/








































































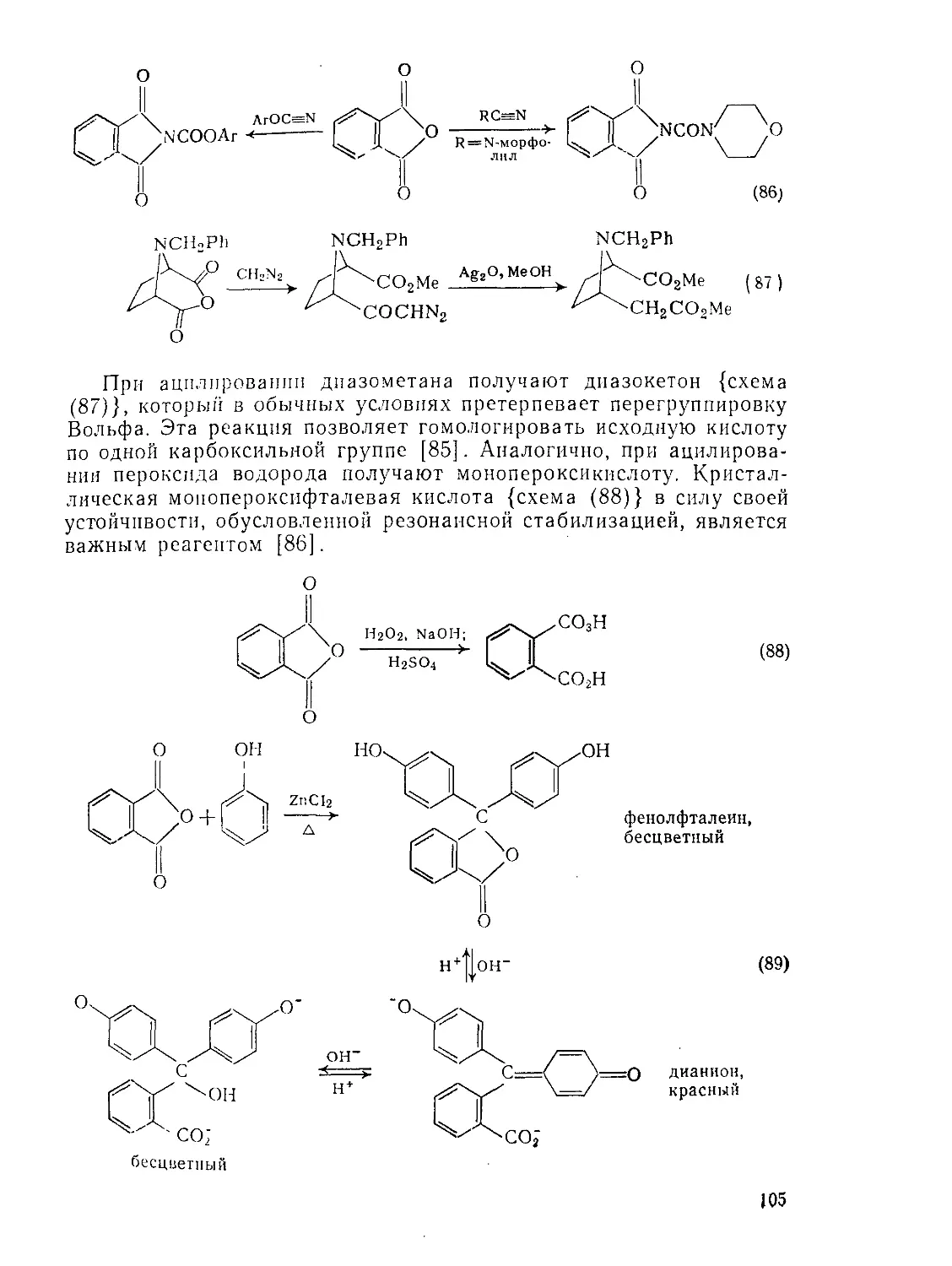
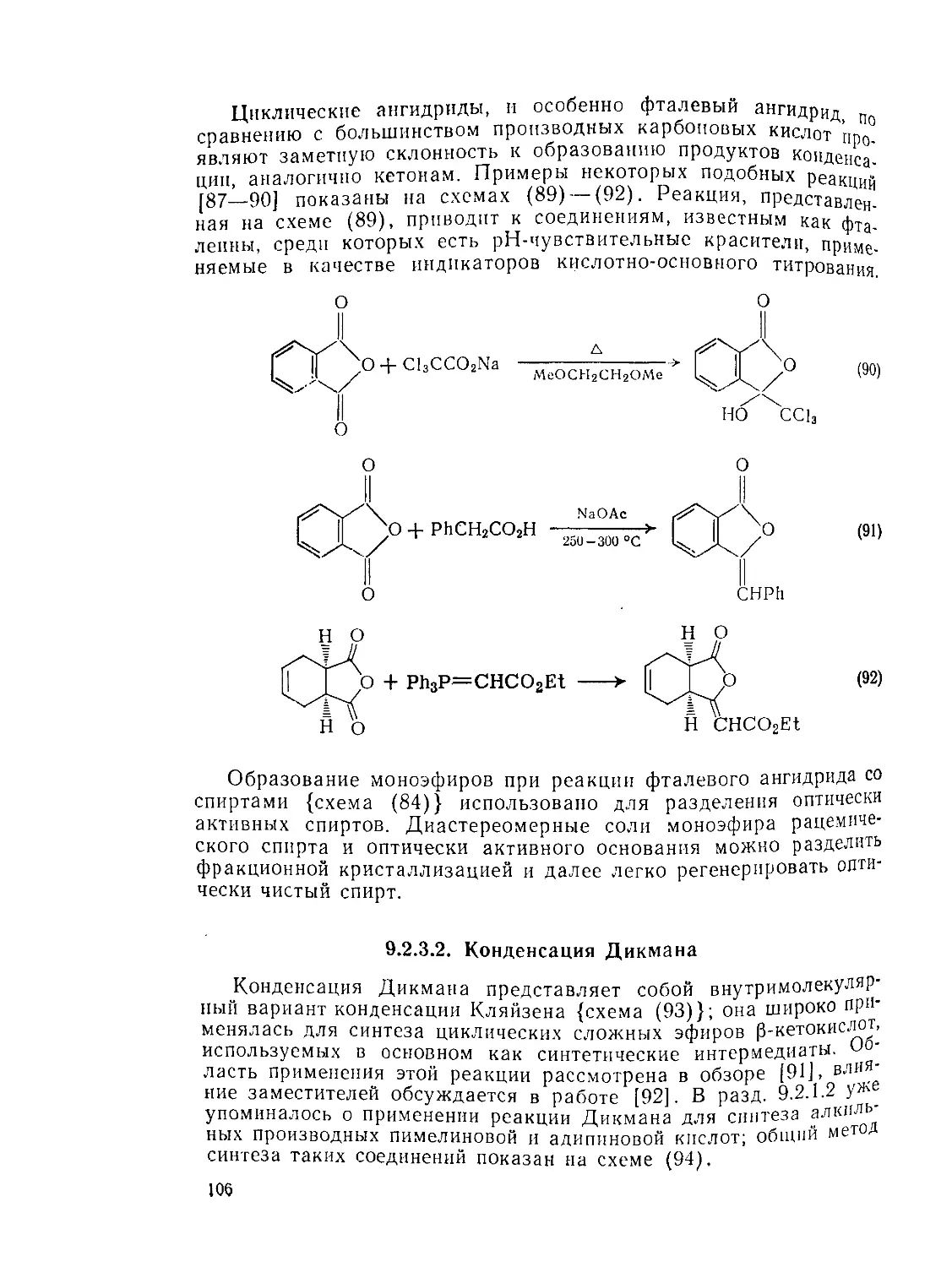
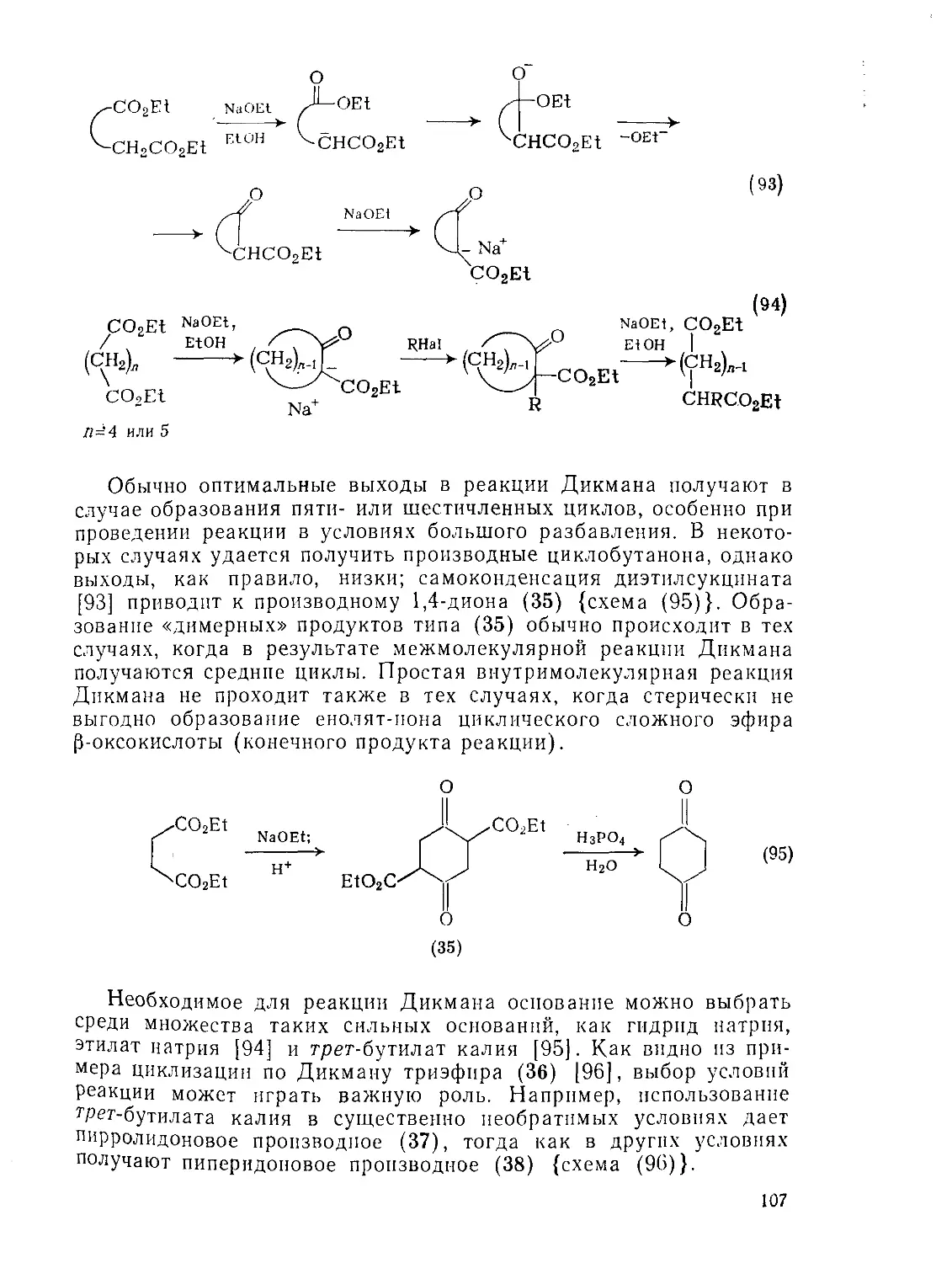
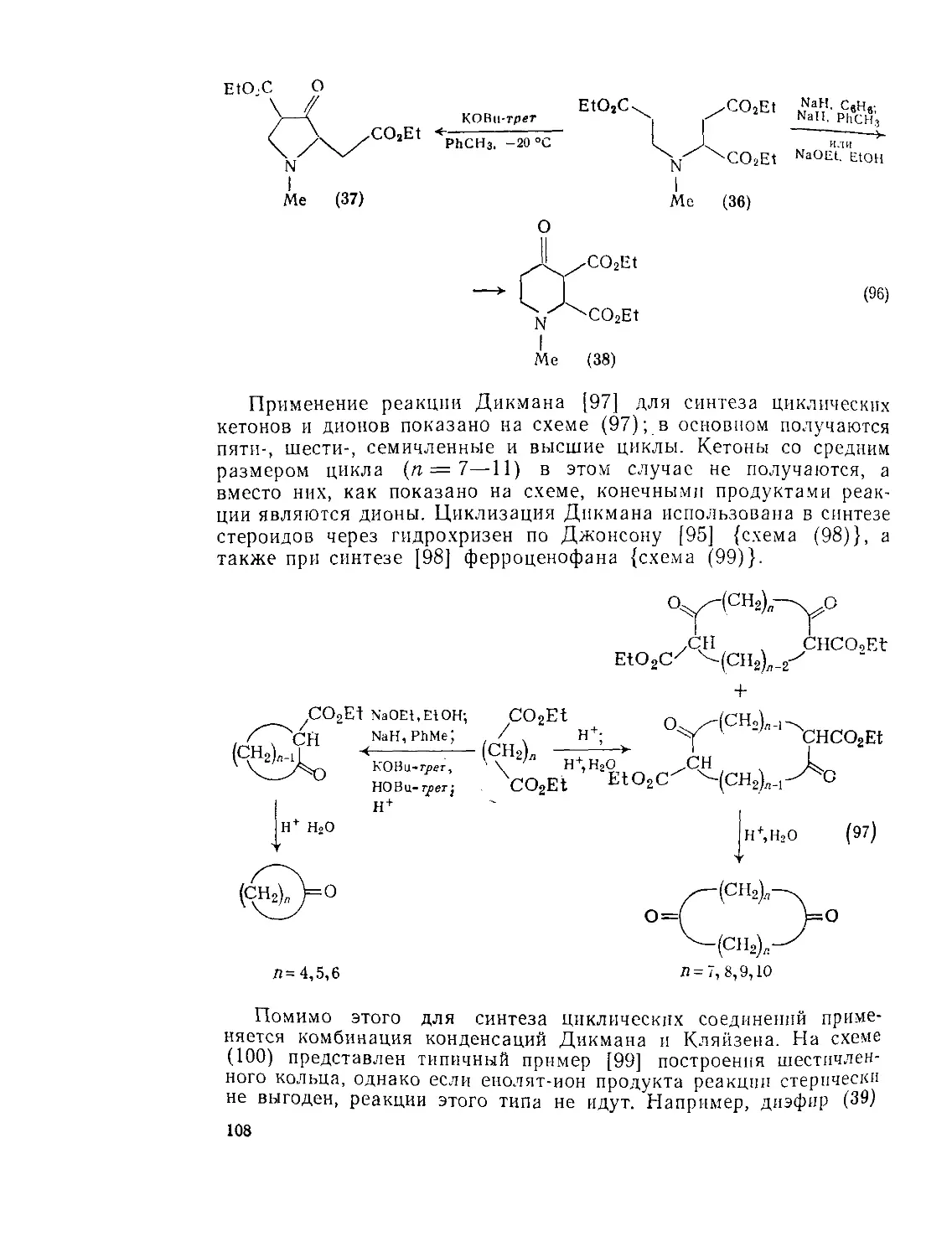







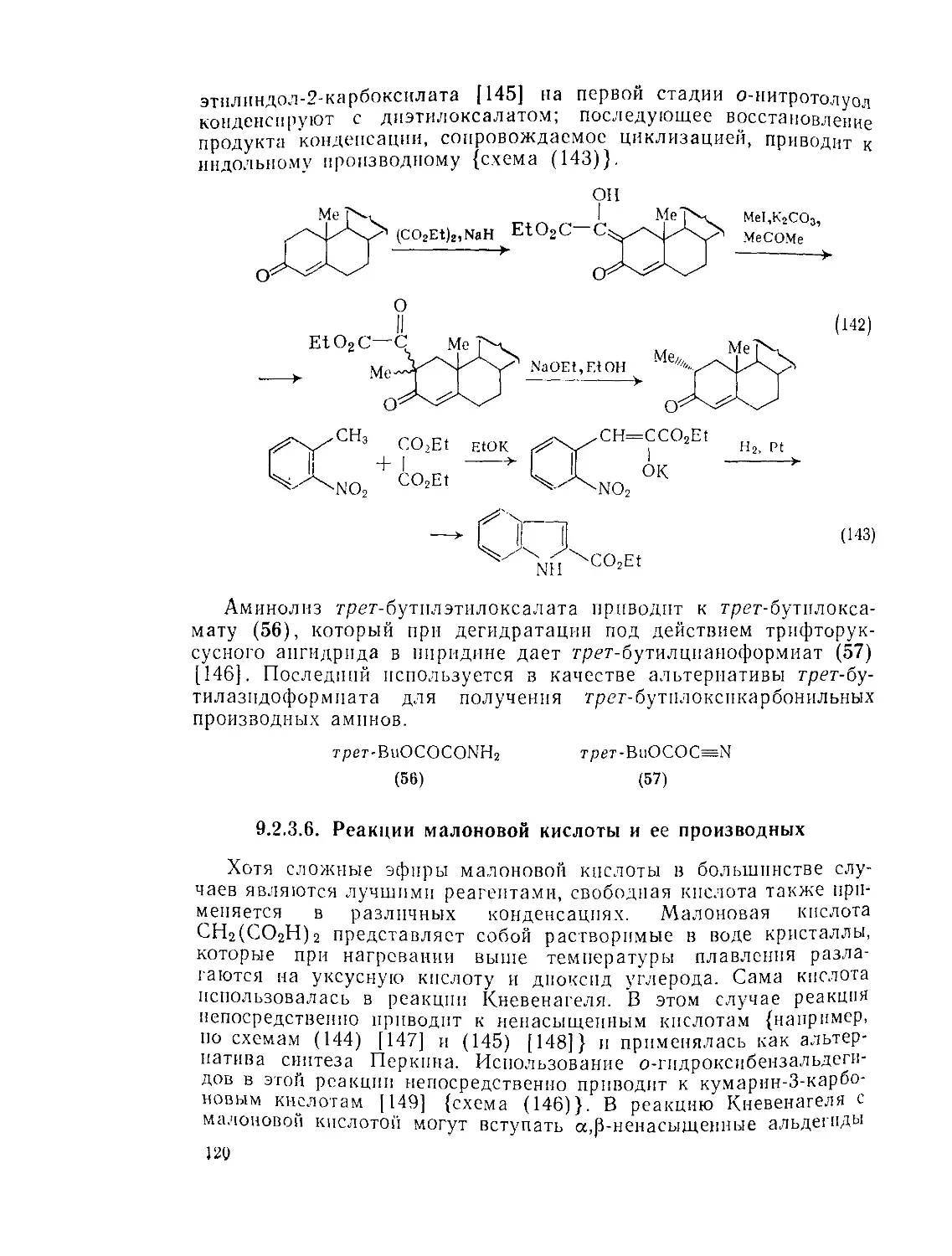


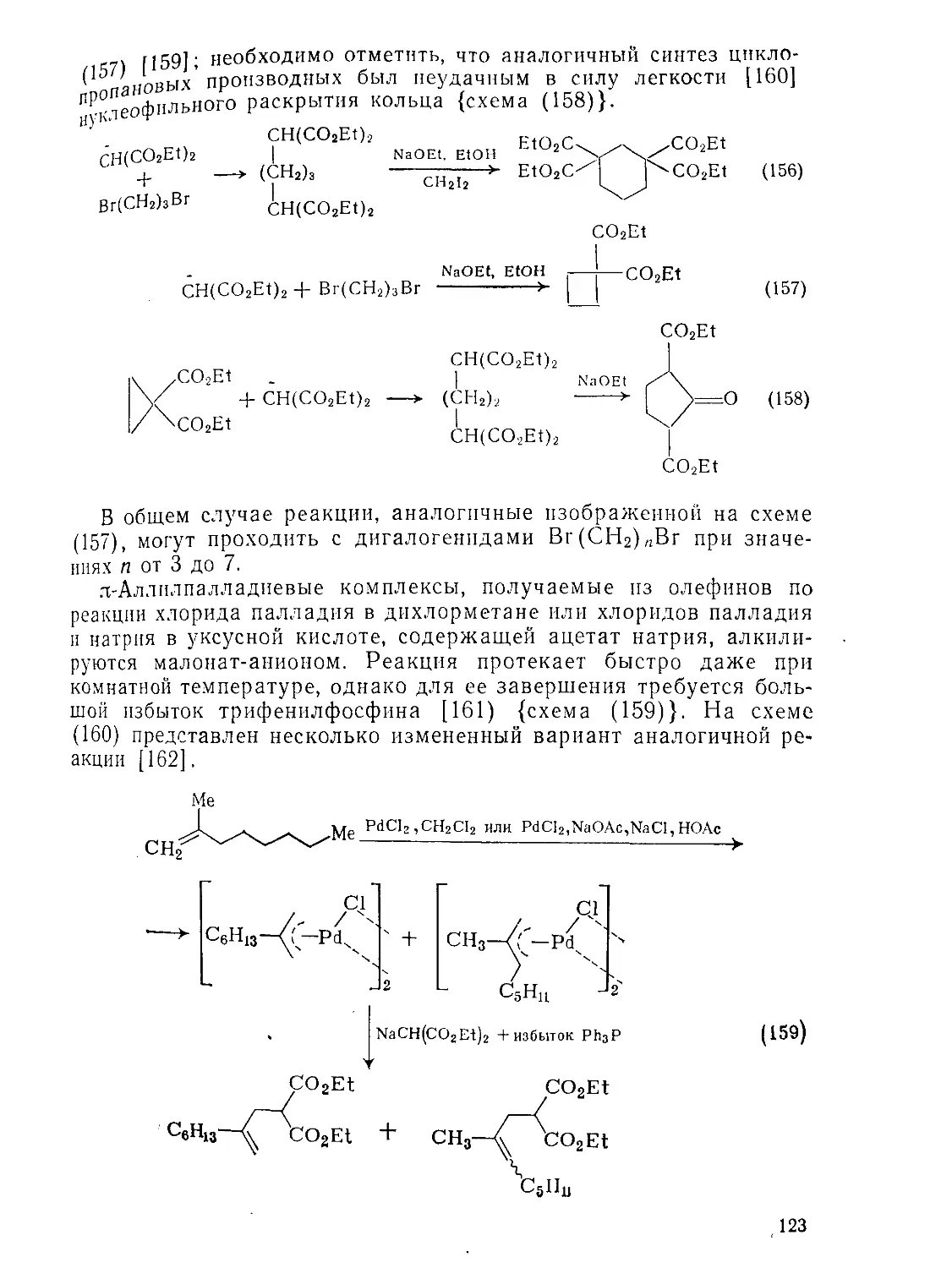

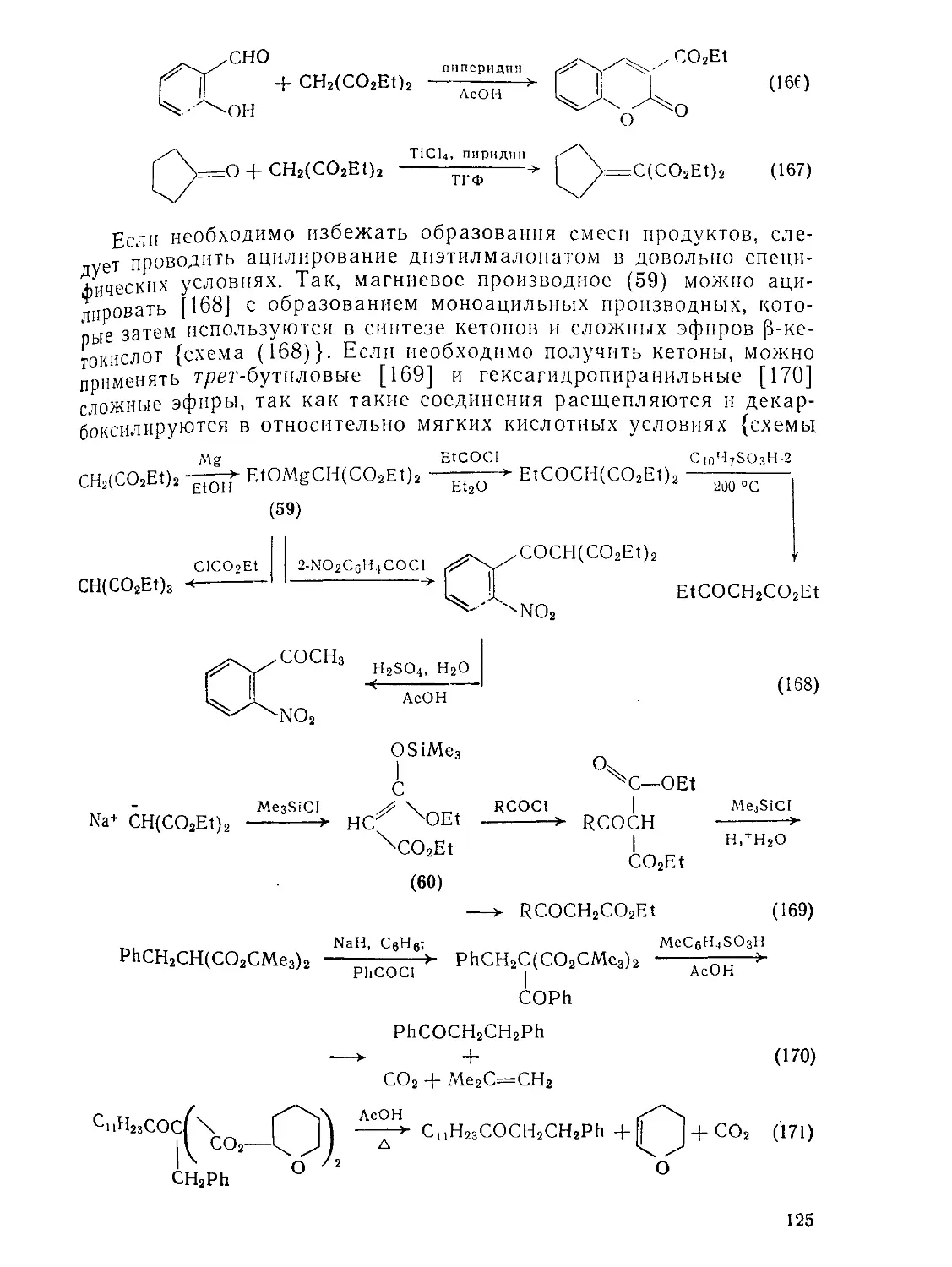
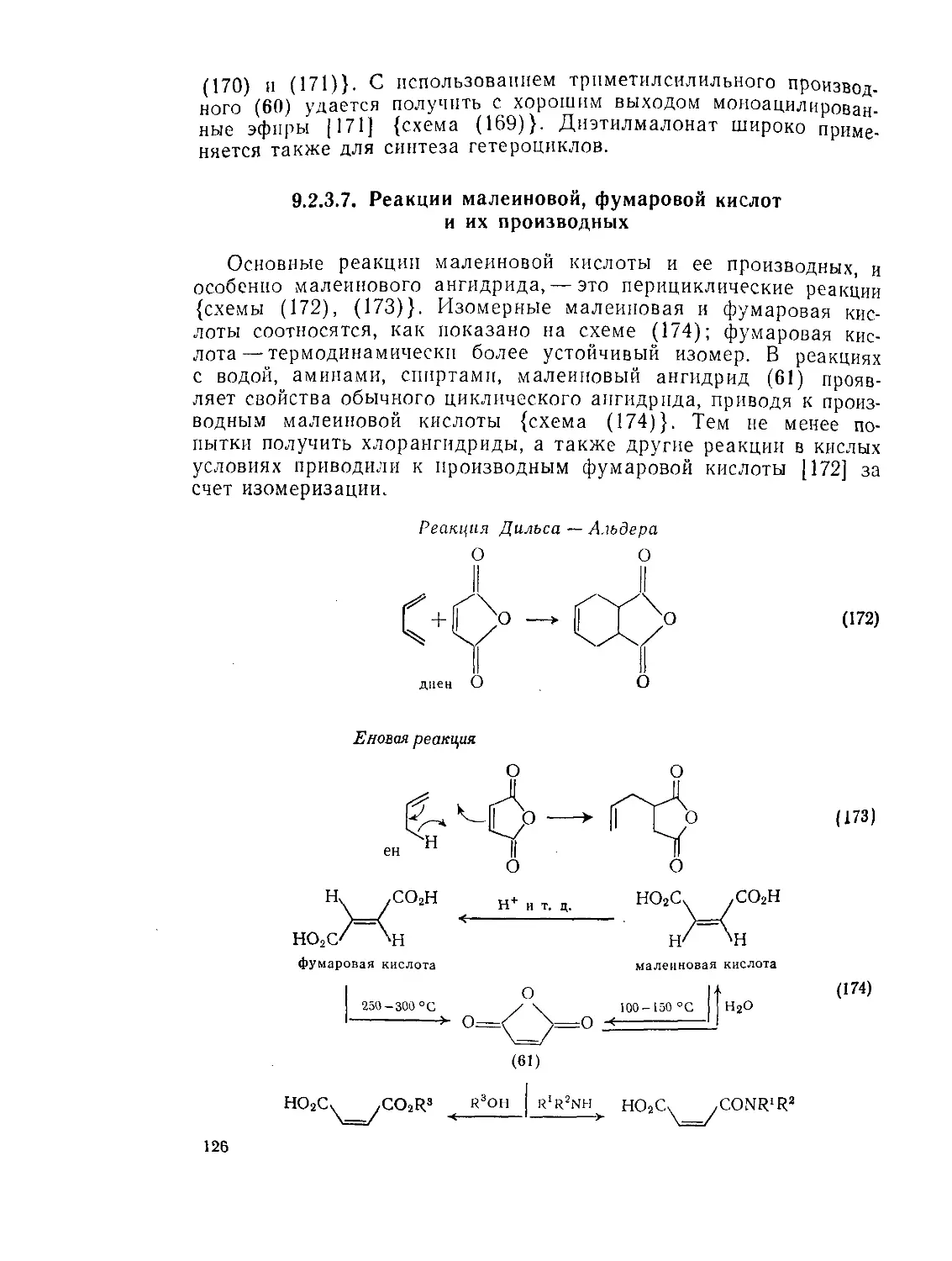
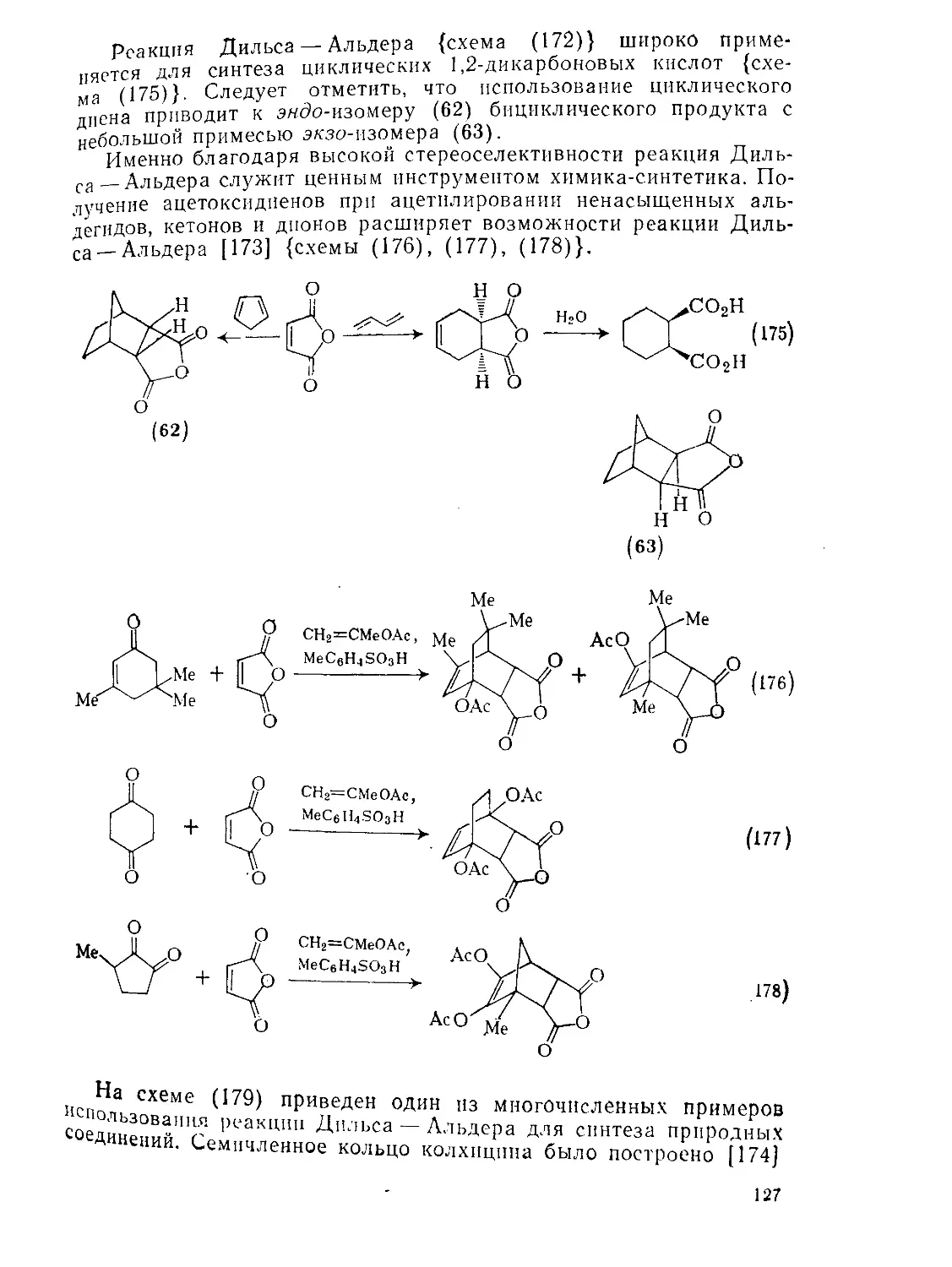
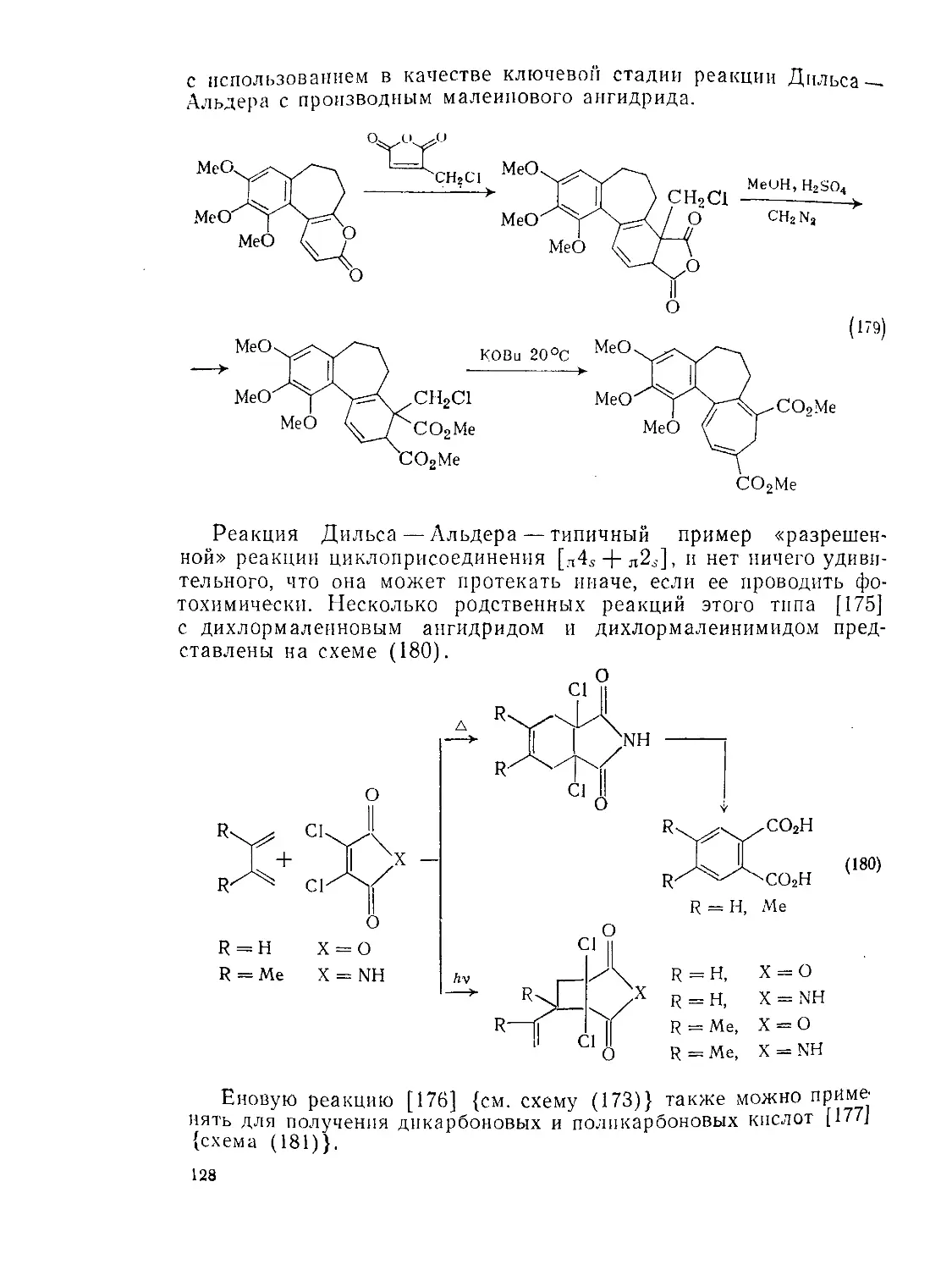

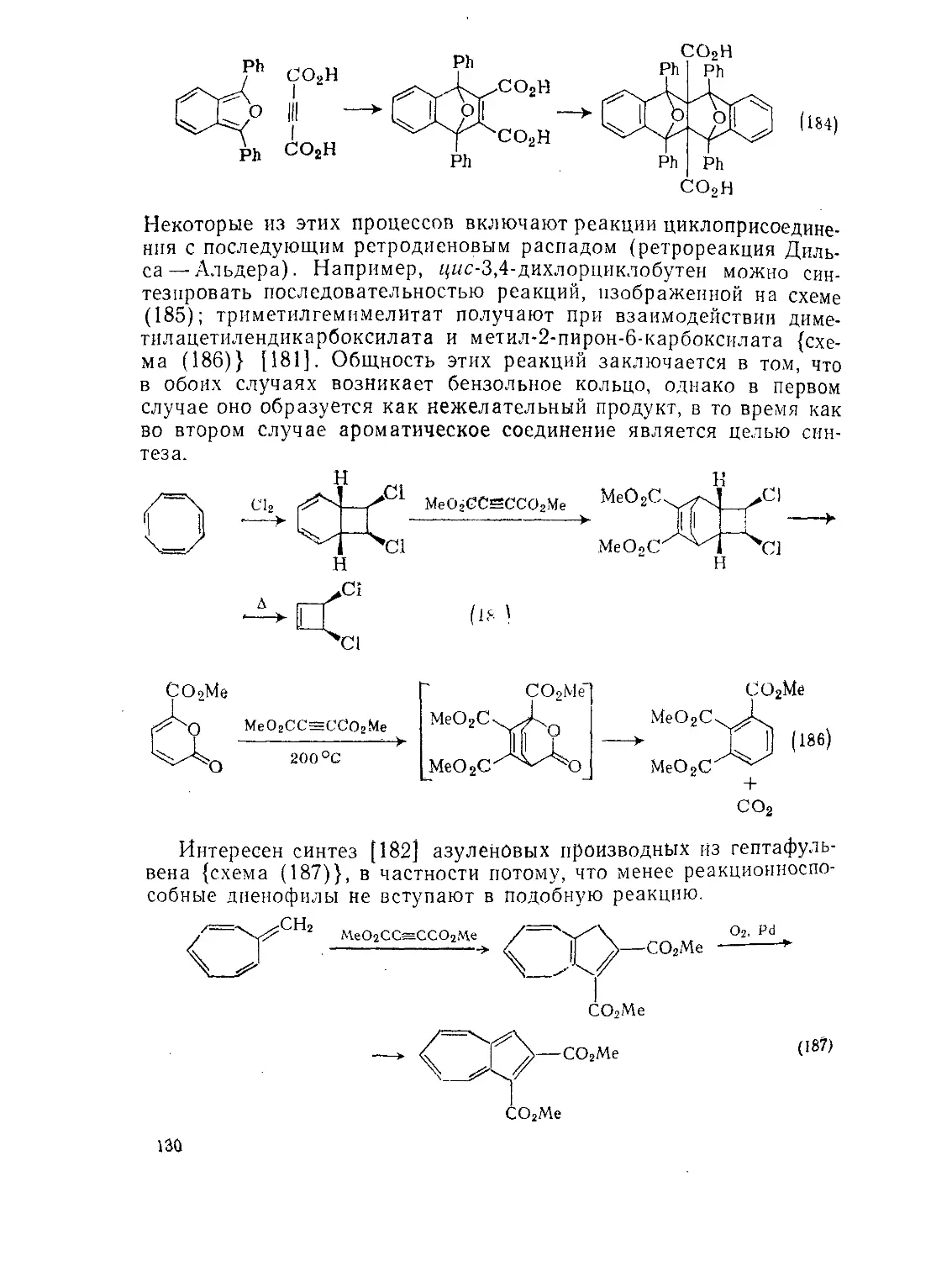








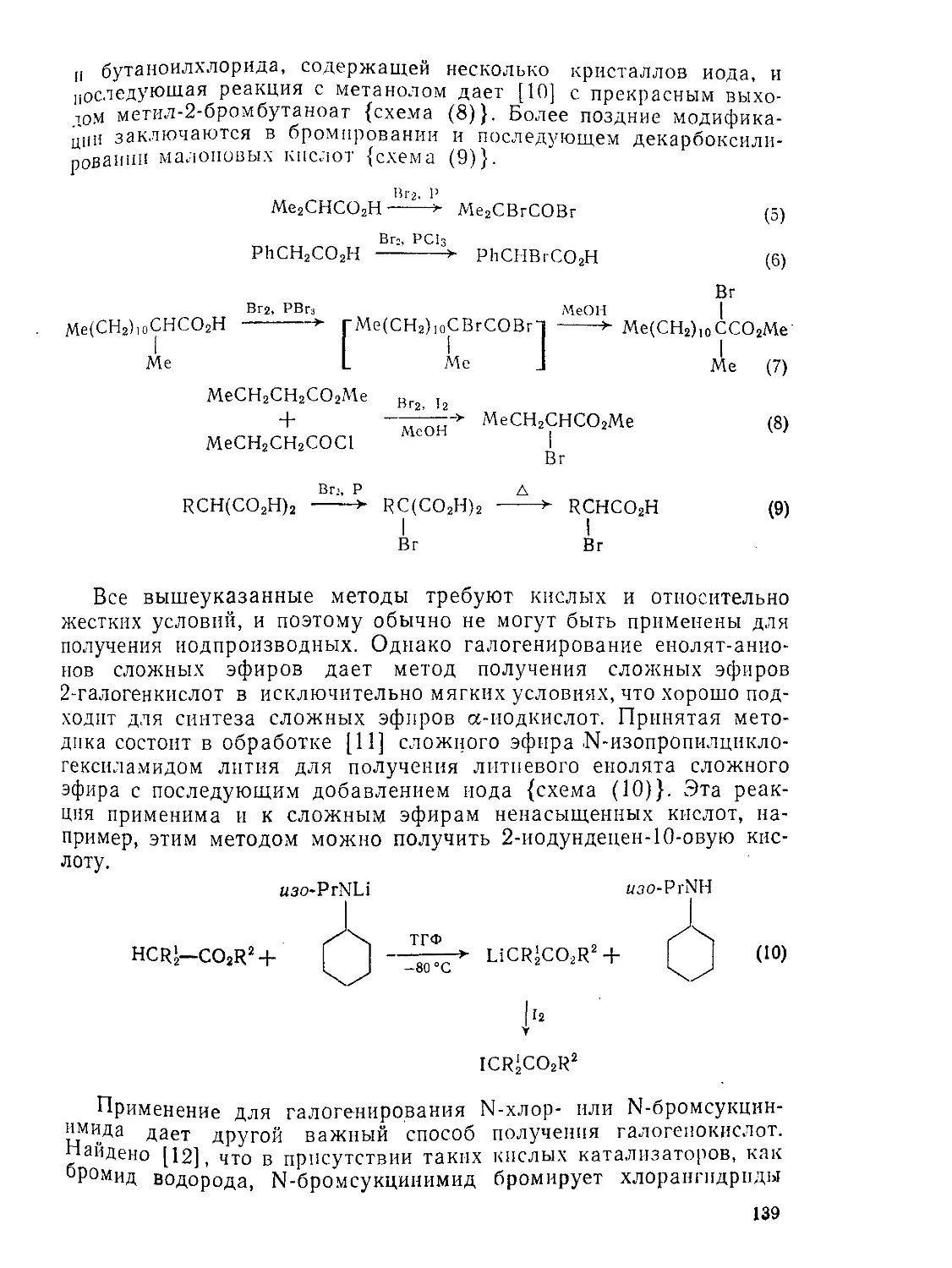










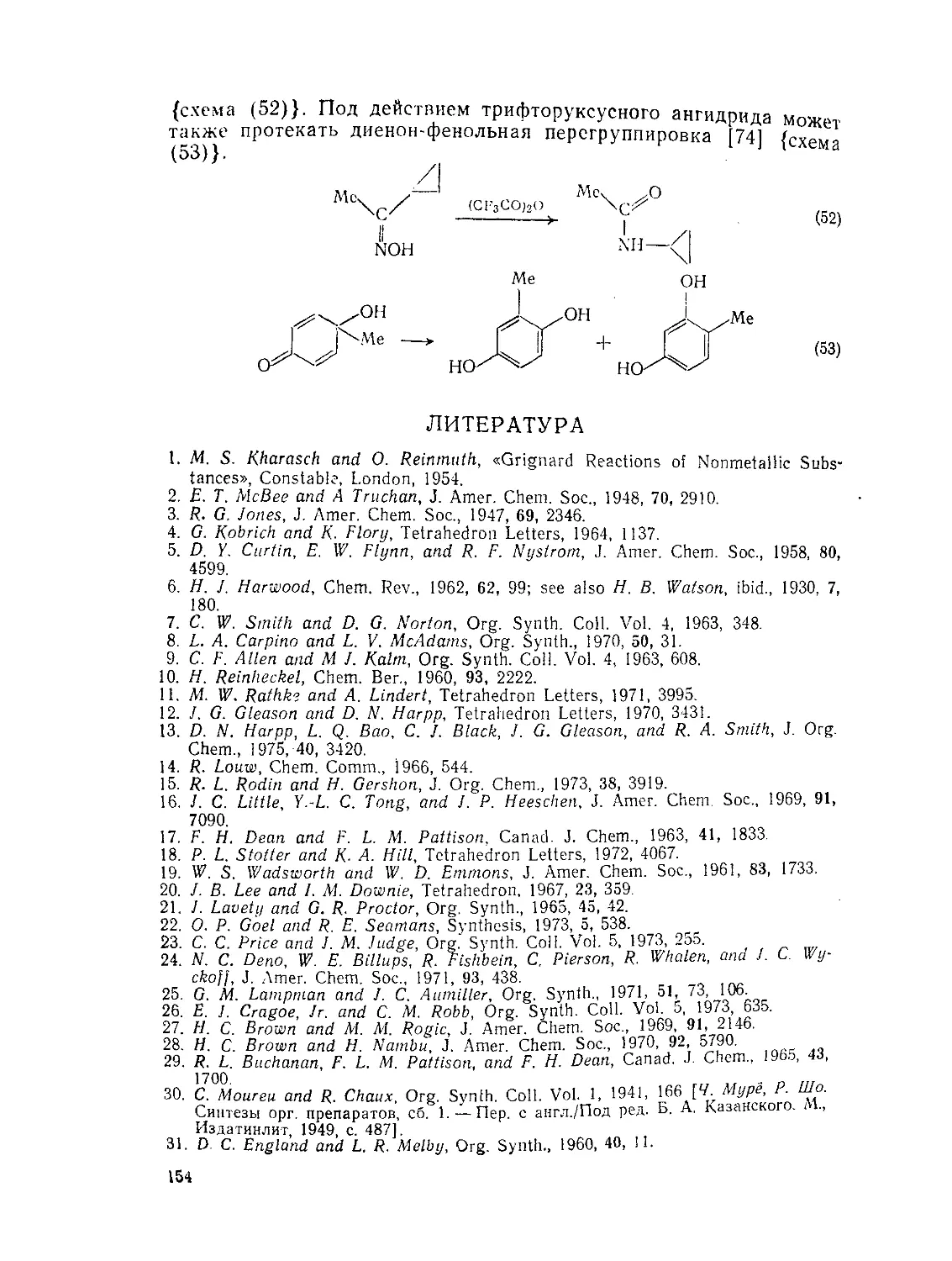
















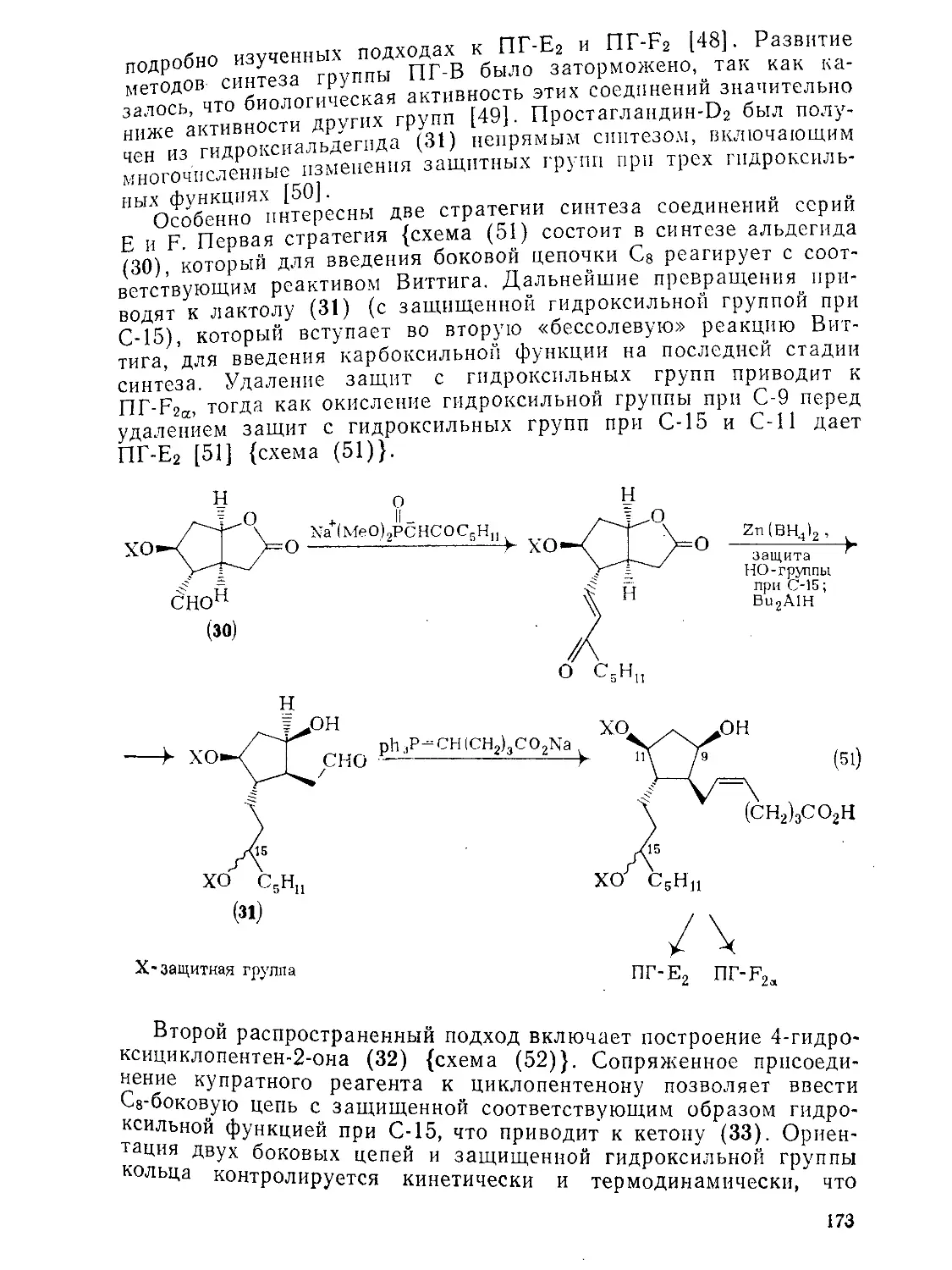
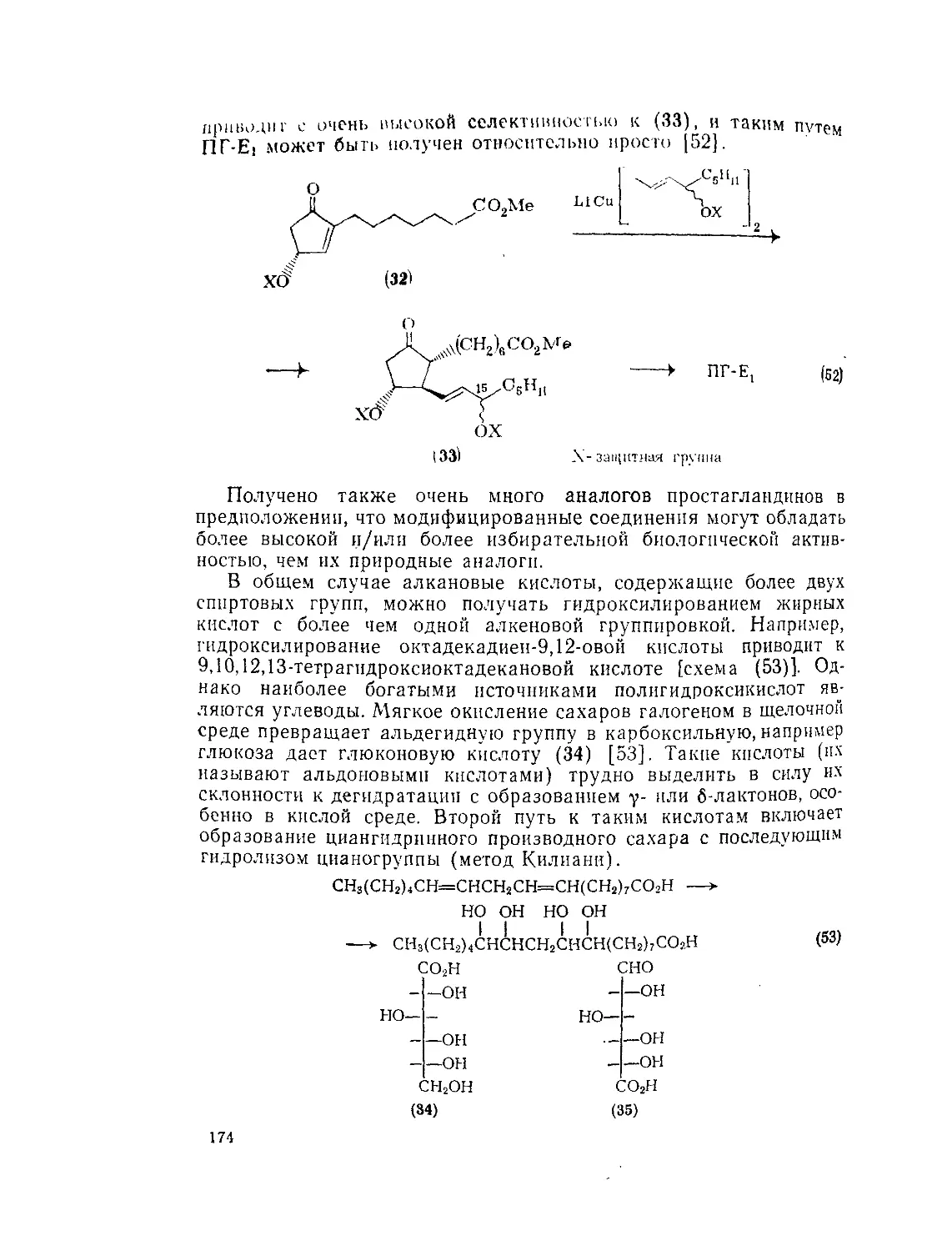






















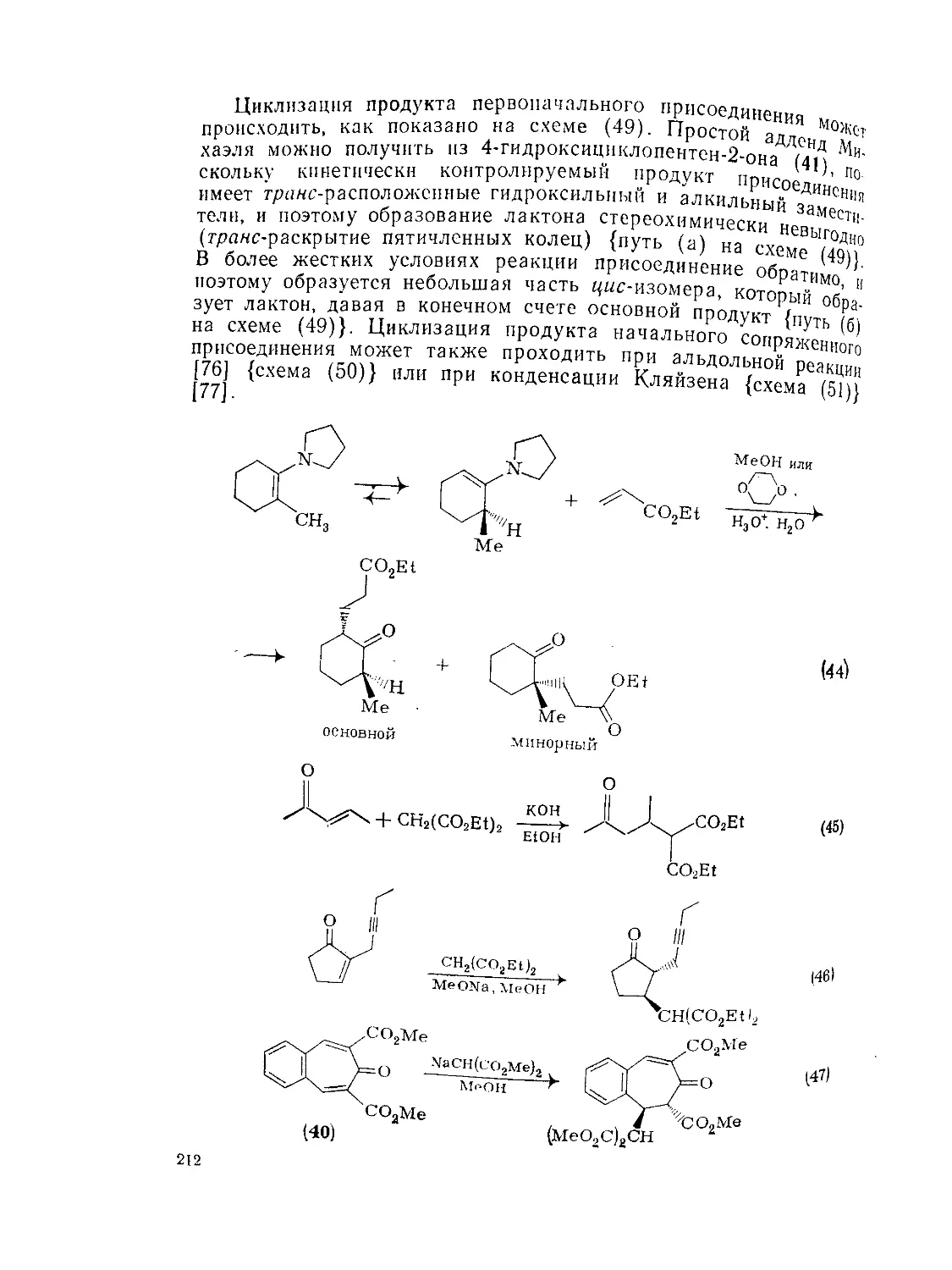
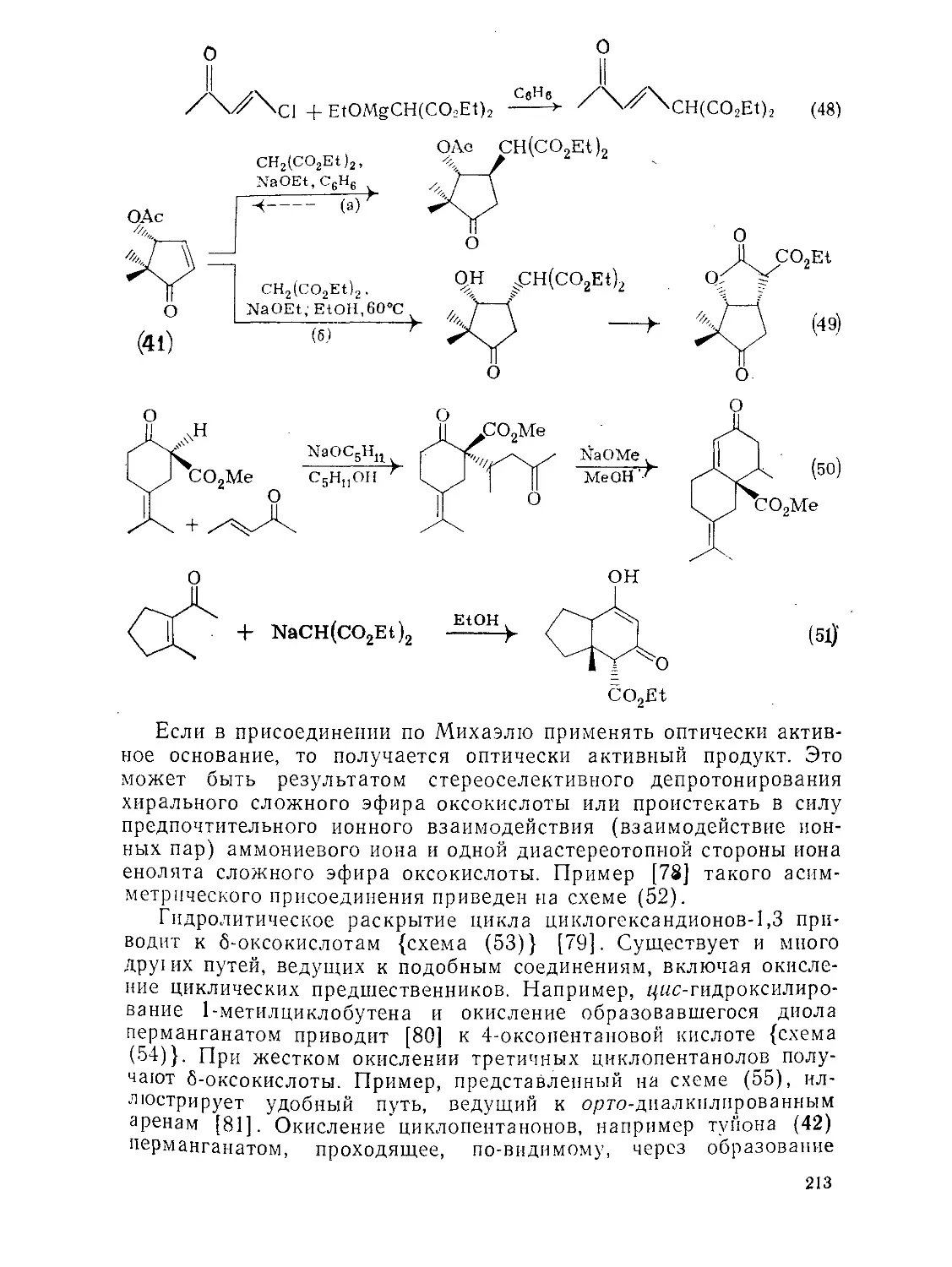
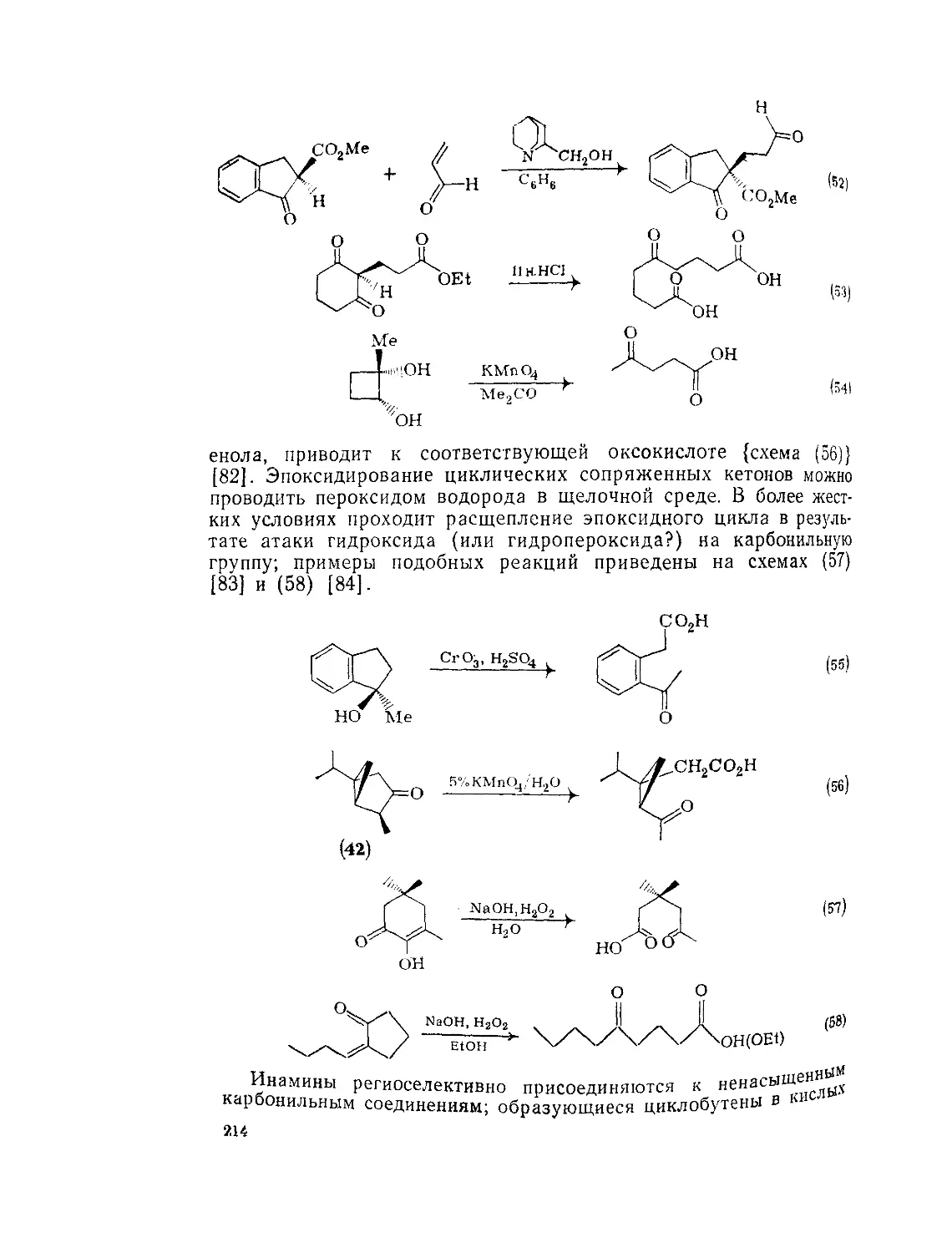


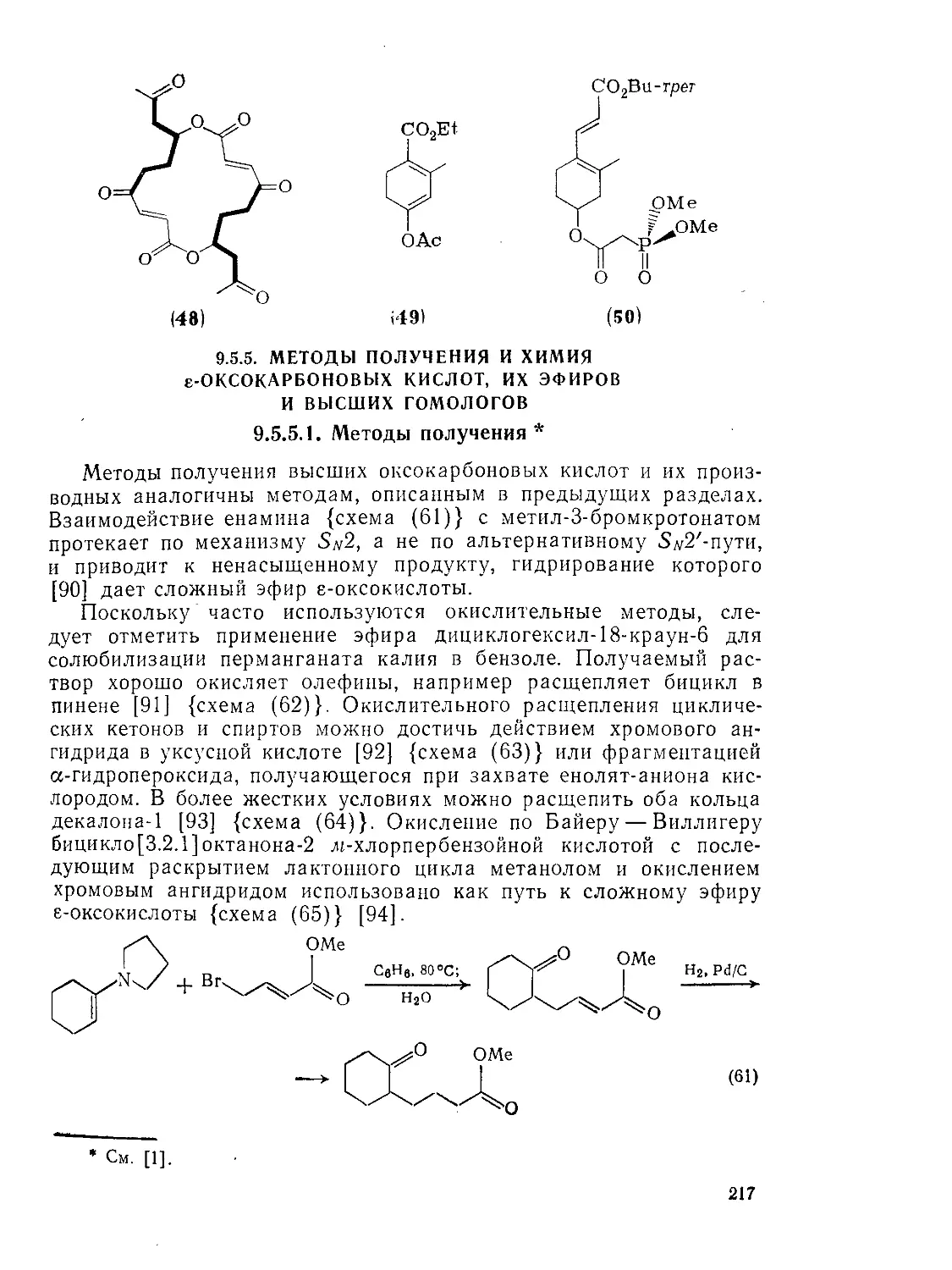


















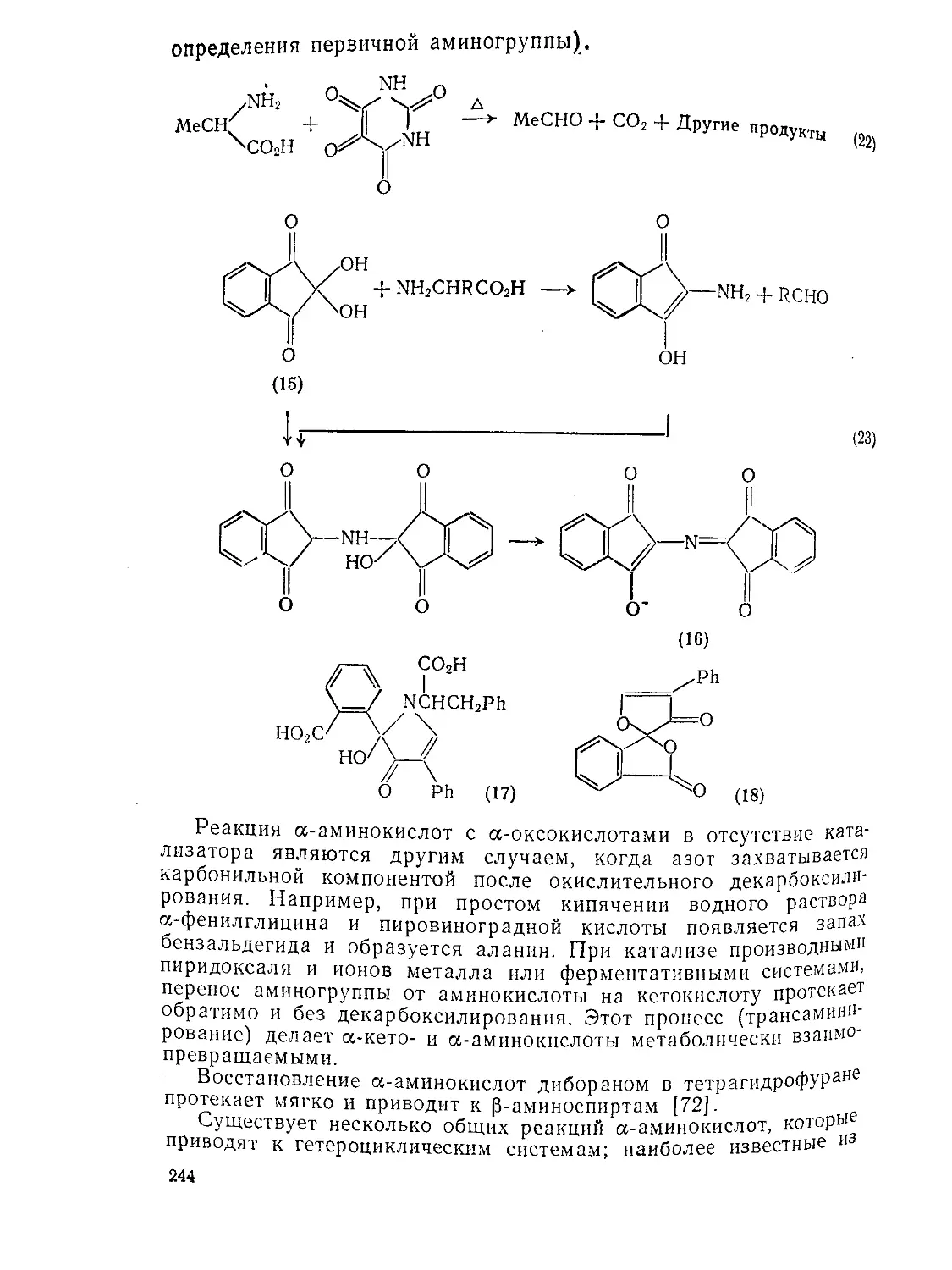
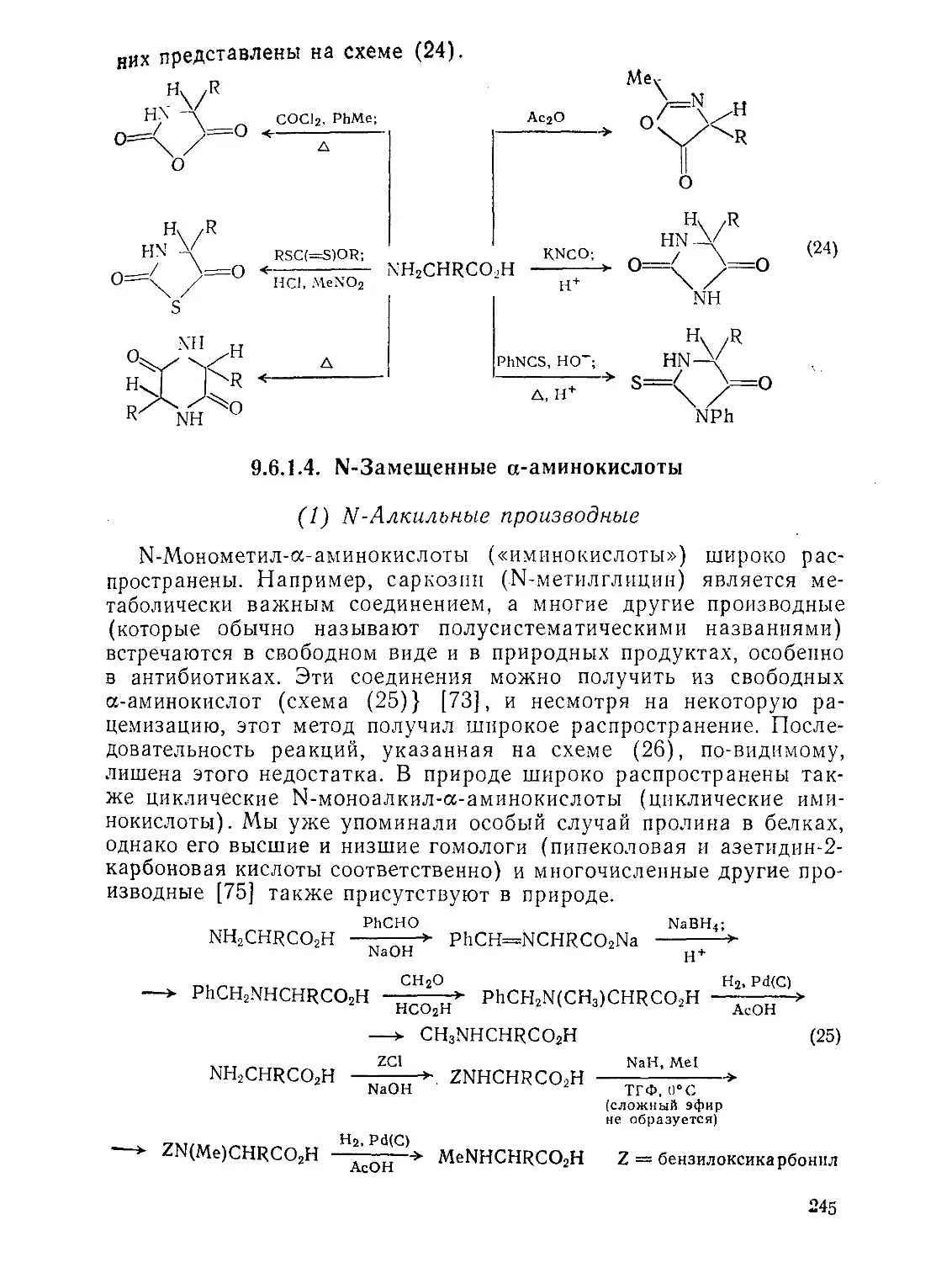

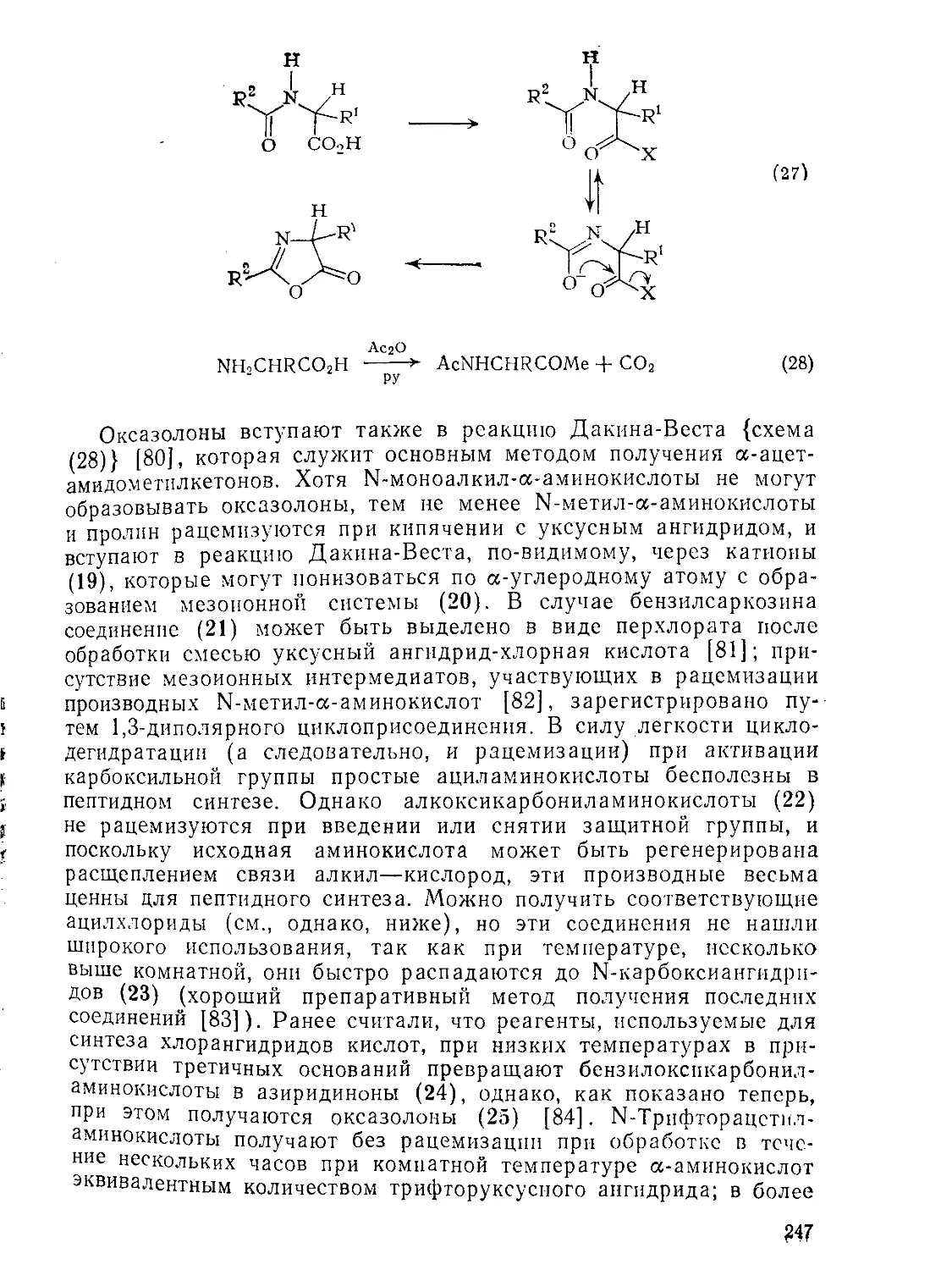




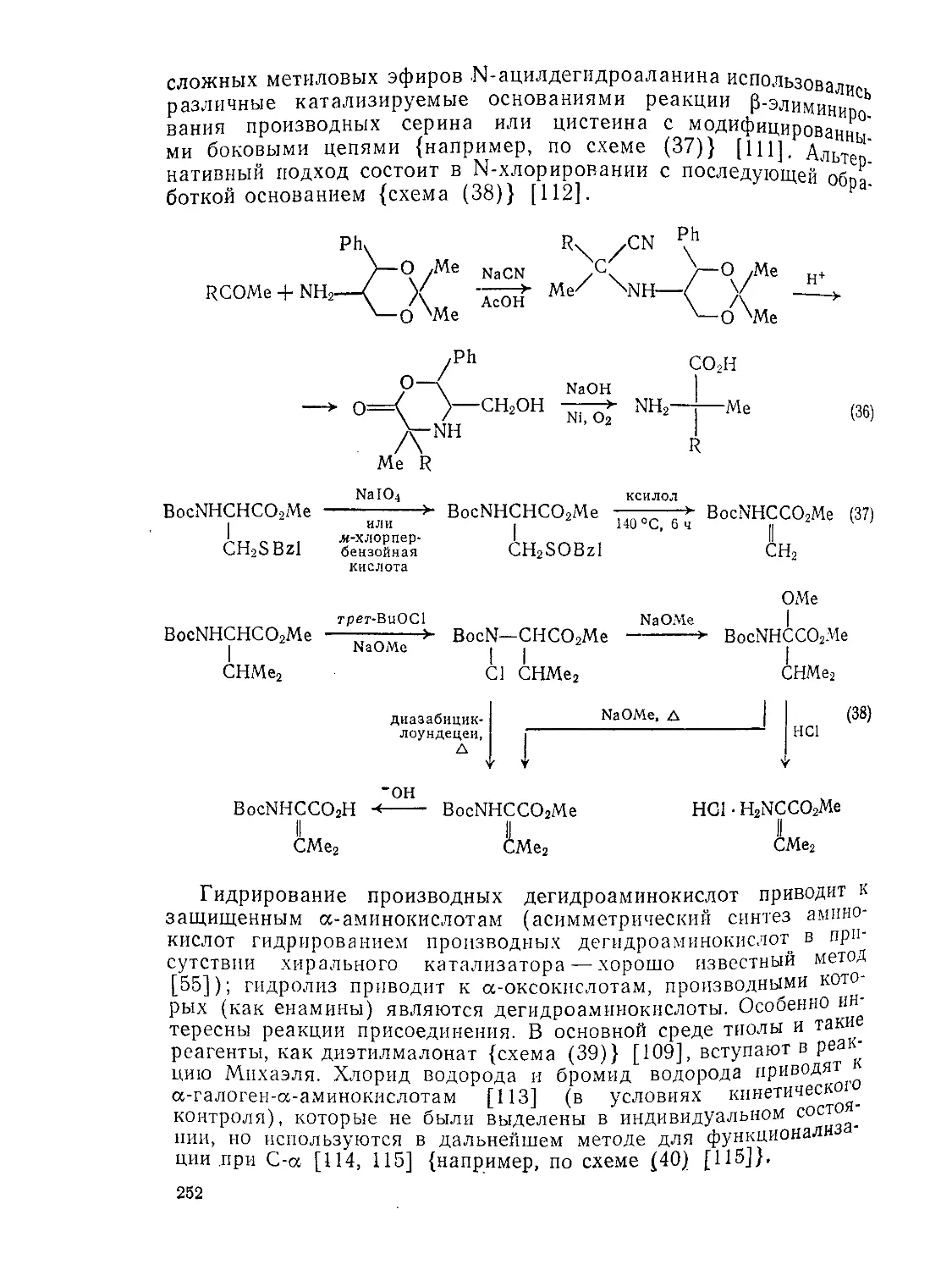















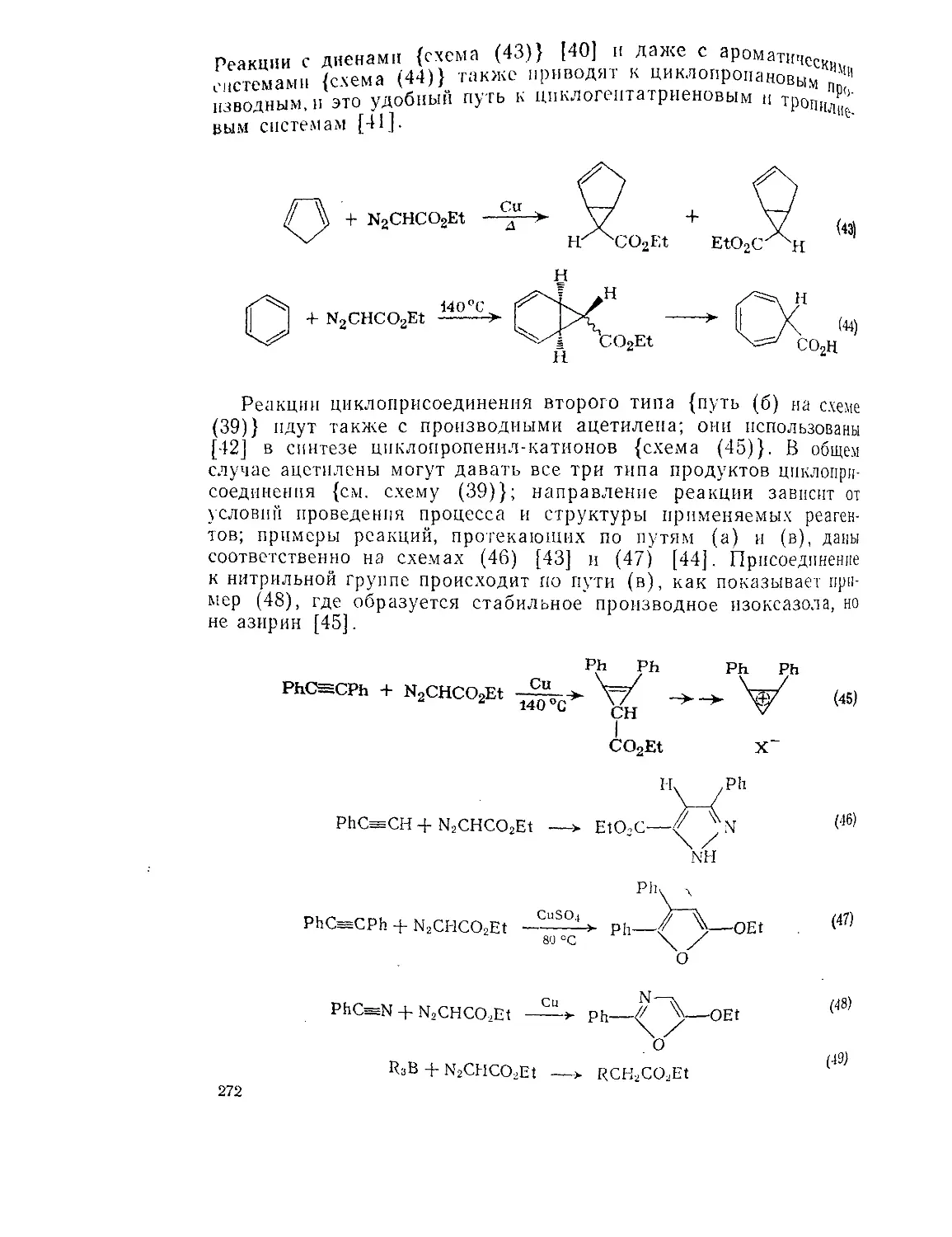


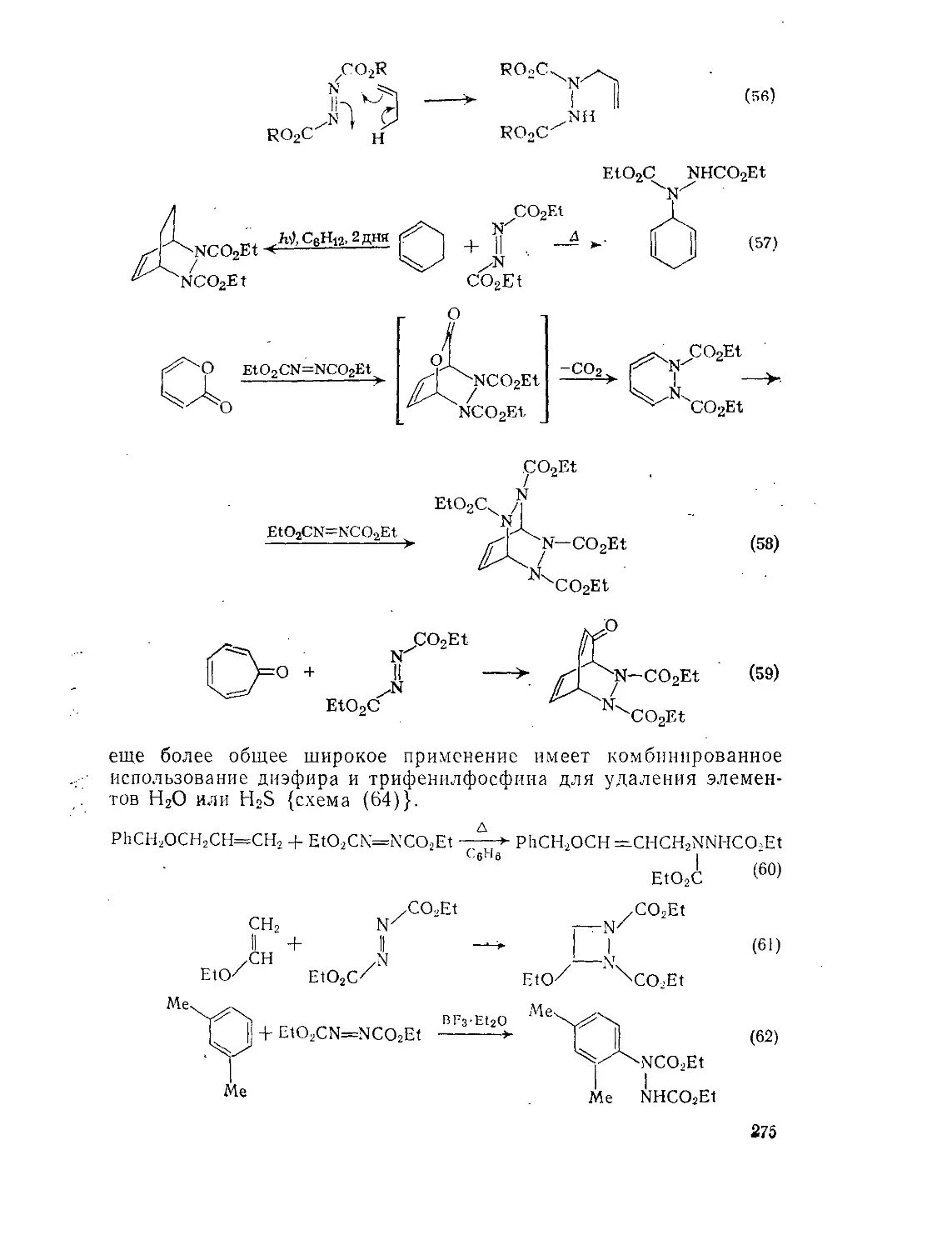
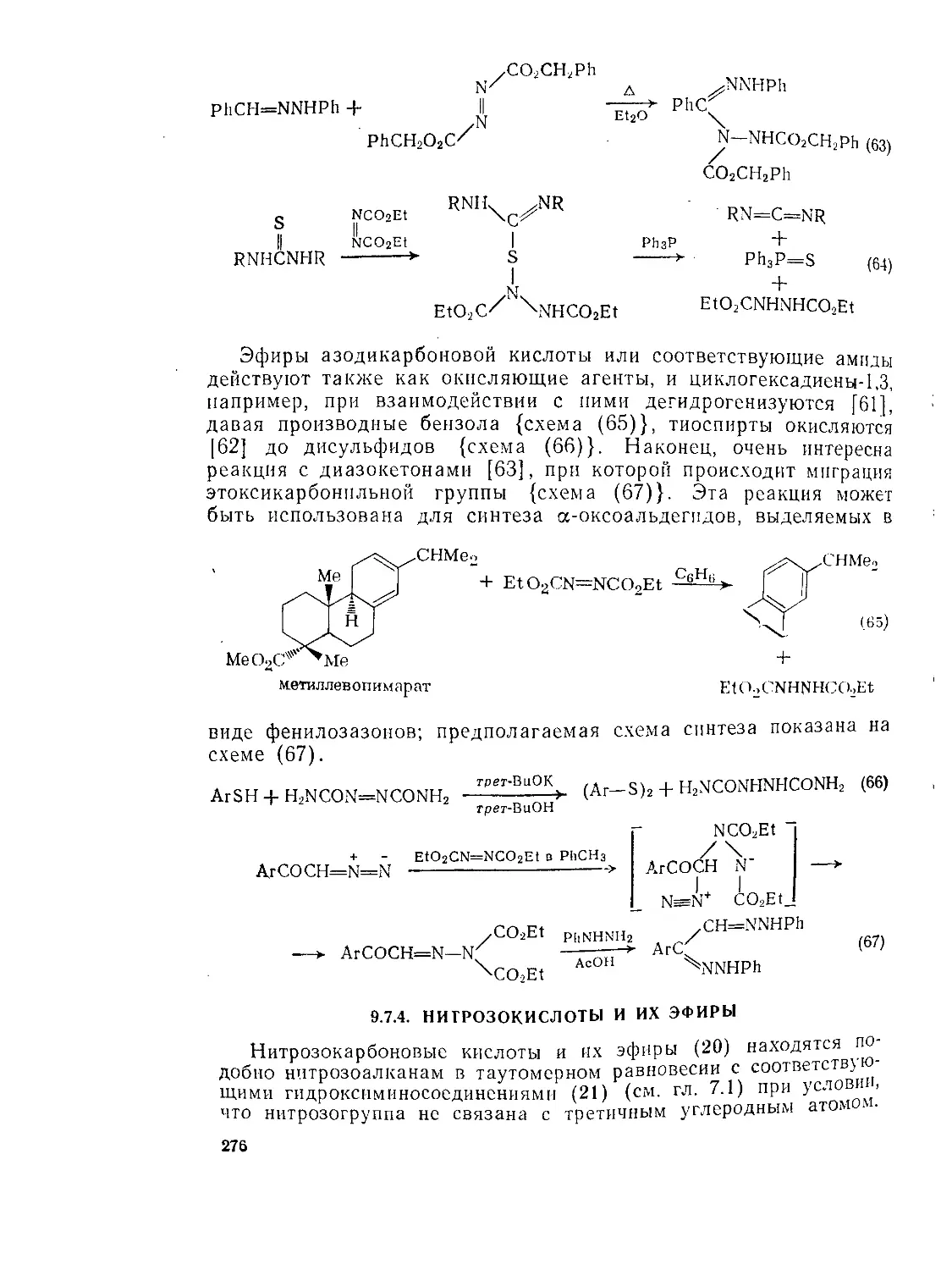
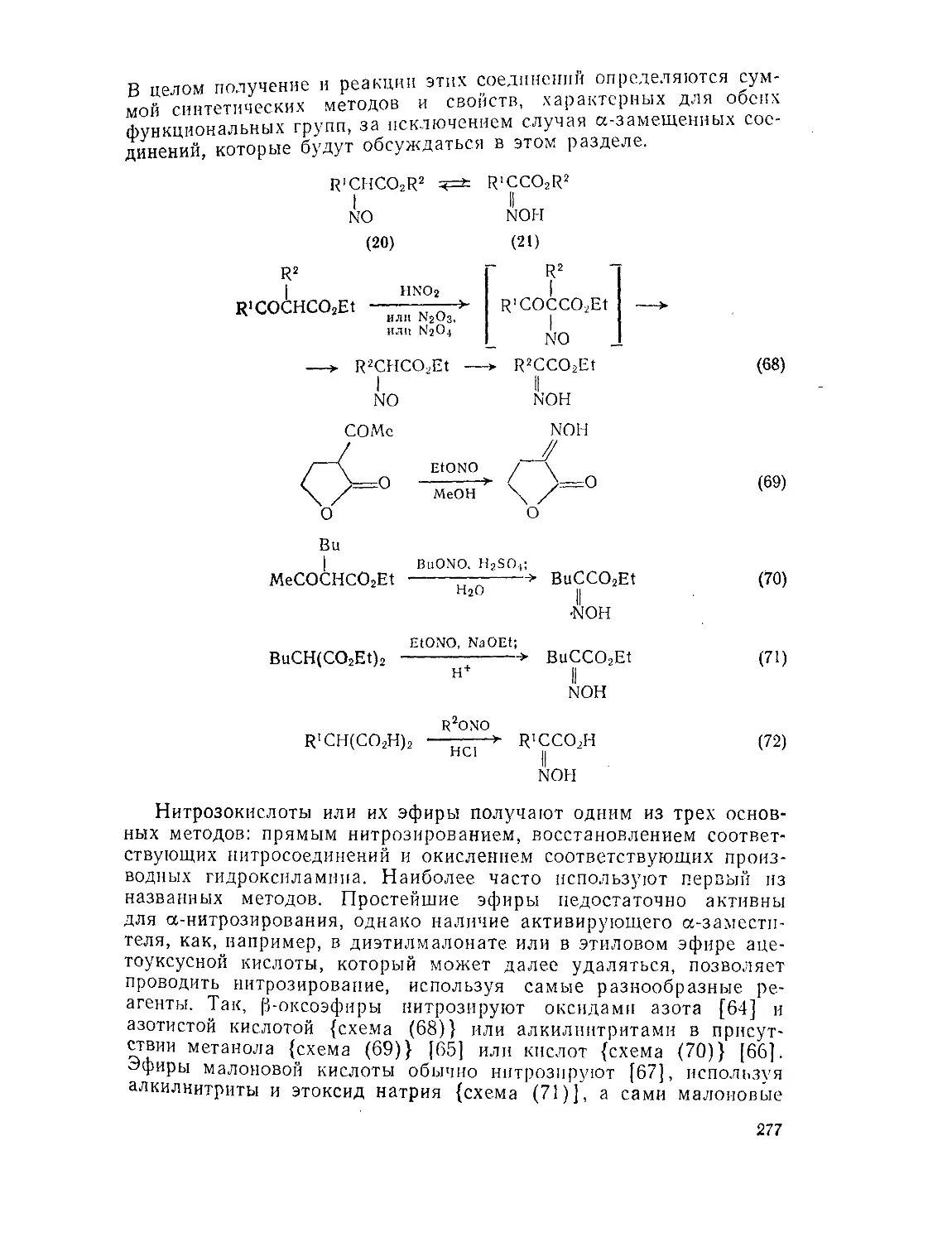















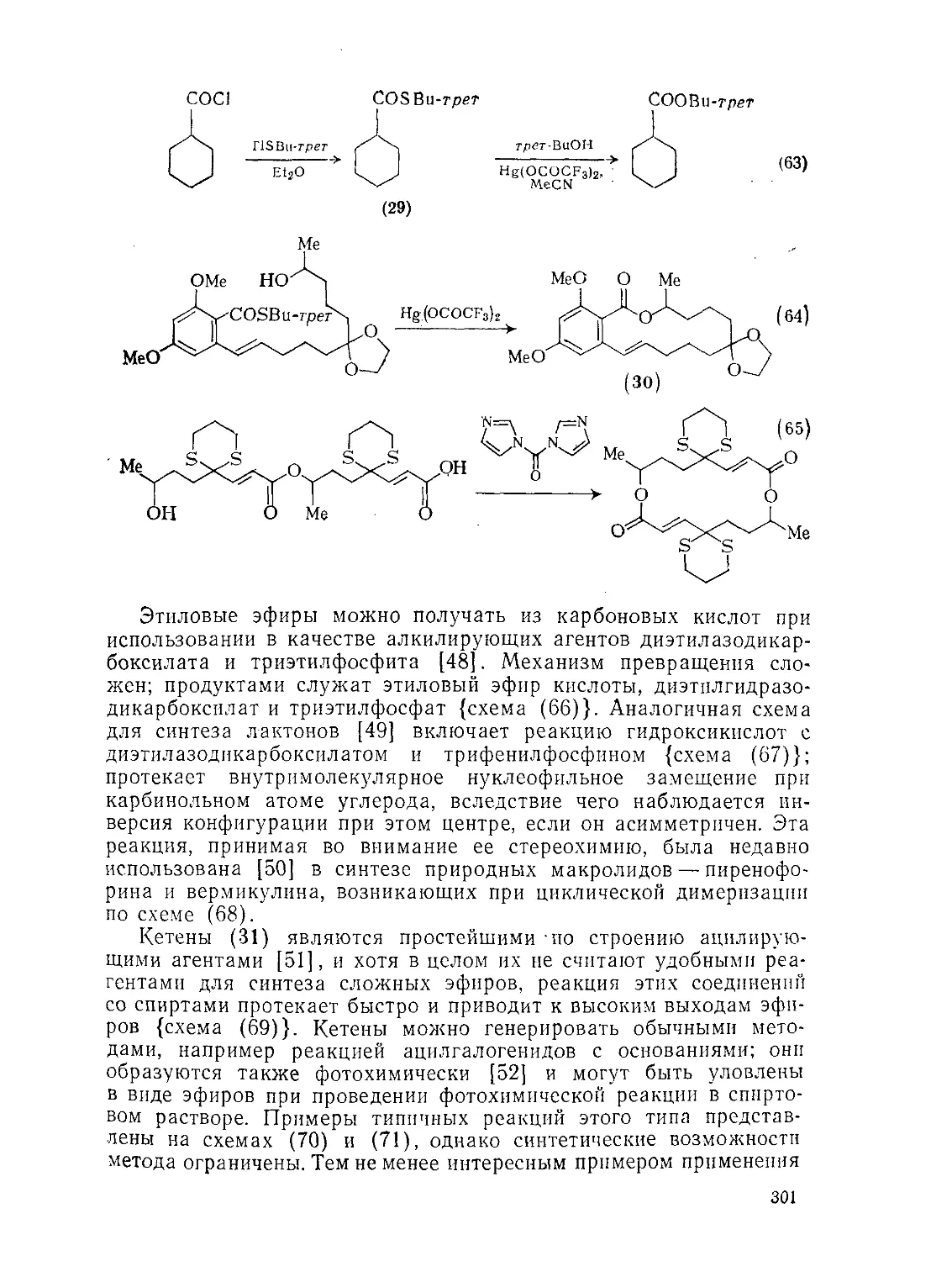
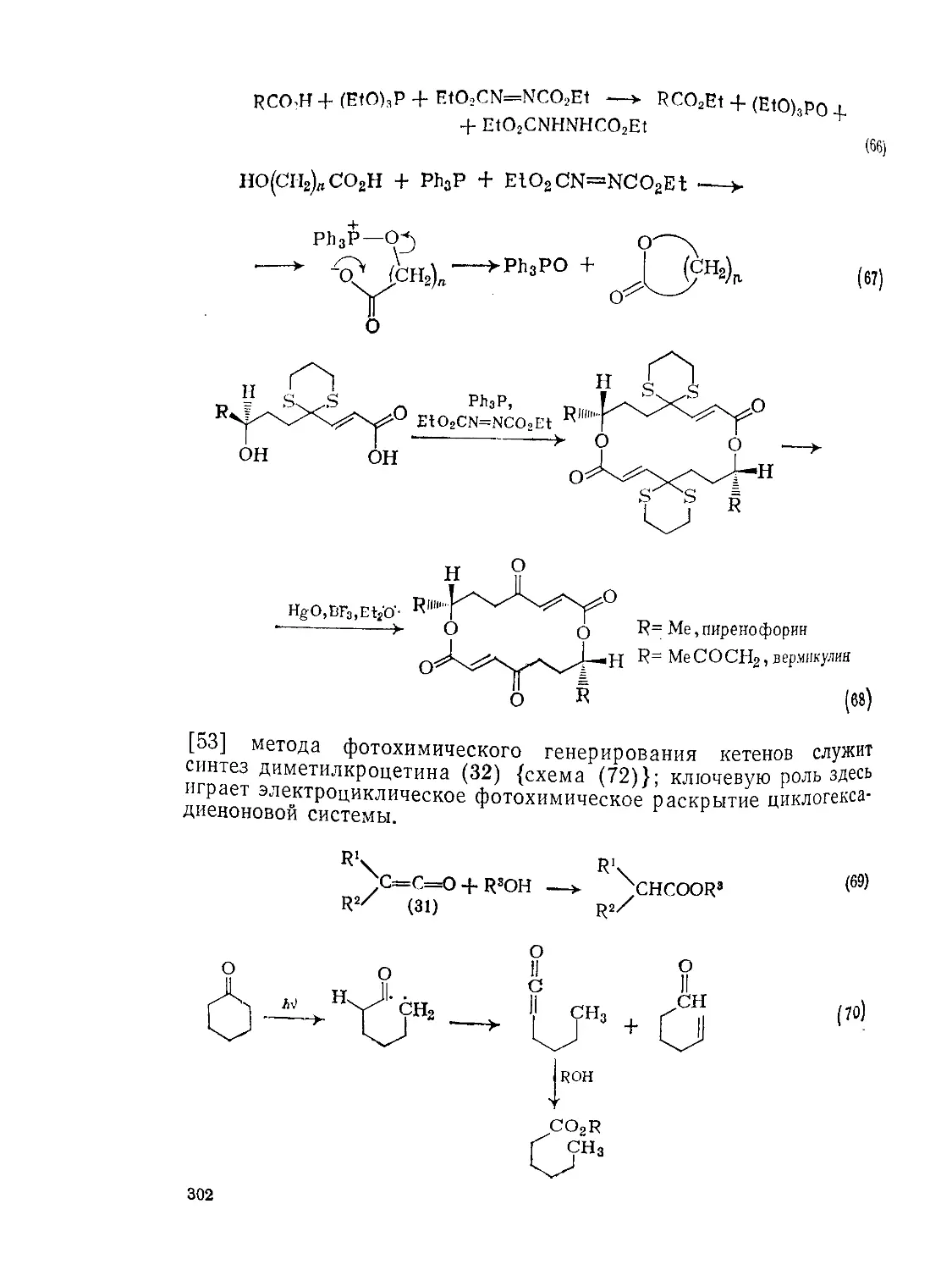
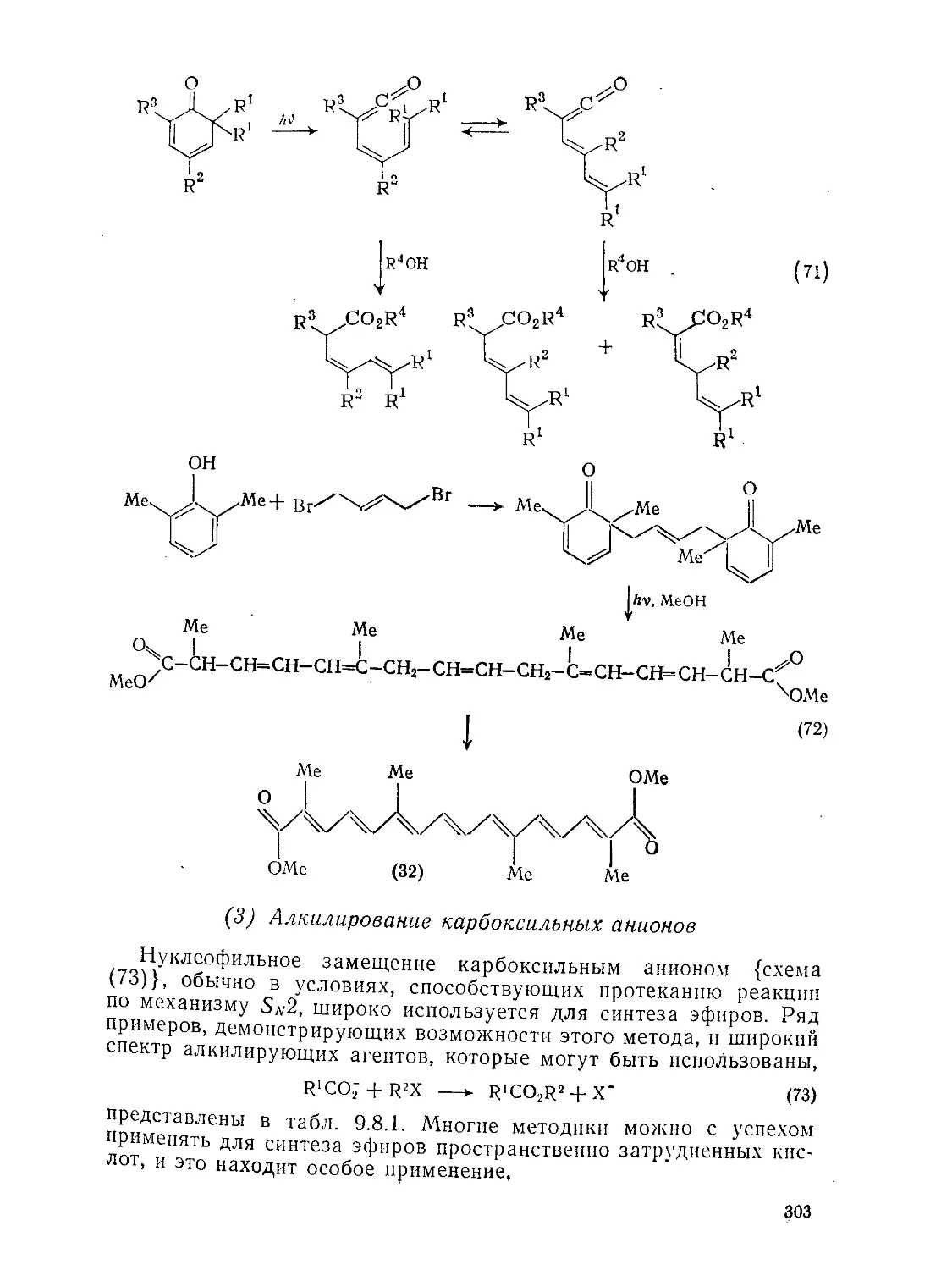




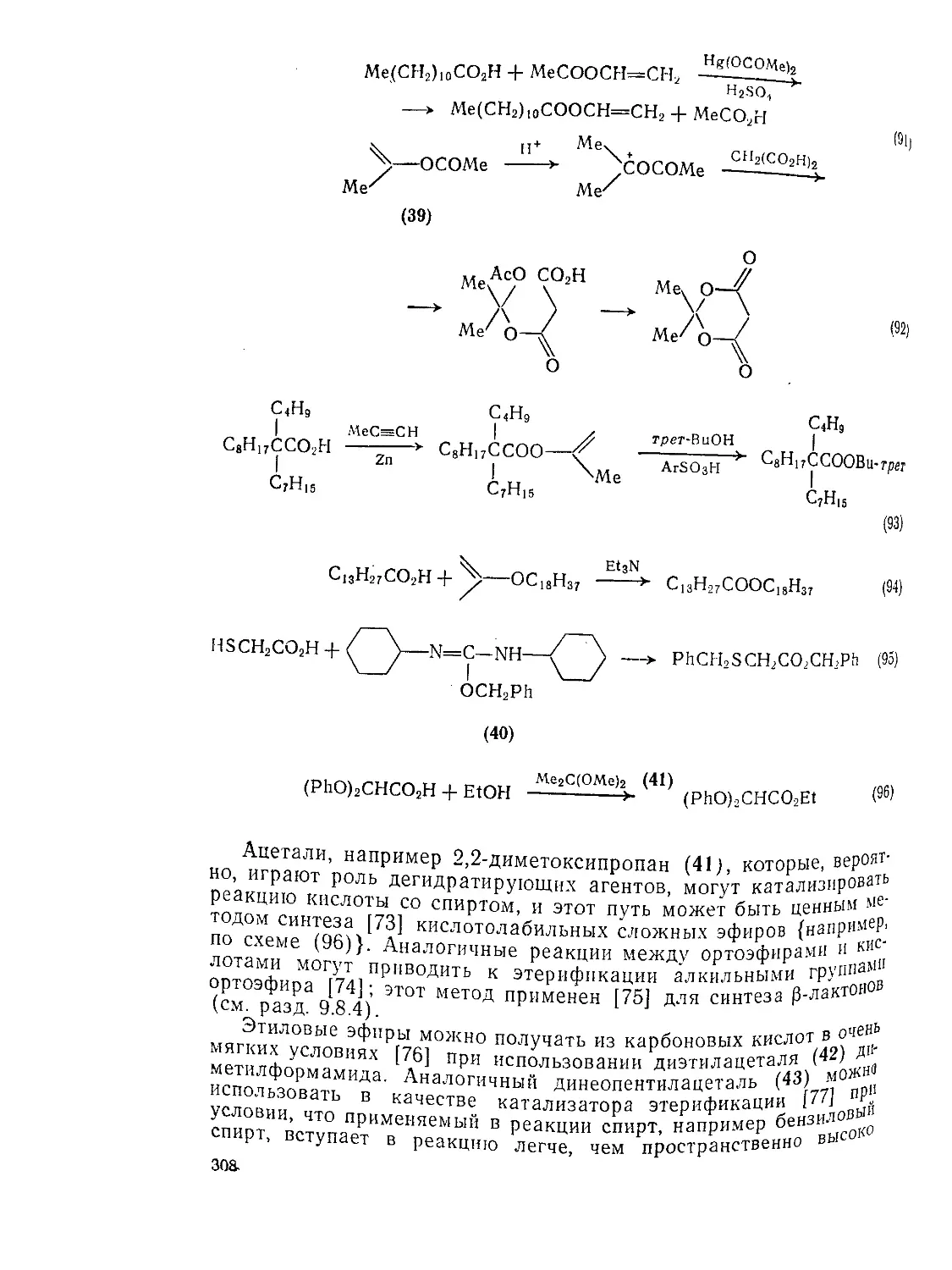


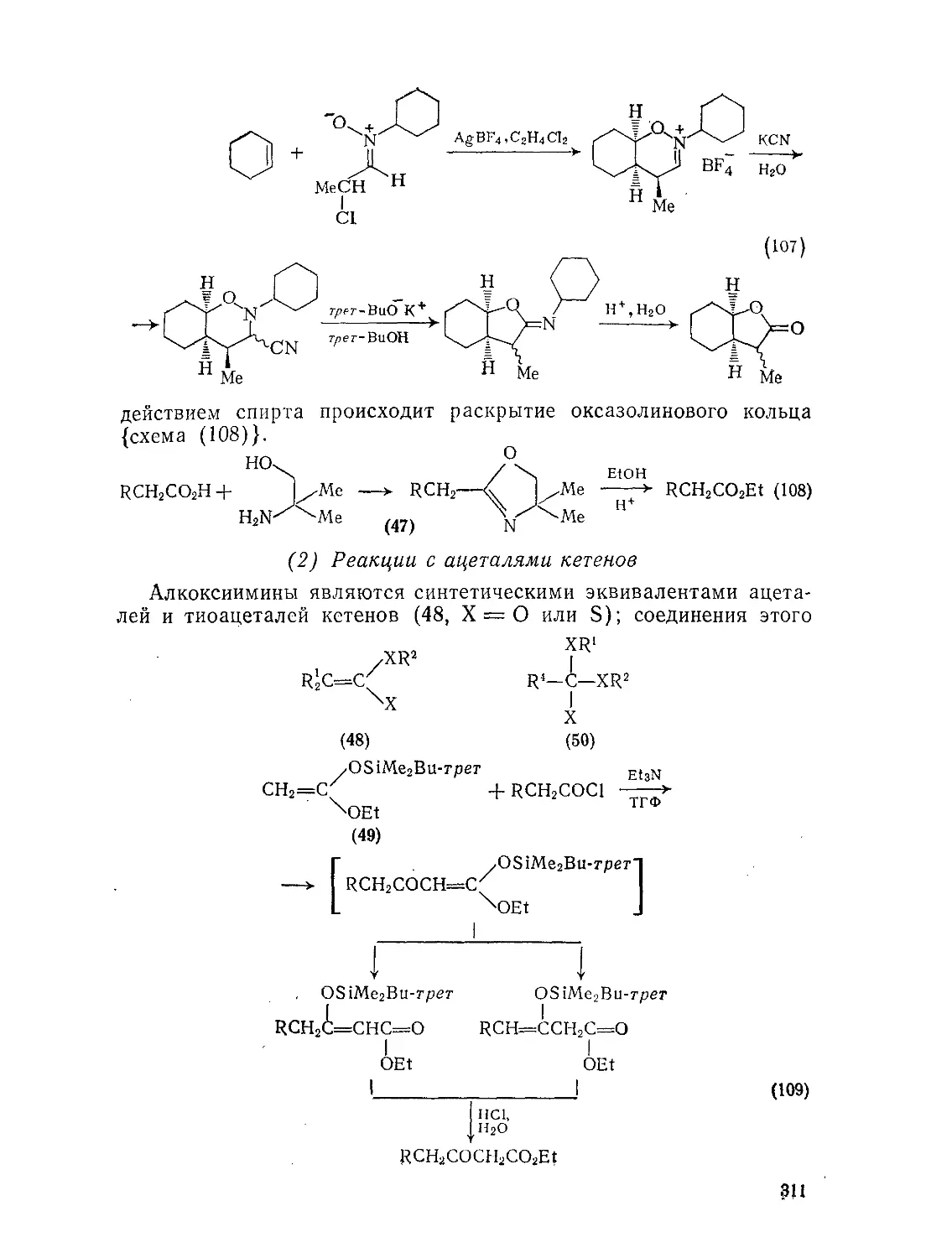
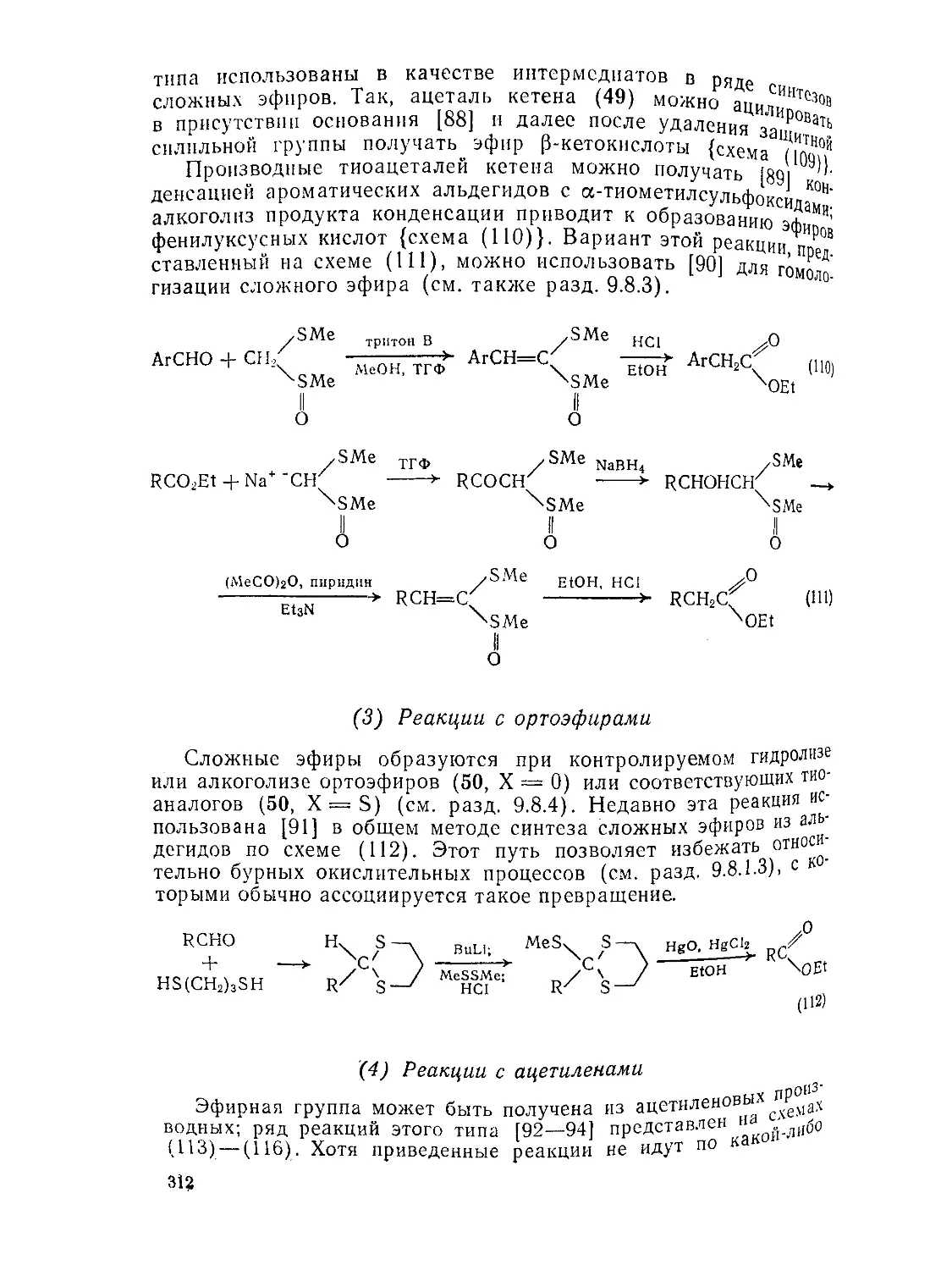




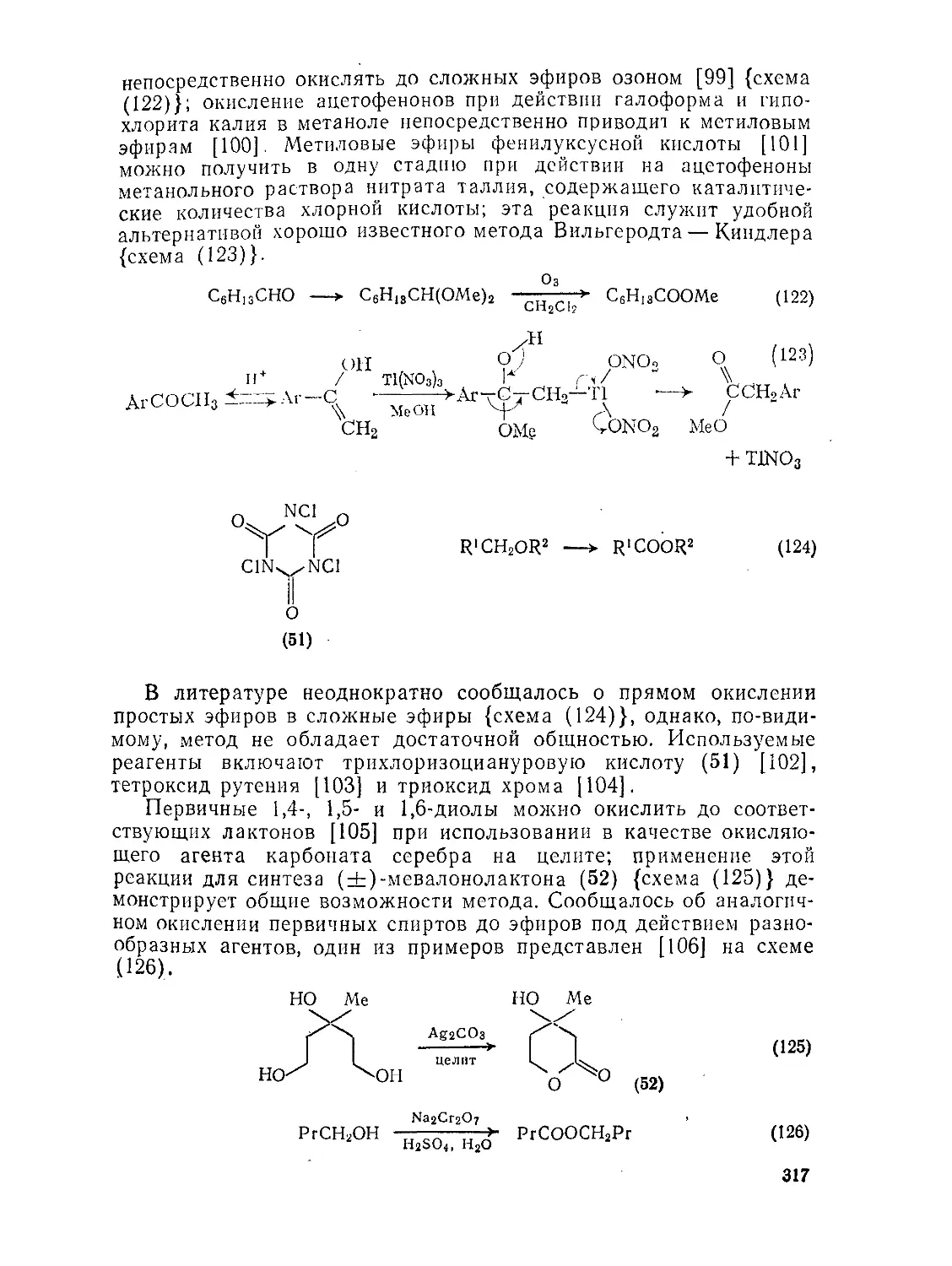


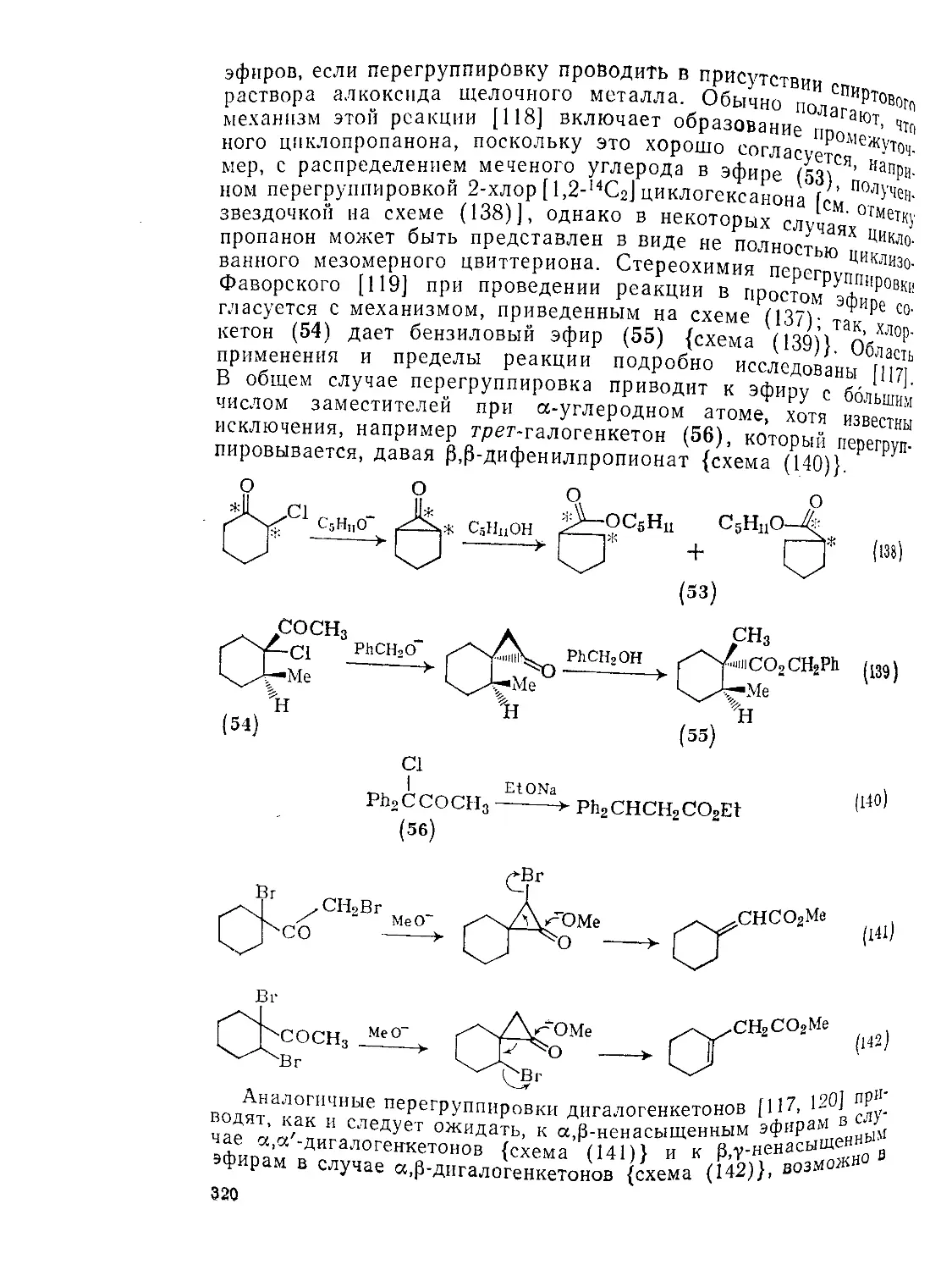

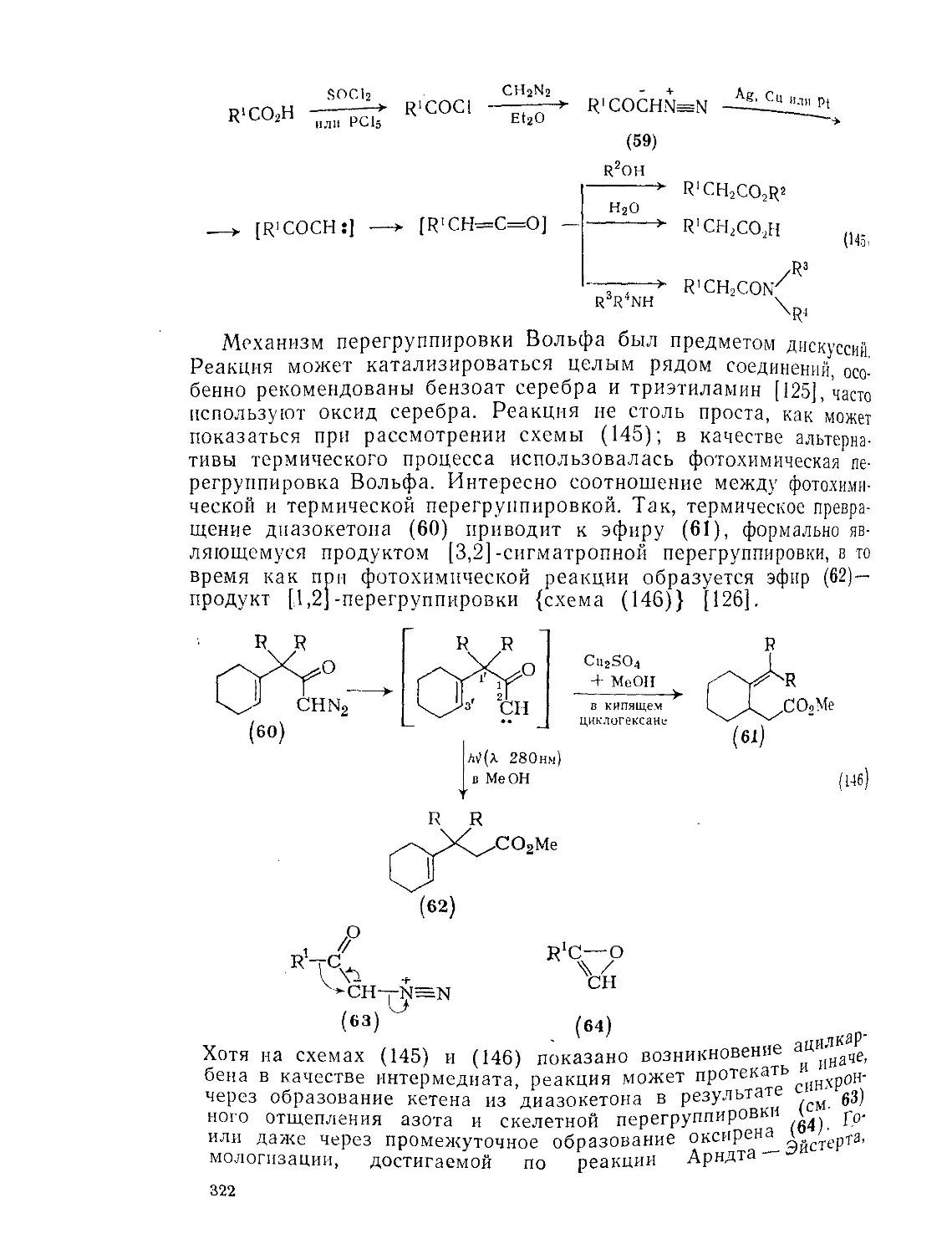
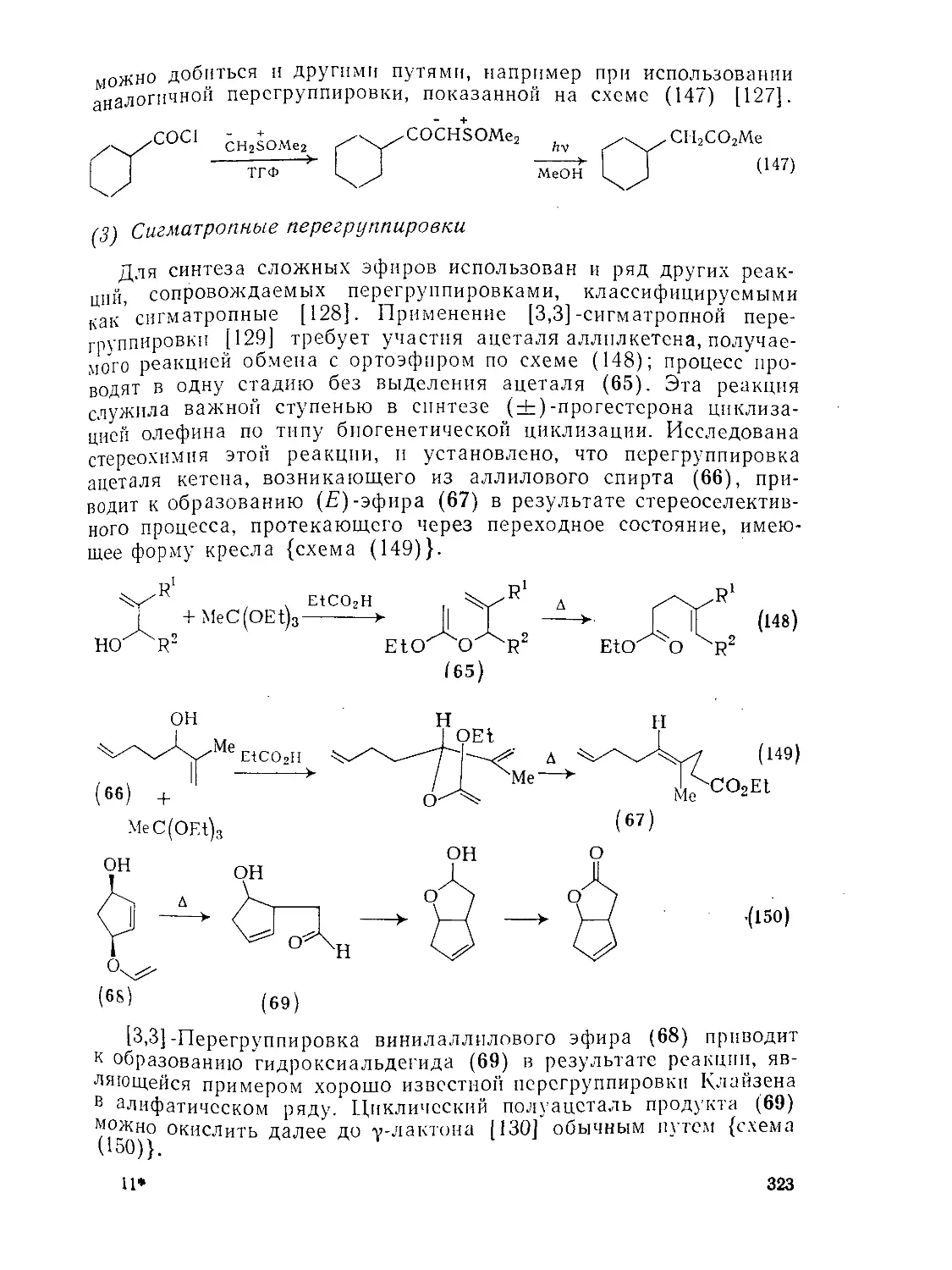




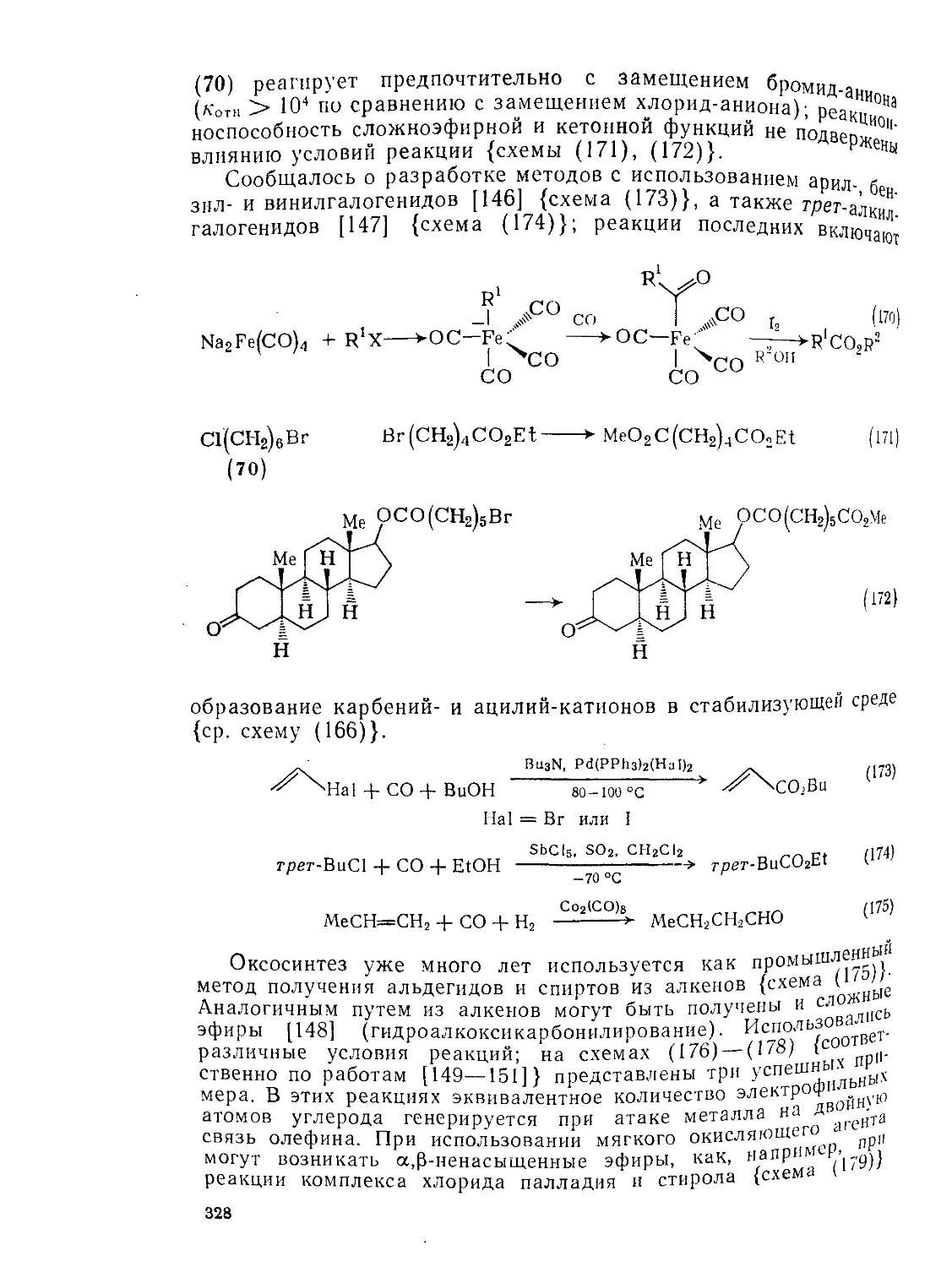


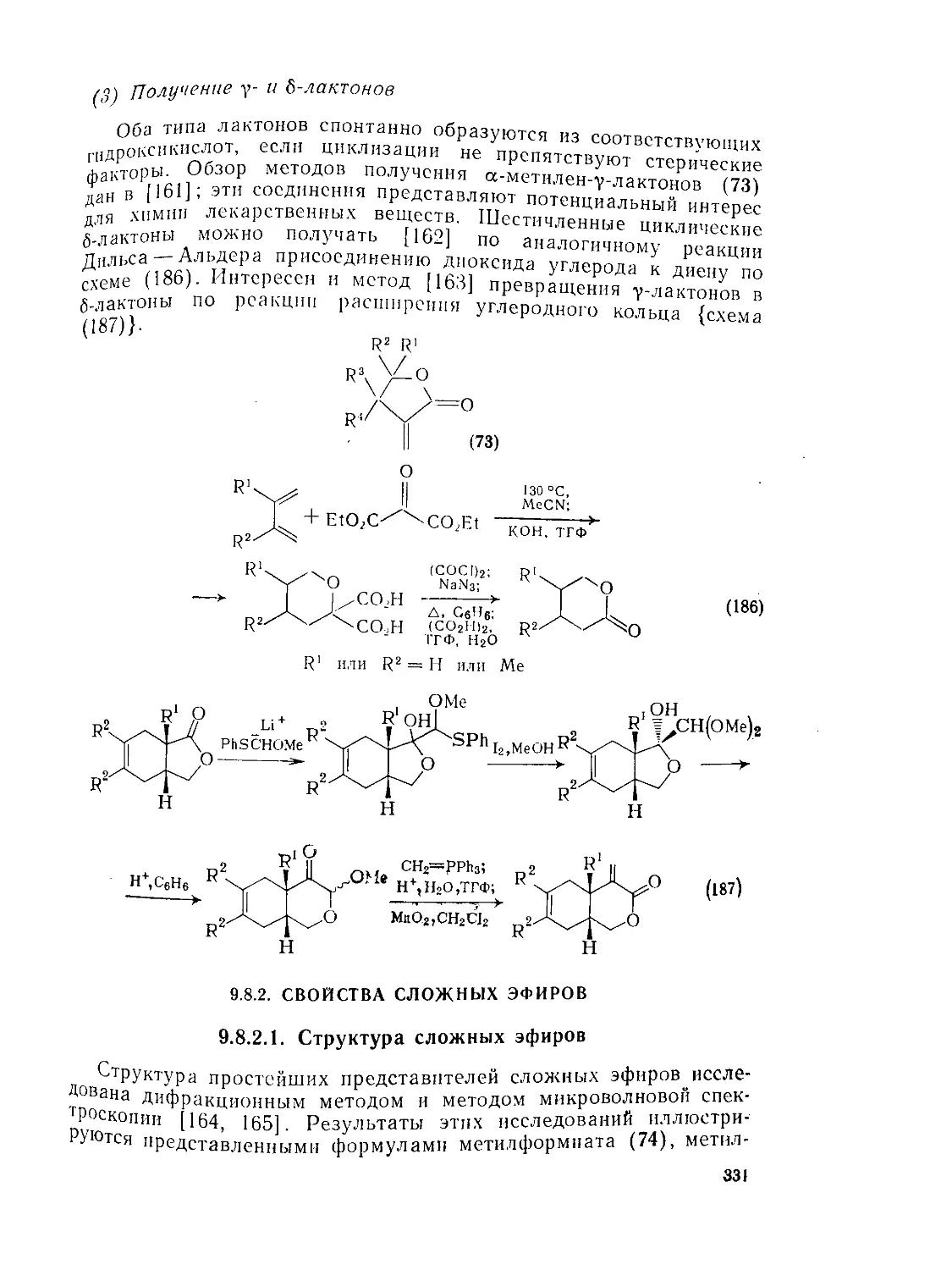
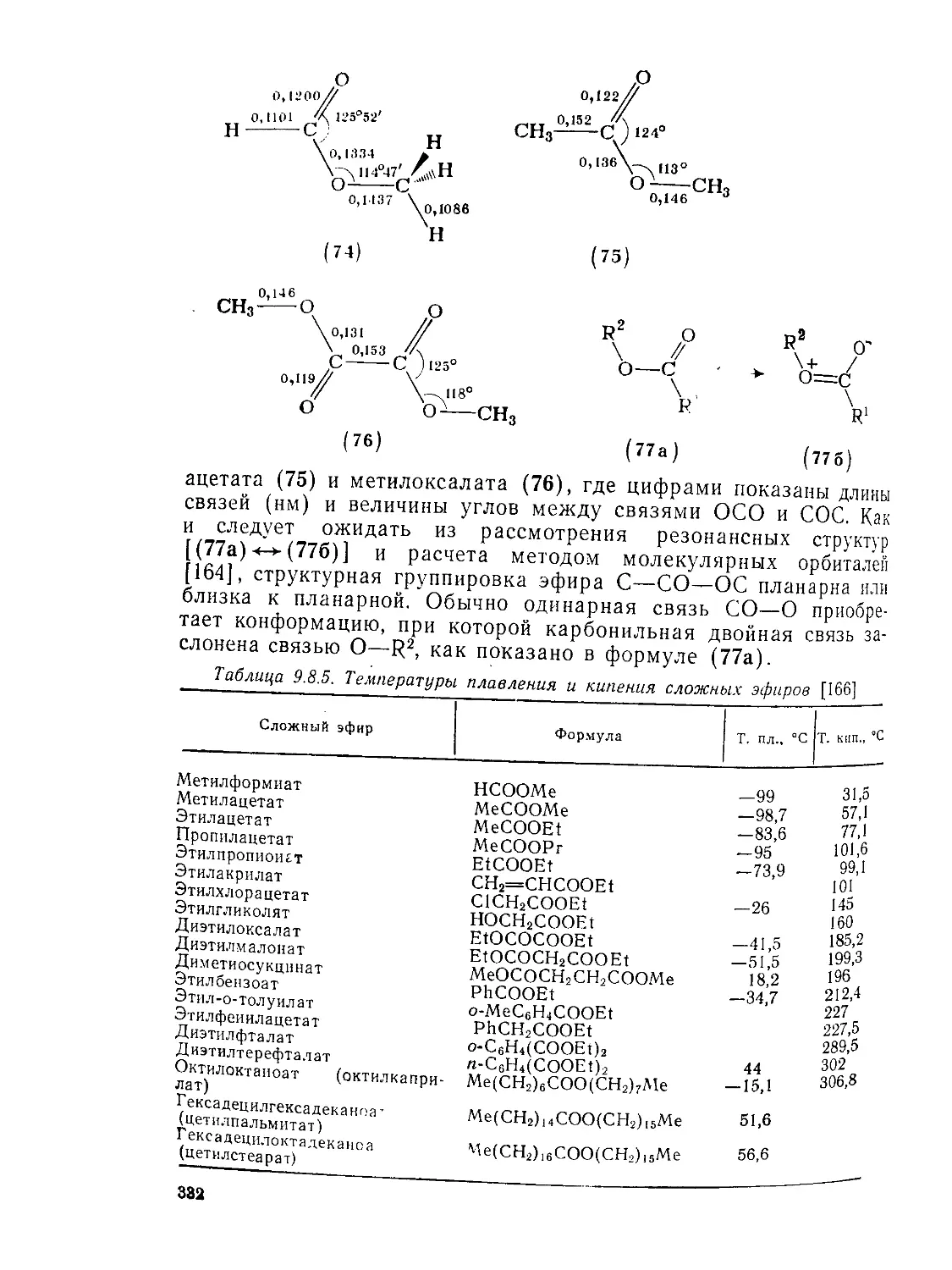







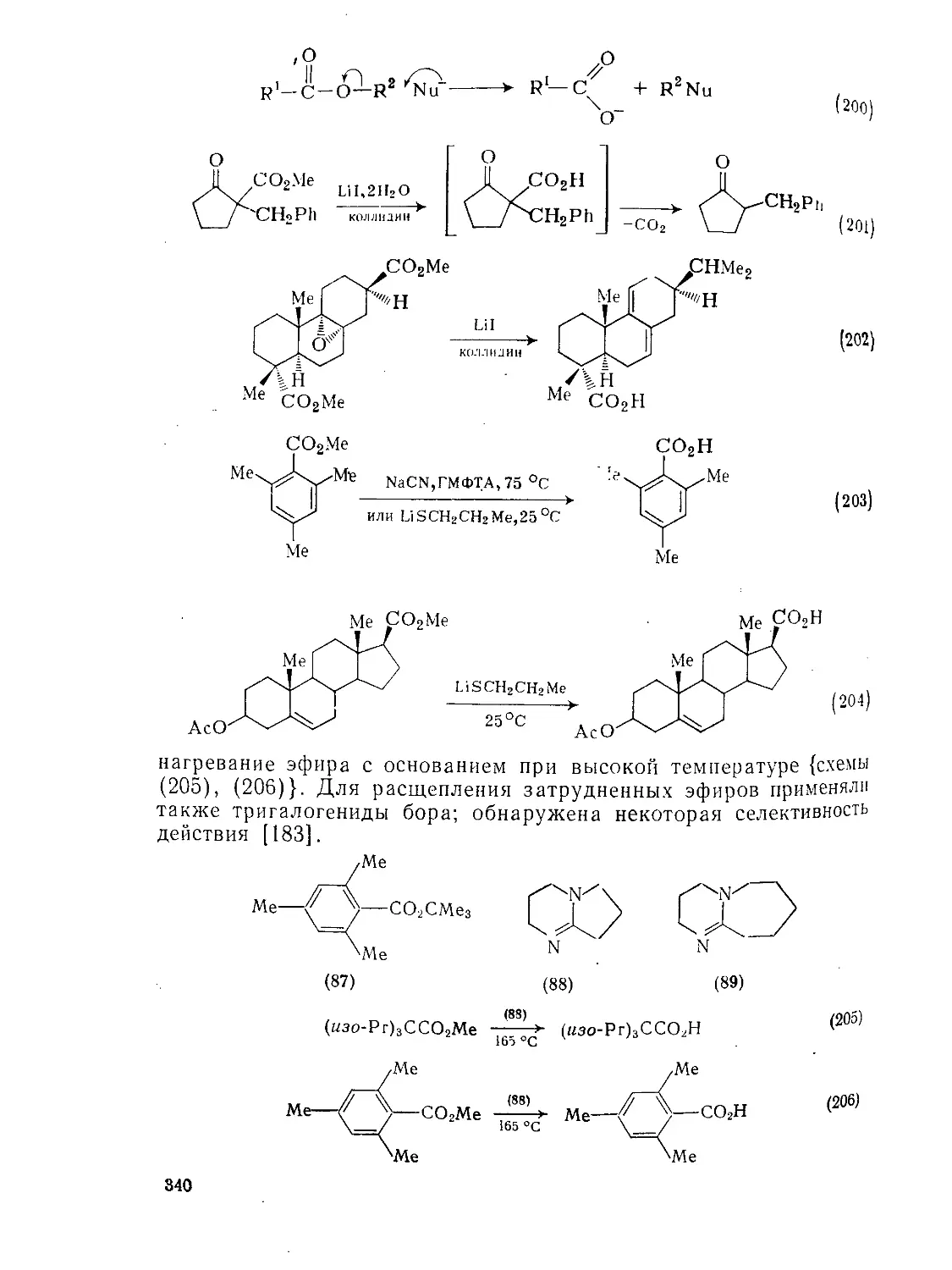



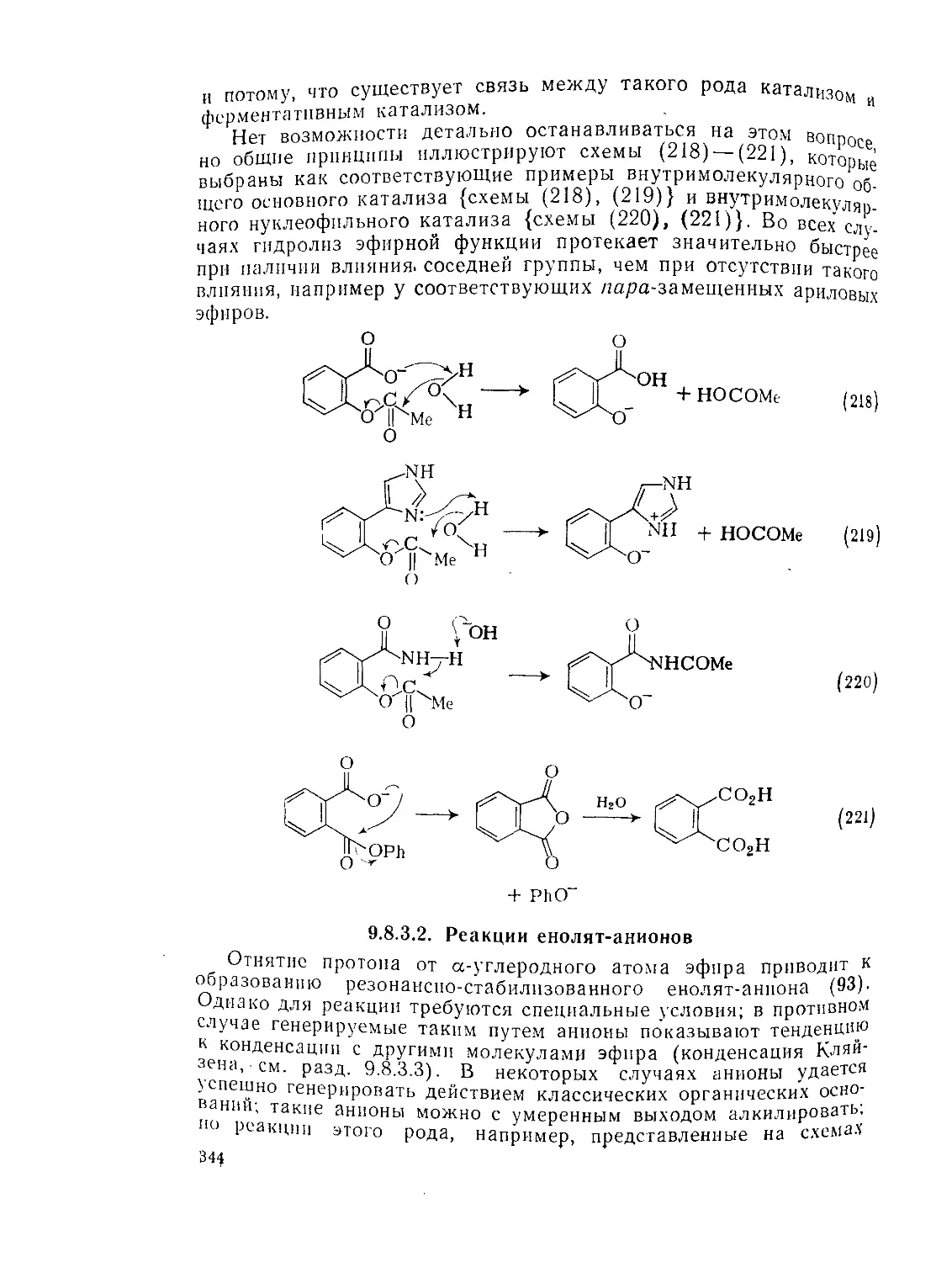

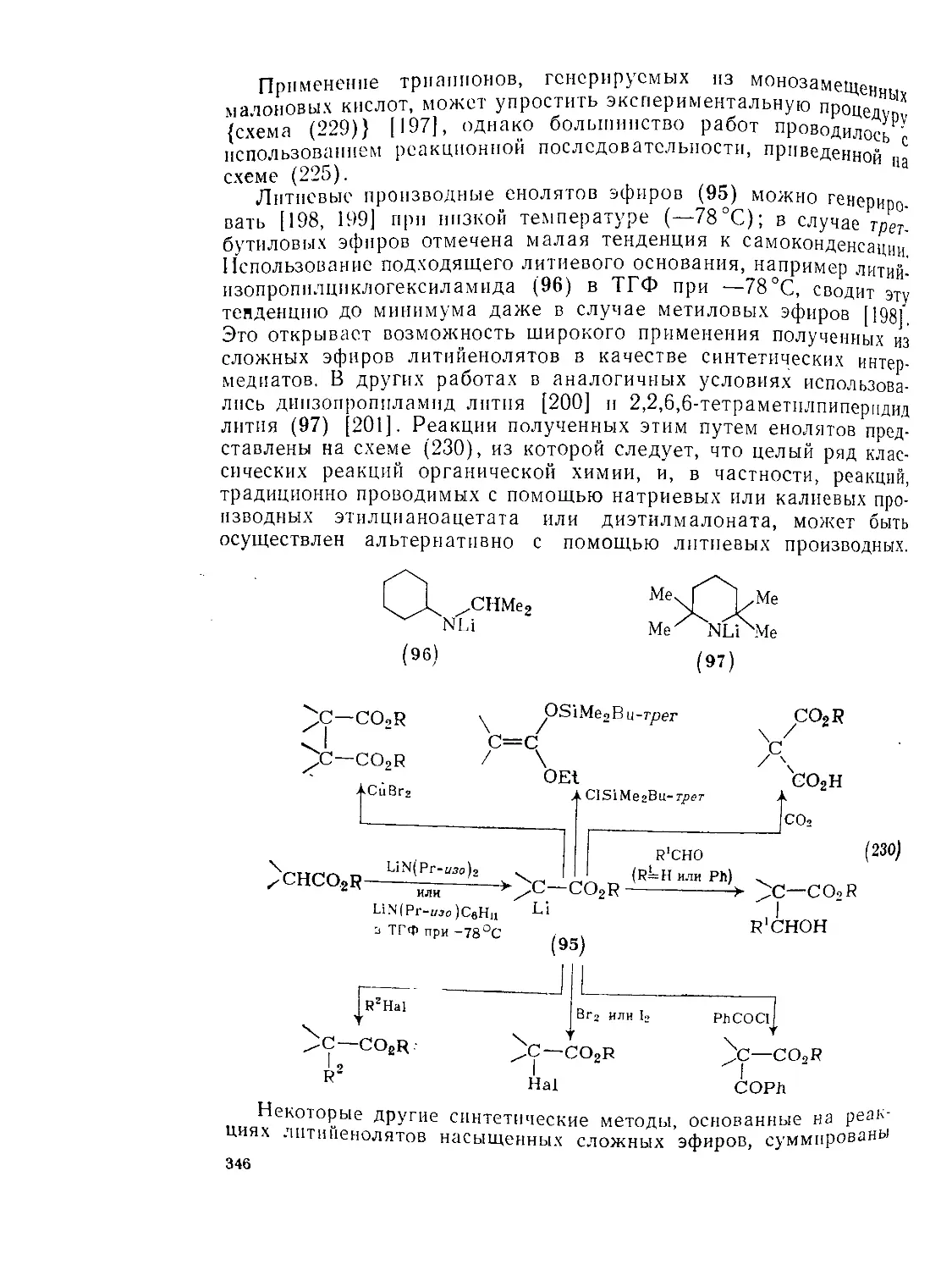


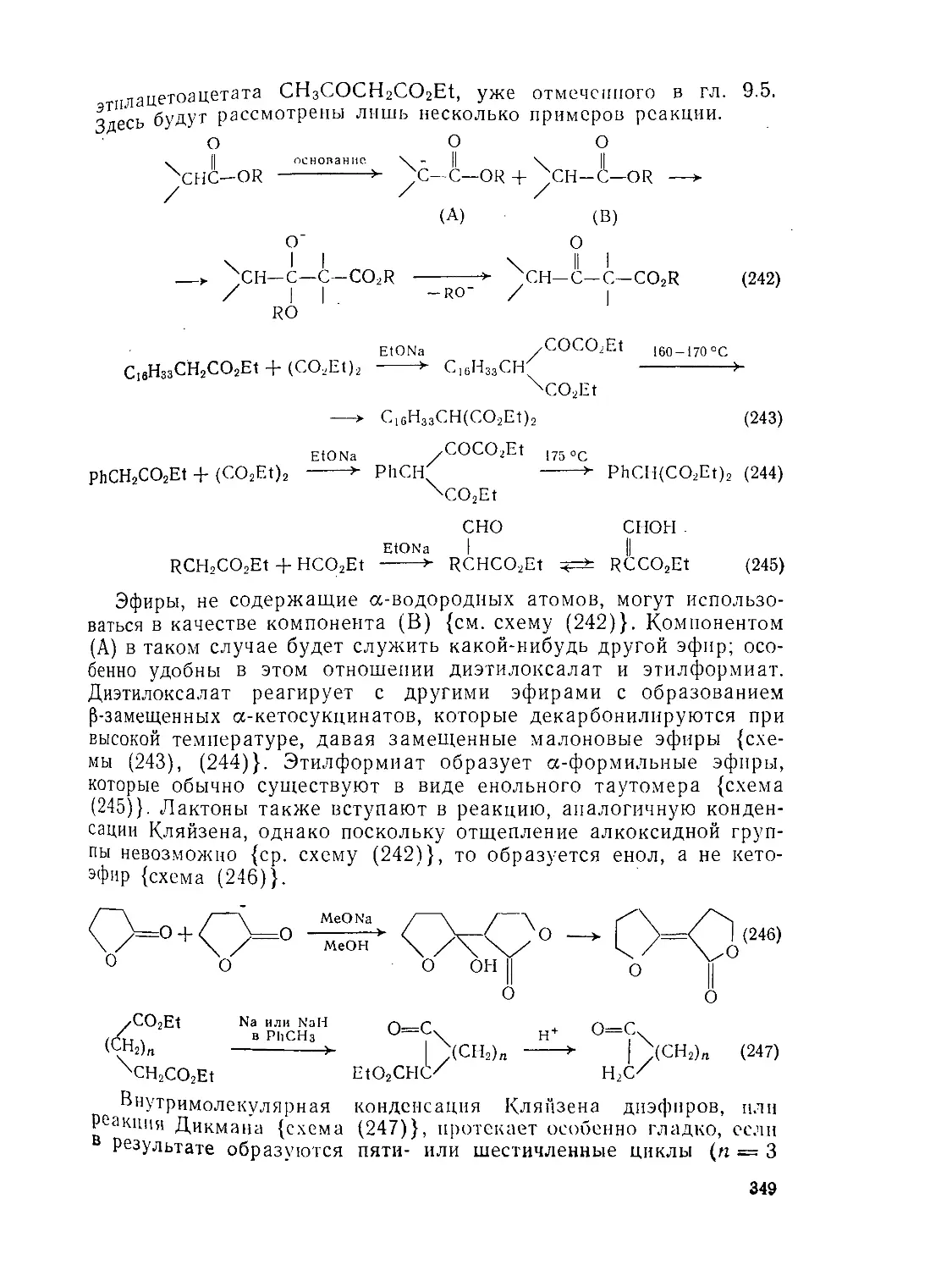


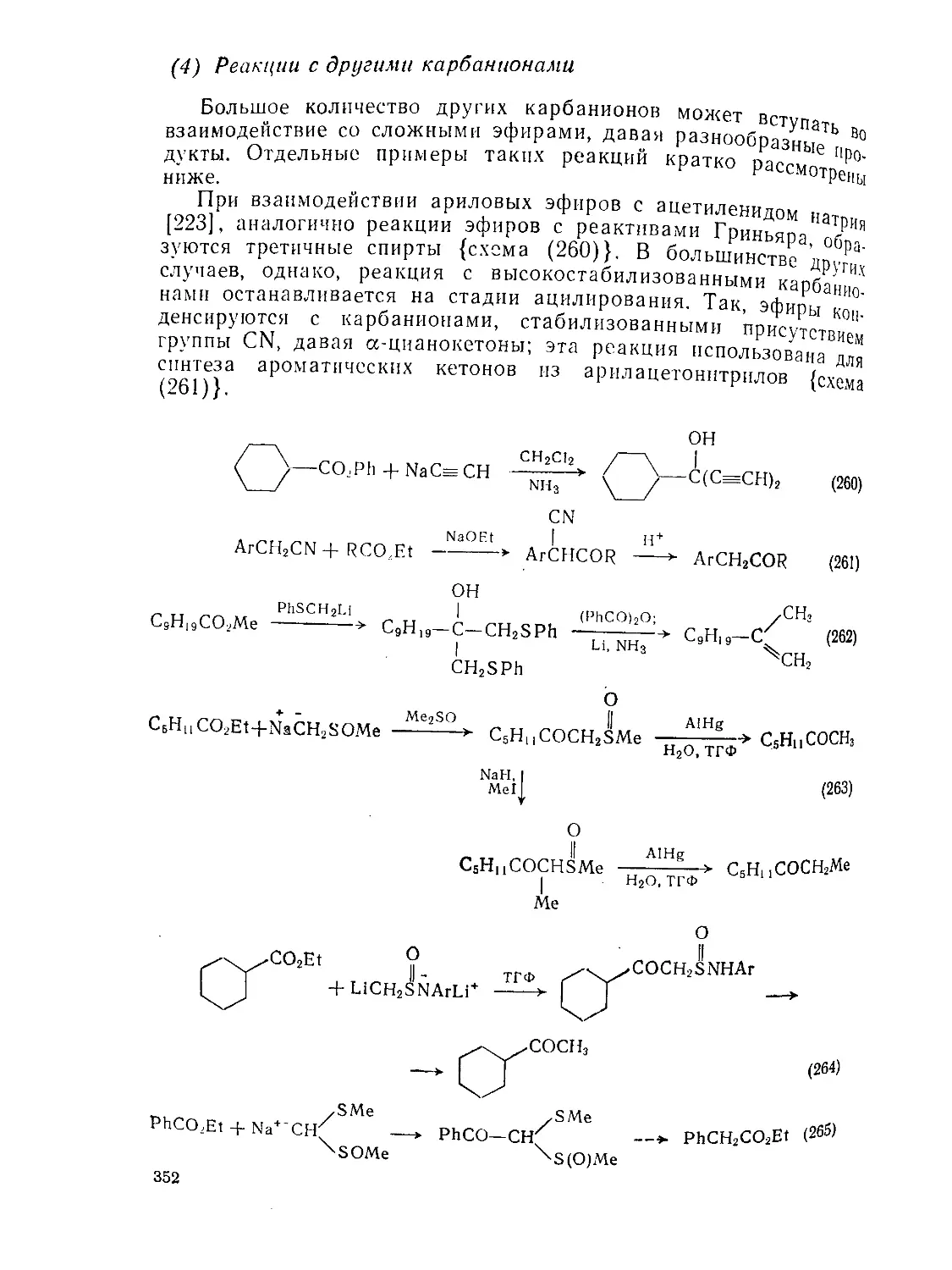






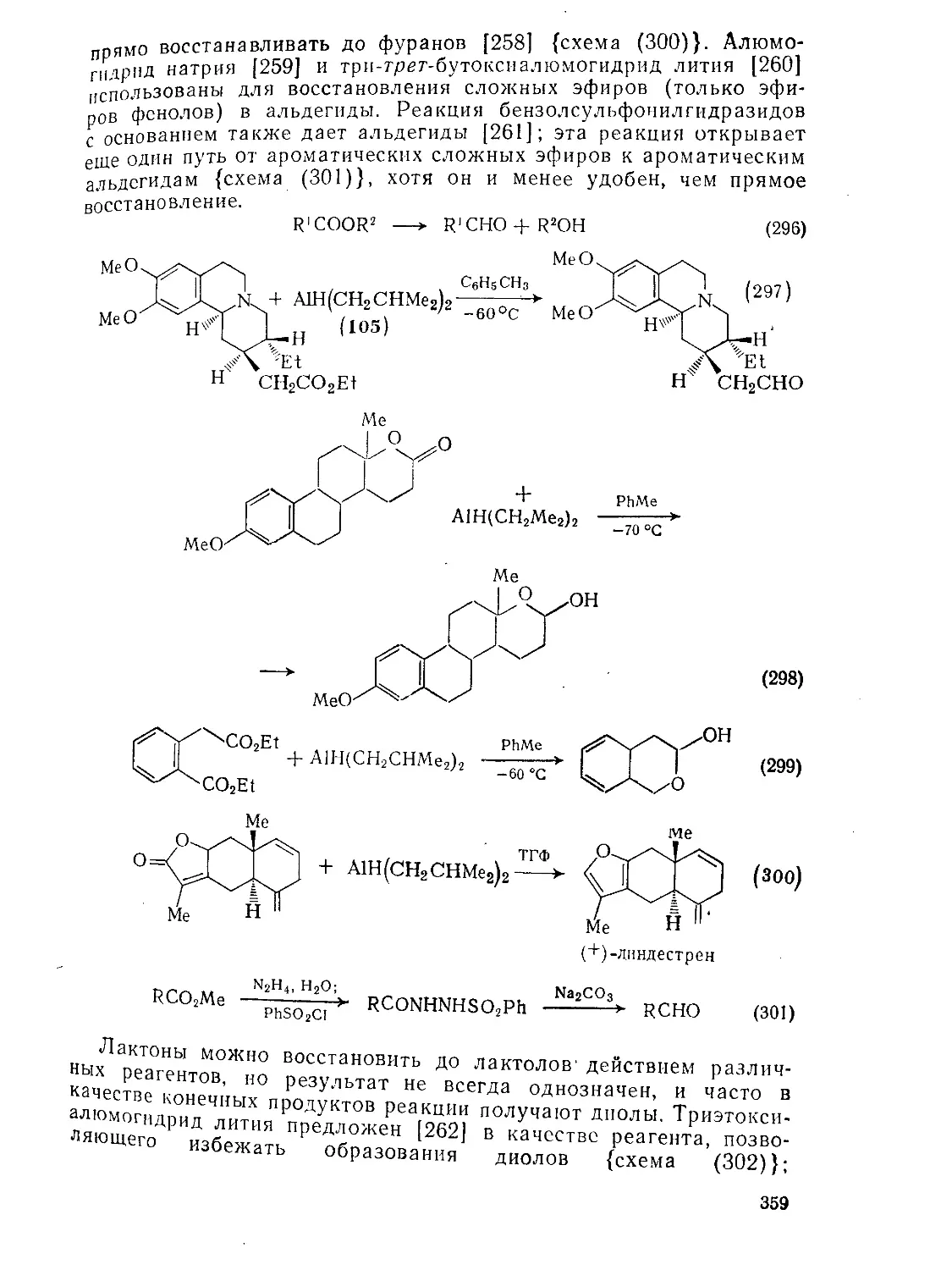


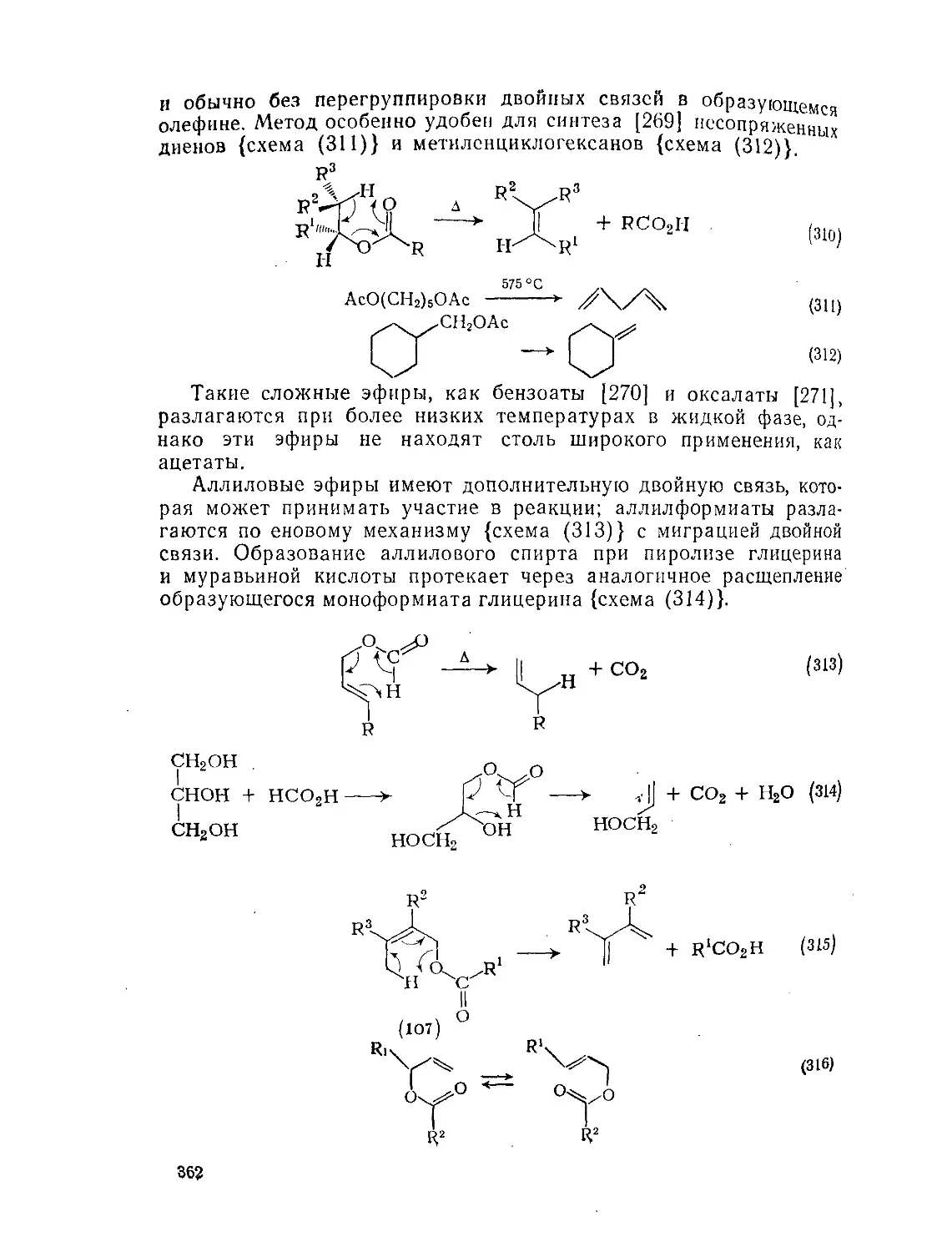
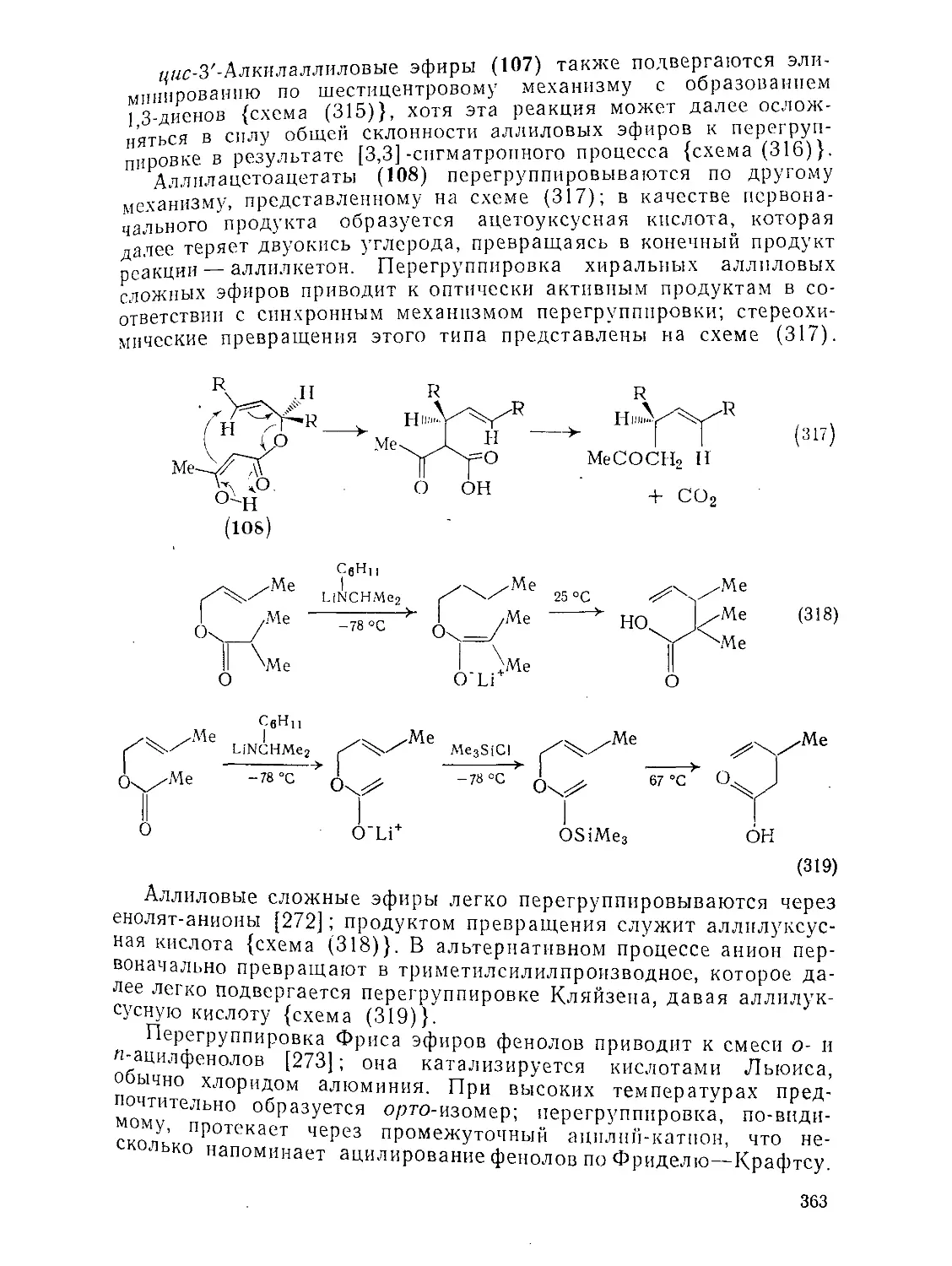





















































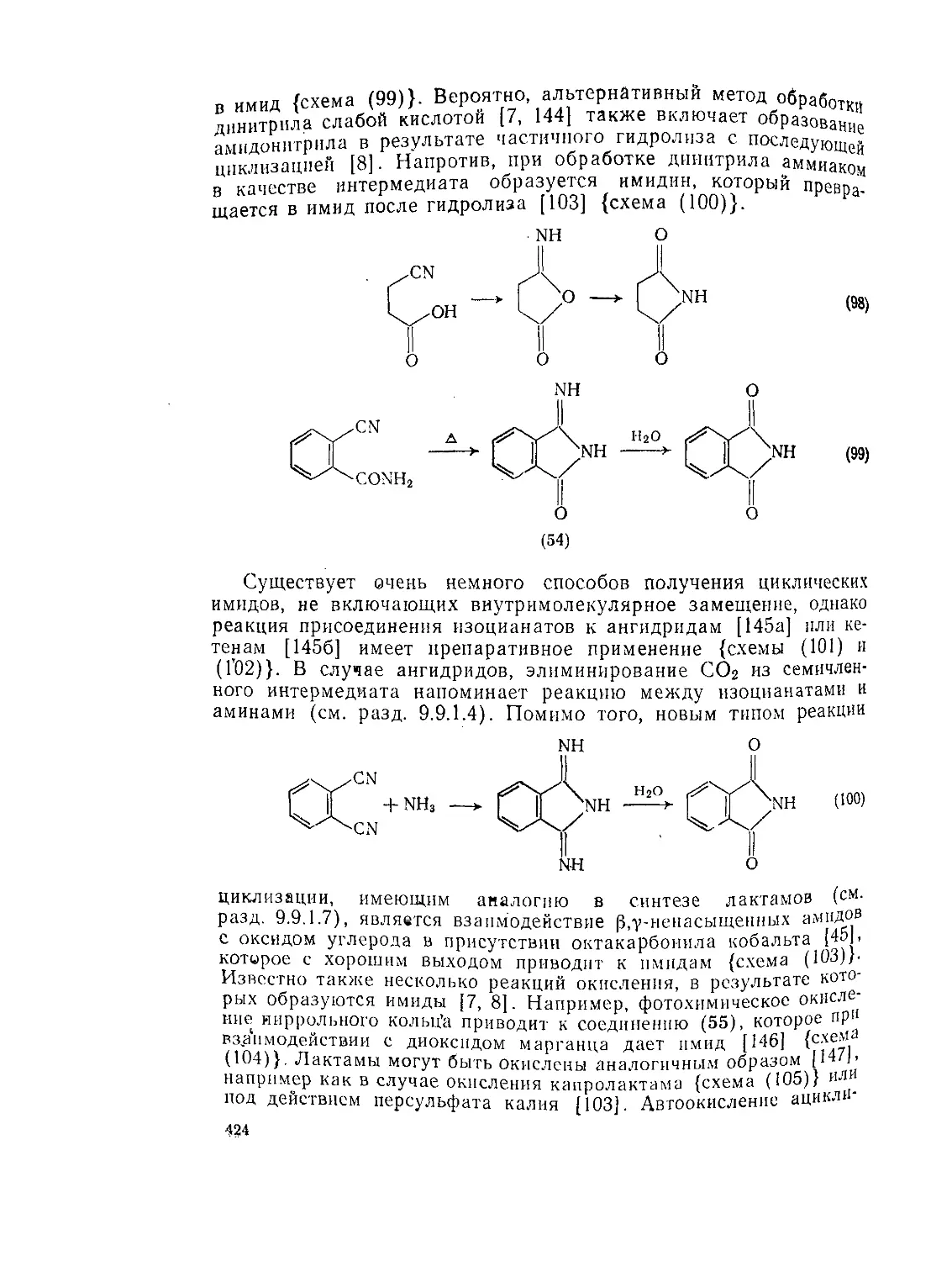

















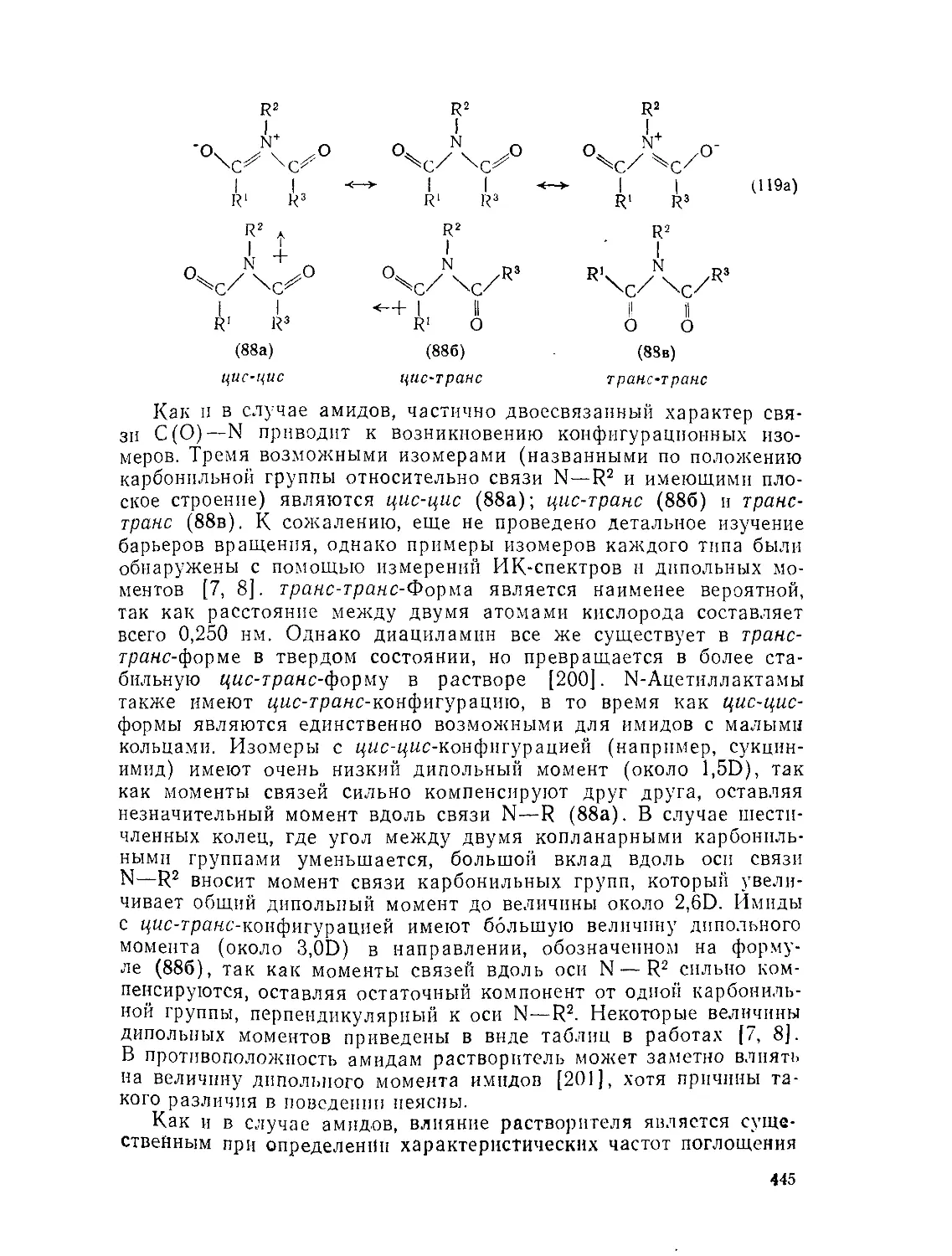







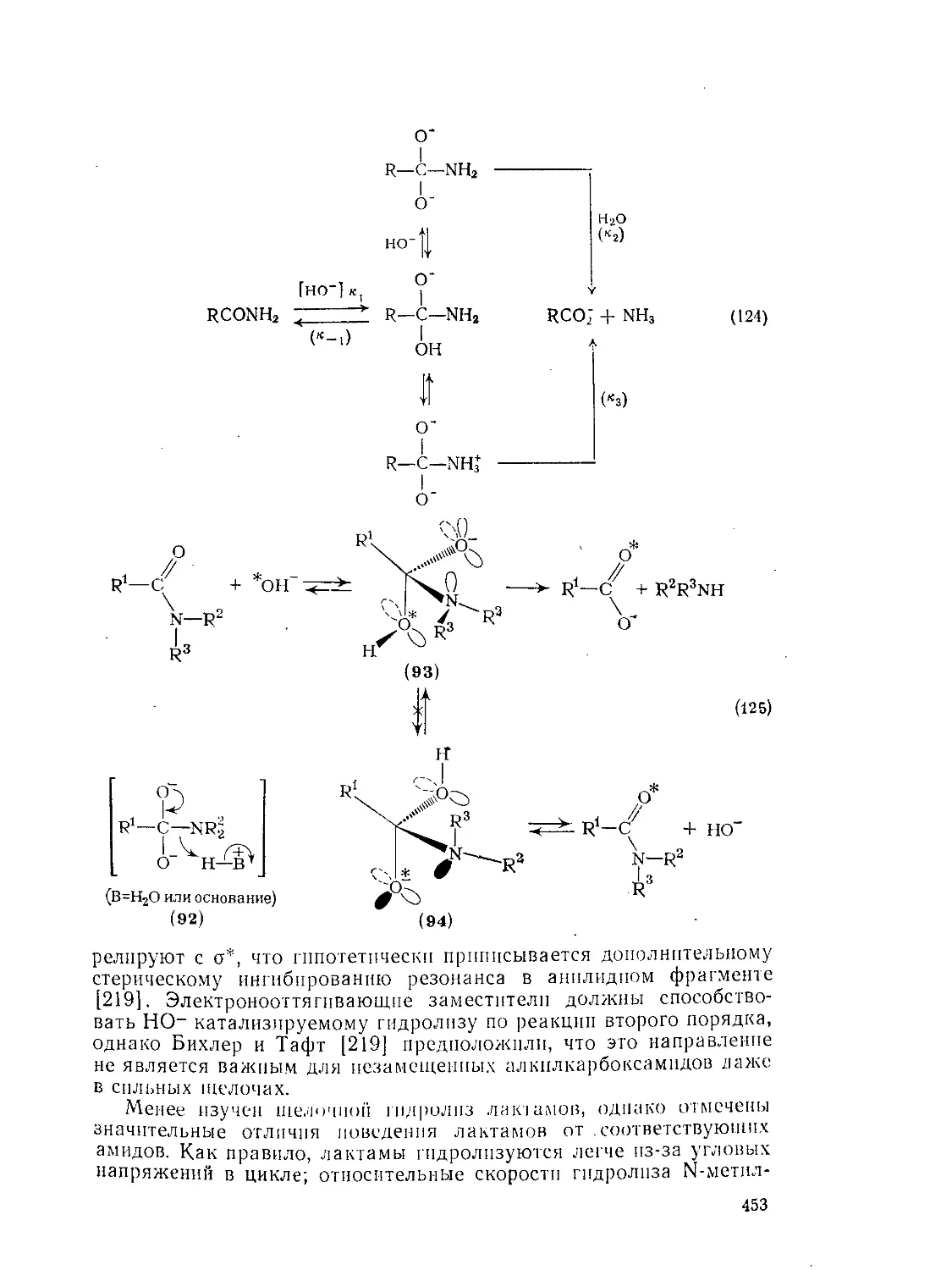




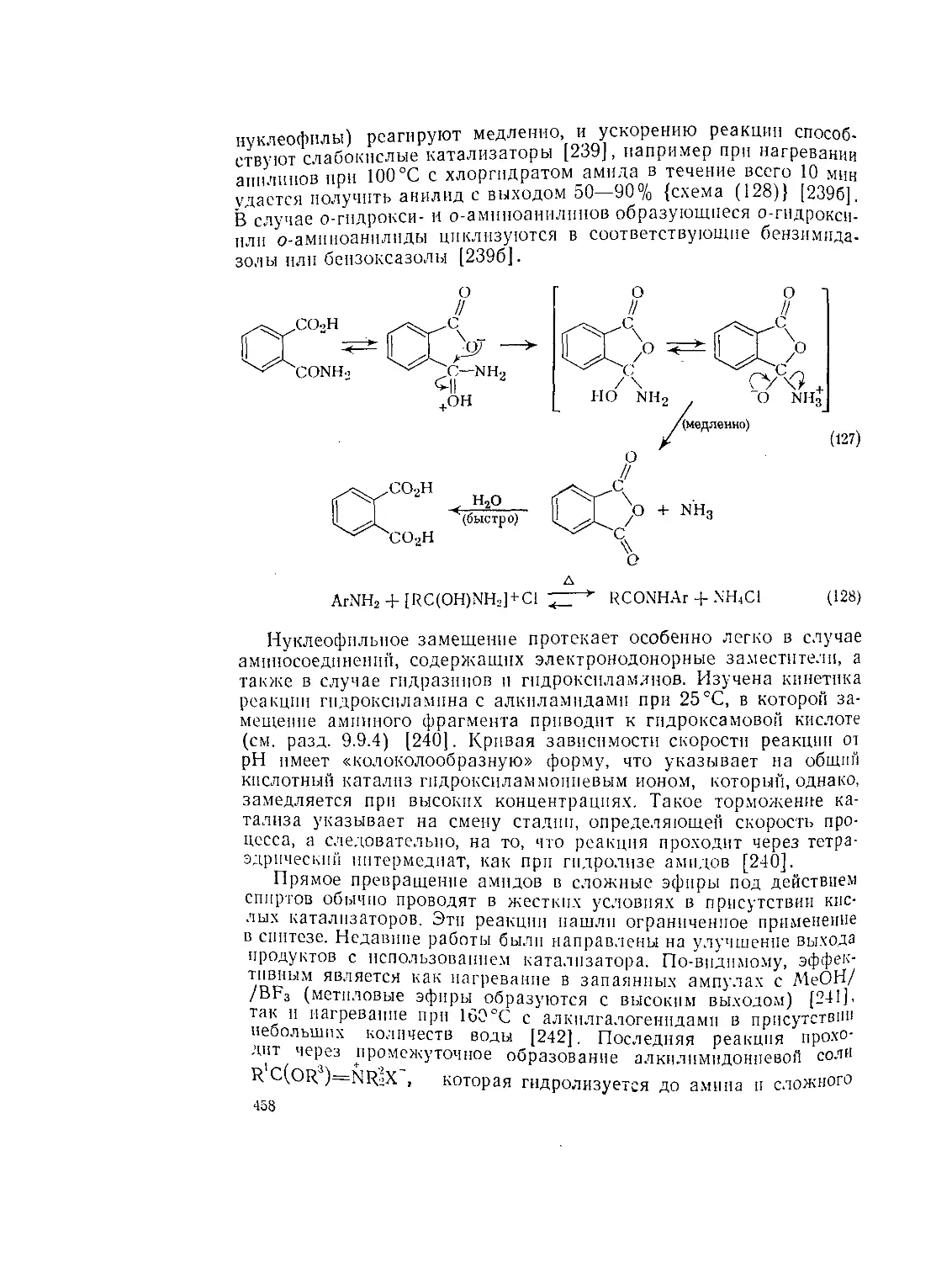






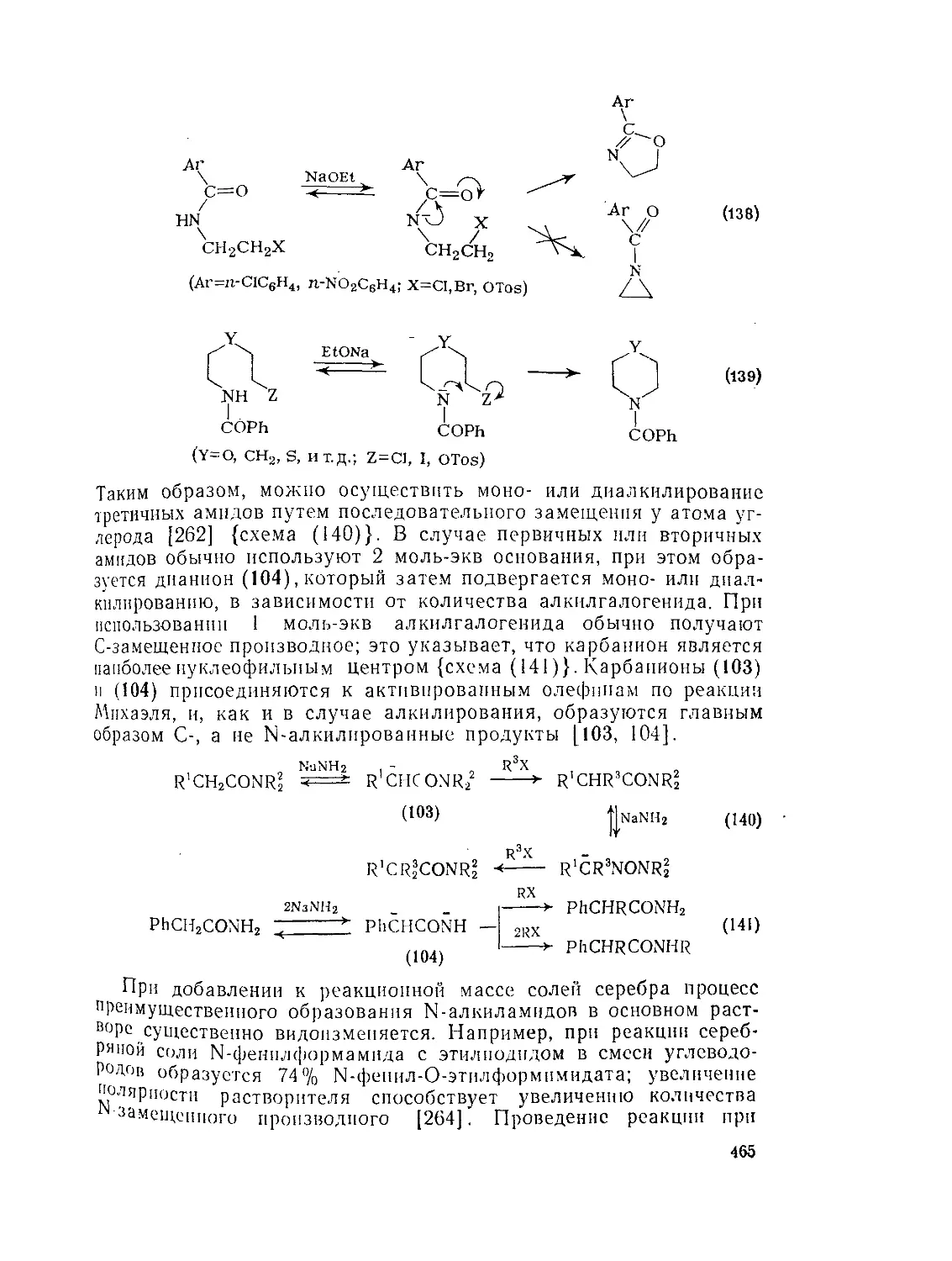








































































































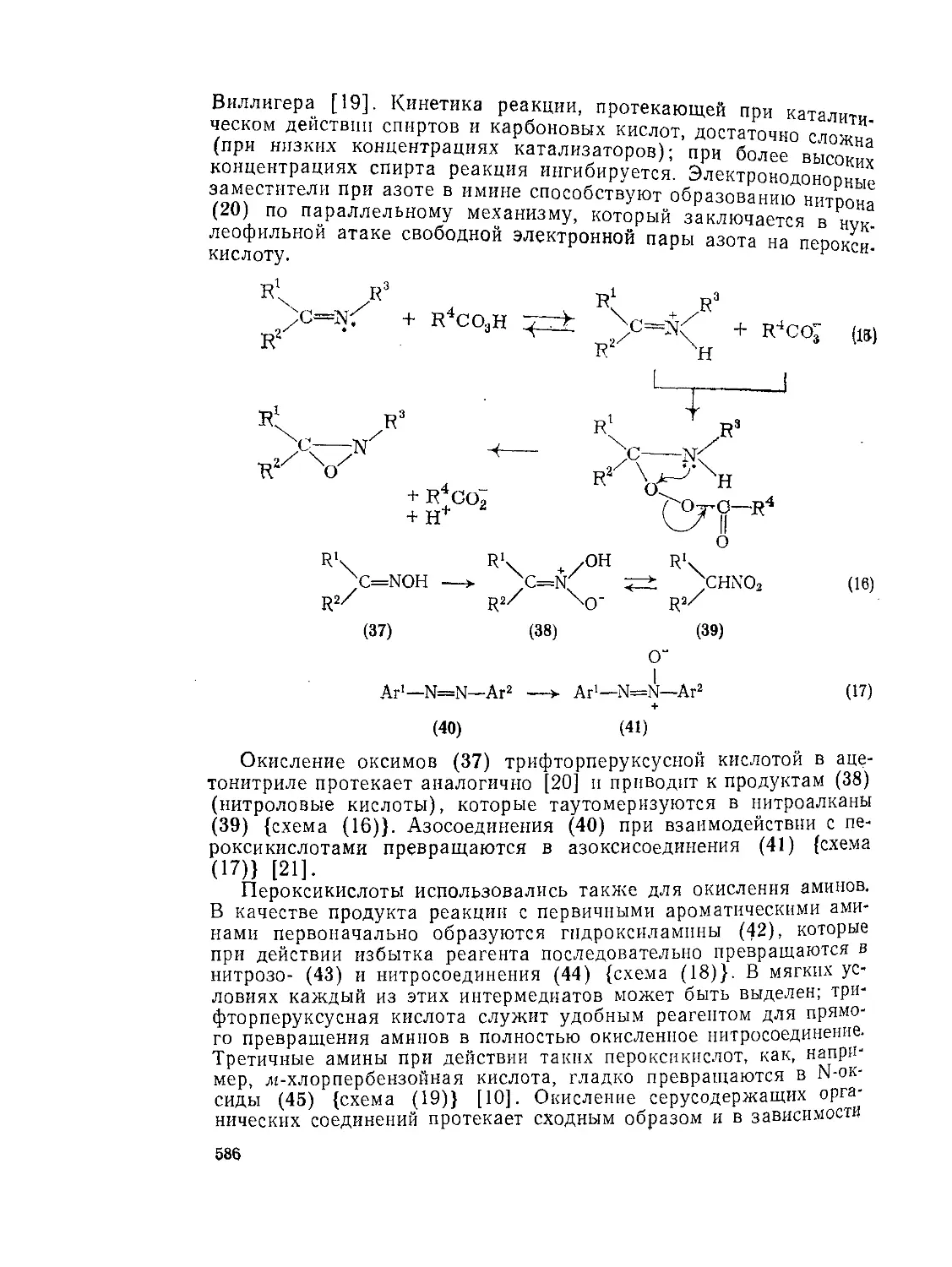
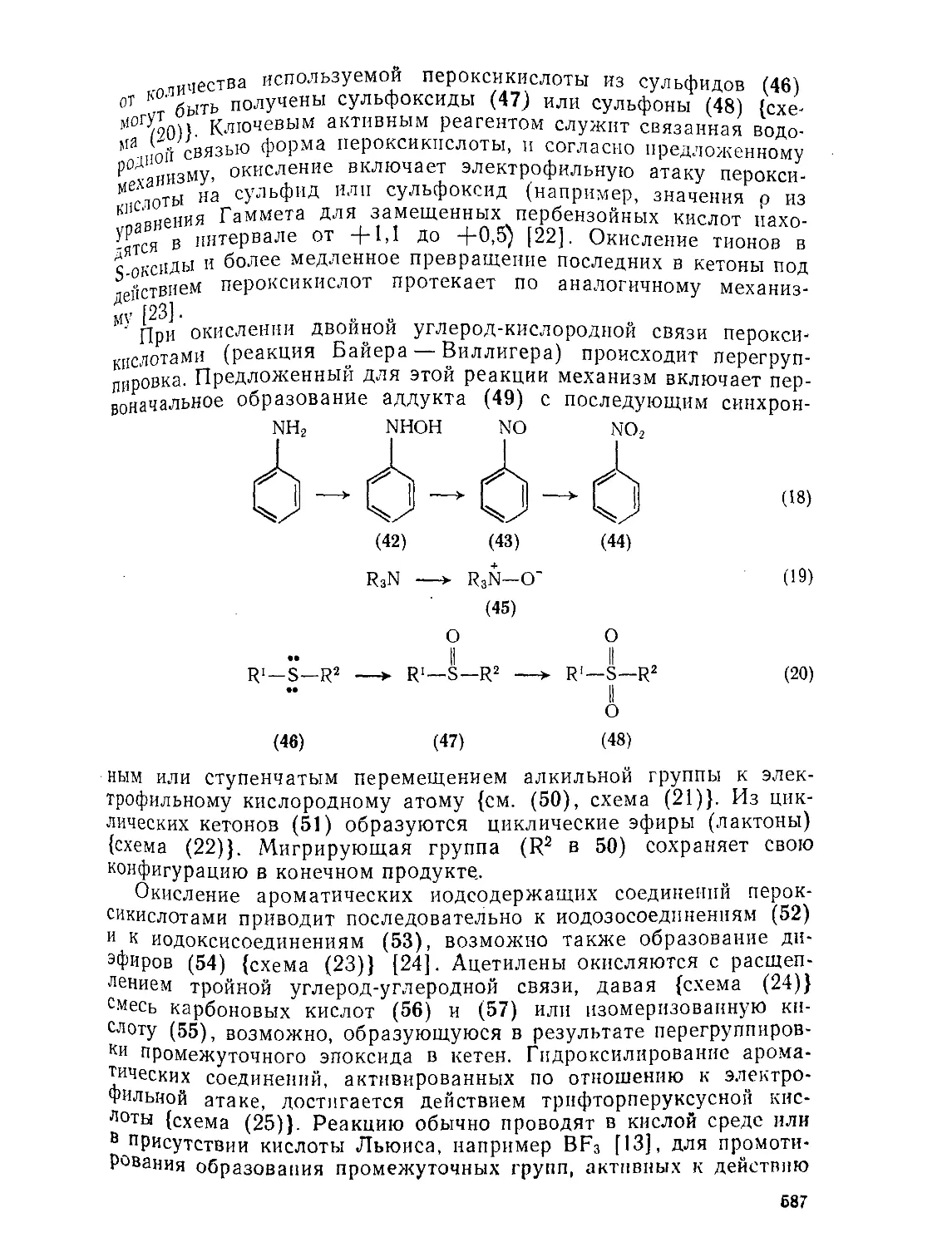


















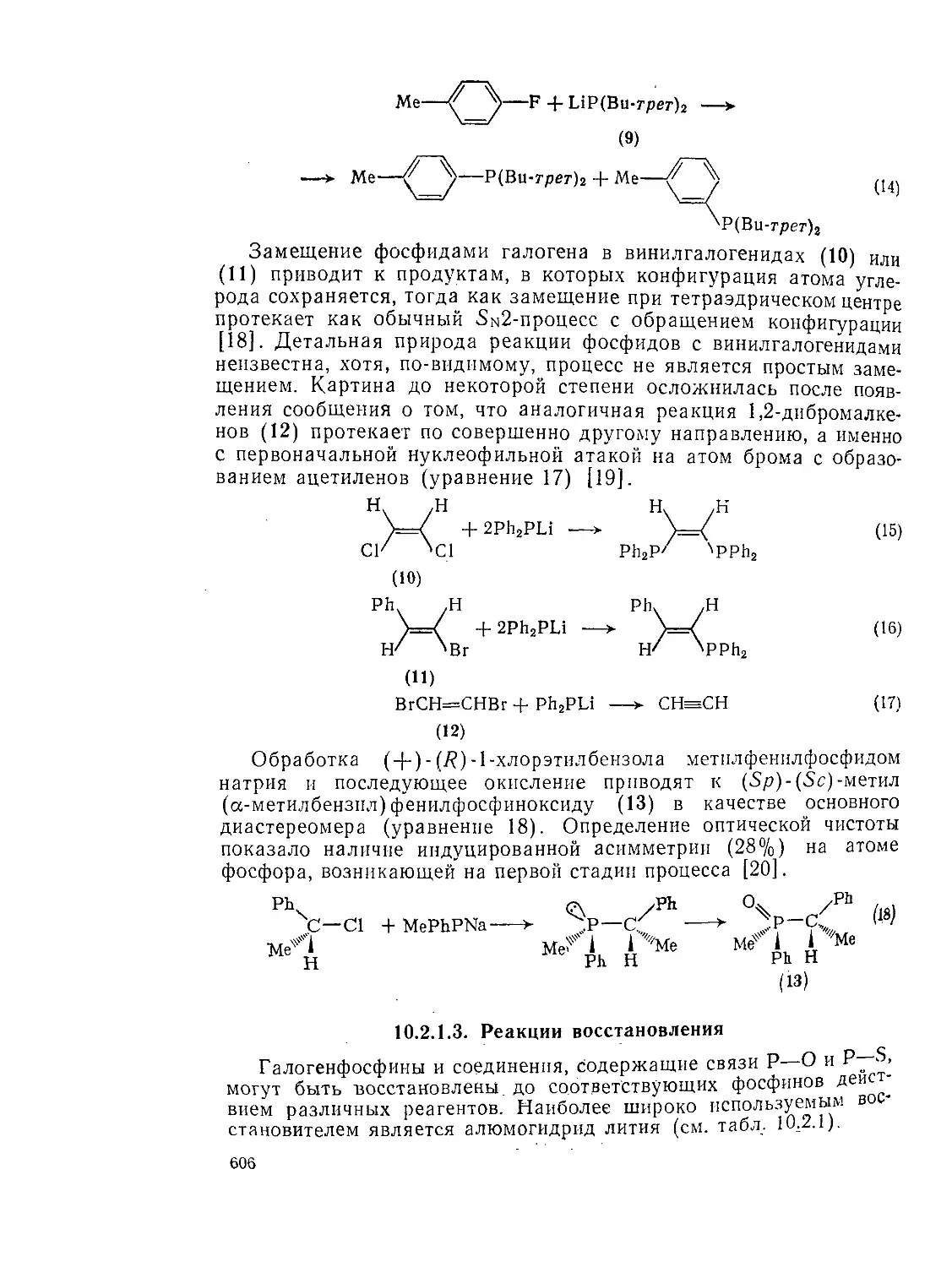



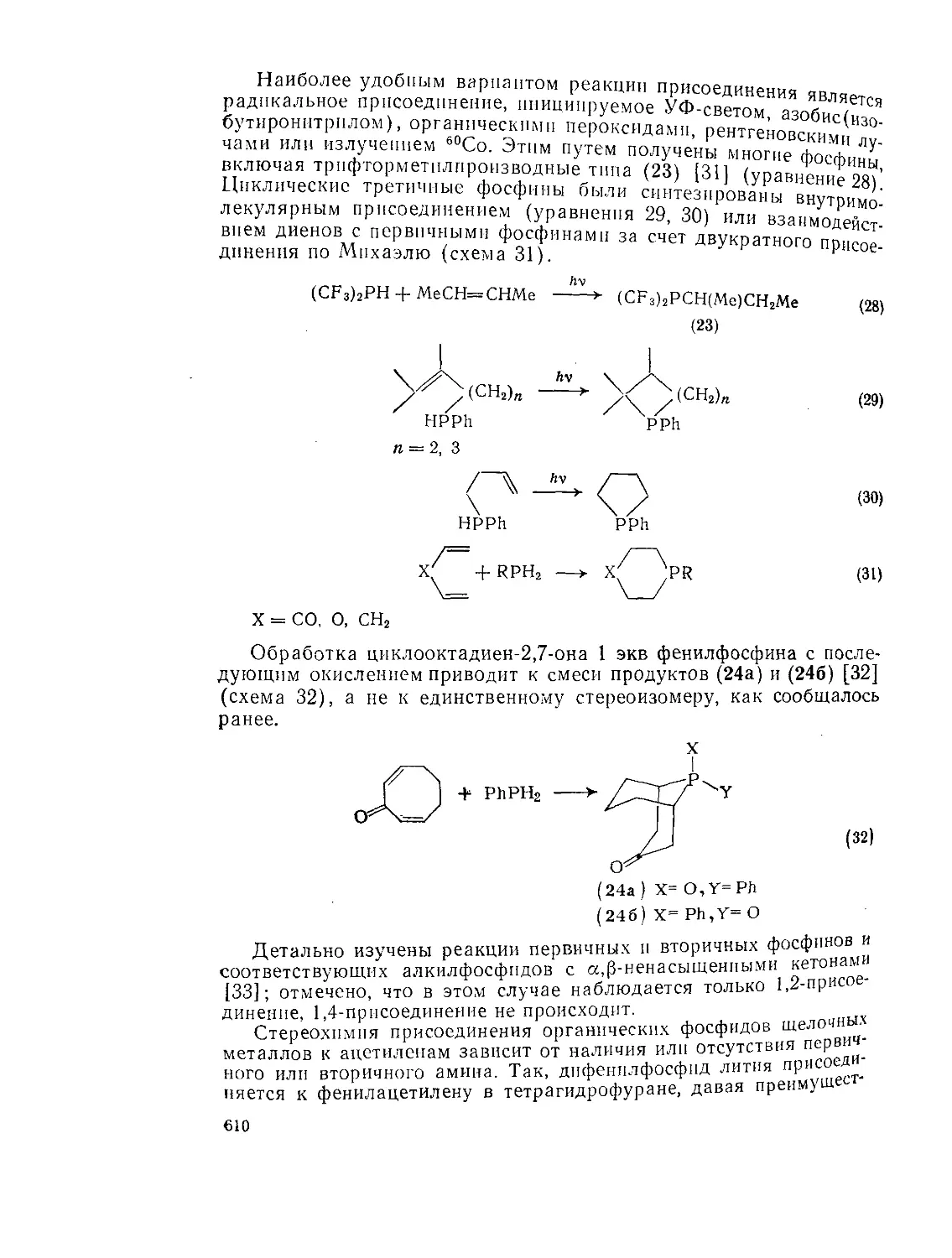


























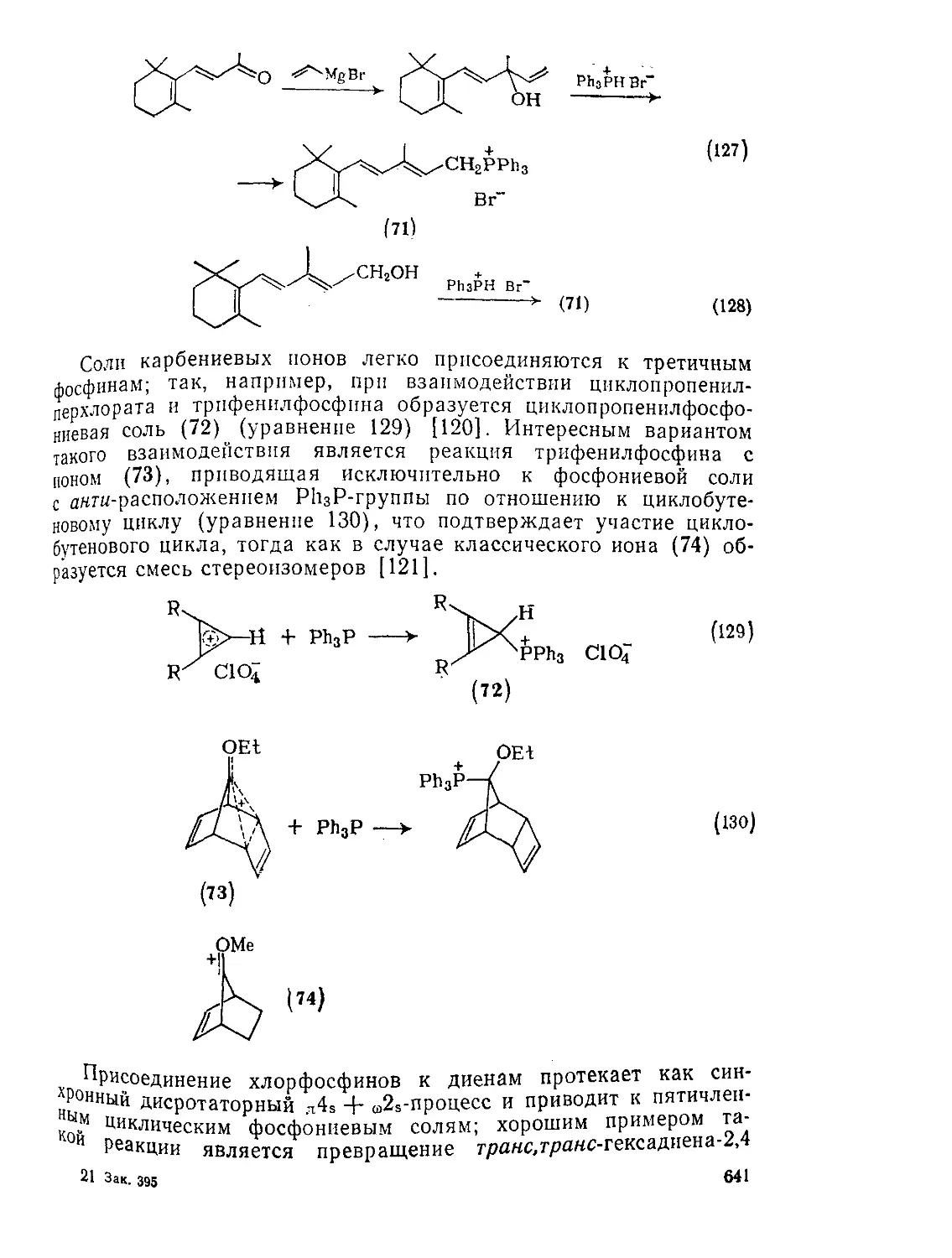







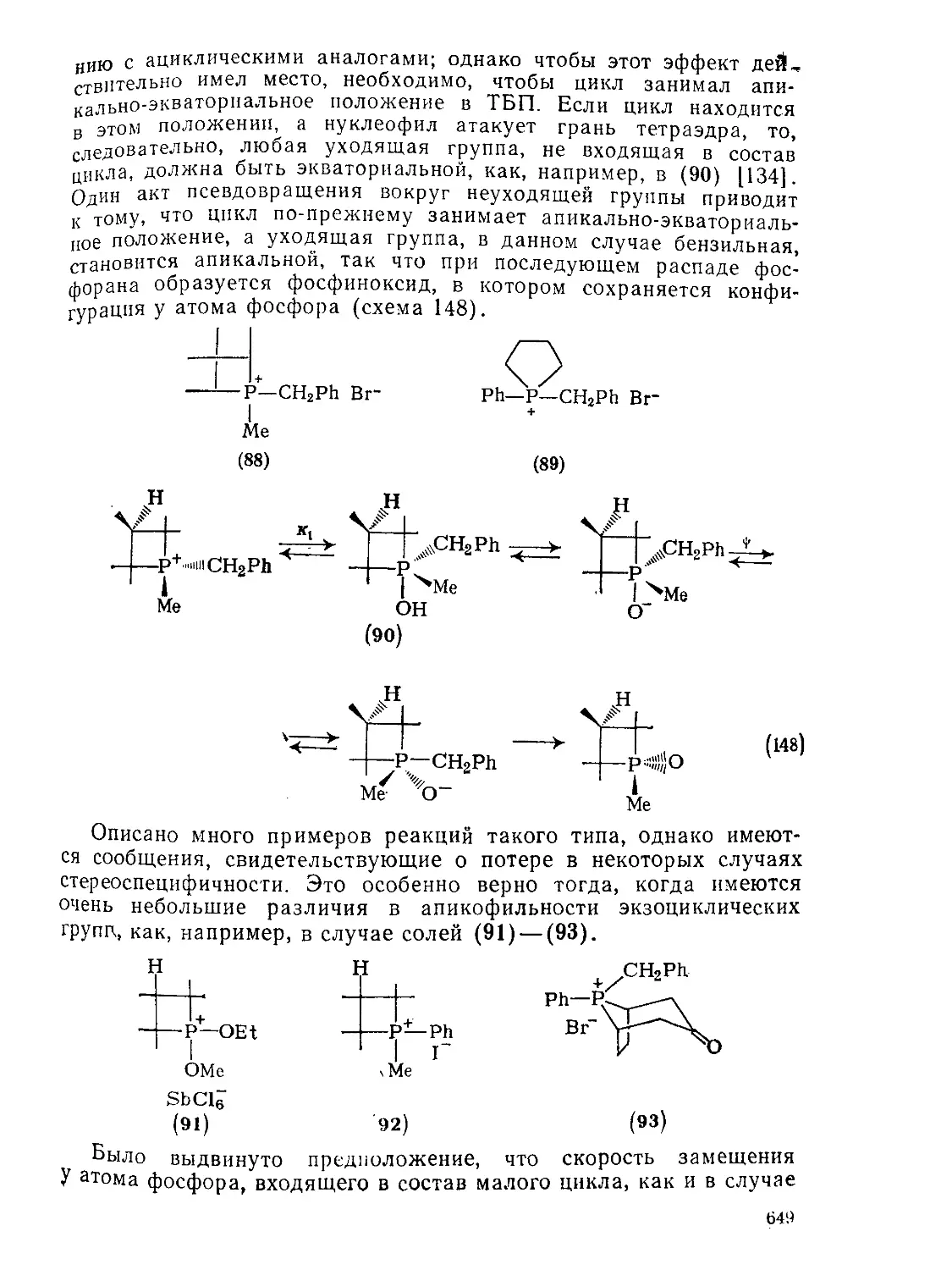

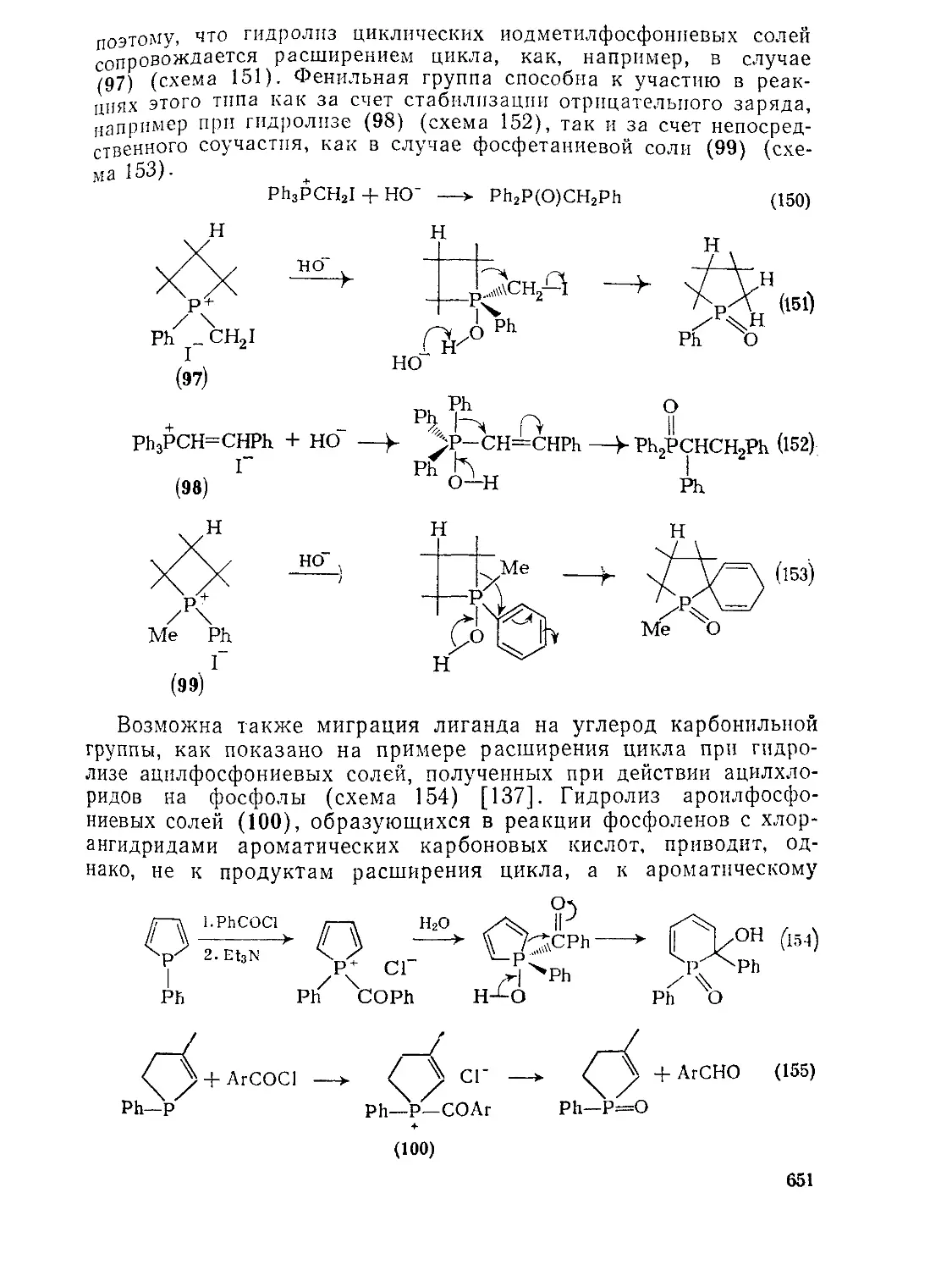

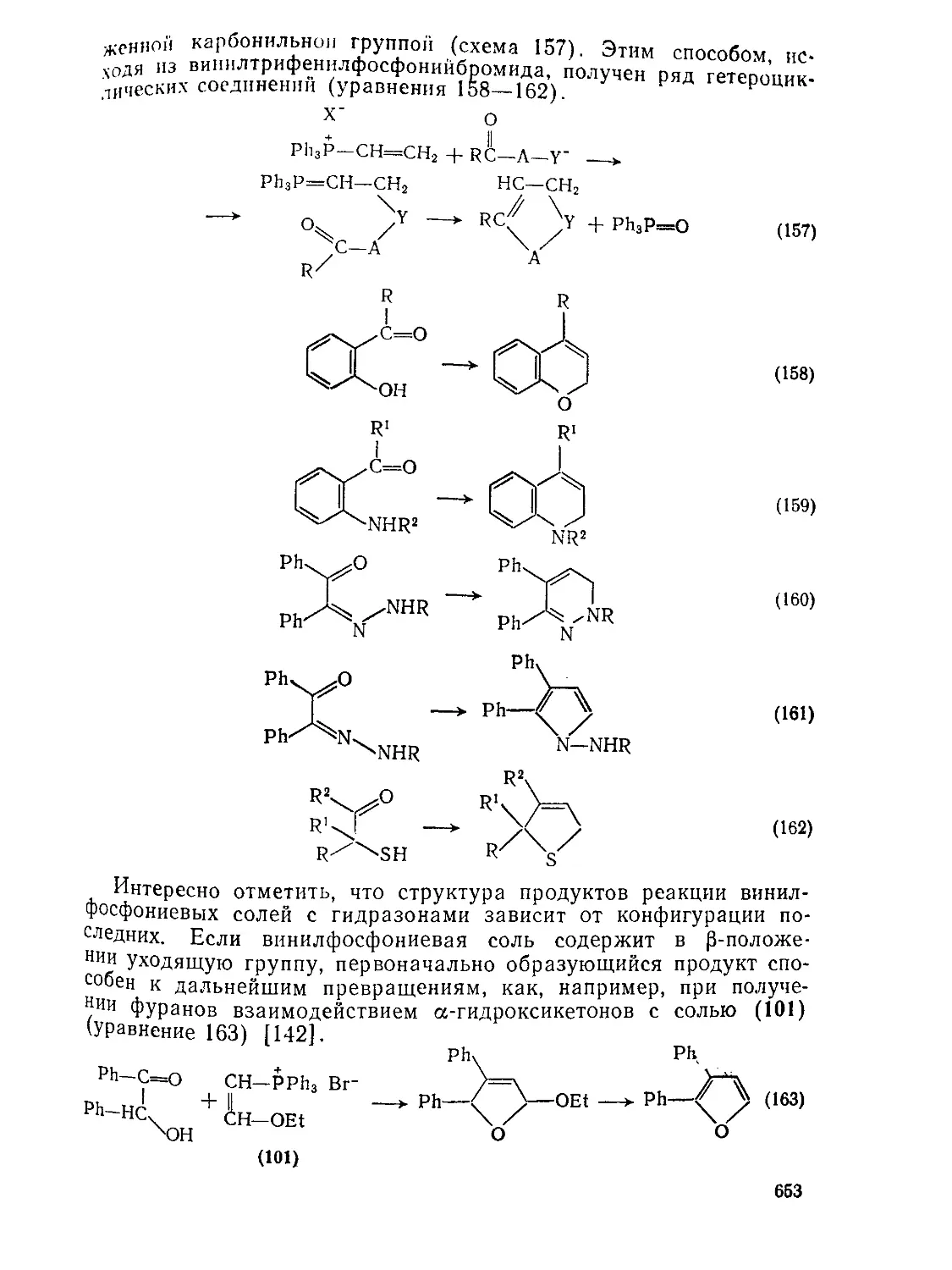
































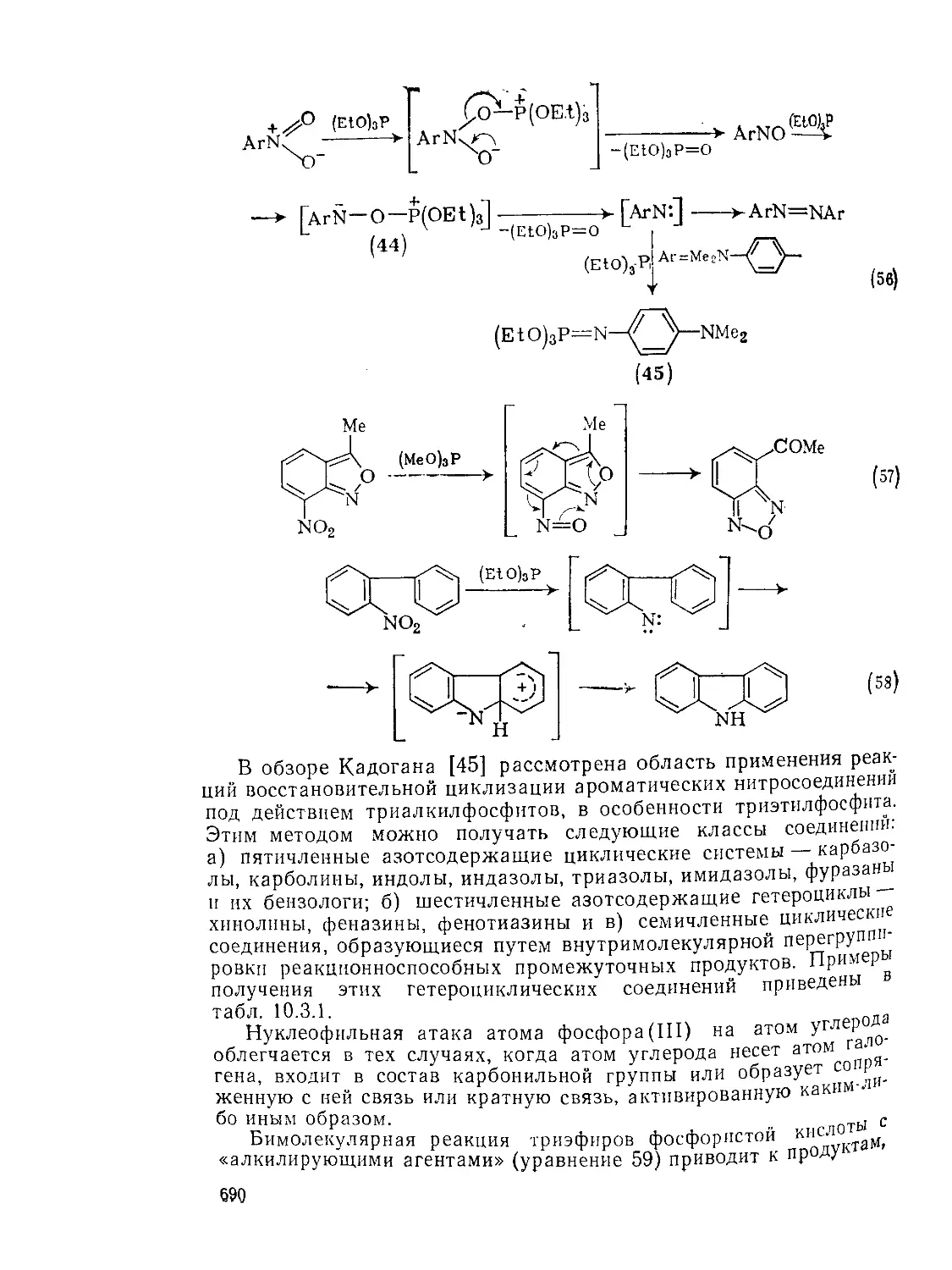
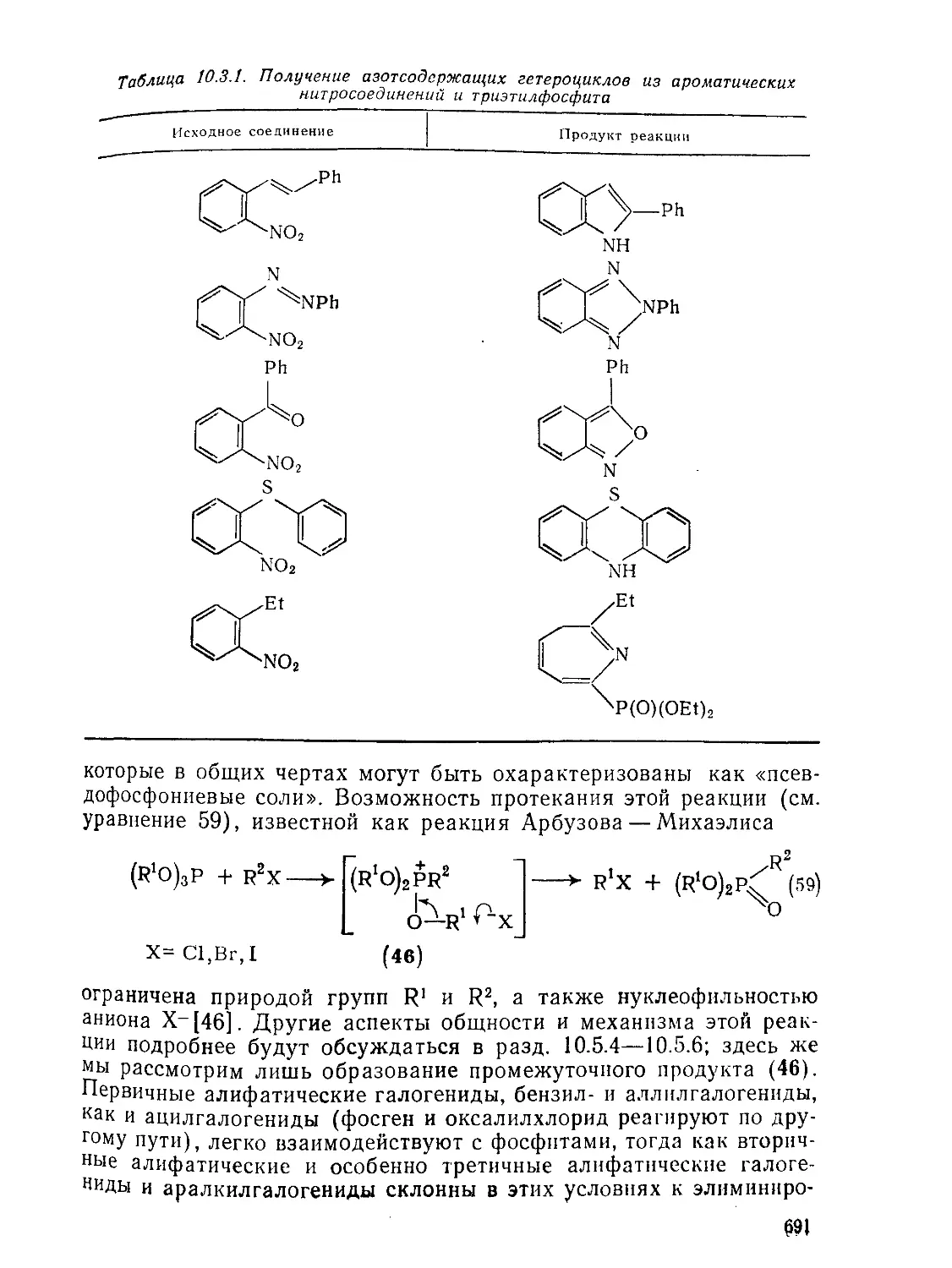







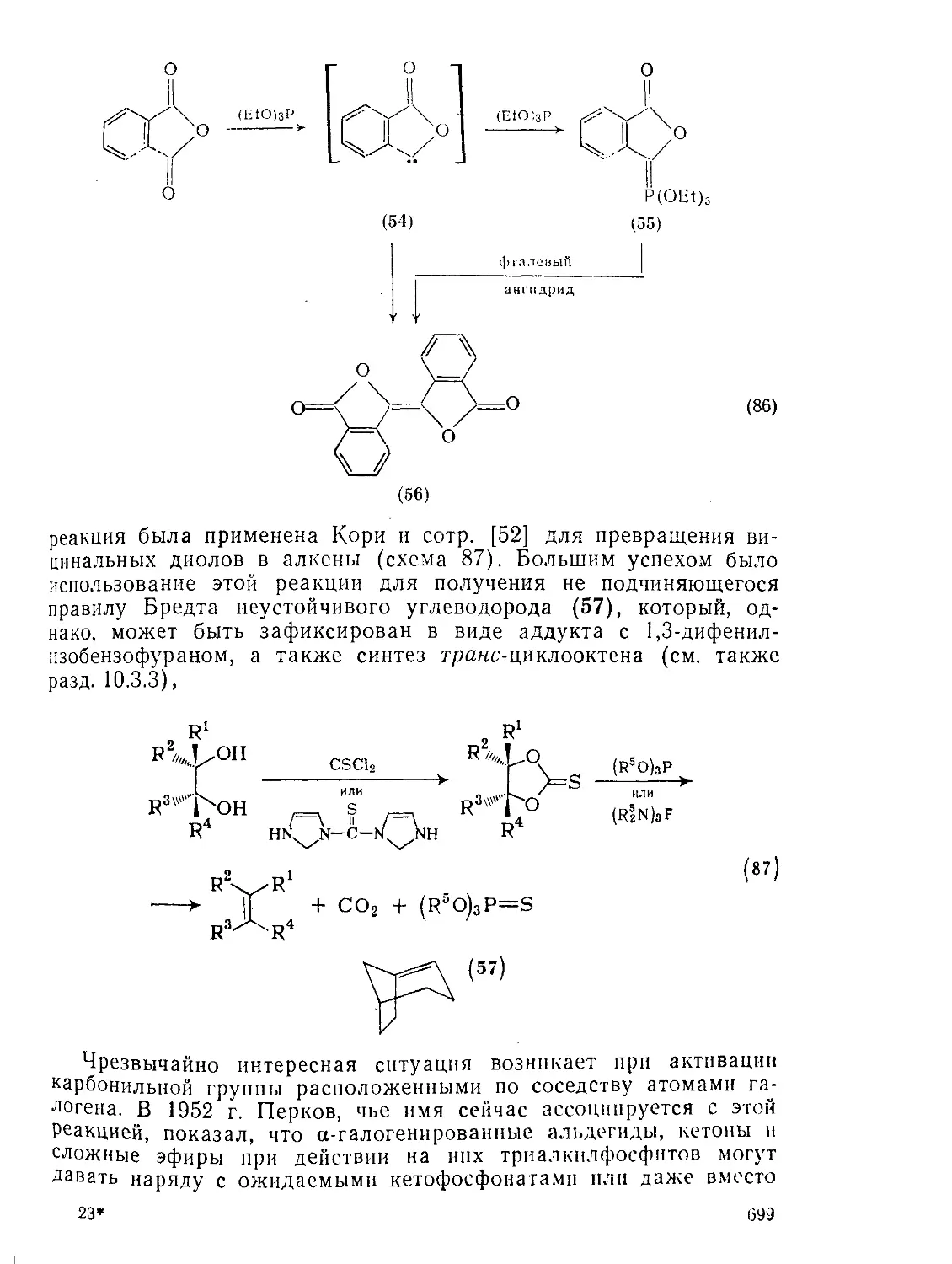





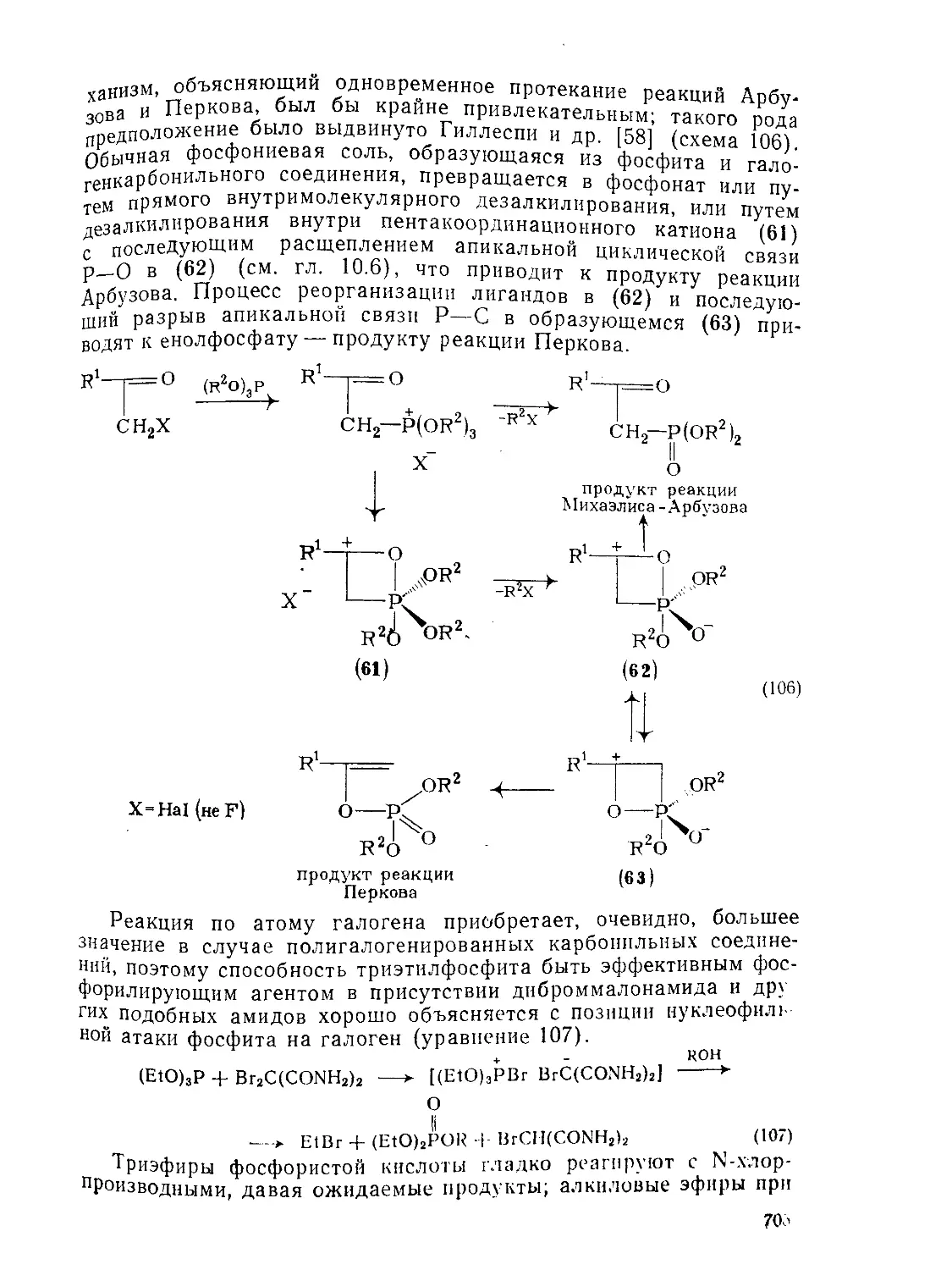




























Текст
COMPREHENSIVE ORGANIC CHEMISTRY
The Synthesis and Reactions of Organic Compounds
CHAIRMAN AND DEPUTY CHAIRMAN OF THE EDITORIAL I3OARD
SIR DEREK BARTON, F.R.S.
W. DAVID OLLIS, F.R.S.
Volume 2 Carboxylic Acids Phosphorus Compounds
Edited by' I. О SUTHERLAND
UNIVERSITY OF LIVERPOOL
PERGAMON PRESS
‘ OXFORD NEW YORK • TORONTO • SYDNEY • PARIS • FRANKFURT
ОБЩАЯ ОРГАНИЧЕСКАЯ ХИМИЯ
ТОМ 4
КАРБОНОВЫЕ КИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ.
СОЕДИНЕНИЯ ФОСФОРА
ПЕРЕВОД С АНГЛИЙСКОГО
канд. хим. наук
В. И. ТОРГОВА и канд. хим. наук Ю. Е. ЦВЕТКОВА
Под редакцией акад.
Н. К. КОЧЕТКОВА, докт. хим. наук
Э. Е. НИФАНТЬЕВА
и канд. хим. наук
М. А. ЧЛЕНОВА
МОСКВА «ХИМИЯ» 1983
УДК 547
Общая органическая химия./Под ред. Д. Бартона и У. Д. Оллиса. Т. 4. Карбоновые кислоты и их производные. Соединения фосфора./Под ред. О. И. Сазерленда. — Пер. с англ./Под ред. Н. К. Кочеткова, Э. Е. Нифантьева и М. А. Членова.— М.: Химия, 1983. — 728 с., ил.
В томе 4 перевода настоящего многотомного издания, являющегося по существу энциклопедией органической химии, рассмотрены карбоновые кислоты и их производные — моно-, ди- и поликарбоновые кислоты, галоген-, гидрокси-, оксо- и азотсодержащие замещенные карбоновых кислот, амиды и родственные соединения, производные диоксида углерода, а также пероксикислоты и пероксиды ацилов. В этот же том включены введение в химию фосфорорганических соединений, а также главы, посвященные фосфинам, фосфористой, фосфо-нистой, фосфинистой кислотам и их производным (гл. 10.1 — 10.3).
Издание предназначено для научных работников, инженеров-химиков, работающих на предприятиях химической, нефтехимической и других отраслей промышленности, преподавателей и аспирантов химических и химико-технологических вузов и факультетов университетов, для биохимиков и биологов.
728 с., 3 рис., 40 табл., 2340 литературных ссылок.
О 1803000000ИП пд ПодПИСНых изд. 1983 г.
050(01)-83
© 1979 Pergamon Press Ltd.
© Перевод на русский язык. Издательство «Химия», 1983 г.
СОДЕРЖАНИЕ
ЧАСТЬ 9. КАРБОНОВЫЕ КИСЛОТЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И
9.1. Карбоновые кислоты. Е. В. Колвин 11
9.1.1. Методы получения карбоновых кислот 11
9.1.1.1. Окислительные методы 11
9.1.1.2. Карбонилирование 19
9.1.1.3. Карбоксилирование 20
9.1.1.4. Гидролитические методы 21
9.1.1.5. Синтезы из малоновой и ацетоуксусной кислот и родственных соединений 23
9.1.1.6. Расщепление гетероциклических соединений 26
9.1.1.7. Использование производных типа уксусной кислоты 30
9.1.1.8. Направленные синтезы ненасыщенных кислот и их производных 32
9.1.1.9. Другие методы . 37
9.1.10. Меченные изотопами и хпральные кислоты 38
9.1.2. Свойства карбоновых кислот 41
9.1.2.1. Структура и геометрия 42
9.1.2.2. Ионизация и димеризация 42
9.1.2.3. Оксокарбениевые ионы 43
9.1.2.4. Мицеллы 44
9.1.2.5. Спектральные свойства 45
9.1.3. Реакции ацилирования 46
9.1.3.1. Прямые реакции 47
9.1.3.2. Активация кислотной компоненты 47
9.1.3.3. Активация спиртовой компоненты 52
9.1.4. Другие реакции (помимо ацилирования) 53
9.1.4.1. Декарбоксилирование 53
9.1.4.2. Галогенирование и окисление 56
9.1.4.3. Восстановление 57
9.1.4.4. Реакции с металлорганическими соединениями 59
9.1.4.5. Реакции ненасыщенных карбоновых кислот 61
9.1.4.6. Реакции ароматических карбоновых кислот 62
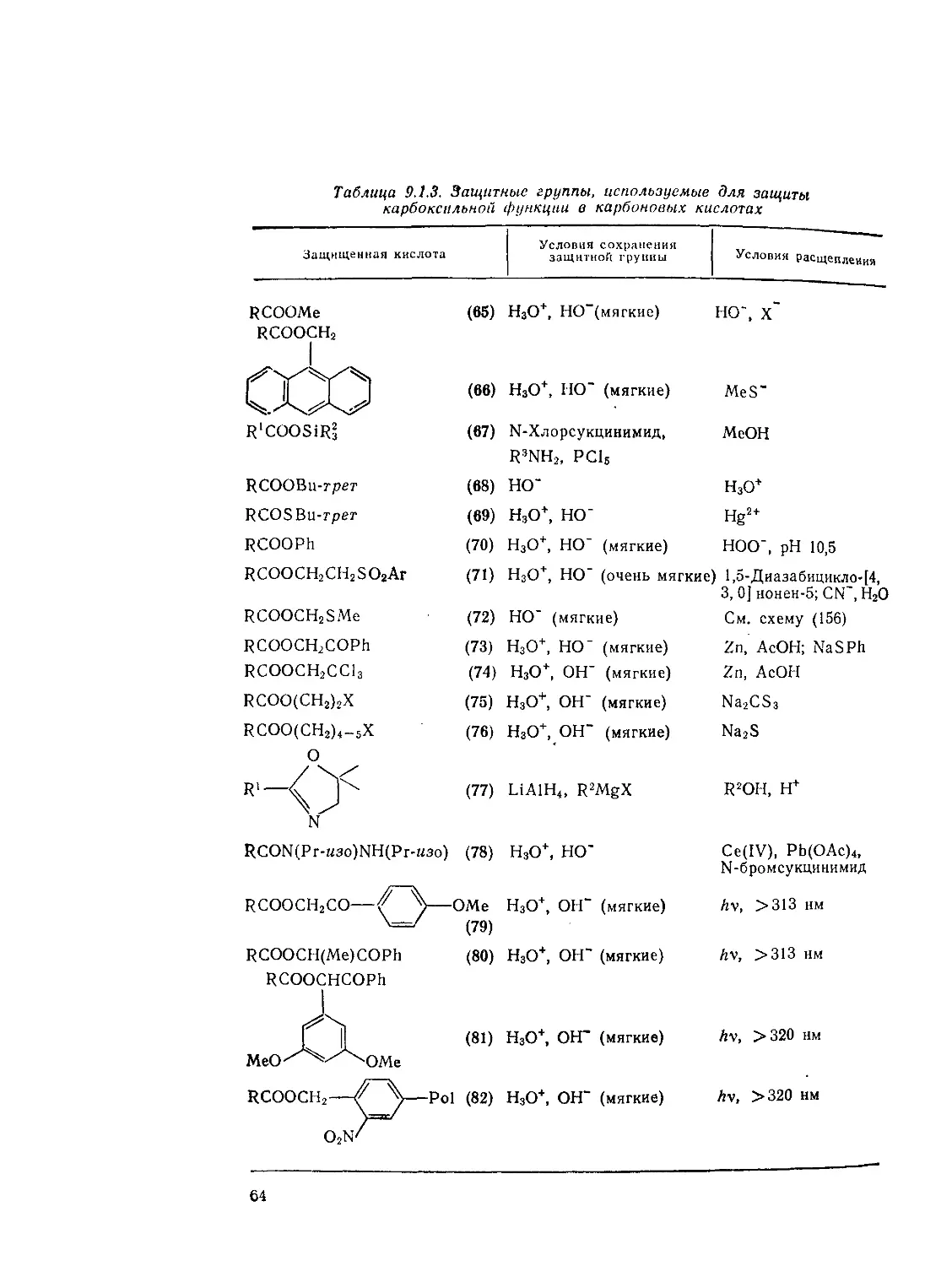
9.1.4.7. Защита карбоксильной группы 63
Литература 67
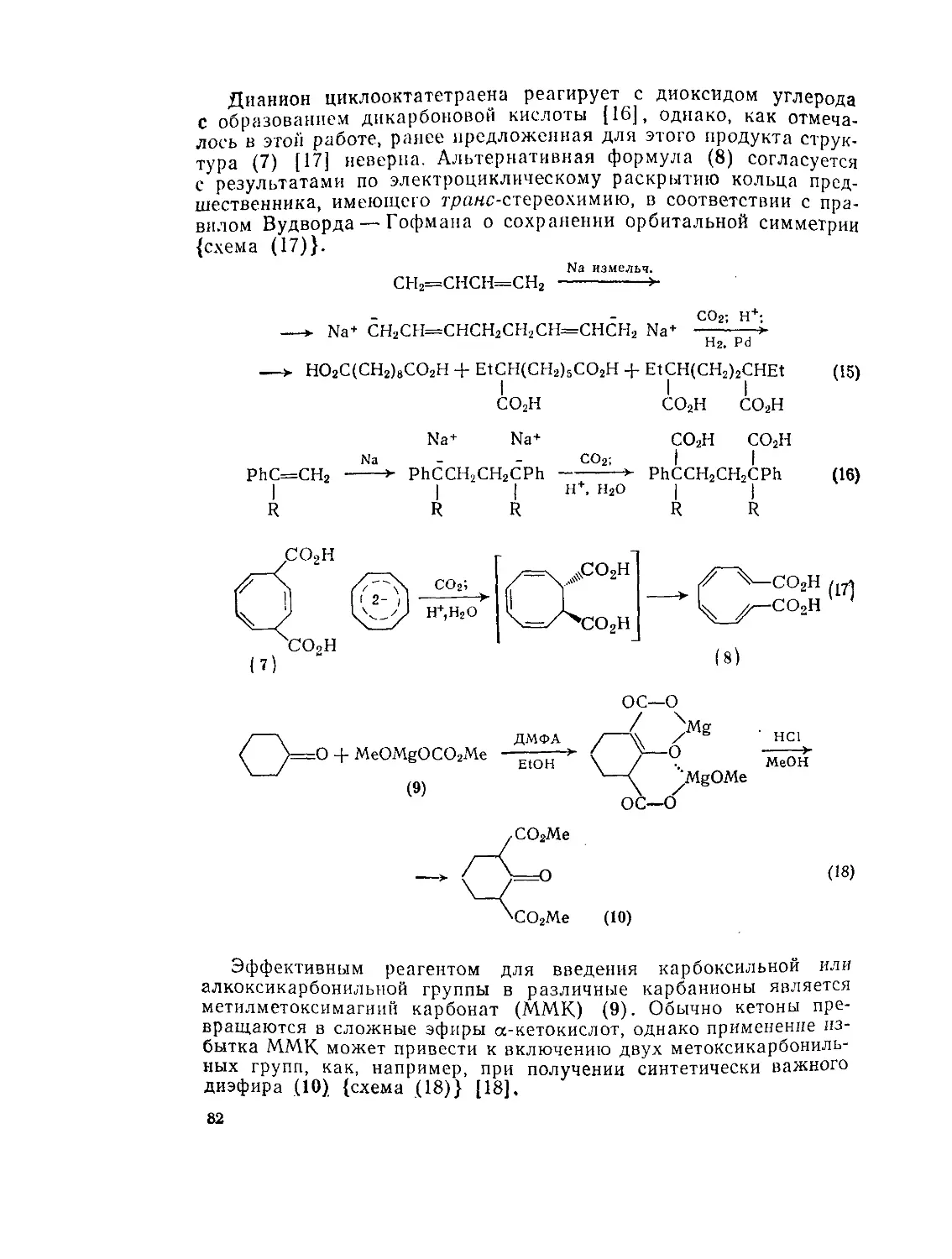
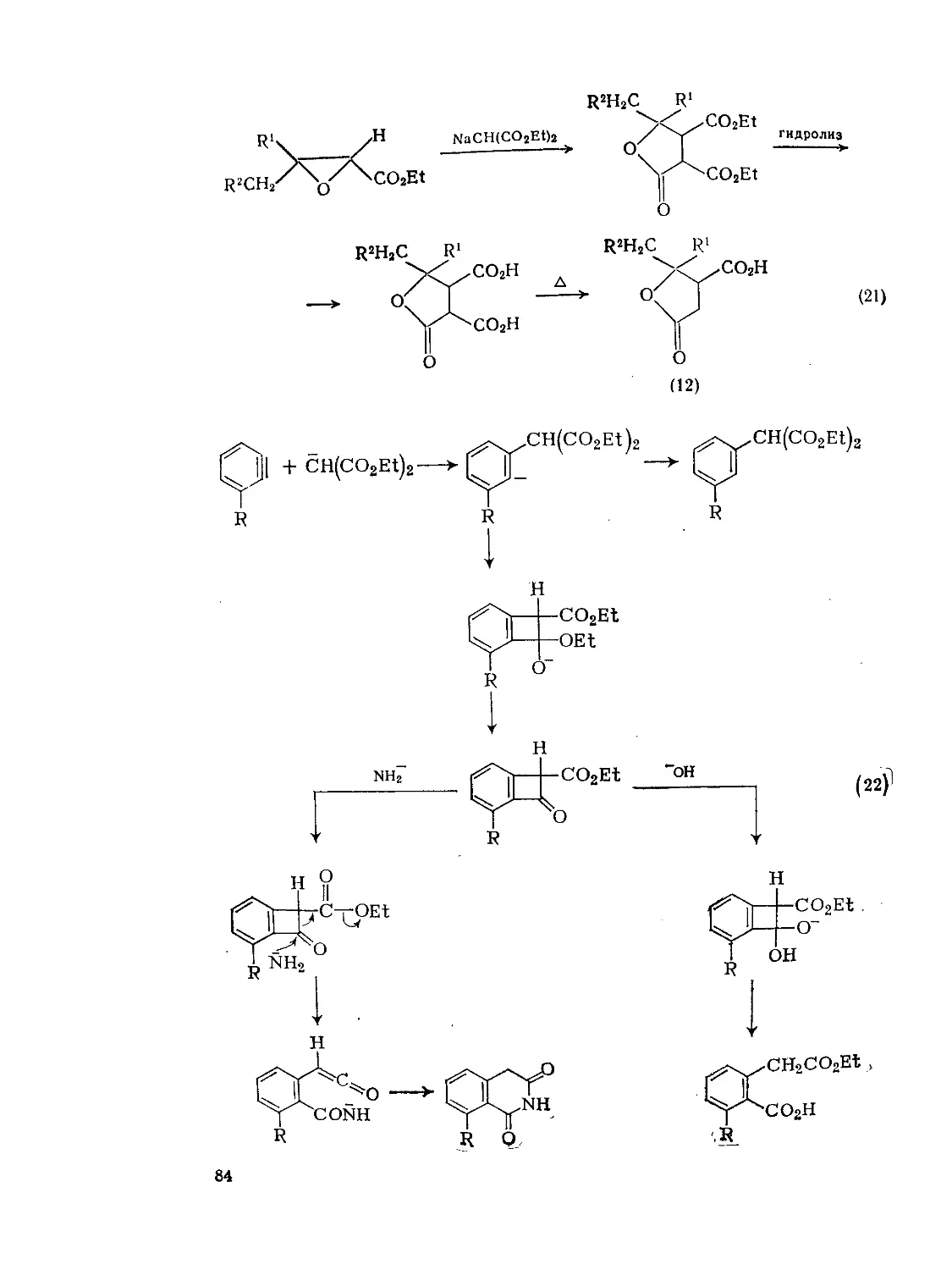
9.2. Дикарбоновые и поликарбоновые кислоты. А. Кокс 77
9.2.1. Методы получения ди- и полнкарбоновых кислот 77
9.2.1.1. Карбоксилирование и алкоксикарбонилирование 78
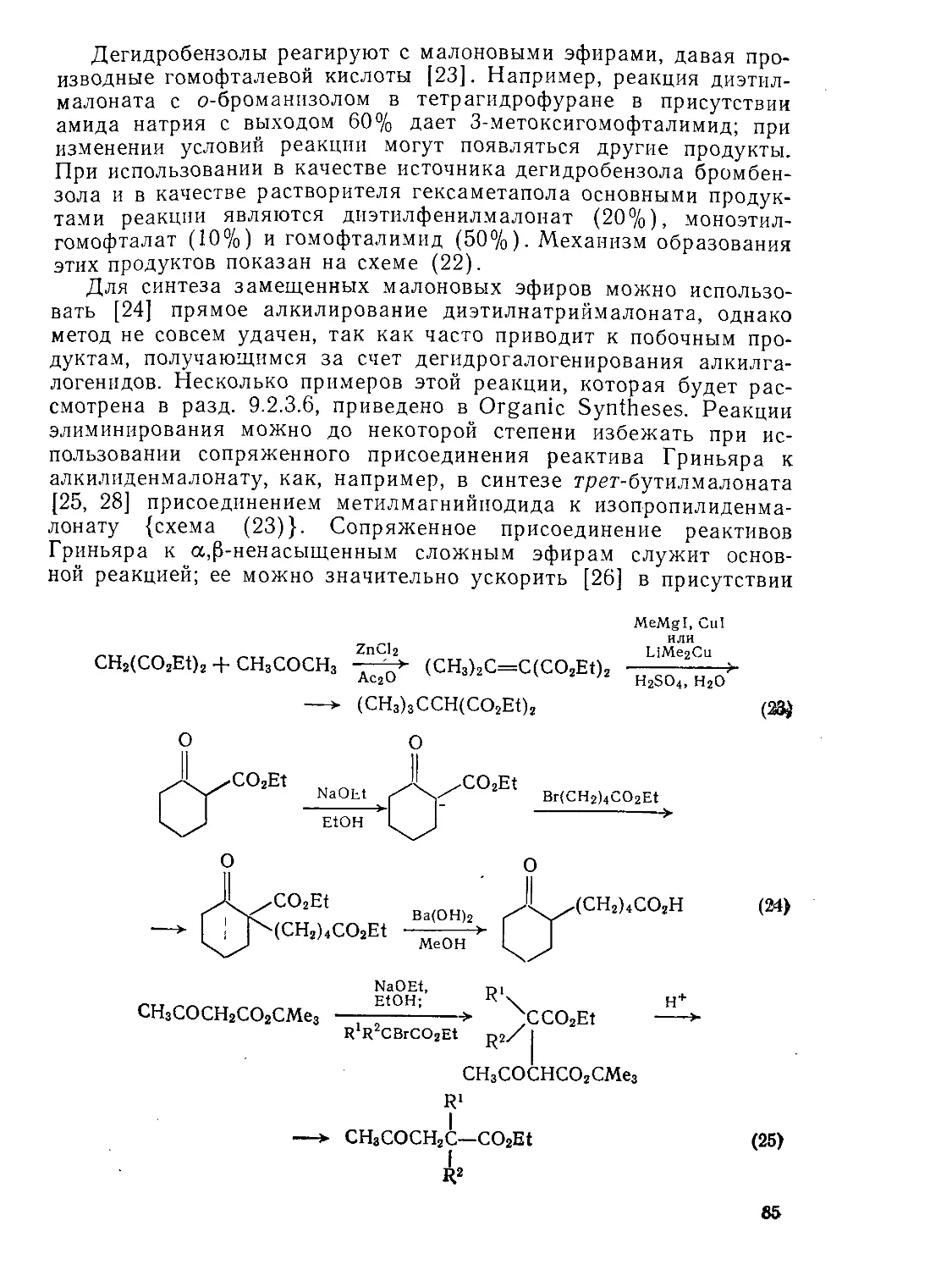
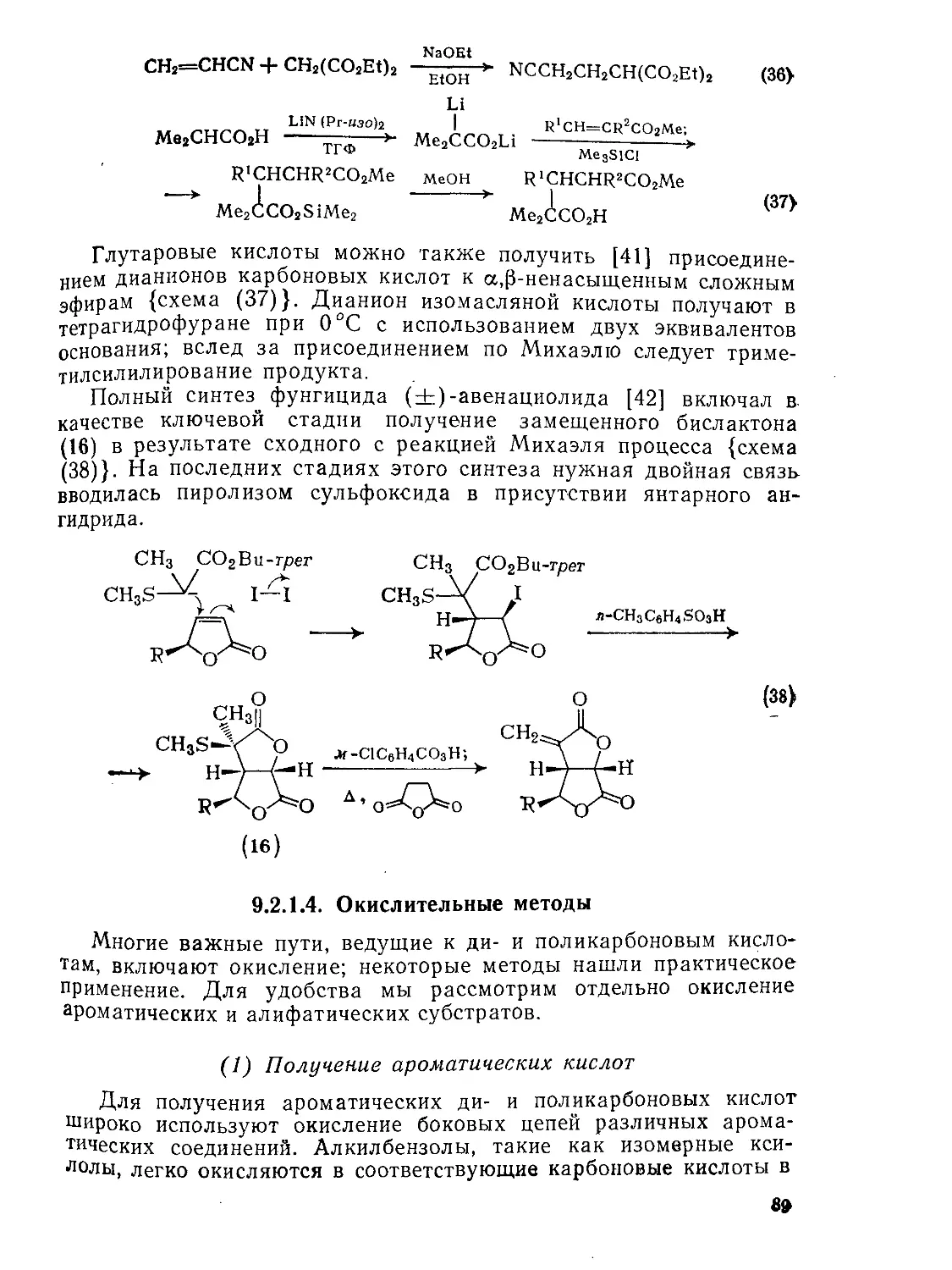
9.2.1.2. Реакции конденсации 83
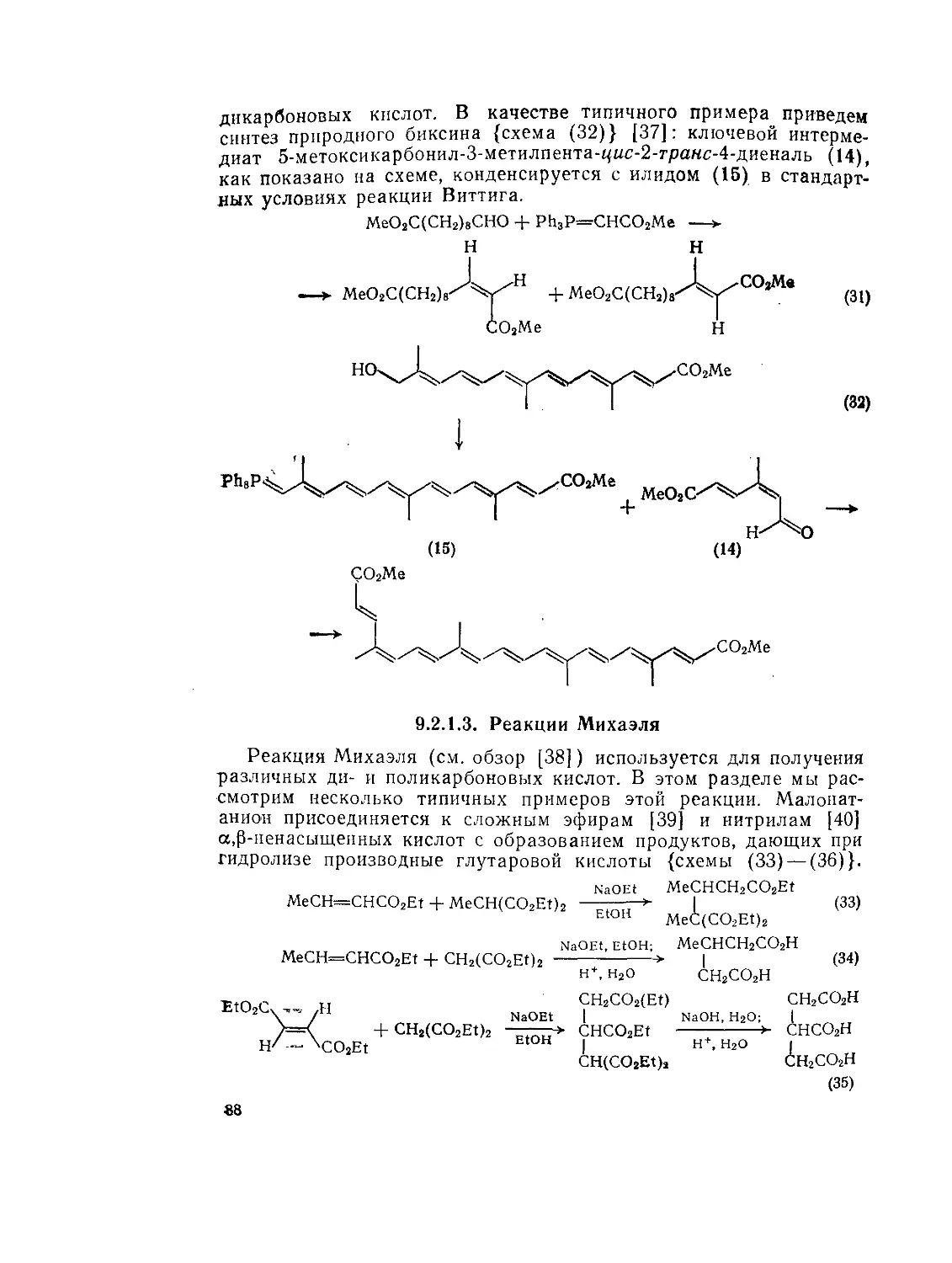
9.2.1.З. Реакции Михаэля 88
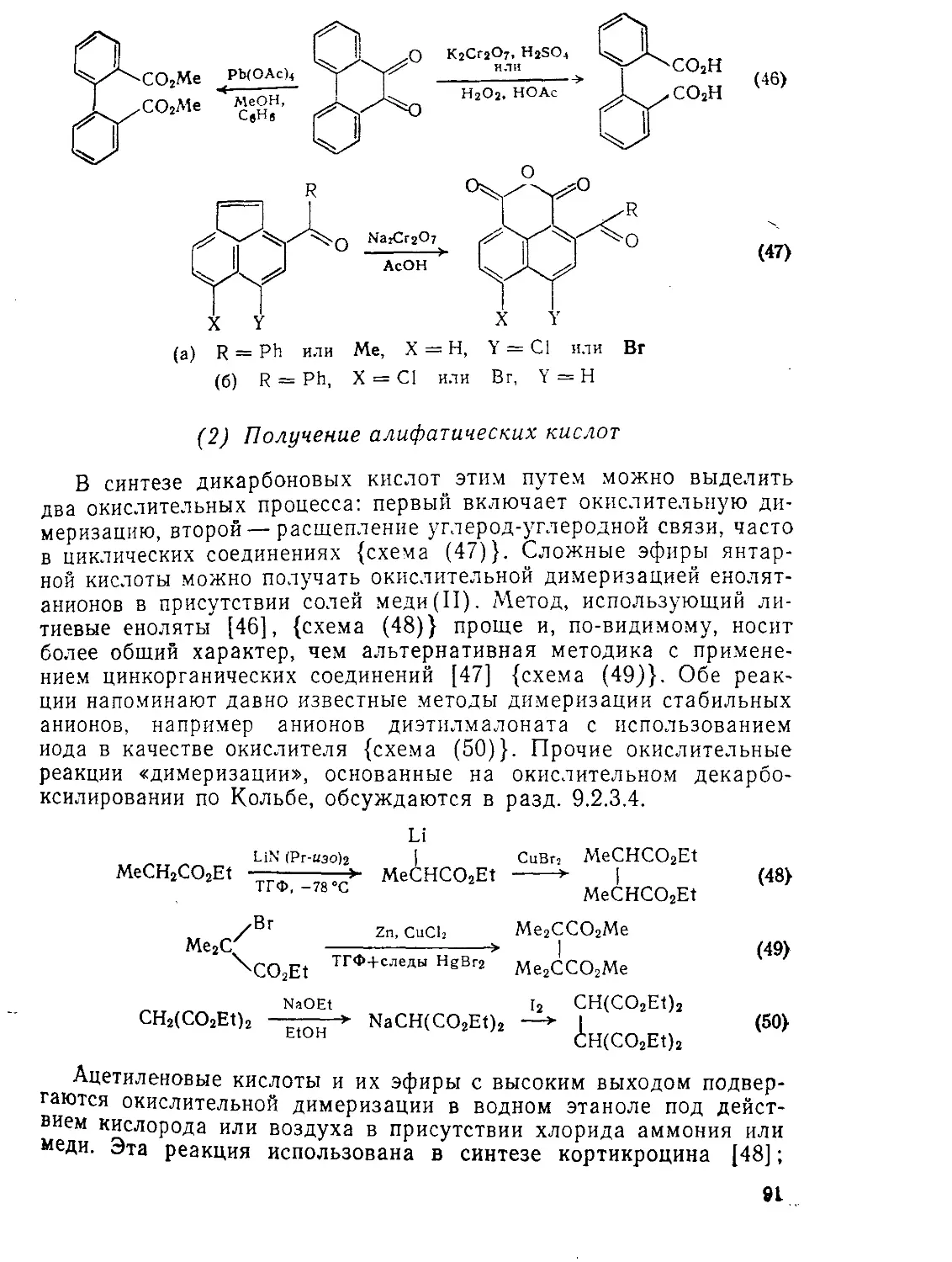
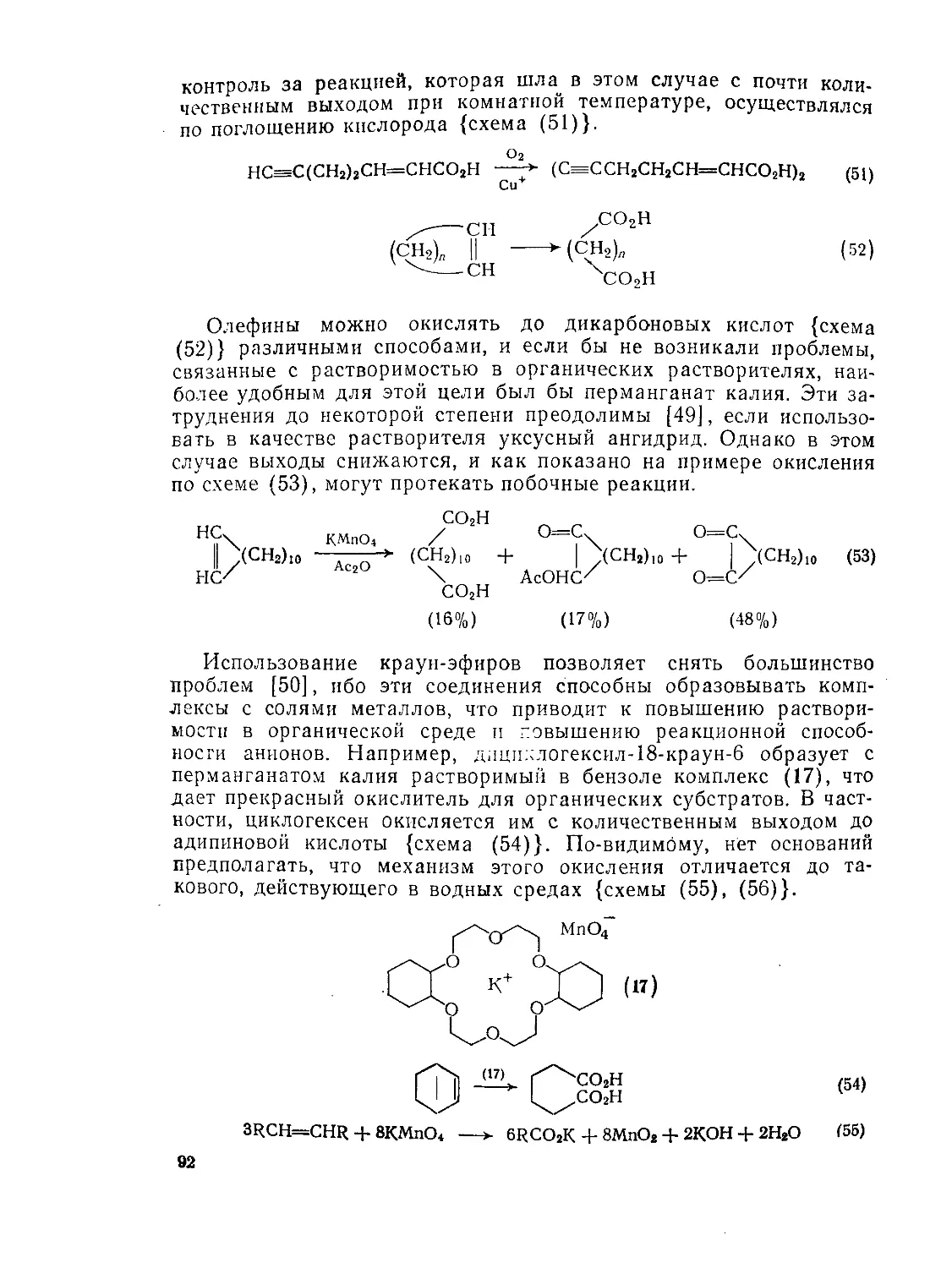
9.2.1.4. ’Окислительные методы 89
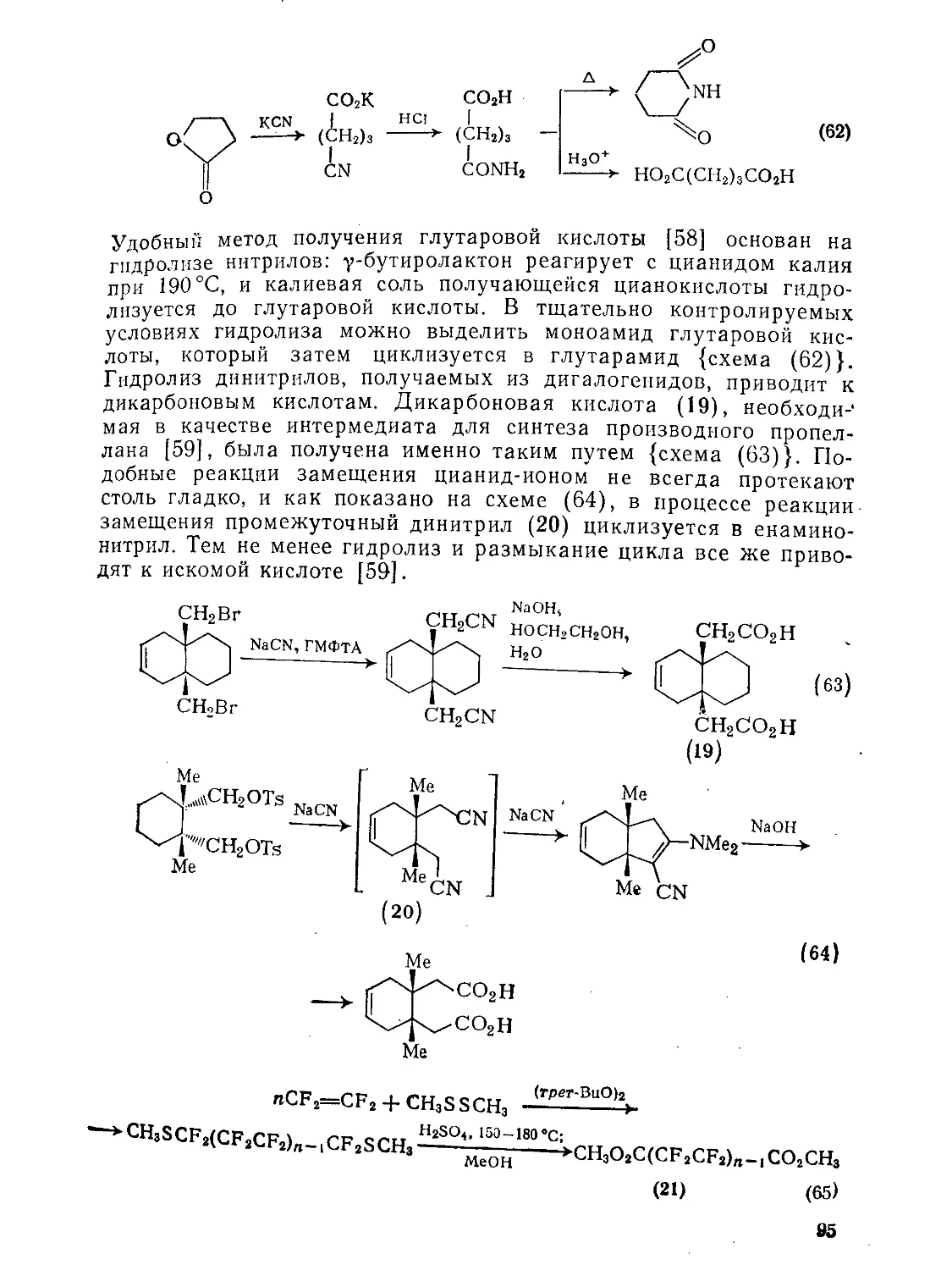
9.2.1.5. Гидролитические методы 94
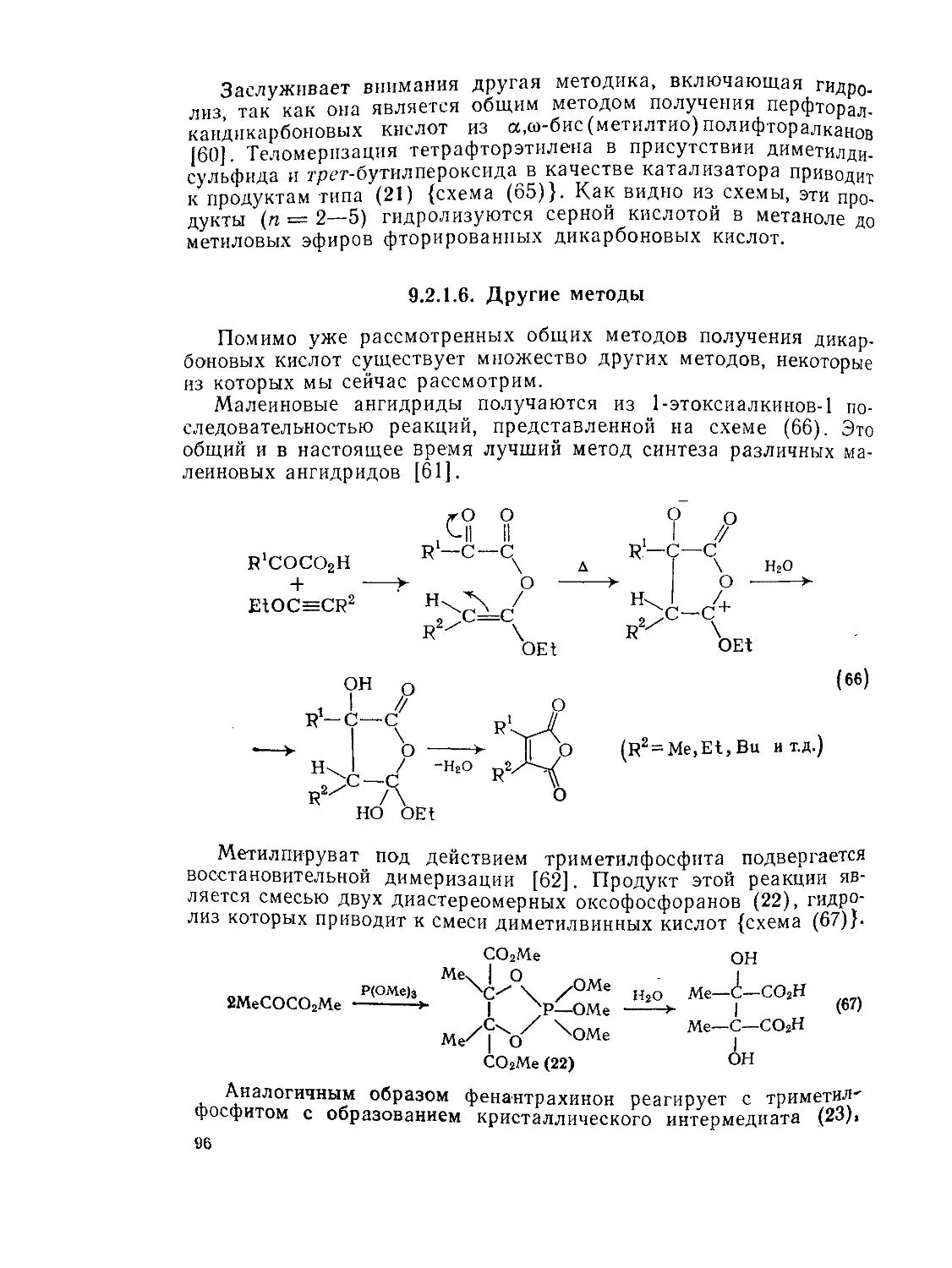
9.2.1.6. Другие методы ’ 96
9.2.2. Свойства ди- и поликарбоновых кислот 98
9.2.3. Реакции ди- и поликарбоновых кислот 101
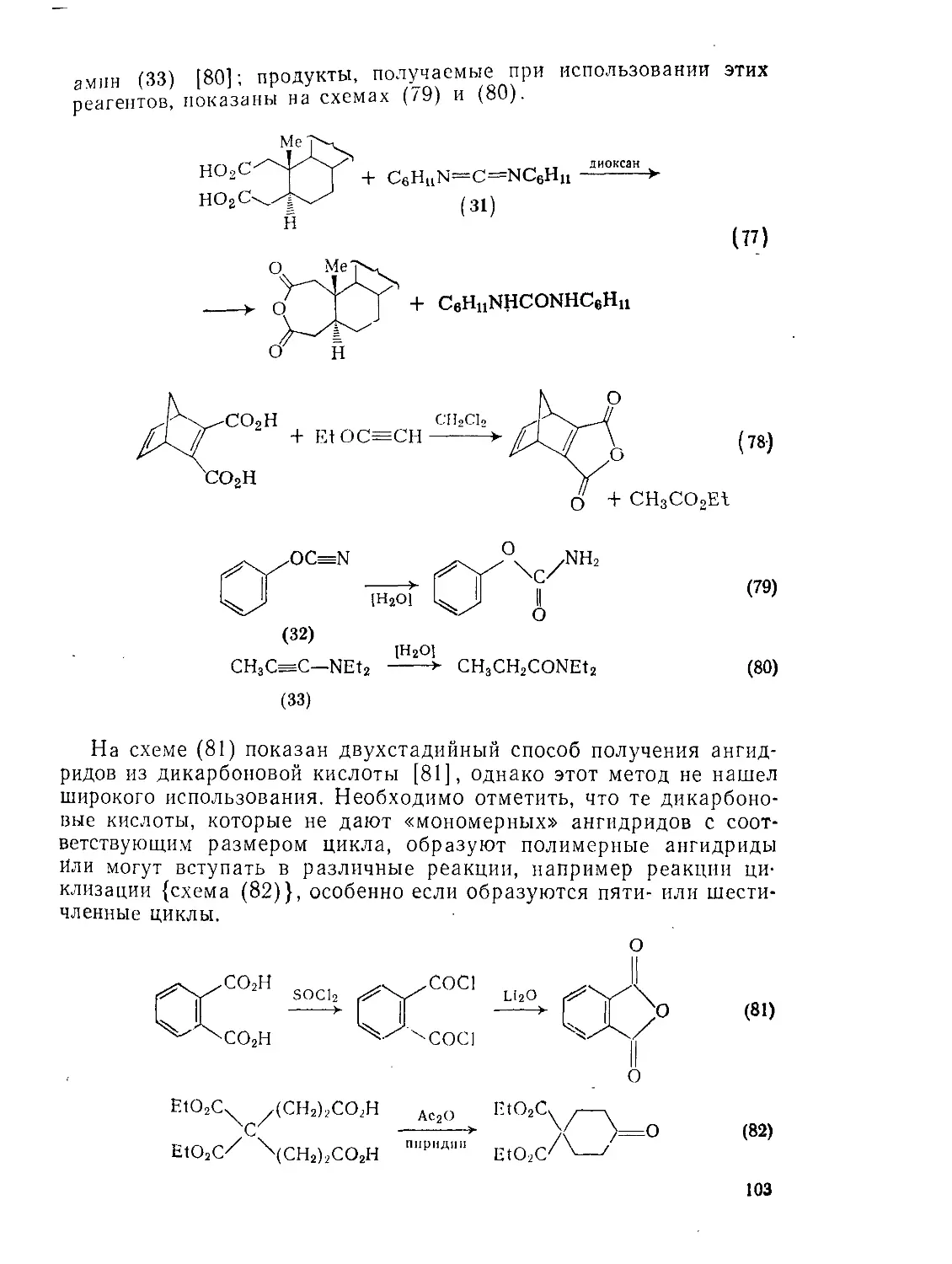
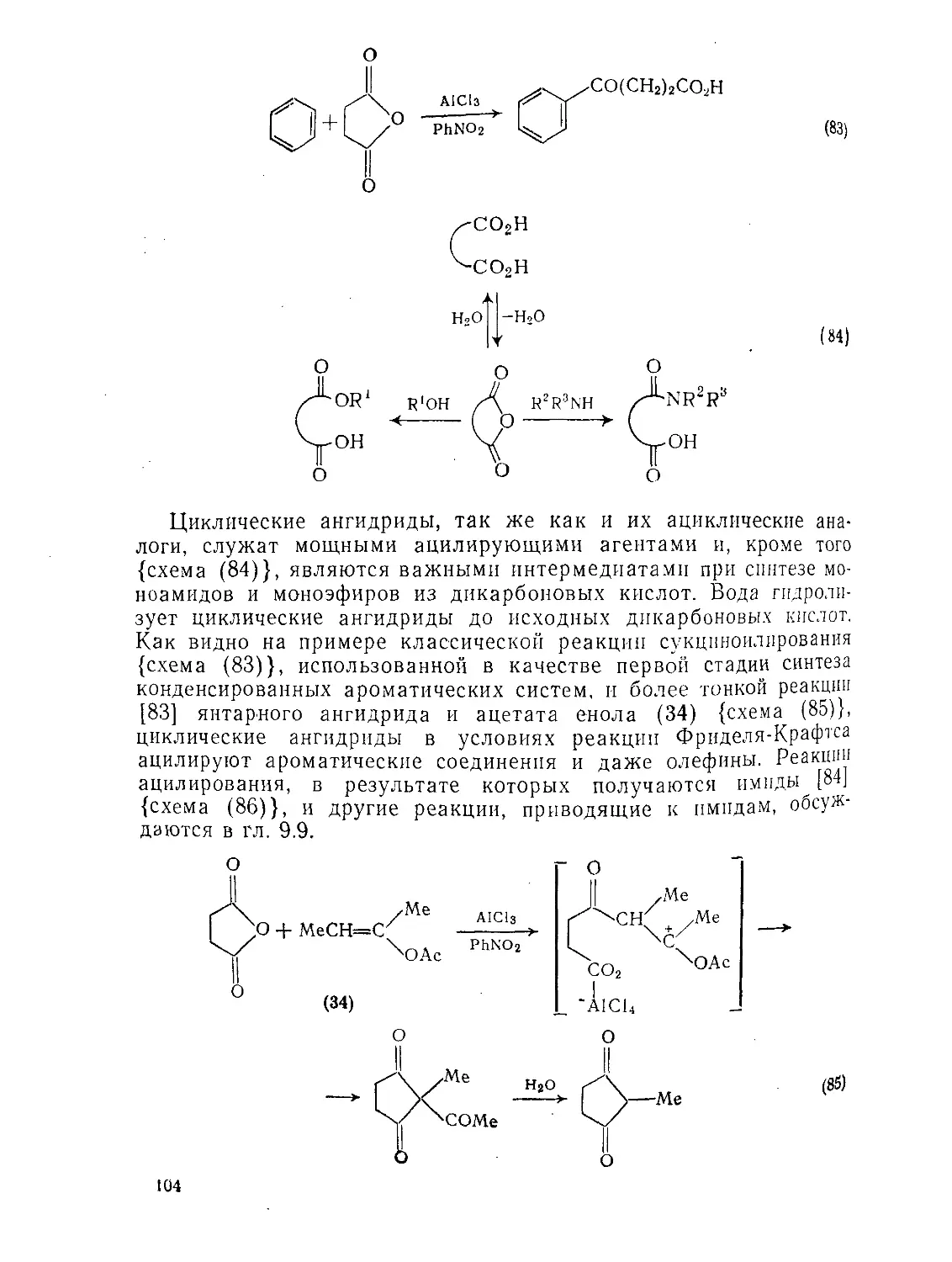
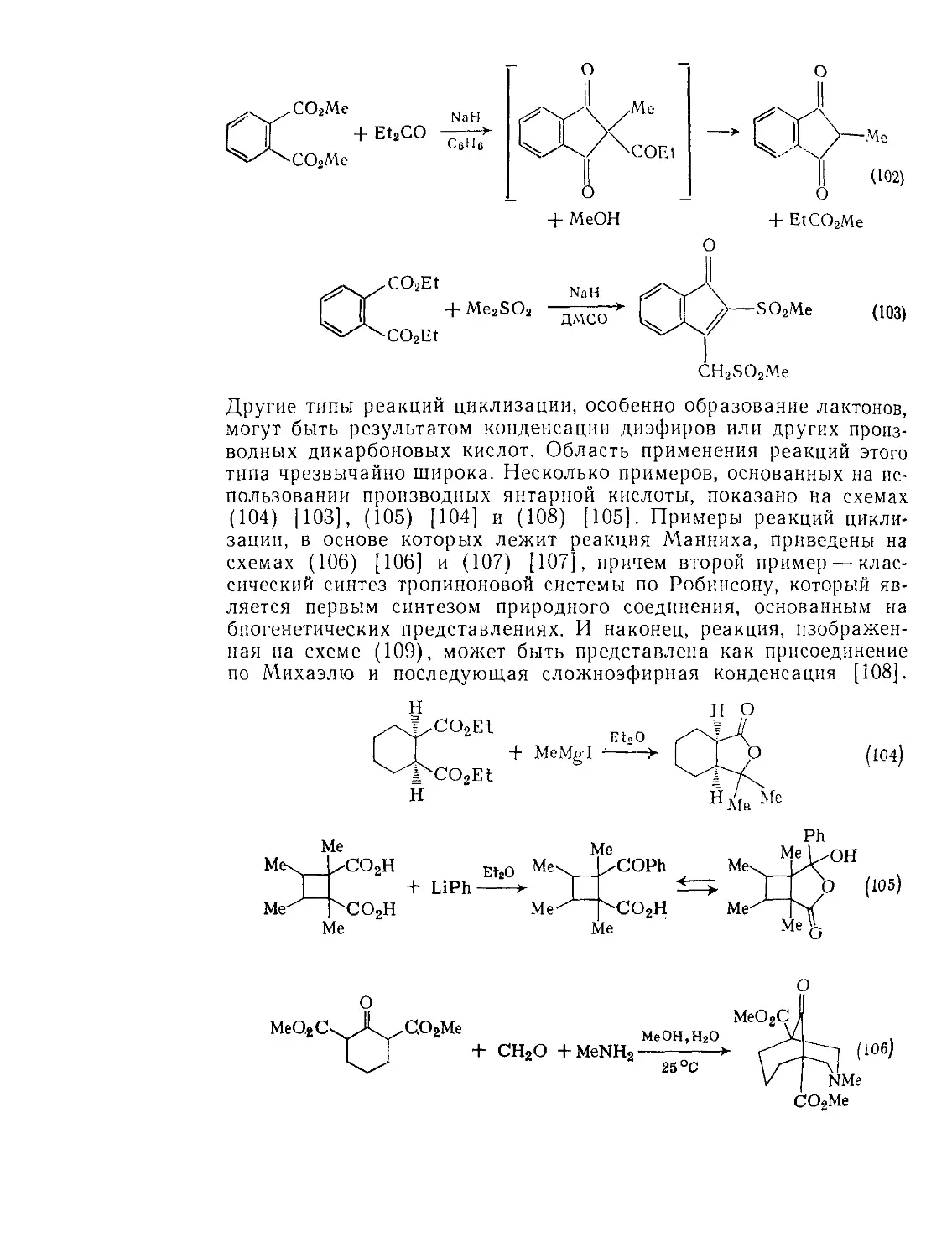
9.2.З.1. Образование и реакции циклических ангидридов 101
9.2.3.2. Конденсация Дикмана 106
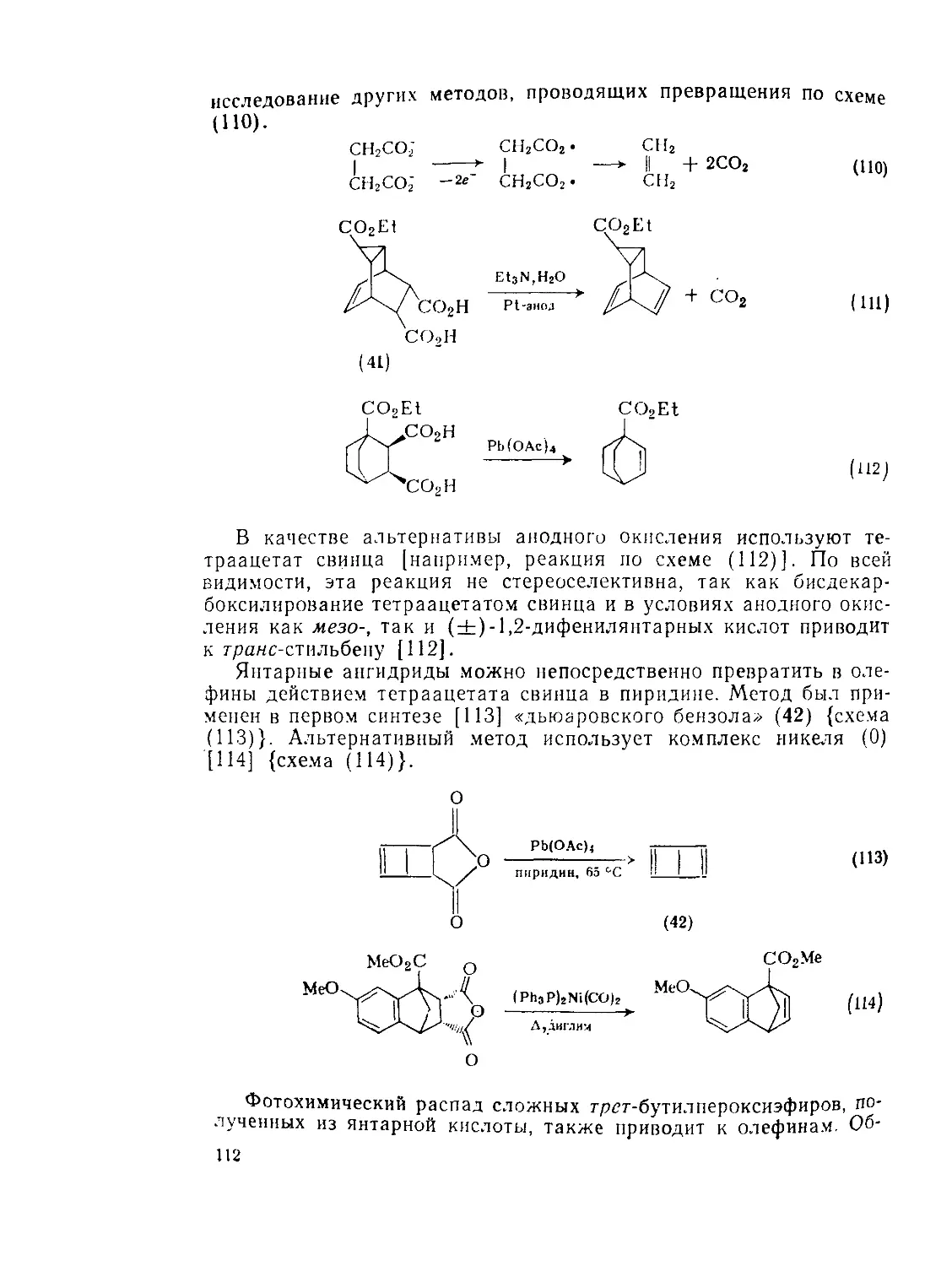
9.2.3.3. Окисление Ш
9.2.3.4. Восстановление И4
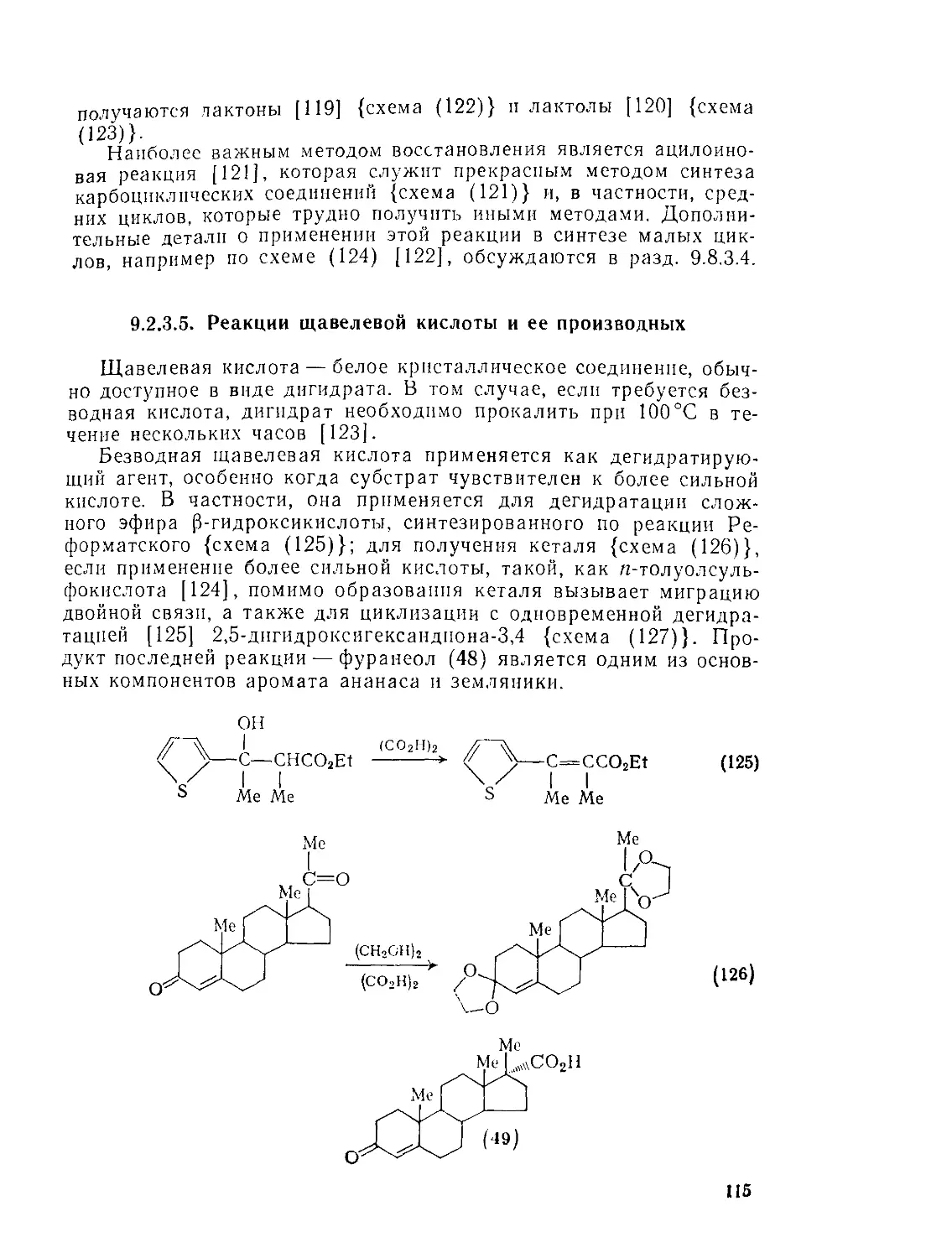
9.23.5. Реакции щавелевой кислоты и ее производных 115
9.2.3.6. Реакции малоновой кислоты и ее производных 120
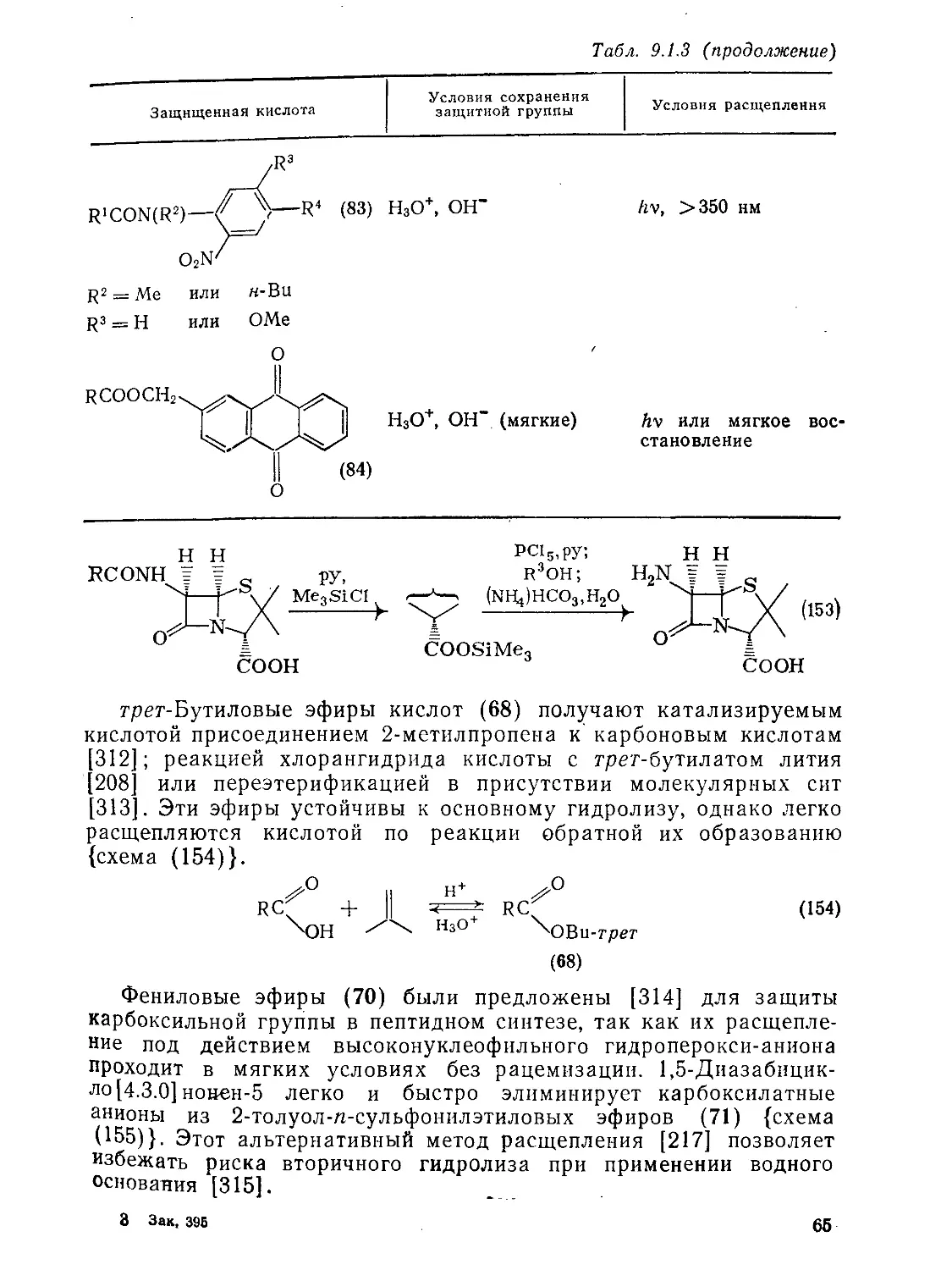
Э.2.3.7. Реакции малеиновой, фумаровой кислот и их производных 126
9.2.3.8. Ацетилендикарбоновая кислота и ее производные 129
Литература 132
5
9.3. Галогенкарбоновые кислоты. А. Кокс
137
9.3.1. Препаративные методы получения галогенкарбоновых кислот 137
9.3.1.1. Карбоксилирование полигалогенсодержащих соединений 137
9.З.1.2. Галогенирование карбоновых кислот и их производных 138
9.3.1.3. Другие методы 143
9.3.2. Свойства галогенкарбоновых кислот 144
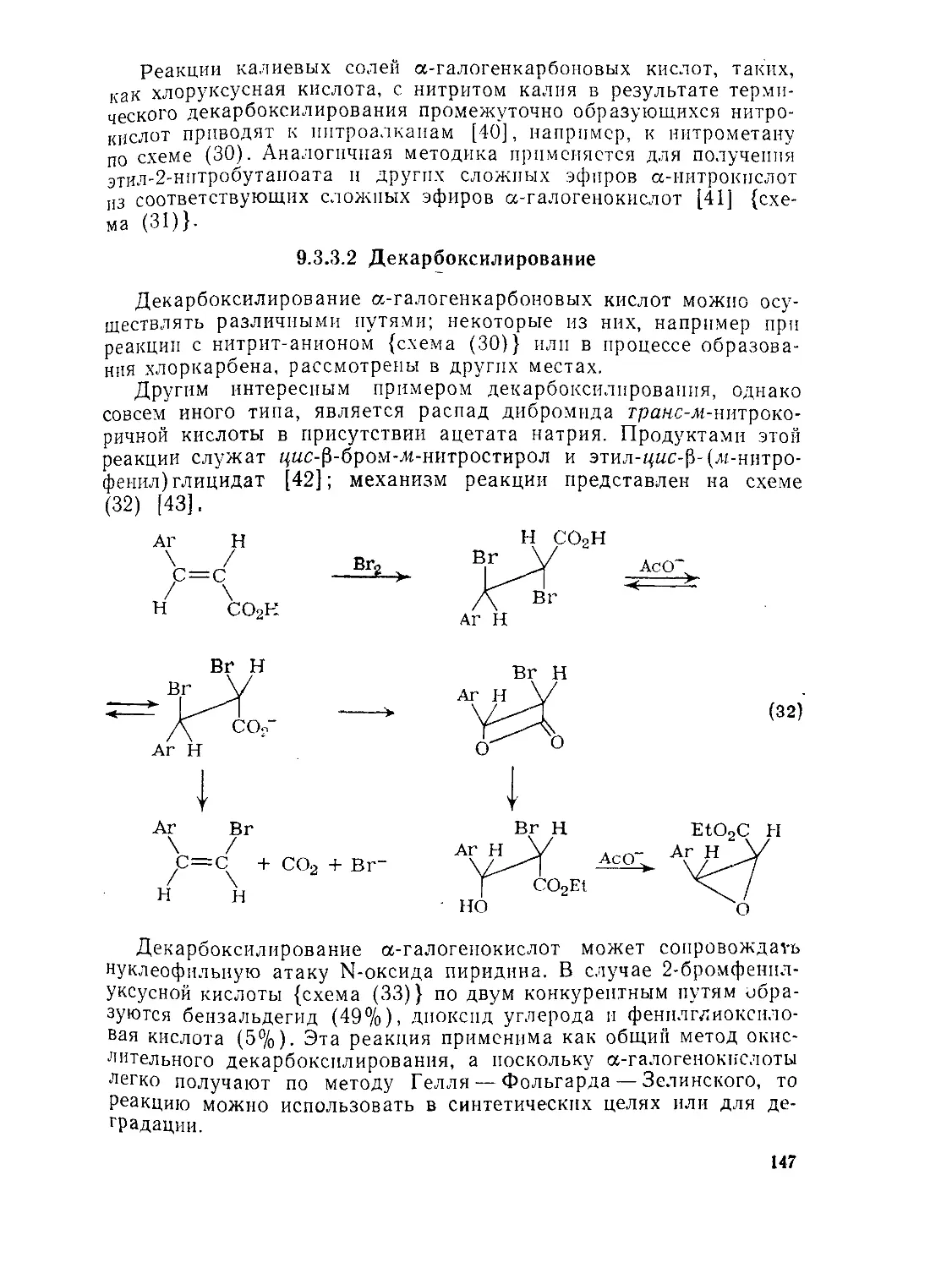
9.3.3. Реакции галогенкарбоновых кислот 146
9.3.3.1. Нуклеофильное замещение 146
9.3.3.2. Декарбоксилирование 147
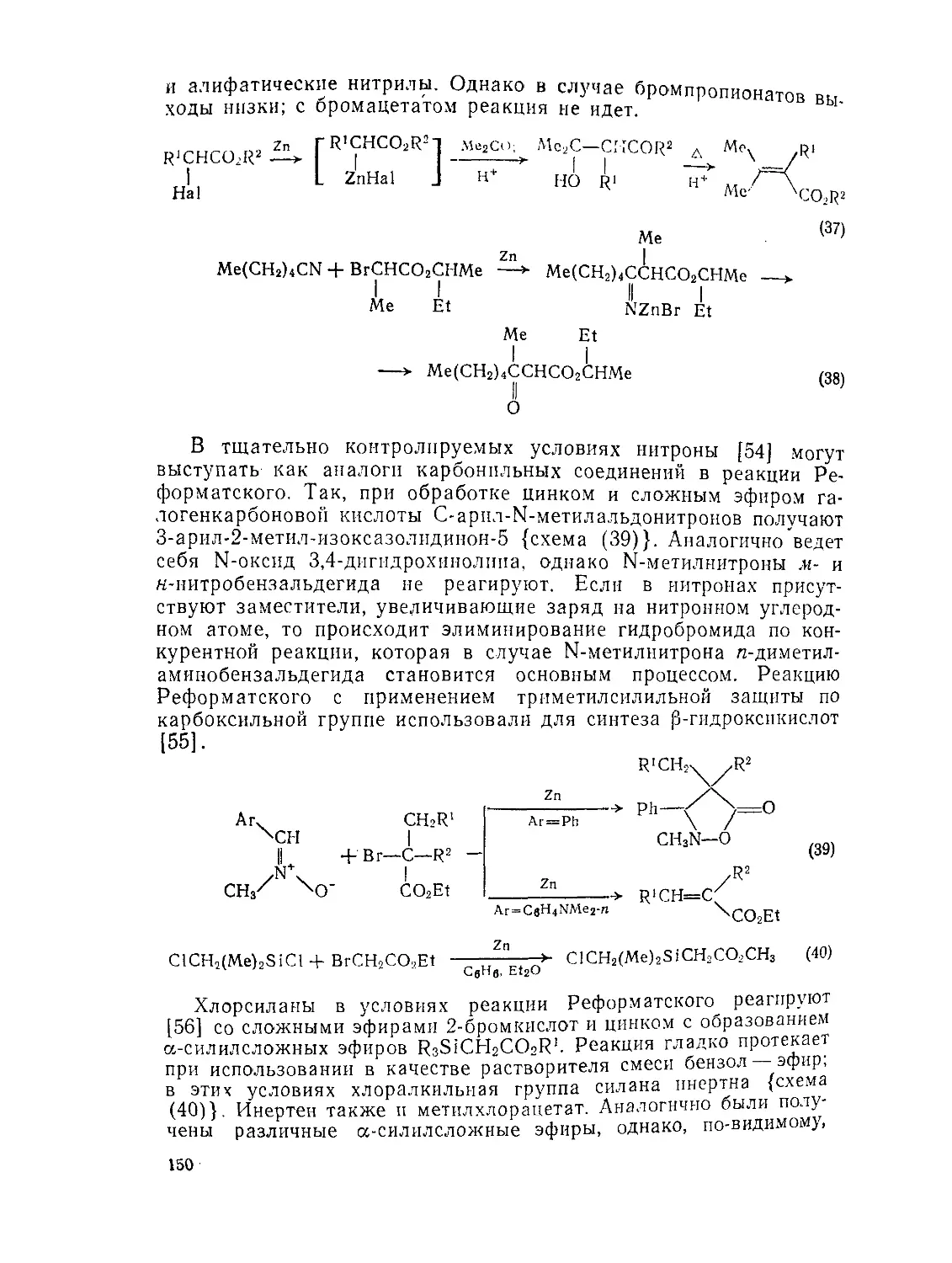
9.3.3.3. Реакции с участием металлорганических интермедиатов 149
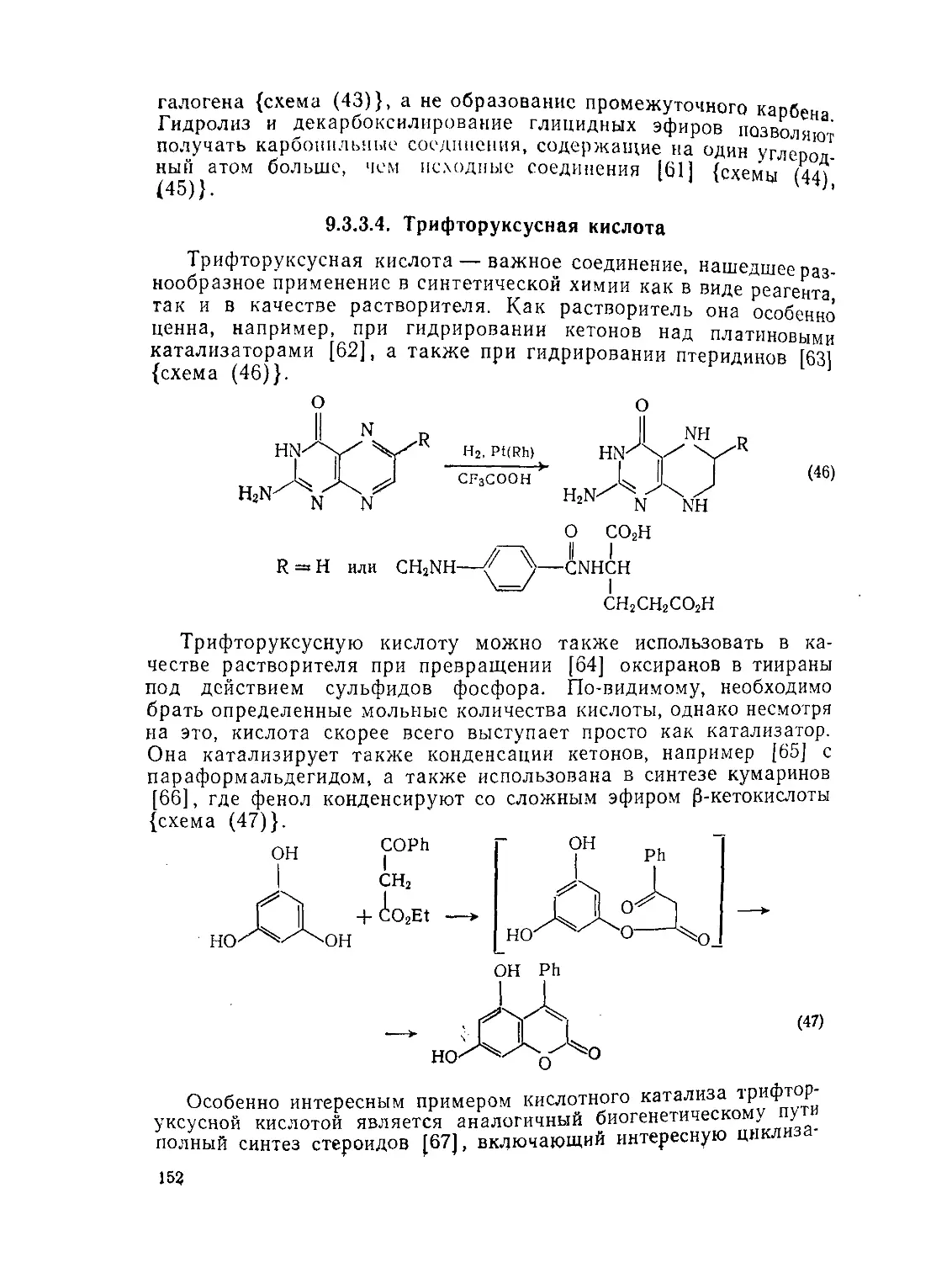
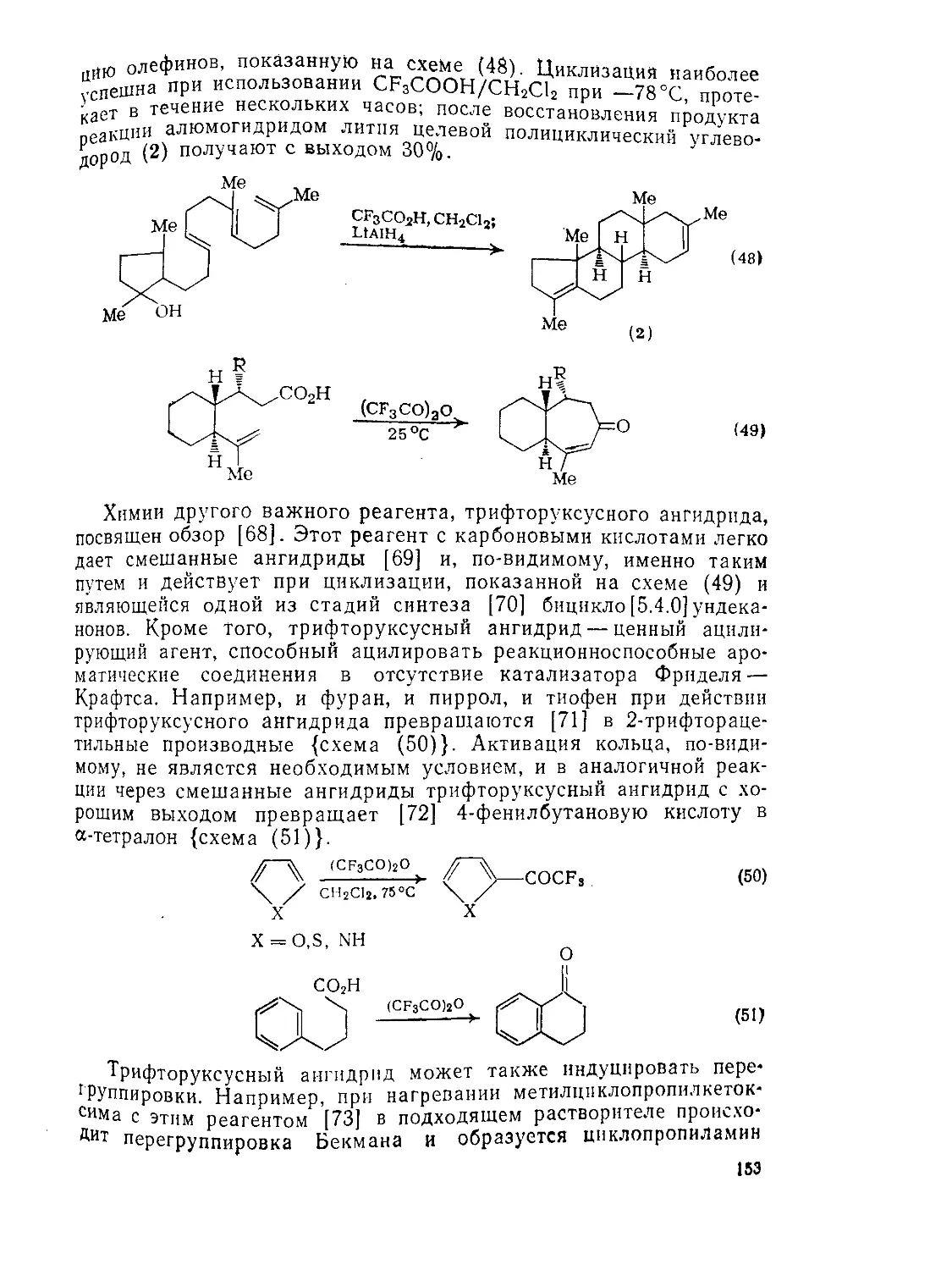
9.3.3.4. Трифторуксусная кислота 152
Литература 154
9.4. Гидрокси- и алкоксикарбоновые кислоты. С. М. Робертс 156
9.4.1. Введение 156
9.4.2. Методы получения гидрокси- и алкоксикарбоновых кислот 157
9.4.2.1. Общие методы получения гидроксиалкаиовых кислот 157
Э.4.2.2. Получение а-гидроксиалкановых кислот 160
9.4.2.3. Получение Р-гидроксиалкановых кислот 163
9.4.2.4. Получение у-, 6- и других гидроксиалкаиовых кислот 165
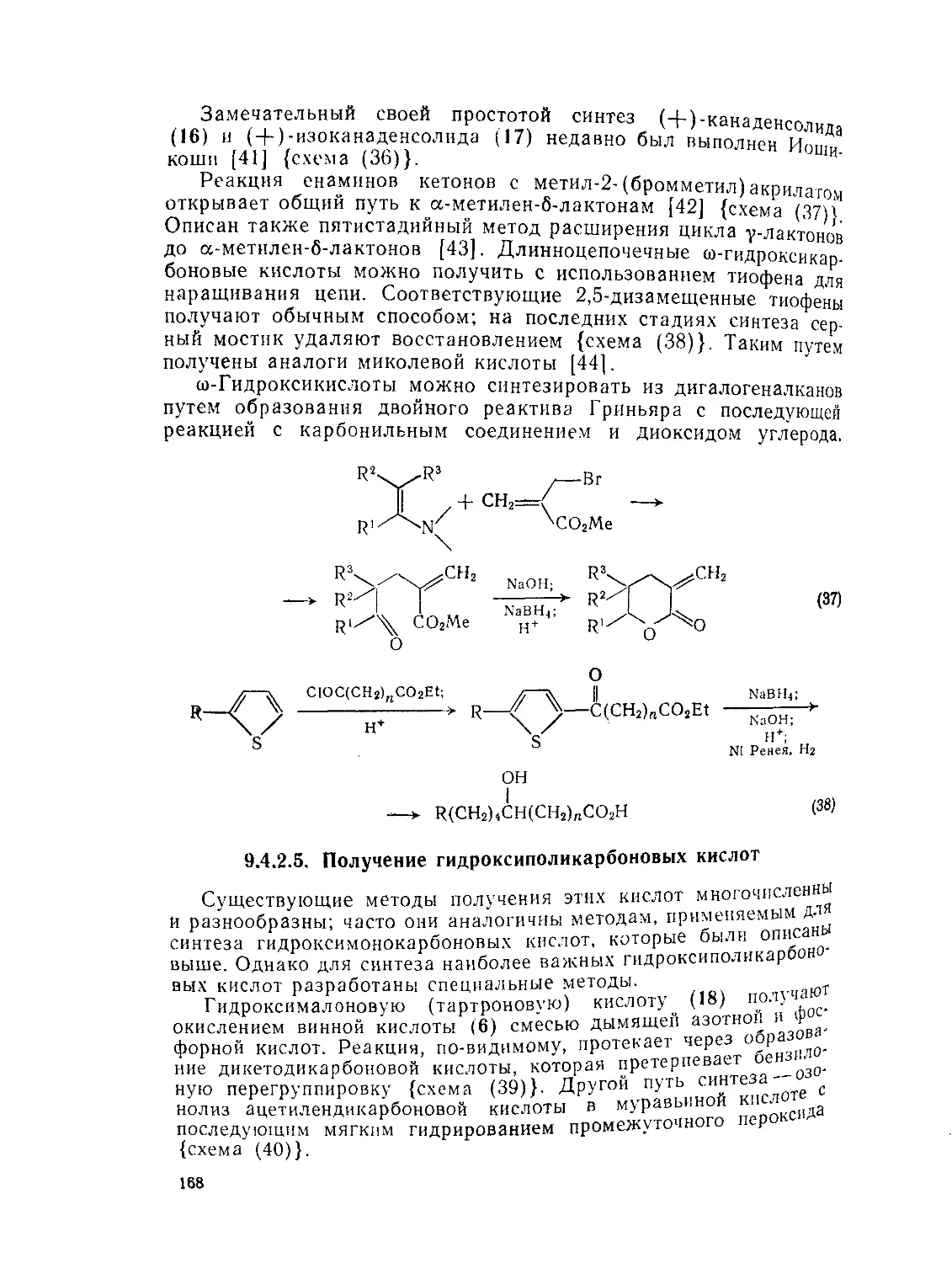
9.4.2.5. Получение гидроксиполикарбоновых кислот 168
9.4.2.6. Получение полигидроксиалкановых кислот 170
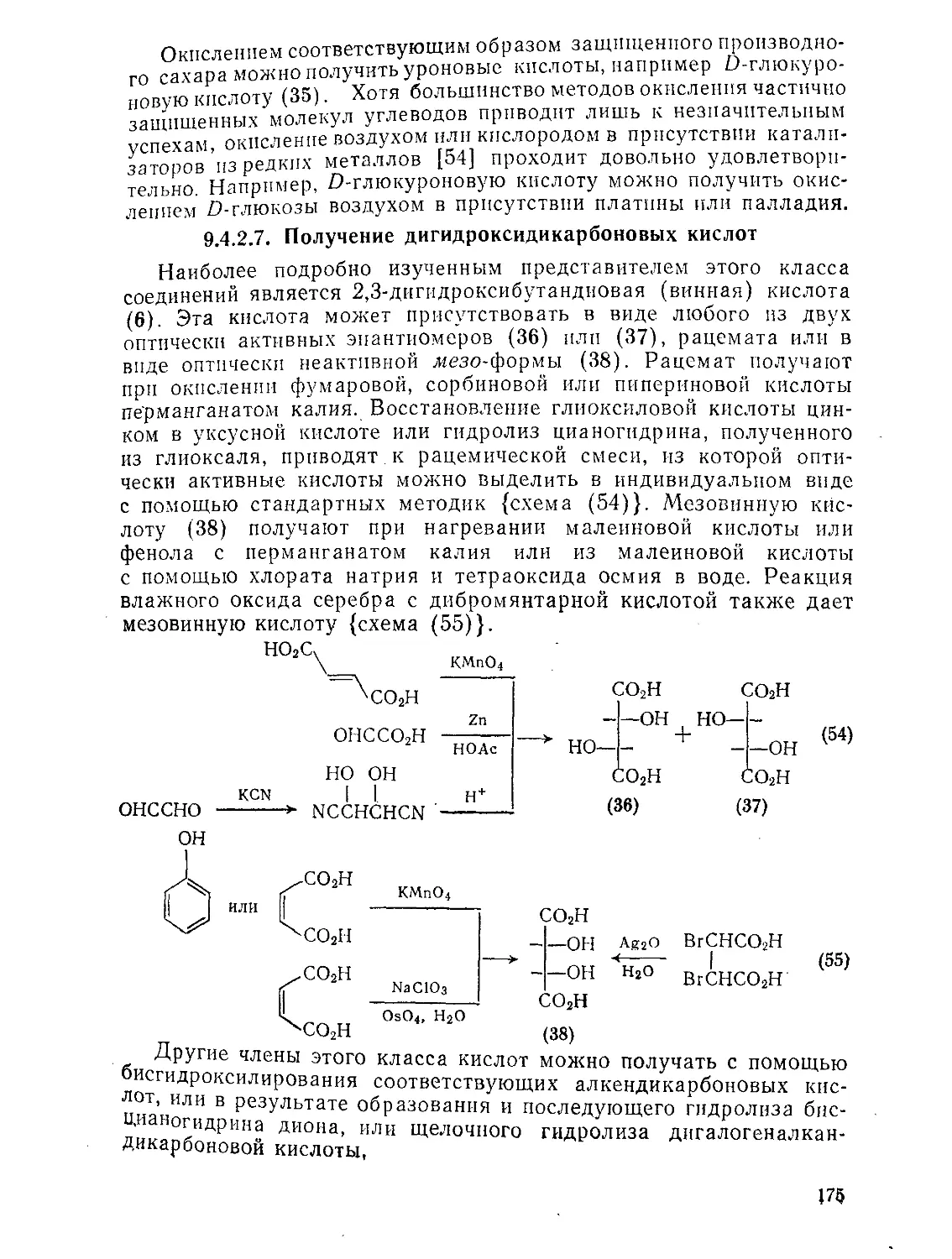
9.4.2.7. Получение дигидроксидикарбоновых кислот 175
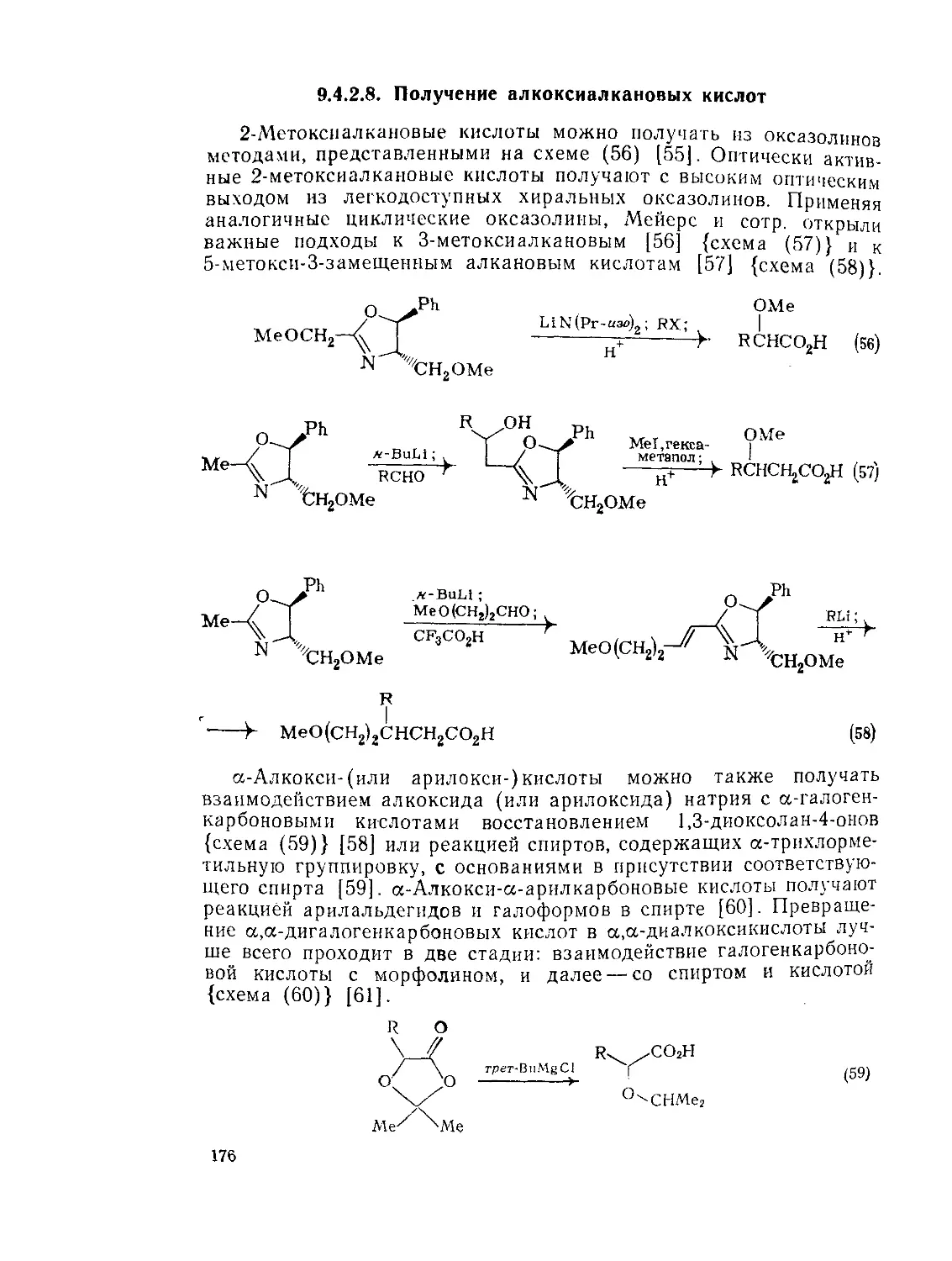
9.4.2.8. Получение алкоксиалкановых кислот 176
9.4.2.9. Получение эпоксикарбоновых кислот 177
9.4.2.10. Получение ненасыщенных гидроксикарбоновых кислот 178
9.4.2.11. Получение алкокси- и гидроксиарилкарбоновых кислот 179
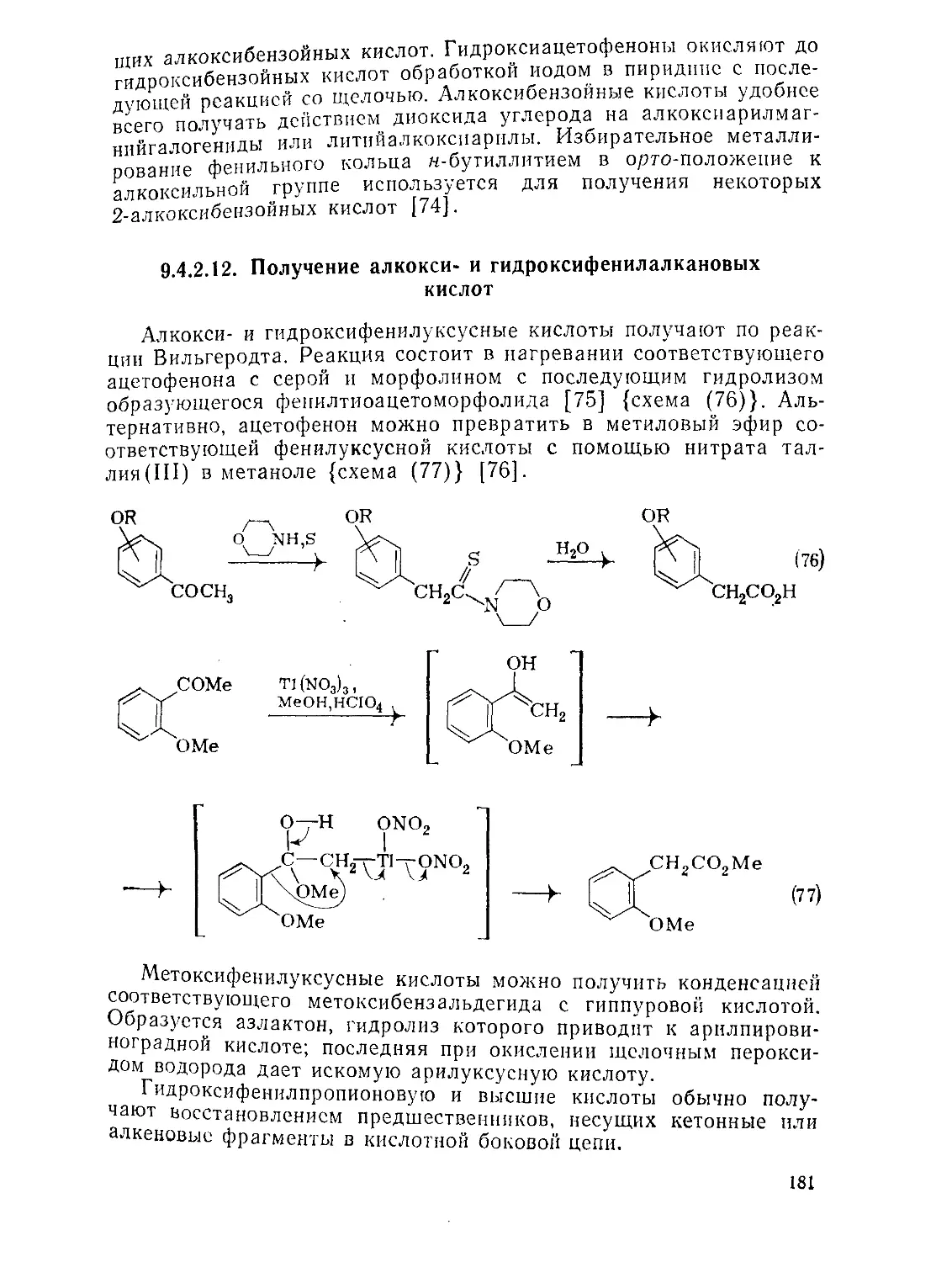
9.4.2.12. Получение алкокси- и гидроксифенилалкановых кислот 181
9.4.3. Свойства гидрокси- и алкоксикарбоновых кислот 182
9.4.4. Реакции гидрокси- и алкоксикарбоновых кислот 183
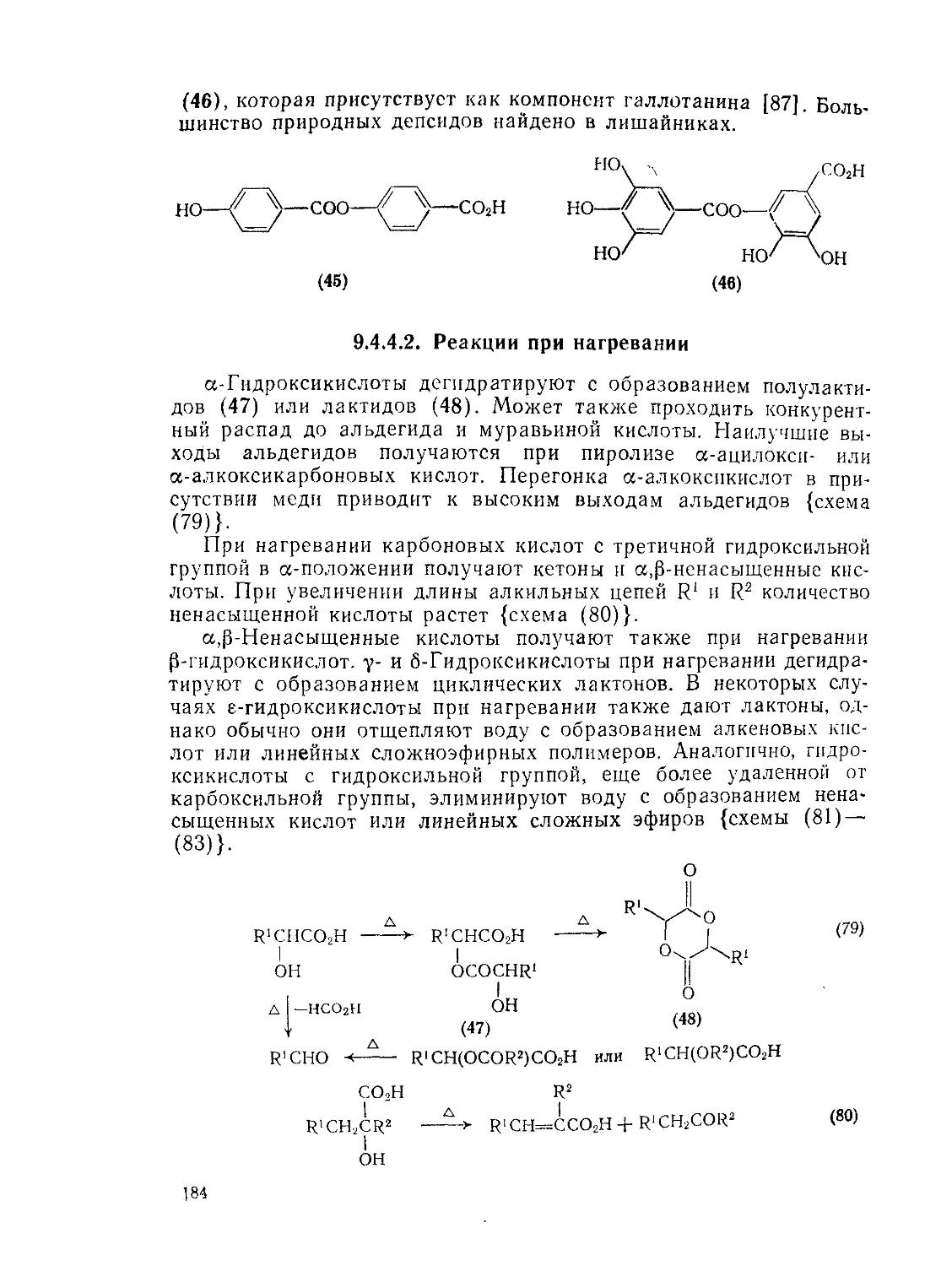
9.4.4.1. Общие реакции 183
9.4.4.2. Реакции при нагревании 184
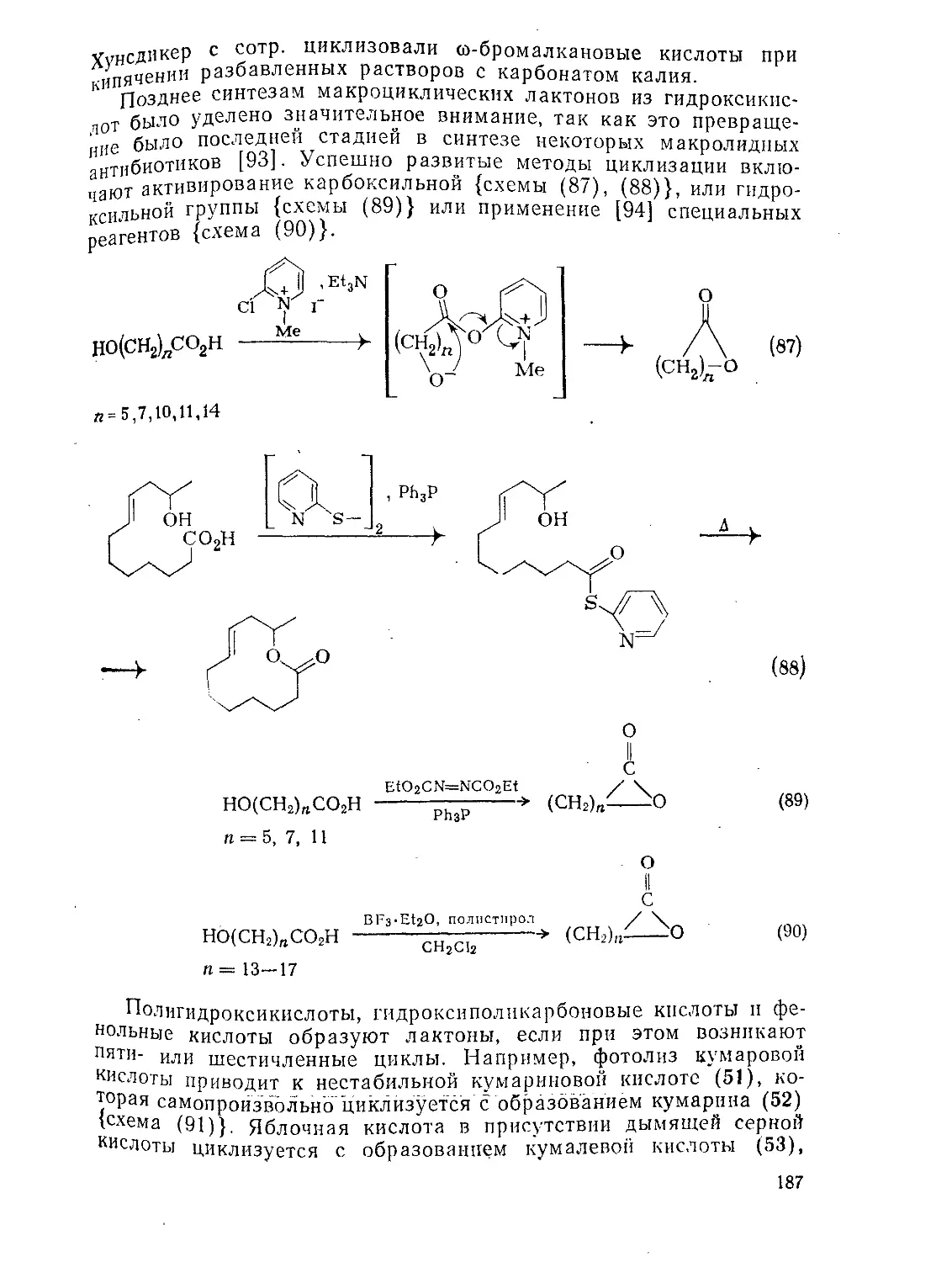
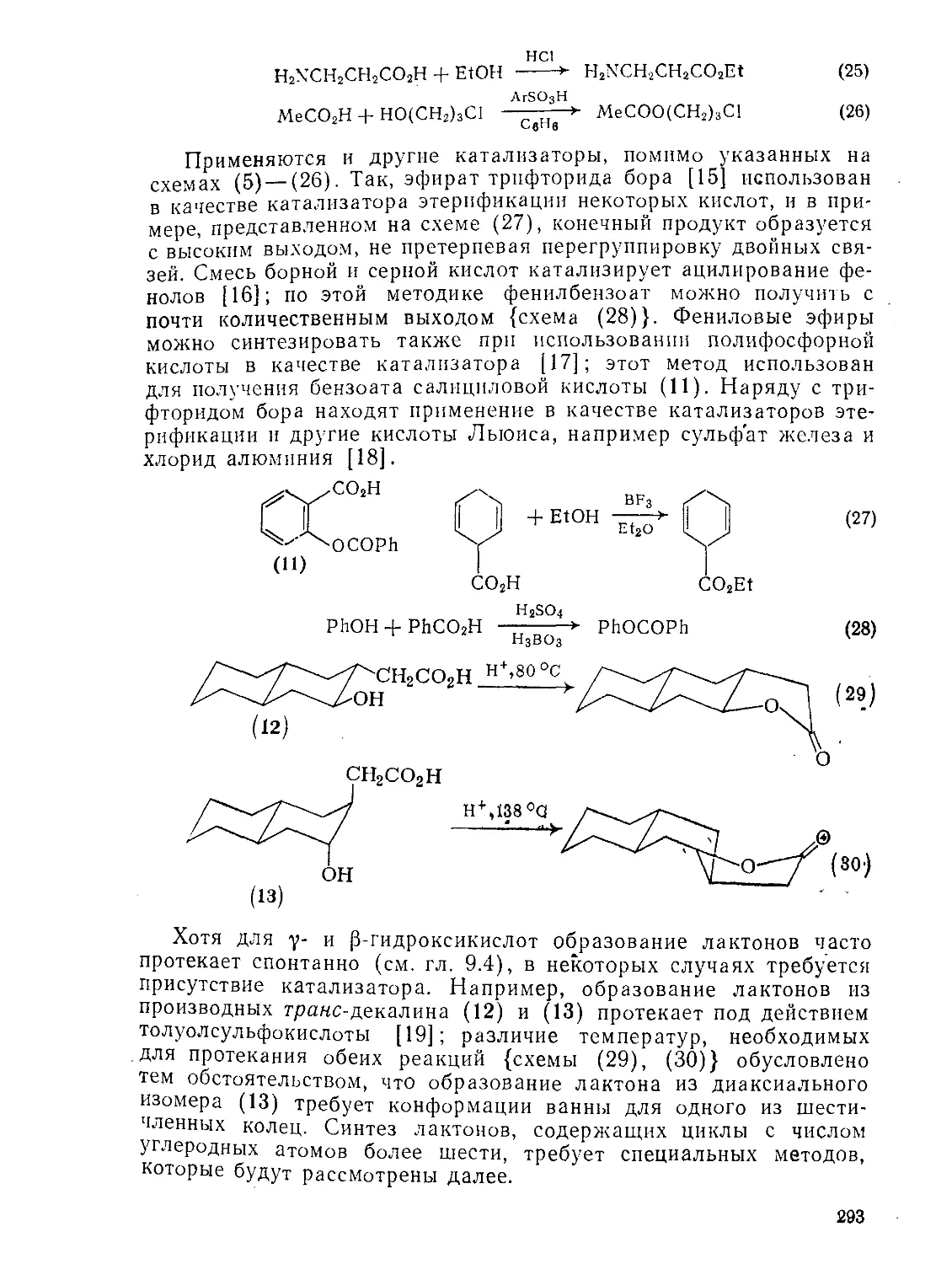
9.4.4.3. Образование лактонов 186
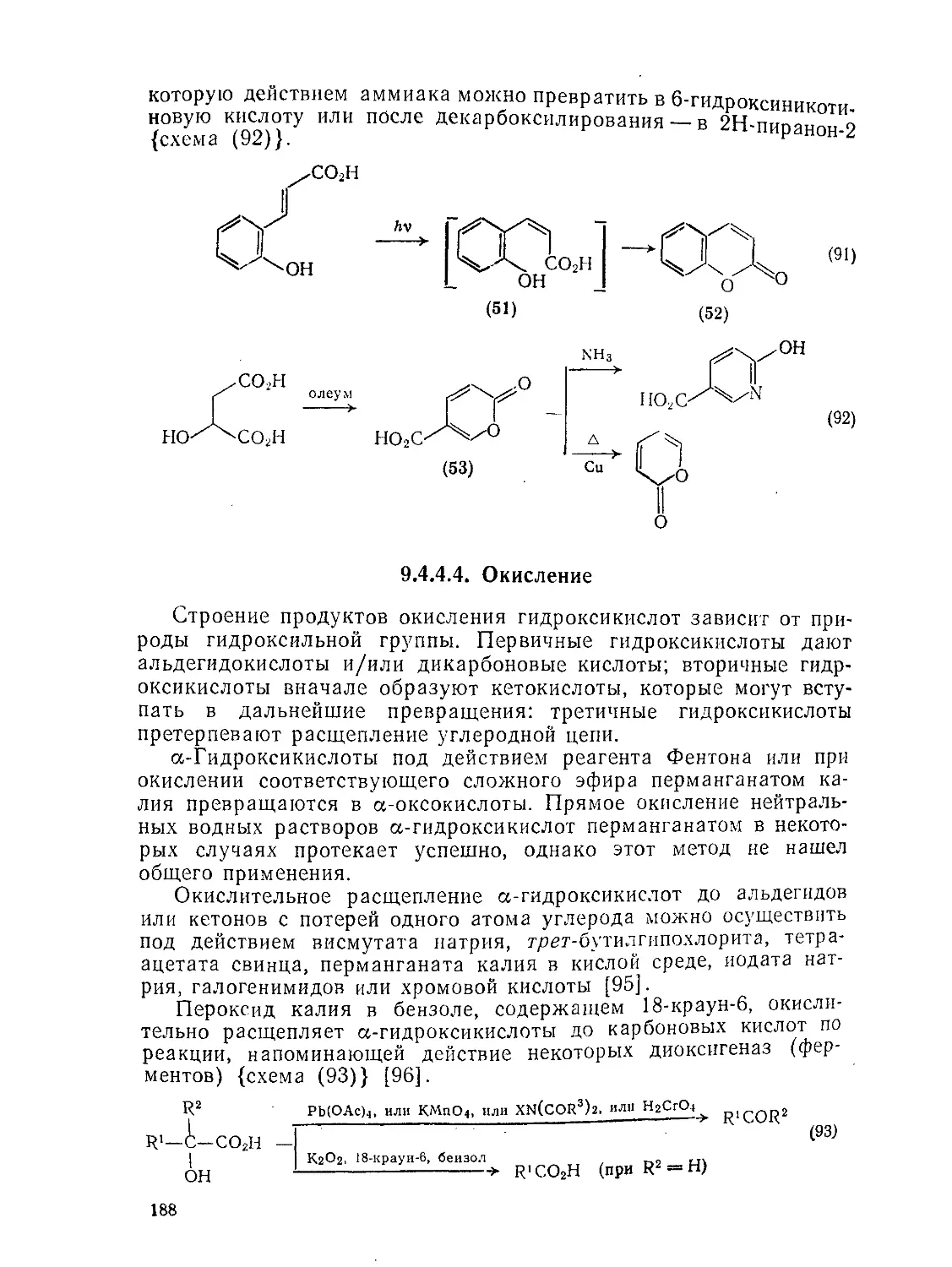
9.4.4.4. Окисление 188
9.4.4.5. Реакции а-гидроксиалкановых кислот 189
9.4.4.6. Реакции Р-гидроксиалкановых кислот 189
Э.4.4.7. Реакции алкоксиалкановых кислот 190
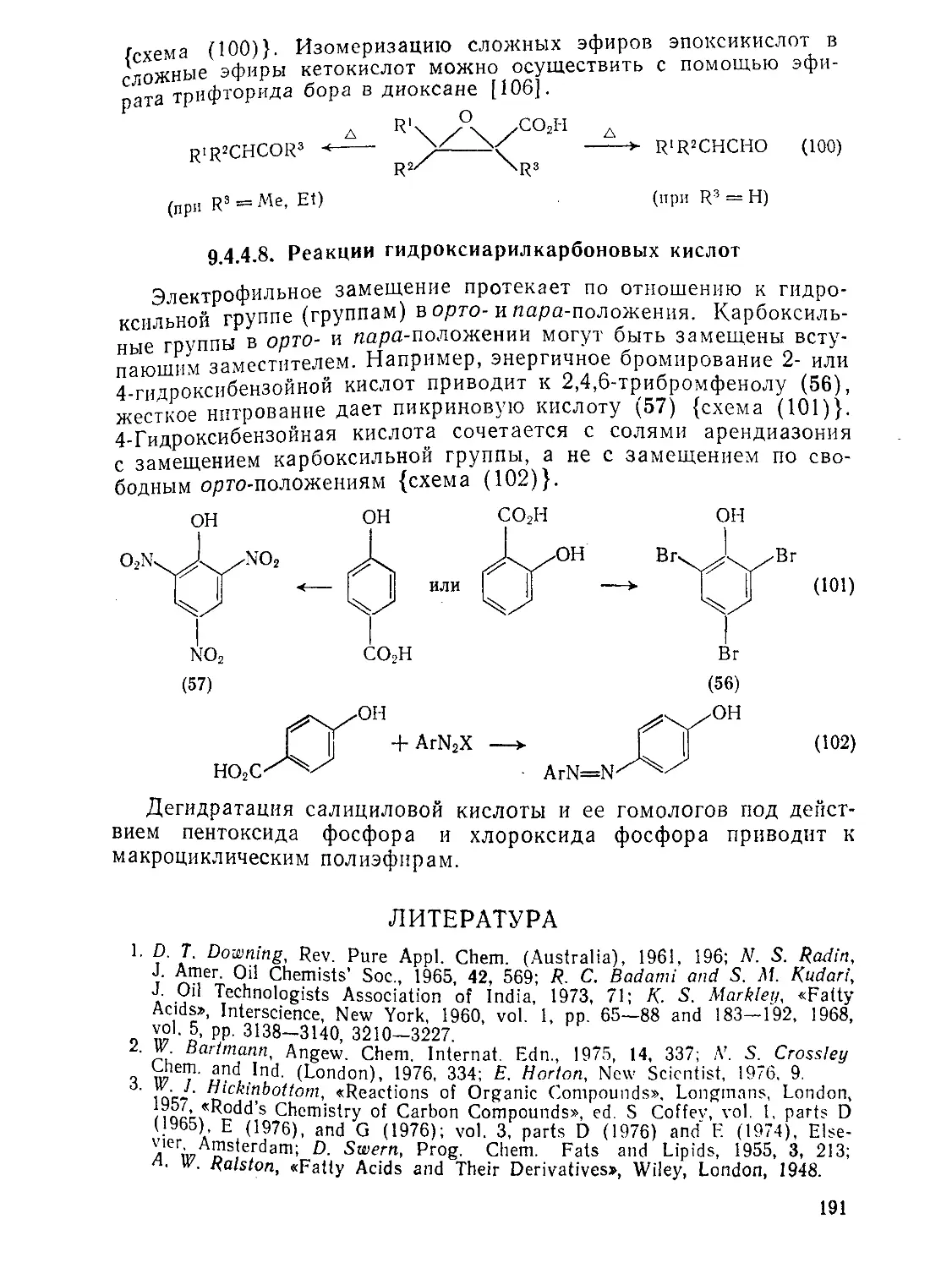
Э.4.4.8. Реакции гидроксиарилкарбоновых кислот 191
Литература 191
9.5. Оксокарбоновые кислоты. Дж. М. Браун 195
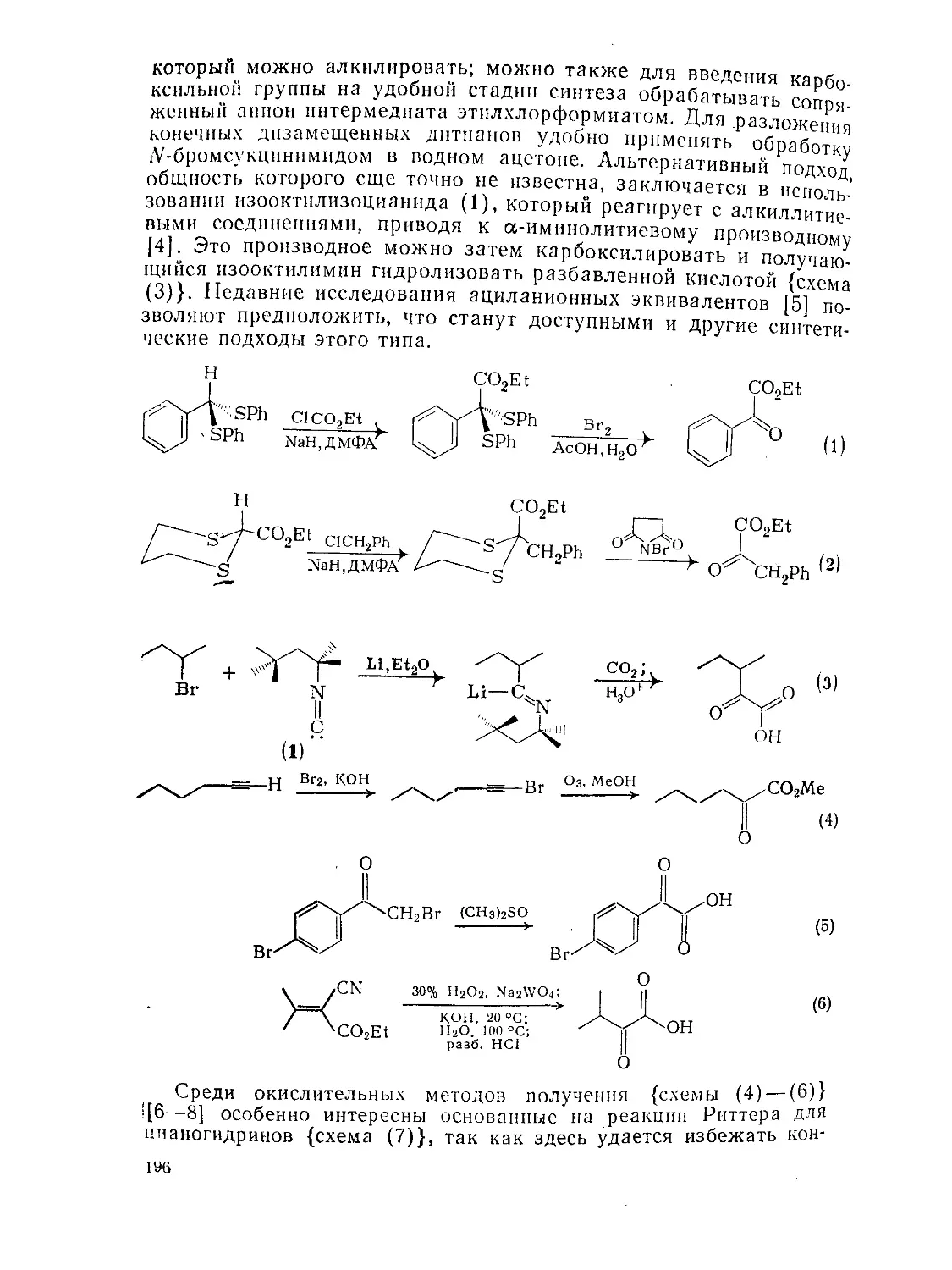
9.5.1. Методы получения и химия а-оксокарбоновых КИСЛОТ и их эфиров 195
9.5.1.1. Методы получения 195
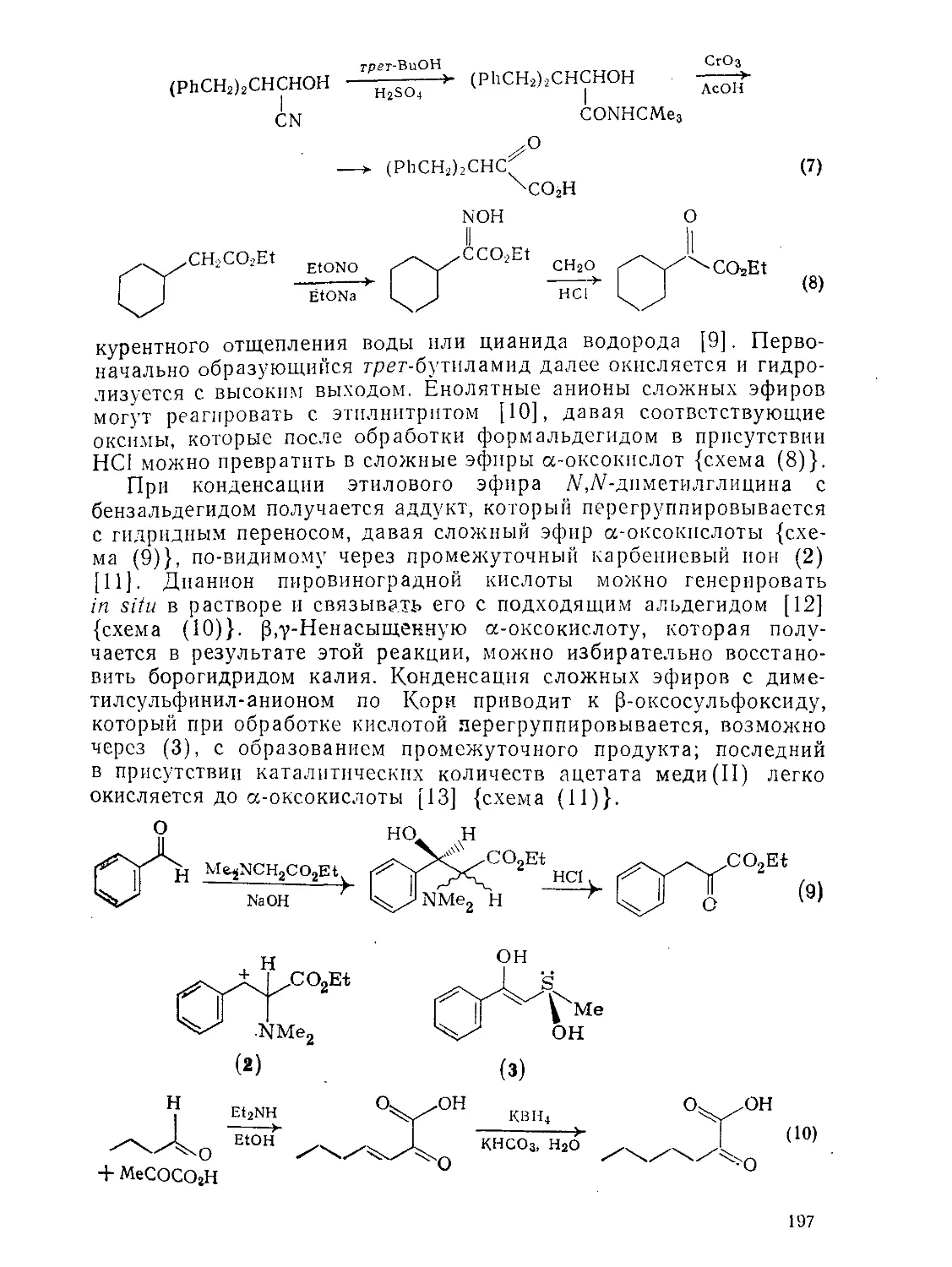
9.5.1.2. Реакции 198
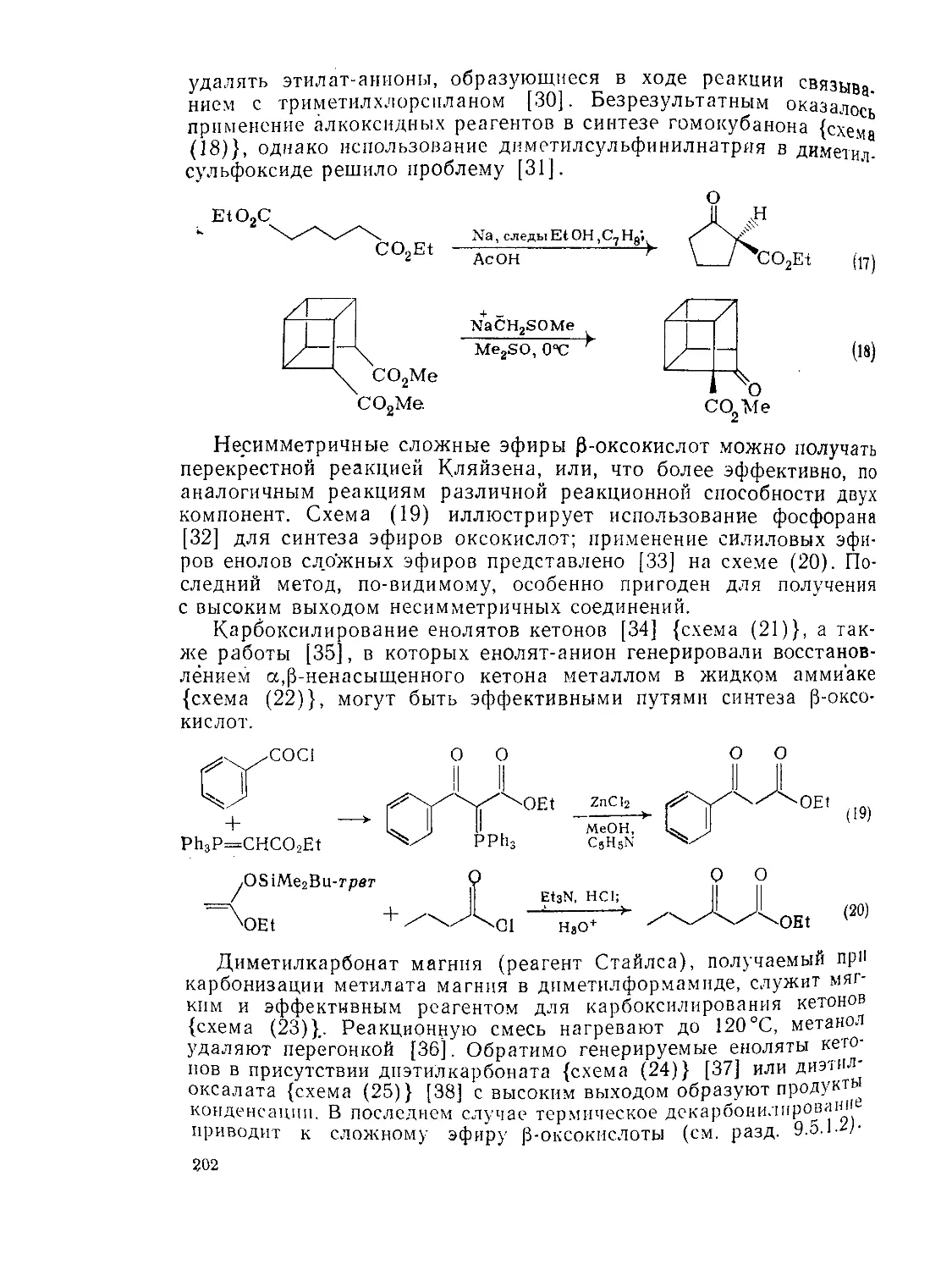
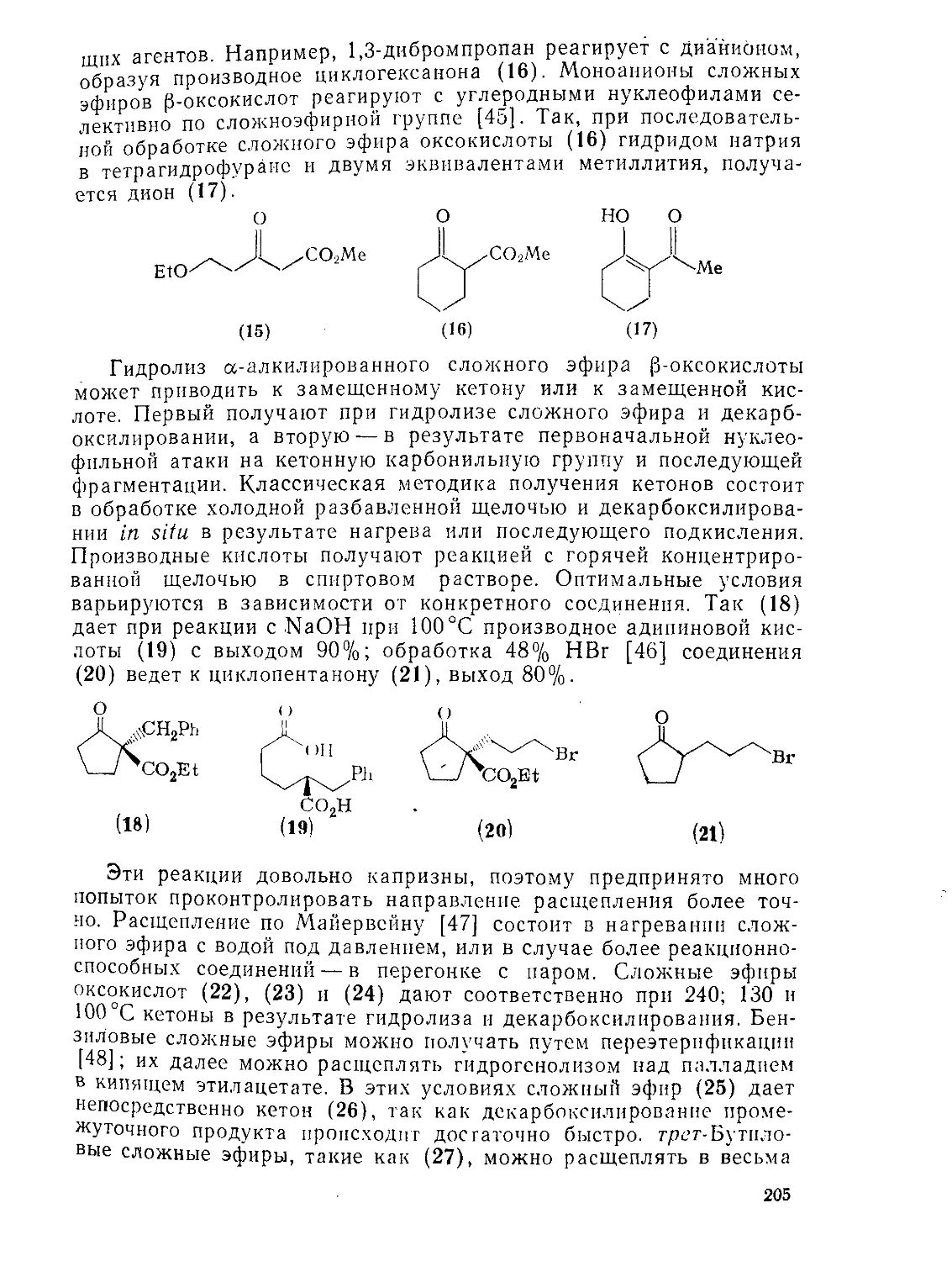
9.5.2. 9.5.2.1. Методы Методы получения получения и химия Р-оксокарбоновых КИСЛОТ и их эфиров 201 201
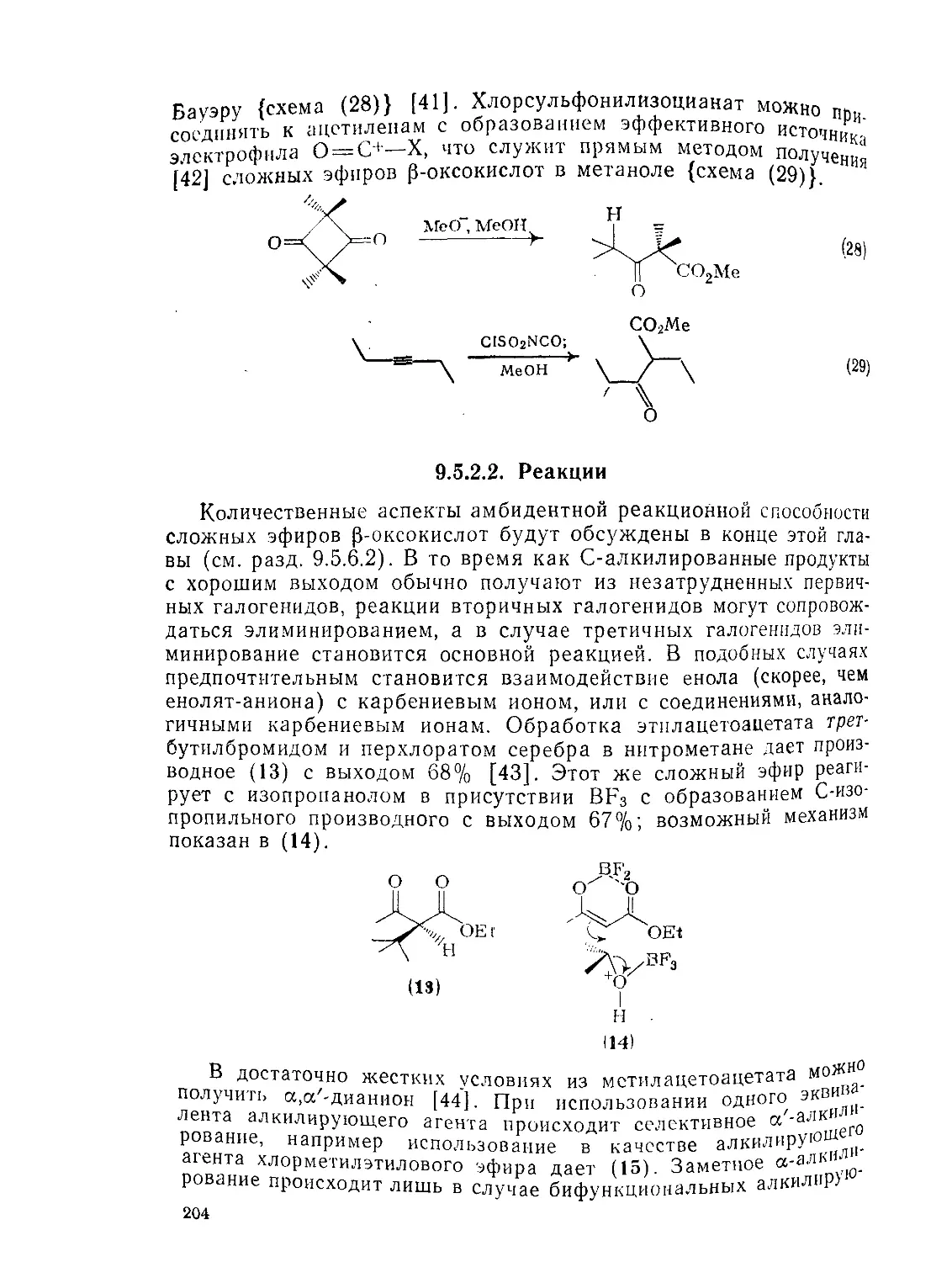
9.5.2.2. Реакции 204
9.5.3. 9.5.З.1. Методы Методы получения получения и ХИМИЯ у-оксокарбоновых кислот и их эфиров 207 207
9.5.3.2. Реакции 209
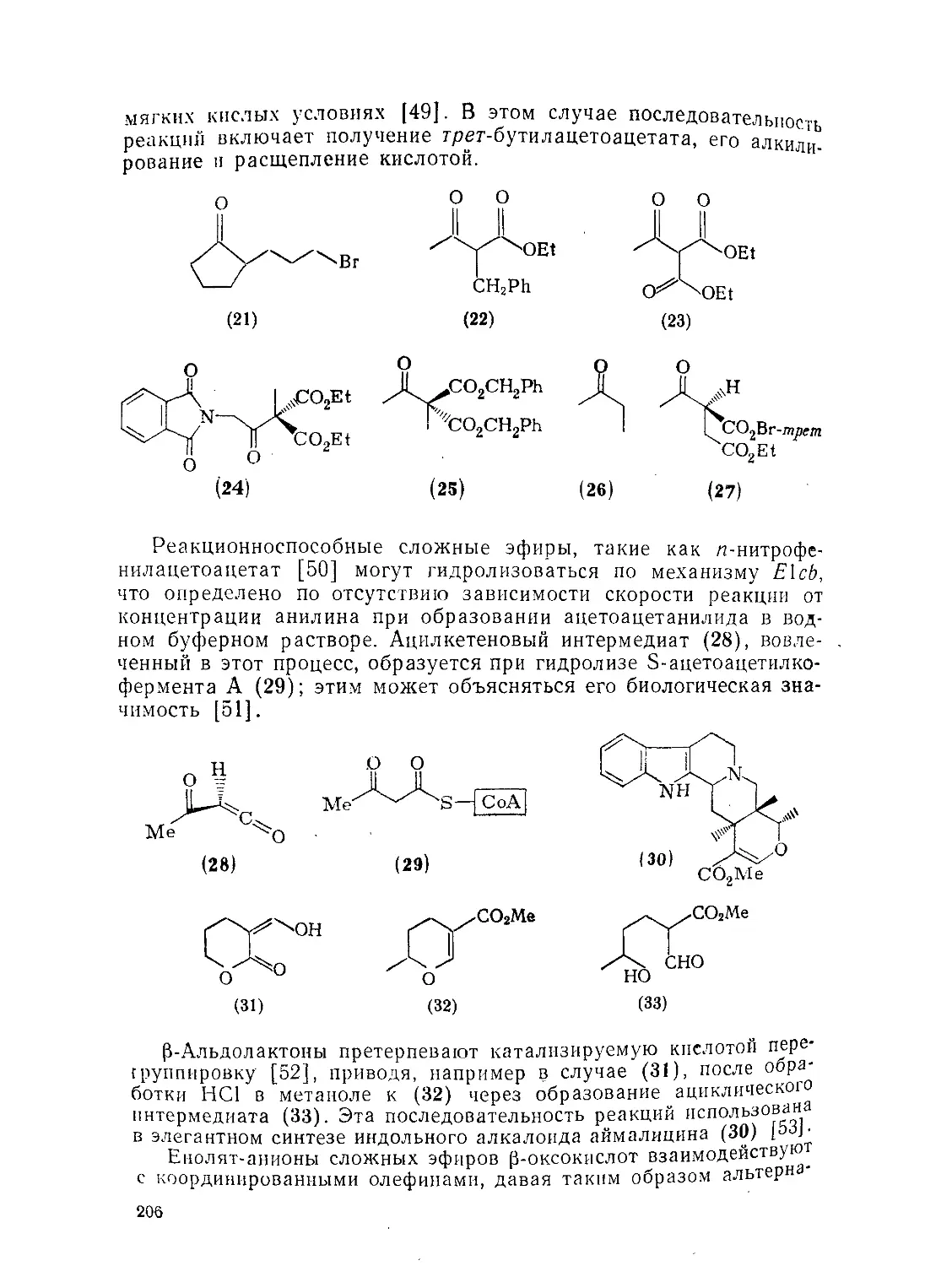
9.5.4. 9.5.4.1. Э.5.4.2. Методы Методы Реакции получения получения и химия б-оксокарбоновых кислот и их эфиров210 210 215
в
9.5.5.
9.5.5.1.
Э.5.5.2.
9.5.6.
9.5.6.1.
Э.5.6.2.
9.5.6.3.
Э.5.6.4.
Методы получения и химия е-оксокарбоновых кислот^ их эфиров и высших гомологов
Методы получения
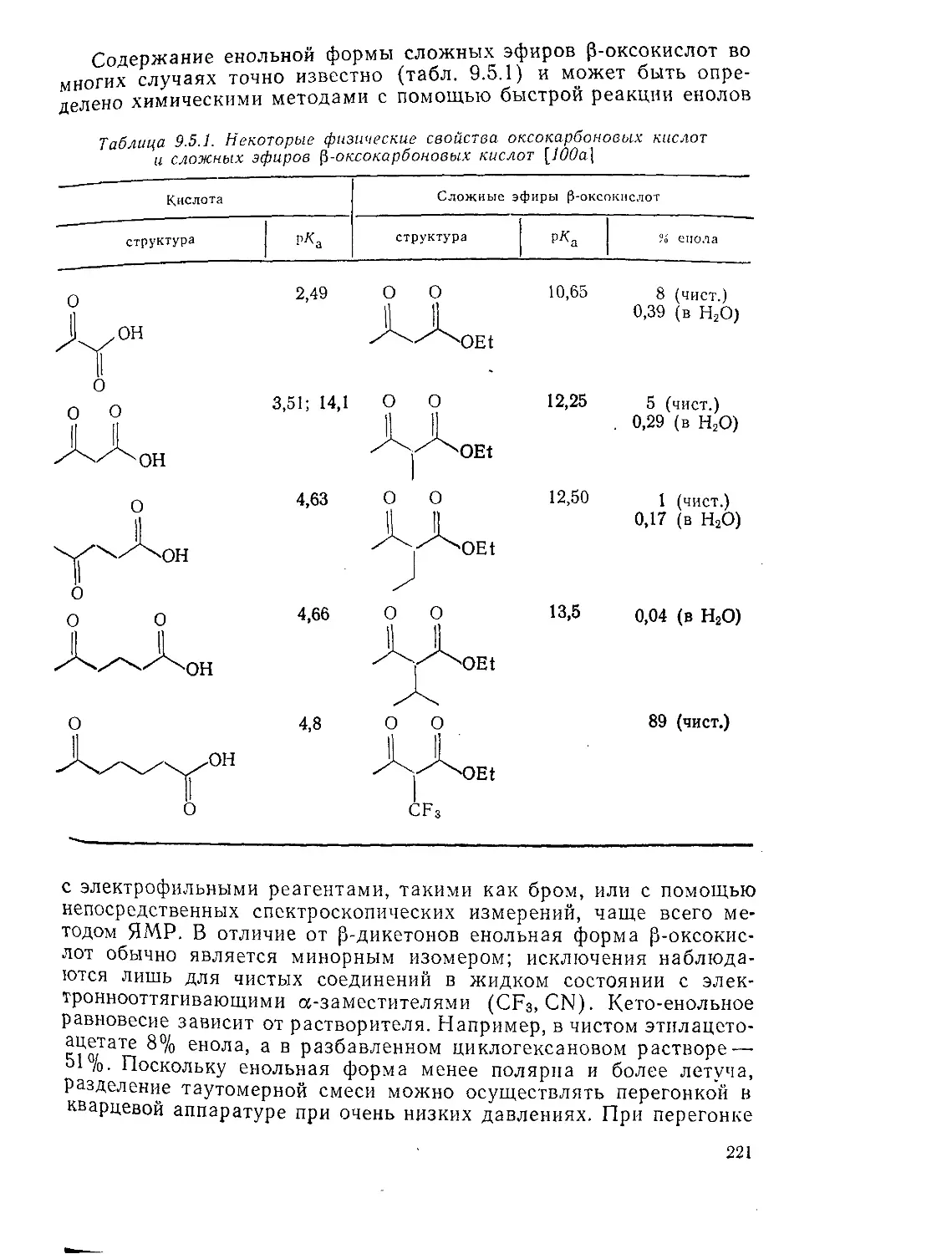
Физико-химические свойства оксокарбоновых кислот 220
Кислотность кислорода и углерода; кето-енольная таутомерия 220
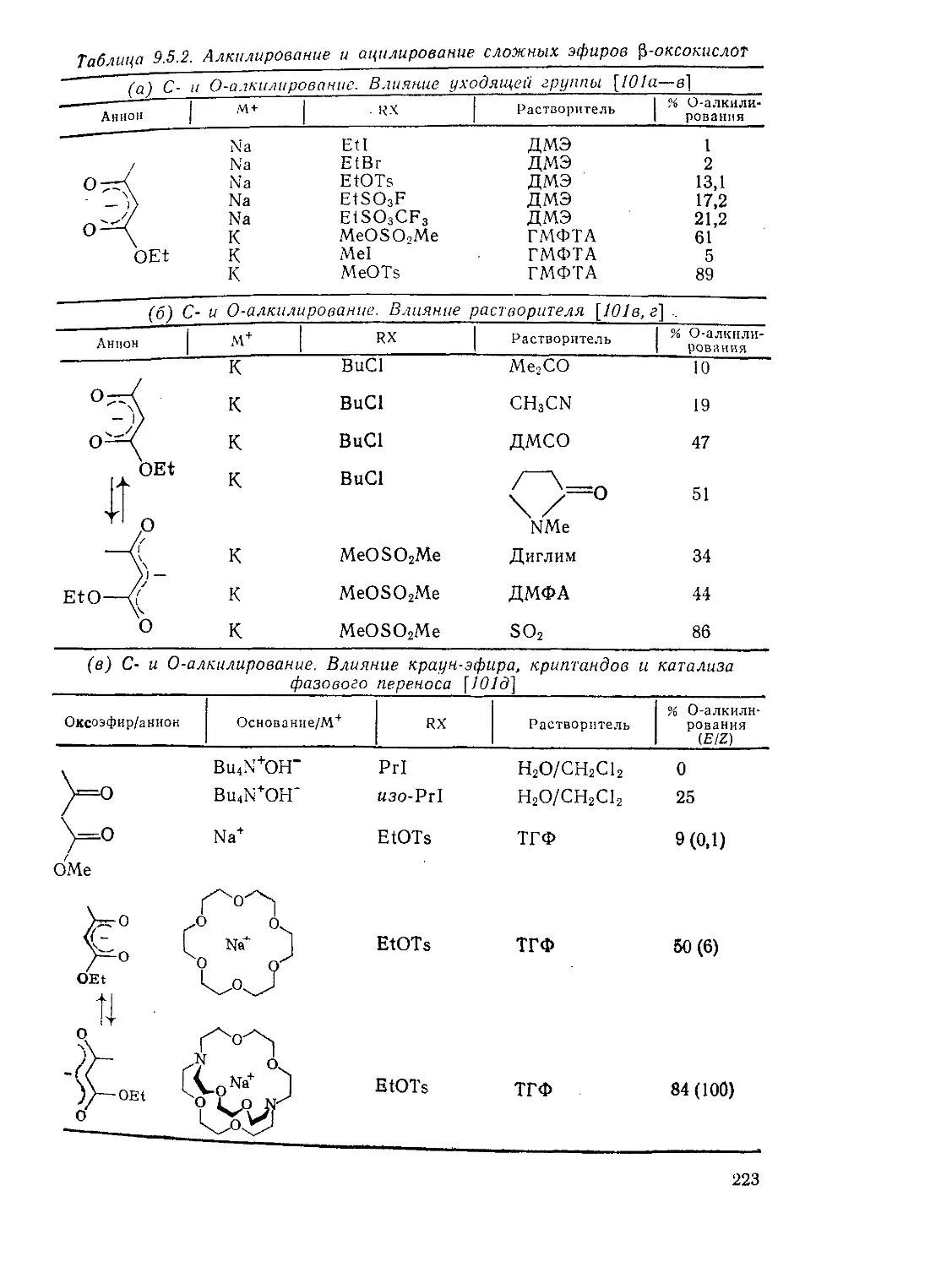
Алкилирование анионов эфиров Р-оксокислот 222
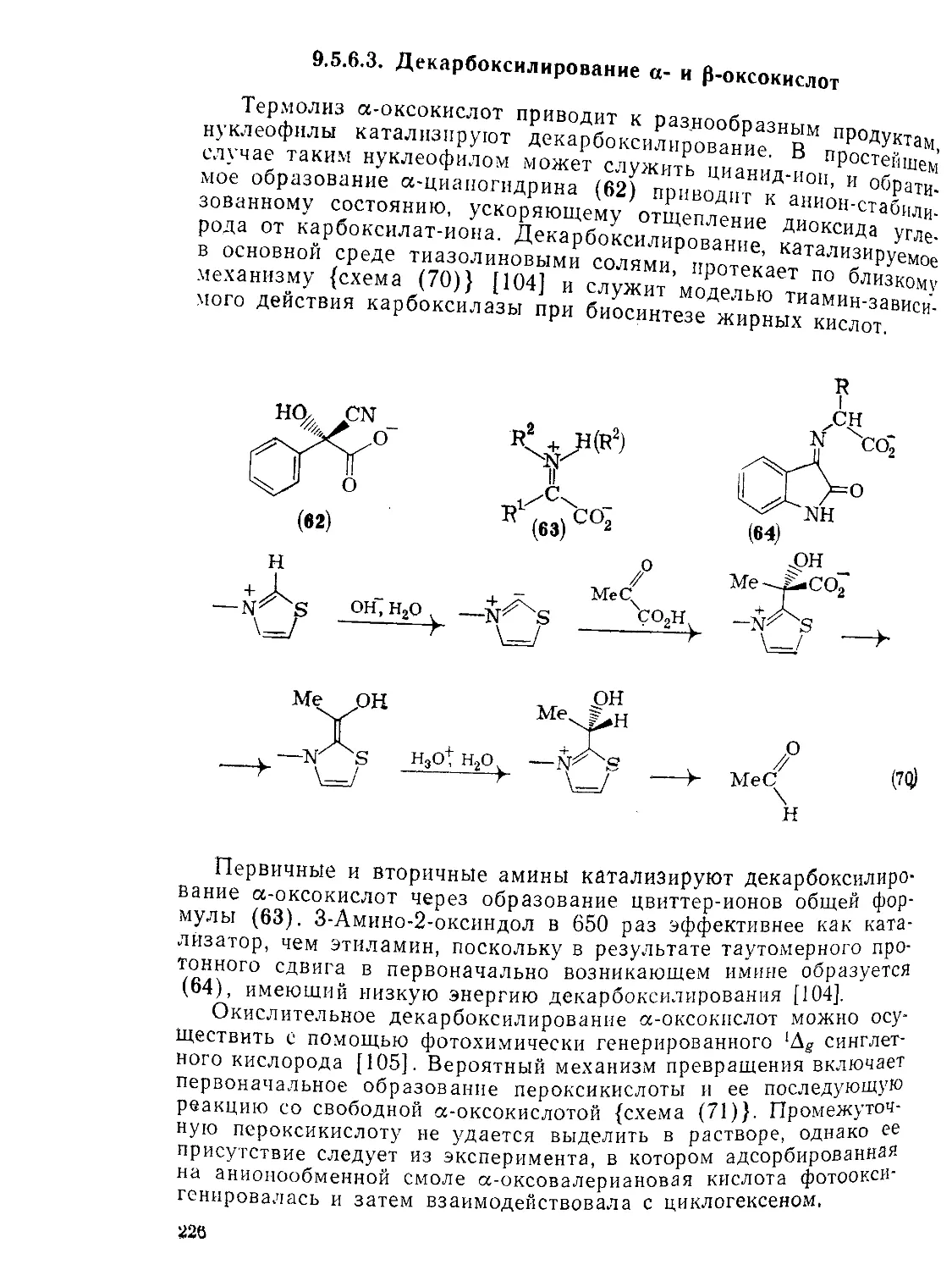
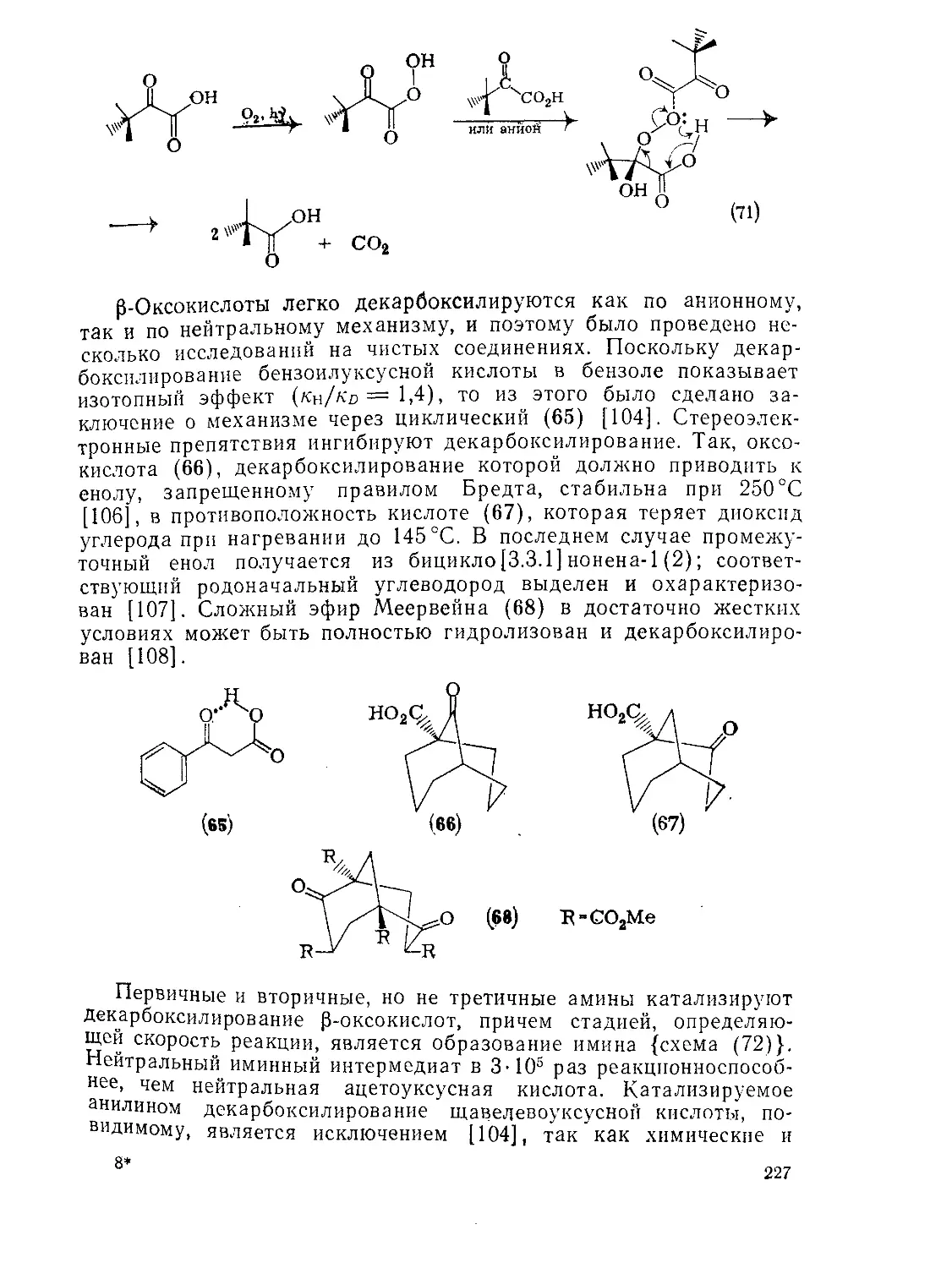
Декарбоксилирование а- и р-оксокислот 226
Внутримолекулярный катализ в химии оксокислот 229
Литература 229
9.6. Аминокислоты. Дж. X. Джонс 233
9.6.1. а-Аминокислоты 233
9.6.1.1. Введение. Структура и конфигурация «-аминокислот 233
9.6.1.2. Методы получения «-аминокислот 237
9.6.1.3. Реакции «-аминокислот 242
9.6.1.4. M-Замещенные а-амннокислоты 245
9.6.1.5. «-Модифицированные а-аминокислоты 250
9.6.1.6. а-Аминокислоты, модифицированные по карбоксильной группе 253
9.6.2. р-Аминокислоты 254
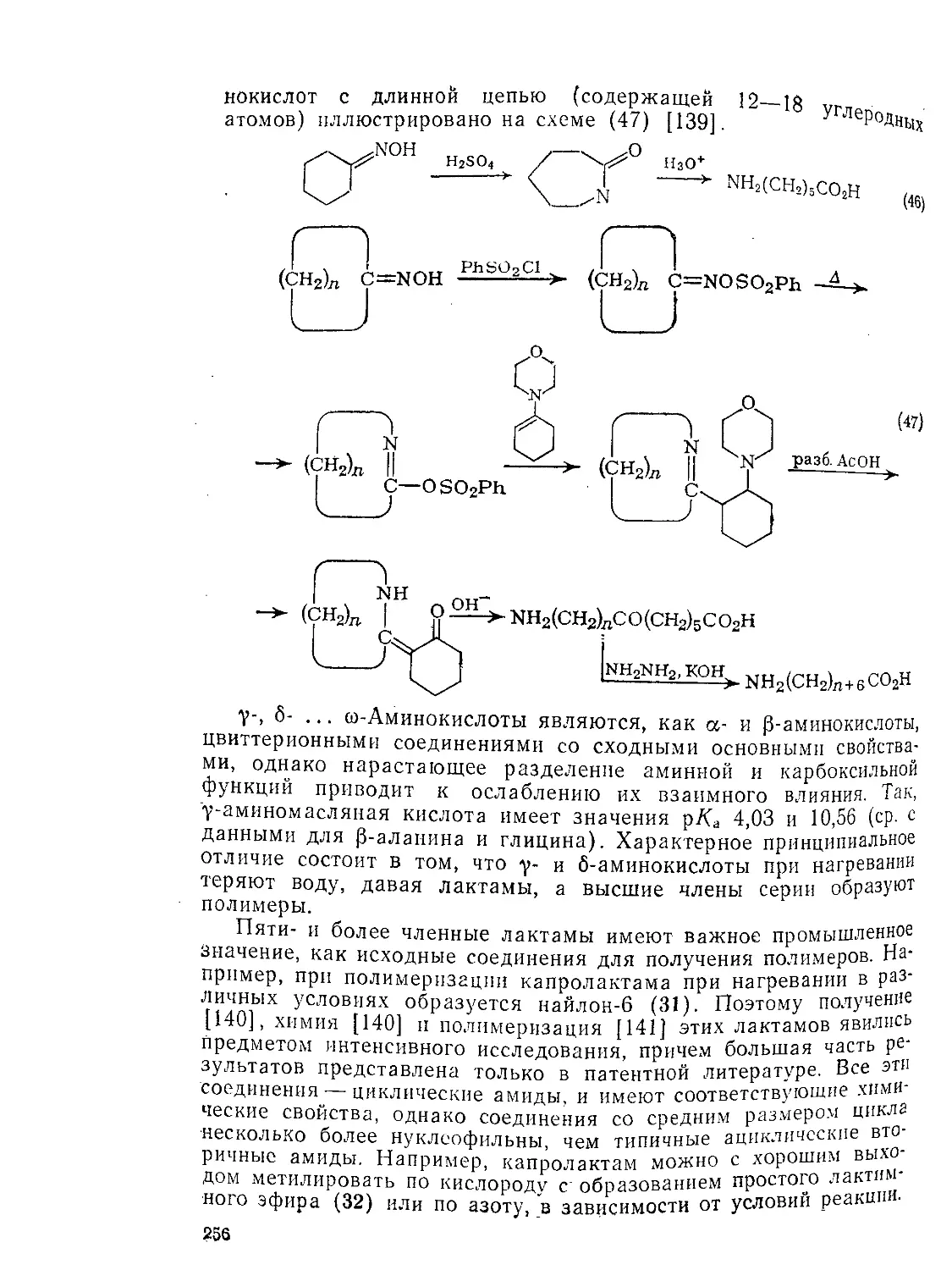
9.6.3. От у- до со-аминокислот 255
9.6.4. Аминоароматические Кислоты 257
Литература . 253
9.7. Карбоновые кислоты с другими азотсодержащими заместителями.
И. О. Сазерленд 261
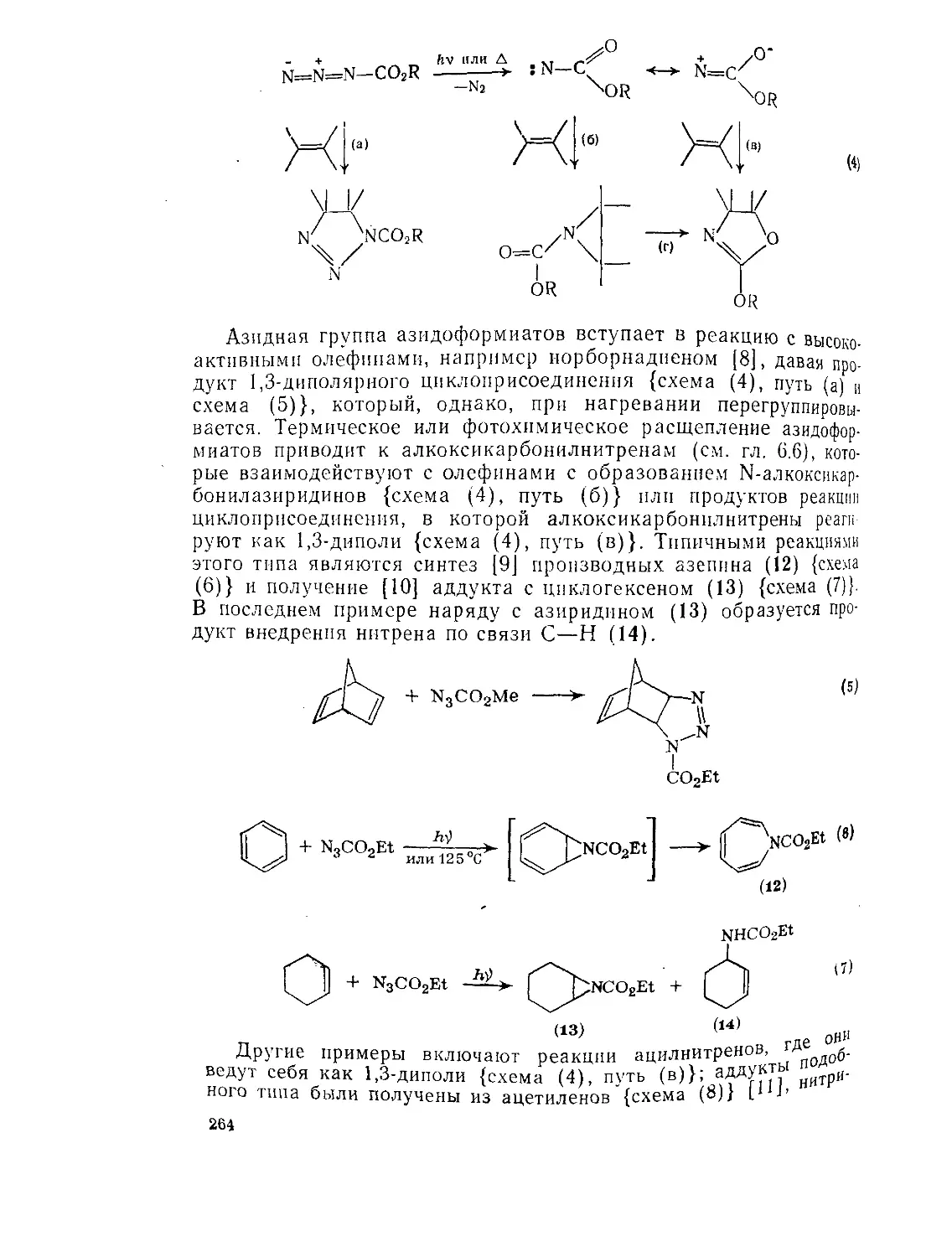
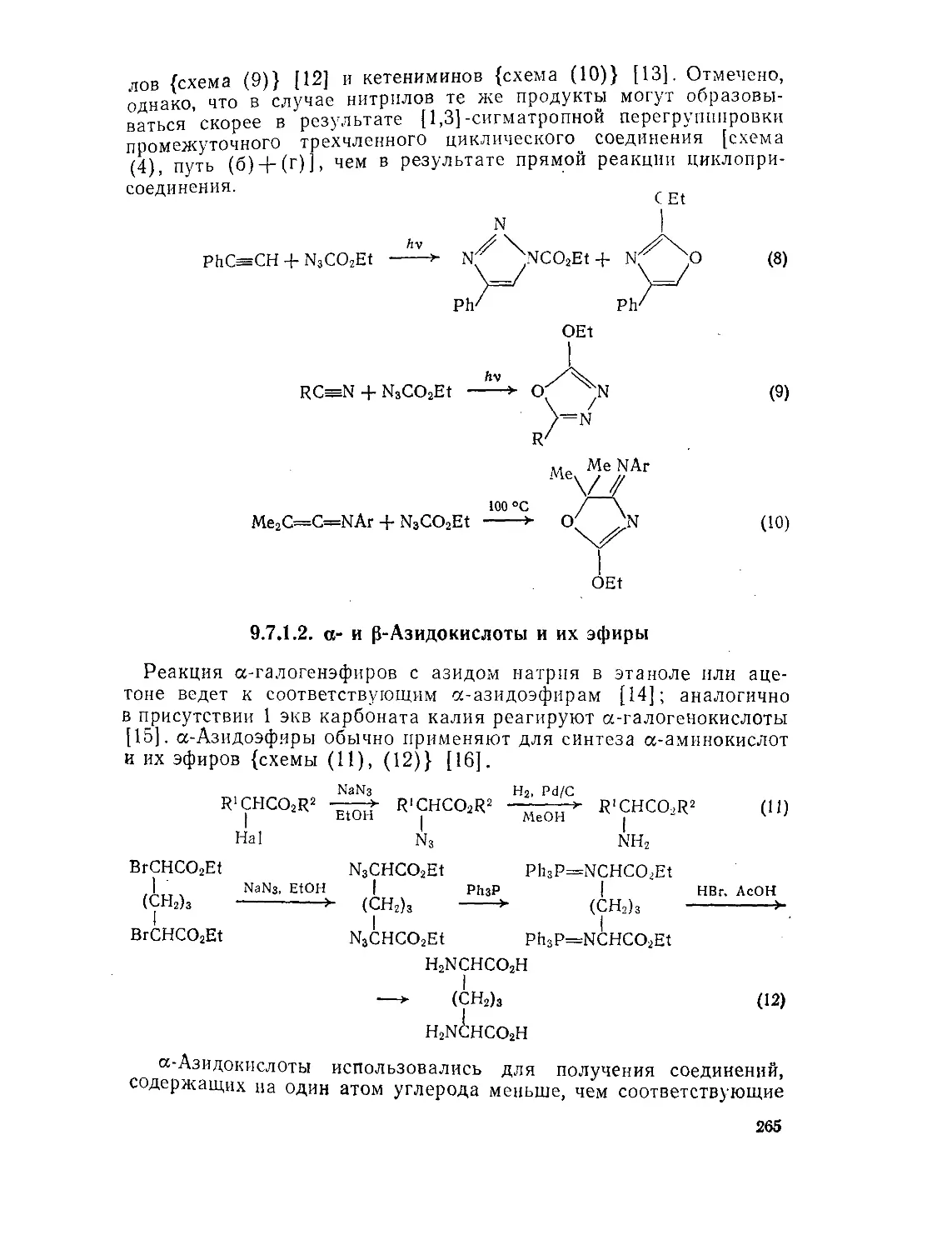
9.7.1. Азидокислоты и их эфиры 262
9.7.1.1. Эфиры азидомуравьиной кислоты 262
9.7.1.2. а- и р-Азидокислоты и их эфиры 265
9.7.2. Эфиры диазокислот 266
9.7.2.1. Методы получения эфиров диазокислот 267
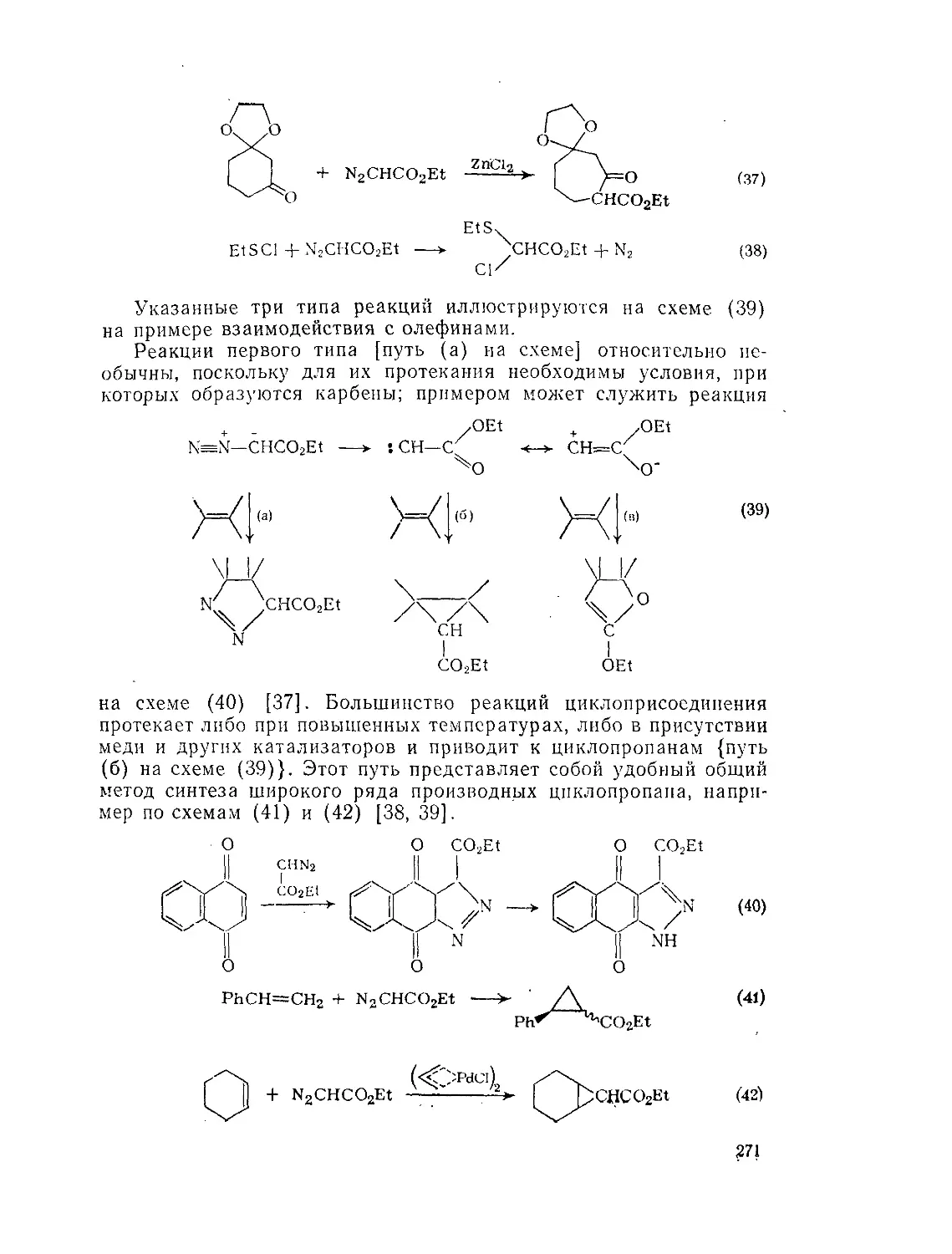
Э.7.2.2. Реакции эфиров диазокислот 269
9.7.3. Азокислоты и их эфиры 273
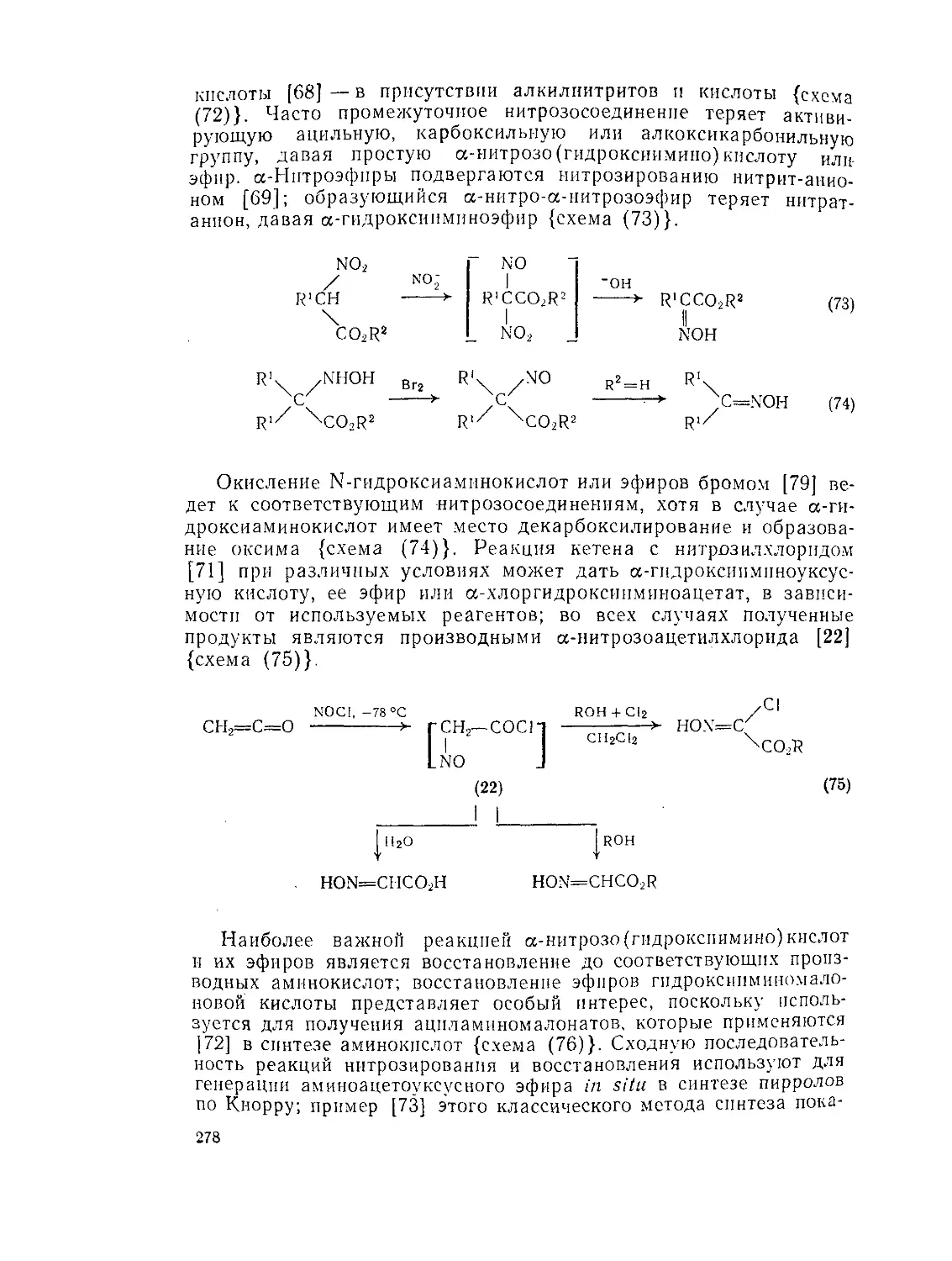
9.7.4. Нитрозокислоты и их эфиры 276
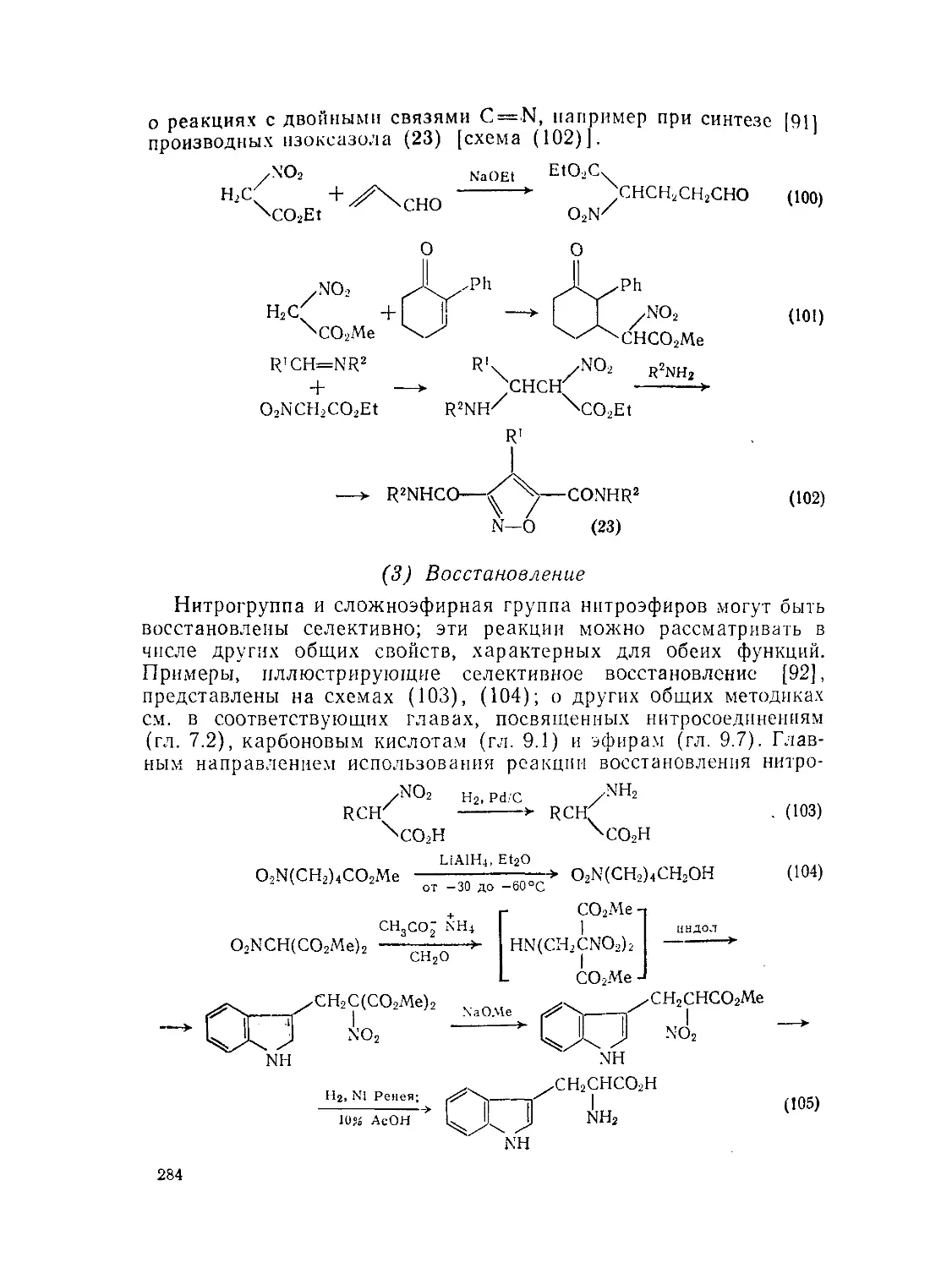
9.7.5. Нитрокислоты и их эфиры 279
9.7.5.1. Методы получения нитрокислот и их эфиров 279
Э.7.5.2. Реакции нитрокислот и их эфиров . 282
Литература 285
9.8. Сложные эфиры. И. О. Сазерленд 288
9.8.1. Методы получения сложных эфиров 290
9.8.1.1. Этерификация 290
9.8.1.2. Алкоголиз 310
9.8.1.3. Окислительные методы 313
9.8.1.4. Перегруппировки 319
9.8.1.5. Алкоксикарбонилирование 324
У.о.1.6. Методы получения лактоиов 329
9.8.2. Свойства сложных эфиров 331
9.8.2.1. А О А л Структура сложных эфиров 331
У.8.2.2. Физические свойства сложных эфиров 333
9.8.2.3. ООП Идентификация сложных эфиров 333
У. о.З. Q Q О 1 Реакции сложных эфиров 337
У.о.3.1. Q Я Q о Гидролиз и другие реакции расщепления 337
(А О а Реакции енолят-анионов 344
9-8.3.3. Нуклеофильное присоединение по карбонильной группе 348
7
9 8 3 4 Восстановление
9 8.3-5. Пиролиз и перегруппировки
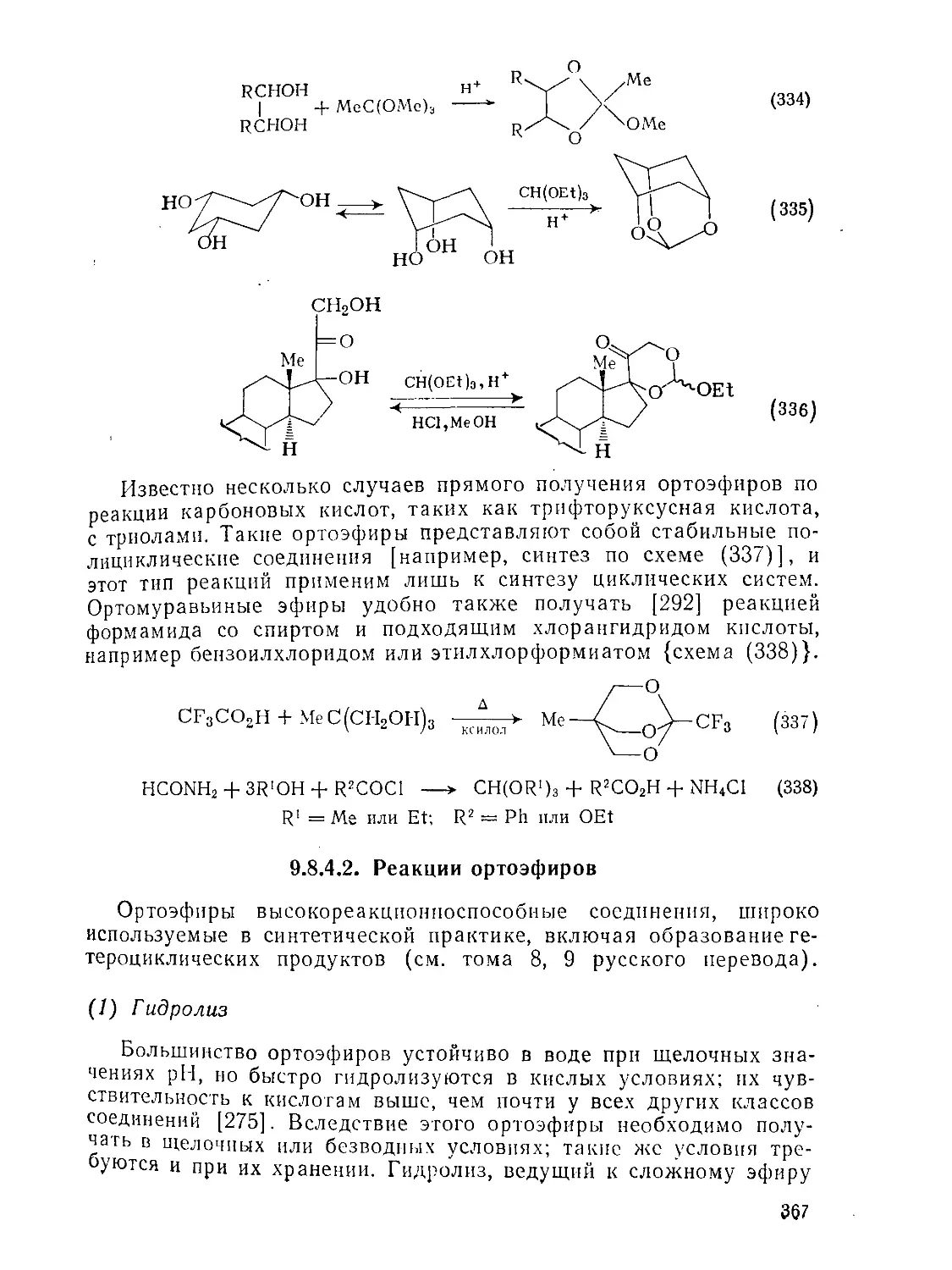
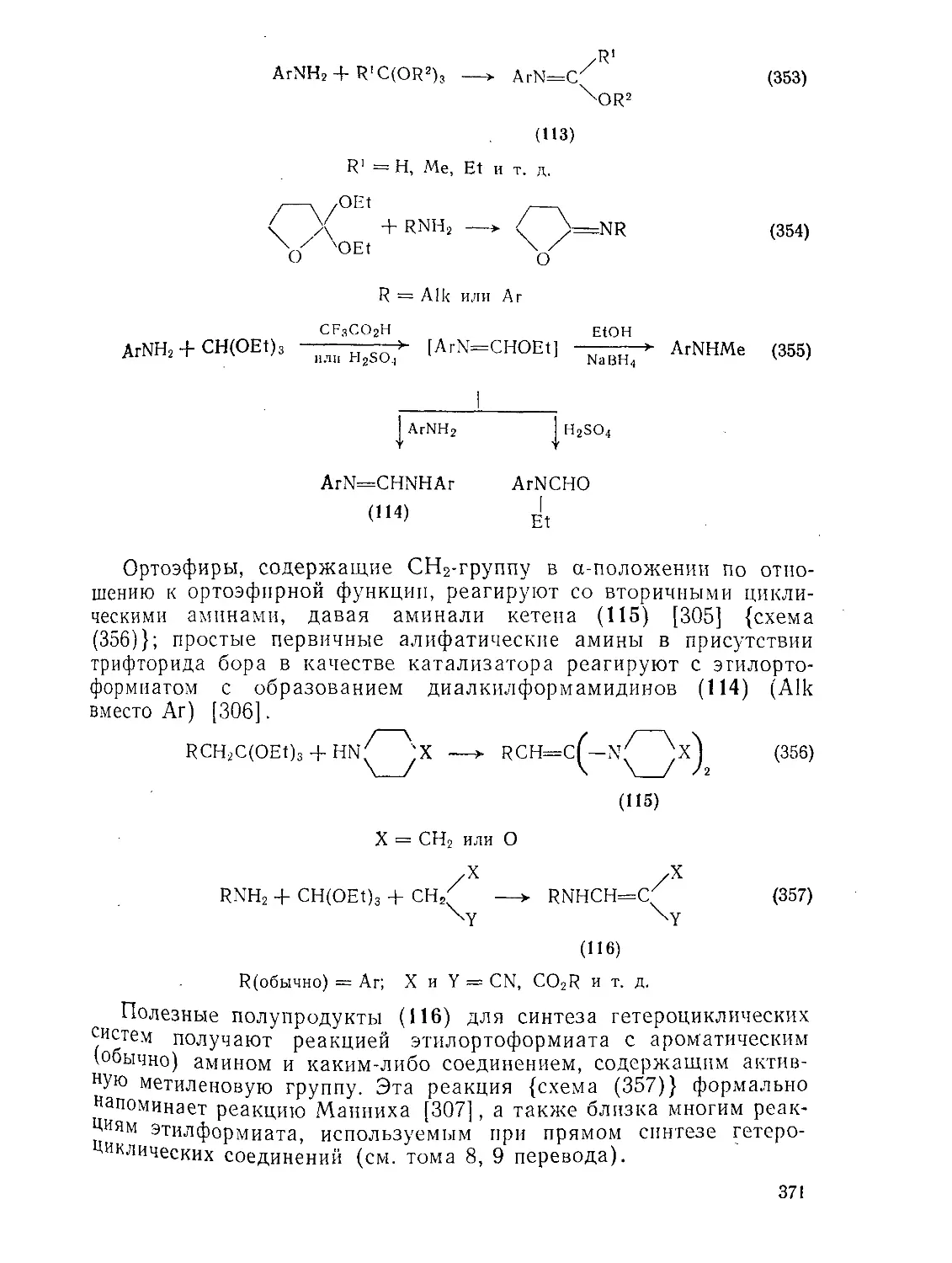
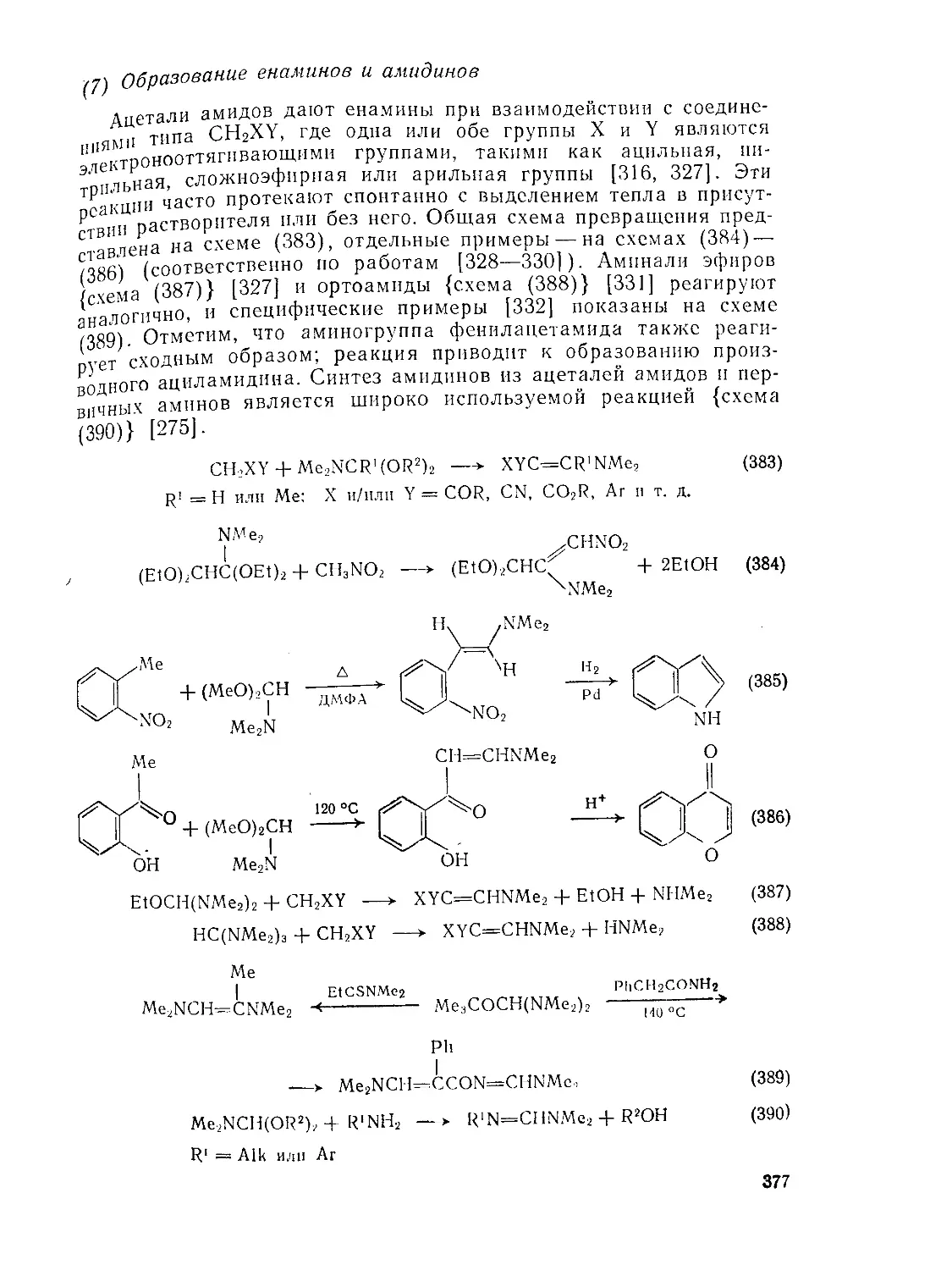
9 8.4. Ортоэфиры
9.8 4.1. Методы получения ортоэфиров
9 8.4.2. Реакции ортоэфнров
д.8.4.3. Ацетали амидов и родственные
Литература
355
360
364
364
367
соединения 373
378
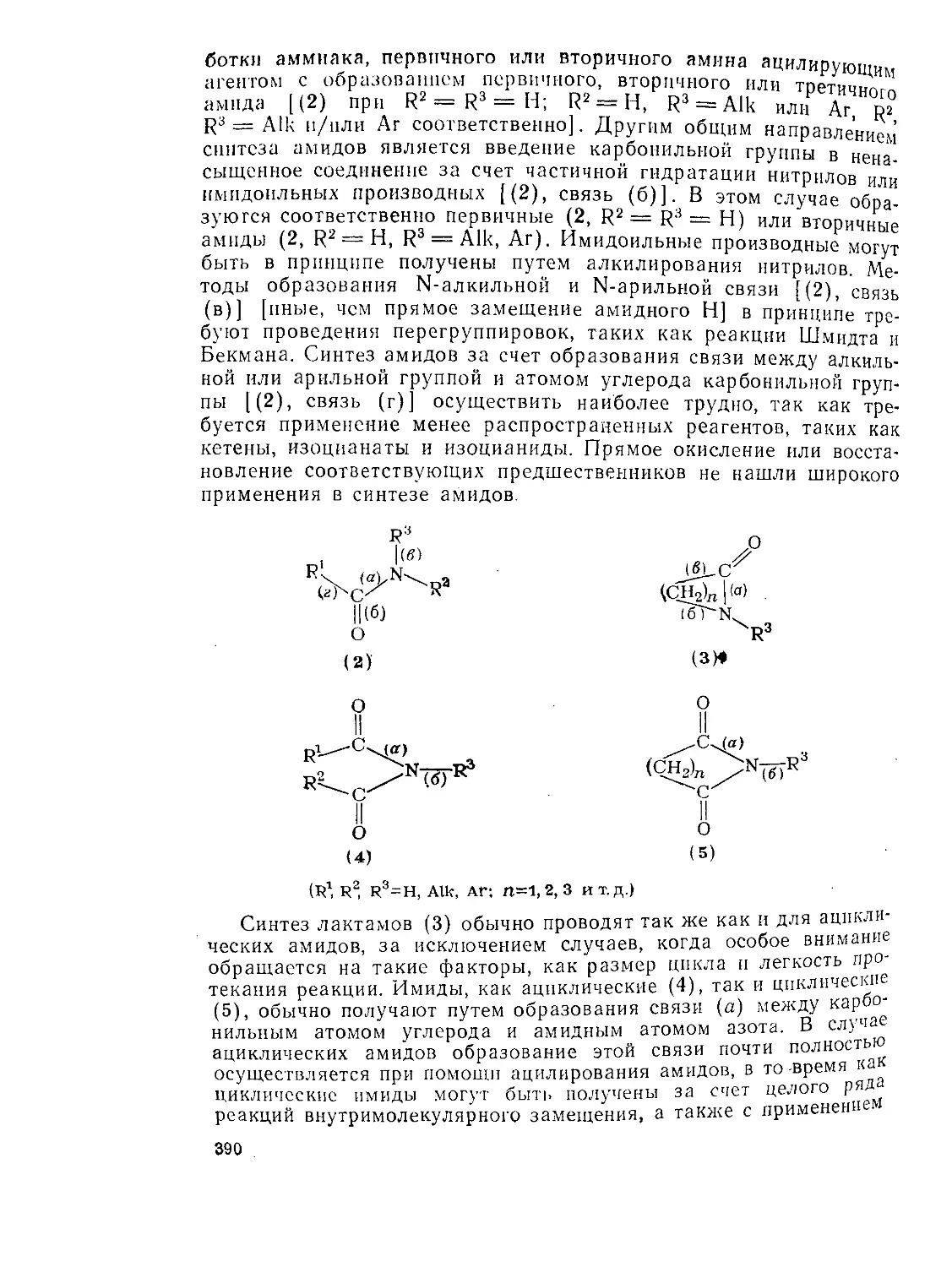
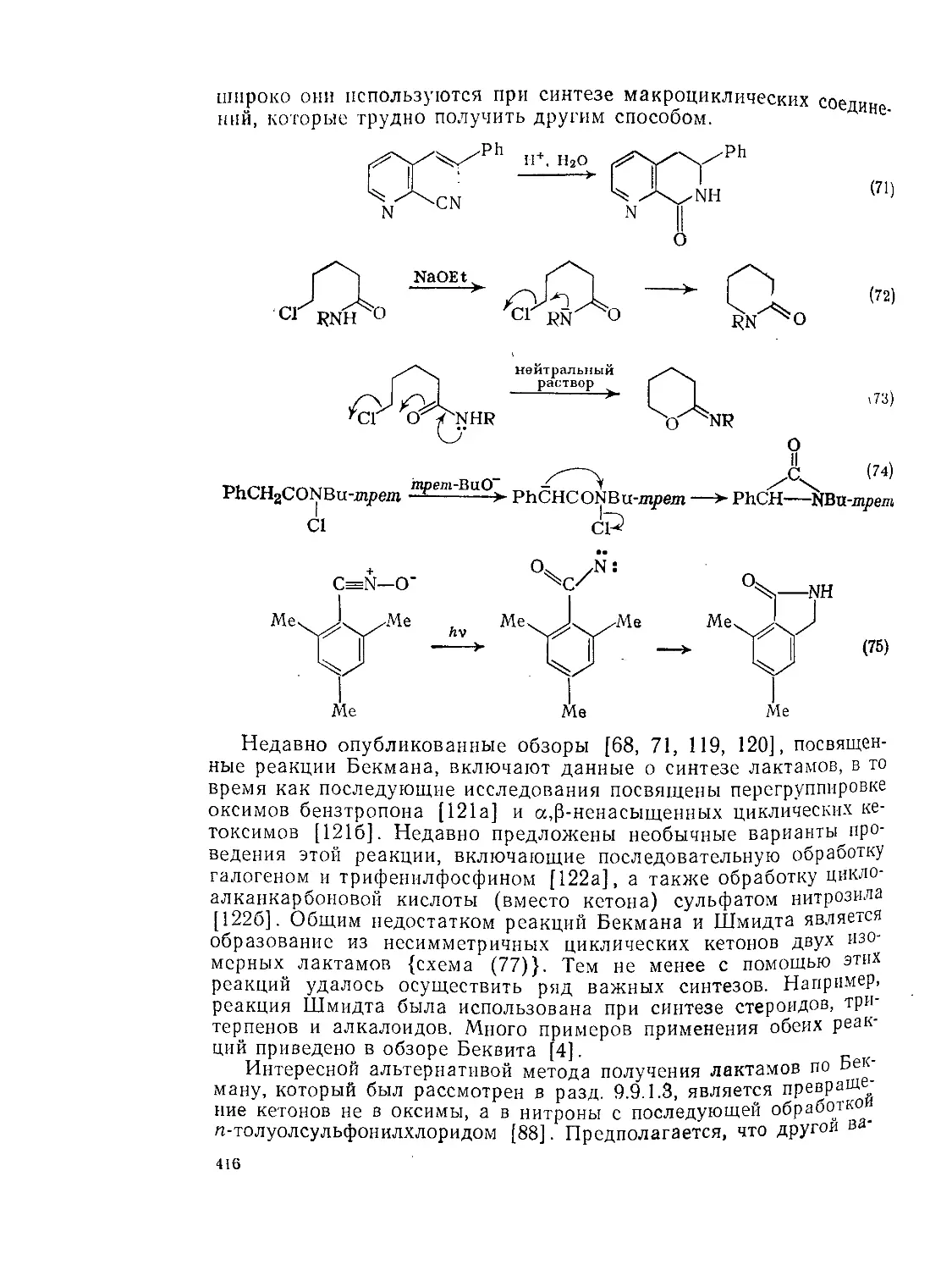
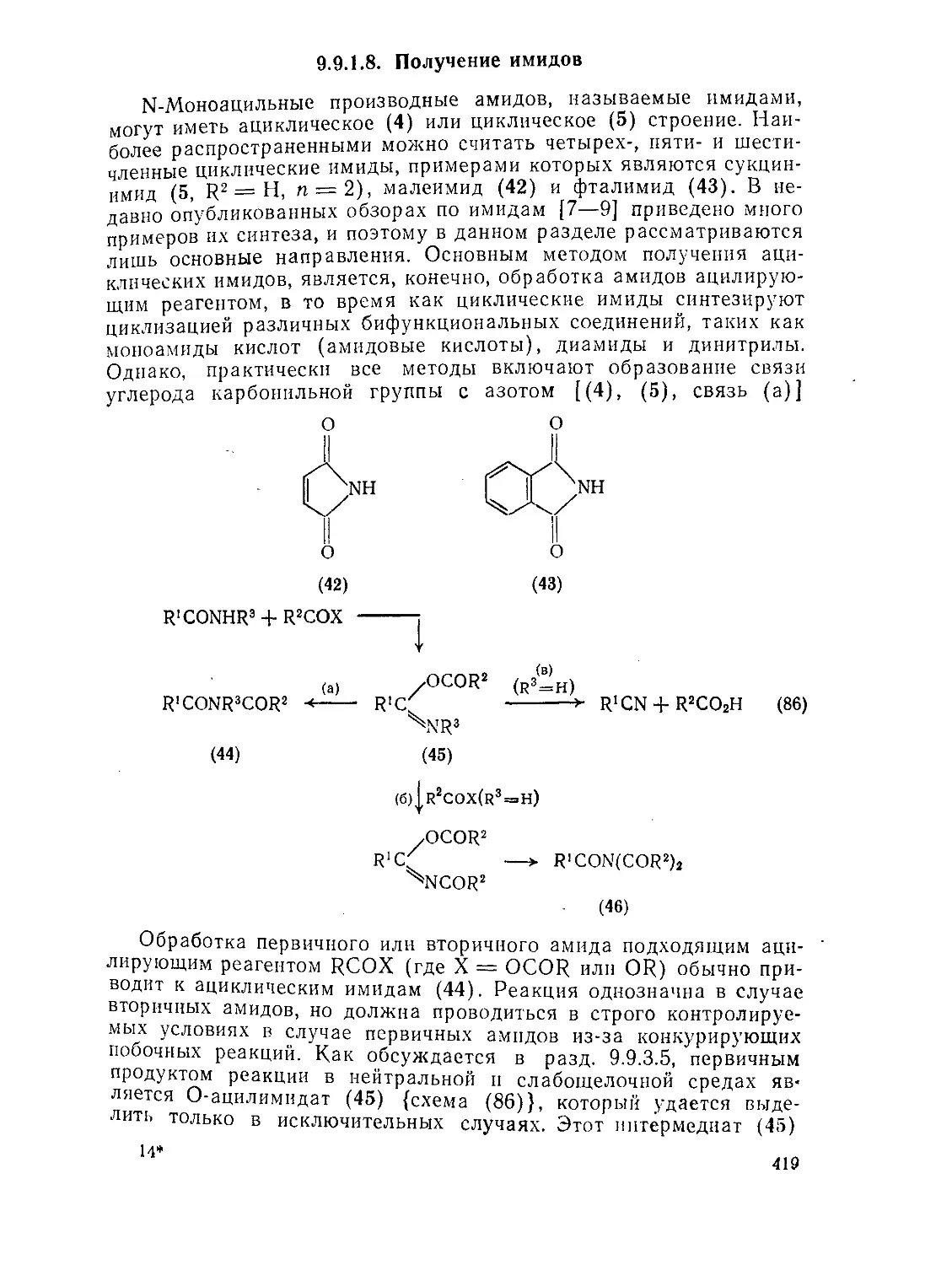
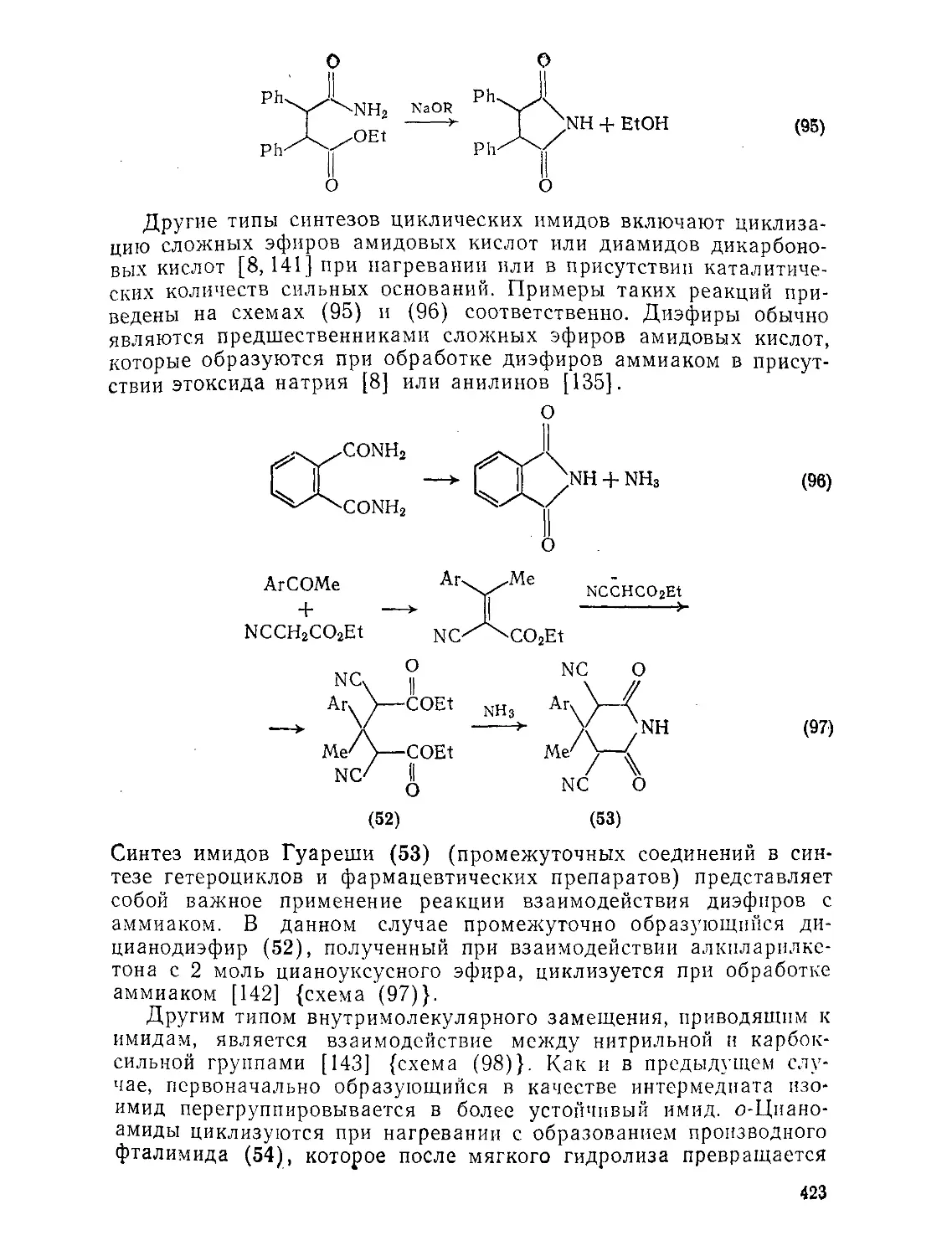
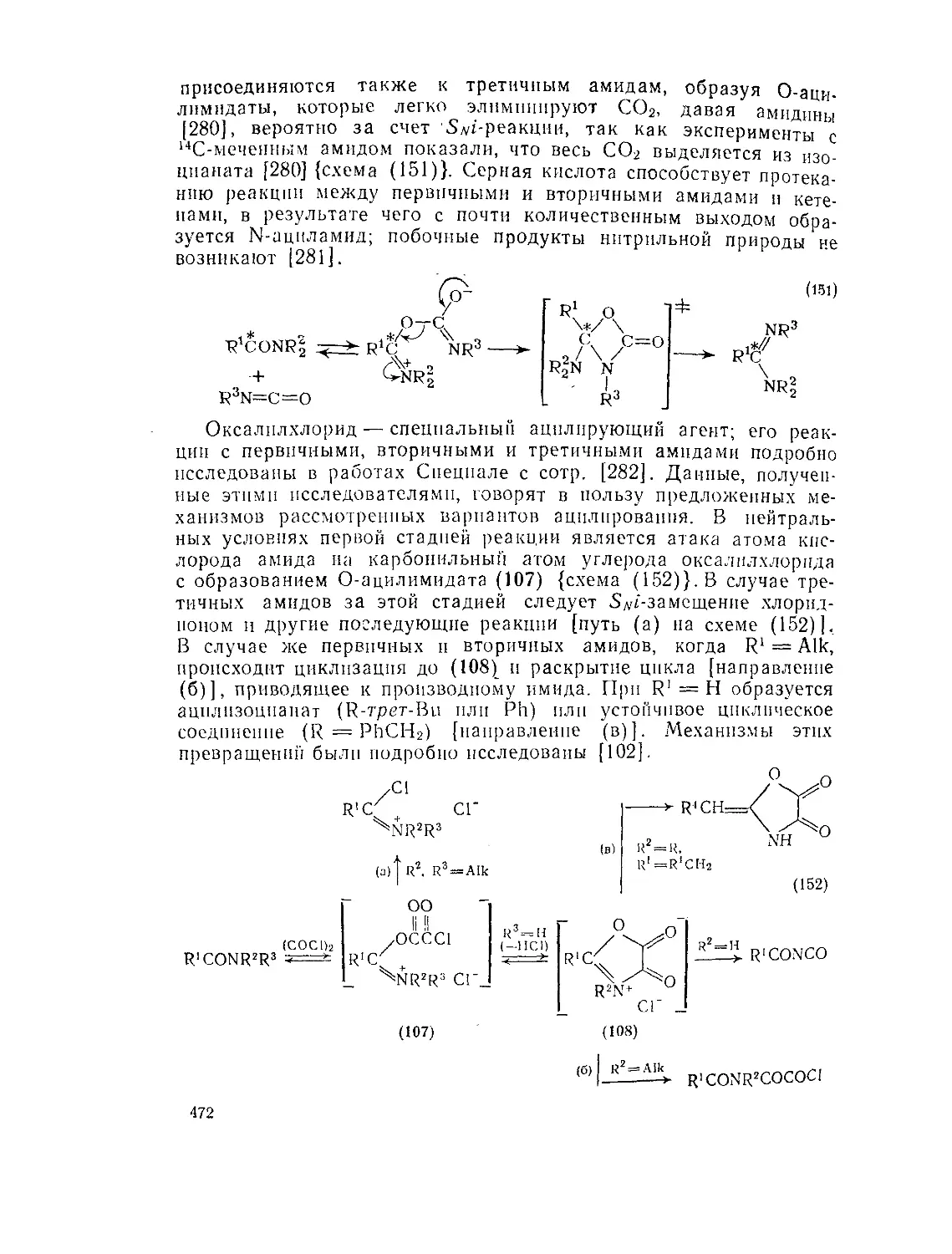
9.9. Амиды и родственные соединения. Б. С. Чаллис и Дж. А. Чаллис 388
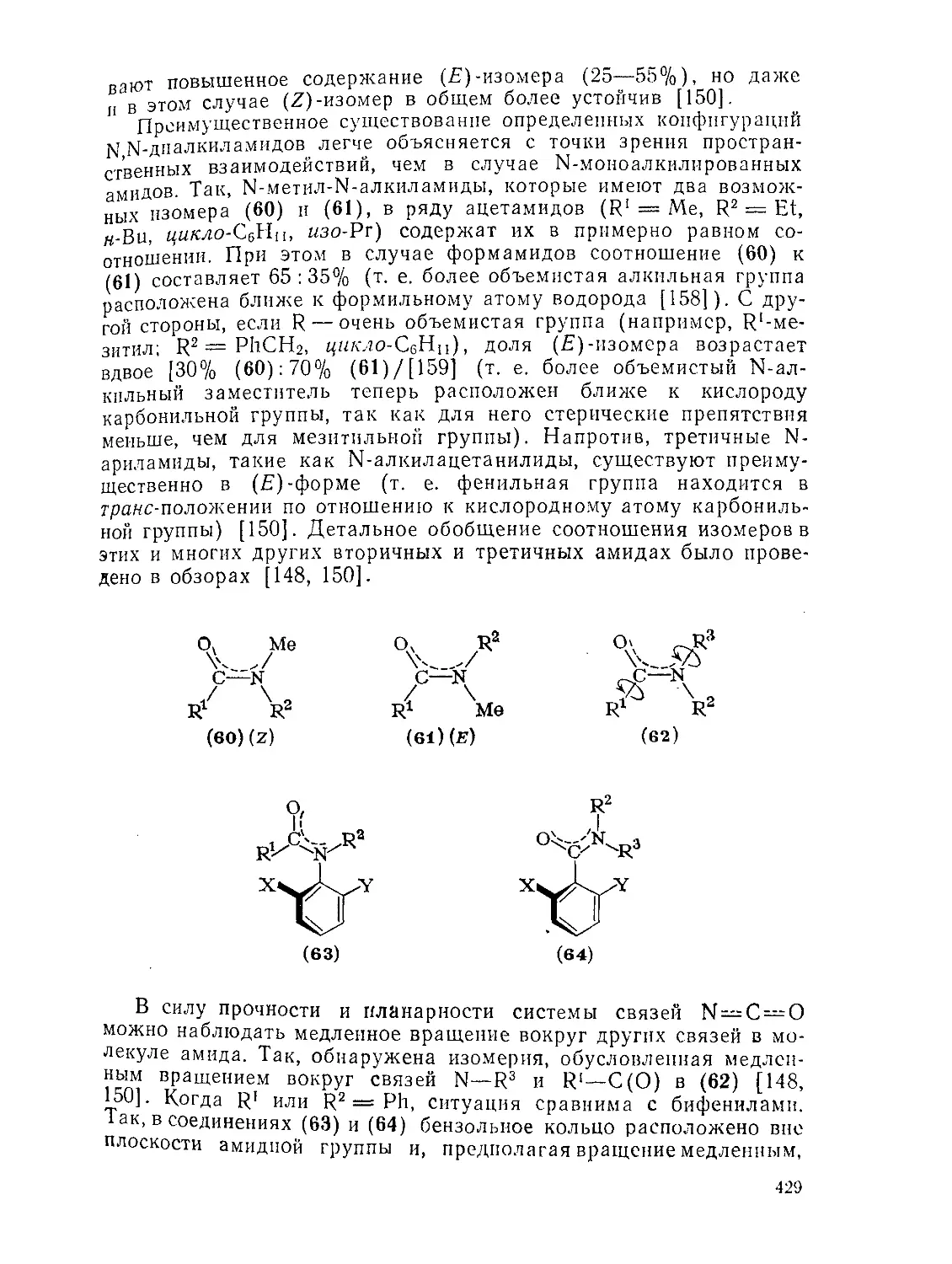
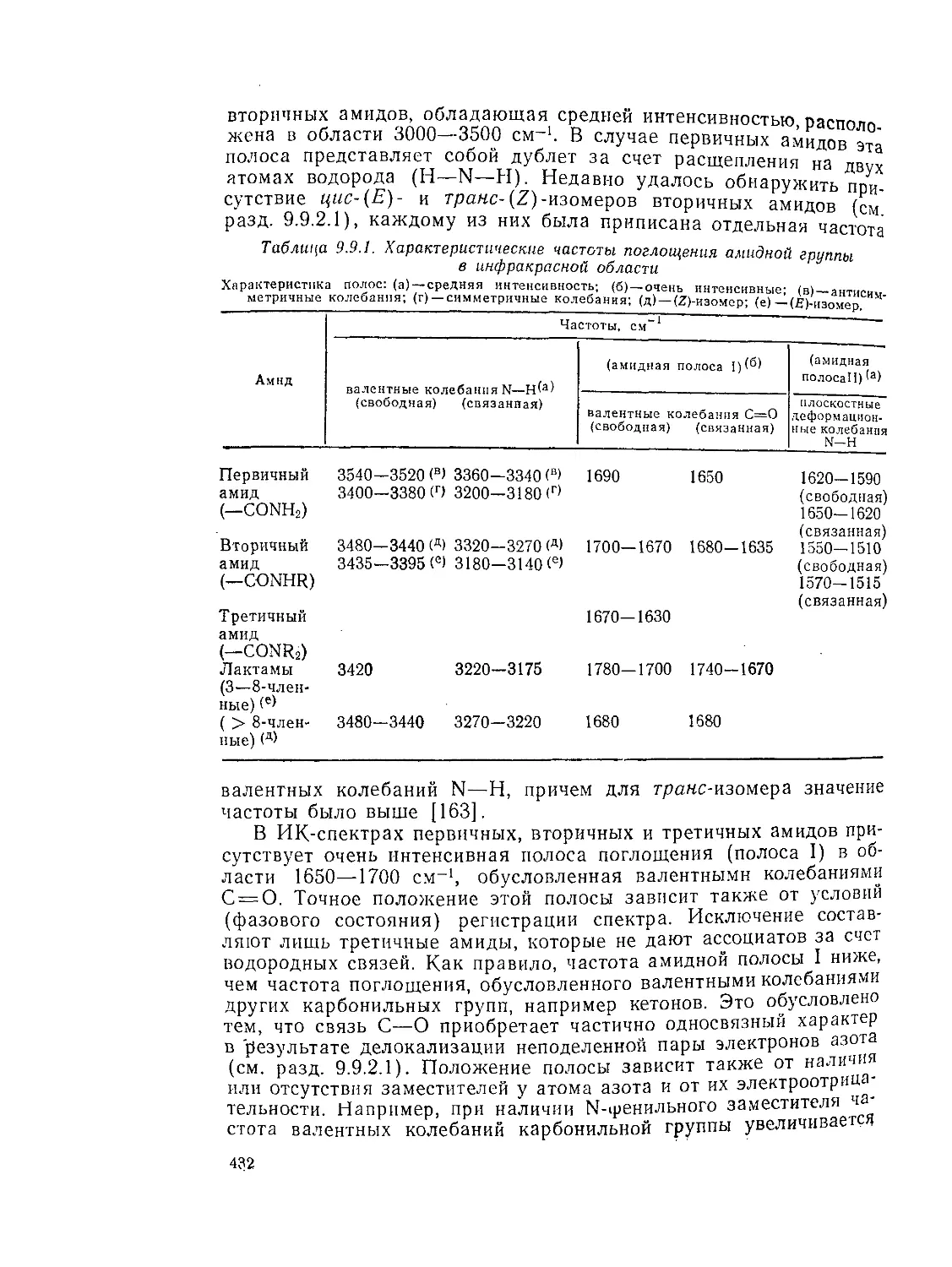
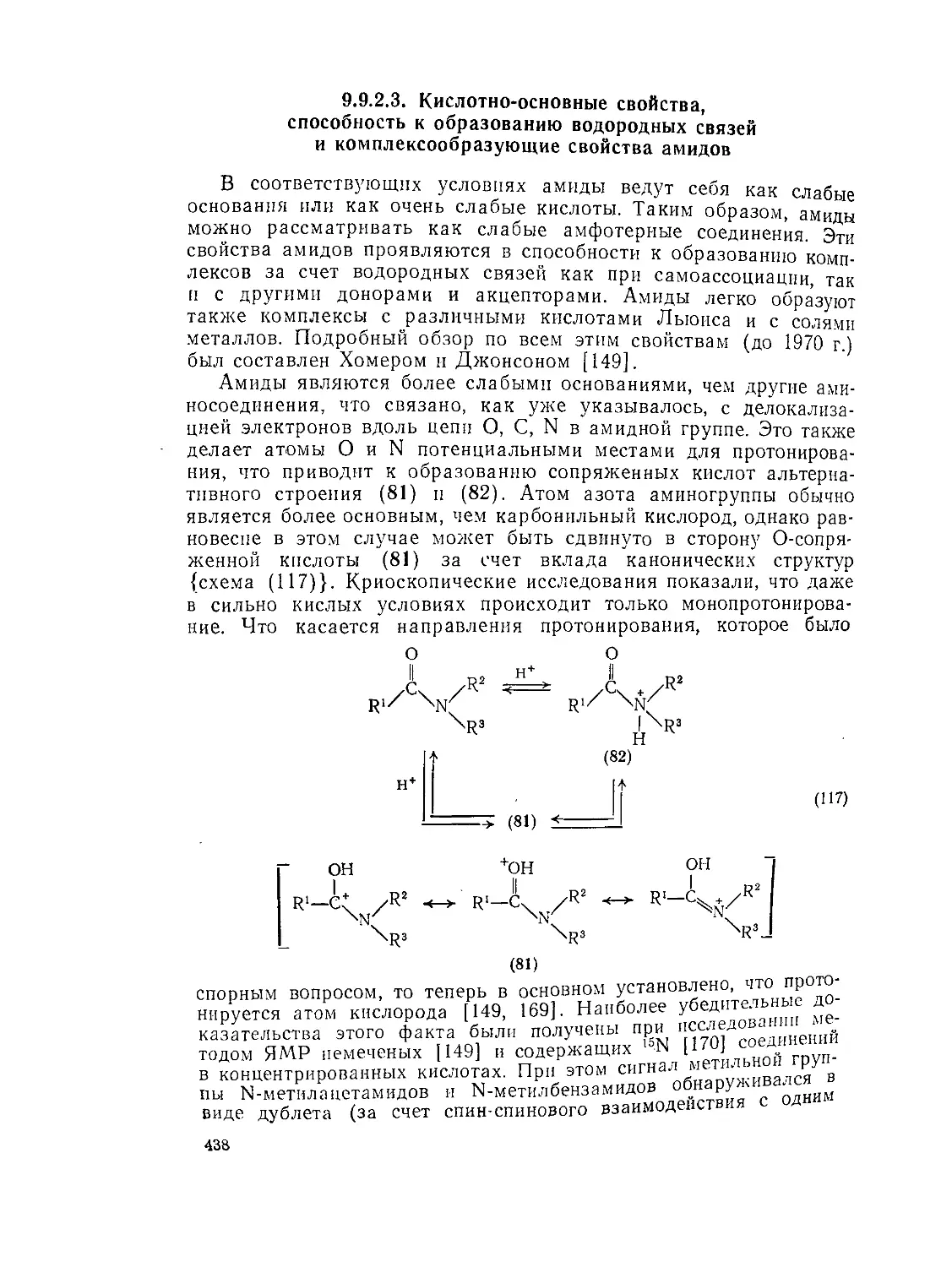
9.9.1. 9.9.1.1. 9.9.1.2. 9.9.1.З. 9.9.1.4. 9.9.1.5. 9.9.1.6. 9.9.1.7. 9.9.1.8. 9.9.2. 9.9.2.1. 9.9.2.2. 9.9.2.3. Методы получения амидов 389 Ацилирование аминов 391 Амиды нз. нитрилов 398 Амиды путем перегруппировок 40Г Реакции изоцианатов, карбамоилгалогенидов и пзоцианидов 407 Алкилирование амидов 410 Окислительные и восстановительные методы 413 Получение лактамов 414 Получение имидов 419 Свойства амидов 425 Структура и стереохимия амидов . 426 Спектральные свойства амидов 431 Кислотно-основные свойства, способность к образованию водородных связей н комплексообразующие свойства амидов 438
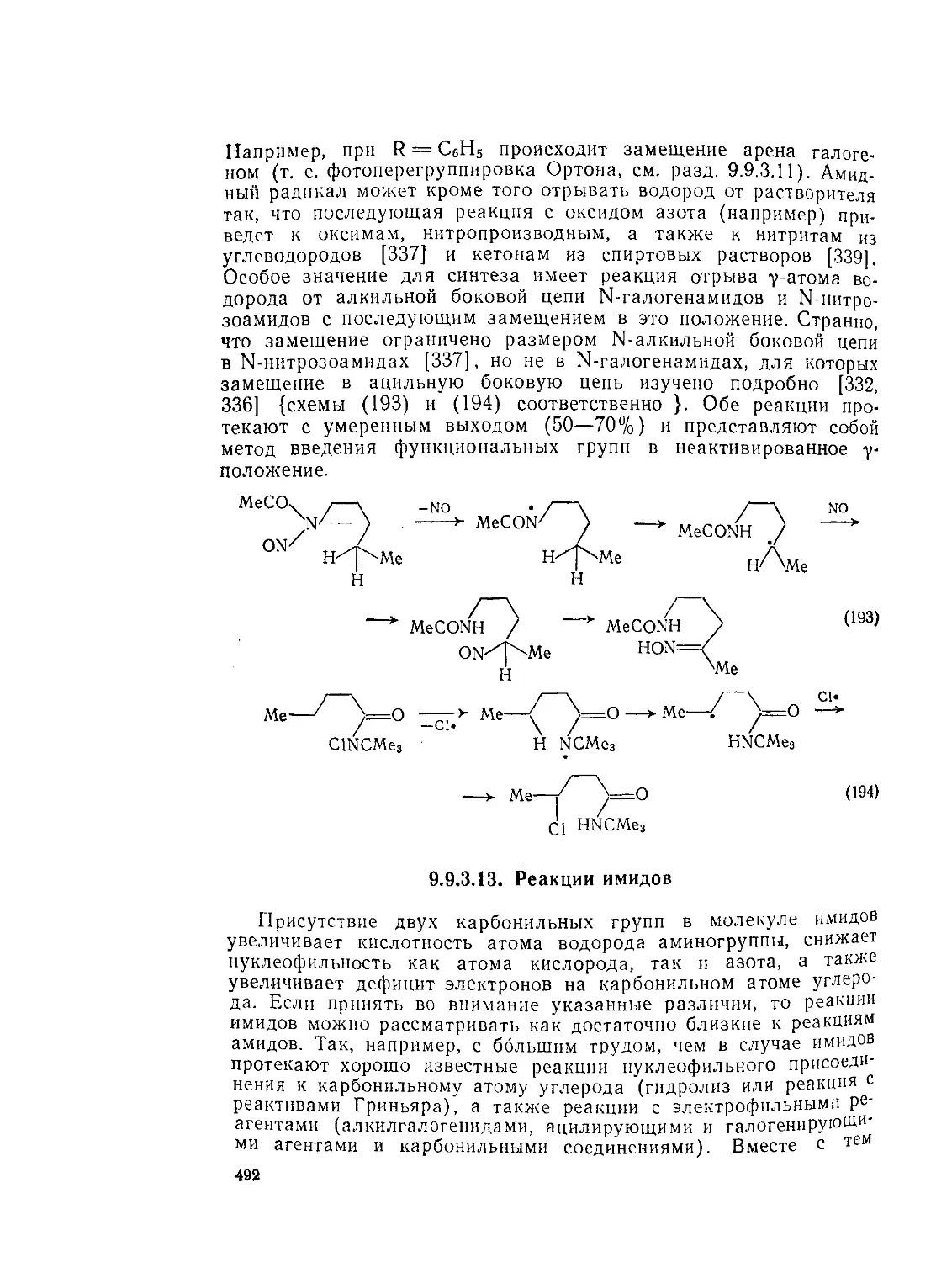
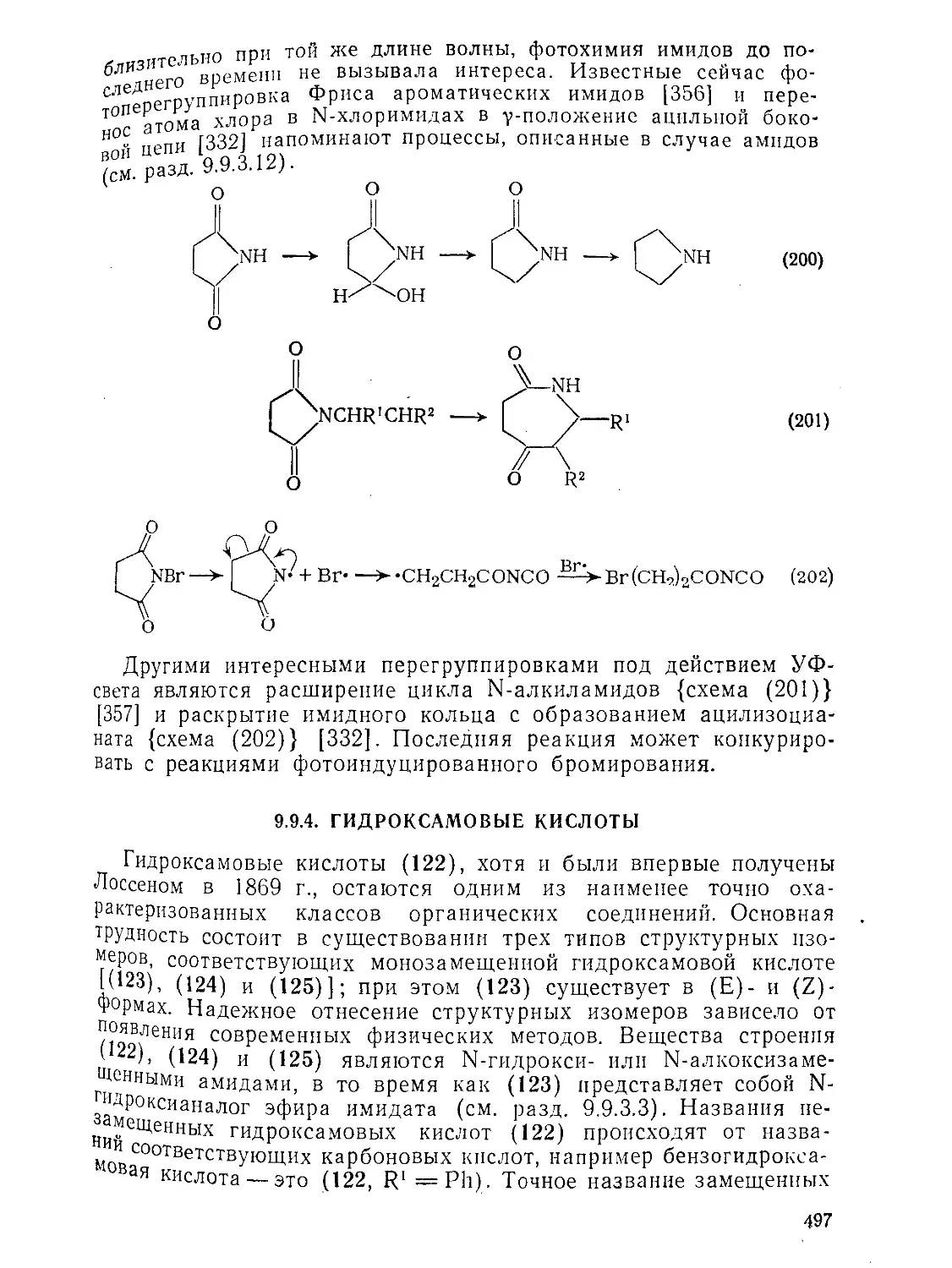
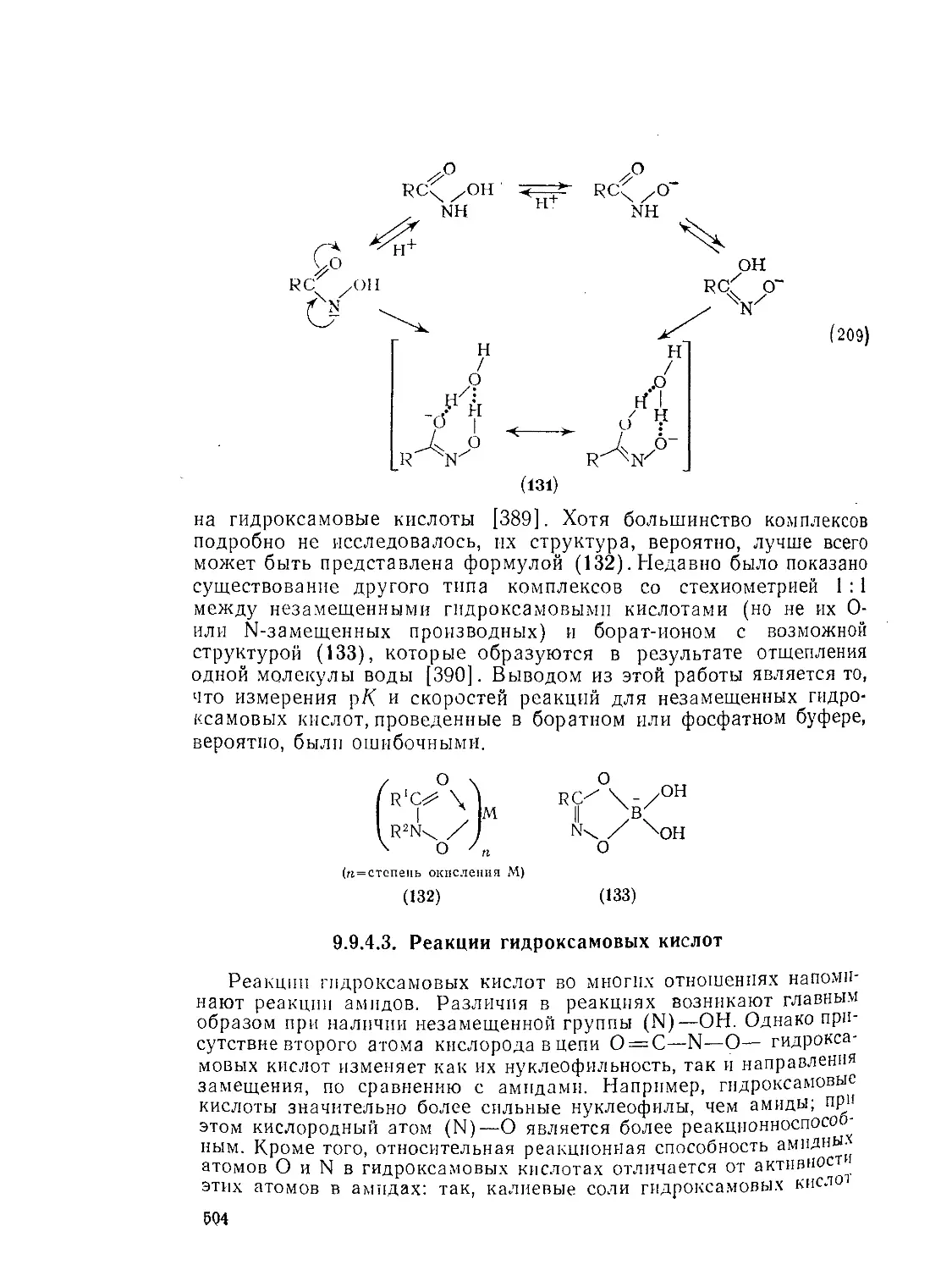
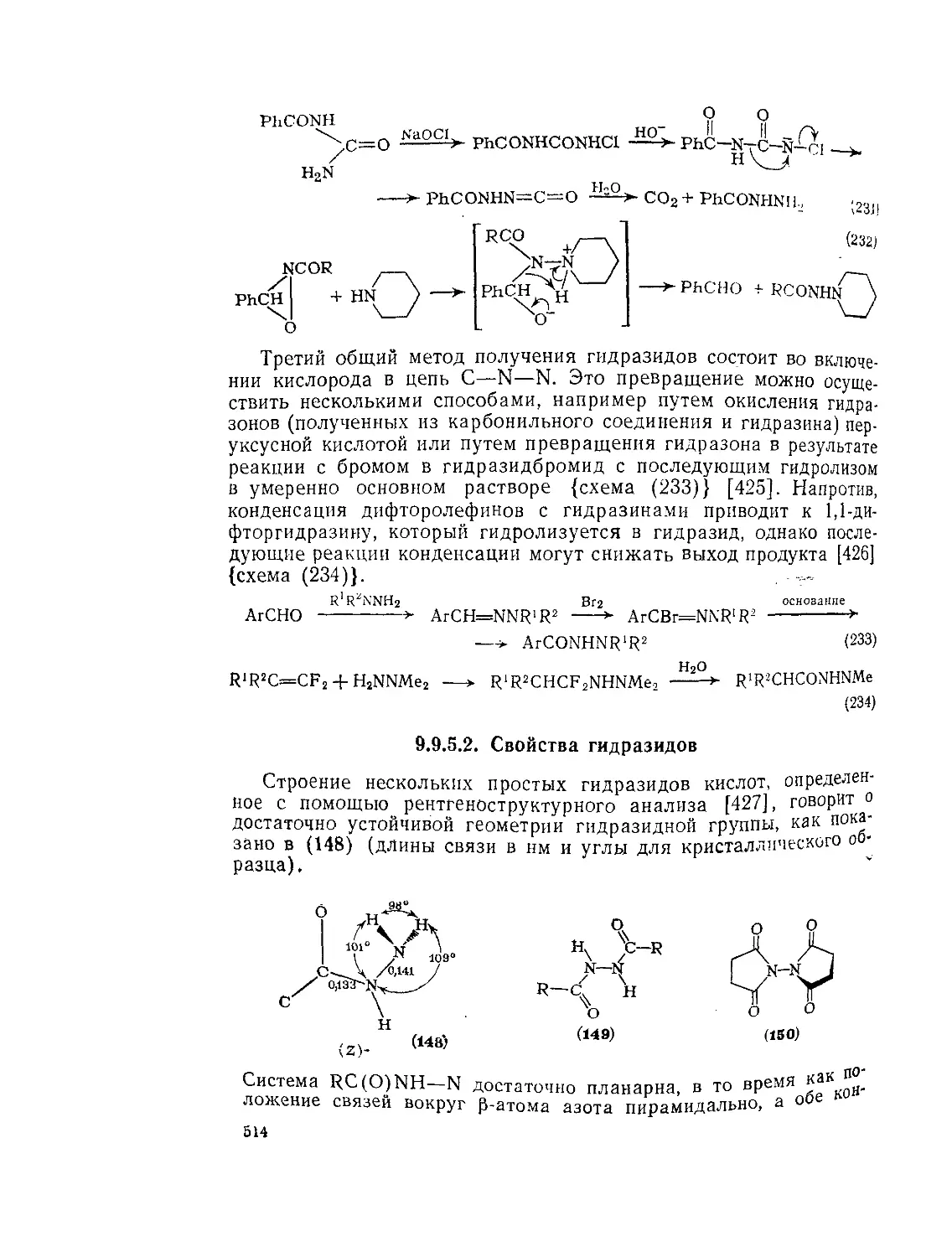
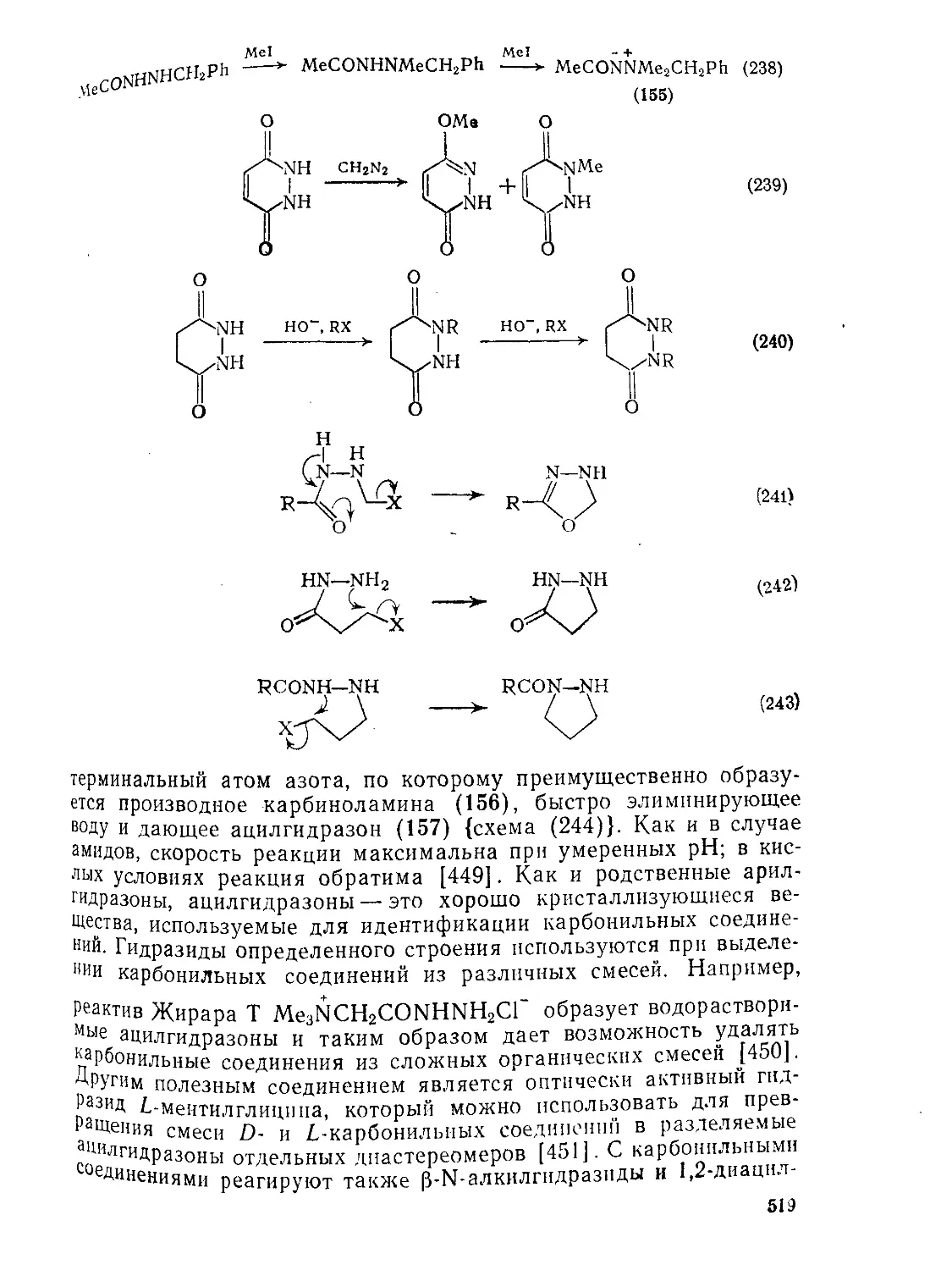
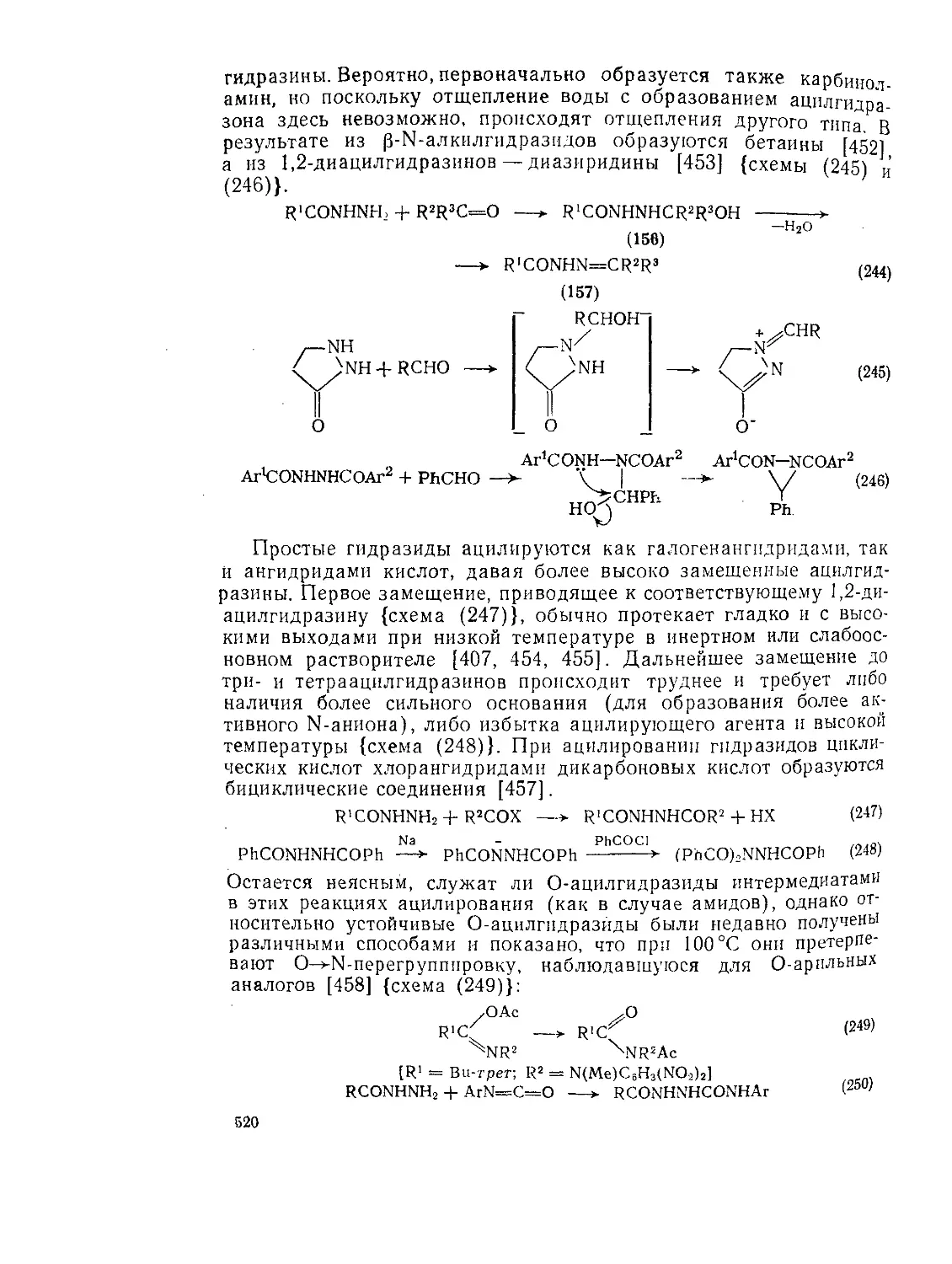
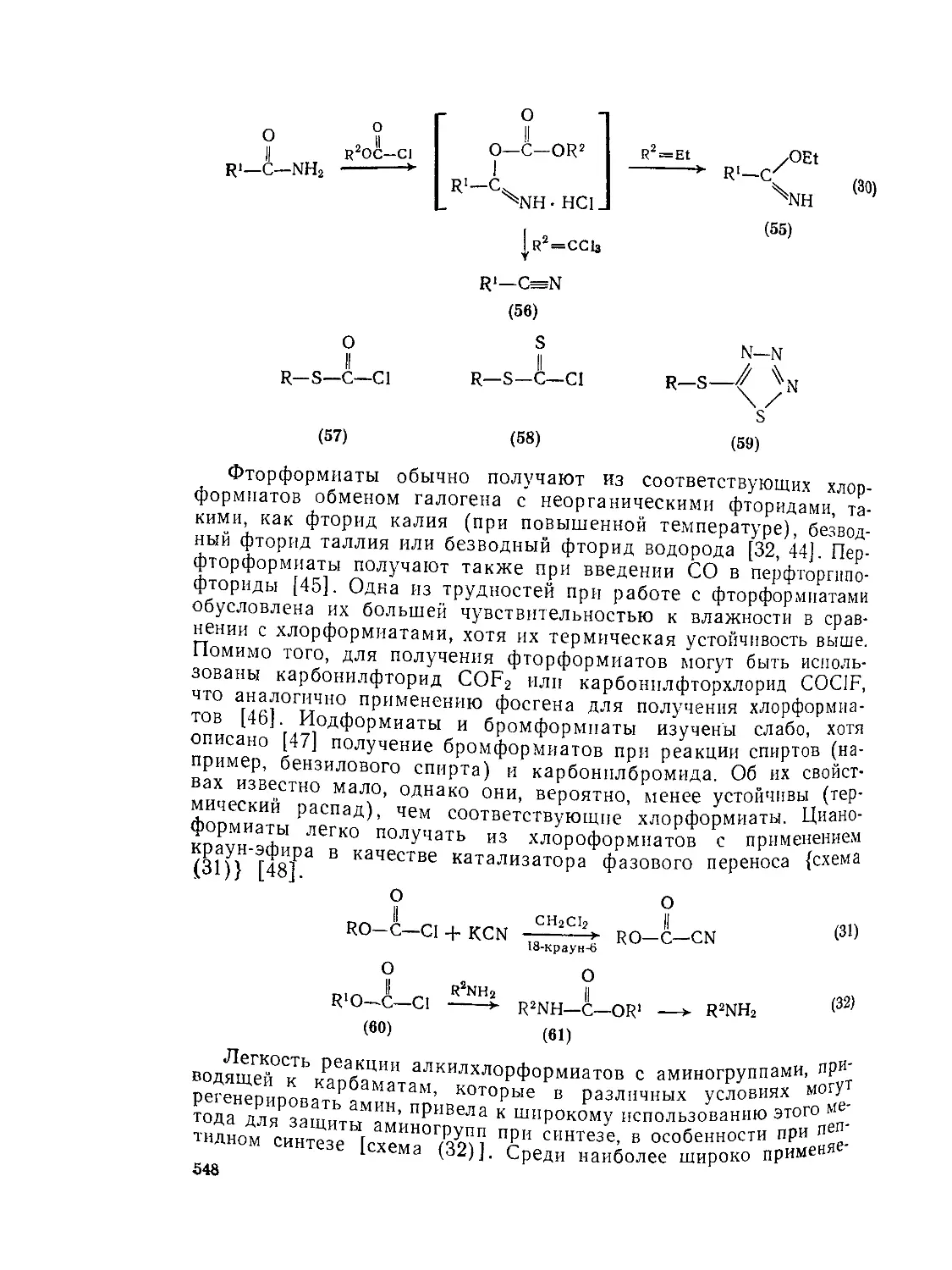
9.9.2.4. 9.9.2.5. 9.9.3. 9.9.3.1. 9.9.3.2. 9.9.3.3. 9.9.3.4. 9.9.3.5. 9.9.3.6. 9.9.3.7. 9.9Д.8. 9.9.3.9. 9.9.3.10. 9.9.3.11. 9.9.3.12. 9.9.3.13. 9.9.4. 9.9- 4.1. 9-9.4.2. 9.9.4.3. 9.9.5. Ш.1. 9-9.52. 9.9.5.3. Структура и спектры имидов 444 Диметилформамид и диметилацетамид как растворители .449 Реакции амидов 450 Гидролиз и сольволиз 451 Присоединение металлорганических реагентов 460 Алкилирование 460 Реакции с альдегидами и кетонами 467 Ацилирование 468 Ннтрозирование и нитрование 473 Галогенирование 475 Реакция с галогенангидридами неорганических кислот 477 Окисление 483 Восстановление 485 Перегруппировки 487 Фотохимия 490 Реакции имидов 492 Гидроксамовые кислоты 497 Методы получения гидроксамовых кислот 499 Свойства гидроксамовых кислот 500 Реакции гидроксамовых кислот 504 Гидразиды 509 Методы получение гидразидов 510 Свойства гидразидов 514 Реакции гидразидов 517 Литература 524
9.10. Производные угольной кислоты. А. Ф. Хегарти
9|0.1. Введение
9.10.21 Э*И0Ы'Г'
9.10.2.2.
9.10.2.3
9.10.2.4.
9.10.2.5.
8
536
536
538
538
539
541
543
544
Эфиры, галогенаигидриды и ангидриды угольной кислоты
Моноэфиры угольной кислоты
Диэфиры угольной кислоты
Циклические эфиры угольной кислоты
Ангидриды угольной кислоты
I алогенангидрнды и азиды угольной кислоты
530
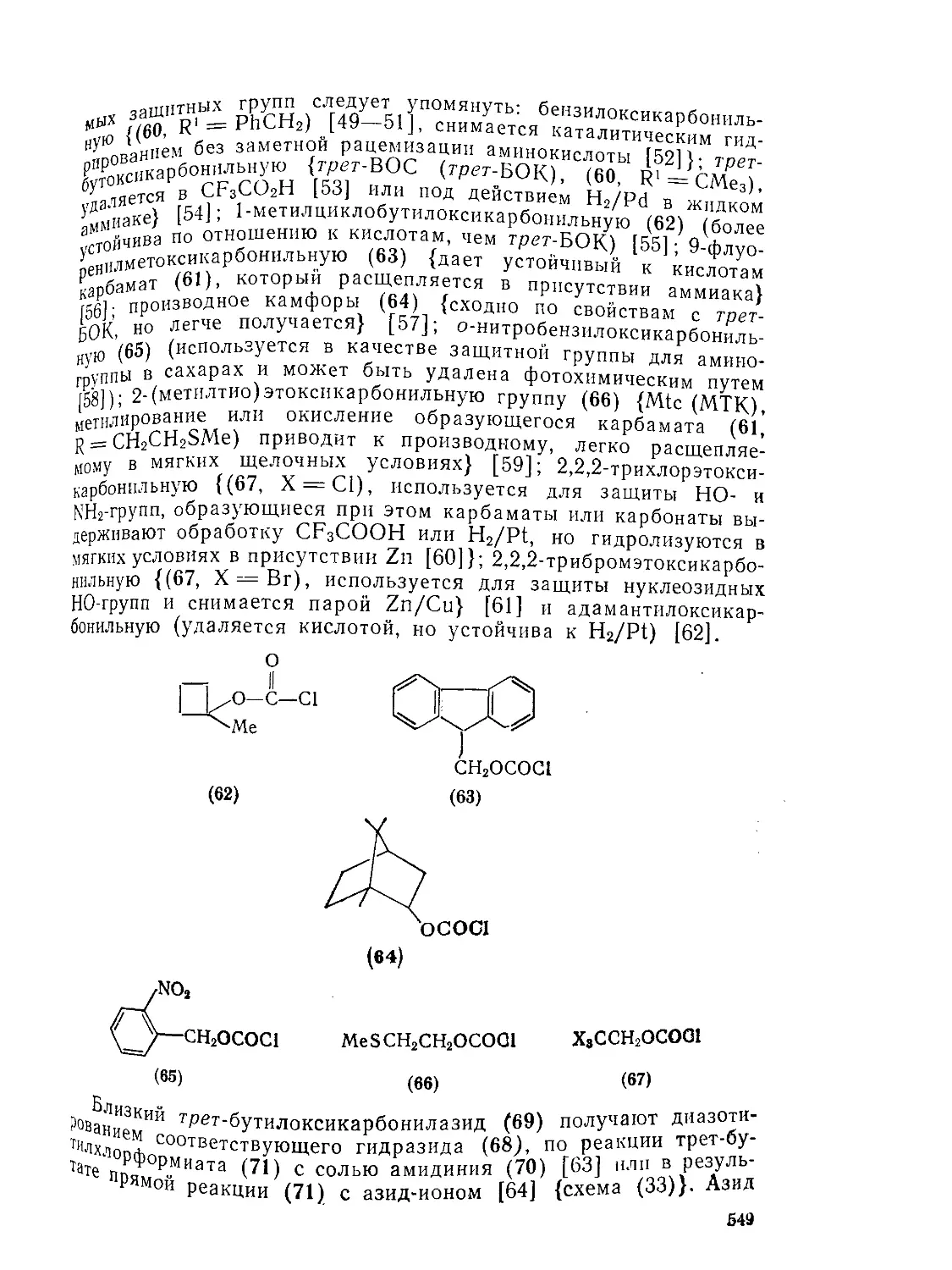
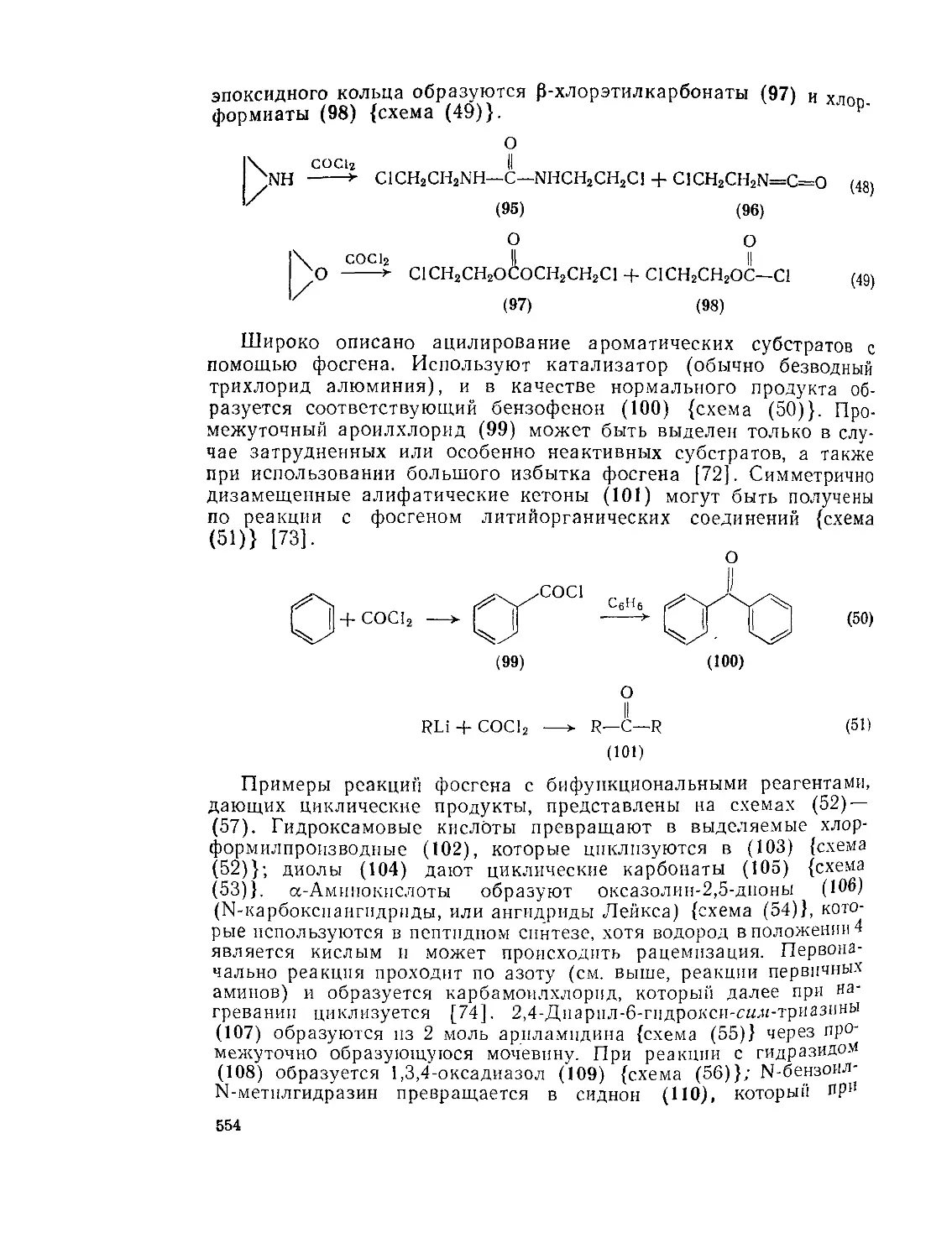
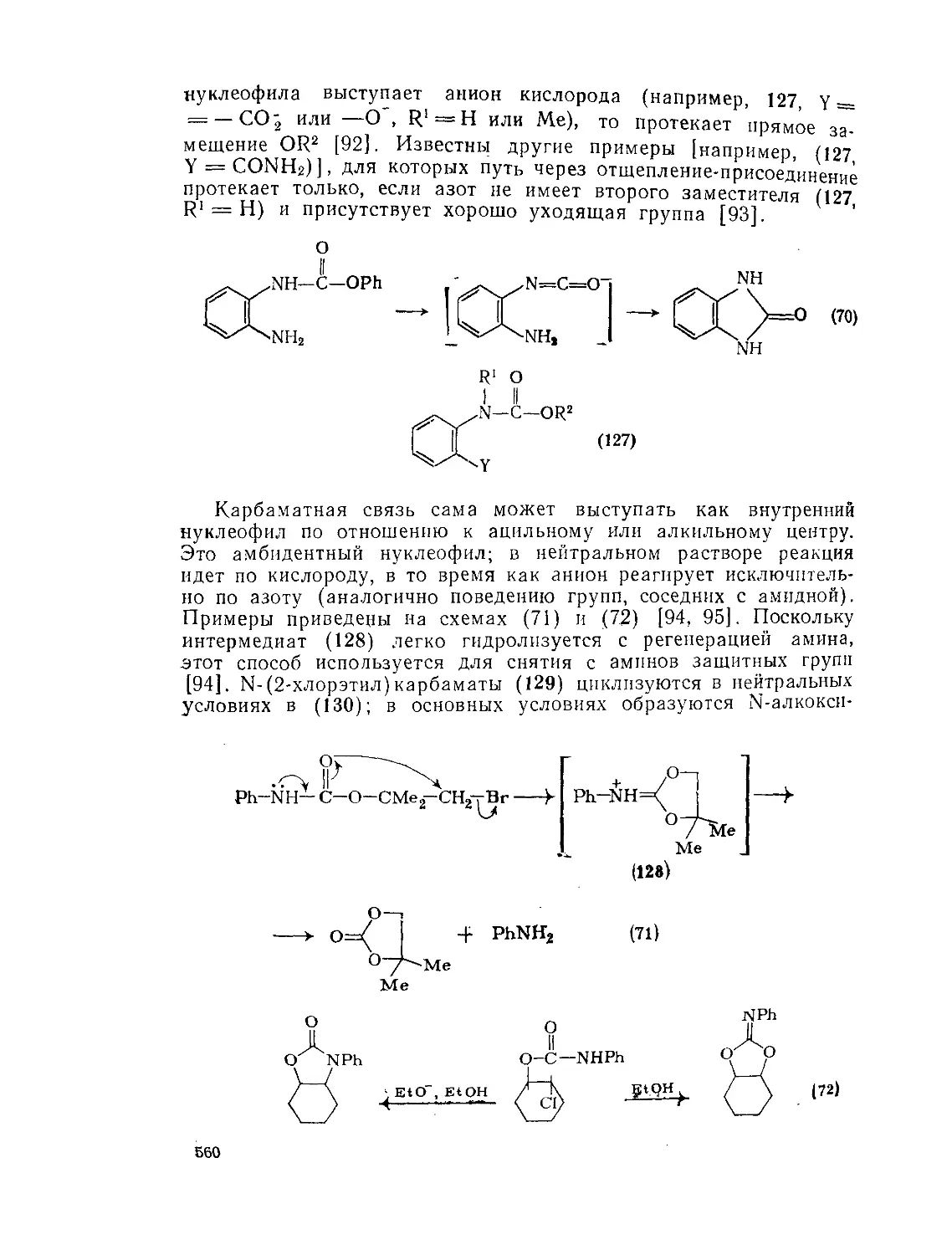
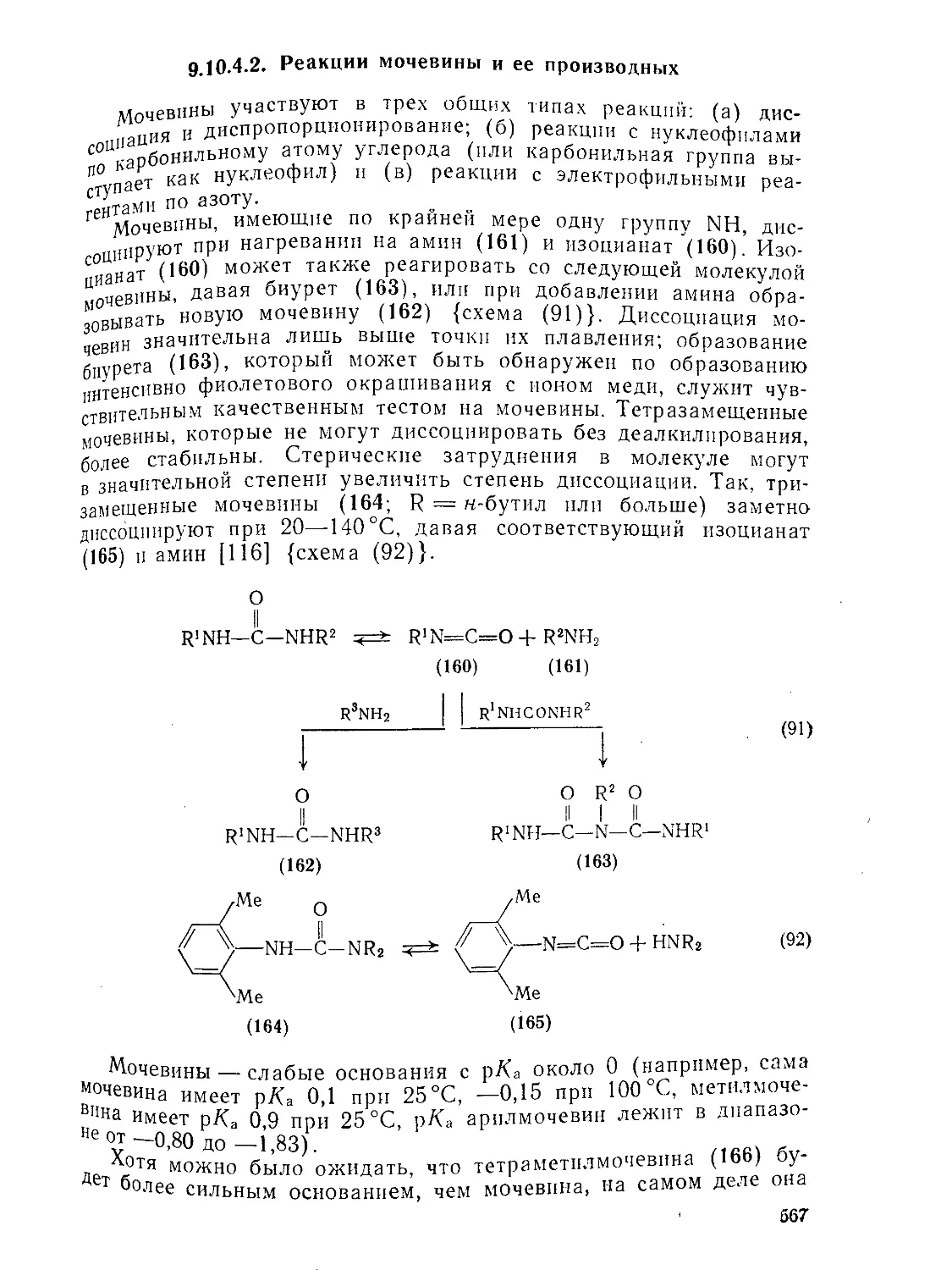
9.Ю.2.6. 9.10.3. 9.Ю.З.1. 9.10.3.2. 9.10.3.3. 9 10.3.4. фосген . Карбаматы (УР^^-х^атов V-Замещениые карбаматы Карбамоилгалогениды и -азиды 556 556 557 561 563 564
9.10.3.5. 9.10.4. 9.Ю.4.1. 9.Ю.4.2. 9.Ю.4.З. Карбаминовые кислоты 565
.ы » ее производных 565 567
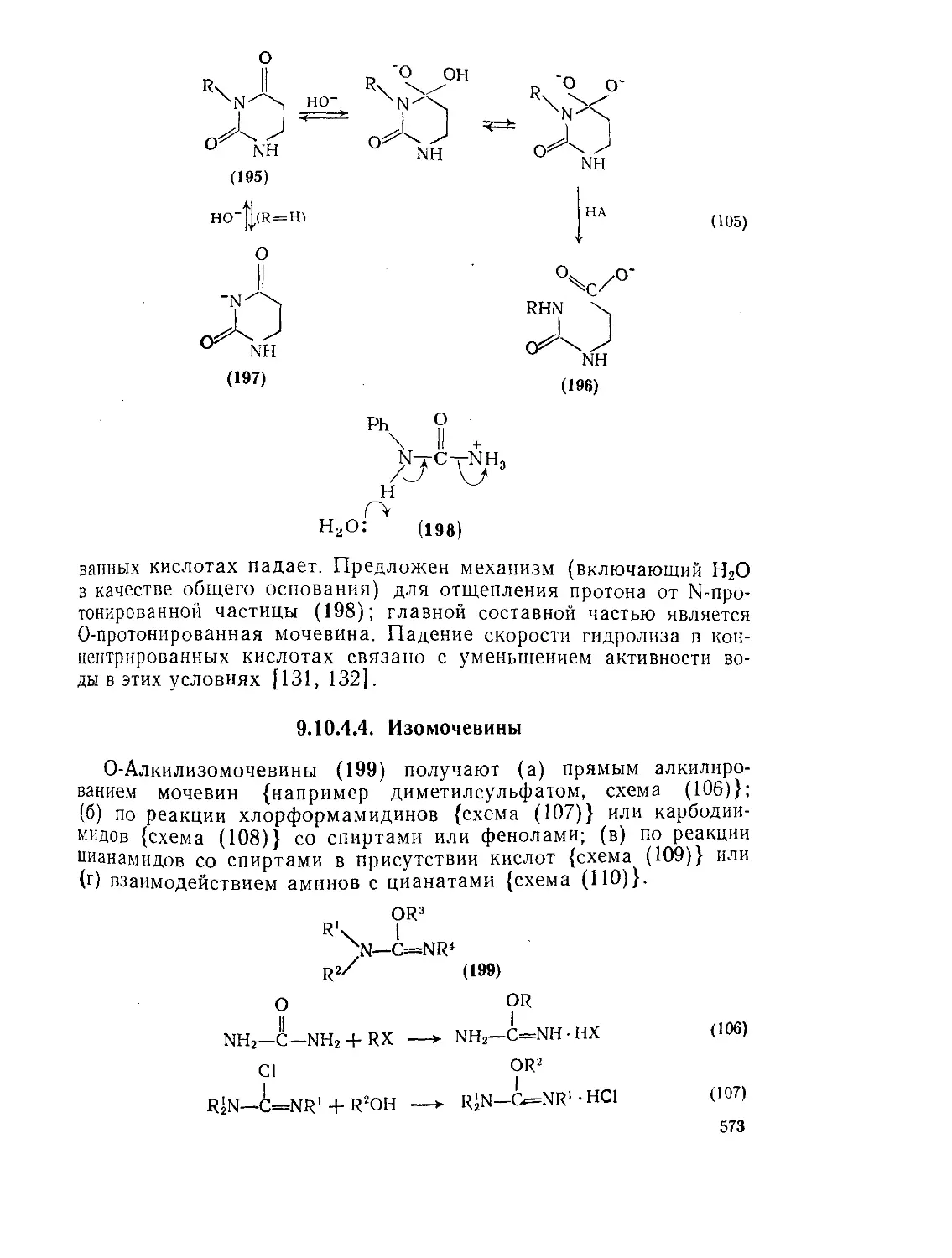
Реакции мочевины и ее производных Гидролиз мочевины и ее производных 571 573
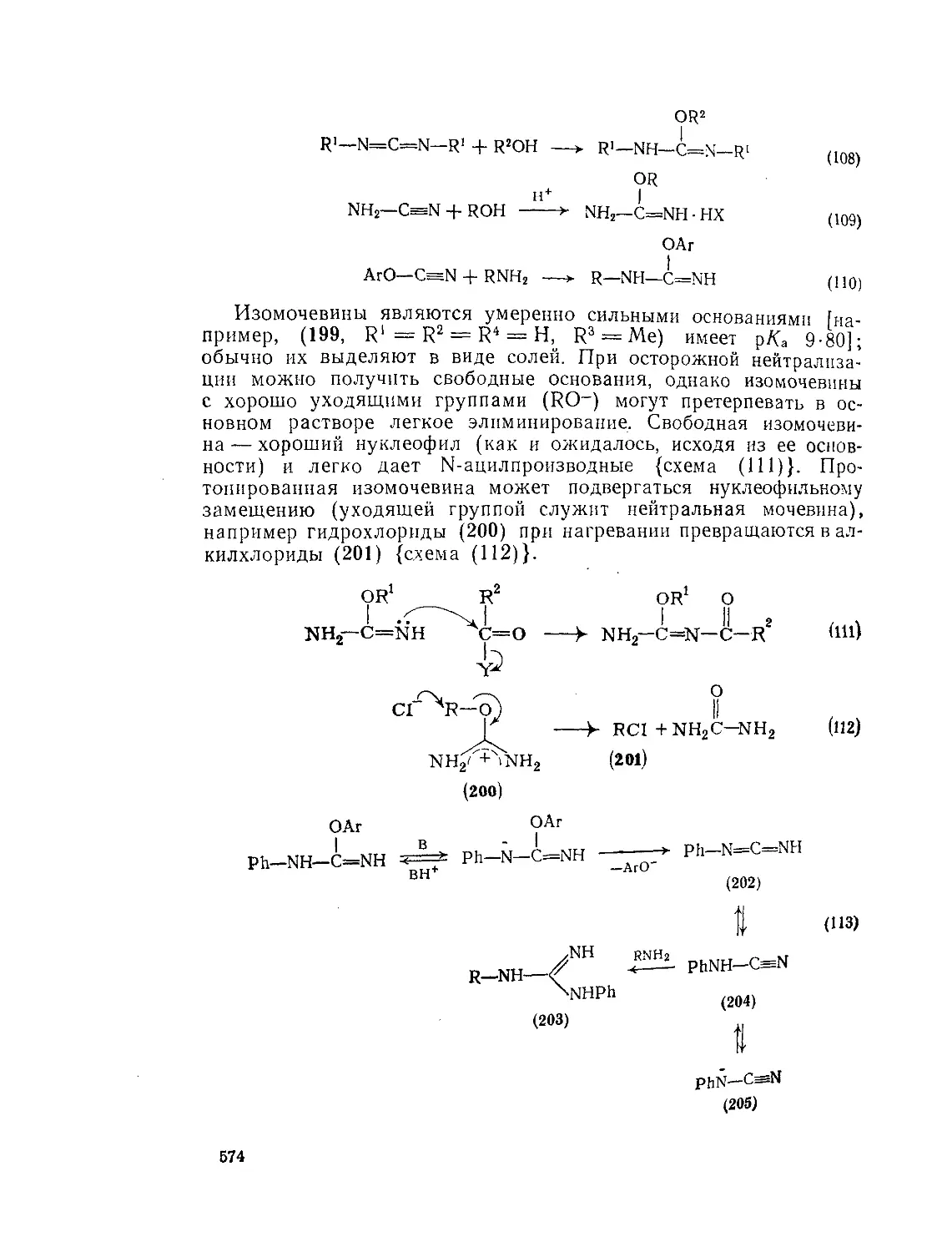
9.Ю.4.4. Изомочёвины 575
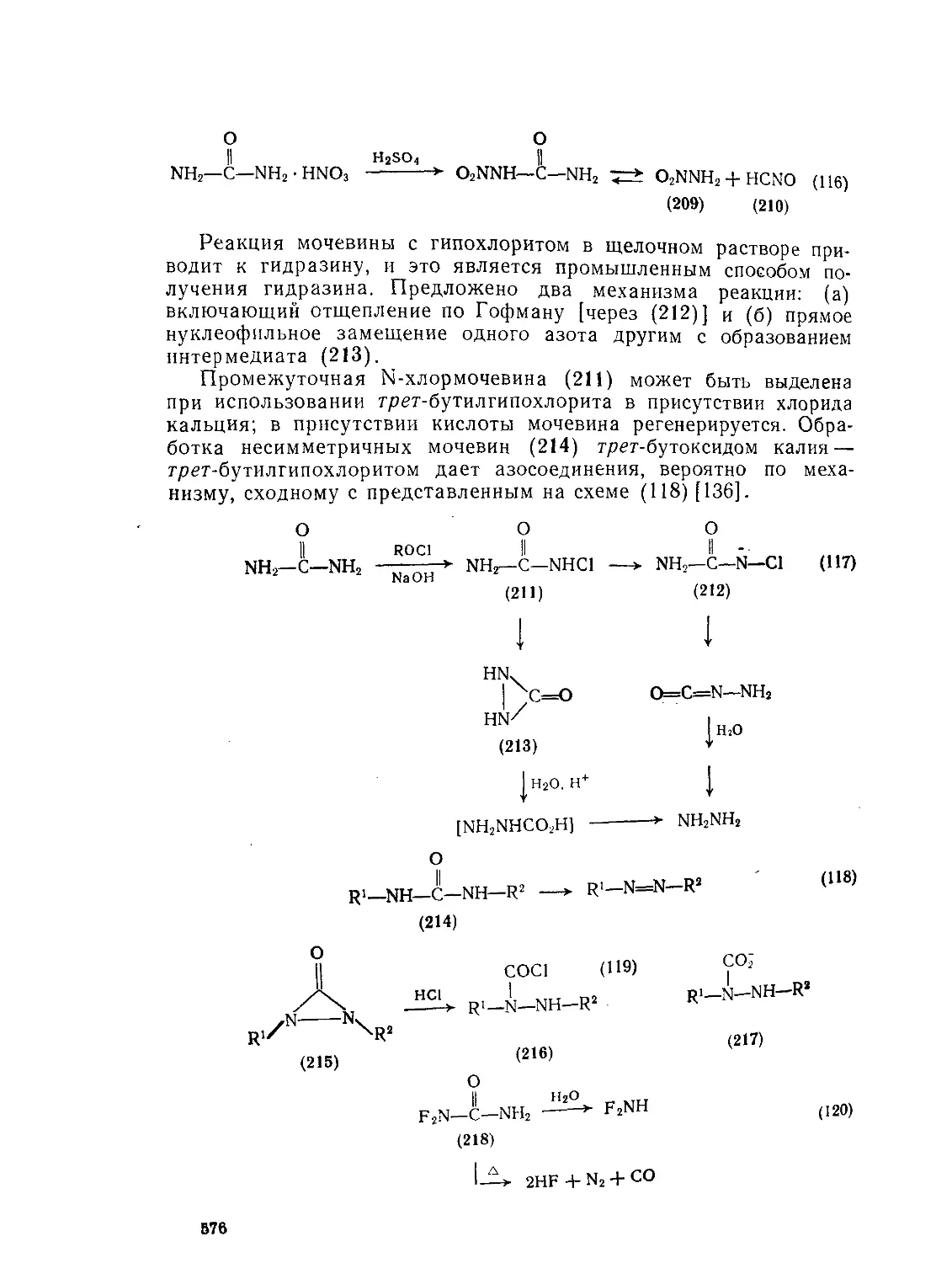
9.10.4.5. N-Замещенные мочевины Литература 577
9.11. Пероксикислоты и пероксиды ацилов. А. Ф. Хегарти 580
580
9.11.1. Введение 581
9.11.2. Получение п свойства 581
9 11 2.1. Методы получения и реакции пероксикислот
9.11.2.2. Методы получения и реакции сложных эфиров пероксикислот и 589
пероксидов ацилов Литература 593
595
ЧАСТЬ 10. СОЕДИНЕНИЯ ФОСФОРА
10.1. Введение. Д. Дж. X. Смит
10.1.1.
10.1.2.
10.1.3.
10.1.4.
Структура и характер связи
Номенклатура фосфорорганических соединений
Спектроскопия соединений фосфора
Синтез фосфорорганических соединений
Литература
10.2. Фосфины, соли фосфония, галогенфосфины. Д. Дж. X. Смит
10.2.1.
10.2.1.1.
10.2.1.2.
10.2.1.3.
10.2.1.4.
10.2.1.5.
10.2.1.6.
10.2.1.7.
10.2.2.
10.2.2.1.
10.2.2.2.
10.2.2.3.
10.2.2 4
10.2.3.
10.2.3.1.
10.2.3.2.
Ю.2.3.3.
Ю.2.3.4.
Ю.2.3.5.
10.2.3.6.
595
596
597
599
601
602
602
602
602
604
606
609
611
612
614
616
616
618
619
621
623
623
625
627
629
633
634
Методы получения фосфинов
Синтез фосфинов с помощью металлорганических реагентов
Синтез фосфинов из металлированных фосфинов
Реакции восстановления
Присоединение фосфинов со связью Р—Н к алкенам
Прочие методы получения фосфинов
Синтез оптически активных фосфинов
Синтез полидентатных фосфорных лигандов
Свойства фосфинов
Основность фосфинов
Нуклеофильность фосфинов
Спектроскопия фосфинов
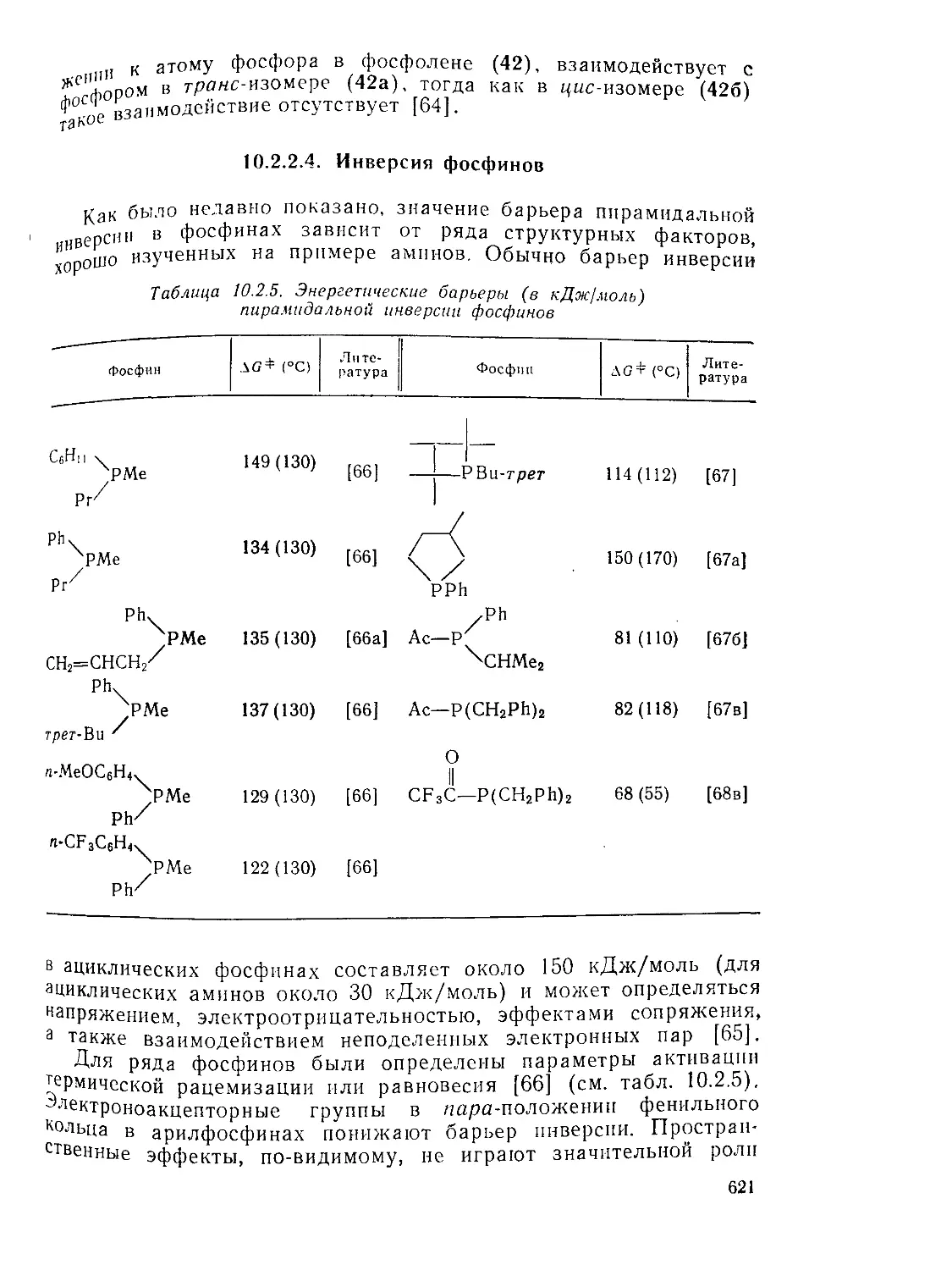
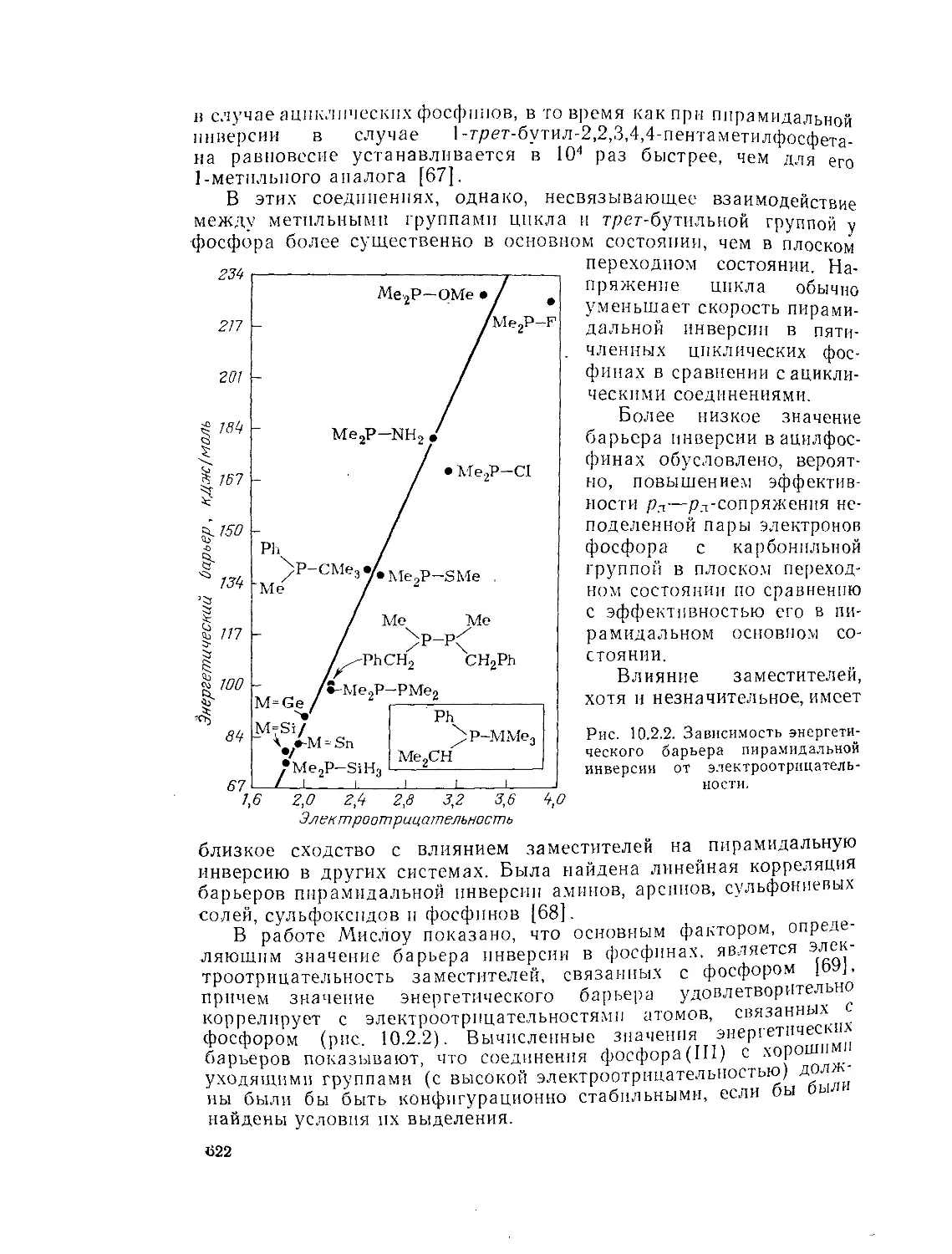
Инверсия фосфинов
Реакции фосфинов
Нуклеофильное замещение при насыщенном атоме углерода
Нуклеофильное замещение в активированных алкенах и ацетиле-нах
Нуклеофильное присоединение к карбонильным соединениям нуклеофильная атака на галоген
Нуклеофильные реакции с кислородом и серой прочие реакции фосфинов
9
10.2.4. Полифосфины 63$
10.2.5. Методы получения фосфониевых солей 63§
10.2.6. Реакции фосфониевых солей 642
10.2.6.1. Щелочной гидролиз 643
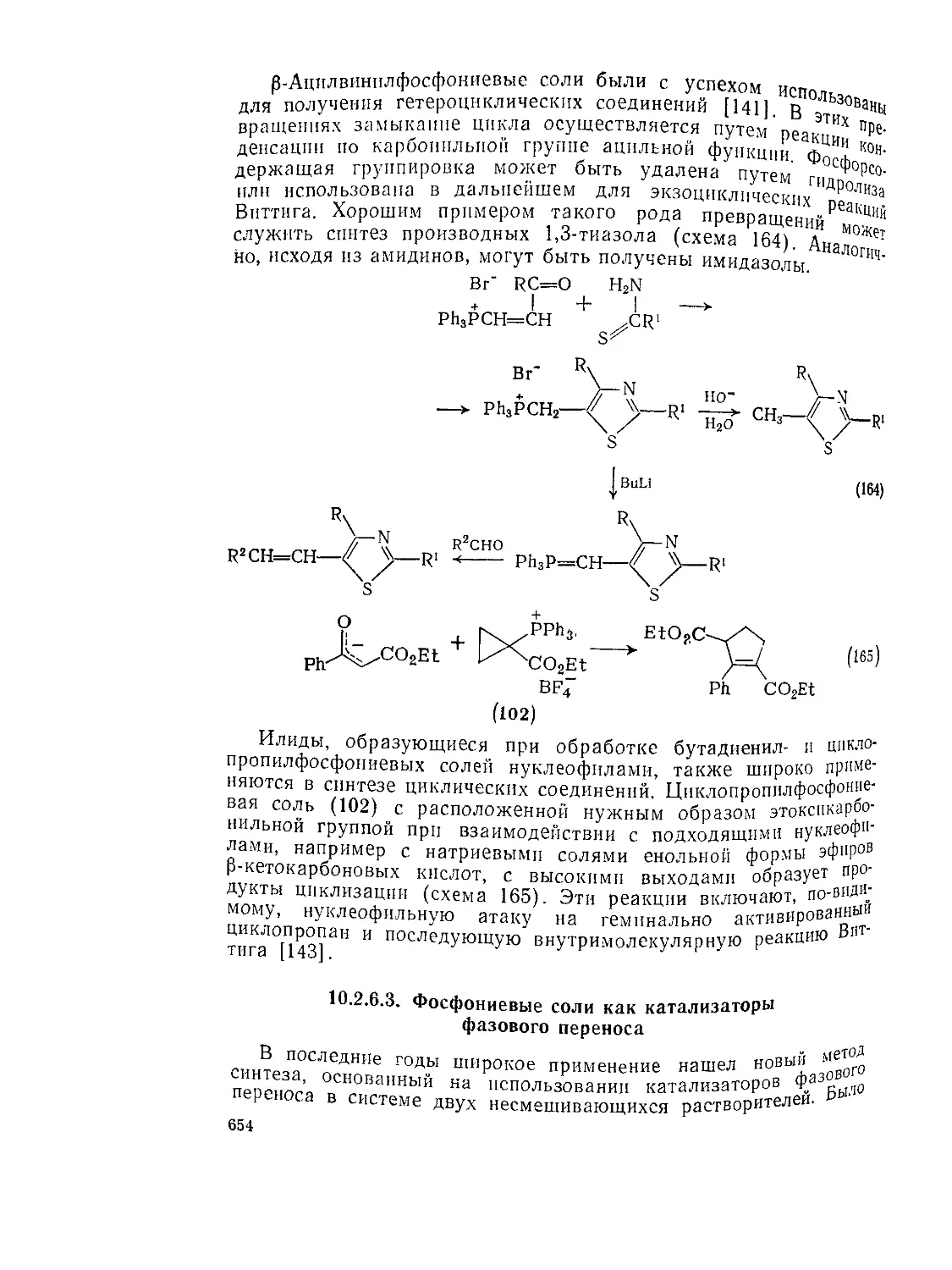
10.2.6.2. Присоединение к винилфосфониевым солям 652
10.2.6.3. Фосфониевые соли как катализаторы фазового переноса 654
10.2.7. Методы получения галогенфосфинов 655
10.2.7.1. Синтез галогенфосфинов из галогенидов фосфора и металлоргани-
ческих соединений 656
10.2.7.2. Синтез галогенфосфинов из ароматических соединений и галоге
нидов фосфора (111) 657
10.2.7.3. Синтез галогенфосфинов из производных Кислот фосфора 657
10.2.7.4. Галогенирование фосфинов 658
10.2.7.5. Реакции обмена галогена 658
10.2.8. Реакции галогенфосфинов 659
10.2.8.1. Нуклеофильная атака по атому фосфора 659
10.2.8.2. Электрофильная атака по атому фосфора 661
10.2.8.3. Бифильные реакции 662
Литература 666
10.3. Производные фосфористой кислоты. Р. С. Эдмундсон 670
10.3.1. Галоген- и азотсодержащие производные фосфористой кислоты 670
10.3.1.1. Синтез галогенфосфитов реакцией тригалогенидов фосфора со
спиртами, фенолами и их тпоаналогами 670
10.3.1.2. Другие пути синтеза галогенфосфитов 673
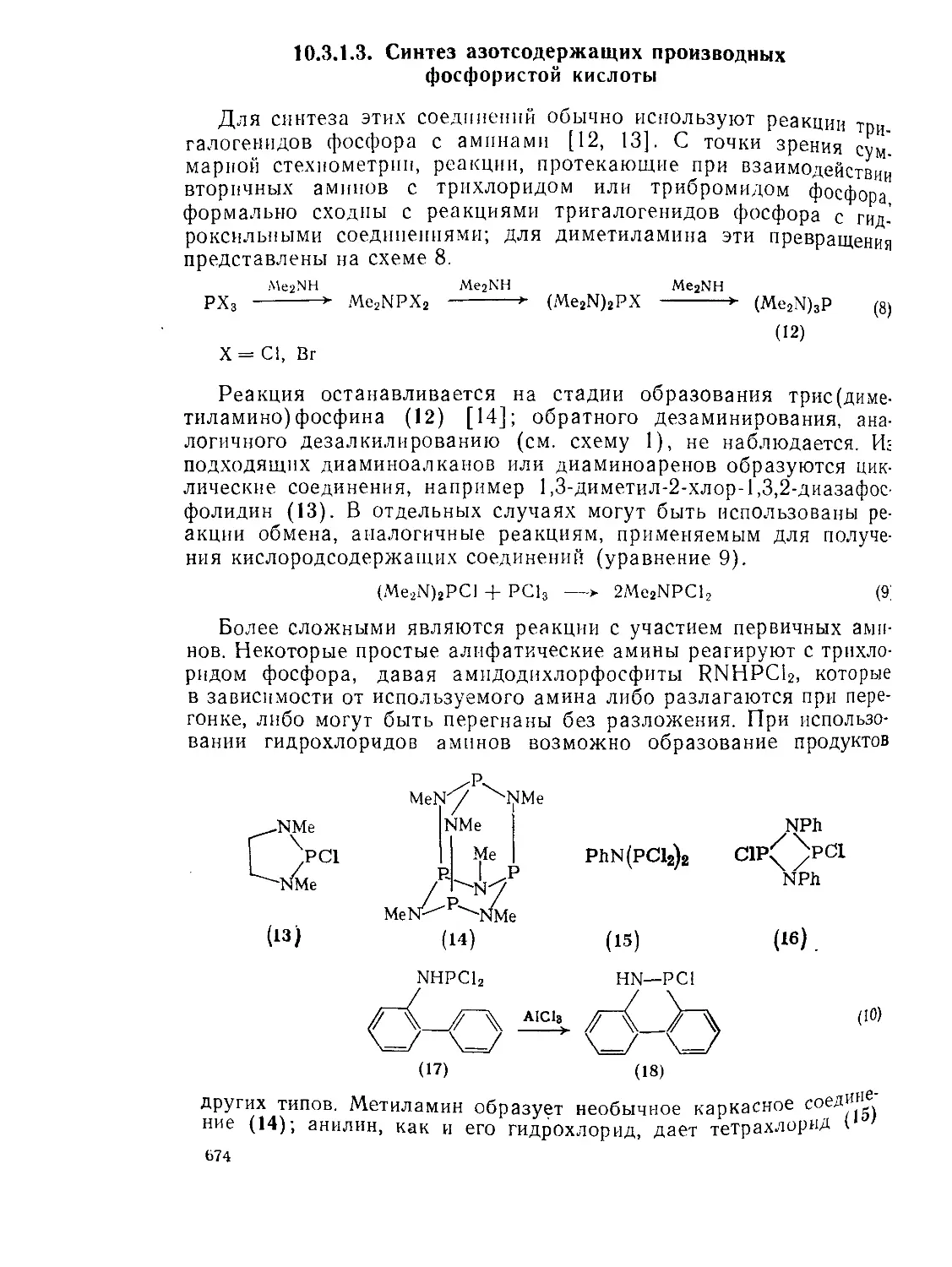
10.3.1.3. Синтез азотсодержащих производных фосфористой кислоты 674
10.3.1.4. Свойства связи фосфор(Ш) —галоген 675
10.3.1.5. Свойства связи фосфор(Ш) —азот 677
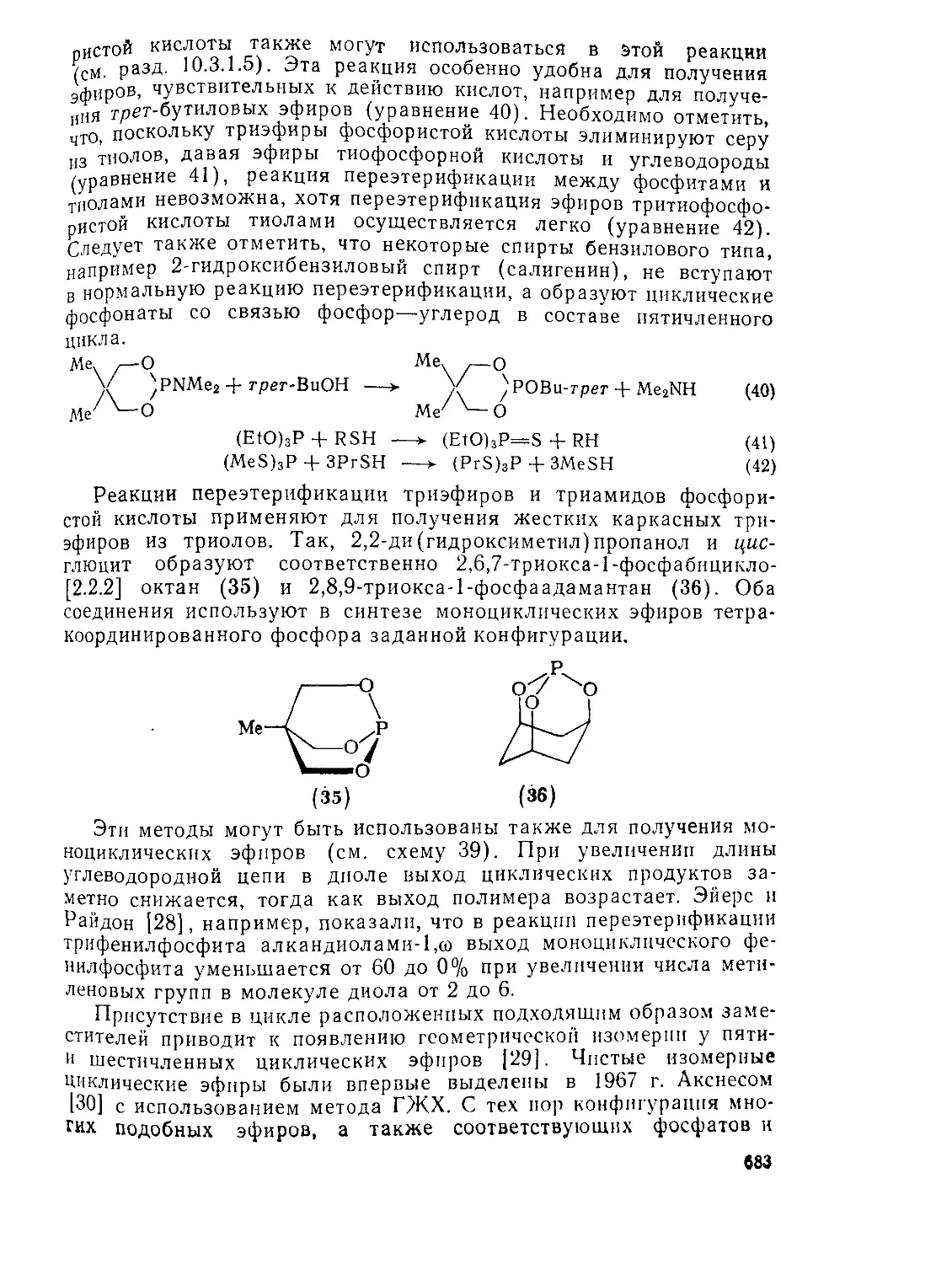
10.3.2. Эфиры фосфористой кислоты 681
10.3.2.1. Электрофильность фосфора в синтезе триэфиров фосфористой кис
лоты 682
10.3.2.2. Электрофильность фосфора в реакциях триэфнров фосфористой
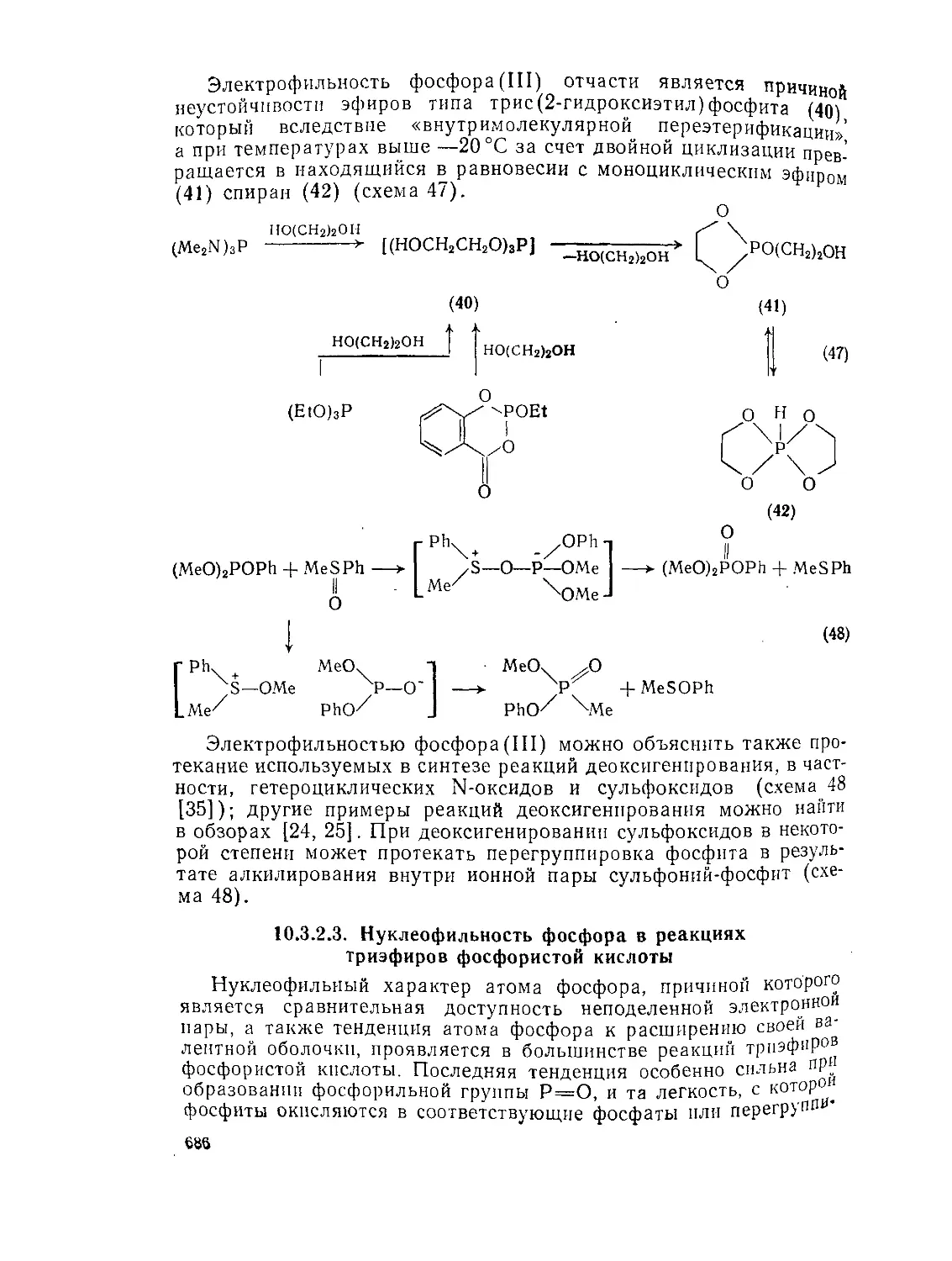
кислоты 684
10.3.2.3. Нуклеофильность фосфора в реакциях триэфиров фосфористой
кислоты 686
10.3.2.4. Вторичные фосфиты 709
10.3.2.5. Сравнительная реакционная способность ациклических и цикличе
10.3.3. ских эфиров и других производных фосфористой кислоты 711
Фосфоиистая и фосфинистая кислоты и их производные 712
Литература 715
Предметный указатель 719
ЧАСТЬ 9.
КАРБОНОВЫЕ КИСЛОТЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ
9.1. КАРБОНОВЫЕ КИСЛОТЫ*
Е. В. Колвин {University of Glasgow)
Данная глава книги посвящена синтезу, физическим свойствам п реакциям простых монофункциональных карбоновых кислот. При рассмотрении первого и последнего вопроса, разд. 9.1.1, 9.1.3 и 9.1.4, были использованы сборники и монографии [1 — 10], которые настоятельно рекомендуются читателю для получения более детальной информации. Была также сделана попытка связать препаративные методы с примерами, взятыми из Organic Syntheses.
9.1.1. МЕТОДЫ ПОЛУЧЕНИЯ КАРБОНОВЫХ КИСЛОТ
9.1.1.1. Окислительные методы
(1) Окисление первичных спиртов и альдегидов
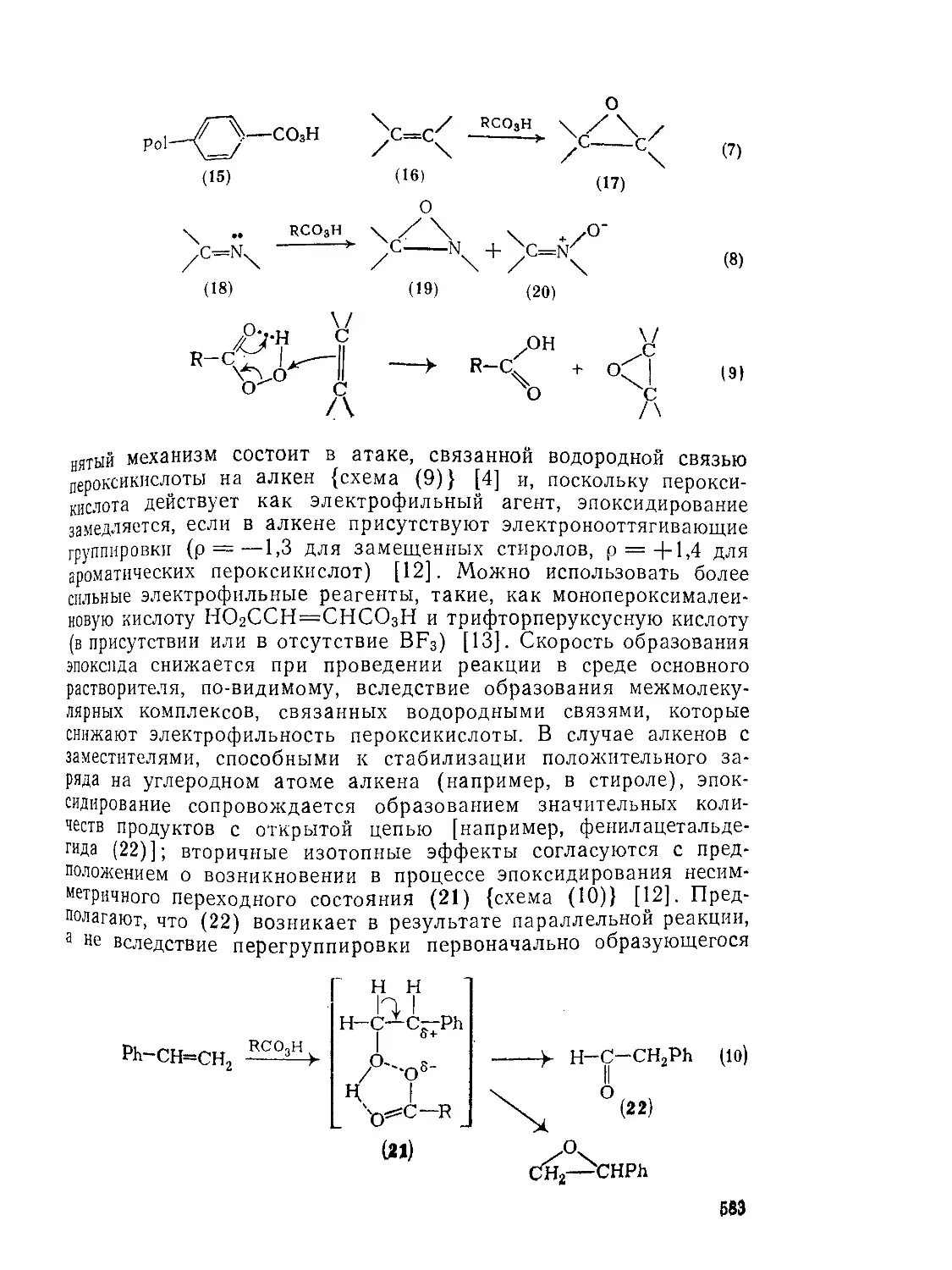
Прямое окисление первичных спиртов до соответствующих кислот может быть осуществлено с помощью хромовой кислоты [И]. Однако эта реакция часто осложняется тем, что альдегид, образующийся в качестве промежуточного соединения, далее окисляется в виде гидрата [12] с относительно небольшой скоростью; кроме того, альдегид и спирт в реакционной смеси могут образовать полуацеталь, который быстро окисляется до сложного эфира {схема (1)}. Образования сложного эфира как побочного продукта окисления можно избежать, если использовать системы диоксид рутения перйодат калия в водном ацетоне [13] или солюбилизован-ныи краун-эфиром перманганат калия в бензоле (так называемый «пурпурный бензол» [14].
Окисление чаще проводят в две стадии, в некоторых случаях выделяя промежуточный альдегид. Для окисления альдегида до
тома* »нг4 русского перевода вошла часть 9 и гл. 10.1-10.3 части 10 второго ома английского издания. — Прим. ред.
11
(1)
он
R—С—ОН I н
R—С
RCH2OH —> R—С
Н
R—С—ОН
I och2r
лэ
R—
X)CH2R
кислоты применяются соединения как хрома, так и марганца, однако наиболее широко используются водные растворы перманганата калия в кислой или щелочной среде {схема (2)} [15]. Среди прочих реагентов, которые могут быть использованы, следует отметить концентрированную азотную кислоту [16] и суспензию оксида серебра [17] в водной щелочи; последний реагент позволяет проводить подобное окисление чрезвычайно мягко и селективно.
КМпО4, H2SO4, Н2О, 20 °с
К-С6Н13СНО ----------------------+ н-С6Н13СООН (2)
(76-78%)
Аллиловые спирты гладко и избирательно окисляются до а,|3-нена-сыщенных кислот с помощью системы цианид ион—оксид се-ребра(П) [18], причем конфигурация двойной связи не затрагивается {схема (3)}. Замена оксида серебра(II) на оксид марганца (IV) в этой системе приводит к образованию сложных эфиров. Этот метод также позволяет проводить окисление как насыщенных, так и а,|3-ненасыщенных альдегидов {схема (4)}.
Альдегиды, в которых отсутствуют а-водородные атомы, под действием сильных оснований вступают в реакцию Канниццаро, давая эквимольные количества кислоты и спирта {схема (5)} [19]. Несмотря на то, что выходы кислот в этой реакции внутримолекулярного окисления-восстановления .не превышают 50%, тем не менее она протекает быстро и достаточно проста.
12
HCN, CN,“. AgO.
HCN. CN . MeOH или ТГФ, H2O
MnO2. MeOH RC00Me RCHO RCOOH (4)
rCHO насыщенный
сопря- или
женный сопряженный
RCH0 ... NaOH' Н2Д- RCH2OH + RCOONa (5)
ОН
NaOH, H2O I
ArCOCOAr -----------*- Аг—C—COONa (6)
Ar
Внутримолекулярный вариант этого процессз прослеживается в перегруппировке ароматических 1,2-дикетонов в бензиловые кислоты [схема (6)].
Озонолиз зцетзлей и простых эфиров тзкже предстзвляется довольно перспективным методом синтезз кислот [21]. Ацетзли альдегидов и тетрагидропираниловые эфиры спиртов глздко окисляются до сложных эфиров, причем в последнем случзе не наблю-дзется образования 6-лактона кзк продукта зльтернзтивного направления рзсщепления, хотя подробные простые эфиры фотохимически [22] преврзщзются в смесь 5-лактона и зциклического сложного эфирз (1) {схемы (7) и (8)}.
Оз
R>CH(OR2)2 -----> RlCOOR2
(2) Окисление кетонов
(7)
(8)
Окисление кетонов до кзрбоновых кислот требует рззрывз связи С—С и поэтому протекзет несколько труднее, чем окисление первичных спиртов или зльдегидов. Тем не менее для проведения этой резкции существует ряд эффективных методов.
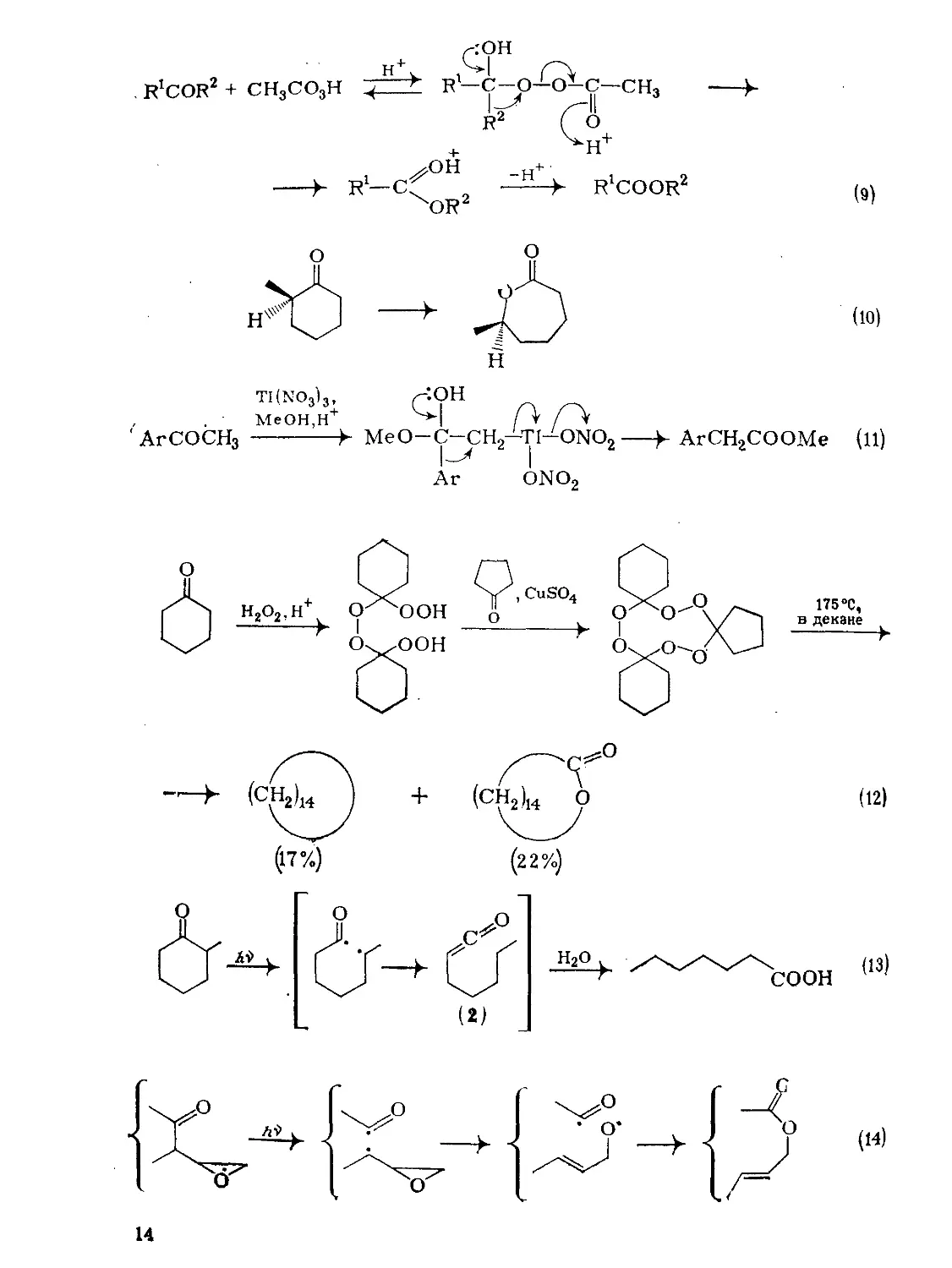
Резкция Бзйерз — Виллигерз [23, 24] с рззличной легкостью идет кзк с зциклическими {схемз (9)}, тзк и с циклическими кетонами {схема (10)}, из которых наиболее реакционноспособны циклобутаноны [25]. Как видно из примера (10), наиболее замещенная из двух а-углеродных групп мигрирует с сохранением стереохимии (см. также разд. 9.8.1).
Арилметилкетоны мягко окисляются до метиловых эфиров арилэтановых кислот нитратом таллия(III) в метаноле в присутствии кислоты {схема (11)} [26],
13
(9)
, R!COR2 +
сн3со3н
-Н
R1—„ ---->- R1COOR2
ХЖ2
'АгСОСНз
TI(NO3)3, МеОН,Н+
----------
г:он
Q|
МеО-С-СН?—TI-ONO.
M l
Ar ONO2
У АгСНгСООМе
(И)
14
Термический распад или фотолиз ди- или трициклоалкилиде-мпных пероксидов, полученных из кетонов, приводит к мопоцпкли-ческим эфирам {схема (12)} [27], и хотя выходы в этом случае умеренны, метод достаточно прост.
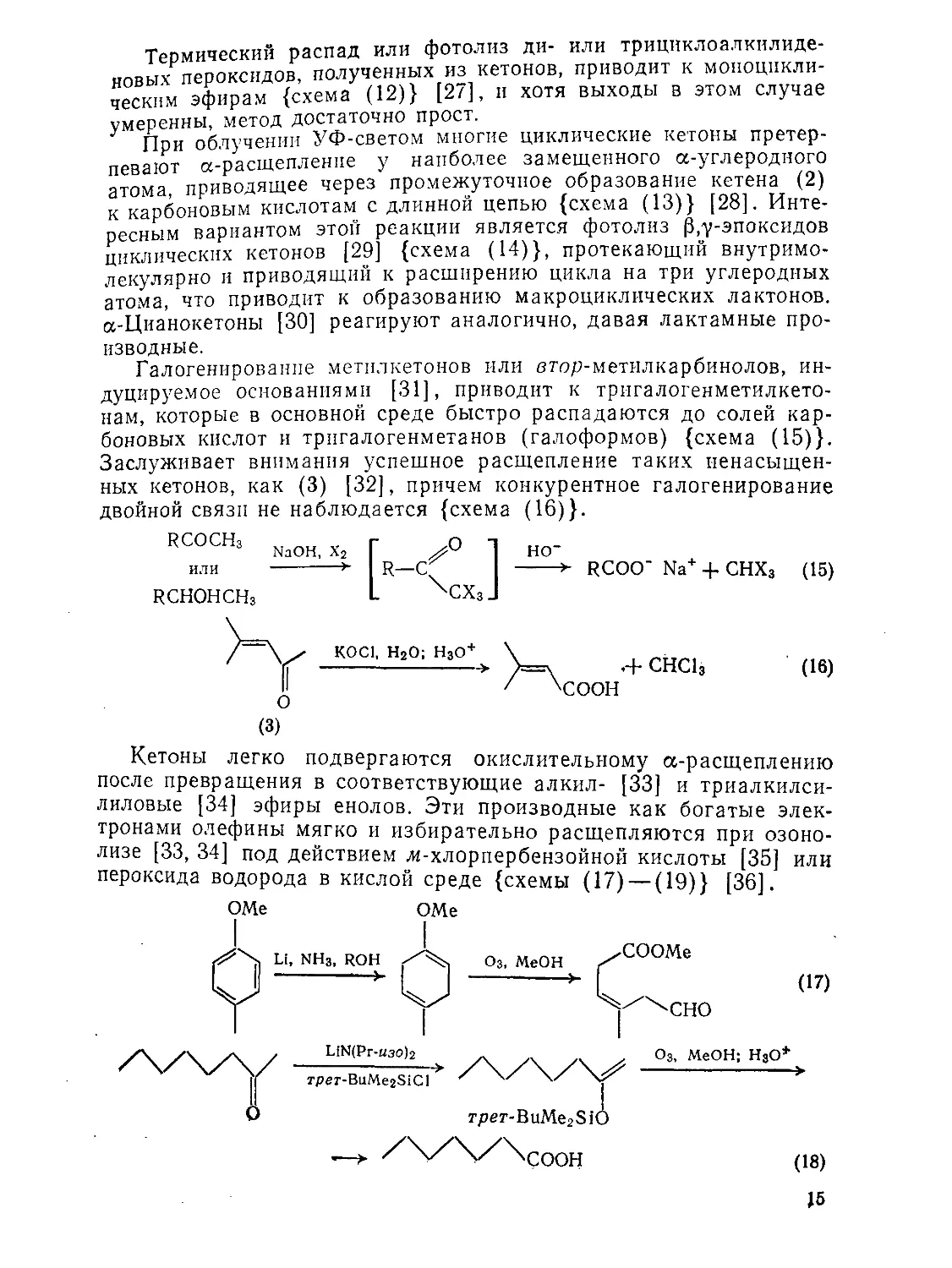
? При облучении УФ-светом многие циклические кетоны претерпевают а-расщепленпе у наиболее замещенного а-углеродного атома приводящее через промежуточное образование кетена (2) к карбоновым кислотам с длинной цепью {схема (13)} [28]. Интересным вариантом этой реакции является фотолиз |3,у-эпоксидов циклических кетонов [29] {схема (14)}, протекающий внутримолекулярно и приводящий к расширению цикла на три углеродных атома, что приводит к образованию макроциклических лактонов. а-Цианокетоны [30] реагируют аналогично, давая лактамные про
изводные.
Галогенирование метплкетонов или етор-метилкарбинолов, индуцируемое основаниями [31], приводит к тригалогенметилкето-нам, которые в основной среде быстро распадаются до солей карбоновых кислот и трпгалогенметанов (галоформов) {схема (15)}. Заслуживает внимания успешное расщепление таких ненасыщенных кетонов, как (3) [32], причем конкурентное галогенирование двойной связи не наблюдается {схема (16)}.
RCOCH3
или
RCHOHCH3
NaOH, Х2 ------->
НО"
---> RCOO' Na+ + СНХз (15)
KOC1, Н2О; Н3О+
СООН
.+ CHCI3
(16)
Кетоны легко подвергаются окислительному а-расщеплению после превращения в соответствующие алкил- [33] и триалкилсилиловые [34] эфиры енолов. Эти производные как богатые электронами олефины мягко и избирательно расщепляются при озонолизе [33, 34] под действием .м-хлорпербензойной кислоты [35] или пероксида водорода в кислой среде {схемы (17) —(19)} [36].
СООН
(18)
15
о
---? силиловых эфиров — Виллигера, причем в
Необходимо подчеркнуть, что использование енотов дополняет перегруппировку Байера этом случае кетон расщепляется с наименее замещенной стороны чеоез кинетически генерируемый енолят-анион.
При взаимодействии силиловых эфиров циклических енолов с аренсульфонилазидами получаются карбоновые кислоты и одновременно происходит сужение цикла [37] {схема (20)}. По-видимому, окислительное расщепление циклических кетонов, приводящее к кетокислотам [38] или двухосновным кислотам [39], также протекает через образование соответствующих енолов.
/OSiMe3 - /OSiMe3 ^СООН
ArSO2N3
(СН2)Я
. \ \'NSO2Ar \
(СН2)„ (СН2)„
(20)
Аддукт кислорода и кобальтоцена [40] вызывает мягкое расщепление а-дикетонов и о-хинонов {схема (21)}.
RCOCOR + [(n-C5H5)2Co]202 —> 2[(л-С5Н5)2Со]+ RCOO’ (21)
HCl Me2SO4
2RCOOH <-------1-------> 2RCOOMe
Кетоны, не дающие енолов, расщепляются под действием сильных оснований [41, 42] до эквимольных количеств углеводорода п соли карбоновой кислоты (реакция Галлера — Бауэра), однако практическое применение этого метода часто ограничено неоднозначностью расщепления. Кетоны, и особенно аралкилкетоны, под действием серы и амина претерпевают примечательную терминаль-КИСпИТеЛЬНуЮ пегРегРУппиРовку. приводящую к амидам на схемах t43D, типичный пример которой показан
s, nhr2
АгСОСНз--------> ArCH2CONR2 (22)
АгСО(СН2)2СН3 —----Ar(CH2)3CONR2 (23)
а-Галогенкетоны в ппиемтсттшы
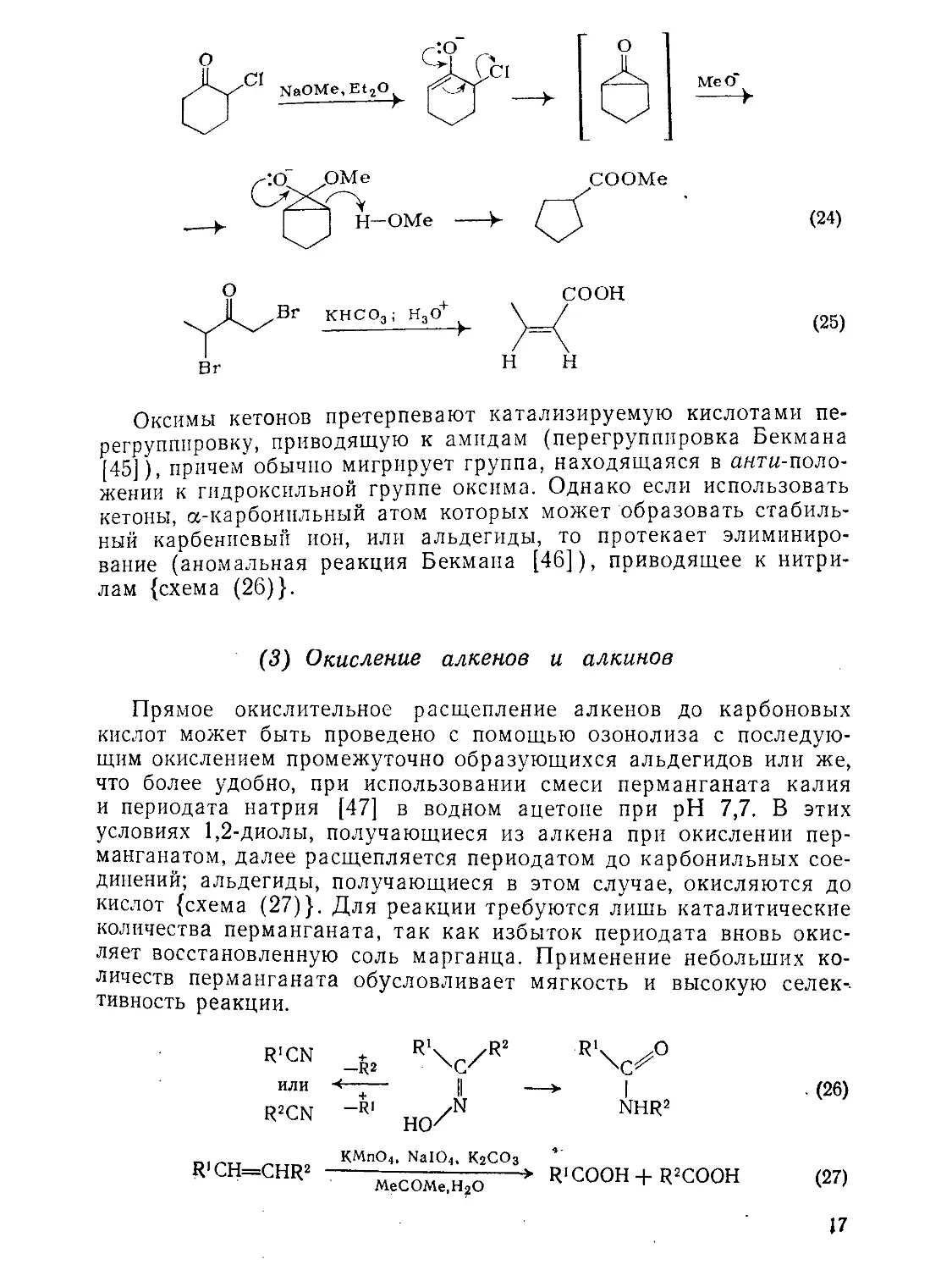
ваются в кислоты присутствии основания перегруппировы-(25)}; см. также раздР дГРу1ппиРовка Фаворского) [44] {схемы (24), 16
Me О* ----►
(24)
соон
(25)
Оксимы кетонов претерпевают катализируемую кислотами перегруппировку, приводящую к амидам (перегруппировка Бекмана [45]), причем обычно мигрирует группа, находящаяся в анти-иоло-женин к гидроксильной группе оксима. Однако если использовать кетоны, а-карбонпльный атом которых может образовать стабильный карбениевый ион, или альдегиды, то протекает элиминирование (аномальная реакция Бекмана [46]), приводящее к нитрилам {схема (26)}.
(3) Окисление алкенов и алкинов
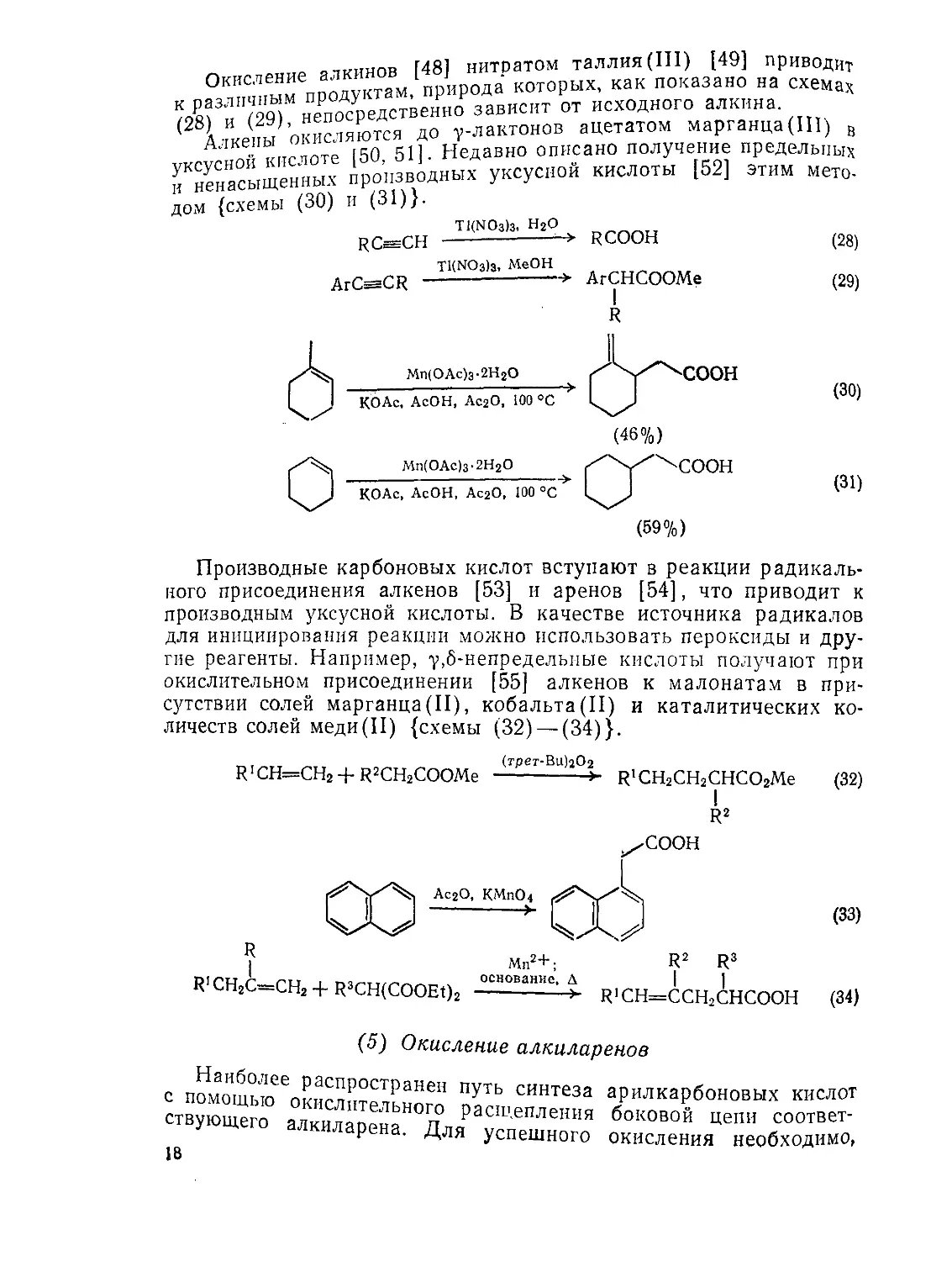
Прямое окислительное расщепление алкенов до карбоновых кислот может быть проведено с помощью озонолиза с последующим окислением промежуточно образующихся альдегидов или же, что более удобно, при использовании смеси перманганата калия и перйодата натрия [47] в водном ацетоне при pH 7,7. В этих условиях 1,2-диолы, получающиеся из алкена при окислении перманганатом, далее расщепляется перйодатом до карбонильных соединений; альдегиды, получающиеся в этом случае, окисляются до кислот {схема (27)}. Для реакции требуются лишь каталитические количества перманганата, так как избыток перйодата вновь окисляет восстановленную соль марганца. Применение небольших количеств перманганата обусловливает мягкость и высокую селективность реакции.
R’CN
ИЛИ
R2CN
R'CH=CHR2
-R2 ч----
+
-R1
R\ /R2
KMnO4, NaIO4, K2CO3 MeCOMe.HgO
I NHR2
R'COOH-f- R2COOH
(26)
(27)
17
„ oTvimnB Г481 нитратом таллия (III) [49] приводит ми™ продуктам, природа которых, как показано на схемах Ж У <29 непосредственно зависит от исходного алкина ( Дзкепы окисляются до у-лактонов ацетатом марганца (III) „ 5 ; пейоте (50 51]. Недавно описано получение предельных ^ненасыщенных производных уксусной кислоты [52] этим мето-дом {схемы (30) И (31)}.
Т1(Ы0з)з. н2о RChsCH ---------------
RCOOH
(28)
T1(NO3)3, МеОН
ArQsCR ’
(29)
Мп(ОАс)з-2Н2О -------------------------> КОАс, АсОН, Ас2О, 100 °C
АгСНСООМе I R
|^уХ'х<ООН
(30)
Мп(ОАс)з-2Н2О
КОАс, АсОН, Ас2О, 100 °C
(31)
(59%)
Производные карбоновых кислот вступают в реакции радикального присоединения алкенов [53] и аренов [54], что приводит к производным уксусной кислоты. В качестве источника радикалов для инициирования реакции можно использовать пероксиды и другие реагенты. Например, у,6-непредельные кислоты получают при окислительном присоединении [55] алкенов к малонатам в присутствии солей марганца(II), кобальта(II) и каталитических количеств солей меди(II) {схемы (32) —(34)}.
, (трет-BuJoOo
RICH=CH2 + R2CH2COOMe -------------> R1CH2CH2CHCO2Me
(32)
R2
Ас2О, КМпО4
R 24-
I Мп2+;
R>CH2C=CH2 + R3CH(COOEt)2 °СН0ВаНИе> Л
R2
(33)
R3
> R'CH=CCH2CHCOOH
(34)
(5) Окисление алкиларенов
с помощьюееокислп?ельногп пУТЬ синтеза аРилкарбоновых кислот ствующего алкиларена л рас1цепления боковой цепи соответ-лкиларена. Для успешного окисления необходимо,
18
чтобы ароматические кольца не содержали гидрокси- или аминогрупп, а также чтобы была хотя бы одна бензильная связь С—Н. Обычные окислители в этом случае: водный раствор перманганата калия, хромовая или азотная кислота [11 j.
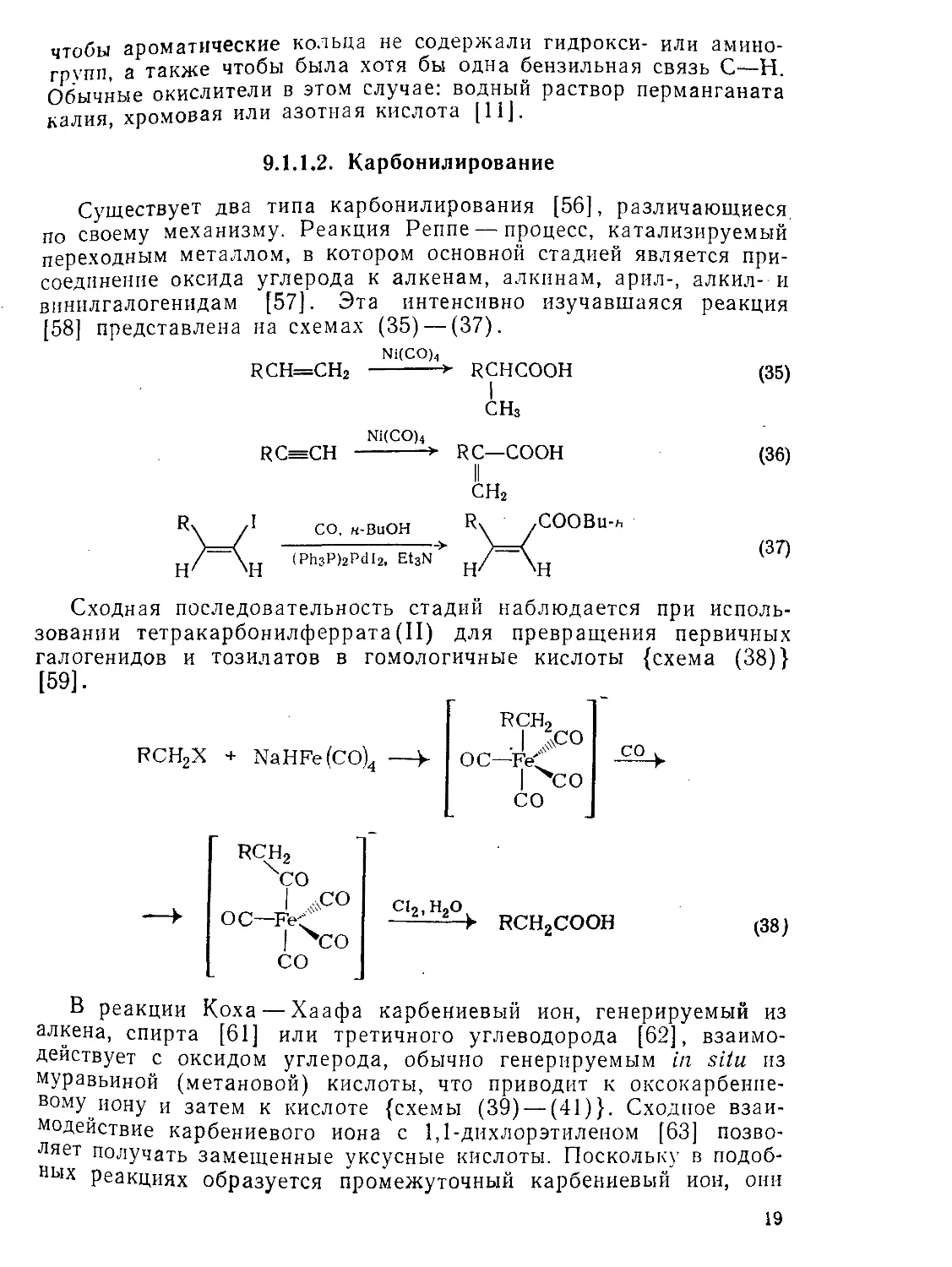
9.1.1.2. Карбонилирование
Существует два типа карбонилирования [56], различающиеся, по своему механизму. Реакция Реппе — процесс, катализируемый переходным металлом, в котором основной стадией является присоединение оксида углерода к алкенам, алкинам, арил-, алкил- и винилгалогенидам [57]. Эта интенсивно изучавшаяся реакция [58] представлена на схемах (35)—(37).
Ni(CO)4
RCH=CH2 ---------* RCHCOOH (35)
СНз
Ni(CO)4
RfeCH --------> RC—COOH (36)
II сн2
CO, н-BuOH
(РЬзР)2Р<112, EtsN
Сходная последовательность стадий наблюдается при использовании тетракарбонилферрата(II) для превращения первичных галогенидов и тозилатов в гомологичные кислоты {схема (38)} [59].
РСН2Х + NaHFe(CO)4 —>-
rch2 . I ОС—FeC
со
СО
RCH2 хсо
I Асо ос—Fe<"
сь.н-о
— RCH2COOH
(38)
В реакции Коха — Хаафа карбениевый ион, генерируемый из алкена, спирта [61] или третичного углеводорода [62], взаимодействует с оксидом углерода, обычно генерируемым in situ из муравьиной (метановой) кислоты, что приводит к оксокарбенпе-вому^иону и затем к кислоте {схемы (39) — (41)}. Сходное взаимодействие карбениевого иона с 1,1-дихлорэтиленом [63] позволяет получать замещенные уксусные кислоты. Поскольку в подоб-НЬ1Х Реакциях образуется промежуточный карбениевый ион, они
19
более подходят для синтеза третичных [64j и разветвленных, а не линейных кислот. В том случае, если карбениевый ион относи-тельно стабилен, реакция заметно обратима.
—)—ОН + СН2=СС12
H2SO4, BF3 ---------->
Н2О
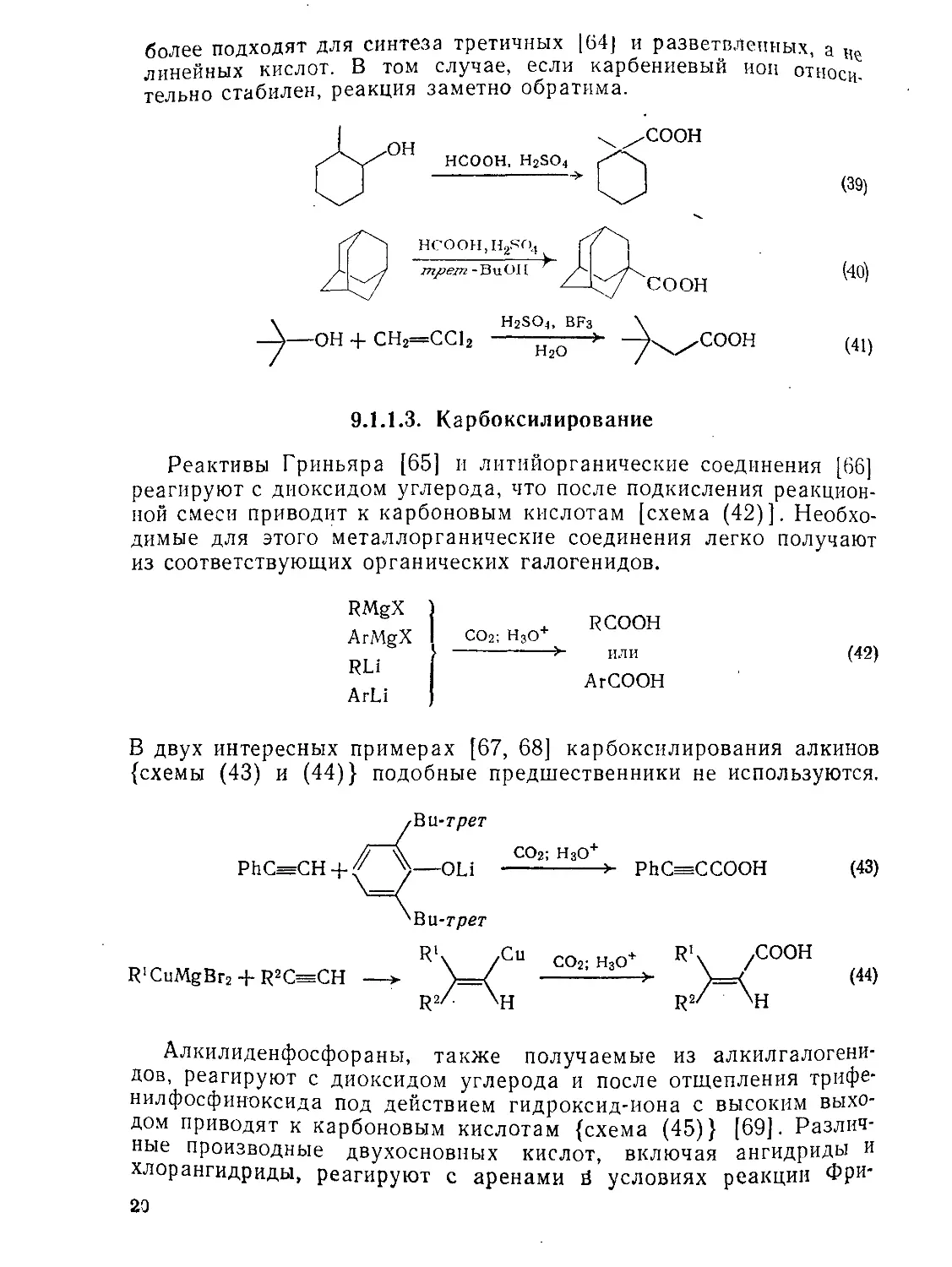
9.1.1.3. Карбоксилирование
Реактивы Гриньяра [65] и литийорганические соединения [66] реагируют с диоксидом углерода, что после подкисления реакционной смеси приводит к карбоновым кислотам [схема (42)]. Необходимые для этого металлорганические соединения легко получают из соответствующих органических галогенидов.
RMgX ArMgX RLi ArLi
+ RCOOH
CO2; H3O+ -------->- ИЛИ
ArCOOH
(42)
В двух интересных примерах [67, 68] карбоксилирования алкинов {схемы (43) и (44)} подобные предшественники не используются.
PhC=CH
Ви-трет
—OLi
Ви-трет
СО2; Н3О+ -------> РЬСе=ССООН
(43)
R'CuMgBr2 + R2CssCH —>
СО2; Н3О+ ---------->
Алкилиденфосфораны, также получаемые из алкилгалогени-дов, реагируют с диоксидом углерода и после отщепления трифе-нилфосфиноксида под действием гидроксид-иона с высоким выходом приводят к карбоновым кислотам {схема (45)} [69]. Различные производные двухосновных кислот, включая ангидриды и хлорангидриды, реагируют с аренами й условиях реакции Фри-23
еля__Крафтса [70]. После восстановления кетогруппы получают
ареналкановые кислоты [схема (46)].
Rk со2
xC=PPh3 ----
R2/
R> HO-; H2O; H3O+ Rl\
\C—PPh3----------------- RCHCOOH (45)
R*/| _ R2/
о
К AICI3; Zn(Hg), HCI
ArH + Ozj:(CH2)n —----------------- Ar(CH2)n+1COOH (46)
о
9.1.1.4. Гидролитические методы
По уменьшению скорости гидролиза простейшие производные кислот можно расположить в следующий ряд: хлорангидриды > > ангидриды > сложные эфиры>амиды>нитрилы {схема (47)}. Только ангидриды и хлорангидриды кислот гидролизуются самопроизвольно и нацело, в большинстве случаев со скоростью, зависящей от их растворимости в водной среде. Однако указанные соединения нельзя рассматривать как исходные соединения для получения карбоновых кислот, так как сами ангидриды и хлорангидриды обычно получают из кислот.
Сложные эфиры и амиды [71] медленно реагируют с водой; в кислых условиях реакция протекает быстрее и приводит к равновесной смеси.
H2o MOH
RCOOH + HX RC/ -----------> RCOO'M+ + HX
^X
(47)
Напротив, гидролиз в щелочной среде проходит до конца, и после подкисления реакционной смеси кислота легко выделяется.
Далее мы рассмотрим только современные методы мягкого и избирательного расщепления амидов и сложных эфиров (см. также разд. 9.8.3 и 9.9.3).
Метиловые эфиры можно избирательно расщепить в присутствии других сложных эфиров, амидов и др. с помощью различных реагентов. Механизм этого довольно редкого типа расщепления [72] О-алкильной связи включает Sn2 атаку нуклеофила на углерод метильной группы с карбоксилат-анионом в качестве уходящей группы. Для проведения этой реакции применяют иодид лития в коллидине [73] или демитилформамиде [74, 75], этантиолат [76] ли пропантиолат лития [77] в гексаметилфосфортриамиде (гек-TD~e), цианиД натрия [78] во влажном гексаметаполе или особ°РИД б°Ра в дихлорметане [79]. Два последних реагента енно удобны для расщепления стерически затрудненных
21
метиловых эфиров {схемы (48) и (49)}; для той же цели пр„ ниют также |(5-диазабицикло[4.3.0]нонен-5 или 1,5-диазабицщ
ло[5.4.0]уидеиеи-5 [80].
П -----h RCOO + CH3-NU
О—СН3^ Nu
СООМе
4- PhCOOEt
NaCN, гексаметапол, НгО;
Н3О+
+ PhCOOEt
(93%)
(85%)
(49)
Расщепление сложных эфиров под действием триметилсилил-иодида [81] протекает по аналогичному механизму и приводит к лабильным силиловым эфирам, которые после расщепления дают кислоты {схема (50)}. Этот метод позволяет избирательно расщеплять сложные трет-бутиловые и бензиловые эфиры, однако он не селективен для простейших сложных эфиров.
/°
R С + Me3SiI у .
SiMe,
I
X)
г
^OR2
,OSiMe3
> R'c^ + R2I (50)
Амиды обычно устойчивы к гидролизу, однако недавно появилось несколько мягких методов их расщепления. В увлекательном обсуждении последних стадий синтеза витамина В12 Вудворд [82] продемонстрировал «дьявольски умный» метод избирательного расщепления амидов в присутствии сложных эфиров. Эшенмозер и сотр. при разработке схемы синтеза применяли высокоэлектрофильный катион (4), получаемый из а-хлорнитрона {схема (51)}-Для той же цели Вудворд рекомендовал использование N2O4 в тетрахлориде углерода в присутствии ацетата натрия.
22
MarvnP пасшепление первичных и вторичных амидов [83] в й„»и о > сред” происходит при взаимодействии ампд-аиионов с ш.суТьфМом углерода {схема (52)). В свою очередь, третичные Д У„« Гпялко гидролизуются при комнатной температуре в двух-Фазной системе. содержащей трет-бутилат калия [84] {схема (53)). Дая мягкого гидролиза амидов рекомендуют применять высоконуклеофильный гидроперокси-анион [85].
, NaH; CS2
2-------
XNHR
KOBu-rper, H2O, Et2O -----------> комнатная темп.
У R^OS
(52)
R2NCS
.О RC^
XNR]
O'
R—C—NR]
OH
RCOO' + R^NH
O’
R—C—NR]
O'
(53)
Нитрилы широко используются как исходные соединения для получения карбоновых кислот, поскольку сами легко получаются из доступных алкилгалогенидов и тозилатов по реакции нуклеофильного замещения.
Амиды легко получаются из нитрилов при действии уксусной кислоты в присутствии хлорида титана (IV) [86] и других мягких реагентов [87, 88].
9.1.1.5. Синтезы из малоновой и ацетоуксусной кислот и родственных соединений
Применение этих соединений для синтеза карбоновых кислот основывается на двух свойствах, наиболее характерных для подобных систем: на относительной легкости образования аниона (и соответственно взаимодействия с электрофилами), а также на простоте декарбоксилирования и (или) расщепления. Однако здесь мы рассмотрим этот метод лишь в общих чертах, так как он хорошо освещен в литературе [1, 3].
Диэтиловый эфир малоновой кислоты (диэтилмалонат) в виде аниона легко алкилируется различными электрофильными реагентами {схема (54)}. Реакция диэтилмалоната с карбонильными оединениями (реакция Кневенагеля [89]) приводит к аф-ненасы-ния*~1М эФиРам Двухосновных кислот и после декарбоксилпрова-вой К одноосновным кислотам. Использование самой малоно-кислоты (реакция Дебнера) [90] позволяет сразу получать
23
IOU„.IP кислоты. Монозамещенные малоновые кислоты
^"алкилировать в виде трианионов |9И первичными галогенида*
RICOR2, .COOEt пиперидин
R’
HO", H2O;
COOEt н3о+, Д
R’
^COOEt
R2'
COOEt
R2,
.COOEt
!—\
'COOEt
HO", H2O; H3o+, A ,_ - -------—
ДМСО, H2O
R3CH2COOH
R3CH2COOEt
(54)
R
I NaOEt;
R3
R4
COOEt
COOEt
HO-, H2O; H3o+, A
ДМСО, H2O
R3R4CHCOOH
R3R4CHCOOEt
л-Аллилпалладиевые комплексы реагируют с анионами производных малоновой кислоты как электрофильные алкилирующие агенты. Если в этом случае использовать хиральные лиганды, то это приводит к продуктам алкилирования с асимметрическим центром, причем оптический выход может достигать 24% {схема (55)} [92].
NaCH(COOEi)2, хиральный лиганд
EtOOC COOEt
(5э)
Замещенные малоновые кислоты легко декарбоксилируются при кислотном катализе или при нагревании; гидролиз приводит к замещенным уксусным кислотам. Сходный процесс деалкоксикарбонилирования эфиров малоновых кислот и родственных соединений ускоряется под действием хлорида' натрия в диметилсульфоксиде (ДМСО) [93], причем применение соли не всегда обязательно [94], поскольку влажный ДМСО является прекрасной средой для этой реакции. Дизамещенные эфиры малоновой кислоты деал-коксикарбонилируются натрием в присутствии триметилсилилхло-рида. В результате реакции получается алкилсилилацеталь кетена (5), который легко гидролизуется до кислоты [95].
R\ zOSiMe3
R' 'ОМе (5)
/S°2Ph _О-снован"е-- RX: ySO2Ph МеО£Щ1РО4 м ,5б)
\гппм R—\---------------------* RCH2COOMe (56)
С00Ме ЧООМе
(6)
24
а-Сульфоновый эфир (6), по-видимому, является более удобным реагентом для получения эфиров замещенной уксусной кислоты Г961 чем диэтилмалонат, так как восстановительное десульфирование протекает в мягких буферных средах {схема (56)}.
Этиловый эфир ацетоуксусной кислоты (этил-3-оксобутаноат) нашел относительно небольшое применение для получения кислот {схема (57)} по двум причинам. Во-первых, довольно часто нельзя быть уверенным в региоспецифичности С-алкилирования [97] амбидентного аниона (в случае диэтилмалоната такая проблема не возникает), и во-вторых, что более важно, дальнейшее расщепление продукта алкилирования до кислоты часто сопровождается гидролизом сложного эфира с последующим декарбоксилированием до кетона. В связи с этим эфиры ацетоуксусной кислоты в основном используются для синтеза кетонов.
NaOEt; RIX COOEt
о
COOEt сильная НО~ ------------> AcOH+RjCH2COOH
(57)
I NaOEt; ]R2X
сильная HO~
----------->
AcOH-f- R'R2CHCOOH
Взаимодействие этилового эфира ацетоуксусной кислоты с изо* пропенилацетатом в кислых условиях всегда приводит к (Z)-ацетату енола, однако если реакцию проводить в триэтиламине в присутствии ацетилхлорида и гексаметапола, то получается (f)-изомер. Это объясняют образованием (Z)-енола и отделенного растворителем (£)-енолят-аниона. Последующее взаимодействие с диалкиллитийкупратом приводит с очень высокой стереоселективностью [98] к а,р-ненасыщенным сложным эфирам {схема (58)}.
Me АсО R
СН2=СНОАс, Н+ I LiCuR2 I
-----------^/%/СООМе -----------^^А^/СОСШе
О
I AcCl, EtsN
’ ----—-------- АсО
гексаметапол
(68)
В качестве высокоэффективного метода восстановления 3-оксо-вкНК1я^И РекоменД°вано «двойное восстановление» метоксиметило* эфиров енольной формы эфиров р-кетокислот (7) [99]; обратка литием в аммиаке приводит в результате восстановления
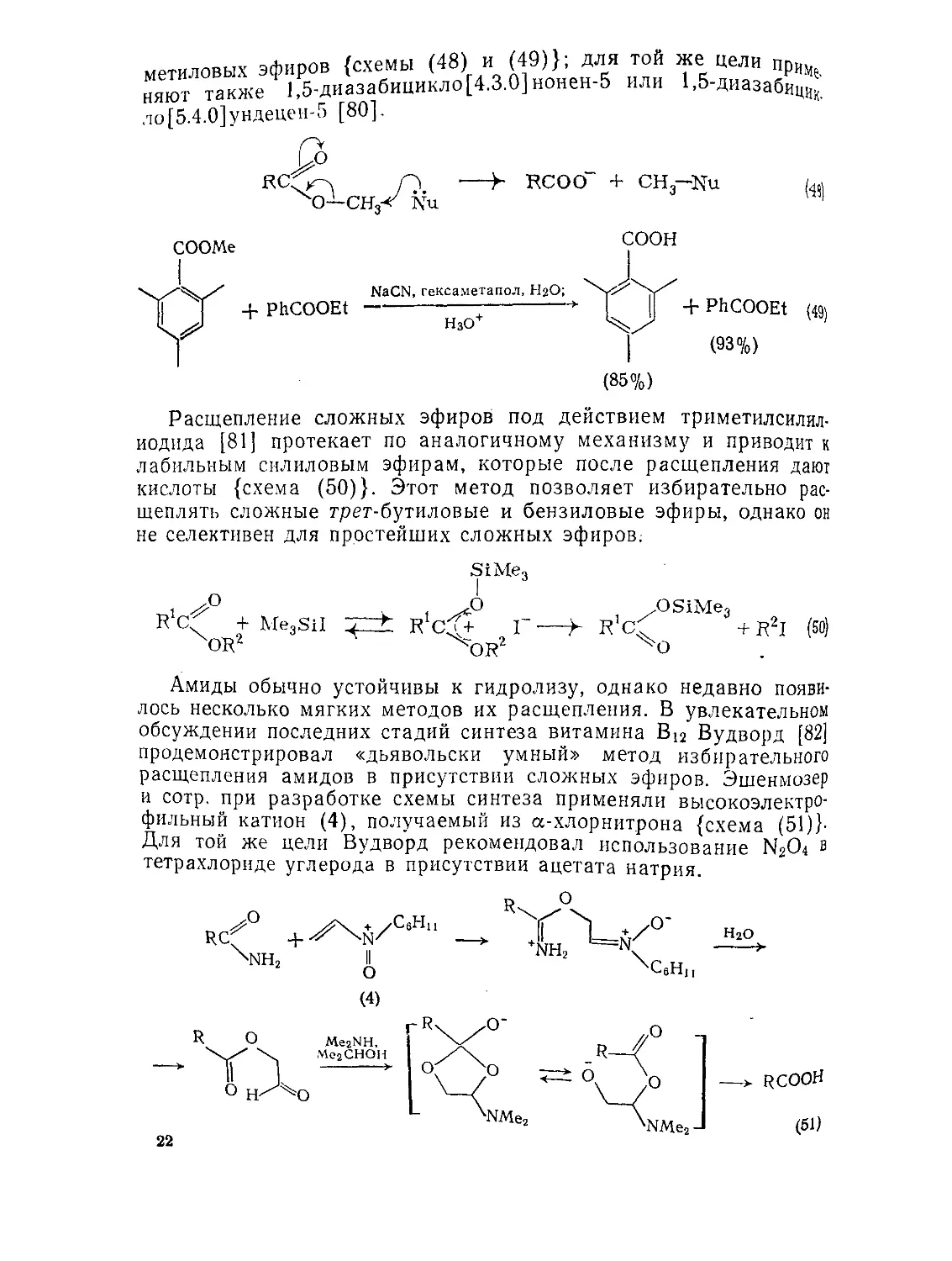
сопряженной двойной связи, отщепления метоксиметанола и дальнейшего восстановления к насыщенному сложному эфиру (8)_ {схема (59)}. Та же процедура столь же эффективна и для самих 3-оксокислот [100].
^^COOR (59)
/Ч/COOR NH3
(7) (8)
С-Алкилирование циклогександиона-1,3 с последующим расщеплением приводит к 5-оксокислотам, которые, в свою очередь, могут быть восстановлены в кислоты с длинной цепью [101]. Альтернативно, расщепление алкилированного циклогександиона, проводимое в условиях восстановления по Кижнеру — Вольфу, приводит непосредственно к насыщенной кислоте {схема (60)}.
! HG1
О О о
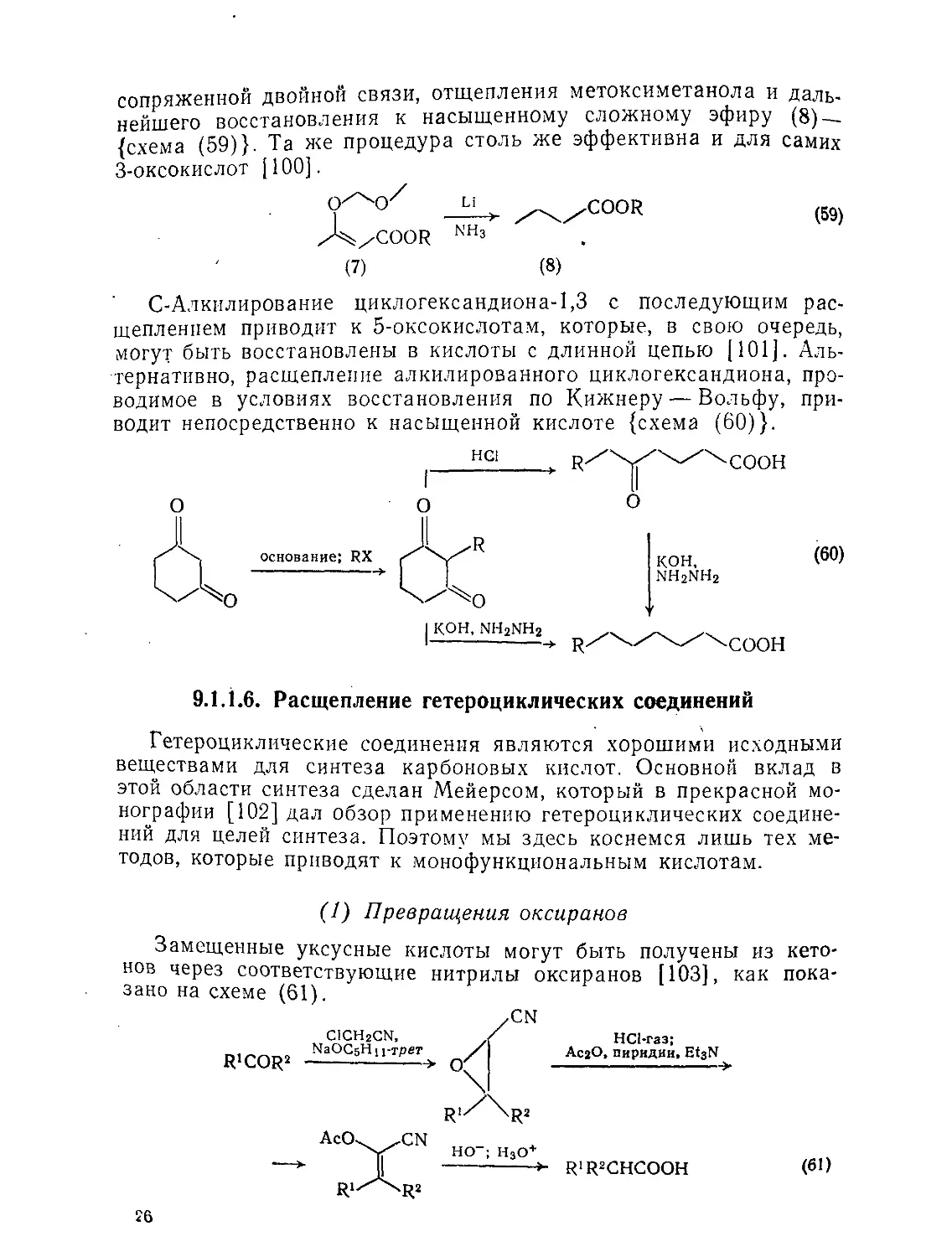
9.1.1.6. Расщепление гетероциклических соединений
Гетероциклические соединения являются хорошими исходными веществами для синтеза карбоновых кислот. Основной вклад в этой области синтеза сделан Мейерсом, который в прекрасной монографии [102] дал обзор применению гетероциклических соединений для целей синтеза. Поэтому мы здесь коснемся лишь тех методов, которые приводят к монофункциональным кислотам.
(1) Превращения оксиранов
Замещенные уксусные кислоты могут быть получены из кетонов через соответствующие нитрилы оксиранов [103], как показано на схеме (61).
C1CH2CN, ГаОСбНп-трет AcCk^.CN R1X^R2 ,CN HCl-газ; / AcjO, пиридин, Et3N -> R1/\R2 HO-; H3O+ >- R'R2CHCOOH (61)
26
(2) Превращения тиофенов
Тиофен действующий как маскированная нуклеофильная по концам бутановая цепочка, может применяться для получения гомологов жирных кислот [104] {схема (62)}.
RCOOH +
РгО5^
(62)
Zn(Hg), Н + ;
Ас2О, Н3РО4;
NaOCl
(3) Превращения 2-оксазолинов и 1,3-оке аз инов
Карбоновые кислоты и сложные эфиры легко превращаются в 2-оксазолины при взаимодействии с производными 2-аминоэта-нола или, что предпочтительнее, 2,2-диметилазиридина [105] {схема (63)}. 2-Алкильная группа таких оксазолинов легко металлируется бутиллитием и затем алкилируется различными электрофильными реагентами [106]. Получающаяся маскированная кислота может быть далее выделена обработкой водной кислотой или выделена в виде эфира после катализируемого кислотой эта-нолиза. Хиральный оксазолин (9), легко получаемый из (—)-аминоспирта (10) [107] известной абсолютной конфигурации, приводит к (S)-2-метилалкановым кислотам {схема (64)}. Преимуществом данного метода являются высокие оптические выходы и возможность регенерации аминоспирта.
RCH2COOH 4-
X?4gN
4- RCH2COOEt
I C6HiiN=C^NC6Hii J
rch2con
(63)
COOH
Применение энантиомерного (+)-аминоспирта приводит к (/?)-кислотам.
Этот способ был модифицирован применительно к асимметрическому синтезу диалкилуксусных кислот [108] {схема (65)}. Первоначальное введение группы с меньшим конфигурационным
Li N(Pr-iz3o)2> r’x;
L i N (Рг-мо) j.F^X; H,OT
СООН Ч-2--------------—
R2
LtN(Pr-uao)2.R2X;
LiN(Pr-«3o)2,R’X;
H3O+
COOH (65)
приоритетом всегда приводит к (5)-кислотам и наоборот. Другие варианты включают различные подходы к синтезу хиральных 3-ал-килалкановых кислот [109].
Гомологичные уксусные кислоты получают [НО] при реакции альдегидов с изоцианометилфосфонатом (11) {схема (66)}. При аналогичной реакции с кетонами необходимо применять литиевую соль фосфоната.
° R °
II •• NaCN (катализ)
RCHO + (EtO)2PCH2N=C : ------------------>
(10 (EtO)2P
II
О
KOBu-трет. ТГФ
Н+
NHCHO НзО+
RCH2COOH
(66)
‘P(OEt)2
II О
28
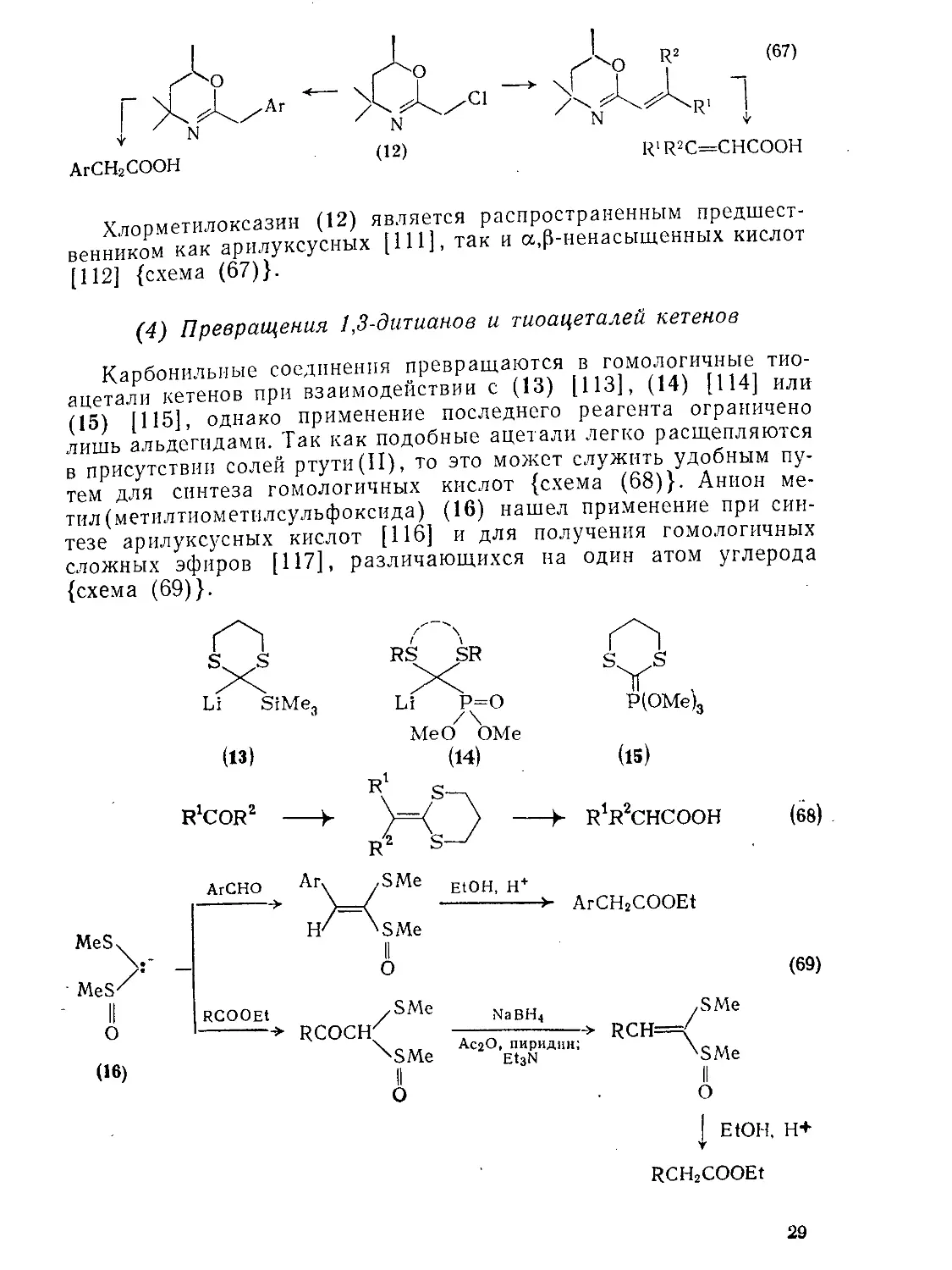
Хлорметилоксазин (12) является распространенным предшественником как арилуксусных [111], так и а,р-ненасыщенных кислот [112] {схема (67)}.
(4) Превращения 1,3-дитианов и тиоацеталей кетенов
Карбонильные соединения превращаются в гомологичные тиоацетали кетенов при взаимодействии с (13) [113], (14) [114] или (15) |115]. однако применение последнего реагента ограничено лишь альдегидами. Так как подобные ацетали легко расщепляются в присутствии солей ртути(II), то это может служить удобным путем для синтеза гомологичных кислот {схема (68)}. Анион метил (метилтиометилсульфоксида) (16) нашел применение при синтезе арилуксусных кислот [116] и для получения гомологичных сложных эфиров [117], различающихся на один атом углерода {схема (69)}.
(13)
^COR2 ------>►
SMe
SMe
R^CHCOOH
АгСНО ------->
EtOH, H + --------►- ArCH2COOEt
RCOOEt ----->
zSMe
RCOCHZ
^SMe
NaBH4
Ac2O, пиридин;
Et3N
SMe
SMe
| ЕЮН, H+
RCH2COOEt
29
(5) Превращения пиразолонов-5
Окисление пиразолонов, обычно получаемых по реакции гидп зима с эфирами 3-оксокислот, хлором [118] или, что нескольк удобнее, нитратом таллня(Ш) [119] {схема (70)}, приводит к эфирам алкиновой кислоты. Сложные эфиры 2-алкил-З-оксокислот аналогично с помощью нитрата таллия(III) превращаются в эфиры алленовой кислоты [120], а при окислении хлором дают эфиры «.^-ненасыщенных кислот [121]. Следует отметить, что при использовании таллиевого реагента отпадает необходимость в выделении промежуточного пиразолона перед проведением окисления"
О
RI^\^C00Et nh2nh2
I кон,
R2 EtOH
Cl2; R2=H
НО-;
H3O+
-------->
r2=#=h
--------->
R'C=CCOOH
R’CH=CCOOH
R2
(70)
R'C=CCOOMe
Т1(.\Оз)з. МеОН
/R1 R2CH=C=(/
^COOMe
9.1.1.7. Использование производных типа уксусной кислоты
До недавнего времени а-алкилирование простейших сложных эфиров под действием оснований [122] казалось невозможным из-за значительной конденсации субстрата с самим собой и, как следствие этого целевые продукты получались с ничтожными выходами. Однако теперь без особых сложностей можно получать анионы сложных эфиров, дианионы кислот и их алкилировать. Этот поворот стал возможным благодаря применению различных литийамидных оснований, из которых наиболее часто употребляют диизопропиламид лития и бис (триметилсилил) амид лития. Нену-клеофильность этих оснований и, по-видимому, связывание до некоторой степени аниона со свободным амином по крайней мере в первом случае [123] и служит причиной относительной устойчивости к самоконденсации. Взаимодействие этих анионов с различными электрофилами, включая альдегиды и кетоны, приводит к «.^-ненасыщенным кислотам через образование производных 3-гидроксикислот. Реакция Реформатского и другие реакции, приводящие к ненасыщенным сложным эфирам и кислотам, будут рассмотрены в следующем разделе.
30
(1) Дианионы кислот
В большинстве случаев наилучшим методом генерации аниона является обработка натриевой соли кислоты диизопропил амидом лития в тетрагидрофуране, содержащим гексаметапол [124]. В менее удобной методике применяют пон-радикальную систему, продуцируемую из лития и нафталина [125]. Дианпоны, получаемые таким образом, гладко реагируют с различными электрофилами, в том числе с алкилгалогенидами [126] и карбонильными соединениями [127, 128], приводя в последнем случае к «^-ненасыщенным кислотам {схема (71)}.
LiNRj _ СН2О; Н3О + ; A J1
R>CH2COO' --------> R'CHCOO' --------------> R'^^COOH (71)
|r2X; H3O+
RiR2CHCOOH
2LiN(Pr-U3O)2i
R1CH2CH=CHCOOH --------------------> R'CH=:CHCHCOOH (72)
E = R2X пли H + ; H3O+ |
E
Простой дианион из бутен-2-овой и бутен-3-овой кислот алкилируется как в а-, так и в у-положении; в случае высших гомологов алкилирование идет только в «-положении [129] из-за сдвига электронной плотности электронодонорной терминальной алкильной группы {пример (72)}.
(2) Анионы сложных эфиров
Литийэтилацетат, получаемый количественно [130] при взаимодействии этилацетата с бистриметилсилиламидом лития при —78°C в ТГФ, быстро реагирует при той же температуре с карбонильными соединениями. Позднее появилось сообщение о том, что изопропилциклогексиламид лития вызывает образование аналогичных енолят-анионов из целой серии сложных эфиров при —78 °C, и в отличие от более ранних методов синтеза эти анионы устойчивы при комнатной температуре [131]. Эти устойчивые анионы можно легко алкилировать в растворе ТГФ или ДМСО {схема (73)}. Литий-трет-бутил ацетат получается [132] в виде белого порошка, умеренно устойчивого на воздухе.
R2COR3; Н3О+ R2\ /R1
L1NR® _ * /=\
R’CHjCOOEt -------► R‘CHCOOEt — R3/ xCOOEt (73)
r4x
------------> R1R4CHCOOEt
(3) Реакции этилдиазоацетата
Взаимодействие диалкилхлорборанов [133] и триалкилборанов
[134] с диазоацетатом приводит к гомологичным сложным эфирам
31
{схема (74)}. С помощью этого метода можно довольно легко вводить объемистые алкильные группы. Более экономично использовать алкил- и арнлдихлорбораны [135], причем арильные производные дают более высокие выходы.
N2CHCOOEt
(RCs-C)3B I R2BCI или RteCCH.COOEt <---------RBE2(RZXr RCH2COOEt (74)
или Ar)
9.1.1.8. Направленные синтезы ненасыщенных кислот и их производных
В данном разделе синтезы объединены в соответствии с положением ненасыщенности.
(1) Получение а,^-ненасыщенных кислот
Классический синтез таких кислот включает реакцию Реформатского [136] {схема (75)}; в современном расширении исследований основное внимание уделено наиболее мягким методам, которые позволяют выделять промежуточно образующийся спирт. Предложенная система растворителей триметилборат-ТГФ [137] позволяет проводить реакцию при комнатной температуре, быстро и с высоким выходом. Триметилборат выполняет функцию слабой кислоты и нейтрализует основные алкоголяты, которые могут взаимодействовать с исходными соединениями, что является главной причиной частых низких выходов в этой реакции.
В качестве другого улучшения описано применение проточной системы с подогреваемой колонной, наполненной гранулированным цинком [138].
zn R’\ /CH2COOEt НзО+ R\
R'COR'2 + BrCH2COOEt ----------------------> )=CHCOOEt (75)
H3O+ R2/ \0H a R2/
Интересен также альтернативный синтез этих соединений из кетена [139]. Последний реагирует с карбонильным соединением в присутствии катализаторов, давая или эфиры аф-ненасыщенных кислот [140], или сложные эфиры [J-гидроксикислот {схема (76)} [141]. Применение других катализаторов Фриделя — Крафтса обычно приводит к (3-лактонам, из которых аф-ненасыщенные кислоты получаются при обработке минеральной кислотой.
ВС|3
R1 COR2 + СН2=С=О --->
R1
R2
СНСОС1
T1(OR3)4
1---------->
R\
\—CH2COORa
ОН
(76)
32
Другой основной путь синтеза а,Р-ненасыщенных кислот и их производных заключается в применении реакции Виттига или одной из ее модификаций, например использование фосфорана (17) или фосфоната (18) [142] {схемы (77), (78)}. Реакция Вит-тига интенсивно использовалась для получения производных не-предельных кислот с двойной связью в различных положениях 11431' R\
R>COR2 + Ph3PCHCOOEt —> )=CHCOOEt (77)
R2/
(17)
? R1
|| 2LlN(Pr-U3o)2; \
R'COR2 + (PhCH2O)2PCH2COOH---------------> )=CHCOOH (78)
H3O R2/
(18)
Использование этого подхода можно показать на примере реакции сложных эфиров а-иод- или а-бромкислот с алкилиденфосфоранами, причем для элиминирования трифенилфосфина из образующейся фосфониевой соли необходимо применение второго эквивалента илида [144] {схема (79)}.
Кетоны и альдегиды можно превратить непосредственно с гомологичные а,|3-ненасыщенные сложные эфиры {схема (80)} с помощью метода [145, 146], основанного на хорошо известном сродстве соответствующим образом расположенных силильных групп к алкоксониевым ионам.
R\ + R\ R1'
PPh3 + ^CHCOOEt —>
R2' X/ R2'
R3
COOEt
Me3Sk .COOR3
R’COR2
R1
R2-
PhP+X~
/R3 R1
( + Ph3P +
'COOEt R2
OLi SiMe3
(79)
(80)
(81)
R'
R2,
COOR3_ NaOAc
ArCHO + Ac2O ----► ArCH=CHCOOH
Д
_R2
а-Анионы а-силилнитрилов аналогичным образом приводят к а,P-ненасыщенным нитрилам [147].
Реакция Перкина [148, 149] обычно применима только к ароматическим альдегидам и протекает при нагревании до относительно высокой температуры смеси альдегида, ангидрида кислоты, содержащей а-метиленовую группу, и такого слабого основання, как щелочная соль органической кислоты {схема (81)}.
2 Зак. 395 яд
Сложные эфиры а,р-алкииовых кислот служат удобными пред шественнпкамп а.Р-иеиредельпых кислот, так как они претерпе вают сопряженное присоединение оргаиокупратов [150], в основ ном с /(«с-стереохимией {схемы (82), (83)}.
LICuR2: R\ /COOR2
R'C==CCOOR2 ---------► )=(
H3o+ JRs/ \pj
LICuR}; R\ /COOR2
R3Ch=CCOOR2 ---------> )=/
НзО+ R[/
(82)
(83)
,R
HC = CCOOEt + Me2CHCMe2-B ---->
4H
R
Me2CHCMe2-В H
H COOEt
Me2CHCMe2-X fr
I >COOEt Br
(84)
—>- F^CH^CHCOOR2
per R3
H ^SeZ
J 2
> r‘CH—CHCOOR2
^CH-jCHjiCOOR2—
—>- r’ch2chcoor2
R1CHj-CnCOOR2
P IO
H sx
<:o<+V
34
Гидроборирование этилового эфира пропионовой кислоты может приводить как к так и к (Z?) -а,Р*ненасыщеиным сложным эфирам {схема (84)}; группа R мигрирует с сохранением стереохимии [151].
Наилучший способ введения аф-двойной связи в доступный насыщенный сложный эфир основан на способности сульфоксидов [152] и селеноксидов [153] легко претерпевать син-1,2-элимини-рование {схема (85)}. Для синтеза исходных а-селенильных и а-сульфенильных соединений существует набор достаточно селективных методов.
(2) Получение ^-ненасыщенных кислот
Простейший способ введения [3,у-двойной связи состоит в несопряженном алкилировании или протонировании [155] доступного аф-ненасыщенного предшественника {схема (86)}. Добавление одного эквивалента гексаметапола (ГМФТА) сильно снижает и без того низкую нуклеофильность основания.
/^/COOEt
LiN (Рг-изо)г, ГМФТА
RX
COOEt
R3B + BrCH2CH=CHCOOEt —> RCH=CHCH2COOEt
(86)
(87)
Реакция борорганических соединений с этил-4-бромбутен-2-оатом служит удобным методом удлинения цепи на четыре углеродных атома {схема (87)} [156].
Различные аллиловые спирты превращаются [157] в гомологичные р,у-ненасыщенные амиды в результате термолиза смешанных ацеталей этих спиртов с ДМФА {схема (88)}. Эта реакция, которая, по-видимому, проходит через [2,3]-сигматропную перегруппировку карбена (19), с хорошими выходами идет со многими аллиловыми спиртами, за исключением у,у-дизамещенных. Перегруппировки, показанные на схемах (89) и (90), дополняют ранее упоминавшиеся превращения аллилгалогенидов [158] и аллиловых тиоэфиров [159].
(88)
[2,31-сдвиг -------------►
(19)
(89)
2*
35
(3) Получение у,8-ненасыщенных кислот
Наилучший способ синтеза этого класса соединений заключается в [3,3]-сигматропной перегруппировке соответствующих аллиловых сложных эфиров через литийеноляты [160] {схема (91)}. В качестве ловушки енолятов часто удобно применять трет-бутилдиметилхлорсилан, так как в этом случае ацеталь (20) образуется стереоселективно и его можно выделить до перегруппировки [161]. Для этой же цели рекомендован триэтилхлорсилан [162]. Альтернативно, цинковый енолят [163] можно получать из соответствующего сложного 2-бромэфира. Сложные эфиры пропинола (пропаргилового спирта) сходным образом перегруппировываются в (3,у- и у,6-алленовые кислоты [163] {схема (92)}.
Если в качестве субстрата использовать оптически активный (Е)-пентен-3-ол-2, то перегруппировка протекает с обращением конфигурации [164]; оптический выход превышает 90%. Данный метод полезен для стереонаправленного введения связи С—С {схема (93)}. Остроумный вариант этой перегруппировки [165],
36
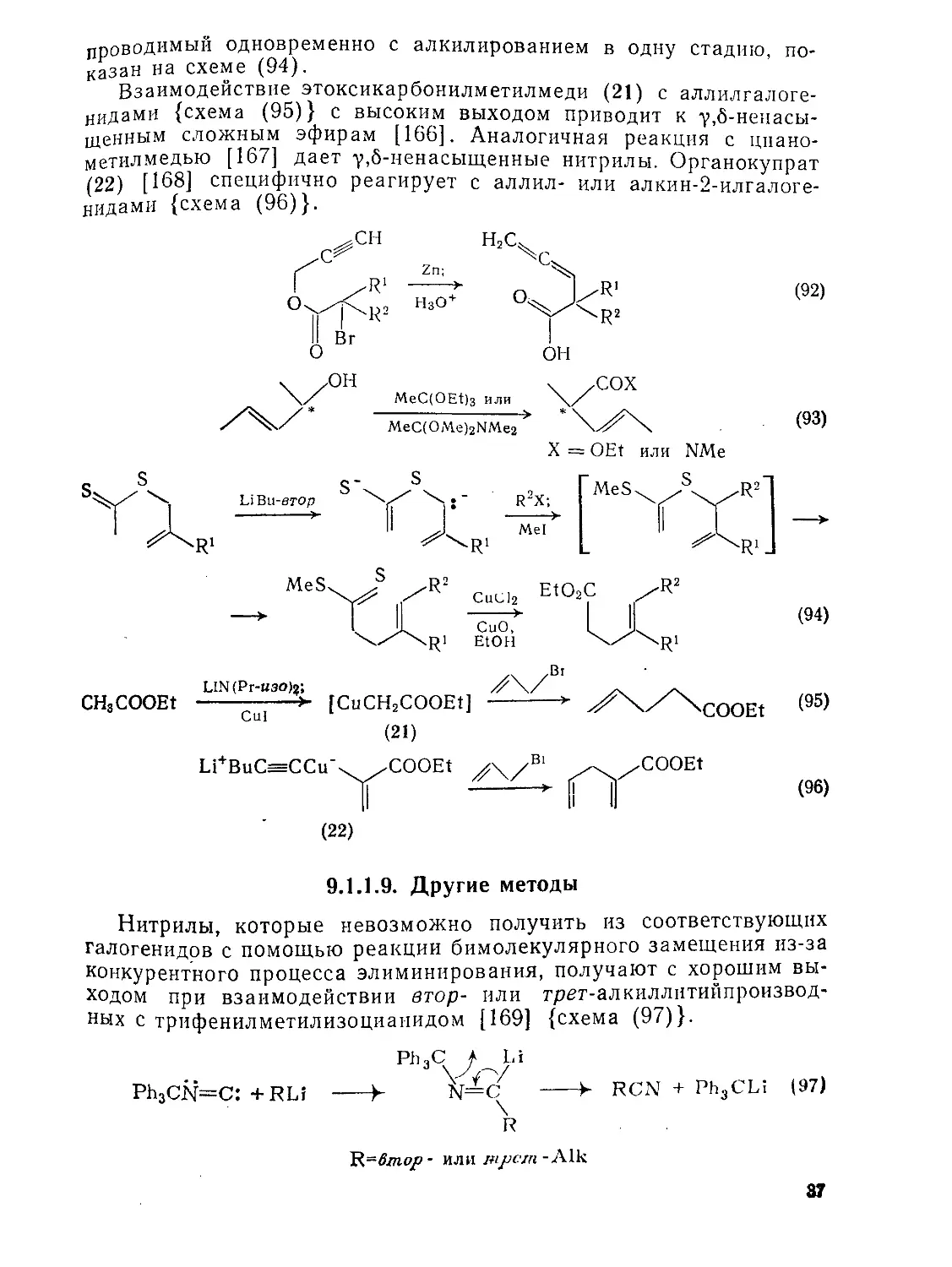
проводимый одновременно с алкилированием в одну стадию, показан на схеме (94).
Взаимодействие этоксикарбонилметилмеди (21) с аллилгалоге-нидами {схема (95)} с высоким выходом приводит к у,б-ненасы-щенным сложным эфирам [166]. Аналогичная реакция с цпано-метилмедью [167] дает у,б-ненасыщенные нитрилы. Органокупрат (22) [168] специфично реагирует с аллил- или алкин-2-илгалоге-нидами {схема (96)}.
(94)
[CuCH2COOEt]
(21)
CH3COOEt
LIN(Pr-U3O)^;
Cui
СиС1г ----->
СиО, EtOH
^^^^COOEt
Li+BuC^CCu
COOEt
COOEt
(95)
(96)
(22)
9.1.1.9. Другие методы
Нитрилы, которые невозможно получить из соответствующих галогенидов с помощью реакции бимолекулярного замещения из-за конкурентного процесса элиминирования, получают с хорошим выходом при взаимодействии втор- или трет-алкиллитийпроизвод-ных с трифенилметилизоцианидом [169] {схема (97)}.
Ph3C
Ph3CN=C: +RLi -------\=C ----> RCN + Ph3CLi (97)
Г?
'R=6mop- или mpcm -Aik
87
Карбонильные соединения можно превратить в гомологичные кис ТОТЫ или нитрилы в результате прямой реакции с арилсульфо, нитизоцнанидом [170, 171] {схема (98)}. Альтернативно, гомологи кислот можно получать перегруппировкой дпазометилкетопов (получаемых из кислот), катализируемой ионами одновалентного серебра {реакция Арндта — Эйстерта [172], схема (99)}.
у-Алкилпрованпе металлированных а,|3-ннамннов предложено [173] как удобный метод наращивания цепи на три углеродных атома {схема (100)}.
KOBu-rper - R'COR2;
PhSO2CH2NC ----——>- PhSO2CHNC —>
н3о+
---->
МеО~
---->
R'R2CHCOOH
R’R2CHCN
(98)
CH2N2 Ag(I) R2OH
► Rlcf ——> R1CH=C=O ---------> R>CH2CO2R2
\ci Vhn2 -N2
H2o I (99)
r>ch2cooh <----1
BuLl R*X;
MeC=CNR2 --------* LiCH2C=CNR2 ------> R1 CH2CH2CONR2 (100)
ТМЭДА H3O+
9.1.1.10. Меченные изотопами и хиральные кислоты
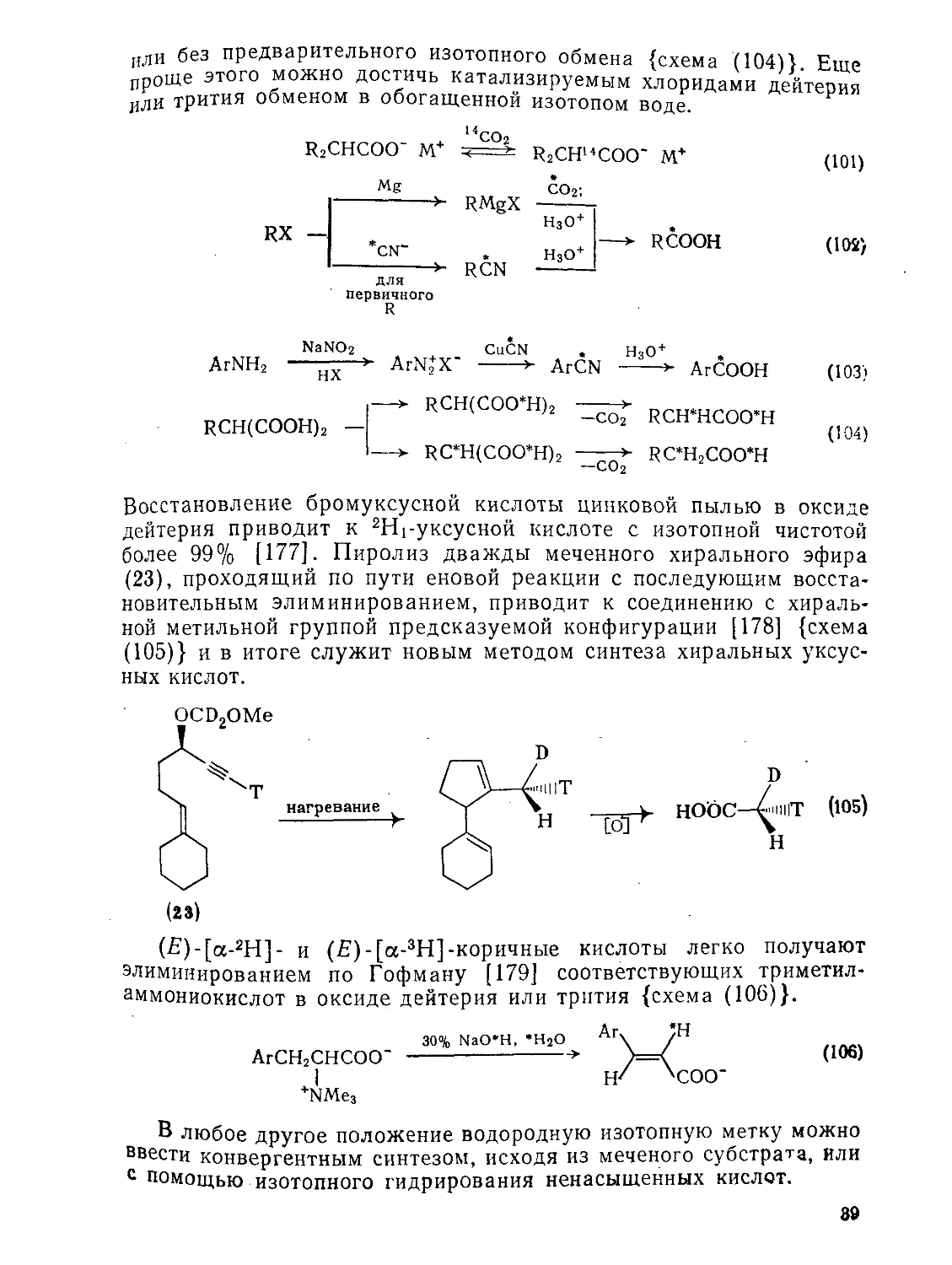
В этом разделе будут рассмотрены только монофункциональные кислоты. Поскольку методы избирательного введения изотопной метки в кислоты подробно описаны в монографии [174], здесь будут представлены лишь обобщенные данные с указанием современных вариантов.
Карбоксильную группу можно пометить радиоактивными изотопами 14С или 3Н, а также с помощью 13С, 18О, 2Н. Углеродная метка {схемы (101) — (ЮЗ)} может быть введена без изменения длины цепи обменной реакцией с меченым диоксидом углерода [175]; реакция, по-видимому, протекает через малонатный интермедиат. Если допустимо или необходимо удлинение углеродной цепи, то метку можно вводить реакцией алкильного или арильного реактива Гриньяра с меченым диоксидом углерода, реакцией бимолекулярного замещения меченого цианид-иона с алкилгалогенп-дом или реакцией диазониевой соли с меченым цианидом меди(1)-Метку в углеродную цепь обычно вводят реакцией диоксида углерода с меченым реактивом Гриньяра. Введение метки по кислороду или по кислотному водороду легко достигается методами изо-топного обмена или гидролиза.
Введение меченого а-водорода можно осуществить декарбоксилированием производных малоновой кислоты с предварительным 38
или без предварительного изотопного обмена {схема (104В Епт? ироше этого можно достичь катализируемым хлоридами или трития обменом в обогащенной
14со2
изотопом воде.
деитерия
R2CHCOO‘ М'
R2CHuCOO' М‘
(101)
RX —
Mg ----->
RMgX
co2;
H3O+
CN~
для первичного R
RCN
H3O
RCOOH
(102)
Na NO 2
ArNH2 ——* ArN2X" HA
—> RCH(COO*H)2
CuCN .
----> ArCN
н3о+
---> АгСООН
(ЮЗ)
RCH(COOH)2
—со2 RCH*HCOO*H
—> RC*H(COO*H)2
—RC*H2COO*H
—со2
(104)
цинковой ПЫЛЬЮ в
оксид
Восстановление бромуксусной кислоты дейтерия приводит к 2Н1-уксусной кислоте с изотопной чистотой более 99% [177]. Пиролиз дважды меченного хирального эфира (23), проходящий по пути еновой реакции с последующим восстановительным элиминированием, приводит к соединению с хираль-ной метильной группой предсказуемой конфигурации [178] {схема (105)} и в итоге служит новым методом синтеза хиральных уксусных кислот.
OCD2OMe
(23)
(£)-[а-2Н]- и (£)-[а-3Н]-коричные кислоты легко получают элиминированием по Гофману [179] соответствующих триметил-аммониокислот в оксиде дейтерия или трития {схема (106)}.
Ai\ ;н
30% NaO*H, *Н2О
АгСН2СНСОО" —--------------
(106)
н-
COO-
+NMe3
в любое другое положение водородную ввести конвергентным синтезом, исходя из .
с помощью изотопного гидрирования ненасыщенных кислот.
изотопную метку можно меченого субстрата, или
39
Хиральные кислоты можно получить в оптически активной фор. ме из оптически активного предшественника [180] или, что делают значительно чаще, путем разделения рацемической смеси. Последний процесс требует превращения смеси рацематов в смесь диастереомерных соединений, компоненты которой можно разделить, и далее индивидуальные энантиомеры порознь регенерируют {схема (107)}.
(/?>, 52)-Кислота + (R2)-PeareHT —> (/?', R2) + (S1, 7?2)
Фракционирование и регенерация
(Я'рКислота
(5')-Кислота
(Ю7)
Для подобных разделений [181] применяют оптически активные спирты и оптически активные амины. Для разделения относительно сильных кислот применяют хиральные амины, включая такие алкалоиды, как хинин, бруцин, стрихнин и т. д.; более сильные основания, такие как (—)-ментиламин, метогидроксид хинина i[ 182] и т. д., используются для разделения слабых кислот. Амины дают соли с кислотами, и после фракционной кристаллизации разделенные кислоты регенерируют обработкой минеральными кислотами или пропусканием раствора соли через колонку с ионообменной смолой в кислотной форме. Применяемые для разделения хиральные спирты включают (—)-ментол, ( + )-борнеол и т. д.; при этом получаются диастереомерные сложные эфиры, которые разделяют фракционной кристаллизацией или фракционной дистилляцией, с последующим гидролизом в каждом случае.
Систематическая техника фракционирования известна под названием «треугольной» кристаллизации и иллюстрируется схемой (108).
^Кристаллы Rf— СООН | (растворитель)
Степень разделения можно контролировать, измеряя оптическое вращение на любом конце основания треугольника: разделение максимально, когда каждая крайняя точка имеет максимальное оптическое вращение. Альтернативно, для контроля степени разделения можно также измерять температуру плавления образцов. Тщательное проведение этой операции утомительно, и поскольку чаще всего требуется получить только один энантиомер, выбирают такой спирт или основание, которые дают искомый диастереомер в кристаллическом виде, для выделения которого достаточно обычной кристаллизации.
Часто после выделения кислоты требуется установить ее абсолютную конфигурацию. Превращение кислоты в оптически активный хлорангидрид (24) и его последующая реакция с двумя эквивалентами рацемического фосфорана (25) приводит к фосфониевой соли (26) с частичным кинетическим разделением. Второй эквивалент энантиомерно обогащенного фосфорана реагирует с солью (26) по реакции трансилидирования, приводя к оптически активной соли (27), которая выпадает в осадок, и диастереомерному ацил-илиду (28), который остается в растворе {схема (109)}.
Me Me Ph СГ
l + -ZPh 1+ I
Ph—Р—CZ + R!R2R3CCOC1 —> Ph—Р—С—COCR‘R2R3 —>
I I ।
н-Pr н-Pr H
(25) (24) (26)
Me Me
(25U ph—P—CH2PhCr + Ph—P—сЙ (109)
I I XCOCR'R2R3
н-Pr н-Рг
(27) (28)
Абсолютные конфигурации хлоридов (27) известны и могут быть скоррелированы с абсолютной конфигурацией исходной кислоты [183].
9.1.2. СВОЙСТВА КАРБОНОВЫХ КИСЛОТ
Согласно определению, карбоновые кислоты RCO2H имеют карбоксильную группу, которая связана с другими группами (схе ма (110)}. Кислотные свойства различных кислот суммированы в табл. 9. Й 1184Ь
Наиболее интересно, с точки зрения выяснения ф з природы, состояние карбоксильной группы, влияние природы радикала R на способность карбоксильной группы к ионизации или к образованию водородных связей и димеризации, образова оксокарбениевых ионов, тенденция солей кислот к агрегаци мицеллообразованию и, наконец, взаимодействие карбоксильной
41
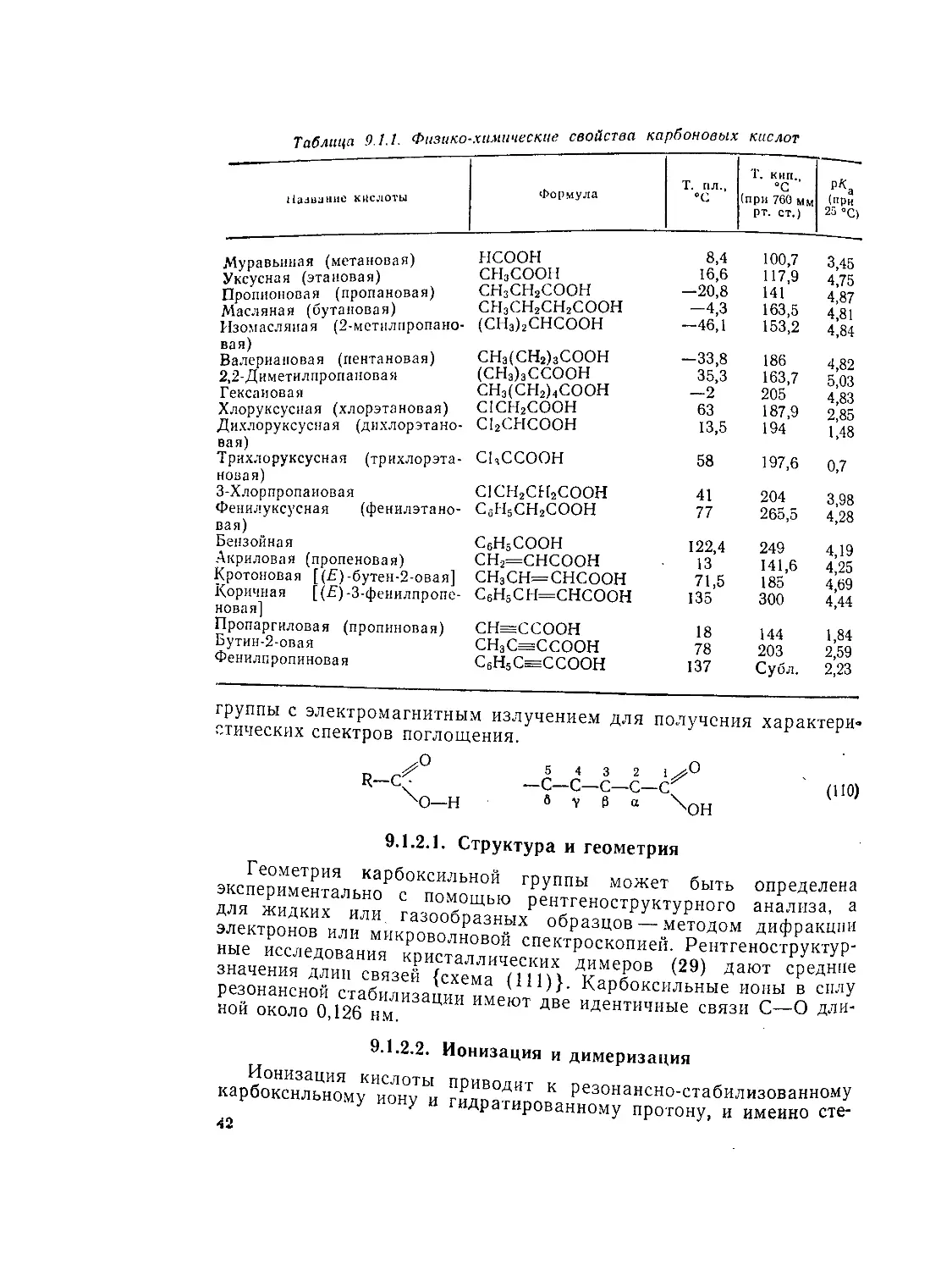
Таблица 9.1.1. Физико-химические свойства карбоновых кислот
Название кислоты Формула Т. пл., °C T. кип., “С (при 760 мм рт. ст.) (при 25 °C)
Муравьиная (метановая) СНзСООН 16,6 Н7Д 475 Лз 111 Масляная (бутановая) ^Ж^НСПОН -46 1 53 2 1’11 Изомасляиая (2-метилпропано- (СН3)2СНСООН 4Ь,1 щз,2 4,84 Валериановая (пентановая) ЯЖ^СООН^ ~35 3 163 7 5 03 Й?»"»."”’0""”'’1 сн^свд?соон Л3 S7 « Х»“у1°У«ая (хаоратаяоаая) CICHjCOOH вз 187 9 2,85 Дихлоруксусная (дихлорэтане- СЬСНСООН 13,о 1У4 1,48 Трихлоруксусная (трихлорэта- CUCCOOH 58 197,6 о,7 З-Хлорпропаиовая CICH2CH2COOH 41 204 3,98 Фенилуксусная (фенилэтано- СбН5СН2СООН 77 265,5 4,28 вая) Бензойная СбН5СООН 122,4 249 4,19 Акриловая (пропеновая) СН2=СНСООН 13 141,6 4,25 Кротоновая [(£)-бутен-2-овая] СН3СН—СНСООН 71,5 185 4,69 Коричная [(Д)-З-феиилпропе- СбН5СН=СНСООН 135 300 4,44 новая] Пропаргиловая (пропиновая) СН=ССООН 18 144 1,84 Бутин-2-овая СНЭС=ССООН 78 203 2,59 Фенилпропиновая С6Н5С^ССООН 137 Субл. 2,23
группы с электромагнитным излучением для получения характеристических спектров поглощения.
54321
R—с< —С—С—С—С—С (110)
ХО—Н в у р а \он
9.1.2.1. Структура и геометрия
Геометрия карбоксильной группы может быть определена экспериментально с помощью рентгеноструктурного анализа, а для жидких или газообразных образцов — методом дифракции электронов или микроволновой спектроскопией. Рентгеноструктурные исследования кристаллических димеров (29) дают средние значения длин связей {схема (111)}. Карбоксильные ионы в силу резонансной стабилизации имеют две идентичные связи С—О длиной около 0,126 нм.
9.1.2.2. Ионизация и димеризация
«•>тч^^ИЗаЦИЯ кислоты приводит к резонансно-стабилизованному кароокснльному иону и гидратированному протону, и именно сте-42
пень ионизации или диссоциации карбоксильной группы определяет термодинамическую кислотность {схема (112)}. В то время как большинство карбоновых кислот имеет значение р/Са около 4 8 любой электроноакцепторный заместитель R стабилизует карбоксильный ион за счет оттягивания электронов и, как видно на примере галогенуксусных кислот, усиливает кислотность и понижает рКа- Однако этот индуктивный эффект быстро ослабевает втоль цепи (ср., например, 3-хлорпропионовую кислоту). Обратный эффект, когда электронодонорный заместитель понижает кислотность и увеличивает p/G, не столь заметен, что видно в ряду муравьиной, уксусной и 2,2-диметилпропановой кислот.
,0-Н—Ок
R—-R
С=О . 0,123 нм
С—О 0,136 нм (111)
ОН -О 0,260-0,270 нм
(29)
122-123°
RC^ + Н2О 7
ОН
^[RCOO~Ih3O+] а [rcoohJ
и Р^а=-1О£1(Л
(30)
В водных растворах карбоновые кислоты показывают тенденцию к димеризации с образованием, однако, не циклической формы (29), как в твердом состоянии, а открытой формы (30). Структурные свойства [185] и способность к ионизации [186] карбоновых кислот детально обсуждены. Сообщалось также о газофазном изучении внутренней кислотности [187] ряда карбоновых кислот [188], и степень кислотности сопоставлена с кор-электронными ионизационными потенциалами [189].
9.1.2.3. Оксокарбениевые ионы
Галогенангидриды и ангидриды кислот дают с кислотами Льюиса комплексы, которым на основании исследований различными '11^?.и1ческими методами [190], и особенно спектроскопии *Н-ЯМР I уИ, приписана структура солей оксокарбениевых ионов (схема
43
(113)}. Предполагается, что катализируемое кислотой ацилиров ние галогенангидридамп и ангидридами кислот включает пром3' жуточное образование оксокарбеииевых ионов как реагирующих частиц.
СООМе
+ H2SO4 конц.
(113)
Образование оксокарбеииевых ионов включают в механизм Адс1 катализируемого кислотой гидролиза сложных эфиров. В качестве примера можно привести быстрый гидролиз метилового эфира 2,4,6-триметилбензойной кислоты [193], когда его раствор в концентрированной серной кислоте выливают на лед {схема (114)}. В то же время это соединение устойчиво к мягкому кислотному гидролизу.
9.1.2.4. Мицеллы
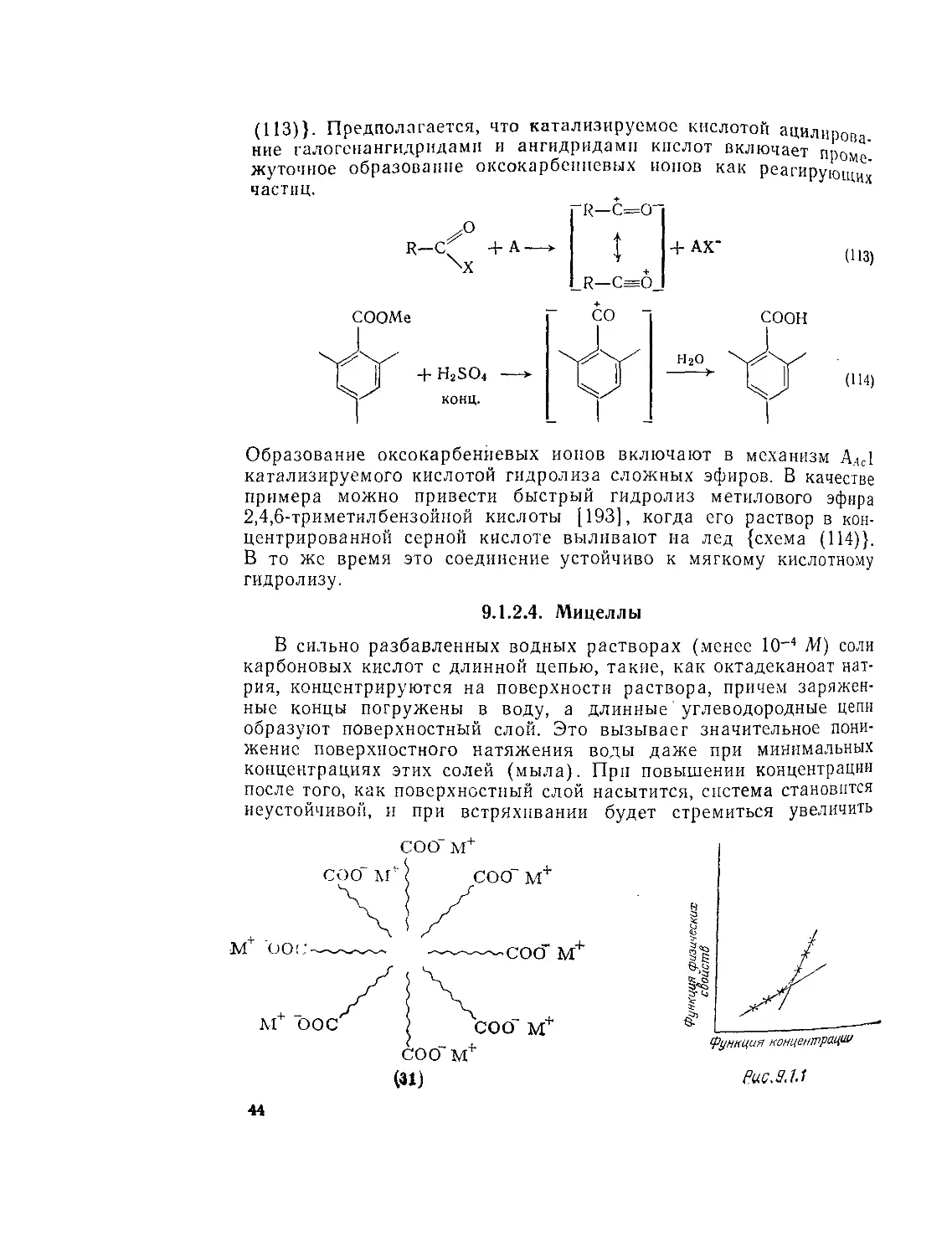
В сильно разбавленных водных растворах (менее 10-4 Л1) соли карбоновых кислот с длинной цепью, такие, как октадеканоат натрия, концентрируются на поверхности раствора, причем заряженные концы погружены в воду, а длинные' углеводородные цепи образуют поверхностный слой. Это вызывает значительное понижение поверхностного натяжения воды даже при минимальных концентрациях этих солей (мыла). При повышении концентрации после того, как поверхностный слой насытится, система становится неустойчивой, и при встряхивании будет стремиться увеличить
СОО м+
(«)
Функция концентпрацш
РШ.З.М
44
свою поверхность за счет пенообразования. Если систему не встряхивать, то при определенной концентрации молекулы соли кислоты агрегируют с образованием мицелл (31). Концентрация, при которой образуются мицеллы (критическая мицельная концентрация) находится в узком интервале значений, а не является точной величиной; она обнаруживается только по заметному отклонению ряда физических свойств раствора (рис. 9.1.1).
К Мицеллы обладают потенциальными каталитическими свойствами, не только образуя неполярные области в водных системах [194], но локализуя также на своих поверхностях высокую плотность заряда. Такие каталитические свойства мицелл заметно увеличивают скорости многих реакций. Эта обширная область исследования явилась предметом очень интересной монографии [195].
9.1.2.5 Спектральные свойства
Здесь мы рассмотрим наиболее характерные особенности; более детальную информацию можно получить из руководств по спектроскопии [196].
(1) Инфракрасная спектроскопия (растворы)
Характеристические полосы поглощения карбоновых кислот суммированы в'табл. 9.1.2.
Таблица 9.1.2. Характеристические частоты инфракрасных спектров карбоновых кислот
Частота.
3000—2500
1760
1710
1610-1550
1400
Пояснения
Характеристическая широкая полоса ОН-группы, связанной водородной связью
Мономер! Свободная ОН-группа при 3550 см-1
Димер j Полоса валентных колебаний С = О; при сопряжении частота понижается примерно на 20 см-1 Характерны для RCOO_M+, появляются вместо трех выше указанных полос
(2) Ульрафиолетовая спектроскопия
Ультрафиолетовая спектроскопия не столь важна для карбоновых кислот, однако она нашла применение для сопряженных кислот после того, как были определены аддитивные правила для заместителей в алкенах [197]. Обычно а,[3-ненасыщенные кислоты оглощают около 215 нм и имеют коэффициент экстинкции порядка 9000.
45
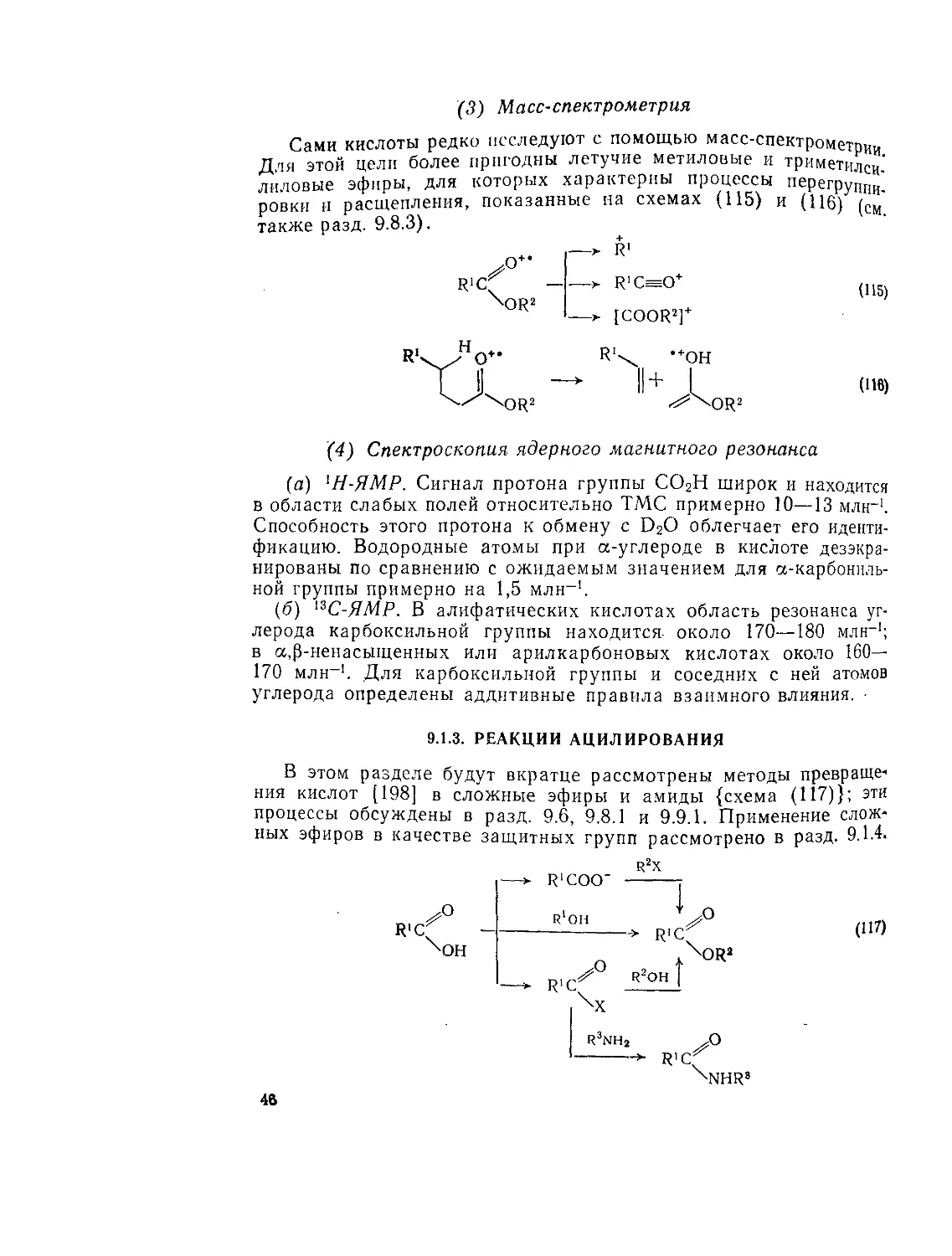
(3) Масс-спектрометрия
Сами кислоты редко исследуют с помощью масс-спектрометрии Дня этой цели более пригодны летучие метиловые и триметилси" лиловые эфиры, для которых характерны процессы перегруппи'_ ровкн и расщепления, показанные на схемах (115) и (116) (см также разд. 9.8.3).
(4) Спектроскопия ядерного магнитного резонанса
(а) 1Н-ЯМР. Сигнал протона группы СО2Н широк и находится в области слабых полей относительно ТМС примерно 10—13 млн-1. Способность этого протона к обмену с D2O облегчает его идентификацию. Водородные атомы при а-углероде в кислоте дезэкра-нированы по сравнению с ожидаемым значением для а-карбониль-ной группы примерно на 1,5 млн-1.
(б) 13С-ЯМР. В алифатических кислотах область резонанса углерода карбоксильной группы находится- около 170—180 млн-1; в а,|3-ненасыщенных или арилкарбоновых кислотах около 160— 170 млн-1. Для карбоксильной группы и соседних с ней атомов углерода определены аддитивные правила взаимного влияния.
9.1.3. РЕАКЦИИ АЦИЛИРОВАНИЯ
В этом разделе будут вкратце рассмотрены методы превращения кислот [198] в сложные эфиры и амиды {схема (117)}; эти процессы обсуждены в разд. 9.6, 9.8.1 и 9.9.1. Применение сложных эфиров в качестве защитных групп рассмотрено в разд. 9.1.4.
R'cf > \он —> R‘C^ R3nh2 to О17) \OR’ н 1 R<° \nhr8
46
Для синтеза сложных эфиров и амидов существует три методологических подхода: прямая катализируемая кислотой реакция спирта с кислотой, предварительное превращение кислоты в реакционноспособное производное и активация спирта или амина по отношению к нуклеофильной атаке карбоксильных ионов. Тетраэдрический интермедиат (32), возникающий при первых двух подходах, интенсивно изучался [199]. Другие менее общие методы синтеза сложных эфиров, такие как реакция Байера — Виллигера или фотохимическое окисление кетонов, рассмотрены в разд. 9.1.1.1. Кетены [200] редко применяют для получения монофункциональных амидов или сложных эфиров.
9.1.3.1. Прямые реакции
Успешное проведение этой обратимой реакции {схема (118)} О'
R1—С—-X NuH = R2OH или R3NH2
Nu
(32)
Н+
R>C^ + R2OH R’C^ +H2O (118)
\0H H3O+ \0R2
зависит от применяемого метода смещения равновесия. Последние модификации этого метода заключаются в использовании «дегидратирующих катализаторов», в качестве которых применяются ионообменная смола кислотного типа в присутствии осушающего агента [201] и полимерно-защищенный трихлорид алюминия [202], который обладает как каталитическими, так и дегидратирующими свойствами. Для этой же цели используют бисульфат графита [203], приготовленный электролитическим методом.
9.1.3.2. Активация кислотной компоненты
(1) Галогенангидриды кислот
Из всех галогенангидридов кислот наиболее часто используются бром- и хлорангидриды, общие методы получения [204] которых представлены на схеме (119). Область применения и ограничения для использования тионилбромида при синтезе броман-гидридов исследованы ранее [205]. Применение реагента трифе-нилфосфин — тетрахлорид углерода [206] представляется важным достижением не только потому, что это позволяет получать хлорангидриды кислот в нейтральных условиях, но и потому, что промежуточное соединение трехвалентного фосфора (33) может непосредственно взаимодействовать [207] с соответствующим нуклеофильным амином. Другие системы с использованием соединении фосфора как промежуточных будут обсуждены далее.
47
PCI3. PGIs или POCI3;
SOCI2 или (СОС1)2» катализ ДМФА
Ph3P> СС14
^Nu
NuH j
R<° /
X)—PPh3 СГ + CHCI3
(33)
(И9
Для получения сложных эфиров или амидов хлорангидрид кислоты после выделения обрабатывают спиртом или амином в присутствии основания в гомогенной или гетерогенной среде. В том случае, если спирт кислотолабилен или стерически затруднен, часто [208] предпочтительнее вводить в реакцию ацилирования не сам спирт, а алкоголят лития, получаемый предварительной обработкой спирта к-бутиллптием.
(2) Ангидриды, кислот
За исключением уксусного ангидрида, другие симметричные ангидриды кислот редко используют для ацилирования, так как выход в расчете на кислоту не превышает 50%. Для создания сложноэфирной и амидной связи [209] большее распространение получили смешанные ангидриды кислот, особенно карбонаты {схема (120)}, причем атака нуклеофила предпочтительно идет по карбонильной группе исходной кислоты. Легко получаемый [210] смешанный ангидрид уксусной и муравьиной кислот применяют для быстрого формилироваппя ряда нуклеофильных субстратов.
О
CICOOEt j, NuH
RC; -р, v~x RC; /Ск —RC; <12°)
XOH 's‘ ^OEt ^Nu
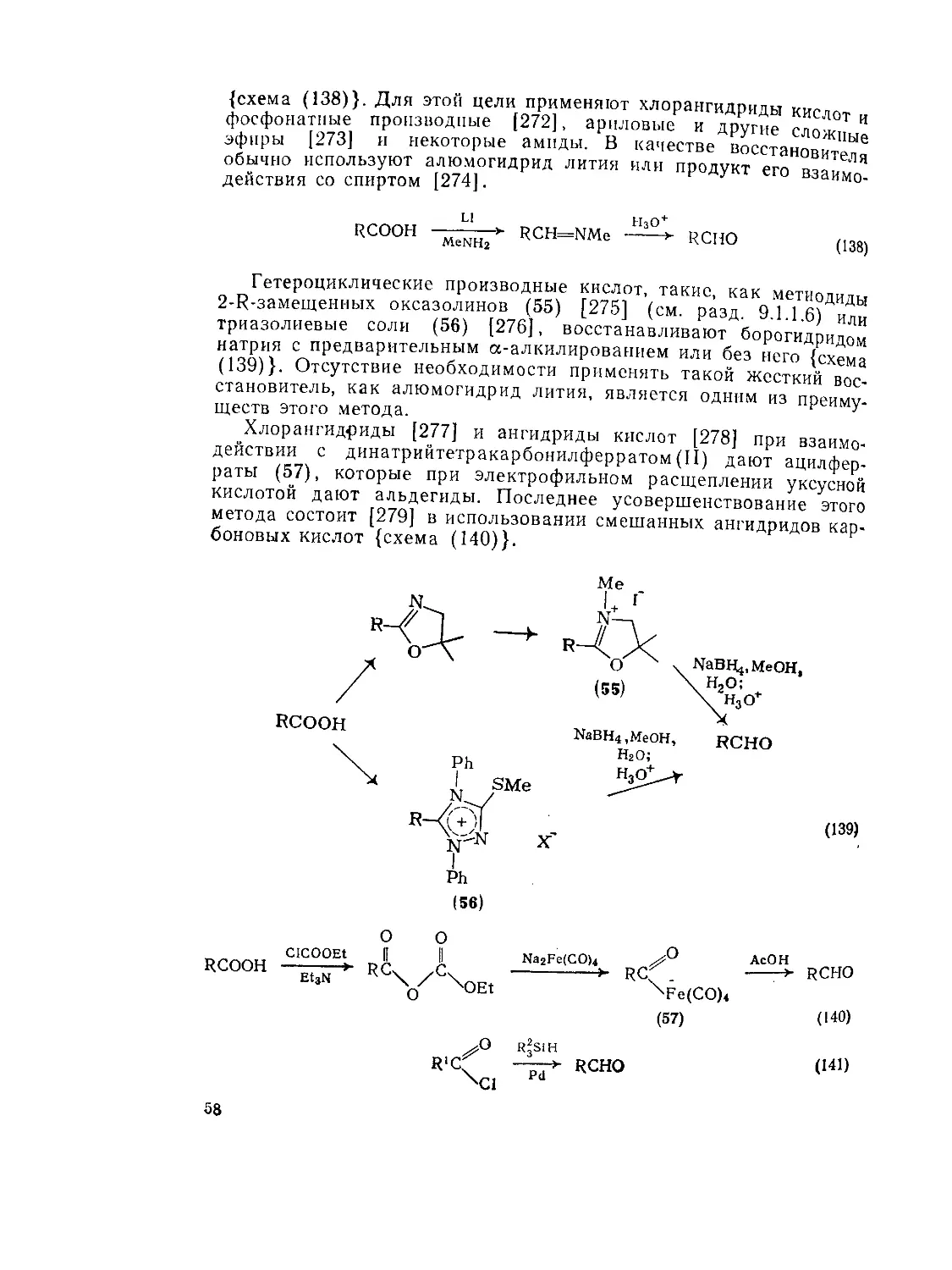
(<?) Реакции в присутствии карбодиимидов
Дициклогексилкарбодиимид (ДЦК) (34), обычно используемый реагент данного типа, взаимодействует [211] с кислотами с первоначальным образованием О-ацилизомочевин (35) {схема (121)}. Эти производные далее могут непосредственно реагировать с нуклеофилами спиртовой или аминной природы пли с дополнительным количеством кислоты, что приводит к симметричным ангидридам, которые, в свою очередь, могут реагировать с нуклеофилами, и так далее. Максимальное вовлечение симметричного ангидрида в общем случае [212] составляет 55—60%, однако в твердой фазе (т. е. реагент привитый к полимеру) ацилирующим реагентом, вероятно, является симметричный ангидрид [213]. Помимо относительных трудностей в очистке продуктов реакции основными не-48
удобствами применения дициклогексилкарбодиимида являются рацемизация кислотного субстрата и конкурентная внутримолекулярная перегруппировка реакционноспособной О-ацилизомочевины в инертную ацилмочевину (36). Тем не менее оба эти неудобства можно в значительной степени свести на нет применением N-ги-дроксисукцинимида или 1-гидроксибензотриазола, которые стабилизуют [214] О-ацилизомочевину.
RC^ + C6H11N=C=NC6H11 х>н
(34)
COR
—> C6H11NHC=NC6H11 —> CeHnNHCNCeHn
I II
OCR О
II
(35) О (36)
RCOOH
(121)
NuH
NuH
(4) Реакции в присутствии N,N'-карбонилдиимидазола
Штааб развил и описал [215] применение этого и других крайне полезных азолидов. Образующиеся ацилимидазолнды [216] можно выделить из реакционной смеси {схема (122)}. Эти соединения спонтанно реагируют с аминами, однако для реакции со спиртами требуется присутствие каталитических количеств основания [217]. Подобные имидазолиды нашли широкое применение для других трансформаций карбоновых кислот, так как они по своей реакционной способности сравнимы с хлорангндридами кислот, однако получаются значительно легче и удобнее в работе.
49
(5) Реакции в присутствии соединений трехвалентного фосфора
Методы, описанные в этом и следующем подразделах, обязаны своим успехом реакции Арбузова (см. разд. 10.6.4).
О
х#° II Et3N
R‘C< + (R2O)2PX ------->
\он
О
II (R2O)2P—OCOR1 + Et3N+ X'
(37)
Nuh|
NuH
4---
(123)
(Me2N)3P—Cl CIO;
(39)
(Me2N)3P—X PFg
(40) X = Br, CN, NCS, N3, CC13, OPh, OC8H4NO2-/i
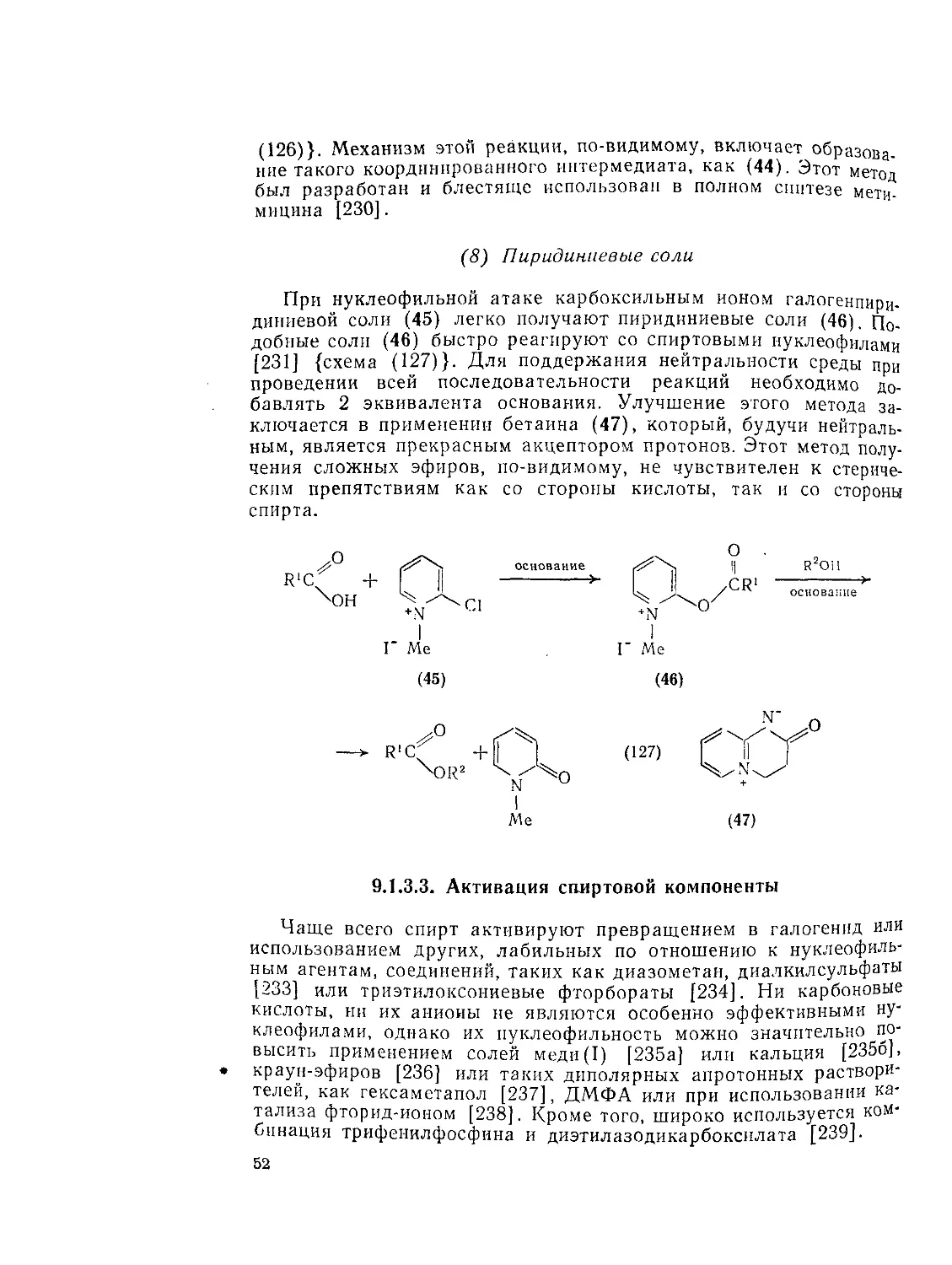
Основной путь этой реакции показан на схеме (123). Интермедиат (37) может непосредственно реагировать с добавленным нуклеофилом или может быть превращен в другое активированное производное (38), например азид кислоты [218], которое затем можно выделить [219]. Аминофосфониевые соли (39) [220] и (40) [221] можно использовать аналогичным образом.
(6) Реакции в присутствии соединений пятивалентного фосфора
Эти реакции представлены на примере использования N-фос-фониевых солей [222] пиридинов {схема (124)}, которые получаются из эфиров фосфорной кислоты и пиридина в отсутствие или при добавлении окисляющего агента, например иода [223]. Последовательность прибавления кислоты и спирта может быть обратной
О
II
HP(OPh)2
II + РУ + Ь —►
HOP(OPh)2
ру+ I 'О—Р—I Pho/ \эри
RCOOH
РУ+ I ’О—Р—OCOR PhC)/ \(3Ph
(124)
50
(7) Тиоэфиры
Сложные эфиры тиопиридона (41), получаемые [225] обработкой кислот 2,2'-дипиридилдисульфидом и трифенилфосфином, чрезвычайно чувствительны к атаке нуклеофильных реагентов; удобнее вести реакцию в присутствии ионов серебра [226]. Этот метод идеально подходит для внутримолекулярной лактонизации [227] {схема (125)}, когда циклизация протекает за счет электростатических сил. Тем не менее бис(1-метилимидазолил-2)дисульфид (42) является наиболее предпочтительным реагентом [228] для синтеза лактонов.
S-трет-бутиловые сложные эфиры (43) обладают
Поскольку
‘ ---- ч-. vj? riрд Di
умеренной устойчивостью, их можно использовать для защиты карбоксильной группы. Однако добавление спирта и трифторацетата ртути(II) вызывает [229] быструю переэтерификацию {схема
TlS(Bu-rper) R2OH
R!C^ -------------> Rl4 ---------->- R’<T
\cn Hg(OCOCFs)2 \лг»9
xSBu-rper XOR2
(43) (126)
О R2OH
R1
Ви-трет
(44)
5/
(126)}. Механизм этой реакции, по-видимому, включает образование такого координированного интермедиата, как (44). Этот метод был разработан и блестяще использован в полном синтезе мети-мицина [230].
(8) Пиридиниевые соли
При нуклеофильной атаке карбоксильным ионом галогенпири-диниевой соли (45) легко получают пиридиниевые соли (46). Подобные соли (46) быстро реагируют со спиртовыми нуклеофилами [231] {схема (127)}. Для поддержания нейтральности среды при проведении всей последовательности реакций необходимо добавлять 2 эквивалента основания. Улучшение этого метода заключается в применении бетаина (47), который, будучи нейтральным, является прекрасным акцептором протонов. Этот метод получения сложных эфиров, по-видимому, не чувствителен к стериче-ским препятствиям как со стороны кислоты, так и со стороны спирта.
(127)
(47)
9.1.3.3. Активация спиртовой компоненты
Чаще всего спирт активируют превращением в галогенид или использованием других, лабильных по отношению к нуклеофильным агентам, соединений, таких как диазометан, диалкилсульфаты [233] или триэтилоксониевые фторбораты [234]. Ни карбоновые кислоты, ни их анионы не являются особенно эффективными ну-клеофилами, однако их нуклеофильность можно значительно повысить применением солей меди(1) [235а] или кальция [2356], краун-эфиров [236] или таких диполярных апротонных растворителей, как гексаметапол [237], ДМФА или при использовании катализа фторид-ионом [238]. Кроме того, широко используется комбинация трифенилфосфина и диэтилазодикарбоксилата [239].
52
9 1 4. ДРУГИЕ РЕАКЦИИ (ПОМИМО АЦИЛИРОВАНИЯ) 9.1.4.1. Декарбоксилирование
Результаты этого общего процесса будут классифицированы в соответствии с образующимися продуктами, причем мы рассмотрим только оптимальные методы получения этих продуктов. Хорошее сопоставимое рассмотрение нескольких типов декарбоксилирования включено в обзор [240] по окислительному декарбоксилированию с применением ацетата свинца (IV). Первые пять процессов, описанных ниже, в основном включают превращение карбоксильной группы с помощью серии одноэлектронных окислительных процессов, приводящих через радикалы к карбениевым ионам или к другим катионам {схема (128)}. Эффективность используемых методов отражает способность радикалов и (или) карбениевых ионов к гибели или превращениям.
/>
RC ---------- RC, > R. —► Продукты
\он -» 2
|-е- |-е- (128)
,0
R<V ——-* R+ ——> Продукты
\о+ -со2
(/) RCOOH-+RR
Этот процесс, известный как реакция Кольбе [241], протекает при электролитическом анодном окислении солей щелочных металлов. Недавние исследования [2421 показали, что интермедиатами служат не ацилокси-катионы (48), как считалось ранее, а более бедные энергией изомерные диоксирильные катионы (49) {схема (129)} [243].
(48)
(129)
—> R+ + CO2
(2) RCOOH->RH
Действие ацетата свинца (IV) в хлороформе эффективно де-ар оксидирует первичные и, в меньшей степени, вторичные кис-“до алканов; Декарбоксилирование третичных карбоновых ynwun пРив°Дит в основном к алкенам. Порошок меди в хинолине чемл гг применять не во всех случаях. Его использование ограни-лотами ;ненасыщенными пли арилсодержащими карбоновыми кис-
, но и в этом случае выходы непостоянны.
53
(,?) RCOOH -> Алкен
Система ацетат свинца(IV)—ацетат ртути(II) эффективно превращает [240] многие кислоты в соответствующие алкены. Декар-боннлпрование хлорапгидридов кислот, имеющих Р-водородный атом, под действием трис (трифенилфосфин) родий (I) хлорида приводит к алкенам [244] по типу расщепления Зайцева. Термодинамические параметры этой реакции предполагают радикальный механизм. р,у-Ненасыщенные кислоты претерпевают термическое декарбоксилирование с миграцией двойной связи {схема (130)},
Изо)
(4) R'COOH —>R'OCOR2
При использовании ацетата свинца(IV) наряду с алкенами обычно получаются сложные эфиры уксусной кислоты, однако выходы последних редко достигают препаративно используемых. Превращение карбоксильной группы в гидроксильную с сохранением конфигурации при углеродном атоме, соседнем с карбоксильным, использовано в синтезе простагландинов [246] и при некоторых трансформациях пенициллинов [247]. В последнем случае спирт получают в виде о-нитробензоата, из которого спирт можно выделить обработкой цинковой пылью {схема (131)}. На основании результатов по конкуренции за кислородные атомы считают, что механизм этой интересной реакции включает рекомбинацию радикальной пары [248].
ClCOQEt.EtjN;
нх 4COOH^ww^
OCOAr
(5) RCOOH-+RX
Декарбоксилирование с замещением на галоген ществить несколькими путями. Для синтеза бромидов в основном применяют реакцию Хунсдиккера [249] {схема (132)}, которая лучше всего проходит с первичными кислотами. Последние улуч-шения состоят в применении солей ртути (II) ]250] и таллия(I). [2э1],
можно осу-
54
о
RCT + Br2 —> RBr + CO2 + AgBr (132)
XOAg
Декарбоксилирование ацетатом свинца (IV) в присутствии хлоридов металлов приводит с хорошими выходами к алкилхлоридам. Основное преимущество этого метода перед реакцией Хунсдиккера заключается в отсутствии структурных изменений [240], что говорит против участия в реакции карбениевых ионов. Этим методом хорошо декарбоксилировать первичные и вторичные кислоты. Третичные карбоновые кислоты реагируют несколько хуже, однако применение N-хлорсукцинимида в качестве донора галогена [252] позволяет повысить выходы в этих случаях.
Иоддекарбоксилирование [253] карбоновых кислот можно провести с помощью облучения раствора кислоты в присутствии ацетата свинца (IV) и иода. Метод дает хорошие выходы продуктов в случае первичных и вторичных кислот.
Хлорангидриды кислот, в которых присутствует хиральный сс-углеродный и отсутствуют p-водородные атомы, в присутствии трис (трифенплфосфин) родий (I) хлорида декарбоксилируются с образованием смеси рацемических алкилхлоридов [244].
(5) RCOOH ->RNH2
Это превращение карбоновых кислот включает образование промежуточного изоцианата (50), который далее гидратируется до карбаминовой кислоты. Последняя самопроизвольно отщепляет диоксид углерода, что приводит к первичному амину {схема (133)}. Сами изоцианаты можно получать двумя путями: пиролизом или фотолизом азида кислоты (реакции Курциуса [254] и Шмидта [255]) или с помощью катализируемой основаниями перегруппировки N-галогенамидов или родственных соединений (перегруппировка Гофмана [256]).
RN=C=O
(50)
(133)
(х= С1,Вг)
[bnhcooh] —RNHg
65
В обоих случаях реакция идет по согласованному механизму [257]' причем ацилнитрены не принимают участия в образовании изоцианата. Последние улучшения реакций Курциуса и Шмидта были направлены па упрощение способов получения необходимых азидов кислот. В частности, введение в практику азидных реагентов (51) [218] и (52) [258] и использование солей (53) [259] н (54) [260] как источников азид-ионов, растворимых в органических растворителях, позволяет проводить реакцию с хлорангидри-дами кислот в гомогенных условиях.
(PhO)2PN3 Me3SiN3 руН+ N3 (h-Bu)4N+ N7
II
О
(51) (52) (53) (54)
NaOMe.MeOH, Br2,-40 до-15"С t 50"C *’
NHCOOMe
(134)
При использовании низкотемпературной неводной модификации [261] перегруппировки Гофмана, как показано на примере реакции по схеме (134), промежуточный изоцианат можно зарегистрировать в виде соответствующего карбамата.
9.1.4.2. Галогенирование и окисление
Этот раздел будет кратким, так как полный обзор этих реакций будет дан в разд. 9.3.1. В общем случае кислоты подвергаются а-галогенированию при обработке соответствующих хлорангидри-дов источником положительно заряженных галогенид-ионов, таким, как N-галогенсукцинимид пли иод [262] {схема (135)}. Фосфор и его тригалогениды применяются для катализа реакции кислот с молекулярным хлором или бромом. Природа интермедиатов в этих реакциях не установлена, но вероятно, это соответствующие производные кетенов.
soci2
RCH2COOH ----->- RCH2COC1
Вг
N-бромсукцинимид I -----------------> RCHCOC1
Cl
N-хлорсукцинимид I ------------------> RCHCOC1
HCI
I h. HI I
------------------> RCHCOC1
(135)
R'CH^OOR2
LiN (Pr-uao)j;
Br2 или I2
R’CHCOOR2
I
X X = Br, I
(136)
и бром реагируют аналогичным образом с анионами слож-ных эфиров [263], приводя к а-галогензамещенным сложным эфи*
56
пам {схема (136)}. а-Фторкислоты можно получать диазотированием [264] а-аминокислот в смеси поли(HF)—пиридин.
Адсорбция кислот с длинной цепью на поверхности раздела тетрахлорид углерода — оксид алюминия приводит к плотной упаковке молекул кислоты перпендикулярно поверхности таким образом что алкильные цепи закрыты, а снаружи находятся лишь терминальные метильные группы. Свободнорадикальное хлорирование в этих условиях резко повышает выход продуктов хлорирования концевой метильной группы [265]. Результат, сходный с хлорированием аминов по Гофману-Лефлеру, наблюдался при свободнорадикальном хлорировании кислот с длинной цепью в сильнокислой среде. Этот процесс, являющийся результатом перегруппировки МакЛафферти в растворе {схема (137)} объясняют [266] образованием кислородных катион-радикалов. В случае гексановой и октановой кислот помимо избирательного хлорирования по С-4, получаются сравнимые количества продуктов терминального хлорирования. Радикальное хлорирование высших кислот и их сложных эфиров, инициируемое катион-радикалом азота [267, 268], проходит с высокой селективностью по —1-положению, причем почти исключительно получается продукт монохлорирования.
Метилоктадеканоат и гомологичные сложные эфиры при окислении смесью хромовый ангидрид-уксусный ангидрид-уксусная кислота превращаются в монокетопроизводные; наблюдается некоторая избирательность [269] к атаке по биологически интересным участкам молекулы.
9.1.4.3. Восстановление
Кислоты можно восстанавливать до альдегидов (см. гл. 5.1 и 5.3) или более полно — до первичных спиртов (см. разд. 4.1.1). В данном разделе будет рассмотрено несколько избирательных методов восстановления.
Для восстановления кислоты до альдегида [270] обычно необходимо превратить кислоту в производное более чувствительное к нуклеофильным реагентам. Единственным исключением является восстановление литием в метиламине [271], когда образующийся альдегид захватывается и защищается растворителем в виде имина
57
< Пля этой цели применяют хлорангидриды кислот и
{схема (138)}.,Цля это! и ариловые и другие сложные фосфонатны р 1нек£ ые ампды. В качестве восстановителя Обычно используют алюмогидрид лития или продукт его взаимодействия со спиртом [274].
RCOOH "mShT” *CH-NMe RCH0 (138)
Гетероциклические производные кислот, такие, как метиодиды 2-И-замещенных оксазолинов (55) [275] (см. разд. 9.1.1.6) или триазолиевые соли (56) [276], восстанавливают борогидридом натрия с предварительным ос-алкилированием или без него {схема (139)}. Отсутствие необходимости применять такой жесткий восстановитель, как алюмогидрид лития, является одним из преимуществ этого метода.
Хлорангидриды [277] и ангидриды кислот [278] при взаимодействии с динатрийтетракарбонилферратом(П) дают ацилфер-раты (57), которые при электрофильном расщеплении уксусной кислотой дают альдегиды. Последнее усовершенствование этого метода состоит [279] в использовании смешанных ангидридов карбоновых кислот {схема (140)}.
R^SiH
-7Г RCH0
58
Хлорангидриды кислот, и особенно ароматических кислот, гидрируются до альдегидов над дезактивированным катализатором (реакция Розенмунда) [280]. Более удобным и, возможно, более эффективным является восстановление триэтилсиланом в присутствии переходных металлов [281], хотя разветвление в a-положении несколько снижает выход {схема (141)}.
Алюмогидрид лития восстанавливает свободные кислоты до первичных спиртов, однако такой мощный восстановитель нс обладает функциональной избирательностью. Борогидрид натрия и другие борогидриды в этом случае лишь приводят к соответствующим солям кислоты. Тем не менее среди большинства функциональных производных карбоновые кислоты наиболее склонны к восстановлению дибораном [282]. Такая реакционная способность объясняется первоначальным образованием триацилоксиборанов (58), которые быстро восстанавливаются до спиртов. Аналогичные интермедиаты, полученные из кислот с длинной цепью, дисмутируют ![283] и, по-видимому, восстанавливаются через ангидриды (59) и (60) {схема (142)}.
Элегантный пример избирательного восстановления карбоновых кислот и сложных эфиров продемонстрирован [284] в синтезе (У?) - и (З)-мевалонолактонов {схема (143)}: борогидрид лития восстанавливает сложноэфирную, а диборан — карбоксильную группы.
ВгНв
RCOOH —(RCOO)3B —> (RCO).O + [(RCOO)2B]2O
(58) (59) (60)
B2He
RCH2OH
в2нв
<------
(142)
МеООС СООМе
эс*гераза печени свиньи
МеООС СООН
9.1.4.4. Реакции с металлорганическими соединениями
Как реактивы Гриньяра [285], так и литийорганические соединения, реагируют с карбоновыми кислотами с образованием соответствующей соли карбоновой кислоты и углеводорода, получающегося из металлорганического соединения. Магниевые соли .карбоновых кислот (61) относительно инертны к дальнейшей реакции, однако проведение реакции в более жестких условиях приводит
59
к третичным спиртам. В отличие от магниевых солей литиевые соли карбоновых кислот (62) легко реагируют с литийорганическими реагентами, что дает чрезвычайно важный путь синтеза кетонов [286], включая винилкетоны [287] (см. гл. 5.2 и 5.4). Успех этого метода обусловлен как большей нуклеофильностью реагента, так и значительной устойчивостью связи Li—О. Как следствие этого литиевая соль карбоновой кислоты (62) становится более ковалентной, и увеличивается ее растворимость в органическом растворителе. Кроме того, возрастает электрофильность карбонильной группы с одновременной стабилизацией дилитпевых производных (63), которые родственны соответствующим магниевым производным {схема (144)}. Значительным недостатком метода является ограничение его применения только к субстратам, не содержащим функциональных групп, или, иными словами, к инертным или защищенным субстратам. Ацилгалогениды легко вступают в реакции замещения [288] с различными медворганическими и органо-купратными реагентами [289] {схема (145)}. Применяемые реагенты достаточно мягки и атакуют только активированную карбонильную группу ацилхлорида, не затрагивая других функциональных групп, включая синтезируемый кетон. Например, а-хлорке-тоны можно получить [290] из хлорангидридов а-хлоркислот.
OMgX ОН
I R2MgX; |
R1—С—OMgX —> R’COR2----------► R1—С—R2 (144)
| H3O+ I
R2 R2
R2Li
R2MgX |
OLi
-----'----- I H3O+
R’COOH+R2M —> R’CX + R2H R’—C—OLi -----> R’COR2
Ndm i
R2
(63)
(61) M — MgX
(62) M = Li
^O
R’C^ + LiCuRf —> R’COR2 045)
^X
RCOOH + AlMe3 —> RCMe3 (146>
Сообщалось [291], что алкилмагнийгалогениды в гексаметаполе реагируют со сложными эфирами алифатических кислот, что приводит с хорошими выходами к соответствующим кетонам, преимущественно в виде их наименее замещенных енолятов. Триметил-алюминий исчерпывающе метилирует [292] карбоксильные группы как арил-, так и алкилкарбоновых кислот до трет-бутильной группы {схема (146)}.
60
9.1.4.5. Реакции ненасыщенных карбоновых кислот
В этом разделе рассмотрены реакции, проходящие по кратным связям алкеновых и алкиновых карбоновых кислот.
(1) Восстановление
Двойную связь в аф-ненасыщенных алкеновых кислотах можно восстанавливать различными реагентами, в том числе и растворами металлов в жидком аммиаке [293, 294] или в гексаметаполе [295], причем эти системы часто позволяют провести селективное восстановление. Другие несопряженные алкеновые кислоты инертны к подобным реагентам, однако могут быть восстановлены каталитически [296].
Как сопряженные, так и несопряженные алкиновые кислоты восстанавливаются сольватированными электронами [293] до (f)-алкеновых кислот; каталитическое гидрирование над катализатором Линдлара [296] приводит к (Z)-геометрическим изомерам.
(2) Окисление и галолактонизация
Алкеновые кислоты окисляются обычным набором алкеновых окислителей с образованием соответствующих фрагментов. Циклический манганатный сложный эфир, который, как предполагалось, образуется при окислении алкенов до диолов, был обнаружен в виде (64) {схема (147)} и определен спектрофотометрически [297].
R\<^X'COOH
R = Me или Ph
(147)
(64)
В то время как аф-ненасыщенные кислоты дают продукты простого присоединения галогенов по двойной связи, соли |3,у- и у,6-ненасыщенных кислот при реакции с галогенами, особенно с иодом и бромом, приводят к галогенолактонам {схемы (148), (149)} [298]. Соли Р,у-алкеновых кислот в качестве кинетических продуктов образуют (3-лактоны [299], которые затем перегруппировываются в термодинамически более устойчивые у-лактоны. Подбирая подходящие условия, особенно используя двухфазные системы и применяя соответствующие карбоксилаты таллия [300], можно легко выделить кинетические продукты этой реакции.
61
X
coo
у,б-Алкеновые кислоты дают у-лактоны как в кинетически, так и в термодинамически контролируемых условиях [300,301], однако некоторые особые стерические эффекты направляют реакцию в сторону образования обычно менее стабильных б-лактонов.
9.1.4.6. Реакции ароматических карбоновых кислот
В этом разделе будут обсуждены восстановление ароматического кольца и замещение в ядро ароматических карбоновых кислот.
(1) Электрофильное ароматическое замещение
В силу того что карбоксильная группа из-за своих электроноакцепторных свойств сильно дезактивирует ароматическое кольцо для последующего замещения, не описано ни одного простого примера алкилирования бензойной кислоты [302]. Ацилирование также довольно трудно осуществить, однако галогенирование, по-видимому, идет успешнее и наблюдается образование ожидаемого продукта .мета-замещения. Ареновые кислоты с такими активирующими заместителями, как гидроксильная группа, значительно реакционноспособнее. Тем не менее большое число замещенных ареновых кислот получают в основном окислением боковой цепи (см. разд. 9.1.1.1) или за счет другого родственного процесса из предварительно синтезированного субстрата.
(2) Восстановление
По той же причине, что указана выше, бензойные кислоты активированы по отношению к восстановлению щелочными металлами в жидком аммиаке [303]. Они быстро восстанавливаются [304], несмотря на образование карбоксилат-аниона, давая в качестве обычных продуктов 1-замещенные 1,4-дигидробензолы, причем промежуточный анион можно алкилировать до протонирования [305] {схемы (150), (151)}. Целый ряд алкилбензойных кислот был восстановлен таким образом, и результаты этих опытов приведены в виде таблиц [306].
62
соон
соон
соон
Бензойные кислоты подвергаются гидрогенизации над родием или никелем Ренея в условиях, сходных с теми, какие необходимы для гидрогенизации алкилбензолов [296]. В этом случае рекомендуют вначале этерифицировать кислоты, так как сложные эфиры гидрируются легче, чем сами кислоты.
9.1.4.7 Защита карбоксильной группы
В табл. 9.1.3 представлен набор групп, применяемых для зашиты карбоксильной группы в карбоновых кислотах.
Простейший метод защиты [307] состоит в превращении карбоновой кислоты в ее метиловый эфир (65) обработкой диазометаном. Метиловые эфиры относительно устойчивы в кислой среде, но легко разрушаются при продолжительной обработке (обычно более 30 мин) водным раствором основания или, если требуется избирательность расщепления в присутствии других сложных эфиров, путем ^2-расщепления [72] подходящим нуклеофилом. 9-Антрилметильная система (66) позволяет защитить карбоксильную группу к действию различных условий; эта защитная группировка быстро снимается анионом метантиолата [308]. Относительная стабильность силиловых эфиров (67) карбоновых кислот {схема (152)} к действию азотистых оснований [309] и к мягким окислителям [310] в совокупности с их быстрым расщеплением в очень мягких условиях обработкой метанолом или этанолом делает их весьма привлекательными защитными группами. Использование подобных сложных эфиров для защиты С-3 карбоксильной группы пенициллина в процессе иминохлоридного расщепления боковой цепи послужило главным, что позволило реально выйти [311] к 6-аминопенициллановой кислоте {схема (153)}. Аналогичный подход позволил значительно улучшить [311] методику, выход и качество продукта при синтезе 7-аминоцефалоспорановой кислоты.
^он
RgSiCl, Et3N -----------> -<---------
МеОН
(152)
(67)
63
Таблица 9.1.3. Защитные группы, используемые для защиты карбоксильной функции в карбоновых кислотах
Защищенная кислота Условия сохранения защитной группы Условия расщепления
RCOOMe RCOOCH2 (05) Н3О+, НО~(мягкие) НО’, х"
oio (66) Н3О+, НО" (мягкие) MeS"
R’COOSiRl (07) N-Хлорсукцинимид, r3nh2, РС15 МеОН
RCOOBu-rper (68) НО" Н3О+
RCOSBu-rper (69) Н3О+, НО’ Hg2+
RCOOPh (70) Н3О+, НО' (мягкие) НОО', pH 10,5
RCOOCH2CH2SO2Ar
RCOOCH2SMe
RCOOCH2COPh RCOOCH2CC13 RCOO(CH2)2X RCOO(CH2)4-5X
(71) Н3О+, НО (очень мягкие) 1,5-Диазабицикло-[4,
3, 0] нонен-5; CN", Н2О
(72) НО' (мягкие) См. схему (156)
(73) Н3О+, НО' (мягкие) Zn, АсОН; NaSPh
(74) Н3О+, ОН' (мягкие) Zn, АсОН
(75) Н3О+, ОН' (мягкие) Na2C S 3
(76) Н3О+, ОН" (мягкие) Na2S
N
(77) LiAlH4, R2MgX R2OH, H+
RCON(Pr-M3o)NH(Pr-«3o) (78) H3O+, HO
Ce(IV), Pb(OAc)4, N-бромсукцинимид
RCOOCHjCO—
OMe (79)
(80)
RCOOCH(Me)COPh RCOOCHCOPh
H3O+, ОН" (мягкие)
H3O+, ОН" (мягкие)
H3O+, ОН" (мягкие)
H3O+, ОН" (мягкие)
Av, >313 нм
Av, >313 нм
Av, >320 нм
Av, >320 нм
64
Табл. 9.1.3 (продолжение)
Защищенная кислота Условия сохранения защитной группы Условия расщепления
Av, >350 нм
R3 = н или ОМе
RCOOCH2
О
Н3О+, ОН" (мягкие)
hv или мягкое восстановление
н н RCONH = =
РУ.
Me3SiCI
рс15.ру; н н
R3OH; H,N = ? (NH4)HCO3)H2O
соон
COOSiMe3
(153)
соон
трет-Бутиловые эфиры кислот (68) получают катализируемым кислотой присоединением 2-метилпропена к карбоновым кислотам [312]; реакцией хлорангидрида кислоты с трет-бутилатом лития [208] или переэтерификацией в присутствии молекулярных сит [313]. Эти эфиры устойчивы к основному гидролизу, однако легко расщепляются кислотой по реакции обратной их образованию {схема (154)}.
II н+.
R<\ + || RC; (154)
\)Н Нз° Х)Ви-трет
(68)
Фениловые эфиры (70) были предложены [314] для защиты карбоксильной группы в пептидном синтезе, так как их расщепление под действием высоконуклеофильного гидроперокси-аниона проходит в мягких условиях без рацемизации. 1,5-Диазабнцик-ло[4.3.0] нонен-5 легко и быстро элиминирует карбоксилатные анионы из 2-толуол-/г-сульфоннлэтиловых эфиров (71) {схема (155)}. Этот альтернативный метод расщепления [217] позволяет избежать риска вторичного гидролиза при применении водного основания [315].
3 Зак, 398
65
Метилтиометиловые эфиры (72) проявляют нормальную устой чивость к действию оснований, однако легко расщепляются в нейтральных или кислых условиях [316] {схема (156)}. Фенациловые эфиры карбоновых кислот (73) получают при реакции фенацил-бромида с солью кислоты [317] или, что более удобно, прямой реакцией, катализируемой фторид-ионом [238]. Эти эфиры расщепляются при комнатной температуре цинком в уксусной кислоте Следует отметить их применение для образования у-гидроксислож-ных эфиров из у-лактонов {уравнение (157)}, иначе говоря, для непрямой трансформации. Трихлорэтиловые эфиры (74) также лабильны по отношению к восстановлению, как элегантно показано в полном синтезе [318] цефалоспорина С. Изучено применение этих сложных эфиров в пептидном синтезе [319].
О—СН2-т-СН—$О2Аг
> RCOO" + CH2=CHSO2Ar
Н
(71)
в:
Дианион тритиокарбоната мягко расщепляет 2-галогенэтило-вые сложные эфиры (75) {схема (158)}, что показывает [320] потенциальную возможность использования таких защитных групп. В сходной реакции [321] сульфид натрия расщепляет ©-хлоралкиловые сложные эфиры (76).
R'COOH
R’COOR2 <156)
R’CONHR3
Sp—COOEt (157) COOCHoCOPh
Детально исследовано [322] использование оксазолинов (77) для защиты кислот от действия реактивов Гриньяра или гидридных восстанавливающих агентов. Для защиты кислот рекомендован также М,М-диизопропилгидразид [323]. Получаемые гидразиды (78) гладко регенерируют исходные кислоты при обработке различными окислителями [324], причем гидразидный фрагмент отщепляется в виде газообразных продуктов. Заслуживает внима-
66
пяя также возможное использование слабоосновных моноацилги-ппазинов (78) в экстракционных процессах.
ДР В качеств'е УФ-фотолабильных защитных групп рекомендованы замещенные фенациловые эфиры (79) [325], (80) [325], (81) 4961 а также сложноэфирная группа на полимерном носителе ИЙ) [327] (см табл. 9.1.3). Сходные свойства были описаны для янилидов (83) [328]. В процессе пептидного синтеза карбоксиль-4vto TDvnnv можно защищать превращением в гидроксиметилан-щахиноновые эфиры (84). Такие эфиры легко расщепляются при фотолизе или при мягком восстановлении [329].
RCOO* Na** (158)
РСОО(СНз)лХ
(75) тг~2
3.
4.
5.
ЛИТЕРАТУРА
1. Н. О. House, «Modern Synthetic Reactions», 2nd edn., W. A. Benjamin, Menlo Park, California, 1972.
2. M. Fieser and L. F. Fieser, «Reagents for Organic Synthesis», Wiley-Inter-science, New York, vols. 1—6, 1967—1977 [27. Физер, M. Физер. Реагенты для органического синтеза. Пер. с англ./Под ред. И. Л. Кнунянца, Р Г Ко-стяновского, т. 1—7. М., Мир, 1970—1978].
С. A. Buehler and D. Е. Pearson, «Survey of Organic Syntheses», Wiley-In-terscience, New York, vol. 1, 1970 and vol. 2, 1977 [К. Бюлер, Д. Пирсон. Орг. синтезы. Пер. с англ. ч. 1—2. М., Мир, 1973].
ши H,ar[lS0n and s- Harrison, «Compendium of Organic Synthetic Methods», Wiley-Intersdence, New York, vol. 1, 1971 and vol. 2, 1974.
Щ70-?079ер°1г1 n °rganic Synthesis», ed. J. McMurry and R. B. Miller, S R WikAnedio?3.B->M1AleinanJ..L- S' Hegedus> 1973; ed L. S. Hegedus and New York ' ed‘ filler and L. G. Wade, 1975, Academic Press, Vo^S?S. Karger BaseL^ Chemistr^ ed‘ W- Theilheimer, up to 1976, «Rod'd’s’ CheJSv гС'РкeS г OrganLc Synthesis», Methuen, London, 1968. and ed M F Ane |CQrbOi Compounds» 2nd edn., ed. S. Coffey, Part Iе, 1973. ’ ’ Anse ’ Supplement to lc/ld, Elsevier, Amsterdam, 1965 and
6.
7.
8.
3*
67
9. F. D. Gunstone, in «Aliphatic Chemistry», volume 1, chapter 3 ed W d i and volume 3, chapter 5, ed. A. McKillop, Specialist Periodical РеппХт^' Chemical Society, London, 1973 and 1975. reports, The
10. E. W. Colvin, in «Aliphatic Chemistry», volumes 1 and 2 ed W Pari^ , mes 3 and 4, ed. A. McKillop, chapter 2, Specialist Periodical Reports'™*» Chemical Society, London, 1973, 1974, 1975, 1976. reports, rhe
11. «Oxidation in Organic Chemistry», part A,’ed. К. B. Wiberg, Academic Pre« New York, 1965. ness,
12. J. Rocek and C. S. Ng, J. Org. Chem., 1973, 38, 3348.
13. P. Eaton and R. H. Mueller, J. Amer. Chem. Soc., 1972, 94, Ю14- «Oxidation in Organic Chemistry», part B, ed. W. S. Trahanovsky, Academic' Press, New York, 1973.
14. D. J. Sam and H. E. Simmons, J. Amer. Chem. Soc., 1972, 94, 4024
15. J. R. Ruhoff, Org. Synth. Coll. Vol. 2, 1943, 315 [Дж. Ру’хоф’ф. Синтезы opr препаратов. Сб. 2 — Пер. с англ./Под ред. Б. А. Казанского. М., Издатин-лит, 1949, с. 571].
16. С. Moureu and R. Chaux, Org. Synth. Coll. Vol. 1, 1941, 166 [С. Муре, P. Шо. Синтезы орг. препаратов, сб. 1, —Пер. с англ./Под ред. Б. А. Казанского. М., Издатинлит, 1949, с. 487].
17. Е. Catnpaigne and \V. М. LeSuer, Org. Synth. Coll. Vol. 4, 1963, 919.
18. E. J. Corey, N. W. Gilman, and В. E. Ganem, J. Amer. Chem. Soc., 1968 90, 5617.
19. T. A. Geissman, Org. Reactions, 1944, 2, 94 [T. А. Гейсман. Opr. реакции, сб. 1. — Пер. с англ./Под ред. А. Я. Берлина. М., Издатиилит. 1948, с. 106].
20. S. Selman and J. F. Eastham, Quart. Rev., 1960, 14, 221.
21. P. Deslongchamps, P. Atlani, D. Frehel, A. Malaval, and C. Moreau, Canad. J. Chem., 1974, 52, 3691 и цитированные там работы.
22. Т. Yamagishi, Т. Yoshimoto, and К. Minami, Tetrahedron Letters, 1971, 2795.
23. С. H. Hassall, Org. Reactions, 1957, 9, 73; J. B. Lee and В. C. Uff, Quart. Rev., 1967, 21, 449.
24. Для теоретической модели см. V. A. Stoute, М. A. Winnik, and I. G. Crizma-dia, J. Amer. Chem. Soc., 1974, 96, 6388.
25. M. J. Bogdanowicz, T. Amberlang, and В. M. Trost, Tetrahedron Letters, 1973, 923; E. J. Corey, Z. Arnold, and J. Hutton, ibid., 1970, 307; E. J. Corey and R. Noyori, ibid., 1970, 311. e
26. A. McKillop, В. P. Swann, and E. C. Taylor, J. Amer. Chem. Soc., 1973, 95, 334°.
27. P. R. Story, B. Lee. С. E. Bishop, D. D. Denson, and P. R. Busch, J. Org. Chem., 1970, 35, 3059 n цитированные там работы; P. R. Story and P. Busch, in «Advances in Organic Chemistry», ed. E. C. Taylor, Wiley-Interscience, 1972, volume 8, p. 67; B. Lee, P. R. Story, and J. E. Sanderson, J. Org. Chem., 197b, 41, 2314.
28. G. Quinkert, Angew. Chem. Internat. Edn., 1965, 4, 211.
29. R. G. Carlson, J. H. A. Huber, and D. E. Henton, J. C. S. Chem. Comm., 19/3,
223
G. K- Chip and T. R. Lynch Canad. J. Chem., 1974, 52, 2249.
R. C. Fuson and B. A. Bull, Chem. Rev., 1934, 15, 275.
L. L Smith, W. W. Prichard, and L. J. Spillane, Org. Synth. Coll. Vol. 3, 19bb, 302
E. ’ J. Corey, J. A. Katzenellenbogen, N. W. Gilman, S. A. Roman, and В. IV Erickson, J. Amer. Chem. Soc., 1968, 90, 5618.
R. D. Clark and С. H. Heathcock, Tetrahedron Letters, 1974, 1713, 2U2/.
I. 1. Borowitz, V. Bandur co, M. Heyman, R. D. G Rigby, and J. Heng, J. Org. Chem., 1973, 38, 1234 и цитированные там работы; A R- Maha/an H. C. Araujo, Synthesis, 1976, 111.
36. J. Becker and G. Ohloff, Helf. Chim. Acta, 1971 54, 2889.
37. R. A. Wohl, Helv. Chim. Acta, 1973, 56, 1826; Tetrahedron Letters 1973, 30 .
38. L R. Schaeffer and A. O. Snoddy, Org. Synth. Coll Vol. 4 1963, 19
39. L Rocek and A. Riehl, J. Amer. Chem. Soc., 1967, 89, 6691; J. Org. Chem.,
30.
31.
32.
33.
34.
35.
1967, 32, 3569.
63
40 H. Корта, S. Takahashi, and H. Hagihara, Tetrahedron Letters, 1973, Л Hamlin and A W Weston, Org. Reactions, 1957, 9, 1 [К. Э. Хэмлин, 4L А. У. Уэстон. Орг. реакции, сб. 9.-Пер. с англ./Под ред. И. Ф. Луценко.
42. Tetrahedron Letters, 1964, 3251 и цитирован-
ий FbievaBr1ownTbSynthesis, 1975, 358; Al. Carmack and M. A. Spielman, Org.
Lpfinnq 1946 3, 83 [Al. Кармак, M. А. Шпильман. Орг. реакции, сб. 3,— Про с англ/Под ред. К. А. Кочешкова. М., Издатинлит, 1951, с. 88].
44 A S Kende, Org. Reactions, 1960, 11, 261 [Э. С. Кенде. Орг. реакции, ' cg fl — Пер. с англ./Под ред. И. Ф. Луценко. М., Мир, 1965, с. 267]; для примеров см. D. W. Goheen and W. R. Vaughan, Org. Synth. CoR Vol. 4, 1963, 594, and C. Rappe, Org. Synth., 1973, 53, 123.
45 L G. Donaruma and W. Z. Heldt, Org. Reactions, 1960, 11, 1; D.H^R.Bar-ton, M. J. Day, R. H. Hesse, and M. M. P echet, Chem. Comm., 1971, 945 [Л.’Г. Донарума, В. 3. Хельдт. Орг. реакции, сб. 11, —Пер. с англ./Под ред. И. Ф. Луценко. М„ Мир, 1965, с. 7].
46. Р. A. S. Smith, in «Molecular Rearrangements», ed. P. de Mayo, Wiley-Inter-science, New York, 1963, volume 1, p. 501.
47. R. U. Lemieux and E. von Rudloff, Canad. J. Chem., 1955, 33, 1701, 1710; E. von Rodloff, ibid., p. 1714; S. W. Pelletier, K. N. Iyer, and C. W. J. Chang, J. Org. Chem., 1970, 35, 3535.
48 A McKHlop, О. H. Oldenziel, R. L. Robey, В. P. Swann, and E. C. Taylor, ’ J.’Amer. Chem. Soc., 1971, 93, 7331.
49. A. McKillop, Pure Appl. Chem., 1975, 43, 463.
50. /. B. Bush and H. Finkbeiner, J. Amer. Chem. Soc., 1968, 90, 5903.
51. E. I. Heiba, R. M. Dessau, and W. J. Koehl, J. Amer. Chem. Soc., 1968, 90, 5905; E. I. Heiba, R. M. Dessau, and P. G. Rodewald, J. Amer. Chem. Soc., 1974, 96, 7977.
M. Okano, Chem. and Ind. (London), 1972, 423.
H.-H. Vogel, Synthesis, 1970, 99.
P. L. Southwick, Synthesis, 1970, 628.
55. G. I. Nikishin, M. G. Vinogradov, and T. M. Fedorova, J. C. S. Chem. Comm., 1973, 693.
56. fl. T. Эйдус, А. Л. Лапидус, К. В. Пузицкий, Б. К- Нефедов.—Усп. химии, 1971, 40, 806; 1973, 42, 442.
57. A. Schoenberg, I. Bartoletti, and R. F. Heck, J. Org. Chem., 1974, 39, 3318; A. Schoenberg and R. F. Heck, ibid., p. 3327.
58. J. Tsuji, «Organic Synthesis by Means of Transition Metal Complexes», Sprin-ger-Verlag, Berlin, 1975; A. P. Kozikowski and H. F. Wetter, Synthesis, 1976, 561; L. Cassar, P. Chiusoli, and F. Guerrieri, ibid., 1973, 509; C. W. Bird, Chem. Rev., 1962, 62, 283; C. W. Bird, «Transition Metal Intermediates in Organic Synthesis», Logos Press, London, 1967; R. F. Heck, Adv. Catalysis, 1977, 26, 323.
59’ ne C°4man’ S. R- Winter, and R. G. Komoto, J. Amer. Chem. Soc., 1973, 95, 249; J. P. Collman, Accounts Chem. Res., 1975, 8, 342.
™ RA Fel1 and F- J- McQuillin, J. C. S. Perkin I, 1972, 2096.
co и Haa^ Or£- synth., 1966, 46, 72.
62. H. Koch and W. Haaf, Org. Synth., 1964, 44, 1.
h a °V’r?'iem’ Ben, 1967, 100, 978; K. Bott and H. Hellman, in «Newer Met-° ‘ rcPa?tive Organic Chemistry», ed. W. Foerst, Academic Press, New York, 1977, vol. 6, p. 67.
r. Sourna H. Sano, and J. lyoda, J. Org. Chem., 1973, 38, 2016.
stance jSascd a!}dP- Rcinmuth, «Grignard Reactions of Nonmetallic Substances», Prentice-Hall, New Jersey, 1954, pp. 913—960.
J' JT Morton> OrS- Reaictions, 1955, 8, 258 [Г. Гилман. Opr.
1956 c 333] ° ^eP- c аигл-/Под ред. IO. А. Арбузова. M., Издатинлит, E- I. Corey and R. H. K. Chen, J. Org. Chem., 1973, 38, 4086.
52.
53.
54.
64.
65.
66.
67.
69 .
68. /. F. Normant, G. Cahiez, C. Chuit, and J. Villieras, J. OrganomebHiTTfi-— 1973, 54, C53. '-’‘ganometailic Chem..
69. H J. Bestmann, Th. Denzel, and H. Salbaum, Tetrahedron Letters 1974 io™ 70. E. Berliner, Org. Reactions, 1949, 5, 229 [3. Берлинер. ОрГ реакции’ К
M„ Издатинлит, 1951, c. 195]; A. G. Peto, in «Friedel Crafts an7 Reactions», ed. G. A. Olah, Wiley-Interscience, 1963, volume III chapter see also G. A. Olah and J. A. Olah, ibid., chapter 39. ₽
71. E. K. Euranto, in «The Chemistry of Carboxylic Acids and Esters» Pd <?
tai, Wiley-Interscience, New York, 1969, chapter 11. ’ ' '
72. J. McMurry, Org. Reactions, 1976, 24, 187.
F. Elsinger, J. Schreiber, and A. Eschenmoser, Helv. Chim. Acta I960 113. *
73.
34;
Pa-
43,
74. P. D. G. Dean, J. Chem. Soc., 1965, 6655.
75. J. McMurry and G. B. Wong, Synth. Comm., 1972, 2, 389.
76. G. I. Feutrill and R. N. Mirrington, Austral. J. Chem., 1972, 25, 1731.
77. P. A. Bartlett and W. S. Johnson, Tetrahedron Letters,' 1970,’ 4459.
78. P. Muller and B. Siegfried, Helv. Chim. Acta, 1974, 57, 987.
79. P. S. Manchand, Chem. Comm., 1971, 667.
80. D. H. Miles and E. J. Parish, Tetrahedron Letters, 1972, 3987; E. J. Parish and D. H. Miles, J. Org. Chem., 1973, 38, 1223.
81. M. E. Jung and M. A. Lyster, J. Amer. Chem. Soc., 1977, 99, 968.
82. R. B. Woodward, Pure Appl. Chem., 1973, 33, 145.
83. I. Shahak and У. Sasson, J. Amer. Chem. Soc., 1973, 95, 3440; об амид-аиио-иах см. также: Е. Н. White, ibid., 1955, 77, 6011.
84. Р. G. Gassman, Р. R. G. Hodgson, and R. J. Balchunis, J. Amer. Chem. Soc., 1976, 98, 1275; для затрудненных эфиров см.: Р. G. Gassman and W. W. Schenk, J. Org. Chem., 1977, 42, 918.
85. H. L. Vaughn and M. D. Robbins, J. Org. Chem., 1975, 40, 1187.
86. T. Mukaiyama, K. Kamio, S. Kobayashi, and H. Takei, Chem. Letters, 1973, 357.
87. S. E. Diamond, B. Grant, G. M. Tom, and H. Taube, Tetrahedron Letters, 1974,
4025.
88. S. Paraskewas, Synthesis, 1974, 574.
89. G. Jones, Org. Reactions, 1967, 15, 204.
90. F. W. Bergstrom, Chem. Rev., 1944, 35, 153.
91. A. P. Krapcho and D. S. Kashdan, Tetrahedron Letters, 1975, 707.
92 В M Trost and T. J. Fullerton, J. Amer. Chem. Soc., 1973, 95, 292;
В. M. Trost and T. J. Dietsche, ibid., p. 8200; В. M. Trost and P. E. Strege,
ibid., 1977, 99, 1649. ,n,„
A. P Krapcho and A. J. Lovey, Tetrahedron Letters, 1973 957
A P Krapcho, E. G. E. Jahngen, A. J. Lovey, and F. W Short Tetrahedron Letters, 1974, 1091; C. Liotta and F. L. Cook, ibid., p. Ю95; D. H. Hunter and R. A. Perry, Synthesis, 1977, 37. r 1071
Y.-N. Kuo, F. Chen, C. Ainsworth, and J. J. Bloomfield, Chem. Comm., 1971, B36M. Trost, H. C. Arndt, P. E. Strege, and T. R. Verhoeven, Tetrahedron Letters, 1976, 3477; В. M. Trost and T. R. Verhoeven, J. Amer. Chem. Soc., 19//, 99 3867
См., однако: H. D. Durst e. a. J. Org. Chem 1974, 39, 3271 Kobayashi, С. P. Casey and D. F. Marten, Tetrahedron Letters 1974, 925, 5. Kocayasm,
93.
94.
95.
96.
97.
98. Г'. UUdey U/m и- I . i’ll*' ---------- 1~Гк-.п 1AO7
H. Takei and T. Mukaiyama, Chem. Letters, 1973 l^y/- 9 Q]
99. R. M. Coates and J. E. Shaw, J. Org. Chem., 1970 35 12597, ^601.
100. К. K. Knutson and J. E. Shaw, J. Org. Chem., 1971, 3b, 1io w Fq_
101. H. Stetter, in «Newer Methods of Preparative Organic Chemist у , erst, Academic Press, New York, 1963, volume 2 p. • Pv-Interscience, New
102. A. /. Meyers, «Heterocycles in Organic Synthesis», Wiley 1
103. ар’, White and D. K. Wu J C. S Chem. Comm., 1974, 988..
104. H. Wynberg and A. Logothetis, J. Amer Chem. H 1979 3031.
Ю5. A. 1. Meyers and D. Haidukewych, Tetrahedron Letters, 1972,
70
1ПА F IF Collinston, Chem. and Ind. (London), 1973, 987; A. I. Meyers and '* E bJtV. Chem. Internal. Edn, 1976 15, 270.
107 A I- Meyers, G. Knaus, and K- Kainata, J. Amer. Chem. Soc., 1974, 96, 108 Д68/ Metiers and G. Knaus, J. Amer. Chem. Soc., 1974, 96, 6508.
09 A J Meuers and K. Kamata, J. Amer. Chem. Soc., 1976, 98, 2290.
ил' I/ ^rhollkovf R. Schroder, and D. Staff orsl, Annalen, 1974, 44.
1 G R Malone and A. I. Meyers, J. Org. Chem., 1974, 39, 618.
112’ G R Malone and A. I. Meyers, J. Org. Chem., 1974, 39, 623.
11ч F A Caret! and A. S. Court, J. Org. Chem., 1972, 37, 1926; P. F. Jones and ' M F Lappert, J. C. S. Chem. Comm., 1972, 526; D. Seebach. В.-Th. Grbbel, A К Beck, M. Braun, and K.-H. Geiss, Angew. Chem. Internal. Edn., 1972, 1 i, 443.
114 M. Mikolajczyk, S. Grzejsxcxak, A. Zatorski, and B. Mlotkowska, Tetrahedron Letters, 1976, 2731.
115. E. J. Corey and G. Markl, Tetrahedron Letters, 1967, 3201.
116. K. Ogura and G. Tsuchihashi, Tetrahedron Letters, 1972, 1383.
117. K. Ogura, S. Furukawa, and G. Tsuchihashi, Chem. Letters, 1974, 659.
118. L. A. Carpino, J. Amer. Chem. Soc., 1958, 80, 599.
119. E. C. Taylor, R. L. Robey, and A. McKillop, Angew. Chem. Internat. Edn., 1972, 11, 48; E. C. Taylor, R. L. Robey, D. K- Johnson, and A. McKillop, Org. Synth, 1976. 55, 73.
120. E. C. Taylor, R. L. Robey, and A. McKillop, J. Org. Chem,, 1972, 37, 2798.
121. L. A. Carpino, J. Amer. Chem. Soc, 1958, 80, 601.
122. A. С. Cope, H. L. Holmes, and H. 0. House, Org. Reactions, 1957, 9, 107 [A. H. Коп, X. Л. Холмс, F. О. Хаус. Орг. реакции, сб. 9. — Пер. с англ./Под ред. И. Ф. Луценко, М, Издатинлит, 1959, с. 125].
123. Р. L. Creger, J. Amer. Chem. Soc, 1970, 92, 1396.
124. P. E. Pfeffer and L. S. Silbert, J. Org. Chem, 1970, 35, 262.
125. B. Angelo. Bull. Soc. chim. France, 1970, 1848; S. Watanabe, K. Suga, T. Fujita, and K. Fujiyoshi, Israel J. Chem, 1970, 8, 731.
126. P. L. Creger, J. Amer. Chem. Soc, 1970, 92, 1397; Org. Synth, 1970, 50, 58.
127. G. W. Moersch and A. R. Burkett, J. Org. Chem, 1971, 36, 1149.
128. P. E. Preffer, E. Kinsel, and L. S. Silbert, J. Org. Chem, 1972, 37, 1256.
129. P. E. Pfeffer, L. S. Silbert, and E. Kinsel, Tetrahedron Letters, 1973, 1163 и цитированные там работы; см. также: J. A. Katzenellenbogen and A. L. Сгит-rine, J. Amer. Chem. Soc, 1974, 96, 5662.
130. M. W. Rathke, J. Amer. Chem. Soc, 1970, 92, 3222; Org. Synth, 1973, 53, 66.
131. M. W. Rathke and A. Lindert, J. Amer. Chem. Soc, 1971, 93, 2318.
132. M. W. Rathke and D. F. Sullivan, J. Amer. Chem. Soc, 1973, 95, 3050.
133. H. C. Brown, M. M. Midland, and A. B. Levy, J. Amer. Chem. Soc, 1972 94, 3662.
134.
135.
136.
137.
138.
139.
J. Hooz and R. B. Layton, Canad. J. Chem, 1972, 50, 1105.
J. Hooz, J. N. Bridson, J. G. Calzada, H. C. Brown, M. M. Midland, and A. B. Levy, J. Org. Chem, 1973, 38, 2574.
R. L. Shriner, Org. Reactions, 1942, 1, 1 [R. Шрайнер. Орг. реакции, сб. 1.— Пер. с англ./Под ред. А. Я. Берлина. М, Издатинлит, 1948, с. 9]; М. W. Rathke, Org. Reactions, 1975, 22, 423.
М. W. Rathke and A. Lindert, J. Org. Chem, 1970, 35, 3966.
E F Ruppert and J. D. White, J. Org. Chem, 1974, 39. 269.
f n ^acep: Adw Org. Chem, 1960, 2, 213; G. Quadbeck, in «Newer Methods Preparative Organic Chemistry», ed. W. Foerst, Academic Press, New York, 1963, volume 2, p. 133.
P. I.Paetzold and S. Kosma, Chem. Ber, 1970, 103, 2003.
,L- and A- Jacot-Guillarinod, Helv. Chim. Acta, 1974, 57, 1703; L. Vui-r’ л labacchi, and A. Jacot-Guillarmod, ibid, p. 1713.
1 n $oppel and M- D- Kinnick, Tetrahedron Letters, 1974, 711.
„ • Bergelson and M. M. Shemyakin, in «Newer Methods of Preparative Or-game Chenustry», ed. W. Foerst, Academic Press. New York, 1968, volume 5, ₽• 1&4; in «The Chemistry of Carboxylic Acids and Esters», ed. S. Patai, Wi-
140.
141.
142.
143.
71
ley-Interscience, New York, 1969, p. 295; №. S Wadsworth
1977, 25, 74. ' ’
144. H. J. Bestmann, H. Dornauer, and R. Rostock, Chem Bnr
2011. ' ”
Org. Reactions, 1970, 103, 685,
145. R. Shimoji, H. Taguchi, R. Ochima, H. Yamamoto, and H Nozahi i л Chem. Soc., 1974, 96, 1620; T. Taguchi, R. Shimoji, H. Yamamoto and HN zaki, Bull. Chem. Soc. Japan, 1974, 47, 2529. штатом, and H. No-
146’ Hartzel1’ D- F- Sulliva", and M. W. Rathke, Tetrahedron Letters 1974 1403; для а,р-ненасыщенных кислот см.: Р. A Grieco С-I t J
/. S. Burke, J С. S. Chem. Comm., 1975, 537. ’ ’ and
1. Ojima, M. Rumagai, and Y. Nagai, Tetrahedron Letters 1974 4005 J. R Johnson, Org. Reactions, 1942, 1, 210 [Дж. P. Джонсон. Орг. реакции сб. 1, —Пер. с англ./Под ред. А. Я. Берлина. М., Издатинлит 1948 с 2671 См. сс. 1, рр. 660—662. ’ ’ ' J‘
147.
148.
149.
150. G. Н. Posner, Org. Reactions, 1972, 19, 1.
151. Е. Negishi, G. Lew, and T. Yoshida, J. Org. Chem., 1974, 39, 2321- E Nemshi and T. Yoshida, J. Amer. Chem. Soc., 1973, 95, 6837, ’ ё
152. К. В. Sharpless and R. F. Lauer, J. Amer. Chem. Soc., 1973, 95, 2697.
153. В. M. Trost and T. N. Salzmann, J. Amer. Chem. Soc., 1973, 95, 6840.
154. В. M. Trost, T. N. Salzmann, and R. Hiroi, J. Amer. Chem. Soc., 1976, 98, 4887 и цитированные там работы.
155. М. №. Rathke and D. F. Sullivan, Tetrahedron Letters, 1972, 4249; J. L. Hermann, G. R. Riesczykowski, and R. H. Schlessiner, ibid., 1973, 2433.
156. H. C. Brown and H. Nambu, J. Amer. Chem. Soc., 1970, 92, 1761.
157. G. Bochi, M. Cushman, and H. Wuest, J. Amer. Chem. Soc., 1974, 96, 5563.
158. J. E. Baldwin and J. A. Walker, J. C. S. Chem. Comm., 1972, 354.
159. G. Andrews and D. A. Evans, Tetrahedron Letters, 1972, 5121.
160. R. T. Arnold and C. Hoffman, Synth. Comm., 1972, 2, 27; о других подходах см.: R. L Trust and R. Ё. Ireland, Org. Synth., 1973, 53, 116.
161. R. E. Ireland, R. H. Mueller, and A. R. Willard, J. Amer. Chem. Soc., 1976,98, 2868; J. Boyd, W. Epstein, and G. Frater, J. C. S. Chem. Comm., 1976,
380.
162. №. C. Still and M. J. Schneider, J. Amer. Chem. Soc., 1977, 99, 948.
163. J. E. Baldwin and J. A. Walker, J. C. S. Chem. Comm., 1973, 117.
164. R. R. Hill, R. Soman, and S. Sawada, J. Org. Chem., 1972, 37, 3737.
165. H. Takahashi, R. Oshima, H. Yamamoto, and H. Nozaki, J. Amer. Chem. Soc.,
1973 95 5803
166. I. Ruwajima and У. Doi, Tetrahedron Letters, 1972. 1163.
167. E. J. Corey and I. Ruwajima, Tetrahedron Letters, 1972, 487.
168. J. P. Marino and D. M. Floyd, J. Amer. Chem. Soc., 1974, 96, 7138; but see P. A. Grieco, C.-L. J. Wang, and G. Majetich, J. Org. Chem., 1976, 41, 726.
169. M. P. Periasamy and H. M. Walborsky, J. Org. Chem., 1974, 39, 611.
170. О. H. Oldenziel and A. M. van Leusen, Tetrahedron Letters, 1974, 163, 16/: ibid., 1973, 1357; D. M. Orere and С. B. Reese, J. C. S. Chem. Comm., 19//,
171.
172.
173.
174.
280
U. Schollkopf, R. Schroder, and E. Blume, Annalen, 1972 766, 130; ^Scholl-kopf and R. Schroder, Angew. Internal. Edn„ 1973, 12 407; see a iso R _ •
№. E. Bachman and №. S. Struve. Org. Reactions, 19421, 38 [В. Бах маh В. Струве. Орг. реакции, сб. 1. — Пер. с англ./ Под ред. А. Я. Р Издатинлит, 1948, с. 53]; V. Lee and М. S. Newman, Org. Sy , 77.
Е. J. Corey and D. E. Cane, J. Org. Chem., 1970 35, 3405 rts [
A. Murray and D. L. Williams, «Organic Syntheses with Isotop , P and II, Wiley-Interscience, New York, 1958; see also M Zielins , pjew
mistry of Corboxylic Acids and Esters», ed. S- Patai, Wiley-Inters York, 1969, p. 453. 1O7, o(i47
175. A. Szabolics, J. Szammer, and L. Noszkd, Tetrahedron 1974 JU, Jo*/-
176. J. L. Garnett, B. Halpern, and R. S. Renyon, J. C. S. Chem. Comm., 1J/A see also A. N. Yeo, Chem, Comm., 1971, 609.
177. D. A. Robinson, J. C. S. Chem. Comm., 1974, 345.
72
178 C A Townsend, Т. Scholl, and D. Arigonl, J. C. S. Chem. Comm., 1975, 921.
179 P Manitto, D. Monti, P. Gramatica, and E. Sabbioni, J. C. S. Chem. Comm., 1 ^73 563
180 См., например, cc. 92, 107—109, 151, 164, 178.
181" IF Theilacker, in Houben-Weyl «Methoden der Organischen Chemie», ed. ’ E Muller, Georg Thieme Verlag, Stuttgart, 1955, volume 4/2, p. 505.
182 R T Major and J. Finkelstein, J. Amer. Chem. Soc., 1941, 63, 1368.
183 H 1. Bestmann, H. Scholtz, and E. Kranz, Angew. Chem. Internat. Edn., 1970, 9 796
184 «Handbook of Chemistry and Physics», 57th edn., ed. R. C. Weast, CRC, Ohio, 1976; о рКа алкиновых кислот см.; G. H. Mansfield and M. C. Whiting, J. Chem. Soc., 1956, 4761.
185 M. Simonetta and S. Carr a, in «The Chemistry of Carboxylic Acids and Esters», ed. S. Patai, Wiley-Interscience, New York, 1969, p. 1.
186. L. Eberson, in «The Chemistry of Carboxylic Acids and Esters», ed. S. Patai, Wiley-Interscience, New York, 1969, p. 211.
187. C. Agami, Bull. Soc. chim. France, 1974, 869.
188. R. Yamdagni, T. B. McMahon, and P. Kebarle, J. Amer. Chem. Soc., 1974, 96, 4035; R. Yamdagni and P. Kebarle, ibid., 1973, 95, 4050.
189. J. s’ Jen and T. D. Thomas, J. Amer. Chem. Soc., 1975, 97, 1265.
190. D. Bethell and V. Gold, «Carbonium Ions», Academic, New York, 1967; «Carbonium Ions», ed. G. A. Olah and P. von R. Schleyer, Wiley-Interscience, New York, 1968, volumes I—IV [Д. Бетел, В. Голд. Карбониевые ионы. — Пер. с англ./Под ред. И. П. Белецкой. М., Мир, 1970]; для деталей по протониро-ванию простейших кислот см.: G. A. Olah, А. М. White, and D. Н. O’Brien, ibid., volume IV, chapter 31.
191. G. A. Olah, K- Dunne, Y. K. Mo, and P. Szilagyi, J. Amer. Chem. Soc., 1972, 94, 4200.
192. G. A. Olah, «Friedel-Crafts Chemistry», Wiley-Interscience, New York, 1973. 193. H. P. Treffers and L. P. Hammett, J. Amer. Chem. Soc., 1937, 59, 1708.
194. L. R. Fisher and D. G. Oakenfull, Chem. Soc. Rev., 1977, 6, 25.
195. J. H. Fendler and E. J. Fendler, «Catalysis in Micellar and Macromolecular Systems», Academic Press, New York, 1975.
196. Для общей информации см., например: D. Н. Williams and I. Fleming, «Spectroscopic Methods in Organic Chemistry», 2nd edn., McGraw-Hill, 1973; S. F. Dyke, A. J. Floyd, M. Sainsbury, and R. S. Theobald, «Organic Spectroscopy», Penguin, London, 1971. Для более специальной информации см.: К. Nakanishi, «Infrared Absorption Spectroscopy», Holden-Day, San Francisco, 1962 [/С. Наканиси. Инфракрасные спектры и строение органических молекул. — Пер. с англ./Под ред. А. А. Мальцева. М., Мир, 1965]; A. I. Scott, «Interpretation of the Ultraviolet Spectra of Natural Products», Pergamon, Oxford, 1964; H. Budzikiewicz, C. Djerassi, and D. H. Williams, «Interpretation of Mass Spectra of Organic Compounds», Holden-Day, San Francisco, 1964 [Г. Будзикевич, К. Джерасси, Д. Уильямс. Интерпретация масс-спектров органических соединений.— Пер. с англ./Под ред. Н. С. Вульфсона. М., Мир, 1966]; I. Н. Веупоп, R. A. Saunders, and А. Е. Williams, «The Mass Spectra of Organic Molecules», Elsevier, Amsterdam, 1968; L. M. Jackman and S. Sternhell, «Applications of N. M. R. Spectroscopy in Organic Chemistry», Pergamon, Oxford, 1969; J. B. Stothers, «Carbon-13 N. M. R. Spectroscopy», Academic Press, New York, 1972, G. C. Levy and G. L. Nelson, «Carbon-13 N. M. R. for Organic Chemists», Wiley-Interscience, New York, 1972 [ Г. Леви, Г. Нельсон. Руководство по ядерному магнит-н°мУ резонансу углерода-13 для химиков-органиков. М., Мир, 1975]; F. W. Wehrli and Т. Wirthlin, «Interpretation of Carbon-13 Spectra», Heyden, London, 1976.
ino’ T- Neilsen> J- Org. Chem., 1957, 22, 1539.
loo d Scdchell, Chem. and Ind. (London), 1974, 683.
УУ d t.sl°ngchamps, Tetrahedron, 1975, 31, 2463; см. также обзоры «Organic Reaction Mechanisms», Wiley-Interscience, New York, начиная c 1965 r. и далее.
73
200 D P N. Satchell and R. S. Satchell, Chem. Soc. Rev., 1975, 4, 231.
201 V I Stenberg and G. F. Vesley, J. Org. Chem., 1971, 36, 2548.
202' E. C. Blossey, L. M. Turner, and D. C. Neckers, Tetrahedron Letters, 1970 1823 '
203. 7. Berlin, И. B. Kagan, J.-L. Luche, and R. Setton, J. Amer. Chem. Soc., 197л
96 8113 *'
204 M.’ F. Ansell, in «The Chemistry of Acyl Halides», ed. S.Patai, Wiley-Inter science, New York, 1972, chapter 2; см. также cc. 3.
205. S. D. Saraf and M. Zaki, Synthesis, 1973, 612.
206. J. B. Lee, J. Amer. Chem. Soc., 1966, 88, 3440.
207. T. Wieland and A. Seeliger, Chem. Ber., 1971, 104, 3992; L. E. Barstow nnri 208.
209.
210.
211.
212.
213.
214.
215.
216.
217.
218.
219.
220.
221.
222.
223.
224.
225.
226.
227.
228.
229.
230.
231.
232.
233.
234.
V. J. Hruby, J. Org. Chem., 1971, 36, 1305.
E. M. Kaiser and R. A. Woodruff, J. Org. Chem., 1970, 35, 1198; idem Ora Synth., 1971, 51, 96. ’ й
. N. F. Albertson, Org. Reactions, 1962, 12, 157 [H. Ф. Альбертсон. Орг. реакции, сб. 12.— Пер. с англ./Под ред. И. Ф. Луценко. М., Мир, 1965, с. 1741' . L. I. Krimen, Org. Synth., 1970, 50, 1.
У. S. Klausner and M. Bodanszky, Synthesis, 1972, 453.
J. Rebek and D. Feitler, J. Amer. Chem. Soc., 1973, 95, 4052; A. F. Hegarty, M. T. McCormack, G. Ferguson, and P. J. Roberts, J. Amer. Chem. Soc 1977’ 99, 2015.
7. Rebek and D. Feitler, J. Amer. Chem. Soc., 1974, 96, 1606.
W. Konig and R. Geiger, Chem. Ber., 1970, 103, 788, 2024.
H. A. Staab, Angew. Chem. Internal. Edn., 1962, 1, 351; H. A. Staab and W. Rohr, in «Newer Methods of Preparative Organic Chemistry», ed. W. Foerst, Academic Press, New York, 1968, volume 5, p. 61.
H. A. Staab and G. Maleck, Chem. Ber., 1966, 99, 2955; 1972, 4747.
См., например: E. W. Colvin, T. A. Purcell, and R. A. Raphael, J. C. S. Perkin I, 1976, 1718.
K. Ninomiya, T. Shioiri, and S. Yamada, Tetrahedron, 1974, 30, 2151; для соответствующего цианида см.: S. Yamada, Y. Kasai, and T. Shioiri, Tetrahedron Letters, 1973, 1595.
Y. S. Klausner and M. Bodanszky, Synthesis, 1974, 549.
B. Castro and J. R. Dormoy, Tetrahedron Letters, 1972, 4747.
B. Castro and J. R. Dormoy, Tetrahedron Letters, 1973, 3243.
N. Yamazaki and F. Higashi, Bull. Chem. Soc. Japan, 1973, 46, 3821; ibid., 1974, 47, 170 и цитированные там работы.
N. Yamazaki, F. Higashi, and S. A. Kazaryan, Synthesis, 1974, 436.
N. Yamazaki, F. Higashi, and Y. Saito, Synthesis, 1974, 495.
T. Mukaiyama, R. Matsueda, and M. Suzuki, Tetrahedron Letters, 1970, 1901; :ТС Mukaiyama, K. Goto, R. Matsueda, and M. Ueki, ibid., p. 5293.
, H. Gerlach and A. Thalmann, Helv. Chim. Acta, 1974, 57, 2661.
. E. J. Corey and К- C. Nicolaou, J. Amer. Chem. Soc., 1974, 96, 5614; E. J. 6o-rey, D. J. Brunelle, and P. J. Stork, Tetrahedron Letters, 1976, 3405.
. E. J. Corey and D. J, Brunelle, Tetrahedron Letters, 1976, 3409.
. S. Masamune, S. Kamata, and W. Schilling, J. Amer. Chem. Soc., 1975, 96 3515.
S. Masamune, H. Yamamoto, S. Kamata and A. Fukuzawa, J. Amer. Chem. Soc., 1975, 97, 3513.
T. Mukaiyama, AL Usui, E. Shimada, and K. Saigo, Chem. Letters, 1975, 1045, T. Mukaiyama, Angew. Chem. Internat. Edn., 1976, 15, 94.
T. Mukaiyama, H. Toda, and S. Kobayashi, Chem. Letters, 1976, 13.
B. Funke and W. Kantlehner, Chem. Ber., 1971, 104, 3711.
---D. J. Raber and P. Gariano, Tetrahedron Letters, 1971, 4741. „
235a. A. H. Lewin and N. L. Goldberg, Tetrahedron Letters, 1972, 491; F. ofl qusa, I. Murase, and Y. Ito, J. Org. Chem., 1973, 38, 1753.
2356. G. Mehta, Synthesis, 1972, 262.
236. H. D. Durst, Tetrahedron Letters, 1974, 2421. „
237. J. E. Shaw and D. C. Kunerth, J. Org. Chem., 1974, 39, 1968; P. B. Pfail11' and L. S. Silbert, J. Org. Chem., 1976, 41, 1373.
74
238 /. В. Clark and J. M. Miller, Tetrahedron Letters, 1977, 599.
239 O. Mitsunobi, T. Sana, and M. Wada, Bull. Chem. Soc. Japan, 1973, 46, 2833; ' H. Loibner and E. Zbiral, Helv. Chim. Acta, 1977, 60, 417.
240 . R. A. Sheldon and J. К Kochi, Org. Reactions, 1972, 19, 279.
241' В. C. L. Weedon, Quart. Rev., 1952, 6, 380; W. H. Sharkey and С. M. Lang-Kammerer, Org. Synth., 1961, 41, 24, and S. Swann and W. E. Garrison, ibid., n 53.
242. G. E. Hawkes, J. H. P. Utley, and G. B. Yates, J. C. S. Chem. Comm., 1973, 305; G. W. Gribble, J. K. Sanstead, and J. W. Sullivan, ibid., p. 735; L. Eberson and G. Ryde-Petterson, Acta Chem. Scand., 1973, 27, 1159.
243. W. F. Maier and M. T. Reetz, J. Amer. Chem. Soc., 1975, 97, 3687.
244. /. K. Stille and M. T. Regan, J. Amer. Chem. Soc., 1974, 96, 1508; I. K. Stille and R. W. Fries, ibid., p. 1514; J. K. Stille, F. Huang, and M. T. Regan, ibid., p 1518; K. S. Y. Lau, Y. Becker, F. Huang, N. Baenziger, and J. K. Stille, ibid., 1977, 99, 5664.
245. D. B. Bigley and J. C. Thurman, J. Chem. Soc. (B), 1968, 436.
246. F. Kienzle, G. W. Holland, J. L. Jernow, S. Kwoh, and P. Rosen, J. Org. Chem., 1973, 38, 3440.
247. D. H. R. Barton, I. H. Coates, and P. G. Sammes, J. C. S. Perkin I, 1973, 599.
248. K. Fujimori and S. Oae, Tetrahedron, 1973, 29, 65.
249. R. G. Johnson and R. K. Ingham, Chem. Rev., 1956, 56, 219; С. V. Wilson, Org. Reactions, 1957, 9, 332 [7. В. Вильсон. Орг. реакции, сб. 9. — Пер. с англ./Под ред. И. Ф. Луценко. М., Издатинлит, 1959, с.445]; N. J. Bunce and N. G. Murray, Tetrahedron, 1971, 27, 5323.
250. N. J. Bunce, J. Org. Chem., 1972, 37, 664; J. Cason and D. M. Walba, ibid., p. 669 и цитированные там работы.
251. A. McKillop, D. Bromley, and E. C. Taylor, J. Org. Chem., 1969. 34, 1172.
252. К. B. Becker, M. Geisel, C. A. Grob, and F. Kuhnen, Synthesis, 1973, 493.
253. D. H. R. Barton and E. P. Serebryakov, Proc. Chem. Soc., 1962, 309; D. H. R. Barton, H. P. Faro, E. P. Serebryakov, and N. F. Woolsey, J. Chem. Soc., 1965, 2438.
254. P. A. S. Smith, Org. Reactions, 1846, 3, 337 [П. А. С. Смит. Орг. реакции, сб. 3. — Пер. с англ./Под ред. К. А. Кочешкова. М., Издатинлит. 1951, с. 322].
255. Н. Wolff, Org. Reactions, 1946, 3, 307 [С. Вольф. Орг. реакции, сб. 3. — Пер. с англ./Под ред. К. А. Кочешкова. М., Издатинлит, 1951, с. 293],
256. Е. S. Wallis and J. F. Lane, Org. Reactions, 1946, 3, 267 [3. С. Уэллис, Дж. Ф. Лэн. Орг. реакции, сб. 3. — Пер. с англ./Под ред. К. А. Кочешкова. М„ Издатинлит, 1951, с. 255].
257. IF. Lwowski, Angew. Chem. Internat. Edn., 1967, 6, 897; T. L. Gilchrist and C. W. Rees, «Carbenes, Nitrenes and Arynes», Nelson, London, 1969.
258. S. Washburne, W. R. Peterson, and D. A. Berman, J. Org. Chem., 1972, 37, 1738; J. H. McMillan and S. Washburne, ibid., 1973, 38, 2982.
259. J. W. van Reijendam and F. Baardmann, Synthesis, 1973, 413.
260. A. Brandstrom, B. Lamm, and I. Palmertz, Acta Chem. Scand., 1974, B28, 699.
261. P. Radlick and L. R. Brown, Synthesis, 1974, 290.
262. J. G. Gleason and D. H. Harpp, Tetrahedron Letters, 1970, 3431; D. N. Harpp, L. Q. Bao, С. J. Black, R. A. Smith, and J. G. Gleason, ibid., 1974, 3235.
263. M. W. Rathke and A. Lindert, Tetrahedron Letters, 1971, 3995.
264. G. A. Olah and J. Welch, Synthesis, 1974, 652.
265. А/. C. Deno, R. Fishbein, and С. E. Pierson, J. Amer. Chem. Soc., 1970,92, 1451.
266. N.?C. Deno, R. Fishbern, and J. C. Wyckoff, J. Amer. Chem. Soc., 1970, 92, 267. F. Minisci, Synthesis, 1973, 1.
268. W. E. Billups, K. C. Deno, R. Fishbein, C. Pierson, R. Whalen, and J. C. Wyckoff, J. Amer. Chem. Soc., 1971, 93, 438; M. C. Deno, R. Fishbein. and ocn C1 Wyckoff, ibid., p. 2065.
97e n %' Eck’ D' J' Bunter, and T. Money, J. C. S. Chem. Comm., 1974, 865.
Z7°- Mosettig, Org. Reactions, 1955, 8, 218 [3. Мозеттинг. Орг. реакции, сб. 8. — Пер. с англ./Под ред. Ю. А, Арбузова. М., Издатинлит, 1956, с .288].
75
271 A. 0. Bedenbaugh, J. H. Bedenbaugh, W. A. Bergin, and J. D. Adkins I ' Chem. Soc., 1970, 92, 5774. ’ ' Anier-
272. L. Horner and H. Roder\ Chem. Ber., 1970, 103, 2984.
273. D. J. Raber and W. C. Guida, Synthesis, 1974, 808; M. R. Johnson
B. Rickborn, Org. Synth., 1971, 51, 11. M and
274. J. Carnduff, Quart. Rev., 1966, 20, 169; E. R. H. Walker, Chem. Soc RP 1976, 5, 23; M. N. Rerick, in «Reduction», ed. R. L. Augustine, Marcel nil’ ker, New York, 1968, chapter 1.
275. I. C. Nordin, J. Heterocyclic Chem., 1966, 3, 531; see also ref. 105.
276. G. Doleschall, Tetrahedron Letters, 1974, 2649; 1975, 681; P L нап n , R. B. Perfetti, J. Org. Chem., 1974, 39, 111.
277. T. Mitsudo, T. Okajima, Y. Takegami, M. Tanaka, Y. Watanabe, and К vn mamoto, Bull. Chem. Soc. Japan, 1971, 44, 2569.
278. Y. Watanabe, M. Yamashita, T. Mitsudo, M. Igami, and У. Takegami Rnii Chem. Soc. Japan, 1975, 48, 2490.
279. Y. Watanabe, M. Yamashita, T. Mitsudo, M. Igami, K. Tomi, and Y. Takegami Tetrahedron Letters, 1975, 1063.
280. E. Mosettig and R. Mozingo, Org. Reactions, 1948, 4, 362 [3. Мозеттинг P. Мозинго. Орг. реакции, сб. 4. — Пер. с англ./Под ред. К. А. Кочешкова’ М., Издатинлит, 1951, с. 337]; A. J. Rachlin, Н. Gurien, and D Р Wagner Org. Synth., 1971, 51, 8. ’
281. J. D. Citron, J. Org. Chem., 1969, 34, 1977; S. P. Dent, C. Eaborn, and A. Pid-cock, Chem. Comm., 1970, 1703.
282. N. M. Yoon, C. S. Pak, H. C. Brown, S. Krishnamurthy, and T. P. Stocky J Org. Chem., 1973, 38, 2786; C. F. Lane, Chem. Rev., 1976, 76, 773.
283. M. G. Hutchings, T. E. Levitt, A. Pelter, and K. Smith, Chem. Comm., 1970, 347.
284. F.-C. Huang, L. F. Hsu Lee, R. S. D. Mittal, P. K. Ravikumar, J. A. Chan, C. J. Sih, E. Caspi, and C. R. Eck, J. Amer. Chem. Soc., 1975, 97, 4144.
285. Cm. cc. 65, pp. 948—951.
286. M. J. Jorgenson, Org. Reactions, 1970, 18, 1; R. E. Levine and M. J. Karten, J. Org. Chem., 1976, 41, 1176.
287. J. C. Floyd, Tetrahedron Letters, 1974, 2877.
288. G. H. Posner and С. E. Whitten, Org. Synth., 1976, 55, 122.
289. G. H. Posner, Org. Reactions, 1975, 22, 253; D. A. Shirley, ibid., 1955, 8, 28 [Д. А. Ширли. Орг. реакции, сб. 8. — Пер. с англ./Под ред. Ю. А. Арбузова. М., Издатинлит, 1956, с. 44]; J. F. Normant, Synthesis, 1972, 63.
290. С. Jallabert, N.-T. Luong-Thi, and H. Riviere, Bull. Soc. chim. France, 1970, 797.
291. F. Huet, G. Emptoz, and A. Jubier, Tetrahedron, 1973, 29, 479.
292. A. Meisters and T. Mole, Austral. J. Chem., 1974, 27, 1665.
293. A. J. Birch and G. Subba Rao, Adv. Org. Chem., 1972, 8, 1; H. Smith, «Organic Reactions in Liquid Ammonia» («Chemistry in Non-aqueous Ionising Solvents»), Wiley-Interscience, New York, 1963; R. G. Harvey, Synthesis, 1970, 161; E. M. Kaiser, ibid., 1972, 391; M. Smith, in «Reduction», ed. R. L. Augustine, Marcel Dekker, New York, 1968, chapter 2.
294. D. Caine, Org. Reactions, 1976, 23, 1. ,
295. H. Normant, Russ. Chem. Rev. 1970, 39, 457; Angew. Chem. Internat._tdn..
1967, 6, 1046. ’ ,
296. R. L. Augustine, «Catalytic Hydrogenation», Edward Arnold, London, IV •
297. D. G. Lee and J. R. Brownridge, J. Amer. Chem. Soc., 1973, 95, 3033; К В. wi berg, C. J. Deutsch, and J. Ro£ek, ibid., p. 3034. ,
298. Как интересный вариант см.: S. Terashima and S. Jew, Tetrahedron Leite , 1977, 1005. , 4 n
299. W. E. Barnett and W. H. Sohn, J. C. S. Chem. Comm., 1972, 472; Tetrahedron Letters, 1972, 1777; W. E. Barnett and J. C. McKenna, Chem. Comm., 551; Tetrahedron Letters, 1971, 2595. , c npr-
300. R. C. Cambie, R. C. Hayward, J. L. Roberts, and P. S. Rutledge, J. C. S- r kin I, 1974, 1864.
301. L. do Amaral and S. C. Melo, J. Org. Chem,. 1973, 38, 800.
76
302. «Friedel-Crafts and Related Reactions», ed. G. A. Olah, Wiley-Interscience, New York, 1963, volumes I—IV; см. также cc. 192.
303. Ref. 293; см. также cc. 1, pp. 199—203.
304. Af. E. Kuehne and B. F. Lambert, Org. Synth., 1963, 43, 22.
305. M. D. Bachi, J. W. Epstein, Y. Herzberg-Minzly, and H. J. E. Loewenthal, J. Org. Chem., 1969, 34, 126.
306. H. van Bekkum, С. B. van den Bosch, G. van Minnen-Pathuis, J. C. De Mos, and A. M. van Wijk, Rec. Trav. chim., 1971, 90, 137 и цитированные там работы.
307. Е. Haslam, in «Protective Groups in Organic Chemistry», ed. J. F. W. McOmie, Plenum Press, London, 1973, chapter 5 [Защитные группы в органической хи-мии/Под ред. Дж. Мак-Оми. — Пер. с англ. М., Мир, 1976, часть 5].
308. N. Kornbluni and A. Scott, J. Amer. Chem. Soc., 1974, 96, 590.
309 A. К. Bose, S. D. Sharma, J. C. Kapur, and M. S. Manhas, Synthesis, 1973, 216.
310. E. J. Corey and C. U. Kim, J. Org. Chem., 1973, 38, 1233.
311. F. M. Huber, R. C. Chauvetfe, and B. G. Jackson, in «Cephalosporins and Penicillins», ed. E. H. Flynn. Academic Press, New York, 1972, chapter 2.
312. A. L. McCloskey, G. S. Fonken, R. W. Kluiber, and W. S. Johnson, Org. Synth. Coll. Vol. 4, 1963, 261.
313. H. van Bekkum, J. W. M. De Graaf, J. A. Hagendoorn, D. P. Roelofsen, and H. M. Verschoor, Rec. Trav. chim., 1970, 89, 193.
314. G. W. Kenner and J. H. Seely, J. Amer. Chem. Soc., 1972, 94, 3259.
315. A. IF. Miller and C. J. M. Stirling, J. Chem. Soc. (C), 1968, 2612.
316. T.-L. Ho and С. M. Wong, J. C. S. Chem. Comm. 1973, 224; Synth. Comm., 1973, 3, 145.
317. J. B. Hendrickson and C. Kandall, Tetrahedron Letters, 1970, 343; J. C. Sheehan and G. D. Davies, J. Org. Chem., 1964. 29, 2006.
318. R. B. Woodward, K. Hausler, J. Gosteli, P. Naegeli, W. Oppolzer, R. Ramage, S. Ranganathan, and H. Vorbruggen, J. Amer. Chem. Soc., 1966, 88, 852; G. Just and K. Grozinger, Synthesis, 1976, 457.
319. V. Marinier, Y. C. Kim, and J.-M. Navarre, Canad. J. Chem., 1973, 51, 208.
320. T.-L. Ho, Synthesis, 1974, 715.
321. T.-L. Ho and С. M. Wong, Synth. Comm., 1974, 4, 307.
322. A. I. Meyers, D. L. Temple, D. Haidukewych, and E. D. Mihelich, J. Org. Chem., 1974, 39, 2787.
323. D. H. R. Barton, M. Girijavallabhan, and P. G. Sammes, J. C. S. Perkin I, 1972 929.
324. T.-L. Ho, H. C. Ho and С. M. Wong, Synthesis, 1972, 562.
325. J. C. Sheehan and K- Umezawa, J. Org. Chem., 1973, 38, 3771.
326. A. W. Oxford, J. C. Sheehan, and R. Wilson, J. Amer. Chem. Soc., 1971, 93, 7222.
327. D. H. Rich and S. K. Gurwara, J. C. S., Chem. Comm., 1973, 610.
328. B. Amit and A. Patchornik, Tetrahedron Letters, 1973, 2205.
329. D. S. Kemp and J. Reczek, Tetrahedron Letters, 1977, 1031.
9.2. ДИКАРБОНОВЫЕ И ПОЛИКАРБОНОВЫЕ КИСЛОТЫ
А. Кокс (University of Warwick)
9.2,1. МЕТОДЫ ПОЛУЧЕНИЯ ДИ- И ПОЛИКАРБОНОВЫХ КИСЛОТ
В данном разделе будет подробно рассмотрен набор общих методов получения ди- и поликарбоновых кислот. Основное внимание будет уделено методам синтеза этих соединений, отличающихся от таковых для получения монокарбоновых кислот.
77
9.2.1.1. Карбоксилирование и алкоксикарбонилирование
Карбоксильная группа может быть введена двумя путями. Пеп-вый путь состоит в применении моноксида углерода в присутствии катализатора, чаще всего металлорганического соединения. Второй путь использует реакцию карбаниона с диоксидом углерода' Оба эти метода мы рассмотрим раздельно.
(1) Карбоксилирование моноксидом углерода
Этому важному методу получения дикарбоновых кислот посвящен обзор [1]. Типичный пример — синтез малеиновых ангидридов при реакции ацетилена с карбонилом железа в водной щелочи {схема (1)}. Продукт реакции (1) при окислении феррицианидом калия или азотной кислотой дает малеиновый ангидрид. Алкоксикарбонилирование органических галогенидов (RHal) карбонилом никеля и алкоксидом щелочного металла разработано Кори с сотр. [2] и другими авторами, и применяется для синтеза сложных эфиров дикарбоновых кислот {схема (2)}. Другие примеры применения этого метода даны в разд. 9.1.1 и 9.8.1.
Fexco)z он
R
ОН о
I р II
. K3Fe(CN)6
\Fe(CO)3 ------—][ О (!)
|^Fe(CO)3 «'if
ОН v ' О
(1)
KOBu-грет
1(СНг)б1 + Ni(CO)4 -------*" тпет-ВиОгСГСНгкСОгВи-трет (2)
трет-ВиОН
Модификацией этого метода получают мононитрилы {схема (3)}. По-видимому, не существует ограничений для использования этой реакции для синтеза динитрилов, хотя в оригинальной работе таких примеров не представлено. Малеинимиды можно получать с высоким выходом [3] по реакции дифенилацетилена, моноксида углерода и ароматического нитросоединения с использованием гексадекакарбонилгексародия {Rh6(CO)16} в качестве катализатора и третичным амином (пиридин, N-метилпирролидин) в качестве растворителя {схема (4)}. Моноксид углерода, по-видимому, выступает в этих реакциях как восстанавливающий и как карбонилирующий агент; механизм реакций сложен.
Алифатические а,(3- и р,у-непредельные амиды кислот взаимо действуют [4] с моноксидом углерода в присутствии подходяще1"® кобальтового катализатора с образованием имидов янтарной нЩ
+ K4Ni2(CN)6
МеОН
KCN
(3)
78
phfeCPh + 4C0 +
(4)
,0
сн2=сн—c;
XNHR
Со-катализатор
CO
(6)
глутаровой кислот. Лучшим катализатором здесь служит Со2(СО)8, хотя и кобальт Ренея, и ацетат кобальта(II) также катализируют эту реакцию. N-Замещенные акриламиды, с высоким выходом дают соответствующие сукцинимиды {схема (5)}. Аналогично, можно использовать и другие производные акриламида.
(2) Карбоксилирование диоксидом углерода
Превращение металлорганических соединений в соли карбоновых кислот при взаимодействии с диоксидом углерода — хорошо известная реакция [5], с помощью которой {схема (6)} можно проводить как моно-, так и дикарбокснлирование. Образование дикарбоновой кислоты зависит от направления реакции первоначально образующейся натриевой соли фенилуксусной кислоты с локальным избытком бензилнатрия, что приводит к динатриевому производному фенилуксусной кислоты.
PhNa
(из PhCl + Na)
РЬСНз -------------->
д
PhCH2Na
<1 со2 ------->
>1 со2 ------->
PhCH2CO2Na (99%)
СО2;
PhCHNaCO2Na --------> PhCH(CO2H)2
н+, Н2О
(6)
толуол
PhCl + Na -----------> PhNa + NaCl
иа холоду
С6Н6 + Me2CHCH2Na •—> PhNa + Ме3СН
(7)
(8)
Получению натрий- и калийорганических соединений посвящен обзор [6], где описаны и детали типичных экспериментальных методик. Эти металлорганические соединения можно получать или прямой реакцией доступных органических соединений (обычно галогенида) со щелочным металлом, или реакцией трансметаллирования, которая в основном является кислотно-основной реакцией. Оба метода показаны на примере получения фенилнатрия {схемы
79
Реакции металлирования, включающие литийорганические соединения рассмотрены также в обзоре [7]. Для получения дикар. боновых ’кислот необходимо использовать бисметаллорганические соединения или металлорганические реагенты, уже содержащие карбоксильную группу. Несмотря на возможность побочных реакций, эти превращения применимы к разнообразным соединениям. Далее мы рассмотрим наиболее важные примеры этой реакции.
При обработке реактивами Гриньяра некоторые алленкарбоновые кислоты можно превратить в металлорганические соединения. Последующее взаимодействие этих соединений с диоксидом углерода {схема (9)} приводит с хорошим выходом к (1-алкилви-нил) малоновым кислотам [8].
/Ви СН2=С=<У
ХСО2Н
RMgBr
R
СН2
Ви
С—CO2MgBr I
MgBr
R
СН2
Ви
^С—С—СО2Н
СО2Н (?)
Алкилмалоновые кислоты с хорошим выходом {схема (10)} получают при реакции алюминийлитиевого производного карбоновой кислоты (2) с диоксидом углерода [9]; в свою очередь, ме-таллорганпческое производное (2), используемое в этой реакции, получают гидроалюминированием алкинов-1. Например, гексин-1 при взаимодействии с 2 моль диизобутилалюминийгидрида приводит (с 85%-ным выходом) к металлорганическому производному (3) {схема (11)}, которое после обработки метиллитие.м дает (4). Это соединение реагирует с диоксидом углерода с образованием малоновой кислоты, причем, как показано на схеме (10), реакция идет через образование интермедиата (2).
О о
II II /
/с-0' Lt+ /С-оаГ СО2.
> RC!R X ------------> RCH(CO2H)2 (10)
XLj н+
(2)
BufeCH
A1HBU2
ТГФ
^A1Bu2 MeLi /Li
BuCH2CH ----> BuCH2CHZ
XA1Bu2 \mBu2
(3) (4)
—> BuCH2CH(CO2H)2
CO2; ________>
H + , H2O
(11)
Аналогично, можно проводить превращение ацетиленов в малоновые кислоты с использованием гелг-борорганических соединении [10] типа (5) {схема (12)}; при использовании 2 моль бутил-лития можно достичь выхода 65—70%. Другой хороший метод {11} интеза производных замещенной малоновой кислоты — реакция 80
а-анионов сложных эфиров с диоксидом углерода. Анионы генерируют с помощью диизопропиламида лития в тетрагидрофуране,
R'C=CH /BRI BuLi /Li СО2;
। —> r'ch2ch; ----------►- R'CHzCH; — ------->•
2R1BH XBRi Vi H - H2°
(5) (12)
zco2h R'CHaCH^
\co2h
R1 = H-C4H9, h-C8H17, CeH5:
R2 = циклогексил
и дальнейшая процедура сводится к пропусканию диоксида углерода в раствор аниона. Последующая обработка приводит к практически чистому продукту {схема (13)}. Прекрасные результаты получены с такими стерически затрудненными сложными эфирами, как этил-2-метилпропионат; в этом случае побочные реакции не наблюдались. Хорошим примером этой реакции служит синтез [12] адамантан-2,2-дикарбоновой кислоты. Метод можно также использовать в гомокубановой серии [13]; сложный эфир (6) можно превратить в соответствующее производное малоновой кислоты {схема (14)} без деградации или перегруппировки «клеточного» каркаса.
Li LiN(Pr-u3O)2 | СО2; /СО2Н
RKHCO2R2 — --------———-> r^cco2r2 — --------------> R^CT (13)
[ИЗ н-BuLi + 2 н+ н о * \
(изо-Рг)2ЫН в ТГФ] ' 2 ХСО2Н
ЫЦ(Рг-йзо)2,ТГФ , -----------------►
со2;
н+;
ch2n2
ОН(СО2Ме)2
(14)
Используя путь, показанный на схеме (15), из бутадиена можно получить набор дикарбоновых кислот [14]. При действии натрия в строго определенных условиях бутадиен димеризуется с образованием динатрийоктадиена. Получающийся делокализованный дианион реагирует с диоксидом углерода, давая смесь трех возможных региоизомерных диеновых дикарбоновых кислот, гидрирование которых приводит к себациновой, 2-этилпробковой и 2,5-диэтиладипиновой кислотам в соотношении 3,5 : 5 : 1 соответственно. Эта важная реакция, распространенная на такие ароматические соединения, как стирол и 2-метилстирол [15], приводит к производным адипиновой кислоты {схема (16)}, причем оба продукта можно гидрировать до соответствующих дициклогексильных производных.
81
Дианион циклооктатетраена реагирует с диоксидом углерода с образованием дикарбоновой кислоты [16], однако, как отмечалось в этой работе, ранее предложенная для этого продукта структура (7) [17] неверна. Альтернативная формула (8) согласуется с результатами по электроциклическому раскрытию кольца предшественника, имеющего тронс-стереохимию, в соответствии с правилом Вудворда — Гофмана о сохранении орбитальной симметрии {схема (17)}.
Na измельч. сн2=снсн=сн2 -------->•
СО2; Н+;
—> Na+ СН2СН=СНСН2СН2СН=СНСН2 Na+ ----------->
Н2, Pd
—> НО2С(СН2)8СО2Н + EtCH(CH2)5CO2H + EtCH(CH2)2CHEt (15)
СО2Н СО2Н СО2Н
Na+ Na+ СО2Н СО2Н
Na - - СО2; | |
РЬС=СН2 -----► PhCCH2CH2CPh---------> PhCCH2CH2CPh (16)
| | | н+, н2о | |
R R R R R
=0 + MeOMgOCO2Me
(9)
(18)
Эффективным реагентом для введения карбоксильной или алкоксикарбонилыюй группы в различные карбанионы является метилметоксимагиий карбонат (ММК) (9). Обычно кетоны превращаются в сложные эфиры а-кетокислот, однако применение избытка ММК может привести к включению двух метоксикарбониль-ных групп, как, например, при получении синтетически важного диэфира (10) {схема (18)} [18].
82
9.2.1.2. Реакции конденсации
Большинство общих подходов к синтезу ди- и поликарбоновых кислот использует реакции конденсации [19]. Эти реакции включают сложноэфирную конденсацию Кляйзена [20] и различные реакции производных малоновой и щавелевой кислот. Некоторые из реакций производных малоновой и щавелевой кислот мы обсудим здесь, часть других реакций будет включена в разд. 9.2.3.5 и 9.2.3.6.
Производные дикарбоновых кислот с длинной цепью получают из доступных производных дикарбоновых кислот в результате сложноэфирной конденсации Кляйзена. Можно использовать, на- . пример, ЫДЧ-диметилсебацамат (11) {схема (19)} [21], так как в конденсацию вовлекаются только сложноэфирная и соседняя с ней а-метиленовая группы.
soci2; НО2С(СН2)8СО2Н —> МеО2С(СН2)8СО2Ме -----> НО2С(СН2)8СО2Ме — ►
ЛИ-21N 1"1
основание
—> Me2NCO(CH2)8CO2Me ----------->
(И)
СО2Ме
I 48% НВг
—> Me2NCO(CH2)8COCH(CH2)7CONMe2 -------->
восстановление
HO2C(CH2)8CO(CH2)8CO2H —----------------> HO:
< по Клемеисену
2C(CH2)17CO2H (19)
Na+
Hal
CCO2Et
ссо2н
/X RCHf
NaOEt
----->• RC
CO2Et ЕЮН '
>CCO2Et
CO2Et
H+, H2o J -----> RCHCO2H
X = CO2Et или CN
Алкилирование анионов, получаемых из эфиров малоновой кислоты или этилцианоацетата, широко используется для синтеза монокарбоновых кислот, и как видно из схемы (20), может также применяться [19] для получения дикарбоновых кислот. Альтернативный способ алкилирования с использованием реакции Михаэля обсуждается в разд. 9.2.1.3. При использовании в качестве алкилирующих агентов соответствующих сложных эфиров галогенокис-лот {схема (20)} этот метод в принципе может позволить получать различные ди- и поликарбоновые кислоты.
Другое применение диэтилмалоната более специфично, так как реакция [22] диэтилнатриймалоната с соответствующим образом защищенными этилглицидатами приводит к а,[3-диэтоксикарбонил-бутиролактонам, которые при последующем гидролизе превращаются в параконовые кислоты (12) {схема (21)}. Обработка параконовых кислот полифосфорной кислотой дает соответствующие циклощентен-2-оны-1, включая дигидрожасмон.
83
NaCH(CO2Et)2
гидролиз
(21)
"ОН
--CO2Et .
О" ОН
84
Дегидробензолы реагируют с малоновыми эфирами, давая производные гомофталевой кислоты [23]. Например, реакция диэтил-малоната с о-броманизолом в тетрагидрофураие в присутствии амида натрия с выходом 60% дает 3-метоксигомофталимид; при изменении условий реакции могут появляться другие продукты. При использовании в качестве источника дегидробензола бромбен-зола и в качестве растворителя гексаметапола основными продуктами реакции являются диэтилфенилмалонат (20%), моноэтилгомофталат (10%) и гомофталимид (50%). Механизм образования этих продуктов показан на схеме (22).
Для синтеза замещенных малоновых эфиров можно использовать [24] прямое алкилирование диэтилнатриймалоната, однако метод не совсем удачен, так как часто приводит к побочным продуктам, получающимся за счет дегидрогалогенирования алкилга-логенидов. Несколько примеров этой реакции, которая будет рассмотрена в разд. 9.2.3.6, приведено в Organic Syntheses. Реакции элиминирования можно до некоторой степени избежать при использовании сопряженного присоединения реактива Гриньяра к алкилиденмалонату, как, например, в синтезе трет-бутилмалоната [25, 28] присоединением метилмагнийиодида к изопропилиденма-лонату {схема (23)}. Сопряженное присоединение реактивов Гриньяра к а,р-ненасыщенным сложным эфирам служит основной реакцией; ее можно значительно ускорить [26] в присутствии
CH2(CO2Et)2 + СН3СОСН3
ZnCl2 (CH3)2C=C(CO2Et)2
(CH3)3CCH(CO2Et)2
MeMgl, Cui или
LiMe2Cu ---------->
H2SO4> H2O
(W
CO2Et
Ва(ОН)2
(CH2)4CO2Et --------
МеОН
NaOEt
EtOH
CO2Et
Br(CH2)4CO2Et
NaOEt,
CH3COCH2CO2CMe3 ---------------
R1R2CBrCO2Et
О
(СН2)4СО2Н
cco2Et
(24)
н
СНзСОСНСО2СМе3
R1
—► СНзСОСН2С—CO2Et
(25)
R2
85
1 % (мол.) хлорида меди(1). В частности [27], такие медьоргани-ческие реагенты, как LiMeCu и MeCuP (С4Н9-Н), селективно присоединяются к p-углеродному атому аф-ненасыщенных кетонов, обеспечивая потенциальное расширение метода по реакциям, аналогичным приведенным на схеме (23).
Для получения производных дикарбоновых кислот можно также использовать алкилирование сложных эфиров р-кетокислот {схемы (24) и (25)}. В общем случае продукты этих реакций подвергаются дальнейшим превращениям или, как это показано на схеме (24), используются для получения кетокислот [29].
Для получения производных сложных эфиров малоновой кислоты можно использовать диэтилоксала г [30], проводя сложно-эфирную конденсацию Кляйзена и последующее термическое декарбонилирование {схема (26)}. Это достаточно общий метод введения этоксикарбонильной группы. Другие примеры, не относящиеся к синтезу производных ди- и поликарбоновых кислот, обсуждаются в разд. 9.2.3.5. Применение сложных эфиров, таких, как диэтилсукцинат {схема (27)}, может приводить к получению а-оксопроизводных дикарбоновых кислот [31] путем гидролиза промежуточного сложного эфира p-оксополикарбоновой кислоты.
NaOEt zCOCO2Et д
C17H35CO2Et + (CO2Et)2 -------> С16н33сн( —
',CO2Et
—> C16H33CH(CO2Et)2 + СО (26)
COCO2Et COCO2H
CH2CO2Et KOEt | HCl 1
1 +(C°2Et)2 FtOT CH2CO2Et ЕЖ CHCO2Et ——> | H2O CH2 1 (27)
CH2CO2Et CH2CO2H
Алкильные производные янтарной кислоты можно получать [32] алкилированием дианиона, в свою очередь полученного из моноэтилсукцината; алкилирование протекает региоспецифично {схема (28)} по соседнему со сложноэфирной группой углеродному атому. Другие а-алкильные производные адипиновой и пи-мелиновой кислот можно получать [33] более сложной последовательностью реакций {схема (29)}, так как в этом случае анионы легко вступают в циклизацию по Дикману.
СН2СО2Н LiNH2
I ------------->
PhCHCO2Et NH3
CH2CO2L.i CH2CO2H
PhCCO2Et PhCH2C‘> Ph—C—CO2Et
Li CH2Ph
(28)
Реакции, аналогичные схеме (28), могут использоваться для синтеза сложных эфиров ненасыщенных дикарбоновых кислот. Например, в результате реакции [34] монолитиевого производного ди-трет-бутилглутарата с различными кетонами с прекрасными
86
выходами получаются сложные эфиры гидроксидикарбоновых кислот (13).
NaOEt
EtO2C(CH2)4CO2Et —
CO2Et
Mel
Me
CO2Et
Me
горячий NaOEt
------> EtO2C(CH2)3CH
EtOH I
EtO2C
(29)
•CH2Ph NaOH, H2O; /CHzPh
’ + ---:-----НО2С(СН2)зСН(
CO2Et н+, н2о \CO2H
V=o
R2Z + Li
7’pe7,-BuO2C(CH2)2CHCO2Bu-7’per
R\ XCOH
R2Z
-—> 7рет-ВиО2С(СН2)2СНСО2Ви-77?ет
(13)
TsOH, ксилол
R1 и/или R2 = Ar
R\
V=CH(CH2)2CO2H
R2Z
(30)
TsOH, КСИЛОЛ ----------->-R1 и R2=#Ar
CO2H „
I zR
HO2CCH2CH2C=(Z \R
2
Гидролиз сложных эфиров (13) с одновременной дегидратацией приводит к ненасыщенным производным глутаровой кислоты, если заместители R1 или R2 не ароматической природы {схема (30)}. Однако если один из этих заместителей ароматический, то гидролиз сопровождается не только дегидратацией, но и декарбоксилированием и приводит к ненасыщенным монокарбоновым кислотам.
Реакция Виттига — важнейший общий метод региоспецифичного синтеза сложных эфиров а,р-ненасыщенных и полиеновых дикарбоновых кислот [35]. В типичном синтезе {схема (31)} [36], как и во многих подобных случаях, продукт реакции является смесью Цис- и транс-изомеров, которые в данном конкретном случае можно разделить дробной кристаллизацией. Особенно широко реакция Виттига применяется в синтезе каротиноидов; в некоторых случаях в этих синтезах используются производные ненасыщенных
87
дикарбоновых кислот. В качестве типичного примера приведем синтез природного биксина {схема (32)} [37]: ключевой интермедиат 5-метоксикарбонил-3-метилпента-цис-2-транс-4-диеналь (14), как показано на схеме, конденсируется с илидом (15) в стандартных условиях реакции Виттига.
МеО2С(СН2)8СНО + Ph3P=CHCO2Me —>
9.2.1.3. Реакции Михаэля
Реакция Михаэля (см. обзор [38]) используется для получения различных ди- и поликарбоновых кислот. В этом разделе мы рассмотрим несколько типичных примеров этой реакции. Малонат-анион присоединяется к сложным эфирам [39] и нитрилам [40] аф-ненасыщенных кислот с образованием продуктов, дающих при гидролизе производные глутаровой кислоты {схемы (33)—(36)}.
NaOEt MeCHCH2CO2Et MeCH=CHCO2Et + MeCH(CO2Et)2 --------> | (33)
EtoH MeC(CO2Et)2
NaOEt, EtOH; MeCHCH2CO2H MeCH=CHCO2Et + CH2(CO2Et)2 -------> | (34)
н+, h2o CH2CO2H
CH2CO2(Et) CH2CO2H
/П NaOEt 1 NaOH, H2O; CHCO2Et >• I H+, H2O AhCO2H 1
/ \ + ’-H2(GU2Lt)2 > Н/ — 'CO2Et EtOH
CH(CO2Et)2 CH2CO2H
(35)
«8
CH2=CHCN + CH2(CO2Et)2
NaOEt
NCCH2CH2CH(CO2Et)2
(36>
LiN (Pr-uso)2
Me2CHCO2H --------—>-
R*CHCHR2CO2Me
Me2^CO2SiMe2
Li I Me2CCO2Li
К‘сН=СК2СО2Ме;
-----—---------->
Me3SlCI
MeOH R1CHCHR2CO2i4e
Me2(!:cO2H
(37>
Глутаровые кислоты можно также получить [41] присоединением дианионов карбоновых кислот к а,р-ненасыщенным сложным эфирам {схема (37)}. Дианион изомасляной кислоты получают в тетрагидрофуране при О С с использованием двух эквивалентов основания; вслед за присоединением по Михаэлю следует триме-тилсилилирование продукта.
Полный синтез^ фунгицида (±)-авенациолида [42] включал в качестве ключевой стадии получение замещенного бислактона (16) в результате сходного с реакцией Михаэля процесса {схема (38)}. На последних стадиях этого синтеза нужная двойная связь вводилась пиролизом сульфоксида в присутствии янтарного ангидрида.
(16)
9.2.1.4. Окислительные методы
Многие важные пути, ведущие к ди- и поликарбоновым кислотам, включают окисление; некоторые методы нашли практическое применение. Для удобства мы рассмотрим отдельно окисление ароматических и алифатических субстратов.
(1) Получение ароматических кислот
Для получения ароматических ди- и поликарбоновых кислот широко используют окисление боковых цепей различных ароматических соединений. Алкилбензолы, такие как изомерные ксилолы, легко окисляются в соответствующие карбоновые кислоты в
в»
жестких условиях. Примеры на схемах (39) —(45) иллюстрируют набор окислительных агентов, которые можно использовать лп этой цели. для
Окисление фенаптрахпнопа {схема (46)} служит удобным ме тодом синтеза как бифенил-2,2'-дпкарбоновой кислоты [431 и ее диметплового эфира [44]. Окисление различных ацил галоген* аценафтенов [45] приводит к соответствующим нафталиновым ан гидридам, хотя существуют заметные различия в легкости обоато вания ангидридов {схема (47)}. 1
О2. Оз ---------------> Co(II), НО Ac
(39)
(44)
(45)
90
(6) R = Ph, X = Cl или Br, Y = H
(46)
(47)
(2) Получение алифатических кислот
В синтезе дикарбоновых кислот этим путем можно выделить два окислительных процесса: первый включает окислительную димеризацию, второй —расщепление углерод-углеродной связи, часто в циклических соединениях {схема (47)}. Сложные эфиры янтарной кислоты можно получать окислительной димеризацией енолят-анионов в присутствии солей меди (II). Метод, использующий литиевые еноляты [46], {схема (48)} проще и, по-видимому, носит более общий характер, чем альтернативная методика с применением цинкорганических соединений [47] {схема (49)}. Обе реакции напоминают давно известные методы димеризации стабильных анионов, например анионов диэтилмалоната с использованием иода в качестве окислителя {схема (50)}. Прочие окислительные реакции «димеризации», основанные на окислительном декарбоксилировании по Кольбе, обсуждаются в разд. 9.2.3.4.
Li LiN (Рг-изо)г | MeCH2CO2Et — _78Oc> MeCHCO2Et
.. Zn> CuCl2
Me2Cz ----------------------->
VqO Ef ТГФ+следы HgBr2
CuBr2 MeCHCO2Et
----* I
MeCHCO2Et
Me2CCO2Me
Me2CCO2Me
NaOEt
CH2(CO2Et)2 —-> NaCH(CO2Et)2 t Юп
I2 CH(CO2Et)2
Ан(СО2Е1)2
(48)
(49)
(50)
Ацетиленовые кислоты и их эфиры с высоким выходом подвергаются окислительной димеризации в водном этаноле под действием кислорода или воздуха в присутствии хлорида аммония или меди. Эта реакция использована в синтезе кортикроцина [48];
91
контроль за реакцией, которая шла в этом случае с почти количественным выходом при комнатной температуре, осуществлялся по поглощению кислорода {схема (51)}.
о2
НС=С(СН2)2СН=СНСО2Н —-> (С=ССН2СН2СН=СНСО2Н)2 (51)
Си '
_______си-----/со2н (СН2)Л II ---->(СН2)Л (52)
- сн СО;||
Олефины можно окислять до дикарбоновых кислот {схема (52)} различными способами, и если бы не возникали проблемы, связанные с растворимостью в органических растворителях, наиболее удобным для этой цели был бы перманганат калия. Эти затруднения до некоторой степени преодолимы [49], если использовать в качестве растворителя уксусный ангидрид. Однако в этом случае выходы снижаются, и как показано на примере окисления по схеме (53), могут протекать побочные реакции.
СО2Н
Кмпо4 / о=сх
(СНг)ю ” (СН2)ю + /(СН2)ю + (СН2)ю (53)
нс/ 20 \ АсОНс/ 0=(/
СО2Н
(16%) (17%) (48%)
Использование краун-эфиров позволяет снять большинство проблем [50], ибо эти соединения способны образовывать комплексы с солями металлов, что приводит к повышению растворимости в органической среде и повышению реакционной способности анионов. Например, длцпхлогексил-18-краун-б образует с перманганатом калия растворимый в бензоле комплекс (17), что дает прекрасный окислитель для органических субстратов. В частности, циклогексен окисляется им с количественным выходом до адипиновой кислоты {схема (54)}. По-видимбму, нет оснований предполагать, что механизм этого окисления отличается до такового, действующего в водных средах {схемы (55), (56)}.
0 3RCH=CHR 4- 8КМпО4 - МпО4 *+ Т J (И) <^СО2Н (54) [ ,со2н 1 -> 6RCO2K + 8MnO2 4- 2КОН 4- 2Н2О 155)
92
(56)
Для окисления алкенов водным перманганатом калия можно с успехом использовать катализ фазового переноса [51]. Реакции субстратов, растворенных в органической фазе, с неорганическими реагентами в водной фазе, которые ингибируются в силу раздела фаз, часто катализируются добавлением следовых количеств растворимых в органической фазе тетраалкиламмониевых или тетраалкилфосфониевых солей. Предполагают, что катализ осуществляется за счет способности катионов, растворимых в органическом растворителе, многократно переносить анионы в органическую фазу в форме, подходящей для реакции. Этот эффект носит название катализа фазового переноса.
Озонолиз олефинов, как правило, проводят в органических растворителях, часто при низких температурах. Образующийся озонид (18), который обычно слишком нестабилен для безопасного выделения, можно окислять до карбоновых кислот. При окислении циклического олефина продуктом реакции служит дикарбоновая кислота {схема (57)}. Этот двухстадийный процесс можно упростить [52], так как было показано, что в благоприятных случаях эмульсии циклических олефинов и щелочного пероксида водорода мягко реагируют с озоном и с хорошими выходами образуют а <о-дикарбоновые кислоты {схема (58)}.
ез
СО2Н
(59)
До днкарбоновых кислот можно окислить и другие карбоциклические соединения. В подходящем растворителе циклические кетоны [53] окисляются молекулярным кислородом до дикарбоновых кислот {схема (59)}. Показано, что многие растворители автоокис-ляются в условиях реакции, однако применение гексаметапола (ГМФТА) сводит эти побочные реакции до минимума и позволяет получать удовлетворительные выходы продуктов. Как правило, окисляется наиболее кислая связь С—Н кетона с образованием нестабильного промежуточного перокси-аниона. Полное окисление, аналогичное схеме (59), достигнуто действием азотной кислоты
[54].
9.2.1.5. Гидролитические методы
Для получения дикарбоновых кислот можно использовать большое число методов, основанных на гидролизе. Часть этих методов мы рассмотрим в данном разделе. Методы, ведущие к сложным эфирам, которые далее можно гидролизовать до ди- или поликарбоновых кислот, рассмотрены в другом месте.
Хотя обычно амиды получают из карбоновых кислот и нитрилов, тиогомофталимиды легко получают из хлорангидридов арилуксусных кислот [55]. Последующий гидролиз тиогомофталимидов служит удобным методом синтеза гомофталевых кислот {схема
СН2СОС1
CH2C0NCS
Pb(NCS)2
сн2со2н
Важным методом синтеза карбоновых кислот является гидролиз нитрилов. В обзоре [56], посвященном малононитрилу, обсуждается его алкоголиз до диэтилмалоната. Синтез янтарной кислоты гидролизом нитрила показан на примере получения [57]; фенилянтарной кислоты {схема (61)}. В этом синтезе использованы конденсация Кневенагеля и последующее присоединение к продукту реакции цианида калия по Михаэлю.
PhCHO + CH2(CO2Et)2 ————-> PhCH=C(CO2Et)2 РпСОгН, C$Hg
PhCHCH2CO2Et KHCO3 PhCHCO2H
—> I ------>- I
CN EtOH CH2CO2H
(61)
94
•О
(СН2),
CN
О
СО2К СО2Н
I HCI I
* (СН2)3
conh2
NH
н3о+
---->- НО2С(СН2)зСО2Н
(62)
з
Удобный метод получения глутаровой кислоты [58] основан на гидролизе нитрилов: у-бутиролактон реагирует с цианидом калия при 190 °C, и калиевая соль получающейся цианокислоты гидролизуется до глутаровой кислоты. В тщательно контролируемых условиях гидролиза можно выделить моноамид глутаровой кислоты, который затем циклизуется в глутарамид {схема (62)}. Гидролиз динитрилов, получаемых из дигалогенидов, приводит к дикарбоновым кислотам. Дикарбоновая кислота (19), необходимая в качестве интермедиата для синтеза производного пропел-лана [59], была получена именно таким путем {схема (63)}. Подобные реакции замещения цианид-ионом не всегда протекают столь гладко, и как показано на схеме (64), в процессе реакции замещения промежуточный динитрил (20) циклизуется в енамино-нитрил. Тем не менее гидролиз и размыкание цикла все же приводят к искомой кислоте [59].
СН2Вг
СН2Вг
NaCN, ГМФтА
Na OH, CH2CN hoch2ch2oh, H2O
ch2cn
СН2СО2Н
>
Me
sLwCh2ots
Т^СНгОТз
Me
(63)
CH2CO2H
(19)
(20)
(64)
Me
CO2H
CO2H Me
nCF2=CF2 + CH3SSCH3
(rper-BuO)2
-----------
~~►CH3SCF2(CF2CF2)„_1CF2SCH3— 4MeOH18° C,>CH3O2C(CF2CF2)n_ICO2CH3
(21) (65)
95
„„„мания Другая методика, включающая гидро-
Заслуживает внимани методом получения перфторал-
лиз, так как она являе а ^-бис(метилтио) полифторалканов
кандикарбоновых кнс о тилена в приСуТСТВии диметилди-[60]. Теломеризация Р сида в качестве катализатора приводит сульфида и трсг-бут ЛемаД/65)р Как видно из схемы, эти про-к продуктам типа < ются серной кислотой в метаноле до
КХв'ьГэфД фторированных днкарбоновых кислот.
9.2.1.6. Другие методы
Помимо уже рассмотренных общих методов получения дикарбоновых кислот существует множество других методов, некоторые из которых мы сейчас рассмотрим.
Малеиновые ангидриды получаются из 1-этоксиалкинов-1 последовательностью реакций, представленной на схеме (66). Это общий и в настоящее время лучший метод синтеза различных малеиновых ангидридов [61].
r’coco2h
EtOCsCR2
R'
о
:сХс
OEt
о р
1 1
R— С—С
I \ н2о
О ----
Н\ I /, 2>с-с+
R OEt
ОН
/
о----:
/ -н£о
(66)
(R2 = Me,Et,Bu ит.д.)
/ I
R
д
R / \ НО OEt
о
Метилпирувар под действием триметилфосфита подвергается восстановительной димеризации [62]. Продукт этой реакции является смесью двух диастереомерных оксофосфоранов (22), гидролиз которых приводит к смеси диметилвинных кислот {схема (67)}-
2МеСОСО2Ме
СО2Ме
Меч I О
'С\
I J^P—OMe
Ме/С^р ^ОМе
СО2Ме (22)
ОН
Н2о Me—С—СО2Н
Me—С—СО2Н
ОН
Аналогичным образом фенантрахинон реагирует с триметил-фосфитом с образованием кристаллического интермедиата (23л* 96
отме-
vT/ячянноб строение, так как при озоно* который, вероятно имее Уосстановлен£е (24) трифенилфосфином лизе дает пероксид дта последовательность реак-
СТема ?б1)ТОальТУериатнвна [63] методике окисления, ценной в разд. 9.2.1.4 {см. схему (46)}.
(23)
(24)
(68)
О селективном алкилировании янтарной и других дикарбоновых кислот уже упоминалось в разд. 9.2.1.2. Эти реакции широко применяются в синтезе; дополнительные детали [32] подчеркивают возможности применения этого метода {схемы (69) — (71)}- Конденсация дианионов, полученных из моноэфнров янтарной кислоты, с кетонами или бензальдегидом также протекает специфично по соседнему со сложноэфирной группой а-углсродному атому, приводя к продуктам замещения по этой метиленовой группе. Ациклические и алициклические карбоновые кислоты могут быть превращены [64] в моноэфиры малоновой кислоты алкоксикарбонилнро-ванием соответствующих дианионов. Этот метод показан на схеме (72); наилучшие выходы получены в случае кислот, имеющих лишь один а-водород. Другие примеры реакций алкоксикарбонилирования описаны в разд. 9.3.1.
СН2СО2Ме Ан2со2н 2NaNH2 СНСО2Мё сн2со; ЩНа1; RCHCO2Me Ан2со2н (69)
NH3
R'CH^R2 1 2NaNH2 R'cco2r2 R3Hal R3 r4co2r2 CH2CO2H (70)
СН2СО2Н NH3 * СН2СО2 Н +
4 Зак. 396
97
R3HaI; R3CHCO2R2 ----—> I
CH2CO2R2 2NaNH2^ CHCO2R2 h R‘CHCO2H
r>chco2h NHa R'CHCO2- R4R5CO; R\ (71)
-------> COH H+ R5/ I
CHCO2R2
r>chco2h
Li CO2R3
LiN(Pr-«3O)2 I CICO2R3 или I
R'R2CHCO2H ---R'R2CCO2Li R‘R2CCO2H (72)
Наращивание цепи дикарбоновых кислот ацилированием енаминов [65] {схема (73)} служит удобной альтернативой электролитическому окислению моноэфиров дикарбоновых кислот по Кольбе.
КОН, N2H4, (НОСН2СН2)2О
—> НО2С(СН2)6СО(СН2)„СО2Н -------------------------->
н+, н2о
—> НО2С(СН2)6СН2(СН2)„СО2Н
(73
9.2.2. СВОЙСТВА ДИ- И ПОЛИКАРБОНОВЫХ КИСЛОТ
Структура и тривиальные названия некоторых дикарбоновых кислот приведены в табл. 9.2.1. Инфракрасная спектроскопия дикарбоновых кислот обсуждается в работе [66]. Интересно отметить, что некоторые кислоты имеют две полосы поглощения в карбонильной области. Например, щавелевая кислота имеет vMaKc при 1710 и 1690 см-1, малоновая кислота — при 1740 и 1710 см-1. Этот эффект взаимодействия уменьшается в случае янтарной кислоты, которая дает слабый пик при 1780 см-1 и основной пик при 1700 см-1. Более высокие члены этого ряда имеют лишь одну полосу поглощения карбонильной группы [67] при 1700 см-1. Если не принимать во внимание первые члены серии, инфракрасные спектры дикарбоновых кислот можно рассматривать как типичные для карбоновых кислот. Фталевая и терефталевая кислоты также имеют обычные для ароматических кислот инфракрасные спектры с полосами поглощения при 1695 и 1690 см-1 соответственно.
Инфракрасные спектры сложных эфиров дпкарбоновых кислот в общем аналогичны спектрам сложных эфиров. Лишь в случае диэтилсукцината наблюдается небольшое уменьшение частоты поглощения (vMaKc 1733 см-1) по сравнению с более типичным значением для диэтиладипата (vMaKc 1739 см-1). Спектры циклических
98
Таблица 9.2.1. Константы диссоциации дикарбоновых кислот [66а]
(а) Влияние длины цепи в НО2С(СН2),,СО2Н_________
Кислота п 1 pKi рК2 К}/К.2
0 1,23 4,19 922
Щавелевая 1 2,83 5,69 734
Малоновая 2 4,19 5,48 19,2
Янтарная 3 4,34 5,42 11,9
Глутаровая 4 4 42 5,41 9,88
Адипиновая 5 4 48 5,42 8,70
Пимелиновая Пробковая (субероновая) Азеланновая 6 7 4^52 4,55 5,40 5,41 7J0 7,30
(б) Влияние алкильного замещения в малоновых кислотах RIR2C(CO2H)2
R1 R* pKi pX2
н н 2,83 5,69 734
Me н 3,05 5,76 510
Me Me 3,17 6,06 783
Et Et 2,21 7,29 121 000
Pr Pr 2,07 7,51 277 000
(в) Влияние ненасыщенности и геометрии
Кислота Формула pKi Г’Щ К1/К2
Малеиновая НО2с/ \сО2Н 1,92 /СО2Н 6,23 20 200
Фумаровая НО2(Х 3,02 4,38 23,2
Ацетилендикарбоновая HO2C&sCCO2H 1,73 4,40 460
Фталевая с О2Н СК ____21^со2н 2,95 5,41 288,4
(г) Влияние размера кольца и конфигурации в алициклических дикарбоновых
кислотах
| /Z/CO2H <Ь-со2н '—'Ч-L _
X Геометрия | PKi РК2 Ki/K2
Ме2С цис 2,34 8,31 92 900
СН2 транс 3,82 5,32 31,8
цис 3,33 6,47 1 380
(СН2)3 транс 3,65 5,13 29,7
цис 4,37 6,51 138
(СН2)4 транс 3,89 5,91 105
цис 4,34 6,76 267
транс 4,18 5,93 56
4*
99
ангидридов полезны при установлении строения. Тем не менее спектры как янтарного (vMaKc 1865 и 1782 см"1), так и глутарового (Умакс 1802 и 1761 см-1) ангидридов существенно отличаются от спектров ациклических ангидридов, например уксусного ангидрида (v-макс 1824 п 1748 см-1). В инфракрасных спектрах ненасыщенных циклических ангидридов также присутствуют характерные полосы поглощения карбонильных групп. Например, малеиновый и фталевый ангидриды имеют поглощение при умаКс 1848, 1790 см~> и 1845, 1775 см-1 соответственно.
ЯМР-Спекгры дикарбоновых кислот аналогичны таковым для карбоновых кислот, причем химические сдвиги как в ‘Н-, так и |3С ЯМР-спектрах находятся в соответствии с обычными аддитивными правилами для эффектов замещения. В обзорах по 13С-ЯМР спектроскопии дикарбоновых кислот [68, 69] приведены примеры экранирования алкильных углеродов. Интересно отметить, что наблюдаемые эффекты экранирования очень близки по своим значениям к предсказываемым при допущении аддитивности параметров химических сдвигов карбоксильной функции по отношению
Таблица 9.2.2. Константы диссоциации ароматических дикарбоновых кислот и их моноэфиров в различных средах [77]
Кислота В CH3CN В ДМСО
РМа) рД(2а) рК(б) Рк(а) рф рК<б>
Бензойная 20,7 11,1 Фталевая 14,2 29,8 19,4 5,7 16,0 9,9 З-Метилфталевая 17,0 25,6 20,1 9,1 15,1 10,4 Изофталевая (мета-) 19,3 23,0 20,0 9,8 12,3 Терефталевая (пара-) 19,7 9,8 12,1 10,1 Нафталин-1,8-дикарбоновая 21,8 24,9 11,0 15,0 2,2/-Дифеновая 15,7 26,1 20,8 7,7 13,9 11,4
Кислота в Н2О В МеОН
рК1а1 р/фо рК(б) pK<a> рК(2а> рК™
Бензойная 4,2 9,4 Фталевая зф 5,40 3,18 7,4 12,1 8,6 3-Ме.тилфталевая 3,1 5,20 8,4 11,7 8,8 Изофталевая (мета-) з’,54 4Д2 3,89 8,7 10,6 8,9 Терефталевая (пара-) 3,54 4,46 8,5 10,3 8.8 Нафталин-1,8-дикарбоновая 9,1 12,7 2,2 -Дифеновая 3,53 5,42 3,96 7,8 12,8 9,6 — -—•
(б) Значения относятся к дикарбоповым кислотам.
Значения относятся к моноэфирам.
100
к соответствующим алканам (С a, -j-20,9; С Р, +2,5, С у, 1.2 2- С —6,+1,0; С —е, +1,2 млн-1) - Обзор по химическим сдвигам карбоксильных углеродов см. в [70].
Значения рКа первой (pKi) и второй (рК2) констант диссоциации некоторых дикарбоновых кислот приведены в табл. 9.2.1..Относительные значения pKj и рК2 (см. табл. 9.2.1) в основном объяснимы с точки зрения электростатического эффекта ингибирования образования дианиона, и как видно, при увеличении расстояния между двумя карбоксильными группами pKi приближается к рК2. Интересные эффекты, связанные с алкильным замещением и геометрией алициклических систем, видны из примеров, представленных в части (б) и (в) табл. 9.2.1. В дополнение к электростатическому эффекту в моноанионах важно наличие водородной связи, особенно в том случае, когда структура оптимальна для образования внутримолекулярной водородной связи между протоном не-ионизованной карбоксильной группы и кислородом соседнего карб-оксилат-аниона. В работах [71—73] детально изучено влияние связывания водорода на относительные значения и рК2, а также оценено влияние структуры кислоты и растворителя. Эффекты растворителя могут быть особенно сильными, как видно из данных по ароматическим дпкарбоновым кислотам и их моноэфирам (табл. 9.2.2). Последние можно использовать в качестве стандарта, в котором электронные эффекты можно оценить без осложнений, связанных с образованием водородной связи. Для более полного обсуждения этих результатов рекомендуем обратиться к оригинальной работе [71].
9.2.3. РЕАКЦИИ ДИ- И ПОЛИКАРБОНОВЫХ КИСЛОТ
Реакции ди- и поликарбоновых кислот, если рассматривать их карбоксильные группы изолированно, сходны с реакциями мо-нокарбоновых кислот. Однако если в процессе реакции происходит взаимодействие между двумя или более карбоксильными группами, то эти реакции будут значительно отличаться от таковых для мо-нокарбоиовых кислот. Реакциям последнего типа в этом разделе будет уделено особое внимание. Кроме того, реакции некоторых дикарбоновых кислот, имеющих большое значение, будут рассмотрены в тех разделах, где описана общая химия индивидуальных соединений. Поскольку получение, свойства и реакции циклических имидов описаны в гл. 9.9 (см. разд. 9.9.1.8; 9.9.2.4 и 9.9.3), здесь они не рассматриваются.
9.2.3.1. Образование и реакции циклических ангидридов
13 подходящих условиях монокарбоновые кислоты образуют ангидриды (25) {схема (74)}; формально реакция сводится к меж-молекулярпой дегидратации. В свою очередь дикарбоновые кис-оты могут реагировать внутримолекулярно с образованием
401
циклических ангидридов (26) {схема (75)} значительно быстрее Реакция протекает особенно легко, если продукт (26) содержит пяти- или шестичлепный цикл, как, например, в случае янтарного (27), малеинового (28), глутарового (29) и фталевого (30) ангидридов.
2RCO2H (RCO)2O (25) (74)
Циклические ангидриды типа (26) легче всего получать из соответствующей дикарбоновой кислоты нагреванием [74] или обработкой уксусным ангидридом [75]. В случае лабильных веществ, например по схеме (76) [76], наилучшим реагентом служит трифторуксусный ангидрид, ибо реакцию с ним можно проводить при низкой температуре.
(CF3CO)2O
Н о
Для этой цели можно применять и другие дегидратирующие агенты. Довольно широко применяют дициклогексилкарбодиимиД (ДЦК, 31), с помощью которого в наиболее удачных случаях получают семичленные циклические ангидриды, например превращение 2,3-секо-5а-холестан-2,3-диовой кислоты в ее ангидрид {схема (77)}. В тех случаях, когда и применение ДЦК не приводит к успеху, как, например, при синтезе [78] ангидрида норборнадиен-2,3-дпкарбоновой кислоты {схема (78)}, используют этоксиацетплен. Для получения ангидридов можно применять и другие реагенты, включая фенилцианат (32) [79] и М,М-диэтиллропин-1-ил-102
амин (33) реагентов,
[80]; продукты, получаемые при использовании этих показаны на схемах (79) и (80).
(33)
На схеме (81) показан двухстадийный способ получения ангидридов из дикарбоновой кислоты [81], однако этот метод не нашел широкого использования. Необходимо отметить, что те дикарбоновые кислоты, которые не дают «мономерных» ангидридов с соответствующим размером цикла, образуют полимерные ангидриды йли могут вступать в различные реакции, например реакции циклизации {схема (82)}, особенно если образуются пяти- или шестичленные циклы.
EtO2Cx Z(CH2)2CO2H
ею2с/ \сн2)2со2н
Ас2О
пиридин
EtO2C
EtO2C
(81)
(82)
ЮЗ
о
Циклические ангидриды, так же как и их ациклические аналоги, служат мощными ацилирующими агентами и, кроме того {схема (84)}, являются важными интермедиатами при синтезе моноамидов и моноэфиров из дикарбоновых кислот. Вода гидролизует циклические ангидриды до исходных дикарбоновых кислот. Как видно на примере классической реакции сукцнноилирования {схема (83)}, использованной в качестве первой стадии синтеза конденсированных ароматических систем, и более тонкой реакции [83] янтарного ангидрида и ацетата енола (34) {схема (85)}, циклические ангидриды в условиях реакции Фриделя-Крафтса ацилируют ароматические соединения и даже олефины. Реакции ацилирования, в результате которых получаются имиды [84] {схема (86)}, и другие реакции, приводящие к имидам, обсуждаются в гл. 9.9.
104
При ацилировании диазометана получают диазокетон {схема (87)}, который в обычных условиях претерпевает перегруппировку Вольфа. Эта реакция позволяет гомологировать исходную кислоту по одной карбоксильной группе [85]. Аналогично, при ацилировании пероксида водорода получают монопероксикислоту. Кристаллическая мопопероксифталевая кислота {схема (88)} в силу своей устойчивости, обусловленной резонансной стабилизацией, является важным реагентом [86].
дианион, красный
(89)
105
Циклические ангидриды, и особенно фталевый ангидрид, Пп сравнению с большинством производных карбоновых кислот про. являют заметную склонность к образованию продуктов конденсации, аналогично кетонам. Примеры некоторых подобных реакций [87—90] показаны на схемах (89) —(92). Реакция, представленная на схеме (89), приводит к соединениям, известным как фталеины, среди которых есть pH-чувствительные красители, применяемые в качестве индикаторов кислотно-основного титрования.
Н О
(91)
р + Ph3P=CHCO2Et
Н О
Образование моноэфиров при реакции фталевого ангидрида со спиртами {схема (84)} использовано для разделения оптически активных спиртов. Диастереомерные соли моноэфира рацемического спирта и оптически активного основания можно разделить фракционной кристаллизацией и далее легко регенерировать оптически чистый спирт.
9.2.3.2. Конденсация Дикмана
Конденсация Дикмана представляет собой внутримолекулярный вариант конденсации Кляйзена {схема (93)}; она широко применялась для синтеза циклических сложных эфиров 0-кетокислот, используемых в основном как синтетические интермедиаты, ласть применения этой реакции рассмотрена в обзоре [91], в‘лиЯ ние заместителей обсуждается в работе [92]. В разд. 9.2.1.2 уже упоминалось о применении реакции Дикмана для синтеза алкиль них производных пимелиновой и адипиновой кислот; общий метод синтеза таких соединений показан на схеме (94).
106
^CO2Et
^-ch2co2ei
о
NaOEt JJ—OEt
EtuH <CHCO2Et
—OEt
CHCO2Et ~OEt"
,O
NaOEt ------->
CO2Et (CH2)„
CO.Et
CHCO2Et
NaOEt, EtOH ------>
(94)
NaOEt, CO2Et EtOH | ----^(СНг)^
CHRCO2Et
л=‘4 или 5
Обычно оптимальные выходы в реакции Дикмана получают в случае образования пяти- или шестичленных циклов, особенно при проведении реакции в условиях большого разбавления. В некоторых случаях удается получить производные циклобутанона, однако выходы, как правило, низки; самоконденсация диэтилсукцината [93] приводит к производному 1,4-диона (35) {схема (95)}. Образование «димерных» продуктов типа (35) обычно происходит в тех случаях, когда в результате межмолекулярной реакции Дикмана получаются средние циклы. Простая внутримолекулярная реакция Дикмана не проходит также в тех случаях, когда стерически не выгодно образование енолят-иона циклического сложного эфира (3-оксокислоты (конечного продукта реакции).
CO2Et
CO2Et
NaOEt;
------->
Н+
(35)
Н3РО4
Н2О
(95)
Необходимое для реакции Дикмана основание можно выбрать среди множества таких сильных оснований, как гидрид натрия, этилат натрия [94] и трет-бутилат калия [95]. Как видно из примера циклизации по Дикману триэфира (36) [96], выбор условий реакции может играть важную роль. Например, использование грет-бутилата калия в существенно необратимых условиях дает пирролидоновое производное (37), тогда как в других условиях получают пиперидоновое производное (38) {схема (96)}.
107
EtOjC C>
<5xxco2Et
N
Me (37)
KOBu-rper
PhCH3, -20 °C
EtO2C\ ^/COjEt
^N^XCO2Et I Me (36)
NaH, c«H«-Nall, Ph’c^, ил и
NaOEt. EtOH
(96)
Применение реакции Дикмана [97] для синтеза циклических кетонов и дионов показано на схеме (97); в основном получаются пяти-, шести-, семичленные и высшие циклы. Кетоны со средним размером цикла (/г = 7—11) в этом случае не получаются, а вместо них, как показано на схеме, конечными продуктами реакции являются дионы. Циклизация Дикмана использована в синтезе стероидов через гидрохризен по Джонсону [95] {схема (98)}, а также при синтезе [98] ферроценофана {схема (99)}.
Q (СНД
СН
CHCOoEt
CO2Et NaOEt, Ei ОН;
NaH, PhMe;
Ч------------
КО В и-грет, НОВц- трет $
/COaEt
(^2)л н+,Н2О СН
XCO2Et EtO2C
О.
(СН2)Л_!^
'• 7 CHCO2Et
(сн^-^о
н+ н2о
71=4,5,6
н+,н2о
Л=7, 8,9,10
Помимо этого для синтеза циклических соединений применяется комбинация конденсаций Дикмана и Кляйзена. На схеме (100) представлен типичный пример [99] построения шестичленного кольца, однако если енолят-ион продукта реакции стернчески не выгоден, реакции этого типа не идут. Например, диэфир (39)
108
Л диметилоксалат не вступают в конденсацию с образованием би-
циклической системы (40) [100].
(98)
(99)
В общем случае конденсация диэфпров с карбанионами, полученными ггз соединений общей формулы CH2XY (где X и/пли Y — активирующие группы, такие как COR, CN, CO2R и др.), приводит к образованию циклических продуктов {схема (101)}. На схемах (102) [101] и (103) [102] приведены отдельные примеры этих реакций. Для подавления побочных реакций, например конкурентной конденсации, необходим тщательный подбор условий и реагентов.
СО2Ме
СО2Ме
СН2СО2Ме + I
СН2СО2Ме
NaOMe
^COCHXY CH2XY-> Г
^СОгР
(101)
109
Другие типы реакций циклизации, особенно образование лактонов, могут быть результатом конденсации диэфиров или других производных дикарбоновых кислот. Область применения реакций этого типа чрезвычайно широка. Несколько примеров, основанных на использовании производных янтарной кислоты, показано на схемах (104) [103], (105) [104] и (108) [105]. Примеры реакций циклизации, в основе которых лежит реакция Манниха, приведены на схемах (106) [106] и (107) [107], причем второй пример — классический синтез тропиноновой системы по Робинсону, который является первым синтезом природного соединения, основанным на биогенетических представлениях. И наконец, реакция, изображенная на схеме (109), может быть представлена как присоединение по Михаэлю и последующая сложноэфирная конденсация [108].
СО2Ме
сио I ч (СН2)з сно
KOBu-трет. трет-BuOH ---------->-или
NaH, EtOH
L—CHCO?Et
CH2CO2Et
(CH2CO2Et)2 + R'COR2
X-BUOH R'\ /CO2Et H+
---------->- C=CZ ---->•
ИЛИ / \ r-ГЛ- H2°
NaH, EtOH R2/ XCH2C(J2
R> /CO2Et
Xc=c/ (108)
R2// \CH2CO2H
9.2.3.3. Окисление
Окисление производных янтарной кислоты может приводить к олефинам в результате процесса окислительного декарбоксилирования, в общем виде изображенного на схеме (НО). Кроме того, пригодным методом для подобных превращений является электролитическое окисление диапиона янтарной кислоты по Кольбе [109], как показано на примере схемы (111) [ИО]. Доступность производных янтарной кислоты [например, (41)] как продуктов реакции Дильса — Альдера (см. разд. 9.2.3) стимулировало интенсивное
Ш
исследование других методов, проводящих превращения по схеме (ИО).
сн2со; сн2со2. сн2
| ----* I —► II + 2СО2 (ид)
СН2СО2 2е СН2СО2 • сн2
В качестве альтернативы анодного окисления используют тетраацетат свинца [например, реакция по схеме (112)]. По всей видимости, эта реакция не стереоселективна, так как бисдекарбоксилирование тетраацетатом свинца и в условиях анодного окисления как мезо-, так и (±)-1,2-дифенилянтарных кислот приводит к транс-стильбену [112].
Янтарные ангидриды можно непосредственно превратить в олефины действием тетраацетата свинца в пиридине. Метод был применен в первом синтезе [113] «дьюаровского бензола» (42) {схема (113)}. Альтернативный метод использует комплекс никеля (0) [114] {схема (114)}.
РЬ(ОАс)4 --------------> пиридин, 65 °C
(42)
(113)
(Ph3P)2Ni(CO)2 --------------->
Д,диглим
(114)
Фотохимический распад сложных трст-бутилпероксиэфиров, полученных из янтарной кислоты, также приводит к олефинам. Об-112
щая процедура этой реакции [115] и характерный ее пример показаны соответственно на схемах (115) и (116).
(44)
Как видно на примере окисления циклобутан-1,3-дикарбоновой кислоты [117] {схема (117)}, анодное окисление (по Кольбе) может приводить к побочным реакциям [116], например к лактонизации. Zj'uc-Дикарбоновая кислота (44) дает бициклобутановое производное {схема (118)}, однако из транс-дпкарбоновой кислоты (43) (а-труксилловая кислота) в качестве основного продукта получается лактон (45). Подобный результат, включающий как сужение цикла, так и лактонизацию, хорошо объясняется промежуточным образованием карбенпевого иона.
Анодное окисление сложных моноэфпров замещенных малоновых кислот [118] открывает путь к эфирам янтарной кислоты, и
/CO2R2
R’CMe2CH/ \зо2'
(46)
С > R1 CMe2CHCO2R2 —> R1 CMe2CHCO2R2
—СО2 I
R'CMe2CHCO2R2 /im.
—е (119)
| (47)
+ R1 CMe2CHCO2R2 —> Небольшие количества продуктов перегруппировки
Ro2c(CH2)nco;
— е ---->
—СО2
RO2C(CH_),t.
RO2C(CII2).„CO2R (121)
из
п плотности трет-алкилзамещенные маловаты (46) с хорошим вы-хоХ приводят к смеси диастереомерных 2,3-ди-трвт-алкилсукци-Swr 147 • одновременно образуются неоольшие количества низ-’ продуктов, которые получаются, по-видимому, по ион-отТхачпзму (схема (119)}. В общем случае электролиз моно. S0B дикарбоновых кислот по Кольбе может применяться как метод [109] получения а.о-дикарбоновых кислот с длинной цепью (схема (120)}.
9.2.3.4. Восстановление
Восстановление диэфиров дикарбоновых кислот в большинстве ечучаев протекает аналогично восстановлению сложных эфиров монокарбоновых кислот (см. разд. 9.8.3.4). Поэтому мы здесь рассмотрим лишь те случаи, когда сближение двух карооксильных групп приводит к иным результатам. „
При использовании подходящих восстановителей и в том случае, когда могут образоваться циклы соответствующего размера,
О II
/COR
(СН2)„
^COR
Na, ксилол ----------->
О"
^СОР (сн2)п
ст
2Na+
----->
-2OR-
IK
получаются лактоны [119] {схема (122)} п лактолы [120] {схема (123)}.
Наиболее важным методом восстановления является ацилоиновая реакция [121], которая служит прекрасным методом синтеза карбоциклических соединений {схема (121)} и, в частности, средних циклов, которые трудно получить иными методами. Дополнительные детали о применении этой реакции в синтезе малых циклов, например по схеме (124) [122], обсуждаются в разд. 9.8.3.4.
9.2.3.5. Реакции щавелевой кислоты и ее производных
Щавелевая кислота — белое кристаллическое соединение, обычно доступное в виде дигидрата. В том случае, если требуется безводная кислота, дигидрат необходимо прокалить при 100°C в течение нескольких часов [123].
Безводная щавелевая кислота применяется как дегидратирующий агент, особенно когда субстрат чувствителен к более сильной кислоте. В частности, она применяется для дегидратации сложного эфира |3-гидроксикислоты, синтезированного по реакции Реформатского {схема (125)}; для получения кеталя {схема (126)}, если применение более сильной кислоты, такой, как п-толуолсуль-фокислота [124], помимо образования кеталя вызывает миграцию двойной связи, а также для циклизации с одновременной дегидратацией [125] 2,5-дигидроксигександпона-3,4 {схема (127)}. Продукт последней реакции — фуранеол (48) является одним из основных компонентов аромата ананаса и земляники.
(126)
(125)
115
МеСН(ОН)СОСОСН(ОН)Ме
НО О
\ // (СО2Н)2 \
-----> Me—" — Me
чо
(127)
(48)
Оксалилхлорид— бесцветная жидкость с удушающим запахом, получается [126] при реакции щавелевой кислоты и пентахлорида фосфора. Этот реагент особенно удобен для превращения карбоновых кислот в соответствующие хлорангидриды. Для этого существует два метода. Первый состоит в прямой реакции оксалил-хлорида и свободной, например стероидной (49) [127], кислоты в бензоле. Во втором методе натриевую или калиевую соль кислоты обрабатывают оксалплхлоридом в присутствии или в отсутствие пиридина. Последний метод применен в полном синтезе ротенона [128] для превращения тубаевой кислоты (50) в ее хлорангидрид {схема (128)}.
Взаимодействие оксалилхлорида с различными нуклеофилами приводит к широкому набору продуктов, в зависимости от стабильности первичного продукта при используемых условиях реакции. Например, со спиртами реакция легко протекает при комнатной температуре и приводит к алкилхлорглпоксилату. Последний в^присутствип пиридина и обычно при нагревании распадается с образованием алкилхлорпда [129] {схема (129)}. В качестве примера этого многообещающего метода синтеза алкилгалогенпдов показано превращение гидроксифульвеиа (51) в соответствующее хлорпропзводиое [130], которое в результате взаимодействия с ди (транс-пропен-1 -ил) купратом приводит к антибактериальному препарату фульвоплюмиерину (52) {схема (130)}.
(СОС1)2 пиридин
ROH ------->- ROCOCOC1 --------» RC1 4-СО2 + СО (129)
А
116
СО2Ме
(51)
Оксалилхлорид применялся также для структурной модифика-нИ [131] Д4-3-кетостеропдов {схема (131)} с целью получения соединений, обладающих анаболико-андрогенной активностью. Эту реакцию использовали для модификации различных стероидов, и в частности прогестерона, ацетатов тестостерона, метилтестосте-рона, кортизона.
(С0С1)2
XNH2 --------> XNHCOCOCI —> XN-=C=O + НС1 + СО (132)
(53)
X = RCO или PhSO2
(131)
Реакция оксалилхлорида с амидами приводит к ацил- [132] или к сульфонил- [133] изоцианатам {схема (132)}, причем в последнем случае можно выделить промежуточный сульфонилокса-моилхлорид (53) (X = PhSO2). 1,1,3-Тризамещенные мочевины в присутствии оксалилхлорида вступают в реакцию циклизации с образованием парабановой кислоты (54) [134] {схема (133)}, Стадия циклизации сопровождается элиминированием олефина и/или алкилгалогенида, аналогично элиминированию по Гофману.
117
Y= CO2Et, OMe, MeJZ= OEt, OH, SCH2CO2Et
(134)
Me2CHN=C=NCHMe2
(COC1)2 -------->
(135)
Близкой является циклизация [135], показанная на схеме (134); она типична для ряда синтезов подобных гетероциклических колец. Вариант этого подхода с применением карбодиимида показан на схеме (135) [136].
Оксалилхлорид в мягких условиях ацилирует ароматические соединения обычным образом, особенно если кольцо активировано. В зависимости от соотношения реагентов получают карбоновые кислоты или а-дикетоны. Например, этим методом можно получить ароматические карбоновые кислоты [137] из алкилбензолов или нафталина {схема (136)}. Бис (диалкил амино) бензилы получают [138] при взаимодействии оксалилхлорида и диалкиланилинов в присутствии хлорида алюминия как катализатора {схема (137)}. Синтез этих соединений обычным путем с помощью бензоиновой реакции осуществить не удалось.
AJCI3. CS2; ------------> н+. Н2О
Me
+ (СОС1)2
+ СО + 2НС1
(136)
(137)
Оксамиды общей формулы R2NCOCONR2 можно легко получить по реакции оксалилхлорида с аминами. Ацилирование по азоту можно также использовать в синтезе гетероциклов. Например, продукт ацилирования фенилгидразона бензальдегида окс-алилхлоридом циклизуется в присутствии хлорида алюминия с образованием изатоинового производного (55), которое под действием горячей щелочи перегруппировывается в З-фенилциннолин-4-карбоповую кислоту {схема (138)} [139]. Эта последовательность реакций открывает важный синтетический путь к циннолиновой группе гетероциклов (синтез Штолле — Бекера).
118
(COClh
AlCIa
О
N=CHPh
CO2H
I N=CHPh
(138)
различных ацилирова-
на схемах
(55)
Сложные эфиры щавелевой кислоты используют в конденсациях. Первый основной тип реакции включает нпе [140] по карбанионному центру общей формулы где X— электроноакцепторная группа {примеры — см.
Ц39) — (141)}. В некоторых случаях продукты этих реакций могут термически декарбонилироваться с образованием эфиров малоновой кислоты {схема (139)} [141] или сложных эфиров р-оксокислот {схема (140)} [142]. Альтернативно, эти продукты можно гидролизовать до а-оксокислот, как в синтезе а-оксоглутаровой кислоты {схема (141)} [143].
NaOEt;
RCH2CO2Et + EtOCOCOOEt --------->
н+, н2о ,CO2Et д
RC< 1зо~~15о °с>' RCH(CO2Et)2 (139)
\СОСО2Ш 130-100 G
О
COCO2Et JI /CO2Et
A Y Y
165-175 °C " I I (140)
Fe, стеклян. порошок
COCO2Et COCO2H
I HCI, H2O I
-> CHCO2Et ---------► CH2 (141)
I Д I
CH2CO2Et CH2CO2H
NaOEt;
| + (CO2Et)2 --------
H+, H2O
CH2CO2Et
I____ + (CO2Et)2
Сходная реакция применялась в ряду стероидов для прямого В нометилирования [144] кольца А холестен-4-она-З {схема (142)}. и ?гом случае оксалильный заместитель является одновременно ?1Яе^0КиРУ,О1Цей и активирующей группой. Диэтнлоксалат приме-ся также в синтезе гетероциклов. Например, при синтезе
Ц9
этнлнндол-2-карбоксилата [145] на первой стадии о-нитротолуол конденсируют с диэтилоксалатом; последующее восстановление продукта конденсации, сопровождаемое циклизацией, приводит к индольному производному {схема (143)}.
Аминолиз трет-бутплэтилоксалата приводит к трет-бутилокса-мату (56), который при дегидратации под действием трифторуксусного ангидрида в пиридине дает трет-бутилцианоформиат (57) [146]. Последний используется в качестве альтернативы трет-бу-тилазпдоформпата для получения трет-бутплокспкарбонильных производных аминов.
трет-ВиОСОСОМНг
(56)
трет-БиОСОС^У!
(57)
9.2.3.6. Реакции малоновой кислоты и ее производных
Хотя сложные эфиры малоновой кислоты в большинстве случаев являются лучшими реагентами, свободная кислота также применяется в различных конденсациях. Малоновая кислота СН2(СО2Н)2 представляет собой растворимые в воде кристаллы, которые при нагревании выше температуры плавления разлагаются на уксусную кислоту и диоксид углерода. Сама кислота использовалась в реакции Кневенагеля. В этом случае реакция непосредственно приводит к ненасыщенным кислотам {например, по схемам (144) [147] и (145) [148]} и применялась как альтернатива синтеза Перкина. Использование о-гндроксибензальдеги-дов в этой реакции непосредственно приводит к кумарин-3-карбо-новым кислотам [149] {схема (146)}. В реакцию Кневенагеля с малоновой кислотой могут вступать а,[3-ненасыщенные альдегиды I2Q
те3 по этому методу диеновой кислоты [150], например, по
11 С1,И /147), служит альтернативой синтезу Виттига.
СХеМе _ ' СНО /СО2Н
пиридин
г о + СН2(СО2Н)2 ----------------->
I I] + пиперидин
^V'^'-'OMe
(144)
OMe
OMe
OMe
(145)
(146)
МеСН=СНСНО + СН2(СО2Н)2
пиридин, 100 °C;
-----------> МеСН=СНСН=СНСО2Н
н+, н2о
(147)
ОН
О
II НОС.
хсн2
СО2Н
(148)
В присутствии подходящего кислотного катализатора малоновая кислота может ацилировать активированные ароматические соединения; в качестве интересного примера такой реакции на схеме (148) приведен синтез флавонов [151].
Замещенные малоновые кислоты можно бронировать; пиролиз продукта реакции приводит к а-бромкислотам [152] {схема (149)}. С другой стороны, незамещенную малонатную систему обычно бронируют в виде соответствующего диэфира [153] {схема (150)}. Эти реакции существенно отличаются от бромирования других Дикарбоновых кислот, которые обычно вводятся в реакцию в виде Дихлорангидридов [154].
RCH(CO2I-;)2 + Вг2 RCBr(CO2H)2 —> RCHBrCO2H (149)
120 1OU
CH2(CO2Et)2 + Br2 BrCH(CO2Et)2 (150)
л Тетраацетат свинца окисляет дизамещепные малоновые кис-ctrW А° ацетатов 1,1-Диолов, которые при гидролизе в прпсут-
ии основания дают кетоны [155] {схема (151)}. При окислении
121
пероксидом водорода в контролируемых условиях [156] образуется малонилпероксиды {схема (152)}.
РЬ(ОАс), КОИ, МеОН
Bu,C(CO2H)2 Bu2C(OAc)2 ----------> Bu2c=o (151)
о
о
I (152)
о
о
О применении моноэфиров малоновой кислоты для получения эфиров янтарной кислоты путем окислительного декарбоксилирования по Кольбе уже упоминалось. Моноэфиры малоновой кислоты легко получают из соответствующих диэфиров гидролизом в контролируемых условиях (см. разд. 9.8.3.1).
Диэфиры малоновой кислоты и ее производных наиболее часто используются в синтезе; их алкилирование — классический подход для образования новых углерод-углеродных связей. Общие принципы синтеза представлены на схеме (153). Моноалкплирование обычно протекает с умеренным выходом, причем для этого можно применять как первичные, так и вторичные алкилгалогениды. Типичная последовательность реакций представлена на схемах (154) [157] и (155) [158]; гидролиз малоновых эфиров обсуждается в разд. 9.8.3.1.
NaOH или КОН,
NaOEt, EtOH; EtOH, Н2О;
CH2(CO2Et)2 ----—--------> R'CH(CO2Et)2 ------------> R'CH2CO2H
RHal H+, H2O, A
R\
^C(CO2Et)2
R2/
NaOH или KOH, EtOH. H2O;
H + , H2O, Д
R*R2CHCO2H
CH2(CO2Et)2
NaOEt, EtOH;
------------->
MeEtCHBr, Д
CHCH(CO2Et)
KOH, H2O;
---------->-
A, H2SO4
Меч
XCHCH2CO2H
(154)
CH3CH(CO2Et)2
NaOEt, EtOH;
C|0H2lBr, A
Me.
XC(CO2Et)
СюН2ц
KOH, H2O;
---------->-
A, H2SO4
Me.
—> XCHCO2H (15S)
C10H2/
Классические синтезы алициклических соединений, использующие диэтилмалонат и дигалогениды, показаны на схемах (156) и
122
[1591; необходимо отметить, что аналогичный синтез цпкло-(157 новых'производных был неудачным в силу легкости [160] ^Геофильного раскрытия кольца {схема (158)}.
CH(CO2Et)2
CH(CO2Et)2
NaOEt, EtOH ^Ю2С
Et0’c
CO2Et
CO2Et
(156)
Вг(СНг)зВг
CH(CO2Et)2
CO2Et
NaOEt, EtOH -----CO,Et
CH(CO2Et)2+ Br(CH2)3Br -----------> | (157)
CO2Et
+ CH(CO2Et)2 —>
CO2Et
CH(CO2Et)2 I
(CH2)2
CH(CO2Et)2
CO2Et
(158)
В общем случае реакции, аналогичные изображенной на схеме (157), могут проходить с дигалогенидами Вг(СН2),гВг при значениях п от 3 до 7.
л-Аллилпалладиевые комплексы, получаемые из олефинов по реакции хлорида палладия в дихлорметане или хлоридов палладия и натрия в уксусной кислоте, содержащей ацетат натрия, алкилируются малонат-анионом. Реакция протекает быстро даже при комнатной температуре, однако для ее завершения требуется большой избыток трифенилфосфина [161) {схема (159)}. На схеме (160) представлен несколько измененный вариант аналогичной реакции [162],
Me
JL Me pdc12,cH2CI2 или PdCI2,NaOAc,NaCI, НОАс
NaCH(CO2Et)2 + избыток Ph2P
(159)
CO2Et CO2Et
CeH13 COaEt + CH3—^CO2Et
V8IIU
123
CO.Et
+ CH2(CO2Et)2 Мп(ОАс)з, Cu(OAc)2 1 -CO2Et (160)
АсОН, 60 °C ' /
EtO3Cx ,Н V/ + CH2(CO2Et); Н/ 'CO2Et МеСН=СНСО2Ме + CH2(CO2Et)2 NaOEt CH2CO2Et CHCO2Et CH(CO2Et)2 CH2CO2Me CHMe CH(CO2Et)2 CH2CO2H HCI |
2 " 2 EtOH NaOEt H2O* CHCO2H CH2CO2H CH2CO2H HCI 1 (161)
EtOif ,, > CHMe H2O | CH2CO2H (162)
Хорошо известно алкилирование дпэтилмалоната в условиях реакции Михаэля; примеры, приведенные на схемах (161) и (162), показывают применимость этой реакции для получения производных глутаровой кислоты. Взаимодействие диэтилмалоната и а,₽-ненасыщснных кетопов приводит к производным циклогександио-на-1,3 [163] {схема (163)}. Многие соединения, содержащие элек-тронодефицптную двойную связь, например а,|3-ненасыщенный сульфоксид (58) па схеме (164) [164], могут служить акцепторами в реакции Михаэля.
Me-
Ме2С=СНСОМе + CH2(CO2Et)2
Me
Me
EtO2C
S—CH=CH2
NaOEt
EtOH
.CHCOMe
Me2C{
О'
Na+
KOH, H2O; ---------->
H+, H2O
CH2(CO2Et)2 ----------->- Me-
CH(CO2Et)2J О
О
(163)
Me
;—SCH2CH2CH(CH2Et)2
О
О
(58)
(164)
Диэтилмалонат легко вступает в реакцию Кневенагеля; типичные условия этой реакции [165] указаны на схеме (165). Конденсация с салициловым альдегидом в довольно близких условиях {схема (166)} приводит к кумарину [166]. Если присутствие сильного основания нежелательно, может оказаться полезным метод с использованием тетрахлорида титана [167] {схема (167)}.
пиперидин Н
ArCHO + CH2(CO2Et)2 + ArCH=C(CO2Et)2
РпСОгН.СвНв П2
—> АгСН=СНСО2Н <165)
124
CHO
пиперидин
+ CH2(CO2Et)2 ---- -
AcOH
OH
CO2Et
(16f)
>=O + CH2(CO2Et)2 —U' пнрнл"» ТГФ
>=C(CO2Et)2
(167)
сле-
Если необходимо избежать образования смеси продуктов, дует проводить ацилирование диэтилмалонатом в довольно специфических условиях. Так, магниевое производное (59) можно ацилировать [168] с образованием моноацильных производных, которые затем используются в синтезе кетонов и сложных эфиров |3-ке-токислот {схема (168)}. Если необходимо получить кетоны, можно применять трег-бутиловые [169] и гексагидропиранильные [170] сложные эфиры, так как такие соединения расщепляются и декарбоксилируются в относительно мягких кислотных условиях {схемы
Mg EtCOCI C)nH,SO,H2
СНИСОЛ), EtOMgCH(CO2Et), - Е.СОСЩСО^), S° A (j
(59)
ClCO2Et
CH(CO2Et)3 <-------
2-NO2C6H4COCl
COCH(CO2Et)2
N02
EtCOCH2CO2Et
COCH3
NO2
H2SO4, H2O <---------
AcOH
(168)
OSiMe3
O:
Me3SiCI
Na+ CH(CO2Et)2 ---------i
\nn, RCOCI
HC xOEt ----------
RCOCH
MejSiCI
H, + H2O
(60)
CO2Et
NaH, C6H6;
PhCH2CH(CO2CMe3)2 v ’ PhCOCI
—> RCOCH2CO2Et
MeC6H4SO3H
PhCH2C(CO2CMe3)2 ---- >
I ACUn
(169)
О
COPh
PhCOCH2CH2Ph
(170)
CO2 + Me2C=CH2
cnH23COC|
AcOH
----> СИН23СОСН2СН2РЬ
o /2
+ co2 (171)
О
CH2Ph
125
/171W С использованием трпметилсилильного произвол-(170) " (\;п^;ся получить с хорошим выходом моноацилирован-ного (60) }ДаеЗс |гхеМа (169)}. Диэтилмалонат широко приме-Z’*Xe л- с'-за -ер’оциклов.
9.2.3.7. Реакции малеиновой, фумаровой кислот и их производных
Основные реакции малеиновой кислоты и ее производных, и особенно малеинового ангидрида,— это перициклические реакции {схемы (172), (173)}. Изомерные малеиновая и фумаровая кислоты соотносятся, как показано на схеме (174); фумаровая кислота— термодинамически более устойчивый изомер. В реакциях с водой, аминами, спиртами, малеиновый ангидрид (61) проявляет свойства обычного циклического ангидрида, приводя к производным малеиновой кислоты {схема (174)}. Тем не менее попытки получить хлорангидриды, а также другие реакции в кислых условиях приводили к производным фумаровой кислоты [172] за счет изомеризации.
Реакция Дильса — Альдера
(172)
Еновая реакция
126
Реакция Дильса — Альдера {схема (172)) широко применяется для синтеза циклических 1,2-дикарбоновых кислот {схема (175)). Следует отметить, что использование циклического диена приводит к эндо-изомеру (62) бициклического продукта о небольшой примесью экзо-изомера (63).
Именно благодаря высокой стереоселективности реакция Лиль са-Альдера служит ценным инструментом химика-синтетика Пп лучение ацетоксидиенов при ацетилировании ненасыщенных'аль' дегидов, кетонов и дпонов расширяет возможности реакции Лиль' са —Альдера {173] {схемы (176), (177), (178)). Р Ц ^иль’
один из многочисленных реакции Дильса — Альдера для синтеза i
На схеме (179) приведен использования Д______ . - 'г
соединений. Семичленное кольцо колхицина было построено [174]
примеров природных
127
с использованием в качестве ключевой стадии реакции Дильса Альдера с производным малеинового ангидрида.
Реакция Дильса — Альдера — типичный пример «разрешенной» реакции циклоприсоединения [,n4s -f- Я2Д, и нет ничего удивительного, что она может протекать иначе, если ее проводить фотохимически. Несколько родственных реакций этого типа [175]
с дихлормалеиновым ангидридом и дихлормалеинимидом представлены на схеме (180).
R = H х=о
R = Me X = NH
(180)
Еловую реакцию [176] {см. схему (173)} также можно применять для получения дикарбоновых и поликарбоновых кислот [177J {схема (181)}.
128
R общем случае малеиновый ангидрид более реакционноспосо* ЛРН как диенофил, чем как енофил; именно поэтому реакции с ]3- п 1,4-диенами протекают достаточно сложно [178] {схемы (182), (183)}. СН^ /СО2Ме
СН2=СНСО2Ме +
CHCO2Et (181)
CHjCOaEt
9.2.3.8 Ацетилендикарбоновая кислота и ее производные
Реакции ацетилендикарбоновой кислоты и ее сложных эфиров весьма интересны, так как эти соединения вступают в очень большое число реакций присоединения. Многие из этих реакций протекают необычно, однако выбор реакций, рассматриваемых в этом разделе, основан на их синтетических возможностях.
Саму ацетиленднкарбоновую кислоту получают действием метанольного раствора гидроксида калия на а,р-дибромянтарную кислоту. Типичный пример поведения этой кислоты как диенофила приведен на схеме (184) [179], причем именно в этом случае из-за высокой реакционной способности диена получается бис(аддукт).
Найдено, что диметиловый эфир ацетилендикарбоновой кислоты, получаемый при взаимодействии кислоты с метанолом, может быть использован в синтезе ряда интересных соединений.
6 Зак. 396 inn
Некоторые из этих процессов включают реакции циклоприсоединения с последующим ретродиеновым распадом (ретрореакция Дильса— Альдера). Например, ^«с-3,4-дихлорциклобутен можно синтезировать последовательностью реакций, изображенной на схеме (185); триметилгемимелитат получают при взаимодействии диме-тилацетилендикарбоксилата и метил-2-пирон-6-карбокснлата {схема (186)} [181]. Общность этих реакций заключается в том, что в обоих случаях возникает бензольное кольцо, однако в первом случае оно образуется как нежелательный продукт, в то время как во втором случае ароматическое соединение является целью син-
теза.
МеО2С
МеО2С
МеО2СС==ССО2Ме --------------—>
200 °C
МеО2С
МеО2С
СО2
Интересен синтез [182] азуленовых производных из гептафульвена {схема (187)}, в частности потому, что менее реакционноспособные диенофилы не вступают в подобную реакцию.
МеО2СС=ССО2Ме
О2,
Pd
(187)
130
Помимо реакций циклоприсоединения диметиловый эфир аце-тендикарбоновой кислоты очень активен по отношению к нукле-т!1'нЛьному присоединению. Реакции этого типа, представленные в °бщем виде на схеме (188), применяются [183] для получения ° юокого набора гетероциклических соединений, включая пирро-UI1, фураны, тиофены, хинолины и тиазолиноны. Пример подобного синтеза представлен на схеме (189).
X-—X --
РО2С—С==С—CO,R
(188)
СО2Ме
PhCO СНСО2Ме
НС^ /С—СО2Ме
Me/ NH
С
СО2Ме
Ph
Me—
СО2Ме
—СО2Ме
(189)
Интересен синтез [184] 1,4,5-триметоксикарбонилазепина {схема (190)} как пример получения гетероциклических соединений с применением диметилацетилендикарбоксилата и последующими разрешенными термическими и фотохимическими перицикличе-скимп реакциями. Первоначальное циклоприсоединение становится необратимым при проведении реакции в присутствии хлорида алюминия. Это дает продукт с выходом 90% по реакции, в которой обычно трудно получить высокие выходы.
(p-OgMe о + L -7 ।
СО2Ме СО2Ме
У — С О2 Me
СО2Ме
—25°С
20 °C /
---—►. MeO2CN
.СО2Ме
‘СО2Ме
(190)
5*
131
Нуклеофильное присоединение к енаминам приводит к важной реакции расширения кольца алициклических кетонов, например при получении восьмичленного цикла по схеме (191) [185]. р
ЛИТЕРАТУРА
1. С. IF. Bird, Chem. Rev., 1962, 62, 283.
2. Е. J. Corey and L. S. Hegedus, J. Amer. Chem. Soc., 1969, 91, 1233
3. A. F. M. Iqbal, Angew. Chem. Internal. Edn., 1972, 11, 634.
4. J. Falbe and F. Korte, Chem. Ber., 1962, 95, 2680.
5. M. Schlosser, Angew. Chem. Internal. Edn., 1964, 3, 287.
6. M. Schlosser, Angew. Chem. Internal. Edn., 1964, 3, 362.
7. H. Gilman and J. W. Morton, Org. Reactions, 1954, 8, 258 [Г. Гилман. Opr. реакции, сб. 8. — Пер. с англ./Под ред. Ю. А. Арбузова. М., Издатинлит, 1956, с. 333].
8. I. Н. Wotiz and Н. Е. Merrill, J. Amer. Chem. Soc., 1958, 80, 866.
9. G. Zweifel and R. B. Steele, Tetrahedron Letters, 1966, 6021.
10. G. Cainelli, G. D. Bello, and G. Zubiani, Tetrahedron Letters, 1965, 3429.
11. S. Reiffers, H. Wynberg, and J. Strating, Tetrahedron Letters, 1971, 3001.
12. S. Reiffers, J. Strating, and H. Wynberg, Tetrahedron Letters, 1971, 2339.
13. A. J. H. blunder and B. Zwanenburg, Tetrahedron Letters, 1972, 2383.
14. С. E. Frank and W. E. Foster, J. Org. Chem., 1961, 26, 303.
15. С. E. Frank, J. R. Leebrick, L. F. Moormeier, J. A. Scheben, and O. Homberg, J. Org. Chem., 1961, 26, 307.
16. T. S. Cantrell, J. Amer. Chem. Soc., 1970, 92, 5480; Tetrahedron Letters, 1968, 5635.
17. W. Reppe, O. Schlichtung, K. Klager, and T. Toepel, Annalen, 1948, 560, 1.
18. M. Stiles, J. Amer. Chem. Soc., 1959, 81, 2598.
19. H. O. House, «Modern Synthetic Reactions», Benjamin, New York, 1972.
20. C. R. Hauser and В. E. Hudson, Org. Reactions, 1942, 1, 266 [У. P. Хаузер, Б. E. Хадсон. Opr. реакции, сб. 1. — Пер. с англ./Под ред. А. Я. Берлина. М., Издатинлит, 1948, с. 345].
21. Н. Cohen and R. Shubart, J. Org. Chem., 1973, 38, 1424.
22. K. Sisido, S. Toril and M. Kawanisi, J. Org. Chem., 1964, 29, 904.
23. M. Guyot and D. Molho, Tetrahedron Letters, 1973, 3433. in-
24. A С. Cope, H. L. Holmes, and H. O. House, Org Reactions, 1957, 9, 1°
[A. 4. Коп, X. Л. Холмс, Г. О. Хаус. Орг. реакции, сб. 9. — Пер. с англ./Н°д nr Ред-и-ф- Луценко. М„ Издатинлит, 1959, с. 125].
25’ LaTPInan< К- Е' Apt, Е. J. Martin, and L. W, Wangen, J. Org. Chem-.
132
or J Munch-Paterson, J. Org. Chem., 1957, 22, 170.
07 H 0. House, W. L. Respess, and G. M. Whitesides, J. Org. Chem., 1966, 31, 3128 H. 0. House and W. F. Fischer, ibid., 1968, 33, 949.
no p [' Eliel, R 0. Hutchins, and Sr. M. Knoeber, Org. Synth., 1970, 50, 38, oo R T Conley and R. T. Czaja, J. Org. Chem., 1962, 27, 1643.
30 D Ё Floyd and S. E. Miller, Org. Synth. Coll. Vol. 4, 1963, 141.
41 ’ I Friedman and E. Kosower, Org. Synth. Coll. Vol. 3, 1955, 510.
32 W G. Kofron and L. G. Wideman, J. Org. Chem., 1972, 37, 555.
чз ₽ Mauer in «Newer Methods of Preparative Organic Chemistry», ed. W. Fo-6 ' erst Academic, New York, 1963, vol. 2, p. 101.
34 S. O. Lawesson, M. Dahlen, and C. Frisell, Acta Chem. Scand., 1962, 16, 1191.
35 S. Trippett, Adv. Org. Chem., 1960, 1, 83; A. Maercker, Org. Reactions, 1965, 14 270 [А. Меаркер. Орг. реакции, сб. 14.— Пер. с англ./Под ред. И. Ф. Луценко. М„ Мир, 1967, с. 286].
36 Е. Truscheit and К. Eiter, Annalen, 1962, 658, 65.
37 . G. Pattenden, J. E. Way, and В. C. L. Weedon, J. Chem. Soc., 1970, 235.
38^ E. D. Bergmann, D. Ginsburg, and R. Pappo, Org. Reactions, 1959, 10, 179 [5. Д. Бергман, Д. Гинзбург, P. Паппо. Орг. реакции, сб. 10. — Пер. с англ/Под ред. И. Ф. Луценко. М., Издатинлит, 1963, с. 181].
39. A. Michael and J. Ross, J. Amer. Chem. Soc., 1930, 52, 4598; H. T. Clarke and T. F. Murray, Org. Synth. Coll. Vol. 1, 1941, 272 [Г. T. Кларк, T. Ф. Мэррей. Синтезы орг. препаратов, сб. 1. —Пер. с англ./Под ред. Б. А. Казанского. М, Издатинлит, 1949, с. 377]; J. Cason, Org. Synth. Coll. Vol. 4, 1963, 630.
40. N.’ F. Albertson and J. F. Fillman, J. Amer. Chem. Soc., 1949, 71, 2818.
41. Y.-N. Kuo, J. A. Yahner, and C. Ainsworth, J. Amer. Chem. Soc., 1971, 93, ' 6321.
42. /. L. Hermann, M. H. Berger, and R. H. Schlessinger, J. Amer. Chem. Soc., 1973, 95, 7923.
43. R. C. Roberts and T. B. Johnson, J. Amer. Chem. Soc., 1925, 47, 1399; F. Bischoff and H. Adkins, ibid., 1923, 45, 1031.
44. L. Canonica, B. Danieli, P. Manitto, and G. Russo, Gazzetta, 1970, 100, 1026.
45. D. V. Nightingale and W. S. Wagner, J. Org. Chem., 1960, 25, 32.
46. M. W. Rathke and A. Lindert, J. Amer. Chem. Soc., 1971, 93, 4605.
47. В. А. Смирнов, А. В. Маркова. — ЖПХ, 1971, 44, 1364.
48. В. L. Shaw and M. C. Whiting, J. Chem. Soc., 1954, 3217.
49. К. B. Sharpless, R. F. Lauer, O. Repic, A. Y. Teranishi, and D. R. Williams, J. Amer. Chem. Soc., 1971, 93, 3303.
50. D. J. Sam and H. E. Simmons, J. Amer. Chem. Soc., 1972, 94, 4024.
51. С. M. Starks, J. Amer. Chem. Soc., 1971, 93, 195.
52. M. 1. Fremery and E. K. Fields, J. Org. Chem., 1963, 28, 2537.
53. T. J. Wallace, H. Pobiner, and A. Schriesheim, J. Org. Chem., 1965, 30, 3768.
54. B. A. Ellis, Org. Synth. Coll. Vol. 1, 1941, 18 [5. А. Эллис. Синтезы орг. пре-
паратов, сб. 1, —Пер. с англ./Под ред. Б. А. Казанского. М., Издатинлит, 1949, с. 15].
55. Р. A. S. Smith and R. О. Kan, Org. Synth., 1964, 44, 62, 91.
56. F. Freeman, Chem. Rev., 1969, 69, 591.
57. C. F. H. Allen and H. B. Johnson, Org. Synth. Coll. Vol. 4, 1963, 804.
58. G. Paris, L. Berlinguet, and R. Gaudry, Org. Synth. Coll. Vol. 4, 1963, 496.
59. L. A. Paquette, R. H. Meisinger, and R. E. Wingard, J. Amer. Chem. Soc., 1973, 95, 2230.
60. R. B. Ward, J. Org. Chem., 1965, 30, 3009.
61. M. S. Newman and W. M. Stalick, J. Org. Chem., 1973, 38, 3386.
62. F. Ramirez, N. B. Desai, and N. Ramanathan, Tetrahedron Letters, 1963, 323.
63. F. Ramirez, N. B. Desai, and R. B. Mitra, J. Amer. Chem. Soc., 1961, 83, 492.
64. A. P. Rrapcho, E. G. E. Jahngen, and D. S. Kashdan, Tetrahedron Letters, 1974, 2721.
65- G. Stork, A. Brizzolara, H. Landesman, J. Sztnuszkovicz, and R. Terrell, J. Amer. Chem. Soc., 1963, 85, 207.
- do^ 1п^гаге(1 Spectra of Complex Molecules», Methuen, Lon-
133
66т Н с Brown, D. Н. McDaniel, and О. Hafliger, in «Determination of Organic-‘ Structures by Physical Methods», ed. E. A. Braude and F. C. Nachod, Acade mic New York, 1955, vol. 1, p. 624—625.
67 P. J Corish and IF. H. T. Davison, J. Chem. Soc., 1955, 2431.
68. J. B. Stothers, «13C. N. M. R. Spectroscopy», Academic, New York, 1972.
69. E. Breitrnaier and W. Voelter, «13C N. M. R. Spectroscopy», Verlag Chemie Weinheim, 1974.
70. E. Lippma and J. Pehk, Kem. Tcollisuus, 1967, 24, 1001.
71. I. M. Kolthoff and M. K. Chartooni, J. Amer. Chem. Soc., 1976, 98, 7465
72. 1. M. Kolthoff and M. K- Chatooni, J. Amer. Chem. Soc., 1976, 98, 5063.
73. L. L. McCoy, J. Amer. Chem. Soc., 1967, 89, 1673.
74. F. A. Mason, J. Chem. Soc., 1930, 700.
75 R V. Volkenburgh, J. R. Olechowski, and G. C. Royston, U. S Pat 3194816/1965/.
76. A. C. Duckworth, J. Org. Chem., 1962, 27, 3146; G. G. Allan and A. N. Neogi Chem. and Ind. (London), 1971, 545.
77. N. J. Doorenbos and W. T. Wu, Chem. and Ind. (London), 1965, 648.
78. J. R. Edman and H. E. Simmons, J. Org. Chem., 1968, 33, 3808.
79. E. Grigat and R. Putter, Chem. Ber., 1965, 98, 1168, 1359.
80. H. G. Viehe, R. Fuks, and M. Reinstein, Angew. Chem. Internal. Edn., 1964 3, 581.
81. A. J. Gordon and R. L. E. Ehrenkaufer, J. Org. Chem., 1971, 36, 44.
82. T. Kutsuma and S. Sugusawa, Tetrahedron, 1958, 3, 175.
83. V. J. Grenda, G. W. Lindberg, N. L. Wendler, and S. H. Pines, J. Org. Chem., 1967 32 1236
84. E. Grigat, Angew. Chem., 1970, 82, 81.
85. E. IF. Della and M. Kendall. J. C. S. Perkin 1, 1973, 2729.
86. H. Bohme, Org. Synth., 1940, 20, 70.
87. G. Drechsler and S. Smagiti, J. prakt. Chem., 1965, 28, 315.
88. A. Winston, J. P. Bederka, W. G. Isner, P. C. Juliano, and J. C. Sharp, J. Org. Chem., 1965, 30, 2784.
89. J, Sam, A. V. Lopez, and R. M. Shaifik, J. Pharm. Sci., 1968, 57, 1755.
90. R. E. Harmon, G. Wellman, and S. K. Gupta, Carbohydrate Res., 1969, 11, 574.
91. R. Mayer, Angew. Chem., 1956, 68, 169; J. P. Schaefer and. J. J. Bloomfield, Org. Reactions, 1967, 15, I.
92. S. Chakravarti, Proc. Inst. Chem. (India), 1961, 33, 261.
93. A. T. Nielsen and W. R. Carpenter, Org. Synth., 1965, 45, 23; W. von E. Doering and А. A. Sayigh, J. Org. Chem., 1961, 26, 1365.
94. M. Fieser and L. F. Fieser, «Reagents for Organic Synthesis», Wiley, New York, 1969, vol. 2, p. 158 [M. Физер, Л. Физер. Реагенты для органического синтеза, т. IV. — Пер. с англ./Под ред. И. Л. Кнунянца, Р. Г. Костянов-ского. М., Мир, 1971, с. 237].
95. W. S. Johnson, В. Bannister, and R. Рарро, J. Amer. Chem. Soc., 1956, 78, 6331.
96. R. L. Augustine, Z. S. Zelawski, and D. H. Malarek, J. Org. Chem., 1967, 32, 2257
97. N. j. Leonard and С. IF. Schimelpfening, J. Org. Chem., 1958, 23, 1708.
98. W. Mock and J. H. Richards, J. Org. Chem., 1962, 27, 4050.
99. G. Jones and R. K. Jones, J. C. S. Perkin I, 1973, 26.
100. P. C. Guha and S. K. Ranganathan, Chem. Ber., 1936, 69, 1199.
101. IF. A. Mosher and R. W. Soeder, J. Org. Chem., 1971, 36, 1561.
102. G. A. Russell, E. 7’. Sabourin, and G. Hamprecht, J. Org. Chem., 1969, 34,
2339
103. J. Wolinsky and AL Setiyek, J. Org. Chem., 1968, 33, 3950,
104. G. Maier and F. Seidler, Chem. Ber., 1966, 99, 1236.
105. IF. S. Johnson and IF. P. Schneider, Org Synth. Coll. Vol. 4, 1963, 132.
106. H. O. House and H. C. Milller, J. Org. Chem., 1962, 27, 4436.
107. A. С. Cope, H. L. Dryden, Jr., and C F. Howell, Org. Synth. Coll. Vol. 4, 1963, 816.
134
ins К. Schank and N. Moell, Chem. Ber., 1969, 102, 71.
109 В. C. L. Weedon, Quart. Rev., 1952, 6, 380; В. C. L. Weedon, Adv. Org. Chem., 1960, 1, 1; Г. E. Сладковская, С. А. Войткевич.— Усп. химии, 1970, 29, 161; Д. Eberson, in «The Chemistry of Carboxylic Acids and Esters», ed. S. Patai, Wiley, New York. 1969, p. 53; S. Wawzonek, Synthesis, 1971, 285.
IЮ H. H- Westberg and H. J. Daubcn, Tetrahedron Letters, 1968, 5123.
Ill C. A. Grob, M. Ohta, E. Rank, and A. Weiss, Helv. Chim. Acta, 1958, 41, 1191. ip/ E. /. Corey and J. Casanova, J. Amer. Chem. Soc., 1963, 85, 165.
113 . К C. van Tamelen and S. P. Pappas, J. Amer. Chem. Soc., 1963, 85, 3297.
114 K. Weisner, P.-T. Ho, R. C. Ian, S. F. Lee, S. Oida, and A. Phillipp, Canad.
' J. Chem., 1973, 51, 1448; K. Wiesner, P.-T. Ho., and H. Ohlani, ibid., 1974, 52, 640.
115 H. H. Westberg and H. J. Dauben, Jr., Tetrahedron Letters, 1968, 5123; ’ E.' N. Cain, R. Vukov, and S. Masamune, Chem. Comm., 1969, 98.
116 . C. G. Overberger and P. Kabasakalian, J. Amer. Chem. Soc., 1957, 79, 3182.
117 A. F. Vellturo and G. W. Griffin, J. Org. Chem, 1966, 31, 2241.
Ц8. L. Eberson and B. Sandberg, Acta Chem. Scand., 1966, 20, 739.
ПЭ' L. A. Paquette and N. A. Nelson, J. Org. Chem., 1962, 27, 2272.
120^ G. Kraiss, M. Povarny, P. Scheiber, and K. Nador, Tetrahedron Letters, 1973, 2359.
121. К- T. Finley, Chem. Rev., 1964, 64, 573; A. C. Cope and E. C. Herrick, J. Amer. Chem. Soc., 1950, 72, 983; N. L. Allinger, Org. Synth. Coll. Vol. 4, 1963, 840.
122. J. J. Bloomfield, Tetrahedron Letters, 1968, 587, 591; U. Schrdpler and R. Ruhl-mann, Chem. Ber., 1964, 97, 1383.
123. E. Bowden, Org. Synth. Coll. Vol. 1, 1941, 424; H. T. Clarke and A. W. Davies, ibid., p. 421.
124 J. J. Brown, R. H. Lenhard, and S. Bernstein, J. Amer. Chem. Soc., 1964, 86, 2183; N. H. Anderson and H.-S. Uh, Synth. Comm., 1973, 3, 125.
125. L. Re, B. Maurer, and G. Ohloff, Helv. Chim. Acta, 1973, 56, 1882.
126. M. S. Kharasch and H. C. Brown, J. Amer. Chem. Soc., 1942, 64, 329.
127. C. R. Engel and G. Just, Canad. J. Chem., 1955, 33, 1515.
128. M. Miyano, J. Amer. Chem. Soc., 1965, 87, 3958.
129. S. J. Rhoads and R. E. Michel, J. Amer. Chem. Soc., 1963, 85, 585.
130. G. Biichi and 'J. A. Carlson, J. Amer. Chem. Soc., 1968, 90, 5336.
131. R. Deghenghi and R. Gaudry, Canad. J. Chem., 1962, 40, 818.
132. A. J. Speziale and L. R. Smith, J. Org. Chem., 1962, 27, 3742.
133. J. E. Franz and C. Osuch, J. Org. Chem., 1964, 29, 2592.
134. P. J. Stoffel, J. Org. Chem., 1964, 29, 2794.
135. R. Jaurin and W. Arnold, Helv. Chim. Acta, 1973, 56, 2569.
136. D. A. Tomalia, T. J. Giacobbe and W. A. Sprenger, J. Org. Chetn,. 1971, 36, 2142.
137. P. E. Sokol, Org. Synth., 1964, 44, 69.
138. C. Tiizun, M. Ogliaruso, and E. I. Becker, Org. Synth., 1961, 41, 1.
139. H. E. Baumgarten and J. L. Furnas, J. Org. Chem,. 1961, 26, 1536.
140. C. R. Hauser, F. W. Swamer, and J. T. Adams, Org. Reactions, 1954, 8, 84, 117 [4. P. Хаузер, Ф. В. Свемер, Дж. Т. Адамс. Орг. реакции, сб. 8. — Пер. с англ./Под ред. Ю. А. Арбузова. М., Издатинлит, 1956, с. 90].
141. R. F. В. Сох and S. М. McElvain, Org. Synth. Coll. Vol. 2, 1943, 272, 279; P- A. Levene and G. M. Meyer, ibid., p. 288; D. E. Floyd and S. E. Miller, Org. Synth. Coll. Vol. 4, 1963, 141.
142. H. R. Snyder, L. A. Brooks, and S. H. Shapiro, Org. Synth. Coll. Vol. 2, 1943, 531 [А. Снайдер, Л. Брукс, C. IHanupo. Синтезы орг. препаратов, сб. 2.— Пер. с англ./Под ред. Б. А. Казанского. М„ Издатинлит, 1949. с. 410]. v М' Pottorf f and L. L. Moore, Org. Synth., 1964, 44, 67.
inc iw Mazur and F. Sondheimer, J. Amer. Chem. Soc., 1958, 80, 5220.
lie г E' Poland and F. J. Baude, Org. Synth.. 1963, 43, 40.
Л7 , u' CarP<no, J. Amer. Chem. Soc., 1960. 82, 2725.
' co, °0’ M- S- Fish, G. N. Walker, and J. Blake, Org. Synth. Coll. Vol. 4, 1963,
135
148 S Raiapolalan and P. V. A. Raman, Org. Synth. Coll. Vol. 3, 1955, 425
149 R A. Hann, J. C. S. Perkin I, 1974, 1379.
150 C F H. Allen and J. van Allan, Org. Synth. Coll. Vol. 3, 1955, 783.
151 L L Woods and J. Sapp, J. Org. Chem., 1964, 29, 3445.
152 C S. Marvel and V. du Vigneaud, Org. Synth. Coll. Vol. 2, 1943, 93 [К. Марвел, В. дю Виньо. Синтезы орг. препаратов, сб. 2. — Пер. с англ./Под ред Б. А. Казанского М„ Издатинлит, 1949, с. 107].
153. С. S. Palmer and Р. W. McWherter, Org. Synth. Coll. Vol. 1, 1941, 245 [V. С. Пальмер, P. У. Мак-Уэртер Синтезы орг. препаратов, сб. I. — Пер. с англ./Под ред. Б. А. Казанского. М., Издатинлит, 1949, с. 541].
154. Р. С. Guha and D. К- Sankaran, Org. Synth. Coll. Vol. 3, 1955, 623.
155. J. J. Tufariello and W. J. Kissel, Tetrahedron Letters, 1966, 6145.
156. A. H. Alberts, H. Wynberg, and J. Strating, Synth. Comm., 1973, 3, 297.
157. C. S. Marvel, Org. Synth. Coll. Vol. 3, 1955, 495; E. B. Vliet, C. S. Marvel and С. M. Hsueh, Org. Synth. Coll. Vol. 2, 1943, 416.
158. C. F. Allen and M. J. Kalnt, Org. Synth. Coll. Vol. 4 1963, 618.
159. W. H. Perkin, Jr., J. Chem. Soc., 1891, 59, 798; R. P. Mariella and R. Raube, Org. Synth. Coll. Vol. 4, 1963, 288.
160 R. W. Kierstead, R. P. Linstead, and В. C. L. Weedon, J. Chem Soc, 1952, 3610, 3616.
161. В. M. Trost and T. J. Fullerton, J. Amer. Chem. Soc., 1973, 95, 292.
162. G. /. Nikishin, M. G. Vinogradov, and T. M. Fedorova, J. C. S. Chem. Comm., 1973, 693.
163. R. L. Shriner and H. R. Todd, Org. Synth. Coll. Vol. 2, 1943, 200 [P. Шрайнер, X. Тодд. Синтезы орг. препаратов, сб. 2 — Пер. с англ./Под ред. Б.А. Казанского. М„ Издатинлит, 1949, с. 220].
164. G. Tsuchihashi, S. Mitamura, S. Inoue, and К. Ogura, Tetrahedron Letters, 1973, 323.
165. C. F. H. Allen and F. W. Spangler, Org. Synth. Coll. Vol. 3, 1955, 377.
166. E. C. Horning, M. G. Horning, and D. A. Dimmig, Org. Synth. Coll. Vol. 3, 1955 165
167. W. Lehnert, Tetrahedron, 1973, 29, 635.
168. H. Lund and A. Voigt, Org. Synth. Coll. Vol. 2, 1943, 594 [X. Лунд, А. Фойгт. Синтезы орг. препаратов, сб. 2. — Пер. с англ./Под ред. Б. А. Казанского. М., Издатинлит, 1949, с. 472]; G. A. Reynolds and С. R. Hauser, Org. Synth. Coll. Vol. 4, 1963, 708.
169. G. S. Fonken and W. S. Johnson, J. Amer. Chem. Soc., 1952, 74, 831.
170. R. E. Bowman and W. D. Fordham, J. Chem. Soc., 1952, 3945.
171. U. Schmidt and M. Schwochau, Tetrahedron Letters, 1967, 4491.
172. P. G. Campbell, G Sumrell, and С. H. Schramm, J. Org. Chem., 1961, 26, 697.
173. С. M. Cimarusti and J. Wolinsky, J. Amer. Chem. Soc., 1968, 90, 113.
174. J. Schreiber, W. Leimgruber, M. Pesaro, P. Schudel, T. Threfall, and A. Es-chenmoser, Helv. Chim. Acta, 1961, 44, 540.
175. H.-D. Scharf and F. Korte, Chem, Ber., 1966, 99, 1299.
176. H. M. R. Hoffmann, Angew. Chem. Internal. Edn., 1969, 8, 556.
177. K. Morita and T. Kobayashi, Bull. Chem. Soc. Japan, 1969, 42, 2732.
178. K. Alder and R. Schmitz-Josten, Annalen, 1955, 595, 1; R. К Hill ana H. J. Barger, Jr., J. Org. Chem., 1965, 30, 2558.
179. J. A Berson, J. Amer. Chem. Soc., 1953, 75, 1240.
180. Л4. Avratn, I. Dinulescu, M. Elian, M. Farcasiu, E. Marcice, G. Mateescu, ana C. D. Nenitzescu, Chem. Ber., 1964, 97, 372.
181. £. Wenkert, D. B. R. Johnston, and K. G. Dave, J. Org. Chem., 1964, 29, 2534.
182. W. von E. Doering and D. W. Wiley, Tetrahedron, 1960, 11, 183.
183. J. B. Hendricks on and R. Rees, J. Amer. Chem. Soc., 1961,83, 1250; J. B. Hendrickson, R. Rees, and J. F. Templeton, J. Amer. Chem. Soc., 1964, 86, Ю7.
184. R. C. Bansal, A. W. McCulloch and A G Mclnnes, Canad. J. Chem., 1969, 4 , 2391.
185. К. C. Brannock, R. D. Burpitt, V. W. Goodlett, and J. G. Thweatt, J. Org-Chem., 1963, 28, 1464; G. A. Berchtold and G. F. Uhlig, ibid., 1963, 28, 1459
9.3. галогенкарбоновые кислоты
А. Кокс {University of Warwick)
9.3.1. ПРЕПАРАТИВНЫЕ МЕТОДЫ ПОЛУЧЕНИЯ галогенкарбоновых кислот
9.3.1.1. Карбоксилирование полигалогенсодержащих соединений
Некоторые галогенкарбоновые кислоты можно получить по реакции соответствующих металлорганическпх производных галогенсодержащего соединения с диоксидом углерода. Для этой цели чаще всего применяют реактивы Гриньяра (экспериментальные методы и их ограничения наиболее полно представлены в монографии [1]). Хотя в том случае, если группа R {схема (1)} содержит атом галогена, возможны побочные реакции, применение низких температур позволяет эффективно их подавлять. В общем случае трифторметильная группа в R не мешает образованию реактива Гриньяра или его карбоксилированию. Это подтверждают примеры в ароматическом и алифатическом ряду. Например [2], эфирный раствор 3,3,3-трифтор-1-хлорпропана обычным образом реагирует с металлическим магнием и дает соответствующий реактив Гриньяра CF3(CH2)2M.gCl, который с хорошим выходом карбоксилн-руется сухим льдом до 4,4,4-трифторбутановой кислоты.
Эта реакция хорошо проходит и в ароматическом ряду: о-, и-бром- и о-иодтрифторметилбензолы можно превратить [3] в соответствующие трифторметилмагнийгалогениды, которые после реакции с диоксидом углерода дают замещенные бензойные кислоты. о-Хлораналоги не реагируют с магнием, однако эту общую для орто-замещенных соединений трудность часто можно обойти, проводя синтез Гриньяра в присутствии этилмагнийбромида.
Известна также карбонизация литиевых производных, где особенно интересны реакции, включающие винилкарбанионы. Например, транс-дихлорэтилен после реакции с бутиллитием в смеси тетрагидрофурана, эфира и гексана можно успешно карбоксилиро-вать диоксидом углерода [4]; при —113 °C реакция проходит с выходом 99% {схема (2)}. Винилхлорид и трихлорэтилен реагируют в аналогичных условиях, однако выходы ниже. Стабильность промежуточных литиевых соединений убывает в порядке, указанном на схеме (3).
со2 НХ'
RMgX -----> RCO2MgX -----> RCO2H + MgXX
H-C4HgLi --------i
H-C4H 1Q
CO2;
----->
H +
Ck ,CO2H
XC=C^ XC1
(1)
(2)
(3)
137
Аналогичный метод можно применить для олефинов, несущИх галогенированные ароматические заместители, однако в этой серии в некоторых случаях возможны перегруппировки литиевого соединения, что значительно снижает выход целевой галогенкарбоновой кислоты. Например, 2-бром-д{ДЩ-1-фенил-1-/г-хлорфенилэтилен после обработки бутиллитием при низкой температуре с последующим присоединением СО2 дает [5] п-хлордифенилацетилен (24%) и ^wc-3-л-хлорфенилкоричную кислоту {схема (4)}.
(4)
9.3.1.2. Галогенирование карбоновых кислот и их производных
Это, несомненно, наиболее важный метод получения а-галоген-карбоновых кислот, для реализации которого существует несколько методических приемов.
Прямое галогенирование карбоновых кислот можно реализовать либо как процесс, катализируемый кислотой (обычно — реакция Гелля — Фольгарда — Зелинского [6]), либо альтернативно, как свободнорадикальный процесс. Первый подход приводит исключительно к а-галогенированным продуктам; второй — менее селективен. Для введения атома галогена в жирную кислоту можно применять различные реагенты, однако в классическом методе по Геллю используется красный фосфор и бром или хлор, что обычно приводит к а-галогенированию. Существует несколько модифицированных методик, включая использование брома в присутствии каталитических количеств трихлорида или трпбромнда фосфора. Типичные примеры показаны на схемах (5) [7] и (6) [8]. Получение метилового эфира 2-бром-2-метилдодекановой кислоты {схема (7)} включает применение брома, трибромида фосфора [9] и последующую этерификацию метанолом промежуточного бромангидрпда cc-бромкислоты. Этот синтез можно также осуществить бромированием метил-2-метилдодеканоата N-бром-сукцинимндом. Другие модификации классических условий реакции включают бромирование эфиров жирных кислот с добавлением небольших количеств соответствующих хлорангидрпД°в кислот, или даже хлорангидридов других кислот, таких, как ацетил-хлорнда. Например, прибавление брома к смеси метилбутапоата 138
п бута нои лх лор и да, содержащей несколько кристаллов иода, и последующая реакция с метанолом дает [10] с прекрасным выходом метил-2-бромбутаноат {схема (8)}. Более поздние модификации заключаются в бромировании и последующем декарбоксилировании малоновых кислот {схема (9)}.
ВГ2. Р
Ме2СНСО2Н------> Ме2СВгСОВг (5)
Вг:, PCi3
PhCH2CO2H --------► PhCHBrCO2H (6)
Br Br2, PBr3 MeOH |
Me(CH2)10CHCO2H -------> ГМе(СН2)10СВгСОВгТ -----> Me(CH2)10CCO2Me
Me L Me J Me (7)
MeCH2CH2CO2Me Br2 ,2
+ MeCH2CHCO2Me (8)
MeCH2CH2COCl I
Br
Br,, P A
RCH(CO2H)2 ------> RC(CO2H)2 ----> RCHCO2H (9)
I I
Br Br
Все вышеуказанные методы требуют кислых и относительно жестких условий, и поэтому обычно не могут быть применены для получения иодпроизводных. Однако галогенирование енолят-анио-нов сложных эфиров дает метод получения сложных эфиров 2-галогенкислот в исключительно мягких условиях, что хорошо подходит для синтеза сложных эфиров а-иодкислот. Принятая методика состоит в обработке [11] сложного эфира N-изопропилцикло-гексиламидом лития для получения литиевого енолята сложного эфира с последующим добавлением иода {схема (10)}. Эта реакция применима и к сложным эфирам ненасыщенных кислот, например, этим методом можно получить 2-иодундецен- 10-овую кислоту.
изо-PrNLi «зо-PrNH
(10)
тгф
-80 °C
LiCR'CO2R2 +
HCR'—CO2R2 +
ICR^CO2R2
Применение для галогенирования N-хлор- или N-бромсукцин-имида дает другой важный способ получения галогенокислот. Найдено [12], что в присутствии таких кислых катализаторов, как бромид водорода, N-бромсукцинимид бромирует хлорангидриды
139
кислот с высоким выходом и за относительно короткое время. Например, в случае фенилацетилхлорида реакция протекает количественно {схема (11)}; продукт реакции удобно превращать в кислоту или сложный эфир обработкой водой или спиртом. Ускорение реакции или прибавлении минеральной кислоты указывает на то, что она протекает по ионному, а не по свободнорадикальному механизму {схемы (11), (12)}.
Вг
NBr I ROH
PhCH2COCl ---------------> PhCHCOCl -----> PhCHBrCO2R (Ц)
o
|| нвг /ОН
RCH2CC1 ----> RCH=C
Br
।
R-CH—+ СГ 421
(12)
Альтернативная методика [13] а-галогенирования ацилгалоге-нидов состоит в применении N-бром-, N-хлорсукцинимида или молекулярного иода и тионилхлорида в качестве растворителя, применение которого обязательно для успешного иодирования. Тио-нилхлорид немедленно реагирует с отщепляющимся иодидом водорода с образованием иода и серы {схема (13)}.
RCH2COC1
RCHICOC1
(13)
X = Cl или Вг
Прямое хлорирование [14] можно также проводить с использованием хлорида меди(II) в инертном растворителе, таком, как сульфолан. Карбоновые кислоты, их ангидриды и хлорангидриды хлорируются этим методом с образованием только а-монохлоркис-лот, причем количество cc-водородов в субстрате не имеет значения. Для получения а-хлоркислот применяют также фотохимическое хлорирование. В частности, хлорангидриды высших и средних жирных кислот при облучении в кипящем тионилхлориде видимым или ультрафиолетовым светом дают продукты, замещенные преимущественно в а-положение [15]. Предполагают, что реакция включает фотолиз тионилхлорида с образованием хлора, который галогенирует енольную форму хлорангидрида кислоты {схема (14)}. Фотохимическое хлорирование можно использовать также для получения монохлорциклогексанкарбоновых кислот [16] в 140
виде сложной смеси, которую можно разделить после ее этерификации.
О О он
II SOC12 II I С12
Ме(СН2)9СН2СОН - hv> Ме(СН2)9СН2СС1^=?Ме(СН2)9СН=СС1 --->
ОН О
—> Ме(СН2)9СНС—С1 —> Ме(СН2)9СНСС1 (14)
С1 С1 С1
Существует несколько различных методов получения галоген-производных карбоновых кислот прямым галогенированием; некоторые из них рассмотрены ниже. Диэтилмонофторсукцинат получают [17] из диэтилсукцината методом {схема (15)}, где стадия галогенирования включает образование енолят-аниона. Другой метод, в котором применяют анион, состоит в последовательном алкилировании и галогенировании подходящего ацетоуксусного эфира; для генерации аниона используют гидрид натрия. Последующее деацилирование легко осуществляют действием суспензии гидроксида бария в спирте {схема (16)}. Этот метод обладает достаточной общностью [18] и, как правило, приводит к довольно чистым продуктам.
CO2Et
CO2Et
(CO2Et)2
rper-BuOK
/COjEt EtO2CCH2CK
\COCO2Et
cio3F ------>
EtOH
^/CO2Et
—> EtO2CCH2CF
\COCO2Et
KHCO3
----—> EtO2CCH2CHFCO2Et (15)
О О ОВг
II NaH; j NaH; || I Ba(OH)2
МеССНгСОгК1 Т"*’ MeCCHCO2R‘ —~> MeCCCOaR1 —г—->
R*X | 0Г2 | ttOH
R2 R2
—► R2CHBrCO2R‘ (16)
Реакция карбанионов сложных эфиров, содержащих а-фосфо-натную группировку, с галогеном с последующей генерацией a-аниона и взаимодействием с альдегидом или кетоном приводит [19] к сложным эфирам а-галогенакриловой кислоты [схема (17)].
_ NaH; NaH;
(EtO)2PCH2CO2Et ► (EtO)2PCHBrCO2Et - у „ > Me2CHCH==CBrCO2Et
|| Br2 11 Me2CHGHO
0 О
(17)
Сложные эфиры а-гидроксикислот можно превратить в хлор-4о°сЬЗВ0Л'НЬ1е в очень мягких условиях [20] действием трифенил-Цюсфина в тетрахлориде углерода. Механизм этой реакции приведен
141
на схеме (18), здесь ROH —сложный эфир а-гидроксикислоты В некоторых случаях эта реакция служит хорошей альтернативой прямому галогенированию; сложные эфиры а-бромкислот получаются при использовании тетрабромида углерода. Эта реакция родственна применению лактонов для получения ш-галогеп-карбоновых кислот. Например, сложные эфиры 4-бромбутановой кислоты можно получить из у-бутиролактона действием бромида водорода и спирта [21]; 4-хлорбутаноилхлорид получают при обработке лактона тионилхлоридом в присутствии хлорида цинка [22] {схема (19)}. у-Бутиролактон можно бромировать также традиционным методом действием брома и красного фосфора [23] {схема (19)}.
Ph3P + CCI4 -> Ph3PCCI3 СГ cr%-Q^PPh3 + СНС13
I (18)
R—Cl + Ph3P=O
Монохлорирование некоторых алифатических карбоновых кислот можно осуществить [24] с помощью азотных катион-радика-лов, получаемых фотолизом R2NCI в 96%-ной серной кислоте. Реакция высокоселективна для монохлорирования в и—1-положение, и поэтому представляет возможное решение проблемы превращения монофункциональных соединений в бифункциональные, в которых функциональные группы значительно удалены друг от друга Стадии роста цепи представлены на схеме (20).
R1NH + R2H —> R2NH2 + • R2
. R2 + R]NHC1 —> R2C1 + R2NH
(20)
CO2H
CO2H
+ SO2C12
(PhCO2)2 -------->
CO2H
CO2H
ci—<^>—c°2H
(21)
CH3COCO2H+SO2C12 —> C1CH2COCO2H+ SO2 + HC1 (22)
Галогенирование карбоновых кислот сульфурилхлоридом, по-видимому, также включает образование свободных радикалов. Два примера синтетического использования этого метода приведены на схемах (21) [25] и (22) [26].
142
9.3.1.3. Другие методы
Ппоизводные галогенкарбоновых кислот можно успешно полу-
1 с помошью реакций борорганических соединений, получаемых чаТЬдобавлении рассчитанных количеств диборана к соответствую-npIIv олефину. Прибавление сложного эфира а,а-дигалогенкар-вой кислоты к борорганическому соединению с последующей боткой трет-бутилатом калия в трет-бутаноле приводит к °бРжным эфирам а-алкилированных а-галокарбоновых кислот to?] как, например, при реакции циклогексена и этилдибромаце-
а (схема (23)}. В довольно сходных условиях борорганическое таТдцненпе реагирует с дихлорацетонитрилом в присутствии 2,6-ди-С° т-бутилфеноксида калия с образованием С-алкилированного ^етонитрила [28], из которого катализируемым кислотой гидро-лпзом получают карбоновую кислоту {схема (24)}.
B2Hg ----->• (С6ни)3в
Br2CHco2Et;
трег-ВиОК. трет-ВиОН
C6HuCHCO2Et
Вг
(23)
C12CHCN +
Et3B
ch3ch2chcn I CI
HCI
—> CH3CH2CHCO2H
H2O I
Cl
(24)
Для получения галогенкарбоновых кислот использовали ряд реакций присоединения. Показано, что 2-монофторалкановые кислоты служат важными интермедиатами при синтезе многих биологически активных фторсодержащих соединений. В связи с этим исследованы основные пути синтеза этих соединений [29]. Среди них: (а) реакция алкенов с фторидом водорода и N-бромацетами-Дом, (б) синтезы с использованием монофтормалоната и (в) прямое фторирование перхлорил фторидом. Первый метод оказался наилучшим способом получения простых незамещенных а-фтор-кислот; на его основе были развиты три пути синтеза {схема (25)}.
R—СН=СН2
не
N-бром-ацетамид
RCHFCH2ONO2 [RCHFCH2OH] —> rchfco2h
*1U АС
КСНРСНгВг -Д2Д. RCHFCIbOAc (RCHFCHjOH) (25)
*
JHCO2Na |
* IINO3 __________I
rchfch2ocho---------------SUXT-"
143
Вицинальные фторбромиды, применяемые в этих синтезах, очень медленно вступают в реакцию нуклеофильного замещения, и потому, хотя превращения просты, они достаточно длительны.
Классический пример использования реакций присоединения — это получение [30] p-хлорпропионовой кислоты из акролеина {схема (26)}. Известны и другие пути синтеза этой кислоты, например гидролиз соляной кислотой этиленциангидрина, однако синтез из акролеина, по-видимому, наиболее удачен. Получение 2,2,3-трифтор-3-хлорпропионовой кислоты в результате присоединения синильной кислоты к хлортрифторэтилену включает применение реакции присоединения [31] {схема (27)}.
НС1 конц. HNO3
CHj—CHCHO С1СН2СН2СНО -----------► С1СН2СН2СО2Н (26)
KCN
Н2О
CHCF2CO2H
(27)
2,2-Дифторянтарная кислота также получена с использованием реакции присоединения, но несколько иного типа, чем указанные выше [32]. Синтез начинается реакцией циклоприсоединения, приводящей к полигалогенциклобутану, который после дегалогенирования и окисления превращается в производное янтарной кислоты {схема (28)}. Этот пример примечателен тем, что здесь проявляются достаточно общие тенденции, ибо многие фторалкены вступают в реакцию циклоприсоединения, в результате которой получаются фторциклобутаны, которые, в свою очередь, являются многоцелевыми интермедиатами. Например, 2,3,3-трифтор-1,1,2-трихлорциклобутан можно превратить в трифторхлорянтарную, трифторянтарную, фтормалеиновую, фторфумаровую, дифтормалеиновую и дифторфумаровую кислоты.
С1
СН2=СС12 +
CF2=CC1F
ХМ’МИ-Ц» z-ч т** ту
NaOH; CF2CO2H
------->- I (28)
h2S°4 ch2CO2H
F F
9.3.2. СВОЙСТВА ГАЛОГЕНКАРБОНОВЫХ КИСЛОТ
В табл. 9.3.1, 9.3.2 и 9.3.3 представлены характерные значения констант диссоциации (выраженные через p^a) для ряда галогенкарбоновых кислот в воде. Галогензаместитель повышает силу кислоты по сравнению с незамещенной; с увеличением расстояния между карбоксильной группой и галоген-заместителем (-заместителями) эффект резко ослабляется. На основании измерений серии жестких бициклических кислот [33] показано, что эффекты заместителей можно объяснить в терминах эффекта поля. Влияние 144
галогензаместителей на силу бензойных кислот интенсивно изучалось; отмечены, кроме того, эффекты растворителя [34]. Эффекты мета- и /гара-заместителей использовались как часть основы для классических констант замещения о Гаммета, и для определения линейного отношения свободных энергий в большом ряду органических реакций и равновесий.
Таблица 9.3.1. Кислотность галогензамещенных уксусных кислот [32а]
X Значения рКд
для CH3CO2H для СНгХСО^Н для СНХ2СО2Н для СХзСО2Н
F 4,76 2,66 1,24 (1,34)* 0,23 (0.50) *
С1 — 2,86 1,29 (1,36) * 0,65 (0,52) *
Вг — 2,86 — 0,66
I — 3,12 — —
CF3 — 3,07 — —
• См. [326].
Таблица 9.3.2. Кислотность галогензамещенных пропионовых и масляных кислот [32а]
Заместитель X Значения рДа
для пропионовых кислот для а-Х масляных кислот Р-Х Y-X
а-Х 0 X
Н 4,88 4,82
С1 2,80 4,08 2,84 4,06 4,52
Вг 2,98 4,02 2,99 — 4,58
I — 4,06 — — <64
CF3 — 4,18 —- 4,49
Таблица 9.3.3. Кислотность бензойных кислот ХСбН4СО2Н [32а]
X Значения рДа
для орто-кнслот для лсета-кислот для пара-кислот
Н 4,20
F 3,27 3,87 4,14
С1 2,94 3,83 3,99
Вг 2,85 3,81 4,00
I 2,86 3,86
CF3 — 3,79 —-
Инфракрасная спектроскопия галогенкарбоновых кислот обсуждена в монографии [35], и обнаружено, что введение а-галогена
145
сдвигает поглощение карбонильной группы в сторону более высоких частот. Например [36], а-хлорпропионовая кислота имеет vMaKc при 1730 см~‘, в то время как для (3-хлорпропионовой кислоты vMaKC лежит при 1710 см-'. Введение дополнительных атомов галогена, как показано в серии от моно- до трихлоруксусной кислоты вызывает возрастание частоты. Тем не менее сдвиги, вызываемые введением второго и третьего атомов галогена, заметно меньше чем при введении первого атома.
Спектры |3С-ЯМР а-хлоркарбоновых кислот обсуждены в общем обзоре [37], и найдено [38], что а-хлорирование приводит к значительному эффекту экранирования карбоксила. Сдвиг в сторону сильных полей связан с возрастанием числа а-атомов хлора и может быть объяснен их индуктивным эффектом. Этот эффект вызывает оттягивание электронов от карбонильного углерода и возрастание порядка связи карбонильной группы в силу уменьшения поляризации л-электронов, индуцированной карбонильным кислородом.
9.3.3. РЕАКЦИИ ГАЛОГЕНКАРБОНОВЫХ КИСЛОТ
9.3.3.1. Нуклеофильное замещение
Реакции нуклеофильного замещения — это одни из наиболее важных реакций галогенкарбоновых кислот и их производных, особенно сс-галогенокислот и сложных эфиров [примеры см. на схеме (29)]. Получение сложных эфиров цианокислот, таких, как этилцианоацетат, замещением цианид-ионом, давно известная реакция; наиболее просто 3-14С-цианоацетат получают [39] реакцией хлоруксусной кислоты с K14CN и последующей этерификацией диметилсульфатом.
ch(co2r2)2
(R2O2C)2CHCH2CO2Ri <-----------
CN
CH2CO2R‘
CN-
NO" r' = H
CH3CO2R1 —> HalCH2CO2R‘----► OsNCHzCCW--------> CH3NO2
|r2O" I r2nH2.„ r2NhcH2CO2R’ r2s~
r2sch2co2r‘ <--------- R2OCH2CO2R‘ 9)
rchco2k+ —kn°2->- RCHCO?K+ -7-* rch2no2 (30) I I л
Hal NO2
RCHCO2Et ------NaNCb---RCHCO2Et (3l)
XV ДМФА или ДМСО ।
Hal xNOj
14Q
Реакции калиевых солей а-галогенкарбоновых кислот, таких, как хлоруксусная кислота, с нитритом калия в результате термического декарбоксилирования промежуточно образующихся нитрокислот приводят к иитроалканам [40], например, к нитрометану по схеме (30). Аналогичная методика применяется для получения этил-2-нптробутаноата и других сложных эфиров а-нитрокпелот из соответствующих сложных эфиров а-галогенокислот [41] {схема (31)}.
9.3.3.2 Декарбоксилирование
Декарбоксилирование а-галогенкарбоновых кислот можно осуществлять различными путями; некоторые из них, например при реакции с нитрнт-анионом {схема (30)} или в процессе образования хлоркарбена, рассмотрены в других местах.
Другим интересным примером декарбоксилирования, однако совсем иного типа, является распад дибромида тра«с-Л4-ннтроко-ричной кислоты в присутствии ацетата натрия. Продуктами этой реакции служат г{ис-Р-бром-л<-нитростирол и этил-^«с-|3-(лг-нитро-фенил)глицидат [42]; механизм реакции представлен на схеме (32) [43].
Вг2
АсОД
< - *
+ СО2
Декарбоксилирование а-галогенокислот может сопровождать нуклеофильную атаку N-оксида пиридина. В случае 2-бромфеннл-уксусной кислоты {схема (33)} по двум конкурентным путям образуются бензальдегид (49%), диоксид углерода и фенил глиоксиловая кислота (5%). Эта реакция применима как общий метод окислительного декарбоксилирования, а поскольку а-галогенокислоты легко получают по методу Гелля — Фольгарда — Зелинского, то реакцию можно использовать в синтетических целях или для деградации.
147
PhCHO + co2 + H+ + c5h5n
(33)
L->- PhCOCO2H + H+ + CeH5N
При электролизе карбоксилат-анионов (реакция Кольбе [44]) происходит декарбоксилирование и образование радикалов, которые затем рекомбинируют. Исследования в ряду ш-галогена’лкано-вых кислот показали [45], что хлоркислоты дают продукты рекомбинации с хорошим выходом, в то время как соответствующие бром- и иодкислоты отщепляют галоген и лишь с небольшим выходом превращаются в дигалогенуглеводороды. Электролиз различных а-хлоркислот в метаноле также не приводит к образованию димера Кольбе [46]. Однако в случае 2-фторкапроновой и 2-фторгептановой кислот такие димеры {схема (34)} получают с хорошим выходом, наряду с небольшими количествами других продуктов.
МеОН
Ме(СН2)3СНСО2Н---------->• Ме(СН2)3СНСО2Ме + Ме(СН2)3СНСО3Ме +
I электролиз I ।
F F Н
/ОМе
+ Ме(СН2)3СН^ + Ме(СН2)3СНСН(СН2)3Ме (34)
'ОМе I I
Перекрестные реакции Кольбе не имеют общего значения для синтеза углеводородов с короткой цепью, однако тем не менее электролиз смесей соответствующих перфторкислот и 2Н3-уксусной кислоты в специальных условиях дает [47] трифтор-2Н3-этан и пентафтор-2Н3-пропан. Однако трифторпропан из трифторуксуснои и пропионовой кислот этим методом получить нельзя: в этом случае получается смесь продуктов [48], которая, по-видимому, является результатом сложной серии реакций, включающих атаку радикала -CF3 на олефины.
(ЗВ)
Cl3CCO3Na
Д,МеО(СН2)г ОМв
[С12С:] + NaCl + СО2
148
2,2,2-Тригалогенкарбоновые кислоты склонны к декарбоксилированию, в основном с образованием карбенов. Действительно, если раствор трихлорацетата натрия кипятить с обратным холодильником в 1,2-диметоксиэтане, содержащем акцепторы дихлор-Карбена, то получаются ожидаемые продукты присоединения. Например, при реакции с циклогексеном образуется [49] 7,7-дихлор-норкаран с выходом 65%; с циклогептатриеном получается 8,8-ДИХЛОрбпцикло [5.1.0] октадиен-2,4, выход 46% {схема (35)}. Наиболее привлекательной особенностью этого подхода является возможность генерации карбенов в нейтральных условиях в отли-чиеот применявшихся ранее способов генерации таких интермедиатов при действии сильного основания на хлороформ. Термический распад хлордифторацетата натрия приводит [50] к образованию ди-фторкарбена, который применяется для удобного превращения альдегидов в 1,1-дифторолефины с помощью модифицированной реакции Виттига. Раствор альдегида, трифенилфосфина и источника карбена нагревают; реакция, по-видимому, отвечает схеме (36). Эта реакция применима к различным альдегидам, включая алифатические, ароматические и гетероциклические. Вместе с тем попытки [51] расширить область применения этой реакции и распространить ее на кетоны имели лишь незначительный успех. Замена трифенилфосфина на трибутилфосфин или замена диглима на N-метилпирролидон позволяет получить 1,1-дифторметиленцик-логексан с выходом 1—2%, однако если одновременно использовать трибутилфосфин и N-метилпирролидон, то выход достигает 46%.
Д Ph3P RCHO
ClCF2CO2Na —> [: CF2] ----> Ph5P=CF2 -------► RCH=CF2 (36)
+ NaCl + CO2
9.3.3.3. Реакции с участием металлорганических интермедиатов
Среди многих реакций галогенкарбоновых кислот, которые протекают под действием металла, по-видимому, наиболее известной и широко применяемой является реакция Реформатского [52]. По этой реакции альдегид или кетон обрабатывают цинком и галогенидом, чаще всего сложным эфиром 2-галогенокислоты или его винилогом. Формально реакцию можно рассматривать как присоединение цинкорганического бромида к карбонильному соединению, в результате чего после гидролиза получают спирт или, если протекает дегидратация, соответствующий олефин {схема (37)}. Важность реакции Реформатского определяется тем обстоятельством, что нельзя получить реактив Гриньяра из сложного эфира галогеикарбоновой кислоты. Помимо карбонильных соединений в качестве электрофилов могут выступать нитрилы [53] {схема (38)}; превращению подвергаются как ароматические, так
149
и алифатические нитрилы. Однако в случае бромпропионатов вк ходы низки; с бромацетатом реакция не идет. вы
R'CHCCW —>
I Hal
rR'CHCOaR2
L ZnHal
Mc2C—CMCOR2
HO Ri
м (37)
Me '
Zn I
Me(CH2)4CN + BrCHCO2CHMe —> Me(CH2)4CCHCO2CHMe __>
II II I
Me Et NZnBr Et
Me Et
—> Me(CH2)4CCHCO2CHMe z381
О
В тщательно контролируемых условиях нитроны [54] могут выступать как аналоги карбонильных соединений в реакции Реформатского. Так, при обработке цинком и сложным эфиром галогенкарбоновой кислоты С-арпл-Ы-метилальдонитронов получают 3-арил-2-метил-изоксазолидинон-5 {схема (39)}. Аналогично ведет себя N-оксид 3,4-дигидрохинолииа, однако N-метилнитроны .и- и н-нитробензальдегида не реагируют. Если в нитронах' присут-
ствуют заместители, увеличивающие заряд на нитронном углеродном атоме, то происходит элиминирование гидробромида по конкурентной реакции, которая в случае N-метилнитрона п-диметил-аминобензальдегида становится основным процессом. Реакцию Реформатского с применением триметилсилильной защиты по карбоксильной группе использовали для синтеза р-гидроксикислот [55].
Аг. CH2R1
ХСН |
|| + Вг—С—R2
/N\ ।
CH3Z Х0‘ CO2Et
Zn
Ar=Ph
Zn
Ar=CeH4NMe2-«
CH3N—О
^COaEt
(39)
ClCH2(Me)2SiCl + BrCH2CO2Et ClCH2(Me)2SiCH2CO2CH3 (40)
Хлорсиланы в условиях реакции Реформатского реагируют [56] со сложными эфирами 2-бромкислот и цинком с образованием а-силилсложных эфиров R3S1CH2CO2R1. Реакция гладко протекает при использовании в качестве растворителя смеси бензол —эфир, в этих условиях хлоралкильная группа силана инертна {схема (40)}. Инертен также и метилхлорацетат. Аналогично были получены различные а-силилсложные эфиры, однако, по-видимому,
150
реакция ограничена монохлорснланами п
посредственно превратить в алкилировя ^РОМкислотЬ! можно не-катализируемои медью реакции фиванные кислоты с помоппю {схема Метод основа/™ ™ Грдкьяр/^П
низких- температурах реактивы Гриньяоа ’ ЧТ°’ ПОС1<ольку При руюг с карбоксил ат-ашюнами, он!т будут ме^енно реагР. модеиствпе с алкнлбромпдами в присутСТВЛ встУпать во взаи-ЭТо различие в реакционной способности Пя1тС°ЛеИ меди- Именно чаТь алкилированные кислоты из оеат» г 03М0Жность полу-ступны растворимые соли последних М₽т'„ „ ГриньяРа> если до-сгву (о-бромкислот; бромуксусная кисло™ РИМеним к больший-исключением.. СЛОта является очевидным
RMgBr + Вг(СНЛ,СОгМга
ТГФ «<<-Н2)„СОгМеС1 + MgBr2 (4|)
формально синтезы по Дарзану включают образование промежуточного карбенонда п состоят в конденсации альдегидов или кетонов со сложными эфирами а-галогенокислот в присутствии таких основании, как этилат или амид натрия. Образуются сложные эфиры аф-эпоксикислот (глицидные эфиры). В типичном примере эту реакцию используют для получения этилфф-пента-метиленглицидата (1) в результате реакции циклогексанона с этилхлорацетатом в присутствии трет-бутилата калия [58] {схема (42)}. Сфера применения этой реакции рассмотрена в обзоре [59]; ее механизм, по-видимому, включает присоединение [60] енолят-аниона сложного эфира галогенокислоты по карбонильной группе кетона или альдегида с последующим нуклеофильным замещением
>=О + ClCH2CO2Et
Cl CI
H1—chco2r2 —> r1—c—co2r2
(42)
R1 CO2P2
A
rper-BuOK ----------> трет-BuOH
(43)
—4—3 - co2
(44)
ClCH2C;o2Bu-mpem
/CHCOR3 или r2—c—cho
R2 R3
151
галогена {схема (43)}, а не образование промежуточного карбе Гидролиз и декарбоксилирование глицидных эфиров позволя*3 получать карбонильные соединения, содержащие на один углепоТ ный атом больше, чем исходные соединения [61] {схемы (441 (45)}.
9.3.3.4. Трифторуксусная кислота
Трифторуксусная кислота — важное соединение, нашедшее разнообразное применение в синтетической химии как в виде реагента так и в качестве растворителя. Как растворитель она особенно ценна, например, при гидрировании кетонов над платиновыми катализаторами [62], а также при гидрировании птеридинов [631 {схема (46)}. 1 J
Н2, Pt(Rh) --------->
CF3COOH
(46)
О СО2Н ---\ II I
R = H или CH2NH—{ \—CNHCH \=/ I
СН2СН2СО2Н
Трифторуксусную кислоту можно также использовать в качестве растворителя при превращении [64] оксиранов в тиираны под действием сульфидов фосфора. По-видимому, необходимо брать определенные мольные количества кислоты, однако несмотря на это, кислота скорее всего выступает просто как катализатор. Она катализирует также конденсации кетонов, например [65] с параформальдегидом, а также использована в синтезе кумаринов [66], где фенол конденсируют со сложным эфиром |3-кетокислоты {схема (47)}.
Особенно интересным примером кислотного катализа ’Р Ф Р уксусной кислотой является аналогичный биогенетическому у полный синтез стероидов [67], включающий интересную ш
152
цйю олефинов, показанную на схеме (48). Циклизация наиболее “спешна при использовании CF3COOH/CH2C12 при -78 °C протекает в течение нескольких часов; после восстановления продукта реакции алюмогидридом лития целевой полициклический углеводород (2) получают с выходом 30%. 5 Лев0
(48)
(49)
Химии другого важного реагента, трифторуксусного ангидрида, посвящен обзор [68]. Этот реагент с карбоновыми кислотами легко дает смешанные ангидриды [69] и, по-видимому, именно таким путем и действует при циклизации, показанной на схеме (49) и являющейся одной из стадий синтеза [70] бицикло [5.4.0] ундеканонов. Кроме Того, трифторуксусный ангидрид — ценный ацилирующий агент, способный ацилировать реакционноспособные ароматические соединения в отсутствие катализатора Фриделя — Крафтса. Например, и фуран, и пиррол, и тиофен при действии трифторуксусного ангидрида превращаются [71] в 2-трифтораце-тильные производные {схема (50)}. Активация кольца, по-видимому, не является необходимым условием, и в аналогичной реакции через смешанные ангидриды трифторуксусный ангидрид с хорошим выходом превращает [72] 4-фенилбутановую кислоту в а-тетралон {схема (51)}.
fA O-C0CF, (50)
\ / CH2CI2.75°С \ /
X X
Трифторуксусный ангидрид может также индуцировать перегруппировки. Например, при нагревании метилцпклопропилкеток-сима с этим реагентом [73] в подходящем растворителе происходит перегруппировка Бекмана и образуется циклопропиламин
153
{схема также (53)}.
(52)}. Под действием трифторуксусного ангидрида протекать диенон-фенольная перегруппировка [74]
может {схема
(СГ3СО)2О
>
(52)
ЛИТЕРАТУРА
1. М. S. Kharasch and О. Reintnuth, «Grignard Reactions of Nonmetallic Substances», Constable, London, 1954.
2. E. T. McBee and A Truchan, J. Amer. Chem. Soc., 1948, 70, 2910.
3. R. G. Jones, J. Amer. Chem. Soc., 1947, 69, 2346.
4. G. Robrich and K. Flory, Tetrahedron Letters, 1964, 1137.
5. D. Y. Curtin, E. W. Flynn, and R. F. Nystrom, J. Amer. Chem. Soc., 1958, 80, 4599.
6. H. J. Harwood, Chem. Rev., 1962, 62, 99; see also H. B. Watson, ibid., 1930, 7, 180.
7. C. W. Smith and D. G. Norton, Org. Synth. Coll. Vol. 4, 1963, 348.
8. L. A. Carpino and L. V. McAdams, Org. Synth., 1970, 50, 31.
9. C. F. Allen and M J. Kalm, Org. Synth. Coll. Vol. 4, 1963, 608.
10. H. Reinheckel, Chem. Ber., 1960, 93, 2222.
11. M. W. Rathke and A. Lindert, Tetrahedron Letters, 1971, 3995.
12. J. G. Gleason and D. N. Harpp, Tetrahedron Letters, 1970, 3431.
13. D. N. Harpp, L. Q. Bao, C. J. Black, J. G. Gleason, and R. A. Smith, J. Org. Chem., 1975, 40, 3420.
14. R. Louw, Chem. Comm., 1966, 544.
15. R. L. Rodin and H. Gershon, J. Org. Chem., 1973, 38, 3919.
16. J. C. Little, Y.-L. C. Tong, and J. P. Heeschen, J. Amer. Chem. Soc., 1969, 91, 7090.
17. F. H. Dean and F. L. Л1. Pattison, Canad. J. Chem., 1963, 41, 1833.
18. P. L. Stotter and K. A. Hill, Tetrahedron Letters, 1972, 4067.
19. W7. S. Wadsworth and W. D. Emmons, J. Amer. Chem. Soc., 1961, 83, 1733.
20. J. B. Lee and J. M. Downie, Tetrahedron, 1967, 23, 359.
21. J. Lavety and G. R. Proctor, Org. Synth., 1965, 45, 42.
22. О. P. Gael and R. E. Seamans, Synthesis, 1973, 5, 538.
23. С. C. Price and J. M. Judge, Org. Synth. Coll. Vol. 5, 1973, 255.
24. N. C. Deno, W. E. Billups, R. Flshbein, C. Pierson, R. Whalen, and J. C Wy ckojj, J. Amer. Chem. Soc., 1971, 93, 438.
25. G. M. Lampman and J. C. Aumiller, Org. Synth., 1971, 51, 73 106.
26. E. J. Cragoe, Jr. and С. M. Robb, Org. Synth. Coll. Vol. 5, 1973 635.
27. H. C. Brown and M. M. Rogic, J. Amer. Chem. Soc., 1969, 91, 2146.
28. H. C. Brown and H. Nambu, J. Amer. Chem. Soc., 1970, 92, 5790.
29. R. L. Buchanan, F. L. M. Pattison, and F. H. Dean, Canad. J. Chem., 19 •, ,
30. C. Moureu and R. Chaux, Org. Synth. Coll. Vol. 1, 1941, 166 [V Wc P- Шо. Синтезы орг. препаратов, co. 1.—Пер. с англ./Под ред. Б. А. Казанское . i •, Издатинлит, 1949, с. 487].
31. D С. England and L. R. Melby, Org. Synth., I960, 40, 11.
154
47.
48.
49.
Silverstein, Tetrahedron Letters, Silverstein, Tetrahedron Letters, [P. Шрайнер. Орг. реакции, сб.
M„ Г
Fuqua, W. G. Duncan, and R. M.
Fugua, W. G. Duncan, and R. M.
Shrincr, Org. Reactions, 1942, 1, 1
с англ./Под ред. А. Я- Берлина. M., Издатинлит, 1948, с. 9];
М. Diaper and A. Kuksis, Chem. Rev., 1959, 59, 89.
56.
57.
58.
59.
60.
61.
62. Г
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
? Raasch and J. E. Castle, Org. Synth., 1962 42 4.4.
32. r Brown, D. H. McDaniel, and O. Haf liger in' Xi • ,•
cd e a ята»
Хч l"m:sis JSO^,1?69C!9'' T-
» Г. Л НЬ. 0.«. %
44 ML rol/!iofl ami AL K. Clianlooni, J. Amer. Chem. Soc., 1971. 93, 3843
L Chem Soc 19°5I 9б"? Mo,e<;“le”. London. 1966.
07' J B.' Stothers, «l3C-NMR Spectroscopy», Academic, New York 1972 oo / в Slathers and P. C. Lauterbur, Canad. J. Chem, 1964 42 1563 oq F Korte and H. Barkemeyer, Chem. Ber., 1957, 90, 392.
40 F C. Whitmore and M. G. Whitmore, Org. Synth. Coll. Vol 1 1941 401 [Ф К. Уитмор, M. F. Уитмор. Синтезы орг. препаратов, сб I — Пер с аНГл./Под ред. Б. А. Казанского. М„ Издатинлит, 1949, с. 303].
41 Л'. Kornblum and R. К. Blackwood, Org. Synth. Coll. Vol. 4, 1963, 454.
49 S 1. Cristol and W. P. Norris, J. Amer. Chem. Soc., 1953, 75, 632.
43' E Grovenstein and D. E. Lee, J. Amer. Chem. Soc., 1953, 75, 2639.
44' В. C. L. Weedon, Quant. Rev. Chem. Soc., 1952, 6, 380; В. C. L. Weedon Adv. Org. Chem., 1960, 1,1.
45. K. Maruyama and K. Murakami, J. Chem. Soc. Japan, 1968, 89, 1288.
4б' P. C. Arora and R. G. Woolford, Canad. J. Chem., 1971, 49, 2681.
R. N. Renaud and D. E. Sullivan, Canad. J. Chem., 1972, 50, 3084.
R. N. Renaul and D. E. Sullivan, Canad. J. Chem., 1973, 51, 772.
W. M. Wagner, Proc. Chem. Soc., 1959, 229.
50. S. A. 1461.
51. S. A. 521. R. L. Пер. D. G.
K. L. Rinehart, Org. Synth. Coll. Vol. 4, 1963, 120.
H. Stamm and J. Hoenicke, Annalen, 1971, 749, 146.
55. A. Horeau, Tetrahedron Letters, 1971, 3227.
R. J. Fessenden and J. S. Fessenden, J. Org. Chem., 1967, 32, 3535.
T. A. Baer and R. L. Carney, Tetrahedon Letters, 1976, 4697.
R. H. Hunt, L. J. Chinn, and W. S. Johnson, Org. Synth., 1954, 34, 54.
At. S. Newman and B. J. Magerlein, Org. Reactions, 1949, 5, 413 [At. С. Ньюмен, Б. Дж. Мейджерлейн. Орг. реакции, сб. 5. — Пер. с англ. Издатинлит, 1951, с. 319].
М Ballester, Chem. Rev., 1955, 55, 283.
/, Scharp, H. Wynberg, and J. Strating, Rec. Trav. chim., 1970, 89, 18.
P. E. Peterson and C. Casey, J. Org. Chem., 1964, 29, 2325.
63. A. Bobst and Al. Viscontini, Helv. Chim. Acta, 1966, 49, 875.
7- H. Chan and J. R. Finkenbine, J. Amer. Chem. Soc., 1972, 94, 2880.
W. C. Lumma and О. H. Ma, J. Org. Chem., 1970, 35, 2391.
C L. Woods and J. Sapp, J. Org. Chem., 1962, 27, 3703.
w. S. Johnson, Accounts Chem. Res., 1968, 1, 1.
'At. Tedder, Chem. Ber., 1955, 55, 787. „
W. D. Emmons, Д. S McCallum, and A. F. Ferris, J. Amer. Chem. Soc., 19o3, 75, 6047
}-A. Marshall, N. H. Andersen, and J. U7. SMicher, J. Org. Chem., 1970, 35. obo,
S- Clementi, F. Genet and G. Marino, Chem. Comm., 1967, 498.
w \FerHer and J. M. Tedder, J. Chem. Soc., 1957, 1435.
D. Emmons, J. Amer. Chem. Soc., 1957, 79, 6322.
b- Hecker and E. Meyer Angew. Chem. Internal. Edn.. 1954. «*, ^yy.
155
1964,
1965,
1,—
52.
53.
54.
9.4. ГИДРОКСИ- и алкоксикарбоновые кислоты
С. М. Робертс (University of Salford)
9.4.1. ВВЕДЕНИЕ
В природе встречаются разнообразные гидрокси- и алкокси карбоновые кислоты [1]. Гидроксиалкановые кислоты были выделены из ланолина, пчелиного воска, коры, корней, воскообразного слоя листьев, семян, бактерий и грибов. Наиболее широко встречаются а-гидроксиалкановые кислоты, а в комбинации со сфингозинами тканей животных или фитосфингозинами растений и микроорганизмов они, очевидно, входят в состав всего живого. Кроме того, а-гидроксикислоты были обнаружены в цереброзидах, выделенных из тканей мозга.
Р-Гидроксикислоты играют важную роль в биосинтезе и катаболизме жирных кислот, а в процессе метаболизма воска насекомых или растений синтезируются из ненасыщенных кислот.
Жирные кислоты с длинной цепью с одной или более гидроксильными группами обнаружены в жирах растительного и животного происхождения. Гидроксильные функции находятся главным образом на некотором расстоянии от карбоксильной группы. Например, 85% от суммы жирных кислот касторового масла составляет 12-гидроксиоктадецен-9-овая кислота (рицинолевая кислота) (1).
Эпоксикарбоновые кислоты были найдены в маслах семян. ^ис-12,13-Эпоксиоктадецен-9-овая кислота (верноловая кислота) (2) присутствует в различных семействах растений и составляет около 70% масла семян vernonia anthelmintica.
о-Гидроксибензойная (салициловая) кислота (3) присутствует в виде метилового эфира во многих эфирных маслах и является основным компонентом масла гаултерии. Известное лекарство аспирин — это О-ацетилпроизводное салициловой кислоты. Во многих растениях присутствуют в свободном состоянии 3,4-дигидрокси-и 3,4,5-тригидроксибензойные и прочие гидроксибензойные кислоты.
Гидроксибутандиовая (яблочная) (4) и 2-гидроксипропан-1,2,3-трикарбоновая (лимонная) (5) кислоты находят во многих плодах. Кроме того, обе кислоты принимают участие в цикле трикарбоновых кислот (цикл Кребса) катаболизма ацетата. 2,3-Z)-(+)-Дигидроксибутандиовая (винная) (6) кислота присутствует в виноградном соке.
Простагландины, сложные гидроксикислоты С20 широко распространены в тканях животных. То обстоятельство, что эти соедине ния проявляют широкий спектр биологической активности, ст -мулировало многочисленные химические и биохимические иссл
3,5-Дигидрокси-3-метилпентановая (мевалоновая) (7) та — служит in vivo важным синтетическим строительнь • ФР ментом. 2,4-Дигидрокси-3,3-диметилбутановая (пантовая/ (, )
156.
лота интересна с точки зрения биологии, так как ее амид с [3-алани-ном является важным ростовым веществом, пантотеновой кислотой.
он
Ме(СН2)6СНСН2СН==СН(СН2)7СО2Н Ме(СН2)4СН—СНСН2СН=СН(СН2)7 СО2Н v
(1)
(3)
НО2ССНСНСО2Н I I но он
(6)
(2) он
НО2ССН2СНСО2Н НО2ССН2ССН2СО2Н
он io2H
(4) (5)
Me Me он
HO2CCH2CCH2CH2OHJ hoch2AchZ
ОН Me
(7) (8)
Большинство природных гидроксикислот оптически активны. Синтез этих соединений из симметричных или рацемических предшественников требует на подходящем этапе синтеза разделения энантиомеров с помощью стандартной техники. В природных соединениях нетерминальные двойные связи чаще всего имеют Z^quc)-конфигурацию.
В следующих разделах мы рассмотрим пути синтеза гидрокси-и алкоксикарбоновых кислот, уделяя основное внимание работам по синтезу природных соединений. Стандартные методы, тщательно разобранные в ранних монографиях [3], здесь будут рассмотрены лишь вкратце; будет подчеркнута современная синтетическая методология. Мы не будем подробно описывать физические свойства многих гидрокси- и алкоксикислот, поскольку их детальное описание можно найти в обзорах [3].
И наконец, рассматриваются реакции гидрокси- и алкоксикарбоновых кислот, причем наиболее тщательно обсуждаются важные, с точки зрения синтеза, реакции в сравнении с реакциями, которые сводятся к простым превращениям одной из функциональных групп и которые относятся к рассмотренным ранее реакциям монофункциональных соединений.
9.4.2. МЕТОДЫ ПОЛУЧЕНИЯ ГИДРОКСИ-И АЛКОКСИКАРБОНОВЫХ КИСЛОТ
9.4.2.1. Общие методы получения гидроксиалкаиовых кислот
Существует несколько методов синтеза простых гидроксиалкановых кислот, не зависящих от расстояния между двумя функциональными группами в углеродной цепи. Тем не менее надо отменить, что выделение у- и б-гидроксикислот в чистом виде часто
157
затруднено в силу склонности этих соединений к внутримолсксляп ной дегидратации и образованию лактонов. В некоторых случая' также трудно получить 0-гидроксикислоты вследствие их тендем ции к дегидратации до а,[3-ненасыщенных кислот. д н'
Гидроксикислоты получают при контролируемом окислении диолов, содержащих по меньшей мере одну первичную гидроксильную группу, или гидроксиальдегидов. Для этого успешно приме няли следующие окислители: хромовую кислоту, бихромат натрия хромит меди, перманганат калия, и (недавно) карбонат серебра на целите [4]. н
Этилоксоалканоаты восстанавливали борогидрндом натрия амальгамами натрия или алюминия до соответствующих этиловых эфиров гидроксиалкановых кислот. Например, (±)-кориномиколе-вую кислоту (9), найденную в дифтерийных бактериях, получают восстановлением соответствующего сложного эфира оксокислоты с последующим омылением и хроматографическим разделением двух рацематов [5] {схема (1)}. Аналогично при восстановлении по Меервейну — Пондорфу— Верлею у-кетокислот образуются у-лактоны.
ОН
NaBH4; |
CH3(CH2)I4COCHCO2Et -----—-------> СН3(СН2)14СНСНСО2Н (1)
| к о н I I
(CH,),SCH, “ (CH.)1SCHS
W
Реактивы Гриньяра реагируют со сложными эфирами кетокислот преимущественно по кетогруппе. Этот путь к сложным эфирам а-гидроксикислот интенсивно изучался, а способность хираль-ной спиртовой компоненты в сложноэфирноп функции к введению асимметричности в ос-положение была исследована и оптимизо-вана МакКензи и Прелогом. Позднее хиральные углеводные остатки, пришитые на полимер, широко применялись в качестве хираль-ной спиртовой компоненты [6] {схема (2)}.
Полимер-^—о
R3MgX он' г
R2
/С—CO2H
R3 । п ОН
(2)
Гидроксикислоты с первичной гидроксильной группой получаются из двухосновных кислот с помощью образования полуэфир пли соответствующего полуэфира полухлорашпдрида и их поел, дующего восстановления (легкость восстановления Ф)нки''1^ ной группы распространенными гидридами убывает в РЯА> - Р . гидрид > альдегид > сложный эфир > карбоно ошью Озоно-Сложные эфиры альдегидокислот получаются с д.
лиза простых эфиров циклических енолов; восстановл Р 158
рядом натрия специфично для альдегидной функции [7] {схема (3)}-
OR OR OR
А Л,
V <_АН2ОН
R=SiMe3 илиМе
Олефиновая двойная связь в алкеновых кислотах гидратируется под действием водной кислоты или щелочи. Наиболее мягкие условия гидратации состоят в гидроборировании двойной связи с последующей обработкой борорганического соединения пероксидом водорода. В результате присоединение воды протекает против правила Марковникова, эта методика использована при синтезе ряда 6-гидроксикислот с выходом к соответствующим б-лактонам [8] {схема (4)}. Альтернативная методика заключается в превращении алкеновой кислоты в эпоксикислоту с последующим восстановительным расщеплением эпоксидного кольца алюмогидридом лития [8] {схема (5)}.
R2BH
R'CH—СН(СН2)п.СО2Н ---->
br2 br2
I I H2O2
—> R’CH2CH(CH4)nCO2H+ R,CHCH2(CH2)riCO2H ----►
OH OH
—-> RICH2CH(CH2)nCO2H +RICHCH2(CH2)„CO2H (4)
о
jw-ClCeH4CO3H / \ ЫА1Н4
RCH=CH(CH2)„CO2H ------------> RCH—CH(CH2)„CO2H ------>
OH OH
—> RCH2CH(CH2)nCO2H + RCHCH2(CH2)„CO2H (5)
Галогенкарбоновые кислоты можно превратить в гидроксикис-Лоты с помощью влажного оксида серебра или кипячением со Щелочью. Этот метод наиболее подходит для получения а- и со-ги-Дроксикислот; (3-галогенкарбоновые кислоты в этих условиях дают «ф-ненасыщенные кислоты.
Обработка аминокислот азотистой кислотой приводит к гидро-ксикарбоновым кислотам. С помощью этого метода удобнее всего получать а-гидроксикислоты из природных сс-аминокислот. Аналогично, диазокарбоновые кислоты дают гидрокспкислоты при взаимодействии с водой или водной кислотой.
Прямое окисление щелочным перманганатом калия карбоновых кислот, имеющих третичные атомы водорода в ос- пли ^-положении, приводит к а- и -у-гидроксикарбоновым кислотам.
139
1-Этоксиалкиновая функция превращается в карбоксильтт группу в результате кислотного гидролиза, и поскольку различи! 1-этоксигидроксиалкины легко синтезировать, соответствуют гидроксикислоты (или лактоны) вполне доступны [9]. Аналоги™!6 реактив Гриньяра (10), полученный из этоксиацетилена, реагирует с альдегидами и кетонами с образованием продуктов, которые Легко гидролизуются до 0-гидроксикислот {схема (6)}. р
2 ?Н он
R'R2CO; | H2SO4 I
EtOQsCMgBr R‘R2CCs=COEt -------> R'R2CCH2CO2H (6)
9.4.2.2. Получение а-гидроксиалкановых кислот
Широко применявшийся метод синтеза а-гидроксикарбоновых кислот включает превращение альдегида или кетона в соответствующий циангидрин с последующим кислотным гидролизом цианогруппы. Реакция литиевого производного N.N-диизопропил-формамида с кетонами дает полезный альтернативный подход к а-гидроксикислотам {схема (7)}, который особенно ценен в тех случаях, когда образование цианогидрина затруднено [10].
ОН он
R!R2CO I NaOH I
LiCON(Pr-U3o)2 ------> RlR2CCON(Pr-U3o)2 ----> R[R2CCO2H (7)
OH OH
I ‘OH ‘OH |
R'CHCOR2 ...-» R'COCOR2 ------> RlR2CCO2H (8)
а-Кетолы в присутствии воздуха и щелочи окисляются до а-дио-йов, которые претерпевают бензиловую перегруппировку, превращаясь при этих условиях в а-гидроксикислоты [11] {схема (8)}-Взаимодействие этилоксалата с легкодоступными цинкоргани-ческими соединениями показано на схеме (9). Поскольку вторая Стадия этой реакции протекает медленнее первой, это и позволяет получать сложные эфиры несимметричных высокозамещенных а-гидроксикислот {см. схему (9)}.
Показано, что монометиловый эфир ацетиляблочнои кисло (11) служит хорошим синтоном для получения а-гидроксикисло путем анодного взаимодействия (реакция Кольбе) с карбоновы кислотами.
OEt 2 R2 + ?Н
RJZn + (CO2Et)2 —> R'ico2Et R‘CCO2Et ” R'R2CCO2Et (9)
iznR1 OZnR1
oac _ он ^он он
НО2ССН2(2НСО2Ме + RCO2H RCH2CHCO2Me RCH2CHCO2H (Ю)
(И)
160
с использованием доступной /.-яблочной кислоты этот метод применялся для установления абсолютной конфигурации некото-рь1Х оптически активных природных а-гидроксикарбоиовых кислот {схема (10)} РаГ ^логично’ конфигурация рицинолсвой кислоты при С-12 определена путем анодного сочетания оптически активной З-ацетоксн-4-этоксикарбонилбутановой кислоты (легко получаемой из лимонной кислоты) с гексановой кислотой {схема (11), искомый хиральный центр помечен звездочкой}
Окисление метнлкетонов до а-гидроксикислот’гипоиодитом натрия в изоытке щелочи позволяет провести превращение без выделения промежуточных продуктов («превращение в одной колбеч>) и служит альтернативой более старому методу, включавшему реакцию днгалогенметилкетонов с основанием Г131 {схема (12)}.и
Вейганд с сотр. показали, что а-диазокетоны быстро реагируют с сульфенилхлоридами. а-Тио-а-хлоркетоны. получаемые по этой реакции, превращаются в а-гидроксикислоты гидролизом в присутствии гидроксида натрия или последовательной обработки ацетатом натрия и гидроксидом натрия {схема (13)).
ОН
I . Ме(СН2)4СО2Н
НО2ССН2С*СН2СО2Н —► —> ею2ссн2снсн2со2н ----------------------->
j । электролиз
СО2Н ОАс
—> Me(CH2)6CHCH2CO2Et
I
ОАс
ОН";
Ас2О, пиридин;
-----------------------> электролиз с
HO2C(CH2)s>C02F.t; ОН'
Ме(СН2)5СН(СН2)|0СО2Н
ОН
I ♦ H2/Pt
— ч— Ме(СНг)5СНСН2СН=СН(СН2)7СО2Н---------
ОН
(1)
ОН
Na 01 NaOH I
R‘R2CHCOCHS ------> R’R2CHCOCHI2 ----> R’R2CHCHCO2H (12)
NaOH
R'COCI
ch2n2
R(cochn2 r2sc}
Cl
R’COCHSR2
NaOH
О Ac OH (13>
NaOAc | NaOH I
------» R’COCHSR2 ----->- R'CHCOaH
Ацилхлорид можно превратить в а-гидроксикислоту реакцией с трпэтил фосфитом п цианидом калия с последующей перегруппировкой интермедиата (12) с образованием фосфата циангидрина (14| {схема (14)).
161 G Зак. 395
а-Ацилоксикарбоновые кислоты получают с высоким втп обработкой исходной кислоты ацетатом таллия; последуют™ [15] . Гидроксильную группу можно ввести в^а-положейиГмХ дилитиевого производного кислоты кис-последующим гидролизом образующегося пероксида
таллия; последующий щелочной гидролиз приводит к соответствующей гидроксикисГоЛ I I 51 I II ЛППКГПЛkWVln rnvnnv Mnwun ,___
новой кислоты обработкой лородом С 1-------------
[16] {схема (15)}.
Моноэтиловые эфиры замещенных малоновых кислот легко реагируют с тетраацетатом свинца; получающиеся сложные эфиры после щелочной обработки превращаются в а-гпдроксикислоты {схема (16)}.
Синтез а-гидроксикислот из сложных эфиров карбоновых кислот, содержащих на один атом углерода меньше ]RCO2Et-^ ->RCH(OH)CO2H], возможен через промежуточное образование Р-кетосульфоксидов [17] {схема (17)}.
Показано, что бис (триметилсилил) ацетали кетенов могут слу
жить многоцелевыми интермедиатами в органическом синтезе. Например, окисление этих соединений ж-хлорпербензойной кислотой приводит к бис (триметилсилильным) производным а-гидроксикислот, гидролиз которых дает а-гидроксикислоты [18] {схема (18)}.
RCOC1
(EtO)3P;
-------->
KCN
- НО OEt -I I R—С—Р==О
NC OEt _
О—P(OEt)2 I II
—> RCH О
CN
НС1 ОН КОНЦ. I ---* RCHCO2H (14)
(12)
Li
LiN(Pr-i<30)2 I
R'R2CHCO2H --------------R'R2CCO2L1
OH
O2; ।
---> R>R2CCO2H
H +
(15)
CO2Et PH
1 РЬЮАСЩ 50 °C; u
RCHCO2H ------:------> RCHCO2H
(16)
MeSOCH“Na+; NaOAc
RCO2Et -----—-----* RCOCH2SOMe Ac2O
RiR2C=C(OSiMe3)2
OAc OH
__> RCHCOSMe RCHCO2H
OSiMe3 + OH
*-С1СаН4СОзН^ R1R2(tCO2SiMe3 R'R^H
(17)
(18)
а-Гидроксиарилуксусные кислоты п°лУча1^дроксид? к^лия ^и в альдегидов с бромоформом в присут [схема (19)}- ?а3'
качестве катализатора — хлорида лития | Jжн0 спнтезиро-личные p-арил-а-гидроксипропионовые кислодь зование окса-
вать из соответствующих арилальдегид Р
1R2
золонов с последующим восстановительным расщеплением гетероцикла [20] {схема (20)}.
АгСНО + СНВгз + ЗКОН —> АгСНСО2Н + 31<Вг + Н2О (19)
Zn/Hg;
НС1
он
I АгСН2СНСО2Н
(20)
9.4.2.3. Получение р-гидроксиалкановых кислот
Галогеногидрины можно превратить в p-гидроксикислоты реакцией с цианидом калия с последующим кислотным гидролизом образующегося р-гидрокеннитрила.
Дилитневые производные карбоновых кислот получают при действии на кислоту динзопроппламида лития или нафталида лития {ср. схему (15)}. Эти дианионы реагируют с альдегидом или кетоном с образованиегл солей, которые легко гидролизуются до Р-гидроксикислот [21] {схема (21)}. Кроме того, хорошо известен
R’CH2CO2H
LiN(Pr-U3o)2 ------------------> или нафталид лития
Li
R’CHCO2Li
r2r3co-. R'CHCO2H
---—> |
н+ R2R3COH
(21)
соответствующий подход к сложным эфирам p-гидроксикислот путем реакции литиевых, натриевых или магниевых енолятов сложных эфиров с кетоном. Этот подход дополнительно ценен тем, что применение хнрального спирта в сложноэфирной функции позволяет вводить асимметрический центр [22].
Несомненно, что наиболее известный и в большинстве случаев самый эффективный метод синтеза р-гидроксикислот — это реакция Реформатского [23]. Метод включает превращение сложного эфира а-бромкислоты in situ в цинковый енолят, который далее реагирует с альдегидом или кетоном. В реакцию вступают простые алкил-, арил-, а,р-олефиновые, а,р-ацетиленовые и полиненасыщен-ные альдегиды и кетоны, а также циклические кетоны {схема (22)}. Известен, внутримолекулярный вариант реакции Реформатского, ряд функциональных групп, например алкокси- и ацилоксигруппы, выдерживают условия реакции. Сложные эфиры
R1 R1 НО R1
_ I . Zn | R3R'CO; I | H +
BrCHCO2R2 ---> BrZnCHCO2R2 ——R3R<C—CHCO2R2 -----------*
П2О
HO R1
I I
—► R3R4C—CHCO2H (22)
Me OH Me
I r’r2co, Zn I I
BrCH2C~=CHCO2Et ------->- R1 R2CCH2C=CHCO2Et (23)
163
6*
у-бром-а,p-ненасыщенных кислот как винилоги применяют в реакции Реформатского {схема (23)}, которая использовалась в синтезах витамина А и других каротиноидов. Основное ограничение получения p-гидроксикислот этим методом связано с легкостью, с какой промежуточные сложные эфиры р-гидроксикислот дегидратируются или претерпевают ретроальдольный распад в условиях гидролиза сложного эфира. Поэтому наиболее подходящими являются те методики получения гидроксикислот, в которых используются трет-бутиловые или триметилсилиловые сложные эфиры {схема (22), R2 = СМе3 или SiMe3}, поскольку расщепление этих сложных эфиров проходит при очень мягкой кислотной обработке [24]. Другим вариантом является применение цинковых солей а-бромкислот [25] {схема (22), R2 = ZnBr}. Применение енолятов, полученных пз хиральных сложных эфиров галогенкарбоновых кислот, приводит к частичному асимметрическому синтезу Р-гидроксикислот. Реакция ахирального енолята и карбонильного соединения в присутствии (—)-спартеина может приводить к введению асимметрии в продукт реакции [26].
Р-Арил-р-гидроксикарбоновые кислоты и их сложные эфиры можно синтезировать, исходя из ароматических альдегидов и О-силилполуанеталей и О-силил-О-алкилацеталеи кетена соответственно [27] {схема (24)}.
ОН
Me. .OSiMes iso °с I
АгСНО + —\ ---АгСНС(Ме)гСО2К (24)
Me' 'OR Н +
Ti(OR‘)4 + 3R2COR3 + ЗН2С=С=О —*
ОН
R\i xCCH2CO2R!
(25)
Тетраалкоксиды титана индуцируют реакцию кетена с различными кетонами (алициклическими, циклическими, алифатическими
F1
Xfe,CH.\te2CB/ Н 2
.R1 Ме2СНМе2СВ^ + HC^CCO,Et >Н
р* н’°
--► Ме2СНМе2Св4^— OEt
, I ।
R-О Н
н CO2Et OR2
r2°»> Me2CHyfe2CB-CHCH2CO2Et
R1
NaOH, i H2O2
ОН r’chch2co2h
164
или ароматическими), приводящую к сложным эфирам ₽-гидрокси-К"С'н?дй ₽“wpoSLo™ был" п"1>'чены с помощью внутрмолекулярной реакция Михаэля боратного комплекса [29] {СХВ?,н,моксибораны служат важными интермедиатами в синтезе сложных тиоэфпров и других производных р-гидроксикнслот [30] {схема (27)}.
R\ /SR1 W /R1 RV 'О—
R5 R3
Rs—С—C—COSR1
HO R‘
R5R6CO;
-------Э
H2O
(27)
9.4.2.4. Получение у-, 6 и других гидроксиалкановых кислот
Общий метод получения этих кислот состоит в окислении циклических кетонов до лактонов с последующим катализируемым щелочью раскрытием лактонного цикла и осторожным подкислением. Стадию окисления (реакция Байера — Виллигера) наиболее часто проводят с использованием персульфата или пероксикислоты, например лгхлорпербензойной кислоты. Применение щелочного пероксида водорода приводит непосредственно к образованию соли гидроксикарбоновой кислоты, это наилучший реагент, если хотят свести к минимуму конкурентное эпоксидирование двойных связей {схема (28)}. Окисление несимметрично замещенных по а- и а'-положениям кетонов по Байеру — Виллигеру приводит к внедрению атома кислорода между карбонильной группой и более электроотрицательным атомом углерода [31].
•v-ClCgH^COgH
НО ; Н+____
ИЛИ №ОН,Н2О2
(28)
Циклобутаноны и циклопентаноны претерпевают расширение цикла, давая у- и б-лактоны соответственно. Соответствующие гидроксикислоты получаются при очень осторожном подкислении солей гидроксикислот, однако эти кислоты часто самопроизвольно дегидратируют и вновь превращаются в лактоны. Более удаленные гидроксикислоты более стабильны, и их можно получать со значительно меньшими трудностями из лактонов с размером цикла более семичленного.
Заслуживают внимания несколько других подходов к у- И -лактонам (и соответственно к у- и 6-гпдрокспкислотам). Напри-тон* -ПРИ электРолизе алкилалкен-2-оаты и кетоны дают у-лак-ы, замещенные у-лактоны получают при фотохимическом
165
присоединении алканолов к сложным эфирам а,Р-ненасыщенных кислот [32]. у-Лактоны получают также при окислении циклических простых эфиров бромом при pH 5, тстроксидом рутения(VIII) Или хромовым ангидридом [33]. С другой стороны, ангидриды карбоновых кислот можно частично гидрировать до лактонов с помощью трис(трифенилфосфии)рутений(II)хлорида [34].
а-Металлированные сложные эфиры, амиды, нитрилы и соли карбоновых кислот реагируют с эпоксидами, давая у-лактоны. Некоторые примеры из многих описанных реакций этого типа приведены на схемах (29) — (32) [35].
Широко применяемым методом получения лактонов является иодлактонизация алкеновых кислот или алкеноатов. у,б-Нена-сыщенные кислоты дают б-галоген-у-лактоны. Однако если образование у-лактона сопровождается появлением значительного напряжения, то образуются у-галоген-б-лактоны. В случае р,у-нена-сыщенных кислот в соответствии с условиями реакции могут образовываться у- или р-лактоны. Рекомендовано использование алкеноатов таллия(I), особенно в тех случаях, когда образующиеся иодлактоны неустойчивы [36]. Помимо того, лактоны можно получить из алкеновых кислот по методике оксимеркурирования-демер-курирования [37].
LiCH2CONMe2 +
Ме(СН2)4С^ССН
Ме(СН2)4С=ССН2
О
Li
Ph2CCN +
R2
R>CCO2Li
Li
В последние годы широко исследовались пути синтеза а-м лен-у-лактонов и а-метилен-б-лактонов, ибо такие блоки, по димому, служат основными ключевыми фрагментами н®кот Р „ природных соединений, обладающих противоопухолевой 'к ностыо. Обзоры [38] дают сводку общих методов построения лактонов; недавние достижения в этой области включают [|0. бициклических а-метилен-у-л актонов из моноциклических <
166
лов {схема (33)} и реакцию л-(2-этоксикарбонилаллил)никельбро-мида с альдегидами и кетонами [39] {схема (34)}.
Обработка литлйпроизводным полуэфира итаконовой кислоты (13), последующие реакции с кетонами и подкисление приводят к а-метилен-у-лактонам {схема (35)}. Этот метод был применен при синтезе протоличестериновой (14), нефростериновой (15) кислот и родственных а-метилен-у-бутиролактонов [40].
СН2
LiN(Pr-«3O)2;
----------->
r'COR2;
Н+;
CF3CO2H
(35)
АгСН2ОСО СО2Н
(13)
(Н) (15)
л-Bu CHCH
Н Н
/CO2R
xco2r
(36)
167
Замечательный своей простотой синтез (+)-канаденсолида (16) и (+)-изоканаденсолида (17) недавно был выполнен Ип>,. коши [41] {схема (36)}. Ноши'
Реакция енаминов кетонов с метил-2-(бромметил)акритатом открывает общий путь к а-метилен-6-лактонам [42] {схема (37)! Описан также пятистадийный метод расширения цикла у-лактонов до а-метилен-6-лактонов [43]. Длинноцепочечные со-гидроксикар-боновые кислоты можно получить с использованием тиофена дня наращивания цепи. Соответствующие 2,5-дизамещенные тиофены получают обычным способом; на последних стадиях синтеза серный мостик удаляют восстановлением {схема (38)}. Таким путем получены аналоги миколевой кислоты [44].
w-Гидроксикислоты можно синтезировать из дигалогеналканов путем образования двойного реактива Гриньяра с последующей реакцией с карбонильным соединением и диоксидом углерода.
ОН I
—> R(CH2)4CH(CH2)nCO2H
9.4.2.5. Получение гидроксиполикарбоновых кислот
Существующие методы получения этих кислот многочисленны и разнообразны; часто они аналогичны методам, применяемым. -синтеза гидроксимонокарбоновых кислот, которые были 0ПЧ. выше. Однако для синтеза наиболее важных гидроксиполикар вых кислот разработаны специальные методы. «гитюТ
Гидроксималоновую (тартроновую) кислоту^ (1°) п0- - ‘ . окислением винной кислоты (6) смесью дымящей азотной , форной кислот. Реакция, по-видимому, протекает через оор л0, ние дикетодикарбоновой кислоты, которая претерпевает ___
ную перегруппировку {схема (39)}. ДРУГОЙ .Х^и ной кислоте с нолиз ацетилендикарбоновой кислоты в муравьин пер0КсиДа последующим мягким гидрированием промежуточного р {схема (40)}.
168
но он
НО2ССНСНСО2Н [НО2ССОСОСО2Н]
н«о
-—► НО2ССНСО2Н + со2 I
он
(39)
(6)
НО2ССгзССО2Н
(18)
ООН ОН
_?? НСОД. НО2ССНСО2Н Pt/eaS-> НО2ССНСО2Н г12
(18)
(40)
Гидроксиянтарную (яблочную) кислоту (4) получают из винной кислоты (6) последовательностью стандартных процедур по схеме (41) Альтернативно, яблочную кислоту можно получить при нагревании малеиновой или фумаровой кислот с водой под давлением в присутствии металлических катализаторов первой или второй групп.
НО ОН АсО ОАс
[ I Ас2О, пиридин; | I SOCI2;
НО2ССНСНСО2Н -----CH2N2—МеО2ССНСНСО2Ме
(6)
ОАс ОН
I "ОН |
—> МеО2ССНСН2СО2Ме --------> НО2ССНСН2СО2Н (41)
(4)
Параконовые (у-лактоновые) кислоты (19), производные а-(гидроксиалкил) янтарных кислот, использовались в синтезе протоли-честериновой кислоты (14) и дигидрожасмона (20) [45]. Один из синтезов параконовой кислоты включает реакцию янтарной кислоты с альдегидом пли кетоном в присутствии основания. Следует отметить, что длительность реакции приводит к распаду сложных эфиров параконовой кислоты в результате необратимого [З-элими-нирования с образованием сложных эфиров а-алкилиденянтарных кислот {схема (42)}. В качестве другого пути может быть использована реакция Реформатского между сложным эфиром бромянтарной кислоты и альдегидом или кетоном {схема (43)}. Другие привлекательные пути синтеза параконовых кислот состоят в раскрытии эпоксидного кольца сложных эфиров ос,0-эпоксикислот под действием енолятов, полученных из диэтилмалоната {схема (44)} или в продолжительном УФ-облучении 2,6-дихлорпиридази-нов {схема (45)}.
_ но-
НО2ССН2СН2СО2Н + R'COR2 ---->
СО2Н
(42)
(19)
169
Наиболее важную гидрокситрикарбоновую кислоту 2-гидрокси-пропан-1,2,3-трикарбоновую (лимонную) кислоту (5) можно получить гидролизом цианогидрина дихлорацетона с последующим замещением атомов хлора на цианогруппы и дальнейшим гидролизом {схема (46)}. Альтернативно, триэтилцитрат получают по реакции Реформатского из этилбромацетата и этилоксалоацетата {схема (47)}.
он он он
С1СН2ССН2С1 —С1СН2ССН2С1 —> NCCH2(3CH2CN —>
CN СО2Н io2H
он
—> но2ссн2ссн2со2н (46)
СО2Н
(5)
BrCH2CO2Et Zn. | (47)
+ ——> EtO2CCH2C— CH2CO2Et
EtO2CCOCH2CO2Et H2°
9Д.2.6. Получение полигидроксиалкановых кислот
Для синтеза этих соединений часто можно нспоЛ^д^сиаЛка' тоды, аналогичные применяемым для синтеза моногидр новых кислот. -л, с не-
присоединение пероксида водорода (непосредственно^^. « пользованием пероксикислот) к алкеновой кислоте р 170
лнгидроксикарбоновой кислоте, в которой гидроксильные группы связаны с соседними углеродными атомами. Этот метод особенно пенен в случае алкениновых кислот, так как тройная связь относительно устойчива к действию пероксида или пероксикислоты 1461" Аналогичное превращение можно осуществить и другими способами": действием высших оксидов металлов, например Os(VIII), V(V), Cr(VI) или Мп(VII) на алкеновые кислоты в трет-бутаноле; эпоксидированием алкеновой кислоты и гидролизом эпоксида; обработкой разбавленным щелочным раствором перманганата калия на холоду; присоединением галогена пли гипогало-кислоты к алкеновой кислоте с последующей реакцией с водной щелочью или оксидом серебра; обработкой кислоты иодбензоатом серебра (реакция Прево) с последующим гидролизом.
Раскрытие цикла в 2-этоксикарбонилтетрагидрофуранах под действием хлорида цинка в уксусном ангидриде приводит к сложным эфирам 2,5-дигидроксиалкановой кислоты {схема (48)}.
CO2Et
ZnCIz, Ас2О ------------>
НО ОН
CO2Et
(48)
При изучении многих биосинтетических процессов, в том числе биосинтеза стероидов, широкое применение нашла меченая мевалоновая кислота. Меченную 14С по любому положению мевалоновую кислоту можно получать по схеме (49). Меченную тритием мевалоновую кислоту можно получить при использовании (Z)- и (Е)-5-бром-3-метилпентеноатов (21) и (22) соответственно. Описан также удобный метод получения оптически активной (/?)-мевалоновой кислоты (в виде соответствующего лактона) из природного терпенового спирта, (S)-линалоола (23) [47] {схема (50)}.
Me
BrCH2CO2Et + МеСОСН2СН(ОМе)2 — "> EtO2CCH2CCH2CH(OMe)2 -В-°--)2>
ОН
Me Me
I 2+
> НО2ССН2ССН2СН(ОМе)2 -----—Н2°’> НО2ССН2ССН2СНО --аВ-^4 >
катионит ।
0Н ОН
Me
—> НО2ССН2ССН2СН2ОН (49)
ОН
(7)
/Ме BrCH2, .Me
ВгСн/ХсцсО.!! . ^СНзССЬК
(21) /9О\
171
Интенсивные попытки синтеза различных представителей класса простагландинов (ПГ) были вызваны разнообразной и ярко выраженной биологической активностью этих соединений [2]. Наиболее важными группами простагландинов являются простагландины А, В, С, D, Е и F (24—29). Отметим, что число двойных связей в боковых цепях обозначают добавлением соответствующего индекса к основному названию, например: ПГ-Ai, ПГ-С2, ПГ-Е3 и т. д.
- СН=СНСНС5 нп или - сн=снснсн2снсн=сн сн2 сн3 бн 8н
в2= — (сн2)всо2н ИЛИ—сн2сн=сн(сн2)3со2н
Поскольку синтетические подходы ко всем простагланД р хорошо описаны, видно, что количество подходов к группам н0, превосходит число подходов к другим группам. Действит в ПГ-А2 и ПГ-С2 были получены с помощью небольших измен
172
подробно изученных подходах к ПГ-Е2 и ПГ-Е2 [48]. Развитие методов синтеза группы ПГ-В было заторможено, так как казалось, что биологическая активность этих соединений значительно ниже активности других групп [49]. Простагландин-В2 был получен из гидроксиальдегида (31) непрямым синтезом, включающим многочисленные изменения защитных групп при трех гидроксильных функциях [50].
Особенно интересны две стратегии синтеза соединений серий Е и F. Первая стратегия {схема (51) состоит в синтезе альдегида (30), который для введения боковой цепочки С8 реагирует с соответствующим реактивом Виттига. Дальнейшие превращения приводят к лактолу (31) (с защищенной гидроксильной группой при С-15), который вступает во вторую «бессолевую» реакцию Виттига, для введения карбоксильной функции на последней стадии
синтеза. Удаление защит с гидроксильных групп приводит к ПГ-Ега, тогда как окисление гидроксильной группы при С-9 перед удалением защит с гидроксильных групп при С-15 и С-11 дает ПГ-Е2 [51] {схема (51)}.
Zn (вн4)2 , защита
НО-группы при С-15; Bu2AIH
(31)
X-защитная группа
пг-е2 пг-е
Второй распространенный подход включает построение 4-гидро ксициклопентен-2-она (32) {схема (52)}. Сопряженное прпсоеди нение купратного реагента к циклопентенону позволяет ввести С8-боковую цепь с защищенной соответствующим образом i идро-ксилъной функцией при С-15, что приводит к кетону (33). Ориентация двух боковых цепей и защищенной гидроксильной группы кольца контролируется кинетически и термодинамически, что
173
приводит с очень высокой селективносиио к (33), и таким путем ПГ-El .может быть получен относительно просто [52].
Получено также очень много аналогов простагландинов в предположении, что модифицированные соединения могут обладать более высокой и/или более избирательной биологической активностью, чем их природные аналоги.
В общем случае алкановые кислоты, содержащие более двух спиртовых групп, можно получать гидроксилированием жирных кислот с более чем одной алкеновой группировкой. Например, гидроксилирование октадекадиен-9,12-овой кислоты приводит к 9,10,12,13-тетрагидроксиоктадекановой кислоте [схема (53)]. Однако наиболее богатыми источниками полигидроксикислот являются углеводы. Мягкое окисление сахаров галогеном в щелочной среде превращает альдегидную группу в карбоксильную, например глюкоза дает глюконовую кислоту (34) [53]. Такне кислоты (их называют альдоновымп кислотами) трудно выделить в силу их склонности к дегидратации с образованием у- или б-лактонов, особенно в кислой среде. Второй путь к таким кислотам включает образование циангидринного производного сахара с последующим гидролизом цианогруппы (метод Килиани).
СН3(СН2)4СН=СНСН2СН=СН(СН2)7СО2Н —>
но он но он
—> СНз(СН2)4СНСНСН2СНСН(СН2)7С02Н
со2н
—он но--
-—он
-—он
сн2он
(34)
оно
-—ОН
-—ОН
-—ОН СО2Н
(35)
174
Окислением соответствующим образом защищенного производного сахара можно получить уроновые кислоты, например D-глюкуроновую кислоту (35). Хотя большинство методов окисления частично защищенных молекул углеводов приводит лишь к незначительным успехам, окисление воздухом или кислородом в присутствии катализаторов ’из редких металлов [54] проходит довольно удовлетворительно Например, D-глюкуроновую кислоту можно получить окислением D-глюкозы воздухом в присутствии платины или палладия.
9.4.2.7. Получение дигидроксидикарбоновых кислот
Наиболее подробно изученным представителем этого класса соединений является 2,3-дигидроксибутандиовая (винная) кислота (6). Эта кислота может присутствовать в виде любого из двух оптически активных энантиомеров (36) или (37), рацемата или в виде оптически неактивной мезо-формы (38). Рацемат получают при окислении фумаровой, сорбиновой или пипериновой кислоты перманганатом калия. Восстановление глиоксиловой кислоты цинком в уксусной кислоте или гидролиз цианогидрина, полученного из глиоксаля, приводят к рацемической смеси, из которой опти-
чески активные кислоты можно выделить в индивидуальном виде с помощью стандартных методик {схема (54)}. Мезовинную кислоту (38) получают при нагревании малеиновой кислоты или фенола с перманганатом калия или из малеиновой кислоты
с помощью хлората натрия влажного оксида серебра с мезовинную кислоту {схема НО2С,
и тетраоксида осмия в воде. Реакция дибромянтарной кислотой также (55)}.
КМпО4
дает
СО2Н
СО2Н
ОНССО2Н
Zn
НОАс
со2н
—он , но--
НО—
НО он
KCN онссно ----
он
> NCCHCHCN '
н+
СО2Н
(36)
-—ОН СО2Н (37)
(54)
СО2Н
КМп04
СО2Н —он —он СО2Н (38) бипА*'Иу1Не члены этого класса кислот можно получать с помощью лотГыДр0КСИЛИр0вания соответствующих алкендикарбоновых кнс-ииано^пп результате образования и последующего гидролиза бис-
СО2Н
СО2Н
ХСО2Н Другие члены этого
NaCIO3
OsO4, Н2О
Ag2o (---
Н2О
ВгСНСО2Н
ВгСНСО2Н
(55)
дикапбп^ппл» диона’ или Щ^очного гидролиза днгалогеналкан-дикарооновои кислоты,
175
9.4.2.8. Получение алкоксиалкановых кислот
2-Метоксиалкановые кислоты можно получать из оксазолинов методами, представленными на схеме (56) [55]. Оптически активные 2-метоксиалкановые кислоты получают с высоким оптическим выходом из легкодоступных хиральных оксазолинов. Применяя аналогичные циклические оксазолииы, Мейерс и сотр. открыли важные подходы к 3-метоксиалкановым [56] {схема (57)} и к 5-метокси-З-замещенным алкановым кислотам [57] {схема (58)}.
О ^Ph OMe
/ "ф LiN(Pr-us4) ; RX; |
МеОСН2—I ------—-------RCHCO2H (5б)
N z/CH2OMe
OMe
RCHCf^CO^ (57)
Me
л-Ви1Л ;
МеО(СНг)2СНО;
CF3CO2H
R
>- MeO(CH2)2CHCH2CO2H (58)
а-Алкокси-(или арилокси-) кислоты можно также получать взаимодействием алкоксида (или арилоксида) натрия с сс-галоген-карбоновыми кислотами восстановлением 1,3-диоксолан-4-онов {схема (59)} [58] или реакцией спиртов, содержащих а-трихлорме-тильную группировку, с основаниями в присутствии соответствующего спирта [59]. а-Алкокси-а-арилкарбоновые кислоты получают реакцией арилальдегидов и галоформов в спирте [60]. Превращение сс,а-дигалогенкарбоновых кислот в сс,сс-диалкоксикислоты лучше всего проходит в две стадии: взаимодействие галогенкарбоновой кислоты с морфолином, и далее — со спиртом и кислотой {схема (60)} [61].
rper-BuMgCl
----------->
СО2Н
°^СНМе2
(59)
176
С12СНСО2Н
—-> (RO)2CHCO2R
ROH ---->-
HC1
(60)
R Алкоксикислоты получают при 1,4-присоединении спирта или алкоксида к сложному эфиру а,₽-ненасыщенной кислоты или по пеакпип Реформатского сложного эфира а-галогенкарбоновой кислоты и ацеталя. Эти традиционные методы недавно дополнены реакцией О-алкил-О-триалкилсилилацеталей кетена с ацеталями или ортоформиатами, катализируемой тетрахлоридом титана и приводящей к сложным эфирам р-алкокси- и р,р-диалкоксикарбо-новых кислот соответственно [62] {схема (61)}. Реакция алкоксида натрия с р-лактонами дает р-алкоксикислоты [63].
R\ /ОМе R3\ Tici4, ch2ci2; R° R
\=/ + )c(OR5)2 ---——> R3-------------(61)
R2' 'OSiMe3 R4 OMe
Обработка сложного эфира гидроксикарбоновой кислоты ал-килиодидом в присутствии оксида серебра приводит к соответствующему сложному эфиру алкоксикислоты. Общий метод получения а,(о-диэтоксидикарбоновых кислот состоит в конденсации ди-бромалкана с диэтилэтоксималонатом в присутствии этилата натрия с последующим гидролизом и декарбоксилированием {схема (62)}.
NaOEt BrCH2(CH2)„CH2Br + EtOCH(CO2Et)2 ------->
Н+
—> (EtO2C)2C(OEt)CH2(CH2)„CH2C(OEt)(CO2Et)2 ---->
—> HO2CCH(OEt)CH2(CH2)nCH2CH(OEt)CO2H (62)
9А.2.9. Получение эпоксикарбоновых кислот
Эпоксикарбоновые кислоты часто можно получать из соответствующих алкеновых кислот обработкой пероксикислотой или присоединением хлорноватистой кислоты с последующим щелочным дегидрогалогенированием.
R’COR2 + R3CHCO2R<
X
R2 R3 R2 R3
NaOEt || h+ | |
-или > R‘C—CCO2R4 -------> R'C---CCO2H
NaNH2 \ /
О О
(63)
еакция Дарзана открывает метод получения а,Р-эпокснкнслот и их сложных эфиров [64]. Она состоит во взаимодействии альде-ида или кетона со сложным эфиром а-галогенкарбоновой кислоты
Ш
в присутствии амида или этилата натрия (схема (63)}. Сложные >фиры а,а-дпгалогенкарбоновых кислот в присутствии амальгамы магния реагируют с карбонильными соединениями аналогичным образом, давая сложные эфиры а,0-эпоксикарбоновых кистот {схема (64)}. Сложные эфиры и амиды трихлоруксусной кислоты реагируют в гексаметилфосфоротриамиде (ГМФТА) с альдегидом давая а-хлор-а,0-эпоксикислоты [65] {схема (65)}.
Mg/Hg R‘\ /R3
R1CX2CO2R2 + R3R’CO -----> V—С/ (64)
R2O2Cz \Z
ГМФТА C1\
C13CCO2R‘ + R2CHO -=---->- V--------CHR2 (65)
«о,с/\о/
Диазометан реагирует co сложными эфирами а-оксокарбоновых кислот, присоединяясь по кетонной карбонильной группе с образованием соответствующих сложных эфиров эпоксикислот.
9.4.2.10. Получение ненасыщенных гидроксикарбоновых кислот
В общем случае синтез алкеновых и алкиновых кислот данного класса не представляет особых трудностей. Действительно, наличие ненасыщенного фрагмента может быть использовано в синтезе тех (о-гидроксикислот, в которых двойная или тройная связи присутствуют между гидроксильной и карбоксильной группами. Два синтеза рицинолевой кислоты (1) {схемы (66, 67)} демонстрируют, как можно использовать центральный ненасыщенный фрагмент для введения гидроксил- или карбоксилсодержащей части молекулы [66].
Дилитиевая соль гидроксилалкинов реагирует с диоксидом углерода, давая гидроксиалкин-2-овые кислоты {схема (68)}; взаимодействие этих солей с бромалкановыми кислотами приводит к гидроксиалкиновым кислотам с длинной цепью [67] {схема (69)}.
Гидроксидииновые кислоты получают реакцией 1-бромгидр-оксилалкина-1 с алкиноатом {схема (70)} (68). Гидроксиальдегиды и карбоксиалкилфосфонийгалогениды реагируют в условиях реакции Виттига с образованием со-гидроксиалкеновых кислот {схема
в u он
НС=С(СН2)7С1 -н > Me(CH2)sCHCH2(=C(CH2)7Cl —*
он
Nal; KCN; KOH; I Z тт
-------------> Ме(СН2)5СНСН2СН=СН(СН2)гСО?Н
Н2/катализатор '
Линдлара
(66)
178
NaH;
У О KCH2)eCI I
Me(CH2)6CHCH2CsCH Ме(СН2)5СНСН2С=С(СН2)6С1
ОН
Nal; NaCH(CO2Et)2; Me(CH2)5CHCH2CH=CH(CH2)7CO2H (67)
Н2/каталнзатор
Линдлара
RCH(CH2);iC=CLi + СО2 —> RCH(CH2)„C^CCO2H (68)
| I
OLi ОН
RCH(CH2)rtC=CLi + BrCH2(CH2),nCO2Li —> RCH(CH2)n.C—CCH2(CH2)mCO2H
I OH <69>
OLi OH
HOCRjCssCBr + HCssC(CH2)2CO2R2 —> HOCR[(C=C)2(CH2)2CO2R2 (70)
RCH(CH2)ftCHO + Ph3PCH(CH2)mCO2 —> RCH(CH2)nCH=CH(CH2)mCO2H (71)
I I
OH OH
9.4.2.11. Получение алкокси- и гидроксиарил карбоновых кислот
Наиболее важным представителем этой группы соединений является о-гидроксибензойная (салициловая) кислота в силу значимости ее О-ацетильного производного (аспирин). Салициловую кислоту и ее гомологи получают карбоксилированием фенолятов (реакция Кольбе — Шмитта) [69]. При повышенном давлении и температуре (130—140°C) фенолят натрия реагирует с диоксидом углерода, давая салицилат натрия {схема (72)}. Щелочные соли гомологов фенола ведут себя аналогично в отношении диоксида углерода, причем электронодонорные заместители облегчают реакцию. Щелочные соли 2-нафтола в зависимости от условий реакции могут образовывать 1-, 3- или 6-карбоксилаты.
ONa
со2 ----►
(72)
Карбоксильную группу в мягких условиях можно ввести в ди-и тригидроксибензолы, например нагреванием концентрированных водных растворов с бикарбонатом щелочного металла при
С,оили нагреванием полигидроксибензола в глицерине при 130—210 °C с бикарбонатом в токе диоксида углерода. Температура реакции влияет на строение конечного продукта. Например, реакция резорцина при 180 °C приводит к [3-резорциловой кислоте
179
(39), в то время как при 130°C образуется исключительно у-резор-циловая кислота (40) {схема (73)}. Аналогично, пирокатехин (41) дает 2,3- и 3,4-дигидроксибензойные кислоты; последнее соединение получают также сплавлением ванилина со щелочью (42) {схема (74)} [70]. 3,4,5-Тригпдрокспбензойная (галловая) кислота (43) образуется при нагревании пирогаллола с бикарбонатом калия {схема (75)} [71].
Основной бензоат меди при слабом нагревании в инертном растворителе дает салициловую кислоту [72].
Гидроксибензойные кислоты получают также при реакции фенола и тетрахлорида углерода в присутствии медного катализатора (реакция Раймера — Тимана) [73]. В этой реакции из фенола получаются салициловая (25%) и 4-гидроксибензойная (35%) кислоты. Другие способы получения салициловой кислоты — сплавление о-сульфобензойной кислоты с гидроксидом натрия или образование дназониевой соли из о-аминобензойной кислоты и превращение ее в салициловую кислоту обычным образом.
Крезолы окисляются до соответствующих гидроксибензойных кислот при простом сплавлении со щелочью или в присутствии оксидов металлов или при нагревании с хлоратом калия.
Простые ариловые эфиры с алкильными заместителями в ароматическом ядре окисляют перманганатом калия до соответствую-180
„гях ачкоксибензойных кислот. Гидроксиацетофеноны окисляют до гидроксибензойных кислот обработкой иодом в пиридине с последующей реакцией со щелочью. Алкоксибензойные кислоты удобнее всего получать действием диоксида углерода на алкоксиарилмаг-нийгалогениды или литийалкоксиарилы. Избирательное металлирование фенильного кольца н-бутиллитием в орто-положение к алкоксильной группе используется для получения некоторых 2-алкоксибензойных кислот [74].
9.4.2.12. Получение алкокси- и гидроксифенил алкановых кислот
Алкокси- и гидроксифенил уксусные кислоты получают по реакции Вильгеродта. Реакция состоит в нагревании соответствующего ацетофенона с серой и морфолином с последующим гидролизом образующегося фепилтиоацетоморфолида [75] {схема (76)}. Альтернативно, ацетофенон можно превратить в метиловый эфир соответствующей фенилуксусной кислоты с помощью нитрата тал-лия(Ш) в метаноле {схема (77)} [76].
T1(NO3)3, МеОН,НСЮ4
О—-Н ON О,
Р I
CX-CH2TTITONO2
\ОМе ОМе
СН2СО2Ме
OMe
(77)
>
Метоксифенилуксусные кислоты можно получить конденсацией соответствующего метоксибензальдегида с гиппуровой кислотой, разуется азлактон, гидролиз которого приводит к арилпировиноградной кислоте; последняя при окислении щелочным пероксидом водорода дает искомую арилуксусную кислоту.
Гидроксифенилпропионовую и высшие кислоты обычно получают восстановлением предшественников, несущих кетонные или алкеновые фрагменты в кислотной боковой цепи.
181
9.4.3. СВОЙСТВА ГИДРОКСИ-И АЛКОКСИКАРБОНОВЫХ КИСЛОТ*
Как свойственно соединениям с двумя функциональными группами, способными образовывать водородные связи, гидроксиалкановые кислоты в основном растворимы в воде. Однако наличие гидрофобных боковых цепей и циклов в молекуле снижает растворимость таких кислот.
Инфракрасные, ЯМР- и (в меньшей степени) масс-спектры многих синтетических и природных гидрокси- и алкоксикислот описаны в связи с установлением строения этих соединений. ЯМР-спектры трпхлорацетилизоцианатных производных гидроксикислот применялись для локализации положения гидроксильной группы относительно карбоксильной функции (а-, |3- или далее) путем оценки сдвига сигналов водородных атомов, находящихся в a-положении к гидроксильной группе, в сторону слабых полей [78]. Изучение инфракрасных спектров a-гидрокси-, а-алкокси- и а-арилоксикислот было предпринято для количественной оценки внутримолекулярной водородной связи, присутствующей в этих молекулах [79].
Для некоторых а- и |3-гидроксикислот измерены круговой дихроизм и описаны спектры дисперсии оптического вращения [80].
Гидроксильная группа оказывает заметное влияние на диссоциацию соседней с ней карбоксильной группы. Для 2-гидроксиал-кановых кислот отмечено возрастание кислотности карбоксильных групп. Этот эффект вызывается двумя факторами: во-первых, индуктивный эффект гидроксильной группы стремится понизить рКа примерно на 0,45 единицы р/( [81], и, во-вторых, происходит стабилизация карбоксилатного иона с помощью внутримолекулярной водородной связи с гидроксильной группой при соседнем углеродном атоме (44). Показано, что последний эффект зависит от взаимной ориентации гидроксильной и карбоксильной групп, (предполагается, что индуктивный эффект не зависит от стереохимии этих групп) [82].
RHC—С=О
Н‘ (44)
Чем дальше отстоит гидроксильная группа от карбоксильной, тем меньше ее влияние на константу диссоциации. Тот факт,что салициловая кислота сильнее бензойной и /г-гпдроксибензойной кислот объясняется образованием внутримолекулярной водородной связи между карбоксилат-анионом и соседней гидроксильной группой [83].
Соседние алкокси- и арилоксигруппы также оказывают влияние на диссоциацию алкановых кислот. Это проистекает в силу
* См. [77].
182
индуктивного эффекта (только), и, как и ожидалось, арилокси-rovnna в большей степени способствует диссоциации карбоксильной функции, чем алкоксигруппа. n-Алкокси- и п-арилоксибензои-нпе кислоты немного слабее, чем бензойная кислота (в силу мезо-мепной стабилизации кислоты заместителями),однако мета- и орто изомеры несколько более сильные кислоты (благодаря индуктивному эффекту заместителей) [84].
9.4.4. РЕАКЦИИ ГИДРОКСИ-ИАЛКОКСИКАРБОНОВЫХ КИСЛОТ
9.4.4.1. Общие реакции
Гидроксиалкановые кислоты ведут себя п как спирты, и как кислоты. Во многих реакциях гидроксильная и карбоксильная функции не влияют друг на друга, особенно если эти две группы удалены друг от друга. Карбоксильную группу можно превратить в сложноэфирную, амидную, цианогруппу или превратить в хлор-ангидрид, используя обычные условия реакции. Этерификацию гидроксикарбоновых кислот, чувствительных к кислым воздействиям, проводят с помощью триалкоксиборана. Обычно гидроксильную группу модифицируют после того, как карбоксильная группа превращена в сложноэфирную. Например, хлоркислоты обычно получаются из гидроксикислот через промежуточное получение сложного эфира гидроксикислоты [85]. В других случаях предварительная этерификация не требуется: так, ацетилирование гидроксильной группы можно проводить на свободной кислоте. Реакция гидроксикислоты с пентахлоридом фосфора приводит к соответствующему хлорацилхлориду, который можно превратить в кетен-имин {схема (78)} [86].
ОН
Ph2CCO2H
С1
PCi5 | ---> Ph2CCOCl
Аг\’Н2;
РС15;
Nal
Ph2C=C=NAr
(78)
Аналогично, гидроксильную группу фенольных кислот можно алкилировать и ацилировать обычными способами. Часто, но не всегда, зеленое окрашивание, возникающее при добавлении хлорида железа, типично для фенольной группы. Салициловую кислоту можно непосредственно этерифицировать. Сложные" эфиры ароматических гидроксикислот с теми же или с другими ароматическими гидроксикислотами известны как ди-депсиды, три-депсиды, и т. д., в соответствии с числом остатков гидрокснкислот в сложном эфире. Например, простой дидепсид (45) получается при конденсации двух молекул н-гидроксибензопной кислоты. Первым тщательно исследованным депсидом была дигалловая кислота
183
(46), которая присутствует как компонент галлотанина [87]. Большинство природных депсидов найдено в лишайниках.
(45)
Э.4.4.2. Реакции при нагревании
а-Гидроксикислоты дегидратируют с образованием полулактидов (47) или лактидов (48). Может также проходить конкурентный распад до альдегида и муравьиной кислоты. Наилучшие выходы альдегидов получаются при пиролизе а-ацилокси- или а-алкоксикарбоновых кислот. Перегонка а-алкокспкислот в присутствии меди приводит к высоким выходам альдегидов {схема (79)}.
При нагревании карбоновых кислот с третичной гидроксильной группой в a-положении получают кетоны и аф-ненасыщенные кислоты. При увеличении длины алкильных цепей R1 и R2 количество ненасыщенной кислоты растет {схема (80)}.
аф-Ненасыщенные кислоты получают также при нагревании Р-гидроксикислот. у- и 6-Гидроксикислоты при нагревании дегидратируют с образованием циклических лактонов. В некоторых случаях е-гидроксикислоты при нагревании также дают лактоны, однако обычно они отщепляют воду с образованием алкеновых кислот или линейных сложноэфирных полимеров. Аналогично, гпдро-ксикислоты с гидроксильной группой, еще более удаленной от карбоксильной группы, элиминируют воду с образованием ненасыщенных кислот или линейных сложных эфиров {схемы (81) — (83)}.
О
А А
R‘CHCO2H ----> r!chco2h ------>
I I
OH OCOCHR1
д — HCO2H ОН
(47)
R’CHO <---- R'CH(OCOR2)CO2H или
О
(48)
R!CH(OR2)CO2H
СО2Н R2
R'CH2CR2 R'CH=CCO2H+ R‘CH2COR2 (80)
I
OH
]84
RCHCH2CO2H I он
RCH(CH2)rtCO2H —~ он
л
> rch=chco2h
RCH—(CH2)rt
(n = 2, 3, 4)
(81)
(82)
RCHCH2(CH2)„CO2H —
OH
RCH=CH(CH2)MCO2H +
R CHCH2(CH2)ftCO—OCH(R)---
I
O—CO(CH2)rtCH2CHR (n>3) .
О----
(83)
При нагревании дигидроксикислот, в которых гидроксильные группы расположены у соседних атомов, в зависимости от времени реакции, температуры и природы катализатора, могут образовываться разнообразные продукты. Например, 9,10-дигидроксистеариновая кислота (49) при кратковременном нагревании в присутствии каталитических количеств n-толуолсульфокислоты образует смесь сопряженных диеновых кислот (43—47%), несопряженных диеновых кислот (31—36%), кетокислот (5—11%), эпоксикислот (7—8%) и гидроксиалкеновых кислот (4—7%).
НО ОН
СН3(СН2)7СНСН(СН2)8СО2Н
(49)
СН2СО2Н I носсо2н
СН2СО2Н
СО2Н (50)
А А
—> ОС(СН2СО2Н)2 ------> СН3СОСН3
Гидроксиполикарбоновые кислоты распадаются аналогичным образом до более простых соединений. Например, лимонная кислота при нагревании около 175 °C дегидратирует до аконитовой кислоты (50), однако при быстром нагревании до более высоких температур протекают два процесса распада, приводящие к ацетону и метилмалеиновому ангидриду {схема (84)}.
185
Фенольные кислоты при нагревании декарбоксилируются; легкость декарбоксилирования возрастает с увеличением числа орто-и пара- (по отношению к карбоксилу) гидроксильных групп. Добавлением патронной извести декарбоксилирование можно ускорить.
9.4.4.3. Образование лактонов
p-Гидроксикислоты можно превратить в [3-лактоны действием дициклогексилкарбодиимида (ДЦК) или через промежуточное образование смешанного ангидрида при реакции с метан- или бензолсульфохлоридом с последующим внутримолекулярным нуклеофильным замещением под действием пиридина или карбоната натрия [88]. Альтернативно, р-лактоны получают встряхиванием водного раствора натриевой соли p-хлоркарбоновой кислоты с хлороформом. Изучено мягкое стереоспецифическое декарбоксилирование р-лактонов, приводящее к алкенам с предсказуемой конфигурацией [89] {схема (85)}. Обработка p-гидроксикислоты пероксидом водорода в серной кислоте приводит к Р-пероксилактону |90].
Как отмечалось выше, у- и 6-гидроксикислоты имеют заметную тенденцию к образованию пяти- и шестичленных лактонов соответственно. Изучение равновесия этих гидроксикпслот с соответствующими лактонами показало, что доля лактона в равновесной смеси очень велика. В тех случаях, когда стерическое напряжение несколько затрудняет замыкание цикла, уксусный ангидрид или ДЦК в пиридине ускоряют процесс лактонизации [91]. С другой стороны, степень лактонизации заметно растет, если гидроксильная и карбоксильная группы жестко сближены [92].
Известно лишь несколько е-лактонов; они легко ооразуются только тогда, когда геометрические факторы благоприятствую замыканию цикла {схема (86)}. Синтез макроциклических л тонов из w-гидроксикпслот весьма затруднен. Ранние исследован показали, что лактоны можно получать с высоким выходом ПРИ_^ гревании гидроксикислоты в сильно разбавленном растворе. » 186
Уунсдикер с сотр. циклизовали со-бромалкановые кислоты при -ипячении разбавленных растворов с карбонатом калия.
К Позднее синтезам макроциклических лактонов из гидроксикис-оТ было уделено значительное внимание, так как это превращение было последней стадией в синтезе некоторых макролидных антибиотиков [93]. Успешно развитые методы циклизации включают активирование карбоксильной {схемы (87), (88)}, или гидроксильной группы {схемы (89)} или применение [94] специальных реагентов {схема (90)}.
но(сн2)лсо2н
Me
О
п = 5,7,10,11,14
и = 5, 7, 11
(88)
(89)
BFa-EtjO, полистирол
НО(СН2)„СО2Н ---------—-----------
(90)
п = 13-17
Полигидроксикислоты, гидроксиполикарбоновые кислоты и фенольные кислоты образуют лактоны, если при этом возникают пяти- или шестичленные циклы. Например, фотолиз кумаровой кислоты приводит к нестабильной кумариновой кислоте (51), которая самопроизвольно циклизуется с образованием кумарина (52) {схема (91)}. Яблочная кислота в присутствии дымящей серной кислоты циклизуется с образованием кумалевой кислоты (53),
187
которую действием аммиака можно превратить в 6-гидроксиникп™ новую кислоту ИЛИ rrrirnn -------- X. икоти-
{схема (92)}.
после декарбоксилирования —в 2Н-пиранон-2
9.4.4.4. Окисление
Строение продуктов окисления гидроксикислот зависит от природы гидроксильной группы. Первичные гидроксикислоты дают альдегидокислоты и/или дикарбоновые кислоты; вторичные гидроксикислоты вначале образуют кетокислоты, которые могут вступать в дальнейшие превращения: третичные гидроксикислоты претерпевают расщепление углеродной цепи.
а-Гидроксикислоты под действием реагента Фентона или при окислении соответствующего сложного эфира перманганатом калия превращаются в а-оксокислоты. Прямое окисление нейтральных водных растворов а-гидроксикислот перманганатом в некоторых случаях протекает успешно, однако этот метод не нашел общего применения.
Окислительное расщепление а-гидроксикислот до альдегидов или кетонов с потерей одного атома углерода можно осуществить под действием висмутата натрия, трет-бутилгипохлорита, тетраацетата свинца, перманганата калия в кислой среде, йодата натрия, галогенимидов или хромовой кислоты [95].
Пероксид калия в бензоле, содержащем 18-краун-6, окислительно расщепляет а-гидроксикислоты до карбоновых кислот по реакции, напоминающей действие некоторых диоксигеназ (ферментов) {схема (93)} [96].
R2
R1—i-CO2H
I ОН
РЬ(ОАс)4. или КМпО4, или XN(COR3)2. или H2CrOi ^ d i qqд 2
: — - (93)
К2О2. 18-крауи-б, бензол --------------------------> ЩСО2Н
(при R2 - Н)
188
Дпгпдроксикислоты, имеющие гидроксильные группы у соседних атомов углерода, легко расщепляются обычным способом до альдегидов под действием тетраацетата свинца, периодной кислоты, висмутата натрия и ацетата марганца.
9.4.4.5. Реакции а-гидроксиалкановых кислот
В силу близости двух функциональных групп эти кислоты обладают рядом характерных реакций. При нагревании с альдегидами в результате катализируемой кислотой дегидратации образуются 1,3-диоксоланоны-4 {схема (94)} [97]. а-Гидроксикислоты легко образуют комплексы с металлами, например с медью (54), включающие обе функциональные группы. Бор, там где возможно, легко образует хелатные циклы с а-гидроксикислотамн. Именно это и было использовано как метод определения конфигурации алициклических гидроксикислот, таких, как 2-гидроксициклопен-танкарбоновая кислота [98].
(55)
«-Гидроксикислоты реагируют с первичными аминами и ди-этилкарбонатом с образованием 2,4-оксазолидинонов (55) {схема
94.4.6. Реакции р-гидроксиалкановых кислот
В подходящих условиях большинство гидрокснкпслот теряет воду с образованием алкеновых кислот, однако р-гидроксп-кислоты особенно склонны к дегидратации, так как при этом поучаются аф-ненасыщенные кислоты. В некоторых случаях
189
отмечено сопровождающее {схема (96)} [99].
дегидратацию Декарбоксилирование
R!R2CCH2CO2H
он
Си
7* R*R2c~ch2 (96)
он
R*CHO + R2R3icHCO2H
R4
о
* О-/
R1—<о )—R4 (97)
R2\3
p-Гидроксикислоты реагируют с альдегидами, образуя 4-оксо-1,3-диоксаны {схема (97)} [100]. Ретроальдольная реакция р-гидр-оксикислот, катализируемая кислотой или щелочью, приводит к кетонам и простым карбоновым кислотам {схема (98)}. Область применения этой реакции, ее условия и ограничения описаны в работе [101].
ОН
I Н+ или ОН"
R‘R2CCHCO2H --------->- R*R2CO + R3CH2CO2H (98)
R3
R Ph R Ph
I I ZnCI2 | I
ArC—CHCO2H ArC=CCOCH3 (99)
I AC2O 4
OH
Некоторые З-арил-З-гидроксикислоты при нагревании с хлоридом цинка в уксусном ангидриде превращаются в а,Р-ненасыщен-ные метилкетоны {схема (99)} [102].
9.4.4.7. Реакции алкоксиалкановых кислот
а-Алкоксикислоты при облучении в присутствии брома превращаются в а-кетокислоты [103], а под действием борогидрида натрия и иода — в гидроксикислоты [104].
Эпоксикислоты чувствительны к реагентам, расщепляющим оксирановые циклы. Гидрогенолиз над палладием в уксусной кислоте приводит к изомерным гидроксикислотам; отмечена небольшая избирательность в направлении раскрытия оксиранового кольца [105]. Кислотный гидролиз эпоксикислот приводит к расщеплению эпоксидного кольца с образованием дигидроксиалкановых кислот, кислый алкоголиз дает алкоксигидроксикислоты, взаимодействие с аминами приводит к аминогидроксикислотам, реакция с алкановыми кислотами дает ацилоксигидроксикислоты. Последние реакции, в основном, не обладают селективностью.
Декарбоксилирование а,р-эпоксикарбоновых кислот в зависимости от природы заместителя приводит к альдегидам или кетонам 190
/схема (ЮО)}. Изомеризацию сложных эфиров эпоксикислот в гожные эфиры кетокислот можно осуществить с помощью эфи-рата трифторида бора в диоксане [106] .
д R'\ /°\ /со=ы д
rir2CHCOR3 *---- X—> R’R2CHCHO (100)
R2/ \r3
(при R8 = Me, Et> (при R3 = Н)
9 4.4.8. Реакции гидроксиарилкарбоновых кислот
Электрофильное замещение протекает по отношению к гидроксильной группе (группам) в орто- и пара-положения. Карбоксильные группы в орто- и пара-положении могут быть замещены вступающим заместителем. Например, энергичное бромирование 2- или 4-гидроксибензойной кислот приводит к 2,4,6-трибромфенолу (56), жесткое нитрование дает пикриновую кислоту (57) {схема (101)}. 4-Гидроксибензойная кислота сочетается с солями арендиазония с замещением карбоксильной группы, а не с замещением по свободным орто-положениям {схема (102)}.
Дегидратация салициловой кислоты и ее гомологов под действием пентоксида фосфора и хлороксида фосфора приводит к макроциклическим полиэфирам.
ЛИТЕРАТУРА
1. D. Т. Downing, Rev. Pure Appl. Chem. (Australia), 1961, 196; N. S. Radin, J. Amer. Oil Chemists’ Soc., 1965, 42, 569; R. C. Badami and S. M. Kudari, J. Oil Technologists Association of India, 1973, 71; K. S. Markley, «Fatty Acids», Interscience, New York, 1960, vol. 1, pp. 65—88 and 183—192, 1968, vol. 5, pp. 3138-3140, 3210-3227.
2. w Hartmann, Angew. Chem. Internat. Edn., 1975, 14, 337; A. S. Crossley 3 U7e?' Hnd -Ind' (London)> 1976, 334> E- Horton, New Scientist, 1976, 9.
‘Lickinbottom, «Reactions of Organic Compounds», Longmans, London, odd’s Chemistry of Carbon Compounds», ed. S Coffev, vol. 1, parts D (1"о5) E (1976), and G (1976); vol. 3, parts D (1976) and F. (1974), Else-4lerii7AnS,terdam; D- Swern< Prog. Chem. Fats and Lipids, 1955, 3, 213;
• w. Holston, «Fatty Acids and Their Derivatives», Wiley, London, 1948.
191
4. /И. Fetizon, М. Golfier, and J.-M. Louis, Tetrahedron, 1975, 31, 171.
5. J. Polonsky and E. Lederer. Bull. Soc. chim. France, 1954, 504.
6. M. Kawana and S. Emoto, Bull. Chem. Soc. Japan, 1974, 47, 160.
7. R. D. Clark and С. H. Heathcock, Tetrahedron Letters, 1974, 1713; R. т n hill, J. Chem. and Eng. Data, 1972, 17, 399.
8. K. Kurata, S. Tanaka, and K. Takahashi, Chem. and Pharm. Bull (Jananl 1976,24,538. ' 1
9. G. Vollema and J. F. Arens, Rec. Trav. chim., 1963, 82, 305.
10. R. R. Fraser and P. R. Hubert, Canad. J. Chem., 1974, 52, 185.
11. H. R. Nace and M. Inaba, J. Org. Chem., 1962, 27, 4024.
12. D. H. S. Horn and Y. Y. Pretorius, J. Chem. Soc., 1954, 1460.
13. M. Charpentier and A. Skrobek, Compt. rend., I960, 251, 1391; J. G. Aston J. D. Newkirk, D. M. Jenkins, and J. Dorsky, Org. Synth. Coll Vol 3 1955’ 538. ‘ '
14. Y. Okamoto, T. Nitta, and H. Sukarai, Kogyo Kagakir Zasshi 1968 71 187 (Chem. Abs., 1968, 69, 35342).
15. E. C. Taylor, H. IF. Altland, G. McGillivray, and A. McKillop, Tetrahedron Letters, 1970, 5285.
16. D. A. Konen, L. S. Sibert, and P. E. Pfeffer, J. Org. Chem., 1975, 40, 3253.
17. S. Jriuchijima, K. Maniwa, and G. Tsuchihashi, J. Amer. Chem. Soc, 1975 97, 596.
18. G. M. Rubottom and R. Marrero, J. Org. Chem., 1975, 40, 3783.
19. E. L. Compere, J. Org. Chem., 1968, 33, 2565.
20. E. C. Bubl and J. S. Butts, J. Amer. Chem. Soc., 1951, 73, 4972.
21. S. Watanabe, K. Suga, T. Fujita, and K. Fujiyoshi, Israel J. Chem., 1970, 8, 731; B. Angelo, Bull. Soc. chim. France, 1970, 1848; G. W. Moersch and A. R. Burkett, J. Org. Chem., 1971, 36, 1149.
22. E. B. Dongala, D. L. Dull, C. Miuskowski, and G. Solladie, Tetrahedron Letters, 1973, 4983; S. Brandange, S. Josephson, and S. Vallen, Acta Chem. Scand., 1973, 27, 1084.
23. M. Gaudemar, Organometallic Chem. Rev. (A), 1972, 8, 183.
24. D. A. Cornforth, A. E. О par a, and G. Read, J. Chem. Soc. (C), 1969, 2799; Y. Maroni-Bamaud, G. Gilard, A. Montalla, M. Perry, and J. E. Dubois, Bull. Soc. chim. France, 1966, 3243; A. Horeau, Tetrahedron Letters, 1971, 3227.
25. M. Bellassoued, R. Couffignal, and Al. Gaudemar, J. Organometallic Chem., 1973, 61, 9.
26. J. D. Morrison and H. S. Mosher, «Asymmetric Organic Reactions», Printice Hall, New Jersey, 1971, p. 142 [Дж. Д. Моррисон, F, С. Мошер. Асимметрические органические реакции. — Пер. с англ. М., Мир, 1973, гл. 3]; Л1. Guette, J. Capillon, and J.-P. Guette, Tetrahedron, 1973, 29, 3659.
27. P. L. Creger, Tetrahedron Letters, 1972, 79.
28. L. Vuitel and A. Jocot-Guillarmod, Synthesis. 1972, 608.
29. E. Negishi and T. Yoshida, J. Amer. Chem. Soc., 1973, 95, 6837.
30. T. Mukaiyama, K. Inomata, and M. Muraki, J. Amer. Chem Soc., 1973, 95, 967; Bull. Chem. Soc. Japan, 1973. 46, 1807.
31. С. H. Hassall, Org. Reactions, 1957, 9, 73; J. B. Lee and В. C. Ujf, Qua1"! Rev., 1967, 21, 429 [4. X. Хассел. Орг. реакции, сб. 9. — Пер. с англ. ПоД ред. И. Ф. Луценко. М., Издатинлит, 1959, с. 83].
32. AL Jtoh, Т. Taguchi, V. V. Chung, М. Tokuda, and A. Suzuki, J. Org. Chem-1972, 37, 2357; Л4. Tokuda, Y. Yokouama, T. Taguchi, A. Suzuki, and M. Hon, ibid., 1972, 37, 1859. ‘ _,a9.
33. L. M. Berkowitz and P. N. Rylander, J. Amer Chem. Soc., 1958, 80, 000 , N. C. Deno and N. H. Potter, ibid., 1967, 89, 3550. Сйй9.
34. L. M. Berkowitz and P. N. Rylander, J Amer. Chem. Soc., 1958, 80, '
N. C. Deno and N. H. Potter, ibid., 1967, 89, 3550; J. E. Lyons, J. C. S. СПеп •
Comm., 1975, 412.
35. R. M. Adams and C. A. VanderWerf, J. Amer. Chem. Soc., 1950; 72, ’
K. Mori, Tetrahedron, 1976, 32, 1101; IF. Sacrow and U. Klein, Chem. :•
1975, 108, 3518; J. Attenburrow, J. Elks, B. A. Hems, and K. N. $РеУе,гМА Chem. Soc,, 1949, 510; K. Iwai, H. Kosugi, and H. Uda, Chem. Letters, 1»' ’
192
„ . c i J Wane and S. D. Burke. J. C. S. Chem. Comm., 1237; P- AnGr,ieCc’a'rL\ 'or/Sem 1972, 37, 1907; see also А. I. Meyers,
kin I, 1974 1864 Roberts, and P. S. Rutledge, J C. S .Per-
37- S' 1’864- H ChHstol, F. Plenat, and J. Revel, Bull. Soc. chmi. Fran- . kin 1, iy<*>
ce, 1971, ,45;J7- • 1975 67; в. Gammill, C. A. Wilson, and T. A. Bry-
38. P. A. Grieco Sunthesis 1У/О, oz, n
S,OflD Svnl I' C0TKmE Shenton and J. Schwartz, Tetrahedron Letters, 1975 51, 39' L. S. H°egedusKSEDS^agner, E. L. Waterman, and K. Siirala-Hansen, J. Org.
40 Carls’on^and^A. R. Oz/Zar, Tetrahedron Letters, 1975 4099
4L M KatoM Kageyama, R Tanaka, K. Kuwahara, and A. Yoshtkoshi, J. Org.
42^ E and P. Weyerstahl, Chem Ber, 1974, 107 2852
43 H. Marshall, F. Vogel, and P. Weyerslahf Chern^Ber., Д974, 107, ’ в'.
44. К
C.
45. E.
K.
46. J.
2852;
5217;
3079;
48.
49.
50.
51.
Marshall F Vogel, and P. weyersiam, cnem. oer., ю/, M Trost and С. H. Miller, J. Amer. Chem. Soc., 1975, 97, 7182.
E Miller, C. R. Haymaker, and H. Gilman, J. Org. Chem., 1961, 26, Malani, D. E. Minnikin, and K. Polgar, J. Chem. Soc., 1965, 5562.
E. van Tamelen and S. R. Bach, J. Amer. Chem. Soc., 1958, 80, Sisido, S. Torii, and M. Kawanisi, J. Org. Chem., 1964, 29, 904.
Grigor, D. M. Maclnnes, J. McLean, and A. J. P. Hogg, J. Chem. Soc., 1955, 1069; F. D. Gunstone and W. C. Russell, ibid., 3782.
47. J. W Cornforth, R. H. Cornforth, G. Popjak, and L. Yengoyan, J. Biol. Chem., 1966, 241, 3970; R. H. Cornforth, J. W. Cornforth, and G. Popjak, Tetrahend-ron, 1962, 18, 1351; /. IF. Cornforth, F. P. Ross, and C. Wakselman, J. C. S. Perkin I, 1975, 429; J. A. Lawson, W. T. Colwelt, J. I. DeGraw, R. H. Peter, R. L. Dehn, and M. Tanabe, Synthesis, 1975, 729.
E. J Corey and G. Moinet, J. Amer. Chem. Soc., 1973, 95, 6831; E. J. Corey and J. Mann, ibid., 1973, 95, 6832; See also J. B. Wiel and F. Rouessac, J. C. S. Chem. Comm., 1976, 446; R. E. Ireland, R. H. Mueller, and A. K- Willard, J. Org. Chem., 1976, 41, 986; P. Crabbe, A. Guzman, and M. Vera, Tetrahedron Letters, 1973, 4730; E. J. Corey and G. Moinet, J. Amer. Chem. Soc., 1973, 95, 7185; E. J. Corey and C. R. Cyr, Tetrahedron Letters, 1974, 1761.
P. Collins. С. J. Jung, and R. Pappo, Israel J. Chem., 1968, 6, 839; J. Katsube and M. Matsui, Agric. and Biol., Chem. (Japan), 1969, 33, 1078; Y. Yura and J Ide, Chem. and Pharm. Bull. (Japan), 1969, 17, 408.
M. Hayashi and T. Tanouchi, J. Org. Chem., 1973, 38, 2115; E. F. Jenny, P. Schdublin, H. Fritz, and H. Fuhrer, Tetrahedron Letters, 1974, 2235.
E. I. Corey, T. K. Schaaf, W. Huber, U. Koelliker, and N. M. Weinshenker J. Amer. Chem. Soc., 1970, 92, 397; E. J. Corey, Z. Arnold, and J. Hutton Tetrahedron Letters, 1970, 307; see also E. J. Corey and R. Noyori, Tetrahedron Letters, 1970, 311; F. Kienzle, G. W. Holland, J. L. Jernow, S. Kwoh and P Rosen, J. Org. Chem., 1973, 38, 3440; E. D. Brown, R. Clarkson, 1. J Leeney, and G. E. Robinson, J. C. S. Chem. Comm., 1974, 642; G. Jones, R A. Raphael, and S. Wright, J. C. S. Perkin I, 1974, 1683; S. Ranganat-ir--- Ranganathan, and A. K. Mehrotra, Tetrahedron Letters, 1975, 1215; дкад JrA2*’ L' Gruber> G- Kovacs, I. Szekely, and V. Simonidesz, ibid., 1976, iook G' Paul’ F- lohns°n, and D. Favara, J. Amer. Chem. Soc., 1976, 98, 52. С I Sih,J B. Heather, G. P. Peruzzotti, P. Price, R. Sood, and L.-F. H. Lee, Soc- 1973- 95> 1676; G- Siork and T- Takahashi, ibid., 1977, 53 Her P\v^nc,atel11 and A- Scettri’ Tetrahedron Letters, 1977, 1131.
’ 1 { что 4". Wl t' nd- and EnS- Chem' (Product Res. and Development), 1972, - 11> «j/U, O/4.
55 A and Paulsen’ Adv. Carbohydrate Chem., 1962, 17, 169.
56’. A j Meuerv n’and % tM-^endal1’ Tetrahedron Letters, 1974, 3495.
• • Meyers and G. Knaus, Tetrahedron Letters, 1974, 1333.
7 Зак, 395
193
57 A 1 Meyers and С. E. Whitten, Tetrahedron Letters, 1976, 1947.
N G. Gaylord and J. R. Benzinger, J. Org. Chem., 1954, 19, 1991.
59 E D Be/gmaun and A. Becker, J. Org. Chem., 1958, 23, 1553.
60' W. Reeve and C. W. Woods, J. Amer. Chem. Soc., 1960, 82, 4062; Ц7. Repria and E. L. Compere, ibid., 1961, 83, 2755.
61. Al. Kerfanto and D. Jegou, Compt. rend., 1965, 261, 2232.
62. K. Saigo, M. Osaki, and T. Mukaiyama, Chem. Letters, 1976, 769.
63* P D. Bartlett and P. N. Rylander, J. Amer. Chem. Soc., 1951, 73, 4273
64. M. S. Newman and B. J. Magerlein, Org. Reactions, 1949, 5, 413 [Af. с. Ньюмен, Б. Дж. Мейджерлейн. Орг. реакции, сб. 5.—Пер. с англ. М, Издатинлит, 1951, с. 318].
65. J. Villieras, G. Lavielle, R. Burgada, and В. Castro, Compt. rend, (c), 1969 268, И 64. ’ ’
66. W. J. Gensler and С. B. Abrahams, J. Amer. Chem. Soc., 1958, 80, 4593 L. Crombie and A. J. Jacklin, J. Chem. Soc., 1955, 1740; E. Crundwell M. A. Pinnegar, and W. Templeton, J. Medicin. Chem., 1965, 8, 41.
67. В Ф. Кучеров, А. И. Кузнецова, M. В. Мавров, Е. Ф. Алексеева. — Изв АН СССР, сер. хим., 1962, 484.
68. А. Р. Держинский, М. В. Мавров, В. Ф. Кучеров. — Изв. АН СССР, сер. хим., 1965, 1265.
69.
70.
71.
72.
73.
74.
75.
Orga-
86.
87.
88.
89.
90.
91,
A. S. Lindsey and Н. Jeskey, Chem. Rev., 1957, 57, 583.
I. A. Pearl, Org. Synth. Coll. Vol. 3, 1955, 745.
A. Critchlow, R. D. Haworth, and P. L. Pauson, J. Chem. Soc., 1951, 1318.
IP. W. Kaeding and A. T. Shulgin, J. Org. Chem., 1962, 27, 3551.
H. Wynberg, Chem. Ber., 1960, 60, 169.
H. Gilman and J. W. Morton, Org. Reactions, 1954, 8, 258 [Г. Гилман. Opr. реакции, сб. 8. — Пер. с англ./Под ред. Ю. А. Арбузова. М, Издатинлит, 1956, с. 333].
М. Carmack and М. A. Spielman, Org. Reactions, 1946, 3, 83 [М. Кармак, М. А. Шпильман. Орг. реакции, сб. 3. — Пер. с англ./Под ред К. А. Кочешкова. М., Издатинлит, 1951, с 88].
76. См. сс. 75; А. МсКШор, В. Р. Swann, and Е. С. Taylor, J. Amer. Chem. Soc., 1971, 93, 4919.
77. R. T. O’Connor, in «Fatty Acids», ed. K. S. Markley, Interscience, New York, 1968, vol. 5, p. 331‘5.
78. F. Will and C. Varsel, Analyt Chim. Acta, 1969, 44, 233.
79. M. Okl and M. Hirota, Bull. Chem. Soc. Japan, 1961, 34, 374, 378; W. O. George, J. H. S. Green, and D. Pailthorpe, J. Mol. Structure, 1971, 10, 297.
80. B. Sjoberg, B. Hansson, and R. Dahlbom, Acta Chem. Scand., 1962, 16, 1057, L. I. Katzin and E. Gulyas, J. Amer. Chem. Soc., 1968, 90, 247.
81. G. Kortiim, W. Vogel, and K. Andrussow, «Dissociation Constants of nic Acids in Aqueous Solutions», Butterworths, London, 1961.
82. J. Sicher, M. Tichy, F. Sipos, M. Svoboda, and J. Jonas, Coll. Czech. Chem. Comm, 1964, 29, 1561.
83. L. Eberson, in «The Chemistry of Carboxylic Acids and Esteps», ed.. S. PaaI> interscience, New York, 1969, p. 284; J Hermans, S. J. Leach, and H. A.ocne-0, Гг8агУ Amer Chem- Soc - 1963> 85, 1390. , in.
84. G. Kohnstam and V. L. H. Williams, in «The Chemistry of the Ether Lin kage», <ed. S. Patai, Interscience, New York, 1967, p. 81. ,nco 1RQ
oe' £>’ ’ Al. •7- Fisk, and T. Prosser, Org. Synth. Coll. Vol. 4, 1963,
Ji ip Sevens and J. C. French, J. Amer. Chem. Soc, 1953, 77, 657.
“ King and T. White, J. Chem. Soc, 1961, 3231.
"Adam, J. Baeza, and J.-C. Liu, J. Amer.Chem. Soc, 1972, 94, 2000.
S Mageswaren and M. U. S. Sultanbawa, J. C. S. Perkin I, 1976, 884 '
7. D. Greene W. Adam, and G. A. Knudsen, J. Org. Chem, 1966, 30, 208Л c N°VakMJKn °' /ilek' B- Kukac, I. Ernest, and M. Protiva, Coll. Czech. Chem C°T 251 2196: R- B- Woodward, F. E. Bader, H. Bickel, A. J- Frey.
Kierstead, Tetrahedron, 1958, 2, 1. eaiS-
D. R. Storm and D. E. Koshland, J. Amer. Chem. Soc, 1972, 94, 5805, 5»101 B. Capon, J. Chem. Soc. (B), 1971, 1207.
92.
194
о„ 117 D Celmer Pure Appl. Chem., 1971, 28, 413; W. Keller-Schierlein Fortschr. 9 Sem. org NatuSoffe 1973, 30, 313; R. K. Boeckman, J. Fayos, and J. Clar-94. Ly’T.S™oti anrZSOC”№K6SySthesis, 19761 738, T. Kurihara, Y. Naka-
45 Venkatasubramanian, In-
95' Ln? Chem 1971’ 9’, 1102; J. M. Bachhawat and N. K- Mathur, ibid., 1971,
Q 1335- К К * Sen Gupta, A. K. Chatterjee, T. Sarkar, Sen Gupta, ibid., 1975, 13 1024- К К- Sen Gupta, D. K. Khemrui, and D. C. Mukherjee, J. Indian Chem Soc 1975, 52, 467; У. Rocker and В. C. Davis, J. Amer. Chem. Soc.,
Q6 ^San^Filippo Ci-1. Chem, and J. S. Valentine, J. Org. Chem., 1976, 40, 1077.
97 ! Soulier and’M. Farines, Compt. rend, (c), 1969, 268, 1546.
98 /. Zemlicka, R. Gasser, J. V. Freisler, and J. P. Hortmitz, J. Amer. Chem. Soc., 1972, 94, 3213; D. S. Noyse, P. A. King, and G. L. Woo, J. Org. Chem., ISfei’, 26, 632; A. Campbell and R. S. MacPherson, J. C. S. Perkin I, 1974, 42; Л4 Vilkas, N. A. Abraham, and J. Candehore, Bull. Soc. chim. France, 1960,
99. P. Ayrds and K- Pihlaja, Tetrahedron Letters, 1970, 4095.
100 R L. Shriner, Org. Reactions, 1942, 1, 14; F. F. Blicke and R. H. Cox, J. Org.
' Chem., 1957, 22, 1741.
101. С. P. Ivanov and D. M. Mandeshka, Tetrahedron, 1973, 29, 1745.
102. H. Gross and J. Freiberg, Chem. Ber., 1967, 100, 3777.
103. G. Odhani and B. Samuelsen, Acta Chem. Scand., 1970, 24, 468.
104. F. J. Julietti, J. F. McGhie, B. L. Rao, and W. A. Ross, Chem. and Ind. (London), 1960, 874; D. R. Howton and R. W. Kaiser, J. Org. Chem., 1964, 29, 2420.
105. H. A. Walens, R. P. Koob, W. C. Ault, and G. Maerker, J. Amer. Oil Chemists’ Soc., 1965, 42, 126.
106. W. Baker, I. B. Harborne, A, J. Price, and A. Rutt, J. Chem, Soc., 1954, 2042.
9.5. ОКСОКАРБОНОВЫЕ КИСЛОТЫ
Дж. M. Браун {University of Oxford)
Химия оксокарбоновы.х кислот имеет удобную классификацию, связанную с взаимным расположением двух карбонильных групп оксо- и карбоксильной функции. В разд. 9.5.1 по 9.5.5 будут рассмотрены отдельно именно те аспекты синтеза и реакций оксокарбоновых кислот, которые зависят от этого расположения. Некоторые общие аспекты химии оксокарбоновых кислот, и особенно физико-химические аспекты все типов оксокарбоновых кислот и сложных эфиров, будут обсуждены в разд. 9.5.6.
9.5.1. МЕТОДЫ ПОЛУЧЕНИЯ И ХИМИЯ а-ОКСОКАРБОНОВЫХ КИСЛОТ И ИХ ЭФИРОВ
9.5.1.1. Методы получения*
Современные методы синтеза а-оксокарбоновых кислот представлены на схемах (1) —(11). Реактив Кори-Зеебаха— наиболее универсальный ацил анионный эквивалент {схемы (1), (2)} [2, 3],
См. [1].
7»
195
который можно алкилировать; можно также для введения капб ксильной группы на удобной стадии синтеза обрабатывать сопоя жснный анион интермедиата этилхлорформиатом. Для разложена конечных днзамещенных днтпанов удобно применять обработку А-бромсукцинимидом в водном ацетоне. Альтернативный подход общность которого еще точно не известна, заключается в испопь зовании изооктнлизоцианида (1), который реагирует с алкиллитие' выми соединениями, приводя к а-иминолитиевому производному [4]. Это производное можно затем карбоксилировать и получающийся изооктилимин гидролизовать разбавленной кислотой {схема (3)}. Недавние исследования ациланионных эквивалентов [51 позволяют предположить, что станут доступными и другие синтетические подходы этого типа.
Вг2
АсОН,Н2О
ClCO2Et .
№Н,ДМФа
(5)
(6)
CN
COjEt
КОН, 20 °C: Н2о. 100 °C; разб. НС1
30% Н2О2, Na2WO4;
Среди окислительных методов получения {схемы (4) — (6)} ;[6—8] особенно интересны основанные на реакции Риттера для цианогидринов {схема (7)}, так как здесь удается избежать кон-
196
трет-ouun _ —‘-о
(PhCH2)2CHCHOH H2SO;->- (PhCH2)2CHCHOH
CN CONHCMe3
—> (PhCH2)2cHc; (7)
XCO2H
курентного отщепления воды или цианида водорода [9]. Первоначально образующийся трет-бутиламид далее окисляется и гидролизуется с высоким выходом. Енолятные анионы сложных эфиров могут реагировать с этилнитритом [10], давая соответствующие оксимы, которые после обработки формальдегидом в присутствии НС1 можно превратить в сложные эфиры а-оксокислот {схема (8)}.
При конденсации этилового эфира АДУ-дпметилглицина с бензальдегидом получается аддукт, который перегруппировывается с гидридным переносом, давая сложный эфир а-оксокислоты {схема (9)}, по-видимому через промежуточный карбениевый ион (2) [11]. Дианион пировиноградной кислоты можно генерировать in situ в растворе и связывать его с подходящим альдегидом [12] {схема (Ю)}. р,у-Ненасыщекную а-оксокислоту, которая получается в результате этой реакции, можно избирательно восстановить борогидридом калия. Конденсация сложных эфиров с диме-тилсульфинил-анионом по Кори приводит к Р-оксосульфоксиду, который при обработке кислотой перегруппировывается, возможно через (3), с образованием промежуточного продукта; последний в присутствии каталитических количеств ацетата меди (II) легко окисляется до а-оксокислоты [13] {схема (11)}.
Et2NH
EtOH
+ МеСОСОгН
197
9.5.1.2. Реакции
(11)
а-Оксокислоты выступают в качестве интермедиатов зависимого от пиридоксаля биосинтеза аминокислот. Последовательность реакций {схема (12)} состоит в конденсации а-карбонильной группы с первичной аминогруппой пиридоксаля, последующем стереоспецифическом ферментативном переносе протона и гидролизе образующегося имина [14]. Можно имитировать стереоспецифический 1,3-протонный перенос в таком амине, как (4), с помощью простого основного катализа [15], однако более близкие модели ферментативного процесса, по-видимому, недоступны. Наиболее эффективное превращение оксокислота — аминокислота включает много стадий {схема (13)}, где асимметрический имин сначала восстанавливается, а затем вновь окисляется. По этой методике преобладание одного энантиомера может достигать 87% [16].
198
Присоединение реактивов Гриньяра к сложным эфирам а-оксо-кислот протекает избирательно по карбонильной группе. Сравнимую селективность демонстрируют и гидридные восстановители. Если боковая цепь сложного эфира хиральна, эти реакции могут протекать с индуцированием асимметрии; механизм процесса интенсивно изучался [17]. Реакции сложных эфиров (5), (6) и (7) с метилмагнийиодидом типичны и приводят соответственно к избытку 48% S-, 23% R- и 71% /^-энантиомера в образующемся атролактиловом сложном эфире. Эти и другие результаты объясняют с использованием модели (8), которая предполагает вытянутую трансоидную геометрию реагирующего сложного эфира. В большинстве изученных систем асимметрическая индукция довольно мала, однако достаточный набор примеров превращения бензоилформиат—>-атролактат позволяет использовать эту реакцию для определения абсолютной конфигурации хиральных спиртов.
199
Сложные эфиры а-оксокислот могут восстанавливаться N-ал-килникотннампдами в присутствии перхлората магния [18] с образованием соответствующих сложных эфиров а-гидроксикислот И хотя асимметрический центр достаточно удален от места реакции, тем не менее при восстановлении этилбензоилформиата амидом (9) получается 19% преобладания /?-энантиомера этилового эфира миндальной кислоты. Механизм введения асимметрии точно не установлен, однако он может включать координацию иона магния двумя карбонильными группами в переходном состоянии, необходимом для гидридного переноса. Сходным образом эфир Ганча (10) [19] подходит для введения асимметрии при катализируемом ионами цинка восстановлении этилпирувата или бензоилфор-миата (в виде аддукта реакции Реформатского из метилбромаце-тата и цинка). Весьма многообещающим альтернативным методом восстановления сложных эфиров а-оксокислот является каталитическое асимметрическое гидроксилирование кетонов в присутствии лиганда (11) и циклоокта-1,5-диенродий(I)хлорида. С помощью этого метода пропилпируват превращали в пропил-(S)-лактат с оптической чистотой 75% [20].
(10)К=(-)-ментил
Гидратация метилпирувата и производных пировиноградной кислоты по сравнению с превращением простых карбонильных соединений термодинамически более предпочтительна. Равновесие исследовано методом 17О-ЯМР и показано, что константа диссоциации К для гидратов составляет 0,32 для метилпирувата, 0,42 для пировиноградной кислоты и 18,5 для пируват-аниона [21]. Эти значения можно сравнить со значениями К, равными 500 для ацетона и 0,7 для ацетальдегида. Гидратация катализируется НзОл основаниями и бычьей карбоангидразой [22].
Термолиз метилпирувата в газовой фазе при 440 °C дает 90/о метилацетата [23], хотя термолиз аллил- и бензилпируватов приводят к продуктам, образующимся при рекомбинации радикалов. В одном из классических примеров [24] применения метки с для выяснения механизма реакций Калвин и Леммон показали что (12) декарбонилируется при высоких температурах с выбро сом немеченого оксида углерода и с образованием 14С-меченного этилацетата. Декарбонилирование катализируется в кипящем то луоле комплексами родия (I) [25]. Использование этой реакди декарбонилирования в синтезе сложных эфиров производных М 200
ппновой кислоты и Р-оксокислот с помощью последовательности реакций, изображенных на схеме (14), описано в другом месте.
\ NaOEt
(CO2Et)2 + СНХ ЕЮН-
/Х
\cOCO2Et
(14)
X = COR или CO2R
О OEt
(12)
9.5.2. МЕТОДЫ ПОЛУЧЕНИЯ И ХИМИЯ
р-ОКСОКАРБОНОВЫХ КИСЛОТ И ИХ ЭФИРОВ
9.5.2.1. Методы получения [1]
Классическим методом получения сложных эфиров р-оксокар-боновых кислот является сложноэфирная конденсация Кляйзена; в простейшем случае для этиловых эфиров неразветвленных кислот примененные условия мало отличались от условий, опубликованных Гейтером в 1863 г. [26]. При использовании эквивалентных количеств натрия реакция по существу необратима [27], так как рКа эфира р-оксокислоты около 10; для этанола рКа равно 16 [схема (15)]. а,а-Дизамещенные сложные эфиры в присутствии этилата натрия в этаноле не вступают в конденсацию Кляйзена, так как в продукте реакции нет кислых атомов водорода в а-поло-жении по отношению к сложноэфирной группе. Однако поскольку кетоны на 4—5 единиц р/< сильнее как кислоты, чем сложные эфиры, то необратимую реакцию можно провести [28], применив достаточно сильное основание {схема (16)} с тем, чтобы начальный продукт превращался в енолят-ион, понижая таким образом свою свободную энергию.
Н
^XCO2Et
(15)
Ph3C" Na+, Et2O
25 °C
(16)
Циклизация а,со-сложных диэфнров по Дикману [29] — важный метод получения сложных эфиров пяти- и шестичленных циклических оксокислот {схема (17)}, однако в других случаях эта реакция „ редко применима. Подобная циклизация может быть серьезной конкурентной реакцией ацилоиновой конденсации диэфиров адипиновой и пробковой кислот в том случае, если не
201
удалять этилат-анионы, образующиеся в ходе реакции связыва-нием с триметилхлорсиланом [30]. Безрезультатным оказалось применение алкоксидных реагентов в синтезе гомокубанона {схема (18)}, однако использование диметилсульфинилнатряя в диметип-сульфоксиде решило проблему [31].
EtO2C
CO2Et
Na, следы Et ОН ,С7 Hg>v Ас ОН
NaCH2SOMe k
Me2SO, 0°С
(17)
Несимметричные сложные эфиры 0-оксокислот можно получать перекрестной реакцией Кляйзена, или, что более эффективно, по аналогичным реакциям различной реакционной способности двух компонент. Схема (19) иллюстрирует использование фосфорана [32] для синтеза эфиров оксокислот; применение силиловых эфиров енолов сложных эфиров представлено [33] на схеме (20). Последний метод, по-видимому, особенно пригоден для получения с высоким выходом несимметричных соединений.
Карбоксилирование енолятов кетонов [34] {схема (21)}, а также работы [35], в которых енолят-анион генерировали восстанов-лёнием аф-ненасыщенного кетона металлом в жидком аммиаке {схема (22)}, могут быть эффективными путями синтеза р-оксо-кислот.
(19)
(20)
при
Диметилкарбонат магния (реагент Стайлса), получаемый карбонизации метилата магния в диметилформамиде, служит мягким и эффективным реагентом для карбоксилирования кетонов {схема (23)}.. Реакционную смесь нагревают до 120°С, метанол удаляют перегонкой [36]. Обратимо генерируемые еноляты кетонов в присутствии диэтилкарбоната {схема (24)} [37] или диэтил-оксалата {схема (25)} [38] с высоким выходом образуют продукту конденсации. В последнем случае термическое декарбонилированп приводит к сложному эфиру 0-оксокислоты (см. разд. 9.5. М/*
202
Рнамины легко реагируют с этплхлорформиатом {схема (26)} [391 Хотя эфиры енолов по отношению к слабым углеродным электрофилам не реакционноспособны, их триметилснлнльные аналоги [40] в жестких условиях конденсируются с фепилизоцианатом, открывая путь к р'-оксо-М-фениламидам {схема (27)}.
тит?1М«рЬ' кетенов гидролизом или алкоголизом можно превра-жрии™р-оксокислоты или их сложные эфиры. Например, напря-метитг™ Пр°ИЗВОДНОе циклобУтанДиона, полученное из димера ди-тена, в мягких условиях расщепляется по Галлеру —
203
Бауэру {схема (28)} [41]. Хлорсульфонилизоцианат можно пьи соединять к ацетиленам с образованием эффективного источник эчектрофила О = С+—X, что служит прямым методом получения [42] сложных эфиров р-оксокислот в метаноле {схема (29)}
9.5.2.2. Реакции
Количественные аспекты амбидентной реакционной способности сложных эфиров р-оксокислот будут обсуждены в конце этой главы (см. разд. 9.5.6.2). В то время как С-алкилированные продукты с хорошим выходом обычно получают из незатрудненных первичных галогенидов, реакции вторичных галогенидов могут сопровождаться элиминированием, а в случае третичных галогенидов элиминирование становится основной реакцией. В подобных случаях предпочтительным становится взаимодействие енола (скорее, чем енолят-аниона) с карбениевым ионом, или с соединениями, аналогичными карбениевым ионам. Обработка этплацетоацетата трет-бутилбромидом и перхлоратом серебра в нитрометане дает производное (13) с выходом 68% [43]. Этот же сложный эфир реагирует с изопропанолом в присутствии BF3 с образованием С-изо-пропильного производного с выходом 67%; возможный механизм показан в (14).
I н
114)
В достаточно жестких условиях из мстил ацетоацетата мо получить а,а'-дианион [44]. При использовании одного экви лента алкилирующего агента происходит селективное а'-алк рование, например использование в качестве алкилирую^ агента хлорметилэтилового эфира дает (15). Заметное а-аль рование происходит лишь в случае бифункциональных алкилир 204
ЩПХ агентов. Например, 1,3-дибромпропан реагирует с Дианибном, образуя производное циклогексанона (16). Моноанионы сложных эфиров р-оксокислот реагируют с углеродными нуклеофилами селективно по сложноэфирной группе [45]. Так, при последовательной обработке сложного эфира оксокислоты (16) гидридом натрия в тетрагидрофуране и двумя эквивалентами метиллития, получается дион (17).
о о но о
(15) (16) (17)
Гидролиз а-алкилированного сложного эфира р-оксокислоты может приводить к замещенному кетону или к замещенной кислоте. Первый получают при гидролизе сложного эфира и декарбоксилировании, а вторую — в результате первоначальной нуклеофильной атаки на кетонную карбонильную группу и последующей фрагментации. Классическая методика получения кетонов состоит в обработке холодной разбавленной щелочью и декарбоксилировании in situ в результате нагрева или последующего подкисления. Производные кислоты получают реакцией с горячей концентрированной щелочью в спиртовом растворе. Оптимальные условия варьируются в зависимости от конкретного соединения. Так (18) дает при реакции с NaOH при 100°С производное адипиновой кислоты (19) с выходом 90%; обработка 48% НВг [46] соединения (20) ведет к циклопентанону (21), выход 80%.
Эти реакции довольно капризны, поэтому предпринято много попыток проконтролировать направление расщепления более точно. Расщепление по Майервсйну [47] состоит в нагревании сложного эфира с водой под давлением, или в случае более реакционноспособных соединений — в перегонке с паром. Сложные эфиры оксокислот (22), (23) и (24) дают соответственно при 240; 130 и 100 С кетоны в результате гидролиза и декарбоксилирования. Бензиловые сложные эфиры можно получать путем переэтерификации [48]; их далее можно расщеплять гидрогенолизом над палладием в кипящем этилацетате. В этих условиях сложный эфир (25) дает непосредственно кетон (26), так как декарбоксилирование промежуточного продукта происходит достаточно быстро, трет-Бутиловые сложные эфиры, такие как (27), можно расщеплять в весьма
205
мягких кислых условиях [49]. В этом случае последовательность реакций включает получение трет-бутилацетоацетата, его алкили рование и расщепление кислотой.
Реакционноспособные сложные эфиры, такие как /?-нитрофе-нилацетоацетат [50] могут гидролизоваться по механизму Е\сЬ, что определено по отсутствию зависимости скорости реакции от концентрации анилина при образовании ацетоацетанилида в водном буферном растворе. Ацилкетеновый интермедиат (28), вовлеченный в этот процесс, образуется при гидролизе S-ацетоацетил кофермента А (29); этим может объясняться его биологическая значимость [51].
(28)
(31)
(29)
Р-Альдолактоны претерпевают катализируемую кислотой перегруппировку [52], приводя, например в случае (31), после обработки НС1 в метаноле к (32) через образование ациклического интермедиата (33). Эта последовательность реакций использована в элегантном синтезе индольного алкалоида аймалицина (30) рЗ|-
Енолят-анионы сложных эфиров р-оксокислот взаимодействую с координированными олефинами, давая таким образом альтерн
206
w
ci 01
Т„ВЮЙ тип образования
ладайднхлорм превращается в функциональное цнклооктеновое производное {схема (30)}.
9.5.3. МЕТОДЫ ПОЛУЧЕНИЯ И ХИМИЯ у-ОКСОКАРБОНОВЫХ КИСЛОТ И ИХ ЭФИРОВ
9.5.3.1. Методы получения*
говоря, 1,4-дикарбонильные соединения труднее полу-
Вообще
чить, чем 1,3- (прямая конденсация) или 1,5- (присоединение по Михаэлю) дикарбонильные соединения; это видно из относительно небольшого числа разработанных синтетических подходов к производным у-оксокислот. Кислотная функция может быть введена реакцией Манниха [55] с последующим получением четвертичного амина, замещением цианогруппой и гидролизом {схема (31)}. Енамины реагируют со сложными эфирами а-бромкислот, образуя соответетвующий замещенный енамин, который можно избирательно гидролизовать [56] с сохранением сложноэфирной функции {схема (32)}. Аналогична по своему принципу реакция илида а-оксопиридиния со сложным эфиром а-бромкарбоновой кислоты. В этом случае [57] получающуюся пиридиниевую соль восстанавливают цинком в уксусной кислоте {схема (33)}. Эквиваленты ацил-анионов сопряженно присоединяются к сложным эфирам аф-ненасыщенных кислот. В качестве примера на схеме (34) приведена реакция, где карбонильная группа активирована и представлена в виде цианогидрина [58]. Альтернативно, ионы цианида и тиазолия можно применять в качестве катализаторов реакции альдегидов (ароматических) со сложными эфирами ненасыщенных кислот, таким путем осуществлено значительное число превращении. На схеме (35) показано применение алкилиденянтарной кислоты для синтеза у-оксокислот [59]
* См- [1],
207
оксокисл^
Хорошими предшественниками сложных эфиров леНИда являются ацетилены. Присоединение терминального а эфИру) ко-этилбромацетату приводит к (3,у-иноатному слоЖНОМ"окСОКислоты торый можно гидратировать до сложного эфира у- прцво-{схема (36)} [60]. Последняя реакция региоспецифи^ эф'
дит к сложным эфирам у-оксокислот, большей чавтЬ1нОд группой фекта дестабилизации карбенневого иона сложноэфир с блй' который способствует протонированию ацетиленовой rpHa;V жайшей к сложноэфирной группе стороны. Взаимоде которь1*1 килборанов [61] с этилдиазоацетатом приводит к 11
Ж
без дополнительной очистки перегруппировывается с миграцией одной алкинильной группы. Протонолиз образующегося борана проводят обычным образом, и полученный иноатный сложный эфир гидратируют {схема (37)} аналогично описанному выше методу {см. схему (36)}. Более сложный подход гидроборирования Пел-тера и сотр. [62] {схема (38)} включает реакцию аниона алкилбо-роната с бромуксусным эфиром. Превуалирующим путем является нуклеофильная атака по |3-углероду; промежуточный илид претерпевает алкильную миграцию, как показано на схеме. Окисление образующегося винилборана пероксидом водорода дает целевой сложный эфир оксокислоты с высоким суммарным выходом.
О
____ HgSO4, H2SO4 II
- \co2Et Et0H, н2о * (36)
о
Катализируемое металлами карбонилирование иногда дает удобный путь синтеза циклических у-оксокислот {схема (39)} путем последовательного внедрения и гидролиза ацилметаллического промежуточного продукта [63].
СО, МеОН, 150 "С, ЮООатм;
' (Ви3Р)2Р<112
(39)
^Н2СО2Ме
9.5.3.2. Реакции
При дегидратации у-оксокислот образуются ненасыщенные лактоны [64]. Реакция может протекать без добавляемого катализатора, и, например, кипячение оксокислоты (34) в толуоле с водяным сепаратором Дина — Старка приводит с высоким выходом к (35). В присутствии кислого катализатора могут происходить
20?
перегруппировки. Например, оксокислота (36) в присутстви лпфосфорноп кислоты перегруппировывается в продукт (37) И По' стадию алкильной миграции промежуточного катиона (3§) ЧеРез
Ароматические у-оксоки,слоты получают реакцией Фриделя -Крафтса янтарного ангидрида или его производных с аренами [65]. Получающиеся соединения служат важными интермедиатами в реакциях аннелирования {например, по схеме (40)}. Карбазол избирательно ацилируется в положение 3 [66]; диацилирование приводит к 3,6-производному. Восстановление по Клеменсену с последующей циклизацией в 80%-ной серной кислоте и ароматизацией дает лишь одно дибензопроизводное, вероятно в силу сте-рических затруднений, препятствующих образованию 3,4; 5,6-изомера и на стадии внутримолекулярного ацилирования.
Zn, HCI; -----------> 80% H2SO4:
Pd, Н2
9.5.4. МЕТОДЫ ПОЛУЧЕНИЯ И ХИМИЯ 6-ОКСОКАРБОНОВЫХ КИСЛОТ И ИХ ЭФИРОВ
9.5.4.1. Методы получения *
Сопряженное присоединение стабилизованных енолят-ани к сложным эфирам непредельных кислот протекает в мягк
* См. [1].
2Ю
вия.х необратимо, как показано на схемах (41) [67] и (42) [68]. При использовании в качестве карбанионной компоненты сложного эфира оксокислоты а-сложноэфпрную группировку можно избирательно отщепить кислотным гидролизом. Аналогично взаимодействуют со сложными эфирами непредельных кислот енамины [69] {схема (43)}. Вначале образуется напряженный дизамещен-ный циклобутан, который при кислотном гидролизе претерпевает раскрытие цикла в результате фрагментации, как показано в (39). Региоселективность этой реакции в случае енаминов, полученных из а-замещенных кетонов, зависит от растворителя. По-видимому, два замещенных енамина находятся в быстром, по сравнению со скоростью реакции конденсации, равновесии, и пропорции конечных веществ отражают их индивидуальные концентрации и константы скорости реакции с этил акрил атом. Четвертичное соединение образуется в значительной степени лишь в диоксане, когда оно составляет 35% продукта {схема (44)} [70].
Часто используют присоединение стабилизованных анионов, полученных из эфиров малоновой кислоты, к ненасыщенным альдегидам и кетонам [71, 72] {схемы (45), (46)}. В последнем примере продукт был далее использован в полном синтезе метилжас-моната [72].
Стабилизованный бензотропон (40) требует более жестких условий для присоединения аниона диэтилмалоната, однако реакция может проходить [73] с хорошим выходом {схема (47)}. Ненасыщенные сложные эфиры 6-оксокислот можно получать сопряженным присоединением малонат-аниона к |3-галоген-а,|3-ненасы-щенным кетонам [74] {схема (48)}
211
Циклизация продукта первоначального присоединения мпх происходить, как показано на схеме (49). Простой адденд м т хаэля можно получить из 4-гидроксицнклопентен-2-она (41) и‘ скольку кинетически контролируемый продукт присоединен0 имеет трп«с-расположепные гидроксильный и алкильный замес"Я телн, и поэтому образование лактона стереохимически невыгод11 (тушнс-раскрытие пятичленных колец) {путь (а) на схеме (49П В более жестких условиях реакции присоединение обратимо ' поэтому образуется небольшая часть гр/оизомера, оор
зует лактон, давая в конечном счете основной продукт {путь (б) на схеме (49)}. Циклизация продукта начального сопряги,Д„. прнсоединения может также проходить при альдольной [76] {схема (50)} или ----
[77].
и который обра-1'”"” г- J сопряженного реакции при конденсации Кляйзена {схема (51)}
^Х"СО2Е1
XfeOH или
О ’
212
(44)
Z\^\ci + EtOMgCH(CO2Et)2 //'\^X\CH(CO2Et)2 (48)
CO2Et
Если в присоединении по Михаэлю применять оптически активное основание, то получается оптически активный продукт. Это может быть результатом стереоселективного депротонирования хирального сложного эфира оксокислоты или проистекать в силу предпочтительного ионного взаимодействия (взаимодействие ионных пар) аммониевого иона и одной диастереотопной стороны иона енолята сложного эфира оксокислоты. Пример [73] такого асимметрического присоединения приведен на схеме (52).
Гидролитическое раскрытие цикла циклогександионов-1,3 приводит к 6-оксокислотам {схема (53)} [79]. Существует и много дру! их путей, ведущих к подобным соединениям, включая окисление циклических предшественников. Например, г{«с-гидроксилиро-вание 1-метилциклобутена и окисление образовавшегося диола перманганатом приводит [80] к 4-оксопентановой кислоте {схема (54)}. При жестком окислении третичных циклопентанолов получают 6-оксокислоты. Пример, представленный на схеме (55), иллюстрирует удобный путь, ведущий к орто-диалкплированным аренам [81]. Окисление циклопентанонов, например туйона (42) перманганатом, проходящее, по-видимому, через образование
213
енола, приводит к соответствующей оксокислоте {схема (56)} [82]. Эпоксидирование циклических сопряженных кетонов можно проводить пероксидом водорода в щелочной среде. В более жест-
ких условиях проходит расщепление эпоксидного цикла в результате атаки гидроксида (или гидропероксида?) на карбонильную группу; примеры подобных реакций приведены на схемах (57) [83] и (58) [84].
СгОд, H2SO4
(55)
NaOH, Н2О2
EtOH
Инамины региоселективно присоединяются к ненасышенн х карбонильным соединениям; образующиеся циклобутены в 1
214
ИЛИ основных условиях легко гидролизуются [85] {схема (59)). Новый асимметрический центр образуется на последней стадии; стереохимический ход реакции зависит от того, применялась ли для гидролиза кислота или щелочь. Механизм кислотного катализа требует медленного протонирования по углероду с наименее стерически затрудненной стороны, последующего гидролиза енам-мониевого иона и расщепления образующегося дикетона. В случае основания промежуточный продукт ретроциклоприсоединения захватывается, и свободное вращение на стадии еноламида вызывает обращение стереохимии протонирования.
Одновременное присутствие удобно расположенных карбонильных групп разной реакционной способности делает сложные эфиры 6-оксокислот важными синтетическими интермедиатами. Вот несколько примеров из химии природных соединений. Так, промежуточный сложный эфир 6-оксокислоты, представленный на схеме (46) [72], — ключевое вещество в синтезе метилжасмоната. Элегантное расширение цикла в реакции Эшенмозера и сотр. [86], приводящее к синтезу (±)-мускона, требует классического присоединения по Михаэлю к этоксикарбонилциклододеканону для генерации предшественника бициклического интермедиата {схема (60)}. Циклизацию проводят восстановлением борогидридом натрия с последующей обработкой полученного сложного эфира 6-гидроксикислоты полифосфорной кислотой.
Важным синтетическим интермедиатом является легко получаемый ненасыщенный бифункциональный сложный эфир Хаге-манна (43). Это соединение можно алкилировать в основной среде, хотя получающийся при этом амбидентный карбанион может реагировать как по а-, так и у-положениям. Например, при алкилировании изопропилбромидом в присутствии этилата натрия получают как (44), так и (45) [87]. Карбонильную группу в (43) можно защитить кетализацией. Эта реакция (но не тиокетализация)
215
сдвигает двойную связь, вызывая ее сопряжение со эфирной группой [88]. В полном синтезе прогестерона Сл°Ж|ю сону продукт (46), являющийся предшественником^ (47)П°б'^>К0н-пользован как компонент кольца A. ' ис-
Цитотоксичный макролид вермикулин (48) получеь
ного эфира Хагсманна (43) через диенол ацетат (49)' Т СЛо>к’ образуется непосредственно из (43) [89]} и далее поевпКОТОрый в интермедиат (50). Циклогексановое кольцо, изначала еНИе‘м ствующее в сложном эфире Хагеманна, последовательно П₽ИСут' ляется с помощью окисления и становится значительнойРасЩеп’ углеродного скелета вермикулина (48). и Частью
(47)
216
(48)
ОАс
i49)
9.5.5. МЕТОДЫ ПОЛУЧЕНИЯ И ХИМИЯ е-ОКСОКАРБОНОВЫХ КИСЛОТ, ИХ ЭФИРОВ И ВЫСШИХ ГОМОЛОГОВ
9.5.5.1. Методы получения *
Методы получения высших оксокарбоновых кислот и их производных аналогичны методам, описанным в предыдущих разделах. Взаимодействие енамина {схема (61)} с метил-3-бромкротонатом протекает по механизму Sw2, а не по альтернативному Sw2/-nyTH, и приводит к ненасыщенному продукту, гидрирование которого [90] дает сложный эфир е-оксокислоты.
Поскольку часто используются окислительные методы, следует отметить применение эфира дициклогексил-18-краун-6 для солюбилизации перманганата калия в бензоле. Получаемый раствор хорошо окисляет олефины, например расщепляет бицикл в пинене [91] {схема (62)}. Окислительного расщепления циклических кетонов и спиртов можно достичь действием хромового ангидрида в уксусной кислоте [92] {схема (63)} или фрагментацией а-гидропероксида, получающегося при захвате енолят-аниона кислородом. В более жестких условиях можно расщепить оба кольца декалона-1 [93] {схема (64)}. Окисление по Байеру — Виллигеру бицикло[3.2.1]октанона-2 лг-хлорпербензойной кислотой с последующим раскрытием лактонного цикла метанолом и окислением хромовым ангидридом использовано как путь к сложному эфиру 8-оксокислоты {схема (65)} [94].
(61)
* См. [1].
217
Реакция
* '-опция, предста п по
Щ^лочмк°ВЫМ расщеплениемЯ В диСХеМе ^66^’ является по существу та ™ЫЫХ условиях [95] пп , Дикетонов; она протекает в мягких в кетоиапРИЙТеТракаРб°нилфеппятаИе металлоРганического реаген-нуклрпЖ В пРис>’тствии сложил^73, П03в0Ляет превратить бромид 5^2 прЯФиЛЬНь1Й Реагент заметяФирИОи ГРУППЫ (96]. Этот высоко сидзРигпВДИ С П0СЛеДУЮщей быр£Т [алоген 0 результате обычной
У лерода. Добавленный стРОи Цис~ л иг а ид ной миграцией ок-218 ОДэтан улавливает ацплтетракарбо-
нилферрат(—1) интермедиат; кетонный продукт образуется при последующем синхронном отщеплении лиганда {схема (67)}.
Производные высших оксокислот можно получать расщеплением циклов или селективным подбором функциональных групп. Окисление моноэпоксида циклооктадиена-1,5 диметилсульфоксидом и последующее расщепление ацилоина тетраацетатом свинца приводят к w-кетоальдегиду {схема (68)}, который далее превращают в (51), половой феромон самки хлопковой совки. Успех этого синтеза, возможно, указывает на вероятный общий метод превращения сложного диэфира а,w-дикарбоновой кислоты в сложный эфир а,со-оксокислоты через образование ацилоина и его расщепление.
.«-CICgKjCOjH
CHCJ3
Me2SO -------->-воздух, r
11О»С
(68)
Pb(OAc). EtOH
-------£-------у
(£)-+ (Z)- OCOMe
(51)
SOCI2; у 11 JI (ch2oh)2
OMe LtCtiMe2, л-TsOH
JS L a U Li
Пример селективного подбора защитных групп в дикарбониль-ном соединении с длинной цепью показан на простом синтезе
219
вещества пчелиной матки [98] {схема (69)}. Кислотную (к, можно превратить в присутствии сложноэфирной груППЬ1 уНКЦ1>Н) ную обработкой тионилхлоридом и диметилкупратом литцКеТОн’ щита кетона в виде диоксолана позволяет ввести двойную** по соседству со сложноэфирной группой с помощью сульфенцпВЯ31> ния а-аниона и термического элиминирования метилсульфиг ’ кислоты из соответствующего сульфоксида. v НоМ
Э.5.5.2. Реакции
Катализируемая кислотой циклизация £-оксокислот протека по-видимому, путем электрофильной атаки ацилиевого иона ил' его эквивалента на енол кетона. Так, (52) реагирует с полифос* форной кислотой в уксусной кислоте при 100 °C, давая (53) [991' В основной среде циклизация может идти по типу конденсации Кляйзена. Синтез перистилана (54) [94] требовал ступенчатого построения периферических пятичленных колец; получение (55) проводилось или по схеме (65), или альтернативным методом включающим присоединение подходящего медьорганического реагента к циклопентен-2-ону. Сложный эфир оксокислоты (55) под действием метилата натрия в метаноле легко циклизуется в (56), однако поскольку реакция легко обратима, наилучший метод выделения этого неустойчивого дикетона состоит в гашении реакционной смеси раствором дигидрофосфата калия.
(54)
9.5.6. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА оксокарбоновых кислот
9.5.6.1. Кислотность кислорода и углерода; кето-енольная таутомерия
Кислотность оксокарбоновых кислот характеризуется мыми закономерностями (табл. 9.5.1), и поскольку анион-стао зующий индуктивный эффект карбонильной группы быстро хает по мере удаления от карбоксильного центра, 5-оксоге вая кислота лишь немного сильнее, чем гексановая.
220
Содержание енольной формы сложных эфиров р-оксокислот во многих случаях точно известно (табл. 9.5.1) и может быть определено химическими методами с помощью быстрой реакции енолов
Таблица 9.5.1. Некоторые физические свойства оксокарбоновых кислот и сложных эфиров fi-оксокарбоновых кислот [1д0а]
Кислота Сложные эфиры р-оксокнслот
структура рда структура Р*а % енола
10,65 8 (чист.)
0,39 (в Н2О)
12,25 5 (чист.)
. 0,29 (в Н2О)
12,50 1 (чист.)
0,17 (в Н2О)
13,5 о,О4 (в Н2О)
89 (чист.)
с электрофильными реагентами, такими как бром, или с помощью непосредственных спектроскопических измерений, чаще всего методом ЯМР. В отличие от р-дикетонов енольная форма р-оксокис-лот обычно является минорным изомером; исключения наблюдаются лишь для чистых соединений в жидком состоянии с элек-троннооттягивающими a-заместителями (CF3, CN). Кето-енольное равновесие зависит от растворителя. Например, в чистом этилацето-г?о;тате 8°/о енола, а в разбавленном циклогексановом растворе — 51 /о. Поскольку енольная форма менее полярна и более летуча, разделение таутомерной смеси можно осуществлять перегонкой в кварцевой аппаратуре при очень низких давлениях. При перегонке
221
при атмосферном давлении метилацетоацетат дает смесь жащую до 15% (£)-изомера (57), хотя (2)-изомер (58)’с в ₽' родной связью термодинамически более устойчив [100]. дей ОДо' тельно, ультрафиолетовый импульсный фотолиз равновесной к’ енольной смеси этилацетоацетата в гептане [101] переводит^0’ бильный (Д)-енол в его (£)-изомер, который вновь изомеризуе? в (Z)-форму с периодом полупревращения, равным нескольк минутам при комнатной температуре. По-видимому, процесс вкл*М чает внутримолекулярный перенос протона с образованием по ' межуточного енольного эфира (59), минуя таким образом стабиль ную кетонную форму. При стабильном облучении, однако, отмечена слабая фотокетонизация (58). Более высокая стабильность (Z)-енола объясняет понижение содержания енольной формы при увеличении стерического объема а-алкильных заместителей (см табл. 9.5.1).
Me О
HO^^OEt
(57)
9.5.6.2. Алкилирование анионов эфиров 0-оксокислот
Перенос протона к а-углеродному атому сложных эфиров р-оксокислот и от него в протонных растворителях протекает медленно; перенос протона к p-карбонильному кислороду и от него — быстро [102]. Тем не менее алкилирование аниона енолята сложного эфира р-оксокислоты алкилгалогенидами проходит преимущественно по атому углерода, хотя более «жесткий» электрофил (имеющий более карбенийионный характер в переходном состоянии по типу Sn2) этилтозилат в диполярных апротонных растворителях [103] приводит преимущественно к О-этилированию. результаты недавних количественных исследований алкилирования представлены в табл. 9.5.2 (ДМЭ — диметоксиэтан, остальные сокращения— обычные).
Ион металла и степень вовлечения аниона в образование ион ных пар или ионных агрегатов играют важную роль lC1 • табл. 9.5.2(г)]. Кажущиеся различия в тенденции к О-алкилир ванию при замене Na+ на К+ с различными субстратами не по. чили четкого объяснения. В эфирах бензолконденсированн циклических [3-оксокислот возрастание степени О-алкилирова совпадает с увеличением размера цикла. Интересное изменение р. гиоселективности при сравнении реакций (60) и (61) с этил Р^_ пропионатом {см. табл. 9.5.2(e)) может отражать повышение _0. пости заряда на кислороде в соединении (61). Хелатное вза
222
Таблица 9.5.2. Алкилирование и ацилирование сложных эфиров $-оксокислот
(а) С- и О-алкилирование. Влияние уходящей группы [101а—в]
Анион | м+ | • RX Растворитель | % О-алкили-рования
Na EtI ДМЭ 1
Na EtBr ДМЭ 2
Na EtOTs ДМЭ 13,1
Na EtSO3F ДМЭ 17,2
Na EtSO3CF3 ДМЭ 21,2
° \ К MeOSO2Me ГМФТА 61
OEt К Mel ГМФТА 5
К MeOTs ГМФТА 89
(б) С- и О-адкилирование. Влияние растворителя [101 в, г] ..
Анион M + RX | Растворитель % О-алкили-рования
К BuCl Me2C0 10
К BuCl CH3CN 19
к BuCl ДМС0 47
OEt к BuCl
q=0 51
NMe
к MeOSO2Me Диглим 34
EtO—« к MeOSO2Me ДМФА 44
0 к MeOSO2Me S02 86
(в) С- и О-алкилирование. Влияние краун-эфира, криптандов и катализа ____________________фазового переноса [ 101 д]
Оксоэфир/анион Основа ние/М+ RX Растворитель % 0-алкилн-рования (EIZ)
\ Bu4N+OH“ PrI H2O/CH2C12 0
7=° Bu4N+OH* ИЗО-PrI H2O/CH2C12 25
У=о Na+ EtOTs ТГФ 9 (0,1)
ОМе
223
Табл' 9 5 2 (одолжены
(г) С- и О-алкилирование. Влияние иона металла [101а\ "
Аннон М+ RX Растворитель % °"алкилип0
аания
°4 Li’ EtI ДМЭ < 1
О—/ Cs+ EtI ДМЭ 2
OEt Bu4N+ ЕН ДМЭ 106
Li+ EtOTs ДМЭ 32
Cs+ EtOTs ДМЭ 37
Bu4N+ ЕЮ 1s ДМЭ уд
(д) Ацилирование этилацетоацетата [lOle]
Анион/оксоэфир M+ RCOX Растворитель Ь/и \£/ZJ
О о Na+ PhCOCl ДМЭ 99:1 (низкое)
р _ 11 Е12О/ГМФТА (2,57)
(1:1)
OEt м + Na+ МеСОС! ДМЭ 28:72
Е12О/ГМФТА (1,27)
(1:1)
Na+ грет-ВиСОС! ДМЭ 4:96 (низкое)
Е12О/ГМФТА (5,67)
(1:1)
Na+ МеСН=СНСОС1 ДМЭ 34:66 (низкое)
О О Е12О/ГМФТА (11,5)
AJU °е‘ (1:1) MeCOCl Пиридин (2,3)
1 ОМе - МеСО2СОМе Пиридин (5,3)
МеС/ , Н3О+> чист. (0,05)
м
(е) Алкилирование сложных эфиров циклических fl-оксокислот [101ж]
% О-алкилн
Анион М4 RX Растворитель ровання
О О Na+ II II У' ДМСО 15
SA- к’ ДМСО 0
Na+ /Х/Хвг ДМСО 14
224
Табл. 9.5.2. (продолжение)
46,5
CO2Et Br
Вг
CO2Et
Вг
| : ДМФА (5:2)
62,1
К’
CO2Et
Br
75,0
CO2Et
85,0
действие ионной пары становится менее предпочтительным из-за уменьшения углов а и а' экзоциклических связей.
Катализ фазового переноса — важный синтетический метод для анион-электрофильных реакций; он применялся при реакции метилацетоацетата с алкилиодидами. В случае первичных субстратов не обнаружено О-алкилирования, хотя для изопропилиодида это была заметная конкурирующая реакция [см. табл. 9.5.2(e), примеры (1) и (2)]. В случае всех первичных иодидов найдены ощутимые количества продуктов диалкилирования.
а Зак. зэа 225
9.5.6.3. Декарбоксилирование а- и р-оксокислот
Тепмолиз а-оксокислот приводит к разнообразным продуктам, игктеоФилы катализируют декарбоксилирование. В простейшем лхчае таким нуклеофилом может служить цианид-иоп, и обрати. МНР образование а-цианогидрина (62) приводит к анион-стабили-чпванному состоянию, ускоряющему отщепление диоксида угле-пХ от карбоксилат-иона. Декарбоксилирование, катализируемое п прионной среде тиазолиновыми солями, протекает по близкому механизму {схема (70)} [104] и служит моделью тиамин-зависи-меланп^пд^______________ „ __3 жирных кислот.
мого действия карбоксилазы при биосинтезе
ваниееРЛпТгп! И втоРичнь1е амины катализируют декарбоксилиро-мулы ГК'П о Г°т LIePe3 образование цвиттер-ионов общей фор-’ -Ами«°-2-оксиндол в 650 раз эффективнее как ката-томмпгп’ ЧеМ Этиламин> поскольку в результате таутомерного про-сДвига в первоначально возникающем имине образуется ), имеющий низкую энергию декарбоксилирования [104].
шее КИСЛИТеЛЬН°е декаРб°ксилирование а-оксокислот можно осу-твить с помощью фотохимически генерированного 1Д5 синглет-го кислорода [105]. Вероятный механизм превращения включает рвоначальное образование пероксикислоты и ее последующую реакцию со свободной а-оксокислотой {схема (71)}. Промежуточной пеРоксикислоту не удается выделить в растворе, однако ее Р сутствие; следует из эксперимента, в котором адсорбированная нионообменной смоле а-оксовалериановая кислота фотоокси-нировалась и затем взаимодействовала с циклогексеном.
22G
Р-Оксокислоты легко декарбоксилируются как по анионному, так и по нейтральному механизму, и поэтому было проведено несколько исследований на чистых соединениях. Поскольку декарбоксилирование бензоилуксусной кислоты в бензоле показывает изотопный эффект (кн/кг> = 1,4), то из этого было сделано заключение о механизме через циклический (65) [104]. Стереоэлек-тронные препятствия ингибируют декарбоксилирование. Так, оксокислота (66), декарбоксилирование которой должно приводить к енолу, запрещенному правилом Бредта, стабильна при 250°C [106], в противоположность кислоте (67), которая теряет диоксид углерода при нагревании до 145 °C. В последнем случае промежуточный енол получается из бицикло [3.3.1] нонена-1 (2); соответствующий родоначальный углеводород выделен и охарактеризован [107]. Сложный эфир Меервейна (68) в достаточно жестких условиях может быть полностью гидролизован и декарбоксилирован [108].
Первичные и вторичные, но не третичные амины катализируют Декарбоксилирование Р-оксокислот, причем стадией, определяющей скорость реакции, является образование имина {схема (72)}. Нейтральный иминный интермедиат в 3-105 раз реакционноспособнее, чем нейтральная ацетоуксусная кислота. Катализируемое анилином декарбоксилирование щавелевоуксусной кислоты, по-видимому, является исключением [104], так как химические и
8*
227
„Р указывают на то, что интермедиатом, при-кинетические данные укал 10В> служит протонированный
водящим к ^^Ферментативное Декарбоксилирование £.ОКсо. каобиноламин (69). промежуточным образованием имина По кислот может протекД™С ная карбонильная группа конденед. механизму, в КОТ°РпИзина в активном центре фермента j: образо-пуется с остатком лизина чае ацетоацетат-декарооксилазы
нтипем 6-нМИНОКИСЛОГЫ. уловить действием боротидрида
[109] этот интермедиат м ингибированию. Такой ые.
натрия, приводящееа^а°иеР цвиттерионного N-протонированного ханпзм включает образован» ование „МИна.АО"»»"™»
нмннокарбоксилата обрати фИ катализирует обменную
подтверждено Кетонной карбонильной группы зце-
реа Ц ___ лnvj
то ацет ат а и воды.
CN
CN
HN+ О
HN
О О NH2
II II ch2cn
CN
CN
HN+
(72)
N О
О
б (R=M<?)
ведевоуксусной кисло-1ЛЛ0В. Соли М3+ эффек-п-1лгЯ и в случае
Декарбоксилирование производных ща1 ты,таких как (70), ускоряется нонами металлов тивнее солей М2+, хотя заметный катализ наблюдался
228
ионов меди [110]. Механизм превращения может быть кинетически сложным; добавление аминов или других лигандов может втиять на скорость реакции. Кинетические исследования [110] Си2+-катализируемого декарбоксилирования (70а) допускают три кинетических события: быстрое декарбоксилирование, сопровождаемое енолизацней, протонированис пирувата меди(II) и медленное декарбоксилирование. Добавление таких аминов, как фенантролин, в концентрациях, сравнимых с Си2+-ионами, ингибирует стадию' енолизации, но ускорение декарбоксилирования подавляется образованием биядерных комплексов.
9.5.6.4. Внутримолекулярный катализ в химии оксокислот *
Кинетический анализ скоростей иодирования оксокислот в буферных растворах обнаруживает вклад внутримолекулярных термов /сна и кх~. В случае 5-оксогексановой кислоты оба терма действуют с максимумом эффективности, которая быстро затухает при увеличении п в МеСО(СН2)«СО2Н. Ускоряемый основаниями терм Яд- действует наиболее эффективно при п =* 2, 3, 4, и поскольку реакция имеет нулевой порядок по иоду (лимитирующая стадия — перенос протона), это указывает на механизм, подобный показанному в (71), эффективность которого максимальна, когда перенос протона проходит через шестичленное переходное состояние. Это подтверждается тем фактом, что соответствующие сложные эфиры не демонстрируют внутримолекулярного катализа. Ацетоуксусная кислота бромируется значительно быстрее этилацетоацетата, хотя в случае ацетоацетат-аниона не отмечено никакого увеличения скорости. Здесь постулируется, что карбоксильная группа действует как общий кислотный катализатор, активирующий карбонильную группу.
ЛИТЕРАТУРА
1. О классических методах синтеза см.: D. Si. С. Black, G. М. Blackburn, and G. A. R. Johnson, In «Rodd’s Chemistry of Carbon Compounds», 2nd edn., ed. S. Coffey, Elsevier, Amsterdam, 1965, chapter 16; I. T. Harrison and S. Harrison, «Compendium of Organic Synthetic Methods», Interscience, New York, 1974, vol. 2.
2> Truce and F- E- Roberts, J. Org. Chem., 1963, 28, 961; E. J. Corey and B. W. Erickson, ibid., 1971, 36, 3553.
U LC,E!i>el and A- A- Hartmann, J. Org. Chem., 1972, 37, 505.
« Walborsky, W. H. Morrison, II, and G. E. Niznik, J. Amer. Chem. Soc., , 1970 92 6675. ibid ( 1969> gi /778.
b. U. Schollkopf and H. Beckhaus, Angew. Chem. Internat. Edn., 1976, 15, 293;
L». Seebach and F. Lehr, ibid., 1976, 15, 505.
? л b^CC7hr1’ L' CagUoti>, and P. Zappelli, J. Org. Chem., 1973, 38, 3653.
o' >; , Ниманис H. О. Салдабол, С. А. Гиллер. Ж. орг. хим., 1972, 8, 2618.
В. М. Igarashi and Н. Midorikawa, J. Org. Chem., 1963, 28, 3088.
* См. [Ш].
229
16.
17.
18.
о J. Anatol and A. Medele, Compt. rend, (c), 1971, 272, 1157; Svnthf.=' 538- Bull. Soc. chim. France, 1972, 189. ^"thesis, i971
10 . R. Fischer and T. Wieland, Chem. Ber., 1960, 93, 1387.
11 L. Horner and E. 0. Renth, Annalen, 1967, 703, 37.
12 C. G. Wermuth, Bull. Soc. chim. France, 1966, 1435.
13' G. A. Russell and G. J. Mikol, J. Amer. Chem. Soc., 1966, 88, 5498
14 . K. R. Hanson and I. A. Rose, Accounts Chem. Res., 1975, 8, 1.
R. D. Guthrie, D. A. Jaeger, W. Meister, and D. J. Cram, J. Amer Chem
1971, 93, 5137; D. A. Jaeger and D. J. Cram, ibid., 1971, 93, 5153 Soc’ S. Yamada, N. Ikota, and K. Achiwa, Tetrahedron Letters, 1976, iooi J. D. Morrison and H. S. Mosher, «Asymmetric Organic Reactions» PrP < Hall, New Jersey, 1971, p. 64 [Дж. Д. Моррисон, Г. Мошер. Асимметпн?' ские органические реакции. — Пер. с англ. М., Мир, 1973]. *
У. Ohnishi, М. Ragami, and A. Ohno, J. Amer. Chem. Soc 1975 Q7 4766. 4 ’
19. К. Nishiyama, N. Baba, J. Oda, and Y. Inouye, J. C. S. Chem. Comm., 19-f
20. I. Ojima, T. Kogure, and Y. Nagai, Tetrahedron Letters, 1974, 1889.
21. P. Greenzaid, Z. Luz, and D. Samuel, J. Amer. Chem. Soc., 1967, 89 749 H. M. Menzel, Ber., Bunsengesellschaft Phys. Chem., 1974, 78, 89.
22. Y. Packer, J. E. Meany, and C. Zadorojny, J. Phys. Chem., 1971, 75, 799 Y. Packer, J. E. Meany, and В. C. Davis, Biochemistry, 1974, 13, 14H.'
23. R. Louw and E. C. Kooyman, Rec. Trav. chim., 1967, 86, 1041.
24. M. Calvin and R. M. Lemmon, J. Amer. Chem. Soc., 1947, 69, 1232.
25. K. Kaneda, H. Azuma, M. Wayaku, and S. Teranishi, Chem. Letters, 1974 215.
26. A. Geuther, Jahresbericht uber die Fortschritte der Chemie, 1863, 323; 1865, 302.
27. C. R. Houser and В. E. Hudson, Jr., Org. Reactions, 1942, 1, 266 [¥. P. Хаузер, Б. E. Хадсон. Орг. реакции, сб. 1. — Пер. с англ./Под ред. А. Я. Берлина. М., Издатинлит, 1948, с. 345].
28. R. Н. Hasek, R. D. Clark, Ё. U. Elam, and R. G. Nations, J. Org. Chem., 1962, 27, 3106.
29. J. P. Schaefer and J. J. Bloomfield, Org. Reactions, 1967, 15, 1; H. 0. House, «Modern Synthetic Reactions», Benjamin, California, 1972, p. 740.
30. K. Ruhlmann, Synthesis, 1971, 236; J. J. Bloomfield, D. C. Owsley, ana J. M. Nelke, Org. Reactions, 1976, 23, 259.
31. C. G. Chin, H. W. Cuts, and S. Masamune, Chem. Comm., 1966, 880.
32. H. J. Bestmann and H. Kolm, Chem. Ber., 1963, 96, 1948.
33. M. W. Rathke and D. F. Sullivan, Tetrahedron Letters, 1973, 1297.
34. J. Sicher, F. Sipos, and M. Tichy, Coll. Czech. Chem. Comm., 1961,..- • 847. * ,
35. T. A. Spencer, T. D. Weaver, R. M. Villarica, R. J. Friary, J. Poster, at M. A. Schwartz, J. Org. Chem., 1968, 33, 712.
36. M. Stiles, J. Amer. Chem. Soc., 1959, 81, 2598. ,, ,ой7 47
37. A. P. Krapcho, J. Diamanti, C. Cayen, and R. Bingham, Org. Synth., 19 >
20
38. H. R. Snyder, L. A. Brooks, and S. H. Shapiro, Org. Synth. Coll. Vol. 2,
531 [A'. CuatWep, Л. Брукс, C. Шапиро. Синтезы орг. препаратов, Пер. с англ./Под ред. Б. А. Казанского. М„ Издатинлит, 1949, с. 41 J-ц j.
39. G. Stork, A. Brizzolara, Н. Landesman, J. Szmuszkovicz, and к.
Amer. Chem. Soc., 1963, 85, 207.
40. 1. Ojima, S. Inaba, and У. Nagai, Tetrahedron Letters, 1973, 4271. gg
41. S.-O. Lawesson, S. Gronwall, and R. Sandberg, Org. Syntm, 19 -• д'
42. E. J. Moriconi, J. G. White, R. W. Franck, J. Jansing, J. F. Nelly, lomone, and Y. Shimakawa, Tetrahedron Letters, 1970, 27. p ^гуп-
43. P. Boldt, A. Ludwieg, and H. Militzer, Chem. Ber., 1970, 103, 1312,
mins and C. R. Hauser. J. Org. Chem., 1967, 32, 2615. „ g Sufi
44. S. N. H tickin and L. Weiler, .1. Amer. Chem. Soc., 1974, 96, 10° ’ and L. Weiler, J. C. S, Chem. Comm., 1977, 91.
230
45 S N- Huckin and L. Weiler, Canad. J. Chem., 1974, 52, 1379.
ifi P Mauer in «Newer Methods of Preparative Organic Chemistry», ed. W Foerst' Academic. New York, 1963, vol. 2, p. 101.
47 H Meerwein. Annalen, 1913, 398, 242; R. Gelin and S. Gelin Compt. rend., 1964 258. 4783: S. M Dixit, S. D. Verma, and J. K- Mehrotra, J. Indian Chem. Soc '1961, 38. 853.
4Я RE Bowman. J. Chem. Soc.. 1950, 325.
19 1 Treibs and K- Hintcrmeicr, Chem. Ber., 19o4, 87, 1163; S.-O. Lawesson, S Gronwall, and R. Sandberg, Org. Synth., 1962, 42, 28; об использовании триметилсилиловых эфиров см.: U. Schmidt and М. Schwochau, Tetrahedron Letters, 1967, 875.
50 A. Williams and К. T. Douglas, Chem. Rev., 1975, 75, 627.
51. N. F. Yaggi and К. T. Douglas, J. Amer. Chem. Soc., 1977, 99, 4844.
52^ F. Korte and К. H. Buchel, Angew. Chem., 1959, 71, 709.
53 E. E. Van Tamelen, C. Placeway, G. P. Schiemenz, and I. G. Wright, J. Amer. Chem. Soc., 1969, 91, 7359.
54. J. Tsuii, Accounts Chem. Res., 1969, 2, 144.
55. F. J. McEvoy and G. R. Allen, Jr., J. Org. Chem., 1973, 38, 4044.
56. G. Stork, A. Brizzolara, H. Landestnan, J. Szmuszkovicz, and R. Terrell, J. Amer. Chem. Soc., 1963, 85, 207.
57. C. A. Henrick, E. Ritchie, and W. C. Taylor, Austral. J. Chem., 1967, 20, 2441.
58. H. Stetter and M. Schreckenberg, Angew. Chem. Internat. Edn., 1973, 12, 81; H. Stetter, ibid., 1976, 15, 639.
59. A. Takeda, K- Takhashi, S. Torii, and T. Moriwake, J. Org. Chem., 1966, 31, 616.
60. J. Hooz and R. B. Layton, Canad. J. Chem., 1972, 50, 1105.
61. H. C. Brown, M. M. Midland, and A. B. Levy, J. Amer. Chem. Soc., 1972, 94, 3662.
62. A. Pelter, C. R. Harrison, and D. Kirkpatrick, Tetrahedron Letters, 1973, 4491.
63. L. Cassar, G. P. Chiusoli, and F. Guerrieri, Synthesis, 1973, 509.
64. E. Piers and M. B. Geraghty, Canad. J. Chem., 1973, 51, 2166.
65. M. E. Scanlon and S. MacKenzie, J. Org. Chem., 1972, 37, 1579.
66. A. Rahman and 0. L. Tombesi, Anales Asoc. quim. argentina, 1974, 62, 109 (Chem. Abs., 1974, 81, 135876).
67. F. Korte and H. Machleidt, Chem. Ber., 1955, 88, 1676.
68. P. Talukdar and P. Bagchi, J. Org. Chem., 1955, 20, 25.
69. G. Stork, A. Brizzolara, H, Landestnan, J. Szmuszkovicz, and R. Terrell, J. Amer. Chem. Soc., 1963, 85, 207.
70. H. 0. House and M. Schellenbaum, J. Org. Chem., 1963, 28, 34; N. F. Firrell and P. W. Hickmott, Chem. Comm., 1969, 544.
71. F. 1. Carroll, G. N. Mitchell, J. T. Blackwell, A. Sobti, and R. Meek, J. Org. Chem., 1974, 39, 3890.
72, G. Buchi and B. Egger, J. Org. Chem., 1971, 36, 2021.
73. B. Fohlisch, E. Schupp, U. Dukek, and G. Schwaiger, Annalen, 1973, 1861.
74. N. K. Kochetkov, L. J. Kudryashov, and В. P. Gottich, Tetrahedron, 1961, 12, 63.
75. A. Ichihara, J. Morita, K, Kobayashi, S. Kagawa, H. Shirahama, and T. Matsumoto, Tetrahedron, 1970, 26, 1331.
76. J. A. Marshall and T. M. Warne, Jr., J. Org. Chem., 1971, 36, 178.
to' d’ G^uanB> C- Al. Mau, and Y. L. Tien, Chem. Ber., 1935, 68, 1946.
то и ^angstrom and G. Bergson, Acta Chem. Scand.. 1973, 27, 3118
cn r ^e^.er. and Rauhut, Chem. Ber., 1958, 91, 2543 и ранние работы.
80. L. Kno], Annalen, 1961, 647, 53; 1962, 656, 183.
qo r? 9>‘ Halford und Weissmann. J Org Chem., 'Ost 18 30.
qq n Fi 9.lark and - G- B- Howes, J. Chem. Soc., 1956, 1152.
RA r Dc le,nfHe' J- Org- Chem., 1970, 35, 1275.
r! t r ’ Guill™ton, Bull. Soc. chim. France, 1969, 2871.
co- J. ticini and A. Krief, Tetrahedron Letters, 1970, 1397,
231
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
T. N. Wheeler,
100.
Brouillard and
102.
103.
A Fschenmoser, D. Felix, and G. Ohloff, Helv. Chim. Acta. 1967 sn n Felix J Schrieber, G. Ohloff, and A. Eschenmoser, ibid., 1971 D. Nasiptiri, G. Sarkar, M. Guha, and R. Roy, Tetrahedron Letters’ ?q96' 927 1
в E McGarry, R. L. Markezich, and W. S. Johnson, J. Amer. Chem s 1973 95, 4416. ' d0C'
У. Fukuyama, C. L. Kirkemo, and J. D. White, J. Amer. Chem. Soc., 1977 qo 646 *
A Chatterjee, Tetrahedron Letters, 1965, 959.
D J Sam and H. E. Simmons, J. Amer. Chem. Soc., 1972, 94, 4024 L. F. Fieser and J. Szmuszkovicz, J. Amer. Chem. Soc., 1948, 70, 3352 G. P. Chiusoli, F. Minisci, and A. Quilico, Gazzetta, 1957, 87, 90.
P. E. Eaton, R. H. Mueller, G. R. Carlson, D. A. Cullison, G. F Coon» T.-C. Chou, and E.-P. Krebs, J. Amer. Chem. Soc., 1977, 99, 2751. ’ '
S. Swaminathan and M. S. Newman, Tetrahedron, 1958, 2, 88.
J. P. Collman, Accounts Chem. Res., 1975, 8, 342; J. P. Collmann, R. Q Finke, J. N. Cawse, and J. I. Brauman, J. Amer. Chem. Soc., 1977, 99, 2515. R. J. Anderson and C. A. Henrick, J. Amer. Chem. Soc., 1975, 97, 4327.
В. M. Trost and T. N. Salztnann, J. Org. Chem., 1975, 40, 148. H. Gerlach and W. Muller, Helv. Chim. Acta, 1972, 55, 2277; C. A. Jackson, and К- H. Young, J. Org. Chem., 1974, 39, 1318. R. Matusch, Angew. Chem. Internal. Edn., 1975, 14, 260.
100a. Beilstein, «Handbuch der Organischen Chemie», and R.
J. E. Dubois, J. Org. Chem., 1974, 39, 1137.
101. A. Veierou, T. Bercovici, E. Fischer, Y. Mazur, and H. Yogev, Soc., 1977, 99, 2723.
101a. F. Guibe, P. Sarthou, and G. Bram, Tetrahedron, 1974, 30, 3, 39.
1016. P. Sarthou, F. Guibe, and G. Bram, J. C. S. Chem. Comm., 1974, 377.
101b. Y. Hara and M. Matsuda, Bull. Chem. Soc. Japan, 1976, 49, 1126.
101r. G. Brieger and W. M. Pelletier, Tetrahedron Letters, 1965, 3555.
101д. C. Cambillau, P. Sarthou, and G. Bram, Tetrahedron Letters, 1976, 281; E. V. Dehtnlow, Angew. Chem. Internat. Edn., 1974, 13, 170.
101 e. R. Gelin, S. Gelin, and A. Galliaud, Bull. Soc. chim. France, 1973, 3416; bG. Ent-enrnann, Tetrahedron Letters, 1975, 4241; cC. P. Casey and D. F. Morten, ibid., 1974, 925; dR. A. Johes, S. Nokkeo, and S. Singh, Synth. Comm.. 1977, 195. ,
101ж. S. J. Rhoads and R. H. Hasbrouk, Tetrahedron, 1966, 22, 3557; bS. J. Rhoads and A. W. Desora, ibid., 1963, 19, 1645; CA. Chatterjee, D. Banerjee, and S. Banerjee, Tetrahedron Letters. 1965, 3851; M. E. Kuehne, J. Org. Chem., 1970, 35, 170.
M. Eigen, Angew. Chem. Internat. Edn., 1964, 3, 1. , ,
G. Bram, F. Guibe, and P. Sarthou, Compt. rend., 1973, 277, 429; Tetrahedron Letters, 1972, 4903; .4. Л. Курц, С. M. Сахеябаева, И. П.
О. А. Реутов. Ж. Орг. хим., 1973, 9, 1553; ДАН СССР, 1973, 210, 144; 19^ 2! 1,602.
М. L. Bender, «Mechanisms of Homogeneous Catalysis from Protons to r, teins», Wiley, New York, 1971 p 169' M. H O’Leary and R. L. Baugnn, Amer. Chem. Soc., 1972, 94, 626. . fl, and
C. IF. Jefford, A. F. Boschung, T. .4. В. M. Bolsman, R. M. Mortar J, B. Melnick, J. Amer. Chem. Soc., 1976 98, 1017.
J. P. Ferris and N. C. Miller, J Amer. Chem. Soc.,. 1966, 88. 3522.
J. A. Marshall and H. Faubl, J. Amer Chem. Soc., 1970, 92, 948; J- K-man and W. A. Pletcher, ibid., p. 956. „=
H. Stetter, H. Held, and A. Schulte-Oestrich, Chem. Ber., 1962, , yojk,
1. Fridovich, «The Enzymes», 3rd edn., ed. P. Boyer, Academic, New
Amer. Chem. Soc., 1977,
R. P. Bell and P. de Maria, Trans. Faraday Soc., 1970, 66, 930, R-and M. A. D. Fluendy, ibid., 1963, 59, 1623.
J. Amer. Chem.
104.
105.
106.
107.
108.
109.
1972, vol. 6, p. 255.
ПО. /V. V. Raghavan and D. L. Leussing, J.
2188. K
111.
232
9.6. АМИНОКИСЛОТЫ
Дж. X. Джонс {University of Oxford)
Химию аминокислот вплоть до 50-х годов охватывают два исчерпывающих обзора [1, 2], а с 60-х годов Special Periodical Reports [31 дают ежегодный обзор. Период 50 60-х годов лишь избранно освещен в Annual Reports [4]. Среди появившихся общих критических исследований лучшее опубликовано в современном исправленном издании монографии Сиджвика (Sidgwick), где также оценена и биологическая значимость аминокислот [5].
Основной упор в данной главе будет сделан на синтезе и химических реакциях аминокислот и их простых производных [но не пептидов и белков, которые рассмотрены в томе 10 (русского издания), части 23 и 24]. Кроме того, мы не будем рассматривать здесь несколько важных разделов, интересующих химиков, а именно технику тонкослойной хроматографии [6], практику автоматического аминокислотного анализа [7], химические последствия облучения аминокислот и их растворов [8], метаболические и исторические аспекты [10—12] и аминокислоты как антиметаболиты [13—14]. Более того, мы сконцентрируем внимание на алифатических аминокислотах, хотя и добавлен короткий обзор об ароматических аминокислотах.
9.6.1. а-АМИНОКИСЛОТЫ
9.6.1.1. Введение
Структура и конфигурация а-аминокислот
«-Аминокислоты, которые несмотря на их цвиттерионную природу (см. ниже), изображают как (1), являются наиболее важными, многочисленными и разнообразными среди всех аминокислот, встречающихся в природе. Набор 19 аминокислот вместе с N-замещенной аминокислотой пролином составляют строительные блоки, из которых под генетическим контролем синтезируются по-липептидные цепи. В белковых гидролизатах (особенно структурных белков) найдено и несколько других аминокислот, однако обычно они являются результатом окислительных процессов, затрагивающих боковые цепи, после завершения создания белкового кора. Общее число а-аминокислот, идентифицированных в свободном или включенном виде в природных соединениях животных, растений [15] и микроорганизмов, исчисляется сотнями, и их число все время растет. Полные таблицы аминокислот можно найти в двух хорошо известных справочниках [16, 17], в которых помимо сведений о наличии той или иной аминокислоты приведены данные по оптическому вращению, константы диссоциации и растворимости. Кроме того, рекомендации комиссии IUPAC ПЛЗ [18] по номенклатуре а-аминокислот включают большие дополнения,
233
объясняющие общепринятые тривиальные названия. Система™, ские названия используются редко, поэтому существует бол™ ' число прочно вошедших тривиальных названий. Вместе с Ое имеется договоренность называть аминокислоты, не имеющие с^' цпальных или обычных тривиальных названий, как производи^' или аналоги тех аминокислот, которые имеют тривиальные нГ звания.
Распространенные «-аминокислоты представляют собой бес цветные кристаллы, не имеющие четких температур плавления и разлагающиеся при температуре около 200°C. Они растворимы в воде, слегка растворимы в этаноле и нерастворимы в большинстве других растворителей. Эти свойства, типичные для сильнополярных соединений, являются следствием того, что в кристаллах «-аминокислоты находятся в виде диполей цвиттерионной структуры (2); цвиттерионы присутствуют также в водных растворах [19, 20]. При титровании раствора «нейтральной» аминокислоты (т. е., например, с недиссоциирующей боковой цепью) в кислой и щелочной области обнаруживаются два ветствующие равновесию, указанному глицин (1, R = Н) имеет р/С 2,35 и 9,78.
~NH2CHRCO2H'
(1)
типа диссоциации, coot-на схеме 1. Например,
NH3CHRCO2H +
nh2chrco; (1)
NH3CHRCO2
(2)
Структура (1), по-видимому, присутствует в растворах аминокислот, однако в очень малых количествах. Помимо того, они присутствуют в парах, образующихся при сублимации аминокислот При высоких температурах, и, например, в случае глицина (1, R = Н) подобное соединение было выделено вымораживанием на аргоновой матрице при 20 К [21]. Для каждой аминокислоты существует характеристическое значение pH, при котором она на ходится в основном в виде цвиттериона (2). Поскольку эта форма в целом электрически нейтральна, то при этом pH, которое назы вают изоэлектрической точкой, молекула не движется в электр) ческом поле и имеет при этом pH минимальную растворимое • Тот факт, что поведение аминокислот при ионизации весьма . рактерно для этого класса соединений с заметными различи * между отдельными представителями класса, сделало ионоо * ную хроматографию главным аналитическим и препаративт методом разделения аминокислот друг от друга, от солей и ДР-°г0 веществ. Вследствие этого классические методы избпрател осаждения солей и комплексов были в значительной степей теснены из лабораторной практики. Для крупномасштабных раторных процедур ионообменная хроматография неудоби , 234
нако такое происходит довольно редко, ибо современные методы позволяют и поощряют работы, проводимые в небольшом масштабе, и, кроме того, все природные аминокислоты, необходимые для работы, коммерчески доступны в очень чистом виде и по разумным ценам.
Все а-аминокислоты, обнаруженные в белках, за исключением глицина, хиральны. Тщательными химическими исследованиями и выборочным определением абсолютной конфигурации показано, что все такие аминокислоты принадлежат к 2,-ряду с абсолютной стереохимией у а-углеродного атома, показанной на проекции Фишера (3) (для удобства читателя изображена и другая проекционная формула). Поскольку /^-терминология глубоко не проникла в биологическую химию, мы будем следовать существующему соглашению и придерживаться DL-системы, тем не менее необходимо отметить, что в общем случае (3) имеет S-конфигурацию. Большинство других а-аминокислот, давно обнаруженных в природе, также имеют L-конфигурацию а-углеродного атома, однако известны многие природные а-аминокислоты D-ряда. Они встречаются в небелковых компонентах растений, грибов и микроорганизмов, необычны для животных, и никогда не встречаются в белках. Некоторые из L-аминокислот (изолейцин, треонин, гидрокси-лизин, гидроксипролин), найденные в белках, имеют два хираль-пых центра. В этом случае тривиальные названия применяются только в одной паре энантиомеров, к другой паре добавляется приставка алло-. Таким образом и названы четыре стереоизомера 2-амино-З-метилвалериановой кислоты (изолейцина) (4) — (7), причем только (4), абсолютная конфигурация которого установлена с помощью рентгеноструктурного анализа, найден в белках. Необходимо отметить, что эпимеризация а-аминокислоты с двумя хиральными центрами по а-углероду дает не энантиомер, а химически отличный диастереомер, и если это происходит при синтезе или гидролизе, например, изолейцинсодержащего пептида, то при последующем анализе аминокислот можно обнаружить алло-изо-лейцин. Номенклатура циклически замещенных пролинов усложняется тем, что относительная стереохимия двух центров может быть определена с помощью цис/трянс-обозначений, и хотя это не используется широко в случае гидроксипролина, это установленный путь называния других пролинов. Поэтому, например, (8) на схеме (3) обычно называют трпнс-З-метил-А-пролином. Для ациклических небелковых а-аминокислот с двумя асимметрическими центрами обычно применяют трео/эритро-обозначения.
СО2Н СОоН
NH2--------н
R ч
R Н
(8)
235
nh2 СНз :о2н и
и
С (4 :н2сн3 )
Л-изолейцин
( :о2н
Т4 МИ
н— —сн3
( (5 ЗН2СН3 )
£>-нзолейцин
( :о2н
мн н
н— —СИз
( (« :н2сн3 )
Л-аддо-изолейцин
( н— CT-L :о2н —nh2
( Г1 ЗН2СН3 7)
D-ал ло-изолейцин
Для определения абсолютной конфигурации производных а-аминокислот существуют три основных подхода, которые включают химическую корреляцию, ферментативные и хирооптические методы. Рентгеноструктурные методы в большинстве своем применяют для исследования ключевых соединений или в таких особенно важных или сложных случаях, как, например, компонент антибиотика стрептолидина [22]. Химические корреляции (ряд примеров приведен в [23]) широко использовались раньше и продолжают применяться и сейчас, однако с меньшей интенсивностью. В качестве примеров на схемах (2) [24] и (3) [25] показаны реакции, использованные для выяснения стереохимии транс-3-ме-тил-Л-пролина при корреляции с изолейцином.
Широко распространено использование ферментов с известной субстратной специфичностью для выбора между конфигурациями а-аминокислот. Например, способность служить субстратом для оксидазы /^-аминокислот считается строгим подтверждением ^-конфигурации аминокислоты. Окислительное отщепление а-во-дорода этим ферментом происходит лишь в том случае, если атом водорода находится в том положении, какое он имеет в серии
NaOH;
нейтрализация и нагревание
смесь цис- и транс -
Me
AcNHCH(CO2Et)2 —> / Yx''C°2Et
+ НСГ \ / >CO2Et
СН3СН=СНСНО 1
Ас
NaOH________
(до прекращения'* р-ии с диастереомером)
EtSH,HCl;
Ni Ренея;
Н3О+
DL-алло-Изолейцин
(2)
H2/Pd; Н3О+
(8) РЬ-траяс-З-метилпролин
236
£)-аминокислот. Именно это и было использовано для выбора [26] между энантиомерными а-монотритиированными глицинами {схема (4), два энантиомера, каждый из которых получен ферментативно, ведут себя противоположным образом, в зависимости от положения трития}.
Одно время для быстрой предварительной оценки конфигурации было популярным использование эмпирических правил сдвига величины оптического вращения при одной длине волны в зависимости от изменения условий или замещения. Наиболее известно правило Клу — Лутца — Иргенсона, которое звучит следующим образом [27]: «Если при добавлении кислоты к водному раствору оптически активной аминокислоты ее молекулярное вращение изменится в положительную сторону, то эта аминокислота имеет L-конфигурацию; если изменение происходит в обратном направлении, то это характеризует £)-аминокислоту».
Me
ci2,cf3co2h,aV;
NaOH
L-изолейцин
Ь-лгуэанс-З-метилпролин
(8)
(з)
Оксидаза D-аминокислот ------------->
(4)
83 % сохранения трития
Современным развитием этого подхода являются измерения кругового дихроизма. Показано, что карбоксильная группа «-аминокислот Е-ряда дает положительный эффект Коттона примерно при 200 нм в воде и 208—210 нм в кислоте, при условии, что нет необычного конформационного напряжения и нет других интерферирующих хромофоров [23]. Альтернативно, можно ввести хромофор химическим путем без затрагивания а-углерода и использовать круговой дихроизм нового хромофора для конфигурационных исследований [28]. Хотя для этой цели предложено множество хромофоров, тем не менее появилось лишь несколько примеров их использования.
9.6.1.2. Методы получения а-аминокислот
Известно большое число принципиально общих методов синтеза а-аминокислот [2, 29—31]. Некоторые из этих методов, включая аминирование а-галогенкарбоновых кислот {схема (5)} [32], синтез Штрекера {схема (6)} [33], подходы через гидантоины {схема (7)} [34], и оксазолоны {схема (8)} [35], появились на заре химии аминокислот, однако они сохранили свою значимость
237
И поныне Длительное время использовались также синтезы, в торых аминогруппа вводилась путем восстановления [36] Или перегруппировки [37].
со2н рс‘3 / \со2н Мсо2н (5:
NaCN
СНзСНО NH4C1
снзсн:
NH2 н3о+ /NH2
----► CH3CHZ
•CN ^СОгН
(6)
RCHO
KCN, (NH4)2CO3
(есть несколько альтернатив для образования кольца)
н3о+
----► RCH
ИЛ1>
ОН-
(7)
PhCONH
НО2ССН2
PhCHO —----->
Ас2О, NaOAc
Hi /NH2
-р-> PhCH2CH'
NCO2H
(8)
Предложено несколько современных улучшений гидантоинового подхода [38], в частности использование хелата магния (9) {схема (9)} [39] дает замечательные выходы продуктов. Оксазолоновый метод также был расширен [38], особенно с помощью реакций, достраивающих боковую цепь в ненасыщенном оксазолоне {схема (Ю)} [40].
магний-метилкар-бонат
---------->
RX
(9)
NaOH;
PhCH2CI
Ва(ОН)2 /NH2
-----•-*- RCH^ + PhCH2NH2 + ВаСО3 (у)
\СО2Н
EtMgBr ------->
Et
н3о+ ----->
Г /NHj PhCHCH^ \со2н (10)
Тем не менее ни один из вышеописанных методов не иСП?л зовался столь часто, как применение ациламиномалонатов iH пример, по схеме (11)} [41]. Несмотря на то, что аминомалонов} кислоту [42] и ее производные можно выделить, эти проД}1' неустойчивы и декарбоксилируются, поэтому аминокислоты м 238
но непосредственно получать в жестких условиях на последней стадии. Диэтилацетамидомалонат (10) легко получают по схеме (12) [431; аналогично можно получать и использовать такие ацил-аминомал’онаты, как (11) [44], (12) [45] и (13) [46]. Эти соединения можно алкилировать {схема (И)} или использовать как нуклеофильные компоненты в реакциях /Михаэля, которые оставляют функциональные группы боковой цепи для дальнейшей модификации. Так, (10) реагирует с акролеином [47], давая (14), который после трансформации боковой цепи можно превратить в различные аминокислоты. Однако иногда использование этого подхода осложняется тем, что ациламиноальдегиды, такие, как (14), в значительной степени существуют в форме циклических таутомеров. Собрана избранная библиография вплоть до 1970 г. [48] по использованию соединений (10) и (11) в синтезе. Последнее значительное достижение [49] ациламиномалонатного подхода объединяет последнюю стадию и разделение D- и L-изомеров, как показано на схеме (13).
NaOEt Н3О+
AcNHCH(CO2Et)2 д ,рн , гп AcNHC(CO2Et)2 ----------------> NH2CHCO2H (11)
Br(CH2)5C(J2b.t । ।
<10) (CH2)5CO2Et (CH2)5CO2H
NaNO2 H2/Pd(C);
CH2(CO2Et)2 . nu -> HON=C(CO2Et)2 - > AcNHCH(CO2Et)2 (12)
АсОН AC2O
(10) (10)
AcNHCH(CN)CO2Et OCHNHCH(CO2Et)2 ZNHCH(CO2Bzl), (H) (12) (13)
AcNHC(CO2Et)2
CH2CH2CHO
(14)
NaOEt KOH
ZNHCH(CO2Me)2 ———> ZNHCR(CO2Me)2--------------------> ZNHCRCO2H —>
KX (до омыления J
одной группы) I
CO2Me А протеаза
ZNHCHRCO2Me ZNHCHRCO2H + ZNHCHRCO2Me (13)
DL- Carlsberg) £. £).
Z = бензилоксикарбонил
Несмотря на. большое разнообразие хорошо проверенных методов, развитие новых общих стратегий синтеза а-аминокислот все еще^ актуально. Характерной чертой трех схем относительно недавней концепции является использование изоцианидов. Этот
подход включает четырехкомпонентную конденсацию [например, показано далее на схеме (35)], карбоксилирование a-металлированных изоцианидов {схема (14)} [50] и общий синтез а-амино-р-гидроксикислот [51] по реакции сложных эфиров изоцианокислот с карбонильными соединениями {схема (15)}. Два других
239
метода использующих «современные» реагенты, и которые в Прин_ пипе достаточно общи, показаны на схемах (16) [52] в (17)
Обсуждение синтезов меченых а-амвнокислот приведено в гл. 23.J,
PhCHCO2H (U)
NH2
znh2 HOCH2CHf (15)
BuLi
PhCH2NC -----PhCHLi
С02. —80°С;
Н3О
CN
NaCN
CNCH2CO2Et
О ,N
гидролиз
СО2Н
CO2Et
zSOMe NaH. H2N, ,SOMe
h2c; —V/
XSMe RCN R/'SMe
Дс2О, ру
(перегруппировка)
NHAc
NHAc
RCCOSMe
MeOH "ыуГ
SMe
NH2
RCH
NHAc
I н3о
Ni Ренея ------> RCH —
CO2Me
RCH2CO2H -> RCH(Li)CO2Li RCH(NH2)CO2H (17)
В 1950-х годах Миллер показал [54], что пропускание электрического разряда через смесь метана, аммиака, воды и водорода приводит к образованию среди прочих продуктов аминокислот. Поскольку, по-видимому, аналогичные условия могли существовать на ранних стадиях истории земли, из этого следует, что уже тогда могли образовываться аминокислоты. С тех пор лабораторное моделирование возможной «предбиологической» химии стало активной областью исследования [3].
Значительные усилия были направлены на развитие методов асимметрического синтеза а-аминокислот (когда необходимая хиральность вводится в ходе синтеза с помощью хиральных реагентов) [55]. На схеме (18) [56] приведен особенно успешный пример перспективного метода (см. также гл. 23.2). Тем не менее асимметрический синтез все еще представляет более академический нежели практический интерес, и синтетическая работа в этой области обычно требует последующего разделения оптических изомеров. Разделение диастереомерных производных и использование избирательных ферментов (особенно ацилаз, которые могут гидролизовать только одну из двух энантиомерных ацил аминокислот) стали классическими методами [57]. Эти методы все еще заслоняют другие подходы, такие как хроматографию на оптически активных средах и предпочтительную кристаллизацию. Среди мно гих приемов, использующих диастереомерные производные, осо юго упоминания заслуживает тирозин-гидразидный метод Р ' (схема (19)}. Естественно, здесь особенно важны критерии оптической чистоты [59]. Как это ни звучит парадоксально, но в тох
RCCO2Me
SMe LiN(Pr-«3O)2
СО2Н
(16)
244
случае, когда нужен только один из энантиомеров, обычно приходится иметь дело одновременно как с методами разделения, так й с методами рацемизации, а побочный энантиомер после рацемизации снова вводить в цикл. Это можно сделать с помощью распространенной кислотной или щелочной обработки, или при нагревании с уксусным ангидридом с последующим гидролизом образующейся ацетил-РА-аминокислоты. Можно использовать также ферментативные и другие методы [60].
Выше мы в основном пытались проиллюстрировать набор доступных лабораторных методов получения оптически активных аминокислот. Однако нельзя забывать и о значимости промышленного производства этих соединений, имеющих разнообразное коммерческое применение помимо производства пищевых продуктов. Используемые для этой цели методы подразделяются на методы выделения из гидролизатов (сейчас имеют не столь большое значение, как раньше, за исключением некоторых особых случаев), ферментативные методы и химический синтез с последующим разделением. Все это является предметом огромной патентной литературы, кроме того, появились две книги [31, 61], посвященные этому вопросу. Например, производство моногидрата Л-глутамата натрия {схема (20)} в Японии исчисляется тысячами тонн в месяц [62].
чаито гы
Z-DL-Ala 4- L-TyrNHNH2
смешивание эквимольиых количеств в МеОН
------------------------>
—> Z-D-Ala, L-TyrNHNH2 + Z-L-Ala, A-TyrNHNH2
осадок остается в растворе (19)
фильтрование, однократная кристаллизация и нейтрализация
(возврат разделяющего агента)
нейтрализация маточников
(возврат разделяющего
агента);
добавление дициклогекснламина (ДЦА) и однократная перекристаллизация
* Z-0-Ala
—> Z-L-Ala, ДЦА
оптически чистый
оптически чистый
Z == бензилоксикарбонил, Д1а — остаток аланина, Туг — остаток тирозина
241
ncch=ch2
водяной газ^ NCCH2CH2CHO оксо-синтез
синтез ШтреКера.
ра зде ление’^^Г'--тельной кристаллизаций (возврат D-изомера после рацемизации?
частичная нейтрализация
/NH2
—> НО2ССН2СН2СН^ XCO2Na
(20)
9.6.1.3. Реакции а-аминокислот
а-Аминокислоты реагируют с простыми спиртами, алкилирую-щими агентами, активными арилирующими агентами, ацилирующими, сульфонил ирующими агентами и т. д. ожидаемым образом какой можно предполагать для двух изолированных функциональных групп [2]. Эти реакции не будут обсуждаться каждая в отдельности, что же касается продуктов этих реакций, то отдельно будут рассмотрены только N-алкил- и N-ацетильные производные. Сильные кислоты и основания образуют с аминокислотами устойчивые соли, которые в некоторых случаях плохо растворимы и поэтому подходят для методов выделения. В случае производных меди(II) образование прочных хелатов с а-аминокислотной функцией можно использовать в качестве временно блокирующих групп, например при получении е-моноациллизинов, необходимых для избирательной защиты в пептидном синтезе. Комплексы кобальта и меди активированы для реакции с альдегидами по а-углеродному атому и, например, конденсация медного комплекса глицина с ацетальдегидом в щелочных условиях {схема (21)} служит промышленным способом получения треонина [63], причем в продукте преобладает природная стереохимия.
NH2CH2CO2H
ионообменная
------------->
смола
ОН
МеСНСН
При нагревании а-аминокислоты образуют диоксопиперазг, вместе с неопределенными полимерными продуктами. С Ра3 * _ ленной азотистой кислотой образуется а-гидроксикислоты ск* чественным выделением азота (это являлось основой важног прошлом аналитического метода); нитрит натрия и концентр ванная соляная кислота в основном приводят к а-хлоркисл _ (с бромидом водорода получают а-бромкислоты, с фторидом 242
рода —а-фторкислоты [64]). Обычно эти реакции протекают с сохранением конфигурации в силу соучастия карбоксильной группы, однако результат'зависит от растворителя; могут соучаствовать также и боковые цепи (см., например, [65]). Формальдегид реагирует с а-аминокпслотами в водном растворе с образованием N-ги-дроксиметильных производных (реагирует также с функциональными боковыми цепями); в этих производных основность атома азота понижена. Эта реакция была использована в классической химии белков для формальдегидного титрования [66]. Другие простые альдегиды и кетоны реагируют с образованием оснований Шиффа, однако эти соединения устойчивы лишь в холодных нейтральных или щелочных растворах, и могут храниться лишь в виде солей. Если основания Шиффа получать в восстановительных условиях, то это приводит к N-алкилпроизводным (см. ниже). В сильно щелочных условиях глицин конденсируется с бензальдегидом по а-метиленовой группе, что служит простым синтезом |3-фенилсе-рина [67].
Большое число различных реагентов вызывает окислительное декарбоксилирование а-аминокислот. Например, использование пиколината серебра(II) [68] количественно дает альдегиды; к тем же изменениям могут приводить и такие более известные окислители, как N-бромсукцинимид и гипохлорит. Эти реакции, по-види-мому, протекают в широком смысле по одному пути через имин, который гидролизуется с образованием аммиака и альдегида. а-Дикарбонильные и родственные соединения реагируют с а-ами-нокислотами с тем же результатом; эта реакция [представленная на схеме (22) ее прототипом ] известна как деградация Штрекера [69]. В этом случае образование основания Шиффа приводит к таутомерному сдвигу и декарбоксилированию, в конечном итоге приводящему к альдегиду, аммиаку и восстановленному карбонильному соединению. Реакция нингидрина (15) с а-аминокисло-тами (эта цветная реакция применяется в автоматическом аминокислотном анализе) относится к этому же классу, однако в этом случае азот удерживается карбонильной компонентой, и в результате последующих реакций образуется пурпурный Руэманна (16). Реакция в основном протекает как показано на схеме (23), однако одновременно протекает несколько других реакций, причем направление этих реакций и выход окрашенных продуктов варьируют от случая к случаю [70]. Для фенилаланина дальнейшая реакция между аминокислотой, образующимся фенилацетальдегидом и нин-гидринным реагентом приводит помимо прочего к флуоресцирующему соединению (17). Обнаружение того, что этот тип структуры может получаться непосредственно при обработке аминокислот соединением (18) («флуорескамин», Флурам) привело к применению этого реагента для обнаружения аминокислот, причем пределы обнаружения значительно ниже, чем для нингидрина [71], (в общем случае это действительно чувствительный реагент для
243
определения первичной аминогруппы). n NH п
/Ш х.^0 д
МеС<согН + оДЛ ~ МеСН0 + С°! + Другие "р°*^
со2н
NCHCH2Ph
но, с
о ph (17)
лизатора являютс^^пмгим0 а'0КС0КИСл°тами в отсутствие ката-карбонильной компонентой АА'ЧЗеМ’ когда азот захватывается рования. Напримеп пм г,после окислительного декарбоксили-а-фенилглицина и ^ировиног^™ „кипячени,] водного раствора бензальдегида и обоаз^ ^но раднои кислоты появляется запах пиридоксаля и ионов мрА” аланИн- ^Ри катализе производными перенос аминогомппы АА ЛЛа или Ферментативными системами, обратимо и без лекяпКлгл минокислоты на кетокислоту протекает рование) делает а-кетп-°КСИЛИрОванпя- Этот процесс (трансамин»-превращаемыми. ' И а‘амин°кислоты метаболически взапмо-
протекает мягко поивсшиА^й07 дибоРаном в тетрагидрофуране
Существует нескольк^, ₽-аминоспиртам [72].
приводят к гстеппггм^г, обц*их реакций а-аминокислот, которые олл ероциклическим системам; наиболее известные из
244
них представлены на схеме (24).
о
СОС12, PhMe;
Ас2О
RSC(=S)OR;
<----------
НС1, MeNO2
nh2chrco,h
PhNCS, НО-;
--------------> д, н+
(24)
NPh
9.6.1.4. N-Замещенные а-аминокислоты
(7) N-Алкильные производные
N-Монометил-а-аминокислоты («иминокислоты») широко распространены. Например, саркозин (N-метилглицин) является метаболически важным соединением, а многие другие производные (которые обычно называют полусистематическими названиями) встречаются в свободном виде и в природных продуктах, особенно в антибиотиках. Эти соединения можно получить из свободных а-аминокислот (схема (25)} [73], и несмотря на некоторую рацемизацию, этот метод получил широкое распространение. Последовательность реакций, указанная на схеме (26), по-видимому, лишена этого недостатка. В природе широко распространены также циклические N-моноалкил-а-аминокислоты (циклические иминокислоты). Мы уже упоминали особый случай пролина в белках, однако его высшие и низшие гомологи (пипеколовая и азетидин-2-карбоновая кислоты соответственно) и многочисленные другие производные [75] также присутствуют в природе.
PhCHO NaBH4;
NH2CHRCO2H ———> PhCH=NCHRCO2Na -------------->
NaOH H+
CH2O H2, Pd(C)
—> PhCH2NHCHRCO2H PhCH2N(CH3)CHRCO2H - ~ --- >
AcOH
—> CH3NHCHRCO2H (25)
ZC1 NaH, Mel
nh2chrco2h ZNHCHRCO2H--------------•>
NaOH 2 ТГФ, 0°C
(сложный эфир не образуется)
► ZN(Me)CHRCO2H -> MeNHCHRCO2H Z = бензилоксикарбонил
245
Основные свойства и химия N-моноалкиламинокислот ан гичны их незамещенным предшественникам, за очевидным ИС1?Л0‘ чением тех реакций, в которых участвуют оба водорода ами?' группы. Так, пролин и другие N-алкил-а-аминокислоты не даН°' типичного пурпурного окрашивания с нингидрином (пролин да^ желтое окрашивание). При действии азотистой кислоты образ? ются М-нитрозо-М-алкил-а-аминокислоты, которые интересны те? что из них можно получать сидноны (см. раздел 20.4), и, крои’ того, в силу беспокойства [76] об их образовании при’ хранении пищевых продуктов (нитрозамины канцерогенны). М,М-Диалкил-а-аминокислоты не имеют большого значения, однако их четвертичные производные достаточно важны в биохимии [77]. N,N,N-Tpn-метилглицин имеет тривиальное название бетаин; это название используется также для обозначения всего этого класса соединений. Сам бетаин хорошо растворим в воде, не растворим в эфире, плавится с разложением около 200°C. Его можно рассматривать как локализованный цвиттерион, лишенный возможности образовать анион, хотя он может протонироваться по карбоксильной группе сильной кислотой с образованием ярко выраженных солей. Бетаины можно получать из соответствующих а-аминокислот с помощью различных методов метилирования, в частности рекомендуется обработка метилиодидом и бикарбонатом натрия в метаноле [78].
(2) N-Ацилъные производные
Реакция а-аминокислот с ангидридами или хлорангидридами простых кислот приводит к N-ацильным производным, однако оптически активные продукты образуются лишь в очень мягких условиях. Например, бензоилирование по Шоттену — Бауману сопровождается некоторой рацемизацией, а кипячение с уксусным ангидридом и последующий гидролиз рацемического N-ацетилпро-изводного — хороший общий метод рацемизации а-аминокислот. Это является следствием дальнейшей реакции N-ацетильного производного, приводящей к образованию смешанного ангидрида, с последующей его циклизацией в оксазолон, который быстрее раце-мизируется, чем происходит раскрытие цикла. Образование оксазолонов из N-ацил-а-аминокислот при энергичном активировании карбоксильной группы {схема (27)} [79] — важная общая Реа& ция, протекающая, например, при получении хлорангидрида и смешанного ангидрида, или при обработке дициклогексилкар диимидом (ДЦК). Генерирование оксазолонов из гиппуров^ (бензоилглицин) или ацетуровой (ацетилглицин) кислот в РезУ‘оВ тате обработки уксусным ангидридом в присутствии альдеги > которые конденсируются с активированной метиленовой гру > служит основой одного из старейших синтезов а-аминоки {см. схему (8)}.
246
и
(27)
Лс2О NH,CHRCO2H ----> AcNHCHRCOMe + СО2 (28)
РУ
Оксазолоны вступают также в реакцию Дакина-Веста {схема (28)} [80], которая служит основным методом получения а-ацет-амидометилкетонов. Хотя N-моноалкил-а-аминокислоты не могут образовывать оксазолоны, тем не менее N-метил-а-аминокислоты и пролин рацемизуются при кипячении с уксусным ангидридом, и вступают в реакцию Дакина-Веста, по-видимому, через катионы (19), которые могут ионизоваться по а-углеродному атому с образованием мезоионной системы (20). В случае бензилсаркозина соединение (21) может быть выделено в виде перхлората после обработки смесью уксусный ангидрид-хлорная кислота [81]; присутствие мезоионных интермедиатов, участвующих в рацемизации 5 производных N-метил-а-аминокислот [82], зарегистрировано пу-i тем 1,3-диполяркого циклоприсоединения. В силу легкости цикло-f дегидратации (а следовательно, и рацемизации) при активации It карбоксильной группы простые ациламинокислоты бесполезны в $ пептидном синтезе. Однако алкоксикарбониламинокислоты (22) 5 не рацемизуются при введении или снятии защитной группы, и поскольку исходная аминокислота может быть регенерирована расщеплением связи алкил—кислород, эти производные весьма ценны для пептидного синтеза. Можно получить соответствующие ацилхлориды (см., однако, ниже), но эти соединения не нашли широкого использования, так как при температуре, несколько выше комнатной, они быстро распадаются до N-карбоксиангидри-дов (23) (хороший препаративный метод получения последних соединений [83]). Ранее считали, что реагенты, используемые для синтеза хлорангидридов кислот, при низких температурах в присутствии третичных оснований превращают бензилокспкарбонил-аминокислоты в азиридиноны (24), однако, как показано теперь, при этом получаются оксазолоны (25) [84]. N-Трифторацстил-аминокислоты получают без рацемизации при обработке в течение нескольких часов при комнатной температуре а-аминокислот эквивалентным количеством трифторуксусного ангидрида; в более
Ж
жестких условиях при избытке ангидрида проходит циклизация В этом случае циклическое производное, по-видимому, существует главным образом в виде псевдо-оксазолонового таутомера (26) гидролиз которого, например, приводит к а-оксокислотам [85]’ Сложности, связанные с образованием реакционноспособных гетероциклов, не сопровождают химию фталоил аминокислот (27), которые получают по Габриэлю или нагреванием а-аминокислот с фталевым ангидридом примерно при 150 °C [86].
сю;
о rocnhchrco2h
(22)
Н\ /R N-Y
PhCH2O—2=0 О
(25)
(3) N-Гидроксипроизводные
N-Гидрокси-а-аминокислоты (28) были известны еще в XIX веке [87], однако лишь недавно они были идентифицированы как компоненты природных соединений и стали интересны как биологически активные вещества. Для их получения существует набор простых методов, в том числе: гидроксиламинолиз а-галогенкарбоновых кислот (однако применение избытка гпдроксиламина приводит к окислению продуктов до а-оксиминокислот [88]), частичное восстановление нитрокарбоновых кислот и модифицированный синтез типа синтеза Штрекера, в котором гидроксициановую кислоту присоединяют к альдоксиму [89, 90]. Однако наиболее успешный метод синтеза включает реакцию аниона бенз альдоксим а (стереоизомер алкилируется по кислороду) со сложными эфирами а-галогенкарбоновых кислот с образованием нитрона (29), который далее гидролизуется {схема (29)} [89]. Нитронную функцию в (29) можно также расщепить без затрагивания сложноэфир-248
ной группы путем обработки солями гидроксиламина, Что дает сложные эфиры N-гидроксиаминокислот [90].
ph. _,Н Ркк /Н +
EtOH Н3о+
Т| + BrCHRCO2Et -------> II ------>
№+5хЫ -o/NCHRCO2Et
(29)
—> HONHCHRCO2H + PhCHO (29)
(28)
Многие обобщения, касающиеся N-гидроксиаминокислот, существовавшие в ранней литературе, противоречивы и неточны. Это было выяснено в 1961 г., когда появилось сообщение о том, что полученные в то время соединения обычно бывали загрязнены «-аминокислотами и другими побочными продуктами синтеза. Кислоты (28)—бесцветные кристаллические соединения с высокими температурами плавления; они не растворимы в большинстве органических растворителей, но растворимы в воде, амфотерны и имеют изоэлектрическую точку в области pH 4 [91]. При нагревании водных растворов этих соединений они медленно разлагаются, диспропорционируя до аминокислоты и оксиминокислоты (которые сами по себе неустойчивы при pH ниже 7) {схема (30)} [91]. Ацилирующие агенты дают смеси продуктов за счет атаки по кислороду и азоту (преобладает N-ацилирование) [92]. Простейшим методом получения производных для идентификации является каталитическое восстановление [93] до а-аминокислот, которое протекает гладко. N-Гидроксиаминокислоты можно окислять {например, при диспропорционировании по схеме (30)}; они восстанавливают Фелингову жидкость [89]. Известны также изомерные а-аминооксикислоты [94].
2HONHCHRCO2H —> nh2chrco2h + o=nchrco2h
I (30)
RCN + CO2 + H2O 4— hon=crco2h
(4) N-Аминопроизводные
N-Амино-а-аминокислоты (а-гидразинокислоты) представляют значительный интерес как биологически активные аналоги аминокислот. Их можно получать восстановлением гидразонов а-оксо-кислот, ^сложных эфиров диазокислот или нитрозопронзводных, реакцией типа синтеза Шгрекера, в котором вместо аммиака применяют гидразин, или ГНдразинолизом а-галогенкарбоновых кислот [95]. В принципе привлекательно получение этих соединений из а-аминокислот путем N-гомологирования, н возможно, этот метод будет развит в дальнейшем. Примером может служить проращение а-амииокислоты в гидразиновый аналог по схеме (31)
249
гок) которое неожиданно оказалось доказательством конфигура. [96], кот°р“„ялога -так как каталитическим гидрогенолизом его ции этого ’в „сходную а-аминокислоту. Непрямое превра.
можно преьр с^от Q дг.аминоПрОИЗводные с полным обращением Snrv’pannn возможно путем дезаминирования (сохранение кон-Д ш) В концентрированной соляной кислоте с последующим ^ппазинолизом (обращение) [97]. Каки предполагалось, а-гидра-ч нокислоты являются цвиттерионными соединениями, легко раскоп мы в воде и не растворимы в большинстве органических рас-тпопителей Простейшие члены эгого класса соединении, по-ви-XoJy быстро плавятся при б0Лее НИЗКИХ ТемПераТУРаХ
150—200 °C) чем простые а-аминокислоты. Ацилирующие агТнты атакуют'а-гидразинокислоты по обоим атомам азота, однако использование защитных групп дает возможность направить Реакцию в желаемое положение [98]. Сложные метиловые эфиры (полеченные при обработке метанольным хлоридом водорода) реа-гиоуют с азотистой кислотой, образуя нитрозопроизводные, которые при обработке кислотой дают сложные эфиры а-азидокислот [99].
НО^ Y он
^/Me ^CO2H NH2
KNCO;
-----—>
Me2SO4
МеО
OMe
/Me
Ме
NaOCI;
со2н
nhconh2
(31)
но^ "у он
nhnh2
9.6.1.5. «-Модифицированные а-аминокислоты
а-Алкил-а-аминокислоты, из которых наибольший интерес представляют а-метильные соединения, наиболее часто получают из кетонов путем соответствующих модификаций методов Штре-кера {например, по схеме (32)} [100] и гидантоинового метода {например, по схеме (33)} [101].
NaCN
Et2CO -----
NH4CI
NH2CEt2CN
HCl ---?- NH2CEt2CO2H
метода
(32)
D Me О
/ //
(NH4)2CO3
*С0Ме —KCN~~
HN' 'NH
Ва(ОН)2
кипячение, 68 ч
Me NH2CCO2H
R
(33)
о
R = 3,4-дкметоксибензил
250
Иногда вследствие стерических препятствий возникают труд-ипети Так происходит, например, при гидролизе некоторых 5,о-ди-"ямешенных гидантоинов, когда могут потребоваться неооычно Жесткие условия реакции. Более мягкой процедурой является распад до аминокислоты через 3-тозильное производное {схема Г34)> [Ю2]. В последнее время разработано множество других методов (см^ [3]); пример, приведенный на схеме (35) [103], представляет, хотя и стерически затрудненный, но успешный случай синтеза.
(Bzl)2CO /Bzl
Bz1NH2 —:► BzlN=C
^Bzl
NaOH Н3О+
----> TosNHCO-------> NH2CR‘ R2CO2H
HO2CCR‘R2NH (34)
AcOH ----->
PhNC
Bzl
| HBr
BzlN—-C—CONHPh ------>-
Ac Bzl
Bzl Bzl
I H2 * ।
—> BzlNHCCO2H pd(C) nh2cco2h
Bzl Bzl
Bzl == бензил (35)
Разделение а-алкиламинокислот (если нужен один из изомеров)— расточительная процедура, поскольку отсутствие а-водо-рода препятствует рацемизации и делает невозможным повторную обработку ненужного изомера. Более того, ферментативные методы, хотя и применимы в некоторых случаях (см., например, [104]), однако также ограничены. В силу этого здесь особенно интересны асимметрические синтезы {например, модифицированный синтез Штрекера, схема (36) [105]}. Альтернативный подход состоит в разделении асимметрических интермедиатов, как, например, в синтезе сх-метилДОФА по Штрекеру [106], где разделение осуществляют на стадии аминонитрила. Поскольку аминонитрил образуется в процессе равновесной реакции, то ненужный изомер после выделения можно рециклизовать.
Производные сс-гидрокси-а-аминокислот можно получать окис-гыт71еМ тетРааи'етатом свинца сложных эфиров N-ацетилсерина [107] или частичным гидролизом галогеноксазолонов [108].
«,р-Ненасыщенные аминокислоты (дегидроаминокислоты) не обнаружены в свободном виде, но известны в природе как компоненты антибиотиков. 2-Ацетамидокоричную кислоту получают частичным гидролизом соответствующего бензилиденового оксазо-лона | 09] ( 2-ацетамидоакриловая кислота образуется при нагревании пировиноградной кислоты с ацетамидом [110]. Для получения
251
сложных метиловых эфиров N-ацилдегидроаланина использовали различные катализируемые основаниями реакции Р-элиминип* вания производных серина или цистеина с модифицировано.0' ми боковыми цепями {например, по схеме (37)} [111]. Альтео' нативный подход состоит в N-хлорировании с последующей обпа боткой основанием {схема (38)} [112]. р '
Ph
СО2Н
NaOH
-CH2°H NH2
Me R
NaIO4
----------
или л-хлорпер-бензойная кислота
Me
(36)
BocNHCHC02Me
CH2SBzl
КСИЛОЛ
BocNHCHC02Me —-------->
। 1 140 °C, 6 ч
CH2SOBzl
BocNHCCO2Me (37)
CH2
R
OMe
BocNHCHCO2Me
трет-BuOCl
---------->
NaOMe
NaOMe
BocN—CHCO2Me--------
BocNHCCO2Me
СНМе2
Cl
CHMe2
CHMe2
диазабицик-лоундецеи,
NaOMe, Д
(38)
HC1
BocNHCC02H
“ОН e--- BocNHCC02Me
HC1 • H2NCCO2Me
CMe2
CMe2
CMe2
Гидрирование производных дегидроаминокислот приводит к защищенным а-аминокислотам (асимметрический синтез аминокислот гидрированием производных дегидроаминокислот, в присутствии хирального катализатора — хорошо известный метод [55]); гидролиз приводит к а-оксокислотам, производными которых (как енамины) являются дегидроаминокислоты. Особенно интересны реакции присоединения. В основной среде тиолы и таки реагенты, как диэтилмалонат {схема (39)} [109], вступают в реа % цию Михаэля. Хлорид водорода и бромид водорода приводят а-галоген-а-аминокислотам [ИЗ] (в условиях кинетическ контроля), которые не были выделены в индивидуальном сост нии, но используются в дальнейшем методе для функционали ции при С-а [114, 115] {например, по схеме (40) [115]}.
252
ЕЮ2Сч /СО2Ме
/СН2 + СН2=СХ
ЕЮ2СУ \\HAc
NaOMe ----->
МеОН
ЕЮ2С. >СО2Ме
/СНСН2СН^
ЕЮ2с/ \NHAc
(39)
,СО2Н сн2=с;
\\IHAc
HCI ---->-АсОН
(гидролиз приводит к глутаминовой кислоте) со2н со2н
I H2s I
СН3—С—Cl -------> СНз—с—SH (40)
NHAc NHAc
9.6.1.6. а-Аминокислоты, модифицированные по карбоксильной группе
Соли сложных эфиров а-аминокислот — в основном стабильные кристаллические соединения. Так, гидрохлориды метилового, этилового и бензилового эфиров могут быть получены при кипячении а-аминокислоты в спирте в присутствии хлорида водорода [116]. Однако в случае гидрохлорида метилового эфира, обработка а-аминокислоты избытком метанола, в который перед этим был добавлен 1 экв тионилхлорида (т. е. кислый метанольный раствор диметилсульфита), значительно более легкая и быстрая процедура, которая часто дает прекрасные результаты [117]. Бензиловые сложные эфиры удобно выделять в виде п-толуолсульфо-натов (после получения нагреванием а-аминокислот с бензиловым спиртом и /г-толуолсульфокислотой в бензоле с азеотропной отгонкой воды) [118]. Соли трет-бутиловых эфиров можно получать прямо [119] из аминокислот, однако наиболее широко применяют катализируемую кислотой реакцию N-бензилоксикарбониламино-кислот с изобутиленом с последующим гидрогенолизом N-a-защи-ты [120]. Простейшие свободные сложные эфиры аминокислот, полученные после нейтрализации солей эфиров, представляют собой жидкости с сильно основными свойствами; они полностью смешиваются с большинством органических растворителей. Метиловые, этиловые, а также бензиловые сложные эфиры быстро разлагаются при нагревании (хотя метиловые и этиловые эфиры можно перегонять с небольшими потерями) и медленно разлагаются даже при хранении в холодильнике, что ведет в основном к диоксоппперазинам. Однако трет-бутиловые сложные эфиры возможно перегонять без потерь, и они могут храниться при комнатной температуре без изменений. При реакциях метиловых и этиловых сложных эфиров с аминами, гидроксиламином, гидразином и т. д., получаются a-аминоамиды, гидроксамовые кислоты, гидразиды и т. д. Восстановление литийалюминийгидридом приводит к аминоспиртам [121]. Аминогруппу можно обычным образом ацилировать, сульфонилировать и т. д.; трансформация функциональных групп дает подход к сложным эфирам с другими азотсодержащими заместителями в a-положении. Например, этиловый эфир глицина можно превратить в этилдиазоаЦетат [122] (обработка
253
азотистой кислотой), этилизоцианатоацетат [1231 (об
геном), этилизотиоцианатоацетат [124] (реакция с аб°ТКа фос углерода с последующей обработкой хлоридом ртути)ДИСульФиДом цианоацетат (формулирование с последующей обработко?^^0’
Гидрохлориды хлорангидридов а-аминокислот nonvu боткой аминокислоты пентахлоридом фосФопа п учают обра. углерода [126]. р в теРахлорИде
9.6.2. ^-АМИНОКИСЛОТЫ
B-Аминокислоты, простейшим и прекрасно известным примером которых служит (3-аланин (30), можно получать различными общими методами [127], включая присоединение азотных нуклеофилов к «^-ненасыщенным кислотам, сложным эфирам, нитрилам {схема (41)} [128], или гомологизацией по Арндту — Эйстерту N-защищенных ос-аминокислот, и особенно фталоильных производных, которые большей частью дают кристаллические интермедиаты {схема (42)} [129]. Сам 0-аланин удобно получать перегруппировкой сукцинимида по Гофману [130]. Циклоприсоединение хлорсульфонилизоцианата к олефинам [131] {например, по схеме (43)} дает в принципе очень удобный подход, так как р-лактамы легко гидролизуются до |3-а минокисл от.
О
(30)
254
S-Аминокислоты — цвиттерионные соединения; их основные физические характеристики сходны с таковыми для а-аминокислот, хотя они несколько более основны и менее кислы (0-аланин имеет пХа 3,55 и 10,24). Реакции 0-аминокислот с простыми ацилирующими'и алкилирующими реагентами и т. д. ничем нс примеча-течьны. При нагревании до 200°C они теряют аммиак и образуют В-ненасыщенные кислоты. Превращениями 0-аминокислот можно подучить шестичленные аналоги некоторых пятичленных гетероциклов, образуемых из а-аминокислот, однако образование цикла протекает уже не всегда столь легко, и при обработке фосгеном, например, N-карбоксиангндриды обычно не получаются. Наиболее важными гетероциклическими производными служат 0-лактамы, однако их нельзя получить непосредственно из 0-аминокислот. Некоторые Н-алкнл-0-аминоэфиры циклизуются при обработке реактивом Гриньяра (схема (44)} [132], однако выходы продуктов невелики, и потому важнее другие подходы [133]. Особая значимость химии высоко реакционноспособных 0-лактамных систем [134] связана с тем, что они присутствуют в пенициллиновых и цефалоспориновых группах антибиотиков которые будут рассмотрены в другом месте (см. гл. 23.5).
PhCH-CH2 EtMgBr PhX—,
I I ---------I (44)
PhCH2NH CO2Et Et2° PhCH2N—40
9.6.3. ОТ у- ДО w-АМИНОКИСЛОТ
у-Аминомасляную кислоту можно получить несколькими путями, исходя из 1,2-дигалогенпропанов {схема (45)} [135], или гидролизом ее лактама — пирролидона, полученного электролитическим восстановлением сукцинимида, или же действием аммиака на у-бутиролактон [136]>
KCN
С1(СН2)3Вг ----►
C1(CH2)3CN
—> NH2(CH2)3CO2H
(45)
Гидролиз лактамов дает важный общий подход к W* ам ин о к и с лот я м ~__ ________________________
1ЫП IXC1IY VI Cl IX 1 Cl IVJL I_>1 J1UV1J naiKJ 1 nvycipj'iin и миопии no 1 оциклических систем. Так, оксим циклогексанона перегруппи-
высшим
так как лактамы получают перегруппировкой
дал Ша^ СЯ“ ПРИ обработке серной кислотой в капролактам; UeiTTn»]1”1 гидР°лиз дзет е-аминокапроновую кислоту {схема )) 1138]. Развитие метода, который является общим для со-ами-
255
нокислот с длинной цепью (содержащей 12______18
атомов) иллюстрировано на схеме (47) [139].
Углеродных
H2SO4 -----—>
Н3О+
----> NH2(CH2)5CO2H (46)
КН2(СН2)ЛСО(СН2)5СО2Н
№^2, КОН, NH2(CH2)n+6CO2H
? , б- ... со-Аминокислоты являются, как а- и 0-аминокислоты, цвиттерионными соединениями со сходными основными свойствами, однако нарастающее разделение аминной и карбоксильной функции приводит к ослаблению их взаимного влияния. Так, Y-аминомасляная кислота имеет значения рДд 4,03 и 10,56 (ср. с данными для 0-аланина и глицина). Характерное принципиальное отличие состоит в том, что у- и 6-аминокислоты при нагревании теряют воду, давая лактамы, а высшие члены серии образуют полимеры.
Пяти- и более членные лактамы имеют важное промышленное значение, как исходные соединения для получения полимеров. Например, при полимеризации капролактама при нагревании в раз-^дп1ЫХ Условиях образуется найлон-6 (31). Поэтому получение [140], химия [140] и полимеризация [141] этих лактамов явились предметом интенсивного исследования, причем большая часть результатов представлена только в патентной литературе. Все эти соединения^ циклические амиды, и имеют соответствующие химические свойства, однако соединения со средним размером цикла несколько более нуклеофильны, чем типичные ациклические вторичные амиды. Например, капролактам можно с хорошим выходом метилировать по кислороду с образованием простого лактимного эфира (32) или по азоту, в зависимости от условий реакции.
256
Легко также протекает ацилирование, рование по азоту.
[-NH(CH2)5CO-]„
галогенирование и нитрозй-
(32)
(31)
9.6.4. АМИНОАРОМАТИЧЕСКИЕ КИСЛОТЫ
о-Аминобензойная (антраниловая) и n-аминобензойная кислоты имеют существенное биологическое значение. Антраниловая кислота служит интермедиатом в различных путях биосинтеза; н-ампнобензойная кислота участвует в комплексе биосинтеза витаминов группы В и служит бактериальным фактором роста, для которого сульфамидные препараты являются антагонистами. Антраниловую кислоту удобно получать перегруппировкой Гофмана фталимида [142]; п- [143] и л/-изомеры получают восстановлением соответствующих нитробензойных кислот. Эти три аминобензойные кислоты слабо растворимы в воде, растворимы в органических растворителях и даже в эфире; они плавятся при более низких температурах (о- 144°C; м- 174°C, п- 186°C), чем алифатические аминокислоты, и возгоняются. Эти свойства подтверждают неполноту превращения в цвиттерионную форму, что не удивительно с точки зрения подавления нуклеофильности,, аминогруппы ароматическим ядром (рАа для о- 2,05, 4,95; для м- 3,07; 4,73; для п-2,30, 4,64). Все три изомера имеют в общем сходные химические свойства, ожидаемые для соединений с ароматическими амино- и карбоксильными группами. Так, антраниловую кислоту можно обычным способом диазотировать {диазониевая соль при взаимодействии с диметиланилином дает метиловый красный (33) [144]}; обработка сульфидом натрия дает после восстановительного расщепления тиосалициловую кислоту [145]. Диазотирование в безводной среде органическим нитритом приводит к цвиттерионному диазониевому карбоксилату (34), термическое разложение которого служит удобным методом генерации дегпдробензола [146]. И наконец, функциональные группы в антраниловой кислоте идеально расположены для образования гетероциклов, и, например, реакция с фосгеном дает изатоевый ангидрид [147] (35).
(34)
О (35)
(33)
9 Зак. 395
257
ЛИТЕРАТУРА
1. J. Р. Greenstem and M. Winitz, «Chemistry of the Amino .Acids» Wil м York, 1961 [Дж. Гринштейн, M. Виниц. Химия аминокислот и’пент^’ ^ew Пер. с англ./Под ред. М. М. Шемякина. М., Мир, 1965/, идов.—
2. Т. Wieland, R. Muller, Е. Niemann, L. Birkhofer, A. Schoberl, A. Wars
H. Soil, in «Methoden der Organischen Chemie» (Houben-We’yl) 4tli лГ’ org Thieme Verlag, Stuttgarf, 1958, vol. XI/2, chapter 2. ’ n’’ Qe‘
3. J. ~H. Jones, in «Amiho-acids, Peptides and Proteins (Specialist Periodical R ports)», The Chemical Society, London, 1969, 1, Г; B. W. Bucrott ibid кип 9 1- ih;H ' !Q7i я 1- mm 1070 л t. n r' 1974 ’ 5 ZU’
2, 1; ibid.) 1971, 3, 1; ibid., 1972, 4, I; P. M. Hardy ibid ’ О. C. Barrett, ibid., 1975, 6, I; ibid., 1976, 7, 1.
4. D. T. Elmore, Ann. Reports, 1961, 58, SOO'; R. C. Sheppard, ibid., 1963 60 448; H. D. Law, ibid., 1965, 62, 389; ibid., 1966, 63, 517; ibid 1967 64B 45 f. ’
5. N. V. Sidgwick, «The Organic Chemistry of Nitrogen», 3rd edn., ed. 1. T. Mil-lar and H. D. Springall, Claredon Press, Oxford, 1966, chapter 6.
6. G. Palaki, «Techniques of ThTn-Layer Chromatography in Amino Acid and Peptide Chemistry», 2nd English edn., Ann Afdor-Hijmphrey Science, Ann Abbr, Michigan, 1969.
7. M. D. Waterfield and J. Bridgen, in «Instrumentation in Amino Acid Sequence Analysis», ed. R. N. Perham, Academic Press, London, 1975.
8. W. M. Garrison, Radiation Res. Rev., 1972, 3, 305.
9. D, A. Bdhde'r, «Amino Acid Metabolism», Wiley, Chichester, 1975.
10. //. B. Vickery, Adv. Profem Chem., 1972, 26, 81.
II. H. B. Vickery and C. L. A. Schmidt, Chem. Rev., 1931, 9, 169.
12. J. S. Friiton, «Molecules and Life'», Wiley-Interscience, New York, 1972, p. 108.
13. L. Lowden, D. Lewis, and H. Tristram, Adv. Ehzymol., 1967, 29, 89.
14. J. P. Scanned and D. L. Pruess, in «Chemistry and Biochemistry of Amino Acids, Peptides and Proteins», ed. B. Weinstein, Marcel Dekker, New York, 1974, vol. 3,'p. 1897
15. L. Fowden, Prog. Phytochem., 1970, 2, 203.
16. «Handbook of Biochemistry; Selected Data for Molecular ed. H. _A. Sober, Chemical Rubber Co., Cleveland, Ohio,
17. «Data for Biochemical Research», 2nd edn., ed. R. M. C. liott, W. H. Elliott, and I\. M. Jones, Clarendon Press,
Biology», 2nd edn., 1970, section B.
Dawson, D. C. El-Oxford, 1969, Sec-
18.
19.
20.
21.
22.
23.
24.
tion 1.
Biochemistry, 1975, 14, 449.
Cm. cc. 1, vol. 1, chapter 4. ,
E. J. Cohn and J. T. Edsall, «Proteins, Amino Acids and Peptides as Ions ana Dipolar Ions», Reinhold, New York, 1943. Q7n
У. Grenie, J. C. Lassegues, and C. Garrigou-Lagrange, J. Chem. Phys., 19/v, 53, 2980. „„ rro
B. W. Bycroft and T. J. King, J. C. S. Chem. Comm 1972, 6o2.
L. Fowden, P. M. Scopes, and R. N. Thomas, J. Chem Soc. (C), , •
A. B. Manger, F. Jrreverre, and B. Witkop, J. Amer. Chem. Soc.,
25. ^Kollonitsch, A. N. Scott, and G. A. Doldouras, J. Amer. Chem. Soc., 1966, 88, 3624. a7.
26. M. Akhtar and P. M. Jordan, Tetrahedron Letters, 1969, »/□.
27. Cm. cc. 1, p. 85.
28. C. Toniolo and A. Signor, Experientia, 1972, 28, 7bo.
29. R. J. Block, Chem. Rev., 1946, 38, 502. . CvnHwfic Methods»,
30. I. T. Harrison and S. Harrison, «Compendium oi Organic Sy nt!
Wiley, New York, 1974, vol. II, p. 248. , T к-meko Y Izu-
31. «Synthetic Production and Utilization of Amino Acids», ed. • x< mi, I. Chibata, and T. Itoh, Wiley, New York, 1974.
32. Org. Synth. Coll. Vol. 3, 1955, 848.
258
„ c rail Vol 1 1941 21 ГЕ. К. Кендалл, Б. Ф. МакКензи. Синтезы 33'S>rg препаратов, сб. L-Пер.’с англ./Под ред. Б. А. Казанского. М., Издатинлит, 1949, с. 20].
34. См. cc. L vol. 1, Р;^&2 1943 489 ГХ. Джиллеспи, X. Снайдер. Синтезы орг. 35' °Г/ппЙтов 9 - Пер. с англ./Под ред. Б. А. Казанского. М. Издатинлит,
ПрСПЗра 1 (Jo, tu. —" г
36 V и. дай* А * { <>»• с?">, 1973Ъ3Ь8’ 822„ „
к Xinomiya, Т. Shioiri, and S. Yamada, Chem. Pharm. Bull.
22, 1398. - ................-
(Japan), 1974, York, 1974.
39.' H. Finkbeine'r, J. Org. Chem., 1965, 30, 3414.
1968, 41, 1266.
46.
47.
48.
49.
37.
oo A 1 Metiers «Heterocycles in Organic Synthesis», Wiley, New 39' H. Finkbeine'r, J. Org. Chem., 1965, 30, 3414.
40 L Horner and H. Schwann, Annalen, 1955, 591, 99.
4L Hase, R. Kiyoi, and S. Sakakibara, Bull. Chem. Soc. Japan,
42 См cc 1 vol. 3, p. 2407.
43 H R Snyder and С. IF. Smith, J. Amer. Chem. Soc., 1944, 66, 350.
44 H. F. Albertson and B. F. Tullar, J. Amer. Chem. Soc., 1945, 67, 502.
45 A. Galat, J. Amer. Chem. Soc., 1947, 69, 965.
H. M. Kissmail and B. Witkop, J. Amer. Chem. Soc., 1953, 75, 1967.
О. A' Moe and D. T. Warner, J. Amer. Chem. Soc., 1948, 70, 2763.
«Diethyl acetamidomalonate and ethyl acetamidocyanoacetate», distributed by Winthrop Laboratories, New York (73 ссылки).
A. Berger, M. Smolarsky, N. Kurn, and H. R. Bosshard, J. Org. Chem., 1973, 38, 457; H. R. Bosshard and A. Berger, Helv. Chim. Acta, 1973, 56, 1838.
IF.’ Vaalburg, J. Strating, M. G. Woldring, and H. Wynberg, Synth. Comm.,
50.
1972, 2, 423.
51. D. Hoppe and U. Schollkopf, Annalen, 1972, 763, 1.
52. K. Ogura and G. Tsuchihashi, J. Amer. Chem. Soc., 1974, 96, 1960.
53. S. Yamada, T. Oguri, and T. Shioiri, J. C. S. Chem. Comm., 1972, 623.
54. S. L. Miller, J. Amer. Chem. Soc., 1955, 77, 2351.
55. /. D. Morrison and H. S. Mosher, «Asymmetric Organic Reactions», Prentice-Hall, Englewood Cliffs, New Jersey, 1971, chapter 7 [Дж. Д. Моррисон, Г. С. Мошер. Асимметрические органические реакции. М., Мир, 1973, гл. 7].
56. Е. J. Corey, R. J. McCaully, and И. S. Sachdev, J. Amer. Chem. Soc 1970 92, 2476.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
Cm. cc. 1, vol. 1, chapter 9.
K. Vogler and P. Lanz, Helv. Chim. Acta, 1966, 49, 1348.
J. IF. Westley, in «Chemistry and Biochemistry of Amino Acids, Peptides and Proteins», ed. B. Weinstein, Marcel Dekker, New York, 1971 vol. 1 p 1 Cm. cc. 31, p. 43.
K. Yamada, S. Kinoshita, T. Tsunoda, and K- Aida, «The Microbial Production of Amino Acids», Wiley, New York, 1972.
Cm. cc. 31, p. 103.
Cm. cc. 31, p. 197.
G. A. Olah and J. Welch, Synthesis, 1974, 652.
K. Koga С. C. Wu, and S. Yamada, Tetrahedron Letters 1971 2283.
D. trench and J. T. Edsall, Adv. Protein Chem, 1945 2 277
См. cc. 1, vol. 3, p. 2589. ’ ’
SocG'(CM970/V8f5//am/’S°rt’ Л B' L R' M°rley’ a'ld B- Scanlon< J- Chem.
4. Schonberg and R. Moubacher, Chem. Rev., 1952, 50, 261
Chem- Rev., 1960, 60, 39.
) CM. c^ki-pnl
TTSih and Lei,ugrubcr, ibid,, 1972, 94, 5927
Kiin I Г974^9Т' P' Cr°°y’ R' De Neys' and L Eliacrs> C. S.
Pl’ RU,&DermotlleabnadChN V..°SleC Helv. Chim. Acta, 1963, 46, 327.
4-B. Mauger andT Witkop,B^ 51’ ^15.
9*
259
76. См. сс. 3, vol. 7, р. 22.
77. См. сс. 16.
80.
78. F. С. М. Chen and N. L. Benoiton, in the press.
79. J. U7. Cornjorth, in «Heterocyclic Compounds» ed R c ri.r. r- >
№» York, 1957. vol 5. p. Зэк R. Rule,. AdV Holoree^l^'S1"'.
75 [Гетероциклические соединении, т. 5. - Пер. с англ./Под ред и ? V1 четкова. М, Издатинлит, 1961, с. 269]. 1 д- К. Ко-
1730’ AUinSer' °’ L Wang' and B- B- Dewhurst, J. Org. Chem., 1974, 39,
81. G. V. Boyd and P. H. Wright, J. C. S. Perkin I, 1972, 909.
82. J. R. McDermott and L. N. Benoiton, Canad. J. Chem 1973 51 очио
83. См. cc. 1, vol. 2, p. 862. " ’ ’ 2bb2-
8-1. J. H. Jones and M. J. Witty, J. C. S. Chem Comm 1977 281
85. F. Weygand, W. Steglich, and H. Tanner, Annalen, 1962 658 128
86. J. C Sheehan, D. Й7. Chapman, and R. W. Roth, J. Amer. Chem, Soc., 1952
74» 3822. ’ *
87. W. von Millar and J. Plocht, Ber., 1893, 26, 1545.
88. A. Hantsch and W. Wild, Annalen, 1896, 289, 285.
89. E. Buehler and G. B. Brown, J. Org. Chem., 1967, 32, 265.
90. T. Polonski and A. Chimlak, J. Org. Chem., 1976, 41, 2092.
91. /. D. Spenser and A. Ahmad, Proc. Chem. Soc., 1961, 375.
92. 0. Neunhoeffer, G. Lehmann, D. Haberer, and G. Steinle, Annalen 1968 7P 208. ’ "
93. 1. Neelakantan and 117. H. Hartung, J. Org. Chem., 1958, 23. 964.
94. E. 1 esta, B. J. R. Nicolaus, L. Mariani, and G. Pagani, Helv. Chim. Acta, 1963,
46, 766.
95. N. Takamura and S. Yamada, Chem. Pharm. Bull. (Japan), 1976, 24, 800.
96. S. Rarady, M. G. Ly, S. H. Pines, and Л1. Sletzinger, J. Org. Chem., 1971, 36, 1949.
97. Л1. Sletzinger, R. A. Firestone, D. F. Reinhold, C. S. Rooney, and W. H. Nicholson, J. Medicin. Chem., 1968, 11, 261.
98. H. Niedrich, Chem. Ber., 1965, 98, 3451.
99. A. Darapsky, J. prakt. Chem., 1936, 146, 219.
100. Org. Synth. Coll. Vol. 3, 1955, 66. ' ,nc. „
101. G. A. Stein, H. A. Bronner, and K. Pfister, HI, J. Amer. Chem. Soc., 195a, 77,
102. /<. Hiroi, K. Achiwa, and S. Yamada, Chem. Pharm. Bull. (Japan), 1968, 16,
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
119.
444
H. L. Maia, B. Ridge, and H. N. Rydon, J. C. S. Perkin I, 1973, 98.
J. Turk, G. T. Pause, and G. R. Marshall, J. Org. Chem, 197o, 40, 9a3.
К. Weinges and B. Stemmle, Chem. Ber., 1973, 106, 2291.
D. F. Reinhold, R. A. Firestone, W. A. Gaines, J. M. Chemerda, and Л1. Me zinger, J. Org. Chem., 1968, 33, 1209.
W. Oettmeler, Chem. Ber., 1970, 103, 2314. Pn-HM and
M. M. Chemiakine, E. S. Tchaman, I. S. Denisova, G. .4. Rmd.i.
W. J. Rodionow, Bull. Soc. chim. France, 1959, 26, эЗО. Г1,нтр,.. 0Dr Пре-
Org. Synth. Coll. Vol. 2. 1943 I (A X^er Д-паратов, сб. 2. — Пер. с англ./Под ред. Б. А. Казанское .
Т. Wieland, G. Ohnacker, and W. Ziegler Chem. j. c. S.
D. H. Rich, J. Tam, P. Mathiaparanam, J. A. Grant, and C.
Chem. Comm., 1974, 897. , F. 1071; 15, 294.
H. Poisel and U. Schmidt, Angew. Chem. internet. Edn.,
A. L. Love and R. K. Olsen, J. Org Chem, 1972, 373401.
G. Lucentre and D. Rossi, Chem. and Inch (Lon 1°")- 1УСр’ш 1973, 38, 126.
S. M. Patel, 1. O. Currie, Jr., and R. K. Olsen, J. Org. cnun.,
Cm. cc. 1, vol. 2, p. 925. , , r,r, ,!(i т 109
M. Brenner and W. Huber, Helv. Chim. Aeta, 9o3, 3b, 1
H. Determann, O. Zipp, and T- Wieland. Annalen, 1962, bob
R. Roeske, J. Org. Chem, 1963, 28, la51.
260
135.
136.
137.
138.
139.
140.
141.
142.
143.
144.
145.
146.
147.
'» j,' н“сыт. а5Г 19«Л2Л0М,Тшт,'р«В’ан.
ные там работы.
If r%Sj£A™ale.°:i9«.M2. 75
124: О and К. Off'rmmn, Angew. Chem.
s j- °rg-с|м"'' I6,
„n L308' c.,m+h roll Vol 3 1943 19 IX. Кларк, Л. Бэр. Синтезы орг. препара-130‘ ?0?'сбУ 2.h-Шр. Vc ан?к/Пад ред. Б. А. казанского. М. Издатинлит, 1949, 131 CJ. ^Rasmussen and A. Hassner Chem. Rev 1976, 76, 389.
132 : R. W7. Holley and A. D. Holley J. Amer Chem. Soc. 1949, 71, 2124.
133 . A. K. Mukerjee and R. C. Srivastava, Synthesis 1973, 327.
134 A K- Mukerjee and A. K- Singh, Synthesis, 1975, 547.
' Org Svnth. Coll. Vol. 2, 1943, 25 [К. Де Витт. Синтезы орг. препаратов, cq 2. — Пер. с англ./Под ред. Б. А. Казанского. Л1., Издатинлит, 1949, с. 37]. £М(}СDonarutna and W. Z. Heldt, Org. Reactions, 1960, 11. 1 [Л. Г. Дона-рума, В. 3. Хельдт. Орг. реакции, сб. 11. —Пер. с англ./Под ред. И. Ф. Луценко. М., Мир. 1965, с. 7].
Org. Synth.. 1952, 32, 13.
S Huenig, П7. Graessmann, V. Metier, E. Lueke, and IV. Brenninger, Chem. Ber., 1967. 100, 3039.
H. Schnell and J. Nentwig, in «Methoden der Organischen Chemie» (Houben-Weyl), 4th edn., Georg Thieme Verlag, Stuttgart, 1958, vol. XI/2, p. 529. H. Rinke and E. Isiel, in «Methoden der Organischen Chemie» (TIouben-Weyl), 4th edn., Georg Thieme Verlag, Stuttgart, 1963, vol. XIV/2, p. 99.
A. I. Vogel, «А Textbook of Practical Organic Chemistry», 3rd edn., Longmans, London, 1956, p. 773.
Ref. 142, p. 1000.
Org. Synth. Coll. Vol. 1, 1941, 374 [A. T. Кларк, В. P. Kupnep, Синтезы opr. препараюв. co. 1. Пер. <• англ./Под ред. Б. А. Казанского. М., Издатинлит 1949, с. 262].
Org. Synth. Coll. Vol. 2, 1943, 580.
Org. Synth. Coll. Vol. 5, 1973, 54.
Org. Synth. Coll. Vol 3, 1955, 488
Пер. с англ./Под ред. Б. А. Казанского. М., Издатинлит.
9.7. КАРБОНОВЫЕ КИСЛОТЫ
С ДРУГИМИ азотсодержащими заместителями
В. О. САЗЕРЛEHД (University oj Liverpool)
кислот naXZV'0'’’*” обсУ>кде»"ю химии карбоновых КОЙ степени окисления' “’дсржащ"ми ат°мы азота в более высо-оксикислоты, например' «ZilrniT'"”1' а-Ам"'°-лоты (2) и гилппэпстА I J’ М-гидроксиаминокис-
И здесь будут \РпоминятпраТЫ 3 уже обсУждались в главе 9.6 реакциях других поончкпп ЛИШЬ * Случае их образования при
ДРНих производных карбоновых кислот. Азидокисло1ы
261
и их эфиры (4), диазокислогы и их эфиры (5) и диэфири карбоновых кислот (азодиэфпры) (6) уже частично оасемпт*0’ вались (см., например, разд. 6.5.3 и 6.5.4); н ‘^три-
yNH2
°\/==° rchco2h rchco2h
NH 1 NHOH 1 nhnh2
(1) (2) (3)
химия этих соединений будет обсуждена лишь вкратце. Более подробно будут рассмотрены нитрозокислоты и их эфиры (7), таутомерные им гидроксииминокислоты (8), а также нитрокислоты и их эфиры (9).
R‘CHCO2R2
N
II
N
II
N'
(4)
R‘CHCO2R2 I
NO 7
(7)
R'CCO2R2
II +
N
II
N"
(5)
R'CCO2R2
II
NOH
(8)
ro2c4
\N
(6)
R‘CHCO2R2
no2
(9)
9.7.1. АЗИДОКИСЛОТЫ И ИХ ЭФИРЫ
Химия эфиров азидомуравьиной кислоты (азидоформиатов) (10) интенсивно исследована и используется, гораздо менее изучена химия других производных азидокарбоновых кислот. Вследствие этого целесообразно -рассматривать оба названных типа соединений раздельно.
9.7.1.1. Эфиры азидомуравьиной кислоты
Саму азидомуравьиную кислоту (Ю, R = Н) полечить не удается, однако соответствующие сложные эфиры (10) можно лучать [2] реакцией эфира хлормуравьинои кислоты с аз а_ металла, например азидом натрия {схема (1)}. или азидом те Р метилгуанидина (11) [3]. Альтернативно, азиды можно с1’^азата) вать из соответствующего алкоксикарбонилгидразина (кар с е. действием азотистой кислоты; на схеме (2) представлен ' довательность превращений [4], ведущая к трет-бу тилов i „
262
.„пявьнной кислоты. Сходный метод использован также при “X” бенз“др»пового эфира азидомуравьиио,, кислоты [о].
ROCOC1 + NaN3
У
RO-C(
Х\;з
(1)
(Ю)
NMe2
С + N3-
Me2N/ XnH2
(Н)
Низшие эфиры азидомуравьиной кислоты (10) представляют собой перегоняемые жидкости, которые иногда при нагревании разлагаются со взрывом (10, R = Et, т. кип. 114 С, 25 С при 2 мм рт. ст.; 10, R — Ви-трит, т. кип. 73—76°С, однако 10, R = «=СНР112, т.’пл. 37—39°C). У этих соединений имеются два типа харак1ерных реакций: в одних они ведут себя как смешанные ангидриды, в других — как азиды или служат источником ацил-нитренов. Примером первого типа реакций может служить получение трет-бутоксикарбонильных производных аминов и аминокислот [6]; эта реакция находит некоторое применение, что связано с легкостью отщепления трет-бутоксикарбонильной группы под действием ,кислот {схема (3)). Наибольшее использование
хинолин
Ме3СОН + PhOCOCl ---> Me3COCOOPh —
CH2CI2 N2H4
пиридин ---Me3COCONHNH2
Me3COH + MeSCOCl ———Me3COCOSMe—
СНЫз NaNO2
MeCO2H
Me3COCON3 (2)
. CO2
Me3CO‘K+ ----MesCOCOjK*
О
(EtO)2PCl ---------->.
|kns
о
Me3COCOOP(OEt)2
реакция получила
lanfin П0ЛУч11ла для защиты аминогрупп в виде
тррг-бутокси-/г-метоксибсн-алкоксикарбо-при использо-
(3)
H2NCHR'CO2R2 <==* грет-BuOCONHCHR'COeR»
(a) rper-BuOCONg „ MgO(R= = H) EtsN (R2„Alk)
< ) KI, MeCO2H или Нвг. МеСО2Н и Et2O, ЕЮАс пли MeNO2
263
о
Азидная группа азидоформиатов вступает в реакцию с высокоактивными олефинами, например норборнадиеном [8], давая продукт 1,3-диполярного циклоприсоединения {схема (4), путь (а) и схема (5)}, который, однако, при нагревании перегруппировывается. Термическое или фотохимическое расщепление азидоформиатов приводит к алкоксикарбонилнитренам (см. гл. 6.6), которые взаимодействуют с олефинами с образованием N-алкоксикар-бонилазиридинов {схема (4), путь (б)} или продуктов реакции циклоприсоединения, в которой алкоксикарбонилнитрены реагируют как 1,3-диполи {схема (4), путь (в)}. Типичными реакциями этого типа являются синтез [9] производных азепина (12) {схема (6)} и получение [10] аддукта с циклогексеном (13) {схема (7)}. В последнем примере наряду с азиридином (13) образуется продукт внедрения нитрена по связи С—Н (14).
+ N3CO2Et
Ар или 125°С
NCO2Et
NCO2Et (8)
(12)
П п где
Другие примеры включают реакции ацилнитренов, дОб-ведут себя как 1,3-диполи {схема (4), путь (в)}; нИтри'
ного типа были получены из ацетиленов {схема (8)} L Т 264
NHCO2Et
пов (схема (9)} [12] и кетениминов {схема (10)} [13]. Отмечено, однако что в случае нитрилов те же продукты могут образовываться ’ скорее в результате [ 1,3]-сигматропной перегруппировки промежуточного трехчленного циклического соединения [схема (4) путь (б) + (г) ], чем в результате прямой реакции циклопри-
соединения.
PhC=CH + N3CO2Et
(8)
RC=N + N3CO2Et
(9)
Me2C=C=NAr + N3CO2Et
100 °c ------>
(10)
9.7.1.2. а- и p-Азидокислоты и их эфиры
Реакция a-галогенэфиров с азидом натрия в этаноле или ацетоне ведет к соответствующим а-азидоэфирам [14]; аналогично в присутствии 1 экв карбоната калия реагируют а-галогенокислоты [15]. a-Азидоэфиры обычно применяют для синтеза а-аминокислот и их эфиров {схемы (11), (12)} [16].
NaN3 Н2> Pd/C
R'CHCCbR2 —> R‘CHCO2R2 R'CHCO.R2
। ttUH । MeOH ।
Hal N3 NH2
(H)
BrCHCO2Et 1 (СН2)3 | N3CHCO2Et Ph3P=NCHCO2Et NaN3, EtOH | Ph3P 1 HBr. AcOH > (CH2)3 > (CH2)3 >
BrCHCO2Et N3CHCO2Et Ph3P=NCHCO2Et
H2NCHCO2H (СН2)з h2n<!:hco2h
(12)
a-Азидокислоты использовались для получения соединений, содержащих на один атом углерода меньше, чем соответствующие
265
карбоновые кислоты [15]. Последовательность реакций ПредСТав. лена на схеме (13); этот путь служит альтернативой методу Барбье —Виланда.
а-Метиленовая группа эфиров а-азидоуксуснои кислоты активирована к образованию карбаниона; сообщалось [17] о реакции конденсации с /т-метоксибензальдегидом, протекающей по схеме (14).
Прочие азидокислоты и эфиры получают из соответствующих галосоединений замещением галогена азид-анионом; другим общим методом синтеза этих соединений служит присоединение [18] азотистоводородной кислоты к а,Р-ненасыщенной кислоте, ведущее к образованию |3-азидокислоты.
NaN3
К2СО3
RCH2CO2H —>
RCHCOoH
Br
RCHCOjH
N3
SOC или
COC12
RCHC0C1
лз
RCHCO2Et
I
Br
MeCk
NaN3
RCHCO2Et
Аз
-HCI
(13) RC=N
4- CO + N2
ХОЛОД
MeCX >
CHO
+ N3CH2CO2Et
NaOEt
EtOH
1V3
CH=CCO2Et
(U)
9.7.2. ЭФИРЫ ДИАЗОКИСЛОТ
Химия эфиров а-дпазокарбоновы.х кислот уже обсуждалась при рассмотрении диазоалканов в гл. 6.5, и здесь мы рассмотрим этот °ПРОС Л11ШЬ кратко. Простые диазоалканы являются высокоак гивными соединениями, получаемыми обычно только в растворе ’ Н0 ЭФПРЫ а-диазокислот стабилизованы сопряженп ~ стпипт<Л»СПЛу ЭТ0Г° их можно легко синтезировать. Низшие пР жидкости Л11азоэФпров представляют собой желтые маслянис хотя hy u’irV°r0PbIe МОГУТ перегоняться при пониженном давле то используют без перегонки (см. разд. 6.5.3.1)-
° О ?"
. . Д i
N=N=C^ \)R2 -/ '
Rl Ч1
R1
(15)
266
9.7.2.1. Методы получения эфиров диазокислот
Наиболее часто для синтеза эфиров диазокарбоновых кислот используют нитрозирование эфиров а-аминокислот {схема (15)}; этиловый эфир диазоуксусной кислоты (16, R1 = Н, R2 = Et) был первым синтезированным алифатическим диазосоединением [19].
R1
Cr\'H3CHCO2R2
NaNOa
EtgO, Н2О
- * /R! N==N=C\ \co2r2
(15)
(16)
Альтернативно, диазогруппа может быть получена через суль-фонилгидразон, как показано на схеме (16); этот метод успешно [20] использован, например, при наличии в группе R двойной связи. В простейшем варианте {схема (17)} гидразон дпэтилмезоксалатг окисляют оксидом серебра, получая диэтилдиазомалонат [21]
TsNHNH2 SO2CI2. ROH
(НО)2СНСО2Н ------> TsNHN=CHCO2H ---------->
Et3N - +
—> TsNHN=CHCO2R -------N=N=CHCO2R (16)
N"
II nnh2 n+
N2H4 II Ag2o II
EtO2CCOCO2Et EtO2C—C—CO2Et EtO2C—C-CO2Et (17)
а-Диазогруппу можно непосредственно вводить в активированные эфиры, такие, как [3-оксоэфиры, используя реакцию л-толуол-сульфонилазида с соответствующим карбанионом. Можно использовать два общие типа реакций: (а) реакции, в которых активирующая группа X отщепляется одновременно с образованием эфира {схема (18)}, и (б) реакции, в которых сначала отщепляется протон, а затем на второй стадии отщепляется активирующая группа X {схема (19)}. Примеры реакций обоих типов приведены на схемах (20) [22], (21) [23] и (22) [24].
R1 R1 1 MeCeH4SO2N3 - + | х с~сад2 ддд—” N=N-C-CO2R2 (18) - \ 1 о*Чif X = Ph3P+, сно X
X—chco2r (X = СОМе) MeCfjHiSC^Ns • + 1 основание - + ГГТП N=N=CCO2R2 ► N==N=CHCO2R2 (19) \ ’ SIX з)
267
RCH2CO2Et
HCOjEt -------
NaOEt
RCHCO2Et
Aho
—RCCO2Et
EtsN || z
ZM» Ph3p=c^
^COjE t
TsNs ------->
CH2C12
„ + ZMe N=N=C^
• CO2Et
TsN3
МеСОСНгСОгВи-грет • EUN> MeCN>
MeCOCCO2Bu-rper
NaOMe
MeOH
—> N=N—СНСО2Ви-трет
Эфиры а-диазокислот можно получать также ацилированием [25] диазоалканов эфиром хлормуравьиной кислоты {схема (23)} или алкилированием [26] серебросодержащих производных этилдиазоацетата бензил- или аллилгалогенидами {схема (24)}.
Et2O + -
ВгСН2СН2ОСОС1 + CH2N2 --->- BrCH2CH2OCOCH—.N=N (23)
N—N—CHCO2Et 4^. n=N=C—CO2Et N=N=C-CO2Et (24)
E12U I I
Ag R
Несопряженные эфиры диазокислот можно синтезировать [27] методами, аналогичными используемым для получения диазоалканов, а именно нитрозированием соответствующего ацилампноэфлр3 с последующей реакцией образующегося нитрозопроизводного с основанием {схема (25)}. Аналогично можно получать [28] эфиры и-диазокислот путем катализируемого основаниями раскрытия кольца N-нитрозолактамов {схема (26)}.
NO
R’CONH(CH2)nCO2R2 „NaN°2’ 11Ci RiCON(CH2)rtCO2R2 —* или N2Og, или N2O4, ССЦ
K2CO3, МеОН(в’==МеО) или Ва(ОН)2, MeOH(R’=Me)
> ^N^HfCHOn-iCOoR-’
R1 = Me, OMe; R2 = Me, Et, CMe3 и т, д.
NaOMe
MeOH
CO2Me
(CH2)4
CH=N=N
268
9.7.2.2. Реакции эфиров диазокислот
Большинство реакций эфиров диазокислот относятся к двум общим типам: а) внедрение производных карбенов по связи X—У и б) реакции циклоприсоединения либо самих эфиров, либо производных карбенов к олефинам и другим ненасыщенным соединениям.
Реакции внедрения по кислотным связям X—Н часто не требуют катализа и могут протекать через образование соли диазо-ния с последующим нуклеофильным замещением {путь (а) на схеме (27)}; другие реакции внедрения протекают лишь в присутствии катализатора, по-видимому, через промежуточное образование карбенов или карбеноидов {путь (б) на схеме (27)}. Примеры реакций внедрения по разным типам связей О—Н даны на схемах (28)—(30) [29, 30]. В нескольких случаях удалось синтезировать а-ампиокислоты путем реакций внедрения по связи N—Н аминов {схема (31)} [31], хотя этот путь и не обладает общей синтетической значимостью
Си
PhOH + N2CHCO2Et -------> PliOCH2CO2Et
+ + R2O +
R2OH + N2CHCO2Et —> R2OCH2CO2Et •-------> R3O + ROCH2CO2Et (30)
x- x- x-
(27)
(28)
(29)
/NH + N2CHCO2Et
NCH2CO,Et
(31)
Большую группу реакций составляют реакции внедрения по различным связям С—X, где X — атомы Н, О, S или С. С хорошим выходом проходят реакции внедрения по связи С—Н таких, иа-рример, соединений, как кетоны, протекающие через триметилсилильные производные соответствующих енолов {схема (32)} [32]. Связи С—О и С—S простых эфиров или тиоэфиров вступают во
269
АПРЙствие с диазоэфирамп в том случае если первоиачально В'аИ^ппЯвшисся илиды подвергаются затем {1,2 -перегруппировке ооразовавш сся i более высокого порядка), как, например, (или перегрхп ‘ и Г1одобпые реакции могут иметь место
п°каза\°Дао0тОЭфиров или ИХ ссрусодержащих аналогов {схема ”ЛиСЛгУ341 Наибольший интерес в препаративном отношении пред-(34)} р4) _ внедрения по связям С—СО кетонов; при тща-ставляют peai u сл0ВИЙ проведения эти реакции проходят тельном с°б; |о амиУпродуктов. Превращение обычно проводят “пвпТутств °и катализатора, фторбората триэтнлоксония м„ LZ ."«аса, ванр..мер хлорида цинка или тр„лор„да а.,юи. ™Яд™"«ны рыкции'н' "дугами типами связей Х-Y. например зЛвХфир Диазоуксусной кислоты внедряется по связи S-d СУЛЙпюств™т'трп тяпТ'реакций циклоприсоединения по кратным евязя) аналогично тому, как это имеет место в случае азидофор. миатов'. Эфиры «-Д1,азокислот могут реагировать, во-первых, как )™чЗо^цил^ Ск“™Р“3"Р”С0^
ским аддуктам (путь (в) на схеме (39)).
_n2CHCO2E^ BuCOCH2CH2CO2Et Cu, 80 °C (32)
Ph
грет-ВиСОСН3 —+ трет-ВиС=СН2
OSiMe3
_ZPh N.CHCO.BC 1—<Ph U. =!<»» C\CHCO,Et W
I I -------------*- LL
° ^CHCOyEt °
XR2 «1C(XR2):! + N2CHCO2Et —> ---CHCOoEt
XR2 XR2
EtsOBF-t
Me2CO + N2CHCO2Et
CHCO2Et
N2CHCO2Et
MeCO^ Me^
Et3OBF^
CH2Cl3
CHCO2Et
270
+ N2CHCO2Et
EtSCl + N2CHCO2Et —>
(37)
(38)
Указанные три типа реакций иллюстрируются на схеме (39) на примере взаимодействия с олефинами.
Реакции первого типа [путь (а) на схеме] относительно необычны, поскольку для их протекания необходимы условия, при которых образуются карбены; примером может служить реакция
на схеме (40) [37]. Большинство реакций циклоприсоединения протекает либо при повышенных температурах, либо в присутствии меди и других катализаторов и приводит к циклопропанам {путь (б) на схеме (39)}. Этот путь представляет собой удобный общий метод синтеза широкого ряда производных циклопропана, например по схемам (41) и (42) [38, 39].
PhCH=CH2 + N2CHCO2Et ---->
CO2Et
(41)
(^J>CHCO2Et
(42)
271
Реакции с диенами {схема (43)} {40] и лаже с ароматическ системами {схема (44)} также приводят к циклопропановым п * взводным, н это удобный путь к циклогептатриеновым и тропц’₽£ ВЫМ системам [41].
Реакции циклоприсоединения второго типа {путь (б) на схеме (39)} идут также с производными ацетилена; они использованы [42] в синтезе цпклопропенил-катионов {схема (45)}. В общем случае ацетилены могут давать все три типа продуктов циклоприсоединения {см. схему (39)}; направление реакции зависит от условий проведения процесса и структуры применяемых реагентов; примеры реакций, протекающих по путям (а) и (в), даны соответственно на схемах (46) [43] и (47) [44]. Присоединение к нитрильной группе происходит по пути (в), как показывает пример (48), где образуется стабильное производное изоксазола, но не азирин [45].
Ph Ph Ph Ph
PhCSCPh + N2CHCO2Et V W (45)
14U Ц сн V
I
CO2Et X~
PhC=CH + N2CHCO2Et —>
РП
PhC==CPh + N2CHCO2Et —--S°- > ph—/ —OEt
so °c \ /
О
PhC=N -f- N2CHCO2Et ---> ph——OEt
О
РзВ + N2CHCO2Et —> RCH2CO2Et
272
Наконец, реакция этилового эфира диазоуксусной кислоты с алкилборанами служит [46] удобным методом синтеза С-алкили-рованных уксусных кислот {схема (49)}.
9.7.3. АЗОКИСЛОТЫ И ИХ ЭФИРЫ
Единственной важной группой соединений этого типа являются диэфиры азокислоты (17), которые уже рассматривались в разд. 6.5.1.1; здесь будет дан лишь очень краткий обзор химии этих соединений.
/CO2R
Nz NHCO2R
II I
ZN NHCO2R
RO2c/
(17) (18)
Na2CO3 HOC!
+ ClCO2Et -------> EtO2CNHNHCO2Et -------—EtO2CN=NCO2Et (50)
ИЛИ rilMUg 4
пиридин
rper-BuOCONs + H2NNHCO2Bu-7,peT----------->
N- бромсукцннимид
—> TpeT-BuO2CNHNHCO2Bu-TpeT ——------------------>
CH2CI2, пиридин
—> 7’p£T-BuO2CN=NCO2Bu-Tpez’
(51)
Диэфиры азодикарбоновой кислоты (17) обычно получают окислением соответствующих производных гидразина (18) (карбаза-тов); типичные примеры представлены на схемах (50) и (51) [47, 48]. Циклические аналоги соединений (17) имеют богатую энергией Rwc-азосвязь в отличие от трянс-азосвязи ациклических производных и характеризуются соответственно более высокой реакционной способностью. Более стабильные ццс-азосоединения получены окислением соответствующих циклических производных гидразина, например по схеме (52) [49]. Менее стабильные и более реакционные щ/с-азосистемы (19) можно генерировать in situ [50], как показано на схеме (53). Подробнее этот вопрос рассмотрен в разд. 6.5.1.1. Сама азодикарбоповая кислота (17, R = Н)
H2NCONHNHCONH2
PhNH2
О
HNK/
rper-BuOCl ----------9 -78 °C
(52)
не может быть выделена и генерируется [51] лишь как источник восстанавливающего агента, диимида NH = NH, например по схеме (54),
273
Наиболее известными реакциями азодиэфиров являются цикло присоединения по типу присоединения Дильса — Альдера {схема (55)} или аналогичные ему еновые реакции {схема (56)}. Циклоприсоединение наблюдается для простых диенов, например дЛя 2,3-дпметилбутадиена или циклопентадиена [52], и явилось первым примером реакции Дильса — Альдера. Такие диены, как иикло-гексадиен-1,3, вступают в реакции обоих типов [53], в зависимости от того, проводится ли реакция термически или фотохимически {схема (57)}. Циклоприсоединение к а-пирону первоначально протекает по Дильсу — Альдеру, однако последующая потеря
КОН АсОН
H2NCON=NCONH2 ------> [KO2CN=NCO2K] -------> PhNHNHPh (54)
PhN=NPh
+ N2 + CO;
диоксида углерода и присоединение еще одной молекулы азодикарбоксилата ведут к более сложному превращению [54], как показано на схеме (58). Реакция с тропоном, однако, идет в соответствии с обычной схемой я4 + л2-прпсоедпнения [55] {схема (59)}.
Взаимодействие с аллиловыми эфирами относится к еновым реакциям [56] {схема (60)}, но с енольными эфирами и родственными им соединениями, содержащими двойную связь с повышенной электронной плотностью, наблюдаются реакции циклоприсоединения -[2 + л2 {схема (61)} [57]. Некоторые ароматические соединения в присутствии кислых катализаторов, например эфирата трифторида бора, могут вступать в еновую реакцию с диэтилазодикарбоксилатом {схема (62)} [58]. Еновые реакции с системами типа фенилгидразонов [59] протекают через таутомерные превра щения {схема (63)} и формально сходны с реакциями с ароматическими углеводородами. Еновая реакция с дпалкилтпомочевинои открывает интересный, путь синтеза многих карбодиимидов [о Ь
/CO2R
•N
(55)
274
zCO2R
N v
,N (
RO2C h
XNH ro2c^
RO2C
>
EtO2C NHCO2Et
CO2Et
/
EtO2C. /Y
N
Ш“С°2И (58)
vCO2Et
(57)
CO2Et
CO2Et
еще более общее широкое применение имеет комбинированное использование диэфира и трифенилфосфина для удаления элементов Н2О или H2S {схема (64)}.
PhCH2OCH2CH=CH2 + EtO2CN=NCO2Et —
C6He
PhCH2OCH =xCHCH2NNHCO2Et
(60)
Me
СН2
сн
N
CO2Et
EtO2C
/CO2Et r—Nz
---N-
(61)
BF3-Et2O
+ EtO2CN=NCO2Et --------->
Me
(62)
NCO2Et
Me
Me NHCO2Et
275
zCO2CH2Ph NZ д
PhCH=NNHPh + II
PhCH2O2c/
^NNHPh
PhC^
N NHCO2CH2Ph (63)
CO2CH2Ph
S
II RNHCNHR
NCOoEt
NCO2Et
RNIk .NR
\c^ I S Jr EtO2CZ \NHCO2Et
RN=C=NR
Ph3P +
---> Ph3P=S (64)
+
EtO2CNHNHCO2Et
Эфиры азодикарбоновой кислоты или соответствующие амиды действуют также как окисляющие агенты, и циклогексадиены-1,3, например, при взаимодействии с ними дегидрогенизуются [61], давая производные бензола {схема (65)}, тиоспирты окисляются [62] до дисульфидов {схема (66)}. Наконец, очень интересна реакция с диазокетонами [63], при которой происходит миграция этоксикарбонпльной группы {схема (67)}. Эта реакция может быть использована для синтеза а-оксоальдегидов, выделяемых в
м.етил лев о п и м ар ат
EtOoCNHNHCCbEt
виде фенилозазонов; предполагаемая схема синтеза показана на схеме (67).
ArSH + H2NCON=NCONH2 грет~Вц0К>. (Ar— S)2 + H2NCONHNHCONH2 грет-BuOH
NCO2Et
+ - EtO2CN=NCO2Et в РИСНз
ArCOCH=N=N ---------------------
N'
CO2Et_
rn Ft , zCH=NNHPh
.CO2h.t PhNHNH2 /
—► ArCOCH=N—Nf —ГД77-*’ ArC^ \cn Ft Ac0H %NNHPh
(67)
9.7.4. НИГРОЗОКИСЛОТЫ И ИХ ЭФИРЫ
Нитрозокарбоновые кислоты и их эфиры (20) находятся по_ добно нитрозоалканам в таутомерном равновесии с соответств) щими гидроксиминосоединениями (21) (см. гл. 7.1) при уело , что нитрозогруппа не связана с третичным углеродным aroiv
276
В целом получение и реакции этих соединений определяются суммой синтетических методов и свойств, характерных для обеих функциональных групп, за исключением случая а-замещенных соединений, которые будут обсуждаться в этом разделе.
R'CHCO2R2
NO
(20)
R’CCO2R2
II NOH
(21)
R2
R!COCHCO2Et
hno2 ---------->. ИЛИ N2O3, ИЛИ N2O4
R2
R,COCCO2Et
NO
R2CHCO2Et
NO
R2CCO2Et
NOH
Bu
I BuONO. H2SO4;
MeCOCHCO2Et -------—------> BuCCO2Et
H2O у
-NOH
EtONO, NaOEt;
BuCH(CO2Et)2 -------------> BuCCO2Et
H+ ||
NOH
R2ONO
RiCH(C02H)2 ——> R'CCOjH HC1 II
NOH
(68)
(69)
(70)
(71)
(72)
Нитрозокислоты или их эфиры получают одним из трех основных методов: прямым нитрозированием, восстановлением соответствующих нитросоединений и окислением соответствующих производных гидроксиламипа. Наиболее часто используют первый из названных методов. Простейшие эфиры недостаточно активны для а-нитрозирования, однако наличие активирующего а-заместп-теля, как, например, в диэтилмалонате или в этиловом эфире ацетоуксусной кислоты, который может далее удаляться, позволяет проводить нитрозирование, используя самые разнообразные реагенты. Так, p-оксоэфнры нитрозируют оксидами азота [64] и азотистой кислотой {схема (68)} или алкилнитритами в присутствии метанола {схема (69)} [65] или кислот {схема (70)} [66]. эфиры малоновой кислоты обычно нитрозируют [67], используя алкилнитриты и этоксид натрия {схема (71)], а сами малоновые
277
кислоты [68]—в присутствии алкилнитритов и кислоты {схема (72)}. Часто промежуточное нитрозосоединение теряет активирующую ацильную, карбоксильную или алкоксикарбонильную группу, давая простую а-нитрозо (гидроксиимино) кислоту или-эфир. а-Нптроэфпры подвергаются нитрозированию нитрит-анио-ном [69]; образующийся а-нитро-а-нитрозоэфир теряет нитрат-анион, давая а-гидроксииминоэфир {схема (73)}.
NO,
R'CH \o2r2
- NO I
R'CCO^R2 I no2
-oh
----> R'CCO.R2
II
NOH
(73)
R' zNHOH
R'/ ^COjR2
ВГ2 ---->
R1
R"
\ ZN0
/ ^CO2R2
R‘\
^C—NOH R'/
(74)
Окисление N-гидроксиаминокислот или эфиров бромом [79] ведет к соответствующим нитрозосоединениям, хотя в случае а-ги-дроксиаминокислот имеет место декарбоксилирование и образование оксима {схема (74)}. Реакция кетена с нитрозилхлоридом [71] при различных условиях может дать а-гпдроксипмпноуксус-ную кислоту, ее эфир или а-хлоргидроксипминоацетат, в зависимости от используемых реагентов; во всех случаях полученные продукты являются производными а-нитрозоацетилхлорида [22] {схема (75)}.
NOCI, —78°С ROH + C12
--------->- гСН2-СОС1-1 —гт HON=C( I CH2CI2 \co,R
Lno J (22) (75)
|ц2о |rOH
hon=chco2h hon=chco2r
Наиболее важной реакцией а-нитрозо(гидрокспимино)кислот и их эфиров является восстановление до соответствующих производных аминокислот; восстановление эфиров гпдроксипмино.мало-новой кислоты представляет особый интерес, поскольку используется для получения ацпламиномалонатов, которые применяются |72] в синтезе аминокислот {схема (76)}. Сходную последовательность реакций нитрозирования и восстановления используют для генерации аминоацетоуксусного эфира in situ в синтезе пирролов по Кнорру; пример [73] этого классического метода синтеза пока-
278
зан на схеме (77) [см. также том 8 (русского издания), гл. 17.1].
Zn, MeCOjH
CH2(CO2Et)2 HON=C(C02Et)2 AcNHCH(CO2Et)2
Pdc.H2: (76)
v
Cl’NH3CH(CO2Et)2
Zn, MeCO2H,
NaNO2 только in situ
MeCOCH2CO2Et M MeCOCCO2Et ------------>
Mellon ||
NOH
Me, /CO2Et
MeCOCHjCOEt / \
.—> rMeCOCHCO2Etl --------------EtO2C——Me (77)
L nh2 J NH
Доступность таких соединений, как диэтилгидроксниминомало-нат {схема (76)} и этилгидроксииминоацетоацетат {схема (77)} и возможность их окисления [74] до диэтилнптромалоната и этилнитроацетата соответственно открывает новый путь синтеза указанных нитроэфиров наряду с другими известными методами их получения {схемы (78), (79)}. Прочие а-гидроксииминоэфиры также могут быть окислены до а-нитроэфиров.
CF3CO3H HON=C(CO2Et)2 -------> O2NCH(CO2Et)2 (78)
CF3CO3H
MeCOCCO2Et -------------> ГМеСОСНСО2Е11 —> O2NCH2CO2Et (79)
|| или Na2Cr2O? 1 I
NOH L NO2 J
9.7.5. НИТРОКИСЛОТЫ И ИХ ЭФИРЫ
9.7.5.1. Методы получения нитрокислот и их эфиров
Существует целый ряд общих методов получения ннтрокислот и их производных. Большинство из них относится к синтезу а-нп-трокислот и их производных.
(1) Нитрование
Прямое нитрование служит важным методом введения а-иптро-группы в эфиры, в которых a-положение активировано присутствием алкоксикарбонильной или ацильной группы. Так, диэтил-нитромалонат приготовлен [45] нитрованием диэтилмалоната азотной кислотой {схема (80)}; для получения моноалкилнрован-ных малоновых эфиров использовали либо азотную кислоту [76], иго приводит к нитромалоиовому эфиру, либо цианогидрпннитрат
279
ацетона и гидрид натрия [77], дающие соответствующий нитроацетат {схемы (81), 82)}. Сообщалось [78], что при взаимодействии моноэтнлового эфира малоновой кислоты и азотной кислоты получен а,а-дннптроацетат {схема (83)}, однако химия динитросоединений не разработана в той степени, в какой изучена химия мононитроэфиров. Реакция алкилированных ацетоацстатов с циа-ногидриннитратом ацетона и гидридом натрия дает нитроацетаты [77] по схеме (84); она явно аналогична реакции с алкилированными малонатами {ср. схема (82)}.
HNO3
CH2(CO2Et)2 ----> O2NCH(CO2Et)2
RCH(CO2Et)2
HNO3
или HNO3, Ac2O, или НИОз, поли-фосфорная кислота
no2
RC(CO2Et)2
✓ONO2
RCH(CO2Et)2 + Me2CZ
^CN
NaH; --------->. NaOH, H2O
/NO2 RCH
\x)2Et
/ 2 HNO3 2‘ \
CH2 --------> CHCO2Et
\со2н O2n/
,ONO2
MeCOCHRCO2Et + Me2CZ Nzn
NaH
---> RCHCO2Et
NO2
(80)
(81)
(82)
(83)
(84)
(2) Нуклеофильное замещение
Замещение галогена в галогеноэфирах нитрит-ионом служит вторым по важности методом синтеза нитроэфиров, в особенности для а-замещенных систем, которые дают высокие выходы нитросоединений. Наиболее удобным реагентом [79] является нитрит натрия, растворенный в диметилформамиде или диметилсульфоксиде с флороглюцином для поглощения избытка нитрита {схема (85)}. Этот метод позволяет получать широкий спектр а-нитроэфи-ров. В ранее применяемом методе [80] использовали нитрит серебра и иодзамещенный эфир; этот путь {схема (86)} может давать более высокие выходы продуктов, если использование нитрита натрия недостаточно эффективно. Если в синтезе применяют га-логенокислоты в виде их натриевых солей, то образуются нитроалканы; такой путь использован для синтеза [81] этил-5-нитро-пентаноата {схема (87)}; это в достаточной степени общий метод. Более специфическим примером реакции замещения является раскрытие кольца (3-пропиолактона под действием нитрита натрия;
2Ь0
при этом образуется Р-нитропрошюновая кислота {схема (88)}.
ДМФА или ДМСО
RCHCO2Et + NaNO2 —— ~ r н -» RCHCO2Et (85)
। 1,3,5-(НО)зСбНз ।
Hal NO2
Et2O
A^eCHCO2Et + AgNO2——> MeCHCO2Et (86)
I I
I NO,
/Вг NaNO2
ЕЮ2С(СН2)зСН' ---------------->- EtO2C(CH2)3CH2NO2 (87)
^CO2Na
(3) Получение из нитроалканов
Нитроалканы вступают во взаимодействие с метоксимагний-метилкарбонатом MeOMgOCOOCH3 [82], который реагирует и как основание, и как карбоксилирующий агент, давая а-нитрокислоты. Последние легко могут быть превращены в а-нитроэфиры, в связи с чем указанный метод представляет удобный путь синтеза а-ни-трокислот и их эфиров, а также родственных им соединений {схема (89)}. Аналогично, анионы, генерируемые из нитроалканов, реагируют с изоцианатами с образованием амидов а-нитро-кислот {схема (90)} [83].
/ОМе R’R2CHNO24-Mg/
Х)СО2Ме
/NO,
—► R’R2C^
\зо2н
ДМФА /NO2 НС1> о оС
------> R’R2C/ ------>
'V.OoMgX
r3oh, н+ /NO,
---------> R'R2C/ , (89)
\CO,R3
R’R2CHNO2 + ArNCO —>
R1 I ArNHCOCNO2
R2
(90)
У-Нитроэфиры можно легко получить по реакции Михаэля пу-м присоединения анионов нитроалканов к а,|3-ненасышснным фирам {схема (91)}; метод [84] в достаточной степени общ и может быть распространен на синтез производных ннтроглутарата, ли в качестве ннтросоедннения использовать а-нитроэфнр. ДикК0Нец’ Реаки'ия нитрометана с гидроксидом калия приводит к алиевой соли а-нитрокислоты [85], которую можно
281
этерифицировать с получением эфиров а-нитроуксусной кислоты {схема (92)}.
NO2 основание I
R'R2CHNO2 + CH2=CHCO2R3 ------------>- R'R2CCH2CH2CO2R3
КОН °\+ ЕЮН
CH3NO2 -----> N=CHCO2 -----------> O2NCH2CO2Et
-О/ н +
(91)
(92)
(4) Окислительные методы
Об окислении а-гидроксииминоэфиров в а-нитроэфиры уже упоминалось в разд. 9.7.4.
9.7.5.2. Реакции нитрокислот и их эфиров
(1) Реакции карбоксильной группы
а-Нитрокнслоты при нагревании легко декарбоксилируются, что затрудняет их этерификацию; тем не менее разработан ряд методов, применяемых для синтеза а-нитроэфиров {см. схемы (89) и (92)}. Так, катализируемая кислотами этерификация при низкой температуре [86] приводит к получению с неплохими выходами метилового и этилового эфиров, как иллюстрирует схема (93) на примере синтеза метил- и этилннтроацетатов.
CH3NO2
кон
--->
О\+
/N=CHCO2
"Си
МеОН, H2SO4, Na2SO4
------:-----——> O2NCH2CO2Me
от -о до —И °C
EtOH, HCI, MgSO4 от -8 до —10 °C ''
(93)
O2NCH2CO2Et
Моно- и Дианионы а-нитрокислот можно генерировать реакцией с основаниями, например с гидроксидом калия; осторожным действием кислоты можно вновь регенерировать нитрокислоты. Учитывая нестабильность а-нитрокислот, легко отщепляющих ССЬ, большинство реакций проводят с использованием а-нитроэфиров. В противоположность а-нитрокислотам, (3-, у- и далее нитрозаме-щенные кислоты проявляют обычные свойства, присущие обеим функциональным группам, и не обнаруживают повышенной склонности к декарбоксилированию.
Получение хлорангидрпдов непосредственно из а-нитрокислот связано со значительными трудностями; однако эти соединения могут быть получены при действии хлора на а-нитрогидразиды {схема (94)}. Гидразиды использованы также [87] в деградации по Шмидту, приводящей к а-нитроизоцианатам {схема (9э)}; 282
гидразиды Р-, у- и далее замещенных нитрокислот также могут быть превращены в соответствующие нитроизоцианаты.
O2NCH2CO2Me
R'IVCCONHNH;, NO2
N2H4 ----►
С12
---> R'l^CCOCl
NO2
O2NCH2CONHNH2
(94)
NaNO2. 11C1;
---------> O2NCH2NCO (95)
(2) Реакции анионов а-нитроэфиров
а-Нитроэфиры легко образуют анионы, которые принимают участие в реакциях Михаэля и в реакциях конденсации с альдегидами и кетонами, однако при действии алкилгалогенидов не удается провести алкилирование по атому углерода. Типичные реакции конденсации с альдегидами [88, 91] представлены на схемах (96) —(98). Если реакцию не проводить в тщательно контролируемых условиях, то возможно осуществление ее обеих стадий; в присутствии тетрахлорида титана в качестве катализатора протекает [89] реакция, аналогичная конденсации по Кновенагслю {схема (99), ср. схемы (96) —(98)}.
Реакция Михаэля с целым рядом а,[3-ненасыщенных карбонильных соединений, акрилонитрилом и даже с нитроалкенами легко протекает в присутствии оснований. Это открывает возможность получения полифункциональных нитросоединений, которые потенциально могут найти применение в синтезе гетероциклов.
/NO, RCHO + H2C^
NcO2Et
HCI, EtOH /NO2
-------> RCHCH
I ^CO.Et
OH
Лс2О;
Na2CO2
хо2
O2NCH2CO2Et ( /NO2 \
-----------> RCH Cl/
\ NCO2Et/2
(96)
/NO:
ArCHO + Н2СХ
2
МеКНзСГ ( /NO2 \
------------> Агсн сн;
NaHCOa, EtOH ^CO R / 2
(97)
zNO2 M«3NCH2Ph, BuO”
=0 + H2C\
^CO2R
/NO2 RCHO + H2CZ
CH(NO2)CO,Et
(98)
Примеры реакций Ми хаэля
CH(NO2)CO2Et
zNO2
TiCl.t. пиридин /
------------> rch=c ТГФ
(99)
, - г__[90] с участием' а-нитроацетата
финов представлены на схемах (100) и (101). Сообщалось
оле-
и также
283
о реакциях с двойными связями C—N, например при синтезе производных изоксазола (23) [схема (102)].
/NO2 NaOEt EtOjCv
H2CZ + ” снсн2сн2сно
[91]
(ЮО)
н2с;
\сО2Ме
R’CH=NR2
сно
о
no2
Ph
o2n
о
Ph /NO, СНСО2Ме R'\ /NO2 r2nh2 ,CHCHZ ------------->
(101)
O2NCH2CO2Et
CO2Et
—> R2NHCO----;
R’
—CONHR2
N—О (23)
(Ю2)
(3) Восстановление
Нитрогруппа и сложноэфирная группа нитроэфиров могут восстановлены селективно; эти реакции можно рассматривать в числе других общих свойств, характерных для обеих функций. Примеры, иллюстрирующие селективное восстановление [92], представлены на схемах (103), (104); о других общих методиках см. в соответствующих главах, посвященных нитросоединениям (гл. 7.2), карбоновым кислотам (гл. 9.1) и эфирам (гл. 9.7). Главным направлением использования реакции восстановления zNO2 н2, Pd/C /NH2 RCHZ -----------------------------> RCHZ
быть
нитро-
- (ЮЗ)
со2н
LiAlH4, Et2O
O2N(CH2)4CO2Me QT _3Q —
O2N(CH2)4CH2OH
(104)
СН3СО" кн4
O2NCH(CO2Me)2 ---- —....>
CO2Me -i
NH
NO2
CH2C(CO2Me)2 NaQMe
H2, Nl Ренея;
10% AcOH
HN(CH2CNO2)2
СО2Ме
Оо
NH .СН2СНСО2Н
индол
СН2СНСО2Ме
NH
NH2
(105)
284
эфиров п нитрокислот является получение аминокислот, примерами чего могут -ужить синтез триптофана [93] по схеме (105) {ср. схему (76)}, а также синтез других гетероциклических систем, например пирролидона [94] {схема (106)}.
Некоторые синтезы гетероциклических соединений основаны на восстановлении и циклизации различных о-ннтрозамещенных ароматических эфиров. Подробное обсуждение подобных реакций не входит в задачу настоящего раздела. Характерные примеры
Me2CHNO2 \aort /---\ i ,
2 Na0Et_> EtO2C/ \/ Hz. Pd /---\ /
z^\cO2Et O2h/\ "ли Zn’ HOAc* o=\ ZX (t06)
NH 4
представлены на схемах (107) и (108); в них нитрогруппа восстанавливается соответственно до амино- [95] и гидроксиаминогруп-пы [96].
n2h( -------------> Pd/C, EtOH
(108)
ЛИТЕРАТУРА
1. Н. Bretschneider and Г. Vetter, Monatsh, 1958, 89, 625.
2. M. О. Forster and H. E. Fieri, J. Chem. Soc., 1908, 93, 81; I. Hoi a, Coll. Czech. Chem. Comm., 1964, 29, 1079.
3. X. Sakai and J.-P. Anselme, J. Org. Chem., 1971, 36, 2387.
4. L. A. Carpino, B. A. Carpino, P. J. Crowley, C. A. Giza and P. H. Ter/y, g. Synth., 1964, 44, 15; M. A. Insalaco and D. S. Tarbell, Org. Synth 1970 50,9.
5. C. Ruchardt, S. -Eichler, and P. Panse, Angew. Chem. Internat. Edn., 1963, z, 619
6. R. Schwyzer, P. Sieber, and H. Kappeler, Helv. Chim. Acta, 19o9, ^'2622, R. A. Boissonnas, Adv. Org. Chem., 1963, 3, 171; К. P. Polzhojer, Tetrahedron Letters, 1969, 2305.
7. F. Weygand and K. Hunger, Chem. Ber., 1962, 95, 7.
8. A. C. Oehlschlager P. Tillman, and L. H. Zalkow, Chem. Comm. 1965 o96
9. Для общего обзора по химии нитренов см.: «Nitrenes», ed. W. J-wowski ley, New York, 1970; K- Hafner and C. Kbnig, Angew. Chem. Internat Edi , 1963, 2, 96; R. J. Cotter and W. F. Beach, J. Org. Chem 1964 29, 751, K. Haf ner, D. Zinser, and К L. Moritz, Tetrahedron Letters, 1964 1 AW.
10. W. Lwowski, T. J. Maricich, and T. W. Mattingly, Jr., J. Amer. Chem. Soc., 1963, 85, 1200.
И. P- Huisgen and H. Blaschke, Tetrahedron Letters, 1964, 1409^
12. ]V. Lwowski, A. Hartensteln, C. de Vita, and R, L. Snuck, Telialiedron Letters, 1964, 2497; R. Huisgen and H, Blachke, Annalen, 1965, 686, 146,
285
13. IT. J. Kauffman, J. Org. Chem., 1970, 35, 4244.
14. AL O. Forster and H. E. Fierz, J. Chem. Soc., 1908, 93, 79; IV F Huber i Amer. Chem. Soc., 1955, 77, 112; A. Vollmar and Al. S'. Dunn J Огр ClJm 1960, 25, 387. ' ' em '
15. A. Hassner, R. J. Isbister, R. B. Greenwald, J. T. Klig, and E C Tatilor Tetra hedron, 1969, 25, 1637. ’ J ' ira’
16. F. Lingens, Z. Naturforsch., 1960, 15b, 811.
17. N. Hemetsberger, D. Knittel, and H. Weidmann, Monatsh., 1969, too 1599. ’ ’
18. J. H. Boyer, J. Amer. Chem. Soc., 1951, 73, 5248.
19. 7’. Curtins, Ber., 1883, 16, 2230; O. Silberrad, J. Chem. Soc. 1902 600-AL E. Searle, Org. Synth. Coll. Vol. 4, 1963, 424, 426.
20. C. J. Blankley, F. J. Sauter, and H. 0. House, Org. Synth., 1969 49
21. E. Ciganek, J. Org. Chem., 1965, 30, 4366.
22. AL Regitz and F. Menz, Chem. Ber., 1968, 101, 2622.
23. G. R. Harvey, J. Org. Chem., 1966, 31, 1587.
24. S.-O. Lawesson, S. Gronwall, and R. Sandberg, Org. Synth., 1962, 42, 28; M. Regitz, J, Hocker, and A. Liedhegener, ibid., 1968, 48, 36.
25. J. Frank and R. Schwuzer, Experientia, 1970, 21, 1207; H. Staudinger, J Becker and H. Hirzel, Ber., 1916, 49, 1978, 1982.
26. U. Schollkopf and N. Rieber, Angew. Chem., 1967, 79, 238; Chem. Ber., 1969, 102, 488.
27. H. Reimlinger and L. Skaltebol, Chem. Ber., 1960, 93, 2163; H. Reimlinger and J. F. Al. Oth, ibid., 1964, 97, 331; E. H. White and R. J. Baumgarten, J. Org. Chem., 1964, 29, 2070.
28. W. Pritzkow and P. Dietrich, Annalen, 1963, 665, 88.
29. K. Shank, Chem. Ber., 1970, 103, 3093.
30. E. Baltazzi, Compt. rend., 1955, 241, 321.
31. T. Saegusa, У. Ho, S. Kobayashi, K. Hirota, and T. Shimizu, Tetrahedron Letters, 1966, 6131.
32. R. Le Goaller and J.-L. Pierre, Compt. rend., 1973, 276, 193.
33. H. Nozaki, S. Morluti, H. Takaya, and R. Noyori, Tetrahedron Letters, 1966, 5239.
34. A. Schonberg and K. Praefcke, Tetrahedron Letters, 1964, 2043; Chem. Ber., 1966, 99, 196. ,, ,, , , . „
35. IV. T. Tai and E. W. Warnhoff, Canad. J. Chem., 1964, 42, 1333; H. Machleidt and V. Hartmann, Annalen, 1964, 679, 15; W. L. Mock and M. E. Hartman, J. Amer. Chem. Soc., 1970, 92, 5767.
36. F. Weygand and H. J. Bestmann, Chem. Ber., 1956, 89, 1912.
37. L. F. Fieser and M. A. Peters, J. Amer. Chem. Soc., 1931, 53, 4080.
38. H. M. Walborsky and L. Plonsker, J. Amer. Chem. Soc., 1961, 83, 2138; C. Kaiser, J. Weinstock, and M. P. Olmstead, Org. Synth., 1970, 50, 94.
39. R. K. Armstrong, J. Org. Chem., 1966, 31, 618.
40. J. Warkentin, E. Singleton, and J. F. Edgar, Canad. J. Chem I960, 43. 34a .
41. C. Grundmanti and G. Ottmann, Annalen, 1953, 582, 163; R. Huisgen an G. Juppe, Chem. Ber., 1961, 94, 2332.
42. R. Breslow, R. Winter, and M. Battiste, J. Org. Chem,. 1959, 24, 415.
43. E. Buchner and L. Lehmann, Ber., 1902, 35, 35.
44. R. Breslow and D. Chipman, Chem. and Ind. (London), 1960, 110a.
45. R. Huisgen, H. J. Sturm, and G. Binsch, Chem. Ber., 1964, 97, 2864.
46. J. Hooz and S. Linke, J. Amer. Chem. Soc., 1968, 90, 6891. _ ..
47. .V. Rabjohn, Org. Synth. Coll. Vol. 3, 1955, 375; J. C. Kaiter, Org. Synth, con.
Vol. 4, 1963, 411.
48. L. A. Carpino and P. J. Crowley, Org. Synth., 1964, 44, 18.
49. R. C. Cookson, S. S. H. Gilani', and I. D. R. Stevens, Tetrahedron Letters, laoz,
50. R. A. Clement, J. Org. Chem., 1960, 25, 1724; 1962, 27, 1115; T. J. Kealy, J. Amer. Chem. Soc., 1962, 84, 966.
286
51. E. J. Corey. D. J. Pasfo, and W. L. Mock, J. Amer. Chem. Soc., 1961, 83, 2957; E. E. van Tamelen, R. S. Dewey, and R. J. Timmons, ibid., 1961, 83, 3725. 3729; E. J. Corey and W. L. Mock, ibid.. 1962, 84, 685.
52 O. Diels, J. H. Blom, and W. Roll, Annalen, 1925, 443, 242, В. T. Gillis and P. E. Beck, J. Org. Chem., 1962, 27, 1947; 1963, 28, 3177.
53. W. A. Thaler and B. Franzus, J. Org. Chem., 1964, 29, 2226; R. Askant, Chem. Ber., 1965, 98, 2551.
54. P. C. Arora and D. MacKay, Chem. Comm,, 1969. 677.
55. У. Kitahara, I. Murata, and T. Nit la. Tetrahedron Letters, 1967, 3003.
56. T.-L. Ho and С. M. Wong, Synth. Comm., 1974, 4, 109.
57. R. W. Hoffmann and H. Hauser, Angew. Chem. Internat. Edn., 1964, 3, 380; J. Firl and S. Sommer, Tetrahedron Leiters, 1969, 1133.
58. R- Htiisgen, F. Jakob, W. Siegel, and A. Cadus, Annalen, 1954, 590, 1; G. Cas-nati, Gazzetta, 1964, 94, 1221; R. B. Carlin and AL S. Moores, J. Amer Chem Soc., 1962, 84, 4107; R. C. Cookson, I. D. R. Stevens, and С. T. Watts, Chem. Comm., 1965, 259.
59. В. T. Gillis and F. A, Daniher, J. Org. Chem., 1962, 27, 4001.
60. N. Rabjohn, Org. Synth. Coll. Vol. 3, 1955, 375.
61. G. Mehta and S. K. Kapoor, Org. Prep. Proc. Internat., 1972, 4, 257.
62. F. J. Smentowski, J. Amer. Chem. Soc., 1963, 85, 3036.
63. E. Fahr and F. Scheckenbach, Annalen, 1962, 655, 86.
64. H. Adkins and E. W. Reeve, J. Amer. Chem. Soc., 1938, 60, 1328; O. Touster, Org. Reactions, 1953, 7, 327 [О. Тоустер. Орг. реакции, сб. 7. — Пер. с англ. М„ Издатинлит, 1956, с. 409].
65 Н. R. Snyder, J. Н. Andreen, G. W7. Cannon, and C. F. Peters. J. Amer. Chem Soc., 1942, 64, 2082.
66. R. H. Barry and W. H. Hartung, J. Org. Chem., 1947. 12, 460.
67. J. C. Shivers and C. R. Hauser, J. Amer. Chem. Soc., 1947, 69, 1264.
68. 0. Touster, Org. Reactions, 1953, 7, 327 [О. Тоустер. Орг. реакции, сб. 7.— Пер, с англ. М., Издатинлит, 1956, с. 409].
69. N. Kornblum and J. Н. Eicher, J. Amer. Chem. Soc., 1956, 78, 1494.
70. W. Pritzkow and W. Rosier, Annalen, 1967, 703, 66.
71. T. W. Rave and D. S. Breslow, J. Org. Chem., 1971, 36, 3813.
72. .4. J. Zambito and E. E. Howe, Org. Synth. Coll. Vol. 5, 1973, 373; W. H. Hartung, J. H. R. Beaujon, and G. Cocolas, ibid., 1973, 376.
73. H. Fischer, Org. Synth. Coll. Vol. 2, 1943, 202.
74. W. D. Emmons and A. S. Pagano. J. Anter. Chem. Soc., 1955, 77, 4557.
75. D. I. Weisblat and D. A. Lyttle, J. Amer. Chem. Soc., 1949, 71, 3079.
76. J. P. Lorand, J. Urban, J. Overs, and Q. A. Ahmed, J. Org. Chem., 1969, 34, 4176; W. Steinkopf and A. Supan, Ber., 1910, 43, 3239; J. P. Kispersky and K. Klager, J. Amer. Chem. Soc., 1955, 77, 5433.
77. №. D. Emmons and J. P. Freeman, J. Amer. Chem. Soc., 1955, 77, 4391.
78. L. Bouveault and A. Wahl, Compt. rend., 1903, 136, 159.
79. N. Kornblum, R. K. Blackwood, and J. №. Powers, J. Amer. Chem. Soc. 1957, 79, 2507; N. Kornblum and R. K. Blackwood, Org. Synth. Coll. Vol. 4, 1963, 454; N. Kornblum, Org. Reactions, 19G2. 12, 101 [H. Корна,vom. Opr. реакции, сб. 12. — Пер. с англ./Под ред. И. Ф. Луценко. М., Мир, 1965, 117 с.].
80. F. L. М. Pattison, №. J. Cott, №. С. Howell, and R. №. White, J. Amer. Chem. Soc., 1956, 78, 3484.
81. F. C. Whitmore and M. G. Whitmore, Org. Synth. Coll. Vol. 1, 1943. 401; №. Treibs and H. Reinheckel, Chem. Ber., 1954, 87, 341.
82. M. Stiles and H. L. Finkbelner, J. Amer. Chem., Soc., 1959, 81, 505; H. L. Fink-beiner and G. №. Wanner J. Org. Chem., 1963, 28, 215.
83. R. N. Boyd and R. Leshin, J. Amer. Chem. Soc., 1953, 75, 2762.
84. O. von Schickh, Angew. Chem., 1950, 62, 554; J. Colonge and J.-M. Pouchol, Bull. Soc. chim. France, 1962, 596.
85. F. Feuer, H. B. Hass, and K. S. Warren, J. Amer. Chem. Soc., 1949, 71, 3078.
86. V. E. Matthews and D. G. Kubler, J. Org. Chem., 1960, 25, 266; S. Umezawa and S. Zen, Bull. Chem. Soc. Japan, 1963, 36, 1143.
287
87 Н. Е. Ungade and I.. II? Kissinger, J. Org. Chem., 1957, 22, 1662; 1959 24 1244. ’ ’
88 4. Dornow and .1. Frese, Annalen, 1952, 578, 122; Д. Dortioim and G. Wiehler ibid., 1952, 578, 113.
89. U?. Lehnert, Tetrahedron Letters, 1970, 4723; Tetrahedron, 1972, 28, 6G3.
90. D. Ginsburg and R. Pappo, J. Chem. Soc., 1951, 938; E. D. Bergmann, D. Ginsburg, and R. Pappo, Org. Reactions, 1959, 10, 179.
91. S. Utnezawa and S. Zen, Bull. Chem. Soc. Japan, 1963, 36, 1143, 1146, 1150
92. H. Feuer and T. J. Kucera, J. Amer. Chem. Soc., 1955, 77, 5740; M. II. Quid and K. Klager, Tetrahedron, 1963, 19, 77.
93. T: Okada, Bull. Chem. Soc. Japan, 1959, 32, 1165.
94 A. A. Patched, F. Hoffman, F. F. Giarrusso, H. Schwann, and G. E. Arth J. Org. Chem., 1962, 27, 3822.
95. C. A. Grob and 0. Weissbach, Helv. Chim. Acta, 1961, 44, 1748.
96. R. T. Coutts, Л1. Hooper, and D. G. Wibberley, J. Chem. Soc., 1961, 5058.
9.8. СЛОЖНЫЕ ЭФИРЫ
И. О. Сазерленд (University of Liverpool}
Сложные эфиры карбоновых кислот (1) представляют важную группу соединений [1], поскольку они используются в целом ряде химических реакций, а также часто встречаются в качестве природных продуктов.
Сложные эфиры можно получать непосредственно из карбоновых кислот, либо в ряде случаев алкоксикарбонильная группа —COOR может быть получена из других, не столь близких функциональных групп. Эти синтетические методы обсуждаются в разд. 9.8.1, где рассматриваются общие методы получения сложных эфиров. Получение некоторых специфических типов эфиров, например эфиров аминокислот, обсуждалось в предыдущих главах.
Физические свойства эфиров кратко описаны в разд. 9.8.2, где, в частности, рассмотрены спектроскопические данные и структура алкоксикарбонильной группы.
Наконец, в разд. 9.8.3 обсуждаются общие реакции сложных эфиров; описание более специфических реакций отдельных типов эфиров можно найти в предыдущих главах части 9. В настоящую главу включен материал о синтезе и реакциях циклических эфиров и лактонов (2).
Сложные эфиры и лактоны широко распространены в природе, и хотя подробное обсуждение природных продуктов выходит за рамки данного тома целесообразно упомянуть здесь несколько важных примеров. По существу все продажные жиры и масла [2] растительного и животного происхождения состоят почти целиком из сложных эфиров глицерина (3), называемых триглицеридами. В этих соединениях глицерин этерпфицирован различными длинно-цепочечными карбоновыми кислотами. Другим важным классом
288
природных веществ, присутствующим в растениях, являются тан-нины [3], которые частично представляют собой смесь различных эфиров галловой кислоты и глюкозы (4). Из лишайников можно выделить сложные эфиры ароматических карбоновых кислот и фенолов, которые известны под названием депсидов. Известны и их циклические аналоги, депсидоны, например диплоицин (5) [4]. Микроорганизмы вырабатывают множество соединений, содержащих эфирные связи, от простых по структуре эфиров до макроциклических лактонов; последние соединения, включающие мак-
10 Зак. 395
289
ролиды [5], например эритромицин (6), и макротетролиды [6],На. пример динактин (7), особенно важны вследствие их бактерицидных свойств. И наконец, следует отметить, природные эфиры, обладающие инсектицидными свойствами, это пиретрины [7], содержа, щиеся в Chrysanthemum cinerariaefolium-, ппретрин I (8) может служить типичным представителем этой группы соединений.
Синтетические полиэфиры [8] являются важнейшими представителями класса полимеров, и в течение многих лет они находят широкое применение в текстильной промышленности; однако рассмотрение этих соединений опять выходит за рамки данного тома.
В настоящей главе в ходе обсуждения материала даются многочисленные отсылки на синтетические методы по данным Organic Syntheses [9], однако отсылки на отдельные статьи этой важной области, как правило, приводиться не будут.
9.8.1. МЕТОДЫ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ
9.8.1.1. Этерификация
(1) Реакция карбоновых кислот и спиртов
Сложные эфиры образуются при взаимодействии карбоновой кислоты (9) и спирта (10), эта реакция получила название этерификации. Обратную реакцию, а именно образование кислоты и спирта из эфира, называют гидролизом или омылением {схема (1)}. Механизм обеих реакций детально исследован [10, 11], и установлен ряд общих закономерностей. Проведение реакции в отсутствие катализатора не представляет практического интереса вследствие малой скорости протекания процесса; значительно более интересны три механизма, предложенные для протекания реакции в присутствии кислого катализатора. Первым и наиболее важным из них является бимолекулярный механизм, состоящий в расщеплении связи ацил-кислород {связь (а) на схеме (1)} и известный как механизм Ллс2, в соответствии с предложенной Ингольдом номенклатурой [11] {схема (2)}. Второй механизм, применимый лишь к реакциям, протекающим в сильно кислой среде, например в серной кислоте, представляет собой мономолекулярное превр щение, включающее расщепление связи ацил-кислород {механиз! Лдс1, схема (3)}. Третий механизм включает расщепление свяа алкил-кислород {связь (б) на схеме (1)}; это мономолекулярна реакция, которая имеет место лишь в тех случаях, когда сПИР R2OH способен давать стабильный карбений-ион, например пр R2 = бензил, аллил или трет-алкил. Этот третий механизм пред ставлен на схеме (4) и известен как по номенклатуре Ингол -да. Существование бимолекулярного аналога этого механиз , Л.4/,2, в принципе возможно, но не представляется доказанным, роятно в силу того, что факторы, способствующие алкильному Ра '
29Q
щеплснню, способствуют также мономолекулярному протеканию
процесса. ° Q
II . (б) этерификация « (а) (б)
r1J^h + r’-oh < ...— > R1-—С—-O—-R2 4-Н2О (1)
/д\ (10) гидролиз
Механизм Аде 2 R’CO2H + II+ R’COOH 2
R'COOH, + R2OH ^Z± R1 COOR2HH2O (2)
R'COOR-II R >COOR2 + H+
Механизм Аас 1
R‘CO2H + H+ ^Z± r‘co6h2
R‘COOH2 ^z± R'CO + R2OH R'CO+ + H2O 7~> R‘COOR2H (3)
R‘COOR2H ^z ± R‘COOR2+H+
Механизм Aal 1
R2OH + H+ ^Z ± R26n2
r26h2 yz± r R2 + R’COOH 2 + H2O R‘COOR2H (4)
R'COOR2H qz ± R!COOR2 + H+
Большинство методик, используемых для прямого получения эфиров из карбоновых кислот и спиртов, может быть соотнесено с одним из приведенных грех механизмов, хотя дополнительно имеется целый ряд других методик ацилирования спиртов, и на них мы остановимся позже в этой главе. Катализируемая кислотами этерификация все еще часто используется для синтеза сложных эфиров, и в особенности этиловых и метиловых эфиров. Возможности метода иллюстрируют схемы (5) — (26), где приведены хорошо изученные реакции, описанные в Organic Syntheses [9]. Отметим, что наиболее часто в качестве кислых катализаторов применяют серную кислоту, хлорид водорода (метод Фишера) [12] и толуолсульфокислоту и что этот метод может быть использован для синтеза ненасыщенных эфиров, кетоэфиров, эфиров галогенсодержащих кислот, гидроксикислот и алкоксиэфиров. В большинстве случаев механизм реакции не исследован, но, как полагают, почти все (а возможно и все) перечисленные реакции протекают в соответствии с обычно наблюдаемым механизмом Аас2 {см. схему (2)}. Интересно также отметить, что образование эфиров из относительно сильных органических кислот, таких, как щавелевая и муравьиная кислоты [13] может идти и в отсутствие кислого катализатора. Реакции, в которых используются растворители, такие, как тетрахлорид углерода, бензол или толуол, обычно проводят
ю*
291
в аппарате Дина и Старка [9], позволяющем с помощью азеотроп-ной отгонки удалять воду из зоны реакции. Это способствует сдвигу равновесия схемы (1) в направлении этерификации; в дру. гих случаях применяют дегидратирующие агенты типа концентрированной серной кислоты, безводного хлорида кальция или молекулярных сит (14]. нс
C12H28CO2H + EtOH CaClg-> C12H26CO2Et (5
ссц
(СО2Н)2 + EtOH -----> (CO2Et)2
H2SO4
(СО2Н)2 + МеОН ---------- (СО2Ме)2 (7;
H2SO4
НО2С(СН2)4СО2Н + EtOH -тодуо7> EtO2C(CH2)4CO2Et (8)
HCI
НО2С(СН2)8СО2Н 4- EtOH -----> HO2C(CH2)8CO2Et
H2SO4
НО2С(СН2)9СО2Н + МеОН ---------- МеО2С(СН2)9СО2Ме
MeCH==CHCO2H + EtCH(OH)Me MeCH==CHCO2C4H9
H2S21^ MeO2C—C=C—CO2Me hci /CN
----> PhCH=C(
\C02Et
CN chZ \CO2Et
HO2C—C=C—CO2H + MeOH -
yCN РНСН=СД + EtOH
^CO2H
zCN
сн2; + EtOH
H2SO4
BrCH2CO2H + EtOH
BrCH2CH2CO2H + EtOH
CO(CH2CO2H)2 + EtOH
H2SO4
C6H«
RSO3H
CC14 HCI
CO(CH2CO2Et)2
BrCH2CO2Et
BrCH2CH2CO2Et
(9)
(10) (И)
(12)
(13)
(14)
(15)
(16)
(17)
MeCOCO2H + MeOH --'S°3H> MeCOCO2Me (18>
PhCOCO2H 4-EtOH — 4-> PhCOCO2Et (,9>
MeCH(OH)CO2H 4- CH2—CHCH2OH ~> MeCH(OH)CO2CH2CH==CH2 (20)
PhCH(OH)CO2H 4- EtOH ‘ > PhCH(OH)CO2Et (21^
EtOCH2CO2H 4-EtOH > EtOCH2CO2Et <22)
MeCH(OH)CO2H 4- Me2CHOH — > MeCH(OH)CO,CHMe2 (23)
CeHe
(Et0)2CHCO2I14-EtOH (Et0)2CHCO2Et (24)
292
НС1
H2NCH2CH2CO2H + EtOH ----->- H2NCH2CH2CO2Et (25)
ArSOjH
MeCO2H + HO(CH2)3C1 MeCOO(CH2)3Cl (26)
Применяются и другие катализаторы, помимо указанных на схемах (5) —(26). Так, эфират трифторида бора [15] использован в качестве катализатора этерификации некоторых кислот, и в примере, представленном на схеме (27), конечный продукт образуется с высоким выходом, не претерпевая перегруппировку двойных связей. Смесь борной и серной кислот катализирует ацилирование фенолов [16]; по этой методике фенилбензоат можно получить с почти количественным выходом {схема (28)}. Фениловые эфиры можно синтезировать также при использовании полифосфорной кислоты в качестве катализатора [17]; этот метод использован для получения бензоата салициловой кислоты (11). Наряду с трифторидом бора находят применение в качестве катализаторов этерификации и другие кислоты Льюиса, например сульфат железа и хлорид алюминия [18].
СО2Н CO2Et
H2SO4
Хотя для у- и p-гидроксикислот образование лактонов часто протекает спонтанно (см. гл. 9.4), в некоторых случаях требуется присутствие катализатора. Например, образование лактонов из производных транс-декалина (12) и (13) протекает под действием толуолсульфокислоты [19]; различие температур, необходимых для протекания обеих реакций {схемы (29), (30)} обусловлено тем обстоятельством, что образование лактона из диаксиального изомера (13) требует конформации ванны для одного из шестичленных колец. Синтез лактонов, содержащих циклы с числом углеродных атомов более шести, требует специальных методов, которые будут рассмотрены далее.
293
Механизм Д.-п?! применим для каталитической этерификации стернческн затрудненных кислот, в особенности для производных 2,6-замещенной бензойной кислоты; примером может служить 2,4,6-триизопронилбснзойпая кислота (14). В подобных случаях хорошего выхода эфира [20] добиваются путем растворения исходной кислоты в концентрированной серной кислоте, где, вероятно происходит образование ацплий-катиона, с последующим добавлением спирта {схема (31)}. Более наглядной демонстрацией механизма zl-tcl может служить [21] первоначальное образование аце-тплгексафторантимоната из фторангндрида уксусной кислоты и пентафгорпда сурьмы; кристаллическая соль ацилпп-катиона вступает далее во взаимодействие с пространственно затрудненными спиртами и с высоким выходом образует ацетаты {схема (32)}.
Сложные эфиры пространственно затрудненных спиртов могут быть получены по аналогичной методике, предполагающей протекание реакции, вероятно, по механизму Дл/,1. Так, трет-бутпловые эфиры, являющиеся удобными синтетическими интермедиатами, можно получать катализируемой кислотами, используя реакцию
CI2CFCF2CI + ROH
MeCOF + SbFs ------------> МеСО SbF; ---------► MeCOOR
b МеХЮ2
HC1O4 -------►
100 °c
(32)
(33)
Ме(СН2)7/ \cH2)7CO;H
Me(CH2)
карбоновых кислот с 2-метилпропеном [22] или, что реже применяется, с дн-трет-бутиловым эфиром [23]. Другие примеры каталитического присоединения карбоновых кислот к олефинам, вероятно, протекают по аналогичным механизмам. В случае внутримолекулярною аналога этой реакции происходит образование Актона, хотя в примере, представленном на схеме (33), помимо Р ования лактона требуется миграция двойной связи [24].
(2) Ацилирующие агенты для спиртов 1пп1па^п|Нм1пЫе кислоты являются относительно мало эффектив-кзвбоппдму РУювдшц агентами, но в общей реакции производных - кислот (RCOX) со спиртами или алкоксидными анно-294
нами {схема (34)} ацилирующая способность возрастает в ряду-X — ОН < NR2 < OR < OCOR < Hal, где стабильность аниона X- в общем повышается с ростом ацилирующей способности. Процессы ацилирования включают переносы протона, и поэтому они могут протекать как под действием кислот {схема (35)}, так и под действием оснований {схема (36)}.
Наиболее широко используемыми ацилирующими агентами в синтезе эфиров несомненно являются галогенангидриды (X = Hal) 0 О" ОН О
Ri J-X + R2OH R’-C-X Z=± R!-t-X R1-1-OR2 + HX (34)
HOR2 OR2
О +OH , OH
II н+ II R0H. I
R!_с—X 4=^ R1— С—X R1— С—X
I
HOR2
ОН О
I II ' '
R1—С—X R1—C—OR2 + HX (35)
OR2
О О' О
II I II
R1—С—X + R2O' R1—С—X R1—C—OR2 + X’ (36)
OR2
и ангидриды (X = OCOR) карбоновых кислот; примеры, приведенные на схемах (37) и (38), свидетельствуют о широте применимости этих методов. Ацилирование пространственно затрудненных спиртов, например трет-бутилового спирта, обычно требует присутствия катализатора; 4-диалкиламинопиридины (15) и (16) рекомендованы [26] как особенно эффективные катализаторы, превосходящие по активности простые третичные амины. Так, даже весьма чувствительные третичные спирты, такие, как линалоол (17), могут ацилироваться ацетангидридом в присутствии основания (16); 2,4,6-триалкилфенолы ацилируют при использовании ацетангидрида и основания (15). Другие методики ацилирования третичных спиртов представлены на схемах (37) — (52); отметим, что эффективными катализаторами могут служить как кислоты, так и основания.
(15) R=Me
Me
Me'
,Z\Z\Z4 Ме/^ОН
(17)
(16) R2 =» (CH2)<
295
Без катализатора
С1СОСНВг(СН;)2СНВгСОС1 + EtOH —> EtOCOCHBr(CH2)2CHBrCOOEt
PhCH=CHCOCl + PhOH —► PhCH=CHCOOPh
PhCOCl 4- HOCH2CH2C1 —> PhCOOCH2CH2Cl C,0H21CMeBrCOBr 4-MeOH —> C10H21CMeBrCOOMe Катализ основаниями
G^OH ' пиридин
4-PhCOCl -------
xCOMe
OCOPh
xCOMe
Me-
Et2O
СОС1 4- LiOBu-трет ------> Me—
COOBu-трет
(37)
(38)
(39)
(40)
(41)
(42)
/СОС1 PhNMe2 /СООВи-трет
CHZ 4- трет-ВиОН --------------> CH2
\гм Et2° \„кт
(43)
. Mg
MeCOCl грет-ВиОН --------*- МеСООВи-трет
Et2o '
Вез катализатора
О
(44)
115 °с
О + СвН13СН(ОН)Ме --------
СО2Н
(45)
СООСНМеСвН13
=0 + MeOH —► HO2C(CH2)2CO2Me О .
Катализ кислотами
(46)
... „ ZnCl2
(MeCO)2O 4- тргт-ВиОН-------> МеСООВи-трет
О
(47)
' ArSO3H
,0 + Et ОН ---------
кСО2Е1
О
'СО2Е1
,, // h2so4
(МеСО)2О + НО—f \--------ОН-------->• МеСОО—1
ОСОМе
(49)
Катализ основаниями
О
Me
О
~ Et3N
ОPh—С—ОН ------
1 90 °C
Me
Et
Et СО2Н
С02—С—Ph
(50) (сс. [25])
296
о
СО2Ме
СО2Ме
(М)
Неочищенная . /,, „
генциобиоза (MeCOjgO
Me2S0i
К2СО3
Имеется и много других соединений типа RCOX, которые являются эффективными ацилирующими агентами и используются для синтеза эфиров. Первая общая группа состоит из смешанных ангидридов карбоновых кислот; смесь ацетангидрида и муравьиной кислоты использована для синтеза эфиров муравьиной кислоты [27]. Трифторацетаты первичных и вторичных спиртов получены
(CF3CO),O +R'CO2H TZt R'COOCOCF3 + CF3CO2H
(53)
|r2°h
r1coor2 + cf3co2h
(54)
297
при использовании ангидрида трифторуксусной кислоты [281 как в присутствии катализатора, так и без него. При смешении ангид-рнда трифторуксусной кислоты с другой кислотой сначала образуется смешанный ангидрид, который служит активным ацилирующим агентом {схема (53)}, поэтому ангидрид трифторуксусной кислоты можно использовать в качестве катализатора в реакциях ацилирования. Хорошим примером его применения служит образование эфира (18) [29] из пространственно затрудненных кислоты и фенола {схема (54)}.
Смешанные ангидриды образуются также при реакции карбоновых кислот с этиловым эфиром хлормуравьиной кислоты [30] и фосгеном [31]; эти ангидриды можно использовать для синтеза эфиров или лактонов гидроксикислот. Получение трет-бутилового эфира производного цефалоспорина {схема (55)} [31] и макроциклического лактона (±)-дидезоксизеаралона (19) [32] {схема (56)} иллюстрируют эти методы.
Различные смешанные ангидриды на основе карбоновых и серу-содержащих кислот также находят применение в синтезах; в некоторых случаях их выделяют перед использованием, в других случаях генерируют in situ. Ацетил-п-толуолсульфонат (20) получают из ацетилхлорида и л-толуолсульфокислоты; при взаимодействии со спиртами он дает ацетаты [33], а также расщепляет простые эфиры, давая ацетаты и n-толуолсульфонаты {например, по схеме (57)}. Сульфоуксусная кислота (21) [34], образующаяся из ацетангидрида и серной кислоты, также служит активным ацетилирующим агентом и может быть использована для синтеза енолацетатов.
MeCOOSOj \^/—Me MeCOOSO2OH R’COOSO.R2
(20) (21) (22)
О
И ''J.p MeCOOSO2— Ме —* MeCOOCH2CH2OSO2——Ме
О “ ~~ (57)
n-Толуолсульфонилхлорид и бензолсульфонилхлорид использованы [35] как катализаторы реакций ацилирования в присутствии третичных оснований, таких, как пиридин {схема (58)}. Метод можно использовать и для синтеза |3-лактонов из |3-гидроксикпслот [36]; реакция, по-видимому, включает образование ангидридов типа (22, R2 = С6Н4Ме или Ph). Аналогичные ангидриды (22, R = CF3) выделены [37] при реакции хлорангидридов кислот с серебряной солью трифторметилсульфокислоты; они, как и ожидалось, обладают прекрасными ацилирующими свойствами. Тионпл-хлорид обычно применяют для превращения карбоновых кислот в соответствующие хлорангидриды, которые далее можно использовать как ацилирующие агенты. Альтернативный метод [38] включает использование тионилхлорида в качестве катализатора
298
образования эфиров аминокислот {схема (59)}; этот путь, как сообщалось, удобнее обычного метода синтеза по Фишеру.
Некоторые N-ацилированные гетероциклы также могут быть использованы в качестве ацилирующих агентов. Наибольшее применение находят, по-видимому, N-ацилпмидазолы [39]; их получение и ацилирующие свойства иллюстрирует схема (60). Сообщалось [40], что производные N-ацетплгидантоина (23) в мягких
ArSOsCI
R>CO2H+R2OH -пир—--/ R’COOR2 (58)
RCHCO2H + МеОН + SOC12 —> RCHCO2Me + SO2 + HCI (59) NH2 +NH3 Cl-
условиях ацетилируют только фенольные гидроксильные группы; эта специфическая реакция может найти обширное применение.
R!COOR2 +
М
(23)
RIN=C=NR1 R!NH—C=NR!
I OCOR2
(24) (25)
R1 = циклогексил
Дициклогексилкарбодиимид ДЦК (24), при взаимодействии с карбоновыми кислотами дает аддукт (25), который служит эффективным ацилирующим агентом для спиртов и аминов. ДЦК, однако, довольно редко используют для получения эфиров [41], хотя широко применяют в синтезе амидов и пептидов. Получение депсида (26) [42] служит одним из немногих примеров применения ДЦК в синтезе эфиров.
Енолацетаты (ср. 25) часто достаточно активны как ацЙЛй. рующие агенты, и производные пиридина (27) и (28) ацетилируют спирты и фенолы [43], однако активные эфиры этого типа чаще используют в синтезах амидов и пептидов.
S-Ацилтиолы также обладают ацилирующей способностью, й хотя они также не нашли широкого применения для синтеза слод<. ных эфиров, эти соединения использованы недавно для получения макроциклических лактонов. В одном из методов [44] использовали пиридин-2-тиоловые эфиры [45] гидроксикислот, которые в кипящем ксилоле циклизуются с образованием с хорошими выходами макроциклических лактонов {схема (61)}. Этот путь был применен для синтеза природных макролидов, таких как зеаралон {схема (62)}, однако в случае девятичленных лактонов {схема (61) п — 7} выходы продуктов низки.
Второй метод [46] основан на использовании S-трег-бутил-тиоатов [например, (29)], получаемых реакцией хлорангидрида кислоты с 2-метилпропан-2-тиолатом таллия, который взаимодействует со спиртами в присутствии трифторацетата ртути с образованием эфиров практически с количественным выходом {схема (63)}. Эти реакции, быстро протекающие при 25°C, использованы для синтеза макролидов {схема (64)}, например производных зеаралона (30), и метимецина. Реакции дают лучшие выходы продуктов в сравнении с использованием ангидрида трифторуксусной кислоты.
Медленное добавление к кипящему ксилолу
(47-80%)
(61)
лактонизация) СН3СО2Н,НгО, ТГФ.60 °C —--------—>
но
(+)-зеаралон
С хорошим выходом лактоны можно получать при иСП0* лобной бисимидазолил-1-кетона [47] {ср. схему (60)}; пример реакции представлен на схеме (65).
300
COCI
COS Ви-трет
(29)
СООВи-трет
трет-ВиОН
Hg(OCOCF3)2,’>
MeCN
(63)
riSBii-rper
Et2O
Этиловые эфиры можно получать из карбоновых кислот при использовании в качестве алкилирующих агентов диэтилазодикарбоксилата и триэтилфосфита [48]. Механизм превращения сложен; продуктами служат этиловый эфир кислоты, диэтплгидразо-дикарбоксилат и триэтилфосфат {схема (66)}. Аналогичная схема для синтеза лактонов [49] включает реакцию гидроксикислот с диэтилазодикарбоксилатом и трифенилфосфином {схема (67)}; протекает внутримолекулярное нуклеофильное замещение при карбинольном атоме углерода, вследствие чего наблюдается инверсия конфигурации при этом центре, если он асимметричен. Эта реакция, принимая во внимание ее стереохимию, была недавно использована [50] в синтезе природных макролидов — пиренофо-рина и вермикулина, возникающих при циклической димеризации по схеме (68).
Кетены (31) являются простейшими -по строению ацилирующими агентами [51], и хотя в целом их не считают удобными реагентами для синтеза сложных эфиров, реакция этих соединений со спиртами протекает быстро и приводит к высоким выходам эфиров {схема (69)}. Кетены можно генерировать обычными методами, например реакцией ацилгалогенидов с основаниями; они образуются также фотохимически [52] и могут быть уловлены в виде эфиров при проведении фотохимической реакции в спиртовом растворе. Примеры типичных реакций этого типа представлены на схемах (70) и (71), однако синтетические возможности метода ограничены. Тем не менее интересным примером применения
301
RCO’H + (ЕЮ)зР + EtO2CN=NCO2Et —> RCO2Et + (EtO) Pq J + EtO2CNHNHCO2Et 3 +
HO(CH2)rtCO2H + Ph3P + EtO2C!N=NC!O2Et —>
[53] метода фотохимического генерирования кетенов служит синтез диметилкроцетина (32) {схема (72)}; ключевую роль играет электроциклическое фотохимическое раскрытие цик диеноновой системы.
R\
V==C==O + R3OH —> R2// (31)
R\
^CHCOOR3
R2/
302
Me Me Me Me
О I I I I
4C-CH-CH=CH-CH=C-CH2-CH=CH-CH2-C«CH-CH=CH-CH-C
MeOZ xOMe
I (72)
OMe (32) Me ме
(3) Алкилирование карбоксильных анионов
Нуклеофильное замещение карбоксильным анионом {схема (73)}, обычно в условиях, способствующих протеканию реакции по механизму Sw2, широко используется для синтеза эфиров, яд примеров, демонстрирующих возможности этого метода, и широки) спектр алкилирующих агентов, которые могут быть использованы, R'COj + R2X —> R'CO2R24-X- (73>
представлены в табл. 9.8.1. Многие методики можно с успехом применять для синтеза эфиров пространственно затрудненных кп лот, и это находит особое применение,
?03
Таблица 9.8.1. Алкилирование карбоксилат-анионов
Соль Алкилирующий агент Растворитель туоа
RCO2‘Na+ RCO2Ag+ rco2-k+ RCO2H + CaO RCO2H + Cu2O 4- C6HUNC RCO2H4-NR3 RCO2H 4- Et3N RCO2H + (MeCHOHCH2)3N rco2h + nr3 RCO2H 4- EtN(CHMe2)2 rco2 rco2 rco2h Mel MeCONMe2 ,co Et2SO4 MeCONMe2 ^al PhCH2Cl EtOH 536] PhCH2Cl Me2SO ?3b1 p-o2nc6h4ch2ci rco2h grl Ph3CBr C6H6 ЙД1 BuOSOCl C6He Et3o+ bf; (МеО)зР=О(б} ” Mel MeCONMe2 53, MeI Et2O PhCH2Cl [53ж] RHal Е1ОН-ГМФТА(1: lVa) [53hi Mel Me2SO Щ RHal C6H6 [53 H П, p Et2SO4 MeOH [53c PhCOCH2Br Me2CO [53т Me2SO4 или Et2SO4 [53c CeHi3Cl Ксилол [53y Et3OBF4’ CH2C12 [53ф] Me4N [53x] Me3NPh [53ц] EtOCH=NMe2 HCONMe2, (EtO)2SO2 [53ч] Eto so;(B)
ГМФТА—гексаметилфосфортриамид.
(6) Избыток триметилфосфита необходим потому, группа,
Образуется этиловый эфир.
что реагирует лишь одна метильная
уЖе упом™ись катализируемые кислотами реакция вопят™ Ь,Х к^СЛот с олефинами, например 2-метилпропеном, при-о . п образованию эфиров по механизму, эквивалентному rnm'jv ГКе примеры этой и аналогичной реакций, с помощью ко-МЯР /'74\°>KI/7Q?0J?y4aTb гРе7,-бутиловые эфиры, приведены на схе-геи ell примеры заимствованы из Organic Syntheses, также из [t>4, 55J.
СН2(СО2Н)2 + Ме2С=СН2 —2—4' Е{2°-> сН2(СО2СМе3)г (74)
ЕЮ2ССН2СО2Н 4- Ме2С=СН2 —— °4t Et2°» ЕЮ2ССН2СО2СМ.е3 <7^ R'CO2H + R2OCMe3 —-——Л- ArSO^H^ j^co2R2 + СН2==СМе2
НО2ССН2СН2СО2Н + Ме2с=сн2 ——--°4 > Ме3СОСОСН.СН2СООСМе3 <77^
диоксан
30^
H+
RCO2H + Me3COCMe3 ---> RCO2CMe3
(78)
(33)
ch2=n=n + rco2h
RCO;CH3—N=N
RCO2CH3 + N2 (79)
CONMeNO
SO2NMeNO
CONMeNO
Me
(34)
(35)
Me. ,NH
,N— c;
RNHN—NAr
(36)
(37)
MeC6H4N=NNHMe + RCO2H
Et2O
----> RCO2Me
(80)
Реакция диазометана (33) с карбоновыми кислотами служит удобным и мягким методом синтеза метиловых эфиров, в особенности пригодным для получения эфиров в небольших количествах. Механизм реакции в сущности представляет собой еще один пример реакции нуклеофильного замещения {схема (79)}. Диазометан можно генерировать в виде эфирного раствора с помощью ряда методов, однако обычно используют действие гидроксида калия на одно из продажных нитрозопроизводных (34), (35) или (36) [56]. Дейтерированный диазометан CD2N2, можно получить дейтеро-водородным обменом с использованием эфирного раствора диазометана и NaOD в О2О. Взаимодействие дейтерированного диазометана с дейтерированными кислотами RCO2D приводит, как и следует ожидать, к тридейтерометиловым эфирам. Для синтеза эфиров использовали и другие диазоалканы {ср. схему (79)}, в том числе диметилдиазометан (СНз)2СЫ2 [57], фенилдиазометан C6H5CHN2 [58] и дифенилдиазометан (C6H5)2CN2 [59]; два последних реагента важны для получения синтетически ценных бензиловых и бензгидриловых эфиров (см. также гл. 6.5).
1-Алкил-З-арилтриазены (37) также реагируют с карбоновыми кислотами с образованием эфиров; типичным примером может служить использование 1-метил-З-ц-толилтриазена для синтеза метиловых эфиров {схема (80)}.
(4) Алкоголиз и ацидолиз сложных эфиров
Существуют три типа обменных реакций эфиров [60]: алкоголиз {схема (81)}, ацидолиз {схема (82)} и перераспределение {схема (83)}; два первых типа наиболее важны в препаративном отношении. Механизмы реакций классифицируются, как обычно [И]: буквы А или В указывают на то, что проне ’с катализируется соответственно кислотами или основаниями (am л. Acid — кислота
305
или Base —основание); индекс АС или AL обозначает ственно ацильное или алкильное расщепление.
КССМ2 4-R3OH R'CO2R3-f-R2OH
R'CO2R2 4- R4CO2H R'CO2H 4- R<CO2R2
R'CO2R24-R4CO2R3 R‘CO2R3 4- R4CO2R2
с°ответ.
(83)
Большинство катализируемых основаниями реакций алкоголи протекает по механизму Вас2, за исключением пространственн3 затрудненных систем, где может иметь место механизм BAL1, КОт° рын ке обязательно требует только основной катализ. Катализируемые кислотой реакции следуют Ллс-механизму в случае алкоголиза эфиров первичных и вторичных спиртов и фенолов и Аль-механизму в случае эфиров третичных спиртов, а также бензилового и аллилового спиртов. Подробный анализ механизмов перечисленных реакций дан в [60], и здесь мы остановимся лишь на синтетических аспектах.
Метиловый и этиловый эфиры р-кетокислот весьма активны [61], и при нагревании с высшими спиртами подвергаются алкоголизу даже в отсутствие катализатора, однако обычно для протекания реакции необходимо присутствие основного или кислого катализатора. Катализируемый кислотами алкоголиз эфиров использовали для синтеза метиловых и этиловых эфиров; в качестве катализаторов применяли серную кислоту, п-толуолсульфокислоту и кислые иониты. Примеры реакций, используемых в препаративных целях, приведены на схемах (84), (85). Кислотный алкоголиз лактонов может приводить к эфирам гидроксикислот [62] или галогенокислот [63] {схема (86)}; алкоголиз полимеризованной молочной кислоты (38) использован для получения эфиров молочной кислоты {схема (87}.
Природный воск (Murica) С13Н27СО2Ме 4-Ci5H3ICO2Me Iе'
H2SO4 метилмиристат метилпальмитат
СН2=СНСО2Ме 4- ВиОН A-"S°--H-> СН2=СНСО2Ви 4- МеОН
PEtOH / \ EtOH
CO2Et ч------ / \--Q ---->
H2so4 \ / НВг
О
ОН
МеСН(ОН)СО2Н —H2S°4 CeHe
Me НО(^НСОО)ЛН
(38)
.он
н+
(85)
(86)
'CO2Et
Br
Me
CH <3
H°/y
о
Катализируемый основаниями алкоголиз применяли ДДЯ С[1Н-чения простейших эфиров; этот метод особенно удобен ль3о-теза [64] rpez-бутиловых эфиров из метиловых эфиров с 306
ванием трет-бутоксида калия в трет-бутиловом спирте, в некоторых случаях с добавлением молекулярных сит [65] для удаления замещаемого метанола {схемы (88), (89)}.
СО2Ме
СО2Ме
СО2Ме
СО2Ме
+ трет
трет-ВиО'К
-ВиОН -----—-----
Сбн6
СОгВи-трет
(88)
СОгВи-трет
R
. т/>ег-ВиО“К+
+ трег-ВиОН--------------
R
СО2Ме
СО2Ви-грет
(89)
| НС1О4, 2Н2О
XNHCHCO2H + трет-ВиОСОМе
(90)
> ХЫНСНСО2Ви-трет + МеСО2Н
трет-Бутиловые эфиры приготовлены также ацидолизом в присутствии трет-бутилацетата и хлорной кислоты в качестве катализаторов. Метод использован для синтеза трет-бутиловых эфиров защищенных аминокислот [66], как показано на схеме (90) (здесь X — соответствующая защитная группа).
(5) Реакции эфиров енолов, ацеталей и ортоэфиров
Ацидолиз винилацетата катализируется ацетатом ртути и серной кислотой; реакция использована для получения широкого спектра виниловых эфиров; пример представлен на схеме (91). Ацидолиз изопропенилацетата (39), катализируемый серной кислотой [67], использован для получения изопропилиденмалоната — в высшей степени активного эфира малоновой кислоты [68]; реакция, по-видимому, протекает по схеме (92).
Алкоголиз виниловых эфиров можно использовать для получения эфиров третичных спиртов. Так, синтез [69] трет-бутилового эфира пространственно затрудненной кислоты по схеме (93) основан на алкоголизе изопропенилового эфира; изопропенилацетат (39) использован аналогичным образом [10] для ацетилирования третичных спиртов.
Сложные эфиры можно получать также ацидолизом сложных виниловых эфиров; реакция, представленная на схеме (94), служит примером [71] применения этого метода. Довольно необычное использование аддукта ДЦК и бензилового спирта (40) для бензилирования групп —СО2Н и —SH тиогликолевой кислоты [72] также основано на этой реакции {схема (95)}.
807
Ме(СН2)10СО2Н + МеСООСН==СН2 -22222221
H2so„
—> Ме(СНа)|оСООСН=СН2 + МеСО2Н
ч гт + ААбх
\—ОСОМе -------> /СОСОМе --------22222|
Me' Me'
(39)
С4Н9 вН17ССОО—
С7Н,5
C4Hg с8н17ссо2н с?н15
МеС=СН
Zn
с4н9
трет-BuOH |
ArSO3H 5 CgHnCCOOBu-jyej
с7н15
(93)
С13Н27СО2Н + —ОС18Н37
Et3N
----► С13Н27СООС18Н37 (94)
HSCH2CO2H +
OCH2Ph
PhCH2SCH2CO2CH2Ph
(95)
(40)
(PhO)2CHCO2H + EtOH _2j2C(OMe)2> (phO)2CHC02Et
реакцию°и Р°ЛЬ ДегиДра,тирующ?г2СЯпропан (4,)> которые, вероят-тодом синте1а°(7Ч1С0 Спирт°м> и этот лутТм М°ГУ2 катализ,1Рова™ по схрмр кислототябипт °Т Пу7ь может быть ме-
лотами могут Аналогичные ^е^кпии СЛ0ЖНЬ1Х эФиРов {например, оРтоэфира Л4,Г1р"в°литЬ к этери*вкЯ1МеЖДУ °РТОЭФИРЗМ1‘ 11 Ю1С' (см. пяЛ по]’ ЭТот метол ппгг» iCp кации алкильными группами Этиловые^’^’ Р еНеН ДЛЯ синтеза )3-лактояов
метнлфоЛм°ВИЯЛ ^761 прНи° испо ЧЙТЬ И3 каРбоновых кислот в очень испотьзовЯаМИДа' Аналогичный71 ^30вании Диэтилацеталя (42) ди-условии Ь в качестве ?ЯЧ Динеопентилацеталь (43) мохно Спирт встТ0 Применяемый в прЯЛИЗатоРа этерификации [77] при
Р > вступает в реаки пп р„еакции спирт, например бензиловый 30& легче, чем пространственно высоко
затрудненный неопентиловый спирт. Эти методы представлены на схемах (97) —(99) (см. также разд. 9.8.4).
Me2NCHO + EhOBFJ —> Me2NCH(OEt)2 (97)
(42)
RCO2H + Me2NCH(OEt)2 —> RCO2Et + EtOH-+Me2NCHO (98) RCO2H “h ArCH2OH -}- Me2NCH(OCH2CMe3)2 —>
(43)
—> RCO2CH2Ar + Me2NCHO + Me3CCH2OH (99)
(6) Методы получения эфиров енолов
Фениловые эфиры получают прямым путем, как отмечалось выше. Синтез эфиров енолов из альдегидов и кетонов, существующих в основном в кето-форме, требует в некоторых случаях специальных методов. Енолацетаты обычно получают реакцией карбонильного соединения с ацетангидридом и хлорной кислотой или n-толуолсульфокислотой, как, например, в случае селективного получения ацетата стероида [78] {схема (100)}. Этот метод представляет собой общий путь синтеза енолацетатов из метилкетонов, однако, как правило, образование продукта определяется термодинамикой процесса, в то время как при использовании в качестве ацилирующего агента изопропенилацетата (39) выход продукта зависит от кинетики [79]. Енолацетаты из альдегидов можно получать катализируемым основаниями ацетилированием [80] {схемы (101) и (102)}. Для синтеза ацетатов из кетонов, способных к енолизации можно также использовать [34] сульфоуксусную кислоту MeCOOSO2OH [81].
MeCO‘Na+
МеСН=СНСНО + (МеСО)2О --------> СН2=СН— СН=СНОАс (102)
Приведенные методы основаны на ацилировании относительно небольших пропорций енольных таутомеров альдегида или кетона. Прямое ацилирование енолят-анионов является эффективным методом синтеза эфиров енолов; примером такого подхода служит получение [82] енолацетата камфоры (44) {схема (103)}. Анало-
309
гичный путь [83], иллюстрируемый схемой (104), ЯВт, роятно, одним из лучших методов синтеза енол ацетатов* 8е’ кетонов. 'галоген.
(+)-камфора (44)
NaH, МеОН;
PhCOCHBrPh - МеСОСГ^ PhC-=CBrPh (104)
ОАс
9.8.1.2. Алкоголиз
(1) Реакции с нитрилами и алкоксииминами
Катализируемый кислотами алкоголиз нитрилов {схема (105)} является хорошо известной альтернативой прямому гидролизу нитрилов, ведущему к карбоновым кислотам (см. также гл. 8). В качестве кислых катализаторов обычно используют серную кислоту, n-толуолсульфокислоту или соляную кислоту; необходимая для завершающей стадии реакции вода {см. схему (105)} может либо присутствовать в реакционной смеси, либо добавляться после первоначального образования соли алкоксииминия (45). Ряд примеров этой реакции описан в Organic Syntheses [9], в основном для получения этиловых эфиров.
н+ + r2oh /OR2 Н2о /ор
R'C=N ----->- R‘C=NH ----> R‘CZ --------► R'C' О05'
н+ %’Н2 н+ чо
(45)
„ HCI /OEt EtOH //OEt
C1CH2CH2CN-----> C1CH2CH2CZ+ ---------> EtOCH2CH2Cx '
Ч\Н,СГ
(46)
2 некоторых cnvo-n '
SmiX соль (нап₽“^46)
метано^ЗИЛ0Вых эФиров [851 ^ользуется довольно редко; полу-Реакции НЫМ Раста«ром боотп^ЖИ реакции первичных амидов с
Синте Р ^Г0Рпда служит примером этои
протекает через образо-
МейепелЗЧеСТВе Реакционных**Ср’ 45 и Широкое примере-Дни пРЛ С0Тр- [871 получип , ермедиатов благодаря работам Реакции они могут £пРп^ ОКсаэо^пны (47). На конечной ста-
310 РаЩаться в сложные эфиры, если иод
действием спирта
{схема (108)}.
происходит раскрытие оксазолинового кольца
RCH2CO2H +
EtOH
RCH2CO2Et (108)
(2) Реакции с ацеталями кетенов
Алкоксиимины являются синтетическими эквивалентами ацеталей и тиоацеталей кетенов (48, Х = О или S); соединения этого
/XR2 r’c=CZ
(48)
XR1
R4—<L—XR2 I
X (50)
>OSiMe2Bu-Tper Et3N
сн2=с/ + RCH2COC1 ——>
'OEt
(49)
,OSiMe2Bu-Tper" rch2coch==c/
, OSiMe2Bu-7’pe7’ rch2A=chc=o
I
OEt
OS iMe2Bu-Tper I rch=cch2c=o
I
OEt
(Ю9)
HCI, ,H2O
RCH2COCH2CO2Et
ЗЦ
типа использованы в качестве интермедиатов в рЯде сложных эфиров. Так, ацеталь кетена (49) можно ацилиИТезов в присутствии основания [88] и далее после удаления 3аИр°Вать силильной группы получать эфир р-кетокислоты {схема
Производные тиоацеталей кетена можно получать [891 денсацией ароматических альдегидов с а-тиометилсульфоКСилК0Н’ алкоголиз продукта конденсации приводит к образованию эёш фенилуксусных кислот {схема (НО)}. Вариант этой реакции пое°В ставленный на схеме (111), можно использовать [90] для гомол гизации сложного эфира (см. также разд. 9.8.3). °'
эфира (см. также разд. 9.8.3).
/SMe
АгСНО + Сн/
\SMe
тритон в уSMe
МеОН,АГСН=С/
НС1 хО
EtOH АгСН2С (pg)
XOEt
.SMe тгф .SMe NaBH4
RCO2Et + Na+‘CH ---------> RCOCHZ --------->
^SMe ^SMe
/SMe RCHOHCH7
'''SMe
(MeCO)2O, пиридин
Et3N *
.SMe
RCH=C\ ^SMe
EtOH, HCI ----------►
RCH2C\ (111) 'OEt
(3) Реакции с ортоэфирами
Сложные эфиры образуются при контролируемом гидролизе или алкоголизе ортоэфиров (50, X — 0) или соответствующих тиоаналогов (50, X = S) (см. разд. 9.8.4). Недавно эта реакция использована [91] в общем методе синтеза сложных эфиров из аль дегидов по схеме (112). Этот путь позволяет избежать относ тельно бурных окислительных процессов (см. разд. 9.8.1.3), с торыми обычно ассоциируется такое превращение.
RCHO
h.
HS(CH2)3SH
BuLi; -------> MeSSMe;
HCI
MeS
/О
HgO. HgClg_ EtOH \)Et
(112)
(4) Реакции с ацетиленами
Эфирная группа может быть получена из ацетиленовыхпр^ водных; ряд реакций этого типа [92—94] представлен н (113) — (116). Хотя приведенные реакции не идут по к 31?
единой схеме, в каждом случае важной стадией является алкоголиз. Na; EtONa
С5НИС==СН „ cn » С5НИС=СС1 -----------> C8HnCH2CO2Et (113)
А Г SO 2'-* *
N2O, MeOH
CsHnfeCH -----~o~c > С8НцСО2Ме (114)
ciso2nco
EtC=CEt ----> EtCOCHEtCO2Me (115)
MeOH
NaOMe
PhCHC=CH » PhCH2CH2CO2Me (116)
। MeOH
N(NO)CO2Me
9.8.1.3. Окислительные методы
(1) Введение ацилоксигрупп
Формальное замещение водорода при углеродном атоме на ацилоксигруппу {схема (117)} может достигаться Несколькими путями. Лучшим из известных методов является окисление аллильной группы {схема (118)}; ряд примеров этой реакции приведен в табл. 9.8.2 (обзор см. [95]).
I I
—СН ► —COCOR (117)
/Сн-сн=с(
ГМ(ОАсЬ \-сн— с( —> /|> |\
н ОАс
)с=сн-С\
ОАс
(118)
Из перечисленных в табл. 9.8.2 окисляющих агентов наиболее удобным, вероятно, является ацетат палладия. Обычно реакция протекает через аллильную перегруппировку; предложен механизм с промежуточным присоединением и элиминированием окисляющего агента {схема (118)}. Помимо того, существуют многочисленные примеры [95] присоединения ацилоксизаместителей по углеродным атомам, активированным соседними карбонильными группами, где может происходить окисление енола, или по бензильному положению, когда более вероятен свободнорадикальный механизм. Типичные примеры реакций второго типа приведены в табл. 9.8.3.
Перечисленные в табл. 9.8.2 и 9.8.3 реакции не включают замещение при ароматических углеродных атомах. Аналогичное окисление ароматических СН-групп можно осуществить при использовании трифторацетата таллия [96]. Реакция протекает через образование соли ароматического соединения с последующим окислением тетраацетатом свинца и трифеннлфосфином {схема (119)}. Образующийся трифторацетилфенол легко гидролизуется до фенола.
313
Таблица 9.8.2. Примеры аллильного окисления
Углеводород
Hg(OAc)2 в МеСО2Н SvO2 в (МеСО)аО
PhCOOOCMe3> Cu+
МеСОООСМе3, Cu2+
Pd(OAc)2
Pb(OAc).,, N-бромсукцинимид; НО'
Окисляющий агент
Pb(OCOCH3)4
С6Н6 + T1(OCOCF3)3 —> C6H5T1(OCOCF3)2 > C6H5OCOCF3 (119)
(СвН5)зР
(2) Окисление по Байеру — Виллигеру
Реакция Байера — Виллигера [97], состоящая в окислении кетонов пероксикислотами, приводит к образованию сложных эфиров
Таблица 9.8.3. Окислительное введение ацилоксигрупп
Соединение Окисляющий агент Продукт Литература
EtCH(CO2Et)2 (PhCOO)2> NaH, С6Н6 EtC(CO2Et)2 [96а]
rch2cho Pb(OAc)4, bf3, С6Н8 Т1(ОАс)3 । OCOPh RCHCHO ОСОМе [966]
Me—— -Me CeIV(NH4)2(NO3)6> МеСО2НМе——СН2ОАс [96г]
o2n— -Ме СгО3, Ас2О O2N__Q-СН2°АС [9бд]
? \ Ме 'NO2 СгО3, Ас2О СН2ОАс Что2 [9бе]
314
по механизму перегруппировки, как показано на схемах (120), (121). В соответствии с этим ациклические кетоны дают сложные эфиры, а циклические кетоны приводят к лактонам. В качестве окислителей использовались разнообразные реагенты, отвечающие общей формуле ROOH, но в большинстве случаев применялись пероксикислоты (R-ацил). Механизм, приведенный на схеме (120), включает [1,2]-спгматропное перемещение группы R1 (с сохранением конфигурации при центре миграции) в направлении к кислородному атому, обедненному электронами; вследствие этого мигрирует та группа, которая обладает лучшей способностью стабилизовать электрондефицитное промежуточное состояние. Из алифатических альдегидов обычно образуются карбоновые кислоты (формально в результате миграции водорода); в случае ароматических альдегидов, особенно с электронодонорными орто- и пара-заместителями, наблюдается миграция арильной группы и образование формиатов фенолов {схема (120), для случая R1 = Н, R2 = Ат}.
(120)
Ряд примеров применения окисления по Байеру — Виллигеру для синтеза эфиров и лактонов дан в табл. 9.8.4. Следует, однако, отметить следующее. В качестве наилучшего реагента для ациклических кетонов рекомендована трифторперуксусная кислота. Другие реагенты (помимо пероксикислот) редко используются для окисления неароматических кетонов, хотя подобные примеры включены в таблицу. Окисление ароматических альдегидов щелочным пероксидом водорода обычно называют окислением по Дакину; образующиеся при этом эфиры муравьиной кислоты гидролизуются до фенола, как правило, уже в процессе реакции.
Реакцию Байера — Виллигера часто используют в качестве синтетического метода; детальное рассмотрение этой реакции выходит за рамки настоящей главы. Упомянем лишь о ее применении в синтезе простагландинов [98].
(3) Другие окислительные методы
Известны и другие методы синтеза сложных эфиров из альдегидов и кетонов или их производных, и некоторые из них потенциально достаточно общи. Так, ацетали альдегидов можно
315
Таблица 9.8.4. Примеры окисления по Байеру — Виллигерц
Карбонильное соединение Реагент
EtCOEt CeHisCOMe Н2О2,(СРзСО)2О,На2НРО4 Н2О2, BFз, Et2O
\—СОМе phCO3H, CHCI3
Ме2СНСН2СОМе Пероксималеиновая кислота, СНС13
.ООН
СзНцСОМе (CF3)2C; , PhNO2 \он
Ме,2СНСН2ОСОМе
С5НиОСОМе
МеО
EtOCOEt С6Н13ОСОМе
Продукт
СОМе
1Лите!1а ’УРа
[97а | [97б|
[97с]
[97д]
НО
,он
МеО
—осно
[97е]
Н2О2> NaOH
ОНО
СОМе
Н2О2, Me4NOH'
НО
[97ж]
[97з]
ж-С1С8Н4СО3Н, СНС1з
И
СОМе
Н
О СОМе
[97и]
Н
Н
н
о
н /°
H2O2) NaOH
О
[97к]
R1
R2
ВНП
МеСОзН
МеСОзН
R1 R2
[97л]
О // ^О
СН2Ч
} ^С=°
(СН2)б/
Псроксималеиновая кислота, СН2С12
СвНп СН2-—С==О | I (СН2)в—°
[97г]
316
непосредственно окислять до сложных эфиров озоном [99] {схема (122)}; окисление ацетофенонов при действии галоформа и гипохлорита калия в метаноле непосредственно приводит к метиловым эфирам [100]. Метиловые эфиры фенилуксусной кислоты [101] можно получить в одну стадию при действии на ацетофеноны метанольного раствора нитрата таллия, содержащего каталитические количества хлорной кислоты; эта реакция служит удобной альтернативой хорошо известного метода Вильгеродта — Киндлера {схема (123)}.
Оз
С6Н13СНО —> С6Н13СН(ОМе)2 г,'"» С6Н13СООМе (122)
СП2С12 '
/Н , ч
ОН ONO3 О (123)
н+ / T1(NO3)3 И' Гч/ \\ тт ,
АгСОСНз ±=^> Аг—С-------------->Ar“rCj-CH2—И > ССН2Аг
СН2 ОМе CONO2 МеО
+ TJNO3
(51)
R’CH2OR2 —> R’COOR2
(124)
В литературе неоднократно сообщалось о прямом окислении простых эфиров в сложные эфиры {схема (124)}, однако, по-видимому, метод не обладает достаточной общностью. Используемые реагенты включают трихлоризоциануровую кислоту (51) [102], тетроксид рутения [103] и триоксид хрома [104].
Первичные 1,4-, 1,5- и 1,6-диолы можно окислить до соответствующих лактонов [105] при использовании в качестве окисляющего агента карбоната серебра на целите; применение этой реакции для синтеза (±)-мевалонолактона (52) {схема (125)} демонстрирует общие возможности метода. Сообщалось об аналогичном окислении первичных спиртов до эфиров под действием разнообразных агентов, один из примеров представлен [106] на схеме
НО Ме
АоСОз
целит
(125)
Na2Cr2<J7
РгСН2ОН ,4 .4 X РгСООСН2Рг (126)
НаоСц, HgO
317
>с=о
(СН2)У I ХСН2
CII(OEt)3. H2SO4; ------------------>
ArSO3H
/С—OEt (CH2)10; ||
XCH
o3;
yCO2Et (CH2)10
XCHO
>CO2Et
(CH2)Z
\CO2Et
c—о
Na, ксилол^ /у Pb(OAc)4, MeOH;
(СН2)б ' —>
\choh Me0H> H2S0<
yCO2Me (ch2)6;
XCH(OMe)2
Озонолиз сложных эфиров енолов, полученных из циклических кетонов [107], служит потенциально удобным методом синтеза ю-альдегидоэфиров {схема (127)}; их можно также получать [108] окислением циклических ацилоиновых производных из а,(о-диэфи-ров по хорошо известной методике {схема (128)} (см. разд. 9.8.3). Простейшие сложные эфиры можно получать озонолизом винил-галогенидов в растворе метанола [109].
Окислительное декарбонилирование ангидридов или карбоновых кислот {схема (129)} также ведет к сложным эфирам. Это может достигаться действием на карбоновую кислоту тетраацетата свинца и иода [ПО] или действием на ангидрид озона, оксида ртути и иода [111]. Отметим, что в принципе сложные эфиры можно получить восстановлением ангидридов (—СООСО—
—СН2ОСО—), и этот путь был действительно использован [112J при синтезе лактонов из циклических ангидридов {схема (130)г
+ н2о
RCOOCOR + [О] ——RCOOR ——> 2RCO2H + [O]
—СО2 -—СО 2
NaBH4
(130)
можно окислить до сложных
> раствора нитрата таллия; Р ы
Р РУ [ можно нревра«Т‘
inur*
таллия поз
Ацетиленовые производные
ров действием метанольного раствора нитрата сопровождается скелетной j
(131),(132)}. Альтернативно,галогенацетилены
в эфиры а-кетокислот озонолизом в спиртовом 1е„егру
в данном случае {схема (133)} не отмечалось скелетной п пировки. Другой метод с использованием нитрата •
318
ляет превращать эфиры |3-кетокислот в эфиры кислот ацетиленового ряда {схема (134)}.
Et
T1(NO3)3 I
PhC=CEt - -— » PhCHCO2Me (131)
MeOH ' '
T1(NO3)3
PhC=CH —> PhC=CBr - > PhCHBrCO2Me (132)
MeOH 2 v '
O3
PhCsCHal -~qh> PhCOCO2R (133)
Prv_
// \ TI(NO3)3
PrCOCH2CO2Et + N2H4 —> N" )=O PrC=CCO2Me (134) \ / MeOH 4
NH
Эфиры а,<о-дикарбоновых кислот, содержащих длинную углеводородную цепочку, традиционно получают по Кольбе электролитическим окислением моноэфиров дикарбоновых кислот {схема (135)}. Эфиры длинноцепочечных монокарбоновых кислот можно получить [116] по Кольбе реакцией смешанного сочетания монокарбоновой кислоты с моноэфиром дикарбоновой кислоты {схема (136), см. также гл. 9.2}.
RO2C(CH2)„CO2M+ -----RO2C(CH2)n(CH2)„CO2R (135)
(М = к или Na, R = Me или Et)
Pt-анод
RCO2‘ + MeO2C(CH2)nCOj -------> R(CH2)„CO2Me (136)
9.8.1.4. Перегруппировки
Окисление по Байеру — Виллигеру и некоторые окислительные методы с использованием нитрата таллия сопровождаются перегруппировками, однако для удобства изложения эти два типа реакций рассмотрены в разд. 9.8.1.3.
(7) Перегруппировка Фаворского
Перегруппировку Фаворского [117] а-галогенкетонов {схема (137)} можно использовать для получения карбоновых кислот, если в качестве основания использовать водный гидроксид, или
319
эфиров, если перегруппировку проводить в присутствии раствора алкоксида щелочного металла. Обычно пс» СПИртовогп механизм этой реакции [118] включает образование Лагаю>г' 'По ного циклопропанона, поскольку это хорошо согласуПР°Ме>Куточ-мер, с распределением меченого углерода в эфире (531^’ Капри' ном перегруппировкой 2-хлор [1,2-14С2] циклогексанона ГСм П°Луче«-звездочкой на схеме (138)], однако в некоторых случая °™еТку пропанон может быть представлен в виде не полность ЯХ Цикл°’ ванного мезомерного цвиттериона. Стереохимия nepernv ЦИклиз°’ Фаворского [119] при проведении реакции в простомэ(ЬПНр0ВО гласуется с механизмом, приведенным на схеме (137V е с°’ кетон (54) дает бензиловый эфир (55) {схема (139)) о/Л0Р' применения и пределы реакции подробно исследованы 1117? В общем случае перегруппировка приводит к эфиру с ббА ™ ' числом заместителей при а-углеродном атоме, хотя изв™ исключения, например трет-галогенкетон (56), который певегоул пировывается, давая 0,0-Дифенилпропионат {схема (140)}
С1
I EtONa /нМ
Ph2CCOCH3—------->Ph2CHCH2CO2Et ,14 '
(56)
СН2СО2Ме
/ДЭМе
В г
L Ьсосн3 Ме ст В г ’
ия*я1’5а^/^Сл_едуеРт ожидатьДвГалогенкет°нов 1П7> 120^ стонов а>Р'неизсыщенным эфирам в
₽-Дигало(“е"а„<И1Р н “
нкетонов {схема (142)}, возможно
] ПР11' в слУ'
.. .ч ^ш-^спауо*^““Г.7ненась1ШеННЬ‘‘а
чае а,а'-дигалогенкетонов {схема (141)} И К/142)1 возмоги0 эфирам в случае а,р-дигалогенкетонов {схема (14^/> 320
результате прямой нуклеофильной атаки на промежуточные гало-гейниклопропаноны.
Если исходным соединением является а,а-дигалогенкетон, не содержащий а-водородного атома, то реакция может протекать без перегруппировки с расщеплением связи СО—СНа12. Это использовано [121] в синтезе сложных эфиров по схеме (143). Продукт (57) такого расщепления можно использовать для получения полпфункциональных эфиров, как показано на схеме; вся последовательность реакций получила название геминального алкилирования. В альтернативном методе [122] избегают стадии галогенирования, используя производное днтпана (58), которое подвергают нуклеофильному расщеплению кольца, как показано на схеме (]44), ведущему к получению функцпонализованного эфира, если нуклеофилом является алкокенд-анион.
AgNOa, MeOH; HCI, Н2О
(143)
ВизЭпН
Nu“ ----►
(144)
(2) Синтез Арндта-Эйстерта и перегруппировка Вольфа
Синтез Арндта-Эйстерта [123] используют для превращения карбоновых кислот в соответствующие гомологичные кислоты, эфиры или амиды по схеме (145). Промежуточный диазокетон (59) можно выделить; его разложение обычно катализируется мелко РазДробленным или коллоидным серебром. Этот шаг приводит к возникновению ацилкарбеиа, который перегруппировывается (перегруппировка Вольфа) с образованием кетена; последний связывается с добавляемым нуклеофильным агентом. Если группа R' хиральна, то она мигрирует с сохранением конфигурации [124]
11 Зах. 395
321
R'CO2H
SOC12
—------> R'COCl
или PCI5
CH2N2
Et2O
R'COCHN==N
[R'COCH :]
—> [R’CH=C=O] —
(59) r2oh
H2O
R‘CH2CO2R2
R’CH2CO2H
(145,
r3r4nh
/R3 R’CFPCON7
\R1
Механизм перегруппировки Вольфа был предметом дискуссий Реакция может катализироваться целым рядом соединений, особенно рекомендованы бензоат серебра и триэтиламин [125], часто используют оксид серебра. Реакция не столь проста, как может показаться при рассмотрении схемы (145); в качестве альтернативы термического процесса использовалась фотохимическая перегруппировка Вольфа. Интересно соотношение между фотохимической и термической перегруппировкой. Так, термическое превращение дпазокетона (60) приводит к эфиру (61), формально являющемуся продуктом [3,2]-сигматропной перегруппировки, в то время как при фотохимической реакции образуется эфир (62)-— продукт [1,2]-перегруппировки {схема (146)} [126].
.О
pt-/7 Н!С—О
Та - К
(63) (64)
Хотя на схемах (145) и (146) показано возникновение ^^аче, бена в качестве интермедиата, реакция может протекать ‘ хр0Н-через образование кетена из диазокетона в результате кого отщепления азота и скелетной перегруппировки I р0-или даже через промежуточное образование оксирена_ ( сгерта, мологизации, достигаемой по реакции Арндта
322
можно добиться и другими путями, например при использовании аналогичной перегруппировки, показанной на схеме (147) [127].
СОС1
CH2SOMe2
ТГФ
COCHSOMe2
hv ---->-MeOH
СН2СО2Ме
(147)
(3) Сигматропные перегруппировки
Для синтеза сложных эфиров использован и ряд других реакций, сопровождаемых перегруппировками, классифицируемыми как'сигматропные [128]. Применение [3,3] -сигматропной перегруппировки [129] требует участия ацеталя аллплкетена, получаемого реакцией обмена с ортоэфиром по схеме (148); процесс проводят в одну стадию без выделения ацеталя (65). Эта реакция служила важной ступенью в синтезе (±)-прогестерона циклизацией олефина по типу биогенетической циклизации. Исследована стереохимия этой реакции, и установлено, что перегруппировка ацеталя кетена, возникающего из аллилового спирта (66), приводит к образованию (Е)-эфира (67) в результате стереоселектив-ного процесса, протекающего через переходное состояние, имеющее форму кресла {схема (149)}.
[3,3]-Перегруппировка винилаллилового эфира (68) приводит к образованию гидроксиальдегида (69) в результате реакции, являющейся примером хорошо известной перегруппировки Клайзена в алифатическом ряду. Циклический полуацеталь продукта (69) можно окислить далее до у-лактона [130] обычным путем {схема
11*
323
9.8.1.5. Алкоксикарбонилирование
Синтез сложных эфиров путем внедрения алкокси карбон ной грсппы {схемы (151), (152)} охватывает широкий ряд и^ь’ ных соединений и реагентов [131]. В общем случае, сслн а д' углерода, по которому должно идти внедрение, обладает НукЛео фильным характером, то в качестве реагентов используют пп0Из водные диоксида углерода {схема (151)}. Если этот атом потен циально электрофилен, то используют спирты или карбонилы ЧР' талла МДСО), {схема (152)}.
ХС—OR + — С-М —> — С—co2r + mx
ROH + МХ(СО)У +-С-Х —> —C-CO2R + MX
(151)
(152)
(1) Алкоксикарбонилирование по нуклеофильному атому углерода
Нуклеофильный углерод обычно предполагает использование в качестве реагентов металлорганических соединений {схема (151), М = металл}; выбор метода генерации металлорганических соединений зависит от кислотности соответствующего соединения /С—Н. Поэтому удобно рассматривать эти реакции в соответствии с типом синтезируемого сложного эфира. 2
Простейшие алифатические и ароматические эфиры R’CChR и ArCO2R можно легко получить алкоксикарбонилированием реактивов Гриньяра, синтезированных из алкил- или арилгалогенидов {схема (153)}. Это наиболее используемая методика, но имеются и вариации, например синтез карбоновой кислоты действием Ди0^ сида углерода на реактив Гриньяра с последующей этерифик цией. В тех случаях, когда присутствие других функционалы^ групп исключает использование высокоактивного реактива 1Рп^о_ яра, для алифатических соединений можно воспользоваться следовательностыо реакций, представленных на схеме ( матические эфиры в этом случае получают реакцией ФриДе-Крафтса [132] {схема (155)}.
(153)
ХСО2К
R’Hal -—> R'MgHal ---------> R'CO2R2
R1 = Aik или Ar; X = OAlk или Hal
R'Hal + KCN
R OH
RiC==N -------> R'CO3R2
l-I +
(155)
(154)
324
Пргг алкоксикарбоннлированни карбанионов, образованных из сильных карбоновых кислот, можно использовать значительно более широкий набор металлоргаппческпх соединений. Так, сложные эфиры и нитрилы можно превратить в эфиры замещенных малоновых и цпаноуксусиых кислот (соответственно) конденса-цией с диэтилкарбонатом [133] в присутствии этоксида натрия, используемого для генерации карбанионов {схемы (156), (157)}. Альтернативная методика включает использование этилхлорфор-миата вместо диэтилкарбоната и таких оснований, как гидрид натрия пли трнтплнатрпй. Кетоны особенно легко превращаются в (3-кстоэфиры; имеются многочисленные примеры подобных синтезов. Для получения высоких выходов продуктов рекомендовано [134, 135] совместное использование гидрида натрия в диоксане и дпметплкарбоната; метод использован на промежуточной стадии синтезов (±)-кариофиллена {схема (158)} и непетовой кислоты [135]. Отмечалось, что реакция идет преимущественно по наименее замещенному а-углеродному атому кетона.
EtONa
RCH2CO2Et 4- CO(OEt)2 -------> RCH(CO2Et)2
EtONa
PhCH2CN 4-CO(OEt)2 --------> PhCH(CN)CO2Et
(156)
(157)
(158)
Me
Me-
4- (MeO)2CO
Метилмагниикарбонат MeOCOOMgOMe, получаемый из метилата магния и диоксида углерода в диметилформамиде [136], служит как основание и как производное диоксида углерода. Образующийся магниевый комплекс [3-кетокислоты легко этернфици-руется метанольным раствором хлорида водорода; примером подобной реакции может служить синтез 2,6-бисметоксикарбонил-Циклогексанона {схема (159)}. Тот же реагент использован для синтеза других а-замещенных кислот и эфиров [131].
826
Этилдиэтоксифосфинилформиат ЕЮСОРО (OEt)2 получен из трпэтилфосфита и этилхлорформиата. Утверждается ч соединение более эффективный реагент, чем диэтилкарбонатТ° Эт° его можно успешно применять для проведения реакций о’”Что мягких условиях {схема (160)}. очень
Ароматические кетоны, не образующие енолят-анионов быть восстановлены до дианионов действием натрия в аммиа^ Такие дианионы вступают в реакцию с диэтилкарбонатом ИЗйГ давая эфиры а-гидроксикислот {схема (161)}. Этот тип восстало вительного алкоксикарбонилирования не нашел общего примене' ния; более широко используются реакции [139] с участием аф-не". насыщенных кетонов. Так, [3-кетоэфиры можно получать из аф-не-насыщенных кетонов в три стадии. Первая стадия состоит в восстановлении кетона до енолята лития действием лития в аммиаке На второй стадии проводят карбоксилирование диоксидом углерода, и завершает синтез этерификация {схема (162)}.
(EtO)2CO
Na, NH3, Et2O;
(161)
EtMgBr; ------->
ClCO2Et
RC=CLi
ClCO2Et
C6H6
(164)
(165)
OCH2C=CH
RC»CH
LiNH2 --------> или LIBu
0 OCH2CsCCO2Et
ClCO2Et ----->- RC=CCO2Et
Ацилирование кетонов можно также проводить при пс 'сдувании енамина вместо енолята металла [140]; в нек0Т?со)} чаях это дает сравнимые выходы p-кетоэфиров {схема 11Ь^{.оДОм
Алкоксикарбонилирование служит также удобным еНты синтеза эфиров ацетиленовых кислот; ацетиленовые Р ‘ 3TOfi Гриньяра или литиевые и натриевые соли используютс
326
реакции (обычно вместе с этилхлорформиатом) в качестве производного диоксида углерода. Типичные методики иллюстрируются схемами (164) [141] и (165) [142].
п) Алкоксикарбонилирование по электрофильному атому углерода
Принципиальная схема процесса показана на схеме (166), однако на практике алкильные и ацильные катионы обычно стабилизованы в виде комплексов металлов, которые промотируют стадию карбонилирования. Установлено [143], что реакция алкил-галогенидов с оксидом углерода и спиртом в присутствии основания и карбонила металла в качестве катализатора открывает возможность прямого синтеза сложных эфиров из галогенидов {ср. схемы (153) и (154)}. Оригинальная методика с использованием натрпйтетракарбонилкобальтата Na+[Co(CO)4]- оказалась приемлемой для синтеза метиловых эфиров из простых галогенидов (с умеренными выходами), например по схеме (167), и получения замещенных малоновых эфиров из а-бромэфиров.
Этот метод был позднее усовершенствован использованием других карбонильных производных металлов. Тетракарбонилни-кель и метиловый спирт вступают в реакцию [144] с винил- и арил-галогенидами при 25—60 °C в присутствии метоксида натрия или калия, давая с хорошими выходами сложные метиловые эфиры
со + r2oh
+R> ----> R'CO -----> R'COOR2 (166)
Na + (Co(CO)4|~, (CgHu)2NEt
C8H17I + CO + MeOH-----------------------------> C8H17CO2Me (167)
° 11 * 1 пихлл r.n or о £. \ j
Ni(CO)4, MeOH, MeONa + ' 1 ' > 2 ч, 25 °C
Ph
H
\ ,СО2Ме /^H
(168)
Me(CH,)6I
Ni(CO)4, трет-BuOH, трет-ВиО~К
24 ч, 50 °C
Me(CH2)6CO2CMe3
(169)
{схема (168)}. Алкилгалогениды менее активны, но тетракарбонил-никель и трет-бутилат калия в трет-бутиловом спирте представляют собой более мощную алкоксикарбонилирующую систему, и в этих условиях алкилиодиды дают хорошие выходы сложных трет-бутиловых эфиров {схема (169)}. Предложена трехстадниная методика, на первой стадии которой алкилгалогенид или п-то-луолсульфонат взаимодействуют с натрийтетракарбоннлферратом Na2Fe(CO)4, образуя анионный комплекс. Последний последовательно реагирует с оксидом углерода, галогеном и спиртом, давая эфир, возможно через промежуточный ацплгалогенид {схема (170)}. Сообщалось, что общий выход в процессе достаточно высок 51 Реакция обладает некоторой селективностью. Так, дигалогенид
327
(70) реагирует предпочтительно с замещением бромид-ан (к0ТН > 104 по сравнению с замещением хлорид-аниона); реаки 3 неспособность сложноэфирной и кетонной функций не подвепи?011’ влиянию условий реакции {схемы (171), (172)}. ржень)
Сообщалось о разработке методов с использованием арил- б зил- и винилгалогенидов [146] {схема (173)}, а также трет-ал^» галогенидов [147] {схема (174)}; реакции последних включают
I >со г, (170>
>ОС—Ее ——>р‘со9Р2
I ^СО R 011
со
R СО
-I со r’x-->ос—Ее. -----
Na2Fe(CO)4
co
В r (CH2)4CO2Et
MeO2C(CH2)4CO2Et
образование карбений- и ацилий-катионов в стабилизующей среде {ср. схему (166)}.
Bu3N, Pd(PPh3)2(HaI)2
Hal + СО + BuOH 80-юо °с Hal = Br или I
SbCI5, so2. CH2C12 p
—> урет’-ВиСОгЫ
* ^\cO>Bu
(173)
трет-BuCl + CO + EtOH
(174)
-70 °C
С°2—-> МеСН2СН2СНО
(175) используется как промышленнь
МеСН==СН2 + СО + Н2
Оксосинтез уже много лет _________..___ . шан-
метод получения альдегидов и спиртов из алкенов {схема I 'ь,е Аналогичным путем из алкенов могут быть получены и сЛ Л11СЬ эфиры [148] (гидроалкоксикарбонилирование). Ис„пя0,ЛЬг?оОТвет-различные условия реакций; на схемах (176) — (178) t . ственно по работам [149—151]} представлены три успешнь мера. В этих реакциях эквивалентное количество электр0<’\‘я“1НуЮ атомов углерода генерируется при атаке металла на Д агеН'та связь олефина. При использовании мягкого окисляющего ‘ могут возникать а,Р-ненасыщенные эфиры, как, наприл реакции комплекса хлорида палладия и стирола {схем t 328
(152]. Эта реакция, однако, по-видимому, не может служить общим методом синтеза а.р-ненасыщенных эфиров.
, ™ <ph3P)2PdCI2
4- СО 4- МеОН ——
60 °
S. Ni(CO)4,HOAc
? + EtOI-I----------------
{/ 50 °C
Me
co2ei
СНСО2Ме
(176)
(177)
H2PtCI6. SnCl2
Ci2H25CH=CH2 4- CO 4- MeOH -----------------
PdCI2;
CO, МеОН. Et3N
С12Н25СН2СН2СО2Ме
СО2Ме
(178)
(179)
(180)
NI(CO)4
RC=CH 4- СО 4- Н2О -------► RCH=CHCO2H
Ненасыщенные эфиры обычно получают этерификацией а,0-не-насыщенных кислот, которые, в свою очередь, можно получать широко используемым родственным синтезом из ацетилена {схема (180)}. Эти реакции являются развитием трудов Реппе и сотр. [153] по карбонилированию; обзор см. [154]. Предпринята попытка аналогичного прямого синтеза а,[3-ненасыщенных эфиров из ацетилена [155], однако реакция сложна и приводит к смешанным продуктам. Тем не менее этот путь использован [156] для синтеза а-метилен-у-лактонов {схема (181)}.
9.8.1.6. Методы получения лактонов
Хотя в разных разделах ранее мы уже касались вопроса получения лактонов, в настоящей главе целесообразно сгруппировать вместе ряд методов, значительно отличающихся от тех, которые могут использоваться для синтеза ациклических эфиров. Получение макролидов обсуждалось в разд. 9.8.1.1 и здесь рассматриваться не будет.
(О Получение а-лактонов
Сообщение о синтезе и свойствах а-лактонов появилось в 1972 г. [157], хотя ранее считали, что эти соединения являются нестабильными интермедиатами,’ возникающими в ряде реакции. Облучение 4,4-дизамещснных 1,2-диоксолан-3,5-дионов (71) при 7 К приводит к диоксиду углерода и незамещенному ос-лактону
329
Разла.
, 1895 см-1) При температурах выше 173 К а-лактоны
гаются с образованием полиэфиров {схема (182)}.
(71)
(2) Получение ^-лактонов
В отличие от высоконапряженных а-лактонов (3-лактоны достаточно стабильны, и их получение и свойства подробно описаны. Синтез [158], основанный на катализируемой ртутью лактонизации фенилтиоэфира [J-гидроксикарбоновой кислоты (72) приводит к прекрасным выходам (3-лактонов {схема (183)}. Опубликованные до 1971 г. методы получения [3-лактонов рассмотрены в обзоре [159]. Сообщалось об усовершенствованной методике получения из [3-гидроксикислот. Последний метод, основанный на этерификации кислотолабильных спиртов и гидроксикислот представлен на схеме (184). Пиролиз [3-лактонов дает с высоким выходом олефин и диоксид углерода с сохранением стереохимии связи С—С. В об-
ратном направлении эту реакцию провести не удается, однако сообщалось [160] об использовании альтернативной реакции циклоприсоединения, иллюстрируемой схемой (185).
R1
R2
+ CH3COSPh
LiN(CHMe2)2.
-78 °C;
—---------->
HCI
Hg(OSO2Me)j MeCN
R1 О
R2—
-C-OH
R3—
-OH
R4
PhSO2Cl пиридин
CH2Ph или Ph Me-
R2__
R3—
R*
(183)
(184)
R' до R’=H, Me,
MeCH=O + C12C=C=O
Cl
:O
Cl
О
О
(185)
330
(3) Получение у-
и b-лактонов
пГа типа лактонов спонтанно образуются из соответствующих сикислот, если циклизации не препятствуют стерические гпДР01'- Обзор методов получения а-метилен-у-лактонов (73) Ев [161]; эти соединения представляют потенциальный интерес ДаН ’ лекарственных веществ. Шестичленные циклические
ЛЛЯ ” оны можно получать [162] по аналогичному реакции п^са — Альдера присоединению диоксида углерода к диену по Д'1ЛЬ (186). Интересен и метод [163] превращения у-лактонов в схеМепш1 по реакции расширения углеродного кольца {схема ^-ЛЗКТипР г
(187)}- р2 R1
О
и
I + EtO2C'x'x-CO>Et
130 °C, MeCN;
-------->
КОН.ТГФ
(186)
(СОС 1)2; NaNs;
А. СбНв: (СО2Н)2, ТГФ, н2о
R1 или R2 = Н или Ме
(187)
9.8.2. СВОЙСТВА СЛОЖНЫХ ЭФИРОВ
9.8.2.1. Структура сложных эфиров
Структура простейших представителей сложных
эфиров иссле-
^нумура простейших ^дом"микроволновой спек-
Дована дифракционным методом i исследований иллюе р
троеко»4 164, 165]. Р«Ульта™ ,р"е™лформ..ата (74),
руются „реАставленчымн формулам» мети, у
РУются представленными
331
СН3
0,146
3
(76)
.О R2 О'
\ / \+ / О—с ' > О=с
\ , \,
R R1
(77а) (776)
ацетата (75) и метилоксалата (76), где цифрами показаны длины связей (нм) и величины углов между связями ОСО и СОС. Как
и следует ожидать из рассмотрения резонансных структур | (77а) —(776)] и расчета методом молекулярных орбиталей [164], структурная группировка эфира С—СО—ОС планарна или близка к планарной. Обычно одинарная связь СО—О приобретает конформацию, при которой карбонильная двойная связь заслонена связью О—R2, как показано в формуле (77а).
Таблица 9.8.5. Температуры плавления и кипения сложных эфиров [166]
Сложный эфир Формула T. пл., °c T. кип.. °c
Метилформиат НСООМе -99 31,5
Метилацетат МеСООМе —98,7 57,1
Этилацетат MeCOOEt —83,6 77,1
Пропилацетат МеСООРг —95 101,6
Этилпропиоигт EtCOOEf -73,9 99,1
Этилакрилат CH2=CHCOOEt 101
Этилхлорацетат ClCH2COOEt -26 145
Этилгликолят HOCH2COOEt 160
Диэтилоксалат EtOCOCOOEt —41,5 185,2
Диэтилмалонат EtOCOCH2COOEt —51,5 199,3
Диметиосукцинат MeOCOCH2CH2COOMe 18,2 196
Этилбензоат PhCOOEt -34,7 212,4
•Этил-о-тол уплат Этилфеиилацетат o-MeCeHiCOOEt PhCH2COOEt 227 227,5
Диэтил фталат Диэтилтерефталат Октилоктаиоат (октилкапри- o-C6H4(COOEt)2 n-C6H4(COOE t)2 Me(CH2)6COO(CH2)7Me 44 -15,1 289,5 302 306,8
Гексадецилгексадеканоа- (цетилпальмитат) Гекс а децилокта декане, а (цетнлстеарат) Me(CH2)14COO(CH2)l5Me 51,6
Me(CH2)16COO(CH2)15Me 56,6
332
9.8.2.2. Физические свойства сложных эфиров
Простейшие низкомолекулярные сложные эфиры представляют собой низкозамерзающие бесцветные жидкости, температура кипения которых не намного выше таковой соответствующих углеводородов. Некоторые типичные значения [166] температур плавления и кипения эфиров даны в табл. 9.8.5.
Более летучие эфиры часто обладают характерным запахом Сложные эфиры длинноцепочечных спиртов с длинноцепочечными кислотами представляют собой воскоподобные вещества и входят в состав некоторых природных восков. Высококипящие сложные эфиры, например дпэтилфталат, находят применение в качестве неподвижных фаз в газо-жидкостной хроматографии, поскольку они относительно мало полярны и обладают высокой термической устойчивостью. Простейшие эфиры используются в качестве растворителей, в особенности этнлацетат и амил (пентил) ацетат С5Н11ОСОСН3. Сложные эфиры обладают значительным дипольным моментом, величина которого колеблется в пределах 1 7_° 0D
[167] . ’
9.8.2.3. Идентификация сложных эфиров
Сложные эфиры можно охарактеризовать химически либо гидролизом до соответствующей кислоты и спирта, либо получением производных ацильного, или алкокспльного остатков [166]. Типичные реакции, используемые для превращения эфиров в подобные производные, приведены на схемах (188) — (192) [150].
R'CO2R2 + NH3 —> R’CONH2-f-R2OH (188)
R3
Et2O;
:—NHMgBr-4-R1 CO,R2 -----> R3-
H +
—NHCOR1
(189)
R3 = H или Me
R1CO2R2 + N2H4-H2o —> R'CONHNH2 + R2OH
(190)
r'co2r2 + h,nch2ch2n:
,0 —> r'conhch2ch2n;
:o
(191)
o2n
o2n
R*CO2R2 +
o2n
1I2SO4
—CO2H---------> R'CO2H4-
O2N
—CO2R2
(192)
Эфиры обычно легко идентифицировать по их характерным спектральным свойствам, важнейшие из которых обсуждены ниже.
Характеристические полосы поглощения в ИК-спсктрс [ <-1, связанные с валентными колебаниями карбонильной ipxnnbi в раз личных типах сложных эфиров приведены в табл. 9.8 6. Валентные колебания связи С—О также дают поглощение в области IIUU
333
I ЧОО см-' n хотя это полосы могут дать некоторую полез,,», /.^формацию, обычно они используются редко.
Табмща 9.8.6. Валентные колебания С = О в ИК-спектрах [168]
Тип сложного эфира Частота, С,Г*
Ненасыщенные 1750-1735
а ^-Ненасыщенные и ариловые 1730-1715
Эфиры енолов и фенолов 1800-1770
0-Лактоны 1825-1820
у-Лактоны 1780-1760
д-Лакгоны 1750-1735
а, Р-Ненасышенные у-лактоны 1760-1740
р, у-Ненасышенные у-лактоны 1805-1785
Таблица 9.8.7. Химические сдвиги сигналов в спектрах ПМР простейших сложных эфиров [169]
Приведены величины химических сдвигов протонов СНз, СНг или СН групп R (показаны в верху каждой колонки в скобках!
Сложный эфир Химический сдзпг, млн 1 относительно Г'ЛС
при R = Me (СНз) при (СН2) R=Et (СНз) при R = Pr при К = Рг-изо при R = CMej (СНз)
(а-сНг) (3 СН2)(СН3) (СН) (СНз)
R—ОСОМе 3,67 4,05 1,21 3,98 1,56 0,97 4,94 1,22 1,45
R-OCOPh 3,88 4,37 1,38 4,25 1,76 1,07 5,22 1,37 1,58
R—СООМе 2,01 2,28 1,12 2,22 1,65 0,98 2,48 1,15 1,16
протонов, находящ^ас^в^а по пп^Иа хаРак’1ернстичны для нои группе или кислополной 50женип п° отношению к карбониль-ЧесЛИх сдвигов простейшихФУ"г2ИИ- ТиП11™е величины хими-табл. 9.8.7. Для более сппжи сложных эфиров приведены в ских сдвигов а-протонои Zr Ххпредставителей величины химиче-стант экранирования; велХУЛб^.0ЦеНены СУММ прованием кон-наличием группы ___гопп ННа химического сдвига, связанная с
ветственно равна 3 13 Пос;ДаВНЭ 1,55> ГРУППЫ —О—COR, — соот-величину сдвига свивай^™ Дняя величина значительно превышает ЗОМ этерификация спиптлн гРУппой —ОН (2,56), и таким обра-фикации протонов нахои^и/ Жет ^ыть попользована для идентп-вруппе. Так, образование В а’пол°жении к гидроксильной
Достигаться просто пои АРиФтоРаПетатов из спиртов, что может зывает сдвиг сигнала ппл^ВЛеНИи тРифторуксусной кислоты, вылей на величину 0 5 Fa Н°В . ГРУППЫ СН—О в сторону слабых Типичные вели 1,4 Млн~’.
протонов В а.Р-неТаХ,?™'4^'» сигналов олефиновых
ХнФТУЛах <78) и П9 К nnF*HpaX И ЭФИР ах енолов приведены пл^НаЛ в очень слабом ггл ^Р°ТОн гРУПпы СН в формиатах дае'г 1ОМУ формиаты могут бЛ6’ Как ПОказано на формуле (80), и 334 у Ыть легко идентифицированы. Химиче-
ские сдвиги, связанные с обоими типами а-протонов в (3, у- и б-лак-Тонах показаны на формулах (81) —(83). Цифры, указанные при формулах (84), (85), показывают влияние заместителей —СО2М.е н _ОСОМе на химические сдвиги протонов бензольного кольца.
Положительные величины сдвигов указывают на смещение в сторону слабых полей; заместители, оттягивающие электроны, такие, как —СО2Ме, в основном вызывают сдвиг сигнала в сторону слабых полей.
OCOR
Н7,37
(79)
(81)
н-с^°
8,03м С
Ч)Ме
(80)
ОСОМе
(85)
Как и следует ожидать, прекрасным методом идентификации сложноэфирных групп является спектроскопия 13С-ЯМР [170], поскольку сигнал углерода 13С, связанный с углеродом карбонила эфирной группы, лежит в области 160—180 млн-1 и обычно отличается от сигнала углеродных атомов карбонильных групп других типов. Величины химических сдвигов изменяются в зависимости от строения; в общем случае для эфира R'COOR2 группа R1 оказывает большее влияние, чем группы R2, а ненасыщенность приводит к смещению полосы в сторону сильных полей. Углеродные атомы групп R1 и R2 дают сигнал, несколько сдвинутый по сравнению с сигналами аналогичных атомов соответствующих углеводородов; этот эффект наиболее заметен в случае а-угле-
Таблица 9.8.8. Химические сдвиги сигналов
в спектрах 1ЭС-ЯМР простейших сложных эфиров [170а]
Сложный эфир Химический сдвиг, млн.-1 относительно ТМС
R (С-2) (С-3) (С-1) (Me)
RCO2Me СН3 с2н5 С3Н7 (СН3)2СН 19,6 27,2 35,8 33,5 9,2 18,6 18,4 13,6 51,0 50,8 50,0
(С-1') (С-2') (С-3') (С-4') Me
ROCOMe С2Н5 60,1 14,3 20,3
C3H7 65,5 22,5 10,2 — 20,5
(СНз)2СН 65,6 20,2 — — 20,0
с4н9 63,8 31,2 19,7 14,1 20,3
335
ТпЛ.,ша 089 Химические сдвиги сигналов в спектрах 13С-ЯМР ненасыщенных и ароматических сложных эфиров [170а]
Сложный эфир -1 Химический сцвиг, млн , относительно ТМС - (С-1)
X (С-1) (С-2) (С-3) |
СН2=СНХ СО2Ме 128,7 129,9 — —-—«
ОСОСНэ 141,7 96,4 — —
х СО2Ме 130,0 128,2 128,2 132,2
6: ОСОСНэ 151,7 122,3 130,0 126,4
родных атомов (табл. 9.8.8). Аналогичные смещения сигнала относительно соответствующих углеводородов наблюдаются также в случае ненасыщенных и ароматических сложных эфиров. Величины этих сдвигов даны в табл. 9.8.9.
Электронные спектры поглощения [171] простейших эфиров и лактонов характеризуются слабым поглощением, соответствующим л*-переходам, около 210 нм, однако эти данные не находят столь широкого применения для исследований с помощью методов дисперсии оптического вращения и кругового дихроизма [172], как данные длинноволнового поглощения кетонов,, связанного с л*-переходами. Тем не менее установлены общие закономерности, касающиеся взаимоотношения между абсолютной конфигурацией и конформацией лактонов, с одной стороны, и знаком и величиной наблюдаемого эффекта Коттона, с другой. Изучены и другие данные по сложным эфирам [172]. а,(3-Ненасыщенные эфиры дают сильную л->л*-полосу около 210 нм; по мере увеличения ненасыщенности наблюдается сдвиг полосы в длинноволновую область. Ароматические эфиры также дают характеристические электронные спектры поглощения, связываемые с л-^л*'пе‘ реходами. В целом, однако, И К- и ЯМР-спектроскоппя оказались более ценными методами идентификации сложных эфиров.
Интенсивно исследована масс-спектрометрия сложных эфиров [173], и установлен ряд общих закономерностей фрагментации эфиров, что можно использовать при определении структуры. 1а < простейшие метиловые эфиры подвергаются а- и р-расщеплению, как показано на схеме (193); особенно важен процесс [3-расшепле ния при условии, что группа R в (86) содержит водородный ато^ в нужном положении. Этиловые эфиры претерпевают дополнительные превращения {схемы (194), (195)}, приводящие к обр ’ зовапию катионов-радикалов исходных карбоновых кислот^ соответствующих им протонированных карбоновых кислот. На • далось также протекание других специфических процессов р 336
ния [173], примером которых могут служить специфические теПЛеРнтации а’лкилзамещенных у- и б-лактонов, показанные на £ма“ (196). (197).
Метиловые эфиры
1 а-расщепление ; у
рЦ-сЦ-ОМе
(86)
В-расщепление (перегруппировка Мак-Лафферти)
Этиловые эфиры
[r1coor2]’+
Тгфе 74
(193)
>[r1co2h]‘+ + r2(-h)
(194)
[r'coor2J'+—>[r'co2h2]'+ + R2(-2H)
m/e 85
mte 99
(195)
(196)
(197)
9.8.3. РЕАКЦИИ СЛОЖНЫХ ЭФИРОВ
Реакции эфиров карбоновых кислот находят широкое применение в обсуждавшихся в главах 9.1—9.7 синтетических методах, и эти реакции будут лишь кратко упомянуты в настоящем разделе.
9.8.3.1. Гидролиз и другие реакции расщепления
Различные аспекты механизма гидролиза сложных эфиров подробно исследованы, результаты обобщены [10, 11, 174]. Оценка различных путей (механизмов) протекания реакции, проведенная нгольдом и сотр. путем перебора имеющихся возможностей, здесь рассматриваться не будет: важно подчеркнуть, что простейшие редставители эфиров (метиловые, этиловые эфиры и т. д.) гидро-уются в соответствии с одним из обычных механизмов [175],
337
классифицируемых [И] как Алс2 {схема (198)} и Влс2 { (199)}; оба пути (механизма) включают образование тетраэДрМиа
При.
(199)}; оба пути (механизма) включают образование -ческого интермедиата и в сущности являются механизм соединения-отщепления. аМи
аас2
О
R1—С—OR2
=± R1—СГ
X9R2
Н20
ОН
R1—С—OR2 +ОН2
ВАС2
О
R1—С—OR2
"ОН
он
I + -Н+
R'-C-OR2H
ОН
.0
R1—+ R2OH
(198)
О'
R'co;
ОН
R2OH
(199)
О
ГИДР°ЛИЗ обычно проводят с использованием сотя-и кислоты или (иногда) серной кислоты, в воде или в водных спиртах, основной гидролиз ведут с использованием гидроксида епип11Я ИЛИ каЛИЯ пли (Реже) гидроксида бария в воде водном оксида ‘множееХ аПР°Т0Н11ЫХ Р-творителях типа днмщил“ nic Syntheses О примеров этих реакций можно найти в Orga-лее хапактрпнкю ОВНОИ гидРОлиз обычно предпочтительнее; наибо-« вакетоТэеЛиповПРИМерЬ1 ТКЦИИ « табл. 9.8.10. Гидро-
часто явпяРтрР ’ а ЦИаноэфиров и производных этилацетоацетата осхшествлрния важнои стаДИей органического синтеза; методики деле этих Реакций обсуждены ниже в настоящем раз-
жен^влиХню^ра3°ВаНИеМ тетРаэдРическ°го интермедиата подвер-эфипе Р’СО р2теРических факторов, особенно если группа R1 в позволяюшиу^п^АТ“еРИЧеСКИ гР0М03дка- Разработан ряд методик, нуклеофильной я°ИТИ ЭТ" тРУдн°сти- Например, для повышения станем гигтпп- ктивностп гидроксид-иона при гидролизе под де,(-гексил-1Я-кпяиСИАа паЛ11я пРедложено [176] использовать дицикло-стерич^скн PrmHa В ЭТИХ Условиях удалось провести гидролиз тернативно мам и° затР>'Днеш1°го грег-бутнлмезитоата (87). Аль-напримеп 6vTot.-\0 11СП0Ль30Вать более сильное основание [177], применяли ио7^ Д КЗЛИЯ В Дпметилсульфоксиде. В ряде случаев [1791- этот пМГИТИЯ В коллидине [178] или диметилформамиде кого замешрниа Т РасЩепляет эфиры по механизму нуклеофиль' рически^^затпуДненНН° Р?С1депллст амиды. Для расщепления сте-
лы напоимрп^п ,НН1м1Х ЭФНа использовались и другие нуклеофи отношению кРгтДТИИПр0ПанТИ0ЛЯТ П80], который эффективен по рически затрудненным эфирам и действует в очень
3$8
9 8.10. Примеры катализируемого основаниями гидролиза сложных Таблиц эфиров
ы взяты из Organic Syntheses. Продуктами реакции (если не отмечено особо) Все прчме. является соответствующая кислота (и спирт)
Сложный эфир Реагент
— F——CO2Et КОН, EtOH
CO,Et NaOH, Н2О
ОзК’/ HCO2Et кон, Н2О КОН, МеОН, Н2О
СбН1, CH=CHCH2CH=CH(CH2)7CO2Et NHCONH2 CH3C=CHCO2Et МеОСН2СНВгСО2Ме сн2 NaOH, EtOH, Н2О NaOH, Н2О NaOH, Н2О
II С1оН21СС02Ви-тр<?г МеО2С(СН2)9СО2Ме no2 КОН, EtOH Ва(ОН)2, МеОН*
1 Ме2ССН2СН2СО2Ме Me Me КОН, Н2О
C4H9CHCH2CO2CHEt КОН, EtOH, Н2О
* Гидролиз до моноэфира.
мягких условиях. Он селективен также в отношении метиловых эфиров. Цианид натрия в гексаметилфосфорамиде также селективен в расщеплении метиловых эфиров, например сообщалось о селективном расщеплении метилмезитоата в присутствии этилбензоата [181]. Расщепление пространственно затрудненных эфиров под действием указанных выше сильных нуклеофильных агентов протекает, по-видимому, по механизму типа включающему расщепление связи алкил-кислород {схема (200)}. Примеры нуклеофильного расщепления стерически затрудненных эфиров даны на схемах (201) —(204).
Высокоосновные амидины, 1,5-диазабицикло [4.3.0] нонен (88) и >5-диазабицикло [5.4.0] ундсцен (89) также применялись для расщепления стерически затрудненных эфиров [182], обычно
339
/9
R2Nu
1 II ' 9 / 1
r’-C-O—R2 rNu
нагревание эфира с основанием при высокой температуре {схемы (205), (206)}. Для расщепления затрудненных эфиров применяли также тригалогениды бора; обнаружена некоторая селективность действия [183].
340
Алкоголиз {схема (207)} можно использовать нс только для теза сложных эфиров (см. разд. 9.3.1.1), но и для их расщеп-с11Н1Я. наиболее активные эфиры, например трет-бутиловые эфиры, ЛеН1гт'рсагпровать со спиртом без катализатора, хотя обычно для М°отекания реакции требуется присутствие кислого или основного ПР ализатора. Ацидолиз {схема (208)} также иногда используют к расщепления эфиров, например муравьиная кислота [184] Д'пи уксусная кислота в сочетании с сильно кислым катализатором И'огут служить удобными реагентами для расщепления трет-бути-ловых эфиров [185] или таких лабильных эфиров, как акрилаты.
Алкоголиз R'CO2R2+R3OH — > R'CO2R3+R2OH Ацидолиз R'CO2R2 + R4CO2H —> R'CO2H + R'CO2R2
(207)
(208)
Н+. Н2О Д
RiR2C(CO2Et)2 --------> R>R2C(CO2H)2 ——> R>R2CHCO2H (209)
или HO-, —CO2
затем H +
HC1, H2O
CH2(CO2Et)2 + CH2O —> CH2[CH(CO2Et)2], CH2(CH2CO2H)2 (210)
R1 R1
I h+, h2o I
R3CO—C—CO2Et ----------> R3CO-C-CO2H > R3COCHR'R2 (211)
I I ~C°2
R2 R2
Гидролиз и декарбоксилирование эфиров замещенных малоновых кислот с образованием уксусных кислот проводят обычно либо действием соляной кислоты, которая сразу приводит к гидролизу и декарбоксилированию, либо действием гидроксида натрия или калия, приводящих первоначально к образованию соли малоновой кислоты, которая затем при нагревании с кислотой подвергается декарбоксилированию {схема (209)}. Синтез глутаровой кислоты [схема (210)] может служить примером этой методики. [З-Кето-эфиры обычно гидролизуются под действием кислот до соответствующих [3-кетокислот, которые самопроизвольно декарбоксилируются с образованием кетонов {схема (211)}; использование кислоты в качестве катализатора позволяет избежать катализируемого основаниями деацилирования. Селективное декарбоксилирование эфиров малоновой кислоты до эфиров уксусной кислоты и превращение |3-кетоэфиров в кетоны может протекать в более мягких и более селективных условиях. Так, нагревание эфира с цианидом натрия в присутствии диметилсульфоксида и контролируемого количества воды рекомендовано [186] и применено [187] для селективного гидролиза синтетического интермедиата (90) (R = CO2Et-> R = Н). Показано [188], что хлорид натрия в ди-метилсульфоксиде служит более эффективным реагентом и что Реакция может протекать даже в присутствии одного влажного Диметилсульфоксида. Примеры реакции этого типа представле-! на схемах (212) и (213); указанный метод применим также
341
Несфпря. миграции
к а-цианоэфирам, дающим нитрилы {схема (214)}, и к женным ненасыщенным эфирам типа диэтилаллилмалоната (gn' который дает этплпентен-4-оат (реакция протекает без двойной связи).
Na Cl, ДМСО
R1R2C(CO2Et)2 ------—-----> R’R2CHCO2Et
H2<J
NaCI, ДМСО
BuCHCN ------— >- BuCH2CN
I H2O
CO2Et
^CHjCHEtCC^Me
CH2=CHCH2CH(CO2Et)2
(91)
(1) Использование эфирных групп в качестве защиты
Защита карбоксильной группы путем превращения ее в сложноэфирную группировку обсуждалась в разд. 9.1.4.7; для регенерации исходной кислоты используют ряд селективных методов расщепления (см. также [189]). Защите гидроксильных групп посвящен обзор [190], и здесь будут рассмотрены лишь несколько примеров использования ацилирования, как метода защиты. Образование простейших эфиров, например ацетатов, таких соединений, как стероиды, сахара, нуклеозиды и циклические спирты, а также бензоатов получило широкое применение; расщепление таких эфиров достигается гидролизом с основанием, аммонолизом и метано-лизом. Очень легко образуются формиаты, которые затем гладко гидролизуются в присутствии водных или спиртовых основании, например бикарбоната натрия в метаноле. Формиаты поэтому можно использовать для селективной защиты в присутствии дрУ' гих этерифицированных гидроксильных функций; пример подобной селективной защиты представлен на схеме (215).
цены, слишком чув?тТвпРтелГаТ°В’ ХОТя И МогуТ быть легК° П0ЛУ’ _ чузствптельны к спиртам и воде и не находят
□42
покого использования в качестве защитных групп. Другие заме-ш1„ные ацетаты, обладающие меньшей активностью, например ^рЫ XCH2CO2R (92) (X = С1, ОМе или OPh), расщепляются д действием нуклеофилов; это расщепление можно вести селективно в присутствии более стабильных эфирных групп, таких, как ацетаты и бензоаты.
Две специфические защитные группы, используемые в химии пцродных соединений, расщепляются в селективных условиях. Это 0-нитробензоильная группа, которую можно удалить восстановительным расщеплением [191] {схема (216)} путем внутримолекулярного нуклеофильного расщепления, и |3-бензоилпропио-нильная группа, которая удаляется действием гидразина [192] опять же за счет внутримолекулярной нуклеофильной атаки {схема (217)}. Кратко мы еще остановимся на внутримолекулярном взаимодействии подобного типа в следующей части этого раздела.
Для защиты фенольных гидроксильных групп редко используют ацилирование в связи с широкими возможностями для применения алкильной защиты, однако иногда прибегают к ацетилированию или бензоилированию; в обоих случаях защитные группы легко снимаются гидролизом или аммонолизом.
PhCOCH2CH2COOR
N2h4 ---->
(217)
N—NH
(2) Гидролиз и внутримолекулярный катализ
Гидролиз сложных эфиров, катализируемый другими группами, содержащимися в молекуле эфира (влияние соседних групп пли внутримолекулярный катализ), подробно исследован [193]; разработанные принципы легли в основу подбора защитных групп как для кислот, так п для спиртов. Эти исследования важны еще
343
И потому, что существует связь между такого рода катализом й ферментативным катализом.
Нет возможности детально останавливаться на этом вопроСР но общие принципы иллюстрируют схемы (218) —(221), которые выбраны как соответствующие примеры внутримолекулярного общего основного катализа {схемы (218), (219)} и внутримолекулярного нуклеофильного катализа {схемы (220), (221)}. Во всех случаях гидролиз эфирной функции протекает значительно быстрее при наличии влияния, соседней группы, чем при отсутствии такого влияния, например у соответствующих /шра-замещенных ариловых эфиров.
+ PhO~
н2о
---->
(221)
9.8.3.2. Реакции енолят-анионов
Отнятие протона от а-углеродного атома эфира приводит к о разованию резонансно-стабилизованного енолят-аниона (93). днако для реакции требуются специальные условия; в противном случае генерируемые таким путем анионы показывают тенденцию к конденсации с другими молекулами эфира (конденсация Кляй-на, см. разд. 9.8.3.3). В некоторых случаях анионы удается генерировать действием классических органических осно-
ИИ>’ такие анионы можно с умеренным выходом алкилировать, реакции этого рода, например, представленные на схемах 344
(222) —(224) (соответственно по ппа
„рименеивя относительно неаитивнь,2^™ ! 194—196]) требует
)с~с( *-> \~сх°~
ХОР
(93)
.. т— phsCNa Et
Me2CHCO2Et---------> м гг eh ।
Е„о м^ссода -----------> Метода (222)
PhCH,CO,Bu-rper Р№1СО1Ви.гр„ pJ“ (Ж)
‘ ‘‘e.rtccjjBu-Tper
.. гигг\ d NaNH, _ Ph
Nte2CHCO2tiu-TpeT------> м Pht?r i
NH3 Me,CCOpBu-^7 -----------» M I
le-’<-CОрВи-треу (224)
Сложные эфиры, например этилцианоацетат (94) (Y = CN) и диэтнлмалонат (94) (Y = CO2Et), которые образуют стабильные карбанионы и не проявляют тенденции к самокопденсацпи, широко используются в качестве синтетических реагентов, как видно из схемы (225). Структурные ограничения реакций этого типа очевидны; типичные примеры, заимствованные из Organic Syntheses, приведены на схемах (226)—(228).
r'r2c=o
YCH2CO2Et —> YCHCO2Et ---------► YCHCO2Et —> YCCO2Et
(94) Y = CO2Et или CN I R‘R2COH CRiR2
R3COX
YCHCO2Et YCHCO2Et I I
R1 COR3
i i t R1 pi
4 \ гидролиз 14 \
VYCO2Et ----------> XCHCO2H
R2/ R2/
/CN Mc-co'NH(* /CN
Et2CO CH2 -~ Et2C=C
\zO2Et ^CO2Et
_ пиперидин
PhCHO + CH2(CO2Et)2 ------------> PhCH=C(CO2Et)2
(225)
(226)
(227)
CH ton Mg(OEt)2 l^2(CO2Et)2 -----> EtOMgCH(CO2Et)2
PliCH2COCl
-------->- PhCH2COCH(CO2Et)2
HO,C.
\hr.
НОас/ ТГФ
o2cx.
;cr>
О2с/ 3Li +
R2X
O2c4
Vr*r2
O2C'
(228)
H*
---► HObCCHR'R2
(229)
345
Г. Millie триаиионов, генерируемых из монозамещеннЫх Пр» , СЛОТ может упростить экспериментальную процедур, малоновых ки^л , аднак0 боль1ПИцство работ проводилось с ^споТьзованнем реакционной последовательности, приведенной на “лдаис производные енолятов эфиров (95) можно север,,р0.
Л (198 1991 при низкой температуре (—78 С); в случае трет-вать luo, ‘У, ‘ 0ТМечена малая тенденция к самоконденсации.
ГТП Лщподходящего литиевого основания, например литий-Хопп ц " ,огеАкспла“.да (96) в ТГФ при -78"С сводит ,ту тенденцию^до минимума даже в случае метиловых эфиров [198]. 9то открывает возможность широкого применения полученных из сложных эфиров литийенолятов в качестве синтетических интер-Тпиатов В других работах в аналогичных условиях использовались днпзопроппламид лития [200] и 2,2,6,6-тетраметплпиперидид пития (97) 201] Реакции полученных этим путем енолятов представлены на схеме (230), из которой следует, что целый ряд классических реакций органической химии, и, в частности, реакции, традиционно проводимых с помощью натриевых или калиевых производных этилцианоацетата или диэтнлмалоната, может оыть осуществлен альтернативно с помощью литиевых производных.
/С~ СО2р
/С—co2r
ACll Вгя
OSixMe2 В и-трег с=с / \
OEt
A CISiMesBu-r/ier
со2р /CZ чсо2н Jco,
zCHCO2R
R’CHO
<rL.H или Ph)^ ^-c—CO2R r'CHOH
|R2Hal ^C—COeR. R2
LiN(Pr-£,J0)2 III
или /О CO2R
LiNfPr-i/joJCeHn Li 71 ТГФ при -78°c ,
(95)
^Br2 или Io o—co2r
Hal
Ph CO CII ^c—co2r
COPh
Циях лптнГ1енолятовенч,рНпеТИ'1еСКИе метоДЬ1> основанные на реак-ь Генных сложных эфиров, суммированы
на схемах (231) (234) (соответственно m л
^-Ненасыщенные эфиры также могут ляЛ работам [202—2051) которые алкилируются в «-положениеДТЬ Ли™йсноляты [2061 эфиры (схема (235)}. При низких гемпе?атапьР’?/'Ненасыщенние быть получены даже еноляты замещение УРЙХ 78 °с) могут лигнйенолят (98) использован в синтезе 207Т''Ж'<СИЭф"Ров: так, {схема (236)}. Литииэтилацетат (99) MoLt а < — )‘камптотенцина пользовании бис(трнметилсилил) амт да т\?олУчен при не-«гве основания. Генер„ро»а„;,ы7" ’п L,N<8(МОэ)2 ‘в ка.
(208) можно применять при синтезе 6 mL угем еполят-аннон служащим альтернативой хорошо ИзвСсп2°К„СИЭ<Р"ров **»««>», СКОРО [210] (схема (237)}. местной реакции Реформат-
Li С8Н, 7CHCO2Et
Li
PhSSPh
R’CHCO2R2
PhSSPh
-78 °C
SPh
R‘CHCO2R2
SPh
CsH|7CHCO2Et
c8h17coch3
(231)
Li
RCH2CHCO2Et 2i2±^
-78 °C
LiCH2CO2Et
SPh
R'CCOiR2 SPh SePh I H2O2
RCH2CHCO2Et -----> RCH=CHCO2Et
z\ MHal
R'COCO2R2
(232)
(233)
CH3
Cui
— Ho °о CuCH2CO2Et-----------
-зо °с
CO2Et
(234)
LjN(Pr-wo)2
ГМФтд,ТГФ
Br CO2Et
(235)
О
MeCH2CHCO2Et -i!N(Pf-“304 | ТГФ, -78 °C
OCO2Et
Li
MeCH2CCO2Et +
OCO2EtJ
(98)
О
N
:O
(—РКамптотецин
(236)
Et CO2Et
CH8C02Et -HN(SiMc3)2
Lici-i2co2Et
-78 °C
(99)
R'\fH
CH2CO2Et
(237)
347
L!N(Pr-ujo)2;
------------>.
CH2O
Лптийеноляты лактонов также используются в синтезе, напри-мер для получения [209] а-метилен-у-лактонов по схеме (238).
а-Галогенэфиры можно использовать и как источник металлоорганических соединений; реакции Реформатского для синтеза Р-гидрокснэфиров [210] {схема (239)} основаны на взаимодействии цинковых производных енолятов эфира с альдегидом. сс-Га-логенэфиры под действием сильных оснований могут проявлять свойства карбеноидов, и в синтезе Дарзана [211] аД-эпоксиэфиров используется реакция карбонильного соединения с а-галогенэфи-ром в присутствии сильных оснований, например амида натрия, трет-бутоксида калия [218], бис (триметилсилил) амида лития [213] или бутиллития [214] {схема (240)}.
Zn, CgHg
R'CHO-p R2CHBrCO2R3 -------R'CHCHCO2R3 (239)
HO R2
R’COR2 + C1CH2CO2R3
основание
---------->
(240)
9.8.3.3. Нуклеофильное присоединение по карбонильной группе
Щелочной гидролиз сложных эфиров по механизму Влс2 протекает через нуклеофильное присоединение по карбонильной группе с образованием тетраэдрического интермедиата (см. разд. 9.3.3.1). Это общая реакция нуклеофилов с карбонильной группой эфира, и различные примеры ее применения будут рассмотрены ниже в настоящем разделе. Взаимодействие с гидрид-ионами приводит к восстановлению, поэтому эта реакция будет обсуждаться вместе с другими реакциями восстановления (см. разд. 9.8.3.4). Взаимодействие эфиров с нуклеофилами обычно приводит к замещению алкокси-группы {схема (241)}.
О О' О
И Nu~ I || /олП
R1—С—OR2 ----> R‘ — С—OR2 —> R1—С—Nu+'OR2 <241)
Nu
(1) Конденсация Кляйзена
Конденсация Кляйзена включает присоединение енолят-аниона эфира по карбонильной группе другой молекулы эфира. Реакцп {схема (242)} приводит к образованию 0-кетоэфиров, например 348
лацетоацетата CH3COCH2CO2Et, уже отмеченного в гл. 9.5. Здесь будут рассмотрены лишь несколько примеров реакции.
О О
о
основание
СНС—OR
С—С—OR + ХН-С—OR
О’
(А)
(В) О
сн—с—с—co2r -----
I I -RO-
co2r
(242)
RO
EtONa zCOCO2Et 160-170 °C
CieH33CH2CO2Et + (CO2Et)2 ---> С16Н33СН( ---------->-
AC02Et
—> C16H33CH(CO2Et)2 (243)
EtONa zCOCO2Et |75 oc
phCH2CO2Et + (CO2Et)2 ---> PhCH ----> PhCH(CO2Et)2 (244)
XZO2Et
CHO CHOH.
EtONa I ||
RCH2CO2Et + HCO2Et -----> RCHCO2Et ч=*= RCCO2Et (245)
Эфиры, не содержащие а-водородиых атомов, могут использоваться в качестве компонента (В) {см. схему (242)}. Компонентом (А) в таком случае будет служить какой-нибудь другой эфир; особенно удобны в этом отношении диэтилоксалат и этилформиат. Диэтилоксалат реагирует с другими эфирами с образованием ^-замещенных а-кетосукцинатов, которые декарбонилируются при высокой температуре, давая замещенные малоновые эфиры {схемы (243), (244)}. Этилформиат образует а-формильные эфиры, которые обычно существуют в виде енольного таутомера {схема (245)}. Лактоны также вступают в реакцию, аналогичную конденсации Кляйзена, однако поскольку отщепление алкоксидной группы невозможно {ср. схему (242)}, то образуется енол, а не кетоэфир {схема (246)}.
(246)
(247)
Внутримолекулярная
Реакция Дикмана {схема Результате образуются
конденсация Кляйзена диэфвров, пли (247)}, протекает особенно гладко, если пяти- или шестичленные циклы (и == 3
349
или 4). Однако и в случае циклов с большим числом
атомов (п= 11 или 12) могут быть получены достаточГЛер°Ань1* выходы продуктов, особенно если использовать техники ВЬ1с°кИе разбавления [215J. Реакция Дикмана широко испоЛч Выс°к°го синтеза производных циклопентанона и циклогексанон АЛя
(2) Образование енолят-анионов из кетонов
В принципе эта реакция представляет собой общий метод синтеза р-дикетонов, однако возможность протекания конкурирующих реакций конденсации Кляйзена и альдольной конденсации накладывает некоторые ограничения, касающиеся выбора групп Rl, R2 и R3 на схеме (248). В качестве эфирных компонентов обычно'используют этилоксалат и этилформиат и особенно диэтилокса-лат [216] для синтеза p-кетоэфиров из кетонов {схема (249]}. Примером может служить классический синтез эквиленина по Бахману [217], который частично представлен на схеме (250). Использование этилформната дает р-кетоальдегиды. Реакция обратима (гидролиз) {схема (251)}, и это используется [218] для защиты а-СН2-групп в циклических кетонах, например при введении ангулярной метильной группы в синтезе стероидов. Другие примеры реакций кетонов со сложными эфирами в рамках общего синтеза Р-дикетонов представлены на схемах (252), (253) и (254)
О'
R'CHCOR2 + R3CO2R4 —> R2COCHR1—С—R3 —> R2COCHR'COR3 (248)
OR4
350
NaOEt, EtOH . ,n(-04
CH3CO2Et + CH3COCH3 --------> CH3COCH2COCH3 (252)
ОСН2Рг-«зо + M3o-PrCH2CO2Et (M3O-PrCH2CO)2CH2 (253)
РЬСОСН2СОСНз + PhCO2Et PhCOCH2COCH2COPh (254)
Взаимодействие с реактивами Гриньяра
С ножные эфиры обычно взаимодействуют с реактивами Гринь-в двустадийном процессе, дающем спирты {схема (255)}. В некоторых случаях, однако, удается выделить промежуточно боазуюшиеся кетоны, например при реакции пространственно затрудненных эфиров типа метплпивалата {схема (256)} [220]. Этилформпат дает вторичные спирты, в то время как другие эфиры приводят к третичным спиртам, которые часто легко дегидратируются до олефинов. Последняя реакция используется при расщеплении эфиров по Барбье — Виланду [221] {схема (257)}.
ОН
R3MgX |
R'COOR2 + R3MgX —R'COR3-----------> R1— C—R3 (255)
R3
тдег-ВиСОгМе + «-PrMgCl —> тдег-ВиСОРг-» (256)
ОН
PhMgBr I MeCO2H O3, EtOAc;
RCH2CO2Me ---------> RCH2CPh2-----------> RCH=CPh, --------------->
H2O2, HOAc «ли
RuO4, NalO4
—> RCO2H + Ph2CO
(257)
Литийалкилы также присоединяются по карбонильной группе эфиров, но и В-этом случае процесс обычно протекает в дв" стадии (255)}; пример такой реакции представлен на схеме (zoo) [222].
аня'?аКТ0НЬ1 также реагируют с реактивами Гриньяра; реакция ппНх°ГИЧНа ВЗаим°Действию с эфирами и приводит к диолам {например, по схеме (259)}. 1
Me
СН2=С—СО2Ме + Ви 13 г
Me ОН Ы. ТГФ | |
-----> СН2=С—СВи2
(258)
=О + MeMgBr
СзН,,
(259)
351
(4) Реакции с другими карбанионами
нпиргтво других карбанионов может вступать Во
Большое коли 1ьк;й эфирами> давая разнообразные !1ро. взаимодействие со сл такПх реаКций кратко рассмотрены
дуКТЫ. Отдельные НрнмсЦ ниже. йгтиии ариловых эфиров с ацетиленидом натрия
При взаимодеист Р э(Ьир0В с реактивами Гриньяра, обра-[223], аналогично Р^ ы гсхСма (260)}, В большинстве других зуются третичные сиу высокостабилизованными карбанио-
случаев, однако, ре: щ адИИ ацилирования. Так, эфиры копнами 0С'ганавЛИВ^1)банИ0НаМи, стабилизованными присутствием денсируются с «аР° нокетОны; эта реакция использована для
ароС«ческ“х кетонов из арвлацетоннтрнлов (схема (261)}-
\—СО.Ph + NaCs СН -
NaOF.t
ArCH2CN + RCO.Et
он
CH2C12^ NH3
CN
ArCHCOR —ArCH2COR
(260)
(261)
OH
PhSCH2I.l I (РЬСОЦО; / 3
C9H19CO,Me -------> C9H19-C-CH2SPh C9H19-C (262)
| LI> 1Nrt3 ^CH2
CH2SPh
О
+ - McgSO l! AlHg Р/оРП
C6HuCO2Et4-NaCH2SOMe ---------> C5HuCOCH2SMe CsHnCOUh
NaH, | (263)
О
A,Hs - C5H,.COCH,M<
C5Hи COCH SMe НгО1 ТГф Me
CO2Et
4- LiCH2SNArLi
ТГФ
О
COCH2SNHAr
о
Q^-C0CH’ (2S4)
/SMe /SMe p (2б6)
PhCO.Et+ Na+ CH/ — PhCO-CH^ —> PhCH2CO2
\-SOMe \s(O)Me
352
нионы различных структурных типов, стабилизованные нем серы, реагируют со сложными эфирами. Эти реак-присУтСТВ случаев можно использовать в синтетических целях; ции в р „приставлены на схемах (262) —(265) (соответственно по
[224-227]). . . .
р гл гты фосфония реагируют с эфирами по одностадийной реак-павая кетоны {схема (266)} [228], или по двухстадийной ни с образованием олефинов {схема (267)} [229]. Альтер-
Pea,<UH0 олефины из сложных эфиров [230] можно получать по наТП (268); эта реакция родственна синтезу олефинов по В;:т
Вит-
J Et2O + NaOH, МеОН
Ft + Ph3P=CH2 ----> PhCOCH2PPh3-------------> PhCOCHs (266)
pnCvsbi ~ °
? Me2SO
piCOR2 + Ph3P=CH, -------> [RCOCH3] —> RC( (267)
XCH3
Me О Me О
| || ТГФ I || NaBH4;
phCO2Me + LiCHP(NMe2)2 -----> PhCOCHP(NMe2)2--------—► PhCH=CHMe
(268)
(5) Реакции с азотсодержащими нуклеофилами
Реакция сложных эфиров с аммиаком и первичными и вторичными аминами, ведущая к амидам {схема (269)}, служит широко применяемым методам синтеза амидов и обсуждается в главе 9.9. Здесь же она будет лишь кратко упомянута.
R'CO2R2 + NHR3R4 —> R1 CONR3R4 + R2OH (269)
NH3
MeCH(OH)CO2Et -----> MeCH(OH)CONH2 (270)
NH4OH
ClCH2CO2Et --------> C1CH2CONH2 (271)
NH4OH EtO2CCH=CHCO2Et ------> H2NCOCH=CHCONH2 (272)
Реакцию метиловых или этиловых эфиров с аммиаком ведут в растворителе, таком, как метанол, обычно без катализатора, ХОТЯ обычно используют основной катализатор (от хлорида аммония ДО”метоксида натрия и других сильных оснований). Примеры этой реакции представлены на схемах (270) —(272); отметим, что из а-хлорэфира образуется не а-аминоэфир, а амид. На ставЗХ (273)~~(275) (соответственно по работам [231—233] пред-
-Нь1 примеРЬ1 аналогичных реакций с участием первичных и таюкеЧНЫХ аминов’ а также ароматических аминов; эти реакции катализируются основаниями, такими, как метоксид натрия.
/Г~\ .. . 175 °с р—\
(273)
<7—SH + PhCH2NH2
ШО2Ме
12 Зак. зуб
SH
CONHCH2Ph
353
BuLl
PhCO2Et 4- (PhCH2)2NH т~PhCON(CH2Ph)2
МеО—CO2Et + PhNH2 Me2-SQ-> Me' + KOH, MeOH
R’CO2R2 + NH3OHC1‘ ------> R'CONHOH
Взаимодействие сложных эфиров с гидроксиламином привод к гидроксамовым кислотам {схема (276)}; прежде эта реакция i пользовалась для обнаружения сложноэфирных группировок гГ скольку гидроксамовые кислоты с раствором хлорида железа даю? характерное красное окрашивание.
Сложные эфиры легко реагируют с гидразином с образованием гидразидов; эта реакция лежит в основе расщепления эфиров д0 аминов по Шмидту [234] {схема (277)}. Расщепление по Шмидту можно также проводить действием азида натрия и серной кислоты что ведет прямо к ацилазиду, как, например, в синтезе циклоундеканона [235] по схеме (278).
N2H4, H2O, EtOH NaNO2, HCI, Н2О
PhCH2CO2Et -----------> PhCH2CONHNH2 --------
CONHPh
A H+, H2O
—> PhCH2CON3 —> [PhCH2CON:] —> PhCH,NCO -----------
— •> PhCH2NHCO2H —> PhCH2NH2
NaN3 H2O /CH2
(ch,),Q —► (C"')s4=0
CO2Me XnH,
nh2
I Me\/—\
Me2CCH2CH2CO2Me —> V )=O
me NH
NH2 NH 0
R’CHCC^R2 —>
zCH (CH2)/ ||
NH R1
Особенно легко протекает реакция внутримолекулярного
(274)
(275)
(276)
(277)
(278)
(279)
(280)
Особенно легко протекает реакция внутримолекулярного аммонолиза, и, например, у-аминоэфиры, циклизуются уже при ком натной температуре или при нагревании, давая пирролидону {схема (279)}. Эфиры а-аминокислот циклизуются с образование! дикетопиперазинов {схема (280)}.
(6) Реакции с кислород- и серусодержащими нуклеофилами
К наиболее важным реакциям с нуклеофильным кислородов1 относятся гидролиз и алкоголиз, атака гидроксида или алкокс обычно приводит к протеканию реакции по механизму nPHC0ZII нения-отщепления (см. разд. 9.8.3.1). Реакция между лакТ0'(ТЬ и этиленгликолем протекает аномально, и может происход 354
он
Апязование ортоэфира [236] в условиях, которые обычно ведут обГ.,когопнзу сложных эфиров; пример такой реакции дан на схеме /280- Однако обычно для образования ортоэфиров требуются специфические условия (см. разд. 9.8.4).
(ЮО)
О=°
о + MeAlS(CH2)2SAlMe2
RCO2Me + Me2AlSCH2Ph —> RCOSCH2Ph
(102)
(284)
Реакции типа, представленного на схеме (282), легче протекают с серусодержащими нуклеофильными реагентами, хотя и требуют тщательного подбора условий. Так, сложные эфиры реагируют с алюминиевыми производными этандитиола-1,2 (100), давая тиоацетали кетена (101), которые можно превратить в кислоту действием оксида ртути и эфирата трифторида бора [237] {схема (282)}. Эта реакция представляет способ защиты сложноэфирных групп в процессе синтеза, поскольку производное (101) устойчиво к действию органических кислот, нуклеофильной атаке оснований, алюмогидрпду лития и даже к металлоорганическим соединениям. Аналогичным образом можно защитить лактоны [237], однако в этом случае первоначально образующийся продукт циклизуется с образованием дитиоортоэфира (102) {схема (283)}. Если в качестве реагента использовать соответствующее производное то-луолтиола, то реакция приводит к ацнлтиолам [237]; соединения этого типа находят применение в качестве мягких ацилирующих агентов {схема (284)}.
9.8.3.4. Восстановление
Восстановление сложных эфиров можно рассматривать еще как ДИП пример реакций карбонильной группы, в данном случае с идрид-анионом. Восстановление первоначально приводит
12*
355
К образованию альдегида в соответствии с обычным механизмов присоединения-отщепления {схема (285)}, но во многих слуЧа ” альдегид восстанавливается далее до первичного спирта, который служит конечным продуктом реакции. Существуют и другие воз можные направления восстановления, включая образование про' стых эфиров {схема (286)} или продуктов превращения анион' радикала (103), образующегося при присоединении одного элск-трона к сложноэфирной группе {схема (287)}.
О 0“
II И" I Н"; 1
R’COR2 ----> R1—С—OR2 RICH0 RCH2OH (285)
н
о
II (286)
R'COR2 —> R‘CH2OR2 1
О О'
II Na I R'COR2 ----> R'C—OR2 —> Продукты (287)
(103)
(1) Восстановление до спиртов
Впервые сложные эфиры были восстановлены до спиртов действием натрия, растворенного в этаноле или в смеси этанол-то-луол; в ранних выпусках Organic Syntheses имеются многочисленные примеры использования этой методики. Можно также использовать каталитическое гидрирование в присутствии хромита меди при повышенных температуре и давлении. Применение в качестве восстанавливающего агента алюмогидрида лития LiAlH4 [238] существенно улучшило выход и надежность процесса; в настоящее время это соединение наиболее широко используется для восстановления эфиров до первичных спиртов. Реакцию с алюмогидри-дом лития обычно проводят с избытком реагента в среде простого эфира, ТГФ или другого подходящего эфирного растворителя, эффективность действия реагента несколько изменяется в зависимости от применяемой температуры и растворителя. Лактоны восстанавливаются до диолов.
Алюмогидрид лития с хорошими выходами восстанавливает аф-пенасыщенпые эфиры до аллиловых спиртов, например п° схеме (288), однако в отдельных случаях может протекать восста-новление сопряженных двойных связей. Во избежание этого осложнения в качестве восстанавливающих реагентов рекомендован этоксиалюмогидрид лития LiAlH3(OEt) [239] или смесь алюмогидрида лития и хлорида алюминия [240]. Для восстановлен! эфиров в спирты использовались и другие производные алю*. гидрида лития, в которых один или более водородных ато 359
заменены алкоксильной группой, в особенности в случаях, когда требуется селективность реакции.
L1AIH4 /ОЯЯ'Ъ
z^\z^'\cO2Et Et2o
Сообщалось об использовании для восстановления сложных эдиров в спирты гидрида алюминия [241]; иногда для тех же цепей применяют диизобутилалюмогидрпд [242], хотя обычно последний реагент используют для селективного восстановления эфиров до альдегидов (см. ниже).
Алюмогидрид лития можно применять лишь в безводных условиях; натрий-, литий- и кальцийборогидрпды, относительно устойчивые в водных и спиртовых растворах, были поэтому исследованы с точки зрения возможности их использования в качестве реагентов для восстановления сложных эфиров. Эти работы часто приводили к отрицательным результатам, тем не менее борогидрид натрия в растворителе, таком, как метанол, неоднократно использовали в различных синтезах {например, по схеме (289)} [243], а получение енолов эфиров р-кетоамидов (104) с последующим их восстановлением борогидридом натрия служит удобным методом [244] получения первичных спиртов из карбоновых кислот {схема (290)}.
К'аВН4
Ph(CH2)2CO2Me МеО~> Ph(CH2)3OH (289)
NaBH4
-----> RCH2OH (290)
Борогидрид натрия с добавками этандитиола-1,2 [245], анилина [246] или хлорида алюминия [247] также использовали в качестве восстановителя, но эти методики не получили широкого распространения. Сообщалось [245] об использовании в качестве селективного восстанавливающего агента триметоксиборогидрида натрия NaBH(OMe)3, который быстрее восстанавливает эфиры, полученные из первичных спиртов, чем эфиры, содержащие остаток вторичного спирта.
Борогидрид лития, полученный из борогидрпда натрия и хлорида лития, обладает промежуточной активностью между борогидридом натрия и алюмогидридом лития; этот реагент восстанавливает до спиртов как сложные эфиры, так и лактоны [249], и ° ладает более высокой селективностью, Чем алюмогидрид лития.
орогидрид кальция, полученный из борогидрпда натрия И юдида или хлорида кальция, рекомендован [250] в качестве ooReKJHBHOr0 Восстанавливающего агента для превращения эфи-мв спирты; Пример его использования дан на схеме (291).
367
o2n
Ca( ВНЩ ---------►
ТГФ
—CH2OH
zCO2Me
О
NCH2Ph
B2He ----->-
(291)
(292)
Большинство функциональных групп, восстанавливаемых действием алюмогидрида лития, можно восстанавливать и дибораном [249, 251]; однако сложные эфиры в отличие от карбоновых кислот относительно инертны к этому реагенту, так что возможно даже селективное восстановление амидов [252] в присутствии эфиров {схема (292)}.
(2) Образование простых эфиров
Восстановление до простых эфиров {схема (293)} ряда сложных эфиров, в особенности трет-бутиловых эфиров {схема (294)}, можно осуществлять действием борогидрида натрия и эфирата трифторида бора в растворителе, таком как диглим [253], или при использовании амомогидрада лития и эфирата трифторида бора в эфире [254]. Лактоны, как полагали вначале, дают в этих условиях циклические простые эфиры [253, 254], однако позднее было показано [255], что в действительности образуются циклические простые эфиры енолов {схема (295)}.
R'COR2
L1AIH4, BF3
;"иГвн4:вр3" R1CH2°R2
NaBH4
RCOOBu-трет —------—---->- RCH2OBu-rper
BF3*Ei2O
(293)
(294)
(295)
NaBH4 --------->
BF3 • Et2O
(296)} значи-
(3) Восстановление до альдегидов
Восстановление сложных эфиров до альдегидов {схема является потенциально важным синтетическим методом; тельное количество различных восстанавливающих агентов испытаны для этой реакции. Лучшим из них оказался диизобутилалю-могидрид (105) [256]; схема (297) иллюстрирует его использова ние на одной из стадий синтеза эметина [257]. Этот Ре3це!(Т оказался также полезным в синтезе простагландинов (см. гл. 9- ) Лактоны восстанавливаются до лактолов {схема (298)}, которы образуются также при восстановлении некоторых диэфиров {схема (299)}. С другой стороны, а,£-ненасыщениые лактоны можн
358
прямо восстанавливать до фуранов [258] {схема (300)}. Алюмогидрид натрия [259] и три-трег-бутоксиалюмогидрид лития [260] использованы для восстановления сложных эфиров (только эфиров фенолов) в альдегиды. Реакция бензолсульфоиилгидразидов с основанием также дает альдегиды [261]; эта реакция открывает eine один путь от ароматических сложных эфиров к ароматическим альдегидам {схема (301)}, хотя он и менее удобен, чем i-----
прямое
восстановление.
R'COOR2 —> R'CHO+R2OH (296)
Drn м N2H4’ Нг0: Na2CO3
RC°2Me ~~PhS02ci > RCONHNHSO2Ph —+ RCHO (301)
Лактоны можно восстановить до лактолов1 действием различных реагентов, ио результат не всегда однозначен, и часто в качестве конечных продуктов реакции получают диолы. Триэтокси-алюмогндрид лития предложен [262] в качестве реагента, позволяющего избежать образования диолов {схема (302)};
359
органоборан (106) также рекомендован [263] как селективны” восстанавливающий реагент {схема (303)}. ыи
R = —СНСНМе2
Me
(4) Ацилоиновая реакция
Ацилоиновая реакция [264] была предложена для синтеза карбоциклических соединений со средними или большими циклами; она оказалась одним из наиболее надежных методов получения этих соединений. Реакция состоит в восстановлении диэфира длинноцепочечной дикарбоновой кислоты натрием в кипящем ксилоле; ее механизм представлен на схеме (304). В общем реакция протекает довольно гладко, если этому не препятствует образование пяти- и шестичленных колец по конкурентной конденсации Дикмана {схема (304), п = 4 или 5}. Добавление к реакционной смеси триметилсилилхлорида [265] приводит к образованию бис (триметилсилильного) производного ацилоина, которое далее гидролизуется кислотой {схема (305)}. Эта модификация метода препятствует циклизации по Дикману и позволяет успешно получать соединения с малыми циклами [266] {схемы (306), (307)}. Сообщалось о распространении этого метода [267] на превращение лиэфира циклической 1,2-дикарбоновой кислоты в циклический дион-1,2. Реакция протекает с расширением цикла. Ключевой стадией в этой реакционной последовательности является электро-циклическое раскрытие кольца промежуточного производного циклобутана {схема (308)}. Ацилоиновую реакцию можно использовать и применительно к эфирам монокарбоновых кислот для синтеза ациклических ацилоинов {схема (309)}.
9.8.3.5. Пиролиз и перегруппировки
Препаративным аспектам и изучению механизма реакций этого типа посвящен обзор [268]. Пиролиз сложных эфиров нашел ши' рокое применение для получения олефинов из спиртов; реакци включает цпс-элиминирование через симметричное шестицентр 360
о
II yxCOR (СН,)Л ^~COR
Na,ксилол MesSiCl
HCI
ТГФ
, * . I’U, RVHVIU
CH, „ I - -------
V_^CHCO2Me MesSiCl
OSiMe
OSiMe
Na
R’CO2R2 —> R’COCH(OH)R2
(308)
(309)
вое переходное состояние {схема (310)} и может проводиться в газовой или жидкой фазе. Так, пиролиз ацетатов обычно ведут в газовой фазе при 500—600°C; элиминирование протекает гладко
361
и обычно без перегруппировки двойных связей в образующемся олефине. Метод особенно удобен для синтеза [269] нссопряженных диенов {схема (311)} и метиленциклогексанов {схема (312)}.
R3
---* Б + нсо211 .
(310)
АсО(СНг)6ОАс 575 Ч\/Х (3|п
аСН2ОАс
—* Г J] (312)
Такие сложные эфиры, как бензоаты [270] и оксалаты [271], разлагаются при более низких температурах в жидкой фазе, однако эти эфиры не находят столь широкого применения, как ацетаты.
Аллиловые эфиры имеют дополнительную двойную связь, которая может принимать участие в реакции; аллилформиаты разлагаются по еновому механизму {схема (313)} с миграцией двойной связи. Образование аллилового спирта при пиролизе глицерина и муравьиной кислоты протекает через аналогичное расщепление образующегося моноформиата глицерина {схема (314)}.
(313)
СН2он .
снон + нсо2н
сн2он
-----> + СО2 + Н2О (314) носн2
362
имс-З'-Алкилаллиловые эфиры (107) также подвергаются элиминированию по шестицентровому механизму с образованием 1 3-диенов {схема (315)}, хотя эта реакция может далее осложняться в силу общей склонности аллиловых эфиров к перегруппировке в результате [3,3] -сигматропного процесса {схема (316)}.
Аллилацетоацетаты (Ю8) перегруппировываются по другому механизму, представленному на схеме (317); в качестве первоначального продукта образуется ацетоуксусная кислота, которая далее теряет двуокись углерода, превращаясь в конечный продукт реакции — аллилкетон. Перегруппировка хиральных аллиловых сложных эфиров приводит к оптически активным продуктам в со-
ответствии с синхронным механизмом перегруппировки; стереохимические превращения этого типа представлены на схеме (317).
O'Li OSiMe3 ОН
(319)
Аллиловые сложные эфиры легко перегруппировываются через енолят-анионы [272]; продуктом превращения служит аллилуксусная кислота {схема (318)}. В альтернативном процессе анион первоначально превращают в триметилсилилпроизводное, которое далее легко подвергается перегруппировке Кляйзена, давая аллилук-сусную кислоту {схема (319)}.
Перегруппировка ФРнса эфиров фенолов приводит к смеси о- и цилфенолов [273]; она катализируется кислотами Льюиса, ппитг^0 Хлорид°м алюминия- При высоких температурах пред-мпм\/ТеЛЬН° обРазУется орто-изомер; перегруппировка, по-види-CKoni’u/IP°TCKaCT чеРез промежуточный ацилпп-катпон, что не-
‘ ко напоминает ацилирование фенолов по Фриделю—Крафтсу.
363
Примеры перегруппировки Фриса представлены на схемах (320) и (321) [274].
Перегруппировка Фриса может также протекать как фотохимический процесс, но синтетическое использование этой реакции вряд ли можно сравнить с каталитическим процессом.
9.8.4. ОРТОЭФИРЫ
Ортоэфиры (109) [275, 276] представляют собой триалкил- или триарилпроизводные нестабильных ортокислот (ПО); ввиду их близкого химического родства со сложными эфирами карбоновых кислот эти соединения рассматриваются в данной главе.
OR2
R'-C-OR3
OR4
(109) R1 = H, Aik или Ar; R2, R2, R4 = Aik или Ar
(110) R2 = R3 = R4 = H
9.8.4.1. Методы получения ортоэфиров
Имеется ограниченное число общих методов получения ортоэфиров, и они будут рассмотрены в настоящем разделе. Информация о других, более специальных методах получения читатель может найти в монографии [275].
Первым и наиболее важным методом синтеза ортоэфиров служит алкоголиз гидрохлоридов имидатов, чаще всего действием метилового или этилового спиртов (синтез Пиннера) [277]. Реакция, представленная на схеме (322), включает первоначальное превращение нитрила в гидрохлоридимидат, который подвергается алкоголизу и дает ортоэфир (111), содержащий три одинаковые алкоксигруппы. Ортомуравьиные эфиры (111) (R = H) можно получать из цианида водорода [278], но синтез из хлороформа, по-видимому, в данном случае удобнее.
R’CN + R2OH R^C=NH2Cr R °Н RiC(OR2)3 (322)
R2(X
(III)
R' » Н, Aik или Ar; R2 = Aik или Аг
864
+ OR3
R1R2C=C(OR3)2 R'R2CH— R.R2CH-C-OR< (323)
°R3 OR3
PhO- трет-ВиОК
CH2^CHOPh —CH2BrCHBrOPh ---------> CH,BrCH(OPh)2 >
/OPh PhOH
___> CH,=C/-----------CH3C(OPh)3 (324) ^OPh
Присоединение спиртов к другим соединениям, содержащим кратные или формально кратные связи, также может быть полезен Так, ортоэфиры образуются при катализируемом кислотами присоединении спиртов к ацеталям кетена {схема (323)}. Примеры таких синтезов представлены на схемах (324) [279] и (325) [280]. Карбонпевые соли, полученные из циклических ацеталей или лактонов, также реагируют со спиртами с получением ортоэфиров; число реакций этого типа, используемых в препаративных целях невелико; два примера для иллюстрации даны на схемах (326) [281] и (327) [282].
сн2он
I
СНОН + С1СН2СНО—>
сн2он
СН2ОН
СН2С1
Me
-.nOCOPh
-."OCOPh
Me
Ra6 BF4 ---------—>
R=Me или Et
bf4~
RONa -----►
bf3 ------►
CHCla
Реакция получения простых эфиров по Вильямсону может быть использована для синтеза ортоэфиров; этот метод носит общий характер, однако наиболее важен он для получения ортомуравь-иных эфиров [283] {схема (328)}, и в особенно сти метиловых и этиловых эфиров. Простые а-галогеналкиловые эфиры также могут быть использованы в синтезах этого типа, например ортому-равьиные эфиры с умеренными выходами можно получать при реакции 1,1-дихлорметилового эфира с алкоксидами или феноксидами натрия {схема (329)} [284]. Арил- и алкилдифенилортофор-eul?' С ХОР°ШИМИ выходами получаются [285] из хлордифеноксимстана по аналогичной реакции {схема (330)}.
365
ROH
CIICI3 + NaOR ----> CH(OR)3
MeOCHCL- + NaOR —-> MeOCH(OR)2 R = Aik или Ar пиридин ^9)
ClCH(OPh)2 + ROH ——--------> ROCH(OPh)2 R = Aik или Ar
I'J’J’J)
Циклические ортоэфиры, содержащие 1,3-диоксолановое коль цо, иногда можно получить по реакции сложных эфиров с эпокси дамп {схема (331)} [286]. Этот простой и привлекательный метод не получил, однако, широкого распространения, хотя синтез спироортоэфиров из эпоксидов и лактонов [287] {схема (332)} служит неоднократно использовавшимся примером этой реакции.
Легкость получения многих простых ортоэфиров из нитрилов (см. выше) делает их удобными реагентами в синтезе других ортоэфиров с помощью переэтерификации {схема (333). Это достигается реакцией со спиртом или фенолом в присутствии подходящего катализатора (обычно кислоты); реакцию ведут до полного замещения исходных алкоксигрупп [275, 288]. Переэтерификация {схема (333)} равновесный процесс; и для его использования в препаративных целях необходимо так подобрать исходный ортоэфир, чтобы образующийся из него спирт R2OH мог бы легко отгоняться из реакционной смеси (обычно R2 = Ме или Et).
Реакция переэтерификации {схема (333)} особенно удобна, и в качестве продуктов образуются пяти- и шестпчленные ди-клическпе соединения {например, по схеме (334)} [289]; в подоб-тпД С:ЧуЧаЯХ Нет необхадпмости вести реакцию до замещения всех
„.г лкокснгРУпп исходного ортоэфира. Реакцию можно псполь-ч(ЬиппОИ ЛЛЯ СИНТ.еза бициклических или полициклических орто-ппрппят’пппгСХеМе (335) [290] представлен типичный пример такого шийЛ Я’ ПРИ этом’ естественно, необходимо, чтобы образую-тсл[-1н\пл '.Г' пРедставлял собой термодинамически предпочти-такж/ пня о циклическую систему. Этот тип реакции применял1 жет civwbtu В1НТЫ подходящих диолов; типичным примером мо-(112) [291] {схсмГрзз'б)1}1 °ИН0В0Й Функции производных стероид 366 ’'
о
Известно несколько случаев прямого получения ортоэфиров по реакции карбоновых кислот, таких как трифторуксусная кислота, с триолами. Такие ортоэфиры представляют собой стабильные полициклические соединения [например, синтез по схеме (337)], и этот тип реакций применим лишь к синтезу циклических систем. Ортомуравьиные эфиры удобно также получать [292] реакцией формамида со спиртом и подходящим хлорангидридом кислоты, например бензоилхлоридом или этилхлорформиатом {схема (338)}.
CF3CO2H + МеС(СИ2ОИ)з —>
(337)
HCONH2 + 3R>OH + R2COCl —> CH(OR>)3 + R2CO2H +NH4C1 (338) R1 = Me или Et; R2 = Ph или OEt
9.8.4.2. Реакции ортоэфиров
Ортоэфиры высокореакционноспособиые соединения, широко используемые в синтетической практике, включая образование гетероциклических продуктов (см. тома 8, 9 русского перевода).
(1) Гидролиз
Большинство ортоэфиров устойчиво в воде при щелочных значениях pH, но быстро гидролизуются в кислых условиях; fix чувствительность к кислотам выше, чем почти у всех других классов соединений [275]. Вследствие этого ортоэфиры необходимо получать в щелочных или безводных условиях; такие же условия тре-уются и при их хранении. Гидролиз, ведущий к сложному эфиру
367
карбоновой кислоты, протекает через образование стабилизированного кислородом карбений-иона, как показано на схеме (339) Обычно все ортоэфиры гидролизуются при действии разбавленных водных минеральных кислот, однако некоторые из них более стабильны, например трифенилортоформиат, который примерно в 106 раз устойчивее к гидролизу, чем триэтилортоформиат.
НА --
R'C(OR2)3 -------
медленно
r1C(°R2)2-^7>R'CO2R + R2°H + H++ (339) + R2OH + А"
которой можно регенерировать исходные
В силу легкости, с i соединения, ортоэфиры предложены [275] в качестве удобных за" щитных групп для карбоновых кислот {схема (340)} и диольных группировок {схема (341)}, например в стероидах и 2',3'-дигидро-
Л“°Н н+
RCO2H + Me С. ' » McC. „CR
V-он н+,н2о О/
х—ОН х—о
/ОН + ^-О Ri
(СН2)Л + r’C(OR2)3-^-> (сн2)л он °R2
л = 2 или 3
(34 О)
Н+ П2О, мягкие условия --------->-
(341)
рн Н+,Н2О / ------> (СН2)Л + R СО2Н
ОН + r2oh /ОН (СН2) OCOR1
ксинуклеозидах. Последнюю реакцию можно также использовать для генерации моноацилированных диолов, если проводить гидролиз в мягких условиях {например, защита 1,3-диола на схеме (336)}.
(2) Образование простых эфиров енолов
В некоторых случаях простые алкиловые или ариловые эфиры можно получать при использовании ортоэфиров, например по схеме (342) [293], однако реакции этого типа обычно не используют в синтезе. Более важным процессом служит получение из ортоэфиров простых эфиров енолов {схема (343)}.
ArOH + CH(OEt)3 —> ArOEt + EtOH + HCO2Et (342)
н+ R4°\ К!СОСНР2Р3 + CH(OR4)3—-* )
--------’--------л R'/
низкие OR4
темп. |
------> R1—C-CHR2R3
C=CR2R3 + R4OH
4 + HCO2R4
H + , A
(343)
I OR4
368
кипя обычно катализируется кислотами, исходит из альдегида кетона и протекает при высоких температурах. При более низ-11Л. температурах из кетонов могут образовываться ацетали, кото-h,Ip могут служить интермедиатами в ходе образования простых Раппов енола [275, 294]. Для кетонов, содержащих два а-водород-атома, отмечена тенденция к низкой региоселективности и Ъгоеоселективности (где это возможно). Однако примеры, предъявленные на схемах (344) и (345) [295], идут селективно. Реак-с я п0 схеме (345) служит ценным методом защиты стероидных енонов п широко используется [275].
о О
(34S)
стероидные ен-4-оны-З
(3) Образование ацеталей
Альдегиды и кетоны в присутствии кислот или кислот Льюиса как катализаторов взаимодействуют с ортоэфирами, образуя равновесную систему, как показано на схеме (346). Эта реакция служит удобным методом получения ацеталей [296]; положение равновесия, которое можно сдвинуть вправо добавлением спирта R3OH, в данном случае гораздо более способствует выделению ацеталя, чем при катализируемой кислотами реакции между кетоном R!R2CO и спиртом R3OH. Реакцию используют также как общий метод получения диоксаланов {схема (347)}, а также для синтеза ацеталей из сс,В-ненасыщенных альдегидов {схема (348)} [297].
RR н+ /°к3
ХС=О + CH(OR3)3 ----* + HCO2R3 (346)
R2/ R2/ \0R3
R'\
;c=o + ho(ch2)2oh + CH(OEt)3 R2/
(347)
NH4NO3
CH2=CHCHO + CH(OEt)3---------> CH2=CHCH(OEt)2 (348)
(4) Этерификация
Карбоновые кислоты при нагревании с этилортоформиатом с хорошим выходом дают сложные этиловые эфиры {схема (349)} [298]. Реакция не требует участия катализатора и поэтому
369
потенциально может рассматриваться как удобный метод noavu ния сложных эфиров из кпслотолабнльных карбоновых кислот " учетом ограничений, связанных с высокой общей реакционной сп ° собностью этилортоформиата. Реакции с ангидридами карбоновь?' кислот или с гидроксикислотами могут приводить к продуктам / структурой, аналогичной ортоэфирам, как показано на схеме (350? [299], что использовано в синтезе р-лактоиов путем полной гидратации р-гидроксикислот.
де-
RCO2H + CH(OEt)3 —* RCO2Et + HCO2Et + EtOH
0
О
/СО2Н Д
Me2C' + CH(OMe)3 —
(349)
+НСО2Ме
;о
(350)
Д
—> Ме2С
О ХН
(5) Реакции с серусодержащими соединениями
Взаимодействие ортоэфиров с тиолами ведет к тиортоэфирам {схема (351)} по реакции, аналогичной переэтерификации спиртами. Реакция с сульфидом водорода, катализируемая хлоридом цинка {схема (352)}, формально аналогична гидролизу {см. схему (339)}; в данном случае продуктом служит и тиоэфир [300].
R3SH R‘C(OR2)3 —R'C(SR3)3
(351)
H2S x?S
PhC(OEt)3 - r| Phcf
ZnG12
(352)
(6) Реакция с аминами
Взаимодействие ортоэфиров с аминами протекает сложно; простые реакции замещения, наблюдаемые для спиртов и тиолов, встречаются редко. Так, ортоэфиры в тщательно контролируемых условиях реагируют с ароматическими аминами [301], давая ими-доэфиры (113) {схема (353)}. Реакция ограничена ароматическими аминами, в то время как циклические ортоэфиры могут реа‘ тировать как с алифатическими, так и с ароматическими ам^р]И с образованием циклических имидоэфиров {схема (354)} [ЗСМР Некатализируемая или катализируемая кислотой реакция орт формиатов с избытком ароматического амина идет дальше, пр водя к образованию формамидинов (114) [303]; эта и ДРУГ реакции имидоэфиров [304] суммарно представлены на схе (355).
37 Q
ArNH2 + R'C(OR2)3
/R1
—> A,-N=C/ XOR2
(353)
(U3)
R1 = H, Me, Et и т. д.
R = Aik или Ar
CF3CO2H EtOH
ArNH2 + CH(OEt)3 H2S^ [ArN=CHOEt] ArNHMe (355)
|ArNH2
ArN=CHNHAr (И4)
|h2so4
ArNCHO I
Et
Ортоэфиры, содержащие СН2-группу в a-положении по отношению к ортоэфпрной функции, реагируют со вторичными циклическими аминами, давая аминали кетена (115) [305] {схема (356)}; простые первичные алифатические амины в присутствии трифторида бора в качестве катализатора реагируют с этилортоформиатом с образованием диалкилформамидинов (114) (Aik вместо Аг) [306].
RCH2C(OEt)3 + HN^ 4)Х —> RCH=cf—(356) \___________/ \ \__/ /2
(Н5)
X = СН2 или О
RNH2 + CH(OEt)3 + CH
zX RNHCH=(/
(357)
(116)
yivicM получают (обычно) амином
Р(обычно) — Ar; X и Y = CN, CO2R и т. д.
Полезные полупродукты (116) для синтеза гетероциклических реакцией этилортоформиата с ароматическим и каким-либо соединением, содержащим активную метиленовую группу. Эта реакция {схема (357)} формально напоминает реакцию Манниха [307], а также близка многим реак-
циям этилформиата, используемым при прямом синтезе гетероциклических соединений (см. тома 8, 9 перевода).
371
(7) Реакции с соединениями, содержащими нуклеофильный углерод
Ортоформиаты реагируют с реактивами Гриньяра, Дава тали альдегидов, которые могут гидролизоваться до а’льдегилпаЦе' удобный синтетический метод, известный под названием пЛ, Бодру —Чичибабина {схема (358)} [308]. Сообщалось [3091 '!ИИ смешанный ортоэфир (117), полученный при реакции 1 экв фе’НпТ0 с этилортоформиатом, особенно гладко реагирует с реактиве3 Гриньяра, давая диэтилацетали альдегидов {схема (359)}. М
н+
CH(OR')3 + R2MgX —> R2CH(OR')2 -----> R2CHO (358)
R1 (обычно) = Me пли Et; R2 = Aik, Ar, винил, алкинпл
PhOH RMgX
CH(OEt)3 -------► PhOCH(OEt)2 --------> RCH(OEt)2 (359)
(117)
ZnX2
R‘C=CH+ R2C(OEt)3 -------> R>C=CCR2(OEt)2 (360)
ZnCI2 /R1
HCN 4- R’C(OR2)3 -----> (R2O)2C/ (361)
\CN
ацети-
и монозамещенными
Реакции ортоэфиров с ацетиленом ленами в присутствии солей цинка в качестве катализатора ведут к ацеталям ацетиленовых альдегидов {схема (360)} [310]; аналогичная реакция с цианидом водорода, также катализируемая солями цинка, дает ацетали сс-цианоальдегидов {схема (361)} [3111-
Реакции ортоэфиров с соединениями, содержащими активные метиленовые группы, обычно протекают по схеме (362). Обзор большого числа реакций этого типа дан в [275]; они особенно важны для производства цианиновых красителей. Менее общая реакция по схеме (363) наблюдалась относительно редко, можны и (365)
(364)
два других направления, представленные схемами [312].
Ас2О 2CH2XY+ RC(OEt)3 --->- XYC=CR—CHXY
X и Y=CN, COR, CO2R и t. д.; R = H, Me, Et
R'CHXY + R2C(OEt)3 —> XYCR1 —CR2(OEt)2
ZnCl2 „ ,
CH2(CO2Et)2 + CH(OEt)3 -> EtOCH=C(C02Et)2
AC2O
с О /CN
PhCOCH2CN + MeC(OMe)3 MeOCMe==C\copI1
372
(8) ДрУгие PeaKtdlU
Восстановление ортоэфпров действием контролированных ко-пичеств алюмогидрида лития при нагревании в эфире дает ацс-‘ альдегидов [313] {схема (366)}. Ацетали кетенов можно поручать из ортоэфиров двумя методами. Первый путь {схема (367)} сводится к катализируемому кислотами или основаниями этиминированию спирта; лучшими катализаторами служат соединения, обладающие как свойствами основания, так и свойствами кислоты Льюиса, например трвт-бутокепд алюминия [314]. Альтернативный метод основан на первоначальном бромировании группы СН, находящейся в a-положении по отношению к функциональной группе ортоэфира, с последующим элиминированием под действием натрия {схема (368)} [315].
0,25 моль IJAIH4
R’C(OR2)3 -----Д) Et2Q -> R‘CH(OR2)2 (366)
R‘\ А1(ОСМез)з R’\ /OR3
;CHC(OR3)3 ------------> zC=C\ <367)
R2Z л R2/ X)R3
Bra
R'CH2C(OR2)3 --------> R'CHBrC(OR2)3
' пиридин
Na
R!CH=C
(368)
9.8.4.3. Ацетали амидов и родственные соединения
Ацетали амидов (118), аминали сложных эфиров (119) и ортоамиды (120) формально образуются при последовательном замещении алкоксигрупп ортоэфира аминогруппами. Эти соединения будут кратко рассмотрены в настоящем разделе в силу их родства с ортоэфирами.
OR2 OR2 NR2R3 |
R1—С—OR3 R1—C—NR3R4 Ri-C—NR4R;
1 NR’R5 1 NR5Re NR eR7
(118) (П9) (120)
(1) Получение ацеталей амидов (1187
Наиболее общий метод синтеза этих соединений состоит в алкилировании амидов по кислороду, приводящему к иону (121); последний далее нейтрализуют действием алкоксида, получая желаемый ацеталь амида {схема (369)} [316, 317]. Модификация этого метода включает промежуточное получение иона (122) по Реакции амида с фосгеном с последующей нейтрализацией и замещением галогенида на алкоксидную группу {схема (370)} [319]. обменная реакция ацеталя амида, синтезированного одним из
373
nvTY выше методов, с относительно нелетучим спиртом ^жеТбыть использована 1275] для получения ацеталя аМида, М° LXro остаток более высокомолекулярного спирта, чем содержаше (371)) Этот путь часто используют в синтезе исходный tex v м' бициКлического амидоацеталя Ж^ полученного [320 обменной реакцией с участием алкокси-фп,кш". и диметпламиногрупны диметилацеталя диметилформ-амида {схема (372)}.
Et3o bf;
Me2NCOR ----------
bf;
Me2N=C
NaOEt
OEt
Me2N—C—R
OEt
(121)
COC12 + /R NaOR'
Me2NCOR -----> Me2N=Cz -------->
\ci
OR1
Me2N—C—R
OR1
(370)
(122)
О V OR3
I I
Me2N-C-R' 4- R3OH —> Me2N— C—R1 + R2OH (371)
OR2 OR3
R2OH должен быть более летучим, чем R3OH
Me2NCH(OMe)2 + HN(CH2CH2OH)2
(123)
(372)
(2) Получение аминалей сложных эфиров (119)
Эти соединения трудно получить в чистом виде [275], поскольку они диспропорционируют в соответствии со схемой (373). Тем не менее их удалось выделить [391] в достаточно чистом виде, хотя и с незначительным выходом, используя путь, представленный на схеме (374).
2(R>R2N)2CHOR3 —> R'R3NCH(OR3)2 + (R’R2N)3CH (373)
Me2SO4 + Me2NH + NaOR
HCONMe2 -------> Me2N=CHOMe --------► Me2N=CHNMe2
MeSO; MeSO;
OR
I Me2NCHNMe2
(374)
374
(3)
Получение ортоамидов (120)
Эти соединения также трудно получить в чистом виде, однако отоамиды удовлетворительной чистоты синтезированы с неплохим уходом по реакциям, представленным на схемах (375) >3991 м В(376) [323].
' HCONMe2 + Ti(NMe2)4 —> TiO2 4-CH(NMe2)3
[322] и
(375)
Me2N=CHNMe2 4" M+NMe2
X’
M — Li, Na, К
-20 °c
------> (Me2N)3CH (376)
^) Реакции (118) — (120)
Целесообразно рассмотреть реакции всех трех типов соединений (Н8) — (120) вместе; часто эти реакции весьма сходны с реакциями ортоэфиров. Свойства ацеталей амидов изучены подробнее, чем свойства аминалей сложных эфиров и ортоамидов.
(5) Гидролиз и алкоголиз
При действии разбавленной кислоты ацетали амидов быстро гидролизуются, давая сложные эфиры и аммонийную соль, в то время как в нейтральной или щелочной среде образуется смесь амида со спиртом [316]. В соответствии с механизмом, представленным на схеме (377), это объясняют разной способностью к отщеплению протонированной и непротонированной диалкиламиногрупп. Пример этих двух типов гидролиза для полициклической амидоацетальной системы [318] показан на схеме (378). В целом гидролиз в водном растворе идет быстро как в кислой, так и в нейтральной или основной среде, и лишь некоторые (хотя и не все) циклические системы [например, соединение (124)] более стабильны. Алкоголиз идет по совершенно иному пути и приводит к ортоэфирам {схема (379)}, если используется простой одноатомный спирт [324]. Однако при взаимодействии с диолами могут протекать обменные реакции, как, например, в случае метил-[3-/)-рибофуранозида {схема (380)}. Эта реакция была предложена в качестве метода защиты вицинальных гидроксильных групп [325]. (а) Катализ кислотой
, Н+ . , ч , ,+ z°R2 н2о
(R o)2cr2nr! ---► (R O)2CR NHR2 —> r о=с/ ------------>
XR’
+ NHR2
.0
—> R’OH 4- R2—4- NH2R1 (377a)
X)R'
375
б) Нейтральные или основные условия
.OR1
(r‘O)2CR-’NR2 —>
\RJ
И2О 3
> R2N-C 4- 2Rl0H (3776)
\R2 >
AcOH,II2O <--------
или
KOH.EtOH
3hHCI,25°C
Л,EtOH
(37g)
Ph
о nr2 (124)
HOCH2 o
COPh
R'C(OR3)2 I 2 nr2
R1OH ----» AcOH
Cl
.nNH2
OH OCOPh
R'C(OR3)2 + NHR^
(379)
OR4
HO OH
OMe
О
(6) Этерификация
Ацетали амидов реагируют с карбоновыми кислотами с образованием сложных эфиров; реакция служит удобным синтетическим методом, особенно удобным для получения в мягких условия, сложных эфиров из пространственно затрудненных карбоновь. кислот (см. разд. 9.8.1.1) {схемы (381), (382)} [326].
R2CO2H + Me2NCH(OR’)2 ----Me2N=CHOR'+ R2COJ
—r’oh
—> Р2СО,Р‘ + HCONMe2 R2CO2H + R'OH + Me2NCH(OCH2CMe3)2 —* —> R2CO2R' -p HCONMe, 4- Me3CCH2OH
375
(7) Образование енаминов и амидинов
Ацетали амидов дают енаминн пп.. „п.,
типа СН2ХУ, где 0Д11а нА „V С°еД"Не'
электронооттягивающими группами таким , Л Y ЯВЛ511°тся тропная, сложноэфириая или арильная r^nnT ГзТб реакции часто протекают спонтанно с выпопр„и “ 1 Ь’ 327Ь Эти СТЯНИ растворителя пли без него Общая схема ТеПЛа В ПРИСУТ’ ставлена на схеме (383), отдельные примеры~ПреВращснИя преД-(386) (соответственно поработай [328—ЗЗОП дСХСМах (384) — (схема (387)) [327] „ ортоампдь, фе„а Д ) ^™ал” *’Р»° ...^тлгчгшп И enennH'>nuor.VtlQ ю)п1 О 1^81 J реагируют
t на схеме
реаги-
аналогично, и специфические примеры [332] показаны’ не (389). Отметим, что аминогруппа фенилацетамида также рует сходным образом; реакция приводит к образованию производного ациламидина. Синтез амидинов из ацеталей амидов и первичных аминов является широко используемой реакцией ' И (390)} [275].
{схема
CH>XY -j- Me2NCR'(ORz)2 —*• XYC—CR'NMes (383)
R1 = H или Me: X и/или Y — COR, CN, CO2R, Ar и т. д.
NMe?
I ,chno2
(EtO)2CHC(OEt)2 + CH3NO2 —> (EtO)2CHC^ + 2ЕЮН (384) ^NMe2
+ (MeO)2CH I
Me2N
+ (MeO),CH ’I
Me2N
EtOCH(NMe2)2-j-CH2XY —> XYC=CHNMe2 + EtOH + NIIMe2 (387) HC(NMe2)3 + CH2XY —> XYC=CHNMe2 + HNMe2 (388)
Me
, I EtCSNMe2 P1iCH2CONH2
Me2NCH=CNMe2 <---------- Me3COCH(NMe2)2 -----------*
Ph
I
—> Me2NCH=CCON=CHNMc, (389)
Me2NCH(OR2)2 + R‘NH2 —> R'N=CHNMe2 + R2OH (390) R1 ~ Aik или Ar
377
Вторичные амины взаимодействуют с ацеталями амидов с обп зованием тетракис (диалкиламино) этиленов {схема (391)} [333^' Эти соединения раньше получали другими методами, одним из ко торых является пиролиз аминалей циклических эфиров [334] (СХр' ма (392)}, формально протекающий через димеризацию бис(дИал^ киламинокарбена).
Me2NCH(OMe)2 + R2NH —> (R2N)2CHOMe —> (R2N)2C=C(NR2)2 (391) + МеОН -|- МеОН
-CH2CHR'CHR' СН2-(СН2)5—,—(СН2)2О(СН2)2—:; ~(CH2)2NMe(CH2)2-
Na или Ca --------->.
Д
OEt
Me—С—NMe2
OEt
CH2=C
(393)
Аминали кетена получают из ацеталей амидов, содержащих а-СН-группу [316, 335] по схеме (393); этот путь аналогичен синтезу ацеталей кетена из ортоэфиров {см. схему (367)}.
ЛИТЕРАТУРА
«The Chemistry of Carboxylic Acids and Esters», ed. S. Patai, Wiley, London, 1969. .
T. P. Hilditch and P. N. Williams, «The Chemical Constitution of Natural Fats», Chapman and Hall, London, 1964; D. Chapman, «The Structure of Lipids», Methuen, London, 1965; W. W. Christie, «Lipid Analysis», Pergamon, Oxford, 1973.
E. Haslam, «Chemistry of Vegetable Tannins», Academic, London, 1966.
C. J. Brown, D. E. Clark, W. D. Ollis, and P. L. Veal, Proc. ChemJ>oc., 1960, 393; W. Karrer, «Konstitution und Vorkommen der Organischen Ptlan-
1.
2.
з.
4.
5.
6.
7.
8.
9.
10.
11.
12.
zenstoffe», Birkhauser Verlag, Basle, 1958. „„
W. Keller-Schierlein, Prog. Chem. Org. Natural Products, 970, 30, W. Keller-Schierlein and H. Gerlach, Prog. Chem. Org. Natura 1968, 26, 161. , t iq I20
L. Crombie and M. Elliot, Prog. Chem. Org. Natural Products, 19°*’ ’ rm.
/. Goodman and J. A. Rhys, «Polvesters, Volume 1, Saturated Polymers», fe, London, 1965. ' yoi 4
Org. Synth. Coll. Vol. 1, 2nd edn. (1941), Coll. Vol. 2 (1943), Co . (1963), and Vols. 40—56 (1960—1977).
E. K. Euranto, См. cc. 1, chapter 11. . . t v» 2nd edn.,
С. K. Ingold, «Structure and Mechanism in Organic Спе у
Bell, London, 1969, chapter 15. _ lQ74 дя 4196.
J. P. Hardy, S. L. Kerrin, and S. L. Manatt, J. Org. Chem.,
378
И F. Cortese and L. Bauman, J. Amer. Chem. Soc., 1935, 57, 1393.
R. L. Stern and E. N. Bolan, Chem. and Ind. (London), 1967, 825.
, г' j /, Marshall, К. C. Erickson, and T. K. Folsom, Tetrahedron Letters, 1970, 4011'- P. K. Kadaba, Synthesis, 1971, 316; 1972, 628; G. Hallas, J. Chem. Soc., 1965,’ 5770.
lfi tv. W7. Lowrance, Jr., Tetrahedron Letters, 1971, 3453.
7’ A. R- Bader and A. D. Kontowicz, J. Amer. Chem. Soc., 1953, 75, 5416.
18 E. C. Blossey, L. M. Turner, and D. C. Neckers, Tetrahedron Letters, 1973,
1823
JO IF. S. Johnson, V. J. Bauer, J. L. Margrave, M. A. Frisch, L. H. Dreger, and
Ц7. N. Hubbard, J. Amer. Chem. Soc., 1961, 83, 606.
20 M. S. Newman, J. Amer. Chem. Soc., 1941, 63, 2431.
21 G. A. Olah, S. J. Kuhn, W. S. Tolgyesi, and E. B. Baker, J. Amer. Chem. Soc.,
1962, 84, 2733.
99 Л L McCloskey, G. S. Fonken, R. W. Kluiber, and IF. $ Johnson, Org. Synth. Coll. Vol. 4, 1963, 263.
23 F A. Derevitskaya, E. M. Klimov, and N. K. Kochetkov, Tetrahedron Letters, 1970, 4269.
24. J. S. Showell, D. Swern, and W. R. Noble, J. Org. Chem., 1968, 33, 2697.
25. A. G. Davies, J. Kenyon, and L. W. F. Salame, J. Chem. Soc., 1957, 3148.
26. G. Hofle and W. Steglich, Synthesis, 1972, 619.
27. IF. Stevens and A. van Es, Rec. Trav. chim., 1964, 83, 1287, 1294.
28. E. J. Bourne, С. E. M. Tatlow, and J. C. Tatlow, J. Chem. Soc., 1950, 1367; A. Lardon and T. Reichstein, Helv. Chim. Acta, 1954, 37, 388, 443; J. M. Tedder, Chem. Rev., 1955, 55, 787.
29. R- C. Parish and L. M. Stock, J. Org. Chem., 1965, 30, 927.
30. R. D. Cowell and J. H. Jones, J Chem. Soc. (C), 1971, 1082.
31. I- G. Wright, C. W. Ashbrook, T. Goodson, G. V Kaiser, and E. M. van Hey-ningen, J. Medicin. Chem., 1971, 14, 420.
32. H. L. Wehrmeister and D. E. Robertson, J. Org. Chem., 1968, 33, 4173.
33. M. H. Karger and У. Mazur, J. Amer. Chem. Soc., 1968, 90, 3878.
34. F. G. Young, F. C. Frostick, Jr., J. J. Sanderson, and C. R. Hauser, J. Amer. Chem. Soc., 1950, 72, 3635.
35. T. Neilson and E. Werstiuk, Canad. J. Chem., 1971, 49, 493; J. H. Brewster and C. 1. Ciotti, J. Amer. Chem. Soc., 1955, 77, 6214.
36. IF. Adam, J. Baeza, and J.-C. Liu, J. Amer. Chem. Soc., 1972, 94, 2000.
37. F. Effenberger and G. Epple, Angew. Chem. Internat. Edn., 1972, 11, 299.
38. M. Brenner and W. Huber, Helv. Chim. Acta, 1953, 36, 1 109; F. C. Uhle and L. S. Harris, J. Amer. Chem. Soc., 1956, 78, 381; R. P. Patel and S. Price, J. Org. Chem., 1965, 30, 3575.
39. H. A. Staab and A. Mannschreck, Chem. Ber., 1962, 95, 1284; H. A. Staab and B. Polenski, Annalen, 1962, 655, 95; J. Vessman, A. M. Moss, M. G. Horning, and E. C. Horning, Analyt. Letters, 1969, 2, 81.
40. 0. 0. Orazl and R. A. Corral, J. Amer. Chem. Soc., 1969, 91, 2162.
41. A. Stempel and F. W. Landgraf, J. Org. Chem., 1962, 27, 4675.
42. S. Neelakantan, R. Padmasani, and T. R. Seshadri, Tetrahedron, 1965, 21, 3531.
43. Y. Ueno, T. Takaya, and E. Imoto, Bull. Chem. Soc. Japan, 1964, 37, 864.
44. E. J. Corey and К. C. Nicolaou, J. Amer. Chem. Soc., 1974, 96, 5614; E. J. Corey, К. C. Nicolaou, and L. S. Melvin, Jr., ibid., 1975,97, 653, 654; E. J. Corey, К. C. Nicolaou, and T. Toru, ibid., 1975, 97, 2287; E. J. Corey, P. Ulrich, and J. M. Fitzpatrick, ibid., 1976, 98, 222.
45. T. Mukaiyama, K. Goto, R. Matsueda, and M. Ueki, Tetrahedron Letters, 1970, 5293.
46. S. Masamune, H. Yamamoto, S. Kamata, and A. Fukuzawa, J. Amer. Chbm. Soc., 1975, 97, 3513; S. Masamune, S. Kamata, and W. Schilling, ibid., 1975, »7, 3515.
^°lv‘n, T. A. Purcell, ami R. A. Raphael, J. C. S. Chem. Comm., 1972, I vO 1 .
4S. 0, Mitsunobu and M, Yamada, Bull, Chem. Soc. Japan, 1967, 40, 2380.
379
49. T. Kurihara, Y. Nakajitna, and O. Mitsunobu, Tetrahedron Letters 1Q7R
50. D. Seebach, B. Seuring, H. O. Kalinowski, W. Lubosch and BRetinf •
gew. Chem. Internat. Edn., 1977, 16, 264, ' Kcn^Cr> An-
51. R. N. Lacey, Adv. Org. Chem., 1960, 2, 213; W. £. Hanford and J С ч Org. Reactions, 1946, 3, 108 [В. E. Хэнфорд и др Орг. реакции сб Пер. с англ./Под ред. К. А. Кочешкова. М., Издатинлит, 1951, с 1161- ~~
/. N. Gardner, Т. L. Popper, F. Е. Carlon, О. Gnoi, and'И. L ’ Herzoa I n Chem., 1968, 33, 3695. UrS-
52. G. Quinkert, Angew. Chem. Internat. Edn., 1965, 4, 21 R P Yates Pur,. a i Chem., 1968, 16, 93. ’ APPL
53. G. Quinkert, Angew. Chem. Internat. Edn., 1975, 14, 790.
53a. A. 1. Parker, Adv. Org. Chem., 1965, 5, 37; F. S. Alvarez and A N Waft i Org. Chem., 1968, 33, 2143. ’
536. A. Ruziska, O. Jeger, A. Grov, and H. Hosli, Helv Chim Acta 1943 26 2283. ’ ’
53b. G. J. Atwell and B. F. Cain, J. Medicin Chem., 1967, 10, 706.
53г. M. Tandar, Kem. Ind., 1970, 19, 578; G. Mehta, Synthesis, 1972 262"
J. E. Shaw, D. C. Kunerth, and J. J. Sherry, Tetrahedron, Letters, 1973, 689. 53д. W. W. Hartmann and E. J. Rahrs, Org. Synth. Coll. Vol. 3, 1955, 650.' 53e. K. D. Berlin, L. H. Gower, J. W. White, D. E. Gibbs, and G. p'. Sturm J
Org. Chem., 1962, 27, 3595.
53ж. G. C. Stelakatos, A. Paganou, and L. Zervas, J. Chem. Soc. (C), 1966, 1191, 53з. M. S. Newman and W. S. Fones, J. Amer. Chem. Soc., 1947, 69, 1046.
53и. О. Yonemitsu, T. Hamada, and Y. Kanaoka, Tetrahedron Letters, 1969, 1819. 53к. M. M. Harris and P. K. Patel, Chem. and Ind. (London), 1973, 1002. 53л. R. K. Olsen, J. Org. Chem., 1970, 35, 1912.
53м. R. B. Wagner, J. Amer. Chem. Soc., 1949, 71, 3214; J. D. Roberts and
H. E. Simmons, Jr., ibid., 1951, 73, 5487; T. Hasselsirom, С. E. Balmer, and
H. W. Coles, ibid., 1952, 74, 3944.
53н. P. E. Pfeffer, T. A. Foglia, P. A. Barr, I. Schmeltz, and L. S. Silbert, Tetra-
hedron Letters, 1972, 4063.
53o. G. Mehta, Synthesis, 1972, 262; J. Grundy, B. G. James, and G. Pattenden, Tetrahedron Letters, 1972, 757.
53п. T. Saegusa and I. Murase, Synth. Comm., 1972, 2, 1.
53p. A. H. Lewis and N. L. Goldberg, Tetrahedron Letters, 1972, 491.
53c. Al. Sheehan and D. J. Cram, J. Amer. Chem. Soc., 1969, 91, 3544;
J. Org. Chem., 1964, 29, 2490.
53т. W. T. Moreland, J. Org. Chem., 1956, 21.. 820.
53y. R. L. Merker and Al. J. Scott, J. Org. Chem., 1961, 26, 5180.
53ф. D. J. Raber and P. Gariano, Tetrahedron Letters, 1971, 4741.
53x. V. Prelog and M. Piantanida, Helv. Chim. Acta, 1936, 56, 244;
and K. G. Allen, Steroids, 1970, 16, 79.
53ц. /. Gan, J. Korth, and B. Halpern, Synthesis, 1973, 494.
53ч. W. Kantlehner and B. Funke, Chem. Ber, 1971, 104, 3711.
54.
F. Stodola,
J. MacGee
Soc., 1944,
Hauser, J. Amer. Chem.
K. Kochetkov, Tetrahedron Letters, M. Fieser, «Handbook of Organic and Al. C. Whiting, J. Chem. Soc. 1964, 86, 658.
Acta, 1948, 31,
55.
56.
57.
58.
59.
60.
61.
D. S. Breslow, E. Baumgarten, and C. R.
66, 1286.
V. A. Derevitskaya, E. Al. Klimov, and N.
1970, 4269.
См. работы в cc. 9, or L. F. Fieser and Reagents», Wiley, New York, 1900.
A. C. Day, P. Haymond, R. M. Southam, (C), 1966, 467.
C. G. Overberger and J.-P. Anselme, J. Amer. Chem. Soc.,
E. Hardegger, Z. El Heweihi, and F. G. Robinet, Helv. Chim.
439; J. B. Miller, J. Org. Chem., 1959, 24, 560. QUart. Rev„
J. Koskikallio, Ref. 1, chapter 3; A. G. Davies and J. К J >
1955, 9, 203; M. L. Bender, Chem. Rev., 1960, 60, 53. chem Soc. 1951,
A. R. Bader, L. O. Cummings, and H. A. Vogel, J. Amer. СПет. эо
73, 4195. a
H. C. Brown and К. A. Keblys, J. Org. Chem., 1906, 31,
62.
380
гз О. Р- Cocl R Е’ Seamans, Synthesis, 1973, 538.
D. P- Roelofsen, J. A. Hagendoorn, and II. van Bekkutn. Chem. and Ind. (London), 1966, 1622.
[). S. Wulfman, B. McGiboney, and B. W. Peace, Synthesis, 1972, 49; д J Birch, J. E. T. Corrie, P. L. Macdonald, and G. Snbba Rao, J. C. S. Perkin J, 1972, 1186.
«6 E. Taschner, C. Wastelewski, and J. F. Biernat, Annalen, 1961, 646, 119;
£. Taschner, A. Chitniak, B. Bator, and T. Sokolowska, ibid., 1961, 646, 134.
«7 A. N. Meldrum, J. Chem. Soc., 1908, 598; D. Davidson and S. A. Bernhard, J Amer Chem. Soc., 1948, 70, 3426.
68 E. J. Corey, J. Amer. Chem. Soc., 1952, 74, 5897.
69 E. S. Rothman, S. S. Hecht, P. E. Pfeffer, and L. S. Silbert, J. Org. Chem., 1972, 37, 3551.
70 A H. Beckett, R. G. Lingard, and A. E. E. Theobald, J. Medicin. Chem., 1969, 12, 563.
71 E S. Rothman, G. G. Moore, J. M. Chirinko, and S. Scrota, J. Amer. Oil Che-' mists Soc., 1972, 49, 376.
72 £. Vowinkel and C. Wolff, Chem. Ber., 1974, 107, 496.
73 N. B. Lorette and J. H. Brown, Jr., J. Org. Chem., 1959, 24, 261.
74 H. Cohen and J. D. Mier, Chem. and Ind. (London), 1965, 349.
75 . R- C. Blume, Tetrahedron Letters, 1969, 1047.
76 H. Vorbriiggen, Angew. Chem. Internat. Edn., 1963, 2, 211; H. Brechbiihler, H. Biichi, E. Hatz, J. Schreiber, and A. Eschenmoser, Helv. Chim. Acta, 1965,
48, 1746.
77. H. Bilchi, K. Steen, and A. Eschenmoser, Angew. Chem. Internat. Edn., 1964, 3, 62; J. A. Webber, E. M. van Heyningen, and R. T. Vasileff, J. Amer. Chem.
Soc., 1969, 91, 5674.
78. D. H. R. Barton, R. M. Evans, J. C. Hamlet, P. G. Jones, and T. Walker, J. Chem. Soc., 1954, 747; D. H. R. Barton, D. A. J. Ives, and B. R. Thomas,
ibid., 1954, 903.
79 . J. Champagne, H. Favre, D. Vocelle, and I. Zbikowski, Canad. J. Chem., 1964, 42, 212; A. J. Liston, J. Org. Chem., 1966, 31, 2105; P. Toft and A. J. Liston, Chem. Comm., 1970, 111.
80 . 0. Wichterle and M. Hudlicky, Coll. Czech. Chem. Comm., 1947, 12, 564.
81 . £. Taschner and C. Wasielewski, Annalen, 1961, 640, 136, 139.
82 . G. C. Joshi, W. D. Chambers, and E. W. Warnhoff, Tetrahedron Letters, 1967, 3613; G. L. Hodgson, D. F. MacSweeney, and T. Money, ibid., 1972, 3683, 83. D. J. Cooper and L. N. Owen, J. Chem. Soc. (C), 1966, 533.
84. C. L. Schilling, J. Org. Chem., 1974, 39, 1770.
85. D. J. Hamilton and Al. J. Price, Chem. Comm., 1969, 414.
86. T. K. DasGupta, D. Felix, U. M. Kempe, and A. Eschenmoser, Helv. Chim. Acta, 1972, 55, 2198.
87. A. I. Meyers and G. Knaus, J. Amer. Chem. Soc., 1974, 96, 6508; A. 1. Meyers and D. L. Temple, Jr., ibid.. 1970, 92, 6644, 6646.
88. Al. W. Rathke and D. F. Sullivan, Tetrahedron Letters, 1973, 1297.
89. K. Ogura and G. Tsuchihashi, Tetrahedron Letters, 1972, 1383.
90 K. Ogura, S. Furukawa, and G. Tsuchihashi, Chem. Letters, 1974, 659.
91. R. A. Ellison, W. D. Woessner, and С. C. Williams, J. Org. Chem., 1972, 37, 2757.
92.
93.
G. D. Buckley and W. J. Levy, J. Chem. Soc., 1951, 3016.
E. J. Moriconl, J. G. White, R. W. Franck, J. Lansing, J. F. Kelly, R. A. Sa-lomone, and У. Shimakawa, Tetrahedron Letters, 1970, 27.
w. Kirmse, A. Engelmann, and L. Heese, J. Amer. Chem. Soc., 1973, 95, 625.
Qt. £• E Rawlinson and G. Sosnovsky, Synthesis, 1973, 567.
, Ereibs, G. Lucius, H. Kogler, and II. Breslauer, Annalen, 1953, 581, 59. qr u®’ Eee and M. J. Price, Tetrahedron, 1964, 20, 1047.
ms. Л1. S. Kharasch, G. Sosnovsky, and N. C. Yang, J. Amer. Chem. Soc, 1959, 81, 5819; K. Pedersen, P. Jakobsen, and S.-O, Lawesson, Org. Synth., 1968, 48, 18,
94.
95.
88)
95r. J. K. Kochi, J. Amer. Chem. Soc., 1962, 84, 774; D. Z Denneii A л„„ ,l
and D. B. Denney, ibid., 1962, 84, 4969. ''PPelbaum,
95д. W. Kitching, Z. Rappoport, S. Winstein, and HZ. G. Young J Ampr ri
Soc., 1966, 88, 2054. ’ ler Cllcm-
95e. D. H. R. Barton, E. F. Lier, and J. F. McGhie, J. Chem. Soc (Cl 19fis mo, 96. E. C. Taylor, H. HZ. Altland, R. H. Danforth, G. McGillivray and А Л1 '
95e. D. Н. R. Barton, E. F. Lier, and J. F. McGhie, < - - - ........................ - - -
lop, J. Amer. Chem. Soc., 1970, 92, 3520.
96a. E. H. Larsen and S.-O. Lawesson, Org. Synth., 1965, 45, 37.
966. Cm. cc. 95.
96в. M. E. Kuehne and T. J. Giacobbe, J. Org. Chem., 1968, 33, 3359.
96r. IR. S. Trahanovsky and L. B. Young, J. Org. Chem.,’ 1966, 31, 2033
96д. T. Nishimura, Org. Synth. Coll. Vol. 4, 1963, 713.
96e. S. M. Tsang, E. H. Wood, and J. R. Johnson, Org. Synth. Coll. Vol. 3, 1955
641. ’
97. С. H. Hassall, Org. Reactions, 1957, 9, 73; J. B. Lee and В C Uff Quart Rev., 1967, 21, 429. W
97a. IR. D. Emmons and G. B. Lucas, J. Amer. Chem. Soc., 1955, 77, 2287.
976. J. D. McLure and P. H. Williams, J. Org. Chem., 1962, 27, 24.
97b. S. L. Friess, J. Amer. Chem. Soc., 1949, 71, 14, 2571.
97r. R. HZ. White and W. D. Emmons, Tetrahedron, 1962, 17, 31.
97д. R. D. Chambers and M. Clark, Tetrahedron Letters, 1970, 2741.
97e. D. G. Crosby, J. Org. Chem., 1961, 26, 1215.
91'a\.H. D. Dakin, Org. Synth. Coll. Vol. 1, 1941, 149.
973. HZ. Baker, J. F. W. McOmie, and T. L. V. Ulbricht, J. Chem. Soc., 1952, 1825; HZ. Baker, H. F. Bondy, J. Giunb, and D. Miles, ibid., 1953, 1615.
97n. J. Meinwaid, J. J. Tufariello, and J. J. Hurst, J. Org. Chem., 1964, 29, 2914.
97k. M. Bogdanowicz, T. Ambelang, and В. M. Trost, Tetrahedron Letters, 1973, 923.
97л. P. S. Starcher and B. Phillips, J. Amer. Chem. Soc., 1958, 80, 4079.
98. E. J. Corey, N. M. Weinshenker, T. K. Schaaf, and W. Huber, J. Amer. Chem. Soc., 1969, 91, 5675; E. J. Corey, T. K. Schaaf, W. Huber, U. Koelliker, and N. M. Weinshenker, ibid., 1970, 92, 397; E. J. Corey, Z. Arnold, and J. Hutton, Tetrahedron Letters, 1970, 307; E. J. Corey and R. Noyori, ibid., 1970, 311.
99. P. Deslongchamps and C. Moreau, Canad. J. Chem., 1971, 49, 2465.
100. R. T. Arnold, R. Buckles, and J. Stoltenberg, J. Amer. Chem. Soc., 1944, 66,
208.
101. A. McKillop, В. P. Swann, and E. C. Taylor, J. Amer. Chem. Soc., 1971, 93,
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
4919.
E. C. Juenge and D. A. Beal, Tetrahedron Letters, 1968, 5819.
L. M. Berkowitz and P. N. Rylander, J. Amer. Chem. Soc., 1958, 80, 6682.
S. J. Angyal and K. James, Carbohydrate Res., 1970, 12, 147.
M. Fetizov, M. Golfier, and J.-M. Louis, Chem. Comm., 1969, 1118.
G. R. Robertson, Org. Synth. Coll. Vol. 1, 1941, 138.
U. Schmidt and P. Grafen, Annalen, 1962, 656, 97.
C. D. Hurd and W. H. Saunders, Jr., J. Amer. Chem. Soc., 1952, 74, b324.
K. Griesbaum and K. Keul, Angew. Chem. Intermit. Edn., 1975, 14, / •
G. B. Bachman and J. HZ. Wittmann, J. Org. Chem., 1963, 28, bb
G. B. Bachman, G. F. Kite, S. Tuccarbasu, and G. M. Tallman, J. Org. Chem., 1970,35,3167. „ nr
D. AL Bailey and R. E. Johnson, J. Org. Chem., 1970, 35, 3o74 nnhpu A. McKillop, О. H. Oldenziel, В. P. Swann, E. C. Taylor, and R. L. Kooej, J. Amer. Chem. Soc., 1973, 95, 1296. Fdn
E. C. Taylor, R. L. Robey, and A. McKillop, Angew. Chem. Int.rnat. 1972, 11, 48. ,n7Q .,x 3fi53
S. Cacchl, I.. Caglioti, and P. Zappelli, J. Org. Chem., 19/ , >
В. C. L. Weedon, Adv. Org. Chem., 1960, 1, 1. , 0 реакции,
A. S. Kende, Org. Reactions, 1960, 11, 261 [AC. Кение up., p с. II. — Пер. с англ./Под ред И. Ф. Луценко. М, Издаз. • >
R. В. Loftfield, J. Amer. Chem. Soc., 1950, 72, Ь32; 1951, >
382
,n G Stork and I. J. Borowitz, J. Amer. Chem. Soc., 1960, 82, 4307; H. O. House
1 9’ and W. F. Gilmore, ibid., 1961, 83, 3972, 3980.
_ R В Wagner and J. A. Moore, J. Amer. Chem. Soc., 1950, 72, 974.
121 В- M. Trost, M. J. Bogdanowicz, and J. Kern, J. Amer. Chem. Soc., 1975, 97,
* 2218.
122 В M. Trost, M. Preckel, and L. M. Leichter, J. Amer. Chem. Soc., 1975, 97, 2224.
1W. E. Bachmann and W. S. Struve, Org. Reactions, 1942, 1, 38.
94 A. B. Wiberg and T. W. Hutton, J. Amer. Chem. Soc., 1956, 78, 1640.
. M S. Newman and P. F. Beal, J. Amer. Chem. Soc., 1950, 72, 5163.
ofi' A- В. Smith, J. C. S. Chem. Comm., 1974, 695.
127' E. E Corey and M. Chaykovsky. J. Amer. Chem. Soc., 1964, 86, 1639.
12я" R. B. Woodward and R. Hoffmann, «The Conservation of Orbital Symmetry», Academic, New York, 1970.
129 W. S. Johnson, L. Werthemann, W. R. Bartlett, T. J. Brocksom, T. Li, D. J- Faulkner, and M. R. Petersen, J. Amer. Chem. Soc., 1970, 92, 741; W. S. Johnson, M. B. Gravestock, and В. E. McCarry, ibid., 1973, 93, 4332.
130 R- Rondo, M. Matsumoto, and F. Mori, Angew. Chem. Internat. Edn., 1975, 14, 103.
131 . R- P- A- Sneeden, См. cc. 1, chapter 4.
13 2' Q. A. Olah and J. A. Olah, «Friedel Crafts and Related Reactions», Wiley, New York, 1964, vol. Ill, chapter 39; W. H. Coppock, J. Org. Chem., 1957, 22, 325.
133 V. H. Wallingford, A. P. Homeyer, and D. Al. Jones, J. Amer. Chem. Soc., 1941, 63, 2056; V. H. Wallingford, D. M. Jones, and A. P. Homeyer, ibid., 1942, 64, 576.
134. E. J. Corey, R. B. Mitra, and H. Uda, J. Amer. Chem. Soc., 1964, 86, 485.
135. E. J. Eisenbraun, P. G. Hanel, K. S. Schorno, Sr. St. F. Dilgen, and J. Osie-cki, J. Org. Chem., 1967, 32, 3010.
136. M. Stiles and H. L. Finkbeiner, J. Amer. Chem. Soc., 1959. 81, 505; M. Stiles, ibid., 1959, 81, 2598; H. L. Finkbeiner and M. Stiles, ibid., 1963, 85, 616.
137. /. Shahak. Tetrahedron Letters, 1966, 2201.
138. S. Selman and J. F. Eastham, J. Org. Chem., 1965, 30, 3804.
139. G. Stork, P. Rosen, N. Goldman, R. V. Coombs, and J. Tsuji, J. Amer. Chem. Soc., 1965, 87, 275.
140. G. Stork, A. Brizzolara, H. Landesman, J. Szmuszkovicz, and R. Terrell, J. Amer. Chem. Soc., 1963, 85, 207.
141. R. N. Warrener and E. N. Cain, Austral. J. Chem., 1971, 24, 785.
142. W. N. Smith and E. D. Kuehn. J. Org. Chem., 1973, 38, 3588; H. H. Schlabach and K. Repenning, Annalen, 1958, 614, 37.
143. R. F. Heck and D. S. Breslow, J. Amer. Chem. Soc., 1963, 85, 2779.
144. E. J. Corey and L. S. Hegedus, J. Amer. Chem. Soc., 1969, 91, 1233.
145. J. P. Collman, S. R. Winter, and R. G. Komoto, J. Amer. Chem. Soc., 1973, 95,
249.
146. A. Schoenberg, I. Bartoletti, and R. F. Heck, J. Org. Chem., 1974, 39, 3318.
147. AL Nojima, K. Tatsuini, and N. Tokura. Bull. Chem. Soc. Japan, 1971, 44,
2001.
148. G. Natta, P. Pino, and R. Ercoli, J. Amer. Chem. Soc., 1952, 74, 4496.
149. K. Bittier, N. von Kutepow, D. Neubauer, and H. Reis, Angew. Chem. Inter-nat. Edn., 1968, 7, 329.
m0. C. W. Bird, R. C. Cookson, J. Hudec, and R. O. Williams, J. Chem. Soc., 1963, 410.
!3’- r L Kehoe and R- A- Schell, J. Org. Chem., 1970, 35, 2846.
1' ‘ - Yukawa and S. Tsutsumi. J. Org. Chem., 1969, 34, 738.
ml jT RePFR Annalen, 1953, 582, 1.
ml , Bird' Chem- Rcv > 1962, 62> 283-
Tsufi. Accounts Chem. Res., 1969, 2, 144.
K7 n Carlson and A. R. Oyler, Tetrahedron Letters. 1975, 4099.
L Chapman. P. W. Wojtkowski, W Adam, O. Rodriquez, and R. Ruckli-schel, j. Amer. Chem. Soc., 1972, 94, 1365,
383
158 S. Masamune, Y. Hayase, W. K. Chan, and R. L. Sohczak, ,J лтог n, Soc, 1976, 98, 7874. ' ner- c-hem.
159 IP. Adam, J. Boeza, and J.-C. Liu, J. Лтег. Chem. Soc., 1972 44
160 . D. Borrmann and R. Wegler, Chem, Ber., 1969, 102, 64. ’ ’ 200°-
161'. P. A. Grieco, Synthesis, 1975, 67.
162. R. A. Ruden and R. Bonjouklian, J. Amer. Chem. Soc., 1975, 97 6oq9
163. В. M. Trost and С. H. Miller, J. Amer. Chem. Soc., 1975, 97, 7182
164 M. Simonetta and S. Carra, cm. cc. 1, chapter 1.
165 «Tables of Interatomic Distances and Configurations in Molecules and I Special Publication No. 11 (Supplement, Special Publication No 18) The A4 mical Society, London, 1958 (1965). ' ’’ ltleChe-
166. Z. Rappoport, «Handbook of Tables for Organic Compound Identifications 9 j edn., CPC Press, Cleveland, 1976. '• Г(1
167. A. L. McClellan, «Tables of Experimental Dipole Moments», Freeman 8 Francisco, 1963. ’ an
168. L. J. Bellamy, «The Infrared Spectra of Complex Molecules». 2nd edn. Methuen, London, 1958 [Л. И. Беллами. ПК-спектры сложных молекул—Пев с англ./Под ред. Пентина Ю. А. М„ Мир, 1971]; A. D. Cross, «Introduction to Practical Infrared Spectroscopy», Butterworths, London, I960 [А. Д. Rpocc Введение в практическую инфракрасную спектроскопию. — Пер. с англ М Издатинлит, 1961]; L. J. Bellamy, «Advances in Infrared Group Fraquen-cies», Methuen, London, 1968.
169. L. M. Jackman and S. Sternhell, «Application of Nuclear Magnetic Resonance Spectroscopy in Organic Chemistry», 2nd edn., Pergamon, Oxford, 1969.
170a. J. B. Stothers, «Carbon-13 NMR Spectroscopy», Academic, New York. 1972, 1706. G. C. Levy and G. L. Nelson, «Carbon-13 Nuclear Magnetic Resonance for Organic Chemists», Wiley, New York, 1972 [Г. Леви, Г. Нельсон. Руководство по ядерному магнитному резонансу углерода-13 для химиков-органиков.— Пер. с англ. М„ Мир, 1975].
171. Е. S. Stern and С. J. Timmons, «Gillam and Stern’s Introduction to Electro-
nic Absorption Spectroscopy in Organic Chemistry», 3rd edn., Arnold, London, 1970 [3. Штерн, К. Тиммонс. Электронная абсорбционная спектроскопия в огранической химии. — Пер. с англ. М., Мир, 1974].
172. Р. Crabbe, in «Determination of Organic Structures bv Physical Methods», ed. F. C. Nachod and J. J. Zuckerman, Academic, New York, 1971, vol. 3, chap-
173.
174.
175.
176.
177.
178.
179.
180.
181.
182.
183.
184.
185.
ter 3.
H. Budzikiewicz, C. Djerassi, and D. H. Williams, «Mass Spectrometry of Organic Compounds», Holden-Day, San Francisco, 1967, chapter 4 [X. Будзике-вич, К. Джерасси, Д. X. Вильямс. Интерпретация масс-спектров органических соединений. — Пер. с англ./Под ред. Н. С. Вульфсона. М„ Мир, 1»о , гл. 4].
D. Р. N. Satchell and R. S. Satchell, Ref. 1, chapter 9.
M. J. Bender, J. Amer. Chem. Soc., 1951, 73, 1626.
C. J. Pedersen, J. Amer. Chem. Soc., 1967, 89, 7017. o h.rf,
F. C. Chang and N. F. Wood, Tetrahedron Letters, 1964, 2969; W. Ro and M. C. Whiting. J. Chem. Soc., 1965, 1290. _ r!-„aer
E. Taschner and B. Liberek, Chem. Abs., 1957. 51, 1039; F-EsinJ' J. Schreiber, and A. Eschenmoser, Helv. Chim. Acta, 1960, 43, 113; W. er and A. S. Levinson. J. Org Chem 1963, 28, 2184; W. Herz and H. J- w borg, ibid.. 1965, 30, 1881.
P. D. G. Dean, J. Chem. Soc., 1965, 6655.
P. A. Bartlett and W. S. Johnson, Tetrahedron Letters, 1970, 4459.
P. Muller and B. Siegfried, Flelv. Chim. Acta, 1974, 57, 987. . pnrish
D. H. Miles and E. J. Parish, Tetrahedron Letters. 1972. 3987; E ,
and D. H. Miles. J. Ore. Chem., 1973, 38, 1223; E. J. Parish, N. r.
P A. Hedin, and D. H. Miles, ibid., 1974, 39, 1592. Bull.
P. S. Manchand. Chem. Comm., 1971 667; S. Kobayashi, Chem. Pnarrr. (.Japan). 1972, 20, 694.
B. Loev, Chem. and Ind. (London), 1964. 193.
B. Halpern and D. E. Nitecki, Tetrahedron Letters, 1967, 3031.
384
. p Krapcho, G. A. Glynn, and B. J. Grenon, Tetrahedron Letters, 1967, 215; / p Krapcho and В. P. Mundy, Tetrahedron, 1970, 26, 5437.
I Harley-Mason and A.-U. Rahman, Chem. and Ind. (London), 1968, 1845. co л P Krapcho and A. J. Lovey, Tetrahedron Letters, 1973, 957; A. P. Krap-18S' riio E. G. E. Jahngen, Jr., A. J. Lovey, and F. W. Short, ibid., 1974, 1091;
C L. Liotta and F. L. Cook, ibid., 1974, 1095.
„ p Haslam, in «Protective Groups in Organic Chemistry», ed. J. F. W. McOmie, 8У' plenum, London, 1973, chapter 5.
mo С B. Reese, cm. cc. 189, chapter 3.
io; D H. R- Barton, I. H. Coates, and P. G. Sammes, J. C. S. Perkin I, 1973, 599.
,q9 p L Letsinger, M. H. Caruthers, P. S. Miller, and К. K. Ogilvie, J. Amer. Chem. Soc., 1967, 89, 7146.
193 IF. Jencks, Prog. Phys. Org. Chem., 1964, 2, 63; G. G. Smith and F. W. Kelly, ibid., 1971, 8, 75; T. C. Bruice and S. J. Benkovic, «Bio-organic Mechanisms», vol. 1, Benjamin, New York, 1966 [T. Брюс, С. Бенкович, Механизмы биоорганическп.х реакций. — Пер. с англ. М, Мир., 1970]; A. J. Kirby and д. R. Ferscht, Prog. Bio-org. Chem., 1971, 1, 1.
194 В. E. Hudson and C. R. Hauser, J. Amer. Chem. Soc., 1940, 62, 2457.
195 IF. G. Kenyon, E. M. Kaiser, and C. R. Hauser, J Org. Chem., 1965, 30, 2937. 19б' IF. IF. Leake and R. Levine, J. Amer. Chem. Soc., 1959, 81, 1627.
197 A. P. Krapcho and D. S. Kashdan, Tetrahedron Letters, 1975, 707.
198. M. IF. Rathke and J. Deitch, Tetrahedron Letters, 1971, 2953; M. IF. Rathke and A. Lindert, ibid., 1971, 3995; J. Amer. Chem. Soc., 1971, 93, 2318.
199. S. Reiffers, H. Wynberg, and J. Strating, Tetrahedron Letters, 1971, 3001.
200. R. J- Cregge, J. L. Herrmann, C. S. Lee, J. E. Richman, and R. H. Schlessin-ger, Tetrahedron Letters, 1973, 2425; J. L. Herrmann and R. H. Schlessinger, ibid, 1973, 2429; J. L. Herrmann, G. R. Kieczykowski, and R. H. Schlessinger, ibid., 1973, 2433.
201. R. A. Olof son and С. M. Dougherty, J. Amer. Chem. Soc., 1973, 95, 581.
202. В. AL Trost, K. Hiroi, and S. Kurozumi, J. Amer. Chem. Soc., 1975, 97,
438.
203. B. AL Trost and T. N. Salzmann, J. Org. Chem., 1975, 40, 148.
204. H. J. Reich, I. L. Reich, and J. M. Renga, J. Amer. Chem. Soc., 1973, 95, 5813.
205. I. Ruwajima and Y. Doi, Tetrahedron Letters, 1972, 1163.
206. J. L. Herrmann, G. R. Kieczykowski, and R. H. Schlessinger, Tetrahedron Letters, 1973, 2433.
207. G. Stork and A. G. Schultz, J. Amer. Chem. Soc., 1971, 93, 4074.
208. AL IF. Rathke, J. Amer. Chem. Soc., 1970, 92, 3222.
209. P. A. Grieco and K. Hiroi, J. C. S. Chem. Comm., 1972, 1317; P. A. Grieco and M. Miyashita, J. Org. Chem., 1974. 39, 120; P. A. Grieco and J. J. Reap, Tetrahedron Letters, 1974, 1097.
210. M. IF. Rathke, Org. Reactions, 1975, 22, 423.
211. M. S. Newman and В. J. Magerlein, Org. Reactions, 1949, 5, 413 [T. С. Ньюмен, В. Дж. Магерлайн. Орг. реакции, сб. 5.— Пер. с англ. М., Издатинлит,
212. IF. S. Johnson, J. S. Belew, L. J. Chinn, and R. H. Hunt, J. Amer. Chem. Soc., 1953, 75, 4995.
о 7 n F- Borch, Tetrahedron Letters, 1972, 3761.
9i? » Ca‘neBL В. Tangari, and A. U. Ronchi, Tetrahedron, 1972, 28, 3009.
N. J. Leonard and C. IF. Schimelpfenig, Jr., J. Org. Chem., 1958, 23, 1708. г более ранние работы N. J. Leonard e. a.
/Г HauSer' F' S~wamec and J. T. Adams, Org. Reactions, 1954, 8, 84 К. P. Хаузер и др. Орг. реакции, сб. 8. — Пер. с англ./Под ред. Ю. А. Ар-217 Т°Ла- М’ Издатинлит, 1956, с. 90].
on.' Bach'Kann, IF. Cole, and A. L. Wilds, J. Amer. Chem. Soc., 1940, 62, o24.
219 ^,nson an(l Posvic, J. Amer. Chem. Soc., 1947, 69, 1361.
220 r Miles, T. M. Harris, and C. R. Hauser, J. Org. Chem., 1965, 30, 1007.
'luet, G. Emptoz, and A. Jubier, Tetrahedron, 1973, 29, 479.
13 Зак. 395
385
221 G. Stork, A. Meisels, and J. E. Davies, J. Amer. Chem. Soc., 1963 84
222' P J- Pe'atce, D. H. Richards, and N. F. Stilly, Chem. Connn. 197(7 П19-
223. R. D. Dillard and D. E. Pavey, J. Org. Chem,. 1971, 36, 749. ' ' ’ 1,fi0-
224' R. L. Sowerby and R. M. Coates, J. Amer. Chem. Soc., 1972, 94, 475g
225 E. J. Corey and M. Chaykovsky, ,1. Amer. Chem. Soc., 1965 87 Hie
P. G. Gassman and G. D. Richmond, J. Org. Chem., 1966, 31, 2355 ’ 'Я5; 226. E. J. Corey and T. Durst, J. Amer. Chem Soc., 1966, 88, 5656; 1968 90 554» 227. K. Ogura, S. Furukawa, and G. Tsuchihashi, Cliem. Letters, I974, 659 4°'
228. H. J. Bestmann and B. Arnason, Chem. Ber., 1962, 95, 1513.
229’ A. P. Uijttewaal, F. L. Jonkers, and A. van der Gen, Tetrahedron I effPrc
1975, 1439. ' S’
230. E. J. Corey and G. T. Kwiatkowski, J. Amer. Chem. Soc., 1966, 88 5653 231. J. C. Grivas and К. C. Navada, J. Org. Chem., 1971, 36, 1520. ’ .....
232. K. W. Yang, J. G. Cannon, and J. G. Rose, Tetrahedron Letters, 1970 1701 233. B. Singh, Tetrahedron Letters, 1971, 321.
234. P. A. S. Smith. Org. Reactions, 1946, 3, 337 [П. А. Смит. Органические реакции, сб. 3. — Пер. с англ./Под ред. К. А. Кочешкова. М., Издатинлит 1951 с. 322].
235. Е. W. Garbisch, Jr., and J. Wohllebe, J. Org. Chem., 1968, 33, 2157.
236. R. A. LeMahieu and R. W. Kierstead, Tetrahedron Letters, 1970, 5111.
2.3J . E. J. Corey and D. J. Beames, J. Amer. Chem. Soc., 1973, 95, 5829.
238. W. G. Brown, Org. Reactions, 1951, 6, 469 [В. Г. Браун. Орг. реакции, сб. 6. — Пер. с англ. М., Издатинлит, 1953, с. 409].
239. R. S. Davidson, W. FL Н. Gunther, S. М. Waddington-Feather, and В. Lyth-goe, J. Chem. Soc., 1964, 4907.
240. E. J. Corey, H. A. Kirst, and J. A. Katzenellenbogen, J. Amer. Chem. Soc., 1970, 92 6314.
241. S. B. Bowlus and J. A. Katzenellenbogen, Tetrahedron Letters, 1973, 1277. 242. S. Warwel, G. Schmitt, and F. Asinger, Synthesis, 1971, 309.
243. M. S. Brown and H. Rapoport, J. Org. Chem., 1963, 28, 3261.
244. P. L. Hall and R. B. Perfetti, J. Org. Cnem., 1974, 39, 111.
245. Y. Maki, K. Kikuchi, H. Sugiyama, and S. Seto, Tetrahedron Letters, 1975, 3295.
246.
247.
248.
249.
250.
251.
252.
253.
254.
255.
256.
257.
258.
259.
260.
261.
262.
263.
264.
265.
Y. Kikugawa, Chem. Letters, 1975, 1029.
H. C. Brown and В. C. Subba Rao, J. Amer. Chem. Soc.. 1956, 78, 2582.
R. A. Bell and M. B. Gravestock, Canad. J. Chem., 1969, 47, 2099
H. C. Brown, «Hydroboration», Benjamin, New York, 1962, p. 245. n
J. Kollonitsch, 0. Fuchs, and V. Gabor, Nature, 1955, 175, 346; L. ken, R. B. Smyth, and K. Mislow, J. Amer. Chem. Soc., 1958, 80, 48b.
H. C. Brown and W. Korytnyk, J. Amer. Chem. Soc., 1960, 82, 38o6.
M. J. Kornet, P. A. Thio, and S. I. Tan, J. Org. Chem., 1968 33 ЗМ/
G. R. Pettit and T. R. Kasturi, J. Org. Chem., 1960, 25, 875; G. R.
D. M. Piatak ibid., 1962, 27, 2127. n. . , , Пг£?
G. R. Pettit, U. R. Ghatak, B. Green, T. R. Kasturi, and D. M. PiataR, J. s-
Chem., 1961, 26, 1685. n Q9 1637;
H. C. Brown, H. Heim, and N. M. Yoon, J. Amer. Chem. Soc., 1970, >
G. R. Pettit and J. R. Dias, J. Org. Chem., 1971, 36, 3485. . ga.
L. I. Zakharkin and I. M. Khorlina, Tetrahedron Letters, 1962, 61У, ran, J. Org. Chem., 1965, 30, 3564. ....
C. Szantay, L. Toke, and P. Kolonits, J. Org. Chem., 1966, 31, 144/.
H. Minato and T. Nagasaki, J. Chem. Soc. (C), 1968, 621. Tr-frahed'
L. 1. Zakharkin, V. V. Gavrilenko, D. N. Maslin, and I. M. Khorlina, ron Letters, 1963, 2087.
P. M. Weissman and H. C. Brown, J. Org. Chem., 1966, 31, 283.
E. Mosettlg, Org. Reactions, 1954, 8, 218.
F. J. McQuillin and R. B. Yeats, J. Chem. Soc., 1965, 4273. rh §oc.« H. C. Brown, D. B. Bigley, S. К Arora, and N. M. Yoon, J. Amer.
1970, 92, 7161.
К. T. Finley, Chem. Ber., 1964, 64, 573.
U. Schrapler and K. Ruhlmann, Chem. Ber., 1964, 97, 1383,
386
266.
267.
268.
269.
270.
271-
272.
273.
274.
275.
276.
277
278.
279.
280.
281.
and A. N. Bolstad, J. Amer. Chem. Soc., 1951, 73, 1988.
С. А. Саркисяну, E. Г. Иванюк, ЖОХ, 1964, 34, 1940.
Borner, О. Fuchs, H. J. Sasse, H. Schrodt, and J. Spille, 89, 2060.
D. L. Nealy, J. Org. Chem., 1963, 28, 3446.
and E. E. Dreger, Org. Synth. Coll. Vol. 1, 1944, 258.
282.
283.
284.
285.
286.
287.
288.
289.
290.
291.
292.
293.
294.
295.
296.
297.
298.
299.
300.
301.
302.
303.
304.
305.
306
307
308
, i Rinnndield Tetrahedron Letters, 1968, 587, 591. ___
' т Morl T Nakahara, and H. Nozaki, Canad .1. Chem., 1969, 47, 3266.
Г И DePuy and R. W. King, Chem. Rev., 1960, 60, 431; G. G Smith and г W Kelln Prog. Phys. Org. Chem., 1971, 8, 75.
IV J Bailey and W. F. Hale, J. Amer. Chem. Soc., 1959, 81, 647, 651.
E L McGinnis, G. D. Meakins, J. E. Price, and M. C. Styles, J. Chem. Soc., 1965, ^a^abafsos and e. L. Krumel, J. Amer. Chem. Soc., 1969, 91, 3324.
p Е Ireland and R. H. Mueller, J. Amer. Chem. Soc., 1972, 94, 5897; p' T Arnold and C. Hoffman, Synth. Comm., 1972, 2, 27.
/ H Blatt, Org. Reactions, 1942, 1, 342.
N M. Cullinane and B. F. R. Edwards, J. Chem. Soc., 1957, 3016; N. M. Cullinane, R. A. Woolhouse, and B. F. R. Edwards, ibid., 1961, 3842.
g H. DeWolfe, «Carboxylic Ortho Acid Derivatives», Academic, New York,
Межевицкий, E. P. Олехнович, Г. H. Дорофеенко, Усп. хим., 1973, 42, 392.
S М. McElvain and С. L. Stevens, J. Amer. Chem. Soc., 1946. 68, 1917;
S M. McElvain and J. P. Schroeder, ibid., 1949, 71, 40; S. M. McElvain and C L. Aldridge, ibid., 1953, 75, 3987; для многих других примеров см. также сс. 275.
J. G. Erickson, J. Org. Chem., 1955, 20, 1573.
S. M. McElvain Б. Г. Ясницкий, Н. Meerwein, Р. Chem. Вег., 1956, С. F. Wilcox and W. Е. Kaufmann „ . . ,
Н Bohme and A. Dbrries, Chem. Ber., 1956, 89, 723; H. Gross and A. Rieche, ibid., 1961, 94, 538.
H. Schliebler and M. Depner, J. prakt. Chem., 1959, 7, 60.
H. Meerwein, Angew. Chem., 1955, 67, 374.
X. Bodenbenner, Annalen, 1959, 623, 183.
R. M. Roberts, T. D. Higgins, and P. R. Noyes, J. Amer. Chem. Soc., 1955, 77, 3801.
M. S. Newman and С. H. Chen, J. Amer. Chem. Soc., 1973, 95, 278.
H. Stetter and К. H. Steinacker, Chem. Ber., 1953, 86, 790.
R. Gardi, R. Vitali, and A. Ercoli, Tetrahedron Letters, 1961, 448.
R. Ohtne and E. Schmitz, Annalen, 1968, 716, 207.
B. Smith, Acta Chem. Scand., 1956, 10, 1006.
R. A. Wohl, Synthesis, 1974, 38.
E. E. Royals and К- C. Brannock, J. Amer. Chem. Soc., 1953, 75, 2050.
H. E. Carswell and H. Adkins, J. Amer Chem. Soc., 1928, 50, 235; G. J. Pfeiffer and H. Adkins, ibid., 1931, 53, 1043.
J. A. van Allan, Org. Synth. Coll. Vol. 4, 1963, 21.
H. Cohen and J. D. Mier, Chem. and Ind. (London), 1965, 349.
R. C. Blume, Tetrahedron Letters, 1969, 1047.
d K°izum{, and G- Tsuchihashi, Tetrahedron Letters, 1968, 2083.
л. M. Roberts, T. D. Higgins, and P. R. Noyes, J. Amer. Chem. Sbc, 1955, Z7’ 380I; R. M. Roberts and P. J. Vogt, Org. Synth. Coll. Vol. 4, 1963, 464.
1. Mukaiyama and K. Sato, Bulk Soc. Chem. Japan, 1963, 36, 99.
• Mizuno and M. Nishimura, J. Pharm. Soc. Japan, 1948, 68, 58.
• R- M Roberts and P. J. Vogt, J. Amer. Chem. Soc., 1956, 78, 4778; R. A. Cro-cnet.Jr. and C. Blanton, Jr., Synthesis, 1974, 55.
Baganz and L. Domaschke, Chem. Ber., 1962, 95, 2095.
Lehmann, H Scefluth, and G. Hilgetag, Chem. Ber., 1964, 97, 299.
i„'i R-.Sny.de'' and R. E. Jones, J. Amer. Chem. Soc, 1946, 68, 1253; С. Г. Whi-1 ehead, ibid., 1953, 75, 671.
G S'£a,chman’ Org. Synth. Coll. Vol. 2, 1943, 323; C. A. Dornfield and Coleman, Org. Synth. Coll. Vol. 3, 1955, 701.
13*
387
309. H. Stetter and E. Reske, Chem. Ber., 1970, 103, 643.
310 В IF. Howk and J. C. Sauer, J. Amer. Chem. Soc., 1958, 80, 4607- R г ,
’ чалин, H. Г. Иванова, ЖОХ, 1961, 31, 3896; R. Epsztein and !m„q-
Bull. Soc. chim. France, 1968, 313. rszak,
311. J. G. Erickson, J. Amer. Chem. Soc., 1951, 73, 1338.
312. J. Pascual and F. Serratosa, Chem. Ber., 1952, 85, 686.
313. C. J. Claus and J. L. Morgenthau, J. Amer. Chem. Soc., 1951, 73, 5005- a d
dig and E. Degener, Chem. Ber., 1953, 86, 1469. ' ' “oe-
314. S. M. McElvain and J. T. Venerable, J. Amer. Chem. Soc., 1950 72 icei.
S. M. McElvain and W. R. Davie, ibid., 1951, 73, 1400. ’ ’
315. 5. M. McElvain, H. 1. Anthes, and S. H. Shapiro, J. Amer. Chem. Soc 1949
64, 2525; S. M. McElvain, R. L. Clarke, and G. D. Jones, ibid., 1942, 64’ 196fi’ 316. H. Meerwein, W. Florian, N. Schon, and G. Stopp, Annalen, 1961, 641 1 317. H. Bredereck, F. Effenberger, and G. Simchen, Angew. Chem.,’ 1961/73 493 318. T. Taguchi and Y. Kawazoe, J. Org. Chem., 1961, 26, 2699.
319. H. Eilingsfeld, M. Seef elder, and H. Weidinger, Chem. Ber., 1963, 96, 2671-
H. H. Bosshard, E. Jenny, and H. Zollinger, Helv. Chim. Acta, 1961, 44, |20з' 320. Z. Arnold and M. Kornilov, Coll. Czech. Chem. Comm., 1964, 29, 645.’ 321. H. Bredereck, G. Simchen, S. Rebsdat, W. Kantlehner, P. Horn, R. Wahl
H. Hoffmann, and P. Grieshaber, Chem. Ber., 1968, 101, 41.
322. H. Weingarten and W. A. White, J. Org. Chem., 1966, 31, 2874.
323. H. Bredereck, F. Effenberger, T. Brendle, and H. Muffler, Chem. Ber 1968 101, 1885.
324. H. Eilingsfeld, M. Seefelder, and H. Weidinger, Chem. Ber., 1963, 96, 2671. 325. S. Hanessian and E. Moralioglu, Canad. J. Chem., 1972, 50, 233.
326. H. Brechbiihler, H. Biichi, E. Hatz, J. Schreiber, and A. Eschenmoser, Angew. Chem. Internat. Edn., 1963, 2, 212; H. Vorbruggen, ibid., 1963, 2, 211; R. Brechbiihler, H. Biichi, E. Hatz, J. Schreiber, and A. Eschenmoser, Helv. Chim. Acta, 1965, 48, 1746.
327. H. Bredereck, F. Effenberger, and H. Botsch, Chem. Ber., 1964, 97, 3397.
328. H. Bredereck, W. Kantlehner, and D. Schweizer, Chem. Ber., 1971, 104, 3475.
329. B. Stanovnik and M. Tisler, Synthesis, 1974, 120.
330. B. Fohlisch, Chem. Ber., 1971, 104, 348.
331. H. Bredereck, F. E. Effenberger, and T. Brendle, Angew. Chem. Internat. Edn., 1966, 5, 132; H. Weingarten and N. K. Edelmann, J. Org. Chem., 1967, 32, 3293
332. H. Bredereck, G. Simchen and B. Funke, Chem. Ber., 1971, 104, 2709.
333. H. E. Winberg, J. E. Carnahan, D. D. Coffman, and M. Brown, J. Amer. Chem. Soc., 1965, 87, 2055.
334. H.-W. Wanzlick, Angew. Chem. Internat. Edn., 1962, 1, 75.
335. H. Bredereck, F. Effenberger, and H. P. Beyerlin, Chem. Ber., 1964, 97, 3081.
9.9. АМИДЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ
Б. С. ЧАЛЛИС И ДЖ. А. ЧАЛЛИС (Imperial College of Science and Technology, University of London)
В этой главе рассматриваются соединения общей формулы (1)« включающие несколько хорошо известных классов органическ веществ, главными из которых являются амиды (1а). Родственнь амидам соединения, такие как имиды (16), гидроксамовые кисло (1в) и гидразиды (1г) заслуживают того, чтобы быть излож ными в отдельных разделах. Другие производные амидов, та как N-нитрозоамиды (1, х == NO) или М-галогенамиды (I> х
ззз
= На1), которые являются предметом меньшего числа исследова ний, включены в раздел 9.9.3 как часть химии амидов.
,0
R1 —С./ уХ
\R2
(la), Х = Н или R3; R1, R2 = Н, Aik, Аг (16), X = COR3; R1, R2 = II, Aik, Аг (1в), X = ОН; R1, R2 = Н, Aik, Аг (1г), X = NR®; R1, R2 = H, Aik, Аг .
Амидная группа является важной составной частью многих биологически активных соединений, и поэтому знание способов получения, свойств и реакций амидов представляется существенным для дальнейшего развития таких областей, как химия полипептидов и белков. Многие имиды, гидроксамовые кислоты и гидразиды проявляют фармакологическую активность, что стимулирует в последнее время интерес к химии этих соединений. Помимо биологических аспектов, амиды представляют интерес для фундаментальных химических исследований, так как сопряжение между неподеленной парой электронов атома азота и л-электронами карбонильной группы находит свое выражение в характерных физических и химических свойствах. Включение в молекулу амида третьего гетероатома в а- или |3-положение к азоту аминогруппы, как в случае гидроксамовых кислот, гидразидов и имидов, усложняет их химические свойства, и здесь для понимания требуется уже применение современных физических методов.
Соединения, рассматриваемые в данной главе, были предметом ряда ранее опубликованных обзоров. Двумя наиболее важными являются двухтомная монография Смита «Ациклические азотсодержащие соединения» [1], опубликованная в 1965—1966 гг., и книга «Химия амидов» под редакцией Забицкого [2], вышедшая в 1970 г. В последней работе не рассматривались гидроксамовые кислоты, однако обзор способов получения этих соединений был составлен Сэндлером и Каро [За], а их структура и реакции обсуждены в обзоре Бауэра и Экснера [36].
9.9.1. МЕТОДЫ ПОЛУЧЕНИЯ АМИДОВ
Группировка R—С( — О)—NR— может быть синтезирована несколькими путями, и это находит свое отражение в наборе методов, используемых для получения амидов (2), их циклических аналогов— лактамов (3) и родственных N-ациламидов, а также имидов (4) и (5).
Обычным способом получения самих амидов является образование связи между карбонильным углеродом группы и аминным азотом [(2) связь (а)]. Это обычно осуществляется путем обра
389
ботки аммиака, первичного или вторичного амина ацилируюши агентом с образованием первичного, вторичного или тоетииЛ М амида 1(2) при R*=R3=H; R2 = H, R’= Aik ала Ar R!° RJ= Aik ll/или Ar соответственно]. Другим общим направлении! синтеза амидов является введение карбонильной группы в йена сыщенное соединение за счет частичной гидратации нитрилов или имидоильных производных [(2), связь (б)]. В этом случае образуются соответственно первичные (2, R2 = R3 = Н) или вторичные амиды (2, R2 = Н, R3 = Aik, Аг). Имидоильные производные могут быть в принципе получены путем алкилирования нитрилов. Методы образования N-алкильной и N-арильной связи [(2), связь (в)] [иные, чем прямое замещение амидного Н] в принципе требуют проведения перегруппировок, таких как реакции Шмидта и Бекмана. Синтез амидов за счет образования связи между алкильной или арильной группой и атомом углерода карбонильной группы [(2), связь (г)] осуществить наиболее трудно, так как требуется применение менее распространенных реагентов, таких как кетены, изоцианаты и изоцианиды. Прямое окисление или восстановление соответствующих предшественников не нашли широкого применения в синтезе амидов.
RJ
И
CbcX * ||(б) о
(2)
о
R3
(3)*
(4)
(R1, R2, R3 = H, Aik, АГ: П=1, 2, 3 ит.д.)
Синтез лактамов (3) обычно проводят так же как и для ациклических амидов, за исключением случаев, когда особое внимание обращается на такие факторы, как размер цикла и легкость про текания реакции. Имиды, как ациклические (4), так и циклически (5), обычно получают путем образования связи (а) между кар о нильным атомом углерода и амидным атомом азота. В сл^чю ациклических амидов образование этой связи почти полность осуществляется при помощи ацилирования амидов, в то-время i циклические имиды могут быть получены за счет целого ря реакций внутримолекулярного замещения, а также с применен!
390
реакций циклоприсоединения. Для всех трех классов соединении: амидов, лактамов и имидов возможна модификация исходного соединения (R3 = Н) путем замещения у атома азота. Экспериментальные подробности и ряд дополнительных примеров вышеупомянутых реакций приводятся в обзорах: Беквита [4] по амидам и лактамам, Шихана и сотр. [5] и Изаака [6] по а- и р-лак-тамам, а также Уилера и Розадо [7], Харгрнвса и др. [8] и Сандлера и Каро [9] по имидам.
9.9.1.1. Ацилирование аминов
Одним из наиболее широко применяемых методов получения амидов является ацилирование аммиака,, первичного или вторичного амина, когда в результате образования связи между ацильной и аминогруппой {схема (1)} сначала возникает моноациламин (т. е. амид) (2). Избыток ацилирующего агента, RCOX, приводит к дальнейшему ацилированию первичных и вторичных амидов с образованием., средн других продуктов, диациламинов (т. е. имидов) (4). Обсуждение этих последовательных реакций будет проведено в разд. 9.9.1.8 и 9.9.3.5. Этот метод нашел широкое применение как из-за легкости протекания реакции, так и в связи с доступностью большого набора ацилирующих агентов и аминов.
R‘COX + R2R3NH —> R‘CONR2R3 + HX (1)
медленно + R2R3NH
R’COX -------> R'CO + X'--------> R1CONR2R3-RHX (2)
O'
(a) I (6)
R'COX + R2R3NH ---->- R1 —С—X ------R'CONR2R3 + HX (3)
HNR2R3_
(6)
Можно сформулировать два общих механизма этих реакций, которые зависят главным образом от природы ацилирующего агента R’COX, где Х-галоген, R’COO, R2O, R2R3N, 5J3, ОН или BF4. Первое направление [уравнение (2)] включает диссоциацию R*COX по механизму £д,1 с последующей быстрой атакой амина образующимся ацил-катионом. Этот путь {схема (2)} применим только к нескольким реагентам, в которых стабильность X- исключительно высока, как в случае ацилтетрафторбората [10]. Ацилирование аминов протекает главным образом по второму механизму {схема (3)}, который формально представляет собой Sv2-3aMcine-ние амина ацилирующим агентом. В простом случае Sn2 замещения структура (6) представляет собой промежуточную высокоэнергетическую структуру па координате реакции (т. е. переходное состояние). Однако во многих случаях (6) может быть значительно устойчивее и существовать как тетраэдрический интермедиат, для которого стадия (а) или (б) может быть определяющей скорость
391
реакции. Подробное рассмотрение различных вариантов механизма приведено в работах [4, 11]. Здесь следует лишь отметить, Что порядок реакционной способности ацилирующих агентов параллелен ряду кислотности соответствующих кислот НХ (т. е. стабильности Х~) н представляет собой последовательность: RCOHal > | RCO^O « RCO2R > RCONR2 > RCOR. Кроме того, скорость реакции увеличивается с ростом нуклеофильности амина (т. е приблизительно с увеличением силы основания), и может осуществляться кислотный или основной катализ, что часто используется для получения высоких выходов амида.
Галогенангидриды реагируют легко, в некоторых случаях даже бурно, с сильно нуклеофильными аминами и аммиаком, образуя амид и галогеноводород {схема (4)}. Обычно используют хорошо доступные хлор- или бромангидриды, хотя в определенных случаях более эффективны фторангидриды. Например, при получении формамидов формилфторид оказался наилучшим реагентом [12]. Для получения высоких выходов амида часто бывает необходимым удалить кислоту либо действием амина, либо добавлением третичного амина или неорганического основания (реакция Шотте-на — Баумана). Примеры использования различных систем растворителей приведены в работе Беквита [4]. Например, N-этил-амид 4-хлормасляной кислоты (4-хлор-М-этилбутирамид) легко получается при обработке при низкой температуре хлорангидрида соответствующей кислоты этиламином {схема (5)}. Амины с низкой нуклеофильностью требуют специальных условий реакции, таких, как добавление п-иридина или триметиламина в качестве катализатора, благодаря чему образуется ациламмониевая соль, R'CONrIX , которая служит высокоактивным ацилирующим агентом. Напротив, можно добавлять очень сильное основание (например, бутиллитий), для того, чтобы получить нуклеофильный анион R2R3N-, который затем легко реагирует с ацилирующим агентом [13]. Легкость взаимодействия аминов с галогенангидридами кислот можно продемонстрировать на примере синтеза амидов из веществ, содержащих другие реакционноспособные группы. При реакции галогенангидридов кислот, содержащих в качестве заместителей галоген [14] или алкоксикарбонильную группу Пф.»? аминами, содержащими гидроксильный или феноксильный [1Щ заместители, взаимодействие происходит главным образом по ацильной или аминогруппе соответственно.
R'COHal + R2R3NH —> R'CONR2R3 + HHal
C1(CH2)4COC1 +EtNH2 —> Cl(CH2)4CONHEt+ HC1 (R1CO)2O+ R2R3NH —> R>CONR2R3 + R‘CO2H
(4)
(5) (6)
Хотя ангидриды карбоновых кислот обычно менее реакционно
способны, чем соответствующие галогенангидриды, они нашли ПР менение при синтезе амидов в тех случаях, когда галогенангидР»^ реагируют слишком бурно. Суммарно такой путь получения мо ' 392
поедставпть схемой (6), где продуктами являются требуемый амид карбоновая кислота. Хотя обычно считают что эта реакция протекает через тетраэдрический интермедиат (7), в недавно опубликованной работе [17а] предполагается, что возможно синхронное замещение через переходное состояние (8). Реакция катализи-pv-ется кислотами или основаниями (обычно используют пиридин [18] или минеральные кислоты [176]), однако она может быть и автокаталитической, ибо в качестве продукта образуется кислота.
Большей реакционной способностью обладают ангидриды с электронооттягивающими заместителями, например трифторуксусный ангидрид, так как присутствие таких заместителей усиливает как активность карбонила, так и стабильность уходящей группы. Как правило, симметричным ангидридам, (RCO)2O, отдают предпочтение перед смешанными ангидридами RCOOCOR' во избежание затруднений, связанных с образованием смешанных продуктов.
О'
R1—С—OCOR1
HNR2R3
Ч)
о
II
Ri—С—OCOR1
HNR2R3
(8)
Однако определенные несимметричные ангидриды типа R'COOY [при Y == R2CO, R2NCO, R2SOs, (R2O)2RO] находят применение (особенно при получении пептидов [19,20]), так как высокая элек
тронная плотность Y направляет реакцию по карбонильной группе, удаленной от заместителя Y. В синтезах используют также сме* шанный ангидрид, получаемый из уксусного ангидрида и муравьиной кислоты; этот ангидрид избирательно взаимодействует с аминами по формильному углероду, с образованием формамидов [21]. Этот реагент используется также для формилирования ами-
ногруппы аминокислот перед пептидным синтезом [19, 22]. Циклические ангидриды, такие как фталевый или янтарный, также реагируют с аммиаком или аминами [4, 23, 24], давая после раскрытия кольца моноамид дикарбоновой кислоты (амидовую кислоту) {схема (7), стадия (а)}. Для выделения амидовой кислоты необходимо тщательно контролировать условия во избежание циклизации в имид {схема (7), стадия (б)}. И действительно, эта реакция является одним из главных методов синтеза имидов разд. 9.9.1.8).
(см.
R'COOR1 + R3R«NH —> R1CONR3R1 + R’ОН
(8)
393
Эфиры карбоновых кислот нашли меньшее применение дЛя теза амидов {схема (8)}, чем галогенангидриды или ангидпиТ' кислот, однако механизм таких реакции достаточно изучен [4 25]. Аммиак и другие нуклеофильные амины реагируют легко- ’ качестве катализатора можно использовать незначительные п & бавки метилата натрия. Более сложным случаем является катализ эквимольным количеством метилата натрия или другого сильного основания, например алюмогидрида лития [26] или натрия [27] когда в качестве интермедиата, по-видимому, образуется сильно нуклеофильный анион амина R3R4N~. Недавно в качестве эффективного катализатора был использован трибромид бора. Более активны некоторые замещенные эфиры типа RCOOX=Y, например виниловые эфиры (9) или аддукты кислого карбодиимида (10), так как отщепляющаяся группа _О—X=Y стабилизуется за счет сопряжения (в противоположность обычным алкоксильным ионам). Такие эфиры широко используются при получении амидов [4], а также находят применение.в пептидном синтезе [19, 20].
О Ме\
И ,С=СН2
RNH
О \ и ;c=nr PhCH2C—СУ
(9)
(Ю)
Аналогичные реакции аммиака или аминов с лактонами прямо приводят к некоторым дизамещенным соединениям за счет раскрытия цикла. Как и при аминолизе ациклических эфиров, большинство реакций с лактонами протекает путем расщепления связи ацильного углерода с кислородом с образованием гидроксиамида, как, например, при аминолизе у-фенилбутиролактона [29] {схема (9)}. Интересным исключением из этого правила является реакция лактона 3-гидрокснпропионовой кислоты (пропиолактона) с некоторыми аминами, например с анилином, когда происходит расщепление связи между алкильной группой и кислородом [30] с образованием аминокислоты {схема (10)}.
Рьсн-сн2
Ан,+
Г о
PhCH—СН2 1
К ।
°-5/СН2 + о HNRJR2
PhCHCH2CH2CONR1R2
ОН
Н2С—С=о
H2i-O + phNH2 —> PhNHCH2CH2CO2H
R‘CO2H + R2R3NH
O'
R1—C—OH
_ HNR2R3 +
R1CONR2R3 + H2°
(II)
394
Ацилирование аминов карбоновыми кислотами является по существу реакцией, обратной гидролизу амидов, и поэтому можно ожидать, что она будет проходить через тетраэдрический интермедиат, ’характерный для реакции гидролиза {схема (И)} (ср. разд. 9.9.3.1). Моравец и Отаки [31] предположили, что активными частицами являются амин и анион карбоновой кислоты, однако более вероятной представляется кинетически неразличимая атака аниона амина на свободную карбоновую кислоту. Применявшаяся ранее стандартная препаративная методика заключалась в нагревании эквимольной смеси кислоты и амина (т. с. алкил- или ариламмониевой соли кислоты) в присутствии или в отсутствие растворителя [32]. Однако использование таких катализаторов, как эфират трифторида бора [33], 2-галоген-1-метилпиридиний-иодид [34] и различные фосфорорганические соединения [35], позволило в последнее время получить хорошие выходы амидов в довольно мягких условиях, а в случае нереакционноспособных кислот устранило необходимость превращения этих кислот в галогенангидриды.
Хотя нуклеофильная атака аммиака или аминов на атом углерода карбонильной группы альдегидов или кетонов происходит достаточно легко, образование амидов протекает только при определенных условиях. Первоначально образуется обычный тетраэдрический продукт присоединения (11), который может распадаться по двум независимым направлениям — до аминоспирта (12) или до требуемого амида {схема (12), соответственно стадии (а) и (б)}. Преобладание одного из направлений зависит от относительной склонности к отщеплению (т. е. стабильности) остатков -R2 и “ОН. Следовательно, в отсутствие сильных электронооттягивающих заместителей у R2, будет предпочтительной миграция протона, приводящая к аминоспирту (12), который в свою очередь после элиминации воды образует основание Шиффа R’R2C = NR3 (при R4==H) {см. схему (12)}. В качестве электроноакцепторного заместителя для получения амидов наиболее широко используется тригалогенметильная группа (R2 = CC13). Например, из трихлорацетальдегида {схема (12), R1 = Н, R2 = СС13} легко образуются формамиды [36], в то время как гексахлорацетон превращается в трихлорацетамиды {схема (12), R1 = R2 = СС13) [37].
II I
R'CR2 —> R>-C-R2
+ I
R3R4NH - HNR3Ri_
(а) /ОН fp = H
---> R1R2C/ -------------->
\nr3r-‘
(12)
(б)
---> R*CONR3Ri + R2H
R1R2C=NR3 +
H2O
(12)
(Н)
Гап^п?Л0^еНа и«теРЛ£ная модификация этих реакций (реакция топай ры Бауэра) [38], приводящая к первичным амидам, в кольцо нуклеофильный амид натрия (вместо аммиака) взап-
395
модействует с неенолизующимися кетонами в апротонном раств0. рителе. Этот метод оказался полезным при получении амидов третичных карбоновых кислот, которые трудно получить другими способами. Типичный пример — синтез амида 2,2,4-триметилпентановой кислоты [39] {схема (13)}.
NaNH2
Me2CHCH2C(Me)2COPh --------> Me2CHCH2C(Me)2CONH2 + PhH (i3)
(NH4)2Sx
PhCO(CH2)2Me -------> Ph(CH2)3CONH2 (14)
Реакция Вильгеродта — чрезвычайно необычный аминолиз кетонов [40], протекающий при обработке диалкил- или алкиларил-кетонов полисульфидом аммония в запаянной ампуле. Несмотря на многочисленные исследования, механизм этой реакции остается неясным [41]. Можно, однако, полагать, что в данном случае происходит окислительно-восстановительное превращение, в котором кислород карбонильной группы мигрирует в конец алкильной цепи с последующим аминолизом до первичного амида {схема (14)}. Атом кислорода мигрирует в конец более короткой алкильной цепи, но не через четвертичный атом углерода, при этом углеродный скелет не изменяется. Несколько примеров такой миграции приведены Беквитом [4]. В условиях реакции Вильгеродта не только кетоны, но и альдегиды, спирты, олефины и а-галокислоты дают амиды [4].
Другим классом ацилирующих агентов, которые легко подвергаются нуклеофильной атаке аминами с образованием амидов, являются азиды кислот {схема (15), направление (в)}. Эти соединения примерно так же активны, как и ангидриды кислот; их реакционная способность обусловлена как отсутствием стерических препятствий, так и большей склонностью к отщеплению остатка Из- Как и предыдущие, эта реакция, вероятно, протекает через тетраэдрический интермедиат. Азиды кислот легко генерируются из галогенангидридов кислот плюс азид-ион. Если стереохимические факторы неважны, то преимущества использования этих соединений по сравнению с прямой реакцией галогенангидридов с аминами невелики. Этот метод особенно ценен при синтезе пептидов [19], так как ацилирование азидами протекает без рацемизации [42]. В ряде случаев полезен альтернативный метод получения азидов кислот, основанный на нитрозировании гидразидов [43] {схема (15), путь (tz)}, так как высокая нуклеофильность гидразина дает возможность получать гидразиды из сложных эфиров или амидов [44], которые в других случаях неактивны по отноше нию к аминам. Главным недостатком получения амидов через азиды является протекание побочной реакции азидов кислот пе регруппировки Курциуса {путь (б) на схеме (15), см. также разд. 6.5.4}. Эта конкурирующая реакция оказывается особенн предпочтительной, во-первых, когда реагирующий амин обладав слабой нуклеофильностью или стерически затруднен, во-вторы 396
когаа стереохимическая или электронная структура R снижает чеХофпльносгь карбонильного атома углерода, а также когда R особенно чувствителен к миграции (ср. с реакцией Бекмана, тчл 9 9 13) Другим недостатком является то обстоятельство, So некоторые гидразиды при обработке азотистой кислотой дают вместо азидов первичные амиды {путь (а) на схеме (15), дополнительные данные об этой реакции см. в разд. 9.9.5}.
R'COCl
r2r3nh
----> R'N=C=O------------> R'NHCONR2R3
(б* (В)
R2R3NH
O'
RICONHNH2
R'CON3
(r) HONO
HONO
Ri-C—N3 hAr2R3_
(15)
——> R’CONR2R3 —HN3
R'CONH2
Кетены — это необычные, ио очень активные ацилирующие агенты, которые легко реагируют с аминами с образованием амидов {схема (16)}. Формально это реакция присоединения, но имеются доказательства первоначальной нуклеофильной атаки амина на атом углерода карбонильной группы [1, 176].
Другим редко используемым ацилирующим агентом является оксид углерода, который способен присоединяться к первичным или вторичным аминам в присутствии метилата натрия или октакарбонила кобальта с образованием формамидов {схема (17)}. В случае третичных алкиламинов обычно происходит деалкилирование с последующим образованием М,Ы-диалкилформамидов. Диалкиланилины составляют исключение, и внедрение карбонильной группы сопровождается миграцией одного из алкильных заместителей [46] {схема (18)}. Недавно предложено проводить реакцию оксида углерода с первичными и вторичными аминами в присутствии арил- или винилгалогенидов и комплекса галогенид палладия— трифенилфосфин [47], что дает возможность получать соответственно вторичные и третичные амиды {схема (19)}.
R‘R2C=C==O + R3R'NH —> R>R2CHCONR3R4 (16)
R>R2NH+CO —> HCONR'R2 (17)
PhNEt2 + CO —> EtCONEtPh (18)
„ R3N, PdX2(PIi3P)2
R'X 4-R2NH2 + CO ------------> R'CONHR2 (19)
R'CONHR2 + R3R‘NH R*CONR3R« + R2NH2 (20)
Сами амиды по отношению к аминам могут действовать как ацилирующие агенты, что приводит к различным амидам [48] (схема (20)}. Если образующийся R2NH2 более летуч, чем исходный амин, то равновесие можно сдвинуть вправо и получить ко-чный продукт с высоким выходом. Как кислоты, так и основания, используются в качестве катализаторов реакции,
397
9.9.1.2. Амиды из нитрилов
Нитрилы являются другим хорошо доступным классом соеди-нений, из которых легко могут быть получены амиды. Известно два метода синтеза — прямая гидратация нитрилов, приводящая к первичным амидам {схема (21)}, и гидратация с алкилированием по реакции Риттера с образованием вторичных амидов {схема (22)} Гидратацию нитрилов, если требуется выделение амида, следует проводить с осторожностью, так как в условиях реакции первичный амид часто подвергается дальнейшему гидролизу до соответствующей карбоновой кислоты и аммиака. В отсутствие катализаторов гидратация нитрилов до амидов протекает медленно и для получения существенных выходов амида необходимо сильное нагревание в автоклаве. Однако как минеральные кислоты, так и щелочи сильно способствуют протеканию реакции. При кислотном катализе реакция протекает за счет атаки воды на протонированную форму нитрила, в то время как при щелочном катализе ион гидроксила взаимодействует со свободным нитрилом {схема (23)}. Фактически любая сильная кислота является активным катализатором в препаративных синтезах; концентрированная серная, соляная и полифосфорная кислоты [49], а также трифторпд бора в уксусной кислоте [49] нашли свое применение. Например, концентрированная соляная кислота использовалась при получении соответствующих амидов из большого набора арилацетонитрилов, таких как бензилцианид [50]. Концентрированная серная кислота нашла применение в особенности при получении стерически затрудненных амидов, например трибутилацетамида и диизопропил-ацетамида [4]. Эта особенность частично обусловлена устойчивостью образующегося амида к гидролизу, что отражает рост стерических ограничений при гидролизе амидов по сравнению с гидратацией нитрилов. Для неразветвленных соединений таких полезных ограничений не найдено, и поэтому для выделения промежуточного образующегося амида необходимо очень тщательно контролировать условия реакции. Другим преимуществом использования концентрированной серной кислоты в качестве катализатора является ее избирательность при гидролизе нитрила в присутствии потенциально активных групп, таких как лактонная, сложноэфир-ная или имидная [51]. Гидратация нитрилов в присутствии щелочных катализаторов используется в меньшей степени, так как обра зующийся амид легче гидролизуется в щелочной среде и, следовательно, его труднее выделить. Однако гидроксид калия был успеш но использован в ряде случаев [52].
RC=N + H2O —> rconh2 (21)
R2OH н+ /22)
R>C=N+ или --------> R'CONHR2 1
алкен
398
[RO=NH
RC=NH]
RC==N
RC=N I OH
H20
RC=NH1 I
OH J
H2o rRC=NH
---J
L oh2
----> RC\ \nh2
(23)
н
Для того чтобы уменьшить трудности, связанные с дальнейшим гидролизом образующегося амида, обращают внимание на методы, в которых гидратация нитрилов проходит легко. В этом отношении представляется эффективным использование в качестве катализатора щелочного пероксида водорода даже для простых алифатических нитрилов [53]. Механизм этой реакции изучен Вибергом [54], и вероятная последовательность реакций, основанная на этих исследованиях и включающая гидридный перенос от второй молекулы пероксида водорода к первоначальному аддукту (13), приведена на схеме (24). В присутствии олефина распад гидропероксиимина происходит по другому пути и приводит к эпоксиду и амиду {схема (25)}. В случае а,^-ненасыщенных нитрилов двойная связь превращается в эпоксидную группу одновременно с образованием амида [55]. Другие недавно разработанные методы гидратации нитрилов включают катализ металлами и их комплексами [56], диоксидом марганца [57] и даже ионообменными смолами [58]. Многочисленные примеры гидратации нитрилов приведены в обзоре Бэквита [4].
RC=N
"ООН
RC=N
ООН
> rconh2
4- (24)
Н2О + 0g
R
+ НОО—C=NH
(25)
(13)
hci R20v д
R’CN + R20H --> ^C=NHjCr ---------> R!CONH2 + R2C1 (26)
R1Z
(14)
Интересной модификацией реакции гидратации является замена воды на спирт или амин. В первом случае при использовании НС1 в качестве катализатора образуется гидрохлорид имидоэфира (14) (синтез Пиннера), который после нагревания превращается в амид
399
[59] {схема (26)}. Во втором случае промежуточно образующийся амидин (15) после мягкого гидролиза дает амид [601 [схема (27)}.
н2о
R’CN + R*R®NH —> R’Cf -------------> R‘CONR2R3
\NR2R3 W
(15)
H+ r2cn + H2O
R'OH или алкен Цщо" R2C=NR' ------>- R2CONHR‘ (28)
(16)
9.9.1
В недавно опубликованном обзоре [61] обсуждается применение реакции гидратации непосредственно для получения вторичных амидов, уже упоминавшаяся реакция Риттера. Эта реакция состоит в алкилировании азота нитрильной группы с последующим или конкурентным образованием карбонильной функции. Карбениевый ион, образующийся из олефина или спирта в присутствии концентрированной серной кислоты в качестве катализатора реагирует с нитрилом с образованием промежуточной нитрилиевой соли (16), которая обычно без выделения гидролизуется до вторичного амида {схема (28)}. Нитрилиевая соль может быть получена в чистом виде при обработке нитрила солями триалкилоксония или диазо-ния, или алкилгалогенидами в присутствии кислот Льюиса [62] и
далее подвергнута гидролизу.
Спирты и олефины, которые при обработке сильными кисло-
тами превращаются в третичные карбениевые ионы, или арилза-мещенные карбениевые ионы (например, бензиловый спирт), образуют амиды с высокими выходами [4]. Спирты и олефины, образующие при обработке сильными кислотами вторичные карбение
вые ионы, вступают в реакцию с большим трудом, чем соединения, дающие третичные ионы, в то время как применение реакции Риттера к первичным спиртам обычно является безуспешным [4]. Однако недавно появилось сообщение [64] об использовании реакции Риттера для первичных спиртов. В этом случае первичные спирты активировались путем обработки гексахлорантимонатом хлордифенилметилия в нитриле как растворителе. Образующийся
С3.1ЯЮТ ТОТО?! р ЭТИ ; замес •1 трет .к отрт
ыи им даек но на! !тезе я е кото ! ацил а ами, яа ре аанащ пособс j^ecca
Й1 ^еры
н*
ион нитрилия далее гидролизуют обычным способом.
Реакция Риттера применима к целому ряду бифункциональных реагентов, таких как ненасыщенные нитрилы и галогеналкены ]4J-Она также использовалась и при синтезе гетероциклических с0^* нений, что явилось темой недавно опубликованного обзора [64] • Кроме спиртов и олефинов, в реакции Риттера могут участвовать и другие соединения, способные давать карбениевые ионы, к ним относятся: разветвленные парафины в присутствии акцептора ГИД' ряда, третичные карбоновые кислоты, алкилгалогениды и арендиа зониевые соли [4]. В некоторых случаях, первоначально образуй• щийся карбениевый ион подвергается перегруппировке до реакц
400
с нитрилом, что может быть использовано при получении ряда амидов [65].
В присутствии галогенов протекает модификация реакции Нит-тера, так как промежуточно образующийся галогснониевый ион (17)’ возникающий при действии галогена на олефин, атакуется нитрилом, что приводит к N-2-галогеналкиламиду {схема (29)}.
[А Г R2CN;
R'CH— CH2J X' ----> R2CONCHR'CH2X (29)
(17)
9.9.1.3. Амиды путем перегруппировок
Амиды являются продуктами ряда хорошо известных перегруппировок, некоторые из них представляют синтетический интерес. Большинство этих реакций связаны с миграцией арильного или алкильного заместителя к азоту, что приводит к образованию вторичного или третичного амида. Мигрирующая группа может нести избыточный отрицательный заряд и, следовательно, смещаться к азоту, имеющему дефицит электронов, как в реакциях Бекмана и Шмидта, или иметь недостаток электронов, и тогда взаимодействовать с неподеленной парой азота, как в перегруппировке Чапмэна.
Вероятно наиболее изученной и нашедшей широкое применение в синтезе является перегруппировка Бекмана [67, 68, 71], в результате которой оксим после последовательной обработки кислотой или ацилирующим агентом, а затем водой превращается во вторичный амид {схема (30)}. Сейчас является общепринятым [71], что эта реакция начинается с протонирования, ацилирования или координации с кислотами Льюиса гидроксила оксииминогруппы, что способствует этой группировке. Подробнее о катализаторах этого процесса — см. в обзоре Беквита [4]; недавно появилось сообщение об использовании в качестве катализатора комплекса триоксид серы — основание Льюиса [69].
R'R2C=NOH
R'CONHR2 прп Х = С, SO, P(OR); Y = Hal (30)
Н+ „ли RX(O)Y; Н2О
R’CONHR2 R’C=NR2
(20)
(31)
401
Изучению механизма этой реакции уделялось много внимания [68, 70, 71], но только недавно были проведены детальные исследования (Шофильд и сотр. [72]). Исследования этих авторов по перегруппировке оксимов ацетофенона в серной кислоте говорят в пользу механизма, представленного на схеме (31), который ранее постулировался, но не был убедительно доказан [4, 68]. После протонирования или ацилирования гидроксильной группы оксима, образуется квази-трехчленное переходное состояние (19), в котором заместитель, находящийся в анти-положении к гидроксильной группе, мигрирует с одновременным удлинением связи N—-О. Затем образуется нитрилиевая соль (20), которая далее гидролизуется с образованием амида. Этот механизм согласуется с данными о влиянии заместителей на скорость перегруппировки ряда О-пикриловых эфиров оксима ацетофенона в 1,4-дихлорбутане [70] и, следовательно, является применимым к большому набору экспериментальных условий. Факт миграции заместителя из анти-, или f-положения [группа R2 в (18)] был установлен ранее по анализу побочных продуктов [71]. При этом наличие аномальных случаев, когда, по-видимому, мигрируют группы из обоих положений, как cuh-(Z) (группа R1), так и анти-(Е) (группа R2), почти наверняка обусловлено быстрым взаимным превращением двух изомеров перед перегруппировкой [4]. Концепция о трехчленном переходном состоянии была привлечена для того, чтобы объяснить наблюдаемое сохранение конфигурации мигрирующей группы R2 [73].
Синтетическое использование этой реакции сильно зависит от заместителей (R1 и R2) в оксиме (18). Как правило, наиболее удобными являются оксимы, в которых один изомер более устойчив, чем другой, и взаимопревращение син- и анти-изомеров затруднено. Этим требованиям отвечают оксимы алкиларилкетонов, в которых объемистая арильная группа занимает анти-положение и, следовательно, предпочтительнее к миграции, чем алкильная группа. Это обобщение имеет противоположный смысл, когда алкильная группа является более объемистой (например, разветвленной), чем арильная, и, следовательно, алкильная группа становится в анти-положение. Оксимы диалкилкетонов обычно дают смесь амидов, так как взаимные переходы син- и анти-изомеров происходят относительно легко. Если могут мигрировать обе алкильные группы, то в большем количестве образуется тот амид, который возникает в результате перемещения более объемистой группы. Оксимы циклических кетонов также подвергаются перегруппировке Бекмана, что является общепринятым методом синтеза макроциклических лактамов (см. разд. 9.9.1.7).
Трудности возникают в случае кетоксимов (18), имеющих труп-пу R1, способную образовывать относительно устойчивый карбение-вый ион, такой как Аг2СН+. Вместо перегруппировки Бекмана эти соединения претерпевают фрагментацию но Бекману [4, 68, 74] с образованием нитрила и карбениевого иона, вероятно, через ста-40?
пню образования обычного иона нитрилпя (20) {схема (-32)}. В отсутствие другого нуклеофила ион +R2 может рекомбинировать с нитрилом по реакции типа реакции Риттера, вновь образуя ион питрилия (20), который при обработке водой дает обычный амид R'CONHR2. Изомеризация иона +R2 в ион с более стабильной конфигурацией может произойти перед рекомбинацией с нитрилом, и тогда исходная конфигурация группы R2 не сохранится, в отличие от обычной перегруппировки Бекмана [65, 75]. Другие оксимы претерпевают разрыв связи N—О до миграции R1 {стадия (б) на схеме (32)}, что приводит к иону иминия (21) и таким образом дает возможность миграции R1 или R2 с образованием соответственно как R2CONHR1, так и его изомера R‘CONHR2 [76]. Оксимы альдегидов в присутствии, например, полифосфорной кислоты в качестве катализатора, обычно дают первичные амиды [77], вероятно по пути (в) на схеме (32). Использование ацетата никеля в качестве катализатора [79] приводит к возможности и других направлений реакции кроме образования нитрилов. Использование других катализаторов, таких как силикагель, также приводит к получению первичных амидов [78].
RJC^N + ‘R2 r’cn + R2OH + н+
л
R’C=NR2 -1,2°> R’CONHR2
I^CONHR1
. (32)
/
Н2о
^c=N+—^rV^nr1
(21)
r2cn —°> R2CONHl
Реакция Шмидта, аналогичная реакция Бекмана, также находит применение в синтезе амидов [71, 80]. В данном случае происходит катализируемое кислотой присоединение HN3 с образованием тетраэдрического интермедиата (22) {схема (33)}, который может распадаться с потерей молекулы азота, образуя вторичный амид [71]. Распад может происходить по двум направлениям, включающим (оба) миграцию алкильной или арильной группы к электронодефицитному атому азота. По первому направлению {путь (а) на схеме (33)} алкильная или арильная группа мигрирует синхронно с отщеплением газообразного азота, и поскольку соединение (22) не имеет преимущественной конфигурации, то
403
относительная миграционная способность R1 и R2 будет зависеть от электронных, а не стерических факторов. Напротив, второй вариант распада {путь (б) на схеме (33)} протекает через стадию дегидратации с образованием промежуточного имина (23). Структура этого имина сравнима с кетоксимом в реакции Бекмана {см схему (31)}, где предполагается предпочтительная конфигурация с более объемистой группой R2 в анти-положении по отношению к уходящей группе, что приводит к миграции только группы R2. Однако в тех случаях, когда наблюдается легкое взаимное превращение син- и онга-изомеров (как и в случае кетоксимов), будет происходить миграция группы (независимо от ее размера), более подходящей с точки зрения электронного строения (т. е. более электронодонорной группы).
Н+
R1—С—R2 Д
HN3
ОН
ОН _ I^CNHR2 —> R^ONHR2
^rU-R*^ +N*
nhnJ
(22>
(33)
(23)
R^ONHR2 R1C^NR2 + N2
Кетоны по своей реакционной способности в реакции Шмидта располагаются в следующем порядке: диалкилкетоны > алкил-арилкетоны > диарилкетоны, причем перегруппировка диалкил-кетонов происходит легче, когда катализатором является концентрированная соляная кислота, в то время как в случаеалкиларил-и диарилкетонов предпочтительным является применение в качестве катализатора серной или полифосфорной кислот [81а]. Перегруппировка диалкилкетонов обычно проходит по второму направлению, представленному на схеме (33), когда мигрирует более объемистая из двух алкильных групп, хотя этот путь редко оказывается полностью специфичным. Одним из преимуществ данной реакции является мягкие условия ее проведения, и поэтому часто удается превратить диалкилкетоны в амиды, не затрагивая другие заместители, имеющиеся в молекуле кетона. Например, р-, у- п б-кетоэфиры превращаются в амидоэфиры без изменения сложноэфирной группировки [816]. В условиях реакции Шмидта, как и при реакции Бекмана, оксимы циклических кетонов легко перегруппировываются в лактамы, что является удобным способом получения последних (см. разд. 9.9.1.7). В случае алкиларилкетонов строение продуктов сильно зависит от природы алкильного заместителя. Так, в присутствии метильной группы предпочтительна миграция арильной группы {путь (б) на схеме (33)}, но при нали-
404
чип алкильных групп с большим числом углеродных атомов доля ft-алкиламида среди продуктов реакции растет по мере увеличения степени разветвленности алкильного заместителя [71]. В соответствии с этим ацетофенон в присутствии полифосфорной кислоты превращается только в N-фенилацетамид {схема (34)}, а фе-нилизопропилкстон дает почти равные количества N-изопропилбен-замида и анилида изомасляной кислоты {изобутиранилида, схема (35)}. В случае диарилкетоиов размер арильной группы практически не влияет на способность к миграции и поэтому можно ожидать образования смеси амидов по путям (а) или (б) схемы (33). Электронные факторы также, по-видимому, неважны, так как наличие различных мета- и /гара-заместителей в бензофеноне не влияет на относительную долю каждого из двух образующихся амидов [81а].
NaNe
PhCOMe -----> MeCONHPh (34)
NaN3
PhCOPr -----> PhCONHPr-f- PrCONHPh (35)
Как и в реакции Бекмана, существует по крайней мере два различных пути распада промежуточного имина (23), и с помощью этих типов фрагментации {схема (36)} можно объяснить образование многих аномальных продуктов в реакции Шмидта. Известно несколько альдегидов, которые в условиях реакции Шмидта должны были бы Дать формамиды {схема (33) при R1 ~ Н}, но как и в реакции Бекмана для альдегидов, вместо формамидов в результате фрагментации по пути (а) {схема (36), R2 = Н} образуются нитрилы [71]. Образующийся в реакции Шмидта промежуточный имин (23) {см. схему (33)} может быть также получен при диазотировании гидразона или семикарбазона исходного ке-
(а)
I---* N2 + +R2+R>CN
R\
'г кт + Н2О
C=N — R!C=NR2 -----> R’CONHR8 (36)
R2/ 4N2 A
(23) |
(6) +
----> n2+ ;c=n+
R2/
+ + H2O
R2CN + R1 —> R2C=NR‘ --------> R’CONHR’
В соответствии с этим амиды с хорошим выходом удается получить при действии нитрита натрия в присутствии серной [83] или
405
иоллфосфорной кислоты [76] па гидразоны или семикарбазоны диалкил- или алкиларилкетонов, содержащих различные заместители [83]. Указанный метод синтеза является ценной альтернативой реакции Шмидта.
R1 ,NH2
ХС=1<
R’C^NR2 (37)
н2о
R^ONHR2
Другой потенциально полезной реакцией является перегруппировка алкил- и арилимидатов (24) в изомерные амиды {схема (38)}. Эта перегруппировка, известная как реакция Чапмена [59, 68, 84], является чисто термической и требует температур порядка 200 °C. В случае арилимидатов кинетические данные соответствуют внутримолекулярному механизму реакции. С другой стороны, алкилимидаты подвергаются перегруппировке за счет межмолекулярных процессов, так как из смеси имидатов образуются смешанные амиды. Хотя последняя реакция и не проходит гладко в отсутствии катализаторов, из формимидатов и первичных имидатов образуются формамидины или нитрилы. Однако недавно на нескольких примерах показано, что применение электрофильных катализаторов, таких как алкилгалогениды, иод и трифторид бора, заметно снижает температуру, необходимую для протекания реакции [85], и поэтому этот метод может оказаться перспективным для синтеза амидов.
Менее доступными исходными веществами для синтеза являются нитроны (25) и изомерные оксазиридины (26), которые могут образовываться при фотолизе нитронов {схема (39)}. Амиды получаются с хорошими выходами из обоих типов соединений: оксазиридины (26) подвергаются термической перегруппировке с образованием R’CONHR2 [86], в то время как альдо- [86, 87] или кетонитроны (25) [88] перегруппировываются в присутствии катализаторов, таких как ацилгалогенпды, окспгалогениды фосфора или n-толуолсульфохлорид {схема (40)}. Перегруппировка кетонитронов дает альтернативный реакции Бекмана путь синтеза лактамов.
/OR2
R'C^
Nqr3
(24)
X)
R’C;
XNR2R3
(38)
408
RICH=NR2 I O'
hv
R'CH—NR2
(39)
(25) (26)
Cl ( ОСОМе Cl RiCH=NR2 MeC°C1> KCjNR2 —> R1C=NR2 R^CONHR2 (40) O' H
9.9.1.4. Реакции изоцианатов, карбамоилгалогенидов и изоцианидов
Применение этих реагентов в синтезе амидов ограничивается их доступностью и доступностью соответствующих исходных веществ. Реакции изоцианатов и карбамоилгалогенидов в принципе похожи и протекают по двум возможным направлениям, в зависимости от структуры субстрата. В случае карбоновых кислот ковалентная связь между карбонильным углеродом и азотом [(2), связь (а)] возникает в результате атаки на ацильный углеродный атом реагента с последующей перегруппировкой, сопровождающейся потерей диоксида углерода. В случае аренов (в условиях реакции Фриделя — Крафтса), олефинов и карбанионов интермедиат, который образуется при нуклеофильной атаке на ацильный атом углерода реагента, не перегруппировывается. Это приводит к возникновению углерод-углеродной связи [(2), связь (г)], образование которой, как и при взаимодействии изоцианидов с карбонильными соединениями, служит лишь примером амидного синтеза с образованием определенных связей.
R1N=C=O
(27)
^СО2Н
R3CO2H
R1R2NCOCI
(28)
—> R3CONR*R2 + С02
(29)
(41)
(₽2=Н для изоцианатов)
Как уже упоминалось выше, при реакции карбоновых кислот с изоцианатами (27) [89] и с М,М-диалкплкарбамоилхлоридами (28) [90] образуется интермедиат, который представляет собой смешанный ангидрид карбоновой и карбаминовой кислот (29). Это соединение самопроизвольно разлагается с потерей диоксида углерода до амида {схема (41)}, в котором присутствует ацильная I Руппа из изоцианата. Распад интермедиата может быть внутримолекулярным, как показано на схеме, или, что более вероятно,
407
может протекать по межмолекулярному цепному механизму. Когда остаток карбоновой кислоты и изоцианатная группа, обычно возникающая in situ, подходящим образом расположены в одной молекуле, легко образуются циклические ангидриды карбаминовой и карбоновой кислот, такие как (30) и (31) [4, 91]. Эти соединения известные как ангидриды Лейкса, достаточно устойчивы, но при нагревании теряют диоксид углерода и дают пептиды ctdvktvdw (—CHRCONH—)л J
(30) (31)
Более реакционноспособные хлорсульфонилизоцианаты легко реагируют с карбоновыми кислотами, давая смешанные ангидриды, которые также элиминируют диоксид углерода. Образующийся амид содержит N-хлорсульфонильную группу, которая легко удаляется мягким гидролизом; при этом возникает первичный амид {схема (42)}.
rco2h
C1SO2NCO -------> [RCO2CONHSO2C1]---->
—СО2
Н2О
—> RCONHSO2C1 -------> RCONH2 (42)
А1Г1 r- 54 S-
RN=C=O A 3> [RN=C—O—AICI3]
ArH
ArCONHR
5+ O — АЫТ^'^
RNHCOC1 —C-^> [RNHC—O—AICI3]
Cl
AICI3 H2O
ArH + CISO.NCO ----> ArCONHSO.Cl -----> ArCONH,
(43)
(44)
Изоцианаты [92] и карбамоилхлорнды (а также их N-алкил и N-арилпроизводные) [93] служат удобными реагентами для синтеза аренкарбоксампдов по Фриделю — Крафтсу {схема (43)}. Хлорсульфонилизоцианаты взаимодействуют также в мягких условиях с образованием N-хлорсульфонпл)производных карбоксамидов аренов. Соответствующий амид можно получить после мягкого гидролиза {схема (44)}.
Среди других нуклеофилов, используемых при получении амидов из изоцианатов, но не из карбамоилхлоридов, следует отметит карбанионы, полученные из реактивов Гриньяра [94], нитроа нов, малонатов [95] и литийалкилов [96] {схема (45)}. Активир
408
ванные олефины, такие как енолы и енамины, реагируют с изоцианатами аналогичным образом {например, по схеме (46)}; эта реакция широко применяется при синтезе гетероциклических соединений. Неактпвированные олефины реагируют только с более реакционноспособным N-хлорсульфонилпзоцианатом; этот путь используется для синтеза 0-лактамов [6, 15] (см. разд. 9.9.1.7). Исключением из этого правила служит, например, реакция внутримолекулярного циклоприсоединения {схема (47)}, где изоцианатная группа и неактивпрованная двойная связь присутствуют в одной молекуле; реакция приводит к производному азепинона [97].
При получении амидов изоцианаты часто генерируют in situ непосредственно перед добавлением соответствующего субстрата Например, в известном варианте перегруппировки Курциуса для ацилазидов [98] добавление карбоновой кислоты к реакционной смеси приводит к образованию амида за счет перехвата промежуточно образующегося изоцианата {схема (48)}. '
RiN=C=O+ R2X+ —> R2CONR‘X+
(45)
н2о
---> R2CONHR1
(46)
Альтернативным вариантом аналогичного подхода служит обработка первичного амида тетраацетатом свинца в присутствии уксусной или пропионовой кислоты [99], что является удобным способом получения замещенных ацетамидов или пропионамидов соответственно {схема (49)}. Механизм образования изоцианатов по этой реакции обсуждается в разд. 9.9.3.8.
д rzco2h
R'CONs R'NCO --------> R’NHCOR2 (48)
РЬ(ОАс)4 r2co2h
R’CONH2 -------> R'NCO ---------> R2CONHR* (49)
(R2 = Me, Et)
OCOMe OH
о MeCO2H | H2O I
KNC + R2CO ---------R'NHCOCRj ---------->- R'NHCOCR2 (50)
(32)
409
R2
+ I
R'N^C—С—-О"
R3 (33)
(34)
NHCOMe MeCOoH I
R’NC + R22CO + NH3 -----> R'NHCOCR2 (51)
В силу повышенной нуклеофильности изоцианидов пути их использования отличны от таковых для изоцианатов и карбамоилгалогенидов. Основной реакцией изоцианидов является взаимодействие с альдегидами или кетонами в присутствии карбоновой кислоты в качестве катализатора. Например, в уксусной кислоте было получено а-ацетоксипроизводное амида (32) [100], которое в мягких условиях можно гидролизовать до соответствующего а-гидро-ксиамида {схема (50)}. Последнее соединение может быть получено и непосредственно, если вместо карбоновой кислоты использовать минеральную кислоту. Механизм рассматриваемой реакции пока не выяснен, но ее первая стадия включает атаку нуклеофильного атома углерода изоцианида на атом углерода карбонильной группы альдегида или кетона с образованием комплексного интермедиата— продукта присоединения (33), который затем превращается в иминооксиран (34). Это соединение после катализируемого кислотой раскрытия цикла дает амид. По этой реакции можно получать также а-амидоамиды, если ее проводить в иных условиях, например, в присутствии аммиака или амина [101] {схема (5.1)}.
9.9.1.5. Алкилирование амидов
Имеется несколько путей модификации амидов путем алкилирования до их более замещенных аналогов. Прямое алкилирование с применением таких реагентов, как алкилгалогениды или сульфонаты в нейтральных условиях, редко приводит только к N-алки-лированным продуктам. Вместо этого получаются или только О-алкилированные продукты (алкилимидаты), или смесь О- и N-алкилированных продуктов {схема (52)}. Причины этого явления обсуждаются в разд. 9.9.3.3. В данном разделе будут упомянуты только те условия реакции или реагенты, которые представляют интерес для синтеза.
RSX /OR3
R'CONHR2 ----> R'CZ + R‘C^ (52>
*4lR2 XNR2R3
R'CONHR2 -NaH> R'CONR2 Na+ * R'CONR2R3 + NaX (53)
основание _ R3CH=CR4X
R'CONHR2 x .......R'CONR2 --------------»-
R‘CONR2CHR3CHR4X
(54)
(X -= RCO, RSO2, —CN и т. д.)
410
NaNH2
RrCH2CONR2 --------
2BuLl
phCH2CONHPh ------>
_ R3X
R'CHCONR3 Na+ -----► R'R3CHCONR^
PhCHCONPh
(35)
RX
---> PhCHRCONHPh
2RX
---> PhCHRCONRPh
(55)
(56)
Надежным методом получения исключительно N-замещенных амидов является превращение амида в высоконуклеофильный анион с помощью таких сильных оснований, как натрий, амид натрия и гидрид натрия, до обработки алкилирующим агентом. Так, вторичные амиды мягко алкилируются в третичные амиды {схема (53)} путем последовательной обработки основанием и алкилгалогенидом [102].
Анион амида может также присоединяться по Михаэлю к замещенному олефину по схеме (54). При наличии подходящего третичного амида обработка сильным основанием может привести к карбаниону в a-положении к амидной карбонильной группе, что после алкилирования дает разветвленный амид [102, 103] {схема (55)}. В случае вторичных амидов образование карбаниона при обработке эквимольным количеством сильного основания происходит только тогда, когда этот анион стабилизован соседними заместителями (например, ацетоацетамид). Алкилирование таких амидов протекает по схеме (55). Иногда при обработке амида 2 моль бутиллития удается получить дианион (35), дальнейшее моно- или диалкилирование которого осуществляется путем добавления необходимого количества алкилгалогенида {схема (56)}. Анионы типа (35) подвергаются также присоединению по Михаэлю к активированным олефинам [104], что приводит к алкилированию по более нуклеофильной карбанионной стороне, чем по аниону азота.
В отличие от обычного алкилирования N-гидроксилалкилиро-вание альдегидами пли кетонами не требует щелочных условий. Механизм этого процесса подробно обсуждается в разд. 9.9.3.4, а здесь будут рассмотрены лишь основные возможные типы реакций. Как первичные, так и вторичные амиды присоединяются к карбонильной группе альдегида или кетона [4, 102, 105] с образованием вначале N-ациламиноспирта (36), который устойчив в нейтральных или слабощелочных растворах {схема (57)}. Например, такие альдегиды как формальдегид или хлораль, имеющий электронооттягивающий заместитель, легко реагируют при 100—150°С, давая аминоспирт (36, R4 = Н). Недавно предложен удобный метод синтеза N-метиламидов, RCONHMe, основанный на алкилировании формальдегидом с последующим восстановлением аминоспирта нос ^2’ R3’ ~ Н) триэтилсиланом или палладием на угле
[Юба]. Кетоны менее реакционноспособны, и известно лишь несколько примеров, таких как гексафторацетон, ацилоины и а-ами-нокетоны, которые присоединяют амиды. В кислых условиях скорость присоединения по карбонильной группе возрастает, ио часто происходит дегидратация амнпоспирта с последующим
411
1 о H
r‘conh2 + r22chcho +=
[r'conh=chchr22
|riCONH2
(r'conh)2chchr^
образованием других продуктов {схема (58)} в количествах висящих от структуры альдегида и амида. ’ За'
ОН
r’ccnr^ror* (5
(36)
он
R'CONHCHCHR2
||н+(-Н2О)
R’CONH— CHCHR2] {58> ||-н+
R'CONHCH=CR|
(37) (38)
Например, в случае первичных амидов присоединение дополнительного моля амида приводит к алкилиден- или арилиденбисамидам (37) [102, 105]. В случае алифатических альдегидов, имеющих по крайней мере один атом водорода в a-положении, отщепление воды из аминоспирта приводит к N-виниламиду (38). В некоторых случаях могут протекать обе реакции, что приводит к смеси продуктов. Известно лишь несколько примеров реакций кетонов в аналогичных условиях.
Гидроксиметилирование в сильнощелочных условиях приводит к замещению не у атома азота, а у углеродного атома, находящегося в a-положении к амидной группе. Это осложнение уже обсуждалось в связи с простым алкилированием, когда возможна конкуренция между карбанионом и анионом азота. Аналогичная ситуация возникает при гидроксиметилировании, когда алкилирующим агентом служит альдегид, а не алкилгалогенид. Так, фенилацетамид превращается в С-гпдроксиалкилированный амид {схема (59)}, в то время как изобутирамид дает N-гидроксиалки-лированный амид {схема (60)}. Отклонение от обычного гидроксиалкилирования происходит, например, в присутствии спиртов, когда получаются N-алкоксиалкильные производные [105] {схема (61)}. Вместе с тем если вместо альдегида в качестве алкилирующего агента используется ацеталь в серной кислоте, то образуются бисамиды, аналогичные (37) [106].
основание „ R2CHO
PhCH2CONHR‘ ( PhCHCONHR1 --------
основание _ Й2СНО
Me2CHCONHR‘ ~ ' "*• Me2CHCONR‘ -----
R‘CONH2 + R2CHO-|- R3OH —> R'CONHCHR2
R2CHOH PhCHCONHR1 (59)
R2CHOH
Me2CHCONRl (60) OR3
412
Недавно сообщалось о гомолитическом алкилировании амидов, 0 может оказаться удобным методом синтеза простых алкилами-лов В большинстве таких реакций происходит присоединение формамида к олефинам, ацетиленам или аренам; реакции индуцируются фотолизом [107], при облучении электронами [102] или трет-бутилпероксидом [102]. Более подробно механизмы этих реакций обсуждаются ниже в разд. 9.9.3.3, но их можно проиллюстрировать на примере взаимодействия формамида с терминальным олефином {схема (62)}. В случае N-алкилзамещенных амидов могут возникать альтернативные радикалы, и поэтому при реакции с олефинами образуется два продукта. Так, при взаимодействии N-метплацетамида с терминальным олефином получено два продукта, как показано на схеме (63). Аналогично пирролидон-2 алкилируется как в 3, так и в 5 положении [108].
hv RCH=CH2 . HCONH2
hconh2 -----> .conh2 --------->- RCHCH2CONH2-------->
—> R(CH2)2CONH2 + . CONH2 (62)
CH3CONHCH3 —
—► .ch2conhch3
+
—> CH3CONHCH2
rch=ch2 ---------> R(CH2)3CONHCH3
rch=ch2
---------> CH3CONH(CH2)3R
(63)
9.9.1.6. Окислительные и восстановительные методы
Известно, несколько реакций восстановления, приводящих к амидам; наиболее важной из них является восстановление гидроксамовых кислот и гидразидов. Так, восстановление гидразидов над никелем Ренея [109] и каталитическое гидрирование гидроксамовых кислот (включая их О- и N-алкильные производные) [ПО] приводит к амидам [уравнения (64) — (66)]. Эти реакции будут рассмотрены далее в разд. 9.9.4 и 9.9.5. Изучены и некоторые другие реакции окисления и восстановления, но они нашли лишь ограниченное применение. Например, гидразиды могут быть окислены до амидов под действием феррицианид-иона [111] и ряда других окислителей (таких как ион перйодата, бром и N-бромсук-Цинимид). Превращение гидроксамовых кислот в амиды [112] происходит в присутствии амина, вероятно через стадию образования нитрозосоединений {схема (67)}. Другими окислительно-восстановительными реакциями, используемыми для синтеза амидов, являются: восстановление изоцианатов до формамидов с помощью гидрида трифенилолова [113а] {схема (68)}, окисление третичных аминов диоксидом марганца [1136] и окислительное аминирование альдегидов или бензилового пли аллилового спир-Т(кап°Д девствием пероксида никеля в аммиаке [НЗв] {схема 169)}.
NI Ренея RlCONHNH2 ------»- R‘CONH2 + Nil, (64)
413
R'CONHOR2
H2 ? катализатор
R'CONH2 + R2OH
H2 R'CONR2OH ------------>• R'CONHR2 + R2OH
иптлличптлп ' • ' v
[01 R*NH
R'CONHOH -------> [R'CONO] ------> R'CONR2
ArNCO + 2Ph3SnH —> ArNHCHO + (Ph3Sn)2
NiO2. NH3
RCHO (или RCH2OH) -----------> RCONH2
(65)
(66)
(67)
(68)
(69)
9.9.1.7. Получение лактамов
Лактамы — это циклические амиды с различным размером цикла от небольших —а-, |3- и у-лактамы (3, п = 1, 2 и 3 соответственно) до значительно больших циклов. Методы получения этих соединений обычно аналогичны уже описанным для ациклических аналогов [4]. Поскольку малые циклы а- и р-лактамов являются напряженными, существует ряд трудностей получения этих соединений обычными методами. Однако недавно опубликовано несколько обзоров [5, 6, 216], в которых подробно описываются специальные методы, применяемые для их синтеза. Основные методы синтеза лактамов включают внутримолекулярное ацилирование аминов [образование связей (а) и (б) в формуле (3)], перегруппировки Бекмана и Шмидта и реакции присоединения к кетенам и изоцианатам. Дальнейшую модификацию полученных лактамов можно проводить алкилированием по атому азота или, в ряде случаев, по атому углерода, находящемуся в a-положении к карбонильной группе, в условиях, аналогичных алкилированию ациклических амидов.
(40)
(70)
(п=2,3,4,5; X=OR, ОН)
Одним из основных методов синтеза лактамов является внутримолекулярное ацилирование аминогруппы соседней карбоксильной или сложноэфирной группой {схема (70)}. Эти реакции в принципе аналогичны внутримолекулярному ацилированию, описанному в разд. 9.9.1.1, но обычно протекают легче, по крайней мере для лактамов с небольшими циклами. Например, обе аминокислоты (39, X = ОН, п = 3, 4) легко циклизуются с образованием пяти- г шестичленных лактамов [пирролидоны и ппперидоны (40, п , соответственно]. Подробно описана также циклизация сложны.
414
эфиров аминокислот (39, X = OR), при которой образуются пяти-, шести- и семичленные лактамы [114]. Эти методы были использованы [Н5] для синтеза биологически важных корринов. Как аминокислоты, так и их сложные эфиры часто генерируют in situ по следующим реакциям: восстановление иминокислот или нитрокис-дот [Нба], восстановительное аминирование кетокислот [1166], аминолпз лактонов [116в], взаимодействие бромзамещенных сложных эфиров с аммиаком или аминами [116г] и восстановление сложных эфиров, имеющих нитро- [114] и нитрильную группы [116д]. Для синтеза четырехчленных лактамов (40, п = 2) в качестве катализатора используется реактив Гриньяра (реакция Бодру) [И7] • Наиболее эффективными катализаторами оказались стерически затрудненные реактивы Гриньяра, например полученные из броммезнтилена. В связи с этим можно полагать, что катализатор действует как сильное основание, образующее более нуклеофильный аминоанион, который циклизуется легче, чем нейтральное соединение. Для лактамов с большим размером цикла роль межмолекулярных реакций возрастает по мере увеличения расстояния между реагирующими функциональными группами. Эти соединения легче синтезируются с помощью методов, основанных на расширении цикла, таких как реакции Бекмана или Шмидта, которые обсуждаются ниже.
В синтезе лактамов нашли применение несколько других реакций циклизации, многие из которых аналогичны соответствующим реакциям при получении ациклических амидов. Например, алке-нилнитрилы подходящей структуры могут замыкаться в цикл за счет внутримолекулярной реакции Риттера [58] (см. разд. 9.9.1.2), как это представлено схемой (71). Циклизация со-галогенамидов также служит важным методом, который позволяет синтезировать, кроме семичленных, трех-, четырех-, пяти- и шестичленные лактамы [4, 29, 102]. В этом случае для получения лактамов реакцию нужно проводить в сильнощелочной среде (амид натрия, гидрид натрия или трет-бутоксид калия в апротонном растворителе) {схема (72)}. При синтезе в нейтральной или слабощелочной среде образуются изомерные лактимы {схема (73)}. Одной из немногих реакций получения а-лактамов является синтез трехчленных лактамов из N-хлорамидов в сильнощелочной среде {схема (74)}. Другой необычный метод образования лактамного цикла, включающий алкилирование атома азота, основан на взаимодействии группировки ацилнитрена, которая генерируется при фотолизе ацилазида [118] или нитрилоксида [112], подходящим образом расположенной неактивированной группой С—Н. Пример подобной реакции представлен на схеме (75).
Другим важным методом синтеза лактамов является реакция расширения цикла циклических кетонов с помощью перегруппировок Бекмана или Шмидта, как это приведено на схеме (76) (ср. разд. 9.9.1.3). Эти реакции используются для получения циклов разного размера от малых до очень больших. Однако наиболее
415
широко они используются при синтезе макроциклических соей ний, которые трудно получить другим способом. Дине-
раствор
(73)
О
нейтральный
PhCHaCONBuWm ^-Л1'ВЦ°> PhCHCONBu-лгрт —> PhCH-^BU-^ew
Cl cP
нь1еНрХВцииОБ^^2ВаННЫе °бз0ры [68’ 71> 119> 120]. посвящен-время как послелуют’ Включают Данные о синтезе лактамов, в то оксимов бензтропонП12ТаС]ЛТавЗ П0Священы перегруппировке токсимов [12161 3 и а'Р'ненасьнценных циклических ке-
ведения этой иЦи^Д "° предложены необычные варианты про-галогеном и тощ,,. \ 8кяюча1°Щие последовательную обработку ал™ "арбоново,? Ф°СФН,НОМ (122аЬ а ™«е обработку цикло-[1226], Общим недоситком пмСТ° -к?,10"а) сульфатом нитрозила образование ич вд°с а к° РеакДии Бекмана и Шмидта является мерных лактамове^ММетРичных циклических кетонов двух изо-реаРквдй удало { ема (77)>- Тсм не менее с помощью этих реакция Шмидта бмУп?еСТВИТЬ ряд важных синтезов. Например, терпенов и алкая™ пап и.дПОЛьзована при синтезе стероидов, три-ний приведено в обзоре' ПДТ’ер°В примеяеяия о6еих реа“' ману^отошяй 11в°й метода получения лактамов по Бек-
пае кетонов ио « ^ СМОТ₽е“ ‘ 9-91-8, является превраше;
п-толуолсупкгЬгш»™СИМЬ1’ а вгНИтроны с последующей обработкой
У Ф хлоридом [88]. Предполагается, что другой ва-416
риант реакции Бекмана, который включает не каталитическую а фотохимическую перегруппировку, протекает через стадию образования оксазиридина в качестве интермедиата (1231 к
Хиноны также реагируют с HN3; при этом может протекать либо р^иреше цикла, как в реакции Шмидта, либо его coSa шепне [124]. Например, бензохиноны типа (41) в условиях neS ц„и Шмидта превращаются в производные азенина которые НО получить Другими методами [125] {схема (78)}. Р РУД’
~н2о
Как и в случае ациклических амидов, интермедиат в реакции Шмидта можно получать путем нитрозирования семикарбазонов циклических кетонов, таких как циклогексанон, ннданон и тетрадой [76]. Это приводит к реакции Шмидта обычного типа, в которой атом азота включается в цикл, образуя лактамы. Реакция расширения цикла совсем другого типа, применяемая для синтеза
14 Зак. 395
417
азепинонов из 2,6-диалкилфенолов и хлорамина [126], приведена на схеме.
Реакции, включающие различные типы несогласованного цикло присоединения, применяются главным образом для синтеза 0-лактамов. Например, присоединение кетенов к иминам, а также изоцианатов к обычным и сопряженным олефинам приводит к р-лактамам [6] {схемы (80) и (81) соответственно}. В обоих случаях реакция облегчается, если заместителем у атома азота является хлорсульфонильная группа (R = SO2C1). Эта группа легко отщепляется с помощью мягкого гидролиза, давая соответствующий лактам (R = H).
r2ch^nri r^h-nr1
СН2—С—О сн2-с=о
R*N=C=O R1N--С=О
1 -----I I I81)
ch2=cr2r3 ch2-cr2r3
Две другие реакции присоединения, приводящие в конечном счете к лактамам, требуют применения специальных реагентов, таких как цианацетилхлорид [127] или оксид углерода [45] {схемы (82) и (83) соответственно}.
ArCH=NR
ArCH—NR
ArCH—NR
CI—cch2cn
С1 с=о ncch2X
Co2(CO)g ch2=chch2nh2 + со ----->
NC
СН—с %
(82)
(83)
О—NaOEt °'- 9\
| ;(сн2)п -> | (сн2)п
2NaH
=О + 2Ме1 --------’
NH
RCI °=9\
—>- | ;(сн2)„
RNZ
СМе2
(84)
(85)
О
ь=О
NMe NMe
Модификацию лактамов алкилированием по атому азота осуществить аналогично тому, как это описано в разд. 9.9.1.4 дл ациклических амидов. Наилучшими условиями реакции являете обработка лактама алкилгалогенидом в сильнощелочнои сРе^ [128] {схема (84)}. Если атом азота лактама уже замещен, i возможно, как и в случае ациклических амидов, алкилиро cc-атома углерода по отношению к карбонильной группе при } вии, что он связан по крайней мере с одним атомом вод р [129] {схема (85)}.
можно
418
9.9.1.8. Получение имидов
N-Моноацильные производные амидов, называемые имидами, могут иметь ациклическое (4) или циклическое (5) строение. Наиболее распространенными можно считать четырех-, пяти- и шестичленные циклические имиды, примерами которых являются сукцинимид (5, R2 = Н, п = 2), малеимид (42) и фталимид (43). В недавно опубликованных обзорах по имидам [7—9] приведено много примеров их синтеза, и поэтому в данном разделе рассматриваются лишь основные направления. Основным методом получения ациклических имидов, является, конечно, обработка амидов ацилирующим реагентом, в то время как циклические имиды синтезируют циклизацией различных бифункциональных соединений, таких как моноамиды кислот (амидовые кислоты), диамиды и динитрилы. Однако, практически все методы включают образование связи углерода карбонильной группы с азотом [(4), (5), связь (а)]
° О
Onh
о о
(42) (43)
R’CONHR3+ R2COX ----1
(a) zOCOR3 (RfiH)
R’CONR3COR2 -«-- R’c' ------------> R’CN + R2CO2H (86)
\nr3
(44) (45)
(6)|r2COX(r3 = H)
zOCOR2
R’C^ —> R’CON(COR2)2
Xncor2
(46)
Обработка первичного или вторичного амида подходящим ацилирующим реагентом RCOX (где X = OCOR или OR) обычно приводит к ациклическим имидам (44). Реакция однозначна в случае вторичных амидов, но должна проводиться в строго контролируемых условиях в случае первичных амидов из-за конкурирующих побочных реакций. Как обсуждается в разд. 9.9.3.5, первичным продуктом реакции в нейтральной и слабощелочной средах яв-
О’ацилимндат (45) {схема (86)}, который удается выде-
1 только в исключительных случаях. Этот интермедиат (45)
может реагировать с первичными амидами (Rd — Н) тремя различными способами, но только один из них приводит к образованию имида {(44), R3 = Н, путь (а)]. Другие направления приво-дят к трпациламнну (46) за счет дальнейшего ацилирования или к нитрилу в результате отщепления карбоновой кислоты {схема (86), пути (б) и (в) соответственно}. Наилучшие выходы имидов из первичных амидов получены, если использовались относительно слабые ацилирующие агенты (например, бутироилхлорид), и реакция проводилась при О °C в присутствии пиридина в качестве катализатора. Образование побочного триациламина (46) становится предпочтительным в присутствии умеренно сильных реагентов (например, n-хлорфенацетилхлорида), при очень низких температурах (—60°C), в то время как образование нитрила облегчается в присутствии сильного ацилирующего агента (например, /г-нитрофен-ацетилхлорида) при 0°С [102]. По аналогии с другими реакциями, где требуется провести замещение у атома азота амидной группы, кажется предпочтительным, если это возможно, использовать сильно кислые или щелочные условия для промотирования прямой реакции взаимодействия с атомом азота [102]. Например, применение в качестве катализаторов НС1 или H2SO4 в реакции пропионового ангидрида с пропионамидом или уксусного ангидрида с бензамидом приводит к образованию с хорошим выходом дипропионамида и N-ацетилбензамида соответственно [130]. Использование сильных оснований, таких как гидрид натрия или реактивы Гриньяра, промотирует реакцию между амидами и сложными эфирами или хлоридангидридами карбоновых кислот {схемы (87) и (88) соответственно} [131].
2NaH - - 2ArCO2Rz
R!CH2CONH2 ----► R'CHCONH -------> ArCOCHR'CONHCOAr (87)
EtMgBr - RCOCI
PhNHCOMe -------> PhNCOMe ------> RCONPhCOMe (88)
,Me H+
R!CONH2+R2COOCZ --------► R’CONHCOR2 + Me2CO (89)
Чн2
R!CONH2 + R2R3C==C=O "-> R1CONHCOCHR2RS (90)
/OCONHR2 R!CONH2 + R2N==C=O —> R'C/ ‘ (91>
^NH -X-> R!CN+R2NHCO;
-----------> RiCONHCONHR2
(47)
Применение специальных ацилирующих реагентов (Уже^Уп°0 мииавшихся в свяви е ацилированием аминов) приводит к особен
420
высоким выходам имидов. Так, изопренильные сложные эфиры [132а] и кетены [1326] при взаимодействии с амидами образуют имиды в присутствии кислых катализаторов, таких как серная или я-толуолсульфоновая кислоты {схемы (89) и (90) соответственно}. При проведении последней реакции в отсутствие кислых катализаторов из первичных амидов вместо имидов образуются нитрилы [102]. Близкие по строению алкилизоцианаты реагируют с амидами с образованием N-ацилмочевин (47), которые являются производными имидов [133]. Алкилизоцианаты более реакционноспособны, чем кетены, но, как ни странно, при взаимодействии с первичными амидами в нейтральных условиях они не дают нитрилов {схема (91)}. Этот факт может быть связан с низкой способностью к отщеплению аниона карбаминовой кислоты.
Хотя циклические имиды могут быть получены с помощью ряда внутримолекулярных реакций, в основе метода их синтеза лежит взаимодействие между амидом и ацилирующим агентом, при этом один или оба этих остатка могут возникать in situ из таких предшественников, как нитрилы или циклические ангидриды кислот и аммиак. Амидовые кислоты (48) представляют собой удобные исходные соединения для рассмотрения этих реакций. Эти вещества легко циклизуются как при нагревании [134], так и при обработке катализаторами, такими, как ацетилхлорид, пентоксид фосфора или уксусный ангидрид [135]. В нейтральных условиях образуются изоимиды (49), которые могут быть выделены, так ка.к они более устойчивы, чем их ациклические аналоги (т. е. О-ацилимидаты). Перегруппировка соединения (49) в термодинамически более устойчивый имид происходит при сильном нагревании [134] или при обработке веществами основного характера, например, карбонатом калия [135, 136] {схема (92)}. Хорошо изучено влияние соседних групп на образование изоимида при гидролизе сложных эфиров (см. разд. 9.9.3.1). В то же время имеется мало данных о механизме катализа галогенангидридами и ангидридами кислот и о влиянии заместителей на эти реакции. Однако известно, что N-замещенный моноамид малеиновой кислоты, имеющий такие электронооттягивающие заместители, как N-н-карбоксифенил или N-этоксикарбонил, не циклизуется, в то время как N-алкил- и N-фенилзамещенный моноамид малеиновой кислоты да.ет соответствующие имиды [138]. Можно полагать, что природа заместителя у атома азота влияет на его нуклеофильность и в связи с этим на легкость циклизации. Синтез четырехчленных циклических имидов, например 2,4-азетидинов (50) из соответствующих амидовых кислот, требует для замыкания цикла применения тионил’хлорида в пиридине [139]. Попытки получить трехчленный оксимид (51) пока не увенчались успехом.
Широко распространенным способом получения имидов является обработка циклических ангидридов кислот аммиаком или первичными аминами. Наиболее вероятными интермедиатами в этих превращениях считаются амидовые кислоты, хотя они обычно не
421
выделяются (ср. разд. 9.9.1.1). Много конкретных примеров приведено Уилером и Розадо [7], а также Беквитом [4]. Типичным превращением является взаимодействие ангидрида янтарной кислоты с аммиаком с образованием сукцинимида (схема 93). Другими предшественниками амидовых кислот являются сами дикарбоновые кислоты и их хлорангидриды, которые при обработке аммиаком или солями аммония легко дают имиды [8]. Интересной модификацией этой реакции является замена амина более нуклеофильными реагентами — гидроксиламином или гидразингидратом. Такая замена приводит к прямому синтезу ацилгидроксамовой кислоты или к 1,1-диацилгидразину соответственно [140] {схема (94)} (см. разд. 9.9.4 и 9.9.5).
(X = он, nh2)
422
о
о
(95)
Другие типы синтезов циклических имидов включают циклизацию сложных эфиров амидовых кислот или диамидов дикарбоновых кислот [8,141] при нагревании или в присутствии каталитических количеств сильных оснований. Примеры таких реакций приведены на схемах (95) и (96) соответственно. Диэфиры обычно являются предшественниками сложных эфиров амидовых кислот, которые образуются при обработке диэфиров аммиаком в присутствии этоксида натрия [8] или анилинов [135].
АгСОМе
+ NCCH2CO2Et
,X.CONH2
(96)
NH3 ---->
Me')—COEt NC' II О
(53)
(97)
(52)
Синтез имидов Гуареши (53) (промежуточных соединений в синтезе гетероциклов и фармацевтических препаратов) представляет собой важное применение реакции взаимодействия диэфиров с аммиаком. В данном случае промежуточно образующийся дицианодиэфир (52), полученный при взаимодействии алкиларилке-тона с 2 моль цианоуксусного эфира, циклизуется при обработке аммиаком [142] {схема (97)}.
Другим типом внутримолекулярного замещения, приводящим к имидам, является взаимодействие между нитрильной и карбоксильной группами [143] {схема (98)}. Как и в предыдущем случае, первоначально образующийся в качестве интермедиата изоимид перегруппировывается в более устойчивый имид. о-Циано-амиды циклизуются при нагревании с образованием производного фталимида (54), которое после мягкого гидролиза превращается
423
в имид {схема (99)}. Вероятно, альтернативный метод ПА динитрила слабой кислотой [7, 144] также включает обп б°ТК1} амидонитрила в результате частичного гидролиза с поел °Вание циклизацией [8]. Напротив, при обработке динитрила в качестве интермедиата образуется имидин котооый Иак°м вдается в имид после гидролиза [103] {схема (100)} И превРа-
(54)
Существует очень немного способов получения циклических имидов, не включающих внутримолекулярное замещение, однако реакция присоединения изоцианатов к ангидридам [145а] пли кетенам [1456] имеет препаративное применение {схемы (101) и (102)}. В случае ангидридов, элиминирование СОг из семичленного интермедиата напоминает реакцию между изоцианатами и аминами (см. разд. 9.9.1.4). Помимо того, новым типом реакции
(Ю0)
циклизации, имеющим амазогн^
разд. 9.9.1.7), является " синтезе лактамов (см.
с оксидом углерода в ппнДМ°АеИСТВИе P’V-ненасыщенных амидов которое с хорошим выходов™111 октакарбонила кобальта [45], Известно также нескошТ ПРИВ°Д'”' к “мидам (схема (103)}. рых образуются имиды 17 «Г и™ окислення, в результате кото-ине ниррольного кольпЧ пп ' МапримеР> фотохимическое окисле-Рздпмодействии с риводит к соединению (55), которое при (104)}. Лактамы могут бы?° оЛ^™"43 ДаеТ "Мид [I46J {с^,а например как в случае пи- Ь Окислень| аналогичным образом [147], под действием пеосульжДТеНИЯ капР°лактама (схема (105)} или . У Ф ча калия [103]. Автоокисление ациклн-
$24
ческих амидов более сложно и является не бом получения имидов (см. разд. 9.9.3.9).
очень удобным спосо-
RtR2C=C=O + R3NCO —>
(102)
CH2=CHCH2CONII2 + СО —>
(ЮЗ)
Как в случае амидов и лактамов, соответствующие имиды могут быть модифицированы путем N-алкилирования, обычно в сильнощелочных растворах. Подобные реакции будут рассмотрены в разд. 9.9.3.13.
9.9.2. СВОЙСТВА АМИДОВ
Физические свойства и строение амидной группы явилась предметом целого ряда недавно опубликованных работ. Обзоры 'по молекулярной и электронной структуре [148], по кислотно-основным и комплексообразующим свойствам [149] и по fl-MP [150] сообщают большую часть новых достижений, включая вопросы
425
формы и стереохимии амидной группы, спектральные свойства и донорно-акцепторные характеристики. Сравнительные аспекты структуры и свойств имидов были рассмотрены Уилером ц р0. задо [7] и Харгривсом с сотр. [8].
Многие физические свойства амидов и имидов могут быть поняты с точки зрения делокализации неподеленной пары электронов азота на л-электроны карбонильной группы. Этот эффект приводит к тому, что связь С (О)—N до некоторой степени имеет свойства двойной связи (кратность связи в амидах ж 1,5, в имидах 1,3). Вместе с тем возникает 1,3-диполь, в котором азот обладает частичным положительным зарядом, а кислород — частичным отрицательным. Планарная природа амидной группы и существование конфигурационных изомеров также являются следствием частично непредельного характера связи. Вместе с тем донорно-акцепторные свойства амидной группы, проявляющиеся в кислотно-основных взаимодействиях, в склонности к комплексообразованию, а также в тенденции к ассоциации, являются следствием ее биполярного строения. Универсальность амидной группы в образовании частичных связей между собой и с многими другими функциональными группами в значительной мере определяет структурное многообразие производных биологически важных белков (см. части 23 и 24).
9.9,2.1. Структура и стереохимия амидов
Молекулярная структура амидной группы в конденсированной и газовой фазе установлена очень детально. Однако эта структура не так проста, как можно было бы предположить исходя из молекулярной формулы, и при интерпретации результатов исследований этих соединений физическими методами следует учитывать несколько особенностей. Первая особенность связана с частичной двоесвязанностыо связи С (О)—N, что возникает в результате делокализации неподеленной электронной пары азота на л-электроны карбонильной группы. Ниже представлены две предельных валентных формы — без сопряжения (56) и с полным сопряжением (57) неподеленной электронной пары азота, обозначены предполагаемые валентные углы и длины связей в нм. В действительности, в кристаллических амидах {как это было определено с помощью рентгеноструктурного анализа [148] и схематически представлено в (57а)} амидная связь имеет плоское строение (по крайней мере для всех атомов, кроме водородных), а длины связей имеют примерно средние значения из двух резонансных форм (56) и (57). В газовой фазе (как показано при изучении дифракции электронов и микроволн [148, 151]) валентные углы практически ие изменяются (хотя атомы водорода в формамиде находятся вис плоскости О—С—N на расстоянии 0,015 нм). Правда, происходит уменьшение длины связи С—О до 0,119—0,121 нм и соответствующее увеличение длины связи С—N до 0,136—0,137 нм. Эти данные
429
ппапят о том что в газовой фазе структура (56) вносит больший Тпад чем в случае кристаллических амидов. Таким образом, мрртся почти обратная зависимость между длинами связей С—О J C—N: одна связь удлиняется, тогда как другая укорачивается ^4другим важным следствием частично двоесвязанного характера связи В амидах является существование конфигурационных изо-с ов —(58) (Е или цис-) и (59) (Z- или транс-), возникающих из-за отсутствия свободного вращения вокруг связи С (О)— N. Существование этих изомеров установлено с помощью данных ядер-ного магнитного резонанса, инфракрасной и Раман спектроскопии а также измерениями дипольного момента [153] (см. также разд. Э.9.2.2). В отдельных случаях один из изомеров был выделен путем кристаллизации или за счет образования пп» ииа.
кой температуре.
комплекса при низ-
NX
R2 (58) цис- (£)
ж
Ji
(59)транс- (Z)
Наблюдая скорость взаимного превращения изомеров с помощью сложной техники ЯМР, например анализом формы линий, удалось определить барьер вращения вокруг связи С (О)— N для многих амидов. Эти значения приведены в виде таблиц в работах [148, 150]; типичная величина для диметилформамида zXG^ ~ «88 кДж-моль-1 при 118°С, в то время как AG#" для М,Ы-ди-метиламидов, равная около 75 кДж-моль-1 при 75 °C, как можно было предсказать, несколько ниже. (Следует отметить, что влияние температуры на AG^ для вращения амидов [150] невелико—• порядка 1 2%, и поэтому сравнение значений, полученных при различных температурах, вполне обоснованно). Увеличение размера N-алкильного или N-арильного заместителя обычно приводит к постепенному снижению барьера вращения. Однако в некого >ых случаях, например для ряда N-метиламидов R'C(O)NMeR2, где
427
R1 п/или R2—-арильные группы, происходит пространственное взаимодействие между R1 и R2, известное как «блокировка» (locking), в свяди с чем барьер вращения заметно возрастает, например на 42 кДж-моль-1 при R1 = 2,4,6-три-трет-бутилфенйл и Р2-бензил [150]. Креме влияния стерпческих факторов, как было недавно показано [154], барьер вращения растет также, если заместителями в бензольном кольце N-алкилформанилидов являются группы /?-МеО или n-Me2N. Вероятно, этот эффект связан с сопряжением неподеленных электронных пар этих групп с л-системой амида [154].
По сравнению с вращением вокруг обычной двойной связи (например, барьер вращения в случае олефинов составляет около 167 кДж-моль-1), вращательный барьер для амидов заметно ниже и, следовательно, период вращения короче. Например, период вращения N.N-диметилформамида или Ы,Ы-диметилацетамида составляет около 0,1 с [148]. Это означает, что если химические реакции протекают при комнатной температуре, существование вращательных изомеров не существенно в силу их быстрого взаимопревращения. Наличие изомеров становится химически значимым только при низких температурах; кроме того, с ним следует считаться при использовании метода ЯМР, где временной диапазон измерений относительно мал.
С помощью ЯМР-спектроскопии возможно оценить относительное содержание двух изомеров (58) и (59) [148, 150]. В случае N-алкил'амидов (58, 5.9, К2-алкил) (Z)-изомер преобладает в такой степени, что обнаружение (Е)-изомера часто затруднительно. Только в случае N-алкилформамидов (58, 59, R1 = Н) возможно наблюдать заметное количество (£)-изомера (58). Например, N-третбутилформамид содержит 18% (Д)-изомера (58, R2-rper-Bu). Однако даж'е в этом случае объемистая трет-бутильная группа, а не менее стерическц затрудненный протон, стремится занять положение, соседнее с карбонильной группой [153]. Заметное преобладание (2)-формы в твердых N-алкиламидах показано также с помощью рентгеноструктурного анализа [155]. За исключением N-алкилформамидов, доля (F)-формы возрастает только тогда, когда очень велико стерическое отталкивание между кислородом карбонильной группы и N-алкнльным заместителем [148], как это происходит, если R содержит сильно электроотрицательный заместитель, например фтор [156], или когда возникают напряжения внутри цикла, как в случае лактамов с малыми циклами. Рассчитано, что переход из (А1)-формы в более стабильную (Z)-форму будет происходить в девятичленных лактамах. Это предположение недавно подтверждено при рентгеноструктурных кристаллографических исследованиях 7—12-членных лактамов [157J. Следует еше выдвинуть обоснованное объяснение повышенной стабильности (Z)-изомера в случае ациклических вторичных М,М-алкиламид°з и макроциклических лактамов. Напротив, N-ариламиды показы-
428
аЮТ повышенное содержание (Е)-изомера (25—55%), но даже и в этом случае (Z)-изомер в общем более устойчив [150].
Преимущественное существование определенных конфигураций N N-дпалкиламидов легче объясняется с точки зрения пространственных взаимодействий, чем в случае N-моноалкилированных амидов. Так, N-метил-М-алкиламиды, которые имеют два возможных изомера (60) и (61), в ряду ацетамидов (R1 — Me, R2 — Et, н-Bu, цикло-С6Нц, изо-Pr) содержат их в примерно равном соотношении. При этом в случае формамидов соотношение (60) к (61) составляет 65:35% (т. е. более объемистая алкильная группа расположена ближе к формильному атому водорода [158]). С другой стороны, если R — очень объемистая группа (например, R'-ме-зитил; R2 = PhCH2, цикло-С,&1Ап), доля (Е)-изомера возрастает вдвое [30% (60): 70% (61)/[159] (т. е. более объемистый N-ал-кильный заместитель теперь расположен ближе к кислороду карбонильной группы, так как для него стерические препятствия меньше, чем для мезитильной группы). Напротив, третичные N-ариламиды, такие как N-алкилацетанилиды, существуют преимущественно в (Е)-форме (т. е. фенильная группа находится в транс-положении по отношению к кислородному атому карбонильной группы) [150]. Детальное обобщение соотношения изомеров в этих и многих других вторичных и третичных амидах было проведено в обзорах [148, 150].
Me
(во) (z)
(62)
Оч В2
ГД Me
(61)(Е)
В силу прочности и планарности системы связей N—С — О можно наблюдать медленное вращение вокруг других связей в молекуле амида. Так, обнаружена изомерия, обусловленная медленным вращением вокруг связей N—R3 и R1—С(О) в (62) [148, 150]. Когда R' или R2 == Ph, ситуация сравнима с бифенилами, ак, в соединениях (63) и (64) бензольное кольцо расположено вне плоскости амидной группы и, предполагая вращение медленным,
429
молекула не будет иметь плоскости симметрии, а значит (631 и (64) будут иметь оптические изомеры. Барьер вращения д,1я различных анилидов (63) составляет AG* — 63—105 кДж-моль*'1 но в случае бензамидов (64), так как карбонильная группа меньше, чем алкил R2 в (63), барьер вращения примерно вдвое меньше (AG* 33—59 кДж • моль-1) [150]. Естественно, что медленное вращение, а следовательно, и изомерия наблюдались также и тогда, когда R1 пли R3 в (62) представляли собой не только фенильные, но и другие группы. Подобные соединения привлекли меньше внимания, однако ряд примеров приведен в работах [148, 160].
Существование стабильных и метастабильных изомеров, образующихся в результате медленного вращения связи, часто делает ЯМР-спектры амидов очень сложными. Другое осложняющее обстоятельство в структурных исследованиях амидов в конденсированной фазе, возникающее в силу их биполярного характера, связано с тенденцией амидов к ассоциации как между собой, так и с растворителем. Поэтому значения наблюдаемых физических параметров, таких как химические сдвиги при ЯМР-спектроскопии, частоты поглощения при ПК-спектроскопии или дипольные моменты, будут зависеть от концентрации амида в растворе и природы растворителя. В литературе хорошо описаны три типа межмолекулярных ассоциатов амидов: димеры и полимеры, образованные за счет водородной связи (65) и (66), а также диполь-дипольные димеры (67) [149]. Первые два типа относятся, конечно, только к первичным и вторичным амидам и образуются вследствие их кислотно-основных свойств (см. разд. 9.9.2.3). Возникновение третьего типа ассоциатов, вероятно, ограничено третичными амидами, для которых образование водородных связей невозможно. Амиды образуют также ассоциаты с молекулами растворителя; особенно легко образуются комплексы с ароматическими растворителями. Эти эффекты ассоциации рассматриваются в разд. 9.9.2.2 и 9.9.2.3.
Г
О—Н—N
R1 R1
\ // N—Н—О
(65)
,з
R2 R'
S- W о—-N
R1—
\\8+ /7
3/N—" 2
R3/l., 5-
R2
(67)
°\ X
С—N Чн \ г С—N Л 4 R1 Н
(66)
Несмотря на тенденцию к ассоциации, значения дипольных моментов вторичных и третичных N-алкиламидов в газообразнол состоянии [148] и в растворителях, таких как бензол, CCU и Дп0К’
430
сан [161], оказались достаточно близки. Среднее значение дипольного момента для этих амидов 3,75 ± 0,1D. Таким образом, суммарное значение дипольного момента практически не зависит ни от природы растворителя, ни от моментов о-связей различных алкильных заместителей.
По аналогии с кето-енольной таутомерией в качестве таутомерной формы первичных и вторичных амидов ранее постулировалась имидовая кислота (иминол) (68). Однако несмотря на значительные усилия, не было обнаружено физических доказательств существования такой структуры. При этом сложные эфиры имидовой кислоты (69) (т. е. алкил- или арилимидаты, иногда называемые эфирами иминолов (69) — хорошо известные соединения, которые могут быть синтезированы с помощью ряда методов, включая алкилирование амидов (см. разд. 8.1.4 и 9.9.3.3). Хотя имидаты превращаются в более стабильные амиды в результате каталитической или термической перегруппировки {схема (106)}, обратная реакция не наблюдается. Это указывает на значительные различия в устойчивости двух изомеров.
,0 /ОН
R1—С/ R1—(/
\n—R2 ч\т—R2
I
H (68)
R3°\ %
V=NR2 —► NR2R3
RI/Z R]/
(106)
(69)
Э.9.2.2. Спектральные свойства амидов
В данном разделе представляется возможным дать лишь самый общий обзор данных об ИК-, УФ- и ЯМР-спектроскопии, а также о масс-спектрометрии данных соединений. За более детальной информацией следует обращаться к прекрасным монографиям и обзорам, которые цитируются в соответствующих разделах.
В инфракрасных спектрах амидов присутствует несколько характерных полос поглощения, используемых для целей идентификации [163]. Точное значение характеристических частот поглощения зависит от ряда факторов, и поэтому в табл. 9.9.1 приведены средние значения.
Во-первых, следует отметить, что положение полосы поглощения сильно зависит от условий регистрации спектра. Так, в конденсированной фазе первичные и вторичные амиды имеют тенденцию к ассоциации за счет образования водородных связей, и поэтому значения наблюдаемых частот заметно ниже, чем в разбавленных растворах и в газовой фазе, где амиды существуют в виде мономеров. Полоса валентных колебаний N — Н первичных и
431
вторичных амидов, обладающая средней интенсивностью располп жена в области 3000—3500 см-1. В случае первичных амидов эта полоса представляет собой дублет за счет расщепления на двух атомах водорода (Н—N—Н). Недавно удалось обнаружить при-сутствие цис-(Е)- и транс- (Z)-изомеров вторичных амидов (см разд. 9.9.2.1), каждому из них была приписана отдельная частота
Таблица 9.9.1. Характеристические частоты поглощения амидной группы в инфракрасной области
Характеристика полос: (а) —средняя интенсивность; (б) —очень интенсивные- (в) — а™., метричные колебания; (г) —симметричные колебания; (д)—(2)-изомер; (е) — (Е)-изомер М'
Амид Частоты, см 1
валентные колебаниям—Н<а> (свободная) (связанная) (амидная полоса !)(б> (амидная полоса!!) *а)
валентные колебания С=О (свободная) (связанная) плоскостные деформационные колебания N-H
Первичный 3540-3520 (в) 3360-3340 (в) 1690 1650 1620-1590
амид (—CONH2) Вторичный 3400—3380 (г) 3480-3440 (д) 3200—3180 (г> 3320—3270 (д) 1700—1670 1680-1635 (свободная) 1650-1620 (связанная) 1550-1510
амид (—CONHR) Третичный амид (-CONR2) Лактамы 3435-3395 (е) 3420 3180-3140 (е) 3220-3175 1670-1630 1780-1700 1740-1670 (свободная) 1570-1515 (связанная)
(3—8-членные) (е> ( > 8-член- 3480-3440 3270-3220 1680 1680
ные) (д>
валентных колебаний N—Н, причем для транс-изомера значение частоты было выше [163].
В ИК-спектрах первичных, вторичных и третичных амидов присутствует очень интенсивная полоса поглощения (полоса I) в области 1650—1700 см-1, обусловленная валентными колебаниями С = О. Точное положение этой полосы зависит также от условий (фазового состояния) регистрации спектра. Исключение составляют лишь третичные амиды, которые не дают ассоциатов за счет водородных связей. Как правило, частота амидной полосы I ниже, чем частота поглощения, обусловленного валентными колебаниями других карбонильных групп, например кетонов. Это обусловлено тем, что связь С—О приобретает частично односвязный характер в 'результате делокализации неподеленной пары электронов азота (см. разд. 9.9.2.1). Положение полосы зависит также от наличи^ или отсутствия заместителей у атома азота и от их электроотрии тельности. Например, при наличии N-Пенильного заместителя ч стота валентных колебаний карбонильной группы увеличивает 432
приблизительно на 10 см-1. Это указывает на частичную делокализацию неподеленной пары электронов азота на л-систему бензольного кольца. В связи с этим имеет место меньшее взаимодействие с карбонильной л-системой, которая поэтому становится более похожей на изолированную двойную связь.
Вторая по значимости для первичных и вторичных амидов полоса II имеет среднюю интенсивность и обусловлена главным образом плоскостными деформационными колебаниями N—Н. Другие полосы поглощения менее характерны. Например, амидная полоса Ш в области 1300—1400 см-1 (связана со смешанными колебаниями— валентные колебания С—N и деформационные колебания N__Н) более надежно регистрируется в Раман-спектрах. Вместе
с тем в области 650—1400 см-1 обнаружены также полосы IV, V, VI [163].
Два фактора влияют на положение полос поглощения лактамов в ИК-областн относительно их ациклических аналогов. Первый фактор связан с размером цикла, и, как правило, чем меньше цикл, тем выше частота колебаний С = О (максимальный сдвиг для трехчленных лактамов +83 см-1) - Аналогичный эффект наблюдался в случае лактонов. Второй фактор связан с изомерией! относительно С(О)—N связи (см. разд. 9.9.2.1). Более стабильной для лактамов с малым размером кольца (от трех- до восьмичленных) является цис-(Е)-форма, в то время как транс- (Z)-конфигурация преобладает в случае лактамов, содержащих кольца с числом звеньев более девяти. В то время как различия в структуре изомеров незначительно влияют на частоту колебаний С = О, влияние на частоту колебаний N—Н существенно больше. Так, в случае от трех до восьмичленных лактамов, характеристическая частота поглощения N—Н близка к цис(Е)-ациклическим амидам, в то время как для макроциклических лактамов эта частота соответствует транс-(Z)-ациклическим амидам [163] (см. табл. 9.9.1), ИК-спек-тры более сложных лактамов, которые относятся к гетероциклическим соединениям, описаны в обзоре Катрицкого [162]. Более подробная трактовка ПК-спектров поглощения амидов и лактамов приведена в книге Беллами [163].
Несколько характеристических полос поглощения амидной группы обнаружено в ультрафиолетовой области в диапазоне 125—250 нм [107, 148]. Эти полосы маркированы (по мере уменьшения длины волны) как W, Ri, V;. R2 и Q. Полосы Ri и R2 обнаружены только в спектрах в газовой фазе, а на полосу Q (в вакуумной УФ-области) часто накладывается поглощение алкильных групп. Таким образом только полосы W и Vi представляют интерес для химиков-органиков; их характеристики приведены в табл. 9.9.2.
Что касается влияния заместителей, имеются лишь данные о том, что наличие N-метильного заместителя приводит к сдвигу поглощения в длинноволновую область с одновременным увеличением интенсивности полосы [164].
433
Недавно из данных по циркулярному дихроизму получена дополнительная информация об электронной структуре амидной группы нескольких оптически активных амидов. Эти данные находятся в соответствии с простым УФ-поглощением; при этом наблюдаются обе полосы поглощения W и Vi. Полоса W, которая часто имеет недостаточную интенсивность для обнаружения в УФ-спектре поглощения, надежно регистрируется этим методом
Исследования амидов методом ядерного магнитного резонанса рассматриваются в двух недавних обзорах [148, 150]. Представляют интерес два главных аспекта в ЯМР-спектрах амидов: во-первых, расщепление между магнитными ядрами 14N и соседними
Таблица 9.9.2. Ультрафиолетовые спектры поглощения амидов в растворе
Полоса Переход Коэффициент экстинкции, моль-1 -см-1 Длина волны, нм
W п-+л* 100 210—220 (а>
Vi л->л* 8000 170—188 (б>
Не очевидно Для третичных амидов из-за наложения других поглощений.
(°* Эта полоса сдвигается в область больших длин волн, когда рядом с амидной группой имеется л-электронная система.
протонами и, во-вторых, различные химические сдвиги одинаковых N-заместителей, обусловленные цис/трамс-изомерией относительно связи С (О)—N.
Наиболее распространенный изотоп азота 14N (99,6%) имеет электрический квадрупольный момент, так как его ядра имеют спин +1. К сожалению, скорость релаксации квадруполя ядра того же порядка, что и временная шкала ЯМР. В связи с этим расщепление между азотом и соседним протоном (/n-h 65 Гц) реализуется в виде широкого сигнала поглощения (10—100 Гц) промежуточной формы между триплетом 1:1:1, ожидаемым при полном расщеплении, и синглетом, наблюдаемым при отсутствии расщепления. Однако с помощью техники подавления спин-спино-вого взаимодействия можно сузить широкое поглощение до синглета. Поглощение, возникающее на ядрах I5N [спин -ф 1/2 и, следовательно, отсутствует квадрупольный момент], имеет более простой характер. Однако в связи с малой распространенностью изотопа 15N (0,37%) для его регистрации необходима специальная техника.
О. /Н (а) СК /СНз (а)
r/ (б) R/ ^СНз (б)
(70) (71)
Затрудненность вращения вокруг связи С(О)“"^ (см-разд. 9.9.2.1) делает неэквивалентными протоны (а) и (б) пер ных амидов (70) и метильные протоны (а) и (б) третичных а\
434
дов (71), что вызвано диамагнитной анизотропией карбонильной группы. Таким образом протоны Н(а) и Н(б) будут иметь различные химические сдвиги, если скорость вращения вокруг связи С (О)— N будет медленной во временном диапазоне ЯМР. В работе [150] рассматривается несколько методов, пригодных для отнесения химических сдвигов протонов Н(а) и Н(б). Достаточно сказать, что более экранированным из двух протонов считается протон, расположенный ближе к карбонильной группе [Н(а)], хотя это правило приобретает обратный смысл при использовании ароматических растворителей. Различные константы сппн-спино-вого взаимодействия ‘Н—ЧЧ и 15N—ЧЧ для ряда формамидов и ацетамидов приведены в виде таблиц в работе [148]. Типичный пример величин: для (70)—7н(а)-Н(б) 2 Гц и Лы-н <а) —95 Гц; для (71) — /н(а)-Н(б) < 0,1 Гц И J1SN-Н (а) Ч~1,2Гц.
При определении химических сдвигов амидов следует учитывать два специфических эффекта влияния растворителя. Первый эффект связан с превращением ассоциатов молекул амидов в мономеры при переходе от концентрированного раствора к разбавленному (подобно эффекту, уже описанному для ИК-спектров). Второй эффект, вероятно обусловленный комплексообразованием, выражается в значительном сдвиге сигналов N-метильных протонов в сторону сильных полей при использовании ароматических растворителей, например бензола [148, 150]. Важными областями применения ЯМР-спектроскопии являются, как уже отмечалось в разд. 9.9.2.1, определение высоты барьера вращения вокруг связи С (О)—N, определение соотношения цис, тушнс-изомеров и обнаружение других изомеров, возникающих за счет медленного вращения вокруг связей N—R и R—С (О). ЯМР нашел также применение при изучении комплексов амидов с кислотами Льюиса, при изучении протонирования и образования комплексов с ароматическими соединениями (см. разд. 9.9.2.3).
Характер фрагментации при масс-спектрометрии алифатических амидов имеют черты, типичные как для карбонильных соединений, так и для аминов [165, 166]. В случае первичных алифатических амидов важнейшим типом фрагментации является разрыв связи, расположенной в a-положении по отношению к карбонильной группе, с образованием нона (72) (т/е44) {схема (107)}. Оказалось, что этот процесс не зависит от того, происходит ли первоначальная ионизация за счет неподеленной электронной пары кислорода или азота. Многие первичные амиды дают также пик средней интенсивности с т/е 72, вероятно соответствующий иону (73), который образуется за счет разрыва связи С—С, находящейся в у-положении к карбонильной группе {схема (108)}. Как и в случае других карбонильных соединений, содержащих по крайней мере три атома углерода в алкильной цепи, перегруппировка МакЛафферти играет важную роль. Она заключается в миграции водорода от у-углеродного атома к атому кислорода карбонильной группы с последующим 0-разрывом
435
связи С—С, как показано на схеме (109). При этом образуется ион (74) (m/e 59), которому обычно соответствует наиболее интенсивный пик в спектрах высших гомологов первичных алифатических амидов.
5 // . г + + т
RjfC --------> R + [o=C=NH2 -<----> O=C-NH2] (107)i
(S.NH3 (72) m/e 44
+ • +
/° /°\
R-^-CH2CH2C\ ------>- CH2 NH2 (108)
nh2 ch2
(73) m/e 72
+ • OH
*- RCH=CH,2. + H2C=C\ (109)
nh3
(74) m/e 59
В случае вторичных и третичных амидов существует дополнительная возможность фрагментации боковой N-алкильной цепи. Эти соединения распадаются по направлениям, уже установленным для алифатических аминов. Первый тип фрагментации вторичных и третичных амидов основан на расщеплении связи у углерода, находящегося в a-положении по отношению к кислороду или азоту {схемы (110) и (111) соответственно}. Второй тип фрагментации связан с расщеплением связи у углерода, расположенного в p-положении к азоту {схема (112)}. Однако ионы (75) и (76) редко удается наблюдать, так как если заместитель R1 имеет атом водорода при а-, (3- или у-углеродных атомах, то протекает перегруппировка, включающая миграцию водорода и элиминирование кетена с последующим образованием ионов (77) и (78) соответственно {схемы (113) и (114)}. Такие превращения хорошо известны для вторичных аминов.
Сравнительно недавно показано, что первичным процессом распада бензамидов и нафтиламидов является отщепление ароматического атома водорода от молекулярного иона, в связи с чем пик А44— 1, как правило, достаточно интенсивен [167J. Исследования амидов специального строения, таких как N-цпклоалкил-амиды и стероидные амиды, проведены Джерасси с сотр. [looj.
Изучены также различные пати- и шестичленные лактамы и их N-алкильные производные UAe, н-Pr и н-Ви) [168]. В случае пирролидона и его N-метильных производных, кроме высших 436
/°
R1c/^
NHR2
> R1C=O+ + R2NH
(HO)
R \.+ * Я + к2
NH—CH2—P2 NH=CH2
(75)
•+/---------.. ? CH2
R1C NH—CHn—CH2—R2 - > r!c—NH | ^CH2
(76)
О
II + +
<>J-NH=CH2 -----> CH2=C=O + nh2=ch2
H2C-^H . (77)m/e30
(ill)
(112)
(из)
II +xCH2 +//CH2
9^TN\ ---CH2=C=O + HNH
Н2С-^ CH2
(78)ffl/e44
(114)
N-алкильных производных, происходит а-разрыв с последующей миграцией водорода и разрывом связи С (О)—N с образованием иона (79) {схема (115)}. Однако для высших N-алкильных производных преобладающим процессом является a-расщепление в N-алкильной боковой цепи с образованием иона (80) {схема (116)}.
437
9.9.2.3. Кислотно-основные свойства, способность к образованию водородных связей и комплексообразующие свойства амидов
В соответствующих условиях амиды ведут себя как слабые основания или как очень слабые кислоты. Таким образом, амиды можно рассматривать как слабые амфотерные соединения. Эти свойства амидов проявляются в способности к образованию комплексов за счет водородных связей как при самоассоциации, так и с другими донорами и акцепторами. Амиды легко образуют также комплексы с различными кислотами Льюиса и с солями металлов. Подробный обзор по всем этим свойствам (до 1970 г.) был составлен Хомером и Джонсоном [149].
Амиды являются более слабыми основаниями, чем другие аминосоединения, что связано, как уже указывалось, с делокализацией электронов вдоль цепи О, С, N в амидной группе. Это также делает атомы О и N потенциальными местами для протонирования, что приводит к образованию сопряженных кислот альтернативного строения (81) и (82). Атом азота аминогруппы обычно является более основным, чем карбонильный кислород, однако равновесие в этом случае может быть сдвинуто в сторону ©-сопряженной кислоты (81) за счет вклада канонических структур {схема (117)}. Криоскопические исследования показали, что даже в сильно кислых условиях происходит только монопротонирование. Что касается направления протонирования, которое было
О О
(81)
спорным вопросом, то теперь в основном установлено,_Д^ы^до-нируется атом кислорода [149, 169]. Наиболее убед_ ‘ ш ые. казательства этого факта были получены при псе соединений тодом ЯМР немеченых [149] и содержащих N [1 I ой в концентрированных кислотах. При этом сигн J ужнвался в
пы N-метилацетамидов и N-метилбензамидов „ с одНим
виде дублета (за счет спин-спинового взаимодействия
43B
Протоном при азоте) вместо триплета, ожидаемого при наличии группировки +NH2Me. Аналогичные заключения сделаны в случае первичных и третичных амидов, а также пиридонов-2 в концентрированной серной и фторсульфоновой кислотах. В пользу этих выводов говорят данные ИК- и УФ-спектров [149], в том числе данные по борфторидам отдельных амидов, полученных из алифатических амидов н безводной HF/BF3 [171]. Возможность образования N-сопряженных кислот продолжает рассматриваться для более разбавленных кислых растворов (когда быстрый обмен и сниженный барьер вращения вокруг связи С—N делают исследования методом ЯМР не однозначными [170]). Однако последняя интерпретация данных УФ-спектроскопии [172], анализ данных по скоростям обмена амидного протона при кислотном катализе [173] и по скоростям кислотного гидролиза амидов [174] (см. разд. 9.9.3.1) говорят в пользу преимущественного образования О-сопряженных кислот даже в разбавленных кислотах.
Хотя соли амидов можно получать с использованием безводной HF/BF3 [171], для значительного протонирования в растворе нужны умеренно или очень сильные кислоты, где требуется применение функций кислотности. Установлено также, что протонирование большинства амидов (первичных, вторичных и третичных) описывается особой функцией кислотности, обозначаемой символом Н:.. Функция Нй была вычислена для нескольких минеральных кислот с использованием замещенных бензамидов в качестве индикаторов. Обобщенные данные по водной H2SO4 и водной НС1 при 25 °C приведены Хомером и Джонсоном [149], а величины На для водной НС1О4 при 25 °C были представлены в работе [175]. Важной характеристикой величин На является их слабая зависимость от кислотности растворителя, что указывает на то, что степень связывания протона амидами с увеличением кислотности значительно ниже, чем для большинства других слабых оснований (например, первичные амины используются для определения Но). Эти различия вытекают в основном из очень сильного высаливания амид-катиона (83), которое можно качественно интерпретировать как доказательство того, что амид-катионы сильно сольватированы [176].
Н-—ОН2
S+ / р—Н-"ОХ R-cf Н——ОН2
\ч5+/Н---ОН2
N
'н*,,ОН2 <83>
Величины основности (р/(а) многих амидов были приблизительно рассчитаны для кислых водных растворов, исходя из величин На в точке 50% протонирования. Обобщение этих данных проведено в работе [149]; позднее были опубликованы значения
439
для замещенных ацетанилидов и N-алкилбензамидов Г1771 гг скольку в случае вторичных и третичных амидов отклонения °’ величин Ня, определенных с использованием первичных ТЛ как индикаторов невелики, значения рХа для всех трех соединении являются достаточно точными. Основность в вол растворе обычно лежит в диапазоне рХа от —03 до — (табж 9.9.3), при этом требуется 10-65% (по объему) H9SO для 50% протонирования. Ацетамид [рХа — 0,35 т е 50°/ пост/ нированпя в примерно 12% (по объему) Н25О4]’-Одно из самых" сильных оснований, тогда как бензамид (рХа—1,5 т е 50°/ ппо тонирования в примерно 35% H2SO4), и ацетанилид'(р/Са—°1,45)" как и ожидалось, слабее. Наличие N-алкильных заместителей обычно увеличивает основность за счет индуктивного эффекта давая последовательность; первичные <Z вторичные~ третичные '
Таблица 9.9.3. Константы основности амидов
Амид -рАа Амид -Рха
Бензамид 1,54 Пропионамид 0,6
п-Метоксибензамид 1,22 Бутирамид 0,75
п-Хлорбепзамид 1,75 N-Метилацетамид 1,0
.и-Нитробензамид 2,10 N-Изобутилацетамид 0,42
п-Нитробензамид 2,3 N-н-Бутилацетамид 0,29
3,5-Динитробензамид 3,06 Ацетанилид 1,43
N-Метил бензамид 1,5 4-Г идроксиацетанилид 0,89
N-Трифторэтилбензамид 3,13 4-Хлорацстанилид 1,63
N-Диметилбеизамид 1,2 4-Нитроацетанилид 2,16
Ацетамид 0,35
Основность амидов определяли в неводных растворителях (главным образом в уксусной кислоте) с помощью титрования и метода индикаторов. Особенно важны эти измерения для алифатических амидов, которые не дают подходящих УФ-спектров. Показано [178], что уравнение (118) описывает связь значений рЛа в уксусной кислоте и в воде для многих органических основании, включая амиды. Однако, Хомер и Джонсон [149] считают, что уравнение (118) завышает значения p/G (Н2О) примерно на , . Методика титрования была применена к лактамам, у которых основность возрастает по мере увеличения размера коль“^’ остается постоянной (р/<а(Н2О) ~ 0,52) для лактамов с в°сь^ ными и с большими циклами [179]. Основность амидо Р хлорная — уксусная кислота определена также непрямым > с помощью индикаторов. По этой методике находят сан0 диссоциации, из которой можно определить рла(н2о), в работе [149]. (118)
РАа (Н2О) = РХа (НОАс) 4" >
Систематическая оценка влияния заместителей осн° с помощью принципа линейного соотношения св 44()
широко не проводилась, за исключением бензамидов и анилидов. Показано f 149J, что константы диссоциации для алкиламидов в уксусной кислоте приблизительно коррелируют с а*(р* — 1,2), хотя отмечен и ряд существенных расхождений. Значения рК;> мета- и пара-замещенных бензамидов (р —0,78) [149], N-этил-бензамидов (р— 0,95) [180] и ацетанилидов (р— 1,35) [177а] лучше согласуются с о, чем с сгН несмотря на О-протонирование, однако рКа М-2,2,2-трифторэтилбензамидов лучше коррелируют са+(р — 0,80) [180].
В противоположность исследованию основных свойств, имеется гораздо меньше количественных данных по способности амидов действовать как слабые кислоты. Для значительной ионизации требуется использование концентрированных щелочных растворов, к которым приложима функция кислотности Н-, характеризующая перенос протона; основные трудности при измерении воз-Таблица 9.9.4. Константы кислотности амидов
Амиды Р*а Амиды рКа
Бензамид > 19,0 Бензанилид 16,53
Ацетанилид 17,59 я-Нитробензанилид 15,85
Фенилацетанилпд 17,31 ч-Бромбензанилид 15,73
Формамид 17,20 Форманилид 15,56
Феноксиацетамнд 17,20 Трихлорацетанилид 9,98
п-Бромбензамид 17,13 Трифторацетанилид 9,51
никают из-за отсутствия подходящего УФ-поглощения у алкиламидов. Однако Ке для равновесия (119) в изопропаноле была измерена [181] при использовании в качестве индикатора 4-нитро-дифениламина. Показано [182], что из этих данных можно получить приближенные значения p/G с помощью соотношения p/G = — 18,31—p/Се- Отдельные величины, полученные таким способом, приведены в табл. 9.9.4, наряду с данными для трихлор- и трифторацетанилида, полученными с помощью более надежного потенциометрического метода.
R'CONHR2 + изо-РгО' азо-РгОН + R'CONR2 (119)
В соответствии с их кислотно-основными свойствами амиды Действуют как доноры или акцепторы протона при образовании водородных связей. Важным проявлением способности к образованию водородных связей является самоассоцпация в концентрированных растворах, когда каждая молекула амида является донором и акцептором при возникновении водородной связи (только Для первичных и вторичных амидов). Этот факт можно легко наблюдать, так как происходит уменьшение Vnu и vc=o при переходе из газовой в конденсированную фазу [183]. Как в случае протонирования н образования комплексов с металлами, умень-
441
шенпе \’с=о является доказательством того, что атом кислооол карбонильной группы действует как акцептор водорода; это пол тверждается для формамида и N-метилацетамида сдвигами Д" ДМР-спектрах на ядрах 14N и >Н [184]. Возможно образование двух типов структур: линейного полимера (66), например из N-ме-тилацетамида, и циклического димера (65), обычного для лактамов и некоторых других амидов. Для образования циклического димера требуется ^«с-конформация N—Н и карбонильной группы относительно связи С—N, в то время как преобладающей формой за исключением лактамов, является транс-. Хотя известно [153]' что только при наличии очень объемистых N-заместителей образуется значительная доля ^«с-изомера, Девис и Томас [185] показали, что тип ассоциации не обязательно определяется преимущественной конфигурацией свободного амида; это означает (как и в случае протонирования), что доля ^uc-формы в равновесии заметно увеличивается при образовании водородной связи. Таким образом, представляется затруднительным выдвигать предполо-жения о типе и степени образования комплекса (за исключением прочных ассоциатов из ^нс-лактамов). Имеются исключения из правил, например N-метилтрихлорацетамид и N-метилацетамид, оба транс-изомеры, образуют соответственно циклический димер и линейный полимер [185]. Кроме того, образование внутримолекулярных водородных связей (84) может снижать олигомеризацию вторичных амидов, имеющих полярные (например, F, МеО) «-заместители [156]. Тем не менее многие константы ассоциации (вместе с термодинамическими данными) измерены и полученные результаты обобщены в обзоре [149]. Подобное образование водородных связей невозможно для третичных амидов, однако предполагается [186], что затрудненность внутреннего вращения в этих соединениях происходит из-за дпполь-дипольного взаимодействия (67), которое аналогично взаимодействию амидов с ароматическими растворителями [187].
О \\ . С—N
F3BO Me К+ /
С—N
Н Me (85) амиды образуют
Н X (84) Хорошо известно также, что ,
другими соединениями, способными образовывать связи. Взаимодействие с фенолами (в особенности), аминам тиолами изучено количественно; константы связывания прав в обзоре [149]. Амиды образуют также водородные свя^” СТВует хлорметаном; процесс конкурирует с димеризациеи “ пречес^.венно связыванию амидов с другими веществами [i»oj. кИС.
степень образования водородной связи можно связать фе.
лотностью донора, так и с основностью акцептора, лапр - р>
комплексы с водородные
442
иолы > амины, при этом Арнет с сотр. [189] обнаружили удовлетворительно линейную зависимость между энтальпией образования водородной связи при взаимодействии п-фторфенола с несколькими амидами и их тепловой протонирования HFSO3; авторы, однако, предупредили, что указанная зависимость является эмпирической [189]. В противоположность поведению амидов в качестве акцепторов водородной связи, много меньше известно о способности амидов выступать в качестве доноров водородной связи, за исключением констант связывания N-метилацетамида с несколькими О, N и S-акцепторами [190].
Хорошо установлено, что амиды и лактамы легко образуют комплексы с кислотами Лыоиса и солями металлов [149]. Спектральные свойства говорят о предпочтительности координации с атомами О, а не с N. Комплексы И амидов (первичных, вторичных и третичных) с BF3, ВС13 и TiCl4 описаны Жераром и др. [191]. Фактическое отсутствие водородной связи (на это указывает постоянство vnh в спектрах в растворе и в нуйоле) по сравнению со свободными амидами указывает на образование О-комплекса. Кроме того в ПМР-спектре комплекса ДМФА— BF3 обнаружено два отдельных сигнала метильной группы, возникших в результате взаимодействия с формильным водородом, как и ожидалось для структуры (85). Получены также аддукты BF3 с замещенными бензамидами, е-капролактамом и N-метил-е-капролактамом, и сделано заключение, что и они представляют собой О-комплексы [149]. Много внимания уделено исследованию взаимодействия иода с третичными амидами в СС14 [192]; понижение vc=o рассматривалось, как доказательство координации по атому кислорода. Это противоречило, однако, данным по комплексам с BF3, где наблюдалось незначительное изменение в vc=o, а уширение дублета N(CH3)2 в ПМР-спектре Ь1,М-диметилпропионамида с 12 с СС14 указывало на дополнительные слабые взаимодействия. Опубликованы также константы равновесия и термодинамические данные для аддуктов этих третичных амидов с иодом [192]. Эти параметры не зависят от структуры амида и значения К = 2,91 моль-1, АН = —15,5 кДж-моль-1, и AS = = —43,5 Дж • K~f • моль-1, для ДМФА — 12 являются типичными. В обзоре [149] описано также большое количество комплексов амидов с металлами (главным образом третичных амидов и N-алкиллактамов). В образовании комплексов могут участвовать как переходные, так и непереходные металлы, включая соли щелочных металлов. Как правило, в ИК-спектрах обнаружено понижение vc=o по сравнению со свободными амидами, что рассматривается как доказательство О-координации. Это подтверждается недавно опубликованными данными о том, что соли Li+ повышают барьер вращения вокруг связи С (О) —N диметнлацетамида (ДМА) [193], а также наличием сдвигов в 14N- и 'Н-ЯМР-спектрах комплексов ZnCl2 с формамидом и N-метнлацетампдом [184]. ДМА, по-видимому, является универсальным лигандом, и Бул с сотр.
443
[194] получил и охарактеризовал его комплексы с 14 различны» металлами, например [Сг(ДМА)6] (С1О4)3 и [7п(ДМА)2] Вг2. Дш также образует октаэдрические комплексы с Ni(II) и Сг(Ць [195а], а в случае Со(II) известны октаэдрические и тетраэдрические комплексы [1956]. Спектральные данные об искажениях в последних могут указывать на слабую дополнительную N-кооп-дпнацшо. Подробно изучено комплексообразование лактамов (главным образом в-капролактама, у-бутиролактама и N-метил-у-бутиролактама) [196]; в ряде случаев получены гексакоордини-рованные октаэдрические комплексы. N-Метил-у-бутиролактам давал также комплексы со многими непереходными металлами включая соли щелочных металлов [196]. Большинство комплексов легко разлагается под действием воды, однако диметилформ-ампдопентаамин-Со(Ш) достаточно устойчив, так что происходит легкий, катализируемый щелочью, гидролиз амидного лиганда [197]. Эта реакция близка по существу к ферментативному гидролизу пептидов (см. разд. 9.9.3.1).
Э.9.2.4. Структура и спектры имидов
Из данных рентгеноструктурных исследований [198] известно,
что кольцо сукцинимида имеет плоское строение (за исключением водородного атома при азоте, который отстоит на 0,015 нм от плоскости), как показано в (86) (указаны углы и длины связей в нм для кристаллического сукцинимида). В то же время по данным диффракции электронов [199] ациклический имид —диацетамид имеет ^ис-транс-конфигурацию с двугранным углом около 36° между двумя карбонильными группами, как показано в (87) для водного диацетамида. Длины связей С=О и С—N близки
для обеих структур (0,121 и 0,140 нм соответственно), а кратность связи С (О)—N составляет примерно 1,38, т. е. эта связь имеет меньшую двоесвязанность, чем в амидах. Это согласуется с тем,
что в имидах неподеленная пара электронов азота распределяется между двумя карбонильными группами. Найденная кратность связи находится в хорошем соответствии с предсказанием, основанном на теории валентных связей, согласно которой кратность связи в трех основных резонансных формах {схема (119а)}, имеющих плоское строение, составляет величину около , .
iiuc-mpanc-(07)
444
Как к в случае амидов, частично двоесвязанный характер связи С (О)—N приводит к возникновению конфигурационных изомеров. Тремя возможными изомерами (названными по положению карбонильной группы относительно связи N—R2 и имеющими плоское строение) являются цис-цис (88а); цис-транс (886) и транстранс (88в). К. сожалению, еще не проведено детальное изучение барьеров вращения, однако примеры изомеров каждого типа были обнаружены с помощью измерений ИК-спектров и дипольных моментов [7, 8]. транс-транс-Форма является наименее вероятной, так как расстояние между двумя атомами кислорода составляет всего 0,250 нм. Однако диациламин все же существует в транстранс-форме в твердом состоянии, но превращается в более стабильную цис-транс-форму в растворе [200]. N-Ацетиллактамы также имеют ^пс-транс-конфигурацию, в то время как цис-цисформы являются единственно возможными для имидов с малыми кольцами. Изомеры с ^«с-^мс-конфигурацией (например, сукцинимид) имеют очень низкий дипольный момент (около 1,5D), так как моменты связей сильно компенсируют друг друга, оставляя незначительный момент вдоль связи N—R (88а). В случае шестичленных колец, где угол между двумя копланарными карбонильными группами уменьшается, большой вклад вдоль оси связи N—R2 вносит момент связи карбонильных групп, который увеличивает общий дипольный момент до величины около 2,6D. Имиды с Ч^с-транс-конфигурацией имеют большую величину дипольного момента (около 3,0D) в направлении, обозначенном на формуле (886), так как моменты связей вдоль оси N — R2 сильно компенсируются, оставляя остаточный компонент от одной карбонильной группы, перпендикулярный к оси N-—R2. Некоторые величины дипольных моментов приведены в виде таблиц в работах [7, 8]. В противоположность амидам растворитель может заметно влиять на величину дипольного момента имидов [201], хотя причины такого различия в поведении неясны.
Как и в случае амидов, влияние растворителя является существенным при определении характеристических частот поглощения
445
имидов в ИК-области, так как имеется сильная тенденция разованию водородных связей как в концентрированных Я К °6' рах, так и в твердом состоянии. В разбавленных раствора^0’ имиды главным образом находятся в мономерной форме н хГДе дается, как правило, уменьшение частоты поглощения что ан °' гично эффекту, наблюдаемому для амидов. Основные характ °' стические частоты в ИК-спектрах имидов (табл. 9.9 5) свяч ' с валентными колебаниями N—Н (3000—3500 см-1)' для неза^ щенных соединений и валентными колебаниями С=О групп6' (1700—1800 см-1) для всех классов имидов. В концентрирован' ных растворах наблюдается широкая полоса поглощения N—Ц В твердом состоянии появляется две полосы поглощения в этой
Таблица 9.9.5. Характеристические частоты поглощения имидов в инфракрасной области
Характеристика полос: (а) —сильная интенсивность; (б) средняя интенсивность-
(в) — резонанс Ферми.
Имиды Частоты, см-1
валентные колебания N—H(a) валентные v(6) сим колебания С=О v(a) уантисим
Ациклические Циклические Пятичленные 3280—3200 1740-1720 1720-1700
(свободная) 3430 1750 1730
(связанная) Шестичленные 3190 3080 (в) 1770 1700
(свободная) (связанная) Фталимид (свободная) 3390 1740 1700 1780—1770 1730 1670 1750-1710
области, одна из которых приписывается резонансу Ферми двух других более низких полос спектра [8, 163]. Однако в разбавленных растворах колебания N—Н регистрируются в виде острой интенсивной полосы поглощения между 3200 и„3425 см . °тли чие от амидов полоса колебаний карбонильной группы в регистрируется в виде дублета. Это наблюдается даже_в у
фталимида, в котором обе карбонильные ГРУ™7Ы нах л пиблета ной плоскости и в одинаковом окружении. ОбРаз°взн^ приписывают механическому взаимодействию двух Р групп, которое соответствует симметричным (более низ.
тота, средняя интенсивность) и анТ11СИММе?риЧН^рхяНИЯМ [163]. кая частота, очень сильная интенсивность) *ол копланарно-Разность частот этих двух линии зависит от степе аЯ
сти карбонильных групп и от угла межд5е разность частот составляет около 70 см , одна у
446
между карбонильными группами или увеличение степени отклонения от копланарности ослабляет взаимодействие, а следовательно, уменьшает и расстояние между линиями. Так как различия в частотах зависят от степени взаимодействия и от влияния заместителей, то трудно делать общие выводы о положении этих полос поглощения, и, следовательно, значения, представленные в табл. 9.9.5, являются очень приблизительными. Информация об ИК-спектрах отдельных соединений! может быть найдена в недавно опубликованных обзорах [7, 8] и в монографии Беллами [163].
Ультрафиолетовые спектры имидов, как и в случае амидов, содержат полосу поглощения средней интенсивности между 176 и
Таблица 9.9.6. Ультрафиолетовые спектры поглощения имидов
Имиды ^макс’ 11 м 8-10-3
Ациклические 178 8
N-Ме-ациклические 207-216
Насыщенные циклические 190-205 12-14
Малеимпд 216 15
223 13
Фталимид 217 40
230 17
238 И
N-Ацетиллактамы 216-219 9-11
225 нм, обусловленную, по-видимому, лл-первходом в двойной связи карбонильной группы. Типичные примеры представлены в табл. 9.9.6; данные по другим соединениям представлены в виде таблиц в двух обзорах [7,8]. Приблизительное удвоение коэффициента мольной экстинкции е по сравнению с амидами приписывается присутствию двух карбонильных групп. Поглощение, обусловленное и—>-л*-переходами, пока не обнаружено. В обзоре [8] приведены кривые дисперсии оптического вращения ряда оптически активных имидов (например, N-замещенных фталимидов, имидов винной и малеиновой кислот).
Как и в случае амидов, на ЯМР-спектры незамещенных имидов оказывает влияние ядерный квадрупольный момент ядер 14N. В связи с этим сигнал N—Н часто широк (около 9 Гц), и иногда его трудно обнаружить [8]. В случае фталимида наблюдается также спин-спинсвое взаимодействие протона N—Н и ядер 15N и 13С (7(15N—’Н) « 93 Гц, /(13С—’Н) 141 Гц) [202]. ПМР-спектры имидов исследовались недостаточно систематически, однако можно отметить некоторые тенденции. Например, константа спин-спино-вого взаимодействия между соседними метиленовыми протонами в циклических имидах составляет 18,5 Гц для пятпчленных колец, но уменьшается до 16,4 Гц для шестнчленных колец. Кроме того, было отмечено, что химический сдвиг протонов в положении 2 и
447
3 циклических имидов, таких как сукцинимид и малеимид, смен на 0,33 млн-1 в сторону сильных полей по сравнению с соотве вующимп сдвигами в циклических ангидридах. Вместе с тем величину химических сдвигов не влияет замещение ^положен”” алкильными группами. Однако введение N-аллильного заместите” вызывает уменьшение сдвига на 0,17 млн-1 [203]. Ароматически” растворители, такие как бензол, вызывают смещение химически сдвигов протонов в сторону сильных полей примерно на 1 Млн_* [204]. Несколько других примеров ЯМР-спектров имидов привел?, но в обзоре [8].
Данные о масс-спектрометрии незамещенных имидов крайне скудны. Среди немногих изученных примеров можно отметить фталимид и малеимид. В обоих случаях распад молекулярного иона протекает по двум направлениям с отщеплением либо HNCO либо СО2 (схема 120) [205]. Такое необычное отщепление С02’ которое отмечается также и в случае N-замещенных фталимидов [205, 206], как полагают, происходит за счет переноса кислорода
через кольцо, а не в результате изомеризации до изоимида перед отщеплением. Другие исследования касались главным образом N-алкилзамещенных имидов, где кроме обычной фрагментации алкильной боковой цепи (ср. амиды, разд. 9.9.2.2) происходит интересный двойной перенос водорода в молекулярном ионе имида с последующим элиминированием радикала олефина (схема (121)} [1686]. В случае N-замещенных малеимидов наряду с обычной фрагментацией боковой цепи наблюдался разрыв имидного цикла с потерей СО, Н и N [207]. Единственная работа по ациклическим имидам посвящена диацетамиду, который под действием электронного удара подвергался скелетной перегруппировке [208].
О+’
—> [АТ — 43]+* + HN=C==O
I > ~
—> [Af - 44]+* + СО2
О
(120)
(R = Н или R—R = С4Н4)
IV^H2 -----------> Г\Н + CH3=CHCHR
О>+ I +он
t^CHR
По СВОИМ КИСЛОТНО-ОСНОВНЫМ свойствам ИМИДЫ ЗНаЧИТиеЛпрЛ0!<а' пчаются от амидов, что является следствием возможной g зации неподеленной пары электронов азота на дополн
44g
карбонильной группе. Так, имиды, как правило, являются более сильными кислотами (рЛ'а 8—10) [8] и, вероятно, более слабыми основаниями, чем амиды, хотя прямые доказательства этого заключения отсутствуют. Атом водорода при азоте в имидах может быть замещен на металл. Образующиеся соли гидролизуются водой, поэтому их получение и реакции должны проводиться в безводных условиях. Фталимид калия является, конечно, наиболее известным примером такой соли, но известны также соли меди, ртути и серебра [8]. Наряду с простейшими солями имиды образуют также комплексные соли с переходными металлами. Например, сукцинимид дает комплексы с медью и щелочными металлами (М) общей формулы (C6H4NO2) СиМ • пН2О, в то время как фталимид вместе с аминами образует смешанные комплексные соли с кобальтом [8]. Вероятно, во всех таких комплексах с переходными металлами имид выступает как бидентатный лиганд. Другой, менее изученный тип комплексов — комплексы сукцинимида с фенолами или пероксидом водорода [209].
9.9.2.5. Диметилформамид и диметилацетамид как растворители
.\Т,\Т-Дпметилформамид (ДМФА) и N.N-днметплацетамид (ДМА) относят к классу растворителей, известных как биполярные апротонные растворители [210]. Этим соединениям свойственны высокие значения диэлектрической проницаемости (например, для ДМФА 37, 6), и поэтому они являются подходящими растворителями для целого ряда полярных соединений. Кроме того, в них растворимы и многие неполярные вещества, что обеспечивает возможность проведения гомогенных реакций для большого набора реагентов и субстратов. Вместе с тем, поскольку эти вещества являются апротонными растворителями (т. е. они не образуют водородные связи с анионами, как, например, этанол), то реакционно-способность растворенных анионов обычно много выше, чем в протонных растворителях. Наряду с этим, поляризуемые заряженные переходные состояния более сольватированы в биполярных апротонных растворителях, чем в протонных растворителях. В результате этого бимолекулярные реакции с участием анионов протекают значительно быстрее в ДМФА и ДМА, чем в протонных растворителях [210].
К другим достоинствам ДМФА и ДМА относятся: их полная смешиваемость с водой, что позволяет легко выделять многие органические продукты; высокая термическая стабильность и высокие точки кипения, что дает возможность, когда это необходимо, проводить реакции при высоких температурах (150—200 °C) [211]. Например, ДМФА и ДМА оказались очень удобными растворителями при нуклеофильном замещении F_, CN-, NO? карбанионами и анионами азота, а также при присоединении галогеповодородов к олефинам [211]. Однако нужно иметь в виду, что ДМФА и ДМА
15 Зак. 395
449
не полностью инертны, а могут реагировать с сильноэлектрофиль ными реагентами (например, с оксихлоридами фосфора^ q разд. 9.9.3.8). Недавно показано, что взаимодействие ДМфД КМ’ растворителя с более слабыми электрофильными реагентами кими как арилгалогениды, приводит к нежелательным побочна продуктам [212J.
9.9.3. РЕАКЦИИ АМИДОВ
Амиды являются многоплановыми органическими соединениями так как все три атома в цепи О—С—N потенциально реакционноспособны. Это частично связано с делокализацией л-электронов по цепи О—С—N и с тем, что структура основного состояния является гибридом двух резонансных форм (89) и (90) (см. разд. 9.9.2.1). Хотя многоплановость часто означает сложность, можно достичь упрощения, признав, что большинство реакций амидов относятся к двум группам. Первая группа реакций включает нуклеофильную атаку атома кислорода (или иногда атома азота) на положительно заряженные или нейтральные реагенты. В этом случае более вероятной реакционноспособной частицей является резонансная форма (90). Ко второй, менее распространенной группе реакций, относится нуклеофильное присоединение к амидной карбонильной группе формы (89). Большинство других реакций амидов, такие как дегидратация, элиминирование, дезаминирование и др., является продолжением этих двух процессов.
R1C/ + \nr2r3 4NR2R3
(89) (90)
Г /°’ 1
L X\r2.
(91)
Электрофильные свойства атома углерода карбонильной группы ослаблены, так как л-электроны делокализованы по цепи О С— ~ В связи с этим амиды менее реакционноспособны, чем сложны^ эфиры, например в реакциях присоединения по карбонильной груш пе. Для протекания таких реакций обычно необходимо усилен» поляризации связи С—О путем протонпрованпя или комплексоо разования у атома кислорода, как, например, в случае гидролиз или реакции с металлорганнческими реагентами. Поскольку ам!1Д являются относительно слабыми основаниями, то при протонир°® нии или комплексообразовании у атома кислорода (см. разд. 9.9.^- / они так и останутся слабыми нуклеофилами, однако атом кисл рода будет наиболее нуклеофильным местом в молекуле. Этим ° ясняется, почему нейтральные амиды реашруют только с на',б0‘у!е. сильными электрофильными реагентами, давая сначала О-за^
450
щенпые производные. Хотя из реакционных смесей обычно выделяют N-замещенные соединения, их возникновение обычно связано с перегруппировкой О-замещенных производных. Следовательно, N-замещенные соединения представляют собой термодинамически устойчивые продукты. Прямое замещение по N происходит только в особых условиях: под действием сильных электрофильных реагентов (таких как диазоалкапы) или когда реакцию проводят в присутствии сильных оснований, где активной частицей является щелочной коныогат амида (91).
Для подробного ознакомления с общими реакциями амидов следует обратиться к обзору [102], перегруппировки рассмотрены в работе [213], фотохимия — в работе [107], а влияние заместителей н соседних групп изучено в работе [214]. Реакции имидов, во многих отношениях близкие к реакциям амидов, рассмотрены в обзорах [7] и [8].
9.9.3.1. Гидролиз и сольволиз
Амиды обычно гидролизуются с разрывом N-ацпльной связи; при этом регенерируется исходная карбоновая кислота и амин {схема (122)}. Поскольку первой стадиен гидролиза является нуклеофильное присоединение по карбонильной группе, а амины (в особенности анионы аминов) являются плохой уходящей группой, то становится понятным, почему эти реакции часто протекают очень медленно. Например, чистая вода инертна по отношению к амидам, и многие амиды могут быть перекристаллизованы из этого растворителя. Гидролиз протекает гораздо легче как в щелочных условиях, где имеется сильный нуклеофил НО-, так и в кислых условиях, где протонированне субстрата (см. разд. 9.9.2.3) способствует нуклеофильной атаке водой и отщеплению амина. В связи с этим интересно, что четвертичные соли амидов R'cONRjX легко гидролизуются холодной водой [215]. Гидролиз лактамов имеет много общего с гидролизом амидов, но, как правило, он протекает легче из-за напряжений в цикле. Амиды и лактамы реагируют не только с водой, но и с другими нуклеофильными растворителями, а именно с аминами, спиртами, гидроксиламинами, карбоновыми кислотами и др. Например, при взаимодействии со спиртами в присутствии кислых или основных катализаторов образуются сложные эфиры {схема (123)}. Ни один из этих вариантов сольволиза не изучен так же хорошо, как гидролиз, и за некоторым исключением эти реакции не имеют большого значения для синтеза.
Н+ или но" R’CONR2R3 + H2O---------> R'CO2II+ R2R3MI (122)
Н+ или НО"
R‘CONR2R3 + R<OH —-------> R'CO2R'+ R2R3NH (123)
За исключением одного примера, гидролиз амидов в щелочном растворе сопровождается разрывом N-ацильной связи, однако
15*
451
амиды гидролизуются труднее, чем соответствующие сложные эЛ ры, так как анион амина R2N“ является плохой уходящей группщ В связи с этим гидролиз неактивированных соединений, таких к алкиламиды, требует температуры около 100 °C. Механизм таю/ реакций был подробно изучен, детали этих исследований рассмо/ рены в работах [102, 216]. Общепринято, что гидролиз, катализ/ руемый НО-, является многостадийным процессом [схема (124)]' в’ котором стадия распада тетраэдрического интермедиата (ста’ дня k2 или £3) до продуктов является стадией, определяющей скорость процесса. Характерными признаками реакции являются быстрый конкурентный обмен между первичными и вторичными амидами и растворителем [следовательно, образование (^) тетраэдрического интермедиата является быстро обратимым], а также наличие члена второго порядка по НО- {т. е. скорость = [амид] . • (я1 [НО-] + (к2[НО-]2)} для анилидов и амидов с электроно-оттягивающимп заместителями. В ряде случаев тетраэдрический интермедиат был выделен, и с помощью ЯМР показано, что он относительно устойчив в углеводородных растворителях. В дополнение к катализу второй группы НО-, распад тетраэдрического интермедиата для нескольких анилидов подвержен общему кислотноосновному катализу, согласно которому протонирование отщепляющейся аминогруппы [как, например, в (92)] является кинетически существенным. Важным недавним достижением является постулат Делоншампа [217] о стереоэлектронном контроле при образовании и расщеплении тетраэдрического интермедиата, что может объяснить некоторые давно известные аномалии по обмену 18О {схема (125)}. Предположив, что конформационные свойства реагирующих веществ сохраняются в тетраэдрическом интермедиате (93) и что быстрое отщепление любого заместителя из (92) требует ан-тппериплапарного расположения орбиталей неподелениых пар в двух других заместителях, Делоншамп успешно объяснил отсутствие конкурентного обмена 18О при щелочном гидролизе третичных амидов и третичных лактамов. Хотя включение 1 О в ре агирующие вещества возможно через (94), превращение (94) (94), вероятно, протекает медленнее, чем распад (93) до реагиру^ ющих веществ или продуктов. Существенно, что предположение конкурентном обмене 18О в случае вторичных амидов 1глав *в образом (Z)-конфигурация], но не в случае вторичных лакта' [главным образом (£)-конфигурация], было подтверждено экс риментально.
Как правило, скорость щелочного гидролиза амидов незнач!^ тельно возрастает при уменьшении электронной плотност! ацильном углероде, но при введении объемистых групп скор гидролиза падает (например, в случае О-бензамидов). Скор реакции первою порядка (т. е. скорость = kl [амид] [НО J 11М впчных алкилкарбоксамидов согласуется с модпфнцпровф-уравнением Тафта, в котором р = 4-2,08 и S(стерическое) । ’ 1218]. Аналогичные данные для алкилкарбоксанилпдов хуже
452
О’
R—С—NH2
1 О'
н2о
н°-|| («г)
Г 1 О' [но-] к, |
rconh2 ч~* r—с—nh2 rco; + NH3
(*~l) он
It (*з)
О’
R—С—NH3+
О'
! // , ,
•*- R1—С + R2R3NH
О’
(125)
(124)
r1—c—nr;
i a X
о H—В’
(B=H2O или основание)
(92)
релпруют с о*, что гипотетически приписывается дополнительному стерическому ингибированию резонанса в анилидном фрагменте [219]. Электронооттягпвающие заместители должны способствовать НО- катализируемому гидролизу по реакции второго порядка, однако Бихлер и Тафт [219] предположили, что эго направление не является важным для незамещенных алкплкарбоксампдов лаже в сильных щелочах.
Менее изучен щелочной гидролиз лакзамов, однако отмечены значительные отличия поведения лактамов от соответствующих амидов. Как правило, лактамы гидролизуются легче из-за угловых напряжений в цикле; относительные скорости гидролиза N-метил-
453
n-нитроацетанплида и N-n-нитрофенил-б-, у- и р-лактамов
1, 15, 14 и НО соответственно [220]. Однако в отличие от ацета лидов в случае N-ариллактамов наблюдается только зависимо^ первого порядка от НО~ даже при высоких значениях pH Что пСТь вело к заключению [220], что образование тетраэдрического инт И м'едиата всегда является стадией, лимитирующей скорость процесса Отсюда следует, что как образование, так и распад тетраэдриче ского интермедиата, должны ускоряться за счет углового напряжения. р
Устойчивость многих амидов и лактамов к щелочному гидролизу стимулировала поиски мягких методов для применения в синтезе* Наилучшие результаты для большинства амидов получены при использовании сильно нуклеофильного НО2 в водном NaOH при нагревании при 50 °C в течение 1—2 ч; выход продукта составил 80% [221]. Только в случае третичных амидов расщепление проводят при комнатной температуре влажным трет-ВнОК [222]. В связи с этим важно отметить факт возрастания скорости расщепления за счет образования хелатов с металлами, что, вероятно, важно при ферментативном катализе. Найдено, например, [223], что НО_-ка-талпзируемое отщепление N-ацпльного остатка в комплексах ДМФА-(амин)5-Со(III) протекает в 104 раз быстрее, чем в случае координационно не связанного ДМФА. Это ускорение полностью связано с энтропийным фактором и активацией за счет координации кислорода карбонильной группы. Однако ускорение гидролиза Н,И-ди (пиридил-2-метил) амидов (95) приписывают координации атома азота с СиСК [224].
При кипячении N-м-фенилазобензилацетамидов и родственных соединений в щелочном КОН происходит разрыв N-алкильнои связи с образованием ацетамида (37%), что является единствен ным исключением для процесса расщепления N-ацильнои свяо
_______________ _п ЮО °C обычно . V iv * j к-'_i kjkj
Несмотря на многочисленные 216], механизм этих реакций остается предметом сследуется вплоть до настоящего времени. Существу сопряженную кислоту г ____________________________________„,.АПП1’ТП В
в щелочных условиях [225].
Амиды гидролизуются также при нагревании в водных Рв^н0 рах минеральных кислот при температуре около 122 - " кт с расщеплением N-ацильной связи. Несмотря на многочислен^ работы [102, 216], механизм этих реакций остается предмет дискуссии и исследуется вплоть до настоящего времени. Существу^ ряд важных фактов, указывающих на то, что стадиен, лпмптиру^ щей скорость, является атака Н2О на сопряженную кислоту амида. К ним относятся: существование максимумов скоро а. концентрированных кислотах, связанных с полным протон»Р х пнем субстрата; инверсия изотопного эффекта в дейтернров
454
тстворителях В области максимумов скоростей; отсутствие элект-онного (но не стерпческого) влияния заместителей и нулевой или чеиь незначительный конкурентный обмен между бензамн-имп н растворителем. Продолжается дискуссия по вопросу о том вкпючает лп эта медленная стадия образование тетраэдрического |111Термедпата из О-сопряженпоп кислоты или происходит прямое |5Л,2) замещение N-соиряженной кислоты, или возможно имеет место комбинация и того в другого. Альтернативные направления приведены на схеме (126). Большая часть из недавно опубликованных фактических данных говорит в пользу направления, в котором участвует О-сопряжеиная кислота. К ним относятся; относительные скорости превращения N-ацетнлппрролпдниа и пиперидина [226], а также структурно близких амидов и эфиров имидатов [227]; наблюдения об очень незначительном, по измеримом конкурентном обмене 13О для бензамидов [228] и данные [229] о низких скоростях реакций N-сопряжепных кислот при исследовании N-ацетплтрпалкиламмонневых солей. Основной, но менее важный аргумент в пользу направления с участием N-сопряженных кислот— это, вероятно, статистический анализ зависимостей кислотности 60 алифатических и ароматических амидов [230]. В этой работе приводятся также данные о ранее опубликованных альтернативных интерпретациях корреляций функций кислотности. Хотя механизм может быть спорным, нет сомнений в том, что скорости гидролиза большинства амидов проходят через максимум, положение которого зависит от структуры кислоты и амида. Этот максимум обычно составляет около 2—5 М для ?/2SO4, что соответствует 100% протонпрования амида, и наблюдаемое уменьшение скорости связано с уменьшением активности воды. Сделано много попыток определить роль воды в этих реакциях путем корреляции скоростей реакции с функциями кислотности (Яд и Яо) и с близкими данными. Такие сопоставления, проведенные в работах [230, 231], ввиду их важности требуют выбора механизма реакции, однако во всех случаях предполагается наличие в высокой степени сольватированного переходного состояния.
ОН
R1—с'+ nr2r3
он
__Н2° . I 2 (быстро)
%------R -с—nr2r3 >
(медленно) i
OH.t
он
r’-c—nhr2r3 I
он
о
R1-/
n-r2
*3 hV •
R1—C—NHR2R3 ^-7H2° >
(медленно)
R’CO2H + R2R3NHt (126)
Rl 8+ I s+
H2O—c -nhr2r “ 11
о
455
Систематические исследования алкиламидов, бензамидов анилидов показывают, что кислотно-катализируемый гидролиз го раздо более чувствителен к стерическому, чем к электронному влиянию заместителей. Это согласуется с обоими направлениями представленными па схеме (126), так как в обоих случаях необходима регпбрпдизация sp2—^sp's карбонильного атома углерода Первичные амиды, как правило, более реакционноспособны, чем вторичные и третичные в силу стерических эффектов. Данные По алкпламидам, собранные в работе [218], указывают на хорошую корреляцию скоростей реакции с модифицированным уравнением Тафта, содержащим члены, характеризующие стерические и гппер-конъюгационные, но не полярные, взаимодействия. Аналогичный подход применен к замещенным бензамидам, и в случае мета-п парс* заместителей обнаружены очень небольшие изменения скорости, в то время как для орто-группировок отмечено большое влияние стерических факторов (например, #о-х’оДл-хо.- 0,03) [232]. В случае ацетанилидов наличие электроиооттягивающих заместителей в гшрй-положении лишь незначительно ускоряло гидролиз в менее чем 70%-ной по массе H2SO4 (р = 1,0) [233], однако гораздо более сильное ускорение происходило при более высокой кислотности вследствие смены механизма (см. ниже). Поскольку скорость кислотного гидролиза в большинстве случаев не зависит от структуры, наиболее важным параметром с синтетической точки зрения является максимум скорости. Это означает, что не всегда самым эффективным будет использование наиболее концентрированной кислой среды, так как, грубо говоря, максимальная скорость будет достигаться в том растворе, для которого значение Н.\ приблизительно равно рКа амида.
В особых условиях кислотный гидролиз может протекать по механизмам, отличающимся от бимолекулярного расщепления N-ацильной связи, о котором упоминалось выше. Так, в случае ацетанилида, имеющего электронооттягивающие заместители в парц-положении, обычный механизм становится мономолекуляр ным в более чем 70%-ной (по массе) H2SO4, что, по-видимому,требует образования N-сопряженной кислоты [233]. Эти реакции характеризуются возрастанием (а не падением) скорости при более высокой кислотности и большим ускорением за счет электроноот гягивающих заместителей в ла/щ-положении (р == + 5,0), [2 ' Другой механизм, включающий расщепление N-алкильной свя > на людался в H2SO4 в случае вторичных амидов, имеющих N-щ алкильные группы [234].’
В дополнение к механизмам, которые обсуждались вь,ше’ Г'*Д МОЖ,ет т„акже происходить по другим направлениям, в - . катал™ Г102И21гГнОТНЬ,й’ общ|,й основвой "ли текаот п гп ’ 2 Ни одна из этих реакций, как правило, Д.
шюннлспл Ул1аС обычнь1Х амидов, но они важны для таки- f лйза ZZ?blX СубстРатов- как N-ацетилимидазол, и для иД^ ’ р 1и катализируется внутримолекулярно. Гидрол*
456
. а„етИлимидазолия и 1 *аиетил’3-метилимидазолия является един-„рНным надежно установленным примером нуклеофильного ката-сТ „ (как следствие оптимальных свойств нейтрального имидазола л Ячестве уходящей группы). Существенно также, что гидролиз Вямого N-ацетплимидазола (т. е. нейтрального субстрата) осуще-твпяется путем общего кислотно-основного катализа [235]. Кроме с 0 исследования гидролиза N-ацетилимидазола указывают на То что склонность к протонированию отщепляющегося амина преобладает над реакциями амидов с водой и что соединения, имеющие плохо отщепляющиеся группы, показывают повышенную чувствительность к катализу.
Наилучшие примеры общего кислотно-основного гидролиза найдены При изучении внутримолекулярного катализа. Эти исследования, которые имеют отношение главным образом к ферментативным процессам, обсуждаются в обзорах [102, 216, 236]. Необычным признаком этих процессов наряду с необходимостью предпочтительной конфигурации взаимодействующих групп является то, что наиболее эффективный катализ обусловлен наличием соседних группировок (таких как карбоксильные и ноны имидазолин), которые выступают в качестве донора протона, и в качестве нуклеофила. Необходимость протона связана как с низкой реакционно-способностыо амидного карбонила к нуклеофильной атаке, так и (или) со слабой способностью к отщеплению аминного фрагмента. Таким образом, здесь речь идет о внутримолекулярном общем кислотном катализе в противоположность внутримолекулярному нуклеофильному катализу, который обычно наблюдается при гидролизе сложных эфиров. Одним из наиболее ярких примеров является тот факт, что полуамид фталевой кислоты гидролизуется в 105—106 раз быстрее, чем бензамид. Зависимость скорости реакции от pH однозначно указывает на участие непонпзованного остатка карбоновой кислоты. Наряду с этим показано промежуточное образование фталевого ангидрида {схема (127)} [237]. Известно много других примеров внутримолекулярного кислотно-основного катализа за счет карбоксильной группы, из которых наибольший интерес представляют результаты по полуамиду малеиновой кислоты [238]. В этом случае, испольуя производное полуамида диизопропилмалеиновой и аспарагиновой кислот, были получены кривые зависимости скорости от pH и скорости реакции, аналогичные пепсину. Хорошо известен также катализ с помощью протонированных имидазольных групп, а также спиртов и алкок-сид-ионов [102, 216].
В соответствующих условиях другие нуклеофильные растворители, кроме воды, такие как амины, спирты и карбоновые кислоты, взаимодействуют с амилами так же, как и в случае гидролиза. При нх нагревании с первичными аминами происходит обмен аминогруппы, часто в отсутствии дополнительного катализатора, но эти Реакции протекают до конца только в том случае, когда образу-Щнися амин летуч. Однако ароматические амины (более слабые
457
нуклеофилы) реагируют медленно, и ускорению реакции способствуют стабокислые катализаторы [239], например при нагревании анилинов при 100 °C с хлоргидратом амида в течение всего 10 мин удается получить анилид с выходом 50—90% {схема (128)} [2396]. В случае о-гидрокси- и о-амнноанилинов образующиеся о-гидроксн-плп о-ампноанилиды циклизуются в соответствующие бензимидазолы или бепзоксазолы [2396].
асо2н
CONHo
/^/СО2Н
Чч^Ч'со2н
Н2о '(быстро)
Д
ArNH2 + [RC(OH)NH2] + C1 RCONHAr + NH4C1 (128)
Нуклеофильное замещение протекает особенно легко в случае ампносоединенип, содержащих электронодонорные заместители, а также в случае гидразинов и гпдроксиламлнов. Изучена кинетика реакции гпдроксплампна с алкпламидамп при 25°C, в которой замещение аминного фрагмента приводит к гидроксамовой кислоте (см. разд. 9.9.4) [240]. Кривая зависимости скорости реакции от pH имеет «колоколообразную» форму, что указывает на общий кислотный катализ гндроксиламмоииевым ионом, который, однако, замедляется при высоких концентрациях. Такое торможение катализа указывает на смену стадии, определяющей скорость про-
цесса, а следовательно, на то, что реакция проходит через тетраэдрический интермедиат, как при гидролизе амидов [240].
Прямое превращение амидов в сложные эфиры под действием спиртов обычно проводят в жестких условиях в присутствии кислых катализаторов. Эти реакции нашли ограниченное применение в синтезе. Недавние работы были направлены на улучшение выхода продуктов с использованием катализатора. По-видпмому, эффек-Т4ШМ является как нагревание в запаянных ампулах с МеОН/ /Вг3 (метиловые эфиры образуются с высоким выходом) [241]. так и нагревание при 100°С с алкплгалогенпдамп в присутствии небольших количеств воды [242]. Последняя реакция проходит через промежуточное образование алкнлимидонпевой соли R C(OR ) NR2X , которая гидролизуется до амина и сложного
458
ы сложных эфиров высоки и достигают 80%, однако фпра. ВыХОДвториЧные амиды подвержены конкурентному N-ал-перв1,чНЬ1е ю [242]. Другим интересным и потенциально полезным 1-цЛ11р°ванИм яВЛЯется фотосольволиз. Так, 1-ацил-1-нитроиндоли-дОстия'еНпе яются с высоким выходом в метиловые и этиловые jjbi nPeBPaUivnbTaTe облучения в смеси метиленхлорида и спирта эфпрь! в ре У'
[243]' Гревании амидов с карбоновыми кислотами происходит Пр11 ильной группы {схема (129)}, и этим объясняется, почему обмен аиН" единення являются неэффективными N-ацилирующими последние разд. 9.9.3.5). Для того чтобы получить хороший реагентам QgbI4H0 необходимо сместить равновесие вправо
0ыхоД ПР ’ и более летучего компонента. Например, удаление с путем отг отгонки уКСуСНой кислоты приводит к получению беп-помошью бензойной КИслоты с удовлетворительными выходами залоД939а1. Тем не менее эти реакции нашли ограниченное приме-[1°2, в синтезе, а их механизм недостаточно изучен. Однако если иейне случае образуется тетраэдрический интермедиат, то он дол-В попадаться через четырехцентровое переходное состояние, так'как амины и ангидриды обнаружить не удалось [239а] {схема (129)}-
r1C0NR1 + R3CO2H.
OH
o—c=o
R3
R3CONR2 4 R1CO2H (129)
(R2=H,Me; p]R3=Alk,Ar)
Наиболее важные исследования механизма сольволиза касаются ацильных производных третичных аминов, таких как пиридин и имидазол, которые нашли широкое применение в качестве ацилирующих агентов. Так, описано [215] получение N-ацилтриалкпл-аммонийфторборатов из третичных аминов и ацилгалогенидов в присутствии HF/BF3, и показано, что эти соединения легко реагируют с аминами, кислотами, спиртами и т. Д. Преимущества от применения пиридина в качестве растворителя при ацилировании хлорангидрядами и ангидридами кислот приписываются его промежуточному. участию в виде ионов N-ацилпиридиння. Будучи очень неустойчивым в любых условиях, кроме безводных, нон N-аадтнлпирндиния, как показано [244], является интермедиатом !‘Ри катализируемом пиридином гидролизе уксусного ашндрида. Измерена также скорость его реакции с целым рядом кислород-11 азотсодержащих нуклеофилов. Ацилпроизводные имидазола к Родственные азолы достаточно устойчивы, и их удается получить 3 виде кристаллических соединений. Применение этих соединении качестве ацилирующих агентов обсуждается в работе I о]. сольволиз этих веществ водой, спиртами, фенолами, тиолами,
459
аминами, карбоновыми кислотами и другими нуклеофилами проходит гладко и быстро, а легкость гидролиза N-ацетилимида-зола в сравнении с целым рядом других амидов уже рассматривалась. Показано [235], что, как и ожидалось, механизм, по которому N-ацетнлнмидазол, его сопряженная кислота и 1-ацетил-З-метилимндазол взаимодействуют в водном растворе с нуклеофилами, аналогично механизму гидролиза. Эти результаты подтверждают, что способность к протопированию отщепляющегося амина оказывает большое влияние на сольволиз амидов, а такж° что амиды с трудно отщепляющимися группами обладают повышенной чувствительностью к реакционной способности атакующих частиц и к общему кислотно-основному катализу.
Проведено несколько сравнительных исследований сольволиза лактамов; изучение аминолиза N-ариллактамов показывает, что внутренние напряжения увеличивают реакционную способность, как и при гидролизе [246].
9.9.3.2. Присоединение металлорганических реагентов
Металлорганические соединения, такие как реактивы Гриньяра или литийалкилы, взаимодействуют с первичными и вторичными амидами, а также с амидами, имеющими протоны у углеродного атома, находящегося в «-положении по отношению к карбонильной группе, как сильные основания, отнимая кислые протоны. Несколько примеров, включающих такие реакции, приведены в разд. 9.9.3.3, 9.9.3.4 и 9.9.3.5. Нуклеофильное присоединение реактивов Гриньяра [247] и литипорганических соединений [248] к карбонильной группе третичных амидов представляет некоторый теоретический, хотя и незначительный, препаративный интерес. Этот процесс, вероятно, включает согласованную атаку фрагментов — атома металла и карбаниона — на атомы кислорода и углерода амида соответственно. При этом образуется устойчивый комплекс присоединения, который может либо гидролизоваться водным раствором кислоты до кетона {направление (а) на схеме (130)}, либо после дальнейшей обработки реактивом Гриньяра [246] приводить к полному восстановительному алкилированию {т. е. к замещению амидного кислорода на 2 алкильные группы, схема (130), путь (б)}.
R'CONMe2 + R2M
/ОМ
R‘R2C^
\\’Ме2
R‘R2C=O + xMe2NH
(130)
R'R22CNMe2
(М = Li или MgX)
9.9.3.3. Алкилирование
Наличие двух потенциально нуклеофильных атомов (О и N) усложняет реакции амидов с алкилирующими агентами. Из первичных или вторичных амидов путем изменения таких параметров 460
пракнни- как температура, pH, тип алкилирующего агента пли Статора можно получить один из двух изомерных npoZ 'dBTL (96) (О-алкилимидат или пмииоэфир) или (97) (N-алХ-ампл) (схема (131)} В сл\чае третичных амидов промежуточный о плекс не может быть стабилизирован за счет отщепления про-она, и поэтому амиды остаются в виде соли (98), которую можно выделить в безводных условиях [249].
в безводных условиях [249]. OR1
О-алкплп-рование
W2R3
(98)
R —II
-HX
/OR1
R’- С7
^\'R2
(96)
\WR3
N-алкилн-ровзине
R1—Cx +
‘ \\TR2R3R<
R3 = H
-НХ
(131)
О
\NR2R1
(97)
о
За исключением взаимодействия с очень реакционноспособным диазометаном и реакций в присутствии солей серебра в качестве катализаторов, условия, при, которых получается определенный тип продукта из первичных и вторичных амидов, можно сформулировать следующим образом: а) в нейтральных условиях при низкой температуре (0—60°C) требуется использование сильных алкилирующих агентов, таких как фторборат триалкилоксо-ния [249], диалкилсульфаты [250] пли трптилхлорид [251], при этом образуются только О-алкилированные продукты (96); б) в сильнокислых или сильноосновных условиях получены только N-алкилированные продукты (97) Однако алкилирование амидов часто проводят в нейтральных условиях под действием мало реакционноспособных агентов, таких как алкилгалогениды, и в связи с этим реакцию проводят при высоких температурах (100— 170wC). В этом случае обычно образуется смесь О- и N-алкилированных изомеров (96) и (97) Попытки объяснить соотношение изомеров, полученное в нейтральных условиях [252], на основании амбидентной природы нейтральной молекулы амида не убедительны [102] Альтернативным и более убедительным объяснением является то, что О-алкилимидат (96) является первичным (кинетическим) продуктом, который, однако, в условиях реакции пРи повышенной температуре частично перегруппировывается в термодинамически более устойчивый N-алкиламид (97), [102]. Факт первоначального образования О-алкпльпого продукта, согласующийся с местом протонирования нейтральных амидов (см.
9-9 2.3), а также рассмотрение двух переходных состоянии 1 9) и (100), образующихся в результате атаки па атом О и N соответственно {схемы (132) и (133)}, предполагают, что (99) ладает более низкой энергией, так как делокализация,неподе-енн°и пары электронов азота уменьшает положительный заряд.
461
Имеются падежные доказательства того, что кинетически контролируемый продукт, О-алкилимидат, перегруппировывается при повышенных температурах в термодинамически устойчивый N-алкплампд. При достаточно высоких температурах (около 180°С) происходит чисто термическая перегруппировка Чапмена [68] {схема (134)}. В случае О-арилимидатов (101, R2 = Ar) перегруппировка Чапмена протекает внутримолекулярно и облегчается электронооттягивающими заместителями в арильной группе. В случае О-алкилимидатов (101, R2 — Aik), термическая перегруппировка, по крайней мере частично, проходит межмолекулярно, что было показано обнаружением продуктов перекрестной реакции из смеси имидатов. Имеются также некоторые указания на то, что реакция редко протекает количественно и может включать свободнорадикальные стадии [253]. Температуры, необходимые для термической перегруппировки, выше тех, которые обычно используются при алкилировании амидов. Однако [254] алкплгало-геииды (пли другие электрофильные реагенты) катализируют
8+ п 8-о—R“—X
nh2
(99)
OR2 OR2
—> R’c(+ X'—> R’c^ + HX
NH2 NH
(132)
4=
О О О
R'c/ —*r’(/ X-—->R’(/ + HX
\8+ о 8- \+ \ .,
NH.,-R‘--x] NH2R“ NHR-
(100)
(133)
R’C^
\nr2r3
(134)
(101)
перегруппировку имндата в амид при более низких температурах, соответствующих реальным условиям алкилирования амидов. Так, О-алкпл-М-метплбеизпмндаты при 138°С в нитробензоле гладко перегруппировываются в М,М-алкилметплбепзамиды {схема (135)}-Реакцпя протекает по двухстадийиому механизму, при этом сначала в качестве интермедиата образуется бензпмпдоиневый ион, который затем деалкилируется иодид ионом до амида. Реакция
Ph С
NMe
R1
^-R Q
PhC(+
NMeR
О // PhC
NMeR
(135)
(R-Me, Et, Pr-н, Pr-uao>
462
ает количественно (за исключением R = изо-Рг, когда эли-пр0Те1ованпе по механизму £2 с образованием пропена конкури-м!,нНР заМешением изопропильной группы) и может быть псполь-РУет в качестве удобного пути синтеза амидов (ср. разд. 9.9.1.5). л0*33' исследовании некоторых внутримолекулярных реакций по-^рИны дополнительные доказательства того, что О-алкилирование ЛуЧяется первой стадией алкилирования амидов в нейтральных ЯВЛ виях. Например, со-бромалкилампды циклизовались при иа-уСЛваннщ давая только продукты О-алкилирования (102) (лак-ГРмы) [255]. Энергетика раскрытия кольца препятствует пере-Т11уппнровке лактима (102) в изомерный N-алкиллактам, который, Г может быть получен при обработке ю-бромамида сильным (схема (136)}.
Доля продуктов О-алкилирования в нейтральных условиях возрастает при катализе оксидом или солями серебра. Этот факт может быть объяснен снижением температуры, необходимой для протекания реакции, например, в присутствии оксида серебра реакция N-феиилформамида с этилиодидом происходит при 40 °C [253], в то же время как некаталитическая реакция требует температуры выше 100°С. Вероятно, оксид серебра повышает электрофильность этилподида за
счет поляризации связи
RNH
(136)
(R = C6HIb Ph)
Другие данные, указывающие на то, что перегруппировка {схема (135)} ингибируется в присутствии солей серебра [254] (так как они осаждают нуклеофильный галогенид-иои, необходимый для протекания О-деалкилирования), также могут объяснять снижение доли продукта N-алкилпровапия. Высоко реакционноспособные диазоалканы являются единственными алкилирующими агентами, действие которых не согласуется с вышеприведенной схемой алкилирования в нейтральных условиях. В этом случае образуется смесь продуктов О- и N-алкилирования даже при комнатной температуре, когда перегруппировка О-изомера в N-изомер исключена. Можно полагать, что либо эти вещества настолько Реакционноспособны, что их атака направлена в равной степени на атомы О и N, либо, напротив, диазоалканы действуют как снования и путем отщепления протона N—Н от амида промоти-РУют N-алкилировапие, как это происходит в сильно основной
463
среде [102] {схема (137)}. Наиболее эффективным способом получения N-алкплампда (97) с хорошим выходом (ср. разд. 99 15) служит обработка исходного амида сильным основанием (напри-мер, амидом натрия, гидридом натрия или грет-бутоксидом калия) в инертном растворителе, что ведет к аннону амида (91) с последующей реакций с алкплгалогспидом {см разд. 9.9.1.5, схема (53)}. ’ Л
R'CONHR2+ R3CHN2 —• > R1 CONR2-J-R3CH2Nj —R'CONR2CH2R3 (137) x Поскольку анион амида (91) может существовать в двух мезомер-пых формах, можно ожидать появления данных о его амбидентной нуклеофильной природе, что сравнимо с аналогичными анионами [257]. Кроме того, образование О- или N-алкилированных продуктов зависит от природы алкилгалогенида (склонность к Sjvl- или 5я2-замещению) и от условий реакции. Из амидов практически всегда получаются только N-алкилировапные продукты, за исключением нескольких примеров внутримолекулярного алкп- ( лирования, когда размер цикла способствует О-, а не N-алкилированию. Однако близкие гидроксамат-анионы, обладая амбидент- р ными свойствами, дают, напротив, продукты как О-, так и N-алки-лирования (см. разд. 9.9.4.3). Другие алкилирующие агенты, такие как активированные олефины [258] и галогенметнловыс эфиры [259], также легко реагируют с анионами амидов, давая соответствующие N-алкилнрованпые продукты, и эти реакции нашли препаративное применение {ср. разд. 9.9.1.5, схема (54)}. Другой * важной препаративной реакцией, проводимой в сильно основных ус- !i ловпях, является внутримолекулярное алкилирование амидов со- f
седней галогеналкильной группой, что является важным способом й
синтеза лактамов (см. разд. 9.9.1.7). Как уже упоминалось, в ре- 1| зультате реакции обычно образуются продукты N-алкилирования {например, по схеме (136)}, за исключением тех случаев, когда ::м( образующийся цикл слишком мал (трехчленный) п алкилирование по кислороду оказывается предпочтительным за счет образования ( большего (пятичленного) цикла {например, по схеме (138)} [260].
Такое различие в размере цикла может возникнуть только тогда, когда галогеиалкильный заместитель находится у азота, а не у карбонильного атома углерода, как на схеме (136). Процесс N-a.i-килирования оказывается предпочтительным по сравнению с О-ал-килировапием, когда при наличии галогсиалкплыюго заместителя у азота, происходит образование шестпчлениого цикла {схема (139)} [261].
В сильно основных условиях у амидов, имеющих одни или более протонов у а-углеродного атома по отношению к карбоиилыюи группе (например, ацетамиды, бутирамиды, а-фенплаиетамиды), появляется другая потенциально нуклеофильная сторона. Такие сильные основания, как амид натрия или бутнллптпй, дают сильно нуклеофильные карбанионы (103), которые легко реагируют с алкилгалогенида ми.
464
А\ c=o
HN
CH2CH2X
NaOEt
Ar
C=O
CH2CH2
(Ar=n-CIC6H4, n-NO2C6H4; X=CI,Br, OTos)
NH Z I .
COPh
EtONa
(138)
(139)
(Y-O, CH2, S, ИТ.Д.; Z=C1, I, OTos)
Таким образом, можно осуществить моно- или диалкилирование третичных амидов путем последовательного замещения у атома углерода [262] {схема (140)}. В случае первичных или вторичных амидов обычно используют 2 моль-экв основания, при этом образуется дианион (Ю4) , который затем подвергается моно- или дпал-килнрованию, в зависимости от количества алкилгалогенида. При использовании 1 моль-экв алкилгалогенида обычно получают С-замещеннос производное; это указывает, что карбанион является наиболее нуклеофильным центром {схема (141)}. Карбанионы (103) и (104) присоединяются к активированным олефинам по реакции Михаэля, и, как и в случае алкилирования, образуются главным образом С-, а не N-алкилироваиные продукты [103, 104].
N-jNH2 . - п R3X
R'CH2CONR22 +=* r'CHCONR22 -------► R CHR3CONR2
(103) ||NaNH2 (HQ)
R3X -
r'cr23conr2 < — r'cr3nonr2
RX
2№NH2 _ _ ----> PhCHRCONH2
PhCH2CONH2 , - PhCHCONH — 2RX (141)
/mn --------->- PhCHRCONHR
(104)
При добавлении к реакционной массе солей серебра процесс преимущественного образования N-алкиламидов в основном растворе существенно видоизменяется. Например, при реакции серебряной соли N-фенплформамида с этилподпдом в смеси углеводородов образуется 74% N-фепил-О-этилформпмидата; увеличение полярности растворителя способствует увеличению количества w замещенного производного [264]. Проведение реакции при
465
низкой температуре исключает возможность перегруппировки НМ1 дата в амид; в этих условиях вероятно можно нродемонстрироват амбидентную природу аниона амида (91), хотя в недавно опуб1-ликованноп работе по пиридонам-2 [265] высказано предположи ние, что ситуация может быть сложнее. Более убедительные доказательства такого амбидентного поведения были получены при алкилировании гидроксамат-иоиов (см. разд. 9.9.4.3).
Реакции алкилирования амидов в кислом растворе менее распространены, так как большинство алкиламидов и некоторые арил-амиды обладают достаточной основностью, чтобы существовать главным образом в виде сопряженных кислот даже в разбавленных (1М) растворах кислот (см. разд. 9.9.2.3). Поэтому и происходит понижение нуклеофильной способности амидов {схема (142)}. Однако реакционная способность некоторых алкилирующих агентов, таких как спирты, ацетали, этилортоформиаты, трет-бутил-ацетат и виниловые эфиры, заметно повышается в присутствии следов минеральных кислот, и эти вещества могут алкилировать амиды при низких температурах, давая только продукты N-алки-лирования [102, 266]. Это не является неожиданным, так как О-алкилпмидаты в присутствии кислот легко перегруппировываются в устойчивые N-алкиламиды [254].
Р + он
* tfef-f- (142)
NH2 NH2
ch2r2 сн2р’-
hconh2+ r1ch=chr2 " + R2CH (143)
conh2 conh2
Свободнорадикальное алкилирование амидов наблюдается редко. Прямое замещение по атому О или N неизвестно, однако алкилирование по атому С может быть осуществлено путем гомолитического присоединения амида к олефину. Реакции фотоприсоединения формамида к олефинам, ацетиленам и ароматическим системам были также детально изучены [107]. Эти реакции которые обычно инициируются такими кетонами, как ацетофенон, состоят в присоединении радикала формамида к ненасыщенной связи с образованием замещенного амида. В случае терминальных олефинов получены высокие выходы амидов {схема (62)}; в случае терминальных ацетиленов главными продуктами являются 2:2 аддукты (105а). При реакции с нетерминальными олефинам
RCH— CHCH=CHR RCH—CHR
II I ।
h2nco conh2 h2nco co\h2
(105a) (1056)
466
пмамнд Дает смесь алкил амидов {уравнение (143)}. в то время нетерминальные ацетилены дают 1 :2 аддукты (1056). В слу-faK ароматических соединений формамид замещает как в ядро, и в алкильную боковую цепь. Аналогичное гомолитическое Экипирование формамида может инициироваться облучением чектронами или обработкой трет-бутилиерокспдом [102]. В случае 'высокозамещенных амидов радикалы могут образовываться 4 на С-алкильных, так и на N-алкильных заместителях. Например оба радикала из N-метилацетамида после присоединения к грпминальным олефинам превращаются в изомерные продукты {схема (63)} [107] [ср. разд. 9.9.1.5].
9.9.3.4. Реакции с альдегидами и кетонами
Первичные и вторичные амиды, так же как и другие нуклеофилы, в кислых, нейтральных или основных растворах присоединяются к карбонильной группе альдегидов и кетонов. Первоначальным продуктом обычно является N-ацилкарбиноламин {схема (144)}, который образуется в результате замещения у амидного азота. Реакция обратима, если не используются сильно кислые условия, когда происходит дегидратация карбиноламина с последующей конденсацией [102, 105]. В отличие от простого алкилирования N-замещенные продукты образуются даже в нейтральной среде, вероятно, в связи с тем, что перегруппировка имидата в амид облегчается в присутствии гндроксиметпленового заместителя. Температура (100—150 °C), необходимая для проведения реакции, также облегчает О—^-перегруппировку. Кетоны, как правило, менее реакционноспособны, чем альдегиды, однако известно несколько примеров их присоединения к амидам [267]. Реакция легче протекает, если кетоны содержат электропооття-гивающие а-заместителп (например, гексафторацетон, ацилоины или а-аминоалкнлкетоиы), хотя в двух последних случаях происходит последующая конденсация.
О
R’COMHR2 + Р)С=О R'CNR2CR32OH (144)
Наиболее интенсивно исследовался процесс присоединения амидов к формальдегиду и глиоксалю [268], который в дальнейшем будет называться «гидрокспметплированпем». Присоединение формальдегида обратимо, однако из нейтральных пли слабо щелочных растворов с хорошим выходом удается выделить N-гид-роксиметиламид {продукт реакции (144) при R3 = Н} [268].Реакция катализируется как кислотами, так и основаниями; скорость Реакции равна к2 [Амид] • [НСНО]. В основных растворах скорость реакции очень быстро растет с увеличением pH, в связи с чем можно предположить, что, по крайней мере частично, ка-5флиз связан с образованием аниона амида RCONH- [102].
ндроксиметилампд может быть синтезирован в результате
467
обработки формальдегидом такого аниона, полученного при ВЗа модействии амида с сильным основанием [269]. Поскольку реак' ция обратима, для получения высокого выхода необходимо оса дить продукт реакции из раствора. Присутствие кислых «-протонов (как в феиилацетампде), как и при алкилировании амидов приводит к образованию наряду с анионом по азоту и карбаниона' В связи с этим необходимо использовать 2 моль основания для получения дпапнона, который с такими карбонильными соединениями, как бензальдегид и бензофенон, реагирует преимущественно по карбаниону {схема (145)} [270].
PhCHO" 2NaNH2 _ . PhCHO I
PhCH2CONHR > PhCIICONR -------> PhCH
I -
CONR CONHR
r2conh2
R'NH2 + HCHO 4=± R'NHCH2OH --------> R'NHCH2NHCOR2 (146)
В кислых условиях скорость присоединения формальдегида показывает примерно первый порядок зависимости от [Н3О+] в ограниченном диапазоне pH; эффекты замещения для замещенных бензамидов и алкиламидов (константы Гаммета р = —1,1; Тафта р* =—2,16 соответственно) [271] указывают на то, что доноры электронов увеличивают скорость реакции. Следовательно, стадией, определяющей скорость реакции, может быть атака нейтрального амида на протонированный формальдегид, однако эти результаты не исключают взаимодействия между протонированным амидом и нейтральным формальдегидом. Отсутствие продуктов О-алкилировання согласуется с процессом алкилирования, так как в кислых условиях следует ожидать быстрой 0->N-nepe-группировкн. Кроме увеличения скорости присоединения карбонильных соединений, кислоты часто облегчают дегидратацию кар-биноламина (36), который превращается либо в енампд (38), либо, после присоединения амида, образует бисамид (37) {схема (58)} [102]. Среди других карбонильных соединений, подвергающихся катализируемому кислотой присоединению, — изомасляныи альдегид, ацетальдегид и циклогексанон [272] Родственная гид-рокснметилированшо реакция Айнхорна, которую можно назвать аминометилпровапнем, состоит в нагревании амида, формальдегида и амина при «70 °C; при этом образуется N-ампнометил-амид. С учетом того, что температура слишком низка, для образования N-гидроксиметиламида возможное направление превращения представлено на схеме (146) [142].
9.9 3.5 Ацилирование
Третичные амиды взаимодействуют с галогенангидридами кис лот, давая солеподобные комплексы состава 1:1, которые удает ся выделить при низкой температуре [273] и которые, вероятно,
2Н+ |
----> PhCH (145)
468
„«еют строение О-ацилов (106) {схема (147)}. Реакционная спорность аци.тгалогеипдов по отношению к днметнлформамнду
Р«У: MeCOCI > PhCOCl > PhSO,CI > ЕЮСОС1, что позволяет предположить важность стерических факторов и прощание реакции по типу S.v2 [273].
oi и иг3
r’conr; т r:’coci кФ.ч + ci ' X' , NR о
(106)
(147)
Первичные и вторичные амиды более реакционноспособны и обычно под действием ацилирующих агентов, таких как хлорангндрн-дЫ и ангидриды кислот, подвергаются N-замещенпю; сложные эфиры карбоновых кислот менее эффективны, а карбоновые кислоты реагируют по иному, присоединяясь к карбонильной группе амида (см. разд. 9.9.3.1). Наиболее трудным оказалось выделение О-адплпмпдатного интермедиата {(45) на схеме (86)}, вероятно потому, что очень легко происходит O-^-N-перегруппировка. В особых случаях, либо когда реакцию проводят при низкой температуре при наличии объемистых электронооттягивающнх заместителей [274] {например, (45), R2 = 2,4-(ЬЮ2)2СбНз, R1 = R3 = = Ph}, либо когда О-ацильная группа является частью кольца [134, 136] {например, в случае изоимпдов (49) на схеме (92)}, эти интермедиаты удается выделить и провести термическую или катализируемую основанием перегруппировку до N-ациламида (имида) {(44) на схеме (86)} (см. разд. 9 9.1.8). Этот процесс аналогичен перегруппировке О-алкилимидата в N-алкнламид, рассмотренный в разд. 9.9.3.3. В случае вторичных амидов R3 = Aik, Аг (O-^-N-перегруппировка {направление (а) на схеме (86)} является единственно возможной реакцией и, следовательно, приводит к непосредственному выделению имида. Такое превращение обычно осуществляют обработкой вторичного амида хлорангид-ридом кислоты в присутствии пиридина [275] (см. разд. 9.9.1 8). В случае первичных амидов (45, R3 — Н) процесс дегидратации До нитрилов {направление (в) на схеме (86)} постоянно конкурирует с перегруппировкой, и, кроме того, интермедиат (45) может подвергаться дальнейшему ацилированию до трпацилампна (46) [276]. Факт образования нитрила в качестве побочного продукта представляет собой важное доказательство того, что О-ацилими-Дат служит интермедиатом при ацилировании амида, п следовательно, реакция по амидному кислороду кинетически предпочтительна при ацилировании, как это имеет место и при алкилировании (см. разд. 9 9.3.3). Относительное содержание каждого npoj Дукта, образующегося из первичного амида зависит от условий Реакции и природы ацилирующего агента [102]. Влияние всех этих Факторов суммировано в табл. 9.9.7. Использование катализатора Не обязательно, если применяют сильные или умеренные ацплп-
469
рующие агенты; третичные основания, такие как пиридин, способствуют удалению кислых побочных продуктов реакции.' При использовании этих агентов протекание реакции настолько облегчается, что ее можно проводить при очень низких температурах (—60°C) с целью избежать образования нитрила. Однако часто проходит более глубокое ацилирование, что приводит к триацил-аминам. Это означает, что О->М-перегруппировка интермедиата (45) [направление (а) на схеме (86)] при низкой температуре протекает очень медленно, в то время как N-ацилирование (45) {направление (б) на схеме (86)} все-таки протекает, что дает три-ацилампиы. Напротив, при комнатной температуре О—>N-neperpyn-пировка (45) протекает легко, и если используется несильный ацилирующий агент [102], дальнейшая реакция с образованием триацил а ми на не происходит.
Таблица 9.9.7. Продукты ацилирования первичных амидов RCONH, Гем схему (86)]
Главный продукт Ацилирующий агент Температура, °C Катализатор
RCN Сильный (например, n-NO2CGH4COCl) > 0 —
RCONHCOR' Слабый (например, РгСОС!) > 0 Пиридин
RCON(COR')2 Средний (например, п-С1С6Н4СОС1) -60 Пиридин
Другим осложняющим обстоятельством ацилирования с помощью галогенангидридов кислот является возможность протекания реакции обмена ацильными группами амида н хлораигид-рпда. Так, в случае третичных амидов устанавливается равновесие {схема (148)}, которое можно сдвинуть, удаляя из сферы реакции более летучий хлорангидрпд [277]. Этот прием используется при превращении диалкилформамидов в другие амиды, так как образующийся формплхлорнд разлагается на оксид углерода и НС1. Обмен отмечен также в случае первичных амидов. Как и при алкилировании амидов, прямое N-ацилирование, ведущее к диацилампну, и другие побочные конкурирующие реакции могут осуществляться при использовании большого избытка пиридина или сильного основания, такого как реактив Гриньяра [131].
R'CONR* — R3COC1 R'COCl + R3CONR* (148>
Ангидриды кислот менее реакционноспособны, чем галогенангидриды, и для проведения ацилирования амидов с помощью первых агентов требуется присутствие катализаторов, таких как серная кислота, сухой хлорид водорода, ацетилхлорпд или даж хлорид аммония [278]. Как и при взаимодействии с хлорангидрп-дами, в случае первичных амидов наблюдается дегидратация д 470
питов и дп-Ы-замещение {ср. co схемой (86)}. Чтобы умень-[11‘тр степень протекания этих побочных реакций, целесообразно ^бявлять умеренное количество (>0,01 экв.) кислого катализа-•'В * 10 а Как и при алкилировании, проведение реакции в кислых Т°повиях способствует N-замещению, вероятно, за счет облегче-о-^К-перегруппировкн.
RWHs
N—Н
Н
OCOR
4 NH
(rco)2nh
(149)
RCN + RCO2H
Внутримолекулярное ацилирование (диспропорционирование) первичных амидов протекает при повышенной температуре; выделение нитрилов, диацпламинов и аммиака вновь предполагает образование О-ацплпмидата в качестве интермедиата {схема (149)} [279]. Неактивнроваиные сложные эфиры реагируют с амидами при 200°C, давая аналогичные продукты [120], однако использование сильно основных катализаторов, например гидрида натрия, которые превращают амид в высоко нуклеофильный анион, способствует образованию имидов [131]. Внутримолекулярные реакции между амидной и соседней сложноэфирной группой в основных условиях служат очень удобным способом получения циклических имидов (см. разд. 9.9.1.8). Кислые катализаторы также способствуют образованию N-замещенных продуктов, и некоторые специально синтезированные сложные эфиры, как, например, изопренильные эфиры, активированные в присутствии кислоты, гладко превращаются в имиды [132а] {уравнение (89)}. Кетоны [1326] и изоцианаты [280] взаимодействуют с первичными и вторичными амидами с образованием продуктов, которые вполне соответствуют рассмотренным выше реакциям для других ацилирующих агентов. Так, первоначальное О-замещение амида в нейтральных условиях дает О-ацилимидат {схема (150)}, который в случае первичных амидов может затем подвергаться как дегидратации до нитрила, так и перегруппировке до диациламина.
|| R' = IT
R'COXHR2 /ОС—ХН
+ R’C' ---------*
Х=С=О >\R2
RICN + IIXCO2H
R'CONR2COXH
/ХН
R'c;
>XR2
(150)
(X = R3CH или R3N)
В случае изоцианатов образование нитрилов до сих пор не отмечено. В реакциях вторичных амидов, имеющих электронодонор-!,1ь'е заместители у атома азота, процесс образования амидина {путь
Iй) па схеме (150)} конкурирует с образованием диациламина
{путь (б) ца тод >ке схеме} пз изоцианатов [280]. Изоцианаты
471
присоединяются также к третичным амидам, образуя О-ацц. лнмндаты, которые легко элиминируют СО2, давая амидины [280], вероятно за счет Длй-реакции, так как эксперименты с 14С-меченным амидом показали, что весь СО2 выделяется из изоцианата [280] {схема (151)}. Серная кислота способствует протеканию реакции между первичными и вторичными амидами н кетенами, в результате чего с почти количественным выходом образуется N-ацпламид; побочные продукты нитрильной природы не возникают [281].
r1conr|
+
R3№=C=O
о-гС ж R'C NR
<^NR“
NR3
X r’c
NR2
(151)
Оксалилхлорид — специальный ацилирующий агент; его реакции с первичными, вторичными и третичными амидами подробно исследованы в работах Специале с сотр. [282]. Данные, получен-
ные этими исследователями, говорят в пользу предложенных механизмов рассмотренных вариантов ацилирования. В нейтральных условиях первой стадией реакции является атака атома кислорода амида на карбонильный атом углерода оксалилхлорида с образованием О-ацилимидата (107) {схема (152)}. В случае третичных амидов за этой стадией следует Здн-замсщение хлорид-поиом и другие последующие реакции [путь (а) на схеме (152)]. В случае же первичных и вторичных амидов, когда R1 = Aik, происходит циклизация до (108) и раскрытие цикла [направление (б)], приводящее к производному имида. При R1 = Н образуется ацплизоцианат (R-трет-Вн или Ph) или устойчивое циклическое соединение (R = PhCH2) [направление (в)]. Механизмы этих превращений были подробно исследованы [102].
оо
II II
/ОССС1
4s=NR2R3 СГ.
R'CONCO
(СОС1)2
R’CONR2R3
к3 = н
(-НС1)
(107) (108)
(б) R2 = Aik^
R'CONRzCOCOCI
472
9.9.3.6. Нитрозирование и нитрование
птистая » азотная кислоты соединяются с соответствую-А3 спабыми основаниями, образуя сильные электрофильные ^'^епты которые можно рассматривать как переносчики ионов реаГе'он11Я NO+ и нитрония NO2 соответственно. Многие из этих я"тр2)Тов взаимодействуют с первичными и вторичными амидами, РеаГкак правило, не реагируют с третичными амидами. В связи Н°’изкой нуклеофильностью амидов все эти реакции протекают С Нчительно труднее, чем в случае аминов. Как при нитрозиро-знан„ так и при нитровании, получаются аналогичные продукты; ва”в„чные амиды подвергаются дезаминированию до карбоновых Пе 1от [схемы (153) и (154)], вторичные амиды дают соответ-
•юшие N-нптрозо- или N-нитропроизводные {схемы (155) и N-арнл-М-нптрозоамнды— высоко активные соединения, которые подвержены как термическому, так и химическому распаду (см. разд. 9.9.3.11 и 9.9.3.12).
[RCONHNO] —> RCO2H + N2 + HX
кислот
CTBJl-
(156)}. N-Алкил- и
фото-
RCONHs + XNO —> (X = C1, RO, BF4, OH) rCONH2 + YNO2 —► [RCONHNO2]
RCO2H + N2O + HY
(153)
(154)
(Y = МеСО2)
основание
R'CONHR2 + XNO ~~ '• — R'CON(NO)R2 + НХ (155)
(Х = ОН, Cl, NO)
основанне
R'CONHR2 + YNO2 -----------R'CON(NO2)R24-HY (156)
Как и в случае аминов, для нитрозироваиня первичных амидов может быть использован ряд реагентов (NOX). Наилучшие выходы (70—90%) карбоновых кислот получены при использовании раствора нптрозных газов в инертном растворителе или при использовании фторбороната нитрозония в ацетонитриле [283]. Применяется нитрит натрия в водном растворе минеральных кислот, однако дезаминирование протекает очень медленно, если точно не установлена соответствующая кислотность среды [284]. Известно, что в равновесии с азотистой кислотой находится несколько активных иитрозлрующих частиц. На основании подробных исследований реакций этих частиц с аминами предложена последовательность изменения их реакционной способности: n2O3 < NOHal < H2ONO+ < NCH [102]. Сама азотистая кне-л°та совершенно неактивна в этих реакциях. Изучение ннтрози-Ропания ацетамида и родственных алкиламидов в НС1 показало, (об Взаим°Дсйствие протекает между нитрозилхлоридом NOCI разующимся на иредравповесиой стадии) и непротонирован-Pvin аМ!1Дом {схема (157)} [285]. Скорости реакций коррелн-
т с параметрами Тафта о*((>* = —3,0), указывая на то, что
473
вклад стерических факторов тот же, что и при гидролизе сложных эфиров. Поскольку в хлорной и серной кислотах соответствующие нптрозильные соли полностью ионизованы, то нитрози-рующими агентами, в порядке увеличения кислотности, могут быть: азотистый ангидрид, протонированная форма азотистой кислоты FbONO1 п ион нитрозония NO+ [102]. Нет доказательств что сам азотистый ангидрид способен реагировать с амидами’ (хотя он реагирует с аминами), что обусловлено, вероятно, его низкой реакционной способностью. Имеются достоверные подтверждения того, что нптрозпрованне бензамида и ацетамида сильно катализируется минеральными кислотами [284], хотя п остается неясным, указывает ли это на реакцию с участием ионов нитрозония и (или) протонированной формы азотистой кислоты.
rconh2
HNO2 + HC1 H,o + C1NO------------------>
(медл.)
RCO2H + N2 + HC1 (157)
Предполагается, что стадией, определяющей скорость процесса, является атака нитрозирующей частицы на непротонпро-вапную форму амида (так как скорость реакции дезаминирования уменьшается в концентрированных минеральных кислотах, где амиды полностью протонированы) [284]. Однако место первоначального замещения неизвестно, хотя по аналогии с другими электрофильными реагентами кажется вероятным, что после О-нитрозирования происходит быстрая перегруппировка до N-нит
розоамида.
Нитрование первичных амидов подробно не исследовано, однако показано, что бензамид, например, в результате обработки ацетилнитратом (образующимся из нитрата меди или азотной
кислоты в уксусном ангидриде) с высоким выходом превращается в бензойную кислоту [286]. Полупериод этой реакции составляет менее 5 мни, т. е. она протекает значительно быстрее, чем обычный гидролиз бензамида (см. разд. 9.9.3.1). Промежуточно образующееся N-нитропроизводное {см. схему (154)}—лишь незначительно устойчивее соответствующего ннтрозоинтермедиата {см. схему (153)}, однако оно может быть получено в кристалличе-
ском состоянии при низкой температуре.
Вторичные амиды превращают в N-нитрозопроизводные под действием тех же реагентов, что используются и для первичных амидов. Как п в случае вторичных аминов, реакция обратима, и для получения высокого выхода продуктов необходимо использовать подходящее основание для удаления ИХ {см. схему (155)}. Наиболее эффективными реагентами являются нптрозилхлорнд пли N2O4 в присутствии слабоосновпого ацетата натрия. Менее успешным оказалось применение нитрита натрия в минеральной или уксусной кислоте [287]. .
Недавно проведенные кинетические исследования [288] оорат пой реакции (т. е. денитрозировапия N-нитрозоамндов) 1схе*4 (158)} позволили выяснить механизм нптрозирования, котор
474
должен включать те же интермедиаты. Установлено, что отшеппс
NO+ протекает через стадию образования м „I 0Т1-чепле-клслоты (109), однако остается неясным, участвует ли^ор!™0” р.нитрозоиитермедиат (ПО). Показано, однако, что при^ито'о' ^рованни амидов катализа хлорид-поном не происходит Р
О NO о о
r’C-NR2 NH R2 (109)
O-N=O r'cTf-
NHR2
(110)
R1CONHR2 + NO+ (158)
[R^Me; R2=Et; R1-R2=(CH2)4]
Вторичные амиды легко превращаются в N-нптропроизводные при обработке слабыми нитрующими агентами, наиболее эффективными из которых являются ацетилнптрат (т. е. нитрат меди или азотная кислота в уксусном ангидриде) [95, 101, 102] пли N2O4 в инертном растворителе [103]. N-Нитроамиды устойчивее соответствующих N-нитрозоамндов, однако оба класса соединений имеют очень близкие свойства [289]. Если вторичный амид содержит ароматическое ядро (например, бензамиды или анилиды), то процесс нитрования ароматического кольца будет конкурировать с замещением по атому азота. Эти аспекты реакции подробно рассмотрены в обзоре [102].
Реакции N-нптрозоамидов и N-нитроампдов— это в основном перегруппировки, индуцируемые фотохимически или под действием основных катализаторов. Эти реакции рассмотрены в разд. 9.9.3.11 и 9.9.3.12.
9.9.3.7. Галогенирование
Молекулярные галогены (за исключением фтора) п гнпогало-гениты при взаимодействии с первичными и вторичными амидами выступают как ионные галогенирующие агенты. При этих реакциях обычно образуются N-галогенампды, хотя в отдельных случаях возникают и С-замещенные производные. Этот факт может быть связан с неустойчивостью N-галогенамидов, а именно с их разложением в кислых растворах с образованием «положительного» атома галогена, который затем может атаковать другие части молекулы амида.
Первичные и вторичные амиды реагируют с иодом, бромом и хлором, давая N-галогенампды {схема (159)} [290]. Реакция обратима; образованию продуктов способствует наличие^полярных растворителей, например воды. Хотя механизм этой реакции мало изучен, можно полагать, что он аналогичен механизму реакции амидов с гипогалогеннтамп и состоит в первичной атаке амидного кислорода на галоген с последующей ©-^-перегруппировкой {схема (160]}. Обратная реакция (т. е. гндролт
475
N-галогенамида) катализируется галогенокислотами, и скорость реакции [2906] пропорциональна [Н+] [Х+], как и в случае перегруппировки Ортона для N-галогенацетанилидов (см. разд. 9.9.3 11) Третичные амиды при взаимодействии с молекулярным иодом [192] образуют комплексы состава 1 : 1 (см. разд. 9.9.2.3).
R'CONHR2 + Ci2 —> R'CONR2Ci+ HCi
R'CONHR2
ci2
>OCi R'C^ Ci" \NHR2
R'C^+ СГ
\NHR2Ci
(159)
-HCI
^z± R'CONR2C1
(160)
Гппогалогениты — более удобные реагенты для получения N-галогенампдов, чем молекулярные галогены, так как в этом случае протекание конкурирующей реакции С-галогеннрования менее вероятно. Гипогалогенокнслоты обычно получают in situ за счет добавления эквимольного количества гидроксида натрия к смеси молекулярного галогена и амида [291] {схема (161)}. Следует избегать избытка щелочи, так как первичные амиды могут подвергаться деградации по Гофману (см. разд. 9.9.3.11), а в случае вторичных амидов возможен катализируемый основанием гидролиз до исходного амида [реакция, обратная представленной уравнением (161)]. Механизм реакции получения замещенных N-метил-М-хлорбензамндов с помощью гипохлорит-понов и их гидролиза водной щелочью изучены в работе [292]. Эти превращения представляют собой прямую и обратную реакцию одной равновесной системы и должны включать общие интермедиаты. Полученные результаты указывают на то, что сначала происходит атака амидного кислорода на гппохлорпт-нон, далее через переходное состояние (111) образуется О-хлорннтермедиат, который затем подвергается О-*М-перегруппировке до N-хлор-амида {схема (162)}.
R'CONHR2
Хг + NaOH НОХ + NaX -------------> R'CONXR2 + Н2О (161)
(X = Cl, Br, I)
ArCONHMe + CIO’
O--C1 1
>OC1 HO’+ArC^ ^NMe
Me
(111)
ArCONMeCl
(162)
N-Галогенированне первичных и вторичных амидов можно также проводить действием эфиров гипогалогенокислот (напри мер, трет-BuOX). Обычная методика состоит в использовании смеси трет-бутилгппохлорпта, пода и амида в CCU. Эффектней реагент трет-бутилгипоподит образуется на предравновеснои дни, и поэтому в этих условиях исключается взаимодеиств! иодмопохлорпдом [29, 293].
476
Взаимодействие газообразного фтора с амидами приводит к смеси С- и N-фторированных продуктов {схема (163)} [294], вероятно в результате свободнорадикальных процессов, хотя эти реакции не изучены подробно и не применялись в синтетических целях. Недавно показано, что при обработке фторокснтрифтор-метаном амиды вначале подвергаются N-фторированию; добавление избытка реагента приводит к разрыву связи С (О)—N и к дальнейшем}' фторированию с образованием среди прочих продуктов дпфторалкплампнов [схема (164)]. Ароматические амиды, однако замещаются главным образом по кольцу [295].
HCON(CH3)2 FN(CF3)2 + (F3C)2NN(CF3)2 (163)
FOCF-, FOCF3 +
R'CONHR2 ------->• R’CONFR2 -------> R‘CONF2R2 F3CO’ —>
—> R'COF + R2NF2 + R‘COOCF3 (164)
Наиболее подробно изучены перегруппировки N-галогенамидов (см. разд. 9.9.3.11), однако исследован также и ряд других превращений. Так, N-бромампды (но не N-хлорамиды) являются эффективными агентами, бронирующими в аллильное положение [296]. При обработке N-хлорамидов диазометаном происходит включение метиленового звена с образованием N-хлорметиламидов, которые могут быть непосредственно замещены нуклеофилами, например морфолином [297] {схема (165)}. Кроме того, N-ra-логензамещенные первичные и вторичные амиды реагируют с трифенилфосфином, давая трифенилфосфиноксид и нитрил- пли нмидоилгалогеннд соответственно [298]. О механизме этих реакций известно мало, однако можно полагать, что неподелеиная пара электронов фосфора первоначально атакует положительный галоген.
морфолин
R‘CONR2C1 + СН,.\Ч —> R1 CONR2CH,C1----------->
R1CONR2CH2N
(165)
9.9.3.8. Реакция с галогенангидридами неорганических кислот
Многие реакции амидов с галогенангидридами неорганических кислот используются в органическом синтезе. Эти реагенты можно подразделить на три класса: а) фосген, тионилхлорпд и пентахлорид фосфора; б) оксихлорид фосфора и аренсульфонилхло-риды; в) фторангидриды кислот. В случае третичных амидов удается выделить некоторые относительно стабильные комплексы, однако в случае вторичных и первичных амидов происходят дальнейшие превращения, такие, как N-деалкилирование и дегпдрата-пия, быстро приводящие к нитрилам. Наименее сложна ситуация с третичными амидами, которые реагируют с реагентами груп-пы (а) при комнатной (фосген) или при несколько более высокой
477
температуре (тпонилхлорид и пентахлорид фосфора) [299]. При взаимодействии любого из трех реагентов образуется имидоил-хлорид (112), возникающий, вероятно, после первоначального образования О-комплекса (113), который затем подвергается быстрому ЗлН-замещеиию галогенид-ионом с синхронным элиминированием нейтрального фрагмента (СО2, SO2 или РОС13 соответственно [схема (166)].
R1CONR2 4- ХС12 •—> (Х=СО, SO, РС13)
(113)
С1
R‘c СГ + ХО (166)
nr22
(112)
Имидоилгалогениды, полученные из третичных амидов, относительно устойчивы и имеют ионную природу. Они могут подвергаться дальнейшему нуклеофильному замещению алкоксидами, карбоновыми кислотами и т. д., а также аренами [300] (этот путь может быть полезной альтернативой ацилированию ароматических соединений по Вильсмейеру — Хааку, которое рассматривается ниже). При взаимодействии фосгена с любым третичным амидом, имеющим атомы водорода у углерода, находящегося в «-положении по отношению к карбонильной группе, происходит дальнейшее замещение, приводящее к а-хлор-|3-хлоркарбониленаминам. При реакциях со второй группой реагентов [(б), оксихлорид фосфора или аренсульфонилхлориды] также образуется комплекс по атому кислорода (114). Однако этот комплекс не столь легко претерпевает S^i-замещение либо потому что выделяющийся фрагмент неустойчив (например, РО2С1),либо потому, что отсутствует внутренний нуклеофил (аренсульфонилхлориды). Таким образом, оба комплекса тина (114) значительно устойчивее комплексов типа (113), однако они более чувствительны к атаке внешними реагентами, такими, как нуклеофильные ароматические соединения или амины {стадия (б) на схеме (167)}. Хлорид-ион комплекса также может выступать в роли нуклеофила, осуществляя перегруппировку {схема (168)} протекание которой снимает противоречие при выборе структуры комплекса (114) или (115) [301,302].
(а) /0Х (б) /NU
R’CONR?-р ХС1----•> R'CZ СГ -д-* R'C' СГ + ХО (167)
Sir] X’Rl
(X = POC12, ArSO2) (114)
/ОХ /Cl
R'C^ СГ •—> R'C^. XO’ (I68)
SxiR? XnRs
(114) X = POC12, (115)
ArSO2
478
к' мплекс оксихлорида фосфора и третичных амидов (114, Х =
' известный как комплекс Вильсмейера, является питер-Р патом при форматировании ароматических соединений ио к^ьсмейеру — Хааку [302]. Эта реакция заключается во взаимо-"ствии активного арена (бензол и нафталин — нереакционно-Дсобны), например многоядерного углеводорода, О-, N- и S-co-СП°жашсго гетероцикла или производного бензола, имеющего Де<ие электронодонорные заместители, как ОН, ОМе NHMe, с Га'мплексом Вильсмейера, получаемым из диметилформамида (или м°метплформанплида, или N-формилпиперидина) и оксихлорида Фосфора при низких температурах (<25°С). Последующий гид-попиз приводит к альдегиду, как показано па схеме (169) [302]. При взаимодействии с другими третичными амидами, помимо формамидов, получаются кето-, а не альдо-производные. Именно таким путем проводят ацилирование ароматического ядра беизо-фурана и некоторых производных индола и пиррола [303]. Как п в случае имидоилгалогенндов (112), многие другие нуклеофильные частицы, за исключением аренов, легко атакуют карбонильный атом углерода комплекса Вильсмейера. Например, алифатические амины или гидразины дают ампдиниевые соли, и даже первичные ароматические амины подвергаются преимущественно N-замещснию, а не формулированию в кольцо {стадия (б) на схеме (167)}. Олефины после гидролиза промежуточного комплекса дают а, (З-ненасыщеиные альдегиды. Другие примеры синтетического использования этого превращения приведены в [302, 304]. Сообщалось о реакции комплекса спиртами (ио не с аренами) [305].
(114, X = ArSO2) с аминами п
он
HCONMe2 + РОС13 —>
О // О-РС12 н4 NMe2
С1
С6Н5ОН
НС-ОРОС1 I NMe2
+ НС1
(169)
2
2
НО.
РО2С12“
CH=NMe2
ноу=\
К СНО + Me2NH
Третья группа реагентов (карбонилфторнд или тетрафторид серы) при взаимодействии с третичными амидами дает ковалентно-связанные дифторалкиламины {схема (170)}, а не ионные имп-Д°илгалогениды. По-видимому, наилучшим реагентом является Карбонилфторид, так как в этом случае выделение продуктов упрощается. С помощью меченых атомов показано, что СО2 выделяется полностью из реагента, а не из амида, что предполагает протекание реакции через стадию образования комплекса по кислороду, как и в случае других галогенапгидридов кислот [306]. стоичивость дифторалкиламина сильно зависит от природы R.
479
Если это первичный радикал, то происходит элиминирование Нр а образующийся енамин подвергается дальнейшему замещении:
R'CONR; + XF3
/OXF
R‘CZ + F“ 4NR22
RiCF2NR5 4-XO (170)
(X = CO, SF2)
Взаимодействие галогенангидридов неорганических кислот с вторичными амидами аналогично реакциям с третичными амидами в том, что, вероятно, п в этом случае происходит образование комплекса по кислороду. Однако эти комплексы выделить не удается, так как протекает их быстрое превращение (главным образом с образованием нитрила). При нагревании многих N-алкилами-дов с SOC12 или с РС15 происходит разрыв N-алкильной связи с образованием нитрила и алкилгалогенида {уравнение (171)}. Эта важная реакция, открытая Пехманом, изучена в дальнейшем Брауном и теперь носит его имя [102, 307].
PhCONHR
R'CONHR2 + РС15 R'CN+R2C1 + РОС13
SOC19
(а)
/С1
PhCZ СГ
\nhr
-НС1
ZC1 > PhCZ
4\R
(б;
(171)
PhCN + RC1
(172)
Сравнительно недавно вместо пентахлорида фосфора в большинстве синтезов начали применять тионилхлорнд, так как в этом случае выделение и очистка продуктов упрощается [308]. На примере бензамида {схема (172)} показано, что в качестве интермедиата образуется имидоилхлорид. Вместе с тем оказалось, что формамиды и некоторые алкилампды практически не активны в этой реакции. В результате термического распада импдоилхло-рида {стадия (б) на схеме (172)} образуются бензонитрил и ал-килгалогенид. Реакция служит удобным препаративным методом превращения алифатических аминов в соответствующие алкил-галогениды. Чтобы объяснить природу продуктов, образующихся из оптически активных субстратов, и влияние заместителей R! и R2, предложено два конкурирующих направления распада ими-доилхлорнда {схемы (173) и (174)}. Первое направление включает гетеролиз N-алкильной связи по механизму, «подобному S.vl», с последующим быстрым выбросом хлорид-иона. Второе направление осуществляется в обратной последовательности, а именно после первоначального отщепления хлорид-иона образуется ион нитрилия, который далее взаимодействует с хлорид-ионом по механизму «подобному Sn2», и эта стадия определяет скорость процесса. Если R1 ароматическая группа, то преобладает процесс S^-2-типа, так как промежуточно образующийся карбениевый ион стабилизуется за счет резонанса. С другой стороны, направление, ооусловленное реакцией S,vl, оказывается предпочтительным Дл^ амидов, в которых замещаемая группа R2 образует стабильный
480
Арниевый ион (например, трет-Ви). Таким образом, в реакции паРсна участвуют те вторичные амиды, R'CONHR2, у которых радикал R1 может стабилизоваться в виде карбениевого иона, 60 R2 образует стабильный карбениевый ион [102]. Интересно, '’п фосген не вступает в реакцию Брауна с вторичными амидами; ЧТякция останавливается на стадии образования промежуточного Рш1доилхлорида. Причина этого неясна. В ряде случаев отмечено образование енамина [102].
CI
J / медленно,
V) „
N-R“
Cl
R4-.
L N~
5^1-механизм
ф
R2
NR2
- P’C^N-R2 + СГ -^J,eH110>. R1CN + R2C1 (174) <5>м2-механизн
Оксихлорид фосфора также взаимодействует с N-алкилами-дами; продуктами реакции обычно являются соответствующие нитрилы {ср. схему (172)}. Реакция не изучена подробно, так как большее синтетическое применение нашли SOC12 и PCI5. Однако известно, что для ее проведения требуется повышенная температура (около 120 °C) и что амиды, имеющие N-алкильный заместитель и дающие относительно устойчивый карбениевый ион, реагируют наиболее легко [309]. В этом случае, вероятно, протекает S.vl-реакция, сходная с представленной выше на схеме (173). Важное различие состоит в том, что алкилгалогенид обычно не является вторым продуктом, и в том, что +R2 реагирует с ароматическим растворителем или теряет протон, давая олефин; однако причины такого различия пока не ясны. Вторичные формамиды в данном случае нетипичны, так как их взаимодействие с оксихлоридом фосфора, аренсульфонилхлоридами или с тионил-хлоридом приводит с высоким выходом к изоцианидам; эта реакция служит удобным методом синтеза этих соединений {схема (175)} [ЗЮ]. Интермедиатом в данной реакции почти наверняка является соединение, аналогичное комплексу Вильсмейера (144), однако внутримолекулярный отрыв протона фосфорильным или сульфонильным кислородом приводит к образованию изоцианида. ф°рмилирование ароматического ядра вторичными амидами п° Вильсмейеру — Хааку в присутствии оксихлорида фосфора яв-Ляется необычным процессом, однако хорошо известный синтез Цзохинолинов по Бишлеру — Напиральскому вполне может вклю-эть такие превращения [102].
взаимодействие вторичных амидов с карбонилфторидом скорее гап°МИНает алкилирование органическими, чем неорганическими огенангидридами кислот (ср. разд. 9.9.3.5). При этом обычно 16 Зак, 395 481
ОРО_С12
HCONHR -9С1з> НС °
W+ NHR
RNC + HOPOC12 (175)
JPC12
-HCI*
H-j-C
NR
образуются N-фторкарбониламид (который частично диспропор-ционирует до уреида) и дифторалкиламин (минорный продукт), который может подвергаться дальнейшему замещению COF2 {схема (176)} [306]. Такое превращение объясняют образованием соли О-ацилимидата, которая далее либо подвергается О—”N-ne-регруппировке, давая N-фторкарбонильное производное направление (а) на схеме], либо распадается за счет Здц-атаки, образуя дифторалкиламин {направление (б) на схеме (176)}.
R'CONHR2 .O-COF
' + F" ^NHR2
COF2
(176)
R'CONR2COF + HF
|r'conr2cof
(R'CONR2)2CO + cof2
аспектов химии амидов — это дегид-до нитрилов в присутствии пен-
R>CF2NHR2 + CO2 |cof2 R1CF2NR2COF + HF Один из самых известных ратация первичных амидов токсида фосфора или почти любого галогенангидрида кислоты. Последний обзор по этим реакциям сравнительно давно опубликован [311], однако последующих достижений в этой области было немного. Использование галогенангидридов органических кислот для этой цели уже обсуждалось в разд. 9.9.3.5. Препаративное значение имеет дегидратация с помощью пентахлорида фосфора, тионилхлорида, фосгена, оксихлорида фосфора [102] и даже фос-фонитрилхлорида [312]. В реакцию вступают как алифатические, так и ароматические субстраты; для удаления кислых продуктов используются основные органические растворители. Наиболее удобным реагентом является, вероятно, тионилхлорид; он менее токсичен, чем фосген, и превращается в летучие продукты. При использовании днметилформамида в качестве растворителя реакция протекает при 0°С с высоким выходом [313]. Только в случае оксихлорида фосфора требуется 0,25—0,5 моль-экв, вероятно в связи с регенерацией оксихлорида фосфора за счет диспропорционирования продукта — дихлорфосфорной кислоты. Как ни странно, но механизм реакции дегидратации мало изучен. В качестве интермедиатов выступают, вероятно, О-ацил- и имидоил-галогениды, аналогичные уже описанным в случае вторичных и третичных амидов.
482
9.9.3.9. Окисление
Очевидно, что при окислении амидов могут протекать два процессов (обычно включающие ^свободнорадикальные прев-тиП11ениЯ), хотя и было изучено воздействие относительно неболь-Ра1~ чпсла окислителей. Один процесс —это отрыв водорода от Ш°ерОда или азота, второй — окислительное замещение у амид-Угл азота. Наиболее типичным процессом в случае вторичных и Н°еТичных N-алкиламидов служит отщепление водорода от угле-ТР находящегося в a-положении к азоту. Этот углерод активируется по отношению к радикальной атаке под влиянием непо-Р"пенной пары электронов азота. Окислительное замещение на-Динает играть важную роль только тогда, когда у N-заместителя ЧтСутствуют атомы водорода (например, для анилидов). Окисле-°йеУ первичных амидов может протекать за счет отрыва водорода иди путем окислительного замещения, причем один или другой процесс обычно преобладает. Строение продуктов зависит как от природы амида, так и от окислителя.
И Значительное внимание уделено также автоокислению амидов, так как этот процесс является нежелательным при производстве найлона. Такое окисление безусловно является свободнорадикальным процессом и может быть индуцировано либо термически (> 100 °C), либо фотохимическим путем при низких температурах как в присутствии, так и без соответствующих инициаторов [314] г Известно три основных направления этой реакции, причем начальные стадии являются общими для всех направлений и включают (схема (177)} отрыв а-водородного атома по отношению к азоту с последующим присоединением кислорода с образованием перокси-радикала (116). Дальнейший распад этого радикала протекает по трем направлениям: (а) отщепление «ОН с образованием имида; (б) разрыв связи С—С, приводящий к имиду, и (в) расщепление связи С—N с образованием первичного амида и карбонильного соединения. Подробнее этот процесс обсуждается в [102]. Сообщалось [315] о взаимодействии О2 с карбанионом амида (образующимся из R1CH2CONR2R3 в сильном основании), приводящем к а-пероксиалкиламиду, который восстанавливается in situ до а-гидроксиалкиламида R’CH (ОН) CONR2R3.
R'CONHCOR2 + НО*
f (177)
(а) разрыв С—Н
R'CONHCH2R2 -?ИЛИ->- R’CONHCHR2 R’CONHCHR2 R’CONHCHO
~Н‘ Ао* С"С +R’O.
(И6)
(п)
И.
разрыв С—N
R’CONH2 + R2CHO
16*
483
Электрофильные свойства пероксисоединений не проявляются в их реакциях с амидами. Продукты реакции с N-алкиламидамн аналогичны продуктам автоокисления, и, вероятно, протекание реакции обусловлено сходными свободнорадикальными процес-сами, так как проведение реакции при высоких температурах спо-собствует гомолизу пероксида. Так, N.N-диалкиламиды реагируют с диацилпероксидами, давая ацилоксизамещенные амиды, по схеме (178) [316]. С другой стороны, применение в качестве катализатора Н2О2 в присутствии Fe3+ вызывает расщепление связи N—С амидов, таких как N-метил- и N-бутилацетамид, давая первичные амиды [317]. Оба процесса можно объяснить по схемам, аналогичным автоокислению, однако какие-либо данные о механизме отсутствуют. Амиды, не имеющие атомов водорода у а-углеродного атома по отношению к азоту, под действием пероксисоединенип претерпевают прямое N-замещение, давая нитросоединения, возможно по схеме (179) [318].
HCONMe2 + (PhCO2)2 —> HCONMeCH-.OCOPh + PhCO2H (178)
H2O2 /0Н Н2О2 Г /0Н‘| Н2О2
RCONHPh------> RCON' •> РЫТ ——> PhNO------> PhXO2
\ph -Rco2h L \ohJ -H2°
(179)
При действии солей пероксидисульфатов лабильность атомов водорода связи (N—С)—Н в реакциях отщепления под действием радикалов также очевидна. Так, вторичные и третичные N-алкил-амиды при обработке пероксидисульфатами при умеренной температуре претерпевают деалкилирование, давая смесь менее замещенного амида и карбонильного соединения [319] {схема (180)}. Полагают, что окисляющей частицей является иоп-радпкал SOF* который отрывает атом водорода от связи (N—С)—Н и в результате вызывает расщепление связи С—N, аналогичное стадии (в) на схеме (177). Интересно, что лактамы превращаются в имиды, а не претерпевают разрыв связи С—N [103] {например, по схеме (105)}. Первичные амиды взаимодействуют с пероксидисульфатом по разному: формамид распадается на СО2 и NH3, вероятно в результате отщепления атома формильного водорода. Взаимодействие с ацетамидом требует присутствия каталитических количеств солей фосфора; здесь быстрое дезаминирование при 30 °C дает с количественным выходом уксусную кислоту [319].
S2O8'
R'CON(CH2R2)2 ------> R2CHO + R'CONHCH2R2
I S2°r
1------> R'CONH2 + R2CHO (180)
При обработке первичных амидов тетраацетатом свинца происходит необычная реакция, включающая окислительную перегруппировку, близкую к реакции Гофмана (ср. разд. 9.9.3.11) [99].
484
аияпьно образуется изоцианат, который удается выделить Первона онном основном растворителе, таком как ДМФ. Ес-только е। ротекает дальше, то взаимодействие с уксусной кис-
ли р„о(5ра3уЮщейся в качестве одного из продуктов, приводит к лотом, м^ну и производному мочевины {схема (181)}. Если в мо-ацетил ется подходящим образом расположенный нуклеофил, лекуле 1 гетепОциклические соединения. Механизм этой ре-в работе [99].
RCONH2 Pb(OAc)2 + 2НОАс + RNCO
I НОАс
1------► RNHAc + RNHCONHR
(181)
9.9.3.10. Восстановление
Амиды, как правило, очень устойчивы к восстановлению, и эффективны здесь только очень сильные восстановители, такие как гидриды металлов, натрий в жидком аммиаке (восстановление по Берчу) и процесс электролиза. Наибольшее применение среди вышеприведенных реагентов нашли гидриды металлов. Восстановление амидов обычно протекает по двум независимым направлениям, в зависимости в первую очередь от реагента и в меньшей степени от типа амида. По первому из этих направлений амидный карбонил восстанавливается до метиленовой группы с образованием амина {направление (а) на схеме (182)}. По второму направлению происходит расщепление связи С (О)—N и образуются амин и альдегид, который может далее восстанавливаться до спирта {направление (б) на схеме (182)}. Оба направления используются в синтезе, и при правильном подборе реагента и условий реакции обычно удается получить высокие выходы основного продукта. Эти данные обобщены в недавно опубликованном обзоре ,[Ю2].
(а)
—> r'ch2nr^
R’CONR* - |—> R’CH2OH
(б)
—> R2NH + R'CHO
(182)
Комплексные гидриды металлов, такие как алюмогидрид лития и алкоксипроизводные, нашли более широкое применение, чем орогидрид натрия [320] или диборан [321]. Большинство этих Реагентов восстанавливают первичные и вторичные амиды до амина без разрыва связи С—N {направление (а) на схеме (182)}. этих реакциях получают очень хорошие выходы, если наряду с Двумя эквивалентами гидрид иона, необходимого для восстанов-випи каРбоНильной группы, добавлять еще один эквивалент гиц-F Д-иона на каждую группу N—Н амида. «Избыток» гидрида
485
необходим, так как он расходуется в качестве основания при отщеплении протонов N—Н (см. ниже). Реакция с первичными амидами протекает медленно, и для полного восстановления при 0°С требуется ж 24 ч. Очевидно, что восстановление в этом случае включает индуцированную основанием дегидратацию до нитрила (следовательно, действительно необходим «избыток» гидрида) с его последующим быстрым восстановлением до амина. Существенно, что если использовать недостаточное количество гидрида, то с высоким выходом удается выделить нитрил. Предложен гипотетический механизм образования нитрила, включающий первоначальное возникновение комплекса за счет атаки карбонильного кислорода на А1Щ с последующим элиминированием 2 моль газообразного водорода [102].
Взаимодействие третичных амидов с комплексными гидридами металлов более сложно. В зависимости от субстрата и реагента, реакция может протекать по каждому из направлений, представленных на схеме (182). Наилучшими реагентами [321] для получения амина без разрыва связи С—N [направление (а) на схеме] являются А1Н3 и ВН3 с продолжительностью реакции « 30 мин при 0 °C. В случае применения борогидрида натрия часто необходимо предварительное превращение амида или в соль О-этили-мидата (при использовании Et3O+BF4, см. разд. 9.9.3.3.) [322], или в комплекс Вильсмейера с использованием РС15 или РОС13 (см. разд. .9.9.3.8) [323]. Менее обычным является восстановительное дезаминирование до альдегида [направление (б) на схеме], однако оно конкурирует с образованием амина при восстановлении алюмогидридом лития. Образование альдегида преобладает в основном в тех случаях, когда: 1) заместитель у азота является объемистым; 2) используется недостаточное количество восстановителя при температурах от —70° до 0°С; 3) когда гидрид добавляют к амиду (обратный порядок добавления) вместо добавления амида к гидриду, что принято в обычных методах восстановления. Помимо того, восстановительное дезаминирование N-гетероароматических амидов обычно приводит к смеси альдегидов и спиртов, независимо от применяемой методики. Наилучшими реагентами, способствующими образованию альдегидов, являются LiAlH(OEt)3 и LiAlH2(ОЕ1)г. При их использовании даже стерически незатрудненные третичные амиды дают альдегиды с выходом 60—90% [324].
Вторичные и третичные амиды превращаются с высоким выходом в альдегиды с помощью хорошо известного восстановления по Берчу, в котором применяется натрий в жидком аммиаке вместе с такими донорами протонов, как уксусная кислота или спирт. Механизм, предложенный Берчем с сотр., включает два последовательных присоединения электрона с последующим присоединением протона {схема (183)}; эта реакция нашла широкое синтетическое применение [325].
486
Н'
е~ -У0’ Н+ /°
p'CONRj R'C< -* R’C< , ~*
Rсиж<! \NR2 \\r) \Nr*
__> R'CH(OH)NR| —-> R'CHO + RiNH (183)
В отличие от реакции Берча, электролитическое восстановле-всегда приводит к амину без расщепления связи С—N {напыление (а) на схеме (182)}. Процесс обычно ведут в кислом paL.Bope (например, в H2SO4), применяя катод из свинца, и он Раск0 протекает, если у атома углерода или азота имеются элек-Лпоиооттягивающие заместители [326]. Первичные стадии процесса вероятно, аналогичны восстановлению по Берчу, однако образующийся аминоспирт восстанавливается далее floR'CH2NR до того, как произойдет расщепление связи {ср. схему (183)}.
д удалось также осуществить каталитическое восстановление амидов до аминов, используя в качестве катализаторов хромит меди и рений. Однако эта реакция не представляет интереса для синтеза в связи с нежелательными побочными реакциями. Для того чтобы осуществить восстановление амидов с помощью обычных катализаторов, таких как никель Ренея или палладий на угле необходимы высокая температура и давление. Это обычно означает, что может происходить восстановление не только амидной группы, но и других активных групп в молекуле.
9.9.3.11. Перегруппировки
Амиды являются относительно устойчивыми соединениями и перегруппировываются только при наличии определенных структурных особенностей. Хорошо известны четыре класса амидов, которые претерпевают перегруппировки [213]. К ним относятся N-ацил- или N-ароилазиридпны (где амидный азот включен в сильно напряженный трехчленный цикл) и такие N-замещенные соединения, как N-галогенамиды, N-нитрозоамиды и N-нитроами-ды. В данном разделе рассматриваются только термические и каталитические перегруппировки этих соединений. Перегруппировки, индуцированные фотохимически, обсуждаются в разд. 9.9.3.12.
Перегруппировка первого класса амидов (т. е. N-ацил- и N-ароилазиридинов) может происходить при повышенной температуре или под действием как нуклеофильных, так и электрофильных реагентов. В зависимости от природы ацильного или ароильного заместителя, а также от структуры азиридинового кольца перегруппировка обычно протекает по одному из двух возможных направлений. Первое направление, дающее оксазолнны (схема (184)}, осуществляется только в случае N-ароплазиридп-и°в, но при всех трех условиях реакции, в то время как второе управление, приводящее к образованию ненасыщенного амида (схема (185)}, вероятно, индуцируется только термически. Дру-м условием протекания второго направления является наличие
487
атома водорода, который расположен таким образом, что может образовывать с амидным кислородом шестичленное переходное состояние типа (117). Детали этих перегруппировок исследованы в обзоре [213].
ArCON
> Ar-^ J (i84)
R2
О
Н R^C^CHCHjjNHCR2 (185)
(117)k3
(R8=Ar, Aik)
Второй класс амидов, N-галогенамиды, претерпевает два основных типа перегруппировок. Первая — это реакция Гофмана (одна из наиболее известных реакций амидов в связи с ее важностью при укорочении длины цепи). В этом случае N-галоген-амид обычно образуется in situ при обработке первичного амида гипогалогенитом (см. разд. 9.9.3.7). Затем происходит отщепление протона группы N—Н под действием основания, которое способствует внутримолекулярной миграции алкильной группы к азоту с синхронным отщеплением галогенид-иона {схема (186)}. Образующийся таким образом изоцианат подвергается дальнейшим превращениям в зависимости от условий реакции. Например, протекает гидролиз с образованием амина или реакция с такими нуклеофилами, как амины или карбоновые кислоты. Более детально эта реакция рассмотрена в [327] При использовании брома в метоксиде натрия получены лучшие выходы продуктов реакции [328].
RCONHHal °с-н-2ванне> R-CON-Qlal ------------------> RNCO + Hal" (186J
Другая перегруппировка М-арпл-М-галогенамидов (перегруппировка Ортона) заключается в миграции галогена от азота к ароматическому кольцу {схема (187)}. Реакция имеет 0ГРа!!о' денное синтетическое применение п, как показано в обзорах [213, 829], некоторые аспекты ее механизма остаются неясными.
С1
MeCON
MeCONH
(187)
пип9пеВИ^Н0, 410 ТРП ГИПа катализагоров способствуют перегруз чппяк^п Ртона: первый — это УФ-облученпе или пероксиды, в зывающие гомолиз связи N-C1. что приводит главным образом
488
)пара-згмешению ароматического кольца; второй — гало-к °Р споты в протонных растворителях, что вызывает орто/пара-геН°Кцение возможно через стадию образования электрофильных 32Мркутярных галогенов (например, С12) и третий — уксусная или М°Л поруксусная кислота в апротонных растворителях, что выдает перегруппировку по более сложному механизму.
ЗЬ'В\’-Арнл- и N-алкил-М-нитрозоампды являются высокоактивны-соединениями, которые претерпевают несколько типов пере-“уппировок, представляющих интерес как с синтетической, так Г с теоретической точек зрения. Следует отметить, однако, что большая часть этих соединений является сильными канцерогенами Перегруппировки могут индуцироваться термически, фотохимически и с помощью основного катализатора. Любопытно, что термическая перегруппировка существенно отличается от фотохимической; в первом случае происходит разрыв связи С(О)—N, во* втором — N—NO. Фотохимические превращения рассматриваются в разд. 9.9.3.12. Первой стадией термической перегруппировки N-нитрОзоамидов является медленное образование азо-эфира (И8) с последующим быстрым распадом до различных продуктов, природа которых зависит от заместителей у азота {схема (188)}. В случае N-алкиламидов перегруппировка является гетеролитической, и в качестве продуктов образуются газообразный азот, олефин и карбоновая кислота или ее сложный эфир [330]. В случае N-арплампдов такие продукты, как биарилы, вовлекаются в свободнорадикальный процесс [331]. Механизм реакции в обоих случаях сложен; подробно он обсуждается в [213, 331]. Реакция служит эффективным способом дезаминирования N-алкпламидов, так как выходы сложных эфиров высоки (80— 90%), когда R2 является первичной алкильной группой, и умеренны (30—60%), когда R2 представляет собой вторичную или третичную алкильную группу.
(R1CO)3O + R2—R2 + No ит.д.
О R2=At>^
[R'C(O)ON=NR4
(118) (188)
R2 R2=AUT^ R^COaR2 >
(R2=AlRAr) или Г + Na
R1CO2H + Алкен}
Катализируемое основаниями разложение Ы-алкил-Ы-ннтро-зоамидов представляет препаративный интерес как удобный способ получения диазоалкапов, хотя при работе с этими соединениями следует проявлять осторожность в связи с их канцерогенностью {схема (189)}. Реакция может протекать через стадию разевания азоэфира, как и при термической перегруппировке, тивЯ“Г1[2Р°ЛИ3’ ка1али3иРУемы“ основаниями, является альтерна-
489
r1con(no)ch2r2 —>
r1coo^-n=ntchr2
—> tfCOa +
(159)
R2CHN?
A
R’CONR2 -------1
R‘COC1 <a> i
R2nh"-NO2 "70°c
O'
R'COON=NR2
(118a)
—> R‘COOR24-N2O
—> R'CO2H 4-Олефин + N20
(190)
N-Нитроамиды более устойчивы, чем соответствующие N-нит-розопроизводные. Они подвержены аналогичной термической перегруппировке, в которой вместо азота образуется оксид азота(1) {схема (190)}. Наилучшим доказательством образования азоксиэфира в качестве интермедиата (118а) является его встречный синтез [330] {направление (а) на схеме (190)}. Как и в случае N-нитрозоамндов, образование сложных эфиров является предпочтительным при наличии у азота первичной алкильной группы, а образование алкенов—при наличии вторичной алкильной группы.
9.9.3.12. Фотохимия
ласти спектраХ(<;250nP01,3B0^HbIe поглощают в УФ-об-Ряда фотохимических преврашенш?°С5бСТВуеТ пРотеканйю иелого пением реакций N нит™, Раи*ен11и. Эти превращения, за исклю-обзорах [107, 332].' рассмотрены в
блюдаемых продукта Т°Г° ЧТо^ы объяснить образование натри типа гомолитическогопростых N-алкиламидов, предложено тельстза расщепления а™ (меыа Д°мза-
основании обнаружения ма-гл [стадия (а)) получены на
котемпературН0РмУ фот °дом ЭПр Радикалов *CONH2 при низ-П07]. ДруГНе направлен» П?РРИЧНЬ1Х формамидов и ацетамидов РЫв атома водород Д *J(6) и <в) на <^ме] включают от-нильной и с аминогруппямиЛек°ДНЫХ атомов, соседних с карбо-этих радикалов типичны и”’ Ьольшннство последующих реакции нпем превращений кптЛпДЛЯ алкильных радикалов, за исключе-Радикал, вероятно ’ подвергается радикал (119). Этот
Р и превращается в пп 1 Рует ДаВая аминорадикал, кото-!прпДНО такпм образомР^УКТЬ1’ типичные для таких частиа-апИНщЫХ И ненасыщенных Ъясня[от образование аминов, wscu-тнп?а^Иоеских амидов в „у„ еВодоР°ДОВ, Н2 и СО при фотолизе (cd 4,20) уЖе Упомина лиг°КСаНе ИЛЙ гексане [333]. Радикал^
Р- хему (177)}, в то ь в связи с автоокислением амидо 490 ремя кап Радикалы типа (121} встреча-
ппи рассмотрении фотоамидированпя олефинов и ацетиле-1ПС\сР. разд. 9-9.3.3) Хромофоры (такие как двойные связи н°в арильные группы), расположенные рядом с карбонильной с аминогруппой, избирательно поглощают излучение, что "'".'яотит к разрыву различных связей. Например, фотолиз ацет-Прбрнзанилидов вызывает разрыв связи С (О)— N с последующим ’’^//щря-замещением бензольного кольца ацильными или аро-^ьными радикалами [334] {схема (192)}. N-Фениллактамы И ввергаются аналогичному гомолизу, однако в этом случае под П°1янием соседней группы происходит орто-замещение бензоль-Joro кольца [335]. ?
R'CH-j-CONHCHR2
H
н
разрыв
(а) С—С(О)
разрыв
<б> с—н
разрыв w с—н
R'CH2- + R=CH2NHCO
(119)
R‘CHCONHCH2R2
(121)
R'CH2CONHCHR2
(120) (191)
R>CH2NH+ СО
Фотолиз а,р-ненасыщенных вторичных амидов может приводить к гомолитическому разрыву связи N—Н с последующим замы
канием цикла и образованию в основном 0-лактамов. При фото-
лизе а- и р-лактамов образуются имины и СО; в случае 4-заме-щенных 0-лактамов — образуются изоцпанид и кетон [107]. Ряд других примеров фотолиза простых амидов приведен в обзоре
Розенталя [107].
hv
RCONHPh -----
(R = Me, Ph)
(192)
COR
При фотолизе двух классов более реакционноспособных амидов (N-галогенамидов и N-нитрозоамидов) происходит гомолитический разрыв связи N—Hal [332, 336] или связи N—NO [337] соответственно, что приводит к радикалу амида RCONR1 (недавно идентифицированному методом ЭПР) [338] и атому галогена или оксиду азота соответственно. Затем протекают типичные радикальные реакции, природа которых зависит от типа имидного заместителя R и R1 и присутствия других субстратов.
491
Например, при R = С6Н5 происходит замещение арена галогеном (т. е. фотоперегруппировка Ортона, см. разд. 9.9.3.11). Амидный радикал может кроме того отрывать водород от растворителя так, что последующая реакция с оксидом азота (например) приведет к оксимам, нитропроизводным, а также к нитритам из углеводородов [337] и кетонам из спиртовых растворов [339]. Особое значение для синтеза имеет реакция отрыва у-атома водорода от алкильной боковой цепи N-галогенамидов и N-нитро-зоамидов с последующим замещением в это положение. Странно, что замещение ограничено размером N-алкильной боковой цепи в N-нитрозоамидах [337], но не в N-галогенамидах, для которых замещение в ацильную боковую цепь изучено подробно [332, 336] {схемы (193) и (194) соответственно }. Обе реакции протекают с умеренным выходом (50—70%) и представляют собой метод введения функциональных групп в неактивированное у-положение.
9.9.3.13. Реакции имидов
Присутствие двух карбонильных групп в молекуле имидов увеличивает кислотность атома водорода аминогруппы, снижает нуклеофильность как атома кислорода, так и азота, а также увеличивает дефицит электронов на карбонильном атоме углерода. Если принять во внимание указанные различия, то реакции имидов можно рассматривать как достаточно близкие к реакциям амидов. Так, например, с большим трудом, чем в случае имидов протекают хорошо известные реакции нуклеофильного присоединения к карбонильному атому углерода (гидролиз или реакция с реактивами Гриньяра), а также реакции с электрофильными реагентами (алкилгалогенидами, ацилирующими и галогенирующими агентами и карбонильными соединениями). Вместе с тем
492
веется несколько примеров реакции с нитрозирующими и питающими агентами, а также с галогенангидридами неограниче-Гких кислот. Имиды достаточно устойчивы к окислению, но легко Останавливаются и подвергаются фотолизу. Большая часть этих реакций рассмотрена в недавно опубликованных обзорах 17, 8].
Гидролиз имидов легко проходит как в кислой, так и в основной среде, при этом основной катализ имеет место при низких
(деЗ), некатализируемый процесс реализуется при pH 2____
3 а кислотный катализ протекает при pH менее 1,5. В результате гидролиза происходит разрыв N-ацильнои связи и имид деацилируется до амида. В случае циклических имидов продуктами являются амидовые кислоты {схема (195)}, которые в зависимости от условий могут подвергаться дальнейшему гидролизу до дикарбоновых кислот и амина или аммиака [8].
Механизм катализируемого основаниями гидролиза сукцини-
мидов относительно прост и включает первоначальную атаку -ОН на карбонильный углерод и последующее образование тетраэдрического интермедиата, который затем распадается с разрывом связи С (О)— N.
Наблюдаемое влияние размера цикла (в случае циклических имидов) и природы заместителей у атома N можно было ожидать, например происходит возрастание скорости реакции при переходе от пятичленных (планарных) к шестичленным циклам вследствие меньшей Стерической затрудненности карбонильного атома углерода, а также при замене N-алкильных групп на N-фенильные [8, 340]. Фталимид проявляет необычные свойства, вероятно, из-за сильной ионизации очень кислой группы N—Н. Скорость реакции зависит от pH, но наличие электронооттягивающего заместителя в положении 4 облегчает гидролиз при pH 7—10; при более высоких pH скорость реакции не зависит от pH и не зависит от природы заместителя [8]. Напротив, скорость гидролиза N-арилфталимидов зависит от pH даже при высоких значения pH. Катализируемый кислотой гидролиз имидов затруднен и обычно требует продолжительного нагревания при 80—Ю0°С. Поэтому промежуточно образующийся амид не выделяют, ио гидролизуют дальше до карбоновой кислоты и аммиака или амина. Поскольку скорость реакции растет с увеличением кислотности [8], ее механизм включает, вероятно, атаку в°Ды на протонированный субстрат. Образуются ли в данном случае О- или N-сопряженные кислоты, точно не установлено.
493
Наблюдалось возрастание скорости гидролиза фталимидов за счет влияния соседних карбоксильных групп [341], а также за счет нуклеофильного катализа соседних имидазольных [342] и аминогрупп [343].
Как п в случае амидов, хорошо известна атака на карбонильный атом углерода имидов таких нуклеофильных реагентов как спирты (сольволиз), амины, гидроксиламин и гидразин, которая приводит в первых двух случаях к раскрытию цикла с образованием амидовых эфиров и диамидов соответственно [8]. В случае гидразина и гидроксиламина происходит последующее замыкание цикла, которое сопровождается включением в имидный цикл более нуклеофильного NH2- и НО-замещенного атома азота с образованием 1,1-диацилгидразинов и N-ацилгидрокса-мовых кислот соответственнно (см. разд. 9.9.4 и 9.9.5).
Имиды, как и амиды, взаимодействуют с карбанионами (полученными из реактивов Гриньяра) по карбонильному атому углерода [7, 8, 344]. Первой стадией реакции является образование замещенного а-гидроксилактама, затем в зависимости от природы реактива Гриньяра может протекать дегидратация или раскрытие цикла {схема (196)}. Конкретные примеры таких реакций рассмотрены в упомянутых выше обзорах. В связи с повышенным по сравнению с амидами дефицитом электронов на карбонильном углероде имиды могут присоединять некоторые менее активные карбанионы, такие как анионы из цинка и а-гало-генэфиров или фосфоранов (реакции Реформатского [345] и Виттига [346]), давая алкилиденовые производные; а также взаимодействовать с ацетиленидом натрия, давая гидроксилактамы [8].
I—> R‘NHCO(CH2)nCOR2
Алкилирование имидов алкилгалогенидами обычно протекает в основных условиях (т. е. с калиевой или серебряной солью имида) при 150—180°С и приводит к N-алкилпроизводным {схема (197)}. Эта реакция составляет основу синтеза аминов по Габриэлю, в котором получение амина состоит в мягком гидролизе имида после алкилирования алкилгалогенидом [8]. Алкилирование в нейтральных условиях более активными солями оксо-ния или алкилсульфатами изучено недостаточно. Однако имеются данные [347] о внутримолекулярном О-алкилировании имида соседней галогеналкильной группой, что приводит к возникновению 494
м ацплгетероцикличсской соли в качестве интермедиата с после-раскрытием цикла и образованием амидового эфира /схема (198)}. Эта реакция аналогична получению лактамов из галогеналкиламидов {см. схему (136)}. Как и в случае амидов, при взаимодействии 2 моль амида натрия с соответствующим имидом (например, глутаримидом), происходит образование карбаниона (в a-положении по отношению к карбонилу) и аниона азота, однако обычно преимущественно алкилируется карбанион, а не азот [348].
н2О •>
RCOO^H^NHCOR1
(198)
Реакции имидов с альдегидами и кетонами очень близки аналогичным реакциям вторичных амидов. Так, формальдегид, ацетон и изобутиральдегид реагируют в нейтральных или основных условиях, давая соответствующие N-a-гидроксиалкильные производные {ср. схему (144)}, которые в случае высших гомологов могут далее дегидратироваться [7, 8].
Ацилирование незамещенных имидов хлорангидридами или ангидридами кислот приводит к триациламинам (RCO)3N. Имеются косвенные доказательства того, что в слабоосновных условиях (например, в пиридине) реакция протекает через промежуточное образование О-ацильных производных [102]. При взаимодействии с более активными ацилирующими агентами, например с кетенами и с изопренильными эфирами, образуются N-замещен-ные продукты, аналогичные получаемым из амидов (см. разд. 9.9.3.5) [349]. В случае N-алкилдиацетамидов дальнейшее замещение по азоту невозможно, однако с помощью 14С-меченого уксусного ангидрида в пиридине продемонстрирован факт трансацилирования [350].
„N-Галогенимиды, получаемые из соответствующего имида Действием молекулярного галогена или гипогалогенит-иона [327], являются хорошо известными источниками атомарного или положительно заряженного галогена. Широкое применение в синтезе нашел главным образом N-бромсукцинимид [353]. Гетеролптпче-ское бромирование под действием N-бромсукцинимида (т. е. через НОВг или Вг-) может происходить: в водной среде, когда '^соединение к олефинам идет согласно правилу Марковникова [352]; в концентрированной H2SO4, где замещение в ароматическом
495
кольце протекает согласно орто/ппрп-ориентации [354], а также в смеси пиридина и трет-бутанола, где вторичные спирты подвергаются а-замещению [352]. Гомолитическое галогенирование под действием N-бромсукцинимида (т. е. через НаЬ) протекает либо при облучении УФ-светом, либо в присутствии инициаторов радикальных реакций, например пероксидов. Типичными реакциями такого типа являются замещение аллильного или бензильного атома водорода (реакция Воля — Циглера), замещение в ароматическом ядре или даже замещение водорода у насыщенного атома углерода [352]. Эти реакции являются цепными радикальными процессами; их детали зависят от природы N-галогенимида [7, 355].
Другими интересными, но менее важными реакциями N-гало-генимидов являются введение СНг-группы между азотом и галогеном по связи Н—Hal [297] (ср. N-галогенамиды), а также катализируемая основаниями перегруппировка N-галогенфталими-да, приводящая к изоцианатам (аналогичная реакция Гофмана для амидов) {схема (199)} [7].
О
Данные по нитрованию и нитрозированию имидов отсутствуют, а количество примеров реакций с галогенангидридами неорганических кислот незначительно. Взаимодействие РС15 с глутаримидом приводит к ди-, три- и тетрахлорированным пиридинам [7], образование которых объясняется процессом, уже рассмотренным в случае амидов (см. разд. 9.9.3.12). РОС13 реагирует аналогичным образом [7].
Имиды очень устойчивы к окислению, н на них не действуют такие реагенты, как оксид азота, оксид хрома и перманганат калия. Это свойство имидов дало возможность применить удобный метод защиты амидных групп (путем превращения в имид) в процессе окисления других фрагментов молекулы [355]. Имиды гладко восстанавливаются с помощью таких стандартных методов, как взаимодействие с комплексными гидридами металлов, каталитическое гидрирование и электрохимическое восстановление. Из циклических имидов обычно образуются а-гпдроксилакта-мы, лактамы и амины {схема (200)}. Электрохимическое восстановление на свинцовом или на амальгамированном цинковоМ катоде, а также восстановление LiAlH4 обычно приводит к получению лактамов. При действии LiAlH4 наличие заместителей в кольце может способствовать образованию а-гидроксилактамов. Каталитическое гидрирование обычно приводит к амидам [7|-Хотя как имиды, так и амиды, поглощают в УФ-области при-496
ельно при той же длине волны, фотохимия имидов до по-близит ВремеПп не вызывала интереса. Известные сейчас фо-СЛеДпегруппировка Фриса ароматических имидов [356] и пере-тоПедТома хлора в N-хлоримидах в у-положение ацильной боко-Н°" ,трпи [332] напоминают процессы, описанные в случае амидов ВОН це11н lQ Q о 1<л (сМ. разд. 9.9.3.12).
о 9
о
NH
NH
(200)
NH —> L/NH ИК^ОН
(201)
^>Br(CH?)2CONCO
(202)
О
Другими интересными перегруппировками под действием УФ-света являются расширение цикла N-алкиламидов {схема (201)} [357] и раскрытие имидного кольца с образованием ацилизоцианата {схема (202)} [332]. Последняя реакция может конкурировать с реакциями фотоиндуцированного бромирования.
9.9.4. ГИДРОКСАМОВЫЕ КИСЛОТЫ
Гидроксамовые кислоты (122), хотя и были впервые получены Лоссеном в 1869 г., остаются одним из наименее точно охарактеризованных классов органических соединений. Основная трудность состоит в существовании трех типов структурных изомеров, соответствующих монозамещеиной гидроксамовой кислоте Н123), (124) и (125)]; при этом (123) существует в (Е)- и (Z)-формах. Надежное отнесение структурных изомеров зависело от /?9В?ения современных физических методов. Вещества строения И22), (124) и (125) являются N-гидрокси- или N-алкоксизаме-Щенными амидами, в то время как (123) представляет собой N-за^РОксианал°г эфира имидата (см. разд. 9.9.3.3). Названия пе-Нийещеннь1х гидроксамовых кислот (122) происходят от назва-М0 С00ТВетствующих карбоновых кислот, например бензогидрокса-
ая кислота — это (122, R1 = Ph). Точное название замещенных
497
производных сопряжено с рядом трудностей, так как протоны групп N—Н и О—Н (122) кислые. Однако так как все реакции монозамещения гидоксамовых кислот приводят к N___OR.
производным (125), их можно рассматривать как эфиры гидрок-саматов и называть О-алкил(арил)гидроксаматы. При этом N-замещенные производные (124) будут называться N-алкил (арил)-гпдроксамовые кислоты. Дальнейшее замещение в соединениях (124) и (125) приводит к (126), т. е. к N-алкил(арил)-О-алкил-(арил) гпдроксаматам. Соединение типа (123) со структурой имина является эфиром таутомерной гидроксимовой кислоты RC(OH)=NOH и, следовательно, будет называться О-алкил-(арил)гидроксимат; двойное замещение приведет к (127), т. е. к 0,0-диалкил(диарил)гидроксимату.
NH
/OR2
R‘C/ /°Н N
(122)
R’C^ /OR2 NH
(125)
(123)
R‘C\ /OR2 N
R3
(126)
R’C/ /ОН N
R2
(124)
/OR2
R'c^ /OR3 N
(127)
Другие аспекты структуры гидроксамовых кислот и их реакционной способности заметно отличаются от таковых для амидов и не могут быть объяснены путем простого развития химии амидов. Так, причины исключительно высокой кислотности всех трех соединений (122), (124) и (125) и сильная нуклеофильность анионов (122) и (124) до сих пор полностью не прояснены. Кроме того, анион (125) показывает большую склонность к амбидентному поведению, чем анион соответствующего амида, так что из гидроксамовых кислот гораздо чаще образуется смесь О- и N-замещенных продуктов, чем из амидов. Подробный обзор работ по химии гидроксамовых кислот, опубликованных до 1944 г., написан Ялем [358], но, к сожалению, некоторые из предложенных структур сейчас уже устарели. Краткие недавно опубликованные обзоры Сэндлера и Каро [За] по синтезу гидроксамовых кислот, Смита [1], а также Бауэра и Экснера [36] по свойствам и реакциям этих соединений дают общее представление о литературе-Гидроксамовые кислоты, являющиеся составной частью гетероциклических соединений, составили предмет отдельных обзоров [359], и химия этих веществ в данном разделе не рассматривается. Данные о физиологически активных гидроксамовых кислотах кратко изложены в обзоре Бауэра и Экснера [36].
498
9 9 4.1. Методы получения гидроксамовых кислот
и в случае амидов, одним из наиболее распространенных в получения гидроксамовых кислот является ацилирование меТОД°силаМина или его О- или N-замещенных производных. ГИДР°егиды можно превратить в гидроксамовые кислоты либо че-^ЛЬстадию образования оксимов, либо обработкой нитрозобензо-Ре3 Относительно малоизученными альтернативными методами "Лучения являются окисление аминов или амидов, а также пе-П°гпуппировка нитроалканов. Как и в случае амидов, алкилиро-^ние гидроксамовых кислот приводит к более высокозамещенным ваоизводным. Примеры применения большинства указанных ме-щдов приведены в обзоре [За] и в работе [360].
Моноацилирование гидроксиламина гладко осуществляется с помощью большинства ацилирующих агентов, однако при этом могут также образовываться ди- и триацетильные производные. Поскольку гидроксиламин является сильным нуклеофилом (а-эффект) [361а], он легко реагирует со сложными эфирами карбоновых кислот, и, как правило, эти реагенты оказываются лучшими [За]. Реакцию проводят обычно в основных условиях {схема (203)}, применяя раствор гидроксида калия в спирте [362] или в пиридине [363]. Растворимые в воде гидроксамовые кислоты выделяют осаждением их медных солей [364]. В отличие от соответствующей реакции с аминами механизм реакции с гидроксиламином не привлек особого внимания, за исключением взаимодействия с у- и б-лактонами [365], при котором образуются о-гидроксиалкилгидроксамовые кислоты.
RCOX + NH2OH —> RCONHOH (X = OR1, Hal, OCOR, NH2)
(203)
Галогенангидриды кислот используются редко, вероятно потому, что трудно предотвратить дальнейшее ацилирование, однако ряд синтезов приведен в работе [360]. Интересное наблюдение [3666] о преимущественном образовании О-ацилгидроксиламннов вместо желаемых N-ацилпроизводных является настолько исключительным, что вряд ли может найти препаративное применение.
Ангидриды кислот также взаимодействуют с гидроксиламином, однако в синтезе применяются только циклические ангидриды. В результате реакции образуются относительно устойчивые Циклические N-гидроксиимиды [367] {схема (204)}, представляющие собой N-ацильные производные гидроксамовых кислот. За-меЩенные гидроксамовые кислоты (126, R2 или R3 = Aik или Аг) можно получать аналогичным образом из соответствующего заеденного гидроксиламина {схема (205) и (206)} и хлорангид-Р да кислоты в пиридине. Вероятность дп-Н-ацплпрования мож-уменьшить использованием только 1 моль-экв хлорангидрида лоты [368]. Гораздо труднее предотвратить диацилирование лкилгидроксиламинов, и поэтому рекомендуется альтернативная
499
/=О + NH2OH —► О=^/ ^>=0 (204)
методика синтеза, где при использовании избытка реагента получают О,П-диацилгидроксиламии {стадия (б) на схеме (206)}, далее в присутствии метоксида натрия избирательно гидролизуемый по О-ацилыюй группе {стадия (в) на схеме (206)} [369].
О NOH
0=
R'COCl +NH2OR2 —> R'CONHOR2 (205)
(б) r'coci
R'COCl + R2NHOH —Д R’CONR2OH у > R'CONR2OCOR‘ (206)
NaOMe
(в)
Менее широко используемыми ацилирующими агентами являются амиды [370], N-ацилимидазолы [371], кетены и изоцианаты [372]. Все эти соединения взаимодействуют с гидроксиламином с образованием гидроксамовых кислот или их производных.
Помимо ацилирования гидроксиламинов существует мало других общих методов синтеза гидроксамовых кислот, хотя ранее были указания на некоторые интересные возможности (358). Два метода превращения альдегидов в гидроксамовые кислоты разработаны недавно. Один из них включает получение оксима и затем гидроксамоилхлорида RCC1=NOH, который далее гидролизуется до гидроксамовой кислоты [373]. Второй метод состоит в обработке альдегида нитрозобензолом в присутствии изопропо-ксида алюминия [374].
Другой новый метод синтеза гидроксамовых кислот (со средним выходом 40—60%) заключается в обработке солен нитроалканов (например, MeCH=NOiNa+) или нитроновых эфиров (МеСН = NOaEt) концентрированной H2SO4 [375]. Окисление подходящих предшественников, таких, как оксимы, амиды и амины, не нашло широкого применения. Модификация гидроксамовых кислот путем О- или N-алкилированпя рассмотрена в разд. 9.9.4.3 [376].
9.9.4.2. Свойства гидроксамовых кислот
Рентгеноструктурные исследования [377] гидрата ацетогид-роксамовой кислоты (128) [указаны длины связей (в нм) и валентные углы] показывают, что атомы С-1, С-2, N и О-2 лежат в одной плоскости, а атом О-l находится на расстоянии
0,0056 нм от этой плоскости. Длины связей и валентные углы в остатке RC(O)N очень близки аналогичным величинам у амидов [ср. (57а), разд. 9.9.2.1], что указывает на частичную делокализацию неподеленной пары электронов азота в направлении я-электронов карбонильной группы; порядок связи С (О)—N« Е5-Следовательно, можно ожидать изомерии относительно этой ча-
500
двойной связи. Рентгеноструктурные данные указывают СТН«е на присутствие молекулы воды, связанной водородными та ями С двумя молекулами гидроксамовой кислоты. Такое не-С^Я[ЧНое связывание, как правило, не наблюдаемое в случае ами-вероятно обусловлено образованием стабильного семичлен-д0 ’ цикла для каждой из двух молекул гидроксамовой кислоты Н° сЧет включения группы О—Н из воды. Это означает, что (Z)-изомер [см. формулу (128)] будет устойчивее в случае гидроксамовой кислоты, чем для амида, если не принимать тщательных мер предосторожности против попадания влаги. Образование (Е)-изомеров, маловероятное уже в случае амидов, представляется еще более затрудненным для гидроксамовых кислот, имеющих свободную группу NOH.
Н
..ьГ I0’195
/4-0)2)
/ \ „ 0,075
/ 0,125 /121
122° IA УО)
(2ГС^ Х\0,090 (128)
^114° '
Н
(Z)-
I о
(129 а)
Н /9
L
Ra (12 9 б)
Существуют и другие, хотя и немногочисленные, физические данные, указывающие на то, что связанная внутримолекулярной водородной связью структура (Z-конфигурация) может существовать также в растворе соединений (122) и (124), но не (125) (в котором отсутствует группа NOH). Так, дипольные моменты бензо- и и-нитробензогидроксамовых кислот в бензоле и диоксане (4,3—4,5 D) [378] существенно больше, чем дипольные моменты аналогичных амидов, что лучше всего объясняется наличием (Z)-конфигурации, как в (128). Трудно провести надежное обобщение всей обширной информации по ИК-спектроскопии гидроксамовых кислот [379—381], так как исследования выполнены в различных фазах. Однако амидная полоса I обнаружена в области 1620—1655 см-1 в нуйоле, в области 1660—1675 см-1 в три-хлорметане и в одном случае ("hCONHOMe) в области 1694см-1 в сухом диоксане. Последняя величина близка к частоте поглощения свободной аминогруппы — амидная полоса I — в простых амидах (см. табл. 9.9.1), в то время как величины в нуйоле и тРихлорметане соответствуют частоте поглощения связанной аминогруппы— амидная полоса I —в простых амидах. Высокое значение частоты поглощения PhCONHOMe в сухом диоксане рассматривается [380] как доказательство отсутствия межмолекулярного взаимодействия или связывания с растворителем, в то время как амидные полосы I в области более низких частот, характерных для PhCONHOH (1640 см-1) и PhCONMeOH
501
(1670 см-1) в сухом диоксане, могут указывать на наличие внутримолекулярной водородной связи. Однако для подтверждения этого вывода необходимы дальнейшие доказательства. Структуру гидроксамовых кислот с внутримолекулярными водородными связями обычно изображают как (129а). Однако включение молекулы воды [как в кристаллической структуре (128)] позволяет осуществить более линейное расположение (1296), которое, как полагают, необходимо для образования водородных связей.
Для арилгидроксамовых кислот и их N- и О-замещенных производных УФ-спектр охарактеризован только в дальней УФ-области [380, 381], хотя, как правило, больший интерес представляет поглощение в более длинноволновой области (220—260 нм), связанное с арильным кольцом. Однако в работе [385] обращалось внимание на существование полосы поглощения (А.макс 205— 210 нм, 8 16 000—25 000), сравнимой с полосой поглощения амидов, наличие которой связывают с л —>-л*-переходом карбонильной группы.
ЯМР-спектроскопия гидроксамовых кислот исследована мало, и основное внимание здесь уделено вопросу о затрудненности вращения группы NOR в замещенных соединениях. Наблюдалось также медленное вращение вокруг связи С (О)— N [т. е. взаимопревращение (Z) и (£)-изомеров] на примере N-бензил-О-бензопл-бензогидроксамата [382] и Ы,О-дизамешенного формогидроксамата HCONROR' [383]. Кроме того, в случае первого соединения [382] и У-изопропоксипиридона-2 [384] наблюдалось медленное вращение вокруг связи N—OR. Эффект, наблюдавшийся для последнего соединения, близок к вращению вокруг связи N—Аг в анилидах (см. разд. 9.9.2.1). Изучены масс-спектры О-алкилгидроксаматов RCONHOR1 {R = Ph, PhCH2, Ph(CH2)2; R1 = H, Me, Et, PhCH2] [385] и предложены два основных направления их фрагментации. Первое направление, найденное также в случае вторичных амидов [ср. схему (НО)], связано с разрывом связи С (О)—N. Второе направление, обнаруженное только в случае гидроксамовых кислот, состоит в отщеплении гидроксильного радикала с образованием характеристического иона М-17 [схема (207)]. Обнаружены и другие типы фрагментации, но их протекание зависит от природы заместителей.
.О /он
R'C^ 7-» R>CZ —> R'Cs=NOR2 + HO« (207)
ЧЧН—OR2 ^N—OR2
,O .OH
RCT —> RCT <208)
\\HOH ^NOH
Гидроксамовые кислоты могут таутомерно превращаться {схема (208)} в гидроксамовые кислоты. Как и в случае амидов, доказательства существования таутомеров гидроксамовой кислоты отсутствуют, хотя соответствующие эфиры R1C(OR2)=NO 602
естны (см. разд. 9.9.4.3). Незамещенные и монозамещенные П.,дроксамовые кислоты напоминают амиды амфотерным поведе-Гием Однако в случае незамещенных соединений ситуация усложняется за счет присутствия двух кислых протонов (NH и NOH). Основные свойства близки к таковым для амидов; р/<а бензогидро-ксамовых кислот « 1,5—2,0 [386]. Протонпрование также подчиняется функции кислотности Н\ (измеренной с помощью амидных индикаторов) и, следовательно, происходит по карбонильному кислороду (130) (как и в случае амидов, см. разд. 9.9.2.3). Напротив, как гидроксамовые кислоты, так и их О- и N-монозамещенные производные RCONROH и RCONHOR — гораздо более сильные кислоты, чем соответствующие амиды (p/G 8—10 по сравнению с примерно 16 для амидов). Это может быть связано, по крайней мере частично, с индукционным оттягиванием электронов от соседнего электроотрицательного гетероатома (О или N); вместе с тем сопряженные основания как из RCONROH, так и из RCONHOH, могут затем стабилизоваться за счет внутримолекулярных водородных связей.
.ОН .ОН
Rc^ /ОН *“*• ГС/ /ОН NH NH
+
(130)
Несмотря на большое число исследований, структура сопряженных оснований из незамещенных гидроксамовых кислот остается спор" ной [360, 387]. Одной из возможных форм является анион, который стабилизуется посредством внутримолекулярных водородных связей с участием одной молекулы воды (131) {схема (209), ср. рентгеноструктурные данные о строении нейтральных гидроксамовых кислот}. Существование этой структуры объясняет, почему при алкилировании или ацилировании аниона незамещенной гидроксамовой кислоты (см. разд. 9.9.4.3) образуются только (N)—О-, а не N-алкилированные производные (необходим ион RCON_OH), а также объясняет влияние заместителей на кислотность бензо-гидроксамовых кислот [360].
Гидроксамовые кислоты и их N-замещенные [но не (Ы)-О-за-мещенные] производные являются бидентантными лигандами по отношению ко многим ионам металлов, таким, как Fe(II), Си(II), А1(Ш), V(V), Mn(II), U(III) и Sn(II) [388]. Образующийся комплекс, как правило, нейтрален и либо осаждается из водного раствора (давая возможность проводить гравиметрический анализ), либо может быть извлечен из него с помощью органических растворителей. Многие комплексы сильно окрашены (от красного До фиолетового), и поэтому очень удобны при колориметрическом анализе ионов металлов. Образование фиолетовой окраски при обработке хлорным железом любого соединения, содержащего группу ~~C(O)N—ОН, является хорошо известным качественным тестом
503
,о
о
на гидроксамовые кислоты [389]. Хотя большинство комплексов подробно не исследовалось, их структура, вероятно, лучше всего может быть представлена формулой (132). Недавно было показано существование другого типа комплексов со стехиометрией 1 :1 между незамещенными гидроксамовыми кислотами (но не их 0-или N-замещенных производных) и борат-ионом с возможной структурой (133), которые образуются в результате отщепления одной молекулы воды [390]. Выводом из этой работы является то, что измерения рК и скоростей реакций для незамещенных гидроксамовых кислот, проведенные в боратном или фосфатном буфере, вероятно, были ошибочными.
(132)
(га=степепь окисления М)
9.9.4.3. Реакции гидроксамовых кислот
nqJ3eaMlilI! гидроксамовых кислот во многих отношениях напомп-nrtn-LреаКЦНН ам,,дов- Различия в реакциях возникают главным ооразом при наличии незамещенной группы (N)—ОН. Однако присутствие второго атома кислорода в цепи О = С—N—О— гидрокса-х кислот изменяет как их нуклеофильность, так и направления
кигпУ^еНИЯ’ П0 сРавненню с амидами. Например, гидроксамовые чтпм ™ значительно более сильные нуклеофилы, чем амиды; при ним kn °Р°ДНЬ,И атом (N)—О является более реакцнонноспособ-атт)пп Г1Ме м0Г0’ относительная реакционная способность амидных этих- пт И N В ^РОксамовых кислотах отличается от активности омов в амидах: так, калиевые соли гидроксамовых кислот
504
ашт более высокую долю О-замещенных продуктов, чем амиды Д ^логичных условиях (см. разд. 9.9.3.3).
варидролиз гидроксамовых кислот до карбоновых кислот и гидр-пксиламииов протекает достаточно легко как в кислой, так и в ще-
чНой среде {схема (210)}. При умеренной кислотности (0,1 — Og М) скорость гидролиза, как правило, пропорциональна концентрации кислоты, но затем она проходит через максимум при более высоких значениях кислотности, соответствующих полному прото-„прованию субстрата [386]. Корреляции функций кислотности мОгуТ означать, что механизмы кислотного гидролиза гидроксамовых кислот и амидов близки (ср. разд. 9.9.3.1). Однако недавние работы указывают на то, что влияние полярных заместителей на гидролиз как замещенных бензогпдроксамовых кислот, так и алифатических гидроксамовых кислот, более важно, чем в случае соответствующих амидов [391а]. О гидролизе, катализируемом основаниями, как и в случае некоторых амидов, известно немного, известна лишь зависимость первого г. второго порядка от концентрации гидроксид-ионов [3916]. Это указывает, что реакция проходит через тетраэдрический интермедиат.
Н2О, Н+ или НО*
RJCONR2OR3 --------------> R'CO2H + R2NHOR3 (210)
2CH2N2
RCONHOH --------> RC(OMe)=NOMe (211)
основание R’X
RC(0)NHOH----------> RC(O)\HO’ ----> RC(O)NHOR' (212)
Нуклеофильные реакции гидроксамовых кислот осложнены тремя возможными направлениями замещения [N, (N)—О и (С)=О]. По поводу алкилирования в нейтральных условиях известно лишь, что обработка избытком диазометана приводит к метилированию обоих атомов кислорода {уравнение (211)} [392]. Большое внимание уделено изучению алкилирования в основных условиях, когда активной частицей является гидроксамат-аннон (131). Его активность по отношению к электрофильным агентам значительно выше, чем у фенолят-ионов той же основности, что приписывается «a-эффекту» соседнего гетероатома (т. е. электронному отталкиванию неподеленной парой электронов азота, что увеличивает доступность электронов на кислороде). Для объяснения повышенной реакционной способности было предложено и альтернативное объяснение, основанное на внутримолекулярном катализе. Независимо от природы эффекта происходит преимущественное замещение у атома (N)—О гидроксамат-иона с образованием у-алкилгидроксамата {схема (212)}. Отсутствие продуктов N-ал-нлирования кажется неожиданным, однако может быть объяс-Но в предположении, что гидроксамат-ион существует только Т(ЛЙДе .истицы, связанной водородной связью (131), где отрица-ьньщ заряд частично расположен на обоих атомах кислорода, но
На азоте. Алкилирование обычно проводят обработкой 605
гидроксамовой кислоты алкилгалогенидом в присутствии NaOMe-с выходом 50—80% образуется продукт моно-О-алкилирования й незначительное количество диалкилпроизводного [379а]. Изучено строение этого диалкилпроизводного [393]. Возможно образование трех продуктов, а именно N-алкилгидроксамата (135), а также (£)- и (/[-изомеров О-алкилгидроксамата (136) и (137). При алкилировании калиевой соли (134) алкилгалогенидом образуются (135) в качестве главного и (136) в качестве побочного продукта-их точное соотношение зависит от природы растворителя. Изомеризация (136) в (135) в условиях реакции не происходит. Гидрок-самат-ион должен обладать амбидентными свойствами (134а)
R‘C(=O)NOR2 ч-> R‘C(d)=NOR2 (134а)
аналогичными свойствами серебряных солей амидов (см. разд. 9.9.3.3). Алкилирование серебряных солей гидроксамовых кислот приводит к увеличению доли (2)-О-алкильного изомера (136), так как и к образованию (£)-О-алкильного изомера (137), но точные причины образования этого дополнительного продукта неясны.
у OR2 NR3
(135)
R3O, /OR2
R' (Z)-
(136)
R3ox /C=N *(£)-x°r2
(137)
Ацилирование незамещенных гидроксамовых кислот [3616, 394] хлорангидридамп и ангидридами кислот и Сложными эфирами также протекает преимущественно по атому (N)—О. Образующийся О-ацилгидроксамат R'CONHOCOR2, если температура достаточно высока или раствор сильно основен, может подвергаться хорошо известной перегруппировке Лоссена (см. ниже). Происходит также и дальнейшее ацилирование, приводящее к O.N-диацилгид-роксамату R‘CON (COR2) OCOR2.
Амбидентная природа гидроксамат-аниона R'CONOR продемонстрирована и при ацилировании, Так, соли О-бензиловых эфиров арил- и алкилгидроксамовых кислот взаимодействуют с уксусным ангидридом в пиридине, давая продукты О- и N-ацилирования (138) и (139) [381]. В случае серебряной и калиевои[солей О-бензиларплгчдроксаматов главным продуктом (>99 /о! является (138), особенно при взаимодействии с субстратами, н сущими электроподонорные пара-заместители. Однако в СЛУ^ О-бензилапетогидроксаматов соотношение О- и N-изомеров си но зависит от противоиона. Так, при реакциях К+-солей прео дает N-ацилирование (139), а А£+-соли дают в основном О-и мер (138). Эффект Ag+ в этом случае аналогичен его влия на процесс алкилирования гидроксимат-ионов
606
ОСОМе
О
RC^NOCH2Ph
(138)
RC
\NOCH2Ph
COMe
(139)
Ппи взаимодействии с гидроксамовыми кислотами менее рас-паириных ацилирующих агентов, таких как изоцианаты, ди-ПРП кетена [395] и ацетали [396] {схемы (213)-(215) соот-мертвенно] первоначально образуются (N)-О-ацилированные веТдУкты. Однако в первых двух случаях продукты легко распа-ПРются при нагревании в результате перегруппировки Лоссена Да ниже), давая мочевины (из изоцианатов); изоцианаты PNCO, ацетон и СО2 (пз димера кетена). Последняя реакция используется как метод синтеза изоцианатов, так как другие продукты легко отделяются.
RCONHOH + PhN=C=O
Н2С=С—О RCONHOH+ | |
Н2С—с=о
RCONHOH + PhCH(OEt)2 —> PhCONHOCOPh
RCONHOCONHPh
(213)
RCONHOCOCH2Ac
(214)
(215)
> R1N=C=O + R2CO2 —> Продукты конденсации (216)
Перегруппировка Лоссена (М)-О-аиилзамещенных мовых кислот является одним из наиболее подробно разделов химии гидроксамовых кислот [36, 71]. Близость механизма этой реакции с перегруппировками Курииуса, Гофмана, Бекмана и Шмидта (обсуждались в разд. 9.9.1 и 9.9.3) состоит в том, что разрыв связи N—О сопровождается синхронной миграцией алкильной группы {схема (216)}. Как и в реакции Бекмана, неорганические ацилирующие агенты (например, фосфорил-или сульфонилхлориды) активно способствуют разрыву связи
О- В связи с этим О-фосфорилированные или О-сульфонили-Рованные интермедиаты выделить не удается, в то время как U-ацильное производное (140) относительно устойчиво. С помощью нового реагента, полученного из эквимольной смеси три-метиламина и диоксида серы, удается синтезировать кристаллический аддукт 1:1 (RCONHOSO3H • NR3), который легко распа-в присутствии основания с образованием изоцианата I °Г N-Гидроксиимиды также подвергаются перегруппировке типа оссена [38], но в этом случае N-фосфорилоксп- пли N-сульфо-локсиинтермедиаты удается выделить, так как они достаточно
гидрокса-изученных
607
устойчивы. Перегруппировка начинается в основном раствопр с гидролиза имидного цикла {схема (217)}, затем промежуточна образующийся изоцианат гидролизуется в условиях реакции ло амина. Описана другая перегруппировка, включающая разпыв [399] связи N—О, где после арилирования MeCONPhO- солями дифенилиодония Ph2I+ и последующего разрыва связи N—CHPh) образуются орто- и пара-замещенные бифенильные производные амида {схема (218)}.
MeCON—O'+ Ph2I+ —> [MeCON(Ph)OPh] —>
(217)
MeCONH-
(218)
(219)
но7
Взаимодействие О-метиларнлгидроксамовых кислот с РС15 и РВг5 дает соответствующий (Е)-гидроксимоилхлорид или -бромид {уравнение (219)} [381]. Такие (Е)-изомеры под действием УФ-облучения могут превращаться в менее устойчивые (Z)-изомеры. Конфигурация этих соединений была установлена с помощью измерений дипольных моментов и затем уточнялась на основании данных ЯМ.Р-спектроскопии.
РС15 С1\
ArCONHOMe -----> \=N4
Аг/ 'OMe (£)-
Механизм образования гидроксимоилхлорида мало изучен, однако он, вероятно, напоминает механизм получения пмидоилхло-ридов из амидов (см. разд. 9.9.3). Реакции гидроксамовых кислот с другими электрофильными реагентами, такими как бромнова-тистая или азотистая кислоты, подробно не исследованы, однако предполагается [358], что продукты реакции неустойчивы и распадаются с разрывом связи С (О)—N.
Гидроксамовые кислоты, а также их О- или N-алкильные производные можно восстанавливать до амидов гидрированием над никелем Ренея или над амальгамой натрия [400] <схе” (220)}. При восстановлении алюмогидридом лития образует , 608
соответствующий амин [401], вероятно за счет дальней-оДНаК°’оСстановления промежуточно образующегося амида.
Ц1еГ0 R'CONR2OR3 - г>1-"-> R'CONHR2 (220)
Ni Ренея 4 ’
(R2, R3 = H, Aik)
гидроксамовых кислот до первич-
Окисление незамещенных
амидов можно проводить с помощью кислоты Каро или пер-нь1Х а водорода [402]. Такие окислители как перйодат, бром 01<С"к-бромсукцин11мид в присутствии вторичного амина дают че-|,лИ промежуточное нитрозосоединение третичные амиды {схема /99П) Д(-Метилзамещенные гидроксамовые кислоты при образке перйодатом подвергаются разрыву связи С (О)— N с обра-анпем карбоновой кислоты и димера нитрозометана {схема (222)} [403]. Показано, что незамещенные пли N-алкилгидрокса-вые кислоты при окислении сульфатом церия в кислой среде 1404а] пли пероксидом никеля [4046] первоначально превращаются в ацил- или ароилнитроксильные радикалы R'CONR2—О-. Эти радикалы обычно неустойчивы, однако при R1 — Ph, R2 = трет-Ви об разуется сравнительно устойчивый ароильный нитроксильный радикал [4046]. При взаимодействии со щелочным феррицианидом, вместо вышеприведенного образуются анион-радикал
RC(O)N—О • [404а].
to]
R'CONHOH ----> R'CONO --------> R'CONR^ + [HNO] (221)
I0; + +
R'CON(Me)OH ----> R'CO2H + MeN=NMe (222)
O' O'
9,9.5. ГИДРАЗИДЫ
Гидразиды карбоновых кислот близки амидам, так как они содержат, по крайней мере, одну амидную связь. Моноацилгид-разины [простые гидразиды (141)] можно рассматривать как N-аминоамиды. Азот, соседний с карбонильной группой, обозначают 1- или а-, терминальный атом азота — соответственно 2- или (3-. Более ацилированные гидразины могут быть замещены симметрично относительно связи N—N как, например, 1,2-диацилгидра-зины (142) или 1,1,2,2-тетраацнлгидразины (143). Известны также М-диацилгидразины (144), т. е. N-аминопроизводные имидов, 11 М,2-триацилгидразины (145). Все гидразиды, кроме простых гидразидов (141), называют как производные гидразина.
п, (в)
R’C=O ,О О. (X
(а) | R'CX ^CR2 R'C^ J)CR3
R2N-—NR3R4 ^N—n' >-n(
rs/ \r4 R5CZ \cr4
4O
(HI) (142) (143)
509
:О
R'C
NR3R4
R2C^
ч0
(144)
О О;
. R'C\ /CR3
J)N—Iv
R2C^ \4
4
(145)
Реакции простых гидразидов аналогичны реакциям гидроксамовых кислот (см. разд. 9.9.4.3), так как терминальный или R. азот является наиболее нуклеофильным атомом вдоль цепочки О—С—N—N. Замещение может также происходить у атомов 0 и a-N, но этот процесс протекает с большим трудом. Каждый более ацилированный гидразин характеризуется свойственной ему реакционной способностью. Многие гидразиды физиологически активны; эти аспекты, наряду со способами получения, основными свойствами и реакционной способностью гидразидов рассмотрены в обзоре [405].
9.9.5.1. Методы получения гидразидов
Наиболее распространенным методом получения гидразидов является ацилирование гидразина или его алкил- и арилпроиз-водных обычными ацилирующими агентами, например ангидридами кислот и сложными эфирами. Реакция сводится к образованию связи С(О)—N [(141), связь (а)] и аналогична получению амидов ацилированием аминов (см. разд. 9.9.1.1). При определенных условиях возможно дальнейшее ацилирование до ди-, три- и тетраацилгпдразинов, если один или оба атома азота образующегося гидразида незамещены [(141), R2, R3 или R4=H], Существует также несколько методов образования специфичной для гидразидов N—N связи [(141), связь (б)] и, наконец, имеется несколько методов введения карбонильного кислорода [(141), связь (в)] в цепь R1—С—N—N^. Как и в случае амидов, исходные гидразиды можно модифицировать алкилированием по азоту с образованием более замещенных производных (см. разд. 9.9.4.3). Примеры менее общих методов получения приведены в уже упомянутом обзоре [405].
Гидразины, как и гидроксиламины, значительно более нуклеофильны, чем амины (а-эффект) [319]. Поэтому ацилирование гидразинов менее активными сложными эфирами карбоновых кислот является лучшим методом получения простых гидразидов, в то время как хлорангидриды и ангидриды кислот представляют собой более подходящие реагенты для ацилирования неактивных гидразинов или для получения ди-, три- и тетраацилгидразинов, как показано на схеме (223).
RCOX RCOX RCOX
H2NNH2 ------► RCONHNH2 -------->- RCONHNHCOR —
—> (RCO)2NNHCOR RCOX > (RCO)2NN(COR)2 (223)
610
жнЫе эфиры карбоновых кислот часто спонтанно реаги-Сл0 гидразингидратом, хотя в отдельных случаях требуется рУ1(7т С нагревание [406]. Реакция подвержена влиянию стериче-сЛ, факторов [407]. Замещение в моноалкилгидразинах может СК11ХлХпить у любого из двух атомов азота, при этом образуются ПРпмерные гидразиды (146) и (147), {схема (224)}. Однако на И3°ктике заметные количества более стерически затрудненного П мера (147) образуются только из формиатов (R2=H). При И3°личении размера остатков R или R1 скорость реакции падает ^увеличивается количество продукта (146) за счет (147) [408]. Т к взаимодействие метилгидразина со сложным эфиром дает лавным образом (146). Напротив, при ацилировании хлорангид-ГидОм кислоты главным продуктом является (147) , так как электронодонорные свойства метильной группы в данном случае являются фактором, определяющим строение продуктов. В случае । j-дизамешенных гидразинов, например 1,1-диметилгидразина, влияние стерических препятствий таково, что реакция со сложными эфирами не идет, за исключением формиатов и активированных эфиров, несущих циано-, нитро- или галогензаместители [409]. Влияние стерических факторов для активированных эфиров исключается, ибо они ацилируются преимущественно по механизму Sjvl. Дальнейшее замещение 1,2-диметилгидразинов под действием эфиров затруднено. В случае монофенилгидразинов изомер (146) является единственным продуктом, преимущественно образующимся с точки зрения электронных и стерических факто-ров[410]. Взаимодействие гидразинов с этилфенилацетатами подвержено общему кислотному и общему основному катализу [411]. Ацилирование гидразинов циклическими эфирами (лактонами) обычно сопровождается разрывом О-ацильной связи с образованием гидроксизамешенного гидразида {схема (225)}. Эта реакция может быть использована для идентификации природных лактонов с помощью реакции с фенилгидразином и образования сталлических фенилгидразидов [412].
R‘NHNH2 + R2COOR3 —► R‘NHNHCOR2 + R‘N(NH2)COR2
(146) (147)
кри-
(224)
О
+ RNHNH2 —> C13CCH(OH)CH2CONHNHR
(225)
С13С
Ацилирование гидразинов галогенангидридами кислот, как правило, проводят в пиридине, в щелочном водном растворе или в органическом растворителе в присутствии Ыа2СОз. Для проведения Реакции с малоактивными реагентами или субстратами необходи-а повышенная температура. Для остановки ацилирования на тадии моноацилгидразпна (141) успешно используются различ* пРиемы, например разбавление реагента инертным раствори-
Лем или добавление гидразина по каплям при низкой температуре
511
[407, 413а]. Нели в реакции вводят несимметрично замещенные гидразины, направление ацилирования зависит от природу местителя. Например, в случае моноалкилгидразинов ацилирова' нию подвергается атом азота, несущий заместитель [412, 4141* в то время как в случае монофенилгидразинов ацилируется незамещенный атом азота [410, 4136] {схемы (226) и (227) соответственно}. Такого поведения можно ожидать, исходя из электронного эффекта этих заместителей. При взаимодействии с галоген-ангидридами кислот стерические эффекты, по-видимому, имеют меньшее значение, за исключением таких объемистых субстратов как гидразобензол, при реакции с которым введения второй ацильной группы не происходит [4136]. Реакции с хлорангидрпдами и ангидридами некоторых дикарбоновых кислот могут приводить к циклическим гидразидам при условии, что образующийся цикл содержит двойную связь. Например, малеиновый и фталевый ангидриды дают циклические гидразиды [415] {схема (228)}, в то время как янтарный ангидрид не дает таковых.
MeNHNH2 + RCOC1 —> MeN(NH2)COR
PhNHNH2 + RCOC1 —> PhNHNHCOR
(226)
(227)
(228)
Другим исключением, по-видимому, является оксалилхлорид который при взаимодействии с гидразином дает неустойчивый четырехчленный 1,2-диазетидон [416]. Ацилирование менее нуклеофильного азота монозамещенного гидразина можно осуществить с помощью различных непрямых методов. Так, предварительное получение гидразин-аниона при обработке сильным основанием (например, натрием) приводит к отщеплению протона от менее нуклеофильного азота, который затем ацилируется [405], однако эта реакция протекает не всегда однозначно. Альтернативным методом, применимым к фенилгидразинам, является получение 1,2-диацилгидразина с последующим частичным кислотным гид релизом, при котором ацильная группа отщепляется от менее за мещенного азота [405] {схема (229)}. Другим методом ацилирования замещенного азота является получение гидразона по к0^ цевой ЫН2-группе, ацилирование и последующее кислотное ° щепление защитной группы, приводящее к искомому гидразИ [417] {схема (230)}.
PhNHNH2 + 2PhCOCl —> PhN(COPh)NHCOPh PhN(NH2)COPhg)
PhNHNH2
Me2CO
r н+. Hs£.
PhNHN==CMe2 — PhN(COR)N=CMe2
—> PhN(NH2)COR •
(230)
512
качестве агентов, ацилирующих гидразин, могут выступать в н0 для проведения реакции требуется более высокая тем-аМ11^Упа и более длительное время, чем в случае сложных эфиров перахдорангидридов кислот [418], и выходы продуктов невелики. ал11 реакция обычно используется для деградации белков путем ^та ува пептидной связи. Протекание подобных реакций можно Pi? егчить либо с помощью гидразин-аниопа (получаемого в силь-х основаниях) только для реакций с третичными амидами (перечные и вторичные амиды ингибируют образование аниона), В”бо применяя очень активный N-бензоилимидазол, легко бензои-эиоуюший гидразингидрат [370]. Для ацилирования гидразинов возможно использование карбоновых кислот. Как и в случае амидов прямое нагревание солей гидразина и карбоновых кислот не нашло широкого применения [419а], но реакцию можно осуществить при комнатной температуре с помощью М,М-дицикло-гекснлкарбодиимида в качестве дегидратирующего агента [4196]. В реакциях такого типа ориентация заместителя в монозамещен-ных гидразинах такая же, как и при использовании хлорангпдридов кислот (т. е. важными являются электронные, а не стерические факторы). Ацилирование очень активными кетенами проходит в мягких условиях и приводит при реакциях с фенилгидрази-нами к количественному выходу 1-ацил-2-фенилгидразинов [420]. Однако метилгидразин при взаимодействии с дифенилкетеном дает 1,2-диацильное производное; при этом моноацильное соединение не выделено [418, 421].
Альтернативным общим методом получения гидразидов является образование связи N—N [(141), связь (б)], что можно осуществить различными способами. Первый способ состоит в обработке моноацилмочевин гипохлоритом натрия [422]. Эта реакция (реакция Шестакова) аналогична распаду амидов по Гофману (см. разд. 9.9.3.11) и, как полагают, проходит через промежуточное образование N-хлормочевины. Обработка этого соединения основанием дает анион по азоту, в котором происходит разрыв связи N—С1 с синхронной миграцией амидной группы. При этом образуется замещенный изоцианат, который после гидролиза дает гидразид {схема (231)}. Другие методы образования связи N—N включают восстановление N-нитроамидов [405] (см. разд. 9.9.3.6) и обработку N-хлорамидов (см. разд. 9.9.3.7) амидом натрия или анионом вторичного амида с образованием моно- и 1,2-дпацнлгид-разинов соответственно [423]. Новым способом образования связи N—N является обработка N-ацилоксазиридина азотистым основанием, например пиперидином; здесь нуклеофильная атака об-Н24^еТСЯ 33 СЧеТ РаскРытия напРяЖенного трехчленного цикла Оксазиридины — легко доступные соединения, и выход продукта при реакции с 3-фенилоксазирпдином достаточно высок. Гид* Разид необходимого строения может быть получен путем подбора оответствующего основания {схема (232)}.
17 Зак. 395
513
PIiCONH
^c=o h2n
Na0C‘> PhCONHCONHCl -££>• PhC-N-rC-N-Qi
*- PhCONHN=C—О CO2+ PhCONHNH,
Третий общий метод получения гидразидов состоит во включении кислорода в цепь С—N—N. Это превращение можно осуществить несколькими способами, например путем окисления гидразонов (полученных из карбонильного соединения и гидразина) пер-уксусной кислотой или путем превращения гидразона в результате реакции с бромом в гидразидбромид с последующим гидролизом в умеренно основном растворе {схема (233)} [425]. Напротив, конденсация дифторолефинов с гидразинами приводит к 1,1-ди-фторгидразииу, который гидролизуется в гидразид, однако последующие реакции конденсации могут снижать выход продукта [426] {схема (234)}.
R'R2iNNH2 Вгг основание
АгСНО ---------> ArCH=NNR‘R2 ------► ArCBr=NNR'R2 --------->
—> ArCONHNR'R2 (233)
RiR2C=CF2 + H2NNMe2 —> R1R2CHCF2NHNMe2 R*R2CHCONHNMe
(234)
9.Э.5.2. Свойства гидразидов
Строение нескольких простых гидразидов кислот, определенное с помощью рентгеноструктурного анализа [427], говорит о достаточно устойчивой геометрии гидразидной группы, как показано в (148) (длины связи в нм и углы для кристаллического о разца),
о
н\ N—N
R-c н
О
(149)
(150)
Система RC(O)NH—N достаточно планарна, в то время как ложение связей вокруг р-атома азота пирамидально, а о
514
связи направлены к атому кислорода (Z-конфигу рация). цеВЬГ^тпуктура очень напоминает незамещенные гидроксамовые Эта j (сМ. разд. 9.9.4); порядок связи С (О)—N примерно 1,5. кисЛоИСТаллическом состоянии водородные связи (N—Н —N и ₽ 'U...O) являются межмолекулярными, что приводит к образо-ию полимерных цепей. Измерения дипольных моментов [428] ваН3ывают на то, что наиболее устойчивой в растворе является ^.конфигурация, однако не известно, обусловлено ли это нали-
м внутримолекулярных водородных связей. Рентгеноструктур-ЧИр исследования замещенных ацилгидразинов указывают на то, Hbl 1 2-диацилгидразины представляют собой центросимметричные плоские молекулы [429] (149), в то время как в тетраацилгидра-зинах, например в М,М-дисукцинимиде (150), каждая половина молекулы плоская, а угол между ними составляет 65° [430].
Частичная двоесвязанность связи С (О)—N приводит к конфигурационным изомерам, близким к изомерам амидов и имидов. Однако, как и в случае гидроксамовых кислот, о барьерах вращения простейших гидразидов известно очень мало. С помощью
flMP-спектроскопии получены доказательства медленного вращения вокруг связи N—N (AG* ~ 84 кДж • моль-1) в случае 1,2-ди-ацил- и тетраацилгидразинов [431]. Эти результаты сопоставимы с данными о медленном вращении вокруг связи N—О, наблюдаемом в случае О-ацилгидроксамовых кислот (см. разд. Э.9.4.2).
Частоты поглощения гидразидов в ИК-спектрах аналогичны обнаруженным в случае амидов [валентные колебания N—Н 3450 (свободная), 3200—3250 (связанная); амидная полоса I 1645— 1695 (свободная), 1625—1670 (связанная); амидная полоса II 1500 (свободная), 1530—1570 см-1 (связанная)], за исключением дополнительной полосы поглощения (1620—1610 см-1), соответствующей деформационным колебаниям МН2-группы незамещенных гидразидов [413а, 432]. Сдвиг в положении полос ИК-поглощения при переходе от твердой фазы или концентрированного раствора к разбавленному раствору сопоставим с аналогичным сдвигом в случае амидов и обусловлен диссоциацией частиц конденсированной фазы, связанных межмолекулярными водородными связями До мономеров в разбавленном растворе.
Можно полагать, что УФ-спектры поглощения гидразидов близки к спектрам амидов и гидроксамовых кислот, хотя данных по этому вопросу крайне мало (см. разд. 9.9.2.2 и 9.Э.4.2). Однако некоторые гидразиды имеют интересные оптические свойства. Например, циклические гидразиды люминол (151) и TV-аминофтали-миды сильно флуоресцируют при окислении в щелочной среде [8, ^3] {схема (235)}. Полагают, что ответственным за флуоресценцию является возбужденный диапион (151а), однако для выяснения деталей этого процесса требуются дополнительные исследования. Гидразиды использовались также при изучении оптического Ращения, в особенности для определения конфигурации альдоно-Ых кислот (правило Хадсона для феннлгидразидов) [434].
17*
515
о
Гидразиды, подобно амидам и гидроксамовым кислотам, амфотерны. Вследствие протонирования ₽-атома азота (т. е. аминного N) они являются более сильными основаниями (р/Са я» 3), чем амиды или гидроксамовые кислоты (где карбонильный кислород является более основным атомом) (ср. разд. 9.9.2.3 и 9.9.4.2), но более слабыми основаниями, чем гидразин (рЛа 8) [435]. Введение p-N-фенильного заместителя в гидразиды настолько снижает их основность, что они перестают растворяться в кислотах. При протонировании гидразидов бензойной кислоты, несущих заместители в кольце (XC6H4CONHNH2), рХа коррелирует с о, давая р = —0,69, по сравнению с р = —1,21 для фенилгидра-зинов [436]. С другой стороны, гидразиды являются слабыми кислотами (рХа гидразида п-нитробензойной кислоты около 11) [437], несколько более слабыми, чем гидроксамовые кислоты (рЛа 8,7), но значительно сильнее, чем амиды (р/Са около 16). Ионизация a-NH кажется предпочтительной по сравнению с p-NH, так как алкилирование в сильно щелочных растворах происходит по a-N-атому (см. разд. 9.9.5.3). Однако возможно также образование циклических семичленных структур, включающих молекулу воды, аналогичных гидроксамат-аниону (131). 1,2-Днацилгпдра-зины также являются слабыми кислотами и образуют моно- и ди-натриевые соли [438]. Как и в случае амидов и гидроксамовых кислот, доказательства существования таутомерной структуры ими-нола отсутствуют, хотя О-алкил- и О-ацплпроизводные этого таутомера хорошо известны (см. разд. 9.9.5.3).
Моно- и длацилгидразины, как и гидроксамовые кислоты, могут выступать в качестве бидентантных лигандов в комплексах с переходными металлами {например, с Си(II), Cd(II), Fe(II), Mn(H)7 [439] и с кислотами Льюиса (например, с SbCls, TiCl4, SnBrd [440] . Типичной структурой комплекса с переходными металлам является (152). Интересное наблюдение о большей устойчивости комплексов с 1,2-диацилгидразинами по сравнению с комплексам
516
паЦилгидразинов [441] может указывать на то, что обе кар-группы участвуют в образовании хелата с менее напря-60Тым семичленным циклом.
ценной"
9.9.5.3. Реакции гидразидов
Гидразиды подвергаются как нуклеофильному присоединению карбонильной группе (например, при гидролизе), так и вза-п одействию с электрофильными реагентами (например, с алкил-И ацилгалогенидами неорганических кислот и с карбонильными соединениями) по всем трем гетероатомам (О, a-N и [3-N). В нейтральных или слабощелочных растворах происходит замещение p-N, а в сильнощелочных условиях, напротив, замещение обычно осуществляется у a-N. Гидразиды легко окисляются и восстанавливаются. Известно немного перегруппировок этих соединений; их фотохимические превращения не привлекли особого внимания. Важным свойством гидразидов является их способность образовывать гетероциклические соединения как путем внутримолекулярной циклизации, так и в результате конденсации с другими полифункциональными реагентами. Рассмотрение этих реакций выходит за рамки данной главы, однако они обсуждаются в обзоре [405] и в томе 8 настоящего издания.
Реакции 1,2-диацилгидразинов в значительной степени напоминают реакции амидов, так как а- и р-атомы азота расположены рядом с карбонильной группой. Однако отличие состоит в том, что в случае 1,2-диацилгидразинов влияние соседних групп и стерических факторов более существенно. 1,1-Диацилгидразины и тетраацилгидразины имеют структурное сходство с имидами, однако данные об их реакциях незначительны.
Простые гидразиды, как правило, устойчивы к действию как кислот, так и оснований, при комнатной температуре. Гидролиз гидразида до гидразина и карбоновой кислоты обычно проходит при нагревании в течение нескольких часов в водных растворах кислот или оснований. Предпочтителен гидролиз в кислых условиях, так как при щелочном гидролизе часто протекают нежелательные окислительно-восстановительные реакции [422]. На основании изучения кинетики гидролиза гидразидов алифатических кислот (уксусной, пропионовой, масляной и изомасляной) полагают, что последовательность превращений, катализируемых кислотами и основаниями, аналогична схеме гидролиза простейших амидов [443].
Алкилирование гидразидов в зависимости от условий реакции протекает по а- или [3-атому азота {схема (236)}. В нейтральных Растворах алкилируется [3-атом азота {направление (а) на схеме}, в то время как в сильных основаниях (Na или NaOMe) место замещения зависит от растворителя: в апротонных растворителях (.эфир, бензол) происходит замещение у а-атома азота [направле-ие (б)], в протонных растворителях (этанол)—у [3-атома {нап-Равление (а)} [444]. Такую необычную зависимость строения
517
продукта от растворителя можно объяснить образованием связанного водородной связью семичленного цикла, включающего молеку лу этанола (153) и стабилизирующего p-N-анион, в то время как в апротонных растворителях такая стабилизация не имеет места и замещение происходит у а-атома азота (154). Отсутствие при обычных условиях продуктов О-алкилирования связано, вероятно (как и в случае амидов), с быстрой O-^N-перегруппировкой. Сообщалось о внутримолекулярной термической перегруппировке О-арнлгидразидов в p-N-арилгидразиды при 200 °C {схема (237)} [445]. Вероятно, указанная перегруппировка происходит легче, чем O-^N-перегруппировка соответствующих амидов (реакция Чапмэна см. разд. 9.9.3.3.), так как она включает пяти-, а не четырехцентровое переходное состояние. О-Алкильные аналоги не подвергаются термической перегруппировке, но, как и в случае амидов, электрофильные катализаторы могут оказаться эффективными.
R'CONHNH2 + R2X—
(а)
—► R'CONHNHR2
(б)
—*• R‘CONR2NH2
(236)
о—н
RCZ ••
P-R
HN-H
R(T NH2 R(/ NH2
Xn/
(153) (154)
/ОАг2 д ,0
Аг1 С"7 —> Аг1 С/ (237>
V\:NHAr3 X'NHNAr2Ar3
Дальнейшее алкилирование p-N-алкилгидразинов приводит к дизамещенным производным и к тризамещенным имидам (155) {схема (238)} [444а, 446]. Нуклеофильные свойства 1,2-диадил-гидразинов близки к таковым для амидов, так как оба азота в первых симметрично замещены. Гидразид малеиновой кислоты при обработке диазометаном превращается в О- и N-метилированные продукты {схема (239)} [447]. Алкилирование алкилгалогенида^ обычно проводят в сильно щелочных растворах, и в данном слу чае происходит прямое N-алкилирование по одному или по ДВУ‘ атомам азота {схема (240)} [448]. Все три нуклеофильных ато гидразида могут участвовать в реакциях внутримолекулярного а^ килирования, образуя цикл оптимального размера. Примеры разования пятичленных гетероциклических соединений привел на схемах (241) — (243). Шестичленные соединения образуют аналогичным образом. Эти превращения, подробно обсужденные обзоре [405], являются одним из двух основных путей пспольз ния гидразидов в синтезе гетероциклических соединений.
Простейшие гидразиды легко реагируют с карбонильными динениями. В нейтральных условиях наиболее активным явля
518
RCONH—NH
RCON—NH
терминальный атом азота, по которому преимущественно образуется производное карбиноламина (156), быстро элиминирующее воду и дающее ацилгидразон (157) {схема (244)}. Как и в случае амидов, скорость реакции максимальна при умеренных pH; в кислых условиях реакция обратима [449]. Как и родственные арил-гидразоны, ацилгидразоны — это хорошо кристаллизующиеся вещества, используемые для идентификации карбонильных соединений. Гидразиды определенного строения используются при выделении карбонильных соединений из различных смесей. Например,
+ и гч- nfinaaveT водораствори-
реактив Жирара Т Me3NCH2CONHN 2 дает возможность Уд?^ять мые ацилгидразоны и таким обР^ ннчесКнх смесей [450].
карбонильные соединения из слоЖ*ь Р 11чески активный гид Другим полезным соединением яВЛ использовать для пре -
Разид L-ментилглицина, который можн ]lufi в разделяемые
Ращения смеси D- и L-карбонильных_сс> Д с карбо„ильИьми адилгидразоны отдельных диастерео Рлг^ра311ДЫ и 1,2-диац -Уединениями реагируют также р- * 51.9
гидразины. Вероятно, первоначально образуется также карбиноч-амин, но поскольку отщепление воды с образованием ацилгидп'а-зона здесь невозможно, происходят отщепления другого типа В результате из p-N-алкилгидразидов образуются бетаины [4521 а из 1,2-диацилгидразинов — диазиридины [453] {схемы (245) и (246)}. >
R'CONHNH, + R2R3C=O —> R‘CONHNHCR2R3OH
(156)
—► RICONHN=CR2R3
(157)
-Н2О
(244)
„ Ar’CONH—NCOAr2
A^CONHNHCOAr2 4- PhCHO —> < |
Hq3CHPh
Ar'cON—NCOAr2
Y (246)
Ph.
Простые гидразиды ацилируются как галогенангидридами, так й ангидридами кислот, давая более высоко замещенные ацилгид-разины. Первое замещение, приводящее к соответствующему 1,2-ди-ацилгидразину {схема (247)}, обычно протекает гладко и с высокими выходами при низкой температуре в инертном или слабоосновном растворителе [407, 454, 455]. Дальнейшее замещение до три- и тетраацилгидразинов происходит труднее и требует либо наличия более сильного основания (для образования более активного N-аниона), либо избытка ацилирующего агента и высокой температуры {схема (248)}. При ацилировании гидразидов циклических кислот хлорангидридами дикарбоновых кислот образуются бициклические соединения [457].
R'CONHNH2 + R2COX —> R'CONHNHCOR2 + HX (247)
PhCONHNHCOPh —>- PhCONNHCOPh ---------> (PhCO)2NNHCOPh (248)
Остается неясным, служат ли О-ацилгидразиды интермедиатами в этих реакциях ацилирования (как в случае амидов), однако относительно устойчивые О-ацилгидразиды были недавно получены различными способами и показано, что при 100 °C они претерпевают О-ИЧ-перегрупппровку, наблюдавшуюся для О-арильных аналогов [458] {схема (249)}:
zOAc
R'Y
4’R2
R
(249)
(R1 = Bu-трет; R2 = N(Me)CeH3(NO2)2l
RCONHNH2 + ArN=C—О —► RCONHNHCONHAr (2&u;
520
Гидразиды легко ацилируются ичлгг,
„„„карбазидов {схема (250)); скорбь "аМИ с °бРМоВа|1[гем концентрации обоих реагентов, Что VtA°f Реа«Цйи зависит Л по механизму SN2 на углеродный я22<азывает на атаку гипп От Прямое N-галогенирование по-вилгЛ3°ЦИаната [459] ДРЭЗИДа
N-галогенпроизводные могут спужит. Му’ Не происходи лешш гидразидов галогенирующ^ми агента^ }ПР” -
окис-
fRCONHNHN—O"i
RCONHNH2
h2ono+ ________
RCONHN=NOHJ
но~ ---->
RCON3
neperpyn. Курциуса --------->.
или A
rconh2 + n2o
RNCO + n2
(251)
Нитрозированпе простых гидразидов является одной из важнейших реакции замещения для данного класса соединений; его обычно проводят обработкой кислым нитритом при низкой температуре [460]. Промежуточно образующийся p-N-нитрозогидразид, который можно выделить, в зависимости от условий реакции распадается по двум направлениям {схема (251)}. В основном растворе первоначально возникают ацилазпды, которые подвергаются известной перегруппировке Курциуса с образованием аминов [71]. Однако при нагревании или при обработке сильными кислотами образуются первичный амид и оксид азота [461]. При взаимодействии 2,2-диалкилгидразидов с кислым нитритом также происходит нитрозированпе по р-атому азота с конкурентным деалкилированием. Успешное деалкилирование приводит в итоге к ацилази-дам [462] {схема (252)}. При реакции с ионами арендиазония также происходит замещение у р-атома азота гидразидов с образованием тетразена, который обычно не выделяют, а после обработки основанием in situ циклизуют в тетразол {схема (253)}. 1,2-Диацилгидразины реагируют аналогично, давая гексаазадиены [463] {схема (254)}.
, h2ono+ h2ono+
R'CONHNR* —------> R’CONHNRNO -------> RCON3 (252)
RCONHNHCOR + 2ArN2 —> ArN=NN(COR)N(COR)N—NAr (254)
° ют с несколькими ra-Как известно, гидразиды взаиМ0Де^т,У но только РеакДдД-|С логенангидридами неорганических ’имИдоилхлорида [4 Ь
фосгеном, приводящая к произв Д Механизм реакц ,
аналогична реакции с амидами {схе ( Ку аТома кислоро по-вндимому, включает первоначаль У
521
гидразида на фосген с последующим отщеплением СО2 по механизму КД (ср. разд. 9.9.3.8). Напротив, реакции с РС15, SOC12 и ArSO2Cl приводят к замещению у 0-атома азота гидразида [4651 Можно предположить, что эти реагенты также сначала реагируют с кислородом гидразида; далее должна происходить внутримолекулярная атака 0-атома азота на более электронодефицитный X {схема (256)}, что предпочтительно по сравнению с БД-процессом с участием хлорида, приводящим к имидоилхлориду {схема (255)}. При реакциях РС15 с N-арилгидразидами, нуклеофильность азота в которых понижена за счет сопряжения неподеленной пары с бензольным кольцом, действительно образуется имидоилхлорид, вероятно по схеме, аналогичной схеме (255) [466]. При взаимодействии избытка РС15 с 1,2-диацилгидразинами также образуются имидоилхлориды RCC1=N—N=CC1R [467], однако при реакции с тпонилхлоридом и оксихлоридом фосфора происходит внутримолекулярная атака на незамещенный атом кислорода карбонильной группы с образованием оксадиазолов {схема (257)} [468].
RCONHNH2
—-V RC(C1)=NNH3 Cl (255)
(X= SO,PC13)
NH—NH
рОСО RC^-^CI
К+ С1
nhnh2
rconhnh2 + XC12 —>
-> RCONHNHXC1 ——^-RCONHN
(256)
SOC12>
N-NH cf R
+ so2
(257)
Гидразиды, имеющие водород у каждого из атомов азота, под действием таких реагентов, как кислород, хлорид железа (Ш), оксид серебра, галогены и тетраацетат свинца легко окисляются до диимидов [99, 469] {схема (258)}. Диимиды обычно можно выделить, однако незамещенные по азоту динмиды значительно менее устойчивы и могут быть использованы как ацилирующие агенты по отношению к гидразидам и аминам. Симметрично замешенные диимиды (R2 = R'CO) нашли применение в синтезе пептидов. Существует п ряд других окислителей, под действием которых происходит разрыв связи С (О)—N в незамещенных гидразидах. Так, феррицианид аммония дает альдегид и газообразный азот (окисление по Кальб-Гроссу) [470], диоксид марганца дает кар-
522
vk? кислоту и азот [470], а молекулярный галоген в присут-бонову огеНОВОдорода приводит к галогенангидриду кислоты и ствИИ (реакция Карлино) [471]. Незамещенные гидразиды при а3°Т^10действии с иодом или с N-бромсукцинимидом дают соли
иазония RCONo [472], в то время как при окислении гидра-aU р (замешенных только по a-N) перманганатом или гипобро-происходит димеризация с образованием тетразенов M'^Vl^NR’NCOR1 [473]. |О1
R'CONHNHR2 —-> R]CON=NR2 (258)
Восстановление гидразидов комплексными гидридами метал-(например, LiAlH4, NaBH4) приводит к гидразинам {схема (259)} [413а, 474], в то время как при каталитическом гидрировании (никель Ренея в ЕЮН) происходит разрыв связи N—N с образованием амидов и аммиака {схема (260)} [475]. В случае 1 ]. и 1,2-диацилгидразинов ---------- -
леннее и выходы ниже.
RCONHNH2
реакция, как правило, протекает мед-
LiAlH4
-------► rch2nhnh2
(259)
Н2
RCONHNH, ------------> RCONH2 + NH3
катализатор
(260)
Перегруппировки неактивированных гидразидов обычно включают миграцию ацильной группы с одного атома азота на другой, преимущественно через диазиридиновый интермедиат. Так, в присутствии кислых катализаторов происходит миграция ацильной группы от более к менее замещенному азоту {схема (261)} [414, 476], в то время как 1,1- и 1,2-диацилгидразины переходят друг в друга в зависимости от pH {схеме (262)} [477]. Активация Р-атома азота гидразидов за счет образования N-илпда (155) или путем замещения хорошо уходящей группой, например, ArSO2, облегчает протекание других типов перегруппировок или отщеплений. Например, илид (155) может претерпевать термическую перегруппировку {схема (263)} [478] или превращение с разрывом связи N—N (аналогично деградации по Гофману) [479] {схема (264)}, а p-N-сульфонилгидразиды подвергаются элиминации с образованием диимида, который распадается до альдегида и азота (реакция Мак Фейдина — Стивенса) [480] {схема (265)}.
523
MeCONNMe2CH2Ph —> MeCON(CH2Ph)NMe2
(263)
(155)
RN=C=O + Me3N
(264)
RCONHNH—SO2R —> RCON=NH —ri- RCHO + N2 (265)
ЛИТЕРАТУРА
1. P. A. S. Smith., «Open-chain Nitrogen Compounds», Benjamin, New York, 1965 vol. 1, 1966, vol. 2.
2. «The Chemistry of Amides», ed. J. Zabicky, Interscience, London, 1970.
3a. S. R. Sandler and W. Karo, «Organic Functional Group Preparations», Academic Press, London, 1972, vol. 3, chapter 12.
36. A. Bauer and 0. Exner, Angew. Chem. Internat. Edn., 1974, 13, 376.
4. A. L. J. Beckwith, in Ref. 2, chapter 2.
5a./. C. Sheehan and E. J. Corey, Org. Reactions, 1957, 9, 388 [Дж. К. Шихан, Э. Дж. Корей. Орг. реакции, сб. 9 — Пер. с англ./Под ред. И. Ф. Луценко. М., Издатинлит, 1959, с. 504].
56./. Lengyel and J. С. Sheehan, Angew. Chem. Internat. Edn., 1968, 7, 25.
6. N. S. Isaacs, Chem. Soc. Rev., 1976, 5, 181.
7. О. H. Wheeler and 0. Rosado, in Ref. 2, chapter 7.
8. M. K. Hargreaves, J. G. Pritchard, and H. R. Dave, Chem. Rev., 1970, 70, 439.
9. См. cc. 3a, chapter 7.
10. G. A. Olah, S. J. Kuhn, W. S. Tolgyesi, and E. B. Baker, J. Amer. Chem. Soc., 1962, 84, 2733.
11. В. C. Challis and A. R. Butler, in «The Chemistry of the Amino Group», ed. S. Patai, Interscience, London, 1968, chapter 6; D. P. N. Satchell, Quart. Rev., 1963, 17, 160.
12. G. A. Olah and S. Kuhn, J. Amer. Chem. Soc., 1960, 82, 2380.
13. G. Casnati, M. R. Langella, F. Piozzi, A. Ricca, and A. Umani-Ronchi, Gaz-zetta, 1964, 94, 1221.
14. B. Acott, A. L. J. Beckwith, and A. Hassanali, Austral. J. Chem., 1968, 21, 185.
15. R. Graf, Annalen, 1963, 661, 111.
16. У. Inouye, K. Onodera, S. Kitaoka, and S. Hirano, J. Amer. Chem. Soc., 1956, 78, 4722.
17a. J. Hipkin and D. P. N. Satchell, J. Chem. Soc. (B), 1966, 345.
176. P. J. Llllford and D. P. N. Satchell, ibid., 1967, 360.
18. A. R. Butler and V. Gold, J. Chem. Soc., 1961, 4362.
19. E. Schroeder and K. Luebke, «The Peptides», Academic Press, New York, 1965, vol. 1 [3. Шредер, К. Любке. Пептиды. — Пер. с англ./Под ред. М. М. Шемякина и Ю. А. Овчинникова. М., Мир. т. I, 1967].
20. N. F. Albertson, Org. Reactions, 1962, 12, 157 [Н. Ф. Альбертсон. Орг. реак-ции. — Пер. с англ./Под ред. И. Ф. Луценко. М., Мир, 1965, сб. 12, с. 174J-21а. Q. R. Clemo and G. A. Swan, J. Chem. Soc., 1945, 603.
216. C. W. Huffman, J. Org. Chem., 1958, 23, 727.
22. R. A. Boissonnas, Adv. Org. Chem., 1963, 3, 159.
23. E. Hoffmann and A. Schiff-Shenhav, J. Org. Chem., 1962, 27, 4686.
24. R. Chiron and Y. Graff, Bull. Soc. chim. France, 1967, 1904.
524
29.
30.
31-
32.
r Qatterthwait and W. P. Jencks, J. Amer. Chem. Soc. 1974 96 701R
» $£ p“i''B^ceandT^ ^ioe ibU lm, j. 5533 » аит'.ромнпыеТм «1’тьг A Sami, A. S Shawah, and S. S. Btechler, ibid., 1967 89, 3020 PoLit and R- Poisson, Compt. rend., 1958, 247, 1628.
26. J- \ stern, Chem. and Ind. (London), 1956, 277.
27- fj Yazawa, K. Tamaka and К Kariyone, Tetrahedron Letters, 1974 3995.
28- E. и R Barton, A. L. J. Beckwith, and A. Goosen, J. Chem Soc 1965 181 К' p Hurd and S. HayaoJ. Amer. Chem. Soc., 1952, 74, 5889; T. L. 'Gresham 1- E- Jansen’ F-oW:oShaver’ R- E- Banke^ and F. T. Fiedorek, J. Amer, rhem Soc., 1951, 73, 3168.
Й Morawetz and P. Otaki, J. Amer. Chem. Soc., 1963, 85, 463.
p' r Shepard, H. D. Porter, J. F. Noth, and C. K. Simmans, J. Org Chem 1952, 17, 568; L. F. Fieser and J. E. Jones, Org. Synth. Coll. Vol. 3, 1955’
590
j Tani, T. Oine, and I. Inoue, Synthesis, 1975, 714.
чГ E Bald, K. Saigo, and T. Mukaiyama, Chem. Letters, 1975, 1163.
Id' s -I Yamada, Y. Kasai, and T. Shiori, Tetrahedron Letters, 1973, 1595; G. Ga-wne, G. W. Kenner, and R. C. Sheppard, J. Amer. Chem. Soc., 1969, 91, 5669.
or D G Gehring, №. A. Mosher, and G. S. Reddy, J. Org. Chem,. 1966, 31, 3436.
37 B. Sukornick, Org Synth., 1960, 40, 103.
38 К. E. Hamlin and A. №. Weston, Org. Reactions, 1957, 9, 1.
39 A. L. J. Beckwith, J. Chem. Soc., 1962, 2248.
40 AL Carmack and M. A. Spielman, Org. Reactions, 1946, 3, 83 [AL Кармак, M, А. Шпильман. Орг. реакции. — Пер. с аигл./Под ред. К. А. Кочешкова. М., Издатинлит, 1951, сб. 3, с. 881; R. Wegler, Е. Kuhle, and №. Schafer, in’«Newer Methods of Preparative Organic Chemistry», ed. W. Foerst, Academic Press, New York, 1964, vol. 3, chapter 1.
41. F. Asinger, №. Schafer, K. Halcour, A. Saus, and H. Triem, Angew. Chem. Internat. Edn., 1964, 3, 19.
42. N. A. Smart, G. T. Young, and M. №. Williams, J. Chem. Soc., 1960, 3902.
43. F. Weygand and K. Hunger, Chem. Ber., 1962, 95, 1; M. Brenner and P. Zimmermann, Helv. Chim. Acta, 1957, 40, 1933; L. Bernardini and O. Goffredo, Gazzetta, 1964, 94, 947.
44. P. A. S. Smith, Org. Synth. Coll. Vol. 4, 1963, 819.
45. J. Falbe and F. Korte, Chem. Ber., 1965, 98, 1928; 1962, 95, 2680.
46. № Reppe, Annalen, 1953, 582, 14.
47. A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327.
48. G. R. Pettit, M. V. Kalnins, T. M. H. Liu, E. G. Thomas, and K. Parent, J. Org. Chem., 1961, 26, 2563.
49. J. F, Wolfe, C. J. Eby, and C. R. Hauser, J. Org. Chem., 1965, 30, 55.
50. № Wenner, Org. Synth. Coll. Vol. 4, 1963, 760.
51. G. Jones, J. Amer. Chem. Soc., 1961, 73, 5610.
“2. № L. Mosby, J. Amer. Chem. Soc., 1953, 75, 3600.
53. С. M. Atkinson and C. J. Sharpe, J. Chem. Soc., 1959, 2858.
54. К. B. Wiberg, J. Amer. Chem. Soc., 1953, 75, 3961.
55. D. H. R. Barton, E. F. Lier, and J. F. McGhie, J. Chem. Soc. (C), 1968, 1031.
56- 8. E. Diamond, B. Grant, G. M. Tom, and H. Taube, Tetrahedron Letters, 1974, 4025; S. Paraskewas, Synthesis, 1974, 574; M. A. Bennett and T. Yoshida, J. Amer. Chem. Soc., 1973, 95, 3030.
Я J. Cook, E. J. Forbes, and G. M. Khan, Chem. Comm., 1966, 121.
I. Al. Bobbitt and R. E. Doolittle, J. Org. Chem., 1964, 29, 2298.
R. Roger and D. G. Neilson, Chem. Rev., 1961, 61, 179.
A. J. Exner, M. J. Hurwitz, and P. L. de Benneville, J. Amer. Chem. Soc., 1955, 6 * 63 64 * *
6i 7’ 1103-
6? n FErimen and D. J. Cola, Org. Reactions, 1969, 17, 213.
°4 P- D. Bartlett, R. E. Pincock, J. H. Rolston, №. G. Schindel, and L. A. Stn-
63 8per’,}: Amer- Chem. Soc., 1965, 87, 2590. c nC
64 n JPhrlson and R. Madronero, Adv. Heterocyclic Chem 1966, 6, 9b
p H- R, Barton, P. D. Magnus, J. A. Garbarino, and R. N. Young, J. C. S.
perkm I, 1974, 2101.
525
к. к. Hill, R. Т. Conley, and О. T. Chortyk, J. Amer. Chem Soc 19fK o, 5646. ” ’ 87’
65.
66. A. Hassner, L. A. Levy, and R. Gault, Tetrahedron Letters, 1966 31 iq. R. H. Andreatta and A. V. Robertson, Austral. J. Chem., 1966, 19, 161’ *9;
67. L. G. Donaruma and W. Z. Heldt, Org. Reactions, 1960, 11, 1 [Л. Г Пона рума, В. 3. Хельдт. Орг. реакции. — Пер. с англ./Под ред. И Ф JIvitpL/ М„ Мир, 1965, сб. 11, с. 7]. ' У1енко-
68. С. G. McCarty, in «The Chemistry of the Carbon-Nitrogen Double Bond» ed S. Patai, Interscience, London, 1970, chapter 9.
69. К. K. Kelly and J. S. Matthews, J. Org. Chem., 1971, 36, 2159.
70. R. Huisgen, J. Witte, H. Walz, and W. Jira, Annalen, 1957, 604, 191.
71. P. A. S. Smith, in «Molecular Rearrangements», ed. P. de Mayo, 1'nterscience London, 1963, part I.
72. B. J. Gregory, R. B. Moodie, and K- Schofield, J. Chem. Soc. (B), 1970, 338
73. A. Campbell and J. Keynon, J. Chem. Soc., 1946, 25.
74. W. Eisele, C. A. Grob, E. Renk, and H. von Tschammer, Helv. Chim Acta 1968, 51, 816.
75. R. K. Hill and О. T. Chortyk, J. Amer. Chem. Soc., 1962, 84, 1064.
76. P. T. Lansbury and N. R. Mancuso, J. Amer. Chem. Soc., 1966, 88, 1205.
77. R. A. Barnes and M. T. Beachem, J. Amer. Chem. Soc., 1955, 77, 5388.
78. J. B. Chattopadhyaya and A. V. Rama Rao, Tetrahedron, 1974, 30, 2899.
79. L. Field, P. B. Hughmark, S. H. Shumaker, and W. S. Marshall, J. Amer. Chem. Soc., 1961, 83, 1983.
80. H. Wolff, Org. Reactions, 1946, 3, 307.
81a. P. A. S. Smith and E. P. Antoniades, Tetrahedron, 1960, 9, 210; R. T. Conley, J. Org. Chem., 1958, 23, 1330.
816. W. Priizkow and K. Dietsch, Chem. Ber., 1960, 93, 1733.
82. K- N. Carter, J. Org. Chem., 1966, 31, 4257.
83. D. E. Pearson, K- N. Carter, and С. M. Greer, J. Amer. Chem. Soc., 1953, 75, 5905
84. J. W. Schulenberg and S. Archer, Org. Reactions, 1965, 14, 1 [Дж. Шуленберг, С. Арчер. Орг. реакции. — Пер. с англ./Под ред. И. Ф. Луценко. М., Мир, 1967, сб. 14, с. 71.
85. В. С. Challis and A. W. Frenkel, J. С. S. Chem. Comm., 1972, 303; J. C. S.
Perkin II, 1978, 193.
86. M. Lamchen, in «Mechanisms of Molecular Migrations», ed. B. S. Thyagara-jan, Interscience, New York, 1968, vol. 1. p. 1.
87. F. Krohnke, Annalen, 1957, 604, 203. f
88. D. H. R. Barton, M. J. Day, R. H. Hesse, and M. M. Pechet, J. C. S. Perkin 1, 1975, 1764. „ .
89. J. H. Saunders and R. J. Slocombe, Chem. Rev., 1948, 43, 203; R. G. Arnot , J. A. Nelson, and J. J. Verbanc, Chem. Rev., 1957, 57, 47.
90. J. K. Lawson and J. A. T. Croom, J. Org. Chem., 1963, 28, 232.
91. A. L. J. Beckwith and R. J. Hickman, J. Chem. Soc. (C), 1968, 2756.
92. J. Schmutz, F. Kunzle, F. Hunziker, and A. Burki, Helv. Chim. Acta, 19оэ, > 336. .
93. G. A. Olah and J. A. Olah, in «Friedel-Crafts and Related Reactions», G.A. Olah, Interscience, New York, 1964, vol. 3, pp. 1262—1267; J- F. K-shire, Austral. J. Chem., 1967, 20, 575.
94. F. F. Blicke and H. Zinnes, J. Amer. Chem. Soc., 1955, 77, 4849.
95. R. N. Boyd and R. Leshim, J. Amer. Chem. Soc., 1953, 75, 2762.
96. N. A. Le Bel, R. M. Cherltick, and E. A. Curtis, Synthesis, 1973, 678.
97. O. Schindler, R. Blaser, and F. Hunziker, Helv. Chim. Acta, 1966, 49,
98. P. A. S. Smith, Org. Reactions, 1946, 3, 337.
99. J. B. Aylward, Quart. Rev., 1971, 25, 407.
100. R. H. Baker and L. E. Linn, J. Amer. Chem. Soc., 1948, 70, 3721; /• dorm and U. Eholzer, Chem. Ber., 1965, 98, 936. . jn-
101. 1. W. McFarland, J. Org. Chem., 1963, 28, 2179: I. Vgi, Angew. Chem-ternat. Edn., 1962, 1, 8.
102. В. C. Challis and J. A. Challis, см. cc. 2, chapter 13.
526
П I/ Nightingale and J. E. Johnson, J. Heterocyclic Chem.,’1967 '4 Ю2 ”• лпН Д Rpnrtirh I Amar C1™™ C,, mrn nr, ~.L
r Needles and R- E. Whitfield L Org. Chem., 1966, 31, 341 989
S'*'• 261 ”W1 s {HpteZaugg and W. B. Martin, Org. Reactions, 1965, 14, 52.
105- 1 Auerbach, McF. Zamore, and S. M. Weinreb, J. Org. Chem., 1976 41 725 ^ uRohnie aad G. Berg, Chem. Ber, 1966, 99, 2127. ’ b’ 725‘
cm. cc. 2, chapter 5.
1°7- Z- cinnreich and D. Elad, Tetrahedron, 1968, 24, 4509.
1°8’ p p Robinson and R. K. Brown, Canad. J. Chem, 1961, 39, 1171.
109. Lj у Nightingale and J. E. Johnson, J. Heterocyclic Chem, 1967,'4 102 H°- д Giner-Sorolla and A. Bendich, J. Amer. Chem. Soc, 1958, 80 3932 H'- в Skralz and A. F. Al-Sayyab, J. Chem. Soc, 1964, 1318; G. Just and 112' Zehetner, Tetrahedron Letters. 1967, 3389.
D H. Lorenz and E. I. Becker, J. Org. Chem, 1963, 28, 1707.
H B. Henbest and M. J. W. Stratford, Chem. and Ind. (London), 1961, 1170.
*14 X Nakagawa, H. Onoue, and K. Minami, Chem. Comm, 1966, 17.
' 7' j' A. Baker and J. F. Harper, J. Chem. Soc. (C), 1967, 2148; R. K. Hill J. Qrg. Chem, 1957, 22, 830; A. A. Patchett, F. Hoffman, F. F. Giarruso, N Schwam, and G. E. Arth, ibid, 1967, 27, 3822.
115 A. Eschenmoser, Pure Appl. Chem, 1963, 7, 297.
116a E. C. Taylor, A. McKillop, and R. E. Ross, J. Amer. Ghem. Soc, 1965, 87, 1984, 1990.
U66 R. B. Moffat, J. Org. Chem, 1949, 14, 862.
116b>. В. Zienty and G. W. Steahly, J. Amer. Chem. Soc, 1947, 69, 715.
цбг. Д. Burger and A. Hofstetter, J. Org. Chem, 1959, 24, 1290.
Пбд.В- C. Bishop and J. F. Cavalla, J. Chem. Soc. (C), 1966, 802.
117. ’ ‘ ‘ " ............"
118.
119.
120.
G. Gignarella, G. F. Cristiani, and E. Testa, Annalen, 1963, 661, 181.
G. T. Tisue, S. Linke, and W. Lwowski, J. Amer. Chem. Soc, 1967, 89, 6303;
1. Brown and О. E. Edwards, Canad. J. Chem, 1967, 45, 2599. Cm. cc. 67.
M. И. Винник, H. Г. Зарахапи. — Усп. хим, 1967, 36, 121a.L. A. Paquette, L. B. Anderson, J. F. Hansen, S. A.
Amer. Chem. Soc, 1972, 94, 4907.
1216. T. Sato, H. Wakatsuka, and K. Amano, Tetrahedron, таги, У. Kila, and M. Terashima, Chem. Pharm. Bull.
122a. AL Ohno and I. Sakai, Tetrahedron Letters, 1965, 4541.
167.
Lang, and H. Berk, J.
1971, 27, 5381; У. (Japan), 1971, 19,
1226. IF. Zeigenbeim and W. Lang, Angew. Chem., 1962, 74, 943; H. Metzger and
123.
124.
125.
Ta-529.
L. Beer, Z. Naturforsch, 1963, 18b, 986.
G. Just and AL Cunningham, Tetrahedron Letters, 1972, 1151.
R. Huisgen, I. Ugi, H. Brade, and E. Rauenbusch, Annalen, 1954, 586, 30. D. Misiti, H. W. Moore, and K. Folkens, Tetrahedron, 1966, 22, 1201; R. W. Rickards and R, M. Smith, Tetrahedron Letters, 1966, 2361; G. R. Bedford, G. Jones, and B. R. Webster, ibid, 1966, 2367.
E A. Paquette, Org. Synth, 1964, 44, 41.
H- Bohme, S. Ebel, and K. Hartke, Chem. Ber, 1965, 98, 1463.
R- M. Moriarty, J. Org. Chem, 1964, 29, 2748; W. B. Dickinson and E C Lang, Tetrahedron Letters, 1967, 3035.
C G Gassman and B. L. Fox, J. Org. Chem, 1966, 31, 982.
j Paris, L. Berlinguet, and R. Gaudry, Org. Synth, 1957, 37, 47.
• F Wolffe and G. B. Triinitsis, J. Org. Chem, 1968, 33, 894.
Pen.,, _ . l _ 5 . _ - 1964, 29( 646
126.
127.
128. 129 130 131 * * * * * * * * * *
129.
130.
131.
149, г ‘A wu4ie ana Ur. b. irtmilsts, J. urg. tnem., i»oo, 00, о»ч.
139A u n’ R°thman, S. Serota, and D. Swern, J. Org. Chem., 1964, 29, 646.
R- Snyder and С. 7. Elston, J. Amer. Chem. Soc., 1954, 76, 3039; R. E. Dun-
133 м nnd G- C- White, J. Org. Chem., 1958, 23, 915.
°- Л4- Davies and W. J. Jones J Chem. Soc., 1958, 955; W. Logetnann and
134 ? r m’’Chem- Ber., 1958, 91, 2574. n ,n„CCQ
• J- Brown, S. С. K. Su, and J. A. Shafer, J. Amer. Chem. Soc, 1966 88,4468;
? J- Cc>tter, С. K. Sauers, and J. M. Whelan, J. Org. Chem., 1961, 26, 10;
1чкп°Еп^ег^е* C- Carraz, A. M. Revol, and J, Dodu, Bull. Soc. chim. France,
500. -
527
135. W. R. Roderick, J. Amer. Chem. Soc., 1957, 79, 17Ю- F i сь .
F. L. Schaeffer, and E. P. Shoyer, ibid., 1928, 50, 471. ' ’ ‘ ^herrdl,
136. R. Pummerer and G. Domiuller, Chem. Ber., 1912, 45, 292.
137. M. L. Bender, У. L. Chow, and F. Chloupek, J. Amer. Chem Soc ючо on 5380. ’ '' ’ 80-
138. A. B. Mehta, A. P. Phillips, F. Lu-Lin, and R. E. Brooks, J. Org. Chem i960
139. E. Testa and L. Fontanella, Annalen, 1962, 660, 118.
140. E. Fucker, Angew. Chem., 1959, 71, 321; H. D. K. Drew and H H Hnft i Chem. Soc., 1937, 16. ’
141. M. B. Vigneron, O. Crooy, F. Kezdy, and A. Bruylants, Bull. Soc. chim Bel ges, 1960, 69, 616; J. A. Shafer and FI. Morawetz, .1. Org. Chem. 1963 2S 1899; R. K. Robins, J. K. Florner, С. V. Greco, C. W. Noell and C G fan tries, ibid., 1963, 28, 3041. ... oea-
142. A. A. Liebmann and F. E. De Gangi, J. Pharm. Sci., 1963, 52 395-S. H. McElvain and D. Fl. Clemens, J. Amer. Chem. Soc., 1958, 80 3915 '
143. С. M. Hendry, J. Amer. Chem. Soc., 1958, 80, 973.
144. E. H. Зильберман, П. С. Пырялова, Жури. орг. хим., 1965, 1, 983.
145а. A. Kato and S. Wade, Nippon Kagaku Zasshi, 1962, 83, 501.
1456. A. C. Poshkus and I. E. Herweh, J. Org. Chem., 1965, 30, 2466.
146. P. de Mayo and S. T. Reid, Chem. and Ind. (London), 1962, 1576.
147. E. H. White and D. IV. Grisley, J. Amer. Chem. Soc., 1961, 83, 1191.
148. M. B. Robin, F. A. Bovey, and H. Basch, см. cc. 2, chapter 1.
149. R. B. Homer and C. D. Johnson, см. cc. 2, chapter 3.
150. W. E. Stewart and T. H. Siddall, Chem. Rev., 1970, 70, 517.
151. M. Kitano and K. Kuchitsu, Bull. Chem. Soc. Japan, 1974, 47, 631.
152. R. Degeilh and R. E. Marsh, Acta Cryst., 1959, 12, 1007.
153. L. A. La Planche and M. T. Rogers, J. Amer. Chem. Soc., 1964, 86, 337.
154. С. H. Yoder, J. A. Sandberg, and 117. S. Moore, J. Amer. Chem. Soc., 1974, 96,
2260.
155. C. J. Brown and D. E. C. Corbridge, Nature, 1948, 162, 72.
156. R. Lumley-Jones, Spectrochim. Acta, 1974, 30A, 1329.
157. J. D. Dunitz and F. K- Winkler, Acta Cryst., 1975, B31, 251.
158. L. A. La Planche and M. T. Rogers, J. Amer. Chem. Soc., 1963, 85, 3728.
159. A. Mannschreck, Tetrahedron Letters, 1965, 1341.
160. G. Helmchen, Tetrahedron Letters. 1974, 16, 1527.
161. W. Walter and J. Voss, см. cc. 2, chapter 8.
162. A. R. Katriizky and C. Ambler, «Physical Methods in Heterocyclic Chemistry», Academic Press, New York, 1963, vol. 2, p. 161 [A. P. Катриукий, С. Амблер. Физические методы в химии гетероциклических соединений. — Пер. с англ., М,-Л. 1966]. „
163. L. I. Bellamy, «The Infrared Spectra of Complex Molecules», Chapman-Hall, London, 1975, vols. 1 and 2.
164. С. M. Lee and W. D. Kumler, J. Amer. Chem. Soc., 1961, 83, 4586.
165. H. Budzikiewicz, C. Djerassi, and D. H. Williams, «Interpretation of Mass Sprectra of Organic Compounds», Holden-Day, San Francisco, 1964, cnap-ter 4 [А. Будзикевич, К. Джерасси, Д. X. Вильямс. Интерпретация масс спектров органических соединений. — Пер. с англ./Под ред. Н. С. Вульф сона. М., Мир, 1966]. , ..
166. J. A. Gilpin, Analyt. Chem., 1959, 31, 935; Z. Pelah, M. A. !<ielcze®z!’ J. M. Wilson, M. Ohashi, H. Budzikiewicz, and C. Djerassi, J. Amer, cn Soc., 1963, 85, 2470. , 1Q74
167. A. M. Duffield, G. De Martino, and C. Djerassi, Org. Mass Spectrometry, 9, 137. 1064
168a. A. M. Duffield, H. Budzikiewicz, and C. Djerassi, J. Amer. Chem. Soc., 86, 5536 . , io65
1686. Ibid., 1955, 87, 2913; F. Komitsky, J. E. Gurst, and C. Djerassi, ibid., 87, 1398- . , InRI 722
169. A. R. Katritzky and R. A. Y. Jones, Chem. and Ind. (London), 1961, 170. M. Liter, J. C. S. Perkin II, 1972, 816; 1974, 71.
528
172.
173.
174.
175.
176.
p R Homer and R. B. Moodie, J. Chem. Soc., 1963, 4377.
K" _ ~.rr ... n Г T Г Q TT m*7E TAO
J r IT II I 1Pf V4 / L • v • • *•• * V ' у V • V-* • • A v 1 | A A | 4 I I .
i W Barnett and C. J. O'Connor, J. C. S. Perkin II, 1973, 1331. 17' • r i4nn f Amer Chem. Soc . 1930. 52. 5115 178. 179.
180.
181.
182.
183.
184.
185.
186.
187.
188.
189.
190.
191.
192.
„ c Hecht and E. S. Rothman, J. Org. Chem., 1973, 38. 395.
w Renderlu and K. Rosenheck, J. C. S. Chem. Comm., 1972, 179.
dr Martin, J. C. S. Chem. Comm., 1972. 793.
Г R Smith and Yates, Canad. J. Chem., 1972, 50, 771; A. J. Kresge, p H Fitzgerald, and Y. Chiang, J. Amer. Chem. Soc.. 1974, 96, 4698.
X Yates, H. Wai, G. Welch, and R. A. McClelland, J. Amer. Chem. Soc., 1073 95, 418.
„ Homer and R. B. Moodie, J. Chem. Soc., 1963, 4377.
r C Giffneu and C. J. O’Connor, J. C. S. Perkin II, 1975, 706.
*’• .-ij -.-J C I т г с TT m-79 ioni
N F. Hall, J. Amer. Chem. Soc., 1930, 52, 5115.
X Huisgen, H. Brade, H. Walz, and I. Glogger, Chem. Ber., 1957, 90, 1437; g Huisgen, H. Brade, and E. Rauenbusch, Annalen, 1954, 586, 30.
£) 117, Farlow and R. B. Moodie, J. Chem. Soc. (B), 1970, 334.
] Hine and M. Hine, J. Amer. Chem. Soc., 1952, 74, 5266.
X Bowden, Chem. Rev., 1966, 66, 119.
X D. McLachlan and R. A. Nyquist, Spectrochim. Acta, 1964, 20, 1397.
H. Saito, Y. Tanaka, and K. Nukada, J. Amer. Chem. Soc., 1971, 93, 1077.
M. Davies and D. K. Thomas, J. Phys. Chem., 1956, 60, 767.
M. Rabinowitz and A. Pines, J. Amer. Chem. Soc., 1969, 91, 1585; H. J. Bilt-rich and D. Kirsch, Z. phys. Chem. (Leipzig), 1975, 256, 808.
J, V. Hatton and R. E. Richards, Mol. Phys., 1960, 3, 253; 1962, 5, 139.
R. L. Adelman, J. Org. Chem., 1964, 29, 1837.
E M. Arnett, E. J. Mitchell, and T. S. S. R. Marty, J. Amer. Chem. Soc., 1974, 96, 3875.
X. R. Bhaskar and C. N. R. Rao, Biochem. Biophys. Acta, 1967, 136, 561.
W. Gerrard, M. F. Lappert, and J. W. Wallis, J. Chem. Soc., 1960, 2141;
W. Gerrard, M. F. Lappert, H. Pyozora, and J. W. Wallis, ibid., 1960, 2144.
R. S. Drago, T. F. Bolles, and R. J. Niedzielski, J. Amer. Chem. Soc., 1966, 88, 2717 и цитированные там работы.
W. Egan, T. E. Bull, and S. Forsen, J. C. S. Chem. Comm., 1972, 1099.
W. E. Bull, S. K- Madan, and J. E. Willis, Inorg. Chem., 1963, 2, 303.
193.
194. . . „ . .
195a./?. S. Drago, D. W. Meek, M. D. Joesten, and L. La Roche, Inorg. Chem., 1963, 2, 124.
1956. В. B. Wayland, R. J. Fitzgerald, and R. S. Drago, J. Amer. Chem. Soc., 1966,
88, 4600.
196. S. K. Madan, Inorg. Chem., 1967, 6, 421; S. K- Madan and J. A. Sturr, J. Inorg. Nuclear Chem., 1967, 29, 1669.
197. D. A. Buckingham, J. MacB. Harrowfield, and A. M. Sargeson, J. Amer. Chem. Soc.. 1974, 96, 1726.
198. R. Mason, Acta Cryst., 1961, 14, 720.
199. K. L. Gallaher and S. H. Bauer, J. C. S. Faraday II, 1975, 1423.
200. T. Uno and K. Machida, Bull. Chem. Soc. Japan, 1962, 35, 1226.
201. С. Al. Lee and W. D. Kumler, J. Amer. Chem. Soc., 1962, 84, 571.
202. G. Binsch, J. B. Lambert, B. W. Roberts, and J. D. Roberts, J. Amer. Chem. Soc., 1964, 86, 5564.
203. T. Matsuo, Kogyo Kaguka Zasshi, 1965, 68, 1422.
204. D. Bryce-Smith and M. A. Hems, J. Chem. Soc. (B), 1968, 812.
205. T. W. Bentley and R. A. W. Johnstone, J. Chem. Soc. (C), 1968, 2354.
206. J. L. Cotter and R. A. Dine-Hart, Org. Mass. Spectrometry, 1968, 1, 915.
207. R. K. Elsworth and S. Aronoff, Arch. Biochem. Biophys., 1968, 124, 358.
208. J. H. Bowie, R. G. Cooks, S. O. Lawesson, P. J. Jakobsen, and G. Schroll, Chem. Comm., 1966, 539.
209. H. Nagami and M. Horioka, Yakuzaigaku, 1960, 20, 55.
2*0. A. J. Parker, Quart. Rev., 1962, 16, 163.
211- «А. Review of Catalytic and Synthetic Applications for DMF and DMA», Du de Nemours, Delaware, 1960 (Supplement, 1961).
2‘2. H. Bader, A. R. Hansen, and F. J. McCarty, J. Org. Chem., 1966, 31, 2319; °- S. Asquith, W. M. Lord, A. T. Peters, and F. Wallace, J. Chem. Soc. (C), 1966, 95.
529
J. F. Bieron and F. J. Dinan, cm. cc. 2, chapter 4.
J. A. Shafer, см. cc. 2, chapter 12.
213.
214.
215.
216.
217.
218.
219.
220.
221.
222.
223.
224.
225.
226.
227.
228.
229.
230.
231.
232.
233.
234.
235.
236.
237.
J. IF. Paukstelis and M.-G. Kim, J. Org. Chem. 1974 39 15()ч C. J. O'Connor, Quart. Rev., 1970, 24, 553.
Deslongchamps, U. 0. Cheriyan, A. Guida, and R. J Taillefer o Chim., 1976, 1, 235; cf. P. Deslongchamps, Tetrahedron 1975 ’ 31 24M D. Bolton, Austral. J. Chem., 1966, 19, 1013. ’ ’ ' 2463-
S. Biechler and R. W. Taft, J. Amer. Chem. Soc., 1957, 79 4927 M. Blackburn and J. D. Plackett, J. C. S. Perkin II, 1’972’ 1366 L. Vaughn and M. D. Robbins, J. Org. Chem., 1975,’40, 1187
G. Gassman, P. K. G. Hodgson, and R. J. Balchunis, J.’Amer Chem Sn„ ’6 OR. 1275 ' O°C.,
P.
J.
P.
S.
G.
H.
P.
1976, 98, 1275.
D. A. Buckingham, J. McB. Harrowfield, and A. M. Sargeson J Amer Ch₽m Soc., 1974, 96, 1726. ’ ’ em'
R. P. Houghton and R. R. Puttner, Chem. Comm,. 1970, 1270.
F. H. Stodola, J. Org. Chem., 1972, 37, 178.
A. F. Kresge, P. H. Fitzgerald, and Y. Chiang, J. Amer. Chem. Soc., 1974, 96, 4698. ’
C. R. Smith and P. Yates, Canad. J. Chem., 1972, 50, 77b J Amer Chem Soc., 1972, 94, 8811.
R. A. McClelland, J. Amer. Chem. Soc., 1975, 97, 5281.
A. Williams, J. Amer. Chem. Soc., 1976, 98, 5645.
C. J. Giffney and C. J. O'Connor, Austral. J. Chem., 1976, 29, 307.
С. H. Rochester, «Acidity Functions», Academic, London, 1970, p. 132.
J. A. Leisten, J. Chem. Soc., 1959, 765; cf. J. T. Edward, H. S. Chang, K. Yates, and R. Stewart, Canad. J. Chem., 1960, 38, 1518, 2271.
C. J. Giffney and C. J. O’Connor, J. C. S. Perkin II, 1975, 1357.
R. N. Lacey, J. Chem. Soc., 1960, 1633.
D. G. Oakenfull and W. P. Jencks, J. Amer. Chem. Soc., 1971, 93, 178;
D. G. Oakenfull, K- Salvesen, and W. P. Jencks, ibid., 1971, 93, 188.
A. J. Kirby and A. R. Fersht, Prog. Bioorg. Chem., 1971, 1, 1.
M. L. Bender, Y.-L. Chow, and F. Chloupek, J. Amer. Chem. Soc., 1958, 80, 5380.
238. M. F. Aldersley, A. J. Kirby, P. W. Lancaster, R. S. McDonald, and C. R. Smith, J. C. S. Perkin II, 1974, 1487; A. J. Kirby, R. S. McDonald, and C. R. Smith, ibid., 1974, 1495.
239a. D. Davidson and M. Karten, J. Amer. Chem. Soc., 1956, 78, 1066.
2396. E. H. Зильберман, A. E. Куликова, H. AL Тепляков, А. А. Руишнская, ЖОХ. 1962 32 3039
240. W. P. Jencks and M. Gilchrist, J. Amer. Chem. Soc., 1964, 86, 5616.
241. D. J. Hamilton and M. J. Price, Chem. Comm., 1969, 414.
242. J. S. Matthews and J. P. Cookson, J. Org. Chem., 1969, 34, 3204.
243. B. Amit, D. A. Ben-Efraim, and A. Patchornik, J. Amer. Chem. Soc., 1976, 98, 843.
244. A. R. Fersht and W. P. Jencks, J. Amer. Chem. Soc., 1970, 92, 5432, 5442.
245. H. A. Staab, Angew. Chem. Internat. Edn., 1962, 1, 351.
246. G. M. Blackburn and J. D. Plackett, J. C. S. Perkin II, 1973, 981
247. J. G. Sharetkin and A. Forschrim, Analyt. Chem., 1963, 35, 1616; B. Castro, Bull. Soc. chim. France, 1967, 1540; R. Lukes, V. Dudek, O. Sedlakova, and J. Koran, Coll. Czech. Chem. Comm., 1961, 26, 1105.
248. E. A. Evans, Chem. and Ind. (London), 1957, 1596; А. И. Зайцева, E. M. панов. К. А. Кочешков. Изв. АН СССР, сер. хим., 1961, 831.
249. Н. Meerwein, W. Florian, N. Schon, and G. Stopp, Annalen, 1961, 641, b R. F. Borch, Tetrahedron Letters, 1968. 61. .
250. H. Bredereck, F. Effenberger, and E. Henseleit, Chem. Ber., 1965, 98, 2/04.
251. H. Bredereck, R. Gompper, and G. Theilig, Chem. Ber., 1954, 87, 537.
252. R. Compper and O. Christmann, Chem. Ber., 1959, 92, 1935.
253. R. Roger and D. G. Neilson, Chem. Rev, 1961, 61, 179. s
254. В. C Challis and A. D. Frenkel, J. C. S. Chem. Comm., 1972, 303; J. V. л Perkin II, 1978, 193,
630
258.
259.
260.
261.
262.
263.
264.
265.
266.
267.
268.
269.
r Z М Stirling, J. Chem. Soc., 1960, 255.
25b. P Levine and W. C. Fernelius, Chem. Rev., 1954, 54, 467.
256- »/ Kornblum, R. A. Smiley, R. K- Blackwood, and D. C. Iff land, J. Amer. 257’ Them Soc., 1955, 77, 6269.
, 4 Bredereck, R. Gompper, H. Herlinger, and E. Woitun, Chem. Ber., 1960, 93,
£ А Арбузов, О. M. Шаншинская. Труды Казанского хим.-технолог, инет., 1962, 30, 22.
и W. Heine, J. Amer. Chem. Soc., 1956, 78, 3708; C. Zioudrou and G L. Schmir, ibid., 1963, 85, 3258.
C J. M. Stirling, J. Chem. Soc., 1962, 3676.
[) N Crouse and D. Seebach, Chem. Ber., 1968, 101, 3113.
p L Gay and C. R. Hauser, J. Amer. Chem. Soc., 1967, 89, 1647; J. F Wolfe and C.-L. Mao, J. Org. Chem., 1967, 32, 1977.
L I. Smith and J. Nichols, J. Org. Chem., 1941, 6, 489.
G C. Hopkins, J. P. Jonak, H. J. Minnemeyer, and H. Tieckelmann J Org Chem., 1967, 32, 4040.
//. E. Johnson and D. G. Crosby, J. Org. Chem., 1962, 27, 2205; H. Bredereck, F. Effenberger, and H. J. Triebe, Chem. Ber., 1963, 96, 1505; E. Schnabel and H. Schiissler, Annalen, 1965, 686, 229; C. Glacet and G. Troude, Bull. Soc. chim. France, 1964, 292.
p. E. Newallis and E. J. Rumanowski, J. Org. Chem., 1964, 29, 3114.
S. L. Vail, R. H. Barker, and С. M. Moran, J. Org. Chem., 1966, 31, 1642 and refs, therein; J. Chupp and A. J. Speziale, ibid., 1963, 28, 2592.
H. Feuer and U. E. Lynch, J. Amer. Chem. Soc., 1953, 75, 5027; И. K. Moce-eu'i, И. А. Арбузова, А. И. Кольцов, ЖОХ, 1968, 38, 1224.
D. M. von Schriltz, E. M. Kaiser, and C. R. Hauser, J. Org. Chem., 1967, 32,
270.
2610.
271. M. Imoto and M. Kobayashi, Bull. Chem. Soc. Japan, 1960, 33, 1651.
272. R. Giger and D. Ben-Ishai, Israel J. Chem., 1967, 5, 253; T. Falbe and H.-J. Schulze-St einen, Brennstoff-Chem., 1967, 48, 136.
273. H. K. Hall, J. Amer. Chem. Soc., 1956, 78, 2717; D. E. Horning and J. M. Mu-chowski, Canad. J. Chem., 1967, 45, 1247.
274. D. У. Curtin and L. L. Miller, J. Amer. Chem. Soc., 1967, 89, 637.
275. R. Tull, R. C. O'Neill, E. P. McCarthy, J. J. Pappas, and J. M. Chemerda, J. Org. Chem., 1964, 29, 2425.
276. Q. E. Thompson, J. Amer. Chem. Soc., 1961, 73, 5841.
277. A. J. Speziale and L. R. Smith., J. Org. Chem., 1962, 27, 4361; G. M. Cop-
pinger, J. Amer. Chem. Soc., 1954. 76, 1372.
278. D. Davidson and H. Skovronek, J. Amer. Chem. Soc., 1958, 80, 376.
279. D. Davidson and M. Karlen, J. Amer. Chem. Soc., 1956, 78, 1066.
280. IF. Logemann and D. Artini, Chem. Ber,. 1958, 91, 2574 и цитированные там работы; С. King, J. Org. Chem., 1960, 25, 352.
281. R. E. Dunbar and G. C. White, J. Org. Chem., 1958, 23, 915.
282. A. J. Speziale, L. R. Smith, and J. E. Fedder, J. Org. Chem., 1965, 30, 4303
и цитированные там работы.
283. G. A. Olah and J. A. Olah, J. Org. Chem., 1965, 30, 2386.
284. M. N. Hughes and G. Stedman, J. Chem. Soc. Suppl. I, 1964, 5840; H. La-denheim and M. L. Bender, J. Amer. Chem. Soc., 1960, 82, 1895.
285. Z. Kricsfalussy and A. Bruylanls, Bull. Soc. chim. beiges, 1964, 73, 96.
286. R. Campbell and C. J. Peterson, J. Org. Chem., 1963, 28, 2294; V. J. Runge and W. Treibs, J. prakt. Chem,. 1962, 15, 223.
287. E. H. White, J. Amer. Chem. Soc., 1955, 77, 6008.
288. В. C. Challis and C. N. Berry, J. C. S. Perkin II, 1974, 1638.
289. E. H. White and D. W. Grisley, J. Amer. Chem. Soc., 1961, 83, 1191.
290a./?. S. Drago, D. A Wenz, and R. L. Carlson, J. Amer. Chem. Soc., 1962, 84, И06; J. D. Park H. J. Gerjovich, W. R. Lycan, and J. R. Lacher, ibid., 1952, 74, 2189.
2906. С. K. Ingold, «Structure and Mechanism in Organic Chemistry», Bell, London, 1953, pp. 604—610 [К. К. Ингольд. Механизмы реакций и строение
531
291.
292.
293.
294.
295.
296.
297.
298.
299.
300.
306.
307.
308.
309.
310.
органических соединений. — Пер. с англ./Под ред И Л к
Я. Ф. Коммисарова, М., Издатинлит, 1959]. ’ У1Я||Ца и
Е. Р. Oliveto and С. Gerald, Org. Synth. Coll. Vol. 4, 1963 104
F. E. Hardy and P. Robson, J. Chem. Soc. (B), 1967 1151
Я S. Neale, N. L. Marcus, and R. G. Schepers, J. Amer. Chem. Soc., I960 88
J. A Attaway, R. H. Groth, and L. A. Bigelow, J. Amer. Chem Soc юзд
81, 3599. ’’ SJ9’
D. H. R. Barton, R. H. Hesse, M. M. P echet, and H T. Toh J C S pPrn
I, 1974, 732. ’ HerKln
A. E. Fuller and W. J. Hickinbottom, J. Chem. Soc., 1965, 3228
0. 0. Orazi, R. A. Corral, and H. Schuttenberg, J. C. S. Perkin I 1074 9n«7
5. Trippett and D. M. Walker J Chem. Soc., 1960, 2976; A. J. Speziale and
L. R. Smith, J. Amer. Chem. Soc., 1962, 84, 1868.
E. Eilingsfeld M. Seefelder, and H. Weidinger, Chem. Ber., 1963, 96 2671
P. Linda, A. Lucarelli, G. Marino, and G. Savelli, J. C. S. Perkin II’ 1974 1610.
301. M. L. Martin, G. Ricolleau, S. Poignant, and G. J. Martin, J. C. S. Perkin II 1976, 182.
302. В. И. Минкин, Г. H. Дорофеенко — Усп. хим., 1960, 29, 1301.
303. D. S. Deorha and Р. Gupta, Chem. Ber. 1964, 97, 616; E. Emili, A J Castro and P A. Westfall, J. Org. Chem , 1965, 30, 339.
304. R. Sciaky, U. Pallini, and A. Consonni, Gazzetta, 1966, 96, 1284; C. Jutz and W. Mueller, Chem. Ber., 1967, 100, 1536.
305. J. D. Albright, E. Benz, A. E. Lanzilotti, and L. Goldman, Chem. Comm., 1965, 413.
311.
312.
313.
314.
315.
316.
317.
318.
319.
320.
321.
322.
323.
324.
325.
326.
327.
328.
329.
F. S. Fawcett, C. W. Tullock, and D. D. Coffman, J. Amer. Chem. Soc., 1962, 84, 4275.
R. Bonnett, Ref. 68, chapter 13.
W. R. Vaughan and R. D. Carlson, J. Amer. Chem. Soc., 1962, 84, 769.
B. Prajsnar and C. Troszkiewicz, Roczniki Chem., 1962, 36, 265, 843, 853, 1029.
H. M. Walborsky and G. E. Niznik, J. Org. Chem., 1972, 37, 187; I. Ugi and R. Meyer, Chem. Ber., 1960, 93, 239; J. Casanova, R. E. Schuster, and N. D. Werner, J. Chem. Soc., 1963, 4280.
D. T. Mowry, Chem. Rev., 1948, 42, 257.
/. C. Graham and D. H. Marr, Canad. J. Chem., 1972, 50, 3857.
J. C. Thurman, Chem. and Ind. (London), 1964, 752.
M. V. Lock and B. F. Sagar, J. Chem. Soc. (B), 1966, 690; G. M. Burnett and
К. M. Riches, ibid., 1966, 1229. Mr.
H. H. Wasserman and В. H. Lipshutz, Tetrahedron Letters, 1975, 1731
С. H. Bamford and E. F. White, J. Chem. Soc., 1960, 4490; W. Walter, M. Steffen, and K. Heyns, Chem. Ber., 1966. 99, 3204. .
E. Mikdajewski, J. E. Swallow, and M. W. Webb, J. Appl. Polymer Sci., 1964, 8, 2067.
H. Ito, Chem. Pharm. Bull. (Japan), 1964, 12, 326, 329.
D. E. Remy, R. E. Whitfield, and H. L. Needles, Chem. Comm., 1967, 681 цитированные там работы. 04g
S. E. Ellzey, С. H. Mack, and W. J. Cornnick, J. Org Chem., 1967, 32, «« H. C. Brown and N. M. Yoon, J. Amer. Chem. Soc., 1966, 88, 1404.
R. F. Borch, Tetrahedron Letters, 1968. 61. refers
Atta-ur-Rahman, A. Basha, N. Waheed, and S. Ahmed, Tetrahedron Letter, 1976, 219. c lnc. 1 nuo
H. C. Brown and A. Tsukamoto, J. Amer. Chem. Soc., 1964, 8b, iuoy.
A. I. Birch and H. Smith, Quart. Rev., 1958, 12, 17.
F. D. Popp and H. P. Schultz, Chem. Rev., 1962, 62, 19 Mnanien and
E. S Wallis and J. F. Lane, Org. Reactions, 1946, 3, 267; E. Magni
R. Baltzly, J. Org. Chem., 1958, 23, 2029.
P. Radlick and L. R. Brown, Synthesis, 1974, 290.
D. V. Banthorpe, cm. Cc. 11, p. 637.
532
330 EH. White and. D. J. Woodcock, cm. cc. 11, chapter 8.
i r G Cadogan, «Chemical Society Monograph for Teachers No. 24» The 331- AJmical Society, London, 1970, p. 71.
n phc Neale, Synthesis, 1971, 1.
332- p F Moore, Polymer, 1963, 4, 493; G. H. Booth and R. G. IP Norrish J
333- Them. Soc., 1952, 188. ’ ’
„. П j Carlsson, L. H. Gan, and D. M. Wiles, Canad. J. Chem., 1975 53 2337 з31 V; Escher, Tetrahedron Letters, 1968, 4295.
RA Johnson and F. D. Greene, J. Org. Chem., 1975, 40, 2186, 2192
33°- у L Chow, J. N. S. Tam, C. J. Colon, and R. S. Pillay, Canad. J Chem 33/- 1973 51, 2469 и цитированные там работы.
8 С. Danen and R. W. Gellert, J. Amer. Chem. Soc., 1972, 94, 6853
33q L p. Kulm, G. G. Kleispehn, and A. C. Duckworth, J. Amer. Chem Soc 1967
89, 3858. ’ ' ’
Qta H K- Hall< M- K- Brandt, and R. M. Mason, J. Amer. Chem. Soc., 1958 80 6420; I- Wadso, Acta Chem. Scand., 1965, 19, 1079. ’ ’
о,, p D. Hoagland and S. W. Fox. J. Amer. Chem. Soc., 1967, 89, 1389.
ЧА9 S С K. Su and J. A. Shafer, J. Org. Chem., 1969, 34, 2911
чл R. M. Peck, J. Org. Chem,. 1962, 27, 2677.
344 W. Flitsch, Annalen. 1965, 684, 141.
345 W. Flitsch and H. Peters, Chem. Ber., 1970, 103, 805.
346 II7- Flitsch and S. R. Schindler, Synthesis, 1975, 685.
347’ D. A. Tomalia and J. ;V. Paige, J. Org. Chem., 1973, 38, 422; N. Gravitz and W. P. Jencks, J. Amer. Chem. Soc., 1974, 96, 489, 499, 507.
348. J. F. Wolfe and T. G. Rogers, Chem. Comm., 1967, 1040,
349. T. Uno and K. Machida, Bull. Chem. Soc. Japan, 1961, 34, 545.
350. " T. Otvos, F. Dutka, and H. Tudos, Acta Chim. Acad. Sci. Hung., 1965, 43, 53.
351. L. Horner and E. H. Winkelmann, Angew. Chem., 1959, 71, 349.
352. R- Filler, Chem. Rev., 1963, 63, 21; «Newer Methods of Preparative Organic Chemistry», ed. W. Foerst, Academic, New York, 1964. vol. 6, p. 151.
353. R. E. Pearson and J. C. Martin, J. Amer. Chem. Soc., 1963, 85, 3142.
354. F. Dewhurst and P. K. J. Shah, J. Chem. Soc. (C), 1969, 1503.
355. T. R. Beebe and F. M. Howard, J. Amer. Chem. Soc., 1969, 91, 3379.
356. R. 0. Kan and R. L. Furey, Tetrahedron Letters, 1966, 2573.
357. У. Kanaoka and У. Hatanaka, J. Org. Chem., 1976, 41, 400.
358. H. L. Yale, Chem. Rev., 1944, 33, 209.
359. R. T. Coutts, Canad. J. Pharm. Sci., 1967, 2, 1, 27; J. B. Bapat, D. St. C. Black, and R. F. C. Brown, Adv. Heterocyclic Chem., 1969, 10, 199.
360. 0. Exner and W. Simon, Coll. Czech. Chem. Comm., 1965, 30, 4078.
361a./. D. Aubort and R. F. Hudson, J. C. S. Chem. Comm., 1970, 937.
3616. Cm. cc. 361 a, p. 938.
362, IP. B. Renfrow and C. R. Hauser, Org. Synth. Coll. Vol. 2, 1943, 67.
363. G. B. Bachman and J. E. Goldmacher, J. Org. Chem., 1964, 29, 2576.
364. В. E. Hackley, R. Plapinger, M. Stolberg, and T. Wagner-Jauregg, J. Amer. Chem. Soc., 1955, 77, 3651.
365. T. C. Bruice and J. J. Bruno, J. Amer. Chem. Soc., 1961, 83, 3494
366a. L. W. Jones and C. D. Hurd, J. Amer. Chem. Soc., 1921, 43, 2422.
3666. W. P. Jencks, ibid., 1958, 80, 4581.
367. К. T. Wang, D. N. Brattesani, and B. Weinstein, J. Heterocyclic Chem., 1966,
3, 98; T. Fujii, С. C. Wu, and S. Yamada, Chem. Pharm. Bull. (Japan), 1967,
15, 345.
368. M. T. W. Hearn and A. D. Ward, Austral. J. Chem., 1969, 22, 161.
369. H. Ulrich and A. A. R. Sayigh, J. Chem. Soc., 1963, 1098; O. Exner and □7n {-Holubek, Coll. Czech. Chem. Comm., 1965, 30, 940.
M. w. p. Jencks and M. Gilchrist, J. Amer. Chem. Soc., 1964, 86, 5616.
47 «o' Л Staab> M- Puking, and F. H. Durr, Chem. Ber., 1962, 95, 1275
3H6.S A. Bernhard, Y. Shalitin, and Z. H. Tashjian, J. Amer. Chem. Soc., 1964, 86, 7 4406.
2’ r' ^urd an-d P- B. Cochran, J. Amer. Chem. Soc., 1923, 45, 515; O. Exner, Coll. Czech. Chem. Comm., 1961, 26, 201.
533
373. V. H. Chiang, J. Org.,Chem., 1971, 36, 2146, 2155.
374. 0. Neunheoffer and E. Ruske, Chem. Ber., 1961, 94, 623.
375. N. Kornblum and R. A. Brown, J. Amer. Chem. Soc., 1965, 87, 1742.
376 E. Shaw, J. Amer. Chem. Soc., 1949, 71, 67; В. С. Смирнов E А Шкла рук — ЖОХ, 1946, 16, 1443, 1687, 1693; S. A. Matlin and P. G. Samme. J. C. S. Chem. Comm., 1972, 1222; G. Just and K. Dahl, Canad J Chem’ 1970, 48, 966.
377. В. H. Bracher and R. W. H. Small, Acta Cryst., 1970, B26, 1705.
378. 0. Exner, Coll. Czech. Chem. Comm., 1964, 29, 1337; 1965, 30, 652.
379a. J. H. Cooley, W. D. Bills, and J. R. Throckmorton, J. Org. Chem I960 25 1734. ’ ’ ’
3796. /. P. Freeman, J. Amer. Chem. Soc., 1958, 80, 5954.
380a. R. E. Plapinger, J. Org. Chem., 1959, 24, 802.
3806. O. Exner, Coll. Czech. Chem. Comm., 1962, 27, 2284.
381. /. E. Johnson, E. A. Nalley, Y. K. Kunz, and J. R. Springfield, J Org Chem 1976, 41, 252.
382. B. J. Price and I. O. Sutherland, Chem. Comm., 1967, 1070.
383. IF. Walter and E. Schaumann, Annalen, 1971, 743, 154.
384. M. Raban and D. Kost, J. Org. Chem., 1972, 37, 499.
385a./. H. Bowie, M. T. W. Hearn, and A. D. Ward, Austral. J. Chem 1969 22 175. ’ ’
3856.//. A. Akers, C. L. Atkin, and J. B. Neilands, Org. Mass. Spectrometry, 1975 10, 259.
386. A. J. Buglass, K. Hudson, and J. G. Tillett, J. Chem. Soc. (B), 1971, 123.
387. G. M. Steinberg and R. Swidler, J. Org. Chem., 1965, 30, 2362; /. Gerstein and W. P. Jencks, J. Amer. Chem. Soc., 1964, 86, 4655.
388. V. C. Bass and J. H. Yoe, Taianta, 1966, 13, 735; T. J. King and P. G. Harrison, J. C. S. Chem. Comm., 1972, 815.
389. F. Feigl, «Spot Tests in Organic Analysis», 7th edn., Elsevier, Amsterdam, 1966, p. 214.
390. A. R. Fields, В. M. Daye, and R. Christian, Taianta, 1966, 13, 929.
391a. D. C. Berndt and I. E. Ward, J. Org. Chem., 1974, 39, 841.
39 16.0. C. Bendt and R. L. Fuller, J. Org. Chem., 1966, 31, 3312.
392. R. Arndt and H. Scholz, Annalen, 1934, 510, 62.
393. L. J. Hayes, 117. C. Cunningham, J. E. Johnson, J. R. Springfield, J. S. Hwang, and D. L. McLaugherty, J. Org. Chem., 1971, 36, 284.
394. O. Exner, Coll. Czech. Chem. Comm., 1962, 27, 2284; E. M. Усова, E. M. Bo-
рошин — ДАН 1956, 113, 1306; 1956, 114, 120.
395. T. Mukaiyama and H. Nohiro, J. Org. Chem., 1961, 26, 782.
396. O. Exner, Coll. Czech. Chem. Comm., 1956, 21, 1500.
397. D. Samuel and B. L. Silver, J. Amer. Chem. Soc., 1963, 85, 1197.
398. F. A. Daniher, J. Org. Chem., 1969, 34, 2908.
399. J. R. Cox and M. F. Dunn, Tetrahedron Letters, 1963, 985.
400. R. M. Gipson, F. H. Pettit, C. G. Skinner, and W. Skive, J Org. Chem., 1963, 28, 1425; M. Masaki and M. Ohtake, Bull. Chem. Soc. Japan, 1965, 38, 1802.
401. F. Winternitz and C. Wlotzka, Bull. Soc. chim. France, 1960, 509.
402. E. Oliveri Mandala, Gazzetta, 1922, 52, I, 107.
403. T. Emery and J. B. Neilands, J. Amer. Chem. Soc., 1960, 82, 4903.
404a. D. F. Minor, W. A. Waters, and J. V. Ramsbottom, J. Chem. Soc. (B), 196/,
180.
4046. M. J. Perkins and P. Ward, J. C. S. Chem. Comm., 1973, 883.
H. Paulsen and D. Stoye, cm. cc. 2, chapter 10. ,r,
O. L. Salerni, В. E. Smart, A. Post, and С. C. Cheng, J. Chem. Soc. ( )>
405.
406.
407.
408.
409.
410.
411.
1968, 1399.
P. A. S. Smith, Org. Reactions, 1946, 3, 366.
H. Dorm, A. Zubek, and K- Walter, Annalen, 1967, 707, 100.
W. J. McKillip and R. C. Slagel, Canad. J. Chem., 1967, 45, 2619; and J. Carriolo, J. Amer. Chem. Soc., 1960, 82, 1778.
P. Bouchet, J. Elguero, and K. Jacquier, Bull. Soc. chim. France, T. C. Bruice and R. G Willis, J. Amer. Chem. Soc., 1965, 87, 531.
W. P. Jencks
1967, 3502.
534
„ w <Hroh and D. Henning, Chem. Ber., 1967, 100, 388.
412. j J. Amer. Chem. Soc., 1956, 78, 1645.
413a. к- % Co/Wn and J. Nicholson, J. Org. Chem., 1965, 30, 1162.
4136'd Hone and L. A. Wiles, J. Chem. Soc. (C), 1967, 2636.
413,3 r' Ainsworth, Canad. J. Chem., 1965, 43, 1607.
414- v Belniak, E. Domagaliva, and H. Hopkala, Roczniki Chem., 1967, 41, 831.
415’ 1C Stowell, J. Org. Chem., 1967, 32, 2360.
416- п M Гривнак, Б. Г. Болдырев — Ж. Орг. хим., 1967, 3, 2160.
4 o' Н Paulsen and D. Stoye, Chem. Ber., 1966, 99, 908.
m П A. Reynolds and J. A. van Allan, J. Org. Chem., 1959, 24, 1478.
4 nr ₽ p' Smith, A. C. Bates, A. J. Battisti, P. G. Byrnes, С. T. Mroz, T. J. Srnea-il96'rine'and F. X. Albrecht, ibid., 1968, 33, 851.
(On z van Alphcn, Rec. Trav. chim., 1924, 43, 823.
loi R Buyle, Helv. Chim. Acta, 1964, 47, 2449.
499’ p. Schestakoff, Chem. Ber., 1912, 45, 3723.
I93' W. F. Short, J. Chem. Soc., 1921, 120, 1445.
E. Schmitz, S. Schramm, and R. Ohme, J. prakt. Chem., 1967, 36, 86.
495 A. F. Hegarty and F. L. Scott, J. Org. Chem., 1968, 33, 753.
496 А. В. Фокин, Ю. H. Студнев, H. А. Прошин. — ЖОХ, 1967, 37, 1725.
427 L. H. Jensen, J. Amer. Chem. Soc., 1956, 78, 3993.
428 H. Lumbroso and J. Barassin. Bull. Soc. chim. France, 1964, 3190.
429 R- Shintani, Acta Cryst., 1960, 13, 609; G. Tomiie, С. H. Koo, and I. Nitta, ibid., 1958, 11, 774.
430. G. S. D. King, J. Chem. Soc. (B), 1966, 1224.
431. В. H. Korsch and N. V. Riggs, Tetrahedron Letters, 1966, 5897; A. Foucaud and R. R. Roudaut, Compt. rend, (c), 1966, 266, 726; G. J. Bishop, B. J. Price, and I. O. Sutherland, Chem. Comm., 1967, 672.
432. H. E. Baumgarten, P. L. Greger, and R. L. Zey, J. Amer. Chem. Soc., 1960, 82, 3977; D. M. Wiles and T. Suprunchuk, Canad. J. Chem., 1968, 46, 701.
433. E. H. White and K. Matsuo, J. Org. Chem., 1967, 32, 1921.
434. C. S. Hudson, J. Amer. Chem. Soc., 1917, 39, 462.
435. C. R. Lindegren and C. Niemann, J. Amer. Chem. Soc., 1949, 71, 1504.
436. H.-H. Stroh and H. Tengler, Chem. Ber., 1968, 101, 751.
437. G. J. Papariello and S. Commanday, Analyt. Chem., 1964, 36, 1028.
438. 0. Makitie, Suomen Kemi B. 1966, 39, 282 (Chem. Abs., 1967, 66, 99067).
439. R. M. Issa, Л1. F. Ec-Shaly, and M. F. Iskander, Z. anorg. Chem., 1967, 354,90.
440. R. C. Paul and S. L. Chadha, Spectrochim. Acta, 1967, 23A, 1249.
441. G. R. Supp, Analyt. Chem., 1968, 40, 981.
442. H. Stetter and H. Spangenberger, Chem. Ber., 1958, 91, 1982; A. Ebnother, F. Jucker, A. Lindenmann, E. Risei, R. Steiner, R. Suess, and A. Vogel, Helv. Chim. Acta, 1959, 42, 533.
443. A. N'Diaye, D. Cabrol, and A. Broche, J. Chim. phys., 1974, 71, 1388.
444a./?, L. Hinman and M. C. Flores, J. Org. Chem., 1959, 24, 660.
4 446.7/. С. Бердинский, E. Ю. Посягина, В. Ф. Посягин, В. Ф. Усть-Качкин-цев. — Ж. орг. хим., 1968, 4, 91.
Shawali and Н. М. Hassaneen, Tetrahedron, 1972, 28, 1299.
446 . Л. А. Овсянникова, Т. А. Соколова, Н. П. Запевалова — Ж. орг. хим., 1968, 4, 459.
E^hen-berger, A. Staehelin, and J. Druey, Helv. Chim. Acta, 1954, 37, 837. ало Inman and R. J. Landborg, J. Org. Chem., 1959, 24, 724.
л. Ronco, B. Prijs, and H. Erlenmeyer, Helv. Chim. Acta, 1956, 39, 1253; 4-„ -4- S. Smith, J. M. Clegg, and J. Lakritz, J. Org. Chem., 1958, 23, 1595.
451 м Grazi’ F- Conconi, and V. Vigi, J. Biol. Chem., 1965, 240, 2465.
4гл' , • 4 Leonard and J. H. Boyer, J. Org. Chem., 1950, 15, 42.
453 r v H°ward' tj. Gever, and P. H.-L. Wei, J. Org. Chem., 1963, 28, 868.
454 т r Plessing, Arch. Pharm., 1964, 297, 240.
F Ishibashi, T. Matsuo, and M. Kojama, J. Org. Chem., 1964, 29, 455 Blasio, A. Ripamonti, and E. Testa, Gazzetta, 1968, 98, 3.
128^' FPeK°a, В. К- Скрипченко, Ж. орг. хим., 1968, 4, 243; 1967, 3, 1251,
535
456. J. A. Young, W. S. Durrell, and R. D. Dresdner, J. Amer. Chem inon 84, 2105. ’ '962,
457. A. Le Berre and J. Godin, Coinpt. rend., 1965, 260, 5296.
458. A. F. Hegarty and M. T. McCormack, J. C. S. Chem, Comm. 1975 irr
459. H.-H. Stroh and H. Beltz, Annalen, 1966, 700, 78; А. П. Г веков’ В R in ченко, Ж. орг. хим., 1967, 3, 1294, ’ ’ Шев'
460. L. Bernardi and О. Goffredo, Gazzetta, 1964, 94, 947.
461. /. Honzl and J. Rudinger, Coll. Czech. Chem. Comm., 1961 26 2333
462. P. A. S. Smith and H. G. Pars, J. Org. Chem., 1959, 24, 1325. ’
463. J. P. Horwitz and V. A. Grakauskas, J. Amer. Chem. Soc., 1957, 79 1249
464. H. Bredereck, B. Fohlisch, and K. Walz, Angew. Chem., 1962, 74, 388’
465. P. Hope and L. A. Wiles, J. Chem. Soc., 1965, 5386; H.-H Stroh and G inh„ chen, Z. Chem., 1968, 8, 24. ' n'
466. G. Lohaus, Chem. Ber., 1967, 100, 2719.
467. C. N. Yiannios, A. C. Hazy, and J. V. Karabinos, J. Org. Chem., 1968 33 2076. ’
468. J. C. Thurman, Chem. and Ind. (London), 1964, 752; А. П. Греков Л. H Кулакова, О. П. Швайка — ЖОХ, 1959, 29, 3054.
469. S. В. Needleman and AL С. Chaing Кио, Chem. Rev., 1962, 62, 405; R В. Kelly, E. G. Daniels, and J. IF. Hinman, J. Org. Chem., 1962, 27, 3229; D. MacKay, U. F. Marx, and W. A. Waters, J. Chem. Soc., 1964, 4793.
470. R. B. Kelly, G. R. Umbreint, and W. F. Liggett, J. Org. Chem 1964 29 1273.
471, L. Carpino, J. Amer. Chem. Soc., 1957, 79, 96.
472. Y. Wolman, P. M. Gallop, A. Patchornik, and A. Berger, J. Amer Chem Soc 1962, 84, 1889.
473. K. Ronco, B. Prijs, and H. Erlenmeyer, Helv. Chim. Acta, 1956, 39, 1253.
474. K. Kratzl and К. P. Berger, Monatsh., 1958, 89, 83; H. Stetter and H. Span-genberger, Chem. Ber., 1958, 91, 1982; H. C. Brown and В. C. Subba Rao J. Amer. Chem. Soc., 1956, 78, 2582.
475. C. Ainsworth, J. Amer. Chem. Soc., 1956, 78, 1636.
476 T. Taguchi, J. Ishibashi, T. Matsuo, and M. Kojima, J. Org. Chem., 1964, 29, 1097.
477. E. Domagalina and I. Kurpiel, Roczniki Chem., 1967, 41, 1241.
478. S. Wawzonek and R. C. Gtteldner, J. Org. Chem., 1965, 30, 3031.
479. R. C. Slagel, J. Org. Chem., 1968. 33, 1374.
480 M S. Newman and I Ungar, J. Org. Chem., 1962, 27, 1238; M. Sprecher, M. Feldkimel, and M. Wilchek, ibid., 1961, 26, 3664.
9.10. ПРОИЗВОДНЫЕ УГОЛЬНОЙ кислоты
А. Ф. ХЕГАРТИ (University College, Cork)
9.10.1. ВВЕДЕНИЕ
В этой главе рассматриваются соединения общей формулы (1), где X и Y — гетероатомы; как правило, кислород, азот или г
правило, кислород, азот или га
R!O-
О II X—С—Y (1) о о II II НО— С—ОН RO— С-ОН (2) (3) О R‘O—С—OR2 (4)
О О II 11 О о О II
II II -С—О—С—OR2 II II R>O— С—О— С—R2 R11 о—с—х Cl-
(5) (6) (7)
О
I-C-C1
(8)
536
,„reK. Эти вещества можно считать производными (эфиры, ами-S талотенангидриды) угольной кислоты (2). Угольная кислота в водных растворах (однако ее можно получить ' безводных условиях) [1], а многие ее простейшие производные пяются высокоактивными веществами, яВютсЯ в синтезе и обладают рядом свойств.
R\ II
XN—С—OR3 r2/ (9)
R’\ ? ZR3
XN—C—N'
R2/ ZR4
(12)
которые широко исполь-интересных химических
Y\ ?
N—С—OR2 R1Z
(10)
OR2 I Ri—N=C~N
R'\ 1° Л—с—х
(11)
V 0
Y\ II /R2
С—NZ
(13)
(14)
В дальнейшем будут рассмотрены следующие типы химических соединений: моно- и диэфпры угольной кислоты (3, 4, R1, R2 — Aik иди Аг); ангидриды угольной кислоты (5) или смешанные ангидриды с карбоновыми кислотами (6); галогенангидриды угольной кислоты (7; X = галоген, и родственные азиды, X = N3); фосген (8) и другие дигалогенкарбонильные соединения; карбаматы (9, Rl, R2 — Н, Aik пли Аг); N-замещенные карбаматы (10, Y = NO, NO2 или галоген); карбамоилгалогениды (11,X = галоген) и карбаминовые кислоты (11, X — ОН); мочевины (12); изомочевины (13) и N-замещенные мочевины (14, Y = NO, NO2 или галоген).
О О" О'
•• II •• + I •• •• I +
X—С—Y Х=С—Y X—C=Y
(15) (16)
Если в соединениях общей формулы (1) отсутствуют объемистые заместители, то их стереохимия определяется расположением четырех атомов в одной плоскости. Кроме того, так как заместители X и Y имеют неподеленные пары электронов, существенный вклад вносят резонансные структуры (15) и (16), что приводит к укорочению связей X—СО (или Y—СО), а также к затруднению вращения вокруг этих связей. Ограничение вращения (обнаруживаемое, как правило, с помощью метода ЯМР) проявляется наиболее захметно, когда заместителем X (или Y) является ззот (в большей степени, чем кислород), однако оно все-таки Несколько меньше, чем в случае простых амидов. Например, по Данным рентгеноструктурного анализа [2, 3], длины связей С N Дя карбаматов (9) составляют 0,135 нм и 0,137 нм в случае мо-евин (12) (по сравнеиию с 0>1з2 нМ для простых амидов). Вме-е с тем отклонения от планарности составляют 5 9 (вплоть
537
до 15° в случае некоторых стернчески затрудненных тетразам щенных мочевин (12)] по сравнению с ^5° у амидов. Характер' стические частоты валентных колебаний карбонильной rnvn в ПК-области также подтверждают этот факт, а именно эти личины больше, чем в случае соответствующих эфиров и амииЛ карбоновых кислот [4]. д в
Простые производные (3) —(14) растворимы в воде с образованием сильных межмолекулярных водородных связей Однако галогенангидриды (7), (8) и (11), некоторые ангидриды и карбаматы (5), (6) и (9) быстро взаимодействуют с водой. Наличие водородных связей в этих веществах в твердом состоянии приводит к необычно высоким температурам плавления для низкомолекулярных производных. Мочевины (14) представляют собой интересный пример, так как замещение водорода на алкильные группы приводит к понижению температуры плавления, несмотря на увеличение молекулярной массы (12, R1 = R2 = R3 = R* = н т. пл. 133 °C; R’=Me, R2 = R3 = R4 = н т пл 102оС. R1 J = R3 = Me, R2 = R4 = Н, т. пл. 106 °C; R1 = R2 = R3 = R« = Me, жидкость, т. кип. 215 °C). Для простых карбаматов наблюдается аналогичная тенденция (9, R1 == R2 == Н, R3 = Et, т. пл. 50°C, т. кип. 184 °C; R1 = Me, R2 = Н, R3 = Et, т. кип. 170°С) [5, 6].
9.10.2. ЭФИРЫ, ГАЛОГЕНАНГИДРИДЫ И АНГИДРИДЫ УГОЛЬНОЙ кислоты
9.10.2.1. Моноэфиры угольной кислоты
При пропускании газообразного СО2 через водные растворы спиртов при высоких значениях pH устанавливается равновесие, которое дает небольшие количества моноэфиров угольной! кислоты (17); Лравн равна примерно 1 для метанола, но 1 для большинства кислых спиртов [7].
Значительные концентрации монокарбонатов в водных растворах могут возникать за счет первоначального быстрого гидролиза соответствующих хлорформиатов при высоких pH. Затем происходит отщепление СО2 от (17) и вновь устанавливается рав новесие согласно схеме (1).
Яравн ,п
ROH + НСОз , ROCO7 + Н2о ( 1
(17)
Расщепление простых алкилмонокарбонатов при высоких зад чениях pH является процессом, не зависящим от pH. ,орме. ловиях алкилмонокарбонаты находятся в ионизованно! т. По стехиометрии реакции требуется участие иона ни тельНых вора; это происходит в результате быстрых последова реакций {схема (2)}.
538
Бренстеду
Скорость отшепления СО2 от монокарбоната сильно зависит структуры R; сообщалось [7], что величина р по ’ ВИСИТ от о
RO-C—О' RO‘ + CO2
со2 + но' нсо;
RO' + Н2О ROH + НО’
HCOJ + HO- —> СО2з'+Н2О о
(2)
Суммарно: RO—С—О + НО —> ROH + СО2'
ДЛЯ уходящей группы RO- равна 1,1. Суммарная реакция (RO' + C02 =*=* ROCO2) имеет константу Бренстеда +1,4, что предполагает большее влияние на равновесие структуры, чем при протонированпи RO~; это, в свою очередь, означает, что карбоксильная группа в этой реакции значительно сильнее оттягивает электроны, чем водород.
Mg
о О до
II II II н>
A/\C\r + (B'O-C-OhMg — 0_/\/\/\^\R —*
о о (,8)
II II
— ho/\/\/\C\r
Магниевые и натриевые соли моноэфиров используются как мягкие карбоксилирующие агенты для енолизуюгцихся кетонов и в этом отношении превосходят диалкиловые эфиры [9]. Пример такой реакции приведен на схеме (3); промежуточный магниевый хелат (18) может быть алкилирован или ацилирован [10].
9.10.2.2. Диэфиры угольной кислоты
Получение симметрично замещенных диэфиров обычно проводят при использовании фосгена. Может добавляться основание (например, третичный амин) в качестве катализатора; интермедиатом служит хлорформиат (19), который можно выделить. Взаимодействие (19) со второй молекулой спирта или фенола дает возможность получать смешанные диэфиры (20) {схема .(4)}. место фосгена можно использовать М.ГАкарбопилдпимидазол { )• Реакция с (21) также ступенчата и поэтому может быть О^ользована для синтеза смешанных эфиров [11] {схема (5)}. ноо Щ'аЛ°СЬ такЖе 0 прямом карбонилировании алкоксидов мо-ксидом углерода в присутствии селена или Си(ОМе)2 [12].
539
Алкилирование алкилгалогенидами карбонат-дианиона в Дм* приводит к симметричным алкиловым диэфирам [13]. Другой к/ тод, исключающий применение фосгена, основан на обработк" эфиров дихлоруксусной кислоты гидридом натрия, с выходом 30—70% образуются симметричные диэфиры [14].
О
О
R'OH + Cl—(!— Cl
О RiQ—A-Cl—
r'oh
о
R'O—C—OR2
r2oh
(19)
(20)
О
R2OH II
------► R2O—C—OR'
(5)
Гидролиз эфиров угольной кислоты может протекать по схеме специфического кислотного или основного катализа, а также по некаталитическому пути. Карбонаты с сильными электронооттягивающими группами (например 23, R1 = R2 — 4-NO2C6H4) гидролизуются не по пути кислотного катализа, поскольку равновесная концентрация протонированной формы (22) низка. Однако даже катализируемый самой водой гидролиз нейтральных карбонатов может быть достаточно быстрым, например скорость некатализируемого гидролиза (23, R1 = R2 — 4-МО2СбН4) 0,17 л-моль-1-мин-1 при 50 °C {схема (6)}. Сообщалось о катализе гидролиза карбонатов различными основаниями; в случае имидазола на основании наблюдений за промежуточным карбаматом показано, что имеет место нуклеофильный катализ.
♦ОН
II
R'O—С—OR2
(22)
О
R'O—С—OR2
(23)
н2о| но~1н2о
I—> R'O—СО j <—1
(6)
R‘OH4-CO2
Арилкарбонаты (24, Аг = 4-NO2CeH4) могут использоваться для этерификации карбоновых кислот [схема (7)]. Реакция в чает промежуточное образование смешанного ангидрида ( ) применима только для реакционноспособных арилкарбонатов [ I 6 J
Алкоксикарбонилированпе аминов (образование карбаматов) служит удобным методом защиты аминогрупп, особенно
540
и II
. n-C—OAr + R—С—ОН > ArO
(24)
О О о
II II ArO" II
R—С—О— С—Аг ------> R—С— ОАг
(25) О О
II 1 II
RiNH2 + R2O—С—-OR3 —> R'NH—С—OR2 + R'NH—С—OR3
(7)
(8)
(26) (27) (28)
кар-
ее пептидного синтеза {схема (8)}. Регенерацию амина из а мата [27] осуществляют при различных условиях (кислых, ос-бавНых электрохимически или фотохимически, см. разд. 9.10.2.5). Использование эфиров угольной кислоты [17, 18] {например, (26, о2 = СбС15, R3 = Трет.-Bu)} с одной плохой и одной хорошей уходяшей группами имеет то преимущество, что образующийся карбамат (28) содержит ту группу, которая хуже отщепляется. Карбаматы (27) с хорошими уходящими группами (R2 = 4.NO2C6H4 или 2,4-(NO2)2C5H3) претерпевают быстрое элиминирование до изоцианатов и не годятся в качестве защитных групп. Развитием этого подхода является применение хлорформиатов (см. разд. 9.10.2.5); селективность реагентов может существенно изменяться при использовании диэфиров угольной кислоты [19]. Для введения в мягких условиях трет-бутоксикарбонильной защитной группы рекомендованы смешанные эфиры {26, R2 = = Ви-трет., R3 = N=C(CN) Ph} [20].
Сообщалось о катализе гидролиза эфиров угольной кислоты за счет участия соседней группы. Так, 2-гидрокси-5-нитрокарбо-нат (29) имеет два «плато» в области pH (0—3 и 7—9), где скорость реакции не зависит от pH (наряду с обычным катализируемым НО- гидролизом при pH >• 10). Наличие плато в обла-
сти высоких значений pH приписывают внутримолекулярному катализу атаки молекулы воды на карбонат с участием соседней ионизованной феноксигруппы (30). Присутствие группы — О-приводит к возрастанию скорости примерно в 50 раз [21].
9.10.2.3. Циклические эфиры угольной кислоты
Этиленкарбонат [31] взаимодействует с такими пуклеофила-а'1’ Как спирты, карбоновые кислоты, тиолы и ароматические (32\HbIr’ Давая соответствующие 2-гидроксиэтильные производные зде 'СХема (9)}. Однако в отличие от ациклических эфиров сь М0'Кет преобладать расщепление связи алкил — кислород,
541
приводящее к образованию (33) [22]. Так, при реакции с галогенидами щелочных металлов в водном этиленгликоле образуются исключительно 2-галогенэтанолы (33, Х = галоген) [23]. При низких температурах при использовании четвертичных галогенидов аммония можно провести 2-гидроксилирование. Сообщалось о применении этих реагентов для алкилирования соединений с активной метиленовой группой, например, при получении (34); предполагаемый! механизм реакции включает 2-галогеноспирт в качестве активной алкилирующей частицы [24].
О
нх
НО—СН2СН2—О—С—X (32)
(9)
Bu2Sn(OR)2 ---------->.
но—СН2СН2—х
(33)
О
II
RO—С—OR +
(Ю)
(31) (35)
В большинстве этих реакций происходит отщепление СО2 от (31), однако при реакции дибутилстаннилдиалкоксида образуется диалкилкарбонат (35) без сопутствующего декарбоксилирования [25]. Эта реакция служит альтернативным способом получения (35) из СО2 и включает получение (31) в результате взаимодействия СО2 с этиленоксидом.
542
Циклические карбонаты (36) используются для защиты ами-так как образующиеся циклические карбаматы (37) {схема устойчивы при всех условиях кроме восстановительной об-ботки [26]. Циклические карбонаты типа (36) вступают в реак-Р2® цИКлоирисоединения по Дильсу — Альдеру схема (12); гид-Ц”лиз дихлоркарбоната (38) приводит к дикетону (39) {схема ??2)}-
9.10.2.4. Ангидриды угольной кислоты
Смешанные ангидриды угольной и карбоновых кислот (40) могут образовываться при мягких условиях, и поэтому они нашли применение в качестве групп, активирующих карбоновые кислоты в реакциях конденсации. Такие ангидриды синтезируют [28] из карбоновой кислоты и хлорформпата в присутствии третичного амина {схема (13)} пли из хлорангидрида карбоновой кислоты и монокарбоната {схема (14)}; возможно использование карбоксилата таллия [29]. Ограниченное применение нашел распад диацилперокспдов (реакция инверсии карбоксила), так как при этом образуется ряд побочных продуктов {схема (15)}.
° з О О
Il r!n II II
R>CO2H+ R2OC—Cl -->- R1—С—О—C—OR2 + R3NH СГ (13)
(40)
о о О О
II II II II
R2O—С—О’ Na++ R1—С—С1 —► R1—С—О—С—OR2 + NaCl (14)
ОО ОО
II II A II II
R1—С—ОО— С— R2 ---> R1—С—О—С—OR2 (15)
Ангидриды (40) могут взаимодействовать с нуклеофилами как по карбонильным группам, так и по алкильной группе [R1]. Однако расщепление связи алкил-кислород происходит достаточно редко и ограничено ангидридами, в которых R1 может образо-
о
о о -> R‘-c-nr^
II II RfNH (41)
R1—С—О—С—OR2 ------
О
—> R2—О—С—NR32
(42)
О О О О
. II II RO" II II
Ar—С-О-С—OR ------► Ar—С—OR + О—С—OR
(43)
(44)
I.—> СО2 4* RO
(16)
(17)
543
вывать стабильный карбокатион (например, R1 = трет-Ви) рас. щепление не наблюдается при R1 = CH2Ph. Вторичные амины реагируют по обеим карбонильным группам, давая амиды (41) и карбаматы (42) {схема (16)}. Образование карбамата предпочтительно для стерически затрудненных аминов, в то время как если одна из групп R1 или R2 объемиста, реакция протекает по другому пути.
Смешанные ангидриды (43) можно также превратить в соответствующие эфиры карбоновых кислот (44) за счет реакции протекающей в присутствии каталитических количеств алкоксид-пона. Для реакции предположен ионно-цепной механизм, две первые стадии которого представлены на схеме (17) [28].
9.10.2.5. Галогенангидриды и азиды угольной кислоты
Твердо установлено, что сольволитическпе реакции алкилхлороформиатов (45, X = О) идут значительно медленнее, чем для ацил- (например, ароил)хлоридов, и представляется доказанным, что диссоциативный механизм здесь маловероятен. Это связывается с двумя факторами, (а) Исходный хлорид стабилизован за счет сопряжения {схема (18)}; эффект ослабляется, если алкильная группа R содержит электронооттягивающие заместители в соответствии с порядком реакционной способности в бимолекулярной реакции с метанолом {схема (20)}. (б) Ион ацилия (46) дестабилизируется за счет оттягивания электронов алкоксигруппой. Этому Противодействует мезомерный электронодонорный эффект [см. каноническую структуру (46)], и механизм Sjvl наблюдается только для хлороформиатов с сильными электронодонорными заместителями [30].
О ’О
-II *1
R—X—С—С1 <-> R—Х=С—С1
(45)
R—X—С=О R—Х=С=О
(40) 0 0 О
II II II
С1СН2СН2О—С—С1 > СН3О—С—С1 > СНзСН2О— С—С1
(18)
(19)
(20)
Имеется несколько примеров, демонстрирующих изменение механизма (от мономолекулярного до бимолекулярного) за счет из менения либо структуры субстрата, либо условий реакции И ’ 32]. Так, простейшие хлорформиаты в 60%-ном водном диоксане или в 70%-ном водном ацетоне гидролизуются по бимолекуляр ному механизму, при этом стадией, определяющей скорость Реа^ ции, является атака растворителя на хлорид. В этих условп фторформиаты гидролизуются быстрее, однако разрыв связи Уг 544
лерод-галоген не может предшествовать стадии, определяющей Ярость процесса [33]. В соответствии с этим выводом нуХ офилы кроме воды повышали скорость реакции арилхло^р и фторформиатов. Ацетат-ион катализирует полный гидролиз (47)-я качестве интермедиата образуется смешанный ангидпид (481 его растепление является стадией, определяющей ного гидролиза {схема (21)}.
9 ° о
и пол-
скорость
Н2О
(47) (48)
—> PhOH + СО2 + СН3СО2Н
О
II
R—О—С—О' RO" + со2
(49)
3
(21>
(22У
О
II
Н-С—ОН R-O-j-C=O
О
II
> НС—ов + СО2 (23}
Гидролиз хлорформиатов (или фторформиатов) (по любому механизму) приводит к алкил- или арилкарбонатам (49), которые, как известно, претерпевают быстрый распад до СОг и алкоксид или феноксид-иона {схема (22), см. также разд. 9.10.2.1}.
При сольволизе оптически активных алкилхлорформиатов [например, (45), Х=О, R=PhCHMe] в смешанных водно-органических растворителях обычно наблюдают более 90% сохранения конфигурации в спиртовом продукте. Однако отмечено более 90% обращения конфигурации при сольволизе в 98%-ной муравьиной кислоте [32, 34], что предполагает 5м2-замещение либо в исходном хлориде, либо в промежуточно образующемся карбоксилий-ионе {схема (23)}.
Образование карбоксилий-иона (46, X = О) может катализироваться ионами Ag4-. Ион (46) формально сходен с ионом диа-зония (RN0 и претерпевает отщепление СОг с образованием очень активного карбокатиона (аналогично отщеплению азота от ионов диазония). Типичная смесь продуктов, образующаяся при Реакции н-пропилхлорформиата с гексафторантимонатом серебра в хлорбензоле (буфером служит тетраметилмочевина) представлена на схеме (24). Получены данные [35], подтверждающие существование карбоксилий-иона (49а) и карбениевого иона (50) квк Дискретных интермедиатов в этой реакции. В частности, особенно интересно, что как и в случае «дедиазонирования», последовательность образования интермедиатов аналогична [36]. меются данные, свидетельствующие о возможности пспользова-Ия ЭТОго пути для получения неустойчивых (например, мости-°вых) карбениевых ионов [35].
18 Зак. 395 545
о
II AgSbFo
СН.СН2СН2О—С—Cl ---------->
PhC
.—> Гсн3сн2сн2о—с==о ——* сн3сн2снЛ сг ------------->
L —СО2 J PI1GI
(49а) (50)
СН2 С1
/ \ I
• > СН3СН=СН2 -f* Н2с-----СН2 4* СН3СНСН3 4* СН3(СН2)2СзН4С1 4-
(47,5%) (3,9%) (11,5%) (2,4%)
+ (СН3)2СНС6Н4С1 (24)
(5,6%)
При нагревании хлорформиаты претерпевают несколько превращений, но основным направлением является отщепление стабильной молекулы СОг и образование соответствующего алкилгалогенида. Для этих реакций предложены следующие механизмы: (а) реакция через ионную пару {схема (25)}; (б) синхронный процесс, в котором замещение и перегруппировка могут
происходить через пяти- или четырехчленное переходное состояние аналогично механизму {схемы (26) и (27)}; (в) бимолекулярное 5м2-замещение, наблюдаемое главным образом при
добавлении хлорид-иона; (г) цис-элимпнпрование, в результате которого могут образовываться непредельные продукты {схема (28)} [37].
О
RC1 4“ СО2
(25)
(51)
Н Н
R / \ °)
Н н ---->- С1СН2СН2Р + со2 (26)
ЧС1
-----RC1 + СО2 <27)
R- НС;-СН2
о> —>• rch=ch2 + НС1 + СО2 (28
о
Обычно наблюдаемое в случае хлорформиатов сохранение кон фигурации алкильной группы говорит в пользу циклического
546
,ляИзма типа (б) или механизма (а), включающего возникновение ионной пары (51), которая быстро распадается с образова-"1еМ подуктов до того, как может произойти диссоциация. Рас-«^а-арилэтилхлорформиатов (52) в толуоле характеризуется величиной р в уравнении Гаммета, равной -3,56, что указывает нЯ образование на а-углеродном атоме в переходном состоянии значительного заряда, аналогично тому, как это происходит при сольволизе (53) по механизму [38]. Это в значительной степени говорит в пользу механизма (а), хотя отсутствуют надежные доказательства того, что оба атома кислорода становятся эквивалентными благодаря возврату нона из интермедиата (51)
О
СНз—СН—О—С—С1
Аг (52)
СНз—CH—Cl
Аг
(53)
Третичные амины катализируют распад хлороформиатов. Поскольку алкилхлориды, образующиеся в этом случае, имеют обращенную конфигурацию [39], реакция, вероятно, включает вытеснение хлорид-иона за счет атаки амина на ацильный центр с последующим 5лг2-замещением хлорид-ионом [механизм (в)]. Эта реакция деалкилирования может быть использована как общий способ получения вторичных аминов из третичных аминов {схема (29)}. Искомый вторичный амин образуется в результате мягкого восстановительного расщепления карбамата (54) [40]. Фе-ннлхлорформиат также рекомендован в качестве подходящего реагента для расщепления третичных аминов [41].
о о
II R3N II + Д
С13ССН2О—С—С1 -----> С13ССН2О—С—NR3 СГ ----->
О
II Zn, НО Ас
—> С13ССН2О—С—NR2 --------R2NH (29)
° или МеОН
(54)
Амиды реагируют с хлорформиатамп с образованием имидатов (55). Однако если в хлорформиате имеются электронооття-тивающие группы, реакция протекает иначе и главными продуктами становятся нитрилы (56) {схема (30)} [42].
При замене кислорода на серу предпочтительным при нуклеофильной атаке становится мономолекулярное превращение в сравнении с бимолекулярным замещением. Так, хлортиоформиат ' ) и хлордитиоформиат (58) легче гидролизуются по меха НИзмУ SjvI, чем (45, Х = О). Однако граница между двумя меха-низмами нерезка, и в присутствии азпд-понов [43] п,1р”СрХ'° « Ухлеофильная атака, и (58) дает исключительно 5-замещеш ТиатРиазол (59).
18*
547
О
Ri_C-NH2
о
R2OC—Cl
0 1
II I
О—C—OR2
R‘-<L
^NH • HCI _ I r2=CC13
R2 = Et
/OEt
R1—% (30)
^NH
(55)
О
II R-S—C—Cl
R1—C=N (56)
S
II
R—S—C— Cl
R—S—-
(58)
N—N
C /n
S
(59)
(57)
Фторформиаты обычно получают из соответствующих хлорформиатов обменом галогена с неорганическими фторидами, такими, как фторид калия (при повышенной температуре), безводный фторид таллия или безводный фторид водорода [32, 44]. Пер-фторформиаты получают также при введении СО в перфторгипо-фториды [45]. Одна из трудностей при работе с фторформпатами обусловлена их большей чувствительностью к влажности в сравнении с хлорформиатами, хотя их термическая устойчивость выше. Помимо того, для получения фторформиатов могут быть использованы карбонилфторид COF2 или карбонплфторхлорид COC1F, что аналогично применению фосгена для получения хлорформиатов [46]. Иодформиаты и бромформпаты изучены слабо, хотя описано [47] получение бромформиатов при реакции спиртов (например, бензилового спирта) и карбонплбромида. Об их свойствах известно мало, однако они, вероятно, менее устойчивы (термический распад), чем соответствующие хлорформиаты. Цианоформиаты легко получать из хлороформиатов с применением в качестве катализатора фазового переноса {схема (31)} [48].
О
™ 1 „ СН2С12
RO—С—Cl + KCN ---------+
18-краун-6
О о
II R2NH2 I)
RiQ—C—Cl ------> R2NH—С—।
(60) (61)
Легкость реакции алкилхлорформиатов с аминогруппами, при водящей к карбаматам, которые в различных условиях могу тпп^Н^ИР°Вать амин> привела к широкому использованию этого м тилмл^п защиты аминогрупп при синтезе, в особенности при п интезе [схема (32)]. Среди наиболее широко приме
548
О
RO—C—CN
•OR1 —> R2NH2
{схема
(31)
(32)
„ашитных Г?УП" слеДУет]помянУть'- бензилоксикарбониль-мЫХ Н60 R‘ = PhCH2) [49—51], снимается каталитическим гид-нУ10 «нём без заметной рацемизации аминокислоты [52В- тоет-Р"Са,№р6онильНук> {трег-ВОС (грег-БОК), (60, R'1 CMe3) буТ Лтся в CF3CO2H [53] или под действием H2/Pd в жидком удзЛ1Яке) [541; 1-метилциклобутилоксикарбонильную (62) (более аММйчива по отношению к кислотам, чем трет-БОК) [55]; 9-флуо-У нлметоксикарбонильную (63) {дает устойчивый к кислотам Р бамат (61), который расщепляется в присутствии аммиака} [ДЕ. производное камфоры (64) {сходно по свойствам с трет-
но легче получается} [57]; о-нитробензилоксикарбониль-b ’/05) (используется в качестве защитной группы для амино-Н\-ппЫ в сахарах и может быть удалена фотохимическим путем Si)’ 2-(метилтио)этокспкарбонильную группу (66) {Mtc (МТК), ,етилирование или окисление образующегося карбамата (61, р = СН2СН2$Ме) приводит к производному, легко расщепляемому в мягких щелочных условиях} [59]; 2,2,2-трихлорэтокси-карбонильную {(67, Х = С1), используется для защиты НО- и МНг-трупп, образующиеся при этом карбаматы или карбонаты выдерживают обработку CF3COOH или H2/Pt, но гидролизуются в мягких условиях в присутствии Zn [60]}; 2,2,2-трибромэтоксикарбо-нильную {(67, Х==Вг), используется для защиты нуклеозидных
адамантилоксикар-к
иильную {(67, X-
НО-групп и снимается парой Zn/Cu} [61] и бонильную (удаляется кислотой, но устойчива
О — II ^О-С—С1 ^Ме
Нг/Pt) [62].
(62)
MeSCH2CH2OCOGl
(66)
XsCCH20C0G1
(67)
получают дмазоти-кции трет-бу-
СН2ОСОС1
(65)
Вл
р°вацИемИй грет-бутилоксикарбонилазид (69)
ТИлхлорфопм ВеТСТВующего гидразида (68), по реа ате пРямой ИЭТа (?!) с С0Лью амидиния (70) [63] нлп в резуль-реакции (71) с азид-ионом [64] {схема (33)}. Азид
549
(69) широко использовался также для введения трет-БОК-г пы, однако появилось сообщение [65], что во время его 'пРУП' гонки происходит детонация, поэтому для введения трет-Бок' группы рекомендовано использовать другие реагенты [20]
О
II
трет-ВиО-С—NHNH2 + HONO
О
трет -BuO-C-N3 (33)
(69)
9.10.2.6. Фосген
Фосген (дихлорид угольной кислоты) получают в промышленности присоединением хлора к оксиду углерода в присутствии активированного угля. Это ядовитый газ с характерным запахом, может оказывать и последействие на человека, подвергшегося его воздействию.
Наиболее характерной реакцией фосгена является нуклеофильное замещение хлорид-иона. Обычно реакцию можно вести ступенчато, так как образующийся при замещении одного хлора интермедиат обычно менее реакционноспособен, чем сам фосген {схема (34)}. Если реагент, участвующий в реакции с фосгеном, также бифункционален, то могут возникать различные пяти- и шестичленные гетероциклы {схема (35)} [66,67].
О о О
II II НХ II V (34)
С1—С—С1 + НХ —> X—С—С1 ---------> х—с—х ( 7
WO ДЩ- RNH-C-NHR
(72) (73)
/73) воз-
С первичными аминами фосген дает изоцианаты и '^орЫй можно через промежуточный карбамоилхлорид (72), к Jaer легко дегалогенизируется {схема (36)} [68]. Реакция пока
Б50
У второго порядка и, вероятно, включает нуклеофильное к,1НеТ ение хлорида амином. Отмечено образование нескольких за^еШцых продуктов, получающихся в основном в результате П° нейшей реакции изоцианата (73) с амином, и поэтому исполь-даЛЬ различные приемы для снижения реакционной способности 3^10да [67]. При реакции фосгена с избытком амина с хорошим аМ1.’оМ образуются симметрично дизамещенные мочевины (74). ВЫ Вторичные амины реагируют с фосгеном с образованием N N-дизамешенных карбамоилхлоридов (75), которые [схема (37] иее активны, чем фосген, и могут быть выделены с хорошими ^ходами. Третичные амины дают нестабильные катионные комплексы (76) или (77), которые при нагревании элиминируют алкил-хлорид (бимолекулярная атака хлорид-ионом) с образованием карбамоилхлоридов [схема (38)]. При реакции фосгена бытком третичного амина при повышенной температуре ственным выделенным продуктом г ~
с из-един-
является мочевина (78).
СОС12
R2NH ------>
О
II
R2N—с—С1
(75)
СОС12
R8N ----->
О
II r2n-c-nr2
(78)
О 1
+ II
R3N—С—C1J СГ
(76)
]
о
II
R2NC—Cl + RC1
Г ° 1
R3N + II + ---> LR3N—С—NR3J 2СГ
(77)
О
II
R2NC—Cl + RC1 + R3N
(37)
(38)
перво-
Фосген присоединяется по связи углерод-азот иминов; начально образующийся аддукт (79) отщепляет одну или две молекулы НС1 в зависимости от типа замещения исходного имина {схема (39)} [69]. Аддукты (79) являются важными синтетическими интермедиатами, так же как и аддукты, образующиеся при реакции фосгена с иминоэфирами (80), енаминами (81), имидокарбонатами (82) и нитрилами (83) {схемы (40) — (43) соответственно}.
В более жестких условиях фосген реагирует также с изоцианатами, с хлорцианом и с карбодиимидами [67].
Первая стадия реакции спиртов и фенолов с фосгеном, дающая хлорформиаты (84), аналогична реакции с вторичными аминами. лорформиаты (84) можно выделять, если избегать избытка спирта и добавлять спирт к фосгену, а не наоборот. Для ускорения Реакции с фенолами, которая в противном случае протекает мед-нн°> Добавляют третичный амин или другой акцептор кислоты.
551
coci2 /Rs
RjR2C—ch=n— r3 ----► r)r2c—ch—nz
I \20cl
(39)
(79)
-hci
|(R2=h) zRS RjC=CH—rZ \coci
—2HC1 (r2=r3=h)
H)
R|C=CH—N=C=O R’R2C-CH-N«C=o
OR2 OR2 о
' C0C12 I II
R1—C=NH ------> R>—C=N—c_Ci
(80)
R'CH=CR2-NR3
COCI2 R‘\ /NRf ----* —r'
CIOC/ \r2
R]O.
/C=NR2
R'CX
(81) О
COC12 II /R2
----> R*O—C—NZ
Voci
(82)
(42)
n, coci2
Ph—C=N -----Д
Cl О
Ph—C=N—C—Cl
гт (83)
чается сп°ЛЬЗовании ^избытка натриевой соли спирта легко
ся симметричный диалкилкарбонат (85, R1 = R2).
сул1 A°nZ. 1применяют также для превращения карбоновых и дуктов этой п^СЛ0Т В хл°РангиДРиды {схема (45)}; выходы про-примеп г реакции сравнимы с выходами при реакциях, на-вяжш» тионилхлоридом или с РС15. Другим синтетически гидратаппя°1^ессом’ включающим применение фосгена, является де-мешений амВДов до нитрилов (91) 170]. В случае N-моноза-Дии обпячлаМИДОВ 86’ R2 — Н) реакция останавливается на ста-одним L вания имВДОилгалогенида (90); эта реакция является единений *м^ШИХ спос°бов получения этого важного класса со с?вии nt.N'3aMeuxeHHbIe Формамиды (86, R' = R2=H) в присут-случае N №ти^Я дают с хорошим выходом изоцианиды (8S^' лена на ст;
агипуют хлориминия (89) [67]. Мочевины Р
Дины (93) и дГ Н°-и аналогнчным образом, давая ХЛОРФ0Рма„ыМ интерм(едЬтом Д°ФаН0ИЛХЛ°Рвдь’ <“ема (47^ ^ГЙто Рая может паппд ^Жит> вероятно, О-ацилизомочевина (92), я
л Ргаться внутримолекулярному хлорированию
полу-
R>OH+COCI2 —> R’O-C-CI R.O-C-OR2
<84> (85)
° О
II COCI2 II
R-C-OH -----> R-C-CI + Co2 + HCI
(44)
(45)
° D, “ OCOC1
II /R I
R1—C—N +COC12 > R1— Cx +
\R3 _ ^NR2R3 Cl"
(86) (87)
(46)
r'=R2 = h
R3—N=C <----------
(88)
Cl
R'—C=NR2R3 СГ
(89)
Cl
Ri—C=N—R3
(90) (91)
O-^N-ацильной миграции. В общем, более замещенные мочевины показывают тенденцию к образованию главным образом хлор-формамидинов [71].
О
II /ОСОС1
RNH— С—NHR + СОС12 —> RNH—С7 \nR
(92)
(47)
Cl
RNH—C=NHR Cl (93)
4-
О COCI
II I
RNH—C—N—R
(94)
Сильно напряженные малые циклы при взаимодействии с фосгеном раскрываются. Так, главным продуктом реакции с азиридином является бис (2-хлорэтил) мочевина (95). Если реакцию проводить в присутствии основания, то удается выделить также изоцианат (96) [схема (48)}. Аналогичным образом при раскрытии
553
эпоксидного кольца образуются 0-хлорэтилкарбонаты (97) и хлпп формиаты (98) {схема (49)}. р'
О
СОС12 II
:NH ---> C1CH2CH2NH—С—NHCH2CH2C1 + C1CH2CH2N=C=O (48)
(95) (96)
О О
О -----> С1СН2СН2ОСОСН2СН2С1 + С1СН2СН2ОС—С1 (49)
(97) (98)
Широко описано ацилирование ароматических субстратов с помощью фосгена. Используют катализатор (обычно безводный трихлорид алюминия), и в качестве нормального продукта образуется соответствующий бензофенон (100) {схема (50)}. Промежуточный ароилхлорид (99) может быть выделен только в случае затрудненных или особенно неактивных субстратов, а также при использовании большого избытка фосгена [72]. Симметрично дизамещенные алифатические кетоны (101) могут быть получены по реакции с фосгеном литийорганических соединений {схема (51)} [73].
о
(99) (100)
О
RLi + СОС12 —> R—С—R (Ю1)
(51)
Примеры реакций фосгена с бифункциональными реагентами, дающих циклические продукты, представлены на схемах (52) — (57). Гидроксамовые кислоты превращают в выделяемые хлор-формилпроизводные (102), которые циклизуются в (103) {схема (52)}; диолы (104) дают циклические карбонаты (105) {схема (53)}. «-Аминокислоты образуют оксазолин-2,5-дионы (106) (N-карбокспангидрпды, или ангидриды Лейкса) {схема (54)}, которые используются в пептидном синтезе, хотя водород в положении 4 является кислым и может происходить рацемизация. Первоначально реакция проходит по азоту (см. выше, реакции первичных аминов) и образуется карбамоилхлорпд, который далее при нагревании циклизуется [74]. 2,4-Диарнл-6-гпдрокси-сыл/-триазины (107) образуются из 2 моль арилампдина {схема (55)} через промежуточно образующуюся мочевину. При реакции с гидразидом (108) образуется 1,3,4-оксадиазол (109) {схема (56)}; N-бензоил-N-метнлгидразин превращается в сиднон (ПО), который при
554
еакции с фосгеном дает 5-фенил-2-хлор-1,3,4-оксади-
R\ хо
(751.
азол (/J,/ о °
11 СОС12 II
r_C-NHOH ------> R-C-NH0-C-C1 —> О.
о
(52)
О
(102) (ЮЗ)
СОС12
но—(СН21Л—он ------У
(104)
(53)
R—сн—со2н
nh2
(54)
(106)
(107)
СОС12
О
R—С—NH—NH2
N—NH
(109)
(108)
/Ме Сос12
Ph-C—N -------►
\nh2
Me
Ph-V О
(111)
(57)
Ph-ЧС^ О (110)
Карбонилфторхлорид COFCI получают Np3 pg]. Как
ванием трифторида мышьяка или сурьм , ф'тОр11Ды, поэтому, и ожидалось, хлориды легче замета ’ иг1Тппиды В-хлоркарбо-например, фторформиаты (112) пли фтор 110!ЦьЮ указанного
новых кислот (ИЗ) легко синтезируются с по*ошгю . чаЮТ Реагента {схема (58)}. Карбонилдифторид СОЬ так* .
из фосгена по реакции обмена с неорганическими фторидами при взаимодействии с HNF2 [77].
или
О
Cl—С—F
(58)
(113)
9.10.3. КАРБАМАТЫ (УРЕТАНЫ)
9.10.3.1. Методы получения карбаматов
Хотя сама карбаминовая кислота NH2CO2H неустойчива и быстро распадается в кислом растворе с образованием аммиака и СО2, она используется в качестве основы удобной системы названий этой группы соединений. Так, эфиры (замещение по кислороду) называют алкил- или арилкарбаматами, в то время как замещение по азоту показывают через N-префикс: например, PhNHCO2Me — это метил-М-фенилкарбамат. Термин уретан используется также для названия этилкарбамата NH2CO2Et, а иногда даже для обозначения всего класса сложных эфиров карбаминовой кислоты; это может приводить к путанице, и поэтому вряд ли целесообразно сохранение подобной номенклатуры. Термин «карбаниловая кислота» может быть использован для N-фенилкарбаминовой кислоты PhNHCO2H; этим указывают, что это моноанилид угольной кислоты.
О
II
R1—N=C=O + R2OH —> R‘NH—С—OR2 (59)
(114) (115)
О О
R2O—С—Cl + RlNH2 —> RlNH—С—OR2 (60)
(П6)
О О
R^N—С—Cl + R2OH —> R2N—C—OR2 <61)
(П7)
ООО о
Rl—С—ОН —> R1—C—CI —> Rl—C—N3 R’NH— C—OR2 (62)
r2oh
о °
u II
II В?з ш—C—OR2 /63)
R1/\+ NHa— C—OR2 -> RI/\/ '
556
Обшне методы синтеза карбаматов поедете™
/59) —(61). Взаимодействие изоцианатов (114> авлены схемами дПТ быстро и количественно (и используется ип°яС?ИрТаМ11 прохо' спиртов); реакция с фенолами медленнее но моЛараКТеристИки вэться третичными аминами. К ДруГ11м используем^ лкаталпзиР°-лучения относятся образование изоцианата in sttu™ ТОдам воровке Курниуса ацнлазлда {схема (62)} пеакпия пРИ перегрУппи-ацетатом свинца [78] или перегруппировка Лоссена дчя°ппС ТеТра' карбаматов, которые не чувствительны к присутствию* П0ЛуЧения M-Незамещенные карбаматы могут быть пЛ сутствию основании, пз циановой кислоты (114, однако Г П° СХеМе <59)
ровать далее со второй молекулой циановой кислоты ЖаГкГ3™* баматы получают при алкилировании [791 этих А *1ЛКар’ памп в присутствии трифторида бора {схема (63)1 рбаМатов алке’
Получение карбаматов пз хлорформиатов (116) и N N-дизаме шейных карбамоилхлорпдов (117) описано в разд 9 10 34 ГТп ускорения этих реакций используется 1 экв огнпВЯц„а ' Д треги-ный .мне, Ам„полиз сложных эфиров угоХй ™сло™(й1) также приводит к карбаматам (схема (В4П , ” '"мог“ 11 ls> к”" ° карТ
’ КТ СП0с0б“ст" -ОК'СГ-ОД°Уе(Тнапр™еТр8Лк5 =
= Ме). Метод неэффективен для получения карбаматов с’ лег™ отщепляемыми группами [например, (119, RI = Ph)l так КЯ1Г карбамат в этих случаях оказывается более активным ходныи карбонат. ‘мивным,
чем ис-
О О
II II
R’O—С—OR2 + R3NH2 —> R3NH—С—OR1 + R2OH (64)
(118) (119)
О
Т1(ЫОз)з II zz.e.
R—N=C + МеОН ---------> RNH—С—OMe (65)
(120)
Интересным общим способом получения карбаматов является прямая реакция нзоцианидов (120) со спиртами в присутствии нитрата таллия (III) {схема (65)}. Этот метод имеет преимущество перед методом, включающим предварительное окисление изоцианнда до изоцианата перед реакцией со спиртом [81].
9.10.3.2. Реакции алифатических и ароматических карбаматов
Наиболее типичной Для карбаматов является реакция с пук леофплами. Суммарно реакция {схема (66)} сводится к за“®^т_ ИИ1° алкокси- пли арнлоксигруппы, например аминами (о { У ся мочевины) пли спиртами (переэтерификация). Реакци
557
способность простых карбаматов (121, R2 = Aik) ближе к ами дам, чем к сложным эфирам, и для протекания этих реакций тпе" буются жесткие условия. Однако при наличии хорошо уходящей группы (121, R2=Ar) вступает в силу альтернативный механизм Для этого направления реакции (обозначаемого ElcB) существенен основной катализ; стадией, определяющей скорость процесса является отщепление арилоксигруппы от аниона карбамата [8^’ 83]. Образующийся промежуточный изоцианат может быть захвачен нуклеофилом, находящимся в растворе, и далее может превратиться в продукт замещения. Механизм Е\сВ очень чувствителен к природе уходящей группы (гамметовская константа
о о
II - II
R'NH—С—OR2 ч=±: R‘N—С—OR2
(121) (122)
О
X- II
------------> RNH—C—X <
р = 2,95 для заместителей в OR2), поэтому карбаматы, имеющие хорошо уходящую группу, в действительности очень быстро реагируют с нуклеофилами в присутствии оснований.
О
- II
R1—N—С—OR2
(122)
(67)
R4C1
R4 О
I II
R1—N—С—OR2
N-Монозамещенные и незамещенные карбаматы сами являются потенциальными нуклеофилами. Однако они (по аналогии с амидами) являются слабыми нуклеофилами и взаимодействуют в качестве таковых только тогда, когда (а) их превращают в анионы или (б) когда карбамат и активный центр расположены рядом (участие соседней группы). Алкилирование или ацилирование аниона (122) будет успешным только тогда, когда карбамат имеет плохо уходящую группу и не происходит быстрого элиминирования -OR2. Анионы (122) можно получать по реакции с натрием или с сильным основанием; р/G простых карбаматов (R1 = R2 = Aik) > 14, в то время как N-n-нитрофенилкарбаматы имеют рАа около 12; N-ацетилкарбаматы имеют рАа в области 10—13 [84, 86]. Алкил- и ацплгалогениды легко реагируют стабильными анионами карбаматов, давая соответствующие N-ал кил- и N-ацилпроизводные [85].
558
Карбаматы, не имеющие заместителей при азоте, взаимодей-Лт С карбонильными соединениями, но реакция медленна, за С ,-'/лечением наиболее активных альдегидов. Например, N-этоксп-'арбоиилимпн (124) образуется при реакции (123) с хлора-186]. Интересным применением аддукта (124) является потение аминокислот (125) при его реакции с реагентами Гринья-S с последующим гидролизом остатков СС13 и карбамата {схема (68)} И-
о
II CI3CCHO + NH2—С—OEt
(123)
О
NEt3 ||
---> C13CCH=N—С—OEt
(124)
|RMgBr (68)
I + NaOH; | ||
О2С—СН—NH3 —— CI3CCH—NH—С—OEt
riCI
(125)
При нагревании карбаматы претерпевают три основные превращения [88]: (а) элиминирование спирта и образование изоцианата (аналогично реакции в растворе); (б) расщепление, приводящее к амину, диоксиду углерода и алкену и (в) отщепление диоксида углерода {схема (69)}. Температура, при которой протекает реакция, может достигать 200 °C, однако арилкарбаматы (126, R2 = Ph) распадаются [по пути (а)] при «150 °C, а трет-бутилкарбаматы (126, R2 = трет-Ви) превращаются уже при 50°C. Аналогичный пиролитический распад характерен для карбонатов, сложных эфиров карбоновых кислот и др., однако карбаматы— здесь наиболее активная группа соединений. Структурные эффекты (например, значения гамметовскпх констант р) сходны в газовой фазе и в растворе, что указывает на относи-
тельную несущественность сольватации в циклическом переходном состоянии при элиминировании [89]. Чувствительность карбаматов к нагреванию используется для генерации при повышенной температуре изоцианатов, применяемых для образования поперечных сшивок [90].
R1—N=C=O + HOR2
Ri-NH-C-OR2 — R'-NH2 + СО2 + \=( (69)
(126) ' Щ3
(В)-> R1—NH—R2 + CO2
Даже когда рядом с карбаматной связью расположен силь-1и нуклеофил, путь через отщепленпе-прпсоединенпе может ока-аться предпочтительным {схема (70)} [91]. Однако если в качестве
559
нуклеофила выступает анион кислорода (например, 127 Y == = — С0'2 или —О', R’=H или Me), то протекает прямое замещение OR2 [92]. Известны другие примеры [например, (127 Y = CONH2)], для которых путь через отщепление-присоединение протекает только, если азот не имеет второго заместителя (127 Ri = Н) и присутствует хорошо уходящая группа [93]. ’ ’
R1 О
(127)
Карбаматная связь сама может выступать как внутренний нуклеофил по отношению к ацильному или алкильному центру. Это амбидентный нуклеофил; в нейтральном растворе реакция идет по кислороду, в то время как анион реагирует исключительно по азоту (аналогично поведению групп, соседних с амидной). Примеры приведены на схемах (71) и (72) [94, 95]. Поскольку интермедиат (128) легко гидролизуется с регенерацией амина, этот способ используется для снятия с аминов защитных групп [94]. N-(2-хлорэтил) карбаматы (129) циклизуются в нейтральных условиях в (130); в основных условиях образуются N-алкокси-
07--
•О 1г
Ph-NH— С-О-СМе2-СН2-гВг
Ph~NH
Me
О—>
> O=( + PhNH2 (71)
О /^Me
Me
^EtO , EtOH
О
II O-C—NHPh
(72)
560
чинны (131). Гидролиз последних служит удобным яобо«йЛПолучения азиридинов (132) {схема (73)} [96].
Особом полу о
С1СН2СН2—NH—С—OR
(129)
\ ~~
N
(130)
'NH
(132)
(73)
:n-c^°
(131)
9.10.3.3. N-Замещенные карбаматы
Нитрозирование карбаматов приводит к N-нитрозопроизвод-ны.м (133), которые обычно неустойчивы и подвергаются перегруппировке и фрагментации с образованием высокоактивных алкилирующих частиц {схема (74)} [97]. По этой причине N-нитро-зокарбаматы высоко канцерогенны; они нашли применение и в химиотерапии некоторых форм рака. Альтернативным (и лучшим) способом получения (133) является нитрование карбамата с последующим восстановлением цинком [98].
О ON О О
II I II д ||
R‘CH2-NH—С—OR2 —> R!CH2—N—С—OR2 --> R‘CH2—N=N—О-С—OR2 (133) ।
| I (74)
Г ° 1 г । 1
CH2-N=N—O' + CO|" LR’CHt N2 *0—c—or2] Lr!ch2 n2 0—c—or2] (134) + R2OH I I
RCHt
RCH2«
(135)
Кат
Дает ДназотТУем,ая основаниями реакция N-нитрозокарбаматов °бщИм мето ТЫ 134) и диазоалканы (135), и эта схема служит ЙИЗм вклЮчаДОм получения диазоалканов. Предполагаемый меха-ПУ (133) с поет атакУ НО- (или алкоксида) на карбонильную груп-следующим отщеплением диазотата [99].
661
N-Нитрокарбаматы (136) получают прямым нитрованием соответствующего карбамата (обычно в уксусном ангидриде в ка« честве растворителя). Восстановление (136) служит другим способом получения N-нитрозокарбаматов; обменная реакция с амином представляет удобный метод синтеза N-нитроаминов (137) {схема (75)}. Незамещенные нитрокарбаматы (136, R1 = Н) являются сильными кислотами и образуют соли. Алкилирование серебряных солей анионов проходит преимущественно по азоту, хотя известно и алкилирование по кислороду. N-Нитрокарбаматы устойчивее N-нитрозоаналогов, но распадаются сходным образом при нагревании {схема (76)}, давая N2O вместо N2 в качестве одного из продуктов [100].
O2N О О
I II r2nh2 II
R1—N—С—OR2 -------► R1—NHNO2 + R3NH—С—OR2 (75)
(136) (137)
О, + /О'
О Г ° 1 О
I II + II К
R1— N---С—OR2 —> LR1 N2O 'О—С—OR2 J —> R‘O—С—OR2 (76)
+ х\2О
N-Галогенпроизводные получают с помощью гипогалогенокис-лот или прямой обработкой карбамата водным раствором галогена [101] {схема (77)}. Главным продуктом, образующимся при хлорировании незамещенного карбамата (138), служит дихлорид (139) {схема (78)}. Последнее соединение является кислым; его натриевую соль можно ацетилировать или вводить в реакцию с формамидами с образованием аллофанатов. Фторирование карбаматов проводят аналогичным образом. Группа NF2 служит псевдогалогеном, поэтому фтораналог (139) реагирует аналогично галогенформиатам. Моногалогенпроизводные (140) присоединяются к олефинам, давая N-(р-галогеналкил)карбаматы {схема (79)}.
О С1 О
II *2, Н2О | II
R—NH—С—OR1 --------> R—N—C=OR> (77)
О
0 0 0 О OR
II Cl2 II II /C-OR
NH2—С—OR ---> C12N—С—OR + C1NH—С—OR + HN( (7»)
V—OR
(138) (139) (140)
О О
II r2ch=ch2 II , /7Cn
C1NH—C—OR1 --------> C1CH—CH2—NH—C—OR1
(140) R2 (141)
562
9.10.3.4. Карбамоилгалогениды и -азиды
м.Монозамещенные карбамоилгалогениды (142) неустойчивы пи нзгрсвзмын или в растворе диссоциируют даже в отсутст-" ° основания па изоцианат и галогенводород {схема (80)}. Об-ая реакция (присоединение галогенводорода к изоцианату) ,1а-я<ит единственным реальным путем синтеза таких галогенидов Заместители в Аг (р = 0,52) не оказывают существенного Сияния на равновесие между (142) и (143), что предполагает согласованное отщепление НС1 [102]. Образование изоцианата, как п ожидалось по аналогии с карбаматами, катализируется основаниями, а нуклеофилы замещают хлорид-ион по механизму отщепления присоединения.
О
(80)
(142) (143)
Если азот имеет два заместителя и механизм отщепления невозможен, то могут реализовываться два различных механизма: (а) присоединение-отщепление или синхронное замещение гало-генид-иона нуклеофилом и (б) мономолекулярное отщепление галогенида {схема (81)}. Дифенилкарбамоилхлорид (144, R = Ph) в водном растворе обычно гидролизуется по пути ионизации [путь (б)], хотя в присутствии сильного аминного нуклеофила [103] механизм изменяется и происходит атака амина на хлорид (144), определяющая скорость процесса. Продукты, образующиеся в этом случае — это соответствующие мочевины, а влияние заместителей (низкая чувствительность к структуре амина) предполагает ранее переходное состояние.
° г» °
R\ II X- R\ I
N—С—Cl ------—----> /N—C—Cl
r/ *’ RZ I
(144)
(б)|—СГ !-C1”
о R\ + X”, быстро R\ II
j;n—c=o ----------/N—c—x
R/ RZ
Дифенилкарбамоилхлорид (144, R = Ph), как известно, специфически инактивирует химотрипсин [104] н холинэстеразу 1*05]; недавно па лабораторных крысах понизано, что дпметил-КаРбамоилхлорпд является потенциальным канцерогеном при инфляции.
Соучастие соседних групп в карбамоилхлоридах (145) приво-Ит к количественному образованию соответствующих со-хлорал 563
килкарбаматов (146) {схема (82)} [106]. Легче всего реакция проходит при п —2 или 3, так как в этом случае возникают интермедиаты с пяти- и шестичленными циклами.
В О (82) | ||
—> cHch^-n-c-or1
(146)
М,1Ч-Дизамещенные карбамоилхлориды почти всегда получают по реакции вторичных аминов с фосгеном (см. разд. 9.10.2.6), Сообщалось также о внедрении СО в хлорамины в присутствии палладиевого катализатора {схема (83)}. Выходы карбамоилхлоридов (147) высоки, вплоть до 99% при R1 = R2 = Me; другие М,1Ч-диалкилхлорамины также вступают в реакцию, но выходы (147) ниже [107].
Pd или PdCI2 R‘\ II
V-Cl -f- CO ----------> ;N-C-C1 (83)
R2/ R2/
(147)
9.10.3.5. Карбаминовые кислоты
Первичные и вторичные амины реагируют в водном растворе с СО2 с образованием солей карбаминовых кислот (148). Соли карбаминовых кислот получают также при осторожном добавлении концентрированного раствора NaOH к раствору изоцианатов в диоксане или ацетонитриле. Хотя соли карбаминовых кислот могут храниться без заметного разложения, свободные кислоты (149) неизвестны, ибо при подкислении происходит быстрое отщепление СО2 и регенерация амина.
О
R\ II
'N— с—О' М' RZ
(148)
О
II
N—С—ОН
R-7
(149)
Механизм декарбоксилирования карбаматов изучен в некоторых деталях [108, 109]; в дальнейшем интерес к реакциям переноса СО2 перешел с модельных систем к выяснению природы связанного с ферментом О- или N-карбоксибпотина, который включен в перенос СО2 от карбоната к соответствующему субстрату, такому как ацетил-СоА, пируват или мочевина [1101-
Механизм, предложенный для декарбоксилирования, представ-леи на схеме (84). Активной частицей служит цвиттерион (15 1 564
н
H2NCO2H 4=±R2NCO2 + н+ 4=±R2N-C<- + СО2 (84)
(150) (151) (152) (153)
пбаминовой кислоты (150) которая, хотя и присутствует в не-КнаРчительной концентрации (при всех pH), является высоко ак-нвной (t'b ~ 10 ~ 10 с) и Распадается с образованием нейрального амина и диоксида углерода. Специфический и общий яслотный катализ наблюдался для превращения (151) в (152) даже при pH > 12. При высоких pH отмечено ингибирование декарбоксилирования; для карбаматов, полученных из слабооснов-нЫх аминов, разрыв связи N—СО2 [превращение (152) в (153)] является стадией, определяющей скорость реакции, в то время как для других карбаматов такой лимитирующей стадией служит перенос протона {превращение (151) в (152)} [108].
9.10.4. МОЧЕВИНА И ЕЕ ПРОИЗВОДНЫЕ
9.10.4.1. Методы получения мочевины и ее производных
Реакция изоцианатов или циановой кислоты с аминами служит общим методом получения моно-, ди- и тризамещенных мочевин. Тетразамещенные мочевины можно получать по реакции вторичных аминов с М,М-дизамещенными карбамоилгалогенидами {схема (85)}. Гидратация цианамида (154) в присутствии кислот также приводит к производным мочевины {схема (86)}, и, хотя этот метод не нашел общего применения в силу трудности получения цианамидов, он используется в промышленности для получения самой мочевины. Другой промышленный способ получения мочевины основан на взаимодействии аммиака с диоксидом углерода при нагревании под давлением [111]. В качестве интермедиата образуется карбамат аммония; его изомеризация в мочевину сыграла важную роль на ранных этапах развития
R4h/R2
R\ II zR8 )n—c—nz
R2' \r<
R\ 11
N—С—C!
R4/
R3NCO
R\ II
xN-c-nhr3
R2/
R—NH—C=N —► RNHCONH2
(85)
(86)
pi 1 Pb(OAc)4 R2NH2 1 мира
K -c-NH2 —' + ri_n=C=O —-----------------* R‘— NH—C—NHR
ГТ АД /Тч A
(87)
(155)
565
органической химии, являясь первым синтезом природного соединения из неорганических веществ.
Удобный метод превращения амидов в мочевины основан на реакции (в ДМФА) с тетраацетатом свинца {схема (87)}; в качестве интермедиата образуется изоцианат (155), который, если реакцию проводить в присутствии амина, нет нужды выделять [112]. Другие реакции, приводящие к изоцианатам (например, перегруппировки Лоссена и Шмидта), также могут быть изменены аналогичным образом для синтеза мочевины и ее производных Смесь амина, монооксида углерода и нитросоединения превращаются в мягких условиях с хорошим выходом в мочевины в присутствии металлоорганических реагентов. Типичные условия реакции включают катализ небольшим количеством соли Pd(II) при 90 °C {схема (88)} [133] или использование реактивов Гриньяра {схема (89)} [114].
О
Рс1(РРЬз)2Х2 II
ArNH2 + ArNO2 + ЗСО---------* ArNH-C—NHAr (88)
Et4N+ СГ
О
R2NO2 II
R'NHMgBr 4-Fe(CO)5 ------> R'NH-C—NHR2 (89)
H +
Механизм образования мочевин при реакции аминов с циановой кислотой достаточно подробно изучен [115]. Вначале образуется цвиттерионный интермедиат (156), который можно уловить за счет переноса протона либо на растворитель, либо от растворителя; при этом возникают ионы (157) пли (158) и в итоге мочевина (159). В случае сильно нуклеофильных аминов образование цвиттериона (156) является стадией, определяющей скорость процесса, в то время как в случае слабоосновных аминов такой лимитирующей стадией служит перенос протона. Поэтому катализ образования мочевины (например, с помощью обычно применяемых третичных аминов) эффективен^ только в реакциях с участием слабоосновных аминов. Активной частицей является нейтральная циановая кислота (рАа 3,29), а не цпанат-ион, и суммарное равновесие сильно сдвинуто в сторону мочевины ([NH2CONH2]/[NH3] [HCNO], А = 1-ю11 л-моль-1).
О /Р
RNH2 4- Н-К=00 —У rnh2—c'f- —У жй-сц-
VNH <+н+) NH
(156) (157)
(90)
° ?
rAh2-c—nh2 rnh-c-nh2
(158) (+H+) (159)
566
9.10.4.2. Реакции мочевины и ее пн» и ее производных
Мочевины участвуют в трех общих тИпя
сОцпация и диспропорционирование- (б) пЛТ реакций: (а) дис_ по карбонильному атому углерода („ли каобон»^ НуклеоФилами „упает как нуклеофил) i, (в) реакции с Ле™ г₽У""а вы-генгамп по азоту. ЭЛектР0ф>1Льиыии пеа-
Мочевины, имеющие по крайней мепе
соципруют при нагревании на амин (1611 Л У груПпУ NH, д„с-цианат (160) может также реагировать со Z“”. <160>- Изо-„отевины. давая биурет (163), „Л11 пиХТ“" молекулой завывать новую мочевину (162) {схема /оп1Ле1™ амина овра-левин значительна лишь выше точки >,Y ^исс°ВДация мо-биурета (163), который может бы™ XaZLT'™ ов₽взован„е интенсивно фиолетового окра шввания 7 Z™ “ ^вованню ствительным качественным тестом на мочев X тА ’ СЛуЖит чув-мочевины, которые не могут диссоциироватьS 1 етРззамещеНные более стабильны. Стерические затруднение, Деал{™фования, в значительной степени увеличить степень п В МОлекУле могут замещенные мочевины (164 R — н 1Ь Диссоциации. Так, три-днссоцпируют при 20—140°C ПЛИ больше) заметно
(165) н амин [116J {схема (92)}. тветствующий изоцианат
О
r3nh2
О r>nh-c-nhr3 (162) /Ме 0
R!N=C=O + R2NH2
(160) (161)
| | r’nhconhr2
(91)
О R2
О
II
R‘NH— С—N—С—NHR1
(163)
/Ме
(92)
100 °C, метил моче-лежит в дпапазо-
лмочевина (166) бу-
I, на самом деле она
667
х \Ме
*(164) 1’65)
М бь.е основания с рК. «коло О^Р^Р;^/-
Мочевины — слабые основам»» 2_q 15 при
мочевина имеет рКа 0,1 при 2 , пппМОчевин
вина имеет рКа 0,9 при 25 °C, рКа арилм не от—0,80 до—1,83). чт0 тетраметп.
Хотя можно было ожидать, мочевпна Ает более сильным основанием, че
является более слабым основанием, что обусловлено стерическим ингибированием резонансных структур, таких как (167) [117].
О
Мех || .Me C-NZ
Me' ^Me
(166)
„ O'
Мех + । .Me
>=C—NZ
(167)
ОН
R—NH— С—NH2
RNH-C—NH2
(168)
ОН
R—NH-C—NH3
(169)
R—NH-C—NH3
(170)
(93)
О
О
+ н
Место (локализация) протонирования в мочевинах было предметом обсуждения, однако сейчас общепринято, что основная присутствующая форма отвечает О-протонированию (168), а не N-протонпрованию (169). В концентрированных водных кислотах или в очень кислых средах отмечено дипротонирование {образование (170), схема (93)}; за счет комбинации стерических и электронных эффектов протонируется наименее замещенный азот [И8].
Алкилирование мочевины протекает по кислороду (по аналогии с реакциями амидов), и образуются О-алкилпзомочевины (171) {схема (94)}, которые представляют собой относительно сильные основания (р/(а около 6) и обычно выделяются в виде солей. N-Алкилирование наблюдается в тех случаях, когда алкилирующим агентом является карбениевый ион, например алкилирование мочевины трет-бутанолом в серной кислоте приводит к (172). N-Алкилирование мочевин можно проводить и непрямыми методами. Например, как это показано на схеме (95), восстановлением метилола (173) трпэтилсиланом в присутствии трифторуксусной кислоты. Метилол (173) получают реакцией мочевины с формальдегидом [119]; активной частицей при восстановлении служит, вероятно, ион иминия (174).
Ацилирование мочевин может также вначале протекать по кислороду, однако выделенные продукты являются N-ацилпроиз-
О OR
Il Ry I
NH2-C—NH2 -----> NH2—C=NH-HY
(171) (94)
О
Me3C+ II
-----► Me3CNH—C—NH,
(172)
668
о о
phNH—С—NH2 + СН2О PhNH—С—NHCH2OH SIL1302”
(173)
ОН 0
II I Et3SIH II
-—> PhNH C N=CH2 ——-> PhNH—C—NH CH3 (174)
(95).
водными (I77) {схема (96)}, что аналогично образованию имидов из амидов. Интермедиат, аналогичный (176), был действительно выделен при ацилировании тиомочевпны и показано, что S^N-миграция ацильной группы происходит внутримолекулярно и чт0 в случае тризамещенных тиомочевин она ограничена медвяной изомеризацией вокруг связи C=N в S-ацильном интермедиате [120]. Тот факт, что N-метилмочевина (175, R1 = Me, R2 = = Н) превращается в (177, R1 — Me, R2 = H), согласуется с образованием О-ацильного интермедиата, хотя вторичный азот должен быть более нуклеофильным при прямой атаке на ацилирующий агент.
R1—NH—С—NH—R2 —С°С1>
(175)
zr3
о=с
R-N=C-NH-R2
О R1 О
, „з II I II
—>R-C-N-C-NH4R2
(96)
(176) J (177)
Альдегиды и кетоны реагируют с мочевинами с образованием обычных аддуктов, каких можно ожидать по аналогии с другими аминами. Однако реакцию часто трудно остановить на начальной стадии, и, поскольку мочевина имеет не один нуклеофильный центр (а более), интермедиаты или продукты могут далее реагировать с мочевиной [121]. Формальдегид сначала превращается в выделяемый карбиноламин (178), который после дегидратации и взаимодействия со следующей молекулой мочевины дает бисад-дукт (180), или при реакции со следующей молекулой формальдегида образуется дикарбиноламин (179) {схема (97)}. В жестких условиях аддукты (179) и (180) подвергаются дальнейшей конденсации, образуя поперечно-сшитый полимерный материал промышленно важные мочевнно-формальдегидиые смолы.
Ацетон дает с мочевиной аддукт «триацетондимочевину», структура которого (181) до недавнего времени оставалась противоречивой [122].
Изучение механизма образования пиримидонов в кислом растворе из мочевины и 1,3-дикетонов показало, что мочевина реагн-РУет с протонированным дикетоном (182), превращаясь в нециклический интермедиат (183), который подвергается быстрой циклизации с образованием (184) {схема (98)} [123].
669'
о О г о
'll II II I
CH>O + NH2CNH2 hoch2nh—с—nh2 Lh2C=N—с—nh2J
(178)
' -
0 ч н °
° г ? ? 1
\ X/NH hoch2nhcnhch2oh Lnh2cnhch2nhcnh2J
/\ А
Me Me Me Me (179) (180)
(181)
(182) (183) (184)
Восстановление мочевин борогидридом натрия в пиридине приводит к формамидинам [124]. В качестве восстанавливающего агента можно использовать также алюмогидрид лития, но только для тризамещенных мочевин.
Сама мочевина под действием азотистой кислоты распадается на азот и диоксид углерода {схема (99)}. Поскольку в этом случае образуются неактивные газообразные продукты, данная реакция используется для разрушения избытка азотистой кислоты при нитрозировании или диазотировании. Аналогичная реакция протекает с гипохлоритом как с окисляющим агентом в отсутствие щелочи (см. также разд. 9.10.3.3).
О
NH2—с—NH2 + 2HNO2 —> 2N2 +СО2+ЗН2О (99)
Тризамещенные мочевины взаимодействуют с фосгеном или с пентахлоридом фосфора с образованием хлорформамидинов (185). Эти вещества являются высокоактивными соединениями,
э
II r2n-c-nhr
о r2n—c-nr2
f1 Г 1+
> r2n—c=nr -------->» Ir2N^C'sNR] (too)
(res) (tee)
r2n-=c=^nr2 СГ
(101)
670
хЛОриД-нон в которых в диссоциативных реакциях, включающих 2ь> (I86)’ леГКО замещается ^лым рядом нуклеофилов. Тетра решенные мочевины также реагируют с хлорирующими агентами, давая соответствующие соли хлорформамидиния (187) {схема (Ю1)}-
9.10.4.3. Гидролиз мочевины и ее производных
в водном растворе мочевины гидролизуются очень медленнообычно гидролиз протекает с измеримой скоростью лишь при
100 °C. Значительная часть информации о механизме гидролиза получена при изучении обратной реакции (образование мочевин) и при использовании специальных субстратов [таких, как (192)]. Выявлены специфические кислотный и основной катализ, однако наиболее важным направлением гидролиза мочевины является независимая от pH реакция, которая преобладает во всем диапазоне pH.
Первоначально Шоу и Уокером был предложен механизм, включающий определяющий скорость реакции внутримолекулярный перенос протона от одного азота к другому и приводящий к первоначальному образованию изоцианата (188) {схема (102)} [125]. Однако подробное исследование [115] обратной реакции показало, что стадия переноса протона не согласуется с разрывом связи С—N и что активной частицей служит цвиттерион (191) {схема (103)}.
О
Н
I /Л
H--N—Н_
Н—N=C=O + NH3 (102).
(188)
н2о
н+
NH3 + СО2 -<--- NH2CO2
Цвиттерион (191) может не находиться
NCO' NH{
в равновесии с мочевиной. (190) при всех pH, поэтому можно наблюдать общий кислотный катализ [протонирование аниона (189)] и общий основной катализ [отщепление протона от (192)]. Цвиттерион (191) быстро превращается (к = 3-104 с-1 при 25 °C), давая изоцианат, так что медленный гидролиз мочевин в значительной степени приписывают низкой равновесной концентрации цвиттериона (Аг^ = 10-’4) в растворе. Мочевина (193) легко гидролизуется в нейтральном растворе и относительно неактивна в кислоте и в осно вании. Благодаря большей основности имидазольнои гр\ ппы в (193), концентрация цвиттериона (194) выше, чем в с^яае [.„.1’ что объясняет высокую реакционную способность (1- ) I I-Однако и (194) быстро расщепляется, поэтому стадиен, опре е ляющей скорость превращения (193) в продукты, в Д
571
pH 4—10 является перенос протона при превращении НЭЪ (194). В присутствии достаточных концентраций кислот и ос В ваний} соединения (193) и (194) находятся в равновесии {схема
О О
II (-н+) - ||
RNH—С—NH2 RN—С—NH2
(190) (189)
Гидролиз мочевины катализируется гидроксид-ионом, однако суммарная скорость реакции достаточно низка (кНабл = 1 X Ю-6 в 1,0 М НО- при 60°C). Продуктами реакции являются аммиак и цианат-ион и предполагаемый механизм [127] включает моно-молекулярную реакцию уреидо-аниона, возможно сопровождающуюся общим кислотным катализом с участием уходящего амидного аниона.
Гидролиз мочевины в мягких условиях катализируется ферментом уреазой; ферментативный гидролиз применяют для аналитического определения концентрации мочевины. Продуктами гидролиза в нейтральных условиях в зависимости от применяемого буфера служат бикарбонат аммония или карбамат аммония [128].
Циклические ацилмочевины, урацилы (195), гидролизуются со скоростью, которая при низких концентрациях гпдроксид-иона пропорциональна [НО-]2, а при высоких значениях pH изменяется до первого порядка (195, R — Me) или нулевого порядка (R == = Н), т. е. пропорциональна [НО-] {схема (105)} [129, 130]. Отметим, что первоначальная атака гидроксида направлена на «амидную», а не на «мочевинную» карбонильную группу, и поэтому механизм гидролиза аналогичен предполагаемому механизму гидролиза амидов. При R = Н неактивный анион (197) образуется при высоком pH (рД'а = 11,66).
Гидролиз, катализируемый кислотой, для мочевин отн0^” тельно несущественен, например фенилмочевины гидролизую с в разбавленной кислоте лишь ж в 10 раз быстрее, чем в не * ральном растворе, и обычно скорость гидролиза в концентрир
572
ванных кислотах падает. Предложен механизм (включающий Н2О в качестве общего основания) для отщепления протона от N-npo-тонированной частицы (198); главной составной частью является О-протонированная мочевина. Падение скорости гидролиза в концентрированных кислотах связано с уменьшением активности воды в этих условиях [131, 132].
9.10.4.4. Изомочевины
0-Алкилизомочевины (199) получают (а) прямым алкилированием мочевин {например диметилсульфатом, схема (106)}; (б) по реакции хлорформамидинов {схема (107)} или карбодиимидов {схема (108)} со спиртами или фенолами; (в) по реакции Цианамидов со спиртами в присутствии кислот {схема (109)} или (г) взаимодействием аминов с цианатами {схема (ПО)}.
OR3
N—C=NR4
(199)
О OR
NH2—С—NH2 + RX —► NH2—C=NH-HX (106)
Cl OR2
R2N—C=NR‘ + R2OH —► R'N—C₽=NR' • HCI (Ю7)
573
OR2
R1—N=C==N—R1 + R2OH —> Ri-NH-C=N-Ri Ппяч \lUoJ
OR h+ I
NH2—CssN + ROH --->- NH2—C=NH-HX (1o9)
OAr
ArO—C=N+RNH2 —> R—NH—C=NH (ц0)
Изомочевины являются умеренно сильными основаниями [например, (199, R1 = R2 = R4 = Н, R3 = Me) имеет pJ<a 9-80]; обычно их выделяют в виде солей. При осторожной нейтрализации можно получить свободные основания, однако изомочевины с хорошо уходящими группами (RO-) могут претерпевать в основном растворе легкое элиминирование. Свободная изомочевина— хороший нуклеофил (как и ожидалось, исходя из ее основности) и легко дает N-ацилпроизводные {схема (111)}. Протонированная изомочевина может подвергаться нуклеофильному замещению (уходящей группой служит нейтральная мочевина), например гидрохлориды (200) при нагревании превращаются в ал-килхлориды (201) {схема (112)}.
OR1 1 .Г nh2— c=nh сг О Аг 1 в ph—NH— C=NH вь R2 OR1 О ХС=О >- NH,—C=N—С—R2 11И) 4-2 Х-Х О 11 < > JT RCI +NH2C-NH2 (112) NH/+'NH2 (201) (200) OAr => ph—N— C—NH ph N~C NH ,+ —ArO 1 (202) (ПЗ) R_NH-/H ^PbNH-C-N \NHPh (204) (203) phN-~CzsN (205)
574
реакция изомочевин с аминами приводит к гуанидинам, потому простые О-алкил- и О-ацилизомочевины служат удобными ^г'н”н“”(203) ИаПр“еР’ "Р" образован,™
^реакция проходит через первоначальное образование карбо-„дама (202) ПУ™ £'сВ‘’™Т|>0ВаШ1я; карбод.н.мнд ,1алее „гто»«Р||3Уется в ооанамид (204) — активное соединение, „ере-юсяшее остаток амидинов. Однако в основном растворе (204) J „активируется, превращаясь в неактивный анион (205). Обмен эрилокснгруппы в О-арплнзомочевине на алкоксигруппу протекает при нагревании в присутствии алкоксид-иона {схема (114)}; реакция, вероятно, протекает по механизму, включающему процесс, сходный с отщеплением-присоединением.
ОАг OR3
I R3O' |
R1—NH—C=N—R2 ------> R1—NH— C=N—R2
(114)
9.10.4.5. N-Замещенные мочевины
N-Нитрозомочевины (206) получают либо прямой реакцией — алкилмочевин с нитрозирующими агентами (например, с азотистой кислотой), либо мягким восстановлением N-нитромочевин (см. ниже). При обработке основаниями N-нитрозомочевины превращаются в диазоалканы (207) {схема (115)} по реакции, аналогичной для N-нитрозокарбаматов (см. разд. 9.10.3.3). Для этой реакции предложено несколько механизмов, включающих атаку гидроксида на амидный карбонил или на нитрозогруппу или прямое отщепление типа EIcB от аниона, генерируемого из (206) [36]. Восстановление N-нитрозомочевины (206) дает семикарбазид (208) [134].
О O==N О
R-NH—С—NH2 —> R—N—С—NH2 Н°-^ R>=N2 + N2 + СО2 + NH3 (115)
(206) (207)
। (R> = R - H)
достигается обработкой мочевины итромочевина легко диссоциирует
ничного продукта, ннтрамида (209), который распадается, давая
H2N О I II R—N—C—NH2 (208)
Нитрование мочевины легко нитратом в серной кислоте. Н...г — в растворе с образованием циановой кислоты (210) и другого п р ннтрамида (209), который распадается, давая °ксид азота (N2O) и воду {схема (116)}. Восстановление нитро-м°чевиц, подобно нитрозомочевпнам, приводит к семикар [135].
к
575
о о
II H2SO4 ||
NH2—С—NH2 • HNO3 -------► O2NNH—С—NH2 O2NNH2 4- HCNO (116)
(209) (210)
Реакция мочевины с гипохлоритом в щелочном растворе приводит к гидразину, и это является промышленным способом получения гидразина. Предложено два механизма реакции: (а) включающий отщепление по Гофману [через (212)] и (б) прямое нуклеофильное замещение одного азота другим с образованием интермедиата (213).
Промежуточная N-хлормочевина (211) может быть выделена при использовании трет-бутилгипохлорита в присутствии хлорида кальция; в присутствии кислоты мочевина регенерируется. Обработка несимметричных мочевин (214) трет-бутоксидом калия — трет-бутилгипохлоритом дает азосоединения, вероятно по механизму, сходному с представленным на схеме (118) [136].
О 0 0
II Roci II II
NH2— С—NH2 пNH2—С—NHC1 —> NH2—с—N—С1 (117)
NaOH
(211) (212)
I I
|н2о. н+ [NH2NHCO2H]
O=C=N—NH2
| НЮ
-> nh2nh2
о
Ri—NH—C—NH—R2 —► R’—N=N—R2
(214)
О
(215)
COCI (119)
-HC1>- —N—NH—R2
(216)
О
II H2° C мм
F2N—c—NH2 ------f2NH
co;
ri__N—NH—R2
(217)
(120)
(218)
2HF 4- N2 + CO
576
лзводное мочевины (214, R1 = R2-rp<?r-Bu) можно М-Хлорпр0'ка сильным основанием дает диалкилдиазири-,деп1’Ть’е°бггоследнее соединение при реакции с НС1 дает хлор-Б„г7он (215);лпазинкарбоновой кислоты (216) и другие кислоты, „гиДРиД Г оаин веооятно через декарбоксилирование промежу-3 >е ГПДа мата (217) {схема (119)}.
Очного каР°янИе мочевин приводит к М,М-дифтормочевинам, ко-т фторнр°в “ сильными окисляющими агентами, дающими при т0Рые яВЛЯ^кторамин. При нагревании (218) распадаются до ДР°Л1’3е «хФпроХ^ {схема (120)} {1371. Тетрафтормочевину газообразных и N^-дифторкарбамоилфторида с фторидом
получа1<)т п0 Р
«алия. ---------------
и
15.
10.
17.
18.
19.
20.
21.
22.
23.
24.
25.
27 р У' o,fscnan ana г. о. uwziec, и. лшч. _____________________
оо' К Еи51‘8<. К Benson, and N. Duty, J. Org. Chem~ 1967, 32, 851.
Res., 1969, 2, 296.
nd A. McKillop, J. Amer. Chem. Soc., 1968. 90.
M. N. Paddon-Row, and K. Preston, Canad. J. Chem.,
3] £ '”• чо. ozz.
r.- «(• Crunden and R. F. Hudson, J. Chem. Soc., 1961, 3748.
The Chemistry of Acyl Halides», ed. S. Patai, Wiley Inter-
ЛИТЕРАТУРА
j g. Gattow and U. Gerwarth, Angew. Chem. Internat. Edn., 1965, 4, 149.
o'у Lepore, G. C. Lepore, P. Ganis, G. Germain, and M. Goodman’ J Or? Chem., 1976. 41, 2134. ’ ’
3 p, Ganis, G. Avitabile, S. Migdal, and M. Goodman, J. Amer. Chem. Soc 1971, 93, 3328.
4 !. S. Byrne, P. F. Jackson, and K. J. Morgan, J. C. S. Perkin II, 1976, 1800
5. ' p. A. S. Smith, «Open-chain Nitrogen Compounds», Benjamin, New York, 1965, vol. 1, p- 259.
6. R. Howe, in «Rodd’s Chemistry of Carbon Compounds», ed. M. F. Ansell, El-sevier, Amsterdam, 1965, vol. Ic, chapter 11.
7. С. K. Sauers, W. P. Jencks, and S. Groh, J. Amer. Chem. Soc., 1975, 97, 5546.
8. J. Katzhendler, L. A. Poles, and S. Sarel, J. Chem. Soc. (B), 1971, 1847.
9. L. Crombie, P. Hemesley, and G. Pattenden, Tetrahedron Letters, 1968, 3021.
10. B. J. Whitlock and H. W. Whitlock, J. Org. Chem., 1974, 39, 3144.
11. H. A. Staab and A. Mannschreck, Chem. Ber., 1962, 95, 1284.
12. K. Kondo, N. Sonoda, and S. Tsutsumi, Tetrahedron Letters, 1971, 4885.
13. K. Fukui, S. Yoneda, H. Takayatna, and H. Kitano, Kogyo Kagaku Zasshi, 1960, 63, 2146 (Chem. Abs., 1963. 58, 2366).
K. R. Fountain and M. Pierschbacher, J. Org. Chem., 1976, 41, 2039.
T. H. Fife and D. M. McMahon, J. Org. Chem., 1970, 35, 3699.
R. Glatihard and M. Matter, Helv. Chim. Acta, 1963, 46, 795.
M. Fujino and C. Hatanaka, Chem. Pharm. Bull. (Japan), 1967, 15, 2015.
IV. Broadbent, J. S. Morley, and В. E. Stone, J. Chem. Soc. (C), 1967, 2632.
P. Sieber and B. Iselin, Helv. Chim. Acta, 1968, 51, 614.
M. Itoh, D. Hagiwara, and T. Kamiya, Tetrahedron Letters, 1975, 4393.
T. H. Fife and J. E. C. Hutchins, j. Amer. Chem. Soc., 1972, 94, 2837.
IV. IF, Carlson and L. H. Cretcher, J. Amer. Chem. Soc., 1947, 69, 1952.
E. D. Bergmann and I. Shahak, J. Chem. Soc. (C), 1966, 899.
К Ishido, H. Tsutsumi, and S. Inaba, J. C. S. Perkin I, 1977, 521.
H. D. Scharf and W. Kusiers, Chem. Ber., 1972, 105, 564.
26. I. C. Sheehan and F. S. Guziec, J. Amer. Chem. Soc.. 1972, 94, 6561. - " --- ina-i io
on Earbell, Accounts Chem. Res., 1969, 2, 296. ....
” ^422 Baylor, 6- ^'cLay, a,-~ -*- ”
30- A Queen, T. A. Nour,
„ >970, 48, 522
D. N, Kevill, in
science, New York, 1972, chapter 12.
34 Q»leen and T- A- Nour’ J- C- S Perkin H’ l9J,6’ 93d 64 645
M. Matzner, R. P. Kurkjy, and R, J. Cotter, Chem. Rev., 1964, 64, 645.
19 Зак. ЗЭ5 $77
35. P. Beak, Accounts Chem. Res., 1976, 9, 230.
36. А. F. Hegarty, in «Chemistry of the Diazonium and Diazo Groups» ed 3 Do tai, Wiley Intcrscience, New York, 1978, chapter 12. ’
37. P. Il7- Clinch and H. R. Hudson, Chem. Comm., 1968, 925.
38. L. P. Hammett, «Physical Organic Chemistry», 2nd’edn., McGraw-Hill, New York, 1970, p. 355 [Л. Гаммет. Основы физической органической химии — Пер. с англ./Под ред. Л. С. Эфроса. М., Мир, 1972].
39. D. N. Kevill, G. Н. Johnson, and W. A. Neubert, Tetrahedron Letters 1966 3727. ’ ’
40. T. A. Montzka, J. D. Matiskella, and R. A. Partyka, Tetrahedron Letters 1974 1325.
41. J. D. Hobson and J. G. McCluskey, J. Chem. Soc. (C), 1967, 2015.
42. F. H. Suydam, IT. E. Greth, and N. R. Langerman, J. Org. Chem 1969 34 292. ’ ’ ’
43. A. Queen, D. M. McKinnon, and A. W. Bell, Canad. J. Chem., 1976, 54, 1906.
44. C. W. Tullock and D. D. Coffman, J. Org. Chem., 1960, 25, 2016.
45. P. J. Aytnonino, Chem. Comm., 1965, 241.
46. H. J. Emeleus and J. F. Wood, J. Chem. Soc., 1948, 2183.
47. A. W. Rosenmund and H. Dooring, Arch. Pharm., 1928, 266, 277.
48. M. E. Childs and W. P. Weber, J. Org. Chem., 1976, 41, 3486.
49. U. Ragnarsson, S. M. Karlsson, В. E. Sandberg, and L. E. Larsson, Org. Synth., 1973, 53, 25.
50. E. Schnabel, H. Herzog, P. Hoffmann, E. Klauke, and I. Ugi, Angew. Chem. Internat. Edn., 1968, 7, 380.
51. G. Bram, Tetrahedron Letters, 1973, 469.
52. A. E. Jackson and R. A. W. Johnstone, Synthesis, 1976, 685.
53. H. Yajitna, N. Fujii, H. Ogawa, and H. Kawatani, J. C. S. Chem. Comm., 1974, 107.
54. K. Kuromizu and J. Meienhofer, J. Amer. Chem. Soc., 1974, 96, 4978.
55. S. F. Brady, R. Hirschmann, and D. F. Veber, J. Org. Chem., 1977, 42, 143.
56. L. A. Carpino and G. Y. Han, J. Org. Chem., 1972, 37, 3404.
57. M. Fujino, S. Shinagawa, O. Nishimura, and T. Fukuda, Chem. Pharm. Bull. (Japan), 1972, 20, 1017.
58. B. Amit, U. Zehavi, and A. Patchornik, J. Org. Chem., 1974, 39, 192.
59. H. Kunz, Chem. Ber., 1976, 109, 3693.
60. F. R. Pfeiffer S. R. Cohen, and J. A. Weisbach, J. Org. Chem., 1969, 34, 2795.
61. A. F. Cook, J. Org. Chem., 1968, 33, 3589.
62. W. L. Hass, E. V. Krumkalns, and K. Gerzon, J. Amer. Chem. Soc., 1966, 88, 1988.
63. K- Sakai and J. P. Anselme, J. Org. Chem., 1971, 36, 2387.
64. H. Yajima and H. Kawatani, Chem. Pharm. Bull. (Japan), 1968, 16, 182.
65. P. Feyen, Angew. Chem., 1977, 89, 119.
66. E. Kiihle, Angew. Chem. Internat. Edn., 1973, 12, 630.
67. H. Babad and A. G. Zeiler, Chem. Rev., 1973, 73, 75.
68. S. Ozaki, Chem. Rev., 1972, 72, 457. .
69. L. I. Samarai, O. W. Wischnewskij, and G. I. Derkatsch, Chem. Ber., 19b9, -> 2972.
70. A. J. Speziale and L. R. Smith, J. Org. Chem., 1963, 28, 1805.
71. H. Ulrich, J. N. Tilley, and A. A. R. Sayigh, J. Org. Chem., 1964, 29, /4Ui-
72. «Friedel Crafts and Related Reactions», ed. G. A. Olah, Wiley Interscie New York, 1963—65, vols. 1—3.
73. Y. Sawa, M. Ryang, and S. Tsutsumi, Chem. Abs., 1966, 65, 10601.
74. A. J. Speziale and L. P. Smith, J. Org. Chem., 1963, 28, 1805.
75. C. Ainsworth, Canad. J. Chem., 1965, 43, 1607.
76. К. O. Christie and A. E. Pavlath, J. Org. Chem., 1965, 30, 1639, 3170.
77. O. Glemser and U. Biermann, Chem. Ber., 1967, 100, 2484. 2g
78 A. Brandstrom, B. Lamm, and I. Paltnertz, Ada Chem. Scand. (t>), iv/ , 699.
79. G. Muller and R. Merten, Chem. Ber., 1965, 98, 1097. .q7q
80. W. M, McLamore, S. Y. P'an and A. Bavley, J. Org. Chem., 1955, 20, 13/у-
578
93.
К. Kirley, J. W. Spiewak, and
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
Tetrahedron Letters, 1972, 1771.
el A. K'uZ’nrtu and L. N. Frost, J. C. S. Chem. Comm., 1972, 500.
fp- J C S. Perkin II, 1972, 808.
J A. Wdha> ’ j j p Calmon, Bull. Soc. chim. France, 1976, 797.
»« I»» 23-278S'
85. F- W lh В Tucker, and A. A. R. Sayigh, J. Org. Chem., 1968, 33, 2887.
86- «Ma У- ^ki, and Y. Omote, J. C. S. Perkin I, 1975, 2511.
87- C- and G C. Wright, J. Amer. Chem. Soc., 1959, 81, 2138.
88. £ "y L nfld M. P. Thorne, J. C. S. Perkin II, 1976, 799.
89. w/ I evine and J. Fech, J. Org. Chem., 1972, 37, 2455.
9°' 1 F Hegarty and L. N. Frost, J. C. S. Perkin II, 1973, 1719.
91- л F Hegarty, L. N. Frost, and D. Cretnin, J. C. S. Perkin II, 1974, 1249.
92- 1 F Hegarty, L. N. Frost, and J. H. Coy, J. Org. Chem., 1974, 39, 1089.
i A Carpino K. N. Parameswaran, R. K. Kirley, J. W. Spiewak, and 94' f Schmitz, J. Org. Chem., 1970, 35, 3291.
p I Scott and D. F. Fenton, Tetrahedron Letters, 1970, 681.
с г A Foglia and D. Swern, J. Org. Chem., 1969, 34, 1680.
- i I G. Cadogan, Accounts Chem. Res., 1971, 4, 186.
no' / Thiele and F. Dent, Annalen, 1898, 302, 245.
qq A F. Hegarty, in «Chemistry of Diazo and Diazonium Groups», ed. S. Patai, 9 Wiley Interscience, New York, 1978, p. 568.
inn E H. White and K. W. Field, J. Amer. Chem. Soc., 1975, 97, 2148.
mi P. Chabrier, Ann. Chim. (France), 1942, 17, 354.
102 R- Bacaloglu and C. A. Bunton, Tetrahedron, 1973, 29, 2721, 2726.
103 S. L. Johnson and H. M. Giron, J. Org. Chem., 1972, 37, 1383.
>04 B. F. Erlanger, A. G. Cooper, and W. Cohen, Biochemistry, 1966, 5, 190.
Юз" H- P. Metzger and I. B. Wilson, Biochemistry, 1964, 3, 926.
106. R. Banks, R. F. Brookes, and D. H. Godson, J. C. S. Perkin I, 1975, 1836.
T. Saegusa, T. Tsuda, and Y. Isegawa, J. Org. Chem., 1971, 36, 858.
S. L. Johnson and D. L. Morrison, J. Amer. Chem. Soc., 1972, 94, 1323.
Af. Caplow, J. Amer. Chem. Soc., 1968, 90, 6795.
J. Knappe, Ann. Rev. Biochem., 1970, 39, 757.
L. F. Hatch, Petrol. Refiner, 1958, 37, 123.
H. E. Baumgarten, H. L. Smith, and A. Staklis, J. Org. Chem., 1975, 40, 3554.
H. A. Dieck, R. M. Laine, and R. F. Heck, J. Org. Chem., 1975, 40, 2819.
M. Yamashita, K. Mitzushima, Y. Watanabe, T.-A. Mitsudo, and Y. Takegatni, J. C. S. Chem. Comm., 1976, 670.
A. Williams and W. P. Jencks, J. C. S. Perkin II, 1974, 1753.
G. R. grow, С. Pyun, C. Leitz, and J. Marakowski, J. Org. Chem., 1974, 39, 2448.
117. C. A. L. Filgueiras and J. E. Huheey, J. Org. Chem., 1976, 41, 49.
118. C. J. Giffney and C. J. O'Connor, J. C. S. Perkin II, 1975, 1206.
ion R Auerbach, McF. Zamore, and S. M. Weinreb, J. Org. Chem., 1976, 41, 725.
oi B RraG and F. C. Bruice, J. Amer. Chem. Soc., 1972, 94, 2823.
oo u Fiahom and A. Hamburger, Ber., 1908, 41, 24.
42. H. H. Hatt, G. D. Lichtenwalter, and G. H. Riesser, Austral. J. Chem., 1970, 23, 561.
ml ^',Builer and E- Leitch, J. C. S. Perkin II, 1976, 832.
19" .•‘yku-gawa and S.-I. Yamada, Tetrahedron Leiters. 1969, 699.
12fi л г 5 Shaw and D- G- Walker, J. Amer. Chem. Soc., 1958, 80, 5337.
127 v d ^art,-L N- Hegarty, and F. L. Scott, J. C. S. Perkin II, 1974. 1258,
128 N n ,Упп' J- P11VS Chem., 1965, 69, 687.
129 F r ’esPersen< J- Amer. Chem. Soc., 1975, 97, 1662.
130' / bander, J. Amer. Chem. Soc., 1969, 91, 3629.
131' C j r°tesVa’ B- E KuHev, and J. G. Pofarlieff, J. Chem. Soc. (B), 1970, 232.
132 h w/ lMn,eR and c- J- O'Connor, J. C. S. Perkin II, 1976, 362.
133 A rurlow and R- B- Moodie, J. Chem. Soc. (B), 1971, 407.
134 . F A „^adH' c H. Hegarty, and F. L. Scott, J. C. S. Perkin 11, 1973, 2054.
• Arndt, Org. Synth. Coll. Vol. 2, 1943, 461,
19»
579
135. A. W. Ingersoll, L. J. Bircher, and M. M. Brubaker, Org. Synth. Coll Vol 1
136. J. S. Fowler, J. Org. Chem., 1972, 37, 510.
137. G. W. Fraser and J. Shreeve, Chem. Comm., 1966, 532.
9.11. ПЕРОКСИКИСЛОТЫ И ПЕРОКСИДЫ АЦИЛОВ
А. Ф. ХЕГАРТИ (University College, Cork)
9.11.1. ВВЕДЕНИЕ
В этой главе рассматривается химия кислот и сложных эфиров, а также некоторых родственных соединений, содержащих пероксидную связь (—О—О—). Эти соединения отвечают общим формулам (1) — (3), где X = алкил, арил (1, 2, соответственно пероксикарбоновые кислоты и пероксиэфиры), алкокси-, арилоксигруппа (1,2, пероксиугольные кислоты и пероксикарбонаты) или аминогруппа (1,2, пероксикарбаминовые кислоты и пероксикарбаматы). В соединениях, отвечающих формуле (3), R = алкил (ацилпероксиды) или арил (ароилпероксиды).
О о о о
II II II II
X—С—О—О—Н X—С—О—О—R R—С—О—О—С—R
(1) (2) (3)
Простые пероксикислоты (например 1, Х=Ме) обычно используются в виде водных растворов (примерно 40—60%), но могут быть получены и в безводной форме. Хотя перуксусная кислота (1,Х = Ме) потенциально взрывоопасна даже при —20°С, она может быть перегнана. Стабильность пероксикислот возрастает с увеличением длины углеводородной цепи, и пероксилаури-новая кислота (1, X = Н-С11Н23, т. пл. 52°С), и щ-хлорпербен-зойная кислота (1, X = л«-С1С6Н4, т. пл. 92 °C) в безводной форме достаточно устойчивы при хранении (разложение не превышает 1% в год). Перуксусная кислота (1, X — Me) плавится при 0 °C, в то время как следующие пероксикислоты ряда имеют т. пл., равную —13°С (1, X = Et), —10°С (1, Х = Рг) и 15 С (1, X = м-пентил).
Строение пероксикислот тщательно изучалось; некоторые из них получены в кристаллической форме [1, 2]. Твердые кислоты связаны прочной внутримолекулярной водородной связью; между двумя смежными молекулами существует также межмолекулярная водородная связь, как показано в (4). Внутримолекулярная водородная связь сохраняется и при растворении пероксикислоты (Уста‘ новлено ИК-спектроскопией) [3], и, как полагают, соединение ( ) представляет собой активный исходный агент для генерирован электрофильного кислорода при реакциях пероксикислот (по кр ней мере в растворителях, не содержащих гидроксильной групп Расчеты с помощью метода молекулярных орбиталей (метод
580
r MINDO/3) согласуются с почти планарной структурой 5^'rol Наличие внутримолекулярной водородной связи и поцн-(5) пезонансная стабилизация иона (6) снижают кислотность я<еннаЯ ^{СЛ0Т (5) схема (1) по сравнению с соответствующими пеР?КС’пьтми кислотами примерно на 3—4 единицы р/<а (напри-кзРбопТ для НСОзН 7,1; для НСО2Н 3,7; для МеСО3Н 7,6; для
о
К,-С
С—R R—С
9.11.2. ПОЛУЧЕНИЕ И СВОЙСТВА
9.11.2.1. Методы получения и реакции пероксикислот
Равновесная реакция карбоновых кислот (7) с пероксидом водорода, ведущая к образованию пероксикислот (8) в отсутствие сильно кислых катализаторов протекает медленно. Для ускорения обычно добавляют 1%-ную серную кислоту, трифторид бора или n-толуолсульфокислоту, однако в случае нерастворимых в такой среде алифатических карбоновых кислот можно использовать более концентрированную кислоту (например, 95%-ную серную кислоту). Реакция равновесна, и ее можно сдвинуть в сторону образования пероксикислоты добавлением концентрированного пероксида водорода. Безводные пероксикислоты получают в неводных растворах с использованием кислых ионообменных смол в качестве катализатора. Вместо свободной кислоты можно применять хлорангидриды (10) или ангидриды (9) обычно в водно-спиртовом растворе в присутствии пероксида натрия {схема (3)}; сообщалось также об использовании пиридина в качестве катализатора реакции с хлорангидридом [5]. Родственной реакцией является сольволиз диацилпероксидов (11), проводимый в присутствии основания; этим методом пероксид бензоила может быть г-ладко превращен в пербензойную кислоту {схема (4)}.
О О
II н+ II
R—С—ОН + Н2О2 R—С—О—ОН 4-Н2О
(8)
О О
II но; II
R—С—О—О—Н -<---- R—С—CI
(Ю)
О О
Ph~С—о—о—С—РЬ —P11-J „о—О—Н + Ph—4—ОМе (И)
(7) О О
Л II Н°2
R—С—О—С—R -+
(9)
О О
(2)
(3)
(1)
581
Пероксикислоты с длинной цепью, например пероксилауриновя кислота, количественно получаются из соответствующей кислоть и 50—65%-ного пероксида водорода. Для трифторуксусной кие лоты равновесие также сильнее сдвинуто в сторону пероксики слоты по сравнению с уксусной кислотой; простой метод синтеза трпфторперуксусной кислоты состоит во взаимодействии 90%-ного Н2О2 с ангидридом трифторуксусной кислоты в среде СН2С12 [61
Окисление альдегидов в условиях, способствующих образованию радикалов, может приводить к пероксикислотам; в качестве катализаторов реакции обычно используют соли кобальта. Механизм реакции включает образование радикалов (12), которые служат переносчиками цепи, и последующую реакцию,’ которая объясняет автоокисление бензальдегида до бензойной кислоты {схема (5)}. Безводная перуксусная кислота легко может быть получена таким путем из ацетальдегида; аддукт МеСО—ООСН(ОН)Ме при нагревании при 70 °C превращается в перуксусную кислоту (т. пл. 0 °C, т. кип. НО °C) и ацетальдегид (т. кип. 20 °C) [7]. Другой метод получения пероксикислот состоит в реакции стабильных озонидов с карбоновыми кислотами, однако этот путь не получил широкого синтетического применения.
О
• О2 II PhCHO
PhCHO —> Ph—С=О Ph—С—00. -------► PhCO3H + PhC=O
(12)
PhCO3H + PhCHO
О ОН
II I
Ph—С—00—CHPh —> 2PhCO2H
(5)
Имеются лишь отрывочные сведения о пероксиугольных кислотах [например (14)]; они могут быть синтезированы из соответствующих хлорформиатов (13). Использование кислоты (14) в качестве окисляющего агента [8] может представлять интерес в тех случаях, когда нежелательно проводить процесс в кислых условиях, поскольку в качестве побочных продуктов реакции образуется не карбоновая кислота, а спирт и диоксид углерода {схема (6)}.
О >0
АгСН2О—С—С1 АгСН2—О—С—ОН —> Аг - СН2 - ОН + С02
(13) (14) (6)
Описано получение пероксикислот с полимерной основой (15), и хотя выходы эпоксида, получающегося при реакции алкенов с таким реагентом, составляли лишь «50%, этот синтетически метод обычно достаточно удобен в силу легкости отделения по бочных продуктов простой фильтрацией [9]. „
Эпоксидирование ненасыщенных соединений типа алкенов ( ) или аминов (18) можно проводить с использованием ряда пер сикислот; реакция проходит в мягких условиях как в полярн ' так и в неполярном растворителе {схемы (7) и (8)} HJ- к
1!
'I 'I
582
нятый механизм состоит в атаке, связанной водородной связью пероксикислоты на алкен {схема (9)} [4] и, поскольку пероксикислота действует как электрофильный агент, эпоксидирование замедляется, если в алкене присутствуют электронооттягивающие группировки (р = —1,3 для замещенных стиролов, р = ф-1,4 для ароматических пероксикислот) [12]. Можно использовать более сильные электрофильные реагенты, такие, как монопероксималеи-новую кислоту НОгССН=СНСОзН и трифторперуксусную кислоту (в присутствии или в отсутствие BF3) [13]. Скорость образования эпоксида снижается при проведении реакции в среде основного растворителя, по-видимому, вследствие образования межмолекулярных комплексов, связанных водородными связями, которые снижают электрофильность пероксикислоты. В случае алкенов с заместителями, способными к стабилизации положительного заряда на углеродном атоме алкена (например, в стироле), эпоксидирование сопровождается образованием значительных количеств продуктов с открытой цепью [например, фенилацетальдегида (22)]; вторичные изотопные эффекты согласуются с предположением о возникновении в процессе эпоксидирования несимметричного переходного состояния (21) {схема (10)} [12]. Предполагают, что (22) возникает в результате параллельной реакции, а не вследствие перегруппировки первоначально образующегося
Ph-CH=CH2 -с°зН >
> H-C-CH2Ph
II
О (22)
О
Н2—'CHPh
583
(17) (23)
О
2 11
R2—COH -------->
(24)
оксирана, поскольку последний, как показано, в условиях реакции устойчив [14].
Оксираны (17) могут найти применение в целом ряде реакций; например, раскрытие кольца в гидроксилсодержащих растворителях приводит к образованию гликолей (23, R=H) или эфиров гликолей (23, R = Aik) {схема (И)}; циклические алкены
путем эпоксидирования и последующего раскрытия цикла могут давать транс-гликоли. При добавлении к эпоксидам карбоновых кислот образуются моноацилгликоли (24), которые легко гидролизуются в (23) (R = H), и поскольку карбоновые кислоты воз-
никают в процессе синтеза оксиранов, последние не должны находиться с ними в контакте. Другие реакции оксиранов обсуждаются в разд. 4.3.2.
Кетенимины (25) реагируют с лг-хлорпербензойной кислотой с образованием аддукта (26), который быстро дает кетоны (27) и изоцианиды (28) вместе с амидами (29), образующимися в результате взаимодействия с лг-хлорбензойной кислотой {схема (12)} [15].
Эпоксиды могут быть получены из алленов (30) лишь при наличии больших заместителей (например, при R! = R2 = R3 = °U‘ трет), в противном случае образуются диоксаспиро[2,2]пентаны (32, R1 = R2 = Me,R3 = Ви-трет) наряду с продуктами последую щей реакции с кислотой {схема (13)} [16].
R4
V=C=N—. R3 + АгСО3Н R2/
(25) (26)
(27) (28) (29)
Б84
(30)
(31)
Присутствующие соседние группы могут влиять на стереохи* образования эпоксисоединения; этот эффект впервые наблю-мп я для аллиловых кислот. В качестве примера можно указать на'образование изомера (34) (90%) при реакции диэфира (33) с ж-хлорпербензойной кислотой {схема (14)}; циклогексадиен-13—особенно удобный субстрат для подобных исследований, поскольку он обладает планарной структурой и в нем нет стериче-ских или конформационных препятствий для вступающей группы. Предполагается, что образование водородных связей с участием входящей группы, как показано в (35), объясняет стереоселективность реакции; влияние соседних групп изменяется [17] в ряду. ОН » СО2Н > CO2R > o2cr.
Реакция иминов с лг-хлорпербензойной кислотой дает оксази-ридины (19) и нитроны (20); оксазиридины могут при нагревании изомеризоваться в нитроны. Инверсия атома азота в оксазириди-новой системе проходит медленно, и в некоторых случаях можно выделить энантиомеры [18]; при R[ большем, чем R2, главным продуктом является диастереомер (36). Простые имины подвергаются f/Z (цис/транс) -изомеризации быстрее, чем протекает реакция с пероксикислотой, так что соотношение диастереомеров оксиазиридина не обязательно отражает стереохимию иминов.
(331 (34)
(HI
Предложены два механизма образования оксазиридинов; первый заключается в синхронном взаимодействии, аналогичном приведенному на схеме (9), а второй предполагает атаку сопряженного снования перокснкислоты на протонированный имин {схема (15)}, аналогично механизму, предложенному для реакции Байера —
585
Виллигера [19]. Кинетика реакции, протекающей при каталит песком действии спиртов и карбоновых кислот, достаточно спп-лги (при низких концентрациях катализаторов); при более выепи концентрациях спирта реакция ингибируется. Электронодоноп™ заместители при азоте в имине способствуют образованию нитоои! (20) по параллельному механизму, который заключается в нуклеофильной атаке свободной электронной пары азота на пеоок™ кислоту. н си‘
(37) (38) (39)
0“
Ar1—N=N—Аг2 —> Ar1— N=N—Аг2 (17)
(40) (41)
Окисление оксимов (37) трифторперуксусной кислотой в ацетонитриле протекает аналогично [20] и приводит к продуктам (38) (нитроловые кислоты), которые таутомеризуются в нитроалканы (39) {схема (16)}. Азосоединения (40) при взаимодействии с пероксикислотами превращаются в азоксисоединения (41) {схема (17)} [21].
Пероксикислоты использовались также для окисления аминов. В качестве продукта реакции с первичными ароматическими аминами первоначально образуются гпдроксилампны (42), которые при действии избытка реагента последовательно превращаются в нитрозо- (43) и нитросоединения (44) {схема (18)}. В мягких условиях каждый из этих интермедиатов может быть выделен; три-фторперуксусная кислота служит удобным реагентом для прямого превращения аминов в полностью окисленное нитросоединение. Третичные амины при действии таких пероксикислот, как, например, л!-хлорпербензойная кислота, гладко превращаются в N-ok-сиды (45) {схема (19)} [10]. Окисление серусодержащих органических соединений протекает сходным образоги и в зависимости
586
тичества используемой пероксикислоты из сульфидов (46) оТ т быть получены сульфоксиды (47) или сульфоны (48) {схе-^^РО)}- Ключевым активным реагентом служит связанная водо-
связью форма пероксикпслоты, и согласно предложенному Р° -ядизму, окисление включает электрофильную атаку перокси-^е(\оты на сульфид или сульфоксид (например, значения р из явнения Гаммета Для замещенных пербензойных кислот нахо--Ттся в интервале от 4-1,1 до 4-0,5) [22]. Окисление тионов в г.оксиды И более медленное превращение последних в кетоны под действием пероксикислот протекает по аналогичному механизму [23] • „ „
При окислении двойной углерод-кислородной связи перокси-кислотами (реакция Байера — Виллигера) происходит перегруппировка. Предложенный для этой реакции механизм включает пер-
лп „ „ ( СИНХр0Н.
^оксиды 11 более медленное превращение последних в кетоны под
ГТ ГА ГЛ TZ П ТЯ ГгТЯГ ППТ ПППТР^ОРТ ПП п ттп ’Т/А-Г1тгт,т^,г.. _
Д61-- л-1 му [23]. ___Л„„Л
кислотами (реакция Байера —
воначальное образование аддукта (49) с последующим
no2
nh2 nhoh no
(18)
R1 —S—R2
(42) (43)
R3N —> R3N—О'
(45)
О
—> R1—S—R2 —?
(44)
(19)
О
R1 — S—R2
О
(20)
(46) (47) (48)
ным или ступенчатым перемещением алкильной группы к электрофильному кислородному атому {см. (50), схема (21)}. Из циклических кетонов (51) образуются циклические эфиры (лактоны) {схема (22)}. Мигрирующая группа (R2 в 50) сохраняет свою конфигурацию в конечном продукте.
Окисление ароматических иодсодержащих соединений пероксикислотами приводит последовательно к иодозосоединениям (52) и к иодоксисоединениям (53), возможно также образование ди-эфиров (54) {схема (23)} [24]. Ацетилены окисляются с расщеплением тройной углерод-углеродной связи, давая {схема (24)} смесь карбоновых кислот (56) и (57) или изомеризованную кислоту (55), возможно, образующуюся в результате перегруппировки промежуточного эпоксида в кетен. Гидроксилирование ароматических соединений, активированных по отношению к электрофильной атаке, достигается действием трифторперуксусной кислоты {схема (25)}. Реакцию обычно проводят в кислой среде или в присутствии кислоты Льюиса, например BF3 [13], для промоти-рования образования промежуточных групп, активных к действию
687
ней. Дальнейшее окисление фенолов в п-бензохиноны (58) и пи карбоновые кислоты (59), происходящее под Действием пеоукДг ной кислоты в более жестких условиях {схема (26)}, анатогичип биологическому окислению этих соединений [25]. ’ ” но
588
о 11 2.2. Методы получения и реакции сложных эфиров у' * пероксикислот и пероксидов ацилов
С ложные эфиры пероксикислот получают почти исключительно Спцей алкилпероксидов (или их солей) с ацилирующими аген-Pcah в качестве которых можно использовать ацилгалогениды, таМприды, ацилимидазолы (60) или кетены (61) {схемы (27)’ /9Я)} При реакции с ацилгалогенидами для удаления образую-гося галогеноводорода используют основания, такие как гид-ще йД натрия или пиридин для сложных эфиров, чувствительных ^сильным основаниям.
Известно также получение эфиров пероксикислот с использованием хлоридов (62) {схема (29)}; пероксикарбаматы (64) поучают действием пероксидов на изоцианаты (65) {схема (31)} *ППИ на N,N-замещенные карбамоилхлориды (63) {схема (30)}.
Пероксиэфиры могут реагировать с нуклеофильными реагентами, например метоксид-ионами {схема (32)}, причем протекает
о о
II II
R1—С—Y + R2O2H —> R1—С—О—О—R2 + HY
(27)
(60)
О
СН2=С=О + R2O2H —> СНз—с—О—О—R2
(61).
0 ?
R1—O—с—Cl + R2O2H —> R’—о—С—О—О—R2
(28)
(29)
(62)
о о
R\ Y R'\ II
с—Cl + R3O2H —> ^N—С—O--O-R3
R2/ R2/
(63) (64)
О
Rl—N=c=o + R2O2H —> Rl—NH—(L—О—о—R2
(30)
(31)
(65)
либо нормальная переэтерификация, либо обмен при алкоксильном кислороде, как показано на схемах (33) и (34). Реакция с Реагентами Гриньяра {схема (33)} служит удобным методом_вве-Дения трет-бутоксигруппы в ароматические субстраты (К г, R2 == Bu-трет); другие карбанионы или соединения с активными метиленовыми группами реагируют аналогично [4]. Потеря
589
пероксиэфирами кислорода, приводящая к сложным эфипям и ооновых кислот пли ангидридам, протекает в присутствии тп2' (34М26]"'а ’ СМТВеТС1в,'и с “«анязмом, показанным „а *
о
О , II
R С О—OR2 4-МеО" —> Ri—С—ОМе + R2OO"
MgX -
Q * КоЗ
о
(32)
R-C-O-OR2
R3MgfX
R’-C
О—О—R2
--У R3OR2 + RJCO2MgX
(33)
о
Ph3P:<> OR2 ------Y Ph3POR2^O-C-R1 Y Ph3PO (34)
VO—COR1 L J + О
2 'I 1
R2 O-C-R1
трет-Алкплпероксиэфиры перегруппировываются в удивительно мягких условиях (без катализатора) с образованием ацилалей (68) {расщепление Криге, схема (35)}. Для реакции предложен внутримолекулярный ионный механизм через переходное состояние (67) или через тесную ионную пару, поскольку реакция чувствительна к диэлектрической проницаемости среды (что указывает на существование заряженных интермедиатов), причем внешние нуклеофилы не включаются в продукт. Интересно, что метка 18О в карбонильной группе пероксиэфира (66) почти полностью сохраняется в карбонильной группе соединения (68), указывая на то, что атомы кислорода «карбоксильной группы» в (67) не становятся эквивалентными [27]. Карбений-ионный характер переходного состояния (67) может быть стабилизован соучастием соседних групп, таких как —S—Аг [28].
Диацилпероксиды также претерпевают подобную реакцию; продуктами перегруппировки являются смешанные ангидриды карбоновой и угольной кислот (69) {схема (36)}. В этом случае также предполагается возникновение полярного переходного состояния (величина р в уравнении Гаммета для заместителя в R2 составляет +0,9). Если присутствует сильная электронооттягивающая группа [как в (70)], то электронодонорные заместители X в другом ацильном остатке способствуют реакции (р от —0,9 до —0,75) [29].
Конкурирующее термическое расщепление стерпчески громозд ких пероксидов (71) протекает через радикальные интермедиаты [схема (37)]. Влияние заместителей на скорость растепления согласуется со структурой переходного состояния (72К в котором группы R1 и OR2 обладают значительным карбоний-ионным и 590
о
! II _ I
R-C-O------C-
4 s' । °C+1
4R2
US'
О—C—
OR2
(35)
II II
CMe2CH2C-O-O-C
(70)
анионным характером соответственно [30]. Например, величина р по Гаммету для R1 равна примерно —1,0, в то время как для корреляции данных используются значения о+ (а не обычные о). Сте-рическое сжатие пероксиэфира (71) повышает скорость термического расщепления [31]. Если группа R1 не может удерживать частичный положительный заряд (например, при R1 — Me), то разложение протекает [32] не синхронно с потерей СО2, а через первоначальное образование радикальной пары [R’CO2- -OR2]. При замене трег-алкильной группы [R2 в (71)] на вторичную или пер-
> -R1 СО2 -OR2
(37)
/Н2 С)=С
+ V
R^O^
(38)
О |
вичную группу термическое расщепление протекает иначе и приводит к кетону и карбоновой кислоте как основным продуктам Реакции {схема (38)}.
Термическое расщепление диароилпероксидов протекает ь мят-КИх условиях, на чем основано широкое использование этих
691
соединений в качестве источника радикалов, инициирующих цепные процессы. Кинетические исследования показывают, что уравнение скорости реакции содержит два члена, что связано с мономолеку лярным протеканием реакции и индуцированным расщеплением соотвественно. Мономолскулярная реакция диароилпероксидов несколько ускоряется наличием электронодонорных заместителей в силу ослабления связи О—О (73). Стерические эффекты, например присутствие орто-заместителей в арильных группах' (73) [30], также могут ускорять расщепление. Данные ХИДПЯ (химически индуцированная динамическая поляризация ядер) [33], полученные на основе спектров ПМР и 13С-ЯМР, использованы для доказательства первоначального образования при расщеплении бензоилпероксида связанной радикальной пары f74), которая, теряя С02, превращается в (75) — радикалы, обнаруживаемые с помощью метода ХИДПЯ. Фенильные радикалы из (75) могут быть захвачены, например метилиодидом; в этом случае в качестве главного продукта образуется эфир (77) {схема (39)}. В случае диацилпероксидов (78, R1 = Аг) влияние заместителей указывает на синхронную фрагментацию в переходном состоянии (79). Если R1 представляет собой группу (например, PhCH2), способную стабилизовать положительно заряженный фрагмент R1 в переходном состоянии, то расщепление ускоряется. Если R1 = R2 = первичный алкил (в 78), то реакция предпочтительно протекает по несинхронному механизму через промежуточное образование ацилокси-радикала (например, СН3СО2-). Меченые эксперименты (180 в ацильной группе) показали, что может происходить рекомбинация радикала внутри «клетки» из молекул растворителя, хотя декарбоксилирование ацетокси-радикалов протекает очень быстро (к = 2,4 • 109 с-')
Аг-V.—Аг _ \о—о/
(73)
2PhCO2- —> PhCO2..Ph PhCO2.«Me —> PhCO2Me
(74) (75) (76) <73 74 * * 77 78^
0 0 Г ° ?
Il l| II II
R1—-С—OO—C—R2 —> «R’C’O—CR2
- О
(78) (79)
В присутствии катализатора, содержащего ление трет-бутилпербензоата (80) приводит
О О -+R’C’O-CR2
ионы Сц+, расшеп-к метилбензоату и
592
.. катализируемое расщепление может сопровождаться об-^11'Т°’?оеакиией, если присутствуют соединения RH, которые при доений лОрОда дают относительно стабильные радикалы (на-
R’HP= R2™*, R2CHO, R2*C = CR2CH3) .Для этих прев-np,L йнй предложен цепной механизм, инициируемый переносом Ра пона от Си+ к пероксиэфиру и приводящий к свободным трет-3 «си-радикалам, вместе со связанным с медью бензоатом [34] Ккал трет-ВиО- реагирует с субстратом (RH), давая свобод-раС пядикал -R; последующий перенос лиганда приводит к наблю-!а«ому эфиру (81) {схема (41)}.
О
II си+
Ph—С—ООВи-трет ------
о
II
PhC—OMe + Me2C=O
(80)
(41)
О
Си+ ||
——Ph—С—OR1 + rper-BuOH
r’h
(81)
ЛИТЕРАТУРА
1. D. Belitskus and G. A. Jeffrey, Acta Cryst., 1965, 18, 458.
2 L. Al. Hjelmeland and G. Loew, Tetrahedron, 1977, 33, 1029.
3. D. Swern, in «Organic Peroxides», ed. D. Swern, Wiley Interscience, New York, 1970, vol. 1, pp. 475—516.
4. S. 0. Lawesson and G. Schroll, in «The Chemistry of Carboxylic Acids and Esters», ed. S. Patai, Wiley Interscience, New York, 1969, chapter 14.
5. J. У. Nedelec, J. Sorba, and D. Lefort, Synthesis, 1976, 821.
6. L. F. Fieser and M. Fieser, «Reagents for Organic Synthesis», Wiley Interscience, New York, 1967, vol. 1, p. 821.
7. Ref. 6, p. 787.
8. R. Af. Coates and !. W. Williams, J. Org. Chem., 1974, 39, 3054.
9. C. R. Harrison and P. Hodge, J. C. S. Perkin I, 1976, 605.
10. D. Swern, in «Organic Peroxides», ed. D. Swern, Wiley Interscience, New York, 1971, vol. 2, pp. 355—533.
11- Fl. 0. House, in «Modern Synthetic Reactions», ed. H. O. House, Benjamin, New York, 1972, pp. 293—337.
12. R. P. Hanzlik and G. O. Shearer, J. Amer. Chem. Soc., 1975, 97, 5231.
13. H. Hart, Accounts Chem. Res., 1971, 4, 337.
14. V. G. Dryuk, Tetrahedron. 1976, 32, 2855.
15- L K. Crandall and L. C. Crawley, J. Org. Chem., 1974, 39, 489.
16- /. K. Crandall, W. W. Conover, J. B. Komin, and W. H. Machleder, J. Org. Chem., 1974, 39, 1723.
io Cerefice and E. K. Fields, J. Org. Chem., 1976, 41, 355.
18- 0. R. Boyd, D. C. Neill, C. G. Watson, and W. B. Jennings, J. C. S. Perkin II, !975, 1813.
on and Sawaki, J. Amer. Chem. Soc., 1973, 95, 4687.
zu. w. d Emmons and A. S. Pagano, J. Amer. Chem. Soc., 1955, 77, 4.»7.
« n Не&аг1У’ in «Chemistry of Hydrazo, Azo and Azoxy Compounds», ed.
• • Patai, Wiley Interscience, New York, 1975, chapter 16.
593
22 Н. И. Szmant, Н. F. Harnsberger, and F. Krahe, J. Amer. Chem. Soc, 1954 76 2185. ’ ’
23 A. Battaglia, A. Dondoni, G. Maccagnani, and G. Mazzanti, J. C. S. Perkin II ’ 1974, 609.
24 J. Boeseken and E. Wicherlink, Rec. Trav. chim., 1936, 55, 936.
25' R. A. Marshall and R. Naylor, J. C. S. Perkin II, 1974, 1242.
26 M A. Greenbaum, D. B. Denney, and A. K. Hoffmann, J. Amer. Chem. Soc, ' 1956, 78, 2563.
27 . 5. Winstein and G. C. Robinson, J. Amer. Chem. Soc., 1958, 80, 169.
28 P. Livant and J. C. Martin, J. Amer. Chem. Soc., 1976, 98, 7851.
29 . R. C. Lamb, L. L. Vestal, G. R. Cipau, and S. Debnath, J. Org. Chem., 1974, 39, 2096.
30 W. H. Richardson and H. E. O' Neal, in «Comprehensive Chemical Kinetics», ed'. С. H. Bamford and C. F. H. Tipper, Elsevier, Amsterdam, 1972, vol. 5, chapter 4.
31 j p Lorand, J. Amer. Chem. Soc., 1974, 96, 2867.
32 ' T Koenig and J. G. Huntington, J. Amer. Chem. Soc., 1974, 96, 594.
ЗЗ’ E. M. Schulman, R. D. Bertrand, D. M. Grant, A. R. Lepley, and C. Walling, J. Amer. Chem. Soc., 1972, 94, 5972.
34 . H. L. Goering and U. Mayer, J. Amer. Chem. Soc., 1964, 86, 3753,
СОЕДИНЕНИЯ ФОСФОРА
10.1. ВВЕДЕНИЕ
Д. ДЖ. X. СМИТ (University of Leicester
Со времени первых систематических исследований Михаэлиса в конце XIX века темпы развития химии органических соединений фосфора постоянно возрастали. Тем не менее можно выделить несколько периодов, когда важные открытия резко повышали интерес к этим соединениям. Открытие Шредером и другими исследователями [1] в 1930 г. токсических п инсектицидных свойств соединений фосфора вызвало к жизни новую отрасль промышленности. Превращение Виттитом карбонильных соединений в алкены и использование гомогенных катализаторов открыло совершенно новую область применения соединений фосфора в синтезе. Совсем недавно получение стабильных соединений пентаковалентного фосфора и изучение Вестхаймером и другими процессов псевдовращения вызвало прилив новых сил в эту область химии.
Большая часть синтезируемых и используемых в крупнотоннажных масштабах соединений фосфора является неорганическими, однако значение органических соединений фосфора постоянно возрастает. Они находят применение в качестве антиоксидантов и стабилизаторов в нефтяной промышленности и производстве полимеров, расширяется их использование в таких областях, как защита от коррозии, производство огнестойких материалов, экстрагентов, комплексообразователей, а также в сельском хозяйстве [2]. Широкое практическое использование наряду с возрастающим применением соединений фосфора в общем органическом синтезе обусловило быстрый рост числа публикаций по химии фосфорорганических соединений.
Настоящая часть разделена на главы в соответствии с типами единений так, как это принято в «Organophosphorus Chemistry» 3ORCla'*S^ Per'°dical Reports [3]. При написании этой части исполь-и ано шеститомное издание «Organic Phosphorus Compounds» [4] соответствующие тома серии «Methoden der Organishen Chemie ШоиЬеп-Weyl)» [5].
595
10.1.1, СТРУКТУРА И ХАРАКТЕР СВЯЗИ
Фосфор имеет электронную конфигурацию ls22s22p63s23n3 и пас полагается почти в центре периодической системы элементов Вследствие такого положения в системе элементов он обладает высоким потенциалом ионизации, поэтому связи, образуемые фосфором, являются, за редкими исключениями, ковалентными [6 71 В настоящее время известно, что в своих соединениях фосфор ’может иметь координационное число 1, 3, 4, 5 и 6. Соединения, в которых атом фосфора связан только с одним атомом другого элемента, нестабильны при обычных условиях, поэтому двухатомные молекулы, такие, как Р2, PN и РО, можно зафиксировать только спектроскопически при высоких температурах.
Как и следует ожидать на основании электронной структуры, фосфор легко образует три о-связи, давая стабильные производные РХ3, аналогичные соединениям азота NX3. Молекулы этого типа являются пирамидальными; углы между связями несколько больше 90°, однако они меньше тетраэдрических валентных углов в молекулах соответствующих соединений азота. Это позволяет считать, что вклад 5р3-гибридизованных орбиталей в случае фосфора имеет меньшее значение.
Примеры соединений, в которых фосфор образует кратные связи, аналогичные кратным связям углерода, азота или кислорода, редки; в этом отношении фосфор сходен с другими элементами второго большого периода. Условия эффективного перекрывания при образовании л-связп, которая легко образуется за счет 2р-ор-биталей соседних атомов, связанных 2р0-связью, с трудом выполняются для Зр-орбиталей, связанных Зр0-связью. Для объяснения этого эффекта предложено много гипотез [7], однако большинство описанных соединений, которые, как полагают, содержат в своем составе трехвалентный фосфор, связанный кратной связью, являются в действительности полимерными продуктами.
Многие соединения фосфора имеют четыре заместителя, расположенные тетраэдрически вокруг 5р3-гибридизованного атома фосфора. Образование четвертой связи можно формально представить как координацию несвязывающей электронной пары трехвалентного фосфора с кислотой Льюиса, в результате чего атом фосфора приобретает формальный положительный заряд. Если ОДИН из заместителей несет отрицательный заряд, прочность связи с срос фором возрастает; этот эффект ярко иллюстрируется увеличением стабильности карбанионов, в том случае когда с карбанионны центром соседствует тетраэдрический атом фосфора. »
Важным фактором является наличие у фосфора •
Считают, что особые свойства тетраэдрического фосфора 0 лены повышением кратности связи за счет подачи несвязы нт_ 2р-электронов с отрицательно заряженного заместителя на ные ЗсЕорбитали атома фосфора [8]. Имеются достат
596
□ательства наличия такого дополнительного связывания [6]. Д°ка уЮщаяся Рл—d.-t-связь является значительно менее прочной, ^лл-связь, из-за относительно высокой энергии и большей 4eLv3HOCTH Зб/-орбиталей, поскольку максимальное перекрывание .орбиталей и образование прочной связи возможно при взапмо-^“ствип орбиталей одного и того же типа. Для рл—с^-связи, од-деП 1Штеграл перекрывания возрастает с ростом положительного HaLna на атоме фосфора, что приводит к несимметричному распре-зенпю электронов, выраженному в сильной поляризации связи
/p+L-Х"). Высокая энергия образования (410 кДж/моль) фосфо-пьной группы (Р = О), являющаяся движущей силой многих Реакций соединений фосфора, обусловлена возникновением второй Р -связи в плоскости, перпендикулярной плоскости первой связи. Две свободные электронные пары кислорода перекрываются с двумя Зб/-орбиталямп фосфора с образованием связи, имеющей симметрию тройной *.
Поскольку тетраэдрический атом фосфора может принимать
электрон от отрицательно заряженного заместителя, возможно существование соединений, в которых фосфор окружен пятью ковалентно связанными группами. Известно много соединений пентако-ординированного фосфора, хотя точная природа связи в них все еще является предметом дискуссии [9]. Большая часть этих соединений, однако, обладает ожидаемой для $р3с?-гибридизованного атома'фосфора тригонально-бипирамидальной конфигурацией, причем, как и было предсказано, аксиальные связи имеют большую длину (см. разд. 10.4).
Присоединение шестого лиганда к пентаковалентному атому фосфора вполне реально, поскольку этот процесс протекает без дополнительной затраты энергии. Известно много анионов шестикоординированного фосфора, имеющих ожидаемую для sp3d2-rn6-ридизации октаэдрическую конфигурацию (см. разд. 10.4.6).
Ю.1.2. НОМЕНКЛАТУРА ФОСФОРОРГАНИЧЕСКИХ СОЕДИНЕНИИ
Наименование соединений фосфора является малоприятным упражнением для большинства химиков. Наибольшая путаница возникает из-за широкого употребления тривиальных названий, в особенности для кислот фосфора и их эфиров, некоторые из которых склонны к таутомерии. Все это требует большой осторожности при работе со старой литературой. Например, соединение R2P (О) ОН называли диалкилфосфиновой и диалкилфосфоновой кислотой ПО], в ранней английской литературе встречается название диал-килфосфонистая кислота, а в справочнике Бейльштейна — диалкил-Фосфинистая кислота.
5u., В части 10 для удобства фосфорильная и тиофосфорильная группы обо-значены соответственно Р = О и Р = S,
597
a
Таблица 10.1.1. Номенклатура фосфорорганических
Формула Название
приведенное в английском тексте принятое в отечественной литературе
R3P
R3PO
(RO)3P
(R2N)3P
(RO)2PC1 ROPC12 (HO)2PSH (ROhPSR1
Trialkylphosphine Триалкилфосфин
Trialkylphosphine oxide Триалкилфосфиноксид
Кислоты трехвалентного фосфора
Производные фосфористой кислоты [(НО)3Р] Trialkyl phosphite Hexa-alkylphosphorous triamide a
Dialkyl phosphorochloridite Alkyl phosnhorodichloridite Phosphorothious acid
S-Alkyl 0,0-dialkyl phosphoro-thioite
Триалкилфосфит
Гексаалкилтриамидофосфит
Диалкилхлорфосфит
Алкилдихлорфосфит
Тиофосфористая кислота
О,О,S-Триалкилтиофосфит
RP(OR')2
rpci2
Производные фосфонистой кислоты [НР(ОН)2]
Dialkyl alkylphosphonite Alkylphosphonous dichloridea
RP(SH)OH
Alkylphosphonothious acid
Диалкилалкилфосфоиит
Алкилдихлорфосфин, алкилдихлорфосфонит
Алкилтиофосфоиистая кислота
R2POR’ R2PC1
Производные фосфинистой
Alkyl dialkylphosphinite
Dialkylphosphinous chloride'
R2PSR>
r2pnr^
Alkyl dialkylphosphinothioit N,N-Dialkyl dialkylphosphinous amide
кислоты [H2POH]
Алкилдиалкилфосфинит Диалкилхлорфосфин, диалкил-хлорфосфинит
Алкилдиалкилтиофосфинит М,М-Диалкиламидодиалкил-фосфннит
фосфора
(RO)3PO
(R2N)3PO
(RO),P(O)C1
(ROP(O)C12 (RO)2P(O)NR.J
(RO2)P(O)SR
Кислоты пятивалентного
Производные фосфорной кислоты [(НО)3РО]
Trialkyl phosphate
Hexa-alkylphosphoric triamide Dialkyl phosphorochloridate Alkyl phosphorodichloridate N.N-Dialkyl dialkylphosphor-amidate
S-Alkyl 0,0-dialkyl phosphoro-thioate
Производные фосфоновой кислоты [HPO(OH)2]
Dialkyl alkylphosphonate
N,N-Tetra-alkyl alkylphosphonic diamide Alkyl alkylphosphonofluoridate
1 О-Alkyl S-alkyl alkylphospho-no thio ate
Триалкилфосфат Гексаалкилтриамидофосфат Диалкилхлорфосфат Алкилдихлорфосфат М,М-Диалкиламидодиалкил-фосфат 0,0,5-Триалкилтиофосфат
(RO)2P(O)R‘
(R2N)2P(O)R’
(RO)R’P(O)F (RO)R‘P(O)SR
Диалкилалкилфосфонат М.М.Й'.М'-Тетраалкилдиами-поалкилфосфонат Алкилалкилфторфосфоиат O,S - Д иалкилалкилтиофосф пат
598
Продолжение
Формула Название
приведенное в английском тексте принятое в отечественной литературе
R,P(WOg' R,p(O)SH r2P(S)SH r2psC1 Производные фосфиновой кислоты [Н2Р(О)ОН] Alkyl dialkylphosphinate Алкилдиалкилфосфинат Dialkylphosphinic amide Амидодиалкилфосфинат Dialkylphosphinothioic acid Диалкилтиофосфиновая кис- лота Dialkylphosphinodithioic acid Диалкилдитиофосфиновая кис- лота Dialkylphosphinothioic chloride Диалкилхлортиофосфинат
Фосфораны
Pentahalophosphorane Iminotrialkylphosphorane Alkylidentrialkylphosphorane
Пентагалогенфосфоран Иминотриалкилфосфоран Алкилидентриалкилфосфоран
RgP=NH RsP=CHR
Phosphetan(e)
Циклы
Фосфетан
Phospholan (е) Фосфолан
Phosphorinan (е)
Фосфоринан
l,3,2-Dioxaphosphorinan(e)
1,3,2-Диоксафосфоринан
а Соединения подобного типа часто называют как производные фосфинов, например (МегКт)зР — трис(диметиламино)фосфин, РЬгРС! — дифенилхлорфосфин.
Номенклатура соединений фосфора стала, однако, приемлемой со времени опубликования правил, совместно выработанных комиссиями Лондонского Химического общества и Американского Химического общества [И]. Основные правила современной номенклатуры фосфорорганических соединений суммированы в табл. 10.1.1 (русский вариант номенклатуры в основном эквивалентен англо-американской номенклатуре).
10.1.3. СПЕКТРОСКОПИЯ СОЕДИНЕНИЙ ФОСФОРА
При исследовании структуры, и реакций соединений фосфора иР°ко применяют все виды спектроскопии [12]. УФ-Спектроско-используют для изучения эффектов сопряжения в соединениях v сфора( особенно в работах советских авторов [13]. Характери-
599
стические колебательные частоты для большинства фосфопсп жащих функциональных групп известны (табл. 10 1 2)- эта д °Дер’ широко применяют при установлении структуры новых фосХ^ппТ ганических соединений [14]. риР’
Таблица 10.1.2. Частоты поглощения фосфорсодержащих грипп в ИК-области спектра У
Группа Частота, см 1 Группа —' 1— Частота, см-1
Р—ОН 2750-2550 Р—F 985-710 (с.)
(широкая и сивная) малоинтен-
Р—н 2450—2240 (ср.) Р—Р 510-390 (сл.) (с.) (с.) (с.) (о. с.)
P=N 1439—1120 (о. с.) Р—С1 590-420
Р=С 1230-1180 (ср-) Р—О—Р 1060-850
Р=О 1320-1200 (о. с.) P=S 750-550
Р—Ph 1450-1425 (с.) Р—О—С 1060—905
875-730 (с.)
Спектроскопия ЯМР представляет собой наиболее важный источник структурной информации в химии фосфора. Кроме данных, которые могут быть получены из спектров ЯМР Н и 13С, особенно ценные сведения о природе атома фосфора дает спектроскопия ЯМР31Р [15]. К сожалению, значительные практические трудности, возникающие при получении спектров достаточно высокого разрешения, ограничивают применение ЯМР 31Р. Многие из этих трудностей были, однако, преодолены с появлением спектрометров с Фурье-преобразователями и можно считать, что сфера использо-
Таблица 10.1.3. Химические сдвигиа в спектрах ЯМР 31Р некоторых фосфорорганических соединений
Соединение Химический сдвиг, млн-1 Соединение Химический сдвиг, млн-1
Рч РН3 Ме3Р Ph3P (Me2N)3P (МеО)зР РС1з + 460 +240 +62 +7 — 122 -140 -219 Ме2РО2Н МеРО(ОМе)2 (МеО)зРО (PNC12)3 Ph3P+Me Br" Ph3P=CH2 pf5 —49 -33 -1 -19 —23 —4 +35
РИзРО —25
С13РО —3 (МеО)зР jf +49
(Me2N)3PO —24 PhPF6 Et2NH2 + 136
а Относительно 85%-иой Н3РО4 в качестве стандарта. Отрицательные значения чаюг нахождение в более слабом поле относительно опорного сигнала»
60Q
пания ЯМР 3,Р будет постоянно
пня ЯМР будут обсуждаться п0Рхо^ИрЯТься- Данные Р разделах, однако для удобства в ТЛУ и^°жения в поР троско' некоторые типичные значения Li бл’ 10-1-3 и ю i 4°следУющих сплн-спинового взаимодействия Мичес*их сдвигов , приведены и констант
Таблица 10.1.4. Константы спин-спинов ого взаиипОо Фыфоусоаержащы
Группа Л Гц Группа J, Гц
\-Н
\(О)Н
\+
-Р-СНз
/Р(О)СН3
/Р-С-СНз
/РОСН2СН3 /Р-ОСНз
150-225
450-725
450-600
0-5
8-18
10-30
0-4
7-15
7-10
12—14
1-2
2—4
12-15
28-32
10.1.4. СИНТЕЗ ФОСФОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Высокая реакционная способность и доступность элементного фосфора позволяет применять его в фосфорорганическом синтезе 116, 17]. Молекула белого фосфора имеет тетраэдрическую структуру и содержит шесть Р—P-связей, которые могут ступенчато разрушаться с образованием простых продуктов. Поэтому реакции фосфора протекают сложно, и индивидуальные соединения получаются обычно с низким выходом. Для промышленности значительный интерес представляет прямое получение фосфорорганических
601
соединений из фосфора, однако большая часть синтезов основана на использовании таких производных фосфора, как РС13 РО 3 P4Sio, которые легко могут быть получены из элементного фосф^
ЛИТЕРАТУРА
1. D. F. Heath, ‘Organophosphorus Poisons’, Pergamon 1961
2- Л ‘Phosphorus Chemistry in Everyday ’ Living’, American Chemi-
cal Society, Washington, 1976.
3. ‘Organophosphorus Chemistry’, Specialist Periodical Reports ed S TrinnPtt Chemical Society, London, 1970—1976. ’ PP >
4. ‘Organic Phosphorus Compounds’, ed. G. M. Kosolapoff and L. Maier Wilev. Interscience, New York, 1972—1974. ’ y
5. K- Sasie, ‘Methoden der Organischen Chemie (Houben Weyl)’ Thieme Stuff gart, 1963—1964, vols. 12/1 and 12/2.
6. R. F. Hudson, ‘Structure and Mechanism in Organophosphorus Chemistry’, Academic Press, London, 1965, pp. 8—83; [P. Хадсон, Структура и механизм реакций фосфорорганических соединений. Пер. с англ. М., Мир, 1967, с. 17—
7. J. /L Van Wazer, ‘Phosphorus and its Compounds’, Interscience, New York, 1958, vol. I, pp. 1—87; [Ван Везер. Фосфор и его соединения. Пер. с англ. М., Издатинлит, 1962. с. 10—78].
8. A. J. Kirby and S. G. Warren, ‘The Organic Chemistry of Phosphorus’, Elsevier, Amsterdam, 1967, pp. 4—8; [А. Кирби, С. Уоррен. Органическая химия фосфора. Пер. с англ. М., Мир, 1971].
9. R. Hoffmann, J. М. Howell, and Е. L. Muetterties, J. Amer. Chem. Soc., 1972, 94, 3047 (и приведенные там ссылки).
10. G. М. Kosolapoff, ‘Organophosphorus Compounds’, Wiley, New York, 1950.
11. ‘Handbook for Chemical Society Authors’, Chemical Society, London, I960, chapter 6.
12. Cm. J. C. Tebby in ‘Organophosphorus Chemistry’, Specialist Periodical Reports, ed. S. Trippett, 1970—1976, chapter 12.
13 E. N. Tsvetkov and M. I. Kabachnik, Russ. Chem. Rev., 1971, 40, 97; [E. H. Цветков, M. И. Кабачник— Усп. химии, 1971, 40, 177].
14. L. C. Thomas, ‘The Interpretation of the Infrared Spectra of Phosphorus Compounds’, Heyden, London, 1974.
15. J. R. Van Wazer, J. H. Letcher, V. Mark, С. H. Dungan, and M. M. Crutchfield, in ‘Topics in Phosphorus Chemistry’, ed. M. Grayson and E. J. Griffith, Interscience, New York, 1968, vol. 5.
16. M. M. Rauhut, Ref. 15, 1964, vol. 1, p. 1.
17. M. Grayson, Pure Appl. Chem., 1964, 9, 193.
10.2. ФОСФИНЫ, СОЛИ ФОСФОНИЯ, ГАЛОГЕНФССФИНЫ
Д. ДЖ. X. СМИТ (University of Leicester)
10.2.1. МЕТОДЫ ПОЛУЧЕНИЯ ФОСФИНОВ [1, 2]
10.2.1.1. Синтез фосфинов с помощью металлорганических реагентов
Замещение хлора в хлорпроизводных соединений фосфора (Ш) до сих пор остается лучшим методом получения третичных Ф0СФ нов. Исходными соединениями могут служить различные три*, Д §02
, моногалогенпроизводные фосфора и подходящие магний или тпйорганические реагенты (уравнения 1—3) магнии- или
Л" PCl3 + 3RMgX —> R3P + 3Mgxci m
RPC12 + 2R’MgX — > RPR«+2MgXCl
RR’PCl + R2Li -> RR-PR2 + UC1
Более высокие выходы третичных фосфинов были получены ппи использовании большого избытка (примерно четырехкратного) аакил- или арилмагниихлорида, причем смешение реагентов производят при возможно более низкой температуре [3]. Другими исследователями было установлено, что применение избытка реактива Гриньяра не приводит к повышению выхода; например, использование 50%-ного избытка и-бромфенплмагнийбромида не улучшает вЫХод фосфина (1) [4].
Вг—;
Br
(4)
(1)
Первичные алкилмагнийгалогениды обычно достаточно успешно применяются в этой реакции, однако при использовании реактивов Гриньяра — производных вторичных и третичных галогенидов третичные фосфины получаются с низкими выходами или вообще не образуются. По-видимому, в этом случае значительную роль играют стерические факторы, поскольку удается выделить продукты частичного замещения, такие, как ди- и моногалогенфосфины, например (2).
Использование литийорганических реагентов становится все более популярным, особенно для образования связи С (арил)—Р. Выходы третичных фосфинов, получаемых этим методом, сравнимы с выходами при использовании реактивов Гриньяра. Применение литийорганических соединений особенно важно при синтезе стери-чески затрудненных фосфинов, таких, как три-трет-бутилфосфин (3) (уравнение 5) [4].
Третичные фосфины с тремя различными заместителями могут быть получены или из несимметричных хлорфосфннов, или, что менее удовлетворительно, пз дигалогенфосфинов и смеси двух различных реактивов Гриньяра [5], как, например, соединение (4) (уравнение 6).
трет-ВиЫ
РС13 -|- T’pe7’-BuMgX(H36.) —► трег-Ви2РС1 k трег-BujP (5)
(2) (3)
PhPCl5+EtMgBr+PhCH2MgCl —> PhPEt2+PhP(Et)CH2Ph+PliP(CH2Ph)2 (6)
(4) (41%)
Несимметричные триарилфосфины могут быть получены последовательной обработкой фенилдихлорфосфина металлорганиче-скими реагентами, обладающими различной реакционной способностью (уравнение 7) [6]. Полезным методом, позволяющим
603
получать несимметричные фосфины в одну стадию, является олн временное присоединение алкплхлорида и лптийорганическоп)' агента к циклополифосфину (уравнение 8) [7]. Ре'
Многие циклические третичные фосфины типа (5) были синтр знрованы взаимодействием дигалогенфосфинов и реактивов “ яра — производных полпметилендигалогенидов (уравнение С1 Аг1
I Аг’ы I
Ph—РС12 + ArZnCl —> Ph—P—Ar -------> ph—P—Ar
(RP)ra + nR'Li + nR2Cl —> RR‘R2P + wLiCl
CH2
RPC12 + XMgCH2(CH2)nCH2MgX —> Rp/ \сн2)п
CH2
Грпнь-
9)
18].
(7)
(8)
О)
(5)
Для получения третичных фосфинов могут быть использованы эфиры кислот фосфора. Трифенилфосфин, например, почти количественно образуется при действии избытка фенилмагнийхлорида на триметил- или трифенилфосфит. Трифенилфосфит применялся в тех случаях, когда синтезы на основе РС13 были безуспешными или давали низкие выходы. Описаны, например, синтез трицпкло-пропилфосфина [91 и усовершенствованная методика получения триметилфосфина [10] с использованием трифенилфосфита и соответственно циклопропил- и метиллития. Алкокси- и арилоксигруппы, являющиеся при низких температурах эффективными защитными группами у атома фосфора, могут замещаться при более высоких температурах. Поэтому третичные фосфины с двумя одинаковыми заместителями могут быть получены из хлорфосфитов типа (RO)2PC1 или (RO)PC12. Хорошим примером такого превращения служит получение пропиннлфосфинов (6) из диэтилхлор-фосфита (уравнение 10) [11].
LiC=CMe RMgX
(EtO)2PCl 7 —>- (EtO)2PfeCMe > R2PCsCMe (Ю) — /о L> и
(6)
Ph2PCl +
(7)
Недавно сообщалось, что взаимодействие дифенплхлорфосфина с цнклопентадиенилталлием дает фосфин (7) с количественнь выходом (уравнение 11) [12].
10.2.1.2. Синтез фосфинов из металлированных фосфинов
Синтез и реакции фосфидов щелочных металлов 11)иК0Кр1,Эдра. лнсь Исслайбом с сотр. и другими исследователями [13J. Ире щенне днфенилфосфидов и других фосфидов с двумя одинаков
604
группами в третичные фосфины под действием алкилирующих агентов широко отражено в литературе. Синтезы на основе фосфидов приобретают все большее значение н для получения первичных и вторичных фосфинов, поскольку были найдены методы, позволяющие провести частичное замещение атомов металла в молекуле исходного фосфида.
Атом водорода в фосфинах R2PH, а также другие лабильные группы, например галоген или алкоксигруппа в производных фос-фоннстой п фосфинистой кислот, могут селективно замещаться на металл в инертных растворителях. Выбор растворителя очень важен, поскольку некоторые растворители, особенно простые эфиры, расщепляются под действием фосфид-ионов. Более гладко реакции протекают при использовании в качестве металлирующих агентов металлорганпческих соединений, например бутил- или фениллития.
Связь арил—Р в третичных фосфинах R2PAr также может расщепляться под действием щелочных металлов с образованием фосфидов R2PM. В этих и в других реакциях восстановления избыток металла_ вызывает .дальнейшее восстановление до анион-ради-кала R2PM , который может быть зафиксирован с помощью спектроскопии ЭПР [14].
Фосфиды щелочных металлов реагируют с самыми различными алкилирующими агентами; типичные примеры таких реакций приведены на схеме 12. Триметилснлилфосфин (8) также использовался в реакциях этого типа (схема 13) [15], причем это соединение является гораздо более мягким реагентом, чем фосфиды щелочных металлов, и особенно удобно при получении ацилфосфинов.
r2ci RR'PR2 <--------
х
Н2С—сн2
RR'P(CH2)2XH <-------
(12)
r2coci
-------> RR'PCOR2
R, R1 = H, Aik, Ar: R1, R2 Aik, Ar; M = Li, Na, К
О О
ArCOCI CICOCOCi II II
Ph2PCOAr *-------- Ph2PSiMe3 --------► Ph2PC—CPPh2
(13)
(8)
Взаимодействие дифенилфосфида лития с арилгалогенпдами протекает как прямое замещение; альтернативный путь через промежуточное образование дегидробензола не реализуется, так как изомерные продукты не образуются [16]. Дегидроарены, однако, получаются при реакции арилфторидов с дн-трет-бутнлфоефндом лития (9), но не с диэтил- н ди-н-бутплфосфидами лития (уравнение 14) [17]. Полагают, что подобное различие обусловлено более высокой основностью соединения (9), однако оно может быть результатом влияния стернческих факторов.
605
Me'
2—F + LiP(Bu-7’pe7’)2 —► (9)
>—P(Bu-7’pe7’)2 + Me—fl 4
(14)
^Р(Ви-трет)2
Замещение фосфидами галогена в винилгалогенидах (10) или (11) приводит к продуктам, в которых конфигурация атома углерода сохраняется, тогда как замещение при тетраэдрическом центре протекает как обычный SN2-nponecc с обращением конфигурации [18]. Детальная природа реакции фосфидов с винилгалогенидами неизвестна, хотя, по-видимому, процесс не является простым замещением. Картина до некоторой степени осложнилась после появления сообщения о том, что аналогичная реакция 1,2-дибромалке-нов (12) протекает по совершенно другому направлению, а именно с первоначальной нуклеофильной атакой на атом брома с образованием ацетиленов (уравнение 17) [19].
Н. ,Н Н. ,Н
СИ
(10) Ph.
Ph2Pz ^PPh2
Ph. ,Н
Н/ 'PPh2
(15)
(16)
(17)
Н' (И) ВгСН=СНВг + Ph2PLi —> СН=СН (12)
Обработка (+)-(/?)-1-хлорэтил бензола метплфенилфосфидом натрия и последующее окисление приводят к (Sp)-(Sc) -метил (а-метилбензил) фенилфосфиноксиду (13) в качестве основного диастереомера (уравнение 18). Определение оптической чистоты показало наличие индуцированной асимметрии (28%) на атоме фосфора, возникающей на первой стадии процесса [20].
Ph /Ph О.
С—Cl + MePhPNa—*- .Р—CZ
МеН МеП Г-'-Ме
н Ph ri
Ph , ,
,..р-с; И
Me*'" i i 'Me Ph Н
(13)
10.2.1.3. Реакции восстановления
Галогенфосфины и соединения, содержащие связи „
могут быть восстановлены, до соответствующих фосфинов Деи вием различных реагентов. Наиболее широко используемым в становителем является алюмогидрид лития (см. табл. 10.2.1).
P—О и p—S’
606
Таблица 10.2.1.- Восстановление соединений фосфора действием LiAIH,
ИсХОД„Ое соединение Продукт восстановления Исходное соединение Продукт восстановления
.— РрС12 , Dp(O)Ch рр(О) (OR Ъ R2PO2H rph2 Rph2 rph2 r2ph R2P(O)OR r2poci r2psci R3PO R2ph R2ph R2ph R3P
Восстановление фосфонатов и фосфинатов является, по-видимому наиболее удобным методом синтеза соответственно первичных и вторнчных фосфинов, поскольку исходные вещества легко доступны но реакции Арбузова Михаэлиса (см. разд. 10.2.7). Метилфосфины, например, удобно получать восстановлением диметилфосфона-та (14) [21] с последующим метилированием (схема 19). Выход фосфинов может быть заметно повышен, если LiAlH4 медленно (около 8 ч) прибавлять к охлажденному (—78°C) раствору восстанавливаемого соединения фосфора [22].
ЫА!Н4 Mel Mel
МеРО(ОМе)2 МеРН2 -----> Ме2РН ----->- Ме3Р (19)
(14)
(15)
(16)
Фосфин (15), полученный восстановлением соответствующего оптически активного фосфнноксида, был первым примером оптически активного соединения трехвалентного фосфора, оптическая активность которого целиком обусловлена хиральностью атома фосфора [23]. В то же время восстановление оптически активного соединения (16) приводит к соответствующему рацемическому фосфину (уравнение 20) [24]; показано, что исходный фосфиноксид рацемизуется под действием ЫА1Н4. Вероятно, в этом случае L1AIH4 присоединяется к фосфиноксиду с образованием фосфорана, который теряет оптическую активность за счет псевдовращения; распад фосфорана приводит к рацемическому фосфиноксиду. В последние годы в качестве восстановителей все более широко используют силаны, такие, как трихлорсилан, фенилсилан и полиметил-зодородсилоксаны. Эти реагенты особенно полезны при получении Фосфинов, которые содержат функциональные группы, чувствитель-нЫе к действию LiAlFh, например в (17) [25] и (18) [26L
[х-О_];РР|”-” ’’"-«О50
(17) X=CN,CO2H, COgMe (18)
607
Фенилсилан является лучшим восстановителем, когда требуется превратить оптически чистые фосфиноксиды в оптически чистые фосфины; с использованием этого реагента фосфиноксиды с высокими выходами и 100%-ным сохранением конфигурации фосфора были превращены в соответствующие фосфины [27].
Мпслоу показал, что стереохимическое направление восстановления бензилметилфенилфосфиноксида трихлорсиланом в присутствии основания зависит от силы основания. В присутствии слабых оснований (рКь > 7) предпочтительно образуется продукт с сохранением конфигурации при атоме фосфора, тогда как в присутствии сильных оснований (р/(ь < 5) восстановление протекает с преимущественной инверсией у атома фосфора [28]. Комплексообразования, по-видимому, в этом случае не наблюдается, но восстановление с инверсией протекает с участием продукта разложения трихлорсилана под действием основания — перхлорполисилана или три-хлорснлил-аниона (уравнения 21, 22). В этой работе показана, таким образом, возможность использования гексахлордпсилана для восстановления ациклических фосфиноксидов с обращением конфигурации. В то же время восстановление фосфетаноксидов гексахлордисиланом протекает с сохранением конфигурации, что является, по-видимому, результатом нуклеофильной атаки по атому фосфора. Отщепление серы от ациклических фосфинсульфпдов протекает с сохранением конфигурации, вероятно, путем атаки трихлор-силил-аниона по атому серы промежуточного фосфониевого иона (19) (уравнение 23).
HSiClg + Et3N «=± SiCl3 + EtsNH (21)
Ph<?l2 . Me ?HiP‘ Cl,sr
Ме-Щ;Р=о + Et3NH------> >P— OH--------->
Ph/ -EbN PhX +
^CH2Ph /^2 Ph
•—> CleSb-тР^Ме +“OH ► :P^-Me + [HOSiC^J (22>
\>h Ph
R*Z₽=S + Si2C16----► R4p~ s—SiCl3 + SiCl3----->
R2 /
R& . f23)
yP: + ci3si—s—sici3
R2
Фосфониевые соли также могут быть восстановлены до фосфинов, причем преимущественно отщепляется группа, образующая оолее стабильный анион, как показано на примере восстановле-
608
LiAIHj
------> MePrPhP + PhCH,
(24)
теряюшего бензильную группу в виде толуола (уравнения п ’восстановление оптически активных фосфониевых солей иле 24 ем LiAlH4 может приводить как к рацематам, так и к опти-действн' вНЫМ продуктам в зависимости от природы фосфониевой цееки а „огнчная ситуация возникает при щелочном гидролизе •25 («•₽•»10-25|>-
MePrPh(PhCH2)P+ Br’
(20)
Электрохимическое восстановление на свинцовом или ртутном оде является наиболее экономным путем превращения фосфо-ка1вЫх солей в фосфины. Материал катода влияет на порядок от-НИппения органических радикалов; преимущественное отщепление кензильной группы на ртутном катоде успешно использовано для лучения большого числа третичных фосфинов [29]. Электрохимическое восстановление протекает стереоспецифично с сохранением конфигурации при атоме фосфора; оно было использовано в первых синтезах оптически активных третичных фосфинов.
10.2.1.4. Присоединение фосфинов со связью Р— Н к алкенам
Первичные и вторичные фосфины при высокой температуре в отсутствие катализаторов присоединяются к алкенам, двойная связь которых активирована присутствующими электроноакцепторными группами; при этом получаются соответственно вторичные и третичные фосфины (уравнение 25). Однако в основном эти реакции проводят с использованием кислотных или основных катализаторов. Для реакций, катализируемых кислотами, постулируется карбенпевоионный механизм, тогда как реакции, катализируемые основаниями, протекают по схеме присоединения Михаэля.
R2PH + МеСН=СНСО2Ме —> R2PCH(Me)CH2CO2Me
(25>
R = Н, Aik, Аг
Интересный вариант этой реакции изучен Кингом, показавшим [30], что присоединение дифенилфосфина к винилнзоцианиду протекает нормально с образованием (21), в то время как фенил-фосфин в этих условиях дает 1,3-азафосфол (22) (уравнения 26,27).
Ph2PH + CH2=CHNC —> Ph2PCH2CH2NC
(26>
PhPH2 + CH2=CHNC —>
(21)
PPh
(27>
(22)
20 Зак. ЗЭ5
609
Наиболее удобным вариантом реакции присоединения «в™ радикальное присоединение, инициируемое УФ-светом азпби /СЯ бутиронитрилом), органическими пероксидами, рентгеновским И3°' чами или излучением 6°Со. Этим путем получены многие фоХиГ включая трнфторметнлпроизводные типа (23) [311 (уравнениГ^’ Циклические третичные фосфины были синтезированы внутпимА’ лекулярным присоединением (уравнения 29, 30) или взаимодейст' вием диенов с первичными фосфинами за счет двукратного поиеоГ дпнения по Михаэлю (схема 31). р Loe’
_ /IV
(CF3)2PH + МеСН=СНМе ---------> (CF3)2PCH(Me)CH2Me
(23)
(28)
(29)
(30)
(31)
X = СО, О, СН2
Обработка циклооктадиен-2,7-она 1 экв фенилфосфина с последующим окислением приводит к смеси продуктов (24а) и (246) [32] (схема 32), а не к единственному стереоизомеру, как сообщалось ранее.
+ PhPH2
(246) Х= Ph,Y= О
Детально изучены реакции первичных и вторичных фосф/ соответствующих алкилфосфпдов с аф-ненасыщенными ^еТ0 [33]; отмечено, что в этом случае наблюдается только 1,2-прис
динеине, 1,4-присоединение не происходит. ,1Х
Стереохимия присоединения органических фосфидов щел металлов к ацетиленам зависит от наличия или отсутствия п Р ного или вторичного амина. Так, дифенплфосфпд лития пр/ г. няется к фенилацетилену в тетрагидрофуране, давая преиму
610
нно т/7анс-пзомер (25), тогда как в присутствии ’аминов обра-веется /{«с-изомер (26) (уравнение 33), вероятно, вследствие комплексообразования между амином и фосфидом. Днгпдрофосфорпны /27) могут быть получены прямым присоединением фенилфосфина 1 ппалкинильным соединениям в присутствии каталитических количеств амида лития в жидком аммиаке [34] (уравнение 34).
Ph2P\ /Н . R2nh Ph2Px zPh
С=С 4— Ph2PLi + HC=CPh ----------> q=q' (33)
H/ XPh fZ \н
(25) (26)
/R
X(C=C—R)2 + PhPH2 —> \ph
ж
(34).
(27)
x = CH2, o, s
10.2.1.5. Прочие методы получения фосфинов
Одним из наиболее простых методов создания Р—С-связи является реакция ароматических соединений с галогенфосфинами в. присутствии катализаторов — кислот Льюиса. При использовании большого избытка ароматического соединения хорошие выходы третичных фосфинов могут быть достигнуты даже с РС13, как показано на примере синтеза фосфина (28) (уравнение 35). Циклические фосфины, например (29), получены циклизацией хлорфосфинов в условиях реакции Фриделя — Крафтса (уравнение 36).
Дикалийциклооктатетраенид реагирует при —78°C с фенил-Дихлорфосфпном с образованием аддукта, который при повышении температуры изомеризуется в бициклический фосфин (30) (схема 37), способный превращаться в другие бициклические системы и в производное кубана (31) (схема 38) [35].
20*
611
(31)
Собственно фосфин РН3 может быть получен различными способами [36], однако наилучшим лабораторным методом является реакция фосфида алюминия с серной кислотой.
10.2.1.6. Синтез оптически активных фосфинов
Все более широкое применение комплексов фосфинов с металлами в гомогенном катализе и возможность асимметрического синтеза на таких катализаторах при использовании оптически активных фосфиновых лигандов вызвало, естественно, большой интерес к синтезу оптически активных фосфинов [37].
Значительный прогресс в этой области был достигнут благодаря изящной работе Хорнера и сотр., впервые получивших оптически активный третичный фосфин [29] расщеплением фосфониевой соли методом Мак-Ивена с последующим электрохимическим восстановлением. Позднее было показано, что фосфониевые соли могут быть превращены в фосфины через промежуточные фосфиноксиды и последующее их восстановление силанами. Использование силанов (см. разд. 10.2.1.3) приобретает все более важное значение, поскольку оптически активные фосфиноксиды в большинстве случаев более доступны [38].
Оптически активный дифосфин (32)—прекрасный лиганд при асимметрическом гидрировании на родиевых катализаторах — был получен восстановлением соответствующего бис(фосфиноксида) трихлорсиланом [39]. Использование в этой реакции в качестве основания трибутиламина вместо бопее распространенного триэтпл-амина позволяет повысить выход и уменьшить образование мез°' (32) (уравнение 39).
«12
Восстановление третичных фосфннсульфидов LiAlH с полным сохранением конфигурации 1401 Alrt4 протекает
вуюших оксидов которые полностью раце^изуются"6 °Т СООТВеТст-
Классическии метод расщепления рацемГтпЛ п
Поупом, —так называемый метод полпКинии ’ предложенный был применен к трпарплфосфпнам (б7Х " / количеств [40а] -формой и (+)-камфор-10-сульфок1,слотой₽ (поГо\ФоОС‘|;"НОВ паРа' осаждение кристаллической фосфониевой сопи п ' вызываег НЫЙ фосфин при этом может быть выделен в, ' Оптическ" актив-а его энантиомер регенерирован из соли (схема 40)°ЧН°Г° раствоРа>
2(±)-Аг‘-Р-Аг2
Аг3
1.(НСНО)Л ---------->
2. RSO3H
Аг2 Г
Ar'-P-CH2OH "O3SR 4- Н-Аг’-Р-Аг2
Аг3 Аг3
EtaN (40)
(+)-Ar’-P—Ar3 + RSO3 +NHEt3
Аг2
Описано частичное расщепление рацематов третичных фосфинов с использованием асимметричных комплексов палладия(II) [41] и платины (II) [42].
Для асимметрического восстановления рацемических фосфино-ксидов (33) и (34) применяли хиральные аминоаланы; этот метод представляется многообещающим, однако оптическое вращение ([«]d) фосфина, полученного из (33), необъяснимо менялось (от —32,1 до —14,6°) в зависимости от условий реакции [43].
O=PPh
’ R*
HN—Al
I Н
(41)
(±)-(33)
н
I
R* = (+)-Ph-C-।
Me
Me
Pr^P=O
Ph
CH2PPh2
613
В катализаторах асимметрического гидрирования часто исппгт зуют фосфиновые лиганды, не содержащие хирального атома *ос фора, ио имеющие хпральные элементы в других частях молекулы' Наиболее распространенным реагентом этого типа является соедп некие (35), получаемое реакцией соответствующего дптозилата с дпфенилфосфидом натрия [44].
Оптически активный ментилдифенилфосфин может быть полу чен реакцией неоментилхлорида с дифенилфосфидом натрия [18] или взаимодействием дифенилхлорфосфина с конфигурационно стабильным реактивом Гриньяра, полученным из ментилхлорида [45]
Описан метод определения оптической чистоты фосфинов, вклю чающий кватерннзацию фосфина оптически активным бромидом с последующим анализом соотношения диастереомеров в пол;/ ченной смеси методом спектроскопии ПМР [47].
10.2.1.7. Синтез полидентачных фосфорных лигандов
Исследования в области катализа, соединений пентакоордини-рованного фосфора и реакций окислительного присоединения ме-таллоргаиических соединений, содержащих фосфиновые лиганды, стимулировали изучение фосфинов с различной донорной основностью и длиной хелатной цепи, а также соединений с различными донорными атомами.
До недавнего времени большую часть полифосфинов получали взаимодействием органических полпгалогенидов с диалкил- или диарилфосфидами щелочных металлов, как показано на примере относительно простого синтеза «дифоса» (36) (уравнение 42) [47] Использование этого метода для получения более сложных три- и тетрафосфинов встречает, однако, некоторые трудности, связанньк-с получением подходящих органических полпгалогенидов или с не~ полнотой реакции этих полпгалогенидов с фосфидами [48]. Взаимодействие моноалкилфосфпдов щелочных металлов с днгалогени-дамп приводит к вторичным дифосфинам (37) только в том случае, когда углеродная цепь между двумя атомами галогена содержит не менее трех атомов углерода (уравнение 43); реакция моноал-килфосфидов натрия с 1,2-дихлоралканами приводит к фосфиранам (38) (уравнение 44) [49].
2Ph2PLi 4-С1СН2СН2С1 —> Ph2PCH2CH2PPh2 '42)
(36)
2RHPM + Х(СН2)„Х —> RPH(CH2)„PHR (43)
(37)
RCHC1CH2C1 ч-2NaPHR> —> RHC СН2 4- R! РН2 4- 2NaCl <4
PR1
(38)
Наилучшим методом получения простых бидентатных ных лигандов является реакция фосфитов или фосфонито
614
анами с последующим восстановлением LiAlH4, как бромал« пимере синтеза 1,2-бис(фосфино)этана (39) (ур зано на
н11е 4t>) L5UJ' О О
II II LiAlH,
2(EtO)3P + BrCH2CH2Br —> (EtO)2PCH2CH2P(OEt)2 ---------*
пока-авне-
—> Н2РСН2СН2РН2
(39)
(45)
Фраг.
образом, благодаря усилиям Кинга и сото [511 рР°СЛа’ Равным чевой стадии они использовали катализномем™^ Качесгве клю-Ц11Ю присоединения фосфинов, содержащих сГязирВа{?ЯМн Реа«-фосфинам, впнилфосфонатам или вини nihnerhu. д’ к ВИНил’ 2ения 46-48). Продукты этих реакций пш 1 лСуЛьфидам <УРав-„аВ.™валНсьД1А1Н,. Введение в синтетичесиугсXT™ тельно устойчивого винилднметилфосфинсулвфид? г/то У Т“’ ценно, поскольку метилированные тпеХипЛ ф Д1 ос°бенно дом получаемые другим/ меТотом/₽™ ” ^""'^Ф""ы' ' тру. ’ Udjin теперь легкодоступны
ph2PH + Ph2PCH=CH? Р.р„_и
0 Ph2PCH2CH2PPh2 (46)
Ph2PH + (изо-ргО)2рсн=сн2 Ph рсн ГИ du
2. LiAiH4 нп2РСН2СН2РН2 (47)
S
РИзРН + Ме.РСН-СН,
2. LiAiH3 ип2НСН2СН2РМе2 (48)
н2рсн2с6н4сн2рн2 + рь2рсн—сн2 _-iper'DUU у
—► (Ph2PCH2CH2)2PCH2C6H4CH2P(CH2CH2PPh2)2 (49)
(40)
I. Br(CH2)nBt ----------------------
2. NaAlH2(OCH2CH2OCH3)2
(50)
О о
PhP~(CH2)m— PPh I i
OPr-азо ОРг-азо
HSiCls
(CH2)m
Php/ /PPh (CH2)„
n 9 Ю, 12; in = 8, 10
(41)
содеп’СФИНЫ Co связью P—H (см. уравнение 47), а также фосфины, к винЖа1ВДе б°лее одной связи Р—Н, могут сами присоединяться илпроизводным фосфора; при этом образуются лиганды.
615
например (40), содержащие большое число атомов фосфора нение 49) [53]. С помощью этого метода можно получать полил В’ финн, содержащие первичные, вторичные и третичные атомы Т°С' Г гы 1 Фос-
фора [54J.
Интересный макроциклический дифосфин (41) был ПОЛУ1 восстановлением соответствующего фосфинокспда, образующее при взаимодействии диизопропилполиметиленфосфинатов с пол^ метилендибромидами в присутствии алюмогидрцдбис(2-метпкг« этокспд) натрия [NaAlH2(ОСН2СН2ОМе)2 Red-al] при высоком разбавлении (схема 50).
10.2.2. СВОЙСТВА ФОСФИНОВ
Фосфины обычно обладают характерным неприятным запахом. Специальных исследований, посвященных токсичности первичных, вторичных и третичных фосфинов, не проводилось, хотя считается, что в этом отношении они сходны с РН3, который чрезвычайно токсичен. Максимально допустимая безопасная концентрация РН3 в воздухе составляет 0,3 • 10_4% [55]; концентрация РН3, равная 0,05 мг/л, при экспозиции в течение 1 ч считается смертельной для взрослого человека. Инсектицидная активность трифенилфосфина не слишком высока в сравнении с паратионом, однако низшие члены ряда фосфинов несомненно могут вызвать отравление, поэтому при работе с ними необходимо соблюдать осторожность.
Фосфины обладают высокой реакционной способностью и весьма легко окисляются. Первичные, вторичные или третичные алкил-фосфпны реагируют с кислородом столь энергично, что обычно воспламеняются при контакте с воздухом. Вследствие этого все операции с фосфинами должны проводиться в атмосфере азота или аргона.
10.2.2.1. Основность фосфинов [56]
В отличие от аминов первичные, вторичные и третичные ФоСФв_ ны сильно различаются по основности, причем основность в0^е стает с увеличением степени замещения (табл. 10.2.2). Хотя зн ния р/<а для Ме3Р и Me3N сопоставимы, основность самого фо Ф значительно меньше основности аммиака. Полагают, что это 0 няется различием в гибридизации атомов фосфора и аз°та- Пазующиеся при протонпрованпи катионы аммония и фосфонп чае дают 5р3-гибридизацией центрального атома, причем в от0. аминов гибридизация азота практически не изменяется пр» 0. нировании, тогда как переход фосфина в фосфониевую сол ^e^cT. вождается значительной регибрпдпзацпей атома фосфора, иноВ вием чего является снижение р/(а. Различие в основност ‘ об-и фосфинов объясняют также различной энергией сольват, аПцл
разующихся катионов аммония и фосфония (энергия со. ызь1вает резко уменьшается с увеличением радиуса катиона); не
616
ения, что энергия сольватации действительно в значительной С°епенп определяет относительные значения р/Са.
С Таблица 10.2.2. Значения р/(а аминов и фосфинов
Фосфин рДа Амин ’«a
,—-— РНз — 14 NH3 9,21
МеРНа —3,2 MeNH, 10,62
Me, PH 3,9 Me2NH 10,64
Ме3Р 8,65 Me3N 9,76
Как и в случае аминов, значения р7<а для данного класса фосфинов достаточно хорошо коррелируют с индуктивными константами заместителя (заместителей) о* [57]. При построении зависимости значений рЛ'а от суммы о*-констант заместителей видно что фосфины, как и амины, по основности подразделяются на
Рис. 10.2.1. Зависимость р/Са (в воде) фосфинов от суммы о*-констант заме-стителсй.
три группы: первичные., вторичные и третичные, причем зависимость р/(;| от суммы о*-коистапт заместителей для этих ipynn изображается в виде трех параллельных прямых (рис. 10.2.1). Наклон прямых в случае аминов больше, чем для фосфинов, что свидетельствует о большей чувствительности аминов к индуктивному
617
влиянию заместителей. Значение р/<а данного фосфина можно рассчитать по уравнению соответствующей прямой, если известны о*-константы заместителей. Зависимости между’этими величинами приведены в табл. 10.2.3.
Таблица 10.2.3. Зависимость между р/(а и с *
Амины '^а Фосфины
Третичные 9,61 — 3,30о* Третичные 7,85 — 2,67сг*
Вторичные 12,13 — 3,23с>* Вторичные 5,13 — 2,610*
Первичные 13,23 — 3,14ог* Первичные 2,46 — 2,64сг‘
Отклонение от предсказанных значений рКа считается результатом пространственных эффектов, как, например, в случае три-изобутилфосфина (см. рис. 10.2.1). Постоянство пространственных эффектов в первичных и вторичных фосфинах приписывается различной степени затрудненности сольватации соответствующих фосфониевых ионов. Необходимо отметить, что фенилфосфины всегда располагаются примерно на 0,5 единицы рДа ниже соответствующей прямой. Причиной этого явления могут быть, с одной стороны, большие пространственные требования фенильной группы по сравнению с алкильными, а с другой — наличие сопряжения неподеленной пары электронов фосфора с фенильной группой. В случае аминов этот эффект выражен гораздо ярче и составляет 2—4 единицы р/(а.
Для корреляции основности арилфосфинов с их структурой использовали также уравнение Гаммета [58]. Полученные результаты свидетельствуют, что пространственные эффекты обычно невелики. Вновь было подтверждено, что основность аминов в большей степени зависит от природы заместителей, чем основность фосфинов: для /шрп-замещенных анилинов р = 3,35, тогда как для ппрп-замещенных фенилдифенилфосфинов в том же растворителе р = 1,1.
10.2.2.2. Нуклеофильность фосфинов
Несмотря на меньшую основность, фосфины, как правило, являются более сильными нуклеофилами, чем амины. Фосфины легко реагируют с электронодефицитными центрами, однако могут взаимодействовать с электрононасыщенными центрами и с центрами с повышенной электронной плотностью.
В отличие от соответствующих реакций аминов скорость взаимодействия фосфинов с алкилгалогенидами, как и значения рКа, резко возрастают с увеличением числа алкильных заместителей: R3P > R2PH > RPH2 > РН3, причем Вп3Р > Ph3P. Обратный порядок изменения скорости, установленный для аминов, является следствием влияния пространственных факторов, которые гораздо менее существенны в случае фосфинов из-за боль-
618
го радиуса атома фосфора. Однако в ряду однотипных фос-Я1ннов пространственные затруднения могут влиять на скорости реакции [59], так, например: Bu3P > Рг3Р > Е1;3Р.
" реакции фосфинов с отрицательно заряженными центрами, например с кислородом или галогенами, также протекают значительно легче, чем в случае аминов. Отчасти это можно объяснить сравнительно низкой энергией отталкивания между фосфином и отрицательно заряженным реакционным центром, однако важнейшим фактором, обусловливающим легкость протекания та-ких реакций, является, несомненно, высокая энергия образующихся связей фосфора (энергии связей Р—О и Р—С1 составляют соответственно примерно 200 и 120 кДж/моль, что значительно выше энергий соответствующих связей азота).
10.2.2.3. Спектроскопия фосфинов
Алифатические фосфины проявляют слабое поглощение (1g с ;де де 2) в области 220—230 нм, тогда как ароматические фосфины имеют сильное поглощение при 250—270 нм. Полоса поглощения трифенилфосфина имеет более высокую интенсивность, чем полосы поглощения трифенилфосфиноксида в той же области спектра. Этот факт в сочетании с наличием слабого батохромного сдвига, сопровождающегося отчетливым гиперхромным эффектом при переходе к растворителю с меньшей полярностью, указывает на существование резонансного взаимодействия между неподелен-ной парой электронов атома фосфора и ароматическим циклом |6О].
Заместители оказывают незначительное влияние на положение полосы поглощения, хотя наличие n-МеО-группы вызывает слабое смещение полосы поглощения в длинноволновую область, а присутствие n-Cl-заместителя вызывает незначительный коротковолновый сдвиг; полагают, что этот эффект обусловлен стабилизацией основного и, соответственно, возбужденного состояний [56].
Колебательная спектроскопия первичных, вторичных и третичных фосфинов детально описана в литературе [61]. Частоты валентных колебаний связи Р—Н первичных и вторичных фосфинов лежат обычно в области 2280+10 см-1. Большая часть полос поглощения фосфинов в ИК-области спектра приходится на область «отпечатков пальцев», и отнесение этих полос представляет собой непростую задачу. Тот факт, что частоты колебаний связей Р—С лежат в области 770—650 см~* ограничивает их значение, поскольку соединения, не содержащие связей Р—С, также поглощают в этом диапазоне. Наличие связи Р—Ме в фосфинах характеризуется присутствием трех полос поглощения при 1420, 1295—1278'и 930 -840 см->, тогда как связь Ph—Р имеет узкие характеристические полосы поглощения при 1450—1425 и 1010 990 см-1.
619
Очень цепным инструментом для идентификации фосфинов оказалась спектроскопия ЯМР. Методы квантовой механики позволяют рассчитывать химические сдвиги 3|Р [62], однако предсказательная ценность этих методов в значительной мере ограничена, поскольку определение параметров, необходимых для расчета, зачастую представляет собой более сложную и утомительную задачу, чем определение самого химического сдвига. Положение сигнала того или иного фосфина в спектре ЯМР 3|р может быть достаточно точно предсказано с использованием приведенных ниже эмпирических уравнений.
Для первичных фосфинов
д = 163,5 — 2,5стр
Обычная область значений составляет 4-110-------[-160 млн-1.
Для вторичных фосфинов
6 = 99— 1,5ог£
Обычная область значений составляет 4-40--------[-100 млн-1.
Для третичных фосфинов
д = 62 — (аР
Обычная область значений составляет 4-5---------[-62 млн-1.
Значения ор для различных групп приведены в табл. 10.2.4.
Таблица 10.2.4. Значения с>р для различных групп
Группа 0Р Группа 0Р
NC —24,5 Et 4-14
Me 0 PhCH2 4-17
МечССН2 4-3 Ph + 18
СН>=СНСН2 4-9 изо-Pr +27
Ви 4-Ю трет-Ви +44
Константы спин-спинового взаимодействия P—Н характерис-
тичны для валентного состояния атома фосфора; так, для соединений фосфора (III) они составляют 180—225 Гц. Эти кон-
станты могут использоваться для стереохимического отнесения геометрических изомеров (см. табл. 10.1.4). Как было недавно показано, константа взаимодействия между ядрами 31Р и 1 С
в некоторых циклических фосфинах зависит от конфигурации атома фосфора. Углерод метильной группы, находящейся в а-поло-
Ме
! ,„Л\н
(42а) X— •> Y— Me; JpccHs-32 Гц
(42б) Х= Me Y= «7рссНз-О
620
ЖС))ПП К атому фосфора в фосфолене (42)
фосфором в транс-изомере (42a), тогда Д,г ВЗаим°Действует с т'аКОе взаимодействие отсутствует [64]. В ^с'изомере (426)
Ю.2.2.4. Инверсия фосфинов
бьпо недавно показано, значение барьера пирамидальной в фосфинах зависит от ряда структурных факторов, инвеРС1"нзу
ценных на примере аминов. Обычно барьер инверсии
Таблица 10.2.5. Энергетические барьеры (в кДж/моль) пирамидальной инверсии фосфинов
Фосфин (°C) Литература Фосфин AG+ (°C) Литература
СбНц х РМе
Рг/
Ph\
ХРМе р/
Ph\ /РМе
СН2=СНСН2/ ph\ /РМе трет-Ви '
п-МеОС6Н4.
/РМе Ph/
n-CF3C6H4x /РМе Ph/
149 (130) ‘
[66] -----PBu-трет 114(112) [67]
134(130) [66] /~Х 150(170) [67а]
PPh zph
135(130) [66а] Ас—р/ 81(110) [676]
\СНМе2
137(130) [66] Ac—P(CH2Ph)2 82 (118) [67в]
О
II
129 (130) [66] CF3C—P(CH2Ph)2 68 (55) [68в]
122(130) [66]
в ациклических фосфинах ^^^ д^/мольГи може^определяться напряжением, ьЮ,
а также взаимодействием неподелеиных ” ы аКтнвацпн
Для ряда фосфинов были определены параме^трь^^
термической рацемизации или равновес! 1 1 и фенильного Электроноакцепторные группы в п р - пнверсии. Простран-кольца в арилфосфинах понижают °ар р зна1ч11тельНОй роли ственные эффекты, по-видимому, не i р
621
в случае ациклических фосфинов, в то время как при пирамидальной инверсии в случае I-трет-бутил-2,2,3,4,4-пентаметилфосфета-на равновесие устанавливается в 104 раз быстрее, чем для его 1-метилыюго аналога [67].
В этих соединениях, однако, несвязывающее взаимодействие между метильными группами цикла и трет-бутильной группой у фосфора более существенно в основном состоянии, чем в плоском
переходном состоянии. Напряжение цикла обычно уменьшает скорость пирамидальной инверсии в пятичленных циклических фосфинах в сравнении с ациклическими соединениями.
Более низкое значение барьера инверсии в ацилфосфинах обусловлено, вероятно, повышением эффективности рл—рд-сопряжения не-поделенной пары электронов фосфора с карбонильной группой в плоском переходном состоянии по сравнению с эффективностью его в пирамидальном основном состоянии.
Влияние заместителей, хотя и незначительное, имеет
Рис. 10.2.2. Зависимость энергетического барьера пирамидальной инверсии от электроотрицательности.
близкое сходство с влиянием заместителей на пирамидальную инверсию в других системах. Была найдена линейная корреляция барьеров пирамидальной инверсии аминов, арсинов, сульфониевых солей, сульфоксидов и фосфинов [68].
В работе Мислоу показано, что основным фактором, определяющим значение барьера инверсии в фосфинах, является электроотрицательность заместителей, связанных с фосфором [6У], причем значение энергетического барьера удовлетворительно коррелирует с электроотрпцательностямп атомов, связанных с фосфором (рис. 10.2.2). Вычисленные значения энергетических барьеров показывают, что соединения фосфора (III) с хорошим! уходящими группами (с высокой электроотрицательностью) д[?лл< ны были бы быть конфигурационно стабильными, если бы бы найдены условия их выделения.
«322
10.23. РЕАКЦИИ ФОСФИНОВ
Многие реакции фосфинов можно описать, по крайней мере ервом приближении, как нуклеофильные реакции. Соединения I 1лора(Ш) помимо необычайно широкого диапазона нуклео-f льных свойств могут проявлять также и л-акцепторные свой-Ф” впервые обнаруженные в случае координационных соедпне-сТ» ’ фосфиновые лиганды, имеющие двойственную реакционную НпИособность, т. е, способные, с одной стороны, выступать о-доно-памн как основания Льюиса, а с другой — быть л-акцепторамн я счет обратной подачи электронов на вакантные Зс/-орбиталп фосфора, были описаны как бифпльные реагенты [70, 71]. Такие пиганды могут подавать электроны субстрату с образованием р-связп и одновременно принимать их от того же реакционного центра с образованием л-связи. Этот тип реакционной способности реализуется в большинстве характерных реакций соединений фосфора (Ш), хотя о-донорная н л-акцепторная стадии могут и не совпадать. Примерами этого могут служить реакции, приведенные в уравнениях 205 и 206, а также реакции фосфинов с дисульфидами. Многие реакции фосфинов, приводящие к образованию фосфорильной группы Р=О, протекают именно за счет процесса подобного типа, причем наблюдаемое направление реакции определяется прочностью образующейся связи.
10.2.3.1. Нуклеофильное замещение при насыщенном атоме углерода
Третичные фосфины обычно реагируют с алкилгалогенидами по механизму S\2 с образованием фосфониевых солей (см. разд. 10.2.5). Хотя этот вопрос подробно не исследован, можно считать, что влияние заместителей в этой реакции аналогично влиянию заместителей для других реакций, протекающих по механизму Sn2. Реакции этого типа легко протекают с первичными и вторичными алкилгалогенидами, однако третичные галогениды обычно в реакцию не вступают. Замещение при хиральном атоме углерода сопровождается обращением конфигурации, что согласуется с бимолекулярным переходным состоянием (схема 51).
R3
FT
R R3
„5+ I S- 4+ /
->r1p---C-—X-----►R3p-C^//R2 х (50
R2 R1 R1
Нейтральные фосфины способны раскрывать эпоксидный цикл с образованием промежуточного бетаина, аналогичного промежуточному продукту в реакции Виттита. Этот интермедиат может Распадаться по двум направлениям: либо с образованием илида 11 альдегида, либо давая фосфппоксид и алкен, причем последний
623
обладает, как правило, конфигурацией, противоположной конфи гурацпп исходного эпоксида (схема 52) [72]. 4
Модификация этой реакции стала удобным методом обращения конфигурации двойной связи в алкенах [73]. Обработка эпоксида дифенилфосфидом лития и метилиодидом приводит к алкену, конфигурация которого противоположна конфигурации исходного эпоксида Дальнейшим усовершенствованием метода явилось окисление первоначально образующегося соединения вместо его кватернизации, поскольку при этом в качестве побочного фосфорсодержащего продукта получается растворимый в воде ди-фенилфосфннат натрия (схема 53) [74].
I.PhsPLi
2. Mel
1. PhoPLi
R1 R2 R3 H
2. H2O2, Ac OH
R1 R2 R3 H
Ph2P=O OH
NaH -----►
ДМФ
Дифенилфосфид лития является достаточно сильным нуклеофилом, способным замещать алкокспгруппу в ненапряженны простых эфирах. Этот реагент особенно активен по отношени
624
цетиловым эфирам, в результате чего защитные метоксигруппы могут быть избирательно удалены в присутствии других эфирных групп даже при использовании четырехкратного избытка фосфида. Селективное удаление метокспгруппы в соединении (43) было использовано в полном синтезе трптерпена альнузенона (уравнение 54) [/5].
10.2.3.2. Нуклеофильное замещение в активированных алкенах и ацетиленах [76]
Как анноны соединений фосфора (III), так и нейтральные фосфины проявляют нуклеофильные свойства по отношению к поляризованным кратным углерод-углеродным связям, однако в случае нейтральных фосфинов кратная связь должна быть активирована электроноакцепторными группами. Первичные и вторичные фосфины пли фосфиды реагируют обычным образом с образованием фосфинов за счет переноса протона или протониро-вания в процессе последующей обработки (см. разд. 10.2.1.4).
Третичные фосфины обратимо присоединяются к алкенам, давая бетаины (44), которые в дальнейшем могут превращаться в плпды путем внутримолекулярного переноса протона, а в присутствии доноров протонов или алкилирующих агентов могут образовывать фосфониевые соли. Если исходный алкен содержит хорошую уходящую группу, то реакция может протекать по схеме присоединения — отщепления (схема 55).
---> R3P=CZCH2Y нх +
---> R3PCHZCH2Y X'
R3P + ZCH=CHY R3PCHZCHY - [ R' (55)
(44) > R3PCHZCHY X'
---> R3PCH=CHY Z*
Y = электроноакцепторная группа; Z = галоген
Детально изучена полимеризация акрилонитрила и эфиров акриловой кислоты в присутствии третичных фосфинов, которые, по-видимому, инициируют анионную полимеризацию указанных мономеров за счет образования интермедиатов бетаинового типа [77]. В некоторых случаях удается остановить реакцию на стадии димера, как, например, при присоединении эфиров фумаровой кислоты к акрилатам, катализируемом трициклогексплфосфи-ном (уравнение 56). Третичные фосфины являются очень эффективными катализаторами в реакциях присоединения по Михаэлю, причем в данном случае они играют роль нуклеофильных агентов, а не оснований [78].
В последние годы значительное внимание привлекают реакции фосфинов с ацетиленами. Предметом многочисленных исследований
625
CH2=CHX+R3P —>
r3pch2chx
cx-ch2pr3 I
—> CHCO2R‘
CH2CO2R‘
, chx-ch2pr3
r‘o2cch=chco2r‘ I
--------------> CHCO2R‘
'CHCO2R‘
CX=CH2
I
—* CHCO2R‘ R3P
CH2CO2R‘
явилась сложно протекающая реакция трифенилфосфина с диметилацетилендикарбоксилатом. Первоначально образующийся аддукт 1 : 1 может быть зафиксирован при действии СО2 (или SO2) в виде бетаина (45) (уравнение 57). При избытке диметил-ацетилендпкарбоксилата основным продуктом является, по-видимому, 2/7-фос (V) фол (46), при этом образуется также циклопен-тенилиденфосфоран (47). Реакция, вероятно, протекает через промежуточный аддукт 2:1 (48), который может быть выделен при —50 °C (схема 58) [79].
про-фосфпноксиду (49) путем гглерод в первона-(уравнение бУ;
Реакция трифенилфосфина с фенплацетиленом и вод^ текает не столь сложно п приводит к (’."] миграции фенильной группы с фосфора па чально образующейся вннилфосфониевой соли
626
[80]. Более подробно эта реакция обсуждается в разд. 10.2.6.1. На основе этой реакции разработан простой и экономичный метод получения 1,2-дпдейтерпрованных алкенов типа (50) взаимодействием дизамещенных алкинов с трифепилфосфином в пписут-ствпи D2O (уравнение 60) [81].
'ОН ° Ph
Н2о + II I
Ph3P + PhC=CH -> Ph3PCH—CHPh -—> Ph2P—CHCH2Ph (59)
(49)
d2o D\ /R1
Ph3P + RC=CR' >- Ph3PO 4- C=C (60)
RZ
(50)
Трифенилфосфин взаимодействует с дегидробензолом с образованием бетаина, который при действии метилиодида превращается в фосфониевую соль (схема 61). В том случае, если в фосфине содержится заместитель, несущий водород на а-углерод-ном атоме, происходит перенос протона с образованием илида (схема 62) [82].
Ph2PMe +
(62)
10.2.3.3. Нуклеофильное присоединение к карбонильным соединениям [83]
Аддукты, образующиеся в результате присоединения фосфинов к несопряженным карбонильным соединениям, мало устойчивы из-за отсутствия возможности делокализации отрицательного заряда, поэтому подобные реакции присоединения, которые теоретически могут протекать как по кислороду, так и по углероду карбонильной группы (уравнение 63), обратимы, причем равновесие сильно смещено в сторону исходных продуктов. Образование стабильных продуктов возможно, однако, в следующих случаях: а) при использовании первичных или вторичных фосфинов, способных к переносу протона; б) при использовании карбонильных соединений, содержащих хорошую уходящую группу, например хлорангидридов кислот; в) если возможна дальнейшая реакция промежуточного бетаина со второй молекулой карбонильного
627
соединения с образованием стабильного пентакоординационного соединения (см. разд. 10.4).
+ I \ + -/
R3P—С—О R3P + /С=О R3P—О— С/ (63)
Соединения фосфора (III), содержащие связь Р—Н, обычно реагируют с альдегидами и кетонами с образованием продуктов содержащих фрагмент Р—С—О, как, например, в реакциях дифенилфосфина с бензальдегидом или кетеном (уравнения 64 65) [84].
он
P112PH+PhCHO Ph2P—СН—Ph (64)
О II
Ph2PH + СН2=С=О —> Ph2PCCH3 (65)
Третичные фосфины обычно атакуют атом углерода карбонильной группы, однако первоначальные Р—С—О-аддукты всегда изомеризуются в соединения с фрагментом Р—О—С. Атака фосфинов по кислороду не осуществляется даже при наличии сильных электроноакцепторных групп у атома углерода. Дифенилфосфин реагирует с гексафторацетоном с образованием Р—С—0-аддукта, т. е. по атому углерода, а не по кислороду, как сообщалось ранее [85]. Образующийся при этом спирт чрезвычайно легко окисляется, давая соединение (51), которое изомеризуется в наблюдавшийся ранее продукт (уравнение 66).
ОН О ОН
I II I
Ph2PH + (CF3)2C=O —> Ph2P—C(CF3)2 —> Ph2P—C(CF3)2 —> (51)
О
—> Ph2P—О—CH(CF3)2 (66)
В цикле работ Исслайба описан синтез циклических соединений фосфора путем катализируемой кислотами конденсации подходящим образом замещенных фосфинов с карбонильными соединениями (уравнение 67) [86]. В случае близкой реакции с фенилизотиоцианатом (уравнение 68) промежуточный продукт (52) удается выделить.
о (СН2)„
PhPH(CH2)„XH 4- RCR1 —> phP\(67)
CRR1
X=N, О, S; п = 2, 3; R = Н, Aik; R1 = Aik, Ph
PhPH(CH2)2SH 4- PhNCS
PhP(CSNHPh)CH2CH2SH
(52)
628
Хотя реакции фосфинов с карбонильными соединениями обычно протекают путем атаки по карбонильному атому углерода, возможны и исключения — когда атака по кислороду приводит к бе-таяНу; анионный центр которого стабилизирован резонансом. Прекрасным примером такого типа взаимодействия является реакция образования бетаина из трифенилфосфина и циклопентади-еН0На (53) (схема 69).
(69)
10.2.3.4. Нуклеофильная атака на галоген [87]
Соединения фосфора (III) энергично взаимодействуют с галогенами с образованием аддуктов, имеющих в кристаллическом состоянии ионную структуру. Более высокая степень диссоциации наблюдается для аддуктов иода; аддукты хлора проявляют низкую электропроводность в ацетонитриле и существуют, по-види-мому, преимущественно в пентакоординационной форме (уравнение 70).
Третичные фосфины быстро окисляются в соответствующие фосфиноксиды при действии алкилгипогалогенитов, причем, как было показано Денни [88], первоначальная атака фосфина направлена на атом галогена. Галогенфосфониевые соли, которые образуются при взаимодействии третичных фосфинов с гипохлоритами, содержащими группу О—С1 в голове моста, перегруппировываются в стабильные алкоксифосфониевые соли. При проведении реакции в метаноле оптически активные фосфины превращаются в фосфиноксиды с обращенной конфигурацией у атома фосфора (схема 71).
R3P + X—X R3P—X X' R3PX2 (70)
R3P + Cl—О—R —> [R3PCI OR] — R3POR СГ (71)
|меОН
R3PO + MeCl + ROH
Известны многочисленные примеры нуклеофильных реакций фосфинов с соединениями, содержащими «положительный галоген», т. е. с такими соединениями, в которых образующийся при катионоидном отрыве галогена карбанион стабилизирован резонансом (например, в случае а-галогенкетонов, сложных эфиров, сульфонов и т. д.) или индукционным эффектом заместителей (полигалогенпроизводные углеводородов, например СС1«). Струк-
629
тура образующихся продуктов зависит от условий проведения акций, поскольку существует несколько возможных путей рас' пада первоначально образующейся ионной пары. Например за' мещение иона галогена при фосфоре карбанионом приводит к фосфониевой соли, идентичной продукту прямого нуклеофильного замещения при атоме углерода (схема 72). Участие ионной пары в таких аномальных реакциях можно продемонстрировать, проводя процесс в водных растворителях, которые вызывают быстрое разложение таких промежуточных продуктов с образованием фос-фпноксида, как это показано на примере взаимодействия трп-арилфосфпнов с а-бромсульфоиами (54) во влажном ДМФ (схр ма 73) [89]. 1
R3P + X-CH2R‘ —> [R3PX CH2R‘] —> R3PCH2R‘ X- (72) Аг1 Аг1
| Н2О, ДМФ + I I
АгзР + XCHSO2Ph -----------> Аг3РХ CHSO2Ph —
медленно _ ‘ gi.
(54)
—> Аг3РО + НХ -ф Ar'CH2SO2Ph (73)
При взаимодействии с а-галогенкетонами образующийся амбидентный енолят-анион может атаковать фосфониевый центр как атомом кислорода, давая енолфосфониевую соль, так и углеродом, давая [3-кетофосфониевую соль, идентичную продукту прямого нуклеофильного замещения у атома углерода (схема 74) [90]. Соотношение продуктов зависит от конкретной реакции, однако решающее влияние оказывает природа галогена. При действии а-бромкетона на оптически активный фосфин продуктом реакции является енолфосфониевая соль с обращенной конфигурацией у атома фосфора, что свидетельствует о ее образовании за счет рекомбинации ионной пары. В аналогичной реакции с а-хлор-кетоном образуется [З-кетофосфонпевая соль, в которой конфигурация у атома фосфора сохраняется, что является результатом прямого замещения у атома углерода.
RsPCH2COR2 +---R3P + XCH2GOR2--► R3PXCH2—CR2----►
---> P3P—o—
^CH2
Реагенты, включающие тетрагалогенметан, обычно CCU и третичный фосфин, находят все более широкое применение: Р
паративной химии в качестве мягкого реагента для галоге Р ния и дегидратации [91, 92]. Общий механизм таких превр оказался, однако, гораздо более сложным, чем п р чально представлялось, поскольку с реакциями «реагента» у «30
стратом конкурируют собственные реакции «реагента». Достаточно надежно установлено, что первоначальная атака направлена на атом галогена, однако фосфониевая соль (55), которая может быть выделена при реакции трифенилфосфнна с СС14, в отсутствие других реагентов быстро взаимодействует со следующей молекулой фосфина, давая стабильный фосфоран (схема 75) [93].
г 8+ S-
РЬ3Р + ссц. > [Ph3p---Cl—-СС13] -----> Ph3PCCl3 СГ—>
(55)
--►Ph3P=CCl2
Ph3P
+ Ph3PCl2------►
Cl I PfloP^C^PPhg
+
СГ
(75)
В настоящее время признано [94], что существуют два направления взаимодействия субстрата с «реагентом». Прямая реакция субстрата RXH, содержащего подвижный водород, с промежуточным биполярным комплексом приводит к хлороформу и фосфониевой соли (56), которая при разложении дает алкилхлорид (схема 76). С этим процессом конкурирует реакция прямого хлорирования субстрата трифенилдихлорфосфораном (уравнение 77). Выделяющийся при этом НС1 реагирует с находящимися в реакционной среде хлор- и дихлорметилентрифенилфосфоранами, давая выпадающие в осадок соли (58) и (57) соответственно (уравнения 78—80). На долю этих последних реакций, в результате которых не образуется хлороформ, приходится более 95% получающегося алкилхлорида. Вследствие этого более высокие выходы целевых алкилхлоридов достигаются при повышении соотношения трифенилфосфин : тетрахлорметан до ~ 1,5 : 1.
б+ 6-
RXH+ [Ph3P—С1—СС13] _снс-^ РЬзР—X—R С1 —>
(56)
—> Ph3P=X 4- RC1 (76)
Ph3PCl?+RXH —> (56)+НС] (77)
Ph3P=CCI2 + НО —> Ph3PCHCI2 Cl* (78)
(57)
(57) + Ph3P —> Ph3PCI2 + Ph3P=CHCl (79)
Ph3P=CHO + HCI —> Ph3PCH2CI СГ (80)
(58)
Подобные реакции протекают в столь мягких условиях, что Даже такие лабильные спирты, как углеводы, могут быть легко превращены в соответствующие хлориды. Применение трпс(днме-тиламино)фосфина существенно упрощает процесс выделения продуктов, поскольку в этом случае в результате реакции образуется
631
водорастворимый фосфиноксид. Реакции синтеза галогенидов сопровождаются обращением конфигурации у атома углерода даже в случае субстратов, обычно склонных к рацемизации. И
С помощью этих реагентов можно осуществить замещение спиртового гидроксила и на другие нуклеофилы [95]. С этой целью промежуточную алкоксифосфониевую соль обычно переводят в более стабильный гексафторфосфат, при обработке которого необходимым нуклеофилом образуется продукт замещения (схема 81). Возможна избирательная активация одной гидроксильной группы в пропандиоле, а также селективная активация различных гидроксильных групп в углеводах. Подобная активация и замещение протекают без перегруппировок даже в случае систем, •склонных к ним, таких, как, например, неопентильная.
RR‘CHC1 + (Me2N)3PO
|C|-
RR’CHOH + (Me2N)3P + СС14 —► RR'CHOP(NMe2)3 —> + x~
—-> RR1 CHOP(NMe2)3 PF6’ ---> RR'CHX + (Me2N)3PO (81)
При введении в реакционную смесь основания для связывания HCI возможно протекание реакций дегидратации. Первым примером таких превращений явилась реакция дегидратации амидов в нитрилы (уравнение 82). Описан ряд процессов подобного типа [91]. получение изоцианидов из монозамещенных формамидов, карбодиимидов из мочевин, азиридинов из p-аминоспиртов и другие. Эти реагенты были использованы также для получения ангидридов из карбоновых кислот и амидов из карбоновых кислот и аминов. Последняя реакция детально изучена с целью ее использования в пептидном синтезе [96].
СГ R
+ - r‘CONH2 + | Et3N
R3P + СС14 —> R3PCI СС13 rR3P—О—C=NH --------------->
—U riU I3
(59)
—> R3PO + R‘C=N <82>
Все реакции этого типа протекают через стадию одновременного присоединения фосфина, тетрахлорметана и третичного азотистого основания к дегидратируемому соединению. Отщепление элементов воды происходит ступенчато. Первоначально о р зуется аддукт, например (59), который в дальнейшем распадает , давая хлористый водород, фосфиноксид и продукт дегидрат •
Недавно была предложена новая модификация этого м^™^.еле использование фосфинов, закрепленных на полимерном " [97]. Поскольку в данном случае процесс выделения Р эт0Т сводится к простым операциям фильтрования и упарп метод может найти широкое применение.
632
10.2.3-5. Нуклеофильные реакции с кислородом и серой [87]
Многие соединения, содержащие кислород п серу, взаимодействуют в мягких условиях с соединениями фосфора (Ш); движущей силой таких реакций является высокая энергия образующихся связей Р=О и P=S. В последние годы эти реакции, и в особенности их применение в синтезе для модификации субстратов путем вытеснения кислорода или серы, привлекают большое внимание п описаны в ряде обзоров [92, 98]. Несколько примеров таких превращений представлено уравнениями 83—90. Более эффективными реагентами по сравнению с фосфинами являются,, однако, фосфиты, поэтому подобные реакции подробнее рассмотрены в разд. 10.3.
R‘O-OR' + R3P —> R’OR1 + R3P=O
C=,8O +
(83)-
(84>
О
Оз —-► R3p/ /О —> Д^О2 oz
ArNO2 —> ArN:
R‘S—SR' + R3P —-> R‘SR, + R3P=S
R1SSO2R'+R3P —> R‘SO2R'+ R3P=S
(85)
(86)
(87)
(88)
(89)
(90)
He вызывает сомнения, что первоначальная атака фосфина направлена на гетероатом; следует отметить возможность взаимодействия промежуточного продукта с дополнительно введенным реагентом, особенно в случае реакций с сульфидами. Так, например, подробно исследованная реакция восстановления дпал-кчлдпсульфндов трифенилфосфипом (уравнение 91) в присутствии воды приводит только к незначительным количествам трифе-нилфосфинсульфида [99].
Возможность реакции такого промежуточного продукта с карбоновыми кислотами позволяет использовать 2,2/-диппрпдилди-сУльфид и трифенилфосфин для конденсации карбоновых кислот
633
Ph3P + RSSR
PlhPSR + RS'
|h2o
(91)
Ph3PO+ RS’ + H+
в мягких условиях [100]. Эти реагенты особенно полезны в пептидном синтезе и для превращения w-гидроксикарбоновых кислот в макроциклические лактоны в нейтральных апротонных условиях. Последняя реакция подробно изучена Кори [101], который показал, что первоначальным продуктом является S-(пиридил-2)-тпокарбоксилат (60), далее циклизующийся в макроциклический лактон (уравнения 92, 93). Эта реакция успешно использовалась им в синтезе макролидных антибиотиков, например брефельдина (61) (уравнение 94). Более эффективным реагентом по сравнению с 2,2'-диш1рндплдисульфидом является, по-видимому, имидазолилдисульфид (62) [101].
10.2.3.6. Прочие реакции фосфинов
Хорошо исследованы реакции соединений фосфора (III) с с° единениями азота. Фосфины обычно реагируют с азидам атому азота, давая фосфазены (уравнение 95), которые в гих случаях могут быть выделены Для стереоспецифичн теза азиридинов использована реакция 2-подалкилазидо
634
тоифенилфосфином (уравнение 96), на первой стадии которой Спотекает, по-видимому, атака фосфина по атому азота азида И02] -
' ArCON3+ РЬзР
Me N3 Me N~ N=NPPh,
„ \ / Ph3P Н///Л ,/
Н//'"-С—С н -----► с—_
/ Ме
ArCON=N—N=PPh3 —> ArCON=PPh3 + \2 (95)
1“^-РРЕ3
А^С~С^ц (96>
Me Me
I
Me
Дпэтилазодикарбоксилат (ДАД) и третичные фосфины образуют комплексы, способные активировать гидроксильные группы. При такой активации реакции замещения гидроксила протекают в мягких условиях п не сопровождаются перегруппировками, как показано на примере взаимодействия частично ацилированного глицерина (64) с карбоновыми кислотами в присутствии трифенилфосфина и ДАД (уравнение 97) [103]. Предложен простой, метод получения простых алкилариловых эфиров [104] путем обработки спирта фенолом (или енолом) в присутствии третичного фосфина и ДАД. Эта реакция протекает с обращением конфигурации у спиртового атома углерода. Комплексы третичный фосфин— ДАД могут использоваться также для активации карбонильной группы; примером может служить катализ реакции переэтерификации, протекающей в мягких нейтральных условиях. В этом случае фосфиновый комплекс действует, по-видимому, как. бифункциональный катализатор (см. схему 98) [105].
СН2ОСОС17Н35
CH2OCOC17H3S
но-с—и
rco2h
-------------------> H-C—OCOR PhaP. EtO2CN=NCO2Et ,
(97).
CH2OCOPh
(64)
CH2OCOPh
CO2Et
+ /N. _ CO2Et + CH2(CO,Etl
Ph3P^ Л
N—CO2Et ( XNCO2Et
,C
EtO2CCH2 I Me
OEt
Ph3? I
~O OMe
ЕЮгССНг"7 OEt
CO2Et
EtOH + CH2
CO2 Me
(98)
Соединения фосфора (III) вступают в разнообразные радикальные реакции [106]. Присоединение к алкенам фосфиновых радикалов, образующихся путем отрыва водорода от первичных или вторичных фосфинов, уже обсуждалось выше (см. разд. . . . )•
Фосфораиильные радикалы, например (65), получаемые при присоединении радикала к соединению фосфора (111), ооы
635
реагируют по одному из двух возможных направлений: или a-разрыв, или p-разрыв с образованием связи P=S) (схема 99).
происходит
Р=о (или
н-Ви3Р + трет-ВиО •
—► н-ВизРОВи-трет
(65)
—> «-Ви.+н-Ви2РОВи-Грет
н-ВизР=О + трет-Ви •
О ₽Li
V- Р-. ---*
Мер/ XCH2Ph
R—Р—CH2Ph
Me Ph
(66)
Ph
Реакции нуклеофильного замещения в третичных фосфинах под действием литийорганическпх реагентов (схема 100) сопровождаются полным обращением конфигурации у атома фосфора. Эти реакции можно, по-видимому, рассматривать как классическое S.vS-замещение, при котором атака нуклеофила направлена со стороны, противоположной уходящей группе, а в предполагаемом анионном переходном состоянии (66) отсутствует псевдовращение [107].
10.2.4. ПОЛИФОСФИНЫ [108]
Соединения, содержащие связи Р—Р, можно разделить на три группы: элементный фосфор, линейные полифосфины и циклические полифосфины. Линейные полифосфнны представлены в основном дифосфинамп; три- и высшие линейные полифосфины известны, однако в большинстве случаев эти соединения нестабильны. Причиной их термической неустойчивости является относительно низкая энергия Р—P-связи (~200 кДж/моль).
Поскольку каждый атом фосфора имеет как неподеленную пару электронов, так и вакантные З^-орбптали, очевидно существует возможность для л-связывания; о наличии некоторой делокализации электронов свидетельствует, по-видимому, интенсивное ультрафиолетовое поглощение этих соединений в УФ-области спектра по сравнению с монофосфпнами. Делались попытки измерения барьера вращения вокруг связи Р—Р в дифосфпнах, поскольку этот параметр более точно отражает природу связи между атомами, однако данные об энергии такого вращения пока отсутствуют [109]. Дифосфнны типа (67) могут существовать в мезо- и (+)-диастереомерных формах, обладающих раз личными спектрами ПМ.Р; однако при повышении температуры эти спектры превращаются в один усредненный спектр. Мия низм взаимопревращения диастереомеров включает инверс конфигурации фосфора и вращение вокруг связи Р Р (сх
636
101). Какая из стадий процесса является медленной, не ясно, однако барьер инверсии в дифосфинах значительно ниже, чем в’слу чае аналогичных монофосфинов, что является, вероятно следст впем стабилизации переходного состояния за счет йд-связы вания [109].
Ме
инверсия
(67)
Ме МС.
R вРаш-ени<-. р Л /'
—Р. ч- * р „
\ /
R
(101)
Наиболее широко применяемыми методами для создания р—P-связи являются дегидрогалогенирование (обычно под действием третичного амина) и конденсация в присутствии активных металлов (щелочные металлы, ртуть или магний) (уравнения 102— ПО). При получении циклополифосфинов реакцией дегидрогалогенирования первичные фосфины могут быть генерированы in situ путем восстановления половинного количества дихлорфосфина действием LiAlH4. Выходы получаемых таким образом продуктов обычно высоки, однако реакция может протекать неоднозначно. Например, при взаимодействии этилфосфина с этилдихлорфосфи-ном одновременно образуются четырех- и пятичленные циклопо-
лифосфины.
Ph2PH + ClPR2 —> Ph2P—PR2 + НС1 (102)
R2PC1 + 2Na —> R2P—PR2 + 2NaCl (103)
180 °C
Ph2P—CONHPh -----> Ph2P—PPh2 + CO + (PhNH)2CO (104)
R2P(S)P(S)R2+Bu3P —> R2P—PR2 + Bu3PS (105)
R2PX + R2PLi —> R2P—PRj + LiX (106)
PhPH2 + PhPCl2 —> (PhP)5 + HC1 (107)
PhPX2 + M —> (PhP)„ + MX (108)
MePCl2 + Li —> (MeP)5 + LiCl (109)
phPH2 + (Me2N)2PPh —> (PhP)n + Me2NH (U0>
Установление строения циклополифосфинов является
достаточно сложной задачей. Циклополифенилфосфин, например, в зависимости от метода получения существует в четырех различных формах, отличающихся, по-видимому, размером цикла; чаще других образуется циклопентафенилфосфин. Структурный анализ таких соединений, однако, значительно упростился после недавнего сообщения об использовании спектроскопии ЯМР ’Р ДлЯ определения размера цикла [НО]. Для тетрамера (РРЦ в спектре Зф—{*Н} содержится единственный сигнал с химическим СД^И' гом в области 4-60----[-70 млн-1, тогда как пентамеры (К /5
имеют сложные спектры ЯМР. 31Р с сигналами в области 1о -г-т- —20 млн-1.
637
Полифосфнны чрезвычайно чувствительны к действию окислителей; низшие члены ряда воспламеняются на воздухе. Цикло-пентафенплфосфпи в твердом состоянии относительно устойчив' однако достаточно быстро окисляется в растворе.
Результатом большинства реакций полифосфинов, например реакций галогенирования, металлирования и гидролиза, является разрыв Р—Р-связн (уравнения 111 — 115). Металлирование цик-лополифосфинов приводит к сложной смеси тетра-, три- дц- и монофосфидов. Обсуждение этих реакций выходит за рамки этой главы; подробные данные можно найти в обзоре Майера [108]
О О
Il II
R2P—PR2 + O2 > R2P—PR2 (Щ)
R2P—PR2 + 2М —> 2R2PM (1I2)
R2P—PR2 + X2 —> 2R2PX (из)
R2P— PR2+R'OH —> R2PH+R2POR> (114)
(RP)n + ^X2 —> ziRPX2 (115)
При нагревании циклопентафосфинов выше 160 °C образуются одновалентные фосфпнпдены RP:—реакционноспособные промежуточные продукты, которые могут быть зафиксированы при действии дисульфидов пли диенов (схема 116) [111].
RP(SEt)2
10.2.5. МЕТОДЫ ПОЛУЧЕНИЯ ФОСФОНИЕВЫХ СОЛЕЙ
кватернизацией в случае обыч-
Большпнство фосфониевых солей получают фосфинов подходящими алкилгалогенидами. Как и ных 5ы2-реакций, скорость взаимодействия возрастает при переходе от хлоридов к йодидам. Чаще всего используют первичные алкилгалогенпды; взаимодействие с низшими галогенидами протекает легко, при увеличении длины алкильной группы скорость реакции уменьшается. Для кватернпзацпи вторичными галогенидами необходимо проводить реакцию в жестких условиях, например, в запаянной трубке при высокой температуре. Полярные растворители, такие, как муравьиная кислота, ДМФ или ацетонитрил, облегчают реакцию. При наличии в алкплгалогепид электроноакцепторных групп возможны осложнения из-за прей мущественного протекания нуклеофильной атаки по атому гало_ гена [112], хотя продукты реакции в этом случае могут и не от личаться от продуктов прямого замещения у атома углеро
638
(см. разд, ю.2.3.4). Характерные примеры реакций получения фосфониевых солеи приведены ниже (уравнения 117—121).
КзР + РЬСНгВг —> R3PCH2Ph Bi-
ll 17)
(118)
Ph3P + C1CO2R —> Ph3PCO2R СГ (119)
РЬзР + RCOC1 —> Pb3PCOR СГ (120)
Ph3P + C1CH2CH=CH2 —> Ph3PCH2CH=CH2 СГ (121)
Фосфониевые соли обычно удобнее получать в инертном растворителе. Однако аллилтрифенилфосфонийхлорид получают кипячением трифенилфосфина в избытке аллилхлорнда, поскольку продукт, образующийся из эквимольных количеств реагентов в кипящем бензоле, склонен к изомеризации в пропенилтрифенилфос-фонийхлорид [ИЗ]. Несмотря на возможность прямой кватерни-зации трифенилфосфина винилбромидами, винилфосфониевые соли удобнее получать обработкой соответствующих хлорэтил-производных триэтиламином [114].
Арилгалогениды, как правило, не взаимодействуют с фосфинами, поэтому для получения тетраарилфосфониевых солей необходимы специальные методы. Наиболее широкое распространение получил метод комплексных солей, основанный на взаимодействии арилгалогенидов с триарилфосфинами в присутствии солей никеля(П) при нагревании (уравнение 122). Кватерниза-ция оптически активных фосфинов этим методом приводит к фосфониевым солям, в которых, несмотря на использование высоких температур, полностью сохраняется конфигурация фосфора [115]. При действии на комплекс (68) трифенилфосфина получается фосфониевая соль и трис (трифенил фосфин) никель, который в присутствии арилгалогенида вступает в реакцию окислительного присоединения, давая (68); таким образом, процесс носит каталитический характер (схема 123) и с высокими выходами приводит к фосфониевым солям [116]. Эта реакция достаточно легко
NiBr2 +
Аг3Р + Аг‘Х --► Аг3РАг' X (122)
I АгХ
X
Ar—Ni(PPh3)2+ 2Ph3P —► ArPPh, X'+ Ni(PPh3), (123) (68)
639
протекает уже в кипящем этаноле, тогда как в первом методе необходимо нагревание до 150—250°C.
Первичные продукты расщепления дифосфинов алкилгалоге-видами в присутствии избытка реагента претерпевают дальнейшую кватернпзацшо. При взаимодействии тетрафенилдифосфпна с а,(о-дибромалкапамп с высокими выходами получаются циклические фосфониевые соли (69) (схема 124). Меркль предположил [117], что образующийся в реакции галогенфосфин способен далее реагировать с избытком дигалогенида, давая дополнительное количество продукта циклизации (69) (уравнение 125).
СН2-(СН,)„
+ Ph2P-PPh2—► Ph2K> CH2Br
Br Br\>-PPh2
(сн2)л
> H2C^ \h2-4^
(ch2)„
> НгС^ ^/CH2 (л=2-4 J (124)
Ph2P+ Br"
(69)
Ph2PBr+BrCH2(CH2)nCH2Br —Ph2P—CH2(CH2)nCH2Br —> (69) (125)
Br
Помимо рассмотренных методов для получения фосфониевых солей используется катализируемая кислотами реакция фосфинов с простыми эфирами или спиртами. Такие реакции особенно ценны в синтезе циклических фосфониевых солей (70), протекающем путем внутримолекулярной конденсации. Соль (70) гладко образуется из со-гидроксифосфинов при обработке их избытком бромистого водорода (схема 126) [118].
, НВг +
PhRP(CH2),OH-----> РЫ?Р(СН2)лВг —"°вание >
н
(126)
NaC104 ------* \/ H =
zp+ С1О7
Ph R (70)
Удобный метод превращения аф-ненасыщенных кетонов в фосфониевые соли, которые могут быть использованы в реакции Виттита, заключается в последовательной обработке кетона впнпл-магнийбромидом и трпфенилфосфонинбромпдом [119] (см., например, схему 127). Фосфониевая соль (71) может быть получена непосредственно из первичного аллилового спирта и трифенпл-фосфонийбромида (уравнение 128).
640
Соли карбениевых ионов легко присоединяются к третичным фосфинам; так, например, при взаимодействии циклопропенилперхлората и трпфенилфосфина образуется циклопропенилфосфониевая соль (72) (уравнение 129) [120]. Интересным вариантом такого взаимодействия является реакция трифенилфосфина с ионом (73), приводящая исключительно к фосфониевой соли с awrw-расположением РИзР-группы по отношению к циклобутеновому циклу (уравнение 130), что подтверждает участие циклобутенового цикла, тогда как в случае классического иона (74) образуется смесь стереоизомеров [121].
R
R
+ Ph3P
сю;
(129)
(130)
Г)Мр
Присоединение хлорфосфинов к диенам протекает как син-Ронный дисротаторный л43 + ш23-процесс и приводит к пятичлен-к У Циклическим фосфониевым солям; хорошим примером та-
Реакции является превращение траке,гракс-гексадиена- ,
21 Зак. 395 641
в соль (75) при действии диметилхлорфосфина [122].
(уравнение 131)
+ Ме2РС1
Н
Ме
СГ
Фосфониевые соли, так же как и аммониевые, имеют тетраэдрическую геометрию, следствием чего является хиральность атома фосфора в фосфониевых солях, имеющих четыре различных заместителя. Для разделения энантиомерных форм используют фракционную кристаллизацию с применением оптически активных анионов, таких, как Д-(+)-камфорсульфонат или D-(~)-дибензоилводородтартрат [123]. Таким способом многие соли были получены в оптически активной форме; все они конфигурационно стабильны в растворе.
В настоящее время более общим методом получения оптически активных фосфониевых солей является кватернизация оптически активных фосфинов (см. разд. 10.2.1.6).
10.2.6. РЕАКЦИИ ФОСФОНИЕВЫХ СОЛЕЙ
Фосфониевые соли вступают в гораздо более широкий круг реакций, чем соответствующие аммониевые соли [124]. Это различие обусловлено, вероятно, способностью фосфора, в отличие от азота, образовывать пентакоордпнационные интермедиаты благодаря его большему атомному радиусу и возможности участия d-орбиталей. Возможные типы превращений фосфониевых солей, которые лучше всего иллюстрируются их реакциями с гидроксид-цоном, можно разделить на четыре группы в зависимости от места атаки в молекуле фосфониевой соли (уравнения 132—135).
НО'+ R'CH2PR3 —> R‘CH2OH + R3p
(132)
НО“ + R'CH2CH2PR3 —> R‘CH=CH2 + R3P + Н2О (133)
HO' + R‘CH2PR3 —> R‘CH=PR3-f-H2O (134)
HO' + R'CH2PR3 —> R‘CH3 + R3P=O (!35>
Известно всего несколько примеров реакций, в которых осуществляется атака нуклеофила на а-углеродный атом (уравнение 132). При нагревании фосфониевые соли разлагаются с образованием третичных фосфинов; соли с двумя или более различными заместителями у атома фосфора способны расщепляться^ по н скольким направлениям, причем способность заместителей к щепленшо в общем случае возрастает в ряду этил < метил < пропил <С фенил < бензил параллельно с увеличением сП
642
обности к нуклеофильному замещению. Эти различия, однако, невелики, поэтому в большинстве случаев образуется смесь продуктов. Реакции подобного типа могут протекать также в том случае, если а-углеродный атом особенно восприимчив к нуклеофильной атаке, как, например, при щелочном гидролизе ацпл-фосфонпевой соли (76); первоначальная атака в этом случае направлена на карбонильную группу, что приводит к образованию фосфина и карбоновой кислоты (уравнение 136) [125].
Все четвертичные аммониевые соли, содержащие в [3-положении атом водорода, при действии оснований претерпевают распад по Гофману с образованием алкена и свободного амина. Напротив, при действии оснований на фосфониевые соли эта реакция в большинстве случаев имеет лишь побочное значение и становится основной только при повышенной кислотности [3-водорода из-за возможности стабилизации возникающего аниона за счет сопряжения, как, например, в случае солей (77) и (78) (уравнения 137, 138). Последняя реакция использовалась для получения оптически активных фосфинов из разделенных на энантиомеры фосфониевых солей.
R‘COPR2Me Х’+НО" —> Р‘СООН+МеРР2 (136)
(76)
R'CCH2CH2PR3 X"НО" —> R‘CCH=CH2 + R3P (137)
II II
о О
(77)
NCCH2CH2PR3 Х'-фНО" —> CH2=CHCN + R3p (138)
(78)
Отщепление в фосфониевых солях а-водорода приводит к плицам (см. уравнение 134); подробно эта важная реакция рассмотрена в гл. 10.6. В щелочной среде гидроксид обычно атакует фосфониевые соли по атому фосфора, в результате чего образуется фосфиноксид и выделяется углеводород (см. уравнение 135). В последние годы большое число работ было посвящено нуклеофильным реакциям у тетраэдрического атома фосфора, в особенности реакциям щелочного гидролиза фосфониевых солей, которые обсуждены в следующем разделе.
10.2.6.1. Щелочной гидролиз
Систематическому исследованию продуктов щелочного гидролиза фосфониевых солей посвящено значительное число работ. Согласно данным, полученным Ингольдом, способность групп к отщеплению уменьшается в следующем ряду: аллил > бензил > фенил > этил. Изучение гидролиза тетраарилфосфониевых солей показало, что арильные группы, содержащие электроноакцепторные заместители, отщепляются легче, чем группы
21*
643
с электронодонорными заместителями, в соответствии с пор чтительным отщеплением групп, образующих более стабил °' анион. Гидролиз фосфониевых солей, содержащих группы н₽ чительно различающиеся по стабильности образуемых а’Ки ЗНа" приводит к смеси углеводородов и фосфиноксидов. Мак-ИвенН°В’ казал, что относительная склонность данной группы к отш.рпП°' нию зависит также от природы других имеющихся групп пй а при гидролизе стерически затрудненных фосфониевых сол могут возникать отклонения от нормальной способности Тех°Леи иных групп к отрыву из-за пространственных эффектов [126] ИЛИ
Согласно кинетическим данным реакции гидролиза обычн имеют общий третий порядок: первый — по концентрации di фониевой соли и второй —по концентрации гидроксид-ионой [124]. На основании этих данных Мак-Ивен предложил механизм реакции (схема 139), который принят в настоящее время в каче стве рабочей гипотезы. Общее дифференциальное уравнение для скорости реакции представлено уравнением 140. В том случае если реакция, характеризуемая &3, является лимитирующей, суммарная скорость процесса будет в значительной мере определяться способностью группы R к отщеплению в виде аниона. Если уходящая группа, например n-нитробензил или 1,4-дифенилбута-диен-1,3-ил, образует очень стабильный анион, как, например, в случае солей (79) и (80), наблюдается ' “ “ -----
реакции (первый — по фосфониевой соли ксид-иону).
+ к. /ОН -
R1PR4-HO" R'PZ ^R
, как, например, общий второй порядок и первый — по
I 7 + Н2О
з \ XR
гидро-
(139)
Вг-
Ph3PCH2
н2о , „
RHНО" <-------- R' + RsP==O
KiKik3 [ri£r] [H0-j2 = *набл [R>pR] [но*]2
,Н
(140)
Н.
Ph—у—Ph
Ph—Р—Me Br"
(80)
>—NO2
(79)
। oq\ соглас}10
Два варианта общего механизма (см. схему 1 'У м нщке)< щихся с кинетическими и стереохимическими данными ( хроНнук) были предложены Мак-Ивеном. Они включают или с и обра-с отщеплением аниона атаку второго гидроксид-иона, г та с зование нестабильного гексакоординационного интерм 141)-двумя гидроксилами, связанными с атомом фосфора (с- мець-Другими словами, схема механизма реакции включает
644
шей мере один пентакоординационный или один пентакоордпна циопный и один гексакоординационнын промежуточные продукты. Стереохимический результат щелочного гидролиза фосфониевых солей будет определяться структурой, временем жизни и реорганизацией лигандов этих промежуточных продуктов.
> R3P=O + R + Н2О
НО"
R4P + ОН
ОН R .
I он
? R,P^_4==
(141)
ОН I ±R2p4 он
Большая часть пентакоординационных фосфоранов имеет геометрию тригональной бипирамиды (ТБП), и, хотя заведомые гидр-окспфосфораны до сих пор не выделены *, можно вполне обоснованно предположить, что любые пентакоординационные промежуточные продукты гидролиза фосфониевых солей также имеют структуру ТБП. Образование ТБП из тетраэдрического атома фосфора может протекать двумя способами: 1) путем нуклеофильной атаки по любому из шести ребер тетраэдра; в этом случае нуклеофил занимает экваториальное положение (экваториальная атака), 2) путем атаки по любой из четырех граней тетраэдра, что приводит к ТБП, в которой нуклеофил занимает апикальное положение (апикальная атака) (схема 142),
апикальная атака .
Теоретически возможно также отщепление уходящей группы как из экваториального, так и из апикального положения ТБП, что приводит к четырем возможным типам замещения, включающего промежуточное образование ТБП. Стереохимические следствия этого представлены в табл. 10.2.6; здесь учитываются продукты, образующиеся из первоначального пентакоординацпоп-иого интермедиата и не принимаются во внимание многочисленные процессы псевдовращеипя.
Гидроксифосфораны в настоящее время получены [166].
645
Таблица 10.2.6. Стереохимия замещения у тетраэдрического атома фосфора с участием промежуточного продукта типа ТБП '
Атака Отщепление СтереохимичеекиП результат
Апикальная Апикальная Экваториальная Экваториальная Апикальное Экваториальное Апикальное Экваториальное Инверсия Сохранение конфигурации То же Инверсия
Апикальные связи имеют большую длину и, следовательно слабее экваториальных, поэтому апикальная атака должна быть предпочтительной, поскольку в этом случае для образования связи не требуется значительного сближения нуклеофила с атомом фосфора. Кроме того, более слабые апикальные связи должны легче разрываться, нежели экваториальные, вследствие чего апикальное отщепление должно быть предпочтительным. Теоретические расчеты относительных энергий для возможных типов атаки фторид-иона на фосфорилфторид показали [128], что апикальная атака через грань FFF выгоднее экваториальной атаки по ребру FF (на 120 кДж/моль). Таким образом, для реакций замещения у тетраэдрического атома фосфора в качестве рабочей гипотезы приняты апикальная атака и апикальное отщепление.
Апикальная атака на тетраэдрический атом фосфора может протекать по четырем различным граням, что приводит к четырем различным фосфоранам. Эти первоначально получающиеся фосфораны могут претерпевать реорганизацию лигандов с образованием при наличии в молекуле пяти различных заместителей 10 энантиомерных пар фосфоранов, каждый из которых может превращаться в продукт реакции (схема 143). Таким образом, результат каждой конкретной реакции замещения будет зависеть от относительной стабильности фосфоранов (см. гл. 10.4) и скорости их превращения. Очевидно, что стереохимический результат щелочного гидролиза фосфониевых солей является прекрасным подтверждением того или иного механизма реакции, поэтому в последние несколько лет в этой области были сосредоточены значительные усилия исследователей.
^р^-рНО' —> [4ТБП] —> [20ТБП] —> Продукт (143)
Щелочной гидролиз ациклических фосфониевых солей обычно протекает с обращением конфигурации, что было изящно прод монстрировано Мак-Ивеном с сотр. [123] на примере оптичес^ активного бензилметилфенилэтилфосфонипиодида (81), гидро которого сопровождается полной инверсией конфигурации, вызывает сомнений, что первоначальная атака гидроксид-протекает со стороны, противоположной бензильной группе,
646
приводит к наиболее стабильному фосфорану (82); последний превращается в фосфиноксид с обращенной конфигурацией (схема 144). Этот эксперимент дает однозначный результат, поскольку бензильная группа имеет наиболее ярко выраженную апико-фпльность по сравнению с другими лигандами у фосфора (см. вл. 10.4). Однако, если способность групп к отщеплению и апикофильность лигандов достаточно близки, как, например, В случае интермедиатов, возникающих при щелочном гидролизе арплфосфониевых солей (83) и (84), может наблюдаться та или иная степень инверсии или сохранения конфигурации [129]. Стереохимический контроль незначителен, следовательно энергетические различия между возможными промежуточными продуктами, которые могут находиться в равновесии за счет процессов псевдовращения, должны быть очень малы. Это еще более подчеркивается тем фактом, что стереохимический результат таких реакций зависит от того, проводятся ли они в гомогенной или гетерогенной среде [130].
CH2Ph
Ме^ C"Et Ph (81)
> Me—Р
CH2Ph I ..>Et
(144)
ОН
(82)
Pr
Me"-V-Ar
P< Br'
Pr
Me hili,,. p~F j
/ Br"
Ph
(S3)
Тщательное исследование соотношения продуктов, получающихся при щелочном гидролизе соли (85), свидетельствует о наличии псевдовращения и возможного стерического контроля образования продуктов, так как атака гидроксид-пона со стороны, противоположной ментоксигруппе, кинетически несколько* более предпочтительна, чем атака со стороны, противоположной метоксигруппе [131]. Таким образом, стереохимия реакций гидролиза определяется, по-видимому, в первую очередь направлением атаки гидроксид-иона.
Несмотря на то что гидролиз бензилфосфониевых солей протекает обычно с инверсией конфигурации, при наличии в молекуле трет-бутильной группы конфигурация сохраняется. Это обусловлено возникновением значительных пространственных препятствий для подхода нуклеофила при образовании промежуточной ТБП с трет-бутильной группой в экваториальном положении; поэтому, вероятно, атака гидроксид-иона протекает со стороны,
647
противоположной трет-бутильной группе, что приводит к со™ нию конфигурации у атома фосфора (схема 145) [1241 Обпапт ' конфигурации, наблюдаемое при гидролизе алкокси-тоет бХ™6 фосфониевой соли (86) приписывается более высокой апикойшн ности уходящей группы (OEt по сравнению с CH2Ph)
экваториальное
ЧТО за-поло-
ставляет трет-бутпльную группу занимать жение (схема 146).
CH2Ph CH2Ph
/Pt, трет -В u—Р "'Же
Hr \'/z/Bu-rper I ^Ph
Me ОН
Br“
HO-
трет-В u ~ _
| ^H2Ph Me—Pf
| 4Ph
OH
Bu-rper
> Me—P—CH2Ph _ / 4
О Ph
О
II
> Лч (145
Р1Л \ ^Bu-rper ' ’ Me
OEt
P+
Ph^ VMe
rper-Bu SbClg
OEt
| ^Bu-rper
>Ph—P^
I -Me OH
Me
\ f>\Bu-rper
I о
(146)
(86)
Гидролиз алкокси (метилтио) фосфониевых солей протекает с замещением метилтиогруппы и сохранением конфигурации, что согласуется с атакой гидроксид-иона со стороны, противоположной алкоксигруппе, с образованием интермедиата (87), который должен отщеплять SMe-группу из апикального положения после одного псевдовращения (схема 147) [132].
Ph OR
Ч +/ но-
Р --------*
R SMe
SbCi;
OR
Л । _
P—SMe
* OH (87)
Ph OR
4. /
(147)
R= Me,Bu-rpe^
Скорость гидролиза фосфониевых солей значительно в0^Ра_ стает, если атом фосфора включен в состав четырех- или п членного цикла, как, например, в случае (88) и (89) [13JE эффект отчасти можно объяснить уменьшением напРЯетоией в цикле при переходе к промежуточному продукту с геом ” ТБП, что должно увеличивать Ki (см. уравнение 140) по р
648
НИЮ с ациклическими аналогами; однако чтобы .
ствительно имел место, необходимо чтобы пшД эФфект деЙ, кально-экваторпальное положение в ТБП рЦ Л занимал апи-в этом положении, а нуклеофил атакует rnfui. ЦИ° нахОДится следовательно, любая уходящая группа Н1Р! тетРаэдра, то, цикла, должна быть экваториальной как' ияп ходящая в состав Один акт псевдовращения вокруг неуходящей г?’ В (90) 11341-к тому, что цикл по-прежнему занимает а^ЯпТУППЫ пРИводят ное положение, а уходящая группа в пЯнио К Л Н0'ЭКват0Риаль-становится апикальной, так что при пос”” М СЛучае бензильная форана образуется фосфиноксид в котоп™УЮЩ6М распаде фос-гурация у атома фосфора (схема 148) Р СОхРаняется конфи-
Р—CH2Ph Br- Ph—Р—CH2Ph Вг-
Ме
(88) (89)
Z1, vL
—Р+...mCHoPh ---------Pf
| I ^Me
Me ОН
(90)
-f—P—CH2Ph Me" ""Ъ~
т-j
•P’"»////VJ
i
Me
(148)
Описано много примеров реакций такого типа, однако имеются сообщения, свидетельствующие о потере в некоторых случаях стереоспецифичности. Это особенно верно тогда, когда имеются очень небольшие различия в апикофильности экзоциклических групп, как, например, в случае солей (91)—(93).
Н
' +
-j—Р—OEt
OMe
SbCle (91)
-Р—Ph I I 'Me
Было выдвинуто предположение, что скорость замещения У атома фосфора, входящего в состав малого цикла, как и в случае
649
ациклических аналогов, зависит от апикофильности замещаемой группы [135]. Все реакции подобного типа протекают, вероятно, через интермедиат (94). Замещение будет сопровождаться сохранением конфигурации фосфора, если X обладает значительно более высокой апикофильностыо, чем Y [как, например' в (88)], поскольку в этом случае псевдовращеиие будет преимущественно приводить к (95) с последующим отрывом уходящей группы из апикального положения. Если X и Y имеют сравнимые электроотрицательности, необходимо учитывать альтернативный процесс псевдовращения, приводящий к (96), а также дальнейшее псевдовращеиие (схема 149). В системах, имеющих близкие по свойствам экзоциклпческие лиганды, связанные с фосфором, конечный состав продуктов будет зависеть от относительного времени жизни различных интермедиатов и способности групп X и Y к отщеплению.
Известен ряд реакций гидролиза фосфониевых солей, сопровождающихся миграцией лиганда на а-углеродный атом, несут111 уходящую группу или заместитель, способный стабилизировать отрицательный заряд. Хеллман и Бадер [136] изучили гидрол1 _ ряда галогеналкилфосфониевых солей и показали, что значптель ное количество продуктов образуется в результате миграции л ганда с фосфора на углерод (уравнение 150). Неудпвитель 650
поэтому, что гидролиз циклических иодметилфосфонпевых солей сопровождается расширением цикла, как, например, в случае (97) (схема 151). Фенильная группа способна к участию в реакциях этого типа как за счет стабилизации отрицательного заряда, например при гидролизе (98) (схема 152), так и за счет непосредственного соучастия, как в случае фосфетаниевой соли (99) (схема 153)• +
Ph3PCH2I + НО’ —> Ph2P(O)CH2Ph (150)
Возможна также миграция лиганда на углерод карбонильной группы, как показано на примере расширения цикла при гидролизе ацилфосфониевых солей, полученных при действии ацилхло-ридов на фосфолы (схема 154) [137]. Гидролиз ароилфосфониевых солей (100), образующихся в реакции фосфоленов с хлор-ангидридами ароматических карбоновых кислот, приводит, однако, не к продуктам расширения цикла, а к ароматическому
(100)
651
альдегиду и фосфиноксиду (схема 155); эта реакция является альтернативой реакции восстановления по Розенмунду [138].
Из рассмотренных реакций видно, что состав продуктов ще. лочного гидролиза фосфониевых солей определяется сочетанием различных факторов. Если атом фосфора не входит в состав малого цикла, гидролиз ацилфосфониевых солей осуществляется путем нуклеофильной атаки на углерод карбонильной группы однако, если фосфор является частью малого цикла, то нуклеофильная атака направлена на атом фосфора вследствие ослабления напряжения в цикле. Когда напряжение цикла велико например в фосфолах или фосфетанах, происходит расширение цикла; при меньшем напряжении промежуточный продукт имеет большее время жизни, достаточное для перемещения обладающей высокой апикофильностью ацильной группы в апикальное положение и последующего ее отщепления.
В настоящее время известно много органических соединений гексакоординированного фосфора (см. гл. 10.4), и вполне вероятно, что такие соединения в некоторых случаях выступают в качестве промежуточных продуктов или переходных состояний в реакциях нуклеофильного замещения у тетраэдрического атома фосфора. Кинетическое изучение катализируемой этоксид-ионами реакции разложения 3-гидроксипропилтрифенилфосфонийхлорида показало наличие гексакоординационного промежуточного продукта или переходного состояния, образующегося при атаке это-ксид-иона на пентакоординационный промежуточный продукт (схема 156) [139].
-Ph
(156)
О
Et2O + Ph2P—7 ОН
-";D
Pn /-
О—ЕИ OEt.
10.2.6.2. Присоединение к винилфосфониевым солям
С момента появления первого сообщения об использовании винилфосфониевых солей в реакции присоединения по Михаэлю [МО] значение этих реагентов в синтезе существенно возросло [141]. Использование этих соединений основано на их способно сти превращаться в илпды при присоединении по двойной связь образующиеся илиды в свою очередь могут вступать во вНУтР молекулярную реакцию Виттига с подходящим образом распо
652
ценной карбонильной группой (схема 157). Этим способом исходя из винилтрифенилфосфонийбромида, получен ряд гетероциклических соединении (уравнения 158—162).
X’ О
PIi3P—СН=СН2 + RC-A-Y- —>
(157)
(158)
(159)
(160)
(161)
(162)
Интересно отметить, что структура продуктов реакции винил-фосфониевых солей с гидразонами зависит от конфигурации последних. Если винилфосфониевая соль содержит в 0-положении уходящую группу, первоначально образующийся продукт способен к дальнейшим превращениям, как, например, при п0^У’1е‘ нии фуранов взаимодействием а-гидроксикетонов с солью (IUI) (Уравнение 163) [142]. ph ph
Ph-c_° CH-PPh, ВГ- pb_)=\ 0Et ph_0 (163)
Ph-НС. CH—OEt \<( C>
XOH °
(101)
653
й-Ацилвинплфосфониевые соли были с успехом использп для получения гетероциклических соединений [141]. в этих Ь1 вращениях замыкание цикла осуществляется путем реакции денсации по карооиплыюн группе ацильной функции. Фосфо^^' держащая группировка может быть удалена путем гидРолРиС0’ или использована в дальнейшем для экзоциклнческих реап -Внттига. Хорошим примером такого рода превращений мож служить синтез производных 1,3-тиазола (схема 164). Аналогий но, исходя из амидинов, могут быть получены имидазолы.
Br" RC=O
Ph3PCH=CH
h2n
/Л1
R.
R.
Br"
—> Ph3PCH2—
R но-
/—R’ TiZ CH-~ s
R1
S
(164)
R.
R2CH=CH——R1
S
О
r2cho
Ph3P=CH——R1
S
Ph CO2Et
ЦИКЛО-приме-
PPh3, EtO?.C ------------> CO2Et bf^ (102)
Илиды, образующиеся при обработке бутадиенил- и пропилфосфониевых солей нуклеофилами, также широко
няются в синтезе циклических соединений. Циклопропилфосфониевая соль (Ю2) с расположенной нужным образом этокспкар о нильной группой при взаимодействии с подходящими нуклеоф^ лами, например с натриевыми солями енольной формы эФ1'? p-кетокарбоновых кислот, с высокими выходами образует Р дукты циклизации (схема 165). Эти реакции включают, по-в мому, нуклеофильную атаку на гемпнально активирова _ циклопропан и последующую внутримолекулярную реакцию тига [143].
10.2.6.3. Фосфониевые соли как катализаторы фазового переноса
В последние годы широкое применение нашел ноВЬ1^а3овог° синтеза, основанный на использовании катализаторов ф переноса в системе двух несмешивающихся растворител
654
обнарУжеио’ что фосфониевые соли, в особенности соли, имею-щнс четвертичный катион с достаточно высокой молекулярной массой, например грибутилгексадецилфосфопийбромид, являются прекрасными катализаторами в реакциях данного типа. Они особенно удобны как катализаторы реакций замещения алкилгало-Iеиидов неорганическими анионами, окисления алкенов пеоман-; анатом и бензоиновой конденсации в водных средах 11441 Поучение дестабилизированных илидов можно проводить в паст юре дихлорметана при депротопировании водным гидроксидом натрия, а поскольку в этом случае исходная фосфониевая соль сама играет роль катализатора фазового переноса, добавление других солей необязательно. В присутствии альдегидов образующийся илид вступает в реакцию Виттига быстрее, чем протекает гидролиз фосфониевой соли в фосфиноксид.
Использование двухфазных систем позволяет осуществить много интересных превращений, которые в некоторых случаях приводят к продуктам, недоступным при проведении реакций в гомогенных условиях. В качестве примера можно привести стереоспецифичный синтез термодинамически менее стабильных производных моносахаридов с жанно-копфнгурацией, например (103), взаимодействием в двухфазной системе соответствующего литропроизводпого с соединением с активированной метиленовой группой (схема 166) [145].
10.2.7. МЕТОДЫ ПОЛУЧЕНИЯ ГАЛОГЕНФОСФИНОВ
В разд. 10.2.7 и 10.2.8 рассмотрены 1МЬК°^0ДД^'ееН„"ЯХ,ш>,я RPX, „ Й2РХ, где R — алкил или арил а т11пы соед„.
галогснфосфорапов рассмотрена в гл. • ,юших разделах пений, содержащих связи р~Х — в ,CC0J“„a ’
• л. 10.3 и 10.5, посвященных кислотам фосф Р • „pv а также
Галогеафосфилы R2PX и дигалогслфосфл ы **Д|1ЫМ„ „е. трогалогсинды фосфора РХ3 являются 1ЧССК„Х спедллс-
яествами в синтезе разнообразных, «НФ»W””"x “!,»<» Ф«сФ'-чий; их использование для получен! я | < .пат11В1Ю важные иов уже обсуждалось выше. Многие npyi i R 1 подробные све-Реакцни рассмотрены в разд. счсрг1ыиающпх обзорах
нения читатель сможет паити н двух 1
Ц46, 147].
055
10 2 7 1 Синтез галогенфосфинов из галогенидов фосфора и металлорганических соединений
Взаимодействие диалкил- или ди арил производных
хлоридом фосфора в запаянной трубке при нагревании С Три’ исторически наиболее важным методом получения алий ЯВИлось и ароматических дигалогенфосфинов (уравнения 167 1бя\Ческих разевание монохлорфосфинов в этой реакции может быт лено использованием избытка трихлорида фосфора ЭтЬ П°Дав' может быть применен и для получения хлорфосфинов с Т выми или различными заместителями.
метод одинако-
R2Hg + РС13 —> RPC12 + RHgCl (167)
RHgCl + РС1з —> RPCl2 + HgCl2 (168)
трег-BuMgCl + РС1з —> трет-ВиРСЬ + MgCl2 (169)
трет-BuMgCl + МеРС12 —> Tper-BuMePCl + MgCl2 (170)
(PhCH2)2Zn + 2РС1з —> 2PhCH2PCl2 + ZnCl2 (171)
Et4Pb + 2РС1з —> 2EtPCl2 + Et2PbCl2 (172)
Me4Pb + 2PhPCl2 —> 2PhMePCl + Me2PbCl2 (173)
реактивов
Взаимодействие литийорганических соединений и Гриньяра с галогенидами фосфора приводит обычно к третичным фосфинам. Тем не менее, если при использовании реакдяонн способных литийорганических соединений задержать реакцию^ промежуточной стадии как правило невозможно, то в СЛУ ( реактивов Гриньяра удается остановить превращение на с^]пе_ дигалогенфосфина или галогенфосфина, используя низкие т^ие ратуры или пространственно затрудненные магнпйоргани соединения, например трет-бутилмагнийхлорид (уравнени 17°). за.
Другой метод контроля степени замещения основан мене реактивов Гриньяра менее реакционноспособными на. или цинкорганическими соединениями. Бензплдихлорфо пример, образуется с прекрасным выходом при взай*1ОДогО сое-трихлорида фосфора и соответствующего цинкорганич динения (уравнение 171). .Qpa тет-
Подробно изучено алкилирование галогенидов Ф°^®10ТрЯ на раалкильными производными свинца. Этот способ, н _geH для токсичность свинецорганических реагентов, весьма У Орфосф11‘ получения некоторых соединений, в частности этилдь текает на. Выходы целевых продуктов очень высоки; Реаки фосФкНУ-ступенчато и приводит в конечном счете к тРет11ЧН0'у несиММеТ' Этот метод с успехом и: "ользуют также для получен , j73)-рично замещенных моногалогенфосфинов (уравнения 656
10.2.7.2. Синтез галогенфосфинов
из ароматических соединений и галогенидов фосфора(Ш)
Прекрасным методом получения арилдигалогенфосфинов и „арплгалогенфосфннов является реакция Фриделя — Крафтса. Подробно изучено превращение трихлорида фосфора в комплексы фенилдихлорфосфина или дифенилхлорфосфина [148]. На состав продуктов решающее влияние оказывает соотношение трихлорида фосфора и бензола (а:1) (схема 174). Несимметричные диарил-галогенфосфины могут быть получены при действии различных аренов на комплексы фенилдихлорфосфина.
П > I п <_ 1
РНН+РС1з + А1С1з ----► РЬРС12-А1С13 -----> Ph2PCl • А1С13
|АгН (174)
ArPhPCl
Основным недостатком этого метода является образование достаточно устойчивых комплексов галогенфосфина с трихлоридом алюминия, что может существенно снижать выход. Для разрушения этих комплексов разработано много приемов; наиболее эффективным оказалось прибавление в реакционную смесь фосфорил-хлорида или пиридина.
Недавно обнаружена другая, более серьезная трудность. Обычно считалось, что в случае монозамещенных аренов замещение идет в лара-положение, однако более тщательные исследования показали, что толуол, например, образует все три возможных изомера.
10.2.7.3. Синтез галогенфосфинов из производных кислот фосфора
Фосфонистая и фосфинистая кислоты существуют обычно в форме тетракоординационных таутомеров RPH(O)OH и R2P(O)H, соответственно, и при прямом хлорировании дают, как правило, хлорангидриды фосфоновой или фосфиновой кислот. Однако при использовании в качестве хлорирующего агента трихлорида фосфора, эти кислоты превращаются в соответствующие хлорпроиз-водные фосфора(III). Этот метод широко используется для синтеза монохлорфосфинов, поскольку вторичные фосфиноксиды R2P(O)H легко могут быть получены из продажных реагентов
РС1з
(RO)2P(O)H+ R'MgX —> R2P(O)H -----> R2PC1 (175)
R'P(NR2)2 + 4HCI —> R*PC12 + 2R2NH2 Cl' (176)
(104)
657
(схема 175) Эфиры фосфонистой и фосфинистой кислот т могут быть переведены в соответствующие хлора нгидрипн*6 действии избытка трихлорида фосфора. и пРи
Широкое применение нашла реакция расщепления ах фосфонистой и фосфинистой кислот безводным хлористым вИД°В родом (уравнение 176). Для получения высоких выходов не^' ходпмо избегать использования избытка хлористого водопо°°' Наилучшим методом синтеза хлорфосфолана (104) является^3’ щепление соответствующего диэтиламииофосфолана (уравнение
10.2.7.4. Галогенирование фосфинов
Наилучшим методом синтеза многих первичных и вторичных фосфинов является восстановление галогенфосфинов. В некоторых случаях, однако, удобным приемом оказывается обратная реакция особенно если фосфин может быть получен другим путем. Так^ например, наиболее удобным способом получения хлорфосфетана (105) является хлорирование соответствующего вторичного фосфина (уравнение 178) [150]; хорошие выходы получены при отсутствии избытка хлора или фосгена.
PH
CI2 —>
РС1
(178)
(Ю5)
10.2.7.5. Реакции обмена галогена
Вследствие доступности хлорфосфинов важным методом получения других галогенидов являются реакции обмена галогена. Моно- и дибромфосфины были получены взаимодействием соответствующих хлоридов с безводным бромистым водородом. Аналогично, при нагревании при высокой температуре дихлорфосфинов с иодистым водородом образуются дииодфосфины. Для проведени реакций обмена галогена также были ” успехом использован тетрабромид германия и пропилтрииодсилан. а.
Для получения фторидов используют реакции с трифтор ми сурьмы и мышьяка, однако эти превращения протекают осложнений лишь с производными, например (106), имениюь . атома фосфора электропоакцепторные группы (уравненп в других случаях за счет окислительно-восстановительного гф по_ са образуются фторфосфораны. Последнюю реакцию мо> ^олее давить, используя в качестве растворителя амии. Однак _ удобным методом обмена на фтор является взаимодействп Ридами щелочных металлов в инертном растворителе.
ЗС13СРС12 + 2SbF3 —> 3Cl3CPF2-f-3SbCl3
(106)
658
10.2.8. РЕАКЦИИ ГАЛОГЕНФОСФИНОВ
Благодаря наличию в галогенфосфинах как нуклеофильного атома фосфора, так и уходящей группы, присоединенной к этому атому, реакции этого класса соединений весьма разнообразны Вследствие этого превращения галогенфосфинов удобно подразделить на реакции, в которых фосфор является электрофилом п реакции, в которых он проявляет нуклеофильные свойства. Такое разграничение не всегда можно провести достаточно четко особенно в случае реагентов, имеющих одновременно электрофильные и нуклеофильные центры. Известно много реакций, в которых фосфор проявляет бифпльные свойства (см. разд. 10.2.8.3); некоторые реакции, в которых фосфор на первой стадии является либо электрофилом, либо нуклеофилом, рассмотрены ниже.
10.2.8.1. Нуклеофильная атака по атому фосфора
При гидролизе трихлорида фосфора образуется фосфористая кислота (уравнение 180), тогда как взаимодействие трихлорида азота с водой приводит к аммиаку и хлорноватистой (гипохлори-стой) кислоте^ (уравнение 181). Для объяснения такого различия в реакционной способности выдвигались различные предположения, однако наиболее вероятной причиной является способность фосфора стабилизировать путем расширения своей валентной оболочки анионное переходное состояние (107) (схема 182), в котором неподеленная пара электронов находится в экваториальной плоскости.
РС13 + ЗН2О —> Р(ОН)Я + ЗНС1 (180)
NCl3 + 3H2O —> NH3 + ЗНОС1 (181)
Переходное состояние (107) предполагает обращение конфигурации у атома фосфора, что согласуется с данными о реакциях замещения в хлорфосфетане (105), всегда сопровождающихся инверсией конфигурации у атома фосфора. В этом случае реакция протекает через переходное состояние или промежуточный продукт типа S\2 с атакующей и уходящей группами в апикальных положениях, несмотря на увеличение пространственного напряжения из-за расположения четырехчленного цикла в диэкватори-альной позиции (схема 183) [150].
(105) + Y“
659
Атомы галогенов, за исключением фтора, легко замещают под действием разнообразных нуклеофильных агентов, что ДелаСЯ галогенфосфппы удобными исходными соединениями в синтеГ многих органических производных фосфора. зе
Превращение спиртов в алкилгалогениды при действии гало генидов фосфора является стандартной, вошедшей в учебники реакцией, протекающей с образованием алкоксифосфониевой соли в качестве промежуточного продукта. Как было продемонстрировано на примере реакции диметилхлорфосфина с метанолом, промежуточная фосфониевая соль (108) при температуре выше —10°С разлагается, давая диметилфосфиноксид и метилхлорид (уравнение 184) [151]. При использовании вместо метанола метоксида натрия фосфониевая соль не образуется, а получается соответствующий эфир (уравнение 185).
Ме2РС1+МеОН —> Ме2РН— ОМе СГ —> Ме2Р(О)Н + МеС1 (184)
(Ю8)
Ме2РС1 + MeONa —> Ме2РОМе + NaCl (185)
о2
RR'CHOH + 2NaH + ClPPh2 ----> RR'C=O + NaCl + Ph2PO2Na (186)
Эфиры, полученные взаимодействием вторичных алкоксидов с дифенилхлорфосфином (уравнение 186), легко окисляются кислородом воздуха в присутствии избытка гидрида натрия [152]. Эта реакция представляет собой удобный метод окисления вторичных спиртов, поскольку проводится без выделения промежуточно образующихся эфиров.
Первичные и вторичные амины также могут замещать галоген в галогенфосфинах, однако в случае первичных аминов для предотвращения дальнейших реакций, например реакции (3-элиминирования (см. гл. 10.6), необходимо соблюдать предосторожности. Аминофосфины, содержащие первичный или вторичный атом азота, труднодоступны; синтез первого алкилдиаминофосфина ° (109) описан лишь недавно (уравнение 187) [152]. При взаимодействии дигалогенфосфинов с бифункциональными нуклеофилами, например с гликолями и аминоспиртами, образуются циклические соединения, например (110) и (111) (уравнения 188, 189). Амбидентные нуклеофилы обычно реагируют с галогенфосфинами центром с наибольшей электронной плотностью. Например, енолят-анионы дают производные енолов (уравнение 190), а фосфит-анионы реагируют по атому кислорода (уравнение 191).
Описаны также реакции галогенидов фосфора с алкенами. Одной из наиболее полезных реакций данного типа является присое-
грег-ВиРС^ + 4NH3 -—> 7’pez,-BuP(NH2)2 + 2NH4CI
(Ю9)
660
Me 0
Me OH RiN
+ C12PR -----
Me OH
.PR + 2R'NH СГ
Me o/
(HO)
(188)
-OH
+ C12PR
NHMe
R’N
/PR 4- 2R'NH СГ
\ме
(Ш)
(189)
q
R2PC1 + Med—CHCO2Et
Me
> R2P—О—C=CHCO2Et
(190)
О
R2PCI + (ЕЮ)2Р=О ---> R2P—О—P(OEt)2
(191)
динение галогенидов фосфора к высокозамещенным алкенам в присутствии кислот Льюиса с образованием производных фосфетана (112) (схема 192) [153]. Механизм этой реакции включает, вероятно, нуклеофильное замещение иона галогена и последующую 1,2-миграцию метильной группы. Стереохимия фосфетаноксидов, образующихся в реакции олефинов с дихлорфосфинами, определяется на стадии гидролиза промежуточного комплекса водой.
R—Р—CI—А1С13 ii
(192)
10.2.8.2. Электрофильная атака по атому фосфора
Реакция галогенфосфинов с алкилгалогенидами в присутствии кислот Льюиса, являющаяся удобным методом получения многих фосфорорганических галогенидов, протекает, как было показано, с образованием комплексов 1 : 1 (113) (уравнение 193) [154].
трет-BuCl 4-МеРС12 + А1С13 —> [трет-ВиР(Ме)С12]+ А1С14 (193)
(ИЗ)
Комплексы (114), образующиеся при взаимодействии с триал-килборанами, могут окисляться кислородом воздуха непосредственно до фосфиноксидов (уравнение 194). Эта реакция представ-Ляет собой простой способ превращения галогенфосфинов в
661
фосфинокспды, сопровождающийся переносом группы с бог>я фосфор [155]. 1 на
R3B + Р112РС1 —> [R3B-PPh2Cl] —> Ph,P(O)R
(114)
(194)
Для ряда галогенфосфинов описаны реакции сопряженного присоединения. Дналкилхлорфосфииы, например, присоединяются к 2-винилпиридину, давая оранжевые аддукты (115), разлагающиеся в присутствии доноров протонов с образованием 2-(р-диал-килфосфинилэтил) пиридинов (схема 195) [156].
RXH
2РС1
X- ?
'jX^CHCH2PR2
(115)
J X psj ''4CH2CH2PR2 (195)
N
10.2.8.3. Бифильные реакции
Одним из наиболее ценных методов получения пятичленных фосфорсодержащих гетероциклов является впервые описанная Мак-Кормаком конденсация 1,3-диенов с галогенфосфинами (уравнение 196) [157]. Эти циклизации обладают всеми особенностями, присущими реакции Дильса — Альдера, и протекают, как было показано, стереоспецифпчно. Взаимодействие транс,транс-гек.-садиена-2,4 с диметилхлорфосфином и дигалогенфосфинами (уравнения 197, 198) приводит к аддуктам, например (116), в которых метильные группы цикла находятся в цис-положении по отношению друг к другу [158].
+ RPC12
(196)
+ С1РМе2
(197)
Согласно данным ЯМР 31Р, первоначально образуют оли> дукты диенов с дигалогенфосфинами представляют со вЫ. а не пентакоординационпые фосфораны. Эти соедпненп Р н0 чайно гигроскопичны и неудобны в обращении, поэтому
662
ыделения переводят гидролизом в фосфиноксиды. В эту ре-бс3 в вступают разнообразные диены и галогепфосфнны (уравне-аК1ПН199—204). Для получения бициклических фосфорсодержа-произволпых используют реакцию присоединения галогенфос 111П'ов к циклическим 1,3-диенам, например к циклогептадиену-1,3
+ МеРС12 —>
Me—Р—С1 СГ
(199)
+ PhPCl2
(200)
СГ
+ PhPCl2
Ph—P—Cl
(201)
Cl"
+ PhPBr2
Ph—P—Cl
(202)
Ph—P—Br
Br-
+ МеРС12 —► —
Me—Р—С1 СГ
(203)
С1
СГ Р.
(204)
Ранее считали, что все аддукты, образующиеся при взаимодействии диенов с галогенфосфинами, являются производными фосфо-лена-3 (см. уравнение 196), однако позже, в особенности в работах Куина и Корте [158, 160], было показано, что некоторые продукты представляют собой в действительности производные фос-фолена-2. Изомеризация зависит от природы галогена, присоединенного к фосфору, а также от характера замещения в исходном ДНене. Метилдихлорфосфин, например, всегда образует пронзвод-ые фосфолена-3, тогда как фенилдихлорфосфин дает фосфоле-^1-2, за исключением реакций с 2,3-дизамешениымп диенами. Фе-Л^'бромфосфин, однако, реагирует с диенами с образованием к ^°'?еиов'3. Вероятно, в тех случаях, когда присоединенные фосфору группы являются достаточно электроотрицательными
663
(фенил и хлор в PhPCl2 в отличие от фенила и брома в РЬРВг или метила п хлора в МеРС12), благодаря активации «-положения становится возможным аллильный сдвиг в первоначально образующемся фосфолене-3.
Для протекания реакции необходимо цисоидное расположение двойных связей в диене, причем геометрия заместителей в положениях 1 и 4 диена оказывает существенное влияние на его реакционную способность. Так, например, тра«с-пентадиен-1,3 взаимодействует с метилдихлорфосфином с образованием соответствующего аддукта, тогда как ^uc-пентадиен-1,3 в тех же условиях в реакцию не вступает [158].
Имеются данные об образовании производных фосфолена-3 при взаимодействии 1,3-диенов с комплексами арилдихлорфосфин—хлорид алюминия [161]. Реакции этих комплексов с несопряженными диенами находят широкое применение в синтезе фосфорсодержащих гетероциклов (уравнения 205, 206). В случае замещенных 1,4-диенов образуются бициклические соединения типа (П7), тогда как незамещенный пента диен-1,4 дает 3,5-дигало-генфосфоринаны (уравнение 205). Из 1,5-диенов неожиданно образуются производные бицикло [2,2,1] гептана в виде смеси эпимеров (уравнение 206) [161].
i.rpx2, . А1С13 ч------
2.НгО (R’=H)
о1 LRPX2’ м р A1CI3 Ме
------->
2.НгО
(R1= Me)
Me
(205)
/7\
О R (117)
Ме k
А1С13
2.НгО
Me
(206)
Галогенфосфины вступают
До
R
Галогенфосфины вступают также в реакцию Дильса—Альдера с а,р-ненасыщенными кетонами, причем в некоторых случаях первоначально образующиеся аддукты, например (118), были выделены [162].
RPC12 + CH2=CHCO2R‘ —> С12Р—СН2—СН=С—OR1 (207)
I
R
O’
(118)
Многие продукты взаимодействия дихлорфосфинов с акри левой кислотой и ее производными образуются, по-видимому, в результате дальнейшего превращения циклических интермеди тов, как показано на примере превращений акриловой кислоты хлорангидрид (119) (схема 208) и акриламида в нитрил ( )
(схема 209) [163].
664
RPC12 + сн2=снсо2н ->
сг
о
II
RPCH2CH2COC1
Cl
(208)
(119)
rPC12 + CH^CHCONH2
СГ
о
II rpch2ch2cn
ci
(209)
(120)
реакции простых альдегидов и кетонов с галогенфосфинами в некоторых случаях приводят к сложным смесям продуктов; имевшиеся данные позволяют считать, что многие из этих реакций также протекают с участием циклических интермедиатов. При взаимодействии ацетона с фенилдихлорфосфином образуется аддукт (121) (уравнение 210), имеющий структуру, аналогичную структуре промежуточного продукта в реакции окиси мезитила с галогенфосфинами. Другие простые метилкетоны реагируют аналогичным образом [164].
О/С МеСМе 4-PhPCl2 —> II Ph/ \
О
(121)
Для объяснения многих реакций альдегидов с монохлорфосфинами также предполагают образование циклического промежуточного продукта. Так, например, при взаимодействии дифенил-хлорфосфина с бензальдегидом с высоким выходом получается кристаллический продукт, которому приписана структура (122). Гидролиз (122) протекает как показано на схеме 211; при нагревании он может быть превращен в а-хлорфосфиноксид [165].
O^Ph
Ph2PCl + 2PhCHO —► Ph2r I X^o
Cl" Ph
(122)
О OH
H2O II I
---> PhCHO + Ph2P—CHPh
A (211)
О Ph С! О C!
Ph2P—CHOCHPh —► Ph2P—-CHPh + PhCHO
665
ЛИТЕРАТУРА
1. Organic Phosphorus Compounds’, ed. G. M. Kosolapoff and L Maier Interscience, New York, 1972-1974. ’ Wl‘ey-
2. K. Sasse, ‘Methoden der Organischen Chemie’, (Houben Weyl) G Th-Stuttgart, 1963—1964, vols. 12/1 and 12/2. ' n!eme.
3. H. D. Kaesz and F. G. A. Stone, J. Org. Chem., 1959, 24, 635.
4. H. Hoffmann and P. Schellenbeck, Chem. Ber., 1967, 100, 692.
5. W E. McEwen, A. Blade-Font, and C. A. Van der Werf, J. Amer Chem s 1962, 84, 677. ' dOC -
6. G. Wittig, H. Braun, and H.-J. Cristau, Annalen, 1971, 751, 17.
7. M. Schmidt and W.-R. Neeff, Angew. Chem. Internat. Edn., 1970, 9, 807
8. K. D. Berlin and D. M. Hellwege, in ‘Topics in Phosphorus Chemistry’,'ed M. Grayson and E. J. Griffith, Interscience, 1969, vol. 6, p. 1.
9. D. B. Denney and F. J. Gross, J. Org. Chem., 1967, 32, 2445.
10. W. Wolfsberger and H. Schtnidbaur, Synth. React. Inorg. Metal-Org Chem 1974, 4, 149.
11. A. M. Aguiar, J. R. S. Irelan, C. J. Morrow, J. P. John, and G. W Prejean J. Org. Chem., 1969, 34, 2684.
12. F. Mathey and J. P. Lampin, Tetrahedron, 1975, 31, 2685.
13. L. Maier, В cc. 1, vol. 1, chapter 2.
14. S. O. Grim and R. P. Molenda, Phosphorus, 1974, 4, 189.
15. H. J. Becher, D. Fenske, and E. Langer, Chem. Ber., 1973, 106, 177.
16. A. M. Aguiar, H. J. Greenberg, and К. E. Rubenstein, J. Org. Chem., 1963, 28, 2091.
17. K. Issleib, A. Tzschach, and H.-U. Block, Chem. Ber., 1968, 101, 2931.
18. A. M. Aguiar, C. J. Morrow, J. D. Morrison, R. E. Burnett, W. F. Masler, and N. S. Bhacca, J. Org. Chem., 1976, 41, 1545.
19. D. G. Gillespie and B. J. Walker, Tetrahedron Letters, 1975, 4709.
20. R. A. Naylor and В. J. Walker, J. C. S. Chem. Comm., 1975, 45.
21. X. D. Crosbie and G. M. Sheldrick, J. Inorg. Nuclear Chem., 1969, 31, 3684.
22. R. C. Taylor, R. Kolodny, and D. B. Walters, Synth. Inorg. Metal-Org. Chem., 1973, 3, 175.
23. /. G. M. Campbell and J. K. Way, J. Chem. Soc.. 1961, 2133.
24. P. D. Henson, K. Naumann, and K. Mislow, J. Amer. Chem. Soc., 1969, 91, 5645.
25. G. P. Schiemenz and H.-U. Siebeneick, Chem. Ber., 1969, 102, 1883.
26. C. Symmes and L. D. Quin, Tetrahedron Letters, 1976, 1853.
27. K. L. Marsi, J. Org. Chem., 1974, 39, 265.
28. X. Naumann, G. Zon, and K. Mislow, J. Amer. Chem. Soc., 1969, 91, 7012, and following papers.
29. L. Horner, Pure AppL Chem., 1964, 9, 225.
30. R. B. King and A. Efraty, J. C. S. Perkin I, 1974, 1371.
31. R. Fields, R. N. Haszeldine, and J. Kirman, J. Chem. Soc. (C), 1970, 19/.
32. J. R. Wiseman and H. O. Krabbenhoft, J. Org. Chem., 1976, «1, 589.
33. K. Issleib and P. Malotki, Phosphorus, 1973, 3, 141. ,Q74
34. M. Shoufs, J. Meijer, P. Vermeer, and L. Brandsma, Rec. Trav. chim., 93 241.
35. E. W. Turnblom and T. J. Katz, J. Amer. Chem. Soc., 1973, 95,,4292
36. R. C. Marriott, J. D. Odom, and С. T. Sears, Inorg. Synth., 1975, o
37a. R. E. Harmon, S. K. Gupta, and D. J. Brown, Chem. Rev., I9/J, '°’ 376. H. B. Kagan, Pure Appl. Chem., 1975, 43, 401.
37. H. B. Kagan, Pure Appl. Chem., 1975, 43, 401. rhr-m Soc.,
38. O. Korpium, R. A. Lewis, J. Chickos, and K- Mislow, J. Amer.
39. W. S. Knowles, M. J. Sabacky, B. D. Vineyard, and D. J. Weinkauff, J- Amen Chem. Soc., 1975, 97, 2567.
40. R. Luckenbach, Tetrahedron Letters, 1971, 2177.
40a. W. J. Pope, S. J. Reachey, J. Chem. Soc. (London), 1899, 75,
666
£ Л. Nakamura. T. Kaao, and K. Tanl. J. Amer. Chem. See.. ,971. 93,
Г. H. Chan. Chem. Comm., 1968, 895.
E. Cernia, G. M. Giongo, F. Marcati, W. Marconi nrm м о ,, Chim. Acta, 1974, 11, 195 ' and N- Palladino, Inorg.
H. B. Kagan and T--P- Dang, J, Amer. Chem. Soc., 1972 94 6490
M. Tanaka and I. Ogata, Bull. Chem. Soc. Japan 1975 48 ina.i
1 P. Casey, R. A. Lewis, and. K. Mislow J Amor fhL c .r,™
J. Chait and R. A. Hart, J. Chem. Soc., I960, 1378 °C" 969’ 9I’ 2789‘
D. Berglind and D. W. Meek, Inorg Chem., 1969, 8, 2602
?969Л25,’ №?°ldWhite' H' KeyZer’ °' °' ’Row^11’ and R. Tang, Tetrahedron,
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
R. C. Taylor and D. B. Walters, Inorg. Synth., 1973 14 10
R, B. King, Accounts Chem. Res., 1972, 5, 177.
R. B. King and J. C. Cloyd, J. Amer. Chem. Soc., 1975, 97, 53.
M- M. T. Khan and A. E. Martell, Inorg. Chem., 1975,'14, 676
R. B. King, J. C. Cloyd, and P. N. Kapoor, J. C. S. Perkin I, 1973 2926 ‘Threshold Limit Values of Airborne Contaminants’, American Conference of Governmental Industrial Hygienists, 1014 Broadway, Cincinnati, Ohio 45202 R. F. Hudson, ‘Structure and Mechanism in Organophosphorus Chemistry; Academic Press, London, 1965, pp. 8—83; [P. Хадсон. Структура и механизм реакций фосфорорганических соединений. Пер. с англ. М./Мир 1967 с 17— 111].
117. A. Henderson and С. A. Slreuli, J. Amer. Chem. Soc., 1960, 82, 5791.
M. 1. Kabachnik and G. A. Balueva, Izvest. Akad. Nauk S. S. S. R., 1962, 536; [M И. Кабачник, Г. А. Балуева, Изв. АН СССР. Сер. хим., 1962, 536].
W, A. Henderson and S. A. Buckler, J. Amer. Chem. Soc., 1960, 82, 5794.
H. H. Jaffe and L. D. Freedman, J. Amer. Chem. Soc., 1952, 74, 1069.
L. C. Thomas, ‘The Interpretation of the Infrared Spectra of Phosphorus Compounds’, Heyden, London, 1974.
J. R. Van Wazer, ‘Phosphorus and its Compounds’, Interscience, New York, 1958, vol. I, pp. 1—87; [Ван Везер. Фосфор и его соединения. Пер. с англ. М„ Издатинлит, 1962, с. 10—78].
63. L. Maier, в сс. 1, vol. 1, рр. 98—99.
64. J. J. Breen, S. I. Featherman, L. D. Quin, and R. C. Stocks, J. C. S. Chem.
Comm., 1972, 657.
65. A. Rauk, L. C. Allen, and K. Mislow, Angew. Chem. Internat. Edn., 1970, 9,
66. R. D. Baechler and K. Mislow, J. Amer. Chem. Soc., 1970, 92, 3090.
66a. R. D. Baechler, W. B. Franham, and K. Mislow, J. Amer. Chem. Soc., 1969,
68.
69.
70.
91, 5686. , _ . ,r . , , , ,
67. S. E. Cremer, R. J. Chorvai, С. H. Chang, and D. W. Davis, Tetrahedron Letters, 1968, 5799. r , „, с Юто no
67a. IF. Egan, R. Tang, G. Zon, and K. Mislow, J. Amer. Chem. Soc., 1970, 92, 1442
676. W. Egan and K. Mislow, J. Amer. Chem. Soc., 1971, 93, 1805. .
67b. ‘Organophosphorus Chemistry’, Specialist Periodical Reports, . . PP > The Chemical Society. London, 1974, vol. 5, p. 137. Chem
R. D. Baechler, J. D. Andose, J. Stackhouse, and K. Mislow, J. Amer. Chem. Soc., 1972, 94, 8060. c lo71 0, 77o
R. D. Baechler and K. Mislow, J. Amer. Chem Soc, 971’ , Elsc.
A J Kirhn and S G Warren ‘The Organic Chemistry of Phosphorus, r. vter, Amsterdam, p Is «““ *“
ttllX a™. I'-*»-
нов, В. Ф. Желтухин. Усп. химии, 1970, 39, 77 ]• _
D. В. Bissing and А. 1. Speziale, J. Amer. Chem Soc „ 2683.
E. Vedejs and P. L. Fuchs, J. Amer. Chem. Soc ’ ’]974 142
A. J. Bridges and G. H. Whitham, J. C. S. Chem. Comm, 1УМ,
667
71.
72.
73.
74.
75 R. E. Ireland, M. I. Dawson, S. C. Welch, A. Hagenbach, J Bordner B. Trits, J. Amer. Chem. Soc., 1973, 95, 7829.
76. Л1. A. Shaw and R. S. Ward, Topics Phosphorus Chem., 1972. 7, 1.
77. K. Morita and T. Kobayashi, Bull. Chem. Soc. Japan, 1969, 42, 2732 and rences therein.
and
rele-
78. D. A. White and M. M. Balzer, Tetrahedron Letters, 1973, 3597.
79. A. E. Waite, D. W. Allen, and J. C. Tebby, Phosphorus, 1971, 1, 139
80. E. M. Richards and J. C. Tebby, J. Chem. Soc. (C), 1971, 1059.
81. E. M. Richards, J. C. Tebby, R. S. Ward, and D. H. Williams J Chem (C), 1969, 1542. ‘ ‘ 0C-
82. D. Seyferth and J. M. Burlitch, J. Org. Chem., 1963, 28, 2463.
83. E. Evangelidou-Tsolis, F. Ramirez, and J. F. Pilot, Phosphorus, 1974, 4, 109
84. R. G. Kostyanovsky, V. V. Yakshin, and S. L. Zimont, Tetrahedron 1968 24
2995. ’ ’
85. A. F. Janzen and О. C. Vaidya, Canad. J. Chem., 1973, 51, 1136.
86. K- Jssleib and K- D. Franze, J. prakt. Chem., 1973, 315, 471.
87. A. J. Kirby and S. G. Warren, ‘The Organic Chemistry of Phosphorus’, Elsevier, Amsterdam, 1967, pp. 4—8; [4. Кирби, С. Уоррен. Органическая ’химия фосфора. Пер. с англ. М., Мир, 1971, с. 115—136].
88. D. В. Denney and J. W. Hannifin, Tetrahedron Letters, 1963, 2177.
89. В. B. Jarvis and B. A. Marten, J. Org. Chem., 1975, 40, 2587.
90. J. J. Borowitz, К. C. Kirby, P. E. Rusek, and E. W. R. Casper, J. Org. Chem, 1971, 36, 88.
91. R. Appel, Angew. Chem. Internat. Edn., 1975, 14, 801.
92. J. J. G. Cadogan and R. K. Mackie, Chem. Soc. Rev., 1974, 3, 87.
93. R. Appel, F. Knoll, W'. Michel, W. Morbach, H.-D. Wihler, and H. Veltmann, Chem. Ber., 1976, 109, 58.
94. /. Tomoskozi, L. Gruber, and L. Radies, Tetrahedron Letters, 1975, 2473.
95a. R. Boigegrain, B. Castro, and C. Selve, Tetrahedron Letters, 1975, 2529.
956. R.-A. Boigegrain, B. Castro, and C. Selve, Bull. Soc. chim. France, 1974, 2623.
96. R. Appel, G. Baumer, and W. Struver, Chem. Ber., 1976, 109, 801.
97. R. Appel, W. Struver, and L. Willms, Tetrahedron Letters, 1976, 905.
98. J. I. G. Cadogan, Accounts Chem. Res., 1972, 5, 303.
99. L. E. Overman and E. M. O'Connor, J. Amer. Chem. Soc., 1976, 98, 771.
100. T. Mukaiyama, R. Matsueda, and M. Suzuki, Tetrahedron Letters, 1970, 1901.
101. E. J. Corey and D. J. Brunelle, Tetrahedron Letters, 1976, 3409.
102. A. Hassner and J. E. Galle, J. Amer. Chem. Soc., 1970, 92, 3733.
103. R. Aneja, A. P. Davies, A. Harkes, and J. A. Knaggs, J. C. S. Chem. Comm., 1974, 963.
104. M. S. Manhas, W. H. Hoffman, B. Lal, and A. K- Bose, J. C. S. Perkin I, 1975, 461.
105. S. Bittner, Z. Barneis, and S. Felix, Tetrahedron Letters, 1975, 3871.
106. J. I. G. Cadogan, Adv. Free-Radical Chemistry, 1968, 2, 203.
107. E. P. Kyba, J. Amer. Chem. Soc., 1976, 98, 4805.
108. L. Maier, в cc. 1, vol, 1, chapter 2. ,п7П
109. J. B. Lambert, G. F. Jackson, and D. C. Mueller, J. Amer. Chem. Soc., 19/0, 92 3093
110. L. R. Smith and J. L. Mills, J. C. S. Chem. Comm., 1974, 808.
111. P. Beck, в cc. 1, vol. 2, chapter 4. „ 747
112. H. Hoffmann and H. J. Diehr, Angew. Chem. Internat. Edn., 1964, 3, io .
113. G. Buehl and H. Wiiest, Helv. Chim. Acta, 1971, 54, 1767.
114. J. M. Swan and S. H. B. Wright, Austral. J. Chem., 1971, 24, 777
115. L. Horner, R. Luckenbach, and W.-D. Balzer, Tetrahedron Letters, 19oo,
116. L. Casser and M. Foa, J. Organometallic Chem., 1974, 74, 75.
117. G. Miirkl, Angew. Chem. Internat. Edn., 1963, 2, 620.
118. IF. R. Ptirdiim and K. D. Berlin, J. Org. Chem., 1975, 40, 2801.
119a. A. G. Anclrewes, G. Borch, and S. Liaaen-Jensen, Acta Chem. beano.,
30B, 214.
1196. В. C. L. Weedon, Chem. Abs., 1975, 83, 97568.
120. D. T. Longone and E. S. Alexander, Tetrahedron Letters, 1908,
668
12Ь Р. SchiPP^r’lQy4BQ6J'4^QQeSSen’ j' W' de Haan, and H. M. Buck, J. Amer.
,-,o 4 Bond, M. Green, and S. C Pearson, J. Chem. Soc. (B) 1968 929
«7 E. McEwen, Topics Phosphorus Chem., 1965, 2, 1. ’
j. 'Emsley and C. D. Hall, ‘The Chemistry of Phosphorus’, Harper and Row,
1 London, 197b.
, к Issleib and E. Priebe, Chem. Ber., 1959, 92, 3183
J. R- Corfield, N. J. De'ath and S Trippett, J. Chem. Soc. (C), 1971 1930
„7 Г. E. McEwen, C. A. Vanderwerf, and M. Zanger, J. Amer. Chem S<oc 1959
81, 3806. . ’ '
128. P- Gillespie, F. Ramires, I. Ugt, and D. Marquarding, Angew. Chem. Internat. Edn., 1973, 12, 91.
,9q R Luckenbach, Phosphorus, 1971, 1, 223.
30 R- Luckenbach, Phosphorus, 1972, 1, 293.
31' R. E. DeBruin and J. R. Petersen, J. Org. Chem., 1972, 37, 2272
132^ M J- De'ath’ Ellis, D. J. H. Smith, and S. Trippett, Chem. Comm, 1971, 714.
133 R- F- Hudson and C. Brown, Accounts Chem. Res, 1972, 5, 204.
134 ft7. Hawes and S. Trippett, Chem. Comm, 1968, 295.
,35' /. R. Corfield, N. J. De'ath, and S. Trippett, Chem. Comm, 1970, 1502.
1З6 H. Hellmann and J. Bader, Tetrahedron Letters, 1961, 724.
137 F. Mathey, Tetrahedron, 1973, 29, 707.
138 . D. G. Smith and D. J. H. Smith, J. C. S. Chem. Comm, 1975, 459.
139' G. Aksnes and .4. I. Eide, Phosphorus, 1974, 4, 209.
140. P. T- Keough and M. Grayson, J. Org. Chem, 1964, 29, 631.
141" E. Zbiral, Synthesis, 1974, 775.
142. M. E. Garst and T. A. Spencer, J. Org. Chem, 1974, 39, 584.
143. P. L. Fuchs, J. Amer. Chem. Soc, 1974, 96, 1607.
144. E. V. Dehmlow, Angew. Chem. Internat. Edn, 1974, 13, 170.
145. T. Sakakibara, M. Yamada, and R. Sudoh, J. Org. Chem, 1976, 41, 736.
146. M. Fild and R. Schmutzler, in Ref. 1, 1972, 4, pp. 75—154.
147. R. H. Tomlinson, in ‘Mellor’s Comprehensive Treatise on Inorganic and Theoretical Chemistry, Longmans, London, 1971, vol. VIII, suppl. Ill, pp. 438— 535.
148. K. A. Petrov and G. Ya. begin, Zhur. obshchei Khim, 1973, 43, 37; [К. А. Пет~ ров, Г. Я. Левин.—ЖОХ, 1973, 43, 37].
149. В. Fell and Н. Bahrmann, Synthesis, 1974, 119.
150. J. R. Corfield, R. K. Oram, D. J. H. Smith, and S. Trippett, J. C. S. Perkin I, 1972, 713.
151. F. Seel and K. D. Velleman, Chem. Ber, 1972, 105, 406.
152. 0. I. Scherer and P. Klusmann, Angew. Chem. Internat. Edn, 1968, 7, 541.
153. S. E. Cremer, F. L. Welti, F. R. Farr, P. W. Kremer, G. A. Gray, and
H. 0. Hwang, J. Org. Chem, 1973, 38, 3199.
154. J. 1. Bullock, N. J. Taylor, and F. W. Parrett, J. C. S. Dalton, 1972, 1843.
155. P. M. Draper, T. H. Chan, and D. N. Harpp, Tetrahedron Letters, 1970, 1687.
156. V. S. Tsivunin, L. N. Krutskii, T. V. Zykova, and G. K. Kamai, Zhur. obshchei Khim, 1969, 39, 2666; [В. С. Цивунин, Л. H. Крутский, T. В. Зыкова, Г. X. Камай.— ЖОХ, 1969, 39, 2666].
57. W. В. McCormack, U. S. Pat, 2663737/1900.
158. L. D. Quin and T. P. Barket, J. Amer. Chem. Soc, 1970, 92, 4303, and references therein.
ten ^werbouch and Y. Kashman, Tetrahedron, 1975, 31, 33.
m Heitkamp and F. Korte, Z. analyt. Chem, 1964, 204, 245.
mo У/ Kashnian and A. Rudi, Tetrahedron Letters, 1976, 2819.
104 К К. Khairullin, G. V. Dmitrieva, and A. N. Pudovik, Izvest. Akad Nauk s- S. S. R, Ser. Khim. 1970, 871; [В. К. Хайруллин, Г. В. Дмитриева, iRq Н- Пудовик, Изв. АН СССР. Сер. хим, 1970, 8/1].
Od- I. Kh. Gazizov, А. Р. Pashinkin, G. V. Dmitrieva, L. L. Tuzova, k. A. rullin, and A. N. Pudovik Zhur. obshchei Khim, 1972, 42, 1730; [Г. X. Гa-
669
зизов, А. П. Пашинкин, Г. В. Дмитриева, Л. Л. Тузова В К
А. Н. Пудовик.-ЖОХ, 1972, 42, 1730]. ' лаиРУМин,
164. S. Kh. Nurtdinov, R. S. Khatrullin. V. S. Tsivunin, T. V. Zykova, G Kh N i dinov, and G. Kh. Kamai, Zhur. obshchei Khim.. 1970, 40, 2377; ’ [С. X H динов, P. С. Хайруллин, В. С. Цивунин, Т. В. Зыкова, Г X НпптЛ.Р1' Г. X. Камай.-ЖОХ, 1970, 40, 2377]. ' JprauH°e,
165. N. J. Death, J. A. Miller, and М. J. Nunn, Tetrahedron Letters, 1973 sjai
166. F. Ramirez, M. Wowakowski, and J. F. Maracek, J. Amer Chem Soc’ 1077 99, 4515. ‘ ’ У//’
10.3. ПРОИЗВОДНЫЕ ФОСФОРИСТОЙ кислоты
P. С. ЭДМУНДСОН (University of Bradford)
10.3.1. ГАЛОГЕН- И АЗОТСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ФОСФОРИСТОЙ КИСЛОТЫ [1—9]
При взаимодействии спиртов с трихлоридом фосфора протекает сложный ряд последовательных и параллельных реакций, в результате которых образуются алкилдихлорфосфиты (1; Х = С1), диалкилхлорфосфиты (2; Х = С1) и, наконец, в специальных условиях— триалкилфосфиты (3) (схема 1).
Трибромид фосфора ведет себя аналогично, однако ни трифторид, ни трииодид так не реагируют; фенолы образуют арильные аналоги соединений (1) — (3); аналогичные ряды производных дают тиолы п амины. Такие галоген- и азотсодержащие производные фосфористой кислоты играют важную роль как промежуточные продукты в синтезе других соединений фосфора (III), а также соединений четырехкоординированного фосфора. В последние годы эти соединения с большим успехом используются в общем органическом синтезе.
ROH ROH ROH НХ
РХз "TT? ROPX2 (RO)2px —тдг (R°)3p ---”
(1) (2) (3)
—> (RO)2P—ОН (RO)2P^ (О
\н
(4) (5)
X = С1, Вг
10.3.1.1 . Синтез галогенфосфитов реакцией тригалогенидов фосфора со спиртами, фенолами и их тиоаналогами
Благодаря проводившимся на протяжении многих лет дета^_ ным исследованиям была установлена основная особенность Р акций, протекающих при смешении трихлорида или триоро! фосфора со спиртами или фенолами. Результаты этих раоот
670
„лно демонстрируют одно важное свойство трехвалентного £ cd>opa, а именно его электрофильность.
результат реакций трпгалогенидов фосфора со спиртами оп-рделяется прежде всего природой галогена, реакционной способностью гидроксильных групп, а также наличием или отсут-СтвИем подходящего реагента для связывания выделяющегося гапогеноводорода. Необходимо подчеркнуть, что старое правило /Ярошенко, 1897 г.), согласно которому только первичные спирты образуют фосфорсодержащие продукты, тогда как вторичные третичные спирты дают соответственно алкены и алкилгалоге-нпДЫ, не является общим. На направление реакции оказывают влияние пространственные факторы в самом гидроксильном соединении, а также порядок смешения реагентов, поскольку он втияет на состав реакционной смеси в каждый данный момент времени. В отсутствие акцептора хлористого водорода при добавлении первичных или вторичных спиртов «средней» реакционной способности к трихлориду или трибромиду фосфора протекает последовательное замещение галогена, в результате чего быстро образуется алкилдигалогенфосфит (1), более медленно превращающийся в диалкилгалогенфосфит (2) (см. схему 1). На обеих этих стадиях выделяется галогеноводород; на третьей стадии выделения галогеноводорода не происходит из-за дезалкилирования триэфира (3) в диалкилфосфит (4), которое протекает чрезвычайно быстро даже при —10 °C, в результате чего выделить триэфир в этих условиях обычно невозможно. Тринеопентил-фосфит является исключением из-за его устойчивости к дезалкилированию под действием хлористого водорода вследствие пространственного влияния объемистых алкильных групп и может быть получен даже в отсутствие акцептора галогеноводорода.
Дихлорфосфиты и хлорфосфиты— производные первичных и большей части вторичных спиртов термически устойчивы; производные спиртов бензильного типа могут разлагаться при высоких температурах. В случае третичных спиртов при отсутствии точного соблюдения экспериментальных условий образуются значительные количества алкилхлоридов.
При взаимодействии .необходимого количества алканднола с трихлоридом фосфора, иногда в растворителе, с высокими выходами образуются циклические хлорфосфиты, например 2-хлор-1,3,2-диоксафосфолан (6) и 2-хлор-1,3,2-диоксафосфоринан (7; 4=1). Соединение типа (7) с наибольшим размером цикла, синтезированное к настоящему времени, имеет п =10. При наличии заместителей у атомов углерода цикла появляется возможность геометрической изомерии таких циклических хлорфосфитов; многие изомеры были выделены, некоторые нз них в стереохимически чистом виде. В большинстве случаев они отличаются конфигурацией атома фосфора; поскольку эпимеризация, по-ви-ДИМому, происходит лишь в кислых условиях, то присутствие
671
акцептора галогеноводорода может влиять на стереоизомерный со став образующихся циклических продуктов.
(7)
При взаимодействии с фенолами вследствие меньшей электронной плотности на атоме кислорода и устойчивости связи С (арил) — —О к нуклеофильным атакам скорости представленных на схеме 1 реакций уменьшаются, что облегчает их контроль. Тем не менее выходы дихлорфосфитов и хлорфосфитов обычно ниже, чем в случае алкильных производных, хотя и могут быть несколько увеличены введением катализаторов — хлоридов металлов, например хлоридов магния или калия. Двухатомные фенолы соответствующей структуры дают циклические хлорфосфиты. Так, например, из пирокатехина получается 2-хлорбензодиоксафосфолан (8), а из 2,2/-дигидроксибифенила — 2-хлордибензо [с, е]-1,3,2-ди-оксафосфепин (9); эти соединения были подробно изучены Ан-шютцем в начале века. Из салициловой кислоты синтезирован 2-хлор-4//-1,3,2-бензодиоксафосфорииаион-4 (10) [10].
РС1
(8)
(9)
При использовании РВг3 более высокая нуклеофильность Вг~ и большая растворимость бромистого водорода в фосфорорганических реагентах и продуктах затрудняют контроль приведенных на схеме 1 реакций. Лишь в сравнительно небольшом числе случаев удается выделить дибромфосфиты (1; Х = Вг) и бромфос-фиты (2; Х = Вг), не загрязненные продуктами дезалкилирования, хотя бы и незначительного. Бутилдибромфосфит, например, устойчив при 100 °C, однако при более высоких температурах распадается, давая бромбутан; дибутилбромфосфит разлагается уже при комнатной температуре. Однако некоторые арильные производные, в том числе бромсодержащие аналоги соединений (8) (10), достаточно устойчивы.
Как и следовало ожидать, иодсодержащие аналоги соединений (1) и (2) еще менее стабильны. Какие-либо прямые данны в пользу реализации схемы 1 при смешении спиртов с Р1з оТ' 672
cvtctbvjot, а немногие известные нодпооизвотнм₽ л,,-, рованы другими методами. Р е ^ЫЛи счнтези-
Как алифатические, так и ароматические тнолп
ры дигалоген- и галогентиофосфористых кислот т бразу1ит ЭФИ’ соединений (1) и (2), а таТ<жеР (6)1 10? 111 ТИОанало™ спиртов в данном случае возможно осуществление" 1 °ГЛИЧИе От дезалкилирования третьей стадии процесса что Н°Г°
приводит к триэфирам (RS)3P. ’ ° НепосРеДственно
10.3.1.2 . Другие пути синтеза галогенфосфитов
Для получения галогенпроизводных фосфористой кислоты были использованы и многие другие реакции, как в силу необходимо СТИ (в случае иодидов), так и по соображениям практического удобства, например чтобы избежать трудностей, возникающих при разделении продуктов с близкими температурами кипения Э™ методы включают алкилирование тригалогенидов фосфоиа' 11 ксидами и реакции обмена галогена (уравнения 2—4) Р Р
\ / 'о"
РС1з+ —> С1СН2СН2ОРС12 ----------► (С1СН2СН2О)2РС1 (2)
N2, 60 °C
PrOPCI2+SbF3----------> PrOPF, + SbCl3 (3)
эфир
(EtO)2PCl H Lil ---► (EtO)2PI + LiCl (4)
Реакции обмена между галогенидами фосфора и триэфирами фосфористой кислоты (уравнения 5, 6) быстро протекают при температурах перегонки.
(ВиО)зР + 2РС13 —> BuOPCl2 + (ВиО)2РС1 (5)
(ВиО)2РС1 + РС13 —> 2ВиОРС12 (6)
Прекрасный метод получения диэтилхлорфосфита, свободного от этилдихлорфосфита, основан на использовании в реакции обмена пентакоординационного трихлорида (11) (схема 7) [И].
’/222 Зак. 395
673
10.3.1.3 . Синтез азотсодержащих производных фосфористой кислоты
Для синтеза этих соединений обычно используют реакции три галогенидов фосфора с аминами [12, 13]. С точки зрения сум' марной стехиометрии, реакции, протекающие при взаимодействий вторичных аминов с трихлоридом или трибромидом фосфора формально сходны с реакциями тригалогенидов фосфора с гид-’ роксильными соединениями; для диметиламина эти превращения представлены на схеме 8.
Me2NH Me2NH Me2NH
РХ3 *" Me2NPX2 * (Me2N)2PX ► (Ме2М)зР (8)
(12)
X = Cl, Br
Реакция останавливается на стадии образования трис(диме-тиламино)фосфина (12) [14]; обратного дезаминирования, аналогичного дезалкилированию (см. схему 1), не наблюдается. И-подходящих диаминоалканов или диаминоаренов образуются циклические соединения, например 1,3-диметил-2-хлор-1,3,2-диазафос-фолидин (13). В отдельных случаях могут быть использованы реакции обмена, аналогичные реакциям, применяемым для получения кислородсодержащих соединений (уравнение 9).
(Me2N)2PCl + РС13 —> 2Me2NPCl2 ф
Более сложными являются реакции с участием первичных аминов. Некоторые простые алифатические амины реагируют с трихлоридом фосфора, давая амидодихлорфосфиты RNHPC12, которые в зависимости от используемого амина либо разлагаются при перегонке, либо могут быть перегнаны без разложения. При использовании гидрохлоридов аминов возможно образование продуктов
других типов. Метиламин образует необычное каркасное соедин ние (14); анилин, как и его гидрохлорид, дает тетрахлорид (
Ь74
или циклическое соединение (16), которое является циклическим димером соединения PhN — PCI, образующегося при отщеплении HCI из ожидаемого дихлсрида PhNHPCl2. Первоначальному продукту взаимодействия трихлорида фосфора с 2-аминобифенилом приписывается структура (17), поскольку в присутствии трихлорида алюминия протекает циклизация с образованием 2-хлор-1,2-дигидродибензо [с,е]-1,2-азафосфорина (18) (уравнение 10).
10.3.1.4 . Свойства связи фосфор (III) — галоген
В настоящее время, по-видимому, общепринято, что механизм нуклеофильной атаки на атом фосфора и разрыва связи фос-фор(Ш) — галоген аналогичен механизму Sx2 у насыщенного-атома углерода и обозначается поэтому как Sn2 (Р)-замещение галогена (схема 11); как уже упоминалось, в этой реакции фосфор-выступает в роли электрофила.
Л)
Nu~ > Р—Hal
8- 0 8-
Nu---р_—На1
Р1'
> Nu—Py-'/z/R1 + Hal \2
(11>
Примерами подобных реакций замещения могут служить реакции образования симметричных или смешанных эфиров (уравнение 12), изоцианато- (уравнение 13) и циано- (уравнение 14) производных, а также взаимодействие с амбидентными нуклеофилами (уравнение 15).
c5h5n
(EtO)jPCl + MeOH > (EtO)2POMe (12)
/ ° /—Q
/ /PC1 + KNCO — \—0 -> ( /PNCO (13)*
Me2N(EtO)PCl +AgCN — Me2N(EtO)PCN (14>
(EtO)2PCl + MeCOCHCO2Et Na+ —> (EtO)2POC(Me)=-CHCO2Et (15>
Реакции замещения галогена под действием металлорганиче-ских реагентов (уравнения 16—18) протекают даже в лучших случаях лишь с умеренными выходами, а реакции с магний- и литий-органическими соединениями осложняются кроме того расщеплением связи Р—О; даже связь Р—N не является в этих условиях.
абсолютно устойчивой [15].
(EtO)2PCl + PhMgBr —> (EtO)2PPh (16>
EtOPCl2 4- Et2Zn —► EtOPEt, (17>
Me2NPCl2 + 2PhLi —> Me2NPPh2 (18)
*/222* 675,
Особенно важным является замещение галогена под действием воды (схема 19): эта реакция гидролиза в зависимости от соотно* шения реагентов приводит пли к вторичным фосфитам (точнее считать эти соединения производными тетракоордпнированного фОс. фора, см. разд. 10.3.2.4)* [16], или к пирофосфитам (19), которые можно рассматривать как ангидриды вторичных фосфитов (4).При использовании вместо воды сероводорода образуются серусодер-жащие аналоги (4) и (19).
н2о н2о
(EtO)2P-O-P(OEt)2 < ~ 2(EtO)2PCl -----> 2(ЕЮ)2Р(О)Н (19)
(19)
Стереохимическое направление производных фосфористой кислоты
замещения галогена в хлор-изучено на примере конфигу-
(20)
н+ ---->
НгО
(23)
* Строго говоря, термины вторичный фосфит и диоргаиофосфит приме лишь к соединениям типа (RO)2P—ОН, содержащим трехкоординированныйi а фосфора. Однако эти соединения практически полностью существуют в ф г д (RO)2P(O)H с тетракоординированным атомом фосфора; соединения под структуры следует называть диоргапофосфонатами (см. разд. 10.1.2). 1ем ' -сЯ нее в дальнейшем соединения типа (RO)2P(O)H в соответствии со сложп® >1И
в отечественной литературе традицией мы будем называть диорганофосф или вторичными фосфитами. — Прим, перев.
676
ранионно стабильных 2-хлор-1,3,2-диоксафосфоринанов [17] а именно в ряду 4,6-диметилпроизводиых (схема 20). Мосбо и Вер-кейд [18] гидролизом хлорфосфита (20) получили стереоизомерные продукты (21) (22) и (23) в соотношении — 70:30 Эти
эпимерные продукты отличаются по химическим сдвигам в спектрах ЯМР 'Н и 3|Р, константам спин-спинового взаимодействия Р Н, а также по данным поглощения в ИК-области спектра где наиболее важными характеристиками являются частоты колебания связей Р=О и Р—Н, занимающих аксиальное или экваториальное положение.
Соединение бр, млн-1 7РН. Гц VP=O' см"' VP—Н, СМ"’
(22) -3,1 664 1283 2406
(23) + 1,2 719 1254 2470
Хотя изомеры (22) и (23) могут переходить друг в друга, этот процесс является очень медленным и для установления равновесия требуется нагревание при 120°C в течение 72 ч. Считается, что образование основного продукта (22) через его таутомер, вторичный фосфит (21), согласуется с механизмом Sn2(P) независимо от природы частицы, атакующей фосфор.
10.3.1.5 . Свойства связи фосфор(Ш) — азот [13, 14, 19]
Связь фосфор (III)—азот особенно чувствительна к действию кислот: в безводных условиях под действием галогеноводородов образуются амидогалогенфосфиты, тогда как водные кислоты дают вторичные фосфиты или их аналоги (уравнения 21, 22). Взаимодействие с карбоновыми кислотами в безводных условиях приводит к амидам карбоновых кислот (уравнение 23); эту реакцию можно использовать для превращения амидов кислот фосфора(III) во вторичные фосфиты (уравнение 24) (см. также разд. 10.3.2,4).
НВг, эфир
----——у (Me2N)2PBr —> Me2NPBr;
(Et2N)3P [ОН, О /PNMe О Н3О+ >- (Et2N)2POH 7Д* (Et2N)2P^ (22) ХН Р + 3PhCO2H —> 3 / NCOPh + Н3РО3 (23) <0\ +° 2 + МеСО2Н —► MeCONMe2+ Р (24) Ч / хн О
22 Зак. 39q
677
Амидофосфиты являются, таким образом, удобными аминирую» ми агентами. Помимо приведенных выше реакций можно 'т.о ЩИ’ превращение эфиров дикарбоновых кислот в амидоэфиры амиды (уравнение 25), а также получение енаминов из к. ных соединений (уравнения 26, 27).
PhCH(CO2Et)2 +
упомянуть ’Г'1 И ди-арбониль-
CO2Et
PhCHCO—N
побочный продукт основной продукт
MeCOCH2CO2Et + (Et2N)3P —>
NEt2
I
MeC=CHCO2Et
(25)
(26)
(27)
Ангидриды карбоновых кислот реагируют с амидофосфитами, давая ацилфосфиты и амиды карбоновых кислот (уравнение 28). Относительно недавно открыта реакция внедрения диоксида углерода по связи Р—N (схема 29); аналогично реагирует сероуглерод. Эти реакции протекают, по-видимому, путем нуклеофильной атаки неподеленной пары электронов атома азота по атому углерода карбонильной группы. (Далее обсуждаются другие возможные реакции амидофосфитов.)
ЕЮ.
(МеСО)2О +EtOP(NEt2)2 —> MeCONEt, + JTOCOMe (28) Et2N/
^Me2N^2 P—NMe2
(MegN)2P—NMe2
"O—c=o
, . cog
---► (MesN)2P0CONMe2------> Me2NP(OCONMe2)2
Ранние данные об образовании амидов карбоновых кислот пр взаимодействии амидофосфитов с карбоновыми кислотами, напр мер получение соответствующих анилидов из соединений (25), позволили разработать метод синтеза пептидов [20]. Р ботка N-фосфор(III)содержащего производного эфира амино ~ лоты подходящей N-защищенной аминокислотой приводит к от лению вторичного фосфита и образованию N-защищенного nei 678
С незначительном в большинстве случаев рацемизацией (уравнение 30).
О
(EtO)2PNHPh ^PNHPh
О
(24) (25)
PhCH2OCONHCH2CO2H + (EtO)2PNHCHCO2Et -----------—►
| —(EtO)2P(O)H
CH2Ph
PhCH2OCONHCH2CONHCHCO2Et
I CH2Ph
(30)
Электрофильный характер трехвалентного фосфора ярко проявляется также в возможности легкого замещения амидной функции в амидофосфитах при нагревании их со спиртами, фенолами, тиолами или даже с другими аминами (уравнения 31, 32). Реакция «переаминирования» легко поддается контролю; возможность ее применения в синтезе хорошо иллюстрируется схемой 33.
(EtO)2PNMe2 + РЬОН —► (EtO)2POPh + Me2NH (31)
(Et2N)3P + 2PhCH2SH —► Et2NP(SCH2Ph)2 + 2Et2NH (32) о o
P(NEt2)3 S' \ EtOH S' \ PrOH
HOCH2CH2NHMe ---------> || ^)PNEt2 ----> f ^POEt ----------*-
NMe NMe
/OPr
—► MeNHCH2CH2Op/
^OEt
(33)
Способ образования экзоциклической связи фосфор—азот в циклических системах — из циклического хлорфосфита и амина или из диола путем «переаминирования» — может влиять на стереоизомерный состав продуктов, особенно часто на конфигурацию У атома фосфора. Хотя в каждом конкретном случае имеются, вероятно, различия в предпочтительности образования одного из эпимеров, преимущественная ориентация экзоциклической связи Р—N в 1,3,2-диоксафосфоринанах отличается от ориентации связи Р—О в том же ряду (см. разд. 10.3.2.1) [21]. Реакция гексаметплтриами-дофосфита с 2-трет-бутилпропандиолом-1,3 приводит к смеси гранс-5-трет-бутил-2-диметиламино- 1,3,2-диоксафосфоринана (26) (6Р 142,4 млн-1, C6D6, относительно 85%-ной Н3РО<) и щ-ш-изомера (27) (дР 135,4 млн-1) в соотношении 3:2, однако через три недели стояния при комнатной температуре (а в хлороформенном растворе— через два дня) это соотношение возрастает до 5:1. Большая термодинамическая стабильность транс-изомера, по-видимому, обусловлена ослаблением вицинального взаимодействия электронных пар фосфора и азота по сравнению с /^ас-изомером.
23*
До сих пор мы рассматривали электрофильные свойства трехвалентного фосфора. Однако в большинстве реакций соединений фосфора(III) отчетливо проявляется его нуклеофильный характер, причем особенно ярко, если фосфор связан с азотом, и менее значительно— в кислородных соединениях, вследствие чего, например, амидофосфиты присоединяют кислород и серу более энергично, нежели триэфиры фосфористой кислоты. Амиды проявляют также более высокую по сравнению с триэфирами реакционную способность по отношению к алкилгалогенидам, образуя при этом псев-дофосфониевые соли (28), и к галогенам, давая галогенфосфониевые соли (29) и (30) [22]. Более высокая стабильность азотсодержащих солей обусловлена, вероятно, отсутствием сколько-нибудь значительной движущей силы для их распада, поскольку энергия связи P=N значительно ниже энергии связи Р=О.
(Me2N)3PMe Г
(28)
(Me2N)3PX Y" (29)
X = Y = Cl
X = Cl, Y = FeCU X = Br, Y = BPh4
(Me2N)3PX Y3'
(30)
X = Y = Cl
Считается, что промежуточные продукты, аналогичные по структуре приведенным выше галогенсодержащим солям, образуются при взаимодействии гексаметилтриамидофосфита с полигалогени-рованными алканами; при обработке таких смесей спиртами ROH образуются алкилгалогениды RC1 (схема 34) [23].
ссц + - ROH +
(Me2N)3P ----> [(Me2N)3PCl СС13] ———* [(Me2N)3POR СГ] —>
—СпС1з
—> RC1 + (Me2N)3P=O (34)
Вследствие повышенной легкости окисления амидов кислот фосфора (III) по сравнению с триэфирами фосфористой кислоты, эти амиды привлекают к себе все возрастающее внимание как реагенты для отрыва кислорода или серы из молекул различных соединений (см., например, уравнение 35) [24, 25]. Недавно гексаметилтри-амидофосфит был использован в новой реакции превращения тиолов в первичные амины с тем же числом углеродных атомов (схема 36).
Вследствие наличия неподеленных электронных пар на соседних атомах в амидах кислот фосфора(III) возникает чрезвычайно интересный вопрос об относительной доступности этих неподелеН 680
RSOaSR’ + (Et2N)8P —► RR'SO2 + RSOR1 + (Et2N)3P=S
(35)
О
О
(Me2N)sP
NSR ---------► (Me2N)3P=S +
О
О
NR
KOH ---------у или NH2NH2
rnh2
(36)
ных пар для электрофильных реагентов. Можно различать три типа реакций между такими амидами и карбонильными соединениями [26]: примеры реакций первого типа [(31)], в которых участвует неподеленная пара азота, уже приводились выше (см. уравнения 23—29). Реакции второго типа [(32)] протекают с участием иепо-целенной пары электронов фосфора; такой тип нуклеофильной атаки обнаружен в реакциях образования пентакоординационных спироциклических молекул из циклических амидофосфитов и 1,2-ди-карбонильных соединений, таких, как диацетил или бензил, хотя наличие связи Р—N в случае других типов амидных реагентов может ингибировать процесс циклизации. Последний вариант [(33)] также включает атаку неподеленной пары атома фосфора; этот тип реакции реализуется, например, при медленном присоединении гексаалкилтриамидофосфита к ароматическим альдегидам, приводящем к эпоксидам (уравнения 37, 38) [27].
R2n:
R2P^O=C^
r2n:
р2р:-'~^С==о
, , < ч медленно
(Me2N)3P PhCH------------J
О
Ph
> , . + I —
> (Me2N)3P-C-O
H
Ph CH I. О
Plu>H
быстро
|| -(Me2N)3P=O PIT
(37)
10.3.2. ЭФИРЫ ФОСФОРИСТОЙ КИСЛОТЫ Г«-8. 101
Как видно из изложенного выше, многие реакции соединений фосфора (III), содержащих связи Р—N, могут быть объяснены с точки зрения электрофильности атома фосфора несмотря н
681
наличие легко доступной неподеленной электронной пары. Эта ха. рактерная особенность атома фосфора иллюстрируется далее на примере реакций синтеза эфиров фосфористой кислоты.
10.3.2.1. Электрофильность фосфора в синтезе триэфиров фосфористой кислоты
Прямая этерификация фосфористой кислоты является во всех отношениях малопригодной реакцией для получения триэфиров фосфористой кислоты; более удобные методы синтеза этих соединений основаны на использовании галогенсодержащих полупродуктов или амидов кислот фосфора (III).
При проведении реакции спиртов с трихлоридом или трибромидом фосфора в присутствии органических оснований (пиридин, триэтиламин или N.N-диалкиланилины), служащих акцепторами выделяющегося галогеноводорода, конечным продуктом является триэфир (3) (см. схему 1). В принципе этот путь пригоден для получения ациклических асимметрических эфиров (34) (схема 39), однако большая часть известных асимметрических триэфиров имеет циклическую структуру с атомом фосфора в цикле.
юон
РС13 -^77* R‘OPC12 RaN
R2OH
RaN
R3OH
---->
R3N
R'Ck
7P—OR3 r2q/
HOCH2(CH2)nCH2OH, RaN
О —V
R‘O-P( \CH2)„ o—7
Некоторые особенности триэфиров фосфористой кислоты (см. разд. 10.3.1.1) могут осложнять их синтез. Так, например, многие триэфиры, полученные из спиртов бензилового типа, термически нестабильны, особенно в присутствии нуклеофильных примесей. Выбор органического основания также может иметь некоторое значение. Так, трп-трет-бутилфосфит, как сообщалось, образуется при взаимодействии трет-бутилового спирта с трихлоридом фосфора в присутствии N, N-диметнланилина, но не в присутствии пиридина, а при использовании триэтиламина получается ди-трет-бутилфос-фит. Галоген- и нитрофенолы могут давать с хорошими выходам» триэфиры и в отсутствие органического основания.
Очень удобный метод получения триэфиров фосфористой кислоты основан на термодинамически контролируемой равновесной реакции между триэфиром, чаще всего триэтил- или трифенилфос' фитом, и необходимым количеством спирта или фенола; подобные процессы катализируются следовыми количествами оснований, на пример этоксида или феноксида натрия, или кислот, например фосфорной. Гексаалкилтриамидофосфиты и другие амиды фосф 682
ристой кислоты также могут использоваться в этой реакции (см. разД- Ю-3.1.5). Эта реакция особенно удобна для получения эфиров> чувствительных к действию кислот, например для получения Грет-бутиловых эфиров (уравнение 40). Необходимо отметить, что, поскольку триэфиры фосфористой кислоты элиминируют серу нз ТИОЛОВ, давая эфиры тиофосфорной кислоты и углеводороды /уравнение 41), реакция переэтерификации между фосфитами и тиолами невозможна, хотя переэтерификация эфиров тритиофосфо-ристой кислоты тиолами осуществляется легко (уравнение 42). Следует также отметить, что некоторые спирты бензилового типа, например 2-гидроксибензиловый спирт (салигенин), не вступают в нормальную реакцию переэтерификации, а образуют циклические фосфонаты со связью фосфор—углерод в составе пятичленного цикла.
Me, j—О Ale, .—о
V ^PNMe2 + трет-ВиОН —> V РОВи-трет + Me2NH (40)
Me' '—О Me' '—О
(ЕЮ)зР + RSH —> (EtO)3P=S + RH (41)
(MeS)3P + 3PrSH —> (PrS)3P+ 3MeSH (42)
Реакции переэтерификации триэфиров и триамидов фосфористой кислоты применяют для получения жестких каркасных триэфиров из триолов. Так, 2,2-ди (гидроксиметил) пропанол и цпс-глюцит образуют соответственно 2,6,7-триокса-1-фосфабицикло-[2.2.2] октан (35) и 2,8,9-триокса-1-фосфаадамантан (36). Оба соединения используют в синтезе моноциклических эфиров тетра-координированного фосфора заданной конфигурации.
(36)
Эти методы могут быть использованы также для получения моноциклических эфиров (см. схему 39). При увеличении длины углеводородной цепи в диоле выход циклических продуктов заметно снижается, тогда как выход полимера возрастает. Эйерс и Райдон [28], например, показали, что в реакции переэтерификации трифенилфосфита алкандиолами-1,со выход моноциклического фе-нилфосфита уменьшается от 60 до 0% при увеличении числа метиленовых групп в молекуле диола от 2 до 6.
Присутствие в цикле расположенных подходящим образом заместителей приводит к появлению геометрической изомерии у пяти-и шестнчленных циклических эфиров [29]. Чистые изомерные Циклические эфиры были впервые выделены в 1967 г. Акснесом [30] с использованием метода ГЖХ. С тех пор конфигурация многих подобных эфиров, а также соответствующих фосфатов и
683
тиофосфатов явилась предметом ряда исследований с применением методов спектроскопии ЯМР ‘Н,31Р и 13С, ИК-спектроскопии, измерения дипольных моментов и дифракционных методов. Целью этих работ было, отчасти, получение модельных соединений с известной устойчивой конфигурацией атома фосфора для использования их при изучении механизмов реакций. Обычно нет необходимости раз-делять"смесь изомеров; соотношение изомеров как в исходных соединениях, так и в продуктах может быть достаточно надежно установлено, особенно с помощью спектроскопии ЯМР 31Р. Типичными являются работы Бодкина и Симпсона [31], исследовавших 4-ме-тил-1,3,2-диоксафосфоринаны, Бентруда и Харгиса [32], посвященная 5-77>ет-бутил-1,3,2-диоксафосфоринану, а также работа Мосбо и Веркейда [33], изучавших 4,6-диметильные аналоги, полученные из шезо-пентандиола-2,4 (схема 43).
РС!3, c5h5n, Et2O
MeOH,Et3N ---------->.
EtzO
(37)
Me
9
>- (37)
Me""7^.OH P(OMe)3
^Z^oh
Me
Поскольку реакция переэтерификации является равновесной и контролируется термодинамически, тогда как алкоголиз хлорфос-фитов представляет собой кинетически контролируемый процесс, следует ожидать различий в стереоизомерном составе продуктов в этих двух случаях, что и наблюдается в действительности. Принято считать, что обычно термодинамически более стабильный хлорфосфит дает менее стабильный метилфосфит, который в безводных условиях в присутствии следов кислоты эпимеризуется в более устойчивый триэфир; в примере, показанном на схеме 43, эта эпимеризация протекает в направлении (38)—>(37). Кинетически контролируемая реакция хлорфосфита приводит к смеси (37) (6р = —129 млн-1) и (38) (5Р = —133 млн-1) в соотношении, которое сильно изменяется от опыта к опыту, однако обычно составляет ~ 1 :3.
10.3.2.2. Электрофильность фосфора в реакциях триэфиров фосфористой кислоты
Помимо реакций замещения галогена в галогенидах трехва лент кого фосфора и процессов переэтерификации, соединения Ф° фора(III) вступают в некоторые интересные с синтетической точк 684
,ре„,,я реакция, в которых проявляете» его электрофильны,", ха. ’’‘"'реакции триэфиров фосфористой кислоты с магний я„ -рГаннческими соединениям,, приводят к фосфоцитам , фосЛннн там (эФ"Г\ КЙ1ЛОТ тРехва“нтного фосфора с одной ,, двумя связями ФОСФ°Р Угл«род соответственно) и, в конечном счете к третичным фосфинам. Иногда, используя обратный порядок при 6авленпя реагентов, удается остановить реакцию на промежуточных стадиях однако выходы эфиров редко бывают высокими (уравнение 44) [10].
(MeO)3P + 3PhMgBr —> PhP(OMe)2 + Ph2POMe + Ph3P (44)
Гидролиз триэфиров фосфористой кислоты в кислых (быстрый) или основных (более медленный) условиях приводит к вторичным фосфитам. Триметилфосфит быстро гидролизуется водой, тогда как другие простые триалкнлфосфиты устойчивы к действию воды даже при 100 °C. Для получения вторичных фосфитов имеются более эффективные методы (см. разд. 10.3.2.4).
Ацнлфосфиты, являющиеся смешанными ангидридами карбоновых и диорганофосфористых кислот, обладают двумя потенциальными электрофильными центрами — атомом фосфора и карбонильным атомом углерода, которые могут конкурировать в реакциях с нуклеофилами (схема 45) [34].
МеЫН2
о
(RO)2PNHMe + МеСО2Н (путь А)
хО (45)
(RO)2P + MeCONHMe (путь Б)
(RO)2POCMe
MeNH2
О О
РОСМе
О
MeNH2
78% по пути Б, 22% no пути A
О
О
MeNH2 „
РОСМе--------* 100% по пути Б
v О
Ацнлфосфиты типа (39) были получены in sitting пирофосфпта и N-защцщенной аминокислоты в одной из реакций получения пептидов (схема 46) [20].
ХО PrHrM RCONHCHRlCOsH^
(ЕЮ)2РС1 + (EtO)2P^ —-5 (EtOhP—о-—P(OEt)2 _<еЮ)2р(О)Н
R1 О (46)
- RCONHCHCOP(OEt), RCONHCHR'CONHCWCCW
(39)
685
Электрофильность фосфора (III) отчасти является причиной неустойчивости эфиров типа трис(2-гидроксиэтил)фосфита (40) который вследствие «внутримолекулярной переэтерификации» а при температурах выше —20°C за счет двойной циклизации прев.’ ращается в находящийся в равновесии с моноциклическпм эфиром (41) спиран (42) (схема 47).
НО(СН2)2ОН (Me2N)3P -------------> [(HOCH2CH2O)3PJ —\ он
4 •““Г1О(СН2)2'-)Н
(40)
НО(СН2)2ОН |
РО(СН2)2ОН
О
(41)
НО(СН2)2ОН
(47)
(EtO)3P
О POEt
р:
Г Ph.
(MeO)2POPh + MeSPh —>
II
О
/oph' S—О—Р—ОМе
\)Ме-
О О
(42)
О
—> (MeO)2POPh + MeSPh
(48)
О Н о
о
РЬч + МеОч
XS—ОМе ХР—О'
Ме-^ Pho/
МеОч А
ХРХ + MeSOPh Ph©/ ^Ме
Электрофильностью фосфора(III) можно объяснить также протекание используемых в синтезе реакций деоксигенирования, в частности, гетероциклических N-оксидов и сульфоксидов (схема 48 [35]); другие примеры реакций деоксигенирования можно найти в обзорах [24, 25]. При деоксигенировании сульфоксидов в некоторой степени может протекать перегруппировка фосфита в результате алкилирования внутри ионной пары сульфоний-фосфит (схема 48).
10.3.2.3. Нуклеофильность фосфора в реакциях триэфиров фосфористой кислоты
Нуклеофильный характер атома фосфора, причиной которого является сравнительная доступность неподеленной электронной пары, а также тенденция атома фосфора к расширению своей валентной оболочки, проявляется в большинстве реакций трпэфиР0® фосфористой кислоты. Последняя тенденция особенно сильна пр* образовании фосфорильной группы Р=О, и та легкость, с которой фосфиты окисляются в соответствующие фосфаты или перегрупп11’ 688
ровываются в фосфонаты (см. разд. 10.5.4) лежит в основе значительной части химии органических соединений фосфора (III)
‘Бсе триэфиры фосфористой кислоты в большей или меньшей сТепени окисляются кислородом воздуха. Этот процесс обычно является осложняющим, а не полезным обстоятельством, поскольку делает необходимым очистку фосфитов перед их использованием-в синтезе эфиров кислот фосфора (V) эта реакция применяется редко. В то же время реакции фосфитов с пероксидом водорода органическими пероксидами (трет-бутилгидропероксид и дибензоилпероксид), а также с другими реагентами, например N2O4, оксидом ртути(II), активным диоксидом марганца, перманганатом калия, тетраацетатом свинца, иодной кислоты (упомянутые реагенты далеко не исчерпывают список окислителей) имеют огромное значение, однако успешное использование этих-реакций зависит от точного соблюдения условий эксперимента и от устойчивости фосфита и соответствующего фосфата в водных средах, необходимых для применения этих реагентов [24, 25]. Показано, что Ы2О4, удобный из-за легкости его обнаружения в органических растворителях, и трет-бутилгидропероксид (см. уравнения 49, 50) стереоспецифично окисляют циклические фосфиты, давая продукты с сохранением конфигурации у атома фосфора.
(RO)3P + N2O4 ---> (RO)3P=O + N2O +O2
(40)
(ЕЮ)зР'. + трет-BuO—ОН---->[(EtO)3POBu -трет "он] —>
---► (EtO)3P—О + rper-BuOH (50)
Реакции окисления с участием трет-бутилгидропероксида и дибензоилпероксида несомненно имеют ионную природу, тогда как образование углеводородов, например дикумила и 2,2,4,4-тетраме-тилбутана в реакциях с ди-а-кумилпероксидом и ди-трет-бутилпер-оксидом, соответственно, ясно указывает на гомолитическую природу последних процессов [36—42].
Фосфиты легко взаимодействуют с озоном; при этом, очевидно, образуется промежуточный комплекс (1 : 1) фосфит озон. Трифенилфосфит, трициклический фосфит (36), а также этильныи аналог соединения (35) дают с озоном аддукты, периоды полураспада которых при 10°C составляют соответственно 0,5, 25 и 76 мин; все три аддукта разлагаются с образованием соответствующего фос Фата и синглетного кислорода, что позволяет использовать их для генерирования синглетного кислорода (уравнение 51).
Нуклеофильный характер фосфора в триалкилфосфитах выра, Жен не столь ярко, как в случае фосфинов. Оба класса соединении отщепляют кислород от эпоксидов, хотя в обоих случаях и не сте реоспецифично, что указывает на атаку фосфора по атомам Угле Р°Да и кислорода, однако реакции с фосфитами тре уют, к
687
о
о
Et~
правило, более жестких условий, а из промежуточного бетаина может образоваться не только алкен, но и фосфонат (схема 52).
Г О" I
Ме.
_^+(EtO)3P —> L(EtO)3PCH2CHMeJ —> / —(ЕЮ)зР=О
О
О
—> (EtO)2PCH2CHMeOEt + МеСН=СН2 (52)
Результат реакций триэфиров фосфористой кислоты с многими обсуждавшимися выше кислородсодержащими соединениями может отличаться от результатов реакций с их сернистыми аналогами. В некоторых случаях это обусловлено очевидными различиями в механизмах реакций, однако иногда причиной может быть потенциальная конкуренция при распаде амбидентного интермедиата, когда образование фосфорильного продукта, как правило, предпочтительнее образования продукта с тиофосфорильной группой. Десульфуризация тииранов соединениями фосфора(III) впервые описана в 1949 г. Отрыв серы под действием триэтилфосфита протекает стереоспецифично (на 97%) и приводит к алкену, конфигурация которого идентична конфигурации исходного соединения, вследствие чего вполне вероятным представляется объяснение, основанное на прямой атаке фосфора по атому серы (уравнение 53); механизм этой реакции резко отличается от механизма деоксигенирования эпоксидов (см. уравнение 52).
Ме хчН
SZ Ме^.Н ..
(ЕЮ)3Р + S<J ------> (EtO)3P=S + ¥ (Э '
Me Н
Дифенил- и дибутилдисульфиды взаимодействуют с триэтил фосфитом, давая S-бутил и S-фенилтиофосфаты (43), посколь У нуклеофильная атака тиолят-аниона направлена на углерод атом, связанный с более электроотрицательным атомом кислоро ’ а не с атомом серы. Дифенилдисульфид и трифенилфосфит, од ’ 688
Ме
не реагируют между собой из-за невочмп^ атак}! тиофенолят-аниона по ароматическому0":1 н^е°ФпльнОй , у центРУ (схема 54).
(EtO)3P
RSSR---------
(РЬО)зР R=Ph
(phO)2P~ SPh
O~Ph pSPh
(EtO)2P—SR
(EtO)2P\ + RSEt SR
(43) R=Ph,Bu
*-> (PhO)2P<^ + Phgph
SPh
(54)
==st ssb=p-
изо
-BuS—P(OMe)2’ O-^-Me
R1 H SSBu-uso
(MeO)3P
Rl ^SMe oJ-NR2
(55)
r'=p1iCh2conh;
CO2CH2CC13
R=H
(67%)
На способности фосфитов к деоксигенированию основаны также реакции синтеза ароматических гетероциклических соединений из ароматических нитропроизводных и триэтилфосфита. Этот подход был развит на основе первоначальных данных об образовании азоксибензола из нитробензола и трифенилфосфина или триэтилфосфита. Веским доводом в пользу промежуточного образования нитрена служит обнаружение фосфинимина (45) в продуктах реакции 4-диметиламинонитрозобензола (схема 56). Вскоре было обнаружено, что ароматические нитросоединения реагируют аналогичным образом, откуда следует, что нитрозопроизводные образуются в данном случае как промежуточные продукты; особенно изящный пример, демонстрирующий это, приведен на схеме 57.
Позднее было отмечено важное значение предшественника нитрена (44) (см. схему 56), однако независимо от природы промежуточного продукта или продуктов, образующихся в дальнейших Реакциях, по меньшей мере из одного из них в результате реакции с ароматическим кольцом образуется азотсодержащий цикл. Одним из первых примеров реакций такого типа было образование в мя1-ких условиях карбазола из 2-нитробифенила (схема 5 ).
689
(EtO)3P -------->
-----...' > ArNO^
~(EtO)3P=O
пин нпеетЯнпп^аД°ГаН“ рассмотрена область применения реак-пол лейгтит а ТеЛЬН°И Циклизации ароматических нитросоединений Этим мртлпПМ триалкилфосфптов, в особенности триэтилфосфита. а) пятичленимрМя°ЖН0 Получать следующие классы соединений: лы капбппиикт зотсодержащие циклические системы — карбазо-и их беналплг ’ ИЦДОЛЫ’ индазолы, триазолы, имидазолы, фуразаны хинолины rhpJn 6 шестичленные азотсодержащие гетероциклы — соединения^пбп!яН^’ Фенотиазины и в) семичленные циклические ровки пеакпио?НпгЮЩИ|СЯ путем внутримолекулярной перегруппи-получения этих г?СРбнЫХ пРомежуточных продуктов. Примеры табл. 10 3 1 гетероциклических соединений приведены в обл?гчает??1ЛЬтНЛЯ аТЭКа атОма ФосФора(Ш) на атом углерода гена входит в РГ1Р?;1''ЧаяхФ'огда атом углерода несет атом сложенную с ней r>ia->CfaB карбонильной группы или образует сопря-бо иным образом Ь ИЛИ кратную связь, активированную каким-ли-«алкилирую'шимйа^гАреаКЦИЯ/ трвэфнров фосфористой кислоты с
РУ и агентами» (уравнение 59) приводит к продуктам
Таблица 10.3.1. Получение азотсодержащих гетероциклов из ароматических
1 нитросоединений и триэтилфосфита
Исходное соединение
Продукт реакции
которые в общих чертах могут быть охарактеризованы как «псев-дофосфониевые соли». Возможность протекания этой реакции (см. уравнение 59), известной как реакция Арбузова — Михаэлиса
(R’o)3P + R2X
Х= С1,Вг,1
(rio)2pr2
О—R1 ^~Х
(46)
1 /r2
> R’X + (К‘О)2Р^ (59) ХО
ограничена природой групп R1 и R2, а также нуклеофильностью аниона X- [46]. Другие аспекты общности и механизма этой реакции подробнее будут обсуждаться в разд. 10.5.4 10.5.6; здесь же мы рассмотрим лишь образование промежуточного продукта (46). Первичные алифатические галогениды, бензил- и аллилгалогениды, как и ацилгалогениды (фосген и оксалилхлорид реагируют по ДРУ' тому пути), легко взаимодействуют с фосфитами, тогда как вторичные алифатические и особенно третичные алифатические галогениды и аралкилгалогениды склонны в этих условиях к элиминиро
091
ванию галогеноводорода с образованием алкенов; триарилметил галогениды, однако, реагируют нормально. Винил- и арилгалогенидь при обычных условиях термического инициирования в реакцию ц вступают, хотя могут реагировать при фотохимическом иницииро^ вании. Соли, образующиеся из триалкплфосфитов, например (4б> обычно неустойчивы, особенно если в качестве X выступает галоген; движущей силой разложения таких солей является образование очень устойчивой фосфорильной группы. Соли, получающиеся из триарилфосфитов, более стабильны из-за затрудненности ароматического нуклеофильного замещения на второй стадии реакции (см. уравнение 59). Анионная группа Х~ может быть представлена С1~, Вг- или 1~ (но не F-), анионом сульфокислоты; в этих случаях соли обычно нестабильны. Если нуклеофильности аниона недостаточно для осуществления второй стадии, соли типа (46) могут быть выделены; типичным примером являются тетрафторбораты, получение которых приведено в уравнениях 60 и 61.
(ЕЮ)3Р + Et3O BF4 —> (EtO)3PEt BF4 + Et2O (60)
(PhO)3P + Ph3C BF4 —* (PhO)3PCPh3 BF4 (61)
Термолиз соли, полученной из трифенилфосфита и циклогексил-иодида, приводит при 200 °C к циклогексилиодиду и дифенилфе-нилфосфонату (уравнение 62); другие алкилтрифеноксифосфоние-вые соли применяются, особенно в случае пространственно затрудненных спиртов, как реагенты для получения алкилхлори-дов, -бромидов и -иодидов (уравнение 63) [47]. Описано использование метилтрифеноксифосфонийиодида в качестве дегидратирующего агента.
(РЬО)зР + С6Нн1 —-> [(РЬО)зРСбНц Г] —> C6HllI + (PhO)2P(O)Ph (62)
[(PhO)aPMe Г] + ROH —> RI + PhOH + (PhO)2P(O)Me (63)
Строение так называемых псевдофосфониевых солей является предметом дискуссии. Тетрафторбораты (см. уравнения 60, 61) в ацетонитриле ведут себя как электролиты типа 1 : 1, тогда как соли, полученные из гипохлоритов, могут наряду с ионной формой содержать находящиеся в равновесии с ней истинные пентакоор-динацпонные тетра алкоксисоединения.
По отношению к эфирам тиофосфористой кислоты алкилгало-гениды ведут себя иным образом. Триэтилтритиофосфит реагирует с алкилхлоридами при 150°C по типу реакций замещения; аналогично ведут себя ацилхлориды. Такое направление реакции обусловлено, очевидно, более быстрым образованием сульфониевои соли, нежели фосфониевой (уравнения 64, 65).
(EtS)3P + RC1 —> (EtS)2PCl + RSEt (64)
(EtS)2PCl + MeCOCl —► EtSPCl2 + MeCOSEt (6
Как хлороформ, так и тетрахлорметан взаимодействуют с три^ этилфосфитом в условиях протекания свободнорадикальных р
692
й /уравнения 66—68). Однако в отличие от хлороформа, который 11 оеагпрует по ионному механизму, тетрахлорметан вступает в Немные реакции активнее многих других галогеналканов, выступая ятио, в качестве источника положительно заряженного галопа (схема 69) [48]. Разнообразие продуктов, образующихся рассматриваемой реакции при проведении ее в присутствии спир-я указывает на возможное существование равновесия между Гппзкородственными ионными парами. На общее направление рякпии может, однако, влиять электронная плотность на атоме Босфора; так, диэтилфенилфосфпт реагирует с тетрахлорметаном, павая этилфенилтрпхлорметплфосфонат, тогда как дифенилметил-фосфпт образует дифенилхлорфосфат.
. СС13 + (RO)3P — -> (RO)3PCC13 —> (RO)2PCC13 + R . R • + CC14 —> RC1 + .CCI3
(66)
(67)
или
О
II (RO)3PCC1, + CCh —--------► [(RO)3PCC13 СГ] _RC1- > (RO)2PCC13
—CCI3 о
II
RC1 4- (RO)2PCC13
(68)
(RO)3P + CCh
(RO)3PC1 СС1з
L(RO)3PCC13 Cl’J
R1OH
(69)
+ /OR'
(RO)2₽f OH
О
II
RC1 + R‘C1 + (RO)3P=O + (RO)2POR‘
Тетрабромметан реагирует с фосфитами гораздо более энергично, чем тетрахлорметан. Из трифенилфосфита при этом образуется дифенилбромфосфат и бромоформ, однако в случае триал-килфосфитов выделить фосфорсодержащие галогенпроизводные нелегко. Другие полигалогенированные соединения могут претерпевать дегалогенирование, по-видимому, за счет освобождения положительного галогена (уравнение 70).
W Г 0
К ,F ||
(ЕЮ)аР 4- BrCF2CFBrCI —> + EtBr + (EtO)2PCl (70)
р/ ХС1
Можно представить два возможных направления взаимодей-ствия триэфиров фосфористой кислоты с альдегидами или кетонами: по атому углерода или атому кислорода карбонильной группы
693
(схема 71); на практике, однако, чрезвычайно трудно различи эти две возможности. Направление реакции зависит, по-видимоИТЬ от природы карбонильного соединения и в некоторой степени можУ’ определяться возможностью делокализации электронов в перехо^ ном состоянии или промежуточном продукте реакции. Нейтраль' ные фосфиты не являются достаточно сильными нуклеофилами' чтобы образовывать стабильные аддукты состава 1 : 1 (аналогии* ные обсуждавшимся выше псевдофосфоииевым солям) с соединениями с нссопряженной карбонильной группой, поскольку стабилизация аддуктов в результате делокализации заряда в дан. ном случае невозможна [49—51].
I. \ + I
(RO)3P—О—С <— (RO)3P + ^С=О —> (RO)3P-C-O- (71)
В реакции трпметилфосфпта с пропионовым альдегидом фосфор атакует карбонильный атом углерода (схема 72), и хотя возможна стабилизация аддукта 1 :1 путем внутримолекулярного дезалкилирования, это направление не реализуется; вместо этого реакция протекает дальше и стереоселективно приводит к аддукту 1:2, который стабилизируется путем циклизации в тетраалкоксифосфоран (47) (схема 72).
О ОМе
, X 'I I (МеО)2Р—CHEt
О
ч EtCHO ч + 1
(МеО)3Р-----> (МеО)3Р—CHEt
EtCHC
(72)
Et СГ
, ч + I I
(МеО)3Р—CHOCHEt -
Et
/'-'О
О | /Же
Et ОМе
(47)
С бензальдегидом триалкилфосфиты, по-видимому, не реагируют, однако при наличии в молекуле ароматического альдегид электроноакцепторных заместителей в орто- или пара- положениях (заместители в .нета-положеппн не оказывают влияния) происходи реакция, в результате которой образуется смесь стереоизомерии аддуктов 1 : 2, которым приписана структура 2,2,2-трпалкоксифо форанов (48) (схема 73). Эта интересная реакция представля^ собой новый подход к созданию углерод-углеродной связи п м0 у найти широкое применение, поскольку образующиеся ПРОД^'еп; способны реагировать далее с потерей фосфора; так, сид аддукт (48) разлагается в кипящем этаноле, давая стильбен (ср. уравнения 37 и 38).
694
(73)
Ar Ш, / 2ArCHO + (RO)3P--->АГХ| *OR
H ° [SOR OR (48)
Простые алифатические и алициклические кетоны, типичными представителями которых являются ацетон и циклогексанон, не взаимодействуют с кипящими триалкилфосфитами, однако 'при нагревании бензофенона с триизопропил фосфитом при 170 °C в течение 72 ч образуются триизопропилфосфат, пропен и диизопропил- (дифенилметил) фосфонат. Реакция с участием флуоренона (схема 74) обычно приводится в качестве доказательства возможности атаки фосфора по карбонильному атому кислорода, а не по атому углерода. Первоначальный продукт (49) реагирует далее с флуореноном, давая пентаоксифосфоран (50); в кипящем триизо-пропилфосфите аддукт состава 1 :2 (50) претерпевает перегруппировку типа пинаколиновой, что приводит к кетону (51).
(74)
Широко изучено влияние сопряжения на реакции триэфиров фосфористой кислоты с алифатическими и ароматическими карбонильными соединениями. Многие реакции, как было показано, приводят к фосфонатам; приведенные ниже примеры (уравнения 75 77) дополняют способы получения фосфонатов, обсуждающиеся в разд. 10.5.4. Необходимо отметить, что выходы фосфонатов
695
могут быть относительно низкими из-за возможности альтернат ны.х путей разложения промежуточного продукта; в тех случа В когда внутримолекулярное дезалкилирование затруднено вследЯХ’ вне неблагоприятной геометрии молекулы, для завершения резки Т' может быть необходимым добавление протонного растворите"" (схема 78). я
(МеО)зР + СН2=СНСНО —> [(МеО)3РС112СН=СНО'| _____►
О II
—> (МеО)2РСН2СН=СНОМе (75)
Г Ph 1 I . I I
(EtO)3P + PhCH=CHCO2H —> L(EtO)3P-CHCH2CO;J —►
О Ph
II I
—> (ЕЮ)2Р—CHCH2CO2Et (76)
О CH2CO2Et
(EtO)3P + HO2CCH=CHCO2H —> (EtO)2P—CHCO2H (77)
Реакции с участием ацетиленовых соединений в большинстве своем протекают аналогично, однако могут сопровождаться элиминированием внутри алкильной группы (уравнения 79, 80).
(EtO)3P + PhC=CCO2H —>
Ph
♦ I
(ЕЮ)3Р—С=С=С
И । r/°H (EtO)2P—C=C=C' XOEt
О
Ph
—> (EtO)2P—C=CHCO2Et <79)
Простые триалкилфосфиты относительно устойчивы к нагрева-нию при умеренных температурах. Триметилфосфит при 200 количественно изомеризуется в диметилметилфосфонат; пронес^ катализируется, вероятно, следами соединений тетракоординиро ванного фосфора, возможно фосфатом, образующимся при оки
696
(EtO)3P + НС—CPh ---> (EtO)2PCH=CPh
Хл ь
H2c—CHo
о
, X II
----> EtO)2PCH=CHPh + CH2=CH2
(80)
Ленин, или диметилфосфитом, появляющимся в результате гидролиза. Для изомеризации триэтилфосфита в диэтилэтилфосфо-нат требуется температура выше 300 °C; триизопропилфосфит при сильном нагревании разлагается с образованием вторичного фосфита, тогда как триаллиловый эфир полимеризуется. Пропинило-вые и аллиловые эфиры, однако, легко перегруппировываются при температурах ниже температуры перегонки, хотя этот процесс может сопровождаться элиминированием внутри алкильной группы (уравнения 81—83).
/О. /Ме А о
/р+п\ 200 °C
' ^2 -j. ^/\Me —---► (EtO)2P—CH2CH=CMe2 (si)
(Eto)2P-*<^—Me ^EtC))2jLH CH2=C-CH=CH2 (82)
H—CH2
о
/POCH2C=SCH
и
Различия в нуклеофильности фосфитов и фосфинов отчетливо проявляются в реакциях с «-бензохиноном и его галогенпроизвод-ными: в то время как трифенилфосфин реагирует, очевидно, по атому углерода карбонильной группы, реакции триметил- или триэтилфосфита протекают по карбонильному атому кислорода с последующей миграцией алкильной группы (схема 84). Для объяснения аномального поведения триизопропнлфосфита в реакции с тетрахлорбензохиноном предположили, что промежуточный продукт нормальной реакции по кислороду, слишком стабилен для осуществления дальнейшего дезалкилирования, поэтому реакция протекает по атомам углерода, несущим галоген (схема 84).
Внутримолекулярная термическая перегруппировка ацилфос* фитов, например перегруппировка диэтилацетилфосфита (52) в ди-этилацетилфосфонат (53), является, в известном смысле, связующим звеном между уже обсуждавшейся внутримолекулярной перегруппировкой аллиловых эфиров и нуклеофильной атакой трехкоординированного фосфора на атом углерода кароониль-
23 Зак. 398 697
(RO)3P ------->
(КО)зР
R1 R1
R1 Ri
R =Me, Et; R1 = H, Cl
R = изо-Pr; Rl = H
О О
II II
(RO)2Px zP(OR)2
~d~'
(RO)2p/ 'P(OR)2
II II
О О
R = изо-Pr; R1 = Cl
(84)
ной группы. В приведенном примере протекает также дальнейшая реакция ацилфосфита с изомерным фосфонатом (схема 85).
О
/ х 11
(EtO)2POCMe
(52)
О О О Me
160-170'°С II || (52) || | +.
-------> (EtO)2P~СМе-----> (EtO)2P— С----P(EtO)2
(53) о-; fo
СОМе t ! (85)
' О Me О
(EtO)3P + MeCOCl (EtO)2P—С—P(OEt)2
ОСОМе
долгого времени считалось, что реакция деоксиге-
В течение д"----- ----
ходящая Як^тоанс°3 3'бНГлДР!1Да В кипящем триэтилфосфите, при-мериХя\а7бена’(56), протекает как ди-атаки фосфита по wifi ’ °°разующегося путем первоначальной время, однако, образоХХХббТ КПслоРода; в ^стоящее реакции Виттигя пп^. 1 е рассматривается как результат Ридом (схема 86) Р ежУточного илида (55) с фталевым ангид-тов) ТО(1алкил4осАитяКСОГ1аНТ1!ОНОв’2 (циклических тионкарбона-реа1<ц„?"Стер^SЕ°Д"Т * ПотеРе “ Рез^ЬТТ НИЮ исходного тион/япй образуется алкен, имеющий конфигуРа реакции рассматоивямтг°НаТ'а' качестве двух возможных путей тиофХорильного^ синхронный отрыв диоксида углерода « синения или же образование карбена.
698
реакция была применена Кори и сотр. [52] для превращения вицинальных диолов в алкены (схема 87). Большим успехом было использование этой реакции для получения не подчиняющегося правилу Бредта неустойчивого углеводорода (57), который, однако, может быть зафиксирован в виде аддукта с 1,3-дифенил-пзобензофураном, а также синтез тра«с-циклооктена (см. также разд. 10.3.3),
R1 R^UOH
R3"""' ХОН
R4
CSCI2
или
/— S t
HN. C-N
S
(R5O)3P
ИЛИ (r52n)3f
(87)
> II + со2 + (r5o)3p=s
R3^-R4
Чрезвычайно интересная ситуация возникает при активации карбонильной группы расположенными по соседству атомами галогена. В 1952 г. Перков, чье имя сейчас ассоциируется с этой реакцией, показал, что а-галогенированные альдегиды, кетоны и сложные эфиры при действии на них триалкилфосфитов могут Давать наряду с ожидаемыми кетофосфонатамп пли даже вместо
23*
699
ппх ненасыщенные эфиры фосфорной кислоты (енолфосфаты) (уравнения 88 и 89). Соотношение обоих продуктов зависит от природы карбонильного соединения и в особенности от природы
О
II
(ЕЮ)зР + СС13СНО —> (EtO)2P-OCH==CCl2 + EtCl (88)
О
II
(ЕЮ)3Р + С1СН2СОМе —> (EtO)2P—СН2СОМе + EtCl {89) галогена, а также от условий проведения реакции (см. табл. 10 3 2) [53].
Образование оксофосфоната включает атаку фосфита по атому углерода, несущему галоген, и разложение «нормальной» фосфониевой соли (58), однако до сих пор не получено никаких данных, подтверждающих возможность изомеризации (58) в соль
Таблица 10.3.2. Влияние природы галогена и температуры на соотношение продуктов реакции триэтилфосфита с галогенкетонами
Галогенкетон Оксофосфонат : енолфосфат Литература
при 130— 150 °C при комнатной температуре (в эфире)
С1СН2СОМе 12:88 — [53а
BrCH2COMe 80:20 20:80 [53а
ICH2COMe — 90: 10 [53а
ClCH,COPIi 0:100 — [536
BrCH.COPh 85 15 64:36 [536
ICH2COPh — 76:24 [536]
(59), которая могла бы быть предшественником енолфосфата (схема 90). Имеющиеся данные о поведении фосфитов в реакциях с карбонильными соединениями позволяют предсказать два различных направления реакции: атаку по атому углерода или по атому кислорода карбонильной группы (схема 91).
R2
(R1O)3P * CHCOR3—> (R1O)3PCHCOR3 X"
R3 (R1O)3POC=CHR2 >
-r‘x
X— Hal (не F)
О Rd
, , JI I
(r1o)2pchcor3
[кетофосфонат;
' (90)
О R3
II I 2 (R1O)2POC=CHR (енол фосфат)
700
ZR3
(r’o)3p)o=cx
chr2x
О R3
R°
(»4poc=UII)> x-J-^r-o^o^chr'
О X
, v ,Q И , I 9 (r‘o)3P + CR3—CHR2 >
X= Hal (не F)
Несмотря на большое количество данных в пользу существования тетраалкоксифосфониевых солей, данные кинетических исследований не подтвердили их образования за счет непосредственной атаки фосфита по карбонильному кислороду, как первоначально полагали Спезиал и Смит при изучении реакции фосфинов с тригалогенацетамидами. Отсутствие доказательств изомеризации С-фосфониевой соли в О-соль (см. схему 91) исключает, по-видимому, также реакцию по атому углерода карбонильной группы. Подробнее механизм реакции Перкова будет рассмотрен позднее.
Дезалкилирование триалкилфосфитов свободными галогенами протекает тем быстрее, чем эффективнее осуществляется подача электронов на атом фосфора и чем выше электроположительность галогена; в результате этого реакция может идти достаточно быстро даже при температурах ниже О °C. Обычно считают, что этот процесс протекает через галогентриалкоксифосфониевую соль, для образования которой необходим «положительный» галоген, однако имеются серьезные доводы в пользу участия в реакции истинных пентакоординационных интермедиатов; вполне вероятно, однако, что обе эти промежуточные частицы находятся в равновесии (уравнение 92).
(RO)3P + х2
Х=На1 (не F)
В зависимости от размера цикла и степени его замещенности циклические фосфиты при взаимодействии с галогенами могут претерпевать раскрытие цикла, причем, как было показано, реакция протекает стереоспецифично с обращением конфигурации алкильного атома углерода, что согласуется с механизмом (уравнения 93—96). Заслуживает внимания выделение промежуточного продукта (60) в одной из таких реакций галогенирования 154 ].
701
ZR
о
II/К
—> С1СН2СН2ОР\ ХС1
С1 сг
R=C6H13; r2=c8h17
Были выделены также аддукты галогенов с триарилфосфитами (Ноак, 1883 г.), хотя вначале не вполне ясно представляли сложность подобных систем. В настоящее время известно, что
Вг2 ----->
[(PhO)3PBr2 (PhO)3PBr Br-]
(PhO)3P
Вг2 ---->
(PhO)2PBr
|(PhO)3P
(PhO)2PBr+ (PhO)4PBr
Br2 +
-----> [(PhO)2PBr2 Br-]
(97)
(98)
(PhO)4PBr+Br' —> [(PhO)4PBr2J <99)
(PhO)2PBr2 + (PhO)3P —> [(PhO)3PBr] + (PhO)2PBr
(100)
PhO v
(PhO)2PBr2+ (PhO)eP“ ч— (PhO)4PBr
702
первоначально образующиеся аддукты далее реагируют с фосфитами, причем различные равновесия устанавливаются столь быстро, что природа выделяемых продуктов определяется в основном их растворимостью и стабильностью, а не скоростью их образования (уравнения 97—100) [55].
В соответствующих условиях такие аддукты с галогенами ведут себя как алкоксифосфониевые соли, образующиеся из алкил-галогенидов, и при действии насыщенных или ненасыщенных спиртов могут давать алкилгалогениды. Эти аддукты можно подучать Ш situ путем прибавления галогена к смеси трифенилфос фита и спирта (уравнение 101) [56, 57]; реакция сопровождается обращением конфигурации группы R, причем степень инверсии особенно высока в случае хлоридов.
О
ROH II
(PhO)3P + Х2 —> (PhO)3PX2 — ► RX + PhOH 4-(PhO)2PX (101) Х = С1, Br
Взаимодействие диарилалкилфосфитов с иодом с высокими выходами приводит к алкилиодидам, и хотя иодфосфаты (см. уравнение 101) выделены не были, на их образование указьг вает получение О,0-диариламидофосфатов при добавлении в реакционную смесь соответствующих аминов. Известно много других примеров, в которых дегалогенирование под действием трналкил-фосфитов начинается с взаимодействия с «положительным» галогеном в результате нуклеофильной атаки на галоген, в том числе реакции с полигалогенциклопентадиенами (уравнение 102) и бром-циклогексадиенонами (уравнения 103, 104).
Рассмотрев взаимодействие эфиров кислот фосфора(III) с «положительным» галогеном, можно возвратиться к обсуждению возможных типов атаки этих эфиров на галогенированные карбонильные соединения (реакция Перкова). В этом случае (схема 105) взаимодействие с «положительным» галогеном должно было бы приводить к ионной паре, содержащей амбидентный енолят-анион.
X-H.Cl, R-Me,Ph
(102)
703
(EtO)3P -------->
Ви-трет
Me—
ИВи-трет =n хВи-трет
—О' BrP(OEt)3
Ви-трет
/Ви-трет 0
—> Ме——OEt + (ЕЮ)2РВг (104)
'Ви-трег
Попытки зафиксировать образование этого иона были, однако, неудачны. Введение протонного растворителя, что, как можно было ожидать, привело бы к получению дегалогенированного карбонильного соединения, не влияло на результат реакции: а-хлор- и ct-бромкетоны в спиртовом растворе с высокими выходами давали соответствующие енолфосфаты; это полностью противоположно результату реакций трифенилфосфина с теми же га-
логенсодержащими соединениями.
(r‘o)3p^ x-Qhr2cor3
о
ат--^ по-£->. (R’o^P-CH^COR^+RbC
(Ю5)
ат-а-к&п-°?->- £(R1O)3POGR3eCHR2
X = Hal(HeF) Предположение о
О
(R’oJjjP-OCR =chr2+ R3X
_________________ „ том, что механизм зависит от природы конкретного карбонильного соединения, маловероятно. Любой ме-
704
ханизм, объясняющий одновременна ™
30Ва и Пер нова был бы крайне привлекав реакций Арбу-предположение было выдвинуто Гиллеспи ЛЬНЫМ’ такого пОдЯ Обычная фосфониевая соль, образующаяся Т ж8] (схема Юб) генкарбонильного соединения, превращаетгя Из ФосФита и гало-тем прямого внутримолекулярного дезами В фосФонат или пу-дезалкилирования внутри пентакоорщХИли "Утем с последующим расщеплением апикт? °Г° катиона (61) Р—О в (62) (см. гл. 10.6), что п Bn t ° Циклической связи Арбузова. Процесс реорганизации л^ганадв * П^°?УКТУ Реакции шин разрыв апикальной связи Р—с в п« (62) и п°следуЮ-водят к енолфосфату - продукту реакции ПерХа^™ (63) При'
R1 (H2O)3P^ R F=o
CH2X
R1-—г=о
сн2—p(or2)3
-r2x
CH2—P(OR2), II о
продукт реакции Михаэлиса - Арбузова
R1—г
? />R2 —PV R2i4OR2 (61)
-r4x
R1
.OR2
R1
R1
---О
| ...Pr2
----Pv
r2o
(62)
OR2
(106)
X=HaI (HeF)
°~K
R2O ° продукт реакции Перкова
o— P. r2o ° (63)
Реакция по атому галогена приобретает, очевидно, большее
значение в случае полигалогенированных карбонильных соединений, поэтому способность триэтилфосфита быть эффективным фосфорилирующим агентом в присутствии диброммалонамида и др) гих подобных амидов хорошо объясняется с позиции нуклеофиль ной атаки фосфита на галоген (уравнение 107). ^он
(EtO)3P + Br2C(CONH2)2 —► [(EtO)3PBr BrC(CONH2)2] ►
О
II
— > EtBr + (EtO)2POR 4 BrCH(CONH2)2 (107)
Триэфиры фосфористой кислоты гладко реагируют с N-хлор-производными, давая ожидаемые продукты; алкиловые эфиры при
703
этом образуют амидофосфаты (уравнения 108, 109). Однако ппо межуточные «фосфониевые» комплексы из триарилфосфитов как н можно было предположить, более устойчивы к нуклеофильной атаке галогена и не распадаются нормальным образом, в резуль тате чего становится возможным протекание необычного дезалкилирования (уравнение НО).
О
(EtO)3P + Et2NCl
—> (EtO)2P—NEt2 4-EtCl
(ВиО)зР 4-
О
О II II
—► (Bi;O)2P—14-BuCl
Q
(108)
(109)
(PhO)3P + Et2NCI --->-
(PhO)3P-NEt Et^C?
_-->(PhO)3P=NEt (110)
Если в рассмотренных реакциях направление атаки вполне однозначно, то в случае реакций с О-галогенпроизводными сделать выбор между атакой фосфита по галогену или кислороду может быть очень сложно, хотя независимо от этого общая реакция протекает как окисление фосфита в фосфат с образованием алкилгалогенида. В ранних работах на основании поведения в реакциях с трифенилфосфитом тетрагидролиналилгипохло-рита (64) и бициклического гипохлорита (65) был предложен механизм 5n2. В случае гипохлорита (64) распад предполагаемого фосфониевого интермедиата приводит к хлориду с обращенной конфигурацией (уравнение 111)х тогда как в случае (65) нор-
Ме
+ (PhO)3P=O
(111)
О
II
+ (PhO)2PO
(65)
—PhOH + НС1
(112)
7Q6
мяльный распад промежуточного nnoivr^
сткой геометрии молекулы, поэтомг УД Нев°зможен из-за направлению (уравнение 112) Г591 реакиия идет по nnvrn^'
Три- и тетраалкокснфосфониевые С07и е У
тетрафторборат- и гексафторантимонат Л алонУКлеофнльными » оказались вполне устойчивым,,; в L "“ам" был,, выделены „ином счете к эфирам фосфорной кислотаТ® “Р^°Днт в ко
1 (Уравнения 113—1 1*п
(MeO)3P + MeOCl + SbCl, -2T1V /лл Л
^hP+-SbCl6 (38%)
(трег-ВиСНаОзР + грет-ВиСНаОС] -~78 °С ,
-> (грег-ВиСН2О)4Р<-СГ -> (трег-ВиСНгО)зР-О4- п
2^зН_О 4- 7-рег-ВаСН2С1
(113)
(МеО)3Р + трет-ВиСН2ОС1 + AgBF4
—78 °C
(114)
О
(МеСРгРОСНгВи-трет
—> (МеО)зРОСН2Ви-7’р^г BF4 ---------->
6 дн.
Финлей и Денни [60] исследовали мерных метилфосфитов (66) и (67) ( (схема 116); один изомер (неизвестной ххип^пгураци] в этих условиях фосфат практически стереоспецифично, в то
(И5) взаимодействие стереоизо-с неопентилгипохлоритом [ конфигурации) образует --------время
(116)
70/
как второй изомер (также с неизвестной геометрией) приводит к смеси стереоизомерных фосфатов. На основании этих данных считают, что наряду со стереоизомерными тетраалкоксифос-фониевымп солями важнейшую роль в этой реакции играют истинные пентакоордннациопные интермедиаты, поскольку только при их участии оба конечных продукта могут быть получены из одного исходного эфира.
Триэфиры фосфористой кислоты, по-видимому, прямо реагируют с сульфенилхлоридами — сернистыми аналогами гипохлоритов. Очевидное различие в поведении трифенил- и триэтилфосфи-тов в этих реакциях обусловлено затрудненностью нуклеофильного замещения у арильного атома углерода; в таких случаях, как н в случае ацетилсульфенилхлорида, наблюдается относительно редкий разрыв связи С(алиф.) — S (уравнения 117—121).
о
+ II
(ЕЮ)3Р + RSC1 —> ](EtO)3PSR СГ] —> (EtO)2PSR + EtCl (117)
(PhO)3P + EtSCl —> [(PhO)3PSEt СГ] —> (PhO)3P=S + EtCl (118)
(PhO)3P+2PhSCl —(PhO)3PCl2 +PhSSPh (119)
^q CICH2 q о
Ме-СпхР 4- PhSCI ------>- У (120)
X-O Me/VO XSPh
(EtO)3P + MeCOSCl ------->
. . + I )
(EiO)3P—S—COMe
^Cl
---b (EtO)3P=S + MeCOCl . 021)
Примеры реакций трифенил- и триэтилфосфитов с ДРУ1™ хлоридами серы представлены ниже (уравнения 122 12У); неко-
о
(ЕЮ)зР + SO2C12 -—> (EtO)2PCl + EtCl + SO2 О22)
(PhO)3P + SO2C12 -—> (PhO)3P=O+SOC12 (123)
° S О
(EtO)3P 4- SOC12 —> (EtO)3P + (EtO)3P + (EtO)2PCl 4-EtCl + SO2 (12^
(PhO)3P + SOC12 -—> (PhO)3P=O + (PhO)3P=S + (PhO)3PCl2 О о
2(EtO)3P 4-SC12 —> (EtO)2PSCl 4- (EtO)2PCl 4-(EtO)3P=S 4- EtCl О23)
2(PhO)3P 4- SC12 -—> (PhO)3P=S 4- (PhO)3PCl2
О
3(EtO)3P 4-PhSO2Cl —> (EtO)2PSPh 4-EtCl 4-2(EtO)3P=O (12>
6(PhO)3P 4-2PhSO2Cl —> (PhO)3P=O 4-PhSSPh 4-PhCl 4-(ph°)2p
708
рые из этих реакций могут протекать в гомолитических усло^ виях, однако и здесь следует отметить различия в поведении этих двух фосфитов.
10.3.2.4. Вторичные фосфиты* [61—66]
Вторичные фосфиты (диорганофосфиты, диалкилводородфос-фонаты) заслуживают отдельного рассмотрения благодаря той важнейшей роли, которую соединения этого класса играют в синтезе фосфорорганических соединений.
Согласно имеющимся данным, для этого класса производных существует истинное таутомерное равновесие между три- и тетра-координационными формами, причем оно сильно смещено в сторону последней, так что содержание фосфитной формы не превышает 10-4%**. Спектроскопические данные полностью соответствуют структуре с фосфорильной группой. В ИК-спектрах вторичных фосфитов имеются полосы поглощения в области 2380— —2450 см-1 (vp_h) и 1260—1300 см-1 (vp=o); полосы поглощения, которые можно было бы отнести к связи Р—ОН, отсутствуют. В спектрах ЯМР константы спин-спинового взаимодействия фосфор— протон составляют 490—760 Гц, а химические сдвиги сигналов фосфора имеют значения от —13 до 4-4 млн-1, что близко к таковым для фосфорной кислоты; во всех случаях точное значение указанных параметров зависит от природы связанных с фосфором групп и от конформации связей фосфора, когда он является частью циклической системы.
Основные свойства вторичных фосфитов проявляются в их способности растворять и связывать галогеноводороды, а кислотные—в способности реагировать с электроположительными металлами или гидридами этих металлов с выделением водорода и в образовании металлических производных при взаимодействии с алкоксидами металлов. Данные ИК- и ПМР-спектроскопии свидетельствуют об отсутствии фосфорильной группы в таких производных, что согласуется с наличием связи металл—кислород, однако заметная растворимость этих соединений во многих неполярных органических растворителях указывает на то, что эта связь является в значительной мере ковалентной.
Вторичные фосфиты получают различными методами: а) гидролизом триэфиров фосфористой кислоты, который, однако, используется редко [67]; б) гидролизом хлорфосфитов (ср. схему 20); в) взаимодействием диэтилфосфита с другими спиртами (переэтерификация); этот метод удобен для получения циклических соединений [68]; г) с помощью различных реакции, дающих
* См. примечание на с. 676. л „„
Единственным надежно установленным исключением из этого правила, применимого ко всем соединениям фосфора (ПТ) подобного типа, является ис-(трифторметил)фосфнннстая кислота (CFslsPOH, которая практически полностью существует в трехкоординационной форме.
709
вторичный фосфит в качестве одного из продуктов (уравнения 130—133); первая из этих реакций может служить также иллюстрацией алкилирующей способности триалкилфосфитов [69]. Наконец, они могут быть получены дезалкилированием эфиров фосфористой кислоты галогеповодородамп, в особенности НВг и HI Отщепление первой алкильной группы протекает быстрее, чем второй; скорость дезалкилирования, однако, зависит также от природы алкильной группы: она очень высока для трет-бутильной группы и очень низка в случае 2,2,2-трихлорэтильного заместителя. Модификация этой реакции приведена в уравнении 134.
(EtO)3P + RCO2H -—> (EtO)2P 4- RCO2Et (130)
Ml
(EtO)2P—O—P(OEt)2 + AcNH2 —> (EtO)2P + (EtO)2PNHAc (131)
Ж
(EtO)2POCOR + MeCO2H —> (EtO)2P^ MMeCO—0—COR (132)
MI
(EtO)2P—O— P(OEt)2 + PhNH2 —a (EtO)2P^ + (EtO)2PNHPh (133) Ж
Me .—о /0
Me2C(CH2OH)2 + MeOH + PC13 —> /\ /\ +MeCl+2HCl (134)
Me' О Ml
Многие наиболее важные реакции вторичных фосфитов приведены ниже (уравнения 135—150). Многие другие реакции, в том числе каталитическое присоединение по кратным углеродуглеродным связям, рассматриваются в разд. 10.5.4, а реакции родственных соединений — в разд. 10.5.5 и 10.5.6.
RCO2H + (ЕЮ)2Р(О)Н —> RCO2Et
Et2NCl + (EtO)2P(O)H —-> (EtO)2P(O)Cl + (EtO)2P(O)NEt2 М-бромсукцинимид (EtO)2P(O)H -------------:--------> (EtO)2P(O)Br
(EtO)2P(OHI + X2 —> (EtO)2P(O)X + EtOP(O) (OH)X
X = Cl или Br
SO2C12 + (EtO)2P(O)H —> (EtO)2P(O)Cl
NOCI + (EtO)2P(O)H (EtO)2P(O)O(O)P(OEt)2
PhNCO + (EtO)2P(O)H —> (EtO)2P(O)COXHPh
(SCN)2 + (EtO)2P(O)H —> (EtO)2P(O)NCS -^4- (Et0)2P(O)NHCSNHR
S2C12 + (EtO)2P(O)H —> (EtO)2P(O)SS(O)P(OEt)2
SC12 + (EtO)2P(O)H —> (EtO)2P(O)SCl —> (EtO)2P(O)O(S)P(OEt)2 EtOCl + (EtO)2P(O)H —> (EtO)3P=O
(136)
(137)
(138)
(139)
(140)
(141)
(142)
(143)
(144)
(145)
710
XCCH2COXHBr + PhCH2OH -f- (EtO)2P(O)H___>.
—> (E1O)2P(O)OH + \CCH2CONH2 + PhCH2Br
RMgX + (EtO)2P(O)H —> (EtO)2P(O)MgX —> R2P(O)MgX
RiX I
R2p(°)R‘ R2P(O)H
N2CHCO2Et + (EtO)2P(O)H —> (EtO)2P(O)CH2CO2Et
N2O4 (или KMnO4) + (EtO)2P(O)H -—> (EtO)2P(O)OH ZNHCHRCO2H + H2NCHR'CO2R2 + (EtO)2P(O)H ___>
—> EtOP(O)(OH)H + ZNHCHRCONHCHR'CCW
(146)
(147)
(148)
(149)
(150)
10.3.2.5. Сравнительная реакционная способность ациклических и циклических эфиров и других производных фосфористой кислоты
Циклические и ациклические эфиры кислот трехкоординиро-ванного фосфора различаются по своей реакционной способности. Однако это продемонстрировано лишь на относительно небольшом числе реакций. Это. различие в реакционной способности иллюстрируется следующими примерами.
Относительные скорости реакций эфиров (68) — (72) с этил-иодидом (в ацетонитриле) возрастают в ряду (69) < (70) < (72); (72) реагирует в 10 раз быстрее, чем (69) [70]; относительные скорости реакций тех же эфиров с диэтилпероксидом (в отсутствие растворителя) возрастают в ряду (70) < (72) а; (71) х (69) < (68) примерно в той же степени [71]. Налицо, таким образом, противоположность между реакцией замещения галогена и образования триалкоксифосфониевой соли, в которой эфир выступает в роли типичного нуклеофила, и реакцией, которая не зависит от нуклеофильности, но зависит от доступности неподеленной электронной пары фосфора (уравнение 151), и в которой, можно сказать, проявляется бифильнын характер фосфора.
(69)
О
POEt
/
о
(70)
(МеО)3Р (изо-РгО)дР
(71) (72)
(RO)3P + EtOOEt —>
/OEtl
(RO)3P4Z [ —> (RO)3P(OEt)2
'OEt J
(151)
В качестве второго примера можно рассмотреть реакционную способность эфиров фосфористой кислоты в реакции восстановительной циклизации 2-нитроарилальдимииов в 2-арилпндазолы
7Н
(уравнение 152) [172]. При 91,5°С относительные скорости пр акций триэтилфосфита и 2-этокси- 1,3,2-диоксафосфолана относят' ся как 7:1; такое соотношение, по-видимому, типично для пеак" ций, в которых трехвалентный фосфор выступает в качестве hvk' леофила. Другие значения ka/k^ для «аналогичных» ациклических (^а) и циклических (Ац) соединений составляют 3—10 (реакция с алкилгалогенидами), 100 (реакция с ацилгалогенидами) 30 (реакция с изоцианатами) и 6(катализируемый кислотами гидролиз); другие примеры рассмотрены Хадсоном и Брауном [73]
Т?
Nu
(74)
Взаимодействие циклического эфира с электрофилом Е+ (схема 153) приводит к увеличению на ~9° валентного угла, образованного атомом фосфора и атомами кислорода цикла, что увеличивает напряжение в цикле; следовательно, можно предсказать, что циклический эфир будет менее реакционноспособным по отношению к электрофилу Е+, чем его ациклический «аналог»; это предсказание согласуется с уже рассмотренными примерами. Наоборот, можно считать, что взаимодействие того же эфира с нуклеофилом Nu~ приводит к (74), имеющему менее напряженный цикл, что вызывает повышение реакционной способности по сравнению с ациклическим эфиром. Одной из трудностей, очень часто возникающей при интерпретации имеющихся данных, является невозможность определения точной конфигурации фосфора в промежуточных продуктах типа (74), которая может в значительной степени зависеть от природы присоединенных к фосфору групп. Следствием этого, по-видимому, являются значительные колебания соотношений скоростей в различных ^реакциях; типичные значения &ц//га составляют: 10—15 (щелочной гидролиз ароматических эфиров), 3000 (щелочной гидролиз метил-фосфитов) и —1150 (реакция амидофосфитов с бензальдегидом).
10.3.3. ФОСФОНИСТАЯ и ФОСФИНИСТАЯ кислоты И ИХ ПРОИЗВОДНЫЕ [6а, 74—761
Производные фосфонистой и фосфинистой кислот представляют собой соединения фосфора(III), имеющие соответственно одну или две связи фосфор—углерод. Родоначальные кислоты (75) и (79) находятся в таутомерном равновесии с тетракоординациоИ" 71?
ними формами (77) и, соответственно гяп /
фиты). Фосфонистые кислоты занимают пппУР- ВтоР™ные фОС-н„е между сполна окисленными фосфоновь^еЖуТОЧнОе положе-разд. Ю.5.4) п первичными фосфинами и Jnn Лотами (см производных, например эфиры (76) „ /78? Г ДаВать ряда нистые кислоты являются промежутпциУ Аналогично, фосЛн фосфиновыми кислотами (см раздУ Н5единениями ^ежду нами; их таутомерные формы (81) ВтоРичными фосфи-
бой вторичные фосфиноксиды. _
Ф' г....-‘ШИ
актически представляют со-
RP(OH)2 —> RP(OR>)3
(75) (76)
R2POR‘
(80)
О О
II II
R—Р—Н —> R—Р—Н
ОН OR1
(77) (78)
R2POH
(81)
Для получения указанных выше типов соединении используют различные методы, однако лишь немногие из них являются общими п достаточно надежными; даже рассмотренные ранее реакции эфиров с металлорганическими соединениями не дают удовлетворительных результатов, однако такие методы иногда могут быть единственно пригодными.
Для получения производных фосфонистых и фосфинистых кислот наиболее широко применяют три общих метода: а) инициируемое пероксидами или облучением присоединение гипофосфори-стой кислоты или ее солей к алкенам [77] (ср. реакции присоединения вторичных фосфитов) и реакции с карбонильными соеди-
О О
II II
RCH=CH2 4- H2PONa —> RCH2CH2PHONa
О RHN О
II I II
Ме2СО 4- Н2РОН 4- RNH2 —> Ме2С—РНОН
(154)
(155)
(156)
(157)
713
нениями (уравнения 154, 155); б) обмен между хлорфосфинами и кремний- или борсодержащими соединениями, протекающий no-i нагревании смесей соответствующих производных [78] (уравне ния 156, 157), в) ступенчатый алкоголиз или аминолиз дихлоп-или монохлорфосфинов приводит к эфирогалогенидам, диэфирам амидогалогенпдам и диамидам кислот фосфора(Ш). ' н
Недавно описан асимметрический синтез оптически активных эфиров фосфинистой кислоты (схема 158); оптическая чистота продукта составила около 10% [79]. Отнесение конфигурации соединений фосфора(III) сделано на основании их превращения в соответствующие фосфиноксиды или -сульфиды с известной конфигурацией. Гидролиз хлорангидридов кислот можно проводить водой или сильно разбавленной щелочью; явным исключением является трихлорметилдихлорфосфин, дающий при обработке водой хлороформ и фосфористую кислоту.
ОМе
EtPhPCl + МеОН
(+)-или(— )-PhCHMeNMe2 Et2O, -70°С
_______________S__________________I
ОМе О О
j, MeI у MeMgl
Et^SMe м/Ге*
(+)-(.у) (-)-(-$) Н-И
Mef
Ниже приведены примеры реакций, указывающих на близкое сходство химии фосфонистых и фосфинистых кислот и их производны^ и химии фосфористой кислоты и ее производных (уравнения 159—168). /159)
Me2NPCl2 + MeMgl —► Me2NPMe2
OBu OBu
PhPCl + CH2=CHCH2MgBr —> PhPCH2CH=CH2 О
PhP(OMe)2 4-BrCH2CH2OAc —> PhPCH2CH2OAc
OMe
(160)
(161)
+ EtOH
PhP(OPh)2 + Mel —> [PhMeP(OPh)2 Г]
О
II
—PhOH-ф EtI-f- PhMePOPh
7Ц
Et2PCl + Н2С——СН2 —> Et2POCH2CH2ci
О
Ph2PH +BuSCI —> Ph2PSBu + НС)
О
Ph2POR + Br? Ph2PBr+RBr
О
Ph2POR + НВг(водн.) Ph.ph + RBr
(163)
(164)
(165)
(166)
(167)
(168)
/NHBu-трет
Ph2PNHBu-TpeT + PhCH2Cl —> Ph2PZ
\cH2Ph
Интересно сравнить реакционную способность производных фосфонистых и фосфинистых кислот с реакционной способностью производных фосфористой кислоты. Вообще говоря, следует ожидать, что по нуклеофильным свойствам эти соединения будут занимать промежуточное положение между фосфинами и производными фосфористой кислоты; такая общая последовательность изменения реакционной способности действительно наблюдалась по отношению к алкилгалогенидам в реакции Арбузова [81] и при восстановительной циклизации 2-нитроаренов. Иллюстрацией высокой реакционной способности диамидофосфонитов может служить реакция (схема 169), в которой диамид (82) использовался в качестве акцептора серы, отщепляющейся из промежуточного соединения (83) при превращении фгс-циклооктена в его транс-изомер (реакция протекает при 30°C и приводит к продукту 96%-ной чистоты) [82].
(83)
ЛИТЕРАТУРА
!• A F. Hudson, 'Structure and Mechanism 1,1 .ОгЛаПу(’(^°®р!(>р\кт\ра и ме-Academic, London, 19С5, chapter 2, 4, 5, and b.И- > • Д Мир, 1967.
ханизм реакций фосфорорганических соединений, llep. • • гл. 2, 4, 5, 61.
715
2. В. С. Saunders, ‘Phosphorus and Fluorine’, Cambridge University Ргрсс iqk-?
3. A. J. Kirby and S. G. Warren, 'The Organic Chemistry of Phosphorus’ Fl’sevier' Amsterdam, 1967, chapter 3 and 8. [А. Кирби, С. Уоррен. Органическая хим™ фосфора. Пер. с англ. M„ Мир, 1971, гл. 3, 8]. тческая химия
4. В. J. Walker, ‘Organophosphorus Chemistry’, Penguin, London, 1972, chapter 2 and 6. > p z
5. J. Emsley and D. Hall, ‘The Chemistry of Phosphorus’, Harper and Row inn don, 1976, chapters 4 and 5.
6. K. Sasse in ‘Methoden der Organischen Chemie’, (Houben-Weyl), ed E Muller, G. Thieme, Stuttgart, (a) 1963, vol. XII/I; (b) 1964, vol. XII/II
7. (a) D. IP. Hutchinson, in ‘Organophosphorus Chemistry’, ed. S. Trippett, Specialist Periodical Reports, The Chemical Society, London, 1970 vol 1 chanter 5; (b) B. J. Walker, ibid., 1971, vol. 2 и сл. P
8. W. Gerrard and H. R. Hudson, in ‘Organophosphorus Compounds’, ed. G. M Ko-solapoff and L. Maier, Wiley-Interscience, New York, 1973, vol. 5 chapter 13.
9. H. R. Hudson, Synthesis, 1969, 112.
10. R. S. Edmundson, Chem. and Ind. (London), 1962, 1770; ibid., 1967, 1809.
11. J. Gloede, Л1. Mikolafczyk, A. Lopusinski, and J. Omelanczuk, J. prakt Chem 1974, 316, 703.
12. E. Flack, in ‘Topics in Phosphorus Chemistry’, ed. M. Grayson and E. J. Griffith, Interscience, New York, 1967, vol. 4, p. 291.
13. M. L. Nielsen, in ‘Developments in Inorganic Nitrogen Chemistry’, ed. С. B. Colburn, Elsevier, Amsterdam, 1966, vol. 1, chapter 5.
14. V. Mark, Org. Synth. Coll. vol. 5, 1973, 602.
15. K. D. Berlin, T. H. Austin, M. Peterson, and M. Nagabhushanam, in ‘Topics in Phosphorus Chemistry’, ed. M. Grayson and Ё. J. Griffith, Interscience, New York, 1964, vol. 1, p. 17.
16. G. O. Doak and L. D. Freedman, Chem. Rev., 1961, 61, 31.
17. C. L. Bodkin and P. Simpson, J. C. S. Perkin II, 1973, 676.
18. J. A. Mosbo and J. G. Verkade, J. Amer. Chem. Soc., 1973, 95, 204.
19. R. Burgada, Ann. Chim. (France), 1966, (1—2), 15; Bull. Soc. chim. France, 1963, 2335.
20. G. W. Anderson, J. Biodinger, R. W. Young, and A. D. Welcher, J. Amer. Chem. Soc., 1952, 74, 5304; G. W. Anderson, J. Biodinger, and R. W. Young, ibid., 1952, 74, 5309; P. C. Crofts, J. H. H. Markes, and H. N. Rydon, J. Chem. Soc., 1958, 4250; ibid., 1959, 3610; A. IP. Miller and P. IP. G. Smith, J. Chem. Soc. (C), 1967, 2140.
21 IP. G. Bentrude and H. IP. Tan, J. Amer. Chem. Soc., 1973, 95, 4666.
22. H. Noth and H. J. Vetter, Chem. Ber., 1965, 98, 1981.
23. /. M. Downie, J. B. Lee, and M. F. S. Matough, Chem. Comm, 1968, 1350.
24. J. I. G. Cadogan, Quart. Rev, 1962, 16, 208.
25. J. 1. G. Cadogan and R. K. Mackie, Chem. Soc. Rev, 1974, 3, 87.
26. R. Burgada and J. Roussel, Bull. Soc. chim. France, 1970, 192.
27. V. Mark, J. Amer. Chem. Soc, 1963, 85, 1884.
28. D. C. Ayres and H. N. Rydon, J. Chem. Soc, 1957, 1109.
29. D. Z. Denny and D. B. Denny, J. Amer. Chem. Soc, 1966, 88, 1830.
30. G. Aksnes, R. Eriksen, and K. Mellingen, Acta Chem. Scand, 196/: 21, tuzo.
31. C. L. Bodkin and P. Simpson, J. Chem. Soc. (B), 1971, 1136
32. IP. G. Bentrude and J. H. Hargis, J. Amer. Chem. Soc, 1970 9_, 71 •
33. J. A. Mosbo and J. G. Verkade, J. Amer. Chem. Soc, 1973 95, 4659.
34. A. Munoz, M.-T. Boisdon, J.-F. Brazier, and R. Wolf, Bull. Soc. chim. 1971, 1424.
35. S. Oae, A. Nakanishi, and S. Kozuka, Tetrahedron, 1972, 28, 549.
36. D. G. Pobedimskii, N. A. Mukmeneva. and P. A. Kirpichtukoy. К яичгкий Rev, 1972, 41, 555; D. G. Pobedimskii, ibid, 1971, 40, 142; Д. ГПобеди^, H. А. Мукменева, П. А. Кирпичников.—Усп. химии, 1972, 41, 1Z4Z, А бедимский, там же, 1971, 40, 254]. rn п г,„„^погкий. - Усп.
37. V. V. Pen kovsku, Russ. Chem. Rev, 1975, 44, 449, [В. В. Пенькове химии, 1975, 44, 969].
716
45.
46.
47.
48.
49.
50.
51.
52.
53.
53.
54.
55.
56.
57.
58.
59.
60.
61.
38. R. F. Hudson, 'Structure and Merh, •
Academic. London, 1965 chanter o-rn'5? ,n Organo-Phosnhr,r
ций фосфорорганических cnrri™ \P- Хадсон. Структспл P, °rUS Chemistry’, 39. A. L Kirby and S. G ”WarrTn C а"гл Я Жр" !^а"ИЗ
vier Amsterdam, 1967, chapter’5; [А £he™stry of Phosphors'1 Else
40 SO71’№reP:Or:'a,n1nhV’ ,971^T’ °PPeH- С)Рга1П|ческая
40. d. J. waiver, Organophosphorus Chemist™' n
41. A Emsley and D. Hall, 'The Chemist™ I/di,PenJuin’ Lor|don, 1972 chanters don, 1976. chapter.9. Clumpy of Phosphorus’, Harper and Row P Lon
42. R. S. Davidson, in OrcranoDho^nhoruc cin • i •
Periodical Reports, The Chemical Society vol's Ь^б^Л'о ^«'Specialist
vol. 6, 197o, et seq, chapter 11 y 1 b’ 1970—1974, chapter 10-
43. M. E. Brennan, Chem. Comm. 1970 96A- a d c «.
yer, J. Org. Chem., 1975, 40, 1185. ’ ’ ' SchaaP’ Rees, and A. L. Tha-
44. D. H. R. Barton, P. G. Sammes, M V Taular г м r> „
В. E. Looker and IT G. E. Underwood, Chen/comm 1971 НзГ
J. I. G. Cadogan, Quart. Rev 1968 99 ooo. c. K' . 'J1"'-
Chem. Res., 1972, 5, 303. " 2’ SYnthesls. 1969, 11; Accounts
R. G. Harvey and E. R. De Sombre, in 'Tonics in Phnsnhnr., rr m- . . J
A/' °C} E'^' нГ'^а~’ lnterscience- New York, 1964 vol 1 ^57’ * ’
H. N. Rydon, Org. Synth., 1971, 51, 44 ’ ’ p' '
urwir **"**•M * °*" - £- '
I. V. Konovalova and A. N. Pudovik, Russ. Chem. Rev., 1972, 41, 41 [• \И В Коновалова, A. H. Пудовик. — Усп. химии, 1972, 41, 799].
N. A. Pudovik and V. K. Khairullin, Russ. Chem, Rev., 1968. 37, 317; [H. А. Пудовик, В. К. Хайруллин. — Усп. химии, 1968, 37, 745],
F. Ramirez. Pure Appl. Chem., 1964, 9, 337; Bull. Soc. chim. France, 1966, 2443;
Accounts Chem. Res., 1968. 1, 168.
E. I. Corey and R. A. E. Winter, J. Amer. Chem. Soc., 1963, 85, 2677; E. J. Corey, Pure Appl. Chem., 1967, 14, 19.
.. F. U7. Lichtenthaler, Chem. Rev., 1961, 61, 607.
53a. A. A'. Pudovik and V. P. Avery'anova, Zhur. obshchei Khim., 1956, 26, 1956;
[A. H. Пудовик, В. П. Аверьянова. — ЖОХ, 1956, 26, 1956].
A. N. Pudovik, Zhur. obshchei Khim., 1955, 25, 2173; [A. H. Пудовик. —ЖОХ, 1955 25 2173]
G. К. McEwen and J. G. Verkade, Chem. Comm, 1971, 668
H. N. Rydon, in ‘Special Publication No. 8’, The Chemical Society 1957 p. 61.
D G Coe, S. R. Landauer, and H. N. Rydon, J. Chem Soc, 1954, 2281.
D. K. Black, S. R. Landor, A. N. Patel, and P. F. Whiter, J. Chem. Soc. (C).
P^GillSpie, F. Ramirez, I. Ugi, and D. Marquarding, Angew. Chem. Internat.
Edn, 1973. 12, 91. , - tnco 84, 4737.
D. B. Denny and R. R. DiLeone, J. Amer C „• .„Cq g2’ 352
Л Emsley ^all, ^Khimis^ of Phokphoros’, Harper and Row, Lon-
don, 1976, chapter 3 1пгГЯгрг1 Snectra of Organophosphorus
L. C. Thomas, ‘Interpretation of the Infrared bpectra
Com/^ndcs;^gen,iLondoOn,s197 Phos horus Chemisttr’, ed. M. Grayson an
E. J. Griffith, Interscience, New York, ’969'Mark, and J.R-
64. M. M. Crutchfield, C. H.DunganJN.L eted M. Grayson and E. J. Griffith, zer, in ‘Topics in Phosphorus Chemist у •
Interscience, New York, vol 5 Qjienl peV„ 1972, 41, 390, Г •
65. В. 1. Ionin and T. N- Tim0^ ' ^72 41, 758]. , F|4evier Amster-
T. H. Тимофеева. -Усп. химии 19 of phosphorus. Usev.e
66. D. E. C. Corbridge The Structurai phosphorus,
dam, 1974, chapter 8, „ г, К McEwen, and J. G. vere
67 R. D Bertrand, H. J. Berwtn, G. Л.
1974, 4, 81. 717
62.
63.
68. A. Ztvierzak, Canad. J. Chem., 1967, 45, 2501.
69. F. W. Hoffmann and H. D. Weiss, J. Amer. Chem. Soc., 1957. 79, 4759
70. G. Aksnes and R. Eriksen, Acta Chem. Scand., 1966, 20, 2463.
71. D. B. Denny and D. H. Jones, J. Amer. Chem. Soc., 1969, 91, 5821.
72. Al. A. Armour, J. I. G. Cadogan, and D. S. B. Grace, J. C. S. Perkin II 1975 1185. ’ '
73. R. F. Hudson and C. Brown, Accounts Chem. Res., 1972, 5, 204.
74. A. W. Frank, Chem. Rev., 1961, 61, 389.
75. A. W. Frank, in ‘Organophosphorus Compounds’, ed G. M. Kosolapoff and L. Maier, Wiley-Interscience, New York. 1972, vol. 4, chapter 10.
76. L. A. Hamilton and P. S. Landis, in ‘Organophosphorus Compounds’, ed. G. M. Kosolapoff and L. Maier, Wiley-Interscience, New York, 1972, vol. 4^ chapter 11.
77. F. IF. Stacey and J. F. Harris, Jr., Org. Reactions, 1963, 13, 150.
78. С. M. Silcox and J. J. Zuckerman, J. Amer. Chem. Soc., 1966, 88, 168; E. IF. Abel, D. A. Armitage and R. P. Bush, J. Chem. Soc., 1965, 7098.
79 M. Mikolafczyk, J. Drabowicz, J. Omelanczuk and E. Flack, J. C. S. Chem Comm., 1975, 382.
80. H. R. Hudson, A. R. Qureshi, and D. Ragoonanan, J. C. S. Perkin I, 1964, 18, 38.
81. G. Aksnes and D. Aksnes, Acta Chem. Scand., 1964, 18, 38.
82. E. J. Corey and J. I. Shulman, Tetrahedron Letters, 1968, 3655.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Авенаии^нД 89
-триокса-1-фосфа- 683
^^^нкарбсик^^-^о™ 20, 81 йпппновая кислота 9.,
‘ 2,5-ДИЭтил- 8^ диэфир 98,
1 ч ДзафосФол - с,-
Йзафосфорин. днбензо- 675
1,z .илвая кислота 99
АзеЛЭ 31 264, 409, 417
Азе пяны 245 421, 512, 689
дзидокислоты 250, 261
Лучение 264’ 56’’ 634
„еакции 487, 553
Азодикарбоновая кислота 273, 301
Азокислоты 273
Азулен, производные 130
Аймалицнн 206
Айнхорна реакция 468
Аконитовая кислота 183
Акриловая кислота 42, 664
2-ацетиламино- 251
нитрил 625, 664
3-фенил- 42
эфиры 332, 336, 339, 625
Акролеин 142
а-Алаиин 241, 244
Р-Аланпн 254, 255
Алкоголиз 305 сл., 310 сл., 339, 354
Алкоксикарбонилирование 324 сл.
Алкоксикарбоновые кислоты 156,
170 сл., 179, 190
Аллильная перегруппировка 313
Аллильное окисление 313, 314
Аллофановая кислота, производные
562
Альдоиовые кислоты 174
Амидовые кислоты 393
Амиды 388 сл.
ацетали 373 сл.
основность 438 сл.
получение 17, 23, 46 сл., 94, 351 сл.,
389 сл., 632
Реакции 21 сл., 78, 79, 450 сл.
спектры 426 сл., 431 сл.
структура 425 сл.
фотохимия 451, 490
Аминокислоты 159, 233 ст
получение 237 сл., 278
Реакции 242 сл. 299
Фнз. свойства 234
А“”нооксикиел°ты 261
5”миак. основность 616, 617
Ap6v-n"M'),ia? K!lwlOTa 180, 257
гтг? А1и.хаэлиса реакция 50,
<>07.691,705
Арндта Эйстерта реакция 38, 321, Аспарагиновая кислота 457
Аспирин 156, 179
Ацетамид 454, 464, 484
N-алкилпроизводные
ассоциация 449
основность 440 сл.
получение 403, 409
реакции 413, 454, 467, 484
спектры 435, 438, 448’
структура 428, 429, 443 444
Ацетанилид!,! 440, 441, 456
Ацетилендикарбоновая кислота 99
129 сл., 168, 626
Ацетогидроксамовая кислота 500
Ацетоуксусная кислота 229
амид 411
эфиры
кислотность 221
получение 349
реакции 23, 204, 206, 223, 229
таутомерия 221, 222
Ацетуровая кислота см. Глицин, N-ацетил-
Ацидолнз эфиров 305 сл., 339 Ацилали 590
Ацилангидриды см. Карбоновые кн. -лоты, производные
Ацилирующие агенты для спирт 294 сл.
Ацилоиновая реакция 115, 360 Ацилпероксиды (Диацилпероксиды) 580, 590 сл.
Байера — Внллигера реакция 13, 165, 314 сл., 587
Барбье — Виланда реакция 351
Бекмана перегруппировка 17, 153, 401 сл.. 415 сл.
фрагментация 17, 402
Бензиловая перегруппировка 160. 168
Бснзогидроксамовая кислота 497, 501, 503, 505
4-питро- 501
Бензоиновая конденсация 655
Бензойная кислота кислотность 42, 100 реакции 62, 183. 293 физ. свойства 42
Бензойная кислота (производные)
амиды
основность 440. 441
получение 405. 420 реакции 457, 471, 180
719
Бензойная кислота (производные) амиды
спектры 436, 438
амино- 180, 257
бром- 145, 441
2-гидроксн- см. Салициловая кислота
4-гидроксн- 180, 191
дигидрокси- 156, 180
3,5-дниитро- 440
под- 145
4-метоксп- 440
иитро- 257, 440, 441
2-сульфо- 180
3,4,5-тригндрокси- 156, 180
2,4,6-трипзопроппл- 294
2,4,6-триметнл- 338
трифторметил- 137
2,4,6-трихлор- 44
4-фенил- 153
фтор- 145
хлор- 145, 440
эфиры 332, 334, 336, 362
Берча восстановление 485, 486
Бетаины 246
Биксин 88
Биурет 467
Бифенилкарбоновые кислоты 90, 100
Бнфталилидеи 698
Бишлера — Напиральского реакция481
Бодр}' реакция 415
Бодру—Чичибабина реакция 372
Бородина — Хунсдиккера реакция 54,
55
Брауна реакция 480, 481
Брефельдин 634
Бензол
дыоаровский 112
«пурпурный» 11
Бутандиовая кислота см. Янтарная кислота
гидрокси- см. Яблочная кислота
2,3-дигидрокси- см. Винная кислота
Бутановая кислота 42, 335, 420
амид 440
4-амиио- 256
бром- 142, 145
гидразид 517
2,4-дигидрокси- см. Пантовая кис-
лота
иод- 145
лактам 44
лактон 142, 255, 444
иитро- 147
4,4,4-трифтор- 137
фтор- 145
хлор- 142, 145, 392
эфиры 138
Бутен-2-овая кислота 31, 42
бром- 35, 217
Бутен-З-овая кислота 31
Бутип-2-овая кислота 42
У-Бутиролактон 142, 255, 444
Валериановая кислота см. Пентана вая кислота
Ваннлии 180
Вермикулин 216, 301
Верноловая кислота 156
Внльгеродта—Киндлера метод 317
Вильгеродта реакция 16, 181, 396
Вильсмеера комплексы 479, 486
Вильсмеера— Хаака реакция 479
Вильямсона синтез 365
Винные кислоты 156, 168 169 174 447 ’
Витамины
А 164
В 22, 257
Виттига реакция 33, 87, 173, 653 сл.
Вольфа перегруппировка 105, 321,322
Воля — Циглера реакция 496
Габриэля синтез 494
Галлера — Бауэра реакция 16, 395
Галловая кислота 289
Галлотонип 184
Галогенкарбоновые кислоты
получение 56, 57, 121, 137 сл., 242 сл.
реакции 66, 146 сл., 159
Гексадекановая кислота 306, 332
3-хлор- 15
Гексановая кислота 42, 57, 161
s-амино- 255
5-оксо- 220, 221, 229
2-фтор- 148
Гелля — Фольгарда — Зелинского реакция 138
Гемимеллитовая кислота 130
Геминальное алкилирование 321
Гептановая кислота
6-оксо- 221, 229
2-фтор- 148
Гептафульвен 130
Гидантоины 237, 251
Гидразиды кислотность 516 получение 65, 422, 510 сл. реакции 413, 517 сл.
спектры 515
структура 509, 514, 515
Гидразин, получение 576
Гидразннкарбоновая кислота 5//
Гидразинокислоты 249, 250, 2о1
Гидроксамовые кислоты получение 422, 458, 499, 500 реакции 413, 466, 504 сл., 55 спектры 501, 502 структура 497, 500, 501, 515
72Q
., Ги-тооксиамниокнслоты 261, 278 i-,,рОксиимниокислоГЫ 262, 278, 279 Г„арокс»карбоиовые кислоты кислотность 182 получение 61, 66, 87, 1о0, 157 сл. реакции 141, 183 спектры 182 физ. свойства 182 рндрокснмовые кислоты 502 Гипофосфорнстая кислота 713
Гпппуровая кислота см. Глицин,
N-бензоил-
рликолевая кислота 332
Глиоксиловая кислота 175 фенил- 147
Глниериды 288, 362
Глинин 234, 235, 237, 242
N-ацетил- 246
N-беизоил- 181, 246
N.N-Диметил-, эфир 197 N-метил- см. Саркозин N-ментил-, гидразид 519
Ы.Ы.М-трнметил- 246 а-фенил- 244 эфиры 197, 253
Глутаминовая кислота 241, 253
Глутаровая кислота 87 сл., 95, 99, 339
ангидрид 100, 102
имид 79, 495, 496
Глюкоза 174, 175
Глюконовая кислота 174
Глюкуроновая кислота 175
Гомокубан, производные 81, 202
Гомофталевая кислота, производные 85, 94
Гофмана—Лефлера реакция 57
Гофмана перегруппировка 55, 56, 476, 488
Гуанидины 575
Гуареши имиды 423
Дакина — Веста реакция 247
Дакина окисление 315
Дарзана реакция 151, 177, 348
Дебнера реакция 23
Делоншампа постулат 452
Депсидоны 289
Депсиды 183, 289 1,3,2-Диазафосфоланы 674
1,2-Диазины 170, 518 653
1,4-Диазпны 242, 245’
Диазокарбоновые кислоты 159, 262 эфиры 266 сл.
1,2- Диазолы 30, 273, 274, 518
1,3- Диазолы см. Имидазолы
Диазоуксусная кислота, эфиры 32
Диацилпероксиды 580, 581, 592
Дигалловая кислота 183
Диеион-фенольная перегруппировка 154
Динмиды 522, 523
Днкарбоновые кислоты
получение 16, 78 сл., 128, 168 сл., 175
реакции 20, 101 сл.
Дикмана конденсация 86, 106 сл., 201, 349
Дильса — Альдера реакция 111, 126 сл., 274 ретро-реакция 103
Динактин 290
1,3-Дн оксапы 190
1,3,2- Диоксафосфепины 672
1,3,2- Диоксафосфолаиы 671, 672, 712
1,3,2- Диоксафосфорипапы 599, 671, 672, 677, 679, 684
1,2-Диоксолапы 330
1,3-Диоксоланы 176, 189, 366, 369, 698
Диплоиции 289
М,М'-Дисукцинимид 515
1,3-Дитианы 29, 196, 321
Днфеновая-2,2 кислота 90, 100
Днциклогексилкарбодиимид 48, 49
Додекановая кислота
2-бром-2-метил- 138
2-метил-138
Енолов эфиры 16, 25, 309, 317, 344 сл.
Жасмон, производные 83, 169, 215
Жирара реактив 519
Зайцева расщепление 54
Защита групп
амино- 249 сл., 263, 540 сл., 548, 549
гидрокси- 340, 366, 375
карбокси- 63 сл., 125, 340, 341
сложноэфирной 355
Зеаралон 300
производные 298, 300
Изатоевып ангидрид 257
Изоимиды 421
Изоксазолы 150, 272
Изолейцип 235, 236
Изомасляпая кислота см. Пропановая кислота, 2-метил-
Изомочевпны 573—575
О-ацИл- 48, 49
Изофталевая кислота 100
Изохииолины 481
Имидазолы 51, 245, 457, 654, 690
N-ациклпроизводиые получение 49, 299, 459 реакции 49, 299, 300, 456, 459, 460, 513, 539
бензо- 458
721
Имидовые кислоты 431
Имиды 418 сл.
получение 104, 389, 390, 393, 418 сл.
реакции 451, 492 сл.
спектры 446 структура 444 сл.
Иминолы 431
Индазолы 690, 711
Ипдолкарбоновая-2 кислота, эфир 120
Индолы 226, 459, 479, 690
Итаконовая кислота 167
Кальб-Гросса окисление 522
Камптотецин 347
Камфора, производные 309, 613
Кападеисолид 168
изо- 168
Канниццаро реакция 12
Каприловая кислота см. Октановая кислота
Капролактам 255 сл., 424, 443, 444
Капроновая кислота см. Гексановая кислота
Карбазаты 273
Карбазолы 210, 689, 690
Карбаматы
получение 540, 544, 548, 556 сл.
реакции 557. сл.
структура 537
физ. свойства 538
Карбаминовая кислота 556, 562
Карбамоилазпды 87, 164
Карбамоилгалогениды 407, 408, 563,
564
Карбаииловая кислота 556
N-Карбоксиангидриды 408, 554
Карболины 690
Карбонаты 537 сл.
Карбонилбромид 548
Карбонилдиимпдазол 49, 539
Карбонилдифторид (Карбонилфто-рид) 479, 481, 548, 555
Карбонилфторхлорид 548, 555
Карбонилхлорид см. Фосген
Карбоновые кислоты ассоциация 43, 44 двухосновные см. Дикарбоновые кислоты
декарбоксилирование 53, 147 защитные группы 63 сл., 125, 340, 341
кислотность 41 сл,, 99, 100, 145, 182 меченые 38 сл.
Полпосновные см. Поликарбоновые кислоты
получение 11 сл., 78 ел., 137 сл., 157 сл.
реакции 46 сл., 101 сл., 138, 146 сл., 183 сл.
спектры 145, 146
Карбоновые кислоты структура 42, 43 хиральность 40, 41
Карбоновые кислоты (производные) азидо- см. Азидокислоты азиды 55, 56 амиды см. Амиды амино- см. Аминокислоты ангидриды 21, 48, 58 сл., 101 сл
галоген- см. Галогенкарбоновые кислоты
галогенангидриды 21, 47 сл., 52 58 сл.
гидрокси- см. Гидроксикарбоновые кислоты
кето- см. Оксокарбоновые кислоты нитро- см. Нитрокислоты нитрозо- см. Нитрозокислоты
оксо- см. Оксокарбоновые кислоты
эпокси- см. Эпоксикарбоновые кислоты
эфиры см. Эфиры сложные Кариофиллен 325 Каротиноиды 87, 164
Карпино реакция 523
Кплланп метод 174
Клу — Лутца — Иргенсона правило 237
Кляйзена конденсация 83, 106, 201 сл., 323, 348 сл.
Кпевенагеля реакция 33
Кнорра синтез 278
Кобальтоцен 16
Колхицин 127
Кольбе окисление 53, 111 сл., 148
Кольбе — Шмитта реакция 179
Кори—Зеебаха реактив 195
Корпномпколевая кислота 158
Коричная кислота 42
2-ацетиламино- 251
2-нитро- 147
3-(4-хлорфенил)- 138
Коррнны 415
Кортизон 117
Кортикроццн 91
Кофермент А 206
Ко.ха — Хаафа реакция
Кребса цикл 156 Кротоновая кислота см кислота
19
Бутен-2-овая
Кроцетин 302
Крюге расщепление 590
Кубан, производные 81, 202, 611
Кумалевая кислота 187
Кумарин 152, 187
Кумаринкарбоновая-3 кислота 120, 124
Кумариновая кислота 187
Кумаровая кислота 187
Курциуса реакция 55, 56, 396, 403
^"учённГзВДсл.', 402, 404. 409,
П0' 5413 сл., 491, 496
пмкшш 424, 451 сл., 484
Р^ктры 428, 433, 436, 447
структура 44э
фотохимия 491
Лактиды 184
Лактимы 463
Лактолы 115
„отучение 15, 300
а- 329, 330
й- 61 166, 184, 186, 293, 298, 308, 330, 370
... 16 61, 62, 158, 16о сл., 174, 184, 186, 209, 293, 310, 323, 331
6- 62 159, 165 ел., 174, 186, 331 с- 115, 186
реакции 165, 356, 358, 359, 394
спектры 334, 337
Лассепа перегруппировка 506 сл.
Лейкса ангидриды 408, 554
Лпзпи, гидрокси- 235
Лимонная кислота 156, 161, 170, 185
Линалоол 171, 295
Люминол 515
МакЛафферти перегруппировка 57,337 МакФенднна—Стивенса реакция 523 Малеиновая кислота 99, 126, 169, 175 ангидрид 78, 96
получение 100, 102
реакции 126 сл., 512
гидразид 518
дифтор- 144
имид 419, 421, 447, 448
полуамнд 457
фтор- 144
Малоновая кислота 80, 98, 99, 120 сл. гидрокси- см. Тартроновая кислота дибром-, диамид 705 дпэтпловын диэфир получение 94
реакции 23, 83 сл., 122 сл„ 345 физ. свойства 332
диэтиловый эфир (производные) 23, 122 сл.
диазо- 267
изопропилиден- 307
нитрил 94
Манниха реакция 110, 207
тасляная кислота см. Бутановая кислота
Мевалолактон 59, 317
Мевалоновая кислота 156 171
Меервейна - Пондорфа — Берлея восстановление 158
^еервейна реакция 205
Мезоксалевая кислота 267
Метапамин 617
М,М-дпметпл- 616, 617
N-метил- 617
Метановая кислота см. Муравьиная кислота
Метиловый красный 257
Метилмпцпп, производное 300
Миколевая кислота 168
Миндальная кислота, эфир 200
Миристиновая кислота 306
Михаэля реакция 88 сл., 165, 239, 281
Молочная кислота, эфир 200, 306
Мочевина 537, 565
производные 117, 538, 565 сл.
Муравьиная кислота 42 сл., 291, 297,
581
азидо-, эфиры 262 сл.
амид
ассоциация 441 сл.
получение 392, 395, 397
реакции 4Ц, 413
структура 426
фотохимия 466, 467
амид, N-производиые 429, 435
Ы,1Ч-димстил- 428, 449, 375, 479
N-фепнл- 463, 465
бром-, эфиры 548
под-, эфиры 548
фтор-, эфиры 544, 545, 548, 555
хлор- 262
эфиры 262, 268, 298, 543 сл., 551
хлортио-, эфиры 547
циано-, эфиры 548
Мускон 215
Мыла 44, 45
Наплои 256, 483
Нафталпндикарбоиовая-1,8 кислота
100
Непстовая кислота 325
Нефростеариновая кислота 167
Никотиновая кислота, 6-гидроксн- 188
Нингидрин 243, 246
Нитрозокислоты 262, 276 сл.
Нитрокпслоты 262, 279 сл.
Норборнадиен 264
Норборпадиеидикарбоновая-2,3 кислота 102
Норкаран, 7,7-дихлор- 149
Оксадиазолы 522, 554, 555
1,3-ОксазппЫ 29
Оксазнриднны 406, 513, 585
Оксазолы
бензо- 458
гидрированные
получение 189, 245, 247, 408, 487, 554
реакции 58, 176, 237, 310
723
Оксалплхлорид 116 сл.
Оксираны 156
получение 177, 190, 583 сл.
реакции 26, 190, 552, 584
Оксокарбоновые кислоты
кислотность 220, 221
получение 26, 82, 86, 106, 119, 125, 188, 195 сл., 201 сл., 207 сл., 217 сл.
реакции 26, 198 сл., 204 сл., 209 сл., 215 сл., 220 сл.
Октадекадиен-9,12-овая кислота 174
9,10-дигидрокси- 185
Октадекановая кислота 44, 57, 332
Октадецен-9-овая кислота
12-гидрокси- см. Рициполевая кислота
12,13-эпокси- см. Верноловая кислота
Октановая кислота 57, 332
тетрагидрокси- 174
Омыление 290, 337
Ортоамиды 373 сл.
Ортокислоты 364
Ортона перегруппировка ^76, 488, 492 Ортоэфира 312, 364 сл.
Пальмитиновая кислота см. Гексадекановая кислота
Пантовая кислота 156
Пантотеновая кислота 157
Парабановая кислота 117
Параконовые кислоты 83, 169
Паратион 616
Пеннциллановая кислота, 6-амино- 63
Пенициллины 54, 255
Пентановая кислота 42
2-амино-З-метил- см. Изолейцин
3,5-дигидрокси-3-метил- см. Мевалоновая кислота
5-нитро- 280
2-оксо- 226
4-оксо- 213, 221, 229
2,2,4-триметпл- 396
Пентеновые кислоты 340
5-бром-З-метпл- 171
Перистилан 220
Перкина реакция 33
Перкова реакция 699 сл., 703 сл.
Пероксиды ацилов (Диацилпероксн-ды) 580, 590 сл.
Пероксикарбаматы 580, 587
Перокснкарбаминовые кислоты 580
Перокспкарбонаты 580
Пероксикислоты 13, 105, 165, 314 сл., 580 сл.
эфиры 112, 580, 587 сл.
Псроксиугольные кислоты 580, 582 Перокспэфиры 112, 580, 587 сл.
Пикриновая кислота 191
Пимелиновая кислота 99
производные 86, 106
Пинен 217
Пиннера синтез 364, 399 сл
Пипеколовая кислота 245
Пипериновая кислота 175
Пиразины 242, 245
Пиразолы см. 1,2-Диазолы
Пираны 275, 653
Пирепофорин 301
Пиретрины 290
Пиридазины 170, 518, 653
Пиридины 51, 52, 152, 479
Пировиноградная кислота 197 221
244, 251
эфиры 96, 200
Пирофосфиты 676
Пирролы
получение 131, 153, 278, 284, 653
реакции 153, 413, 479
Поликарбоновые кислоты 77 сл.,
101 сл., 128
Полифосфины 636—638
Прево реакция 171
Пробковая кислота 99, 201
2-этил- 81
Прогестерон, производные 323
Пролин 245—247 гидрокси- 235 3-метил- 235, 236
Пропановая кислота 42 амид 409, 420, 440, 443 бром- 145 гидразид 517 гидроксифенил- 18' 2,2-дпметил- 42, 43 иод- 145 2-метил- 42, 89 амид 405, 412 гидразид 517 эфиры 81 нитро- 281 3,3,3-трифтор- 145 2,2,3-трифтор-3-хлор- 144 2-хлор- 145, 146 3-хлор- 42, 43, 144 сл эфиры 35, 332, 334, 335
Пропантрикар боновая-1,2,3,
117, 216,
2-гпдр-
окси- см. Лимонная Пропаргиловая кислота см. Пр вая кислота
Пропеллан, производное 95 Пропен-2-овая кислота см. Акри.
кислота „
3-фенил- см. Коричная кислота
Пропиновая кислота 42 фенил- 42
Пропнолактон 394
724
Пропионовая кислота см. Пропановая кислота
Простагландины 54, 156, 172 сл., 358
Протолнчестериновая кислота 167
Г 169
Птеридины 152
Пчелиной матки вещество 220
Раймера — Тимана реакция 180
резорннловыс кислоты 179, 180
Реппе реакция 19
Реформатского реакция 32, 115
149 сл., 163 сл., 177
Риттера реакция 196, 398, 403, 415
Рпцпполевая кислота 156, 161, 178
Ротенон 116
Руэманна пурпурный 243
Салициловая кислота 156
получение 179, 180
реакции 183, 191, 293, 672
Саркозин 245, 247
Себацпновая кислота 81, 83
Семикарбазиды 521, 575
Серин 252
N-ацетнл-, эфир 251
Сигматропные перегруппировки 323
Сорбиновая кислота 175
Спартеин 164
Стайлса реагент 202
Стеариновая кислота см. Октадекано-
вая кислота
Стрептолпдин 236
Сукцинимид 419
основность 449
получение 78, 79, 422
реакции 254
спектры 448
структура 444, 445
Сукцинимид (производные) 515
N-бром- 495, 496
Тартроиовая кислота 168
Таннины 289
Терефталевая кислота 98, 100, 332
Тестостерон, производные 117
Тетразолы 521
Тиазолы 131, 245, 654
Тиирапы 688
Тиогликолевая кислота 308
Тиофены
получение 131, 153, 168, 653
реакции 27, 153, 168
о-Толуиловая кислота 332
Трансаминирование 244
Треонин 235, 242
Триазолы 58, 690
Триптофан 284
Тропилня производные 272
'Руксиловая кислота 113
Тубаевая кислота 116
Туйон 213
Угольная кислота 537
производные 537 сл.
Ьксусная кислота 42 сл., 148 294 581
азидо- 266 ’ > 081
аллил- 363
амид см. Ацетамид
ангидрид 100
трифтор- 153
бензил- 227
бром- 39, 145, 151
бромфенил- 147
гидразид 517
а-гндроксиимино- 278
диазо-, эфиры 267, 268, 270, 273
дибром- 143
дипитро- 280
ди(фтор)хлор- 149
дихлор- 42, 148, 540
иод- 145
нитро- 282, 283
сульфо- 298, 309
трифтор- 152
кислотность 581
реакции 148, 297, 298, 367
спектры 334, 441
трихлор- 42, 146, 441
фенил-
кнелотность 42, 441, 468
получение 79, 317
реакции 79, 412, 464, 465
физ. свойства 42, 332
фенокси- 441
фтор- 145
хлор- 42, 145, 146, 332
циано- 345, 346, 423
эфиры 332 сл., 362
Упденен-10-овая кислота, 2-иод- 139
Урацилы 572
Уретаны 556
УронЬвые кислоты 175
Фаворского перегруппировка 16 319 сл.
Фазового переноса катализ 93. 225, 654
Феназины 690
Фенаитрахиноп 90, 96
Фенилаланин 243
Фенол, 2,4,6-трпнптро- 191
Фенолфталеин 105
Феиотпазниы 690
Феромон хлопковой совки 219
Фсрроценофан 108
Флавоны 121
Флуоренон 695
флуорескамнн 243
Флурам 243
725
Фосген 477, 521, 550 сл.
Фосфаты 598, 600
Фосфетаны 599, 622, 651 сл., 658, 659
Фосфиды металлов 604 сл.
Фосфин 600, 612, 616, 617 '
Фосфин (производные, см. также
Фосфины) бензилдихлор- 656 бспзилфенплэтил- 603 бром- 658 бутилднхлор- 656 дпамнно-трет-бутил- 660 ди (бензил) фенил- 603 дибром- 658
ди (бром) фенил- 663, 664
диметнл- 617
диметилхлор- 660
дифенил- 609
дифенилхлор- 604, 614, 657, 660,
662, 665
дифенилциклопентадиенпл- 604
дп(хлор)этил- 637, 654
ментплдпфенил- 614
метил- 617
метилдихлор- 661 сл.
метилфенплхлор- 656
трибром- 670,сл.
трибутил- 603, 618
триметил- 600, 604, 607
(триметилсилил) дифенил- 605
трнпропил- 619
трнфенпл-
получение 604
реакции 276, 626 сл., 631, 635,
639, 641, 697
спектры 600, 619
токсичность 616
трихлор- 600, 604, 611, 657, 670,
671, 674, 675 трициклогекспл- 625 трицпклопропил- 604 триэтил- 619 фенил- 609 фенилдихлор- 611 феннлдиэтпл- 609, 657, 663 сл.
Фосфинаты 599
Фосфинидены 638
Фосфинистые кислоты 598, 685, 712
эфиры см. Фосфиниты
Фосфиниты 598, 657, 658, 712 сл.
Фосфиновые кислоты 599
эфиры см. Фосфинаты
Фосфиноксид 598
бензилметилфенпл- 609
диметил- 660
метил (а-метилбензил) фенил- 606
трнфенил- 600, 619
трихлор- 600
Фосфинсульфиды 615, 633
Фосфины 598, 602 сл галоген- 655 сл. основность 616 сл. поли- 636-—638 получение 602 сл. реакции 623 сл. структура 621 сл. физ. свойства 616 Фосфираны 614 Фосфит(ы) 600, 604, 670 сл амидо- 600, 674 , 678 сл. ацетилдиэтил- 697 бромдибутил- 672 вторичные 709—711 гексаметилтриамидо- 600, 631 674 680 ’ ’
дибутил- 682 ди(фтор)пропил- 673 ди(хлор)бутил- 673 ди(хлор)этил- 675 диэтил- 709 иоддиэтил- 673 триаллил- 697 трибутил- 682, 706 триметил- 600, 604, 685, 694, 697 сл. тринеопентил- 671 трипропил- 695, 697 трис(гидроксиэтил)- 684 трифенил- 604, 682, 687, 692 693, 702 сл., 708
триэтил- 682, 689 сл., 692, 696 сл., 705 сл., 712
фенилдиэтил- 693 хлордибутил- 673 хлорди (|3-хлорэтил) - 673 хлордиэтил- 675
Фосфолы 652
гидрированные 599, 658, 663, 664 Фосфонаты 598, 693 Фосфонистые кислоты 598, 685 эфиры см. Фосфониты
Фосфоннты 657, 658, 712 сл.
Фосфония соли 50
получение 638, 701 псевдо- 691, 692 реакции 642, 707 Фосфоновые кислоты 599 эфиры см. Фосфонаты
Фосфора
оксихлорид 477 сл., 486, 496 пентагалогениды 477 сл., 484, 496, 508, 522
Фосфораны 33, 87, 96, 494, 631 Фосфоринаны 599 Фосфористая кислота 598 тио- 692 эфиры см. Фосфиты
Фосфорная кислота 598 эфиры см. Фосфаты
Фриделя — Крафтса реакция 657
726
ьписа перегруппировка 363, 364, 497
I,Lлевая кислота 98 сл.
згшглриД ЮО, Ю2, 106, 393, 512
имид см. Фталимид
3-мстил- ЮО
полvaмил 457
эфиры 332, 333
фталеины 106
фталимид 419
основность 449
попечение 423
реакции 257. 493, 495, 496
спектры 446—448
фульвен, гидрокси- 106
фульвоплюмеприн 116
фумаровая кислота 99. 126, 169, 175,
’' 625
фторпропзводные 144
фуразаны 690
фуранеол 115
фураны
получение 131, 153, 272, 653
реакции 153, 171, 479
Хагемаипа эфир 215
Хадсона правило 515
Хинолины 131, 690
Хлопковой совки феромон 219
Холестандновая-2,3 кислота 102
Хризен, производное 108
Цефалоспориновая кислота, 7-амино-63
Цефалоспорины 66, 255, 298
Цианамиды 565
ЦпангпдрннныЛ синтез 160
Циановая кислота 557, 566
Циклобутандикарбоновая-1,3 кислота,
2,4-дифеннл- ИЗ
Циклогексанонкарбоновая кислота
1-метил- 20
хлор- 140
Циннолины 118
Цистеин, производные 252
Чапмена реакция 406, 462
Шестакова реакция 352, 513
Шмидта реакция 55, 56, 403, 404, 415 сл.
Шоттена — Баумана реакция 246, 392
Штолле—Бекера синтез 118
Штреккера
деградация 243
синтез 237
Щавелевая кислота 98, 99, 115, 291
Дихлорангилрнд см. Оксалплхлорид эфиры
реакции 86, 119, 120, 160, 349
350, 362
структура 332
физ. свойства 332
Щавелевоуксусная кислота 228
Эквилеиин 350
Эметин 358
Эпокспкарбоиовые кислоты 177, 190
Эпоксисоедпиення см. Оксираны
Эритромицин 290
Этап, 1,2-бпс(фосфино) - 615
Этановая кислота см. Уксусная кислота
Этерификация 290 сл.
Эфиры сложные
амнналп 373 сл.
кислотность 100
получение 26 сл., 46 сл., 63 сл., 78,
86, 87, 106, 290 сл., 544
реакции 21, 22, 44, 56, 114, 337
спектры 46, 98, 334
структура 331, 332
Эшенмозера реакция 215
Яблочная кислота 156, 161, 169, 187
Янтарная кислота 98, 99
ангидрид
получение 102, 422
реакции 104, 393, 512
спектры 100
дибром- 129, 175
2,2-дифтор- 144
имид см. Сукцинимид
трифтор- 144
трифторхлор- 144
фенил- 94
фтор- 141
эфиры
получение 97, 113
реакции 86, 97, 113
спектры 98
физ. свойства 332
ОБЩАЯ
ОРГАНИЧЕСКАЯ
ХИМИЯ
ТОМ 4
КАРБОНОВЫЕ КИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ.
СОЕДИНЕНИЯ ФОСФОРА
Редакторы
О. И. СЛУЦКИЙ, М. Н. ПАСТУШЕНКО
Художник
И. В. НОСОВ
Технический редактор
Г. И. КОСАЧЕВА
Корректор
О. Б. ИВАНИЦКАЯ
И Б № I486
Сдано в наб. 26.10.83. Подп. в печ. 21.06.83. Формат бумаги бОХЭО'Лв- Бумага кн.-журн. i№ 2. Гарнитура литературная. Печать высокая. Усл. печ. л. 45,5. Усл. кр.-отт. 45,5. Уч.-изд. л. 52,31. Тираж 15 000 экз. Зак. 395.
Цена 4 р. Изд. № 2058а
Ордена „Знак Почета'1 издательство „Химия", Ю7076, Москва, Стромынка, 13.
Ленинградская типография № 2 головное предприятие ордена Трудового Красного Знамени Ленинградского объединения «Техническая книга» нм. Евгении Соколовой Союзполиграфпрома при Государственном комитете СССР по делам издательств, полиграфии н книж ой торговли. 198052, г. Ленинград, Л-52, Измайловский проспект, 29.
ОБЩАЯ ОРГАНИЧЕСКАЯ
ХИМИЯ