/






























































































































































































































































































































































































Автор: Васильев В.П.
Теги: аналитическая химия химия физическая химия
ISBN: 978-5-358-03522-5
Год: 2007




Текст
Высшее образование
В.П. Васильев
АНАЛИТИЧЕСКАЯ
ХИМИЯ
КНИГА 2
Физико-химические
методы анализа
ДРОФА
УДК 543.06(075.8)
ББК 24.4я73
В19
Рецензент:
д-р хим. наук, проф.
С. Я. Штыков
(Саратовский государственный университет)
Васильев, В. П.
В19 Аналитическая химия. В .2 кн. Кн. 2 : Физико-химические
методы анализа : учеб. для студ. вузов, обучающихся по химико-технол. спец. / В. П. Васильев. — 6-е изд., стереотип. — М. :
Дрофа, 2007. — 383, [1]с. : ил.
ISBN 978-5-358-03522-5 (кн. 2)
ISBN 978-5-358-03520-1
Во второй книге изложены теоретические основы физико-химических
(инструментальных) методов анализа: спектральных, электрохимических, хроматографических и др. Указаны возможности использования методов в
химико-аналитических целях, области практического применения, их значение и ограничение,
средства и методы оперативного аналитического контроля, применение
тест-методов и сенсоров в анализе.
В конце каждой главы приведены контрольные вопросы.
Для студентов вузов, обучающихся по химико-технологическим
специальностям. Может быть использован студентами энергетических,
сельскохозяйственных, медицинских, металлургических, педагогических и других вузов,
а также сотрудниками научно-исследовательских, заводских и экологических
лабораторий, связанных с проведением химических анализов,
УДК 543.06(075.8)
ББК 24.4я73
ISBN 978-5-358-03522-5 (кн. 2)
ISBN 978-5-358-03520-1 © ООО «Дрофа», 2002
Предисловие
Учебник составлен в соответствии с Государственным образо-
вательным стандартом и программой по аналитической химии
для студентов химико-технологических специальностей.
Учебник состоит из двух книг: «Титриметрические и грави-
метрический методы анализа» и «Физико-химические методы
анализа».
В первой книге изложены современные теоретические пред-
ставления о реакциях в растворе, их некоторые термодинамиче-
ские характеристики и соответствующие методы анализа.
Вторая книга посвящена физико-химическим (инструмен-
тальным) методам анализа: спектральным, электрохимическим,
хроматографическим и др. В ней рассмотрены возможности ис-
пользования физико-химических свойств веществ и характерис-
тик процессов в химико-аналитических целях. Вместе с тем в
книге достаточно подробно представлены области практического
применения физико-химических методов анализа, их значение,
достоинства и ограничения.
Второе издание (1-е ^ 1989 г.) учебника существенно перера-
ботано. Почти все главы дополнены новым современным мате-
риалом; совместно с профессором С. Н. Штыковым написана гла-
ва «Средства и методы оперативного аналитического контроля.
Применение тест-методов и сенсоров в анализе».
Все критические замечания будут приняты с благодарностью.
Автор
глава 1
Общая характеристика
физико-химических методов анализа
1.1. Особенности и области применения
физико-химических методов анализа
Все методы анализа основаны на использовании зависимости
физико-химического свойства вещества, называемого аналитиче-
ским сигналом или просто сигналом, от природы вещества и его со-
держания в анализируемой пробе. В классических методах химиче-
ского анализа в качестве такого свойства используются или масса
осадка (гравиметрический метод), или объем реактива, израсходо-
ванный на реакцию (титриметрический анализ). Однако химиче-
ские методы анализа не в состоянии были удовлетворить многооб-
разные запросы практики, особенно возросшие как результат науч-
но-технического прогресса и развития новых отраслей науки и
техники. Наряду с черной и цветной металлургией, машиностро-
ением, энергетикой, химической промышленностью и другими тра-
диционными отраслями большое значение для промышленно-энер-
гетического потенциала страны стали иметь атомная энергетика,
ракетостроение и освоение космоса, прогресс полупроводниковой
промышленности, электроники, широкое применение чистых и
сверхчистых веществ в технике. Развитие этих и других отраслей
поставило перед аналитической химией задачу снизить предел обна-
ружения до 10~5-—10~10%. Только при содержании так называемых
«запрещенных» примесей не выше 10"5% жаропрочные сплавы со-
храняют свои свойства. Примерно такое же содержание примеси
гафния допускается в цирконии при использовании его в качестве
конструкционного материала ядерной техники. (Вначале цирконий
был ошибочно забракован как конструкционный материал этой от-
расли именно из-за загрязнения гафнием.) Еще меньшее содержа-
ние загрязнений (до 10~10%) допускается в материалах полупровод-
никовой промышленности (кремнии, германии и др.). Существенно
изменяются свойства металлов, содержание примесей в которых
находится на уровне 10~б% и меньше. Например, хром и бериллий
становятся ковкими и тягучими, вольфрам и цирконий —■ пластич-
ными, а не хрупкими. Определение столь малых содержаний грави-
метрическим или титриметрическим методом практически невоз-
можно, и только применение физико-химических методов анализа,
4
обладающих гораздо более низким пределом обнаружения, позволя-
ет решать аналитические задачи такого рода.
Другой важной особенностью физико-химических методов
анализа является их экспрессностъ, высокий темп получения ре-
зультатов. Современные автоматические квантометры позволяют
получать результаты буквально через несколькр минут после пос-
тупления пробы в лабораторию. Своевременная информация о со-
ставе сырья, о степени химического передела и т. д. дает возмож-
ность технологу активно вмешиваться в ход технологического
процесса и вводить необходимые коррективы. Весьма существен-
ное значение имеет экспрессность анализа и в металлургическом
производстве, где корректировать состав стали можно по ходу
плавки в зависимости от результатов анализа. Сокращение време-
ни плавки, нередко зависящее от быстроты анализа, дает большой
экономический эффект, снижая энергетические и другие затраты.
Физико-химические методы позволяют проводить дистанци-
онный анализ^ т. е. анализ на расстоянии. Яркими примерами
являются анализ лунного грунта, выполненный рентгенофлу-
оресцентным устройством непосредственно на луноходе, опреде-
ление состава атмосферы, окружающей планету Венера, и т. д.
Важное практическое значение имеет дистанционный анализ в
земных условиях, например, когда анализируются препараты
высокой радиоактивности, токсичности, а также при анализе
морских вод на больших глубинах и решении других аналогич-
ных аналитических задач.
Многие приборы/ используемые в физико-химических мето-
дах анализа, позволяют автоматизировать сам процесс анализа
или некоторые его стадии. Автоматические газоанализаторы
контролируют состав воздуха в шахтах. В металлургической про-
мышленности широко применяют высокоавтоматизированные
оптические и рентгеновские квантометры. В значительной сте-
пени автоматизирован газовый хроматографический анализ в
нефтехимической, коксохимической и других отраслях промыш-
ленности. Нередко приборы физико-химических методов анали-
за используют непосредственно в производстве в качестве датчи-
ков соответствующих сигналов, например при регулировании
рН растворов или корректировке концентрации компонентов.
Анализ с помощью некоторых физико-химических методов
может быть выполнен недеструкционным анализом, т. е. без
разрушения анализируемого образца, что имеет большое значе-
ние для некоторых отраслей промышленности, а также для кри-
миналистики, медицины и т. д. Недеструктивный анализ может
быть выполнен рентгенофлуоресцентным, радиоактивационным
5
и некоторыми другими методами. Часто практический интерес
представляет не общее содержание какого-либо элемента в пробе,
а его распределение по поверхности образца — так называемый
локальный анализ — определение элемента в данной «точке» об-
разца. Этот анализ имеет значение в металловедении и других об-
ластях, где состав отдельных включений определяет качество ма-
териала, а также в минералогии, петрографии, криминалистике,
археологии ит. д. Выполняется локальный анализ рентгено-
сцектральным методом. Электроны собирают в очень тонкий пу-
чок диаметром 1 мкм и меньше (электронный зонд) и направля-
ют его в интересующую точку образца. По характеристикам
возникающего рентгеновского излучения судят о содержании
элементов в «точке». Для целей локального анализа использует-
ся также техника лазерной микроспектроскопии.
Использование компьютеров в аналитической химии очень
ценно не только для расчета результатов анализа и статистиче-
ской обработки, но и для решения других аналитических задач.
С помощью компьютеров можно более надежно выделять анали-
тический сигнал, проводить более четкое разрешение перекры-
вающихся сигналов и т. д. Компьютеры, встроенные в спектро-
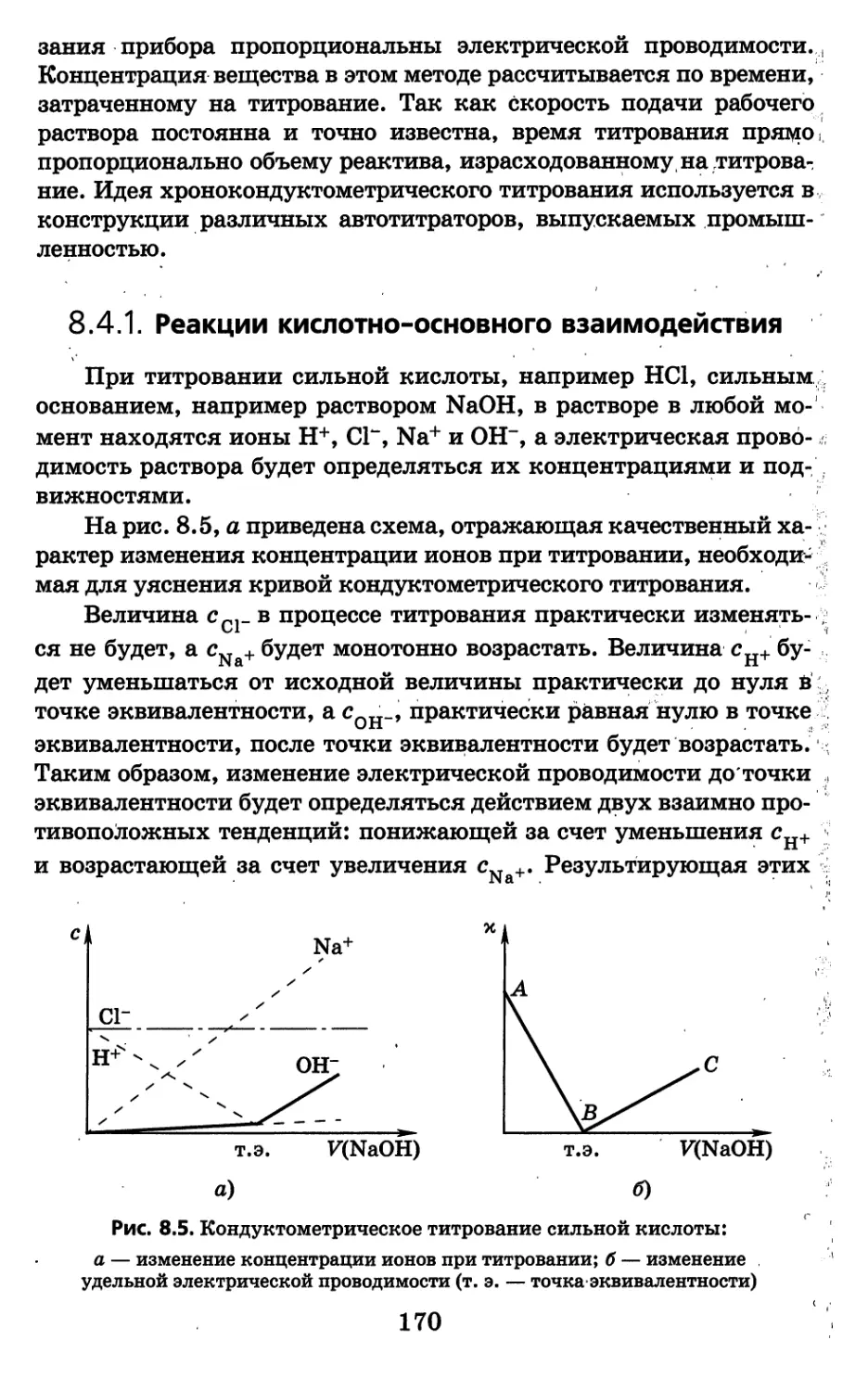
фотомер и в другие аналитические приборы, значительно расши-
ряют возможности этих приборов.
, Погрешность анализа физико-химическими методами состав-
ляет в среднем 2—5%, что превышает погрешность классических
методов анализа. Однако такое сравнение погрешностей не вполне
корректно, так как относится к разным концентрационным облас-
тям. При небольшом содержании определяемого компонента (по-
рядка 10"3% и менее) классические химические методы анализа
вообще непригодны,' при больших концентрациях физико-химиче-
ские методы успешно соперничают с химическими, а такие методы
анализа, как кулонометрия, даже превышают их по точности.
Следует отметить также, что погрешность анализа физи-
ко-химическими методами имеет тенденцию снижаться за счет
конструирования прецизионных аналитических приборов и раз-
работки более совершенных аналитических методик.
Однако химические методы анализа своего значения не поте-
ряли. Они незаменимы там, где при высоком содержании требу-
ется высокая точность и нет серьезных ограничений по времени
(например, анализ готовой продукции, арбитражный анализ, из-
готовление эталонов).
Существенным недостатком большинства физико-химических
методов является то, что для их практического применения требу-
ются эталоны, стандартные растворы и градуировочные графики.
6
1.2. Основные физико-химические методы;
лл^г: анализа
В группе физико-химических методов анализа иногда выде-
ляют физические методьь Однако достаточно строгого и одно-
значного критерия для этого нет, поэтому выделение физических
методов принципиального значения не имеет.
Общее число физико-химических методов анализа довольно
велико — оно составляет несколько десятков/Наибольшее прак-
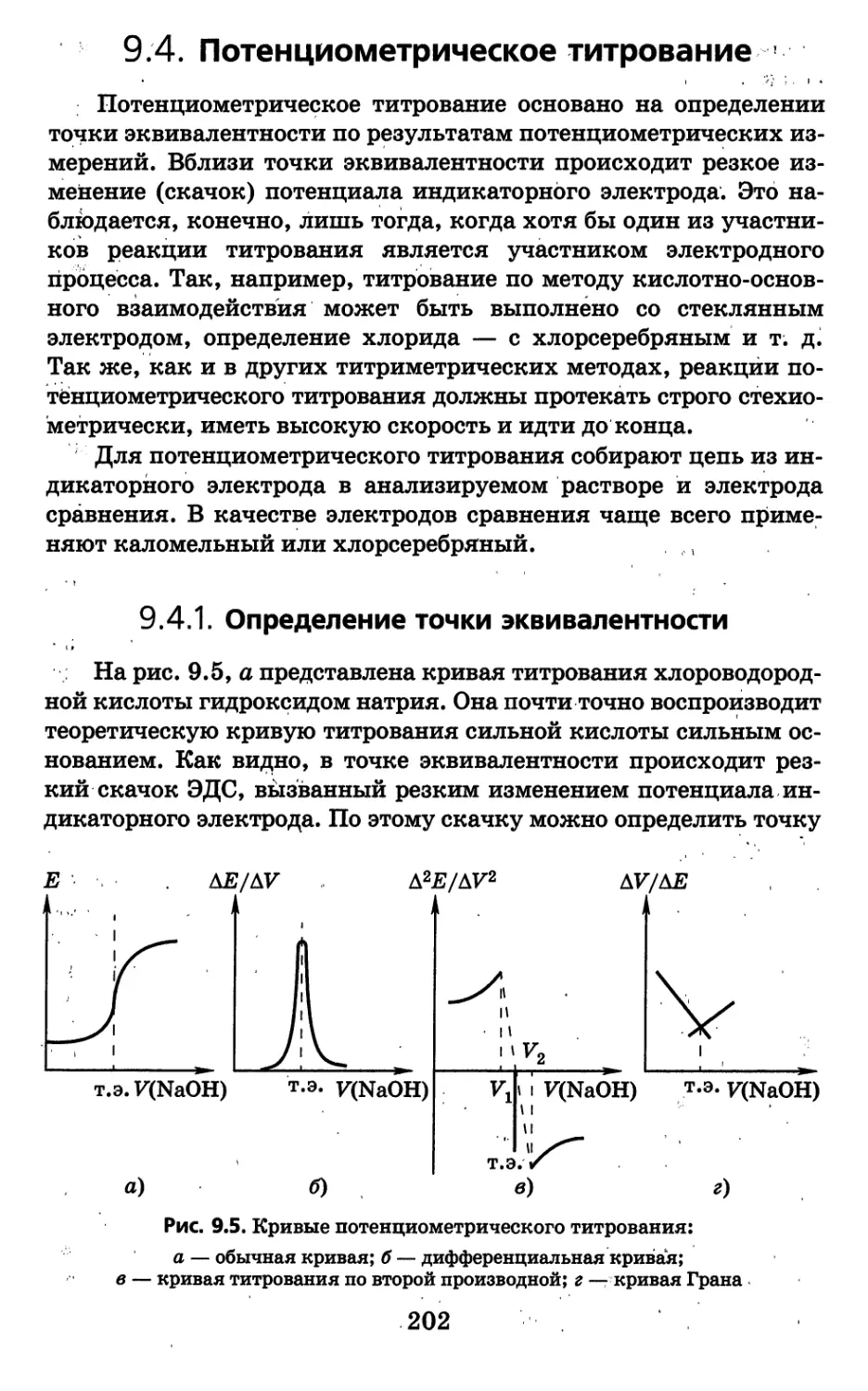
тическое значение среди них имеют следующие:
1) спектральные и другие оптические методы;
2) электрохимические методы;
3) хроматографические методы анализа.
Среди указанных трех групп наиболее обширной по числу ме-
тодов и важной по практическому значению является группа
спектральных и других оптических методов анализа. Она включа-
ет методы эмиссионной атомной спектроскопии, атомно-абсорб-
ционной спектроскопии, инфракрасной спектроскопии, спектро-
фотометрии, люминесценции и другие методы, основанные на из-
мерении различных эффектов, возникающих при взаимодействий
вещества и электромагнитного излучения.
Группа электрохимических методов анализа, основанная на
измерении электрической проводимости, потенциалов й других
свойств, включает методы кондуктометрии, потенциометрйи,
вольтамперометрии и т. д.
В группу хроматографических методов входят методы газо-
вой и газожидкостной хроматографии, жидкостной распредели-
тельной, тонкослойной, ионообменной и других видов хрома-
тографии. Перечень групп является далеко не полным, так как
сюда не вошли многие методы (радиометрические, масс-спёкт-
ральные и др.). Эти методы будут рассмотрены отдельно, что,'ко-
нечно, ни в коей мере нельзя считать признаком их второстепен-
ности. '.
1.3. Основные приемы, используемые
в физико-химических методах анализа
Почти во всех физико-химических методах анализа приме-
няются два основных методических приема: метод прямых изме-
рений и метод титрования (метод косвенных измерений).
Прямые методы. В этих методах используется зависимость
аналитического сигнала от природы анализируемого вещества
7
.лл.л/и
и его концентрации. Свойством,
зависящим от природы вещества*
является, например, длина волны
спектральной линии в эмиссион-
р ной спектроскопии, потенциал по-
Рис. 1.1. Взаимосвязь луволны в полярографии, а коли-
качественной и количественной чественной характеристикой слу-
характеристик компонента пробы жит интенсивность сигнала -
интенсивность спектральной ли-
ний в первом случае, сила диффузионного тока — во втором. В не-
которых методах связь аналитического сигнала с природой вещест-
ва установлена строго теоретически. Например, линии в спектре
атома водорода могут быть рассчитаны по теоретически выведен-
ным формулам с использованием фундаментальных констант (по-
стоянная Планка, заряд электрона и т. д.). Взаимосвязь качествен-
ной и количественной характеристик приведена на рис. 1.1. По оси
абсцисс отложены однородные характеристики Р, например длины
волн спектральных линий в порядке их возрастания, а по ордина-
те — интенсивность аналитического сигнала /. При качественном
анализе наблюдают сигнал, например, какая из ожидаемых длин
волн появится в спектре пробы, а при количественном измеряют
интенсивность сигнала.
Таким образом, аналитический сигнал — это величина,
функционально связанная с содержанием определяемого компо-
нента. Зависимость аналитического сигнала или его преобразо-
ванной величины от содержания определяемого компонента на-
зывают градуировбчной характеристикой. Она может быть
представлена в виде графика, формулы или таблицы. Значение
первой производной традуировочной функции при данном содер-
жании называют коэффициентом чувствительности или прос-
то чувствительностью и обозначают буквой в.
Область значений определяемых содержаний, предусмотрен-
ная данной методикой, составляет диапазон определяемых содер-
жаний. Наименьшее значение определяемого содержания назы-
вают нижней границей определяемых концентраций или ниж-
ним пределом сн. Наибольшее значение — верхней границей или
верхним пределом св. Наименьшее содержание, при котором по
данной методике можно обнаружить присутствие определяемого
компонента с заданной доверительной вероятностью Р (обычно
Р — 0,95), называют пределом обнаружения и обозначают ст1п р
(например, с^р = 0,95). Предел обнаружения находят по вели-
чине минимально обнаруживаемого аналитического сигнала.
8
Связь интенсивности аналитического сигнала / с концентра-
цией вещества имеет различный характер. Часто эта зависимость
выражается простым линейным соотношением
/ = Ас, (¿.1)
где А — константа; с — концентрация.
В аналитической практике наибольшее распространение по-
лучили следующие методы прямого количественного определе-
ния с помощью физико-химических измерений: 1) метод граду-
ировочного графика; 2) метод молярного свойства; 3) метод доба-
вок. Все они основаны на использовании стандартных образцов
или стандартных растворов.
Метод градуировочного графика. В этом методе измеряет-
ся интенсивность аналитического сигнала / у нескольких стан-
дартных образцов или нескольких стандартных растворов и стро-
ится градуировЬчный график обычно, в координатах / = /(с), где
с — концентрация определяемого компонента в стандартном об-
разце или стандартном растворе. Затем в тех же условиях изме-
ряется интенсивность сигнала у анализируемой пробы и по гра-
дуировочному графику находится концентрация анализируемого
вещества. Интервал концентраций на градуировочном графике
должен охватывать предполагаемую область анализируемых
концентраций, а состав стандартного образца или раствора дол-
жен быть близок к составу анализируемого.
Метод молярного свойства. Здесь также измеряется ин-
тенсивность аналитического сигнала у нескольких стандартных
образцов или растворов и рассчитывается молярное свойство1 А,
т. е. интенсивность аналитического сигнала, пропорциональная
1 моль вещества: А = 1/сСТ. Затем в тех же условиях измеряется
интенсивность сигнала у анализируемой пробы и по соотноше-
нию сх = I/А рассчитывается концентрация анализируемого ком-
понента. Метод предполагает строгое соблюдение соотношения
(1.1), по крайней мере, в области анализируемых концентраций.
Метод добавок. В этом методе сначала измеряется интен-
сивность аналитического сигнала пробы, затем в пробу вводится
известный объем стандартного раствора до концентрации с6т и
снова измеряется интенсивность сигнала. Если 1Х ■— интенсив-
ность аналитического сигнала пробы, а 1Х + ст — интенсивность
сигнала после добавки стандартного раствора, то, очевидно,
~ Асх> 1х + СТ—А(сх + сст),
откуда
с^с^: 1х .. (1.2)
Метод также предполагает строгое соблюдение соотношения
(1.1). Уравнение (1.2) нередко решается графически.
Методы титрования. В этих методах в ходе титрования изме-
ряется интенсивность аналитического сигнала / и строится кри-
вая титрования в координатах / - V, где У — объем добавленного
титранта, мл. Точка эквивалентности находится по кривой тит-
рования. Виды кривых титрования весьма многообразны, так
как интенсивность аналитического сигнала может быть связана с
концентрацией определяемого вещества, титранта или продукта
реакции.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Каковы основные особенности физико-химических методов ана-
лиза?
2. Что представляет собой недеструктивный и локальный анализ?
3. Каковы области практического применения физических методов
анализа?
4. Какие свойства вещества практически реализуются в качестве
аналитического сигнала? .■'!;.
5. Какие основные приемы анализа используются в качестве пря-
мых методов определения вещества?
6. В чем состоит метод градуировочного графика? Каковы его до-
стоинства и недостатки?
7. В чём заключаются достоинства и недостатки метода добавок?
г л а в а 2
Эмиссионный
спектральный анализ
2.1. Основные характеристики
электромагнитного излучения
Спектральные и другие оптические методы анализа основаны на
использовании различных явлений и эффектов, возникающих при
взаимодействии вещества и электромагнитного излучения.
Поскольку свет имеет двойственную природу — волновую и
корпускулярную, для его описания используют два вида харак-
10
теристик — волновые и квантовые. К волновым характеристи-
кам относятся частота колебаний, длина волны й волновое числом
к квантовым— энергия квантов.
Частота колебаний v показывает число колебаний в 1с, изме-
ряется в герцах (Гц). Высокие частоты измеряются в килогерцах
(1 кГц — 103 Гц), мегагерцах (1 мГц■= 106 Гц) и т. д. Например,-
красный свет характеризуется частотой 4 • 1014 Гц, зеленый —
6 • 1014 Гц.
Длина волны X показывает наименьшее расстояние между
точками, колеблющимися в одинаковых фазах. Это линейная
единица, измеряется в СИ в метрах (м) и его долях — сантимет-
рах (см), миллиметрах (мм), нанометрах (1 нм = 10~9 м) и т. д.*
Например, зеленый свет представляет собой электромагнитные
колебания с длиной волны Х = 500—550 нм, или 5 • 10~5 — 5,5 х
х 10"5 см. В зависимости от длины волны в электромагнитном
спектре обычно выделяют следующие участки:
Интервал длин волн Участок спектра
Ю-4—0,1 нм, или Ю-13—10"10 м у-Излучение
10"2—10 нм, или 10"11—10"8 м Рентгеновское излучение
10—400 нм, или 10~8—4 • 10~7 м Ультрафиолетовое излучение
400—760 нм, или 4 • 10"7—7,6 • 10~7м Видимый свет
760—106 нм, или 7,6 • 10"7— 10"3м Инфракрасное излучение
10"3 м—1м Микроволны или сверхвысо-
кие частоты
Х> 1 м Радиоволны
Длина волны и частота колебаний связаны между собой соот-
ношением
v = cA, .
где с — скорость света.
Если скорость света выражена в см/с, а длина волны — в см,
то v = 3 • 1010Д> где у выражена в Гц.
Например, для зеленого света А, = 500 нм = 5 • 10"5 см, часто-
Tav=2jJ^ = 6 • Ю14Гц.
5 • Ю-5
Величину, обратную длине волны, называют волновым чис-
лом v' и выражают обычно в обратных сантиметрах (см"1).
Например, для зеленого света v' = = 2 • ■104см~1.
* До введения СИ длину волны выражали в миллимикронах (1 тпцs
- 1 нм * 10~9 м) и в ангстремах (1А - ОД нм 10"10 м).
11
Энергия электромагнитного излучения определяется соот-
ношением
Е = Ъ,у,
где й — постоянная Планка, равная 6,62 • 10"34 Дж • с. Чтобы
получить энергию 1 моль, необходимо это значение умножить на
число Авогадро:
Е = 6,62 • 10"34 • 6,02 • 1023у = 3,99 • 1(У10%
где £ выражено в Дж/моль.
2.2. Теоретические ОСНОВЫ эмиссионной
спектроскопии
2.2.1. Спектральные термы
Методы эмиссионного спектрального анализа основаны на
измерении длины волны, интенсивности и других характеристик
света, излучаемого атомами й ионами вещества в газообразном
состоянии. Возникновение спектрального анализа как метода оп-
ределения химического состава вещества относится к 1860 г.,
когда была опубликована работа Кирхгофа и Бунзена: «Химиче-
ский анализ с помощью наблюдения спектра».
Испускание света атомами происходит за счет изменения
энергии атомов. Атомы могут обладать только строго определен-
ными дискретными запасами внутренней энергии: Е0, Ег, Е2
и т. д. Это означает также, что атомы не могут иметь энергию,
промежуточную между Е0 и Ег или между Ег иЕ2 и т. д. В невоз-
бужденном, т. е. нормальном, состоянии атомы обладают мини-
мальной энергией Е0. При подведении энергии, например при
столкновении с быстро летящими электронами, энергия которых
достаточна для возбуждения, атомы возбуждаются, т. е. перехо-
дят на более высокий энергетический уровень: Е19 Е2 и т. д.
Через очень короткое время (~10~8 с) атом самопроизвольно
возвращается в нормальное или какое-то более низкое возбуж-
денное состояние. Освобождающаяся при этом энергия АЕ излу-
чается в виде светового кванта Ау:
Частота излучения V определяется соотношением
у = АД -еа*~еа _ ЕА* _ Е (21)
12
где ЕА* и ЕА — энергия атома в возбужденном и нормальном со-
стояниях.
Или, характеризуя излучение волновым числом (у', см"1),
у, = £а^£а = £а-_£ (2;2)
где с — скорость света.
Совокупность излучаемых частот связана с энергетическими
состояниями атома.
Энергетическое состояние простейшего одноэлектронного
атома водорода определяется энергетическим состоянием его
единственного электрона и может быть найдено с помощью урав-
нения Шредингера. При решении уравнения Шредингера полу-
чают набор трех квантовых чисел: главное квантовое число п,
побочное квантовое число I и магнитное квантовое число тх.
Необходимое для полной характеристики электрона четвертое
квантовое число /п8, называемое спиновым или просто спином,
с уравнением Шредингера не связано.
Главное квантовое число п характеризует удаленность элект-
рона от ядра, это номер электронной оболочки; -йГ-оболочке соот-
ветствует п = 1, Х-оболочке — п = 2 и т. д. Орбитальное, или по-
бочное, квантовое число / характеризует подоболочки, из кото-
рых состоят оболочки, и орбитальный момент количества
движения электрона; оно приближенно определяет и форму элек-
тронного облака. При главном квантовом числе п побочное кван-
товое число /может принимать значения 0, 1, 2,п - 1, всего п
значений. В спектроскопии побочное квантовое число принято
обозначать буквой:
Числовое значение I ... 0 1 2 3 и т. д.
Символ ............. V Р й £ ит. д.
Магнитное квантовое число //^характеризует проекцию маг-
нитного момента движущегося электрона на направление внеш-
него магнитного поля. В соответствии с правилами простран-
ственного квантования проекция может принимать только
целочисленные значения. При данном I оно принимает значения
0, ±1, ±1, всего (21 + 1) значений.
Спиновое квантовое число //^характеризует собственный
момент электрона и принимает значения 1/2 и-1/2.
Полный момент электрона у является геометрической сум-
мой векторов/и в:
у иногда называют внутренним квантовым числом. Например,
для й электрона (/ = 2) внутреннее квантовое число у = 2 ± 1/2 =
13
= 5/2(8 = 1/2) или; — 2 - 1/2 = 3/2(в = —1/2). В одноэлектронном
атоме водорода энергетическое состояние электрона полностью
определяется главным квантовым числом п. Решение уравнения
Шредингера для такой системы приводит к следующему выраже-
нию для энергии электрона:
Подставляя уравнение (2.3) в соотношения (2.1) и (2.2), полу-
чаем:
■ 2п2те4 |
Л'8
2п2те4
с/*3
где пг и /г* — главное квантовое число электрона в основном и
возбужденном состояниях атома соответственно; Л — константа
Рйдберга, объединяющая фундаментальные физические посто-
янные.
Уравнения (2.4) и (2.5) сыграли большую роль в развитии теории
атомных спектров и теории строения вещества. Впервые соотношение
типа (2.5) было получено в 1885 г. Бальмером как чисто эмпирическое.
Длины волн известных тогда четырех линий в спектре водорода описы-
вались формулой Бальмера с очень высокой точностью. Уже это наводи-
ло на мысль, что формула Бальмера является не просто эмпирическим
соотношением, а скорее отражением какого-то еще не известного закона
природы. Теоретическое значение константы Рйдберга впервые было по-
лучено Бором в 1913 г. на основании предложенной им знаменитой мо-
дели атома, в которой постулировались квантовые уровни энергии
электрона. В настоящее время формулы (2.4) и (2.5) получаются как
следствие квантовр-механических представлений, опирающихся на
уравнение Шредингера.
Волновое число спектральной линии в соответствии с уравне-
нием (2.5) может быть представлено разностью двух величин:
у'=4-4 (2.6)
Эти величины получили название спектральных термов Т:
Т^-^; Г*(п*)-£. (3.7)
Следовательно,
у' - Т^) - П(/п).
14
Термы атома водорода могут быть непосредственно вычис-
лены по соотношениям (2.7). Приняв в формуле (2.7), например,
пг = 1, получим Т^п 1} = Д/1 = 109678,76 см"1.
Если принять пх = 2, а и* > 3, то получим частоты линий, об-
разующих серию Балъмера; при пг = 3 и и* > 4 — серию Лишена
и т. д. Такими сериальными формулами полностью описывается
спектр атомарного водорода, а при учете заряда ядра также
спектры других одноэлектронных частиц — ионизированного ге-
лия Не+, дважды ионизированного лития Ы2+, иона Ве3+ и др.
С учетом заряда ядра г формула (2.6) принимает вид
ч'=Щ-2Ц* (2.8)
Однако в атомах и ионах, содержащих более одного электро-
на, взаимодействие частиц имеет более сложный характер и рас-
четы по формуле (2.8) уже не дают вполне удовлетворительных
результатов. Ридберг показал, что спектральные термы атомов
щелочных металлов могут быть выражены формулой
Т(п) = 1122/(п + А)2.
Поправка Д зависит от побочного квантового числа, сумму (п 4 Д)
иногда называют эффективным квантовым числом. С увеличе-
нием числа внешних электронов формула усложняется.
Взаимодействие электронов в атомах элементов с небольшим
порядковым номером (30—35 и меньше) приводит к тому, что
энергетическое состояние такого атома может быть охарактери-
зовано суммарным орбитальным моментом и суммарным спи-
ном. Поскольку магнитные моменты пропорциональны соответ-
ствующим квантовым числам, можно говорить о суммарном ор-
битальном и суммарном спиновом квантовых числак атома. Для
их обозначения применяют те же, но не строчные, а! прописные
буквы латинского алфавита* какие используют для обозначения
квантовых чисел отдельных электронов. Так, при общем обозна-
чении орбитального квантового числа атома пишут Ь (вместо I
для отдельного электрона), при £ = 0 квантовое число обозначают
буквой 5, при Ь = 1 — буквой Р, при Ь = 2 — буквой Он т. д. Сум-
марный спин атома обозначают буквой 5, а геометрическую сум-
му Ь + в буквой е7.
Группа энергетических состояний, характеризуемая одними и те-
ми же величинами ± и Я имеет близкую энергию и образует один
Терм.
15
В этом смысле и употребляется термин «терм» в современной
спектральной систематике. При записи символа терма прежде
всего указывают его основную характеристику: квантовое число
суммарного орбитального момента Ь. Слева в виде верхнего ин-
декса у символа терма записывают его мультиплетность, пока-
зывающую число близких по энергии состояний, которые образу-
ют данный терм. Мультиплетность М равна
М = 25 + 1,
где в — суммарный спин атома.
Очевидно, мультиплетность на единицу превышает число не-
спаренных электронов в атоме. Термы с М = 1 называют одиноч-
ными или синглетными, термы с М = 2 — двойными или дублет-
ными и т. д. Например, терм 21) называют как дублет 2). Отдель-
ные компоненты терма Ь + 5; Ь +5-1; X - в записывают в
виде правого нижнего индекса терма. Если, например, суммар-
ный спин какого-либо атома или иона равен 1 (в = 1), а суммар-
ный орбитальный момент равен 3 (Ь = 3), то символ терма имеет
вид 3.Р4 з 2 (триплет 2^).
У замкнутых оболочек векторы Ь, Б и J равны нулю, что су-
щественно упрощает суммирование, однако полный вывод систе-
мы термов для данной электронной конфигурации остается до-
вольно трудоемким. Терм основного, состояния обычно определя-
ют, пользуясь правилами Хунда:
1. Основное, т. е. низшее, энергетическое состояние имеет наи-
большее значение суммарного спина (мультиплетности).
2. Среди термов с максимальной мультиплетностью низшим
является тот, у которого наибольшее значениеX.
3. Низшим компонентом мультиплета будет с/ = X - в, если
уровень заполнен меньше чем наполовину, и J = Ь + в, если он
заполнен больше чем наполовину.
Вывод системы термов для заданной электронной конфигу-
рации имеет большое значение в теоретической спектроскопии.
Каждая спектральная линия отражает переход электрона с
одного энергетического уровня на другой й волновое число любой
спектральной линии может быть представлено как разность тер-
мов. Однако не любая комбинация термов соответствует реально
наблюдаемой спектральной линии. Существуют определенные
правила отбора, указывающие, какие комбинации термов воз-
можны и какие невозможны. Эти правила имеют квантово-меха-
ническое обоснование. Переходы, возможные по этим правилам,
называются разрешенными, а невозможные.— запрещенными.
Основные правила отбора:
16
V. Разрешены переходы, при которых терм меняется на еди-
ницу^,т. е. разрешены, например, Р — 5- или В — Р-переходы, но
не разрешены Р — Р-, В - £>- или 5 — £>-переходы.
2. Внутреннее квантовое число J может меняться только на ±1
или совсем не меняться. Запрещены переходы, при которых Д = ±2.
3. Разрешены переходы без изменения мультиплетности.
Найдем разрешенные переходы, например, в атоме натрия, имею-
щем электронную структуру 1822в22р6381. Первые две оболочки (п = 1,
п = 2) в атоме натрия заполнены полностью, поэтому его термы будут оп-
ределяться единственным электроном, который в основном состоянии
находится на уровне Зз. Терм этой конфигурации будет 251/2. Дублет
здесь показывает формальную мультиплетность, в действительности же
все термы 5 являются одиночными (синглетными). При возбуждении
атома натрия электрон с уровня 3$ будет переходить на уровни р, <1 и т. д.
и термами атома в возбужденном состоянии, очевидно, будут 2Рз/2, 1/2 >
2£>5/2, з/2 и т- Д. Энергетические уровни Ыа графически представлены на
рис. 2.1, где также показаны некоторые из разрешенных переходов.
Линии, соответствующие Р — ^-переходам, двойные, так как при этих
переходах комбинируют двойные и одиночные термы 2Рз/2 — 2&1/2>
2Р\/2 — 2^1/2' В первом из этих переходов Де7 = 1, а во втором Д«7 = 0.
Запрет по мультиплетности не нарушается, так как формальная мульти-
плетность терма 5 в данном случае остается равной 2.
Несколько сложнее Б — Р-переходы (рис. 2.2). Термы 21)5/2 и 21>з/2
очень близки; в спектре появляются дублеты, соответствующие перехо-
дам 22>5/2,3/2 — 2^3/2 й 2^3/2 — 2^1/2* ^ приборах высокого разрешения
можно обнаружить триплет, так как появляется линия, соответствую-
щая переходу 2-Е>5/2 — 2^з/2> сливающаяся ранее с линией 22^2 — 2-^з/2*
Переход 22)5/2 — 2Рх/2 запРеЩен» так как ^ ^ 2.
Наиболее яркой в спектре будет линия, отвечающая переходу
с первого возбужденного уровня на основной. Линию, отвечаю-
щую этому переходу, называют резонансной. Например, у натрия
Рис. 2.1. Энергетические Рис. 2.2. Схема
уровни (термы) Э — Р-переходов
атома натрия влтоме натрия
2- 7831
резонансными являются переходы 2Р3/2— 251/2 и 2Р1/2 — 251/2,
им отвечают линии с длиной волны 588,996 и 589,593 нм. Это из-
лучение, в частности, окрашивает пламя горелки в желтый цвет
при введении солей натрия.
Изоэлектронные атомы дают сходные спектры, поэтому мож-
но говорить, например, о спектрах щелочных или спектрах ще-
лочно-земельных металлов. Сходство проявляется в наличии в
спектрах родственных элементов одинаковых групп линий (на-
пример, дублетов у щелочных металлов).
Спектр атома любого элемента существенно отличается от
спектра его иона в связи с изменением числа оптических электро-
нов при ионизации. Поэтому в таблицах спектральных линий ря-
дом с символом химического элемента приводят римскую цифру,
по которой можно судить о кратности ионизации атома. Цифра I
относится к нейтральному атому (например, Ге(1) относится к
Ре), цифра II— к однократно ионизированному атому (например,
Ре(П) относится к Ге+) ит. д.
2.2.2. Интенсивность спектральных линий
Интенсивность спектральной линии 1кг приближенно опреде-
ляется выражением
/Лг=ад^у,г, (2.9)
где Ык — число атомов в возбужденном состоянии 1г;Акг — веро- <
ятность перехода из возбуждённого состояния к в более низкое
состояние г; \кг — частота, соответствующая этому переходу; А —
постоянная Планка.
Для термически равновесной плазмы распределение атомов
по степеням возбуждения определяется законом Болъцмана:
Щ = Н8£ ехр(-§), , (2.10)
где Ы; число атомов в плазме; #А и #0 — статистические веса
возбужденного и нормального состояний; Ек — энергия возбуж-
дения А-го уровня, к — постоянная Больцмана.
При сочетании уравнений (2.9) и (2.10) получаем для интен-
сивности спектральной линии
/,г = АГА,г|Лу,гехр(^). (2.11)
Как видно, интенсивность спектральной линии зависит от .
температуры.
18
При .постоянстве температуры и других условий возбужде-
ния уравнение (2.11) переходит в
: 4=aW, (2.12)
где а' объединяет все сомножители в уравнении (2.11), кроме N.
Если режим работы источника возбуждения достаточно ста-
билен и скорость подачи вещества в плазму постоянна, наступает
некоторое стационарное состояние, при котором число атомов
элемента в плазме оказывается пропорциональным концентра-
ции этого элемента в пробе:
N = a"c, / . (2.13)
где с — концентрация вещества в пробе; а" — коэффициент про-
порциональности.
Подставляя соотношения (2.12) й (2.13) в (2.11), получаем
Ikr = a!a"c = ac. (2.14)
Если условия разряда не меняются при изменений концент-
рации, то коэффициент а остается постоянным и уравнение
(2.14) выполняется достаточно хорошо. Коэффициент а зависит
от параметров разряда, условий поступления вещества в плазму
и констант, характеризующих возбуждение и последующие пере-
ходы.
Однако не все кванты, испускаемые возбужденными части-
цами, достигают приемника света. Квант света может быть по-
глощен невозбужденным атомом и, таким образом, не будет за-
фиксирован приемником излучения. Это так называемое само-
поглощение. С увеличением концентрации вещества самопогло-
щение возрастает.
Самопоглощение учитывается в уравнении Ломакина—Шай-
бе, которое хорошо описывает концентрационную зависимость
интенсивности спектральной линии:
: I = ас\ (2.15)
где коэффициент а зависит от режима работы источника возбуж-
дения* его стабильности, температуры и т. д.; Ь — коэффициент
самопоглощения, учитывающий поглощение квантов света не-
возбужденными атомами.
При логарифмировании уравнения (2.15) получаем
lg I - lg а + big с. (2.16)
Линейная зависимость lg / от Ige? очень удобна для постро-
ения градуировочного графика. Уравнение (2.16) является осно-
вой количественного спектрального анализа.
2*
19
2.2.3. Ширина спектральных линий
Важной характеристикой спектральной линии является ее
ширина. Как известно, спектральная линия — это оптическое
изображение щели спектрального прибора, и чем шире щель, тем
шире спектральная линия. Тем не менее, хотя все спектральные
линии в данном спектре являются изображением одной и той же
щели и, казалось бы, должны иметь одинаковую ширину, она на
самом деле различна. Это кажущееся противоречие вызывается
несколькими причинами. Наиболее существенны из них следую-
щие.
1. Реальное излучение в обычных условиях эмиссионной
спектроскопии не бывает строго монохроматичным (его энергия
распределена в некотором интервале длин волн), и чем больше
этот интервал, тем шире спектральная линия. Это так называе-
мая естественная ширина спектральной линии, она составляет
величину порядка10~3 нм. При решении большинства аналити-
ческих задач с этим уширением практически можно не считать-
ся, так как оно значительно меньше уширения, вызываемого
другими причинами. <
2. Если светящаяся частинд движется вдоль линии наблюде-
ния, то излучаемая ею длина волны испытывает некоторое сме-
щение, приводящее в условиях эмиссионной спектроскопии при
большом числе излучающих частиц к уширению спектральных
линий. Это допплеровское уширение. Оно возрастает с уменьше-
нием атомной массы излучающего атома и повышением темпера-
туры. Для элементов середины периодической системы и темпе-
ратуры 5000 °С допплеровское уширение в видимой части спект-
ра составляет примерно 0,001—0,002 нм.
3. В электрическом или магнитном поле энергетические
уровни атома расщепляются на ряд подуровней. Это явление изг
вестно как эффект Штарка (расщепление в электрическом по-
ле) или эффект Зеемана (расщепление в магнитщиц поле). Поле,
обусловленное заряженными частицами в плазме, оказывается
достаточным, чтобы вызвать ущирение спектральных линий, ко-
торое доступно наблюдению на обычных приборах.
4. С увеличением концентрации элемента в пробе возрастает
самопоглощение, что приводит к уменьшению интенсивности
центральной части линии и ее уширению.
Очень широкие и очень узкие спектральное линии менее
пригодны для спектрального анализа, чем линии средней ши-
рины.
20
2.3 Основные узлы спектральных приборов
Прибор для проведения спектрального анализа имеет следую-
щие основные узлы: источник возбуждения, диспергирующий
элемент и приемник света. Кроме этих основных узлов в любом
спектральном приборе есть оптическая система, предназначенная
для получения параллельного пучка света, его фокусировки, из-
менения хода лучей и т. д.
В источнике возбуждения вещество атомизируется и возбуж-
денные атомы или ионы испускают свет, который диспергирую-
щим элементом разделяется в пространстве на отдельные состав-
ляющие, а приемник света их фиксирует;
2.3.1. Источники возбуждения
Источники возбуждения переводят пробу из конденсирован-
ной фазы в парообразную и возбуждают вещество в этой фазе;
В большинстве источников возбуждения эти функции совмещают-
ся, однако в некоторых случаях применяют два устройства: одно
для получения газовой фазы, другое для возбуждения. При ана-
лизе, например, биологических объектов или некоторых изделий
металлургической промышленности, когда особый интерес вызы-
вает локальный анализ, для перевода избранного участка пробы в
парообразное состояние, с успехом используется лазерная техника.
Возбуждение атомов происходит главным образом при пере-
даче энергии быстролетящими частицами, чаще всего электрона-
ми, если их энергия достаточна для возбуждения. Если кинетиче-
ская энергия летящих частиц меньше энергии возбуждения пер-
вого возбужденного уровня, при столкновении произойдет лишь
перераспределение энергии, как при ударе упругих шаров, но воз-
буждения не произойдет. Это так называемые упругие соударе-
ния. Чтобы атом перешел в возбужденное состояние, необходима
энергия, по меньшей мере равная энергии резонансного уровня
атома/Соударения, сопровождающиеся возбуждением атома, на-
зываются неупругими ударами первого рода.
Источник возбуждения должен обеспечивать необходимую
яркость спектра по сравнению с фоном и быть достаточно стабиль-
ным, т. е. интенсивности спектральных линий должны оставаться
постоянными по крайней мере за время измерения. Современные
успехи количественного спектрального анализа в значительной
степени достигнуты в связи с созданием источников возбуждения
высокой стабильности; Наибольшее применение в качестве источ-
ников возбуждения получили пламя, дуга и искра.
21
К источнику возбуждения часто относят и устройство для
введения анализируемой пробы, вид и конструкция которого за-
висят от характера, агрегатного состояния и физических свойств
пробы. Анализируемые металлические образцы в электрических
источниках возбуждения обычно служат электродами разрядно-
го промежутка. Растворы вводят в источник возбуждения с по-
мощью распылителей, порошкообразные пробы — с помощью
специальных устройств или при использовании угольных элект-
родов, в которых высверливается канал для набивки порошкооб-
разной пробы. Применяют также брикетирование анализируемо-
го порошка с добавкой металлов, их оксидов или графита. Изго-
товленный брикет затем становится электродом.
Пламя. Это известный еще со времен Бунзена и Кирхгофа ис-
точник света в спектральном анализе. Пламя дает достаточно яр-
кий и стабильный спектр. Простота регулировки и надежность
работы пламенных источников обусловили, по сути дела, второе
рождение пламенно-фотометрических методов, применяемых
очень широко. Возбуждение спектров в пламени имеет в основ-
ном термический характер. Температура пламени зависит от со-
става горючей смеси. Пламя обычной газовой горелки имеет тем-
пературу примерно 900 °С Смесь водорода с воздухом дает 2100 °С,
водорода с кислородом 2800 °С, ацетилена с кислородом — около
3000 °С
С помощью пламенных источников определяют свыше 40
элементов (M.gr Си, Мп, Т1, щелочные элементы, щелочно-земель-
ные и т. д.). В пламени не возбуждаются так называемые трудно-
возбудимые элементы и общая картина спектра является более
простой, чем дугового или искрового. Анализируемое вещество
вводится в пламя в виде раствора с помощью специального рас-
пылителя, обеспечивающего равномерное поступление вещества.
Дуга. Электрическая дуга — это электрический разряд при
сравнительно большой силе тока (5—7 А) и небольшом напряже-
нии (50—80 В). Разряд поддерживается за счет термоэлектрон-
ной эмиссии с раскаленной поверхности катода. Разряд пропус-
кают между электродами из анализируемого образца или между
образцом и электродом, не содержащим определяемых элемен-
тов. Температура дуги достигает 5000—6000 °С. Введение в
электроды примесей, обладающих более низким, чем основной
элемент пробы, потенциалом возбуждения понижает температу-
ру дуги. Таку в присутствии солей калия температура дуги между
угольными электродами падает с 7000 до 4000 °С. Это открывает
возможность регулировать температуру дуги и поддерживать ее
постоянной путем введения в зону разряда элемента с низким по-
22
тенциалом возбуждения так называемого спектроскопического
буфера. Обычно это соли натрия или калия в достаточном коли-
честве; В присутствии спектроскопического буфера устанавлива-
ется определенная температура плазмы, практически не завися-
щая от состава анализируемой пробы.
При анализе тугоплавких металлов и сплавов электроды ду-
ги делают из анализируемого образца. Для анализа легкоплав-
ких металлов и сплавов, а также руд, минералов, стекол, шлаков
и других непроводящих материалов электродами служат обычно
графитовые или угольные стержни — так называемые спект-
ральные угли. Анализируемая проба помещается в канал одного
из электродов и испаряется в плазму при работе дуги.
В дуге удается получить спектр почти всех элементов. Ис-
пользуется дуга постоянного и переменного тока. Для обеспече-
ния непрерывности горения и стабилизации процесса разряда
применяют специальные дуговые генераторы. Яркость дугового
спектра достаточно велика; а иногда чрезмерна, что может явить-
ся недостатком, так как значительно увеличивает фон. Не всегда
достаточная воспроизводимость условий возбуждения в дуге ог-
раничивает применение дуговых спектров в основном качествен-
ным и полуколичественным анализом. Существенным недостат-
ком дуги является также значительное разрушение анализируе-
мого образца. Повышение напряжения обычно улучшает
стабильность дуги, что приводит к повышению точности анали-
за. Высоковольтная дуга питается напряжением в несколько ты-
сяч вольт.
В практике спектрального анализа применяют также плаз-
менную горелку или плазмотрон (рис. 2.3). Плазмотрон пред-
ставляет собой камеру с двумя графитовыми электродами. В ка-
мере между анодом 1 и катодом 3. зажигается дуга при силе тока
20—30 А по трубке, расположенной по касательной к стенке, по-
дается инертный газ 2 при 150—200 кПа.
В аноде имеется отверстие, через которое
инертный газ выходит. Вихревые потоки
газа в камере охлаждают плазму снару-
жи, что приводит к сжатию разрядного
шнура и увеличению в нем плотности то-
ка. Сжатая плазма вместе с газом выбра-
сывается через отверстие анода в виде
струи длиной 10—15 мм, которая светит- ЩШ V
ся над поверхностью анода. Температура 4 $
в плазме достигает 5000—10 000 °С и вы-
ше. Анализируемый раствор 4 подается в Рйс. 2.3. Плазмотрон
23
плазму специальным распылителем. При анализе твердых образ-
цов пробы могут помещаться в катод или также вводиться в плаз-
му распылителем. Высокая температура и интенсивность свече-
ния делают плазмотрон весьма перспективным источником воз-
буждения, особенно для анализа трудноиспаряющихся и труд-
новозбудимых веществ. Большое аналитическое применение на-
ходит также высокочастотный плазмотрон с индукционной ка-
тушкой, питаемый ВЧ-генератором (индукционно-связанная
плазма).
Плазма содержит значительное количество ионизированных
атомов и молекул, поэтому ее можно считать газообразным про-
водником. Взаимодействие с магнитным полем позволяет поэто-
му индуктивно связать ее с источником высокочастотной энер-
гии. Полученная индуктивно связанная плазма (ИСП) является
современным универсальным источником возбуждения в эмисси-
онной спектроскопии. Принципиальная схема получения ИСП
показана на рис. 2.4. Три концентрические кварцевые трубки со-
ставляют плазменную горелку. По центральной трубке 2 га-
зом-носителем (аргоном) вводится мелкодисперсный аэрозоль
анализируемого раствора. По внешней трубке 4 аргон подается в
качестве охладителя, термически изолируя плазму, а средняя
трубка 3 предназначена для промежу-
точного плазменного потока. Горелка
помещается внутри медной индукци-
онной катушки 2, подсоединенной к
радиочастотному генератору. Под- к
жиг плазмы производится автомати-
чески «затравочными» электронами,
которые инжектируются в область
магнитного поля с помощью специ- .
ального разряда. При установленных
газовом потоке и уровне мощности ге-
нератора формируется самоудержи-
вающая аргоновая плазма с темпера-
турой 6000—9000 К.
Основными достоинствами ИСП яв-
ляются высокая долговременная ста-
бильность и воспроизводимость усло-
вий возбуждения. За счет высокой
температуры и длительности пребы-
вания в реакционной зоне в ИСП воз-
буждаются такие тугоплавкие эле-
менты, как бор, цирконий, вольфрам
охлажденный
газ
1- промежуточный
поток
-газ-носитель
с аэрозолем
Рис. 2.4. Индуктивно
связанная плазма
24
и др. Самопоглощение в ИСП практически ничтожно, так как
число возбужденных частиц в периферийной области относитель-
но мало.
Искра. Для получения искры используют специальные иск-
ровые генераторы. Принципиальная схема генератора включает
вторичную обмотку повышающего трансформатора, которая при-
соединяется параллельно к емкости и последовательно к катуш-
ке самоиндукции и искровому промежутку. Пробивное напряже-
ние более постоянно в управляемых схемах.
Так, в дуге Райского введен вспомогательный разрядный
промежуток, задающий и поддерживающий на постоянном уров-
не пробивное напряжение основного разрядного промежутка.
При горении искры развивается температура 7000—10 ООО °С и
происходит возбуждение всех элементов. При необходимости
температура искры может быть повышена до 12 ООО °С и выше.
Для проведения локального микроспектрального анализа при-
меряют микроискровой метод, в котором используют игольча-
тые электроды (например, медные) и устанавливают малое меж-
электродное расстояние. Микроискровой метод дает возможность
выявить локальное распределение элементов по поверхности в
сталях, железе и других образцах с локальностью 0,3—0,5 мм2.
Техника микроискрового анализа применяется также в методе
переноса* когда в результате микроискрового разряда небольшое
количество вещества с поверхности образца переносится на вспо-
могательный угольный электрод, спектр которого в дальнейшем
возбуждается и исследуется обычным методом.
Яркость искрового спектра недостаточна для визуального
анализа. Основное достоинство искры составляет большая ста-
бильность условий разряда и, следовательно, необходимая в ко-
личественном анализе стабильность условий возбуждения. Рабо-
та с искрой практически не вызывает разрушения образца, что
выгодно отличает искру от дуги.
Перспективным высокочувствительным источником света
является также полый катод, в котором могут возбуждаться эле-
менты с высоким потенциалом возбуждения.
2.3.2. Диспергирующий элемент
Диспергирующий элемент разлагает излучение в спектр. Это
наиболее важная часть спектрального прибора, в значительной
степени определяющая его аналитические возможности и основ-
ные характеристики: линейную дисперсию и разрешающую спо-
25
собность. Диспергирующий элемент характеризуется угловой дис-
персией, которую определяют какугловое расстояние Дер между
двумя лучами с близкими длинами волн Хг и Х2, отнесенное к ин-
тервалу АХ = Хг - Х2, т. е. И = Щ = ^ . От угловой дисперсий дис-
пергирующего элемента зависит линейная дисперсия спектраль-
ного прибора X), = = $1, где А1 — линейное расстояние в фо-
Да ак
кальной плоскости прибора между двумя лучами с близкими
длинами волн Хг и Х2, отнесенное к разности АХ = Хг- Х2. В прак-
тике часто используется величина, обратная линейной дисперсии:
£> = 1/Х>г. Она обычно находится в пределах от 0,1 до 10,0 нм/мм.
Разрешающей способностью спектрального прибора называют его
способность давать раздельное изображение двух спектральных
линии с близкими длинами волн.
Количественной характеристикой разрешающей способности
прибора является отношение 1? = Х/АХ, где АХ = Х1 - Х2— интер-
вал, в котором линии Хг и Х2 наблюдаются раздельно, а X = ^х * ^2 —-
средняя длина волны. У обычных спектральных приборов разре-
шающая способность составляет величину от 5000 до 50 ООО.
В качестве диспергирующего элемента используют призмы,
дифракционные решетки и интерференционные устройства.
Большое распространение в аналитической практике получили 1-
призменные спектральные приборы и приборы с дифракционной ^
решёткой.
Призмы для спектральных аппаратов изготовляют из стекла
или кварца, так как эти материалы достаточно прозрачны в ши-
рокой области длин волн. Стеклянные призмы имеют более высо-
кую угловую дисперсию и более доступны по сравнению с кварце-
выми, поэтому для работы в видимом и ближнем инфракрасном
участках спектра обычно используют стеклянные призмы. Для
исследования ультрафиолетовой области спектра применяют
призмы из кварца. •
Дифракционные решетки в качестве диспергирующего эле-
мента имеют существенные достоинства. Дисперсия света в диф-
ракционной решетке не зависит от длины волны и разрешающая
способность решетки значительно выше, чем призмы. Спект-
ральный интервал, доступный для исследования, достаточно ши-
рок (от 200 до 1000 нм).
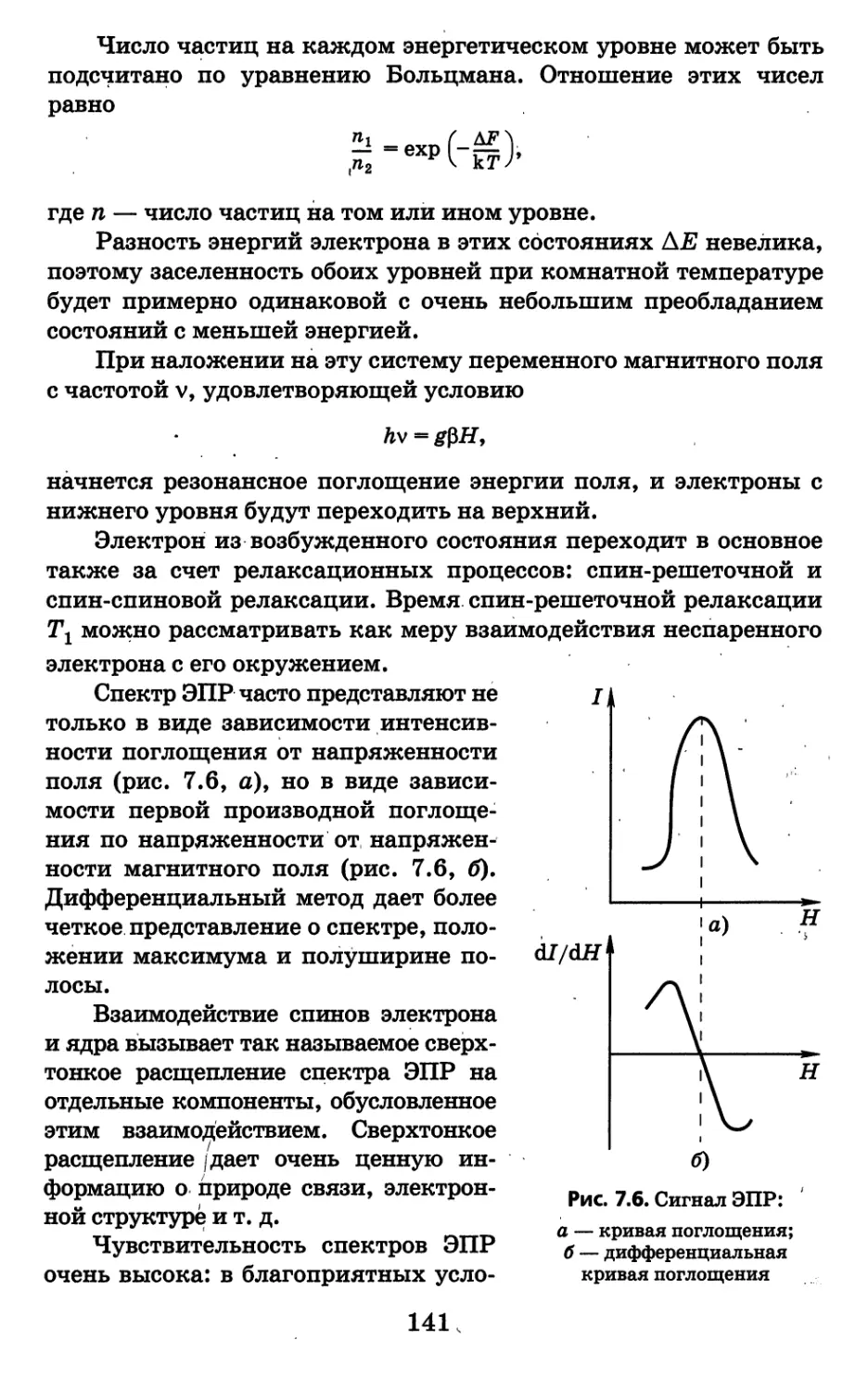
26
2.3.3. Приемники света
Приемники света характеризуются спектральной чувстви-
тельностью, т. е. способностью воспринимать излучение различ-
ной длины волны, и интегральной чувствительностью; которая
измеряется действием неразложенного в спектр излучения.
Глаз человека чувствителен к свету в области спектра при-
мерно от 400 до 760 им. Чувствительность глаза максимальна к
желто-зеленому свету (550 нм) и убывает от него в обе стороны —
и к красной, и к фиолетовой. Возможности глаза как измеритель-
ного прибора ограничены также и тем, что он очень приближенно
оценивает разность или отношение интенсивностей световых по-
токов. С достаточной точностью он устанавливает лишь равенст-
во или неравенство интенсивностей световых потоков одного цве-
та. На этом свойстве основаны все приемы визуальных методов.
Более универсальными приемниками света являются фото-
пластинки и фотоэлементы.
Фотопластинка. Светочувствительный слой фотопластин-
ки — это мелкие кристаллы галогенидов серебра, равномерно
распределенные в тонком желатиновом слое. При освещении фо-
топластинки в светочувствительном слое образуется скрытое
изображение как результат фотолиза бромида серебра под дей-
ствием кванта света:.■ АиВг ■+ Ну - Ag + Вг. На освещенных
местах фотопластинки появляются кристаллы металлического
серебра. Скрытое изображение проявляют путем обработки фо-
топластинки специальным проявителем, который завершает
процесс восстановления серебра на освещенных участках и по-
зволяет получить видимое изображение. Полученное изображе-
ние закрепляют (фиксируют) с помощью раствора тиосульфа-
та натрия (закрепителя или фиксажа), который растворяет
кристаллы бромида серебра, не подвергшиеся действию света:
А§Вг + 2820|" = Ag(S20з)!~ + Вг~. После такой обработки на фо-
топластинке остается изображение спектра в виде спектральных
линий.
Если / и /0 — интенсивность света,
прошедшего соответственно через затем-
ненный (засвеченный) участок фотоплас-
тинки и через незасвеченный (рис. 2.5),
то почернение (или плотность почерне-
ния) 5 равно Рис. 2.5. Фотопластинка:
3 — *—эмульсия;
/ 2 — подложка
27
с
F _
Почернение фотопластинки за-
висит от экспозиции, или количест-
ва освещения Ну которое прибли-
женно определяется формулой
Я = Et,
(2.17)
lgtf
где Е — освещенность; t — время ос-
вещения.
Зависимость почернения от ко-
личества освещения изображается
характеристической кривой фото-
пластинки (рис 2.6). Участок АВ на-
зывают областью недодержек, участок CD — областью передер-
жек. На участке ВС, называемом областью нормальных почерне-
ний, величина почернения линейно зависит от логарифма
экспозиции. Рис. 2.6 показывает, что
Рис. 2.6. Характеристическая
кривая фотопластинки
У ga EF lgtf-lgtf/
(2.18)
где у — фактор контрастности.
В эмиссионной спектроскопии используют контрастные фо-
топластинки, так как чем выше фактор контрастности у, тем
большее почернение будет вызывать одно и тоже количество ос-
вещения. Продолжение прямолинейного участка характеристи-
ческой кривой пересекает ось абсцисс в точке которая опре-
деляет инерцию фотопластинки.
Для прямолинейного участка характеристической кривой в
соответствии с уравнением (2.18) получаем
Я=у1е#-у1£#„
или (так как у и Нх для данной пластинки постоянны)
/■Я-у1*Я-1.- (2.19)
Подстановка в уравнение (2.19) значения Н из соотношения
(2.17) дает
Я-у^Я*--/. (2.20)
Это основное уравнение фотопластинки. Его применимость огра-
ничена прямолинейным участком характеристической кривой.
Другим важным свойством фотопластинки является ее чув-
ствительность. Её определяют как величину, обратную количе-
ству освещения (экспозиции), необходимого для получения по-
чернения, на 0,2 превышающего почернение вуали при освеще-г
нии белым светом. Для спектрального анализа более интересной
28
характеристикой является спектральная чувствительность, кото-
рую обычно представляют графически как в == /(А,), где X •— длина
волны падающего света.
Обычно фотопластинки имеют чувствительность в спектраль-
ном диапазоне от 230 до 500 нм. Эти пределы чувствительности
могут быть значительно расширены сенсибилизацией пластинок.
В настоящее время фотопластинки успешно применяют в широкой
спектральной области от короткого ультрафиолета до 1000 нм.
К основным достоинствам фотопластинок как приемников из-
лучения в спектральном анализе относят их способность интегри-
ровать интенсивность света, высокую чувствительность, достаточ-
но широкий спектральный интервал, документальность анализа, а
также возможность длительное время сохранять информацию, за-
ложенную в спектре. По сфотографированным спектрам, даже
спустя длительное время после их получения, можно, в частности,
проверить содержание различных элементов в пробе, включая и
те, которые ранее не определялись. Точность методов анализа с
применением фотопластинки достаточно высока. При этом следует
отметить наряду с методами точного фотометрирования возмож-
ности визуальной оценки интенсивности спектральных линий.
Одним из основных недостатков фотопластинок является не-
равномерность их эмульсии, представляющая дополнительный
источник погрешности анализа, а также длительность и трудоем-
кость операций по химической обработке фотоматериалов.
Фотоэлементы. Фотоэлементами называют устройства, преоб-
разующие световую энергию в электрическую. Действие фотоэле-
ментов основано на использовании фотоэффекта. Различают
внешний ^внутренний фотоэффекты. При внешнем фотоэффек-
те поглощение света приводит к отрыву электрона с облучаемой
поверхности. Внутренний фотоэффект характеризуется увеличе-
нием электрической проводимости ве-
щества под действием света. Если внут-
ренний фотоэффект проявляется вбли-
зи граничного слоя между двумя полу-
проводниками или полупроводником и
металлом, то возникает фотоЭДС Это
явление иногда выделяют в особый вид
фотоэффекта и называют фотогальвани-
ческим эффектом или эффектом запор-
ного (запирающего) слоя.
Фотоэлемент с внешним фотоэф-
фектом (рис. 2.7) состоит из фотокатода рис 2.7. Фотоэлемент
1 и анода 2, помещенных в стеклянную с внешним эффектом
29
Рис. 2.8. Фотоумножитель:
1 — катод; 2 — эмиттер;
3 —анод
колбу 3. Если колба эвакуирована, фо-
тоэлемент называют вакуумным. При
действии света на катод (обычно кис-
лородно-цезиевый или сурьмяно-це-
зиевый) из него вырываются электро-
ны, которые, попадая на анод, замыка-
ют цепь: гальванометр показывает
наличие тока. Фотоэлементы с внеш-
ним фотоэффектом чувствительны в
широкой области спектра, имеют ли-
нейную световую характеристику и
практически безынерционны. Чувст-
вительность их невысока, однако боль-
шое внутреннее сопротивление позво-
ляет включать эти фотоэлементы в
усилительные схемы. Среди недостат-
ков у элементов этого типа необходимо отметить наличие темно-
вого тока, хрупкость конструкции.
Значительно более чувствительными приемниками света яв-
ляются фотоумножители (рис. 2.8), действие.которых основано
на внешнем фотоэффекте и вторичной электронной эмиссии.
Расположение электродов и фокусирующее поле выбирают так,
чтобы первичный электронный поток, попадая на первый эмит-
тер, вызывал вторичную электронную эмиссию, электроны вто-
ричной эмиссии направлялись на следующий эмиттер и т. д. Уси-
ление подчиняется закону геометрической прогрессии:
где / — сила тока на выходе прибора; /0 — начальная сила тока;
а — коэффициент вторичной электронной эмиссии; т число
каскадов усиления.
Фотоумножители дают усиление в 105—106 раз. Они нашли
широкое применение в измерительной технике, в телевидении,
передачах из космоса, при исследовании ядерных и космических
излучений и в других областях науки и техники.
В фотоэлементах с запирающим слоем используют внутрен-
ний фотоэффект полупроводника и вентильный эффект запи-
рающего слоя, который образуется на границе между полупровод-
ником и металлом или двумя полупроводниками. Запирающий
слой пропускает электроны практически лишь в одном направле-
нии и не пропускает в другом. Таким образом, возбужденные
электроны могут проходить через запирающий слой, создавая
30
Ну
УН НІН.
Рис. 2.9. Фотоэлемент,
с запирающим слоем:
1 — контактное кольцо;
2 — запирающий слой
разность потенциалов. В селено-
вом фотоэлементе (рис. 2.9) элект-
роны, находящиеся в селене, под
действием света возбуждаются и
через запирающий слой проходят
в полупрозрачную пленку золота,
являющуюся покрывным элект-
родом. Обратному переходу элект-
ронов препятствует запирающий
слой. Это приводит к тому, что по-
крывной электрод заряжается от-
рицательно, а слой селена поло- ,
жительно. При замыкании такой системы во внешней цепи появ^
ляется ток. Характерным свойством фотоэлементов с запирающим
слоем является возникновение тока под действием света без учас-
тия постороннего источника напряжения. Аналогично устроены
кремниевый и многие другие фотоэлементы с запирающим слоем.
Достоинствами фотоэлементов с запирающим слоем является
высокая чувствительность, широкий спектральный интервал и
простота конструкции. Основные недостатки: нелинейность све-
товой характеристики, инерционность и заметная температурная
зависимость фототока.
2.4. Конструкции спектральных приборов
Конструкции спектральных приборов весьма разнообразны.
Они различаются по типу диспергирующего элемента, способу
регистрации спектра и т. д.
В приборах для визуального спектрального анализа — раз-
личных стилоскопах и стилометрах — диспергирующим элемен-
том являются стеклянные призмы, приемником света служит
глаз наблюдателя, На рис. 2.10 представлена оптическая схема
Рис. 2.10. Оптическая схема стилоскопа С Л-11
31
одного из распространенных отечественных приборов — стило-
скопа СЛЛ1.
Свет от источника возбуждения 1 через оптическую систему
2 попадает на входную щель 3 постоянной ширины 0,02 мм и по-
воротной призмой 4 через объектив 5 направляется на дисперги-
рующую систему из двух призм 6 и 7. Покрытый серебром катет
призмы 7 отражает лучи, которые вновь проходят диспергирую-
щую систему и через объектив 5 и поворотную призму 8 попада-
ют на зеркало 9 и далее в окуляр 11, служащий для наблюдения
спектра. Фотометрический клин 10 позволяет ослаблять интен-
сивность выбранной спектральной линии и по специальной шка-
ле, связанной с клином, оценивать ее относительную интенсив-
ность. Призма 7 может вращаться, что приводит к перемещению
спектра в поле зрения, а угол поворота призмы показывает по
шкале, к какой области длин волн относится наблюдаемый учас-
ток спектра. Стилоскоп предназначен для работы в спектральной
области от 390 до 700 нм, для возбуждения спектра обычно ис-
пользуется дуговой генератор. Элементарный фотометрический
клин позволяет повысить точность анализа по сравнению с обыч-
ными стилоскопами. Юстировка оптической системы делается на
заводе и заданное положение оптических деталей сохраняется
благодаря жесткому монтажу.
Для выполнения экспрессных аналитических работ вне лабо-
ратории применяют переносной стилоскоп типа СЛП-2, оптиче-
ская схема которого лишь немногим отличается от оптической
схемы СЛ-11.
Более сложную, чем у стилоскопов, оптическую схему ц уст-
ройство имеют сти л ометры. Фотометрическая система такого
прибора позволяет независимо ослаблять интенсивность двух
спектральных линий и количественно характеризовать их отно-
сительную интенсивность, а также сближать в поле зрения ана-
литическую пару линий, что создает удобство в работе и повыша-
ет точность анализа.
Наиболее широко распространенными приборами спектраль-
но-аналитических лабораторий являются кварцевые спектрогра-
фы, позволяющие фотографировать спектры в области длин волн
200—600 нм. Возрастает выпуск приборов с дифракционной ре-
шеткой в качестве диспергирующего элемента и фотографиче-
ской или фотоэлектрической регистрацией спектра (ДФС-13,
ДФС-10М и др.). Существенным достоинством фотоэлектри-
ческих приборов является экспрессность получения результа-
тов при сохранении точности. Так, с помощью квантомет]^
ДФС-10М определяют 11 элементов в одном образце за 6—8 мин. I
32
2.5. Качественный спектральный анализ
Основой качественного спектрального анализа является
свойство каждого химического элемента излучать характерный
линейчатый спектр. Задача качественного спектрального анали-
за сводится к отысканию линий определяемого элемента в спект-
ре пробы. Принадлежность линии данному элементу устанавли-
вается по длине волны и интенсивности линии. Однако общее
число линий в спектре многих элементов очень велико: напри-
мер, спектр тория насчитывает свыше 2500 линий, а спектр ура-
на — более 5000. Нет необходимости, конечно, определять длины
волн всех спектральных линий в спектре пробы. Для целей каче-
ственного анализа необходимо установить наличие или отсутст-
вие в спектре так называемых аналитических или последних ли-
ний.
При уменьшении содержания элемента в пробе интенсив-
ность линий этого элемента в спектре будет уменьшаться, некото-
рые линии исчезнут и число линий уменьшится. При какой-то
очень малой концентрации останется всего несколько линий. Это
и есть последние линии, по которым обычно проводится качест-
венный анализ. Последние линии хорошо изучены, их длины
волн и характеристику интенсивности можно найти в специаль-
ных таблицах и атласах спектральных линий. Это обычно, но не
всегда, резонансные линии. В таблицах их часто отмечают индек-
сами Uv U2 и т. д. или Vv V2 и т. д. Индекс и± показывает, что
при возбуждении спектра в дуге эта линия исчезает последней,
линия с индексом U2 исчезает предпоследней и т. д. Индексы V{>
V2 и т. д. относятся к этой же последовательности исчезновения
линий в искровом спектре. Однако эта последовательность не аб-
солютна. В зависимости от условий возбуждения и состава пробы
она может несколько изменяться.
Для расшифровки спектра и определения длины волны ана-
лизируемой линии пользуются спектрами сравнения, в которых
длины волн отдельных линий хорошо известны. Чаще всего для
этой цели используют спектр железа, имеющий характерные
группы линий в разных областях длин волн.
Спектр анализируемого вещест- Хх ^
ва обычно фотографируют над спект-
ром железа. На рис. 2.11 представле-
но схематическое изображение не-
большого участка спектра железа и
спектра исследуемой пробы/Для оп- Рис. 2.11. Определение длины
ределения длины волны заданной ли- волны спектральной линии
3 - 783 , 33
Х1 Х2
нии Хх измеряют расстояние аг от этой линии до ближайшей к ней
линии спектра железа, длина волны которой Хг точно известна, и
расстояние а2 от линии Хх до другой линии с известной длиной
волны Х2. В небольшом спектральном интервале дисперсия прибо-
ра Остается постоянной, поэтому можно записать пропорцию
\0
а2
После простых преобразований получаем формулу для расче-
та длины волны неизвестной линии:
ах + а2
..>•' Расстояние между линиями можно измерить, например, с
помощью измерительного микроскопа МИР-12, спектропроекто-
ра ПС-18 или компаратора. Надежность анализа возрастает, ког-
да встык ^ пробы фотографируют спектры подозревае-
мых элементов, как это можно видеть на рис. 2.12. Анализ этих
спектров показывает, например, что в железе содержится неболь-
шое количество марганца (линия 294,92 нм) и алюминия (линия;
281,61 нм). В спектре алюминиево-магниевого сплава (спектр 2)
л.^гко обнаруживается железо по групце лцний 275,57—274,65 нм,
медь по линии 282,43 нм ц довольно большое, количество март
ганца (группа линий 294,92—293,31 нм). Четкр видны линии
магния (спектр 3) в спектре алюминиево-магниевого сплава,
(сдектр 2). При проведении качественного спектрального анали-
за часто цользуются атласом спектральных линий. На планшетах (
этого атласа нанесены линии спектра железа и аналитические
лцнии различных элементов. Совместив изображение спектра
Мп(П)
сч1
со тН >
00 со
см сч1
со
тН
00
.00
С<1
сч1
5д
со тН
т£ со
С\Г тН
00 00,
¥е(11)
ъ± ню
ю о ю
1
2
3
Рис. 2,12. Участок спектра:
1 — железа; 2 — алюминиевое марганцевого сплава; 3 т- магния
'Я
.д.
34
яселеза, полученное с помощью спектропроектора ПС-18, с ли-
ниями планшета, отмечают, с какой аналитической линией на
планшете совпадает линия анализируемого спектра.
Отсутствие последней линии определяемого элемента в
спектре гарантирует отсутствие других линий этого элемента.
Однако наличие линии с длиной волны, характерной для послед-
ней линии какого-либо элемента, еще не означает, что линия дей-
ствительно принадлежит именно этому элементу. Основной при-
чиной ошибок является так называемое наложение спектраль-
ных линий, связанное с недостаточной дисперсией рядовых,
спектральных приборов. Таблицы спектральных линий показы-
вают, например, что длина волны последней линии любого эле-
мента в пределах ±0,05 нм совпадает с длиной волны линий мно-
гих других элементов. Часть элементов почти всегда можно иск-
лючить, основываясь на данных о происхождении пробы или
имея в виду интенсивность линии и условия возбуждения. Одна-
ко и после этого нередко остается несколько элементов, которым
эту линию можно приписать. Окончательную идентификаций
проводят, проверяя последние линии всех «подозреваемых» эле-
ментов. '-3 ■ -
Спектральным анализом качественно можно определить бо-
лее 80 элементов. Предел обнаружения методам;и качественного
спектрального анализа колеблется для разных элементов в очень
широких пределах: от 10"2 СЙ£, Об, и и др.) до 10"5% (Ыа, В, В1
и др.). Следует отметить, что отсутствие линии какого-либо эле-
мента в спектре означает лишь, что его концентрация в пробе
меньше предела обнаружения. В связи с низким пределом обна-
ружения методами спектрального анализа нередко «переоткры-
вают» те или иные элементы, попавшие в пробу в результате слу-
чайных загрязнений.
2.6. Количественный спектральный анализ
Попытки использовать зависимость интенсивности спект-
ральных линий от концентрации элемента в пробе для количест-
венного определения долгое время оставались безуспешными.
Даже в начале XX в. возможности количественного спектрально-
го анализа оценивались очень невысоко.
Одной из основных причин неудач была недостаточная ста-
бильность условий' возбуждения. Интенсивность спектральной
линии при прочих равных условиях определяется количеством
в°збужденных атомов в источнике возбуждения, которое зависит
35
не только от концентрации элемента в пробе, но и от условий воз-
буждения. Перевод компонента твердой пробы в плазму связан с
протеканием процессов плавления, испарения и возгонки. На со-
став плазмы, таким образом, оказывают влияние температура и
теплота плавления компонентов пробы, их коэффициенты диф-
фузии, давление пара, температура источника возбуждения и
многие другие факторы, поэтому состав вещества в плазме источ-
ника возбуждения существенно отличается от состава исходной
конденсированной пробы. Недостаточная стабильность условий
возбуждения вызывала изменения в составе и температуре плаз-
мы, что приводило к изменению интенсивности спектральных
линий и, как следствие, к колебаниям в результатах анализа.
Связь между интенсивностью спектральной линии и кон-
центрацией элемента в пробе при стабильной работе источника
возбуждения приближенно устанавливается уравнением (2.14).
Зависимости, наблюдаемые на опыте, хорошо описываются урав-^
нением Ломакина—Шайбе (2.15). Коэффициент самопоглощения;
Ь в этом уравнении зависит от концентрации, однако в некото-
ром, иногда довольно широком интервале концентраций он оста-
ется постоянным.
В практике количественного спектрального анализа обычно
используют интенсивность не отдельной линии, а отношение ин-
тенсивностей двух спектральных линий, принадлежащих разг
ным элементам. Таким образом, в качестве свойства, связанного
с концентрацией элемента, используется отношение интенсив-
ности линии определяемого элемента к интенсивности линии
другого элемента в этом же спектре. Такая методика позволяет
снизить требования к постоянству условий возбуждения и реги-г
страции спектров.
Линию определяемого элемента обычно называют аналити-
ческой линией, и ее интенсивность обозначают 1& или называют,
линией примеси, и обозначают интенсивность /пр. Вторую ли-
нию, обычно называемую линией сравнения, выбирают так, что-
бы отношение интенсивностей зависело только от концентрации
определяемого элементарно не от условий возбуждения и регист-
рации спектра. Иногда в анализируемую пробу специально вво-
дят так называемый внутренний стандартI т. е. элемент, ли-
нию которого используют в качестве линии сравнения. При ана-
лизе проб, содержащих большое количество какого-то элемента,
в; качестве линии сравнения обычно выбирают линию эуого эле-
мента. Например, при анализе сталей это бывает линия спектра .
железа. Интенсивность линии сравнения обозначают /с или'у если
36
линия принадлежит основе, называют линией основы и ее интен-
сивность обозначают/осн. ^
Уравнение Ломакина—Шайбе (2 Л 5) для аналитической ли-
нии и линии основы имеет вид
их отношение
*пр и спр ' /осн и сосн »
К=^<=><. (2.21)
ОСИ
Уравнение (2.21) показывает, что отношение интенсивностей
также пропорционально концентрации элемента в пробе. Это ос-
новное уравнение методов количественного спектрального анали-
за. Методы различаются лишь способом оценки относительной
интенсивности. При выборе пары линий для количественного
анализа руководствуются рядом требований к энергиям возбуж-
дения спектральных линий, их длинам волн и интёнсивностям,
Выполнение этих требований существенно уменьшает зависи-
мость относительной интенсивности от условий возбуждения.
Согласно уравнению (2.11) интенсивность аналитической ли-
нии равна
Аналогично выражается интенсивность линии основы:
Возьмем отношение интенсивностей этих линий:
*щ> - ^щА^ЛУщ, с::р (_Et - Ek\
Обозначим произведение множителей, практически не зависящих
от температуры, через А и при постоянстве Nnp и N0CH получим
&-А-ёж»(-Щ)1 (2.22)
Из уравнения (2.22) следует, что даже при постоянной кон-
центрации компонентов в плазме относительная интенсивность
спектральной линии не остается постоянной — она зависит от
температуры. Уравнение (2.22) показывает также, что чем мень-
ше Д2£, тем меньше относительная интенсивность спектральной
линии зависит от температуры. В практике спектрального анали*
37
за обычно подбирают линии, которым соответствуют сравнитель-
но небольшие АЕ (не более 1 эВ) и элементы с близкими потен-
циалами ионизации.
Существенно также, чтобы интенсивности выбранных линий
не слишком резко отличались между собой. Обычно выбирают
линии, отношение интенсивностей которых не превышает 10;^
т. е. находится в пределах 0,1 < 1пр/10СН.< 10, так как в противном1
случае точность определений уменьшается. Кроме того, обе ли-
нии должны быть в одном участке спектра в пределах примерно
10 нм (А,пр - Хоси = 10 нм), чтобы не сказывалась зависимость по-
казаний приемника излучения от длины волны. Таким образом,
основными требованиями к выбранной паре будут:
ДД<1эВ;
1 :Лф - ^осн < 10 нм;
Пара линий, удовлетворяющая этим требованиям, называется го-
мологической парой. Относительная интенсивность линии гомо-
логической пары обладает малой чувствительностью к условиям
возбуждения и регистрации спектра.
Принципиальная схема аналитического процесса включает
следующие основные операции (этапы):
1. Устанавливают режим источника возбуждения (пламени,
дуги, искры или ИСП) и вводят анализируемую пробу (или эта-
лон) в виде раствора, порошка или твердого образца. Происходит
атомизация вещества и возбуждение газообразных атомов.
2. Излучаемый возбужденными атомами свет проходит через
щель спектрального прибора и диспергирующий элемент (приз-
му, дифракционную решетку), где происходит пространственное
разделение света на отдельные спектральные компоненты, обра-
зующие соответствующие спектральные линии, которые попада-
ют на фотопластинку, фотоэлемент или фиксируются визуально.
3. Интенсивность спектральных линий оценивается по по-
чернению фотопластинки, силе тока или визуально.
4. По спектрам эталонов определяют градуировочную характе-
ристику и рассчитывают содержание определяемого компонента.
В зависимости от способа оценки интенсивностей различа-
ют следующие методы количественного спектрального анализа:
1) визуальные; 2) фотографические; 3) фотоэлектрические.
При классификации методов спектрального анализа в от-
дельную группу выделяют так называемый полу количественный
спектральный анализ.
38
2.6.1. Полуколичественный спектральный анализ
Обычная погрешность полуколичественных спектральных ме-
тодов составляет 10% или более. Однако когда простота и экспрес-
сность, важнее точности, эти методы применяют очень широко.
Оценку) интенсивности спектральных линий в полуколичествен-
ном анализе производят визуально, наблюдая спектр или непо-,
средственно в окуляре спектрального прибора, или на фотоплас-
тинке. , _ \ \
Наиболее распространенным методом полуколичественного,
анализа является метод гомологических пар, или, как его иног-
да называют, метод однородных дублетов. Для проведения ана-
лиза этим методом предварительно подбирают пару линий (гомо-
логическую пару или однородный дублет) и устанавливают, при
какой концентрации определяемого элемента их интенсивности
одинаковы. Например, при разработке методики спектрального
определения свинца в олове интенсивность линии олова (нм) А^п =
= 276,11 сравнивалась с интенсивностью линий свинца Хръ =
= 280,20 (1), А,РЬ ^282,32 (2) и А,РЬ = 287,32 (3). Оказалось, что ер-
ли (орь = 0,1%, то^8п = /Хрь(1), если ШрЬ = 0,6%, то/^п = /^(3), а
если сэРЬ = 1,3%, то 1Х = 1Х (2). Для оценки содержания свинца
в сплаве неизвестного состава на той же основе сравнивают изу-
ченные ранее линии олова и свинца и находят линии одинаковой
интенсивности. Таблицы, связывающие относительную интен-
сивность двух линий с концентрацией элемента в пробе, мог^т
быть составлены и при неодинаковой интенсивности линий дуб-
лета. Такие соотношения найдены, например, для определения
марганца в стали цо линиям ЯМп = 478,43 и ХРе = 477,94 нм. Если
интенсивность линии марганца существенно меньше интенсив-
ности линии железа (7^ ), то содержание марганца в стали,
близко к 0,02%, при небольшом различии в интенсивностях (/^Мп <
< 1^ ) содержание? Марганца составляет 0,04%, равенство йнтен-*
сивностей = 1^ , наблюдается при содержаний маргайца око-
ло 0,06%, а при .содержании 0,08% Мп,интенсивность линии
марганца превышает интенсивность линии железа.
Этот же прием; используют в работе со стилоскопом при непо-
средственном наблюдений; спектра в окуляре прибора и определе-
нии других компонентов стали (N1, Сг, У? и т. д.). Аналитические
таблицы, связывающие относительные интенсивности спект-
ральных линий и концентрацию элемента, составляются заранее
и обычно прилагаются к стилоскопу. Анализ с помощью стилог
39
скопа на 6—7 элементов у опытного спектроскописта занимает
2—3 мин, предел обнаружения составляет обычно 0,01—0,10%,
погрешность около ±20% (относительных). Успешно применяет-
ся стилоскоп, например, при сортировке сталей, контроле плав-
ки и выполнении других аналогичных анализов.
Более высокая точность результатов достигается с помощью
стилометра. Стилометр снабжается устройством, позволяющим
плавно изменять интенсивности сравниваемых линий, добиваясь
их уравнивания. Таким же устройством снабжаются последние
модели стилоскопов.
Из фотографических методов полуколичественного анализа
применяют метод спектров сравнения и метод появления, или ис-
чезновения чувствительных линий. В методе спектров сравнения
на одной пластинке фотографируют спектры нескольких эталонов
и спектр пробы, а затем сравнивают на глаз почернение линий оп-
ределяемого элемента в полученных спектрах. Такое сравнение по-
зволяет установить пределы концентраций анализируемого элемен-
та. В методе появления или исчезновения линий сначала с помощью
эталонов устанавливают, при каких концентрациях появляются
или исчезают те или иные чувствительные линии элементов, а за-
тем, используя полученные данные, находят ориентировочное со-
держание элемента в пробе.
Полуколичественный экспрессный метод фотометрического
интерполирования основывается на визуальной оценке интенсив-
ности спектра, снятого через девятиступенчатый ослабители.
Ступенчатый ослабитель представляет собой кварцевую или
стеклянную пластинку, на которой нанесены узкие параллель-
ные полоски пластины (ступеньки) возрастающей толщины и,
следовательно, убывающей пропускаемости. Если ослабитель по-
местить в спектральный прибор, на фотопластинке получится не-
сколько параллельных изображений спектра убывающей интен-
сивности в соответствии с убывающей пропускаемостью ступеней
ослабителя.
Каждая ступень Ь характеризуется своей пропускаемостью = 1^1^
где /0 — интенсивность падающего света, а 11 —- интенсивность света,
прошедшего через 1-ю .ступень. Пропускаемость указывается в паспор-
те ослабителя. Если при анализе такого спектра будет найдено, что ин-
тенсивность линии примеси на ступеньке г(/пр(г)) будет равна интенсив-
ности линии основы на ступеньке р(1осп(р))9 то очевидно, что
1щ><*>г-1ос*а>Р> (2.23)
где <хг и ар — пропускание ступенек гир ослабителя.
40
Логарифмируя уравнение (2.23):
Іосв ar
и подставляя посіле логарифмирования уравнение (2.21), получаем
lg%=lga + blgcnp. (2.24)
По 1б ^ и известной концентрации спр в эталонах строят градуиро-
вочный график, который не зависит от свойств фотопластинки. Опреде-
ление <?пр в неизвестных образцах по этому графику проводится обычным
путем.
Метод фотометрического интерполирования характеризуется отно-
сительно высокой экспрессностью. Он позволяет получить результат че-
рез несколько минут после окончания химической обработки фотоплас-
тинки.
2.6.2. Фотографические методы количественного анализа
При рассмотрении свойств фотопластинки было установлено,
что почернение £ связано с освещенностью £ уравнением (2.20).
В условиях спектрального анализа Е = /. Подставляем это в урав-
нение (2.20): , -
5-у ^/^.у1* (2.25)
При постоянной выдержке последние члены правой части этого
уравнения постоянны, поэтому
£ = уІ£/ + соп8і. (2.26)
Запишем уравнение (2.26) для почернения линии примеси и
линии основы:
Snp = ylgInp + const;
и вычтем одно из другого:
£Пр -Soch - AS = у lg . (2.27)
*och
При логарифмировании уравнения (2.21) имеем
1е1вр/1жш = 1ша + Ыесвр. (2.28)
41
Объединяя уравнений (2.27) и (2.28), получаем
Д5 = у.І£а + уЬІ£ спр. .
(2.29)
Это основное уравнение фотографических методов количест-
венного спектрального анализа.
? Анализ уравнения (2.29) позволяет оценить ожидаемую точ-
ность фотографических методов эмиссионной спектроскопии.
Для этого найдем сПр из уравнения (2.29):
и продифференцируем (2.30), считая а, Ъ и у постоянными:
ал0с - dcnp _ d(AS)y& .-d(AS)
ё °Р 2,3спр (уЬ)2 у&
(2.30)
или
(2.31)
Формула (2.31) показывает, что относительная ошибка в
концентрации не зависит от абсолютного значения Дв и умень-
шается при увеличении у и Ь. Для ориентировочных подсчетов
в области оптимальных почернений примем Ь = 0,7, с1(Д5) = 0,02,
у 1,3 и подставим эти значения в уравнение (2.31):
dcnp _ Асдр _ 2,3
1,3 • 0,7
0,002 -100-^ = 5,0%,
т. е. относительная погрешность при этих условиях составляет
~5%. Дальнейший расчет показывает, что при увеличении Ъ до
1,0 погрешность уменьшается до 3,5%. Погрешность также сни-
жается при уменьшении (1(Д5), т. е. при увеличении точности из-
мерения почернений фотопластинки.
Наиболее распространённым из фо-
тографических методов количественно-
го спектрального анализа является ме-
тод трех эталонов. Сущность его за-
ключается в следующем. На одной
пластинке фотографируются спектры
анализируемого образца и трех этало-
нов. Для анализа массовых проб (стали,
сплавы ит. д.) применяют специальные
наборы эталонов, кбторыё выпускает
лаборатория стандартаых образцов. По
спектрам эталонов строят трассиро-
вочный график (рис. 2.13). Для повы-
AS
AS
lgcx
lg с
Рис. 2.13. Градуировочный
график в методе трёх
эталонов
42
шения точности спектры эталонов и образца фотографируют 2—
3 раза и берут средние значения AS. В соответствии с уравнением
(2.29) градуировочный график следует ожидать линейным, и для
его построения, казалось бы, будет достаточно двух эталонов.
Опыт показывает, однако, что нередко зависимость AS от lg с
оказывается нелинейной, и третий эталон позволяет получить бо-
лее надежный график. В некоторых случаях применяют четыре и
более эталонов. Существенным недостатком этого наиболее точ-
ного метода спектрального анализа является его длительность и
необходимость получать для каждой пластинки свой градуиро-
вочный график.
В методе постоянного графика удается согласовать измере-
ния, сделанные на различных пластинках, и достроить постоян-
ный график, не зависящий от свойств пластинки. Согласование
достигается при учете коэффициента контрастности. Придадим
уравнению (2.29) вид
^ =lga + Mgcnp. (2.32)
Уравнение (2.32) показывает, что график в координатах AS/y -
- lgcnp от свойств пластинки уже не зависит. Коэффициент
контрастности у можно определить по характеристической кри-
вой фотопластинки или, проще, если на каждой пластинке сфо-
тографировать один или несколько спектров через трехступенча-
тый ослабитель. Если Sx и S2— почернения двух ступенек, т. е.
очевидно, что
S^ylg (аг10);
S2 = y\g (а2/0).
При вычитании получаем
S1-S2 = ASlt2 = ylg (2.33)
а2
откуда
lgax/ttg '
Необходимая для расчета у величина ASX> 2 определяется экс-
периментально, á lg a1/a2 известен по паспорту ступенчатого ос-
лабителя. По этим данным (у и AS) в соответствии с уравнением
(2.32) строят постоянный график, пригодный для любых пласти-
нок, для которых известен у.
■ 43
Этот метод можно упростить и отказаться от использования
ступенчатого ослабителя. Запишем уравнение (2.33) для двух
пластинок:
а2
А«"1,2 = У"1е? '
а2
и возьмем их отношение:
Д^Ь у" Л
Коэффициент А называют переводным множителем. Он по-
зволяет пересчитать разность почернений одной пластинки на
разность почернений другой. Для определения к можно исполь-
зовать следующий прием.
На двух пластинках фотографируют спектр одного и того же
образца в одинаковых условиях й находят разность почернений
одних и тех же аналитических линий. Если ДЗ^ — разность по-
чернений на одной пластинке, Д52 —-разность почернений на
другой, то
и, очевидно,
Отсюда
Д52 = у2(&^с + ^ а)
Д^-ЛД^. '•■ (2.35)
Для определения к необязательно измерять почернения ана-
литических линий. Для этого можно использовать две линий ос-
новного вещества в той же области спектра, в которой находится
аналитическая пара, определить разность их почернений на всех
пластинках и по формуле (2.34) вычислить переводный множи-
тель по отношению к пластинке, выбранной за стандарт. Пользу-
ясь этим коэффициентом, можно по формуле (2.35) пересчитать
почернения аналитических линий на разных пластинках на по-
чернения стандартной пластинки и получить согласованные ве-
личины Дв, пригодные для построения градуировочного графика
и определения концентрации. Этот метод иногда называют мето-
дом переводного коэффициента.
Опыт показал, что неконтролируемые изменения условий
возбуждения вызывают сдвиг градуировочного графика/Так как
44
сдвинутый график, как правило, оказывается параллельным
первоначальному, то для учета сдвига одновременно со спектром
образца фотографируют спектр контрольного эталона. Через точ-
ку, соответствующую этому эталону, проводят прямую, парал-
лельную первоначальному графику, которая и будет истинным
градуировочным графиком для образцов на данной пластинке.
Это твердый график и метод называют методом твердого графи-
ка или контрольного эталона.
В современных методах спектрального анализа^используют
преобразованные значения почернений, полученные с помощью
р- или /-преобразования. Этот прием расширяет область почерне-
ний, пригодную для анализа, которая обычно ограничена линей-
ным участком характеристической кривой фотопластинки.
В практике спектрального анализа применяют также метод
добавок, особенно при анализе немассовых проб сложного состава
и исследованных чистых и сверхчистых веществ. При анализе по
этому методу пробу обычно переводят в раствор и Делят его на не-
сколько частей. Затем в каждую часть добавляют разное, но точ- ;
но известное количество определяемого элемента, снимают
спектр и определяют интенсивность спектральных линий. Для
расчета результатов анализа используют несколько приемов.
В простейшем случае строят график, основываясь на том!, что
в области малых концентраций достаточно хорошо выполняется
линейная зависимость /пр//осн = ас, так как с уменьшением кон-
центрации самопоглощение существенно падает. При этом, если
с — неизвестная концентрация, а сст — концентрация введенного
стандарта, то
Inp/ICT = a(cx + cJ. (2.36)
Откладывая на графике /пр//осн как функцию сст, получают пря-
мую, пересекающую ось ординат при сст = 0 (рис. 2.14). Экстрапо-
ляция этой прямой до пересечения с осью абсцисс дает отрезок на
оси, равный —сх. Действительно, при
^пр/^осн = О ИЗ уравНёНИЯ (2.36) Следует, ^пр/^осн
что -сст = с,.
В более сложных случаях (& ^ 1) ис-
пользуют метод последовательных при-
ближений на основе уравнения Лома-
кина—Шайбе или другой наиболее оп-
тимальный способ обработки.
Метод добавок является надежным : Рис. 2.14. Градуировочный
контрольным методом для установле- график в методе добавок
45
ния правильности анализа. Действительно, если после анализа
добавить в пробу известное количество определяемого элемента и
анализ повторить, то его результат с учетом возможной погреш-
ности должен дать сумму первоначально найденного содержания
и введенной добавки. Если сумма не получилась, результат ана-
лиза следует признать неверным. Метод добавок применяют для
повышения чувствительности спектральных определений тогда,
когда почернение аналитической линии является слишком сла-
бым и не может быть измерено. Введение добавки приводит к
увеличению интенсивности спектральной линии, позволяя про-
водить измерение с более высокой точностью.
2.6.3. Фотоэлектрические методы
Наиболее существенным недостатком фотографических мето-
дов спектрального анализа является большая длительность опре-?
делений. Для получения результата необходимо сфотографировать
спектр, обработать фотопластинку и провести фотометрирование.
Значительно более быстрыми являются спектрометрические мето-
ды, основанные на прямом фотометрическом определении интен-
сивности спектральных линий. Спектрометрические методы ха-
рактеризуются более высокой экспрессностыо и меньшей погреш-
ностью, чем фотографические, поскольку в спектрометрических
Методах исключаются длительные операции обработки фотоплас-
тинки и последующего фотометрирования линий и связанные с
этим погрешности.
Установки спектрометрического и спектрографического ана-
лиза аналогичны, за исключением устройства их рецепторной'
части. В фотоэлектрических установках свет после диспергирую-
щего элемента через специальные щели в фокальной плоскости
попадает на фотоэлемент или фотоумножитель, соединенный с на-
копительным конденсатором и далее с регистрирующим потенцио-
метром. Одна из щелей в приборах с фиксированными приемни-
ками света предназначена для линии сравнения, а остальные ^~
для линий анализируемого элемента или элементов. В приборах
этого типа для каждой линии предусмотрен свой фотоэлектриче-
ский приемник. В сканирующих спектрометрах измерение ин-
тенсивности линии определяемого элемента производится фото-
электрическим приемником, который передвигается вдоль
спектра по специальной программе. Фотоэлектрический измери-
тельный блок может также использоваться в качестве приставки
к спектроскопу или спектрографу. Такой блок, состоящий из
входной щели и фотоэлемента или фотоумнржителя с измери-
46
тельным устройством, устанавливается на место кассеты для фо-
топластинки; Сконструированный таким образом простой спект-
рометр может быт^ эффективно применен для анализа проб с Не-
сложным спектром.
Отечественной промышленностью выпускается несколько
установок такого типа: многоканальная установка ДФС-10М,
квантометры МФС-3, МФС-10 и др. Их аналитические возмож-
ности весьма различны. Например, квантометр ДФС-10М имеет
12 щелей и соответственно 12 накопительных конденсаторов, ра-
ботающих одновременно. С помощью этбго прибора мбжйр одно-
временно определить в пробе 11 элементов (12-й канал служит
для определения интенсивности линии сравнения, основы или
внутреннего стандарта).
2.6.4. Химико-спектральный анализ г
Существенно, расширяет возможнрсти эмиссионной спектро-
скопии применение химических методов обработки пробы. Хи-
мическая обработка и. концентрирование позволяют повысить
чувствительность определения на два порядка и более и во мно-
гих случаях упростить спектральную методику, включая эталр-
нировани?, так как состав получаемых концентратов в опреде-
ленных предела^ нетрудно регулировать. Известные методики
химико-спектрального анализа позволяют определять примеси в
веществах высокой чистоты при содержании 10"5—10"7%.
Обычным приемом обогащения является извлечение микро-
примесей из анализируемого материала, хотя иногда применяют
химический метод отделения основного компонента пробы путем
его осаждения. Например, при анализе высокочистрго свинца
осаждают сульфат свинца, при анализе металлического висмута
осаждают иодид висмута и т. д. Примеси при этрм остаются в рас-
творе и после упаривания или другой обработки могут быть опре-
делены спектральным методом. v, ; , « •
Более широко применяется метод извлечения микропримесей.
Здесь используется осаждение органическими реактивами, досаж-
дение с коллектором, экстрагирование, хроматография, электро-
лиз и другие методы. Предварительное обогащение применяется
при анализе различных биологических объектов, почвы, воды, ве-
ществ ьысокбй степени чистоты и т. д. Нельзя не отметить, одна-
ко, что обработка проб с целью обогащения: предъявляет повышен-
ные требования к чистоте используемых реактивов, воды, раство-
рителей и посуды.' л»;
47
2.7. Фотометрия пламени
Появление специализированных пламенных эмиссионных
спектрометров привело в какой-то степени к обособлению метода
фотометрии пламени (пламенная эмиссионная спектроскопия) и
приданию ему известной самостоятельности, хотя, конечно, фо-
тометрия пламени осталась одним из методов эмиссионного
спектрального анализа.
Как и любой другой прибор эмиссионной спектроскопии, фо-
тометр для фотометрии пламени имеет источник возбуждения
(пламенная горелка), диспергирующий элемент (обычно свето-
фильтр) и приемник света — рецептор (обычно фотоэлемент).
В спектрофотометрах для пламени вместо светофильтров приме-
няют призмы и дифракционные решетки. Анализируемый рас-
твор в пламя горелки вводится в виде аэрозоля. При этом раство-
ритель испаряется, а соли металлов диссоциируют на атомы,
которые при определенной температуре возбуждаются. Возбуж-
денные атомы, переходя в нормальное состояние, излучают свет
характерной частоты, который выделяется с помощью диспер-
гирующих элементов, и его интенсивность измеряется фотоэле-
ментом.
Количественные определения проводят методом градуировоч-
ного графика или методом добавок. Методы фотометрии пламени
характеризуются низким пределом обнаружения (¿0 0,001 мкг/мл
для щелочных металлов и 0,1 мкг/мл для других) при погреш-
ности 1—3%. Этим методом могут быть определены 1л, Ыа, К,
Иэ, Се, Бг, Ва, Са, 1п, Ag и другие элементы. Одним из достоинств
метода фотометрии пламени является также высокая производи-
тельность.
Спектры, получаемые в пламени, более просты, чем дуговые
или искровые, так как температура пламени ниже, чем в элект- '
ричёских источниках возбуждения. Это облегчает анализ, но
вместе с тем сужает возможности метода в отношении числа оп-
ределяемых элементов.
2.8. Практическое применение
Методы эмиссионного спектрального анализа используются
во многих областях науки и техники ив различных отраслях
промышленности. Этим методом выполняется значительная
часть анализов в металлургической промышленности. Анализи-
руется исходное сырье и готовая продукция. Осббое значение
48
имеет спектрально-аналитический контроль за ходом плавки, на
основании которого вносятся оперативные изменения в ход тех-
нологического процесса, например по содержанию легирующих
и других добавок. Визуальный спектральный анализ оказался
очень удобным методом сортировки вторичного сырья металлур-
гического производства, позволяя за несколько минут устано-
вить тип сплава или марку стали, что необходимо при составле-
нии или корректировке шихты.
Очень эффективным оказалось применение спектральных
методов при анализе разного рода геологических проб при поиске
полезных ископаемых, а также для контроля технологического
процесса на горно-обогатительных и гидрометаллургических
предприятиях. Спектральным анализом контролируется качест-
во поступающей руды, степень извлечения полезных и мешаю-
щих компонентов, а нередко и качество продукта.
Существенную роль играет спектральный анализ природных
и сточных вод, почвы, атмосферы и других объектов окружаю-
щей среды, а также в медицине и биологии. Важное значение
имеет спектральный анализ чистых материалов в электронной
технике и других областях, анализ реактивов и т. д. Успешно ис-
пользуется спектральный анализ в космических исследованиях.
Процесс совершенствования методов эмиссионной спектроско-
пии продолжается.
2.9. Общая характеристика метода
При общей оценке методов эмиссионной спектроскопии необ-
ходимо прежде всего отметить их низкий предел обнаружения,
точность, быстроту выполнения анализов и универсальность.
Средний предел обнаружения методами эмиссионной спектроско-
пии составляет 10~3—10~4% (до 10"5%), а при использований
приемов обогащения он снижается до 10~5—10"7%./ Погрешность
определения характеризуется в среднем величиной 1—2%. В свя-
зи с экспрессностью, точностью и другими достоинствами эмис-
сионный спектральный анализ широко используют в практике.
Значительная часть определений в металлургической и машино-
строительной промышленности выполняется с помощью спект-
рального анализа; Многочисленные применения нашел спект-
ральный анализ и в других отраслях промышленности и техники
(геологии, химической промышленности, сельском хозяйстве,
космохимииит. д.).
4-7831
49
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Почему атомные спектры имеют линейчатый характер?
2. Какими квантовыми числами описывается сортряние электрона
в атоме? Что характеризуют квантовые числа?
3. Какие электронные переходы называют разрешенными, запрет
щенными, резонансными?
4. Каковы достоинства и недостатки средств возбуждения: а) пла-
мени; б) электрической дуги; в) конденсированной искры; г) ин-
дуктивно-связанной плазмы?
5. Какие приемники спектра (рецепторы) используют в эмиссион-
ной спектроскопии?
6. Каковы достоинства и недостатки фотопластинки как рецептора?
7. Что называют «последними» линиями? Для чего они служат?
8. Как выполняется качественный спектральный анализ?
9. На чем основаны методы количественного спектрального ана-
лиза? ■•,>;..,
10. Как зависит интенсивность спектральных линий от условий воз-
буждения?
11. Какие требования предъявляются к гомологической паре ли-
12. Какие величины входят в уравнение Ломакина^-Шайбе?
13. В чем сущность метода трех эталонов, одного эталона?
14. Что представляет собой фотометрия пламени? Каковы достоин-
ства и недостатки этого метода?
глава 3
Абсорбционная спектроскопия
3.1. Основной закон светопоглощения
Атом, ион или молекула, поглощая квант света, переходит в
более высокое, энергетическое состояние. Обычно это бывает пе-
реход с основного, невозбужденного уровня на один из более вы-
соких, чаще всего на первый возбужденный уровень. Вследствие
поглощения излучения при прохождении его через слой вещест-
ва интенсивность излучения уменьшается и тем больше, чём вы-
ше концентрация светопоглощающего вещества. >
Закон Бугера—Ламберта—Бера (основной закон светопогло-
щения) связывает уменьшение интенсивности света, прошедшего
50 . ". ■
Раствор^>
^> Н20 >
Рис. 3.1. Прохождение света
через окрашенный раствор
и растворитель
через слои светопоглощающего ве-
щества, с концентрацией вещества и
толщиной слоя. Чтобы учесть поте-
ри света на отражение и рассеяние,
сравнивают интенсивности света,
прюшедшего через исследуемый рас-
твор и растворитель (рис. 3.1). При
одинаковой толщине слоя в кюветах
из одинакового материала, содержа-
щих один и тот же растворитель, по-
тери на отражение и рассеяние света
будут примерно одинаковы у обоих
пучков и уменьшение интенсивности света будет зависетргбт кон-
центрации вещества.
Уменьшение интенсивности света, прошедшего через рас-
твор, характеризуется коэффициентом пропускания (или просто
пропусканием) Т:
где I и /0 — соответственно интенсивности света, прошедшего че-
рез раствор и растворитель.
Взятый с обратным знаком логарифм Г называется оптиче-
ской плотностью А:
Уменьшение интенсивности света при прохождении его че-
рез раствор подчиняется закону Бугера—Ламберта—Вера:
или
или
/=70 • 10-£*,
///0 =10-^,
(3.1)
где 8 — молярный коэффициент поглощения; I — толщина свето-
поглощающего слоя; с — концентрация раствора.
Физический смысл е становится ясным, если принять I = 1 см
и с = 1 моль/л, тогда Д = 8. Следовательно, молярный коэффици-
ент поглощенияравен оптической плотности одномолярного рас-
твора при толщине слоя 1 см.
Оптическая плотность раствора, содержащего несколько ок-
рашенных веществ, обладает свойством аддитивности, которое
иногда называют законом аддитивности светопоглощения. В со-
ответствии с этим законом поглощение света каким-либо вещест-
51
вом не зависит от присутствия в растворе других веществ. При на-
личии в растворе нескольких окрашенных веществ каждое из них
будет давать свой аддитивный вклад в экспериментально опреде-
ляемую оптическую плотность А:
А=А1+А2 + ...+Ак9
где А19 А2 и т. д. — оптическая плотность вещества 1, вещества 2
ит. д.
При учете уравнения (3.1) получаем
А1(&1с1 + е2с2 + ... + еЛсЛ).
3.1.1. Ограничения и условия применимости
закона Бугера—Ламберта—Бера
В соответствии с уравнением (3.1) зависимость оптической
плотности от концентрации графически выражается прямой ли-
нией, выходящей из начала координат. Опыт показывает, одна-
ко, что линейная зависимость наблюдается не всегда. При прак-
тическом применении закона Бугера—Ламберта—-Вера необхо-
димо учитывать следующие ограничения:
1. Закон справедлив для монохроматического света. Чтобы
отметить это ограничение, в уравнение (3.1) вводят индексы и за-
писывают его в виде
Ах = ех1с. ' (3.2)
Индекс А, указывает, что величины А и 8 относятся к моно-
хроматическому излучению с длиной волны X.
2. Коэффициент 8 в уравнении (3.1) зависит от показателя
преломления среды.
Более точное уравнение закона Бугера—Ламберта—Бера
имеет вид:
А = 8 * Ас,
(п2 + 2)2 . , :
щеп — показатель преломления.
Если концентрация раствора сравнительно невелика, его по-
казатель преломления остается таким же, каким он был у чисто-
го растворителя, и отклонений от закона по этой причине не на-
блюдается.
Изменение показателя преломления в высококонцентриро-
ванных растворах может явиться причиной отклонений от основ-
ного закона светопоглощения. ,
52
3. Температура при измерениях Должна оставаться постоян-
ной хотя бы в пределах нескольких градусов.
4. Пучок света должен быть параллельным.
5. Уравнение (3.1) соблюдается только для систем, в которых
светопоглощающими центрами являются частицы лишь одного
сорта. Если при изменении концентрации будет изменяться при-
рода этих частиц вследствие, например, кислотно-основного
взаимодействия, полимеризации, диссоциации и т. д., то зависи-
мость А от с не будет оставаться линейной, так как молярный ко-
эффициент поглощения вновь образующихся и исходных частиц
не будет в общем случае одинаковым.
Например, при разбавлении раствора дихромата калия происходит
не просто уменьшение концентрации иона дихромата, а протекают про-
цессы химического взаимодействия:
Сг26|" + Н20= 2НСЮ; - 2СгО£~ + 2Н+
Вместо дихромат-ионов в растворе появляются гидрохромат- и хро-
мат-ионы. Так как 8Сг202- и еСг0|- различны, зависимость оптической
плотности от общей концентрации хрома в растворе не будет линейной.
6. Интенсивность рассеянного света, возникающего в оптиче-
ской; системе прибора, должна быть сведена до минимума за счет
ограничений при, изменении ширины щели в разных участках
спектра^
3.2. Спектры поглощения
Свет поглощается раствором избирательно: при некоторых
длинах волн светопоглощение происходит интенсивно, а при не-
которых свет не поглощается. Интенсивно поглощаются кванты
света, энергия которых Ау равна энергии возбуждения частицы и
вероятность их поглощения больше нуля. Молярный коэффици-
ент поглощения при этих частотах (или длинах волц) достигает
больших значений.
Распределение. по частотам (или по длинам волн) значений мо-
лярного коэффициента поглощения называется спектром погло-
щения.
Обычно спектр поглощения выражают в виде графической
зависимости оптической плотности А или молярного коэффици-
ента поглощения 6 от частоты у или длины волны X падающего
света. Вместо А или & нередко откладывают их логарифмы.
53
Кривые в координатах Лё А - X, как показывает рис. .3.2, при
изменении концентрации или толщины слоя перемещаются по ор-
динате вверх или вниз параллельно самим себе, в то время как
кривые в координатах А - X (рис 3.3) этим свойством не обладают.
Эту особенность легко понять, если сравнить производные
дАк д\пАх
—- и .
ех зх
Действительно, при дифференцировании уравнения (3.2) по-
лучаем
дх. ех 9
т. е. спектральная зависимость оптической плотности пропорци-
ональна 1и с.
При логарифмировании уравнения (3.2) и последующем диф-
ференцирований имеем:
д\пАх _ д\пех
дх
дХ
т. е. спектральная зависимость логарифма оптической плотности
оцределяется только величиной 1п є и не зависит от / и с.
Существенное значение имеет, эта особенность для качествен-
ного анализа.
Кроме того, дифференциальная зависимость дает воз-
дХ-
можность более детально проанализировать спектр и обнаружить
максимумы, скрытые при обычном представлении спектра, как
зависимости Ах = /(Я,).
Рис. 3.2. Зависимость 1&Аот'Х:
1 — раствор концентрации с
в кювете толщиной / (см);
2 — раствор концентрации }/лс
или в кювете толщиной }/л1 (см)
Рис. 3.3. Зависимость А от X:
1—раствор концентрации с
в кювете толщиной I (см);
2— раствор концентрации 1/4с
или в кювете толщиной 1/л1 (см)
54
' * При изучении инфракрасных спектров на графике обычно
откладывают процент светопропускания как функцию V' или V.
Отдельные полосы в спектрах поглощения математически можно
описать гауссовыми кривыми:
_(У'-Утах)21п2
е = бтахв *2 , (3.3)
где 8 и Етах — молярный коэффициент поглощения при данной длине
волны X (или волновом числе у') и при длине волны А^дх (или волновом
числе ч'тйХ), отвечающей точке максимума; V' и v,meiX — данное волновое
число и волновое число в точке максимума; 8 характеризует ширину по-
лосы поглощения.
Действительно, при V' = у{/2 молярный коэффициент поглощения
равен половине максимального 81/2 = 1/2етах> и вместо уравнения (3.3)
имеем " ;; :
При упрощении и логарифмировании уравнения (3.4) получаем
откуда
Как видно, величина 5 равна полуширине полосы на половине ее
высоты.
Для несимметричных полос обычно указывают ^ах(А;тах)и е^. Дл!я
решения структурных, термодинамических или иных вопросов несим-
метричные полосы нередко разлагают на индивидуальное гауссовы ком-
поненты, каждая из которых описывается уравнением (3.3).
Таким образом, наибольший интерес представляют следую-
щие характеристики спектра: число максимумов (или полос по-
глощения) и их положение по шкале длин волн (или частот); вы-
сота максимума; форма полос поглощения.
7 ' "А. ' " Ч ' V." :
3.2.1. Происхождение спектров поглощения
Появление полос поглощения обусловлено дискретностью
энергетических состояний поглощающих частиц; и квантовой
природой электромагнитного излучения. Интенсивно поглоща-
ются кванты света, которые соответствуют энергии возбуждения
частицы. При поглощении квантов света происходит увеличение
внутренней энергии частицы, которая складывается из эйёртии
55
вращения частицы как целого, энергии колебания атомов и дви-
жения электронов:
Е = Евр + Екол + Еэд, (3.5)
где Евр — вращательная; Екол— колебательная; Еэл — электрон-
ная энергия.
Уравнение (3.5) должно включать также слагаемые энергии
тонкой и сверхтонкой структуры, связанные с электронным и
ядерным спином, поправку на приближенность аддитивной схе-
мы и некоторые другие слагаемые, которыми в первом прибли-
жении можно пренебречь.
По энергии вращательное, колебательное и электронное движе-
ние различаются весьма существенно, причем £вр ^ Екол <С Еэл. Их
числовые значения относятся примерно как 1:102 :103.
Каждый вид внутренней энергии молекулы, как уже отмеча-
лось, имеет квантовый характер и может быть охарактеризован
определенным набором энергетических уровней или термов и со-
ответствующих квантовых чисел.
3.2.2. Вращательные спектры
Вращательную энергию молекул обычно рассматривают с по-
мощью модели жесткого ротатора, который представляет со-
бой две массы, находящиеся одна от другой на фиксированном
расстоянии.
Возбуждение вращательных уровней энергии происходит
уже при поглощении далекого инфракрасного (ИК) и микровол-
нового излучения, имеющего длину волны Л. > 102 мкм или вол-
новое число V' <102 см"1. Энергия квантов в этой области спектра
равна:;Евр = 2,8 • 10~3 у' = 1,2 кДж/моль и меньше. Это значение
соизмеримо с энергией теплового движения кГ> поэтому уже при
комнатной температуре часть вращательных уровней заселена.
В настоящее время чисто вращательные спектры в аналити-
ческих целях почти не используют. Их применяют главным об-
разом для исследования строения молекул, определения межъ-
ядерных расстояний и т. д.
3.2.3. Колебательные спектры
Цолосы, связанные с возбуждением колебательных уровней
энергии, расположены в области спектра примерно от 200—300
до 4000—5000 см"1, что соответствует энергии квантов от 3 до
56
бОкДж/моль. Поэтому при обыч-
ных температурах энергетическое
состояние молекул, как правило,
характеризуется основным колеба-
тельным уровнем. Простейшей мо-
делью, которая используется при
рассмотрении колебаний двухатом-
ной молекулы, является модель гар-
монического осциллятора. Это сис-
тема из двух масс, связанных упру-
гой силой. Кривая потенциальной
энергии гармонического осциллято-
ра обычно аппроксимируется пара-
болой (рис. 3.4, кривая 1). Приме-
нение квантовой теории к такой
системе показывает, что ее энергия
может быть найдена по уравнению
ЯколИ^+ 1/2)Н> (3-6)
где V — колебательное квантовое число, принимающее значения
О, 1,2,3,... • .л- ■
Уравнение (3.6) показывает, что в основном состоянии при
У= 0 осциллятор обладает энергией так называемых нулевых ко-
лебаний. Это существенный результат квантового подхода.
Правилами отбора в таких системах разрешены переходы,
для которых
Д7= ±1. (3.7)
Из сравнения уравнений (3.6) и (3.7) видно, что расстояние
между двумя соседними энергетическими уровнями гармониче-
ского осциллятора постоянно и равно Ау0. Это означает, что при
всех разрешенных переходах будут поглощаться или излучаться
кванты энергии только частоты у0 и в спектре, таким образом, бу-
дет наблюдаться только одна полоса. Частота, характеризующая
эту полосу, может быть вычислена по уравнению
где М — приведенная масса, определяемая как = — + —
< М т2
(тг ит2 — массы ядер); К — силовая постоянная.
Силовую постоянную иногда связывают с прочностью связи
в молекуле.
Е
Рис. 3.4. Кривые
потенциальной энергии
и уровни энергии двухатомной
молекулы
57
Реальные колебания молекул ангармоничныи кривые потенци-
альной энергии молекул имеют более сложный характер (рис. 3.4,
кривая 2). Энергию ангармонического осциллятора рассчитывают
по формуле
^кол = (^+1/2)^о-^(^+1/2)2» (3.8)
где!) — энергия диссоциации молекулы.
Энергетические уровни ангармонического осциллятора, как
это следует из уравнения (3.8), с увеличением квинтового числа
сближаются. Для ангармонического осциллятора разрешенны-
ми являются переходы между любыми уровнями.
Наиболее интенсивной в спектре является первая полоса, воз-
никающая при переходе сУ=0на^=1. Этой полосе соответствует
основная, или фундаментальная, частота. Менее интенсивные по-
лосы дают обертоны, т. е. частоты, характеризующие переход с
уровня V = 0 на уровень V = 2 (первый обертон или вторая гармони-
ка); на уровень V— 3 (второй обертон или третья гармоника) и т. д.
Следует ответить лишь, что колебательными инфракрасны-
ми спектрами обладают нё всё молекулы, а только те, у которых
при колебании происходит изменение ихэлектрйческогб диполь-
ного; момента. ИК-спектрами обладают, например, молекулы
НС1, НВги т. д., но не Н2, 02 й др. ^ ; 1
г Сравнительная простота* колебательных или колёбатель-
но-'вращательных спектров двухатомных молекул обусловлена
тем, что колебания происходят только вдоль линии, соединяющей
ядра. В многоатомной молекуле происходят колебания всех ато-
мов. Число колебательных степеней свободы у нелинейной моле-
кулы, состоящей из N атомов, равно ЗЛГ - 6, а у линейной ЗЛГ - 5.
''"'[ГСл6^фо картину колебаний в многоатомной молекуле обыч-
но ^представляют как суперпозицию так называемых нормальных
кШёбанмй; Частбты нормальны* колебаний характеризуются по-
лйжёниёй полос в ЙК-сйёктр'ах, а амплитуда колебаний опрёдёлй-
'интенсивность>*их' ЙолоЙ Число нормальных
видно, будет равно числу колебательных степеней свободы; та-
ким образок, общая форма молекулярных * колебаний ьбудет
суперпозицией ЗЫ - 6 (или ЗЩ)- 5) нормальных колебаний.
При классификаций нормальных колебаний обычно разли-
чают валентные й неваленпЬные, или деформационные, колеба-
ния. Колебание называют валентным, если происходит измене-
ние длины связи без существенного изменения углов между свя-
зями. Валентные колебания обозначают буквой V. Колебания с
изменением углов между связями называют невалентными или
деформационными и обозначают буквой 8. На рис <3.5 можно ви-
58
/
Y
а) б) в)
Рис. 3.5. Формы нормальных колебаний молекулы Н20
деть формы нормальных колебаний молекулы воды. Так как Н20
является нелинейной трехатомной молекулой, то число колебав
тельных степеней свободы, равное числу нормальных колебаний,
будет ЗЛГ - 6 = 3 • 3 - 6 = 3. Стрелки на рис. 3.5 показывают нат
правление смещения ядер из равновесного положения. На рис,
3.5, а, б можно видеть, что в результате колебаний происходит
изменение длины валентной связи О-^-Н при практически посто-
янной величине угла НОН. Это валентные колебания й v2. На
рис. 3.5, в колебание атомов вызывает изменение угла НОН. Та-
кое колебание называют деформационным 8,
Колебательные спектры многоатомных молекул интерпрети-
руют на основе учения о симметрии молекул и теории групп.
Математический аппарат теории групп позволяет вычислить чис-
ло частот и правила отбора для молекул различной симметрии.
Такая информация, чрезвычайно ценная для определения моле-
кулярных констант, изучения строения молекул и т. д., находит
сравнительно малое применение для решения химико-аналити-
ческих задач. Для решения этих задач используются так1 назы-
ваемые характеристические частоты^
Анализ ИК^спектров показал, что некоторые из наблюдае-
мых частот можно привести в соответствие с колебаниями от-
дельных атомов или групп атомов. Так, например, было найдено;
что в спектрах всех молекул, содержащих связи С-^Н, имеются
частоты в области 2800—3000 см"1, связь С=С характеризуется
частотой 1650 см"1, а связь С=С —. частотой 2100 см"1. Такие
частоты назвали характеристическими. Они широко использу-
ются в практической спектроскопии для определения строения
молекул и проведения качественного анализа по ИК-спектрам.
59
3.2.4. Электронные спектры
Верхней энергетической границей колебательного спектра
обычно считают энергию фотонов примерно в 5000 см"1, или око-
ло 60 кДж/моль. Дальнейшее увеличение энергии облучающих
квантов чаще всего будет приводить к возбуждению электронов и
появлению в спектре полос, характеризующих электронные пе-
реходы, хотя, конечно, указанная граница может несколько сме-
щаться в ту или другую сторону в зависимости от свойств изучае-
мого соединения.
Интерпретация электронных спектров может быть сделана на
основе квантово-механических представлений, например метода
молекулярных орбиталей (МО). В соответствии с основными по-
ложениями этого метода электроны в молекуле могут находиться
на связывающих, несвязывающих и разрыхляющих орбиталях.
Схема относительного расположения энергетических уровней, со-
ответствующих разным МО, показана на рис. 3.6. Различные
электронные переходы требуют неодинаковой энергии, поэтому
полосы поглощения располагаются при разных длинах волн.
Наибольшей энергии требует а-»а*-переход, связанный с воз-
буждением внутренних электронов. Он соответствует поглощению в
далекой ультрафиолетовой области (X < 200 ям, Е> 600 кДж/моль).
Такие переходы характерны, например, для метана, этана и других
насыщенных углеводородов. Переход п-^а* связан уже с меньшими
затратами энергии. Полосы, вызванные этим переходом, располо-
жены в обычном (невакуумном) ультрафиолете (X = 200—300 нм).
Еще меньшая энергия требуется для перехода на разрыхляющие
я*-орбитали. Переходы п-+п* и 7с-+я* встречаются в молекулах соеди-
нений с сопряженными связями и молекулах ароматических соеди-
нений. Этим же переходом можно объяснить, например, интен-
сивную окраску ионов МпО^ и СгО|~
(переход с несвязьюающей орбита^
ли кислорода). Теоретический расчет
энергии разных МО громоздок и свя-
зан с большими трудностями, поэтому
существенное значение имеют различ-
ные эмпирические закономерности,
связывающие спектр поглощения со
строением и свойствами вещества.
Введение в молекулу различных
замезтителей или изменение внеш-
них условий, например растворите-
ля, обычно вызывает сдвиг полосы
в
со
71-»71*
о-» а
Рис. 3.6, Схема электронных
уровней й энергия возможных
электронных переходов
60
поглощения. Если полоса поглощения смещается в сторону более
длинных волн, говорят о батохромном смещении или углубле-
нии окраски (красное смещение), а если полоса сдвигается в сто-
рону более коротких волн, эффект называют гипсохромным сдви-
гом или повышением окраски (голубое или синее смещение).
Кроме переходов внутри валентной оболочки, известны так назы-
ваемые переходы Ридберга, связанные с изменением главного
квантового числа. Полосы, соответствующие этим переходам,
расположены в дальней ультрафиолетовой области.
Электронный переход обычно осложняется наложением ко-
лебательных, а иногда и вращательных переходов, так как каж-
дое электронное состояние молекулы обладает набором колеба-
тельных (и вращательных) уровней (рис. 3.7). Основному элек-
тронному состоянию (I) отвечает система колебательных
уровней, характеризуемая колебательными квантовыми числами
^= 0,1, 2,... . Возбужденное электронное состояние (II) обладает
системой колебательных уровней с квантовыми числами V == О,
1, 2, ... .У каждого колебательного состояния есть система вра-
щательных уровней, энергия которых пропорциональна враща-
тельному квантовому числу у.
При обычных условиях большинство молекул находится в
основном электронном и основном колебательном состояниях.
Переходы увр в этих условиях (см. рис. 3.7) характеризуют чисто
вращательные спектры, линии которых появляются в самой да-
лекой длинноволновой части ИК-спектра и области микроволн.
Чисто колебательные переходы укол (см. рис. 3.7) происходят
при неизменном электронном состоянии молекулы. В случае
разреженных газов наблюдаются колебательно-вращательные
спектры молекул укол + увр, харак-
теризующие переходы между вра-
щательными уровнями различ-
ных колебательных состояний.
Электронные переходы явля-
ются наиболее сложными в связи
с наложением колебательных, а
при определенных условиях и
вращательных переходов. Нало-
жение большого числа колеба-
тельных переходов часто приво-
дят к существенному уширению
полос электронных спектров, так
как колебательная структура не
всегда разрешается. Известны и
■ ]
; • ,. 1
(П)
[
7еее
■]^±11 1 Т;
1р-
Г = 3
Г = 2
г = о
У = 3
У = 2
V - 1
V = 0
(I)
Рис. 3.7. Схема энергетических
уровней молекулы в основном (I)
и первом возбуждённом (II)
электронных состояниях
61
7=0
: Рис. 3.8. Электронно-
колебательный переход
согласно принципу
Франка—Кондона:
1 — основное и
2 возбужденное
состояния
другие причины, вызывающие ушире-
ние полосы поглощения в электронных
спектрах, связанные, например, с вре-
менем жизни возбужденного состояния.
Изменение свойств электронной
оболочки в результате электронного пе-
рехода приводит к изменению потенци-
альной энергии системы, поэтому кри-
вая потенциальной энергии, например,
двухатомных молекул в разных элек-
тронных состояниях не будет одинако-
ва. В возбужденном состоянии обычно
происходит увеличение равновесного
межъядерного расстояния, уменьшение
энергии диссоциации и изменение дру-
гих свойств по сравнению со свойствами
в основном состоянии. Обычные кри-
вые потенциальной энергии молекулы в
основном и возбужденном состояниях
показаны на рис. 3.8.
Существенное значение в электрон-
ной спектроскопии имеет принцип
Франка—Кондона. При взаимодействии молекулы с квантом све-
та электронная оболочка столь быстро переходит в возбужденное
состояние, что положение ядер измениться не успевает. Таким
образом, за время перехода молекулы в возбужденное состояние
межъядерное расстояние меняться не будет. Однако в новом
электронном состоянии равновесное межъядерное расстояние
обычно отличается от того, которым обладала молекула в основ-
ном состоянии, и колебательные уровни возбужденного состоя-
ния также будут другими. Переход на колебательные уровни воз-
бужденного электронного состояния дает информацию о колеба-
тельных уровнях этого состояния.
Неизменность меясъядерного расстояния во время электрон-
ного перехода означает, что на кривых потенциальной энергии
этот переход должен изображаться вертикальной линией, как это
представлено на рис. 3.8. Колебательная структура спектра зави-
сит от относительного смещения кривых потенциальной энергии.
Так как наиболее вероятны переходы без изменения межъядер-
ного расстояния, ими будут отвечать наиболее'интенсивные по-
лосы в спектрах поглощения. Принципу Франка—Кондона мож-
но придать количественную форму. Использование принципа*
Франка—Кондона в спектроскопии многоатомных молекул так-
62
же приводит к важным результатам, дозволяя решить вопрос р
типе колебаний и структуре молекул. В случае многоатомных мо-
лекул картина становится значительно более сложной, так как
потенциальная энергия многоатомной молекулы может быть
представлена уже не просто кривой, а поверхностью потенциаль-
ной энергии в тг-мерном пространстве. (Если зафиксировать опре-
деленные координаты, потенциальную поверхность можно изо-
бразить и в трехмерном пространстве.) :
3.2.5. Интенсивность поглощения
Интенсивность полос в спектре поглощения характеризуют
интегралом поглощения, или силой осциллятора, определяемой
выражением
/=^*, ' (3-9)
где В — коэффициент Эйнштейна, характеризующий вероят-
ность перехода.
Коэффициент Эйнштейна связан с электрическим дипрлъ-
ным моментом перехода и некоторыми другими величинами. Он
может быть также рассчитан из экспериментальных данных по
УраВНеНИЮ /:,;/^^^.;::'Ч'Ч,!-/^г: ,. ' г,,,
где 1Г объединяет несколько величин, а интеграл I е(у) с1у называ-г
ют интегралом поглощения.
Сочетание уравнений (3.8) и (3.10) и подстановка числовых;
значений фундаментальных констант приводят к формуле п
/ = 4,33 • 10"9 ] е(у) (IV. .'. 5
Соотношение (3.9) показывает* что ч;ем более вероятным яв-
ляется переход, тем больше сила осциллятора. Так, разрешенные
переходы характеризуются величиной Д близкой к 1, как, это наЛ:
блюдается у интенсивно;окрашенных красителей, а у запрещен^
ных переходов, например у переходов о изменением мультиплет-.
ности терма, сила осциллятора составляет примерно 10~7. г
Для аналитической характеристики соединений имеет значе-г
ние не столько интегральное поглощение, сколько светопоглощет,
ние при определённой длине водны. Важными аналитическими,
характеристиками являются молярный коэффициент поглощения»
63
Рис. 3.9. Полоса поглощения
в точке максимума етак и полуши-
рина полосы поглощения 8 = Д^/2
(рис. 3.9).
Наибольшей интенсивностью
в спектрах поглощения обладают
полосы переноса заряда, т. ё. по-
лосы, обусловленные переносом
электрона от одного атома к друго-
му.Часто эти полосы связаны с пе-
реносом электрона с р-орбйтали
лиганда на й-орбиталь централь-
ного иона и наоборот. Более пра-
вильно следует говорить о переносе электрона между молекуляр-
ными орбиталями, локализованными у разных атомов. Интен-
сивные полосы в спектре, появившиеся в результате такого рода
переходов, имеют молярный коэффициент поглощения порядка
104 и более. К ним относятся многие 7с-тс*-переходы. Переносом
заряда объясняют, например, интенсивную окраску ионов МпО£,
СгО|", окраску тиоцианатных комплексов железа, кобальта, мо-
либдена, сульфосалицилатных комплексов железа, фанантроли-
новых комплексов и многих других.
Значительно менее интенсивны полосы, связанные с внутри-
атомными й—й- или /—/-переходами. Эти, вообще говоря, запре-
щенные переходы (ДХ, = 0) дают полосы с молярным коэффици-
ентом поглощения от 1 до величин, редко превышающих 100.
Снятие запрета с й—^-переходов в комплексах чаще всего объяс-
няют «частичным смешиванием» й- и р-орбиталей и переходом
электрона уже со смешанных й-, р-орбиталей на й-орбитали, что
не запрещено. Успешно интерпретирует спектры й- и /-элементов
теория кристаллического поля.
Спектры окрашенных соединений в растворе обычно характе-
ризуются довольно широкими полосами поглощения. Уширение
полос связано с сильным влиянием молекул растворителя на энер-
гетические уровни электронов, ответственных за светопоглощение,
и наложением колебательных переходов на электронный переход.
Почти всегда очень широкие полосы наблюдаются в спектрах пере-
носа заряда. Ионы лантаноидов имеют узкие полосы поглощения,
так как их внутренние 4/-электроны, ответственные за светопогло-
щение, экранированы внешними 5«-, 5р-электронами.
, Очевидно, чем выше молярный коэффициент поглощения и
меньше ширина полосы, тем более ценными химико-аналитиче-
скими свойствами обладает соединение, так как эти характерис-
64
тики полосы определяют такие важные показатели, как предел
обнаружения и селективность.
3.2.6. Фотохимические реакции
Взаимодействие лучистой энергии с веществом далеко не
всегда ограничивается только возбуждением поглощающей свет
частицы и последующим ее переходом на основной или более низ-
кий уровень без изменения структуры частицы. Кванты света —
фотоны — можно рассматривать как участников реакции, спо-
собных вызывать, как и любой другой участник, химический
процесс (фотохимическую реакцию). Химические превращения,
происходящие с возбужденной частицей, являются основой каче-
ственного и количественного фотохимического анализа. Качест-
венный анализ основан на образовании под действием света окра-
шенных продуктов, как, например, фотохимическим методом
можно обнаружить 0,015 мкг золота в капле раствора. Фотохи-
мические методы позволяют определять свыше 40 элементов и
многих органических веществ.
Однако фотохимические реакции могут служить источником
ошибок в фотометрических методах анализа, хотя негатившжу
влиянию света на реакции образования окрашенных соединений
и реактивов уделялось явно недостаточное внимание. На раство-
ры и протекание реакций в растворе могут влиять рассеянный
и прямой солнечный свет, а также свет искусственных источни-
ков. Наибольшее влияние оказывает прямой солнечный свет, в
спектре которого значительная часть принадлежит УФ-излуче-
нию. Под действием света изменяются концентрации растворов
КМп04, Ыа2820з, КвСЫ, AgNOз, К2Сг207, Н2С204 и многих дру-
гих. К действию света чувствительны многие органические со-
единения, используемые в качестве реагентов или индикаторов —
малахитовый зеленый, мурексид, эриохром черный Т, ксилено-
ловый оранжевый и др. Под действием света на растворы вин-
ной и лимонной кислот происходит их фотолиз с образованием
Н2, С02, СО, углеводородов, альдегидов, кетонов и т. д. Под воз-
действием рассеянного света обесцвечивается тиоцианатный комп-
лекс Ге3+ вследствие восстановления Ге3+, ослабляется окраска его
салицилатного комплекса, разрушается комплекс с ЭДТА, свето-
чувствительные комплексы Ге(Ш) с оксикислотами. Свет ускоряет
Разрушение оксихинолината алюминия, разрушает дитизон и ди-
тизонаты металлов.
Уже приведенные иллюстративные примеры показывают не-
обходимость учета и предупреждения возможных последствий
5 - 7831
65
негативного влияния света на фотометрические и иные системы.
Литературные источники обычно ограничиваются указанием на
необходимость защиты раствора от действия света. Иногда реко-
мендуется работать при ослабленном искусственном или красном
свете. Частично решают эту проблему лабораторные склянки из
темного стекла. Известен способ стабилизации раствора к дейст-
вию света путем введения в систему окислителя.
Более общее значение имеет применение УФ-абсорберов — ве-
ществ, способных поглощать УФ часть спектра и тем самым пред-
отвращать от фотолиза другие вещества. Молекулы УФ-абсорбе-
ров рассеивают полученную энергию в безызлучательных перехо-
дах главным образом в виде теплоты. Для аналитических целей в
качестве УФ-абсорберов используются производные бензтриазола
и бензофенона.
3.3. Основные узлы приборов
абсорбционной спектроскопии
При всем многообразии схем и конструктивных особенностей,
приборов абсорбционной спектроскопии в каждом из них имеет-
ся несколько основных узлов, функции которых примерно оди-
наковы в разных приборах. Такими узлами являются: источник
света, монохроматизатор света, кювета с исследуемым вещест-
вом, рецептор (приемник света).
; К этим основным узлам следует добавить оптическую систе-
му, состоящую из линз, призм и зеркал; которая служит для со-
здания параллельного пучка света, изменения направления и фо-
кусировки света, а также систему для уравнивания интенсивнос-
ти световых потоков (диафрагмы, оптические клинья и т. д.). ■'■
В приборах абсорбционной спектроскопии свет от источника
освещения проходит через монохроматизатор и падает на кювету
с исследуемым веществом. Интенсивность монохроматического
света, прошедшего 'через кювету, измеряется приемником света
(рецептором). Практически обычно определяют отношение ин-;
тенсивностей монохроматического света, прошедшего через ис-
следуемый раствор и через растворитель или специально выбран-
ный раствор сравнения, и
3.3.1. Источники света н
Основными источниками освещения в абсорбционной спектрог
скопии являются вольфрамовые лампы накаливания, газонапол-
ненные лампы (водородная, ртутная), штифт Нернста и гдобар.
66
В лампе накаливания светящаяся вольфрамовая спираль да-
ет свет в широком спектральном интервале. Однако стекло про-
пускает свет лишь в интервале длин волн 350—1000 нм, т. е*. в
видимой части спектра и самых ближних ультрафиолетовой и
инфракрасной областях. В водородной лампе происходит свече-
ние водорода при разряде. Условия возбуждения подбирают т&к,
что возникает практически сплошное излучение в области 200—
400 нм. В ртутной лампе разряд происходит в парах ртути. Воз-
бужденные атомы ртути испускают линейчатый спектр, в кото-
ром преобладает излучение с длиной волны 254, 302, 334 нм.
Штифт Нернста представляет собой столбик, спрессованный
из оксидов редкоземельных элементов. При накаливании путем
пропускания электрического тока он дает ИК-излучение в облас-
ти 1,6—2,0 или 5,6—6,0 мкм. Глобар-штифт из карборунда &С
дает излучение в интервале 2—16 мкм также при пропускании
электрического тока.
3.3.2. Монохроматизаторы
Монохроматйзаторами или монохроматорами называют уст-
ройства для получения света с заданной длиной волны. Однако
термин «монохроматизатор» является более предпочтительным,
так как под названием монохроматор подразумевают специаль-
ные спектральные приборы. При конструировании монохромати-
заторов используют разные оптические явления: поглощение све-
та, интерференцию, дисперсию и т. д. Наибольшее распростране-
ние в практике абсорбционной спектроскопии имеют приборы;,, в
которых в качестве монохроматизаторов применяются свето-
фильтры, призмы и дифракционные решетки.
Известно несколько типов светофильтров. В зависимости от
вида оптического явления, используемого для монохроматиза-
ции света, конструируют абсорбционные, интерференционные
или интерференционно-поляризационные светофильтры. >
Действие абсорбционных светофильтров основано на том, что
при прохождении света через тонкий слой вследствие ПОГЛОЩвг
ния происходит изменение величины и спектрального состава
проходящего светового потока. Абсорбционные светофильтры
имеют небольшую прозрачность (Т = 0,1) и довольно широкую
полосу пропускания (АХ, = 30 нм и более). Характеристики интер-
ференционных светофильтров значительно лучше. Светофильтр
состоит из двух тончайших полупрозрачных слоев серебра, меж-
ду которыми находится слой диэлектрика. В результате интёрфе-
5* ^ 67
ренции света в проходящем пучке остаются лучи с длиной вол-
ны, равной удвоенной толщине диэлектрического слоя. Прозрач-
норть интерференционных светофильтров составляет Т = 0,3—-
0,8. Эффективная ширина пропускания обычно не превышает
5—-10 нм. Для еще большего сужения полос пропускания иногда
пользуются системой двух последовательных интерференционт
ных светофильтров.
Наиболее универсальными монохроматизаторами являются
призмы, изготовленные из кварца, стекла и некоторых других
материалов, и дифракционные решетки. Для инфракрасной
спектроскопии используют призмы из 1А¥, ЫаС1, КВг и других
галогенидов щелочных и щелочно-земельных металлов. Эти же
материалы применяют для изготовления кювет. Призмы и диф-
ракционные решетки позволяют получать свет высокой монохро-
матичности в широкой области длин волн. В конструкциях МНО-
ГИХ современных спектрофотометров в качестве монохроматора
используются дифракционные решетки. V
3.3.3. Приемники света
> В качестве приемников света (рецепторов) в приборах абсорб-
ционной спектроскопии используют главным образом фотоэлеЦ
менты, фотоумножители, а иногда интенсивность света оценива-
ется на глаз — визуально. Для измерения интенсивности инф-
ракрасного излучения применяют фотоэлементы, термоэлементу
и болометры. Приемники света характеризуются спектральной
чувствительностью — способностью воспринимать излучение
различной длины волны — и интегральной чувствительностью^
которая измеряется по действию на рецептор не разложенного в
спектр излучения.
В термоэлементах используемся термоЭДС, возникающая
при изменении температуры спая между металлами или сплава-
ми под действием инфракрасного излучения. Широко применя-
ются для этих целей термопары медь—константан, серебро—вис-
мут и др.
1 Принцип действия болометра основан на изменении электро-
сопротивления материала при нагревании. ТермочувствительЦ
ный элемент, представляющий собой зачерненную платиновую^
сурьмяную или другую тонкую металлическую пластинку, вклкФ
чают в мостовую схему. Инфракрасное излучение вызывает на*'
гревание термочувствительного элемента и разбаланс моста, пдхк
порциональный интенсивности падающего излучения. I
68
Промышленностью выпускаются различные приборы абсорб-
ционной спектроскопии: фотометры, фотоэлектроколориметры,
спектрофотометры и т. д., в которых используют различные ком-
бинации осветителей, монохроматизаторов и приемников свети.
Широко применяют в аналитических лабораториях различ-
ные ФЭКи (фотоэлектроколориметры) — ФЭК-56-2, ФЭК-56М
и др. В последних моделях приборов серии КФК предусмотрена
микропроцессорная система, позволяющая непосредственно по-
лучать концентрацию вещества, а колориметр КФК-МП снаб-
жен, кроме того, термопечатающим устройством.
3.4. Качественный анализ
Наибольший интерес с точки зрения качественного анализа
представляют колебательные (вернее колебательно-вращатель-
ные) спектры. Они весьма характерны, и в иностранной литера-
туре их нередко называют finger print, т. е, отпечатки пальцев,
имея в виду неповторимость инфракрасного спектра соединения.
Экспериментальные исследования колебательно-вращатель-
ных спектров показали, что полосы при некоторых частотах
можно привести в соответствие с колебаниями определенных
групп атомов или отдельных атомов в молекуле. Такие частоты
назвали характеристическими. Различные молекулы, содержа-
щие одну и ту же связь или одну и ту же атомную группировку,
будут давать в ЙК-спектре полосы поглощения в области одной
и той же характеристической частоты. Это и является основой
качественного анализа по инфракрасным спектрам. Характерис-
тические частоты дают возможность установить по спектру на-
личие определенных групп атомов в молекуле и тем самым поз-
воляют судить о качественном составе вещества й строении
молекул. Например, полосы в области 3000—3600 см"1 могут
быть приписаны только О—Н- или N—Н-связям, и отсутствие
полос в этой области, спектра однозначно свидетельствует об
отсутствии ОН- и NH-групп в анализируемом веществе. Приме-
ры такого роди исследований весьма многообразны. С помощью
инфракрасных сцектров было установлено строение многих оле-
финов, ароматических соединений, карбонильных соединений,
аминокислот и других групп веществ. Было выяснено, напри-
мер, что большинство аминокислот существует в растворе в ио-
низированном состояний, которое можно представить форму-
69
лой К—СН— С. (а), но не И—СН—(б). Действитель-
| ^о- | ^ОН
но, в спектре нейтрального раствора глицина в В20 были найде-
ны полосы поглощения, характерные для ионизированной кар-
боксильной группы (1600 и 1400 см"1). Введение в раствор кисло-
ты БС1 вызвало их замену на полосы при 1710 см"1, а добавление
щелочи опять их восстанавливало.
Полосы валентных колебаний №Н в области 3300—3500 см"1,
где они обычно проявляются, у аминокислот не обнаруживаются,
+
а в области около 3070 см"1, характерной для группы —ЫН3, они
есть. Образование соли аминоуксусной кислоты вызывает появ-
ление полосы вблизи 3300—3500 см"1, характерной для
—ЫН2-группы. Сопоставим эти результаты:
Группа — ЫН2 — ЫНз —СООН —СОО"
Волновое число, см"1 3300; 3500 3070 1710 1600; 1400
Нейтральный раствор ... - + - +
Кислый раствор ........ - + + -
Щелочной раствор ...... + - - ' +
Эти данные совершенно однозначно показывают, что в водном
растворе глицин существует в виде цвиттер-иона ЫН3СН2СОО".
Отсутствие в инфракрасном спектре полосы, связанной с какой-то
характеристической частотой, является обычным надежным до-
казательством отсутствия определенной группы в молекуле. В тр
же время наличие полосы в некотором участке спектра представ-
ляет собой менее доказательное свидетельство, т. е. оно еще не
всегда или не полностью доказывает присутствие предполагаемой
группы в молекуле в связи с возможным наложением полос.
К настоящему времени изучены и сведены в соответствующие
атласы и таблицы инфракрасные спектры более чем 20 000 соеди-
нений, что существенно облегчает практическое проведение анали-
за. Для получения первых ориентировочных данных часто пользу-
ются так называемой картой Колтупа, на которой указаны спект-
ральные области появления многих характеристических частот
их возможное отнесение. Для окончательных выводов обычно тре-
буется более тщательный анализ спектра. Иногда задача качест-
венного анализа может быть решена простым сопоставлением,
спектра анализируемого вещества и «подозреваемого» соединения.
70
Инфракрасная спектроскопия с успехом используется и в
анализе неорганических веществ. Известно, например, что ха-
рактеристическая частота СО|~ составляет 1450 см"1, вО|~ —
ИЗО, N0^ — 1380, ЫН£ — 3300 см"1 и т. д. В связи с этим из инф-
ракрасных спектров минералов получают весьма ценную инфор-
мацию 6 химическом составе.
Электронные спектры поглощения для целей качественного
анализа используются значительно реже, чем колебательные,
так как они обычно бывают представлены небольшим числом
широких полос поглощения, которые часто накладываются одна
на другую и полностью или частично перекрываются. Однако по
электронным спектрам поглощения иногда удается провести до-
статочно эффективный анализ качественного состава, например
в нефтехимии.
3.5. Количественный анализ
Методы количественного анализа основаны на законе Бугера^
Ламберта—Вера, выраженном уравнением (3.2). В связи с тем
что значения коэффициента пропускания Т находятся в преде-
лах от 0 до 1 (1 > Т > 0), оптическая плотность раствора А = Ч£ Т
может принимать, казалось бы, любые положительные значения
от нуля до бесконечности, т. е. оо > А > 0. Однако эксперимен-
тальному определению с необходимой точностью доступны дале-
ко не любые значения А. Так, например, значения А < 0,01 не оп-
ределяют в связи с большой погрешностью их измерения.
Уравнение (3.2) показывает, что основными параметрами фо-
тометрического определения являются длина волны, при кото-
рой производится измерение, оптическая плотность, толщина
кюветы и концентрация окрашенного раствора. Существенное
влияние оказывают различные химические факторы, связанные
с полнотой и условиями протекания фотометрической реакции,
концентрацией окрашенных и других реактивов, их устойчиво-
стью ит. д, В зависимости от свойств анализируемой системы и
характеристик применяемого фотометрического прибора выби-
рают те или иные условия анализа. с
Основными операциями количественных методик абсорбци-
онной спектроскопии являются следующие:
1. Растворение пробы и перевод определяемого компонента в
окрашенное (свётопоглощающее) соединение.
2; Разложение монохроматором света от источника освеще-
ния на отдельные спектральные компоненты, направляемые на
71
кювету с определяемым веществом или раствором сравнения.
Выбор оптимальных условий фотометрического определения
(участка спектра или длины волны, оптической плотности, тол-
щины слоя).
3. Определение оптической плотности анализируемого и
стандартных растворов. Построение градуировочного графика и
использование других приемов для определения интересующего
компонента.
Рассмотрим эти операции.
Основные приемы, используемые для растворения жидких и
твердых проб, обсуждались в первой книге (гл. 3). Для растворе-
ния навески анализируемого вещества чаще всего применяется
обработка пробы минеральными кислотами или их смесью (на-
пример, «царской водкой») при нагревании на водяной или пес-
чаной бане. При анализе некоторых горных пород, огнеупоров
и т. д. кислотной обработки бывает недостаточно — в таких слу-;
чаях используется сплавление пробы с кислыми или щелочными
плавнями и последующее выщелачивание. При проведении рас-
творения необходимо перевести в раствор все определяемые ком-;
поненты и не допустить их потерь за счет уноса при нагревании!
или выполнении других операций, связанных с растворением. Из
полученного многокомпонентного раствора путем операций раз-
деления (осаждения, экстракции и т. д.) выделяют анализируе-
мый компонент и переводят его в окрашенное соединение.
3.5.1. Концентрационные условия проведения
фотометрической реакции
В уравнение основного закона светопоглощения входит кон-
центрация окрашенного (светопоглощающего) соединения, по-
этому превращение определяемого компонента в такое соедине-
ние является одной из важнейших операций, в значительной сте-
пени определяющей точность анализа. Окрашенные соединения
в растворе получают в результате, главным образом, реакций
окисления—восстановления и комплексообразования. Окисли-
тельно-восстановительные реакции, применяемые в фотометрии,
например окисление марганца до МпО^, протекают, как прави-
ло, практически полностью до конца.
Значительно более сложным является вопрос о концентраци-
онных условиях протекания в растворе реакций комплексообра-
зования. Осложняющее влияние здесь могут оказать процессы
72
ступенчатого комплексообразования, протолитические равнове-
сия, недостаточная устойчивость образующегося комплекса, соб-
ственная окраска реагента и т. д. Действие большинства этих
факторов можно предвидеть, если равновесия в интересующей
системе достаточно подробно изучены и константы соответствую-
щих равновесий известны (константы устойчивости координаци-
онных соединений, диссоциации реагентов и т. д.). Используя
эти данные, можно рассчитать, например, при каких значениях
рН и концентрации реагента будет достигнута необходимая пол-
нота реакции, как будут влиять сопутствующие элементы и т. д.
Если окрашенное соединение образовано анионом сильной
кислоты, как, например, при определении висмута в виде иодид-
ного комплекса или железа в виде тиоцианатного, то реакцию
обычно проводят при постоянной концентрации реактива и в до-
вольно кислой среде, обеспечивающей подавление гидролитиче-
ских процессов. Концентрация аниона в таких системах от кис-
лотности среды не зависит.
Найдем, например, условия получения тиоцианатного комп-
лекса железа. При простейшем рассмотрении ограничимся уче-
том двух равновесий:
+ ЭСК" = РеВСМ* рРе8Сы2+ = ,
откуда
[РеБС^Т = рРе8Сн2+[Ре3+][8СЫ-];
Ре3+ + НОН = РеОН2+ + Н+, Кг = [*е°™щн+],
откуда
[РеОНП-*^,
Полнота образования у комплекса РевСЫ24* будет равна:
[Ре8С№+] = [Ге8С№+]
1 с|е [ГевСЫ2*] + |ТеОН2+] + [Ре8+]
= ^р[Ге3+][8СМ-] -р[8СК-][Н+]
Р[РвМ[ВСК-]+^1^а+[Рв»+] Э[ВС№][Н+]+^+[Н+]'-
При подстановке числовых значений констант получаем:
• ■ 1 - 103[8СЫ-][Н+]
У 1 • 10*[8СКГ-][Н+] +1 • Ю-2 + [Н+] '
73
Результаты некоторых расчетов по этому уравнению приве-
дены в табл. 3.1.
Таблица 3.1. Полнота образования РевС№+ в зависимости
от концентрации Н+ и БСМ"
[Н+]
Полнота реакции у
[вС]*-] = 0,1
[8СЫ-] = 0,2
[БСК-] = 0,3
0,01
0,05
0,10
0,980
0,988
0,989
0,990
0,994
0,995
0,993
0,996
0,996
Как видно из табл. 3.1, полнота образования комплекса воз-
растает с увеличением кислотности раствора и концентрации тио-
цианата. Расчет показывает, что во всех стандартных и в анали-
зируемых растворах следует поддерживать одну и ту же кислот-
ность, гарантирующую полноту реакции, например 0,1 М, и одну
и ту же концентрацию тиоцианата, например 0,3 М. Вполне по-
нятно, что при грубом несоблюдении этих условий градуировоч-
ный график не будет линейным. Существенные осложнения в
случае непостоянства концентрации тиоцианата будут связаны
также со сдвигом ступенчатых равновесий, поскольку молярные
коэффициенты светопоглощения; ГевС№+, Ге(8СН)|, Ре(8СМ)3
различаются в 2—3 раза.
При использовании в качестве реагента слабой кислоты, на-
пример, при определении железа в виде сульфосалицилатного
комплекса, рН раствора должен соответствовать слабокислой об-
ласти, в которой диссоциация кислоты достаточна и концентра-:
ция реактива постоянна. Особое внимание должно быть уделено
постоянству рН во всех исследуемых растворах. Для выяснения
оптимальных условий фотометрического определения каждая
система требует специального физико-химического исследования
для установления состава образующихся соединений, определе-
ния констант равновесия и т.д.
В случае образования, например, салицилатного комплекса
железа для выяснения условий протекания реакций будем учи-
тывать три равновесия: '
Ге3+ + 8а12~ ='Рё8а1+, В = [!е8а1+1 V
[Гез+Ква!2-]^
откуда
[Ге8а1+] = р[Гё3+][8а12-];
Ге3+ + НОН = ¥еОН2++В.+, К^3+
__ [ГеОН2+][Н+]
[Ге3+] '
74
откуда
откуда
[FeOH-]^,^;
Полнота образования у салицилатного комплекса будет
равна:
_ [FeSal*] [FeSal+]
с|е [FeSal+]+ [FeOH2+] + [Fe3+]
P[Fe3+][Sal2-]
P^H2sai[H2Sal]
P[Fe3+][Sal2-] + K¿I*^ + [Fe3-] P*H2sai[H2Sal] + *Fe[H+] + [H+]2
[H+]
При подстановке числовых значений констант получаем:
у = 2,2 • 1015 • 2,8 • 10"17
[H2Sal]
6,2 • 10-2[H2Sal] + 10"2[Н+] + [Н+]2
Результаты некоторых расчетов по этому уравнению приве-
дены в табл. 3.2.
Таблица 3.2. Полнота образования FeSal+ в зависимости
от концентрации Н+ и H2Sal
[Н+]
Полнота реакции у
[H2Sal] - 0,01
[H2Sal] = 0,02
[H2Sal] == 0,05
0,01
1 • 10"3
1 • 10"4
0,756
0,984
0,998
0,925
0,992
0,999
0,939
0,995
0,999
Как видно из табл. 3.2, при рН2,0 концентрация салицило-
вой кислоты даже в 0,05 М не гарантирует полноты реакции, а
при рНЗ,0 практическая полнота достигается уже в 0,02 М рас-
творах кислоты. При наличии данных о константах равновесия
реакций аналогичные расчеты можно сделать для любой сис-
темы и найти таким образом оптимальные условия проведения
реакции.
75
3.5.2. Оптимальные условия
фотометрического определения
Длина волны. При определении в растворе одного светопо-
глощающего вещества аналитическую длину волны, как прави-
ло, выбирают на максимуме полосы поглощения. Если в спектре
имеется несколько полос, выбор обычно останавливают на наибо-
лее интенсивной, так как работа в области максимума светопо-
глощения обеспечивает наиболее высокую чувствительность оп-
ределения. Плоские максимумы более предпочтительны, так как
при этом меньше сказывается погрешность в установлении дли-
ны волны, чем в случае острых максимумов или крутоспадаю-
щих участков кривой. Желательно также, чтобы чувствитель-
ность приемника излучения в области аналитической длины вол-
ны была максимальна. Однако практическая реализация этого
условия затруднена, так как конструкция обычных фотометри-
ческих приборов предусматривает не более двух фотоэлементов.
Выбор аналитической длины волны при наличии в растворе не-
скольких светопоглощающих веществ значительно сложнее. Он
будет рассмотрен при выборе условий анализа смеси окрашенных
веществ.
Светопропускание (оптическая плотность). Измерительное
устройство фотометрического прибора обычно имеет постоянную
ошибку АТ в величине коэффициента пропускания Т во всем ин-:
тефвалё его значений. Ошибка в единицах оптической плотности
ДА в связи с этим во всем интервале не будет одинакова. Поэтому
при решении некоторых задач удобнее оперировать с коэффици-,
ентом пропускания, а не с оптической плотностью. Рис. 3.10 по-
казывает, что при одной и той же абсолютной погрешности АТ аб-
солютная погрешность определяемой концентрации Ас существен-
но возрастает с увеличением концентрации раствора (Ас2 > Асг,
хотя АТ2 = АТ,).
Относительная ошибка Ас/с бу-
дет уменьшаться с ростом концентра-
ции и возрастать с увеличением абсо-
лютной ошибки Ас. Ответ на вопрос,
при каких значениях Т относительная
ошибка Ас/с будет минимальна, дает
небольшой математический анализ.
Из уравнения (3.1) следует, что
С = -)М1 Г (з и)
Рис. 3.10. Зависимость Т от с е1
76
При дифференцировании уравнения (3.11) получаем
dT
2,гте1
Сочетание уравнений (3.11) и (3.12) дает
dc _ йт&1 _ <хт
с е1 • 2,ЗТ^Т Т\пТ
или, переходя к конечным приращениям,
ДГ
(3.12)
Ас
с
Т\пТ'
(3.13)
Задаваясь разными числовыми значениями Т при фиксирован-
ной величине ДТ, по уравнению (3.13) можно рассчитать отнбси-
тельную погрешность Ас/с для всего интервала значений Г от 0 до 1.
Результаты такого расчета представлены графически на рис. 3.11,
из которого видно, что относительная погрешность резко возраста-
ет при очень малых и очень больших значениях Т. В области сред-
них значений Т кривая проходит через минимум. Для нахождения
точки минимума продифференцируем уравнение (3.13) по Т при
ДГ = const и производную приравняем нулю:
V с ) \ TJ _.-(1п!Г + 1)АГ .
0.
Ас/с
(Т1пТ)2 (ПпТ)2
Так как ДГ* 0, то, очевидно, 1п Т + 1 = 0. Откуда 1п Т = 2,3 1б Г =
- -1 и-1в Т = А = 0,435. При этом значении оптической плотнос-
ти будет достигаться наибольшая точность измерения.
Однако в проведенном расчете не учитывалась погрешность
за счет других источников, как, например, погрешность при ус-
тановлении прибора на нулевое и полное пропускание. Более
строгое теоретическое рассмотрение и
опыт показали, что оптимальная опти-
ческая плотность находится при более
высоком значении, чем 0,435, и состав-
ляет примерно 0,6—0,7 или несколько
выше. Расчеты и опыт показали также,
что фотометрическое исследование рас-
творов, имеющих 0,03 > А > 2,0, харак-
теризуется брлыпими погрешностями.
Эффективным приемом при анализе
интенсивно окрашенных растворов яв-
ляется применение методов дифферен-
циально^фотометрии.
0,37 Т
Рис. 3.11. Зависимость
относительной погрешности
от пропускания раствора
77
Толщина светопоглощающего слоя. Уравнение закона Буге-
ра—Ламберта—Бера показывает, что чем больше толщина слоя,
тем больше оптическая плотность и, следовательно, тем более
чувствительным будет определение при прочих равных услови-
ях. Однако с увеличением толщины слоя (длины оптического пу-
ти) возрастают потери на рассеяние света, особенно при работе с
растворами. Кюветы с толщиной слоя большей чем 5 см для фо-
тометрии растворов обычно не применяются.
Чувствительность и точность метода. Минимальную кон-
центрацию, которую можно определить фотометрическим мето-
дом, обычно рассчитывают по соотношению
Cmin=Anin/(80.
Если для ориентировочных расчетов принять, что Amin =
= 0,01,1 = 1 см и s — 103, то
с . = -ML = 10"5 моль/л.
min ... 1 i
Это не минимальная концентрация фотометрического мето-
да, так как е может быть больше, однако значение е = Ю3 свойст-
венно многим цветным соединениям, и, таким образом, оно в ка-
кой-то степени характеризует метод. Иногда в качестве показате- „
ля чувствительности фотометрической реакции указывают
просто величину е, известны и другие характеристики чувстви-
тельности. Точность фотометрических методов зависит от инди-
видуальных особенностей фотометрической реакции, характе-
ристик применяемого прибора и других факторов и изменяется в
довольно широких пределах. Обычная погрешность фотометри-
ческих методов составляет примерно 1—2% (относительных).
3.5.3. Основные приемы фотометрических измерений
Метод градуировочного графика. В соответствии с законом
Бугера—Ламберта—Бера график в координатах оптическая
плотность—концентрация должен быть линеен и прямая долж-
на проходить через начало координат. Для построения такого
графика достаточно, вообще говоря, одной экспериментальной
точки. Однако градуировочный график обычно строят не менее
чем по трем точкам, что повышает точность и надежность опреде-
лений.
При отклонениях от закона Бугера—Ламберта—Бера, т. е.
при нарушении линейной зависимости А от с, число точек на гра-
78
фике должно быть увеличено. Применение градуировочных гра-
фиков является наиболее распространенным и точным методом
фотометрических измерений. Основные ограничения метода свя-
заны с трудностями приготовления эталонных растворов и уче-
том влияния так называемых третьих компонентов, т. е. компо-
нентов, которые находятся в пробе, сами не определяются, но на
результат влияют. •
Метод молярного коэффициента поглощения. При работе по
этому методу определяют оптическую плотность нескольких
стандартных растворов (Аст), для каждого раствора рассчитыва-
ют е = Аст/(1сст) и полученное значение г усредняют. Затем изме-
ряют оптическую плотность анализируемого раствора (Ах) и рас-
считывают концентрацию сх по формуле
сх=Ах/(е1).
Ограничением метода является обязательное подчинение
анализируемой системы закону Бугера—Ламберта—Бера, по
крайней мере, в области исследуемых концентраций.
Метод добавок. Этот метод применяют при анализе растворов
сложного состава, так как он позволяет автоматически учесть
влияние «третьих» компонентов. Сущность его заключается в
следующем. Сначала определяют оптическую плотность Ах ана-
лизируемого раствора, содержащего определяемый компонент
неизвестной концентрации сх, а затем в анализируемый раствор
добавляют известное количество определяемого компонента (сст)
и вновь измеряют оптическую плотность Ах + ст.
Оптическая плотность Ах анализируемого раствора равна
Ах = е1сх, (3.14)
а оптическая плотность анализируемого раствора с добавкой
стандартного
Ах + ст = е1(сх + ссг). (3.15)
Сравнение уравнений (3.14) и (3.15) дает
А* — с*
А*х + СТ сх сст
или
АДС* + Сст) = Ас + СТСХ*
Отсюда находим концентрацию анализируемого раствора:
X С* СстА _А «...
\^ лж + ст Ах
■'. ' 79
Концентрацию анализируемого вещества в методе добавок
можно найти также по графику в координатах Ах + ст = /(сст).
Уравнение (8.15) показывает, что если откладывать Ах + ст как
функцию сст, то получится прямая, экстраполяция которой до пе-
ресечения с осью абсцисс даст отрезок, равный -сст. Действитель-
но, при Ах + ст = 0 из уравнения (3.15) следует, что -сст = сх.
Метод дифференциальной фотометрии. Фотометрирование
интенсивно окрашенных растворов успешно осуществляется ме-
тодом дифференциальной фотометрии. В обычной фотометрии
сравнивается интенсивность света 1Х, прошедшего через анализи-
руемый раствор неизвестной концентрации, с интенсивностью
света /0, прошедшего через растворитель. Коэффициент пропус-
кания такого раствора будет равен отношению интенсивностей
(рис. 3.12):
В дифференциальной фотометрии второй луч света проходит
не через растворитель, а через окрашенный раствор известной
концентрации — так называемый раствор сравнения концентра-
ции сср. Его интенсивность обозначим как /ср. Интенсивность све-
та, прошедшего через анализируемый раствор, по-прежнему
пусть будет 1Х. Отношение интенсивностей 1х/1ср называется ус-
ловным коэффициентом пропускания Т'х:
Отношение /ср к /0 характеризует коэффициент пропускания
раствора сравнения:
^ср ^ ^ср/^0*
Так как
^ср = ^срЛ)»
а) б)
Рис. 3.12. Схема обычной и дифференциальной фотометрии:
а — обычная; б — дифференциальная
80
то
1Х/1^ТХ^ТХ/Т,
ср
или, переходя от коэффициентов пропускания к оптическим
плотностям,
Аср»
Ах е1сх Аср,
(3.16)
где А'х — относительная оптическая плотность.
Уравнение (3.16) показывает, что относительная оптическая
плотность, также как и истинная, пропорциональна концентра-
ции окрашенного вещества, однако прямая А'х - сх не проходит
через начало координат (рис. 3.13). Пусть анализируемый рас-
твор имеет оптическую плотность А = 4,6, что методом обычной
фотометрии достаточно точно измерить нельзя. Взяв вместо рас-
творителя раствор Аср = 3,0, получаем относительную оптиче-
скую плотность А'х = Ах - Аср = 4,0 - 3,0 = 1,0, что можно изме-
рить уже с необходимой точностью.
Таким образом, дифференциальная фотометрия существенно
расширяет область концентраций, доступную для точных фото-
метрических измерений. Кроме того, точность некоторых мето-
дик дифференциальной фотометрии превышает точность мето-
дик обычной фотометрии. Найдем, при каких значениях А' или
V относительная погрешность определения методом дифферен-
циальной фотометрии будет минимальной. Для этого из уравне-
ния (3.16) находим сх:
8/
е1
(3.17)
и дифференцируем полученное выражение, считая Гср, е и I по-
стоянными:
йс
2,зг;е/
(3.18)
При сочетании уравнений (3.17) и
(3.18) получаем
6.Т'
с 2,ЗТх\ёТ'х + (\шТср)
или, переходя к конечным приращениям,
Ас _,
2,ЗTilgTІ + (lg!Гcp),
(3.19)
Рис. 3.13.
Градуировочный график
дифференциальной
фотометрии
6- 7831
81
Ае/с
Рис. 3.14. Относительная
погрешность при разном
пропускании раствора сравнения:
1
На рис 3.14 графически пред-
ставлены результаты некоторых
расчетов, выполненных по уравне-
нию (3.19). Кривые на рис. 3.14 по-
казывают, что с уменьшением ко-
эффициента пропускания раствора
сравнения (Тср) относительная по-
грешность определения концентра-
ции падает. И чем меньше коэффи-
циент пропускания Гср, тем мень-
ше относительная погрешность.
Как видно, при Тср = 1 диффе-
Тср=1,0;2-Тср = 0,1;
3 -Гср = 0,01; 4-
1ср
■10"3
ренциальныи метод превращается
в метод прямой фотометрии и, та-
ким образом, обычную фотомет-
рию можно рассматривать как ча-
стный случай дифференциальной. Это иллюстрируется кривой 1
на рис. 3.14, которая по сути дела воспроизводит кривую на рис.
3.11. Кривые, характеризующие погрешность методов дифферен-
циальной фотометрии, как видно, находятся ниже первой кри-
вой, т. е. при прочих равных условиях относительная погреш-
ность определения концентрации методом дифференциальной
фотометрии меньше, чем соответствующая погрешность обычной
фотометрии.
Чтобы найти оптическую плотность, при которой относи-
тельная погрешность определения минимальна, продифференци-
руем уравнение (3.19) по Тх при постоянных АТ й Гср и прирав-
няем производную нулю:
Очевидно,
или
и
.а(^) дг[(іпті+іпгср) + гіі]
ЧтГ = ;'[2.8г;(1вг; + ^тср)]2
1п Т'х +' 1п Тср 4-1 = 0
^Т; + ^Гср = -^ =-0,435
= 0.
А; = 0,435-Аср., * (3.20)
При Аср = 0 это соотношение, как частный случай, переходит в
Ах = 0,435, т. е. Ах принимает оптимальное значение, ранее уста-
новленное для обычной фотометрии.
82
Увеличение оптической плотности раствора сравнения (Аср)
будет вызывать в соответствии с уравнением (3.20) уменьшение
оптической плотности, при которой достигается максимальная
точность результатов. При Аср = 0,435 относительная оптическая
плотность будет равна нулю. Таким образом, точные измерения
будут достигаться при высокой оптической плотности раствора
сравнения и минимальной относительной оптической плотности.
Этот вывод иллюстрируется также кривыми рис 3.14.
Это не единственный способ дифференциальной фотометрии.
Еще более точным является так называемый метод предельной
точности. В этом методе используются два раствора сравнения —
в одном из них концентрация меньше, чем в анализируемом, а в
другом — больше. Первый из них характеризует систему с Т = 0%,
а второй с Т = 100%.
Количественный анализ по инфракрасным спектрам. Ана-
лиз по ИК-спектрам также основан на применении закона Буге-
ра—-Ламберта—Бер&. Чаще всего здесь используется метод гра-
дуировочного графика. Существенно затрудняется применение
метода молярного коэффициента поглощения в ИК-спектроско-
пии тем, что из;-за рассеяния, сплошного поглощения и,других
эффектов часто бывает невозможно, определить положение линии
100% -го пропускания, т.е. определить интенсивность света, прот
шедшего через образец без анализируемого компонента (10).
Многие трудности количественной ЙК-спектроскопии ус-
пешно преодолеваются с помощью метода базовой линии, кото-
рый получил большое распространение в практике. Сущность
его легко понять из рис. 3.15, на котором приведен участок
ИК-спектра с двумя полосами поглощения (их волновые числа уа
и ув). Базовая линия проводится в основании полосы поглощения
(она изображена пунктиром). Коэф-
фициент пропускания определяется
в этом методе как отношение ТА =
= 1А/10{А) или Тв = 1ъ/10{ву По полу-
ченным данным вычерчивают граду-
ировочный график и производят оп-
ределения^
Экстракционно-фотометрические
методы. Экстракционные методы при-
меняют в аналитической химии очень
широко, причем определение анализи-
руемого компонента в экстракте мо-
жет проводиться как (^отометриче-
А , УВ
Рис. 3.15. Метод базовой линии
88
ским, так и любым другим методом: полярографическим, спект-
ральным и т. д. Вместе с тем существуют некоторые группы эк-
стракционных методов, в которых фотометрическое окончание яв-
ляется наиболее эффективным, обеспечивая необходимую быстроту
и точность определения. Эти методы обычно называют экстракцион-
но-фотометрическими.
Весьма распространённой является методика, по которой оп-
ределяемый микрокомпонент переводят в растворимое в воде ок-
рашенное соединение, экстрагируют его и экстракт фотометриру-
ют. Содержание обычно определяют методом градуировочного
графика. Такая методика позволяет устранить мешающее влия-
ние посторонних компонентов и увеличивает чувствительность
определения, так как при экстракции происходит концентриро-
вание микропримесей. Например, определение примесей железа
в солях кобальта или никеля проводят экстракцией его тиоциа-
натных комплексов амиловым спиртом.
Окрашенное соединение определяемого компонента не обяза-
тельно должно быть растворимо в воде. Например, дитизон и
многочисленные дитизонаты металлов практически нераствори-
мы в воде, но растворимы во многих органических растворите-
лях. В этом случае к анализируемому раствору добавляют рас-
твор дитизона в подходящем органическом растворителе (напри-
мер, в тетрахлориде углерода), получают окрашенный экстракт,
который и фотометрируют. Иногда бывает удобнее экстракцию
проводить одним реагентом, а окрашенное соединение определяе-
мого элемента получать в экстракте уже с помощью другого.
3.5.4. Определение смеси светопоглощающих веществ
Спектрофотометрический метод, в принципе, позволяет опреде-
лить несколько светопоглощающих веществ в одном растворе без
предварительного разделения. Большое практическое значение име-
ет частный случай такой системы — анализ смеси двух окрашенных
веществ. В соответствии с законом аддитивности светопоглощения
для такой смеси веществ, например А и В, можно записать:
-АХ1авг(?А,Х^А + 8В,Х1св);
1^а$х2са +ев,х2св)-
(3.21)
Решение этой системы уравнений при I = 1 см дает:
_ Ачевд2~А28вд1 .
(3.22)
^Д^ВДа ~ еАД^8ВД1
84
Длины волн и Х29 при которых
следует проводить измерения опти-
ческой плотности, выбирают по
спектрам поглощения веществ А и В,
применяя различные приемы. Хоро-
шие результаты дает, например, ме-
тод максимальных разностей. При
использований этого метода сначала
снимают спектры поглощения ком-
понентов А и В (рис 3.16, а), а затем
строят график зависимости еА - ев
или ев - еА от длины волны и находят
области максимума и минимума
(рис. 3.16, б). Длины волн, соответст-
вующие этим областям, выбирают в
качестве аналитических, учитывая,
что абсолютные значения гА и 8В в
В
В
б)
Рис 3.16. Спектр
поглощения вещества А и В (а)
и зависимость еА - ев от длины
волны (б)
этих областях тоже должны быть до-
статочно велики. Часто, но не всегда
эти области совпадают с областью
максимального поглощения каждо-
го из компонентов. Молярные коэффициенты поглощения опреде-
ляются заранее, поэтому анализ сводится к определению оптиче-
ской плотности при двух выбранных длинах волн.
Особый интерес представляют спектральные участки, в кото-
рых одно из веществ свет не поглощает, а другое обладает интен-
сивным светопоглощением. Если, например, ев^ = 0, то вместо
(3.22) будем иметь:
4е
1
ад2
Как видно, при наличии такой области упрощаются расчеты и
повышается точность определения интенсивно поглощающего
вещества. Этот случай реализуется, например, при определении
хрома и марганца. В области длин волн 550 нм поглощает только
MnOj и он может быть определен по оптической плотности рас-
твора при этой длине волны. При 430 нм свет поглощают оба ком-
понента (MnOj и CrOf").
Довольно распространенным примером такого анализа явля-
ется также определение с помощью реагента, имеющего собст-
венную окраску. Этот метод может быть распространен и на более
сложные многокомпонентные смеси. При подчинении светопо-
глощения отдельных компонентов закону Бугера—Ламберта—
85
Бера и соблюдении закона аддитивности светопоглощения число
слагаемых в уравнениях типа (3.21) увеличивается пропорци-
онально числу определяемых компонентов и соответственно воз-
растает число уравнений. Для решения систем таких уравнений с
успехом используются компьютеры.
3.5.5. Фурье-спектроскопия
Существенным недостатком изложенного, так называемого
традиционного способа регистрации спектра является его инерци-
онность. Аналитический сигнал рецептора непрерывно записыва-
ется при изменении частоты (длины волны) падающего света в те-
чение довольно длительного времени. Этого недостатка лишен ме-
тод Фурье-спектроскопии, позволяющий записывать спектр в
широкой области частот нередко в течение нескольких секунд.
В методе Фурье-спектроскопии используется то, что излучению
каждой длины волны соответствует определенная интерференцион-
ная кривая (интерферограмма). Период такой функции зависит
только от длины волны. Кривая получается с помощью интерферо-
метра. Излучение со множеством частот — интерферограмма —
представляет собой сумму кривых, соответствующих каждой часто-
те (длине волны) в спектре излучения. Преобразование Фурье (гар-
монический анализ) позволяет преобразовать интерферограмму в
спектр, т. е. выделить кдждую частоту. Эта сложная математиче-
ская процедура выполняется с помощью компьютера.
Таким образом, в Фурье-спектрометрах спектр получается в
два этапа. Сначала регистрируют интерферограмму, представ-
ляющую собой наложение отдельных компонент спектра (гармо-
нических составляющих). Частоты этих составляющих связаны с
волновыми числами, а амплитуды — с интенсивностью линий.
Затем с помощью комцьютера проводят разложение интерферо-
граммы на частотные составляющие путем обратного преобразо-
вания Фурье.
Достоинством Фурье-спектроскопии является быстродейст-
вие — интерферограмма записывается в запоминающее устройство
компьютера в течение примерно одной секунды. В Фурье-спектро-
скопии нет щелей и не требуется фокусировка света, необходимая
при обычном способе регистрации спектра. Прибор характеризует-
ся высокой светосилой, поскольку через него проходит все излуче-
ние и высокой разрешающей способностью при одновременной ре-
гистрации большого числа спектральных линий. Эти достоинства
наиболее полно проявляются в ИК-спектроскопии при исследова-
нии процессов как поглощения, так и излучения.
86
3.5.6. Фотометрическое титрование
В методе фотометрического титрования точка эквивалент-
ности определяется с помощью фотометрических измерений.
В ходе такого титрования измеряется светопоглощение раствора.
Естественно, реализация этого метода возможна, если имеется
подходящий индикатор или хотя бы один из компонентов реак-
ции титрования поглощает свет. Например, при титровании же-
леза(П) дихроматом кривая фотометрического титрования имеет
вид, изображенный на рис. 3.17. До точки эквивалентности (т. э.)
оптическая плотность раствора практически не изменяется, а
после точки эквивалентности она линейно возрастает пропорци-
онально объему добавленного дихромата. Точку эквивалентности
находят графически. Для этого достаточно иметь несколько то-
чек, характеризующих недотитрованный раствор, и несколько
точек для перетитрованного раствора.
При титровании разбавленных или слабоокрашенных рас-
творов кривая титрования не имеет резкого минимума. Для на-
хождения точки эквивалентности в таких системах приходится
применять более сложные графические построения или специ-
альную математическую обработку.
Основным достоинством метода фотометрического титрова-
ния является возможность анализа слабоокрашенных и разбав-
ленных растворов, которые часто невозможно оттитровать други-
ми методами, а также возможность автоматизации самого про-
цесса титрования.
В ходе титрования стандартный раствор выдавливается из бю-
ретки 1 поршнем, приводимым в движение мотором 2 (рис 3.18).
Интенсивность света, прошедшего через светофильтр 6 и раствор 5,
фиксируется фотоэлементом 4, соединенным с мотором через реле 3.
При заданном значении оптической плотности по сигналу фотоэле-
мента реле срабатывает и выключает мотор, связанный с поршнем
бюретки, прекращая титрование. Результаты анализа рассчитывают
обычным путем по объему раствора, израсходованного на титрова-
Рис. 3.17. Кривая фотометрического ' Рис 3.18. Принципиальная схема
титрования железа(П) дихроматом ♦ фотометрического автотитратора
87
ние. Известны также приборы, в которых вычерчивается полная
кривая титрования, включая перетитрованные растворы, и диффе-
ренциальная кривая титрования в координатах |~ как функция V.
3.5.7. Определение неокрашенных соединений
Неокрашенные соединения, имеющие полосу поглощения в
ультрафиолете (например, молибдат-ион), могут быть определе-
ны по светопоглощению в этой области длин волн, используя
приведенные методики. Однако некоторые бесцветные соедине-
ния могут быть определены и без привлечения УФ-спектров с
применением обычной техники фотометрических измерений в
видимом участке спектра.
Одним из таких методов является использование растворимос-
ти малорастворимых соединений с окрашенными или образующим
окрашенное соединение анионом, таким, как СгО|~, РО |"и др. На-
пример, хлорид-ион может быть определен по реакции
Ад2Сг04 + 2С1- = 2АёС1 + СгО|"
При соблюдении необходимых условий в растворе появится
эквивалентное содержанию хлорида количество СгО|~-ионов, ко-
торые и фотометрируются. Константа этого равновесия
[СгО|г] _ ПРА?2Сг04 _^ 1,1 .ю-12 • 7
[С1"]2 ПР1,С1 (1,8 • 10-Ю)2 \
гарантирует протекание реакции в 5 • 10~3 М растворе хлорида
не менее, чем на 99,9%.
Аналогично можно провести реакцию с фосфатом:
А£3Р04 + ЗС1- = ЪАёЫ + РО|~
Выделившийся фосфат фотометрически определяют по реакции с
молибдатом.
Другим приемом анализа бесцветных соединений является
реакция бесцветного иона с окрашенным комплексным соедине-
нием, например обесцвечивание тиоцианатного комплекса желе-
зами) под действием фторид-ионов. Простейшая схема этого про-
цесса без учета ступенчатых равновесий имеет вид:
Ре8С*Г2+ + Р" = ¥е¥2+ + вСК-
Большое значение при проведении такого типа реакций име-
ет рН и устойчивость окрашенного комплекса. С салицилатным
или сульфосалицилатным комплексом Ре(Ш) реакция, очевидно,
не пройдет.
88
3.5.8. Фотометрический метод
исследования реакций в растворе
Простота и достаточная точность фотометрических измере-
ний привели к широкому использованию фотометрических мето-
дов для исследования реакций в растворе и особенно цветных ре-
акций, имеющих химико-аналитическое значение. Для опреде-
ления состава соединений часто применяется метод изомолярных
серий. При использовании этого метода готовят серию растворов,
в которых отношение концентрации центрального иона к кон^
центрации лиганда (см : сь) изменяется от 9 : 1 до 1 : 9, а суммар-
ная концентрация (см + сь) остается одинаковой во всех раство-
рах (изомолярные серии). Затем измеряют оптическую плотность
растворов и строят график зависимости оптической плотности от
концентрационного отношения см: сь. Максимум на этом графи-
ке указывает состав комцлекса. Метод изомолярных серий имеет
ограничения и недостатки, однако является одним из наиболее
широко применяемых в практике.
Устойчивость соединений по данным фотометрических изме-
рений может быть рассчитана графическими или аналитически-
ми методами. Если, например, удается определить молярный ко-
эффициент поглощения е комплекса МЬ, то константа устойчи-
вости может быть рассчитана по соотношению
в - А/(г1)
Рмь [с^- (А/(е1))][с1- (пА/(в1))]9
где А — оптическая плотность раствора; и с£ — исходные кон-
центрации растворов центрального иона и лиганда.
При этом предполагается, что кислотно-основные равновесия
в исследуемых растворах осложнений не вызывают, т. е. централь-
ный ион не гидролизован и лиганд не взаимодействует с протоном.
Однако столь идеальные условия при исследовании реаль-
ных систем выполняются довольно редко, да при современном со-
стоянии вычислительной техники они существенного значения
не имеют. Современные методы расчета констант устойчивости
комплексных соединений используют специальные программы,
реализуемые с помощью персональных компьютеров. Програм-
мы обычно основаны на принципе максимального правдоподобия
и учитывают как ступенчатые кислотно-основные равновесия в
растворах многоосновных кислот, так и процессы комплексооб-
разования. Константы протолитических равновесий, в которых
центральный ион участия не принимает, как правило, определя-
ются независимо. Выбор спектральной области для исследования
также выбирается по программе, учитывая возможные наложе-
ния полос поглощения и другие осложнения.
89
3.6. Практическое применение
Фотометрические и спектрофотометрические методы анали-
за применяются для определения многих (более 50) элементов пе-
риодической системы, главным образом металлов. Методами аб-
сорбционной спектроскопии анализируются руды, минералы и
иные природные объекты, продукты переработки обогатитель-
ных* и гидрометаллургических предприятий. Эффективно ис-
пользуются эти методы в металлургической, электронной, хими-
ческой и других отраслях промышленности, в медицине, биоло-
гии и т. д. Большое значение они имеют в аналитическом
контроле загрязнений окружающей среды и решении экологиче-
ских проблем. Значительно расширились области практического
применения методов абсорбционной спектроскопии благодаря бо-
лее широкому использованию инфракрасной области спектра,
Фурье-спектроскопии и приборов со встроенным компьютером.
Это позволило разработать методы анализа сложных многоком-
понентных систем без их химического разделения.
Успешно развиваются спектрофотометрические методы ана-
лиза неметаллов и органических соединений. Разработаны фото-
метрические методы определения аминного и общего азота, бора,
фосфора, кремния, мышьяка, кислорода, серы, галогенидов и
других элементов. Большое практическое значение имеет фото-
метрический анализ органических соединений; который часто
применяется в сочетании разделением сложных смесей методами
экстракции или распределительной хроматографии. Здесь следу-
ет назвать анализ аминокислот, лекарственных препаратов, кра-
сителей ит. д. Методы абсорбционной спектроскопии продолжа-
ют развиваться и совершенствоваться.
3.7. Общая характеристика метода
Методы абсорбционной спектроскопии имеют высокую чувстви-
тельность (низкий предел обнаружения), они избирательны и точ-
ны. Методы могут; быть применены для анализа больших и малых
содержаний, но особенно ценной их особенностью является возмож-
ность определения примесей (до 10~5—10_6%). Важное значение
имеет избирательность многих фотометрических методов, позво-
ляющая проводить определения элементов в сложных пробах без
химического разделения компонентов. Погрешность фотометриче-
ских методов обычно составляет 3—5%, уменьшаясь в благоприят-
ных случаях до 1—2% и нередко до 0,5—1,0%. Методы абсорбцион-
90
ной спектроскопии используют в химической, металлургической,
металлообрабатывающей и других отраслях промышленности, гор-
ном деле, сельском хозяйстве, медицине й т. д.
Простые, быстрые и точные фотометрические методы анализа
применяются для контроля производства, определения примесей
и решения многих других важных вопросов в заводских и науч-
но-исследовательских лабораториях. Большое значение имеют
эти методы для исследования различных реакций, установления
состава и устойчивости образующихся соединений. Успехи химии
координационных соединений и достижения приборостроения да-
ют все основания ожидать дальнейшего повышения точности и
чувствительности этих методов.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Что называют коэффициентом пропускания Т и оптической
плотностью А? В каких пределах изменяются эти величины?
2. Какими уравнениями выражается основной закон светопоглоще-
ния Бугера—Ламберта—Бера?
3. Что означает свойство аддитивности оптической плотности?
4. Действие каких факторов может привести к нарушению линейной
зависимости оптической плотности от концентрации раствора?
5. Каков физический смысл молярного коэффициента поглощения?
Какие факторы на него влияют: а) длина волны проходящего света;
б) температура; в) концентрация раствора; г) природа вещества?
6. Что называют спектром поглощения вещества и в каких коорди-
натах его можно представить?
7. Какие величины входят в уравнение, характеризующее полосу
поглощения?
8. Какова природа светопоглощения в ультрафиолетовом, видимом
и инфракрасном участках спектра?
9. При каких оптимальных значениях Т и А обеспечивается на-
именьшая относительная погрешность измерения?
10. В чем сущность метода градуировочного графика и каковы его
особенности?
11. Какова сущность метода добавок? Как рассчитывается концентра-
ция определяемого вещества этим методом с помощью графика?
12. В каких случаях используют метод дифференциальной фотомет-
рии и каковы особенности этого метода?
13. На чем основано фотометрическое определение смеси окрашен-
ных веществ без их предварительного разделения?
14. Каковы особенности инфракрасных спектров? Какова природа
поглощения в инфракрасном участке спектра?
15. В чем сущность количественного анализа в ИК-спектроскопии
по методу базовой линии?
91
глава 4
Атомно-абсорбционный
спектральный анализ
4.1. Теоретические основы метода
Атомно-абсорбционный спектральный анализ предложен
Уолшем в 1955 г. Метод сразу получил признание. ,
При поглощении кванта света Н\ свободный атом А перехо-
дит в возбужденное состояние А*:
I А + = А*
где Л — постоянная Планка; у — частота, определяемая условием
частот Бора,
ЕА* и ЕА — энергия атома в возбужденном и основном состояни-
ях соответственно.
Наиболее вероятным изменением энергетического состояния
атома при возбуждении является его переход на уровень, бли-
жайший к основному энергетическому состоянию, т. е. резонанс-
ный переход. Если на невозбужденный атом направить излуче-
ние с частотой, равной частоте резонансного перехода, кванты
света будут поглощаться атомами и интенсивность излучения бу-
дет уменьшаться. Использование этих явлений составляет физи-
ческую основу атомно-абсорбционной спектроскопии. Таким об-
разом, если в эмиссионной спектроскопии концентрация вещест-
ва связывалась с интенсивностью излучения, которое было
прямо пропорционально числу возбужденных атомов, то в атом-
но-абсорбционной спектроскопии аналитический сигнал (умень-
шение интенсивности излучения) связан с числом невозбужден-
ных атомов.
Заселенность энергетических уровней так же, как и в эмис-
сионной спектроскопии, определяется уравнением (2.10). Расче-
ты показывают, что число атомов в возбужденном состоянии не-
значительно по сравнению с числом атомов на основном (ниж-
нем) уровне и не превышает 1—2% от общего числа атомов. Это
выгодно отличает атомно-абсорбционный анализ от эмиссионно-
го, так как при прочих равных условиях величина аналитическо-
го сигнала оказывается связанной с большим числом атомов, ч^м
92
в эмиссионной спектроскопии, и, следовательно, величина сигна-
ла в меньшей степени будет подвержена влиянию случайных ко-
лебаний в режиме работы различных узлов ^томно-абсорбцион-
ного спектрофотрметра.
4.2. Основные узлы приборов
для атомно-абсорбционного анализа
Принципиальная схема установки атомно-абсорбционной
спектроскопии приведена на рис. 4.1.
Источником излучения является обычно лампа с полым като-
дом) содержащим определяемый элемент. Катод такой лампы из-
готовляют в виде металлического стаканчика, в котором происхо-
дит испарение .вещества и возбуждение атомов элементов при
электрическом разряде в атмосфере инертного газа под неболь-
шим давлением (~102 Па). Катоды, изготовленные из элементов с
относительно низкими температурами плавления, легко разруша-
ются. Для определения таких элементов используют графитовые
катоды, пропитанные солями определяемых элементов. Анод в
виде металлического стержня размещают рядом с катодом и оба
электрода помещают в стеклянный баллон со стеклянным или
кварцевым окошком. Лампа питается током от высокоточного вы-
прямителя-стабилизатора, дающего напряжение 500—600 В с ко-
лебаниями, не превышающими сотых долей процента.
Пары материала катода и других веществ* находящихся на
внутренней поверхности катода, попадают в плазму вследствие
катодного распыления и испарения в процессе разряда при 200—
300 В и 5—30 мА, В спектре свечения при температуре около 800
К в полом катоде наблюдаются резонансные частоты этих элемен-
тов. Применяются также лампы с СВЧ-возбуждением (СВЧ-лам-
пы) для определения, например, мышьяка, сурьмы, висмута,
Рис 4.1. Схема атомно-абсорбционного спектрофотометра:
1—источник излучения; 2 — пламя; 3 — монохроматизатор;
4 —' приёмник света; 5 — анализируемый раствор
93
свинца и некоторых других элементов. Анализируемое вещество в
виде раствора подается в пламя горелки, где при 2000—3000 °С
происходит испарение растворителя й атомизация пробы.
Поскольку уменьшение интенсивности излучения пропорци-
онально толщине светопоглощающего слоя, горелки имеют спе-
циальную конструкцию, обеспечивающую постоянную и доста-
точно большую длину поглощающего слоя пламени (5—10 см).
В последнее время широкое распространение в атомно-аб-
сорбцйбнной спектроскопии получили непламенные электротер-
мические атомизаторы. Хотя электропечи для атомизации при-
меняются сравнительно давно, начало практическому использо-
ванию их в атомно-абсорбционной спектроскопии было положено
Б. В. Львовым в 1961 г. Графитовая кювета Львова представляет
собой трубчатую печь с графитовым электродом в центре. Проба в
виде раствора или порошка наносится на торец графитового
электрода за счет мощного дугового разряда при большой силе
тока и низком напряжении мгновенно испаряется. Термиче-
ски-временной режим работы в современных конструкциях элек-
тротермических атомизаторов устанавливается с помощью
компьютеррв по заранее заданной программе; что значительно
повысило воспроизводимость условий атомизации, и, как следст-
вие, привело к повышению точности определений. Применение
электротермических атомизаторов значительно расширило воз-
можности атомно-абсорбционной спектроскопии и привело к
улучшению химико-аналитических характеристик метода: бо-
лее, чем на 2—3 порядка снизился предел обнаружения, стало
возможным применение проб меньшего объема, расширился
спектральный интервал измерений за счет вакуумного ультрафи-
олета. Известны и другие конструкции электротермических ато-
мизаторов.
В качестве монохроматизатбров применяют призмы или
дифракционные решетки. В качестве приемника света использу-
ют фотоэлементы или фотоумножители.
Комплектные приборы для атомно-абсорбционной спектро-
скопии выпускаются во многих странах. В настоящее время из-
вестно свыше 50 моделей таких спектрофотометров.
В нашей стране выпускаются пламенные спектрофотометры
под общим названием «Спектр» (последние модели «Спектр-5» и
«Спектр-5-1»), Более совершенным является прибор КАС-120, в
состав которого входят спектрофотометр и компьютер с набором
программ, позволяющим оптимизировать условия измерения.
Результат анализа в атомно-абсорбционных методах зависит
главным образом от числа невозбужденных атомов, которое в из-
94
вестных пределах сравнительно мало изменяется с температу-
рой/Это уменьшает эффекты взаимного влияния компонентов
пробы на аналитический сигнал. В эмиссионной спектроскопии
результат анализа определяется, в основном, числом возбужден-
ных атомов, доля которых невелика и существенно зависит даже
от небольших колебаний температуры. Поэтому требования ста-
бильности условий работы источника возбуждения, в эмиссион-
ной спектроскопии выступающее на первый план, в атомно-аб-;
сорбционной спектроскопии решающего значения не имеют* хо-
тя, конечно, стабилизация условий необходима.
В атомно-абсорбционной спектроскопии практически полно-
стью исключена возможность наложения линий различных эле-
ментов, так как в условиях атомнр-абсорбционного анализа чис-
ло линий в спектре значительно меньше, чем в эмиссионной
спектроскопии.
4.3. Количественные определения
Уменьшение интенсивности резонансного излучения в усло-
виях атомно-абсорбционной спектроскопии подчиняется экспо-
ненциальному закону убывания интенсивности в зависимости от
длины слоя и концентрации вещества, аналогичному закону Бу-
гера—Ламберта—Вера. Если /0 — интенсивность падающего мо-
нохроматического свфта, а / — интенсивность этого света, про-
шедшего через пламя, то величину ^ (10/1) можно назвать опти-
ческой плотностью. Концентрационная зависимость оптической
плотности выражается уравнением
1ё(16/1)=А = Ыс, (4.1)
где к — коэффициент поглощения; / — толщина светопоглощаю-
щего слоя (пламени); с — концентрация.
Постоянство толщины светрпогл бщающего слоя, т. е. пламе-
ни, достигается с цомощью горелок специальной конструкции.
Оптическая плотность согласно уравнению (4.1) прямо про-
порциональна концентрации вещества. Опыт показывает, что за-
висимость оптической плотности от концентрации часто оказы-
вается не строго линейной. Отклонения от линейности вызы-
ваются несколькими причинами, среди которых наиболее суще-
ственное значение имеют такие, К£К нестабильность работы раз-
личных узлов спектрофотометра (источника возбуждения и др.),
немонохроматичность линий испускания, вызванная сверхтон-
кой структурой, образование в пламени различных соединений
95
определяемых элементов с кислородом или сопутствующими эле-
ментами и т. д. В практике анализа обычно применяют метод
градуировочного графика и метод добавок.
Количественные определения методом атомной абсорбции
проводятся по следующей схеме:
1. Растворение пробы (параллельно подготовка стандартного
раствора) и введение растворов в виде аэрозоля в пламя горелки
или испарение в графитовом анализаторе с целью получения све-
топоглощающего атомарного пара.
2. Облучение атомарного пара источником света.
3. Разложение света, прошедшего через атомарный слой и
выделение линии поглощения.
4. Оценка степени поглощения света выделенной длины волны
(оптической плотности) стандартных и анализируемых растворов.
5. Определение градуировочной характеристики и расчет
концентрации определяемого компонента.
В методе градуировочного графика измеряют оптическую плот-
ность нескольких стандартных растворов и строят график в коорди-
натах оптическая плотность-^концентрация. Затем в тех же усло-
виях определяют оптическую плотность анализируемого раствора и
по градуировочному графику находят его концентрацию.
При работе по методу добавок сначала измеряют оптиче-
скую плотность анализируемого раствора (Ад.), затем вводят в
анализируемый раствор определенный объем стандартного рас-
твора и снова измеряют оптическую плотность (АХ + СТ). Если сх —
концентрация анализируемого раствора, а сст — стандартного, то
■ Ах = Ысх;
Ах + ст = Ь1(сх + сст).
Учитывая, чтоки1 одинаковы, получаем
Ах.. ._ .. Сх
Ах + ст Сх + сст ' <* '
И окончательно
Ах + ст л*
Метод применим для систем, подчиняющихся в исследуемой
области концентраций уравнению (4.1). Можно использовать
также графический метод нахождения сх на основе уравнения
(4.2), откладывая на графике Ах + ст как функцию сст. Щ>иАх + ст = О
96
4.4. Практическое применение
Методы атомно-абсорбционной спектроскопии могут быть ис-
пользованы или используются в анализе практически почти любого
технического или природного объекта, особенно там* где необходи-
мо определять небольшие содержания элементов. Методики атом-
но-абсорбционного определения разработаны более чем для 70 эле-
ментов (Мб, гп, Си, Са, РЬ, Ре, Ag, №, Hg, СИ,- В1 и др.). Из тех-
нических объектов методами атомно-абсорбционной спектроскопии
анализируют металлы, сплавы, продукты гидрометаллургической
переработки руд, различные концентраты и т. д. Например, в зо-
лоте определяют серебро, свинец, медь и цинк при содержании
10~4%. Примерно такие же концентрации кадмия й свинца нахо-
дят в цирконии. Успешно применяются атомно-абсорбционные ме-
тодики для определения цинка, железа, магния, меди и некоторых
других элементов в почвах, удобрениях, растениях и других агро-
химических материалах при содержании порядка 10~4 или 10~5%.
Атомно-абсорбционный метод используется также в клинических и
различных биологических анализах (кровь, сыворотка и т. д.) ?на
свинец, ртуть, висмут и другие элементы. Дальнейшего расшире-
ния областей практического применения атомно-абсорбционной
спектроскопии можно ожидать при более широком использовании
графитовой кюветы и высокотемпературных пламен.
4.5. Общая характеристика метода
Атомно-абсорбционный спектральный анализ получил широ-
кое распространение в практике вследствие многих своих досто-
инств. Важным достоинством атомно-абсорбционного метода яв-
ляется наличие менее жестких требований, чем в эмиссионной
спектроскопии, к условиям получения поглощающей плазмы, по-
скольку аналитический сигнал зависит от числа невозбужденных
атомов, которое сравнительно мало меняется при небольших коле-
баниях температуры. Существенно также, что число линий- в
спектре в условиях атомно-абсорбционного анализа невелико, по-
этому наложения аналитических линий практически не происхо-
дит, хотя неселективное поглощение остаётся значительным. Пре-
дел обнаружения с помощью атомно-абсорбционного анализа при
электротермической атомизации составляет величину (мкг/мл)
порядка 10~5 (Са, Се, Аё, Мп и др.) или 10~6 (Ве, Zn, Mg9 и др.).
При пламенной ионизации для большинства элементов он увели-
чивается на 2—3 порядка (Ag, СсЦ Яп, Си, Ре, N1 и др.) и еще боль-
ше (А1, РЬ, Ва, Т1, V и др.). Погрешность определения обычно со-
7-7831
97
ставляет примерно 5% и в зависимости от различных условий из-?
меняется в пределах от 3 до 10% .
{Метод имеет также ряд ограничений. Атомно-абсорбцион-
ным методом не определяются элементы, резонансные линии ко-
торых лежат в далеком ультрафиолете (углерод, фосфор, галогет
ны и др.). Необходимость растворения, .пробы также можно рас-?/
сматривать как недостаток, поскольку эта операция удлиняет?
ацализ. Однако работа с растворами упрощает эталонирование и
обеспечивает высокую воспроизводимость результатов. К сущест-
венным недостаткам метода относится невозможность одновре-
менного определения нескольких элементов, хотя для этого име-
ются все предпосылки. Необходимо отметить также, что помимо
чисто аналитического применения атомно-абсорбционная спект-
роскопия используется для определения силы осциллятора, ко-
эффициентов диффузии, давления насыщенных паров и т. д.
контрольные вопросы
1. В чем сущность атомно-абсорбционного анализа? Что является ,
аналитическим сигналом в этом методе?
2. Какие достоинства и недостатки характерны для методов атом-
но-абсорбционного анализа и эмиссионной спектроскопии?
• 3. Почему величина аналитического сигнала в методе атомно-аб-
?! сорбционного анализа меньше подвержена влиянию случайных
колебаний в работе прибора, чем в эмиссионной спектроскопии?
. 4, Какие особенности имеют источники излучения в методе атом-
. <т но-абсорбционнрго анализа? ,
5. Чем можно объяснить нелинейную зависимость поглощения от
концентрации в атомно-абсорбционном анализе?
6. Что ограничивает применение метода атомно-абсорбционного
анализа в целях качественного анализа?
г л а в а 5
Люминесцентный анализ
5.1. Спектры люминесценции
Согласно определению С. Ц. Вавилова, люминесценцией на-
зывают свечение, избыточное над температурным и обладающее
длительностью не менее чем 10~*° с, что превышает период свето-
98
вых колебаний. От излучения нагретых тел она отличается своей
неравновесностью: люминесценция практически не использует
тепловую энергию излучающей системы, поэтому ее часто назы-
вают холодным светом. Это определение отличает люминесцен-
цию также от всех другик видов неравновесного свечения — рас-
сеяния и отражения света,* комбинационного рассеяния, излуче-
ния Вавилова—Черенкова и т. д.
Люминесценция возникает в результате электронного пере-
хода при возвращении частиц из возбужденного состояния в нор-
мальное. Таким образом, молекула преобразует поглощенную
энергию в собственное излучение. Этим люминесценция также
отличается от процессов несобственного излучения — рассеяния
и отражения света. Люминесцирующие вещества могут нахо-
диться в любом агрегатном состоянии.
В возбужденное состояние частицы люминесцирующего ве-
щества могут переходить под действием света и тогда люминес-
ценцию называют фотолюминесценцией (флуоресценцией или
фосфоресценцией)у иод действием рентгеновского излучения —
рентгенолюминесценцией, в результате химической реакции —
хемилюминесценциейИТ. д.
Происхождение люминесцентного излучения поясняется
схемой на рис. 5.1. Здесь схематически изображены основной и
возбужденные {синзлетный и триплетный) электронные уров-
ни молекулы. На каждый электронный /уровень накладываются
колебательные подуровни с квантовыми числами 0, 1,2, 3 и т. д.
При поглощении кванта света электрон переходит с основного
У = 3
У = 2
У = 1
= 0 ■
К=3
У= 1
^=0-
Синглетнйй уровень
Триплетный уровень
■Vм = 3
'V" = 2
■ V" = 1
•V" - 0
Рис. 5.1. Схема энергетических уровней молекулы, поясняющая
: возникновение люминесцентного излучения:.
Е0, Е1 ^ электронные уровни; V, V — колебательные
99
уровня на более высокий, соответствующий возбужденным синг-
летному (антипараллельные спины) и триплетному (параллель-
ные спины) состояниям. Прямой переход основного синглетного
в возбужденное триплетное состояние запрещен по спину и прак-
тически не наблюдается. Энергия триплетного состояния не-
сколько меньше, чем синглетного. Триплетные уровни могут за-
полняться за счет интеркомбинационной конверсии (волнистая
стрелка У0 = 0 —►У = 2). При комнатной температуре молекулы
обычно находятся в основном состоянии и почти все электронные
переходы при поглощении света происходят с нижнего (основно-
го) колебательного подуровня на различные колебательные под-
уровни возбужденного синглетного состояния. На рис. 5.1 эти
переходы изображены стрелками: V = 0 V7 = 0;-У — 0 —► У .'— 1;
У=0 —У = 2ит. д.
Возбужденная молекула за счет так называемой колебатель-
ной релаксации при столкновении с окружающими молекулами
очень быстро по сравнению с временем электронного перехода те-
ряет избыточную колебательную энергию и переходит на основ-
ной колебательный уровень возбужденного электронного (синг-
летного) состояния. На рис .5.1 этот процесс изображен вол-
нистой стрелкой: У = 3 У = 0; У = 2 -+У = 0; У = 1 У = 0
и т. д. .
При переходе с основного колебательного подуровня возбуж-
денного синглетного состояния на какой-либо колебательный под-
уровень основного электронного (тоже синглетного) состояния про-
исходит излучение кванта света. Этот процесс называют флуорес-
ценцией. На рис. 5.1 ему соответствуют переходы = 0 У = 0;
УЬ = 0 -» У= 1; Уо = 0 -» У = 2 и т. д. Время затухания флуорес-
ценции составляет 10~9—10~7 с. Дезактивация возбужденной мо-
лекулы может происходить также за счет безызлучательных пе-
реходов внутренней конверсии. В триплетном так же, как и в
возбужденном синглетном состоянии, происходит колебательная
релаксация, и электрон переходит на нижний колебательный уро-
вень триплетного состояния (волнистая стрелка У1 — 2-V! — 0;
V = 1 -» V = 0). Запрещенный по спину излучательный три-
плет-синглетный переход (V = 0 У = 0; V = 0 -* у = 1 и т. д.)
называют фосфоресценцией. Время жизни триплетного состоя-
ния велико (10"3—102 с). Переход от триплетного состояния в ос-
новное синглетное происходит также при столкновении возбуж-
денной частицы с окружающими молекулами за счет безызлуча-
тельных процессов внутренней конверсии, вероятность которых
при комнатной температуре очень велика. По этой причине, что-
бы наблюдать фосфоресценцию и использовать ее в аналитиче-
100
ских целях, пробу обычно замораживают, часто при температуре
жидкого азота (77 К), что сводит до минимума вероятность без-
ызлучательного перехода. Спектр фосфоресценции сдвинут в длин-
новолновую сторону на величину, пропорциональную энергии ко-
лебательной релаксации триплетного состояния.
Так как в зависимости от частоты облучающего излучения
частица переходит в энергетически разные возбужденные состоя^
ния, можно было ожидать связь спектра флуоресценции со спект-
ром возбуждения источника. Однако в действительности эта зави-
симость обычно не наблюдается. Независимость спектра флуорес-
ценции от длины волны падающего света, в основном, связана с
тем, что возбужденные молекулы в результате колебательной ре-
лаксации успевают растратить колебательную энергию за время,
значительно меньшее, чем время жизни возбужденного состоя-
ния, и перейти на основной колебательный уровень возбужденно-
го электронного состояния. Переход такой системы в невозбуж-
денное состояние характеризуется испусканием одних и тех же
квантов света, т. е. наблюдается один и тот же спектр флуоресцед-
ции. В то же время в некоторых случаях это расширяет возмож-
ности люминесцентного анализа, давая возможность селективно-
го возбуждения определенных соединений.
Из рис. 5.1 видно, что между спектром поглощения вещества
и его спектром флуоресценции можно ожидать определенного
сходства, так как и тот и другой определяются, в сущности, одни-
ми и теми же электронными переходами./Сходство, действитель-
но, есть, причем, как установлено законом Стокса—Ломмеля,
спектр излучения в целом и его максимум всегда сдвинуты в сто-
рону более длинных волн по сравнению со спектром поглощения
и его максимумом.
Эта закономерность легко объясняется схемой, изображен-
ной на рис. 5.1, в соответствии с которой энергия квантов флу-
оресценции должна быть меньше энергии поглощенных квантов
на величину энергетических потерь при колебательной релакса?
ции в возбужденном электронном состоянии и перехода на неос-
новной колебательный подуровень основного электронного со-
стояния. Спектры люминесценции большинства веществ харак-
теризуются широкими полосами излучения (100—200 нм). Если,
однако, вещество исследуют в условиях сильного охлаждения (до
температур жидкого азота 77 К или водорода 20 К), спектры ста-
новятся квазилинейчатыми с разрешенной колебательной струк-
турой (эффект Шпольского). Этот эффект проявляется не у всех
веществ, так как у многих соединений остаются вклады за счет
внутримолекулярного и других видов взаимодействия.
101
5,1.1. Энергетический и квантовый
выходы люминесценции
Эффективность преобразования энергии поглощенного света
в энергию люминесценции характеризуется энергетическим и
квантовым выходами люминесценции. Отношение излучаемой
энергии люминесценции к энергии поглощенного света называют
энергетическим выходом люминесценции, а отношение числа из-
лучаемых квантов к числу поглощенных называют квантовым
выходом люминесценции.
Если Вэн —энергетический, а Вкв — квантовый выход люми-
несценции, Ел и Ес — соответственно энергия люминесценции и
энергия поглощенного света, а Ыл и Nc — число испускаемых и
поглощенных квантов, то очевидно, что
Связь между Вэн и Вкв легко установить, если учесть, что
энергия N квантов равна Е = Nhv:
эн Nchvc KBv/
Зависимость энергетического выхода люминесценции от дли-/
ны волны возбуждающего света подчиняется закону Вавилова.
В соответствии с этим законом энергетический выход люминес-
ценции с увеличением длины волны возбуждающего света снача-
ла возрастает пропорционально длине волны, затем остается по-
стоянным и после достижения некоторой граничной длины вол-
ны резко падает.
Учитывая пропорциональность энергетического выхода дли-
не волны возбуждающего света
и соотношение X = c/v, получаем
Вкв = В9а^ = А^=А^=хХл = const, ¿1)
ул ул ул
т. е. пропорциональность энергетического выхода длине волны
поглощенного света (5.1) означает постоянство квантового выхо-
да люминесценции в этом спектральном интервале. Очевидно,
чем больше квантовый выход люминесценции, тем меньшее ко-
личество люминесцирующего вещества может быть обнаружено
по его свечению. ' ч
102
5.îi2. Интенсивность люминесценции
Интенсивность люминесценции /л пропорциональна числу
излучаемых квантов А^л:
. I^kN^kB^, (5,2)
где х — коэффициент пропорциональности.
Число поглощенных квантов Nc пропорционально интенсив-
ности поглощенного света:
JVc = x'(J0-/), (5.3)
где /0 — интенсивность падающего света; 7 — интенсивность све-
та, прошедшего через раствор; х' ■— коэффициент пропорци-
ональности.
Величины I и /0 связаны уравнением закона Бугера—Лам-
берта—Вера:
/ = /010-^. (5.4)
Сочетание уравнений.(5.3) и (5.4) приводит к соотношении*
ЛГс = х,/0(1-10-^). (5.5)
Подставляя уравнение (5.5) в (5.2), получаем
. /л = хх,Вкв/0(1-Ю-^). (5.6)
Разложение 10"е/с в ряд дает !
10-" = 1 - 2,3eZc + <2'30е/с>2 - . (5.7)
¿1 о!
При sic < 10~2 вклад третьего и последующих членов разло-
жения становится пренебрежимо малым. Ограничиваясь при
этом условии двумя первыми членами (5.7) и подставляя резуль-
тат разложения в уравнение (5.6), находим
/л = 2,Зхх,Вкв1об/с
или, объединяя постоянные величиныj
1л = Ъс. (5.8)
Линейная зависимость интенсивности люминесценции от
концентрации будет соблюдаться при постоянстве таких факто-
ров, как квантовый выход, интенсивность возбуждающего света
и т. д. Также существенным является условие низкой концентра-
ции люминесцирующего вещества.
103
С увеличением концентрации условие е1с < 10~2 будет нару-
шаться и зависимость интенсивности люминесценции рт кон-
центрации будет отклоняться от линейной. При достаточно боль-
шой концентрации интенсивность люминесценции вообще может
уменьшаться, т. е. может начаться так называемое концентраци-
онное тушение люминесценции. В этой связи верхний предел
концентрации раствора в люминесцентном анализе обычно не
превышает 10"3—10~4 моль/л.
5.1.3. Тушение люминесценции
Концентрационное тушение люминесценции по С. И. Вави-
лову в широком интервале концентраций во многих случаях опи-
сывается уравнением
В = В0 ехр [-Цс - с0)],
где В и В0 — выход люминесценции при концентрации сис^О;,
с0 —- пороговая концентрация, по достижении которой развива-
ется концентрационное тушение; к — постоянная.
,'. Однако это уравнение оказывается применимым не всегда.
Существенное значение имеет температурное тушение лю-:
минесценций, т. е. уменьшение выхода свечения с повышением
температуры. В той или иной степени это явление свойственно
всем люминесцируюнщм веществам. Оно объясняется тем, что с
повышением температуры увеличивается колебательная энер-
гия молекул и возрастает вероятность безызлучательных перехо-
дов, а также вероятность диссоциаций возбужденных частиц,
происходящая без излучения квантов света.
Отрицательное влияние на выход люминесценции оказыва-
ют многие примеси. Тушение люминесценции лежит в основе не-
скольких методик их определения.
5.1.4. Люминесценция кристаллофосфоров
Кристаллофосфорами называют сложные неорганические
кристаллы, способные люминесцировать. К ним относятся, на-
пример, кристаллы на основе сульфидов кальция, стронция и их
смесей, сульфата или вольфрамата кальция, бромида или иодида
натрия и других соединений.
Некоторые неорганические кристаллы приобретают способ-
ность люминесцировать при внедрении в их решетку посторон-
них элементов, так называемых активаторов.
104
Люминесценция кристаллофосфоров зависит от многих факто-
ров. При повышении температуры их свечение уменьшается. Это
тоже проявление температурного тушения люминесценции. Силь-
ное тушащее действие на свечение кристаллофосфатов оказывают
ничтожные (10"~б—10"6%) примеси многих веществ (например, со-
единения железа, никеля, кобальта и др.). Эти вещества часто на-
зывают люминесцентными ядами. Однако при некоторых услови-
ях и концентрационных соотношениях люминесцентные яды ста-
новятся активаторами свечения.
Зависимость интенсивности свечения кристаллофосфора от
концентрации используется в анализе.
5.1.5, Атомная флуоресценция
I Атомной флуоресценцией называют фотолюминесценцию газооб-
| разных атомов/возникающую при облучений атомов потоком света.
Если поток света содержит кванты, соответствующие воз-
буждению первого уровня, облучаемый атом перейдет в возбуж-
денное состояние, а затем при возвращений в основное будет ис-
пускать свет с длиной волны, соответствующей резонансному пе-
реходу. Этот процесс часто называют также резонансной
флуоресценцией.
Интенсивность атомной резонансной флуоресценции /фл за-
висит главным образом от трех основных факторов: интенсивнос-
ти возбуждающего излучения /с, числа атомов N в основном со-
стоянии в единице исследуемого объема пламени и квантового
выхода люминесценции Бкв:
/фл = *1^Бкв, '
где к — коэффициент пропорциональности, учитывающий гео-
метрические факторы и спектральные характеристики линии
(ширину, коэффициент поглощения и др.).
Хотя явление атомной флуоресценции изучал еще Вуд в 1905 г.,
аналитическое применение этот факт нашел лишь в 1964 г. в рабо-
тах Вайнфорднера.
5.1.6. Хемилюминесцентный анализ
Хемилюминесцентный анализ основан на измерений спектра
и интенсивности хемилюминесценции, т. е. свечения, возникаю-
щего за счет энергии химической реакции. В ходе экзотермиче-
105
ской реакции внутренняя энергия системы может быть выделена
не только в виде теплоты, но частично превращена в энергию воз-
буждения продуктов реакции. Излучение, испускаемое возбужг
денными таким образом частицами, получило название хемилюг
минесценции. Возбуждение хемилюминесценции в видцмом уча-
стке спектра происходит, если тепловой эффект реакции не менее
160 кДж/моль. Это обычно окислительно-восстановительные ре-
акции с участием свободных радикалов.
В хемилюминесцентном анализе используется окисление ве-
ществ, дающих яркую люминесценцию — окисление люминола,
лкщигенина и других соединений пероксидом водорода, гипохло-
ритом, бромом и другими окислителями. Разработаны методы
анализа, основанные на каталитическом или ингибиторном эф-
фекте различных веществ. Содержание ионов железа, кобальта,
м;еди, титана, циркония, платиновых металлов и других опреде-
ляется с пределом обнаружения 10"9 • Ю"8 г/мл, однако селек-
тивность методик невелика. ,
ь Высокой чувствительностью и большой селективностью об-
ладают, хемилюминесцентные методики газового анализа. Опре-
деление аммиака, оксидов азота и серы, сероводорода и других
имеет предел обнаружения порядка 10"7%. Изучение хемилюми-
несценции не требует внешних источников возбуждения и моно-
хроматора: излучение регистрируется обычным фотоэлектрон-;
ньгм умножителем.
5.2. Схема прибора
для люминесцентного анализа
; Схема прибора для проведения люминесцентного анализа
представлена на рис. 5.2. Свет от источника освещения! проходит
через светофильтр 2 и падает на кювету 3
с исследуемым раствором. Приемник све-
та 5 измеряет люминесцентное излучение
под прямым углом к направлению воз^;
буждающего света. Светофильтр 4 про-
4 5 пускает свет люминесценции и погло-
щает рассеянный свет от источника воз-
буждения.
В практике люминесцентного ана-
лиза исследуемое вещество обычно осве-
Рис. 5.2. Схема щают ультрафиолетовыми лучами. Наи-
флуориметра большее распространение среди различ-
(4
106
ных источников освещения, вызывающих люминесценцию, по-
лучили газоразрядные лампы, чаще всего ртутно-кварцевые и
ксеноновые. Приемником люминесцентного излучения может
служить глаз человека. В современных; приборах для количест-
венного анализа в качестве приемника излучения используют
фотоумножители.
5.3. Качественный анализ
Очень чувствительны люминесцентные качественные реак-
ции, когда добавление некоторых органических реагентов к рас-
твору неорганических веществ вызывает яркую люминесценцию.
Например, интенсивную люминесценцию вызывает добавление
салициловой кислоты к раствору соли цинка, что может быть ис-
пользовано для его качественного открытия. Для обнаружения
лития и алюминия люминесцентным методом предложен 8-окси-
хинолин, для открытия бериллия, циркония и других элементов
используют морин и т. д. Качественный люминесцентный анализ
основан на способности исследуемого вещества в соответствую-^
щих условиях люминесцировать или, реже, гасить люминесцен-
цию. Возникновение или исчезновение люминесценции обычно
наблюдается визуально.
Достоинством люминесцентных реакций является их исклю-
чительно низкий предел обнаружения. Например, ярко-зеленая
люминесценция соединения лития с 8-оксихинолином возникает
при содержании 0,5 мкг 1л в 5 мл, медь открывают по яркой синей
люминесценции ее соединения с салицилалазином при концент-
рации 0,255 мкг в 5 мл и т. д. Низким пределом обнаружения и
селективностью обладают и так называемые реакции получения
кристаллофосфоров (например, реакции образования перлов буры
и фосфатов).
Большое значение имеет люминесцентный анализ в биологии
и медицине для диагностики заболеваний рака, малярии и др.,
контроля за качеством лекарственных препаратов, анализа био-
логически активных веществ — витаминов, антибиотиков и т. д.
В сельском хозяйстве и пищевой промышленности люминес-
центный анализ используют для определения жизнеспособности
семян, анализа пищевых продуктов и т. д. Например, жизнеспо-
собные семена дают желтую люминесценцию, а нежизнеспособ-
ные — коричневую. По цвету люминесценции можно установить
сорт муки: чем больше в ней отрубей, тем интенсивнее свечение.
Люминесценция позволяет легко обнаружить начальную стадию
загнивания различны* овощбй и фруктов.
107
5.4. Количественный анализ
Практически определение компонента, образующего люйинёс-
цирующее соединение в растворе, проводят по следующей схеме:
1. Растворение пробы и перевод определяемого компонента в
люминесцирующее соединение.
2. Облучение пробы источником ультрафиолетового излучения.,
3. Определение интенсивности люминесценции анализируе-
мого и стандартных растворов (по току фотоумножителя, почер-
нению фотопластинки и т. д.).
V 4. Определение градуировочной характеристики и расчет со-
держания определяемого компонента.
Люминесцентное определение кристаллофосфоров, включая
использование низких температур, отличается от приведенной
схемы лишь методом перевода вещества в люминесцирующее со-
стояние.
Количественный люминесцентный анализ основан на исполь-
зовании соотношения (5.8), связывающего интенсивность флуорес-
ценции с концентрацией флуоресцирующего вещества. В практике
количественного люминесцентного анализа обычно применяется ;
метод градуировочного графика на основе этого уравнения. К на- •
стоящему времени разработаны методы количественного люминес-;
центного определения почти всех элементов при содержании в
среднем 10"5%.
Большой интерес вызвало применение люминесцентных ин-
дикаторов в титриметрическ.их методах. Люминесцентные инди-
каторы (а-нафтиламин, акридин и др.) изменяют цвет или интен-
сивность люминесценции в зависимости от свойств участников
реакции, рН раствора или присутствия окислителя. Используя в
качестве люминесцентного индикатора, например, морин, мож-
но с погрешностью 5—10% титримётрически определять алюми-
ний, галлий, цирконий и другие элементы при содержании 1—
10 мкг. Медь можно титровать флуорексоном в присутствии ни-
келя, кобальта, железа, марганца и некоторых других элементов
в растворах, содержащих 0,01—0,1 мкг Си/мл. Применение лю-^
минесцентных индикаторов позволило решить ряд сложных ана-
литических задач, связанных, в частности, с анализом мутных и
окрашенных сред (фруктовые соки, вина и другие напитки).
Существенно увеличивается интенсивность люминесценции
при замораживании растворов. Это явление используется для ко-
личественного определения свинца, висмута и сурьмы в виде га-
логенидных комплексов. Например, при охлаждении до -196 °С
раствор, содержащий свинец в концентрированной НС1, дает фи-
108
олетовую люминесценцию. Люминесценция наблюдается также
при замораживании органических веществ и комплексов метал-
лов с органическими лигандами.
Известны некоторые обобщения по использованию химиче-
ских реагентов в люминесцентном анализе для определения катио-
нов. Как правило, флуоресцируют соединения, катионы которых
образуют с данным реагентом бесцветные соединения. Так, напри-
мер, флуоресцируют комплексы бериллия, магния, кальция, алю-
миния, цинка и других катионов с многими реагентами. Наоборот,
комплексы с окрашенными катионами, такими, как хром, железо,
никель, медь и другие флуоресцируют редко. Причиной этого яв-
ляется низкое расположение энергетических уровней этих ионов,
при котором более вероятными становятся безызлучательная дез-
активация возбужденных уровней комплекса с люминесцентным
реагентом. Однако такие комплексы часто обладают способностью
фосфоресцировать. Так, медь образует ряд фосфоресцирующих
комплексов, но почти не образует флуоресцирующих.
Реагент для люминесцентного определения элемента должен
интенсивно люминесцировать, вообще говоря, только после реак-
ции с анализируемым веществом. На практике такое требование
соблюдается не всегда. Этому требованию удовлетворяют вещест-
ва, молекулы которых имеют не жесткую и не плоскую аромати-
ческую структуру с системой сопряжённых мостиковых связей.
Сюда можно отнести основания Шиффа, азосоединения (ализа-
рин и другие производные диоксибензолсульфокислоты), окси-
хинолин, ß-дикетоны, родамины и др.
5.5. Практическое применение
В практике люминесцентных методов значительное место за-
нимает анализ обнаружения. По люминесценции фосфоров и друг
гих веществ обнаруживают инфракрасное, ультрафиолетовое,
рентгеновское и у-излучение, регистрируют потоки протонов,
нейтронов, электронов, а-частиц. Известны люминесцентные спо-
собы диагностики различных заболеваний. В оптико-механиче-
ской промышленности люминесцентный анализ обнаружения ис-
пользуют для маркировки различных сортов стекла, в резиновой
промышленности для контроля состава шихты, в бумажной —
для установления качества целлюлозы, в алмазодобывающей про-
мышленности по характерному свечению отбирают алмазы и т. д.
Методы люминесцентного анализа успешно используют в
анализе лантаноидов, соединений урана и ряда других элемен-
109
тов. Люминесцентной способностью обладают многие органиче-
ские соединения: бензол, нафталин и их многочисленные произ-
водные, биологически активные вещества (витамины, антиби-
отики, гормоны), многие пигменты и т. д. Благодаря низкому
пределу обнаружения и простоте применяемой аппаратуры лю-
минесцентный анализ успешно развивается и является одним из
перспективных методов.
5.6. Общая характеристика метода
Важнейшей особенностью люминесцентного метода анализа
является его применяемость к определению микропримесей. По-
грешность метода составляет 5—7%, Применимость люминес-
центного анализа очень широка. Он может быть использован для
определения почти любого элемента, многих органических, био-
логически активных и других веществ.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1 1. Какова природа люминесцентного излучения?
: 2. Чем можно объяснить смещение максимума спектра люминес-
ценции в область больших длин волн по сравнению со спектром
поглощения? /
3. На чем основан качественный люминесцентный анализ? Как/
проводят качественные определения методом люминесценции в
технике, сельском хозяйстве, медицине и т. д.?
4. От чего зависит интенсивность люминесцентного излучения?
Как она связана с концентрацией?
5. Каковы достоинства и недостатки люминесцентного анализа?
гл а в а 6
Рентгеноспектральные
методы анализа
6.1. Рентгеновские спектры
Быстролетящйе частицы, Например электроны, могут вызы-
вать возбуждение или йойййацйю атомов не только в газообраз*
йом состояний. При стелкйовбййй электрона с какой-либо Фвер-
110
дой поверхностью может также произойти возбуждение или
ионизация атомов. Если энергия летящего электрона достаточна,
происходит выбивание электрона из внутренних Кг или Х-оболо-
чек атомов вещества, подвергающегося бомбардировке. На освб-
бодившееся место в^- или Х-оболочке переходит электрон с более
высокого энергетического' уровня, что сопровождается характе-
ристическим рентгеновским излучением*. 1
Кроме характеристического в этом процессе возникает рент-
геновское излучение с непрерывным спектром, связанное с час-
тичным превращением энергии тормозящихся электронов при
столкновении с мишенью в энергию излучения. Это излучение
называют тормозным. Максимальная частота непрерывного
рентгеновского излучения утах связана с напряжением ^ на рент-
геновской трубке соотношением
• •• • уг- \ #*утах ч •■, ... ■..;
Таким образом, эмиссионный рентгеновский спектр пред-
ставляет собой непрерывный фон, перекрытый линиями характе*
ристического излучения. Характеристическое рентгеновское из-
лучение наблюдается не только при бомбардировке электронами,
оно возникает такз£се при облучении поверхности электромагнит-
ным излучением большой энергии, достаточной для выбивания
внутренних электронов из атомов. Излучение непрерывного
спектра при этом не происходит, и характеристический спектр,
полученный таким способом, называется флуоресцентным или
вторичным.
Следует отметить, однако, что внутриатомный переход
электрона с верхнего энергетического уровня на К- или Х-уровень
не всегда сопровождается характеристическим рентгеновским
излучением. Возможен и безызлучательный переход. При этом
происходит перестройка электронноц оболочки и один из внеш-
них электронов отрывается от атома. Этот процесс известен как
эффект Оже, а электроны, отрывающиеся от атома, называют
оже-электронами. Вероятность проявления эффекта Оже во мно-
гих случаях очень велика, а у легкй^ элементов она даже боль-
ше, чем вероятность рентгеновского излучения, что вызывает оп-
ределенные трудности в рентгеноспектральном анализе легких
элементов. ;; ;
* В иностранной литературе это излучение часто называют Х-луча-
ми, сохраняя оригинальный термин Рентгена. <ч п
111
Рентгеновские термы. Линии характеристического рентге-
новского излучения соответствуют разности энергетических
уровней внутренних электронных оболочек атома. Частоты ха-
рактеристического излучения атомов данного элемента могут
быть рассчитаны по уравнению
¥-!^(і..і).д1(іД)..
(6.1)
Здесь ¥ — эффективный заряд ядра, равный
где Z — заряд ядра, равный порядковому номеру элемента в пе-
риодической системе Д. И. Менделеева; а — постоянная экрани-
рования.
Вместо уравнения (6.1) можно записать
Величину
,_Д(£-с)2 _Д(г-а)2-Т _Т
Т^ щг - о)2
называют рентгеновским термом.
Символика, используемая для обозначения и классификации
линий рентгеновского спектра, схематично приведена на рис.
6.1. Для учэта подуровней у символа линии ставят числовой ин-
декс, например, Каі, Ьр2 и т. д. Число линий в рентгеновском
спектре сравнительно невелико. В соответствии с законом Мозлщ
квадратный корень из волнового числа (или частоты) первой
1Г-линии связан с порядковым номером элемента в периодичес-
кой системе линейной зависимо-
стью:
ІЇ-КІЇ-г а). (6.2)
График зависимости £ от л/у пред-
ставляет собой прямую. С откры-ти-
ем закона Мозли порядковый номер
элемента в периодической системе
получил четкое физическое истол-
кование как заряд ядра. Поскольку
характеристические линии рент-
геновского спектра при переходе
от одного элемента к другому сме-
щаются на одну и ту же величину,
■д -3,-
л-2-
71=1-
-М
-К
Рис. 6.1. Схема электронных
переходов рентгеновского спектра:
п = 1, 2, 3, 4 — главные квантовые
числа; К> Ь, М, ІУ — электронные
оболочки
112
это свойство позволяет установить, имеется ли между двумя эле-
ментами периодической системы какой-то третий, еще неизвест-
ный, и предсказать его рентгеновский спектр. Замечательным
практическим применением закона Мозли было его использова-
ние при открытии предсказанных Д. И. Менделеевым элементов
периодической системы (гафния и рения).
Интенсивность линий рентгеновского спектра зависит от рас-
пределения бомбардирующих электронов по скоростям или от
распределения интенсивности в спектре возбуждающего излуче-
ния в случае флуоресцентных спектров. Цри одинаковых услови-
ях интенсивность характеристических линий спектр>а макси-
мальна, когда максимальная интенсивность источника возбуж-
дения соответствует энергии возбуждения данной линии.
Интенсивность спектра зависит также от числа излучающих ато-
мов, вероятности излучательного перехода и некоторых других
факторов. Точная оценка величин, оказывающих влияние на ин-
тенсивность спектральной линии* очень сложна. Более надежны
данные, так же как и в оптической эмиссионной спектроскопии,
полученные по относительной интенсивности двух спектральных
линий, находящихся в одной и той же области длин волн. Пря-
мая пропорциональность между интенсивностью линии и кон-
центрацией элемента в пробе наблюдается довольно часто.
6.2. Поглощение рентгеновского излучения
Ослабление рентгеновского излучения при прохождении че-
рез пробу подчиняется закону светопоглощения:
1-/0 • 10-*>',
где 70и/ — интенсивность падающего и прошедшего через пробу
рентгеновского излучения соответственно; |х — массовый коэффи-
циент поглощения; р — плотность вещества; I — толщина слоя.
Зависимость массового коэффициента поглощения от атом-
ного номера элемента и длины волны падающего излучения в
первом приближении выражается соотношением
ц = Л3. V
% 6.3. Основные узлы
рентгеноспектральных приборов
Конструкции приборов, используемых в рентгеноспектраль-
ных методах анализа, включают следующие основные узлы: ис-
точник возбуждения; диспергирующий элемент; приемник излу-
чения (рецептор).
8 -7831
113
б;3;1. ИСТОЧНИК ВОЗбуЖДеНИЯ ^ < 3'
. Первичное излучение в рентгеноспектральных, методах полу-
чают с помощью рентгеновской трубки и реже радиоактивного
излучателя.
Рентгеновская трубка. Конструкции трубок весьма многооб-
разны. Принцип их действия; иллюстрируется на рис. 6.2. В ва-
куумированном сосуде под постоянным напряжением в десятки
киловольт находятся анод 1 и раскаленный катод 2, мейсду кото-
рыми пропускается ток 50—100 мА. Раскаленный током катод
испускает электроны, которые ускоряются электрическим полем
и специальным фокусирующим устройством направляются на
анод. Бомбардирующий электронный пучок выбивает электроны
из внутренней оболочки атомов вещества, пошедшего на изготов-
ление анода. Остальная часть кинетической энергии электронов
расходуется на так называемое тормозное излучение и нагрева-
ние анода. Возникающий рентгеновский спектр наряду со сплошг
ным фоном тормозного излучения содержат характеристическое
излучение элементов, входящих в состав анода. Через выходное
окно 3 рентгеновское излучение направляется на диспергирую-
щий элемент или на аияализируемую пробу в зависимости от вы-
бранной схемы анализа. /
В методах анализа по первичным спектрам анализируемую
пробу помещают непосредственно на анод и подвергают действию
электронного пучка. Вполне понятно, что при этом собственно
анод не должен содержать анализируемых элементов в связи со
сложностью введения поправки. В такого
+ ' " рода анализах используются разборные
рентгеновские трубки. Применяется так-
же сфокусированный электронный пучок
(электронный зонд), имеющий размеры
1—2 мкм2. Он предназначен для проведе-
ния лотса^ анализа, например от-
дельных зерен шлифа или распределения
одного или нескольких элементов ПО ПО-/
верхности пробы и т. д.
При анализе по вторичным, или флу-
оресцентным, спектрам в качестве источ-
ника излучения используют рентгенов-
ские, лучи на выходе из рентгеновской
трубки.
Рис. 6.2. Рентгеновская Радиоактивные излучатели. Они ис-
. трубка пускают у-кванты или у-квант^1, и 0-части-
114
цы. Эти излучатели используются непосредственно для бомбар-
дировки пробы или для облучения мишени, испускающей под
действием радиоактивного излучения характеристический рент-
геновский спектр.
6.3.2. Диспергирующий элемент
В качестве диспергирующего элемента в рентгеноспектраль-
ных приборах используют главным образом кристаллы, являю-
щиеся своеобразными дифракционными решетками. ,Их назы-
вают кристалл-анализаторами. Дифракция рентгеновских лу-
чей в кристалле происходит в соответствии с законом Вульфа-—
Брэгга:
пХ = 2d sin 0, (6.3)
где п.— целое число, показывающее порядок спектра (обычно ог-
раничиваются рассмотрением спектров первого порядка); <¿ —
кратчайшее расстояние между соседними плоскостями кристал-
ла; 0 — угол падения параллельного пучка рентгеновского излу-
чения на плоскость кристалла (его называют углом скольжения).
От плоскости кристалла под углом 0 будет отражаться излу-
чение с длиной волны X, удовлетворяющей условию Вульфа-—
Брэгга (6.3). Излучение, не удовлетворяющее этому условию,
рассеивается й частично поглощается кристаллом.
Таким образом, в зависимости от угла скольжения данный
кристалл будет отражать лучи с разной длиной волны, удовлет-
воряющей соотношению (6.3). Угол скольжения изменяют пово-
ротом плоскости кристалла-анализатора. На этом принципе раз-
работаны многочисленные схемы диспергирующих устройств,
используемых в практике.
Формула.. (6.3) является основой рентгеноструктурного и
качественного рентгеноспектрального анализа. Если известна
длина волны падающего излучения, то по синусу угла 0 можно
найти постоянную решетки d, что используется в рентгенострук-
турном анализе. Если d известно, то по sin 0 рассчитывают длину
волныу^ и проводят качественный, а затем и количественный
рентгеноспектральный анализ.
Выбор кристалла-анализатора определяется свойствами
предполагаемого объекта исследования и целью работы. Для про-
ведения, например, рентгеноспектрального элементного анализа
желательно иметь яркий спектр йе обязательно высокого разре-
шения. Такие óiieK'rpbi получаются ó помощью кристаллов ка-
** 116
менной соли, алюминия и др. Высокой разрешающей способно-
стью обладают кристаллы из кварца, кальцита, а также слюды,
флюорита и некоторых других веществ. При выборе учитывается
также предполагаемая область длин волн, поскольку в соответстг
вии с уравнением (6.3) при одном и том же угле скольжения 0
длина волны «отраженного» излучения зависит от межплоскост-
ного расстояния в кристалле-анализаторе. Соответствующие ха-
рактеристики кристаллов хорошо изучены и сведены в специаль-
ные таблицы. Например, у кальцита dCáC0 = 0,302945 им, у
з -
флюорита dcaF2 = 0,3145 нм, у каменной соли dNaC1 = 0,281400 нм
йт. д. У кварца в зависимости от выбора кристаллических плос-
костей dSi0 принимает значения 0,424602; 0,333626; 0,245144;
0,117762 и 0,101275 нм.
В области длин волн, превышающих 1,5—-2,0 нм, применя-
ются дифракционные решетки.
6.3.3. Приемники излучения
В качестве приемников рентгеновского излучения могут;*
быть использованы фотоматериалы и счетчики рентгеновских'
квантов: ионизационные и сцинтилляционные. Эти же счетчики
применяют для регистрации радиоактивного излучения. |
В рентгеноспектральном анализе используют специальные
рентгеновские пленки, часто двуслойные. Для повышения чувст-
вительности к рентгеновскому излучению в фотоэмульсию рент-
геновских плёнок вводят повышенное по сравнению с обычными
фотопластинками, содержание бромида серебра.
Ионизационные счётчики. Схе-
ма ионизационного счетчика пред-
ставлена на рис. 6.3. Счётчик пред-
ставляет собой устройство из двух
электродов: цилиндрического като-
да и анода в виде металлической
нити, натянутой вдоль оси цилинд-
ра. Пространство в трубке между
электродами заполнено газом (на-
пример, аргоном) при пониженном
давлении. В зависимости от режима
работы это устройство может быть
ионизационной камерой, пропорци-
ональным счетчиком или счетчи-
ком Гейгера—Мюллера.
РИС. 6.3.
Ионизационный счетчик:
1 — стеклянная колба;
2 — катод; 3 — анодная нить;
4 — источник высокого
напряжения
ив
Действие счетчика основано на ионизации газообразного на-
полнителя. При небольшом напряжений ток через счётчик не
идет (рис. 6.4). Под действием рентгеновского излучения атом
аргона ионизирует
Аг + Лу = Аг+ + е"
а образовавшийся электрон при столкновении вызывает иониза-
цию других атомов аргона. Под действием приложенного напря-
жения ионы Аг+ будут двигаться к катоду, а электроны — к ано-
ду. Однако при небольшом напряжении скорость движения неве-
лика, и значительная часть ионов успевает рекомбинировать до
достижения электродов. Повышение напряжения примерно до У1
(см. рис. 6.4) приводит к увеличению скорости ионов и уменьше-
нию вероятности рекомбинации. ПриУг наступает «насыщение».
Дальнейшее увеличение напряжения уже не вызывает увеличе-
ния силы тока. При этом напряжении все образовавшиеся ионы
доходят до электродов, и рекомбинация практически не происхо-
дит. Очевидно, при напряжениях V < Уг прибор для измерения
интенсивности рентгеновского излучения использован быть не
может. В области напряжений от уг д,оУ29 т. е. в области насыще-
ния, прибор работает в режиме ионизационной камеры. Иониза-
ционная камера служит для измерения рентгеновского излуче-
ния сравнительно большой интенсивности (вызывающей 105—
106 импульсов в минуту).
Возрастание напряжения на электродах счетчика приводит к
увеличению скорости электронов, что вызывает ударную иониза-
цию. Происходит «газовое усиление» и лавинообразное увеличе-
ние числа ионов. Амплитуда импульса (скачок потенциала) в этих
условиях меняется пропорционально энергии фотона и составляет
10"4—10"2 В. Прибор, работающий в этой области, называется
пропорциональным счетчиком. Область напряжений от У3 до У4
щ
Г 1 1
1 II - ^
1 ^-*<**^П^ 1
^1—А- '
р 1
^-<Т 1 1
| 1 I
1 11
200 500 1000
1500 2000 2400
V
Рис. 6.4. Зависимость импульса от напряжения
117
называют областью ограниченной пропорциональности. Вэтом
интервале ионизационные приборы не используются. : ?
В области от У4 доУ5 попадание в счетчик фотона вызывает
лавинообразную ионизацию, не зависящую от энергии фотона.
Это гейгеровская область. Прибор, работающий в этом режиме,
называют счетчиком Гейгера—Мюллера.
Электроны в счетчике движутся к нити, а положительно за-
ряженные ионы — к цилиндру. Вблизи нити напряженность
электрического поля возрастает до таких значений, при которых
происходит ударная ионизация и образуется довольно большое
число электронов и положительных ионов. Электроны в течение
очень короткого промежутка времени, порядка 10"7 с, собирают-
ся на нить счетчика. За столь короткое время положительные
ионы не могут сколько-нибудь заметным образом сдвинуться с
места. Их поле экранирует поле нити, благодаря чему теряется
возможность ударной ионизации. По мере удаления слоя поло-
жительных ионов от нити их экранирующее действие будет осла-
бевать и способность счетчика фиксировать появление ионов бу-
дет восстанавливаться. Промежуток времени, в течение которого
импульс не может быть зарегистрирован, называют «мертвым»
временем счетчика. Он имеет длительность примерно 10"4 с. /
Через 10~5 с положительные ионы доходят до катода и разря-
жаются. Разряд этих ионов может сопровождаться ультрафиоле-
товым излучением и образованием электронов, которые* в свою
очередь, генерируют в электрическом поле новые электроны. Та-
ким образом, в счетчике возникает лавинный разряд. Попадание
извне новых рентгеновских квантов в такой «горящий» счетчик
не может заметно изменить силу тока и, следовательно, не будет
зарегистрировано.
В самогасящихся счетчиках к основному наполнителю арго-
ну добавляют некоторое количество (до 10%) паров многоатом-
ных соединений, таких, как этиловый спирт, ксилол и др. Мно-
гоатомные молекулы поглощают фотоны и разрушаются без вы-
свечивания, что практически сводит к нулю фотоэффект на
катоде. Кроме того, многоатомные молекулы легко отдают свои
электроны положительным ионам аргона при столкновениях,
так как потенциал ионизации аргона значительно выше:
С2НбОН'+ Аг+ - С2НбОН+ + Аг
Кинетическая энергия крупных многоатомных ионов неве-
лика, поэтому выбивания электронов на катоде они не вызывают.
Самогашенйе счетчика достигается, как видно; за счет разруше-
118
ния и диссоциации многоатомного соединения. Это, естественно,
ограничивает срок службы самогасящихся счетчиков.
В последнее время в качестве добавки, вызывающей эффект
самогашения в счетчиках, используют хлор и бром. Счетчики с
галогенным наполнением работают при низком напряжении (до
400 В) и имеют практически неограниченный срок службы, так
как процесс диссоциации молекулы галогена на атомы обратим.
Сцинтилляционный счетчик. Действие сцинтилляционных
счетчиков основано на измерении сцинтилляций — световых
вспышек, появляющихся в сцинтилляторе под действием рентге-
новского излучения (рис. 6.5). В качестве сцинтилляторов ис-
пользуются вещества, молекулы которых под действием рентгвг
новского излучения возбуждаются и, переходя в нормальное
состояние, дают вспышку света, которая фиксируется фото-
электронным умножителем (ФЭУ). Сцинтилляторами могут
быть, например, Nal, ZnS, антрацен и многие другие вещества.
Большой интерес к сцинтилляционным счетчикам вызван их
более высокой чувствительностью ко всем видам излучений по
сравнению с ионизационными, их большой разрешающей способ-
ностью (до 10~9 с), так как у них нет «мертвого» времени. Кроме
того, сцинтилляционные счетчики позволяют измерять энергию
ИЗЛучеНИЯ. ,
Другие приемники рентгеновского излучения., Рентгенов-
ское излучение можно регистрировать также непосредственно
фотоэлектронными умножителями (ФЭУ) и фотоэлементами, с
помощью кристаллического счетчика и калориметрическим ме-
тодом. Некоторые металлы и сплавы (например, тантал, сплав
меди с бериллием и др.) после специального поверхностного ак-
тивирования могут быть использованы в качестве катодов ФЭУ
для прямого измерения интенсивности рентгеновского излуче-
1
2
3
4
5
6
10
9
8
7
Рис 6.5. Сцинтилляционный счетчик:
1 — сцинтиллятор; 2 — фотокатод; 3 — фотоэлектронной умножитель;;
;^ 4, 5^6, 8, 9,10 — эмиттеры; 7 — анод -
119
ния. У ФЭУ такого типа окошко открыто, что имеет особую цен-
ность при работе в области мягкого рентгеновского излучения.
Чувствительность обычных фотоэлементов (например, селе-
новых) к рентгеновскому излучению примерно в 1000 раз мень-
ше, чем к излучению в видимом участке спектра. Для повышения
чувствительности поверхность фотоэлемента покрывают соста-
вом, способным люминесцировать под действием рентгеновского
излучения. Устройства такого типа с успехом применяют в анали-
тической практике.
Кристаллическим счетчиком называют полупроводниковый
монокристалл типа, например, Сей, который при освещении
рентгеновским излучением обнаруживает значительное умень-
шение сопротивления. Эти счетчики весьма перспективны, так
как обладают чувствительностью ионизационных, но не требуют
для питания стабилизированного высокого напряжения.
6.4. Конструкции рентгеновских
спектральных приборов
Конструкции приборов, применяемых в рентгеновском/
спектральном анализе, различают по типу источников возбужде^
ния, характеристикам диспергирующего элемента и свойствам
приемника излучения. Если, например, спектр регистрируется с
помощью фотопленки, прибор называют рентгеновским спект-
рографом, если регистрация ионизационная, — спектрометром.
В зависимости от используемой спектральной области приборы
подразделяют на длинноволновые и коротковолновые. Сконстру-
ированы приборы, предназначенные для работы как с эмиссион-
ными рентгеновскими спектрами, так и по поглощению рентге-
новского излучения.
Анализ по первичному рентгеновскому излучению; т. е. по
излучению, полученному при электронной бомбардировке анода
рентгеновской трубки, в последнее время в значительной степени /
теряет свое значение; Основную роль играют методы, использую-
щие вторичное (флуоресцентное) излучение. Особое место зани-
мают рентгеновские квантомётры и рентгеновские микроанали-
заторы (электронный микрозонд).
Квантометры. Квантометрами называют спектрометры, в ко-
торых производится одновременная регистрация нескольких
длин волн флуоресцентного излучения. Используют конструк-
ции с прямыми и изогнутыми кристалл-анализаторами, с иони-
120
зационными и сцинтилляционными счетчиками. Особо эффект
тивно; применение квантометров для экспрессного определения
нескольких заданных элементов в серии однотипных образцов.
Успешно применяется, например, восьмиканальный квантометр
для анализа на семь компонентов. Продолжительность анализа
составляет 2,5 мин.
Рентгеновские микроанализаторы. В рентгеновских микро-
анализаторах электронно-оптическая система (электронная пуш-
ка) формирует электронный луч-зонд диаметром 1—2 мкм2, на^-
правляемый на анализируемый образец, вернее в какую-то точку
на анализируемом образце или «зерно». Флуоресцентное излуче-
ние элементов, входящих в состав зерна, кристалл-анализатором
разлагается в спектр, а детектором определяется интенсивность от-
дельных линий. Применение электронного зонда позволило ре-
шить ряд важнейших задач теоретического и практического ха-
рактера: найти распределение данного элемента по поверхности об-
разца, определить состав отдельных участков поверхности и т. д.,
что имеет особое значение в металлургической промышленности,
электровакуумной технологии, геохимии, биологии ит. д.
Спектральные приборы абсорбционного анализа. В прибо-
рах этого типа измеряется интенсивность рентгеновского излуче-
ния, прошедшего через анализируемую пробу, вернее уменьше-
ние интенсивности излучения, связанное с поглощением рентге-
новского излучения. Конструкции абсорбционных приборов
различаются взаимным расположением анализируемого образца
и кристалл-анализатора. В одних приборах после рентгеновской
трубки помещен кристалл-анализатор и через анализируемую
пробу проходит монохроматическое излучение. В приборах дру-
гой конструкции анализируемая проба помещается между рент-
геновской трубкой и кристалл-анализатором и таким образом в
спектр разлагается излучение, прошедшее через анализируемую
пробу.
6.5. Качественный
рентгеноспектральный анализ
Так же как и в эмиссионной спектроскопии, качественный
анализ рентгеноспектральным методом проводят путем опреде-
ления длины волны интересующих линий и их последующей
идентификации. Длину волны рентгеновской линии в спектре
обычно определяют с помощью известных опорных линий, яв-
.4 121
ляющихся своеобразными стандартами. В качестве такого стан-
дарта может быть использована или «основа» пробы, как это час-
то делается в эмиссионной спектроскопии, или известное вещест-
во, специально вводимое в анализируемую пробу. Нередко для
этого рядом со спектром анализируемой пробы фотографируют
спектр известного стандартного вещества. Методика определения:
длины волны в этих условиях практически не отличается от той,
которая используется в эмиссионной спектроскопии.
Расшифровка рентгеновских спектров также в принципе не
отличается от соответствующей методики эмиссионной спектро-
скопии. Существенно облегчается эта работа благодаря наличию
подробных таблиц линий рентгеновского спектра. Хотя рентге-
новские спектры намного проще эмиссионных, что заметно упро- ;
щает задачу идентификации линий, все же определение их при- '
надлежности тому или иному элементу остается далеко не прос-
тым. Осложнения вызывают главным образом спектры разных
порядков, что приводит к наложению линий. Для надежности \
определения находят длину волны и оценивают интенсивность не
одной, а нескольких спектральных линий. Вполне понятно* что
среди них должна находиться наиболее интенсивная линия ана-
лизируемого элемента (обычно это Ка- или 1^-линия). Чувстви-
тельность рентгеноспектрального анализа (предел обнаружения)
составляет в среднем 0,05—0,1%, для некоторых элементов (№.,
Си и др.) он снижается до 5 • 10~"3%, для других (например, ред-
коземельных элементов) повышается до 0,1—0,2%.
Закон Мозли (6.2) связывает атомный номер элемента с час-
тотой рентгеновского излучения. Особенно примечательным при-
мером использования закона Мозли является открытие новых
элементов периодической системы: гафния в 1922 г. и рения в
1925 г; Вывод о наличии этих элементов в соответствующих кон-
центратах был сделан по их характеристическим рентгеновским
спектрам.
Рёнтгеноспёктральный метод имеет ряд существенных досто-
инств й'преимуществ перед другими методами анализа. Рентге-
новские спектры малочувствительны к химическому окружению
элемента и практически не зависят от того, в виде какого соеди-
нения находится анализируемый элемент в пробе! Рентгено-
спёктральным методом легко обнаруживаются галогены, сера и
другие элементы, анализ которых методом эмиссионной спектро-
скопии не проводится. Большим достоинством рентгенофлуорёс-
центного метода является возможность анализа образца без его
разрушения, что особенно ценно при анализе уникальных из-
делий. '
122
6.6. Количественный
рентгеноспектральный анализ
Для проведения количественного анализа может быть ис-
пользовано как первичное рентгеновское излучение, так и вто-
ричное (флуоресцентное). При использовании первичного излу-
чения порошкообразную пробу обычно втирают в рифленую по-
верхность анода. Если анализируется металлическая проба,
анодом служит анализируемый образец.
Рентгеноспектральный анализ по вторичному (флуоресцент-
ному) излучению имеет существенные преимущества по сравне-
нию с анализом по первичному рентгеновскому излучению. Ана-
лиз по флуоресцентному излучению имеет более высокую чувст-
вительность, так как при этом отсутствует фон непрерывного
рентгеновского спектра. Немаловажное значение имеет также
упрощение экспериментальной методики, поскольку анализи-
руемый образец находится вне вакуумной системы рентгенов-
ской трубки. Правда, интенсивность вторичных спектров мень-
ше, чем первичных, и поэтому, например, фотографическая ре-
гистрация здесь не применяется. Однако достаточно высокая'
чувствительность счетчиков рентгеновских квантов обеспечивает
быстрое и точное измерение интенсивности линий.
Количественные определения основаны на пропорциональ-
ности между интенсивностью линии характеристического излу-
чения и концентрацией элемента в пробе. На абсолютную интен-
сивность линий влияют условия возбуждения и другие факторы,
а также химический состав пробы, что приходится учитывать се-
рией специальных измерений и теоретическими расчетами. За-
висимость интенсивности линий рентгеновского спектра от кон-
центрации элемента имеет более сложный характер, чем кон-
центрационная зависимость интенсивности линий в эмиссионной
спектроскопии.
В методах внутреннего стандарта сравнивается интенсив-
ность линий определяемого элемента с линией стандартного, спе-
циально введенного в пробу элемента в точно известном количе-
стве. Сравниваемые линии должны иметь близкие потенциалы
возбуждения, т. е. близкие длины волн и не слишком сильно раз-
личаться по интенсивности. Удобным стандартным элементом
является соседний с определяемым элемент периодической сис-
темы. Относительная интенсивность линий значительно меньше
зависит от состава пробы, условий получения и регистрации
спектров и других факторов, чем их абсолютная интенсивность.
Отношение интенсивностей линий определяемого элемента и эле-
123
мента-стандарта предполагается пропорциональным их концент-
рации:
где 1Х и /ст — интенсивности линий определяемого и стандартно-
го элементов; сх и сст — их концентрации.
Ввиду невозможности точного учета всех факторов коэффи-
циент к определяется эмпирически по интенсивности линий
стандартных образцов. Уравнение (6.4) является также основой
градуировочного графика.
В анализе по методу внешнего стандарта интенсивность ли-
нии определяемого элемента сравнивается с интенсивностью этой
линии в спектрах стандартных образцов с известным содержанием.
Отношение интенсивностей линий принимается равным отноше-
нию концентраций элемента. Точные результаты получаются при
условии, что состав анализируемой пробы и стандартных образцов ;
по основным компонентам был достаточно близким, так как интен-
сивность линий зависит от общего состава пробы, в особенности от
наличия так называемых мешающих элементов. Успешно приме- •
няется метод добавок, например, при анализе лантаноидов. ч
Современные приборы для рентгенофлуоресцентного анализа
полностью автоматизированы, имеют встроенный компьютер и |
позволяют быстро получать достаточно точные результаты. у'}.
Несколько менее широкими возможностями обладает рентге-
новская абсорбционная спектроскопия. Поглощение рентгенов-
ского излучения подчиняется уравнению, сходному с уравнением
закона Бугера—Ламберта—Вера:
А = 1* ^ - 0,43*(цпрРпр + ЦоенРоен),
где I и10 — интенсивность падающего и прошедшего через обра-
зец рентгеновского излучения; / — толщина поглощающего слоя,
см; цпр — массовый коэффициент поглощения примеси; цосн —
коэффициент поглощения основы, см2 • г"1; рпр — концентрация
примеси, г • см""3; р^ — концентрация основы, г •см"3. Массо7
вый коэффициент поглощения зависит от атомного номера £ эле-
мента и длины волны А,:
и = кг2х3.
Вполне естественно, что длина волны падающего излучения
должна быть меньше, чем характеристическая длина волны оп-
ределяемого элемента. В справочной литературе приводится свы^
ше 2500 значений ц для элементов и некоторых соединений в об-
ласти длин волн от 0,008 до 0,10 нм.
124
Определение концентрации анализируемого элемента прово-
дят методом градуировочного графика в координатах А - р^, как
и в абсорбционной спектроскопии.
6.7. Практическое применение
Рентгеноспектральные методы анализа имеют обширные и
разнообразные области применения. В геологии, горном деле, ме-
таллургии этим методом определяют состав минералов, руд, гор-
ных пород и продуктов их переработки — шлаков, концентратов
и т. д., устанавливают состав легированных сталей и сплавов, в
химических отраслях промышленности (электрохимии, нефте-
химии и т. д.) анализируют исходное сырье и готовую продук-
цию, в ядерной технике контролируют изменения в составе за-
медлителей, теплоносителей ит. д. Широко используются рент-
геноспектральные методы для анализа керамики, стекла,
пластмасс, абразивов, катализаторов и других материалов слож-
ного химического состава. Весьма эффективным оказалось при-
менение рентгеноспектрального флуоресцентного анализа для
контроля за загрязнением окружающей среды (определение со-
держания различных элементов в аэрозолях, почвах, воде, расти-
тельных и животных тканях и т. д.). Например, в аэрозолях.
определяют до 40 элементов от натрия до свинца. Рентгеносцект-
ральный анализ показал, в частности, что источником загрязне-
ния атмосферы кальцием служит цементная промышленность,
свинцом и бромом — выхлопные газы автомобилей и т. д.
Широкое применение нашел рентгеноспектральный метод
определения толщины покрытии — тонкого слоя, нанесенного на
основной материал, как, например, цинка на оцинкованном же-
лезе, слоя ферропорошка на магнитофонной ленте и т. д. Метод
основан на использовании градуировочных графиков, показы-
вающих зависимость интенсивности спектральной линии от тол-
щины покрытия. Градуировочный график строится по стандар-
там с известной толщиной слоя.
Очень эффективным оказалось применение рентгеновского флу-
оресцентного метода в анализе космических объектов. С помощью
спектрометрической аппаратуры РИФМА (рентгеновский изотоп-
ный флуоресцентный метод анализа), установленной на «Лунохо-
де-1»* было определено содержание основных породообразующих
элементов непосредственно на поверхности Луны. Для возбуждения
флуоресцентного излучения применялись радиоактивные источни-
ки, характеризующиеся высокой стабильностью и не нуждающиеся
125
в электрической энергии. В качестве детектора использовались про-
порциональные счетчики. Электрический импульс счетчика преоб-
разовывался и по радио передавался на Землю.
Широко известны и весьма важны применения рентгенов-
ского излучения во многих других областях науки и техники (оп-
ределение структуры кристаллов, рентгеновская дефектоскопия
изделий, диагностика заболеваний и т. д.).
6.8. Общая характеристика метода
Методами рентгеноспектрального анализа определяют состав
различных сплавов, руд, минералов, цементов, пластмасс и мно-
гих других изделий, устанавливают характер загрязнений окру-
жающей среды, анализируют космические объекты и т. д. Его ис-
пользуют для определения больших содержаний (десятки про-
центов) й небольших примесей (10~2—10~3%).
Предел обнаружения рентгеноспектральными методами в об-
щем ограничивается величинами порядка 10~2—І0~3%. Сочета-
ние с химическими методами обработки позволяет его значитель-
но снизить. Средняя квадратичная погрешность методов состав-
ляет примерно 2—5%, при благоприятных условиях она
снижается до ±0,5%. Рентгёнбспектральный анализ легко авто-
матизируется.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. В результате каких процессов возникает тормозное и характе-
ристическое рентгеновское излучение?
2. Как возникает флуоресцентное или вторичное излучение?
3. Как рассчитывается частота характеристического рентгеновско-
го излучения?
4. Какое теоретическое и практическое значение имеет закон Мозли?
5. Для решения каких основных задач применяется уравнение за-
кона Вульфа—Брэгга?
6. Какие особенности имеет рентгеновский микроанализатор (элек-
тронный микрозонд)?
7. На чем основан рентгеноструктурный анализ?
8. Как проводится качественный рентгеноспектральний анализ?
9. Какие элементы были открыты методом рентгеноспектрального
анализа?
10. Какие основные приемы используют в количественном рентгено-
спектральном анализе?
11. Каковы достоинства и недостатки рентгеноспектрального анализа?
126
•.,: глав а / ' •. , 7..
Другие спектральные
и оптические методы анализа
7.1. Анализ по спектрам
комбинационного рассеяния
Комбинационное\ рассеяние света было открыто в 1928 г.
отечественными физиками Л. И. Мандельщтамрм и Г. О. Ландс-
бергом и одновременно индийским физиком В. Раманом*.
7.1.1. Спектры комбинационного рассеяния
Взаимодействие электромагнитного излучения с веществом
не всегда проявляется в поглощении квантов энергии и переходе
молекулы на новый квантованный энергетический уровень. Дру-
гие эффекты наблюдаются, когда частота облучающего электрот
магнитного излучения значительно отличаетря от частоты элек-
тронного перехода в спектре данного вещества. Упрощенная энер-
гетическая схема этого взаимодействия представленана рис. 7.1.
При взаимодействии электромагнитного, излучения с вещест-
вом происходит поляризация молекулы, ее деформация в поле
электромагнитной волны, когда электроны молекулы сдвигают-
2
Е
У2
Ух'
а)
б)
. Рис. 7.1. Энергетическая схема происхождения
спектра комбинационного рассеяния:
IУ0, У1 и У2 — колебательные уровни энергии;
Е — энергия падающего излучения Лу; v — частота
* В иностранной литературе комбинационное рассеяние света назы-
вают эффектом Рамана.
127
ся в одну сторону, а положительно заряженные ядра —- в,другую.
Молекула приобретает энергию Ё, но это не значит, что она пере-
ходит в какое-то возбужденное состояние в квантовом смысле
этого слова. Этот процесс на рис. 7.1 обозначен как 1 и Г. Про-
цесс 1 относится к взаимодействию с электромагнитным излуче-
нием молекулы, находящейся на основном колебательном уровйе
У0, а процесс V — к взаимодействию молекулы, находящейся на
возбужденном уровне Уг. Таким образом, индуцированный ди-
поль осциллирует и молекула из поляризованного состояния воз-
вращается в обычное. Этому процессу соответствует переход 2
(рис. 7.1, а) или 2' (рис. 7.1, б). Возвращение молекулы на тот же
самый колебательный уровень, на котором она находилась ранее
(У0 в процессе 2 или Уг в процессе 2% сопровождается испускани-
ем электромагнитного излучения с частотой V, совпадающей с
частотой падающего излучения. Процессы 2и2' характеризуют
классическоерэлеевское рассеяние.
При комбинационном рассеянии молекула из поляризован-
ного состояния возвращается не на первоначальный, а на ка-
кой-то другой энергетический уровень. Энергия осцилляции ди-
поля в этом случае будет отличаться от Ё. Если сначала молекула
находилась на уровне У0, а возвратилась на уровень Уг (рис. 7.1,
а, процесс 3), то очевидно, что энергия осцилляции Е8 будет
меньше Е на столько же, на сколько энергия возбужденного
уровня Уг (Еу^ отличается от энергии основного уровня У0 (Еу0):
ЕУі~ Еуо= АЕ = Е-Е8 (7.1)
или
Ъ,\у=Ъ,\Е-Ь,\8 (7.2)
и
">Ъ='Уя-V (7.3)
Частота комбинационного рассеяния меньше, чем частота
возбуждающего излучения. Эту частоту (или линию в спектре)
называют стоксовой или красным спутником.
На рис. 7.1, б показана схема взаимодействия с электромаг-
нитным излучением молекулы, находящейся на возбужденном
колебательном уровне Уг, а после осцилляции возвращающейся
на основной уровень У0 (процесс 3'). Путем аналогичных рассуж-
дений можно установить, что
АЕ = Ё8-Е
и , -у ;
ч!з=*Е-Чу. (7.4)
128
Рис. 7.2. Схема спектра
комбинационного
рассеяния света
Линия комбинационного рассеяния в
этом случае будет находиться в области
больших частот, чем падающего излуче-
ния. Это анмистоксова частота (ли-
ния)^ или фиолетовый спутник.
' Разность частот в формулах (7.3) и
(7.4) одинакова по абсолютной величине и
отличается по знаку, поэтому линии ком-
бинационного рассеяния будут находить-
ся слева и справа на одинаковом расстоя-
нии от линии падающего излучения. Схе-
ма взаимного расположения линий в спектре комбинационного
рассеяния показана на рис. 7.2. Очень важной особенностью
спектров комбинационного рассеяния является независимость
разности уЕ±\у от частоты возбуждающего излучения.
При комнатной температуре большая часть молекул нахо-
дится на основном невозбужденном уровне, поэтому процессы 1 и
3 (см. рис. 7.1, а) более вероятны, чем Г и 3' (см. рис. 7.1, б). В со-
ответствии с этим линия комбинационного рассеяния с частотой
\Е - Уу (стоксова) будет более интенсивна, чем линия с частотой
уе + уу (антистоксова). С повышением температуры число воз-
бужденных молекул увеличивается и интенсивность антистоксо-
вых линий возрастает.
I Интенсивность линий комбинационного рассеяния также пропор-
циональна интенсивности падающего света.
Эффект комбинационного рассеяния может быть вызван не
только колебательными переходами, но также вращательными и
электронными, однако для аналитических целей обычно исполь-
зуют колебательные переходы.
Для наблюдения спектров комбинационного рассеяния кювету
с исследуемой жидкостью или раствором освещают источником
линейчатого спектра и под прямым углом изучают спектральный
состав рассеянного света. Нужный спектральный участок для осве-
щения пробы обычно выделяют с помощью светофильтра. К воз-
буждающей спектральной линии осветителя предъявляется не-
сколько требований: она не должна находиться в области спект-
ральной полосы поглощения анализируемого вещества, не должна
вызывать его флуоресценцию и т. д. В значительной степени этим
требованиям отвечает излучение различных видов ртутных ламп,
которые и стали наиболее употребительными источниками осве-
щения в спектроскопии комбинационного рассеяния. Чаще всего
работа ведется с использованием яркой синей линии в спектре рту-
9- 7831
129
ти, имеющей длину волны 435,8 нм. В последнее время значитель-
но расширяется применение в качестве источников освещения га-
зовых и иных лазеров — гёлиево-неонового (632,8 нм), аргонового
(438,0 и 514,5 нм) и т. д. Применение лазеров для этой цели имеет
ряд достоинств: оно позволяет, например, существенно увеличить
интенсивность комбинационного излучения и уменьшить массу ве-
щества для анализа до нескольких миллиграммов.
Спектральный состав рассеянного света исследуется с по-
мощью различных спектральных приборов, имеющих достаточ-
но большую светосилу и дисперсию при малом количестве пара-
зитного света. Часто применяют, например, трехпризменный
спектрограф ИСП-51 с фотографической регистрацией спектра.
Для исследования спектров комбинационного рассеяния и анали-
тических определений также используют спектральные приборы
с дифракционной решеткой в качестве диспергирующего элемен-
та и фотоэлектрическим измерением интенсивности излучения.
7.1.2. Качественный анализ по спектрам КР
Как и в эмиссионном спектральном анализе, в спектроскопии
комбинационного рассеяния наличие вещества в анализируемом
растворе устанавливается по характерным линиям в спектре.
Принадлежность линии тому или иному веществу определяется
по разности волновых чисел рассматриваемой и возбуждающей
линий, которая не зависит от волнового числа возбуждающего из-
лучения, и по интенсивности линии комбинационного рассеяния.
Данные по характеристикам спектра КР (комбинационного рас-
сеяния) многих веществ имеются в соответствующих таблицах.
Чувствительность анализа по спектрам КР не очень велика и не
относится к числу преимуществ метода. Основным достоинством
спектроскопии КР является возможность анализа сложных мно-
гокомпонентных смесей и веществ, близких по строению и соста-
ву. Методами спектроскопии КР анализируют смеси парафино-
вых и ароматических углеводородов, нафтенов, олефйнов и т. д.
7.1.3. Количественный анализ по спектрам КР
Методы количественного анализа по спектрам КР основыва-
ются на использовании линейной зависимости между интенсив-
ностью линии I и концентрацией вещества с:
1 = кс. , . V ;
130
Если, например, анализируется бинарная смесь веществ А и
В, то очевидно, что
: ^в = Лв^в> (7.5)
где X — доля вещества А или В.
При строгой стандартизации условий получения спектра и
обработки данных коэффициент пропорциональности можно оп-
ределить по интенсивности линий чистых исходных веществ. Из
отношения уравнений (7.5) при ХА = 1 и Хв = 1 находим отноше-
ние коэффициентов пропорциональности:
Отношение интенсивностей (1&/1^)х для анализируемой сме-
си находится экспериментально. Оно связано с отношением кон-
центраций
откуда
Кроме того, для бинарной смеси
ХА + ХВ=1. (7.7)
Совместное решение уравнений (7.6) й (7.7) дает состав би-
нарной смеси:
ХА= (/а//в) . (7.8)
Такой прием можно применить при анализе и более сложных
смесей, чем бинарные. Для этого следует попарно скомбини-
ровать отдельные компоненты смеси, чтобы найти коэффициен-
ты пропорциональности, и после экспериментального определе-
ния интенсивностей выбранных линий в спектре пробы решить
совместно уравнения типа (7.6) и (7.7). Коэффициент пропор-
циональности к часто определяют по интенсивности линий в
спектрах1 эталонных проб известного состава. Используется так-
же метод добавок и метод градуировочного графика. Погреш-
ность анализа по спектрам КР обычно не превышает ±3% относи-
тельных.
9«
131
7.2. Радиоспектроскопия
Интенсивное применение наиболее длинноволновой части
электромагнитного спектра — микроволн и радиоволн в физи-
ко-химических исследованиях и аналитической химии — нача-
лось сразу после открытия явлений электронного и ядерного маг-
нитного резонанса. Эти явления отражают взаимодействие мо-
лекулы с магнитным полем. Электронный парамагнитный
резонанс (ЭПР) характеризует взаимодействие с магнитным по-
лем магнитного момента электрона. Явление ядерного магнитно-
го резонанса (ЯМР) отражает взаимодействие с полем магнитного
момента ядра. Оба явления основаны на эффекте Зеемана, заклю-
чающемся в расщеплении спектральных линий или уровней
энергии в магнитном поле на отдельные компоненты.
7.2.1. Ядерный магнитный резонанс
Явление ЯМР открыли в 1946 г. американские физики
Ф. Блох и Е. Переел. Если элемент обладает нечетным порядко-
вым номером или изотоп какого-либо (даже четного) элемента
имеет нечетное массовое число, ядро такого элемента (изотопа)
обладает спином, отличным от нуля. Очевидному изотопов чет-
ных элементов с четным массовым числом спин от нуля не отли-
чается. Например, изотоп углерода 12С с массовым числом 12
спином не обладает, а изотоп 13С имеет спин, равный 1/2. Нали-
чие неспаренного спина у 13С вызывает появление у него ядерно-
го магнитного момента, в то время как ядра изотопов 12С магнит-
ного момента не имеют. В соответствии с этим внешнее магнит-
ное поле не будет оказывать влияния на хаотическое
распределение по энергии ядер 12С, но будет влиять на распреде-
ление ядер 13С, снимая вырождение энергетических уровней.
Спин ядра, равный 1/2> соответствует двум возможным ори-
ентациям вектора магнитного момента ядра в магнитном поле —
по полю {т1 = х/2) и против поля (тп7 = -1/2); при этом состояние
щ = —1/2 обладает во внешнем поле несколько более высокой
энергией, чем состояние т1 — х/2. Энергия перехода между этими
состояниями равна
АЕ = 2цЯ0,
где р — магнитный момент ядра; Н0 — напряженность внешнего
магнитного поля.
132
Число частиц на каждом из этих уровней может быть подсчи-
тано по закону распределения Больцмана:
: (-¥)• ■■■■■■■ >•*>
где пг и п2 — число частиц, находящихся на нижнем и верхнем
энергетических уровнях и обладающих энергией соответственно
Ег и Е2. Расчет по уравнению (7.9) показывает, что отношение
/г1//г2 в области обычных температур начинает отличаться от еди-
ницы только в шестом знаке после запятой. Таким образом, при
обычной температуре заселенность обоих уровней будет пример-
но одинакова с очень небольшим преобладанием состояний,
имеющих меньшую энергию. Если такую систему, находящуюся
в магнитном поле напряженностью Н0, поместить в переменное
электромагнитное поле с частотой у0, чтобы энергия кванта Ау0
совпадала с энергией перехода 2цД"0, т. е. чтобы
_ ку0 = 2цН0, (7.10)
то вследствие поглощения энергии поля ядра с нижнего энерге-
тического уровня будут переходить на верхний.
Важной характеристикой свойств ядер является гиромаг-
нитное отношение у, равное отношению магнитного момента яд-
ра к его механическому вращательному моменту:
у = 4яц/Л. (7.11)
Сочетание уравнений (7.10) и (7.11) дает
у=_о или ч0 = уш.
Ядра с гиромагнитным отношением у, находящиеся в однород-
ном магнитном поле напряженностью под воздействием пере-
менного поля с частотой у0 переходят в состояние с более высокой
энергией. Этот переход называют магнитным резонансом или
магнитным резонансным переходом. Гиромагнитное отношение
характеризует частоту и напряженность поля, удовлетворяющие
условию резонанса. Частоту у0 называют резонансной.
Из возбужденного состояния в нормальное ядра могут возвра-
щаться, передавая энергию возбуждения окружающей сред$ —
«решетке», под которой в данном случае понимаются электроны
или атомы другого сорта, чем исследуемые. Этот механизм пере-
дачи энергии называют спин-решеточной релаксацией, его эф-
фективность может быть охарактеризована постоянной Тг, назы-
ваемой временем спин-решеточной релаксации.
133
Возбужденное ядро может также передать энергию возбуж-
дения ядру такого же сорта, находящемуся в низшем энергетиче-
ском состоянии. Этот процесс называют спин-спиновой релакса-
цией и характеризуют величиной Т2 —временем спин-спиновой
релаксации. Цоследний процесс не приводит к изменению числа
возбужденных ядер, однако этот механизм передачи энергии
очень важен для понимания некоторых явлений. Кроме постоян-
ных Т1 и Т2 в практике используют и другие характеристики ре-
зонансного поглощения.
Совокупность сигналов ЯМР, т. е. зависимость интенсивности
поглощения от напряженности магнитного поля (или от частоты),
называют обычно спектром ЯМР. Основными его характеристи-
ками являются высота (максимальная интенсивность) и ширина,
измеренная на половине максимальной высоты сигнала.
Конечная ширина сигнала ЯМР показывает, что резонансное
поглощение происходит не строго при одной фиксированной час-
тоте, как можно было ожидать на основании (7.10), а захватыва-
ет некоторый интервал частот. Ширина сигнала зависит от инди-
видуальных особенностей, структуры, агрегатного состояния ве-
щества и других факторов.
Теория ЯМР связывает ширину сигнала Ду с временем
спин-решеточной и спин-спиновой релаксации следующим соот-
ношением:
2пТ1 2пТ2
Кроме того, на ширину сигнала ЯМР влияет неоднородность маг-
нитного поля в разных точках образца, что предъявляет соответ-
ствующие требования к измерительному прибору.
Химический сдвиг. Величина Н0 в уравнении (7.10) характе-
ризует резонансное поглощение по сути дела свободного ядра, ли-
шенного электронной оболочки. Однако ядра одного и того же эле-
мента в разных молекулах при наложении одной и той же частоты
у0 показывают резонансное поглощение при различной напряжен-
ности поля. Если резонансное поглощение частоты у0 ядрами ка-
кой-то молекулы наблюдается при напряженности поля Н,то
Ь,\0 = 2цЛ0 = 2\хН(1 - о). (7.12)
Здесь Н0 следует рассматривать как резонансное магнитное поле
свободного ядра. Величину а называют константой экранирова-
ния. Она характеризует сдвиг резонансной частоты или поля, вы-
званный ближайшим окружением ядра, т. е. так называемый хи-
мический сдвиг. Химический сдвиг практически измеряют по от-
134
ношению к некоторому стандарту. Если Нх и Нст — напряжен-
ности поля, при которых происходит резонансное поглощение
ядрами исследуемого и стандартного веществ соответственно, то
уравнение (7,12) будет иметь вид:
#0 = Яст(1-аст).
Почленное деление этих уравнений дает
(7.13)
При вычитании цо единице из обеих частей уравнения (7.13)
после небольших преобразований получаем
Ъ1^-Ъ^*™. (7.14)
Так как ах 1, то вместо уравнения (7.14) можно записать
а,-аст = 5 = ^9^-т. (7.15)
Величину 8 называют обычно химическим сдвигом и выра-
жают в условных единицах — миллионных долях магнитного по-
н ~ н
ля (м. д.), умножая ст на 10е, т. е.
8 = Я*~Яст106 м. д. (7.16)
Если измеряется не поле, а частота, вместо (7.16) имеем
•8= ^1^106м. д., Г"
где'уж и'у^т — резонансные частоты исследуемого образца и стан-
дарта.
Как видно из уравнения (7.15), исследуемый химический
сдвиг является разностью постоянных экранирования исследуе-
мого вещества и стандарта. Точные измерения химического сдви-
га требуют введения различных поправок, например, на разницу
диамагнитных восприимчивостей исследуемого образца и эта-
лона, на растворитель и т. д. Хотя метод ЯМР можно использо-
вать для изучения очень многих ядер, наиболее важное значение
имеют исследования на ядрах ХН, 19¥ и 31Р, к которым относится
большинство выполненных экспериментальных работ. Наиболее
подробно изучен ЯМР протонов, входящих в различные соедине-
ния. Название ЯМР протонов часто сокращают как ПМР — про-
тонный магнитный резонанс.
135
Схема ЯМР спектрометра. Уравнение (7.10) показывает, что
резрнансное поглощение может быть достигнуто или изменением
напряженности магнитного поля Н0 при постоянной частоте, или
изменением наложенной частоты в постоянном магнитном поле.
Достоинством обычно применяемых приборов, в которых усло-
вия резонанса достигаются за счет изменения напряженности
магнитного поля, является удобство и простота работы, так как
стабилизировать частоту проще, чем поле. Тем не менее иногда
предпочитают изменять частоту при постоянном поле, так как
это позволяет перекрывать более широкую область энергии и ре-
шать другие задачи. Принципиальная схема спектрометра для
наблюдения ЯМР представлена на рис. 7.3.
Ампулу с исследуемым веществом помещают в катушку ра-
диочастотного генератора, которая находится между полюсами
электромагнита. В приборах со стабилизированной частотой и
переменным магнитным полем изменение магнитной индукции
осуществляется генератором (на схеме не указан). При выполне-
нии условия (7.10), т. е. при поглощении энергии поля, детектор
регистрирует некоторое изменение напряжения в контуре, кото-
рое записывается в виде сигнала ЯМР на самопишущем потенцио-
метре или наблюдается на экране осциллографа.
Спектрометр ЯМР содержит сложный набор электронных
устройств, предназначенных для обеспечения высокой стабиль-
ности и точности задаваемых параметров поля. Для успешной ра-
боты прибора необходимо поддерживать частоту и напряжен-
ность магнитного поля с погрешностью порядка 10~6—10"7%.
Качественный анализ и структурные исследования методом
ЯМР. На рис. 7.4 приведен спектр ЯМР протонов этилового спир-
Рис. 7.3. Схема
ЯМР-спектрометра:
Рис. 7.4. Спектр
ЯМР этанола
І —
— магнит; 2 — исследуемое
вещество; 3 — детектор;
— генератор радиочастоты
4 —
136
та. Как видно, протоны —ОН-, =СН2-, —СН3-групп различаются
очень четко. Таким образом, по табличным значениям резонанс-
ных сдвигов или по данным предварительной калибровки мож-
но установить наличие тех или иных атомных группировок в ис-
следуемой молекуле, т. е. получить информацию о ее структуре,
а по площади пика определить число ядер. Применение метода
ЯМР позволило установить структуры многих сложных соеди-
нений. Это один из основных методов исследования в органиче-
ской химии и химии координационных соединений. Методом
ЯМР исследуется также структура кристаллов, кинетика быст-
рых реакций и многие другие свойства веществ и характеристи-
ки реакций.
Количественный анализ методом ЯМР. При количествен-
ном анализе растворов площадь пика может быть использована
как мера концентрации в методе градуировочного графика или
методе добавок. Известны также методики, в которых градуиро-
вочный график отражает концентрационную зависимость хими-
ческого сдвига. .
Применение метода ЯМР в неорганическом анализе основано
на том, что в присутствии парамагнитных веществ происходит
укорочение времени ядерной релаксации. Скорость релаксации
ядер в присутствии парамагнитных веществ выражается уравне-
ниями /
их = А^с; и2 = £2с, (7.17)
где и и2 — скорость соответственно спин-решеточной и
спин-спиновой релаксации данных ядер; кх и к2 — коэффициен-
ты релаксационной эффективности парамагнитных частиц по от-
ношению к данным ядрам, причем индекс «1» относится к
спин-решеточной, а индекс «2» — к спин-спиновой релаксации;
с — концентрация парамагнитных частиц в растворе.
Коэффициенты ^ и к2 зависят от природы анализируемых
парамагнитных частиц и изучаемых ядер, от растворителя, тем-
пературы и некоторых других факторов. По физическому смыслу
коэффициенты релаксационной эффективности соответствуют
скорости релаксации ядер при концентрации анализируемых па-
рамагнитных частиц в 1 моль/л, так как при с = 1 моль/л из
уравнений (7.17) следует, что кх и и2.= к2. Числовые значе-
ния обоих коэффициентов для парамагнитных аква-ионов при-
мерно одинаковы и изменяются от нескольких десятков до 104
(например, АСи2+= 103; АМп2+= 104).
137
Опыт показал, что температурная зависимости коэффициен-
тов релаксационной эффективности невелика и в области темпе-
ратур 15—30 °С не превышает 1—2% на градус, оставаясь во
многих случаях меньше этого значения. Так же незначительно;
влияет на коэффициенты кг и к2 присутствие в растворе диамаг-
нитных солей. Это существенно упрощает разработку аналитиче-
ских методик, позволяя, например, обходиться без химического
отделения диамагнитных примесей.
Измерение скорости релаксации может быть выполнено не-
сколькими методами. Надежным и универсальным является, на-
пример, импульсный вариант метода ЯМР, или, как его обычно
называют, метод спинового эха. При измерениях по этому методу
на'исследуемый образец в магнитном поле через определенные
промежутки времени накладывают кратковременные радиочас-
тотные импульсы в области резонансного поглощения. В прием-
ной катушке появляется сигнал спинового эха, максимальная;
амплитуда которого связана с временем релаксации простым со-
отношением. ;
Для проведения обычных аналитических определений нет
необходимости находить абсолютные значения скоростей релак-
сации. В этих случаях можно ограничиться измерением к&-
кой-либо пропорциональной им величины, например амплитуды
сигнала резонансного поглощения. Измерение амплитуды может
быть выполнено на простой, более доступной аппаратуре.
Существенным достоинством метода ЯМР является широкий
интервал значений измеряемого параметра. С помощью установ-
ки спинового эха можно определять время релаксации от 10~5
до 100 с с погрешностью 3—5%. Это позволяет определять кон-
центрацию раствора в очень широком интервале: от 1—2 до
10~6—10"7 моль/л. Наиболее часто используемым аналитическим
приемом является метод градуировочного графика.
В настоящее время разработаны методики прямого аналитиче-
ского определения многих парамагнитных ионов по скорости ре-/
лаксации протонов и ядер фтора 19Р (ионы элементов середины
четвертого периода таблицы Д. И. Менделеева, лантаноиды и т. д).
Особый интерес представляет применение релаксационного метода
для определения концентрации парамагнитных ионов в движу-
щейся жидкости и для дистанционного определения парамагнит-,
ных веществ в растворе, что позволяет контролировать концентра-
цию парамагнитных веществ в ходе, например, технологического
процесса.
138
В
ф
»
Т.Э.
т.э. Объем
титранта
Рис 7,5, Типы кривых
магнитно-релаксационного
титрования:.
1 — титрование
. меди купферроном;
2 —перманганатометрическое
определение ¥е2+
Помимо прямых определений, ос-
нованных на уравнении (7.17), маг-
нитно-релаксационный метод успеш-
но используется для разработки титри-
метрических методов. В этих методах
используется линейная зависимость
скорости магнитной релаксации ядер
от концентрации парамагнитных ве-
ществ в растворе/Кривая титрования
будет представлять, очевидно, зависи-
мость скорости релаксации ядер (в ус-
ловных единицах) от объема добав-
ленного титранта. Некоторые типы
кривых титрования представлены на
рис. 7.5.
Кривая 1 на этом рисунке отражает изменение скорости ре-
лаксации, когда в результате реакции титрования парамагнит-
ный ион осаждается, образуя диамагнитное соединение. Такой
вид имеет, например, кривая титрования иона меди(П) раство-
ром купферрона. Точка эквивалентности соответствует излому на
кривой титрования.
Вид кривой титрования практически также не изменится, ес-
ли определяемый парамагнитный ион при титровании будет об-
разовывать не осадок, а диамагнитное комплексное соединение в
растворе. Иллюстрацией такого определения может быть, напри-
мер, титрование Ре3+раствором ЭДТА.
Кривая 2 на рис. 7.5 показывает возрастание скорости релак-
сации в ходе титрования. Этот случай реализуется, например,
при титровании иона Ре2+ перманганатом:
5Ре2+ + МпО^ + 8Н+ = 5Ре3+ + Мп2+ + 4Н20
В результате этого титрования образуются ионы Ре3+ и Мп2+, об-
ладающие более высоким коэффициентом релаксационной эф-
фективности, чем вступающие в реакцию ионы ¥е2+. После до-
стижения точки эквивалентности скорость релаксации остается
постоянной. Разработаны также методики титриметрического
определения диамагнитных веществ парамагнитным титрантом
и другие оригинальные методики.
Основное достоинство метода ЯМР — возможность определять
концентрации в широком интервале (от 10~5—10"6 до 1—2 моль/л
и выше), используя для анализа небольшие объемы раствора (0,1—
139
0,5 мл). Погружения каких-либо датчиков в анализируемый рас-
твор не требуется. Измерение ЯМР дает возможность контролиро-
вать концентрацию неустойчивых парамагнитных частиц в рас-
творе, образующихся, например, в результате какой-либо реак-
ции. Большой интерес представляют перспективы автоматизации
контроля с помощью ЯМР, так как метод позволяет проводить
дистанционные определения в движущейся жидкости без отбора
проб анализируемого раствора. Ценной особенностью метода явля-
ется возможность анализа интенсивно окрашенных и мутных рас-
творов в присутствии кислот, щелочей, поверхностно-активных
веществ и т. д.
Однако при всех его Многочисленных достоинствах магнит-
но-релаксационный метод еще недостаточно используется в ана?
литической практике главным образом в связи со сложностью и;
малой доступностью аппаратуры.
7.2.2. Электронный парамагнитный резонанс
Электронный парамагнитный резонанс (ЭПР) открыт в 1944 г.
отечественным физиком Б. К. Завойским. В методе ЭПР, так
же как и в ЯМР, используется резонансное поглощение электро-
магнитных волн веществом в постоянном магнитном поле. Од-
нако ЭПР связан уже с магнитными свойствами электрона. Маг-
нитное поле электрона примерно на три порядка превышает поле
ядра. В отсутствие магнитного поля спиновые энергетические со-
стояния электрона вырождены. При наложении магнитного поля
вырождение снимается и появляются два энергетических уров-
ня: верхний уровень, имеющий спин т8 = 1/2, й нижний со спи-
ном т8 = _1/2* Разность энергий АЕ в этих состояниях составляет
АЕ - gp#,
где#-— фактор спектроскопического расщепления,называемый
обычно g-фактором; 0 ■— магнетон Бора; Н — напряженность
магнитного поля.
Магнетоном называют единицу измерения магнитного мо-
мента, которую иногда рассматривают как «квант» магнитного
момента системы. Магнетон Бора используется при описании
магнитных свойств электрона. У свободного электрона магнит-
ный момент равен одному магнетону Бора, а ^-фактор равен
2,0023.
140
Число частиц на каждом энергетическом уровне может быть
подсчитано по уравнению Больцмана. Отношение этих чисел
равно
-г = ехр
«п2
V кт)'
где п — число частиц на том или ином уровне.
Разность энергий электрона в этих состояниях АЕ невелика,
поэтому заселенность обоих уровней при комнатной температуре
будет примерно одинаковой с очень небольшим преобладанием
состояний с меньшей энергией.
При наложении на эту систему переменного магнитного поля
с частотой у, удовлетворяющей условию
начнется резонансное поглощение энергии поля, и электроны с
нижнего уровня будут переходить на верхний.
Электрон из возбужденного состояния переходит в основное
также за счет релаксационных процессов: спин-решеточной и
спин-спиновой релаксации. Время спин-решеточной релаксации
Тг можно рассматривать как меру взаимодействия неспаренного
электрона с его окружением.
Спектр ЭПР часто представляют не
только в виде зависимости интенсив-
ности поглощения от напряженности
поля (рис. 7.6, а), но в виде зависи-
мости первой производной поглоще-
ния по напряженности от напряжен-
ности магнитного поля (рис. 7.6, б).
Дифференциальный метод дает более
четкое представление о спектре, поло-
жении максимума и полуширине по-
лосы.
Взаимодействие спинов электрона
и ядра вызывает так называемое сверх-
тонкое расщепление спектра ЭПР на
отдельные компоненты, обусловленное
этим взаимодействием. Сверхтонкое
расщепление /дает очень ценную ин-
формацию о природе связи, электрон- рис> 7 б Сигнал эпр.
ной структуре и т. д.
__ а — кривая поглощения;
Чувствительность спектров ЭПР б _ дифференциальная
очень высока: в благоприятных уело- кривая поглощения
141 <
виях может быть зарегистрировано до 10~12 г вещества и обнару-
жено в растворе до 10~9 моль/л примесей. Это характеризует ЭПР
как весьма перспективный аналитический метод. Однако чисто
аналитическое применение ЭПР пока невелико. ЭПР является
уникальным методом исследования кинетики и механизма реак-
ций, в которых принимают участие парамагнитные вещества,
ценнейшим методом изучения процессов с участием радикалов,
процессов комплексообразования в растворе, включая решение
вопроса о дентатности лигандов, строения комплексных соедине-
ний, влияния рН ит. д.
7.3. Рефрактометрические методы анализа
Исследование преломления света при прохождении луча че-
рез границу раздела прозрачных однородных сред (рефракто-
метрия) является, по-видимому, старейшим из оптических мето-
дов, известным еще по работам И. Ньютона, Л. Эйлера, М. В. Ло-
моносова и др.
В 80-е годы XIX в. рефрактометры начали использовать в
практике работы заводских лабораторий и значение рефракто-
метрических методов стало быстро возрастать. Рефрактометриче-
ский метод сохранил свое значение и в настоящее время как ме-
тод анализа сложных смесей, исследования свойств веществ и
взаимодействия в химических системах.
7.3.1. Показатель преломления
и полное внутреннее отражение
При падении луча света на границу раздела двух прозрачных
сред происходит частичное отражение света от поверхности раз-
дела и частичное распространение света в
другой среде (рис. 7.7). Направление луча во
второй среде изменяется в соответствии с за-
коном преломления:
■>7К77777777777777777,
«24
_ вша!
(7.18)
РИС 7.7.
Преломление света
на границе двух сред
Величину л2(отн) называют относитель-
ным показателем (коэффициентом) прелом-
ления второй среды шх отношению к пер-
вой. Показатель преломления по отноше-
142
нию к вакууму называют абсолютним показателем преломле-
ния: ..
п
2(абс) '
8ша2
Но так как для первой среды также можно записать
'і(абс) "
то очевидно, что
*2(отн)"
втаї
в1Па2 81ПавакуумП1(абс) ^і(абс)
(7.19)
т. е. относительный показатель преломления равен отношению
абсолютных показателей преломления. Из уравнения (7.19) по-
лучаем другую форму записи закона преломления:
' ^(абс) б1п а1 = Л2(абс) 81п а2-
Относительный показатель преломления по отношению к
воздуху называют просто показателем преломления п:
абс ^,абс(воздух)^,Л
При атмосферном давлении и комнатной температуре тгабс(воздуха) =
= 1,00027, поэтому
лабс= 1,00027/1. (7.20)
В прецизионных измерениях учитывают зависимость яабс(воздуха) от
давления, температуры и влажности. Однако в подавляющем боль-
шинстве случаев формула (7.20) оказывается вполне пригодной.
Опыт показывает, что если свет переходит, например, из воз-
духа в какую-то конденсированную, более преломляющую среду,
то угол падения всегда больше угла преломления. При переходе
из среды, более преломляющей, в среду, менее преломляющую,
угол преломления а2 оказывается больше угла падения 04. Если
угол преломления а2 = 909 (рис. 7.8), то, очевидно, преломления
Рис. 7.8. Принципиальная схема рефрактометра
143
вообще не произойдет, и поскольку sin 90° = 1, формула (7.18) пе-
реходит в
"2(отн) = Sin а1- (7.21)
Угол а19 при котором преломление не происходит, называют уг-
лом полного внутреннего отражения, а также предельным или
критическим углом. Например, при переходе светового луча из
стекла в воздух под углом в 40° угол преломления составляет 90°
и, следовательно, при угле падения аг > 40° преломления не про-
исходит, а свет будет полностью отражаться от поверхности раз-
дела. Уравнение (7.21) показывает, что по условию полного внут-
реннего отражения можно рассчитать показатель преломления.
Это соотношение часто используют в практике рефрактометрии.
На показатель преломления оказывают влияние как физи-,
ко-химические свойства вещества, так и многие внешние усло-
вия. Волновая теория света связывает показатель преломления
со скоростью света в вакууме сив данной среде V2:
n2=^c/V2.
Показатель преломления зависит от длины волны падающе-
го света, температуры и некоторых других внешних условий.
Температуру и длину волны света, при которой производится из-
мерение, обычно указывают у символа п. Например, запись л||9
означает, что показатель преломления измерен при 25 °С для
желтой D-линии натрия с длиной волны 589 нм. В качестве ниж-
него индекса у п вместо длины волны часто указывают только
буквенный символ линии (D для желтой линии натрия 589 нм,
С для красной линии водорода 656 нм и т. д.) и вместо, например,
пЦ9 пишут яjj5. Величину /lo,, характеризующую показатель пре-
ломления при бесконечно большой длине волны, находят экстра-
поляцией зависимости п = f(X) на X °°.
Показатель преломления и плотность вещества изменяются
симбатно, т. е. с ростом плотности происходит увеличение пока-
зателя преломления. Теоретическими и экспериментальными ис-
следованиями было установлено, что некоторая функция показа-
теля преломления fin) прямо пропорциональна плотности веще-
ства р:
f(n) = гр.
Коэффициент пропорциональности г назвали удельной рефракци-
ей. При умножении г на молярную массу M получают молярную
рефракцию R:
R = Mr.
144
Для выражения функции f{n) и, следовательно, для расчета
рефракции было предложено несколько уравнений. Наибольшее
распространение получила теоретически обоснованная формула
Лоренц—Лорентца:
R_ п2-1М
п2 + 2 р '
Величина рефракции, найденная по этой формуле, практически
не зависит от внешних условий (температуры, давления и т. д.).
В органической химии широко применяется правило адди-
тивности молярных рефракций, в соответствии с которым мо-
лярная рефракция соединения равна сумме атомных рефракций
элементов, образующих это соединение, а рефракция смеси равна
сумме молярных рефракций ее составных частей. Молярную
рефракцию растворов можно поэтому рассматривать как линей-
ную функцию их состава, выраженного в молярных долях. Рас-
считывались также рефракции связей и некоторые другие реф-
рактометрические константы, пользуясь которыми можно опре-
делять рефракции сложных соединений без проведения
экспериментальных измерений. Эти величины представляют ин-
терес и в настоящее время для идентификации органических со-
единений, определения их структуры и проведения различных
физико-химических расчетов. Можно отметить также, что мо-
лярная рефракция по Лоренца-Лорентцу Дл__л является мерой
поляризуемости молекул а:
Дл_л = 2,52 • 1024а. (7.22)
Поляризуемость, как известно, является характеристикой де-
формируемости молекул под действием электрического ПОЛЯ.
Зависимость показателя преломления от длины волны па-
дающего света называют дифракционной дисперсией или просто
дисперсией. Обычно мерой дисперсии считают разность показате-
лей преломления при двух длинах волн. Относительное измене-
ние дисперсии, например, в ряду гомологических соединений
обычно превышает изменение показателя преломления.
7.3.2. Приборы для определения
показателя преломления
Для определения показателя преломления в наиболее широ-
ко применяемых приборах используют измерение угла полного
внутреннего отражения. Принципиальная схема измерительного
ю-7831
145
устройства представлена на рис. 7.8. Основной частью прибора
является измерительная призма 3 из оптического стекла с точно
известным показателем преломления N. Источником света слу-
жит натриевая лампа или газоразрядная трубка (водородная, ге-
лиевая или ртутная), дающая линейчатый спектр. В рефрактог
метре Аббе освещение производится белым (немонохроматич-
ным) светом, однако благодаря призме Амичи, пропускающей;
желтые лучи без изменения, показатель преломления в этих при-
борах относится к D-линии натрия.
Луч света 1 падает на кювету 2, находящуюся на входной
грани АВ измерительной призмы 3. Входная грань находится в
оптическом контакте с исследуемой жидкостью и служит грани-
цей раздела, на которой происходит преломление и полное внут-
реннее отражение. Луч, соответствующий предельному углу ф,
называют предельным лучом. После преломления на границе вы-
ходная грань призмы ВС— воздух он образует с нормалью к гра-
ни ВС угол р. Если угол ф близок к предельному, поле зрения 5
трубки 4 оказывается разделенным на светлую (освещенную) и
темную (неосвещенную) части. В этом состоянии отсчетное уст-
ройство измерительного прибора показывает точную (до десятых
долей градуса) величину угла р. Показатель преломления п ис-
следуемой жидкости рассчитывают по формуле
п = sin a Jn2 - sin2p,
где а —г преломляющий угол измерительной призмы; N — ее по-
казатель преломления.
Вполне понятно, что при измерениях по схеме, изображен-
ной на рис. 7.8, должно соблюдаться условие п < N, т. е. показа-
тель преломления исследуемого вещества должен быть меньше
показателя преломления измерительной призмы. Это, естествен-
но, ограничивает интервал значений тг, доступных для исследова-
ния с данной призмой. В комплект рефрактометра поэтому обыч-
но входит несколько призм, позволяющих работать в различных
диапазонах значений показателя преломления.
Наиболее известны конструкции рефрактометров типа
Пульфриха и типа Аббе. Кроме метода предельного угла для из-
мерения показателя преломления используется метод призмы,
а также иммерсионный, интерференционные и некоторые другие
методы.
Показатель преломления в иммерсионном методе находят
при качественном сравнении исследуемого вещества с эталонны-
'■' , ' 146 Ч
ми средами. Чтобы найти показатель преломления, например,
каких-либо минеральных зерен или кристаллов, их последова-
тельно рассматривают под микроскопом в жидкостях с известны-
ми показателями преломления. С помощью полоски Бекке или
других эффектов определяют, большую или меньшую величину
показателя преломления имеет исследуемое вещество по сравне-
нию с эталонной средой. Полоска Бекке появляется при слабом
нарушении фокусировки микроскопа как тонкая светлая поло-
ска на границе двух сред вследствие преломления света. Для оп-
ределения показателя преломления используется свойство по-
лоски Бекке переходить при поднятии тубуса микроскопа на сре-
ду с более высоким показателем преломления, а при опускании
тубуса — на среду с более низким значением этой величины.
Минимальный размер зерна, при котором обнаруживается этот
эффект, составляет 1—2 мкм. С помощью полоски Бекке улав-
ливают разницу между показателями преломления на 0,001.
Иммерсионный набор для определения показателя преломления
состоит из 50—100 жидкостей с разными показателями прелом-
ления. • ■
Иммерсионный метод широко используется в практике ми-
нералого-петрографических исследований. Большое значение он
имеет для определения состава. бинарных изоморфных смесей
карбонатов, сульфатов и др., при исследовании продуктов хими-
ческой технологии и т. д. Этот метод позволяет определять состав
совместно кристаллизующихся отдельных твердых фаз, отли-
чать двойные соли от механической смеси солей, различать изо-
меры и различные модификации веществ одинакового состава и
решать другие задачи. Он очень удобен при анализе взрывчатых
и ядовитых веществ, так как для анализа требуются ничтожно
малые пробы (миллиграммы) вещества/Однако несмотря на ряд
бесспорных достоинств, иммерсионный метод сравнительно ред-
ко применяется в практике аналитических лабораторий.
7.3.3. Основные рефрактометрические
методики анализа
Разработаны многочисленные рефрактометрические методики
определения составных частей в двухкомпонентных растворах (вод-
ные растворы спиртов, сахара, глицерина, кислот, солей и т. д.).
Чем больше разность показателей преломления компонентов, тем
более высокой будет точность анализа. Показатели преломления
147
многих технически важных смесей сведены в специальные табли-
цы, облегчающие проведение рефрактометрического анализа. Для
определения концентрации раствора обычно используется метод
градуировочного графика, который строится в координатах показа-
тель преломления—концентрация раствора.
Анализ тройных систем значительно сложнее, так как наря^
ду с измерением показателя преломления обычно приходится оп-
ределять еще какое-либо свойство системы (плотность, вязкость
и т. д.). Большое распространение получил рефрактоденсимет-
рическийметод, основанный на измерении показателя прелом-
ления и плотности. При анализе по этому методу строится тре-
угольная диаграмма составов, на которую по эксперименталь-
ным данным для стандартных растворов наносится сетка
изорефракт (линий одинакового показателя преломления) и изо-
денс (линий одинаковой плотности). По измеренным значениям
показателя преломления и плотности анализируемого раствора
на треугольнике составов находят точку, соответствующую этим
величинам. Координаты этой точки прямо указывают на состав
анализируемого раствора. В настоящее время этим методом ана-
лизируется несколько десятков тройных систем, таких, напри-
мер, как метанол-^—этанол—вода, метанол—этанол—ацетон и т. д.
Совершенно необходимым условием успешного анализа является
существенная разница в величине хотя бы одного из измеряемых
свойств.
Менее трудоемки такие методы анализа тройных систем, как
дисперсиометрический и метод извлечения, основанные на ис-
пользовании только рефрактометрических измерений. В диспер-
сиометрическом методе измеряют показатель преломления при
двух длинах волн и на треугольнике составов наносят сетку из
изорефракт и линий одинаковой дисперсии. Дальнейшие опе-
рации не отличаются от приемов рефрактоденсиометрического
метода.
При работе по методу извлечения определяют показателе
преломления тройной системы и один из компонентов количест-
венно удаляют подходящим реагентом. Так, например, анилин
из смеси с толуолом и метилциклогексаном удаляют хлорово-
дородной кислотой, метилэтилкетон из смеси с циклогексаном и
бензолом извлекают водой и т. д. Оставшуюся двойную смесь
можно проанализировать обычным методом градуировочного
графика и далее по треугольной диаграмме установить состав ис-
ходного тройного раствора.
148
Определение всех компонентов в смеси более сложных, чем
тройные, рефрактометрически невозможно/Однако вовсе не ред-
костью является определение в таких смесях одного-двух компо-
нентов.
Например, в растворах типа морской воды рефрактометриче-
ски определяют один-два компонента и сумму остальных. Най-
денная таким образом «соленость» является важной характерис-
тикой морской воды. К этому типу рефрактометрического анали-
за относится определение концентрации жиров и масел в
органических растворителях, анализ полупродуктов и растворов
в сахарном производстве, изготовлении фруктовых соков и дру-
гих напитков, производстве джема и т. д. По типу тройных сме-
сей анализируют лекарственные препараты, кондитерские изде-
лия, косметику ит. д. Широко применяются различные методы
предварительного разделения сложных смесей ■— фракционная
перегонка, экстракция и т. д. — с последующим рефрактометри-
ческим анализом фракций. Специальные рефрактометрические
методы разработаны для анализа нефтяных фракций. Измерение
показателя преломления может быть использовано в титримет-
рическом анализе для построения кривой титрования и обнару-
жения точки эквивалентности. Однако рефрактометрическое
титрование большого распространения не получило.
7.3.4. Рефрактометрические исследования
химического взаимодействия, строения
и других свойств соединений
Работы по применению рефрактометрии к исследованию хи-
мического взаимодействия показали, что по измерениям показа-
теля преломления можно обнаружить только достаточно сильное
взаимодействие, поэтому рефрактометрический метод исследо-
вания химических процессов большого распространения не по-
лучил.
Интенсивное развитие рефрактометрии в начале XX в. в зна-
чительной степени связано с ее применением для исследования
структуры и свойств химических соединений. Данные по моляр-
ной рефракции и дисперсии привлекали внимание как величи-
ны, характеризующие внутренние свойства молекул и практиче-
ски не зависимые от температуры, давления и других внешних
условий/Были установлены некоторые эмпирические закономер-
ности, связывающие рефрактометрические константы со стро-
149
ением соединений. Оказалось, например, что молярная рефрак-
ция /прайс-соединений всегда выше, чем цис-изомеров. В [гомо-
логических рядах рефракции соседних членов отличаются поч-
ти точно на одно и тоже значение и т, д. Рефракция применяет-
ся для исследования поляризуемости, а также электрических,
термических и других свойств веществ. Так, например, по пока-
зателю преломления и диэлектрической проницаемости можно
рассчитать электрический дипольный момент. Для малополяр-
ных жидкостей успешно используется упрощенное уравнение
Онзагера:
V= 10-™Je-ni,
где р — электрический дипольный момент; 8 — диэлектрическая
проницаемость.
По рефрактометрическим данным можно рассчитывать ради-
усы молекул, так как довольно точно соблюдается пропорци-
ональность
ос = kr3,
где а — поляризуемость; г. — радиус молекулы; к — эмпириче-
ский коэффициент, сохраняющий постоянное значение в опреде-
ленных группах веществ.
Поляризуемость рассчитывается из рефрактометрических
данных по уравнению (7.22). Известны и другие применения
рефрактометрии.
7.4. Поляриметрия
Вращение плоскости поляризации было открыто Д. Aparo
(1811) при исследовании кристаллического кварца и Ж. Био
(1815) при исследовании растворов. Поляриметрические измере-
ния, основанные на определении угла вращения, являются обще-
принятыми, официально утвержденными методами анализа в
различных отраслях промышленности, особенно в сахарной.
7.4.1. Вращение плоскости поляризации света
У обычного естественного луча колебания световой волны
происходят во всех плоскостях, перпендикулярных направлению
света. Луч, у которого эти колебания происходят только в ка-
150
кой-то одной плоскости, называют поляризованным, а плоскость,
в которой происходят колебания, — плоскостью колебаний.
Плоскость, перпендикулярная ей, называется плоскостью поля-
ризации. Некоторые кристаллы обладают способностью пропус-
кать свет одного определенного колебания/После прохождения
такого кристалла луч света становится поляризованным. Вещест-
ва, способные изменять плоскость поляризации, называют опти-
чески активными веществами, а неспособные — оптически не-
активными. При прохождении поляризованного света через оп-
тически активное вещество происходит поворот плоскости
поляризации на некоторый угол, называемый углом вращения
плоскости поляризации. Вращение называют правым и считают
положительным (+), если оно происходит по часовой стрелке,
когда смотрят навстречу лучу, и левым и считают отрицатель-
ным (-), если оно происходит против движения часовой стрелки.
Перед названием или химической формулой правовращающего
соединения обычно ставят букву й, а левовращающего — букву I.
Оптически неактивную эквимолекулярную смесь право- и лево-
вращающих изомеров называют рацемическим соединением. Пе-
ред их названием помещают обе буквы, например рацемат яблоч-
ной кислоты называется сй-яблочной кислотой. Прописные бук-
вы В и £ перёд названием или формулой оптически активного со-
единения (обычно моносахарида или а-аминокислоты)
указывают на его принадлежность к стерическим рядам В- или
Ь-глицеринового альдегида, который выбран как соединение
сравнения. К 2):ряду относят соединения, которые можно полу-
чить из £>-формы глицеринового альдегида, а к Х-ряду — из его
Ь-формы:
СНО СНО
н—С—ОН но—с—н
СН2ОН СН2ОН
£>-СН2ОНСНОНСНО £-СН2ОНСНОНСНО
Вращение плоскости поляризации кристаллическими веще-
ствами является важной характеристикой кристалла, которая
широко используется в технике микроскопии ив кристалло-
химии. Оптическая активность газообразных молекул или рас-
творенных веществ связана с особенностями строения моле-
кул (например, отсутствием у них центра и плоскости симметрии
ит. д.). -V
151
Угол вращения плоскости поляризации а связан с концент^
рацией оптически активного вещества в растворе с (г/мл) й тол^
щиной слоя раствора I (дм) соотношением
а:
= аудгс,
(7.23)
где ауд — удельное вращение плоскости поляризации.
Оно зависит от природы вещества, длины волны поляризуе-
мого света, растворителя и температуры. Символ означает,'
что удельное вращение плоскости поляризации относится к 20 °С
и желтой В-линии натрия. Уравнение (7.23) лежит в основе коли-
чественных поляриметрических методов. Молярное вращение
плоскости поляризации Ф равно произведению ауд на молярную
массу М:
Ф = аудМ.
Особый интерес представляет зависимость удельного или мо-
лярного вращения плоскости поляризации от длины волны све-
та. Эту зависимость называют дисперсией оптического вращения
(ДОВ).
Оптическое вращение растет с уменьшением длины волны.
В области полосы спектра поглощения оно достигает максимума
и затем быстро падает до минимума, после которого медленно
возрастает (эффект Коттона). Кривая эффекта Коттона пред-
ставлена на рис. 7.9.
Величину
ф
100
называют амплитудой, а расстояние по оси длин волн Ъ — шири-
ной эффекта Коттона.
Специальный аналитический ин-
терес вызывает область спектра, в ко-
торой удельное или молярное враще-?
ние изменяет свой знак. Длину волны
света, при которой вращения плоскос-
ти поляризации не происходит, назы-
вают длиной волны нулевого враще-
ния. Она находится как точка пересев
чения кривой зависимости удельного?
Рис. 7.9. Кривая дисперсии
оптического вращения
или молярного вращения от длины
волны с осью длин волн. ■■••у"Д
Плоскополяризованная волна, какі
известно, состоит из двух циркулярно
152
поляризованных компонент (левовращающей Ь и правовращаю-
щей Л), каждая из которых обладает своим показателем прелом-
ления пь и пк в данной среде и своим молярным коэффициентом
поглощения ех и 8Д. Разность молярных коэффициентов поглоще-
ния характеризует круговой (циркулярный) дихроизм:
Дб = 8£( - 8Д.
Он может быть также выражен молярной эллиптичностью 0:
9 = 2,303^^ - 8Д) = 3300 Де.
Характеристики эффекта Коттона, кривая ДОВ и зависи-
мость кругового дихроизма от длины волны дают ценную инфор-
мацию о структуре, стереохимии и конформации органических и
координационных соединений.
7.4.2. Приборы для поляриметрических измерений
В любом приборе для поляриметрического анализа (поляри-
метре) есть поляризатор и анализатор, между которыми находит-
ся трубка с анализируемым раствором. Если поляризатор и ана-
лизатор установлены так, что их плоскости поляризации парал-
лельны между собой, то в отсутствие анализируемого вещества
свет будет беспрепятственно проходить через оба устройства й на-
блюдаться в зрительную трубу. Если в отсутствие анализируемо-
го вещества анализатор повернуть на 90°, т. е. ориентировать
так, что его плоскость поляризации будет перпендикулярна
плоскости поляризатора, то, очевидно, поляризованный свет че-
рез анализатор проходить не будет. Это положение «на темно-
ту» . При введении между поляризатором и анализатором опти^
чески активного анализируемого раствора в зрительной трубе
появится свет. Чтобы вновь добиться «темноты», анализатор не-
обходимо повернуть на некоторый угол, равный углу вращения
плоскости поляризации анализируемым веществом. Величина
угла вращения может быть непосредственно прочитана на отсчет-
ном устройстве зрительной трубы.
В качестве поляризатора и анализатора обычно используют
призму Николя (или просто николь), изготовляемую из исланд-
ского шпата (СаС03). Осветителем часто служит натриевая лампа.
Оптическая система поляриметра включает также устройство для
повышения точности установки на «темноту». Это могут быть до-
полнительные призмы Николя или так называемые пластинки
бикварца. Пластинка бикварца состоит из лево- и правовращаю-
153
щего кварца и помещается после поляризатора перед трубкой с
анализируемым раствором. При предварительной установке на:
«темноту», когда николи взаимопараллельны, и в отсутствие ана-
лизируемого раствора пластинка бикварца окрашивает поле зри-
тельной трубы в сплошной серо-фиолетовый цвет. Введение ана-1
лизируемого раствора вызывает резкий цветовой эффект: одна по*
ловинка поля становится красной, другая — синей. Поворотом
анализатора восстанавливают первоначальную серо-фиолетовую
окраску всего поля и по углу поворота анализатора находят угол
вращения плоскости поляризации анализируемым раствором.
Интересным видоизменением поляриметра является сахари-
метр, применяемый специально для анализа растворов сахара.
В отличие от обычного поляриметра, осветителем в котором слу-
жит натриевая лампа или другой источник монохроматического »
света, в сахариметре для этой цели используется белый немонохрог \
магический свет. Применение такого осветителя оказалось воз-^
можным вследствие случайного совпадения вращательной диспер- (
сии кварца и растворов сахара. Раствор сахара вызывает правое
вращение плоскости поляризации. Это вращение в сахариметрах
компенсируют введением в луч света клина из левовращающего-
кварца. Вследствие равенства дисперсии оптического вращения»
кварца и раствора сахара компенсация происходит при всех дли- ,
нах волн, что и позволяет использовать для освещения сахаримет-
ров белый свет. Определения на сахариметре характеризуются вы- ,
сокой точностью, так как толщину клина можно измерить очень: [
точно. Клином называют устройство из двух клинообразных плас- "
тинок левовращающего кварца и плоской пластинки правовра-
щающего. Положение клина часто калибруют в единицах кон- *
центрации, или так называемых международных сахарных граду-1"
сах (0в). Величине сто сахарных градусов (100 °8) соответствует
раствор сахарозы, содержащий 26 г в 100 мл раствора при 20 °С и 4
длине трубки 2 дм. ^
Вращение плоскости поляризации при различных длинах
волн (дисперсию оптического вращения) исследуют с помощью,*
спектрополяриметра, осветитель которого дает монохроматиче*
ский свет заданной длины волны в широком спектральном ин~ ^
тервале обычно с помощью кварцевой диспергирующей призмы. ^
В новейших конструкциях поляриметров и спектрополяри? ^
метров для измерения интенсивности света применяют фотоэле* |
менты и фотоумножители, нередко соединенные с электронным ц
записывающим потенциометром. Особую ценность они имеют '
для исследований в ультрафиолетовом участке спектра, недо* -
ступном для визуальных наблюдений. ч №
' 154 . . .;,!
..: ■-. : ... ■. ■ м
7.4.3. Поляриметрические методики
Основу количественных поляриметрических методик состав-
ляет уравнение (7.23), связывающее угол вращения плоскости
поляризации с концентрацией раствора. Однако непосредствен-
ный расчет по уравнению (7.29) производится сравнительно ред-
ко, так как удельное вращение плоскости поляризации ауд также
зависит от концентрации. Наиболее часто в практике использует-
ся метод градуировочного графика в координатах угол вращения
а—концентрация с Особенно широко применяют поляриметри-
ческие методики анализа в сахарной промышленности и некото-
рых других отраслях пищевой промышленности (масложировой
и т. д.), в фармацевтических производствах, парфюмерии и т. д.
Смесь оптически активных веществ может быть проанализи-
рована спектрополяриметрическим методом, т. е. измерением уг-
ла вращения при разных длинах волн. Методика этого анализа
очень близка к методике спектрофотометрического определения
смеси двух окрашенных веществ. Расчетные формулы сохраняют
свою применимость и в спектрополяриметрическом методе, если
молярные коэффициенты поглощения 6 заменить удельными
вращениями ауд, учесть длину трубки и переход к другой кон-
центрационной шкале. Точность анализа возрастает, если одно
из анализируемых соединений (а еще лучше — оба) в исследуе-
мой области спектра имеет длину волны нулевого вращения, так
как при этой длине волны угол вращения будет определяться
концентрацией только второго компонента.
Спектрополяриметрические измерения дают ценную инфор-
мацию также о структуре и других свойствах органических и ко-
ординационных соединений. Изменение стереохимического рас-
положения отдельных групп и другие структурные особенности
соединений находят отражение в основных характеристиках
кривой эффекта Коттона. Как правило, спектрополяриметриче-
ские данные рассматриваются совместно со спектрофотометриче-
скими, так как такое сопоставление показывает, какая полоса в
спектре поглощения ответственна за эффект Коттона. Кроме то-
го, теорема Кронига—Крамера дает возможность по спектру по-
глощения предсказать кривую дисперсии оптического вращения
и наоборот. При интерпретации спектрополяриметрических дан-
ных используют также и другие эмпирические обобщения,
связывающие спектрополяриметрические, спектрофотометриче-
ские, структурные и другие физико-химические характеристики
и свойства веществ.
155
7.5. Нефелометрия и турбидиметрия
Нефелометрический и турбидиметрический методы приме-
няют для анализа суспензий, эмульсий, различных взвесей и
других мутных сред. Интенсивность пучка света, проходящего,
через такую среду, уменьшается за счет рассеивания и других
процессов взаимодействия света со взвешенными частицами.
Нефелометрический метод определения концентрации осно-
ван на измерении интенсивности света, рассеянного взвешенны-
ми частицами, а турбидиметрический — на измерении интенсив-
ности света, прошедшего через эту среду.
7.5.1. Рассеяние света
Рассеяние света частицами, размеры которых больше длины
волны облучающего света, называют рассеянием Ми по фамилии
ученого, разработавшего теорию этого явления (1908).
Интенсивность рассеянного света этими частицами» подчиня-
ется закону Рэ лея: ,
1 = 1о^^чгШ(1 + 0082 Р)> '; (7-24)
где пх и п2 — показатели преломления частиц и среды соответ-
ственно; N — общее число светорассеивающих частиц; У1 —'■■
объем данной частицы; X — длина волны падающего света; г —-
расстояние до приемника рассеянного света; р — угол между па-
дающим и рассеянным светом.
В присутствии крупных частиц, диаметр которых измеряет-
ся, например, десятками нанометров, закон Рэлея нарушается,
однако это не вызывает больших затруднений в аналитической
работе, так как связь концентрации с интенсивностью устанавли-
вают по градуировочным графикам. При исследовании заданной
системы показатели преломления % и п2 остаются постоянными,
величины г и р определяются конструкцией прибора и тоже не
меняются. В этих условиях уравнение (7.24) переходит в
1 = М0^. (7.25)1
Множитель 1Д4 указывает на быстрое возрастание интенсивнос-
ти рассеянного света с уменьшением длины волны падающего г
света.. "
156
Концентрация, по определению, характеризует число частиц
в единице объема:
с - Л- , (7.26)
NAV . v '
где V —- объем суспензии; NA — постоянная Авогадро.
Подставляя уравнение (7.26) в (7,25), получаем
I-kI0^^l. (7.27)
При строгом соблюдении условий приготовления объемы суспен-
дированных частиц получаются примерно одинаковыми и их раз-
меры вполне удовлетворительно воспроизводятся от опыта к опы-
ту. При постоянных F, Vt, X уравнение (7.27) принимает вид
I = k'I0c
или
I/I0 = k'c. (7.28)
Уравнение (7.28) показывает, что отношение интенсивности
рассеянного света к интенсивности падающего пропорционально
концентрации взвешенных частиц. Градуировочный график в
координатах I/IQ как функция с будет линеен.
Из уравнения (7.28) следует, что
Л^^Ч^с-^^ (7.29)
т. е. кажущаяся оптическая плотность уменьшается с рос-
том концентрации, так как с увеличением концентрации увели-
чивается число рассеивающих частиц и интенсивность рассеян-
ного света возрастает.
В соответствии с уравнением (7.29) график в координатах
Акаж — lg с будет линеен в противоположность графику в коорди-
натах^^ - с.
При достаточном разбавлении раствора интенсивность света
Iv прошедшего через суспензию или другую мутную среду, под-
чиняется уравнению, имеющему при постоянстве некоторых ус-
ловий вид, аналогичный уравнению закона Бугера—Ламберта—
Вера:
lg £ ■- -Ыс, (7.30)
0
где I — толщина слоя, а k иногда называют молярным коэффици-
ентом мутности раствора.
157
Уравнение (7.30) справедливо в условиях строгого постоянст^
ва условий получения суспензии. 1
7.5.2. Приборы для нефелометрических
и турбидиметрических определений
Пучок света интенсивностью /0 от электрической лампы на*
каливания падает на кювету с анализируемой суспензией или
эмульсией и частично рассеивается взвешенными частицами?
Интенсивность рассеянного света равна /, интенсивность света;
прошедшего через кювету, 1Г Рассеянный свет наблюдается
обычно под прямым углом к направлению падающего света. Ин-*
тенсивность рассеянного света и света, прошедшего через анали-
зируемую смесь, может быть измерена с помощью фотоэлементов
или визуально. В выпускаемом промышленностью нефелометре
НФМ интенсивность рассеянного света измеряется визуально.
Для измерения интенсивности света, прошедшего через взвесь,
успешно используются фотоэлектроколориметры. Количествен-
ные определения обычно проводятся методом градуировочного
графика. В случае нефелометрических измерений в соответствии
с уравнением (7.28) или (7.29) график строится в координатах
1/10 - с или - 1ц с, а при турбидиметрических определениях —
в координатах А - с. Известны также методики турбидиметриче-
ского титрования, основанные на реакциях образования осадков
малорастворимых соединений. При титровании, например, магт
ния фосфатом оптическая плотность в ходе титрования возраста?
ет, так как увеличивается концентрация взвешенных частиц ■
фосфата магния, а по достижении точки эквивалентности остает-
ся постоянной.
Основным достоинством нефелометрических и турбидиметри-
ческих методов является их высокая чувствительность, что осо-
бенно ценно по отношению к элементам или ионам, для которых
отсутствуют цветные реакции и не разработаны колориметриче-
ские (фотометрические) методы. В практике широко применяют
нефелометрическбё определение хлорида и сульфата в природных
водах и аналогичных объектах. По точности турбиДиметрия и не-
фелометрия уступают фотометрическим методам, что связано?
главным образом, с трудностями получения суспензий, обладаю-
щих одинаковыми размерами частиц, стабильностью во времени
ит. д. К обычным сравнительно небольшим погрешностям фото-
метрического определения добавляются ошибки, связанные с не-
158
достаточной воспроизводимостью химико-аналитических свойств
суспензий.
7.6. Практическое применение
Спектры комбинационного рассеяния света используют в
анализе продуктов, главным образом, органической химии. Ме-
тодом ЯМР анализируются органические и неорганические веще-
ства. Магнитно-релаксационный метод, основанный на зависи-
мости скорости релаксации ядер от концентрации парамагнит-
ных соединений, успешно применяют в анализе многих веществ
(например, ионов Ре2+, Со2+, №2+ и др.) в широком интервале
концентраций. Помимо прямых определений магнитно-релакса-
ционный метод используют в титриметрическом анализе.
Рефрактометрический метод анализа применяют в техноло-
гическом контроле в пищевой промышленности, в клинических
медицинских исследованиях, при анализе кондитерских изде-
лий, молока, масла, различных жиров и т. д. Нередко показатель
преломления включается в ГОСТ как характеристика качества
вещества (например, стирола). Содержание сахара, жиров, белка
и т.п. измеряют также поляриметрическим методом. Этот же ме-
тод используют при анализе лекарственных препаратов, напри-
мер пенициллина. Нефелометрию и турбидиметрию используют
в анализе взвесей, эмульсий и т. п. гетерогенных систем. Нередко
такие системы получают специально для определения ионов
(обычно анионов), не образующих окрашенных соединений (на-
пример, С1~, вО|" и т. д.).
77. Общая характеристика методов
Рассмотренные в настоящей главе методы успешно применя-
ются в анализе многих систем, имеющих большое практическое
значение, и часто позволяют получать информацию о составе
анализируемого вещества наиболее простьщ и быстрым путем.
Особую ценность и значение имеют некоторые из этих методов в
анализе объектов пищевой, фармацевтической и иных отраслей
промышленности. Однако методы этой главы используются,
главным образом, для решения специфичных задач и по масшта-
бам практического применения они уступают многим другим, бо-
лее универсальным и распространенным методам.
159
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Какое происхождение имеют спектры комбинационного рассеяния?
2. На чем основан качественный анализ по спектрам комбинацион-
ного рассеяния?
3. На чем основаны методы количественного анализа по спектрам
комбинационного рассеяния?
4. В чем сущность ядерного магнитного резонанса (ЯМР)?
5. Какие ядра обладают парамагнитными и какие диамагнитными
свойствами?
6. Как рассчитывается химический сдвиг, что он характеризует?
7. Какие требования предъявляются к стандартам в ЯМР? Какие
вещества используются в качестве стандартов при снятии спект-
ров ЯМР?
8. В чем сущность качественного и структурного анализа по спект-
рам ЯМР?
9. Что такое время ядерной релаксации, какова связь его с кон-
центрацией парамагнитного вещества?
10. Какие методы количественного анализа используют в ЯМР?
11. В чем сущность магнитно-релаксационного титрования?
12. Какие величины входят в уравнение Рэлея?
13. Как зависит интенсивность рассеянного света: а) от бпектраль-
ной характеристики падающего излучения; б) от размера рассе-
ивающих частиц?
14. Какой вид имеют графики зависимости^^ от с; Гкаж от си^
от^с?
15. В чем сущность: а) метода эталонной шкалы; б) метода градуиро-.
вочного графика; в) метода добавок?
16. Какое свойство используется в нефелометрических методах ана-
лиза: а) поглощение света атомами; б) рассеяние света частица-
ми; в) излучение света молекулами и ионами?
17. Какой свет рассеивается в наибольшей степени частицами, на-
ходящимися в растворе во взвешенном состоянии: а) желтый;
б) синий; в) зеленый; г) красный?
глав а 8
Кондуктометрия
8.1. Электрическая проводимость растворов
Электрической проводимостью называют способность вещества'
. проводить электрический ток под действием внешнего электриче-
ского поля. Ее единицей измерения является сименс (См).
160
Перенос электричества в проводниках первого рода — метал-
лах — осуществляется движением электронов по проводнику в
направлении от отрицательного полюса источника тока к поло-
жительному. В проводниках второго рода — растворах электро-
литов —- перенос электричества осуществляется движением
ионов. Анионы движутся к аноду, катионы — к катоду.
Закон Ома остается справедливым и для растворов электро-
литов:
где Е — разность потенциалов между электродами, В; / — сила
тока, А; Л — сопротивление, Ом; р — удельное сопротивление,
Ом • см; I ■— расстояние между электродами, см; в — сечение
(площадь поверхности), см2.
Величина х = 1/р, обратная удельному сопротивлению элект-
ролита, называется удельной электрической проводимостью.
Удельная электрическая проводимость равна электрической про-
водимости 1 см3 раствора, находящегося между параллельными
электродами площадью 1 см2 при расстоянии между ними 1см,
или, другими словами, — это электрическая проводимость столба
раствора длиной 1 см и площадью поперечного сечения 1 см2. Ее
единицей измерения является См/см.
В разбавленных растворах удельная электрическая проводи-
мость с увеличением концентрации растет, при некоторой доста-
точно высокой концентрации достига-
ет максимума и затем уменьшается.
На рис. 8.1 приведены типичные при-
меры этой зависимости. Электриче-
ская проводимость слабого электроли-
та уксусной кислоты значительно ни-
же соответствующей величины для
растворов НС1 или КОН. Возрастание
электрической проводимости с ростом
концентрации в растворах умеренно
высоких концентраций происходит
вследствие увеличения числа ионов с
концентрацией. Однако в концентри-
рованных растворах возникают и дру-
гие эффекты, приводящие уже к
уменьшению электрической проводи-
мости. В концентрированных раство-
рах возрастают силы межионного
взаимодействия, вследствие чего про-
х, См/см
10 с, моль/л
Рис. 8.1. Электрическая
проводимость:
1 — НС1; 2 — КОН;
3 — СНоСООН
Н-7831
161
исходит образование межионных ассоциатов или ионных пар, уве-
личивается вязкость раствора и проявляются другие эффекты,
снижающие скорость движения ионов и вызывающие уменьшение
электрической проводимости. Как суммарный результат действия
этих факторов, на кривой электрической проводимости возникает
максимум. Для аналитических измерений обычно используется,
участок кривой с возрастающей удельной электрической проводит
мостью, т. е. область разбавленных и умеренно концентрирован-
ных растворов. :
Эквивалентной электрической проводимостью называют проводи-
мость раствора, содержащего 1 моль эквивалента вещества и на-
ходящегося между двумя параллельными электродами, расстоя-
ние между которыми 1 см. Ее единицей измерения является
См • см2/(моль экв).
Очевидно, площадь электродов такова, что между ними по-
мещается раствор, содержащий 1 моль эквивалентов вещества.
Удельная и эквивалентная проводимости связаны соотноше-
нием
Д = 1000х/с, (8.1)
где с — молярная концентрация эквивалента, моль/л. ■<:-
В области сравнительно невысоких концентраций эквива-
лентная электрическая проводимость электролитов обычно рас-
тет с уменьшением концентрации раствора и повышением темпе-
ратуры.
У полностью диссоциированных (так называемых сильных)'
электролитов в области разбавленных растворов (0,001 М и мень-
ше) концентрационная зависимость проводимости выражается і
уравнением
2,
\
X = X0-aJc, (8.2) 'I
где А,0 — предельная эквивалентная ,
электрическая проводимость силь- ;
~ : ного электролита при бесконечном'й
v ^ разведении; а — константа. Эта за* |
_——2^ висимость представлена на рис. 8.2
• 4с прямой 1. VT
Рис. 8.2. Зависимость С Уменьшением концентрации,
эквивалентной электрической электролита эквивалентная элект^
проводимости от концентрации: рическая проводимость возрастает и
1 — сильного; 2 — слабого в области бесконечно больших ра,3г
электролитов бавлений стремится к предельному;;
162
значению Х0, характеризующему электрическую проводимость
гипотетического бесконечно разбавленного раствора.
Константа а уравнения (8.2) нашла истолкование в теории
Дебая—Хюккеля, развитой Онзагером. Уменьшение эквивалент-
ной электрической проводимости теория Дебая—Онзагера объяс-
няет эффектами электрофоретического и релаксационного тор-
можения. Оба эффекта связаны с существованием вокруг иона
ионной атмосферы из противоположно заряженных ионов.
Электрофоретический эффект вызывается тем, что центральный
ион под действием электрического поля движется в одном на-
правлении, а ионная атмосфера, имеющая противоположный за-
ряд, — в противоположном и тормозит движение иона. Релакса-
ционное торможение обусловлено процессами разрушения и
формирования ионной атмосферы при движении иона. Концент-
рационная зависимость эквивалентной электрической проводи-
мости в этой теории дается уравнением
Х = Х0 + (АХ0 + В)*/с,
где А — коэффициент, характеризующий электрофоретический
эффект, а В — релаксационный. Величины Ал В определяются
температурой, вязкостью и диэлектрической проницаемостью
растворителя и вычисляются теоретически.
Предельная эквивалентная электрическая проводимость Х0
может быть представлена суммой предельных электрических
проводимостей, или предельных подвижностей ионов:
~ ^о(+) ^о(-)» (8.3)
где А,0(+) и ^о(-) — предельная эквивалентная электрическая про-
водимость, или предельная подвижность соответственно катиона
и аниона.
Закон аддитивности электрической проводимости растворов
электролитов при бесконечном разведении, выражаемый уравне-
нием (8.3), установлен Ф. Кольраушем в 1879 г. еще до появле-
ния теории электролитической диссоциации. Соотношение (8.3)
называют также законом независимого движения ионов.
Числовые значения подвижностей ионов в водном рас-
творе при комнатной температуре находятся в пределах 30—
70 См • см2/(моль экв) и лишь у ионов Н+ и ОН" они су-
щественно превышают эти значения (XQ (Н+) = 350; XQ (НГ) =
= 199 См • см2/(моль экв), что связано с особым (эстафетным) ме-
ханизмом перемещения этих ионов в электрическом поле, сущ-
ность которого заключается в следующем.
11*
163
Ион водорода Н+ в водном растворе находится в виде гидро-
ксоний-иона Н30+, который обменивается протонами с соседни-;
мй молекулами воды. При наложении электрического поля пере-
ход протона от Н30+ к Н20 будет проходить преимущественно в
направлении поля, как это можно представить схемой: (
Н Н Н Н
I I I I
н—о—н + о--нн—о +н—о—н
Н30+ Н20 Н20 Н30+
Наряду с обычной миграцией ионов Н30+, как и других,
ионов, электричество будет переноситься протонами по эстафет-
ному механизму, что и приводит к более высокой подвижности
Н30+ по сравнению с подвижностью других ионов. Аналогичным
образом объясняется высокая подвижность ионов ОН"", Интерес-.
но отметить, что в ацетоне, нитробензоле и других растворите-;
лях, не содержащих гидроксид-ионов, подвижность Н+ примерно
такая же, как и у других однозарядных ионов.
Концентрационная зависимость электрической проводимости |
слабых электролитов имеет более сложный характер (см. рис. 8.2,
кривая 2), чем у сильных электролитов. Это объясняется техл, что
на электрическую проводимость слабых электролитов влияют не я
только электрофоретический и релаксационный эффекты, как это ;
наблюдается у сильных электролитов, но и увеличение степени
диссоциации электролита с разбавлением раствора, вызывающее;;
в области разбавленных растворов более быстрое, чем у сильных |
электролитов, увеличение электрической проводимости. Данные
по электрической проводимости растворов слабых электролитов*
часто используются для расчета констант диссоциации. ;^
Электрическая проводимость неводных растворов имеет ряд С
особенностей. Существенное влияние на электрическую проводи/
мость оказывает диэлектрическая проницаемость растворителя,,
Концентрационная зависимость электрической проводимости в?;>
растворах с высокой диэлектрической проницаемостью анало- *
гична соответствующей зависимости для водных растворов. На "
кривой электрической проводимости растворов с малой диэлект- *
рической проницаемостью растворителя — хинолице, пиридине
и т. д. .— образуются минимумы и максимумы, что объясняется, •
главным образом, сложным характером взаимодействия ионов ей
растворителем и между собой. Одним из наиболее широко приме*
няемых неводных растворителей в аналитической химии являет-
■ 164. , ' Ш
ся диоксан, имеющий низкую диэлектрическую проницаемость
(-2) и смешивающийся с водой в любых отношениях.
Электрическая проводимость растворов с ростом температу-
ры повышается. В водных растворах повышение составляете—
2,5% на градус. Температурную зависимость предельной под-
вижности ионов часто выражают уравнением
^0(0 " ^0(25°) И1 + <*)(* ~ 25)]>
где а — эмпирический коэффициент, зависящий от природы
ионов и растворителя.
Электролит в поле тока высокой частоты. Токи, имеющие
частоту порядка мегагерц и десятков мегагерц, называют токами
высокой частоты. При таких частотах в растворе начинают иг-
рать роль эффекты молекулярной, или деформационной, иориен-
тационной поляризации.
Под действием электрического поля электроны любой моле-
кулы будут оттягиваться в сторону положительного электрода, а
ядра — в сторону отрицательного. Это явление получило название
молекулярной, или деформационной, поляризации. Полярные
молекулы в электрическом поле обладают также ориентационной
поляризацией, стремящейся ориентировать дипольные молекулы
вдоль поля. Поляризация обоих типов вызывает кратковремен-
ный электрический ток (ток смещения). Кроме того, поляризация
молекул приводит к существенному изменению диэлектрической
и магнитной проницаемости раствора, что открывает новую воз-
можность исследования свойств раствора при титровании.
Полная проводимость цепей (к), имеющих емкость или ин-
дуктивность, как известно из электротехники, состоит из актив'
ной и реактивной А,реакт составляющих:
^ ~ ^акт ^ Ареакт»
при этом реактивная компонента проводимости, зависящая от
емкости, равна
Ч(реакт) ^ юс>
а зависящая от индуктивности
"1, (реакт)
где со — частота; с— емкость; Ь — индуктивность; у = 4~Л.
Таким образом, за изменением в составе раствора, например,
при титровании можно следить по изменению проводимости и
емкости или по изменению проводимости и индуктивности.
165
8.2. Схема установки для определения
электрической проводимости
Принципиальная схема для проведения кондуктометриче-
скйх измерений представлена на рис. 8.3. Это обычный мостик
Уитстона, питаемый переменным током от генератора 2. Посто-
янный ток нежелателен, так как вызывает электролиз раствора.
В то же время применение моста переменного тока приводит к
появлению так называемого реактивного сопротивления вслед-
ствие конечной величины емкости измерительной ячейки Rx в
цепи, что особенно заметно при работе с растворами, имеющими
большое сопротивление. По этой причине, кстати, нельзя свести
к нулю силу тока в диагонали моста гв. Сопротивление Дм извест-
но (магазин сопротивлений). Положение передвижного контакта
в подбирается таким образом, чтобы нуль-инструмент 1 не пока-
зывал ток (или ток был минимальным). Тогда сопротивление
ячейки Rx можно рассчитать по формуле
где 1г и 12 — длины плеч реохорда при компенсации, пропорци-
ональные сопротивлениям Rx (ав) и R2 (вб).
Для питания моста обычно используется ток частотой при-
мерно 1000 Гц, вырабатываемый звуковыми генераторами типа
ЗГ-1,ЗГ-10, ЗГ-33 и др. В качестве эталонного сопротивления
включают магазины сопротивлений типа Р-517 М, Р-58 и т. д.
Нуль-инструментом может быть телефон, гальванометр или ос-
циллограф. Широко применяется для этой цели осциллографиче-
ский индикатор нуля типа ИНО-ЗМ, а также электронный инди-
катор нуля Ф-510 и аналогичные устройства.
Промышленность выпускает комплектные приборы для оп-
ределения электрической проводимости растворов — мосты перег
менного тока и кондуктометры. Некоторые из них, например
кондуктометр «Импульс» К Л 1-2, имеют цифровой отсчет пока-
заний в единицах удельной электрической проводимости.
Конструкции измерительных ячеек весьма разнообразны.
В прямой кондуктометрии обычно применяют ячейки с жестко
закрепленными в них электродами (рис. 8.4, а). В методах кон-
дуктометрического титрования наряду с ячейкой этого типа час-
то используют так называемые погружные электроды (рис. 8.4,
б), позволяющие проводить титрование в любых сосудах, в котФ
рых можно разместить электроды. и
166
Рис. 8.3. Мостик Уитстона Рис. 8.4. Ячейки
для кондуктометрических измерений:
а — ячейка с жестко закрепленными
электродами; б — погружные электроды
Экспериментально измеряемая величина сопротивления рас-
твора зависит не только от размера электродов и расстояния меж-
ду ними, но и от их формы и взаимного расположения, объема
раствора и других факторов, не всегда поддающихся точному
учету, так как токопроводящим является не только тот объем
раствора, который заключен между электродами. Действитель-
ная электрическая проводимость раствора, конечно, не зависит
от формы или взаимного расположения электродов или ка-
ких-либо других факторов, а определяется лишь концентрацией
раствора, природой компонентов и температурой. Истинная
электрическая проводимость раствора х пропорциональна экспе-
риментально измеренной величине х':
х = ктс ,
где к — константа сосуда.
Это очень важная характеристика ячейки. Она зависит от
площади электродов, расстояния между ними, от формы сосуда и
объема раствора, проводящего ток. Константу сосуда находят
экспериментально по электрической проводимости стандартных
растворов с хорошо известными значениями х в широкой области
температур и концентраций. Обычно в качестве стандартных ис-
пользуют водные растворы хлорида калия.
О', - . " : - •
р 8.3. Прямая кондуктометрия
д...;; Методы прямой кондуктометрии основаны на том, что в об-
ласти разбавленных и умеренно концентрированных растворов
электрическая проводимость растет с увеличением концентра-
г
ции электролита. В практической работе обычно используют за-
ранее построенную градуировочную кривую зависимости элект-
рической проводимости раствора от концентрации тех или иных
электролитов. В связи с относительно близкими значениями под-
вижностей ионов кондуктометрические измерения дают инфор-
мацию, главным образом, лишь об общей концентрации ионов в
растворе. Малая селективность кондуктометрического метода яв-
ляется одним из его существенных ограничений.
Кондуктометрическое определение физико-химических свойств
и характеристик веществ. Данные по электрической проводи-
мости разбавленных растворов послужили в свое время экспери-
ментальным фундаментом теории электролитической диссоци-
ацииАррениуса.
Если слабая кислота НЬ в разбавленном растворе концентра-
ции диссоциирует по схеме
нь^н+ + ь-
то степень ее диссоциации а может быть найдена по электриче-
ской проводимости раствора:
где X — экспериментальное значение эквивалентной электриче-
ской проводимости; Х0 — эквивалентная электрическая проводи-
мость при бесконечном разбавлении, рассчитываемая по таблич-
ным данным как сумма подвижностей ионов.
Константа диссоциации кислоты рассчитывается по уравнению
к - [Н+][1г] а* о _ X* .о (*л)
Если табличные данные о подвижностях ионов отсутствуют,
величины Кнъ иХ0 можно найти из экспериментальных данных
по электрической проводимости растворов исследуемой кислоты
при нескольких концентрациях. Для этого придадим уравнению
(8.4) вид
и обе части поделим на КщХ§Х, После небольших упрощений бу-
дем иметь
1 _ г в Ьс£ъ
168
В координатах - - Хс^ь это будет прямая с угловым коэффициен-
том * , отсекающая на ординате отрезок, равный 1/Х0. После-
дующее развитие теории растворов электролитов позволило в
значительной степени усовершенствовать расчет, учесть коэффи-
циенты активности, й в настоящее время кондуктометрические
измерения довольно широко используют для расчета констант
равновесия.
Данные по электрической проводимости растворов применя-
ют также для определения растворимости малорастворимых со-
единений. Концентрацию насыщенного раствора малораствори-
мого полностью диссоциированного соединения состава 1:1 рас-
считывают по формуле
я = Ю00хнас
где хнас— удельная электрическая проводимость насыщенного
раствора малорастворимой соли.
Кондуктометрическйм методом можно исследовать также
кинетику химических реакций, когда участниками или продук-
тами реакции являются ионы.
8.4. Кондуктометрическое титрование
Измерения электрической проводимости растворов широко
применяют в титриметрическом анализе для определения точки
эквивалентности (кондуктометрическое титрование). В методах
кондуктометрического титрования измеряют электрическую
проводимость раствора после добавления небольших определен-
ных порций титранта и находят точку эквивалентности графиче-
ским методом с помощью кривой в координатах х - V (титранта).
Практически в этом методе могут быть использованы такие хи-
мические реакции, в ходе которых достаточно заметно изменяет-
ся электрическая проводимость раствора или происходит резкое
изменение (обычно возрастание) электрической проводимости
после точки эквивалентности (реакции кислотно-основного взаи-
модействия, осаждения и т. д.).
В методе хронокондуктометрического титрования тйтрант
подается в анализируемый раствор непрерывно или небольшими
одинаковыми дозами через строго определенные промежутки вре-
мени. Одновременно на диаграммной ленте самописца производит-
ся соответственно непрерывная или точечная запись кондуктометр
рической кривой в координатах показание прибора—время. Пока-
169
зания прибора пропорциональны электрической проводимости.
Концентрация вещества в этом методе рассчитывается по времени,
затраченному на титрование. Так как скорость подачи рабочего
раствора постоянна и точно известна, время титрования прямо
пропорционально объему реактива, израсходованному на титрова-
ние. Идея хронокондуктометрического титрования используется в
конструкции различных автотитраторов, выпускаемых промыш-
ленностью.
8.4.1. Реакции кислотно-основного взаимодействия
При титровании сильной кислоты, например HCl, сильным
основанием, например раствором NaOH, в растворе в любой мо-
мент находятся ионы Н+, СГ~, Na+ и ОН", а электрическая прово-
димость раствора будет определяться их концентрациями и под-
вижностями.
На рис. 8.5, а приведена схема, отражающая качественный ха-
рактер изменения концентрации ионов при титровании, необходи-
мая для уяснения кривой кондуктометрического титрования.
Величина сС1_ в процессе титрования практически изменять-
ся не будет, а cNa+ будет монотонно возрастать. Величина бу-
дет уменьшаться от исходной величины практически до нуля в
точке эквивалентности, а cQH_, практически равная нулю в точке
эквивалентности, после точки эквивалентности будет возрастать.
Таким образом, изменение электрической проводимости доточки
эквивалентности будет определяться действием двух взаимно про-
тивоположных тенденций: понижающей за счет уменьшения сн+
и возрастающей за счет увеличения ск+. Результирующая этих
с
Na+
С
т.э.
V(NaOH)
т.э.
F(NaOH)
а)
Рис. 8.5. Кондуктометрическое титрование сильной кислоты:
а — изменение концентрации ионов при титровании; б — изменение .
удельной электрической проводимости (т. э. — точка эквивалентности)
170
вкладов АВ (рис. 8.5, б) показывает резкое уменьшение электри-
ческой проводимости до точки эквивалентности. Падение прово-
димости вызывается уменьшением концентрации иона Н+, имею-
щего при 25 °С подвижность 350 См • см2/(моль экв), что намного
превышает подвижность иона Ыа+ [50 См • см2/(моль экв)].
После точки эквивалентности начинается резкий подъем
электрической проводимости (ветвь БС), так как в растворе будет
нарастать концентрация ионов Ыа+ и ОН", подвижность которых
составляет 199 См • см2/(моль экв). Однако возрастание по ВС
будет более пологим, чем уменьшение по АВ, так как подвиж-
ность иона ОН" почти в 2 раза меньше подвижности иона водо-
рода.
Точка эквивалентности в кондуктометрическом титровании
обычно находится путем графического построения. Как видно из
рис. 8.5, б, экспериментальные значения электрической прово-
димости раствора в непосредственной близости от точки эквива-
лентности особого значения не имеют. Для построения использу-
ются области недотитрованных и перетитрованных растворов.
В некоторых случаях, например при титровании очень разбав-
ленных растворов, обе ветви кривой титрования сглаживаются и
для определения точки эквивалентности приходится применять
более сложные построения.
На рис. 8.6 схематично показано изменение концентрации
реагирующих частиц при титровании слабой кислоты НЬ силь-
ным основанием ЫаОН
НЬ + ЫаОН = ЫаЬ + Н20
и изменение удельной электрической проводимости раствора при
этом титровании.
В.
У(ШОВ)
т. э.
б)
7(ЫаОН)
Рис. 8.6. Кондуктометрическое титрование слабой кислоты:
а — изменение концентрации частиц; б — изменение
удельной электрической проводимости при титровании
171
Концентрация недиссоцииро-
ванных молекул кислоты НЬ моно-
тонно падает практически до нуля.
Концентрация анионов 1г, равная в,
начале титрования концентрации
ионов Н+, в ходе титрования воз-
^7 т.э. 7(^аОН) Растает, а после точки эквивалент-
ности остается практически неиз-
Рис. 8.7. Кривая менной. Концентрация ионов Н+
кондуктометрического ,
титрования смеси сильной падает, ионов Ка+ возрастает.
и слабой кислот Удельная электрическая прово-
димость раствора до точки эквива-
лентности (рис. 8.6, б, ветвь АВ) несколько возрастает, так как при
титровании происходит увеличение концентрации ионов Ыа+ и!".'
Концентрация ионов Н+ в растворе слабой кислоты и ее соли, полу-
чившейся при титровании, невелика и уменьшение [Н+] в ходе
титрования не вызывает столь резкого падения электрической про-
водимости, какое наблюдалось при титровании сильной кислоты.
При титровании кислот средней силы на кривой АВ может
возникнуть минимум электрической проводимрсти, отражаю-
щий суммарный эффект уменьшения концентраций ионов Н+ и
возрастания концентрации ионов Ыа"1" и 1г. После точки эквива->
лентности наблюдается резкий рост электрической проводимос-
ти, вызванный появлением в растворе ионов ОН" с высокой под-
вижностью (рис. 8.6, б, ветвь ВС).
' На рис. 8.7 приведена кривая титрования смеси сильной и
слабой кислот. Кривая имеет два излома, соответствующие двум
точкам эквивалентности: первая показывает объем щелочи, по-
шедшей на титрование сильной кислоты, а вторая дает общий
объем щелочи, израсходованный на титрование обеих кислот.
С увеличением константы диссоциации слабой кислоты первый
излом будет становиться менее резким, а второй — более четким,
и, наоборот, чем слабее кислота, тем резче будет первый излом,
и более закругленным второй.
8.4.2. Реакции осаждения
Вид кривой кондуктометрического титрования по методу1
осаждения зависит от концентрации и подвижности ионов и рас-*
творимости образующегося соединения. Чем меньше ПР продукт
та реакции, тем резче выражен излом кривой титрования в точкф
эквивалентности. Результаты анализа 0,1 М растворов бывают
172
вполне удовлетворительными, если у бинарного соединения ПР <
< 10~5. У более растворимых соединений точка эквивалентности
устанавливается с трудом, так как кривая титрования плавно за-
кругляется. Излом на кривой титрования становится менее чет-
ким также с уменьшением концентрации раствора, и, например,
при анализе 10~3 М растворов произведение растворимости про-
дукта реакции должно быть уже не больше чем 10~9. Введение в
анализируемый водный раствор органического растворителя по-
нижает растворимость, поэтому излом на кривой титрования ста-
новится более резким.
Влияние подвижности ионов проявляется в наклоне кривой
титрования до точки эквивалентности. После точки эквивалент-
ности проводимость во всех случаях будет расти, так как увели-
чивается концентрация ионов в растворе.
Титрование, например, растворимой соли бария сульфатом
происходит по уравнению реакции
Ва(Ж>3)2 + Ка2804 = Ва804| + 2КаЖ)3
До точки эквивалентности электрическая проводимость раствора
будет несколько падать, так как вместо Ва(М03)2 (^о (Ва2+) ^
= 63,6) в растворе появится эквивалентное количество ЫаЖ)3
(Х0 (Ка2+)== .5.0,1), т. ё. в растворе появятся катионы с меньшей ве-
личиной подвижности (Ыа"1" вместо Ва2+). Первая же капля рас-
твора Ыа2804 после точки эквивалентности вызовет резкое увели-
чение электрической проводимости благодаря возрастанию кон-
центрации электролита в растворе. Электрическая проводимость
перетитрованных растворов, естественно, также будет возрас-
тать. Кондуктометрическое титрование по методу осаждения не
лишено и обычных ошибок титрования, характерных для метода
осаждения — ошибок за счет соосаждения или адсорбции, за счет
немгновенной скорости установления равновесия между осадком
и раствором и т. д.
8.4.3 Реакции комплексообразования
Для кондуктометрического титрования катионов в качестве
титрантов могут быть использованы растворы различных кислот
и оксикислот (щавелевой, винной, лимонной и др.), комплексо-
нов и других лигандов. Наибольшее практическое значение име-
ет кондуктометрическое титрование катионов двузамещенной
солью этилендиаминтетрауксусной кислоты (ЭДТА).
173
т.э. Г(Ыа2Н2Ь)
Рис 8.9. Кривая кондуктометрического
титрования Са2+ раствором ЭДТА
в буферном растворе (рН ~ 10)
При титровании, например, Ге3+ раствором ЭДТА (У4-) про-
текает реакция ;-
Ге3++ Н2У2" = ГеУ-+ 2Н+
в результате которой выделяются ионы Н+ и растет электриче-
ская проводимость раствора. После точки эквивалентности,
электрическая проводимость раствора падает, так как выделив-
шиеся ионы Н+связываются анионом Н2У2_
Н+ + Н2У2" - н3у-
■ ' \
Кривая такого титрования представлена на рис. 8.8.
Несколько иной вид имеет кривая титрования катиона в бу-
ферном растворе (рис. 8.9). Выделяющиеся ионы Н+ в этом слу-
чае взаимодействуют с протоноакцепторным компонентом буфер-
ной системы и не дают поэтому столь заметного вклада в элект-
рическую проводимость раствора. До точки эквивалентности
электрическая проводимость раствора несколько увеличивается,
что связано главным образом с увеличением концентрации ионов
Ыа+, вводимых с титрантом, а после точки эквивалентности рез-
ко возрастает, так как увеличивается концентрация титранта.
8.4.4. Реакции окисления—восстановления
Окислительно-восстановительные реакции сравнительно ред-
ко используются в практике кондуктометрического титрования.
Возможности кондуктометрии здесь несколько сужаются в связи
с тем, что реакцию титрования нередко приходится проводить в,
присутствии большого количества электролитов в сильнокислой
т.э. F(Na2H2L)
Рис. 8.8. Кривая
кондуктометрического
титрования Fe3+ раствором ЭДТА
174
среде и т. д. В таких растворах не всегда удается с достаточной
точностью определить изменение электрической проводимости,
связанное с протеканием реакции титрования. Известные ограни-
чения накладывает и скорость некоторых реакций окисления-
восстановления, не всегда достаточно высокая при обычной тем-
пературе. Повышение температуры оказывается малоэффектив-
ным, так как становится крайне необходимым термостатирование
ячейки, а существенное увеличение электрической проводимости
раствора затрудняет установление точки эквивалентности.
8.5. Высокочастотное титрование
Установки для высокочастотного титрования во многом от-
личаются .от установок обычной низкочастотной кондуктомет-
рии. Ячейка с анализируемым раствором при высокочастотном
титровании помещается или между пластинками конденсатора,
или внутри индукционной катушки (рис. 8.10). Соответственно
этому в первом случае ячейку называют конденсаторной, или ем-
костной, или С-ячейкой, а.во втором — индуктивной или Ь-ячей-
кой. В ячейках высокочастотного титрования электроды не со-
прикасаются с исследуемым раствором, что! является одним из
существенных достоинств метода.
Изменения в ячейке, происходящие в результате реакций
титрования, вызывают изменения в режиме работы высокочас-
тотного генератора. Индуктивная Ь-ячейка с анализируемым рас-
твором включается в цепь колебательного контура (помещается
внутрь катушки индукции). Изменение состава раствора при тит-
ровании в такой ячейке вызывает изменение индуктивности, что
легко фиксируется микроамперметром через несложную схему.
В конденсаторных С-ячейках при
титровании раствора вследствие из-
менения диэлектрической прони-
цаемости происходит сдвиг рабочей
частоты генератора, что устанавли-
вается с помощью измерительного
конденсатора. При построении кри-
вой титрования показания прибора
откладывают как функцию объема
добавленного титранта. Промыш-
ленностью выпускаются стандарт-
ные высокочастотные титраторы.
а)
б)
Рис. 8.10. Ячейка
для высокочастотного
титрования:
а — емкостная (С-ячейка);
б — индуктивная (Ь-ячейка)
175
8.6. Практическое применение
Прямое измерение электрической проводимости является
наиболее эффективным методом контроля качества дистиллиро-
ванной воды в лабораториях, технической воды в так называемых
тонких химических или фармацевтических производствах, в тех-
нологии водоочистки и оценке загрязненности сточных вод, теп-
лотехнике (питание котлов) и т. д. Кондуктометрические датчики
с успехом применяются в автоматизированных схемах контроля
производства в некоторых отраслях химической, текстильной и
пищевой промышленности, гидроэлектрометаллургии и т. д. Раз-
работана методика кондуктометрического определения малых ко-
личеств углерода (10~2—10~3%) в сталях и металлах. Методика
включает сожжение образца в токе кислорода, поглощение С02
раствором Ва(ОН)2 и измерение его электрической проводимости.
Содержание углерода находят по градуировочной кривой.
Методы прямой кондуктометрии используют для контроля
качества молока, различных напитков и пищевых продуктов.
Простота и высокая точность кондуктометрических измере-
ний, возможность использования полученных данных в автома-
тизированных схемах контроля и управления и другие достоин-
ства метода электрической проводимости вызывают большой ин-
терес к этому методу и в настоящее время. Однако прямее
кондуктометрические измерения весьма чувствительны к влия-
нию примесей, особенно примесей кислотно-основного характера
в связи с резким различием подвижностей ионов Н+ и ОН" по
сравнению с подвижностями других ионов.
Обширную область применения имеет кондуктометрическое
титрование. Сильные минеральные кислоты в водном растворе
(НС104, НС1, НЖ)3 и др.) титруются щелочью при больших и до-
статочно малых концентрациях (до 10~4 моль/л). Так же титруют-
ся сильные основания (ЫаОН, КОН и др.) сильными кислотами.
Легко титруются муравьиная, уксусная и другие кислоты средней
силы. Кривые кондуктометрического титрования ряда органиче-
ских кислот (янтарной, адипиновой и др.) при титровании слабым
основанием имеют более резко выраженный излом в точке эквива-
лентности, чем кривые титрования сильным основанием. Эти кис-
лоты титруют раствором аммиака, причем в реакцию вступают
оба протона. Слабые основания могут титроваться сильными и
слабыми кислотами. Легко титруются, например, этаноламиньх
растворами уксусной кислоты. Практическое значение имеет кон-
дуктометрическое титрование солей аммония и других слабых ос-
нований растворами щелочей и титрование солей слабых кислот
176
(ацетатов, фенолятов и др.) сильными кислотами. Аминокислоты
(глицин, аланин, валин и др.) титруются сильными основаниями.
Крндуктометрически могут быть оттитрованы смеси слабых
кислот, смеси слабых оснований, а также смеси кислот или осно-
ваний с солями слабых кислот или слабых оснований.
Особенно широкие возможности титрования различных элект-
ролитов и их смесей открывает применение органических и вод-
но-органических растворителей: водно-диоксанового, водно-ацето-
нового, водно-спиртовых, ледяной уксусной кислоты и др. В этих
растворах анализируются трех-, четырех- и пятикомпонентные
смеси (например, смеси НС1 + СН3СООН + ЫН4С1 + СбН5ОН;
ЫаОН + ЫаВ02 + С2Н5ЫН2 + СН3СООЫа + ЫаЖ>2 и др.).
Методом кондуктометрического титрования определяют мно-
гие катионы и анионы. Нитратом серебра титруют хлорид, бро-
мид, иодид, цианид, тиоцианат, оксалат, ванадат, тартрат, сали-
цилат и некоторые другие анионы. Титрованием в среде 90%-го
спирта определяют С1~ в природных водах при содержании поряд-
ка 10 мкг. Содержание I" и С1~ в смеси может быть определено без
предварительного разделения. Титрование ацетатом или хлори-
дом бария применяют для определения сульфата, хромата, карбо-
ната, оксалата, цитрата и других анионов обычно при добавлении
в анализируемый раствор спирта. Сульфаты таким методом опре-
деляют в природных водах и аналогичных объектах.
Кондуктометрическое титрование раствором ЭДТА применя-
ется для определения ¥е3+, Си2+, №2+, Со2+, Ъп2\ РЬ2+, Сс12+,
Са24", Mg2+ и других катионов. Катионы, образующие очень ус-
тойчивые комплексы, как, например, ¥е3+, Си2+, Ш2+, Со2+ и не-
которые другие, титруются в нейтральном или слабокислом рас-
творе. Некоторые смеси катионов могут быть проанализированы
прямым кондуктометрическим титрованием без предварительно-
го химического разделения. Например, ионы Ге3+ могут быть оп-
ределены в присутствии 2п2+, Со2+, Сс12+, ¥е2+,Мп2+и других ка-
тионов. Кондуктометрическим титрованием можно определить
общую жесткость воды.
В методе высокочастотного титрования может быть исполь-
зована практически любая химическая реакция: кислотно-основ-
ного взаимодействия, осаждения и т. д. в водном и неводном рас-
творах. Кривые высокочастотного титрования имеют такой же
вид, как и кривые обычного кондуктометрического титрования.
Концентрационная область титрования слабых кислот и основа-
ний высокочастотным методом остается примерно такой же, как
и в обычном кондуктометрическом титровании.
12-7831
177
Широкие возможности для развития новых методов анализа
открывает использование неводных растворителей. Высокочастот-
ное титрование проводят в ледяной уксусной кислоте, диметилформ-
амиде, смесях диоксан—вода, ацетон—вода и других смешанных
растворителях. В ледяной уксусной кислоте методом высокочастот-
ного титрования можно определить НС104 в присутствии ЬШ03 по
взаимодействию с раствором пиридина, серную кислоту можно тит-
ровать в присутствии 20-кратного количества фосфорной, что пред-
ставляет практический интерес в производстве фосфатов. В ледя-
ной уксусной кислоте титруют также алкалоиды, антибиотики и
другие продукты фармацевтической промышленности.
Образование малорастворимых сульфата бария, галогенидов
серебра и других осадков составляет основу определения анионов
в природных водах и промышленных стоках методом высокочас-
тотного титрования.
8.7. Общая характеристика метода
Кондуктометрические методы характеризуются высокой экс-
прессностью, простотой и доступностью измерительных прибо-
ров, удобством работы и достаточной точностью. Ценной особен-
ностью кондуктометрических методов является возможность про-
ведения автоматического и Дистанционного анализа. Прямые
кондуктометрические измерения имеют погрешность 1—2%, при
соблюдении специальных условий она снижается до 0,2%.
Погрешность кондуктометрического титрования без термо-
статирования растворов обычно оценивается величиной пример-
но в ±(2—3)%. Особое значение вообще для кондуктометриче-
ских измерений имеет температура в связи с довольно большим
температурным коэффициентом электрической проводимости:
изменение температуры на 1° вызывает изменение электриче-
ской проводимости на 2—-3%. Термостатирование растворов су-
щественно увеличивает точность метода.
Основным достоинством метода высокочастотного титрова-
ния является возможность анализа любых агрессивных сред, так
как электроды с анализируемым раствором не соприкасаются.
Электроды можно поместить, например, с наружной стороны
трубопровода, по которому протекает жидкость, и получать та-'
ким образом информацию о составе раствора в любой момент вре-
мени. Методом высокочастотного титрования с успехом могут
быть проанализированы различного рода мутные раствору, взве-
си, эмульсии, окрашенные растворы и т. д.
178
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. В чем сущность кондуктометрических методов анализа?
2. Какие величины входят в уравнение закона Кольрауша?
3. Как влияет на электрическую проводимость: а) природа электро-
лита и растворителя; б) концентрация электролита (сильного,
слабого); в) температура?
4. В каких областях применяют методы прямой кондуктометрии?
5. Каковы особенности кондуктометрического титрования?
6. Какие эффекты возникают в растворе электролита под действи-
ем тока высокой частоты?
7. В чем сущность высокочастотного титрования и каковы особен-
ности этого метода анализа?
8. Каковы достоинства и недостатки кондуктометрического метода
анализа?
глава
Потенциометрия
9.1. Электродный потенциал.
Уравнение Нернста
Потенциометрические методы основаны на измерении элек-
тродвижущихсил (ЭДС):
Е — Е-^ ~~.Е29
где Ё —электродвижущая сила; Ег и Е2 — потенциалы электро-
дов исследуемой цепи.
Потенциал электрода Е связан с активностью и концентраци-
ей веществ, участвующих в электродном процессе, уравнением
Нернста:
Е = Е° + Щ 1п = Е° + Щ 1п г[6*3/ох , (9.1)
аТей пР [геа]уге(1
где Е° — стандартный потенциал редокс-систёмы; В, — универ-
сальная газовая постоянная, равная 8,312 ДжДмоль • К); Т — аб-
солютная температура, К; ^ — постоянная Фарадея, равная
96485 Кл/моль; п — число электронов, принимающих участие в
электродной реакции; аох, аге& —- активности соответственно окис-
ленной и восстановленной форм редокс-системы; [ох], [ге<1] —- их
молярные концентрации; уох, уге& — коэффициенты активности.
1?* 179
Е =\Е° при аох = ared = 1, причем имеется в виду гипотетиче-
ский стандартный 1 М раствор, в котором коэффициент актив-
ности каждого растворенного вещества равен 1, а чистые вещест-
ва находятся в наиболее устойчивом физическом состоянии при
данной температуре и нормальном атмосферном давлении.
Подставляя Т = 298,15 и числовые значения констант в урав-
нение (9 Л), получаем для 25 °С
£ = jgo+Mgg ig.^ ^o+Qiggg ig Еох^о* . (9.2)
n ared n [red]yred
Однако потенциал отдельного электрода экспериментально
определить невозможно. Относительные значения электродного
потенциала находят, комбинируя данный электрод со стандарт-
ным водородным электродом, который является общепринятым
международным стандартом. Потенциал стандартного водородно-
го электрода принят равным нулю при всех температурах, поэто-
му потенциал данного электрода — это, в сущности, ЭДС элемен-
та, состоящего из данного и стандартного водородного электрода.
Конструктивно стандартный водородный электрод представ-
ляет собой платинированную платиновую пластинку, омываемую
газообразным водородом при давлении 1,013 • 105 Па (1 атм) и
погруженную в раствор кислоты с активностью ионов Н+, равной
единице. При работе водородного электрода протекает реакция
Н2 (г) = 2Н+ + 2е~
В практической работе вместо хрупкого и нередко капризно-
го водородного электрода применяют специальные, более удоб-
ные в работе стабильные электроды сравнения, потенциал кото-
рых по отношению к стандартному водородному электроду точно
известен.
Уравнение (9.2) можно переписать
где
Я = Я°С) +0,069 lg Joxl ^ (9.3)
п [red]
'Е°('Уш. е° + 0,059 lg 1^
п Yred
Величину Е0^ называют формальным потенциалом. Как вид-у
но, формальный потенциал характеризует систему, в которой кон-j
центрации (а не активности) всех участников равны 1,0 моль/л. г
Формальный потенциал включает в себя коэффициенты активное- \
ти, т. е. зависит от ионной силы раствора. Если у = 1, то Е°^ = Е°,
180
т. е. формальный потенциал совпадает со стандартным. Точность та-
кого приближения для многих расчетов оказывается достаточной.
Природа возникновения потенциала различна. Можно выде-
лить следующие три основные класса потенциалов, которые не
исчерпывают, конечно, всего многообразия. Это:
1. Электродные потенциалы.
2. Редокс-потенциалы.
3. Мембранные потенциалы.
Хотя под термином «электродный потенциал» нередко име-
ют в виду любой потенциал, независимо от механизма его воз-
никновения, в более узком понимании —- это потенциал непо-
средственно связанный с материалом электрода. Например, цин-
ковый электрод:
гп2+ + 2е~ =
Егп2+/гп в Егп2+/гп + ^1п агп*+ = Егп2+/гп + а^2+ *
Активность свободного металла принимается равной едини-
це. Электродные потенциалы отличаются от редокс-потенциалов,
для которых материал электрода не имеет значения, так как они
химически инертны по отношению ко всем веществам в растворе,
и от мембранных, для которых разность потенциалов на мембра-
не измеряется с помощью пары других (в принципе, возможно,
одинаковых) электродов.
Потенциометрические методы анализа известны с конца про-
шлого века, когда Нернст вывел (1889) известное уравнение
(9.1), а Беренд сообщил (1883) о первом потенциометрическом
титровании. Интенсивное развитие потенциометрии в послед-
ние годы связано, главным образом, с появлением разнообразных
типов ионоселективных электродов, позволяющих проводить
прямые определения концентрации многих ионов в растворе, и
успехами в конструировании и массовом выпуске приборов для
потенциометрических измерений.
Потенциометрические методы анализа подразделяют на пря-
мую потенциометрию (ионометрию) и потенциометрическое тит-
рование. Методы прямой потенциометрии основаны на прямом
применении уравнения Нернста (9.1) для нахождения активнос-
ти или концентрации участника электродной реакции по экспе-
риментально измеренной ЭДС цепи или потенциалу соответст-
вующего электрода. При потенциометрическом титровании точ-
ку эквивалентности определяют по резкому изменению (скачку)
потенциала вблизи точки эквивалентности.
' '.л ■ 181 ;/■'
9.2. Схема установки
для потенциометрических измерений
Для проведения потенциометрического анализа обычно соби-
рают гальванический элемент, на одном из электродов которого
протекает электрохимическая реакция с участием определяемого
иона или иона, реагирующего с определяемым. При схематичном
изображении различных гальванических элементов или электро-
дов используют условную запись, достаточно полно отражающую
состав и характерные особенности элемента. Форма и символика
схематического изображения гальванических элементов установ-
лены решением ИЮПАК. По этим правилам формулы веществ, на-
ходящихся в одном растворе, записывают через запятую, а грани-
цу между электродом и раствором или между разными растворами
обозначают вертикальной чертой |. Двойная вертикальная черта ||
показывает, что так называемый диффузионный потенциал, воз-
никающий на поверхности раздела растворов разного состава, све-
ден к минимуму или элиминирован с помрщью солевого мостика.
Так, например, гальванический элемент, состоящий из водородно-
го и хлорсеребряного электродов, условно может быть изображен
схемой Р1;, (Н2) 10,1 М Н2в04 || 0,1 МКС11 AgCl; Ag. Электродвижу-
щая сила (ЭДС) такой гальванической цепи обычно измеряется
компенсационным методом, когда ЭДС исследуемого элемента точ-
но компенсируется внешним источником напряжения и через эле-
мент тока практически не проходит, и, следовательно, в системе не
протекают процессы, ведущие к заметным химическим или кон-
центрационным изменениям за счет электролиза.
Потенциометрическое титрование можно проводить также не-
компенсационным методом или с применением биметаллических
электродов, не взаимодействующих с
Я определяемым веществом.
П"~ " Принципиальная схема потен-
циометрической установки изобра-
жена на рис 9.1. Реохорд (линей-
* ный делитель напряжения) АВ пита-
ется током через реостат Д, которым
регулируют подаваемое напряжение.
Подвижный контакт С позволяет по-
давать в цепь напряжение, необхо-
1
Г
1
1
С
" ^
1 ,
димое для компенсации электродви-
жущей силы стандартного (£ст) или
Рис 9.1 Схема /т? \ ^ V;
потенциометрической исследуемого (^) элемента. Вклюг
установки чение Яст или Ех в п;епь производит-,
182
ся переключателем К1У а кратковременное замыкание цепи клю-
чом К2* В.момент компенсации ЭДС гальванометр б тока не пока-
зывает.
Падение напряжения на концах реохорда АВ (Едд) по закону
Ома равно
Еав-Шав- (9.4)
где /сила тока; — сопротивление реохорда. Между точка-
ми А и С падение напряжения составляет
ЕАС = тАС. (9.5)
Сопротивление реохорда пропорционально его длине:
Вав 888 Ыав>
^ас ~ ЫаС> '
где к — коэффициент пропорциональности.
Вместо уравнений (9.4) и (9.5) можно поэтому записать:
Е^кП^у (9.6)
ЕАс = ШАС. (9.7)
При совместном решении уравнений (9.6) и (9.7) получаем
Если переключатель Кг замкнут на Ех, уравнение (9.8) при-
нимает вид
Е^Е^, (9.9)
а если на ЕСТ, то
Е^Е^. (9.10)
Ыв
Деление уравнения (9.9) на (9.10) и небольшое упрощение дает
ЕХ = ЕСТ1^. (9.11)
В качестве стандартного используется элемент Вестона, обла-
дающий постоянным й воспроизводимым значением ЭДС. По
уравнению (9.11) и экспериментальным данным рассчитывается
ЭДС исследуемого элемента. Для удобства работы на реохорд АВ
обычно подается такое напряжение через реостат Д, чтобы отно-
183
шение ЕСТ/1АС в уравнении (9.11)
ст
было целочисленным, например
равным 2 мВ/деление. Тогда зна-
чение ЭДС исследуемой цепи полу-
чают умножением показаний ре-
охорда 1АС на установленную цену
деления.
Эта принципиальная схема ком-
пенсационных измерений сохраня-
ется ив современных потенциомет-
рах, использующих вместо реохор-
да шунтирующие декады (рис 9.2).
Декады аЪ и с<1 состоят каждая из
десяти катушек одинакового сопро-
Рис. 9.2. Принципиальная • тивления. На клеммы аЪ так же,
схема шунтирующих декад как на КОНЦы реохорда, подается на-
пряжение Е, тогда на клеммах ей
падение напряжения составит 0,1Е независимо от положения пере-
ключателя (1—2, 2—3 ит. д.). Если падение напряжения на каж-
дой катушке декады аЪ составляет, например, 0,1 В, то на каждой
катушке декады ей оно будет 0,01 В, и в положении на рис. 9.2 на
выходных клеммах В потенциометра оно составит 0,24 В.
Таким образом, потенциометр из четырех декад дает возмож-
ность определить падение напряжения с точностью 0,0001 В.
Специальная цепь со стандартным элементом позволяет подавать
на первую декаду потенциометра напряжение точно в 1 В. При
работе потенциометра ЭДС исследуемого элемента непосредст-
венно отсчитывается по цифрам, показывающим положение пе-
реключателя на каждой декаде. При работе со стеклянным'
электродом, исследовании неводных растворов и т. д. обычный
потенциометр оказывается неприменимым, так как внутреннее
сопротивление таких гальванических элементов очень велико.
В таких установках применяют электронные потенциометры, в
которых гальванометр заменен на электронную схему с высоким
входным сопротивлением.
9.2.1. Стандартный гальванический элемент
В качестве стандартного элемента в потенциометрии исполь-
зуется нормальный элемент Вестона, электродвижущая сила
которого обладает строго постоянным воспроизводимым значени-
ем, сохраняющимся в течение многих лет, и незначительным ;
184
температурным коэффициентом. Электродами элемента Вестона
являются ртуть (положительный полюс) и насыщенная амальга-
ма кадмия, содержащая 12,5% Cd (отрицательный полюс), а
электролитом — насыщенный водный раствор по отношению к
CdS04 • 8/3Н20 и Hg2S04. Чтобы обеспечить существование насы-
щенного раствора, в элемент в контакте с электролитом вводят
кристаллы этих соединений. Электрохимическая цепь элемента
Вестона имеет вид
- Cd(Hg) | Cd2+, SOf", Hg22+ | Hg +
При его работе протекает электрохимическая реакция
Cd + Hgf + ^ 2Hg + Cd2+
Электродвижущая сила элемента Вестона может быть рас-
считана по уравнению
Е = 1,01830 - 4,06 • 10-5(^ - 20) - 9,5 • 10"7(* - 20)2.
В обычной аналитической работе квадратичным членом обычно
пренебрегают.
9.2.2. Исследуемый гальванический элемент
Исследуемый гальванический элемент обычно состоит из ин-
дикаторного электрода и электрода сравнения, индикаторным
называю^ потенциал которого зависит от концентра-
ции (активности) определяемого иона. Потенциал электрода
сравнения должен оставаться постоянным независимо от проте-
кания каких-либо реакций в анализируемом растворе. Электро-
движущая сила исследуемого элемента выражается как разность
между потенциалом электрода сравнения (2?ср) и потенциалом
индикаторного электрода СЕИНД):
Е = Еср- Еияд + Яд,
где £д '.— диффузионный потенциал, или потенциал жидкостного
соединения.
9.2.3. Индикаторные электроды
Потенциал индикаторного электрода связан уравнением
Нернста (9.2) с концентрацией (активностью) определяемого
иона. Индикаторный электрод должен удовлетворять ряду требо-
185
Необходимо, чтобы его потенциал был воспроизводим и уста-
навливался достаточно быстро. При исследовании потенциала
металлического электрода в растворе его соли, и в некоторых
других случаях индикаторный электрод должен быть обратим.
Электрод должен обладать также определенной химической ус-
тойчивостью, чтобы не реагировать с другими компонентами ана-
лизируемого раствора. В потенциометрии в качестве индикатор-
ных црименяют металлические и мембранные электроды.
Металлические электроды первого рода представляют собой
металлическую пластинку или проволоку, погруженную в рас-
твор хорошо растворимой соли этого металла. Электроды из се-
ребра, ртути, кадмия и некоторых других металлов обратимы и
дают воспроизводимые результаты. Например:
А&+ + е' * М; Е = Е°ыу^ + 0,059 \ш аА^,;
Учтем связь активности и концентрации: а = сАё+УАё+'> Е = ^Аё+ +
+ 0,0591ёсАё^Аё+ или Е = £<><'> + 0,059 .1* с^уЕ^ = Ё^+/^ +
+ 0,059'1& УАё+> где ЕАё+/Ае — формальный потенциал. .
Однако электроды из таких металлов, как хром, кобальт и
другие в качестве индикаторцых не исцользуются, так как они не
дают, достаточно воспроизводимых результатов. У многих элект-
родов воспроизводимость значительно улучшается, если исполь^
зовать не просто металл, а его амальгаму. Это [амальгамные
электроды.
Особое место среди индикаторных электродов занимают ре-
докс-электроды, служащие для измерения окислительно-восста-
новительного потенциала системы. В качестве редокс-электродов
используются благородные металлы: платина, золото, иридий
или графит. Потенциал таких электродов зависит от отношения
концентраций (активностей) окисленной и восстановленной
форм редокс-пары и не зависит от материала электрода.
Иногда недостаточно высокая скорость реакций окисления-—
восстановления в растворе приводит к медленному дрейфу ре-
докс-потенциала во времени. К этому же может приводить и взаи-
модействие компонентов раствора с кислородом воздуха.
Второе нежелательное явление у редокс-электродов — сме-
шанный потенциал, возникающий в растворе в присутствии не-
скольких редокс-систем, находящихся в равновесии друг с другом.
Электроды второго рода состоят из металла, покрытого сло-
ем малорастворимого соединения этого металла и погруженного
в раствор хорошо растворимого соединения с тем; же анионом .V
К ним относятся хлорсеребряный, каломельный и некоторое
.186.'
другие электроды. Электроды второго рода обычно применяют
как электроды сравнения, хотя их применение в качестве инди-
каторных также не исключено.
В работе мембранных электродов используется не электро-
химическая реакция с переносом электрона, а разность потен-
циалов, возникающая на границе раздела фаз, и равновесие об-
мена ионов между мембраной и раствором. В обычных конструк-
циях мембранных электродов мембрана разделяет исследуемый
раствор и вспомогательный внутренний раствор. Наиболее широ-
ко применяемым электродом этого типа является стеклянный
электрод. Известны также фторидный, сульфидный и многие
другие мембранные электроды.
9,2.4. Электроды сравнения
Электроды сравнения должны обладать устойчивым во вре-
мени воспроизводимым потенциалом, не меняющимся при про-
хождении небольшого тока. Чаще всего в качестве электродов
сравнения применяют электроды второго рода: хлорсеребряный
и каломельный.
Хлорсеребряный электрод представляет собой серебряную
проволоку или пластинку, покрытую слоем AgCl и помещенную
в раствор KCl. Активность ионов серебра в таком растворе равна
+ = — Agci^ Подставляем эту величину в уравнение Нернста
f асг ь
для серебряного электрода:
po _ ро I RT ] — IT° -4-. In '^•^AgCl _
AAgVAg ~ ^AgVAg + T 1П aAg+ ~ ^AgVAg + T 1П ~
= ^/A8+7lnIIPAjC,- XlnßCl- (9Л2)
Первые два слагаемых в уравнении (9.12) зависят только от тем-
пературы:
При сочетании этого уравнения с (9.12) получаем
^Ag+/AgCl = EAg+/Ag ~ у- 1пас1- *
Как видно, потенциал хлорсеребряного электрода определяется
активностью хлорид-иона в растворе/Обычно используется насы-
щенный раствор KCl. Потенциал электрода второго рода, вообще
187
говоря, зависит от активности аниона малорастворимого соединен
ния, входящего в состав электрода. <
Каломельный электрод состоит из металлической ртути, ка?
ломели и раствора KCl. Его потенциал также зависит от актив-
ности хлорид-ионов:
EHgf+/Hg2Cl2 * EHg|+/Hg2Cl2 ~ ~fln асг >
где .! ■
EHg|VHg2ci2 = EHgf^Hg + 2^lnnPHg2ci2 •
Потенциалы хйорсеребряного, каломельного и ряда других
электродов сравнения изучены при различных концентрацион-
ных и температурных условиях и их величины по отношению к
стандартному водородному электроду хорошо известны. Стан-
дартный потенциал хлорсеребряного электрода при разных тем-
пературах может быть рассчитан по уравнению
EV/Agcis=0'2224-6,4 • 10-4(*-25)-3,2 • 1(Н(*-25)2.
Для каломельного электрода уравнение имеет вид
^^2-/нв2С12 = 0,2415 - 7,6 • 10-«(* - 25).
Точное значение потенциала электрода сравнения для мно-
гих потенциометрических измерений часто не требуется, важно
лишь его постоянство. Однако его значение необходимо при вы-;
полнении таких измерений, когда представляет интерес не толь-
ко ЭДС, но и потенциал индикаторного электрода. Если нет осо-
бых оговорок, потенциал индикаторного электрода обычно пере-
считывается и относится к стандартному водородному электроду.
Эта величина, конечно, уже не зависит от выбранного электрода
сравнения.
9.2.5. Диффузионный потенциал
Диффузионный потенциал, или потенциал жидкостного
соединения, возникает на границе между растворами двух раз-
ных электролитов или растворов разной концентрации одного
электролита. Возникновение диффузионного потенциала обус*
ловлено неравномерным распределением катионов и анцонов
вдоль границы раздела растворов вследствие различия в скорор-
тях диффузии ионов через поверхность раздела. Различие в ско-
ростях диффузии вызывается разницей в подвижностях ионов
или градиентом концентраций. В простейших случаях, напримеру!
188
при соприкосновении растворов одного электролита разной кон-
центрации, диффузионный потенциал может быть приближенно
рассчитан, если известны подвижности ионов и концентрации
растворов. Однако в практических условиях анализа состав ана-
лизируемого раствора бывает неизвестным (в противном случае
анализ не требуется) и теоретическая оценка диффузионного по-
тенциала невозможна.
В зависимости от заряда ионов, их подвижностей, концент-
рации раствора, природы растворителя и других факторов диф-
фузионный потенциал изменяется в очень широких пределах: от
долей милливольта до десятков милливольт и более. На практике
диффузионный потенциал стремятся элиминировать с помощью
так называемого солевого мостика. Основным,шßш^ш&ЖJeгg
является осуществление электролитического контакта, однако с
целью элиминирования диффузионного потенциала для солевого
мостика применяют концентрированный раствор электролита с
приблизительно одинаковыми подвижностями катиона и ани-
она. Очень часто в качестве электролита для солевого мостика ис-
пользуют насыщенный раствор KCl, применяют также растворы
NH4N03, KN03 и др. Солевой мостик помещают между раствора-
ми* заменяя таким образом границу раздела и существенно
уменьшая диффузионный потенциал. При работе с неводными
растворами для солевого мостика используют спиртовые раство-
ры Nal и KSCN.
9.3 .Прямая потенциометрия
Методы прямой пбтенциометрии (ионометрии) основаны на
непосредственном применении уравнения Нернста для нахожде-
ния активности или концентрации участника электродной реак-
ции по экспериментально измеренной ЭДС цепи или потенциалу
электрода. Наибольшее распространение среди прямых потенцио-
метрических методов получил метод определения pH, хотя созда-
ние в последнее время надежно работающих ионоселективных
электродов значительно расширило практические возможности
прямых методов. Прямые потенциометрические методы часто
стали называть ионометрическими методами анализа или ионо-
метриеи. Эта группа методов интенсивно развивается в связи с
успехами в конструировании и улучшении качества ионоселек-
тивных электродов, позволяющих проводить быстрее и точнее
определение концентрации или активности ионов и обладающих
рядом других достоинств.
189
9.3.1. Определение рН
Понятие о водородном показателе введено в 1909 г. Зёренсе-
ном, который под водородным показателем понимал отрицатель^
НЫЙ ДеСЯТИЧНЫЙ ЛОГарифм МОЛЯРНОЙ КОНЦеНТраЦИИ ИОНОВ ВОДО!
рода: рН = [Н+]. Для определения числового значения рН Зё*
ренсен предложил использовать измерение ЭДС элемента:
; Pt,H2|HCl,л:||KCl;0,lM|Hg2Cl2,Hg 1
В то время считалось, что ЭДС элемента зависит от концентр
рации вещества, а не от его активности, как это принято в на-
стоящее время:
Я = Я£онц- §1п[Н+].
Величина -Б^онц определялась экспериментально, причем кон-
центрация ионов водорода [Н+] в стандартном растворе вычисля-
лась по формуле
[Н+] = аснс1, (9.13)
где а — степень диссоциации НС1, рассчитанная по электриче-
ской проводимости; сНС1 — молярная концентрация раствора
НС1. . . ■
При этих условиях и 25 °С Я£онц = 0,3376 В и р^Н =
= Е л пкоо76' символ р^Н означает единицу рН в шкале Зёренсена.
Как видно, эта величина не является достаточно строгой мерой ни
концентрации ионов водорода, ни их активности. Действительно,
величина [Н+], рассчитанная по формуле (9,13), не является актив-
ностью ионов водорода, так как ан+ = [Н+]ун+, и не равна их кон-
центрации, поскольку в разбавленных растворах хлороводородной
кислоты [Н+] = сНС1, ввиду полной диссоциации НС1.
В соответствии с современными представлениями ЭДС эле-
мента Зёренсена выражается уравнением
Е = Е°^ "Ш \пая+аС1 + Ед9 (9.14)
где Ел — диффузионный потенциал.
В настоящее время рН считается характеристикой активнос-
ти ионов водорода:
рН = 4$ ан+ = -\ё [Н+]ун+ = раН.
Цоэтому иногда в символ рН вводят нижний индекс «а»: раН. ^
190
В соответствии с определением рН из уравнения (9.14) находим
еН = ^Й^ + 1**с, (9-15)
Уравнение (9.15) показывает, что для точного определения рН
необходимы данные по диффузионным потенциалам и по актив-
ности иона С1" в 0,1 М КС1. Ни одна из этих величин не может
быть получена совершенно строго, в связи с чем найденная экспе-
риментально величина рН также не является вполне строгой. Эти
трудности были преодолены путем введения соответствующего
Государственного стандарта на шкалу рН.
Принятая в России по стандарту 8.134—74 шкала рН основа-
на на воспроизводимых значениях рН нескольких растворов:
1) 0,1 моль/кг Н20 раствор НС1 (рН 1,10 при 0 °С и 1,14 при
150 °С);
2) 0,05 моль/кг Н20 раствор тетраоксалата калия КНС204 •
• Н2С204 • 2Н20 (рН 1,666 при 0 °С и 1,806 при 95 °С);
3) 0,01 моль/кг Н20 раствор натрия тетрабората Na2B407 •
• 10Н2О (рН 9,464 при 0 °С и 8,68 при 150 °С);
4) насыщенный при 25 °С раствор гидроксида кальция
Са(ОН)2 (рН 13,423 при 0 °С и 11,449 при 60 °С).
В стандарте приводятся значения рН этих и других растворов
в интервале температур от 0 до 95 °С или 150 °С с шагом в 5°.
Шкала рН обладает внутренней согласованностью, т. е. экспери-
ментально измеренная величина рН не зависит от того, какой из
растворов был выбран в качестве стандартного. Набор первичных
стандартных растворов используется также и в других странах —-
США, Великобритании, Японии и т. д.
Значения рН стандартных растворов устанавливают путем из-
мерения ЭДС цепей без переноса. Для этого чаще всего используют
цепь типа : \
Pt(H2) | буферный раствор, КС11 AgCl, Ag
В таких системах хотя и сохраняются трудности, связанные
с оценкой коэффициентов активности отдельных ионов, но отпа-
дает необходимость учета диффузионного потенциала.
Если рНх и рНст — значения рН исследуемого и стандартного
растворов, аЕхиЕст — ЭДС элементов типа
Pt(H2) | НА || КС1, р-р | AgCl, Ag
с исследуемым и стандартным растворами, то .
Ех = Е° + 2,зоздгрН^ дт;1п ^ + (9 1б)
Д„-Е° + 2'30/Д7рНст- *flnасг + Ед(ст). (9.17)
191
При вычитании уравнения (9.17) из (9.16) по-
лучаем
РН, = рНст +
(Ех - Ест + Еі
Д(ст)
2,303Д!Г
Для экспериментального определения рН мо-
гут быть использованы различные индикаторные
электроды: водородный, хингидронный, стеклян-
ный и др. Наибольшее практическое применение в
последнее время нашел стеклянный электрод,
используемый в широком интервале рН и в при-
сутствии окислителей.
Стеклянный электрод (рис. 9.3) представляет
собой тонкостенный стеклянный шарик 1, запол-
ненный раствором НС1 или каким-либо буферным;
раствором 2. Внутрь шарика помещают хлорсе-
ребряный электрод 3. Это устройство обычно за-
крывают защитной трубкой 4.
Перед работой стеклянный электрод некото-
рое время (несколько суток) вымачивают в 0,1 М
НС1. При этом ионы водорода из раствора обмени-
ваются на йоны натрия из стеклянной мембраны и в системе ус-
танавливается некоторое равновесие. Подготовленный таким об-
разом электрод, в котором протоны поверхности стекла находят-
ся в равновесии с протонами раствора, может быть использован
для определения рН.
Схематически это устройство может быть представлено сле-
дующим образом:
Рис 93.
Стеклянный
электрод
Внешний
раствор
ак+(х)
Стекл.
мембрана
Внутренний
раствор
%+(1)
Ионы водорода, адсорбированные на обеих сторонах поверх-
ности мембраны, на схеме не указаны. Таким образом, электрод-
ная реакция на стеклянном электроде сводится к обмену ионами
водорода между раствором и стеклом.
Н+(р) = Н+(стекло)
т. е. она не связана с переходом электронов.
Ионы водорода на поверхности внешней стороны мембраны
находятся в равновесии с ионами водорода в исследуемом раство-
ре и на границе раздела возникает потенциал
Н+(1)
192
где ан+^ — активность ионов водорода в исследуемом растворе;
ан+(1) -— активность ионов водорода на внешней поверхности
мембраны.
Аналогично на границе раздела внутренней поверхности
мембраны возникает потенциал
Е2 = Е\+^ 1п^Ш.,
F аН+(2)
где ан+(2) и а,==н+(2) — активность ионов водорода соответствен-
но во внутреннем растворе и на внутренней поверхности мемб-
раны.
Суммарный потенциал стеклянной мембраны будет равен
Е^Ег-Е2 = El -Е°2+*£ 1п ан^)а^(2) .
* аН+(1)^Н+(2)
При постоянных значениях a'n+(iy а'н+(2) и ан+(2) УРавнение
принимает вид
Ям = const + ^ In а н+(ж),
т. е. потенциал мембраны характеризует рН исследуемого раствора.
Измерение рН со стеклянным электродом сводится к измере-
нию ЭДС цепи:
Hg, Hg2Cl2 IКС11 с H+(JC) I стекло IHC11 AgCl, Ag
ЭДС этой цепи равна
E = E1-E2, (9.18)
где •
^1 = ^Hg2Cl2/Hg ~ jflnac\-(i)~ flnflHW
E2 = #AgCl/Ag — ^■lnaCl-(2)"" Щ-ln аН+(ст)-
Подставляя последние соотношения в (9.18), получаем
Е = [^Hg2Cl2/Hg - ^AgCl/Ag + + ^"1паН+(ст)]"" ^•1паН+(дг)
= ^2текл " ^ 1п аН+(ст)
ИЛИ
F
Я = Ястекл + 2,303^- рН.
13-7831 193
г В величину -Е^текл входит и так называемый потенциал асим-
метрии, представляющий собой разность потенциалов между двумя
сторонами стеклянной мембраны. Она возникает из-за несовпадения
свойств разных сторон мембраны и может быть измерена экспери-
ментально, если по обе стороны мембраны поместить один и тот же
раствор. Величина Е1текл зависит, также от константы равновесия
Н+ (р-р) *=> Н+ (стекло), характеризующей сорт стекла и некоторые
другие свойства стеклянного электрода. Стандартный потенциал
стеклянного электрода Е1текл обычно не определяют. При использо-
вании заводских рН-метров эта операция заменяется настройкой
приборов по стандартным буферным растворам, так как шкала
рН-метров проградуирована непосредственно в единицах рН.
Основными достоинствами стеклянного электрода являются
простота работы, применимость в широкой области рН, быстрое
установление равновесия и возможность определения рН в окис-
лительно-восстановительных системах. К недостаткам относят
хрупкость их конструкции и усложнение работы при переходе к
сильно щелочным и сильно кислым растворам. Тем не менее стек-
лянный электрод в настоящее время наиболее широко применяет-
ся: для измерения рН. Этому способствует также широкий выпуск
промышленностью рН-метров со стеклянным электродом.
9.3.2. Ионоселективные электроды
м Решающее влияние на развитие и успехи ионометрического
метода оказало удачное конструирование ионоселективных
электродов на основе различных мембран. Стеклянные ионосе-
лективные электроды чувствительны к ионам щелочных метал-
лов Li+, Na+, К+, Rb+, Cs+, а также к ионам Ag+, Т1+ и NHJ. Их
устройство и принцип действия такие же, как и стеклянного
рН-электрода. Наиболее существенным отличием является со-
став стекла, из которого готовятся'мембраны. Установлено, в ча-
стности, что введение А1203 в стекло положительно влияет на се-
лективность мембраны к ионам металлов, но не к Н+..
Селективный электрод, например, с натриевой функцией на-
ходится в равновесии с ионами натрия в растворе:
Na+ (стекло) Na+ (р)
В кислом растворе это равновесие может осложняться за счет
взаимодействия с ионами водорода:
Na+ (стекло) + Н+ (р) = Na+ (р) + Н+ (стекло)
поэтому в кислых растворах показания такого электрода искажа-
ются. • ■ ' ' ' .'■л'*- : ]
194
Важной характеристикой ионоселектйвнрго электрода явля-
ется его коэффйцйёнтсёлёктивйости, показывающий, во сколько
раз электрод более чувствителен к данным ионам, чем к посто-
ронним (мешающим). Например, если коэффициент селектив-
ности натриевого электрода по отношению к ионам калия состав-
ляет 1000, т. е. К^&+ к+ = Ю3, то это означает, что данный элект-
род в 1000 раз чувствительнее к ионам натрия, чем к ионам
калия. Другими словами, если электрод имеет потенциал Е при
концентрации ионов натрия равной 10~3 моль/л, то для достиже-
ния этого потенциала потребуется концентрация ионов калия в
1000 раз большая, т. е. 1 моль/л.
В техническом паспорте электрода обычно указывают крат-
ность превышения концентрации, при которой электрод сохра-
няет селективность. Например, в паспорте электрода на калий
марки ЭМ-К-01 указано, что электрод селективен в присутствии
ионов Ыа+ и ЫН£ при превышении их концентрации соответ-
ственно в 200 и 20 раз по сравнению с ионом К+. V
Время отклика, т. е. время достижения постоянного значе-
ния потенциала после погружения электрода в раствор, находит-
ся в пределах от нескольких секунд до нескольких минут и зави-
сит от природы мембраны, методики работы и т. д. Естественно,
чем меньше время отклика, тем лучше.
Стеклянные ионоселективные электроды широко использу-
ют для определения катионов щелочных металлов в различных
биологических пробах — крови, плазме, сыворотках и т. д., в
объектах окружающей среды — водах, растениях, различных
экстрактах и т. д. Определения с помощью ионоселективных
электродов успешно конкурируют с пламенно-фотометрически-
ми методами по точности и нередко превосходят их по скорости.
Твердые ионоселективные электроды. В твердых мембран-
ных электродах ионочувствительный элемент изготовляется из
малорастворимого кристаллического вещества с ионным харак-
тером проводимости. Перенос заряда в таком кристалле происхо-
дит за счет дефектов кристаллической решетки. Вакансии могут
заниматься ионом только определенного размера и заряда, что
обусловливает высокую селективность кристаллических мемб-
ран. Конструктивно такие электроды сходны со стеклянными: в
обоих электродах мембрана разделяет исследуемый раствор и
раствор сравнения, в котором находится электрод сравнения
(обычно хлорсеребряный). Из электродов этого типа широко при-
меняется фторидный электрод, в котором мембраной является
монокристалл Ъ*а¥3, имеющий чисто фторидную проводимость; с
добавкой ЕиГ2 для увеличения электрической проводимости.
Чувствительность фторидного электрода позволяет проводить из-
мерения равновесной концентрации фторид-ионов Г" в широкой
области концентраций от 10~6 до 1 моль/л. В этой области откло-
нений от уравнения Нернста не наблюдается.. Селективность
электрода очень высока — даже тысячекратный избыток посто-
ронних ионов (галогенид-, нитрат-, сульфат-ионов и др.) по срав-
нению с фторид-ионом не мешает определению ¥~и только в при-
сутствии ОН-ионов селективность падает (ОН" является мешаю-
щим ионом). Работа фторидного электрода ухудшается также в
присутствии лигандов, образующих с ионом Ьа3+ прочные ко-
ординационные соединения в растворе (цитрат-, оксалат-ионы
и др.). Вполне понятно также, что с увеличением кислотности сре-
ды равновесная концентрация фторид-ионов Р~ в растворе умень-
шается за счет образования молекул НГ. Таким образом, показа-
ния фторидного электрода в кислой области будут существенно
зависеть от рН. В щелочной области на поверхности электрода
может образоваться осадок Ьа(ОН)3, что также вызовет искаже-
ние показаний электрода. Точные границы рН, в которых пока-
зания фторидного электрода от рН зависят несущественно, при-
вести трудно, так как с уменьшением концентрации фторид-иона
эта область также уменьшается. Для растворов с концентрацией
фторид-иона п • 10""4 моль/л и более этот интервал охватывает
область значений рН примерно от 4—5 до 8—9.
Фторидный электрод используется для определения фто-
рид-ионов Р" в питьевой воде, различных биологических пробах,
витаминах, при контроле за загрязнением окружающей среды
ит, д. Он широко применяется также для исследования процес-
сов образования фторидных комплексов в растворе и других ре-
акций с участием фторид-иона.
Практическое значение имеет ионоселективный электрод с
мембраной из сульфида серебра, пригодный для измерения кон-
центрации (активности) и Ag+- и в2""-ионов. Подвижными в мемб-
ране А§28 являются ионы А§+. Серебро этим электродом может
быть определено в интервале концентраций от 1 до 10""7 моль/л, а
в некоторых условиях до 10~12 моль/л и ниже. В столь же низких
концентрациях могут быть определены 82"-ионы.
На основе сульфида серебра конструируются также различ-
ные галогенидные и металлочувствительнзде электроды. Для это-
го в сульфид серебра вводят галогениды серебра или сульфиды
меди, кадмия, свинца и некоторых других металлов. Электроды
"... '■' ' 196
на основе сульфида серебра с добавкой соответствующего галоге-
нида серебра чувствительны к ионам С1~, Вг~, I", СЫ" и др. Введе-
ние в сульфид серебра сульфидов других металлов позволяет по-
лучить электрод, чувствительный к ионам металла, внесенного
со вторым сульфидом (С(12+, РЬ2+, Си2+ и др.). Используются так-
же электроды на основе сульфида или селенида меди, чувстви-
тельные к ионам Си2"1".
В гетерогенных твердых электродах активное вещество сме-
шивается с инертйой матрицей. В качестве ионочувствительного
активного вещества используются различные малорастворимые
соединения, такие, как сульфат бария, оксалат кальция и т. д., а
в качестве инертной матрицы эпоксидные смолы, поливинилхло-
рид, силиконовый каучук и др. Потенциал таких электродов не-
достаточно устойчив и селективность невелика.
Интересны ферментные электроды, в которых на ионочувёт-
вительную мембрану нанесен фермент, катализирующий опреде-
ленную реакцию, продукты которой вызывают отклик стеклян-
ного электрода. Так определяют различные неорганические Йе-
щества (фосфат, мочевину и др.).
Жидкостные ионоселективные электроды. В электродах с
жидкой мембраной раствор сравнения отделен от анализируемо-
го тонким слоем органической жидкости, содержащей жидкий
ионит, не смешивающийся с водой, но селективно реагирующий
с определяемым ионом. Слой ионочувствительной органической
жидкости получается пропиткой этой жидкостью пористой. гид-
рофобной мембраны из пластика. Схема жидкостного ионоселек-
тивного электрода показана на рис. 9.4. Внутренний хлорсереб-
ряньга электрод 1 погружен в раствор МС12, где М — определяе-
мый катион. Пористая мембрана 3 одной стороной соприкасается
с раствором сравнения хлорсеребряно-
го электрода, другой — с анализируе- ^ ^ 1
мым раствором. Ионочувствительная
жидкость в резервуаре 2, пропитывают
щая мембрану, состоит из жидкого ор-
ганического ионита, имеющего кислот-
ные, основные или хелатообразующие
функциональные группы, растворен-
ного в подходящем растворителе, кото-
рый с водой не смешивается.
Электрод такого типа для опреде-
ления кальция содержит в качестве
жидкого ионита кальциевую соль ал- Рис 9.4. Схема жидкостного
К1| л фосфорной кислоты, растворенную ионоселективного электрода
.'.= ■ 197 • , 'V ,'
в диалкилфенилфосфонате, или аналогичную композицию. Рас-
твор сравнения внутреннего хлорсеребряного электрода 1 содер-
жит в этом случае СаС12.. С каждой стороны ионоселективной
мембраны устанавливается равновесие:
CaR2 (орг) ^ 2R- (орг)+ Са2+ (водн)
Концентрация (и активность) иона Са2+ в растворе сравнения по-
стоянна, поэтому потенциал электрода будет зависеть только от
концентрации (активности) иона Са2+ в анализируемом растворе.
Эта зависимость передается уравнением Нернста:
Я = Дмембр-0,029 lgaCa2+. (9.19)
Уравнение (9.19) соблюдается в области концентраций (ак-
тивностей) от 10~б до 1 моль/л в области рН от 6,0 до 11,0. При
более высоких значениях рН возможно образование осадка
Са(ОН)2, а в кислой области равновесие органического ионита с
кальцием осложняется участием иона водорода.
В практике применяют также ионоселективные Мембранные
электроды на ионы калия, натрия, аммония и некоторые другие.
Сконструированы газочувствительные мембранные электроды
для определения NH3, NO и других газов. В пленочных электро-
дах вместо жидкой мембраны используют тонкую пленку. У пле-
ночных электродов такой же механизм действия, что и у мемб-
ранных, но они долговечнее и более удобны в работе.
9.3.3. Основные приемы ионометрического анализа
Во всех приемах прямой потенциометрии используется зави-
симость потенциала индикаторного (обычно ионоселективного)
электрода от активности или концентрации определяемого веще-
ства. Активность определяемого иона может быть рассчитана по
уравнению
AgaM 2.303ДТ ' (9^0)
где 2£инд —- экспериментально найденный потенциал индикатор-
ного электрода.
Величину 2£инд находят по ЭДС исследуемого элемента:
^инд = £ср-^ + ^д, (9.21)
где Е — ЭДС элемента; Еср — потенциал электрода сравнения; Ед—
диффузионный потенциал.
198
Сочетание уравнений (9.20) и (9.21) дает <
е м г.ЗОЗДГ
ИЛИ "'"
üa s=4ffa . [Е-(ЕСР- E^+EA)]nF
Р*м ^ам 2,303ДГ V
Так как ам = смум, где ум — коэффициент активности; см — кон-
центрация иона М, то
PCM = -lgcM= [£-(^2;3ggr+^"f -lgyM. (9.22)
Уравнение (9.22) лежит в основе нескольких аналитических
приемов. Трудности, связанные с неопределенностью коэффици-
ента активности отдельного иона, при определении рН были пре-
одолены путем разработки шкалы рН на основе стандартных бу-
ферных растворов. Для других ионов таких стандартных раство-
ров нет, поэтому трудности оценки коэффициентов активности
отдельных ионов остаются.
/Активности ионов и иных растворенных частиц используют-
ся в расчете равновесий, стандартных электродных потенциалов
и т. д., но результаты аналитического определения, как правидо,
должны показывать концентрацию вещества. Прямой расчет
концентрации по активности иона не всегда можно сделать с не-
обходимой точностью, так как ионная сила анализируемого рас-
твора, от которой зависят коэффициенты активности/обычно не-
известна. Введение в анализируемый раствор так называемых
фоновых или поддерживающих электролитов обеспечивает по-
стоянное значение ионной силы и коэффициентов активности.
В практике используется несколько приемов ионометрии.
Метод градуировочного графика. При постоянной и одина-
ковой в обоих полуэлементах ионной силе и неизменном составе
коэффициенты активности ионов будут одинаковы, а диффузион-
ный потенциал пренебрежимо мал. В этих условиях уравнение
(9.22) принимает вид
рсм = аЕ + Ь, (9.23)
где а и Ъ —- константы, смысл которых становится ясным при
сравнении уравнений (9.22) и (9.23).
В соответствии с уравнением (9.23) график в координатах
ЕlgcM должен быть линеен. Для построения градуировочного
графика измеряют ЭД С элемента при нескольких концентрациях
199
определяемого иона и постоянной ионной силе. Затем определя-
ют ЭДС, помещая индикаторный электрод в анализируемый рас-
твор, и по ЭДС и градуировочному графику находят концентра-
цию иона в анализируемом растворе. Перед определением ЭДС в
анализируемом растворе стремятся создать такой же солевой фон
и ионную силу, какие были в стандартных растворах, использо-
ванных для построения градуировочного графика. Для этого в
анализируемый раствор вводят определенный объем концентри-
рованного раствора фонового электролита.
Успешно применяется метод градуировочного графика при
анализе слабо минерализованных растворов, поскольку в таких
пробах можно легко создать заданное значение ионной силы.
Практически используется этот метод, например, при определе-
нии фторид-иона в питьевой воде с помощью фторселективного
электрода.
Метод концентрационного элемента. Идея метода заключа-
ется в том, что составляют концентрационный элемент с двумя
одинаковыми индикаторными электродами и помещают один из
них в раствор с известной концентрацией определяемого иона, а
другой — в анализируемый раствор. ЭДС такого элемента будет
равна
Е= Щ. +я, , (9.24)
ров; уст и ух — коэффициенты активности определяемого иона в
стандартном и исследуемом растворах; Ел — диффузионный по-
тенциал.
Если состав и ионная сила обоих растворов примерно одина-
ковы за счет одного и того же фонового электролита, то диффузи-
онный потенциал становится пренебрежимо малым, а коэффици-
енты активности уст и ух примерно одинаковыми. Тогда уравне-
ние (9.24) принимает вид
Е~ ^1п^.
с
При Е = 0, очевидно, сст = сх, так как при этом 1п — =0. Вы-
сх
полнить условие Е = 0 можно разбавлением стандартного раство-
ра раствором фонового электролита или добавлением более кон-
центрированного стандартного раствора.
200
Вместо экспериментального выравнивания концентраций
можно добавить несколько порций разбавителя или стандартного
раствора в заведомом избытке и после каждого добавления изме-
рить ЭД С. Таким образом можно получить значения ЭДС до и
после Е = 0. По этим данным строят кривую линейного титрова-
ния в координатах Е — 1ц сст и находят точку ее пересечения с
осью абсцисс, которая и соответствует равенству сст = сх, так как
в этой точке Е = 0. Таким методом определяют фторид-ион, ион
серебра и другие, для которых сконструированы ионоселектив-
ные электроды.
о Метод добавок. Для применения этого метода необходимо до-
статочно точно знать электродную функцию по отношению к оп-
ределяемому иону. Обычно ее находят заранее по стандартным
растворам. При осуществлении метода добавок сначала измеря-
ют ЭДС цепи с анализируемым раствором (Ех)9- затем добавляют к
нему определенный объем стандартного раствора и снова измеря-
ют ЭДС.
До добавления стандартного раствора ЭДС цепи равна
У Ех = Е°+^1псхух + Ел,
а после добавления
Ех + ст = Е°+^1п(сх + АсУух + Ед,
где Ас — прирост концентрации определяемого иона за счет вве-
дения стандартного раствора.
Разность этих ЭДС равна
Введение добавки стандартного раствора практически не измени-
ло ионной силы раствора, поэтому ух и Ед остались теми же самы-
ми и при вычитании сократились.
Из уравнения (9.25) получаем концентрацию определяемого
иона:
10* -1
Метод добавок, как уже отмечалось, автоматически учитыва-
ет влияние третьих компонентов и позволяет находить концент-
рацию очень разбавленных растворов, '
201
9.4. Потенциометрическое титрование
Потенциометрическое титрование основано на определении
точки эквивалентности по результатам потенциометрических из-
мерений. Вблизи точки эквивалентности происходит резкое из-
менение (скачок) потенциала индикаторного электрода. Это на-
блюдается, конечно, лишь тогда, когда хотя бы один из участни-
ков реакции титрования является участником электродного
процесса. Так, например, титрование по методу кислотно-основ-
ного взаимодействия может быть выполнено со стеклянным
электродом, определение хлорида — с хлорсеребряным й т. д.
Так же, как и в других титриметрических методах, реакции по-
тенциометрического титрования должны протекать строго стехио-
метрически, иметь высокую скорость и идти до конца.
Для потенциометрического титрования собирают цепь из ин-
дикаторного электрода в анализируемом растворе и электрода
сравнения. В качестве электродов сравнения чаще всего приме-
няют каломельный или хлорсеребряный.
9.4.1. Определение точки эквивалентности
: На рис. 9.5, а представлена кривая титрования хлороводород-
ной кислоты гидроксидом натрия. Она почти точно воспроизводит
теоретическую кривую титрования сильной кислоты сильным ос-
нованием. Как видно, в точке эквивалентности происходит рез-
кий скачок ЭДС, вызванный резким изменением потенциала ин-
дикаторного электрода. По этому скачку можно определить точку
АЕ/АУ
А2Е/АУ2
т.э.^(КаОН)
т.э.
7(МаОН)
АУ/АЕ
\\
п
и .
л\У0
| ^(ЫаОН)
т.э.
т.э.
7(ЫаОН)
а) б) в) г)
Рис. 9.5. Кривые потенциометрического титрования:
а — обычная кривая; б — дифференциальная кривая;
в — кривая титрования по второй производной; г — кривая Грана
202
эквивалентности и потом рассчитать содержание хлороводород-
ной кислоты.
Для нахождения точки эквивалентности часто строят диффе-
АЕ
ренциальную кривую в координатах — - ^(рис. 9.5, б). На точку
эквивалентности указывает максимум полученной кривой, а от-
счет по оси абсцисс, соответствующий этому максимуму, дает
объем титранта, израсходованного на титрование до точки экви-
валентности. Определение точки эквивалентности по дифферен-
циальной кривой значительно точнее, чем по простой зависимос-
ти^-К
Поскольку производная функции, имеющей максимум, в
точке максимума равна нулю, вторая производная потенциала по
объему (ДЯЕ/ДУ2) в точке эквивалентности будет равна нулю. Это
свойство также используется для нахождения точки эквивалент-
ности (рис. 9.5, в).
В простом и удобном методе Грана точка эквивалентности
определяется по графику в координатах - V. Перед точкой эк-
вивалентности и после нее кривая Грана линейна, а сама точка
эквивалентности находится как точка пересечения этих прямых
(рис 9.5, г). Достоинства и удобства метода Грана особенно за-
метны при анализе разбавленных растворов, позволяя опреде-
лить точку эквивалентности с достаточной точностью вследствие
линейности графика.
Кроме рассмотренного классического метода потёнциометри-
ческого титрования нередко используют его новые варианты: не-
компенсационные методы и методы титрования под током.
В некомпенсационном методе потенциометрического титро-
вания измеряют не ЭДС, а ток, возникающий в гальваническом
элементе. В начальный момент титрования ЭДС элемента ком-
пенсируют внешней ЭДС и ток в цепи отсутствует. В процессе
титрования компенсация нарушается, в цепи возникает ток, при-
чем в области точки эквивалентности ток резко возрастает про-
порционально скачку ЭДС в этой области. Точку эквивалентнос-
ти можно фиксировать непосредственно по резкому возрастанию
тока или найти графически по зависимости ^ - V, где Д/ — при-
ращение силы тока при добавлении в раствор ДУ титранта.
К некомпенсационным методам можно отнести и потенцио-
метрическое титрование с биметаллической парой электродов.
Этот метод основан на том, что некоторые инертные металлы с
разной скоростью отзываются на изменение потенциала системы.
Например, платина быстро реагирует на изменение отношения
203
концентраций окисленной и восстановленной формы, а вольфрам
медленно. Поэтому, если опустить платиновый и вольфрамовый
электроды в титруемый раствор, содержащий окислительно-вос-
становительную систему, и измерять разность потенциалов меж-
ду ними в ходе титрования, то до точки эквивалентности она бу-
дет близка к нулю, а в точке эквивалентности резко возрастает.
Скорость некоторых окислительно-восстановительных реак-
ций, особенно с участием органических веществ, оказывается не-
достаточной для использования их в титриметрических методах.
В этих случаях электроды поляризуют путем пропускания не-
большого тока (10~5 А), что резко увеличивает скорость установ-
ления равновесных потенциалов и существенно — скачок титро-
вания. Титрование под током позволяет проводить анализ систем,
которые в отсутствие тока потенциометрически не титруются.
9.4,2. Виды потенциометрического титрования
Кислотно-основное титрование. В кислотно-основном титро-
вании в качестве индикаторного обычно используют стеклянный
электрод, как правило, входящий в комплект серийно выпускае-
мых промышленностью рН-метров.(Потенциометрический метод
позволяет провести количественное определение компонентов в
смеси кислот, если константы диссоциации различаются не ме-
нее чем на три порядка. ^Например, при титровании смеси, содер-
жащей хлороводородную и уксусную кислоты ,f на кривой титро-
вания обнаруживается два скачка. Первый свидетельствует об
окончании титрования HCl, второй скачок наблюдается при от-
титровывании уксусной кислоты. Также несколько скачков име-
ют кривые титрования многоосновных кислот, константы диссо-
циации которых существенно различаются (хромовая, фосфор-
ная и др.).
Широкие возможности анализа многокомпонентных смесей
без разделения открывает применение неводных растворителей.
Например, определение содержания хлороводородной и монохлор-
уксусной кислот в смеси титрованием водного раствора является
сложной задачей в связи с трудностью обнаружения двух скач-
ков титрования. При титровании в ацетоне оба скачка выражены
достаточно четко и содержание каждой кислоты в смеси может
быть рассчитано.
Комплексонометрическое титрование. Потенциометрическое
титрование катионов комплексоном III (ЭДТА) можно проводить
с использованием в качестве индикаторного электрода соответст-
вующего металла: титрование солей меди с медным электродом,
204
солеи цинка — с цинковым и т. д. или подходящего ионоселек-
тивного электрода. Однако многие металлические индикаторные
электроды необратимы, а число ионоселективных электродов не-
велико.
Для комплексонометрических титрований может быть ис-
пользован универсальный электрод
Hg | HgY2" или Au(Hg) I HgY2-
где Au(Hg) — амальгамированное золото; HgY2" — комплекс рту-
ти с анионом этилендиаминтетрауксусной кислоты. При титрова-
нии, например, ионов кальция собирают цепь типа
Hg | Hg2Cl2, КС11 Са2+, HgY2" (Ю-4) | Hg
Устойчивый комплексонат ртути (lgPHgY2- = 21,8) в очень не-
большой степени диссоциирует:
HgY2- = Hg2+ + Y4- (9.26)
что определяет величину потенциала ртутного электрода:
E*g*+\ug = Ене*+\н8 + °>029^tH^2+]» \Г:'
или, учитывая диссоциацию HgY2" по схеме (9.26),
^н^|нв=^|нг + 0.029 lgfI§£^, ^ (9.27)
где PHgY2-— константа устойчивости HgY2".
Образующийся при титровании комплексонат CaY2" характе-
ризуется константой устойчивости PCaY2-:
Из уравнения (9.28) находим [Y4-] и подставляем в (9.27):
£Hg*+|Hg - ^Hg^iHg + ' « [CaY2-]pHgY2. '
ИЛИ
^Hg-|Hg = ^Hg-|Hg + 0,029 lg|gr^!: +0)0291g[Can (9.29)
Концентрации HgY2" и CaY2" при приближении к точке эквива-
лентности изменяются очень незначительно. Объединяя первые
два слагаемых правой части уравнения (9.29) в const, получаем
:EHg2+lHg^ const + 0,029 lg[Ca2+]. (9.30)
205
Уравнение (9.30) показывает,«что потенциал ртутного электрода
JigY2~ | Б£ чувствителен к ионам Са2+. В области точки эквива-
лентности, очевидно, произойдет резкое изменение потенциала и
будет наблюдаться скачок титрования. С помощью ртутного
электрода этого типа могут быть оттитрованы любые ионы, кото-
рые образуют с У4" комплексы с константой устойчивости, не
превышающей РН£у2_. Это, например, ионы Mg2+, Са2+, Со2+,
№2+, Си2+, гп2+и др.
Титрование по методу осаждения. Индикаторными электро-
дами в методах потенциометрического титрования, использую-
щих реакции осаждения, служат металлические или мембран-
ные электроды, чувствительные к определяемому иону или
иону-осадителю. Практически по методу осаждения могут быть
определены катионы серебра, ртути, цинка, свинца, анионы хло-
ра, брома, иода и некоторые другие. Смесь галргенидов, напри-
мер I" и СГ, может быть оттитрована без разделения нитратом се-
ребра. Серебряный электрод позволяет фиксировать два скачка в
ходе такого титрования. Первый скачок свидетельствует об от-
титровывании иодид-иона и может быть использован для расчета
содержания этого иона, второй скачок относится к окончанию
осаждения хлорид-иона. По второму скачку можно рассчитать
суммарное содержание галогенидов или концентрацию хло-
рид-иона, если концентрация иодйд-иона будет известна из дан-
ных по титрованию до первого скачка.
Окислительно-восстановительное титрование. Кривые окис-
лительно-восстановительного титрования могут быть построены
в координатах или рМ - У (титранта) или Е - V (титранта), если
рМ =? -1ц [М] ([М] — концентрация участника реакции, Е — по-
тенциал системы, V (титранта) — объем титранта. Кривые титро-
вания первого типа представляют практический интерес, когда
имеется индикаторный электрод, чувствительный к М. Кривые
второго типа имеют более общее значение, так как любое окисли-
тельно-восстановительное титрование может быть проведено по
измерению Е с использованием индикаторного электрода из бла-
городного металла, чаще всего платины.
9.4.3. Автоматическое титрование
Сравнительная легкость автоматизации потенциометриче-
ских измерений позволила создать ряд автоматических приборов
для титрования — автотитраторов* Отечественный автотитра-
тор или блок автоматического титрования типа ВАТ-15 выцуска-
206
■7
ется в комплексе с бюреткой и
рН-метром — милливольтметром,
имеющим диапазон измерений в
единицах рН от -1 до +14 (100 +
+ 1400 мВ). Схема установки для
автоматического титрования пред-
ставлена на рис. 9.6.
В емкость с анализируемым
раствором 1 вводится дозирующая
трубка 2 для подачи титранта, ин-
дикаторный электрод 4 (стеклян-
ный или платиновый) и хлорсереб-
ряный электрод сравнения 3. Пода-
ча титранта в раствор регулируется
клапаном 9. Напряжение Ех> про-
порциональное ЭДС системы, с вы-
хода рН-метра -—милливольтметра
5 подается на вход БАТ-15, где
сравнивается с заранее заданным напряжением Е0 на задатчике
конечной точки титрования 6. Разность заданной и эксперимен-
тально наблюдаемой величины Ех - Е0 через усилитель 7 подает-
ся на бесконтактное электронное реле 8, которое управляет рабо-
той электромагнитного клапана 0, открывающего или закрываю-
щего подачу титранта. При Ех = Е0 клапан прекращает подачу
титранта. Объем рабочего раствора отсчитывают по бюретке. По-
грешность титрования на этой установке не превышает ±1%.
Рис. 9.6. Схема установки
для автоматического титрования
9.5. Потенциометрическое определение
физико-химических свойств веществ
Потенциометрические измерения часто использует не 1-олько
в химико-аналитических целях для определения концентраций
вещества или установления точки эквивалентности. Их широко
применяют для исследования реакций в растворе, определения
констант равновесия и различных характеристик вещества. По
данным потенциометрических измерений вычисляют константы
диссоциации кислот и оснований, константы устойчивости коор-
динационных соединений, произведение растворимости и т. д.,
рассчитывают тепловые эффекты и другие термодинамические
характеристики процессов в растворе.
Широкое распространение потенциометрического метода
связано с тем, что он с высокой точностью дает эксперименталь-
207
ные значения равновесных концентраций (активностей) ионов в
исследуемых системах и для обработки потрнциометрических
данных имеется соответствующий математический аппарат, ос-
нованный на теории ступенчатых равновесий. Изложение этих
методов можно легко найти в специальной литературе. Здесь бу-
дет рассмотрено лишь несколько простых примеров.
Константу диссоциации слабой кислоты можно определить
по экспериментально измеренным рН ее растворов известной
концентрации или по кривой титрования кислоты щелочью.
Диссоциация кислоты НА происходит по схеме
НА = Н+ + А-
Если раствор кислоты имеет концентрацию с§А, то равновесные
концентрации продуктов диссоциации будут равны: [Н+] = [А-] =
== Ю"РН, а [НА] = с§А " Ю~рН« Для простоты пренебрегаем разницей
между концентрацией и активностью и подставляем равновесные
концентрации в выражение для константы диссоциации кислоты:
^_[Н+][А]_ (Ю-рн)2 9
К [НА] сЙа-10-рн- }»м>.
Как видно, для расчета константы диссоциации необходимо экс-
периментально определить рН раствора.
При титровании кислоты щелочью в точке, где прореагиро-
вала половина исходного количества кислоты, концентрации
частиц НА и А" одинаковы, и из уравнения (9.31) следует
.йГ-[Н+]1/2 = 10"рН1/2.
Здесь рН1/2 относится к точке, где кислота прореагировала напо-
ловину.
Произведение растворимости малорастворимой полностью
диссоциированной соли МА может быть рассчитано по уравнению
ПРма^ЕМНАЗ-ЕМ^^еа]2,
где [М] и [А] — концентрации катиона М и аниона А в насыщен-
ном растворе соли, определяемые потенциометрически с по-
мощью ионоселективных электродов.
Произведение растворимости можно определить также по
экспериментальному значению равновесной концентрации кати-
она или аниона в растворе, содержащем избыточное количество
противоиона и осадок. Для определения по этому методу произ-
ведения растворимости Вав04 в соответствующий объем 0,1 М
Ш2804 вводят каплю раствора ВаС12. Концентрация суль-
208
фат-иона при этом практически не меняется, а осадок BaS04 об-
разуется. Равновесную концентрацию ионов бария в таком рас-
творе при наличии осадка определяют с помощью ионоселектив-
ного электрода и рассчитывают произведение растворимости как
ПРВа804 = [Ва2+] • 0,1, где 0,1 — концентрация исходного раство-
ра сульфата натрия, практически равная равновесной концентра-
ции ионов SOf ~ после добавления капли раствора ВаС12.
Для расчета констант устойчивости координационных соеди-
нений и констант диссоциации многоосновных кислот обычно ис-
пользуют специальный математический аппарат, учитывающий
одновременное протекание нескольких процессов ступенчатого
комплексообразования или ступенчатой диссоциации кислот.
9.6. Ионоселективные полевые транзисторы
Термином транзистор (от transfer— преобразователь и
resistor — сопротивление) в 1948 г. Д. Бардин и В. Бриттон
(США) назвали первый точечный полупроводниковый триод. По-
левой транзистор —■ это полупроводниковый усилитель, управ-
ляемый электрическим полем. Электрод, на который подается
электрический сигнал, используемый дЛя управления величиной
тока, называют затвором. Идея ионоселективного полевого тран-
зистора — заменить затвор полевого транзистора ионочувстви-
тельной мембраной так, чтобы силой тока управляла концентра-
ция (активность) потенциал определяющих ионов в растворе.
Первый ионоселективный полевой транзистор был чувствителен
к концентрации ионов натрия в растворе.
Современная микроэлектронная технология позволяет раз-
рабатывать электроды миллиметрового размера с малым време-
нем отклика и низким сопротивлением (датчики состава). Извес-
тен микровариант датчика.в виде иглы диаметром 30 мкм и дли-
ной 5 мм. Такой датчик предложен для непрерывного измерения
рН и концентрации карбонат-ионов. На поверхность затвора мо-
жет быть нанесена любая ионоселективная мембрана. Ионоселек-
тивные полевые транзисторы, по-видимому, следует рассматри-
вать как ионоселективные электроды третьего поколения.
9.7. Практическое применение
Большое практическое значение имеют потенциометриче-
ские методы определения рН раствора со стеклянным и другими
электродами, а также прямые потенциометрические определе-
14-7831
209
ния концентрации (активности) других ионов с помощью ионосе-
лективных электродов (ионометрия). Сконструированные ионо-
селёктивные электроды на ионы Си^ Аё*, Са24у Ыа+, К+, СГ, Г~,
в2", N0^ и др. успешно применяют в анализе различных техноло-
гических растворов, объектов окружающей среды и т. д. Потен-
циометрические датчики на основе ионоселективных электродов
позволяют следить за ходом технологического процесса.
Во многих областях находит практическое применение каль-
циевый ионоселёктивный электрод. Помимо традиционного ана-
лиза воды, различных растворов и т. д. большое практическое
значение кальциевый электрод имеет в медико-биологических
исследованиях, клинической медицине и т. д., поскольку кон-
центрация (активность) ионов кальция влияет на многие процес-
сы жизнедеятельности и физиологические процессы (нервная де-
ятельность, функция ферментов ит. д.). Известен мембранный
ионоселёктивный электрод, позволяющий определять жесткость
воды, так как он имеет примерно одинаковую чувствительность
на оба иона (кальций и магний).
Другой важной областью применения потенциометрических
методов является потенциометрическое титрование кислот, основа-
ний; солей и других веществ, где также эффективно используют
ионосёлективные электроды. Потенциометрические методы успеш-
но применяют в анализе мутных и окрашенных растворов и в ана-
лизе растворов на основе смешанных и неводных растворителей.
9.8. Общая характеристика метода
Основными достоинствами потенциометрического метода яв-
ляются его высокая точность, высокая чувствительность и воз-
можность проводить титрования в более разбавленных растворах,
чём это позволяют визуальные индикаторные методы. Необходи-
мо отметить также возможности определения этим методом не-
скольких; веществ Б бдном растворе без предварительного разделе-
ния й тйтрЪванйя в мутных и окрашенных средах. ЗначительноV
расширяется область практического применения потенциометри-
ческого титрования при использовании неводных растворителей.
Они позволяют, например, найти содержание компонентов; кото-
рые в водном растворе раздельно не титруются, провести анализ
веществ, нерастворимых или разлагающихся в воде и т. д. Нема-
ловажным достоинством потенциометрии явлкется также воз-
можность автоматизировать процесс титрования. Промышлен-
ность выпускает несколько типов автотитраторов, использующих
потенциометрические датчики.
210
К недостаткам потенциометрического титрования можно от-
нести не всегда быстрое установление потенциала после добавле-
ния титранта ц необходимость во многих случаях делать при тит-
ровании большое число отсчетов.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. На чем основаны потенциометрические методы анализа?
2. Какая зависимость выражается уравнением Нернста?
3. Какие функции выполняют индикаторные электроды и какие —
электроды сравнения?
4. Как устроен стеклянный электрод? Как с его помощью определя-
ют рН раствора? Какие достоинства и недостатки он имеет?
5. Каковы основные типы ионоселективных электродов? Как они
устроены? Какие имеют характеристики?
6. В чем сущность и области применения методов прямой потенцио-
метрии?
7. В чем сущность метода концентрационного элемента?
8. Какие ограничения в работе имеет фторидный электрод?
9. В каких координатах строят кривые потенциометрического тит-
рования?
глава 10
Вольтамперометрия
10.1. Кривая «ток—потенциал»
Вольтамперометрия основана на изучении поляризационных
или вольтамперных кривых (кривых зависимости силы тока от
напряжения), которые получаются, если при электролизе раство-
ра анализируемого вещества постепенно повышать напряжение и
фиксировать при этом силу тока. Электролиз следует проводить с
использованием легко поляризуемого электрода с небольшой по-
верхностью, на котором происходит электровосстановление или
электроокисление вещества. V
Применение вольтамперных кривых в аналитических целях
началось с разработки в 1922 г. чешским ученым Я. Гейровским
полярографического метода анализа. За открытие и развитие это-
го метода Я. Гейровскому в 1959 г. была присуждена Нобелев-
14'
2І1
екая премия. Я. Гейровский проводил электролиз на ртутном ка-
пающем электроде и вольтамперометрию, связанную с использо-
ванием ртутного капающего электрода, вслед за Я. Гейровским
стали называть полярографией.
Рассмотрим электролиз в системе, где катодом служит ртут-
ный капающий электрод, а анодом является практически непо-
ляризуемый каломельный электрод. Изменение внешней ЭДС в
такой системе будет полностью идти на изменение потенциала
катода. Если в растворе нет веществ, способных восстанавливать-
ся под действием электрического тока, сила тока / будет пропор-
циональна приложенному напряжению Е (закон Ома):
/ = Я/Д,
где Л — сопротивление.
В присутствии веществ, способных восстанавливаться на
ртутном электроде в области исследуемых напряжений, вид кри-
вой зависимости тока от напряжения существенно изменится. По
достижении потенциала восстановления ионы начнут разряжать-
ся на ртутном катоде нередко с образованием амальгамы:
Мл++ пе~ + Не = М(Н£) (10.1)
Потенциал ртутного катода, на котором протекает обрати-
мый процесс (10.1), выражается уравнением Нернста:
Е = Е0+Щ\пая*СмУм, (10.2)
где са — концентрация амальгамы; уа — ее коэффициент активное-;
ти; см -—концентрация восстанавливающихся ионов в приэлект-
родном слое (заряд лона для простоты опущен); ум — его коэффи-
циент активности; аНй — активность ртути в амальгаме; Е° —
стандартный потенциал электрода (10.1).
В результате процесса (10.1) сила тока в цепи начнет возрас-
тать и концентрация восстанавливающихся ионов у поверхности
ртутной капли уменьшится. Однако за счет диффузии из массы
раствора к поверхности капли доставляются новые порций
ионов. Сила тока в цепи будет зависеть от скорости диффузии,
которая пропорциональна разности концентраций в массе рас-
твора (Сод) и в приэлектродном слое (см). Сила тока I будет про-
порциональна этой разности:
(Ю.З)
Вклад других, недиффузионных механизмов поступления
ионов в прикатодный слой в условиях большого избытка индиф-
212
ферентного фонового электролита пренебрежимо мал. Основное
значение среди недиффузионных процессов имеет миграция
ионов к катоду под действием электрического поля. Если не уст-
ранить вызываемый этим процессом миграционный ток, общий
ток окажется неконтролируемым. Подавление миграционного
тока достигается введением в раствор в достаточной концентра-
ции так называемого индифферентного, т. е. не принимающего
участия в электродной реакции, или фонового, электролита со
значительно более отрицательным потенциалом выделения, чем
у анализируемого иона. Катионы фонового электролита экрани-
руют электрод, уменьшая тем самым движущую силу миграции
под действием электрического поля практически до нуля.
При некоторомчпотенциале катода концентрация ионов у по-
верхности ртутной капли см уменьшится до ничтожно малой по
сравнению с концентрацией в массе раствора, и скорость разряда
ионов на катоде станет равной скорости диффузии.
Концентрация восстанавливающегося иона в глубине раство-
ра постоянна, так как электролиз идет при очень небольшой силе
тока (порядка 10"6 А), а концентрация в прикатодном слое близ-
ка к нулю. Поэтому разность концентраций, определяющая ско-
рость диффузии при данной температуре, будет постоянна, что и
приводит к постоянной скорости поступления ионов к катоду.
Наступившее состояние равновесия будет характеризоваться по-
стоянной силой тока, не изменяющейся при дальнейшем увели-
чении напряжения. Этот постоянный ток, контролируемый диф-
фузией, называют диффузионным и обозначают 1а. Выражение
для силы диффузионного тока получается из уравнения (10.3)
при см = 0:
1а = ЬмС&. (Ю.4)
Сила диффузионного тока прямо пропорциональна концентра-
ции восстанавливающегося иона в массе раствора. При сочетании
уравнений (10.3) и (10.4) получаем
или' : .
%=ПГ^ (Ю.5)
Концентрация амальгамы, образовавшейся в результате процес-
са (10.1), пропорциональна силе тока:
: сйч;/^, (Ю.6)
213
Соотношения (10.5) и (10.6) подставляем в уравнение (10.2):
Е = Е° + Щ; 1п
пР
(10.7)
Некоторые величины в этом уравнении постоянны или зависят
только от температуры. Так, амальгама, образующаяся при
электролизе на ртутном катоде, очень разбавлена, поэтому актива
ность ртути в амальгаме (аНё) практически равна активности чис-
той ртути, т. е. величина постоянная. Коэффициент активности
ионов (ум) при постоянной ионной силе, которая создается фоно-
вым электролитом, остается постоянным, так же как коэффици-
ент активности уа и коэффициенты кш и к&. Выделим в уравнении
(10.7) величины, зависящие только от температуры, и придадим
ему вид
Я = + — 1п + 51-1п
пР
*мУа
ИЛИ
где
Е = Е°1/2+ Щ 1п
1/2 пР
пР
Е°1/2 = Е0 +Щ\па^УмК.
(10.8)
(10.9)
(10.10)
Уравнение (10.9) — это уравнение полярографической волны*
а величину -Е1/2 называют потенциалом полуволны.
Типичная зависимость силы тока от приложенного напряже-
ния дана на рис. 10.1. Это полярографическая волна (полярограм-
ма). Из рисунка видно, что в начале процесса при небольшом по-
тенциале катода сила тока медленно увеличивается с возраста-
нием потенциала — это так называемый остаточный ток, его
величина имеет порядок 10~7 А. По до-
стижении потенциала восстановле-
нияна.-катоде начинается разряд ионов
и сила тока резко возрастает, стремясь
к предельной величине диффузионно-
го тока. При / = 1/2/^ уравнение (10.9)
переходит в
Это соотношение, так же как и (10.10),
показывает независимость потенциала
полуволны от силы тока и, следова-
тельно, от концентрации восстанавли-
РЙС. 10.1 Поляррграмма:
1 остаточный ток;
2 — диффузионный ток
214
вающейося.' иона; Потенциал полуволны является, таким обра-
зом, качественной характеристикой иона в растворе данного
фонового электролита и определение потенциала полуволны со-
ставляет основу качественного полярографического анализа.
; Однако потенциал полуволны существенно зависит от среды,
природы и концентрации фонового электролита. Особое значение
имеет наличие в растворе веществ, способных к комцлексообра-
зованию с определяемым ионом. Присутствие в исследуемом рас-
творе лиганда смещает потенциал полуволны в отрицательную
область, что используется для определения состава и констант ус-
тойчивости координационных соединений. Сдвиг потенциала по-
луволны при введении в раствор лиганда значительно расширяет
возможности полярографического анализа, позволяя создавать
условия для определения нескольких компонентов в одном рас-
творе без их предварительного разделения. Например, в 1 М KCl
ионы свинца(П) и таллия(1) имеют потенциалы полуволны, соот-
ветственно, -0,435 и -0,483 В и на этом фоне их раздельное опре-
деление неосуществимо. В 1 М NaOH потенциал полуволны свин-
ца становится равным—0,755 В, а у таллия остается практически
без изменений, поэтому в щелочном растворе эти ионы могут
быть определены при совместном присутствии.
Если в растворе находится несколько веществ, потенциалы по-
луволны которых различаются на 100 мВ и больше, то на поляро-
грамме будет не одна волна, а несколько — по числу восстанавли-
вающихся ионов (рис. 10.2), а возможно и больше, так как при сту-
пенчатом восстановлении один ион может давать две волны. На-
пример, две волны дает ион Си2+ в присутствии 1 М NH3: первую
при потенциале полуволны -0,20 В и вторую цри -0,48 В. Можно
получить таким образом полярографический спектр ионов, а затем
по этим данным и измеренному потен-
циалу полуволны идентифицировать не-
известное вещество. Вполне понятно,
что" положение элемента в таком спект-
ре будет зависеть от фонового электро-
лита: его природы и концентрации.
Полярограмма, изображенная на
рис. 10.1, несколько идеализирована,
так как на ней не видны осцилляции
тока, вызванные периодическим отры-
вом капель ртути. Иногда эти осцилля-
ции очень затрудняют работу, особен-
но в области малых концентраций оп-
ределяемого элемента.
I
• е1/2(А) El/2(ßj е1/2(С) Е
Рис. 10.2. Полярограмма
при наличии в растворе
восстанавливающихся
веществ А, ВиС
215
Кроме того, на подпрограммах нередко возникают максиму-
мы различной формы, мешающие определению истинного потен-
циала полуволны и силы тока. Различают максимумы I и II рода.
Теория связывает их появление с гидродинамическими явления-
ми в растворе, вызываемыми каплями ртути, и адсорбционными
процессами. Для подавления максимумов в полярографируемый
раствор обычно вводят поверхностно-активные вещества: жела-
тин, агар-агар и др. Подавление максимумов поверхностно-ак-
тивными веществами лежит в основе нескольких чувствитель-
ных (до 10"9 моль/л) аналитических методик определения этих
веществ в растворе.
Связь диффузионного тока с концентрацией иона см и дру-
гими величинами передается уравнением Ильковича:
1а = 6052г2)1/2т2/з^1/бСм> (10.11)
где 2 — заряд иона; Ъ — коэффициент диффузии; т — масса рту-
ти, вытекающей из капилляра в 1 с, мг; * — время образования
капли (период капания).
Среди величин, входящих в это уравнение, труднее всего
поддается экспериментальному определению коэффициент диф-
фузии I), а использование соответствующих справочных данных
не всегда возможно. Поэтому коэффициент пропорциональности
между концентрацией вещества и силой диффузионного тока
обычно устанавливают с помощью стандартных растворов. Дей-
ствительно, при постоянных условиях полярографирования I), т
и £ постоянны и уравнение (10.11) переходит в
1а = ксм. (10.12)
В связи с этим в работах по полярографии всегда указывается так
называемая характеристика капилляра, вычисляемая как
т2/з^1/бв Линейная зависимость (10.12) является основой количе-
ственного полярографического анализа.
10.2. Схема полярографической установки
Принципиальная схема полярографической установки пред-
ставлена на рис. 10.3. Анализируемый раствор 2 находится в
электролизере 3, на дне которого имеется слой ртути 1, являю-
щийся анодом. Часто в качестве анода используют насыщенный
каломельный электрод (НКЭ). Катодом служит ртутный капаю-
216
Рис. 10.3. Схема
полярографической
установки
щий электрод 4, соединенный с ре-
зервуаром ртути 5. Внешнее нап-
ряжение, подаваемое на электроды,
можно плавно менять с помощью ре-
охорда или делителя напряжения 7
и измерять при этом гальванометром
6 силу тока, проходящего через рас-
твор.
Как уже отмечалось, напряже-
ние, которое подается на электроды,
будет практически целиком опреде-
лять потенциал катода (капающего
ртутного электрода).
В вольтамперометрии с успехом
применяют также твердые микро-
электроды, изготовляемые из благо-
родных металлов (платины, золота
и др.) или графита. Основными достоинствами твердых электро-
дов являются возможность работы в более положительной облас-
ти потенциалов (до 1,3 В), чем с ртутным электродом (ртутный
капающий электрод используется в области примерно от 0,3 до
-2,0 В), и их нетоксичность (пары ртути, как известно, чрезвы-
чайно ядовиты и работа с ртутным электродом требует строгого
соблюдения специальных правил техники безопасности).
Однако использование твердых электродов также имеет свои
трудности, связанные, главным образом, с обновлением поверх-
ности электродов. Стационарные твердые электроды не нашли
широкого применения в практике из-за медленности установле-
ния предельного тока, невысокой чувствительности и других не-
достатков.
Значительно более широкое применение имеют вращающие-
ся и вибрирующие платиновые микроэлектроды, на которых ус-
тойчивая сила тока устанавливается быстро. При работе таких
электродов раствор непрерывно перемешивается, благодаря чему
к поверхности электрода ионы доставляются не только за счет
диффузии, но и за счет механического перемешивания. Это зна-
чительно (в 10—20 раз) увеличивает предельный ток по сравне-
нию с диффузионным. По точности методы с применением твер-
дых электродов часто уступают методам, использующим ртут-
ный капающий электрод, однако применение вращающегося
платинового микроэлектрода позволяет существенно расширить
анодную область потенциалов, пригодную для полярографиче-
217
ских измерений до 1,4 В по сравнению с областью, в которой
обычно применяется ртутный капающий электрод (до 0,3 В).
Тем не менее ртутный капающий электрод сохраняет свое
большое практическое значение, так как на твердых электродах
ограничены катодные процессы из-за небольшого перенапряже-
ния водорода на платине — из кислых растворов на платине он
начинает выделяться при потенциале около -0,ГВ, а: на ртути
только при -2,0 В. Промышленностью выпускаются полярогра-
фы нескольких марок, которые пригодны для выполнения ана-
литических работ и проведения научных исследований (ПЭ-312,
КАП-22бу, ППТ-1 и др.).
10.3. Прямая полярография
Методы прямой полярографии основаны на непосредствен-
ном применении уравнения полярографической волны (10.9) и
уравнения Ильковича (10.11) или (10.12). Потенциал полуволны
не зависит от концентраций и является качественной характе-
ристикой вещества. Обычно потенциал полуволны определяют
графическим методом. Уравнение (10.9) показывает, что ^ 1(1 1
является линейной функцией Еу и, следовательно, если на гра-
фик нанести 1$ 1(1 т 1 как функцию Е9 то получится прямая, кото-
рая пересекает ось абсцисс в точке, где Е — Е1/2> т. е. когда
0 (рис. 10.4).
Для идентификации неизвестного вещества можно этим
методом определить потенциал полуволны и, пользуясь табли-
цей потенциалов полуволны или по-
лярографическим спектром, устано-
вить наиболее вероятный элемент.
Однако чаще это свойство использу-
ют для выбора фонового электроли-
та. Зная качественный состав про(5ы,
подбирают по табличным данным та-
кой фон, на котором полярографиче-
ская волна определяемого элемента
может быть получена без каких-ли-
бо искажений за счет волны кёпшр-
щегр элемента или иного электродно-
го процесса.
Рис. 10.4. Графическое
определение потенциала
полуволны
218
10.3.1. Количественный полярографический анализ
Наиболее широко в количественном полярографическом ана-
лизе применяется метод градуировочного графика на основе
уравнения (10.12). График строят по данным полярографирова-
ния нескольких стандартных растворов. На оси ординат откла-^
дывается пропорциональная силе диффузионного тока высота по-
лярографической волны, а по оси абсцисс — концентрация ана-
лизируемого вещества. В соответствии с уравнением (10.12)
градуировочный график должен представлять прямую линию,
проходящую через начало координат. Метод дает точные резуль-
таты при условии строгой идентичности условий полярографиро-
вания стандартных растворов и неизвестной пробы. К условиям
полярографирования относят условия работы капилляра, темпе-
ратуру и среду (фоновой электролит). Метод градуировочного
графика является наиболее трудоемким, но и наиболее точным.
При анализе некоторых хорошо изученных систем, для кото-
рых применимость уравнения (10.12) установлена вполне надеж-
но, часто применяется менее трудоемкий метод стандартных
растворов. В этом методе в строго одинаковых условиях снимают
по л программы стандартного и анализируемого растворов и из
пропорции, основанной на уравнении (10.12), рассчитывают не-
известную концентрацию сх:
сх = Сс*г> (10ЛЗ)
где сх — концентрация стандартного раствора; кх и Аст — высота
волны при полярографировании соответственно анализируемого
и стандартного растворов.
Метод применим также только в условиях строгой стандар-
тизации условий полярографирования.
Широко распространен в количественной полярографии ме-
тод добавок. Пусть при полярографировании исследуемого рас-
твора сила диффузионного тока равна
1Х = ксх. (10.14)
Добавим к этому раствору известное количество стандартного
раствора и снова определим диффузионный ток:
', + «-*(«, + *«)• (10.15)
При почленном делении уравнений (10.14) и (10.15) получаем
г^=-^Г, откуда>;=сст—^—. (10.16)
хх + ст сх ' сст .х+ст*
219
~СХ
Рис. 10.5. Градуировочный
график в методе добавок
По соотношению (10.16) находим
концентрацию анализируемого рас-
твора. Можно использовать также
графический метод. В этом случае по-
лученные данные наносят на график
зависимости I
X + ст
от сст (рис 10.5).,
При 1Х + СТ = 0, как показывает уравне-
ние (10.16), сх = -сст, т. е. при экстра-
поляции прямая на этом графике при
1Х + ст ~ 0 отсекает на оси абсцисс вели-
чину, равную концентрации определяемого вещества. В методе до-
бавок автоматически учитывается влияние фона итак называемых
третьих компонентов, что является важным достоинством метода,
позволяющим применять его при анализе сложных смесей. ;
Если в анализируемом растворе присутствует несколько ве-
ществ, восстанавливающихся на ртутном катоде, на полярограм-
ме, как уже говорилось, появится несколько волн (см. рис. 10.2).
По величине потенциала полуволны определяют качественный
состав, а по силе диффузионного тока — концентрацию каждого
из компонентов. Так, например, полярограмма на рис. 10.2 со-
стоит из трех волн, каждая из которых характеризует один из!
компонентов смеси — компонент А имеет потенциал полуволны 1
-Е1/2(Д) и диффузионный ток 1а(А)У У компонента В потенциал по-
луволны равен Е1/2(в) и ток ^(в) и т» Д. Этот метод успешно при-
меняется в практике, например, для определения меди и цинка'в
рудах по одной полярограмме. с
10.3.2. Дифференциальная полярография
Для анализа смесей, содержащих ионы или вещества с близ-
кими потенциалами полуволны, применяют методы дифферен-
лт
циальной полярографии, использующие кривые — - Е. Необхо-
а£ .
димые соотношения можно получить на основании уравнения
(10.9). Придадим ему вид
771 771 ЛТ
и после потенцирования
КГ/п*
220
решим относительно /:
<Н/<1Е
Для удобства дальнейшего диффе-
ренцирования объединим постоянные:
г
а1/2
Е
1+е(Е-Е1/2)к
и продифференцируем по Е:
АЕ
Рис. 10.6. Полярограмма
в методе дифференциальной
полярографии
1лк
I _е(Е-Е1/2)к
(10.17)
а/
Зависимость — от Е графически представлена на рис. 10.6. Что-
бы найти положение максимума, продифференцируем уравнение
(10.17) еще раз по Е и приравняем производную нулю. Тогда по-
лучим, что в точке максимума
(Я)тах = Я:
1/2 >
(10.18)
т. е. что потенциал точки, соответствующей максимуму кривой
на рис. 10.6, является потенциалом полуволны/Величину орди-
наты в этой точке находим при подстановке соотношения (10.18)
в уравнение (10.17):
1дк
1/2 (1 + 1)2
П1?
4ЕТ'
Следовательно, ордината в точке максимума пропорциональна
силе диффузионного тока и является, таким образом, мерой кон-
центрации вещества. Это значение можно использовать, напри-
мер, при построении градуировочных графиков.
Получать дифференциальные полярограммы можно графи-
ческим дифференцированием обычных полярограмм или с по-
мощью специальной электрической схемы, позволяющей непо-
средственно записывать дифференциальную кривую во время по-
лярографирования.
Дифференциальная полярография имеет значительно более
высокую разрешающую способность, позволяя определять в од-
ном растворе ионы с близкими потенциалами полуволны. Напри-
мер, этим методом могут быть определены свинец и таллий, у ко-
торых потенциалы полуволны на фоне 2М КЖ)3 различаются
221
Е
Е
а)
Рис. 10.7. Полярогракма раствора^ содержащего РЬ(М03)2 и Т1Ж>3
только на 0,06 В. На интегральной полярограмме оба иона обра-
зуют одну общую волну (рис. 10.7, а), а на дифференциальных
кривых четко видны два максимума (рис. 10.7, б). Кроме того,
методы дифференциальной полярографии более точны, так как
фиксировать положение максимума и измерять его высоту мож-
но с более высокой точностью, чем определять аналогичные ха-
рактеристики в методе обычной полярографии.
10.3.3. Хроноамперометрия
с линейной разверткой потенциала
Новые возможности в анализе и изучении электродных про- ;
цессов открывает хроноамперометрия с линейной разверткой
потенциала. Обычная скорость изменения потенциала в этом ме-
тоде составляет до 50 мВ/с вместо 2^-3 мВ/с в классической по--
лярографии. Для измерения силы тока здесь вместо гальваномет-6
ра используют безынерционный осциллограф.
Полярограмма, получаемая в этом методе, представлена на
рис. 10.8. При достижении потенциала восстановления ток резко
возрастает и достигает максимума, превышая величину 1^ клас-
сической полярографии, поскольку происходит электровосста-
новленйе практически всех ионов приэлектродного слоя, и затем "
падает, так как приэлектродный слой обедняется ионами, а ско-
рость диффузии недостаточна, чтобы пополнить дефицит за столь
короткое время. Потенциал максимума Етах на этой кривой яв-
ляется качественной характеристикой иона, а высота максимума
Атах пропорциональна концентрации иона. Хроноамперометрия с/
линейной разверткой потенциала имеет более низкий предел об-
наружения, чем обычная вольтамперометрия.
Развитие полярографии в последние годы привело к появле-
нию новых полярографических методик, таких, как импульсная:
222
Рис. 10.8. Рис. 10.9.
Осциллографическая Кривая анодного
полярограмма растворения
полярография, векторная полярография и др., Во многих из них
используется наложение на обычную медленную постоянйотоко^
вую полярографическую развертку потенциала переменного на-
пряжения. Наложение переменного напряжения понижает пре-
дел обнаружения и улучшает разрешающую способность. Пере-
меннотоковая вольтамперная кривая также является кривой с
максимумом.
1ОД.4. Инверсионная вольтамдерометрия
Существенное увеличение чувствительности дает инверсион-
ная вольтамперометрия.
Идея метода инверсионной полярографии состоит в выделе-
нии определяемого элемента из очень разбавленного раствора на
ртутной капле или тонкой пленке ртути на графитовом электроде
или просто на графитовом электроде электролизом с последую-
щим анодным растворением полученной амальгамы. Процесс на-
копления происходит при потенциале, соответствующем пре-
дельному току. Зависимость силы тока от напряжения при анод-
ном растворении имеет вид характерного пика (рис. 10.9),
глубина которого Ъ, пропорциональна концентрации .определяем
мого иона, а потенциал минимума Ет1л определяется природой
иона. Предел обнаружения в методике инверсионной вольтампе-
рометрии на 2—3 порядка ниже предела обнаружения в обычных
полярографических методиках. Чем больше продолжительность
накопительного электролиза, тем большее количество металла
перейдет из раствора в ртутную каплю и тем больше возрастет
чувствительность анализа. Например, при анализе растворов,
в которых концентрация определяемого элемента составляет
10"9 моль/л, время электролиза доходит до 1ч.
223
10.3.5. Анализ органических соединений
Объектами вольтамперометрического анализа являются не
только неорганические вещества или ионы, но й многие органи-
ческие вещества, способные к электрохимическим превращени-
ям. В электродной реакции органического вещества обычно при-
нимают участие ионы водорода:
К + пК+ + пе~ = ЯЯп
Анализ в связи с этим проводится в буферных растворах с доста-
точно высокой буферной емкостью.
К вольтамперометрически активным группировкам относят-
ся, например, /СНО, ^>С=М, —Ж)2, —О—О—, —в—в— и мно-
гие другие. В условиях вольтамперометрического анализа легко
восстанавливаются и могут быть определены многие альдегиды;
кетоны, азо- и нитросоединения, органические пероксиды. Орга-
нические кислоты (малеиновая, фумаровая и др.) и эфиры окис-
ляются на платиновом электроде.
10.3.6. Полярографическое исследование
реакций комплексообразования
Зависимость потенциала полуволны от свойств фонового
электролита эффективно используется в химии и термодинамике ;
координационных соединений для установления состава и опре-;
деления констант устойчивости образующихся комплексов. Чем ц
выше устойчивость образующихся комплексных соединений и:
концентрация лиганда, тем больше сдвигается потенциал полу- \
волны в отрицательную область. При образовании одного соеди-
нения эта зависимость при 25 °С выражается уравнением
л™ _ 0,059 0,059 „1„ ■ -
где — сдвиг потенциала полуволны при данной концентра-с.1
ции лиганда сь по сравнению с потенциалом полуволны в отсутст-
вие лиганда; Р — константа устойчивости комплексного соедине-/
ния; р — координационное число.
Величины и р определяются графически, используя зави-
симость■ АЕ{/2 от 1£ сь*
224
10.4. Амперометрическое титрование
В процессе амперометрического титрования после прибавле-
ния отдельных порций реактива отмечают силу тока при напря-
жении, соответствующем величине предельного тока. По этим
данным строят кривую амперометрического титрювания в коор-
динатах сила тока-—объем титранта и графически находят точку
эквивалентности. В качестве индикаторного электрода в амперо-
метрическом титровании обычно применяются вращающиеся
платиновыёлграфитовые и другие твердые электроды.
10.4.1. Кривые амперометрического титрования
Вид кривой амперометрического титрования зависит от того,
какой компонент реакции титрования вступает в электродную ре-
акцию — определяемое вещество, титрант или продукт реакции.
Например, если при титровании серебра иодидом используется про-
цесс восстановления серебра на платиновом вращающемся катоде,
кривая титрования имеет вид, изображенный на рис. 10.10. Если
в этом титровании используется процесс анодного окисления
иодид-иона (титранта), кривая титрования имеет вид, изображен-
ный на рис 10.11. В первом случае в ходе титрования сила тока
уменьшается, так как концентрация серебра в растворе падает в ре-
зультате образования осадка Agi, а после достижения точки экви-
валентности остается постоянной. Во втором случае, когда исполь-
зуется анодное окисление иодид-иона, концентрация его после точ-
ки эквивалентности увеличивается и это отражается в возрастании
силы тока. Точка эквивалентности в обоих случаях находится гра-
фически как точка пересечения соответствующих прямых.
На рис. 10.12 приведена кривая амперометрического титро-
вания по диффузионному току, обусловленному концентрацией
Tl II Ii
т.э. V
Рис 10.10.
Амперометрическое
титрование катиона,
восстанавливающегося
на катоде Ag+
т.э. V
Рис. 10.11.
Амперометрическое
титрование Ag+
по окислению
на аноде Г"
т.э. V
Рис. 10.12.
Амперометрическое
титрование мышьяковой
кислоты иодидом
калия
15 — 7831
225
образовавшегося продукта реакции титрования. Этот случай ре-
ализуется, например, при титровании мышьяковой кислоты
иодидом калия:
НАвО|- + 21" + 2Н+ = НАвО|- + 12 + Н20
По мере титрования концентрация свободного иода, выделяющее
гося как продукт реакции, будет возрастать до точки эквивалент-
ности, после чего останется постоянной.
10.4.2. Основные типы реакций
амперометрического титрования
В методах амперометрического титрования используют реак-
ции осаждения, комплексообразования и окисления—восстанов-
ления. Необходимо, чтобы реакция, протекающая при амперо-
метрическом титровании удовлетворяла тем требованиям, кото-
рые предъявляются к реакциям в титриметрических методах в
отношении полноты и скорости их протекания. Многие анионы:
СГ, Вг", I", ЭО| ~, МоО|" и др. титруются солью свинца при потен-
циале -0,4 В, когда на ртутном капающем электроде происходит
восстановление иона РЬ2+. Окисление ферроцианида Ре(СЫ)|" на
вращающемся платиновом электроде при 0,7—1,0 В исцользует-
ся для амперометрического титрования Ъъ2*, Си2+, Сс12+, Са2+ и
других катионов. В методиках амперометрического титрования
часто применяется осаждение органическими реагентами: 8-ок-
сихинолином, купфероном, диметилглиоксимом и др., причему
титрование можно проводить как по току восстановления кати-
она, так и по току органического реагента.
Если в растворе находятся два иона, способных образовывать
малорастворимые соединения с титрантом, и их ПР различаются
существенно, а электрохимические свойства системы позволяют
получать кривую титрования с двумя изломами, то становится
возможным амперометрическое титрование каждого компонента
в одном растворе без предварительного химического разделения.
Так титруют, например, смесь никеля и меди рубеановодородной
кислотой.
Широко используется в амперометрическом титровании ре-
акция образования этилендиаминтетраацетатных комплексов
различных элементов. С помощью этой реакции определяют де-
сятки катионов, способных к электрохимическому ворстановле-
нию в условиях анализа: В13+, Те3+, ¥е2+, №2+, РЬ2"1", гп?+, ри*+,
226
C62+, Gd2+ и др. При изменении рН создаются условия для титро-
вания: этим методом нескольких катионов и таким образом ана-
лизировать смеси катионов без их химического разделения. Так
титруют, например, раствор, содержащий висмут и цинк: при рН
1,0—2,0 определяют висмут, затем при рН 4,7—5,0 — цинк. Раз-
работаны также методики амперометрического титрования» ос-
нованные на анодном окислении ЭДТА на платиновом микро-
электроде. > г
При амперометрическом титровании, с исцользованием реак-
ций окисления—восстановления в качестве титрантов использу-
ют K2Cr207, Ce(S04)2, KBrOg, 12 и др.'— 'для определения восста-
новителей; FeS04, Na2S203 и др. — для определения окислителей.
Практическое применение нашли также некоторые органиче-
ские реагенты — хлорамин Б как окислитель, аскорбиновая кис-
лота как восстановитель и другие вещества. Если в растворе име-
ется два окислителя или два восстановителя с существенно
разными окислительно-восстановительными потенциалами, воз-
можно последовательное амперометрическое титрование оборх
компонентов без предварительного химического разделения.
10.4.3. Титрование с двумя >
индикаторными электродами
В последнее время широкое распространение получил метод
амперометрического титрования с двумя индикаторными элект-
родами. Иногда его называют также методом мертвой конечной
точки (dead-stop end point). При определениях по этому методу в
анализируемый раствор вводят два платиновых или иных инерт-
ных электрода под небольшим постоянным напряжением (поряд-
ка 10~2 В) и в ходе титрования отмечают силу тока. До начала
титрования сила тока между электродами или очень мала, или
вообще не наблюдается, так как в отсутствие окислительно-вос-
становительной пары при столь малой разности потенциалов
электродные процессы йе происходят. Введение титранта вызы-
вает появление в анализируемом растворе двух окислитель-
но-восстановительных пар, причем до точки эквивалентности* в
растворе в заметнь!х количествах будут находиться компоненты
пары, образованной титруемым веществом, а после точки эквива-
лентности компоненты, образованные титрантом. Вид кривой
титрования будет определяться, в основном, электрохимической
обратимостью этих пар.
15'
227
> Если обе окислительно-восстановительные пары обратимы,
как, например, в реакции титрования железа(П) солью це-
рия(ІУ):
і Ге2+ + Се4+ = Ге3+ + Се3+
кривая титрования имеет вид, изображенный на рис. 10.13, а.
Добавление первых же порций соли Се4+ вызовет образова-
ние в растворе окислительно-восстановительной пары Ге3+/Ее2+ и
в цепи появится ток, так как восстановление Ге3+ на катоде и
окисление Ге2+ на аноде вследствие высокой обратимости систе-
мы происходит при минимальной разности потенциалов. Сила
тока будет возрастать, пока не прореагирует примерно половина
Ее2+, затем начнет уменьшаться и упадет почти до нуля в точке
эквивалентности. После точки эквивалентности на катоде будет
восстанавливаться Се4+, а на аноде окисляться Се3+ и ток в цепи
снова появится.
Если система, образуемая определяемым веществом, обрати-
ма, а титрантом — необратима, как» например, при титровании
Рё2+ перманганатом калия, кривая титрования до точки эквива-
лентности не будет отличаться от кривой на рис. 10.13, а, так каїс
в обоих случаях сила тока в системе до точки эквивалентности
контролируется электрохимически обратимой системой. Однако
после точки эквивалентности возрастания силы тока не произой-
дет, так как при этой разности потенциалов Мп2+ на аноде окис-
ляться не будет.
Если титруется электрохимически необратимая системна, а
титрант образует обратимую окислительно-восстановительную
пару, то до точки эквивалентности тока не будет, а после точки
эквивалентности он резко возрастает (рис. 10.13, б). Такой вид
т.э. V т.э. V
о) б)
Рис. 10.13. Кривая амперометрического титрования
с двумя индикаторными электродами:
а — обе пары обратимы; б —-необратимая система титруется обратимой
228
имеет кривая титрования, например, перманганата раствором со-
ли Мора или тиосульфата раствором иода.
Метод амперометрического титрования с двумя индикатор-
ными электродами достаточно точен и чувствителен: он пригоден
для анализа растворов с концентрацией определяемого вещества
10~5 моль/л и менее. В аппаратурном отношении он проще, чем
метод с одним индикаторным электродом. При титровании по
этому методу часто отпадает необходимость в построении кривой
титрования, так как точка эквивалентности может быть опреде-
лена по резкому прекращению или появлению тока.
10.5. Практическое применение
Вольтамперометрический метод применяют для определения
многих металлов. Кадмий, кобальт, медь, свинец, марганец, ни-
кель, олово, цинк, железо, висмут, уран, ванадий и многие дру-
гие могут быть определены в рудах, концентратах, сплавах и
иных природных и технических объектах. При достаточно раз-
личающихся потенциалах полуволны (АЕ1/2 0,10 В) возможно
количественное определение нескольких элементов без предвари-
тельного разделения. Например, в аммиачном буферном растворе
можно полярографировать смесь кадмия (Д££/2 = -0,81 В) и ни-
келя (ДЕ|/2 == -1,10 В). Существенное практическое значение
имеет вольтамперометрическое определение хромат-, иодат-, м;о-
либдат-ионов и некоторых других, а также многих органических
соединений: альдегидов, кетонов, азо- й нитросоединений и т. д.
Широко испольйуют полярографический метод для анализа био-
логически важных материалов: крови, сыворотки и т. д.
Интенсивно развиваются также современные вольтамперо-
метрические методы: с быстрой разверткой потенциала, перемен-
нотоковая, инверсионные, импульсные и т. д.
Амперометрическое титрование применяется для определе-
ния катионов и анионов в различных технических и природных
объектах, минеральном сырье и продуктах его переработки, при-
родных водах, промышленных растворах, продуктах металлур-
гии и т. д., а также в анализе многих органических веществ. Ис-
пользование реакций различного типа (осаждения, комплексооб-
разования и окисления—восстановления) позволяет подбирать
условия амперометрического титрования для большинства эле-
ментов периодической системы. Значительно расширились воз-
можности амперометрического титрования в связи с применени-
ем органических реагентов, аналитические достоинства которых
229
(селективность, чувствительность) хорошо известны. Многие из
них способны к электрохимическим превращениям на электро-
дах, что еще больше повышает их ценность, так как позволяет
проводить амперометрическое титрование по току титранта. Для
амперометрического титрования характерна экспрессность, его
можно проводить в разбавленных растворах (до 10~5 моль/л и
меньше) и анализировать мутные и окрашенные растворы.
10.6. Общая характеристика метода
Вольтамперометрический метод достаточно универсален и
применим к многочисленному кругу объектрв. Основными досто-
инствами метода являются быстрота анализа, возможность опре-
деления нескольких веществ в смеси без предварительного разде-
ления, достаточно высокая точность и применимость к анализу
небольших содержаний определяемого элемента. Погрешность
полярографического анализа в обычных условиях составляет
±2% для растворов концентрации порядка 10"3—10"4 моль/л и
около ±5% для более разбавленных. При сочетании вольтамперо-
метрии с методами экстракции, хроматографии и т. д. предел об-
наружения снижается еще больше.
; Амперометрическое титрование характеризуется более высо-
кой точностью и более высокой чувствительностью, чем методы
прямой вольтамперометрии. Аппаратурное оформление устано-
вок амперометрического титрования несложно, особенно просты
установки для титрования с двумя индикаторными электродами.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. В чем сущность полярографических методов анализа?
2. Как взаимосвязаны потенциал полуволны и предельный (диффу-
зионный) ток? '
3. Как рассчитать потенциал полуволны на основании вольтампёр-
ной кривой?
4. Отчего зависит величина предельного тока?
5. На чем основан качественный полярографический анализ? ^
6. Какие величины входят в уравнение Ильковича? Каково практи-
ческое применение этого уравнения?
7. Какие аналитические приемы используют в количественной по-
лярографии? , .
8. Что представляет собой инверсионная полярография?
9. В чем сущность амперометрического титрования? V "'
10. Какой вид имеют кривые амперометрического титрования?
230
" глава 11 : :
Электролиз и кулонометрия
11.1. Законы электролиза
Электролизом называют химическое разложение вещества
под действием электрического тока. На катоде (отрицательно за-
ряженном электроде) происходит восстановление:
¥е3+ + е- = ¥е2+
Си2+ + 2е- = Си (к)
а на аноде (положительно заряженном электроде) окисление:
2СІ" - 2е~ = С12 (г)
При электролизе растворов сульфатов, фосфатов и некото-
рых других солей на аноде происходит окисление не 80|~- или
РО|"-, а ОН--ИОНОВ: >
20Н" ~ 2е~ = 1/202 + Н20
поскольку ОН" легче отдают свои электроны, чем вО|" или Р0|'~,
даже в кислом растворе. На аноде может происходить окисление
не только анионов, но и катионов. Например, ионы РЬ2+ образу-
ют диоксид: - 4>;.
РЬ2+ + 2Н20 = РЬ02 + 4Н+ + Не-
основные законы электролиза установлены еще Фарадеєм.
Масса вещества, выделившаяся при электролизе, пропорциональ-
на количеству электричества, прошедшего через раствор.
При прохождении через раствор одного и того же количества
электричества на электродах выделяется одно и то же количество
вещества эквивалента.
Эти законы выражаются формулой
/ т --. ЯМ _: им т п
т 96485 96485'
где т — масса вещества, выделившегося при электролизе; (І —
количество электричества; М — молярная масса эквива^^а ве-
щества; 96 485 ~ число Фарадея, равное количеству электриче-
ства, которое требуется для выделения молярной массы эквива-
лента вещества; / — сила тока; і — время электролиза.
231
Важной характеристикой процесса электролиза является вы-
ход по току 9 равный отношению количества выделившегося веще-
ства к тому количеству вещества, которое должно было выделить-
ся по закону Фарадея, т. е. в соответствии с уравнением (11.1).
11.2. Потенциал разложения и перенапряжения
Потенциалом разложения называют минимальную величину
внешней ЭДС, при которой в данных условиях начинается непре-
рывный электролиз.
Потенциал разложения превышает величину обратимой ЭДС
гальванического элемента, образованного электродами системы,
в которой происходит электролиз. Превышение вызывается дей-
ствием нескольких факторов. Одним из них является сопротив-
ление ячейки Д, поскольку в соответствии с законом Ома
или
где I — сила тока; Е'общ — напряжение, наложенное на электро-
ды; Ек.— ЭДС обратимого элемента, рассчитанная по уравнению
Нернста.
Для протекания электролиза обычно требуется некоторое уве-
личение напряжения, называемое перенапряжением П, и тогда
Добщ = Дн + Ш + П, (11.2)
где Еобщ — действительная величина приложенной ЭДС, при ко-
торой происходит электролиз в данной системе. <~
Перенапряжение зависит от свойств электродов и участников
электрохимической реакции, состояния поверхности электродов,
условий проведения процесса (плотности тока, температуры) и т. д.
Установлено, что на гладком электроде перенапряжение больше,
чем на шероховатом, а перенапряжение при выделении металлов
значительно меньше, чем при выделении газов, й т. д.
Основной причиной перенапряжения является необратимость
процессов на электродах при проведении электролиза. В случае га-
зообразных продуктов электролиза дополнительный эффект вызы-
вается замедленностью стадии образования двухатомных молекул
таза. Напряжение, подаваемое на электроды при электролизе,
представляет разность потенциалов анода и катода: >
232
Е^щ = (Е& + Пй)-(Ек-Пк) + т, (ІЬЗ)
где Е& — потенциал анода; Ек — потенциал катода; Па и Пк — пе-
ренапряжение на аноде и на катоде соответственно.
Потенциалы Ей и Ек могут быть рассчитаны по уравнению
Нернста (9.2).
11.3. Электрогравиметрический анализ
11.3.1. Схема установки для электролиза
В электрогравиметрическом анализе анализируемое вещест-
во количественно выделяют из раствора электролизом и по массе
выделившегося металла или его оксида рассчитывают содержа-
ние определяемого элемента в пробе. Схема установки для прове-
дения электролиза показана на рис. 11.1. Для получения посто-
янного тока обычно используют выпрямитель переменного тока
или батарею аккумуляторов 1. Скользящий контакт 2 позволяет
регулировать подаваемое напряжение, которое измеряют вольт-
метром. Сила тока контролируется амперметром. При выделении
металлов катод 5 обычно изготовляют из пла-
тиновой сетки, анод 4 — из платиновой спи-
рали или пластинки. При выделении оксидов
знаки электродов меняются: платиновая сет-
ка становится анодом, а спираль — катодом.
Раствор перемешивается механической или
магнитной мешалкой 3.
Электрогравиметрический анализ можно
рассматривать как один из видов гравиметри-
ческого анализа, в котором в качестве осади-
теля выступают электроны. Электролитиче-
ски получаемые осадки металлов и оксидов
являются и формой осаждения, и гравимет-
рической формой.
Важнейшими требованиями к форме
осаждения являются ее малая растворимость
и чистота, т. е. отсутствие загрязнений. Эти
требования в элёктрогравиметричёском мето- рис 1<и Схема
де выполняются идеально, так как больший- установки
ство металлов и указанные оксиды практиче- для проведения
ски нерастворимы в воде, а при электролити- электролиза
233
ческом выделении металлов или оксидов соосаждение не
происходит или его почти всегда можно предупредить путем вы-
бора условий электролиза. Полученный осадок металла или ок-
сида удобен для промывания и взвешивания.
11.3.2. Электрогравиметрические разделения
Если необходимо проанализировать раствор, содержащий
смесь ионов без их предварительного разделения, электролиз ве-
дут при строго контролируемом потенциале, вводя в схему допол-
нительный электрод с постоянным потенциалом (например, ка-
ломельный электрод) и регулирующее электронное устройство,
связывающее рабочий и дополнительный электроды. Для этого
используют специальные приборы — потенциостаты.
Заключение о возможности или невозможности разделения двух ме-
таллов методом электролиза можно сделать на основании следующих
расчетов. Пусть, например, требуется установить, можно ли провести
электролитическое разделение меди и цинка из 0,1 М растворов их суль-
фатов. Значения стандартных Электродных потенциалов равны 2££и2+/Си =
= 0,345 В, Е^п2+^п = ~0,764 В. Потенциалы, необходимые для выде-
ления этих металлов из раствора, можно рассчитать по уравнению
Нернста. Для выделения меди требуется, чтобы потенциал катода был
ЯСд2+/Си = °>345 + Ъ-0»1 0,316В,
а для выделения цинка
^гп^/гп^-0'764^ ^^0,1 = -0,793 В.
Выделение меди будем считать количественным, если концентрация
ионов Си2+в растворе будет 10"6 моль/л или меньше. Потенциал катода
в этом случае будет равен
Лу+/Си-М45 + 10-6 = 0,171 В.
При этом значении потенциала осаждение цинка не начнется, поэтому
на вопрос о возможности электролитического выделения меди в присут-
ствии цинка из сернокислых растворов следует отвечать утвердительно.
Можно найти концентрацию ионов Си2+ при потенциале выделения
цинка: ,
-0,793 = 0,345 +0,029 ^[Си2+], ". /Я-
откуда
^ [Си2*] = "°'7^3"^345 = -39, т. е. [Си2+] = 10"39 моль/л,
234 .'■
что еще раз подтверждает возможность разделения этих элементов; На
аноде, очевидно, будет выделяться кислород: У
Н20-2е-= 1/202 + 2В+ ^
и кислотность раствора будет увеличиваться.
Потенциал анода при [Н+] = 1 моль/л можно также рассчитать по
уравнению Нернста
Яо2/н2о= !'23 + °»058 1ё 1 = 1.23 В.
Перенапряжение кислорода на гладкой платине при плотности тока
0,001 А/см2 составляет 0,78 В и с увеличением плотности тока возраста-
ет. Потенциал анода с учетом перенапряжения кислорода будет равен:
1,23 + 0,78 = 2,01 В.
Разность потенциалов, которую необходимо поддерживать для про-
текания процесса и полного выделения меди, можно рассчитать по урав-
нению (11.3): 4
Еобщ = 2,01 -0,171 -1,84 В.
(" ■ . ■ • ■ ' ■ •
Можно также определить, при каком потенциале начнется выделе-
ние водорода. Поскольку на платиновом электроде выделилась медь, по
справочнику находим перенапряжение водорода на медном электроде
(-0,94 В) и рассчитываем разность потенциалов по уравнению (11.3), не-
обходимую для выделения водорода (электролиза воды):
Еобщ = 2,01-(-0,94) = 2,95 В.
Следовательно, в условиях количественного осаждения меди
(Е — 1,8—2,0 В) выделение водорода происходить не будет.
Чтобы предотвратить выделение водорода в результате слу-
чайных изменений потенциала, особенно в конце процесса или в
результате каких-либо других причин, в анализируемый раствор
при определении меди вводят небольшое количество азотной кис-
лоты. На катоде N0^ может восстанавливаться до N0^ (25°0_/ыо_ =
= 0,94 В) или N111 (£^0./кн+ = 0,87 В). Восстановление нитрата
будет происходить при разности потенциалов
.Д^-2,01-0,9-1,1В.
Следовательно, пока в растворе будет находиться ни-
трат-ион, водород на катоде выделяться не будет. Вещества, кото-
рые вносят в раствор для предотвращения нежелательных элект-
родных процессов, называют деполяризаторами. Обычно они
претерпевают электрохимическое изменение по достижении не-
которого определенного потенциала. В данном случае при опре-
делении меди нитрат-ион является деполяризатором, предотвра-
щающим выделение водорода.
Результаты аналогичных расчетов показывают, что разделе-
ние металлов методом электролиза возможно, если их стандарт-
235
ные электродные потенциалы различаются на 0,2—0,3 В. Этот
критерий позволяет по значениям стандартных электродных по-
тенциалов предсказать возможность или невозможность разделе-
ния любой заданной пары ионов. Так, например, серебро
^Аё+/Аё = 0,7994 В) можно выделить в присутствии В13+
СЕ£.2+/Ш = 0,23 В), меди №си2+/си в 0,345 В> и многих ДРУГИХ ме-
таллов, но нельзя отделить от ртути СЕ^2+/Н£ = 0,850 В). Медь
можно легко электролитически осадить в присутствии цинка
СЕ°п2+/2п - -0,764 В), кадмия (Я£а2+/са = -0,403 В), свинца
(2£рЬ2+д>ь — -0,126 В) и других элементов, нов присутствии вис-
мута сделать это можно только при строго контролируемом по-
тенциале катода.
Изменение рН или введение в раствор комплексообразующих
добавок весьма существенно сказывается на величине потенци-
ала. Использование этих факторов значительно расширяет воз-
можности выделения и разделения металлов методом электроли-
за. Например, электроосаждению никеля, кобальта и других
элементов, у которых Е° < 0, из кислых растворов мешает выде-
ление водорода. С понижением кислотности выделение водорода
уменьшается и осаждение металлов, в принцице, становится воз-
можным. Найдем, при каком рН возможно количественное выде-
ление никеля из раствора. Полнота осаждения никеля будет до-
стигнута при потенциале
Е = -0,228 + 0,029 1£ (Ю"6) = -0,402 В.
Чтобы при этом потенциале не происходило выделения водорода,
концентрация ионов водорода должна удовлетворять уравнению
-0,402 = 0 + 0,058 ^ [Н+],
откуда >
т. е. при рН > 7 водород выделяться не будет и никель может быть
количественно осажден на катоде. Практически электролиз нике-
ля ведут из аммиачных растворов. Однако образование аммиач-
ных комплексов никеля вызывает и отрицательный эффект, так
как электродный потенциал никеля в результате комплексообра-
зования понижается и для предотвращения выделения водорода
необходимо дальнейшее увеличение рН. Стандартный электрод-
ный потенциал никеля в аммиачном растворе будет равен
?кккн3)|^1 " Е^/т ~ 0,029 1§рт^Нз)|+ = -0,288 -0,029 • 8,01 =
= 0,460 В. ■ ■ ;,
236
Полнота выделения никеля из аммиачного раствора будет до-
стигнута при потенциале
Е - -0,460 + 0,029 1$ Ю-6 = -0,634 В.
Отсюда находим концентрацию ионов водорода, при которой
электролиз будет происходить без выделения водорода:
1* [Н+] - —§т8§щ - "п или РН > П.
В 1 М ЫН4ОН рН составляет примерно 11,5. Следовательно, в
этом растворе выделение водорода происходить не будет и коли-
чественное выделение никеля осуществимо практически.
Изменение электродного потенциала при комплексообразова-
нии является одним из эффективных приемов управления элек-
трохимическими реакциями. Тонкая регулировка рН раствора и
концентрации комплексообразующих веществ позволяет созда-
вать условия ^разделения металлов с близкими значениями стан-
дартных электродных потенциалов. Например, известна методика
определения в одном растворе Си2+, В13+, РЬ2+ и вп2+. В нейтраль-
ном тартратном растворе при потенциале -0,20; -0,40 и -0,60 В
поочередно выделяется на катоде и взвешивается медь, висмут и
свинец, а затем раствор подкисляется для разрушения тартратного
комплекса й выделяется олово.
Значительно проще электрогравиметрические установки, не
требующие строгого поддержания потенциала. Обычно они при-
меняются для определения в растворе только одного иона. Ме-
шающие ионы должны быть отделены или связаны в прочный
комплекс, не поддающийся электрохимическому восстановле-
нию в условиях анализа. Электрогравиметрическим методом оп-
ределяется медь из сернокислых растворов в присутствии азот-
ной кислоты, серебро и кадмий из цианидного раствора, никель
из аммиачного раствора и другие элементы. >
11.3.3. Электролиз на ртутном катоде
Наиболее существенными особенностями электролиза на
ртутном катоде являются большая величина перенапряжения во-
дорода и образование амальгамы многими металлами. Перенап-
ряжение водорода на ртутном катоде превышает 1 В, поэтому при
электролизе кислых растворов происходит выделение многих ме-
таллов, не выделяющихся на платиновом или других катодах. >
При электролизе кислых растворов на ртутном катоде выделя-
ются висмут, кобальт^ ^ железо, молибден, никель, ос-
мий, свинец, палладий, платина и многие другие, всего более 20 эле-
ментов, но не выделяются алюминий, ванадий, уран, титан и неко-
237
торые другие. Таким образом, электролиз на ртутном катоде
позволяет отделить большие содержания железа, хрома, меди от
вацадия, титана и других, что часто существенно, упрощает и уско-
ряет анализ сложных объектов.— минералов, руд, концентратов,
сплавов и т. д.
11.3.4. Внутренний электролиз
В методе внутреннего электролиза внешнего источника тока не
требуется. Здесь используется способность металлов с более поло-
жительным электродным потенциалом выделяться в свободном ви-
де из растворов их солей под действием металлов с меньшим значе-
нием стандартного потенциала (менее благородного). Пластинка
менее благородного металла, являющаяся анодом, соединяется с
платиновым катодом и, таким образом, выделение анализируемого
благородного металла происходит на платине. При небольшом со-
держании определяемого элемента осаждение металла на платино-
вом катоде происходит без каких-либо осложнений, но при боль-
ших концентрациях наряду с осаждением на катоде может проис-
ходить некоторое выделение металла на аноде. Чтобы исключить
этот процесс, анод покрывают тонкой пленкой из коллодия или ка-
тодное и анодное пространство разделяют пористой перегородкой.
Одним из важных достоинств метода внутреннего электроли-
за является возможность проведения тонких химических разде-
лений, так как на платиновом катоде выделяются металлы толь-
ко более благородные, чем металл анода.
Если, например, в качестве анода используется свинцовая
пластинка, то на катоде будут выделяться только те металлы, по-
тенциал которых превышает потенциал пары РЬ2+/РЬ, и не будут
выделяться металлы с более отрицательным потенциалом. Меняя
анод, можно создавать условия разделения металлов с близкими
потенциалами. Существенным преимуществом метода является
также чрезвычайная простота аппаратурного оформления, позво-
ляющая использовать метод практически в любой лаборатории.
После выделения из раствора методом электролиза осадок
можно взвесить и по массе осадка рассчитать содержание опреде-
ляемого элемента. Если масса осадка очень мала и не может быть
взвешена с достаточной точностью, его растворяют и определяют
концентрацию вещества в растворе любым подходящим методом.
Таким образом, электролиз больших объемов раствора позволяет
наряду с разделением провести концентрирование определяемого
элемента* так как осадок, полученный при электролизе, можно
растворить в небольшом объеме растворителя.
• /
238
11.4. Кулонометрия
Принцип и теоретические основы кулонометрии были из-
вестны давно, однако до 1938 г. этому методу достаточного вни-
маний не уделялось. С 40-х годов кулонометрия начинает широ-
ко применяться в аналитической химии и для решения различ-
ных физико-химических задач.
|В кулонометрических методах определяют количество электриче-
ства, которое расходуется в ходе электрохимической "реакций.
Различают два основных вида кулонометрических определе-
ний: .прямую кулонометрию и кулонометрическое титрование.
В методах прямой кулонометрии анализируемое вещество непо-
средственно подвергается электрохимическому превращению в
кулонометрической ячейке. В методе кулонометрического титро-
вания определяемое вещество реагирует с титрантом, который
получается! кулонометрической ячейке при электролизе специ-
ально подобранного раствора.
11.4.1. Кулонометрии при постоянном
контролируемом потенциале
Потенциостатическйе или кулонометрические методы при
постоянном контр9лируемом потенциале широко применяются в
прямой кулонометрии. Принципиальная схема установки для
потенциобтатической кулонометрии приведена на рис. 11.2.
Напряжение с аккумуляторной батареи 1 через делитель на-
пряжения 2 подается на рабочий электрод 4 кулонометрической
Рис. 11.2. Схема установки для потенциостатической кулонометрии
239
ячейки 5. Потенциал электрода определяется милливольтметром
или потенциометром, сила тока — амперметром. Количество из-
расходованного электричества измеряется кулонометром 6. В сов-
ременных установках в качестве источника стабилизированного
напряжения обычно используют специальные электронные прибо-
ры — потенциостаты, поддерживающие заданный потенциал с точ-
ностью примерно ±10 мВ в интервале от -2,5 до 2,5 В. Потенциал
рабочего электрода устанавливают с помощью поляризационной
кривой (/—^-кривой) в области, где достигается предельный ток.
Рабочим электродом кулонометрической ячейки обычно слу-'
жит платиновая пластинка или ртуть, хотя иногда используют
также золотые, серебряные или графитовые электроды. Вспомо-
гательный электрод изготовляется из тех же материалов. Элект-
родные пространства рабочего и вспомогательного электродов
разделены. Контакт между ними осуществляется через пористую
перегородку. В качестве электрода сравнения 3 (рис. 11.2) обыч-
но выбирают каломельный или хлорсеребряный. Количество
электричества, израсходованное на протекание электрохимиче-
ской реакции, может быть измерено с помощью интеграторов то-
ка или кулонометров, а также определено расчетным методом.
Принцип действия кулонометров основан на том, что через по-
следовательно включенный прибор в цепи протекает такой же ток,
какой проходит через анализируемый раствор, и, следовательно, за
некоторый промежуток времени через анализируемый раствор и
через прибор пройдет одно и то же количество электричества. В по-
следовательно включенном кулонометре со 100%-м выходом проте-
кает хорошо известная электрохимическая реакция, и измерение
количества электричества сводится, таким образом, к определению
количества вещества, полученного в результате этого процесса. '
В зависимости от способа измерения объема или массы веще-
ства различают газовые, электрогравиметрические, титрацион-
ные и другие кулонометры. В газовых кулонометрах определяет-
ся объем газа, выделившегося в результате электрохимического
процесса. В электрогравиметрических кулонометрах определяет-
ся масса вещества. Например, в медных кулонометрах находят
массу металлической меди, выделившейся при электролизе суль-
фата меди, в серебряных —- массу серебра, полученного при
электролизе нитрата серебра, и т. д.
Операцию взвешивания катода с выделившейся медью или се-
ребром иногда заменяют анодным растворением металла с этих
электродов при постоянной силе тока. Зная длительность прбцесса
и силу тока по формуле (11.1), рассчитывают массу выделившегося
металла или сразу количество электричества. Полученную систему
240
не совсем удачно называют кулонометрическим кулонометром.
Этот метод значительно экономит время, не ухудшая точности.
При прямом кулонометрическом определении, например,
олова(ГУ) или железа(Ш) в ячейке происходит восстановление
вп(ГУ) до 8п(П) или Ре(Ш) до Ге(И). По мере уменьшения кон-
центрации восстанавливающегося иона сила тока в цепи падает.
Эта зависимость приближенно выражается уравнением
Л = <1Ь4)
где 1г — сила тока в момент времени Р> 10 — сила начального то-
ка; к константа, включающая коэффициент диффузии, пло-
щадь электрода и другие величины.
Уравнение (11.4) может быть использовано для определения
количества электричества ф, израсходованного на электрохими-
ческое превращение вещества, поскольку
оо
Я= 17,(1*. (11.5)
Подставляем уравнение (11.4) в (11.5) и интегрируем:
6= //^Л-/,!"-^-* (11.6)
о
Величина к находится графически. При логарифмировании
(11.4) получаем
1п— 1п10-Ы
или
Как видно, в координатах \% 1г - * зависимость (11.7) представля-
ть
ет прямую с угловым коэффициентом - . При экстраполя-
2,о0о
ции эта прямая отсекает на ординате отрезок, равный /0.
В практических условиях длительность процесса редко превы-
шает 30 мин. Окончание восстановления обычно фиксируется по
прекращению изменения силы тока в течение некоторого време-
ни: сила тока при этом уменьшается почти до нуля. В некоторых .
случаях, например, при большом остаточном токе применяют
химические или физико-химические способы индикации. Массу
определяемого вещества рассчитывают по формуле
т = —М.
96 485
Поправку на остаточный ток вводят, зная его силу и время элект-
ролиза.
16-7831 ... 241
11.4.2. Кулонометрия при постоянной
контролируемой силе тока
(кулономе7рическое титрование)
В методе кулонометрического титрования используют уста-
новки с постоянной силой тока. Так как титрант генерируется в
количестве, точно эквивалентном содержанию анализируемого
вещества, то по количеству электричества, израсходованного йа
генерацию титранта, можно рассчитать содержание определяе-
мого вещества. Блок-схема установки для кулонометрического
титрования приведена на рис. 11.3. Пульт-переключатель 4 пита-
ется током стабилизированного напряжения от аккумуляторной
батареи 1 через сопротивление 2 и амперметр 3. Постоянство си-
лы тока в генераторной цепи 7 контролируется потенциометром
6 по падению напряжения на стандартном сопротивлении. Пуск
секундомера 5 и включение генераторной цепи 7 производится
через пульт одновременно (8 и 8' — генераторные электроды). Ко-
нец реакций фиксируется с помощью индикаторных электродов
9 и измерительного потенциометра 10. Титрант генерируется в
результате электролиза на электроде 8 (рабочий генераторный
электрод). Вторым электродом схемы ген так на-
зываемый вспомогательный электрод #\ Его обычно изолируют
от раствора анализируемого вещества, помещая в трубку с дном
из пористого стекла, так как продукт реакций на вспомогатель-
ном электроде нередко мешает кул биометрическому определе-
нию. Индикаторными электродами могут быть два платиновых
электрода, если для индикации применяются; амперометриче-
скйй метод, или платиновый и каломельный, если используется
потенциомётрическая индикациями т. д. Может быть использо-
Рис. 11.3. Блок-схема установки для кулонометрического титрования
, • 242
ван также спеьстррфотометричеекий или какой-либо другой спо-
соб определения точки эквивалентности. Ячейки с внешней гене-
рацией титранта, когда титрант генерируется в отдельной камере
и затем добавляется к анализируемому веществу, применяются
реже, однако иногда они бывают крайне необходимы, например
кулонометрическое определение кислот основано на генерирова-
нии ОН~-ионов при электролизе воды. На катоде происходит вос-
становление воды
2Н20 + 2е~ - Н2 + 20Н-
а на аноде ее окисление
2Н20 = 02 + 4Н+■+ 4е~
Чтобы не допустить случайного попадания ионов Н+ из анод-
ного пространства в рабочий объем, применяются ячейки с внеш-
ней генерацией ОН"\
Кулонометрическое титрование имеет некоторые преимуще-
ства перед обычными титриметрическими методами. Наиболее
существенным достоинством кулонометрического титрования яв-
ляется то, что рабочий раствор в этом методе не готовят и не стан-
дартизируют: титрант генерируется электрохимически непосред-
ственно в присутствии анализируемого вещества и в количестве,
необходимом только для данного титрования. Это позволяет ис-
пользовать для титрования малоустойчивые или легколетучие
вещества, такие, как, например, С12, Вг2 или соединения Си(1),
Сг(П) ит. д., и обходиться в работе небольшими навесками веще-
ства, так как, регулируя силу тока, можно точно дозировать
очень небольшое количество титранта. Достоинствами кулоно-
метрического титрования являются также универсальность мето-
да приготовления титранта (один и тот же источник тока можно
использовать для генерирования различных титрантов) и воз-
можность легкой автоматизации процесса титрования.
"11.5. Практическое применение
Электрогравиметрический анализ характеризуется высокой
точностью: погрешность определения составляет 0,1-^-0,2%. До-
стоинствами его является также возможность проведения анали-
за во многих случаях без предварительного разделения и сравни-
тельно простая аппаратура. Ограничением метода является его
применимость к относительно небольшому числу элементов и
для анализа сравнительно больших содержаний, а также дли-
тельность анализа.
16'
243
Перспективными направлениями развития электрогравимет-
рических методов анализа является поиск условий для определе-
ния элементов в сложных смесях без разделения, совершенствова-
ние методов внутреннего электролиза и расширение возможнос-
тей практического приложения к различным объектам анализа.
Практическая применимость метода потенциостатической
кулонометрии достаточно широка. Известны методики аналити-
ческого определения сурьмы, мышьяка, висмута, кадмия, меди и
многих других элементов. Разработаны методики определения
нескольких элементов при совместном присутствии, как, напри-
мер, определение малых содержаний кадмия в присутствии ме-
ди, что является вообще сложной аналитической проблемой.
Анализ галогенидов является примером кулонометрического
определения неэлектроактйвных веществ, т. е. веществ, не изме-
няющихся в данных условиях под действием тока. Галогениды Х~
осаждают ионами Аё+, которые генерируются на А§-электроде:
А£ + Х-=^Х + е-
Кулонометрическим методом определяется также ряд орга-
нических веществ (пикриновая кислота, аспарагиновая кислота,
хинон, хлорбензолы и фенолы, азокрасители, нитрозосоедине-
ния и т. д.). Например, пикриновая кислота на ртутном катоде
легко восстанавливается до триаминофенола:
Ш2 +18Н+ + 18е- - ЩЯ-т^^г-ТЯЩ + 6Н20
Успешно применяется кулонометрический метод в фазовом и
металлографическом анализе, для исследования кинетики хими-
чески^ реакций, механизма электроокисления и восстановления
органических и неорганических веществ, изучения коррозии и
решения многих других вопросов.
В кулонометрическом титровании используют химические
реакции самого различного типа: кислотно-основного взаимодей-
ствия, окисления—-восстановления, комплексообразования и т. д.
Сильные и слабые кислоты могут быть точно нейтрализованы
гидроксидом, образующимся при электровосстановлении воды
на платиновом катоде. Различные восстановители [Те(П), 8п(П),
8Ь(Ш),. А8(Ш) и др.] могут быть оттитрованы, например, дихро-
матом. При анодном растворении хрома в серной кислоте полу-
244
чается дихромат, который может быть использован для этого
титрования. В кулонометрическом титровании применяется так-
же, например, свободный бром, генерируемый на платиновом
аноде из солянокислого раствора бромида калия. Бромом тит-
руют, например, гидразин, тиоцианат (вОТ" + ЗВг2 ■+ 4Н20 =
= Н2в04 + НСЫ + 6Вг" + 5Н+), фенол, различные метал л ооргани-
ческие соединения, АвСШ), вЬСЩ), Ге(П), Т1(1) и многие другие
восстановители. Определение выполняется с высокой точностью,
так как неизбежные трудности при титровании раствором брома,
возникающие в обычных титриметрических методах, здесь отпа-
дают в связи с мгновенным расходом брома на реакцию титрова-
ния. По этой же причине в методиках кулонометрического тит-
рования часто используются вещества крайне неустойчивые при
обычных условиях хранения, например соединения Си(1), Сг(П),
ТЦШ) и т. д. Возможность использования таких веществ в прак-
тике количественных определений является очень ценной осо-
бенностью кулонометрического титрования. Многие катионы оп-
ределяются Нитрованием этилендиаминтетраацетатом, который
генерируется из соответствующих комплексов ртути или кадмия
по схеме
11.6. Общая характеристика метода
Кулонометрический метод позволяет определять очень неболь-
шое содержание вещества с высокой точностью (0,1—0,05%), пре-
восходя в этом отношении многие другие методы. Кулонометрия
характеризуется также высокой селективностью (избирательно-
стью), позволяя определять многие вещества в растворе без предва-
рительного химического разделения. Избирательность обеспечива-
ется обоснованным выбором рабочего потенциала электрода и под-
держиванием его постоянного значения с высокой точностью во
все время электролиза. Кулонометрический анализ не требует ка-
кой-либо предварительной градуировки измерительных приборов
по концентрации или построения градуировочных графиков, свя-
зывающих свойство вещества с его концентрацией, и в этом смыс-
ле кулонометрию следует считать абсолютным методом.
Метод кулонометрического титрования характеризуется вы-
сокой чувствительностью и точностью (0,1—0,05%), позволяя
прямым титрованием определять вещества в растворе при кон-
центрации до 10~6 моль/л, что намного превышает возможности
других титриметрических методов. Он не требует предваритель-
245
ного приготовления, стандартизации и хранения; стандартных
растворов. Кулонометрическое титрование может быть легко ав-
томатизировано. Области применения методов кулонометриче-
ского титрования непрерывно расширяются.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. В чем сущность электрогравиметрического метода анализа?
2. Какие законы лежат в основе методов электрогравиметрии и ку-
лономётрии?
3. Что такое электрохимический эквивалент вещества?
4. Какие особенности имеет электролиз на ртутном катоде и как их
используют в анализе?
5. Какие аналитические цели достигаются с помощью электролиза?
6. Что представляет собой метод внутреннего электролиза и каковьг
возможности этого метода?
7. По какому закону изменяется сила тока в ходе прямого кулоно-
метрического определения? /
8. Каковы преимущества кулонометрического анализа при конт-
ролируемой силе тока?
г л а в а 1 2
Радиометрические
методы анализа
12.1. Типы радиоактивного распада
и радиоактивного излучения
! Открытие радиоактивности относится к 1896 г., когда А. Бек-
керёль обнаружил, что уран самопроизвольно испускает излучение,
названное им радиоактивным (от лат. radio ^излучаю и activas —
действенный).
Радиоактивное излучение возникает при самопроизвольном
распаде атомного ядра. Известно несколько типов радиоактивно-
го распада и радиоактивного излучения.
а-Распад. Распад ядра с выделением а-частиц, которые явля-
ются ядрами Не2+. Например,
92U~* 90Th + 2a Г:
246
В соответствии с законом радиоактивного смещения, при а-рас-
падё пблучается атом, порядковый номер которого на две едини-
цы, а атомная масса на четыре единицы меньше, ч!ем у исходного
атома.
Р -Распад. Различают несколько видов Р-распада: электрон-
ный Р-распад; позитронный Р-распад; К-захват. При электрон-
ном Р-распаде, например,
' , ^Р-^в + р- > ' |
нейтрон внутри ядра превращается в протон. При испускании от-
рицательно заряженной Р-частицы порядковый номер элемента
возрастает на единицу, а атомная масса практически не меняется.
При позитронном Р-распаде из атомного ядра выделяется по-
зитрон (Р+-частица), а протон внутри ядра превращается в нейт-
рон. Например:
^Ка-^е + р+
Продолжительность жизни позитрона невелика, так как при
столкновении его с электроном происходит аннигиляция, сопро-!
вождающаяся испусканием у-квантов.
При ТТ-захвате ядро атома захватывает электрон из близле-
жащей электронной оболочки (из ^-оболочки) и один из прото-
нов ядра превращается в нейтрон. Например,
40» . 40 А , ,
19К + е = 18Аг + лу
На свободное место в ^-оболочке переходит один из электронов
внешней оболочки, что сопровождается испусканием жесткого
рентгеновского излучения.
Спонтанное деление. Оно характерно для элементов пери-
одической системы Д. И. Менделеева с 2! > 90. При спонтанном
делении тяжелые атомы делятся на осколки, которыми обычно
являются элементы середины таблицы Д. И, Менделеева,, Спон-
танное деление и а-распад ограничивают получение новых тран-
сурановых элементов. • - ^а:
Поток а- и р-частиц называют соответственно а- и Р-излу??е-
нием. Кроме того, известно у-излучение. Это электромагнитные
колебания с очень короткой длиной волны. В принципе, у-излу-
чение близко к жесткому рентгеновскому и отличается от него
своим внутриядерным происхождением. Рентгеновское излуче-
ние возникает при переходах в электронной оболочке атома, а
у-излучение испускает возбужденные атомы, получившиеся в ре-
зультате радиоактивного распада (а или Р).
247
i В результате радиоактивного распада получаются элементы,
которые по заряду ядер (порядковому номеру) должны быть по-
мещены в уже занятые кЛетки периодической системы элемента-
ми с таким же порядковым номером, но другой атомной массой:
Это так называемые изотопы. По химическим свойствам их при-
нято считать неразличимыми, поэтому смесь изотопов обычно
рассматривается как один элемент* Неизменность изотопного со-
става в подавляющем большинстве химических реакций иногда
называют законом постоянства изотопного состава. Напри-
мер, калий в природных соединениях представляет собой смесь
изотопов, состоящую на 93,259% из 39К, на 6,729% из 41К и на
0,0119% из 40К (If-захват и Р-распад). Кальций насчитывает
шесть стабильных изотопов с массовыми числами 40, 42, 43, 44,
46 и 48. В химико-аналитических и очень многих других реакци-
ях это соотношение сохраняется практически неизменным, по-
этому для разделения изотопов химические реакции обычно не
применяются. Чаще всего для этой цели используются различ-
ные физические процессы — диффузия, дистилляция или элект-
ролиз. <
Единицей активности изотопа является беккерель (Бк), рав-
ный активности нуклида в радиоактивном источнике, в котором
за время 1 с происходит один акт распада.
12.2. Закон радиоактивного распада
Скорость радиоактивного распада --=- пропорциональна чис-
лу имеющихся ядер ЛГ:
где,Х--; постоянная распада.
., ?При:интегрировании получаем - ••; : г
с и- ••. -inisr =г^ + const; : *?'•; / . ■ ■ •
еслиJ -=пQ^tq К. == N0 и, следовательно, const = ^lg N0. Оконча-
тельно
>N = N0e-u . (12.1)
или \ ' . - •
(12.2)
где A — активность в момент времени t; Aq — активность при
248
^Уравнения (12.1) и (12*2) характеризуют закон радиоактив-
ного распада. В кинетике они известны как уравнения реакции
первого^ порядка; / -
В качестве характеристики скорости радиоактивного распада
обычно указывают период полураспада Г1/2, который так же,
как и Х9 является фундаментальной характеристикой процесса,
не Зависящей от количества вещества.
Периодом полураспада называют промежуток времени, в течение
которого данное количество радиоактивного вещества уменьшает-
ся наполовину. •
Таким образом, в момент времени Т1/2 отношение Ы/Ы0 =
- У2<г^1/2 - 7^ откуда -ХГ1/2 * -1п2 и Т1/2 т = С уче-
том этого соотношения уравнение (12.2) можно переписать:
Период полураспада различных изотопов существенно раз-
личен. Он находится в пределах примерно от 1010 лет до ничтож-
ных долей секунды. Конечно, вещества, имеющие период полу-
распада 10—15 мйй й меньше* использовать в лаборатории труд-
но. Цзотопы с очень большим периодом полураспада также
нежелательны в лаборатории, так как при случайном загрязне-
нии этими веществами окружающих предметов потребуется „спе-
циальная работа по дезактивации помещения и приборов.
12.3. Взаимодействие радиоактивного
излучения с веществом и счетчики излучения
В результате взаимодействие радиоактивного излучения с
веществом происходит ионизация и возбуждение атомов и моле-
кул вещества, через которое оно проходит. Излучение -произво-
дит также световое, фотографическое, химическое и биологиче-
ское действие. Например, первичным результатом действия ра-
диоактивного излучения на воздух является появление ионов: -
02 + Н\ = 0£ + е" 02 + <г - 02
N2 += N£4-«г Ы2 +-ег = Щ
Н20 + Ну - Н20+ + е~ Н20 + е- = Н20-
Н20+ = Н+ + ОН* Н20-нч он-
249
у Образующиеся при протекании эт&их процессов радикалы Н#
и ОН* обладают сильным физиологическим действием — при
больших дозах они являются одной из причин лучевой болезни,
малокровия и т. д., так как энергично взаимодействуют с фер-
ментами и составными частями крови. Опасность радиоактивно-;
го воздействия возрастает вследствие того, что организм не обла-
дает болевыми реакциями на действие радиоактивного излуче-
ния., быстрое превращение этих частиц в безопасные для
человеческого организма является одним из эффективных прие-
мов борьбы с лучевой болезнью.
Радиоактивное излучение вызывает большое число химиче-
ских реакций в газах, растворах, твёрдых веществах. Их обычно
объединяют в группу радиационно-химических реакций. Сюда от-
носятся, например, разложение (радиолиз) воды с образованием
водорода, пероксида водорода и различных радикалов, вступаю-
щих: в окислительно-восстановительные реакции с растворенны-
ми веществами. Радиоактивное излучение вызывает разнообраз-
ные радиохимические превращения различных органических со-
единений , .—г. аминокислот, кислот, спиртов, \ эфиров и т. д.
Интенсивное радиоактивное излучение вызывает свечение стек-
лянных трубок и ряд других эффектов в твердых телах. На изуче-
ний взаимодействия радиоактивного излучения с веществом осно-
ваны различные способы обнаружения и измерения радиоактив-
ности. • < • с
В зависимости от принцица действия счетчики радиоактив-
ных излучений подразделяют на несколько групп.
< Ионизационные счетчики. Их действие основано на возник-
новении ионизации или газового разряда, вызванного ионизаци-
ей при попадании в счетчик радиоактивных частиц или у-кван-
то'в. Среди десятков приборов, использующих ионизацию, типич-
ными являются; .ионизационная камера и счетчик Гейгера-
Мюллера, который получил наибольшее распространение в хи-
мико-аналитических и радиохимических лабораториях. Схема
устройства, принцип его работы и некоторые технические харак-
теристики счетчика указывались при рассмотрении рентгено-
спектрального анализа (см. гл. 6, ріаздел 6.3.3). г
; Для радиохимических и других лабораторий промышленно-
стью выпускаются специальные счётные установки.
Сцинтилляционные счетчики. Действие этих счетчиков осно-
вано на возбуждении атомов сцинтиллятора у-квантами или радио-
активной частицей, проходящей через счетчик. Возбужденные •
атомы, переходя в нормальное состояние, дают вспышку света. *
250
В начальный период изучения ядерных процессов визуаль^
ный счет сцинтилляций сыграл большую роль, однако в дальней-
шем он был вытеснен более совершенным счетчиком Гейгера-
Мюллера. В настоящее время сцинтилляционный метод вновь
стал широко применяться уже с использованием фотоумножите-
ля (см. гл. 6, раддел 6.3.3). у,
Черенковские счетчики. Действие этих счетчиков основано
на использовании эффекта Черенкова, который состоит в излуче-
нии света при движении заряженной частицы в прозрачном ве-
ществе, если скорость частиц превышает скорость света в данной
среде. Факт сверхсветовой скорости частицы в данной среде, ко-
нечно, не противоречит теории относительности, поскольку ско-
рость света в какой-либо среде всегда меньше, чем в вакууме.
Скорость движения частицы в веществе может быть больше ско-
рости света в этом веществе, оставаясь в то же время меньше ско-
рости света в вакууме в полном соответствии с теорией относи-
тельности. Счетчики Черенкова применяются для исследователь-
ских работ с очень быстрыми частицами, для исследований в
космосе и т. д., поскольку с их помощью может быть определен
ряд других важных характеристик частиц (их энергия, направ-
ление движения и др.). *
12.4. Ядерная химия
и искусственная радиоактивность
При бомбардировке вещества потоком а-частиц, протонов,
нейтронов и т. д. происходит превращение атомов. Ядерные пре-
вращения можно, представить с помощью уравнений, напоми-
нающих уравнения обычных, химических реакций., Например,
27А1 под действием протонов превращается в ?4Mg: г
27А1 + 1р = 24м?+^а ' ' ;а
Под действием нейтронов 27А1 превращается в 27М£:
Часто используется краткая запись этих уравнений. Вместо
первого уравнения пишут 27А1 (р, а) 24Mg, а вместо второго —
27А1 (п, р) 27М£. При такой записи сначала указывают символ ис-
ходного атома, далее в скобках через запятую приводят бомбард
дирующую и выбиваемую частицы^ после чего записывают сим-
вол образующегося атома. Один и тот же элемент под действием
251
различных бомбардирующих частиц может претерпевать множе-
ство разнообразных превращений.
В 1934 г. И. Кюри и Ф. Жолио открыли искусственную ра-
диоактивность. Они обнаружили, что излучение, возникающее
при бомбардировании алюминия а-частицами, подчиняется за-
кону радиоактивного распада (12.1). Оказалось, что при бомбар-
дировке алюминия образуется неустойчивый радиоактивный
изотоп фосфора: 27А1 + 4а = 30Р 4- 1п, который в результате радио-
активного распада превращается в кремний: 30Р —► 3081 ■+ р+. В на-
стоящее время известно большое число искусственно получен-
ных радиоактивных изотопов. С помощью ядерных реакций уда-
лось получить элементы, в природе не существующие, но для
которых в таблице Д. И. Менделеева имелись соответствующие
клетки под номерами 43, 61, 85, 87. Это технеций, прометий, ас-
тат и франций.
12.5. Методики анализа, основанные
на измерении радиоактивности
12.5.1. Использование естественной радиоактивности
в анализе
Элементы, имеющие естественную радиоактивность, могут
быть определены по этому свойству количественно. Это и, ТЪ,
Ка, Ас, К и др., всего более 20 элементов. Например, калий мож-
но определить по его радиоактивности в растворе при концентра-
ции 0,05 М. Определение различных элементов по их радиоак-
тивности обычно проводят с помощью градуировочного графика,
показывающего зависимость активности от содержания (%) оп-
ределяемого элемента или методом добавок.
Большое значение имеют радиометрические методы в поис-
ковой работе геологов, например при разведке месторождений
урана. ,
12.5.2. Активационный анализ
При облучении нейтронами, протонами и другими частица-
ми высокой энергии многие нерадиоактивные элементы стано-
вятся радиоактивными. Активационный анализ основан на изме-
рении этой радиоактивности. Хотя в принципе для облучения
могут быть использованы любые частицы, наибольшее практиче-
252
ское значение имеет процесс облучения нейтронами. Применение
для этой цели заряженных частиц связано с преодолением более
значительных технических трудностей, чем в случае нейтронов.
Основными источниками нейтронов для проведения активацион-
ного анализа являются атомный реактор и так называемые пор-
тативные источники (радиевобериллиевый и др.). В последнем
случае а-частицы, получившиеся при распаде какого-либо а-ак-
тивного элемента (11а, ш1 и т. д.), взаимодействуют с ядрами бе-
риллия, выделяя нейтроны:
9Ве + 4Не ^ 12С + п
Нейтроны вступают в ядерную реакцию с компонентами анали-
зируемой пробы, например
б5Мп + п = б6Мп или Мп (п, у) 56Мп
Радиоактивный 56Мп распадается с периодом цолураспада
2,6 ч: ■ ,
56Мп — 56Ре + е-
Для получений информации о составе образца некоторое вре-
мя измеряют его радиоактивность и анализируют полученную
кривую. При проведении такого анализа необходимо располагать
надежными данными о периодах полураспада различных изото-
пов, с тем чтобы провести расшифровку суммарной кривой.
На практике обычно используют относительный метод ана-
лиза, когда в одинаковых условиях облучают анализируемый об-
разец и эталон с известным содержанием определяемого элемен-
та, что существенно упрощает анализ. Во многих случаях обра-
зец после облучения переводят в раствор, производят химическое
выделение интересующих компонентов путем экстракции, хро-
матографии, осаждения или другим методом и определяют ак-
тивность продуктов разделения. Операции химического разделе-
ния значительно расширяют возможность метода, повышают се-
лективность, однако это имеет реальное значение лишь для
изотопов с не слишком малым периодом полураспада.
Другим вариантом активацйонного анализа является метод
у-спектроскопии, основанный на измерении спектра у-излучения
образца. Энергия у-излучения является качественной, а скорость
счета —- количественной характеристикой изотопа. Измерения
производят с помощью многоканальных у-спектрометров со
сцинтилляционными или полупроводниковыми счетчиками.
Это значительно более быстрый и специфичный, хотя и нескольг
ко менее чувствительный метод анализа, чем радиохимический.
253
В настоящее время различные варианты активационного ана-
лиза получили широкое и разнообразное применение: Наиболь-
шее значение имеет анализ веществ высокой чистоты, используе-
мых, например, в полупроводниковой и атомной промышленнос-
ти, ракетной технике и т. д. Он с успехом применяется для
анализа воды и многих геохимических объектов, для определения'
состава различных биологических и космохимических материа-
лов и во многих других областях науки и промышленности, свя-
занных, главным образом, с новой техникой.
Важным достоинством активационного анализа является его
низкий предел обнаружения. С его помощью может быть обнару-
жено при благоприятных условиях до 10~13—10~15 г вещества.
В некоторых специальных случаях удавалось достигнуть еще бо-
лее низких пределов обнаружения. Например, с его помощью
контролируют чистоту кремния и германия в промышленности
полупроводников, обнаруживая содержание примесей до 10~8—
10"9%. Такие содержания никаким другим Методом, кроме акти-
вационного анализа определить невозможно. При получении тя-
желых элементов периодической системы, таких, как менделе-
вий и курчатовий, исследователям удавалось считать почти каж-
дый атом полученного элемента.
Основным недостатком активационного анализа является
громоздкость источника нейтронов, а также нередко длитель-
ность самого процесса получения результатов.
Применение активационного анализа позволило решить мно-
гие геохимические и общенаучные проблемы, например, связан-
ные с установлением возраста минералов и образцов органиче-
ского происхождения. Например, хорошо известный радиомет-
рический способ определения возраста древних предметов и
изделий органического происхождения основан на том, что в ат-
мосфере в результате ядерной реакции постоянно образуется ра-
диоактивный углерод 14С с периодом полураспЗДа 5300 лет:
"'" 14К + п = 14С + р
Нейтроны генерируются в атмосфере при взаимодействии косми-
ческого излучения с веществом, а азот является составной частью
атмосферы. Радиоактивный Я4С образует радиоактивный 14С02,
который вступает в биологический круговорот. Т^ким образом,
вещества и организмы, участвующие в этом круговороте (расте-
ния, животные и т. д.), будут иметь примерно постоянную радио-
активность, пропорциональную содержанию 14С. В организмах,
которые выпадают из круговорота в результате гибели, количест-
254
во радиоактивного 14С не пополняется и их активность уменьша-
ется. .Следовательно, если измерить активность какого-либо древ-
него изделия (предмета из дерева, кожи и т. д.), то зная период по-
лураспада углерода 14С и общее содержание углерода в образце,
можно рассчитать промежуток времени, прошедший с момента
гибели организма. Этот промежуток будет характеризовать при-
мерный возраст изделия. Анализ этим методом позволил уточ-
нить даты многих важных событий далекой древности.
Интересным примером применения активационного анализа
является предложенный недавно быстрый Способ обнаружения
взрывчатки. Как известно, в качестве взрывчатых веществ обыч-
но используют различные нитросоединения. Способ обнаруже-
ния основан на том, что взрывчатка наряду с 14№ содержит неко-
торое количество 15Ы, который при облучении нейтронами пре-
вращается в 16КГ. Этот изотоп имеет период полураспада 7 с и при
его распаде кроме Р-частйц происходит испускание у-квантон оп-
ределенной энергии. Появление Р-квантов с энергией; характер-
ной для распада 16Ы после облучения вещества нейтронами, яв-
ляется сигналом о наличии азотсодержащего вещества, возмож-
но взрывчатки. Очень ценные результаты дает активационный
анализ в криминалистике, судебной химии й т. д.
12.5.3. Метод изотопного разбавления
Его идею лучше всего рассмотреть на конкретном примере.
При определении свинца методом изотопного разбавления в ана-
лизируемый раствор, содержащий х (г) свинца, вводят неболь-
шое известное количество радиоактивного изотопа РЬ*, в резуль-
тате чего раствор приобретает активность А0. Какое-то количе-
ство полученного раствора осаждают раствором сульфата и полу-
чают т (г) осадка РЬЭ04 с активностью А. Если радиоактивней
изотоп вводился в анализируемый раствор без носителя, то из
пропорции
; х~Ао дг-тп^ф, ( (а)
тр-А ^ А •••
где 2^ — фактор пересчета РЬв04 на РЬ.
Если изотоп вводился с носителем, то
х + т1 -Ац
т¥-А
х= — А ^ а ~шу (б)
где «г' — масса радиоактивного препарата с носителем,
255
При т* = 0 (изотоп без носителя) соотношение (б) переходит
в (а). Как видно, пропорциональность активности изотопа коли-
честву определяемого компонента позволяет получить точный
результат и без достижения полноты осаждения, что является су-
щественным достоинством метода изотопного разбавления.
Определение активности осадка часто нежелательно из-за ад-
сорбции, самопоглощения и других явлений, искажающих ис-
тинную величину активности, поэтому обычно определяют ак-
тивность раствора после отделения осадка. Если Аг и Уг — удель-
ная активность и объем раствора до осаждения, А2иУ2 — после
него, то, очевидно, активность осадка А будет равна
А-А^-А^.
Применение метода изотопного разбавления предполагает,
что закон постоянства изотопного состава элементов соблюдает-
ся, химические свойства радиоактивного и неактивного изотопов
неразличимы и реакции изотопного обмена радиоизотопа с
«третьими» компонентами смеси не происходят.
Метод изотопного разбавления открывает новые возможнос-
ти в анализе сложных смесей и элементов, близких по своим хи-
мико-аналитическим свойствам. Например, при анализе смесей
цирконий—гафний или ниобий—-тантал можно получить чистый
осадок одного из компонентов, но осаждение не будет полным.
Если добиться полного осаждения, то полученный осадок будет
загрязнен элементом-аналогом. В методе изотопного разбавления
проводят неполное осаждение и, используя измерения активное^
ти, находят содержание анализируемого элемента с достаточной
точностью. Аналогичный прием используется также при анализе
различных смесей органических веществ.
Для выделения анализируемого компонента в методе изотоп-
ного разбавления наряду с осаждением используются также экс-;
тракция, хроматография и другие методы разделения, а для оп-
ределения количества; выделенного компонента применяют
спектральные, электрохимические и другие методики.
12.5.4. Радиометрическое титрование
_ ;
При радиометрическом титровании индикаторами являются
радиоактивные изотопы элементов. Например, при титровании.
фосфата магнием в анализируемый раствор вводят небольшое ко-*
личество фосфата, содержащего радиоактивный Р.*.
256
При титровании протекает реакция
Мв2++ НР*0|" = МвНР*04
Изменение активности в ходе этого титрования можно видеть на
рис. 12.1, а. Здесь же показано графическое определение точки
эквивалентности (т. э.). До т.э. активность раствора будет резко
убывать, так как радиоактивный НР*0|~ из раствора будет пере-
ходить в осадок. После т.э. активность раствора будет оставаться
практически постоянной и очень небольшой.
Как видно из рис. 12.1, б, добавление гидрофосфата к раство-
ру Mg2+ до т.э. практически не будет вызывать увеличения ак-
тивности раствора, так как радиоактивный изотоп Р* будет пере-
ходить в осадок М§НР*04. После т.э. активность раствора начи-
нает возрастать пропорционально концентрации гидрофосфата.
Реакции радиометрического титрования должны удовлетво-
рять требованиям, обычно предъявляемым к реакциям титри-
метрического анализа (скорость и полнота протекания реакции,
постоянство состава продукта реакции и т. д.). Очевидным усло-
вием применимости реакции в данном методе является также пе-
реход продукта реакции из анализируемого раствора в другую
фазу, с тем чтобы устранить помехи при определении активности
раствора. Этой второй фазой часто является образующийся оса-
док. Известны методики, где продукт реакции экстрагируется
органическим растворителем. Например, при титровании многих
катионов дитизоном в качестве экстрагента применяют хлоро-
форм или тетрахлорид углерода. Применение экстракции позво-
ляет более точно установить точку эквивалентности, так как в
этом случае для ее определения можно измерять активность обе-
их фаз.
т.э. V т.э. V
а) б)
Рис. 12.1. Типы кривых радиометрического титрования:
а — изменение активности раствора фосфата, содержащего Р *
при титровании раствором Mg2+; б — изменение активности раствора Ъ/lg2+
при титровании фосфатом, содержащим Р *
17—7831
257
12.5.5. Эффект Мессбауэра
• Эффект открыт в 1958 г. Р. П. Мессбауэром. Под этим назва-
нием часто объединяют явления испускания, поглощения и рас-
сеяция у-квантов ядрами атомов без затраты энергии на отдачу,
ядер. Обычно исследуется поглощение у-излучения, поэтому Эфтт
фект Мессбауэра часто называют также у-резонансной спектро-^
скопией (ГРС).
При испускании у-квантов ядро атома возвращается в нор-
мальное состояние. Однако энергия испускаемого излучения бу-
дет-определяться не только разностью энергетических состояний
ядра в возбужденном и нормальном состояниях. Вследствие зако-
на сохранения импульса ядро испытывает так называемую отда-
чу,. Это приводит к тому, что в случае газообразного атома энер-
гия испускаемого излучения будет меньше, чем в случае, когда
излучатель будет находиться в твердом теле. В последнем случае
потери энергии на отдачу уменьшаются до пренебрежимо малого
значения. Таким образом, у-кванты излучения, испускаемого без
отдачи, могут поглощаться невозбуждёнными атомами того же
элемента. Однако различие в химическом окружении ядра-излу-
чателя и ядра-поглотителя вызывает некоторую разницу в энер-
гетических состояниях ядер, достаточную, чтобы резонансное по-
глощение у-квантов не происходило. Разницу в энергетических
состояниях ядер количественно компенсируют с помощью эф-
фекта Допплера, в соответствии с которым частота излучения
(в данном случае энергия у-квантов) зависит от скорости движе-
ний. При некоторой скорости движения излучателя (или погло-
тителя, так как значение имеет лишь их относительная скорость
перемещения) наступает резонансное поглощение. Зависимость;
интенсивности поглощения у-квантов от скорости движения на-
зывают спектром Мессбауэра. Типичный мессбауэровский спектр
представлен на рис. 12.2, где в качестве меры интенсивности пог-
лощения отложена обратно пропорци-
ональная ей скорость счета. Скорость
перемещения образца или излучателя
обычно не превышает нескольких сан-
тиметров в секунду. Спектр Мессбауэра
является очень важной характеристик
^ кой вещества. Он^ позволяет судить Ох
1 Скорость природе химической связи в исследуе^
источника, см/с мых соединениях, их электронной?
Рис 12.2. Мессбауэровский структуре и других особенностях и свош
спектр поглощения ствах.
«••в
а!
о т:
О ш
.Б
АЛ*"
258
12.6. Практическое применение
Применение радиоактивности в аналитической химии весьма
многообразно. Измерение радиоактивности широко применяют
также в научно-исследовательских целях: для исследования меха-
низмов химических реакций, определения растворимости малора-
створимых соединений, исследования процессов разделения и для
решения многих других задач, включая определение важнейших
физико-химических констант (констант устойчивости координа-
ционных соединений, констант ионообменных процессов ит. д.).
Эффект Мессбауэра в аналитической химии имеет ограничен-
ное применение. Он используется, например, для открытия при-
месных атомов, если атомы основной решетки не мешают прохож-
дению резонансного у-излучения. Мессбауэровская спектроскопия
имеет большие потенциальные аналитические возможности как ме-
тод неразрушающего анализа и как один из эффективных методов
Определения валентного состояния и степени окисления элементов.
12.7. Общая характеристика метода
Основными достоинствами аналитических методов, основан-
ных на измерении радиоактивного излучения, являются низкрй
порог обнаружения анализируемого элемента и широкая универ-
сальность. Радиоактивационный анализ имеет абсолютно ни-
зший порог обнаружения среди всех других аналитических мето-
дов (Ю-15 г). Достоинством некоторых радиометрических мето-
дик является анализ без разрушения образца, а методов,
основанных на измерении естественной радиоактивности, — бы-
строта анализа. Ценная особенность радиометрического метода
изотопного разведения заключена в возможности анализа смеси
близких по химико-аналитическим свойствам элементов, таких,
как цирконий—гафний, ниобий—тантал и др.
Дополнительные осложнения в работе с радиоактивными пре-
паратами обусловлены токсичными свойствами радиоактивного
излучения, которые не вызывают немедленной реакции организ-
ма и тем самым осложняют своевременное применение необходи-
мых мер. Это усиливает необходимость строгого соблюдения тех-
ники безопасности при'работе с радиоактивными препаратами.
В необходимых случаях работа с радиоактивными веществами
происходит с помощью так называемых манипуляторов в специ-
альных камерах, а сам аналитик остается в другом помещении,
надежно защищенном от действия радиоактивного излучения. ■>
259
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Какие существуют типы радиоактивного излучения?
2. Что такое постоянная радиоактивного распада?
3. В каких пределах должна находиться величина периода полу-
распада для изотопов, используемых в аналитических целях, и
почему?
4. На чем основаны методы обнаружения радиоактивного излуче-
ния?
5. В чем состоит принцип работы счетчика Гейгера—Мюллера и
сцинтилляционного счетчика?
6. Как используется естественная радиоактивность в аналитиче-
ских целях? Какие элементы можно определить по их естествен-
ной радиоактивности? ,
7. В чем сущность метода изотопного разбавления?
8. В чем сущность активационного анализа?
9. На чем основано радиометрическое определение «возраста» древ-
них рукописей и других предметов органического происхожде-
:'' ния?
глава 13
Масс-спектрометрия
13.1. Теоретические ОСНОВЫ
масс-спектрометрии
Масс-спектральный анализ основан на способности газооб-
разных ионов разделяться в магнитном поле в зависимости от от-
ношения т/е, где т — масса, е — заряд иона. Ионизация моле-
кул в газе происходит под действиемупотока электронов. Наибо-
лее вероятными являются процессы образования однозарядных
положительных ионов:
М + е~ = М+ + 2е~
Образование двух- и более высокозаряженных ионов, а так-
же захват электрона с образованием отрицательных ионов явля-
ются менее вероятными процессами. По величине т/е определя-
ют массовое число иона, а по интенсивности соответствующего
сигнала судят о концентрации ионов.
260
Принципиальная схема масс-
спектрометра приведена на рис. 13.1.
В камере анализируемое вещество
переводится в газообразное состоя-
ние при давлении 10"2—10"3 Па. Ре-
жим работы камеры устанавливает-
ся в зависимости от того, трудно- Рис 13.1. Принципиальная схе-
или легколетучие соединения вхо- ма масс-спектрометра:
дят в состав анализируемого образ- 'Т^Я^^
ца. При анализе газообразных проб 4 — ускоряющие пластинки;
СТаДИЯ ИСПареНИЯ, естественно, ОТ- 5 — магнитное поле; 6 — детектор
падает. Далее молекулярный пучок
ионизируется. Нередко для анализа твердых проб применяются и
источники с поверхностной ионизацией, в которых процессы ис-
парения и ионизации не разделены.
Ионизация молекулярного пучка газообразной пробы может
быть вызвана фотонами, ионами, электрическим полем, элек-
тронным ударом и другими способами. Наибольшее распростра-
нение в аналитической практике получили приборы, в которых
ионизация осуществляется электронной или ионной бомбарди-
ровкой пробы либо искровым разрядом. Для ионизации элек-
тронным ударом используется стабилизированный пучок элект-
ронов, перпендикулярный потоку пробы. Энергия электронов не-
велика и обычно составляет 10—100 эВ. При бомбардировке
молекул или атомов электронами одновременно происходит не-
сколько процессов. В условиях масс-спектрального анализа, как
показывает опыт, образуются преимущественно положительные
однозарядные ионы и частично ионы с более высоким зарядом.
Если энергия бомбардирующих электронов достаточно велика,
чтобы вызвать разрыв химических связей, происходит фрагмен-
тация молекул и в потоке появляются так называемые ионы-
осколки.
Образовавшиеся положительно заряженные ионы проходят
через ускоряющие пластины, разность потенциалов между кото-
рыми достаточно высока (несколько тысяч вольт). Здесь они при-
обретают энергию еУ, а их скорость возрастает до v. Энергия ёУ,
очевидно, будет равна кинетической энергии ионов ти2/2, поки-
дающих ионный источник со скоростью!;:
еУ=ти2/2. (13.1)
После ускорения в электрическом поле ионы под прямым уг-
лом пересекают магнитное поле напряженностью Н, подверга-
261
ясь, таким образом, действию силы Ней, направленной перпен-
дикулярно движению иона. Поэтому траекторией движения ио-
нов будет окружность радиуса г.
Приравнивая силы Неи = то2/г9 находим
т -.
Подставляем эту величину в уравнение (13.1):
Отсюда получаем радиус окружности:
Т-А/^.' ~ (13.2)
Ионы, описывающие дугу радиуса г, попадают в детектор. Де-
тектирование ионов производится фотографическим или электри-
~. ческим способом. При фотографическом детектировании пучок
ионов попадает на фотопластинку, вызывая почернение, пропорци-
ональное числу ионов. В электрических детекторах масс-спектро-
метров ионный ток измеряется электрометром, электронным умно-
жителем или другим аналогичным устройством. Сигналы обычно
регистрируются быстродействующим потенциометром. В последнее
время разработаны устройства, передающие информацию с детек-
тора на компьютер, что позволяет значительно ускорить обработку
данных.
Перепишем уравнение (13.2) в виде м. • .
т/е = г2Н2/2У. (13,3)
Для получения масс-спектра, в принципе, можно менять любую
переменную правой части этого уравнения (г, Н или У) и изме-
рять почернение фотопластинки или ионный ток. Радиус г обыч-
но не меняется, он задается конструкцией прибора и остается по-
стоянным для всех ионов. Таким образом, меняя Н при постоян-
ном V или меняя V при постоянном Н, можно направлять на
детектор ионы с разной величиной т/е. Масс-спектр представля-
ют зависимостью в виде спектрограммы или таблицы, содержа-
щей величины т/е и соответствующие им интенсивности. Про-
порциональность между экспериментально измеренным V и от-
ношением т/е можно найти путем градуировки по веществу с
известным масс-спектром. ,
262
13.2. Качественный анализ
Качественный масс-спектромётрический анализ основан на изт
мерении массы ионов. Идентификация масс проводится по положе-
нию линии на фотопластинке, которое фиксируют, измеряя расстоя-
ние между линиями с известной массой и анализируемой линией.
Масс-спектры многих веществ изучены достаточно подробно
и составлены специальные атласы. При использовании таких ат-
ласов учитывается, например, что двухзарядный ион с массой 56
дает такую же линию в спектре, как и однозарядный ион с мас-
сой 28, а также условия получения спектра — температуру ион-
ного источника, энергию электронов и т. д.
13.3. Количественный анализ
Количественные измерения в масс-спектрометрии проводят
по току, фиксируемому детектором, или по почернению фото-
пластинки. В первом случае расчеты основаны на том, что пик
ионного тока / пропорционален содержанию компонента или его
парциальному давлению:
■•■ .-.^ •'. / = ксг= кр,
где А, х — коэффициенты пропорциональности; с — концентра-
ция; р — давление. Почернение фотопластинки измеряется по от-
ношению к почернению линий внешнего стандарта.
13.4. Практическое применение
Практическое применение масс-спектрометрии весьма мно-
гообразно. Большую роль сыграли измерения масс-спектров при
изучении изотопного состава различных веществ. Основные све-
дения о стабильных изотопах фактически получены с помощью
масс-спектрометра.
Масс-спектрометрический метод применяют для анализа
твердых, жидких и газообразных проб. Значительное распрост-
ранение он получил в органической химии для анализа многих
классов соединений, в нефтехимии, где масс-спектрометриче-
ским методом анализируют сложные многокомпонентные смеси
углеводородов, в технологии неорганических веществ и других
областях химической промышленности. Небольшой объем газа,
требующийся для анализа, возможность определения всех кЬм-
понентов смеси без разделения и другие достоинства масс-спект-
ромётрйи позволили успешно использовать ее для определения
газов в металлах (после вакуумного плавления). Метод применим
263
для анализа металлов, полупроводников и других неорганиче-
ских и органических веществ. Он позволяет определять примеси
на поверхности и по всему объему пробы. Большие перспективы
открывает метод, сочетающий хроматографическое разделение и
масс-спектрометрическое определение полученных продуктов.
Масс-спектрометрия применяется также для установления
строения молекул, определения термодинамических характерис-
тик газообразных веществ и т. д.
13.5. Общая характеристика метода
Масс-спектральный метод характеризуется высокой универ-
сальностью. Он применим для определения почти всех элементов
периодической системы со средним пределом обнаружения 10"3—
10"4%, а при благоприятных условиях и до 10~7%. Одним из до-
стоинств метода является возмоясность одновременного определе-
ния нескольких элементов и использование в работе небольших
навесок (1 мг и меньше). Погрешность метода составляет 5—10%.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Какую зависимость называют масс-спектром вещества?
2. На чем основан масс-спектрометрический анализ?
3. Какие свойства ионов приводят к их разделению в масс-спектро-
метре?
4. На чем основан качественный масс-спектрометрический анализ?
5. На чем основан количественный масс-спектрометрический анализ?
6. Каковы области практического применения, достоинства и не-
достатки масс-спектрометрического метода?
г л а в а 14
Кинетические методы анализа
14.1. Основные методы обработки
кинетических данных
В кинетических методах анализа измеряемым свойством сис-
темы, на основании которого делают выводы о концентрации ве-
щества, является скорость химической реакции. Пусть, напри-
264
мер, вещества А и В реагируют между собой, образуя продукт ре-
акции X:
А + В = Х
В начальный момент времени концентрации веществ А и В
будут равны соответственно а и Ъ. Концентрация продукта реак-
ций в этот момент, естественно, будет равна нулю. В какой-то мо-
мент времени после начала реакции концентрация образующего-
ся продукта X будет равна х, а концентрации исходных веществ
будут равны (а - х) и (Ъ - х) соответственно. Скорость химической
реакции при данной температуре, как известно, пропорциональ-
на произведению концентраций реагирующих веществ и нередко
в степени, отличной от единицы:
^• = Ца-х)(Ъ-х)9 (14.1)
где к — константа скорости реакции.
Очень быстрые и очень медленные реакции в химико-анали-
тических целях являются малопригодными. Хотя трудно устано-
вить какие-либо жесткие правила или критерии, ограничиваю-
щие применение тех или иных реакций в кинетических методах
анализа, все же некоторые пределы применимости можно отме-
тить. Почти повсеместно принято считать, что аналитическая
реакция (в кинетических методах ее часто называют индикатор-
ной реакцией) должна продолжаться не менее 1 мин и не более 2 ч.
Более быстрые реакции, как, правило, не применяются потому,
что при использовании обычного оборудования химико-аналити-
ческой лаборатории скорость таких реакций трудно измерить с
достаточной точностью. При наличии специального оборудова-
ния эти затруднения отпадают и соответствующее ограничение
снимается. Реакции, продолжающиеся более 2 ч, нежелательны
из-за большой длительности анализа. Оптимальным временем
для измерения скорости реакции считается 10—15 мин. Указан-
ные пределы в значительной степени условны, поскольку можно
в достаточно широких пределах регулировать скорость химиче-
ской реакции, например, изменением температуры, концентра-
ции реагирующих веществ или введением в раствор катализато-
ров (или ингибиторов),
Хотя в кинетических методах анализа, в принципе, могут
быть использованы любые реакции, скорость которых может
быть измерена достаточно точно, все же наиболее часто применя-
ются так называемые каталитические реакции, скорость кото-
рых зависит от концентрации катализатора. При наличии в
265
растворе катализатора в кинетическом уравнении (14.1) появля-
ется соответствующий сомножитель:
=кск(а-х)(Ь-х), (14.2)
гдеск — концентрация катализатора.
Концентрацию одного из участников реакции, например ве-
щества В, можно взять заведомо в большом избытке, так что его
убыль в результате протекания реакции будет пренебрежимо ма-
ла и, следовательно, Ъ - х - Ъ. Тогда, объединяя кЬ = х, вместо
(14.2) получаем:
^=хск(а-х). (14.3)
На рис. 14.1 приводятся типичные кинетические кривые. Гра-
фик показывает возрастание во времени концентрации вещества
X. Кривая 1 в начальный момент времени имеет линейный учас-
ток, т. е. в начальный момент времени угловой коэффициент кине-
тической кривой постоянен. На кривой 2 линейный участок даже в
начальный момент времени отсутствует. Различный характер ки-
нетических кривых вызывает разные способы их обработки.
Рассмотрим сначала кривую 1. Уравнение (14.3) показывает,
что величина ^ может быть постоянной только при условии
постоянства а - х9 а это, в свою очередь, будет возможно, если
а - х• — а, т. е. если концентрация вещества А в ходе реакции су-
щественно меняться не будет. Кинетическое уравнение для этого
промежутка времени принимает вид:
37 =^ (14.4)
Это уравнение является основой различных вариантов кинетиче-
ского метода, названных дифференциальными. Интегрирование
. дает:
х = хска£. (14.5)
Несколько сложнее обработка данных,
представленных кривой 2. Здесь нет облас-
ти, в которой а - х — а, поэтому приходится
интегрировать кинетическое уравнение
(14.3). Разделив переменные и проинтегри-
ровав, получаем.
Рис. 14.1.
Кинетические I &х _ |хс ^
кривые а - х к
266
ИЛИ;
-In (а- х) = xcKt + const.
Постоянную интегрирования находим из начальных условий:
при t = 0, х = 0 и, следовательно, -In а = const. Окончательно
можно записать:
In = XcKt. (14.6)
а-х к
Методы анализа, основанные на применении этого уравне-
ния, называют интегральными.
14.2. Основные приемы
кинетических методов анализа
Уравнения (14.4)—(14.6) в явном виде связывают кинетиче-
ские характеристики реакции с концентрацией катализатора.
Как видно, концентрация катализатора может быть найдена или
непосредственно по скорости реакции, или по времени ее проте-
кания, или по концентрации образующихся продуктов. В зависи-
мости от того, какое свойство или какая характеристика реакции
используется для определения концентрации, выделяют методы
тангенсов, фиксированного времени, фиксированной концент-
рации.
Известны, кроме того, и другие методы, имеющие более част-
ный характер, например методы индукционного периода, непо-
средственногодифференцирования и т. д.
Метод тангенсов. В методе тангенсов измеряют скорость ре-
акции обычно по возрастанию концентрации одного из образую-
щихся продуктов и строят график, аналогичный изображенному
на рис 14,1. Если кинетическая кривая в начальный период про-
текания реакции имеет линейный характер, применяют диффе-
ренциальный вариант метода тангенсов. Уравнение (14.4) пока-
зывает, что в этом случае скорость реакции характеризуемая
at
тангенсом угла наклона кинетической кривой, пропорциональна
концентраций катализатора.
График в координатах тангенс угла наклона—концентрация
определяемого вещества (отсюда название «метод тангенсов»)
обычно линеен. При анализе неизвестного раствора измеряют
скорость реакции в тех же условиях, в каких она определялась
для построения градуировочного графика, определяют tgа и по
градуировочному графику находят концентрацию анализируемо-
го компонента сх.
267
Если кинетическая кривая имеет вид кривой 2 (см. рис 14.1),
т. ё. линейный участок отсутствует, применяется интегральный
вариант метода тангенсов. В соответствии с уравнением (14.6) ки-
Тангенс угла наклона прямой в этих координатах, как показывает
уравнение (14.6), пропорционален концентрации катализатора и,
следовательно, градуировочный график будет также линеен.
Для измерения текущей концентрации очень удобны фото-
метрические методы, так как оптическая плотность раствора
прямо пропорциональна концентрации вещества. При постро-
ении кинетической кривой на оси ординат вместо концентрации
можно откладывать оптическую плотность: тангенс угла наклона
для построения градуировочного графика можно вычислять как
АА/дА. Метод тангенсов с успехом применяется для самых раз-
личных реакций, по точности определения он превосходит все ос-
тальные варианты кинетических методов.
Метод фиксированного времени. В методе фиксированного
времени определяют концентрацию продукта реакции или кон-
центрацию одного из участников реакции за строго определен-
ный промежуток времени. Если, например, продукт реакции ок-
рашен, через определенный промежуток времени измеряют опти-
ческую плотность раствора. При небольшой глубине протекания
реакции применяют дифференциальный вариант. Из уравнения
(14.5) получаем
где £ — заданный промежуток времени.
Выражение в скобках постоянно, так как а и £ фиксированы.
Его числовое значение может быть определено по стандартному
раствору и использовано для последующих расчетов ск по изме-
ренной величине x.
Можно построить градуировочный график в координатах ск -х,
где ск — концентрация определяемого вещества (катализатора), а
х .— концентрация продукта реакции, образовавшегося за фикси-
рованный отрезок времени. Вполне понятдо, что фиксированный
отрезок времени сохраняется один и тот же как при построении
градуировочного графика, так и при анализе неизвестного рас-
твора. При большой глубине протекания реакции применяется
интегральный вариант, основанный на решении уравнения (14.6)
относительно ск. Метод фиксированного времени, как видно, про-
ще метода тангенсов, однако по точности он ему уступает.
нетическую кривую следует строить в координатах ^
а
а - х
268
Метод фиксированной концентрации. В методе фиксирован-
ной концентрации измеряют время, в течение которого концент-
рация продукта реакции или одного из реагирующих веществ до-
стигает определенного, заранее заданного значения. Этот метод
по сути близок методу фиксированного времени. Если глубина
протекания реакции невелика, используют дифференциальный
вариант, так же как и в методе фиксированного времени, осно-
ванной на решении уравнения (14.5) относительно ск:
с«=^=(£)? <14-7>
где х — заданная концентрация продукта реакции.
Выражение в скобках постоянно, поскольку постоянны х и а,
его числовое значение может быть определено по стандартному
раствору. Градуировочный график в методе фиксированной кон-
центрации, как показывает уравнение (14.7), следует строить в
координатах ск - 1/*, где ск — определяемая концентрация, а I —
время, необходимое для достижения заданной концентрации
продукта реакции.
При более глубоком протекании реакции используется ин-
тегральный вариант, основанный на решении уравнения (14.6):
Градуировочный график, как видно, следует строить также в ко-
ординатах ск - 1/£.По точности метод фиксированной концент-
рации близок к методу фиксированного времени и уступает мето-
ду тангенсов.
Метод добавок. Этот метод обладает известными достоинст-
вами. Принципиальная сущность его уже рассматривалась при
изложении спектрального анализа, фотометрии, вольтамеро-
метрии и др. При выполнении анализа по методу добавок необхо-
димо провести два измерения: сначала определить скорость реак-
ции в анализируемом растворе, а затем скорость реакции в этом
же растворе с добавкой стандартного раствора.
Метод каталиметрического титрования. Сущность этого ме-
тода можно пояснить на примере церий-арсенитной реакции, ко-
торая катализируется осмием, рутением и иодидом:
2Се4+ + НАв02 + Н20 - 2Се3+ + НАвОд + 2Н+ (14.8)
Так как иодид является катализатором реакции церия с арсени-
том, скорость этой реакции будет возрастать с увеличением кон-
центрации иодида. Ионы Ag+ обладают ингибиторным эффек-
том, блокируя катализатор. При введении в раствор ионов Ag+
269
происходит образование Agi, и концентрация I" падает, вызывая
соответствующее уменьшение скорости церий-арсенитной реак-
ции. При каталиметрическом титровании к анализируемому рас-
твору, содержащему иодид-ион I" в церий-арсенитной смеси, не-
сколькими порциями добавляют титрованный раствор AgN03 и
определяют скорость реакции. Точку эквивалентности находят
из графика в координатах объем раствора AgN03 — скорость ре-
dx
акции, характеризуемая угловым коэффициентом —. Этим мето-
at
дом можно оттитровать иодид в очень разбавленных растворах
(10~6 моль/л и меньше).
14.3. Практическое применение
Кинетические методы анализа характеризуются высокой
чувствительностью й поэтому часто используются при определе-
нии малых и ультрамалых содержаний (до 10~8—-1Ô"6 мкг). Осо-
бенно эффективным оказалось применение кинетических мето-
дов для определения микропримесей в чистых и сверхчистых ве-
ществах и материалах.
В настоящее время известны десятки каталитических реак-
ций, с помощью которых могут быть определены свыше 40 эле-
ментов периодической системы. Например, реакция иодида с пе-
роксидом водорода без катализатора идет очень медленно:
Н202 4-2Г + 2Н+= 12 + 2Н20
В присутствии следов молибдена, вольфрама, циркония, гафния,
ниобия, тантала и других элементов она проходит за несколько
минут. Скорость реакции легко определяется по возрастанию оп-
тической плотности иодкрахмального раствора в единицу време-
ни. Чувствительность реакции достаточно высока. Например, с
ее цомощью можно определить 0,01 мкг/мл W, 0,02 мкг/мл Mo,
0,1 мкг/мл Zr и Hf.
Интересны каталитические эффекты в реакции (14.8) взаимо-
действия мышьяковистой кислоты с солями церия(ГУ), которая
катализируется осмием, рутением и иодидом. Исследование пока-
зало, что скорость этой реакции, катализируемой осмием, не за-
висит от концентрации церия, но увеличивается с ростом кон-
центрации мышьяковистой кислоты. Если в качестве катализато-
ра выступает рутений, скорость реакции (14.8) перестает зависеть
от концентрации мышьяковистой кислоты, но увеличивается с
концентрацией церия. Эти особенности церий-арсенитной реак-
270
ции открывают возможность определения осмия и рутения в од-
ном растворе без предварительного разделения. По скорости реак-
ции в растворах с увеличивающейся концентрацией церия можно
определить рутений, а по скорости реакции при возрастающей
концентрации мышьяковистой кислоты найти содержание осмия.
С помощью кинетических методов анализа в большинстве слу-
чаев определяется не общая, а равновесная концентрация реаги-
рующих веществ. В связи с этим кинетические методы успешно
применяются для изучения различных равновесий в растворах
(комплексообразование, кислотно-основное взаимодействие и др.).
14.4. Общая характеристика метода
Наиболее ценной особенностью кинетических методов яв-
ляется возможность определения элементов при их содержании
10"8—10~6 мкг, что превосходит соответствующую характеристи-
ку спектрального, спектрофотометрического, потенциометриче-
ского и многих других методов анализа. Анализ с помрщью кине-
тических методов выполняется быстро и просто, без применения
сложных приборов. Кинетическим методом можно определить
свыше 40 элементов периодической системы с погрешностью, не
превышающей ±10% (отн.). В некоторых случаях кинетические
методы обладают достаточной специфичностью, однако, как пра-
вило, их специфичность не высока. Специфичность каталитиче-
ских реакций повышают путем маскировки каталитически ак-
тивных примесей при введений в анализируемый раствор соот-
ветствующих реагентов.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Что является аналитическим сигналом в кинетических методах
анализа?
2. Какие реакции называют индикаторными? Какие требования
предъявляются к таким реакциям?
3. Что представляет собой дифференциальный вариант кинетиче-
ских методов анализа?
4. В каких случаях применяется интегральный вариант кинетиче-
ских методов анализа? '
5. Какие, основные приемы используют в кинетических методах
анализа: а) методе тангенсов; б) методе фиксированной концент-
рации; в) методе фиксированного времени?
6. Каковы области практического применения, достоинства и не-
достатки кинетических методов анализа?
271
г л а в а 1 5
Термометрическое титрование
15.1. Тепловой эффект реакции
как аналитический сигнал
Методы термометрического, или энталъпийного, титрова-
ния основаны на измерений теплового эффекта реакции титрова-
ния или величин, пропорциональных этому тепловому эффекту.
Тепловой эффект некоторых реакций может не отличаться от ну-
ля, однако специально рассматривать этот случай нет необходи-
мости в связи с его сравнительно редкой реализуемостью в прак-
тических условиях титрования.
Если реакция титрования вещества А титрантом В протекает
в соответствии с уравнением
А + В-АВ
и изменение энтальпии при титровании пА (моль) вещества А
равно АНТИтр, а изменение энтальпии в расчете на 1 моль вещест-
ва А равно АН, то
Д#титр = иД#. (15.1)
В то же время ДЯ"титр связано с теплоемкостью системы Ср и
изменением температуры Д Т вследствие теплового эффекта реак-
ции соотношением
ДЯтитр = СрДТ. (15.2)
Сочетание уравнений (15.1) и (15.2) дает
АГ=^т1А. (15.3)
р
При соблюдении некоторых условий (постоянство ионной си-
лы и небольшое изменение температуры) величина АН в ходе
титрования будет оставаться постоянной, а существенное увели-
чение концентрации титранта по сравнению с концентрацией ве-
щества А обеспечит достаточное постоянство теплоемкости систе-
мы Ср, так как исходный раствор разбавляться практически не
будет. Тогда отношение АН/Ср также останется постоянным и
вместо уравнения (15.3) можно записать
АТ = ЫА. (15.4)
272
Уравнение (15.4) показывает, что изменение температуры
при титровании прямо пропорционально количеству вещества А,
вступившего в реакцию титрования.
В первых работах по термометрическому титрованию темпе-
ратуру измеряли термометром Бекмана и после каждого добавле-
ния стандартного раствора систему выдерживали до достижения
термического равновесия. Стандартный раствор добавляли из
бюретки небольшими порциями каждые 5—10 мин, и таким об-
разом весь процесс титрования продолжался примерно 1 ч. Впол-
не понятно, что такая методика не имела заметных преимуществ
перед обычными методиками титриметрического анализа.
Практическое значение термометрического титрования зна-
чительно возросло с применением в качестве датчиков темпера-
туры термисторов и использованием термостатированных порш-
невых бюреток или специальных устройств для автоматизиро-
ванной подачи титранта в анализируемый раствор.
Сопротивление термистора очень чувствительно к измене-
нию температуры. Увеличение температуры на один градус вы-
зывает уменьшение сопротивления термистора на 1—5%. Инер-
ция термистора невелика: он реагирует на изменение темпера-
туры в течение 3—5 с. Эти ценные качества термисторов и позво-
лили разработать современные методы термометрического тит-
рования. Изменение температуры раствора вызывает изменение
сопротивления термистора, что приводит к «разбалансу» моста,
который может быть зарегистрирован на самопишущем потенцио-
метре.
В дифференциальных установках измеряется разность тем-
ператур между двумя сосудами, один из которых содержит ана-
лизируемый раствор, а другой — раствор сравнения, и в оба сосу-
да с одинаковой скоростью поступает титрант. Такая схема по-
зволяет автоматически исключить влияние теплоты разбавления
титранта, тепловыделения при перемешивании раствора, эффект
Джоуля при прохождении тока через термисторй другие эффек-
ты, затрудняющие измерение теплоты собственно реакции титро-
вания.
15.2. Кривые термометрического титрования
Кривые термометрического титрования показывают изменег
ние температуры в ходе титрования в зависимости от объема до-
бавленного титранта. Типичный график изменения температуры
18-7831
273
У=0 т.э
Рис. 15.1. Кривые
термометрического
титрования:
- реакция экзотермическая;
- реакция эндотермическая
Рис. 15.2. Кривые
термометрического титрования
при разной температуре титранта:
1 -
2
гр > т
л титранта ■* раствора'
т —т
л титранта л раствора»
о гр ■ : < Т
■ титранта х раствора
в ходе титрования приведен на рис, 15.1. Отрезок АВ (или А!В')>
показывающий температуру системы перед началом титрования,
иногда называют основной линией; ВС (или В'С) характеризует
изменение температуры раствора в ходе титрования, а отрезок СВ
(или СП) показывает изменение температуры после достижения
точки эквивалентности С (или С", если реакция эндотермична).
Практически получаемые кривые в той или иной степени от-
личаются от идеализированной, так как экспериментально изме-
ряемый тепловой эффект наряду с изменением энтальпии в реак-
ции титрования включает эффект разбавления титранта, тепло-
выделение при перемешивании раствора и т. д. Изменение темпе-
ратуры при титровании может быть вызвано также разностью тем-
ператур анализируемого раствора и титранта, выделением теплоты
при прохождении через термистор электрического тока и т. д. Не
все эти факторы действуют отрицательно. Иногда температуру
титранта специально устанавливают ниже или выше температуры
анализируемого раствора (в зависимости от знака теплового эф-
фекта), чтобы более ч;етко фиксировать точку эквивалентности.
Как показывает рис. 15.2, если реакция титрования экзотер-
мична, то применение титранта с более низкой температурой,
чем у анализируемого раствора; позволяет более четко фиксиро1
вать точку эквивалентности (кривая 3). При одинаковой темпе-
ратуре обоих растворов (кривая 2) или при более высокой темпё^-
ратуре титранта (кривая 1) точка эквивалентности фиксируется
менее четко. Однако в большинстве своем посторонние тепловые
эффекты оказывают неблагоприятное воздействие, вызывая, на-
274
&(АТ)/о\У1
В
т.э. V
Рис. 15.3. Дифференциальная
кривая термометрического
титрования
т.э.(1) т.э.(И)
Рис. 15.4. Кривая
титрования смеси ионов
Са2+иМг2+
пример, плавное закругление кривой в конце титрования, что ос-
ложняет определение точки эквивалентности и является допол-
нительным источником погрешности. При закруглении кривой
точку эквивалентности определяют графической экстраполяцией
линейных участков кривой титрования. Более точно точку экви-
валентности можно определить при построении дифференциаль-
ной кривой титрования. Типичная дифференциальная кривая
термометрического титрования представлена на рис. 15.3. Точка
эквивалентности на этой кривой фиксируется более четко.
Термометрическое титрование успешно применяется при оп-
ределении компонентов смеси без предварительного химического
разделения. Так анализируют, например, технически важную
смесь кальция и магния титрованием ЭДТА. Реакция образова-
ния комплекса кальция СаУ2" экзотермична, а комплекса маг-
ния М£У2~ эндотермична. Кривая титрования этой смеси пред-
ставлена на рис. 15.4. Погрешность определения кальция состав-
ляет 0,5%, а магния 2%.
15.3 Практическое применение
В термометрическом титровании могут быть использованы
реакции кислотно-основного взаимодействия, окисления—вос-
становления и любые другие, тепловые эффекты которых доста-
точно велцки, чтобы произвести точные измерения. Важным до-
стижением термометрических методов является возможность
ррямого титрования слабых кислот с высокой точностью. Напри-
мер, борная кислота титруется в водном растворе без добавления
маннита. Также могут быть оттитрованы некоторые слабые орга-
18*
275
нические кислоты, аминокислоты, слабые основания и другие ве-
щества. Термометрическое титрование различных восстановите-
лей дихроматом показывает более высокую точность, чем титро-
вание с применением дифениламина в качестве индикатора.
Известны методы термометрического титрования, основанные на
реакциях осаждения сульфатов, галогенидов, оксалатов и других
малорастворимых соединений, методы, основанные на образова-
нии этилендиаминтетраацетатных и других комплексов и т. д.
Разработаны методы анализа смесей путем последовательного
титрования компонентов без предварительного химического раз-
деления. Термометрическим методом титруются также различ-
ные вещества в неводных растворителях (ледяная уксусная кис-
лота, ацетонитрил и др.) и в расплавах солей, например в расплав-
ленной эвтектике, нитритов лития и калия аргентометрически
титруется хлорид.
Методики термометрического титрования используют во
многих отраслях промышленности: в металлургической, хими-
ческой, электрохимической, нефтехимической и др., в анализе
фармацевтических материалов и во многих других областях. ^
15.4. Общая характеристика метода
Существенным достоинством метода термометрического тит-
рования является его универсальность. Один и тот же термомет-
рический прибор может быть использован для самых различных
определений, так как здесь не нужны специфические индикато-
ры, селективные электроды ит. д. В настоящее время термомет-
рическое титрование квалифицируется как один из наиболее пер-
спективных методов анализа: он обладает высокой точностью,
универсальностью и легко может быть автоматизирован.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. На чем основаны методы термометрического титрования?
2. Как связано количество вещества, вступившего в реакцию, с из-
менением температуры раствора?
3. В каких координатах строятся кривые термометрического тит- г
рования? ^
4. На чем основан анализ смеси кальция и магния в растворе без ,
предварительного химического разделения?
5. Каковы достоинства и недостатки метода термометрического '
титрования? Где практически используется этот метод? '
276
глава 16
Экстракция
16.1. Распределение вещества
между двумя жидкостями
При соприкосновении водного раствора вещества А с ка-
ким-либо неводным растворителем, не смешивающимся или ог-
раниченно смешивающимся с водой, растворенное вещество А
будет распределяться между обоими растворителями и через не-
которое время в такой системе установится равновесие
где Ад и А0 — вещество А в воде и в органическом растворителе
соответственно.
Процесс переноса растворенного вещества из одной жидкой
фазы в другую, с ней несмешивающуюся или ограниченно сме-
шивающуюся жидкую фазу, называют жидкость-жидкостным
распределением или распределением между двумя жидкостями.
Количественно этот процесс характеризуется законом распреде-
ления Нернста—Шилова, в соответствии с которым отношение
концентраций растворенного вещества в обеих фазах при посто-
янной температуре постоянно и не зависит от общей концентра-
ции растворенного вещества:
1> = (16.1)
где jD — коэффициент распределения; [А]0 — аналитическая, т. е.
суммарная концентрация всех форм вещества А в органической
фазе; [А]в — то же, в водной фазе.
Величина I) сохраняет постоянство лишь в отсутствие про-
цессов диссоциации, ассоциации, полимеризации и других пре-
вращений растворенного вещества.
При подстановке в уравнение (16.1) активностей вещества А
в органической фазе и в водном растворе вместо их концентраций
коэффициент распределения будет оставаться постоянным в ши-
рокой области концентраций, поскольку процессы ассоциации и
другие будут формально учтены коэффициентами активности.
Однако при небольших концентрациях вещества А коэффициент
распределения I) и без этого сохраняет удовлетворительное пос-
тоянство и часто используется как основная характеристика рас-
пределения вещества.
277
Иногда выражению закона распределения придают вид
в котором п подбирают эмпирически.
В простейшем случае п характеризует степень полимериза-
ции растворенного вещества в органическом растворителе. Отно-
шение концентрации (точнее, активности) вещества в одной оп-
ределенной форме (например, МЬД) в фазе органического раство-
рителя к его концентрации (активности) в той же форме в водной
фазе называют константой распределения К£:
у0САмья)0 _ [МЬД]0(Умьв)0 _ у (Умьй)0 пао\
^(^в"[МЬл]в(уМЬп)в "^(^в- (16-2)
Коэффициент и константа распределения связаны с раство-
римостью вещества. В простейшем случае, когда вещество в обе-
их фазах существует в одной и той же форме (например, в виде
недиссоциированных мономерных молекул), константа и коэффи-
циент распределения равны отношению растворимостей вещест-
ва в органическом растворителе й в воде. Действительно, если в
систему из воды и несмешивающегося в ней органического рас-
творителя ввести твердое вещество до насыщения, то концентра-
ция вещества в каждой фазе будет равна его растворимости в со-
ответствующем растворителе и, следовательно,
(5А)В'
где (5А)0 — растворимость вещества в органическом растворите-
ле; (5А)В — растворимость в воде.
16.2. Основные количественные
характеристики экстракции
Экстракция представляет частный случай жидкость-жидко-
стного распределения, когда из водного раствора вещество извле-
кается в несмешивающийся с водой органический растворитель.
Реагент, который образует экстрагирующееся соединение, назы-
вают экстракционным реагентом, а органический раствори-
тель, используемый для экстракции, или раствор экстракцион-
ного реагента в органическом растворителе, называют экстра-
гентом. Для улучшения физических (плотности, вязкости и др.)
или экстракционных свойств экстрагента в него нередко добавля-,
ют разбавитель инертный органический растворитель или ис-
пользуют смесь растворителей. •/{
278
Важной характеристикой экстракции является фактор (или
степень) извлечения
(16.3)
где п(А) — количество вещества в органической фазе; п(А)И — ис-
ходное его количество в водном растворе.
Очевидно, что
/1(А) = [А]0У0; (16.4)
п(А)И = е%уш = [А]0^0 + [А]ВГВ, (16.5)
где Сд — концентрация вещества А в исходном водном растворе.
Подставим соотношения (16.4) и (16.5) в уравнение (16.3) и
разделим числитель и знаменатель полученного выражения на
[А]ВК0:
"д- [А]°У° = п = (16.6)
[А]07Ь + [А]ВГВ' Л+Г,/Гв • 1> + 1/г
где X) определяется соотношением (16.1), а г = У0/Уъ. Уравнение
(16.6) относится к однократной экстракции и остается справед-
ливым при многократном повторении этой опер
После первой экстракции будем иметь : ; .
где индекс «1» — номер экстракции.
Подставляем сюда уравнение (16.1) и решаем относительно
[Ах]в:
с^з^^А^ + ЕА^,
[Ах!
iJb DV0 + Vb Dr + 1
После второй экстракции аналогично:
ГА У - [Ai1b - С°А
1 2Jb 57+1, (Dr + 1)2'
а после т экстракции:
[Ат]я = С°А . (16:7)
Отсюда
с°а
лу, т = [Ат1в . (16.8)
По уравнению (16.8) можно рассчитать число экстракций,
необходимое для достижения заданной степени извлечения. На-
пример, для экстракции 99% вещества из водного раствора необ-
279
2 *■
ходимо сделать т = — экстракций; при Пг = 1 это составив
\giDr +1) а
т = 2/0,3 = 7 операций, а при 1>г = 5 т = рг— = 2,6 ~ 3 операции.
О,7о
Степень извлечения при тп-кратной экстракции нахо-
дим путем сочетания уравнений (16.3) и (16.7):
Д - с<кУ* ~}^т]У* - 1 ^ =1--—! . (16.9)
т с%Ув (Вг + 1)тсАУв (Лг + 1)т
При т = 1иг = 1 оно переходит в
Возможность разделения двух катионов А и В до недавнего
времени связывалась с коэффициентом разделения х, равным от-
ношению коэффициентов распределения этих ионов х = 1)А/Х)В.
При х = 1 разделение невозможно. Чем больше х отличается от 1,
тем ближе к оптимальным будут условия разделения. Более об-
щей характеристикой возможности разделения является фактор
обогащения Б, показывающий, во сколько раз отношение коли-
честв разделяемых веществ в фазе экстр>агента превышает это от-
ношение в исходном растворе до разделения. Другими словами,
эта величина, на которую следует умножить отношение исход-
ных количеств разделяемых катионов, чтобы получить отноше-
ние их количеств после разделения (в органической фазе):
п(В)/7г(А) = 5в/А71(В)0/д(А)0,
откуда фактор обогащения
5В/А = ИШ^2^ ДВ/ДА. (16.10)
В/А л(В)0 п(А) ...... /
Зависимость фактора обогащения от коэффициентов распреде-
ления и других величин находим путем подстановки (16.9) в (16.10):
д _[(Ръг + 1Г-1](РАг + 1Г (16 11)
При т = 1иг=1
^в/а-дв(да + 1)> (16.12)
Если, например, £>А = 104, а 2)в = 0,1, то коэффициент разде-
ления х = 2>А/Х)В = 104/0,1 = 105 имеет довольно большое значе-
ние. Однако это не обеспечивает удовлетворительного разделе-
ния, так как хотя элемент А извлекается на 99,99%, но и эле-
280
мент В переходит в органическую фазу тоже в значительных
количествах ( ~10%). Фактор обогащения для этой системы в со-
ответствии с уравнением (16.12) равен
В/А 104(0,1 + 1)
Если при том же значении коэффициента разделения х = 105 ко-
эффициенты распределения будут 1>А = 102 и Х>в = 10"3, то фактор
обогащения составит Яв/А = 10 3^102 + = 10~3, т. е. значение на
В/А ю2(10-3 + 1)
два порядка меньше, чем в первом случае. Элемент А перейдет в
органический растворитель на 99%, а элемент В — только на
0,1%. Таким образом, коэффициент 5В/А более реально, чем х,
характеризует возможности разделения элементов. Опыт пока-
зывает, что в органическую фазу из водного раствора вещество
переходит в виде электронейтральных частиц: координационных
соединений, ионных ассоциатов и т. д. С увеличением температу-
ры экстракция, как правило, ухудшается.
16.3. Экстракция хелатов
Анион внутрикомплексной соли является полидентатным
лигандом, образующим с катионом один или несколько циклов.
Внутрикомплексная соль, как правило, малорастворима в воде и
хорошо растворима в органических растворителях, что для экс-
тракции имеет особое значение. Типичными экстракционными
реагентами этого класса являются 8-оксихинолин, дитизон, аце-
тилацетон и др. Обычно они обладают свойствами слабой кисло-
ты, поэтому кислотность раствора относится к числу наиболее
важных факторов, определяющих полноту экстракции.
Существенное значение имеет распределение самого экстрак-
ционного реагента между неводным растворителем и водой. Кис-
лотно-основное равновесие в растворе реагента НЬ
НЬ + Н20 = Н30++ Ь"
в кислой среде может быть осложнено протонированием
НЬ + Н30+> Н2Ь++ Н20
как это происходит, например, в кислых растворах 8-оксихино-
дина. Коэффициент распределения реагента будет равен
" п^±Йь)о=[Н2^]о + [НЬ]о + [Ь-]0 6
НЬ (Снь). [Н2Ь+] + [НЬ] + [1г]
281
Константы распределения относятся к каждой из форм су-
ществования реагента:
тго _ (aH2L+)0 _ [H2L4]0Yhl^(o) _ jr Vio), пллл\
tro _ (ань)0 _ [HL]0yHL(o) v Yhl(o), /11* i к\1
^(НЬ)^^Г"' [hl]Yhl ■"^<HL)l^r' (16Л5);
— Sbiísi"_-£М^ы122 —-jht^^..íbiísa. (16.16)
^ > <*L- [L-]yL. ^ > YL-
Концентрации в уравнении (16.13) можно выразить с по-
мощью констант диссоциации HL и H2L+ и констант распределе-
ния (16.14)—(16.16):
n ^2)(H2L+)[H2L+] + ^I>(HL)[HL1 + ^D(L-)[L~]
нь= IÍ^ + tHL]+i,H™
AZ)(H2L-)^— +Ai)(HL)+AD(L-)— ,
[H+] + 1+ £hl.
Это общее уравнение распределения реагента между раствот
рителями. В достаточно кислой или щелочной области уравнение
обычно упрощают, так как в таких средах некоторыми членами
можно пренебречь. Например, в щелочной области уравнение
(16.17) принимает вид
Если в органическую фазу экстрагируются только молекулы,
HL, то
- ; .. i+irHL/[H+]
^Процессэкстракции катионаМп+ обычно представляют схемой:
M^ + nHL(0) = MLn(0) + nH+
где HL — экстракционный реагент; индекс (о) указывает на орга-
ническую фазу. Константа этого процесса — константа экс-
тракции Кех:
Kl . *s*£k , [ml ] гш^гъ=Еуш^; (16Л8)1:
вХ 0M„+OgL(o) [M"+][HL]» ум„.ТЙьи) ехУм-УЙь(0)
282
При постоянной ионной силе раствора
к'*~ [м»*][ныг- (16Л9)|
Если равновесие экстракции в заметной степени не осложне-
но кислотно-основным взаимодействием, ступенчатым комплек-
сообразованием, ассоциацией и другими процессами, то
[МЬ„]В + [М]В
± ■- ±+ [М]»
Подставим сюда отношениеиз уравнения (16.19):
. (16.20)
Ям Кв ^ех[МЩ . 1 . ■
При Кв ^> £>м величиной 1/-Кд пренебрегают и уравнение
(16.20) переходит в
_1_ _ [Н+]»
или •
•°м = ^ех^|- (16.21)
При логарифмировании уравнения (16.21) получаем
1* £>м - \ё Кех + п \ё [НЬ]0 + прН. (16.22)
Уравнение (16.22) и более точное (16.20) показывают, что коэф-
фициент распределения тем больше, чем выше рН раствора и
концентрация реагента НЬ. Это не означает, однако, что экстрак-
цию во всех случаях следует проводить из щелочного раствора.
Для каждой системы существуют оптимальные значения рН и
концентрации реагента. Уравнению (16.22) можно придать Эйд:
=рн- + 1£[НЬ]0. (16.23)
л п
,у При Ьм, = 1 и равных объемах водной и органической фаз
экстрагируется 50% металла и в этом случае уравнение (16.23^
переходит в
(ь, -щ™ =рн1/2 + 1&[нь]0.
283
Если [НЬ]0 = 1, то - ех равен значению рН, при котором
экстрагируются 50% металла:
№1/2)1,0-
Величина (рН1/2)1>0 является одной из практически важных
характеристик экстракции, позволяющей предвидеть возмож-
ности и условия экстракционных разделений.
Константа экстракции связана с константой устойчивости об-
разующегося координационного соединения, константой диссо-
циации реагента и другими константами:
_[МЬД]0[НЧ» [Ь-]»[МЬЛ][НЬ]" _ Рмья^(мьд)^йь (162лу
ех [М"Ч[НЬ]» [Ь-]«[МЬП][НЬ]« ЩНЪ) 1 • '
Произведение Рмь„^1)(мьп) часто называют двухфазной конс-
тантой устойчивости. Как видно из уравнения (16.24), чем вы-
ше устойчивость соединения МЬ„, больше константа диссоци-
ации реагента НЬ и меньше его коэффициент распределения, тем
больше константа экстракции и, следовательно, тем эффективнее
будет экстракция.
Существенное влияние на экстрагируемость и полноту экс-
тракции оказывают и другие факторы: растворимость соедине-
ния МЬЛ, природа органического растворителя, наличие маски-
рующих агентов и т. д. Наибольшей экстрагируемостыо облада-
ют внутрикомплексные соединения, малорастворимые в воде, но
легко растворимые в органических растворителях (ацетилацето-
наты железа, галлия, индия и др.), а соединения, растворимые в
обеих фазах, экстрагируются лишь частично (ацетилацетонаты
цинка, кобальта, никеля и др.).
Влияние природы органического растворителя на экстрак-
цию имеет ряд характерных особенностей. При переходе от одно-
го растворителя к другому коэффициенты распределения КщЫЬп)
и КВ(:аъ) изменяются симбатно и существенного влияния природы
растворителя на константу экстракции (16.24) не наблюдается.
Однако такие растворители, как трибутилфосфат, спирты и дру-
гие, образуют с экстрагируемым металлом комплексы, которые
менее гидрофильны, чем аква-ионы, и их экстракция неполяр-
ными растворителями улучшается.
Во многих случаях смесь экстрагентов более эффективно экс-
трагирует соединения из водного раствора, чем каждый из ком-
понентов в отдельности. Это так называемый синергизм. Напри-
. • ■ ■ • ■ ■■ ■ •''•'!'..
284
мер, при экстракции урана(VI) раствором теноилтрифторацетона
в циклогёксане коэффициент распределения составляет 0,063, а
раствором окси-3-н-бутилфосфата — 38,5. У смешанного экстра-
гента, состоящего из этих трех компонентов, коэффициент рас-
пределения достигает 1000.
164. Экстракционные хелатные системы
Широкое применение в практике экстракции нашли 8-окси-
хинолин и его производные, дитизон и его аналоги, Р-дикетоны,
оксимы, дитиокарбаминаты, алкилфосфорные кислоты и многие
другие соединения. Рассмотрим свойства некоторых наиболее из-
вестных экстрагентов.
Ъ-Оксихинолин I I I (или оксин) малорастворим в воде
• , : • ОН .
(3,6 • 10~3 моль/л при комнатной температуре) и эфире, но хоро-
шо растворим в спирте, бензоле, хлороформе и других органи-
ческих растворителях. Для экстракций обычно используются
хлороформные растворы, содержащие 1—5% оксихинолина. Кон-
станта распределения 8-оксихинолина между водой и хлорофор-
мом при 25 °С составляет -500. Оксихинолин является одним
из самых универсальных реагентов: он взаимодействует более
чем с 50 элементами (Р(1, Мо, /\У, V, Т1, ¥е, Ъг, ва, Си, /И, 1п,
В1, N1 и др.). Для повышения избирательности разделений с
помощью 8-оксихинолина наряду с регулированием рН широко
используются различные маскирующие реагенты (цианид-ион,
1,10-фенантролин, ЭДТА и др.). Например, ионы Ее, Си, № и Мо
маскируются цианидом. Алюминий, кобальт, железо и некото-
рые другие элементы в присутствии ЭДТА при рН < 8 не экстра-
гируются, хотя на экстракции урана, вольфрама и молибдена
присутствие ЭДТА не сказывается. Многочисленные галогено- и
другие замещенные оксихинолины также обладают экстракци-
онными свойствами, но, по-видимому, они менее универсальны;
Например, 2-метил-8-оксихинолйн не экстрагирует алюминий.
" Широко используются в качестве экстрагентов Р-дикетоны,
особенно ацетилацетон и теноил трифторацетон.
, л' Ацетилацетон СН3— С — СН«— С — СН3 смешивается с хло-
11 н .
роформом, бензолом, эфиром и другими органическими раство-
285
рителями, егр растворимость в воде составляет 172 г/л при 20 °Ся
Константа распределения ацетилацетона между водой и бензоо
лом составляет 5,8, между водой и хлороформом 25. Для экс?
тракции можно использовать непосредственно сам ацетилацетона
который в таком случае является и реагентом, и растворителем; а*
также растворы ацетилацетона в бензоле, хлороформе, тетрахлот!
риде углерода или другом органическом растворителе. Ацетил-:
ацетон образует соединения более чем с 60 элементами (РЬ, Т1^
Ее, Ри, и, ва, Си, Бс, А1, 1п, ТЬ, №, Ьа и др.). Ацетилацетонаты
хорошо растворимы в органических растворителях и имеют вы-
сокую термическую устойчивость: многие из них могут возго-
няться без разложения.
Теноилтрифторацетон ^~}^С — СЩ—С — СР3 в воде
8 О О
растворим незначительно, в органических растворителях значи-
тельно лучше. Константа распределения между водой (подкислена
ной) и бензолом равна 40. С помощью теноилтрифторацетона воз-
можна экстракция; металлов из кислых растворов, что является
его важным достоинством. Очень широко применяют теноилтрйф^
торацетоц для выделения и разделения актинидов. Соединения;
многих металлов (урана, меди, железа и др.) с теноилтрифтораце-;
тоном окрашены, поэтому нередко органическая фаза, содержат
щая теноилтрифторацетонаты этих металлов, может фотометриро-
ваться.
К числу реагентов, наиболее часто применяемых в экстрак-
ции, относятся дитизон й его аналоги.
,N11—ЫН—( )
Дитизон (или дифенилтиокарбазон) 8=С^ /г—\~^
(сокращенно Н2Бг) в воде практически не растворим, хорошо рар-,
творим в хлороформе и тетрахлориде углерода. Константа распре-
деления дитизона между водой и хлороформом равна 2 • Ю5,*
между водой и тетрахлоридом углерода 1 • 104. По реакции с ди-
тизоном определяют: Рс1, Аи, 'H.g, Ag, Си, В1, Р1;, 1п, гп, С(1, Сою
другие элементы. Для повышения селективности экстракцию^
проводят в разных областях рН в присутствии комплексообразую-г
щих добавок. Например, экстракцией висмута в слабокислой срек
де можно отделить его от цинка, кадмия, свинца и других элемент
тов. В присутствии цианид-иона дитизон экстрагирует тольког
286
цинк и олово. Эффективно использование в качестве комплексо-
образующйх добавок тиоцианата, тиосульфата, ЭДТА и др. Сам
дитизон и дитизонаты металлов интенсивно окрашены, что позво-
ляет проводить чувствительные фотометрические определения не-
посредственно в органической фазе после экстракции. Находят
применение также различные аналоги дитизона (метил-, фенил-,
хлор-, бром- и другие производные), реагирующие с меньшим
числом ионов и, следовательно, более селективные, чем дитизон.
Диэтилдитиокарбаминат натрия ^-^^ ^ — С \ д-^ —
важный представитель реагентов группы дитиокарбаминатов. Он
хорошо растворим в воде, несколько хуже в спирте, в виде ди-
этилдитиокарбаминрвой кислоты растворим в хлороформе, бен-
золе, тетрахлорйде углерода и родственных растворителях. Конс-
танта распределения между органической и водной фазами равна
340 для тетрахлорида углерода и 2360 для хлороформа. Водные
растворы диэтилдитиокарбаминовой кислоты неустойчивы. Ди-
этилдитиокарбаминат реагирует с несколькими десятками эле-
ментов (Н§, Аё, Си, Т1,№^В1,РЬ, Сс1, и др.). В присутствии
ЭДТА селективность экстракции увеличивается. Некоторые ди-
этилдитиокарбаматы имеют окраску: комплекс висмута — жел-
тую, кобальта — зеленую, меди■— коричневую и т. д., что позво-
ляет проводить фотометрические определения, используя непо-
средственно органические экстракты.
16.5. Экстракция ионных ассоциатов
Экстрагирующиеся системы ионных ассоциатов могут обра-
зовываться в результате взаимодействия заряженного хелата или
другого крупного иона с противоионом в растворе. Например,
тетрафениларсоний Аз(С6Н5)£ с перманганатом и перренатом об-
разует ассоциаты, которое экстрагируются хлороформом. Экс-
тракция в таких системах практически не отличается от экстрак-
ции хелатов. V
Более сложный характер имеют процессы, когда ассоциаты
образуются в результате ступенчатого комплексообразования,
например ионов металлов с галогенйдами. Типичным примером
таких систем является экстракция из солянокислых растворов
железа(Ш), сурьмы(У), кобальта(Н) и др. При взаимодействии,
например, ионовТе3+ и С1~ в растворе в зависимости от концент-
рации хлорида возможно образование комплексов различного со-
287
става от РеС12+ при небольшой концентрации С1~ до РеС1^ при вы-
сокой.
В органическую фазу могут извлекаться ассоциаты типа
Н+РеС1^, цричем сольватные оболочки ионов Н+ и ?еС1^ могут
наряду с водой содержать органический растворитель.
16.6. Экстракционные галогенидные
и тиоцианатные системы
Большое применение в практике экстракции нашла хлорид-
ная система. Значительная часть металлов экстрагируется в виде
ассоциатов типа ¥еС1±, БЬС^, СоС1|~ с ионом Н+ или в виде моле-
кул НбС12, СеС14 и др. В качестве экстрагентов используют црос-
тые и сложные эфиры (диэтиловый, этилацетат), спирты (бутило-
вый, амиловый), фосфорорганические соединения (трибутилфост
фат, трибутилфосфиноксид) и др. Экстракция хлоридных
комплексов широко применяется в аналитической химии, радио-
химии и некоторых гидрометаллургических производствах. Из-
вестна хорошая экстрагйруемость из хлоридных растворов Аи,
Т1, ЭЬ, ва,Те, Мо, N1), вс и других элементов.
Бромидные и иодидные комплексы также экстрагируются
кислородсодержащими растворителями, но практическое значе-
ние этих систем значительно меньше, чем хлоридных. В виде
фторидных комплексов экстрагируются лишь очень немногие;;
металлы (Та, №>, Ые и др.). Л
Тиоцианатные комплексы, как правило, экстрагируются;
лучше, чем галогенидные. В качестве экстрагента обычно ис-
пользуют тиоцианат калия или аммония в присутствии разбав-
ленной серной или хлороводородной кислоты. В виде тиоцианат-
ных комплексов экстрагируются В1, Ре, Со, 1п и другие элементы
трибутилфосфатом, диэтиловым эфиром, изоамиловым спиртом
и иными растворителями.
16.7. Скорость экстракции
Распределение вещества между фазами является результа-
том многих физико-химических процессов, протекающих в обе?
их фазах и на границе между ними. Скорость экстракции опреде^;
ляется главным образом скоростью образования экстрагирующе-
гося соединения и скоростью его распределения между фазами.
Лимитирующей стадией в разных системах может быть как тот,
288
так и другой процесс.Обычно считается, что если скорость экс-
тракции не зависит от интенсивности перемешивания, то лими-
тирующей стадией является скорость образования экстрагирую-
щегося соединения. Соединения типа ионных ассоциатов образу-
ются быстро и равновесие экстракции в таких системах также
устанавливается с высокой скоростью за 3—5 мин. Это же наблю-
дается во многих хелатных системах, и, таким образом, в боль-
шинстве случаев равновесие устанавливается довольно быстро.
Однако образование некоторых хелатов происходит медленно и
химическая реакция становится стадией, определяющей ско-
рость экстракции. Например, равновесие при экстракции дити-
зонатов таллия и цинка устанавливается за 1—3 ч. Небольшая
скорость образования координационных соединений хрома, пла-
тиновых и некоторых других металлов обусловливает и малую
скорость экстракции этих элементов. Для ускорения процесса
иногда анализируемый водный раствор нагревают вместе с ре-
агентом, чтобы прошло комплексообразование, а затем после ох-
лаждения экстрагируют.
Существенное влияние на скорость экстракции оказывает
природа органического растворителя. Отмечено, в частности, что
во многих случаях быстрее экстрагируют те растворители, в ко-
торых растворимость реагента меньше. Например, раствори-
мость ацетилацетона в тетрахлориде углерода почти на порядок
меньше, чем в хлороформе. Равновесное распределение ацетила-
цетоната железа(Ш) в системе СС14—Н20 достигается примерно
за 30 мин, а в системе СНС13—Н20 — за 3 ч. Различие в скорости
экстракции используется для разработки методик разделения эле-
ментов. Известно, например, что дитизонат ртути экстрагируется
хлороформом очень быстро (за 1—2 мин), а дитизонат меди —
медленно. Это различие в скоростях экстракции составило основу
методики их экстракционного разделения.
16.8. Практическое применение
С помощью экстракционных методик проводят разделение
элементов, относительное концентрирование цримесей и очистку
основного компонента от примесей. Для проведения экстракци-
онных разделений в аналитических лабораториях обычно ис-
пользуют делительные воронки. В препаративной работе приме-
няется также методика противоточной экстракции.
Для разделения элементов, реагирующих с данным экстра-
гентом, используются различия в константах экстракции, в зна-
19-7831
289
чениях рН, при которых происходит экстракция, применяют
введение специальных веществ, образующих комплексы с компот
нентами смеси, и т. д. Удобной характеристикой, позволяющей
предвидеть и количественно оценить врзможность разделения,
является коэффициент обогащения. »1,т
Различия в рН, при котором происходит экстракция разных,
элементов, используются, например, при анализе смеси Ре3+,
А13+, Мп2+. Все эти ионы экстрагируются раствором 8-оксихино-
лина в хлороформе при разных значениях рН. Ион Ре3+ экстраги-
руется в широкой области рН (от 2,0 до 10,0), ион А13+ — в облас-
ти рН 4,5—11 и Мп2+ — при рН 6,5—10,0. Практически ион Ре3+
извлекается при рН 2,5—3,0, ион А13+ — при рН 5,0 и ион Мп2+ —
при рН 10,0. Аналогично проводят разделение при экстракции
дитизоном, ацетилацетоном и другими реагентами.
При разделении в галогенидньгх системах используют зави-
симость экстрагируемости от концентрации галогенида. Так, на-
пример, в хлоридной среде Со2+ в виде СоС1|~ легко отделяется от
№2+, хлоридныё комплексы которого малоустойчивы.
Своеобразен Щт6& промежуточного элемента, когдк в ана-
лизируемую систему вводят специальный элемент, экстрагирую-
щийся хуже, чем определяемый, но лучше, чем мешающий. По
этой методике отделяют, в частности, олово от нескольких десят-
ков мешающих элементов введением цинка, который препятст-
вует экстракции более чем 60 элементов, но не влияет на экстрак-
цию олова.
Нередко эффективным оказывается применение реэкстрак-
ции, т. е. процесса обратного извлечения вещества из органиче-
ской фазы в водную. Водные растворы для реэкстракции подби-
рают таким образом, чтобы извлечение из органической фазы бы-
ло селективным. Например, при обработке раствора дитизонатов
свинца, цинка, железа и меди в СС14 0,4 М раствором НС1 из орга-
нической фазы извлекается только цинк, при обработке 4 М рас-
твором НС1 в водную фазу переходит и свинец. Железо и медь ос-
таются в органической фазе.
Широко применяется экстракция и для абсолютного й отно-
сительного концентрирования микропримесей. Абсолютное кон-
центрированиедостигается за счёт меньшего объема органиче-
ской фазы по сравнению с исходным объемом водного раствора.
Под относительным концентрированием понимают увеличение
концентрации примесей по отношению к содержанию основного
компонента. Оно сводится к селективной экстракции примесей
или (реже) экстракции макрокомпонента.
290
Существенное значение имеет методика последующей работы
с экстрактом. Особый интерес вызывают аналитические методи-
ки* в которых органическая фаза используется непосредственно
для количественного определения. Концентрацию окрашенного
компонента в экстракте можно определить фотометрически, со-
держание радиоактивного элемента — по его радиоактивности.
Используют также полярографию, эмиссионную спектроскопию,
атомно-абсорбционные методы и т. д.
16.9. Общая характеристика метода
Экстракция является весьма эффективньщ методом разделе-
ния и концентрирования, особенно при отделении микроком-
понента смеси от больших количеств других веществ. Сущестг
венным достоинством экстракционных методов является их бы-
строта. Для проведения разделения обычно бывает достаточно
нескольких минут и, как правило, кроме делительной воронки
никакой другой аппаратуры не требуется. Широкий выбор экст-
ракционных реагентов и растворителей позволяет подобрать оп-
тимальные условия селективного выделения того или иного ком-
понента, практически из любой смеси. Нередко экстрагированное
соединение окрашено, что позволяет непосредственно использо-
вать экстракт для количественных фотометрических определе-
ний микрокомпонента. Известно также эффективное использова-
ние экстракционных методов в технологии цветных и других ме-
таллов.
Крупные успехи в анализе микрокомпонентов достигнуты в
значительной степени благодаря применению экстракционных
методик, которые продолжают интенсивно развиваться и совер-
шенствоваться.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Чем характеризуется распределение вещества между несмеши-
вающимися растворителями?
2. Что называют коэффициентом распределения* константой рас-
пределения, степенью извлечения?
3. Какие особенности характеризуют экстракционные хелатные
системы?
4. Какие факторы влияют на скорость экстракции?
5. Какие химико-аналитические вопросы решаются с помощью
экстракции?
6. В чем сущность экстракционно-фотометрических определений? '
19*
291
глава 17
Хроматография
, ' ■' •. "V-"). '.
17.1. Абсорбция вещества —
основа хроматографии
1 Хроматографическии метод анализа разработан русским бо-
таником М. С. Цветом в 1903 г. В первых же работах с помощью
этого метода М. С. Цвет установил, что считавшийся однородным
зеленый пигмент растений:— хлорофилл — на самом деле состо-
им из нескольких веществ. При пропускании экстракта зеленого
листа через колонку, заполненную порошком мела, и промыва-
нии петролейным эфиром, он получил несколько окрашенных
зон* что с несомненностью говорило о наличии в экстракте не-
скольких веществ. Впоследствии это было подтверждено други-
ми исследователями. Этот метод он назвал хроматографией (от
грёч. хрдматос -—цвет), хотя сак же указал на возможность раз-
деления и бесцветных веществ/Работы М. С. Цвета довольно дол-
гое время оставались забытыми й не привлекали внимания, что в
известной степени было связано с отрицательной оценкой этих
работ, которую дали некоторые авторитеты того времени. Замет-
ное развитие хроматографических методов началось в 30-е годы
XX в., когда возникла острая потребность в новом методе разде-
ления смесей и очистке веществ, разлагающихся при нагрева-
нии. Хроматография продолжает бурно развиваться и в настоя-
щее время является одним из перспективных методов анализа.
О значимости хроматографии говорит тот факт, что за работы,
выполненные с применением хроматографических методов, было
црисуждено 14 Нобелевских премий.
Хроматографию можно определить как процесс, основанный
на многократном повторении актов сорбции и десорбции вещест-
ва при перемещении его в потоке подвижной фазы вдоль непо-
движного сорбента..
Сорбцией (от лат. — вогЬео — поглощаю) называют процесс
поглощения твердым телом или жидкостью (сорбентом) газооб-
разного или растворенного вещества (сорбата), обратный процесс
называют десорбцией. Сорбцию подразделяют на адсорбцию —
поглощение вещества (адсорбата) поверхностью твердого или
жидкого адсорбента и абсорбцию — поглощение вещества (абсор-
бата) поверхностью абсорбента.
292
Поглощение вещества сорбентом с об-
разованием химических соединений назы-
вают хемосорбцией (химической сорбцией).
Вещество подвижной фазы непрерыв-
но вступает в контакт с новыми участка-
ми сорбента и частично сорбируется, а со-
рбированное вещество контактирует со с
свежими порциями подвижной фазы и Рис 17.1.
частично десорбируется. Изотерма адсорбции
При постоянной температуре адсорб-
ция увеличивается с ростом концентрации раствора или давле-
ния газа. Зависимость количества поглощенного вещества от
концентрации раствора или давления газа при постоянной темпе-
ратуре называют изотермой адсорбции. Типичная изотерма ад-
сорбции приведена на рис 17.1 • Математически эта зависимость
может быть выражена уравнением Лэнгмюра
п = Поо-2£-, (17.1)
1 +Ъс
где п — количество адсорбированного вещества при равновесии;
/loo — максимальное количество вещества, которое может быть
адсорбировано на данном адсорбенте; Ъ — постоянная, с г— кон-
центрация.
По Лэнгмюру на поверхности твердого тела имеется некото-
рое число мест с минимальной энергией, расположенных через
определенные интервалы по' всей поверхности. Их число равно
Лоо. На этих местах могут адсорбироваться молекулы из раствора
или газа. В области небольших концентраций изотерма линейна.
Действительно, при Ъс <ЗС 1 знаменатель (17.1) становится равным
единице, и уравнение (17.1) переходит в
n^nJbc^Tc. (17.2)
Это уравнение линейной адсорбции. Оно соответствует урав-
нению Генри (Г — коэффициент Генри). Область линейной адсорб-
ции иногда называют также областью Генри. При высокой кон-
центрации Ъс >> 1 и уравнение (17.2) принимает вид п = п^, что
соответствует так называемому насыщению: изотерма адсорбции
выходит практически на прямую, параллельную оси абсцисс.
Однако известны случаи^ когда зависимость количества ад-
сорбированного вещества от концентрации раствора или давле-
ния газа существенно отличается от изображенной на рис 17.1.
Изотерма адсорбции может быть, например, вогнутой или
S-образной. Это может быть вызвано образованием на поверхно-
сти адсорбента не moho-, а полимолекулярного слоя, что не пред-
293
усматривается теорией Лэнгмюра, а также тем* что поверхность
реальных твердых тел неоднородна, и другими причинами,; Не^,
смотря на некоторые существенные ограничения, применимость
уравнений (17.1) и (17.2) в теории хррщгографических процес-
сов остается довольно широкой* но
При адсорбции двух или нескольких веществ уравнение
(1$1) для 1-го компонента принимает вид
1 +IV*
• ¿-1 •
Следует отметить также, что количество адсорбированного ве-
щества будет определяться не только егр концентрацией, но и срод-
ством к адсорбенту. При адсорбции нескольких веществ проявле-
ние сродства особенно заметно, так как возможно вытеснение одних
сорбированных веществ другими, обладающими большим сродст-
вой, хотя имеющими, может быть, и меньшую концентрацию.
17.2. Классификация методов хроматографии
Различные методы хроматографии можно классифицировать
по агрегатному состоянию фаз, способу их относительного пере-
мещения, аппаратурному оформлению процесса и т. д. По агре-
гатному состоянию фаз хроматографические методы обычно
классифицируют следующим образом (табл. 17.1).
Таблица 17.1.* Классификация хроматографических методов
по агрегатному состоянию фаз
Неподвижная
Подвижная фаза
фаза
газообразная '
жидкая \
Твердая
Газовая адсорбционная
хроматография
Жидкостная адсорбцион-
ная, ионообменная, ион-
ная , тонкослойная, оса-
дочная хроматографа
Жидкая
Газожидкостная рас-
пределительная хрома-
тография, капилляр-
ная
Жидкостная распредели-
тельная, высокоэффектив-
ная жидкостная, гельхро-
матография ■
* Таблица имеет иллюстративный характер, и некоторые виды хро-
матографии в нее не включены.
294
^ ^По способу относительного перемещения фаз. различают
фронтальную, проявительную, или элюэнтную, и вытеснитель-
ну^ Хроматографию.
Фронтальный метод: Это простейший по методике вариант
хроматографии. Он состоит в том, что череаЗколонку с адсорбен-
том непрерывно пропус1кают анализируемую смесь, например,
компонентов А и В в растворителе 8о1у. В растворе, вытекающем
из колонки, определяют концентрацию каждого комйонента и
строят график в координатах концентрация вещества—объем
раствора, прошедшего через колонку. Эту зависимость обычно и
называют хроматограммой или выходной кривой (рис. 17,2).
Вследствие сорбции веществ А и В сначала из колонки бу^ет
вытекать растворитель ЭоЬг, затем растворитель и,менее сорби-
рующийся компонент А, а затем и компонент В и, таким обра-
зом, через некоторое время состав раствора при прохождении ^е-:
рез колонку меняться не будет. Фронтальный метод исцользует-
ся сравнительно редко. Он применяется, например, для очистки
раствора от примесей, если они,сорбируются существенцо лучше,
чем основной компонент, или для выявления из смеси наиболее
слабо сорбирующегося вещества.
Проявительный (элюэнтный) метод. При работе по этому ме^
тоду в колонку вводят анализируемую смесь, содержащую ком-
поненты А и В в виде порции раствора или газа и колонку непре-
рывно промывают газом-носителем или растворителем во1у. При
этом компоненты анализируемой смеси разделяются на зоны: хо-
рошо сорбирующееся вещество В занимает верхнюю часть колон-
ки, а менее сорбирующийся компонент А будет занимать ниж-
нюю часть. Типичная выходная кривая изображена на рис. 17.3.
В газе или растворе, вытекающем из колонки, аначала появ-
ляется компонент А, далее — чистый растворитель, а затем ком-
с
%РЙс. 17.2. Выходная кривая Рис. 17.3. Выходная кривая
фронтального анализа проявительного анализа
295
понент В. Чем больше концентрация компонента, тем выше пик
и больше его площадь, что составляет основу количественного
хррматографического анализа. Проявительный метод дает воз-
можность разделять сложные смеси, он наиболее часто применя-
ется в практике. Недостатком метода является уменьшение кон-
центрации выходящих растворов за счет разбавления раствори-
телем (газом-носителем).
Вытеснительный метод. В этом методе анализируемую смесь
компонентов А и В в растворителе Эо1у вводят в колонку и про-
мывают раствором вещества Б (вытеснитель), которое сорбирует-
ся лучше, чем любой из компонентов анализируемой смеси.
; Концентрация раствора при хроматографировании не умень-
шается в отличие от проявительного метода. Существенным не-
достатком вытеснительного метода является частое наложение
зоны одного вещества на зону другого, поскольку зоны компонен-
тов в этом методе не разделены зоной растворителя.
17.3. Хроматографический пик
и элюционные характеристики
В хроматографии чаще всего используют методику прояви-
тельного (элюэнтного) анализа, при которой газ или раствор,
выходящий из колонки, анализируется непрерывно. Типичная
выходная кривая (хроматограмма) проявительного анализа при-
ведена на рис. 17.3. Рассмотрим ее более подробно (рис. 17.4).
Если точка А' соответствует вводу анализируемой пробы,
А—- появлению на выходе какого-то несорбирующегося компо-
нента, а В — появлению анализируемого вещества, то линию
А'АВ и ее продолжение ВЕ называют нулевой линией. Кривую
ВЭ¥ называют хроматографическим пиком и характеризуют вы-
сотой, шириной и площадью. С удов-
летворительной точностью контур пи-
ка опцсывается уравнением Гаусса:
(У-У0)2
" 2ц?т
(17.3)
Рис. 17.4. Кривая
проявительного анализа
(хроматографический пик)
где V —-объем подвижной фазы; У0 —
объем подвижной фазы, соответст-
вующий стах; рСТ — стандартное от-
клонение, равное полуширине пика
при = .'. ■■■-^г^::^^^
296
с
"Действительно, при условии = е1/2 из уравнения (17.3)
Следует, ЧТО 72 в (Г2ц10) ИЛИ Цст = - ^0>2 И 1^ст = У " ^0> Т- е-
цри указанном условии ^ равна полуширине пика.
Высотой пика считает либо величину Л, либо Л' (см. рис.
17.4). Последняя равна расстоянию от нулевой линии до точки
пересечения касательных к кривой в точках перегиба. Шириной
пика называют расстояние между точками контура на половине
его высоты (СЕ = Ц0,б) или на какой-то другой отметке по высоте,
либо расстояние между точками перегиба (\хп) или между точка-
ми пересечения нулевой линии с касательными к кривой в точ-
ках перегиба (ВТ' = цк = ю). Соотношения между этими величи-
нами хорошо известны:
ц0,5 - 2,355цст; цп = 0,850ц0>5 = 2цст;
цк-1,700ц0>б-ш-4цст. (17.4)
Связь, например, цст с ц0>5 можно установить следующим об-
разом. По определению ц0>5 соответствует расстоянию между точ-
с
ками контура при -Н^ = 2. Тогда из уравнения (17.3) получаем
(у _ у \2
^ = 1п 2, где У1/2 абсцисса точки контура при половин-
ном значении стл„.
Таким образом, У1/2 ~ Уо = цстл/21п2. Но по определению
Ио,б = 2(^/2" ^о)и? следовательно, ц0>5 = цст2л/21п2 = 2,335цст.
Важной хроматографической характеристикой системы явля-
ется время удерживания или пропорциональный ему удерживае-
мый объем. На рис. 17Л приведенному удерживаемому объему со-
ответствует отрезок Ав, а общий удерживаемый объем характери-
зуется отрезком А!С
Если длину отрезка А'С обозначить /, то время удерживания гг
будет равно
гг = 1/и> (17.5)
где II.— скорость движения ленты самописца.
Удерживаемый объем Уг пропорционален времени удержива-
ния ^г:
Уг = *&1 (17.6)
где со — объемная скорость газа-носителя.
297
v Приведенный удерживаемый объем У'г, соответствующий от-
резку Лб, определяется соотношением
у г уг у0> : { .
где У0 пропорционален отрезку АА', длина которого /0. !
Величина У0 характеризует удерживаемый объем несорби-
рующегрс^Е газа, или мертвый объем колонки.
Приведенному удерживаемому объему соответствует приве-
денноевремя удерживания ^:
*г ~ *г "" *0>
где пропорциональное величине /0, характеризует время удер-
живания несорбирующегося таза:.
Полнота разделения двух компонентов количественно может
быть выражена критерием разделения К:
АУ
, ц0,б(1) + Мо,5(2) Ш>б,0,5(1) + Иоб(1),0,б(2)
(17.7)
где Д/ или ДТ^. — расстояние между максимумами пиков разде-
ляемых элементов ;ц0 5 — полуширина хроматографического пи-
ка первого (1) и второго (2) компонентов на половине высоты, а
нижний индекс «об» указывает на объемные единицы измере-
ния.
При1Г = 1 разделение бывает достаточно полным.
Если допустить, что ширина хроматографического пика обо-
их компонентов примерно одинакова, т. е. рг «V ц2, уравнение
(17.7) принимает вид
, . :- ■•; АК , ■■'■-V;..
' •■ ' 2цоб,0>б v
При взаимном перекрывании пиков определение ширины зр-
ны каждого пика становится невозможным (рис. 17.5). В таких
случаях рассматривают степень разделе
ния Ч?:
' ^ = (^-Лтіп)/^ (17.8)
где А2 — высота пика вещества, имеюще-
го меньшую концентрацию; — высо-
та минимума. ^
Значение тех или иных элюцион-
ных характеристик меняется в зависи-
і Рис. 17.5. Определение мости от цели анализа. В качественном
ртепени разделения ¥ анализе основное внимание уделяется
298
определению характеристик удерживания и устранению искаже-
ний этих величин за счет второго компонента. В количественном
анализе важно, чтобы четкость разделения обеспечивала доста-
точную точность определения площади или высоты.хроматогра-
фического пика. нк
17,4. Теоретические основы хроматографии
Известно несколько теорий хроматографического процесса.
Существенное значение имеют метод теоретических тарелок и
кинетическая теория.
В методе теоретических тарелок Мартина и Синджа хрома-
тографическая колонка мысленно делится на ряд элементарных
участков— «тарелок» и предполагается, что на каждой тарелке
очень быстро устанавливается равновесие между сорбентом- и
подвижной фазой. Каждая новая порция газа-носителя вызывает
смещение этого равновесия, вследствие чего часть вещества пере-
носится на следующую тарелку, на которой, в свою очередь, уста-
навливается новое равновесное распределение и происходит переч
нрс вегцества на последующую тарелку. В результате ; этих
процессов хроматографируемое^ вещество распределяется на не-
скольких тарелках, причем на средних тарелках его концентра-
ция оказывается максимальной по сравнению с соседними тарел-
ками. Распределение вещества вдоль слоя сорбента подчиняется
уравнению
" (*-*0)2 "
с = стахе~ «* , (17,9)
где х — расстояние от начала колонки до точки, в которой кон-
центрация равна с; х0 — координата центра полосы; Н — высота,
эквивалентная теоретической тарелке (ВЭТТ); / — длина слоя со-
рбента; на которой произведено.поглощение'и п теоре-
тических тарелок, при этом
ь п±1/Н. (17.10)
Если числитель дробного показателя степени в уравнениях
(17.3) и (17.9) выразить в одних и тех же единицах, то при срав-
нении этих уравнений получим
$1 —'• '■'
Число теоретических тарелок будет равно
ц :2 (i7.li)
Нет
299
или, учитывая уравнение (17.4),
л--5,5б(^)2-1б(±У. (17.12)
Эффективность колонки тем выше, чем меньше высота, экви-
валентная теоретической тарелке, и больше число теоретических
тарелок.
Таким образом, теория тарелок позволяет рассчитать-важ-
ные количественные характеристики хроматографического про-,
цесса. Однако теория тарелок, основанная на допущении ступен-
чатого характера хроматографического процесса, по существу
формальна, так как реальный процесс протекает непрерывно.
Значение высоты, эквивалентной теоретической тарелке, и число
тарелок являются характеристиками размытости зон. Эти вели-
чины сохраняют свое значение и в кинетической теории хрома-
тографии, учитывающей скорость миграции вещества, диффу-
зию и другие факторы.
Кинетическая теория хроматографии основное внимание
уделяет кинетике процесса, связывая высоту, эквивалентную те-
оретической тарелке, с процессами диффузии, медленным уста-
новлением равновесия и неравномерностью процесса. Высота, эк-
вивалентная теоретической тарелке, связана со скоростью потока
уравнением Ван-Деемтера:
■Н=А + £ + Си,' ' (17.13)
где А, В и С — константы; и — скорость подвижной фазы.
Константа А связана с действием вихревой диффузии, кото-
рая зависит от размера частиц и плотности заполнения колонки,
величина В связана с коэффициентом диффузии молекул в под-
вижной фазе, это слагаемое учитывает действие продольной диф-
фузии, а С характеризует кинетику процесса сорбция-десорбция,
массопередачу и другие эффекты. Влияние каждого слагаемого
уравнения (17.13) на величину Н в за-
висимости от скорости подвижной фа-
зы показано на рис. 17.6. Первое сла-
гаемое дает постоянный вклад в Н.
*^~си Вклад второго слагаемого существен
при небольшой скорости потока. С уве-
личением скорости подвижной фазы
влияние третьего слагаемого возраста-
ет, а доля второго уменьшается. Сум-
Рис. 17.6. Зависимость ВЭТТ марная кривая, характеризующая за-
от скорости подвижной фазы висимость Н от скорости потока, пред-
300
ставляет собой гиперболу. При небольшой скорости потока высота,
эквивалентная теоретической тарелке, уменьшается, а затем начи-
нает возрастать. Поскольку эффективность колонки тем выше, чем
меньше высота, эквивалентная теоретической тарелке, оптималь-.
ная скорость подвижной фазы будет равна Скорости, соответствую-
щей точке минимума этой кривой. Чтобы найти эту точку, продиф-
ференцируем уравнение (17.13) и производную приравняем нулю:
откуда
Подставляя эту величину в уравнение (17.13), находим опти-
мальную высоту, эквивалентную теоретической тарелке:
Нопт=А+2«/ВС. (17:14)
Таким образом, динамическая теория дает основу для опти-
мизации хроматографического процесса. ,
17.5. Основные узлы приборов
для хроматографического анализа
Отечественная промышленность и зарубежные фирмы вы-
пускают большое число хроматографов самых различных типов.
Однако сложные хроматографическиё установки требуются не
всегда. Для проведения хроматографического разделения мето-
дами бумажной, тонкослойной и некоторыми другимц видами
хроматографии используются простые установки, которые могут
быть собраны в лхобой химической лаборатории. Независимо от
сложности устройства основными узлами хроматографической
установки являются дозатор (система ввода пробы), хрЬматогра-
фическая колонка и детектор. Кроме того, в установке имеются
устройства для подачи газа-носителя или растворителя, для пре-
образования импульса детектора в соответствующий сигнал и не-
которые другие.
Дозатор предназначен для точного количественного отбора
пробы и введения ее в хроматографическую колонку. Одним из
основных требований к дозатору является воспроизводимость
размера пробы и постоянство условий ее введения в колонку.
Кроме того, введение пробы не должно вызывать резкого измене-
ния условий работы колонки и других узлов хроматогр>афиче-
ской установки, а внутренняя поверхность дозатора не должна.
301
обладать каталитической или адсорбционной активностью по от-
ношению к пробе.
Газообразные и жидкие пробы обычно вводят с помощью спе-
циальных шприцев, прокалывая в месте ввода пробы каучуко-
вую мембрану. Применяются газовые шприцы для газообразных
проб и микрошприцы для жидких. Микрошприцы позволяют
вводить в хроматограф пробы объемов от долей до десятков мик-
ролитров. Нередко в лабораторной практике в качестве дозатора
применяется медицинский шприц.
Твердые пробы вводятся в хроматограф или после перевода
их в раствор, или непосредственным испарением пробы в нагре-
том дозаторе, куда она вводится с помощью игольного ушка. Из-
вестны и другие устройства.
В хроматографической колонке происходит разделение ком-
понентов. Колонки весьма различны по форме, размерам и конст-
рукционным материалам. Применяются прямые, спиральные и
другие колонки длиной от 1—2 м и менее до нескольких десятков
метров. Внутренний диаметр колонок составляет обычно не-
сколько миллиметров. В зависимости от свойств анализируемой ,
системы в качестве конструкционных материалов для колонок
чаще всего используют сталь, латунь, медь, стекло и др. Матери-
ал колонки должен обладать определенной химической инертно-
стйю по отношению к компонентам пробы, например медные ко-
лонки будут непригодны при разделении ацетиленсодержащих
сйесей.
Адсорбент, наполняющий колонку, должен обладать рядом
свойств: необходимой селективностью, достаточной механиче-
ской прочностью, химической инертностью к компонентам смеси
й быть доступным. Практически в качестве адсорбентов исполь-
зуются оксид алюминия, силикагели, активированные угли, по-
ристые полимеры на основе стирола, дивинилбензола и т. д. и
синтетические цеолиты. Широко используют модифицирован-
ные адсорбенты > которые получают обработкой исходных адсор-
бентов растворами кислот, щелочей, неорганических солей и т. д.
Выбор адсорбента зависит от агрегатного состояния фаз, методи-
ки хроматографирования и других факторов.
Большое влияние на сорбируемость вещества оказывает тем-
пература, поэтому хроматографические колонки, как правило,
термостатируются, используя обогрев жидкостью или парами кц-
пящёй жидкости, воздушное термостатирование или какой-либо
другой прием. Обычно термостатирование производится при тем-
пературах, значительно превышающих комнатные, однако в т~
которых случаях создаются температуры ниже 0 бС (при разде-
302 .'■ .":
лении низкокипящих газов). В бумажной, тонкослойной и неко-
торых других видах хроматографии функцию колонки выполняет
хроматографическая бумага, тонкий слой сорбента на подложке
ит. д. ,>•:/:■...'••-'
Детектор предназначен для обнаружения изменений в соста-
ве газа или раствора, прошедшего через колонку. Показания де-
тектора обычно преобразуются в электрический сигнал и переда-
ются фиксйруйЙ^еМ^ или записывающему прибору, например на
ленту электр6йЙо1,о потенциометра. Основными характеристика-
ми детектора являются чувствительность, пределы детектирова-
ния, инерционность и диапазон линейной зависимости между
концентрацией и величиной сигнала. Детекторы подразделяются
на дифференциальные, которые отражают мгновенное изменение
концентрации, и интегральные, суммирующие изменение кон-
центрации за некоторый отрезок времени.
В интегральных детекторах анализируемый газ на выходе из
колонки поглощается каким-либо раствором, а затем анализиру-
ется; или поглощающий раствор или оставшийся непоглощенный
газ. Есди носителем является диоксид углерода, то после колонки
газ барботируется через раствор щелочи и измеряется объем газа,
не поглощенного этой жидкостью. Достоинствами интегральных
детектрров являются их простота и широкая область линейной за-
висимости, показаний детектора от количества вещества. К недос-
таткам относятся значительная инерционность и низкая чувстви-
тельность, в связи с чем такие детекторы в настоящее время при-
меняются редко.
К группе дифференциальных относятся детекторы по тепло-
проводности (катарометр), по плотности, по электрической прово-
димости, пламенный, пламенно-ионизационный (ПИД) и другие
ионизационные детекторы, термохимический, пламенно-фотомет-
рический цт. д. Детектор выбирают в зависимости от свойств изу-
чаемой ристемы, агрегатного достояния фаз и других особенностей.
17.6. Газовая хроматография
Подвижной фазой в газовой хроматографии является газ или
пар. В зависимости от состояния неподвижной фазы газовая хро-
матография подразделяется на газоадсорбционную, когда непо-
движной фазой является твердый адсорбент, и газожидкостную,
когда неподвижной фазой является жидкость, а точнее пленка
жидкости на поверхности частиц твердого сорбента.
Как уже отмечалось, в качестве дозаторов в газовой хрома-
тографии используют шприцы и микрошприцы.
303
17.6.1. Хроматографические колонки и детекторы
Колонки. В газоадсорбционной хроматографии колонки за-
полняются твердым сорбентом. Адсорбция газа на твердом со-
рбенте подчиняется уравнению изотермы адсорбции (17.1). Стро-
ение и свойства сорбентов весьма многообразны. По классифика- -
ции Киселева к первому типу относятся неспецифические
сорбенты, не имеющие на поверхности^ каких-либо функциональ-
ных групп (например, угли), ко второму типу принадлежат со-,
рбенты, имеющие на поверхности заряды (например, гидроксид-
ные группы силикагеля), и к третьему — адсорбенты, имеющие
на поверхности связи или группы атомов с сосредоточенной элек-
тронной плотностью (например, полимеры с нитрильными груп-
пами).
Активированные угли неполярны. Их высокая удельная по-
верхность (1 ООО-—1700 м2/г) обусловливает большую силу взаи-
модействия с анализируемым веществом, что ограничивает об-
ласть применение углей анализом легких газов. ' ,
Адсорбционная активность силикагеля в основном связана с
поверхностными ОН-группами. Это полярный адсорбент. Поляр-
ным адсорбентом является также оксид алюминия.
Цеолиты часто рассматривают как молекулярные сита: при-
родными л и синтетические. Своё название молекулярных*сит они
получили потому, что их поры имеют размеры, близкие к разме-
рам молекул (от 0,4 до 1,0 нм в зависимости от марки), и адсорб-
ция на них является своеобразным «просеиванием». Действи-
тельно, на молекулярных ситах могут сорбироваться только те
вещества, молекулы которых могут проникнуть в поры. Напри-
мер, на молекулярных ситах с малыми размерами пор могут со- ^
рбироваться только легкие вещества, размеры молекуд которых
невелики, а тяжелые вещества, у которых размеры модекул пре-
вышают размеры пор, естественно, адсорбироваться не могут. На
обычных адсорбентах* как известно, более эффективно адсорби-
руются тяжелые вещества, поэтому предпочтительная адсорбция
легких веществ на цеолитах сначала казалась непонятной и по- '
лучила объяснение лишь после раскрытия роли размера пор в
процессе поглощения цеолитом. В качестве молекулярных сит
используются также пористые стекла. Существенными достоин-
ствами обладают модифицированные адсорбенты: у них в более
широкой области концентраций наблюдается линейная изотерма
адсорбции, теряется каталитическая активность и т. д. -:\
Возможности хроматографического определения вещества в
газовой фазе значительно возросли с открытием в 1952 г. метода
304
газожидкостной хроматографии. При анализе по этому методу
анализируемая газовая смесь проходит через колонку, наполнен-
ную твёрдым носителем, на поверхность которого нанесен тон-
кий слой жидкой фазы. Таким образом, с компонентами пробы
здесь взаимодействует уже вещество жидкой пленки, хотя в ре-
альных условиях газожидкостной хроматографии компоненты
смеси частично взаимодействуют и с твердым адсорбентом.
Появление жидкой пленки привело к изменению природы
физико-химических процессов в хроматографической колонке.
Вместо процесса адсорбции газа на твёрдом адсорбенте в колонке
стал происходить процесс растворения таза в тонкой пленке, на-
ходящейся на твердом носителе. Эффективность разделения ста-
ла определяться не процессами адсорбции-десорбции газа, как
это было в адсорбционной газовой хроматографии, а процессами
растворения газа в жидкой пленке и его выделения. Различней
растворимости газов оказалось более существенным, чем разли-
чие в их абсорбционных свойствах, поэтому газожидкостная хро-
матография открыла более широкие возможности в разделении и
анализе многокомпонентных смесей. Очень важным преимуще-
ством газожидкостной хроматографии является возможность ра-
боты в области линейной изотермы в более широкой области кон-
центраций, чем в газовой адсорбционной хроматографии, что
обеспечивает получение практически симметричных хроматогра-
фических пиков.
Эффективность разделения в газожидкостной хроматограф
фии зависит главным образом от правильности выбора жидкой
фазы. Строго обоснованных теоретически способов выбора жид-
кой фазы не существует в связи со сложностью протекающих
процессов и недостаточной разработанностью теории растворов.
Однако требования к жидкой фазе предъявляются совершенно
определенные. Жидкая фаза должна обладать достаточно высо-
кой селективностью, т. е. способностью разделять смеси компо-
нентов, быть химически инертной по отношению к компонентам
смеси и к твердому носителю, оставаться термически устойчи-
вой, не растворять газ-носитель, иметь з^алую вязкость и быть не-
летучей (или иметь летучесть незначительную).
При выборе жидкой фазы полезным оказалось старое прави-
ло — «подобное растворяется в подобном». В соответствии с этим
правилом для разделения смеси двух веществ выбирают жидкую
фазу, близкую по химической природе одному из компонентов.
Ограниченность такого подхода для веществ с близкими свойст-
вами или смесями сложного состава очевидна.
20 -7831
305
Накопленный экспериментальный материал по хроматогра-
фированию различных систем и установленные закономерности
часто дают возможность предвидеть поведение отдельных компо-
нентов в ходе хроматографирования и обоснованно выбирать оп-
тимальные условия разделения сложных смесей. <
Эффективным оказалось применение колонок, содержащих
несколько неподвижных фаз или сложные сорбенты. В состав-
ных колонках анализируемую смесь последовательно пропуска-
ют через несколько колонок с разными жидкими фазами. Колон-
ка , со смешанным сорбентом содержит равномерно перемешан-
ный сорбент с неодинаковыми жидкими фазами. При нанесении
на один твёрдый носитель смеси несколько неподвижных фаз по-
лучают колонки со смешанной фазой.
В качестве жидкой фазы в газожидкостной хроматографии
используют вазелиновое масло, силиконовое масло, фталаты (ди-
бутил, дйоктил и др.)* дйметилформамид, трикрезилфосфат и др.
Специфические особенности проявляют жидкостные кристаллы,
например а^кёйэфиры типа
Нематические жидкие кристаллы, взятые в качестве жидкой'
фазы, проявляют селективное сродство к линейным молекулам,
по-видимому, в связи с тем, что в нематической фазе молекулы
могут перемещаться только в параллельных плоскостях. Количе-
ство жидкой фазы зависит от свойств системы и составляет от 1
до 30—50% от массы твердого носителя. Пленка жидкости быва-
ет очень тонкой, поэтому внешний вид носителя с пленкой обыч-
но остается таким же, каким он был у носителя без пленки. *
;. В качестве твердых носителей применяют инертные вещест-
ва с развитой поверхностью, но малой микропористостью, чтобы
по возможности исключить адсорбцию газа на поверхности. На-
ибольшее распространение полумили носители на основе кизель-
гура или диатомита. Применяют также стеклянные микрошарит
ки. Для разделения реакционноспособных веществ используется
ТефЛОН.;.: '•<:
Существенно повышается эффективность разделения в кат
пиллярной хроматографии. Название метода связано с тем, чтр в
качестве хроматографической колодки используется капилляр с
внутренним диаметром 0,1—0,5 мм и длиной в несколько десят*
ков метров. Жидкая фаза наносится непосредственно на стенку
этбго капилляра, которая в данном случае играет родь носителя.;
зов . . :^
В капиллярной колонке существенно уменьшается сопротивле-
ние потоку газа, поэтому появляется возможность увеличить
длину колонки и повысить таким образом эффективность разде-
ления. Значительно уменьшается в капиллярной хроматографии
объем пробы для анализа и сокращается длительность анализа:
метод становится близким к экспрессному.
Проба для анализа в капиллярной хроматографии уменьша-
ется в 1000 раз и больше по сравнению с обычной методикой на
насадочных колонках, что вызывает определенные эксперимен-
тальные трудности и требует разработки специальной методики.
Нередко приходится более 99,9% вводимой пробы выпускать в
атмосферу через специальное ответвление и лишь 0,01—0,05%
взятого количества направлять в колонку. Детектирование столь
малых количеств требует применения высокоэффективных де-
текторов, например, типа пламенно-ионизационного.
Внутреннюю поверхность капилляра можно покрывать не
только неподвижной жидкостью, но и тонким слоем твердого ве-
щества. Этот вариант называют твердослойной капиллярной
хроматографией.
Основными достоинствами капиллярной хроматографии яв-
ляются высокая эффективность при анализе многокомпонент-
ных смесей, особенно с применением кварцевых капилляров, вы-
сокая чувствительность и малая длительность анализа. Недостат-
ки связаны, главным образом, с трудностями дозировки и
детектирования, а также с недостаточной, селективностью слабо
сорбирующихся веществ в условиях капиллярной хроматогра-
фии.
Детекторы. Одним из наиболее распространенных дифферен-
циальных детекторов является катарометр. Принцип его работы
основан на измерении сопротивления нагретой платиновой или
вольфрамовой нити, которое зависит от теплопроводности омы-
вающего газа. Количество теплоты, отводимое от нагретой нити
при постоянных условиях, зависит от состава газа. Чем больше
теплопроводности определяемых компонентов смеси будет отли-
чаться от теплоцроводности газа-носителя, тем большей чувстви-
тельностью будет обладать катарометр. Наиболее подходящим
газом-носителем с этой точки зрения является водород, тепло-
проводность которого значительно превышает соответствующую
характеристику большинства других газов. Однако в целях тех-
ники безопасности чаще применяется гелий, теплопроводность
которого тав;же достаточно высока. В последнее время металли-
ческие нити в катарометре успешно заменяются, термисгорами,
имеющими боле^высОкий, чем у металлов, температурный коэф-
20* 307
фициент электрической проводимости. Достоинствами катаро-
метра являются простота, достаточная точность и надежность в
работе. Однако из-за сравнительно невысокой чувствительности
он не применяется для определения микропримесей.
В основе работы термохимического детектора лежит измере-
ние сопротивления платиновой проволоки, которое меняется?
вследствие изменения температуры при сгорании горючих газов.
Газы на выходе из хроматографической колонки каталитически
сжигаются на нагретой платиновой проволоке, которая одновре-
менно является плечевым сопротивлением мостовой схемы. Чув-
ствительность термохимического детектора выше, чем катаро-
метра; однако платиновая нить требует частой калибровки и за-
мены. Применимость термохимического детектора ограничена
горючими веществами.
Принцип действия пламенного детектора основан на том, что
температура водородного пламени горелки изменяется при попада-
нии в него органических веществ. Наибольшей чувствительностью
обладают ионизационные детекторы, например пламенно-иониза-
ционный (ПИД), позволяющие обнаруживать до 10~12 г. В этих де-
текторах измеряют электрическую проводимость пламени водо-
родной горелки. Чисто водородное пламя обладает очень низкой
электрической проводимостью. При появлении в водороде приме-
сей органических соединений происходит ионизация пламени,
пропорциональная концентрации примеси, что легко может быть
измерено. Высокая чувствительность детекторов этого типа обус-
ловила их широкое применение. Однако высокая чувствитель-
ность ПИД проявляется по отношению только к органическим со-
единениям, а к неорганическим, таким, как аммиак, сероводород,
оксиды серы, кислород, азот и т. д., чувствительность детектора
резко падает.
Очень высокой чувствительностью обладает аргонный детек-
тор, ионизация в котором происходит при столкновении молекул
определяемого вещества с метастабильными атомами аргона, об-
разующимися под действием радиоактивного Р-излучения.
В термоионном детекторе в пламя горелки вводят соли ще-
лочных металлов. При попадании в такое пламя соединений фос-
фора появляется ионный ток, пропорциональный содержанию,
атомов фосфора. Это селективный фосфорный детектор высокой
чувствительности.
Известны и другие типы детекторов: ультразвуковые^ ди$-
фрагменные, микрокулонометрические и т. д. Для проведение
количественных расчетов детектор обычно градуируется.
■ ■ ; .':\ . . зов."
17.6.2. Качественный анализ
Качественный состав вещества может быть установлен с по-
мощью хроматографической методики по характеристикам полу-
ченной хроматограммы или по результатам анализа компонентов
смеси после прохождения хроматографической колонки подходя-
щим химическим или физикогхимическим методом. Типичная
хроматограмма приведена на рис. 17.7. Как видно, хроматографи-
ческое разделение смеси из семи компонентов цроведено вполне
успешно. Каждому компоненту смеси отвечает свой пик и после-
довательность появления пиков на хроматограмме закономерна:
она соответствует последовательности кислот в гомологическом
ряду. Собственно хроматографичёский качественный анализ ос-
нован на использовании характеристик удерживания —. времени
удерживания или пропорционального ему удерживаемого объема
и индексов удерживания. Для этой цели применяются относи-
тельные удерживаемые объемы [см. формулу (17.6)], которые в
значительно меньшей степени, чем абсолютные величины, под-
вержены действию случайных факторов. Идентификация иссле-
дуемых веществ проводится сравнением полученных и табличных
данных. Условия опыта, естественно, не должны отличаться от
тех, в которых были получены табличные данные. Идентифика-
ция вещества по его хроматограмме может быть выполнена также
методом тестеров, когда сравнивают объем или время удержива-
ния компонента анализируемой смеси и эталона, найденные в од-
них и тех же условиях опыта. Иногда эталонное вещество, нали-
чие которого предполагается в анали-
зируемой смеси, специально вводят в
пробу и сравнивают высоту и площадь
пиков на хроматограммах, получен-
ных до и после введения эталона. Уве-
личение 'высоты или площади пика
рассматривается как указание на при-
сутствие предполагаемого компонента
в пробе. Однако этот метод не вполне
надежен, так как удерживаемые объ-
емы довольно многих веществ близки
между собой. Трудности идентифика-
ции преодолеваются, например, хро-
матографированием пробы на колон-
ках с разными сорбентами. Получение
одинаковых результатов при исполь-
зовании разных сорбентов увеличива-
ет надежность анализа. В настоящее
Рис 17.7. Хроматограммы
смеси воды и кислот (115 °С):
1 — вода;
2 — муравьиная кислота;
3 — уксусная кислота;
4 — пропионовая кислота;
5 — изомасляная кислота;
6 — «-масляная кислота;
7 — изовалериановая кислота
309
время разработаны и успешно применяются также различные
многоступенчатые схемы. В таких схемах после разделения смеси
на первой колонке полученные фракции подаются на колонки
второй ступени, где достигается более тонкое разделение и более
точная идентификация вещества, так как имеют дело с более
простыми смесями, чем на первой колонке.
В тазожидкостной хроматографии для качественного анали-
за часто используют индексы удерживания Ковача I:
где —приведенное время удерживания; п — число атомов уг-
лерода в алкане; г -—определяемое вещество.
Стандартом при определении индекса удерживания являют-
ся два соседних нормальных алкана (насыщенных углеводорода),
один из которых элюируется до, а второй после исследуемого со-
единения, т. е. <^г1< tr (Л + !)• При программировании темпе-
ратуры индекс удерживания рассчитывается по температуре
удерживания Тг:
Л г(п + 1) 1 г,п
Идентификация вещества по индексу удерживания произвол
дйтся путем хроматографирования соединения с последующим
хроматографированием в тех же условиях двух соседних алка-
нов, выбранных в качестве стандарта. Результаты анализа по ин-
дексу удерживания оказываются более надежными, чем по удер-|
живаемому объему, так как индекс удерживания является более\
индивидуальной характеристикой вещества. Индексы удержива-
ния многих веществ при определенных температурах имеются в
соответствующих справочных таблицах, что облегчает проведе-%
ние качественного анализа.
Накопленный экспериментальный материал позволил обна^
руйсить некоторые закономерности и установить определенные
зависимости между хроматографическими характеристиками й ;
физико-химическими свойствами вещества. Оказалось, напри-
мер, что индексы удерживания и удерживаемые объемы связаны
простой зависимостью с числом атомов углерода у членов гомоло-
гического ряда, с температурами кипения гомологов и др^и^и
характеристиками и свойствами вещества. Эти зависимости^
существенно расширяют возможности хроматографии. Соответ^
ствующие графики* например* зависимости удерживаемого рбъ-1
310
ема от температуры кипения почти линейны и их широко ис-
пользуют для идентификации компонентов смеси. Если прина-
длежность компонента к гомологическому ряду известна, то
найденная по такому графику температура кипения или другое
свойство достаточны для идентификации компонента. Установ-
лено, что индексы удерживания соседних членов в любом гомо-
логическом ряду различаются примерно на 100. Отмечается, что
Д/ изомеров равна пятикратной разнице их температур кипения,
т. е. Д/ = 5ДГКИП. Известны и другие закономерности в индексах
удерживания.
- Эффективным оказалось применение независимой аналити-
ческой идентификации продуктов хроматографического разделе-
ния и сочетание газовой хроматографии с другими методами ис-
следования: ИК-спектроскопией и масс-спектрометрией, а также
использование селективных и последовательно работающих де-
текторов. Методом масс-спектрометрии можно проводить непре-
рывный качественный анализ компонентов смеси и для анализа
бывает достаточно самых небольших количеств вещества; Такой
комбинированный метод получил название хроматомасс-спект-
рометрии. Возможно использование также методов ядерного
магнитного резонанса, пламенной фотометрии, абсорбционной
спектроскопии и других, включая химические методы.
17.6.3. Количественный анализ
Количественный хроматографический анализ основан на из-
мерении различных параметров пика, зависящих от концентра-
ции хроматографируемых веществ — высоты, ширины, площади
и удерживаемого объема — илц произведения удерживаемого
объема на высоту пика. При достаточной стабильности условий
хроматографирования и детектирования определяющим пара-
метром пика можно считать его высоту. Расчет по площади пика
позволяет несколько снизить требования к стабильности условий
хроматографирования по сравнению с расчетом по высоте пика,
однаьсО самр измерение площади вызывает появление новых ис-
точников ошибок. В случае узких пиков некоторые преимущест-
ва имеет измерение произведения удерживаемого объема на вы-
соту пика. При неполном разделении пиков ошибки возрастают
из-за наложения и искажения контуров пика. При работе с таки-
ми хррматограммами используют специальные приемы, опираю-
щиеся, главным образом, на измерение высоты пиков.
■ ■ 311 .
Основными в количественной хроматографии являются ме-
тоды: простой нормировки, нормировки с калибровочными коэф->
фициентами, внутренней стандартизации и абсолютной калиб^
ровки. - •
При использовании метода простой нормировкипринимав
ют сумму каких-либо параметров пиков, например сумму высот
всех пиков или сумму их площадей, за 100%. Тогда отношение
высоты отдельного пика к сумме высот или отношение площади
одного пика к сумме площадей, умноженное на 100, будет харак-
теризовать массовую долю (%) компонента в смеси. Вполне по-
нятно, что такой метод предполагает существование одинаковой
зависимости величины измеряемого параметра от концентрации
для всех компонентов смеси.
В методе нормировки с калибровочными (градуировочными)
коэффициентами за 100% принимается сумма параметров пиков
с учетом чувствительности детектора. Различие в чувствитель-
ности детектора учитывается с помощью поправочных коэффи-
циентов для каждого компонента. Один из преобладающих ком-
понентов смеси считают сравнительным и поправочный коэффи-
циент для него принимают равным единице. Калибровочные
(градировочные) коэффициенты 1Г рассчитывают по формуле
1" ТГ7~9
1Нсст
где Пст — параметр пика (высота, площадь и т. д.) стандартного
вещества; П^ — параметр пика определяемого компонента; с —
концентрация.
За 100% принимается сумма исправленных параметров 2ГД
и результат анализа рассчитывается так же, как и в методе прос-;
той нормировки.
Наиболее точным является метод абсолютной калибровки.
В этом методе эксцериментально определяют зависимость высо-
ты или площади пика от концентрации вещества и строят градут
ировочные графики. Далее определяют те же характеристики пит
ков в анализируемой смеси и по градуировочному графику нахо- ,
дят концентрацию анализируемого вещества. Этот простой и
точный метод является основным методом определения микро?
примесей. Кроме того, метод не требует^разделения всех КОМПО*
нентов смеси, а ограничивается лишь теми, определение которые;;
необходимо в данном конкретном случае. о
Метод внутреннего стандарта основан на введении в анаь|
лизируемую смесь точно известного количества стандартного вё-
щества. В качестве стандартного выбирают вещества, близкое по
физико-химическим свойствам к компонентам смеси, но не обя-
зательно являющееся ее компонентом. После хроматографировд-
ния измеряют параметры пиков анализируемого компонента и
стандартного вещества. Если стандартное вещество не входит в
достав анализируемой смеси, массовую долю компонента (%) рас-
считывают по формуле
©,-^100*
где П^ и Пст — параметры пиков анализируемого компонента и
стандарта соответственно; г' — отношение массы внутреннего
стандарта к массе пробы.
17.6.4. Влияние температуры
Температура оказывает существенное влияние на хроматог-
рафический процесс. Изменение температуры вызывает измене-:
ние элюацирнных и других характеристик: удерживаемого объ-
ема, коэффициента Генри и т. д. Зависимость удельного, удержи-
ваемого объема Ут от температуры Т передается уравнением
Щ Ут 2,ЗДГ 2,ЗД ЩА\
где АН° и Дв0 — изменение стандартных энтальпии и энтропии
при абсорбции соответственно; А — коэффициент пропорцио-
нальности между Г и Ут.
Так как обычно ДйГ0 < 0, с повышением температуры вели-
чина удерживаемого объема уменьшается, что приводит к сокра-
щению длительности анализа и может вызвать изменение поряд-
ка выхода компонентов из колонки. При понижении температу-
ры длительность анализа увеличивается, но при этом улучшается
степень разделения. Низкие температуры используются, напри-
мер, в процессах разделения изотопов водорода. Хроматографи-
рование таких систем производится при температурах жидкого
азота (-196 °С).
При выборе оптимальной температуры хроматографирова-
ния приходится учитывать, таким образом, несколько факторов,
нередко действующих в противоположных направлениях. Вы-
бранная оптимальная температура является важной особенно-
стью хроматографической методики и для поддержания темпера-
турного режима хроматографическую колонку помещают в тер-
мостат. . :..••? г '
313
Однако газовая хроматография может проводиться не только
при постоянной температуре, но и в условиях программирован-:
ного изменения температуры. Температура колонки в этом случ
чае может меняться по разным режимам: ступенчато, непрерыв-
но с постоянной скоростью (линейно) иг непрерывно с переменной
скоростью (нелинейно). Каждый из этих способов имеет свои до*
стоинства, недостатки и области применения. Если, например,
основную часть смеси составляют легкокипящие компоненты,
целесообразно применять метод нелинейного программирования
температуры: вначале температуру повышать медленно, а затем
этот процесс ускорить.
Изменение температуры открывает новые возможности в
разработке хроматографических методов анализа. В методе ста-
ционарной хроматермографии разделяемая смесь подвергается
действию переменного температурного поля. При осуществле-
нии этой методики электрическая печь движется вдоль колонки
в одном направлений с движением газа-носителя. Если скорость
движения пЬлосы адсорбированного газа превышает скорость
движения температурного поля, то полоса из высокотёмператур-;
ной зоны попадает в низкотемпературную и ее скорость движе-1
ния замедляется. Но при замедлении движения зона оказывается
в области высоких температур и её скорость увеличивается. Та-
ким образом, в некоторой температурной области скорости дви-;
жения полосы газа и печи становятся одинаковыми. Температу-
ру, при которой это происходит, называют характеристической
Г^ар. Она связана с энтальпией адсорбции АН и условиями хро-
матографическогр опыта соотношением
"• • _ АН ■'.-''•/'А
хар и)9 I П
где го — скорость движения температурного поля; V — линейная .
скорость газа-носйтёля. :
При фиксированных условиях опыта разность Тхар двух ком-
понентов! смеси, а следовательно, и возможность разделения опт;
ределяются разностью их энтальпий сорбции. При равенстве эн-.
тальпий разницу в !Гхар следует создавать за счет изменения усл:оф
вий опыта, т. е. меняя скорость газа-носителя или скоррсты
движения электропечи. В оптимальных условиях хроматографи-э
ческого опыта каждый компонент анализируемой смеси будет
двигаться при своей характеристической температуре со скоро->
стью, равной скорости движения печи. Это может быть использо-
вано для идентификации веществ по наличию или отсутствию
пика при данной температуре.
Существенным достоинством метода стационарной хроматер-
мографии является сжатие полосы, что улучшает разделение и
повышает концентрацию компонентов (особенно важно при ана-
лизе примесей). К недостаткам метода относится уменьшение
расстояния между пиками, что может затруднить разделение.
17.6.5. Аналитическая реакционная
газовая хроматография
Суть этого метода состоит в изменении химического состава
пробы до хроматографирования с целью получения веществ лег-
че разделяющихся при хроматографировании или легче детекти-
руемых. Можно использовать также поглощение части компо-
нентов молекулярными ситами или другими поглотителями.
Оба црюцесса — химический и хроматографический — осу г
ществляются в одной установке аналитической реакционвюй га-
зовой хроматографии. Все или некоторые из компонентов анали-
зируемой смеси реагируют в реакторе, а продукты реакции вмес-
те с компонентами, не принимающими участия в реакции,
потоком газа-носителя уносятся в хроматографическую колонку.
В практике реакционной газовой хроматографии используется
несколько схем взаимного расположения реактора, колонки и дет
тектора. Реактор может быть помещен перед колонкой или после
колонки перед детектором или между двумя детекторами. Разра-
ботаны и более сложные схемы с применением нескольких реак-
торов и колонок.
Типы используемых химических реакций самые разнообраз-
ные. Органические кислоты этерификацией (взаимодействием со
спиртами) превращают в сложные эфиры, обладающие более ни-
зкими температурами кипения, чем соответствующие кислоты.
Микропримеси паров воды определяют при пропускании анали-
зируемой смеси через реактор с литий-алюминий-гидридом, ре-
агирующим с водой с образованием водорода, который легко
фиксируется катарометром. Если реактор заполнен карбидом
кальция, вода образует ацетилен, наличие которого устанавлива-
ется с помощью пламенно-ионизационного детектора. Алифати-
ческие сульфиды можно гидрогенизировать на никеле Ренея до
соответствующих углеводородов.
315
17.6.6. Практическое применение
Широкое применение и большое значение газовой хроматог-
рафии в практике вызвано тем, что с ее помощью можно иден-
тифицировать отдельные компоненты сложных газовых смесей
и определять их количественно, выполнение анализа не требу-,
ет больших. затрат времени, и метод является достаточно уни-
версальным. Эффективно используется газовая хроматография
в препаративных целях, в физико-химических исследованиях
и других областях.
Методом газовой хроматографии анализируют нефтяные и
рудные газы, воздух, продукцию основной химии и промышлен-
ности органического синтеза, нефть и продукты ее переработки,
многочисленные металлоорганические соединения и т. д. Методы
газовой хроматографии пригодны для разделения изотопов неко-
торых элементов, например водорода. Хроматография газов ис-
пользуется в биологии и медицине, в технологии переработки
древесины, в лесохимии и пищевой промышленности, в техноло-
гии некоторых высокотемпературных процессов и многих дру-
гих. Газовая хроматография может быть применена для анализа
жидкостей после перевода их в пар в условиях работы хромато-
графической колонки.
Необходимо отметить применение газовой хроматографии
для автоматизации производственных процессов. Датчик про-
мышленного хроматографа используется не только как регистри-
рующий прибор, но и как регулирующее устройство, подающее
сигналы непосредственно исполнительным механизмам/Таким
образом, промышленный хроматограф может контролировать и
регулировать важнейшие параметры технологического процесса: ;
температуру, давление, расход сырья и т. д.
Газовая хроматография широко используется также в физи-
ко-химических исследованиях. Хроматографическая методика
позволяет сравнительно легко определить константу сорбционно-
го равновесия при нескольких температурах и по этим данным;
рассчитать термодинамические характеристики сорбции: изме-
нение энтальпии и энтропии в этом процессе. Хроматографиче-
ским методом определяют также коэффициенты активности,
диффузии и другие характеристики вещества, изучают кинетику
реакций и т. д.
Аналитическая реакционная газовая хроматография приме-
няется для анализа сложных многокомпонентных смесей, опре^
деления микропримесей, анализа нелетучих соединений (напри- •
мер, различных полимеров), для элементного анализа и т. д.
316 ■'■>".:';;::г-;,|
17.7; Жидкостная
адсорбционная хроматография
До конца 50-х годов XX в. практическое значение жидкост-
ной адсорбционной хроматографии было сравнительно невелико
и ее развитие происходило медленно. Однако с появлением в кон-
це 50-х годов высокочувствительных методов детектирования и
новых селективных адсорбентов на основе полимеров жидкост-
ная адсорбционная хроматография стала высокочувствительным
и селективным методом анализа многокомпонентных смесей в
растворах. Ее практическое значение возросло еще больше; когда
стали применять высокие давления.
17-7.1. Теоретические основы
жидкостной хроматографии
Жидкостная адсорбционная хроматография основана на те-
ории адсорбции из раствора. Адсорбционное равновесие между
раствором и адсорбентом подчиняется уравнению изотермы ад-
сорбции Лэнгмюра (17.1), в области разбавленных растворов изо-
терма линейна (17.2). Селективность адсорбции зависит от при-
роды сил взаимодействия между адсорбирующимся веществом и
адсорбентом. Эффективность хроматографической колонки зави-
сит главным образом от процессов диффузии и массопередачи в
обеих фазах и определяется, как и в газовой хроматографии, вы-
сотой Н эквивалентной теоретической тарелки (ВЭТТ). С линей-
ной скоростью подвижной фазы и и некоторыми другими вели-
чинами ВЭТТ связана уравнением
Я = 2ДГ(1-ДГ)£^, (17.15)
причем
где 1т — среднее время между десорбцией и повторной адсорбци-
ей молекулы вещества, когда она движется со скоростью подвиж-
ной фазы и; 48 — среднее время пребывания молекулы в непо-
движной фазе.
Уравнение (17.15) показывает, что с ростом линейной скорос-
ти движения подвижной фазы ВЭТТ возрастает и, следовательно;
эффективность колонки падает. Зависимость ВЭТТ от Лг имеет
довольно сложный характер, но хотя с ростом ^величина Лг
уменьшается, ВЭТТ может расти, так как ts входит в уравнение
(17.15) в качестве множителя.
317
Время пребывания молекулы в неподвижной фазе ^связано
с глубиной пор й и коэффициентом диффузии В уравнением
Эйнштейна:
<*2==22)*3. (17.16)
При подстановке уравнения (17.16) в (17.15) получаем
-_дг(1-Дг)ЕМа--
• "—
Чем больше й, тем больше Ни, следовательно, меньше эф-
фективность колонки. Большая глубина пор у адсорбентов клас-
сической жидкостной адсорбционной хроматографии была одной
из основных причин ее низкой эффективности.
В современной хроматографии широко применяются поверх-
ностно-пористые адсорбенты (ППА). Это твердые, не обладающие
пористостью сферические зерна, на поверхность которых нанесен
тонкий слой адсорбента с высокой пористостью толщиной при-
мерно 1 мкм. У этих адсорбентов нет глубоких пор, что приводит
к уменьшению Н и значительному увеличению эффективности
колонки. Применение поверхностно-пористых адсорбентов по-
зволило также значительно повысить скорость жидкостной ад-
сорбционной хроматографии.
В последнее время большой интерес вызывает жидкбстная
хроматография при высоких давлениях, позволяющая проводить
сложные разделения. Этот метод получил название высокоэф-
фективной жидкостной хроматографии (ВЭЖХ).
17.7.2. Основные узлы приборов
жидкостной хроматографии
Как и в газовой, в жидкостной адсорбционной хроматогра-
фии основными узлами хроматографической установки являют-
ся колонка, дозатор и детектор.
Колонки и дозаторы. Выбор оптимальных длины и внутренне-
го диаметра колонки является компромиссным, так как приходит-
ся учитьюать селективность и эффективность колонки, длитель-
ность анализа, удобство работы и другие факторы. Учитывается,
например, что критерий разделения увеличивается пропорции
онально лишь корню квадратному из длины колонки, а продолжив
тельность анализа пропорциональна ее длине. Уменьшение диа-
метра колонки вызывает трудности ее заполнения, а увеличение
диаметра приводит к уменьшению скорости движения подвижной ,
фазы. В жидкостной адсорбционной хроматографии используют-
318
ся колонки длиной от 15—20 см до 1,5—2,0 м и не более 10 м
с внутренним диаметром 1—6 мм и не более 12 мм. Изготовляют-
ся колонки из толстостенного стекла или нержавеющей стали и
плотно и равномерно заполняются адсорбентом. При плотном за-
полнении создаются условия для более постоянной скорости по-
тока жидкости. '
К адсорбентам в современной жидкостной адсорбционной хро-
матографии предъявляются требования не только селективности и
эффективности, но и высокой скорости хроматографирования, что
определяется, главным образом, структурой поверхности. Отлич-
ными структурными свойствами обладают поверхностно-порис-
тые адсорбенты (ППА), у которых большая механическая проч-
ность и отсутствуют глубокие поры. В качестве активного слоя на
поверхность стеклянных шариков в ППА наносят силикагель,
оксид алюминия или некоторые полимеры. Для современной вы-
сокоскоростной жидкостной адсорбционной хроматографии это
наиболее подходящий адсорбент. Применяют также объемно-по-
ристые мелкозернистые адсорбенты на основе силикагеля,. адю-
МОГеЛЯ И ^руГИХ ВещеСТВ. , %Лч...; ч..(;. '.. \ •■■. I ' ^ ^
Определенные требования предъявляются к подвижной фазе
растворителю. Растворитель в жидкостной адсорбционной хро-
матографии должен хорошо растворять все компоненты анализи-
руемой смеси, обладать химической инертностью по отношению
к растворенным веществам, адсорбенту и кислороду воздуха,
быть маловязким, не содержать примесей, не вызывать отклоне-
ний в работе детектора и быть доступным.
Хотя изотермы адсорбции растворенных веществ, как и га-
зов, описываются одним и тем же уравнением (17.1), все же про-
цесс адсорбции из раствора осложняется участием растворителя.
Подвижная фаза часто принимает участие в адсорбционном про-
цессе и, таким образом, может влиять на селективность колонки.
Эта особенность жидкостной адсорбционной хроматографии
очень важна, так Как открывает еще одну возможность регулиро-
вания хроматографи^еского процесса и выбора оптимальных ус-
ловий разделения.
Для элюирования смеси обычно применяют, не индивидуаль-
ные растворители, а раствор одного или: нескольких веществ в
растворителе, который сам адсорбируется слабо, введенные же
вещества адсорбируются сильнее нескольких, а возможно и всех
Компонентов анализируемой смеси. Состав цодвижной фазы
Можно изменять так, что ее вытесняющая способность будет нет
прерывно возрастать. Это градиентная хроматография. :
■■/•■■■'■■. 319 "'
Элюирующую способность растворителей при адсорбции на
неорганических веществах часто оценивают по шкале Гильдебран-
да, основанной на энергии поляризации растворителей. Состав-
ленный по возрастанию энергии поляризации так называемый
элюотропный ряд растворителей содержит растворители в поряд-
ке возрастания их элюирующей способности. Например, для окси-
да алюминия элюотропный ряд имеет вид: бензол < хлороформ <
ацетон — диоксан < ацетонитрил < этанол < метанол. Для неполяр-
ных адсорбентов (полиамиды, активированные угли и др.) порядок
растворителей меняется. До подачи в систему растворитель под-
вергают дегазации, так как выделение пузырьков газа при повы-
шении температуры в колонке или детекторе нежелательно. Дега-
зация проводится нагреванием или вакуумированием.
Процесс жидкостной адсорбционной хроматографии идет под
высоким давлением. Ввод пробы в колонку, в основном, произво-
дится с помощью шприца через самоуплотняющуюся резиновую
прокладку и с помощью кранов. С помощью шприца объем пробы ;
легко регулируется и проба может быть подана непосредственно
на насадку, однако при высоких давлениях метод становится не-
пригодным из-за неплотностей в поршне шприца. Система с кра-
нами позволяет работать при высоких давлениях и может быть
автоматизирована.
Детекторы. Для определения концентрации вещества на вы- '
ходе из хроматографической колонки можно или последователь-
но отбирать отдельные пробы и их затем анализировать, или про-
водить непрерывный анализ. Методика непосредственного ана-
лиза с автоматической записью концентраций имеет бесспорные
достоинства. Создание чувствительных детекторов непрерывного
действия в значительной степени обусловило современный уро-
вень жидкостной адсорбционной хроматографии и ее успехи в •
разделении сложных многокомпонентных смесей. Практическое
применение нашли детекторы, реагирующие на свойства раство-
ра, растворенного вещества и на свойство вещества после удале-
ния растворителя.
Рефрактометрический детектор непрерывно измеряет раз-
ность показателей преломления между чистым растворителем и;
раствором после прохождения колонки. Чувствительность детек-
тора достигает 3 мкг/мл. Он универсален, однако для получения
надежных данных необходимо достаточно тонкое термостатиро-
вание (±0,001 °С).
Спектрометрические детекторы основаны на применении зако-
на Бугера—Ламберта—Вера. Обычно используется светопоглоще-,
ние в ультрафиолетовом участке спектра, реже в инфракрасном.
320
В детекторах транспортного типа раствор после хроматографи-
ческой колонки попадает на непрерывно движущуюся транспорт-
ную ленту, которая подается в печь, где происходит испарение
элюента. Остаток на ленте переносится в реактор, где превращает-
ся в летучее соединение, которое далее анализируется методами га-
зовой хроматографии.
17.7.3. Качественный и количественный анализ
В жидкостной адсорбционной хроматографии, как и в газо-
вой, идентификация веществ производится по характеристикам
удержания, а количественный анализ основан на измерении вы-
соты или площади хроматографического пика. Используется
также анализ фракций раствора после хроматографической ко-
лонки различными химическими или физико-химическими ме-
тодами.
Жидкостная адсорбционная хроматография часто применя-
ется в органической химии: в технологии и анализе. Этим мето-
дом весьма успешно изучают, например, состав нефти, керосина,
углеводородов, эффективно разделяют транс- и цмс-изомеры, ал-
калоиды ит, д. Особенно большую роль она сыграла в разработке
методов разделения, анализа и исследования нелетучих и неста-
бильных соединений. Очень эффективно применение жидкост-
ной хроматографии при высоком давлении для разделения непо-
лярных соединений и соединений со средней полярностью.
17.8. Тонкослойная хроматография
Метод тонкослойной хроматографии (ТХС), получивший в
настоящее время широкое распространение, был разработан
Н. А. Измайловым и М. С. Шрайбер еще в 1938 г.
В методе ТСХ неподвижная твердая фаза тонким слоем нано-
сится на стеклянную, металлическую или пластмассовую плас-
тинку. В 2—-3 см от края пластинки на стартовую линию вносят
пробу анализируемой жидкости и край пластинки погружают в
растворитель, который действует как подвижная фаза жидкост-
ной адсорбционной хроматографии. Под действием капиллярных
сил растворитель движется вдоль слоя сорбента и с разной скоро-
стью переносит компоненты смеси, что приводит к их простран-
ственному разделению. Диффузия в тонком слое происходит в
продольном и поперечном направлениях, поэтому процесс следу-
ет рассматривать как двумерный;
21 -7831
321
17.8.1. Основные характеристики ТСХ
Схема ТСХ и экспериментально определяемые величины, ко-
торые характеризуют этот процесс, приведены на рис. 17.8. ; '>■.
Сорбционные свойства системы в ТСХ характеризуются под-
вижностью Щ, которая рассчитывается из экспериментальных
данных (рис. 17.8) по уравнению
Лг = ^, (17.17)
где хг — расстояние от стартовой линии до центра зоны: х^ — рас-
стояние, пройденное за это же время растворителем.
Строго говоря, при определении подвижности вместо расстоя^
ний хх и х^ следовало бы учитывать скорость движения соответст-
вующих веществ в тонком слое, так как в теории ТСХ принято счи-
тать, что скорость движения центра зоны хроматографируемого ве-
щества составляет строго определенную долю скорости движения
растворителя. Однако измерение этих величин затруднительно, а
значения рассчитанные,по соотношению (17.17), также харак-
теризуют подвижность. По смысловому определению как свойств
во, характерное для данной системы, не должно зависеть от кон-
центрации и других факторов. Опыт показывает, однако, что восп-
роизводимость и постоянство значений В? не всегда достаточны,
особенно при анализе неорганических ионов. На влияет качество
и активность сорбента, его влажность, толщина слоя, качество раст
творителя и другие факторы, не всегда
поддающиеся достаточному контро-
лю. На практике часто пользуются от-
носительной величиной
д - дл*
. отн р '
где Дд ст также рассчитывается по
уравнению (17.17).
Стандартное вещество (свидетель)
в том же растворителе наносится на
стартовую линию рядом с анализи-
руемой пробой и, таким образом, хро-
матографируется в тех же условиях.
Число теоретических тарелок пв
методе ТСХ рассчитывается по соота
ношению
ь
ь
а.
щ
а 1
Н
ял
Рис. 17.8. ТСХ-схема:
а—а — стартовая линия;
Ъ—Ъ — граница фронта
растворителя в конце опыта;
заштрихованные пятна
характеризуют положение
компонентов в тонком слое
в конце опыта
322
Значения /н и 1П определяются в соответствии с рис. 17.8.
Тогда высота Н, эквивалентная теоретической тарелке, будет
равна
7 72 "
Коэффициент разделения в тонком слое связан с числом
теоретических тарелок и подвижностями Щ уравнением
где Д, , Д, — подвижности соседних компонентов смеси.
Теоретический анализ показывает, что при небольших значе-
ниях Д^ и уменьшении длительности анализа размывание зойы
вещества на сорбенте бывает максимальным, следовательно, кон-
центрация вещества будет максимальна и чувствительность ана-
лиза увеличится. Уменьшение диаметра зерна в тонком слое при-
водит к увеличению продолжительности анализа и усиливает
диффузное размывание! ^
г Подложки для сорбента (пластинки) обычно изготовляют из
стекла, алюминиевой фольги или полиэфирной плёнки. Одно из
достоинств пленки сострит в том, что она прозрачна примерно гдо
320 нм и это позволяет проводить прямое фотометрирование мно-
гих веществ непосредственно в слое.
Сорбент может быть нанесен на подложку в виде пасты (за-
крепленный слой), тонкого порошка (незакрепленный слой) или
быть приготовленным при обжиге силикагеля на стеклянной
пластине.
В качестве сорбента в ТСХ применяют силикагели, оксид
алюминия, крахмал, целлюлозу и некоторые другие вещества с
высокой адсорбционной способностью. Для характеристики со-
рбционной активности оксида алюминия часто используется шка-
ла Брокмана, составленная по Д^ стандартного набора красителей.
Эффективно применяются в ТСХ в качестве сорбентов жидкие
иониты и другие вещества, обладающие ионообменными свойст-
вами и свойствами молекулярных сит (например, сефадексы).
Выбор растворителя зависит от природы сорбента и свойств
анализируемых соединений. Часто применяют смеси растворите-
лей из двух или трех компонентов-. Например, при хроматогра-
фировании аминокислот используют смесь л-бутанола с уксусной
кислотой и водой, при анализе неорганических ионов — водные
буферные растворы, создающие постоянное значение рН,
21* 323
В восходящей хроматографии растворитель поднимается
снизу вверх под действием капиллярных сил, в нисходящей
растворитель передвигается по слою вниз под действием и капил-
лярных, и гравитационных сил. Горизонтальная хроматогра-
фия выполняется в виде круговой и со свободным испарением
растворителя. В круговой хроматографии в центр горизонтально
установленной пластинки вносят каплю анализируемой смеси
и непрерывно подают растворитель, который под действием каг
пиллярных сил движется в радиальном направлении от центра?.
Компоненты смеси располагаются в слое в виде концентрических
колец.
По окончании хроматографирования зоны на хроматограмме
проявляют химическим или физическим способом. При химиче-
ском способе пластинку опрыскивают раствором реактива, взаи-
модействующего с компонентом смеси. При обработке, напри-
мер, парами иода четко проявляются непредельные соединения.
В физических способах проявления используется способность, не-
которых веществ флуоресцировать под действием ультрафиоле-
тового излучения, часто при добавлении флуоресцирующего ин-
дикатора, взаимодействующего с компонентами смеси. Вещест-
ва, обладающие радиоактивным излучением, обнаруживаются
методом радиоавтографии с помощью различных фотоматериа-
лов (бумаги или пленки). После проявления хроматограммы при-
ступают к идентификации веществ и дальнейшему анализу.
178.2. Качественный анализ
Проще всего идентификация вещества может быть сделана,
если пятно определяемого вещества имеет характерную окраску
или определяемое вещество образует на хроматограмме окрашен-
ное пятно под действием специального реактива. Однако число
таких веществ, особенно органических, невелико, поэтому метод
имеет ограниченное применение.
, Наиболее общий подход к качественному анализу основан на
значениях Щ. Хроматографическая подвижность является чувст-
вительной характеристикой вещества, однако она существенно
зависит от условий определения. Эта трудность преодолевается
путем проведения опыта в строго фиксированных стандартных,
условиях, которые регламентируют размер пластин, толщину
слоя сорбента, объем пробы, длину пути фронта растворителя и
другие факторы. При соблюдении стандартных условий получа-
ются воспроизводимые значения Л^, которые можно исподьзб-
324
вать в аналитических целях при сравнении с табличными, если
они получены в тех же условиях опыта.
Самым надежным является метод свидетелей, когда на старто-
вую линию рядом с пробой наносятся индивидуальные вещества,
соответствующие предполагаемым компонентам смеси. Влияние
различных факторов на все вещества будет одинаковым, поэтому
совпадение Rf компонента пробы и одного из свидетелей дает осно-
вания для отождествления веществ с учетом возможных наложе-
ний. Несовпадение интерпретируется более однозначно: оно
указывает на отсутствие в пробе соответствующего компонента,
Интересные результаты получаются при сочетании ТСХ с
другими методами. При сочетаний ТСХ с газовой хроматографи-
ей пластинка становится своеобразным детектором. Выходящий
из колонки газ направляется на стартовую линию цластинки и
затем хроматографируется по методике ТСХ выбранным раство-
рителем. Анализ тонкослойных хроматограмм позволяет незави-
симым методом идентифицировать компоненты смеси, что уве-
личивает надежность анализа. Хроматографирование вещества
методом ТСХ после прохождения газовой колонки может дать до-
полнительную информацию о составе смеси, в частности о компо-
нентах, разделение которых методом газовой хроматографии бы-
ло неполным. Сочетание ТСХ и газовой хроматографии позволя-
ет также установить, все ли компоненты смеси вымываются из
колонки, происходят ли химические изменения при хроматогра-
фировании, и решить некоторые другие вопросы.
Сочетание ТСХ с электрофорезом расширяет возможности
разделения, в частности, неорганических ионов и значительно
ускоряет процесс разделения. ТСХ комбинируется также с экст-
ракцией и другими методами анализа.
■ 17.8.3. Количественный анализ
Количественные определения в ТСХ могут быть сделаны или
непосредственно на пластинке, или после удаления вещества с
пластинки. При непосредственном определении на пластинке из-
меряют тем или иным методом площадь пятна (например, с по-
мощью миллиметровой кальки) и по заранее построенному гра-
дуировочнрму графику находят количество вещества.
Применяют также прямое спектрофотометрирование плас-
тинки с помощью спектроденситометров. Для количественных
расчетов также предварительно строят градуировочный график,
используя оптическую плотность в центре пятна.
325
Наиболее точным считается метод, в котором вещество после
разделения удаляется с пластинки и анализируется спектрофото-:
метрическим или иным методом. Удаление вещества с пластинки
обычно производят механическим путем, хотя иногда применяв
ют вымывание подходящим растворителем. я
В настоящее время ТСХ является одним из важных методов
аналитической химии. Это непревзойденный метод анализа слож-
ных смесей. Он прост по методике выполнения и аппаратуре, экс-
прессен, не требует для анализа больших количеств веществ. <
17.9. Жидкостно-жидкостная
распределительная хроматография
Жидкостно-жидкостная хроматография по сути близка к га-
зожидкостной хроматографии. На твердый носитель также нано-
сится пленка жидкой фазы и через колонку, наполненную таким
сорбентом, пропускают жидкий раствор. Этот вид хроматографии
называют жидкостно-жидкостной распределительной хроматог-
рафией или просто распределительной хроматографией. Жид-
кость, нанесенную на носитель, называют неподвижной жидкой
фазой, а растворитель, передвигающийся через носитель, — под-
вижной жидкой фазой. Жидкостно-жидкостная хроматография
может проводиться в колонке (колоночный вариант) и на бумаге
(бумажная хроматография, или хроматография на бумаге).
17.9.1. Колоночный вариант
Разделение смеси веществ в жидкостно-жидкостной хрома-
тографии основывается на различии коэффициентов распределе-
ния вещества между несмешивающимися растворителями. Коэф-
фициент распределения вещества равен
где списн - концентрация в подвижной и неподвижной фазах.
Для членов одного гомологического ряда установлены неко-
торые закономерности в величинах Ки н. Известна, например, за-
висимость Кп н в данном гомологическом ряду от числа атомов
углерода. ;•
Поиск несмешивающихся фаз, обеспечивающих разделение,
обычно производится эмпирически на основе эксперименталь?
ных данных. Широкое применение в жидкостно-жидкостной
326
хроматографии получили тройные системы, состоящие из двух
несмешивающихся растворителей и третьего растворимого в обе-
их фазах. Такие системы позволяют получать набор несмеши-
вающихся фаз различной селективности. В качестве примера
можно привести систему из несмешивающихся между собой геп-
тана и воды, в которую введен этанол, растворяющийся в обоих
растворителях. , ■
Хотя в качестве подвижной и неподвижной фаз выбираются
растворители, не смешивающиеся между собой, все же во многих
системах наблюдается некоторая взаимная растворимость. Что-
бы предотвратить процессы взаимного растворения жидкостей в
ходе хроматографирования, подвижную жидкую фазу предвари-
тельно насыщают неподвижной. Для сохранения неизменного со-
става фаз применяют также метод химического закрепления не-
подвижной фазы на сорбенте. При этом используют взаимодейст-
вие растворителя с группами ОН" на поверхности носителя.
Адсорбенты с закрепленной на их поверхности жидкой фазой вы-
пускаются промышленностью.
Эффективность колонки связана с вязкостью, коэффициен-
том диффузии и другими физическими свойствами жидкостей.
С уменьшением вязкости подвижной фазы сокращается продол-
жительность анализа, но с увеличением вязкости несколько воз-
растает эффективность. В практике обычно используют маловяз-
кие растворители, так как возрастание эффективности колонок
при увеличении вязкости не очень велико.
Носитель неподвижной фазы должен обладать достаточно
развитой поверхностью, быть химически инертным, прочно удер-
живать на своей поверхности жидкую фазу и не растворяться в
применяемых растворителях. В качестве носителей используют
вещества различной химической природы: гидрофильные носи-
тели — силикагель, целлюлоза и др. и гидрофобные — фторо-
пласт, тефлон и другие полимеры. Успешно развивается приме-
нение в жидкостно-жидкостной распределительной хроматогра-
фии высокого давления (ВЭЖХ).
17.9.2 Распределительная хроматография на бумаге
Кроме обычных носителей, используемых для заполнения
колонок, в распределительной хроматографии применяют специ-
фический носитель, позволяющий обходиться вообще без колон-
ки. Таким носителем является специальная хроматографическая
бумага, а методика, основанная на ее применении, получила на-
звание распределительной хроматографии на бумаге или рас-
327
пределительной бумажной хроматографии. Во многом она сход-
на с хроматографией в тонком слое (ТСХ).
Важной характеристикой в бумажной распределительной
хроматографии, так же как и в ТСХ, является Щ = х/Хр где л: —
смещение зоны компонента; х^ — смещение фронта растворите-
ля. Методика определения Щ в бумажной хроматографии не от-
личается от соответствующей методики в ТСХ, основанной на из-
мерениях в соответствии с рис 17.8. В начальный момент време-
ни хроматографируемая проба наносится на начальную
(стартовую) линию бумажной полоски и подвергается действию
подвижной фазы (растворителя). Если компоненты окрашены, че-
рез некоторое время на хроматограмме можно будет видеть отдель-
ные цветные пятна. Первый компонент будет иметь = хг/Хр
второй — = я2/Лу и т. д.
При идеальных условиях коэффициент Щ определяется толь-
ко природой вещества, параметрами бумаги и свойствами раство-
рителей, но не зависит от концентрации вещества и присутствия
других компонентов. В действительности коэффициент Щ в неко-
торой степени оказался зависящим от этих факторов й техники !
эксперимента. Тем не менее при достаточном постоянстве уело-
вий опыта и не слишком больших колебаниях в составе смеси эти v
коэффициенты остаются вполне постоянными и могут быть ис-
пользованы для идентификации компонентов смеси.
Хроматографическая бумага должна быть химически чис-
той, нейтральной, инертной по отношению к компонентам рас-
твора и подвижному растворителю и быть однородной по плот-
ности. Имеют значение также такие свойства, как структура мо-
лекул целлюлозы в бумаге, набухаемость, ориентация волокна и
другие, влияющие на скорость движения растворителя и на иные
характеристики процесса.
В атмосфере водяных паров бумага поглощает значительное
количество влаги (до 20—-25% своей массы), поэтому когда непо-
движной жидкой фазой является вода, никакого, дополнительно- ■;
го увлажнения бумаги не делают. При выборе в качестве непо-
движной фазы некоторых органических веществ, гидрофильную
бумагу превращают в гидрофобную, пропитывая ее растворами
различных гидрофобных веществ (парафина, растительного мас-
ла и др.). ,
В выбранных растворителях компоненты пробы должны
иметь разную растворимость, иначе разделения вообще не про-
изойдет. В растворителе, являющемся подвижной фазой, раство-
римость каждого компонента должна быть меньшей, чем в рас-
328
творителе неподвижной фазы, но все же составлять вполне за-
метное значение. Это ограничение связано с тем, что если раство-
римость вещества будет очень велика, вещество будет двигаться
вместе с фронтом растворителя, а если растворимость будет очень
мала, вещество останется на начальной линии.
Для разделения водорастворимых веществ в качестве под-
вижной фазы обычно берут органический растворитель, а в каче-
стве неподвижной — воду. Если вещество растворимо в органиче-
ских растворителях, вода используется уже в качестве подвиж-
ной фазы, а органический растворитель является неподвижной
фазой. Это так называемый метод обращенных фаз.
К растворителям обычно предъявляются следующие требова-
ния: растворители подвижной и неподвижной фаз не должны
смешиваться, состав растворителя в процессе хроматографирова-
ния не должен изменяться, растворители должны легко удалять-
ся с бумаги, быть недефицитными и безвредными для человека.
Индивидуальные растворители в распределительной хрома-
тографии используются относительно редко. Чаще для этой цели
употребляют смеси веществ, например бутилового шга амилового
спирта с метиловым или этиловым, насыщенные водные раство-
ры фенола, крезола и др., смеси бутилового спирта с уксусной
кислотой, аммиаком и т. д. Применение различных смесей рас-
творителей позволяет плавно изменять Щ и тем самым создавать
наиболее благоприятные условия разделения.
По технике выполнения различают следующие виды бумаж-
ной хроматографии: одномерную, двумерную, круговую и элек-
трофоретическую. Для получения двумерных хроматограмм
хроматографирование производят дважды во взаимно противопо-
ложных направлениях: после обработки пробы одним раствори-
телем хроматограмму поворачивают на 90° и хроматографируют
вторично уже другим растворителем. Такая методика позволяет
производить более тонкие разделения компонентов смеси.
Весьма эффективным оказалось сочетание хроматографии и
электрофореза, т. е. получение электрофоретических хромато-
грамм на бумаге. Воздействие электрического поля можно ис-
пользовать-или одновременно с хроматографированием, или по-
следовательно, т. е. сначала провести электрофорез, а затем хро-
матографирование.
Качественный состав пробы в методе бумажной распредели-
тельной хроматографии так же, как и в ТСХ, может быть уста-
новлен или по специфической окраске отдельных пятен на хро-
матограмме, или по числовому значению Щ каждого компонента.
329
' Количественные определения в распределительной, так же
как ив тонкослойной хроматографии, выполняются или по хро^
матографическим характеристикам (площади пятна на хромав
тограмме и интенсивности его окраски), или по методу вымыва-
ния. Нередко хроматограмму разрезают на отдельные части по
числу пятен, каждое пятно обрабатывают соответствующим экст
трагентом и определяют количество экстрагированного вещества
любым подходящим методом: фотометрическим, полярографиче-
ским и т. д.
Методики, основанные на использовании хроматографиче-
ских характеристик, довольно многообразны. Количество веще-г
ства можно определить методом шкалы, когда на одной и той же
хроматограмме получают пятно раствора с неизвестной концент-
рацией и пятна нескольких стандартов. Концентрацию исследуе-
мого раствора находят прямым сравнением площади пятна и
интенсивности его окраски у исследуемого и у стандартного рас-
творов.
Более точным является метод, основанный на применении
градуировочного графика в - ^с, где 5 — площадь пятна, с —
концентрация. В указанных координатах график линеен. Исполь-
зуется также. измерение интенсивности окраски пятна, которая ;
пропорциональна концентрации. Методами распределительной
жидкостной хроматографии успешно анализируют смеси катио-
нов в неорганическом качественном анализе, смеси аминокислот
и других органических кислот, смеси красителей и т. д.
17.10. Гель-хроматография
Это совершенно своеобразный вид хроматографии, основан-
ный на использовании различия в размерах молекул. Его назы-
вают также гель-фильтрацией или ситовой хроматографией.
Неподвижной фазой в гель-хроматографйи является раствори-
тель, находящийся в порах геля, а подвижной — сам раствори-
тель, т. е. и подвижную и неподвижную фазы составляет одно и
то же вещество или одна и та же смесь вещества. Гель готовят на
основе, например, декстрана, полиакрил амид а или других при-
родных и синтетических соединений.
В процессе гель-хроматографирования могут быть отделены
крупные молекулы, которые гелем не сорбируются, так как их
размеры превышают размеры пор, от мелких, которые проника-
ют в поры, а затем могут быть элюированы. Проводятся и более
тонкие разделения, так как размеры пор можно регулировать,
330
изменяя, например, состав растворителя и, как следствие, набу-
хаемость геля. Гель-хроматография может быть выполнена в ко-
лоночном варианте и в тонкослойном.
Применяемые на практике гели обычно подразделяют на
•мягкие, полужесткие и жесткие. Мягкими гелями являются
высокомолекулярные органические соединения с незначитель-
ным числом поперечных связей. Фактор емкости, равный отно-
шению объема растворителя внутри геля к его объему вне геля, у
них равен 3. При набухании они значительно увеличивают собст-
венный объем. Это сефадексы или декстрановые гели, агарочные
гели, крахмал и др. Они применяются для разделения смесей
низкомолекулярных веществ, часто в тонкослойном варианте.
Хроматографирование на мягких гелях называют гель-фильтра-
цией.
Полужесткие гели получают путем полимеризации. Большое
распространение получили стирогели — продукты сополимери-
зации стирола и дивинилбензола с большим числом поперечных
связей. Фактор емкости полужестких гелей лежит в пределах
0,8—1,2, их объем при набухании увеличивается не очень значи-
тельно (в 1,2—1,8 раза). Хроматографирование на полужестких
гелях называют гель-проникающей хроматографией.
К жестким гелям относят силикагели и часто пористые стек-
ла, хотя они и не являются гелями. Жесткие гели имеют неболь-
шой фактор емкости (0,8—1,1) и фиксированный размер пор. Эти
материалы используют в гель-хроматографии при высоком дав-
лении. .
Растворители гель-хроматографии должны растворять все
компоненты смеси, смачивать поверхность геля и не адсорбиро-
ваться на ней.
Практическое применение гель-хроматографии связано, глав-
ным образом, с разделением смеси высокомолекулярных соедине-
ний, хотя нередко они используются для разделения и низкомоле-
кулярных, так как разделение этим методом возможно при ком-
натной температуре.
17.11. Ионообменная хроматография
Ионообменная хроматография основана на обратимом стехио-
метрическом обмене ионов, находящихся в растворе, на ионы,
входящие в состав ионообменника. Хотя явление, известное в на-
стоящее время как ионный обмен, фактически было известно с
середины XIX в., широкое применение ионообменных процессов
331
в практике началось после создания синтетических ионорбмен-
никое — так называемых ионообменных смол или ионитрв. Ис-
пользуемые ранее естественные ионообменники (различнее алю-
мосиликаты и другие соединения) не обладали достаточной восп-
роизводимостью свойств, не были химически устойчивыми и т. Д.
и поэтому существенного практического значения не имели.:
Применяемые в настоящее время синтетические ионообмён-
ники лишены многих недостатков, присущих естественным
ионообменникам, и обладают рядом важных достоинств: они
имеют высокую обменную емкость и воспроизводимые ионооб-
менные и другие свойства, устойчивы к действию кислот и осно-
ваний, не разрушаются в присутствии многих окислителей и вос-
становителей и т. д. Обычно синтетический ионообменник пред-
ставляет собой высокополимер, например поперечно-сшитый
полистирол, содержащий различные функциональные группы,
которые и определяют наиболее характерные свойства смол. Из-
вестны также синтетические неорганические иондты, например
различные пермутиты, активированный оксид алюминия, гели
на основе соединений железа или соединений циркония и т. д.
Однако органические ионообменные смолы имеют намного боль-
шее практические применение.
17.11.1. Типы ионообменных смол
В зависимости от знака заряда функциональных групп ионо-
обменные смолы являются катионитами или анионитами. Ка-
тиониты содержат кислотные функциональные группы [—вО^;
—СОО"; —РОд; —КГ(СН2С02)], поэтому каркас катионита, несу-
щий фиксированные отрицательные заряды, заряжен отрица-
тельно. Отрицательные заряды каркаса компенсируются поло-
жительными зарядами противоионов, так что в целом катионит
остается электронейтральным. Однако противоионы, в данном
случае катионы, в отличие от функциональных групп каркаса об-
ладают подвижностью и могут переходить в раствор в обмен на
эквивалентное количество ионов из раствора. Этот обмен приво-
дит к установлению подвижного равновесия между ионами, на-"
ходящимися в фазе смолы, и ионами в растворителе. "Наиболее
распространенными катионитами являются сульфокислоты, об-
разованные сульфированными продуктами сополимеризации
стирола и дивинилбензола. Это отечественные смолы КУ-2,
СДВ-3 и др., иностранные дауэкс-50, амберлит 1К-120 и др. Суль-
фокатиониты характеризуются высокой химической стойкостью
332
и механической прочностью, большой скоростью установления
ионообменного равновесия.
Функциональными группами каркаса анионов являются чет-
вертичные ^Ш^", третичные —ЫН2Н+ или первичные —ам-
мониевые, пиридиновые или другие основания, а в качестве под-
вижных протй&Йионов выступают анионы. Анионообменные смо-
лы получаютс^также путем проведения реакций полимеризации
или поликонденсации с использованием различных аминосоеди-
нений (фенилендиамина, полиэтиленполиамина и т. п.), форм-
альдегида и др. Так были получены аниониты АН-1, АН-2Ф, ам-
берлит. Широкое распространение получил полифункциональ-
ный анионит ЭДЭ-10П, содержащий амины различной степени
замещения, включая четвертичные. Амфотерные иониты или ам-
фолиты способны осуществлять одновременный обмен катионов
и анионов. Биполярным, или амфотерным, ионитом является
продукт поликонденсации диэтилентриамина, фенола и форм-
альдегида, так как в его состав наряду с аминогруппами входят
слабокислотные фенольные группы. Амфотерными и комплексо-
образующими свойствами обладают смолы, содержащие остатки
комплексонов, например ЭДТА.
Важной характеристикой ионообменника является его об-
менная емкость, определяемая в первом приближении числом
функциональных групп каркаса и степенью их ионизации при
данном рН раствора. Обменную емкость ионита численно можно
выразить количеством молей эквивалента противоиона на едини-
цу массы или объема смолы. В аналитической химии емкость
ионита обычно выражают количеством молей эквивалента обме-
нивающегося иона на 1 г сухой смолы в Н+-форме для катйонита
и СГ- или ОН"-форме для анионита. Оговорка относительно су-
хой смолы необходима, так как в контакте с водой смола набуха-
ет в 1,5—2 раза, а некоторые виды смол — в 5 раз и бойее. Ве-
личина обменной емкости характеризуется несколькими (3—:5
до 1(3) доюль экв иона на 1 г смолы. Емкость, найденную в статиче-
ских условиях, когда навеску смолы помещают .в'раствор насы-
щающего иона достаточной концентрации и выдерживают при
встряхивании до полного насыщения, называют статической
обменной емкостью (СОЕ). Величину емкости, полученную в ди-
намических условиях при пропускании насыщающего раствора
через колонку с ионитом, называют динамической обменной ем-
костью (ДОЕ). Эта емкость ионита, определяемая по первому по-
явлению (до проскока) насыщающего иона в вытекающем раство-
ре. Полная динамическая обменная емкость (ПДОЕ) находится
по полному насыщению ионита данным ионом.
333
17.11.2. Ионообменное равновесие
Взаимодействие ионообменной смолы с раствором электроли-
та включает несколько сложных процессов, наиболее важными
из которых являются собственно ионный обмен, физическая аб-
сорбция ионов и молекул на смоле и набухание смолы за счет по-
глощения растворителя и проникновения электролита внутрь
смолы.
Процесс собственно ионного обмена протекает стехиометри-
чески. Если, например, катионит в водородной форме RH ввести
в раствор, содержащий ионы Са2+, в системе установится равно-
весие:
; 2HR + Са2+= CaR2 + 2Н+
т. е. в растворе появятся ионы водорода, а эквивалентное количе-
ство ионов Са2+будет поглощено катионитом.
Аналогичный процесс обмена имеет место при взаимодейст-
вии раствора, содержащего, например, хлорид, с анионитом
ROH:
ROH + Cl- = RC1 + ОНг
Распределение каждого иона между смолой и раствором
можно охарактеризовать коэффициентом распределения Р:
P _ lCaR2l или р = IRC1I
• *Са*+ |c^2Tf ИЛИ * q\- JcTT'
Концентрация иона в растворе обычно выражается в моль/л,
а в фазе ионита — молярными долями.
.Более строго это равновесие характеризуется константой
ионного рбмена:
_ а^+аСлщ
а г— ,
aCa2+aHR
где а — активности частиц.
Уравнение ионного обмена нередко записывают так же, как
Н+ + У 2Са2+ = 72Са2++ н+ • .
где горизонтальная черта показывает принадлежность иона к фа-
зе ионита. 5
По Никольскому константа ионного обмена имеет вид
334
Во многих случаях оказываются достаточными концентра-
ционные константы равновесия:
. _ [H+][CaR2]
HVCa2+ [Ca2+][HR]2V
Константы обмена связаны с коэффициентом распределения
соотношением
ЛН+/Са2+ "рГ~ •
Хотя количественный подход к ионообменным равновесиям
на основании закона действующих масс, когда константы равно-
весия выражены через концентрации, а не через активности, яв-
ляется приближенным, все же он оказывается весьма полезным.
На основе так называемых концентрационных констант ионного
обмена могут быть, например, построены ряды сродства катионов
к данной смоле, позволяющие предвидеть возможности ионооб-
менных разделений. и
К настоящему времени установлено несколько эмпирических
закономерностей, связывающих константы ионного обмена со
свойствами ионов. Так, в частности, найдено, что с ростом заряда
сродство ионов к смоле увеличивается и, например, в ряду Na^" <
< Са2+ < А13+ < Th4+ оно возрастает.
С повышением температуры избирательность поглощения
несколько уменьшается, хотя этот эффект не очень велик.
Введение в раствор веществ, способных образовывать комп-
лексные соединения с присутствующими ионами, сдвигает рав-
новесие ионного обмена, так как в результате комплексообразо-
вания уменьшается равновесная концентрация иона в растворе.
17.11.3. Практическое применение
Методы ионообменной хроматографии используются пре-
имущественно для разделения ионов. Количественные определе-
ния компонентов после разделения могут быть выполнены лю-
бым подходящим методом.
Простейшая методика ионообменного разделения состоит в
поглощении компонентов смеси ионитом и последовательном
элюированйи каждого компонента подходящим растворителем.
Например, катионы щелочных металлов легко элюируются
разбавленным раствором хлороводородной кислоты (0,1 М HCl).
335
Выходная кривая на рис. 17.9 пока-
зывает эффективность этого разделе-
ния. Как видно, элюирование раство-
ром 0,1 М НС1 позволяет легко разде-
лить Ыа+ иК+.
В качестве другого примера можно
указать на методику разделения ионов
циркония(ГУ) и гафния(ГУ). Для разде-
ления эти катионы сначала переводят
в анионные сульфатные комплексы,
которые поглощают анионитом. При
последующем элюировании 1 М рас-
твором Н2804, содержащим сульфат натрия, происходит их полное
разделение: сначала вымывается гафний, а затем цирконий.
Применение ионообменной хроматографии к анализу смеси
лантаноидов с использованием в качестве элюентов растворов лак-
тата, цитрата, ЭДТА и других привело к разработке эффективного
способа разделения этих элементов. На основе полученных данных
была предложена и успешно осуществлена на практике технологи-
ческая схема промышленной переработки руд лантаноидов.
Известны ионообменные методики даже для разделения изо-
топов. Например 14Ы и 15Ы могут быть разделены на сульфосмоле
в виде №Н|, так как оказалось, что 14№ЕЦ" сорбируется хуже и по-
том вымывается раньше, чем 15ЫН£.
Ионообменные методы применяют для определения суммар-
ного содержания катионов или анионов в растворе и для анализа
растворов чистых солей. При пропускании через катионит в
Н+-форме раствора, например, соли натрия в результате ионооб-
менного процесса
НЫ + Ыа+ = ЫаК + Н+
в растворе появляется эквивалентное количество Н+-ионов. Кон-
центрация ионов Н+ в этом растворе может быть определена, на-
пример, титрованием и, таким образом, найдена концентрация
Ыа+в исходном растворе.
Большое практическое значение имеет основанный на ион-
ном обмене процесс деминерализации воды. Сущность его заклю-
чается в следующем. Солевой раствор или вода, предназначенная
для деминерализации, обрабатывается одновременно катиони-
том в Н+-форме й анионитом в ОН"-форме, В результате обмена
на катионите в растворе появятся Н+-ионы:
НИ + М+ = МИ + Н+
V(HCl)
Рис 17.9.
Элюирование поглощенных
на катионите ионов Na+ и К+
0,1 М раствором НС1
336
а в результате обмена на анионите ОН--ионы:
ЫОН + х-= КХ + он-
Но Н+ и ОН" в растворе взаимодействуют Н+ 4- ОН" = Н20, сдви-
гая равновесие ионного обмена до полного извлечения ионов из
раствора и получения чистой деминерализованной воды. Такая
вода используется в лабораториях вместо дистиллированной.
Ионообменные процессы применяются также для переведе-
ния в раствор малорастворимых соединений. Для этого взвесь ма-
лорастворимой соли МХ следует обработать ионитом НЕ до на-
ступления равновесия:
МХ + НЕ = МИ + № + X"
и десорбировать М+ с ионита подходящим растворителем. Воз-
можность растворения будет, очевидно, определяться сродством
М к К и растворимостью МХ. В настоящее время известны мето-
дики растворения с помощью ионного обмена таких осадков, как
Вав04, AgCl и др.
Интенсивно разрабатываются электрохимические аспекты
применения ионообменных смол. Специально для электрохими-
ческих целей изготавливают так называемые ионообменные
мембраны. Их получают в виде листов из ионообменной смолы,
поэтому они обладают одновременно и свойствами ионообменни-
ка, способного к ионному обмену, и свойствами мембраны как по-
лупроницаемой перегородки. Однако ионная проницаемость
ионообменных мембран селективна — мембраны из катионита
проницаемы только для катионов, а мембраны из анионита —
для анионов. Это открывает широкие возможности их практиче-
ского использования.
Так, например, с помощью ионообменных мембран можно
приготовить растворы чистых ЫаОН и Н2804 при электролизе
раствора Ыа2804 в ячейке, разделенной двумя ионообменными
мембранами (рис. 17.10).
Н2а МК И А д02
"N¡12864 ~
+
Рис. 17.10. Электролиз раствора Ыа2804 в ячейке с ионообменными мембранами
22 - 7831 33 7
Катодное пространство отделено от раствора Ыа2804 катиони-
том МЕ, пропускающие ионы Ыа+, но задерживающим ионы
80£~, а анодное пространство отделено мембраной из анионита
ЕА, проницаемой для вО|" и непроницаемой для иона Ыа+.
При электролизе водного раствора в катодном пространстве
происходит восстановление воды:
2Н20 + 2е~:- Н2 + 20Н~
а в анодном пространстве идёт окисление воды:
Н2р - 1/20а + 2Н+ + 2«г
Таким образом, в катодном пространстве образуется чистый
ЫаОН, в анодном — чистая Н2804, а в центральном отделении,
в конце концов, остается деминерализованная вода. В принципе,
такое устройство может быть использовано для водоочистки или
ббессолйванйя морской воды.
Рассмотренные способы применения ионообменных смол, ко-
нечно, Нв) исчерпывают всего многообразия, однако они показы-
вают широкие возможности, которые открывает использование
ионитов в аналитической химии и технологии.
17.12. Ионная хроматография
Это один из вариантов разделения на ионитах, характери-
зующийся более высокоэффективной техникой, чем обычная
ионообменная хроматография. В методе ионной Хроматографии
используются поверхностно-слойные сорбенты с небольшой ем-
костью (10""2—10"1 ммоль экв/г) и небольшим размером частиц
(5—50 мкм), повышенное давление на входе в колонку (2—
5 МПа) и высокочувствительные детекторы с автоматической за-
писью сигнала. Для ионной хроматографии характерны экспрес-
сность, удобство работы й более высокая разделительная способ-
ность. _
Детекторы ионообменных разделений должны регистриро-
вать концентрацию анализируемых ионов в элюате в присутст-
вии ионов элюента. Особенно широко в ионной хроматографии
используют кондуктометрический детектор, являющийся уни-
версальным, так как он реагирует на все ионы в растворе. Приме-
няют также селективные детекторы: электрохимические (поля-
рографический, кулонометрический и т. д.) и спектрофот^мёт-
,338 -:: '■' ■/.
рический. В кондуктометрическом детекторе должны быть пред-
усмотрены какие-либо средства для компенсации фонового сиг-
нала, который может на 2—3 порядка превышать аналитический
сигнал. Фоновый сигнал может быть скомпенсирован с помощью
электронной схемы. Элюат можно обработать также на второй
колонке, чтобы ослабить электрическую проводимость элюента и
усилить проводимость образца.
17.12.1. Методы ионной хроматографии
В ионной хроматографии используют два основных метода:
одноколоночный и двухколоночный.
В одноколоночной анионной хроматографии применяют элюен-
ты с низкой электрической проводимостью, например, разбавлен-
ные растворы (Ю-3—10~4 ммоль/л) бензоата или фталата у щелочно-
го металла или бензойной кислоты [Хп н 0 = 32 См • см2/(мольэкв)].
Электрическая проводимость хлорида, нитрата и многих других
анионов существенно превышает электрическую проводимость
бензоата или фталата [А,§0 = 71 См • см2/(моль экв)], что вызы-
вает при элюировании увеличение электрической проводимости
и на хроматограмме регистрируется соответствующий пик. В ка-
честве элюента используют также разбавленные растворы щелоч-
ных гидроксидов. В этом случае регистрируются так называемые
отрицательные пики, которые появляются вследствие уменьше-
ния электрической проводимости при замене гидроксид-иона в
растворе на какой-либо анион.
В катионной хроматографии элюирование проводят разбав-
ленным раствором азотной или другой минеральной .кислоты.
Здесь также получаются отрицательные пики дкак результат зат
мены ионов Н+, имеющих высокую подвижность, на какоц^либр
Катион...., ••/,:..Г;. .•;.; ,:• г V."
В двухколоночном методе используются две колонки: разде-
лительная и компенсационная. Для разделения применяют ко-
лонки с малой емкостью: в качестве элюентов при анализе анио-
нов используют гидроксиды щелочных металлов.и соли слабых
кислот, а при анализе катионов— азотную или другую мине-
ральную кислоту. В анионной хроматографии компенсационная
колонка заполняется сильнокислотным катионитом, при пропус-
кании через который щелочной раствор нейтрализуется. Б кати-
онной хроматографии компенсационную колонку с той же целью
339
заполняют сильноосновным анионитом. Электрическая проводи-г
мость раствора на выходе из компенсационной колонки опреде-
ляется главным образом ионами анализируемого образца.
17.12.2. Практическое применение
Методами ионной хроматографии определяют очень многие
анионы в питьевой и технической воде, в продуктах технологиче-
ской переработки в пищевой, фармацевтической и других отрас-
лях промышленности. Известны методики определения галоге-
нидов, нитрата, нитрита, сульфата, ацетата и т. д., всего свыше
70 анионов неорганических и органических кислот. Число катио-
нов значительно меньше. Методами ионной хроматографии опре-
деляют главным образом катионы щелочных и щелочно-земель-
ных металлов, а также органические катионы замещенных солей
аммония. Определение многих других катионов оказывается не-
надёжным, так как они выпадают в осадок в компенсационной
колонке с сильноосновной смолой. Ионная хроматография ус-
пешно применяется в анализе объектов окружающей среды (ат-
мосферы, воды и т. д.), в клинических исследованиях и многих
отраслях промышленности.
17.13. Осадочная хроматография
Разделение смеси в осадочной хроматографии основано на по-
следовательном образовании малорастворимых соединений (осад-
ков). Процесс разделения может быть проведен в колонке, на бума-
ге или в тонком слое. В ходе образования осадочной хроматограм-
мы происходит многократное повторение процесса образования и
растворения осадка, подобно тому, как в адсорбционной хроматог-
рафии многократно повторяется процесс сорбции десорбции. По-
рядок расположения осадков на хроматограмме зависит от ПР об-
разующихся соединений и соотношения концентраций компонен-
тов в анализируемом растворе. Вполне понятно, что чем больше
разница в растворимости осадков, тем более четким будет разделе-
ние на зоны при прочих равных условиях.
Колонками в осадочной хроматографии служат стеклянные
трубки диаметром 4—5мм и длиной 10—15 см. В качестве носи-
теля используются вещества, обладающие хорошей фильтруемо-
стью (силикагель, оксид алюминия и др.) и светлой окраской.
Осадителями являются реагенты, образующие малорастворимые
340 С
осадки анализируемого вещества. Однако в некоторых случаях
возможно образование осадка и без введения специального осади-
теля. Например, при заполнении колонки оксидом алюминия
кислотность хроматографируемого раствора будет понижаться и
при наличии в растворе ионов Ге3+, Сг3+ и др. возможно образова-
ние соответствующих гидроксидов или основных солей. Для луч-
шего разделения зон первичную хроматограмму промывают чис-
тым растворителем.
Качественный состав анализируемой пробы устанавливают
по цвету осадков и порядку их расположения в колонке. В случае
бесцветных осадков в колонку вводят раствор реактива, образую-
щего окрашенное соединение с осадком. В количественных опре-
делениях используют зависимость длины зоны осадка от кон-
центрации.
17.14. Общая характеристика метода
Хроматография является эффективным методом разделения
и анализа сложных по составу газообразных и жидких смесей.
Твердые вещества могут быть проанализированы после перевода
их в жидкое (растворенное) или газообразное состояние. Качест-
венный и количественный анализ проводится по характеристи-
кам удерживания. Универсальность газовой и газожидкостной
хроматографии значительно возрастает при сочетании хромато-
графического разделения и анализа компонентов масс-спект-
ральным или иным подходящим методом. Жидкостная распреде-
лительная хроматография особенно эффективна при разделе-
нии веществ, близких по химическим свойствам:, например ами-
нокислот.
В органическом и биохимическом анализе большое значение
имеет бумажная хроматография — простейший вариант хро-
матографическогр метода, обладающий высокой чувствитель-
ностью. Более воспроизводимые результаты дает тонкослойная
хроматография, широко применяемая в анализе лекарств, био-
химических проб и различных природных объектов. Ионооб-
менная хроматография ценна как метод разделения сложных
смесей ионов и как метод концентрирования микропримесей.
Успешно развиваются также новые хроматографические мето-
ды, например высокоэффективная жидкостная ионная хромато-
графия.
341
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. В чем сущность хроматографического процесса?
2. Как классифицируют методы хроматографии по агрегатному со-
стоянию фаз и по методике проведения эксперимента?
3. В чем состоит проявительный (элюэнтный) анализ?
4. Что такое: а) высота хроматографического пика; б) ширина хро-
матографического пика; в) общий удерживаемый объем; г) при-
веденный удерживаемый объем?
5. Какие достоинства и недостатки газовой адсорбционной и газо-
жидкостной хроматографии?
6. В чем состоит метод теоретических тарелок в хроматографии?
7. Что представляет собой кинетическая теория хроматографии?
8. Какие основные величины входят в уравнение Ван-Деемтера?
9. Какие особенности капиллярной хроматографии?
10. На чем основан качественный хроматографический анализ?
11. В чем состоит метод хромато-масс-спектрометрии?
12. В чем сущность основных методов количественной хроматографии:
а) метода нормировки; б) нормировки с калибровочными коэффи-
циентами; в) абсолютной калибровки; г) внутреннего стандарта?
13. Как влияет температура на хроматографический процесс?
14. Какое практическое значение имеет газовая хроматография?
15. Какие особенности имеет жидкостная абсорбционная хромато-
графия?
16. В чем сущность тонкослойной хроматографии (ТСХ)? Как прово-
дится качественный и количественный анализ методом ТСХ?
17. Какие варианты используются в жидкостно-жидкостной распре-
делительной хроматографии?
18. Чем характеризуется ионообменное равновесие?
19. Чем отличается ионная хроматография от обычной ионообменной?
глава 18
Математическое планирование
эксперимента в аналитической химии
18.1. Основные понятия и определения
Традиционный классический метод изучения сложных хими-,
ческих процессов и определения оптимальных условий проведе-
ния реакций состоит в том, что исследуется влияние одного пара-
метра при постоянстве остальных. Например, при выявлении опти-
342
Рис. 18.1. Модель объекта
исследования в виде
«черного ящика»:
х19 х2у хп — входы, факторы
или параметры; у = /(л^у л:2, хп) ~"
выход, функция отклика или ';
оптимизируемый параметр
мальных условий фотометрического
определения обычно изучают влия-
ние концентрации реагентов, рН рас-
твора, времени развития окраски и
других факторов на оптическую плот-
ность раствора в некотором спектраль-
ном: интервале. Изучение влияния
каждого фактора при постоянстве ос-
тальных требует больших затрат вре-
мени и средств, а порядок изучения
факторов определяется, в основном,
личным опытом и интуицией исследователя. Современные статис-
тические методы планирования экспериментапозволяютотыски-
вать оптимальные условия проведения реакции при значительном
сокращении числа опытов.
Метод анализа, являющийся в данном случае объектом иссле-
дования, можно представить в виде «черного ящика», как это
обычно делается в кибернетике (рис. 18.1). Независимые величи-
ны хг, х2 ит. д., влияющие на протекание реакции, называют
факторами, параметрами или входами. Их можно фиксировать
с определенной точностью. Величину у, являющуюся характерис-
тикой реакции, называют выходом, оптимизируемым парамет-
ром или функцией отклика.
При разработке, например, фотометрического метода анализа
факторами, или параметрами, являются концентрация раствора,
его рН, концентрация реагента и др. Функцией отклика или опти-
мизируемым параметром будет оптическая плотность раствора.
Координатное пространство, по осям которого отложены л^, #2
ит. д., называют факторным пространством. Функция отклика
в этом пространстве может быть изображена в виде поверхности
отклика. При геометрической интерпретации поверхности от-
клика обычно пользуются двумерным сечением, т. е. оставляют
постоянными все факторы, кроме двух, действие которых изуча-
ется в данном сечении. Поверхность отклика может иметь вид
«вершины», «хребта», «оврага»* «седла» и т. д.; встречаются и бо-
лее сложные конструкции. v
Систему уравнений, связывающих функцию отклика с факторами,
называют математическим описанием процесса или математиче-
ской моделью., - ;
Математические методы планирования эксперимента дают
возможность получить математическую модель процесса даже
при отсутствии сведений о его механизме или о физически обо-
снованных математических зависимостях между факторами и
функцией отклика.
343
*02
Математическую модель процесса
можно получить методом полного фак-
торного эксперимента или методом
дробных реплик. В методе полного фак-
торного эксперимента исследуется ло-
кальная область факторного простран-
ства вблизи выбранной точки с коорди-
натами х01, х02, х0п (рис. 18.2), т. е;.
зависимость функции отклика (пара-
метра оптимизации) от всех факторов
во всех возможных комбинациях. В ме-
тоде дробных реплик проводится толь-
ко некоторая часть полного факторного
эксперимента.
Начало координат факторного пространства перенесем в вы-
бранную точку, окрестности которой исследуются, и введем но-
4-1
+1
-І0
V -1-
1
1
1^
^*"|
Л01
Рис. 18.2. Введение
кодированных переменных
вую переменную Х£:
Х01
Ах1
(18.1)
где х1 — координата в старой системе, т. е. естественное, или на-
туральное, значение фактора I вблизи исследуемой точки; х01 —
координата выбранной точки, в окрестности которой ведется ис-
следование, в старой системе, т. е. натуральное значение фактора
I, взятое за нуль отсчета в новой системе; Ах1 — интервал варьи-
рования или масштаб по оси хг.
Величину Х1 обычно называют кодированной переменной.
В ходе полного факторного эксперимента в аналитической химии
все факторы варьируют на двух уровнях: верхнем и нижнем. Ес-
ли, например, нужно исследовать влияние рН на оптическую
плотность раствора в области рН от 5,0 до 10,0, то, очевидно, хь—':
= рН, верхний уровень х[ = 10,0, нижний х" = 5,0, а интервал
варьирования в данном случае составляет Дяг= 2,5. Выбранная
точка лежит в середине исследуемого интервала: х01 = 7,5. Коди-
рованное значение Х1 по уравнению (18.1) будет равно Х[ =
= 10,о = +1 для верхнего предела и Х'[ — 5,0 ~ 7,5 = -1 для.
¿,0 2,5
нижнего. Очевидно, натуральное значение фактора на верхнем
уровне кодируется всегда как 4-1, а на нижнем как -1. Таблицу
кодированных значений факторов называют матрицей планиро-
вания эксперимента. Вполне понятно, что она кодирует, в сущ-
ности, условия проведения опытов, предусматривающие в пбл-
ном факторном эксперименте все возможные комбинации факто-
344
ров. В качестве примера в табл. 18.1 приведены натуральные
значения факторов, составленные для изучения влияния рН и
концентрации растворов на оптическую плотность. В табл. 18.2
дана матрица планирования эксперимента, которая получена по
натуральным значениям факторов из табл. 18.1.
Таблица 18.1. Натуральные значения факторов
и интервал варьирования
Факторы
Основной
Интервал
Уровни
уровень
варьиро-
верхний
нижний
вания
*1 = РН
7,5
2,5
10
5
х2 = концентрация раствора
0,6
0,2
0,8
0,4
Та.б л и ц а 18.2. Матрица планирования при полном
двухфакторном эксперименте
Опыт
Факторы
Функция
отклика у
*1
Х2
1
-1
-1
У\
2
+1
-1
У2
3
-1
+ 1 .
Уз
4 •
+1
+1
У 4
Общее число опытов N в матрице планирования равно
ЛГ = 2", (18.2)
где п — число факторов.
В табл. 18.3 представлена матрица трехфакторного экспери-
мента.
Таблица 18.3. Матрица планирования при полном
трехфакторном эксперименте
Опыт
Факторы
Функция
отклика у
хг
х2
хз
хг~ х1х2
1
-1
-1
-1
+1
Ух
2
+1
-1
-1
-1
У2
Уз
3
-1
+1
-1
-1
4
+1
+1
-1
+1
У4
5
-1
-1
+1
—
Уъ
Уб
6
+1
-1
+1
—
7
-1
+1
+1:
—
Уч
8
+1
+1
+1
Уъ
345
При составлении матрицы полного факторного эксперимента
уровень варьирования первого фактора изменяется от опыта к
опыту, уровень варьирования второго фактора изменяется с час-
тотой, уже в 2 раза меньшей, чем первого. Как общее правило,
можно отметить, что уровень варьирования каждого последующе-
го фактора в матрице изменяется с частотой вдвое меньшей, чем
предыдущего. Это правило четко проявляется в матрицах в табл.
18.2 и 18.3. Например, при трехфакторном эксперименте^(табл.
18.3) третий фактор х3 остается постоянным (поддерживается на
нижнем уровне) во всех первых четырех опытах и опять остается
постоянным (на верхнем уровне) в последних четырех опытах.
Матрица планирования имеет следующие свойства:
где N — число опытов полного факторного эксперимента; у — но-
мер опыта; i, I, т — номера факторов.
18.2. Уравнение регрессии
и регрессионный анализ
Математическое описание процесса по данным полного фак-
торного эксперимента находят в виде уравнения регрессии:
у-Ь0 + ЪгХг + Ъ2Х2 + ... + ЬпХп + Ъ12ХгХ2 + ... + Ь(п1)пХп_1Хп + ЬпХ1 +
+ Ъ22Х% + ... + « + ...,
где Ь0—константа; bv b2, bn — коэффициенты, характери-
зующие линейные эффекты; blv b22,Ъпп — квадратичные эф-
фекты; Ь12 Ъ(П_ 1)л — эффектны взаимодействия:
Коэффициенты этого уравнения называют коэффициента-
Ми регрессии.
Коэффициенты регрессии рассчитываются по следующим
формулам: !
^.iviv,. '..Po^'ßrM ,1; (18.3)
v-J.l1* (18-4)
Ь1т=^ХХ^^щт1^т. .(18.5)
346
Для решения задач аналитической химии в уравнении рег-
рессии можно ограничиться лишь линейными членами. Тогда
расчетные формулы для коэффициентов регрессии, например,
при полном двухфакторном эксперименте в развернутой форме
будут иметь вид:
ь _ У\ + У 2 + Уз + У 4 .
0 ~ ——"1——~'
и _ (-1)У1 + (+1)уд + (-1)Уа + (+1)у4..
~~ 4 '
Ь _ (-1)У1 +(-1)Й +(+1)Уа +(+1)У4
В соответствии с требованиями регрессионного анализа дис-
персии строк матрицы должны быть однородными. Для неодно-
родных дисперсий математические методы планирования экспе-
римента неприменимы. Однородность дисперсий проверяется по
критерию Кохрена б:
С?=^р^, , (18.6)
где Щтах)— наибольшая из дисперсий, рассчитанных как
£ *2
5 = — , если (I — отклонение от среднего; к — число парал-
к-1
лельных.
Дисперсия считается однородной, если рассчитанное по урав-
нению (18.6) значение критерия Кохрена удовлетворяет условию
<2<(2табл,
где Стабл — табличное значение критерия Кохрена при данном N и
числе степеней свободы /, причем / = к - 1, если к, как и ранее,
число параллельных опытов. Табличные значения С?табл, соответст-
вующие доверительной вероятности 0,95, приведены в табл. 18,4.
Однородную дисперсию нескольких серий можно усреднить
и найти дисперсию воспроизводимости в* по соотношению
«»- I (18.7)
Число степеней свободы у 5| равно / = Щк - 1).
347
Таблица 18.4. Критерий Кохрена
N
/=1
/=2
/ = 3
/ = 4
/ = 5
/ = 6
/=7
/ = 8
/ = 9
/=10
/-16
Р =
0,95
2
0,9985
0,9750
0,9392
0,9057
0,8772
0,8534
0,8332
0,8159
0,8010
0,7880
0,7341
3
0,9669
0,8709
0,7977
0,7457
0,7071
0,6771
0,6530
0,6333
0,6167
0,6025
0,5466
4
0,9065
0,7679
0,6841
0,6287
0,5895
0,5598
0,5365
0,5175
0,5017
0,4884
0,4366
5
0,8412
0,6838
0,5981
0,5441
0,5065
0,4783
0,4564
0,4387
0,4241
0,4118
0,3645
6
0,7808
0,6161
0,5321
0,4803
0,4447
0,4184
0,3980
0,3817
0,3682
0,3568
0,3135
7
' 0,7271
0,5612
0,4800
0,4307
0,3974
0,3726
0,3535
0,3384
0,3259
0,3154
0,2756
8
0,6798
0,5157
0,4377
0,3910
0,3595
0,3362
0,3185
0,3043
0,2926
0,2829
0,2462
9
0,6385
0,4775
0,4027
0,3584
0,3286
0,3067
0,2901
0,2768
0,2659
0,2568
0,2226
10
0,6020
0,4450
0,3733
0,3311
0,3029
0,2823
0,2666
0,2541
0,2439
0,2353
0,2032
12
0,5410
0,3924
0,3264
0,2880
0,2624
0,2439
0,2299
0,2187
0,2098
0,2020
0,1737
15
0,4709
0,3346
0,2758
0,2419
0,2195
0,2034
0,1911
0,1815
01736
0,1671
0,1429
20
0,3894
0,2705
0,2205
0,1921
0,1735
0,1602
0,1501
0,1422
0,1357
0,1303
0,1108
Значимость коэффициентов регрессии устанавливается по
данным о погрешностях их определения в& и критерию Стьюден-
та £р. Если выполняется условие
Ь > 56*а или гр < (18.8)
то коэффициент регрессии значим, т. е. отличается от нуля. По-
грешности в определении коэффициентов регрессии одинаковы и
рассчитываются по формуле
(18.9)
Числовые значения критерия Стьюдента приведены в табл.
18.5. Слагаемые с незначащими коэффициентами из уравнения
регрессии исключаются. Полученное после этого уравнение рег-
рессии проверяется на адекватность, т. е. на точность описания
поверхности отклика. Проверка производится с помощью крите-
рия Фишера. Для этого сначала находят расчетное значение
функции отклика по уравнению регрессии и определяют диспер-
сию адекватности по формуле
тгН %<уГу№ (18-ю)
где В — число коэффициентов в уравнении регрессии, включая
свободный член; у? и у? — экспериментальное и рассчитанное по
уравнении} регрессии значения функции отклика в ;-м опыте.
Число степеней свободы у равно= N:- В.
Таблица 18.5. Коэффициенты Стьюдента (РР>^
/
Р =
Р =
Р =
Р =
Р =
/
р=
Р =
Р =
Р =
*=0,75
= 0,90
= 0,95
= 0,98
= 0,99
= 0,75
= 0,90
= 0,95
= 0,98
= 0,99
1
2,41
6,31
12,71
31,82
63,66
13
1,20
1,77
2,16
2,65
3,01
2
1,60
2,92
4,30
6,97
9,92
14
1,20
1,76
2,14
2,62
2,98
3
1,42
2,35
3,18
4,54
5,84
15
1,20
1,75
2,13
2,60
2,95
4
1,34
2,13
2,78
3,75
4,60
16
1,19
1,75
2,12
2,58
2,92
5
1,30
2,01
2,57
3,37
4,03
17
1,19
1,74
2,11
2,57
2,90
6
1,27
1,94
2,45
3,14
3,71
18
1,19
1,73
2,10
2,55
2,88
7
1,25
1,89
2,36
3,00
3,50
19
1,19
1,73
2,09
2,54
2,86
8
1,24
1,86
2,31
2,90
3,36
20
1,18
1,73
2,09
2,53
2,85
9
1,23
1,83
2,26
2,82
3,25
30
1Д7
1,70
2,04
2,46
2,75
10
1,22
1,81
2,23
2,76
3,17
40
1Д7
1,68
2,02
2,42
2,70
11
1,21
1,80
2,20
2,72
3,11
60
1,16
1,67
2,00
2,39
2,66
12
1,21
1,78
2,18
2,68
3,05
120
1,16
1,66
1,98
2,36
2,62
оо
1,15
1,64
1,96
2,33
2,58
349
Расчетное значение критерия Фишера вычисляют по формуле
(18.11)
Если полученное по этому соотношению значение критерия
Фишера не превышает табличной величины СРтабл) при данном
числе степеней свободы, уравнение регрессии является адекват-
ным и условие адекватности имеет вид ¥ < ^табл* Табличные
значения критерия Фишера приведены в табл. 18.6.
Таблица 18.6. Р-критерий при различной вероятности появления Р
и
./і-1
/і-2.
/г-8-
^ = 4
А-б
/і-в
/і-8
/і = Ю
/і = 12
/і = 20
Р = 0,95
1
161
200
216
225
230
234
239
242
244
241
2
18,51
19,00
19,16
19,25
19,30
19,33
19,37
19,39
19,41
19,44
3
10,13
9,55
9,28
9,12
9,01
8,94
8,84
8,78
8,74
8,66
4
7,71
6,94
6,59
6,39
6,26
6,16
6,04
5,96
5,91
5,80
5
6,61
5,79
5,41
5,19
5,05
4,95
4,82
4,74
4,68
4,56
6
5,99
5,14
4,76
4,53
4,39
4,28
4,15
4,06
4,00
3,87
7
5,59
4,74
4,35
4,12
3,97
3,87
3,73
3,63
3,57
3,44
8
5,32
4,46
4,07
3,84
3,69
3,58
3,44
3,34
3,28
3,15
9
5,12
4,26
3,86
3,63
3,48
3,37
3,23
3,13
3,07
2,93
10
4,96
4,10
3,71
3,48
3,33
3,22
3,07
2,97
2,91
2,77
В качестве примера найдем уравнение регрессии, связывающее оп-
тическую плотность раствора тиомочевинных комплексов олова(ІУ) (у) с
временем развития окраски концентрацией тиомочевины (х2) и кон-
центрацией хлорной кислоты (яг3). Полный трехфакторный эксперимент
проводился в окрестностях точки факторного пространства с координа-
тами х01 = 40 мин, #02 = 1,7 моль/л и хог = 2,5 моль/л. Условия проведе-
ния полного факторного эксперимента приведены в табл. 18.7.
Введем кодированные переменные:
■у _ х1 ~ х01-у _ х2 ~ х02л у хз7хог
Таблица 18.7. Характеристики плана экспериментов
Характеристика
х19 мин
д:2(ТЬЮ),
. моль/л.
х3 (НС104),
моль/л.
Основной уровень
40
1,7
2,5
Интервал варьирования
10
0,2
0,5 '
Верхний уровень
50
1,9
3,0
Нижний уровень
. 30
1,5
2,0
Матрица планирования и результаты эксперимента представлены в
табл. 18.8.
350
Таблица 18.8. Матрица планирования и результаты полного трехфакторного эксперимента
(опыты дублирования)
Опыт
*2
*3
Х1>
МИН
Х2
(ТЫО),
моль/л
*з
(НСЮ4),
моль/л
Оптическая
плотность
(А - А)2
Я2
"^расч
А -^расч
(А-А^)2
¿1
А
1
-1
-1
-1
30
1,5
2,0
0,395
0,388
0,392
1,6-10"5
3,2 .-10-е
0,395
-0,003
9-10"6
2
+1
-1
-1
50
1,5
2,0
0,438
0,420
0,429
8,1*10 5
1,62-10~4
0,426
0,003
9-Ю"6
3
-1
+1
-1
30
1,9
2,0
0,513
0,548
0,531
3,24-Ю"4
6,48-Ю"4
0,536
-0,005
2,5-10~5
4
+1
+.1
-1
50
1,9
2,0
0,558
0,584
0,571
1,69-Ю"4
3,38-О"4
0,567
0,004
1,6 -Ю"5
5
-1
-1
+1
30
1,5
3,0
0,533
0,491
0,512
4,41 • 10"4
8,82-Ю"4
0,505
0,007
4,9-10"5
6
+1
-1
+1
50
1,5
3,0
0,538
0,518
0,528
1-Ю"4
2-Ю"4
0,536
-0,008
6,4-Ю-5
7
-1
+1
+1
30
1,9
3,0
0,640
0,652
0,646
3,6 • 10"5
7,2-Ю"5
0,646
0,000
0,0000
8
+1
+1
+1
50
1,9
3,0
0,682
0,673
0,678
2,5-10"5
5,0 -Ю-5
0,677
0,001
ыо-6
1 = 2,384 • 10"3
2=1,73 -Ю"4
Для проверки однородности дисперсий рассчитываем по уравнению
(18.6) критерий Кохрена:
5£ах = 8,82 - 10~4 в о,3700
£5? 2,384 • Ю-3
й сравниваем это значение с табличным. Из табл. 18.4 для / = 1, Р = 0,95
и N = 8 находим Отабл = 0,6798. Табличное значение превышает рассчи-
танные из экспериментальных данных (0,37), поэтому дисперсии следу-
ет квалифицировать как однородные.
Дисперсия воспроизводимости по уравнению (18.7) будет равна
и дисперсия среднего
аь . 2,384 ^ = 2,98.10-4
о
£2 = 2'98 • 10-4 = 1,49 • 10"4.
Число степеней свободы дисперсии воспроизводимости составляет
/ = ЩИ - 1) = 8(2 - 1) = 8. Теперь по формулам (18.3)—-(18.5) рассчитыва-
ем коэффициенты уравнения регрессии:
Ъ0 = 1/8(0,392 + 0,429 + 0,531 + 0,571 + 0,512 + 0,528 + 0,646 + 0,678) =
= 0,5359 = 0,536;
Ьг = 1/8(-0,392 + 0,429 -0,531+0,571 - 0,512 + 0,528 - 0,646 + 0,678) =
.= 0,01563 = 0,0156;
Ь2 = 1/8(-0,392 - 0,429 + 0,531 + 0,571 - 0,512 - 0,528 + 0,646 + 0,678) = '-
= 0,07063 = 0,0706;
Ьг = 1/8(-0,392 - 0,429 - 0,531 -0,571 + 0,512 + 0,528 + 0,646 + 0,678) =
= 0,05513 = 0,0551.
Значимость полученных коэффициентов оцениваем по критерию Стью-
дента. Для этого сначала определим погрешность коэффициентов по форму-
ле(18.9): •
^= /5; 5Ь= /2>98'10"4 = 4,31 >10-з.
Затем составляем ^-отношение для всех коэффициентов уравнения
регрессии:
t = 12
0,536 =124,4;
0 в,, 4,31 • 10-»
°'°156 =3,62;
1 4,31 • Ю-3
Ъг= 0,0706 16)38.
2 Я» 4,31 • Ю-»
&з = 0,0551 12>78.
3 в» 4,31 • Ю-3
352
Для Р = 0,95 и / — 8(2 - 1) = 8 табличное значение критерия Стыо-
дента равно ^95. 8 = 2,31. Это удовлетворяет условию (18.8), следова-
тельно, все коэффициенты уравнения регрессии значимы и уравнение
регрессии имеет вид
Арасч = 0,536 + 0,0156Х1 + 0,0706Х2 + 0,0551Х3. (18.12)
'V ' •
Найденные по этому уравнению значения оптической плотности Арасч
приведены в табл. 18.8.
Адекватность уравнения эксперименту проверяем по критерию Фи-
шера. Дисперсию адекватности рассчитываем по соотношению (18.10) и
данным табл. 18.6:
= 1ГЪ ^ - А-ао,)2 = !'73 *■ 10-4 = 8'65 • 10"6-
По формуле (18.11) находим ^-отношение:
>=2,98 ' 10"4 =3,45
8,65 • Ю-5
(в числитель ^-отношения ставят большую из дисперсий). Из табл. 18.6
находим, ,что для Р = 0,95, 2^ — 4 и ¥г — 8 критерий Фишера равен
^о,9б(4,8) ™ Тв е* ^табл > Следовательно, уравнение регрессии (18.12)
адекватно эксперименту.
18.3. Дробный факторный эксперимент
Уравнение (18.2) показывает, что с увеличением числа фак-
торов резко возрастает объем полного факторного эксперимента.
Если в уравнении регрессии можно ограничиться только линей-
ными членами, число опытов уменьшают с помощью дробного
факторного эксперимента или методом дробных реплик от пол-
ного факторного эксперимента. В качестве реплики берут пол-
ный факторный эксперимент для меньшего числа факторов. При
этом число опытов должно остаться превышающим или равным
числу неизвестных в уравнении регрессии. Например, в полном
трехфакторном эксперименте, как показывает табл. 18.3, дол-
жно быть проведено восемь опытов. Можно, однако, ограничить-
ся четырьмя опытами, если в матрице планирования в качестве
плана для Х3 использовать произведение Х1Х2* Такой сокращен-
ный план называют полурепликой полного факторного экспери-
мента. Используют также четверть реплики и более дробные
реплики. Если парные взаимодействия относятся к статистиче-
ски значимым, то соответствующие коэффициенты регрессии не
будут равны нулю и, следовательно, полученные в методе дроб-
23-7831
353
ных реплик коэффициенты регрессии будут смешанные, отра-
жающие эффект фактора и парного взаимодействия. Вид плани-
рования эксперимента, когда некоторые факторы приравнивают-
ся к произведению факторов, называют планированием со сме-
шиванием и обозначают как 2п~р> где п тт- общее число факторов,
р — число факторов, которые приравниваются произведению.
Соотношение типа
' Хг = Х1Х2 (18.13)
называют генерирующим соотношением. Оно показывает* с ка-
ким эффектом смешан данный эффект.
При умножении соотношения (18.13) на Х"3 получаем ^
но Х"^ = 1, поэтому
- 1-ВДХ3. (18.14)
Произведение (18.14) называют определяющим контрас-
том. С его помощью определяют смешанные элементы. Для это-
го его умножают поочередно на Х2 и Х3:
Х1 — Х^Х2Х3 = Х2Х3; Х2 = Х^Х^*, Х$ = Х^Х2.
Отсюда получаем систему смешанных оценок:
> &1 = + Ргз5 &2 = Рг + Р13? &з ^ Рз + Р12'
где Р — истинные коэффициенты ряда Тейлора.
Статистическую значимость имеют не все парные взаимодей-
ствия, поэтому не все коэффициенты регрессии являются сме-
шанными. Составление уравнения регрессии и его анализ в мето-
де дробных реплик не отличаются от соответствующих операций
в полном факторном эксперименте.
18.4. Оптимизация
по методу крутого восхождения
Положительное значение коэффициентов уравнения регрес-
сии (18.12) показывает, что оптическая плотность раствора будет
возрастать с увеличением всех факторов: времени развития окра-
ски, концентрации тиомочевины и концентрации хлорнбй кисло-
ты. Поскольку количественной мерой влияния каждого из фактов
ров на параметр оптимизации является коэффициент регрессии^
наиболее существенное влияние на оптическую плотность раство-
ра при выбранных интервалах варьирования будет оказывать кон-
354
центрацйя тиомочевины. С увеличением интервалов варьирова-
ния абсолютное значение коэффициентов уравнения регрессии
также возрастает, что может привести к изменению относитель-
ного вклада каждого из факторов. Это обстоятельство играет су-
щественную роль при выборе интервалов варьирования.
Задача оптимизации ев данном случае заключается в поиске
условий или значений факторов, при которых оптическая плот-
ность раствора заданной концентрации по олову будет макси-
мальной. Вполне понятно, что при изменении независимых пере-
менных пропорционально коэффициентам регрессии функция
отклика будет эффективно отражать суммарное действие всех
факторов. Для получения максимального значения функции от-
клика необходимо увеличивать те переменные, которые входят в
уравнение регрессии с положительным знаком, и уменьшать те,
которые входят с отрицательным. Бокс и Уилсон разработали
прием достижения максимума в функции отклика, получивший
название крутого восхождения. Техника расчета по этой мето-
дике заключается в следующем. Один из факторов, например Х2,
выбирают как базовый и вычисляют для него! произведения
коэффициента регрессии Ъ2 на интервал варьирования ДХ2, т. е.
Ь2ДХ2, и определяют шаг движения по градиенту ДХ*. Выбор
АХ* является очень важным элементом расчета. Чрезмерно ма-
лый шаг потребует очень большого числа опытов, а при слишком
большом шаге могут быть не замечены важные особенности сис-
темы или будет очень быстро превышен предел физически воз-
можных значений фактора (например, концентрация раствора
превысит растворимость соединения и т. д.). Величина шага
должна, конечно, существенно превышать погрешность измере-
ния фактора. Обычно принимают АХ* < АХ. Затем вычисляют
отношение
У=г^_. (18.15)
Величину шага для других факторов рассчитывают по формуле
АХ^уЪ^Хи ,
Движение начинают из центра плана путем прибавления
АХ* к основному уровню и последующим значениям факторов.
В данном примере выбираем шаг АХ2 = 0,1 и по уравнению
(18.15) находим пересчетный множитель у:
.■^^/■..г..---:.у = 0Д = 7,08. , •
1 0,0706 -0,2
23'
355
Тогда шаг по времени развития окраски будет равен
= ДХХ = 7,08 • 0,0156 • 10 = 1,1 - 1
и шаг по концентрации хлорной кислоты АХ* = ybbB AXS = 7,08 х
х 0,0551 - 0,5 = 0,2.
Для удобства расчетов значение шага обычно округляют. Pe-í
зультаты расчетов по методу крутого восхождения приведены в
табл. 18.9. Значение Арасч получено по уравнению регрессии
(18.12). Это результат так называемых мысленных опытов. При
адекватной модели в экспериментально изученной области вли-
яющих факторов результаты мысленных опытов эксперимен-
тально обычно не проверяются. В рассматриваемом примере
(табл. 18.9) это относится только к опытам с первого по третий.
Опыт четвертый и следующие по концентрации тйомочевины
уже выходят за пределы экспериментально изученной области,
цоэтому результаты мысленных опытов, начиная с четвертого,.
следует проверить экспериментально. Одндко возможности экс-
периментальной проверки ограничиваются растворимостью тй-
омочевины в воде, составляющей при 25 °С примерно 2 моль/л.
Можно зафиксировать АХ2 примерно на этом уровне и еще раз
рассчитать крутое восхождение с учётом изменения только Хг и
Х3. Однако более целесообразным представляется проведение
полного факторного (двухфакторного) эксперимента, зафиксиро- ,
вав постоянную концентрацию тйомочевины в 2 моль/л и взяв в
качестве центра плана оптимальные условия, например условия
второго опыта. i
Т а б л и ц а 18.9. Расчет крутого восхождения
X Характеристика
и номер опыта г
xv мин
*2
(ThiO),
моль/л
Ч
(НС104),
моль/л
Кодированные
значения
•^расч
*1
Основной уровень
(центр плана)
40
1,7
2,5
0
0
0
0,536
Интервал варьирования
10
0,2
0,5
1
1
1
Шаг движения
1
0,1
0,2
0,1
0,5
0,4
—
Опыт 1
41
1,8
2,7
0,1
0,5
0,4
0,595
V 2
42
1,9
2,9
0,2
1,0
0,8
0,654
3
43
2,0
3,1
0,3
1,5
1,2
0,713
- » 4
44
2,1
3,3
0,4
2,0
1,6
0,772
» 5
45
2,2
3,5
0,5
2,5
2,0
0,831
, » 6
46
2,3
3,7
0,6
3,0
2,4
0,889
356
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Каким методом можно получить математическую модель про-
цесса?
2. Что называют матрицей планирования эксперимента?
3. Какие величины входят в уравнение регрессии?
4. По какому критерию определяется однородность дисперсии?
5. По каким данным устанавливается значимость коэффициентов
регрессии?
6. По какой формуле вычисляют критерий Фишера?
глава 19
Средства и методы оперативного
аналитического контроля.
Применение тест-методов
и сенсоров в анализе
19.1. Особенности тест-методов и сенсоров
Тест-методы ц сенсоры ..— это группа аналитических уст-
ройств для прямого, селективного, максимально автомагазинов
ванного в случае сенсоров, а также простого и быстрого контроля
или диагностики разнообразных объектов. Они не заменяют
обычных методов анализа, а позволяют получать оперативные
данные о состоянии объекта и принимать решение о необходи-
мости отбора пробы и последующего лабораторного анализа.
Появление и применение в качестве инструмента химическо-
го анализа тест-методов и сенсоров связано с несколькими причи-
нами. Одной из основных является накопление большого количе-
ства техногенных веществ в окружающей среде, вызвавшее необ-
ходимость постоянной оценки уровня загрязненности больших
территорий. Отбор огромного числа проб и их анализ в стаци-
онарных лабораториях потребовал бы резкого увеличения чис-
ленности аналитиков, количества проведенных анализов и, соот-
ветственно, не всегда оправданных материальных затрат. Ис-
пользуя' тест-методы и сенсоры, оценку содержания того или
357
иного вещества в воздухе, воде, почве, растениях и т. д. прямо на
месте может производить не только химик-аналитик, но и гео-
лог, эколог, биолог ИТ. д.
Другая область применения тест-методов и сенсоров — конт-
роль появления ядовитых или отравляющих и других опасных
веществ в воздухе рабочей зоны, населенных пунктах, а также
состояния среды в труднодоступных условиях, например при из-
вержении вулканов, или в организме человека.
19.2. Тест-методы
В тест-методах используются упрощенные приемы и приспо-
собления для быстрого обнаружения и полуколичественной оцен-
ки содержания химических веществ в различных объектах без
отбора пробы, т. е. в полевых условиях. Тест-методы обычно не
требуют сложного лабораторного оборудования и специального
обучения персонала. Тест-методы могут быть химическими, био-
химическими и биологическими. Конструктивно они выполня-
ются в виде индикаторных лент, полосок, трубок или специаль-
ных наборов растворов реагентов с вспомогательными приспособ-
лениями для проведения реакций. Химические и биохимические
тест-наборы, как правило, снабжаются стандартными цветовыми
шкалами, по которым оценивают приближенное содержание оп-
ределяемого компонента в объекте.
Химические тест-методы используют реакции с окрашенны-
ми неорганическими или органическими реагентами, которые
при контакте с определяемым компонентом изменяют цвет. Ок- :
рашенный реагент наносят на твердую матрицу, в качестве кото-
рой используют впитывающую целлюлозную бумагу, полимер-
ные материалы или неорганические сорбенты. Закрепление
реагентов на матрице может быть адсорбционным или ко-
валентным.
в Адсорбционное закрепление осуществляется пропиткой мат-
рицы раствором соответствующего реагента. Матрица может до-
полнительно пропитываться буферными растворами, маскирую-
щими, эмульгирующими, закрепляющими соединениями, ката-
лизаторами, стабилизаторами и т. д. Примером такого типа
тест-методов являются обычные индикаторные бумажки для оп-
ределения рН раствора. Недостатком этого способа закрепления
реагента является возможность его вымывания водой и загрязне-
■ ' 358' " 1
ция исследуемого р<аствора. Поэтому подобные тесты предназна-
чены для однократного использования.
Другой способ нанесения реагента, устраняющий недостатки
предыдущего, заключается в его иммобилизации на матрице пу-
тем многостадийного синтеза с использованием вспомогательных
реагентов .В результате реагент ковалентно связывается с мате-
риалом матрицы. Такой способ закрепления реагентов приводит
к тому, что тесты можно длительно хранить и многократно при-
менять, разрушая образующиеся продукты реакции промывани-
ем водой или кислотой. Если матрицей является бумага, то такие
тесты называют реактивными индикаторными бумагами
(РИБ). При проведении анализа интенсивность возникающей ок-
раски можно оценивать не только по стандартной цветовой шка-
ле, но и измерять количественно с помощью денситометров по
спектрам отражения продукта реакции.
Устройства, используемые в тест-методах, несмотря на их
простоту, могут обладать многофункциональным действием. Они
позволяют производить предварительное разделение и концент-
рирование веществ, их окисление или восстановление и в связи с
этим содержать несколько слоев. Измерение концентрации мо-
жет проводиться сравнением полученного эффекта с приложен-
ной шкалой, а также по длине окрашивающейся или обесцвечи-
вающейся зоны. Контакт с анализируемым объектом осуществ-
ляется прямым опусканием РИБ в жидкость или прокачиванием
воздуха через индикаторную трубку.
В биохимических тест-методах на матрице иммобилизируют
различные ферменты, антигены или антитела. Биотесты позво-
ляют контролировать содержание болезнетворных микроорга-
низмов в природных и питьевых водах.
Тест-методы применяют для качественного и количественно-
го экспресс-анализа природных и сточных вод, технологических
растворов, биологических жидкостей, воздуха и других газовых
сред при оценке экологической ситуации, санитарном и техноло-
гическом контроле, медицинском обследовании. С помощью РИБ
определяют ионы меди, никеля, цинка, кобальта, ртути в степе-
ни окисления +2, железа(П,Ш) с пределами обнаружения
0,001^-0,1 мг/л, ацетон, глюкозу в крови и моче, кислотность
(рН) среды, формальдегид, нитраты, нитриты. Для определения
газов и отравляющих веществ чаще используют индикаторные
трубки. Наборы для тестового определения веществ выпускаются
как зарубежными, так и отечественными фирмами.
359
19.3. Сенсоры
Сенсорами (от лат. зепзив — чувство) называют чувствитель-
нее элементы небольших размеров, генерирующие аналитиче-
ский сигнал, интенсивность которого зависит от концентрации
определяемого вещества в объекте. В отличие от тест-методов
сенсоры позволяют проводить не визуальное, а инструменталь-
ное количественное измерение содержания вещества, связанное с
предварительной градуировкой прибора по стандартам.
Сенсоры (сенсорные анализаторы) являются по сути дела ос-
новными элементами нового поколения аналитических прибо-
ров, включающих устройство для ввода пробы, чувствительный"
элемент, обработку аналитического сигнала и выдачу конечно-
го результата о концентрации компонента. Для них характерны
малая масса (редко превышающая 200 г) и габариты примерно
100 х 50 х 20 мм, автономный, автоматизированный режим рабо-
ты и малый расход энергии.
г Существует три типа сенсоров: физические, химические и
биосенсоры. В физических сенсорах какие-либо реакций отсутст-
вуют, а под влиянием анализируемого вещества изменяются
электрические, тепловые, магнитные или спектральные характе-
ристики. ;
Например, для определения концентрации С02 используют-
ся кондуктометричёские сенсоры, основанные на измерении
электрической проводимости водного раствора диоксида углеро-
да. Проводимость анализируемого раствора определяется кон-
центрацией ионов Н+ и НСО3, Которая в свою очередь зависит от
парциального давления С02. Увеличение электрической проводи-
мости анализируемого раствора по сравнению с холостым (без
С02) является аналитическим сигналом.
Отличительный признак химических сенсоров и биосенсбров —
наличие рецептора — слоя молекул или частиц вещества, участ-
вующего в химических, биохимических или биологических про-
цессах, протекающих при контакте сенсора с определяемым ком-
понентом объекта. Другим необходимым элементом таких сенсо-
ров является преобразователь энергии (трансдъюсер) указанных
аналитических процессов в электрический или световой сигнал.
Далее этот сигнал обрабатывается в электронном блоке и подает-
ся на дисплей.
В химических сенсорах роль рецептора играют различные
реагенты, которые изменяют оптические, электрохимические
или иные характеристики при изменении рН раствора, взаимо-
360
действии с катионами, анионами или молекулами газов в жид-
ких и газообразных объектах промышленного производства, ок-
ружающей среды, медицины.
Селективность электрохимического сенсора в значительной
степени определяется материалом электрода. Например, опреде-
ление Ы02 в выхлопных газах двигателей осложняется присутст-
вием в смеси 02, а также СО, Н2 и углеводородов. Платиновый
электрод в качестве индикаторного не подходит из-за малой се-
лективности. Наиболее подходящим является электрод, модифи-
цированный фталоцианином кобальта, на котором вступает в ре-
акцию только Ж)2.
Химические сенсоры дают прямую информацию о составе
среды без отбора пробы и ее какой-либо предварительной подго-
товки. Они могут работать автономно и обычно связаны с систе-
мой накопления информации и ее обработки.
Для повышения избирательности химических сенсоров пе-
ред химически чувствительным слоем обычно размещают ионо-
обменные и иные мембраны, селективно пропускающие частицы
определяемого вещества.
В бйосенсорах рецепторами являются ферменты, антитела,
антигены, биологические мембраны (биохимические сенсоры),
или микроорганизмы (биологические сенсоры). Основная область
применения биосенсоров — анализ жидких сред в медицине, био-
технологии, химической, пищевой промышленности и окружаю-
щей среде. О содержаний определяемого вещества в объекте час-
то судят по концентрации продуктов биохимической реакции,
например пероксида водорода, кислорода и т. д. Преимущества-
ми биосенсоров являются высокая селективность и чувствитель-
ность определений, недостатками — невысокая стабильность,
трудность получения биоорганическогр материала постоянного
состава.
Трансдьюсерами в биосенсорах могут быть электрохимиче-
ские и оптические преобразователи, калориметрические системы
ИТ. Д.. '.. . ./О,:'
Наибольшее развитие получили ферментные и клеточные
биосенсоры.
Сочетание ферментативных и электрохимических реакций
позволило разработать многие биосенсоры для определения ами-
нокислот, глюкозы, мочевины и других продуктов жизнеде-
ятельности.
Развитие методов включения живых: клеток в полимеры и
твердые носители привело к созданию соответствующих биосен-
361
соров с использованием живых иммобилизированных Клеток, об-
ладающих удивительными свойствами.
Применения биосенсоров весьма многообразны, известны
биосенсоры для определения фенолов, пролина, тирозина и дру-
гих аминокислот, глюкозы, молочной и аскорбиновой кислот
и т; д. Уникален экспресс-анализ качества воды и сточных вод на
основании определения ВПК (биологическое потребление кисло-
рода). Традиционный метод анализа занимает несколько дней,
биосенсор с иммобилизированными клетками дает те же резуль-
таты в течение нескольких минут.
Основной областью приложения биосенсоров является ана-
лиз жидких сред в медицине, пищевой и химической промыш-
ленности, биотехнологии. Применение биосенсоров в качестве
биологического компонента ДНК может радикально изменить
диагностику заболеваний, особенно на ранних стадиях. Интен-
сивно ведутся работы для ранней диагностики сердечно-сосудис-
тых заболеваний, предрасположенности к раку и т. д.
Другая классификация сенсоров основана на способе регист-
рации аналитического сигнала. Сенсоры делят на электрические,
электрохимические (вольтамперометрические, потенциометри-
ческие, кондуктометрические, кулонометрические), оптические
(фотометрические (УФ, ИК)), люминесцентные, оптотермиче-
скце), сенсоры, чувствительные к изменению массы, ИТ. д.
К электрическим сенсорам относят полупроводниковые уст-
ройства с электронной проводимостью на основе оксидов Sn, Zn,
Cd, Cr, Ti, W, V, органических полупроводников (xeлаты фтало-
цианинов, порфиринов и другие органические соединения), поле-
вых транзисторов. Измеряемыми величинами являются проводи-
мость, заряд, емкость, разность потенциалов, изменяющиеся при
адсорбции или ином воздействии определяемого вещества. Наи-
более распространены и перспективны полевые транзисторы, в
которых металлический контакт затвора (управляющего фгёкт-
рбда) заменен химически чувствительным слоем и электродом
сравнения. Главными достоинствами полевых транзисторов яв-
ляются малые размеры (1—2 мм2) и масса, быстродействие и пле-
нарная модульная технология изготовления. г
В электрохимических сенсорах химическое превращение и
генерация аналитического сигнала протекают в миниатюрной
электрохимической ячейке, выполняющей роль йоноселёктивно-
го Электрода с жидкой или твердой мембраной. Наиболее распро-
странены потенциометрические и амперометрическиё сенсоры,
мембраны которых могут содержать как химические; Так й био-
химические компоненты. С помощью электрохимических сёнСо-
362
ров определяют ионные и нейтральные соединения органической
и неорганической природы, а также газы и биологически актив-
ные вещества в широком диапазоне концентраций (2—4 порядка).
Действие оптических сенсоров (оптодов — оптических
электродов, оптодов) основано на измерении поглощения и отра-
жения падающего светового потока, люминесценции или тепло-
вого эффекта, сопровождающего поглощение света рецептором.
В волоконно-оптических сенсорах фоточувствительный реагент
может быть иммобилизован на поверхности волокна световода,
изготовленного из кварца или других видов стекол, позволяю-
щих работать в УФ-, видимой и ИК-области спектра. Разработа-
ны оптические сенсоры для определения рН, ионов металлов,
анионов, глюкозы, мочевины, пероксида водорода, газов, некото-
рых органических соединений в объектах окружающей среды,
медицине, промышленности.
Сенсоры предназначены для прямого определения конкрет-
ного вещества в заданном диапазоне концентраций при фиксиро-
ванных способах ввода пробы и обработки полученной информа-
ции. Они могут входить в состав более сложных аналитических
приборов. Для повышения избирательности определения на вхо-
де в сенсорное устройство иногда помещают селективные мембра-
ны. Мембрана может также концентрировать определяемый ком-
понент и способствовать повышению чувствительности его опре-
деления. Основное преимущество сенсоров — малые размеры,
способность работать в автоматическом, автономном и непрерыв-
ных режимах.
На основе сенсоров конструируют сенсорные анализаторы,
представляющие собой батарею отдельных сенсоров, каждый из
которых дает информацию о содержании отдельного компонента.
Подключенная к компьютеру, такая батарея обеспечивает анализ
сложных многокомпонентных смесей.
Сенсорные анализаторы широко используются в различных
областях промышленности, энергетике, транспорте, медицине,
экологии, сельском хозяйстве. Большое значение имеет своевре-
менное обнаружение взрывчатых, горючих, вредных веществ и т. д.
Сенсоры дают возможность оптимизации работы двигателей по
составу выхлопных газов. В медицине с их помощью проводится
экспрессное определение состава крови и других физиологиче-
ских жидкостей, исследуется поведение лекарств, химически
вредных веществ, наркотиков и т. д. Очень важно обнаружение
взрывчатых, химически вредных веществ, утечки ракетного топ-
лива и т. д.
363
Сенсорные анализаторы контролируют содержание многих
органических и неорганических примесей в воздухе, природных
и сточных водах.
Применение сенсоров в анализе является новым, интенсивно
развивающимся направлением.
КОНТРОЛЬНЫЕ ВОПРОСЫ
Для каких целей используют тест-методы и сенсоры?
Какие используют тест-методы и из чего они выполняются?
Каким действием обладают устройства, используемые в тест-ме-
тодах?
Для каких целей применяют сенсоры? В чем отличие сенсоров от
тест-методов?
Чем отличаются различные типы сенсоров?
Каково преимущество сенсоров?
С п и с ok лит ер ату р ы
Айвазов Б. В. Введение в хроматографию. — М.: Высшая школа,
1983.
Алесковский В. Б., Бардин В. В., Бойчинова Е. С и др. Физико-хи-
мические методы анализа. Практическое руководство. — Л.: Химия,
1988.
Аналитическая химия. Физические и физико-химические методы
анализа / Под ред. О. М. Петрухина. — М.: Химия, 2001.
Булатов М. И., Калинкин И. П. Практическое руководство по фото-
колориметрическим методам анализа.— Л.: Химия, 1986.
Васильев В. П. Термодинамические свойства растворов электроли-
тов. — М.: Высшая школа, 1982.
Вяхирев Д. А., Шушунова А. Ф. Руководство по газовой хромато-
графии. — М.: Высшая школа, 1987.
Лурье Ю. Ю. Справочник по аналитической химии. — М.: Химия,
1989.
Основы аналитической химии / Под ред. академика Ю. А. Золотова. —
М.: Высшая школа, 2002. Кн. 1, 2.
Петере Д., Хайес Д., Хифтье Г. Химическое разделение и измере-
ние. Теория и практика аналитической химии. — М.: Химия, 1987. Т. 1,2.
Пилипенко А. Т., Пятницкий И. В. Аналитическая химия. —:М.:
Химия, 1990. Т. 1, 2.
Скуг Д., Уэст Д. Основы аналитической химии. — М.: Мир, 1979.
Т. 1,2.
Терек Т., Мика И., Гегуш Э. Эмиссионный спектральный анализ. —
М.: Мир, 1982. Т. 1, 2.
Чарыков А. К. Математическая обработка результатов химического
анализа. — Л.: Химия, 1984.
Юинг Д. Инструментальные методы химического анализа. — М.:
Мир, 1989.
П Р И Л О ЖЕ НИЕ
1. Стандартные окислительно-восстановительные потенциалы
Электродная реакция
Электродная реакция
я°,в •
Ag+ + е~ = Ag
0,7991
Си2+ + е~ = Си+
0,167
AgBr + е~ = Ag + Br-
0,073
¥2 + е- = 2¥-
2,65
AgCl + е~ = Ag + С1"
0,222
¥е3+ + 3е~ = ¥е
-0,036
Ag2S + 2e- = Ag + S2-
-0,71
¥е2+ + 2е~ = ¥е
-0,441
А13+ + 3£Г = А1
-1,67
¥е(СЯ)§- + е~ =
= ¥е(СЯ)%-
0,36
А1Г|- + Зе" = А1 + 6¥~
-2,07
¥ег+ + е~ = ¥е2+
0,771
Н3Ав04 + 2Н+ + 2е" =
= НдАзОд + Н20
0,559
2Н+ + 2е" = Н2
0,00
АвО|- + 2Н20 + 2е~ = '
= Ав02 + 40Н"
-0,71
2Н+ + (10"7М) + 2е~ =
-н2
-0,413
Аи3+ + 3в" = Аи
1,50
4Н+ + 02 + 4е~ = 2Н20
1,229
Ва2+ + 2<г = Ва
-2,9
4Н+ + (10-7М) + 4е~ =
= 2Н2
0,82
В13+ + Зв~ = В1
0,3172
Н202 + 2Н+.+ 2е~=
= 2Н20
1,77
ЫаВЮ3 + 6Н++Зе- =
- Вг*+ + Ыа+ + ЗН20
>1,8
21^2+ + 2е-:-
0,920
Вг2(ж) + 2е~ = 2Вг~
1,0652
^2+ + 2<г = Н£
0,789 .
366
Окончание табл. 1
Электродная реакция
Я°,В
Электродная реакция
д°,в
ВгОд + 6Н+ + Зе~ =
= Вг- + ЗН20
1,44
Нё2С12 + 2е~ = 2^ +
'+ 2С1"(1н.КС1)
0,283
2С02 + 2Н+ + 2е~ -
= Н2С204 (водн)
-0,49
Н§2С12 + 2е~ = 2Н&+
+ 2С1"(аС1_.- 1)
0,267
Са2+.+ 2*- = Са
-2,87
12(т) + 2в- = 21-
0,534
С(12+ + 2е- = С(1
-0^402
12 + 2е- = 2Г
0,621
Се4+ + е=Се3+
1,61
Юз + 6Н+ + 6е- = 1- +
+ ЗН20
1,085
С12 + 2е- = 2С1-
1,3595
К+ + «г = К
-2,934 ,
НСЮ; + Н+-Ь 2е~ =
= С1- + Н20
1,49
ЫОд * 4Н+ + Зе- =
= N0 + 2Н20
0,96 с
СЮз + ЗН20 + 6<Г =
= С1- + 60Н-
0,62
№2+ + 2е~ = №
-0,250
СЮ 1+ 2Н+ + 2<г
= С10з- + Н20
1,19
РЬ2+ + 2е- = РЬ
-0,126 ,
Со3+ + <г = Со2+
1,82
РЬ02 + 4Н+ + 2(Г =
= РЬ2+ + 2Н20
1,455
Со2+ + 2е- = Со
-0,277
840|" + 2е" = 2820|-
0,09/
Со(МНз)3++>- =
= Со(КН3)62+
0,1 !
80|- + 4Н++ 2е~ -
= Н2803 + Н20
0,17
Сг3+ + Зе~ = Сг
-0,71
вп4++ 2е" ^ 8п2+
0,15
Сг26|-+ 14Н++ бе" =
= 2Сг3++7Н20
1,33
Ш|++ 4Н++ 2е" =
= и4+ + 2Н20
0,33
№'+¿- = 03
-3,02
УО£ + 2Н++"в~ = "
= У02+ + 2Н20
1,00
Си+ + 2е~ - Си
0,345
гп2+ + 2е- = гп
-0,762
367
2. Подвижности ионов при 25 °С
Ион
См • см2/моль
Ион
См • см2/моль
Ион
См • см2/моль
Аё+
61,9
1/3 Ьа3+
69,7
Вг"
78,4
1/3 Ags+
63
Ы+
38,7
ВгОз
55,8
1/2 Ва2+
63,7
1/2 ]У^2+
53,1
СНдСОО"
41
1/2 Ве2+
45
1/2 Мп2+
53,5
С1-
76,3
1/2 Са2+
59,5
Ыа+
50,1
сю^
63,7
1/2 са2+
54
1/3 Ш3+
64,3
сы-
78
1/3 Се3+
67
73,7
1/2 СО|"
69,3
1/2 Со2+
54
1/2 №2+
54
1/2 СгО|"
85
1/3 Сг3+
67
1/2 РЬ2+
70
Б1"
55,4
Сз+
76,8
1/3 Рг3+
65,4
Г
76,9
1/2 Си2+
56,6
1/2 Ка2+
66,8
Юз
41
1/3 Еи3+
67,8
ИЬ+
77,5
1/2 Мп(^
62,8
1/2 ¥е2+
53,5
1/3 Бт3*
65,8
1/2 МоО|~
74,5
1/3 Ге3+
68
1/2 вг2+
59,5
N02
72
Н+
349,7
Т1+
74,9
N03
71,4
1/2 ОД*
68,6
1/3 УЬ3+
65,2
всы-
66,5
1/2 Яg2+
63,8
1/2гп2+
53,5
1/2 80|-
72
К+
73,5
1/2 БО%-
79,8
3. Молярный коэффициент поглощения
Металл
Реагент
Длина волны
X, нм
6 -ю-3
А1
Ксиленоловый оранжевый
540
10,3
В1
Ксиленоловый оранжевый
545
5,2.
са
Дитизон
518
79,0
368
Окончание табл. 3
Металл
Реагент
Длина волны
X, нм
е-10"3
Со
ПАН*
640
36,5
Сг
Дифенилкарбазид
540
29,0
ш
Ксиленоловый оранжевый
530
15,7
Ш
Мети лтимоловый синий
570
18,7
1п
ПАН
545
24,5
Мо
н2о2
415
0,44
N(1
Ксиленоловый оранжевый
535
16,0
№)
н2о2
340
0,90
Родамин Б
565
71,8
Бс
Ксиленоловый оранжевый
535
27,5
вп
Пирокатехиновый синий
555
62,0
Та
н2о2
285
0,955
ТЬ
Эриохром черный Т
700
35,0
Т!
Купферрон
350
6,2
Т1
Ксиленоловый оранжевый
502
14,9
Тг
н2о2
410
0,70
ТЬ
Эриохром черный Т
700
35,0
и
ПАН
570
23,0
V
Ксиленоловый оранжевый
562
24,0
V
н2о2
500
0,32
ЛАГ
н2о2
295
0,56
ПАН
560
27,8
Ъг
Ксиленоловый оранжевый
535
24,2
Метилтимоловый синий
560
21,7
* ПАН — (2-пиридилазо) - 2-нафтол.
24-7831 369
4. Значения потенциалов полярографических полуволн
на ртутном капельном электроде ^
(жел. — желатин)
Определяемый
элемент
Состав раствора (фон)
Изменение
валентности
Потенциал
полуволны, Е1/2
А1ш
0,5 н. ВаС12 (не исключено,
3 - 0(?)
-1,7
что выделяется Н2)
Са11 и другие
В растворах солей тетраме-
щелочно-
тиламмония
2-0
-2,22
земельные
металлы
одн.на
2-0
-0,60
6 н. НС1
2-0
-0,79
lH.NH4cn-lH.NH3
2-0
-0,81
Сг1П
0,1 н. КС1
3-2
-0,81
2—0
-1,50
СгУ1
1 м кон
6-3
-1,03
Ре11
1 М Ш2С204
3<=±2
-0,24
1М№2С204
3^2
-0,24
1 М НС104, рН = 0—2
2-0
-1,37
Те111
0,5М^Н4)2С4Н4О5 +
(3-2
-0,98
+ 1 М 1Ш3+ 0,005% жел.
[2-0
-1,53
Мп11
0,5 МКН3■ + 0,5 н. Ш14С1
2-0
-1,54
2 М К&ОН.+ 5% 1ШаС4Н406
|2-3
-0,4
[2-0
-1,7
НС104, рН = 0—2; 1 н. КС1
2—0
-1,1
1 М,Щ3 + 0,2 М ЫН4С1 +
+ 0,005% жел.
2-0
-1,Д6
Р(1п
1 М ЫН3 + 1 М N^01 +
+ 0,001 %. Метиловый
красный
2-0
-0,72
2МЙаОНйлиКОН
2-0
-1;41
2 МНС1
3-0
-0;22
1 М ЫаОН
|3.-5
-0,45
13-0
-1Д5.
2МНС1
5
-0(?)
-0,24
370
П р е дметный
У КA3 ATE Л Ь
Абсорбционная спектроскопия 50
Адсорбция 292, 294
Активационный анализ 252
Амперометрическое титрование 225
Аниониты 332
Атомно-абсорбционная спектроско-
пия 92
Болометр 68
Вольтамперометрия 211
Время удерживания 298
Высота, эквивалентная теоретиче-
ской тарелке 299
Выход люминесценции 102
Гальванический элемент
стандартный 184
исследуемый 185
Гейгера—Мюллера счетчик 118
Генераторы
— дуги 23
— звуковой 166
<— иск*ры конденсированной 25
Гомологическая пара 38
Детектор(ы) 303, 307
— пламенно-ионизационный 308
— рефрактометрический 320
— спектрометрический 320
— па теплопроводности (катаро-
метр) 307
— термоионный 308
— термохимический 308
Дисперсия 145
— адекватности 349
— воспроизводимости 347
— однородности 347
— оптического вращения 152
— спектральных приборов 34
Дифракционная решетка 26, 67
Дихроизм круговой (циркуляр-
ный) 153
Дозатор 301V ^
Дуга электрическая 23
Емкость обменная 333
Закон(ы)
— Больцмана 18
— Бугера—-Ламберта—Бера 50
— Вавилова 102
— Вул ьфа-^Брэгга 115
— Кольрауша 163
— Мозли 112, 122
1 —преломления 142
' — радиоактивного распада 248
— распределения 277
— Рэлея, 156
— Стокса—«Доммеля 101
— Фарадея ¿31
Зонд электронный 6, 114, 121
Извлечения степень 280
Излучение рентгеновское 110
Изотерма адсорбции 293
Изотопного разбавления метод 255
Иммерсионный метод 146
24*
371
Индекс удерживания 309
— — Ковача 310
Ионйты типы 332
Ионометрия 198
Ионообменное равновесие 334
Искра высоковольтная 21, 25
Катализатор 265
Катарометр 307
Катиониты 332
Квантовое число
главное 13
магнитное 13
побочное 13
спиновое 13
Квантометры 5,120
Кинетические методы анализа 264
— теория в хроматографии 299, 300
Колонки хроматографические 301,
302
Колтупа карта 70
Кондуктометрйя 160
— аппаратура 166
— высокочастотная 165
— практика 176
Константа
— ионного обмена 334
— распределения 278
— скорости реакции 265
— экстракции 282
Коэффициент(ы)
— поглощения молярный 51
— преломления 142
— пропускания 51
— разделения 280
— распределения 277
Критерий
— разделения 298
— Фишера 349
Крутое восхождение 354
Кулонометрия 239
— амперостатическая 242
— потенциостатическая 239 '
— практика 243
Кулонометры 240
Лазер 21
Лампа с полым катодом 93
Линия
— базовая 83
— аналитическая 33, 36
— комбинационного рассеяния 128
Линии спектральные 18, 34, 36
Люминесцентный анализ 98
аппаратура 106
качественный 107
— — количественный 108
— — теория 98
Люминесценция 98 ^
— интенсивность 103
— спектры 98
Масс-спектр 260
Масс-спектрометр 261
Масс-спектрометрический метод
анализа 263
Матрица планирования экспери-
мента 345
Метод
— градуировочного графика 9, 78,
96,200
— дифференциальной фотометрии
80
-г- добавок 9, 45, 79, 96,124, 201
— молярного свойства 9
— постоянного графика 43
— твердого графика 45
— трех эталонов 42
— фотоэлектрический 46
Модель математическая 343
Монохроматизаторы 67
Мультиплетность 16
Нефелометрия 156
Определение рН 190
Орбитали молекулярные 60
Ослабитель ступенчатый 40
Осциллятор
— ангармонический 58
— гармонический 57
Отбора правила 16
Отклика поверхность 343
372
Пара гомологическая 38
Параметр оптимизируемый 343
Переменная кодированная 344
Переход
— запрещенный 16
— разрешенный 16 '
— резонансный 17
— — магнитный 133
Период полураспада 249
Пламена 22
Планирование эксперимента мате-
матическое 343
Плоскость поляризации 151
Плотность оптическая 51
Подвижность ионов 163
Показатель преломления 52
Полуволны потенциал 214
Поляризация молекулы 127,146
Поляриметрический метод
— аппаратура 153
— теория 150
Полярограмма 214
Полярографический метод
анализа органических соеди-
нений 224
аппаратура 216
количественный 219
практика 216, 229
теория 211
Полярография 212
— инверсионная 223
— осциллографическая 222
Постоянная
— Планка 12
— распада радиоактивного 248
— Ридберга 14
— силовая 57
— сосуда 167
— экранирования 112
Потенциал(ы)
— полуволны 214
— стандартный 179
Потенциометр 182, 184
Потенциометрический метод 179
Потенциостаты 240
Почернение фотопластинки 27
Преломление света 142
Предел обнаружения 4, 8, 126
Пространство координатное 343
Радиоспектроскопия 132
Радиометрические методы анализа
246
Рассеяние света
— комбинационное 129
— рэлеевское 128
Регрессии уравнение 346
Рентгеновская трубка 114
Рентгеноспектральный анализ 115
аппаратура 120
количественный 123
практика 125
Рефрактометр 143
Рефрактометрический метод 142
Рефракция 144
Ротатор жесткий 56
Самопоглощение 19
Светофильтры 67
Сдвиг химический
Сила осциллятора 63
Смолы ионообменные 332
Спектр
вращательный 56
— инфракрасный 70 ~
— колебательный 56
— комбинационного рассеяния 127
— Мессбауэра 258
— поглощения 53
— полярографический 215
— рентгеновский 110
— электронный 60
— эмиссионный 12
— ЭПР140
— ЯМР 134
Спектрометр ЯМР 136
Спектрофотометр
— атомно-абсорбционный 93
Спин
— электрона 140
— ядра 132
Спиновое эхо 138
373
Степень разделения 298
Стилометры 31, 32, 40
Стилоскопы 31, 40
Стьюдента критерий 349
Счетчики
— Гейгера—Мюллера 116,118, 250
— ионизационные 116, 250
— кристаллические 120
— самогасящиеся 118
— сцинтилляционные 119, 250
— черенковские 251
Температура удерживания 310
Теоретических тарелок метод 299
Терм
— рентгеновский 112
— спектральный 12
Термометрическое титрование 273
— практика 273
— теория 272
Титраторы автоматические 87,175,
206
Титрование
— амперометрическое 225
— высокочастотное 175
— каталиметрическое 269
— кондуктометрическое 169
— кулонометрическое 242
— потенциометрическое 18, 206
— радиометрическое 256
— термометрическое 273
— турбидиметрическое 158
— фотометрическое 87
— хронокондуктометрическое 169
Ток диффузионный 213
Турбидиметрический метод 158
Удерживания
— время 298
— объем 297
— температура 310
У итстона мостик 166
Уравнение
— Ван-Деемтера 300 ,
— Ильковича 216
— Ломакина—Шайбе 19
— Лорентца— Лоренца 14{5
— Лэнгмюра 293 ,
— Нернста 179, 212
— Онзагера 150
— полярографической волны 214
— Шредингера 13
Фактор
— натуральное значение 344
— извлечения 279
— контрастности пластинки 28
— обогащения 280
Флуоресценция 100
— атомная 105
— выход 102
— интенсивность 103
— спектры 98
Флуорометры 106
Фосфоресценция 100
Фотометрический анализ
аппаратура 66
методы 78
экстрационно-фотометри-
ческий 83
Фотометрия пламени 48
Фотопластинка 27
Фотоумножители 30
Фотоэлектроколоримётры 69
Фотоэлементы 29
— с внешним фотоэффектом 29
— с внутренним фотоэффектом 29
— с запирающим слоем 30
Фотоэффект 29
Функцця отклика 343
Хроматограмма 295
Хроматографические методы
качественного анализа 309
— — количественного анализа 311
абсолютной калибровки 312
внутренней стандартизации
312
нормировки 312
— —* нормировки с калибровоч-
ным коэффициентом 312
Хроматографический пик 296
374
Хроматография 292
— аналитическая реакционная га-
зовая 315
— бумажная 327
— восходящая 324
— газовая 303
— влияние температур 314
— газожидкостная 305
— гель 330
— жидкостная адсорбционная 317
— жидкостно-жидкостная распре-
делительная 326
— ионная 338
— ионообменная 331
—- капиллярная 306
— круговая 324
— нисходящая 324
;—- теоретические представления
299
— тонкослойная 321
— электрофоретическая 329
Хроматермография 314
Хроматомасс-спектрометрия 311
Частота(ы)
— антистоксова 129
— резонансная 133
— нормальных колебаний 58
— характеристическая 59, 69
— стоксова 129
Число
— волновое 11
— квантовое 13
— степеней свободы 58
— теоретических тарелок 299
Шкала рН 191
Экстрационные системы
— галогенидные и тиоцианатные
288
— хелатные 285
Экстракция 277
Электрическая проводимость 160
предельная 163
удельная 161
эквивалентная 162
Электрогравиметрические разделе-
ния 233 1
Электрод(ы)
— амальгамные 186
— вращающийся 217
— второго рода 186
— индикаторный 185
— ионоселективный 194
— — жидкостной 197
— — твердый 195
— каломельный 188
— первого рода І86
— редокс 186
— ртутный капающий 217
— сравнения 187
— стеклянный 192
— хлорсеребряный 187
Электролиз
— аппаратура 233
— внутренний 238 ":
— на ртутном катоде І37
Электромагнитный спектр 11
Электронный парамагнитный резо-
нанс 140
Элюционные характеристики 296
Энергия
— вращательная 56
— колебательная 56
— электромагнитного излучения 12
— электронная 60 ; ! (
ЭффеКТ °! [■.',
— Допплера 20, 258 г fг
— Зеемана 20
— Коттона 152
— Мессбауэра 258
— Оже 111
— Шпольского 101
— Штарка 20
От Л А В Л Е Н И Е
Предисловие ... ........ 3
глава 1
Общая характеристика
физико-химических методов анализа 4
1.1. Особенности и области применения
физико-химических методов анализа .............. 4
1.2. Основные физико-химические методы анализа ... 7
1.3. Основные приемы, используемые
в физико-химических методах анализа ............. 7
глава 2
Эмиссионный спектральный анализ 10
2.1. Основные характеристики
электромагнитного излучения .............. . . .... 10
2.2. Теоретические основы
эмиссионной спектроскопии . ............ 12
2.2.1. Спектральные термы ...................... 12
2.2.2. Интенсивность спектральных линий ...... . . . . 18
2.2.3. Ширина спектральных линий ............... 20
2.3. Основные узлы спектральных приборов 21
2.3.1. Источники возбуждения ........... . ........ 21
2.3.2. Диспергирующий элемент . 25
2.3.3. Приемники света . . . 27
2.4. Конструкции спектральных приборов ........... 31
2.5. Качественный спектральный анализ 33
2.6. Количественный спектральный анализ 35
2.6.1. Полуколичественный спектральный анализ .... 39
2.6.2. Фотографические методы
количественного анализа .................... .. . . 41 :
2.6.3. Фотоэлектрические методы ..... .... . ... . . . . * 46
2.6.4. Химико-спектральный анализ . . . . . . , . . . . . . . . 47 ,
2.7. Фотометрия пламени ... ....... . . 48
2.8. Практическое применение 48
2.9. Общая характеристика метода 49
Глава 3
Абсорбционная спектроскопия 50
3.1. Основной закон светопоглощения 50
3.1.1. Ограничение и условия применимости
закона Бугёра—Ламберта—Вера .................. 52
3.2. Спектры поглощения 53
3.2.1. Происхождение спектров поглощения ......... 55
3.2.2. Вращательные спектры . . . . . . ..... 56
3.2.3. Колебательные спектры .................... 56
3.2.4. Электронные спектры . . . ......... . ... . . .... 60
3.2.5. Интенсивность поглощения .............. . 63
3.2.6. Фотохимические реакции ............ . .. . . . . . 65
3.3. Основные узлы приборов
абсорбционной спектроскопии .................... 66
3.3.1. Источники света . . . . ... . . ... . . . . .. . . . . .. . . . 66
3.3.2. Монохроматизаторы .... .... ... .... ... ... 67
3.3.3. Приемники света ... ......... ... .... ...... 68
3.4. Качественный анализ . . .... 69
3.5. Количественный анализ . . .... .......... 71
3.5.1. Концентрационные условия проведения
фотометрической реакции . . . . .... . . .... ....... . . 72
3.5.2. Оптимальные условия
фотометрического определения .... . . ... ........ ... 76
3.5.3. Основные приемы фотометрических измерений ... 78
3.5.4. Определение смеси светопоглощающих веществ ... 84
3.5.5. Фурье-спектроскопия ..... . . ..... . . . . ... . . . . 86
3.5.6. Фотометрическое титрование ........ . . . . . ... 87
3.5.7. Определение неокрашенных соединений . . . . . . . 88
3.5.8. Фотометрический метод
исследования реакций в растворе 89
3.6. Практическое применение . ...... . . ......... . . 90
3.7. Общая характеристика метода ................ 90
Глава 4
Атомно-абсорбционный спектральный анализ ... . 92
4.1. Теоретические основы метода . . . . . . ........... 92
4.2. Основные узлы приборов
для атомно-абсорбционного анализа . .............. 93
4.3. Количественные определения . . . . . . . . ... ... . . . 95
377
4.4. Практическое применение .............. . . . . . . 97
4.5. Общая характеристика метода ... . . .... 97
Глава 5
Люминесцентный анализ .......................... 98
5.1. Спектры люминесценции ...... . ... .é . ... . ... . . 98
5.1.1. Энергетический и квантовый
выходы люминесценции ......................... 102
5.1.2. Интенсивность люминесценции . . . ....... . . . . 103
5.1.3. Тушение люминесценции ......... ... 104
5.1.4. Люминесценция кристаллофосфоров . . . . . . .... 104
5.1.5. Атомная флуоресценция . •.... . . . . . . . . . .. 105
5.1.6. Хемилюминесцентныйанализ ... .... ... ..... . ... 105
5.2. Схема прибора для люминесцентного анализа . ... 106
5.3. Качественный анализ ........ 107
5.4. Количественный анализ . . ... ........ .... ..... 108
5.5. Практическое применение ..... ...... .. .. . . . . . 109
5.6. Общая характеристика метода . . . . ...... . . .... 110
Г л а в а б
Рентгеноспектральные методы анализа ................. 110
6.1. Рентгеновские спектры .. . .......... . .... .... 110
(6.2. Поглощение рентгеновского излучения ..... . . . . 113
6.3. Основные узлы рентгеноспектральных приборов . . . 113
6.3.1. Источник возбуждения .. . . . . . . . . .. . . . . .. ... 114
6.3.2. Диспергирующий элемент ...... ..... ...... . 115
6.3.3. Приемники излучения . . ..... . . ..... ... .... 116
6:4. Конструкции рентгеновских
спектральных приборов . . . ..... . . . . .. . ... ..... . . 120
6.5. Качественный рентгеноспектральный анализ . . . . . 121
6.6. Количественный рентгеноспектральный анализ . . . 123
• 6.7. Практическое применение ....... . . . .... . ..... 125
V } 6.8. Общая-характеристика метода . . . . ... . ... . . . . , 126
Г л а в а 7
Другие спектральные и оптические методы анализа .. > 127
7.1. Анализ по спектрам комбинационного рассеяния . . 127
7.1.1. Спектры комбинационного рассеяния .... ... ..127
7.1.2. Качественный анализ по спектрам КР . . . . . . ... 130
7.1.3. Количественный анализ по спектрам КР . . . ... . . 130
7.2. Радиоспектроскопия ..... . ...... . . ... .... . 132
7.2.1. Ядерный магнитный резонанс . . . . . ...... . > . . 132
7.2.2. Электронный парамагнитный резонанс . . .... .. 140
7.3. Рефрактометрические методы анализа ... ... .... 142
378
- 7.3.1. Показатель преломления
и полное внутреннее отражение ... . . 142
7.3.2. Приборы для определения
показателя преломления 145
7.3.3. Основные рефрактометрические
методики анализа 147
7.3.4. Рефрактометрические исследования
химического взаимодействия, строения
и других свойств соединений . . . ...... . . ... ,Л . ... . 149
7.4. Поляриметрия .... ...... . .... . , . . . . . . ...... 150
7.4.1. Вращение плоскости поляризации света .". . .... 150
7.4.2. Приборы для поляриметрических измерений ... 153
7.4.3. Поляриметрические методики . . . ... . . ... .... . 155
7.5. Нефелометрия и турбидиметрия . . . ......... . ;. 156
7.5.1. Рассеяние света . . . ............... ... ...... 156
7.5.2. Приборы для нефелометрических
и турбидиметрических определений ...... . . ... . . . . 158
7.6. Практическое применение. . . .... . . ........ 159
7.7. Общая характеристика методов . ... 159
Глав а 8
Кондуктометрия ................................... 160
8.1. Электрическая проводимость растворов .... . . ... 160
8.2. Схема установки для определения
электрической проводимости . ..... .... . .'. . . . .... ,166
8.3. Прямая кондуктометрия .................. . . . 167
8.4. Кондуктометрическое титрование . . . .......... . 169
8.4.1. Реакции кислотно-основного взаимодействия . . . 170
8.4.2. Реакции осаждения ..... . . .......... . . . , . . 172
8.4.3. Реакции комплексообразования ...... ... . . ... 173
8.4.4. Реакции окисления—восстановления . . . . . . . . . 174
8.5. Высокочастотное титрование . . . . .............. 175
8.6. Практическое применение . . .... . V. ; . . ........ 176
8.7. Общая характеристика метода .......... . v. ... 178
Глав а 9.
Потенциометрия ....... . . .................. • • 179
9.1. Электродный потенциал. Уравнение Нернста . . ..... 179
9.2. Схема установки
для потенциометрических измерений. .... . ..... . ... 182
9.2.1. Стандартный гальванический элемент . . . . . . . . . 184
9.2.2. Исследуемый гальванический элемент .... .... 185
9.2.3. Индикаторные электроды . . . ... . ... .... ..... 185
9.2.4. Электроды сравнения ... . . . . . . ...... ... .... 187
379
9.2.5. Диффузионный потенциал . ... . . . . . .... .... . . 188
9.3. Прямая потенциометрия .. . . . ..... ... 189
9.3.1. Определение рН . . . . . ... 190
9.3.2. Ионоселективные электроды . .... ... . . . . . . . . . 194
9.3.3. Основные приемы ионометрического анализа ... 198
9.4. Потенциометрическое титрование ........ . . .... 202
9.4.1. Определение точки эквивалентности , . ........ 202
9.4.2. Виды потенциометрического титрования ... .... 204
9.4.3. Автоматическое титрование ...... . . . . ...... . 206
9.5. Потенциометрическое определение
физико-химических свойств веществ .......... . .... 207
9.6. Ионоселективные полевые транзисторы . . ..... . , 209
9.7. Практическое применение ,, ... ... . . ..... . :.Y. . 209
9.8. Общая характеристика метода ...... ........ . . 210
Глава 10
Вольтамперометрия ............................... 211
10.1. Кривая «ток—потенциал» . . .. .... ...... . 211
10.2. Схема полярографической установки ...... . . . . 216
10.3. Прямая полярография .... . .......... ... . ... 218
10.3.1. Количественный полярографический анализ .. . 219
10.3.2. Дифференциальная полярография . .... . . ... . 220
10.3.3. Хроноамперометрия с линейной разверткой
потенциала ..... . . . .... .... .... .... 222
10.3.4. Инверсионная вольтамперометрия . . . .... .... 223
10.3.5. Анализ органических соединений , . , .. . .... . 224
10.3.6. Полярографическое исследование реакций
комплексообразования .. .............. . . . . • . .. ... 224
10.4. Амперометрическое титрование .. ..... ....... 225
10.4.1. Кривые амперометрического титрования . .... . 225
10.4.2. Основные типы реакций
амдеррметрического титрования .... . . . . . . . , . . . . . . 226
10.4.3. Титрование с двумя
индикаторными электродами .... . ................ 2Я7
10.5. Практическое применение ........... . . .... . . 229
10.6. Общая характеристика метода ............... 230
Г л а в а 11
Электролиз и кулонометрия ........................ 231 .
11.1. Законы электролиза . . . . , . . . . . 231
11.2. Потенциал разложения и перенапряжения . . . . . 232
11.3. Электрогравиметрический анализ . ........ ^. . , 233
11.3.1. Схема установки для электролиза . . . 233
11.3.2. Электрогравиметрййческие разделения . у ... . . .234
V 380 /І
11.3.3.дЭлектролиз на ртутном катоде ........ . ..... 237
11.3.4.Внутреннийэлектролиз . . 238
11.4. Кулонометрия . ..... . . 239
11.4.1. Кулонометрия при постоянном
контролируемо^ потенциале . . . . . . 239
11.4.2. Кулонометрия при постоянной контролируемой
силе тока (кулонометрическое титрование) .......... 242
11.5. Практическое применение ..... ... .'. . . ... .... 243
11.6. Общая характеристика метода ...... ..... .... 245
Г Л А В' А І2^:^^
Радиометрические методы анализа 246
12.1. Типы радиоактивного распада , ,
и радиоактивного излучения ....... . . . . . .'. 246
12.2. Закон радиоактивного распада . . . . ... .... . 248
12.3. Взаимодействие радиоактивного излучения
с веществом й счетчики излучения .,. ..... ......... 249
12.4. Ядерная химия
и искусственная радиоактивность 251
12.5. Методики анализа, основанные
на измерении радиоактивности ". • • * • • • • 252
12.5.1. Использование естественной радиоактивности
(в анализе) ....... ............................ 252
12.5.2. Активационный анализ ...... ..... ... .,. . . 252
12.5.3. Метод изотопного разбавления .............. 255
12.5.4. Радиометрическое титрование ... .. . .. ... ... 256
12.5.5. Эффект Мёссбауэра • • . . 258
12.6. Практическое применение ....... ... .... . . ... 259
12.7. Общая характеристика метода •............. •.. 259
■ ■ -і " ' ' •' ,. ■' ■ :" '■' ^
ГЛАВ А 13
Масс-спектрометрия .............................. 260
13.1. Теоретические основы маос-спектрометрии ..... 260
13.2. Качественный анализ ,..,... ..........263
13.3. Количественный анализ ........ч. .. . . ;. . ... • 263
13.4. Практическое применение ........... . ... .. 263
13.5. Общая характеристика метода ........ . . ..... 264
Г Л А В А 14
Кинетические методы анализа ............ 264
14.1. Основные методы обработки г
кинетических данных . . . . . . . ... ........ . . . 264
14.2. Основные приемы кинетических методов анализа . , 267
■ ' 381
14.3. Практическое применение v . , ..... 270
14.4. Общая характеристика метода7 . . . . . . .• • • - • . . .'. 271
Г л а в а 15 . Чч ,:; ^^7#^;./
Термометрическое титрование .......:......... .^/т. ; • 272
15.1. Тепловой эффект реакции ^ !
как аналитический сигнал .. . , . .. ......... 272
. 15.5. Кривые термометрического титрования^;. .... . . . 273
15.3. Практическое применение . . . .... ... . 275
15.4. Общая характеристика метбда . . . . . . .Л ., ? 1 . . . . 276
Г л а в а 16 . ' ;
Экстракция .". . л.. .V;............ 277
16.1. Распределение вещества ; ^
между/ДВУМЯ ЖИДКОСТЯМИ . . . .••.•;■л.>.;■'. . . 1. . . 277
16.2. Основные колич^ствённы(В л
характеристики экстракции 278
16.3. Экстракция хелатов . 281
16.4. Экстракционные хе|іатцьіе системы . . . . . . ... . 285
Іб.ІЗ^ ....... ... , 287
16.6. Экстракционные галбгенидные :Л
и тиоцианатные системы .> .. . .... ..... .... .'.,>.. . . . 288
16.7. Скорость экстракции .... . ........•./..,. 288
16.8. Практическое применение . . . . ....... . . . .... . 289
16.9. Общая характеристика метода . ...... ... . . ... 291
г лав а 17 . , -^^л^і:^ ' •
Хроматография 292
17.1. Адсорбция вещества — оснора хроматографии . . . 292
17.2. Классификация м:етодов хроматографии . . . . . . . .294
17.3. Хроматографический пц*с
и элюционные характеристики ? 296
17.4. Теоретические основы хроматографии ......... 299
17.5. Основные узлы приборов
для хроматографического анализа . . . . ... . . . Збі
17.6. Газовая хроматография . . . . . . . . . ..... 303
, 17.6.1. Хроматографические колонки и детекторы . .у. 304
17.6.2. Качественный анализ . . . . .... . . . . . . > 309
17.6.3. Количественный анализ .... . . . .... 311
17.6.4. Сияние температуры 313
17.6.51 аналитическая реакционная
газрвая^оматография ..... ........ . . ..... ...... 315
17.6.6д Практическое применение . . . ... ..... ... .ч. . . 316
17.7 .Жидкостная адсорбционная хроматография ... . . 317
17.7.1. Теоретические оснрвы ,
жидкостной хроматографии .., . 317
17.7.2. Основные узлы приборов
жидкостной хроматографии . . . . .. .. 318
17.7.3.^Качественный и количественный анализ 321
17.85Тонк0(рлШная хроматография .. . ..... 321
17.8.1. Основные характеристики ТСХ ............. 322
17.8.2. Качественный анализ .. . . . . . . ... 324
17.8.3. ^Количественный анализ . .... . .... . ... ..... 325
17.9.ЖІІдкрстно-жидкрстная
распределительная хроматография ... . ... . . ........ 326
17.9.1. Колоночный вариант .. ......... ......... . 326
17.9.2. Распределительная хроматография наЩъ&аге .. 327
17.10. Гель-хроматография . . ... . . Й;.^,. . . 1;;. I . . . . . 330
17.11. Ионообменная хроматография .... ... .1.. ... . 331
17.11.1. Типы ионообменных смол ... .. . ... ... . . . . . 332
^17.11.2. Ионообменное равновесце . .. . .... .. 334
17.11.3. Практическое применение . .. . .... ........ 335
17.12. Ионная хроматография .. ..... .......... I.; 338,
17.12.1. Методы ионной хроматографии . ."^^339:;?
17.12.2. Практическое применение ........... ,... . 340]
17.13. Осадочная хроматография . ...... . .;. . .... . .340
17.14. Общая характеристика метода • .;>...; ...... 341
Г Л А В А 18 •
Математическое планирование, эксперимента
в аналитической химии ........................... 342
18.1. Основные понятия и определения ............. 342
18.2. Уравнение регрессии и регрессионный іанализ . . . 346
- 18.3. Дробный факторный эксперимент . . ........ . . . 353
18.4. Оптимизация по методу крутого восхождения .. . 354
Г ЛАВА 19 ;
Средства и методы ; л
оперативного аналитического контроля. ;
Применение тест-методов и сенсоров в анализе ...... 357
19.1. Особенности тест-методов и сенсоров ...... . . . . ; 357
19.2. Тест-методы . . ... . . ...... ... . . .... .... ...... 358
19.3. Сенсоры ....... ... ........,..... .... . . . 1. 360
Список литературы ........ .... .. .. . .. ..•. * .365
Приложение -. ................ 366
Предметный указатель .... ..... ..... • • • • • • • • • • 371
Учебное издание
Васильев Владимир Павлович
АНАЛИТИЧЕСКАЯ ХИМИЯ
Физико-химические методы анализа
Учебник для студентов вузов,
обучающихся по химико-технологическим
специальностям
Зав. редакцией Н. Е. Рудомазина
Ответственный редактор В. Н. Бораненкова
Художественное оформление Л. Л. Копачева
Технический редактор Н. И. Герасимова
Компьютерная верстка А. В. Маркин
Корректор И. Т. Белугина
Санитарно-эпидемиологическое заключение
№ 77.99.02.953.Д.006315.08.03 от 28.08.2003.
Подписано клечати 22.06.07. Формат 60 х 9071б. Бумага типографская.
Гарнитура «Школьная». Печать офсетная. Усл. печ. л. 24,0.
Тираж 3000 экз. Заказ № 7831.
ООО «Дрофа». 127018, Москва, Сущевский вал, 49.
По вопросам приобретения продукции
издательства «Дрофа» обращаться по адресу: '
127018, Москва, Сущевский вал, 49.
Тел.: (495) 795-05-50, 795-05-51. Факс: (495) 795-05-52. 4
Торговый дом «Школьник». ^
109172, Москва, ул. Малые Каменщики, д. 6, стр. 1А.
Тел.: (495) 911-70-24, 912-15-16, 912-45-76.
Магазины «Переплетные птицы».
127018, Москва, ул. Октябрьская, д. 89, стр. 1.
Тел.: (495) 912-45-76;
140408, Московская обл., г. Коломна, Голутвин,
ул. Октябрьской революции, 366/2.
Тел.: (495) 741-59-76.
Интернет-магазин: http://www.drofа.ru
Отпечатано а готовых диапозитивов в ОАО ордена «Знак Почета»
«Смоленская областная типография им. В. И. Смирнова».
214000, г. Смоленск, проспект им. Ю. Гагарина, 2.
Высшее образование
в. п. Васильев
АНАЛИТИЧЕСКАЯ
ХИМИЯ
КНИГА 2
Физико-химические
методы анализа
6-е издание, стереотипное
Рекомендовано
Министерством образования
Российской Федерации в качестве учебника
ДЛЯ студентов высших учебных заведений,
обучающихся по химико-технологическим
специальностям
ДРОФА
МОСКВА • 2007
Для студентов
высших учебных заведений
химико-технологических
и технологических
специальностей
УЧЕБНИК
В.п.Васильев
АНАЛИТИЧЕСКАЯ
ХИМИЯ
КНИГА 1
Титриметрические
и гравиметрический
методы анализа
УЧЕБНИК
в. п. Васильев
АНАЛИТИЧЕСКАЯ
ХИМИЯ
КНИГА 2
Физико-химические
методы анализа
ПОСОБИЕ
АНАЛИТИЧЕСКАЯ
ХИМИЯ
Сборник вопросов,
упражнений и задач
Под редакцией В. П. Васильева
ПОСОБИЕ
АНАЛИТИЧЕСКАЯ
ХИМИЯ
Лабораторный
практикум
Под редакцией В. П. Васильева