.3.6). Во-
вторых, изменения, обусловленные
возможной ассоциацией растворен-
ных молекул. Корреляционная функ-
ция для последнего случая перестает
описываться одной экспонентой, пос-
кольку в нее вносятся вклады, связан-
ные с диффузией мономеров, димеров
и больших агрегатов.
Наличие в растворе частиц разного
размера приводит к задаче о вычислении
распределения частиц по размерам из
сложной многоэкспоненциальной кор-
реляционной функции. Подобная задача
нам уже встречалась в параграфе Г4.5.2,
посвященном анализу гетерогенных сис-
тем методом скоростного аналитического
центрифугирования. Как и ранее, реше-
ние такой задачи в методе ДРС приводит
к проблеме решения интегрального урав-
нения Фредгольма первого рода:
С(,’(<7,т) = ]с(Г)ехр(-СО«/Г, (Г10.10)
О
где G(l)(<7, t) — нормализованная вре-
менная автокорреляционная функция
электрического поля первого поряд-
ка, G (Г) — функция распределения по
частотам, которая должна быть опреде-
лена из экспериментальной функции
G(1>(c/, t) (комментарий Г1().(>). В том
случае, если экспериментальная функ-
ция G(1)(<7, t) описывается одной экспо-
нентой, то решение уравнения Г10.6 не
представляет труда и может быть прове-
дено стандартным метолом наименьших
квадратов. Однако анализ мультиэкс-
поненциального сигнала представляет
большие трудности и становится вообще
невозможным, когда константы времени
отличаются на фактор 2 или меньше.
Г10.4. Динамическое
рассеяние света в условиях
гауссовой статистики
При интерпретации G (1)(£) или
G(2\t) делается несколько допуще-
ний. Предполагается, что движение
молекулы как целого и внутренние
движения в ней (если последние при-
сутствуют) не зависят друг от друга,
растворы существенно разбавлены, а
все виды релаксаций, включая посту-
пательное и вращательное движение,
имеют экспоненциальный спад. При
таких упрощениях для корреляцион-
Комментарий Г10.6. Интегральное
уравнение Фредгольма первого
рода
Эксперименты, в которых используется
: метод ДРС, служат примером физических
j экспериментов, в которых функция-след- :
| ствие измеряется с целью определения
: функции-причины. Для решения такого
класса задач используется математическая
, модель, основанная на использовании ин-
| тетрального уравнения Фредгольма пер-
| вого рода. Уравнение Г10.6 устанавли-
! вает связь между функцией-следствием
(известной корреляционной функцией
G^'\q, f)) и функцией-причиной (неизвес-
тное распределение экспонент G(T) в ней).
, Общеизвестно, что решение уравнения
; ПО.6 неустойчиво в отношении небольших
I изменений в G11 (д, t) и, строго говоря, не
; существует, поскольку измерения функции
Gm(q, t) проводятся в реальных условиях
шума. Эти задачи относятся к классу «не-
корректно поставленных» задач. Для пере-
вода таких задач в корректно поставленные
используются различные подходы, полу-
чившие название регуляризационных.
ной функции первого порядка спра-
ведливо выражение:
G(1)(<7,t) = zlexp(-£>,t7?t)[l +
+ ХВ,ехр(-Г/т7)][1+ (Г10.11)
j
+Сехр(-г/ткор)].
Первый экспоненциальный член
в этом выражении описывает поступа-
тельную диффузию молекул с коэффици-
ентом диффузии £>т, второй ~ вращение
вокругу’-й оси молекулы (В есть соответс-
твующие компоненты анизтропии поля-
ризуемости). Последний член описывает
спад корреляционной функции, обуслов-
ленный внутренней релаксацией частиц с
характерным корреляционным временем
т . Константы /1, В и С суть амплитуды
соответствующих флуктуаций.
коммен। арий Г 10.7. Быстрота
спада корреляционной функции для
больших и малых частиц i
Напомним, что в соответствии с урав-
нением Эйнштейна-Стокса (уравнение
Г3.24) D. =^7'/(6лт]()7?()). Отсюда следует,
что для водных растворов коэффициент по- |
стунательной диффузии Z) при комнатной
температуре Dt~‘2 х 1О |3//?о. Для видимой
области, X - 5000 А, и для угла рассеяния 0
- 90', абсолютная величина вектора рассея- '
ния q = (4лтг/Х) sin (0/2) (секция Г10.3) рав-
на около 105 см-1. Отсюда следует, что q!D,
- 10-3/7?0, а корреляционное время т про-
порционально -7?()х Ю3, так что измеренная :
корреляционная функция будет падать
более медленно для больших частиц. Так,
для сфер радиусом 0,01 мкм, т - 10-3 сек.
Предварительная оценка этого времени
полезна для правильного выбора времени ।
выборки коррелятора.
Из формул Г10.2 и Г10.3 видно, что
по спаду корреляционной функции
G(1)(?) можно вычислить коэффициент
поступательной диффузии, а по спаду
функции G его удвоенное значение
( KOMMl'II l.ipin'i 1'1 ().; ).
Когда система состоит из смеси N
частиц с коэффициентами диффузии £).,
G(1) (?) и G(2) (?) становятся суммой спа-
дов экспонент с параметрами q2D, т. е.
G(1)(t) = ехр(-£)//2т), (ПО. 14)
1=1
(7(2>(т) = constanta +
* , (Г10.15)
+2/(ехр(-О/п).
1=1
Г10.4.2. Жесткие частицы,
размеры которых сравнимы
с длиной волны света
Фундаментальной проблемой в ана-
лизе данных ДРС для сложных сис-
тем является разложение уравнения
(Г10.11) на составные части, т. е. нахож-
дение вкладов индивидуальных компо-
нентов по измеренным G(1)(?) или G(2,(?).
Эта проблема будет обсуждаться ниже.
Г10.4.1. Частицы, размеры которых
малы по сравнению с длиной
волны света
В качестве первого примера рассмот-
рим рассеяние света частицами, малы-
ми по сравнению с длиной волны света.
В этом случае коэффициенты В. и С рав-
ны нулю и в уравнении (ПО.7) сохраня-
ется только первый член:
G (1)(т) = а, ехр (-£>с<72т), (ПО.12)
G <2>(т) = 1 +
Q (ПОЛЗ)
+ ах ехр (-2/)(<72т).
Если интенсивность рассеянного света
молекулами зависит от их ориентации в
отношении лабораторной системы коор-
динат, то появляется возможность опре-
деления коэффициента вращательной
диффузии 0 молекулы. Для этого име-
ются два экспериментальных подхода.
Первый основан на использовании поля-
ризованного света, тогда как второй — на
использовании деполяризованного света.
Поляризованные компоненты
рассеянного света
Если частица имеет несферическую фор-
му и размер, сравнимый с длиной волны
света, то интенсивность рассеяния зави-
сит от ориентации, благодаря эффекту
интерференции между светом, рассеи-
ваемым различными частями молекулы.
Например, если сравнить интенсивность
света, рассеиваемого в обратном направ-
лении длинной палочкообразной час-
тицей длиной L, то мы найдем, что при
перпендикулярном положении молеку-
лы к лучу света, она рассеивает свет в
большей степени, чем при ее параллель-
ном положении к лучу, поскольку в пер-
вом случае все рассеянные длины волн
находятся в фазе, в то время как во вто-
ром имеется определенное количество
деструктивной интерференции. Теория
показывает, что для простейшего случая,
когда вращательное и поступательное
движение разобщены, корреляционная
функция G(1) (т) будет равна:
G(1)(t) = £ В,ехрх
четные (ПО. 16)
х(-П,<72+/(/ + 1)0)т,
где / имеет значения, равные 0, 2, 4...
aBt зависит от qL, как показано на ри-
сунке 10.9. Из рисунка видно, что для
малых значений qL, (малые углы рас-
сеяния) можно определить константы
поступательной диффузии для больших
молекул, причем, только Во является
значимым коэффициентом. Для боль-
ших значений qL вклад В2 становится
существенным и появляется вторая
экспонента в G(1)(t), из которой можно
вычислить коэффициент вращательной
диффузии 0. Стандартная процедура
анализа данных состоит в экстрапо-
ляции одной экспоненты, полученной
при малых углах, к значениям больших
углов. По мере увеличения угла будет
все более проявляться вклад второй эк-
споненты, так что данные по всем углам
могут быть описаны одной экспонентой
с временной константой: D^q2 + 60.
Деполяризованная компонента
рассеянного света
Этот подход используется, когда по-
ляризуемость рассеивающей молеку-
лы зависит от относительной ориента-
Рис. Г 10.9. Относительная амплитуда компо-
нентов корреляционной функции, приведен-
ной в уравнении (ПО.12), которая получается
при рассеянии света палочкообразными моле-
кулами длиной L. Кривая В является суммой
всех Вл и, следовательно, относится ко всей ин-
тенсивности рассеянного света (Ресога, 1968)
ции электрического поля падающего
света и оси молекулы. В этом случае
поляризуемость анизотропна и в рас-
сеянном свете появляется компонента
(обычно небольшая), поляризованная
в направлении, перпендикулярном к
молекуле. Иными словами свет стано-
вится деполяризованным и его корре-
ляционная функция первого порядка,
G(1)(r), содержит только одну экспо-
ненту:
G(”(T) = exp-(60)T. (Г10.17)
Этот результат примечателен тем,
что позволяет определить 0 из моно-
экспоненциального анализа деполяри-
зованной компоненты рассеяния света.
Такой анализ проще, чем вычисление 0
из уравнения Г10.16.
К сожалению, деполяризованное
рассеяние обычно является очень сла-
Комментарий Г 10.8.
। Деполяризованная компонента для
1 вируса табачной мозаики (ВТМ)
I Возьмем в качестве примера вирус та-
! бачной мозаики. Его деполяризационное
отношение, определяемое как: pv = Я./Г,
где Я и V обозначают интенсивность рас-
I сеяния горизонтальной и вертикальной
составляющей при вертикальной поляри-
I зации падающего света составляет очень
: маленькую величину (-0,0030). Она пока-
зывает, что степень амплитудной модуля-
I ции рассеянного света броуновским движе-
I нием составляет приблизительно 1 /300 от
общей интенсивности.
Концентрация олигонуклеотидов (мг/мл)
Рис. ГК). 10. Коэффициенты вращатель-
ной диффузии для трех олигонуклеотидов
(приведенные к воде и 20 °C), как функция
концентрации. Обозначения на графике: о =
олигонуклеотиды длиной 20 пар оснований,
• = 12 пар оснований, и □ = 8 пар основа-
ний (Eimer and Ресога, 1991). Заметим, что
эти значения использованы нами при пост-
роении зависимости коэффициентов враща-
тельной диффузии ДНК от молекулярной
массы в широком интервале значений пос-
ледней (см. таб. I. Г(>,2).
бым (коммен rapnii I'lO.S). Несмотря
па это, коэффициент вращательной
диффузии вируса табачной мозаики,
полученный с применением этой тех-
ники, 350 сек *, находится в хорошем
согласии с другими данными. В случае
лизоцима, его молекулярные размеры,
вычисленные из 0 и De с использовани-
ем формул Перрена Г.2.29 и Г.2.22 для
коэффициентов вращательной и посту-
пательной диффузии, соответственно,
близки к таковым, полученным из крис-
таллографических данных. Анализируя
эти данные, надо помнить, что они были
получены в конце 1960-х гг., с использо-
ванием очень простого волнового ана-
лизатора, дающего частотный спектр
рассеянного света и имеющего намного
меньшую точность, чем современные
цифровые корреляторы.
В начале 1990-х Р. Пекора с кол-
легами применил эту технику для
измерений коэффициентов враща-
тельной диффузии ряда небольших
олигонуклеотидов. Частотный анализ
рассеянного под углом 90‘ света в VH-
геометрии, где V и Н — вертикальная и
горизонтальная плоскости рассеяния,
соответственно, был проведен при его
пропускании через пьезоэлектрический
интерферометр Фабри-Перо. Интер-
ферометр был оборудован набором
конфокальных 750 MHz зеркал, позво-
ляющих анализировать процессы сла-
бой релаксации с разрешением порядка
наносекунд. Именно в этом временном
интервале находятся коэффициенты
вращательного трения для маленьких
белков (М~ 10 кДа) и коротких нуклео-
тидов. На рисунке ПО. 10 показана кон-
центрационная зависимость деполяри-
зованной компоненты для трех ДНК
олигонуклеотидов, состоящих из 8, 12,
и 20 пар оснований (см. табл. Г6.2). От-
метим также, что объединение методов
деполяризованного и поляризованного
ДРС обеспечивает возможность полу-
чения гидродинамических размеров и
асимметрии коротких палочко-образ-
ных частиц (уравп. Г2.34).
П 0.4.3. Гибкие частицы, размеры
которых сравнимы с длиной
волны света
Кроме изучения поступательного и вра-
щательных движений жестких частиц,
обсужденных в предыдущих парагра-
фах, особый интерес представляет изу-
чение времен релаксации гибких моле-
кул, чьи размеры сравнимы с длиной
волны падающего света. Для таких мо-
лекул дополнительный вклад в значе-
ние корреляционной функции вносят
изменения, обусловленные эффектом
интерференции между длинами волн
рассеянного света от различных сегмен-
тов одной и той же молекулы.
В течение последних 30 лет боль-
шой объем исследований был проделан
для получения данных о временах внут-
ренней релаксации для гибких молекул,
и, в частности, для ДНК .методом ДРС.
Сформировались три подхода для по-
лучения такой информации. В первом
их них допускается, что для длинных
молекул (более 200 нм) корреляцион-
ная функция описывается двумя экспо-
ненциальными вкладами:
С")(т) = £>1ф’ + 2/т1, (Г10.18)
где rt — время релаксации первой нор-
мальной моды колебания молекулы
(см. уравнение Г9.5). Как показала
практика, отделение вклада внутренних
движений от вклада поступательного
движения для гибких молекул является
трудной задачей, поскольку нахожде-
ние каждой экспоненты по отдельности
возможно на практике только для очень
Комментарий Г10.9. Разделение
эффектов внутренней релаксации
и поступательного движения
Прежде всего заметим, что разделение
эффектов внутренней релаксации и посту-
пательного движения возможно для моно-
дисперсных образцов с оптимальной моле-
i кулярной массой и только в ограниченной
• области значений q. Лело в том, что линии
частот для поступательного и внутреннего
движения становятся шире при высоких
значениях q. С другой стороны, в области
низких значений q амплитуда внутрен-
них движений довольно мала, что создает
трудности для вычисления ее значения из
। корреляционной функции. Например, для
I линейной риС19-ДНК (2760 пар основа-
' ний) при угле рассеяния равном 45' час-
। тоты линий, относящиеся к внутренним
: движениям, отличаются от линий частот,
! соответствующих поступательному дви-
жению в 4 раза, тогда как величина амп-
i литуды внутренних движений составляет
всего 10% от суммарной корреляционной
функции. При увеличении угла рассеяния
до 120° вклад внутренних движений увели-
чивается до 75 %, но теперь частоты отлича-
ются в 1,7 раза. Такое различие в частотах
не позволяет выделить их из суммарной
I корреляционной функции никакими про-
I цедурами подгонки. Отсюда следует, что
для р11С19-ДНК (молекулярная масса
1,8 х 10е Да) при ионной силе 0,15 М NaCl '
i (R -150 нм) внутреннее движение мож-
i но отличить от поступательного только в
очень небольшой области углов, близких к '
90'. Именно в этой области углов для моле- ;
кул таких размеров создаются условия для j
: выделения внутренних движений: разли- :
чие в частотах составляет около 2,2, а ам-
плитуда внутренних движений около 60%
(Seils and Dorfmuller, 1991).
i. ______________
ограниченного интервала значений q,
который заранее неизвестен (к<>мм<-н-
rapnii ГН).!)).
Второй подход базируется на опре-
делении значения Dt при низких зна-
чениях q. Тогда экстраполяция к q -> 0
Рис. Г10.1 1. Зависимость кажущегося коэф-
фициента диффузии гибкого полимера в ДРС
эксперименте как функция квадрата вектора
рассеяния ^2(Langovski et al., 1992)
дает значение наибольшего времени
внутренней релаксации т(.
В третьем подходе кажущееся зна-
чение коэффициента диффузии £>каж
определяется путем аппроксимации
корреляционной функции через при-
нудительную подгонку данных одной
экспонентой. В этом случае наблю-
дается рост величины £>каж с повыше-
нием q (рис. ПО. 11). Область кривой
при низких значениях q соответствует
коэффициенту поступательной диф-
фузии Dt, область кривой для средних
значений q соответствует вкладу посту-
пательного и вращательного движения,
в то время как плато, соответствующее
Комментарий Г10.10. Сдвиг |
Доплера
По аналогии со звуковой волной, сдвиг
Доплера на оптической частоте света ра-
вен:
и
Av = -v0, (А)
I
i где v0 — исходная частота источника света,
взятая в качестве эталона, а с — скорость |
света для рассеянного света. Если источник I
i света движется под некоторым углом по от- I
ношению к детектору, то значение сдвига 1
। Доплера уменьшается на фактор sin 0.
области высоких значений q, отражает
движение наименьших по размеру не-
зависимых сегментов полимера. Из из-
ложенного следует, что для выделения
внутренних мод движения в любом гиб-
ком полимере необходимо выполнение
двух условий. Первое, молекула долж-
на иметь размеры, равные или большие,
чем длина волны падающего света. Вто-
рое, корреляционная функция должна
быть измерена в максимально большом
интервале углов рассеяния с целью раз-
деления эффектов движения молекулы
как целого и внутренних мод движе-
ния.
Г10.4.4. Электрофоретическое
рассеяние света (ЭРС)
Рассеяние света от движущихся частиц
получило название частотного эффекта
Доплера (ко.м.меп rupnii I 10.10):
Av- —qu. (Г10.19)
2л
Уравнение ПО. 19 показывает, что,
измеряя Av, можно получить значение
скорости частиц и, поскольку значение
вектора рассеяния q определяется уг-
лом рассеяния света. Электрофорети-
ческое рассеяние света позволяет из-
мерить скорость электрофоретической
подвижности по сдвигу Доплера рас-
сеянного света с большой скоростью
( коммсн lapiiii 1'10.11). Принципиаль-
ным преимуществом этого метода яв-
ляется его способность охарактеризо-
вать электрофоретические свойства
многих частиц одновременно. Когда
раствор заряженных частиц находится
в электрическом поле, то частицы дви-
жутся к электродам с противополож-
ной полярностью, с определенной ско-
ростью. Эффект электрического поля
как дополнительный член должен быть
включен в уравнение корреляционной
функции, считая, что переориентация
частиц и химические реакции отсут-
ствуют:
<=(’>= Лехр(-С,9Ч)х (ПО2О)
х exp(z'(7p£cos0/2),
где Е — значение приложенного элект-
рического поля и р — электрофорети-
ческая подвижность.
Уравнение Г10.20 показывает, что
ширина пика, обусловленного диффу-
зией, пропорциональна квадрату амп-
литуды вектора рассеяния q, в то время
как пик, относящийся к сдвигу Допле-
ра, линейно пропорционален абсолют-
ному значению q. Таким образом, раз-
решающая способность техники ЭРС
увеличивается при работе под малыми
углами рассеяния, поскольку при таких
углах уменьшается вклад диффузии
в ширину пика.
На рисунке ПО. 12 представлены
теоретический вид корреляционной
функции и результирующий спектр.
Ожидается, что в электрическом поле
корреляционная функция будет про-
модулирована косинусоидальной фун-
кцией, чья частота относится к элект-
Комментарий ПО. 11. Скорость
: определения электрофоретической .
| подвижности по сдвигу Доплера
1 В типичном ЭРС-эксперименте значе- |
\ ния сдвига Доплера накладывают ограни-
, чения на продолжительность одного изме-
рения. Например, если частица находится
под воздействием достаточно сильного элек-
трического поля, то определяемый рассеян-
ный свет от частицы имеет величину сдвига
Доплера около 100 Hz по отношению к час- ;
тоте действующего света. Если задаваемая ’
, точность равна 0,5 % (т. е. 0,5 Hz), то частица !
[ должна находиться в электрическом поле,
! по крайней мере, 0,5 Hz-1 = 2,0 сек, и, следо-
| вательно, интенсивность рассеянного света
от этой частицы должна быть зарегистриро-
вана за этот же период времени.
рофоретической подвижности частиц,
а скорость затухания определяется
коэффициентом диффузии. В нулевом
электрическом поле спектральная фун-
кция является Лоренцианом; в присутс-
твии электрического поля спектральная
функция также является Лоренцианом,
но сильно сдвинутой и асимметричной;
ее ширина подобна ширине гетеродин-
ного пика, центрированного на нулевой
частоте.
0 = 0,
Полуширина ок2
на половине =
максимума
_ Смещение К-у
частоты ~ 2л
Рис. ПО.12. Схематическая диаграмма спектральных форм и корреляционных функций, на-
блюдаемых в электрофоретическом рассеянии света
Комментарий Г10.12. Лазерный ;
велосиметр Доплера 1
В лазерном велосиметре Доплера падаю-
щий свет освещает поток движущихся час- 1
тип. На практике, один луч лазера, расщеп- |
ленный на два равных по интенсивности,
фокусируется в одной точке в потоке поля.
Интерференционная картинка формируется I
! в точке пересечения лучей и создает измеря- |
емый объем. Частицы, проходя через такой I
объем, создают вспышки света. Результиру-
| ющая частота, определяемая фотодетекто- |
ром, напрямую связана со скоростью частиц. ;
Экспериментальная геометрия
Экспериментальную ячейку для электро-
форетического рассеяния можно рассмат-
ривать как видоизменение таковой для
лазерного велосиметра Доплера и ДРС
(комментарий Г10.12). Однако примене-
ние внешнего электрического поля и от-
носительно медленно перемещающиеся
в ЭРС частицы требуют использования
специальной светорассеивающсй камеры
для образца, отличной от применяемых
в ДРС- экспериментах (рис. Г10.13).
Свет основного луча лазера, рас-
сеиваемый частицами под некоторым
углом 0, попадает на фотодетектор од-
новременно с нерассеянным светом от
опорного луча этого же лазера. Рассе-
янный свет имеет смещение по частоте
но отношению к опорному лучу из-за
эффекта Доплера, равное:
Av = — q-u = —sin(0/2). (Г10.21)
2л Ло
Следует заметить, что, когда вектор
и перпендикулярен вектору рассеяния
q, смещение частоты (сдвиг Доплера)
не наблюдается. Область значений
частот Av, с которой мы сталкиваемся
в экспериментах по электрофоретиче-
скому рассеянию равна приблизитель-
но 100 Hz.
Метод ЭРС может быть применен
в исследованиях электрокинетических
свойств любых заряженных частиц в
растворе, но на практике его приме-
нение ограничено размерами частиц.
Так, например, этот метод очень слож-
но применить к малым глобулярным
белкам, таким как лизоцим, в основном
из-за существенного вклада диффузии
в ширину пиков ЭРС (рис. ПО. 14 <7).
Разрешающая способность метода су-
щественно повышается с увеличением
молекулярной массы. Спектр электро-
форетической подвижности препара-
тов БСА показан на рисунке ПО.14 б.
По крайней мере, пять различных вкла-
дов в электрофоретическую подвиж-
ность различимы на этом графике.
Рис. I 10.13. Геометрия лучей в ЭРС-эксперименте. Е и и являются векторами, обозначающими
электрическое поле и скорость частиц, соответственно; 0 — угол рассеяния (Langley, 1992)
а б
Рис. 1’10.14. Спектральные частоты в ЭРС: а — молекулы лизоцима: расширение диффузии
«размазывает» ник, кроме двух наименьших углов рассеяния; б — мультимеры БСА: три частот-
ных спектра были получены одновременно при трех различных рассеивающих углах в одном
эксперименте. Результаты представлены в виде графика на одной и той же оси подвижности
(Langley, 1992)
ПО.5. Динамическое
рассеяние света в условиях
негауссовой статистики
Г10.5.1. Рассеяние малым числом
частиц
Давайте представим, что мы проводим
эксперимент по контролю за числом
частиц в большом объеме. При большом
числе частиц в объеме контролировать
их число наиболее просто по флуктуа-
циям, определяемым методом ДРС.
Поскольку общая интенсивность рас-
сеянного света, регистрируемая детек-
тором, является суммой интенсивности
рассеяния от каждой частицы. Измене-
ния интенсивности, обусловленные чис-
лом флуктуаций, обычно малы именно
в силу большого числа частиц в объеме
(коммеп гарий I' 10.13).
Теперь представим, что мы прово-
дим тот же эксперимент, наблюдая за
концентрацией частиц, растворенных
в малом объеме, находящемся внутри
большого объема образца в растворе,
и что этот объем имеет проницаемые
стенки. Эксперименты такого типа
i Комментарий Г10.13. Флуктуации
: В системах, состоящих из сравнительно
небольшого числа частиц, возможны зна-
чительные отклонения некоторых физиче-
ских величин, характеризующих системы,
от их средних значений. Такие отклонения
называются флуктуациями. Термодинами-
ческая теория флуктуаций дает величину
таких отклонений в терминах различия
между среднеквадратичным отклонением ;
<Д7У?> и его средним значением <У>. Угло-
вые скобки означают, что указанные вели-
, чины усреднены по времени. Мерой флук-
туации числа частиц У является средняя
величина квадрата разности ДУ, которая
называется квадратичной флуктуацией:
<(д№)> = <<у - <у>)2 >. (А) ;
Абсолютной флуктуацией называется I
. величина V<A№>, также характеризующая I
I отклонения Уот <У>.
Относительной флуктуацией 8М назы-
вается отношение абсолютной флуктуации
к среднему значению <У>
-/<ДУ2>/<У> = 1/УУ. (Б)
Из последней формулы видно, что отно-
сительная величина флуктуаций обратно
пропорциональна корню квадратному из •
числа частиц. При N = Ух — постоянная
Авогадро — 8М имеет величину порядка
10 |2.
были выполнены в начале XIX в. с ис-
пользованием микроскопа. Целью та-
ких исследований было определение
среднего значения числа частиц <N>
(в этом случае коллоидальных) в еди-
нице объема. Если бы мы могли полу-
чить определенное число «мгновенных
кадров» объема как функции времени,
то обнаружили бы, что число подсчи-
танных частиц изменяется во времени
из-за диффузии, имеющей место как
внутри, так и снаружи определяемого
объема.
Для нашего случая важными яв-
ляются два вклада во временную за-
висимость интенсивности рассеяния.
Один из них связан с входом и выхо-
дом частиц из рассеивающего объема,
что вызывает временные флуктуации.
Другой связан с диффузией частиц
внутри рассеивающего объема на рас-
стояниях порядка длины волны света,
которая также вызывает временные
флуктуации. Если предположить, что
эти два времени не коррелированы
друг с другом, тогда для гомодинно-
го детектирования нормализованную
корреляционную функцию можно
представить как:
+BN~ ехр(-2£>,<7'Г)].
Первый член в правой части этого
уравнения связан флуктуацией обще-
го числа частиц внутри рассеивающе-
го объема, а второй обусловлен вкла-
дом диффузии. В уравнении Г10.22
No — это среднее число частиц в рас-
сеивающем объеме; у — характерное
время, необходимое для диффунди-
рования частицы через рассеивающий
объем и которое имеет величину по-
рядка L2/2^NQDt (где L — характерный
размер рассеивающего объема), А и
В — константы.
Рис. ПО. 15. Корреляционная функция времени интенсивности рассеянного света раствором
вируса птичьего миелобластоза (Af = 4 х 108 Да, s = 680S, Dt = 0,31 х 107см2сек ') в очень низкой
концентрации (2хЮ7 частиц на см3). Рассеивающий объем равен около 10-3 см3. Присутствие по-
логой базовой линии, смещенной по отношению к нулевой линии коррелятора, является мерой
числа флуктуаций, ожидаемых при низких концентрациях (Salmeen et al., 1975)
Очевидно, что при высоких концен-
трациях второй член в уравнении Г10.22
доминирует и по экспериментально на-
блюдаемой константе спада вычисляет-
ся константа диффузии. При очень низ-
ких концентрациях, когда No сравнимо
с BNU2, оба компонента вносят вклад в
наблюдаемую корреляционную функ-
цию. На рисунке ПО.15 представлена
корреляционная функция для образца
вируса птичьего миелобластоза, в кото-
рую флуктуации от числа частиц вносит
уже существенный вклад, поскольку в
рассеивающем объеме содержится все-
го около 2000 частиц. Однако рассеяние
при такой концентрации и такой моле-
кулярной массе вируса (М = 4х108Да)
является настолько слабым, что точное
значение коэффициента диффузии ви-
руса определить не представляется воз-
можным.
Г10.5.2. Кросс-корреляция (метод
двух детекторов).
Как следует из изложенного в пре-
дыдущем параграфе, наблюдение за
флуктуациями числа частиц методом
ДРС, возможно только в том случае,
если когерентная часть рассеяния су-
щественно уменьшена или устранена
совсем. Такое устранение может быть
достигнуто разными способами. Во-
первых, можно перейти от рассеяния
к флуоресценции, которая по своей
природе является некогерентным яв-
лением. Такой переход мы будем об-
суждать в Главе Г.11. Другой способ
устранения пространственной коге-
рентности основан на использовании
двух детекторов и построения кросс-
корреляционной функции (см. параг-
раф Г10.3.1).
На рисунке Г К). 16 а показана
кросс-корреляционная функция, обус-
ловленная вращательным движением
вируса табачной мозаики вокруг его
короткой оси симметрии при различ-
ных углах рассеяния. Видно, что кросс-
коррреляционная функция не зависит
от угла рассеяния. В отличие от кор-
реляции, обусловленной релаксацией
вращательной диффузии жестких мо-
лекул, измеренная кросс-корреляци-
онная функция для плазмидной ДНК
E.coli, обусловленная внутренними
степенями свободы, зависит от вектора
рассеяния (рис. Г10.16 б).
Г10.5.3. Рассеяние одной частицей
Основное явление, которое наблю-
дается при динамическом рассеянии
света на растворах частиц, связано с
диффузионным уширением. Это дает
возможность определять коэффици-
енты поступательной диффузии час-
тиц с высокой точностью, но не позво-
ляет судить о динамических процессах
в исследуемой частице. Как следует
из формулы ПО.18, вклад трансля-
ционной диффузии уменьшается с
уменьшением числа частиц в рассеи-
вающей системе. Этот вклад устраня-
ется полностью, если мы переходим
к рассеянию одной частицей. В этом
случае спектр рассеянного света со-
держит информацию только о внут-
ренних процессах, проявляющихся в
изменении форм-фактора или внут-
ренней динамики частицы. Обычно,
форм-фактор частицы с размерами,
равными или меньшими длины вол-
ны света, слабо флуктуирует относи-
тельно своего среднего значения, по-
этому отношение сигнал-фон в таких
экспериментах невелико. Однако для
частицы микронных размеров такие
исследования становятся возможны-
ми с помощью метода, получившего
Рис. ГК). 16: а — кросс-корреляционные функции, измеренные для вируса табачной мозаики.
Подобранное корреляционное время равно 240 мксек и не зависит от угла рассеяния. Кор-
реляционные функции, измеренные при 90' и 60‘, сдвинуты для наглядности вдоль ордина-
ты на 0,25 и 0,5 единиц, соответственно; б — кросс-корреляционная функция рассеяния для
плазмидных ДНК Е. coli. Подобранные времена корреляции равны 400, 120, и 80 мксек при
углах рассеяния 60°, 90° и 120°, соответственно. Корреляционная функция для углов 90° и
60° сдвинута для наглядности вдоль ординаты на 0,25 и 0,5 единиц, соответственно (Kam and
Rigler, 1982).
название метода локализованного
динамического рассеяния света. Его
основной идеей является выполнение
ДРС-эксперимента на одной частице в
сильно неоднородном световом поле,
рассеивающий свет от которой соби-
рается с помощью микроскопа. Неод-
нородность светового поля приводит
к появлению специфической зависи-
мости интенсивности рассеяния от
угла рассеяния, которая регистриру-
ется датчиком (тонкое стекловолок-
но), через который свет попадает на
фотодетектор для последующего вы-
числения корреляционной функции
интенсивности. Такие эксперименты
позволяют вычислить спектр динами-
ки объекта, флуктуирующего в широ-
ком интервале времен вплоть до нано-
секунд. Реальные возможности этого
подхода выявляются при измерении
силовых констант одной бусинки или
пары бусинок, захваченных лазерны-
ми пинцетами (твизерами) (параграф
Д5.2.1).
Г10.6. Перечень ключевых идей
• Все термодинамические величины в растворе флуктуируют относительно
средних значений; поскольку такие флуктуации очень медленны по сравнению
с частотой падающего света, то для их определения требуется спектроскопия
очень высокого разрешения.
• Динамическое рассеяние света относится к когерентным явлениям, в которых
измеряются временные флуктуации интенсивности рассеянного света; этим
оно принципиально отличается от классического статического рассеяния, где
измеряется интенсивность, усредненная по времени.
• Определение коэффициентов трансляционной диффузии биологических мак-
ромолекул требует применения спектроскопии очень высокого разрешения,
которое не может быть обеспечено обычными методами спектроскопии; однако
последние адекватны для определения вращательных диффузионных коэффи-
циентов небольших глобулярных белков и коротких спиралей ДНК.
• Временная автокорреляционная функция показывает: насколько два значения
флуктуирующего процесса коррелируют во времени; кросс-корреляционная
функция показывает: насколько два флуктуирующих процесса коррелируют
во времени.
• В лабораторных условиях трансформация флуктуирующего сигнала в корреляци-
онную функцию происходит с помощью приборов, называемых корреляторами.
• Для молекул, малых по сравнению с длиной волны света, подавляющий вклад
в корреляционную функцию вносит трансляционная диффузия; что обеспечи-
вает быстрый (2-3 мин) и точный (1-2 %) способ определения коэффициентов
поступательной диффузии биологических макромолекул.
• Для молекул, размеры которых сравнимы с длиной волны света, вращательная
диффузия и внутренние движения (для гибких частиц) вносят дополнитель-
ный вклад в корреляционную функцию.
• Фундаментальная проблема анализа данных динамического рассеяния света
состоит в разложении сложной корреляционной функции на индивидуальные
составляющие (экспоненты).
• В стандартном ДРС-экспсрименте молекулы исследуются в макрообъеме, кото-
рый содержит большое число молекул и поэтому такие эксперименты носят назва-
ние экспериментов в условиях гауссовой статистики (большое число событий).
• Если рассеивающий! объем содержит относительно небольшое число молекул
(около 1000), то ДРС-эксперпменты носят название экспериментов в условиях
негауссовой статистики (небольшое число событий); в этих условиях наравне
с вкладом диффузии проявляется вклад флуктуации числа частиц в корреля-
ционную функцию.
• Если рассеивающий объем содержит всего несколько частиц, то основной вклад
в корреляционную функцию вносит флуктуация числа частиц.
Литература для дальнейшего чтения
Исторический обзор и введение в биологические проблемы
Вете B.J. and Pecora R. Dynamic Light Scattering. New York: Wiley. DLS as a
spectroscopy of very high resolution, 1976.
Benedek G. B. Optical mixing spectroscopy in physics, chemistry, biology and
engineering. In Polarization: Matiere and Rayonnement. Pp. 4-84. Paris: Les Presses
Universitaries de France, 1969.
Dubin S. B., Lunacek J. H. and Benedek G. B. Observation of the spectrum of light
scattered by solutions of biological macromolecules. Proc. Natl. Acad. Sci USA, 1967.
57, 1164-1171.
Doherty J. V. and Clarke H. R. Noisy solutions: a source of valuable kinetic information,
Sci. Prog. Oxf„ 1980. 66, 385-419.
Динамическое рассеяние в условиях гауссовой статистики
Chu В. Laser Light scattering. Basic Principles and Practice. San Diego: Academic
Press, 1991.
Wada A., Suda N., Tsuda T. and Soda K. Rotary-diffusing broadening of Rayleigh
lines scattered from optically anisotropic macromolecules in solution. J. Chem. Phys.,
1968. 50,31-35.
Dubin S. B., Clark N. A. and Benedek G. B. Measurment of the rotational diffusion
coefficient of lysozyme by depolarized light scattering: configuration of lysozyme in
solution. J. Chem. Phys., 1971. 54, 5158-5164.
Seils J. and Dorfmuller T. H. Internal dynamics of linear and superhelical DNA as
studied by photon correlation spectroscopy. Biopolymers, 1991. 31, 813-825.
Langowski J., Kremer W. and Kapp U. Dynamic light scattering for study of
solution conformation and dynamics of superhelical DNA. Meth. Enzymol., 1992.
211, 431-448.
Eimer W. and Pecora R. Rotational and translational diffusion of short rodlike
molecule in solution: Oligonucleotides. J. Chem Phys., 1991. 94, 2324-2329.
Ware B. R. and Haas D. D. Electrophoretic light scattering in fast methods in
physical biochemistry and cell Biology, eds. R. I. Sha’afi and S. M. Fernandez, Elsevier
Sci. Publishers, 1983.
Langley К. H. Developments in electrophoretic laser light scattering and some
biochemical application. In Laser Scattering in Biochemistry, eds. S. E. Harding,
D. B. Sattelle and V. A. Bloomfield. Cambridge: Royal Society of Chemistry.
1992.
Динамическое рассеяние в условиях негауссовой статистики
Bar-Ziv R., Metier A. et al. Localized dynamic light scattering: probing single particle
dynamics at the nanoscale. Phys. Rev. Lett., 1997. 78, 154-157.
Metier A., Bar-Ziv R. et al. Localized dynamic light scattering: a new approach to
dynamic measurements in optical microscopy, Biophys. J., 1998. 74, 1541-1548.
Weissman M., Schindler II. and Feher G. Determination of molecular weights by
fluctuation spectroscopy: application to DNA. PNAS, 1976. 73, 2776-2780.
Kam Z. and RiglerR. Cross-correlation laser scattering, Biophys J., 1982.39,7-13.
Глава Г11
ФЛУОРЕСЦЕНТНАЯ КОРРЕЛЯЦИОННАЯ
СПЕКТРОСКОПИЯ
Г11.1. Исторический обзор
1916
М. вон Смолуховски (М. von
Smoluchowski) создал термодинами-
ческую теорию флуктуационных про-
цессов в равновесных диффузионных
системах.
1972-1974
Д. Магде, Л. Элсон и В. В. Вебб
(D. Magde, L. Elson and W. W. Webb)
дали строгое описание метода флуорес-
центной корреляционной спектроско-
пии (ФКС), различных вариантов его
практического применения и предсказа-
ли большие возможности метода для ис-
следования релаксационных процессов.
1990
Р. Риглер (R. Rigler) и соавторы
осуществили серьезный прорыв в разви-
тии метода флуоресцентной корреляци-
онной спектроскопии. Скомбинировав
оборудование для ФКС с конфокаль-
ным микроскопом, они существенно
улучшили соотношение «сигнал-шум»
и впервые достигли уровня детектиро-
вания одиночных молекул. Четкой фо-
кусировкой луча лазера и установкой
точечной диафрагмы в плоскости изоб-
ражения они добились существенного
улучшения латерального и аксиального
разрешения.
1994
М. Эйген и Р. Риглер (М. Eigen and
R. Rigler) продолжили дальнейшее раз-
витие идей ФКС, предложив метод двух-
цветной флуоресцентной кросс-корре-
ляции. Метод позволил удобно и просто
разделить вклады продуктов, имеющих
«одноцветную метку» от продукта реак-
ции, имеющего «двухцветную метку», что
привело к существенному повышению
специфичности узнавания продуктов ре-
акции. В 1997 г. П. Швайль (Р. Schwille)
успешно применила этот метод для на-
блюдения за реакцией гибридизации
двух меченых различным флуорофорами
олигонуклеотидов, а в 2000 г. К. Г. Хайнц
(К. G. Heinze) — за энзиматическим рас-
щеплением ДНК эндонуклеазой.
2000 и но настоящее время
Метод ФКС стал базой для созда-
ния ряда родственных методов, в осно-
ве которых лежит принцип флуорес-
центного анализа флуктуаций. Метод
двухцветной флуоресцентной кросс-
корреляции обеспечивает простой и
удобный способ слежения за работой
молекулярных ферментов в режиме ре-
ального времени.
Г11.2. Введение
в биологические проблемы
Хотя первое описание флуктуационных
процессов в терминах амплитуд и вре-
Коммешарий Г11.1. Определение
молекулярных масс ДНК методом
ФКС
В середине 1970-х гг. техника ФКС
успешно применялась для определения
молекулярных масс молекул ДНК. Эти
эксперименты выполнялись в раство-
рах, содержащих около 1000 молекул,
что обеспечивало удовлетворительное
соотношение «сигнал-шум». Переписав
уравнение (В) в комментарии ПО.12, мы
получим:
где С — концентрация молекул. Меньшему i
числу молекул соответствует большая ам-
плитуда флуктуаций. Следовательно, для
данной концентрации <О масштаб флук- :
туаций повышается с ростом молекулярной
массы. Измеряя эти флуктуации через па- ।
раметры, чувствительные к концентрации,
можно из уравнения (А) определить число
молекул У в данном флуктуационном объ- i
еме V. Зная среднюю концентрацию <С>,
можно определить молекулярную массу по
уравнению: :
M = <C>[^-pW1. (Б)
С использованием этой формулы были
определены значения молекулярных масс
для ДНК из различных источников: для
Т2 фага — 1,14 х Юа Да; реплицирован-
ная ДНК из Е. coli — 3,9 х Ю9 Да. Моле-
кулярные массы ядерных и индивиду-
альных хромосомных ДНК из Drosophila
melongaster были равны 3 х 10'’Да и 4 х
1010 Да, соответственно (Weissman et al.,
1976). Найденные молекулярные массы
находятся в разумном согласии с точными
значениями молекулярных масс, опреде-
ленными гораздо позже из их нуклеотид-
ной последовательности < '> '' : I >.
мен затухания было сделано М. Смо-
луховским почти столетие назад, после
этого в течение долгого времени не было
техники с адекватной чувствительно-
стью для определения флуктуаций от
небольшого числа частиц, где эффекты
выражены особенно отчетливо. Страте-
гия метода флуоресцентной корреляци-
онной спектроскопии была предложена
в 1970-х гг. для измерения кинетики
химических процессов и молекулярной
диффузии с использованием анализа
флуктуаций концентрации в малых ан-
самблях (-1000 частиц), находящихся в
равновесном состоянии. Сильные сто-
роны этой техники были продемонс-
трированы при измерении диффузии
и кинетики связывания флуоресциру-
ющего соединения, этидиум-бромида,
с молекулой ДНК и с определением
молекулярного веса ДНК гигантских
фагов (комментарий Г1 1.1). Эти иссле-
дования позволили в дальнейшем дать
математическое описание метода ФСК,
что обеспечило основу для анализа ско-
ростей процессов диффузии и процес-
сов химической кинетики.
С недавних пор метод ФКС пре-
терпел существенные обновления.
Благодаря последним инновациям
(прецизионная фокусировка света в
пространстве, имеющем геометрию
песочных часов, точечная диафрагма
в плоскости изображения) появилась
возможность измерять флуоресценцию
в небольших, менее чем фемтолитр,
микрообъемах (см. Главу Д4). Если
учесть, что при концентрации, равной
1 иМ, в таком объеме будет в среднем
находиться менее одной молекулы, то
появляется уникальная возможность
работы на уровне нескольких молекул.
Это делает метод ФКС крайне привле-
кательным для исследования биологи-
ческих систем.
Г11.3. Теория и практика
метода
Флуоресцентная корреляционная спек-
троскопия может рассматриваться
как ветвь динамического рассеяния в
негауссовых условиях эксперимента,
поскольку флуоресценция является
фундаментально некогерентным яв-
лением. ФКС является методом, в ко-
тором интенсивность флуоресценции,
исходящая от небольшого числа флу-
оресцентных молекул в очень малом
объеме, коррелируется для получения
информации о процессах, которые вы-
зывают флуктуации флуоресценции.
Одним из важных процессов, который
приводит к стохастическому появле-
нию и исчезновению флуоресценции в
малом наблюдаемом объеме, является
их броуновское движение. Относитель-
ная амплитуда флуктуации обратно
пропорциональна числу одновременно
наблюдаемых молекул (уравнение (А)
комментария I I l.l). Поэтому присутс-
твие большого числа молекул в макро-
скопической среде, подавляет эффект
таких флуктуаций. Этот простой аргу-
мент показывает, что для ФКС-измере-
ний желательно наличие в микрообъеме
очень небольшого числа интенсивно
светящихся молекул.
Основные принципы метода ФКС
продемонстрированы на рисун-
ке Г11.1. Флуктуации сигнала флуо-
ресценции вызываются или диффузи-
ей флуоресцирующих молекул через
объем, или изменением сигнала во
времени из-за химических реакций.
По записанному сигналу флуктуа-
ции флуоресценции (горизонтальные
стрелки) вычисляется автокорре-
ляционная функция (вертикальные
стрелки). Из функции автокорреля-
ции могут быть получены среднее
Комментрий Г11.2.
. Автокорреляционные функции
: в ФКС
I
Часто в методе ФКС используется не-
сколько другое определение корреляни-
I онной функции, чем в главе ПО. Если мы
! определим 54(г)как
' 84(г)н. 1(г)- </!)>. (А),
которое есть отклонение мгновенного зна-
i чения от среднего, то легко показать, что
(Б)
(В)
уравнс-
(Г)
<84(0)84(т)> =
= <4(0)4(т)>-<4>’’
, и, следовательно:
<842> = <54(0)84(0) > =
= [<4‘>-<4>"]-
Комбинируя уравнение Г10.3 с
. пнями (Б) и (В), получаем:
1 <84(0)84(т)> =
= <84’>схр——.
C.p
I Величину 8А(г) принято называть
«флуктуацией», поскольку она опреде-
ляет отклонение свойства от среднего.
Отметим, что корреляционная функция
I флуктуаций имеет более простой вид, чем
1 автокорреляционная функция, определя-
: емая уравнением ПО. 1. так как инвариант
<4>2 при этом удаляется.
время жизни (т ) и число молекул
(А) в объеме (см. ниже и коммента-
рий П 1.2). По этим данным и разме-
ру флуоресцирующего объема можно
определить коэффициент диффузии
D флуоресцирующей молекулы. Зна-
чение D может быть использовано для
оценки формирования молекулярных
комплексов, поскольку коэффициент
диффузии частицы в изолированном
состоянии (закрашено зеленым) будет
больше, чем коэффициент диффузии
молекулярного комплекса (закраше-
но красным и зеленым цветом) (см.
рис. Г11.1).
t сек
Рис. Г1 1.1. Основные принципы ФКС (см. текст) (Bastiaens and Pepperkok, 2000)
П1.3.1. Трехмерная и двумерная
диффузия
Если лазерный пучок с гауссовым про-
филем используется для возбуждения,
а флуоресценция собирается через кон-
фокальную точечную диафрагму (см.
параграф Д 1.1.2), то генерируемый объ-
ем обладает профилем, который может
быть описан в хорошем приближении
трехмерной гауссовой функцией. Если
г и / характеризуют радиальные и акси-
альные размеры объема (т. е. когда га-
уссова функция падает до 1/е2 от своего
максимального значения), то отношение
между автокорреляционной функцией А
(т) и временем диффузии т равно:
Время диффузии т характеризует
среднее значение времени, которое не-
обходимо для диффундирования моле-
кулы через радиальную часть наблюда-
емого в микроскоп объема.
Это время зависит от размеров на-
блюдаемого объема, от длины волны
лазера и свойств инструментальной
оптики. Взаимосвязь между временем
диффузии и коэффициентом диффузии
выражается как
W
Тер
(Г11.2)
Полное описание трехмерной диф-
фузии может быть получено при ис-
пользовании оптики с двухфотонным
возбуждением (параграф ДЗ.3.2). В этом
случае время диффузии уменьшается
в два раза: r2/8D.
При высоком значении г/I или дву-
мерной диффузии уравнение Г11.1 уп-
рощается до:
(Г11.3)
Г11.3.2. Различные виды движений
Биологические макромолекулы в клетке
находятся в условиях, далеких от тако-
вых в разбавленных растворах. Поэто-
Время (мсек)
Рис. 1 1 1.2. Модельная автокорреляционная кривая для частиц с различной подвижностью:
(1) трех- и (2) двухмерная диффузия; (3) активный транспорт и (4) аномальная диффузия (см.
текст) (Hausten and Schwille, 2003)
му коэффициенты диффузии, описыва-
ющие их поведение в клетке, сильно
отличны от таковых, рассмотренных
нами в секции Г2.3. Кривые модельных
корреляционных функций, показываю-
щие различные способы движения био-
логических макромолекул, приведены
на рисунке Г11.2. Из рисунка видно, что
корреляционная функция для активно-
го транспорта имеет самый крутой спад,
в то время как кривая для аномальной
диффузии уменьшается довольно мед-
ленно (сравните с рис. ГЗ. 10). Частицы,
диффундирующие внутри биологиче-
ской мембраны, могут демонстрировать
большое разнообразие видов движения,
в зависимости от локального окружения
для каждого индивидуального хромофо-
ра (см. параграф ГЗ.5.2 и рис. Г11.3).
Г11.3.3. Динамический интервал
времен
Динамический интервал времен в ме-
тоде ФКС простирается от 1 микросе-
кунды до 100 милисекунд. Для более
удобного построения кривых в таком ши-
роком интервале времен обычно исполь-
зуется логарифмическая шкала времени
(рис. Г11.4). В зависимости от состава ок-
ружающей среды величина диффузион-
ной подвижности молекул может изме-
няться на несколько порядков, начиная
от 3 х 10"fi см2 • сек 1 для свободного кра-
сителя в водном буферном растворе до
10“10 см2 • сек-1 для больших рецепторов в
клеточной мембране. Отметим, что коэф-
фициент поступательной диффузии, оп-
ределенный методом ФСК, для молекулы
красителя (Alexa 488, М -0,6 кДа) в воде
равен D = 30 х Ю“7 см2 • сек1, что очень
близко к коэффициентам диффузии са-
харов соответствующего молекулярного
веса (см. рис. П 1.4). Это позволяет отнес-
ти этот краситель к категории «средних»
по размерам молекул (параграф ГЗ.7.5).
Напоминаем, что для таких частиц вы-
числение гидродинамического радиуса
из коэффициента диффузии может при-
вести к неверным результатам.
ческого движения in vitro и in vivo при помощи
корреляционных функций. Фокальный объем,
т (ысе«)
который приблизительно равен 0,2 фемтолитра,
может быть наблюдаем как вне клетки (в чистом
буфере), так и внутри нее (в цитоплазме или
в плазме клеточной мембраны)
Поскольку временная разреша-
ющая способность метода ФКС равна
около 100 нсек, то метод ФКС in vivo
лучше всего применим для исследова-
ния медленно протекающих процессов.
При анализе медленно движущихся
молекул, например тех, которые взаи-
модействуют с цитоскелетом, для ядер-
ных ДНК и для мембран, необходимым
условием проведения эксперимента
является отсутствие фотовыцветания
красителя во время их движения через
фокальный объем (параграф ГЗ.5.2).
Г11.3.4. Исследование
многокомпонентных систем
В многокомпонентных системах оценка
корреляционных кривых происходит
Рис. Г1 1.4. Диффузия различных биологиче-
ских макромолекул в разном окружении:
а — маленькая молекула красителя (Alexa 488,
М -0,6 кДа) в воде, D = 3 х 10'6см2 • сек б— зе-
левый флуоресцирующий белок (eGFP) в вод-
ном растворе(М -27 кДа) Л = 9х 10’7см2 сек ');
в — этот же белок в цитоплазме клеток M-HeLa
D = Зх10-7 см2 • сек1; г — большой белковый
комплекс (кальмодулин-зависимая киназа
II и eGFP белок) в цитоплазме клеток линии
НЕК, Л = 3х Ю-8 см2 • сек д — двумерная
диффузия флуоресцирующего зонда (длин-
ная цепь карбоцианинового красителя «dif
С18’») в плазме мембраны клеток линии НЕК-,
D = 6 х 10 В 9 см2 сек-1 (Bacia and Schwille, 2003)
при допущении, что диффузионная сис-
тема описывается моделью из двух или
трех компонент, с рутинным подбором
решения методом наименьших квадра-
тов. Однако для получения коэффици-
ентов диффузии или соответствующих
других параметров в большинстве слу-
чаев требуются обширные калибровоч-
ные измерения для всех образцов.
Г11.4. Двухцветная
флуоресцентная
кросс-корреляционная
спектроскопия
Двухцветная кросс-корреляционная схе-
ма дает возможность исключить предва-
рительный этап калибровки, поскольку
в ней используются две флуоресцентные
метки, различимые спектрофотометри-
чески. Это позволяет одновременно из-
мерять количество изолированных учас-
тников реакции и продукты, полученные
в результате реакции.
В этом методе используются два
отдельных лазера для возбуждения
двух молекул, каждая из которых име-
ет метку своего цвета, зеленую и крас-
ную (флуорофоры, испускающие свет в
двух областях спектра) и два независи-
мых детектора для измерения зеленой
и красной флуоресценции (рис. 1'11.5).
Затем корреляция сигналов измеря-
ется между двумя каналами (отсюда
название — кросс-корреляция). Моле-
кулы, несущие зеленую метку, будут
регистрироваться только детектором,
измеряющим зеленую флуоресцен-
цию, молекулы, несущие красную мет-
ку, — только детектором, измеряющим
красную флуоресценцию, а продукт
реакции будет регистрироваться обои-
ми детекторами (см. рис. И1.5). Таким
образом, величина кросс-корреляцион-
ного сигнала будет мерой количества
продуктов реакции, т. е. мерой комплек-
собразования. В противоположность
методу одноцветной ФКС, интерпре-
тация данных которого неизбежно
связана с математической обработкой
корреляционной функции, метод двух-
цветной ФКС, в принципе, дает ответ
типа «да» или «нет» на вопрос о ком-
плексообразовании между молекулами.
Рис. Г11.5. Схематическое изображение принципа измерения: «зеленый» и «красный» лазеры
освещают образец, который содержит два сорта диффундирующих частиц, «красные» (К) и «зеле-
ные» (3), а также комплекс между ними (КЗ). «Красный» фотодетектор регистрирует (К) и (КЗ)
частицы; «зеленый» фотодетектор — (3) и (ЗК) частицы (Haustein and Schwille, 2003)
Рис. П 1 .(>. Временная зависимость амплиту-
ды кросс-корреляционной функции, описыва-
ющая энзиматическое расщепление молекулы
ДНК, один конец которой мечен флуорофо-
ром, излучающим в зеленой области, а другой
конец — флуорофором, излучающим в крас-
ной области (Haustein and Schwille, 2003)
На сегодня выполнено большое ко-
личество экспериментов с применением
метода двухцветной ФКС в анализе ре-
акций между биологическими макромо-
лекулами. Приведем два примера таких
реакций. Первый связан с энзиматиче-
ским расщеплением ДНК эндонукле-
азой (рис. Г11.6). Концы ДНК были ме-
чены двумя разными флуорофорами и
за скоростью расщепления наблюдали
по изменению кросс-корреляционной
амплитуды в реальном режиме времени.
Как видно из рисунка, максимальная
амплитуда наблюдается в начале реак-
ции, которая затем уменьшается с тече-
нием времени прямо пропорционально
количеству расщепленной ДНК.
Другой пример — наблюдение за
протеканием реакции гибридизации
двух комплементарных молекул ДНК
(рис. П 1.7), каждая из которых была
Рис. 1'11.7. Временная зависимость ампли-
туды кросс-корреляционой функции реакции
гибридизации ДНК в интервале 8-120 мин.
Один конец ДНК мечен флуорофором, излу-
чающим в зеленой области, а другой конец —
флуорофором, излучающим в красной облас-
ти. Время инкубации: светлые точки — 8 мин;
темные точки — 15 мин; короткие штрихо-
вые — 25 мин; длинные штриховые линии —
60 мин; сплошная линия — 120 мин (Schwille
et al., 1997)
флуоресцентно мечена с одного конца
зеленым родамином, с другого - красите-
лем CY-5. За кинетикой реакций гибри-
дизации наблюдали за ростом амплиту-
ды корреляционной функции в реальном
режиме времени. Как видно из рисунка,
минимальная амплитуда сигнала наблю-
дается в начале реакции, которая затем
возрастает прямо пропорционально кон-
центрации гибридизоваипой ДНК.
Эти два примера ясно показывают
основные преимущества метода двух-
цветной ФКС: кросс-корреляционный
сигнал существует только тогда, когда
в растворе имеются молекулы, несущие
иа своих концах оба флуорофора (еще
не все молекулы ДНК разрезаны, как
Рис. Г11.8. Эмиссионный спектр родамина
зеленого (1) и CY-5 (2), а также трансмисси-
онные характеристики пропускания дихро-
ичного зеркала (3), эффективно отделяющего
один спектр от другого (Schwille et al., 1997)
в первом примере, пли часть или все
молекулы ДНК гибридизованы, как во
втором примере). В случае же, когда все
молекулы ДНК разрезаны или они еще
не вступили в реакцию гибридизации,
такой сигнал отсутствует.
В заключение заметим, что эффек-
тивность метода ФКС сильно зависит
от того, насколько эффективно спект-
ры флуорофоров разнесены по длинам
волн. Рисунок Г. 11.8 демонстрирует, что
пара флуорофоров (родамин зеленый н
CY-5) наиболее подходит для реализа-
ции возможностей метода двухцветной
флуоресцентной корреляционной спек-
троскопии.
Г11.5. Перечень ключевых идей
• В методе флуоресцентной корреляционной спектроскопии анализируется сто-
хастическое появление и исчезновение флуоресцирующих молекул в малень-
ком (микроскопическом) объеме.
* Флуоресцентная корреляционная спектроскопия является некогерентным яв-
лением и не может быть использована для измерения подвижности в ансамбле
молекул; наилучшие условия ее наблюдения — наличие всего нескольких мо-
лекул в микрообъеме.
9 В методе двухцветной флуоресцентной кросс-корреляционной спектроскопии
используются два отдельных лазера для возбуждения двух молекул, каждая из
которых имеет метку своего цвета, зеленую и красную (флуорофоры, испуска-
ющие свет в двух областях спектра) и два независимых детектора для измере-
ния зеленой и красной флуоресценции.
9 В противоположность методу одноцветной ФКС, интерпретация данных кото-
рого неизбежно связана с математической обработкой корреляционной функ-
ции, метод двухцветной ФКС, в принципе, дает ответ типа «да» или «нет» на
вопрос о комплексообразовании между молекулами.
Литература для дальнейшего чтения
Исторический обзор и введение в биологические проблемы
Bastianes Р. I. Н. and Pepperkok R. Observing proteins in their natural habitat: the
living cell. TIBS, 2000. 25, 631-637.
Elson E. L. and Magde D. Fluorescence correlation spectroscopy. I. Conceptual basis
and theory. Biopolymers, 1974.13, 1-27.
Haustein E. and Schwille P. Ultrasensitive investigations of biological systems by
fluorescence correlation spectroscopy. Methods, 2003. 29, 153-166.
Основные принципы метода ФКС
Magde D., Elson E. L. and Webb W. W. Thermodynamic fluctuations in a reacting
system. Measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett., 1972.
29, 705-708.
Eigen M. and Rigler R. Sorting single molecules: application to diagnostics and
evolutionary biothechnology. Proc. Natl. Acad Sci. USA, 1994. 91, 5740-5747.
Bacia K. and Schwille P. A dynamic view of cellular processes by in vivo fluorescence
auto- and cross-correlation spectroscopy. Methods, 2003. 2974-85.
Schwille P. and Kettling U. Analyzing single protein molecules using optical methods.
Cwt. Opin. Biotech., 2001.12, 382-386.
Двухцветная флуоресцентная кросс-корреляционная спектроскопия
SchwilleP., Meyer-AlmesF.J. and Rigler R. Dual-colour fluorescence cross-correlation
spectroscopy for multicomponent diffusional analysis in solution. BiophysJ., 1997. 72,
1878-1886.
Koltermann A., Kettling U., Stephan J., Winkler T. and Eigen M. Dual-color confocal
flourescence spectroscopy and its application in biotechnology. In Fluorescence
Correlation Spectroscopy — Theory and Application, eds. R. Rigler and E. Elson.
Heidelberg: Springer — Verlag, 2001.
Stephan J., Dorre K. et al. Towards a general procedure for sequencing single
DNA molecules./. Biotechnol., 2001. 86, 255-267.