/































































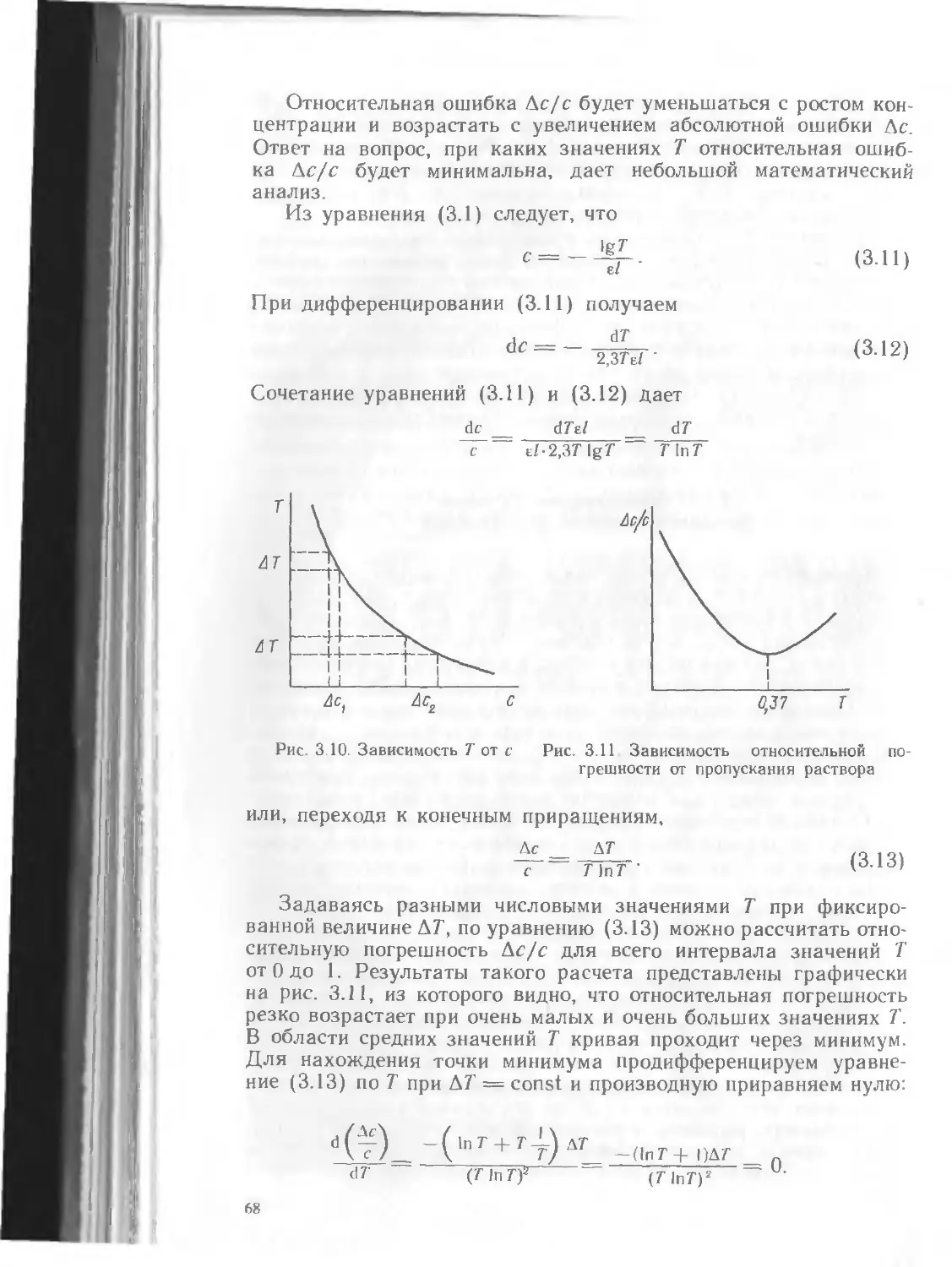























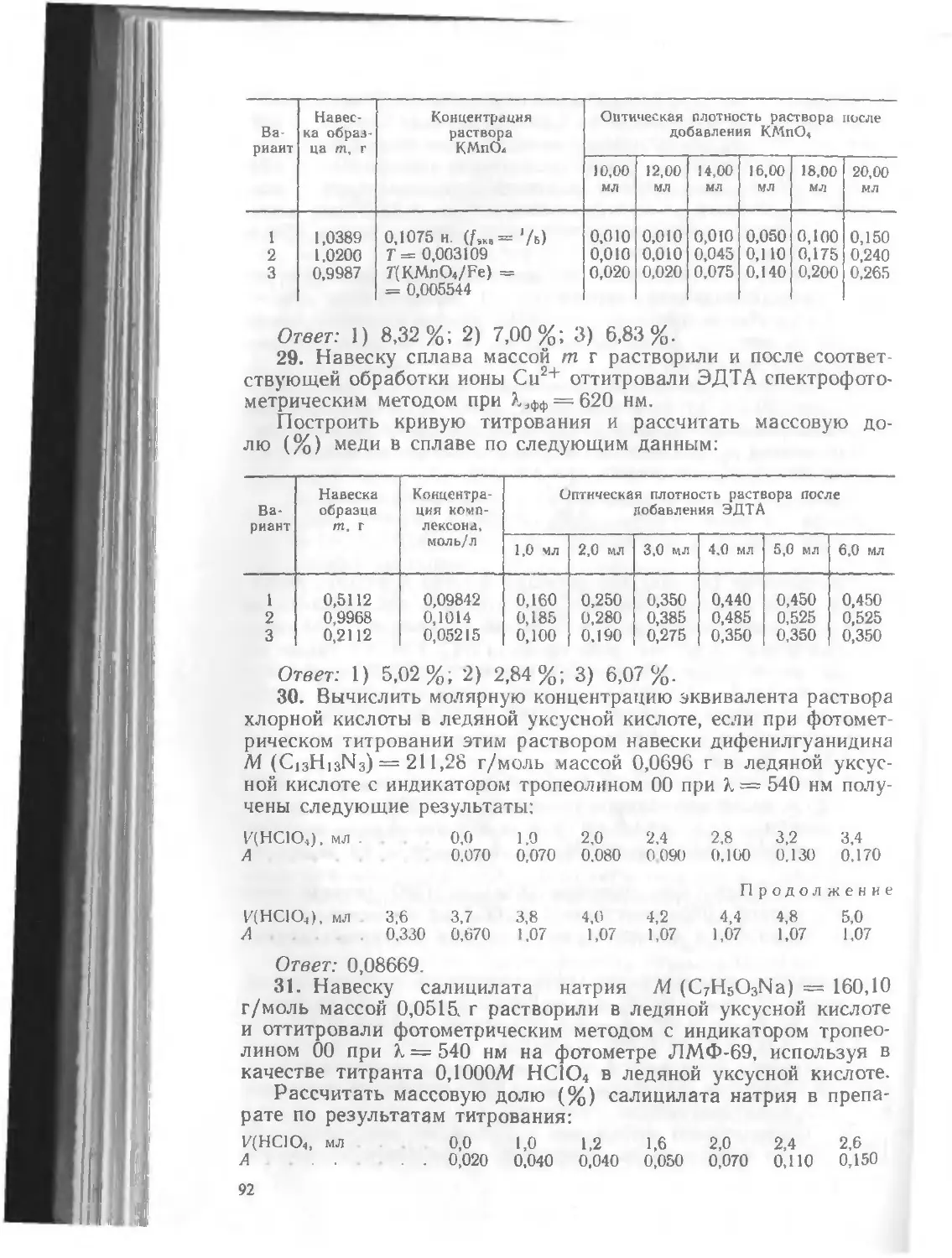

































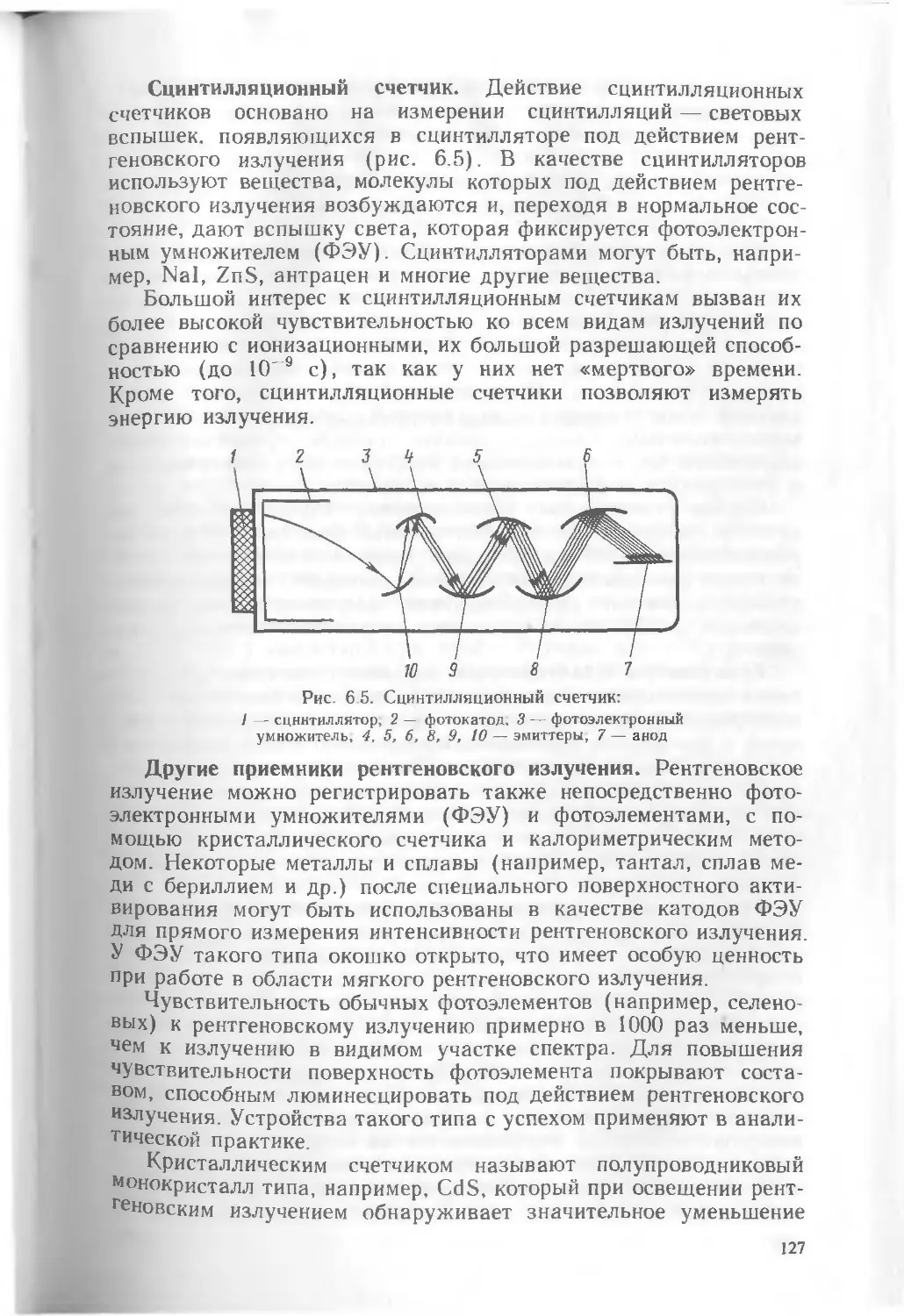























































































































































































































































Текст
В. П. ВАСИЛЬЕВ
АНАЛИТИЧЕСКАЯ
ХИМИЯ
В ДВУХ ЧАСТЯХ
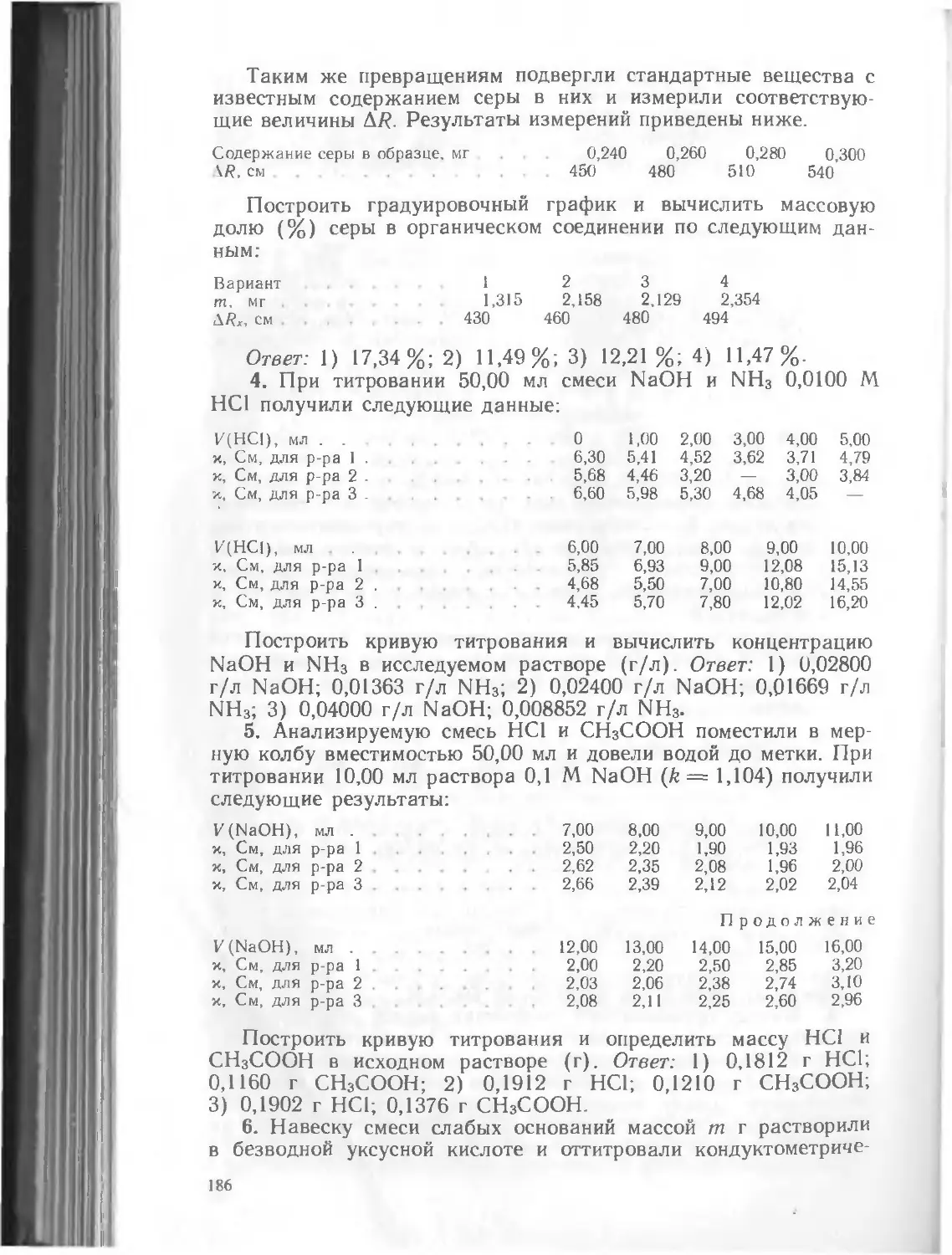
Физико-химические методы анализа
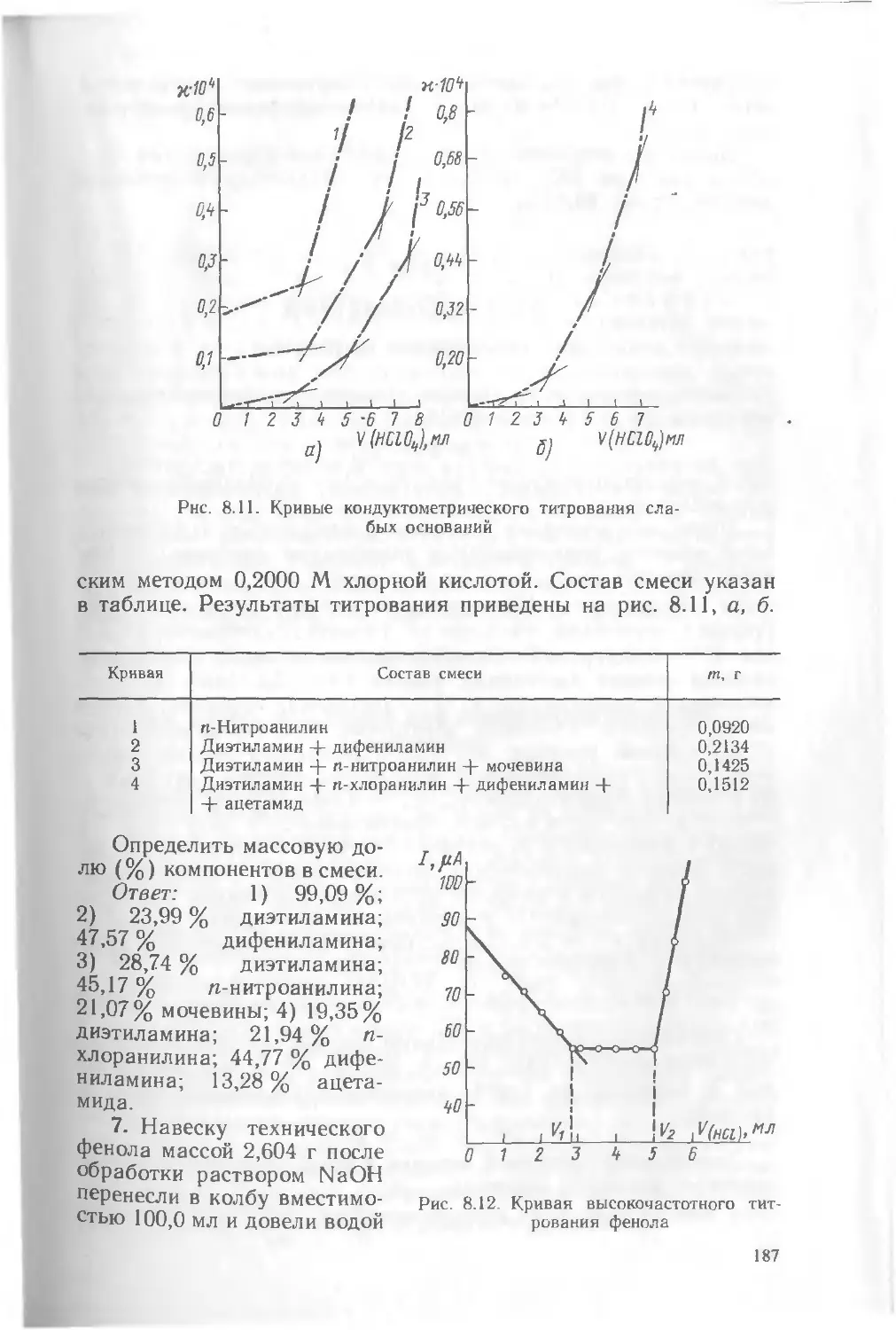
Допущено Государственным комитетом СССР по народному образованию в качестве учебника для студентов химико-технологиче-4 гких специальностей высших учебных заведений
' I
Москва „Высшая школа“1989
ББК 24.4
В19
УДК 543
Рецензенты: кафедра аналитической химии Ленинградского технологического института им Ленсовета (зав кафедрой проф. В В. Бардин), проф. Э И Исаев (Московский институт нефти и газа им И. М Губкина)
Васильев В. П.
В19 Аналитическая химия. В 2 ч. Ч. 2. Физико-химические методы анализа: Учеб, для химико-технол. спец, вузов. — М.: Высш, шк., 1989. — 384 с.: ил.
ISBN 5-06-000067-2
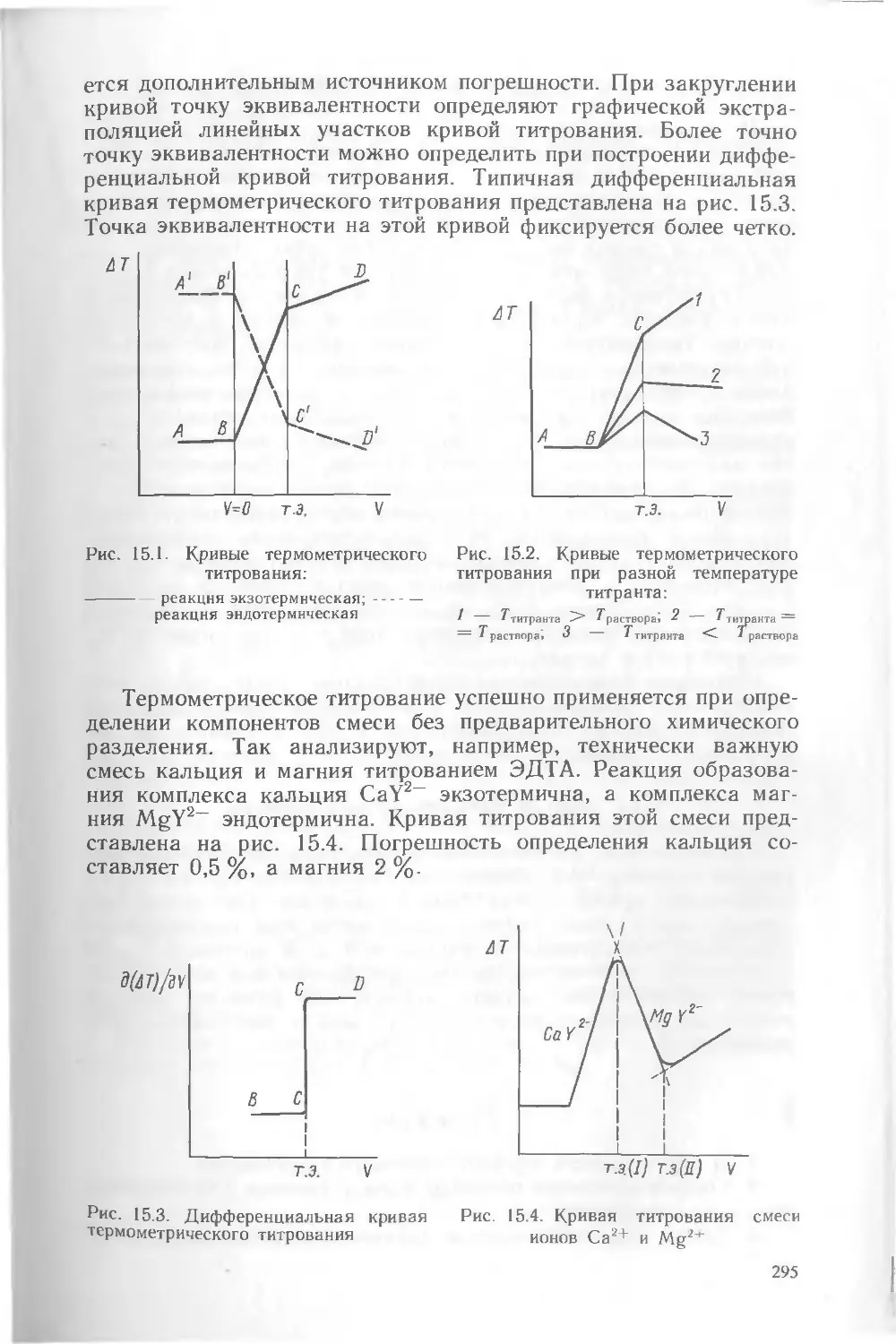
Во второй части учебника изложены основы физико-химических методов анализа Даны принципиальные схемы основных установок н приборов Рассмотрены условия и области практического применения методов, их достоинства и недостатки, ограничения, перспективы развития и другие особенности и характе^ ристики Изложены математические методы плвиирования эксперимента в анали тическоЙ химии
В конце каждой главы приведены вопросы, задачи и решения типовых задач
„ 1707000000(4309000000)-372 оп " ББК 24.4
В---------001(01)—89----------И6~89 543
i Б И Б Л И О ТЕ К н" ание С Л, СК ОГО
ир
ь уиьзйрснтет^
АНАЛИТИЧЕСКАЯ ХИМИЯ
В двух частях
Честь вторая ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
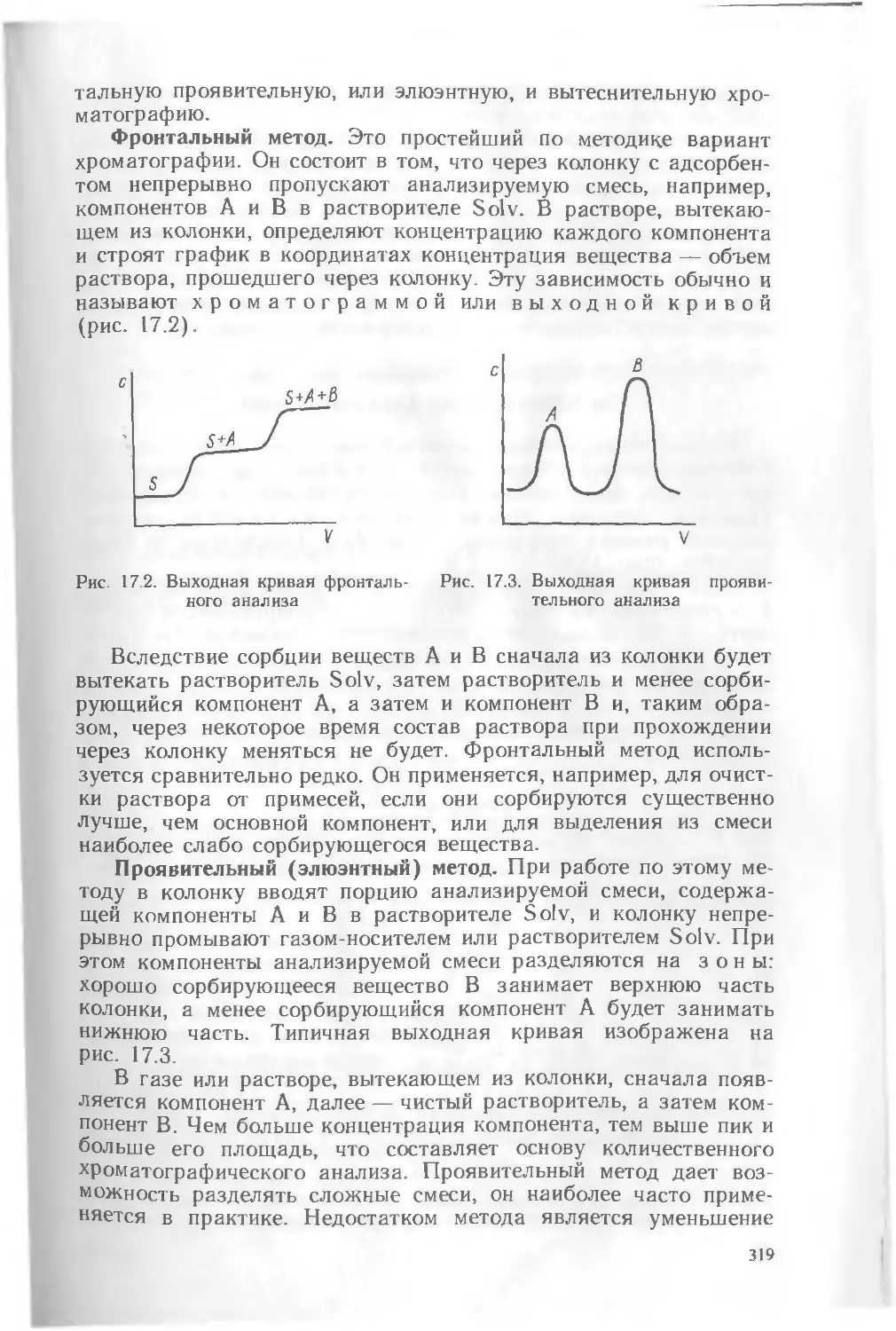
Зав редакцией С Ф Кондрашкова Редактор В Н Бораненкова Мл редакторы Е Н Хорошева, С М Ерохина, Л С Макаркина Художник В В Гарбузов Художест венный редактор Е Д Косырева Технический редактор 3 А Муслимова Корректор Р К Косинова
ИБ № 8058
Изд № Хнм —790 Сдано в набор 12 08 88 Подл в печать 30 05 89 Формат 60X90'/ie Ьум тип № 1 Гарнитура литературная Печать офсетная Объем 24.0 усл печ л ?4 0 усл кр отт 24,45 уч нзд л Тираж 40 000 экз Зак № 1533 Цена 1 р 10 к Издательство «Высшая школа», 101430 Москва, ГСП 4 Неглннная ул д 29/14
Ярославский полиграфкомбинат Госкомиздата СССР 150014 Ярославль, ул Свободы 97
ISBN 5-06-000067-2
ISBN 5-06-001519-Х
(Ч 2)
© Издательство «Высшая школа», 1989
ПРЕДИСЛОВИЕ
Вторая часть учебника посвящена физико-химическим, или, как их иногда называют, инструментальным, методам анализа. Химические и физико-химические методы анализа взаимно дополняют друг друга, составляя в целом предмет аналитической химии.
Основное внимание в учебнике уделено теории физико-химических методов анализа, которая опирается на фундаментальные законы физики и химии. В первую очередь рассмотрена возможность их использования в химико-аналитических целях. Вместе с тем в книге дается достаточно подробное изложение вопросов практического применения различных методов анализа, их значения, возможностей и ограничений.
Так же, как и в первой части учебника, в конце каждой главы приведены вопросы и задачи, работа над которыми должна стимулировать более глубокое изучение материала.
Описание лабораторных работ будет дано в специальном «Практикуме по аналитической химии», а вопросы использования ЭВМ будут рассмотрены в отдельном пособии «Применение ЭВМ в химико-аналитических расчетах».
Все критические замечания будут приняты с благодарностью.
Автор
Глава 1
ОБЩАЯ ХАРАКТЕРИСТИКА ФИЗИКО-ХИМИЧЕСКИХ МЕТОДОВ АНАЛИЗА
1.1. ОСОБЕННОСТИ И ОБЛАСТИ ПРИМЕНЕНИЯ ФИЗИКО-ХИМИЧЕСКИХ МЕТОДОВ АНАЛИЗА
Все методы анализа основаны на использовании зависимости физико-химического свойства вещества, называемого аналитическим сигналом или просто сигналом, от природы вещества и его содержания в анализируемой пробе. В классических методах химического анализа в качестве такого свойства используются или масса осадка (гравиметрический метод), или объем реактива, израсходованный на реакцию (титриметриче-ский анализ). Однако химические методы анализа не в состоянии были удовлетворить многообразные запросы практики, особенно возросшие как результат научно-технического прогресса и развития новых отраслей науки, техники и народного хозяйства в целом. Наряду с черной и цветной металлургией, машиностроением, энергетикой, химической промышленностью и другими традиционными отраслями большое значение для промышленноэнергетического потенциала страны стали иметь освоение атомной энергии в мирных целях, развитие ракетостроения и освоение космоса, прогресс полупроводниковой промышленности, электроники и ЭВМ, широкое применение чистых и сверхчистых веществ в технике. Развитие этих и других отраслей поставило перед аналитической химией задачу снизить предел обнаружения до 10-5 ... Только при
содержании так называемых «запрещенных» примесей не выше 10-5 % жаропрочные сплавы сохраняют свои свойства. Примерно такое же содержание примеси гафния допускается в цирконии при использовании его в качестве конструкционного материала ядерной техники. (Вначале цирконий был ошибочно забракован как конструкционный материал этой отрасли именно из-за загрязнения гафнием). Еще меньшее содержание загрязнений (до 10“'° %) допускается в материалах полупроводниковой промышленности (кремнии, германии и др.). Существенно изменяются свойства металлов, содержание примесей в которых находится на уровне 10-5 % и меньше. Например, хром и бериллий становятся ковкими и тягучими, вольфрам и цирконий становятся пластичными, а не хрупкими. Определение столь малых содержаний гравиметрическим или титриметрическим методом практически невозможно, и только применение физико-химических методов анализа, обладающих гораздо более низким пределом обнаружения, позволяет решать аналитические задачи такого рода.
4
Другой важной особенностью физико-химических методов анализа является их экспрессност ь, высокий темп получения результатов. Современные автоматические квантометры позволяют получать результаты буквально через несколько минут после поступления пробы в лабораторию. Своевременная информация о составе сырья, о степени химического передела и т. д. дает возможность технологу активно вмешиваться в ход технологического процесса и вводить необходимые коррективы. Весьма существенное значение имеет экспрессность анализа и в металлургическом производстве, где корректировать состав стали можно по ходу плавки в зависимости от результатов анализа. Сокращение времени плавки, нередко зависящее от быстроты анализа, дает большой экономический эффект, снижая энергетические и другие затраты.
Физико-химические методы позволяют проводить анализ на расстоянии. Яркими примерами являются анализ лунного грунта, выполненный рентгенофлуоресцентным устройством непосредственно на луноходе, определение состава атмосферы, окружающей планету Венера, и т. д. Важное практическое значение имеет дистанционный анализ в земных условиях, например, когда анализируются препараты высокой радиоактивности, токсичности, а также при анализе морских вод на больших глубинах и решении других аналогичных аналитических задач.
Многие приборы, используемые в физико-химических методах анализа, позволяют автоматизировать сам процесс анализа или некоторые его стадии. Автоматические газоанализаторы контролируют состав воздуха в шахтах. В металлургической промышленности широко применяют высокоавтоматизированные оптические н рентгеновские квантометры. В значительной степени автоматизирован газовый хроматографический анализ в нефтехимической, коксохимической и других отраслях промышленности. Нередко приборы физико-химических методов анализа используют непосредственно в производстве в качестве датчиков соответствующих сигналов, например, при регулировании pH растворов или корректировке концентрации компонентов.
Анализ с помощью некоторых физико-химических методов может быть выполнен без разрушения анализируемого образца (недеструктивный анализ), что имеет большое значение для некоторых отраслей промышленности, а также для криминалистики, медицины и т. д. Недеструктивный анализ может быть выполнен рентгенофлуоресцентным, радиоактива-ционным и некоторыми другими методами. Часто практический интерес представляет не общее содержание какого-либо элемента в пробе, а его распределение по поверхности образца — так называемый локальный анализ — определение элемента в данной «точке» образца. Этот анализ имеет значение в металловедении и других областях, где состав отдельных включений определяет качество материала, а также в минералогии, петро
5
графин, криминалистике, археологии и т. д. Выполняется локальный анализ рентгеноспектральным методом. Электроны собирают в очень тонкий пучок диаметром 1 мкм и меньше (электронный зонд) и направляют его в интересующую точку образца. По характеристикам возникающего рентгеновского излучения судят о содержании элементов в «точке». Для целей локального анализа используется также техника лазерной микроспектроскопии.
Перспективным является использование ЭВМ в аналитической химии не только для расчета результатов анализа и статистической обработки, но и для решения других аналитических задач. С помощью ЭВМ можно более надежно выделять аналитический сигнал, проводить более четкое разрешение перекрывающихся сигналов и т. д. ЭВМ, встроенные в спектрофотометр и в другие аналитические приборы, значительно расширяют возможности этих приборов.
Погрешность анализа физико-химическими методами составляет в среднем 2...5 %, что превышает погрешность классических методов анализа. Однако такое сравнение погрешностей не вполне корректно, так как относится к разным концентрационным областям. При небольшом содержании определяемого компонента (порядка 10-3 % и менее) классические химические методы анализа вообще непригодны, при больших концентрациях физико-химические методы успешно соперничают с химическими, а такие методы анализа, как кулонометрия, даже превышают их по точности.
Следует отметить также, что погрешность анализа физико-химическими методами имеет тенденцию снижаться за счет конструирования прецизионных аналитических приборов и разработки более совершенных аналитических методик.
Однако химические методы анализа своего значения не потеряли. Они незаменимы там, где при высоком содержании требуется высокая точность и нет серьезных ограничений по времени (например, анализ готовой продукции, арбитражный анализ, изготовление эталонов).
Существенным недостатком большинства физико-химических методов является то, что для их практического применения требуются эталоны, стандартные растворы и градуировочные графики.
1.2. ОСНОВНЫЕ ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
В группе физико-химических методов анализа иногда выделяют физические методы. Сднако достаточно строгого и однозначного критерия для этого нет, поэтому выделение физических методов принципиального значения не имеет.
Общее число физико-химических методов анализа довольно велико — оно составляет несколько десятков. Наибольшее практическое значение среди них имеют следующие:
6
1) спектральные и другие оптические методы;
2) электрохимические методы;
3) хроматографические методы анализа.
Среди указанных трех групп наиболее обширной по числу методов и важной по практическому значению является группа спектральных и других оптических методов анализа. Она включает методы эмиссионной атомной спектроскопии, атомно-абсорб ционной спектроскопии, инфракрасной спектроскопии, спектрофотометрии, люминесценции и другие методы, основанные на измерении различных эффектов при взаимодействии вещества с электромагнитным излучением.
Группа электрохимических методов анализа, основанная на измерении электрической проводимости, потенциалов и других свойств, включает методы кондуктометрии, потенциометрии, вольтамперометрии и т. д.
В группу хроматографических методов входят методы газовой и газожидкостной хроматографии, жидкостной распределительной, тонкослойной, ионообменной и других видов хроматографии.
Перечень групп является далеко не полным, так как сюда не вошли многие методы (радиометрические, масс-спектральные и др.). Эти методы будут рассмотрены отдельно, что, конечно, ни в коей мере нельзя считать признаком их второстепенности.
1.3. ОСНОВНЫЕ ПРИЕМЫ, ИСПОЛЬЗУЕМЫЕ В ФИЗИКО-ХИМИЧЕСКИХ МЕТОДАХ АНАЛИЗА
Почти во всех физико-химических методах анализа применяется два основных методических приема: метод прямых измерений и метод титрования (метод косвенных измерений).
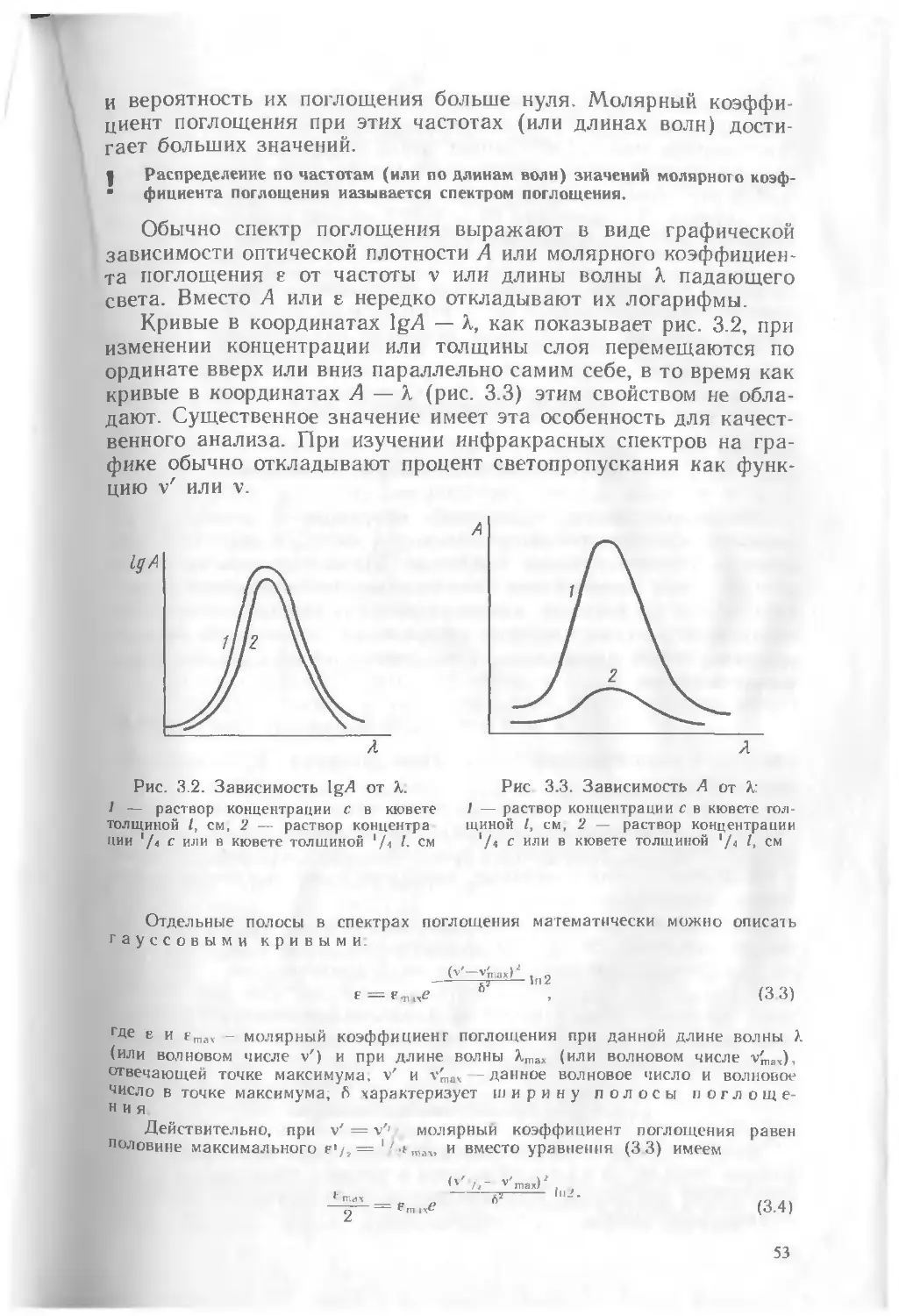
Прямые методы. В этих методах используется зависимость аналитического сигнала от природы анализируемого вещества и его концентрации. Свойством, зависящим от природы вещества, является, например, длина волны спектральной линии в эмиссионной спектроскопии, потенциал полуволны в полярографии, а количественной характеристикой служит интенсивность сигнала — интенсивность спектральной линии в первом случае, сила диффузионного тока — во втором. В некоторых методах связь аналитического сигнала с природой вещества установлена строго теоретически Например, линии в спектре атома водорода могут быть рассчитаны по теоретически выведенным формулам с использованием фундаментальных констант (постоянная Планка, заряд электрона и т. д.). Взаимосвязь качественной и количественной характеристик приведена на рис. 1.1. По оси абсцисс отложены однородные характеристики Р, например длины волн спектральных линий в порядке нх возрастания, а по ординате — интенсивность аналитического сигнала I. При качественном анализе наблюдают сигнал, например, какая из ожидаемых длин Волн появится в спектре пробы, а при количественном измеряют интенсивность сигнала.
7
Связь интенсивности аналитического сигнала 1 с концентрацией вещества имеет различный характер. Часто эта зависимость выражается простым линейным соотношением
Рис 1.1. Взаимосвязь качественной и количественной характеристик компонента пробы где А — константа; с — концентрация.
В аналитической практике наибольшее распространение получили следующие методы прямого количественного определения с помощью физико-химических измерений: 1) метод градуировочного графика; 2) метод молярного свойства; 3) метод добавок. Все они основаны на использовании стандартных образцов или стандартных растворов.
Метод градуировочного графика. В этом методе измеряется интенсивность аналитического сигнала I у нескольких стандартных образцов или нескольких стандартных растворов и строится градуировочный график обычно в координатах 1 = f(c), где с — концентрация определяемого компонента в стандартном образце или стандартном растворе. Затем в тех же условиях измеряется интенсивность сигнала у анализируемой пробы и по градуировочному графику находится концентрация анализируемого вещества. Интервал концентраций на градуировочном графике должен охватывать предполагаемую область анализируемых концентраций, а состав стандартного образца или раствора должен быть близок к составу анализируемого.
Метод молярного свойства. Здесь также измеряется интенсивность аналитического сигнала у нескольких стандартных образцов или растворов и рассчитывается молярное свойство А, т. е. интенсивность аналитического сигнала, пропорциональная 1 моль вещества: А = 1/с. Затем в тех же условиях измеряется интенсивность сигнала у анализируемой пробы и по соотношению с = 11А рассчитывается концентрация анализируемого компонента. Метод предполагает строгое соблюдение соотношения (1.1), по крайней мере, в области анализируемых концентраций.
Метод добавок. В этом методе сначала измеряется интенсивность аналитического сигнала пробы, затем в пробу вводится известный объем стандартного раствора до концентрации сст и снова измеряется интенсивность сигнала. Если 1Х — интенсивность аналитического сигнала пробы, а 1х + „—интенсивность сигнала после добавки стандартного раствора, то, очевидно,
Л = Асх, А + ст — А(сх 4“ Сет ) ,
откуда
Метод также предполагает строгое соблюдение соотношения (1.1). Уравнение (1.2) нередко решается графически.
Методы титрования. В этих методах в ходе титрования измеряется интенсивность аналитического сигнала I и строится кривая титрования в координатах I — V, где V — объем добавленного титранта, мл. Точка эквивалентности находится по кривой титрования. Виды кривых титрования весьма многообразны, так как интенсивность аналитического сигнала может быть связана с концентрацией определяемого вещества, титранта или продукта реакции.
Глава 2
ЭМИССИОННЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
2.1. ОСНОВНЫЕ ХАРАКТЕРИСТИКИ ЭЛЕКТРОМАГНИТНОГО ИЗЛУЧЕНИЯ
| Спектральные и другие оптические методы анализа основаны на использо-* ваиии различных явлений и эффектов, возникающих при взаимодействии вещества и электромагнитного излучения.
Поскольку свет имеет двойственную природу — волновую и корпускулярную, для его описания используют два вида характеристик — волновые и квантовые. К волновым характеристикам относятся частота колебаний, длина волны и волновое число, к квантовым — энергия квантов.
Частота колебаний v показывает число колебаний в 1 с, измеряется в герцах (Гц). Высокие частоты измеряются в килогерцах (1 кГц= 103 Гц), мегагерцах (1 мГц=106 Гц) и т. д. Например, красный свет характеризуется частотой 4-Ю14 Гц, зеленый 6-Ю14 Гц.
Длина волны к показывает наименьшее расстояние между точками, колеблющимися в одинаковых фазах. Это линейная единица, измеряется в СИ в метрах (м) и его долях — санти метрах (см), миллиметрах (мм), нанометрах (1 нм=10-9 м) и т. д.* Например, зеленый свет представляет собой электромагнитные колебания с длиной волны X = 500...550 нм, или 5-10-5 ... ...5,5 • 10-5 см. В зависимости от длины волны в электромагнитном спектре обычно выделяют следующие участки:
* До введения СИ длину волны выражали в миллимикронах (1тц = 1 нм = = 10~9 м) н в ангстремах (1А = 0,1 нм = 10 10 м)
9
Интервал длин волн
10 0,1 нм, или м
10 . 10 нм, или 10-11. .10“® м
10 400 нм, или 1О’й.4-1О ’ и
400 760 нм, или 4-10-7. .7,6-1СГ м
760. 10° нм, или 7,6-10~7. .10 J м
10 * м 1м
л > 1 м
Участок спектра
у-Изл учение
Рентгеновское излучение
Ультрафиолетовое излучение
Видимый свет
Инфракрасное излучение
Микроволны или сверхвысокие частоты
Радиоволны
Длина волны и частота колебаний связаны между собой соотношением
v = с/к,
где с — скорость света.
Если скорость света выражена в см/с, а длина волны — в см, то v = 3 - Ю|0/Х, где у выражена в Гц.
Например, для зеленого света X = 500 нм = 5-10“5 см, частота v = -|1|2_== 6. ю14 Гц.
Величину, обратную длине волны, называют волновым числом у' и выражают обычно в обратных сантиметрах (см-1).
Например, для зеленого света v' = 5 = 2-104 см-1.
Энергия электромагнитного излучения определяется соотношением
Е — hv,
где h — постоянная Планка, равная 6,62 -10-34 Дж-с. Чтобы получить энергию 1 моль, необходимо это значение умножить на число Авогадро:
Е = 6,62- 10-34-6,02- 1023v = 3,99-10-'%,
где Е выражено в Дж/моль.
2.2. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ЭМИССИОННОЙ СПЕКТРОСКОПИИ
2.2.1. Спектральные термы
Методы эмиссионного спектрального анализа основаны на измерении длины волны, интенсивности и других характеристик света, излучаемого атомами и ионами вещества в газообразном состоянии. Возникновение спектрального анализа как метода определения химического состава вещества относится к 1860 г., когда была опубликована работа Кирхгофа и Бунзена: «Химический анализ с помощью наблюдения спектра».
Испускание света атомами происходит за счет изменения энергии атомов. Атомы могут ооладать только строго определенными дискретными запасами внутренней энергии: Ер, Е\, Е% и т. д. Это означает также, что атомы не могут
ю
иметь энергию, промежуточную между Ео и Е\ или между Е\ и £2 и т. д. В невозбужденном, т. е. нормальном, состоянии атомы обладают минимальной энергией £0. При подведении энергии, например при столкновении с быстролетящими электронами, энергия которых достаточна для возбуждения, атомы возбуждаются, т. е. переходят на более высокий энергетический уровень: £ь £2 и т. д.
Через очень короткое время (~10-8 с) атом самопроизвольно возвращается в нормальное или какое-то более низкое возбужденное состояние. Освобождающаяся при этом энергия Д£ излучается в виде светового кванта hv:
АЕ — hv.
Частота излучения v определяется соотношением
Е&* Е^. _Еа* Е& >2 h____________________________________h h h ’ ' ’ '
где £д* и £д — энергия атома в возбужденном и нормальном состояниях.
Или, характеризуя излучение волновым числом (vz, см-1),
^z f А* ^А f А* Е& |2 2)
ch ch ch ’ ' ’ 1
где с — скорость света.
Совокупность излучаемых частот связана с энергетическими состояниями атома.
Энергетическое состояние простейшего одноэлектронного атома водорода определяется энергетическим состоянием его единственного электрона и может быть найдено с помощью уравнения Шредингера. При решении уравнения Шредингера получают набор трех квантовых чисел: главное квантовое число п, побочное квантовое число I и магнитное квантовое число mi. Необходимое для полной характеристики электрона четвертое квантовое число ms, называемое спиновым или просто спином, с уравнением Шредингера не связано.
Главное квантовое число п характеризует удаленность электрона от ядра, это номер электронной оболочки; К—оболочке соответствует п = 1, L — оболочке — л = 2 и т. д. Орбитальное, или побочное, квантовое число / характеризует подоболочки, из которых состоят оболочки, и орбитальный момент количества движения электрона; оно приближенно определяет и форму электронного облака. При главном квантовом числе п побочное квантовое число / может принимать значения 0, 1,2, ..., п—1, всего п значений. В спектроскопии побочное квантовое число принято обозначать буквой:
Числовое значение / . О 1 2 3 и т д.
Символ.................... . s р <f f ит д
II
Магнитное квантовое число mt характеризует проекцию магнитного момента движущегося электрона на направление внешнего магнитного поля. В соответствии с правилами пространственного квантования проекция может принимать только целочисленные значения. При данном / оно принимает значения 0, ±1; .., ±/, всего (2/+ 1) значений.
Спиновое квантовое число ms характеризует собственный момент электрона и принимает значения 1/2 и —1/2.
Полный момент электрона / является геометрической суммой векторов I и s:
/==/ + s,
/ иногда называют внутренним квантовым числом. Например, для d электрона (/ = 2) внутреннее квантовое число j = 2+1/2 = 5/2(s = 1/2) или / = 2-1/2 = 3/2(s =—1/2). В одноэлектронном атоме водорода энергетическое состояние электрона полностью определяется главным квантовым числом п. Решение уравнения Шредингера для такой системы приводит к следующему выражению для энергии электрона:
Подставляя уравнение (2.3) в соотношения (2.1) и (2.2), получаем:
(2-4)
где П| и п* — главное квантовое число электрона в основном и возбужденном состояниях атома соответственно; R — константа Ридберга, объединяющая фундаментальные физические постоянные.
Уравнения (2.4) и (2.5) сыграли большую роль в развитии теории атомных спектров и теории строения вещества. Впервые соотношение типа (2.5) было получено в 1885 г Бальмером как чисто эмпирическое. Длины волн известных тогда четырех линий в спектре водорода описывались формулой Бальмера с очень высокой точностью. Уже это наводило на мысль, что формула Бальмера является не просто эмпирическим соотношением, а скорее отражением какого-то еще не известного закона природы. Теоретическое значение константы Ридберга впервые было получено Бором в 1913 г на основании предложенной им знаменитой модели атома, в которой постулировались квантовые уровни энергии электрона. В настоящее время формулы (2.4) и (2.5) получаются как следствие квантово-мехачических представлений, опирающихся на уравнение Шредингера.
Волновое число спектральной линии в соответствии с уравнением (2.5) может быть представлено разностью двух величин:
(2-6)
12
Эти величины получили название спектральных термов Т:
Т,(п,) = Лг-. (2.7)
Следовательно, v' = Т\{п\) —
Термы атома водорода могут быть непосредственно вычислены по соотношениям (2.7). Приняв в (2.7), например, ni = 1, получим Гц,,, „ |) = R/1 = 109678,76 см-1.
Если принять ni = 2, а п* 3, то получим частоты линий, образующих серию Бальмера; при п\ = 3 и и. 4 — серию Пашена ит. д. Такими сериальными формулами полностью описывается спектр атомарного водорода, а при учете заряда ядра также спектры других одноэлектронных частиц — ионизированного гелия Не4, дважды ионизированного лития Li24", иона Be34 и др.
С учетом заряда ядра z формула (2.6) принимает вид
Однако в атомах и ионах, содержащих более одного электрона, взаимодействие частиц имеет более сложный характер и расчеты по формуле (2.8) уже не дают вполне удовлетворительных результатов. Ридберг показал, что спектральные термы атомов щелочных металлов могут быть выражены формулой
Т(п) = Яг2/(п + Д)2.
Поправка Д зависит от побочного квантового числа, сумму (п -|- Д) иногда называют эффективным квантовым числом. С увеличением числа внешних электронов формула усложняется.
Взаимодействие электронов в атомах элементов с небольшим порядковым номером (30 ... 35 и меньше) приводит к тому, что энергетическое состояние такого атома может быть охарактеризовано суммарным орбитальным моментом и суммарным спином. Поскольку магнитные моменты пропорциональны соответствующим квантовым числам, можно говорить о суммарном орбитальном и суммарном спиновом квантовых числах атома. Для их обозначения применяют те же, но не строчные, а прописные буквы латинского алфавита, какие используют для обозначения квантовых чисел отдельных электронов. Так, при общем об» значении орбитального квантового числа атома пишут L (вместо I для отдельного электрона), при L = 0 квантовое число обозначают буквой S, при L = 1 — буквой Р, при L = 2 — буквой D и т. д. Суммарный спин атома обозначают буквой S, а геометрическую сумму L -|- S буквой J.
13
| Группа энергетических состояний, характеризуемая одними и теми же вели-‘ чинами L и S, имеет близкую энергию и образует один терм.
В этом смысле и употребляется термин «т е р м» в современной спектральной систематике. При записи символа терма прежде всего указывают его основную характеристику: квантовое число суммарного орбитального момента L. Слева в виде верхнего индекса у символа терма записывают его мультиплетност ь, показывающую число близких по энергии состояний, которые образуют данный терм Мультиплетность М равна
М = 25 + 1,
где 5 — суммарный спин атома.
Очевидно, мультиплетность на единицу превышает число неспаренных электронов в атоме. Термы с М — 1 называют одиночными или синглетными, термы с М = 2 — двойными или дублетными и т. д. Например, терм 2D называют как дублет D. Отдельные компоненты терма L + 5; L + 5— 1; ...; L — S записывают в виде правого нижнего индекса терма. Если, например, суммарный спин какого-либо атома или иона равен 1 (5 = 1), а суммарный орбитальный момент равен 3(Л = 3), то символ терма имеет вид 3/+з, 2 (триплет F).
У замкнутых оболочек векторы L, S и J равны нулю, что существенно упрощает суммирование, однако полный вывод системы термов для данной электронной конфигурации остается довольно трудоемким. Терм основного состояния обычно определяют, пользуясь правилами Хунда:
1. Основное, т. е. низшее, энергетическое состояние имеет наибольшее значение суммарного спина (мультиплетности).
2. Среди термов с максимальной мультиплетностью низшим является тот, у которого наибольшее значение L.
3. Низшим компонентом мультиплета будет J = L — S, если уровень заполнен меньше чем наполовину, и J = L + 5, если он заполнен больше чем наполовину.
Вывод системы термов для заданной электронной конфигурации имеет большое значение в теоретической спектроскопии.
Каждая спектральная линия отражает переход электрона с одного энергетического уровня на другой и волновое число любой спектральной линии может быть представлено как разность термов. Однако не любая комбинация термов соответствует реально наблюдаемой спектральной линии. Существуют определенные правила отбора, указывающие, какие комбинации термов возможны и какие невозможны. Эти правила имеют квантово-механическое обоснование. Переходы, возможные по этим правилам, называются разрешенными, а невозможные — запрещенными. Основные правила отбора:
1 Разрешены переходы, при которых терм меняется на единицу, т. е. разрешены, например, Р — S- или D — P-переходы, но не разрешены Р — Р-, D — D- или 5 — £)-переходы.
14
2. Внутреннее квантовое число J может меняться только на ±1 или совсем не меняться. Запрещены переходы, при которых Д7 = ±2.
3. Разрешены переходы без изменения мультиплетности.
Найдем разрешенные переходы, например, в атоме натрия, имеющем электронную структуру ls22s22p63s‘. Первые две ооолочки (п = 1, п = 2) в атоме натрия заполнены полностью, поэтому его термы будут определяться единственным электроном, который в основном состоянии находится на уровне 3s Терм этой конфигурации будет 23./г. Дублет здесь показывает формальную муль-типлетность, в действительности же все термы 5 являются одиночными (синглетными) При возбуждении атома натрия электрон с уровня 3s будет переходить на уровни р, d и т. д и термами атома в возбужденном состоянии, очевидно, будут 2Р1/2 !Z1, 2О»/2 ’„ит д Энергетические уровни Na графически представлены на рис 2.1, где также показаны некоторые из разрешенных переходов Линии, соответствующие Р — S-переходам, двойные, так как при этих переходах комбинируют двойные и одиночные термы 2PS/1—2Siz„ 2Р/г—25'/,. В первом из этих переходов AJ = 1, а во втором Д/ = 0 Запрет по мультиплетности не нарушается, так как формальная мультиплетность терма 5 в данном случае остается равной 2.
Рис. 2.1. Энергетические уровни (термы) ато- Рис. 2.2. Схема D — Р-перехо-ма натрия дов в атоме натрия
Несколько сложнее D— P-переходы (рис 2.2). Термы 2D5/ и 2D'/2 очень близки; в спектре появляются дублеты, соответствующие переходам 2D5, 3„ — — 2Р3/ и 2D3„ — 2Р(/ . В приборах высокого разрешения можно обнаружить трипле’г, так как появляется линия, соответствующая переходу 2D5„ — 2^K/->’ сливающаяся ранее с линией 2D3f?—2Р3(,2. Переход 2Е>5/2— 2Pj^2 запрещен, так как Д/ = 2.
Наиболее яркой в спектре будет линия, отвечающая переходу с первого возбужденного уровня на основной. Линию, отвечающую этому переходу, называют резонансной Например, у натрия резонансными являются переходы 2Р3/2 — и 2Р|, — 2Si/2, им отвечают линии с длиной волны 588,99b и 589,593 нм. Это излучение, в частности, окрашивает пламя горелки в желтый цвет при введении солей натрия.
Изоэлектронные атомы дают сходные спектры, поэтому можно говорить, например, о спектрах щелочных или спектрах щелочноземельных металлов. Сходство проявляется в наличии в спектрах родственных элементов одинаковых групп линий (например, дублетов у щелочных металлов).
Спектр атома любого элемента существенно отличается от
15
спектра его иона в связи с изменением числа оптических электронов при ионизации. Поэтому в таблицах спектральных линий рядом с символом химического элемента приводят римскую цифру, по которой можно судить о кратности ионизации атома. Цифра I относится к нейтральному атому (например, Fe(I) относится к Fe), цифра II — к однократно ионизированному атому (например, Fe(II) относится к Fe+) и т. д.
2.2.2. Интенсивность спектральных линий
Интенсивность спектральной линии hr приближенно определяется выражением
hr = NkAkrhvkr, (2.9)
где Nk — число атомов в возбужденном состоянии Л; Акг — вероятность перехода из возбужденного состояния k в более низкое состояние г; хкг — частота, соответствующая этому переходу; h — постоянная Планка.
Для термически равновесной плазмы распределение атомов по степеням возбуждения определяется законом Больцмана:
<2Ю)
где N — число атомов в плазме; gk и go — статистические веса возбужденного и нормального состояний; Ек — энергия возбуждения Л-го уровня.
При сочетании уравнений (2.9) и (2.10) получаем для интенсивности спектральной линии
Ikr=NAkr-^hNkrew{—(2.11)
Как видно, интенсивность спектральной линии зависит от температуры.
При постоянстве температуры и других условий возбуждения уравнение (2.11) переходите
hr=a'N, (2.12)
где а' объединяет все сомножители в уравнении (2.11), кроме N.
Если режим работы источника возбуждения достаточно стабилен и скорость подачи вещества в плазму постоянна, наступает некоторое стационарное состояние, при котором число атомов элемента в плазме оказывается пропорциональным концентрации этого элемента в пробе:
N=a"c, (2.13)
где с — концентрация вещества в пробе; а" — коэффициент пропорциональности.
Подставляя соотношения (2.12) и (2.13) в (2.11), получаем
hr = a'a"c — ac. (2.14)
б
Если условия разряда не меняются при изменении концентрации, то коэффициент а остается постоянным и уравнение (2.14) выполняется достаточно хорошо. Коэффициент а зависит от параметров разряда, условий поступления вещества в плазму и констант, характеризующих возбуждение и последующие переходы.
Однако не все кванты, испускаемые возбужденными частицами, достигают приемника света. Квант света может быть поглощен невозбужденным атомом и, таким образом, не будет зафиксирован приемником излучения. Это так называемое с а м о-поглощение. С увеличением концентрации вещества само-поглощение возрастает.
Самопоглощение учитывается в уравнении Ломакина, которое хорошо описывает концентрационную зависимость интенсивности спектральной линии:
1=ась, (2.15)
где коэффициент а зависит от режима работы источника возбуждения, его стабильности, температуры и т. д.; b — коэффициент самопоглощения, учитывающий поглощение квантов света невозбужденными атомами.
При логарифмировании уравнения (2.15) получаем
lg/=lga 4- b Igc. (2.16)
Линейная зависимость 1g/ от Igc очень удобна для построения градуировочного графика. Уравнение (2.16) является основой количественного спектрального анализа.
2.2.3. Ширина спектральных линий
Важной характеристикой спектральной линии является ее ширина. Как известно, спектральная линия — это оптическое изображение щели спектрального прибора, и чем шире щель, тем шире спектральная линия. Тем не менее, хотя все спектральные линии в данном спектре являются изображением одной и той же щели и, казалось бы, должны иметь одинаковую ширину, она на самом деле различна. Это кажущееся противоречие вызывается несколькими причинами. Наиболее существенны из них следующие.
1. Реальное излучение в обычных условиях эмиссионной спектроскопии не бывает строго монохроматичным (его энергия распределена в некотором интервале длин волн), и чем больше этот интервал, тем шире спектральная линия. Это так называемая естественная ширина спектральной линии, она составляет величину порядка 10~4 нм. При решении большинства аналитических задач с этим уширением практически можно не считаться, так как оно значительно меньше уширения, вызываемого другими причинами.
2. Если светящаяся частица движется вдоль линии наблюде-
|БИБ Л ИС'Тс К Я - 17
ОМСКОГО •
У Государственного S
ния, то излучаемая ею длина волны испытывает некоторое смещение, приводящее в условиях эмиссионной спектроскопии при большом числе излучающих частиц к уширению спектральных линий. Это допплеровское уширение. Оно возрастает с уменьшением атомной массы излучающего атома и повышением температуры. Для элементов середины периодической системы и температуры 5000 °C допплеровское уширение в видимой части спектра составляет примерно 0,001...0,002 нм.
3. В электрическом или магнитном поле энергетические уровни атома расщепляются на ряд подуровней. Это явление известно как эффект Штарка (расщепление в электрическом поле) или эффект Зеемана (расщепление в магнитном поле). Поле, обусловленное заряженными частицами в плазме, оказывается достаточным, чтобы вызвать уширение спектральных линий, которое доступно наблюдению на обычных приборах.
4. С увеличением концентрации элемента в пробе возрастает самопоглощение, что приводит к уменьшению интенсивности центральной части линии и ее уширению.
Очень широкие и очень узкие спектральные линии менее пригодны для спектрального анализа, чем линии средней ширины
2.3. ОСНОВНЫЕ УЗЛЫ СПЕКТРАЛЬНЫХ ПРИБОРОВ
Прибор для проведения спектрального анализа имеет следующие основные узлы: источник возбуждения, диспергирующий элемент и приемник света. Кроме этих основных узлов в любом спектральном приборе есть оптическая система, предназначенная для получения параллельного пучка света, его фокусировки, изменения хода лучей и т. д.
В источнике возбуждения вещество атомизируется и возбужденные атомы или ионы испускают свет, который диспергирующим элементом разделяется в пространстве на отдельные составляющие, а приемник света их фиксирует.
2.3.1. Источники возбуждения
Источники возбуждения переводят пробу из конденсированной фазы в парообразную и возбуждают вещество в этой фазе. В большинстве источников возбуждения эти функции совмещаются, однако в некоторых случаях применяют два устройства: одно для получения газовой фазы, другое — для возбуждения. При анализе, например, биологических объектов или некоторых изделий металлургической промышленности, когда особый интерес вызывает локальный анализ, для перевода избранного участка пробы в парообразное состояние с успехом используется лазерная техника.
Возбуждение атомов происходит главным образом при передаче энергии быстролетящими частицами, чаще всего электронами, если их энергия достаточна для возбуждения. Если кине
18
тическая энергия летящих частиц меньше энергии возбуждения первого возбужденного уровня, при столкновении произойдет лишь перераспределение энергии, как при ударе упругих шаров, но возбуждения не произойдет. Это так называемые упругие соударения. Чтобы атом перешел в возбужденное состояние, необходима энергия, по меньшей мере равная энергии резонансного уровня атома. Соударения, сопровождающиеся возбуждением атома, называются неупругими ударами первого рода.
Источник возбуждения должен обеспечивать необходимую яркость спектра по сравнению с фоном и быть достаточно стабильным, т. е. интенсивности спектральных линий должны оставаться постоянными по крайней мере за время измерения. Современные успехи количественного спектрального анализа в значительной степени достигнуты в связи с созданием источников возбуждения высокой стабильности. Наибольшее применение в качестве источников возбуждения получили пламя, дуга и искра.
К источнику возбуждения часто относят и устройство для введения анализируемой пробы, вид и конструкция которого зависят от характера, агрегатного состояния и физических свойств пробы. Анализируемые металлические образцы в электрических источниках возбуждения обычно служат электродами разрядного промежутка. Растворы вводят в источник возбуждения с помощью распылителей, порошкообразные пробы — с помощью специальных устройств или при использовании угольных электродов, в которых высверливается канал для набивки порошкообразной пробы. Применяют также брикетирование анализируемого порошка с добавкой металлов, их оксидов или графита. Изготовленный брикет затем становится электродом.
Пламя. Это известный еще со времен Бунзена и Кирхгофа источник света в спектральном анализе. Пламя дает достаточно яркий и стабильный спектр. Простота регулировки и надежность работы пламенных источников обусловили, по сути дела, второе рождение пламенно-фотометрических методов, применяемых очень широко. Возбуждение спектров в пламени имеет в основном термический характер. Температура пламени зависит от состава горючей смеси. Пламя обычной газовой горелки имеет температуру примерно 900°C. Смесь водорода с воздухом дает 2100°C, водорода с кислородом 2800°C, ацетилена с кислородом — около 3000°C.
С помощью пламенных источников определяют свыше 40 элементов (Mg, Си, Мп, Т1, щелочные элементы, щелочно-земельные и т. д.). В пламени не возбуждаются так называемые трудновозбудимые элементы и общая картина спектра является более простой, чем дугового или искрового. Анализируемое вещество вводится в пламя в виде раствора с помощью специального распылителя, обеспечивающего равномерное поступление вещества.
19
Дуга. Электрическая дуга — это электрический разряд при сравнительно большой силе тока (5...7 А) и небольшом напряжении (50...80 В). Разряд поддерживается за счет термоэлектронной эмиссии с раскаленной поверхности катода. Разряд пропускают между электродами из анализируемого образца или между образцом и электродом, не содержащим определяемых элементов. Температура дуги достигает 5000...6000 °C. Введение в элект- j роды примесей, обладающих более низким, чем основной элемент I пробы, потенциалом возбуждения понижает температуру дугц^* Так, в присутствии солей калия температура дуги между угольными электродами падает с 7000 до. 4000 °C. Это открывает возможность регулировать температуру дуги и поддерживать ее постоянной путем введения в зону разряда элемента с низким потенциалом возбуждения — так называемого х ще ктроскоп и-ч е с ко г о буфера. Обычно это соли натрия или калия в~^о-'“-статочном количестве. В присутствии спектроскопического буфера устанавливается определенная температура плазмы, практически не зависящая от состава анализируемой пробы.
При анализе тугоплавких металлов и сплавов электроды дуги делают из анализируемого образца. Для анализа легкоплавких металлов и сплавов, а также руд, минералов, стекол, шлаков и других непроводящих материалов электродами служат обычно графитовые или угольные стержни — так называемые спектральные угли. Анализируемая проба помещается в канал одного из электродов и испаряется в плазму при работе дуги.
В дуге удается получить спектр почти всех элементов. Используется дуга постоянного и переменного тока. Для обеспечения непрерывности горения и стабилизации процесса разряда применяют специальные дуговые генераторы. Яркость дугового спектра достаточно велика, а иногда чрезмерна, что может—явитьея—недостатком, так как значительно увеличивает фон. Не всегда достаточная воспроизводимость условий возбуждения в дуге ограничивает применение дуговых спектров в ос-_,дшвцом качествошпям и подуколичественным анализом, |Сущест-венным недостатком дуги является также значительное разрушение анализируемого образца. Повышение напряжения обычно улучшает стабильность дуги, что приводит к повышению точности анализа. Высоковольтная дуга питается напряжением в несколько тысяч вольт.
В практике спектрального анализа применяют также плазменную горелку или плазмотрон (рис. 2.3). Плазмотрон представляет собой камеру" С двумя графитовыми электродами. В камере между анодом 1 и катодом 3 зажигается дуга при силе тока 20...30 А и по трубке, расположенной по касательной к стенке, подается инертный газ 2 при 150...200 кПа. В аноде имеется отверстие, через которое инертный газ выходит. Вихревые потоки газа в камере охлаждают плазму снаружи, что приводит к сжатию разрядного шнура и увеличению в нем плотности тока. Сжатая плазма вместе с газом выбрасывается через отверстие анода в
20
виде струи длиной 10... 15 мм, которая светится над поверхностью анода. Температура в плазме достигает 5ООО...1ОООО°С и выше. Анализируемый раствор 4 подается в плазму специальным распылителем. При анализе твердых образцов пробы могут помещаться в катод или также вводиться в плазму распылителем. Высокая температура и интенсивность свечения делают плазмотрон весьма перспективным источником возбуждения, особенно для анализа трудноиспаряющихся и трудновозбудимых веществ. Большое аналитическое применение находит также высокочастотный плазмотрон с индукционной катушкой, питаемый ВЧ-ге-нератором.
Рис. 2 3. Плазмотрон
Рис. 2.4. Принципиальная схема искрового генератора
Искра. Для получения искры используют специальные искровые генераторы. Принципиальная схема генератора (рис. 2.4) включает вторичную обмотку повышающего трансформатора I, которая присоединяется параллельно к емкости 2 и последовательно к катушке самоиндукции 3 и искровому промежутку 4. Пробивное напряжение более постоянно в управляемых схемах. Так, в дуге Райского введен вспомогательный разрядный промежуток, задающий и поддерживающий на постоянном уровне пробивное напряжение основного разрядного промежутка. При горении искры развивается температура 7000...10000 °C и происходит возбуждение всех элементов. При необходимости температура искры может быть повышена до 12 000 °C и выше. Для проведения локального микроспектрального анализа применяют микроискровой метод, в котором используют игольчатые электроды (например, медные) и устанавливают малое межэлектродное расстояние. Микроискровой метод дает возможность выявить локальное распределение элементов по поверхности в сталях, железе и других образцах с локальностью 0,3...0,5 мм2. Техника микроискрового анализа применяется также в методе переноса, когда в результате микроискрового разряда небольшое количество вещества с поверхности образца переносится на вспомогательный угольный электрод, спектр которого в дальнейшем возбуждается и исследуется обычным методом.
21
Яркость искрового спектра недостаточна для визуального анализа. Основное достоинство искры составляет большая стабильность условий разряда и, следовательно, необходимая в количественном анализе стабильность условий возбуждения. Работа с искрой практически не вызывает разрушения образца, что выгодно отличает искру от дуги.
Перспективным высокочувствительным источником света является также полый катод, в котором могут возбуждаться элементы с высоким потенциалом возбуждения.
2.3.2. Диспергирующий элемент
Диспергирующий элемент разлагает излучение в спектр. Это наиболее важная часть спектрального прибора, в значительной степени определяющая его аналитические возможности и основные характеристики: линейную дисперсию и разрешающую способность. Диспергирующий элемент характеризуется угловой дисперсией, которую определяют как угловое расстояние А<р между двумя лучами с близкими длинами волн X, и Х2, отнесенное к интервалу AX=Xi — Х2, т. е От угловой
дисперсии диспергирующего элемента зависит линейная дисперсия спектрального прибора где А/ —
Да оЛ
линейное расстояние в фокальной плоскости прибора между двумя лучами с близкими длинами волн Xi и Х2, отнесенное к разности АХ=Х,— Х2. В практике часто используется величина, обратная линейной дисперсии: D = \/Di. Она обычно находится в пределах от 0,1 до 10,0 нм/мм.
| Разрешающей способностью спектрального прибора называют его способ-* ность давать раздельное изображение двух спектральных линий с близкими длинами волн.
Количественной характеристикой разрешающей способности прибора является отношение R = k/\k1 где АХ—Xi— Х2— интервал, в котором линии Xi и Х2 наблюдаются раздельно, а Х= =2±i2± — средняя длина волны. У обычных спектральных приборов разрешающая способность составляет величину от 5000 до 50 000.
В качестве диспергирующего элемента используют призмы, дифракционные решетки и интерференционные устройства. Большое распространение в аналитической практике получили призменные спектральные приборы и приборы с дифракционной решеткой.
Призмы для спектральных аппаратов изготовляют из стекла или кварца, так как эти материалы достаточно прозрачны в широкой области длин волн. Стеклянные призмы имеют более высокую угловую дисперсию и более доступны по сравнению с кварце
22
выми, поэтому для работы в видимом и ближнем инфракрасном участках спектра обычно используют стеклянные призмы Для исследования ультрафиолетовой области спектра применяют призмы из кварца.
Дифракционные решетки в качестве диспергирующего элемента имеют существенные достоинства. Дисперсия света в дифракционной решетке не зависит от длины волны и разрешающая способность решетки значительно выше, чем призмы. Спектральный интервал, доступный для исследования, достаточно широк (от 200 до 1000 нм).
2.3.3. Приемники света
Приемники света характеризуются спектральной чувствительностью: способностью воспринимать излучение различной длины волны и интегральной чувствительностью, которая измеряется действием неразложенного в спектр излучения.
Глаз человека чувствителен к свету в области спектра примерно от 400 до 760 нм. Чувствительность глаза максимальна к желто-зеленому свету 7550 нм) и убывает от него в обе стороны — и к красной и к фиолетовой. Возможности глаза как измерительного прибора ограничены также и тем, что он очень приближенно оценивает разность или отношение интенсивностей световых потоков. С достаточной точностью он устанавливает лишь равенство или неравенство интенсивностей световых потоков одного цвета. На этом свойстве основаны все приемы в и-зуальных методов.
Фотопластинка. Светочувствительный слой фотопластинки — это мелкие кристаллы галогенидов серебра, равномерно распределенные в тонком желатиновом слое. При освещении фотопластинки в светочувствительном слое образуется скрытое изображение, как результат фотолиза галогенида серебра под действием кванта света: AgBr-|-/rv=Ag-(-Br На освещенных местах фотопластинки появляются кристаллы металлического серебра. Скрытое изображение проявляют путем обработки фотопластинки специальным проявителем, который завершает процесс восстановления серебра на освещенных участках и позволяет получить видимое изображение. Полученное изображение закрепляют (фиксируют) с помощью раствора тиосульфата натрия (закрепителя или фиксажа), который растворяет кристаллы галогенида серебра, не подвергшиеся действию света: AgBr-(-4-2520з~=Ag(S2O3)2~ + Вг-. После такой обработки на фотопластинке остается изображение спектра в виде спектральных линий.
Если / и /0 — интенсивность света, прошедшего соответственно через затемненный (засвеченный) участок фотопластинки и через незасвеченный (рис. 2.5), то почернение (или плотность почернения) S равно
23
S=lg-y-.
Почернение фотопластинки зависит от экспозиции, или количества освещения//, которое приближенно определяется формулой
H=Et, (2.17)
где Е — освещенность; t — время освещения.
Зависимость почернения от количества освещения изображается характеристической кривой фотопластинки (рис. 2.6). Участок АВ называют областью недодержек, участок CD — областью передержек. На участке ВС, называемом областью нормальных почернений, величина почернения линейно зависит от логарифма экспозиции. Рис. 2.6 показывает, что
EF \gH-\gH,’ (2-18)
где у — фактор контрастности.
Рис. 2.5. Фотопластинка:
I — эмульсия, 2 — подложка
Рис 2 6. Характеристическая кривая фотопластинки
В эмиссионной спектроскопии используют контрастные фотопластинки, так как чем выше фактор контрастности у, тем большее почернение будет вызывать одно и то же количество освещения. Продолжение прямолинейного участка характеристической кривой пересекает ось абсцисс в точке 1g//., которая определяет инерцию фотопластинки.
Для прямолинейного участка характеристической кривой в соответствии с уравнением (2.18) получаем
S = ylgtf — ylg/7„
или (так как у и И. для данной пластинки постоянны)
S=ylg// — /. (2.19)
24
Подстановка в уравнение (2.19) значения Н из (2.17) дает
S=y\gEt — I. (2.20)
Это основное уравнение фотопластинки. Его применимость ограничена прямолинейным участком характеристической кривой.
Другим важным свойством фотопластинки является ее чувствительность. По ГОСТу чувствительность определяют как величину, обратную _количеству освещения (экспозиции), необходимого для подуцация почерТОТПТя^ на 0,2 превышающего почернение вуали при освещении белым счетом. Для спектрального анализа более интересной характеристикой является спектральная чувствительность, которую обычно представляют графически как S=f(X), где X — длина волны падающего света.
Обычные фотопластинки имеют чувствительность в спектральном диапазоне от 230 до 500 нм. Эти пределы чувствительности могут быть значительно расширены сенсибилизацией пластинок. В настоящее время фотопластинки успешно применяют в широкой спектральной области от короткого ультрафиолета до 1000 нм.
К основным достоинствам фотопластинок как приемников излучения в спектральном анализе относят их способность интегрировать интенсивность света, высокую чувствительность, достаточно широкий спектральный интервал, документальность анализа, а также возможность длительное время сохранять информацию, заложенную в спектре. По сфотографированным спектрам, даже спустя длительное время после их получения, можно, в частности, проверить содержание различных элементов в пробе, включая и те, которые ранее не определялись. Точность методов анализа с применением фотопластинки достаточно высока. При этом следует отметить наряду с методами точного фотометриро-вания возможности визуальной оценки интенсивности спектральных линий.
Одним из основных недостатков фотопластинок является неравномерность их эмульсии, представляющая дополнительный источник погрешности анализа, а также длительность и трудоемкость операций по химической обработке фотоматериалов.
Фотоэлементы. Фотоэлементами называют устройства, преобразующие световую энергию в электрическую Действие фотоэлементов основано на использовании фотоэффекта. Различают внешний и внутренний фотоэффекты. При внешнем фотоэффекте поглощение света приводит к отрыву электрона с облучаемой поверхности. Внутренний фотоэффект характеризуется увеличением электрической проводимости вещества под действием света. Если внутренний фотоэффект проявляется вблизи граничного слоя между двумя полупроводниками или полупроводником и металлом, то возникает фотоЭДС. Это явление иногда выделяют в особый вид фотоэффекта и называют фотогальваническим эффектом или эффектом запорного (запирающего) слоя.
Фотоэлемент с внешним фотоэффектом (рис. 2.7) состоит из
25
фотокатода 1 и анода 2, помещенных в стеклянную колбу 3. Если колба эвакуирована, фотоэлемент называют вакуумным При действии света на катод (обычно кислородно-цезиевый или сурьмяно-цезиевый) из него вырываются электроны, которые, попадая на анод, замыкают цепь: гальванометр показывает наличие тока. Фотоэлементы с внешним фотоэффектом чувствительны в широкой области спектра, имеют линейную световую характеристику и практически безынерционны. Чувствительность их невысока, однако большое внутреннее сопротивление позволяет включать эти фотоэлементы в усилительные схемы. Среди недостатков у элементов этого типа необходимо отметить наличие темнового тока, хрупкость конструкции.
Рис. 2.7. Фотоэлемент с внешним эф фектом
Рис 2.8 Фотоумножитель:
/ — катод, 2 — эмиттер, 3 — анод
Значительно более чувствительными приемниками света являются фотоумножители (рис. 2.8), действие которых основано на внешнем фотоэффекте и вторичной электронной эмиссии. Расположение электродов и фокусирующее поле выбирают так, чтобы первичный электронный поток, попадая на первый эмиттер, вызывал вторичную электронную эмиссию, электроны вторичной эмиссии направлялись на следующий эмиттер и т. д. Усиление подчиняется закону геометрической прогрессии:
/=/оот.
где / — сила тока на выходе прибора; /о — начальная сила тока; а — коэффициент вторичной электронной эмиссии; т — число каскадов усиления.
Фотоумножители дают усиление в 105...106 раз. Они нашли широкое применение в измерительной технике, в телевидении, передачах из космоса, при исследовании ядерных и космических излучений и в других областях науки и техники.
В фотоэлементах с запирающим слоем используют внутренний фотоэффект полупроводника и вентильный эффект
26
Fe - подложка
HIIUUI /
2
Рис 2.9 Фотоэлемент с запирающим слоем
I — контактное кольцо, 2 — запирающий слой
возбуждаются и
запираю-
запирающего слоя, который образуется на границе между полупроводником и металлом или двумя полупроводниками Запирающий слой пропускает электроны практически лишь в одном направлении и не пропускает в другом. Таким образом, возбужденные электроны могут проходить через запирающий слой, создавая разность потенциалов. В селеновом фотоэлементе (рис. 2.9) электроны, находящиеся в селене, под действием света
щий слой проходят в полупрозрачную пленку золота, являющуюся покрывным электродом. Обратному переходу электронов препятствует запирающий слой. Это приводит к тому, что покрывной электрод заряжается отрицательно, а слой селена положительно При замыкании такой системы во внешней цепи появляется ток. Характерным свойством фотоэлементов с запирающим слоем является возникновение тока под действием света без участия постороннего источника напряжения. Аналогично устроены кремниевый и многие другие фотоэлементы с запирающим слоем.
Достоинствами фотоэлементов с запирающим слоем является высокая чувствительность, широкий спектральный интервал и простота конструкции. Основные недостатки, нелинейность световой характеристики, инерционность и заметная температурная зависимость фототока
2.4. КОНСТРУКЦИЯ СПЕКТРАЛЬНЫХ ПРИБОРОВ
Конструкции спектральных приборов весьма разнообразны. Они различаются по типу диспергирующего элемента, способу регистрации спектра и т. д.
В приборах для визуального спектрального анализа — различных стилоскопах и стилометрах — диспергирующим элементом являются стеклянные призмы, приемником света служит глаз наблюдателя. На рис. 2.10 представлена оптическая схема одного из распространенных отечественных приборов — стило-скопа СЛ-11.
Свет от источника возбуждения 1 через оптическую систему 2 попадает на входную щель 3 постоянной ширины 0,02 мм и поворотной призмой 4 через объектив 5 направляется иа диспергирующую систему из двух призм 6 и 7. Покрытый серебром катет призмы 7 отражает лучи, которые вновь проходят диспергирующую систему и через объектив 5 и поворотную призму 8 попадают на зеркало 9 и далее в окуляр //, служащий для наблюдения спектра. Фотометрический клин 10 позволяет ослаблять ин
27
тенсивность выбранной спектральной линии и по специальной шкале, связанной с клином, оценивать ее относительную интенсивность. Призма 7 может вращаться, что приводит к перемещению спектра в поле зрения, а угол поворота призмы показывает по шкале, к какой области длин волн относится наблюдаемый участок спектра. Стилоскоп предназначен для работы в спектральной области от 390 до 700 нм, для возбуждения спектра обычно используется дуговой генератор. Элементарный фотометрический клин позволяет повысить точность анализа по сравнению с обычными стилоскопами. Юстировка оптической системы делается на заводе и заданное положение оптических деталей сохраняется благодаря жесткому монтажу.
Для выполнения экспрессных аналитических работ вне лаборатории применяют переносной стилоскоп типа СЛП-2, оптическая схема которого лишь немногим отличается от оптической схемы СЛ-11.
Рис. 2.10. Оптическая схема стилоскопа СЛ-11
Более сложную, чем у стилоскопов, оптическую схему и устройство имеют стилометры, например стилометр СТ-7. Фотометрическая система этого прибора позволяет независимо ослаблять интенсивность двух спектральных линий и количественно характеризовать их относительную интенсивность, а также сближать в поле зрения аналитическую пару линий, что создает удобство в работе и повышает точность анализа.
Наиболее широко распространенными приборами спектрально-аналитических лабораторий являются кварцевые спектрографы ИСП-22 и ИСП-28, позволяющие фотографировать спектры в области длин волн 200...600 нм. Оптическая схема спектрографа ИСП-28 представлена на рис. 2.11. От источника излучения 1 свет проходит оптическую систему 2 и через щель 3 попадает на зеркальный объектив 4, который отражает падающие лучи параллельным потоком на диспергирующую призму 5 и далее через объектив 6 на фотопластинку 7. На пластинке может быть сфотографирована также постоянная миллиметровая шкала, облегчающая расшифровку спектра. Спектрограф ИСП-28 удобен, прост и надежен в работе.
Модификацией ИСП-28 является спектрограф ИСП-30, име-
28
ющий реле времени и некоторые другие дополнительные устройства. Возрастает выпуск приборов с дифракционной решеткой в качестве диспергирующего элемента и фотографической или фотоэлектрической регистрацией спектра (ДФС-13, ДФС-10М и др.) Существенным достоинством фотоэлектрических приборов явля
ется экспрессность получения результатов при сохранении точности. Так, с помощью квантометра ДФС-10М определяют 11 элементов в одном образце за 6...8 мин.
2.5. КАЧЕСТВЕННЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
Основой качественного спектрального анализа является свойство каждого химического элемента излучать характерный линейчатый спектр. Задача качественного спектрального анализа сводится к отысканию линий определяемого элемента в спектре пробы. Принадлежность линии данному элементу устанавливается по длине волны и интенсивности линии. Однако общее число линий в спектре многих элементов очень велико: например, спектр тория насчитывает свыше 2500 линий, а спектр урана — более 5000. Нет необходимости, конечно, определять длины волн всех спектральных линий в спектре пробы. Для целей качественного анализа необходимо установить наличие или отсутствие в спектре так называемых аналитических или последних линий.
При уменьшении содержания элемента в пробе интенсивность линий этого элемента в спектре будет уменьшаться, некоторые линии исчезнут и число линий уменьшится. При какой-то очень малой концентрации останется всего несколько линий. Это и есть последние линии, по которым обычно проводится качественный анализ. Последние линии хорошо изучены, их длины волн и характеристику интенсивности можно найти в специальных таблицах и атласах спекгральных линий. Это обычно, но не всегда резонансные линии. В таблицах их часто отмечают индексами U\, (А и т д. или Vi, V2 и т д. Индекс (7, показывает, что при возбуждении спектра в дуге эта линия исчезает последней, линия с индексом U2 исчезает предпоследней и т. д. Индексы Vi, V2 и т. д. относятся к этой же последовательности исчезновения линий в искровом спектре. Однако эта последователь-
29
Рис 2 12. Определение длины волны спектральной линии
ность не абсолютна. В зависимости от условий возбуждения и состава пробы она может несколько изменяться.
Для расшифровки спектра и определения длины волны анализируемой линии пользуются спектрами сравнения, в которых длины волн отдельных линий хорошо известны. Чаще всего для этой цели используют спектр железа, имеющий характерные областях длин волн.
группы линий в разных
Спектр анализируемого вещества обычно фотографируют над спектром железа. На рис. 2.12 представлено схематическое изображение небольшого участка спектра железа и спектра исследуемой пробы. Для определения длины волны заданной линии X* измеряют расстояние ai от этой линии до ближайшей к ней линии спектра железа, длина волны которой Z, точно известна, и расстояние а? от линии X* до другой линии с известной длиной волны Л2. В небольшом спектральном интервале дисперсия прибора остается постоянной, поэтому можно записать пропорцию
Х|——Kg
а, а2
После простых преобразований получаем формулу для расчета длины волны неизвестной линии:
К = + (>-2 — Х|) •
Расстояние между линиями можно измерить, например, с помощью измерительного микроскопа МИР-12, спектропроектора ПС-18 или компаратора. Надежность анализа возрастает, когда встык со спектром пробы фотографируют спектры подозреваемых элементов, как это можно видеть на рис. 2.13. Анализ этих спектров показывает, например, что в железе содержится небольшое количество марганца (линия 294,92 нм) и алюминия (линия 281,61 нм). В спектре алюминиево-магниевого сплава (спектр 2) легко обнаруживается железо по группе линий 275,57....274,65 нм, медь по линии 282,43 нм и довольно большое количество марганца (группа линий 294,92...293,31 нм). Четко видны линии магния (спектр 3) в спектре алюминиево-магниевого сплава (спектр 2). При проведении качественного спектрального анализа часто пользуются атласом спектральных линий. На планшетах этого атласа нанесены линии спектра железа и аналитические линии различных элементов. Совместив изображение спектра железа, полученное с помощью спектропроектора ПС-18, с линиями планшета, отмечают, с какой аналитической линией на планшете совпадает линия анализируемого спектра.
Отсутствие последней линии определяемого элемента в спектре гарантирует отсутствие других линий этого элемента. Однако наличие линии с длиной волны, характерной для последней линии
зо
Рис. 2.13. Участок спектра:
1 — железа, 2 — алюминиево-марганцевого сплава, 3 — магния
какого-либо элемента, еще не означает, что линия действительно принадлежит именно этому элементу. Основной причиной ошибок является так называемое наложение спектральных линий, связанное с недостаточной дисперсией рядовых спектральных приборов. Таблицы спектральных линий показывают, например, что длина волны последней линии любого элемента в пределах ± 0,05 нм совпадает с длиной волны линий многих других элементов. Часть элементов почти всегда можно исключить, основываясь на данных о происхождении пробы или имея в виду интенсивность линии и условия возбуждения. Однако и после этого нередко остается несколько элементов, которым эту линию можно приписать. Окончательную идентификацию проводят, проверяя последние линии всех «подозреваемых» элементов.
Спектральным анализом качественно можно определить более 80 элементов. Предел обнаружения методами качественного спектрального анализа колеблется для разных элементов в очень широких пределах: от 10~2 (Hg, Os, U и др.) до 10~5 % (Na, В, Bi и др.). Следует отметить, что отсутствие линии какого-либо элемента в спектре означает лишь, что его концентрация в пробе меньше предела обнаружения. В связи с низким пределом обнаружения методами спектрального анализа нередко «перестирывают» те или иные элементы, попавшие в пробу в результате случайных загрязнений.
2.6. КОЛИЧЕСТВЕННЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
Попытки использовать зависимость интенсивности спектральных линий от концентрации элемента в пробе для количественного определения долгое время оставались безуспешными. Даже в начале XX в. возможности количественного спектрального анализа оценивались очень невысоко.
Одной из основных причин неудач была недостаточная стабильность условий возбуждения. Интенсивность спектральной линии при прочих равных условиях определяется количеством возбужденных атомов в источнике возбуждения, которое зависит не только от концентрации элемента в пробе, но и от условий
31
возбуждения. Перевод компонента твердой пробы в плазму связан с протеканием процессов плавления, испарения и возгонки. На состав плазмы, таким образом, оказывают влияние температура и теплота плавления компонентов пробы, их коэффициенты диффузии, давление пара, температура источника возбуждения и многие другие факторы, поэтому состав вещества в плазме источника возбуждения существенно отличается от состава исходной конденсированной пробы. Недостаточная стабильность условий возбуждения вызывала изменения в составе и температуре плазмы, что приводило к изменению интенсивности спектральных линий и, как следствие, к колебаниям в результатах анализа.
Связь между интенсивностью спектральной линии и концентрацией элемента в пробе при стабильной работе источника возбуждения приближенно устанавливается уравнением (2.14). Зависимости, наблюдаемые на опыте, хорошо описываются уравнением Ломакина (2.15). Коэффициент самопоглощения b в этом уравнении зависит от концентрации, однако в некотором, иногда довольно широком интервале концентраций он остается постоянным.
В практике количественного спектрального анализа обычно используют интенсивность не отдельной линии, а отношение интенсивностей двух спектральных линий, принадлежащих разным элементам. Таким образом, в качестве свойства, связанного с концентрацией элемента, используется отношение интенсивности линии определяемого элемента к интенсивности линии другого элемента в этом же спектре. Такая методика позволяет снизить требования к постоянству условий возбуждения и регистрации спектров.
Линию определяемого элемента обычно называют аналитической линией и ее интенсивность обозначают I в или называют линией примеси и обозначают интенсивность 1пр. Вторую линию, обычно называемую линией сравнения, выбирают так, чтобы отношение интенсивностей зависело только от концентрации определяемого элемента, но не от условий возбуждения и регистрации спектра. Иногда в анализируемую пробу специально вводят так называемый внутренний стандарт, т. е. элемент, линию которого используют в качестве линии сравнения. При анализе проб, содержащих большое количество какого-то элемента, в качестве линии сравнения обычно выбирают линию этого элемента. Например, при анализе сталей это бывает линия спектра железа. Интенсивность линии сравнения обозначают Iс-или, если линия принадлежит основе, называют линией основы и ее интенсивность обозначают /осн.
Уравнение Ломакина (2.15) для аналитической линии и линии основы имеет вид
пр О С пр, I осн — О £осн,
32
их отношение
= (2-21)
1 ОСН u %сн
уравнение (2.21) показывает, что отношение интенсивностей также пропорционально концентрации элемента в пробе. Это основное уравнение методов количественного спектрального анализа. Методы различаются лишь способом оценки относительной интенсивности. При выборе пары линий для количественного анализа руководствуются рядом требований к энергиям возбуждения спектральных линий, нх длинам волн и интенсивностям. Выполнение этих требований существенно уменьшает зависимость относительной интенсивности от условий возбуждения.
Согласно уравнению (2.11) интенсивность аналитической линии равна
/пр= ^„pAf^/iVn^xp^ кт")’
Аналогично выражается интенсивность линии основы:
I огн A Й-V ос^ехр к/7") *
Возьмем отношение интенсивностей этих линий:
Ср ____ ^npAtgigohVnp /______________Е,___Ek \
/осн кТ )
Обозначим произведение множителей, практически не зависящих от температуры, через А и при постоянстве Апр и N осн получим
/ пр
/ осн
(2.22)
Из уравнения (2.22) следует, что даже при постоянной концент
рации компонентов в плазме относительная интенсивность спектральной линии не остается постоянной — она зависит от температуры. Уравнение (2.22) показывает также, что чем меньше ДЕ, тем меньше относительная интенсивность спектральной линии зависит от температуры. В практике спектрального анализа обычно подбирают линии, которым соответствуют сравнительно небольшие ДЕ (не более I эВ) и элементы с близкими потенциалами ионизации.
Существенно также, чтобы интенсивности выбранных линий не слишком резко отличались между собой. Обычно выбирают линии, отношение интенсивностей которых не превышает 10, т. е. находится в пределах 0,1 /п₽//оси^ 10, так как в противном
случае точность определений уменьшается. Кроме того, обе линии должны быть в одном участке спектра в пределах примерно 10 нм (1Пр— 1осн = Ю нм), чтобы не сказывалась зависимость показаний приемника излучения от длины волны. Таким образом, основными требованиями к выбранной паре будут:
2-1533
ЬЕ < 1 эВ;
^пр Ю НМ,
0,1 </Пр//осн^ 10.
Пара линий, удовлетворяющая этим требованиям, называется гомологической парой. Относительная интенсивность линий гомологической пары обладает малой чувствительностью к условиям возбуждения и регистрации спектра.
В зависимости от способа оценки интенсивностей различают следующие методы количественного спектрального анализа: 1) визуальные; 2) фотографические; 3) фотоэлектрические.
При классификации методов спектрального анализа в отдельную группу выделяют так называемый полуколичестве н-ный спектральный анализ.
2.6.1. Полуколичественный спектральный анализ
Обычная погрешность полуколичественный спектральных методов составляет 10 % или более. Однако когда простота и экс-прессность важнее точности, эти методы применяют очень широко. Оценку интенсивности спектральных линий в полуколичест-венном анализе производят визуально, наблюдая спектр или непосредственно в окуляре спектрального прибора, или на фотопластинке.
Наиболее распространенным методом полуколичественного анализа является метод гомологических пар, или, как его иногда называют, _м е год однородных дубле-д_р в- Для проведения анализа этим методом предварительно 'подбирают пару линий (голомогическую пару или однородный дублет) и устанавливают, при какой концентрации определяемого элемента их интенсивности одинаковы. Например, при разработке методики спектрального определения свинца в олове интенсивность линии олова (нм) Z.sn= 276,11 сравнивалась с интенсивностью линий свинца >.рь = 280,20 (1), ZPb = 282,32 (2) и Z.pb= 287,32 (3). Оказалось, что если сРЬ=0,1%, то 7Xsn = = ЛРЬ (1). если СрЬ= 0,6 %, то /xSn= Лрь (3), а если сРь = 1,3 %, то ^sn= Дрь (2). Для оценки содержания свинца в сплаве неизвестного состава на той же основе сравнивают изученные ранее линии олова и свинца и находят линии одинаковой интенсивности. Таблицы, связывающие относительную интенсивность двух линий с концентрацией элемента в пробе, могут быть составлены и при неодинаковой интенсивности линий дублета. Такие соотношения найдены, например, для определения марганца в стали по линиям Я,Мп = 478,43 и Z.Fe= 477,94 нм. Если интенсивность линии марганца существенно меньше интенсивности линии железа (ДМп«С Д.а), то содержание марганца в стали близко к 0,02 %, при небольшом различии в интенсивностях (ЛМп^ Л.Р() содер
34
жание марганца составляет 0,04 %, равенство интенсивностей наблюдается при содержании марганца около 0,06%, а при содержании 0,08 % Мп интенсивность линии марганца превышает интенсивность линии железа.
Этот же прием используют в работе со стилоскопом при непосредственном наблюдении спектра в окуляре прибора и определении других компонентов стали (Ni, Cr, W и т. д.). Аналитические таблицы, связывающие относительные интенсивности спектральных линий и концентрацию элемента, составляются заранее и обычно прилагаются к стилоскопу. Анализ с помощью стилоскопа на 6...7 элементов у опытного спектроскописта занимает 2...3 мин, предел обнаружения составляет обычно 0,01.. ...0,10 %, погрешность около ±20% (относительных). Успешно применяется стилоскоп, например, при сортировке сталей, контроле плавки и выполнении других аналогичных анализов.
Более высокая точность результатов достигается с помощью стилометра. Стилометр снабжается устройством, позволяющим плавно изменять интенсивности сравниваемых линий, добиваясь их уравнивания. Таким же устройством снабжаются последние модели етилоскопов.
Из фотографических методов полуколичественного анализа применяют метод спектров сравнения и метод появления или исчезновения чувствительных линий. В методе спектров сравнения на одной пластинке фотографируют спектры нескольких эталонов и спектр пробы, а затем сравнивают на глаз почернение линий определяемого элемента в полученных спектрах. Такое сравнение позволяет установить пределы концентраций анализируемого элемента. В методе появления или исчезновения линий сначала с помощью эталонов устанавливают, при каких концентрациях появляются или исчезают те или иные чувствительные линии элементов, а затем, используя полученные данные, находит ориентировочное содержание элемента в пробе.
Полуколичественный экспрессный метод фотометрического интерполирования основывается на визуальной оценке интенсивности спектра, снятого через девятиступенчатый ослабитель.
Ступенчатый ослабитель представляет собой кварцевую или стеклянную пластинку, на которой нанесены узкие параллельные полоски платины (ступеньки) возрастающей толщины и, следовательно, убывающей пропускаемое™. Если ослабитель поместить в спектральный прибор, на фотопластинке получится несколько параллельных изображений спектра убывающей интенсивности в соответствии с убывающей пропускаемостью ступеней ослабителя.
Каждая ступень i характеризуется своей пропускаемостью at = hflt>, где '° интенсивность падающего света, а /, — интенсивность света, прошедшего через г-ю ступень Пропускаемость а, указывается в паспорте ослабителя Если пРи анализе такого спектра будет найдено, что интенсивность линии примеси на ступеньке г(/„р(г)) будет равна интенсивности линии основы на ступеньке Pvoc„(p)), то
очевидно, что
2*
35
А|рЩ ОСН^Р) (2.23)
где а, и ар — пропускание ступенек г и р ослабителя. Логарифмируя уравнение (2.23):
и подставляя после логарифмирования уравнение (2.21), получаем
lg-^- = Iga + b lgc„p. (2.24)
По lg~ и известной концентрации спр в эталонах строят градуировочный график, который не зависит от свойств фотопластинки. Определение с„р в неизвестных образцах по этому графику проводится обычным путем. Если не удается подобрать ступени, точно удовлетворяющие условию (2.23), то интерполяцией на глаз между ступенями ар и аи-н находят необходимую поправку, выражая ее долей отношения 1g ар+—. С учетом этой поправки уравнение (2.24) принимает
Ctp
ВИД
(ga^ /^tLy=|gc+ftlgCnpj (225)
Ur \ Up /
где a — доля 1g a''tl, определяемая интерполяцией на глаз. При a = 0 уравнение (2.25) переходит в (2.24), а при а= 1 оно превращается в
lgjwi=lgc + fcigCiip
Ur
Метод фотометрического интерлолирования характеризуется относительно высокой экспрессностью. Он позволяет получить результат через несколько минут после окончания химической обработки фотопластинки
2.6.2. Фотографические методы количественного анализа
При рассмотрении свойств фотопластинки было установлено, что почернение S связано с освещенностью Е уравнением (2.20). В условиях спектрального анализа Е — I. Подставляем это в уравнение (2.20):
S = ylg/ + ylgt — i.
При постоянной выдержке последние члены правой части этого уравнения постоянны, поэтому
X — ylg/ -J- const. (2.26)
Запишем уравнение (2.26) для почернения линии примеси и линии основы:
Snp= у lg/„p+ const;
•Soch = ylg/осн + const и вычтем одно из другого:
S„p-Soc„ = AS = ylg-^- (2.27)
• осн
36
При логарифмировании уравнения (2.21) имеем lg/пЛОСН lga+ big £ пр- (2.28)
Объединяя уравнения (2.27) и (2.28), получаем
AS = у Iga + у b lgcnp. (2.29)
Это основное уравнение фотографических методов количественного спектрального анализа.
Анализ уравнения (2.29) позволяет оценить ожидаемую точность фотографических методов эмиссионной спектроскопии. Для этого найдем спр из уравнения (2.29):
lgrnp = AS~v'gc (2.30)
УЬ
и продифференцируем (2.30), считая а, b и у постоянными:
л lar — dc"P ___ d(AS)?6 d(AS)
ИЛИ
±^=^.d(AS).
Спр
(2.31)
Формула (2.31) показывает, что относительная ошибка в концентрации не зависит от абсолютного значения AS и уменьшается при увеличении у и Ь. Для ориентировочных подсчетов в области оптимальных почернений примем b — 0,7, d(AS) = 0,02, у = 1,3 и подставим эти значения в уравнение (2.31):
dcnp Дспр
£др
С пр
^0,02.100 = ^-=5.0%
т. е. относительная погрешность при этих условиях составляет ~5 %. Дальнейший расчет показывает, что при увеличении b до 1,0 погрешность уменьшается до 3,5 %. Погрешность также снижается при уменьшении d(AS), т. е. при увеличении точности измерения почернений фотопластинки.
Наиболее распространенным из фотографических методов количественного спектрального анализа является метод трех эталонов. Сущность его заключается в следующем. На одной пластинке фотографируются спектры анализируемого образца и трех эталонов. Для анализа массовых проб (стали, сплавы и т. д.) применяют специальные наборы эталонов, которые выпускает лаборатория стандартных образцов. По спектрам эталонов строят градуировочный график (рис. 2.14). Для повышения точности спектры эталонов и образца фотографируют 2...3 раза и берут средние значения AS. В соответствии с урав-нением (2.29) градуировочный график следует ожидать линейным, и для его построения, казалось бы, будет достаточно двух эталонов. Опыт показывает, однако, что нередко зависимость AS от Igc оказывается нелинейной, и третий эталон позволяет полу-
37
Рис 2.14 Градуировочный график в методе трех эталонов
стирается при учете уравнению (2.29) вид
чить более надежный график. В некоторых случаях применяют четыре и более эталонов. Существенным недостатком этого наиболее точного метода спектрального анализа является его длительность и необходимость получать для каждой ма-стинки свой градуировочный график.
В методе постоянного графи-к а удается согласовать измерения, сделанные на различных пластинках, и построить постоянный график, не зависящий от свойств пластинки. Согласование до-коэффициента контрастности. Придадим
-у-= Iga + b lgcnp. (2.32)
Уравнение (2.32) показывает, что график в координатах AS/y — IgCnp от свойств пластинки уже не зависит. Коэффициент контрастности у можно определить по характеристическсй кривой фотопластинки или, проще, если на каждой пластинке сфотографировать один или несколько спектров через трехступенчатый ослабитель. Если Si и S2— почернения двух ступенек, т. е. очевидно, что
Si = у lg(ai/o);
S2 = у lg(a2/o),
где у — коэффициент контрастности. При вычитании получаем
Si — S2 = ASii2 = у ig—, (2.33)
И2
откуда
AS j 2 у =-------Z— .
lgai/az
Необходимая для расчета у величина ASi,2 определяется экспериментально, а lgai/a2 известен по паспорту ступенчатого ослабителя. По этим данным (у и AS) в соответствии с уравнением (2.32) строят постоянный график, пригодный для любых пластинок, для которых известен у.
Этот метод можно упростить и отказаться от использования ступенчатого ослабителя Запишем уравнение (2.33) для двух пластинок:
ASf,2 = y'ig^-;
ASf'.2 = y"lg^-
38
и возьмем их отношение:
AS[ 2 у'
AS" 2 Г
Коэффициент k называют переводным множителем. Он позволяет пересчитать разность почернений одной пластинки на разность почернений другой. Для определения k можно использовать следующий прием.
На двух пластинках фотографируют спектр одного и того же образца в одинаковых условиях и находят разность почернений одних и тех же аналитических линий. Если AS । — разность почернений на одной пластинке, AS2 — разность почернений на другой, то
AS, = yi(61gc + Iga);
AS2 = y£b Iga + Iga)
и, очевидно,
AAi ^_= k
ASg
(2-34)
Отсюда
AS i = /sAS2.
(2.35)
Для определения k необязательно измерять почернения аналитических линий. Для этого можно использовать две линии основного вещества в той же области спектра, в которой находится аналитическая пара, определить разность их почернений на всех пластинках и по формуле (2.34) вычислить переводный множитель по отношению к пластинке, выбранной за стандарт. Пользуясь этим коэффициентом, можно по формуле (2.35) пересчитать почернения аналитических линий на разных пластинках на почернения стандартной пластинки и получить согласованные величины AS, пригодные для построения градуировочного графика и определения концентрации. Этот метод иногда называют методом переводного коэффициента.
Опыт показал, что неконтролируемые изменения условий возбуждения вызывают сдвиг градуировочного графика. Так как сдвинутый график, как правило, оказывается параллельным первоначальному, то для учета сдвига одновременно со спектром образца фотографируют спектр контрольного эталона. Через точку, соответствующую этому эталону, проводят прямую, параллельную первоначальному графику, которая и будет истинным градуировочным графиком для образцов на данной пластинке. Это твердый график и метод называют методом твердого графика или контрольного эталона.
В современных методах спектрального анализа используют преобразованные значения почернений, полученные с помощью р- или /-преобразования. Этот прием расширяет область почернений, пригодную для анализа, которая обычно ограничена
39
Рис 2 15 Градуировочный график в методе добавок
линейным участком характеристической кривой фотопластинки.
В практике спектрального анализа применяют также метод добавок, особенно при анализе немассовых проб сложного состава и исследованных чистых и сверхчистых веществ. При анализе по этому методу пробу обычно переводят в раствор и делят его на несколько частей. Затем в каждую часть добавляют разное, но точно известное количество определяемого элемента, снимают спектр и определяют интенсивность спектральных линий. Для расчета результатов анализа используют несколько приемов.
В простейшем случае строят график, основываясь на том, что в области малых концентраций достаточно хорошо выполняется линейная зависимость /пр//0Сн = ас, так как с уменьшением концентрации самопоглощение существенно падает. При этом, если с—неизвестная концентрация, а сст—концентрация введенного стандарта, то
/пР//ст = а(сх + гст). (2.36)
Откладывая на графике /пр//осн как функцию с„, получают прямую, пересекающую ось ординат при гст=0 (рис. 2.15). Экстраполяция этой прямой до пересечения с осью абсцисс дает отрезок на оси, равный —сх. Действительно, при IП[>/1 ОсН = 0 из уравнения (2.36) следует, что — с„=сх.
В более сложных случаях (Ь 1) используют метод последовательных приближений на основе уравнения Ломакина или другой наиболее оптимальный способ обработки.
Метод добавок является надежным контрольным методом для установления правильности анализа. Действительно, если после анализа добавить в пробу известное количество определяемого элемента и анализ повторить, то его результат с учетом возможной погрешности должен дать сумму первоначально найденного содержания и введенной добавки. Если сумма не получилась, результат анализа следует признать неверным. Метод добавок применяют для повышения чувствительности спектральных определений тогда, когда почернение аналитической линии является слишком слабым и не может быть точно измерено. Введение добавки приводит к увеличению интенсивности спектральной линии, позволяя проводить измерение с более высокой точностью.
2.6.3. Фотоэлектрические методы
Наиболее существенным недостатком фотографических методов спектрального анализа является большая длительность определений. Для получения результата необходимо сфотографиро
40
вать спектр, обработать фотопластинку и провести фотометриро-вание. Значительно более быстрыми являются спектрометрические методы, основанные на прямом фотометрическом определении интенсивности спектральных линий. Спектрометрические методы характеризуются более высокой экспрессностью и меньшей погрешностью, чем фотографические, поскольку в спектрометрических методах исключаются длительные операции обработки фотопластинки и последующего фотометрирования линий и связанные с этим погрешности.
Установки спектрометрического и спектрографического анализа аналогичны, за исключением устройства их рецепторной части. В фотоэлектрических установках свет после диспергирующего элемента через специальные щели в фокальной плоскости попадает на фотоэлемент или фотоумножитель, соединенный с накопительным конденсатором и далее с регистрирующим потенциометром. Одна из щелей в приборах с фиксированными приемниками света предназначена для линии сравнения, а остальные — для линий анализируемого элемента или элементов. В приборах этого типа для каждой линии предусмотрен свой фотоэлектрический приемник. В сканирующих спектрометрах измерение интенсивности линии определяемого элемента производится фотоэлектрическим приемником, который передвигается вдоль спектра по специальной программе. Фотоэлектрический измерительный блок может также использоваться в качестве приставки к спектроскопу или спектрографу. Такой блок, состоящий из входной щели и фотоэлемента или фотоумножителя с измерительным устройством, устанавливается на место кассеты для фотопластинки. Сконструированный таким образом простой спектрометр может быть эффективно применен для анализа проб с несложным спектром.
Отечественной промышленностью выпускается несколько установок такого типа: многоканальная установка ДФС-10М, квантометры МФС-3, МФС-10 и др. Их аналитические возможности весьма различны. Например, квантометр ДФС-10М имеет 12 щелей и соответственно 12 накопительных конденсаторов, работающих одновременно. С помощью этого прибора можно одновременно определить в пробе 11 элементов (12-й канал служит Для определения интенсивности линии сравнения, основы или внутреннего стандарта).
2.6.4. Химико-спектральный анализ
Существенно расширяет возможности эмиссионной спектроскопии применение химических методов обработки пробы. Химическая обработка и концентрирование позволяют повысить чувствительность определения на два порядка и более и во многих случаях упростить спектральную методику, включая эталонирование, так как состав получаемых концентратов в определенных пределах нетрудно регулировать. Известные методики химико-
41
спектрального анализа позволяют определять примеси в веществах высокой чистоты при содержании 10-5...10~7%.
Обычным приемом обогащения является извлечение микропримесей из анализируемого материала, хотя иногда применяют химический метод отделения основного компонента пробы путем его осаждения. Например, при анализе высокочистого свинца осаждают сульфат свинца, при анализе металлического висмута осаждают иодид висмута и т. д. Примеси при этом остаются в растворе и после упаривания или другой обработки могут быть определены спектральным методом.
Более широко применяется метод извлечения микропримесей. Здесь используется осаждение органическими реактивами, осаждение с коллектором, экстрагирование, хроматография, электролиз и другие методы. Предварительное обогащение применяется при анализе различных биологических объектов, почвы, воды, веществ высокой степени чистоты и т. д. Нельзя не отметить, однако, что обработка проб с целью обогащения предъявляет повышенные требования к чистоте используемых реактивов, воды, растворителей и посуды.
2.7. ФОТОМЕТРИЯ ПЛАМЕНИ (ПЛАМЕННАЯ ЭМИССИОННАЯ СПЕКТРОСКОПИЯ)
Появление специализированных пламенных эмиссионных спектрометров привело в какой-то степени к обособлению метода фотометрии пламени и приданию ему известной самостоятельности, хотя, конечно, фотометрия пламени осталась одним из методов эмиссионного спектрального анализа.
Как и любой другой прибор эмиссионной спектроскопии, фотометр для фотометрии пламени имеет источник возбуждения (пламенная горелка), диспергирующий элемент (обычно светофильтр) и приемник света — рецептор (обычно фотоэлемент). В спектрофотометрах для пламени вместо светофильтров применяют призмы и дифракционные решетки. Анализируемый раствор в пламя горелки вводится в виде аэрозоля. При этом растворитель испаряется, а соли металлов диссоциируют на атомы, которые при определенной температуре возбуждаются. Возбужденные атомы, переходя в нормальное состояние, излучают свет характерной частоты, который выделяется с помощью светофильтров, и его интенсивность измеряется фотоэлементом.
Количественные определения проводят методом градуировочного графика или методом добавок Методы фотометрии пламени характеризуются низким пределом обнаружения (до 0,001 мкг/мл для щелочных металлов и 0,1 мкг/мл для других) при погрешности 1...3%. Этим методом могут быть определены Li, Na, К, Rb, Cs, Sr, Ba, Ca, In, Ag и другие элементы. Одним из достоинств метода фотометрии пламени является также высокая производительность.
Спектры, получаемые в пламени, более просты, чем дуговые
42
или искровые, так как температура пламени ниже, чем в электрических источниках возбуждения. Это облегчает анализ, но вместе с тем сужает возможности метода в отношении числа определяемых элементов.
2.8. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Методы эмиссионного спектрального анализа используются во многих областях науки и техники и в различных отраслях народного хозяйства. Этим методом выполняется значительная часть анализов в металлургической промышленности. Анализируется исходное сырье и готовая продукция. Особое значение имеет спектрально-аналитический контроль за ходом плавки, на основании которого вносятся оперативные изменения в ход технологического процесса, например по содержанию легирующих и других добавок. Визуальный спектральный анализ оказался очень удобным методом сортировки вторичного сырья металлургического производства, позволяя за несколько минут установить тип сплава или марку стали, что необходимо при составлении или корректировке шихты.
Очень эффективным оказалось применение спектральных методов при анализе разного рода геологических проб при поиске полезных ископаемых, а также для контроля технологического процесса на горно-обогатительных и гидрометаллургических предприятиях. Спектральным анализом контролируется качество поступающей руды, степень извлечения полезных и мешающих компонентов, а нередко и качество продукта.
Существенную роль играет спектральный анализ природных и сточных вод, почвы, атмосферы и других объектов окружающей среды, а также в медицине и биологии. Важное значение имеет спектральный анализ чистых материалов в электронной технике и других областях, анализ реактивов и т. д. Успешно используется спектральный анализ в космических исследованиях. Процесс совершенствования методов эмиссионной спектроскопии продолжается.
2.9. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
При общей оценке методов эмиссионной спектроскопии необходимо прежде всего отметить их низкий предел обнаружения, точность, быстроту выполнения анализов и универсальность. Средний предел обнаружения методами эмиссионной спектроскопии составляет 10“3...10“4% (до 10-5%), а при использовании приемов обогащения он снижается до 1O*F’...1O 7%. Погрешность определения характеризуется в среднем величиной 1...2%. В связи с экспрессностью, точностью и другими достоинствами эмиссионный спектральный анализ широко используют в практи ко. Значительная часть определений в металлургической и машиностроительной промышленности выполняется с помощью
43
спектрального анализа. Многочисленные применения нашел спектральный анализ и в других отраслях народного хозяйства и техники (геологии, химической промышленности, сельском хозяйстве, космохимии и т. д.).
! Вопросы
1. Какова природа и происхождение атомных эмиссионных спектров? Почему атомные спектры имеют линейчатый характер?
2. Чем характеризуется энергетическое состояние электронов и атома в целом?
3. Назвать основные правила отбора. Указать разрешенные переходы для термов: a) 2S, б) 2Р, в) 'Р, г) 2D, д) 3D. Сколько линий можно ожидать в спектре при соблюдении правил отбора в каждом случае?
4. От чего зависит интенсивность спектральных линий?
5. Чем определяется ширина спектральной линии? В чем состоят эффекты Допплера, Штарка и Зеемана?
6. Дать сравнительную характеристику призмы и дифракционной решетки как диспергирующих устройств.
7. Что происходит при действии света на фотопластинку? Какие основные химические процессы идут при проявлении и закреплении фотопластинки? Написать уравнения реакций.
8. Указать сравнительные достоинства и недостатки фотоэлементов с внешним и фотогальваническим эффектом, фотоэлектронных усилителей.
9. На чем основан качественный спектральный анализ? Какие приборы используются для проведения качественного анализа?
10. От каких факторов зависит интенсивность спектральных линий?
И. Каким требованиям должна удовлетворять гомологическая пара линий?
12. Что такое относительная интенсивность спектральной линии? Как она зависит от условий возбуждения?
13. Дать общую характеристику фотоэлектрических методов эмиссионного спектрального анализа. Привести принципиальную схему соответствующей установки.
14. Дать общую характеристику метода фотометрии пламени. Какие основные приемы работы используются в методе фотометрии пламени? Какие достоинства и недостатки имеет этот способ?
Задачи
1. Для определения длины волны интересующей линии X, были выбраны две линии в спектре железа с известными длинами волн: Xi = 325,436 и ?-2=328,026 нм. На измерительной шкале
44
микроскопа были получены следующие отсчеты: bi =9,12; b2 = 10,48; Ьх = 10,13 мм. Какова длина волны искомой линии в спектре анализируемого образца?
Так как выбранные линии железа — М и находятся соответственно слева и справа от интересующей линии, для расчета X, используем уравнение
К = U -----(Х2 _ X,).
п, + а2
Сначала находим значения oi и а2 по данным отсчета:
О] = Ьх — 61 = 10,13 — 9,12 = 1,01 мм;
а2 = 62 — Ь„= 10,48 — 10,13 = 0,35 мм.
Подставляем соответствующие числовые значения в уравнение и находим значение Хх:
X, = 325,436 + , (328,026 - 325,436) = 327,360 нм.
1 ,U 1 _р U,oD
2. При анализе стали на хром по методу трех эталонов было измерено почернение S линий гомологической пары (ХСг= 279,216 и Хре—279,388 нм) в спектрах эталонов и в исследуемом спектре. Найти массовую долю (%) хрома по следующим данным:
Параметр Эталон Аналнзнруе-мый образец
1 2 3
И'Сг, % 0,50 1,23 4,17 ?
Scr 0,07 0,37 0,86 0,61
Spe 0,27 0,23 0,27 0,25
В методе трех эталонов используется зависимость разности почернений AS линий гомологической пары от логарифма концентрации определяемого элементе
По показателям измерительной шкалы микрофотометра находим значения AS(AS = SCr — Sre) для трех эталонов:
AS, = 0,07 — 0,27 = —0,20; AS2 = 0,37 — 0,23 = 0,14;
AS3 = 0,86 •- 0,27 = 0,59.
Определяем логарифмы массовых долей (%) : lgwt = —0 30, lgw2 = 0,09; lgw3 = 0,62 и строим градуировочный график в координатах AS — Igw (рис. 2.16).
Находим значение AS для анализируемого образца: Д5х = 0,61 — — 0,25=0,36 и по градуировочному графику определяем Igw(Cr) и w (Сг) в образце: 1g w (Сг) = 0.35 и W (Сг) =2,24 %.
3. Для построения постоянного (или твердого) графика при определении олова в бронзе на одной пластине были сфотографированы спектры четырех эталонов и получены следующие результаты:
Рис. 2.16. Определение w (Сг) методом трех эталонов
45
Эталон
AS = Ss„ — SCli w(Sn), %
1 2 0,690 0.772 6,23 8,02
3 4
0,831 0,910
9,34 11,63
Рис 2.17 Определение w(Sn) методом твердого графика
Спектр одного из эталонов был снят через трехступенчатый ослабитель; при этом для линии Sn с длиной волны 286,332 нм разность' почернений двух ступеней (Д5ступ) оказалась равной 1,065.
Спектр анализируемого образца был снят на другой пластинке также через трехступенчатый ослабитель и были получены следующие результаты:
Д5'туп= 0,925 — разность почернений двух ступеней линии Sn;
ASX = 0,695 — .разность почернений пары Sn—Си.
Определить массовую долю (%) олова в образце.
Находим логарифм массовой доли Sn
[gw, =0,795; lgwz = 0,904, lgws = 0,970, lgw.)= 1,068
и строим градуировочный график в координатах AS — Igw (рис. 2.17).
Определяем переводной множитель k, позволяющий использовать постоянный график для анализа образцов, спектры которых получены на разных пластинках.
ASCTyll у
ASCTyn у
где у и у' — коэффициенты контрастности пластинок со спектрами эталонов и анализируемого образца соответственно.
Подставляем числовые значения: &S/k= 1,065/0,925= 1,151. Находим значение разности почернений линий гомологической пары анализируемого образца с учетом переводного множителя:
Д5Л = feASJ = 1,151 • 0,695 = 0,800
По градуировочному графику находим
lg= 0,933 и w(Sn)=8,57%
4. Для определения длины волны заданной линии в спектре анализируемого образца были выбраны две линии в спектре железа с длинами волн Х| и Ха. Отсчеты по шкале измерительного микроскопа для этих линий были равны соответственно Ь\ и Ь%. Определить длину волны заданной линии, если отсчет по шкале для нее равен bt.
Вариант Длины волн выбранных линий железа, нм Отсчеты по шкале микроскопа, мм
м Х,2 61 £>2 Ь,
1 373,713 374,556 5,08 6,14 5,62
2 360,885 361,877 2,06 3,’ 8 3,06
3 486,370 487,130 13,36 15,23 14,17
4 248,327 249,064 8,22 9,48 8,76
5 325,436 328,026 9,12 10,49 10,13
46
Ответ: 1) 374,143 нм, 2) 361,771 нм; 3) 486,699 нм; 4) 248,643 нм; 5) 327,345 нм,
5. При качественном анализе цветного сплава было исследовано несколько участков спектра этого сплава. Наиболее интенсивные линии были затем идентифицированы (см. таблицу). Указать, какой элемент может составлять основу сплава.
Вариант 1 Вариант 2 Вариант 3 Вариант 4
К, нм (/) М X, нм (/) M X» нм (/) M X, нм (/) M
213,85 (8) Zn 259,81 (3) Sb 259 85 (3) Si 261,42 (2) Pb
326,11 (1) Cd 261,42 (6) Pb 256,30 (6) Al 263,00 (5) Cu
326,23 (3) Sn 265,12 (3) Ge 267,75 (5) Ta 266,12 (3) Sn
328,23 (3) Zn 266,32 (5) Pb 266,95 (5) Al 276,64 (4) Cu
330,26 (4) Zn 268,51 (3) Ta 273,39 (3) Pt 281,36 (5) Sn
334,50 (6) Zn 279,83 (4) Mn 279,55 (4) Mg 282,44 (6) Cu
340,51 (4) Co 280,20 (6) Pb 308,22 (8) Al 235,11 (3) Sb
Ответ: 1) Zn; 2) Pb; 3) А1; 4) Си.
6. При определении содержания марганца в стали по методу трех эталонов на микрофотометре было измерено почернение линий гомологической пары: Z,Fe = 293,690, Z,Mn = 293,306 нм. Определить массовую долю (%) марганца в стали по данным:
Вариант Параметр Эталон Исследуемый образец
1 2 3
ш(Мп), % 0,33 0,89 3,03 ?
1 Spe 1,33 1,24 1,14 1,08
^Mn 0.95 1.06 1,20 0,96
w(Mn), % 0,10 0,38 1,90
2 Spe 0,98 0,94 0,99 0,75
0,71 0,90 1,24 0,61
ш(Мп), % 0,05 0,21 0,30 ?
3 Spe 0,72 0,74 0,78 0,73
^Mn 0,53 0,90 1,03 0,91
Ответ: 1) 1,20%; 2) 0,22%; 3) 0,23 %.
7. Определить массовую долю (%) хрома в стали, если при фотометрировании по методу трех эталонов были получены следующие результаты:
Параметр Эталон Анализируемый образец
1 2 3
w(Cr), % 0,50 1,23 4,17
Scr 0,07 0,29 0,86 0,73
Spe 0,27 0,15 0,27 0,33
Ответ: 2,57 %.
47
8. Определить массовую долю (%) меди в алюминии, если при фотометрировании по методу трех эталонов были получены следующие результаты:
Параметр Эталон Анализируемый образец
l 2 3
w(Cu), % 0,25 0,80 1,20 ?
SCu 0,25 0,54 0,61 0,41
Sai 0,42 0,44 0,41 0,45
Ответ: 0,66%.
9. Определить массовую долю (%) кремния в сплаве, если при фотометрировании по методу трех эталонов были получены следующие результаты:
Параметр Эталон Анализируемый образец
l 2 3
w(Si), % 0,63 1,65 3,86
Ss. 0,186 0,414 0,614 0,514
Sai 0,184 0,184 0,184 0,184
Ответ: 2,50 %.
10. Определить массовую долю (%) вольфрама в стали, если при фотометрировании по методу трех эталонов были получены следующие результаты:
Параметр Эталон Анализируемый образец
1 2 3
w(W),% 0,87 1,37 2,19 ?
Sw 0,388 0,634 0,774 0,782
SFe 0,462 0,464 0,424 0,452
Ответ: 2,04 %.
11. Для построения градуировочного графика при определении олова в бронзе были получены следующие результаты:
w(Sn), % 6,23 8,02 9,34 11,63
AS . 0,690 0,772 0,831 0,910
Фотометрирование спектра, снятого через трехступенчатый ослабитель, показало, что для двух ступеней линии Sn ASCTyn = = 1,055. При фотометрировании спектра образца бронзы, сфотографированного на другой пластинке, для двух ступеней той же линии Sn получено ASCTyn (см. выше), а для аналитической
48
пары Sn—Си в спектре образца на той же пластинке — &S'X. Определить массовую долю (%) олова в образцах:
I Образец
ДХ{туп \ ДХ£
1 2 3 4 5
0.930 0,920 0,890 0,900 0,910
0,635 0,663 0,746 0,725 0,694
Ответ: 1) 6,80%; 2) 7,60%; 3) 11,00%; 4) 10,00%; 5) 8,60%.
12. Для построения градуировочного графика при определении никеля в бронзе были получены следующие результаты:
w(Ni), % bS
3,00 3,42 3,84 4,24 0,580 0,662 0,740 0,821
Для двух линий меди на основной пластинке AS осн = 0,828. При фотометрировании спектра образца бронзы для той же пары линий меди получено AS'0C„, а для аналитической пары Ni—Си в спектре образца на той же пластинке — AS£. Определить массовую долю (%) никеля в образцах:
Образец
ДХ ОСН дх;
1 2 3 4 5
0,765 0,828 0,822 0,804 0,790
0,566 0,708 0,764 0,820 0,538
Ответ: 1) 3,20%; 2) 3,82%; 3) 4,03 %; 4) 4,52%; 5) 2,98%.
13. Навеску стекла массой 0,1000 г растворили в смеси H2SO4 и HF, раствор выпарили, остаток смочили НС1 и перенесли в мерную колбу вместимостью 250 мл. Полученный раствор фотометрировали в пламени так же, как и стандартные растворы, приготовленные из NaCL Данные фотометрировании приведены в таблице (/отн — относительная интенсивность излучения):
Параметр Стандартный раствор Образец
1 2 3 1 2 3
Cn«, мг/л 10,0 20,0 30,0 ? ?
^отн 16,0 31,5 47,5 24,0 35,0 42,5
Построить градуировочный график и определить массовую Долю (%) натрия в образце. Ответ: 1) 3,75 %; 2) 5,50%; 3) 6,75%.
14. Навеску удобрения массой 2,0000 г обработали при кипячении насыщенным раствором оксалата аммония; после охлаждения раствор разбавили в мерной колбе вместимостью 500,0 мл и профильтровали. Аликвоту фильтрата объемом 5,00 мл разбавили до 250,0 мл. Полученный раствор фотометрировали в пламени так же, как и стандартные растворы, приготовленные из КС1. Данные фотометрировании приведены в таблице:
49
Параметр Стандартный раствор Образец
1 2 3 1 2 3
w(K),% отн 5,00 8,0 10,00 15,0 15,00 24,2 12,7 18,4 ? 20,8
Построить градуировочный график и определить массовую долю (%) калия в удобрении. Ответ: 1) 10,00%; 2) 14,40%; 3) 16,25%.
15. Порцию исследуемой воды объемом 25,00 мл разбавили дистиллированной водой в мерной колбе вместимостью 500,0 мл и фотометрировали в пламени так же, как и стандартные растворы, приготовленные из СаСЬ- Результаты фотометрирования приведены в таблице:
Параметр Стандартный раствор Образец
1 2 3 4 1 2 3
ССа, мг/л I отн 10,0 16,0 30,0 47,6 50,0 80,2 70,0 111,0 32,0 76,9 101,8
Построить градуировочный график и определить концентрацию кальция (мг/л) в исследуемой воде. Ответ: 1) 400,0 мг/л; 2) 980,0 мг/л; 3) 1300,0 мг/л.
Глава 3
АБСОРБЦИОННАЯ СПЕКТРОСКОПИЯ
3.1. ОСНОВНОЙ ЗАКОН СВЕТОПОГЛОЩЕНИЯ (ЗАКОН БУГЕРА — ЛАМБЕРТА — БЕРА]
Атом, ион или молекула, поглощая квант света, переходит в более высокое энергетическое состояние. Обычно это бывает переход с основного, невозбужденного уровня на один из более высоких, чаще всего на первый возбужденный уровень. Вследствие поглощения излучения при прохождении его через слой вещества интенсивность излучения уменьшается и тем больше, чем выше концентрация светопоглощающего вещества.
Закон Бугера — Ламберта — Бера связывает уменьшение интенсивности света, прошедшего через слой светопоглощающего вещества, с концентрацией вещества и толщиной слоя. Чтобы учесть потери света на отражение и рассеяние, сравнивают интенсивности света, прошедшего через исследуемый раствор и растворитель (рис. 3.1). При одинаковой толщине слоя в кюветах из одинакового материала, содержащих один и тот же раство-
50
ритель, потери на отражение и рассеяние света будут примерно одинаковы у обоих пучков и уменьшение интенсивности света будет зависеть от концентрации вещества.
Уменьшение интенсивности света, прошедшего через раствор, характеризуется коэффициентом пропускания (или просто пропусканием) Т:
Т = 1/10,
где I и /0 — соответственно интенсивности света, прошедшего через раствор и растворитель.
Взятый с обратным знаком логарифм Т ческой плотностью Л:
Рис. 3.1 Прохождение света через окрашенный раствор и раство ритель
называется о п т и-
_lg7 = _lg^.= lgA = A
Уменьшение интенсивности света при прохождении его через раствор подчиняется закону Бугера — Ламберта — Бера:
/ = /0. ю~б'с,
или
1/10 = Ю~е'с, или
— \gT = A = elc, (3.1)
где е — молярный коэффициент поглощения; / — толщина светопоглощающего слоя; с — концентрация раствора.
Физический смысл е становится ясным, если принять I — 1 см и с = 1 моль/л, тогда А — е. Следовательно, молярный коэффициент поглощения равен оптической плотности одномолярного раствора при толщине слоя 1 см.
Оптическая плотность раствора, содержащего несколько окрашенных веществ, обладает свойством аддитивности, которое иногда называют законом аддитивности с в е-топоглощения. В соответствии с этим законом поглощение света каким-либо веществом не зависит от присутствия в растворе других веществ. При наличии в растворе нескольких окрашенных веществ каждое из них будет давать свой аддитивный вклад в экспериментально определяемую оптическую плотность А:
А = Ai Д2 4- 4- Ah,
гДе Д1, А2 и т. д. — оптическая плотность вещества 1, вещества 2 и т. д.
При учете уравнения (3.1) получаем
А = /{е(С( ^2^2 4“ “Б в^сл).
51
3.1.1. Ограничения и условия применимости закона Бугера — Ламберта — Бера
В соответствии с уравнением (3.1) зависимость оптической плотности от концентрации графически выражается прямой линией, выходящей из начала координат. Опыт показывает, однако, что линейная зависимость наблюдается не всегда. При практическом применении закона Бугера — Ламберта — Бера необхо димо учитывать следующие ограничения.
I. Закон справедлив для монохроматического света. Чтобы отметить это ограничение, в уравнение (3.1) вводят индексы и записывают его в виде
А = eJc. (3.2)
Индекс X указывает, что величины А и е относятся к монохроматическому излучению с длиной волны X.
2. Коэффициент е в уравнении (3.1) зависит от показателя преломления среды. Если концентрация раствора сравнительно невелика, его показатель преломления остается таким же, каким он был у чистого растворителя, и отклонений от закона по этой причине не наблюдается.
Изменение показателя преломления в высококонцентрированных растворах может явиться причиной отклонений от основного закона светопоглощения.
3. Температура при измерениях должна оставаться постоянной хотя бы в пределах нескольких градусов.
4. Пучок света должен быть параллельным.
5. Уравнение (3.1) соблюдается только для систем, в которых светопоглощающими центрами являются частицы лишь одного сорта. Если при изменении концентрации будет изменяться природа этих частиц вследствие, например, кислотно-основного взаимодействия, полимеризации, диссоциации и т. д., то зависимость /1 от с не будет оставаться линейной, так как молярный коэффициент поглощения вновь образующихся и исходных частиц не будет в общем случае одинаковым.
Например, при разбавлении раствора дихромата калия происходит не просто уменьшение концентрации иона дихромата, а протекают процессы химического взаимодействия'
Сг2О?” + Н2О = 2НСгОГ = 2СгО?- + 2Н+
Вместо дихромат-ионов в растворе появляются гидрохромат- и хромат-ионы Так как еСг!О?~ и еСгО; различны, зависимость оптической плотности от общей концентрации хрома в растворе не будет линейной
3.2. СПЕКТРЫ ПОГЛОЩЕНИЯ
Свет поглощается раствором избирательно: при некоторых длинах волн светопоглощение происходит интенсивно, а при некоторых свет не поглощается. Интенсивно поглощаются кванты света, энергия которых Av равна энергии возбуждения частицы
52
и вероятность их поглощения больше нуля. Молярный коэффициент поглощения при этих частотах (или длинах волн) достигает больших значений.
f Распределение по частотам (или по длинам волн) значений молярного коэф-* фициента поглощения называется спектром поглощения.
Обычно спектр поглощения выражают в виде графической зависимости оптической плотности А или молярного коэффициента поглощения е от частоты v или длины волны X падающего света. Вместо А или е нередко откладывают их логарифмы.
Кривые в координатах lg.A — X, как показывает рис. 3.2, при изменении концентрации или толщины слоя перемещаются по ординате вверх или вниз параллельно самим себе, в то время как кривые в координатах А — Л (рис. 3.3) этим свойством не обладают. Существенное значение имеет эта особенность для качественного анализа. При изучении инфракрасных спектров на графике обычно откладывают процент светопропускания как функ цию v' или V.
Рис. 3.2. Зависимость lg/1 от X.
1 — раствор концентрации с в кювете толщиной I, см, 2 —- раствор концентра ни и '/4 с или в кювете толщиной ‘/4 I. см
Рис 3.3. Зависимость А от X:
1 — раствор концентрации с в кювете толщиной I, см, 2 — раствор концентрации ‘/4 с или в кювете толщиной 1/4 I, см
Отдельные полосы в спектрах поглощения математически можно описать гауссовыми кривыми.
- б ,[12
Е==гт„е й , (33)
где в и Fma, - молярный коэффициент поглощения при данной длине волны X (или волновом числе v') и при длине волны Хп1а, (или волновом числе v(„ax), отвечающей точке максимума, v' и гЦ, -данное волновое число и волновое число в точке максимума, 6 характеризует ширину полосы поглощения
Действительно, при v' = v'1 молярный коэффициент поглощения равен половине максимального е'/,= и вместо уравнения (3 3) имеем
*х /, - v max)2 ,
-^=е,„|Хе (3.4)
53
При упрощении и логарифмировании (3.4) получаем
_ ig2 = —
откуда
$ - Т|/2
Как видно, величина 6 равна полуширине полосы на половине ее высоты.
Для несимметричных полос обычно указывают (Хтах) и вта. Для решения структурных, термодинамических или иных вопросов несимметричные полосы нередко разлагают на индивидуальные гауссовы компоненты, каждая из которых описывается уравнением (3 3)
Таким образом, наибольший интерес представляют следующие характеристики спектра: число максимумов (или полос поглощения) и их положение по шкале длин волн (или частот); высота максимума; форма полос поглощения.
3.2.1. Происхождение спектров поглощения
Появление полос поглощения обусловлено дискретностью энергетических состояний поглощающих частиц и квантовой природой электромагнитного излучения. Интенсивно поглощаются кванты света, которые соответствуют энергии возбуждения частицы. При поглощении квантов света происходит увеличение внутренней энергии частицы, которая складывается из энергии вращения частицы как целого, энергии колебания атомов и движения электронов:
Е = Евр + Екол + Е„, (3.5)
где £вр—вращательная, £кол—колебательная, Еэл — электронная энергия.
Уравнение (3.5) должно включать также слагаемые энергии тонкой и сверхтонкой структуры, связанные с электронным и ядерным спином, поправку на приближенность аддитивной схемы и некоторые другие слагаемые, которыми в первом приближении можно пренебречь.
По энергии вращательное, колебательное и электронное движение различаются весьма существенно, причем £пр С £кол С <С Еэ~. Их числовые значения относятся примерно как 1 : 102: 103.
Каждый вид внутренней энергии молекулы, как уже отмечалось, имеет квантовый характер и может быть охарактеризован определенным набором энергетических уровней или термов и соответствующих квантовых чисел.
3.2.2. Вращательные спектры
Вращательную энергию молекул обычно рассматривают с помощью модели жесткого ротатора, который представляет собой две массы, находящиеся одна от другой на фиксированном расстоянии.
54
Возбуждение вращательных уровней энергии происходит уже при поглощении далекого инфракрасного (ПК) и микроволнового излучения, меющего длину волны 102 мкм или волновое число v' 102 см-1. Энергия квантов в этой области спектра равна: — 2,8-10-3 v' — 1,2 кДж/моль и меньше. Это значе-
ние соизмеримо с энергией теплового движения кТ, поэтому уже при комнатной температуре часть вращательных уровней заселена.
В настоящее время чисто вращательные спектры в аналитических целях почти не используют. Их применяют главным образом для исследования строения молекул, определения межъядерных расстояний и т. д.
3.2.3. Колебательные спектры
Полосы, связанные с возбуждением колебательных уровней энергии, расположены в области спектра примерно от 200...300 до 4000...5000 см-1, что соответствует энергии квантов от 3 до 60 кДж/моль. Поэтому при обычных температурах энергетическое состояние молекул, как правило, характеризуется основным колебательным уровнем. Простейшей моделью, которая используется при рассмотрении колебаний двухатомной молекулы, является модель гармонического осциллятора. Это система из двух масс, связанных упругой силой. Кривая потенциальной энергии гармонического осциллятора обычно аппроксимируется параболой (рис. 3.4, кривая /). Применение квантовой теории к такой системе показывает, что ее энергия может быть найдена по уравнению
£ko« = (V+ 72)/iv0, (3.6)
где V — колебательное квантовое число, принимающее значения 0, 1,2, 3, ...
Уравнение (3.6) показывает, что в основном состоянии при
V = 0 осциллятор обладает энергией колебаний. Это существенный результат квантового подхода.
Правилами отбора в таких системах разрешены переходы, для кото-
ДК=±1. (3.7)
Из сравнения уранений (3.6) и (3.7) видно, что расстояние между Двумя соседними энергетическими уровнями гармонического осциллятора постоянно и равно hv0. Это означает, что при всех разрешенных переходах будут поглощаться или излучаться кванты энергии только частоты vo и в спектре, таким образом, будет наблюдаться
так называемых нулевых
Рис 3.4 Кривые потенциальной энергии и уровни энергии двухатомной молекулы
55
только одна полоса. Частота, характеризующая эту полосу, может быть вычислена по уравнению
где М — приведенная масса, определяемая как ——F
-|—— (mi и m2 — массы ядер); К — силовая постоя н-н а я.
Силовую постоянную иногда связывают с прочностью связи в молекуле.
Реальные колебания молекул анга р монич ны и кривые потенциальной энергии молекул имеют более сложный характер (рис. 3.4, кривая 2). Энергию ангармоничного осциллятора рассчитывают по формуле
EKO. = (V+'/2)hv0-^{V+l/2)2, (3.8)
где D — энергия диссоциации молекулы.
Энергетические уровни ангармонического осциллятора, как это следует из уравнения (3.8), с увеличением квантового числа сближаются. Для ангармонического осциллятора разрешенными являются переходы между любыми уровнями.
Наиболее интенсивной в спектре является первая полоса, возникающая при переходе с V = 0 на V = 1. Этой полосе соответствует основная, или фундаментальная, частота. Менее интенсивные полосы дают обертоны, т. е. частоты, характеризующие переход с уровня V = 0 на уровень V = 2 (первый обертон или вторая гармоника); на уровень V = 3 (второй обертон или третья гармоника) и т. д.
Следует отметить лишь, что колебательными инфракрасными спектрами обладают не все молекулы, а только те, у которых при колебании происходит изменение их электрического дипольного момента. ИК-спектрами обладают, например, молекулы НС1, НВг и 1. д , но не Н2, О2 и др.
Сравнительная простота колебательных или колебательновращательных спектров двухатомных молекул обусловлена тем, что колебания происходят только вдоль линии, соединяющей ядра. В многоатомной молекуле происходят колебания всех атомов. Число колебательных степеней свободы у нелинейной молекулы, состоящей из N атомов, равно 3N — 6, а у линейной 3N — 5, так как у них отсутствует одна вращательная степень свободы.
Сложную картину колебаний в многоатомной молекуле обычно представляют как суперпозицию так называемых нормальных колебаний. Частоты нормальных колебаний характе-
56
Рис. 3 5 Формы нормальных колебаний молекулы НгО
ризуются положением полос в ИК-спектрах, а амплитуда колебаний определяет интенсивность этих полос. Число нормальных колебаний, очевидно, будет равно числу колебательных степеней свободы, и, таким образом, общая форма молекулярных колебаний будет суперпозицией 3N — 6 (или ЗА— 5) нормальных колебаний.
При классификации нормальных колебаний обычно различают валентные и невалентные, или д е-формационные колебания. Колебание называют валентным, если происходит изменение длины связи без существенного изменения углов
между связями. Валентные колебания обозначают буквой v. Колебания с изменением углов между связями называют невалентными или деформационными и обозначают буквой 6. На рис. 3.5 можно видеть формы нормальных колебаний молекулы воды. Так как Н2О является нелинейной трехатомной молекулой, то число колебательных степеней свободы, равное числу нормальных колебаний, будет 3N— 6=3-3 — 6=3. Стрелки на рис. 3.5 показывают направление смещения ядер из равновесного положения. На рис. 3.5, а, б можно видеть, что в результате колебаний происходит изменение длины валентной связи О—Н при практически постоянной величине угла НОН. Это валентные колебания vi и v2. На рис. 3.5, в колебание атомов вызывает изменение угла НОН. Такое колебание называют деформационным 6.
Колебательные спектры многоатомных молекул интерпретируют на основе учения о симметрии молекул и теории групп. Математический аппарат теории групп позволяет вычислить число частот и правила отбора для молекул различной симметрии. Такая информация, чрезвычайно ценная для определения молекулярных констант, изучения строения молекул и т. д., находит сравнительно малое применение для решения химикоаналитических задач. Для решения этих задач используются так называемые характеристические частоты.
Анализ ИК-спектров показал, что некоторые из наблюдаемых частот можно привести в соответствие с колебаниями отдельных атомов или групп атомов. Так, например, было найдено, что в спектрах всех молекул, содержащих связи С—Н, имеются частоты в области 2800...3000 см-1, связь С=С характеризуется частотой 1650 см~’, а связь CsC — частотой 2100 см-1. Такие частоты назвали характеристическими. Они широко используются в практической спектроскопии для определения строения молекул и проведения качественного анализа по ИК-спектрам.
57
3.2.4. Электронные спектры
Верхней энергетической границей колебательного спектра обычно считают энергию фотонов примерно в 5000 см-1, или около 60 кДж/моль. Дальнейшее увеличение энергии облучающих квантов чаще всего будет приводить к возбуждению электронов и появлению в спектре полос, характеризующих электронные переходы, хотя, конечно, указанная граница может несколько смещаться в ту или другую сторону в зависимости от свойств изучаемого соединения.
Интерпретация электронных спектров может быть сделана на основе квантово-механических представлений, например метода молекулярных орбиталей (МО). В соответствии с основными положениями этого метода электроны в молекуле
Рис. 3.6. Схема электронных уровней и энергия возможных электронных переходов
Рис. 3.7. Схема энергетических уровней молекулы в основном (А) и первом возбужденном (Б) электронных сос-
тояниях
могут находиться на связывающих, несвязывающих и разрыхляющих орбиталях. Схема относительного расположения энергетических уровней, соответствующих разным МО, показана на рис. 3.6. Различные электронные переходы требуют неодинаковой энергии, поэтому полосы поглощения располагаются при разных длинах волн.
Наибольшей энергии требует о -► о*-переход, связанный с возбуждением внутренних электронов. Он соответствует поглощению в далекой ультрафиолетовой области [X 200 нм, Е ^ 600 кДж/моль]. Такие переходы характерны, например, для метана, этана и других насыщенных углеводородов. Переход п -► о* связан уже с меньшими затратами энергии. Полосы, вызванные этим переходом, расположены в обычном (невакуумном) ультрафиолете (X = 200...300 нм). Еще меньшая энергия тре
58
буется для перехода на разрыхляющие л*-орбитали. Переходы п — л* и л —» л* встречаются в молекулах соединений с сопряженными связями и молекулах ароматических соединений. Этим же переходом (и—» л*) можно объяснить, например, интенсивную окраску ионов МпО? и СгО|_ (переход с несвязывающей орбитали кислорода). Теоретический расчет энергии разных МО громоздок и связан с большими трудностями, поэтому существенное значение имеют различные эмпирические закономерности, связывающие спектр поглощения со строением и свойствами вещества.
Введение в молекулу различных заместителей или изменение внешних условий, например растворителя, обычно вызывает сдвиг полосы поглощения. Если полоса поглощения смещается в сторону более длинных волн, говорят о батохромном смещении или углублении окраски (красное смещение), а если полоса сдвигается в сторону более коротких волн, эффект называют гипсохромным сдвигом или повышением окраски (голубое или синее смешение). Кроме переходов внутри валентной оболочки, известны так называемые переходы Ридберга, связанные с изменением главного квантового числа. Полосы, соответствующие этим переходам, расположены в дальней ультрафиолетовой области.
Электронный переход обычно осложняется наложением колебательных, а иногда и вращательных переходов, так как каждое электронное состояние молекулы обладает набором колебательных (и вращательных) уровней (рис. 3.7).
Основному электронному состоянию А отвечает система колебательных уровней, характеризуемая колебательными квантовыми числами V = 0, 1, 2, ... Возбужденное электронное состояние Б обладает системой колебательных уровней с квантовыми числами V = 0, 1, 2, ... У каждого колебательного состояния есть система вращательных уровней, энергия которых пропорциональна вращательному квантовому числу /.
При обычных условиях большинство молекул находится в основном электронном и основном колебательном состояниях. Переходы vop в этих условиях (рис. 3.7) характеризуют чисто вращательные спектры, линии которых появляются в самой далекой длинноволновой части ИК-спектра и области микроволн.
Чисто колебательные переходы vKM (рис. 3.7) происходят при неизменном электронном состоянии молекулы. В случае разреженных газов наблюдаются колебательно-вращательные спектры молекул Ткол + vBp, характеризующие переходы между вращательными уровнями различных колебательных состояний.
Электронные переходы являются наиболее сложными в связи с наложением колебательных, а при определенных условиях и вращательных переходов. Наложение большого числа колебательных переходов часто приводит к существенному уширению полос электронных спектров, так как колебательная структура не всегда разрешается. Известны и другие причины, вызывающие
59
Рис. 3.8. Электронно-колеба тельный переход согласно принципу Франка — Кондо на-
1 — основное и 2 — возбужден ное состояния
уширение полосы поглощения в электронных спектрах, связанные, например, с временем жизни возбужденного состояния.
Изменение свойств электронной оболочки в результате электронного перехода приводит к изменению п о-тенциальной энергии системы, поэтому кривая потенциальной энергии, например, двухатомных молекул в разных электронных состояниях не будет одинакова. В возбужденном состоянии обычно происходит увеличение равновесного межъядерного расстояния, уменьшение энергии диссоциации и изменение других свойств по сравнению со свойствами в основном состоянии. Обычные кривые потенциальной энергии молекулы в основном и возбужденном состояниях показаны иа рис. 3.8.
Существенное значение в электронной спектроскопии имеет принцип Франка — Кондона. При взаимодействии молекулы с квантом света электронная оболочка столь быстро пе
реходит в возбужденное состояние, что положение ядер измениться не успевает. Таким образом, за время перехода молекулы в возбужденное состояние межъядерное расстояние меняться не будет. Однако в новом электронном состоянии равновесное межъядерное расстояние обычно отличается от того, которым обладала молекула в основном состоянии, и колебательные уровни возбужденного состояния также будут другими.
Неизменность межъядерного расстояния во время электронного перехода означает, что на кривых потенциальной энергии этот переход должен изображаться вертикальной линией, как это представлено на рис. 3.8. Так как наиболее вероятны переходы без изменения межъядерного расстояния, им и будут отвечать наиболее интенсивные полосы в спектрах поглощения. Использование принципа Франка — Кондона в спектроскопии многоатомных молекул также приводит к важным результатам, позволяя решить вопрос о типе колебаний и структуре молекул. В случае многоатомных молекул картина становится значительно более сложной, так как потенциальная энергия многоатомной молекулы может быть представлена уже не просто кривой, а поверхностью потенциальной энергии в n-мерном пространстве. (Если зафиксировать определенные координаты, потенциальную поверхность можно изобразить и в трехмерном пространстве.)
60
3.2.5. Интенсивность поглощения
Интенсивность полос в спектре поглощения характеризуют интегралом поглощения, или силой осциллято-р а, определяемой выражением
f = 0-9)
где В — коэффициент Эйнштейна, характеризующий вероятность перехода.
Коэффициент Эйнштейна связан с электрическим дипольным моментом перехода и некоторыми другими величинами. Он может быть также рассчитан из экспериментальных данных по уравнению
оо
В = e(v)dv, (3.10)
о
где К объединяет несколько величин, а интеграл Je(v)dv называют интегралом поглощения.
Сочетание уравнений (3.9) и (3.10) и подстановка числовых значений фундаментальных констант приводят к формуле
f = 4,33-10“9 fe(v)dv.
О
Соотношение (3.9) показывает, что чем более вероятным является переход, тем больше сила осциллятора. Так, разрешенные переходы характеризуются величиной /, близкой к 1, как это наблюдается у интенсивно окрашенных красителей, а у запрещенных переходов, например у переходов с изменением мультиплет-ности терма, сила осциллятора составляет примерно 10~ .
Для аналитической характеристики соединений имеет значение не столько интегральное поглощение, сколько светопоглощение при определенной длине волны. Важными аналитическими характеристиками являются молярный коэффициент поглощения в точке максимума е1Пах и полуширина полосы поглощения 6=Av' г (рис. 3.9).
Наибольшей интенсивностью в спектрах поглощения обладают полосы, обусловленные переносом электрона от одного атома к Другому (полосы переноса заряда). Часто эти полосы связаны с переносом электрона с р-орбитали лиганда на d-орбиталь центрального иона и наоборот. Более правильно следует говорить о переносе электрона между молекулярными орбиталями, локализованными у разных атомов. Интенсивные-полосы в спектре, появившиеся в результате такого рода переходов, имеют молярный коэффициент поглощения порядка 104 и более. К ним относятся многие л — л*- и п -► л*-переходы. Переносом заряда объясняют, например, интенсивную окраску ионов МпОг, CrOl-, окраску тиоцианатных комплексов железа, кобальта, молибдена.
61
Рис 3 9 Полоса поглощения
сульфосалицилатных комплексов железа, фанантролиновых комплексов и многих других.
Значительно менее интенсивны полосы, связанные с внутриатомными d— d- или f — f-переходами. Эти, вообще говоря, запрещенные переходы (ДА = 0) дают полосы с молярным коэффициентом поглощения от 1 до величин, редко превышающих 100 Снятие запрета с d — d-переходов в комплексах чаще всего объясняют «частичным смешиванием» d- и р-орбиталей и переходом электрона уже со смешанных d-, р-орбиталей на d-орбитали, что не запрещено. Успешно интерпретирует спектры d- и f-элементов теория кристаллического поля.
Спектры окрашенных соединений в растворе обычно характеризуются довольно широкими полосами поглощения. Уширение полос связано с сильным влиянием молекул растворителя на энергетические уровни электронов, ответственных за светопоглощение, и наложением колебательных переходов на электронный переход. Почти всегда очень широкие полосы наблюдаются в спектрах переноса заряда. Ионы лантаноидов имеют узкие полосы поглощения, так как их внутренние 4/-электроны, ответственные за светопоглощение, экранированы внешними 5^-, 5р-электронами.
Очевидно, чем выше молярный коэффициент поглощения и меньше ширина полосы, тем более ценными химико-аналитическими свойствами обладает соединение, так как эти характеристики полосы определяют также важные показатели, как предел обнаружения и селективность.
3.3. ОСНОВНЫЕ УЗЛЫ ПРИБОРОВ АБСОРБЦИОННОЙ СПЕКТРОСКОПИИ
При всем многообразии схем и конструктивных особенностей приборов абсорбционной спектроскопии в каждом из них имеется несколько основных узлов, функции которых примерно одинаковы в разных приборах. Такими узлами являются: источник света, мо-
62
нохроматизатор света, кювета с исследуемым веществом, рецептор (приемник света).
К этим основным узлам следует добавить оптическую систему, состоящую из линз, призм и зеркал, которая служит для создания параллельного пучка света, изменения направления и фокусировки света, а также систему для уравнивания интенсивности световых потоков (диафрагмы, оптические клинья и т. д.).
В приборах абсорбционной спектроскопии свет от источника освещения проходит через монохроматизатор и падает на кювету с исследуемым веществом. Интенсивность монохроматического света, прошедшего через кювету, измеряется приемником света (рецептором). Практически обычно определяют отношение интенсивностей монохроматического света, прошедшего через исследуемый раствор и через растворитель или специально выбранный раствор сравнения.
3.3.1. Источники света
Основными источниками освещения в абсорбционной спектроскопии являются вольфрамовые лампы накаливания, газонаполненные лампы (водородная, ртутная), штифт Нернста и глобар. В простейших приборах в качестве источника освещения используется дневной свет.
В лампе накаливания светящаяся вольфрамовая спираль дает свет в широком спектральном интервале. Однако стекло пропускает свет лишь в интервале длин волн 350... 1000 нм, т. е. в видимой части спектра и самых ближних ультрафиолетовой и инфракрасной областях. В водородной лампе происходит свечение водорода при разряде. Условия возбуждения подбирают так, что возникает практически сплошное излучение в области 200...400 нм. В ртутной лампе разряд происходит в парах ртути. Возбужденные атомы ртути испускают линейчатый спектр, в котором преобладает излучение с длиной волны 254, 302, 334 нм.
Штифт Нернста представляет собой столбик, спрессованный из оксидов редкоземельных элементов. При накаливании путем пропускания электрического тока он дает ИК-излучение в области 1,6...2,0 или 5,6...6,0 мкм Глобар-штифт из карборунда SiC дает излучение в интервале 2... 16 мкм также при пропускании электрического тока.
3.3.2. Монохроматизаторы (монохроматоры)
Монохроматизаторами или монохроматорами называют Устройства для получения света с заданной длиной волны. Однако термин «монохроматизатор» является более предпочтительным, так как под названием монохроматор существуют специальные спектральные приборы. При конструировании монохроматизато-Р°в используют разные оптические явления: поглощение света, интерференцию, дисперсию и т. д. Наибольшее распространение
63
в практике абсорбционной спектроскопии имеют приборы, в которых в качестве монохроматизаторов применяются светофильтры и призмы.
Известно несколько типов светофильтров. В зависимости от вида оптического явления, используемого для монохроматизации света, конструируют абсорбционные, интерференционные или интерференционно-поляризационные светофильтры.
Действие абсорбционных светофильтров основано на том, что при прохождении света через тонкий слой вследствие поглощения происходит изменение величины и спектрального состава проходящего светового потока. Абсорбционные светофильтры имеют небольшую прозрачность (Т = 0,1) и довольно широкую полосу пропускания (АХ — 30 нм и более). Характеристики интерференционных светофильтров значительно лучше. Светофильтр состоит из двух тончайших полупрозрачных слоев серебра, между которыми находится слой диэлектрика. В результате интерференции света в проходящем пучке остаются лучи с длиной волны, равной удвоенной толщине диэлектрического слоя. Прозрачность интерференционных светофильтров составляет Т = 0,3...0,8. Эффективная ширина пропускания обычно не превышает 5... 10 нм. Для еще большего сужения полос пропускания иногда пользуются системой двух последовательных интерференционных светофильтров.
Наиболее универсальными монохроматизаторами являются призмы, изготовленные из кварца, стекла и некоторых других материалов. Для инфракрасной спектроскопии используют призмы из LiF, NaCl, КВг и других галогенидов щелочных и щелочно-земельных металлов. Эти же материалы применяют для изготовления кювет. Призмы позволяют получать свет высокой монохроматичности в широкой области длин волн.
3.3.3. Приемники света (рецепторы)
В качестве рецепторов в приборах абсорбционной спектроскопии используют главным образом фотоэлементы, фотоумножители, а иногда интенсивность света оценивается на глаз. Для измерения интенсивности инфракрасного излучения применяют фотоэлементы, термоэлементы и болометры. Приемники света характеризуются спектральной чувствительностью — способностью воспринимать излучение различной длины волны — и интегральной чувствительностью, которая измеряется по действию на рецептор не разложенного в спектр излучения.
В термоэлементах используется термоЭДС, возникающая при изменении температуры спая между металлами или сплавами под действием инфракрасного излучения. Широко применяются для этих целей термопары медь — константан, серебро — висмут и др-Принцип действия болометра основан на изменении электросопротивления материала при нагревании. Термочувствительный элемент, представляющий собой зачерненную платиновую, сурь-64
мяную или другую тонкую металлическу пластинку, включают в мостовую схему. Инфракрасное излучение вызывает нагревание термочувствительного элемента и разбаланс моста, пропорциональный интенсивности падающего излучения.
Промышленностью выпускаются различные приборы абсорбционной спектроскопии- колориметры, фотометры, фотоэлектроколориметры, спектрофотометры и т. д , в которых используют различные комбинации осветителей, монохроматизаторов и приемников света.
3.4. КАЧЕСТВЕННЫЙ АНАЛИЗ
Наибольший интерес с точки зрения качественного анализа представляют колебательные (вернее колебательно-вращательные) спектры. Они весьма характерны, и в иностранной литературе их нередко называют fingerprint, т. е. отпечатки пальцев, имея в виду неповторимость инфракрасного спектра соединения
Экспериментальные исследования колебательно-вращательных спектров показали, что полосы при некоторых частотах можно привести в соответствие с колебаниями определенных групп атомов или отдельных атомов в молекуле. Такие частоты назвали характеристическими. Различные молекулы, содержащие одну и ту же связь или одну и ту же атомную группировку, будут давать в ИК-спектре полосы поглощения в области одной и той же характеристической частоты. Это и является основой качественного анализа по инфракрасным спектрам. Характеристические частоты дают возможность установить по спектру наличие определенных групп атомов в молекуле и тем самым позволяют судить о качественном составе вещества и строении молекул. Например, полосы в области 3000...3600 см-1 могут быть приписаны только О—Н- или N—Н-связям, и отсутствие полос в этой области спектра однозначно свидетельствует об отсутствии ОН- и NH-групп в анализируемом веществе. Примеры такого рода исследований весьма многообразны. С помощью инфракрасных спектров было установлено строение многих олефинов, ароматических соединений, карбонильных соединений, аминокислот и других групп веществ. Было выяснено, например, что большинство аминокислот существует в растворе в ионизированном состоянии, которое можно предстало .О
вить формулой R—СН—(а), но не R— СН—(б). I х)- । хин
+NH3 nh2
Действительно, в спектре нейтрального раствора глицина в D2O были найдены полосы поглощения, характерные для ионизированной карбоксильной группы (1600 и1400 см-1). Введение в раствор кислоты DC1 вызвало их замену на полосы при 1710 см-1, а добавление щелочи опять их восстановливало.
Полосы валентных колебаний NH в области 3300...3500 см-1, гДе они обычно проявляются, у аминокислот не обнаруживаются, 3~-1533
4-
а в области около 3070 см-1, характерной для группы —NH3, они есть. Образование соли аминоуксусной кислоты вызывает появление полосы вблизи 3300...3500 см-1 , характерной для —МНг-груп-пы. Сопоставим эти результаты:
Группа — nh2 - NH3 —соон — СОО-
Волновое число, см-1 3300, 3500 3070 1710 1600 1400
Нейтральный раствор — + — +
Кислый раствор — + + '—
Щелочной раствор + — — +
Эти данные совершенно однозначно показывают, что в водном растворе глицин существует в виде цвиттер-иона МНзСНгСОО-. Отсутствие в инфракрасном спектре полосы, связанной с какой-то характеристической частотой, является обычным надежным доказательством отсутствия определенной группы в молекуле. В то же время наличие полосы в некотором участке спектра представляет собой менее доказательное свидетельство, т. е. оно еще не всегда или не полностью доказывает присутствие предполагаемой группы в молекуле.
К настоящему времени изучены и сведены в соответствующие атласы и таблицы инфракрасные спектры более чем 20000 соединений, что существенно облегчает практическое проведение анализа. Для получения первых ориентировочных данных часто пользуются так называемой картой К о л т у п а, на которой указаны спектральные области появления многих характеристических частот и их возможное отнесение. Для окончательных выводов обычно требуется более тщательный анализ спектра. Иногда задача качественного анализа может быть решена простым сопоставлением спектра анализируемого вещества и «подозреваемого» соединения.
Инфракрасная спектроскопия с успехом используется и в анализе неорганических веществ. Известно, например, что характеристическая частота СОз- составляет 1450 см-1, SO®-—1130, NO3 — 1380, NH^ — 3300 см-1 и т. д. В связи с этим из инфракрасных спектров минералов получают весьма ценную информацию о химическом составе.
Электронные спектры поглощения для целей качественного анализа используются значительно реже, чем колебательные, так как они обычно бывают представлены небольшим числом широких полос поглощения, которые часто накладываются одна на другую и полностью или частично перекрываются. Однако по электронным спектрам поглощения иногда удается провести достаточно эффективный анализ качественного состава, например в нефтехимии
3.5. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
Методы количественного анализа основаны на законе Бугера — Ламберта — Бера, выраженном уравнением (3.2). В связи с тем что значения коэффициента пропускания Т находятся в пределах
бб
от 0 до 1 (1^7>0), оптическая плотность раствора Д = — IgZ’ может принимать, казалось бы, любые положительные значения от нуля до бесконечности, т. е. оо А ^0. Однако экспериментальному определению с необходимой точностью доступны далеко не любые значения А Так, например, значения А^0,01 не определяют в связи с большой погрешностью их измерения.
Уравнение (3.2) показывает, что основными параметрами фотометрического определения являются длина волны, при которой производится измерение, оптическая плотность, толщина кюветы и концентрация окрашенного раствора. Существенное влияние оказывают различные химические факторы, связанные с полнотой и условиями протекания фотометрической реакции, концентрацией окрашенных и других реактивов, их устойчивостью и т. д. В зави симости от свойств анализируемой системы и характеристик применяемого фотометрического прибора выбирают те или иные условия анализа.
3.5.1. Оптимальные условия фотометрического определения
Длина волны. При определении в растворе одного светопоглощающего вещества аналитическую длину волны, как правило, выбирают на максимуме полосы поглощения. Если в спектре имеется несколько полос, выбор обычно останавливают на наиболее интенсивной, так как работа в области максимума светопоглоще-ния обеспечивает наиболее высокую чувствительность определения. Плоские максимумы более предпочтительны, так как при этом меньше сказывается погрешность в установлении длины волны, чем в случае острых максимумов или крутоспадающих участков кривой. Желательно также, чтобы чувствительность приемника излучения в области аналитической длины волны была максимальна. Однако практическая реализация этого условия затруднена, так как конструкция обычных фотометрических приборов предусматривает не более двух фотоэлементов. Выбор аналитической длины волны при наличии в растворе нескольких светопоглощаю-Щих веществ значительно сложнее. Он будет рассмотрен при выборе условий анализа смеси окрашенных веществ.
Светопропускание (оптическая плотность). Измерительное устройство фотометрического прибора обычно имеет постоянную ошибку АТ в величине коэффициента пропускания Т во всем интервале его значений. Ошибка в единицах оптической плотности ДД в связи с этим во всем интервале не будет одинакова. Поэтому при решении некоторых задач удобнее оперировать с коэффициентом пропускания, а не с оптической плотностью. Рис 3.10 показывает, что при одной и той же абсолютной погрешности АТ абсолютная погрешность определяемой концентрации Ас существенно возрастает с увеличением концентрации раствора 'Лс2>-Дс1, хотя Д7,2 = АГ1).
з*
67
Относительная ошибка Дс/с будет уменьшаться с ростом концентрации и возрастать с увеличением абсолютной ошибки Ас. Ответ на вопрос, при каких значениях Т относительная ошибка \с/с будет минимальна, дает небольшой математический анализ.
Из уравнения (3.1) следует, что
е/
При дифференцировании (3.11) получаем
, dT
d 2,37в/ '
(3.11)
(3.12)
Рис. 3.10. Зависимость 7 от с
Рис. 3.11 Зависимость относительной погрешности от пропускания раствора
или, переходя к конечным приращениям.
Ас __ АТ
с Т \пТ '
(3.13)
Задаваясь разными числовыми значениями Т при фиксированной величине ДГ, по уравнению (3.13) можно рассчитать относительную погрешность Дс/с для всего интервала значений Т от 0 до 1. Результаты такого расчета представлены графически на рис. 3.11, из которого видно, что относительная погрешность резко возрастает при очень малых и очень больших значениях Т. В области средних значений Т кривая проходит через минимум. Для нахождения точки минимума продифференцируем уравнение (3.13) по Т при Д7’ = const и производную приравняем нулю:
d(v)_ -(>п7- + 4)аТ
<17 ‘ (Т In 7)2
-(1пГ+ 1)А7
(7 1пТ)
= 0.
68
jaK как АГ =/= О, то, очевидно, 1п7"+1=0. Откуда InT = == 2,3lgТ = — 1 и—lg7 = А = 0,435. При этом значении оптической плотности будет достигаться наибольшая точность измерения.
Однако в проведенном расчете не учитывалась погрешность за счет других источников, как, например, погрешность при уста-, новлении прибора на нулевое и полное пропускание. Более строгое теоретическое рассмотрение и опыт показали, что оптимальная оптическая плотность находится при более высоком значении, чем 0,435, и составляет примерно 0,6...0,7 или несколько выше. Расчеты и опыт показали также, что фотометрическое исследование растворов, имеющих 0,03 А 2,0, характеризуется большими погрешностями. Эффективным приемом при анализе интенсивно окрашенных растворов является применение методов дифференциальной фотометрии.
Толщина светопоглощающего слоя. Уравнение закона Бугера—Ламберта—Бера показывает, что чем больше толщина слоя, тем больше оптическая плотность и, следовательно, тем более чувствительным будет определение при прочих равных условиях. Однако с увеличением толщины слоя (длины оптического пути) возрастают потери на рассеяние света, особенно при работе с растворами. Кюветы с толщиной слоя большей чем 5 см для фотометрии растворов обычно не применяются.
Концентрационные условия проведения фотометрической реакции. В уравнение основного закона светопоглощения входит концентрация окрашенного (светопоглощающего) соединения, поэтому превращение определяемого компонента в такое соединение является одной из важнейших операций, в значительной степени определяющей точность анализа. Окрашенные соединения в растворе получают в результате, главным образом, реакций окисления — восстановления и комплексообразования. Окислительно-восстановительные реакции, применяемые в фотометрии, например окисление марганца до МпОГ, протекают, как правило, практически полностью до конца.
Значительно более сложным является вопрос о концентрационных условиях протекания в растворе реакций комплексообразования. Осложняющее влияние здесь могут оказать процессы ступенчатого комплексообразования, протолитические равновесия, недостаточная устойчивость образующегося комплекса, собственная окраска реагента и т. д. Действие большинства этих факторов можно предвидеть, если равновесия в интересующей системе достаточно подробно изучены и константы соответствующих равновесий известны (константы устойчивости координационных соединений, диссоциации реагентов и т. д.). Используя эти данные, можно рассчитать, например, при каких значениях pH и концентрации реагента будет достигнута необходимая полнота реакции, как будут влиять сопутствующие элементы и т. д.
Если окрашенное соединение образовано анионом сильной кислоты, как, например, при определении висмута в виде иодидного
69
комплекса, то реакцию обычно проводят при постоянной концентрации реактива и в довольно кислой среде, обеспечивающей подавление гидролитических процессов. Концентрация аниона в таких системах от кислотности среды не зависит. При использовании в качестве реагента слабой кислоты, например при определении железа в виде сульфосалицилатного комплекса, pH раствора должен соответствовать слабокислой области, в которой диссоциация кислоты достаточна и концентрация реактива постоянна. Особое внимание должно быть уделено постоянству pH во всех исследуемых растворах. Для выяснения оптимальных условий фотометрического определения каждая система требует специального физико-химического исследования для установления состава образующихся соединений, определения констант равновесия и т. д.
Чувствительность и точность метода. Минимальную концентра цию, которую можно определить фотометрическим методом, обычно рассчитывают по соотношению
min == Д mm/f^O- ।
Если для ориентировочных расчетов принять, что A min = 0,01, 1=1 см и е = 10s, то
cm.n = = 10-5 моль/л.
Это не минимальная концентрация фотометрического метода, так как е может быть на несколько порядков больше, однако значение е= 103 свойственно многим цветным соединениям, и, таким образом, оно в какой-то степени характеризует метод. Иногда в качестве показателя чувствительности фотометрической реакции указывают просто величину е, известны и другие характеристики чувствительности. Точность фотометрических методов зависит от индивидуальных особенностей фотометрической реакции, характеристик применяемого прибора и других факторов и изменяется в довольно широких пределах. Обычная погрешность фотометрических методов составляет примерно 1...2 % (относительных).
3.5.2. Основные приемы фотометрических измерений
Метод градуировочного графика. В соответствии с законом Бугера — Ламберта — Бера график в координатах оптическая плотность — концентрация должен быть линеен и прямая должна проходить через начало координат. Для построения такого графика достаточно, вообще говоря, одной экспериментальной точки. Однако градуировочный график обычно строят не менее чем по трем точкам, что повышает точность и надежность определений.
При отклонениях от закона Бугера—Ламберта — Бера, т. е. при нарушении линейной зависимости А от с, число точек на графике должно быть увеличено. Применение градуировочных графиков является наиболее распространенным и точным методом
70
фотометрических измерений. Основные ограничения метода связаны с трудностями приготовления эталонных растворов и учетом влияния так называемых третьих компонентов, т. е. компонентов, которые находятся в пробе, сами не определяются, но на результат влияют.
Метод молярного коэффициента поглощения. При работе по этому методу определяют оптическую плотность нескольких стандартных растворов Дст, для каждого раствора рассчитывают е = Ат/(/Сст) и полученное значение е усредняют. Затем измеряют оптическую плотность анализируемого раствора Ах и рассчитывают концентрацию сх по формуле
Сх=Дх/(е/).
Ограничением метода является обязательное подчинение анализируемой системы закону Бугера—Ламберта — Бера, по крайней мере, в области исследуемых концентраций.
Метод добавок. Этот метод применяют при анализе растворов сложного состава, так как он позволяет автоматически учесть влияние «третьих» компонентов. Сущность его заключается в следующем. Сначала определяют оптическую плотность Ах анализируемого раствора, содержащего определяемый компонент неизвестной концентрации сх, а затем в анализируемый раствор добавляют известное количество определяемого компонента (сст) и вновь измеряют оптическую плотность Дк+ст.
Оптическая плотность Ах анализируемого раствора равна
Ах=е1сх, (3.14)
а оптическая плотность анализируемого раствора с добавкой стандартного
Дх+ст=е/(Сх + Сст). (3.15)
Сравнение уравнений (3.14) и (3.15) дает
Ах______ сх
Ar-j-ст Сх + Сс,
или
Ах(сх Д- с ст)—А, ст сх.
Отсюда находим концентрацию анализируемого раствора:
Концентрацию анализируемого вещества в методе добавок можно найти также по графику в координатах Ах :т=Кс<Л Уравнение (3.15) показывает, что если откладывать Лх+ст как У-экцию сст, то получится прямая, экстраполяция которой до пересечения с осью абсцисс даст отрезок, равный —сх. Действительно, при т4л+ст==0 из уравнения (3.15) следует, что —с„=сх
Метод дифференциальной фотометрии. Фотометрирование интенсивно окрашенных растворов успешно осуществляется мето-
71
Н20 сх
о
Рис 3.12 Схема обычной и дифференциальной фотометрии: а — обычная, б — дифференциальная
дом дифференциальной фотометрии. В обычной фотометрии сравнивается интенсивность света Л, прошедшего через анализирумый раствор неизвестной концентрации, с интенсивностью света /о, прошедшего через растворитель. Коэффициент пропускания такого раствора будет равен отношению интенсивностей (рис. 3.12):
ТХ=1Х/1О.
В дифференциальной фотометрии второй луч света проходит не через растворитель, а через окрашенный раствор известной концентрации — так называемый раствор сравнения концентрации сср. Его интенсивность обозначим как /ср. Интенсивность света, прошедшего через анализируемый раствор, по-прежнему пусть будет 1Х. Отношение интенсивностей Л/ДР называется условным коэффициентом пропускания Т'х.
Тх= I х/1 ср.
Отношение /ср к /о характеризует коэффициент пропускания раствора сравнения:
т Гср=/Ср//о.
Так как
1х = Тх1о',
1ср=Т с[/о,
ТО
Л/Лр=Т'=7\/Лр
или, переходя от коэффициентов пропускания к оптическим плотностям,
Ах=Ах—АСр',
Ах=е1^-Ахр, (3.16)
где А'х — относительная оптическая плотность.
Уравнение (3.16) показывает, что относительная оптическая плотность, так же как и истинная, пропорциональна концентрации окрашенного вещества, однако прямая А'х—сх не проходит через начало координат (рис. 3.13). Пусть анализируемый раствор имеет оптическую плотность Д=4,0, что методом обычной фотометрии достаточно точно измерить нельзя. Взяв вместо растворителя раствор Дср=3,0, получаем относительную оптическую плотность АХ=АХ—Дср=4,0— 3,0=1,0, что можно измерить уже с необходимой точностью.
Таким образом, дифференциальная фотометрия существенно расширяет область концентраций, доступную для точных фотометрических измерений. Кроме того, точность некоторых методик
72
дифференциальной фотометрии превышает точность методик обычной фотометрии. Найдем, при каких значениях А' или Т относительная погрешность определения методом дифференциальной фотометрии будет минимальной. Для этого из уравнения (3.16) находим сх.
+ . 1g Т'х + Т'ср Z4 1-7,
Сх—4 н е/ l'!
и дифференцируем полученное выражение, считая Гер, ей/ постоянными:
Рис 3 13 Градуировочный график дифференциальной фонометрии
dcx
dr;
2,зг;Ег ‘
При сочетании уравнений (3.17) и (3.18) получаем
de __ dr;
С — 2,3TK(igT;+igTcp)
или, переходя к конечным приращениям,
Де____ дг;
~~ 2,3T;(igr;-(-igTLp)
(3.19)
На рис. 3.14 графически представлены результаты некоторых расчетов, выполненных по уравнению (3.19). Кривые на рис. 3.14 показывают, что с уменьшением коэффициента пропускания раствора сравнения (Тср) относительная погрешность определения концентрации падает. И чем меньше коэффициент пропускания 7\р, тем меньше относительная погрешность.
Как видно, при 7\Р= 1 дифференциальный метод превращается в метод прямой фотометрии и, таким образом, обычную фотометрию можно рассматривать как частный случай дифференциальной. Это иллюстрируется кривой 1 на рис. 3.14, которая по сути дела воспроизводит кривую на рис. 3.11. Кривые, характеризующие погрешность методов дифференциальной фотометрии, как видно, находятся ниже первой кривой, т. е. при прочих равных условиях относительная погрешность определения концентрации методом дифференциальной фотометрии меньше, чем соответствующая погрешность обычной фотометрии.
Чтобы найти оптическую плотность, при которой относительная погрешность определения минимальна, продифференцируем Уравнение (3.19) по Т'х при постоянных АГ и Т<.р и приравняем производную нулю:
d 0*\т [(in г; + in ггр) + г;-1-]
—drT~= [2,3rxigr; + igrtp)]2 =0 ‘
73
Очевидно,
In Т'х + 1п Гер-)- 1 = 0 или
'х + lg rcp=—Jg-= -0,435 и Ал—0,435 — Аср.
При А сР = 0 это соотношение, как частный случай, переходит в Ах = 0.435, т. е. Ах принимает оптимальное значение, ранее установленное для обычной фотометрии.
Увеличение оптической плотности раствора сравнения (Atp) будет вызывать в соответствии с уравнением (3.20) уменьшение оптической плотно
Рис 3.14. Относительная погрешность при разном пропускании раствора сравнения:
I — Тср= 1,0, 2 - 7’сР = 0,1, 3 - Тср = 0,01, 4 - Т™ = = 10 3
сти, при которой достигается максимальная точность результатов. При Дср = 0,435 относительная оптическая плотность будет равна нулю. Таким образом, точные измерения будут достигаться при высокой оптической плотности раствора сравнения и минимальной относительной оптической плотности. Этот вывод иллюстрируется также кривыми рис. 3.14.
Количественный анализ по инфракрасным спектрам. Анализ по ИК-спектрам также основан на применении закона Бугера — Ламберта — Бера. Чаще всего здесь используется метод градуировочного графика. Существенно затрудняется применение метода молярного коэффициента поглощения в ИК-спектроскопии тем, что из-за рассеяния, сплошного поглощения и других эффектов часто бывает невозможно определить положение линии 100%-ного пропускания, т. е. определить интенсивность света, прошедшего через образец без анализируемого компонента (/о).
Многие трудности количественной ИК-спектроскопии успешно преодолеваются с помощью метода базовой линии, который получил большое распространение в практике. Сущность его легко понять из рис. 3.15, на котором приведен участок ИК-спектра с двумя полосами поглощения (их волновые числа и ve). Базовая линия проводится в основании полосы поглощения (она изображена пунктиром). Коэффициент пропускания определяется в этом методе как отношение ТА=IА/1или Тв= = / /;//(>(/;) По полученным данным вычерчивают градуировочный график и производят определения.
Экстракционно-фотометрические методы. Экстракционные методы применяют в аналитической химии очень широко, причем определение анализируемого компонента в экстракте может проводиться как фотометрическим, так и любым другим методом: полярографическим, спектральным и т. д. Вместе с тем существуют некоторые группы экстракционных методов, в которых фотометрическое окончание является наиболее эффективным, обес-
74
печивая необходимую быстроту и точность определения. Эти методы обычно называют экстракционно-фотометрическими.
Весьма распространенной является методика, по которой определяемый микрокомпонент переводят в растворимое в воде окрашенное соединение, экстрагируют его и экстракт фотометрируют. Содержание обычно определяют методом градуировочного графика. Такая методика позволяет устранить мешающее влияние посто
ронних компонентов и увеличивает
чувствительность определения, так как Рис 3 15 Метод базовой ли-при экстракции происходит концентри-
рование микропримесей. Например, оп-
ределение примесей железа в солях кобальта или никеля проводят экстракцией его тиоцианатных комплексов амиловым
спиртом.
Окрашенное соединение определяемого компонента не обязательно должно быть растворимо в воде. Например, дитизон и многочисленные дитизонаты металлов практически нерастворимы в воде, но растворимы во многих органических растворителях. В этом случае к анализируемому раствору добавляют раствор дитизона в подходящем органическом растворителе (например, в тетрахлориде углерода), получают окрашенный экстракт, который и фотометрируют. Иногда бывает удобнее экстракцию проводить одним реагентом, а окрашенное соединение определяемого элемента получать в экстракте уже с помощью другого.
Определение смеси светопоглощающих веществ. Спектрофотометрический метод, в принципе, позволяет определить несколько светопоглощающих веществ в одном растворе без предварительного разделения. Большое практическое значение имеет частный случай такой системы — анализ смеси двух окрашенных веществ. В соответствии с законом аддитивности светопоглоще-ния для такой смеси веществ, например А и В, можно записать:
А,,— /(е А >C А+ е в Х)С в);
А х2= а х2с а+ е в ;рв)-
Решение этой системы уравнений при /=1 см дает:
еА ? 2 еА AjeBAi
а, -^л,еА > ЕА2 еВ £А >2еВ>
(3-21)
(3.22)
Длины волн А, и Аг, при которых следует проводить измерения оптической плотности, выбирают по спектрам поглощения ве-
75
Рис. 3 16 Спектр поглощения вещества А и В (а) и зависимость еА — ев от длины волны (б)
ные коэффициенты поглощения анализ сводится к определению выбранных длинах волн.
ществ А и В, применяя различные приемы. Хорошие результаты дает, например, метод максимальных разностей. При использовании этого метода сначала снимают спектры поглощения компонентов А и В (рис. 3.16, а), а затем строят график зависимости еа—ев или ев — еА от длины волны и находят области максимума и минимума (рис. 3.16,6). Длины волн, соответствующие этим областям, выбирают в качестве аналитических, учитывая, что абсолютные значения еА и ев в этих областях тоже должны быть достаточно велики. Часто, но не всегда эти области совпадают с областью максимального поглощения каждого из компонентов. Моляр-определяются заранее, поэтому оптической плотности при двух
Особый интерес представляют спектральные участки, в кото рых одно из веществ свет не поглощает, а другое обладает интенсивным светопоглощением. Если, например, еВ) =0, то вместо (3.22) будем иметь:
Как видно, при наличии такой области упрощаются расчеты и повышается точность определения интенсивно поглощающего вещества. Этот случай реализуется, например, при определении хрома и марганца. В области длин волн 550 нм поглощает только МпОг и он может быть определен по оптической плотности раствора при этой длине волны. При 430 нм свет поглощают оба компонента (МпО? и СгО?-).
Довольно распространенным примером такого анализа является также определение с помощью реагента, имеющего собственную окраску. Этот метод может быть распространен и на более сложные многокомпонентные смеси. При подчинении свето-поглощения отдельных компонентов закону Бугера — Ламберта — Бера и соблюдении закона аддитивности светопоглощения число слагаемых в уравнениях типа (3.21) увеличивается пропорционально числу определяемых компонентов и соответственно возрастает число уравнений. Для решения систем таких уравнений с успехом используются вычислительные машины.
76
Фотометрическое титрование. В методе фотометрического титрования точка эквивалентности определяется с помощью фотометрических измерений. В ходе такого титрования измеряется светопоглощение раствора. Естественно, реализация этого метода возможна, если имеется подходящий индикатор или хотя бы один из компонентов реакции титрования поглощает свет. Например, при титровании железа (II) дихроматом кривая фотометрического титрования имеет вид, изображенный на рис. 3.17. До точки эквивалентности (т. э.) оптическая плотность раствора практически не изменяется, а после точки эквивалентности она линейно возрастает пропорционально объему добавленного дихромата. Точку эквивалентности находят графически. Для этого достаточно иметь несколько точек, характеризующих недотитро-ванный раствор, и несколько точек для перетитрованного раствора.
Рис. 3.17 Кривая фотометрическо Рис 3 и Принципиальная схема фото-го титрования железа (11) дихро- метрического автотитратора
матом
При титровании разбавленных или слабоокрашенных растворов кривая титрования не имеет резкого минимума. Для нахождения точки эквивалентности в таких системах приходится применять более сложные графические построения или специальную математическую обработку.
Основным достоинством метода фотометрического титрования является возможность анализа слабоокрашенных и разбавленных растворов, которые часто невозможно оттировать другими методами, а также возможность автоматизации самого процесса титрования.
В ходе титрования стандартный раствор выдавливается из бюретки 1 поршнем, приводимым в движение мотором 2 (рис. 3-18). Интенсивность света, прошедшего через светофильтр 6 и раствор 5, фиксируется фотоэлементом 4, соединенным с мотором через реле 3. При заданном значении оптической плотности по сигналу фотоэлемента реле срабатывает и выключает мотор, связанный с поршнем бюретки, прекращая титрование. Результаты анализа рассчитывают обычным путем по объему раствора, израсходованного на титрование. Известны также приборы, в которых вычерчивается полная кривая титрования, включая пере-
77
титрованные растворы, и дифференциальная кривая титрования dj4 ,< 1 г
в координатах как функция к.
Определение состава и устойчивости комплексных соединений в растворе. Простота и достаточная точность фотометрических измерений привели к широкому использованию фотометрических методов для исследования реакций в растворе и особенно цветных реакций, имеющих химико-аналитическое значение. Для определения состава соединений часто применяется метод изо-молярных серий. При использовании этого метода готовят серию растворов, в которых отношение концентрации центрального иона к концентрации лиганда (cM:cL) изменяется от 9:1 до 1:9, а суммарная концентрация (cM+cL) остается одинаковой во всех растворах (изомолярные серии). Затем измеряют оптическую плотность растворое и строят график зависимости оптической плотности от концентрационного отношения cM:cL. Максимум на этом графике указывает состав комплекса. Метод изо-молярных серий имеет ограничения и недостатки, однако является одним из наиболее широко применяемых в практике.
Устойчивость соединений по данным фотометрических измерений может быть рассчитана графическими или аналитическими методами. Если, например, удается определить молярный коэффициент поглощения е комплекса ML„, то константа устойчивости может быть рассчитана по соотношению
о =___________А/(£1)______
1 ML [^-Л/(е/)][С“-пЛ/(Е/)Г ’
где А — оптическая плотность раствора; с°м и c°L — исходные концентрации растворов центрального иона и лиганда.
При этом предполагается, что кислотно-основные равновесия в исследуемых растворах осложнений не вызывают, т. е. центральный ион не гидролизован и лиганд не взаимодействует с протоном. При протекании процессов кислотно-основного взаимодействия расчет константы устойчивости усложняется.
3.6. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Фотометрические и спектрофотометрические методы анализа применяются для определения многих (более 50) элементов периодической системы, главным образом металлов. Методами абсорбционной спектроскопии анализируются руды, минералы и иные природные объекты, продукты переработки обогатительных и гидрометаллургических предприятий. Эффективно используются эти методы в металлургической, электронной, химической и других отраслях промышленности, в медицине, биологии и т. д. Большое значение они имеют в аналитическом контроле загрязнений окружающей среды и решении экологических проблем. Значительно расширились области практического применения методов абсорбционной спектроскопии благодаря более широко
78
му использованию инфракрасной области спектра и приборов со встроенной ЭВМ. Это позволило разработать методы анализа сложных многокомпонентных систем без их химического разделения. Методы абсорбционной спектроскопии продолжают успешно развиваться и совершенствоваться.
3.7. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Методы абсорбционной спектроскопии имеют высокую чувствительность (низкий предел обнаружения), они избирательны и точны. Методы могут быть применены для анализа больших и малых содержаний, но особенно ценной их особенностью является возможность определения примесей (до 10-5... 10-6 %). Важное значение имеет избирательность многих фотометрических методов, позволяющая проводить определения элементов в сложных пробах без химического разделения компонентов. Погрешность фотометрических методов обычно составляет 3...5%, уменьшаясь в благоприятных случаях до 1—2% и нередко до 0,5...1,0 %. Методы абсорбционной спектроскопии используют в химической, металлургической, металлообрабатывающей и других отраслях промышленности, горном деле, сельском хозяйстве, медицине и т. д.
Простые, быстрые и точные фотометрические методы анализа применяются для контроля производства, определения примесей и решения многих других важных вопросов в заводских и научно-исследовательских лабораториях. Большое значение имеют эти методы для исследования различных реакций, установления состава и устойчивости образующихся соединений. Успехи химии координационных соединений и достижения приборостроения дают все основания ожидать дальнейшего повышения точности и чувствительности этих методов.
Вопросы
1. Как изменится оптическая плотность и пропускание раствора КМпС>4, если его концентрация уменьшится в 2 раза?
2. Как изменится оптическая плотность и пропускание раствора при увеличении толщины светопоглощающего слоя'*
3. Какие условия следует соблюдать, чтобы при фотометрировании растворов аммиаката меди (II) получить линейную зависимость А— ecu?
4- Будет ли сохраняться линейная зависимость А — с, если пРи разбавлении раствора происходит диссоциация:
Fe(SCN)3^Fe(SCN)^ + SCN~
5. При изменении кислотности раствора происходит сдвиг Равновесия: 200?“+ 2Н+^Сг2О7- + Н2О. Будет ли в этом слу
79
чае соблюдаться основной закон светопоглощения Бугера — Ламберта— Бера?
6. Фотометрическое определение Р2О3 в виде фосфоромолиб-деновованадиевой кислоты выполняют при X—400 нм. Используемые реагенты — смесь метаванадата и молибдата аммония — поглощают в этой же области длин волн. Как в этом случае, выбирают условия фотометрирования?
7. Молярные коэффициенты светопоглощения ионов FeSCN2+ е453=5000, Fe(SCN)^ е485=9800. Объяснить, почему е одного из ионов почти вдвое больше другого.
8. Оптические плотности трех исследуемых растворов равны 0,10; 0,44; 0,80. В каком случае относительная ошибка измерения будет наименьшей?
9. Какие из соединений имеют полосы поглощения в УФ-области 200...400 нм; NaNO3, Na2CO3, СН3—СНг—СН2—СН3, СН2=СН—СН=СН2? С какими энергетическими переходами связано это поглощение?
10. Какие из указанных соединений имеют спектры поглощения в видимой области: NaCl, КМпО4, СС14,
ноос
11. Какие из указанных соединений имеют полосы поглощения в ИК-области: О2, N2, КВг, СС14, СНС13, С6Нб\О2? С какими энергетическими переходами связано это поглощение?
12. Почему спектр поглощения бензола в УФ-области имеет вид более широких полос по сравнению с ИК-спектром?
13. Ион Мп (Н2О)б+ имеет е4?5 — 5, поглощение МпО4-иона характеризуется е528=2400. С какими типами переходов связано поглощение этими ионами? На каком поглощении основано фотометрическое определение Мп в стали?
14. Какое соединение Co(SCN)4~ (е62о==1О3) или Со(Н2О)в+ (е53о=Ю) следует выбрать для определения «следов» (~10~4 моль/л) кобальта (II)?
15. Фотометрирование растворов висмута (III) с тиомочевиной можно проводить при Х=470 нм (е=9-103) и при Х=322 нм (е=35-103). В каком случае предел обнаружения будет ниже?
16. При определении никеля с диметилглиоксимом можно использовать методы прямой и дифференциальной фотометрии. Какой метод предпочтительнее, если исследуемый раствор, содержащий никель и диметилглиоксим, имеет оптическую плотность >1,0?
17. Студент построил две градуировочные прямые, измерив оптическую плотность стандартных растворов КМпО4 относительно воды и относительно раствора сравнения с известной концентрацией КМпО4 (со). В каких случаях следует использовать первую и в каких случаях вторую зависимость?
80
18. Предложите вариант фотометрического метода определения железа (III) с сульфосалициловой кислотой в солях ZnCla, CdCU или А1С13, учитывающий влияние фона.
19. Предложите оптимальные условия (интервал I и с) для фотометрического определения титана с Н2О2 (e4io=72O).
20. Какой вариант фотометрического метода следует выбрать, если главным требованием к анализу элемента является: а) быстрота выполнения анализа; б) высокая точность при достаточно большом содержании элемента; в) высокая точность при оптимальном содержании элемента; г) учет влияния фона (третьего компонента)?
21. Каким образом выбирают условия (X) фотометрического определения смеси веществ, если их спектры поглощения полностью или частично накладываются друг на друга?
22. Какой вид имеет кривая фотометрического титрования, если: а) определяемый компонент и титрант не поглощают, а продукт реакции поглощает свет; б) определяемый компонент поглощает, а титрант и продукт реакции не поглощают свет; в) поглощает только титрант; г) определяемый компонент и титрант поглощают, а продукт реакции не поглощает свет.
23. Три студента выполняли измерения на разных приборах: на визуальном фотометре со стеклянным светофильтром, на фотоэлектроколориметре с интерференционным светофильтром и на спектрофотометре. Оцените полученные ими результаты с точки зрения правильности, воспроизводимости и избирательности.
Задачи
I. После растворения 0,2500 г стали раствор разбавили до 100,0 мл. В три колбы вместимостью 50,00 мл поместили по 25,00 мл этого раствора и добавили: в первую колбу стандартный раствор, содержащий 0,50 мг Ti, растворы Н2О2 и Н3РО4, во вторую — растворы Н2О2 и Н3РО4, в третью — раствор Н3РО4 (нулевой раствор). Растворы разбавили до метки и фотометриро-вали два первых раствора относительно третьего. Получили значения оптической плотности: Ах+Ст=0,650, Ах=0,250.
Рассчитать массовую долю (%) титана в стали.
Находим концентрацию титана, добавленного со стандартным раствором
1.00- J0-2 мг/мл,
где 0 50 мг — масса добавленного титана; 50,00 мл — объем раствора. Вычисляем концентрацию титана по формуле
А П 9^0
Сг= 1,00- А оНл-==6-25' 10-3 МГ/МЛ-
Определяем массу титана во взятой навеске-
6,25-10"3-50.00-100,0 19К,п-з.
т ---------—-----------— 1 25 мг — 1 25-10 г
25.00
81
Рис. 3.19. Определение никеля методом дифференциальной фотометрии
и рассчитываем массовую долю (%):
w(Ti) =
I 25-КГ3-100 0,2500
= 0,50 % Ti.
2. Из навески стали массой 0,2542 г после соответствующей обработки получили 100,0 мл раствора, содержащего диметилглиоксимат никеля. Оптическая плотность этого раствора относительно раствора
сравнения, содержащего 6,00 мг Ni в 100,0 мл, равна 0,440. Для построения градуировочного графика взяли три стандартных раствора с содержанием 4,00; 8,00; 10,0 мл никеля в 100,0 мл и получили при тех же условиях относительные оптические плотности соответственно: —0,240;
0,240; 0,460.
Вычислить массовую долю (%) никеля в стали.
РсЛИ Сх~^> ^сравнения, ТО Ал^>^сравнения 11 Аотн Ах ^сравнения ПОЛОЖИТеЛЬНЗ. При сх<ссравневия относительная оптическая плотность отрицательна.
Строим градуировочный график в координатах: значения относительных оптических плотностей — концентрации растворов никеля в мг/100 мл (рис. 3.19). По графику находим сх = 9,80 мг Ni/ЮОмл, соответствующую А,. = 0,440, и рассчитываем массовую долю (%) никеля в стали:
ffi'(Ni)
9,80-10“3-100 0,2542
3,86% Ni.
3. После соответствующей обработки навески стали массой 0,2025 г получили 100,0 мл раствора, содержащего ионы МпОГ и СггО?-, и измерили оптическую плотность этого раствора при светофильтрах Хэфф=533 нм и Хэфф=432 нм. Для построения градуировочных графиков в мерные колбы вместимостью 100,0 мл поместили 10,00; 15,00; 20,00 мл стандартного раствора перманганата (7’(Мп)=0,0001090) или дихромата(Т(Сг)=0,001210) и разбавленные до метки растворы фотометрировали при тех же светофильтрах.
Рассчитать массовую долю (%) марганца и хрома в стали по следующим данным:
Параметр Стандартные растворы Исследуемый раствор
КМпО, К2СГ2О7
V, мл Абзз 10,00 0,230 15,00 0,350 20,00 0,470 10,0 0 15,0 0 20,0 0 0,320
А 432 0,100 0,140 0,180 0,430 0,600 0,780 0,720
82
Рассчитываем концентрации стандартных растворов после разбавления: Мл TV, 0,0001090-10,00 С| — 100 — 100,0
0,0001090-15,00 ,
100,0 0,0001090-20,00 100,0 0,001210-10,00 100,0 0,001210-15,00 100,0 0,001210-20,00 оло ,
------—--------=2,42-10 г Сг/мл.
Строим градуировочные графики в координатах Д— с, используя данные по светопоглощению перманганата при 533 и 432 нм и дихромата только при 432 нм (рис. 3.20).
Так как при 533 нм Дс„~ДМл, то по градуировочному графику (кривая /) находим: сМл= 1,50-10 5 г/мл. Массовая доля (%) марганца в стали равна ... . 1,50-10~5-100,0-100,0
w(Mn) =----------гОжк---------= 0,74 % Мп-
1,09-10~5 г Мп/мл;
с?п
cP
..Сг
С1
С?
rfr
1,64-10 5 г Мп/мл;
2,18-10 5гМп/мл;
1,21-10 4 г Сг/мл;
1,82-10 4 г Сг/мл;
0,2025
При 432 нм Ас„ = ДСг + АМл. По градуировочному графику (кривая 2) находим ДМл, используя значение сМл = 1,50-10 5 г/мл: ДМл=0,130. Вычисляем А Сг=А см - А Мл = 0,720 - 0,130 = 0,590.
По градуировочному графику (кривая 3) находим концентрацию хрома сСг= 1,77-10 4 г/мл и рассчитываем массовую долю (%) хрома:
,г . 1,77-IO”4-100,0-100
w(Cr)=---------0ДО5--------=8’74 % СГ-
4. Молярные коэффициенты светопоглощения моноэтиламина при 785 и 728 см-1 составляют e785,i = l,67 и e728,i =0,0932, а ди-этиламина е785,2=0,0446 и е728,2= 1,17.
Вычислить концентрации (моль/л) моно- и диэтиламина в техническом триэтиламине, если измеренные при тех же условиях значения оптической плотности при /=1,0см равны: Л785=0,525, -4 728 = 0,715.
Оптические плотности смеси моно-и диэтиламина равны:
Д785 =£785 |С| + £785,2^2
Л?28= £728 1Й + £728 2^2,
Л /?/ -
и
гДе С) и с2 — концентрации моно-Диэтиламина.
Решаем систему уравнений относительно с, и с2:
0,2
„ £785,2^728 — £728.2^785 С|------------------------------
£728 1^785 2—£728 2С785.1
0,0
2,0 с^пК5,г/мл
с е728 1^785 ~~ Е 785 1^728 £728,1 £785 2 — £728,2£785 I
Рис 3.20. Определение хрома и марганца при совместном присутствии
83
Вычисляем концентрацию моно- и диэтиламина:
0,0446-0,715— 1,17-0,525
0,0932-0,0446— 1,17-1,67
0,298 моль/л;
Сг
0,0932-0,525— 1,67-0,715
0,0932-0,0446— 1,17-1,67
= 0,586 моль/л.
5. На рис. 3.21 представлена полоса поглощения циклогексана при Х=11,6мкм. Общая интенсивность в данной области поглощения равна I в. Данные по светопоглощению при этой длине волны исследуемого раствора wx и трех стандартных растворов циклогексана иц_з представлены в таблице:
Определить массовую долю (%) циклогексана в исследуемой смеси.
В соответствии с методом базовой линии пропускание Тд определяется отношением ТЛ = 1Д/1К (см. рис. 3.21), а величина оптической плотности
А = - lg Т = - 1g /л//я= lg/B/fA
Вычисляем оптические плотности стандартных растворов (А|_з) и исследуемого раствора Ах:
A, = lg. 1^|=о,О55; A2 = lg-|||=0,100: /3=lg-^-=0.142; A,= |g±£=0,084.
По данным значениям А _з и «'i-з строим градуировочный график (рис. 3 22! по которому находим иц = 78,70%.
6. Молярный коэффициент светопоглощения дитизоната меди (П) в тетрахлориде углероца при МФФ— 550 нм равен е=4,52Х ХЮ4. Какую массовую долю (%) меди можно определить с дитизоном, если из навески образца сплава массой 1,00 г получают 25,00 мл раствора дитизоната в ССК и измеряют минимальную оптическую плотность 0,020 в кювете I—5,0 см. Ответ: 1,4-105%.
84
7. Молярный коэффициент светопоглощения комплекса MoO(SCN)s~ в изоамиловом спирте при Х,фф=475 нм равен е= 1,50-104. Вычислить минимальную массовую долю (%) молибдена в почве, которую можно
Рис. 3.22. Градуировочный график для определения циклогексана
определить этим методом, если из навески почвы массой 20,00 г извлекают молибден в 200,0 мл ок-
салатного буферного раствора.
Отбирают 150,0 мл фильтрата и после соответствующей обработки экстрагируют образующийся MoO(SCN)i- 15,00 мл изоамилового спирта. Экстракт фотометрируют в кювете I = 3,0 см. Минимальную оптическую плотность принимают 0,020. Ответ: 4Х ХЮ“6%-
8. Для определения никеля с диметилглиоксимом навеску стали растворяют и разбавляют раствор до 100,0 мл. К 5,00 мл раствора добавляют необходимые реактивы, разбавляют водой до 50,00 мл и фотометрируют при /=1,0см, Хэфф=470 нм (е= —1,30- 104). Вычислить массу навески стали для анализа, если оптимальное значение оптической плотности 0,435 и приблизительная массовая доля (%) никеля в стали равна: 1) 0,5%; 2) 1,0%; 3) 5,0%. Ответ: 1) 0,3928 г; 2) 0,1964 г; 3) 0,0393 г.
9. Для определения железа (III) в концентрированной серной кислоте в виде сульфосалицилата навеску помещают в колбу вместимостью 100,0 мл, добавляют необходимые реактивы и доводят до метки водой. Измеряют оптическую плотность при ХЭфф= = 420 нм (е=6,0-103) и толщине кюветы I см.
Рассчитать массу навески кислоты для анализа, если оптимальное значение оптической плотности равно 0,435.
Вариант w(Fe(III)), % Z, см
1 2 3
0,001 0,005 0,010
5 3 1
Ответ: 1) 8,0978 г; 2) 2,6993 г; 3) 4,0489 г.
10. В две мерные колбы вместимостью 100,0 мл поместили по V мл сточной воды. В одну колбу добавили 10,00 мл стандартного раствора CuSO4 (Т(Сн) = 0,001000). В обе колбы ввели растворы аммиака, рубеановодородной кислоты и разбавили до метки водой. При фотометрировании растворов получили оптические плотности Ах И Лл+ст-
Определить концентрацию (г/л) меди в сточной воде для следующих вариантов:
Вариант . 12 3 4
v. мл [0,00 20,00 30.00 40,00
ф 0,240 0,280 0,320 0,400
л-+ст 0.380 0.420 0.460 0,540
Отв^т: 1) 1.71 г/л; 2) 1,00 г/л; 3) 0,76 г/л; 4) 0,71 г/л.
85
11. Навеску стали массой т г растворили в колбе вместимостью 50,00 мл. Две пробы по 20,00 мл поместили в колбы вместимостью 50,00 мл. В одну колбу добавили раствор, содержащий 0,003000 г ванадия. В обе колбы прилили пероксид водорода и довели водой до метки.
Вычислить массовую долю (%) ванадия в стали, если при фотометрировании растворов получили следующие оптические плотности Ах и Лх+ст:
Вариант т, г Лх
Лл+„ .
1 2 3 4
0 5000 0,6572 0,7468 0,9580
0,200 0,230 0,250 0,280
0,480 0,510 0,530 0,560
Ответ: 1) 1,07 %; 2) 0,94 %; 3) 0,90 %; 4) 0,78 %.
12. Навеску стали массой m г растворили в колбе вместимостью 50,00 мл. В две мерные колбы вместимостью 50,00 мл отобрали аликвоты по 20,00 мл. В одну колбу добавили раствор, содержащий 0,001000 г Ti. В обе колбы поместили раствор пероксида водорода и довели растворы до метки.
Вычислить массовую долю (%) титана в стали, если при фотометрировании растворов получили следующие оптические плотности Ах и Лх+ст:
Вариант т, г
Ах
А г-^СТ
1 2 3 4
0,4600 0,4828 0,5000 0,6150
0,200 0,190 0,220 0,250
0,420 0,410 0,440 0,470
Ответ: 1) 0,49 %; 2) 0,45 %; 3) 0,50 %; 4) 0,46 %.
13. Содержание антрацена в растворе определяли по собственному поглощению при Хэфф = 253 нм. Относительная оптическая плотность стандартного раствора, содержащего 35,0 мг/л антрацена, равна Лот„.ст= 0,412. У исследуемого раствора эта величина равна Лотн.тссл= 0,396. В кювете сравнения в обоих случаях был раствор с содержанием 30,0 мг/л антрацена.
Вычислить концентрацию (мг/л) антрацена в исследуемом растворе. Ответ: 34,3 мг/л.
14. Относительная оптическая плотность раствора сульфоса-лицилатного комплекса железа (III) равна Лотн.х = 0,290 (/= = 5 см).
Вычислить концентрацию (мг/мл) железа, если раствор сравнения содержал 0,0576 мг Fe в 50,00 мл, а молярный коэффициент светопоглощения сульфосалицилатного комплекса железа (III) равен 3000. Ответ: 2,231 • 10~3 мг/мл.
15. Для приготовления стандартного раствора циркония навеску ZrOC12-8H2O массой 0,3533 г растворили в 100,0 мл соляной кислоты. В мерные колбы вместимостью 50,00 мл поместили 1,00; 1,20; 1,50; 1,70; 2,00 мл стандартного раствора, прибавили галлоцианин МС (цирконии) и довели до метки водой. Измерили оптические плотности относительно первого раствора. Получили
86
величины Лотн для второго, третьего, четвертого и пятого растворов соответственно: 0,100; 0,235; 0,330; 0,470. Навеску циркониевого сплава массой т г растворили в 100,0 мл смеси кислот. В мерную колбу вместимостью 50,00 мл отобрали аликвоту 2,00 мл, добавили цирконии и довели до метки. Измерили относительную оптическую плотность ЛОТ(1,Й как при построении градуировочного графика.
Вычислить массовую долю (%) циркония в сплаве.
Вариант т, г
Д ОТН.х
1 2 3
0,1120 0,1242 0,1460
0,320 0.380 0,440
Ответ: 1) 74,78%; 2) 72,46 %; 3) 65,92 %.
16. Для построения градуировочного графика в координатах А о™—смп в мерные колбы вместимостью 250,0 мл поместили 11,0; 12,0; 13,0; 14,0; 15,0 мл стандартного раствора, содержащего 1,25 мг/мл Мп, и окислили марганец до перманганата. Оптическую плотность измерили относительно раствора, содержащего 12,5 мг Мп в 250,0 мл, и получили:
уст, мл
Дотн
11,0
0,200
12,0 0,400
13,0 0,600
14,0
0,800
15,0
1,01
Навеску руды массой 0,5000 г растворили и раствор разбавили до 1000,0 мл. В 50,00 мл фильтрата окислили марганец до перманганата и разбавили раствор до 250,0 мл. Измерили относительную оптическую плотность, как при построении градуировочного графика.
Вычислить массовую долю (%) марганца в образцах руды, если для них получены величины ЛОТН1: 1) 0,320; 2) 0,420; 3) 0,560. Ответ: 1) 58,20%; 2) 60,50%; 3) 64,00%.
17. Навеску стандартного алюминиевого сплава, содержащего 12,50 % Si, массой 0,3000 г растворили в колбе вместимостью 500,0 мл. Для построения градуировочного графика в мерные колбы вместимостью 100,0 мл поместили 5,00; 5,20; 5,40; 5,60; 5,80; 6,00 мл этого раствора, добавили реактивы — растворы молибдата аммония и сульфата железа (II) и довели до метки. Измерили оптические плотности относительно первого раствора и получили:
уст, мл 5.20 5,40 5,60 5,80 6.00
Дотн 0,105 0,215 0,330 0,440 0,550
Навеску анализируемого сплава массой 0,2500 г растворили в колбе вместимостью 500,0 мл. Пробу 5,00 мл поместили в мерную колбу вместимостью 100,0 мл, добавили реактивы и измерили относительную оптическую плотность, как при построении градуировочного графика.
Вычислить массовую долю (%) кремния в образцах по следующим данным: 1) Лотн х = 0; 270; 2) Лот„ х = 0,360; 3) Лотн.х = 0,400. Ответ: 1) 16,48%; 2) 16,96%; 3) 17,20%.
87
18. Навеску NazHPCh- 12НгО массой 0,5046 г растворили в хлорной кислоте и разбавили до 1000,0 мл. Для построения градуировочного графика в мерные колбы вместимостью 50,0 мл поместили 10,0; 20,0; 25,0; 30,0; 35,0 мл этого раствора, добавили смесь молибдата и метаванадата аммония и разбавили водой до метки. Измерили оптическую плотность относительно первого раствора и получили:
мл 20,0 25,0 30,0 35,0
Лот’н 0,18b 0,285 0.380 0,475
Навеску нитроаммофоски массой 0,3000 г растворили в соляной кислоте и разбавили раствор до 250,0 мл. Аликвоту 20,00 мл поместили в колбу вместимостью 50,00 мл и выполнили те же операции, как при построении градуировочного графика.
Вычислить массовую долю (%) Р2О5 в нитроаммофоске, если для различных образцов получили: 1) Аотн х = 0,365; 2) Аотн х= 0,415; 3) Аот„ ,= 0,455. Ответ: 1) 12,19%; 2) 13,29 %; 3) 14,17%.
19. Стандартный раствор церия приготовили растворением 0,0500 г СеОа в 250,0 мл смеси хлорной и соляной кислот. В колбы вместимостью 25,00 мл поместили: 2,50; 3,00; 3,50; 4,00; 4,50; 5,00 мл стандартного раствора, добавили аммиак, пероксид водорода, нитрилотриуксусную кислоту и разбавили до метки водой. При фотометрировании относительно первого раствора получили:
У„, мл 3.00 3,50 4,00 4,50 5,0
Лт„ 0,135 0,270 0,405 0,545 0,680
Навеску оксидов лантаноидов массой 0,0500 г растворили, добавили необходимые реактивы, разбавили до 25,00 мл и фото-метрировали, как при построении градуировочного графика.
Вычислить массовую долю (%) СеОг в образце оксидов лантаноидов по следующим данным: 1) Лт„ v = 0,345; 2) Аотн х = = 0,440; 3) ДО7Н..=0,510; Ответ: 1) 1,51 %; 2) 1,65%; 3) 1,75 %.
20. Из навески руды массой m г, содержащей ниобий, после соответствующей обработки получили 100,0 мл раствора. В мерные колбы вместимостью 25,00 мл поместили 2,00; 3,00 и 3,00 мл полученного раствора и в третью колбу добавили 1,00 мл стандартного, содержащего 10,0 мкг/мл ниобия. В каждую колбу прилили ЭДТА, винную и серную кислоты, люмогаллион ИРЕА, нагрели для ускорения реакции комплексообразования, охладили и довели до метки. Оптические плотности второго (Аотн 2) и третьего (Ао1и3) растворов измерили относительно раствора в первой колбе.
Вычислить массовую долю (%) ниобия в руде для следую-
ших вариантов: Вариант 1 2 3
т, г 0,1000 0,1500 0.2500
А отн.2 0.325 0,310 0,245
А птн 3 ... 0,510 0,495 ОчЗО
Ответ: 1) 1,76%; 2) 1,12 %; 3) 0,53%.
88
21. Из навески стали массой 0,2000 г после соответствующей обработки приготовили 100,0 мл раствора, содержащего МпОГ и СГ2О7-, и измерили его оптическую плотность при светофильтрах с Хэфф=533 нм и Х,фф=432 нм (Л533, Л432). В шесть мерных колб вместимостью 100,0 мл поместили 5,00; 8,00; 10,00 мл стандартного раствора перманганата (Т (Мп) =0,0001090) или дихромата (Г (Сг) = 0,001210), разбавили водой до метки и фо-тометрировали при тех же условиях.
Определить массовую долю (%) Мп и Сг в стали для следующих вариантов:
Параметр Стандартный раствор Исследуемый раствор
КМпО, К2Сг2О7 I 2 3
У, мл ДбЗЗ Л 432 5,00 0,230 0,095 8,00 0,365 0,150 10,0 0,460 0,190 5,00 0,430 8,00 0,640 10,0 0,780 0,280 0,820 0,330 0,760 0,370 0,720
Ответ: 1) 0,34; 5,45%; 2) 0,39; 4,73%; 3) 0,44; 4,20%.
22. Для приготовления стандартных растворов титана (IV) и ванадия (V) навеску стали, не содержащей ванадий и титан, растворили в азотной кислоте. В шесть мерных колб вместимостью 50,00 мл поместили одинаковые аликвоты полученного раствора, добавили 0,50; 1,00; 1,50 мл раствора, содержащего 0,20 мг/мл титана (IV) или ванадия (V), пероксид водорода, и довели до метки. Измерили оптические плотности пероксидных комплексов титана (IV) при 400 нм и ванадия (V) при 400 и 619 нм относительно раствора сравнения, содержащего все компоненты, кроме Н2О2. Получили следующие данные для построения градуировочного графика:
Параметр
Ванадий (V)
Титан (IV)
V, мл 0,50 1,00 1,50 0,50 1,00 1,50
0,165 0,340 0,510 0,290 0,575 0,860
^619 0,060 0,120 0.185 0 0 0
Из навески стали массой m г после соответствующей обработки приготовили 100,0 мл раствора. Аликвоту 10,00 мл отобрали в колбу вместимостью 50,00 мл, добавили растворы Н2О2, HNO3 и довели до метки. Измерили оптическую плотность при 400 и 619 нм относительно аликвоты исследуемой смеси, не со-
держащей Н2О2.
Вычислить массовую долю (%) ванадия и титана в стали для следующих вариантов: вариант 1 2 3
ГМ, г Д«оо Дб19 .
0,2000 0,2500 0,3000
0,920 0,940 0,900
0,115 0,105 0,180
Ответ: 1) 0,95; 1,05%; 2) 0,70; 0,90%; 3) 0,97; 0,47 %.
84
23. При конденсации с анилином фурфурол С4Н3ОСНО образует соединение красного цвета С4Н3ОСН(СбН4МН2)2, молярный коэффициент светопоглощения которого при 518 нм равен е£18 = 6,20-104.
фурфурол и его производные при конденсации с бензидином образуют соединения желтого цвета. При 413 нм молярные коэффициенты светопоглощения продуктов конденсации равны: е4)3 = 2,00-102 (для фурфурола), e4i3 = 1,00-103 (для метилфур-фурола и других производных).
На анализ взяли V мл сточной воды производства пластмасс, отогнали фурфурол и его производные в колбу вместимостью 500,0 мл и довели до метки водой. К аликвоте объемом 5,00 мл добавили приготовленную смесь реактивов, содержащую анилин, и разбавили водой до 20,00 мл. Измерили оптическую плотность А518 при 518 нм и кювете I = 1,0 см. К другой аликвоте объемом 50,00 мл добавили солянокислый раствор бензидина и разбавили до 100,0 мл. Измерили оптическую плотность A4i3 при 413 нм и кювете I — 5,0 см
Вычислить концентрацию (мг/л) фурфурола (М (С4Н3ОСНО) = = 96,0854 г/моль) и его производных в расчете на метилфур-фурол (М (С5Н5ОСНО) = 110,1158 г/моль) в сточных водах для следующих вариантов:
Вариант Объем пробы сточной воды V, мл Asia ^413
1 100 0,525 0,450
2 200 0,480 0,410
3 250 0,410 0.350
Ответ: 1) 16,27, 95,37 мг/л; 2) 7,44; 43,44 мг/л; 3) 5,08; 29,67 мг/л.
24. В две делительные воронки поместили по 50,00 мл стандартных растворов кобальта (0,30 мкг/мл) и никеля (0,10 мкг/мл), добавили N, /У'-диэтилдитиокарбамат и экстрагировали образовавшиеся комплексы СС14. Экстракты собрали и разбавили до 10,00 мл СС14. Измерили оптическую плотность обоих экстрактов при 367 и 328 нм в кюветах 1= 1,0 см и получили: А367 Со = = 0,365; А 328,со = 0,100; А367 Ni = 0,186; A32g ni — 0,300.
Аликвоту анализируемого раствора объемом 50,00 мл, содержащего кобальт и никель, поместили в делительную воронку и добавили все реактивы, как для стандартных растворов. Оптическую плотность полученного экстракта (10,00 мл СС14) измерили при 367 и 328 нм, 1=1,0 см.
Вычислить концентрацию (мкг/мл) кобальта и никеля в анализируемом растворе по приведенным результатам измерений: 1) А зет = 0,385; А328 = 0,310; 2) А367 = 0,410; А328 = 0,320; 3) А367 = 0,515; А328 = 0,425. Ответ: 1) 0,191, 0,0821 мкг/мл; 2) 0,210; 0,0834 мкг/мл; 3) 0,249; 0,114 мкг/мл.
90
25. Молярные коэффициенты светопоглощения 8-оксихиноля-тов кобальта (II) и никеля (II) в растворе соляная кислота — ацетон равны при ХЭфф—365 нм еСо=3530, eNl=3230. При Х,фф = = 700 нм свет поглощает только оксихинолят кобальта еСо = 429.
Из 10,00 мл исследуемого раствора получили осадки окси-хинолятов кобальта и никеля, растворили их в 25,00 мл смеси НС1 — ацетон и измерили оптическую плотность при 365 и 700 нм в кювете I = 1,0 см.
Вычислить концентрацию (мкг/мл) кобальта и никеля в растворе для следующих вариантов: 1) Д365 = 0,820; Д700 = = 0,083; 2) Л365 = 0,860; Д700 = 0,050; 3) Д365 = 0,920; Д700 = = 0,075. Ответ: 1) 28,5; 6,23 мкг/мл; 2) 17,2; 20,4 мкг/мл; 3) 25,8; 13,8 мкг/мл.
26. Для построения градуировочного графика в колбы вместимостью 50,00 мл поместили 5,00; 8,00; 10,00 мл раствора железоаммонийных квасцов с Т (Fe) = 0,0000500, добавили соляную кислоту и тиоцианат калия, довели до метки водой и измерили оптическую плотность при 496 нм:
V7, мл 5,00 8,00 10,00
А 0,365 0,595 0,750
Исследуемый раствор, содержащий железо и никель, разбавили в колбе вместимостью 100,0 мл, отобрали две аликвоты по 20,00 мл в колбы вместимостью 50,00 мл. Одну пробу разбавили до метки водой, ко второй пробе прилили НО, KSCN и также довели до метки водой. Оптическая плотность первой пробы при 496 нм равна ДМ1 = 0,080. Оптические плотности второй пробы для разных вариантов равны: 1) 0,460; 2) 0,530; 3) 0,660.
Рассчитать массу (мг) железа в растворе. Ответ: 1) 1,30 мг; 2) 1,53 мг; 3) 1,88 мг.
27. Оптическую плотность стандартных растворов кислотного красного и кислотного синего, содержащих 0,0600 мг/мл красителя, измерили при 1= 1,0 см ХЭфф=500 нм: Д500. Крас = 0,250, 5оо,син== 0,540 и при ХЭфф= 440 нм: Д440.крас = 0,740, Д44о,скн = = 0,380.
Порцию красильного раствора объемом 10,00 мл разбавили водой в мерной колбе вместимостью 100,0 мл и измерили оптическую плотность в кювете с 1= 1,0 см при указанных длинах волн.
Вычислить концентрацию (г/л) красителей для следующих вариантов: 1) Двое == 0,320; Д44о = 0,650; 2) Двое 0,430; Д44о == = 0,560; 3) Двое = 0,530; Д440 = 0,720. Ответ: 1) 0,452; 0,146 г/л; 2) 0,274; 0,351 г/л; 3) 0,369; 0,418 г/л.
28. Навеску руды массой m г растворили и после соответствующей обработки оттитровали ионы Fe2+ раствором перманга-ната калия -фотометрическим методом.
Построить кривую титрования и рассчитать массовую долю железа в образце по следующим результатам измерений:
91
Ва риаит Навеска образца т, г Концентрация раствора KMnOz Оптическая плотность раствора после добавления КМпО4
10,00 мл 12,00 мл 14,00 мл 16,00 мл 18,00 мл 20,00 мл
1 1,0389 0,1075 н. (Ак.= 7б) 0,010 0,010 0,010 0,050 0,100 0,150
2 1,0200 Т= 0,003109 0,010 0,010 0,045 0,110 0,175 0,240
3 0,9987 7(KMnO«/Fe) = = 0,005544 0,020 0,020 0,075 0,140 0,200 0,265
Ответ: 1) 8,32%; 2) 7,00%; 3) 6,83%.
29. Навеску сплава массой m г растворили и после соответ ствующей обработки ионы Си2+ оттитровали ЭДТА спектрофотометрическим методом при 1 ,фф — 620 нм.
Построить кривую титрования и рассчитать массовую долю (%) меди в сплаве по следующим данным:
Вариант Навеска образца т. г Концентрация комплексона, моль/л Оптическая плотность раствора после добавления ЭДТА
1,0 мл 2,0 мл 3,0 мл 4.0 мл 5,0 мл 6,0 мл
1 0,5112 0,09842 0,160 0,250 (,350 0,440 0,450 0,450
2 0,9968 0 1014 0,185 0,280 0,385 0,485 0,525 0,525
3 0,2112 0,05215 0,100 0.190 0,275 0,350 0,350 0,350
Ответ: 1) 5,02%; 2) 2,84%; 3) 6,07 %.
30. Вычислить молярную концентрацию эквивалента раствора хлорной кислоты в ледяной уксусной кислоте, если при фотометрическом титровании этим раствором навески дифенилгуанидина М (Cj3Hi3N3) = 211,28 г/моль массой 0,0696 г в ледяной уксусной кислоте с индикатором тропеолином 00 при X = 540 нм получены следующие результаты:
И(НС1О4), мл А 0,0 0,070 1.0 0,070 2,0 0,080 2.4 0 090 2,8 0,100 3,2 0,130 3,4 0,170
П р о д о л ж е н и е
И(НС1О4), мл 3,6 3,7 3,8 4,0 4,2 4,4 4,8 5,0
А 0,330 0,670 1.07 1,07 1,07 1,07 1,07 1 07
Ответ: 0,08669.
31. Навеску салицилата натрия М (CyHsCbNa) = 160,10 г/моль массой 0,0515. г растворили в ледяной уксусной кислоте и оттитровали фотометрическим методом с индикатором тропеолином 00 при X = 540 нм на фотометре ЛМФ-69, используя в качестве титранта 0,10007И НС1О4 в ледяной уксусной кислоте.
Рассчитать массовую долю (%) салицилата натрия в препарате по результатам титрования:
Г(НСЮ4, мл 0,0 1,0 1,2 1,6 2,0 2,4 2,6
А . . . 0,020 0,040 0,040 0,050 0,070 0,110 0,150
92
Продолжение
(/(HCIOj. мл 2,8 3,0 3,1 3,2 3,4 3,6 4.0 4,4
д 0,230 0,440 0,680 1,04 1,07 1,07 1,07 1,07
Ответ: 99,48%.
32. Из 100,00 мл сточной воды экстрагировали гербицид — которан хлороформом. Экстракт упарили, перенесли в кювету и оттитровали при Л = 290 нм уксуснокислым раствором HCIO4 (Г (НС1О4/которан) = 0,000300).
Вычислить концентрацию (г/мл) которана в воде по следую щим результатам:
Вариант Оптическая плотность раствора после добавления НС1О4
0 мл 0,4 мл 0,8 мл 1,2 мл 1,6 мл 2,0 мл 2,4 мл 2,8 мл 3,2 мл 3,6 мл 4,0 мл
1 0,315 0,215 0,125 0,060 0,035 0,030 0,020 0,015
2 0,345 0,290 0,240 0,190 0,146 0,095 0,070 0,050 0,040 0,030 0,020
3 0,375 0,335 0,300 0,265 0,225 0,190 0,155 0,125 0,100 0,085 0.070
Ответ: 1) 3,45 мг/л; 2) 6,45 мг/л; 3) 9,45 мг/л.
33. Навеску образца, содержащего смесь аминов и индифферентных примесей, массой 0,0250 г растворили в безводной уксусной кислоте и оттитровали уксуснокислым раствором 0.0800М НСЮ4.
Построить кривую титрования и рассчитать массовую долю (%) аминов в смеси: 1) п-броманилин и л-нитроанилин; 2) диметиланилин и л«-нитроанилин; 3) п-нитрозодиметилани-лин и л«-нитроанилин; 4) 4-метил-2-нитроанилин и о-нитроанилин.
Вариант Оптическая плотность раствора после добавления титранта НСЮ4
0,4 мл 0,6 мл 0,8 мл 1,0 мл 1,2 мл 1,4 мл 1,6 мл 1,8 мл 2,0 мл 2,2 мл 2,4 мл
1 0,680 0,680 0,680 0,680 0,550 0,410 0,280 0,150 0,040 0,040 0,040
2 0,300 0,300 0,300 0,300 0,220 0,140 0,050 0,040 0,040 0,040 0,040
3 0,330 0,260 0 190 0,160 0,140 0.125 0,100 0,075 0,060 0,060 0,060
4 0,800 0,680 0,570 0,500 0 480 0,470 0,450 0,430 0,420 0,420 0,420
Ответ: 1) 55,05; 43.10%, 2) 39,94; 26,52%; 3) 41,33; 47,74%, 4) 45,04; 47,52 %
34. Из навески метиленхлорида CH2CI2 массой 20,00 г приготовили 100,0 мл раствора в хлороформе для построения градуировочного графика. В мерные колбы вместимостью 50,0 мл внесли по V мл этого раствора и довели до метки хлороформом. Методом базовой линии при 7,98 мкм нашли значения А —
— 1g приведенные ниже:
К мл
А
2,00 4,00 6,00 8,00 10,00
0,085 0.155 0,225 0,300 0,375
93
Вычислить концентрацию (г/л) метиленхлорида в исследуемом растворе, если после разбавления 25,0 мл его хлороформом до 50,0 мл найдены следующие значения А: 1) 0,325; 2) 0,280; 3) 0,125. Ответ: 1) 70 г/л; 2) 60 г/л; 3) 25 г/л.
35. Содержание нормального бутана и изобутана в смеси с индифферентными примесями определили по полосам поглощения при 10,3 мкм (поглощает только н-бутан) и при 8,5 мкм (поглощает только изобутан). Для стандартных смесей получили следующие данные:
Поглощение при 10,3 мкм, % 18,0 28 5 39,0 49,0 54,0
w (н-бутана}, % . 18,0 36,0 58,0 79,0 92,0
Поглощение при 8,5 мкм, %-. 22,5 41,0 53,0 63,0 69,0
w (изобутана), % 15,0 42,0 61.0 83,0 95,0
Вычислить массовую долю (%) н-бутана и изобутана в смеси, если для них получены в тех же условиях следующие результаты:
Вариант Поглощение при 10,3 мкм, % Поглощение при 8,5 мкм, %
1 20,0 60,0
2 28,0 52,0
3 34,0 45,0
4 42,0 32,0
5 51,0 18,0
Ответ: 1) 22 и 75 %; 2) 34 и 60 %; 3) 46 и 48 %; 4) 63 и 29 %; 5) 84 и 10%.
36. Из навески смеси 2-метилбутанола-2 и 2-метилбутена-2 и индифферентных примесей массой m г приготовили 100,0 мл раствора в СС14. Для построения градуировочных графиков и анализа смеси использованы полосы поглощения при 930 см-1 (поглощает 2-метилбутанол-2) и при 2740 см-1 (поглощают оба соединения). Для стандартных растворов получены данные:
2' метилбутанол-2 2-метилбутен-2
с, г/100 мл ^ЭЗОсм 1 Л 1 л2740 см с, г/100 мл 4 2740 см ’
2,5 0,280 0,050 0,300 0,220
5,5 0,620 0,110 0,600 0,450
9.0 1,000 0,180 1,000 0,750
Вычислить массовую долю (%) 2-метилбутанола-2 и 2-метил бутена-2 в смеси, если при анализе в аналогичных условиях получены результаты:
94
Вариант т. г
А 930 см 1
Л 2740 см
I 3,20 0,300 0,470
2 3 4 5
5,10 8,50 8,80 10,00
0,480 0,860 0,720 0,980
0,320 0,320 0,800 0,860
Ответ: 1) 78,13 и 17,50%; 2) 84,31 и 6,27%; 3) 90,59 и 2,59%; 4) 72,07 и 10,23%; 5) 88,00 и 8,60%.
37. Для определения циклогексанона в циклогексане по поглощению при 1718 см-1 в мерные колбы вместимостью 50,0 мл поместили m г циклогексанона (х.ч.) и довели до метки циклогексаном. Полученные стандартные растворы фотометрировали по отношению к чистому циклогексану и получили следующие
данные: т, г
0,0625 0,125 0,250 0,500 1,000
0,060 0,085 0,140 0,265 0,480
Vx мл исследуемой смеси разбавили циклогексаном до 250,0 мл и полученный раствор фотометрировали в тех же условиях.
Определить концентрацию г/100 мл циклогексанона в исходном растворе, если Ах и Vx составляют: 1) 0,345; 100,0 мл; 2) 0,07; 10,0 мл; 3) 0,160; 50,0 мл; 4) 0,258; 100,0 мл. Ответ: 1) 3,5 г/100 мл; 2) 4,5 г/100 мл; 3) 2,9 г/100 мл; 4) 2,5 г/100 мл.
38. Навеску 1,2-дибромпропана массой 1,00 г смешали с 9,00 г 1,3-дибромпропана и m г смеси еще раз смешали с (10 — — т) г 1,3-дибромпропана (оба соединения являются тяжелыми маслами). Последнюю смесь фотометрировали при 7,29 мкм, где поглощает только 1,2-дибромпропан, и получили следующие данные:
т, г . 1,00 2.00 3,00 4,00 5,00
А при А =
= 7,29 мкм 0,040 0,090 0,130 0,170 0,220
В аналогичных условиях фотометрировали исследуемые смеси и получили Ах: 1) 0,19; 2) 0,15; 3) 0,08.
Вычислить массовую долю 1,2-дибромпропана в анализируемых образцах 1,3-дибромпропана. Ответ: 1) 4,3%; 2) 3,4%; 3) 1,9%.
39. Для контроля состава смеси в производстве триацетата целлюлозы, состоящей из бензола, уксусной кислоты и уксусного ангидрида, на содержание уксусной кислоты измерили поглощение при 1,92 мкм стандартных смесей при разной толщине кюветы (1, мм), и разной массовой доле (%) уксусной кислоты (вд, %) и получили:
1, мм Показания прибора а, %, при
<»! = 10,0% = 30,0 % = 40,0 %
1.00 28,5 35,0 38,0
2,00 34,5 45,0 50,5
3,00 38,0 53,5 61,5
4,00 40,5 57,0 62,0
95
Выбрать толщину кюветы (/, мм), обеспечивающую максимальную чувствительность анализа, и определить массовую долю (%) уксусной кислоты в смеси, если при Л получены следующие показания прибора: 1) 42,5 %; 2) 51,0%; 3) 58,0 % Поглощением других компонентов смеси пренебречь. Ответ: /Л = 3 мм; 1) 15,0%; 2) 27,0%; 3) 35,0%.
40. При анализе смеси бензола, уксусной кислоты и уксусного ангидрида используются полосы поглощения бензола при 1,1 мкм и уксусной кислоту при 1,92 и 1,1 мкм. Поглощение при этих длинах волн стандартных растворов составило:
Параметр Бензол Уксусная кислота
w, % Поглощение при At i % 30,0 40,0 50,0 10,0 30,0 40,0
35,0 41,0 47,0 1,0 13,0 19,0
Поглощение при А, <jj, % — —- — 27.0 48,0 58.0
Определить массовую долю по следующим данным: Вариант Поглощение при A, i, % Поглощение при А| 92, % (%) бензола 1 45 34 и уксусной кислоты 2 3 49 62 44 56
Ответ: 1) 16,0 и 40,0%; 2) 26,0 и 36,0 %; 3) 38,0 и 45,5 %.
41. Для выбора условий анализа водно-метанольных смесей было измерено поглощение при 1,44 и 1,94 мкм четырех растворов:
Щ(Н2О). % . 26,0 29,0 32,0 34,0
А | 44 0,130 0,145 0,180 0,080
A f <14 0,470 0,500 0.538 0.560
На основании этих данных выберите аналитическую полосу X* для анализа смеси метанол — вода и определите массовую долю (%) воды и метанола в анализируемых образцах по следующим данным: 1) 0,570; 2) Л>, = 0,540; 3) Л;,—0,455.
Ответ: = 1,94 мкм; 1) 34,6 и 65,4 %; 2) 32,0 и 68,0 %; 3) 25,0 и 75,0 %.
42. Установлено, что выходной сигнал ИК-анализатора при-X = 1,68 мкм является аддитивной функцией концентрации аммиака и карбоната аммония, а при X = 2,19 мкм — функцией концентрации только аммиака, стандартные растворы NH3 и (NH4)2CO3 имеют следующие значения оптической плотности:
Параметр Аммиак Карбонат аммония
• , г/л 5,0 12,5 20,0 280 360 410
7^2.19 0,150 0,330 0,510 — — —
^1 68 0,075 0,150 0,220 0 но 0,270 0,540
96
Определить концентрацию (г/200 мл) (NH4)2CO3 и NH3 в исследуемых растворах по следующим данным: 1) Л219 = 0,450-д168 = 0,380; 2) Д2.19 = 0,300; Л,.68 = 0,450; 3) Л2.19 = 0,210; Д168 = 0,350. Ответ: 1) 63,0 г/200 мл и 3,5 г/200 мл; 2) 77,0 г/200 мл и 2,2 г/200 мл; 3) 70,0 г/200 мл и 1,5 г/200 мл.
Глава 4 АТОМНО-АБСОРБЦИОННЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
4.1. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДА
Атомно-абсорбционный спектральный анализ предложен Уолшем в 1955 г. Метод сразу получил признание.
При поглощении кванта света hv свободный атом А переходит в возбужденное состояние А*:
А + hv = А*,
где h — постоянная Планка; v — частота, определяемая условием частот Бора,
__ £д- Еа h
Ед* и Ед — энергия атома в возбужденном и основном состояниях соответственно.
Наиболее вероятным изменением энергетического состояния атома при возбуждении является его переход на уровень, ближайший к основному энергетическому состоянию, т. е. резонансный переход. Если на невозбужденный атом направить излучение с частотой, равной частоте резонансного перехода, кванты света будут поглощаться атомами и интенсивность излучения будет уменьшаться. Использование этих явлений составляет физическую основу атомно-абсорбционной спектроскопии. Таким образом, если в чмиесионной спектроскопии концентрация вещества^ свяяыпалась^с интенсивностью излучения, которое было прямо пропорционально числу Возбужденных~~атомов, то в-втом но-аЬсорбпилмной спектроскопии аналитический сигнал (уменьшение игиенетгвностм-юлльч ия) связан с числом невозбужденных атомов.
Заселенность энергетических уровней так же, как и в эмиссионной спектроскопии, определяется уравнением (2.10). Расчеты показывают, что число атомов в возбужденном состоянии незначительно по сравнению с числом атомов на основном (нижнем) уровне и не превышает 1...2% от общего числа атомов. dTo выгодно отличает атомно-абсорбционный анализ от эмис-си°нного, так как при прочих равных условиях величина аналитического сигнала оказывается связанной с большим числом 4'1533
97
атомов, чем в эмиссионной спектроскопии, и, следовательно, величина сигнала в меньшей степени будет подвержена влия нию случайных колебаний в режиме работы различных узлов атомно-абсорбционного спектрофотометра.
4.2. ОСНОВНЫЕ УЗЛЫ ПРИБОРОВ ДЛЯ АТОМНО-АБСОРБЦИОННОГО АНАЛИЗА
Принципиальная схема установки атомно-абсорбционной спектроскопии приведена на рис. 4.1.
Источником излучения является обычно лампа с полым катодом, содержащим определяемый элемент. Катод такой лампы изготовляют в виде металлического стаканчика, в котором происходит испарение вещества и возбуждение атомов элементов при электрическом разряде в атмосфере инертного газа под небольшим давлением (~ 102 Па). Катоды, изготовленные из элементов с относительно низкими температурами плавления, легко разрушаются. Для определения таких элементов используют графитовые катоды, пропитанные солями определяемых элементов. Анод в виде металлического стержня размещают рядом с катодом и оба электрода помещают в стеклянный баллон со стеклянным или кварцевым окошком. Лампа питается током от высокоточного выпрямителя — стабилизатора, дающего напряжение 500...600 В с колебаниями, не превышающими сотых долей процента.
Пары материала катода и других веществ, находящихся на внутренней поверхности катода, попадают в плазму вследствие катодного распыления и испарения в процессе разряда при 200...300 В и 5...30 мА. В спектре свечения при температуре около 800 К в полом катоде наблюдаются резонансные частоты этих элементов. Применяются также лампы с СВЧ-возбужде-нием (СВЧ-лампы) для определения, например, мышьяка, сурьмы, висмута, свинца и некоторых других элементов. Анализируемое вещество в виде раствора подается в пламя горелки, где при 2000...3000°C происходит испарение растворителя и атомизация пробы.
Поскольку уменьшение интенсивности излучения пропорционально толщине светопоглоща-
Рис 4 1. Схема атомно-абсорбционного спектрофотометра:
1 - источник излучения 2 — пламя, 3 — монохроматизатор, 4 — приемник света, 5 — анализируемый раствор
ющего слоя, горелки имеют специальную конструкцию, обеспечивающую постоянную и достаточно большую длину поглощающего слоя пламени (5„ ...10 см).
В последнее время широкое распространение в атомно-абсорбционной спектроскопии получили непламенные электротермические атомизаторы. Хотя
98
электропечи для атомизации применяются сравнительно давно, начало практическому использованию их в атомно-абсорбционной спектроскопии было положено Б. В. Львовым в 1961 г. Графитовая кювета Львова представляет собой трубчатую печь с графитовым электродом в центре. Проба в виде раствора или порошка наносится на торец графитового электрода и за счет мошного дугового разряда мгновенно испаряется. Известны и другие конструкции электротермических атомизаторов. В качестве монохроматизаторов применяют призмы или дифракционные решетки. В качестве приемника света используют фотоэлементы или фотоумножители.
Комплектные приборы для атомно-абсорбционной спектроскопии выпускаются во многих странах. В настоящее время известно свыше 50 моделей таких спектрофотометров. В нашей стране выпускается атомно-абсорбционный спектрофотометр типа «Спектр-4», «Сатурн» и др.
Спектрофотометр «Сатурн» является одним из наиболее совершенных отечественных приборов атомно-абсорбционной спектроскопии. Он может работать как по двух-, так и по однолучевой схемам. Осветительная система спектрофотометра сконструирована на основе лампы с полым катодом, атомизация пробы происходит в пламени горелки, хотя в комплект прибора входит графитовая кювета Львова. В качестве монохроматизатора используется дифракционная решетка, а интенсивность излучения измеряется фотоэлектронным умножителем.
Результат анализа в атомно-абсорбционных методах зависит главным образом от числа невозбужденных атомов, которое в известных пределах сравнительно мало изменяется с температурой. Это уменьшает эффекты взаимного влияния компонентов пробы на аналитический сигнал. В эмиссионной спектроскопии результат анализа определяется, в основном, числом возбужденных атомов, доля которых невелика и существенно зависит даже от небольших колебаний температуры. Поэтому требования стабильности условий работы источника возбуждения, в эмиссионной спектроскопии выступающее на первый план, в атомно-абсорбционной спектроскопии решающего значения не , имеют, хотя, конечно, стабилизация условий необходима.
В атомно-абсорбционной спектроскопии практически полностью исключена возможность наложения линий различных элементов, так как в условиях атомно-абсорбционного анализа число линий в спектре значительно меньше, чем в эмиссионной спектроскопии.
4.3. КОЛИЧЕСТВЕННЫЕ ОПРЕДЕЛЕНИЯ
Уменьшение интенсивности резонансного излучения в условиях атомно-абсорбционной спектроскопии подчиняется экспоненциальному закону убывания интенсивности в зависимости от Длины слоя и концентрации вещества, аналогичному закону Угера — Ламберта — Бера. Если /0 — интенсивность падающе-4*
99
го монохроматического света, а I — интенсивность этого света, прошедшего через пламя, то величину 1g (/о//) можно назвать оптической плотностью. Концентрационная зависимость оптической плотности выражается уравнением
1g (/о//) = A = klc, (4.1)
где k — коэффициент поглощения; I — толщина светопоглощающего слоя (пламени); с — концентрация.
Постоянство толщины светопоглощающего слоя, т. е. пламени, достигается с помощью горелок специальной конструкции
Оптическая плотность согласно уравнению (4.1) прямо пропорциональна концентрации вещества. Опыт доказывает, что зависимость оптической плотности от концентрации часто оказывается не строго линейной. Отклонения от линейности вызываются несколькими причинами, среди которых наиболее существенное значение имеют такие, как нестабильность работы различных узлов спектрофотометра (источника возбуждения и др.), немонохроматичность линий испускания, вызванная сверхтонкой структурой, образование в пламени различных соединений определяемых элементов с кислородом или сопутствующими элементами и т. д. В практике анализа обычно применяют метод градуировочного графика и метод добавок.
В методе градуировочного графика измеряют оптическую плотность нескольких стандартных растворов и строят график в координатах оптическая плотность — концентрация. Затем в тех же условиях определяют оптическую плотность анализируемого раствора и по градуировочному графику находят его концентрацию.
При работе по методу добавок сначала измеряют оптическую плотность анализируемого раствора (Лх), затем вводят в анализируемый раствор определенный объем стандартного раствора и снова измеряют оптическую плотность (Дх+ст). Если сх — концентрация анализируемого раствора, а сст — стандартного, то
Ах = klcx\
4 х 4- ст М(СХ 4- Сст).
Учитывая, что k и I одинаковы, получаем
А, _____с±__
А , 4 ,, сх -|- с„
И окончательно
С^е (4.2)
X + СТ 2* к
Метод- применим для систем, подчиняющихся в исследуемой области концентраций уравнению (4.1). Можно использовать также графический метод нахождения сх на основе уравнения (4.2), откладывая на графике Лх + сг как функцию сст. При А х ст — О Сх — с ст.
100
4.4. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Методы атомно-абсорбционной спектроскопии могут быть использованы или используются в анализе практически почти любого технического или природного объекта, особенно там, где необходимо определять небольшие содержания элементов. Методики атомно-абсорбционного определения разработаны более чем для 70 элементов (Mg, Zn, Си, Са, Pb, Fe, Ag, Ni, Hg, Cd, Bi и др.). Из технических объектов методами атомно-абсорбционной спектроскопии анализируют металлы, сплавы, продукты гидрометаллургической переработки руд, различные концентраты и т. д. Например, в золоте определяют серебро, свинец, медь и цинк при содержании 10-4%. Примерно такие же концентрации кадмия и свинца находят в цирконии. Успешно применяются атомно-абсорбционные методики для определения цинка, железа, магния, меди и некоторых других элементов в почвах, удобрениях, растениях и других агрохимических материалах при содержании порядка 10-4 или 10~5%. Атомно-абсорбционный метод используется также в клинических и различных биологических анализах (кровь, сыворотка и т. д.) на свинец, ртуть, висмут и другие элементы. Дальнейшего расширения областей практического применения атомно-абсорбционной спектроскопии можно ожидать при более широком использовании графитовой кюветы и высокотемпературных пламен.
4.5. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Атомно-абсорбционный спектральный анализ получил широкое распространение в практике вследствие многих своих достоинств. Важным достоинством атомно-абсорбционного метода является наличие менее жестких требований, чем в эмиссионной спектроскопии, к условиям получения поглощающей плазмы, поскольку аналитический сигнал зависит от числа невозбужденных атомов, которое сравнительно мало меняется при небольших колебаниях температуры. Существенно также, что число линий в спектре в условиях атомно-абсорбционного анализа невелико, поэтому наложения аналитических линий практически не происходит, хотя неселективное поглощение остается значительным. Предел обнаружения с помощью атомно-абсорбцион-ного анализа для многих элементов характеризуется величиной порядка 10~5 или 10-6%. Погрешность определения обычно составляет примерно 5 % и в зависимости от различных условий изменяется в пределах от 3 до 10%.
Метод имеет также ряд ограничений. Атомно-абсорбционным методом не определяются элементы, резонансные линии которых ^еЖат в далеком ультрафиолете (углерод, фосфор, галогены ДР)- Необходимость растворения пробы также можно рас-атРнвать как недостаток, поскольку эта операция удлиняет ализ- Однако работа с растворами упрощает эталонирование
101
и обеспечивает высокую воспроизводимость результатов. К существенным недостаткам метода относится невозможность одновременного определения нескольких элементов, хотя для этого имеются все предпосылки. Необходимо отметить также, что помимо чисто аналитического применения атомно-абсорбционная спектроскопия используется для определения силы осциллятора, коэффициентов диффузии, давления насыщенных паров и т. д.
2 Вопросы
1. Получена зависимость атомного поглощения хлорида цезия от концентрации растворов нелинейной формы. Объяснить возможные причины подобной зависимости в методе атомно-абсорбционной спектроскопии. Предложить другой, более предпочтительный метод определения цезия.
2. Элементы подгруппы меди (Cu, Au, Ag) можно определить методами атомно-абсорбционной (ААС) и эмиссионной спектроскопии (ЭС). Объяснить, почему метод ААС имеет более низкий предел обнаружения.
3. Необходимо определить ванадий методом атомно-абсорбционной спектроскопии. Предложить различные варианты этого метода, если: а) исследуется чистый раствор VOCU; б) исследуемый раствор VOCb содержит избыток посторонних солей (КО, NaCl и др.); в) исследуемый образец содержит термически устойчивые формы ванадия (например, образец нефти).
4. Какой атомизатор (пламенный или непламенный) предпочтительнее использовать при анализе органических растворителей, масел и пр.?
5. Какой из методов атомно абсорбционной или пламенноэмиссионной спектроскопии предпочтительнее использовать при определении: К, Са, Pb, V, Ti?
6. Какой из методов предпочтительнее при проведении полного качественного анализа: метод атомно-абсорбционной или эмиссионной спектроскопии?
Задачи
1. При определении марганца в сплаве методом добавок навеску массой 0,5000 г растворили и разбавили раствор до 200,0 мл. Отобрали четыре одинаковые порции раствора и к каждой порции добавили равные объемы стандартных растворов марганца, содержащих 0; 2; 4; 6 мкг/мл Мп.
На атомно-абсорбционном спектрофотометре измерили оптическую плотность для аналитической линии 279,48 нм, распыляя растворы в пламени ацетилен — воздух. Получили соответственно 0,225; 0,340; 0,455; 0,570.
Вычислить массовую долю (%) марганца в сплаве.
Принимаем концентрацию исследуемого раствора за с„ Тогда концентрации измеряемых растворов равны: с„/2, (сж/2) + 1, (сх/2)-|-2, (сх/2)-(-3 мкг/мл. На
102
оси абсцисс произвольно выбираем точку с,/2 и откладываем от нее точки (сх/2) Д--|- 1, (Сх/2) + 2, (Сх/2) + 3. Для построения градуировочного графика по оси ординат откладываем соответствующие точкам значения оптической плотности А
Считая, что зависимость А — с линейна, находим положение начала коор динат, экстраполируя построенную по четырем точкам прямую до пересечения с осью абсцисс, как это показано на рис. 4 2.
Длина отрезка 0 — с,/2 соответствует сх/2 = 2,0 мкг/мл Следовательно, сх = 4,0 мкг /мл
Вычисляем массовую долю (%) Мп в сплаве-
4,0-200,0-10“6-100 % П1г0,
w (Мп) =—--------Нге--------—= 0,10%Мп
0,5
Рис 4.2. Определение марганца методом добавок
2. Два образца нефти, стандартный и анализируемый, массой по 1,000 г разбавили в 10 раз метилизобутилкетоном и распылили в пламени атомно-абсорбционного спектрофотометра. Оптическая плотность линии ванадия для образца с известным содержанием ванадия w„ составила и для образца с неизвестным содержанием Ах.
Вычислить массовую долю (%) ванадия для следующих образцов нефти:
Вариант ™СТ, % Оптическая плотность
Лст Ах
1 0,01 0,740 0,520
2 0,05 0,370 0,440
3 0,02 0,148 0,270
Ответ: 1) 0,70-10“2%; 2) 0,59-10~2 %; 3) 0,36-10“2%.
3. При определении платиновых элементов (Pd, Pt, Rh, Ir) навеску образца массой 1,000 г после разложения и соответствующей обработки перевели в раствор объемом 10,00 мл. Пробы по 100,0 мкл полученного раствора поместили в электротермический атомизатор (графитовую трубку) автоматического атомно-абсорбционного спектрофотометра и на диаграммной ленте самописца записали сигнал поглощения аналитической линии элемента в виде пика высотой hx мм.
Для построения градуировочного графика в атомизатор вводили последовательно по 100,0 мкл стандартных растворов солей платиновых металлов и измеряли величины h мм пиков.
Сст (Pd), мкг/мл ft(Pd), мм Со (Pt), мкг/мл ft(pt), мм (Rh), мкг/мл ft (Rh), мм Сст (1г), мкг/мл Л (1г), ММ
0,01 8,5 0,04 170 0,01 12,0 0,10 12,0
0,05 45,0 0,08 34,5 0,05 59,0 0,20 24,0
0,10 90,0 0,12 52,0 0,10 118,0 0,40 48,0 103
Вычислить концентрацию (г/т) палладия, платины, родия и иридия в образцах для следующих вариантов:
Вариант fl, мм, для линии
Pd Pt Rh Ir
1 “ 28,0 31,0 79,6 21,0
2 44,0 39,0 74,0 35,0
3 63,0 46,0 55,0 42,0
Ответ: 1) 0,31; 0,72; 0,67; 1,75 г/т; 2) 0,49; 0,90; 0,63; 2,90 г/т; 3) 0,70; 1,06; 0,47; 3,50 г/т.
4. Образец животной ткани массой ш г сожгли в муфельной печи, остаток растворили в HCI и разбавили раствором соли лантана до 5,00 мл для устранения мешающего влияния фосфат -ионов.
К четырем равным порциям анализируемого раствора добавили равные объемы стандартных растворов кальция, содержащих Со = 0; Ci = 2,0; с2 = 4,0; Сз = 8,0 мкг/мл Са и такое же количество соли лантана, как в анализируемом растворе. На атомно-абсорбционном спектрофотометре измерили оптическую плотность аналитической линии кальция (422,67 нм).
Вычислить концентрацию кальция (мг/кг) в следующих образцах, используя построение градуировочного графика по методу добавок:
Вариант m, г Атомное поглощение
Лх/2 Л(х+с|)/2 ^(х+с2)/2 ^(х+ед/г
1 0,15 0,050 0,116 0,185 0,320
2 0,20 0,075 0,150 0,226 0,380
3 0,25 0,110 0,185 0,260 0,410
Ответ: 1) 50,0 мг/кг; 2) 50,0 мг/кг; 3) 60,0 мг/кг.
Глава S ЛЮМИНЕСЦЕНТНЫЙ АНАЛИЗ
5.1. СПЕКТРЫ ЛЮМИНЕСЦЕНЦИИ
Согласно определению С. И. Вавилова люминесценцией называют свечение, избыточное над температурным и обладающее длительностью не менее чем Ю-10 с, что превышает период световых колебаний. От излучения нагретых тел она отличается своей неравновесностью: люминесценция практически не использует тепловую энергию излучающей системы, поэтому ее часто называют холодным светом. Это определение отличает люми
104
несценцию также от всех других видов неравновесного свечения — рассеяния и отражения света, комбинационного рассеяния, излучения Вавилова — Черенкова и т. д.
Люминесценция возникает в результате электронного перехода при возвращении частиц из возбужденного состояния в нормальное. Таким образом, молекула преобразует поглощенную энергию в собственное излучение Этим люминесценция также отличается от процессов несобственного излучения — рассеяния и отражения света. Люминесцирующие вещества могут находиться в любом агрегатном состоянии.
В возбужденное состояние частицы люминесцирующего вещества могут переходить под действием света и тогда люминесценцию называют фотолюминесценцией (флуоресценцией или фосфоресценцией), под действием рентгеновского излучения — рентгенолюминесценци-е й, в результате химической реакции — хемилюминесценцией и т. д.
Рис 5.1. Схема энергетических уровней молекулы, поясняющая возникновение люминесцентного излучения: Eq. Ei — электронные уровни, V. V' — колебательные
Происхождение люминесцентного излучения поясняется схемой на рис. 5.1. Здесь схематически изображены основной и возбужденные (синглетный и триплетный) электронные уровни молекулы. На каждый электронный уровень накладываются колебательные подуровни с квантовыми числами о. 1, 2, 3 и т. д. При поглощении кванта света электрон переходит с основного уровня на более высокий, соответствующий возбужденным синглетному (антипараллельные спины) и триплетному (параллельные спины) состояниям. Прямой переход основ-ногосинглетного в возбужденное триплетное состояние запрещен по спину и практически не наблюдается. Энергия триплетного состояния несколько меньше, чем'синглетного: Триплетные уров-и могут заполняться за счет интеркомбинационной
105
конверсии (волнистая стрелка V6 = 0 -> И' = 2). При комнатной температуре молекулы обычно находятся в основном состоянии и почти все электронные переходы при поглощении света происходят с нижнего (основного) колебательного подуровня на различные колебательные подуровни возбужденного синглетного состояния. На рис. 5.1 эти переходы изображены стрелками: V'= 0 —V''’= 0; У = 0 —V" = 1; V = 0V" = 2 и т. д.
Возбужденная молекула за счет так называемой колебательной релаксации при столкновении с окружающими молекулами очень быстро по сравнению с временем электронного перехода теряет избыточную колебательную энергию и переходит на основной колебательный уровень возбужденного электронного (синглетного) состояния. На рис. 5.1 этот процесс изображен волнистой стрелкой: V' = 3~^V' = 0; V' — = 2 V" = 0; V = 1 V = 0 и т. д.
При переходе с основного колебательного подуровня возбужденного синглетного состояния на какой-либо колебательный подуровень основного электронного (тоже синглетного) состояния происходит излучение кванта света. Этот процесс называют флуоресценцией. На рис. 5. 1 ему соответствуют переходы 1/0 = 0 —V' = 0; V6 = 0 -> V = 1; V'o — O-^V — 2 и т. д. Время затухания флуоресценции составляет 10~9...10~7 с. Дезактивация возбужденной молекулы может происходить также за счет безызлучательных переходов внутренней конверсии. В триплетном так же, как и в возбужденном синглетном состоянии, происходит колебательная релак сация и электрон переходит на нижний колебательный уровень триплетного состояния (волнистая стрелка V" — 2 V" — 0; V" — 1 -> V" = 0). Запрещенный по спину излучательный три-плет-синглетный переход (V" = 0 —I/= 0; V" — 0 V = 1 и т. д.) называют фосфоресценцией. Время жизни триплетного состояния велико (10-3...102 с). Переход из триплет ного состояния в основное синглетное происходит также при столкновении возбужденной частицы с окружающими молекулами за счет безызлучательных процессов внутренней конверсии, вероятность которых при комнатной температуре очень велика. По этой причине, чтобы наблюдать фосфоресценцию и использовать ее в аналитических целях, пробу обычно замораживают, часто при температуре жидкого азота (77 К), что сводит до минимума вероятность безызлучательного перехода. Спектр фосфоресценции сдвинут в длинноволновую сторону на величину, пропорциональную энергии колебательной релаксации триплетного состояния.
Так как в зависимости от частоты облучающего излучения частица переходит в энергетически разные возбужденные состояния, можно было ожидать связь спектра флуоресценции со спектром возбуждения источника. Однако в действительности эта зависимость обычно не наблюдается. Независимость спектра
106
флуоресценции от длины волны падающего света, в основном, связана с тем, что возбужденные молекулы в результате колебательной релаксации успевают растратить колебательную энергию за время, значительно меньшее, чем время жизни возбужденного состояния, и перейти на основной колебательный уровень возбужденного электронного состояния. Переход такой системы в невозбужденное состояние характеризуется испусканием одних и тех же квантов света, т. е. наблюдается один и тот же спектр флуоресценции. В то же время в некоторых случаях это расширяет возможности люминесцентного анализа, давая возможность селективного возбуждения определенных соединений.
Из рис. 5.1 видно, что между спектром поглощения вещества и его спектром флуоресценции можно ожидать определенного сходства, так как и тот и другой определяются, в сущности, одними и теми же электронными переходами. Сходство, действительно, есть, причем, как установлено законом Стокса — Ломмеля, спектр излучения в целом и его максимум всегда сдвинуты в сторону более длинных волн по сравнению со спектром поглощения и его максимумом.
Эта закономерность легко объясняется схемой, изображенной на рис. 5.1, в соответствии с которой энергия квантов флуоресценции должна быть меньше энергии поглощенных квантов на величину энергетических потерь при колебательной релаксации в возбужденном электронном состоянии и переходе на неосновной колебательный подуровень основного электронного состояния. Спектры люминесценции большинства веществ характеризуются широкими полосами-излучения (100...200 нм). Если, однако, вещество исследуют в условиях сильного охлаждения (до температур жидкого азота 77 К или водорода 20 К), спектры становятся квазилинейчатыми с разрешенной колебательной структурой (эффект Шпольского). Этот эффект проявляется не у всех веществ, так как у многих соединений остаются вклады за счет внутримолекулярного и других видов взаимодействия.
5.1.1. Энергетический и квантовый выходы люминесценции
Эффективность преобразования энергии поглощенного света в энергию люминесценции характеризуется энергетическим и квантовым выходами люминесценции отношение излучаемой энергии люминесценции к энергии поглощенного света называют энергетическим выходом люминесценции, а отношение числа излучаемых квантов к числу поглощенных называют квантовым выходом люминесценции.
Если Вэн — энергетический, а Вкв— квантовый выход люминесценции, £л и £с — соответственно энергия люминесценции и и энеРгия поглощенного света, a Nn и 7VC — число испускаемых поглощенных квантов, то очевидно, что
107
Ес ’ Ькв Nc '
Связь между В9Н и Вкв легко установить, если учесть, что энергия N квантов равна E = Nhv:
г> ___ _________ R v„
Оэн--- M L ----- dkb *
Nchvc vc
Зависимость энергетического выхода люминесценции от длины волны возбуждающего света подчиняется закону Вавилова. В соответствии с этим законом энергетический выход люминесценции с увеличением длины волны возбуждающего света сначала возрастает пропорционально длине волны, затем остается постоянным и после достижения некоторой граничной длины волны резко падает.
Учитывая пропорциональность энергетического выхода длине волны возбуждающего света
ВЭц-- kvc
и соотношение А = c/v, получаем
Вкв= В,н —=/г —= хАл= const, (5.1) V, V„ V»
т. е. пропорциональность энергетического выхода длине волны поглощенного света (5.1) означает постоянство квантового выхода люминесценции в этом спектральном интервале. Очевидно, чем больше квантовый выход люминесценции, тем меньшее количество люминесцирующего вещества может быть обнаружено по его свечению.
5.1.2. Интенсивность люминесценции
Интенсивность люминесценции /л пропорциональна числу излучаемых квантов N л:
In= = xBkb/Vc, (5.2)
где к — коэффициент пропорциональности.
Число поглощенных квантов Nc пропорционально интенсивности поглощенного света:
7Vc = xVo-/), (5.3)
где /0 — интенсивность падающего света; / — интенсивность света, прошедшего через раствор; v.' — коэффициент пропорциональности.
Величины 1 и /о связаны уравнением закона Бугера — Ламберта — Бера:
/ = /о1О-е,с. (5.4)
Сочетание (5.3) и (5.4) приводит к соотношению
7Vc=z70(l-10-e'c). (5.5)
108
Подставляя (5.5) в (5.2), получаем
/л = хх'ВД(1-Ю-рЧ (5.6)
Разложение 10 tic в ряд дает
10-е,с= 1 — 2,3е/с + —------(2.|е1с)3 . (5.7)
При е/с^ 10-2 вклад третьего и последующих членов разложения становится пренебрежимо малым. Ограничиваясь при этом условии двумя первыми членами (5.7) и подставляя результат разложения в (5.6), находим
1п — 2,Зхх/Вкв/оь/с
или, объединяя постоянные величины,
In=kc. (5.8)
Линейная зависимость интенсивности люминесценции от кон центрации будет соблюдаться при постоянстве таких факторов, как квантовый выход, интенсивность возбуждающего света и т. д. Также существенным является условие низкой концентрации люминесцирующего вещества.
С увеличением концентрации условие е/с 10-2 будет нарушаться и зависимость интенсивности люминесценции от концентрации будет отклоняться от линейной. При достаточно большой концентрации интенсивность люминесценции вообще может уменьшаться, т. е. может начаться так называемое концентрационное тушение люминесценции. В этой связи верхний предел концентрации раствора в люминесцентном анализе обычно не превышает 10 ...10-4 моль/л.
5.1.3. Тушение люминесценции
Концентрационное тушение люминесценции по С. И. Вавилову в широком интервале концентраций во многих случаях описывается уравнением
В — Воехр[— k(c — Со)],
где В и Во — выход люминесценции при концентрации с и с->0; Со — пороговая концентрация, по достижении которой развивается концентрационное тушение; k — постоянная.
Однако это уравнение оказывается применимым не всегда.
Существенное значение имеет температурное тушение люминесценции, т. е. уменьшение выхода свечения с повышением температуры. В той или иной степени это явление свойственно всем люминесцирующим веществам. Оно объясняется тем, что с повышением температуры увеличивается колебательная энергия молекул и возрастает вероятность безызлучательных переходов, а также вероятность диссоциации возбужденных частиц, происходящая без излучения квантов света.
109
Отрицательное влияние на выход люминесценции оказывают многие примеси. Тушение люминесценции лежит в основе нескольких методик определения примесей.
5.1.4. Люминесценция кристаллофосфоров
Кристаллофосфорами называют сложные неорганические кристаллы, способные люминесцировать. К ним относятся, например, кристаллы на основе сульфидов кальция, стронция и их смесей, сульфата или вольфрамата кальция, бромида или иодида натрия и других соединений.
Некоторые неорганические кристаллы приобретают способность люминесцировать при внедрении в их решетку посторонних элементов, так называемых активаторов.
Люминесценция кристаллофосфоров зависит от многих факторов. При повышении температуры их свечение уменьшается Это тоже проявление температурного тушения люминесценции. Сильное тушащее действие на свечение кристаллофосфоров оказывают ничтожные (10-5...10-6%) примеси многих веществ (например, соединения железа, никеля, кобальта и др.). Эти вещества часто называют люминесцентными ядами. Однако при некоторых условиях и концентрационных соотношениях люминесцентные яды становятся активаторами свечения.
Зависимость интенсивности свечения кристаллофосфора от концентрации используется в анализе.
5.1.5. Атомная флуоресценция
| Атомной флуоресценцией называют фотолюминесценцию газообразных ато-* мов, возникающую при облучении атомов потоком света.
Если поток света содержит кванты, соответствующие возбуждению первого уровня, облучаемый атом перейдет в возбужденное состояние, а затем при возвращении в основное будет испускать свет с длиной волны, соответствующей резонансному переходу. Этот процесс часто называют также р е-зонансной флуоресценцией.
Интенсивность атомной резонансной флуоресценции /фл зависит главным образом от трех основных факторов: интенсивности возбуждающего излучения /с, числа атомов N в основном состоянии в единице исследуемого объема пламени и квантового выхода люминесценции Вкв:
7 фл kl В кв.
где k — коэффициент пропорциональности, учитывающий геометрические факторы и спектральные характеристики линии (ширину, коэффициент поглощения и др.).
Хотя явление атомной флуоресценции изучал еще Вуд в 1905 г., аналитическое применение этот эффект нашел лишь в 1964 г. в работах Вайнфорднера.
по
\
\ 5.2. СХЕМА ПРИБОРА
ДЛЯ ЛЮМИНЕСЦЕНТНОГО АНАЛИЗА
Схема прибора для проведения люминесцентного анализа представлена на рис. 5.2. Свет от источника освещения 1 проходит через светофильтр 2 и падает на кювету 3 с исследуемым раствором. Приемник света 5 измеряет люминесцентное излучение под прямым углом к направлению возбуждающего света. Светофильтр 4 пропускает свет люминесценции и поглощает рассеянный свет от источника возбуждения.
В практике люминесцентного анализа исследуемое вещество обычно освещают ультрафиолетовыми лучами. Наибольшее распространение среди различных источников освещения, вызывающих люминесценцию, получили газоразрядные лампы, чаще всего ртутно-кварцевые и ксеноновые. Приемником люминесцентного излучения может служить глаз человека. В современных приборах для количественного анализа в качестве приемника излучения используют фотоумножители. Для проведения люминесцентного анализа используются флуориметры («Квант», ФЛ-1, ЭФ-ЗМЛ и др.) и спектрофлуориметры (СДЛ 2 и др.).
5.3. КАЧЕСТВЕННЫЙ АНАЛИЗ
Очень чувствительны люминесцентные качественные реакции, когда добавление некоторых органических реагентов к раствору неорганических веществ вызывает яркую люминесценцию. Например, интенсивную люминесценцию вызывает добавление салициловой кислоты к раствору соли цинка, что может быть использовано для его качественного открытия. Для обнаружения лития и алюминия люминесцентным методом предложен 8-окси-хинолин, для открытия бериллия, циркония и других элементов используют морин и т. д. Качественный люминесцентный анализ основан на способности исследуемого вещества в соответ-
ствующих условиях люминесцировать или, реже, гасить люминесценцию. Возникновение или исчезновение люминесценции обычно наблюдается визуально.
Достоинством люминесцентных реакций является их исключительно низкий предел обнаружения. Например, ярко-зеленая люминесценция соединения лития с 8-оксихинолином возникает
при содержании 0,5 мкг Li в 5 мл, медь открывают по яркой синей люминесценции ее соединения с салицилалазином при концентрации 0,25 мкг в 5 мл и т. д. Низким пределом обнаружения и селективностью обладают и так называемые реакции получения кристаллофос-форов (например, реакции образования перлов оуры и фосфатов).
Большое значение имеет люминесцентный анализ в биологии и медицине для диагностики аболеваний рака, малярии и др., контроля за
Рис 5 2 Схема флуориметра
Ill
качеством лекарственных препаратов, анализа биологически активных веществ — витаминов, антибиотиков и т. д.
В сельском хозяйстве и пищевой промышленности люминесцентный анализ используют для определения жизнеспособности семян, анализа пищевых продуктов и т. д. Например, жизне способные семена дают желтую люминесценцию, а нежизнеспособные — коричневую. По цвету люминесценции можно установить сорт муки: чем больше в ней отрубей, тем интенсивнее свечение. Люминесценция позволяет легко обнаружить начальную стадию загнивания различных овощей и фруктов.
5.4. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
Количественный люминесцентный анализ основан на использовании соотношения (5.8), связывающего интенсивность флуоресценции с концентрацией флуоресцирующего вещества. В практике количественного люминесцентного анализа обычно применяется метод градуировочного графика на основе этого уравнения. К настоящему времени разработаны методы количественного люминесцентного определения почти всех элементов при содержании в среднем 10~5%.
Большой интерес вызвало применение люминесцентных индикаторов в титриметрических методах. Люминесцентные индикаторы (а-нафтиламин, акридин и др.) изменяют цвет или интенсивность люминесценции в зависимости от свойств участников реакции, pH раствора или присутствия окислителя. Используя в качестве люминесцентного индикатора, например, морин, можно с погрешностью 5...10 % титриметрически определять алюминий, галлий, цирконий и другие элементы при содержании 1...10 мкг. Медь можно титровать флуорексоном в присутствии никеля, кобальта, железа, марганца и некоторых других элементов в растворах, содержащих 0,01...0,1 мкг Cu/мл. Применение люминесцентных индикаторов позволило решить ряд сложных аналитических задач, связанных, в частности, с анализом мутных и окрашенных сред (фруктовые соки, вина и другие напитки).
Существенно увеличивается интенсивность, люминесценции при замораживании растворов. Это явление используется для количественного определения свинца, висмута и сурьмы в виде галогенидных комплексов. Например, при охлаждении до — 196°С раствор, содержащий свинец в концентрированной НС1, дает фиолетовую люминесценцию Люминесценция наблюдается также при замораживании органических веществ и комплексов металлов с органическими лигандами.
5.5. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
В практике люминесцентных методов значительное место занимает анализ обнаружения. По люминесценции фосфоров и других веществ обнаруживают инфракрасное, ультрафиолетовое,
112
рентгеновское и у-излучение, регистрируют потоки протонов, нейтронов, электронов, а-частиц. Известны люминесцентные способы диагностики различных заболеваний. В оптико-механической промышленности люминесцентный анализ обнаружения используют для маркировки различных сортов стекла, в резиновой промышленности для контроля состава шихты, в бумажной — для установления качества целлюлозы, в алмазодобывающей промышленности по характерному свечению отбирают алмазы и т. д.
Методы люминесцентного анализа успешно используют в анализе лантаноидов, соединений урана и ряда других элементов. Люминесцентной способностью обладают многие органические соединения: бензол, нафталин и их многочисленные производные, биологически активные вещества (витамины, антибиотики, гормоны), многие пигменты и т. д. Благодаря низкому пределу обнаружения и простоте применяемой аппаратуры люминесцентный анализ успешно развивается и является одним из перспективных методов.
5.6. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Важнейшей особенностью люминесцентного метода анализа является его применяемость к определению микропримесей. Погрешность метода составляет 5...7%. Применимость люминесцентного анализа очень широка. Он может быть использован для определения почти любого элемента, многих органических, биологически активных и других веществ.
I Вопросы
1. Какова природа люминесцентного излучения? Объяснить происхождение спектров люминесценции.
2. Чем можно объяснить смещение максимума спектра люминесценции в область больших длин волн по сравнению со спектром поглощения?
3. На чем основан качественный люминесцентный анализ? Привести примеры качественных определений методом люминесценции в технике, сельском хозяйстве, медицине и т. д.
4. От чего зависит интенсивность люминесцентного излучения? Как она связана с концентрацией?
5. В чем сущность следующих приемов люминесцентного анализа: а) метода шкалы; б) метода градуировочного графика; в) метода добавок?
6. Привести примеры количественных определений элементов и веществ методом люминесценции: а) по их собственной люминесценции; б) по свечению их кристаллофосфоров; в) по люминесценции их комплексных соединений с органическими лигандами.
из
7. Как определяют содержание урана по люминесценции его перлов с помощью: а) дозирования пробы; б) окунания перлов?
8. Как определяют содержание свинца: а) по люминесценции замороженных растворов; б) по всплеску люминесцении при размораживании растворов его хлоридных комплексов?
9. Каковы особенности метода люминесцентного титрования? Что представляют собой люминесцентные индикаторы и в каких случаях они применяются? Привести примеры комплексонометрического титрования с использованием металлофлуоресцентных и хемилюминесцентных индикаторов (Zr, Са, Си).
10. В чем состоит сущность определения веществ: а) флуори-метрическим методом (Zr, В); фосфориметрическим методом (Be); в) экстракционно-флуориметрическим методом (Al, Ga, In)?
’ j. Задачи
1. При анализе пробы массой 0,9816 г на содержание кобальта хемилюминесцентным фотографическим методом на одну пластинку снимали свечение пробы анализируемого раствора, стандартов и холостого опыта. В ячейки кюветы помещали по 0,5 мл раствора кобальта, прибавляли салицилат натрия (для устранения мешающего действия катионов Си и Fe) и Н2О2. Затем кювету выдерживали до полного прекращения свечения; пластинку фотометрировали на МФ-2. Значения AS для четырех стандартных растворов, содержащих 4, 8, 12 и 16 мкг Co/мл, составили 0,17; 0,28; 0,40 и 0,53 соответственно. Вычислить массовую долю (%) Сов пробе, если ASX = 0,20.
В хемилюминесцентном фотографическом методе используется зависимость разности почернений пятен в их центрах и фона вблизи холостой пробы (AS) от концентрации определяемого элемента При соблюдении определенных условий эта зависимость близка к линейной
Строим градуировочный график в координатах AS — с (рис 5 3) и по графику определяем сСо= 5,0 мкг/мл Массовую долю (%) кобальта в пробе находим по
формуле
Рис 5.3 Определение кобальта хе милюминесцентным фотографиче ским методом
ч ш(Со)10-4 0.5-5,0
w(Co> =------?п---= -^816
10 ' =
= 2,55-10-*%.
где т(Со) — масса Со, мкг, т — масса навески пробы, г
2. Для определения рибофлавина (витамина В2) методом добавок навеску пищевого продукта массой 0,2000 г растворили и после соответствующей обработки измерили интенсивность люминесценции полученного раствора: /х = 30. После добавления стандартного раствора, содержащего
114
\
40 мкг витамина В2, интенсивность люминесценции увеличилась ' д0 /л+д=80. Определить массовую долю витамина В2 в продукте, если интенсивность люминесценции холостого раствора равна 5.
Результаты люминесцентного анализа по методу добавок рассчитывают по формуле
tnJJx — /о)1О-4
W = 4r-~7^T-
где w— массовая доля определяемого вещества, %; т„ — масса добавки стан дартного раствора определяемого вещества, mki, 1х, 1х + ± и —интенсивность люминесценции исследуемого раствора, того же раствора с добавкой стандарта и холостого раствора соответственно, т — масса навески анализируемого образ
ца, г
Подставляя числовые значения, получаем
w
40(30—5)10V4 (80 — 30)0,2000
0,01 %
3. Содержание самария в диоксиде церия определяли методом добавок. Стандартный раствор нитрата самария приготовили растворением 115,8 мг 5т20з в азотной кислоте. Сухой остаток после упаривания растворили в воде и разбавили до 100,0 мл (раствор А). Затем 0,10 мл раствора А разбавили водой до 100,0 мл (раствор Б).
Навеску СеО2 массой т мг смешали с сульфатом натрия и разделили на две равные части. В одну из них ввели 0,5 мл раствора Б; смесь высушили и прокалили. Интенсивность люминесценции полученных фосфоров (высота пиков, мм) оказалась равной:
Вариант ш, мг 1 200 2 170 3 190
hx. мм 50 40 30
й»+д, мм . 70 60 55
Вычислить массовую долю (%) самария в диоксиде церия. Ответ: I) 6,24^10-4%; 2) 5,87 • 10-4%; 3) 3,15-Ю-4 %.
4. Навеску нефелинового сиенита массой tn г обработали смесью фтороводородной и азотной кислот и разбавили до 100,0 мл.
К аликвоте 10,00 мл прибавили твердый нитрат кальция и при pH 3 ввели избыток ЭДТА для удержания примесей — гасителей в растворе Уран отделили экстракцией раствором трибу-тилфоссЬата в тетрахлориде углерода и реэкстрагировали его из органической фазы в водную раствором триметафосфата натрия.
Для построения градуировочного графика использовали водные растворы нитрата уранила, содержащие 1,0; 3,0; 5,0; 7,0 и 9,0 мкг U/мл соответственно, подвергая их экстракции и ре-экстракции по той же методике. Интенсивность люминесценции стандартных растворов оказалась равной 20, 55, 90, 125, 160
Определить массовую долю (%) урана в минерале, если интенсивность люминесценции анализируемого раствора (Д) соста
115
Вариант т, г . !к .
1 2 3
2,021 1,931 1,899
35 75 120
Ответ: 1) 9,90-10-4%; 2) 2,18-10~3%; 3) З,16-1О~3%
5. При определении свинца в соляной кислоте V мл анализируемого раствора (с = 1,19 г/мл) заморозили жидким азотом, затем записали всплеск люминесценции /* при размораживании раствора.
Аналогично зарегистрировали всплеск люминесценции анализируемого раствора с добавкой 0,40 мл раствора свинца, содержащего 0,02 мкг Pb/мл (Л + д).
Вычислить массовую долю (%) свинца в анализируемой пробе по следующим данным:
Вариант Г, мл...........
/*, делений шкалы . !>+\, делений шкалы
12 3
0,2 0,3 0,4
20 30 25
40 50 60
Ответ: 1) 3,36-10~6%; 2) 3,36-10~6%; 3) 1,20-10“6%.
6. При определении свинца в этиловом спирте анализируемый раствор массой tn г выпарили, сухой остаток растворили в соляной кислоте и измерили интенсивность всплеска люминесценции при размораживании раствора (Л). В аналогичных условиях определили интенсивность I всплеска люминесценции стандартных растворов свинца.
Навеску свинца массой 0,1020 г растворили в концентрированной НС1 и довели объем раствора до 100,0 мл; 1,0 мл этого раствора разбавили до 100,0мл (раствор А). Затем в мерные колбы вместимостью 100,0 мл внесли 0; 0,1; 0,2; 0,3; 0,4; 0,5 мл раствора А,довели до метки и получили для стандартных растворов следующие значения I: 11, 20, 29, 40, 49.
Вычислить массовую долю (%) свинца в спирте по следующим данным, если в кювету для измерений вносили по 1,00 мл раствора:
Вариант 1 2 3
т, г . 0,9082 1,034 1,544
А 15 24 28
Ответ: 1) 1,65- IO”6 %; 2) 2,42-10~6 %; 3) 1,88-10~6 %.
7. При определении циркония по флуоресценции цирконий моринового комплекса после сплавления 0,25 г руды с содой и выщелачивания плава водой к раствору добавили 6 н. НС1 и довели объем до 25,00 мл. В мерную колбу вместимостью 25,00 мл отобрали 2,00 мл анализируемого раствора, добавили тиоглико-левую кислоту для восстановления Fe(III), концентрированную НС1, спиртовой раствор морина и довели водой до метки. Таким же образом приготовили стандартный раствор с содержанием 2 мкг циркония в 25,00 мл. Измерили интенсивность флуоресценции этих растворов: А, = 35 и / = 70. В анализируемом и стандартном растворах растворили одинаковую навес
пб
ку ЭДТА — гасителя флуоресценции только цирконий-мориноьо-го комплекса и вновь измерили интенсивности флуоресценции растворов /Лг = 3,0 и h = 4,0.
Вычислить массовую долю (%) циркония в руде. Ответ: 4,85-10-3 %.
8. Для построения градуировочного графика при флуоресцентном определении магния с люмомагнезоном ИРЕА навеску Mg(NO3)2-6H2O массой 0,1054 г растворили в мерной колбе вместимостью 100,0 мл. Затем 1,00 мл этого раствора разбавили в колбе вместимостью 100,0 мл (раствор А).
Для приготовления четырех стандартных растворов в мерные колбы вместимостью 50,00 мл внесли 1,5; 2,5; 3,5 и 5,0 мл раствора А и раствор люмомагнезона ИРЕА и довели объем до метки. Интенсивность флуоресценции стандартных растворов оказалась равной 37, 56, 75 и 103 соответственно.
Исследуемый раствор объемом 10,00 мл разбавили в колбе вместимостью 100,0 мл. Затем 5,00 мл этого раствора внесли в колбу вместимостью 50,00 мл, добавили раствор люмомагнезона ИРЕА и довели объем до метки (раствор Б).
Построить градуировочный график и определить концентрацию магния (мг/мл) в анализируемом растворе, если интенсивность флуоресценции раствора Б равна /л:
Вариант 1234
1Х 45 71 95 63
Ответ: 1) 3,8-10~3 мг/мл; 2) 6,7-10-3 мг/мл; 3) 9,4Х ХЮ-3 мг/мл; 4) 5,8-10~3 мг/мл.
9. Для люминесцентного определения алюминия в латуни навеску латуни массой tn г растворили и после соответствующей обработки довели объем до 500,0 мл. Затем 10,00 мл этого раствора перенесли в колбу вместимостью 100,0 мл, прибавили раствор кислотного хром-сине-черного и довели объем до метки. Интенсивность люминесценции полученного раствора оказалась равной 1Х. Интенсивность четырех стандартных растворов, содержащих в 100,0 мл 10,0; 20,0; 30,0 и 40,0 мкг алюминия, равна 60, 82, 104 и 126 соответственно.
Построить градуировочный график и определить массовую долю (%) алюминия в латуни по следующим данным:
Вариант 1234
т- г 0,1000 0,0800 0.1600 0,1500
О 72 56 109 91
'о-
Ответ: 1) 0,80%; 2) 0,53%; 3) 1,02%; 4) 0,82%.
Ю. При анализе сточных вод на содержание бериллия пробу объемом 100,0 мл упарили до 1,00 мл и добавили растворы ацетатного буфера и дибензоилметана. После введения в раствор изоамилового спирта отделили органическую фазу и сняли спектр фосфоресценции полученного комплекса бериллия при температуре жидкого азота.
117
Параллельно приготовили такую же пробу с введением 0,2 мкг бериллия и сняли спектр ее фосфоресценции; измерили высоты пиков для обеих проб h, и hBe и получили следующие данные:
Вариант 1 2 3
Лве, ММ . 100 90 75
hx, мм 60 70 50
Вычислить концентрацию бериллия в анализируемом растворе (мкг/мл). Ответ: 1) 3-10~3 мкг/мл; 2) 7-10~3 мкг/мл; 3) 4-10-3 мкг/мл.
11. Навеску диоксида кремния массой tn разложили фтороводородной кислотой, добавили раствор родамина 6Ж и бензола. Интенсивность флуоресценции экстракта измерили по отношению к раствору холостого опыта, проведенного в тех же условиях (Л-/о).
Для построения градуировочного графика к раствору, содержащему 0,05; 0,1; 0,2; 0,3; 0,4 и 0,5 мкг Ta2Os, прибавили родамин 6Ж и экстрагировали бензолом. Измерили флуоресценцию экстрактов по отношению к флуоресценции нулевого раствора и получили (/Ст— /о): 4, 8, 16, 24, 33, 41 соответственно.
Вычислить массовую долю (%) тантала в SiO2 по следующим данным:
Вариант ш, г .
Л - /о
1 2 3
2,356 2,031 1,992
28 20 12
Ответ: 1) 5,21-10-6%; 2) 1,01 -10~5%; 3) 1,44-10“5%.
12. Две навески SiHCls массой по 10,00 г, в одну из которых ввели 0,01 мкг Та2С>5, разложили фтороводородной кислотой, добавили раствор родамина 6Ж и бензол. Интенсивность флуоресценции бензольных экстрактов измерили по отношению к нулевому раствору, содержащему все компоненты, кроме тантала.
Вычислить массовую долю (%) тантала в SiHCb по следующим данным:
Вариант Л - /о Л+л — 7о
1 2 3
12 18 8
18 24 13
Ответ: 1) 61,64-10“7%; 2) 2,46- 1С)-7%; 3) 1,42-10”7%.
13. При анализе технического продукта на содержание Н2О2 хемилюминесцентным методом навеску массой m г растворили в 100,0 мл и в ячейки кюветы отобрали по 0,20 мл исследуемого и стандартных растворов. Кювету поместили в оправку с фотопластинкой и прибавили в каждую ячейку смесь щелочного раствора люминола и сульфата меди. Пластинку фотометриро-вали на МФ-2. Значения AS для четырех стандартных растворов, содержащих 0,4; 0,6; 0,8 и 1,0 мкг Н2О2, составили 0,35; 0,55; 0,77 и 1,00 соответственно.
118
Вычислить массовую долю (%) по следующим данным:
пероксида водорода в пробе
Вариант т, г -Lsv
1 2
1,010 1,082
0,50 0,60
з
1,178
0,70
Ответ: 1) 2,67-1С) 2%; 2) 2,96-1(Г2%; 3) 3,14-1С)-2%.
14. Навеску марганцевого концентрата массой 0,2792 г разложили действием концентрированной соляной кислоты, раствор нейтрализовали аммиаком и разбавили до 25,00 мл. С целью микрофотометрического определения железа к аликвоте объемом 5,00 мл прибавили избыток иодида калия, оттитровали в ультрафиолете 0,01 М NagSgOs и получили следующие данные:
(/(NasSaC))), мл .
Показания шкалы
0,1 0,2 0,3 0,4 0,5 0,6 0,7
1 3 11 18 22 23,5 24
Построить кривую титрования и определить массовую долю (%) железа в концентрате, если единственным веществом, поглощающим при X = 365 мкм, является иод. Ответ: 0,44%.
15. Навеску бронзы массой 0,4612 г растворили в азотной кислоте и провели цементацию меди и других мешающих элементов на металлическом алюминии. Из фильтрата осадили гидроксид железа; осадок растворили в соляной кислоте и довели до метки в мерной колбе вместимостью 25,00 мл.
Определение железа выполнили методом микрофотометрического титрования при облучении ультрафиолетом К 5,00 мл солянокислого раствора соли железа (III), прибавили избыток иодида калия, оттитровали 0,02 М Na2S2C>3 и получили следующие данные:
V(Na2S2O3), мл
I. мкА
0,2 0,4 0,6 0,8 1,0 1,2 1,4
1 7 14 21 24 25 25
Построить кривую титрования И вычислить лю (%) железа в бронзе. Ответ: 1,14%.
массовую до-
Глава 6 РЕНТГЕНОСПЕКТРАЛЬНЫЕ МЕТОДЫ АНАЛИЗА
6.1. РЕНТГЕНОВСКИЕ СПЕКТРЫ
Быстролетящие частицы, например электроны, могут вызывать возбуждение или ионизацию атомов не только в газообразном состоянии. При столкновении электрона с какой-либо твердой поверхностью может также произойти возбуждение или ионизация атомов. Если энергия летящего электрона достаточна, происходит выбивание электрона из внутренних /С- или L-оболочек атомов вещества, подвергающего бомбардировке. На освободившееся место в К- или L-оболочке переходит электрон с более
119
высокого энергетического уровня, что сопровождается характеристическим рентгеновским излучением*.
Кроме характеристического в этом процессе возникает рентгеновское излучение с непрерывным спектром, связанное с частичным превращением энергии тормозящихся электронов при столкновении с мишенью в энергию излучения. Это излучение называют тормозным. Максимальная частота непрерывного рентгеновского излучения vma, связана с напряжением V на рентгеновской трубке соотношением
eV hv niax-
Таким образом, эмиссионный рентгеновский спектр представляет собой непрерывный фон, перекрытый линиями характеристического излучения. Характеристическое рентгеновское излучение наблюдается не только при бомбардировке электронами, оно возникает также при облучении поверхности электромагнитным излучением большой энергии, достаточной для выбивания внутренних электронов из атомов. Излучение непрерывного спектра при этом не происходит, и характеристический спектр, полученный таким способом, называется флуоресцентным или вторичным.
Следует отметить, однако, что внутриатомный переход электрона с верхнего энергетического уровня на К- или L-уровень не всегда сопровождается характеристическим рентгеновским излучением. Возможен и безызлучательный переход. При этом происходит перестройка электронной оболочки и один из внешних электронов отрывается от атома. Этот процесс известен как эффект Оже, а электроны, отрывающиеся от атома, называют оже-электронами. Вероятность проявления эффекта Оже во многих случаях очень велика, а у легких элементов она даже больше, чем вероятность рентгеновского излучения, что вызывает определенные трудности в рентгеноспектральном анализе легких элементов.
Рентгеновские термы. Линии характеристического рентгеновского излучения соответствуют разности энергетических уровней внутренних электронных оболочек атома. Частоты характеристического излучения атомов данного элемента могут быть рассчитаны по уравнению
v = ДД = rf2(^--4) • (6.1)
ft \ nf niJ \ nf ni /
Здесь F — эффективный заряд ядра, равный
F = Z - о,
где Z — заряд ядра, равный порядковому номеру элемента в периодической системе Д. И. Менделеева; о — постоянная экранирования.
* В иностранной литературе это излучение часто называют X-лучами, сохраняя оригинальный термин Рентгена
120
Вместо уравнения (6.1) можно записать
____Я(2-а)2 7?(Z-o)2
V nt ---------------
= Л - т2.
п =4 п=3
п=2
п=1
Величину
-----------оу---------Рис. 6.1. Схема электронных переходов ' —--------_ рентгеновского спектра:
и = I, 2, 3, 4 — главные квантовые числа; называют рентгеновским К. L. М. N — электронные оболочки термом.
Символика, используемая для обозначения и классификации линий рентгеновского спектра, схематично приведена на рис. 6.1. Для учета подуровней у символа линии ставят числовой индекс, например Kav Lp2 и т. д. Число линий в рентгеновском спектре сравнительно невелико. В соответствии с законом Мозли квадратный корень из волнового числа (или частоты) первой К-линии связан с порядковым номером элемента в периодической системе линейной зависимостью:
yV=K(Z-<r). (6.2)
График зависимости Z от /у” представляет собой прямую. С открытием закона Мозли порядковый номер элемента в периодической системе получил четкое физическое истолкование как заряд ядра. Поскольку характеристические линии рентгеновского спектра при переходе от одного элемента к другому смещаются на одну и ту же величину, это свойство позволяет установить, имеется ли между двумя элементами периодической системы какой-то третий, еще неизвестный, и предсказать его рентгеновский спектр. Замечательным практическим применением закона Мозли было его использование при открытии предсказанных Д. И. Менделеевым элементов периодической системы (гафния и рения).
Интенсивность линий рентгеновского спектра зависит от распределения бомбардирующих электронов по скоростям или от распределения интенсивности в спектре возбуждающего излучения ь случае флуоресцентных спектров. При одинаковых условиях интенсивность характеристических линий спектра максимальна, koi да максимальная интенсивность источника возбуждения соответствует энергии возбуждения данной линии. Интенсивность спектра зависит также от числа излучающих атомов, вероятности излучательного перехода и некоторых других факторов. Точная оценка величин, оказывающих влияние на интенсивность спектральной линии, очень сложна. Более надежны данные, ак же как и в оптической эмиссионной спектроскопии, получен-е по относительной интенсивности двух спектральных линий,
121
находящихся в одной и той же области длин волн. Прямая пропорциональность между интенсивностью линии и концентрацией элемента в пробе наблюдается довольно часто.
6.2. ПОГЛОЩЕНИЕ РЕНТГЕНОВСКОГО ИЗЛУЧЕНИЯ
Ослабление рентгеновского излучения при прохождении через пробу подчиняется закону светопоглощения
где /о и I — интенсивность падающего и прошедшего через пробу рентгеновского излучения соответственно; р, — массовый коэффициент поглощения; I — толщина слоя; g — плотность вещества.
Зависимость массового коэффициента поглощения от атомного номера элемента и длины волны падающего излучения в первом приближении выражается соотношением
р, = fez3X3.
6.3. ОСНОВНЫЕ УЗЛЫ РЕНТГЕНОСПЕКТРАЛЬНЫХ
ПРИБОРОВ
Конструкции приборов, используемых в рентгеноспектральных методах анализа, включают следующие основные узлы: источник возбуждения; диспергирующий элемент; приемник излучения (рецептор).
6.3.1. Источник возбуждения
Первичное излучение в рентгеноспектральных методах получают с помощью рентгеновской трубки и реже радиоактивного
Рис. 6.2. Рентгеновская трубка
излучателя.
Рентгеновская трубка. Конструкции трубок весьма многообразны. Принцип их действия иллюстрируется на рис. 6.2. В вакуумированном сосуде под постоянным напряжением в десятки киловольт находятся анод 1 и раскаленный катод 2, между которыми пропускается ток 50... 100 мА. Раскаленный током катод испускает электроны, которые ускоряются электрическим полем и специальным фокусирующим устройством направляются на анод. Бомбардирующий электронный пучок выбивает электроны из внутренней оболочки атомов вещества, пошедшего на изготовление анода. Остальная часть кинетической энергии электронов расходуется на так называемое тормозное излучение и нагревание анода. Возникающий рентгеновский спектр наряду со
122
сплошным фоном тормозного излучения содержит характеристическое излучение элементов, входящих в состав анода. Через выходное окно 3 рентгеновское излучение направляется на диспергирующий элемент или на анализируемую пробу в зависимости от выбранной схемы анализа.
В методах анализа по первичным спектрам анализируемую пробу помещают непосредственно на анод и подвергают действию электронного пучка. Вполне понятно, что при этом собственно анод не должен содержать анализируемых элементов в связи со сложностью введения поправки. В такого рода анализах используются разборные рентгеновские трубки. Применяется также сфокусированный электронный пучок (электронный зонд), имеющий размеры 1...2 мкм2. Он предназначен для проведения локального анализа, например анализа отдельных зерен шлифа или распределения одного или нескольких элементов по поверхности пробы и т. д.
При анализе по вторичным, или флуоресцентным, спектрам в качестве источника излучения используют рентгеновские лучи на выходе из рентгеновской трубки.
Радиоактивные излучатели. Они испускают у-кванты или у-кванты и 0-частицы. Эти излучатели используются непосредственно для бомбардировки пробы или для облучения мишени, испускающей под действием радиоактивного излучения характеристический рентгеновский спектр.
6.3.2. Диспергирующий элемент
В качестве диспергирующего элемента в рентгеноспектральных приборах используют главным образом кристаллы, являющиеся своеобразными дифракционными решетками. Их называют кристалл-анализаторами. Дифракция рентгеновских лучей в кристалле происходит в соответствии с законом Вульфа — Брэгга:
пк — 2rfsin0, (6.3)
где п — целое число, показывающее порядок спектра (обычно ограничиваются рассмотрением спектров первого порядка); d — кратчайшее расстояние между соседними плоскостями кристалла; 0 — угол падения параллельного пучка рентгеновского излучения на плоскость кристалла (его называют углом скольжения).
От плоскости кристалла под углом 0 будет отражаться излучение с длиной волны X, удовлетворяющей условию Вульфа — Брэгга (6.3). Излучение, не удовлетворяющее этому условию, Рассеивается и частично поглощается кристаллом.
Таким образом, в зависимости от угла скольжения данный кристалл будет отражать лучи с разной длиной волны, удовлетворяющей соотношению (6.3). Угол скольжения изменяют поворотом плоскости кристалла-анализатора. На этом принципе раз-
123
работами многочисленные схемы диспергирующих устройств, используемых в практике.
Формула (6.3) является основой рентгеноструктур ного и качественного ренгено спектра льно г о анализа. Если известна длина волны падающего излучения, то по синусу угла 0 можно найти постоянную решетки d, что используется в рентгеноструктурном анализе. Если d известно, то по sin 0 рассчитывают длину волны X и проводят качественный, а затем и количественный рентгеноспектральный анализ.
Выбор кристалла-анализатора определяется свойствами предполагаемого объекта исследования и целью работы. Для проведения, например, рентгеноспектрального элементного анализа желательно иметь яркий спектр не обязательно высокого разрешения. Такие спектры получаются с помощью кристаллов каменной соли, ачюминия и др. Высокой разрешающей способностью обладают кристаллы из кварца, кальцита, а также слюды, флюорита и некоторых других веществ. При выборе учитывается также предполагаемая область длин волн, поскольку в соответствии с уравнением (6.3) при одном и том же угле скольжения 0 длина волны «отраженного» излучения зависит от межплоскостного расстояния в кристалле-анализаторе. Соответствующие характеристики кристаллов хорошо изучены и сведены в специальные таблицы. Например, у кальцита скасо3= 0,302945 нм, у флюорита <Д„р2= 0,3145 нм, у каменной соли dNaci = = 0,281400 нм и т. д У кварца в зависимости от выбора кристаллических плоскостей dSl02 принимает значения 0,424602; 3,33626; 0,245144, 0,117762 и 0,101275 нм.
В области длин волн, превышающих 1,5...2,0 нм, применяются дифракционные решетки.
6.3.3. Приемники излучения
В качестве приемников рентгеновского излучения могут быть использованы фотоматериалы и счетчики рентгеновских квантов: ионизационные и сцинтилляционные. Эти же счетчики применяют для регистрации радиоактивного излучения.
В рентгеноспектральном анализе используют специальные рентгеновские пленки, часто двуслойные. Для повышения чувствительности к рентгеновскому излучению в фотоэмульсию рентгеновских пленок вводят повышенное по сравнению с обычными фотопластинками содержание бромида серебра.
Ионизационные счетчики. Схема ионизационного счетчика представлена на рис. 6.3. Счетчик представляет собой устройство из двух электродов: цилиндрического катода и анода в виде металлической нити, натянутой вдоль оси цилиндра. Пространство в трубке между электродами заполнено газом (например, аргоном) при пониженном давлении. В зависимости от режима работы это устройство может быть ионизационной камерой, пропорциональным счетчиком или счетчиком Гейгера — Мюллера.
124
Действие счетчика основано на ионизации газообразного наполнителя. При небольшом напряжении ток через счетчик не идет (рис. 6.4). Под действием рентгеновского излучения атом аргона ионизирует
Аг + hv = Аг+ (- е~
а образовавшийся электрон при столкновении вызывает ионизацию других атомов аргона. Под действием приложенного напряжения ионы Аг+ будут двигаться к катоду, а электроны — к аноду. Однако при небольшом напряжении скорость движения невелика, и значительная часть ионов успевает рекомбинировать до достижения электродов. Повышение напряжения примерно до Vi (рис. 6.4) приводит к увеличению скорости ионов и уменьшению вероятности рекомбинации. При Vi наступает «насыщение». Дальнейшее увеличение напряжения уже не вызывает увеличения силы тока. При этом напряжении все образовавшиеся ионы
Рис. 6.3. Ионизационный счетчик:
/ — стеклянная колба, 2 — катод, 3 — анодная нить, 4—источник высокого напряжения
Рис. 6.4. Зависимость импульса от напряжения
доходят до электродов, и рекомбинация практически не происходит. Очевидно, при напряжениях V < Vi прибор для измерения интенсивности рентгеновского излучения использован быть не может. В области напряжений от Vi до V2, т. е. в области насыщения, прибор работает в режиме ионизационной камеры. Ионизационная камера служит для измерения рентгеновского излучения сравнительно большой интенсивности (вызывающей 105...106 импульсов в минуту).
Возрастание напряжения на электродах счетчика приводит к увеличению скорости электронов, что вызывает ударную ионизацию. Происходит «газовое усиление» и лавинообразное увеличение числа ионов. Амплитуда импульса (скачок потенциала) в этих условиях меняется пропорционально энергии фотона и составляет 10-4...10~2 В. Прибор, работающий в этой области, называется пропорциональным счетчиком. Область напряжений от V3 до называют областью ограниченной пропорциональности. В этом интервале ионизационные приборы не используются.
125
В области от V4 до Vs попадание в счетчик фотона вызывает лавинообразную ионизацию, не зависящую от энергии фотона. Это гейгеровская область. Прибор, работающий в этом режиме, называют счетчиком Гейгера — Мюл-л е дэ а.
Электроны в счетчике движутся к нити, а положительно заряженные ионы — к цилиндру. Вблизи нити напряженность электрического поля возрастает до таких значений, при которых происходит ударная ионизация и образуется довольно большое число электронов и положительных ионов. Электроны в течение очень короткого промежутка времени, порядка 10~7 с, собираются на нить счетчика. За столь короткое время положительные ионы не могут сколько-нибудь заметным образом сдвинуться с места. Их поле экранирует поле нити, благодаря чему теряется возможность ударной ионизации. По мере удаления слоя положительных ионов от нити их экранирующее действие будет ослабевать и способность счетчика фиксировать появление ионов будет восстанавливаться. Промежуток времени, в течение которого импульс не может быть зарегистрирован, называют «мертвым» временем счетчика. Он имеет длительность примерно НГ4 с.
Через 10~5 с положительные ионы доходят до катода и разряжаются. Разряд этих ионов может сопровождаться ультрафиолетовым излучением и образованием электронов, которые, в свою очередь, генерируют в электрическом поле новые электроны. Таким образом, в счетчике возникает лавинный разряд. Попадание извне новых рентгеновских квантов в такой «горящий» счетчик не может заметно изменить силу тока и, следовательно, не будет зарегистрировано.
В самогасящихся счетчиках к основному наполнителю аргону добавляют некоторое количество (до 10%) паров многоатомных соединений, таких, как этиловый спирт, ксилол и др. Многоатомные молекулы поглощают фотоны и разрушаются без высвечивания, что практически сводит к нулю фотоэффект на катоде. Кроме того, многоатомные молекулы легко отдают свои электроны положительным ионам аргона при столкновениях, так как потенциал ионизации аргона значительно выше:
С2Н5ОН + Аг+ = С2Н5ОН+ + Аг
Кинетическая энергия крупных многоатомных ионов невелика, поэтому выбивания электронов на катоде они не вызывают. Самогашение счетчика достигается, как видно, за счет разрушения и диссоциации многоатомного соединения. Это, естественно, ограничивает срок службы самогасящихся счетчиков.
В последнее время в качестве добавки, вызывающей эффект самогашения в счетчиках, используют хлор и бром. Счетчики с галогенным наполнением работают при низком напряжении (до 400 В) и имеют практически неограниченный срок службы, так как процесс диссоциации молекулы галогена на атомы обратим.
126
Сцинтилляционный счетчик. Действие сцинтилляционных счетчиков основано на измерении сцинтилляций — световых вспышек, появляющихся в сцинтилляторе под действием рентгеновского излучения (рис. 6.5). В качестве сцинтилляторов используют вещества, молекулы которых под действием рентгеновского излучения возбуждаются и, переходя в нормальное состояние, дают вспышку света, которая фиксируется фотоэлектронным умножителем (ФЭУ). Сцинтилляторами могут быть, например, Nal, ZnS, антрацен и многие другие вещества.
Большой интерес к сцинтилляционным счетчикам вызван их более высокой чувствительностью ко всем видам излучений по сравнению с ионизационными, их большой разрешающей способностью (до 1(Г9 с), так как у них нет «мертвого» времени. Кроме того, сцинтилляционные счетчики позволяют измерять энергию излучения.
Рис. 6 5. Сцинтилляционный счетчик:
I — сцинтиллятор, 2 — фотокатод. 3 — фотоэлектронный умножитель, 4, 5, б, 8, 9, 10 — эмиттеры, 7 — анод
Другие приемники рентгеновского излучения. Рентгеновское излучение можно регистрировать также непосредственно фотоэлектронными умножителями (ФЭУ) и фотоэлементами, с помощью кристаллического счетчика и калориметрическим методом. Некоторые металлы и сплавы (например, тантал, сплав меди с бериллием и др.) после специального поверхностного активирования могут быть использованы в качестве катодов ФЭУ для прямого измерения интенсивности рентгеновского излучения. У ФЭУ такого типа окошко открыто, что имеет особую ценность при работе в области мягкого рентгеновского излучения.
Чувствительность обычных фотоэлементов (например, селеновых) к рентгеновскому излучению примерно в 1000 раз меньше, чем к излучению в видимом участке спектра. Для повышения чувствительности поверхность фотоэлемента покрывают составом, способным люминесцировать под действием рентгеновского излучения. Устройства такого типа с успехом применяют в аналитической практике.
Кристаллическим счетчиком называют полупроводниковый монокристалл типа, например, CdS, который при освещении рент-геновским излучением обнаруживает значительное уменьшение
127
сопоставления. Эти счетчики весьма перспективны, так как обладают чувствительностью ионизационных, но не требуют для питания стабилизированного высокого напряжения.
6.4. КОНСТРУКЦИИ РЕНТГЕНОВСКИХ СПЕКТРАЛЬНЫХ ПРИБОРОВ
Конструкции приборов, применяемых в рентгеновском спектральном анализе, различают по типу источников возбуждения, характеристикам диспергирующего элемента и свойствам приемника излучения. Если, например, спектр регистрируется с помощью фотопленки, прибор называют рентгеновским спектрографом, если регистрация ионизационная, — спектрометром. В зависимости от используемой спектральной области приборы подразделяют на длинноволновые и коротковолновые. Сконструированы приборы, предназначенные для работы как с эмиссионными рентгеновскими спектрами, так и по поглощению рентгеновского излучения.
Анализ по первичному рентгеновскому излучению, т. е. по излучению, полученному при электронной бомбардировке анода рентгеновской трубки, в последнее время в значительной степени теряет свое значение. Основную роль играю"" методы, использующие вторичное (флуоресцентное) излучение. Особое место занимают рентгеновские квантометры и рентгеновские микроанализаторы.
Квантометры. Квантометрами называют спектрометры, в которых производится одновременная регистрация нескольких длин волн флуоресцентного излучения. Используют конструкции с прямыми и изогнутыми кристалл-анализаторами, с ионизационными и сцинтилляционными счетчиками. Особо эффективно применение квантометров для экспрессного определения нескольких заданных элементов в серии однотипных образцов. Успешно применяется, например, восьмиканальный квантометр для анализа на семь компонентов. Продолжительность анализа составляет 2,5 мин.
Рентгеновские микроанализаторы (электронный микрозонд). В рентгеновских микроанализаторах электронно-оптическая система формирует электронный луч-зонд диаметром 1...2 мкм2, направляемый на анализируемый образец, вернее в какую-то точку на анализируемом образце или «зерно». Флуоресцентное излучение элементов, входящих в состав зерна, кристалл-анали-затором разлагается в спектр, а детектором определяется интенсивность отдельных линий. Применение электронного зонда позволило решить ряд важнейших задач теоретического и практического характера: найти распределение данного элемента по поверхности образца, определить состав отдельных участков noj верхности и т. д., что имеет особое значение в металлургической промышленности, электровакуумной технологии, геохимии, биологии и т. д.
128
Спектральные приборы абсорбционного анализа. В приборах этого типа измеряется интенсивность рентгеновского излучения, прошедшего через анализируемую пробу, вернее уменьшение интенсивности излучения, связанное с поглощением рентгеновского излучения. Конструкции абсорбционных приборов различаются взаймным расположением анализируемого образца и кристалл-анализатора. В одних приборах после рентгеновской трубки помешен кристалл-анализатор и через анализируемую пробу проходит монохроматическое излучение. В приборах другой конструкции анализируемая проба помещается между рентгеновской трубкой и кристалл-анализатором и таким образом в спектр разлагается излучение, прошедшее через анализируемую пробу.
6.5. КАЧЕСТВЕННЫЙ РЕНТГЕНОСПЕКТРАЛЬНЫЙ АНАЛИЗ
Так же, как и в эмиссионной спектроскопии, качественный анализ рентгеноспектральным методом проводят путем определения длины волны интересующих линий и их последующей идентификации. Длину волны рентгеновской линии в спектре обычно определяют с помощью известных опорных линий, являющихся своеобразными стандартами. В качестве такого стандарта может быть использована или «основа» пробы, как это часто делается в эмиссионной спектроскопии, или известное вещество, специально вводимое в анализируемую пробу. Нередко для этого рядом со спектром анализируемой пробы фотографируют спектр известного стандартного вещества. Методика определения длины волны в этих условиях практически не отличается от той, которая используется в эмиссионной спектроскопии.
Расшифровка рентгеновских спектров также в принципе не отличается от соответствующей методики эмиссионной спектроскопии. Существенно облегчается эта работа благодаря наличию подробных таблиц линий рентгеновского спектра. Хотя рентгеновские спектры намного проще эмиссионных, что заметно упрощает задачу идентификации линий, все же определение их принадлежности тому или иному элементу остается далеко не простым Осложнения вызывают главным образом спектры разных порядков, что приводит к наложению линий. Для надежности определения находят длину волны и оценивают интенсивность не одной, а нескольких спектральных линий. Вполне понятно, что среди них должна находиться наиболее интенсивная линия анализируемого элемента (обычно это Ка- или £а-линия). Чувствительность рентгеноспектрального анализа (предел обнаружения) составляет в среднем 0,05...0,1 %, для некоторых элементов (Ni, Си и др.) он снижается до 5-10-3 %, для других (например, Редкоземельных элементов) повышается до 0,1...0,2 %.
Закон Мозли (6.2) связывает атомный номер элемента с частотой рентгеновского излучения. Особенно примечательным примером использования закона Мозли является открытие новых ^изз „о
элементов периодической системы: гафния в 1922 г. и рения в 1925 г. Вывод о наличии этих элементов в соответствующих концентратах был сделан по их характеристическим рентгеновским спектрам.
Рентгеноспектральный метод имеет ряд существенных достоинств и преимуществ перед другими методами анализа. Рентгеновские спектры малочувствительны к химическому окружению элемента и практически не зависят от того, в виде какого соединения находится анализируемый элемент в пробе. Рентгеноспектральным методом легко обнаруживаются галогены, сера и другие элементы, анализ которых методом эмиссионной спектроскопии не проводится. Большим достоинством рентгенофлуоресцентного метода является возможность анализа образца без его разрушения, что особенно ценно при анализе уникальных изделий.
6.6. КОЛИЧЕСТВЕННЫЙ РЕНТГЕНОСПЕКТРАЛЬНЫЙ АНАЛИЗ
Для проведения количественного анализа может быть использовано как первичное рентгеновское излучение, так и вторичное (флуоресцентное). При использовании первичного излучения порошкообразную пробу обычно втирают в рифленую поверхность анода. Если анализируется металлическая проба, анодом служит анализируемый образец.
Рентгеноспектральный анализ по вторичному (флуоресцентному) излучению имеет существенные преимущества по сравнению с анализом по первичному рентгеновскому излучению. Анализ по флуоресцентному излучению имеет более высокую чувствительность, так как при этом отсутствует фон непрерывного рентгеновского спектра. Немаловажное значение имеет также упрощение экспериментальной методики, поскольку анализируемый образец находится вне вакуумной системы рентгеновской трубки. Правда, интенсивность вторичных спектров меньше, чем первичных, и поэтому, например, фотографическая регистрация здесь не применяется. Однако достаточно высокая чувствительность счетчиков рентгеновских квантов обеспечивает быстрое и точное измерение интенсивности линий.
Количественные определения основаны на пропорциональности между интенсивностью линии характеристического излучения и концентрацией элемента в пробе. На абсолютную интенсивность линий влияют условия возбуждения и другие факторы, а также химический состав пробы, что приходится учитывать серией специальных измерений и теоретическими расчетами. Зависимость интенсивности линий рентгеновского спектра от концентрации элемента имеет более сложный характер, чем концентрационная зависимость интенсивности линий в эмиссионной спектроскопии.
130
В методах внутреннего стандарта сравнивается интенсивность линий определяемого элемента с линией стандартного, специально введенного в пробу элемента в точно известном количестве. Сравниваемые линии должны иметь близкие потенциалы возбуждения, т. е. близкие длины волн и не слишком сильно различаться по интенсивности. Удобным стандартным элементом является соседний элемент периодической системы. Относительная интенсивность линий значительно меньше зависит от состава пробы, условий получения и регистрации спектров и других факторов, чем их абсолютная интенсивность. Отношение интенсивностей линий определяемого элемента и элемента-стандарта предполагается пропорциональным их концентрации:
~T=k^ (6-4)
1 ст 15 ст
где 1Х и /ст — интенсивности линий определяемого и стандартного элементов; сх и сст — их концентрации.
Ввиду невозможности точного учета всех факторов коэффициент k определяется эмпирически по интенсивности линий стандартных образцов. Уравнение (6.4) является также основой градуировочного графика.
В анализе по методу внешнего стандарта интенсивность линии определяемого элемента сравнивается с интенсивностью этой линии в спектрах стандартных образцов с известным содержанием. Отношение интенсивностей линий принимается равным отношению концентраций элемента. Точные результаты получаются при условии, когда состав анализируемой пробы и стандартных образцов по основным компонентам достаточно близок, так как интенсивность линий зависит от общего состава пробы, в особенности от наличия так называемых мешающих элементов.
Успешно применяется метод добавок, например, при анализе лантаноидов.
Разработаны также методы анализа, основанные на поглощении рентгеновского излучения. В основе определения лежит метод градуировочного графика.
6.7. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Рентгеноспектральные методы анализа имеют разнообразные области применения. В геологии, горном деле, металлургии и гидрометаллургии этим методом определяют состав минералов, руд, и продуктов их переработки — шлаков, концентратов и т. д., Устанавливают состав легированных сталей и сплавов, в химических отраслях промышленности (электрохимии, нефтехимии и т. д.) анализируют исходное сырье и готовую продукцию, в ядерной технике контролируют изменения в составе замедли-телей, теплоносителей и т. д. Широко используются рентгеноспектральные методы для анализа керамики, стекла, пластмасс, разивов, катализаторов и других материалов сложного хими
5*
131
ческого состава. Весьма эффективным оказалось применение рентгеноспектрального флуоресцентного анализа для контроля за загрязнением окружающей среды (определение содержания различных элементов в аэрозолях, почвах, воде, растительных и животных тканях и т. д.). Например, в аэрозолях определяют до 40 элементов от натрия до свинца. Рентгеноспектральный анализ показал, в частности, что источником загрязнения атмосферы кальцием служит цементная промышленность, свинцом и бромом — выхлопные газы автомобилей и т. д.
Широкое применение нашел рентгеноспектральный метод определения толщины покрытий — тонкого слоя, нанесенного на основной материал, как, например, цинка на оцинкованном железе, слоя ферропорошка на магнитофонной ленте и т. д. Метод основан на использовании градуировочных графиков, показывающих зависимость интенсивности спектральной линии от толщины покрытия. Градуировочный график строится по стандартам с известной толщиной слоя.
Очень эффективным оказалось применение рентгеновского флуоресцентного метода в анализе космических объектов. С помощью спектрометрической аппаратуры РИФМА (рентгеновский изотопный флуоресцентный метод анализа), установленной на «Луноходе-1», было определено содержание основных породообразующих элементов непосредственно на поверхности Луны. Для возбуждения флуоресцентного излучения применялись радиоактивные источники, характеризующиеся высокой стабильностью и не нуждающиеся в электрической энергии. В качестве детектора использовались пропорциональные счетчики. Электрический импульс счетчика преобразовывался и по радио передавался на Землю.
Широко известны и весьма важны применения рентгеновского излучения во многих других областях науки и техники (определение структуры кристаллов, рентгеновская дефектоскопия изделий, диагностика заболеваний и т. д.).
6.8. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Методами рентгеноспектрального анализа определяют состав различных сплавов, руд, минералов, цементов, пластмасс и многих других изделий, устанавливают характер загрязнений окружающей среды, анализируют космические объекты и т. д. Он используется для определения больших содержаний (десятки процентов) и небольших примесей (10-2...10-3 %).
Предел обнаружения рентгеноспектральными методами в общем ограничивается величинами порядка 10-2...10~3 %. Сочетание с химическими методами обработки позволяет его значительно снизить. Средняя квадратичная погрешность методов составляет примерно 2...5 %, при благоприятных условиях она снижается до ±0,5%. Рентгеноспектральный анализ легко автоматизируется.
132
| Вопросы
1. Назвать основные узлы рентгеноспектральных приборов и указать их назначение.
2. Привести схему устройства рентгеновской трубки н объяснить принцип ее работы.
3. Записать уравнение закона Вульфа — Брэгга. Для решения каких основных задач оно применяется?
4. Записать уравнение закона Мозли. Указать его теоретическое и практическое применение.
5. Какие приемники излучения используются в рентгеноспектральном анализе?
6. Пояснить принцип работы ионизационного счетчика.
7. Какие особенности имеет рентгеновский микроанализатор (электронный микрозонд) ?
8. Как проводится качественный рентгеноспектральный анализ?
9. Какие основные приемы используются в количественном рентгеноспектральном анализе?
10. Какое практическое применение имеет рентгеноспектральный анализ?
И. Охарактеризовать достоинства и недостатки рентгеноспектрального анализа.
Глава 7
ДРУГИЕ СПЕКТРАЛЬНЫЕ И ОПТИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
7.1. АНАЛИЗ ПО СПЕКТРАМ КОМБИНАЦИОННОГО РАССЕЯНИЯ
Комбинационное рассеяние света было открыто в 1928 г. советскими физиками Л. И. Мандельштамом и Г. С. Ландсбергом и одновременно индийским физиком В. Раманом*.
7.1.1. Спектры комбинационного рассеяния
Взаимодействие электромагнитного излучения с веществом не всегда проявляется в поглощении квантов энергии и переходе молекулы на новый квантованный энергетический уровень. Другие эффекты наблюдаются, когда частота облучающего электромагнитного излучения значительно отличается от частоты электронного перехода в спектре данного вещества. Упрощенная и. В иностранной литературе комбинационное рассеяние света называют эффектом Рамана.
133
энергетическая схема этого взаимодействия представлена на рис. 7.1.
При взаимодействии электромагнитного излучения с веществом происходит поляризация молекулы, ее деформация в поле электромагнитной волны, когда электроны молекулы сдвигаются в одну сторону, а положительно заряженные ядра — в другую. Молекула приобретает энергию £, но это не значит, что она переходит в какое-то возбужденное состояние в квантовом смыс-
Рис 7 1 Энергетическая схема пронсхожде ния спектра комбинационного рассеяния Го. Г, и V2 — колебательные уровни энергии, Е — энергия падающего излучения hv. г — частота
ле этого слова. Этот процесс на рис. 7.1 обозначен как 7 и 1'. Процесс 1 относится к взаимодействию с электромагнитным излучением молекулы, находящейся на основном колебательном уровне Vo, а процесс Г — к взаимодействию молекулы, находящейся на возбужденном уровне Vi. Индуцированный таким образом диполь осциллирует и молекула из поляризованного состояния возвращается в обычное. Этому процессу соответствует переход 2 (рис. 7.1, а) или 2' (рис. 7.1, б). Возвращение молекулы на тот же самый колебательный уровень, на котором она находилась ранее (Vo в процессе 2 или Vi в процессе 2'), сопровождается испусканием электромагнитного излучения с частотой v, совпадающей с частотой падающего излучения. Процессы 2 и 2' характеризуют классическое рэлеев
ское рассеяние.
При комбинационном рассеянии молекула из поляризованного состояния возвращается не на первоначальный, а на какой-то другой энергетический уровень. Энергия осцилляции диполя в этом случае будет отличаться от Е. Если сначала молекула находилась на уровне Vo, а возвратилась на уровень Vi (рис. 7.1, а, процесс <?), то очевидно, что энергия осцилляции Es будет меньше Е на столько же, на сколько энергия возбужденного уровня Vi (£vi) отличается от энергии основного уровня Vo(Erc):
или
и
Е Vl — Е v0 — АЕ = Е — Е$ hw= hvE — hvs
vs = ve— Vv-
(7.1)
(7.2)
(7.3)
Частота комбинационного рассеяния v$ меньше, чем частота возбуждающего излучения. Эту частоту v$ (или линию в спектре) называют стоксовой или красным спутником.
134
На рис. 7 1,6 показана схема взаимодействия с электромагнитным излучением молекулы, находящейся на возбужденном колебательном уровне К, а после осцилляции возвращающейся на основной уровень (процесс 3'). Путем аналогичных рас-суждений можно установить, что
= Es - Е
и
v's = Ve— Vv. (7.4)
Линия комбинационного рассеяния в этом случае будет находиться в области больших частот, чем падающего излучения. Это антистоксова частота (линия),или фиолетовый спутник.
Разность частот в формулах (7.3) и (7.4) одинакова по абсолютной величине и отличается по знаку, поэтому линии комбинационного рассеяния будут находиться слева и справа на одинаковом расстоянии от линии падающего излучения. Схема взаимного расположения линий в спектре комбинационного рассеяния показана на рис. 7.2. Очень важной особенностью спектров комбинационного рассеяния является независимость разности V£± vy от частоты возбуждающего излучения.
При комнатной температуре большая часть молекул находится на основном невозбужденном уровне, поэтому процессы 1 и 3 (см. рис. 7.1, а) более вероятны, чем 1' и 3' (см. рис. 7.1, 6). В соответствии с этим линия комбинационного рассеяния с частотой v£—vy (стоксова) будет более интенсивна, чем линия с частотой Vf+vy (антистоксова). С повышением температуры число возбужденных молекул увеличивается и интенсивность антистоксовых линий возрастает.
| Интенсивность линий комбинационного рассеяния также пропорциональна интенсивности падающего света.
Эффект комбинационного рассеяния может быть вызван не только колебательными переходами, но также вращательными и электронными, однако для аналитических целей обычно используют колебательные переходы.
7-1.2. Схема установки
Для наблюдения спектров комбинационного рассеяния кювету с исследуемой жидкостью или раствором освещают источником линейчатого спектра и под прямым углом изучают спектральный состав рассеянного света. Нужный спектральный участок для освещения пробы обычно выделяют с помощью свето
i)-9 >) 1+i> 9
£ V Е E V
Рис. 7 2 Схема спектра комбинационного рассеяния света
135
фильтра. К возбуждающей спектральной линии осветителя предъявляется несколько требований: она не должна находиться в области спектральной полосы поглощения анализируемого вещества, не должна вызывать его флуоресценцию и т. д. В значительной степени этим требованиям отвечает излучение различных видов ртутных ламп, которые и стали наиболее употребительными источниками освещения в спектроскопии комбинационного рассеяния. Чаще всего работа ведется с использованием яркой синей линии в спектре ртути, имеющей длину волны 435,8 нм. В последнее время значительно расширяется применение в качестве источников освещения газовых и иных лазеров — гелиево-неонового (632,8 нм), аргонового (438,0 и 514,5 нм) и т д. Применение лазеров для этой цели имеет ряд достоинств: оно позволяет, например, существенно увеличить интенсивность комбинационного излучения и уменьшить массу вещества для анализа до нескольких миллиграммов.
Спектральный состав рассеянного света исследуется с помощью различных спектральных приборов, имеющих достаточно большую светосилу и дисперсию при малом количестве паразитного света. Часто применяют, например, трехпризменный спектрограф ИСП-51 с фотографической регистрацией спектра. Для исследования спектров комбинационного рассеяния и аналитических определений также используют спектральные приборы с дифракционной решеткой в качестве диспергирующего элемента и фотоэлектрическим измерением интенсивности излучения (спектрометры типа ДФС-12, ДФС-24 и т. д.).
7.1.3. Качественный анализ по спектрам КР
Как и в эмиссионном спектральном анализе, в спектроскопии комбинационного рассеяния наличие вещества в анализируемом растворе устанавливается по характерным линиям в спектре. Принадлежность линии тому или иному веществу определяется по разности волновых чисел рассматриваемой и возбуждающей линий, которая не зависит от волнового числа возбуждающего излучения, и по интенсивности линии комбинационного рассеяния. Данные по характеристикам спектра КР (комбинационного рассеяния) многих веществ имеются в соответствующих таблицах. Чувствительность анализа по спектрам КР не очень велика и не относится к числу преимуществ метода. Основным достоинством спектроскопии КР является возможность анализа сложных многокомпонентных смесей и веществ, близких по строению и составу. Методами спектроскопии КР анализируют смеси парафиновых и ароматических углеводородов, нафтенов, олефинов и т. д.
136
7.1.4. Количественный анализ по спектрам КР
Методы количественного анализа по спектрам КР основываются на использовании линейной зависимости между интенсивностью линии / и концентрацией вещества с:
1 - kc.
Если, например, анализируется бинарная смесь веществ А и В, то очевидно, что
I а= k/Л а;
ZB=kBXB, (7‘5)
где X — доля вещества А или В.
При строгой стандартизации условий получения спектра и обработки данных коэффициент пропорциональности можно определить по интенсивности линий чистых исходных веществ Из отношения уравнений (7.5) при ХА= 1 иХв=1 находим отношение коэффициентов пропорциональности:
Отношение интенсивностей (/а/7в)х для анализируемой смеси находится экспериментально. Оно связано с отношением концентраций
откуда
*а \ = _1_ (ЬЛ хъ) k \1ЪГ
(7.6)
Кроме того, для бинарной смеси
Ха+Хв=1. (7.7)
Совместное решение уравнений (7.6) и (7.7) дает состав бинарной смеси:
v (/a/Zb)
А (/д//в)+Л ’
(7-8)
Такой прием можно применить при анализе и более сложных смесей, чем бинарные. Для этого следует попарно скомбинировать отдельные компоненты смеси, чтобы найти коэффициенты пропорциональности, и после экспериментального определения интенсивностей выбранных линий в спектре пробы решить совместно уравнения типа (7.6) и (7.7). Коэффициент пропорциональности k часто определяют по интенсивности линий в спектрах эталонных проб известного состава. Используется также метод добавок и метод градуировочного графика. Погрешность анализа по спектрам КР обычно не превышает ± 3 % относительных.
137
7.2. РАДИОСПЕКТРОСКОПИЯ
Интенсивное применение наиболее длинноволновой части электромагнитного спектра — микроволн и радиоволн в физико-химических исследованиях и аналитической химии — началось сразу после открытия явлений электронного и ядер-н ого магнитного резонанса. Эти явления отражают взаимодействие молекулы с магнитным полем. Электронный парамагнитный резонанс (ЭПР) характеризует взаимодействие с магнитным полем магнитного момента электрона. Явление ядер-ного магнитного резонанса (ЯМР) отражает взаимодействие с полем магнитного момента ядра. Оба явления основаны на эффекте Зеемана, заключающемся в расщеплении спектральных линий или уровней энергии в магнитном поле на отдельные компоненты.
7.2.1. Ядерный магнитный резонанс (ЯМР)
Явление ЯМР открыли в 1946 г. американские физики Ф. Блох и Е. Переел. Если элемент обладает нечетным порядковым номером или изотоп какого-либо (даже четного) элемента имеет нечетное массовое число, ядро такого элемента (изотопа) обладает спином, отличным от нуля. Очевидно, у изотопов четных элементов с четным массовым числом спин от нуля не отличается. Например, изотоп углерода |2С с массовым числом 12 спином не обладает, а изотоп |3С имеет спин, равный ’/2. Наличие неспаренного спина у |3С вызывает появление у него ядерно-го магнитного момента, в то время как ядра изотопов |2С магнитного момента не имеют. В соответствии с этим внешнее магнитное поле не будет оказывать влияния на хаотическое распределение по энергии ядер |2С, но будет влиять на распределение ядер |3С, снимая вырождение энергетических уровней.
Спин ядра, равный 1 /г, соответствует двум возможным ориентациям вектора магнитного момента ядра в магнитном поле — по полю (т.]= ‘/2) и против поля (ш/=— '/г)'. при этом состояние т,=— ‘/2 обладает во внешнем поле несколько более высокой энергией, чем состояние т,= ‘/2. Энергия перехода между этими состояниями равна
ДЕ = 2|i/7q,
где |i — магнитный момент ядра; Но — напряженность внешнего магнитного поля.
Число частиц на каждом из этих уровней может быть подсчитано по закону распределения Больцмана:
^ = ехр(-^)=.хр(-», (7.9>
где п\ и п2 — число частиц, находящихся на нижнем и верхнем энергетических уровнях и обладающих энергией соответственно
138
£4 и £2- Расчет по уравнению (7.9) показывает, что отношение П1/П2 в области обычных температур начинает отличаться от единицы только в шестом знаке после запятой. Таким образом, при обычной температуре заселенность обоих уровней будет примерно одинакова с очень небольшим преобладанием состояний, имеющих меньшую энергию. Если такую систему, находящуюся в магнитном поле напряженностью Но, поместить в переменное электромагнитное поле с частотой v0, чтобы энергия кванта hv0 совпадала с энергией перехода 2цН0, т. е. чтобы
hv0 = 2nH0, (7.10)
то вследствие поглощения энергии поля ядра с нижнего энергетического уровня будут переходить на верхний.
Важной характеристикой свойств ядер является гиромагнитное отношение у, равное отношению магнитного момента ядра к его механическому вращательному моменту:
у = 4лц//г. (7.11)
Сочетание уравнений (7.10) и (7.11) дает
Y = или vo = -
Ядра с гиромагнитным отношением у, находящиеся в однородном магнитном поле напряженностью Яо, под воздёйствием переменного поля с частотой v0 переходят в состояние с более высокой энергией. Этот переход называют магнитным резонансом или магнитным резонансным переходом. Гиромагнитное отношение характеризует частоту и напряженность поля, удовлетворяющие условию резонанса. Частоту Vo называют резонансной.
Из возбужденного состояния в нормальное ядра могут возвращаться, передавая энергию возбуждения окружающей среде — «р е ш е т к е», под которой в данном случае понимаются электроны или атомы другого сорта, чем исследуемые. Этот механизм передачи энергии называют спин-решеточной релаксацией, его эффективность может быть охарактеризована постоянной Т1, называемой временем спин-решеточной релаксации.
Возбужденное ядро может также передать энергию возбуждения ядру такого же сорта, находящемуся в низшем энергетическом состоянии. Этот процесс называют спин-спиновой Релаксацией и характеризуют величиной — временем спин-спиновой релаксации. Последний процесс не приводит к изменению числа возбужденных ядер, однако этот механизм передачи энергии очень важен для понимания некоторых явлений. Кроме постоянных Т\ и Г2 в практике используют и другие характеристики резонансного поглощения.
Совокупность сигналов ЯМР, т. е. зависимость интенсивности °тлощения от напряженности магнитного поля (или от часто
139
ты), называют обычно спектром ЯМР. Основными его характеристиками являются высота (максимальная интенсивность) и ширина, измеренная на половине максимальной высоты сигнала.
Конечная ширина сигнала ЯМР показывает, что резонансное поглощение происходит не строго при одной фиксированной частоте, как можно было ожидать на основании (7.10), а захватывает некоторый интервал частот. Ширина сигнала зависит от индивидуальных особенностей, структуры, агрегатного состояния вещества и других факторов.
Теория ЯМР связывает ширину сигнала Av с временем спин-решеточной и спин-спиновой релаксации следующим соотношением:
л 1 । I
Av=-----------.
2ПТ| 2ЯТ2
Кроме того, на ширину сигнала ЯМР влияет неоднородность магнитного поля в разных точках образца, что предъявляет соответствующие требования к измерительному прибору.
Химический сдвиг. Величина Но в уравнении (7.10) характеризует резонансное поглощение по сути дела свободного ядра, лишенного электронной оболочки. Однако ядра одного и того же элемента в разных молекулах при наложении одной и той же частоты vo показывают резонансное поглощение при различной напряженности поля. Если резонансное поглощение частоты v0 ядрами какой-то молекулы наблюдается при напряженности поля И, то
Ьо==2ц/7о=2|1Я(1-а). (7.12)
Здесь Но следует рассматривать как резонансное магнитное поле свободного ядра. Величину а называют константой экранирования. Она характеризует сдвиг резонансной частоты или поля, вызванный ближайшим окружением ядра, т. е. так называемый химический сдвиг. Химический сдвиг практически измеряют по отношению к некоторому стандарту. Если Нх и НСт напряженности поля, при которых происходит резонансное поглощение ядрами исследуемого и стандартного веществ соответственно, то уравнение (7.12) будет иметь вид:
Но=Нх(1—ах);
Яо=ЯсХ1~Ост).
Почленное деление этих уравнений дает
1 Пет Их
1 Ох Н сх
(7.13)
При вычитании по единице из обеих частей уравнения (7.13) после небольших преобразований получаем
Ох Ост И х Нхх /у 14)
1 -Ох ~ Н„ • V
140
Так как то вместо (7.14) можно записать
О*-Оет=6= Н’Т,Н" (7.15)
г? ст
Величину 6 называют обычно химическим сдвигом и выражают в условных единицах — миллионных долях магнитного поля (м.д.), умножая на 10®, т. е.
” ст
6=—~Яст106м.д. (7.16)
/7 ст
Если измеряется не поле, а частота, вместо (7.16) имеем
- V-T10® м. д.,
Vct
где vx и vCT— резонансные частоты исследуемого образца и стандарта.
Как видно из уравнения (7.15), исследуемый химический сдвиг является разностью постоянных экранирования исследуемого вещества и стандарта. Точные измерения химического сдвига требуют введения различных поправок, например, на разницу диамагнитных восприимчивостей исследуемого образца и эталона, на растворитель и т. д. Хотя метод ЯМР можно использовать для изучения очень многих ядер, наиболее важное значение имеют исследования на ядрах *Н, ,9F и 31Р, к которым относится большинство выполненных экспериментальных работ. Наиболее подробно изучен ЯМР протонов, входящих в различные соединения. Название ЯМР протонов часто сокращают как ПМР — протонный магнитный резонанс.
Схема ЯМР спектрометра. Уравнение (7.10) показывает, что резонансное поглощение может быть достигнуто или изменением напряженности магнитного поля Но при постоянной частоте, или изменением наложенной частоты в постоянном магнитном поле. Достоинством обычно применяемых приборов, в которых условия резонанса достигаются за счет изменения напряженности магнитного поля, является удобство и простота работы, так как стабилизировать частоту проще, чем поле. Тем не менее иногда предпочитают изменять частоту при постоянном поле, так как это позволяет перекрывать более широкую область энергии и решать другие задачи. Принципиальная схема спектрометра для наблюдения ЯМР представлена на рис. 7.3.
Ампулу с исследуемым веществом помещают в катушку радиочастотного генератора, которая находится между полюсами электромагнита. В приборах со стабилизированной частотой и переменным магнитным полем изменение магнитной индукции осуществляется генератором (на схеме не указан). При выполнении условия (7.10), т. е. при поглощении энергии поля, детектор регистрирует некоторое изменение напряжения в контуре.
141
которое записывается в виде сигнала ЯМР на самопишущем потенциометре или наблюдается на экране осциллографа.
Спектрометр ЯМР содержит сложный набор электронных устройств, предназначенных для обеспечения высокой стабильности и точности задаваемых параметров поля. Для успешной работы прибора необходимо поддерживать частоту и напряженность магнитного поля с погрешностью порядка 10-6...10“7%
Качественный анализ и структурные исследования методом ЯМР. На рис. 7.4 приведен спектр ЯМР протонов этилового спирта. Как видно, протоны —ОН-, =СН2-, —СНз-групп различаются очень четко. Таким образом, по табличным значениям резонансных сдвигов или по данным предварительной калибровки можно установить наличие тех или иных атомных группировок в исследуемой молекуле, т. е. получить информацию о ее структуре, а по площади пика определить число ядер. Применение метода ЯМР позволило установить структуры многих сложных соединений. Это один из основных методов исследования
Рис. 7.3 Схема ЯМР-спектрометра: 1 — магнит, 2 — исследуемое вещество, 3 — детектор. 4 — генератор радиочастоты
Сила поля
Рис 7 4 Спектр ЯМР
этанола
в органической химии и химии координационных соединений. Методом ЯМР исследуется также структура кристаллов, кинетика быстрых реакций и многие другие свойства веществ и характеристики реакций.
Количественный анализ методом ЯМР. При количественном анализе растворов площадь пика может быть использована как мера концентрации в методе градуировочного графика или методе добавок. Известны также методики, в которых градуировочный график отражает концентрационную зависимость химического сдвига.
Применение метода ЯМР в неорганическом анализе основано на том, что в присутствии парамагнитных веществ происходит укорочение времени ядерной релаксации. Скорость релаксации ядер в присутствии парамагнитных веществ выражается уравнениями
v(=fe(c; V2=k^c, (7.17)
где Vi и V2 — скорость соответственно спин-решеточной и спин-спиновой релаксации данных ядер; ki и k2 — коэффициенты релаксационной эффективности парамагнитных частиц по отно
142
шению к данным ядрам, причем индекс «1» относится к спин-решеточной, а индекс «2» — к спин-спиновой релаксации; с — концентрация парамагнитных частиц в растворе.
Коэффициенты /г( и /г2 зависят от природы анализируемых парамагнитных частиц и изучаемых ядер, от растворителя, температуры и некоторых других факторов. По физическому смыслу коэффициенты релаксационной эффективности соответствуют скорости релаксации ядер при концентрации анализируемых парамагнитных частиц в 1 моль/л, так как при с=1 моль/л из уравнений (7.17) следует, что Vi = ki и v2 = k2. Числовые значения обоих коэффициентов для парамагнитных аква-ионов примерно одинаковы и изменяются от нескольких десятков до 104 (например, /гСц2+= 103; ^Мг,!+— 104).
Опыт показал, что температурная зависимость коэффициентов релаксационной эффективности невелика и в области температур 15...30 °C не превышает 1...2% на градус, оставаясь во многих случаях меньше этого значения. Так же незначительно влияет на коэффициенты k\ и й2 присутствие в растворе диамагнитных солей. Это существенно упрощает разработку аналитических методик, позволяя, например, обходиться без химического отделения диамагнитных примесей.
Измерение скорости релаксации может быть выполнено несколькими методами. Надежным и универсальным является, например, импульсный вариант метода ЯМР, или, как его обычно называют, метод спинового эха. При измерениях по этому методу на исследуемый образец в магнитном поле через определенные промежутки времени накладывают кратковременные радиочастотные импульсы в области резонансного поглощения. В приемной катушке появляется сигнал спинового эха, максимальная амплитуда которого связана с временем релаксации простым соотношением.
Для проведения обычных аналитических определений нет необходимости находить абсолютные значения скоростей релаксации. В этих случаях можно ограничиться измерением какой-либо пропорциональной им величины, например амплитуды сигнала резонансного поглощения. Измерение амплитуды может быть выполнено на простой, более доступной аппаратуре.
Существенным достоинством метода ЯМР является широкий интервал значений измеряемого параметра. С помощью установки спинового эха можно определять время релаксации от 10-5 до ЮО с с погрешностью 3...5 %. Это позволяет определять концентрацию раствора в очень широком интервале от 1...2 до 10-6..
10 7 моль/л. Наиболее часто используемым аналитическим приемом является метод градуировочного графика.
В настоящее время разработаны методики прямого аналитического определения многих парамагнитных ионов по скорости Релаксации протонов и ядер фтора (19F) (ионы элементов середины IV периода таблицы Д. И. Менделеева, лантаноиды и т. д). собый интерес представляет применение релаксационного мето-
143
да для определения концентрации парамагнитных ионов в движущейся жидкости и для дистанционного определения парамагнитных веществ в растворе, что позволяет контролировать концентрацию парамагнитных веществ в ходе, например, технологического процесса.
Помимо прямых определений, основанных на уравнении (7.17), магнитно-релаксационный метод успешно используется для разработки тит-риметрических методов. В этих методах используется линейная зависимость скорости магнитной релак-
Рис 7 5. Типы кривых магнитно-релаксационного титрования. / — титрование меди купферроном, 2 — перманганатометрическое определение Fe2+
сации ядер от концентрации парамагнитных веществ в растворе. Кривая титрования будет представлять, очевидно, зависимость скорости релаксации ядер (в условных единицах) от объема добавленного титранта. Некоторые типы кривых титрования представлены на рис. 7.5.
Кривая 1 на этом рисунке отражает изменение скорости релаксации, когда в результате реакции титрования парамагнитный ион осаждается, образуя диамагнитное соединение. Такой вид имеет, например, кривая титрования иона меди (II) раствором купферрона. Точка эквивалентности соответствует излому на кривой титрования.
Вид кривой титрования практически также не изменится, если определяемый парамагнитный ион при титровании будет образовывать не осадок, а диамагнитное комплексное соединение в растворе. Иллюстрацией такого определения может быть, например, титрование Fe3+ раствором ЭДТА.
Кривая 2 на рис. 7.5 показывает возрастание скорости релаксации в ходе титрования. Этот случай реализуется, например, при титровании иона Fe2+ перманганатом:
5Fe2+ + МпОг + 8Н+=5Fe3+ + Мп2+ + 4Н2О
В результате этого титрования образуются ионы Fe3+ и Мп2+, обладающие более высоким коэффициентом релаксационной эффективности, чем вступающие в реакцию ионы Fe2+, что и вызывает увеличение скорости релаксации протонов в ходе титрования После достижения точки эквивалентности скорость релаксации остается постоянной. Разработаны также методики титри-метрического определения диамагнитных веществ парамагнитным титрантом и другие оригинальные методики.
Основное достоинство метода ЯМР — возможность определять концентрации в широком интервале от 10“5...10-6 до 1-...2 моль/л и выше, используя для анализа небольшие объемы раствора (0,1...0,5 мл). Погружения каких-либо датчиков в анализируемый раствор не требуется. Измерение ЯМР дает воз
144
можность контролировать концентрацию неустойчивых парамагнитных частиц в растворе, образующихся, например, в результате какой-либо реакции. Большой интерес представляют перспективы автоматизации контроля с помощью ЯМР, так как метод позволяет проводить дистанционные определения в движущейся жидкости без отбора проб анализируемого раствора. Ценной особенностью метода является возможность анализа интен сивно окрашенных и мутных растворов в присутствии кислот, щелочей, поверхиостно-активных веществ и т. д.
Однако при всех его многочисленных достоинствах магнитнорелаксационный метод еще недостаточно используется в аналитической практике главным образом в связи со сложностью и малой доступностью аппаратуры.
7.2.2. Электронный парамагнитный резонанс (ЭПР)
Электронный парамагнитный резонанс (ЭПР) открыт в 1944 г. советским физиком Е. К. Завойским. В методе ЭПР, так же как и в ЯМР, используется резонансное поглощение электромагнитных волн веществом в постоянном магнитном поле. Однако ЭПР связан уже с магнитными свойствами электрона. Магнитное поле электрона примерно на три порядка превышает поле ядра. В отсутствие магнитного поля спиновые энергетические состояния электрона вырождены. При наложении магнитного поля вырождение снимается и появляются два энергетических уровня: верхний уровень, имеющий спин ms=x/2, и нижний со спином ms= — ’Д. Разность энергий ДЕ в этих состояниях составляет
ДЕ=£0//,
где g — фактор спектроскопического расщепления, называемый обычно g-фактором; 0 — магнетон Бора; Н — напряженность магнитного поля.
Магнетоном называют единицу измерения магнитного момента, которую иногда рассматривают как «квант» магнитного момента системы. Магнетон Бора используется при описании магнитных свойств электрона. У свободного электрона магнитный момент равен одному магнетону Бора, а g-фактор равен 2,0023.
Число частиц на каждом энергетическом уровне может быть подсчитано по уравнению Больцмана. Отношение этих чисел равно
«I / дг \
— = ехр( -——), «2 \ кТ /’
где п — число частиц на том или ином уровне.
Разность энергий электрона в этих состояниях ДЕ невелика, поэтому заселенность обоих уровней при комнатной температуре будет примерно одинаковой с очень небольшим преобладанием состояний с меньшей энергией.
145
I
I
I
Рис 7.6 Сигнал ЭПР:
а — кривая поглощения, б — дифференциальная кри вая поглощения
При наложении на эту систему переменного магнитного поля с частотой v, удовлетворяющей условию
/iv = g (ЗЛ7, начнется резонансное поглощение энергии поля, и электроны с нижнего уровня будут переходить на верхний.
Электрон из возбужденного состояния переходит в основное также за счет релаксационных процессов: спин-решеточ-ной и спин-спиновой релаксации. Время спин-решеточной релаксации Tt можно рассматривать как меру взаимодействия неспаренного электрона с его окружением.
Спектр ЭПР часто представляют не только в виде зависимости интенсив
ности поглощения от напряженности поля (рис. 7.6, а), но в виде зависимости первой производной поглощения по напряженности от напряженности магнитного поля (рис. 7.6, б). Дифференциальный
метод дает более четкое представление о спектре, положении
максимума и полуширине полосы.
Взаимодействие спинов электрона и ядра вызывает так называемое сверхтонкое расщепление спектра ЭПР на отдельные компоненты, обусловленное этим взаимодействием. Сверхтонкое расщепление дает очень ценную информацию о природе связи, электронной структуре и т. д.
Чувствительность спектров ЭПР очень высока: в благоприятных условиях может быть зарегистрировано до 10—12 г вещества и обнаружено в растворе до 10-9 моль/л примесей. Это характеризует ЭПР как весьма перспёктивный аналитический метод. Однако чисто аналитическое применение ЭПР пока невелико. ЭПР является уникальным методом исследования кинетики и механизма реакций, в которых принимают участие парамагнитные вещества, ценнейшим методом изучения процессов с участием радикалов и т. д.
7.3. РЕФРАКТОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Исследование преломления света при прохождении луча через границу раздела прозрачных однородных сред (р е ф-рактометрия) является, по-видимому, старейшим из оптических методов, известным еще по работам И. Ньютона, Л. Эйлера, М. В. Ломоносова и др.
В 80-е годы XIX в. рефрактометры начали использовать в практике работы заводских лабораторий и значение рефрактометрических методов стало быстро возрастать. Рефрактометри
146
ческий метод сохранил свое значение и в настоящее время как метод анализа сложных смесей, исследования свойств веществ и взаимодействия в химических системах.
7.3.1. Показатель преломления и полное внутреннее отражение
При падении луча света на границу
Рис. 7 7. Преломление света на границе двух сред
раздела двух прозрачных сред происходит частичное отражение света от поверхности раздела и частичное распространение света в другой среде (рис. 7.7). На-
правление луча во второй среде изменяется в соответствии с
законом преломления:
«2(отн)
S ГШ1
Sinci2 '
(7.18)
Величину М2(отн) называют относительным показателем (коэффициентом) преломления второй среды по отношению к первой. Показатель преломления по отношению к вакууму называют абсолютным показателем преломления:
«2(абс)
Sin (Хвакууад
Sin СС2
Но так как для первой среды также можно записать
ft 1(абс)
Sin (1Вакуум sin си
то очевидно, что
__ Sin Cti _ _ SinaBaKyyMn2(a6c) _ ^2(абс)
712 (отн) “— ----
S1H (12 SIH (ХваКуЛМ/1 [ (абс) 71] (абс)
(7.19)
т. е. относительный показатель преломления равен отношению абсолютных показателей преломления. Из уравнения (7.19) получаем другую форму записи закона преломления:
и 1 (абс) sin Cll = H2(a6c)Sina2.
Относительный показатель преломления по отношению к воздуху называют просто показателем преломления «:
«абс== «абс(воздух)«-
При атмосферном давлении и комнатной температуре «абс(воздухе)= ==1 >00027, поэтому
па6с= 1,00027«, (7.20)
Е>
прецизионных измерениях учитывают зависимость пабс(воздУх) от Давления, температуры и влажности. Однако в подавляющем
147
большинстве случаев формула (7.20) оказывается вполне пригодной.
Опыт показывает, что если свет переходит, например, из воздуха в какую-то конденсированную, более преломляющую среду, то угол падения всегда больше угла преломления. При переходе из среды, более преломляющей, в среду, менее преломляющую, угол преломления аг оказывается больше угла падения а(. Если угол преломления аг=90° (см. рис. 7.8), то, очевидно, преломления вообще не произойдет, и поскольку sin90°=l, формула (7.18) переходит в
^2(отн)=== sinaj. (7.21)
Угол ai, при котором преломление не происходит, называют углом полного внутреннего отражения, а также предельным или критическим углом. Например, при переходе светового луча из стекла в воздух под углом в 40° угол преломления составляет 90° и, следовательно, при угле падения aiZ>40° преломления не происходит, а свет будет полностью отражаться от поверхности раздела Уравнение (7.21) показывает, что по условию полного внутреннего отражения можно рассчитать показатель преломления. Это соотношение часто используют в практике рефрактометрии.
На показатель преломления оказывают влияние как физико-химические свойства вещества, так и многие внешние условия Волновая теория света связывает показатель преломления со скоростью ci ста в вакууме сив данной среде Уг:
П2=с/Уг.
Показатель преломления зависит от длины волны падающего света, температуры и некоторых других внешних условий. Температуру и длину волны света, при которой производится измерение, обычно указывают у символа п. Например, запись пЦд означает, что показатель преломления измерен при 25°C для желтой £)-линии натрия с длиной волны 589 нм. В качестве нижнего индекса у п вместо длины волны часто указывают только буквенный символ линии (£) для желтой линии натрия 589 нм, С для красной линии водорода 656 нм и т. д.) и вместо, например, nils пишут «д5. Величину «оо, характеризующую показатель преломления при бесконечно большой длине волны, находят экстраполяцией зависимости «=/(/) на X— со.
Показатель преломления и плотность вещества изменяются симбатно, т. е. с ростом плотности происходит увеличение показателя преломления. Теоретическими и экспериментальными исследованиями было установлено, что некоторая функция показателя преломления f(n) прямо пропорциональна плотности вещества р:
f(n)=rp.
148
Коэффициент пропорциональности г назвали удельной рефракцией. При умножении г на молярную массу М получают молярную рефракцию
R—Mr.
Для выражения функции f(n) и, следовательно, для расчета рефракции было предложено несколько уравнений. Наибольшее распространение получила теоретически обоснованная формула Лоренц — Лорентц а:
Величина рефракции, найденная по этой формуле, практически не зависит от внешних условий (температуры, давления и т. д.).
В органической химии широко применяется правило аддитивности молярных рефракций, в соответствии с которым молярная рефракция соединения равна сумме атомных рефракций элементов, образующих это соединение, а рефракция смеси равна сумме молярных рефракций ее составных частей. Молярную рефракцию растворов можно поэтому рассматривать как линейную функцию их состава, выраженного в молярных долях. Рассчитывались также рефракции связей и некоторые другие рефрактометрические константы, пользуясь которыми можно определять рефракции сложных соединений без проведения экспериментальных измерений. Эти величины представляют интерес и в настоящее время для идентификации органических соединений, определения их структуры и проведения различных физико-химических расчетов. Можно отметить также, что молярная рефракция по Лореиц—Лорентцу 7?л-л является мерой поляризуемости молекул а:
/?л_л=2,52*1024а. (7.22)
Поляризуемость, как известно, является характеристикой деформируемости молекул под действием электрического поля.
Зависимость показателя преломления от длины волны падающего света называют дифракционной дисперсией или просто дисперсией. Обычно мерой дисперсии считают разность показателей преломления при двух длинах волн. Относительное изменение дисперсии, например, в ряду гомологических соединений обычно превышает изменение показателя преломления.
7.3.2. Приборы для определения показателя преломления
Для определения показателя преломления в наиболее широко применяемых приборах используют измерение угла полного внутреннего отражения. Принципиальная схема измерительного устройства представлена на рис. 7.8. Основной частью прибора
149
ГЧ
является измерительная призма 3 из оптического стекла с точно известным показателем преломления N. Источником света служит натриевая лампа или газоразрядная трубка (водородная, гелиевая или ртутная),
Рнс. 7.8. Принципиальная схема рефрак- дающая линейчатый спектр ТПМР.ТПЯ _ . * . '
тометра
В рефрактометре Аббе освещение производится белым
(немонохроматичным) светом, однако благодаря призме Амичи, пропускающей желтые лучи без изменения, показатель преломления в этих приборах относится к £)-линии натрия.
Луч света 1 падает на кювету 2, находящуюся на входной грани АВ измерительной призмы 3. Входная грань находится в оптическом контакте с исследуемой жидкостью и служит границей раздела, на которой происходит преломление и полное внутреннее отражение. Луч, соответствующий предельному углу <р, называют предельным лучом. После преломления на границе выходная грань призмы ВС — воздух он образует с нормалью к грани ВС угол 0. Если угол <р близок к предельному, поле зрения 5 трубки 4 оказывается разделенным на светлую (освещенную) и темную (неосвещенную) части. В этом состоянии отсчетное устройство измерительного прибора показывает точную (до десятых долей градуса) величину угла |3. Показатель преломления п исследуемой жидкости рассчитывают по формуле
п = sina — sin2p ,
где a — преломляющий угол измерительной призмы; N — ее показатель преломления.
Вполне понятно, что при измерениях по схеме, изображенной на рис. 7.8, должно соблюдаться условие п < N, т. е. показатель преломления исследуемого вещества должен быть меньше показателя преломления измерительной призмы. Это, естественно, ограничивает интервал значений п, доступных для исследования с данной призмой. В комплект рефрактометра поэтому обычно входит несколько призм, позволяющих работать в различных диапазонах значений показателя преломления.
Наиболее известны конструкции рефрактометров типа Пуль-фриха и типа Аббе. Кроме метода предельного угла для измерения показателя преломления используется метод призмы, а так же иммерсионный, интерференционные и некоторые другие методы.
Показатель преломления в иммерсионном методе находят при качественном сравнении исследуемого вещества с эталонными средами. Чтобы найти показатель преломления, например, каких-либо минеральных зерен или кристаллов, их последовательно рассматривают под микроскопом в жидкостях с известными пока
150
зателями преломления. С помощью полоски Бекке или других эффектов определяют, большую или меньшую величину показателя преломления имеет исследуемое вещество по сравнению с эталонной средой. Полоска Бекке появляется при слабом нарушении фокусировки микроскопа как тонкая светлая полоска на границе двух сред вследствие преломления света. Для определения показателя преломления используется свойство полоски Бекке переходить при поднятии тубуса микроскопа на среду с более высоким показателем преломления, а при опускании тубуса — на среду с более низким значением этой величины. Минимальный размер зерна, при котором обнаруживается этот эффект, составляет 1...2 мкм. С помощью полоски Бекке улавливают разницу между показателями преломления на 0,001. Иммерсионный набор для определения показателя преломления состоит из 50... 100 жидкостей с разными показателями преломления.
Иммерсионный метод широко используется в практике минералого-петрографических исследований. Большое значение он имеет для определения состава бинарных изоморфных смесей карбонатов, сульфатов и др., при исследовании продуктов химической технологии и т. д. Этот метод позволяет определять состав совместно кристаллизующихся отдельных твердых фаз, отличать двойные соли от механической смеси солей, различать изомеры и различные модификации веществ одинакового состава и решать другие задачи. Он очень удобен при анализе взрывчатых и ядовитых веществ, так как для анализа требуются ничтожно малые пробы (миллиграммы) вещества. Однако несмотря на ряд бесспорных достоинств, иммерсионный метод сравнительно редко применяется в практике аналитических лабораторий.
7.3.3. Основные рефрактометрические методики анализа
Разработаны многочисленные рефрактометрические методики определения составных частей в двухкомпонентных растворах (водные растворы спиртов, сахара, глицерина, кислот, солей и т. д.)_ Чем больше разность показателей преломления компонентов, тем более высокой будет точность анализа. Показатели преломления многих технически важных смесей сведены в специальные таблицы, облегчающие проведение рефрактометрического анализа. Для определения концентрации раствора обычно используется метод градуировочного графика, который строится в координатах показатель преломления — концентрация раствора.
Анализ тройных систем значительно сложнее, так как наряду с измерением показателя преломления обычно приходится определять еще какое-либо свойство системы (плотность, вязкость и т- Д.) Большое распространение получил рефрактоденси-Метрический метод, основанный на измерении показателя преломления и плотности. При анализе по этому методу
151
строится треугольная диаграмма составов, на которую по экспериментальным данным для стандартных растворов наносится сетка изорефракт (линий одинакового показателя преломления) и изоденс (линий одинаковой плотности). По измеренным значениям показателя преломления и плотности анализируемого раствора на треугольнике составов находят точку, соответствующую этим величинам. Координаты этой точки прямо указывают на состав анализируемого раствора. В настоящее время этим методом анализируется несколько десятков тройных систем, таких, например, как метанол — этанол — вода, метанол — этанол — ацетон и т. д. Совершенно необходимым условием успешного анализа является существенная разница в величине хотя бы одного из измеряемых свойств.
Менее трудоемки такие методы анализа тройных систем, как дисперсиометрический и метод извлечения, основанные на использовании только рефрактометрических измерений. В дисперсиометрическом методе измеряют показатель преломления при двух длинах волн и на треугольнике составов наносят сетку из изорефракт и линий одинаковой дисперсии. Даль нейшие операции не отличаются от приемов рефрактоденсиомет-рического метода.
При работе по методу извлечения определяют показатель преломления тройной системы и один из компонентов количественно удаляют подходящим реагентом. Так, например, анилин из смеси с толуолом и метилциклогексаном удаляют соляной кислотой, метилэтилкетон из смеси с циклогексаном и бензолом извлекают водой и т. д. Оставшуюся двойную смесь можно проанализировать обычным методом градуировочного графика и далее по треугольной диаграмме установить состав исходного тройного раствора.
Определение всех компонентов в смеси более сложных, чем тройные, рефрактометрически невозможно. Однако вовсе не редкостью является определение в таких смесях одного-двух компонентов.
Например, в растворах типа морской воды рефрактометрически определяют один-два компонента и сумму остальных. Найденная таким образом «соленость» является важной характеристикой морской воды. К этому типу рефрактометрического анализа относится определение концентрации жиров и масел в органических растворителях, анализ полупродуктов и растворов в сахарном производстве, изготовлении фруктовых соков и других напитков, производстве джема и т. д. По типу тройных смесей анализируют лекарственные препараты, кондитерские изделия, косметику и т. д. Широко применяются различные методы предварительного разделения сложных смесей — фракционная перегонка, экстракция и т. д. — с последующим рефрактометрическим анализом фракций. Специальные рефрактометрические методы разработаны для анализа нефтяных фракций. Измерение показателя преломления может быть использовано в титриметри-
152
ческом анализе для построения кривой титрования и обнаружения точки эквивалентности. Однако рефрактометрическое титрование большого распространения не получило.
7.3.4. Рефрактометрические исследования химического взаимодействия, строения и других свойств соединений
Работы по применению рефрактометрии к исследованию химического взаимодействия показали, что по измерениям показателя преломления можно обнаружить только достаточно сильное взаимодействие, поэтому рефрактометрический метод исследования химических процессов большого распространения не получил.
Интенсивное развитие рефрактометрии в начале XX в. в значительной степени связано с ее применением для исследования структуры и свойств химических соединений. Данные по молярной рефракции и дисперсии привлекали внимание как величины, характеризующие внутренние свойства молекул и практически не зависимые от температуры, давления и других внешних условий. Были установлены некоторые эмпирические закономерности, связывающие рефрактометрические константы со строением соединений. Оказалось, например, что молярная рефракция транс-соединений всегда выше, чем г{нс-изомеров. В гомологических рядах рефракции соседних членов отличаются почти точно на одно и то же значение и т. д. Рефракция применяется для исследования поляризуемости, а также электрических, термических и других свойств веществ. Так, например, по показателю преломления и диэлектрической проницаемости можно рассчитать электрический дипольный момент. Для малополярных жидкостей успешно используется упрощенное уравнение Онзагера:
р = 10“18 у/е — пд ,
где р, — электрический дипольный момент; е — диэлектрическая проницаемость.
По рефрактометрическим данным можно рассчитывать радиусы молекул, так как довольно точно соблюдается пропорциональность
а = /гг3, где а — поляризуемость; г — радиус молекулы; k — эмпирический коэффициент, сохраняющий постоянное значение в определенных группах веществ.
Поляризуемость рассчитывается из рефрактометрических данных по уравнению (7.22). Известны и другие применения рефрактометрии.
7.4. ПОЛЯРИМЕТРИЯ
Вращение плоскости поляризации было открыто Д. Араго (1811) при исследовании кристаллического кварца и Ж. Био (1815) при исследовании растворов. Поляриметрические измере
153
нии, основанные на определении угла вращения, являются общепринятыми, официально утвержденными методами анализа в различных отраслях промышленности, особенно в сахарной.
7.4.1. Вращение плоскости поляризации света
У обычного естественного луча колебания световой волны происходят во всех плоскостях, перпендикулярных направлению света. Луч, у которого эти колебания происходят только в какой-то одной плоскости, называют поляризованным, а плоскость, в которой происходят колебания, — плоскостью колебаний. Плоскость, перпендикулярная ей, называется плоскостью поляризации. Некоторые кристаллы обладают способностью пропускать свет одного определенного колебания. После прохождения такого кристалла луч света становится поляризованным. Вещества, способные изменять плоскость поляризации, называют оптически активными веществами, а неспособные — оптически неактивны-м и. При прохождении поляризованного света через оптически активное вещество происходит поворот плоскости поляризации на некоторый угол, называемый углом вращения плоскости поляризации. Вращение называют правым и считают положительным (+), если оно происходит по часовой стрелке, когда смотрят навстречу лучу, и левым и считают отрицательным (—), если оно происходит против движения часовой стрелки. Перед названием или химической формулой правовращающего соединения обычно ставят букву d, а левовращающего —- букву I. Оптически неактивную эквимолекулярную смесь право- и левовращающих изомеров называют рацемическим соединением. Перед их названием помещают обе буквы, например рацемат яблочной кислоты называется ^/-яблочной кислотой. Прописные буквы D и L перед названием или формулой оптически активного соединения (обычно моносахарида или а-аминокислоты) указывают на его принадлежность к стерическим рядам D- или L-глицеринового альдегида, который выбран как соединение сравнения. К D-ряду относят соединения, которые можно получить из О-фсрмы глицеринового альдегида, а к L-ряду — из его L-формы:
сно
н—<!;—он
<!:н2он
П-СНгОНСНОНСНО
сно
I
НО—С— Н
I
СН2ОН
L СНгОНСНОНСНО
Вращение плоскости поляризации кристаллическими веществами является важной характеристикой кристалла, которая широко используется в технике микроскопии и в кристаллохимии. Оптическая активность газообразных молекул или растворенных
154
веществ связана с особенностями строения молекул (например, отсутствием у них центра и плоскости симметрии и т. д.).
Угол вращения плоскости поляризации а связан с концентрацией оптически активного вещества в растворе с (г/мл) и толщиной слоя раствора / (дм) соотношением
а = ауд/с, (7.23)
где ауд — удельное вращение плоскости поляризации.
Оно зависит от природы вещества, длины волны поляризуемого света, растворителя и температуры. Символ аЬ° означает, что удельное вращение плоскости поляризации относится к 20 °C и желтой D-линии натрия. Уравнение (7.23) лежит в основе количественных поляриметрических методов. Молярное вращение плоскости поляризации Ф равно произведению ауд на молярную массу М:
Ф = аудЛ4.
Особый интерес представляет зависимость удельного, или молярного, вращения плоскости поляризации от длины волны света. Эту зависимость называют дисперсией оптического вращения (ДОВ).
Оптическое вращение растет с уменьшением длины волны. В области полосы спектра поглощения оно достигает максимума и затем быстро падает до минимума, после которого медленно возрастает (эффект Коттона). Кривая эффекта Коттона представлена на рис. 7.9.
Величину
-- Ф max Фп.„.
100
называют амплитудой, а расстояние по оси длин волн b — шириной эффекта Коттона.
Специальный аналитический интерес вызывает область спектра, в которой удельное или молярное вращение изменяет свой знак. Длину волны света, при которой вращения плоскости поля
ризации не происходит, называют длиной волны нулевого вращения. Она находится как точка пересечения кривой зависимости удельного или молярного вращения от длины волны С осью длин волн.
Плоскополяризованная волна, как известно, состоит из двух Циркулярно поляризованных компонент (левовращающей L и правовращающей R), каждая из которых обладает своим показате-
Рис. 7.9. Кривая дисперсии оптического вращения
155
лем преломления щ и «к в данной среде и своим молярным коэффициентом поглощения е£ и ед. Разность молярных коэффициентов поглощения характеризует круговой (циркулярный) дихроизм:
Де = е£— е R.
Он может также выражен молярной эллиптичностью 0:
0 = 2,303-^(е£ - ед) = ЗЗООДе.
Характеристики эффекта Коттона, кривая ДОВ и зависимость кругового дихроизма от длины волны дают ценную информацию о структуре, стереохимии и конформации органических и координационных соединений.
7.4.2. Приборы для поляриметрических измерений
В любом приборе для поляриметрического анализа (поляриметре) есть поляризатор и анализатор, между которыми находится трубка с анализируемым раствором. Если поляризатор и анализатор установлены так, что их плоскости поляризации параллельны между собой, то в отсутствие анализируемого вещества свет будет беспрепятственно проходить через оба устройства и наблюдаться в зрительную трубу. Если в отсутствие анализируемого вещества анализатор повернуть на 90°, т. е. ориентировать так, что его плоскость поляризации будет перпендикулярна плоскости поляризатора, то, очевидно, поляризованный свет через анализатор проходить не будет. Это положение «на темнот у». При введении между поляризатором и анализатором оптически активного анализируемого раствора в зрительной трубе появится свет. Чтобы вновь добиться «темноты», анализатор необходимо повернуть на некоторый угол, равный углу вращения плоскости поляризации анализируемым веществом. Величина угла вращения может быть непосредственно прочитана на отсчетном устройстве зрительной трубы.
В качестве поляризатора и анализатора обычно используют призму Николя (или просто николь), изготовляемую из исландского шпата (СаСО3). Осветителем часто служит натриевая лампа. Оптическая система поляриметра включает также устройство для повышения точности установки на «темноту». Это могут быть дополнительные призмы Николя или так называемые пластинки бикварца. Пластинка бикварца состоит из лево- и правовращающего кварца и помещается после поляризатора перед трубкой с анализируемым раствором. При предварительной установке на «темноту», когда николи взаимопараллельны, и в отсутствие анализируемого раствора пластинка бикварца окрашивает поле зрительной трубы в сплошной серо-фиолетовый цвет. Введение анализируемого раствора вызывает резкий цветовой эффект: одна половинка поля становится красной, другая — синей. Поворотом
156
анализатора восстанавливают первоначальную серо-фиолетовую окраску всего поля и по углу поворота анализатора находят угол вращения плоскости поляризации анализируемым раствором.
Интересным видоизменением поляриметра является сахариметр, применяемый специально для анализа растворов сахара. В отличие от обычного поляриметра, осветителем в котором служит натриевая лампа или другой источник монохроматического света, в сахариметре для этой цели используется белый немонохроматический свет. Применение такого осветителя оказалось возможным вследствие случайного совпадения вращательной дисперсии кварца и растворов сахара. Раствор сахара вызывает правое вращение плоскости поляризации. Это вращение в сахариметрах компенсируют введением в луч света клина из левовращающего кварца Вследствие равенства дисперсии оптического вращения кварца и раствора сахара компенсация происходит при всех длинах волн, что и позволяет использовать для освещения сахариметров белый свет. Определения на сахариметре характеризуются высокой точностью, так как толщину клина можно измерить очень точно. Клином называют устройство из двух клинообразных пластинок левовращающего кварца и плоской пластинки правовращающего. Положение клина часто калибруют в единицах концентрации, или так называемых международных сахарных градусах (°S). Величине сто сахарных градусов (100°S) соответствует раствор сахарозы, содержащий 26 г в 100 мл раствора при 20 °C и длине трубки 2 дм.
Вращение плоскости поляризации при различных длинах волн (дисперсию оптического вращения) исследуют с помощью спектрополяриметра, осветитель которого дает монохроматический свет заданной длины волны в широком спектральном интервале обычно с помощью кварцевой диспергирующей призмы.
В новейших конструкциях поляриметров и спектрополяриметров для измерения интенсивности света применяют фотоэлементы и фотоумножители, нередко соединенные с электронным записывающим потенциометром. Особую ценность они имеют для исследований в ультрафиолетовом участке спектра, не доступном для визуальных наблюдений.
7.4.3. Поляриметрические методики
Основу количественных поляриметрических методик составляет Уравнение (7.23), связывающее угол вращения плоскости поляризации с концентрацией раствора. Однако непосредственный расчет по уравнению (7.29) производится сравнительно редко, так как удельное вращение плоскости поляризации ауд также зависит от концентрации. Наиболее часто в практике используется метод градуировочного графика в координатах угол вращения а концентрация с. Особенно широко применяют поляриметрические методики анализа в сахарной промышленности и некоторых Других отраслях пищевой промышленности (масложировой
157
и т. д.), в фармацевтических производствах, парфюмерии и т. д.
Смесь оптически активных веществ может быть проанализирована спектрополяриметрическим методом, т. е. измерением угла вращения при разных длинах волн. Методика этого анализа очень близка к методике спектрофотометрического определения смеси двух окрашенных веществ Расчетные формулы сохраняют свою применимость и в спектрополяриметрическом методе, если молярные коэффициенты поглощения е заменить удельными вращениями ауд, учесть длину трубки и переход к другой концентрационной шкале. Точность анализа возрастает, если одно из анализируемых соединений (а еще лучше — оба) в исследуемой области спектра имеет длину волны нулевого вращения, так как при этой длине волны угол вращения будет определяться концентрацией только второго компонента.
Спектрополяриметрические измерения дают ценную информацию также о структуре и других свойствах органических и координационных соединений. Изменение стереохимического расположения отдельных групп и другие структурные особенности соединений находят отражение в основных характеристиках кривой эффекта Коттона. Как правило, спектрополяриметрические данные рассматриваются совместно со спектрофотометрическими, так как такое сопоставление показывает, какая полоса в спектре поглощения ответственна за эффект Коттона. Кроме того, теорема Кронита — Крамера дает возможность по спектру поглощения предсказать кривую дисперсии оптического вращения и наоборот. При интерпретации спектрополяриметрических данных используют также и другие эмпирические обобщения, связывающие спектрополяриметрические, спектрофотометрические, структурные и другие физико-химические характеристики и свойства веществ.
7.5. НЕФЕЛОМЕТРИЯ И ТУРБИДИМЕТРИЯ
Нефелометрический и турбидиметрический методы применяют для анализа суспензий, эмульсий, различных взвесей и других мутных сред. Интенсивность пучка света, проходящего через такую среду, уменьшается за счет рассеивания и других процессов взаимодействия света со взвешенными частицами.
Нефелометрический метод определения концентрации основан на измерении интенсивности света, рассеянного взвешенными частицами, а турбидиметрический — на измерении интенсивности света, прошедшего через эту среду.
7.5.1. Рассеяние света
Рассеяние света частицами, размеры которых больше длины полны облучающего света, называют рассеянием Ми по фамилии ученого, разработавшего теорию этого явления (1908).
Интенсивность рассеянного света этими частицами подчиняется закону Рэлея:
158
I = lo V ^‘(1+cos2P), (7.24)
где Mi и n2 — показатели преломления частиц и среды соответственно; N — общее число светорассеивающих частиц; V, — объем данной частицы; X — длина волны падающего света; г — расстояние до приемника рассеянного света, р — угол между падающим и рассеянным светом.
В присутствии крупных частиц, диаметр которых измеряется, например, десятками нанометров, закон Рэлея нарушается, однако это не вызывает больших затруднений в аналитической работе, так как связь концентрации с интенсивностью устанавливают по градуировочным графикам. При исследовании заданной системы показатели преломления щ и м2 остаются постоянными, величины г и р определяются конструкцией прибора и тоже не меняются. В этих условиях уравнение (7.24) переходит в
l = (7.25)
Множитель 1 /X4 указывает на быстрое возрастание интенсивности рассеянного света с уменьшением длинр! волны падающего света.
Концентрация, по определению, характеризует число частиц в единице объема:
где V — объем суспензии; Na—постоянная Авогадро.
Подставляя уравнение (7.26) в (7.25), получаем
I = klo-^^. (7.27)
При строгом соблюдении условий приготовления объемы суспендированных частиц получаются примерно одинаковыми и их размеры вполне удовлетворительно воспроизводятся от опыта к опыту. При постоянных V, Ё, X уравнение (7.27) принимает вид
/ = k'loc
или
I/Io = k'c. (7.28)
Уравнение (7.28) показывает, что отношение интенсивности рассеянного света к интенсивности падающего пропорционально концентрации взвешенных частиц. Градуировочный график в координатах 1/10 как функция с будет линеен.
Из уравнения (7.28) следует, что
А каж= — Igc — IgA/, (7.29)
т- е- кажущаяся оптическая плотность А Еаж уменьшается с ростом концентрации, так как с увеличением концентрации увеличивается
159
число рассеивающих частиц и интенсивность рассеянного света возрастает.
В соответствии с уравнением (7.29) график в координатах — Igc будет линеен в противоположность графику в координатах Лкаж — с.
При достаточном разбавлении раствора интенсивность света Л, прошедшего через суспензию или другую мутную среду, подчиняется уравнению, имеющему при постоянстве некоторых условий вид, аналогичный уравнению закона Бугера — Ламберта — Бера:
1g ,Л = — klc, /о
(7.30)
где I — толщина слоя, a k иногда называют молярным коэффициентом мутности раствора.
Уравнение (7.30) справедливо в условиях строгого постоянства условий получения суспензии.
7.5.2. Приборы для нефелометрических и турбидиметрических определений
Пучок света интенсивностью /0 от электрической лампы накаливания падает на кювету с анализируемой суспензией или эмульсией и частично рассеивается взвешенными частицами. Интенсивность рассеянного света равна I, интенсивность света, прошедшего через кювету, Л. Рассеянный свет наблюдается обычно под прямым углом к направлению падающего света. Интенсивность рассеянного света и света, прошедшего через анализируемую смесь, может быть измерена с помощью фотоэлементов или визуально. В выпускаемом промышленностью нефелометре НФМ интенсивность рассеянного света измеряется визуально. Для измерения интенсивности света, прошедшего через взвесь, успешно используются фотоэлектроколориметры. Количественные определения обычно проводятся методом градуировочного графика. В случае нефелометрических измерений в соответствии с уравнением (7.28) или (7.29) график строится в координатах ///о — с или А каж—Igc, а при турбидиметрических определениях — в координатах А — с. Известны также методики турбидиметрического титрования, основанные на реакциях образования осадков малорастворимых соединений. При титровании, например, магния фосфатом оптическая плотность в ходе титрования возрастает, так как увеличивается концентрация взвешенных частиц фосфата магния, а по достижении точки эквивалентности остается постоянной.
Основным достоинством нефелометрических и турбидиметрических методов является их высокая чувствительность, что особенно ценно по отношению к элементам или ионам, для которых отсутствуют цветные реакции и не разработаны колориметриче-
160
ские (фотометрические) методы. В практике широко применяют нефелометрическое определение хлорида и сульфата в природных водах и аналогичных объектах. По точности турбидиметрия и нефелометрия уступают фотометрическим методам, что связано, главным образом, с трудностями получения суспензий, обладающих одинаковыми размерами частиц, стабильностью во времени и т. д. К обычным сравнительно небольшим погрешностям фотометрического определения добавляются ошибки, связанные с недостаточной воспроизводимостью химико-аналитических свойств суспензий.
7.6. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Спектры комбинационного рассеяния света используют в анализе продуктов, главным образом, органической химии. Методом ЯМР анализируются органические и неорганические вещества. Магнитно-релаксационный метод, основанный на зависимости скорости релаксации ядер от концентрации парамагнитных соединений, успешно применяют в анализе многих веществ (например, ионов Fe2+, Co2+, Ni2+ и др.) в широком интервале концентраций. Помимо прямых определений магнитно-релаксационный метод используют в титриметрическом анализе.
Рефрактометрический метод анализа применяют в технологическом контроле в пищевой промышленности, в клинических медицинских исследованиях, при анализе кондитерских изделий, молока, масла, различных жиров и т. д. Нередко показатель преломления включается в ГОСТ как характеристика качества вещества (например, стирола). Содержание сахара, жиров, белка и т. п. измеряют также поляриметрическим методом. Этот же метод используют при анализе лекарственных препаратов, например пенициллина. Нефелометрию и турбидиметрию используют в анализе взвесей, эмульсий и т. п. гетерогенных систем. Нередко такие системы получают специально для определения ионов (обычно анионов), не образующих окрашенных соединений (например, С1~, SO2- и т. д.).
7.7. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДОВ
Рассмотренные в настоящей главе методы успешно применяются в анализе многих систем, имеющих большое практическое значение, и часто позволяют получать информацию о составе анализируемого вещества наиболее простым и быстрым путем. Особую ценность и значение имеют некоторые из этих методов в анализе объектов пищевой, фармацевтической и иных отраслей промышленности. Однако методы этой главы используются, главным образом, для решения специфичных задач и по масштабам практического применения они уступают многим другим, более универсальным и распространенным методам.
6-1533 161
1
Вопросы
1. Какое происхождение имеют спектры комбинационного рассеяния?
2. Каким образом производится наблюдение спектров комбинационного рассеяния? Какие требования предъявляются к осветителям при получении спектров комбинационного рассеяния?
3. На чем основан качественный анализ по спектрам комбинационного рассеяния?
4. На чем основаны методы количественного анализа по спектрам комбинационного рассеяния?
5. В чем сущность ядерного магнитного резонанса (ЯМР)?
6. Какие ядра обладают парамагнитными и какие диамагнитными свойствами?
7. Сформулировать условие резонансного поглощения в магнитном поле с частотой Но Гц.
8. Как рассчитывается химический сдвиг, что он характеризует?
9. Какие требования предъявляются к стандартам в ЯМР? Какие вещества используются в качестве стандартов при снятии спектров ЯМР?
10. В чем сущность качественного и структурного анализа по спектрам ЯМР?
11. Что такое время ядерной релаксации, какова связь его с концентрацией парамагнитного вещества?
12. Какие методы количественного анализа используют в ЯМР?
13. В чем сущность магнитно-релаксационного титрования?
14. Начертить принципиальную схему прибора для проведения поляриметрических измерений.
15. Привести основной закон светорассеяния (уравнение Рэлея) и охарактеризовать величины, входящие в это уравнение.
16. Как зависит интенсивность рассеянного света: а) от спектральной характеристики падающего излучения; б) от размера рассеивающих частиц?
17. Какой вид имеют графики зависимости ДКаж от с; Ткаж от с и А
каж от Igc?
18. В чем сущность: а) метода эталонной шкалы; б) метода градуировочного графика; в) метода добавок?
19. Какое свойство используется в нефелометрических методах анализа: а) поглощение света атомами; б.) рассеяние света частицами; в) излучение света молекулами и ионами?
20. Какой свет рассеивается в наибольшей степени частицами, находящимися в растворе во взвешенном состоянии: а) желтый; б) синий; в) зеленый; г) красный?
21. Для каких целей при приготовлении суспензии необходимо вводить в рабочие растворы стабилизирующие реагенты, соблюдать определенный порядок смешения компонентов, постоянную температуру и проводить измерения через строго определенное 162
время: а) чтобы число частиц осадка во всех растворах было одинаковым; б) чтобы не протекали побочные реакции; в) чтобы частицы осадка во всех растворах имели одинаковый размер?
Л Задачи
1. Определить положение полос поглощения в спектре ЯМР уксусной кислоты СН3СООН в шкале химических сдвигов 6 и частот Av для прибора с vreH = 40 МГц. Указать возможное число пиков и соотношение их относительных интенсивностей.
В соответствии с формулой уксусной кислоты протоны, резонансное поглощение энергии которыми вызывает появление спектров ЯМР, входят в состав двух группировок: а) СН3—С=О и б) НООС—R. В табличных данных значения 6 для указанных группировок равны: а) б] = 2,6...2,1 м.д., б) б2 = 12,2.. 11 м.д.
Таким образом, в спектре ЯМР уксусной кислоты будут наблюдаться два пика с 61 = 2,6...2,1 м.д. и б2 = 12,2...11 м д. и с относительной интенсивностью 3:1, пропорциональной числу резонирующих протонов в группировках.
Соответствующие значения Av находим по формулам: Av, = 6ivre„; Av, = = (2,6...2,1)40 = 104...84 Гщ Av2 = 62vre„; Av2 = (12,2...11)40 = 488...440 Гц.
2. На рис. 7 10 приведен спектр ЯМР соединения, которое содержит 62,1 % С; 10,4 % Н и 27,5 % О. Определить строение этого соединения, указать, какой атомной группировке соответствует каждая из четырех полос поглощения с относительной интенсивностью соответственно 3:1,5:1:0,5 (указаны в виде ступенчатой (интегральной) линии на рис. 7.10).
По спектру ЯМР на рис. 7.10 определяем положение четырех максимумов поглощения в миллионных долях (м.д.) шкалы и записываем в виде таблицы:
Полоса поглощения б, м.д. Относительная интенсивность Число протонов в группировке
1 1,25 3 6
2 2,2 1,5 3
3 2,6 1 2
4 4,0 0,5 1
Соотношение интенсивностей 3:1,5:1:0,5 пропорционально числу протонов в атомных группировках, например 6:3:2:1.
Полоса поглощения ^набл> м. Д Атомная группировка ^табл» М. Д.
1 1,25 СНз—С—ОН 1 1,9 .1,2
2 2,2 СНз—с=о 2,6...2,1
3 2,6 1 СНз—сн2—сно 2,46
4 4,0 ОН—R 5,2...3,0
6‘
163
По величине 6 с помощью 6та6л протонов находим возможные атомные группировки
Рис. 7 10. Спектр ЯМР
Согласно относительной интенсивности пика поглощения для первой группировки в ней должно быть две СНз-группы, следовательно, структура соединения должна быть следующей,-
(1)
СНзч >С— СН2—с— СНз
CIV I ||
ОН о
(4) (3) (2)
Валовая формула СбО2Н|2 соответствует указанному содержанию (%) С, О и Н в соединении
3. На рис. 7.11 приведен спектр C6HsCH2COOH, Ступенчатая линия является интегральной кривой, показывающей соотношение интенсивностей пиков 1:2:3 как 2,0.5,0:1,0. Определить, к какой группировке относятся протоны каждого пика. Каким частотам соответствуют эти пики, если спектр ЯМР снят на приборе с vre„ = 60 МГц? Ответ: a) R—СН2—СООН, Av = 210 Гн; б) С6Н5—С^, Av = 432 Гц; в) R—СООН, Av = 720 Гц.
4. Построить ожидаемый спектр ЯМР бензальдегида СеНвСНО. Указать положение пиков поглощения в шкале 6 (м.д.) и в шкале частот (Гц) при vreH = 40 МГц, указать относительные интенсивности пиков поглощения. Ответ: В спектре два пика с относительной интенсивностью пиков 5:1. Для С6Н5—С^ = 7,5 м.д., Av = 300 Гц, для —СНО, 62 = 10,0...9,7 м.д., Av2 = 404...384 Гц.
5. На спектрометре с vreH = 100 МГц пик поглощения в спектре ЯМР проявляется при частоте 722 Гц (стандарт ТМС). Определить
164
_Jl7'g ' ।--------------1----'----1----'----1--------L
12 10 8 6 4 2
J 2 1
2,0
j_____
0 S, м.д.
Рис. 7.11 Спектр ЯМР C6H6CH2COOH
частоту (Гц), при которой проявится этот пик на приборе с vreH = = 220 МГц, выразить значение химического сдвига в шкале 6, найти б, если в качестве стандарта использовать бензол, воду. Ответ: v = 1588,4 Гц, 6 = 7,22 м.д. (стандарт ТМС), 6 = 0 м.д. (стандарт бензол), 6 = 2,47 м.д. (стандарт вода).
6. Спектр соединения СвНю имеет два пика поглощения с частотой 92 и 282 Гц (в качестве стандарта принят тетраметилсилан, vГен =40 МГц). Определить, в каких группировках находятся резонирующие протоны. Указать интегральные относительные интенсивности пиков поглощения. Ответ: В соединении две группы СНз и одна группа СвН4^; интегральные относительные интенсивности пиков поглощения 6:4.
7. Построить спектр ЯМР бензойной кислоты, указать положение полос поглощения в шкале 6 м.д. и в шкале частот (Гц) при vrc„=60 МГц. Указать интегральные, относительные интенсивности пиков поглощения. Ответ: В спектре два пика с относительной интенсивностью поглощения 5:1; 61 = 7,8...7,2 м. д., 62 = = 12,2... 11 м.д.; Av। = 468...432 Гц, Av2 = 732...660 Гц.
8. На рис. 7.12 приведен спектр соединения С2Н4О2, прибор имел vreH=40 МГц. Определить структуру соединения, выразить значения химических сдвигов в шкале 6 (м.д., стандарт тетраме-тилсилан). Как изменятся значения 6, если за стандарт будет взят циклогексан? Ответ: СНзСООН; СНз—6| = 2,6...2,1 м.д. (стандарт тетраметилсилан); Н—ООС—62=12,2...П м д. (стандарт тетраметилсилан); 6| = 1,43 м.д. (стандарт циклогексан), 62= 10,77 м.д (стандарт циклогексан).
9. Для определения содержания никеля в солях кобальта взяли навеску Со5О4'7Нг О массой m г, перевели в раствор, добавили необходимые реактивы и оттитровали 0,1000 М раствором ди-метилглиоксима магнитно-релаксационным методом, измеряя из-
165
-I__________I_________I__________I__________I__________I___________I_____
600 500 400 300 200 100 0 Н,Гц
2 1
Рис. 7.12. Спектр ЯМР С2Н4О8
менение скорости релаксации. Результаты титрования представлены в виде таблицы:
V(диметилглиоксима), мл 0,20 0,40 0,60 0,80 1,00 1,20 1,40 1,60 1,80 2,00
А, усл. ед. при 82,0 70,0 59,0 58,0 37,0 26,0 26,0 26,0 26,0 26,0
m = 1,304 г А, усл. ед. при 84,0 73,0 63,0 54,0 44,0 34,0 26,0 26,0 26,0 26,0
m = 1,600 г А, усл. ед. при m = 1,800 г 88,0 77,0 67,0 56,0 46,0 36,0 26,0 19,0 19,0 19,0
Рассчитать массовую долю (%) никеля в исследуемой соли. Ответ: 1) 0,27%; 2) 0,22%; 3) 0,25 %. ,
10. Раствор парамагнитной соли Ni(NOs)2 (7 = 0,003314) использовали в качестве титранта в магнитно-релаксационном титровании 25,00 мл раствора диамагнитной соли K4Fe(CN)6.
По результатам титрования определить, какая масса (г) K4Fe(CN)6 содержится в 200,0 мл растворов 1, 2 и 3:
ITNilNOsls), мл 1,00 2 00 3,00 4,00 5,00 6,00 7,00 8,00 9,00
А, усл. ед. для р-ра 1 19,0 19,0 19,0 19,0 19,0 22,0 37,0 52,0 67,0
А, усл. ед. для р-ра 2 15,0 15,0 15,0 15,0 15,0 15,0 23,0 35,0 47,0
А, усл. ед. для р-ра 3 12,0 12,0 12,0 12,0 12,0 12,0 14,0 26,0 40,0
Ответ: 1) 0,4824 г; 2) 0,5240 г; 3) 0,5739 г.
11. 10 л воздуха производственного помещения с содержанием хлороводорода пропускали через 20,0 мл воды в поглотительном сосуде, после чего 5,00 мл полученного раствора отобрали в пробирку для нефелометрирования методом шкалы. Одновременно приготовили серию эталонных растворов, добавляя в такие же пробирки по V мл раствора КС1 (7 = 0,01023 мг/мл). Во все пробирки добавили одинаковый объем HNO3, AgNO3, довели объем до 10,0 мл, перемешали и через 10 мин сравнили помутнения исследуемого и эталонных растворов.
166
Определить концентрацию НС1 (мг/м3), если равенство помутнений наблюдалось при следующих величинах V:
Вариант 1 2 3
V, мл 0,50 1,50 3,00
Ответ: 1) 1,0 мг/м3; 2) 3,0 мг/м3; 3) 6,0 мг/м3,
12. Для нефелометрического определения серы в каменном угле использовали в качестве стандартного 0,01000 М H2SO4. При этом 2,50 мл его разбавили до 1000,0 мл, из V мл полученного раствора в колбе вместимостью 100,0 мл приготовили суспензии BaSO4 и после доведения до метки измерили кажущуюся оптическую плотность. По полученным данным построили градуировочный график:
У, мл 20,0 15,0 12,0 8,0 4,0 2,0
Дмж 0,210 0,330 0.420 0,600 0,800 0,920
Из навески каменного угля массой tn г приготовили 100,0 мл раствора, 20,0 мл его поместили в мерную колбу вместимостью 250,0 мл, приготовили суспензию BaSO4 и довели до метки.
Определить массовую долю (%) серы в каменном угле, если кажущаяся оптическая плотность составила:
Вариант т, г . .
каж -
1 2 3
1,832 3,348 5,04
0,690 0,460 0,300
Ответ: 1) 3,34%; 2) 3,29%; 3) 3,10%.
13. При определении NaCl в растворе гидроксида натрия приготовили серию стандартных растворов. Для этого V мл раствора NaCl (Т = 0,100 мг/мл) перенесли в мерные колбы вместимостью 25,0 мл, добавили реактивы, необходимые для получения суспензии AgCl, измерили кажущиеся оптические плотности с помощью нефелометра и получили:
V, мд д каж
0,30 0,50 0,80 1,50
0,670 0.550 0,390 1.150
Навеску анализируемого раствора массой 21,74 г нейтрализовали HNO3, перенесли в мерную колбу и приготовили 50,0 мл раствора, Vi мл которого использовали для приготовления 25,0 мл суспензии AgCl.
Определить массовую долю (%) NaCl по следующим данным:
Вариант Hi, мл Л
/хкаж -
1 2 3
2,0 5,0 5,0
0,480 0,300 0,420
Ответ: 1) 0,0072%; 2) 0,0046 %; 3) 0,0035 %.
14. При турбидиметрическом определении хлорид-иона для построения градуировочного графика в мерную колбу вместимостью 100,0 мл поместили 20,0 мл раствора КС1 (7'=1,051 мг/мл). •Затем в мерных колбах вместимостью 50,0 мл, содержащих V мл
167
этого раствора, приготовили суспензии AgCl, довели водой до метки, измерили их оптические плотности:
V мл
А
2,0
0 220
4,0 0,470
6,0
0 700
80
0,940
Пробу Vi мл анализируемого раствора разбавили до 100,0 мл, затем 5,00 мл этого раствора перенесли в колбу вместимостью 50,0 мл и приготовили в ней суспензию AgCL
Определить концентрацию (мг/л) хлорид-иона в анализируемом растворе, если измеренные оптические плотности были равны:
Вариант
Vi, мл
А
1 2 3
15,0 25,0 50,0
0,380 0,560 0,820
Ответ: 1) 433 мг/л; 2) 388 мг/л; 3) 280 мг/л.
15. Для определения серебра в сплаве методом фототурби-диметрического титрования навеску анализируемого металла массой 3,17 г растворили в кислоте и довели до 100,0 мл водой. В мерные колбы вместимостью 100,0 мл поместили по 10,0 мл этого раствора, 5 мл желатина, 5 мл 0,1 М HNO3 и V мл раствора КС! (Т = 0,0080 г/мл) Оптическая плотность этих растворов составила:
ИКС1), мл А
2,0 4,0
0,120 0,330
6,0 8,0
0,560 0,670
10,0 12,0
0,680 0,670
Построить кривую титрования и определить массовую долю (%) серебра в сплаве. Ответ: 25,42 %
Глава 8 КОНДУКТОМЕТРИЯ (АНАЛИЗ ПО ЭЛЕКТРИЧЕСКОЙ ПРОВОДИМОСТИ)
8.1. ЭЛЕКТРИЧЕСКАЯ ПРОВОДИМОСТЬ РАСТВОРОВ f Электрической проводимостью называют способность вещества проводить электрический ток под действием внешнего электрического поля.
Измерение электрической проводимости веществ в различных растворителях было предметом многочисленных исследований еще в прошлом веке. Не потеряли своего значения кондуктометрические измерения и в настоящее время.
Единицей электрической проводимости является проводимость проводника сопротивлением 1 Ом. В СИ эта единица получила название сименс (См), Электрическая проводимость раствора выражается в единицах или удельной, или эквивалентной электрической проводимости. Удельная электрическая проводимость х измеряется в См/м и представляет собой электрическую проводимость 1 м3 раствора, находящегося
168
между параллельными электродами площадью 1 м2 каждый при расстоянии между ними 1 м. Более удобной единицей объема для практического использования в лаборатории является дольная единица измерения, такая, как кубический сантиметр (см3) или кубический дециметр (дм3). Тогда удельная электрическая проводимость будет измеряться в См/см и представлять собой электрическую проводимость столба жидкости длиной 1 см и поперечным сечением 1 см2.
Рис 8 1 Электрическая проводимость:
1 — НС1, 2 - КОН, 3 — СНзСООН
В разбавленных растворах удельная электрическая проводимость с увеличением концентрации растет, при некоторой достаточно высокой концентрации достигает максимума
и затем уменьшается. На рис. 8.1 приведены типичные примеры этой зависимости. Электрическая проводимость слабого электролита уксусной кислоты значительно ниже соответствующей величины для растворов HCI или КОН. Возрастание электрической
проводимости с ростом концентрации в растворах умеренно высоких концентраций происходит вследствие увеличения числа ионов с концентрацией. Однако в концентрированных растворах возникают и другие эффекты, приводящие уже к уменьшению электрической проводимости. В концентрированных растворах возрастают силы межионного взаимодействия, вследствие чего происходит образование межионных ассоциатов или ионных пар, увеличивается вязкость раствора и проявляются другие эффекты, снижающие скорость движения ионов и вызывающие уменьшение электрической проводимости Как суммарный результат действия этих факторов на кривой электрической проводимости возникает максимум. Для аналитических измерений обычно используется участок кривой с возрастающей удельной электрической проводимостью, т. е. область разбавленных и умеренно концентрированных растворов.
| Эквивалентной электрической проводимостью называют проводимость ра-* створа, содержащего 1 моль эквивалента вещества и находящегося между двумя параллельными электродами, расстояние между которыми 1 см. Ее единицей измерения является См-см2/моль экв.
Удельная и эквивалентная проводимость взаимосвязаны соот-
ношением
X = 1000х/с,
(8.1)
где с — молярная концентрация эквивалента, моль/л.
В области сравнительно невысоких концентраций эквивалентная электрическая проводимость электролитов обычно растет с Уменьшением концентрации раствора и повышением температуры.
169
У полностью диссоциированных (так называемых сильных) электролитов в области разбавленных растворов (0,001 М и меньше) концентрационная зависимость проводимости выражается уравнением
К=Ко — а /с. (8.2)
Ус где Хо — предельная эквивалентная
Рис 8.2. Зависимость эквивалентной электрической проводимости от концентрации-1 — сильного; 2 — слабого электролитов
электрическая проводимость сильного электролита при бесконечном разведении; а — константа.
Эта зависимость представлена на рис. 8.2 прямой 1. С уменьшением кон
центрации электролита эквивалентная электрическая проводимость возрастает и в области бесконечно больших разбавлений стремится к предельному значению Хо,
характеризующему электрическую проводимость гипотетического бесконечно разбавленного раствора.
Константа а уравнения (8.2) нашла истолкование в теории Дебая — Хюккеля, развитой Онзагером. Уменьшение эквивалентной электрической проводимости теория Дебая — Онзагера объясняет эффектами электрофоретического и релаксационного торможения. Оба эффекта связаны с существованием вокруг иона ионной атмосферы из противоположно заряженных ионов. Электрофоретический эффект вызывается тем, что центральный ион под действием электрического поля движется в одном направлении, а ионная атмосфера, имеющая противоположный заряд, — в противоположном и тормозит движение иона. Релаксационное торможение обусловлено процессами разрушения и формирования ионной атмосферы при движении иона. Концентрационная зависимость эквивалентной электрической проводимости в этой теории дается уравнением
— Хц -р (ДХц -р Д)д/с ,
где А — коэффициент, характеризующий электрофоретический эффект, а В — релаксационный. Величины А и В определяются температурой, вязкостью и диэлектрической проницаемостью растворителя и вычисляются теоретически.
Предельная эквивалентная электрическая проводимость может быть представлена суммой предельных электрических проводимостей, или предельных подвижностей ионов:
= ^о(+) + ^о(-), (8.3)
где Хоц-) и Хо(_)—предельная эквивалентная электрическая проводимость, или предельная подвижность соответственно катиона и аниона.
Закон аддитивности электрической проводимости растворов электролитов при бесконечном разведении, выражаемый уравне
но
нием (8.3), установлен Ф. Кольраушем в 1879 г. еще до появления теории электролитической диссоциации. Соотношение (8.3) называют также законом независимого движения ионов.
Числовые значения подвижностей ионов в водном растворе при комнатной температуре находятся в пределах 30...70 См-см2/(моль экв) и лишь у ионов Н+ и ОН- они существенно превышают эти значения (Х.о = 350; к0 (ОН-) = 199 См-см2/ (моль экв), что связано с особым механизмом перемещения этих ионов в электрическом поле.
Концентрационная зависимость электрической проводимости слабых электролитов имеет более сложный характер (см. рис. 8.2, кривая 2), чем у сильных электролитов. Это объясняется тем, что на электрическую проводимость слабых электролитов влияют не только электрофоретический и релаксационный эффекты, как это наблюдается у сильных электролитов, но и увеличение степени диссоциации электролита с разбавлением раствора, вызывающее в области разбавленных растворов более быстрое, чем у сильных электролитов, увеличение электрической проводимости. Данные по электрической проводимости растворов слабых электролитов часто используются для расчета констант диссоциации.
Электрическая проводимость неводных растворов имеет ряд особенностей. Существенное влияние на электрическую проводимость оказывает диэлектрическая проницаемость растворителя. Концентрационная зависимость электрической проводимости в растворах с высокой диэлектрической проницаемостью аналогична соответствующей зависимости для водных растворов. На кривой электрической проводимости растворов с малой диэлектрической проницаемостью растворителя — хинолине, пиридине и т. д. — образуются минимумы и максимумы, что объясняется, главным образом, сложным характером взаимодействия ионов с растворителем и между собой. Одним из наиболее широко применяемых неводных растворителей в аналитической химии является диоксан, имеющий низкую диэлектрическую проницаемость (~ 2) и смешивающийся с водой в любых отношениях.
Электрическая проводимость растворов с ростом температуры повышается. В водных растворах повышение составляет 2...2,5 % на градус. Температурную зависимость предельной подвижности ионов часто выражают уравнением
^о(/) = A-о (25°)[(1 + a (t — 25)], где а — эмпирический коэффициент, зависящий от природы ионов и растворителя.
Электролит в поле тока высокой частоты. Токи, имеющие частоту порядка мегагерц и десятков мегагерц, называют токами высокой частоты. При таких частотах в растворе начинают играть роль эффекты молекулярной, или деформационной, и ориентационной поляризации.
Под действием электрического поля электроны любой молекулы
171
будут оттягиваться в сторону положительного электрода, а ядра — в сторону отрицательного. Это явление получило название молекулярной, или деформационной, поляризации. Полярные молекулы в электрическом поле обладают также ориентационной поляризацией, стремящейся ориентировать дипольные молекулы вдоль поля. Поляризация обоих типов вызывает кратковременный электрический ток (ток смещения) Кроме того, поляризация молекул приводит к существенному изменению диэлектрической и магнитной проницаемости раствора, что открывает новую возможность исследования свойств раствора при титровании.
Полная проводимость цепей (к), имеющих емкость или индуктивность, как известно из электротехники, состоит из активной Лакт и реактивной Лревкг составляющих:
к -- какт -f" / pea кт,
при этом реактивная компонента проводимости, зависящая от емкости, равна
кс (реакт) — (ОС,
а зависящая от индуктивности
кь (реакт) == ~
где (о — частота; с — емкость; L — индуктивность; ) == д/ —1 .
Таким образом, за изменением в составе раствора, например, при титровании можно следить по изменению проводимости и емкости или по изменению проводимости и индуктивности.
8.2. СХЕМА УСТАНОВКИ ДЛЯ ОПРЕДЕЛЕНИЯ ЭЛЕКТРИЧЕСКОЙ ПРОВОДИМОСТИ
Принципиальная схема для проведения кондуктометрических измерений представлена на рис. 8.3. Это обычный мостик Уитстона, питаемый переменным током от генератора 2. Постоянный ток нежелателен, так как вызывает электролиз раствора. В то же время применение моста переменного тока приводит к появлению так называемого реактивного сопротивления вследствие конечной величины емкости измерительной ячейки Rx в цепи, что особенно заметно при работе с растворами, имеющими большое сопротивление. По этой причине, кстати, нельзя свести к нулю силу тока в диагонали моста гв. Сопротивление /?м известно (магазин сопротивлений). Положение передвижного контакта в подбирается таким образом, чтобы нуль-инструмент / не показывал ток (или ток был минимальным). Тогда сопротивление ячейки Rx можно рассчитать по формуле
R — R ______ R к
*\х - А ---- А м ,
где к и li — длины плеч реохорда при компенсации, пропорциональные сопротивлениям Ri (ав) и R2 (вб).
172
Для питания моста обычно используется ток частотой примерно 1000 Гц, вырабатываемый звуковыми генераторами типа ЗГ-1,ЗГ-10, ЗГ-ЗЗ и др. В качестве эталонного сопротивления /?м включают магазины сопротивлений типа Р-517 М, Р-58 и т. д. Нуль-инструментом может быть телефон, гальванометр или осциллограф. Широко применяется для этой цели осциллографический индикатор нуля типа ИНО-ЗМ, а также электронный индикатор нуля Ф-510 и аналогичные устройства.
Промышленность выпускает комплектные приборы для определения электрической проводимости растворов — мосты переменного тока и кондуктометры Некоторые из них, например кондуктометр «Импульс» КД 1-2, имеют цифровой отсчет показаний в единицах удельной электрической проводимости.
Рис 8 3 Мостик Уитстона
Рис. 8 4 Ячейки для кондуктометрических измерений:
а — ячейка с жестко закрепленными элек тродами, б — погружные электроды
Конструкции измерительных ячеек весьма разнообразны. В прямой кондуктометрии обычно применяют ячейки с жестко закрепленными в них электродами (рис. 8.4, а) В методах кондуктометрического титрования наряду с ячейкой этого типа часто используют так называемые погружные электроды (рис. 8.4,6), позволяющие проводить титрование в любых сосудах, в которых можно разместить электроды.
Экспериментально измеряемая величина сопротивления раствора зависит не только от размера электродов и расстояния между ними, но и от их формы и взаимного расположения, объема раствора и других факторов, не всегда поддающихся точному учету, так как токопроводящим является не только тот объем раствора, который заключен между электродами. Действительная электрическая проводимость раствора, конечно, не зависит от формы или взаимного расположения электродов или каких-либо других факторов, а определяется лишь концентрацией раствора, природой компонентов и температурой Истинная электрическая проводимость раствора х пропорциональна экспериментально измеренной величине и':
х — /гх', где k — константа сосуда.
173
Это очень важная характеристика ячейки. Она зависит от площади электродов, расстояния между ними, от формы сосуда и объема раствора, проводящего ток. Константу сосуда находят экспериментально по электрической проводимости стандартных растворов с хорошо известными значениями х в широкой области температур и концентраций. Обычно в качестве стандартных используют водные растворы хлорида калия.
8.3. ПРЯМАЯ КОНДУКТОМЕТРИЯ
Методы прямой кондуктометрии основаны на том, что в области разбавленных и умеренно концентрированных растворов электрическая проводимость растет с увеличением концентрации электролита. В практической работе обычно используют заранее построенную градуировочную кривую зависимости электрической проводимости раствора от концентрации тех или иных электролитов. В связи с относительно близкими значениями подвижностей ионов кондуктометрические измерения дают информацию, главным образом, лишь об общей концентрации ионов в растворе. Малая селективность кондуктометрического метода является одним из его существенных ограничений.
Кондуктометрическое определение физико-химических свойств и характеристик веществ. Данные по электрической проводимости разбавленных растворов послужили в свое время экспериментальным фундаментом теории электролитической диссоциации Аррениуса.
Если слабая кислота HL в разбавленном растворе концентрации Chl диссоциирует по схеме
HL Н+ + L-
то степень ее диссоциации а может быть найдена по электрической проводимости раствора:
где к — экспериментальное значение эквивалентной электрической проводимости; — эквивалентная электрическая проводимость при бесконечном разбавлении, рассчитываемая по табличным данным как сумма подвижностей ионов.
Константа диссоциации кислоты рассчитывается по уравнению
К„, — [H+l IL ] __ о2 ,.о ___ V о /е 4)
Л L [HL] l-aCHL— Хо(%о-МС
Если табличные данные о подвижностях ионов отсутствуют, величины Ahl и >-о можно найти из экспериментальных данных по электрической проводимости растворов исследуемой кислоты при нескольких концентрациях. Для этого придадим уравнению (8.4) вид
К шЛо — Л' hiAqA, = Z.2Chl
174
и обе части поделим на ЛнгЛоА.. После небольших упрощений будем иметь
1. _ 1 Mil Хо АоЛ’нк
В координатах — Kchl это будет прямая с угловым коэффициен-
том отсекающая на ординате отрезок, равный 1/?.о По-
следующее развитие теории растворов электролитов позволило в значительной степени усовершенствовать расчет, учесть коэффициенты активности, и в настоящее время кондуктометрические измерения довольно широко используют для расчета констант равновесия.
Данные по электрической проводимости растворов применяют также для определения растворимости малорастворимых соединений. Концентрацию насыщенного раствора малорастворимого полностью диссоциированного соединения состава 1:1 рассчитывают по формуле
1000и„ас /-о
где Кнас—удельная электрическая проводимость насыщенного раствора малорастворимой соли.
Кондуктометрическим методом можно исследовать также кинетику химических реакций, когда участниками или продуктами реакции являются ионы.
8.4. КОДУКТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Измерения электрической проводимости растворов широко применяют в титриметрическом анализе для определения точки эквивалентности (кондуктометрическое титрование). В методах кондуктометрического титрования измеряют электрическую проводимость раствора после добавления небольших определенных порций титранта и находят точку эквивалентности графическим методом с помощью кривой в координатах х—V (титранта). Практически в этом методе могут быть использованы такие химические реакции, в ходе которых достаточно заметно изменяется электрическая проводимость раствора или происходит резкое изменение (обычно возрастание) электрической проводимости после точки эквивалентности (реакции кислотно-основного взаимодействия, осаждения и т. д.).
В методе хронокондуктометрического титрования титрант подается в анализируемый раствор непрерывно или небольшими одинаковыми дозами через строго определенные промежутки времени. Одновременно на диаграммной ленте самописца производится соответственно непрерывная или точечная запись кондуктометрической кривой в координатах показание прибора — воемя. Показания прибора пропорциональны
175
электрической проводимости. Концентрация вещества в этом методе рассчитывается по времени, затраченному на титрование. Так как скорость подачи рабочего раствора постоянна и точно известна, время титрования прямо пропорционально объему реактива, израсходованному на титрование. Идея хронокондуктомет рического титрования используется в конструкции различных автотитраторов, выпускаемых промышленностью.
8.4.1. Реакции кислотно-основного взаимодействия
При титровании сильной кислоты, например НС1, сильным основанием, например раствором NaOH, в растворе в любой момент находятся ионы Н+, Cl-, Na+ и ОН-, а электрическая проводимость раствора будет определяться их концентрациями и подвижностями.
На рис. 8.5, а приведена схема, отражающая качественный характер изменения концентрации ионов при титровании, необходимая для уяснения кривой кондуктометрического титрования.
Величина Со- в процессе титрования практически изменяться не будет, a cNa+ будет монотонно возрастать. Величина сн+ будет
Рис. 8.5. Кондуктометрическое титрование сильной кислоты:
а — изменение концентрации ионов при титровании; б — изменение удельной электрической проводимости (т а — точка эквивалентности)
уменьшаться от исходной величины практически до нуля в точке эквивалентности, а Сон-, практически равная нулю в точке эквивалентности, после точки эквивалентности будет возрастать. Таким образом, изменение электрической проводимости до точки эквивалентности будет определяться действием двух взаимно противоположных тенденций: понижающей за счет уменьшения Сн‘ и возрастающей за счет увеличения cNa+. Результирующая этих вкладов АВ (рис. 8.5, б) показывает резкое уменьшение электрической проводимости до точки эквивалентности. Падение проводимости вызывается уменьшением концентрации иона Н+, имеющего при 25 °C подвижность 350 См*см2/(моль-экв), что намного превышает подвижность иона Na+ [50 См-см2/(моль-экв)[
176
После точки эквивалентности начинается резкий подъем электрической проводимости (ветвь ВС), так как в растворе будет нарастать концентрация ионов Na+ и ОН-, подвижность которых составляет 199 См-см2/(моль-экв). Однако возрастание по ВС будет более пологим, чем уменьшение по Л В, так как подвижность иона ОН- почти в 2 раза меньше подвижности иона водорода.
Точка эквивалентности в кондуктометрическом титровании обычно находится путем графического построения. Как видно из рис 8.5, б, экспериментальные значения электрической проводимости раствора в непосредственной близости от точки эквивалентности особого значения не имеют. Для построения используются области недотитрованных и перетитрованных растворов. В некоторых случаях, например при титровании очень разбавленных растворов, обе ветви кривой титрования сглаживаются и для определения точки эквивалентности приходится применять более сложные построения.
На рис. 8.6 схематично показано изменение концентрации реагирующих частиц при титровании слабой кислоты HL сильным основанием NaOH:
HL -I- NaOH = NaL + H2O
и изменение удельной электрической проводимости раствора при этом титровании.
Рис. 8.6. Кондуктометрическое титрование слабой кислоты: а — изменение концентрации частиц; б — изменение удельной электрической проводимости при титровании
Концентрация недиссоциированных молекул кислоты HL монотонно падает практически до нуля. Концентрация анионов L-, равная в начале титрования концентрации ионов Н+, в ходе титрования возрастает, а после точки эквивалентности остается практически неизменной. Концентрация ионов Н+ падает, ионов Na+ возрастает.
Удельная электрическая проводимость раствора до точки эквивалентности (рис. 8.6,6, ветвь АВ) несколько возрастает, так как при титровании происходит увеличение концентрации ионов Na и L-. Концентрация ионов Н+ в растворе слабой кислоты и ее соли, получившейся при титровании, невелика и уменьшение [Н+] в ходе титрования не вызывает столь резкого падения элект-
177
Рис 8 7 Кривая кондуктометрического титрования смеси сильной и слабой кислот
рической проводимости, какое наблюдалось при титровании сильной кислоты.
При титровании кислот средней силы на кривой АВ может возникнуть минимум электрической проводимости, отражающий суммарный эффект уменьшения концентрации ионов Н+ и возрастания концентрации ионов Na+ и L~. После точки эквивалентности наблюдается резкий рост электрической проводимости, вызванной появле
нием в растворе ионов ОН- с высокой подвижностью (рис. 8.6, б, ветвь ВС).
На рис. 8.7 приведена кривая титрования смеси сильной и слабой кислот. Кривая имеет два излома, соответствующие двум
точкам эквивалентности: первая показывает объем щелочи, пошедшей на титрование сильной кислоты, а вторая дает общий объем щелочи, израсходованный на титрование обеих кислот. С увеличением константы диссоциации слабой кислоты первый излом будет становиться менее резким, а второй — более четким, и, наоборот, чем слабее кислота, тем резче будет первый излом, и более закругленным второй.
8.4.2. Реакции осаждения
Вид кривой кондуктометрического титрования по методу осаждения зависит от концентрации и подвижности ионов и растворимости образующегося соединения. Чем меньше ПР продукта реакции, тем резче выражен излом кривой титрования в точке эквивалентности. Результаты анализа 0,1 М растворов бывают вполне удовлетворительными, если у бинарного соединения ПР 10-5. У более растворимых соединений точка эквивалентности устанавливается с трудом, так как кривая титрования плавно закругляется. Излом на кривой титрования становится менее четким также с уменьшением концентрации раствора, и, например, при анализе 1СГ3 М растворов произведение растворимости продукта реакции должно быть уже не больше чем 10-9. Введение в анализируемый водный раствор органического растворителя понижает растворимость, поэтому излом на кривой титрования становится более резким.
Влияние подвижности ионов проявляется в наклоне кривой титрования до точки эквивалентности. Если подвижность осаждаемых ионов больше подвижности ионов осадителя, проводимость раствора до точки эквивалентности будет понижаться, при равенстве подвижностей проводимость меняться не будет, а если подвижность ионов осадителя будет больше подвижности осаждаемых ионов, электрическая проводимость до точки эквивалентности будет возрастать. После точки эквивалентности проводи
178
мость во всех случаях будет расти, так как увеличивается концентрация ионов в растворе.
Титрование, например, растворимой соли бария сульфатом происходит по уравнению реакции
Ba(NO3)2 + Na2SO4 = BaSO4| + 2NaNO3
До точки эквивалентности электрическая проводимость раствора будет несколько падать, так как вместо Ba(NO3)2 P-о(ва2п = 63,6) в растворе появится эквивалентное количество NaNO3 (Z.o(Na+) = = 50,1), т. е. в растворе появятся катионы с меньшей величиной подвижности (Na+ вместо Ва2+). Первая же капля раствора Na2SO4 после точки эквивалентности вызовет резкое увеличение электрической проводимости благодаря всзрастанию концентрации электролита в растворе. Электрическая проводимость пере-титрованных растворов, естественно, также будет возрастать. Кондуктометрическое титрование по методу осаждения не лишено и обычных ошибок титрования, характерных для метода осаждения — ошибок за счет соосаждения или адсорбции, за счет немгновенной скорости установления равновесия между осадком и раствором и т. д.
8.4.3. Реакции комплексообразования
Для кондуктометрического титрования катионов в качестве титрантов могут быть использованы растворы различных кислот и оксикислот (щавелевой, винной, лимонной и др.), комплексонов и других лигандов. Наибольшее практическое значение имеет кондуктометрическое титрование катионов двузамещенной солью этилендиаминтетрауксусной кислоты (ЭДТА).
При титровании, например, Fe3+ раствором ЭДТА (Y4-) протекает реакция
Fe3+ + H2Y2~ = FeY- + 2Н+
в результате которой выделяются ионы Н+ и растет электрическая проводимость раствора. После точки эквивалентности электрическая проводимость раствора падает, так как выделившиеся ионы Н+ связываются анионом H2Y2-
Н+ + H2Y2“ = H3Y-
Кривая такого титрования представлена на рис. 8.8.
Несколько иной вид имеет кривая титрования катиона в буферном растворе (рис. 8.9). Выделяющиеся ионы Н+ в этом случае взаимодействуют с протоноакцепторным компонентом буферной системы и не дают поэтому столь заметного вклада в электрическую проводимость раствора. До точки эквивалентности электрическая проводимость раствора несколько увеличивается, что связано главным образом с увеличением концентра-
179
Рис. 8.8 Кривая кондуктометрического титрования
Fe3- ЭДТА
Рис 8 9 Кривая кондуктометрического титрования Саг+ ЭДТА в буферном растворе (pH ~ 10)
ции ионов Na+, вводимых с титрантом, а после точки эквивалентности резко возрастает, так как увеличивается концентрация титранта.
8.4.4. Реакции окисления — восстановления
Окислительно-восстановительные реакции сравнительно редко используются в практике кондуктометрического титрования. Возможности кондуктометрии здесь несколько сужаются в связи с тем, что реакцию титрования нередко приходится проводить, в присутствии большого количества электролитов, в сильнокислой среде и т. д. В таких растворах не всегда удается с достаточной точностью определить изменение электрической проводимости, связанное с протеканием реакции титрования. Известные ограничения накладывает и скорость некоторых реакций окисления — восстановления, не всегда достаточно высокая при обычной температуре. Повышение температуры оказывается малоэффек тивным, так как становится крайне необходимым термостатиро-вание ячейки, а существенное увеличение электрической проводимости раствора затрудняет установление точки эквивалентности.
8.5. ВЫСОКОЧАСТОТНОЕ ТИТРОВАНИЕ
Установки для высокочастотного титрования во многом отличаются от установок обычной низкочастотной кондуктометрии Ячейка с анализируемым раствором п'ри высокочастотном титровании помещается или между пластинками конденсатора, или внутри индукционной катушки (рис. 8.10). Соответственно этому в первом случае ячейку называют конденсаторной или емкостной, или С - я ч е йк о й, а во втором — индуктивной или L-я ч е й к о й. В ячейках высокочастотного титрования электроды не соприкасаются с исследуемым раствором, что является одним из существенных достоинств метода.
Изменения в ячейке, происходящие в результате реакции титрования, вызывают изменения в режиме работы высоко-
го
частотного генератора. Индуктивная L-ячейка с анализируемым раствором включается в цепь колебательного контура (помещается внутрь катушки индукции). Изменение состава раствора при титровании в такой ячейке вызывает изменение индуктивности, что легко фиксируется мик-
Рис 8.10. Ячейка для высокочас-
роамперметром через несложную схему. В конденсаторных С-ячей-ках при титровании раствора
тотного титрования.
а — емкостная (С-ячейка), б—индуктивная (/.-ячейка)
вследствие изменения диэлектри-
ческой проницаемости происходит сдвиг рабочей частоты генера-
тора, что устанавливается с помощью измерительного конденсатора. При построении кривой титрования показания прибора откладывают как функцию объема добавленного титранта. Про-
мышленностью выпускаются стандартные высокочастотные тит-
раторы.
8.6. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Прямое измерение электрической проводимости является наиболее эффективным методом контроля качества дистиллированной воды в лабораториях, технической воды в так называемых тонких химических или фармацевтических производствах, в технологии водоочистки и оценке загрязненности сточных вод, теплотехнике (питание котлов) и т. д. Кондуктометрические датчики с успехом применяются в автоматизированных схемах контроля производства в некоторых отраслях химической, текстильной и пищевой промышленности, гидроэлектрометаллургии и т. д. Разработана методика кондуктометрического определения малых количеств углерода (10~2. ,10~3%) в сталях и металлах. Методика включает сожжение образца в токе кислорода, поглощение СОг раствором Ва(ОН)г и измерение его электрической проводимости. Содержание углерода находят по градуировочной кривой.
Методы прямой кондуктометрии используют для контроля качества молока, различных напитков и пищевых продуктов.
Простота и высокая точность кондуктометрических измерений, возможность использования полученных данных в автоматизированных схемах контроля и управления и другие достоинства метода электрической проводимости вызывают большой интерес к этому методу и в настоящее время. Однако прямые кондуктометрические измерения весьма чувствительны к влиянию примесей, особенно примесей кислотно-основного характера в связи с Резким различием подвижностей ионов Н+ и ОН- по сравнению с подвижностями других ионов.
Обширную область применения имеет кондуктометрическое титрование. Сильные минеральные кислоты в водном растворе
181
(HC104, НС1, HNO3 и др.) титруются щелочью при больших и достаточно малых концентрациях (до 10-4 моль/л). Так же титруются сильные основания (NaOH, КОН и др.) сильными кислотами. Легко титруются муравьиная, уксусная и другие кислоты средней силы. Кривые кондуктометрического титрования ряда органических кислот (янтарной, адипиновой и др.) при титровании слабым основанием имеют более резко выраженный излом в точке эквивалентности, чем кривые титрования сильным основанием. Эти кислоты титруют раствором аммиака, причем в реакцию вступают оба протона. Слабые основания могут титроваться сильными и слабыми кислотами. Легко титруются, например, этаноламины растворами уксусной кислоты. Практическое значение имеет кондуктометрическое титрование содей аммония и других слабых оснований растворами щелочей и титрование солей слабых кислот (ацетатов, фенолятов и др.) сильными кислотами. Аминокислоты (глицин, аланин, валин и др.) титруются сильными основаниями.
Кондуктометрически могут быть оттитрованы смеси слабых кислот, смеси слабых оснований, а также смеси кислот или оснований с солями слабых кислот или слабых оснований.
Особенно широкие возможности титрования различных электролитов и их смесей открывает применение органических и водно-органических растворителей: водно-диоксанового, водно-ацетонового, водно-спиртовых, ледяной уксусной кислоты и др. В этих растворах анализируются трех-, четырех- и пятикомпонентные смеси (например, смеси НС1 4- СН3СООН 4- NH4C1 4-4- С6Н5ОН; NaOH 4- NaBO2 4- C2H5NH2 4- CH3COONa4-NaNO2 и др.).
Методом кондуктометрического титрования определяют многие катионы и анионы. Нитратом серебра титруют хлорид, бромид, иодид, цианид, тиоцианат, оксалат, ванадат, тартрат, салицилат и некоторые другие анионы. Титрованием в среде 90 %-но-го спирта определяют С1— в природных водах при содержании порядка 10 мкг. Содержание I- и С1~ в смеси может быть определено без предварительного разделения. Титрование ацетатом или хлоридом бария применяют для определения сульфата, хромата, карбоната, оксалата, цитрата и других анионов обычно при добавлении в анализируемый раствор спирта. Сульфаты таким методом определяют в природных водах и аналогичных обьектах.
Кондуктометрическое титрование раствором ЭДТА применяется для определения Fe3+, Cu2+, Nr*-, Со2+, Zn2+, Pb2 , Cd2+, Ca2+, Mg2+ и других катионов. Катионы, образующие очень устойчивые комплексы, как, например, Fe3+, Cu2+, Ni2+, Со2+ и некоторые другие, титруются в нейтральном или слабокислом растворе. Некоторые смеси катионов могут быть проанализированы прямым кондуктометрическим титрованием без предварительного химического разделения. Например, ионы Fe3+ могут быть определены в присутствии Zn2+, Со2+, Cd2+, Fe2+, Mn2+
182
и других катионов. Кондуктометрическим титрованием можно определить общую жесткость воды.
В методе высокочастотного титрования может быть использована практически любая химическая реакция: кислотно-основного взаимодействия, осаждения и т. д. в водном и неводном растворах. Кривые высокочастотного титрования имеют такой же вид, как и кривые обычного кондуктометрического титрования. Концентрационная область титрования слабых кислот и оснований высокочастотным методом остается примерно такой же, как и в обычном кондуктометрическом титровании.
Широкие возможности для развития новых методов анализа открывает использование неводных растворителей. Высокочастотное титрование проводят в ледяной уксусной кислоте, диме-тилформамиде, смесях диоксан — вода, ацетон — вода и других смешанных растворителях. В ледяной уксусной кислоте методом высокочастотного титрования можно определять НСЮ4 в присутствии HNO3 по взаимодействию с раствором пиридина, серную кислоту можно титровать в присутствии 20-кратного количества фосфорной, что представляет практический интерес в производстве фосфатов. В ледяной уксусной кислоте титруют также алкалоиды, антибиотики и другие продукты фармацевтической промышленности.
Образование малорастворимых сульфата бария, галогенидов серебра и других осадков составляет основу определения анионов в природных водах и промышленных стоках методом высокочастотного титрования.
8.7. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Кондуктометрические методы характеризуются высокой экс-прессностью, простотой и доступностью измерительных приборов, удобством работы и достаточной точностью. Ценной особенностью кондуктометрических методов является возможность проведения автоматического и дистанционного анализа. Прямые кондуктометрические измерения имеют погрешность 1...2%, при соблюдении специальных условий она снижается до 0,2 %.
Погрешность кондуктометрического титрования без термоста-тирования растворов обычно оценивается величиной примерно в ±(2-]-3) %. Особое значение вообще для кондуктометрических измерений имеет температура в связи с довольно большим температурным коэффициентом электрической проводимости: изменение температуры на 1° вызывает изменение электрической проводимости на 2...3 %. Термостатирование растворов существенно увеличивает точность метода.
Основным достоинством метода высокочастотного титрования является возможность анализа любых агрессивных сред, так как электроды с анализируемым раствором не соприкасаются. Электроды можно поместить, например, с наружной стороны трубопровода, по которому протекает жидкость, и получать таким об
183
разом информацию о составе раствора в любой момент времени. Методом высокочастотного титрования с успехом могут быть проанализированы различного рода мутные растворы, взвеси, эмульсии, окрашенные растворы и т. д.
I Вопросы
1. В чем сущность кондуктометрических методов анализа?
2. От каких факторов: а) температура; б) концентрация иона; в) природа иона, г) присутствие фонового электролита; д) скорость перемешивания раствора зависит подвижность иона в растворе?
3. Как с помощью прямых кондуктометрических измерений определяют содержание: а) С, N и S в металлах и сплавах; б) С, Н, О, S в органических соединениях?
4. Какая зависимость положена в основу метода кондуктометрического титрования: а) сила тока — объем титранта; б) эквивалентная электрическая проводимость — концентрация определяемого вещества; в) удельная электрическая проводимость — объем титранта; г) сила тока — электрическое сопротивление раствора; д) подвижность определяемого иона — концентрация определяемого иона?
5. Какой вид имеют кривые кондуктометрического титрования для реакций: а) кислотно-основного взаимодействия; б) осаждения; в) комплексообразования?
6. В каких растворах: а) НС1 + H2SO4, б) НС1 + СНзСООН; в) H2SO4 + CuSO4; г) H2SO4 + Na2SO4 можно определить содержание обоих компонентов методом кондуктометрического титрования раствором NaOH?
7. В каких растворах: a) NaOH + NH4OH; б) NaOH -f- КОН, в) NaOH + NaCl можно определить содержание обоих компонентов методом кондуктометрического титрования раствором НС1? Какой вид имеют кривые титрования смесей^
8. Какие из перечисленных достоинств следует отнести к методу кондуктометрического титрования: а) высокая точность, б) высокая чувствительность; в) возможность титрования мутных и окрашенных растворов; г) возможность анализа смесей двух веществ без предварительного разделения; д) возможность титрования в присутствии посторонних электролитов?
9. В чем сущность высокочастотного титрования? Каковы особенности измерительной аппаратуры высокочастотного титрования: Z-, Q- и /^-метров?
Задачи
1. В навеске органического соединения массой т г кислород количественно перевели в СОг. Диоксид углерода растворили в электролитической ячейке, наполненной разбавленным раство
184
ром щелочи, и определили Дхх — уменьшение электрической проводимости поглотительного раствора
Таким же превращениям подвергли стандартные вещества, содержащие от 200 до 1000 мкг кислорода, и измерили соответствующие величины Ах. Результаты измерений представлены ниже:
Содержание кислорода в об-
разце, мкг . . 200 400 600 800 1000
Лх-106, См 80 150 220 285 355
Построить градуировочный график и вычислить массовую долю (%) кислорода в органическом соединении по следующим данным:
Вариант 1 2 3
т, мг 2,299 2,161 2,091
Дх-106, См . ’ 300 210 260
Ответ: 1) 36,10%; 2) 26,84%; 3) 34,43 %.
2. Навеску органического соединения массой m г сожгли в токе кислорода. В газообразных продуктах пиролиза воду выморозили, а диоксид углерода пропустили в ячейку для измерения электрической проводимости, содержащую раствор Ва(ОН)г. Сопротивление раствора в ячейке до и после опыта изменилось на А/?х.
В аналогичных условиях сожгли стандартные вещества, содержащие от 200 до 1000 мкг углерода, и также измерили для каждого образца значение АТ? Результаты измерений представлены ниже:
Содержание углерода в образ-
це. мкг 200 400 600 800 1000
1Я-10-6, Ом 80 150 220 285 355
Построить градуировочный график и вычислить массовую долю (%) углерода в органическом соединении по следующим данным:
Вариант 1 2 3
т, мг 1,221 0,9982 0 9919
1Ял-Ю-6,Ом 90 115 250
Ответ: 1) 16,38%; 2) 30,05%; 3) 70,57 %.
3. Навеску органического соединения массой m г подвергли пиролизу в условиях, когда находящаяся в веществе сера переходит в сероводород. Газообразные продукты распада после хроматографического отделения циана пропустили в кондуктометрическую ячейку, содержащую раствор нитрата ртути. Со-противпение раствора в ячейке в результате этого возросло на АТ?Х.
185
Таким же превращениям подвергли стандартные вещества с известным содержанием серы в них и измерили соответствующие величины А/?. Результаты измерений приведены ниже.
Содержание серы в образце, мг 0,240 0,260 0,280 0 300
\Я. см 450 480 510 54о’
Построить градуировочный график и вычислить массовую долю (%) серы в органическом соединении по следующим данным:
Вариант т. мг \Rx, см
12 3 4
1,315 2,158 2,129 2,354
430 460 480 494
Ответ: 1) 17,34%; 2) 11,49%; 3) 12,21 %; 4) 11,47%.
4. При титровании 50,00 мл смеси NaOH и NH3 0,0100 М НС1 получили следующие данные:
l^HCl), мл . . х, См, для р-ра 1 . х, См, для р-ра 2 . х, См, для р-ра 3 . 0 6,30 5,68 6,60 1,00 5,41 4,46 5,98 2,00 3 00 4,00 4,52 3,62 3,71 3,20 — 3,00 5,30 4,68 4,05 5,00 4,79 3,84
И(НС1), мл 6,00 7,00 8,00 9,00 10,00
х. См, для р-ра 1 5,85 6,93 9,00 12,08 15,13
х, См, для р-ра 2 . 4,68 5,50 7,00 10,80 14,55
х, См, для р-ра 3 . 4,45 5,70 7,80 12,02 16,20
Построить кривую титрования и вычислить концентрацию NaOH и NH3 в исследуемом растворе (г/л). Ответ: 1) 0,02800 г/л NaOH; 0,01363 г/л NH3; 2) 0,02400 г/л NaOH; 0,01669 г/л NH3; 3) 0,04000 г/л NaOH; 0,008852 г/л NH3.
5, Анализируемую смесь НС1 и СН3СООН поместили в мерную колбу вместимостью 50,00 мл и довели водой до метки. При титровании 10,00 мл раствора 0,1 М NaOH (k = 1,104) получили
следующие результаты:
V(NaOH), мл 7,00 8,00 9,00 10,00 11,00
X, См, для Р-ра 1 2,50 2,20 1,90 1,93 1,96
х, См, для Р-ра 2 2,62 2,35 2,08 1,96 2,00
х, См, для Р-ра 3 2,66 2,39 2,12 2,02 2,04
П р О д о л жени
V(NaOH), мл 12,00 13,00 14,00 15,00 16,00
х, См, для Р-Ра 1 2,00 2,20 2,50 285 3,20
х, См, для Р-ра 2 2,03 2,06 2,38 2,74 3,10
х. См, для Р-Ра 3 2,08 2,11 2,25 2,60 2,96
Построить кривую титрования и определить массу НС1 и СН3СООН в исходном растворе (г). Ответ: 1) 0,1812 г НС1; 0,1160 г СНзСООН; 2) 0,1912 г НС1; 0,1210 г СН3СООН; 3) 0,1902 г НС1; 0,1376 г СН3СООН.
6. Навеску смеси слабых оснований массой m г растворили в безводной уксусной кислоте и оттитровали кондуктометриче-
186
Рнс. 8.11. Кривые кондуктометрического титрования слабых оснований
ским методом 0,2000 М хлорной кислотой. Состав смеси указан в таблице. Результаты титрования приведены на рис. 8.11, а, б.
Кривая Состав смеси tn, г
1 п-Нитроанилин 0,0920
2 Диэтил амин + дифениламин 0,2134
3 Диэтиламин -|- n-нитроанилин -|- мочевина 0,1425
4 Диэтиламин п-хлоранилин + дифениламин -р 0,1512
-р ацетамид
Определить массовую долю (%) компонентов в смеси.
Ответ: 1) 99,09 %;
2) 23,99 % диэтиламина;
47,57 % дифениламина;
3) 28,74 % диэтиламина;
45,17 % п-нитроанилина; 21,07% мочевины; 4) 19,35% диэтиламина; 21,94% п-хлоранилина; 44,77 % дифениламина; 13,28 % ацетамида.
7. Навеску технического фенола массой 2,604 г после обработки раствором NaOH перенесли в колбу вместимостью 100,0 мл и довели водой
Рис. 8.12. Кривая высокочастотного титрования фенола
187
до метки. При высокочастотном титровании пробы объемом 1,00 мл 0,1180 М НС1 получили следующие результаты (рис. 8.12).
Вычислить массовую долю (%) фенола в образце, если 1Л — объем раствора НС1, пошедший на нейтрализацию свободной щелочи. Ответ: 93,82 %.
Глава 9 ПОТЕНЦИОМЕТРИЯ
9.1. ЭЛЕКТРОДНЫЙ ПОТЕНЦИАЛ
Потенциометрические методы основаны на измерении электродвижущих сил (ЭДС):
Е = Е] - Е2,
где Е — электродвижущая сила; £i и £2 — потенциалы электродов исследуемой цепи.
Потенциал электрода £ связан с активностью и концентрацией веществ, участвующих в электродном процессе, уравнением Нернста:
£ = £» + _«11п
а°» _ £0 I RT |п [ох]ута «red nF [red]yred ’
(9.1)
где £° — стандартный потенциал редокс-системы; £ — универсальная газовая постоянная, равная 8,312 Дж/(моль-К); Т — абсолютная температура, К; £ — постоянная Фарадея, равная 96500 Кл/моль; п — число электронов, принимающих участие в электродной реакции; аох, агеА — активности соответственно окисленной и восстановленной форм редокс-системы; [ox], [red] — их молярные концентрации; у0Х1 yred— коэффициенты активности.
£ — Е° при aox=ared= 1, причем имеется в виду гипотетический стандартный 1 М раствор, в котором коэффициент активности каждого растворенного вещества равен 1, а чистые вещества находятся в наиболее устойчивом физическом состоянии при данной температуре и нормальном атмосферном давлении.
Подставляя Т — 298,15 и числовые значения констант в уравнение (9.1), получаем для 25 °C
£ __ £0 I 0-059 | Дох _ £0 । 0-050 ]„ 1ох1 Ym (9.2) ' п ё artd п в [redlv.ed
Окислительно-восстановительную систему представляют также многие металлы (Ag, Zn, Cd, Hg, Sn, Pb и др.), погруженные в растворы их солей, например металлический цинк в растворе:
Zn2+ + 2е = Zn
Электродный потенциал металла в такой системе также подчиняется уравнению Нернста:
Ezrf+/Zn— ^Zn2+/Zn +
flZn8+ aZn
(9.3)
188
Активность свободного металла постоянна, она принимается равной единице, и уравнение (9.3) переходит в
г, __ ро [ RT ।
^Zn7+/Zn—cZn!+/zriT “27^|пагп,‘ —
г-о । RT <
R1 Zn2 /Zn “Г ^^Zn2+Vzn2T*
Потенциометрические методы анализа известны с конца прошлого века, когда Нернст вывел (1889) известное уравнение (9.1), а Беренд сообщил (1883) о первом потенциометрическом титровании Интенсивное развитие потенциометрии в последние годы связано, главным образом, с появлением разнообразных типов ионоселективных электродов, позволяющих проводить прямые определения концентрации многих ионов в растворе, и успехами в конструировании и массовом выпуске приборов для потенциометрических измерений.
Потенциометрические методы анализа подразделяют на прямую потенциометрию (ионометрию) и потенциометрическое титрование. Методы прямой потенциометрии основаны на прямом применении уравнения Нернста (9.1) для нахождения активности или концентрации участника электродной реакции по экспериментально измеренной ЭДС цепи или потенциалу соответствующего электрода. При потенциометрическом титровании точку эквивалентности определяют по резкому изменению (скачку) потенциала вблизи точки эквивалентности.
9.2. СХЕМА УСТАНОВКИ ДЛЯ ПОТЕНЦИОМЕТРИЧЕСКИХ ИЗМЕРЕНИИ
Для проведения потенциометрического анализа обычно собирают гальванический элемент, на одном из электродов которого протекает электрохимическая реакция с участием определяемого иона или иона, реагирующего с определяемым. При схематичном изображении различных гальванических элементов или электродов используют условную запись, достаточно полно отражающую состав и характерные особенности элемента. Форма и символика схематического изображения гальванических элементов установлены решением ИЮПАК. По этим правилам формулы веществ, находящихся в одном растворе, записывают через запятую, а границу между электродом и раствором или между разными растворами обозначают вертикальной чертой |. Двойная вертикальная черта || показывает, что так называемый диффузионный потенциал, возникающий на поверхности раздела растворов разного состава, сведен к минимуму или элиминирован с помощью солевого мостика. Так, например, гальванический элемент, состоящий из водородного и хлорсеребряного электродов, условно может быть изображен схемой Pt, (НгЦОДМ. H2SO4II II0.1M KClIAgCl, Ag. Электродвижущая сила (ЭДС) такой галь-
189
Рис 9 1 Схема потенциометрической установки
ванической цепи обычно измеряется компенсационным методом, когда ЭДС исследуемого элемента точно компенсируется внешним источником напряжения и через элемент тока практически не проходит, и, следовательно, в системе не протекают процессы, ведущие к заметным химическим или концентрационным изменениям за счет электролиза.
Потенциометрическое титрование можно проводить также некомпенса
ционным методом или с применением биметаллических электродов, не взаимодействующих с определяемым веществом. Принципиальная схема потенциометрической установки изображена на рис. 9.1.
Реохорд (линейный делитель напряжения) АВ питается током через реостат R, которым регулируют подаваемое напряжение. Подвижный контакт С позволяет подавать в цепь напряжение, необходимое для компенсации электродвижущей силы стандартного (£ст) или иойтедуемого (£х) элемента. Включение £ст или Ех в цепь производится переключателем К\, а кратковременное замыкание цепи ключом /Сг- В момент компенсации ЭДС гальванометр G ток не показывает.
Падение напряжения на концах реохорда Д£(£ЛВ) по закону Ома равно
Е АВ IRaB>
(9-4)
где / — сила тока; RAB—сопротивление реохорда. Между точками А и С падение напряжения составляет
Вдс = 1Вас- (9-5)
Сопротивление реохорда пропорционально его длине:
Е ав— Ь1ав>
Е ас = Мао
где k — коэффициент пропорциональности.
Вместо уравнений (9 4) и (9.5) можно поэтому записать:
EAB=kIlAB, (9.6)
EAC=kIlAc. (9.7)
При совместном решении уравнений (9.6) и (9.7) получаем
Еас=Еав^-- (9-8)
1АВ
Если переключатель Ki замкнут на Ех, уравнение (9.8) принимает вид
190
ех = еав-±
*АВ
а если на £ст, то
„ ^АС
<7 __ г лист
ст— Сав~Т^~-
(9.9)
(9.10)
Деление уравнения (9.9) на (9.10) и небольшое упрощение дает 1ЛС
Ех^Ест-'-^. (9.11)
‘лсст
По уравнению (9.11) и экспериментальным данным рассчитывается ЭДС исследуемого элемента. Для удобства работы на реохорд АВ обычно подается такое напряжение через реостат R, чтобы отношение Е„/1АСст в уравнении (9.11) было целочисленным, например равным 2 мВ/делеиие. Тогда значение ЭДС исследуемой цепи получают умножением показаний реохорда lACt на установленную цену деления.
Эта принципиальная схема компенсационных измерений со
храняется и в современных потенциометрах, использующих вместо реохорда шунтирующие декады (рис. 9.2). Декады ab и cd состоят каждая из десяти катушек одинакового сопротивления. На клеммы ab так же, как на концы реохорда, подается напряжение Е, тогда на клеммах cd падение напряжения составит ОДЕ независимо от положения переключателя ef (1—2, 2—3 и т. д.). Если падение напряжения на каждой катушке декады ab составляет, например, 0,1 В, то на каждой катушке декады cd оно будет 0,01 В, и в положении на рис. 9.2 на выходных клеммах В потенциометра оно составит 0,24 В.
Таким образом, потенциометр из четырех декад дает возможность определить падение напряжения с точностью 0,0001 В. Специальная цепь со стандартным
элементом позволяет подавать на первую декаду потенциометра напряжение точно в 1 В. При работе потенциометра ЭДС исследуемого элемента непосредственно отсчитывается по цифрам, показывающим положение переключателя на каждой декаде. При работе со стеклянным электродом, исследовании неводных растворов и т. д. обычный потенциометр оказывается неприменимым, так как внутреннее сопротивление таких гальванических элементов очень велико. В таких установках применяют электронные потенциометры, в которых гальванометр заменен на электронную схему с высоким входным сопротивлением.
Рис 9.2 Принципиальная схема шунтирующих декад
191
9.2.1. Стандартный гальванический элемент
В качестве стандартного элемента в потенциометрии используется нормальный элемент Вестона, электродвижущая сила которого обладает строго постоянным воспроизводимым значением, сохраняющимся в течение многих лет, и незначительным температурным коэффициентом. Электродами элемента Вестона являются ртуть (положительный полюс) и насыщенная амальгама кадмия, содержащая 12,5 % Cd (отрицательный полюс), а электролитом — насыщенный водный раствор по отношению к CdSCU-Vs Н2О и Hg2SOi. Чтобы обеспечить существование насыщенного раствора, в элемент в контакте с электролитом вводят кристаллы этих соединений. Электрохимическая цепь элемента Вестона имеет вид
-Cd(Hg)|Cd2+, sor, Hgi+|Hg +
При его работе протекает электрохимическая реакция
Cd + Hgi+^2Hg + Cd2+
Электродвижущая сила элемента Вестона может быть рассчитана по уравнению
Е = 1,01830 - 4,06- 10-5(f - 20) - 9,5-10~7(1 - 20)2.
В обычной аналитической работе квадратичным членом обычно пренебрегают.
9.2.2. Исследуемый гальванический элемент
Исследуемый гальванический элемент обычно состоит из индикаторного электрода и электрода сравнения. Индикаторным называют электрод, потенциал которого зависит от концентрации (активности) определяемого иона. Потенциал электрода сравнения должен оставаться постоянным независимо от протекания каких-либо реакций в анализируемом растворе. Электродвижущая сила исследуемого элемента выражается как разность между потенциалом электрода сравнения (Еср) и потенциалом индикаторного электрода (Ет1):
Е = Еср— ЕННд+ Ед, где Ед—диффузионный потенциал, или потенциал жидкостного соединения.
9.2.3. Индикаторные электроды
Потенциал индикаторного электрода связан уравнением Нернста (9 2) с концентрацией (активностью) определяемого иона. Индикаторный электрод должен удовлетворять ряду требований.
Необходимо, чтобы его потенциал был воспроизводим и уста-
192
навливался достаточно быстро. Иногда, когда, например, исследуется потенциал металлического электрода в растворе его соли и при исследовании некоторых других систем, индикаторный электрод должен быть обратим. Электрод должен обладать также определенной химической устойчивостью, чтобы не реагировать с другими компонентами анализируемого раствора.
В потенциометрии в качестве индикаторных применяют металлические и мембранные электроды.
Металлические электроды первого рода представляют собой металлическую пластинку или проволоку, погруженную в раствор хорошо растворимой соли этого металла. Электроды из серебра, ртути, кадмия и некоторых других металлов обратимы и дают воспроизводимые результаты. Однако для многих металлов таких, как хром, кобальт и других, это не характерно и электроды из этих металлов в качестве индикаторных не используются, так как не дают достаточно воспроизводимых результатов. У многих электродов воспроизводимость значительно улучшается, если использовать не просто металл, а его амальгаму. Это амальгамные электроды. Особое место среди индикаторных электродов занимают редокс-электро-ды, служащие для измерения окислительно-восстановительного потенциала системы. В качестве редокс-электродов используются благородные металлы: платина, золото, иридий или графит. Потенциал таких электродов зависит от отношения концентраций (активностей) окисленной и восстановленной форм редокс-пары.
Электроды второго рода состоят из металла, покрытого слоем малорастворимого соединения этого металла и погруженного в раствор хорошо растворимого соединения с тем же анионом. К ним относятся хлорсеребряный, каломельный и некоторые другие электроды. Электроды второго рода обычно применяют как электроды сравнения.
В работе мембранных электродов используется не электрохимическая реакция с переносом электрона, а разность потенциалов, возникающая на границе раздела фаз, и равновесие обмена ионов между мембраной и раствором. В обычных конструкциях мембранных электродов мембрана разделяет исследуемый раствор и вспомогательный внутренний раствор. Наиболее широко применяемым электродом этого типа является стеклянный электрод. Известны также фторидный, сульфидный и многие другие мембранные электроды.
9.2.4. Электроды сравнения
Электроды сравнения должны обладать устойчивым во времени воспроизводимым потенциалом, не меняющимся при прохождении небольшого тока. Чаще всего в качестве электродов сравнения применяют электроды второго рода: хлорсеребряный и каломельный.
Хлорсеребряный электрод представляет собой серебряную
7-1533
193
проволоку или пластинку, покрытую слоем AgCl и помещенную в раствор КС1. Активность ионов серебра в таком растворе равна аА + = ПРавС| . Подставляем эту величину в уравнение СС1
Нернста для серебряного электрода:
рО _____ го । RT । __
Ag+/Ag CAg + /Ag I —^inaAg4
_ рО I КТ | ПРАеС| _
^Ag^/AgT" р 111 qc|
= E0Ar-Ag + lnnPAgc> - 4^lnac> (9.12)
Первые два слагаемых в уравнении (9.12) зависят только от температуры:
F° । НПР = F°
CAg’/Ag I р 111114 AgCI Ag+/AgCI-
Прй сочетании этого уравнения с (9.12) получаем
Р __ ро КТ ।
£ Ag+/AgCl С Ag+/AgCl -р - 1Пас1-.
Как видно, потенциал хлорсеребряного электрода определяется активностью иона хлора в растворе. Обычно используется насыщенный раствор КС1. Потенциал электрода второго рода, вообще говоря, зависит от активности аниона малорастворимого соединения, входящего в состав электрода.
Каломельный электрод состоит из металлической ртути, каломели и раствора КС1. Его потенциал также зависит от активности хлорид-ионов:
>7 __ г; О RT 1
с Hgi+zHgzCh— с Hgi+/Hg,ci2 — -р- шас1 ,
где рО __ рО I RT 1пПР
cHg2*/Hg2ci2—^Hgi /HgT- 2F 1111 ‘^HgzCh-
Потенциалы хлорсеребряного, каломельного и ряда других электродов сравнения изучены при различных концентрационных и температурных условиях и их величины по отношению к стандартному водородному электроду хорошо известны. Стандартный потенциал хлорсеребряного электрода при разных температурах может быть рассчитан по уравнению
EAg< AgC| = 0,2224 - 6,4 -10“4(/ - 25) - 3,2-10“6(/ - 25)2.
Для каломельного электрода уравнение имеет вид £Hgi+/Hg2cl2= 0,2415 - 7,6- 10-V - 25).
Точное значение потенциала электрода сравнения для многих потенциометрических измерений часто не требуется, важно лишь
194
его постоянство. Однако его значение необходимо при выполнении таких измерений, когда представляет интерес не только ЭДС, но и потенциал индикаторного электрода. Если нет особых оговорок, потенциал индикаторного электрода обычно пересчитывается и относится к стандартному водородному электроду. Эта величина, конечно, уже не зависит от выбранного электрода сравнения.
9.2.5. Диффузионный потенциал
Диффузионный потенциал, или потенциал жидкостного соединения, возникает на границе между растворами двух разных электролитов или растворов разной концентрации одного электролита. Возникновение диффузионного потенциала обусловлено неравномерным распределением катионов и анионов вдоль границы раздела растворов вследствие различия в скоростях диффузии ионов через поверхность раздела. Различие в скоростях диффузии вызывается разницей в подвижностях ионов или градиентом концентраций. В простейших случаях, например при соприкосновении растворов одного электролита разной концентрации, диффузионный потенциал может быть приближенно рассчитан, если известны подвижности ионов и концентрации растворов. Однако в практических условиях анализа состав анализируемого раствора бывает неизвестным (в противном случае анализ не требуется) и теоретическая оценка диффузионного потенциала невозможна.
В зависимости от заряда ионов, их подвижностей, концентрации раствора, природы растворителя и других факторов диффузионный потенциал изменяется в очень широких пределах: от долей милливольта до десятков милливольт и более. На практике диффузионный потенциал стремятся элиминировать с помощью так называемого солевого мостика. Основным назначением его является осуществление электролитического контакта, однако с целью элиминирования диффузионного потенциала для солевого мостика применяют концентрированный раствор электролита с приблизительно одинаковыми подвижностями катиона и аниона. Очень часто в качестве электролита для солевого мостика используют насыщенный раствор КС1, применяют также растворы NH4NO3, KNO3 и др. Солевой мостик помещают между растворами, заменяя таким образом границу раздела и существенно уменьшая диффузионный потенциал. При работе с неводными растворами для солевого мостика используют спиртовые растворы Nal и KSCN.
9.3. ПРЯМАЯ ПОТЕНЦИОМЕТРИЯ
Методы прямой потенциометрии основаны на непосредственном применении уравнения Нернста для нахождения активности или концентрации участника электродной реакции по экспери-7*
195
ментально измеренной ЭДС цепи или потенциалу электрода. Наибольшее распространение среди прямых потенциометрических методов получил метод определения pH, хотя создание в последнее время надежно работающих ионоселективных электродов значительно расширило практические возможности прямых методов. Прямые потенциометрические методы часто стали называть ионометрическими методами анализа или ионометрией. Эта группа методов интенсивно развивается в связи с успехами в конструировании и улучшении качества ионоселективных электродов, позволяющих проводить быстро и точно определение концентрации или активности ионов и обладающих рядом других достоинств.
9.3.1. Определение pH
Понятие о водородном показателе введено в 1909 г. Зёренсе-ном, который под водородным показателем понимал отрицательный десятичный логарифм молярной концентрации ионов водорода: pH = — 1g [Н+]. Для определения числового значения pH Зёренсен предложил использовать измерение ЭДС элемента:
Pt, Н2|НС1, х||КС1; 0,lM|Hg2Cl2, Hg
В то время считалось, что ЭДС элемента зависит от концентрации вещества, а не от его активности:
Е = Е?о„ц-^-1п [Н+].
Величина Е°онц определялась экспериментально, причем концентрация ионов водорода [Н+] в стандартном растворе вычислялась по формуле
[Н+] — аст, (9.13)
где а — степень диссоциации НС1, вычисленная по электрической проводимости; сНС[ — молярная концентрация раствора НС1.
При этих условиях и 25 °C Е°онц= 0,3376 В и р<Н = Е__л 3376 25
= — 05’92—; символ psH означает единицу pH в шкале Зёрен-сена. Как видно, эта величина не является достаточно строгой мерой ни концентрации ионов водорода, ни их активности. Действительно, величина [Н+], рассчитанная по формуле (9.13), не является активностью ионов водорода, так как ан+ = [Н+] ун+> и не равна их концентрации, поскольку в разбавленных растворах соляной кислоты [Н+] — сНС1 ввиду полной диссоциации НС1.
В соответствии с современными представлениями ЭДС элемента Зёренсена выражается уравнением
Е = Е° - -gl 1пан+аС| + Еа (9.14)
где —диффузионный потенциал.
196
В настоящее время величина pH считается характеристикой активности ионов водорода:
pH = - lgoH+ = - 1g [Н+]Тн+ = раН.
Поэтому иногда в символ pH вводят нижний индекс «а»: раН.
В соответствии с определением pH из уравнения (9.14) находим
Рн - £J + [g°a- <915)
Уравнение (9.15) показывает, что для точного определения pH необходимы данные по диффузионным потенциалам и по активности иона С1_ в 0,1М КС1. Ни одна из этих величин не может быть получена совершенно строго, в связи с чем найденная экспериментально величина pH также не является вполне строгой. Эти трудности были преодолены путем введения соответствующего ГОСТа на шкалу pH.
Принятая в СССР по ГОСТ 8.134—74 (СТ СЭВ 629—77) шкала pH основана на воспроизводимых значениях pH следующих растворов:
1) 0,1 моль/кг Н2О раствор НС1 (pH 1,10 при 0 °C и 1,14 при 150 °C), 2) 0,05 моль/кг Н2О раствор тетраоксалата калия КНС2О4-Н2С2О4-2Н2О (pH 1,666 при 0 °C и 1,806 при 95 °C),
3) насыщенный при 25 °C раствор кислого тартрата калия С4Н5О6К (pH 3,557 при 25 °C и 3,90 при 150 °C);
4) 0,05 моль/кг Н2О раствор кислого фталата калия С8Н5О4К (pH 4,003 при 0 °C и 4,227 при 95 °C);
5) 0,025 моль/кг Н2О раствор КН2РО4 и 0,025 моль/кг Н2О раствор Na2HPO4 (pH 6,984 при 0 °C и 7,14 при 150 °C),
6) 0,08695 моль/кг Н2О раствор КН2РО4 и 0,03043 моль/кг Н2О раствор Na2HPO4 (pH 7,534 при 0 °C и 7,367 при 50 °C);
7) 0,01 моль/кг Н2О раствор натрия тетрабората Na2B4O7-ЮН2О (pH 9,464 при 0 °C и 8,68 при 150 °C),
8) насыщенный при 25 °C раствор гидроксида кальция Са(ОН)2 (pH 13,423 при 0 °C и 11,449 при 60 °C).
В ГОСТе приводятся значения pH этих растворов в широком интервале температур от 0 до 95 °C или 150 °C с шагом в 5 °. Шкала pH обладает внутренней согласованностью, т. е. экспериментально измеренная величина pH не зависит от того, какой из растворов был выбран в качестве стандартного. Набор первичных стандартных растворов используется также и в других странах — США, Великобритании, Японии и т. д.
Значения pH стандартных растворов устанавливают путем измерения ЭДС цепей без переноса. Для этого чаще всего используют цепь типа Pt(Hz) |буферный раствор, KCl|AgCl, Ag. В таких системах хотя и сохраняются трудности, связанные с оценкой коэффициентов активности отдельных ионов, но отпадает необходимость учета диффузионного потенциала.
Если pH* и рНст—значения pH исследуемого и стандартного растворов, а Ех и Е„— ЭДС элементов типа Pt(H2)|НА||КС1, P-plAgCl, Ag с исследуемым и стандартным растворами, то
р __ р0 । 2,303/? 7 тт RT * 1 р zn , с
Ех — Е —р—pH* —-р~ 1паС1- -г Сд(*), (9.16/
г> ___ r?o i 2,303/?Г тт RT i_ 1/7 /о 171
EC[— E “| p pHCT -тт- In Gci ~H Eд (ct)« (9.17)
г г
При вычитании уравнения (9.17) из (9.16) получаем
рНх — рНст
I (£* 4~ £д (ст)
' 2,303/?/'
Для экспериментального определения pH могут быть использованы различные индикаторные электроды: водородный, хингидронный, стеклянный и др. Наибольшее практическое применение в последнее время нашел стеклянный электрод, используемый в широком интервале pH и в присутствии окислителей.
Стеклянный электрод (рис. 9.3) представляет собой тонкостенный стеклянный шарик 1, заполненный раствором НС1 или каким-либо буферным раствором 2. Внутрь шарика помещают хлорсеребряный электрод 3. Это устройство обычно закрывают защитной трубкой 4.
Перед работой стеклянный электрод некоторое время вымачивают в 0,1 М НС1. При этом ионы водорода из раствора обмениваются на ионы натрия из стеклянной мембраны и в системе устанавливается некоторое равновесие. Подготовленный таким образом электрод, в котором протоны поверхности стекла находятся в равновесии с протонами раствора, может быть использован для определения pH.
Таким образом, электродная реакция на стеклянном электроде сводится к обмену ионами водорода между раствором и стеклом:
Н+ (р-р) Н+ (стекло),
т. е. она
Ионы
Рис 9.3. Стеклянный электрод
не связана с переходом электронов, водорода
на поверхности внешней стороны мембраны
находятся в равновесии с ионами водорода в исследуемом растворе и на границе раздела возникает потенциал
Ех = Е° 4—1п-^ ,
где aH+w — активность ионов водорода в исследуемом растворе; а'н+()) — активность ионов водорода на внешней поверхности мембраны.
Аналогично на границе раздела внутренней поверхности мембраны возникает потенциал
Е2 = Е° 1п-^’ , F он (2)
где ан (2j и Qh+i2)— активность ионов водорода соответственно во внутреннем растворе и на внутренней поверхности мембраны.
198
Суммарный потенциал стеклянной мембраны будет равен
Ем= Ех - Е2 = Е°\ - Е°2 + In ,
г аН* (I )аН' (2)
При постоянных значениях а'н+(2) и ан^(2) уравнение
принимает вид
Ек = const | lnaH+(x),
т. е. потенциал мембраны характеризует pH исследуемого раствора.
Измерение pH со стеклянным электродом сводится к измерению ЭДС цепи:
Hg, Hg2Cl2|KCl|cH^ стекло |НС1| AgCl, Ag
ЭДС этой цепи равна
Е = Е1-Е2, (9.18)
где
с1 со RT 1 RT 1
с 1 — с н₽2с12|нв —у in dci-(i)--у Ш ан+(4
с- c-о RТ । RT .
£2 tAgCHAg у lnaci-(2)-------р- 1П <2н+(ст>
Подставляя последние отношения в (9.18), получаем
С __ Г Е’О Е’О | RT |л GCI (2) | RT „ "1
Е — [_EhB2ci2|hb — CAgCliAg + -у1п а (1) + —у |п Gh+ictJ —
RT । e-о RT „
-p— Ш ^H+(x)*"— стекл p иц+(л)
ИЛИ
f = ДРтекл -|-2,303-y-PH
В величину Естекл входит и так называемый потенциал асимметрии, представляющий собой разность потенциалов между двумя сторонами стеклянной мембраны. Она возникает из-за несовпадения свойств разных сторон мембраны и может быть измерена экспериментально, если по обе стороны мембраны поместить один и тот же раствор. Величина Е°„екл зависит также от константы равновесия Н+ (р-р) Н (стекло), характеризующей сорт стекла и некоторые другие свойства стеклянного электрода. Стандартный потенциал стеклянного электрода обычно не определяют. При использовании заводских pH-метров эта операция заменяется настройкой приборов по стандартным буферным растворам, так как шкала pH-метров проградуирована непосредственно в единицах pH.
Основными достоинствами стеклянного электрода являются простота работы, применимость в широкой области pH, быстрое установление равновесия и возможность определения pH в экисли-
199
тельно-восстановительных системах. К недостаткам относят хрупкость их конструкции и усложнение работы при переходе к сильно щелочным и сильно кислым растворам. Тем не менее стеклянный электрод в настоящее время наиболее широко применяется для измерения pH. Этому способствует также широкий выпуск промышленностью pH-метров со стеклянным электродом.
9.3.2. Ионосепективные электроды
Решающее влияние на развитие и успехи ионометрического метода анализа оказало удачное конструирование ионоселективных электродов на основе различных мембран. Стеклянные ионоселективные электроды чувствительны к ионам щелочных металлов Li+, Na+, К , Rb+, Cs+, а также к ионам Ag+, Т1+ и NHt. Их устройство и принцип действия такие же, как и у стеклянного pH-электрода. Наиболее существенным отличием является состав стекла, из которого готовятся мембраны. Установлено, в частности, что введение А120з в стекло положительно влияет на селективность мембраны к ионам металлов, но не к Н+.
Селективный электрод, например, с натриевой функцией находится в равновесии с ионами натрия в растворе: Na+ (стекло) Na+ (р-р).
В кислом растворе это равновесие может осложняться за счет взаимодействия с ионами водорода:
Na+ (стекло) + Н+ (р-р) = Na+ (р-р) + Н+ (стекло), поэтому в кислых растворах показания такого электрода искажаются.
Важной характеристикой ионоселективного электрода является его коэффициент селективности, показывающий, во сколько раз электрод более чувствителен к данным ионам, чем к посторонним (мешающим). Например, если коэффициент селективности натриевого электрода по отношению к ионам калия составляет 1000, т. е. Лг^ к+= 103, то это означает, что данный электрод в 1000 раз чувствительнее к ионам натрия, чем к ионам калия. Другими словами, если электрод имеет потенциал Е при концентрации ионов натрия равной 10~3 моль/л, то для достижения этого потенциала потребуется концентрация ионов калия в 1000 раз большая, т. е. 1 моль/л.
В техническом паспорте электрода обычно указывают кратность превышения концентрации, при которой электрод сохраняет селективность. Например, в паспорте электрода на калий марки ЭМ-К-01 указано, что электрод селективен в присутствии ионов Na+ и NH^ при превышении их концентрации соответственно в 200 и 20 раз по сравнению с ионом К+.
Стеклянные ионоселективные электроды широко используют для определения катионов щелочных металлов в различных биологических пробах — крови, плазме, сыворотках и т. д.,
200
в объектах окружающей среды — водах, растениях, различных экстрактах и т. д. Определения с помощью ионоселективных электродов успешно конкурируют с пламенно-фотометрическими методами по точности и нередко превосходят их по скорости.
Твердые ионоселективные электроды. В твердых мембранных электродах ионочувствительный элемент изготовляется из малорастворимого кристаллического вещества с ионным характером проводимости. Перенос заряда в таком кристалле происходит за счет дефектов кристаллической решетки. /Вакансии могут заниматься ионом только определенного размера и заряда, что обусловливает высокую селективность кристаллических мембран. Конструктивно такие электроды сходны со стеклянными: в обоих электродах мембрана разделяет исследуемый раствор и раствор сравнения, в котором находится электрод сравнения (обычно хлорсеребряный). Из электродов этого типа широко применяется фторидный электрод, в котором мембраной является монокристалл LaF3, имеющий чисто фторидную проводимость, с добавкой E11F2 для увеличения электрической проводимости. Чувствительность фторидного электрода позволяет проводить измерения равновесной концентрации фторид-ионов F- в широкой области концентраций от 10~6 до 1 моль/л. В этой области отклонений от уравнения Нернста не наблюдается. Селективность электрода очень высока — даже тысячекратный избыток посторонних ионов (галогенид-, нитрат-, сульфат-ионов и др.) по сравнению с фторид-ионом не мешает определению F- и только в присутствии ОН-ионов селективность падает (ОН~ является мешающим ионом). Работа фторидного электрода ухудшается также в присутствии лигандов, образующих с ионом La3+ прочные координационные соединения в растворе (цитрат-, оксалат-ионы и др.). Вполне понятно также, что с увеличением кислотности среды равновесная концентрация фторид-ионов F" в растворе уменьшается за счет образования молекул HF. Таким образом, показания фторидного электрода в кислой области будут существенно зависеть от pH. В щелочной области на поверхности электрода может образоваться осадок Ьа(ОН)з, что также вызовет искажение показаний электрода. Точные границы pH, в которых показания фторидного электрода от pH зависят несущественно, привести трудно, так как с уменьшением концентрации фторид-иона эта область также уменьшается. Для растворов с концентрацией фторид-иона п-10""4 моль/л и более этот интервал охватывает область значений pH примерно от 4...5 до 8...9.
Фторидный электрод используется для определения фторид-иоцов F- в питьевой воде, различных биологических пробах, витаминах, при контроле за загрязнением окружающей среды и т. д. Он широко применяется также для исследования процессов образования фторидных комплексов в растворе и других реакций с участием фторид-иона.
201
Практическое значение имеет ионоселективный электрод с мембраной из сульфида серебра, пригодный для измерения концентрации (активности) и Ag+- и S2--hohob. Подвижными в мембране Ag2S являются ионы Ag+ Серебро этим электродом может быть определено в интервале концентраций от 1 до 10-7 моль/л, а в некоторых условиях до 10~12 моль/л и ниже. В столь же низких концентрациях могут быть определены 52--ионы.
На основе сульфида серебра конструируются также различные галогенидные и металлочувствительные электроды. Для этого в сульфид серебра вводят галогениды серебра или сульфиды меди, кадмия, свинца и некоторых других металлов. Электроды на основе сульфида серебра с добавкой соответствующего галогенида серебра чувствительны к ионам С1-, Вг Д I~, CN- и др. Введение в сульфид серебра сульфидов других металлов позволяет получить электрод, чувствительный к ионам металла, внесенного со вторым сульфидом (Cd2+, Pb2+, Си2+ и др.). Используются также электроды на основе сульфида или селенида меди, чувствительные к ионам Си2+.
В гетерогенных твердых электродах активное вещество смешивается с инертной матрицей. В качестве ионочувствительного активного вещества используются различные малорастворимые соединения, такие, как сульфат бария, оксалат кальция и т. д., а в качестве инертной матрицы эпоксидные смолы, поливинилхлорид, силиконовый каучук и др. Потенциал таких электродов недостаточно устойчив и селективность невелика.
Жидкостные ионоселективные электроды. В электродах с жидкой мембраной раствор сравнения отделен от анализируемого тонким слоем органической жидкости, содержащей жидкий ионит, не смешивающейся с водой, но селективно реагирующий с определяемым ионом. Слой ионочувствительной органической жидкости получается пропиткой этой жидкостью пористой гидрофобной мембраны из пластика. Схема жидкостного ионоселективного электрода показана на рис. 9.4. Внутренний хлорсеребряный электрод 1 погружен в раствор МС12, где М —
определяемый катион. Пористая мембрана 3 одной стороной соприкасается с раствором сравнения хлорсеребряного электрода, другой — с анализируемым раствором. Ионочувствительная жидкость в резервуаре 2, про-
Рис 9.4 Схема жид костного ионоселективного электрода
2 питывающая мембрану, состоит из жидкого органического ионита, имеющего кислотные, основные или хелатообразующие функциональные группы, растворенного в подходя-
- щем растворителе, который с водой не смешивается.
Электрод такого типа для определения кальция содержит в качестве жидкого ионита кальциевую соль алкилфосфорной кисло-
202
ты, растворенную в диалкилфенилфосфонате, или аналогичную композицию Раствор сравнения внутреннего хлорсеребряного электрода 1 содержит в этом случае СаС12. С каждой стороны ионоселективной мембраны устанавливается равновесие:
CaRz (орг) 2R- (орг) + Са2+ (водн)
Концентрация (и активность) иона Са2+ в растворе сравнения постоянна, поэтому потенциал электрода будет зависеть только от концентрации (активности) иона Са2+ в анализируемом растворе. Эта зависимость передается уравнением Нернста:
Е = Е°ембр — 0,0291g аСаг+. (9.19)
Уравнение (9.19) соблюдается в области концентраций (активностей) от 10-5 до 1 моль/л в области pH от 6,0 до 11,0. При более высоких значениях pH возможно образование осадка Са(ОН)2, а в кислой области равновесие органического ионита с кальцием осложняется участием иона водорода.
В практике применяют также ионоселективные мембранные электроды на ионы калия, натрия, аммония и некоторые другие. Сконструированы газочувствительные мембранные электроды для определения NH3, NO и других газов. В пленочных электродах вместо жидкой мембраны используют тонкую пленку. У пленочных электродов такой же механизм действия, что и у мембранных, но они долговечнее и более удобны в работе.
9.3.3. Основные приемы ионометрического анализа
Во всех приемах прямой потенциометрии используется зависимость потенциала индикаторного (обычно ионоселективного) электрода от активности или концентрации определяемого вещества. Активность определяемого иона может быть рассчитана по уравнению
lgaM= (£73;45Hj)nf , (9.20)
где Еиид—экспериментально найденный потенциал индикаторного электрода.
Величину Еиид находят по ЭДС исследуемого элемента:
Еиид= Еср—Е + Ед, (9.21)
где Е — ЭДС элемента; Еср—потенциал электрода сравнения; Ед—диффузионный потенциал.
Сочетание уравнений (9.20) и (9.21) дает
i„ п _ (£сР — Е — Е°тл + E^nF
ё Ым 2,303/? Т
ИЛИ
пп ___ _ [£ — (£Ср — Евал +E7)]nF
Р - - 1 g Ом --------^303/?f------ -
203
Так как ам = СмТм, где ?м— коэффициент активности; — концентрация иона М, то
nr _ __ E£-(£cp-EU + Ei)]nf /ооо\
рем— — lgCM=----------2303/? Г---- >g?M. (9.22)
Уравнение (9.22) лежит в основе нескольких аналитических приемов. Трудности, связанные с неопределенностью коэффициента активности отдельного иона, при определении pH были преодолены путем разработки шкалы pH на основе стандартных буферных растворов. Для других ионов таких стандартных растворов нет, поэтому трудности оценки коэффициентов активности отдельных ионов остаются.
Активности ионов и иных растворенных частиц используются в расчете равновесий, стандартных электродных потенциалов и т. д., но результаты аналитического определения, как правило, должны показывать концентрацию вещества. Прямой рас чет концентрации по активности иона не всегда можно сделать с необходимой точностью, так как ионная сила анализируемого раствора, от которой зависят коэффициенты активности, обычно неизвестна. Введение в анализируемый раствор так называемых фоновых или поддерживающих электролитов обеспечивает постоянное значение ионной силы и коэффициентов активности. В практике используется несколько приемов ионометрии.
Метод градуировочного графика. При постоянной и одинаковой в обоих полуэлементах ионной силе и неизменном составе коэффициенты активности ионов .будут одинаковы, а диффузионный потенциал пренебрежимо мал. В этих условиях уравнение (9.22) принимает вид
рс1Л=аЕ + Ь, (9.23)
где а и b — константы, смысл которых становится ясным при сравнении уравнений (9.22) и (9.23).
В соответствии с уравнением (9.23) график в координатах £ — 1g См должен быть линеен. Для построения градуировочного графика измеряют ЭДС элемента при нескольких концентрациях определяемого иона и постоянной ионной силе. Затем определяют ЭДС, помещая индикаторный электрод в анализируемый раствор, и по величине ЭДС и градуировочному графику находят концентрацию иона в анализируемом растворе. Перед определением ЭДС в анализируемом растворе стремятся создать такой же солевой фон и ионную силу, какие были в стандартных растворах, использованных для построения градуировочного графика. Для этого в анализируемый раствор вводят определенный объем концентрированного раствора фонового электролита. Успешно применяется метод градуировочного графика при анализе слабо минерализованных растворов, поскольку в таких пробах можно легко создать заданное значение ионной силы. Практи чески используется этот метод, например, при определении фто
204
рид-иона в питьевой воде с помощью фторселективного электрода.
Метод концентрационного элемента. Идея метода заключается в том, что составляют концентрационный элемент с двумя одинаковыми индикаторными электродами и помещают один из них в раствор с известной концентрацией определяемого иона, а другой — в анализируемый раствор. ЭДС такого элемента будет равна
£ |п+ £'- (9-24)
где сст и сх — концентрации стандартного и исследуемого растворов; уст и ух — коэффициенты активности определяемого иона в стандартном и исследуемом растворах; £д—диффузионный потенциал.
Если состав и ионная сила обоих растворов примерно одинаковы за счет одного и того же фонового электролита, то диффузионный потенциал становится пренебрежимо малым, а коэффициенты активности уст и ух примерно одинаковыми. Тогда уравнение (9.24) принимает вид
£ = -^1п-£^. nF сх
При £=0, очевидно, сст—сх, так как при этом In —= 0. Вы-
Сх
полнить условие Е = 0 можно разбавлением стандартного раствора раствором фонового электролита или добавлением более концентрированного стандартного раствора.
Вместо экспериментального выравнивания концентраций можно добавить несколько порций разбавителя или стандартного раствора в заведомом избытке и после каждого добавления измерить ЭДС. Таким образом можно получить значения ЭДС до и после £ = 0. По этим данным строят кривую линейного титрования в координатах £ — 1g сст и находят точку ее пересечения с осью абсцисс, которая и соответствует равенству сст= сх, так как в этой точке £ = 0. Таким методом определяют фторид-ион, ион серебра и другие, для которых сконструированы ионоселективные электроды.
Метод добавок. Для применения этого метода необходимо достаточно точно знать электродную функцию по отношению к определяемому иону. Обычно ее находят заранее по стандартным растворам. При осуществлении метода добавок сначала измеряют ЭДС цепи с анализируемым раствором (£*), затем добавляют к нему определенный объем стандартного раствора и снова измеряют ЭДС.
До добавления стандартного раствора ЭДС цепи равна
£х = £° +-^1псхУх + £д,
205
а после добавления
£х + ет= Е° +-^ 1П (Сх + Дс)Ь + Ед,
где Ас — прирост концентрации определяемого иона за счет введения стандартного раствора.
Разность этих ЭДС равна
£<+ст -ЕХ = \Е In — = /г 1g с* + Дс . (9.25)
Введение добавки стандартного раствора практически не изменило ионной силы раствора, поэтому ух и Ед остались теми же самыми и при вычитании сократились.
Из уравнения (9.25) получаем концентрацию определяемого иона:
Метод добавок, как уже отмечалось, автоматически учитывает влияние третьих компонентов и позволяет находить концентрацию очень разбавленных растворов.
9.4. ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Потенциометрическое титрование основано на определении точки эквивалентности по результатам потенциометрических измерений. Вблизи точки эквивалентности происходит резкое изменение (скачок) потенциала индикаторного электрода. Это наблюдается, конечно, лишь тогда, когда хотя бы один из участников реакции титрования является участником электродного процесса. Так, например, титрование по методу кислотно-основного взаимодействия может быть выполнено со стеклянным электродом, определение хлорида — с хлорсеребряным и т. д. Так же, как и в других титриметрических методах, реакции потенциометрического титрования должны протекать строго стехиометрии е-ски, иметь высокую скорость и идти до конца.
Для потенциометрического титрования собирают цепь из индикаторного электрода в анализируемом растворе и электрода сравнения. В качестве электродов сравнения чаще всего применяют каломельный или хлорсеребряный.
9.4.1. Определение точки эквивалентности
На рис. 9.5, а представлена кривая титрования соляной кислоты гидроксидом натрия. Она почти точно воспроизводит теоретическую кривую титрования сильной кислоты сильным основанием. Как видно, в точке эквивалентности происходит резкий скачок ЭДС, вызванный резким изменением потенциала индикаторного электрода. По этому скачку можно определить точку
206
Рис. 9.5. Кривые потенциометрического титрования: а — обычная кривая, б — дифференциальная кривая, в — кривая титрования по второй производной, г—кривая Грана
эквивалентности и потом рассчитать содержание соляной кислоты.
Для нахождения точки эквивалентности часто строят дифференциальную кривую в координатах ЛЕ/ЛГ—V (рис. 9.5, б). На точку эквивалентности указывает максимум полученной кривой, а отсчет по оси абсцисс, соответствующий этому максимуму, дает объем титранта, израсходованного на титрование до точки эквивалентности. Определение точки эквивалентности по дифференциальной кривой значительно точнее, чем по простой зависимости Е — V.
Поскольку производная функции, имеющей максимум, в точке максимума равна нулю, вторая производная потенциала по объему (Д2£/ДГ2) в точке эквивалентности будет равна нулю. Это свойство также используется для нахождения точки эквивалентности (рис. 9.5, в).
В простом и удобном методе Грана точка эквивалентности определяется по графику в координатах — V. Перед точкой эквивалентности и после нее кривая Грана линейна, а сама точка эквивалентности находится как точка пересечения этих прямых (рис. 9.5, г). Достоинства и удобства метода Грана особенно заметны при анализе разбавленных растворов, позволяя определить точку эквивалентности с достаточной точностью вследствие линейности графика.
Кроме рассмотренного классического метода потенциометрического титрования нередко используют его новые варианты: некомпенсационные методы и методы титрования под током.
В некомпенсационном методе потенциометрического титрования измеряют не ЭДС, а ток, возникающий в гальваническом элементе. В начальный момент титрования ЭДС элемента компенсируют внешней ЭДС и ток в цепи отсутствует. В процессе титрования компенсация нарушается, в цепи возникает ток, при-
207
чем в области точки эквивалентности ток резко возрастает пропорционально скачку ЭДС в этой области. Точку эквивалентности можно фиксировать непосредственно по резкому возрастанию тока или найти графически по зависимости ------И, где
Д/ — приращение силы тока при добавлении в раствор ДУ титранта.
К некомпенсационным методам можно отнести и потенциометрическое титрование с биметаллической парой электродов. Этот метод основан на том, что некоторые инертные металлы с разной скоростью отзываются на изменение потенциала системы. Например, платина быстро реагирует на изменение отношения концентраций окисленной и восстановленной формы, а вольфрам медленно. Поэтому, если опустить платиновый и вольфрамовый электроды в титруемый раствор, содержащий окислительно-восстановительную систему, и измерять разность потенциалов между ними в ходе титрования, то до точки эквивалентности она будет близка к нулю, а в точке эквивалентности резко возрастает.
Скорость некоторых окислительно-восстановительных реакций, особенно с участием органических веществ, оказывается недостаточной для использования их в титриметрических методах. В этих случаях электроды поляризуют путем пропускания небольшого тока (10-5 А), что резко увеличивает скорость установления равновесных потенциалов и существенно — скачок титрования. Титрование под током позволяет проводить анализ систем, которые в отсутствие тока потенциометрически не титруются.
9.4.2. Виды потенциометрического титрования
Кислотно-основное титрование. В кислотно-основном титровании в качестве индикаторного обычно используют стеклянный электрод, как правило, входящий в комплект серийно выпускаемых промышленностью pH-метров. Потенциометрический метод позволяет провести количественное определение компонен тов в смеси кислот, если константы диссоциации различаются не менее чем на три порядка. Например, при титровании смеси, содержащей соляную и уксусную кислоты, на кривой титрования обнаруживается два скачка. Первый свидетельствует об окончании титрования НС1, второй скачок наблюдается при оттитро-вывании уксусной кислоты. Также несколько скачков имеют кривые титрования многоосновных кислот, константы диссоциации которых существенно различаются (хромовая, фосфорная и др.).
Широкие возможности анализа многокомпонентных смесей без разделения открывает применение неводных растворителей. Например, определение содержания соляной и монохлоруксусной кислот в смеси титрованием водного раствора является сложной задачей в связи с трудностью обнаружения двух скачков титро
208
вания. При титровании в ацетоне оба скачка выражены достаточно четко и содержание каждой кислоты в смеси может быть рассчитано.
Комплексонометрическое титрование. Потенциометрическое титрование катионов комплексоном (III) (ЭДТА) можно проводить с использованием в качестве индикаторного электрода соответствующего металла: титрование солей меди с медным электродом, солей цинка — с цинковым и т. д. или подходящего ионоселективного электрода. Однако многие металлические индикаторные электроды необратимы, а число ионоселективных электродов невелико.
Для комплексонометрических титрований может быть использован универсальный электрод Hg | HgY2~ или Au(Hg) | HgY2-, где Au(Hg) —амальгамированное золото, HgY2- — комплекс ртути с анионом этилендиаминтетрауксусной кислоты. При титровании, например, ионов кальция собирают цепь типа
Hg | Hg2Cl2, КС11 Ca2+, HgY2" (10-4) | Hg
Устойчивый комплексэнат ртути (IgPHgY2 -== 21,8) в очень небольшой степени диссоциирует:
HgY2- = Hg2+ + Y4- (9.26)
что определяет величину потенциала ртутного электрода:
Енег+1 Hg = Днв2+1 Hg + 0,0291g [Hg2+]
или, учитывая диссоциацию HgY2- по схеме (9.26),
fHg2+,Hg = £Hga+1Hg + 0,0291g , (9.27)
где ₽HgY=--константа устойчивости HgY2~.
Образующийся при титровании комплексонат CaY2~ характеризуется константой устойчивости PcaY2 :
tcg‘iY|v4- (9'28)
Из уравнения (9.28) находим [Y4~] и подставляем в (9.27): г- гЧ) , [HgY2-] [Ca2+]₽CaY2-
£нв’+1 Hg = £Hg2+1 Hg + 0,0291g-[CaY2-]₽HgY2- ’
или
£Hg2+1 Hg - £°Hg-1 Hg + 0,0291g }^ДРНС;;: - + 0.0291g [C a2+].
(9.29) Концентрации HgY2- и CaY2- при приближении к точке эквивалентности изменяются очень незначительно. Объединяя первые Два слагаемых правой части уравнения (9 29) в const, получаем
£нв2-1 Hg — const + 0,929lg[Ca2+]. (9 30)
209
Уравнение (9.30) показывает, что потенциал ртутного электрода HgY2- | Hg чувствителен к ионам Са2+. В области точки эквивалентности, очевидно, произойдет резкое изменение потенциала и будет наблюдаться скачок титрования. С помощью ртутного электрода этого типа могут быть оттитрованы любые ионы, которые образуют с Y4- комплексы с константой устойчивости, не превышающей ₽hky!-- Это, например, ионы Mg2+, Са2+, Со2+, Ni2+, Cu2+, Zn2+ и др.
Титрование по методу осаждения. Индикаторными электродами в методах потенциометрического титрования, использующих реакции осаждения, служат металлические или мембранные электроды, чувствительные к определяемому иону или иону-осадителю. Практически по м,етоду осаждения могут быть определены катионы серебра, ртути, цинка, свинца, анионы хлора, брома, иода и некоторые другие. Смесь галогенидов, например 1~ и С1“, может быть оттитрована без разделения нитратом серебра. Серебряный электрод позволяет фиксировать два скачка в ходе такого титрования. Первый скачок свидетельствует об оттитровывании иодид-иона и может быть использован для расчета содержания этого иона, второй скачок относится к окончанию осаждения хлорид-иона. По второму скачку можно рассчитать суммарное содержание галогенидов или концентрацию хло-рид-иона, если концентрация иодид-иона будет известна из данных по титрованию до первого скачка.
Окислительно-восстановительное титрование. Кривые окислительно-восстановительного титрования могут быть построены в координатах или рМ— К(титранта) или Е— К(титранта), если рМ = — 1g [М] ([М] — концентрация участника реакции, Е — потенциал системы, V(титранта) — объем титранта). Кривые титрования первого типа представляют практический интерес, когда имеется индикаторный электрод, чувствительный к М. Кривые второго типа имеют более общее значение, так как любое окислительно-восстановительное титрование может быть проведено по измерению Е с использованием индикаторного электрода из благородного металла, чаще всего платины.
9.4.3. Автоматическое титрование
Сравнительная легкость автоматизации потенциометрических измерений позволила создать ряд автоматических приборов для титрования — автотитраторов. Отечественный автотитратор или блок автоматического титрования типа БАТ-15 выпускается в комплекте с бюреткой и рН-метром — милливольтметром, имеющим диапазон измерений в единицах pH от — 1 до —(-14 (100 —1400 мВ). Схема установки для автоматического титрования представлена на рис. 9.6.
В емкость с анализируемым раствором 1 вводится дозирующая трубка 2 для подачи титранта, индикаторный электрод 4
210
(стеклянный или платиновый) и хлорсеребряный электрод сравнения 3. Подача титранта в раствор регулируется клапаном 9. Напряжение Ех, пропорциональное ЭДС системы, с выхода рН-метра — милливольтметра 5 подается на вход БАТ-15, где сравнивается с заранее заданным напряжением Ео на задатчике конечной точки титрования 6. Разность заданной и экспериментально наблюдаемой величины Ех — Ео через усилитель 7 подается на бесконтактное электронное реле 8, которое управляет работой электромагнитного клапана 9, открывающего или
Рис 9 6 Схема установки для автоматического титрования
закрывающего подачу титранта. При Ех = Ео клапан прекращает подачу титранта. Объем рабочего раствора отсчитывают по бюретке. Погрешность титрования на этой установке не превышает ч- 1 °/ ± 1 /о-
9.5. ПОТЕНЦИОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ФИЗИКО-ХИМИЧЕСКИХ СВОЙСТВ ВЕЩЕСТВ
Потенциометрические измерения часто используют не только в химико-аналитических целях для определения концентрации вещества или установления точки эквивалентности. Их широко применяют для исследования реакций в растворе, определения констант равновесия и различных характеристик вещества. По данным потенциометрических измерений вычисляют константы диссоциации кислот и оснований, константы устойчивости координационных соединений, произведение растворимости и т. д., рассчитывают тепловые эффекты и другие термодинамические характеристики процессов в растворе.
Широкое распространение потенциометрического метода связано с тем, что он с высокой точностью дает экспериментальные значения равновесных концентраций (активностей) ионов в исследуемых системах и для обработки потенциометрических данных имеется соответствующий математический аппарат, основанный на теории ступенчатых равновесий. Изложение этих методов можно легко найти в специальной литературе. Здесь будет рассмотрено лишь несколько простых примеров.
Константу диссоциации слабой кислоты можно определить по экспериментально измеренным pH ее растворов известной концентрации или по кривой титрования кислоты щелочью. Диссоциация кислоты НА происходит по схеме
НА = Н+ + А~
211
Если раствор кислоты имеет концентрацию Сна, то равновесные концентрации продуктов диссоциации будут равны: [Н+] = = [А~] — 10 рН, а [НА] = Сна — 10 “рН. Для простоты пренебрегаем разницей между концентрацией и активностью и подставляем равновесные концентрации в выражение для константы диссоциации кислоты:
[Н+] [А-] _ (10~рН)2 [НА] ~ ?нд-10^’
Как видно, для расчета константы диссоциации необходимо экспериментально определить pH раствора.
При титровании кислоты щелочью в точке, где прореагировала половина исходного количества кислоты, концентрации частиц НА и А- одинаковы, и из уравнения (9.31) следует
К = [Н+] ,/2 = 10_рН1/2.
здесь pH|,2 относится к точке, где кислота прореагировала наполовину.
Произведение растворимости малорастворимой полностью диссоциированной соли МА может быть рассчитано по уравнению
ПРМЛ=[М] [А] = [М]2 —[А]2,
где [М] и [А] — концентрации катиона М и аниона А в насыщенном растворе соли, определяемые потенциометрически с помощью ионоселективных электродов.
Произведение растворимости можно определить также по экспериментальному значению равновесной концентрации катиона или аниона в растворе, содержащем избыточное количество противоиона и осадок. Для определения по этому методу произведения растворимости BaSO4 в соответствующий объем 0,1 М Na2SO4 вводят каплю раствора ВаС12. Концентрация сульфат -иона при этом практически не меняется, а осадок BaS'O4 образуется. Равновесную концентрацию ионов бария в таком растворе при наличии осадка определяют с помощью ионоселективного электрода и рассчитывают произведение растворимости как nPBaSo, = [Ва2+]-0,1, где 0,1—концентрация исходного раствора сульфата натрия, практически равная равновесной концентрации иоиов SO2- после добавления капли раствора ВаС12.
Для расчета констант устойчивости координационных соединений и констант диссоциации многоосновных кислот обычно используют специальный математический аппарат, учитывающий одновременное протекание нескольких процессов ступенчатого комплексообразования или ступенчатой диссоциации кислот.
212
9.6. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Большое практическое значение имеют потенциометрические методы определения pH раствора со стеклянным и другими электродами. а также прямые потенциометрические определения концентрации (активности) других ионов с помощью ионоселектив ных электродов (ионометрия). Сконструированные ионоселективные электроды на ионы Сч5Л Ag2+, Ag+, Са2+, Na+, К+, С1“, F-, S2-, NOr и др. успешно применяют в анализе различных технологических растворов, объектов окружающей среды и т. д. Потенциометрические датчики на основе ионоселективных электродов позволяют следить за ходом технологического процесса.
Во многих областях находит практическое применение кальциевый ионоселективный электрод. Помимо традиционного анализа воды, различных растворов и т. д. большое практическое значение кальциевый электрод имеет в медико-биологических исследованиях, клинической медицине и т. д., поскольку концентрация (активность) ионов кальция влияет на многие процессы жизнедеятельности и физиологические процессы (нервная деятельность, функция ферментов и т. д.). Известен мембранный ионоселективный электрод, позволяющий определять жесткость воды, так как он имеет примерно одинаковую чувствительность на оба иона (кальций и магний).
Другой важной областью применения потенциометрических методов является потенциометрическое титрование кислот, оснований, солей и других веществ, где также эффективно используют ионоселективные электроды. Потенциометрические методы успешно применяют в анализе мутных и окрашенных растворов и в анализе растворов на основе смешанных и неводных растворителей.
9.7. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Основными достоинствами потенциометрического метода являются его высокая точность, высокая чувствительность и возможность проводить титрования в более разбавленных растворах, чем это позволяют визуальные индикаторные методы. Необходимо отметить также возможности определения этим методом нескольких веществ в одном растворе без предварительного разделения и титрования в мутных и окрашенных средах. Значительно расширяется область практического применения потенциометрического титрования при использовании неводных растворителей. Они позволяют, например, найти содержание ком понентов, которые в водном растворе раздельно не титруются, провести анализ веществ, нерастворимых или разлагающихся в воде и т. ц. Немаловажным достоинством потенциометрии является также возможность автоматизировать процесс титрования. Промышленность выпускает несколько типов автотитрато-ров, использующих потенциометрические датчики.
213
К недостаткам потенциометрического титрования можно отнести не всегда быстрое установление потенциала после добавления титранта и необходимость во многих случаях делать при титровании большое число отсчетов.
! Вопросы
1. На чем основаны потенциометрические методы анализа?
2. Какая зависимость выражается уравнением Нернста? Пояснить смысл входящих в него величин.
3. Что представляют собой электроды I и II рода? Привести примеры этих электродов.
4. Какие функции выполняют индикаторные электроды и какие — электроды сравнения? Указать требования, которые к ним предъявляются.
5. Привести схему установки для потенциометрических измерений.
6. Зачем нужен стандартный гальванический элемент? Какой элемент обычно используется в потенциометрии в качестве стандартного?
7. Что такое солевой мостик и каково его назначение?
8. В чем сущность потенциометрического определения pH раствора? Какие индикаторные электроды могут быть использованы для определения pH?
9. Как устроен стеклянный электрод? Как можно определить стандартный потенциал этого электрода? Указать достоинства и недостатки стеклянного электрода.
10. Каковы основные типы ионоселективных электродов? Как они устроены? Какие имеют характеристики?
11. Указать достоинства, недостатки и области применения метода прямой потенциометрии.
12. В каких координатах строят кривые потенциометрического титрования? Чем обусловливается выбор координат?
13. В чем сущность некомпенсационного метода потенциометрического титрования и титрования под током?
14. Назвать соответствующие пары электродов и привести примеры потенциометрического титрования с использованием: а) реакций кислотно-основного взаимодействия; б) реакций осаждения; в) реакций комплексообразования; г) реакций окисления — восстановления.
15. Каковы особенности потенциометрического титрования в неводных средах? Какие требования предъявляются к неводному растворителю?
16. Привести примеры потенциометрических определений в среде ледяной уксусной кислоты по методу: а) прямого титрования; б) обратного титрования; в) замещения.
17. Назвать достоинства и области применения потенциометрического титрования в неводных средах.
214
Задачи
1. Рассчитать pH раствора по следующим данным:
Вариант Электрохимическая цепь (элемент) ЭДС, в t. °C
1 Водородный электрод I СН3СООН [[0,1 М KCL, Hg2Cl2 I Hg 0,498 30
2 Хингидронный электрод | N2H4 + Н2О || нас. КС1, Hg2Cl21 Hg 0,004 18
3 Водородный электрод | НСООН II 0,1 М КС1, AgCl 1 Ag 0,580 20
4 Хингидронный электрод | HCOONa || 1 М КС1, AgCl I Ag 0,006 20
5 Водородный электрод I Н3ВО3 || 1 М КС1, Hg2Cl21 Hg 0,594 25
6 Водородный электрод | акриловая кислота || 1 М КО, Hg2Cl21 Hg 0,467 30
Ответ-. 1) 2,69; 2) 7,83; 3) 4,99; 4) 7,91; 5) 5,27; 6) 3,08.
2. В стандартных растворах CdSO4 с различной активностью Cd2+ были измерены электродные потенциалы кадмийселектив-ного электрода относительно хлорсеребряного электрода:
acd, моль/л. 1,0-10-' 1,0-10“2 1,0-Ю-3 1,0-Ю-4 1,0-Ю-6
—£, мВ . 75,0 100 122 146 170
По этим данным построили градуировочный график в координатах Е — paCd.
Исследуемый раствор соли кадмия объемом 10,00 мл разбавили водой до 50,00 мл в мерной колбе и измерили электродный потенциал кадмийселективного электрода в полученном растворе (Ех):
Вариант 1 2 3 4
—Ех, мВ . 94,0 116 130 159
Определить активность исследуемого раствора соли кадмия (моль/л). Ответ: 1) 8,90-10-2 моль/л; 2) 1,00-10-2 моль/л; 3) 2,55-10-3 моль/л; 4) 1,58-10“4 моль/л.
3. В стандартных растворах соли калия с концентрацией с(К+) были измерены электродные потенциалы калийселектив-ного электрода относительно хлорсеребряного электрода:
с(К+), моль/л 1,0-10-' 1,0-Ю-2 1,0-Ю-3 1,0-Ю-4
мВ 100 46.0 —7,00 —60,0
По этим данным построили градуировочный график в координатах Е — рс(К+).
Навеску образца массой 0,2000 г, содержащего калий, растворили в воде и объем довели до V мл. Затем измерили электродный потенциал калийселективного электрода (Ех) в получен-
ном растворе:
вариант 1 2 3 4
У, мл . 100,0 250,0 500,0 1000
Ех, мВ . . 60,0 34,0 10,0 —30,0
215
Вычислить массовую долю (%) калия в образце. Ответ. 1) 34,80%; 2) 27,47%; 3) 19,55%; 4) 6,94 %.
4. Анализируемый раствор НС1 разбавили в мерной колбе до 100,0 мл и аликвоту объемом 20,00 мл оттитровали потенциометрически 0,1000 М NaOH.
Построить кривые титрования в координатах pH — V и ЛрН/дг— V и определить массу НС1 в растворе (мг) по следующим данным:
17 (NaOH), мл рн Вариант 1 2,00 6,98 2,02 9,95 2,05 10,53 2,10 10,65
1,50 2,64 1,80 3,05 1,90 3,36 1,95 3,64 1,98 4,05
Ва р и а н т 2
Г(NaOH), мл 1,30 1,50 1,60 1,65 1,68 1,70 1,72 1,74 1,80
pH . 1,78 3,03 3,34 3,64 4,03 6,98 9,96 10,36 10,66
Ответ: 1) 36,5 мг; 2) 31,0 мг.
5, Построить кривые потенциометрического титрования в координатах pH—V и ДрН/дг—V и определить концентрацию раствора СНзСООН (г/л), если при титровании 10,00 мл этой кислоты 0,1000 М КОН получили следующие результаты:
Вариант 1
V(KOH), мл 15,00 18,00 19,00 19,50 19,90 20,00 20,10 20,50 21,00
pH 5,22 5,71 6,04 6,35 7,05 8,79 10,52 11,22 11,51
Вариант 2
Г(КОН), мл 10,00 13,00 14,00 14,50 14,90 15,00 15,10 15,50 16,00
pH 5,05 5,56 5.88 6,19 6,92 8,82 10,59 11,29 11,58
Ответ: 1) 12,01 г/л; 2) 9,01 г/л.
6. Анализируемый раствор метиламина CH3NH2 объемом 20,00 мл разбавили в мерной колбе до 100,0 мл, затем 10,00 мл полученного раствора оттитровали потенциометрически 0,1000 М НС1.
Построить кривые титрования в координатах pH — V и ЛрН/Д-И— V и определить концентрацию исходного раствора метиламина (моль/л) по следующим данным:
Вариант 1
Г(НС1), мл 10,60 12,00 14,00 14,50 14,90 15,00 15,10 15,50 16,00
pH . 10,40 10,12 9,56 9,28 8,42 6,02 3,52 2,85 2,55
Вариант 2
Г(НС1), мл 12,00 15.00 17,00 17,50 17,80 18.00 18.20 18,50 19,00
pH 10,36 9.96 9,43 9.17 8,28 5.99 3,28 2.89 2,58
Ответ: 1) 0,3750 моль/л; 2) 0,4500 моль/л.
216
7. Смесь соляной и борной кислот оттитровали потенциометрически 0,1000 М NaOH последовательно: сначала оттитровали НС1 (израсходовав объем титранта Vi), затем прибавили к раствору глицерин и оттитровали Н3ВО3 по первой ступени (получив суммарный объем титранта Vs).
Построить кривые титрования в координатах pH — V и ДрН/дк— К определить объемы Vt и Vs и рассчитать концентрацию НС1 и Н3ВО3 (г/л), если для анализа было взято 20,00 мл смеси кислот и при титровании получены следующие данные:
V'(NaOH), мл . рн
У(NaOH), мл . pH
Вариант 1
0,00 0,20 0,30 0,40 0,46 0,50 0,55 0,60 1 прибавили
2,60 2,84 3,02 3,40 3,95 5,58 7,03 7,38 J глицерин
0,80 1,00 1,20 1,40 1,45 1,50 1,55 1,60 1,70
5,95 6,25 6,55 7,04 7,28 7,73 8,55 9,10 9,55
У(NaOH), мл рн
К(NaOH), мл pH
Вариант 2
. 0,00 0,40 0,60 0,80 1,00 1,05 1,10 1,20 1
. 2,45 2,61 2,72 2,85 3,42 5,82 6,98 7,32 J
1,40 1,60 1,80 2,00 2,10 2,15 2.20 2,25
5,88 6,26 6,70 7,28 7,67 8,10 9,03 9,75
прибавили глицерин
2,40
10,28
Ответ: 1) 0,0912 г/л НС1 и 0,3246 г/л Н3ВО3; 2) 0,1914 г/л НС1 и 0,3555 г/л Н3ВО3.
8. Навеску серебряного сплава массой 2,157 г растворили и после соответствующей обработки довели объем раствора до 100,0 мл.
Построить кривые потенциометрического титрования в координатах Е— V и Л£/дг— V и определить массовую долю (%) серебра в сплаве, если при титровании 25,00 мл приготовленного раствора 0,1200 М NaCl получили:
V'(NaCl), мл 16,00 18,00 19,00 19,50 19,90 20,00 20,10 20,50 21,00
Е, мВ 689 670 652 634 594 518 440 401 383
Ответ: 48,00 %.
9. Построить кривые потенциометрического титрования в координатах Е — V и А£/дг—И и рассчитать концентрацию СаС12 в растворе (г/л), если при титровании 20,00 мл анализируемого раствора 0,0500 н. Hg2(NOs)2 (/экв = */2) получили: nHg2(NO3)2), мл . . 10,00 15,00 17,00 17,50 17,90 18,00 18,10 18,50 19,00 Е, мВ . 382 411 442 457 498 613 679 700 709
Ответ: 2,50 г/л.
10. Построить кривые потенциометрического титрования в координатах Е— V и А£/ду — V и рассчитать концентрацию MgBr2 в растворе (г/л), если при титровании 20,00 мл раствора 0,1000 н. Hg(NOs)2 (f3KB='/2) получили:
'z(Hg(NO3)2), мл . 10,00 15,00 18,00 19,00 19,50 19,90 20,00 20,10 20,50
Е. мВ . 501 526 552 570 589 629 704 737 757
Ответ: 9,21 г/л.
217
11. Навеску медного сплава массой 0,7500 г растворили, объем раствора довели до 250,0 мл и 20,00 мл приготовленного раствора оттитровали потенциометрически раствором тиосульфата натрия с титром по меди T(Na2S2O3/Cu) = 0,01664 г/мл.
Построить кривые титрования в координатах Е — Ии &Е/ду— V и рассчитать массовую долю (%) меди в сплаве по следующим данным:
y(Na2S2O3), мл . 1,50 1,90 2,00 2,05 2,08 2,10 2,12 2,15 2 20
Е, мВ . 475 445 424 405 382 305 232 186 162
Ответ: 58,24 %.
12. Навеску стали массой 1,200 г растворили, железо перевели в Fe (II) и оттитровали потенциометрически 1,000 М Ce(SO4).
Построить кривые титрования в координатах Е — V и Д£/ду — — V и вычислить массовую долю (%) железа в сплаве по данным:
V(Ce(SO4)2), мл 2,00 10,00 18,00 19,80 20,00 20,20 22,00
Е, мВ 712 771 830 889 1110 1330 1390
Ответ: 93,08 %.
13. Навеску руды массой 0,0800 г растворили, уран перевели в уран (IV) и оттитровали потенциометрически 0,0100 н. КМпО4 (Мв= 7б):
2МпОГ + 5U4+ + 2Н2О 2Мп2+ + 5UOi+ + 4Н+
Построить кривые титрования в координатах Е — V и д£7ду— V и вычислить массовую долю (%) урана в руде по следующим данным:
У(КМпО4), мл 2,00 10,00 16,00 17,80 18,00 18,20 20,00
Е, мВ 301 330 359 389 1170 1480 1500
Ответ: 26,78 %.
14. Навеску образца массой 2,040 г растворили и таллий (I) оттитровали потенциометрически 0,1000 н. КВгОз (/эм=1/6) в солянокислой среде:
ВгОг + ЗТ1+ + 6Н+ Вг~ + ЗТ13+ + ЗН2О
Построить кривые титрования в координатах Е—V и д£/ду — — F и вычислить массовую долю (%) таллия в сплаве по следующим данным:
У (КВгОз), мл 2,00 10,00 17,00 18,80 19,00 19,20 21,00
Е, мВ . . 1250 1280 1310 1340 1410 1430 1430
Ответ: 9,52%.
15. Навеску стали массой 2,000 г растворили в азотной кислоте и после соответствующей обработки ванадий оттитровали потенциометрически 0,1000 М FeSO4:
VO2+ + Fe2+ + 2Н+ VO2+ + Fe3+ + Н2О
Построить кривые титрования в координатах Е — V и
218
&E/kV — V и вычислить массовую долю (%) ванадия в стали по следующим данным:
V(FeSO4),Ma. 2,00 12,00 19,00 20,80 21,00 21.20 23,00
Е, мВ 1060 1000 940 901 885 841 830
Ответ: 5,35 %.
16. Навеску установочного вещества массой m г растворили в мерной колбе вместимостью 50,00 мл и довели раствор до метки ледяной уксусной кислотой. При потенциометрическом титровании 5,00 мл полученного раствора хлорной кислотой в безводной уксусной кислоте получили следующие результаты:
Вариант 1 (гидрофталат калия KHCeH4O4, m = 0,3996 г)
V(HC1O4), мл 1,20 1,40 1,60 1,80 2,00 2,20 2,40 2 60
Е, мВ . 383 389 398 420 509 547 562 568
Вариант 2 (дифенилгуанидин CisHisNs.m = 0,4003 г)
V(HCIO4), мл 1,20 1,40 1,60 1,80 2,00 2,20 2,40 2,60 2,80
£, мВ 382 389 402 419 473 594 622 636 644
Вариант 3 (карбонат натрия NasCOs, tn = 0,2500 г)
F(HC1O4), мл 3,60 3,80 4,00 4,20 4,40 4,60 4,80 5,00 5,20
Е, мВ . . 431 439 450 465 486 523 550 566 575
Построить кривые потенциометрического титрования в координатах Е — V и ЕЕ/др—V и вычислить молярную концентрацию эквивалента раствора хлорной кислоты по: 1) гидрофталату калия; 2) дифенилгуанидину; 3) соде.
Ответ: 1) 0,1030 моль/л; 2) 0,09028 моль/л; 3) 0,1046 моль/л.
17. Навеску технического салициловокислого натрия массой 0,8606 г растворили в мерной колбе вместимостью 50,00 мл и раствор довели до метки ледяной уксусной кислотой. При потенциометрическом ’титровании 5.00 мл полученного раствора 0,1 М НСЮ4 в ледяной уксусной кислоте (Д' = 1,030) получили следующие результаты:
Г(НСЮ4), мл .4,00 4,20 4,40 4,60 4,80 5,00 5,20 5,40
Е, мВ 440 447 458 474 512 595 615 625
Построить кривые титрования и вычислить массовую долю (%) индифферентных примесей в препарате. Ответ: 6,12%.
18. Навеску пролина массой 0,0480 г (Л4 = 115,13 г/моль) марки «ч» растворили в ледяной уксусной кислоте и оттитровали 0,1 М (К =1,035) НСЮ4 в безводной СНзСООН Результаты потенциометрического титрования приведены ниже:
1/(НС1О4), мл
Е. мВ
3,00 3,20 3,40 3,60 3,80 4,00 4,20 4,40
136 145 157 176 279 325 339 345
Построить кривые титрования и вычислить массовую до-лю (%) основного вещества в образце. Ответ: 97,98 %.
19. Навеску смеси аланина массой m г (М = 89,09 г/моль) и Фенилаланина (М = 165,2 г/моль) растворили и раствор довели
219
до метки ледяной уксусной кислотой в мерной колбе вместимостью 50,00 мл При титровании аликвоты объемом 5,00 мл 0,1 М НС1О4 (К = 0,828) в безводной уксусной кислоте получили следующие результаты:
Г (НС1О/), мл
Г, мВ, при т = 0,3953 г
Е, мВ, при т = 0,3702 г
3,00 3.20 3,40 3,60 3,80 4,00 4,20
432 444 466 580 624 640 650
434 445 470 556 596 612 624
Вычислить массовую долю (%) компонентов смеси. Ответ: 1) 28,76 % аланина; 71,24% фенилаланина; 2) 45,40 % аланина; 54,60 % фенилаланина.
20. Навеску лекарственного препарата массой m г, содержащего амидопирин (М = 231,3 г/моль) и индифферентные примеси, растворили в безводной уксусной кислоте, добавили дихлорэтан и оттитровали 0,1 М (К = 0,9109) НС1О4 в ледяной уксусной кислоте.
Вычислить массовую долю (%) основного вещества в препарате по следующим результатам потенциометрического титрования:
1/(НС1О4), мл
F, мВ, при m = 0,1260 i
Е, мВ, при m = 0,1256 г
3,80 4,00 4,20 4,40 4,60 4,80 5,00 5,20
355 360 382 523 579 597 611 620
344 357 381 516 587 604 617 —
Положение точки эквивалентности Грана. Ответ: 1) 71,90%; 2) 72,13%.
определить по методу
Глава 10 ВОЛЬТАМПЕРОМЕТРИЯ
10.1. КРИВАЯ ТОК — ПОТЕНЦИАЛ
Вольтамперометрия основана на изучении поляризационных или вольтамперных кривых (кривых зависимости силы тока от напряжения), которые получаются, если при электролизе раствора анализируемого вещества постепенно повышать напряжение и фиксировать при этом силу тока. Электролиз следует проводить с использованием легко поляризуемого электрода с небольшой поверхностью, на котором происходит электровосстановление или электроокисление вещества.
Применение вольтамперных кривых в аналитических целях началось с разработки в 1922 г. чешским ученым Я. Гейровским полярографического метода анализа. За открытие и развитие этого метода Я. Гейровскому в 1959 г. была присуждена Нобелевская премия. Я. Гейровский проводил электролиз на ртутном капающим электроде и вольтамперометрию, связанную с использованием ртутного капающего электрода, стали называть п о-лярографией.
220
Рассмотрим электролиз в системе, где катодом служит ртутный капающий электрод, а анодом является практически неполяризуемый каломельный электрод. Изменение внешней ЭДС в такой системе будет полностью идти на изменение потенциала катода. Если в растворе нет веществ, способных восстанавливаться под действием электрического тока, сила тока I будет пропорциональна приложенному напряжению Е (закон Ома):
/ = E/R,
где R — сопротивление.
В присутствии веществ, способных восстанавливаться на ртутном электроде в области исследуемых напряжений, вид кривой зависимости тока от напряжения существенно изменится. По достижении потенциала восстановления ионы начнут разряжаться на ртутном катоде нередко с образованием амальгамы:
М"+ + пе~ Hg = М (Hg) (10.1)
Потенциал ртутного катода, на котором протекает обратимый процесс (10.1), выражается уравнением Нернста:
Е = Е° + Дг 1п , (10.2)
' nF CaYa ' '
где св—концентрация амальгамы; уа—ее коэффициент активности; См—концентрация восстанавливающихся ионов в при-электродном слое (заряд иона для простоты опущен); ум—его коэффициент активности; анБ — активность ртути в амальгаме; /^ — стандартный потенциал электрода (10.1).
В результате процесса (10.1) сила тока в цепи начнет возрастать и концентрация восстанавливающихся ионов у поверхности ртутной капли уменьшится. Однако за счет диффузии из массы раствора к поверхности капли доставляются новые порции ионов. Сила тока в цепи будет зависеть от скорости диффузии, которая пропорциональна разности концентраций в массе раствора (с^) и в приэлектродном слое (см) Сила тока / будет пропорциональна этой разности:
I = — См)- (10.3)
Вктад других, недиффузионных механизмов поступления ионов в прикатодный слой в условиях большого избытка индифферентного фонового электролита пренебрежимо мал. Основное значение среди недиффузионных процессов имеет миграция ионов к катоду под действием электрического поля. Если не Устранить вызываемый этим процессом миграционный т о к, общий ток окажется неконтролируемым. Подавление миграционного тока достигается введением в раствор в достаточной концентрации так называемого индифферентно-г о, т. е. не принимающего участия в электродной реакции, или фонового, электролита со значительно более от
221
рицательным потенциалом выделения, чем у анализируемого иона. Катионы фоновою электролита экранируют электрод, уменьшая тем самым движущую силу миграции под действием электрического поля практически до нуля.
При некотором потенциале катода концентрация ионов у поверхности ртутной капли См уменьшится до ничтожно малой по сравнению с концентрацией в массе раствора, и скорость разряда ионов на катоде станет равной скорости диффузии.
Концентрация восстанавливающегося иона в глубине раствора постоянна, так как электролиз идет при очень небольшой силе тока (порядка 10-5 А), а концентрация в прикатодном слое близка к нулю. Поэтому разность концентраций, определяющая скорость диффузии при данной температуре, будет постоянна, что и приводит к постоянной скорости поступления ионов к катоду. Наступившее состояние равновесия будет характеризоваться постоянной силой тока, не изменяющейся при дальнейшем увеличении напряжения. Этот постоянный ток, контролируемый диффузией, называют диффузионным и обозначают 1а. Выражение для силы диффузионного тока получается из уравнения (10.3) при см = 0:
/d = ^мСм- (10.4)
Сила диффузионного тока прямо пропорциональна концентрации восстанавливающегося иона в массе раствора. При сочетании уравнений (10.3) и (10.4) получаем
I — Id —
ИЛИ
(10.5) «м
Концентрация амальгамы, образовавшейся в результате процесса (10.1), пропорциональна силе тока:
Ca=k'J=^-. (10.6)
Соотношения (10.5) и (10.6) подставляем в уравнение (10.2)’
£ е° I К? In Qh^‘i ~ (10.7)
‘ nF ЙмГУа
Некоторые величины в этом уравнении постоянны или зависят только от температуры. Так, амальгама, образующаяся при электролизе на ртутном катоде, очень разбавлена, поэтому активность ртути в амальгаме 4ZHg практически равна активности чистой ртути, т. е. величина постоянная. Коэффициент активности ионов ум при постоянной ионной силе, которая создается фоновым электролитом, остается постоянным, так же как коэффициент активности уа и коэффициенты и ks. Выделим в урав
222
нении (10.7) величины, зависящие только от температуры, и придадим ему вид
ИЛИ £ £/,+ nF In \ , (ю.9)
где £ /2= £° +-^ 1п-^м-^ . (10.10) Пг ^мТа
Уравнение (11.9) передает зависимость силы тока от приложенного напряжения при обратимом электродном процессе. Это уравнение полярографической волны, а величину £>/2 называют потенциалом полуволны.
Типичная зависимость силы тока от приложенного напряжения дана на рис. 10.1. Это полярографическая волна (полярограмма). Из рисунка видно, что в начале процесса при небольшом потенциале катода сила тока медленно увеличивается с возрастанием потенциала — это так называемый остаточный ток, его величина имеет порядок 10~7 А. По достижении потенциала восстановления на катоде начинается разряд ионов и сила тока резко возрастает, стремясь к предельной величине диффузионного тока. При 1 = = 1/rid уравнение (10.9) переходит в
£ = £ /,
Это соотношение, так же как и (10.10), показывает независи-
мость потенциала полуволны от силы тока и, следовательно, от концентрации восстанавливающегося иона. Потенциал полуволны является, таким образом, качественной характеристикой иона в растворе данного фонового электроли-
та и определение потенциала полуволны составляет основу качественного полярографического анализа.
Однако потенциал полуволны существенно зависит от среды, природы и концентрации фонового электролита. Особое значение имеет наличие в растворе веществ, способных к комплексообразованию с определяемым ионом. Присутствие в исследуе-
мом растворе лиганда смещает потенциал полуволны в отрицательную область, что используется для определения состава и констант устойчивости координационных соединений. Сдвиг потенциала полуволны при введении в раствор лиганда значительно расширяет возможности полярографического анализа, позволяя создавать условия для определения нескольких компонентов в одном растворе без их предварительного разделения. Например, в 1 М К.С1 ионы свинца (П) и таллия (I) имеют потенциалы полуволны, соответственно, —0,435 и —0,483 этом фоне их раздельное определение неосуществимо.
б!аОН потенциал полуволны свинца становится
В и на
В 1 М равным
223
— 0,755 В, а у таллия остается практически без изменений, поэтому в щелочном растворе эти ионы могут быть определены при совместном присутствии.
Если в растворе находится несколько веществ, потенциалы полуволны которых различаются на 100 мВ и больше, то на полярограмме будет не одна волна, а несколько — по числу восстанавливающихся ионов (рис. 10.2), а возможно и больше, так как при ступенчатом восстановлении один ион может давать две волны. Например, две волны дает ион Сп2+ в присутствии 1 М NH3: первую при потенциале полуволны —0,20 В и вторую при — 0,48 В. Можно получить таким образом полярографический спектр ионов, а затем по этим данным и измеренному потенциалу
Рис. 10.1. Полярограмма:
1 — остаточный гок; 2 — диффузионный ток
Рис. 10.2. Полярограмма при наличии в растворе восстанавливающихся веществ А, В и С
полуволны идентифицировать неизвестное вещество. Вполне понятно, что положение элемента в таком спектре будет зависеть от фонового электролита: его природы и концентрации.
Полярограмма, изображенная на рис. 10.1, несколько идеализирована, так как на ней не видны осцилляции тока, вызванные периодическим отрывом капель ртути. Иногда эти осцилляции очень затрудняют работу^ особенно в области малых концентраций определяемого элемента.
Кроме того, на полярограммах нередко возникают максимумы различной формы, мешающие определению истинного потенциала полуволны и силы тока. Различают максимумы I и II рода. Теория связывает их появление с гидродинамическими явлениями в растворе, вызываемыми каплями ртути, и адсорбционными процессами. Для подавления максимумов в по-лярографируемый раствор обычно вводят поверхностно-активные вещества: желатин, агар-агар и др. Подавление максимумов поверхностно-активными веществами лежит в основе нескольких чувствительных (до 10~9 моль/л) аналитических методик определения этих веществ в растворе.
Связь диффузионного тока с концентрацией иона См и другими величинами передается уравнением Илько-в и ч а:
224
Id = 605zD'/1m/4'l'‘CMa (10.11)
где z — заряд иона; D — коэффициент диффузии; т — масса ртути, вытекающий из капилляра в 1 с, мг; t — время образования капли (период капания).
Среди величин, входящих в это уравнение, труднее всего поддается экспериментальному определению коэффициент диффузии D, а использование соответствующих справочных данных не всегда возможно. Поэтому коэффициент пропорциональности между концентрацией вещества и силой диффузионного тока обычно устанавливают с помощью стандартных растворов. Действительно, при постоянных условиях полярографирования D, т и t постоянны и уравнение (10.11) переходит в
Id=^kcfA. (10.12)
В связи с этим в работах по полярографии всегда указывается так называемая характеристика капилляра, вычисляемая как mW*. Линейная зависимость (10.12) является основой количественного полярографического анализа.
10.2. СХЕМА ПОЛЯРОГРАФИЧЕСКОЙ УСТАНОВКИ
Принципиальная схема полярографической установки представлена на рис. 10.3. Анализируемый раствор 2 находится в электролизере 5, на дне которого имеется слой ртути 1, являющийся анодом. Часто в качестве анода используют насыщенный каломельный электрод (НКЭ). Катодом служит ртутный капающий электрод 4, соединенный с резервуаром ртути 5. Внешнее напряжение, подаваемое на электроды, можно плавно менять с помощью реохорда или делителя напряжения 7и измерять при этом гальванометром 6 силу тока, проходящего через раствор.
Как уже отмечалось, напряжение, которое подается на электроды, будет практически целиком определять потенциал катода (капающего ртутного электрода).
В вольтамперометрии с успехом применяют также твердые микроэлектроды, изготовляемые из благородных металлов (платины, золота и др.) или графита. Основными достоинствами твердых электродов являются возможность работы в более положительной области потенциалов (до 1,3 В), чем с ртутным электродом (ртутный капающий электрод используется в области
Рис 10.3. Схема полярографической установки
8'1533
225
примерно от 0,3 до —2,0 В), и их нетоксичность (пары ртути, как известно, чрезвычайно ядовиты и работа с ртутным электродом требует строгого соблюдения специальных правил техники безопасности).
Однако использование твердых электродов также имеет свои трудности, связанные, главным образом, с ооновлением поверхности электродов. Стационарные твердые электроды не нашли широкого применения в практике из-за медленности установления предельного тока, невысокой чувствительности и других недостатков.
Значительно более широкое применение имеют вращающиеся и вибрирующие платиновые микроэлектроды, на которых устойчивая сила тока устанавливается быстро. При работе таких электродов раствор непрерывно перемешивается, благодаря чему к поверхности электрода ионы доставляются не только за счет диффузии, но и за счет механического перемешивания. Это значительно (в 10...20 раз) увеличивает предельный ток по сравнению с диффузионным. По точности методы с применением твердых электродов часто уступают методам, использующим ртутный капающий электрод, однако применение вращающегося платинового микроэлектрода позволяет существенно расширить область потенциалов, пригодную для полярографических измерений до 1,4 В по сравнению с областью, в которой обычно применяется ртутный капающий электрод (до 0,3 В).
Тем не менее ртутный капающий электрод сохраняет свое большое практическое значение, так как на твердых электродах ограничены катодные процессы из-за небольшого перенапряжения водорода на платине — из кислых растворов на платине он начинает выделяться при потенциале около —0,1 В, а на ртути только при —2,0 В. Промышленностью выпускаются поляро-графы нескольких марок, которые пригодны для выполнения аналитических работ и проведения научных исследований (ПЭ-312, КАП-225у, ППТ-1 и др.).
10.3. ПРЯМАЯ ПОЛЯРОГРАФИЯ
Методы прямой полярографии основаны на непосредственном применении уравнения полярографической волны (10.9) и уравнения Ильковича (10.11) или (10.12). Потенциал полуволны не зависит от концентрации и является качественной характеристикой вещества. Обычно потенциал полуволны определяют графическим методом. Уравнение (10.9) показывает, Что 1g1 является линейной функцией Е, и, следовательно, если на график нанести lg как функцию Е, то получится прямая, которая пересекает ось абсцисс в точке, где Е = Е*/2, т. е. когда lg-;-j ' = 0 (рис. 10.4).
226
Для идентификации неизвестного вещества можно этим методом определить потенциал полуволны и, пользуясь таблицей потенциалов полуволны или полярографическим спектром, установить наиболее вероятный элемент. Однако чаще это свойство используют для выбора фонового электролита. Зная качественный состав пробы, подбирают по табличным данным такой фон, на котором полярографическая волна определяемого элемента может быть получена без каких-либо искажений
Рис 10.4 Графическое определение потенциала полуволны
за счет волны мешающего элемента или иного электродного процесса.
10.3.1. Количественный полярографический анализ
Наиболее широко в количественном полярографическом анализе применяется метод градуировочного графика на основе уравнения (10.12). График строят по данным полярографирова-ния нескольких стандартных растворов. На оси ординат откладывается пропорциональная силе диффузионного тока высота полярографической волны, а по оси абсцисс — концентрация анализируемого вещества. В соответствии с уравнением (10.12) градуировочный график должен представлять прямую линию, проходящую через начало координат. Метод дает точные результаты при условии строгой идентичности условий полярографи-рования стандартных растворов и неизвестной пробы. К условиям полярографирования относят условия работы капилляра, температуру и среду (фоновой электролит). Метод градуировочного графика является наиболее трудоемким, но и наиболее точным.
При анализе некоторых хорошо изученных систем, для которых применимость уравнения (10.12) установлена вполне надежно, часто применяется менее трудоемкий метод стандартных растворов. В этом методе в строго одинаковых условиях снимают полярограммы стандартного и анализируемого растворов и из пропорции, основанной на уравнении (10.12), рассчитывают неизвестную концентрацию сх.
сх = с„^~, (10.13)
Пст
где с„— концентрация стандартного раствора; hx и йст— высота волны при полярографировании соответственно анализируемого и стандартного растворов.
Метод применим также только в условиях строгой стандартизации условий полярографирования.
Широко распространен в количественной полярографии м е-я*
227
тод добавок. Пусть при полярографировании исследуемого раствора сила диффузионного тока равна
ix = kcx. (10.14)
Добавим к этому раствору известное количество стандартного раствора сст и снова определим диффузионный ток:
Д+ст= k(cx + сст). (10.15)
При почленном делении уравнений (10.14) и (10.15) получаем
= СС1Г - откуда сх = сст -1х_- . (10.16)
f X-f-СТ СХ I- ^ст 'х+ст *х
По соотношению (10.16) находим концентрацию анализируемого раствора. Можно использовать также графический метод. В этом случае полученные данные наносят на график зависимости Ix+li от сст (рис. 10.5). При /х+ст, как показывает уравнение (10.16), сх=—Сст, т е. при экстраполяции прямая на этом графике при /х+ст = 0 отсекает на оси абсцисс величину, равную концентрации определяемого вещества. В методе добавок автоматически учитывается влияние фона и так называемых третьих компонентов, что является важным достоинством метода, позволяющим применять его при анализе сложных смесей.
Если в анализируемом растворе присутствует несколько веществ, восстанавливающихся на ртутном катоде, на полярограмме, как уже говорилось, появится несколько волн (см. рис. 10.2). По величине потенциала полуволны определяют качественный состав, а по силе диффузионного тока — концентрацию каждого из компонентов. Так, например, полярограмма на рис. 10.2 состоит из трех волн, каждая из которых характеризует один из компонентов смеси — компонент А имеет потенциал полуволны Е /2(А) и диффузионный ток ld(A), У компонента В потенциал полуволны равен Еунв) и ток /d(B) и т. д. Этот метод успешно применяется в практике, например, для определения меди и цинка в рудах по одной полярограмме.
10.3.2. Дифференциальная полярография
Рис. 10.5 Градуировочный график в методе добавок
Для анализа смесей, содержащих ионы или вещества с близкими потенциалами полуволны, применяют мето ды дифференциальной полярографии, использующие кривые -----Е. Необ-
ходимые соотношения можно получить на основании уравнения (10.9). Придадим ему вид
228
и после потенцирования
еЕ-Е j._( Id _ ^KT/nF
решим относительно I: [ =-----------------------------11_____
I е(Е -E2)nF/RT '
Для удобства дальнейшего дифференцирования объединим постоянные:
1 + e(£-£1/Pft
и продифференцируем по Е:
— . — -----Idk pfE-E/Jk (10 17)
Зависимость от E графически представлена на рис. 10.6.
Чтобы найти положение максимума, продифференцируем (10.17) еще раз по Е и приравняем производную нулю. Тогда получим, что в точке максимума
(Е)тах=Е./и (10.18)
т. е. что потенциал точки, соответствующей максимуму кривой на рис. 10.6, является потенциалом полуволны. Величину ординаты в этой точке находим при подстановке соотношения (10.18) в уравнение (10.17):
/ dZ IAk _ г nF
\ &е) Е = Еч, (1 + I)2 d 4RT ’
Следовательно, ордината в точке максимума пропорциональна силе диффузионного тока и является, таким образом, мерой концентрации вещества. Это значение можно использовать, например, при построении градуировочных графиков.
Получать дифференциальные полярограммы можно графическим дифференцированием обычных полярограмм или с помощью специальной электрической схемы, позволяющей непосредственно записывать дифференциальную кривую во время полярографирования.
Дифференциальная полярография имеет значительно более высокую разрешающую способность, позволяя определять в одном растворе ионы с близкими потенциалами полуволны. Например, этим
Рис. 10 6 Полярограмма в методе дифференциальной полярографии
229
Рис. 10.7. Полярограмма раствора, содержащего Pb(NO3)2 и TINO3 на фоне 2MKNO3
методом могут быть определены свинец и таллий, у которых потенциалы полуволны на фоне 2М KNO3 различаются только на 0,06 В. На интегральной полярограмме оба иона образуют одну общую волну (рис. 10.7, а), а на дифференциальных кривых четко видны два максимума (рис. 10.7, б). Кроме того, методы дифференциальной полярографии более точны, так как фиксировать положение максимума и измерять его высоту можно с более высокой точностью, чем определять аналогичные характеристики в методе обычной полярографии.
10.3.3 . Хроноамперометрия с линейной разверткой потенциала
»
Новые возможности в анализе и изучении электродных процессов открывает хроноамперометрия с линейной разверткой потенциала. Обычная скорость изменения потенциала в этом методе составляет до 50 мВ/с вместо 2...3 мВ/с в классической полярографии. Для измерения силы тока здесь вместо гальванометра используют безынерционный осциллограф.
Полярограмма, получаемая в этом методе, представлена на рис. 10.8. При достижении потенциала восстановления ток резко возрастает и достигает максимума, превышая величину Id классической полярографии, поскольку происходит электровосстановление практически всех ионов приэлектродного слоя, и затем падает, так как приэлектродный слой обедняется ионами, а скорость диффузии недостаточна, чтобы пополнить дефицит за столь короткое время. Потенциал максимума на этой кривой Етах является качественной характеристикой иона, а высота максимума hmax пропорциональна концентрации иона. Хроноамперометрия с линейной разверткой потенциала имеет более низкий предел обнаружения, чем обычная вольтамперометрия.
Развитие полярографии в последние годы привело к появ-
230
Рис 10.8 Осциллографическая полярограмма
Рис 10.9 Кривая анодного растворения
лению новых полярографических методик, таких, как и м-пульсная полярография, векторная полярография и др. Во многих из них используется наложение на обычную (медленную постояннотоковую полярографическую развертку потенциала переменного напряжения. Наложение переменного напряжения понижает предел обнаружения и улучшает разрешающую способность. Переменнотоковая вольтамперная кривая также является кривой с максимумом.
10.3.4 . Инверсионная вольтамперометрия
Существенное увеличение чувствительности дает инверсионная вольтамперометрия.
Идея метода инверсионной полярографии состоит в выделении определяемого элемента из очень разбавленного раствора на ртутной капле или тонкой пленке ртути на графитовом электроде или просто на графитовом электроде электролизом с последующим анодным растворением полученной амальгамы. Процесс накопления происходит при потенциале, соответствующем предельному току. Зависимость силы тока от напряжения при анодном растворении имеет вид характерного пика (рис. 10.9), глубина которого h пропорциональна концентрации определяемого иона, а потенциал минимума Етт определяется природой иона. Предел обнаружения в методике инверсионной вольтамперометрии на 2...3 порядка ниже предела обнаружения в обычных полярографических методиках. Чем больше продолжительность накопительного электролиза, тем большее количество металла перейдет из раствора в ртутную каплю и тем больше возрастет чувствительность анализа. Например, при анализе растворов, в которых концентрация определяемого элемента составляет 1СГ9 моль/л, время электролита доходит до 1 ч.
10.3.5 . Анализ органических соединений
Объектами вольтамперометрического анализа являются не только неорганические вещества или ионы, но и многие органические вещества, способные к электрохимическим превраще
231
ниям. В электродной реакции органического вещества обычно принимают участие ионы водорода:
R + nH+ + пе~~ = RHn
Анализ в связи с этим проводится в буферных растворах с достаточно высокой буферной емкостью.
К вольтамперометрически активным группировкам относятся, например, ^СНО, ^C=N, —NO2, —О—О, —S—S— и многие другие. В условиях вольтамперометрического анализа легко восстанавливаются и могут быть определены многие альдегиды, кетоны, азо- и нитросоединения, органические пероксиды. Органические кислоты (малеиновая, фумаровая и др.) и эфиры окисляются на платиновом электроде.
10.3.6 . Полярографическое исследование реакций комплексообразования
Зависимость потенциала полуволны от свойств фонового электролита эффективно используется в химии и термодинамике координационных соединений для установления состава и определения констант устойчивости образующихся комплексов. Чем выше устойчивость образующихся комплексных соединений и концентрация лиганда, тем больше сдвигается потенциал полуволны в отрицательную область. При образовании одного соединения эта зависимость при 25 °C выражается уравнением
. с 0,059 , о 0,059 , 1g ₽--------------7~plgCb
где Д£'/2 — сдвиг потенциала полуволны при данной концентрации лиганда cL по сравнению с потенциалом полуволны в отсутствие лиганда; 0 — константа устойчивости комплексного соединения; р — координационное число.
Величины 1g 0 и р определяются графически, используя зависимость Д£'/2 от 1g cL.
10.4. АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
В процессе амперометрического титрования после прибавления отдельных порций реактива отмечают силу тока при напряжении, соответствующем величине предельного тока. По этим данным строят кривую амперометрического титрования в координатах сила Тока — объем титранта и графически находят точку эквивалентности. В качестве индикаторного электрода в амперометрическом титровании обычно применяются вращающиеся платиновые, графитовые и другие твердые электроды.
232
10.4.1. Кривые амперометрического титрования
Вид кривой амперометрического титрования зависит от того, какой компонент реакции титрования вступает в электродную реакцию — определяемое вещество, титрант или продукт реакции. Например, если при титровании серебра иодидом используется процесс восстановления серебра на платиновом вращающемся катоде, кривая титрования имеет вид, изображенный на рис. 10.10. Если в этом титровании используется процесс анодного окисления иодид-иона (титранта), кривая титрования имеет вид, изображенный на рис. 10.11. В первом случае в ходе титрования сила тока уменьшается, так как концентрация серебра в растворе падает в результате образования осадка Agl, а после достижения точки эквивалентности остается постоянной. Во втором случае, когда используется анодное окисление иодид-иона, концентрация его после точки эквивалентности увеличивается и зТо отражается в возрастании силы тока. Точка эквивалентности в обоих случаях находится графически как точка пересечения соответствующих прямых.
Рис 10 10. Амперомет рическое титрование катиона, восстанавливающегося на катоде
Ag+
Рис 10.11 Амперометрическое титрование Ag+ по окислению на аноде 1
Рис. 10 ’2 Амперометри ческое титрование мышьяковой кислоты иодидом калия
На рис. 10.12 приведена кривая амперометрического титрования по диффузионному току, обусловленному концентрацией образовавшегося продукта реакции титрования. Этот случай реализуется, например, при титровании мышьяковой кислоты йодидом калия:
HAsOi“ + 21“ + 2Н+ = HAsOi- + 12 + Н2О
По мере титрования концентрация свободного иода, выделяющегося как продукт реакции, будет возрастать до точки эквивалентности, после чего останется постоянной.
233
10.4.2. Основные типы реакций в амперометрическом титровании
В методах амперометрического титрования используют реакции осаждения, комплексообразования и окисления — восстановления. Необходимо, чтобы реакция, протекающая при амперометрическом титровании удовлетворяла тем требованиям, которые предъявляются к реакциям в титриметрических методах в отношении полноты и скорости их протекания. Многие анионы СГ, Вг“, I', SO2-, МоОГ и др. титруются солью свинца при потенциале — 0,4 В, когда на ртутном капающем электроде происходит восстановление иона РЬ2+. Окисление ферроцианида Fe(CN)6 на вращающемся платиновом электроде при 0,7...1,0 В используется для амперометрического титрования Zn2+, Cu2+, Cd2+, Са2+ и других катионов. В методиках амперометрического титрования часто применяется осаждение органическими реагентами: 8-оксихинолином, купфероном, диметилглиоксимом и др., причем титрование можно проводить как по току восстановления катиона, так и по току органического реагента.
Если в растворе находятся два иона, способных образовывать малорастворимые соединения с титрантом, и их ПР различаются существенно, а электрохимические свойства системы позволяют получать кривую титрования с двумя изломами, то становится возможным амперометрическое титрование каждого компонента в одном растворе без предварительного химического разделения. Так титруют, например, смесь никеля и меди рубеановодородной кислотой.
Широко используется в амперометрическом титровании реакция образования этилендиаминтетраацетатных комплексов различных элементов. С помощью этой реакции определяют десятки катионов, способных к электрохимическому восстановлению в условиях анализа: Bi3+, Fe2+, Fe3+, Ni2+, Pb , Zn2+, Cu2+, Co2+, Cd2+ и др. При изменении pH создаются условия для титрования этим методом нескольких катионов и таким образом анализировать смеси катионов без их химического разделения. Так титруют, например, раствор, содержащий висмут и цинк: при pH 1,0...2,0 определяют висмут, затем при pH 4,7...5,0 — цинк Разработаны также методики амперометрического титрования, основанные на анодном окислении ЭДТА на платиновом микроэлектроде.
При амперометрическом титровании с использованием реакций окисления — восстановления в качестве титрантов используют К2СГ2О7, Ce(SO4)2, КВгОз, 12 и др. — для определения восстановителей; FeSO4, Ма252Оз и др. — для определения окислителей. Практическое применение нашли также некоторые органические реагенты — хлорамин Б как окислитель, аскорбиновая кислота как восстановитель и другие вещества. Если в растворе имеется два окислителя или два восстановителя с существенно разными окислительно-восстановительными потенциалами, воз
234
можно последовательное амперометрическое титрование обоих компонентов без предварительного химического разделения. При этом возможно титрование как без изменения потенциала электрода, так и с изменением потенциала при переходе от одного компонента к другому.
10.4.3. Титрование с двумя индикаторными электродами
В последнее время широкое распространение получил метод амперометрического титрования с двумя индикаторными электродами. Иногда его называют также методом мертвой конечной точки (dead — stop end point). При определениях по этому методу в анализируемый раствор вводят два платиновых или иных инертных электрода под небольшим постоянным напряжением (порядка 10-2 В) и в ходе титрования отмечают силу тока. До начала титрования сила тока между электродами или очень мала, или вообще не наблюдается, так как в отсутствие окислительно-восстановительной пары при столь малой разности потенциалов электродные процессы не происходят. Введение титранта вызывает появление в анализируемом растворе двух окислительно-восстановительных пар, причем до точки эквивалентности в растворе в заметных количествах будут находиться компоненты пары, образованной титруемым веществом, а после точки эквивалентности компоненты, образованные титрантом. Вид кривой титрования будет определяться, в основном, электрохимической обратимостью этих пар. Если обе окислительно-восстановительные пары обратимы, как, например, в реакции титрования железа (II) солью церия (IV):
Fe2+ + Се4+ = Fe3+ + Се3+
кривая титрования имеет вид, изображенный на рис. 10.13, а.
Добавление первых же порций соли Се4+ вызовет образование в растворе окислительно-восстановительной пары Fe3+/Fe2+ и в цепи появится ток, так как восстановление Fe3+ на катоде
и окисление Fe2+ на аноде вследствие высокой обратимости системы происходит при минимальной разности потенциалов. Сила тока будет возрастать, пека не прореагирует примерно половина Fe , затем начнет уменьшаться и упадет почти до
Рис 10.13 Кривая
амперометрического
титрования с двумя индикаторными элек тродами
а — обе пары обратимы, б — необратимая система титруется обратимой
нуля в точке эквивалентности. После точки эквива
лентности на катоде будет восстанавливаться Се4+, а на аноде окисляться Се3+ и ток в цепи снова появится
235
Если система, образуемая определяемым веществом, обратима, а титрантом — необратима, как, например, при титровании Fe2+ перманганатом калия, кривая титрования до точки эквивалентности не будет отличаться от кривой на рис. 10.13, а, так как в обоих случаях сила тока в системе до точки эквивалентности контролируется электрохимически обратимой системой. Однако после точки эквивалентности возрастания силы тока не произойдет, так как при этой разности потенциалов Мп2+ на аноде окисляться не будет.
Если титруется электрохимически необратимая система, а титрант образует обратимую окислительно-восстановительную пару, то до точки эквивалентности тока не будет, а после точки эквивалентности он резко возрастает (рис. 10.13,6). Такой вид имеет кривая титрования, например, перманганата раствором соли Мора или тиосульфата раствором иода.
Метод амперометрического титрования с двумя индикаторными электродами достаточно точен и чувствителен: он пригоден для анализа растворов с концентрацией определяемого вещества 10-5 моль/л и менее. В аппаратурном отношении он проще, чем метод с одним индикаторным электродом. При титровании по этому методу часто отпадает необходимость в построении кривой титрования, так как точка эквивалентности может быть определена по резкому прекращению или появлению тока.
10.5. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Вольтамперометрический метод применяют для определения многих металлов. Кадмий, кобальт, медь, свинец, марганец, никель, олово, цинк, железо, висмут, уран, ванадий и многие другие могут быть определены в рудах, концентратах, сплавах и иных природных и технических объектах. При достаточно различающихся потенциалах полуволны (ДЕ^ 0,10 В) возможно количественное определение нескольких элементов без предварительного разделения. Например, в аммиачном буферном растворе можно полярографировать смесь кадмия (£?/,=— 0,81В) и никеля (£?/,= —1,10 В). Существенное практическое значение имеет вольтамперометрическое определение хромат-, иодат-, мо-либдат-ионов и некоторых других, а также многих органических соединений: альдегидов, кетонов, азо- и нитросоединений и т. д. Широко используют полярографический метод для анализа биологически важных материалов: крови, сыворотки и т. д.
Интенсивно развиваются также современные вольтамперометрические методы: с быстрой разверткой потенциала, переменнотоковая, инверсионная, импульсная и т. д.
Амперометрическое титрование применяется для определения катионов и анионов в различных технических и природных объектах, минеральном сырье и продуктах его переработки, природных водах, промышленных растворах, продуктах металлургии и т. д„ а также в анализе многих органических веществ.
236
Использование реакций различного типа (осаждения, комплексообразования и окисления — восстановления) позволяет подбирать условия амперометрического титрования для большинства элементов периодической системы. Значительно расширились возможности амперометрического титрования в связи с применением органических реагентов, аналитические достоинства которых (селективность, чувствительность) хорошо известны. Многие из них способны к электрохимическим превращениям на электродах, что еще больше повышает их ценность, так как позволяет проводить амперометрическое титрование по току титранта. Для амперометрического титрования характерна экспрессность, его можно проводить в разбавленных растворах (до 10-5 моль/л и меньше) и анализировать мутные и окрашенные растворы.
10.6. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Вольтамперометрический метод достаточно универсален и применим к многочисленному кругу объектов. Основными достоинствами метода являются быстрота анализа, возможность определения нескольких веществ в смеси без предварительного разделения, достаточно высокая точность и применимость к анализу небольших содержаний определяемого элемента. Погрешность полярографического анализа в обычных условиях составляет ±2 % для растворов концентрации порядка 10-3...10-4 моль/л и около ±5 % для более разбавленных. При сочетании вольтамперометрии с методами экстракции, хроматографии и т. д. предел обнаружения снижается еще больше.
Амперометрическое титрование характеризуется более высокой точностью и более высокой чувствительностью, чем методы прямой вольтамперометрии. Аппаратурное оформление установок амперометрического титрования несложно, особенно просты установки для титрования с двумя индикаторными электродами.
Вопросы
1. Как рассчитать потенциал полуволны на основании вольт-амперной кривой?
2. Будет ли одинаков потенциал полуволны кадмия (II) в средах: 1 М КС1 и 1 М NH3 + 1 М NH4NO3?
3. Как изменится сила диффузионного тока раствора CdSO4 при уменьшении концентрации CdSO4?
4. Объяснить возникновение ошибок в вольтамперных измерениях при несоблюдении термостатирования ячейки.
5. Почему в вольтамперометрии сила тока достигает предельного значения? От каких факторов зависит величина предельного тока?
6. Смесь Fe2+ и Fe3+ в щавелевокислом растворе дает катод но-анодную волну с£ ,= —0,24 В. Предложить вольамперомет-
237
рический метод определения ионов Fe2+ и Fe3+ при совместном присутствии.
7. Раствор содержит ионы индия, кадмия, олова, сурьмы на фоне 6 М НС1. Какой вид имеет вольтамперная кривая этого раствора?
8. Как можно использовать табличные данные по £|/2 для выбора оптимальных условий определения Pb, Си, Cd в металлическом цинке?
9. При анализе каких систем можно вести амперометрическое титрование в катодной области? в анодной области?
10. Какой вид имеет кривая амперометрического титрования висмута (III) раствором ЭДТА, если на катоде при Е = —0,2 В (отн. НКЭ) восстанавливается только висмут (III)?
11. Какой вид имеет кривая амперометрического титрования магния (II) оксихинолином, если на катоде при Е = —1,6 (отн. НКЭ) восстанавливается только титрант?
12. Какой вид имеет кривая амперометрического титрования свинца (II) дихроматом при £ = —0,8 В (отн. НКЭ), если при этом восстанавливаются и свинец (II) и титрант?
13. Какой вид имеет кривая амперометрического титрования-раствора h мышьяком (III) с двумя индикаторными электродами (Pt — Pt) и биамперометрическим фиксированием конечной точки?
14. Как изменится сила тока пика анодного растворения кадмия (метод инверсионной вольтамперометрии): а) при изменении концентрации кадмия в анализируемом растворе; б) при изменении времени электролиза?
15. Какой метод вольтамперометрического анализа следует выбрать, если: а) выполняют серийное определение однотипных образцов (например, латуни на содержание Си и Zn); б) выполняют определение малого содержания Со (II) в солях никеля (II); в) для точных вольтамперометрических измерений нет достаточных условий: термостатирование, инертный газ отсутствуют?
ZA Задачи
1. Различие в потенциалах полуволны Ni и Со на фоне HCI и пиридина позволяет проводить определение никеля в кобальтовых солях. Навеску сульфата кобальта массой 2,500 г растворили, добавили необходимые реактивы — НС1, желатину, пиридин — и разбавили до 100,0 мл.
Аликвоту раствора объемом 50,00 мл полярографировали и получили диффузионный ток (1,35 мкА). Затем в полярографическую ячейку добавили 5,00 мл стандартного раствора, содержащего 1,00-10-2 моль/л NiClz, и получили диффузионный ток 3,80 мкА. Вычислить массовую долю (%) Ni в препарате.
238
В соответствии с уравнением Ильковича
А = kci, /2 = kCi,
где /|, /2 — диффузионные токи до и после прибавления стандартного раствора с, — начальная концентрация никеля; сг — концентрация никеля после добавле ния стандартного раствора
Если сст — концентрация стандартного раствора, V — начальный объем раствора в полярографической ячейке, Гст— объем прибавленного стандартного раствора, то
cctVct + С1 V С 2
V+ VCT
г /г / ci(V + Ест)
Отношение /i//2 = ci/c2 =-------———тт- преобразуем относительно
£ст » ст + C,v
______________________ ИЛИ С| СгтУстА A(V + Ver) - AV_______(А - /1)Е-+ /2РСТ
Подставляем числовые значения.
1 • 10_2-5-1,35 ]П_4
С1^ (3,80 - 1,35)50 + 3,80-5 4,77’° моль/л
Массовая доля (%) никеля (.A(Ni) =58,70) равна
,К1 4,77-10^4-100,0-58,70-100
W (N,) =---------1000^500--------
0,11 %
2. Используя зависимость |£(//Д — 7) —ЕУ вычислить потенциал полуволны и число электронов, участвующих в катодном процессе, по следующим данным:
Pb(NO3)2 в 1 М NaOH TlNOs в растворе ЭДТА Бензальдегид СеН5СНО в фосфатном буферном растворе, pH 2,75 1пС13 в 1 М KCI
— Е ,В I, мкА — Е , В /, мкА — Е , В /, мкА — Е , В 1, мкА
0,645 0,30 0,500 0 0,800 0 0,550 0
0,695 0,30 0,650 1,00 0,900 0 0,562 0,64
0,720 0,46 0,675 2,70 0,950 0,35 0,575 2,36
0,745 1,14 0,700 6,90 1,000 1,26 0,587 8,41
0,770 2,35 0,725 15,0 1,050 2,20 0,600 20,5
0,795 2,89 0,750 26,8 1,100 2,44 0,612 30,2
0,820 3,00 0,775 35,8 1,150 2,50 0,625 33,7
0,870 3,00 0,800 41,0 1,200 2,50 0,650 35,0
0,920 3,00 0,850 45,0 — -— 0,675 35,0
— — 0,900 45,0 — — — —
Ответ: 1) —0,755 В; 2; 2) —0,742 В; 1; 3) —1,00 В; 1; 4) —0,597 В; 3.
3. При полярографировании стандартных растворов свинца (II) получили следующие результаты:
срь‘ Ю6, г/мл 0,50 1,00 1,50 2,00
й, мм 4,0 8,0 12,0 16,0
239
Навеску алюминиевого сплава массой т г растворили и раствор разбавили до 50,00 мл. Высота полярографической волны свинца в полученном растворе оказалась равной hx.
Вычислить массовую долю (%) свинца в анализируемых образцах:
Вариант т, г . й, мм
1 2 3
2,500 5,134 5,300
6,0 9,0 11,0
Ответ: 1) 1,52-1СГ3%; 2) 1,09 10~3%; 3) 1,ЗО-1О-3%.
4. Для построения градуировочного графика записали полярограммы четырех стандартных растворов меди (II) и измерили высоту волны hx мм:
Ccu-l О3 г/мл hx, мм
0,50 1,00 I 50 2,00
9,0 17,5 26,2 35,0
Навеску латуни массой пг г растворили и раствор разбавили до 50,00 мл.
Вычислить массовую долю (%) меди в анализируемых образцах, если высота волны на полярограммах оказалась равной hx:
Вариант т, г /г,, мм
1 2 3
0,0690 0,1000 0,1200
11,0 18,0 23.0
Ответ: 1) 44,93%; 2) 52,00%; 3) 55,00%.
5. В 0,1 М KCI (рН7) D-рибофлавин C17H20N4O6, тиамин C12H.7N4OSCl.HCl и никотиновая кислота C5H4NCOOH имеют потенциалы полуволны Е'/г соответственно: —0,35; —1,25; — 1,17 В. Для построения градуировочных графиков записали полярограммы четырех стандартных растворов этих веществ с концентрацией 0,000400; 0,000600; 0,000800 и 0,001000 моль/л и измерили силу диффузионного тока 1а при соответствующих условиях (—0,6; —1,5; —1,8 В):
Стандартное вещество ld, мкА
D-Рибофлавин 2,40 3,60 4,80 6,00
Тиамин 3,60 5,40 7,20 9,00
Никотиновая кислота 2,20 3,30 4,40 5,50
Вычислить концентрации (мг/мл) £)-рибофлавина, тиамина и никотиновой кислоты в анализируемом растворе, если полученная полярограмма имела при соответствующих потенциалах три волны и диффузионные токи 1а были равны:
240
Вещество Id, мкА, для вариантов
1 2 3
£)-Рибофлавин 2,80 5,20 4,90
Тиамин 7,50 3,80 3,90
Никотиновая кислота 4,10 4,80 2,80
| Ответ: 1) 0,177; 0,283; 0,0923 мг/мл; 2) 0,324; 0,145; 0.108 мг/мл; 3) 0,309; 0,148; 0,0628 мг/мл.
6. При полярографировании на фоне диметилформамида стандартных растворов диаллиламиноэтилметакрилата (ДАМА) и диаллиламиноэтилакрилата (ДАА) с концентрацией 0,40 мг/мл каждый получили диффузионный ток /di соответственно 6,90 и 4,32 мкА. Затем в полярографические ячейки прибавили равные объемы раствора фенола и измерили силу диффузионного тока /d2J получив 4,08 мкА для ДАМА и 4,34 мкА для ДАА. При условиях измерений фенол неэлектроактивен.
Навеску исследуемого образца массой m г растворили в 20,00 мл смеси диметилформамида с водой, поместили в ячейку и измерили Id,. Добавили 20,00 мл раствора фенола и измерили 1а?
Рассчитать массовую долю (%) ДАМА и ДАА в исследуемых образцах:
Вариант т, г la,, мкА ld3, мкА
1 0,0205 10,45 10,22
2 0,0195 15,25 9,92
3 0,0232 16,15 8,92
Ответ: 1) 3,81; 88,32 %; 2) 77,69; 20,74 %; 3) 62,46; 12,16%.
7. Для определения свинца в цинковой руде методом добавок навеску руды массой m г растворили в смеси кислот, восстановили железо (III), добавили желатину и разбавили раствор до 200,0 мл. Аликвоту объемом 20,00 мл поместили в электролизер и измерили высоту hi полярографической волны при Е = ~ —0,45 В (НКЭ). При этих условиях ионы меди, цинка, кадмия не мешают определению свинца. После добавления в электолизер стандартного раствора VrT мл 0,0020 М РЬ(\Оз)г получили высоту волны h-г.
Рассчитать массовую долю Щих вариантов: (%) свинца в руде для следую-
Вариант 1 2 3 4
т, г . 1,000 2,266 2,268 3,073
hi, мм 22,0 25,0 26,5 28,5
Vct. МЛ 10,00 5,00 5,00 2,00
hi, мм 42,0 35,0 36,5 32,5
Ответ: 1) 2,22 %; 2) 1,22%, 3) 1,27 %, 4) 2.65 %.
241
8. Для определения кадмия в сплаве методом добавок навеску сплава массой т г растворили в смеси кислот и раствор разбавили до 250,0 мл. Аликвоту объемом 20,00 мл полярографи-ровали и измерили высоту полярографической волны кадмия. Другие компоненты сплава в этих условиях не мешали опреде-1 лению кадмия. После добавления в электролизер стандартного
раствора VCT мл 0,0300 М CdSOi высота волны увеличилась до Л2.
Определить массовую долю дующих вариантов: (%) кадмия в сплаве для еле-
Вариант 1 2 3 4
т, г 3,542 4,130 3,746 3,508
Л|, мм 19,0 20,5 18,5 16,5
Г,,, мл 10,00 2,00 5,00 5,00
А12. ММ 29,0 22,5 23,5 21,5
Ответ: 1) 9.23%; 2) 9,85% ; 3) 9,57%; 4) 9,56 %.
9. После соответствующей обработки навески биологического материала массой т мг получили 20,00 мл щелочного раствора, содержащего билирубин (2И (СззНзвОвКД = 584,67 г/моль). Измерили диффузионный ток катодного восстановления билирубина Idl. В электролизер добавили 5,00 мл стандартного раствора билирубина с концентрацией 5,00-10“4 моль/л и измерили lds при тех же условиях.
Вычислить концентрацию (мг/г) билирубина в образцах биологических материалов:
Вариант т, мг Диффузионный ток, мкА
1 20,0 0,40 0,93
2 15,0 0,35 0,88
3 35,0 0,50 1,03
Ответ: 1) 38,34 мг/г; 2) 45,47 мг/г; 3) 26,52 мг/г.
10. Навеску биологического материала массой m мг подвергли гидролизу, к полученному гидролизату протеина добавили растворы хлорида кобальта, аммонийного буферного раствора и разбавили до 50,00 мл. Полярографическая волна этого раствора вызвана каталитическим выделением водорода на катоде в присутствии образующегося комплексного соединения кобальта (II) с цистином, содержащимся в гидролизате протеина. Измерили высоту каталитической волны 7dl при Е = — 1,6 В. К раствору добавили 5,00 мл стандартного раствора цистина [Л4( (HOOCCH(NH2)CH2S)2) = 240,291 г/моль] с концентрацией 2,00-10~5 моль/л. Увеличение высоты каталитической волны при том же потенциале A7d составило 7,20 мкА.
Вычислить концентрацию (мг/кг) цистина в следующих биологических материалах:
242
Вариант Биологический материал т, мг /dj. мкА Вариант Биологический материал т, мг мкА
1 Шерсть 50,0 14,5 4 Мука 45,0 7,80
2 Альбумин 25,0 7,30 5 Г ормоны 15,0 5,10
3 Белок 35,0 12,5 6 Волосы 15,0 4,20
Ответ: 1) 743,7 мг/кг; 2) 811,2 мг/кг; 3) 935,9 мг/кг;
4) 478,7 мг/кг; 5) 969,1 мг/кг; 6) 806,7 мг/кг.
11. Определить концентрацию цинка (мг/л) в исследуемом растворе, если при амперометрическом титровании 10,00 мл этого раствора раствором K-iFe(CN)6 с /{K-iFefCNJe/Zn) = 0,00244 получили следующие результаты:
HK4Fe(CN)6), мл /d, мкА, для вариантов
1 2 3 4 5 6 7 8 9 10
0 30,0 20,0 20,0 30,0 30,0 30,0 44,0 60,0 75,0 75,0
0,20 30,0 20,0 20,0 31,0 29,0 30,0 45,0 60,0 75,0 75,0
0,40 31,0 31,0 21,0 30,0 31,0 30,0 45,0 60,0 75,0 75,0
0,50 40,0 40,0 22,0 32,0 32,0 30,0 45,0 61,0 75,0 75,0
1,00 94,0 94,0 60,0 80,0 32,0 31,0 46,0 61,0 75,0 120
1,50 146 146 120 200 60,0 32,0 46,0 120 75,0 165
2,00 200 200 180 310 137 120 46,0 176 75,0 210
2,50 — — — — 220 210 175 230 120 255
3,00 — — — — 300 300 300 285 300 300
Ответ: 1) 98 мг/л; 2) 73 мг/л; 3) 165 мг/л; 4) 195 мг/л; 5) 323 мг/л; 6) 366 мг/л; 7) 488 мг/л; 8) 244 мг/л; 9) 586 мг/л; 10) 122 мг/л.
12. В ячейку для амперометрического титрования поместили 50,00 мл раствора, содержащего медь (II) и кальций (II), и титровали 0,0100 М ЭДТА при Е = —0,25 В (НКЭ) в аммонийной буферной среде. При этих условиях восстанавливается только аммиачный комплекс меди (II). После достижения точки эквивалентности установили потенциал Е = 0,00 В и продолжили титрование, измеряя диффузионный ток ЭДТА.
У (ЭДТА), мл Id, мкА, при Е == — 0,25В для вариантов У(ЭДТА), мл /d, мкА, при Е — 0,00 В для вариантов
1 2 3 1 2 3
0,00 22,5 17,0 23,5 3,00 —0,50 —0,50 —0,50
0,50 16,0 11,0 17,7 3,50 —0,50 —0,50 —0,50
1,00 10,0 5,00 11,7 4,00 —0.50 —0,50 -0,50
1,50 3,75 0,50 5,75 4,50 —0,50 —2,25 —0,50
2,00 0,50 0,50 0,50 5,00 —0,50 —5,00 —2,00
2,50 0,50 0,50 0,50 5,50 — 1,50 —7,50 —4,50
3,00 0,50 0,50 0,50 6,00 —3,75 — 10,0 —6,75
6,50 —5,75 — 12,2 —9,25
243
Построить кривые титрования и рассчитать концентрацию (мг/л) меди и кальция в исследуемом растворе:
Ответ: 1) 22,56; 27,86 мг/л; 2) 17,48; 22,24 мг/л; 3) 24,78; 22,24 мг/л.
Глава 11 ЭЛЕКТРОЛИЗ И КУЛОНОМЕТРИЯ
11.1. ЗАКОНЫ ЭЛЕКТРОЛИЗА
Электролизом называют химическое разложение вещества под действием электрического тока. На катоде (отрицательно заряженном электроде) происходит восстановление:
Fe3+ + е~ = Fe2+, Cu2+ + 2е~ = Си (к)
а на аноде (положительно заряженном электроде) окисление:
2СГ - 2е~ = С12 (г)
При электролизе растворов сульфатов, фосфатов и некоторых других солей на аноде происходит окисление на SO2-- или РОГ-, а ОН~-ионов:
2ОН- - 2е~ = */2О2 + Н2О
поскольку ОН~ легче отдают свои электроны, чем SO2- или РО?~, даже в кислом растворе. На аноде может происходить окисление не только анионов, но и катионов. Например, ионы РЬ2+ образуют диоксид:
Pb2+ + 2Н2О = РЬО2 + 4Н+ + 26-
Основные законы электролиза установлены еще Фарадеем.
| Масса вещества, выделившаяся при электролизе, пропорциональна количе-* ству электричества, прошедшего через раствор.
При прохождении через раствор одного и того же количества электричества на электродах выделяется одно и то же количество вещества эквивалента. Эти законы выражаются формулой
где пг — масса вещества, выделившегося при электролизе; Q — количество электричества; М — молярная масса эквивалента вещества; 96500 — число Фарадея, равное количеству электричества, которое требуется для выделения молярной массы эквивалента вещества; I — сила тока; t — время электролиза.
Важной характеристикой процесса электролиза является выход по току, равный отношению количества выделив-
244
\
\
. шегося вещества к тому количеству вещества, которое должно ' было выделиться по закону Фарадея, т. е. в соответствии с уравнением (Н.1).
11.2. ПОТЕНЦИАЛ РАЗЛОЖЕНИЯ И ПЕРЕНАПРЯЖЕНИЯ
| Минимальную величину внешней ЭДС, при которой в данных условиях на-* чинается непрерывный электролиз, называют потенциалом разложения.
Потенциал разложения превышает величину обратимой ЭДС гальванического элемента, образованного электродами системы, в которой происходит электролиз. Превышение вызывается действием нескольких факторов. Одним из них является сопротивление ячейки R, поскольку в соответствии с законом Ома
Г _ ^обш
£'о6щ— £н 4~ IR,
где I — сила тока; £'о6ш—напряжение, наложенное на электроды; £н — ЭДС обратного элемента, рассчитанная по уравнению Нернста.
Для протекания электролиза обычно требуется некоторое увеличение напряжения, называемое перенапряжением П, и тогда
£о6щ=£н + «£ + П, (11.2)
где £о6щ — действительная величина приложенной ЭДС, при которой происходит электролиз в данной системе.
Перенапряжение зависит от свойств электродов и участников электрохимической реакции, состояния поверхности электродов, условий проведения процесса (плотности тока, температуры) и т. д. Установлено, что на гладком электроде перенапряжение больше, чем на шероховатом, а перенапряжение при выделении металлов значительно меньше, чем при выделении газов, и т. д.
Основной причиной перенапряжения является необратимость процессов на электродах при проведении электролиза. В случае газообразных продуктов электролиза дополнительный эффект вызывается ’замедленностью стадии образования двухатомных молекул газа. Напряжение, подаваемое на электроды при электролизе, представляет разность потенциалов анода и катода:
£ общ = £а £к + IR или
£овщ = (£а-|-Па) — (£к 4- Пк) + //?, (11.3)
где £а—потенциал анода; £к — потенциал катода; Па и Пк — перенапряжение на аноде и на катоде соответственно
Потенциалы £а и £к могут быть рассчитаны по уравнению Нернста (9.2). У
/ 245
11.3. ЭЛЕКТРОГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ
11.3.1. Схема установки для электролиза
В электрогравиметрическом анализе анализируемое вещество количественно выделяют из раствора электролизом и по массе выделившегося металла или его оксида рассчитывают содержание определяемого элемента в пробе. Схема установки для проведения электролиза показана на рис. 11.1. Для получения постоянного тока обычно используют выпрямитель переменного тока или батарею аккумуляторов 1. Скользящий контакт 2 позволяет регулировать подаваемое напряжение, которое измеряют вольтметром. Сила тока контролируется амперметром. При выделении металлов катод 5 обычно изготовляют из платиновой сетки, анод 4 из платиновой спирали или пластинки. При выделении оксидов знаки электродов меняются: платиновая сетка становится анодом, а спираль — катодом. Раствор перемешивается механической или магнитной мешалкой 3.
Электрогравиметрический анализ можно рассматривать как один из видов гравиметрического анализа, в котором в качестве осадителя выступают электроны. Электролитически получаемые осадки металлов и оксидов являются и формой осаждения и гравиметрической формой.
Важнейшими требованиями к форме осаждения являются ее малая растворимость и чистота, т. е. отсутствие загрязнений. Эти требования в электрогравиметрическом методе выполняются идеально, так как большинство металлов и указанные оксиды
Рис 11 1 Схема установки для проведения электролиза
практически нерастворимы в воде, а при электролитическом выделении металлов или оксидов соосаждение не происходит или его почти всегда можно предупредить путем выбора условий электролиза. Полученный осадок металла или оксида удобен для промывания и взвешивания.
11.3.2. Электрогравиметрические разделения
Если необходимо проанализировать раствор, содержащий смесь ионов без их предварительного разделения, электролиз ведут при строго контролируемом потенциале, вводя в схему дополнительный электрод с постоянным потенциалом (например, каломельный электрод) и регулирующее электронное устройство, связывающее рабочий и дополнительный электроды. Для этого используют специальные приборы — потенциостаты.
246
• Заключение о возможности или невозможности разделения двух металлов методом электролиза можно сделать на основании следующих расчетов Пусть, например, требуется установить, можно ли провести электролитическое разделение меди и цинка из 0,1 М растворов их сульфатов Значения стандартных электродных потенциалов равны £cu2+/Cu= 0,345 В, E'zn2+/Zn = — 0,764 В Потенциалы, необходимые для выделения этих металлов из раствора, можно рассчитать по уравнению Нернста Для выделения меди требуется, чтобы потенциал катода был
£Cu2t/Cu = 0,345 +-2^8-igoj =0,316 В,
а для выделения цинка
£Zn-/Zn = -0,764 + -»igo,l =-0,793 в.
Выделение меди будем считать количественным, если концентрация ионов Сн2+ в растворе будет 10-6 моль/л или меньше Потенциал катода в этом случае будет равен
£Cu2+/Cu = 0,345-ф—1g 10 6 = 0,171 ВJ
При этом значении потенциала осаждение цннка не начнется, поэтому на вопрос о возможности электролитического выделения медн в присутствии цинка из сернокислых растворов следует отвечать утвердительно.
Можно найти концентрацию ионов Си2+ при потенциале выделения цинка
— 0,793 = 0,345 + 0,029 1g [Cu2+],
откуда
lg[Cu2+] =---~’7^ЭО290’345 = ~39' т е IClj2+l = 10-39 моль/л,
что еще раз подтверждает возможность разделения этих элементов На аноде, очевидно, будет выдепяться кислород-
Н2О - 2е“ = */2О2 + 2Н+
и кислотность раствора будет увеличиваться
Потенциал анода при [Н+] = 1 моль/л можно также рассчитать по уравнению Нернста
^/ню^ '-23 + 0.0581g 1 = 1.23 В
Перенапряжение кислорода на гладкой платине при плотности тока 0,001 А/см2 составляет 0,78 Вне увеличением плотности тока возрастает Потенциал анода с учетом перенапряжения кислорода будет равен 1,23 4- 0,78 = 2,01 В
Разность потенциалов, которую необходимо поддерживать для протекания процесса и полного выделения меди, можно рассчитать по уравнению (113)
£обш= 2,01 — 0,171=1,84 В
Можно также определить, при каком потенциале начнется выделение водорода Поскольку на платиновом электроде выделилась медь, по справочнику находим перенапряжение водорода на медном электроде (— 0,94 В) и рассчитываем разность потенциалов по (113), необходимую для выделения водорода (электролиза воды)
£о(5ш = 2.01 —(—0,94) = 2,95 В
Следовательно, в условиях количественного осаждения меди (£ ~ 1,8 2,0 В) выделение водорода происходить не будет
Чтобы предотвратить выделение водорода в результате случайных изменений потенциала, особенно в конце процесса или в
ТА!
результате каких-либо других причин, в анализируемый раствор при определении меди вводят небольшое количество азотной кислоты. На катоде NO3” может восстанавливаться до NO2 (^NOr/MOj- — 0,94 В) или NH<+ (£°NO3-/nh<+=0,87 В). Восстановление нитрата будет происходить при разности потенциалов
£общ = 2,01 - 0,9= 1,1 В.
Следовательно, пока в растворе будет находиться нитрат-ион, водород на катоде выделяться не будет. Вещества, которые вносят в раствор для предотвращения нежелательных электродных процессов, называют деполяризаторами. Обычно они претерпевают электрохимическое изменение по достижении некоторого определенного потенциала. В данном случае при определении меди нитрат-ион является деполяризатором, предотвращающим выделение водорода.
Результаты аналогичных расчетов показывают, что разделение металлов методом электролиза возможно, если их стандартные электродные потенциалы различаются на 0,2...0,3 В. Этот критерий позволяет по значениям стандартных электродных потенциалов предсказать возможность или невозможность разделения любой заданной пары ионов. Так, например, серебро (£L+/Ag= 0,7994 В) можно выделить в присутствии Bis+ (£в,з+/В1 = 0,23 В), меди (£°иг+/Си = 0,345 В) и многих других металлов, но нельзя отделить от ртути (£нвг+/Нц = 0,850 В). Медь можно легко электролитически осадить в присутствии цинка (E°Zn^/Zn = —0,764 В), кадмия (£°d2+/C(1= —0,403 В), свинца (£pb2+/pb=—0,126 В) и других элементов, но в присутствии висмута сделать это можно только при строго контролируемом потенциале катода.
Изменение pH или введение в раствор комплексообразующих добавок весьма существенно сказывается на величине потен циала. Использование этих факторов значительно расширяет возможности выделения и разделения металлов методом электролиза. Например, электроосаждению никеля, кобальта и других элементов, у которых £°< 0, из кислых растворов мешает выделение водорода. С понижением кислотности выделение водорода уменьшается и осаждение металлов, в принципе, становится возможным. Найдем, при каком pH возможно количественное выделение никеля из раствора. Полнота осаждения никеля будет-достигнута при потенциале
£ = — 0,228+0,029 lg(l О-6) = -0,402 В.
Чтобы при этом потенциале не происходило выделения водорода, концентрация ионов водорода должна удовлетворять уравнению
— 0,402 = 0 + 0,0581g [Н+], откуда
248
т. е. при pH 7 водород выделяться не будет и никель может быть количественно осажден на катоде. Практически электролиз никеля ведут из аммиачных растворов. Однако образование аммиачных комплексов никеля вызывает и отрицательный эффект, так как электродный потенциал никеля в результате комплексообразования понижается и для предотвращения выделения водорода необходимо дальнейшее увеличение pH. Стандартный электродный потенциал никеля в аммиачном растворе будет равен ^N1(NH3)?+/N.= f Ni2+/Ni — 0,029 lgPN| (NHi)S- =
=—0,228 —0,029-8,01 =—0,460 В.
Полнота выделения никеля из аммиачного раствора будет достигнута при потенциале
Е= —0,460+0,029 1g 10-6 = —0,634 В.
Отсюда находим концентрацию ионов водорода, при которой электролиз будет происходить без выделения водорода:
lg[H+] = —= — 11 или рН> 11.
В IM NH4OH pH составляет примерно 11,5. Следовательно, в этом растворе выделения водорода происходить не будет и количественное выделение никеля осуществимо практически.
Изменение электродного потенциала при комплексообразовании является одним из эффективных приемов управления электрохимическими реакциями. Тонкая регулировка pH раствора и концентрации комплексообразующих веществ позволяет создавать условия разделения металлов с близкими значениями стандартных электродных потенциалов. Например, известна методика определения в одном растворе Си2+, Вг+, РЬ2+ и Sn2+. В нейтральном тартратном растворе при потенциале —0,20; —0,40 и —0,60 В поочередно выделяется на катоде и взвешивается медь, висмут и свинец, а затем раствор подкисляется для разрушения тартратного комплекса и выделяется олово.
Значительно проще электрогравиметрические установки, не требующие строгого поддержания потенциала. Обычно они применяются для определения в растворе только одного иона. Мешающие ионы должны быть отделены или связаны в прочный комплекс, не поддающийся электрохимическому восстановлению в условиях анализа. Электрогравиметрическим методом определяется медь из сернокислых растворов в присутствии азотной кислоты, серебро и кадмий из цианидного раствора, никель из аммиачного раствора и другие элементы.
11.3.3. Электролиз на ртутном катоде
Наиболее существенными особенностями электролиза на ртутном катоде являются большая величина перенапряжения водорода и образование амальгам многими металлами. Перенапря
249
жение водорода на ртутном катоде превышает 1 В, поэтому при электролизе кислых растворов происходит выделение многих металлов, не выделяющихся на платиновом или других катодах.
При электролизе кислых растворов на ртутном катоде выделяются висмут, кобальт, хром, медь, железо, молибден, никель, осмий, свинец, палладий, платина и многие другие, всего более 20 элементов, но не выделяются алюминий, ванадий, уран, титан и некоторые другие Таким образом, электролиз на ртутном катоде позволяет отделить большие содержания железа, хрома, меди от ванадия, титана и других, что часто существенно упрощает и ускоряет анализ сложных объектов — минералов, руд, концентратов, сплавов и т. д.
11.3.4. Внутренний электролиз
В методе внутреннего электролиза внешнего источника тока не требуется. Здесь используется способность металлов с более положительным электродным потенциалом выделяться в свободном виде из растворов их солей под действием металлов с меньшим значением стандартного потенциала (менее благородного). Пластинка менее благородного металла, являющаяся анодом, соединяется с платиновым катодом и, таким образом, выделение анализируемого благородного металла происходит на платине. При небольшом содержании определяемого элемента осаждение металла на платиновом катоде происходит без каких-либо осложнений, но при больших концентрациях наряду с осаждением на катоде может происходить некоторое выделение металла на аноде. Чтобы исключить этот процесс, анод покрывают тонкой пленкой из коллодия или катодное и анодное пространство разделяют пористой перегородкой.
Одним из важных достоинств метода внутреннего электролиза является возможность проведения тонких химических разделений, так как на платиновом катоде выделяются металлы только более благородные, чем металл анода.
Если, например, в качестве анода используется свинцовая пластинка, то на катоде будут выделяться только те металлы, потенциал которых превышает потенциал пары РЬ2+/РЬ, и не будут выделяться металлы с более отрицательным потенциалом. Меняя анод, можно создавать условия разделения металлов с близкими потенциалами. Существенным преимуществом метода является также чрезвычайная простота аппаратурного оформления, позволяющая использовать метод практически в любой лаборатории.
После выделения из раствора методом электролиза осадок можно взвесить и по массе осадка рассчитать содержание определяемого элемента. Если масса осадка очень мала и не может быть взвешена с достаточной точностью, его растворяют и определяют концентрацию вещества в растворе любым подходящим методом. Таким образом, электролиз больших объемов раствора
250
позволяет наряду с разделением провести концентрирование определяемого элемента, так как осадок, полученный при электролизе, можно растворить в небольшом объеме растворителя.
__ 11.4. КУЛОНОМЕТРИЯ
Принцип и теоретические основы кулонометрии были известны давно, однако в 1938 г. этому методу достаточного внимания не уделялось. С 40-х годов кулонометрия начинает широко применяться в аналитической химии и для решения различных физико-химических задач.
у В кулонометрических методах определяют количество электричества, которое расходуется в ходе электрохимической реакции.
Различают два основных вида кулонометрических определений: прямую кулонометрию и кулонометрическое титрование. В методах прямой кулонометрии анализируемое вещество непосредственно подвергается электрохимическому превращению в кулонометрической ячейке. В методе кулонометрического титрования определяемое вещество реагирует с титрантом, который получается в кулонометрической ячейке при электролизе специально подобранного раствора.
11.4.1. Кулонометрия при постоянном контролируемом потенциале
4
Рис. 11.2. Схема установки для потенциостатической кулонометрии
Потенциостатические или кулонометрические методы при постоянном контролируемом потенциале широко применяются в прямой кулонометрии. Принципиальная схема установки для по-тенциостатической кулонометрии приведена на рис. 11.2.
Напряжение с аккумуляторной батареи I через делитель напряжения 2 подается на рабочий электрод 4 кулонометрической ячейки 5. Потенциал электрода определяется милливольтметром или потенциометром, сила тока — амперметром. Количество израсходованного электричества измеряется кулонометром 6. В современных установках в качестве источника стабилизированного напряжения обычно используют специальные электронные приборы — потенциостаты, поддерживающие заданный потенциал с точностью примерно ±10 мВ в интервале от —2,5 до 2,5 В. Потенциал рабочего электрода устанавливают с помощью поляризационной кривой (/ — У-кривой) в области, где достигается предельный ток.
Рабочим электродом кулоно-
251
метрической ячейки обычно служит платиновая пластинка или ртуть, хотя иногда используют также золотые, серебряные или графитовые электроды. Вспомогательный электрод изготовляется из тех же материалов. Электродные пространства рабочего и вспомогательного электродов разделены. Контакт между ними осуществляется через пористую перегородку. В качестве электрода сравнения 3 (рис. 11.2) обычно выбирают каломельный или хлорсеребряный. Количество электричества, израсходованное на протекание электрохимической реакции, может быть измерено с помощью интеграторов тока или кулонометров, а также определено расчетным методом.
Принцип действия кулонометров основан на том, что через последовательно включенный прибор в цепи протекает такой же ток, какой проходит через анализируемый раствор, и, следовательно, за некоторый промежуток времени через анализируемый раствор и через прибор пройдет одно и то же количество электричества. В последовательно включенном кулонометре со 100%-ным выходом просекает хорошо известная электрохимическая реакция, и измерение количества электричества сводится, таким образом, к определению количества вещества, полученного в результате этого процесса.
Е зависимости от способа измерения объема или массы вещества различают газовые, электрогравиметрические, титрацион-ные и другие кулонометры. В газовых кулонометрах определяется объем газа, выделившегося в результате электрохимического процесса. В электрогравиметрических кулонометрах определяется масса вещества. Например, в медных кулонометрах находят массу металлической меди, выделившейся при электролизе сульфата меди, в серебряных — массу серебра, полученного при электролизе нитрата серебра, и т. д.
Операцию взвешивания катода с выделившейся медью или серебром иногда заменяют анодным растворением металла с этих электродов при постоянной силе тока. Зная длительность процесса и силу тока по формуле (11.1), рассчитывают массу выделившегося металла или сразу количество электричества. Полученную систему не совсем удачно называют кулонометрическим кулонометром. Этот метод значительно экономит время, не ухудшая точности.
При прямом кулонометрическом определении, например, олова (IV) или железа (III) в ячейке происходит восстановление Sn (IV) до Sn (II) или Fe (III) до Fe (II) По мере уменьшения концентрации восстанавливающегося иона сила тока в цепи падает. Эта зависимость приближенно выражается уравнением
Л = (11.4)
где It — сила тока в момент времени t\ 10 — сила начального тока; k — константа, включающая коэффициент диффузии, площадь электрода и другие величины.
Уравнение (11.4) может быть использовано для определения
252
количества электричества Q, израсходованного на электрохимическое превращение вещества, поскольку 00
(11.5) о
Подставляем уравнение (11.4) в (11.5) и интегрируем: ОО оо
Q= $/ое-^ = /о| —= (Н.6)
о о й й
Величина k находится графически. При логарифмировании (11.4) получаем
In It = Io — kt
или
lg Z, = lg Zo — 2^-- (11.7)
Как видно, в координатах IgA — t зависимость (11.7) представляет прямую с угловым коэффициентом —. При экстрапо-ляции эта прямая отсекает на ординате отрезок, равный /о. В практических условиях длительность процесса редко превышает 30 мин. Окончание восстановления обычно фиксируется по прекращению изменения силы тоКа в течение некоторого времени: сила тока при этом уменьшается почти до нуля. В некоторых случаях, например, при большом остаточном токе применяют химические или физико-химические способы индикации. Массу определяемого вещества рассчитывают по формуле
Поправку на остаточный ток вводят, зная его силу и время электролиза.
11.4.2. Кулонометрия при постоянной контролируемой силе тока (кулонометрическое титрование)
В методе кулонометрического титрования используют установки с постоянной силой тока. Так как титрант генерируется в количестве, точно эквивалентном содержанию анализируемого вещества, то по количеству электричества, израсходованного на генерацию титранта, можно рассчитать содержание определяемого вещества. Блок-схема установки для кулонометрического титрования приведена на рис. 11.3. Пульт-переключатель 4 питается током стабилизированного напряжения от аккумуляторной батареи 1 через сопротивление 2 и амперметр 3. Постоянство силы тока в генераторной цепи 7 контролируется потенцио
253
метром 6 по падению напряжения на стандартном сопротивлении. Пуск секундомера 5 и включение генераторной цепи 7 производится через пульт одновременно (8 и 8' — генераторные электроды). Конец реакции фиксируется с помощью индикаторных электродов 9 и измерительного потенциометра 10. Титрант генерируется в результате электролиза на электроде 8 (рабочий генераторный электрод). Вторым электродом схемы генерации является так называемый вспомогательный электрод (8'). Его обычно изолируют от раствора анализируемого вещества, помещая в трубку с дном из пористого стекла, так как продукт реакции на вспомогательном электроде нередко мешает кулонометрическому определению. Индикаторными электродами могут быть два платиновых электрода, если для индикации применяются амперометрический метод, или платиновый и каломельный, если используется потенциометрическая индикация, и т. д. Может
Рис 11.3. Блок-схема установки для кулонометрического титрования
быть использован также спектрофотометрический или какой-либо другой способ определения точки эквивалентности. Ячейки с внешней генерацией титранта, когда титрант генерируется в отдельной камере и затем добавляется к анализируемому веществу, применяются реже, однако иногда они бывают крайне необходимы, например кулонометрическое определение кислот основано на генерировании ОН -ионов при электролизе воды. На катоде происходит восстановление воды
2Н2О + 2е~ = Нг + 2ОН~
а на аноде ее окисление
2Н2О= О2 + 4Н+ + 4е-
Чтобы не допустить случайного попадания ионов Н+ из анодного пространства в рабочий объем, применяются ячейки с внешней генерацией ОН-
Кулонометрическое титрование имеет некоторые преимущест
254
ва перед обычными титриметрическими методами. Наиболее существенным достоинством кулонометрического титрования является то, что рабочий раствор в этом методе не готовят и не стандартизируют: титрант генерируется электрохимически непосредственно в присутствии анализируемого вещества и в количестве, необходимом только для данного титрования. Это позволяет использовать для титрования малоустойчивые или легколетучие вещества, такие, как, например, С12, Вг2 или соединения Cu (I), Сг (II) и т. д., и обходиться в работе небольшими навесками вещества, так как регулируя силу тока, можно точно дозировать очень небольшое количество титранта. Достоинствами кулонометрического титрования являются также универсальность метода приготовления титранта (один и тот же источник тока можно использовать для генерирования различных титрантов) и возможность легкой автоматизации процесса титрования.
11.5. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Электрогравиметрический анализ характеризуется высокой точностью: погрешность определения составляет 0,1...0,2 %. Достоинствами его является также возможность проведения анализа во многих случаях без предварительного разделения и сравнительно простая аппаратура. Ограничением метода является его применимость к относительно небольшому числу элементов и для анализа сравнительно больших содержаний, а также длительность анализа.
Перспективными направлениями развития электрогравиметрических методов анализа является поиск условий для определения элементов в сложных смесях без разделения, совершенствование методов внутреннего электролиза и расширение возможностей, практического приложения к различным объектам анализа.
Практическая применимость метода потенциостатической кулонометрии достаточно широка. Известны методики аналитического определения сурьмы, мышьяка, висмута, кадмия, меди и многих других элементов. Разработаны методики определения нескольких элементов при совместном присутствии, как, например, определение малых содержаний кадмия в присутствии меди, что является вообще сложной аналитической проблемой.
Анализ галогенидов является примером кулонометрического определения неэлектроактивных веществ, т. е. веществ, не изменяющихся в данных условиях под действием тока. Галогениды Х~ осаждают ионами Ag+, которые генерируются на Ag-электроде: Ag -j- X := AgX-f-e .
Кулонометрическим методом определяется также ряд органических веществ (пикриновая кислота, аспарагиновая кислота, хинон, хлорбензолы и фенолы, азокрасители, нитрозосоединения
255
и т. д.). Например, пикриновая кислота на ртутном катоде легко восстанавливается до триаминофенола:
Успешно применяется кулонометрический метод в фазовом и металлографическом анализе, для исследования кинетики химических реакций, механизма электроокисления и восстановления органических и неорганических веществ, изучения коррозии и решения многих других вопросов.
В кулонометрическом титровании используют химические реакции самого различного типа: кислотно-основного взаимодействия, окисления — восстановления, комплексообразования и т. д. Сильные и слабые кислоты могут быть точно нейтрализованы гидроксидом, образующимся при электровосстановлении воды на платиновом катоде. Различные восстановители (Fe(II), Sn(II), Sb (III), As (III) и др.) могут быть оттитрованы, например, перманганатом, который легко генерируется из MnSO4 в ячейке с платиновым анодом. При анодном растворении хрома в серной кислоте получае~ся дихромат, который также может быть использован для этого титрования. В кулонометрическом титровании применяется также, например, свободный бром, генерируемый на платиновом аноде из солянокислого раствора бромида калия. Бромом титруют, например, гидразин, тиоцианат (SCN“ 4- ЗВг2 + 4Н2О = H2SO4 + HCN + 6Вг~ + 5Н+) фенол, различные металлорганические соединения, As (III), Sb (Ш), Fe (II), Т1 (I) и многие другие восстановители. Определение выполняется с высокой точностью, так как неизбежные трудности при титровании раствором брома, возникающие в обычных титриметрических методах, здесь отпадают в связи с мгновенным расходом брома на реакцию титрования. По этой же причине в методиках кулонометрического титрования часто используются вещества крайне неустойчивые при обычных условиях хранения, например соединения Си (I), Cr (II), Ti (III) и т. д. Возможность использования таких веществ в практике количественных определений является очень ценной особенностью кулонометрического титрования. Многие катионы определяются титрованием этилендиаминтетраацетатом, который генерируется из соответствующих комплексов ртути или кадмия по схеме
НеЭДТА2 +2е =ЭДТА4~ + Нр (ж)
256
11.6. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Кулонометрический метод позволяет определять очень небольшое содержание вещества с высокой точностью (0,1...0,05%), превосходя в этом отношении многие другие методы. Кулонометрия характеризуется также высокой селективностью (избирательностью), позволяя определять многие вещества в растворе без предварительного химического разделения. Избирательность обеспечивается обоснованным выбором рабочего потенциала электрода и поддерживанием его постоянного значения с высокой точностью во все время электролиза. Кулонометрический анализ не требует какой-либо предварительной градуировки измерительных приборов по концентрации или построения градуировочных графиков, связывающих свойство вещества с его концентрацией, и в этом смысле кулонометрию следует считать абсолютным методом.
Метод кулонометрического титрования характеризуется высокой чувствительностью и точностью (0,1...0,05%), позволяя прямым титрованием определять вещества в растворе при концентрации до 10-6 моль/л, что намного превышает возможности других титриметрических методов. Он не требует предварительного приготовления, стандартизации и хранения стандартных растворов. Кулонометрическое титрование может быть легко автоматизировано. Области применения методов кулонометрического титрования непрерывно расширяются.
1 Вопросы
1. В чем сущность электрогравиметрического метода анализа?
2. В чем различие методов прямой кулонометрии и кулонометрического титрования?
3. Какие законы лежат в основе методов электрогравиметрии и кулонометрии? Сформулировать их.
4. Что представляет собой выход по току и каким образом его учитывают в расчетах?
5. На чем основаны электрогравиметрические разделения?
6. Назвать основные узлы установок для электрогравиметрического анализа.
7. Какие особенности имеет электролиз на ртутном катоде и как они используются в анализе?
8. Что представляет собой метод внутреннего электролиза и каковы возможности этого метода анализа?
9. Каковы особенности метода кулонометрии при контролируемом потенциале?
10. Каковы преимущества кулонометрического анализа при контролируемой силе тока?
11. Привести принципиальную схему установки для кулонометрического титрования.
*~I533 7S7
12. По какому закону изменяется сила тока в ходе прямого кулонометрического определения? Привести примеры прямых кулонометрических определений.
13. Назвать наиболее распространенные способы фиксирования точки эквивалентности в кулонометрическом титровании.
14. Привести примеры кулонометрического титрования: а) электрогенерированными окислителями; б) электрогенериро-ванными восстановителями; в) с использованием реакций осаждения и комплексообразования; г) с использованием реакций кислотно-основного взаимодействия.
15. В чем сущность определения воды методом кулонометрического титрования?
16. Указать достоинства и недостатки кулонометрических методов анализ?
Задачи
1. Навеску сплава массой 0,6578 г растворили и через полученный раствор в течение 20,0 мин пропускали ток силой 0,200 А, в результате чего на катоде полностью выделилась медь. Определить массовую долю (%) меди в сплаве, если выход по току составлял 80,0%.
В соответствии с законом Фарадея
m(Cu)
_ /Ш(Си)т]
— Ап-100
где m(Cu) — масса выделенной меди, г; / — сила тока, A; t — время электролиза, с; М(Си) —молярная масса меди, г/моль; F— постоянная Фарадея; п — число электронов, участвующих в электрохимическом процессе; т; — выход по току, %
Массовая доля (%) меди в сплаве равна
m(Cu)100 /Ш(Си)т)100 0,200-20-60-63,55-80,0
т(спл) /•'пт(спл) 100 96 500-2-0,6578
= 9,5 %.
2. Навеску вольфрамовой присадки массой 0,6000 г перевели в раствор и выделили алюминий в виде оксихинолината. Осадок после очистки растворили в небольшом объеме концентрированной НС1 и выделившийся 8-оксихинолин оттитровали в кулонометрической ячейке бромом, генерируемым из КВг. Конец реакции определили биамперометрически.
Вычислить массовую долю (%) алюминия в исследуемом образце, если титрование велось при постоянной силе тока 8,0 мА в течение 125 с (выход по току принят равным 100%).
Вычисления производят на основе закона Фарадея:
. т(А1)100 _ /Ш (А1) 100
т(спл) Fnm (обр)
Генерированный бром реагирует не с алюминием, а с эквивалентным количеством 8-оксихннолина. Последний связан с алюминием в окснхинолинате в стехиометрическом соотношении 3:1.
258
По уравнению реакции
C9H6NOH +2Br2 C9H4Br2NOH +2Н+ +2Вг"
на 1 моль 8-оксихинолина приходится 2 моль брома. Следовательно, число электронов, участвующих в электрохимическом превращении, в расчете на 1 моль алюминия составляет п = 3-4 = 12.
Подставляем числовые значения в формулу и находим массовую долю (%) алюминия в образце:
w(Al)
0,008-125-26,98-100
96 500-12-0,6000
3,9-10-3 %.
3. Определить массовую долю (%) индифферентных примесей в образце медного купороса, если после растворения его навески массой m (CUSO4 • 5НгО) г в азотной кислоте и электролиза полученного раствора выделено на платиновом катоде m(Cu) г:
Вариант . . . т (CuSO4-5Н2О), m(Cu), г
12 3 4
0,4556 0,5237 0,6274 0,5807
0,1145 0,1322 0,1586 0,1463
Ответ: 1) 1,26 %; 2) 0,82%; 3) 0,68%; 4) 1,01 %.
4. При электролизе раствора нитрата свинца на аноде выделилось т(РЬОг) г. Определить молярную концентрацию эквивалента Pb(NO3)2 (/экв = 1/2)> если для анализа взяли V мл этого раствора:
Вариант I/, мл .
m (РЬО2), г
12 3 4
20,00 25,00 40,00 10,00
0,2506 0,1945 0,2136 0,1670
Ответ: 1) 0,1048 моль/л; 2) 0,0651 моль/л; 3) 0,0446 моль/л; 4) 0,1396 моль/л.
5. Навеску цветного сплава массой m г растворили и путем электролиза при постоянной силе тока / за время t выделили полностью на катоде медь и на аноде свинец в виде РЬО2.
Определить массовую долю (%) меди и свинца в сплаве, если выход по току составлял 100%:
Вариант т, г .
/, А .
t, мин
1,525 0,200
45,0
2
1.442
0,150
50,0
3
1,811
0,220
40,0
4
1,621
0,180
38,0
Ответ: 1) 11,66% Си и 38,02 % РЬ; 2) 10,28% Си и 33,51 % РЬ; 3) 9,60% Си и 31,30% РЬ; 4) 8,34 % Си и 27,18 % РЬ.
6. Определить время, теоретически необходимое для полного выделения на катоде кадмия из И мл раствора CdSO4 указанной концентрации, если электролиз проводился при силе тока 0,100 А и выход по току составлял 100 %:
Вариант V, мл . с('/2 CdSO4), моль/л
12 3 4
20,00 40,00 50,00 25,00
0,0622 0,0466 0,0435 0,0945
Ответ: 1) 20 мин; 2) 30 мин; 3) 35 мин; 4) 38 мин.
9*
259
7. Из анализируемого раствора, содержащего ионы трехвалентного металла, в результате электролиза при силе тока 1,000 А за время t было выделено на катоде т г металла:
Вариант т, г t, мин
1 2 3
0,3772 0,6497 0,5047
35,0 15,0 20.0
Определить, какой металл был в растворе, если т] = 100%. Ответ: 1) Сг; 2) Bi; 3) Sb.
8. Навеску цинковой руды массой m г перевели в раствор и полностью выделили из него цинк путем электролиза при силе тока 1,000 А в течение времени t.
Рассчитать массу выделившегося цинка (г) и массовую долю (%) ZnO в руде (выход по току составлял 100 %):
Вариант /, мин т, г .
12 3 4
10,0 13,0 16,9 20,0
1,250 1,400 1,550 1,700
Ответ: 1) 0,2033 г; 20,24 %; 2) 0,2642 г; 23,49%; 3) 0,3252 г; 26,12%; 4) 0,4065 г; 29,76%.
9. Навеск) руды массой m г растворили и восстановили железо до Fe , а затем в кулонометрической ячейке количест венно окислили его на платиновом аноде при контролируемом потенциале. Количество затраченного электричества определили кулонометром, состоящим из платинового анода, который погружен в раствор KI. На титрование иода, выделившегося при прохождении тока, потребовалось V мл 0,0500 М Na2S2O3.
Вычислить массовую долю (%) железа в руде:
Вариант т, г . V, мл
12 3 4
0,7905 0,5710 0,5886 0,4324
22,30 20,45 18 75 19,50
Ответ- 1) 7,88%; 2) 10,00%; 3) 9,25%; 4) 12,11 %.
10. Пикриновую кислоту полностью восстановили в кулонометрической ячейке по уравнению реакции
С6Н2(ОН) (NO2)3 +18Н++ 18е- = С6Н2(ОН) (1ЧН2)з + 6Н2О
Количество затраченного электричества установили по количеству выделившегося в иодидном кулонометре иода, на титрование которого потребовалось V мл 0,0200 М Na2S2O3.
Определить массу пикриновой кислоты в растворе (мг) по следующим данным:
Вариант 12 3 4
V. мл 16,50 18,70 21,15 23.35
Ответ: 1) 4,20 мг; 2) 4,76 мг; 3) 5,38 мг; 4) 5,94 мг.
11*. Для титрования иодид-ионов использовали ионы МпОГ, которые генерировались в анодном пространстве кулонометриче
* В задачах 11 —19 выход по току принят равным 100 %.
260
ской ячейки в сернокислой среде. Точку эквивалентности установили потенциометрически.
Вычислить массу (мг) иодида в растворе, если титрование продолжалось t с при постоянной силе тока I мА.
Вариант t, с 1, мА
12 3 4
272 240 225 308
18,0 16,0 14,0 20,0
Ответ: 1) 6,44 мг; 2) 5,05 мг; 3) 4,14 мг; 4) 8,10 мг.
12. Раствор дихромата калия объемом V мл оттитровали ионами железа (II), генерируемыми при силе тока / Ав течение времени t мин, конец реакции фиксировался по фотометрическим данным:
Вариант V, мл /, А /, мин
1 ! 3 4
20,00 25,00 30,00 40,00
0,200 0,250 0,250 0,300
15,0 20,0 35,0 33,0
Определить молярную концентрацию эквивалента К2СГ2О7 (fSKH= '/б)-
Ответ: 1) 0,0933 моль/л; 2) 0,1244 моль/л; 3) 0,1813 моль/л; 4) 0,1539 моль/л.
13. Навеску сурьмы массой m г перевели в раствор и содержащийся в качестве примеси алюминий выделили в виде оксихи-нолината. После очистки и растворения осадка в соляной кислоте провели кулонометрическое титрование оксихинолина бромом, генерируемым из КВг при постоянной силе тока 8,5 мА Точка эквивалентности, найденная биамперометрически, соответствовала времени электролиза t.
Рассчитать массовую долю (%) алюминия в сурьме по следующим данным:
Вариант 12 3 4
т, г 0.5000 0,5346 0.7015 0,6712
I, с 114 102 92 176
Ответ: 1) 4,52-10“3 %; 2) 3,78-10~3 %; 3) 2,60-10~3 %; 4) 5,20-10~3 %.
14. Навеску алюминия массой т г растворили и содержащиеся в виде примеси ионы Fe3+ оттитровали ионами Sn2+, получаемыми при постоянной силе тока 4,0 мА. Точка эквивалентности фиксировалась потенциометрическим методом, время электролиза t определялось хронометром.
Определить массовую долю (%) железа в алюминии по следующим данным:
Вариант т, г t, с
1
1,000
100
2 3 4
1,224 1,513 1,358
80 60 176
Ответ: 1) 2,32-10'2 %; 2) 4) 3,00-10 ~2 %.
1,51 -10 2 %;
3) 9J8-10~3 %;
261
15. Кальций в растворе оттитровали ионами этилечдиамин-тетраацетата, которые генерировались в кулонометрической ячейке из соответствующего ртутного комплекса. Конец реакции установили потенциометрически.
Вычислить массу (мкг) кальция в растворе, если титрование велось при силе тока / мА в течение времени / с (в генерировании 1 моль ЭДТА участвует два электрона):
Вариант 1234
/. мА 3.0 4,0 5,0 6,0
/, с . 65 80 100 120
Ответ-. 1) 40,5 мкг; 2) 66,5 мкг: 3) 103,7 мкг; 4) 149,5 мкг.
16. Раствор 8-оксихинолина титровали бромом, генерируемым из КВг в присутствии H2SO4 при постоянной силе тока I. Конец реакции определили биамперометрически.
Определить массу 8-оксихинолина (мг), если время электролиза составило t, а для окисления 1 моль оксихинолина требуется 2 моль Вг2:
Вариант 1234
/, мА 6,0 5,0 7,0 8,0
/, с . 75 68 112 125
Ответ-. 1) 0,169 мг; 2) 0,128 мг; 3) 0,295 мг; 4) 0,376 мг.
17. Пиридин в растворе определили кулонометрическим титрованием с помощью ионов Н , образующихся при электролизе воды. Окончание реакции установили потенциометрически.
Определить массу пиридина (мг), если электролиз проходил при силе тока I мА в течение времени t мин по следующим данным:
Вариант
/, мА t, мин
1 2 3 4
50,0 100,0 125 0 75,0
10,0 12,0 6,5 4,0
Ответ-. 1) 24,6 мг; 2) 59,0 мг; 3) 40,0 мг; 4) 14,8 мг.
18. В анодную камеру кулонометрической ячейки ввели раствор SO2 в пиридине и раствор KI в метаноле. Установив силу тока генерационной цепи I мА, провели генерирование иода из KI до достижения заданного значения тока в индикационной цепи. Затем внесли в ячейку пробу органического растворителя массой m г, включили секундомер и провели титрование воды до получения такого же значения индикаторного тока, измерив время / мин:
Вариант Вещество 1 СНзСООН 2 СгН5ОН 3 С6Н6 4 ССЦ 5 (СНз)гМСОН
т, г 0,8021 0,4608 0,5106 0,3300 0,3020
/, мА 50,0 75,0 40,0 35,0 80,0
t, мин 2,0 4,5 2,5 4,0 5,0
Вычислить массовую долю (%) воды в органическом растворителе, если на 1 моль воды при взаимодействии с получаемым 262
при генерировании реактивом Фишера (SO2, I2, C5H5N) приходится два электрона:
C5H5N-I2 + C6H5N-SO2 + C5H5N + Н2О^ 2C5H5N-HI + + C5H5N • SO3
Ответ: 1) 0,07%; 2) 0,41 %; 3) 0,11 %; 4) 0,24 %; 5) 0,74 %.
19. Пробу сточной воды объемом V мл, содержащей фенол, оттитровали бромом, генерируемым из КВг по уравнению реакции
СеН5ОН -|- ЗВг2 СбН2ОНВг3 -р ЗВг -р ЗН"*"
Рассчитать концентрацию фенола в сточной воде (мкг/мл), если электролиз длился в течение времени t при силе тока 0,035 А по следующим данным:
Вариант 1234
V, мл 100,0 50,00 200,0 250,0
I 4 мин 35 с 2 мин 42 с 7 мин 4 с 6 мин 16 с
Ответ: 1) 15,64 мкг/мл; 2) 18,43 мкг/мл; 3) 12,06 мкг/мл;
4) 8,53 мкг/мл.
Глава 12
РАДИОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
12.1. ТИПЫ РАДИОАКТИВНОГО РАСПАДА И РАДИОАКТИВНОГО ИЗЛУЧЕНИЯ
Открытие радиоактивности относится к 1896 г., когда А. Беккерель обнаружил, что уран самопроизвольно испускает излучение, названное им радиоактивным (от лат. radio — излучаю и activas — действенный).
Радиоактивное излучение возникает при самопроизвольном распаде атомного ядра. Известно несколько типов радиоактивного распада и радиоактивного излучения.
а-Распад. Распад ядра с выделением а-частиц, которые являются ядрами Не2+. Например,
292U —► 29оТЬ -р 2а
В соответствии с законом радиоактивного смещения, при а-рас-паде получается атом, порядковый номер которого на две единицы, а атомная масса на четыре единицы меньше, чем у исходного атома.
Р-Раслад. Различают несколько видов Р-распада: электронный Р-распад; позитронный p-распад; А-з а х в а т. При электронном p-распаде, например,
?iP -> 3>26s + р-
263
нейтрон внутри ядра превращается в протон. При испускании отрицательно заряженной 0-частицы порядковый номер элемента возрастает на единицу, а атомная масса практически не меняется. При позитронном 0-распаде из атомного ядра выделяется позитрон (0+-частица), а протон внутри ядра превращается в нейтрон. Например,
ffNa ?gNe |- 0+
Продолжительность жизни позитрона невелика, так как при столкновении его с электроном происходит аннигиляция, сопровождающаяся испусканием у-квантов.
При К -захвате ядро атома захватывает электрон из близлежащей электронной оболочки (из К-оболочки) и один из протонов ядра превращается в нейтрон. Например,
*9 К + в — igAr -|- hv
На свободное место в K-оболочке переходит один из электронов внешней оболочки, что сопровождается испусканием жесткого рентгеновского излучения.
Спонтанное деление. Оно характерно для элементов периодической системы Д. И. Менделеева с Z > 90. При спонтанном делении тяжелые атомы делятся на осколки, которыми обычно являются элементы середины таблицы Д. И. Менделеева. Спонтанное деление и а-распад ограничивают получение новых трансурановых элементов.
Поток а и 0-частиц называют соответственно а- и 0-излуче-нием. Кроме того, известно у-излучение. Это электромагнитные колебания с очень короткой длиной волны. В принципе, у-излу-чение близко к жесткому рентгеновскому и отличается от него своим внутриядерным происхождением. Рентгеновское излучение возникает при переходах в электронной оболочке атома, а у-излучение испускают возбужденные атомы, получившиеся в результате радиоактивного распада (а или 0).
В результате радиоактивного распада получаются элементы, которые по заряду ядер (порядковому номеру) должны быть помещены в уже занятые клетки периодической системы элементами с таким же порядковым номером, но другой атомной массой. Это так называемые изотопы. По химическим свойствам их принято считать неразличимыми, поэтому смесь изотопов обычно рассматривается как один элемент. Неизменное!ь изотопного состава в подавляющем большинстве химических реакций иногда называют законом постоянства изотопного состава. Например, калий в природных соединениях представляет собой смесь изотопов, состоящую на 93,259 % из 3Ж, на 6,729% из 41К и на 0,0119% из 40К (/(-захват и 0-распад). Кальций насчитывает шесть стабильных изотопов с массовыми числами 40, 42, 43, 44, 46 и 48. В химико-аналитических и очень многих других реакциях это соотношение сохраняется практически неизменным, поэтому для разделения изотопов хи
264
мические реакции обычно не применяются. Чаще всего для этой цели используются различные физические процессы — диффузия, дистилляция или электролиз.
Единицей активности изотопа является беккерель (Бк), равный активности нуклида в радиоактивном источнике, в котором за время 1 с происходит один акт распада.
12.2. ЗАКОН РАДИОАКТИВНОГО РАСПАДА
Скорость радиоактивного распада----пропорциональна
числу имеющихся ядер N-.
dW 1 к, ----
где X — постоянная распада. При интегрировании получаем
— InW = W + const, если t = 0, то N — No и, следовательно, const = — IgA/o- Окончательно
N=h’oe~>l (12.1)
или A=Aoe^f, (12.2)
где А — активность в момент времени /; Ао — активность при t = 0.
Уравнения (12.1) и (12.2) характеризуют закон радиоактивного распада. В кинетике они известны как уравнения реакции первого порядка.
В качестве характеристики скорости радиоактивного распада обычно указывают период полураспада Т1/2, который так же, как и к, является фундаментальной характеристикой процесса, не зависящей от количества вещества.
| Периодом полураспада называют промежуток времени, в течение которого * данное количество радиоактивного вещества уменьшается наполовину
Таким образом, в момент времени Т1г2 отношение N/No = '/2 откуда — ЬТ1/2= - |п2 и Г1/2 = -^- = °^. С учетом этого соотношения уравнение (12.2) можно переписать: А = Ле~МЙЗ,/7|/2.
Период полураспада различных изотопов существенно различен. Он находится в пределах примерно от 1О10 лет до ничтожных долей секунды. Конечно, вещества, имеющие период полураспада Ю... 15 мин и меньше, использовать в лаборатории трудно. Изотопы с очень большим периодом полураспада также нежелатель-
265
ны в лаборатории, так как при случайном загрязнении этими веществами окружающих предметов потребуется специальная работа по дезактивации помещения и приборов.
12.3. ВЗАИМОДЕЙСТВИЕ РАДИОАКТИВНОГО ИЗЛУЧЕНИЯ С ВЕЩЕСТВОМ И СЧЕТЧИКИ ИЗЛУЧЕНИЯ
В результате взаимодействия радиоактивного излучения с веществом происходит ионизация и возбуждение атомов и молекул вещества, через которое оно проходит. Излучение производит также световое, фотографическое, химическое и биологическое действие. Например, первичным результатом действия радиоактивного излучения на воздух является появление ионов:
О2 + hv = Ot + е~ О2 + е~ = ОГ
N2 hv — N2 -j- e~ N2 -j- e~ = N2
H2O + hv = H2O+ + e~ H2O + e~ = H2O-
H2O+ = H+ + OH’ H2O- = НЧ- OH-
Образующиеся при протекании этих процессов радикалы Н‘ и ОН' обладают сильным физиологическим действием — при больших дозах они являются одной из причин лучевой болезни, малокровия и т. д., так как энергично взаимодействуют с ферментами и составными частями крови. Опасность радиоактивного воздействия возрастает вследствие того, что организм не обладает болевыми реакциями на действие радиоактивного излучения. Быстрое превращение этих частиц в безопасные для человеческого организма является одним из эффективных приемов борьбы с лучевой болезнью.
Радиоактивное излучение вызывает большое число химических реакций в газах, растворах, твердых веществах Их обычно объединяют в группу радиационно-химических реакций. Сюда относятся, например, разложение (радиолиз) воды с образованием водорода, пероксида водорода и различных радикалов, вступающих в окислительно-восстановительные реакции с растворенными веществами. Радиоактивное излучение вызывает разнообразные радиохимические превращения различных органических соединений — аминокислот, кислот, спиртов, эфиров и т. д. Интенсивное радиоактивное излучение вызывает свечение стеклянных трубок и ряд других эффектов в твердых телах. На изучении взаимодействия радиоактивного излучения с веществом основаны различные способы обнаружения и измерения радиоактивности.
В зависимости от принципа действия счетчики радиоактивных излучений подразделяют на несколько групп.
Ионизационные счетчики. Их действие основано на возникновении ионизации или газового разряда, вызванного ионизацией при попадании в счетчик радиоактивных частиц или у-квантов. Среди десятков приборов, использующих ионизацию, типичными 266
являются ионизационная камера и счетчик Гейгера — Мюллера, который получил наибольшее распространение в химико-аналитических и радиохимических лабораториях. Схема устройства, принцип работы его и некоторые технические характеристики счетчика указывались при рассмотрении рентгено-спектрального анализа.
Для радиохимических и других лабораторий промышленностью выпускаются специальные счетные установки.
Сцинтилляционные счетчики. Действие этих счетчиков основано на возбуждении атомов сцинтиллятора у-квантами или радиоактивной частицей, проходящей через счетчик. Возбужденные атомы, переходя в нормальное состояние, дают вспышку света.
В начальный период излучения ядерных процессов визуальный счет сцинтилляций сыграл большую роль, однако в дальнейшем он был вытеснен более совершенным счетчиком Гейгера — Мюллера. В настоящее время сцинтилляционный метод вновь стал широко применяться уже с использованием фотоумножителя.
Черенковские счетчики. Действие этих счетчиков основано на использовании эффекта Черенкова, который состоит в излучении света при движении заряженной частицы в прозрачном веществе, если скорость частиц превышает скорость света в данной среде. Факт сверхсветовой скорости частицы в данной среде, конечно, не противоречит теории относительности, поскольку скорость света в какой-либо среде всегда меньше, чем в вакууме. Скорость движения частицы в веществе может быть больше скорости света в этом веществе, оставаясь в то же время меньше скорости света в вакууме в полном соответствии с теорией относительности. Счетчики Черенкова применяются для исследовательских работ с очень быстрыми частицами, для исследований в космосе и т. д„ поскольку с их помощью может быть определен ряд других важных характеристик частиц (их энергия, направление движения и др.).
12.4. ЯДЕРНАЯ ХИМИЯ И ИСКУССТВЕННАЯ РАДИОАКТИВНОСТЬ
При бомбардировке вещества потоком а-частиц, протонов, нейтронов и т. д. происходит превращение атомов. Ядерные превращения можно представить с помощью уравнений, напоминающих уравнения обычных химических реакций. Например, 27А1 под действием протонов превращается в 24Mg:
27А1 + 'р = 24Mg + 4а
Под действием нейтронов 27А1 превращается в 27Mg:
27А1 + ‘п = 27Mg + ’р
267
Часто используется краткая запись этих уравнений. Вместо первого уравнения пишут 27А1 (р, a) 24Mg, а вместо второго — 27А1 (п, р) 27Mg. При такой записи сначала указывают символ исходного атома, далее в скобках через запятую приводят бомбардирующую и выбиваемую частицы, после чего записывают символ образующегося атома. Один и тот же элемент под действием различных бомбардирующих частиц может претерпевать множество разнообразных превращений.
В 1934 г. И. Кюри и Ф. Жолио открыли искусственную радиоактивность. Они обнаружили, что излучение, возникающее при бомбардировании алюминия а-частицами, подчиняется закону радиоактивного распада (12.1). Оказалось, что при бомбардировке алюминия образуется неустойчивый радиоактивный изотоп фосфора: 27А1 + 4а = ЗПР +’и, который в результате радиоактивного распада превращается в кремний: 30Р—► ->30Si 4* Р+- В настоящее время известно большое число искусственно полученных радиоактивных изотопов. С помощью ядерных реакций удалось получить элементы, в природе не существующие, но для которых в таблице Д. И. Менделеева имелись соответствующие клетки под номерами 43, 61, 85, 87. Это технеций, прометий, астат и франций.
12.5. МЕТОДИКИ АНАЛИЗА, ОСНОВАННЫЕ НА ИЗМЕРЕНИИ РАДИОАКТИВНОСТИ
12.5.1. Использование естественной радиоактивности в анализе
Элементы, имеющие естественную радиоактивность, могут быть определены по этому свойству количественно. Это U, Th, Ra, Ас, К и др., всего более 20 элементов. Например, калий можно определить по его радиоактивности в растворе при концентрации 0,05 М. Определение различных элементов по их радиоактивности обычно производят с помощью градуировочного графика, показывающего зависимость активности от процентного содержания определяемого элемента или методом добавок.
Большое значение имеют радиометрические методы в поисковой работе геологов, например при разведке месторождений урана
12.5.2. Активационный анализ
При облучении нейтронами, протонами и другими частицами высокой энергии многие нерадиоактивные элементы становятся радиоактивными. Активационный анализ основан на измерении этой радиоактивности. Хотя в принципе для облучения могут быть использованы любые частицы, наибольшее практическое значение имеет процесс облучения нейтронами. Применение для этой цели заряженных частиц связано с преодолением более
268
значительных технических трудно-стей, чем в случае нейтронов. Основными источниками нейтронов для проведения активационного анализа являются атомный реактор и так называемые портативные источники (радиевобериллиевый и др.). В последнем случае а-частицы, получившиеся при распаде какого-либо а активного элемента (Ra, Rn и т. д.),
взаимодействуют с ядрами берил-Рис 12, Уменьшение
радиоактив-
лия, выделяя нейтроны, ности во времени
9Ве + 4Не ,2С + п
Нейтроны вступают в ядерную реакцию с компонентами анализируемой пробы, например
55Мп п = 56Мп или Мп (п, -у) 56Мп
Радиоактивный 56Мп распадается с периодом полураспада 2,6 ч:
56Mn -> 56Fe + е~
Для получения информации о составе образца некоторое время измеряют его радиоактивность и анализируют полученную кривую (рис. 12.1). При проведении такого анализа необходимо располагать надежными данными о периодах полураспада различных изотопов, с тем чтобы провести расшифровку суммарной кривой.
На практике обычно используют относительный метод анализа, когда в одинаковых условиях об. |учают анализируемый образец и эталон с известным содержанием определяемого элемента, что существенно упрощает анализ. Во многих случаях образец после облучения переводят в раствор, производят химическое выделение интересующих компонентов путем экстракции, хроматографии, осаждения или другим методом и определяют активность продуктов разделения. Операции химического разделения значительно расширяют возможность метода, повышают селективность, однако это имеет реальное значение лишь для изотопов с не слишком малым периодом полураспада.
Другим вариантом активационного анализа является метод у-с п е к т р о с к о п и и, основанный на измерении спектра у-из-лучения образца. Энергия у-излучения является качественной, а скорость счета — количественной характеристикой изотопа. Измерения производят с помощью многоканальных у-спектро-метров со сцинтилляционными или полупроводниковыми счетчиками. Это значительно более быстрый и специфичный, хотя и несколько менее чувствительный метод анализа, чем радиохимический.
269
В настоящее время различные варианты активационного анализа получили широкое и разнообразное применение. Наибольшее значение имеет анализ веществ высокой чистоты, используемых, например, в полупроводниковой и атомной промышленности, ракетной технике и т. д. Он с успехом применяется для анализа воды и многих геохимических объектов, для определения состава различных биологических и космохимических материалов и во многих других областях науки и народного хозяйства, связанных, главным образом, с новой техникой.
Важным достоинством активационного анализа является его высокая чувствительность. С его помощью может быть обнаружено при благоприятных условиях до 1013... 10-15 г вещества. В некоторых специальных случаях удавалось достигнуть еще более высокой аналитической чувствительности. Например, с его помощью контролируют чистоту кремния и германия в промышленности полупроводников, обнаруживая содержание примесей до 10-8...10-в %. Такие содержания никаким другим методом, кроме активационного, определить невозможно. При получении тяжелых элементов периодической системы, таких, как менделевий и курчатовий, исследователям удавалось считать почти каждый атом полученного элемента.
Основным недостатком активационного анализа является громоздкость источника нейтронов, а также нередко длительность самого процесса получения результатов.
Применение активационного анализа позволило решить многие геохимические и общенаучные проблемы, например, связанные с установлением возраста минералов и образцов органического происхождения. Например, хорошо известный радиометрический способ определения возраста древних предметов и изделий органического происхождения основан на том, что в атмосфере в результате ядерной реакции постоянно образуется радиоактивный углерод 14С с периодом полураспада 5300 лет:
UN + п = 14С + р
Нейтроны генерируются в атмосфере при взаимодействии космического излучения с веществом, а азот является составной частью атмосферы. Радиоактивный |4С образует радиоактивный 14СО2, который вступает в биологический круговорот. Таким образом, вещества и организмы, участвующие в этом круговороте (растения, животные и т. д ), будут иметь примерно постоянную радиоактивность, пропорциональную содержанию |4С. В организмах, которые выпадают из круговорота в результате гибели, количество радиоактивного |4С не пополняется и их активность уменьшается. Таким образом, если измерить активность какого-либо древнего изделия (предмета из дерева, кожи и т. д.), то, зная период полураспада углерода 14С и общее содержание углерода в образце, можно рассчитать промежуток времени, прошедший с момента гибели организма Этот промежуток будет характеризовать примерный возраст изделия. Анализ этим мето
270
дом позволил уточнить даты многих важных событий далекой древности.
Интересным примером применения активационного анализа является предложенный недавно быстрый способ обнаружения взрывчатки. Как известно, в качестве взрывчатых веществ, обычно используют различные нитросоединения. Способ обнаружения основан на том, что взрывчатка наряду с 14N содержит некоторое количество 15N, который при облучений нейтронами превращается в 16N. Этот изотоп имеет период полураспада 7 с и при его распаде кроме р-частиц происходит испускание у-квантов определенной энергии. Появление р-квантов с энергией, характерной для распада 16N после облучения вещества нейтронами, является сигналом о наличии азотсодержащего вещества, возможно взрывчатки. Очень ценные результаты дает активационный анализ в криминалистике, судебной химии и т. д.
12.5.3. Метод изотопного разбавления
Его идею лучше всего рассмотреть на конкретном примере. При определении свинца методом изотопного разбавления в анализируемый раствор, содержащий х г свинца, вводят небольшое известное количество радиоактивного изотопа РЬ*, в результате чего раствор приобретает активность Ло- Какое-то количество полученного раствора осаждают раствором сульфата и получают т г осадка PbSO4 с активностью А. Если радиоактивный изотоп вводился в анализируемый раствор без носителя, то из пропорции
х — Ло| mF — ТУ
х = mF-^~, А
(а)
где F — фактор пересчета PbSO4 на РЬ.
Если изотоп вводился с носителем, то
х t т'л~ Ло I X = mFA°~Am' = mF^- - т', (б)
mF — А' А А '
где т' — масса радиоактивного препарата с носителем.
При т' = 0 (изотоп без носителя) соотношение (б) переходит в (а). Как видно, пропорциональность активности изотопа количеству определяемого компонента позволяет получить точный результат и без достижения полноты осаждения, что является существенным достоинством метода изотопного разбавления.
Определение активности осадка часто нежелательно из-за адсорбции, самопоглощения и других явлений, искажающих истинную величину активности, поэтому обычно определяют активность раствора после отделения осадка. Если At и 1Л— удельная активность и объем раствора до осаждения, а А2 и Vi — после него, то, очевидно, активность осадка А будет равна
А = Ai Vi — А 2 Кг-
271
Применение метода изотопного разбавления предполагает, что закон постоянства изотопного состава элементов соблюдается, химические свойства радиоактивного и неактивного изотопов неразличимы и реакции изотопного обмена радиоизотопа с «третьими» компонентами смеси не происходят.
Метод изотопного разбавления открывает новые возможности в анализе сложных смесей и элементов, близких по своим химико-аналитическим свойствам. Например, при анализе смесей цирконий — гафний или ниобий — тантал можно получить чистый осадок одного из компонентов, но осаждение не будет полным. Если добиться полного осаждения, то полученный осадок будет загрязнен элементом-аналогом. В методе изотопного разбавления проводят неполное осаждение и, используя измерения активности, находят содержание анализируемого элемента с достаточной точностью. Аналогичный прием используется также при анализе различных смесей органических веществ.
Для выделения анализируемого компонента в методе изотопного разбавления наряду с осаждением используются также экстракция, хроматография и другие методы разделения, а для определения количества выделенного компонента применяют спектральные, электрохимические и другие методики.
12.5.4. Радиометрическое титрование
При радиометрическом титровании индикаторами являются радиоактивные изотопы элементов. Например, при титровании фосфата магнием в анализируемый раствор вводят небольшое количество фосфата, содержащего радиоактивный Р*.
При титровании протекает реакция
Mg2+ + НР*ОГ = MgHP*O4
Изменение активности в ходе этого титрования можно видеть на рис. 12.2, а. Здесь же показано графическое определение точки эквивалентности (т. э.). До т. э. активность раствора будет резко убывать, так как радиоактивный НР*О2- из раствора будет переходить в осадок. После т э. активность раствора будет оставаться практически постоянной и очень небольшой.
Как видно из рис. 12.2, б, добавление гидрофосфата к раствору Mg2 *" до т. э. практически не будет вызывать увеличения активности раствора, так как радиоактивный изотоп Р* будет переходить в осадок MgHP*O4. После т. э. активность раствора начинает возрастать пропорционально концентрации гидрофосфата.
Реакции радиометрического титрования должны удовлетворять требованиям, обычно предъявляемым к реакциям титри-метрического анализа (скорость и полнота протекания реакции, постоянство состава продукта реакции и т. д.). Очевидным условием применимости реакции в данном методе является также
272
Рис 12 2. Типы кривых радиометрического титрования
а — изменение активности раствора фосфата, содержащего Р* при титровании раствором Mg2+, б—изменение активности раствора Mg2+ при титровании фосфатом, содержащим Р*
переход продукта реакции из анализируемого раствора в другую фазу, с тем чтобы устранить помехи при определении активности раствора. Этой второй фазой часто является образующийся осадок. Известны методики, где продукт реакции экстрагируется органическим растворителем. Например, при титровании многих катионов дитизоном в качестве экстрагента применяют хлороформ или тетрахлорид углерода. Применение экстракции позволяет более точно установить точку эквивалентности, так как в этом случае для ее определения можно измерять активность обеих фаз.
12.5.5. Эффект Мессбауэра
Эффект открыт в 1958 г. Р. П. Мессабауэром. Под этим названием часто объединяют явления испускания, поглощения и рассеяния у-квантов ядрами атомов без затраты энергии на отдачу ядер. Обычно исследуется поглощение у-излучения, поэтому эффект Мессбауэра часто называют также у-резонансной спектроскопией (ГРС).
При испускании у-квантов ядро атома возвращается в нормальное состояние. Однако энергия испускаемого излучения будет определяться не только разностью энергетических состояний ядра в возбужденном и нормальном состояниях. Вследствие закона сохранения импульса ядро испытывает так называемую отдачу. Это приводит к тому, что в случае газообразного атома энергия испускаемого излучения будет меньше, чем в случае, когда излучатель будет находиться в твердом теле. В последнем случае потери энергии на отдачу уменьшаются до пренебрежимо малого значения. Таким образом, у-кванты излучения, испускаемого без отдачи, могут поглощаться невозбужденными атомами того же элемента. Однако различие в химическом окружении ядра-излучателя и ядра поглотителя вызывает некоторую разницу в энергетических состояниях ядер, достаточную, чтобы резонансное поглощение у-квантов не происходило. Разницу в энер-
273
Скорость источника, см/с
Рис 12 3 Мессбауэровский спектр поглощения
гетических состояниях ядер количественно компенсируют с помощью эффекта Допплера, в соответствии с которым частота излучения (в данном случае энергия у-квантов) зависит от скорости движения При некоторой скорости движения излучателя (или поглотителя, так как значение имеет лишь их относительная скорость перемещения) наступает резонансное поглощение. Зависимость интенсивности поглощения
•р-квантов от скорости движения называют спектром Мессбауэра. Типичный мессбауэровский спектр представлен на рис. 12.3, где в качестве меры интенсивности поглощения отложена обратно пропорциональная ей скорость счета. Скорость перемещения образца или излучателя обычно не превышает нескольких сантиметров в секунду. Спектр Мессбауэра являетсся очень важной характеристикой вещества. Он позволяет судить о природе химической связи в исследуемых соединениях, их электронной структуре и других особенностях и свойствах.
12.6. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Применение радиоактивности в аналитической химии весьма многообразно. Области практического использования отдельных аналитических методов, основанных на измерении радиоактивности, охарактеризованы в разделе 12.5. Там же описаны принципы некоторых наиболее важных аналитических методик, их погрешности и пределы обнаружения. Измерение радиоактивности широко применяют также в научно-исследовательских целях: для исследования механизмов химических реакций, определения растворимости малорастворимых соединений, исследования процессов разделения и для решения многих других задач, включая определение важнейших физико-химических констант (констант устойчивости координационных соединений, констант ионообменных процессов и т. д.).
Эффект Мессбауэра в аналитической химии имеет ограниченное применение. Он используется, например, для открытия примесных атомов, если атомы основной решетки не мешают прохождению резонансного у-излучения. Мессбауэровская спектроскопия имеет большие потенциальные аналитические возможности как метод неразрушающего анализа и как один из эффективных методов определения валентного состояния и степени окисления элементов.
274
12.7. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Основными достоинствами аналитических методов, основанных на измерении радиоактивного излучения, являются низкий порог обнаружения анализируемого элемента и широкая универсальность. Радиоактивационный анализ имеет абсолютно низший порог обнаружения среди всех других аналитических методов (1СГ 1S г). Достоинством некоторых радиометрических методик является анализ без разрушения образца, а методов, основанных на измерении естественной радиоактивности, — быстрота анализа. Ценная особенность радиометрического метода изотопного разведения заключена в возможности анализа смеси близких по химико-аналитическим свойствам элементов, таких, как цирконий Д- гафний, ниобий 4- тантал и др.
Дополнительные осложнения в работе с радиоактивными препаратами обусловлены токсичными свойствами радиоактивного излучения, которые не вызывают немедленной реакции организма и тем самым осложняют своевременное применение необходимых мер. Это усиливает необходимость строгого соблюдения техники безопасности при работе с радиоактивными препаратами. В необходимых случаях работа с радиоактивными веществами происходит с помощью так называемых манипуляторов в специальных камерах, а сам аналитик остается в другом помещении, надежно защищенном от действия радиоактивного излучения.
Вопросы
1. Привести кинетическое уравнение радиоактивного распада. Как изменяется радиоактивность во времени?
2. Что называется периодом полураспада? В каких пределах должна находиться эта величина для изотопов, используемых в аналитических целях, и почему?
3. На чем основаны методы обнаружения радиоактивного излучения?
4. Каково устройство и принцип работы основных счетчиков радиоактивного излучения: а) ионизационной камеры; б) счетчика Гейгера — Мюллера; в) счетчика Черенкова; г) сцинтилляционного счетчика?
5. Как используется естественная радиоактивность в аналитических целях? Какие элементы можно определить количественно по их естественной радиоактивности?
6. В чем сущность активационного анализа? Привести примеры аналитических определений.
7. Каковы особенности относительного метода активационного анализа и метода у-спектроскопии?
8. На чем основано радиометрическое определение «возраста» образцов органического происхождения (древних рукописей, бытовых предметов и т. д.)?
275
9. В чем сущность метода изотопного разбавления? Привести примеры аналитических определений.
10. Какой радиометрический метод анализа эффективен при анализе сложных смесей и элементов с близкими аналитическими свойствами: Zr и Hf; Nb и Та?
11. Какой вид имеют кривые радиометрического титрования, если: а) радиоактивное вещество титруется нерадиоактивным реагентом; б) нерадиоактивное вещества титруется радиоактивным реагентом; в) радиоактивное вещество титруется радиоактивным реагентом?
12. Как с помощью осадительного радиометрического титрования выполняется анализ двух- и трехкомпонентных систем,
таких, как Л1+ +Т1+; Hg2+ 4- Zn2+; Са2+ 4- Mg2+; Cd2+4-+ Zn2+; Cu2++ Hg2++ Zn2+?
13. Как можно определить методом радиометрического тит-
рования ионы: Ва2+, РЬ2+, Zn2+, Hg2+, Cu2+, НРО2 ,
SO1~?
14. Как определяют растворимость малорастворимых соединений с помощью радиоактивных изотопов?
15. Как используются радиоактивные изотопы для изучения процессов соосаждения и для разработки гравиметрических методов анализа? Привести примеры.
Задачи
1. В раствор аминокислот ввели 0,1000 г глицина, меченного радиоактивным углеродом 14С с удельной активностью 19 000 имп/ (мин-г). Из этого раствора частично выделили глицин, активность* которого составила 800 имп/мин на 1 г вещества.
Вычислить массу глицина (г) в исследуемом растворе
Если удельная активность взятого препарата S\ = A\/m и удельная активность выделенного осадка Ss, то очевидно:
S\tn = Ss(m + х),
где т — масса носителя; х — масса глицина в исследуемом растворе. Следовательно, масса определяемого компонента равна
Подстазляем числовые значения в уравнение
= 2.275 г.
2. Вычислить массовую долю (%) калия в исследуемом образце, если активность чистого КС1 1800 имп/мин,-а активности исследуемых образцов (Лх) следующие:
* Под активностью имеется в виду пропорциональная ей величина скорости счета (имп/мин).
Образец .
Ах, имп/мин
12 3 4
220 398 616 798
Ответ: 1) 6,41 %; 2) 11,60 %; 3) 17,95 %; 4) 23,25%.
3. К 1 л исследуемого раствора прибавили m г радиоактивного свинца с удельной активностью 432 ими/(мин-мг). Затем из раствора выделили PbSO«, имеющий удельную активность Sx.
Определить концентрацию свинца в растворе (мг/л) по следующим данным:
Вариант т, мг . Sx, имп/(мин-мг)
12 3 4
10 20 30 40
12 23 34 54
Ответ: 1) 350 мг/л; 2) 356 мг/л; 3) 351 мг/л; 4) 350 мг/л.
4. К образцу гидролизованного протеина добавили m г чистого глицина, меченного 14С с удельной активностью 25 000 имп/(мин-г). Выделенный из смеси глицин имел удельную активность 5Л.
Вычислить массу глицина (г) в гидролизованном протеине по следующим данным:
Вариант т, г .
Sx, имп/(мин-мг)
12 3 4
0,1000 0,2000 0,3000 0,4000
442 840 1240 1625
Ответ: 1) 5,56 г; 2) 5,75 г; 3) 5,75 г; 4) 5,75 г.
5. Навеску чистого пенициллина, меченного |4С с активностью 4200 имп/(мин*мг), массой m г прибавили к исследуемому образцу. Из смеси выделили часть кристаллического пенициллина с удельной активностью Sx.
Рассчитать массу пенициллина (г) в исходном образце по следующим данным:
Вариант т, мг . Sx, имп/(мин-мг)
12 3 4
10,0 12,0 14,0 16,0
405 354 312 277
Ответ: 1) 0,0937 г; 2) 0,1304 г; 3) 0,1745 г; 4) 0,2266 г.
6. Навеску известняка массой тоср г перевели в раствор, добавили пренебрежимо мало радиоактивного 45Са и измерили общую активность раствора Лобш. После этого осадили CaSO4, взвесили осадок (т«) и измерили активность осадка Лос.
Определить массовую долю (%) СаСОз в известняке по следующим данным:
Вариант ^обр, г 1 0,3023 2 0,3560 3 0.3410 4 0,4105
^Гос. Г 0,2546 0.2872 0,2905 0,3178
^общ, имп/мин 870 911 950 925
71ос, имп/мин 816 868 846 886
Ответ: 1) 66,02%; 2) 62,46 %; 3) 70,33 %; 4) 59,42 %.
277
7. Для определения микропримесей сурьмы в свинце применили метод двукратного изотопного разбавления. Навеску свинца массой т г растворили в смеси азотной и винной кислот. Полученный раствор разделили на две равные части и в каждую из них ввели различные добавки радиоактивной сурьмы (mi и т2).
Свинец осадили в виде РЬС12, а сурьму окислили до Sb (V). К каждой порции раствора добавили субстехиометрическое количество метилвиолета, связанную в комплекс сурьму экстрагировали толуолом и измерили активность каждой порции (Ai и А2). Вычислить массовую долю (%) сурьмы в свинце, если:
Вариант 1 2 3
пг, г . . 1 5 10
ГП\, мкг 12 6 6
7712, МКГ 24 12 12
Л|, имп/мин . 282 35 380
Дг, имп/мин . 386 42 450
Ответ: 1) 1,402-10'3 %; 2) 6,00-10 6 %; 3) 2,71-10 6 %.
8. Навеску бронзы массой m г растворили в азотной кислоте, перенесли раствор в мерную колбу вместимостью 25,00 мл, нейтрализовали аммиаком и довели водой до метки. Для определения бериллия отобрали 5,00 мл полученного раствора, добавили ЭДТА для маскирования мешающих элементов, ацетатный буфер и оттитровали 0,1000 М (А = 0,913) раствором двухзамещенного фосфата аммония, меченного радиоактивным 32Р. Результаты титрования приведены ниже:
Г(№2НРО4), мл............
А, имп/мин, при m = 0,8120 г
А, имп/мин, при m = 0,7981 г
А, имп/мин, при m = 0,9002 г .
1,00 2,00 3,00 4,00 5,00 6,00 7,00
60 60 60 60 60 840 1600
50 50 50 50 300 1200 —
60 60 60 60 180 500 800
Определить массовую долю (%) бериллия в бронзе. Ответ: 1) 2,58%; 2) 2,47%; 3) 2,10 %.
9. В стакан для радиометрического титрования внесли 1,00 мл исследуемого раствора, соль цинка, содержащую 0,238 мг изотопа 65Zn, и разные объемы (от 0,20 до 1,60 мл) 0,0287 н. К.4ре(СМ)б (}эхв= */з) - После прибавления каждой порции титранта и перемешивания отбирали пробу фильтрата, измеряли активность и возвращали пробу обратно в стакан. Полученные результаты представлены в таблице:
V (K4Fe(CN)b), мл .
А, имп/мин, для варианта 1
А, имп/мин, для варианта 2
А, имп/мин, для варианта 3
0,20 0,40 0,60 0,80
950 820 680 570
1030 910 800 680
930 720 520 310
1,00 1,20 1,40 1,60
420 280 150 0
550 440 330 200
100 0 0 0
Построить кривую титрования и вычислить концентрацию цинка в растворе (мг/мл), если активность раствора до начала титрования составляла 1150 имп/мин. Ответ: 1) 1,263 мг/мл; 2) 1,572 мг/мл; 3) 0,794 мг/мл.
10. Анализ раствора смеси таллия (III) и цинка (II), содер-
278
жашего 204Т1, проводили методом осадительного титрования 0,1 М раствором 1-дитиокарбокси-3-доетил-5-фенилпиразолина CnHioN2S2Na, меченного 35S.
В центрифужную пробирку вводили анализируемый раствор, хлорид аммония, тартрат натрия и NaOH до pH 13,0. Затем из бюретки порциями по 0,25 мл прибавляли раствор титранта. После отделения осадка центрифугированием отбирали 0,01 мл раствора, наносили на фильтр, высушивали и измеряли активность.
После достижения первой точки эквивалентности pH раствора доводили до 7,0 и продолжали титрование*.
Построить кривые титрования и вычислить массу таллия и цинка (г) в анализируемом растворе по следующим данным:
ElCuHjoNsSzNa), мл 0,25 0,50 0,75 1,00 1,1 1,25 1,50 1,75 2,00 2,25 2,50 А, имп/мин . 1500 1000 550 125 150 350 750 100 100 450 800
Ответ: 7,153-10“3 г и 3,187- 1(Г3 г.
Глава 13
МАСС-СПЕКТРОМЕТРИЯ
13.1. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МАСС-СПЕКТРОМЕТРИИ
Масс-спектральный анализ основан на способности газообразных ионов разделяться в магнитном поле в зависимости от отношения m/е, где т — масса, е — заряд иона. Ионизация молекул в газе происходит под действием потока электронов. Наиболее вероятными являются процессы образования однозарядных положительных ионов:
М + = М+ + 2е~
Образование двух- и более высокозаряженных ионов, а также захват электрона с образованием отрицательных ионов являются менее вероятными процессами. По величине т/е определяют массовое число иона, а по интенсивности соответствующего сигнала судят о концентрации ионов.
Принципиальная схема масс-спектрометра приведена на рис. 13.1. В камере анализируемое вещество переводится в газообразное состояние при давлении 10-2...10-3 Па. Режим работы камеры устанавливается в зависимости от того, трудно- или легколетучие соединения входят в состав анализируемого образца. При анализе газообразных проб стадия испарения, естественно, отпадает. Далее молекулярный пучок ионизируется. Нередко для анализа твердых проб применяются источники с поверхностной
* Таллий полностью осаждается из тартратных растворов при pH >13,0, цинк — только при более низких значениях pH (~ 7,0).
279
1
ионизацией, в которых процессы испарения и ионизации не разделены.
Ионизация молекулярного пучка газообразной пробы может быть вызвана фотонами, ионами, электрическим полем, электронным ударом и другими способами. Наибольшее распространение в аналитической практике получили приборы, в которых ионизация осуществляется электронной или ионной бомбардировкой пробы либо искровым разрядом. Для ионизации электронным ударом используется стабилизированный пучок электронов, перпендикулярный потоку пробы. Энергия электронов невелика и обычно составляет 10...100 эВ. При бомбардировке молекул или
Рис. 13.1. Принципиальная схема масс-спектрометра
/ — газообразная проба, 2 — катод, 3 — анод, 4 — ускоряющие пластинки, 5 — магнитное поле, 6 — детектор
атомов электронами одновременно происходит несколько процес-сов. В условиях масс-спектрального анализа, как показывает опыт, образуются преимущественно положительные однозарядные ионы и частично ионы с более высоким зарядом. Если энергия бомбардирующих электронов достаточно велика, чтобы выз вать разрыв химических связей, происходит фрагментация молекул и в потоке появляются так называемые ионы-осколки.
Образовавшиеся положительно заряженные ионы проходят через ускоряющие пластины, разность потенциалов между которыми достаточно высока (несколько тысяч вольт). Здесь они приобретают энергию eV, а их скорость возрастает до v. Энергия eV, очевидно, будет равна кинетической энергии ионов mv2/2, покидающих ионный источник со скоростью v:
eV=mv2/2. (13.1)
После ускорения в электрическом поле ионы под прямым углом пересекают магнитное поле напряженностью Н, подвергаясь, таким образом, действию силы Hev, направленной перпендикулярно движению иона. Поэтому траекторией движения ионов будет окружность радиуса г.
Приравнивая силы Hev — mv2/r, находим
280
Подставляем эту величину в уравнение (13.1):
2ггг
Отсюда получаем радиус окружности:
r = (13.2)
Ионы, описывающие дугу радиуса г, попадают в детектор. Детектирование ионов производится фотографическим или электрическим способом. При фотографическом детектировании пучок ионов попадает на фотопластинку, вызывая почернение, пропорциональное числу ионов. В электрических детекторах масс-спектрометров ионный ток измеряется электрометром, электронным умножителем или другим аналогичным устройством. Сигналы обычно регистрируются быстродействующим потенциометром. В последнее время разработаны устройства, передающие информацию с детектора на ЭВМ, что позволяет значительно ускорить обработку данных.
Перепишем уравнение (13.2) в виде
m/e = r2H2/2V. (13.3)
Для получения масс-спектра, в принципе, можно менять любую переменную правой части этого уравнения (г, Н или V) и измерять почернение фотопластинки или ионный ток. Радиус г обычно не меняется, он задается конструкцией прибора и остается постоянным для всех ионов. Таким образом, меняя Н при постоянном V или меняя V при постоянном Н, можно направлять на детектор ионы с разной величиной т/е. Масс-спектр представляют зависимостью в виде спектрограммы или таблицы, содержащей величины т/е и соответствующие им интенсивности. Пропорциональность между экспериментально измеренным V и отношением т/е можно найти путем градуировки по веществу с известным масс-спектром.
13.2. КАЧЕСТВЕННЫЙ АНАЛИЗ
Качественный масс-спектрометрический анализ основан на измерении массы ионов. Идентификация масс проводится по положению линии на фотопластинке, которое фиксируют, измеряя расстояние между линиями с известной массой и анализируемой линией.
Масс-спектры многих веществ изучены достаточно подробно и составлены специальные атласы. При использовании таких атласов учитывается, например, что двухзарядный ион с массой 56 дает такую же линию в спектре, как и однозарядный ион с массой 28, а также условия получения спектра—температуру ионного источника, энергию электронов и т. д.
281
13.3. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
Количественные измерения в масс-спектрометрии проводят по току, фиксируемому детектором, или по почернению фотопластинки. В первом случае расчеты основаны на том, что пик ионного тока I пропорционален содержанию компонента или его парциальному давлению:
I = kc = хр, где k, х — коэффициенты пропорциональности; с — концентрация; р — давление.
13.4. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Практическое применение масс-спектрометрни весьма многообразно. Большую роль сыграли измерения масс-спектров прн изучении изотопного состава различных веществ. Основные сведения о стабильных изотопах фактически получены с помощью масс-спектрометра.
Масс-спектрометрический метод применяют для анализа твердых, жидких и газообразных проб. Значительное распространение он получил в органической химии для анализа многих классов соединений, в нефтехимии, где масс-спектрометрическим методом анализируют сложные многокомпонентные смеси углеводородов, в технологии неорганических веществ и других областях химической промышленности. Небольшой объем газа, требующийся для анализа, возможность определения всех компонентов смеси без разделения и другие достоинства масс-спектрометрии позволили успешно использовать ее для определения газов в металлах (после вакуумного плавления). Метод применим для анализа металлов, полупроводников и других неорганических и органических веществ. Он позволяет определять примеси на поверхности и по всему объему пробы. Большие перспективы открывает метод, сочетающий хроматографическое разделение и масс-спектрометрическое определение полученных продуктов.
Масс-спектрометрия применяется также для установления строения молекул, определения термодинамических характеристик газообразных веществ и т. д.
13.5. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Масс-спектральный метод характеризуется высокой универсальностью. Он применим для определения почти всех элементов периодической системы со средним пределом обнаружения 10* ...10“4 %, а при благоприятных условиях и до 10“7%. Одним из достоинств метода является возможность одновременного определения нескольких элементов и использование в работе небольших навесок (1 мг и меньше). Погрешность метода составляет 5...10%.
282
Вопросы
1
1. На чем основан масс-спектрометрический анализ?
2. Начертить схему масс-спектрометра и пояснить назначение отдельных частей прибора.
3. Вывести уравнение зависимости радиуса дуги, которую описывают ионы в масс-спектрометре, от характеристик масс-анали-затора и т/е иона.
4. На чем основан количественный масс-спектральный анализ?
5. Указать области практического применения, достоинства и недостатки масс-спектрометрического метода.
Глава 14
КИНЕТИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
14.1. ОСНОВНЫЕ МЕТОДЫ ОБРАБОТКИ КИНЕТИЧЕСКИХ ДАННЫХ
В кинетических методах анализа измеряемым свойством системы, на основании которого делают выводы о концентрации вещества, является скорость химической реакции. Пусть, например, вещества А и В реагируют между собой, образуя продукт реакции X:
А + В = Х
В начальный момент времени концентрации веществ А и В будут равны соответственно а и Ь. Концентрация продукта реакции в этот момент, естественно, будет равна нулю. В какой-то момент времени после начала реакции концентрация образующегося продукта X будет равна х, а концентрации исходных веществ будут равны (а — х) и (Ь — х) соответственно. Скорость химической реакции при данной температуре, как известно, пропорциональна произведению концентраций реагирующих веществ и нередко в степени, отличной от единицы:
-^ = k(a - x)(b - х), (14.1)
где k — константа скорости реакции.
Очень быстрые и очень медленные реакции в химико-аналитических целях являются малопригодными. Хотя трудно установить какие-либо жесткие правила или критерии, ограничивающие применение тех или иных реакций в кинетических методах анализа, все же некоторые пределы применимости можно отметить Почти повсеместно принято считать, что аналитическая реакция (в кинетических методах ее часто называют индикаторной реакцией) должна продолжаться не менее 1 мин и не более 2 ч. Более быстрые реакции, как правило,
283
не применяются потому, что при использовании обычного оборудования химико-аналитической лаборатории скорость таких реакций трудно измерить с достаточной точностью. При наличии специального оборудования эти затруднения отпадают и соответствующее ограничение снимается. Реакции, продолжающиеся более 2 ч, нежелательны из-за большой длительности анализа. Оптимальным временем для измерения скорости реакции считается 10...15 мин. Указанные пределы в значительной степени условны, поскольку можно в достаточно широких пределах регулировать скорость химической реакции, например, изменением температуры, концентрации реагирующих веществ или введением в раствор катализаторов (или ингибиторов).
Хотя в кинетических методах анализа, в принципе, могут быть использованы любые реакции, скорость которых может быть измерена достаточно точно, все же наиболее часто применяются так называемые каталитические реакции, скорость которых зависит от концентрации катализатора. При наличии в растворе катализатора в кинетическом уравнении (14.1) появляется соответствующий сомножитель:
-^-=/гск(о — ~ *)> (14.2)
где ск—концентрация катализатора.
Концентрацию одного из участников реакции, например вещества В, можно взять заведомо в большом избытке, так что его убыль в результате протекания реакции будет пренебрежимо мала и, следовательно, Ь — х^Ь. Тогда, объединяя kb = х, вместо (14.2) получаем
~ = хск (а — х). (14.3)
На рис. 14.1 приводятся типичные кинетические кривые. График показывает возрастание во времени концентрации вещества X Кривая 1 в начальный момент времени имеет линейный участок, т. е. в начальный момент времени угловой коэффициент кинетической кривой постоянен. На кривой 2 линейный участок
Рис. 14 1. Кинетические кривые
даже в начальный момент времени отсутствует. Различный характер кинетических кривых вызывает разные способы их обработки.
Рассмотрим сначала кривую 1. Уравнение (14.3) показывает, что величина может быть постоянной только при at
условии постоянства а—х, а это, в свою очередь, будет возможно, если а — х~а, т. е. если концентрация вещества А в ходе реакции существенно меняться не бу дет. Кинетическое уравнение для этого промежутка времени принимает вид
284
dx di
xc„a.
(144)
Это уравнение является основой различных вариантов кинетического метода, названных дифференциальными. Интегрирование дает
х = хс„а/. (14.5)
Несколько сложнее обработка данных, представленных кривой 2. Здесь нет области, в которой а —- х ~ а, поэтому приходится интегрировать кинетическое уравнение (14.3). Разделив переменные и проинтегрировав, получаем
S^t=S^
или
— 1п(а — х) = хск^+ const.
Постоянную интегрирования находим из начальных условий: при t = 0, х — 0 и, следовательно, — Ina = const. Окончательно можно записать:
In—-—= хск/. (14.6)
Методы анализа, основанные на применении этого уравнения, называют интегральными.
14.2. ОСНОВНЫЕ ПРИЕМЫ КИНЕТИЧЕСКИХ МЕТОДОВ АНАЛИЗА
Уравнения (14.4) ... (14.6) в явном виде связывают кинетические характеристики реакции с концентрацией катализатора. Как видно, концентрация катализатора может быть найдена или непосредственно по скорости реакции, или по времени ее протекания, или по концентрации образующихся продуктов. В зависи мости от того, какое свойство или какая характеристика реакции используется для определения концентрации, выделяют метод тангенсов, метод фиксированного времени, метод фиксированной концентрации.
Известны, кроме того, и другие методы, имеющие более частный характер, например метод индукционного периода, метод непосредственного дифференцирования ит. д.
Метод тангенсов. В методе тангенсов измеряют скорость реакции обычно по возрастанию концентрации одного из образующихся продуктов и строят график, аналогичный изображенному на рис. 14.1. Если кинетическая кривая в начальный период протекания реакции имеет линейный характер, применяют дифференциальный вариант метода тангенсов. Уравнение (14.4) по
285
казывает, что в этом случае скорость реакции характеризуемая тангенсом угла наклона кинетической кривой, пропорциональна концентрации катализатора.
График в координатах тангенс угла наклона — концентрация определяемого вещества (отсюда название «метод тангенсов») обычно линеен. При анализе неизвестного раствора измеряют скорость реакции в тех же условиях, в каких она определялась для построения градуировочного графика, определяют tga и по градуировочному графику находят концентрацию анализируемого компонента сх.
Если кинетическая кривая имеет вид кривой 2 (рис. 14.1), т. е. линейный участок отсутствует, применяется интегральный вариант метода тангенсов. В соответствии с уравнением (14.6)
о кинетическую кривую следует строить в координатах 1g ——-— — t. Тангенс угла наклона прямой в этих координатах, как показывает уравнение (14.6), пропорционален концентрации катализатора и, следовательно, градуировочный график будет также линеен.
Для измерения текущей концентрации очень удобны фотомет-^ рические методы, так как оптическая плотность раствора прямо пропорциональна концентрации вещества. При построении кинетической кривой на оси ординат вместо концентрации можно откладывать оптическую плотность: тангенс угла наклона для построения градуировочного графика можно вычислять как d/1/d/. Метод тангенсов с успехом применяется для самых различных реакций, по точности определения он превосходит все остальные варианты кинетических методов.
Метод фиксированного времени. В методе фиксированного времени определяют концентрацию продукта реакции или концентрацию одного из участников реакции за строго определенный промежуток времени. Если, например, продукт реакции окрашен, через определенный промежуток времени измеряют оптическую плотность раствора. При небольшой глубине протекания реакции применяют дифференциальный вариант. Из уравнения (14.5) получаем
где t — заданный промежуток времени.
Выражение в скобках постоянно, так как а и t фиксированы. Его числовое значение может быть определено по стандартному раствору и использовано для последующих расчетов ск по измеренной величине х.
Можно построить градуировочный график в координатах Ск—х, где ск — концентрация определяемого вещества (катализатора) , а х — концентрация продукта реакции, образовавшегося за фиксированный отрезок времени. Вполне понятно, что фик
286
сированный отрезок времени сохраняется один и тот же как при построении градуировочного графика, так и при анализе неизвестного раствора. При большой глубине протекания реакции применяется интегральный вариант, основанный на решении уравнения (14 6) относительно ск. Метод фиксированного времени, как видно, проще метода тангенсов, однако по точности он ему уступает.
Метод фиксированной концентрации. В методе фиксированной концентрации измеряют время, в течение которого концентрация продукта реакции или одного из реагирующих веществ достигает определенного, заранее заданного значения. Этот метод по сути близок методу фиксированного времени. Если глубина протекания реакции невелика, используют дифференциальный вариант, так же как и в методе фиксированного времени, основанной на решении уравнения (14.5) относительно ск:
где х — заданная концентрация продукта реакции.
Выражение в скобках постоянно, поскольку постоянны х и а, его числовое значение может быть определено по стандартному раствору. Градуировочный график в методе фиксированной концентрации, как показывает уравнение (14.7), следует строить в координатах ск — 1 //, где ск — определяемая концентрация, a t — время, необходимое для достижения заданной концентрации продукта реакции.
При более глубоком протекании реакции используется интегральный вариант, основанный на решении уравнения (14 6):
ск = (1п-^----L) J_.
Градуировочный график, как видно, следует строить также в координатах ск — 1Ц. По точности метод фиксированной концентрации близок к методу фиксированного времени и уступает методу тангенсов.
Метод добавок. Этот метод обладает известными достоинствами. Принципиальная сущность его уже рассматривалась при изложении спектрального анализа и фотометрии. При выполнении анализа по методу добавок необходимо провести два измерения: сначала определить скорость реакции в анализируемом растворе, а затем скорость реакции в этом же растворе с добавкой стандартного раствора.
Метод каталиметрического титрования. Сущность этого метода можно пояснить на примере церий-арсенитной реакции, которая катализируется осмием, рутением и иодидом:
2Се4+ + HAsO2 + Н2О = 2Се3+ + HAsO3 + 2Н+ (14.8) Так как иодид является катализатором реакции церия с арсенитом, скорость этой реакции будет возрастать с увеличением кон
287
центрации иодида. Ионы Ag+ обладают ингибиторным эффектом, блокируя катализатор. При введении в раствор ионов Ag+ происходит образование Agl, и концентрация 1у падает, вызывая соответствующее уменьшение скорости церий-арсенитной реакции. При каталиметрическом титровании к анализируемому раствору, содержащему иодид-ион 1~ в церий-арсенитной смеси, несколькими порциями добавляют титрованный раствор AgNO3 и определяют скорость реакции. Точку эквивалентности находят из графика в координатах объем раствора AgNCb — скорость реак-d х
ции, характеризуемая угловым коэффициентом Этим методом можно оттитровать иодид в очень разбавленных растворах (10~6 моль/л и меньше).
14.3. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Кинетические методы анализа характеризуются высокой чувствительностью и поэтому часто используются при определении малых и ультрамалых содержаний (до 10“8...10~6 мкг). Особенно эффективным оказалось применение кинетических методов для определения микропримесей в чистых и сверхчистых веществах и материалах.
В настоящее время известны десятки каталитических реакций, с помощью которых могут быть определены свыше 40 элементов периодической системы. Например, реакция иодида с пероксидом водорода без катализатора идет очень медленно:
Н2О2 + 21- + 2Н+ = 12 + Н2О
В присутствии следов молибдена, вольфрама, циркония, гафния, ниобия, тантала и других элементов она проходит за несколько минут. Скорость реакции легко определяется по возрастанию оптической плотности иодкрахмального раствора в единицу времени. Чувствительность реакции достаточно высока. Например, с ее помощью можно определить 0,01 мкг/мл W, 0,02 мкг/мл Мо, 0,1 мкг/мл Zr или Hf.
Интересны каталитические эффекты в реакции (14.8) взаимодействия мышьяковистой кислоты с солями церия (IV), которая катализируется осмием, рутением и иодидом.
Исследование показало, что скорость этой реакции, катализируемой осмием, не зависит от концентрации церия, но увеличивается с ростом концентрации мышьяковистой кислоты Если в качестве катализатора выступает рутений, скорость реакции (14.8) перестает зависеть от концентрации мышьяковистой кислоты, но увеличивается с концентрацией церия. Эти особенности церий-арсенитной реакции открывают возможность определения осмия и рутения в одном растворе без предварительного разделения. По скорости реакции в растворах с увеличивающейся концентрацией церия можно определить рутений, а по скорости
288
реакции при возрастающей концентрации мышьяковистой кислоты найти содержание осмия.
С помощью кинетических методов анализа в большинстве случаев определяется не общая, а равновесная концентрация реагирующих веществ. В связи с этим кинетические методы успешно применяются для изучения различных равновесий в растворах (комплексообразование, кислотно-основное взаимодействие и др.).
14.4. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Наиболее ценной особенностью кинетических методов является возможность определения элементов при их содержании 10-'8..-10-6 мкг, что превосходит соответствующую характеристику спектрального, спектрофотометрического, потенциометрического и многих других методов анализа. Анализ с помощью кинетических методов выполняется быстро и просто, без применения сложных приборов. Кинетическим методом можно определить свыше 40 элементов периодической системы с погрешностью, не превышающей ±10% (отн.). В некоторых случаях кинетические методы обладают достаточной специфичностью, однако, как правило, их специфичность не высока. Специфичность каталитических реакций повышают путем маскировки каталитически активных примесей при введении в анализируемый раствор соответствующих реагентов.
! Вопросы
1. В чем сущность кинетических методов анализа?
2. Какую реакцию называют индикаторной? Какие требования предъявляются к таким реакциям?
3. Назвать наиболее распространенные способы измерения скорости химических реакций, используемых в кинетических методах анализа.
4. Что представляет собой дифференциальный вариант кинетических методов анализа?
5. Что представляет собой интегральный вариант кинетических методов анализа?
6. Охарактеризовать основные приемы кинетических методов анализа: а) метода тангенсов; б) метода фиксированной концентрации; в) метода фиксированного времени; г) метода добавок.
7. Указать факторы, влияющие на точность аналитических определений с помощью кинетических методов анализа.
8. Назвать индикаторную реакцию, способ измерения скорости реакции и метод обработки данных при определении: a) Mo (VI); б) Со (II); в) W(VI); г) V (V); д) Си (II); е) Pd (II); ж) Ag (I); з) Hg (II); и) ионов I-.
10-1533
289
9. В чем сущность каталиметрического титрования? Привести примеры.
10. Назвать области применения, достоинства и недостатки кинетических методов анализа.
ДА Задачи
1. При определении меди с помощью реакции окисления н-фе-нилендиамина пероксидом водорода, катализируемой соединениями Си (II), был использован метод добавок.
В две мерные колбы вместимостью 50,00 мл внесли по 5,00 мл исследуемого раствора. В одну из колб прибавили необходимые реагенты, довели объем до метки и измерили время tx, необходимое для достижения заданного значения оптической плотности. В другую колбу ввели те же реагенты, добавили 1,00 мл стандартного раствора меди с Г(Си) = 2,50-10-5 г/мл, довели объем до метки и измерили время /х + ст, необходимое для достижения того же значения оптической плотности.
Определить концентрацию меди в исследуемом растворе (мкг/мл), если tr_ = 12,5 мин, a G + £T=9,5 мин.
Рассчитываем концентрацию стандартного раствора меди после разбавления.
Л Си )lzc,
2,5-10-5 • 1,00
50,00
5,0-10 7 г/мл.
Определяем концентрацию меди в анализируемом растворе:
1/6 Ик 7 1/12,5 50,00
1/6+ст- 1/6 П, ’ 1/9,5-1/12.5’ 5,00
= 5,00-10 7-3,167 —- = 15,835-10 6 г/мл = 15,8 мкг/мл.
5,00
2. Для определения молибдена использовали реакцию окисления иодид-иона пероксидом водорода, катализируемую соединениями Mo (VI).
В три мерные колбы вместимостью 50,00 мл внесли 5,00; 10,00 и 15,00 мл раствора молибдата аммония с ^((NH^zMoCU/Mo) = 4,45-10-8 г/мл. Исследуемые растворы также поместили в мерные колбы вместимостью 50,00 мл. После добавления реагентов (KI, НС1, крахмал, Н2О2) и доведения объемов до метки измерили оптическую плотность А в зависимости от времени t для всех растворов:
Стандартные растворы (tga) Исследуемые растворы (tgax)
1 2 3 1 2 3 4
0,750 1,000 1,250 0,950 1,100 0,850 1,210
290
Построить градуировочный график в координатах tga — сМо и определить массу молибдена (г) в исследуемых растворах. Ответ: 1) 4,0-10“7 г; 2) 5,2-10'7 г; 3) 3,2-11Н г; 4) 6,1-10“7 г.
3. Для определения палладия в растворе методом фиксированной концентрации использовали реакцию окисления а-на-фтиламина нитрат-ионами, катализируемую соединениями Pd (II).
Определить концентрацию палладия (мкг/мл) в исследуемом растворе, если при измерении времени t, необходимого для достижения фиксированной оптической плотности, получили следующие результаты:
Параметры Стандартные растворы Исследуемые растворы
1 2 3 4 5 1 2 3 4
Ср<), мкг/л 1,0 2,3 28 3,5 4,5 — — —
/, мин 18,2 7,7 6,3 5,1 4,0 12,0 7,1 4,4 5,5
Ответ: 1) 1,50 мкг/мл; 2) 2,50 мкг/мл; 3) 4,05 мкг/мл; 4) 3,25 мкг/мл.
4. Для определения тория в растворе методом фиксированной концентрации использовали реакцию окисления иодид-иона пероксидом водорода, катализируемую соединениями тория. Взяли 2,00 мл исследуемого раствора, разбавили до 100,0 мл в мерной колбе и после введения необходимых реагентов раствор разбавили еще в 10 раз.
Определить концентрацию тория (моль/л) в исследуемом растворе, если при измерении времени t, необходимого для достижения фиксированной оптической плотности, получили следующие результаты:
Параметры Стандартные растворы Исследуемые растворы
1 2 3 4 5 1 2 3 4
cTh-106. моль/л 3,0 4,5 6,0 8,2 12,0 — — —
/, мин 10,0 6,7 5,0 3,7 2,5 4,3 8,3 2,9 Гб,1
Ответ: 1) 3,5-10-3 моль/л; 2) 1,8-10 3 моль/л; 3) 5,2Х ХЮ'3 моль/л; 4) 2,5-10—3 моль/л.
5. Для определения рения в руде методом фиксированного времени использовали реакцию восстановления сульфосалицилат-ного комплекса железа (III) хлоридом олова (II), катализируемую перренат-ионами. Оптические плотности стандартных растворов, измеренные через 30,0 мин после начала реакции, оказались равными:
cRe-Ю2, мкг/л . 1,5 3,0 4,5 7,5
А ............. 0,42 0,39 0,36 0,30
10*
291
Из навески руды массой т г после соответствующей обработки получили 100,0 мл раствора; 10,00 мл этого раствора разбавили в мерной колбе вместимостью 50,00 мл.
Определить массовую долю (%) рения в руде, если значения оптической плотности исследуемых растворов через 30,0 мин после начала реакции составили:
Вариант т, г 4
12 3 4
0,1280 0,0920 0,0654 0,0758
0,33 0,40 0,38 0,35
Ответ: 1) 0,023 %; 2) 0,014 %; 3) 0,027 %; 4) 0,033 %.
6. Для определения иодид-ионов использовали реакцию окисления арсенита йодатом калия, катализируемую ионами 1~. Был применен метод добавок при фиксированной концентрации продукта реакции.
Навеску биологического материала массой m г после минерализации и соответствующей обработки растворили в 25,00 мл воды. В две колбы вместимостью 25,00 мл поместили по 5,00 мл полученного раствора, в одну из них добавили 1,00 мл стандартного раствора иодида с концентрацией 0,5 мкг/мл. В обе колбы при одинаковых условиях добавили растворы реагентов (КЮз, NasAsOa, цитратный буфер), объемы довели до метки водой и из полярографических данных определили время окончания реакции в каждой колбе: tx и ^х+ст.
Рассчитать концентрацию иодида в биологическом материале (мг/кг) по вариантам:
Вариант 1 2 3 4
мин . 19,6 14,7 11,1 16,3
G+ст. мин . 9,1 8,2 5,7 7,8
т, г 1,000 1,200 1,500 1,700
Ответ: 1) 2,16 мг/кг; 2) 2,62 мг/кг; 3) 1,76 мг/кг
4) 1,35 мг/кг.
Глава 15 ТЕРМОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
15.1. ТЕПЛОВОЙ ЭФФЕКТ РЕАКЦИИ КАК АНАЛИТИЧЕСКИЙ СИГНАЛ
Методы термометрического, или энтальпийного, титрования основаны на измерении теплового эффекта реакции титрования или величин, пропорциональных этому тепловому эффекту. Тепловой эффект некоторых реакций может не отличаться от нуля, однако специально рассматривать этот случай нет необходимости в связи с его сравнительно редкой реализуемостью в практических условиях титрования.
292
АТ ДЯ Д/ = —т~ Па.
'-'Р
Если реакция титрования вещества А титрантом В протекает в соответствии с уравнением
А + В = АВ
и изменение энтальпии при титровании пА моль вещества А равно Д//Титр , а изменение энтальпии в расчете на 1 моль вещества А равно А//, то
Д/7 ™тр = пкН. (15.1)
В то же время Д//титр связано с теплоемкостью системы Ср и изменением температуры ДТ вследствие теплового эффекта реакции соотношением
Д/7титр = СРкТ. (15.2)
Сочетание уравнение (15.1) и (15.2) дает
(15.3)
При соблюдении некоторых условий (постоянство ионной силы и небольшое изменение температуры) величина Д// в ходе титрования будет оставаться постоянной, а существенное увеличение концентрации титранта по сравнению с концентрацией вещества А обеспечит достаточное постоянство теплоемкости системы Ср, так как исходный раствор разбавляться практически не будет. Тогда отношение к.Н/Ср также останется постоянным и вместо уравнения (15.3) можно записать
\T=knK. (15.4)
Уравнение (15.4) показывает, что изменение температуры при титровании прямо пропорционально количеству вещества А, вступившего в реакцию титрования.
В первых работах по термометрическому титрованию температуру измеряли термометром Бекмана и после каждого добавления стандартного раствора систему выдерживали до достижения термического равновесия. Стандартный раствор добавляли из бюретки небольшими порциями каждые 5... 10 мин, и таким образом весь процесс титрования продолжался примерно 1 ч. Вполне понятно, что такая методика не имела заметных преимуществ перед обычными методиками объемного анализа.
Практическое значение термометрического титрования значительно возросло с применением в качестве датчиков температуры термисторов и использованием термостатированных поршневых бюреток или специальных устройств для автоматизированной подачи титранта в анализируемый раствор.
Сопротивление термистора очень чувствительно к изменению температуры. Увеличение температуры на один градус вызывает уменьшение сопротивления термистора на 1...5%. Инерция термистора невелика: он реагирует на изменение температуры в течение 3...5 с. Эти ценные качества термисторов и позволили
293
разработать современные методы термометрического титрования, Изменение температуры раствора вызывает изменение сопротивления термистора, что приводит к «разбалансу» моста, который может быть зарегистрирован на самопищущем потенциометре.
В дифференциальных установках измеряется разность температур между двумя сосудами, один из которых содержит анализируемый раствор, а другой — раствор сравнения, и в оба сосуда с одинаковой скоростью поступает титрант. Такая схема позволяет автоматически исключить влияние теплоты разбавления титранта, тепловыделения при перемешивании раствора, эффект Джоуля при прохождении тока через термистор и другие эффекты, затрудняющие измерение теплоты собственно реакции титрования.
15.2. КРИВЫЕ ТЕРМОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
Кривые термометрического титрования показывают изменение температуры в ходе титрования в зависимости от объема добавленного титранта. Типичный график изменения температуры в ходе титрования приведен на рис. 15.1. Отрезок АВ (или А'В'), показывающий температуру системы перед началом титрования, иногда называют основной линией; ВС (или В'С') характеризует изменение температуры раствора в ходе титрования, а отрезок CD (или C'D') показывает изменение температуры после достижения точки эквивалентности С (или С', если реакция эндотермична).
Практически получаемые кривые в той или иной степени отличаются от идеализированной, так как экспериментально измеряемый тепловой эффект наряду с изменением энтальпии в реакции титрования включает эффект разбавления титранта, тепловыделение при перемешивании раствора и т. д. Изменение температуры при титровании может быть вызвано также разностью температур анализируемого раствора и титранта, выделением теплоты при прохождении через термистор электрического тока и т. д. Не все эти факторы действуют отрицательно. Иногда температуру титранта специально устанавливают ниже или выше температуры анализируемого раствора (в зависимости от знака теплового эффекта), чтобы более четко фиксировать точку эквивалентности.
Как показывает рис. 15.2, если реакция титрования экзотер-миина, то применение титранта с более низкой температурой, чем у анализируемого раствора, позволяет более четко фиксировать точку эквивалентности (кривая 3). При одинаковой температуре обоих растворов (кривая 2) или при более высокой температуре титранта (кривая /) точка эквивалентности фиксируется менее четко. Однако в большинстве своем посторонние тепловые эффекты оказывают неблагоприятное воздействие, вызывая, например, плавное закругление кривой в конце титрования, что осложняет определение точки эквивалентности и явля
294
ется дополнительным источником погрешности. При закруглении кривой точку эквивалентности определяют графической экстраполяцией линейных участков кривой титрования. Более точно точку эквивалентности можно определить при построении дифференциальной кривой титрования. Типичная дифференциальная кривая термометрического титрования представлена на рис. 15.3. Точка эквивалентности на этой кривой фиксируется более четко.
Рис. 15.1. Кривые термометрического титрования: -------реакция экзотермическая,----- реакция эндотермическая
Рис. 15.2. Кривые термометрического титрования при разной температуре титранта:
1 Ттитранта Траствора, 2 Ттитранта
Т раствора, 3 Ттитранта * раствора
Термометрическое титрование успешно применяется при определении компонентов смеси без предварительного химического разделения. Так анализируют, например, технически важную смесь кальция и магния титрованием ЭДТА. Реакция образования комплекса кальция СаУ2- экзотермична, а комплекса магния MgY2 эндотермична. Кривая титрования этой смеси представлена на рис. 15.4. Погрешность определения кальция составляет 0,5 %, а магния 2 %.
Рис. 15.3. Дифференциальная кривая термометрического титрования
г.3.(1} т.з(Д) V
Рис. 15.4. Кривая титрования смеси ионов Са2+ и Mg2’*'
295
15.3. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
В термометрическом титровании могут быть использованы реакции кислотно-основного взаимодействия, окисления—восстановления и любые другие, тепловые эффекты которых достаточно велики, чтобы произвести точные измерения. Важным достижением термометрических методов является возможность прямого титрования слабых кислот с высокой точностью. Например, борная кислота титруется в водном растворе без добавления маннита. Также могут быть оттитрованы некоторые слабые органические кислоты, аминокислоты, слабые основания и другие вещества. Термометрическое титрование различных восстановителей дихроматом показывает более высокую точность, чем титрование с применением дифениламина в качестве индикатора. Известны методы термометрического титрования, основанные на реакциях осаждения сульфатов, галогенидов, оксалатов и других малорастворимых соединений, методы, основанные на образовании этилендиаминтетраацетатных и других комплексов и т. д. Разработаны методы анализа смесей путем последовательного титрования компонентов без предварительного химического разделения. Термометрическим методом титруются также различные вещества в неводных растворителях (ледяная уксусная кислота, ацетонитрил и др.) и в расплавах солей, например в расплавленной эвтектике нитритов лития и калия аргентометрически титруется хлорид.
Методики термометрического титрования используют во многих отраслях промышленности: в металлургической, химической, электрохимической, нефтехимической и др., в анализе фармацевтических материалов и во многих других областях.
1S.4. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Существенным достоинством метода термометрического титрования является его универсальность. Один и тот же термометрический прибор может быть использован для самых различных определений, так как здесь не нужны специфические индикаторы, селективные электроды и т. д. В настоящее время термометрическое титрование квалифицируется как один из наиболее перспективных методов анализа: он обладает высокой точностью, универсальностью и легко может быть автоматизирован.
Вопросы
1. В чем сущность термометрического титрования?
2. Охарактеризовать основные узлы установки для термометрического титрования.
3. Какой вид имеют кривые термометрического титрования?
296
4. Какое практическое применение имеет метод термометрического титрования?
5. Дать общую оценку метода термометрического титрования.
Задачи
1. К 25,0 мл раствора соли стронция в калориметрическом стаканчике добавляли порции титранта 1,0 М ЭДТА (А = = 0,9872) и измеряли тепловой эффект реакции. Отклонения шкалы прибора в делениях шкалы составили:
У(титранта), мл . 0,5 1,0 1,5 2,0 2,5 3,0
А, деления шкалы . 5,5 11,0 17,0 17,5 17,5 17,5
Вычислить концентрацию стронция (г/л) в исследуемом растворе. Ответ: 5,536 г/л.
2. Из навески двойного карбоната пВаСОз-тСаСОз массой 0,6524 г приготовили 200,0 мл раствора. Из 20,0 мл этого раствора барий выделили в виде BaSO4. Для растворения этого осадка на фильтре использовали избыток раствора Na4 ЭДТА [25,00 мл 0,1 М (А = 0,9616)]. Фильтрат и промывные воды собрали в мерную колбу вместимостью 100,00 мл и довели до метки дистиллированной водой. Аликвоту объемом 10,00 мл полученного раствора титровали термометрически 0,1 М MgSO4 (/< = 0,9721). Отклонения прибора, измеряющего тепловой эффект реакции, составили:
V(MgSO4), мл . 0,2 0,4 0,6 0,8 1,0 1,2
А, деления шкалы . 13 26 38 42 42 42
Определить массовую долю (%) ВаСОз в исследуемом двойном карбонате. Ответ: 53,54 % ВаСОз.
3. При термометрическом титровании 20,00 мл раствора, содержащего Са2+ и Mg2+, раствором 1,0 М ЭДТА (К = 0,9521) наблюдали сначала повышение температуры, а затем уменьшение. Максимальному подъему температуры (титруется Са2+) соответствовал объем титранта 0,12 мл минимальному значению температуры (титруются и Са2+ и Mg2+) соответствовал объем 0,26 мл титранта.
Вычислить концентрацию (г/л) Са и Mg в исследуемом растворе. Ответ: 0,229 г/л Са и 0,162 г/л Mg.
।
Глава 16
ЭКСТРАКЦИЯ
16.1. РАСПРЕДЕЛЕНИЕ ВЕЩЕСТВА МЕЖДУ ДВУМЯ ЖИДКОСТЯМИ
При соприкосновении водного раствора вещества А с каким-либо неводным растворителем, не смешивающимся или ограниченно смешивающимся с водой, растворенное вещество А будет распределяться между обоими растворителями и через некоторое время в такой системе установится равновесие
Ав^ Ао
где Ав и Ао— вещество А в воде и в органическом растворителе соответственно.
Процесс переноса растворенного вещества из одной жидкой фазы в другую, с ней несмешивающуюся или ограниченно смешивающуюся жидкую фазу называют жидкость-жидкостным распределением или распределением между двумя жидкостями. Количественно этот процесс характеризуется законом распределения Нернста — Шилова, в соответствии с которым отношение концентраций растворенного вещества в обеих фазах при постоянной температуре постоянно и не зависит от общей концентрации растворенного вещества:
(16.1)
где D — коэффициент распределения; [А] о—аналитическая, т. е. суммарная концентрация всех форм вещества А в органической фазе; [А] в — то же, в водной фазе.
Величина D сохраняет постоянство лишь в отсутствие процессов диссоциации, ассоциации, полимеризации и других превращений растворенного вещества.
При подстановке в уравнение (16.1) активностей вещества А в органической фазе и в водном растворе вместо их концентраций коэффициент распределения будет оставаться постоянным в широкой области концентраций, поскольку процессы ассоциации и другие будут формально учтены коэффициентами активности. Однако при небольших концентрациях вещества А коэффициент распределения D и без этого сохраняет удовлетворительное постоянство и часто используется как основная характеристика распределения вещества.
Иногда выражению закона распределения придают вид
pt___ [А] о
[A]S ’
в котором п подбирают эмпирически.
298
В простейшем случае п характеризует степень полимеризации растворенного вещества в органическом растворителе. Отношение концентрации (точнее, активности) вещества в одной определенной форме (например, MLn) в фазе органического растворителя к его концентрации (активности) в той же форме в водной фазе называют константой распределения
I/O fc*.LB)o [MLn]('уf-U n) о I/ (*Yо / 1 П 91
Д Ими). [ML„]„ (Yml)b — Лс (Vml.)b '
Коэффициент и константа распределения связаны с растворимостью вещества. В простейшем случае, когда вещество в обеих фазах существует в одной и той же форме (например, в виде недиссоциированных мономерных молекул), константа и коэфи-циент распределения равны отношению растворимостей вещества в органическом растворителе и в воде. Действительно, если в систему из воды и несмешивающегося с ней органического растворителя ввести твердое вещество до насыщения, то концентрация вещества в каждой фазе будет равна его растворимости в соответствующем растворителе и, следовательно,
Г) _ С$а)о
(Sa), ’
где (Sa)o—растворимость вещества в органическом растворителе; (SA)B — растворимость в воде.
16.2. ОСНОВНЫЕ КОЛИЧЕСТВЕННЫЕ ХАРАКТЕРИСТИКИ ЭКСТРАКЦИИ
Экстракция представляет частный случай жидкость-жид костного распределения, когда из водного раствора вещество извлекается в несмешивающийся с водой органический растворитель Реагент, который образует экстрагирующееся соединение, называют экстракционным реагентом, а органический растворитель, используемый для экстракции или раствор экстракционного реагента в органическом растворителе, называют экстрагентом. Для улучшения физических (плотности, вязкости и др.) или экстракционных свойств экстрагента в него нередко добавляют разбавитель — инертный органический растворитель или используют смесь растворителей.
Важной характеристикой экстракции является фактор (или степень) извлечения
где п (А) — количество вещества в органической фазе; п (А) „ — исходное его количество в водном растворе.
299
Очевидно, что
n(A) = [A]oV<i (16.4)
п (А) „ = = [А] „К, + [ AJ „14, (16.5)
где — концентрация вещества А в исходном водном растворе.
Подставим соотношения (16.4) и (16.5) в уравнение (16.3) и разделим числитель и знаменатель полученного выражения на [A] BVO:
р________[А] <V° __ D — В (16 6^
Л [A]0V0+ [A)BVB D + VB/VB D + \/r' 1
где D определяется соотношением (16.1), a r — V0/VB. Уравнение (16.6) относится к однократной экстракции и остается справедливым при многократном повторении этой операции.
После первой экстракции будем иметь dVB = [A,] OVO +
где индекс «1» — номер экстракции.
Подставляем сюда уравнение (16.1) и решаем относительно [AjJ в:
dVB=D [A,]BVO+ [A,] BVB;
ГД I __ СдУ в ____ СД
L ,Jb DV„+ Vb Dr \ •
После второй экстракции аналогично:
г д 1 [Al) в сд 1А21 в рг 1 (Dr + 1)2 ’
а после т экстракций:
[Ат] в (Dz. + .
(16.7)
Отсюда
lg _£1_
/77 = f [А,,] lg(Dr 4- 1)
(16.8)
По уравнению (16.8) можно рассчитать число экстракций, необходимое для достижения заданной степени извлечения. Например, для экстракции 99 % вещества из водного раствора не-
обходимо сделать т — , .=--
lg (Dr + 1)
составит m=2/0,3=7 операций,
экстракций; при Dr = 1 это а при Dr = 5 /71 = у|^- =
= 2,6 ~ 3 операции.
Степень извлечения при m-кратной экстракции {Rm) находим путем сочетания уравнений (16.3) и (16.7) :
Р СаУу [Ат] У в ._____________СдУв_________ ___ I___________1______ /16 9)
Кт СдУв (Dr + 1)гас°АУв (Dr + 1)т ( ’
300
При т = 1 и г = 1 оно переходит в
Возможность разделения двух катионов А и В до недавнего времени связывалась с коэффициентом разделения х, равным отношению коэффициентов распределения этих ионов х = DbJDv. При х=1 разделение невозможно. Чем больше х отличается от 1, тем ближе к оптимальным будут условия разделения. Более общей характеристикой возможности разделения является фактор обогащения S, показывающий, во сколько раз отношение количеств разделяемых веществ в фазе экстрагента превышает это отношение в исходном растворе до разделения. Другими словами, эта величина, на которую следует умножить отношение исходных количеств разделяемых катионов, чтобы получить отношение их количеств после разделения (в органической фазе):
п (В)/п (А) = Sb/ап (В)о/п (А) о.
откуда фактор обогащения
= «»/«.. пело
Зависимость фактора обогащения от коэффициентов распределения и других величин находим путем подстановки (16.9) в (16.10):
С КДвГ + I)"1 П (ДдГ + I)"1 I | /? | \
5в/А [(ПдГ + If - 1] (DBr + I)"1 • (lb.ll)
При m = 1 и 7 = 1
Если, например, ДА= 104, а Дв = 0,1, то коэффициент разделения х = Dк/DB — 104/0,1 = 105 имеет довольно большое значение. Однако это не обеспечивает удовлетворительного разделения, так как хотя элемент А извлекается на 99,99 %, но и элемент В переходит в органическую фазу тоже в значительных количествах (~10 %) Фактор обогащения для этой системы в соответствии с уравнением (16.12) равен
е _ 0,1 (104 + 1) ~ .
•Эв/А Ю* (0,1 + 1) — '
Если при том же значении коэффициента разделения х = 105 коэффициенты распределения будут £>д = Ю2 и Ов= 10“3, то фактор обогащения составит £В/д = = Ю-3, т. е.
значение на два порядка меньше, чем в первом случае. Элемент А перейдет в органический растворитель на 99 %, а элемент В —
301
только на 0,1 %. Таким образом, коэффициент SB/a более реально, чем х, характеризует возможности разделения элементов. Опыт показывает, что в органическую фазу из водного раствора вещество переходит в виде электронейтральных частиц: координационных соединений, ионных ассоциатов и т. д. С увеличением температуры экстракция, как правило, ухудшается.
16.3. ЭКСТРАКЦИЯ ВНУТРИКОМПЛЕКСНЫХ СОЕДИНЕНИЙ (ХЕЛАТОВ)
Анион внутрикомплексной соли является полидентатным лигандом, образующим с катионом один или несколько циклов. Внутрикомплексная соль, как правило, малорастворима в воде и хорошо растворима в органических растворителях, что для экстракции имеет особое значение. Типичными экстракционными реагентами этого класса являются 8-оксихинолин, дитизон, ацетилацетон и др. Обычно они обладают свойствами слабой кислоты, поэтому кислотность раствора относится к числу наиболее важных факторов, определяющих полноту экстракции.
Существенное значение имеет распределение самого экстракционного реагента между неводным растворителем и водой. Кислотно-основное равновесие в растворе реагента HL
HL + Н2О = Н3О+ + L~ в кислой среде может быть осложнено протонированием
HL + НзО+ = Н2Ь+ + Н2О
как это происходит, например, в кислых растворах 8-оксихино-лина. Коэффициент распределения реагента будет равен
D _ г Chl)„ [H2l+]o+ [HL]„ + [L ]
HL (Chl)b [H2L+] + [HL] + [L 1
(16.13)
Константы распределения относятся к каждой из форм существования реагента:
„0 __ (aH2L+) о _ [H2L+] oTh2L+(o)
Ao(h2l ) ~ ~ ГТ, =
°Н21.1 Ih2l1vh2l
_ Th2L+(o)
~Ло(НЛ+)^Г~’
•о _____ (“hl)o __ [HL]o7hl(o) __ THL(o) .
(O(HL)------ [HI 1/'O(HL) — ;
aHL lnLJ THL THL
__ lo _ 1 <Vi (о) Vi io)
'D(L I ai [L ]VL > ?L ~
(16.14)
(16.15)
(16.16)
Концентрации в уравнении (16 13) можно выразить с помощью констант диссоциации HL и H2L+ и констант распределения (16.14)...(16.16):
302
*O(h2l+)[h*l+] +K £>(HL) [HL] + Kdil ) [L |
HL---
[H+] fHLl X [HI 1 X К
(16.17)
Это общее уравнение распределения реагента между растворителями. В достаточно кислой или щелочной области уравнение обычно упрощают, так как в таких средах некоторыми членами можно пренебречь. Например, в щелочной области уравнение (16.17) принимает вид
£»hl=^<l) + -^[H+].
Если в органическую фазу экстрагируются только молекулы HL, то
п ___ KO(HL)
HL 1+/WIH+] '
Процесс экстракции катиона М"+ обычно представляют схемой
Мп+ + nHL(o) = ML„(0) + пН+, где HL — экстракционный реагент; индекс (о) указывает на органическую фазу. Константа этого процесса — константа экстракции
0 _ ОДГп(о)ащ _ [ML„]o[H+]" Tml„(0)Yh-_ aMn+a"HL(0) [M"+][HL]o Tm»xV"hL(o)
= ТмьП(0)Тщ (16 18)
ТМ“ THL (о)
При постоянной ионной силе раствора
ипи д- __ [ML„] (О)[Н+]П (16 19)
ИЛИ Лех [М”+] [HL]" ' '
Если равновесие экстракции в заметной степени не осложнено кислотно-основным взаимодействием, ступенчатым комплексообразованием, ассоциацией и другими процессами, то
ГЧ ___ ____[М-Ln] о___
м [ML„]B+ [М].
1 _ 1 . [М]в
KD "Г [ML„] „ ‘
Подставим сюда отношение
[М]в
[ML„] о
из уравнения (16.19):
1 _ 1 । [Н+]"
DK KD "Г Xex[HL]"0 •
(16.20)
303
При D,.. величиной l/KD пренебрегают и уравнение (16.20) переходит в
1 _ [Н+]"
Dh XM[HL]"
ИЛИ
йм=/<ех-1|^. (16.21)
При логарифмировании уравнения (16.21) получаем
!g^M= lg^ex + nig [HL] 0 + npH. (16.22)
Уравнение (16.22) и более точное (16.20) показывают, что коэффициент распределения тем больше, чем выше pH раствора и концентрация реагента HL. Это не означает, однако, что экстракцию во всех случаях следует проводить из щелочного раствора. Для каждой системы существуют оптимальные значения pH и концентрации реагента. Уравнению (16.22) можно придать вид
-_1^=рН - + lg [HL] о. (16.23)
При D м= 1 и равных объемах водной и органической фаз экстрагируется 50 % металла и в этом случае уравнение (16.23) переходит в
--^=pH-A+lg[HL]0.
Если [HL] о= 1, то---lg^~ равен значению pH, при котором
экстрагируются 50 % металла:
-^=(рн72)1.0.
Величина (рН72)1>0 является одной из практически важных характеристик экстракции, позволяющей предвидеть возможности и условия экстракционных разделений.
Константа экстракции связана с константой устойчивости образующегося координационного соединения, константой диссоциации реагента и другими константами:
к _ [ML„]0[H+]" [L-]"[ML„] [HL]" _ ех [М"+] [HL]"0 [L~]"[ML„] [HL]"
₽MuAd(ML„)^HL Zicoaas
=--------------• (16.24)
aD(HL)
Произведение РЧ1 КО(МЬ) часто называют двухфазной константой устойчивости. Как видно из уравнения (16.24), чем выше устойчивость соединения MLn, больше константа диссоциации реагента HL и меньше его коэффициент распределения, тем больше константа экстракции и, следовательно, тем эффективнее будет экстракция.
304
Существенное влияние на экстрагируемость и полноту экстракции оказывают и другие факторы: растворимость соединения MLn, природа органического растворителя, наличие маскирующих агентов и т. д. Наибольшей экстрагируемостью обладают внутрикомплексные соединения, малорастворимые в воде, но легко растворимые в органических растворителях (ацетил-ацетонаты железа, галлия, индия и др.), а соединения, растворимые в обеих фазах, экстрагируются лишь частично (ацетил-ацетонаты цинка, кобальта, никеля и др.).
Влияние природы органического растворителя на экстракцию имеет ряд характерных особенностей. При переходе от одного растворителя к другому коэффициенты распределения KD(Mbl и Xo(HL) изменяются симбатно и существенного влияния природы растворителя на константу экстракции (16.24) не наблюдается. Однако такие растворители, как трибутилфосфат, спирты и другие, образуют с экстрагируемым металлом комплексы, которые менее гидрофильны, чем аква-ионы, и их экстракция неполярными растворителями улучшается.
Во многих случаях смесь экстрагентов более эффективно экстрагирует соединения из водного раствора, чем каждый из компонентов в отдельности. Это так называемый синергизм. Например, при экстракции урана (VI) раствором теноилтрифтор-ацетона в циклогексане коэффициент распределения составляет 0,063, а раствором окси-3-м-бутилфосфата — 38,5. У смешанного экстрагента, состоящего из этих трех компонентов, коэффициент распределения достигает 1000.
16.4. ЭКСТРАКЦИОННЫЕ ХЕЛАТНЫЕ СИСТЕМЫ
Широкое применение в практике экстракции нашли 8-окси-хинолин и его производные, дитизон и его аналоги, (J-дикетоны, оксимы, дитиокарба^инаты, алкилфосфорные кислоты и многие другие соединения. Рассмотрим свойства некоторых наиболее известных экстрагентов.
8-0 ксихинолин
или оксин) малорастворим
в воде (3,6-10-3 моль/л при комнатной температуре) и эфире, но хорошо растворим в спирте, бензоле, хлороформе и других органических растворителях. Для экстракции обычно используются хлороформные растворы, содержащие 1...5% оксихинолина. Константа распределения 8-оксихинолина между водой и хлороформом при 25 °C составляет 500. Оксихинолин является одним из самых универсальных реагентов: он взаимодействует более чем с 50 элементами (Pd, Mo, W, V, Tl, Fe, Zr, Ga, Си, Ti, In, Bi, Ni и др.). Для повышения избирательности разделений с помощью 8-оксихинолина наряду с регулированием pH широко
305
используются различные маскирующие реагенты (цианид-ион, 1,10-фенантролин, ЭДТА и др.). Например, ионы Fe, Си, Ni и Мо маскируются цианидом. Алюминий, кобальт, железо и некоторые другие элементы в присутствии ЭДТА при pH < 8 не экстрагируются, хотя на экстракции урана, вольфрама и молибдена присутствие ЭДТА не сказывается. Многочисленные галогено-и другие замещенные оксихинолины также обладают экстракционными свойствами, но, по-видимому, они менее универсальны. Например, 2-метил-8-оксихинолин не экстрагирует алюминий.
Широко используются в качестве экстрагентов fJ-д и к е т о-н ы, особенно ацетиланетон и теноилтрифторацетон.
Ацетилацетон сн3—С—СН2—С—СН3 смешивается с
II II
О о
хлороформом, бензолом, эфиром и другими органическими растворителями, его растворимость в воде составляет 172 г/л при 20 °C. Константа распределения ацетилацетона между водой и бензолом составляет 5,8, между водой и хлороформом 25. Для экстракции можно использовать непосредственно сам ацетилацетон, который в таком случае является и реагентом, и растворителем, а также растворы ацетилацетона в бензоле, хлороформе, тетрахлориде углерода или другом органическом растворителе Ацетилацетон образует соединения более чем с 60 элементами (Pb, Tl, Fe, Pu, U, Ga, Си, Sc, Al, In, Th, Pb, Ni, La и др.). Аце-тилацетонаты хорошо растворимы в органических растворителях и имеют высокую термическую устойчивость: многие из них могут возгоняться без разложения. _______
Теноилтрифторацетон (Г 1—с—СН2—С—CF3 в воде
II II
о о
растворим незначительно, в органических растворителях значительно лучше. Константа распределения между водой (подкисленной) и бензолом равна 40. С помощью теноилтрифторацетона возможна экстракция металлов из кислых растворов, что является его важным достоинством. Очень широко применяют теноилтрифторацетон для выделения и разделения актинидов. Соединения многих металлов (урана, меди, железа и др.) с теноилтри-фторацетоном окрашены, поэтому нередко органическая фаза, содержащая теноилтрифторацетонаты этих металлов, может фо-тометрироваться.
К числу реагентов, наиболее часто применяемых в экстракции, относится дитизон и его аналоги.
Дитизон (или дифенилтиокарбазон)
(сокращенно H2Dz) в воде практически
306
не растворим, хорошо растворим в хлороформе и тетрахлориде углерода. Константа распределения дитизона между водой и хлороформом равна 2-105, между водой и тетрахлоридом углерода 1 • 10 . По реакции с дитизоном определяют: Pd, Au, Hg, Ag, Си, Bi, Pt, In, Zn, Cd, Co и другие элементы. Для повышения селективности экстракцию проводят в разных областях pH в присутствии комплексообразующих добавок. Например, экстракцией висмута в слабокислой среде можно отделить его от цинка, кадмия, свинца и других элементов. В присутствии цианид-иона дитизон экстрагирует только цинк и олово. Эффективно использование в качестве комплексообразующих добавок тиоцианата, тиосульфата, ЭДТА и др. Сам дитизон и дитизонаты металлов интенсивно окрашены, что позволяет проводить чувствительные фотометрические определения непосредственно в органической фазе после экстракции. Находят применение также различные аналоги дитизона (метил-, фенил-, хлор-, бром- и другие производные), реагирующие с меньшим числом ионов и, следовательно, более селективные, чем дитизон
Важным представителем реагентов группы дитиокарбамина-тов является диэтилдитиокарбаминат натрия С2Н6\ я
>N—Он хорошо растворим в воде, несколько хуже, C2HS/ ^SNa
в спирте, в виде диэтилдитиокарбаминовой кцслоты растворим в хлороформе, бензоле, тетрахлориде углероДа и родственных растворителях. Константа распределения между органической и водной фазами равна 340 для тетрахлорида углерода и 2360 для хлороформа. Водные растворы диэтилдитиокарбаминовой кислоты неустойчивы. Диэтилдитиокарбаминат реагирует с несколькими десятками элементов (Hg, Ag, Си, Tl, Ni, Bi, Pb, Cd, Sb и др.) В присутствии ЭДТА селективность экстракции увеличивается. Некоторые диэтилдитиокарбаматы имеют окраску: комплекс висмута — желтую, кобальта — зеленую, меди — коричневую и т. д., что позволяет проводить фотометрические определения, используя непосредственно органические экстракты.
16.5 ЭКСТРАКЦИЯ ИОННЫХ АССОЦИАТОВ
Экстрагирующиеся системы ионных ассоциатов могут образовываться в результате взаимодействия заряженного хелата или другого крупного иона с противоионом в растворе. Например, тетрафениларсоний AsfCeHs)^- с перманганатом и перренатом образует ассоциаты, которые экстрагируются хлороформом. Экстракция в таких системах практически не отличается от экстракции хелатов.
Более сложный характер имеют процессы, когда ассоциаты образуются в результате ступенчатого комплексообразования, например ионов металлов с галогенидами. Типичным примером таких систем является экстракция из соляно-кислых растворов железа (III) сурьмы (V). кобальта (II) и др. При взаимодей
307
ствии, например, ионов Fe3+ и С1~ в растворе в зависимости от концентрации хлорида возможно образование комплексов различного состава от FeCl2+ при небольшой концентрации С1_ до FeClr при высокой.
В органическую фазу могут извлекаться ассоциаты типа H+FeClr, причем сольватные оболочки ионов Н+ и FeClr могут наряду с водой содержать органический растворитель.
16.6. ЭКСТРАКЦИОННЫЕ ГАЛОГЕНИДНЫЕ И ТИОЦИАНАТНЫЕ СИСТЕМЫ
Большое применение в практике экстракции нашла хлоридная система. Значительная часть металлов экстрагируется в виде ассоциатов типа FeClF, SLCltT, CoCi2- с ионом Н+ или в виде молекул HgCl2, GeCl4 и др. В качестве экстрагентов используют простые и сложные эфиры (диэтиловый, этилацетат), спирты (бутиловый, амиловый), фосфорорганические соединения (трибутилфосфат, трибутилфосфиноксид) и др. Экстракция хлоридных комплексов широко применяется в аналитической химии, радиохимии и некоторых гидрометаллургических производствах. Известна хорошая экстрагируемость из хлоридных растворов Au, Tl, Sb, Ga, Fe, Mo, Nb, Sc и других элементов.
Бромидные и иодидные комплексы также экстрагируются кислородсодержащими растворителями, но практическое значение этих систем значительно меньше, чем хлоридных. В виде фторидных комплексов экстрагируются лишь очень немногие металлы (Та, Nb, Re и др.).
Тиоцианатные комплексы, как правило, экстрагируются лучше, чем галогенидные. В качестве экстрагента обычно используют тиоцианат калия или аммония в присутствии разбавленной серной или соляной кислоты. В виде тиоцианатных комплексов экстрагируются Bi, Fe, Со, In и другие элементы трибутилфосфа том, диэтиловым эфиром, нзоамиловым спиртом и иными растворителями.
16.7. СКОРОСТЬ ЭКСТРАКЦИИ
Распределение вещества между фазами является результатом многих физико-химических процессов, протекающих в обеих фазах и на границе между ними. Скорость экстракции определяется главным образом скоростью образования экстрагирующегося соединения и скоростью его распределения между фазами. Лимитирующей стадией в разных системах может быть как тот, так и другой процесс. Обычно считается, что если скорость экстракции не зависит от интенсивности перемешивания, то лимитирующей стадией является скорость образования экстрагирующегося соединения. Соединения типа ионных ассоциатов образуются быстро и равновесие экстракции в таких системах также устанавливается с высокой скоростью за 3...5 мин. Это же
308
наблюдается во многих хелатных системах, и, таким образом, в большинстве случаев равновесие устанавливается довольно быстро. Однако образование некоторых хелатов происходит медленно и химическая реакция становится стадией, определяющей скорость экстракции. Например, равновесие при экстракции ди-тизонатов таллия и цинка устанавливается за 1...3 ч. Небольшая скорость образования координационных соединений хрома, платиновых и некоторых других металлов обусловливает и малую скорость экстракции этих элементов. Для ускорения процесса иногда анализируемый водный раствор нагревают вместе с реагентом, чтобы прошло комплексообразование, а затем после охлаждения экстрагируют.
Существенное влияние на скорость экстракции оказывает природа органического растворителя. Отмечено, в частности, что во многих случаях быстрее экстрагируют те растворители, в которых растворимость реагента меньше. Например, растворимость ацетилацетона в тетрахлориде углерода почти на порядок меньше, чем в хлороформе. Равновесное распределение ацетил-ацетоната железа (III) в системе СС14—Н2О достигается примерно за 30 мин, а в системе СНОз—Н2О — за 3 ч. Различие в скорости экстракции используется для разработки методик разделения элементов. Известно, например, что дитизонат ртути экстрагируется хлороформом очень быстро (за 1...2 мин), а дитизонат меди — медленно. Это различие в скоростях экстракции составило основу методики их экстракционного разделения.
16.8. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
С помощью экстракционных методик проводят разделение элементов, относительное концентрирование примесей и очистку основного компонента от примесей. Для проведения экстракционных разделений в аналитических лабораториях обычно используют делительные воронки. В препаративной работе применяется также методика противоточной экстракции.
Для разделения элементов, реагирующих с данным экстрагентом, используются различия в константах экстракции, в значениях pH, при которых происходит экстракция, применяют введение специальных веществ, образующих комплексы с компонентами смеси, и т. д. Удобной характеристикой, позволяющей предвидеть и количественно оценить возможность разделения, является коэффициент обогащения.
Различия в pH, при котором происходит экстракция разных элементов, используются, например, при анализе смеси Fe3+, Al3+, Мп2+. Все эти ионы экстрагируются раствором 8- жсихино-лина в хлороформе при разных значениях pH. Ион Fe3+ экстрагируется в широкой области pH (от 2,0 до 10,0), ион А13+ — в области pH 4,5... И и Мп2+ — при pH 6,5...10,0. Практически ион Fe3+ извлекается при pH 2,5...3,0, ион А13+ — при pH5,0 и ион Мп2+ — при pH 10,0. Аналогично проводят разделение при
ЗОР
экстракции дитизоном, ацетилацетоном и другими реагентами.
При разделении в галогенидных системах используют зависимость экстрагируемости от концентрации галогенида. Так, например, в хлоридной среде Со2+ в виде СоС12- легко отделяется от N?+, хлоридные комплексы которого малоустойчивы.
Своеобразен метод промежуточного элемента, когда в анализируемую систему вводят специальный элемент, экстрагирующийся хуже, чем определяемый, но лучше, чем мешающий. По этой методике отделяют, в частности, олово от нескольких десятков мешающих элементов введением цинка, который препятствует экстракции более чем 60 элементов, но не влияет на экстракцию олова.
Нередко эффективным оказывается применение реэкстракции, т. е. процесса обратного извлечения вещества из органической фазы в водную. Водные растворы для реэкстракции подбирают таким образом, чтобы извлечение из органической фазы было селективным. Например, при обработке раствора дитизонатов свинца, цинка, железа и меди в СС14 0,4 М раствором НС1 из органической фазы извлекается только цинк, при обработке 4 М раствором НС1 в водную фазу переходит и свинец. Железо и медь остаются в органической фазе.
Широко применяется экстракция и для абсолютного и относительного концентрирования микропримесей. Абсолютное концентрирование достигается за счет меньшего объема органической фазы по сравнению с исходным объемом водного раствора. Под относительным концентрированием понимают увеличение концентрации приместей по отношению к содержанию основного компонента. Оно сводится к селективной экстракции примесей или (реже) экстракции макрокомпонента.
Существенное значение имеет методика последующей работы с экстрактом. Особой интерес вызывают аналитические методики, в которых органическая фаза используется непосредственно для количественного определения. Концентрацию окрашенного компонента в экстракте можно определить фотометрически, содержание радиоактивного элемента — по его радиоактивности. Используют также полярографию, эмиссионную спектроскопию, атомно-абсорбционные методы и т. д.
16.9. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Экстракция является весьма эффективным методом разделения и концентрирования, особенно при отделении микрокомпонента смеси от больших количеств других веществ. Существенным достоинством экстракционных методов является их быстрота. Для проведения разделения обычно бывает достаточно нескольких минут и, как правило, кроме делительной воронки никакой другой аппаратуры не требуется. Широкий выбор экстракционных реагентов и растворителей позволяет подобрать опти
310
мальные условия селективного выделения того или иного компонента практически из любой смеси. Нередко экстрагированное соединение окрашено, что позволяет непосредственно использовать экстракт для количественных фотометрических определений микрокомпонента. Известно также эффективное использование экстракционных методов в технологии цветных и других металлов.
Крупные успехи в анализе микрокомпонентов достигнуты в значительной степени благодаря применению экстракционных методик, которые продолжают интенсивно развиваться и совершенствоваться.
! Вопросы
1. В чем сущность метода экстракции? Записать уравнение, которому подчиняется распределение вещества между несмеши-вающимися растворителями.
2. Дать определения понятий: коэффициент распределения, константа распределения, константа экстракции, степень (фактор) извлечения, коэффициент разделения, фактор обогащения.
3. Вывести уравнение для расчета числа экстракций, необходимого для достижения заданной степени извлечения.
4. Назвать органические растворители, наиболее часто используемые в методах экстракции.
5. Что характеризует возможность разделения двух веществ методом экстракции?
6. Назвать основные типы соединений, в виде которых экстрагируются ионы металлов. Привести примеры.
7. Указать, какие элементы чаще всего экстрагируются в виде: а) ацетилацетонатов; б) дитизонатов; в) диэтилдитиокарбаматов; г) купферонатов; д) оксихинолинатов.
8. Какие факторы оказывают влияние на экстрагируемость и полноту экстракции?
9. Факторы разделения муравьиной и серной кислот составляют 15, 648 и 3864 соответственно в растворителях «-амиловом спирте, диэтил овом эфире и метил из обутилкетоне. Какой из растворителей обеспечит более полное разделение указанной смеси кислот?
10. Из растворов азотной кислоты равным объемом диэтилового эфира экстрагируется 80 % Се (IV) и 0 % Th (IV) в 4 М HNO3; 95 % Се (IV) и 18 % Th (IV) в 6 М HNO3; 96 % Се (IV) и 35 % Th (IV) в 8 М HNO3. При какой концентрации HNO3 обеспечивается отделение экстракцией Се (IV) от Th (IV)?
11. Известно, что ионные пары более устойчивы и растворимы в среде с низкой диэлектрической проницаемостью е. Какой растворитель обеспечит лучшую экстрагируемость ионной пары: а) вода, е = 80; б) нитробензол, е = 35; в) изоамиловый спирт, е= 15; г) диэтиловый эфир, е = 4?
311
12. Для разделения двух ионов металлов А и В, образующих с одним и тем же лигандом HL комплексы АЬг и ВЬг, константы их устойчивости должны различаться в I06 раз и более. Какова роль pH и маскирующих агентов в обеспечении этого условия?
13. В чем сущность методов концентрирования: а) с экстракцией микропримесей; б) с экстракцией макрокомпонента?
14. В чем сущность реэкстракции определяемого вещества в водную фазу?
15. Каковы достоинства и недостатки методов экстракции?
.Д Задачи
1. Определить степень извлечения и понижение концентрации в водной фазе за одну экстракцию при встряхивании 50,0 мл раствора 0,5 М по FeCU и 5 М по НС1 с 5,0 мл диэтилового эфира, если коэффициент распределения FeCK в системе эфир — вода в этих условиях составляет 17,6. Рассчитать число последовательных экстракций, необходимых для доведения концентрации Fe3+ в водной фазе до 5-10~3 моль/л.
Степень извлечения R и снижение концентрации FeCI3 в водной фазе за одну экстракцию (с,) вычисляем по формулам:
г, О ]/в
R = D + VB/V„ ис' = со ув + V3KcD
Подставляя данные условия задачи, получаем
R =---ЮО % = 63,8 %;
17,6+
О
50
С| = 0.5 = 0,18 моль/л.
50 + 5-17,6
Число экстракций т, необходимое для понижения концентрации Fe3+ до 5-10-3 моль/л, определяем по уравнению
__ __1g Гнач 1g Гкон
- lg(VB+ FaKcDl-IgF. ’
= 1g 0,5 — 1g (5-10~3) = (1,0-0,7)-(-3 + 0,7) = 2,6 = =
Ig (50 + 5-17,6) - 1g 50 2,14 — 1,70 0,44
= ~ 6 экстракций.
2. Определить степень извлечения пикриновой кислоты из водного 0,05 М раствора при трехкратной экстракции бензолом при Г = У6енз/УВОдн= 1:10. Коэффициент распределения пикриновой кислоты в системе бензол — вода составляет 35.
Какова остаточная концентрация пикриновой кислоты в водном растворе?
Степень извлечения пикриновой кислоты при трехкратной экстракции Rz вычисляем по формуле
1 V
Лз = 1 ~ (Dr + I)3 ’ ГДе Г = ТГ = °’1;
312
Rs 1 (35.од _|_ !)3 1 91 1 1 0,011 — 0,989,
или 98,9 %.
Остаточная концентрация пикриновой кислоты в водной фазе составляет 0,05-0,011 = 5,5-10~4 моль/л.
3. Можно ли добиться 99 %-ного извлечения растворенного вещества с константой распределения 20 в результате: а) однократной обработки 100,0 мл водного раствора этого вещества 25,0 мл бензола; б) трехкратной такой же обработки? Ответ: а) нет; Ri = 83 %; б) да; R3 = 99,5 %.
4. Определить степень извлечения Ri и Rm, а также остаточную концентрацию в водной фазе диметилглиоксимата никеля из Ув мл водного раствора при pH 8,0 и m-кратном встряхивании с Уо мл хлороформа, если коэффициент распределения D = 410, а начальная концентрация с°в моль/л:
Вариант VB, мл Vo, мл m Св, моль/л
1 100 2,0 2 0,10
2 50 5,0 2 0,25
3 50 2,0 3 0,50
Ответ: 1) 89,1 %; 98,9%; 1,1 • 10“3 моль/л; 2) 97,6%; 99,94%; 1,5-10"4 моль/л; 3) 94,2 %; 99,98%; 1,0-КГ4 моль/л.
5. Определить степень извлечения Ri и Rm купфероната олова (IV) и остаточную концентрацию в водной фазе при pH 2 из Ув мл водного раствора при m-кратном встряхивании с Уо мл бензола, если коэффициент распределения 0=350, а начальная концентрация с° моль/л:
Вариант Vs, мл Vo, мл m Св, моль/л
1 10 2,0 1 0,2
2 20 2,0 3 0,5
3 25 2,0 2 0,5
Ответ: 1) 98,59%; 98,59%; 2,8-10~3 моль/л; 2) 97,22%; 99,998 %; 1,0-10-5 моль/л; 3) 96,55 %; 99,88 %; 6,0-10-4 моль/л.
6. Определить степень извлечения и снижение концентрации в водной фазе за одну экстракцию и число последовательных экстракций, необходимое для понижения концентрации алюминия (III) в водной фазе до конечной концентрации с', если Увмл 0,05 М А1С1з встряхивают с Уо мл ацетилацетона при pH 2,0, а коэффициент распределения составляет 23:
Вариант VB, мл Vo, мл с', моль/л
1 10,0 2,0 1-10~’
2 25,0 5,0 5-Ю-3
3 50,0 5,0 1-ю-3
313
Ответ: 1) 82,14%; 8,9-Ю а моль/л; 4; 2) 82,14 %; 1,1-10~2 моль/л; 2; 3) 69,7 %; 1,5-10~2 моль/л; 4
7. Из VB мл водных 0,1 М растворов солей извлекают в виде оксихинолятов трехкратной экстракцией хлороформом; а) лантан, б) самарий. Коэффициенты распределения равны Z)La=370, DSm=280. Рассчитать минимальный расход растворителя для извлечения каждого из элементов, чтобы концентрация его в водном растворе понизилась до конечной концентрации с':
Вариант мл Vfm, мл с', моль/л
1 250 200 1-10-3
2 50 25 5-IO”4
3 100 100 1-ю-4
Ответ: 1) а) 7,50 мл, б) 7,86 мл; 2) а) 1,98 мл, б) 1,32 мл; 3) а) 7,29 мл, б) 9,63 мл.
8. Навеску никель-цинкового феррита, содержащего медь, массой m г растворили и получили 100,0 мл раствора. Аликвоту объемом Кал мл поместили в делительную воронку, добавили необходимые реактивы и экстрагировали медь тремя порциями по 10,0 мл этилацетата. Объединенный экстракт довели до 50,00 мл этилацетатом, фотометрировали и получили значения оптической плотности Ах.
Определить массовую долю (%) меди в феррите, используя для построения градуировочного графика следующие данные, полученные в аналогичных условиях экстрагирования и фотомет-рирования (Кст мл стандартного раствора CuSO4, концентрация меди в котором составляла 2 мкг/мл):
Вариант т, г
Ах - .
ал> М«Л .
VCT, мл
А ст
1 2 3
0,2042 0,5216 0,982
0,210 0,415 0,580
20,0 10,0 20,0
10,0 15,0 20,0
0,250 0,370 0,500
Ответ: 1) 4,2-10“2 %; 2) 6,3-10 ~2 %; 3) 3,9-10“2 %.
9. Из навески почвы массой m г взбалтыванием с 50,00 мл разбавленного раствора НС1 извлекли медь; из 25,0 мл фильтрата медь экстрагировали 10,0 мл раствора диэтилдитиокарбамата натрия в тетрахлориде углерода и измерили оптическую плотность органической фазы.
Аналогично экстрагировали медь из четырех стандартных растворов, приготовленных смешением Кст мл рабочего раствора CuSO4, содержащего 10,0 мкг в 1,0 мл и (25,0 — V\T) мл раствора НС1, и вновь измерили оптические плотности органических фаз А ст:
V„, мл . 0,2 0,5 1,0 2,0
Л,.,........................ 0,025 0,065 0,120 0,245
314
Определить концентрацию меди в почве (мг/кг), если для исследуемых образцов почвы найдены значения Ах:
Вариант т, г . А*
I 2 3
10,06 9,85 8,95
0,072 0,080 0,125
Ответ: 1) 11,9 мг/кг; 2) 13,2 мг/кг; 3) 22,8 мг/кг.
10. Из 250,0 мл сточной воды отогнали анилин с водяным паром и отгон выпарили досуха в присутствии кислоты, остаток растворили в V мл буферного раствора с pH 3,5, Гал мл полученного раствора экстрагировали 4,0 мл хлороформа в присутствии бромкрезолового пурпурового и измерили оптическую плотность органического слоя Ах.
Для построения градуировочного графика воспользовались стандартным раствором, содержащим 0,30 г/л анилина. Для этого 10,0 мл его разбавили до 500,0 мл водой, отобрали четыре пробы по VCT мл, каждую упарили досуха, остаток растворили в 20,0 мл того же буферного раствора, 2,0 мл полученного раствора экстрагировали и измерили Лст аналогично исследуемым пробам:
мл
Л с.
5,0 10,0 15,0 20,0
0,080 0,165 0,245 0,325
Вычислить концентрацию анилина кмкг/л)
в сточных водах
по следующим данным.
Вариант V, мл .
Va„, МЛ . Л,
Ь,0 10,0 1э,0
2,0 5,0 5,0
0,130 0,195 0,310
Ответ: 1) 46,0 мкг/л; 2) 57,6 мкг/л; 3) 132,0 мкг/л.
11. Навеску руды массой m г перевели в раствор и довели до 50,0 мл. Из полученного раствора отобрали две пробы объемом 20,00 мл. К одной из проб прибавили 10,00 мл стандартного раствора рения (Т=0,02 мг/мл), к другой—10,00 мл воды. Из обеих проб рений многократно экстрагировали толуолом в присутствии метилового фиолетового. Экстракты собрали в две колбы вместимостью 50,0 мл, довели объем толуолом до метки и измерили оптические плотности Ах и Лх+ст.
Определить массовую долю (%) рения в руде по следующим данным:
Вариант т, г .
Ах ^х+ст
1 2 3
1,250 2,000 2,500
0.210 0,340 0,420
0,450 0,560 0,700
Ответ: 1) 3,36-10~2 %; 2) 3,86-10'2 %; 3) 3,00-10~2 %.
12. Две пробы производственной воды, по V мл каждая, перенесли в делительные воронки. К одной из проб добавили 10,0 мл раствора с концентоацией сст мг/л ксантогената, к другой —
05
10,0 мл воды. В каждую пробу добавили сульфата никеля и необходимые реактивы и экстрагировали несколько раз ксантогенат никеля толуолом по 1,5 мл. Экстракты собрали и довели объемы до 250,00 мл толуолом. Полученные растворы фотометрировали и получили Ах и Ах+ст.
Вычислить концентрацию (мг/л) ксантогената в промышленной воде по следующим данным:
Вариант V, мл С ст» м Г J Л А„ ^«4-ст
1 100 50 0,180 0,560
2 200 5 0 300 0,630
3 25 300 0,280 0,740
Ответ: 1) 2,50 мг/л; 2) 3,86 мг/л; 3) 7,64 мг/л.
13. В делительную воронку поместили V мл раствора, содержащего пикраминовую и пикриновую кислоты, прибавили необходимые реактивы и в том числе раствор красителя катионного красно-фиолетового, позволяющего экстрагировать пикриновую кислоту в слой бензола. При экстрагировании 2,00 мл бензола и измерении оптической плотности органического экстракта полу чили Ах.
Вычислить концентрацию пикриновой кислоты в растворе (мг/л) по следующим данным:
Вариант V, мл . Ах
12 3
10,0 15,0 20,0
0,480 0,660 0,840
Для построения градуировочного графика следует воспользоваться данными, полученными при экстрагировании в таких же условиях раствора, содержащего 1,10 мкг/мл пикриновой кислоты:
. 1,50 3,50 5,50 7,50 9,50
Дст . 0,180 0,360 0,620 0,780 1,00
Ответ: 1) 0,50 мг/л; 2) 0,47 мг/л; 3) 0,45 мг/л.
Глава 17 ХРОМАТОГРАФИЯ
17.1. АДСОРБЦИЯ ВЕЩЕСТВА — ОСНОВА ХРОМАТОГРАФИИ
Хроматографический метод анализа разработан русским ботаником М. С. Цветом в 1903 г. В первых же работах с помощью этого метода М. С. Цвет установил, что считавшийся однородным зеленый пигмент растений хлорофилл на самом деле состоит из нескольких веществ. При пропускании экстракта зеленого листа через колонку, заполненную порошком
316
мела, и промывании петролейным эфиром он получил несколько окрашенных зон, что с несомненностью говорило о наличии в экстракте нескольких веществ. Впоследствии это было подтверждено другими исследователями. Этот метод он назвал хроматографией (от греч. хроматос — цвет), хотя сам же указал на возможность разделения и бесцветных веществ. Заметное развитие хроматографических методов началось в 30-е годы, когда возникла острая потребность в новом методе разделения смесей и очистки веществ, разлагающихся при нагревании. Хроматография продолжает бурно развиваться и в настоящее время является одним из наиболее перспективных методов анализа.
f Хроматографию можно определить как процесс, основанный на многократ-* ном повторении актов сорбции и десорбции вещества при перемещении его в потоке подвижной фазы вдоль неподвижного сорбента.
Вещество подвижной фазы непрерывно вступает в контакт с новыми участками сорбента и частью сорбируется, а сорбированное вещество контактирует со свежими порциями подвижной фазы и частично десорбируется.
При постоянной температуре адсорбция увеличивается с ростом концентрации раствора или давления газа. Зависимость количества поглощенного вещества от концентрации раствора или давления газа при постоянной температуре называют изотермой адсорбции. Типичная изотерма адсорбции приведена на рис. 17.1. Математически эта зависимость может быть выражена уравнением Лэнгмюра:
1 (17.1)
1 + be х 7
где п — количество адсорбированного вещества при равновесии; Па,— максимальное количество вещества, которое может быть адсорбировано на данном адсорбенте; b — постоянная; с — концентрация.
По Лэнгмюру на поверхности твердого тела имеется некоторое число мест с минимальной энергией, расположенных через определенные интервалы по всей поверхности. Их число равно п^. На этих местах могут адсорбироваться молекулы из раствора или газа. В области небольших концентраций изотерма линейна. Действительно, при 6с<С1 знаменатель (17.1) становится равным единице и уравнение (17.1) переходит в
п=Поо6с=Гс. (17.2)
Это уравнение линейной адсорбции. Оно соответствует уравнению Генри (Г — коэффициент Генри). Область линейной адсорбции иногда называют также областью Генри.
Однако известны случаи, когда зависимость количества адсорбированного вещества от концентрации раствора или давления газа существенно отличается от изображенной на рис. 17.1.
317
Рис 17 I Изотерма ад сорбции
Изотерма адсорбции может быть, например, вогнутой или S-образной. Это может быть вызвано образованием на поверхности адсорбента не моно-, а поли-молекулярного слоя, что не предусматривается теорией Лэнгмюра, а также тем,
что поверхность реальных твердых тел неоднородна, и другими причинами. Несмотря на некоторые существенные ограничения, применимость уравнений (17.1) и (17.2) в теории хроматографических процессов остается довольно широкой.
При адсорбции двух или нескольких веществ уравнение (17.1) для z-ro компонента принимает вид
Ь,с, n = naQ---------------------
«= т 1 + 2 ь,с,
Таким образом, количество адсорбированного вещества будет определяться не только его концентрацией, но и сродством к адсорбенту. При адсорбции нескольких веществ проявление сродства особенно заметно, так как возможно вытеснение одних сорбированных веществ другими, обладающими большим сродством, хотя имеющими, может быть, и меньшую концентрацию.
17.2. КЛАССИФИКАЦИЯ МЕТОДОВ ХРОМАТОГРАФИИ
Различные методы хроматографии можно классифицировать по агрегатному состоянию фаз, способу их относительного перемещения, аппаратурному оформлению процесса и т. д. По агрегатному состоянию фаз хроматографические методы обычно классифицируют следующим образом (табл. 17.1).
По способу относительного перемещения фаз различают фрон-
Таблица 171* Классификация хроматографических методов по агрегатному состоянию фаз
Неподвижная фаза Подвижная фаза
газообразная жидкая
Твердая Жидкая Газо адсорбционная хроматография Распределительная газо-жидкостная хроматография Жидкостно-адсорбционная колоночная, тонкослойная, ионообменная, осадочная Распределительная жидкостно-жидкостная хроматография
* Таблица имеет иллюстративный характер и не включает поэтому некоторые виды хроматографии, например гель-хроматографию и др
318
тальную проявительную, или элюэнтную, и вытеснительную хроматографию.
Фронтальный метод. Это простейший по методике вариант хроматографии. Он состоит в том, что через колонку с адсорбентом непрерывно пропускают анализируемую смесь, например, компонентов А и В в растворителе Solv. В растворе, вытекающем из колонки, определяют концентрацию каждого компонента и строят график в координатах концентрация вещества — объем раствора, прошедшего через колонку. Эту зависимость обычно и называют хроматограммой или выходной кривой (рис. 17.2).
Рис. 17.2. Выходная кривая фронтального анализа
V
Рис. 17.3. Выходная кривая проявительного анализа
Вследствие сорбции веществ А и В сначала из колонки будет вытекать растворитель Solv, затем растворитель и менее сорбирующийся компонент А, а затем и компонент В и, таким образом, через некоторое время состав раствора при прохождении через колонку меняться не будет. Фронтальный метод используется сравнительно редко. Он применяется, например, для очистки раствора от примесей, если они сорбируются существенно лучше, чем основной компонент, или для выделения из смеси наиболее слабо сорбирующегося вещества.
Проявительный (элюэнтный) метод. При работе по этому методу в колонку вводят порцию анализируемой смеси, содержащей компоненты А и В в растворителе Solv, и колонку непрерывно промывают газом-носителем или растворителем Solv. При этом компоненты анализируемой смеси разделяются на зон ы: хорошо сорбирующееся вещество В занимает верхнюю часть колонки, а менее сорбирующийся компонент А будет занимать нижнюю часть. Типичная выходная кривая изображена на рис. 17.3.
В газе или растворе, вытекающем из колонки, сначала появляется компонент А, далее — чистый растворитель, а затем компонент В. Чем больше концентрация компонента, тем выше пик и больше его площадь, что составляет основу количественного хроматографического анализа. Проявительный метод дает возможность разделять сложные смеси, он наиболее часто применяется в практике. Недостатком метода является уменьшение
319
концентрации выходящих растворов за счет разбавления растворителем (газом-носителем).
Вытеснительный метод. В этом методе анализируемую смесь компонентов А и В в растворителе Solv вводят в колонку и промывают раствором вещества D (вытеснитель), которое сорбируется лучше, чем любой из компонентов анализируемой смеси.
Концентрация раствора при хроматографировании не уменьшается в отличие от проявительного метода. Существенным недостатком вытеснительного метода является частое наложение зоны одного вещества на зону другого, поскольку зоны компонентов в этом методе не разделены зоной растворителя.
17.3. ХРОМАТОГРАФИЧЕСКИЙ пик И ЭЛЮЦИОННЫЕ ХАРАКТЕРИСТИКИ
В хроматографии чаще всего используют методику проявительного (элюэнтного) анализа, при которой газ или раствор, выходящий из колонки, анализируется непрерывно. Типичная выходная кривая (хроматограмма) проявительного анализа приведена на рис. 17.3. Рассмотрим ее более подробно (рис. 17.4).
Если точка А' соответствует вводу анализируемой пробы, А — появлению на выходе какого-то несорбирующегося компонента, а В — появлению анализируемого вещества, то линию А'А В и ее продолжение BF называют нулевой линией. Кривую BDF называют хроматографическим пиком и характеризуют высотой, шириной и площадью. С удовлетворительной точностью контур пика описывается уравнением Гаусса:
(Г-УсУ
с=стахе , (17.3)
где V — объем подвижной фазы; Ко — обьем подвижной фазы, соответствующий стах; р,ст — стандартное отклонение, равное полуширине пика при с™ах = е1/2.
Рис. 17.4. Кривая проявительного авали за (хроматографический пик)
Высотой пика считают либо величину h, либо h' (см. рис. 17.4). Последняя равна расстоянию от нулевой линии до точки пересечения касательных к кривой в точках перегиба. Шириной пика называют расстояние между точками контура на половине его высоты (СЕ= = цо5) или на какой-то другой отметке по высоте, либо расстояние между точками
320
перегиба (цп) или между точками пересечения нулевой линии с касательными к кривой в точках перегиба (B'F' = цк = .&у). Соотношения между этими величинами хорошо известны:
Цо 5— 2,36цстт цп— 0,850цо,5— 2р,ст;
р,к=1,700ц05=ау=4цст. (17.4)
Важной хроматографической характеристикой системы является время удерживания или пропорциональный ему удерживаемый объем. На рис. 17.4 приведенному удерживаемому объему соответствует отрезок AG, а общий удерживаемый объем характеризуется отрезком A'G.
Если длину отрезка A'G обозначить /, то время удерживания tr будет равно
tr===l/U,
где U — скорость движения ленты самописца.
Удерживаемый объем Уг пропорционален времени удерживания tr’
V,=tra>, где со — объемная скорость газа-носителя.
Приведенный удерживаемый объем V', соответствующий отрезку AG, определяется соотношением
V'r=Vr—Vo,
где Ео пропорционален отрезку АА', длина которого /о.
Величина Уо характеризует удерживаемый объем несорби-рующегося газа, или мертвый объем колонки.
Приведенному удерживаемому объему соответствует приведенное время удерживания t'r:
t'r=tr — to, где to, пропорциональное величине to, характеризует время удерживания несорбирующегося газа.
Произведение приведенного удерживаемого объема У' на коэффициент сжимаемости j называют эффективным удерживаемым объемом УЭфф:
У эфф- У г/’
Коэффициент сжимаемости
з (pi/po)2—1 7 2 (pi/po)3 — 1 ’
где pi и ро — соответственно давление на входе в колонку и на выходе из нее.
Важной характеристикой является абсолютный удельный удерживаемый объем Ут, рассчитываемый по формуле
у — ^фф 273-16 (17 5)
т т Гк ’ (17-5)
где т — масса адсорбента; Тк—температура колонки.
П-1533 32J
Величина Vm не зазисит от геометрических параметров колонки и может быть использована для качественной характеристики системы адсорбент — адсорбированный газ. Однако на эту величину заметное влияние оказывают случайные факторы. В значительно меньшей степени они влияют на относительный удерживаемый объем, равный отношению абсолютно удельного удерживаемого объема исследуемого вещества Vm., к соответствующему объему вещества, принятого за стандарт, V„CT:
VOTH=-^. (17.6)
Значения относительных удерживаемых объемов приводятся в справочных таблицах.
Полнота разделения двух компонентов количественно может быть выражена критерием разделения Л':
К=------, (17.7)
H0.5(l) I И0.5(2) Ио5 J 5(1) Т Иоб(1) 0.5(2)
где А/ или А К — расстояние между максимумами пиков разделяемых элементов; ц0,5—полуширина хроматографического пика первого (1) и второго (2) компонентов на половине высоты, а нижний индекс «об» указывает на объемные единицы измерения.
При /<=1 разделение бывает достаточно полным.
Если допустить, что ширина хроматографического пика обоих компонентов примерно одинакова, т. е. уравнение (17.7)
принимает вид
ДИ,
2Иоб 0.5
При взаимном перекрывании пиков определение ширины зоны каждого пика становится невозможным (рис. 17.5). В таких случаях рассматривают степень разделения Ф
'F=(ft2-ftmin)/ft2, (17.8)
где h2 — высота пика
t
Рис 17.5 Определение степени разделения У
вещества, имеющего меньшую концентрацию; hmn—высота минимума.
Значение тех или иных элюционных характеристик меняется в зависимости от цели анализа. В качественном анализе основное внимание уделяется определению характеристик удерживания и устранению искажений этих величин за счет второго компонента. В количественном анализе важно, чтобы четкость разделения обеспечивала достаточную точность определения площади или высоты хроматографического пика.
322
17.4. ТЕОРЕТИЧЕСКИЕ ПРЕДСТАВЛЕНИЯ В ХРОМАТОГРАФИИ
Известно несколько теорий хроматографического процесса. Существенное значение имеют метод теоретических тарелок и кинетическая теория.
В методе теоретических тарелок Мартина и Синджа хроматографическая колонка мысленно делится на ряд элементарных участков — «тарелок» и предполагается, что на каждой тарелке очень быстро устанавливается равновесие между сорбентом и подвижной фазой. Каждая новая порция газа-носителя вызывает смещение этого равновесия, вследствие чего часть вещества переносится на следующую тарелку, на которой, в свою очередь, устанавливается новое равновесное распределение и происходит перенос вещества на последующую тарелку. В результате этих процессов хроматографируемое вещество распределяется на нескольких тарелках, причем на средних тарелках его концентрация оказывается максимальной по сравнению с соседними тарелками. Распределение Вещества вдоль слоя сорбента подчиняется уравнению
(х — хо)2
(17.9)
где х — расстояние от начала колонки до точки, в которой концентрация равна с; Хо — координата центра полосы; Н — высота, эквивалентная теоретической тарелке (ВЭТТ); I — длина слоя сорбента, на которой произведено поглощение и размещено п теоретических тарелок, при этом
п^1/Н. (17.10)
Если числитель дробного показателя степени в уравнениях (17.3) и (17.9) выразить в одних и тех же единицах, то при сравнении этих уравнений получим
, 2
LJ Ист
I •
Число теоретических тарелок будет равно
n=(v-)! <1711’
Или, учитывая уравнение (17.4),
Эффективность колонки тем выше, чем меньше высота, эквивалентная теоретической тарелке, и больше число теоретических тарелок.
Таким образом, теория тарелок позволяет рассчитать важные
и*
323
количественные характеристики хроматографического процесса Однако теория тарелок, основанная на допущении ступенчатого характера хроматографического процесса, по существу формальна, так как реальный процесс протекает непрерывно. Значение высоты, эквивалентной теоретической тарелке, и число тарелок являются характеристиками размытости зон. Эти величины сохраняют свое значение и в кинетической теории хроматографии, учитывающей скорость миграции вещества, диффузию и другие факторы.
Кинетическая теория хроматографии основное внимание уделяет кинетике процесса, связывая высоту, эквивалентную теоретической тарелке, с процессами диффузии, медленным установлением равновесия и неравномерностью процесса. Высота, эквивалентная теоретической тарелке, связана со скоростью потока уравнением Ван-Деемтера:
H=A+-~-+CU, (17.13)
где А, В и С — константы; U — скорость подвижной фазы.
Константа А связана с действием вихревой диффузии, которая зависит от размера частиц и плотности заполнения колонки, величина В связана с коэффициентом диффузии молекул в подвижной фазе, это слагаемое учитывает действие продольной диффузии, а С характеризует кинетику процесса сорбция-десорбция, массопередачу и другие эффекты. Влияние каждого слагаемого уравнения (17.13 на величину И в зависимости от скорости подвижной фазы показано на рис. 17.6. Первое слагаемое дает постоянный вклад в И. Вклад второго слагаемого существен при небольшой скорости потока. С увеличением скорости подвижной фазы влияние третьего слагаемого возрастает, а доля второго уменьшается. Суммарная кривая, характеризующая зависимость Н от скорости потока, представляет собой гиперболу. При небольшой скорости потока высота, эквивалентная теоретической тарелке, уменьшается, а затем начинает возрастать. Поскольку эффективность колонки тем выше, чем меньше высота, эквивалентная теоретической тарелке, оптимальная скорость подвижной фазы будет равна скорости, соответствующей точке минимума u этой кривой. Чтобы найти эту
\\ точку, продифференцируем урав-
\\ нение (17.13) и производную при-
'Л — равняем нулю:
\\
XX— ------------------/ ___я с—о
и Откуда
Рис 17 6. Зависимость ВЭТТ от скорости подвижной фазы
Попг= уГв/С.
324
Подставляя эту величину в уравнение (17.13), находим оптимальную высоту, эквивалентную теоретической тарелке:
7/Опт=Д+2/ВС. (17.14)
Таким образом, динамическая теория дает основу для оптимизации хроматографического процесса.
17.5. ОСНОВНЫЕ УЗЛЫ ПРИБОРОВ ДЛЯ ХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА
Отечественная промышленность и зарубежные фирмы выпускают большое количество хроматографов самых различных типов. Однако сложные хроматографические установки требуются не всегда. Для проведения хроматографического разделения методами бумажной, тонкослойной и некоторыми другими видами хроматографии используются простые установки, которые могут быть собраны в любой химической лаборатории. Независимо от сложности устройства основными узлами хроматографической установки являются дозатор (система ввода пробы), хроматографическая колонка и детектор. Кроме того, в установке имеются' устройства для подачи газа-носителя или растворителя, для преобразования импульса детектора в соответствующий сигнал и некоторые другие.
Дозатор предназначен для точного количественного отбора пробы и введения ее в хроматографическую колонку. Одвдм из основных требований к дозатору является воспроизводимость размера пробы и постоянство условий ее введения в колонку. Кроме того, введение пробы не должно вызывать резкого изменения условий работы колонки и других узлов хроматографической установки, а внутренняя поверхность дозатора не должна обладать каталитической или адсорбционной активностью по отношению к пробе.
Газообразные и жидкие пробы обычно вводят с помощью специальных шприцев, прокалывая в месте ввода пробы каучуковую мембрану. Применяются газовые шприцы для газообразных проб и микрошприцы для жидких. Микрошприцы позволяют вводить в хроматограф пробы объемом от долей до десятков микролитров. Нередко в лабораторной практике в качестве дозатора применяется медицинский шприц.
Твердые пробы вводятся в хроматограф или после перевода их в раствор, или непосредственным испарением пробы в нагретом дозаторе, куда она вводится с помощью игольного ушка. Известны и другие устройства.
В хроматографической колонке происходит разделение компонентов. Колонки весьма различны по форме, размерам и конструкционным материалам. Применяются прямые, спиральные и другие колонки длиной от 1...2 м и менее до нескольких десятков метров. Внутренний диаметр колонок составляет обычно несколько миллиметров. В зависимости от свойств анализируемой
325
системы в качестве конструкционных материалов для колонок чаще всего используют сталь, латунь, медь, стекло и др. Материал колонки должен обладать определенной химической инертностью по отношению к компонентам пробы, например медные колонки будут непригодны при разделении ацетиленсодержащих смесей.
Адсорбент, наполняющий колонку, должен обладать рядом свойств: необходимой селективностью, достаточной механической прочностью, химической инертностью к компонентам смеси и быть доступным. Практически в качестве адсорбентов используются оксид алюминия, силикагели, активированные угли, пористые полимеры на основе стирола, дивинилбензола и т. д. и синтетические цеолиты. Широко используют модифицированные адсорбенты, которые получают обработкой исходных адсорбентов растворами кислот, щелочей, неорганических солей и т. д. Выбор адсорбента зависит от агрегатного состояния фаз, методики хроматографирования и других факторов.
Большое влияние на сорбируемость газа оказывает температура, поэтому хроматографические колонки, как правило, термо-статируются, используя обогрев жидкостью или парами кипящей жидкости, воздушное термостатирование или какой-либо другой прием. Обычно термостатирование производится при температу рах, значительно превышающих комнатные, однако в некоторых случаях создаются температуры ниже О °C при разделении низко-кипящих газов. В бумажной, тонкослойной и некоторых других видах хроматографии функцию колонки выполняет хроматографическая бумага, тонкий слой сорбента на подложке и т. д.
Детектор предназначен для обнаружения изменений в составе газа, прошедшего через колонку. Показания детектора обычно преобразуются в электрический сигнал и передаются фиксирующему или записывающему прибору, например на ленту электронного потенциометра. Основными характеристиками детектора являются чувствительность, пределы детектирования, инерционность и диапазон линейной зависимости между концентрацией и величиной сигнала. Детекторы подразделяются на дифференциальные, которые отражают мгновенное изменение концентрации, и интегральные, суммирующие изменение концентрации за некоторый отрезок времени.
В интегральных детекторах анализируемый газ на выходе из колонки поглощается каким-либо раствором, а затем анализируется или поглощающий раствор или оставшийся непоглощенный газ. Если носителем является диоксид углерода, то после колонки газ барботируется через раствор щелочи и измеряется объем газа, не поглощенного этой жидкостью. Достоинствами интегральных детекторов являются их простота и широкая об ласть линейной зависимости показаний детектора от количества вещества. К недостаткам относятся значительная инерционность и низкая чувствительность, в связи с чем такие детекторы в настоящее время применяются редко.
326
К группе дифференциальных относятся детекторы по теплопроводности (катарометр), по плотности, по электрической проводимости, пламенный, пламенно-ионизационный (ПИД) и другие ионизационные детекторы, термохимический, пламенно-фотометрический и т. д. Детектор выбирают в зависимости от свойств изучаемой системы, агрегатного состояния фаз и других особенностей.
17.6. ГАЗОВАЯ ХРОМАТОГРАФИЯ
Подвижной фазой в газовой хроматографии является газ или пар. В зависимости от состояния неподвижной фазы газовая хроматография подразделяется на газо-адсорбционную, когда неподвижной фазой является твердый адсорбент, и газожидкостную, когда неподвижной фазой является жидкость, а точнее пленка жидкости на поверхности частиц твердого сорбента.
Как уже отмечалось, в качестве дозаторов в газовой хроматографии используются шприцы и микрошприцы.
17.6.1. Хроматографические колонки и детекторы
Колонки. В газо-адсорбционной хроматографии колонки заполняются твердым сорбентом. Адсорбция газа на твердом сорбенте подчиняется уравнению изотермы адсорбции (17.1). Строение и свойства сорбентов весьма многообразны. По классификации Киселева к первому типу относятся неспецифические сорбенты, не имеющие на поверхности каких-либо функциональных групп (например, угли), ко второму типу принадлежат сорбенты, имеющие на поверхности заряды (например, гидроксидные группы силикагеля), и к третьему — адсорбенты, имеющие на поверхности связи или группы атомов с сосредоточенной электронной плотностью (например, полимеры с нитрильными группами).
Активированные угли неполярны. Их высокая удельная поверхность (1000...1700 м2/г) обусловливает большую силу взаимодействия с анализируемым веществом, что ограничивает область применение углей анализом легких газов.
Адсорбционная активность силикагеля в основном связана с поверхностными ОН-группами. Это полярный адсорбент. Полярным адсорбентом является также оксид алюминия.
Цеолиты часто рассматривают как молекулярные сита: природные или синтетические. Свое название молекулярных сит они получили потому, что их поры имеют размеры, близкие к размерам молекул (от 0,4 до 1,0 нм в зависимости от марки), и адсорбция на них является своеобразным «просеиванием». Действительно, на молекулярных ситах могут сорбироваться только те вещества, молекулы которых могут проникнуть в поры. Например, на молекулярных ситах с малыми размерами пор могут сор
327
бироваться только легкие вещества, размеры молекул которых невелики, а тяжелые вещества, у которых размеры молекул превышают размеры пор, естественно, адсорбироваты я не могут. На обычных адсорбентах, как известно, более эффективно адсорбируются тяжелые вещества, поэтому предпочтительная адсорбция легких веществ на цеолитах сначала казалась непонятной и получила объяснение лишь после раскрытия роли размера пор в процессе поглощения цеолитом. В качестве молекулярных сит используются также пористые стекла. Существенными достоинствами обладают модифицированные адсорбенты: у них в более широкой области концентраций наблюдается линейная изотерма адсорбции, теряется каталитическая активность и т. д.
Возможности хроматографического определения вещества в газовой фазе значительно возросли с открытием в 1952 г. метода газо-жидкостной хроматографии. При анализе по этому методу анализируемая газовая смесь проходит через колонку, наполненную твердым носителем, на поверхность которого нанесен тонкий слой жидкой фазы. Таким образом, с компонентами пробы здесь взаимодействует уже вещество жидкой пленки, хотя в реальных условиях газо-жидкостной хроматографии компоненты смеси частично взаимодействуют и с твердым адсорбентом.
Появление жидкой пленки привело к изменению природы физико-химических процессов в хроматографической колонке. Вместо процесса адсорбции газа на твердом адсорбенте в колонке стал происходить процесс растворения газа в тонкой пленке, находящейся на твердом носителе. Эффективность разделения стала определяться не процессами адсорбции-десорбции газа, как это было в адсорбционной газовой хроматографии, а процессами растворения газа в жидкой пленке и его выделения. Различие в растворимости газов оказалось более существенным, чем различие в их адсорбционных свойствах, поэтому газо-жид костная хроматография открыла более широкие возможности в разделении и анализе многокомпонентных смесей. Очень важным преимуществом газ о-жидкости ой хроматографии является возможность работы в области линейной изотермы в более широкой области концентраций, чем в газовой адсорбционной хроматографии, что обеспечивает получение практически симметричных хроматографических пиков.
Эффективность разделения в газь-жидкостной хроматографии зависит главным образом от правильности выбора жидкой фазы. Строго обоснованных теоретически способов выбора жидкой фазы не существует в связи со сложностью протекающих процессов и недостаточной разработанностью теории растворов. Однако требования к жидкой фазе предъявляются совершенно опреде ленные. Жидкая фаза должна обладать достаточно высокой селективностью, т. е. способностью разделять смеси компонентов, быть химически инертной по отношению к компонентам смеси и к твердому носителю, оставаться термически устойчивой, не рас-328
творять газ-носитель, иметь малую вязкость и быть нелетучей (или иметь летучесть незначительную).
При выборе жидкой фазы полезным оказалось старой правило — «подобное растворяется в подобном». В соответствии с этим правилом для разделения смеси двух веществ выбирают жидкую фазу, близкую по химической природе одному из компонентов. Ограниченность такого подхода для веществ с близкими свойствами или смесями сложного состава очевидна
Накопленный экспериментальный материал по хроматографированию различных систем и установленные закономерности часто дают возможность предвидеть поведение отдельных компонентов в ходе хроматографирования и обоснованно выбирать оптимальные условия разделения сложных смесей.
Эффективным оказалось применение колонок, содержащих несколько неподвижных фаз или сложные сорбенты. В составных колонках анализируемую смесь последовательно пропускают через несколько колонок с разными жидкими фазами. Колонка со смешанным сорбентом содержит равномерно перемешанный сорбент с неодинаковыми жидкими фазами. При нанесении на один твердый носитель смеси несколько неподвижных фаз получают колонки со смешанной фазой.
В качестве жидкой фазы в газо-жидкостной хроматографии используют вазелиновое масло, силиконовое масло, фталаты (дибутил, диоктил и др-), диметилформамид, трикрезилфосфат и др. Специфические особенности проявляют жидкие кристаллы, например азоксиэфиры типа RO——N=N——OR. Нематические жидкие кристаллы, взятые в качестве жидкой фазы, проявляют селективное сродство к линейным молекулам, по-видимому, в связи с тем, что в нематической фазе молекулы могут перемещаться только в параллельных плоскостях. Количество жидкой фазы зависит от свойств системы и составляет от 1 до 30...50 % от массы твердого носителя. Пленка жидкости бывает очень тонкой, поэтому внешний вид носителя с пленкой обычно остается таким же, каким он был у носителя без пленки.
В качестве твердых носителей применяют инертные вещества <с развитой поверхностью, но малой микропористостью, чтобы по возможности исключить адсорбцию газа на поверхности. Наибольшее распространение получили носители на основе кизельгура или диатомита. Применяют также стеклянные микрошарики. Для разделения реакционноспособных веществ используется тефлон.
Существенно повышается эффективность разделения в капиллярной хроматографии. Название метода связано с тем, что в качестве хроматографической колонки используется капилляр с внутренним диаметром 0,1...0,5 мм и длиной в несколько десятков метров. Жидкая фаза наносится непосредственно на стенку этого капилляра, которая в данном случае
329
играет роль носителя. В капиллярной колонке существенно уменьшается сопротивление потоку газа, поэтому появляется возможность увеличить длину колонки и повысить таким образом эффективность разделения. Значительно уменьшается в капиллярной хроматографии объем пробы для анализа и сокращается длительность анализа: метод становится близким к экспрессному.
Проба для анализа в капиллярной хроматографии уменьшается в 1000 раз и больше по сравнению с обычной методикой на насадочных колонках, чт о вызывает определенные экспериментальные трудности и требует разработки специальной методики. Нередко приходится более 99,9 % вводимой пробы выпускать в атмосферу через специальное ответвление и лишь 0,01...0,05 % взятого количества направлять в колонку. Детектирование столь малых количеств требует применения высокоэффективных детекторов, например, типа пламенно-ионизационного.
Внутреннюю поверхность капилляра можно покрывать не только неподвижной жидкостью, но и тонким слоем твердого вещества. Этот вариант называют твердослойной капиллярной хроматографией.
Основными достоинствами капиллярной хроматографии являются высокая эффективность при анализе многокомпонентных смесей, особенно с применением кварцевых капилляров, высокая чувствительность и малая длительность анализа. Недостатки связаны, главным образом, с трудностями дозировки и детектирования, а также с недостаточной селективностью слабо сорбирующихся веществ в условиях капиллярной хроматографии.
Детекторы. Одним из наиболее распространенных дифференциальных детекторов является катарометр. Принцип его работы основан на измерении сопротивления нагретой платиновой или вольфрамовой нити, которое зависит от теплопроводности омывающего газа. Количество теплоты, отводимое от нагретой нити при постоянных условиях, зависит от состава газа. Чем больше теплопроводность определяемых компонентов смеси будет отличаться от теплопроводности газа-носителя, тем большей чувствительностью будет обладать катарометр. Наиболее подходящим газом-носителем с этой точки зрения является водород, теплопроводность которого значительно превышает соответствующую характеристику большинства других газов. Однако в целях техники безопасности чаще применяется гелий, теплопроводность которого также достаточно высока. В последнее время металлические нити в катарометре успешно заменяются термисторами, имеющими более высокий, чем у металлов, температурный коэффициент электрической проводимости. Достоинствами катарометра являются простота, достаточная точность и надежность в работе. Однако из-за сравнительно невысокой чувствительности он не применяется для определения микропримесей.
В основе работы термохимического детектора лежит измерение сопротивления платиновой проволоки, которое меняется вследствие изменения температуры при сгорании горючих газов.
ззо
Газы на выходе из хроматографической колонки каталитически сжигаются на нагретой платиновой проволоке, которая одновременно является плечевым сопротивлением мостовой схемы. Чувствительность термохимического детектора выше, чем катарометра, однако платиновая нить требует частой калибровки и замены. Применимость термохимического детектора ограничена горючими веществами.
Принцип действия пламенного детектора основан на том, что температура водородного пламени горелки изменяется при попадании в него органических веществ. Наибольшей чувствительностью обладают ионизационные детекторы, например пламенно-ионизационный (ПИД), позволяющие обнаруживать до 10-12 г. В этих детекторах измеряют электрическую проводимость пламени водородной горелки. Чисто водородное пламя обладает очень низкой электрической проводимостью. При появлении в водороде примесей органических соединений происходит ионизация пламени, пропорциональная концентрации примеси, что легко может быть измерено. Высокая чувствительность детекторов этого типа обусловила их широкое применение. Однако высокая чувствительность ПИД проявляется по отношению только к органическим соединениям, а к неорганическим, таким, как аммиак, сероводород, оксиды серы, кислород, азот и т. д., чувствительность детектора резко падает.
Очень высокой чувствительностью обладает аргонный детектор, ионизация в котором происходит при столкновении молекул определяемого вещества с метастабильными атомами аргона, образующимися под действием радиоактивного Р-излучения.
В термоионном детекторе в пламя горелки вводят соли щелочных металлов. При попадании в такое пламя соединений фосфора появляется ионный ток, пропорциональный содержанию атомов фосфора. Это селективный фосфорный детектор высокой чувств ител ьности.
Известны и другие типы детекторов: ультразвуковые, диафрагменные, микрокулонометрические и т. д. Для проведения количественных расчетов детектор обычно градуируется.
17.6.2. Качественный анализ
Качественный состав вещества может быть установлен с помощью хроматографической методики по характеристикам полученной хроматограммы или по результатам анализа компонентов смеси после прохождения хроматографической колонки подходящим химическим или физико-химическим методом Типичная хроматограмма приведена на рис. 17.7. Как видно, хроматографическое разделение смеси из семи компонентов проведено вполне успешно. Каждому компоненту смеси отвечает свой пик и последовательность появления пиков на хроматограмме закономерна: она соответствует последовательности кислот в гомологическом ряду. Собственно хроматографический качественный анализ
331
Рис 17 7. Хроматограммы смеси воды и кислот (115 °C).
1 — вод.а3 2 — муравьиная кислота, 3 — уксусная кис лота, 4 — пропионовая кис лота; 5 — изомасляная кис лота, 6 — н масляная кисло та, 7 — изовалериановая
кислота
основан на использовании характеристик удерживания — времени удерживания или пропорционального ему удерживаемого объема и индексов удерживания. Для этой цели применяются относительные удерживаемые объемы [см. формулу (17.6)], которые в значительно меньшей степени, чем абсолютные величины, подвержены действию случайных факторов. Идентификация исследуемых веществ производится сравнением полученных и табличных данных. Условия опыта, естественно, не должны отличаться от тех, в которых были получены табличные данные. Идентификация вещества по его хроматограмме может быть выполнена также методом тестеров, когда сравнивают объем или время удерживания компонента анализируемой смеси и эталона, найденные в одних и тех же условиях опыта. Иногда эталонное вещество, наличие которого предполагается в анализируемой смеси, специально вводят в пробу и сравнивают высоту и площадь пиков на хроматограммах, полученных до и после введения эталона. Увеличение высоты или площади пика рассматривается как указание на присутствие предполагаемого компонента в пробе. Однако этот метод не вполне надежен, так как удерживаемые объемы довольно многих веществ близки между собой. Трудности идентификации преодолеваются, например, хроматографированием пробы на колонках с разными сорбентами. Получение одинаковых результатов при использовании разных сорбентов увеличивает надежность анализа. В настоящее время разработаны и успешно применяются также различные многоступенчатые схемы. В таких схемах после разделения смеси на первой колонке полученные фрации подаются на колонки второй ступени, где достигается более тонкое разделение и более точная идентификация вещества, так как имеют дело с более простыми смесями, чем на первой колонке.
В газо-жидкостной хроматографии для качественного анализа часто используют индексы удерживания Ковача Г.
/=100
lg ig(<;
+ lOOn,
где t'r — приведенное время удерживания; п — число атомов углерода в алкане; i — определяемое вещество.
Стандартом при определении индекса удерживания являются два соседних нормальных алкана (насыщенных углеводорода), один из которых элюируется до, а второй после исследуемого соединения, т. е. t',n < t'r., < t'r, („+1). При программировании
332
температуры индекс удерживания рассчитывается по температуре удерживания Тг:
, Тг,-Тгп
Идентификация вещества по индексу удерживания производится путем хроматографирования соединения с последующим хроматографированием в тех же условиях двух соседних алканов, выбранных в качестве стандарта. Результаты анализа по индексу удерживания оказываются более надежными, чем по удерживаемому объему, так как индекс удерживания является более индивидуальной характеристикой вещества. Индексы удерживания многих веществ при определенных температурах имеются в соответствующих справочных таблицах, что облегчает проведение качественного анализа.
Накопленный экспериментальный материал позволил обнаружить некоторые закономерности и установить определенные зависимости между хроматографическими характеристиками и физико-химическими свойствами вещества. Оказалось, например, что индексы удерживания и удерживаемые объемы связаны простой зависимостью с числом атомов углерода у членов гомологического ряда, с температурами кипения гомологов и другими характеристиками и свойствами вещества. Эти зависимости существенно расширяют возможности хроматографии. Соответствующие графики, например, зависимости удерживаемого объема от температуры кипения почти линейны и их широко используют для идентификации компонентов смеси. Если принадлежность компонента к гомологическому ряду известна, то найденная по такому графику температура кипения или другое свойство достаточны для идентификации компонента. Установлено, что индексы удерживания соседних членов в любом гомологическом ряду различаются примерно на 100. Отмечается, что Л/7 изомеров равна пятикратной разнице их температур кипения, т. е. Л/г = бЛТкнп. Известны и другие закономерности в индексах удерживания.
Эффективным оказалось применение независимой аналитической идентификации продуктов хроматографического разделения и сочетание газовой хроматографии с другими методами исследования: ИК-спектроскопией и масс-спектрометрией, а также использование селективных и последовательно работающих детекторов. Методом масс-спектрометрии можно проводить непрерывный качественный анализ компонентов смеси и для анализа бывает достаточно самых небольших количеств вещества. Такой комбинированный метод получил название х р о м а т о -масс-спектрометрии. Возможно использование также методов ядерного магнитного резонанса, пламенной фотометрии, абсорбционной спектроскопии и других, включая химические методы.
ззз
17.6.3. Количественный анализ
Количественный хроматографический анализ основан на измерении различных параметров пика, зависящих от концентрации хроматографируемых веществ — высоты, ширины, площади и удерживаемого объема — или произведения удерживаемого объема на высоту пика. При достаточной стабильности условий хроматографирования и детектирования определяющим параметром пика можно считать его высоту. Расчет по площади пика позволяет несколько снизить требования к стабильности условий хроматографирования по сравнению с расчетом по высоте пика, однако само измерение площади вызывает появление новых источников ошибок. В случае узких пиков некоторые преимущества имеет измерение произведения удерживаемого объема на высоту пика. При неполном разделении пиков ошибки возрастают из-за наложения и искажения контуров пиков. При работе с такими хроматограммами используют специальные приемы, опирающиеся, главным образом, на измерение высоты пиков.
Основными в количественной хроматографии являются методы: нормировки, нормировки с калибровочными коэффициентами, внутренней стандартизации и абсолютной калибровки.
При использовании метода нормировки принимают сумму каких-либо параметров пиков, например сумму высот всех пиков или сумму их площадей, за 100 %. Тогда отношение высоты отдельного пика к сумме высот или отношение площади одного пика к сумме площадей, умноженное на 100, будет характеризовать массовую долю (%) компонента в смеси. Вполне понятно, что такой метод предполагает существование одинаковой зависимости величины измеряемого параметра от концентрации для всех компонентов смеси.
В методе нормировки с калибровочными (градуировочными) коэффициентами за 100% принимается сумма параметров пиков с учетом чувствительности детектора. Различие в чувствительности детектора учитывается с помощью поправочных коэффициентов для каждого компонента. Один из преобладающих компонентов смеси считают сравнительным и поправочный коэффициент для него принимают равным единице. Калибровочные (градуировочные) коэффициенты К рассчитывают по формуле
где Пст—параметр пика (высота, площадь и т. д.) стандартного вещества; П/ — параметр пика определяемого компонента
За 100 % принимается сумма исправленных параметров КЛ; и результат анализа рассчитывается так же, как и в методе нормировки.
Наиболее точным является метод абсолютной к а-
334
либровки. В этом методе экспериментально определяют зависимость высоты или площади пика от концентрации вещества и строят градуировочные графики. Далее определяют те же характеристики пиков в анализируемой смеси и по градуировочному графику находят концентрацию анализируемого вещества. Этот простой и точный метод является основным методом определения микропримесей. Кроме того, метод не требует разделения всех компонентов смеси, а ограничивается лишь теми, определение которых необходимо в данном конкретном случае.
Метод внутреннего стандарта основан на введении в анализируемую смесь точно известного количества стандартного вещества. В качестве стандартного выбирают вещество, близкое по физико-химическим свойствам к компонентам смеси," но не обязательно являющееся ее компонентом. После хроматографирования измеряют параметры пиков анализируемого компонента и стандартного вещества. Если стандартное веществ'о не входит в состав анализируемой смеси, массовую долю компонента (%) рассчитывают по формуле
w, = -&- 100г,
Чсст
где Qi и QCT—параметры пиков анализируемого компонента и стандарта соответственно; г — отношение массы внутреннего стандарта к массе пробы.
17.6.4. Влияние температуры
Температура оказывает существенное влияние на хроматографический процесс. Изменение температуры вызывает изменение элюционных и других характеристик: удерживаемого объема, коэффициента Генри и т. д. Зависимость удельного удерживаемого объема Vm от температуры Т передается уравнением
ig vm — 23RT + 23R g ,
где A/f° и AS0 — изменение стандартных энтальпии и энтропии при абсорбции соответственно; А — коэффициент пропорциональности между Г и Vm.
Так как обычно А//° < 0, с повышением температуры величина удерживаемого объема уменьшается, что приводит к сокращению длительности анализа и может вызвать изменение порядка выхода компонентов из колонки. При понижении температуры длительность анализа увеличивается, но при этом улучшается степень разделения. Низкие температуры используются, например, в процессах разделения изотопов водорода. Хроматографирование таких систем производится при температурах жидкого азота (—196°C).
335
При выборе оптимальной температуры хроматографирования приходится учитывать, таким образом, несколько факторов, нередко действующих в противоположных направлениях. Выбранная оптимальная температура является важной особенностью хроматографической методики и для поддержания температурного режима хроматографическую колонку помещают в термостат.
Однако газовая хроматография может проводиться не только при постоянной температуре, но и в условиях программированного изменения температуры. Температура колонки в этом случае может меняться по разным режимам: ступенчато, непрерывно с постоянной скоростью (линейно) и непрерывно с переменной скоростью (нелинейно). Каждый из этих способов имеет свои достоинства, недостатки и области применения. Если, например, основную часть смеси составляют легкокипящие компоненты, целесообразно применять метод нелинейного программирования температуры: вначале температуру повышать медленно, а затем этот процесс ускорить.
Изменение температуры открывает новые возможности в разработке хроматографических методов анализа. В методе станционарной хроматермографии разделяемая смесь подвергается действию переменного температурного поля. При осуществлении этой методики электрическая печь движется вдоль колонки в одном направлении с движением газа-носителя. Если скорость движения полосы адсорбированного газа превышает скорость движения температурного поля, то полоса из высокотемпературной зоны попадает в низкотемпературную и ее скорость движения замедляется. Но при замедлении движения зона оказывается в области высоких температур и ее скорость увеличивается. Таким образом, в некоторой температурной области скорости движения полосы газа и печи становятся одинаковыми. Температуру, при которой это происходит, называют характеристической Гхар. Она связана с энтальпией адсорбции Д7/ и условиями хроматографического опыта соотношением
т _ ДЯ ‘ и₽— w ’ Я1пЛ —
где w — скорость движения температурного поля: U — линейная скорость газа-носителя.
При фиксированных условиях опыта разность Ткяр двух компонентов смеси, а следовательно, и возможность разделения определяются разностью их энтальпий сорбции. При равенстве энтальпий разницу в Ткар следует создавать за счет изменения условий опыта, т. е. меняя скорость газа-носителя или скорость движения электропечи. В оптимальных условиях хроматографического опыта каждый компонент анализируемой смеси будет двигаться при своей характеристической температуре со скоростью, равной скорости движения печи. Это может быть исполь
336
зовано для идентификации веществ по наличию или отсутствию пика при данной температуре.
Существенным достоинством метода стационарной хроматер-мографии является сжатие полосы, что улучшает разделение и повышает концентрацию компонента (особенно важно при анализе примесей). К недостаткам метода относится уменьшение расстояния между пиками, что может затруднить разделение.
17.6.5. Аналитическая реакционная газовая хроматография
Суть этого метода состоит в изменении химического состава пробы до хроматографирования с целью получения веществ легче разделяющихся при хроматографировании или легче детектируемых. Можно использовать также поглощение части компонентов молекулярными ситами или другими поглотителями.
Оба процесса — химический и хроматографический — осуществляются в одной установке аналитической реакционной газовой хроматографии. Все или некоторые из компонентов анализируемой смеси реагируют в реакторе, а продукты реакции вместе с компонентами, не принимающими участия в реакции, потоком газа-носителя уносятся в хроматографическую колонку. В практике реакционной газовой хроматографии используется несколько схем взаимного расположения реактора, колонки и детектора. Реактор может быть помещен перед колонкой или после колонки перед детектором или между двумя детекторами. Разработаны и более сложные схемы с применением нескольких реакторов и колонок.
Типы используемых химических реакций самые разнообразные. Органические кислоты этерификацией (взаимодействием со спиртами) превращают в сложные эфиры, обладающие более низкими температурами кипения, чем соответствующие кислоты. Микропримеси паров воды определяют при пропускании анализируемой смеси через реактор с литий-алюминийгидридом, реагирующим с водой с образованием водорода, который легко фиксируется катарометром. Если реактор заполнен карбидом кальция, вода образует ацетилен, наличие которого устанавливается с.помощью пламенно-ионизационного детектора. Алифатические сульфиды можно гидрогенизировать на никеле Ренея до соответствующих углеводородов.
17.7. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Широкое применение и большое значение газовой хроматографии в практике вызвано тем, что с ее помощью можно идентифицировать отдельные компоненты сложных газовых смесей и определять их количественно, выполнение анализа не требует больших затрат времени, и метод является достаточно универсальным. Эффективно используется газовая хроматография в
337
препаративных целях, в физико-химических исследованиях и других областях.
Методом газовой хроматографии анализируют нефтяные и рудничные газы, воздух, продукцию основной химии и промышленности органического синтеза, нефть и продукты ее переработки, многочисленные металлорганические соединения и т. д. Методы газовой хроматографии пригодны для разделения изотопов некоторых элементов, например водорода. Хроматография газов используется в биологии и медицине, в технологии переработки древесины, в лесохимии и пищевой промышленности, в технологии некоторых высокотемпературных процессов и многих других. Газовая хроматография может быть применена Для анализа жидкостей после перевода их в пар в условиях работы хроматографической колонки.
Необходимо отметить применение газовой хроматографии для автоматизации производственных процессов. Датчик промышленного хроматографа используется не только как регистрирующий прибор, но и как регулирующее устройство, подающее сигналы непосредственно исполнительным механизмам. Таким образом, промышленный хроматограф может контролировать и регулировать важнейшие параметры технологического процесса: температуру, давление, расход сырья и т. д.
Газовая хроматография широко используется также в физико-химических исследованиях. Хроматографическая методика позволяет сравнительно легко определить константу сорбционного равновесия при нескольких температурах и по этим данным рассчитать термодинамические характеристики сорбции: изменение энтальпии и энтропии в этом процессе. Хроматографическим методом определяют также коэффициенты активности, диффузии и другие характеристики вещества, изучают кинетику реакций и т. д.
Аналитическая реакционная газовая хроматография применяется для анализа сложных многокомпонентных смесей, определения микропримесей, анализа нелетучих соединений (например, различных полимеров), для элементного анализа и т. д.
17.8. ЖИДКОСТНАЯ АБСОРБЦИОННАЯ ХРОМАТОГРАФИЯ
До конца 50-х годов XX в. практическое значение жидкостной адсорбционной хроматографии было сравнительно невелико и ее развитие происходило медленно. Однако с появлением в конце 50-х годов высокочувствительных методов детектирования и новых селективных адсорбентов на основе полимеров жидкостная адсорбционная хроматография стала высокочувствительным и селективным методом анализа многокомпонентных смесей в растворах. Ее практическое значение возросло еще больше, когда стали применять высокие давления.
338
17.8.1. Теоретические представления
Жидкостная адсорбционная хроматография основана на теории адсорбции из раствора. Адсорбционное равновесие между раствором и адсорбентом подчиняется уравнению изотермы адсорбции Лэнгмюра (17.1), в области разбавленных растворов изотерма линейна (17.2). Селективность адсорбции зависит от природы сил взаимодействия между адсорбирующимся веществом и адсорбентом. Эффективность хроматографической колонки зависит, главным образом, от процессов диффузии и мас-сопередачи в обеих фазах и определяется, как и в газовой хроматографии, высотой эквивалентной теоретической тарелки (ВЭТТ) Н. С линейной скоростью подвижной фазы U и некоторыми другими величинами ВЭТТ связана уравнением
Z7 = 27?r(l -Rr)Uts, (17.15)
причем
Т ts
где tm — среднее время между десорбцией и повторной адсорбцией молекулы вещества, когда она движется со скоростью подвижной фазы U; ts — среднее время пребывания молекулы в неподвижной фазе.
Уравнение (17.15) показывает, что с ростом линейной скорости движения подвижной фазы ВЭТТ возрастает и, следовательно, эффективность колонки падает. Зависимость ВЭТТ от Rr имеет довольно сложный характер, но хотя с ростом ts величина Rr уменьшается, ВЭТТ может расти, так как ts входит в уравнение (17.15) в качестве множителя.
Время пребывания молекулы в неподвижной фазе ts связано с глубиной пор d и коэффициентом диффузии D уравнением Эйнштейна:
d2 = 2Dts. (17.16)
При подстановке уравнения (17.16) в (17.15) получаем
„ КД1 - Rr)Ud2
И =-------D-----'
Чем больше d, тем больше Н и, следовательно, меньше эффективность колонки. Большая глубина пор у адсорбентов классической жидкостной адсорбционной хроматографии была одной из основных причин ее низкой эффективности.
В современной хроматографии широко применяются поверхностно-пористые адсорбенты (ППА). Это твердые, не обладающие пористостью сферические зерна, на поверхность которых нанесен тонкий слой адсорбента с высокой пористостью толщиной примерно 1 мкм. У этих адсорбентов нет глубоких пор, что приводит к уменьшению Н и значительному увеличению эффек
339
тивности колонки. Применение поверхностно-пористых адсорбентов позволило также значительно повысить скорость жидкостной адсорбционной хроматографии.
В последнее время большой интерес вызывает жидкостная хроматография при высоких давлениях, позволяющая проводить сложные разделения.
17.8.2. Основные узлы приборов жидкостной хроматографии
Как и в газовой, в жидкостной адсорбционной хроматографии основными узлами хроматографической установки являются дозатор, колонка и детектор.
Колонки и дозаторы. Выбор оптимальных длины и внутреннего диаметра колонки является компромиссным, так как приходится учитывать селективность и эффективность колонки, длительность анализа, удобство работы и другие факторы. Учитывается, например, что критерий разделения увеличивается пропорционально лишь корню квадратному из длины колонки, а продолжительность анализа пропорциональна ее длине. Уменьшение диаметра колонки вызывает трудности ее заполнения, а увеличение диаметра приводит к уменьшению скорости движения подвижной фазы. В жидкостной адсорбционной хроматографии используются колонки длиной от 15...20 см до 1,5...2,0 м и не более Юме внутренним диаметром 1...6 мм и не более 12 мм. Изготовляются колонки из толстостенного стекла или нержавеющей стали и плотно и равномерно заполняются адсорбентом. При плотном заполнении создаются условия для более постоянной скорости потока жидкости.
К адсорбентам в современной жидкостной адсорбционной хроматографии предъявляются требования не только селективности и эффективности, но и высокой скорости хроматографирования, что определяется, главным образом, структурой поверхности. Отличными структурными свойствами обладают поверхностнопористые адсорбенты (ППА), у которых большая механическая прочность и отсутствуют глубокие поры. В качестве активного слоя на поверхность стеклянных шариков в ППА наносят силикагель, оксид алюминия или некоторые полимеры. Для современной высокоскоростной жидкостной адсорбционной хроматографии это наиболее подходящий адсорбент. Применяют также объемно-пористые мелкозернистые адсорбенты на основе силикагеля, алюмогеля и других веществ.
Определенные требования предъявляются к подвижной фазе — растворителю. Растворитель в жидкостной адсорбционной хроматографии должен хорошо растворять все компоненты анализируемой смеси, обладать химической инертностью по отношению к растворенным веществам, адсорбенту и кислороду воздуха, быть маловязким, не содержать примесей, не вызывать отклонений в работе детектора и быть доступным.
340
Хотя изотермы адсорбции растворенных веществ, как и газов, описываются одним и тем же уравнением (17.1), все же процесс адсорбции из раствора осложняется участием растворителя. Подвижная фаза часто принимает участие в адсорбционном процессе и, таким образом, может влиять на селективность колонки. Эта особенность жидкостной адсорбционной хроматографии очень важна, так как открывает еще одну возможность регулирования хроматографического процесса и выбора оптимальных условий разделения.
Для элюирования смеси обычно применяют не индивидуальные растворители, а раствор одного или нескольких веществ в растворителе, который сам адсорбируется слабо, введенные же вещества адсорбируются сильнее нескольких, а возможно и всех компонентов анализируемой смеси. Состав подвижной фазы можно изменять так, что ее вытесняющая способность будет непрерывно возрастать. Это градиентная хроматография.
Элюирующую способность растворителей при адсорбции на неорганических веществах часто оценивают по шкале Гильдебранда, основанной на энергии поляризации растворителей. Составленный по возрастанию энергии поляризации так называемый элюотропный ряд растворителей содержит растворители в порядке возрастания их элюирующей способности. Например, для оксида алюминия элюотропный ряд имеет вид: бензол < хлороформ < ацетон ~ диоксан < <ацетонитрилОтанол<;метанол. Для неполярных адсорбентов (полиамиды, активированные угли и др.) порядок растворителей меняется. До подачи в систему растворитель подвергают дегазации, так как выделение пузырьков газа при повышении температуры в колонке или детекторе нежелательно. Дегазация производится нагреванием или вакуумированием.
Процесс жидкостной адсорбционной хромографии идет под высоким давлением. Ввод пробы в колонку, в основном, производится с помощью шприца через самоуплотняющуюся резиновую прокладку и с помощью кранов. С по.мощью шприца объем пробы легко регулируется и проба может быть подана непосредственно на насадку, однако при высоких давлениях метод становится непригодным из за неплотностей в поршне шприца. Система с кранами позволяет работать при высоких давлениях и может быть автоматизирована.
Детекторы. Для определения концентрации вещества на выходе из хроматографической колонки можно или последовательно отбирать отдельные пробы и их затем анализировать, или проводить непрерывный анализ. Методика непрерывного анализа с автоматической записью концентраций имеет бесспорные достоинства. Создание чувствительных детекторов непрерывного действия в значительной степени обусловило современный уровень жидкостной адсорбционной хроматографии и ее успехи в разделении сложных многокомпонентных смесей. Практическое применение нашли детекторы, реагирующие на свойства раствора, раство
341
ренного вещества и на свойство вещества после удаления растворителя.
Рефрактометрический детектор непрерывно измеряет разность показателей преломления между чистым растворителем и раствором после прохождения колонки. Чувствительность детектора достигает 3 мкг/мл. Он универсален, однако для получения надежных данных необходимо достаточно тонкое термостатирование (±0,001 °C).
Спектрометрические детекторы основаны на применении закона Бугера — Ламберта — Бера. Обычно используется светопо-глощение в ультрафиолетовом участке спектра, реже в инфракрасном.
В детекторах транспортного типа раствор после хроматографической колонки попадает на непрерывно движущуюся транспортную ленту, которая подается в печь, где происходит испарение элюента. Остаток на ленте переносится в реактор, где превращается в летучее соединение, которое далее анализируется методами газовой хроматографии.
17.8.3. Качественный и количественный анализ
В жидкостной адсорбционной хроматографии, как и в газовой, идентификация веществ производится по характеристикам удержания, а количественный анализ основан на измерении высоты или площади хроматографического пика. Используется также анализ фракций раствора после хроматографической колонки различными химическими или физико-химическими методами.
Жидкостная адсорбционная хроматография часто применяется в органической химии: в технологии и анализе. Этим методом весьма успешно изучают, например, состав нефти, керосина, углеводородов, эффективно разделяют транс- и qwc-изомеры, алкалоиды и т. д. Особенно большую роль она сыграла в разработке методов разделения, анализа и исследования нелетучих и нестабильных соединений. Очень эффективно применение жидкостной хроматографии при высоком давлении для разделения неполярных соединений и соединений со'средней полярностью.
17.9. ТОНКОСЛОЙНАЯ ХРОМАТОГРАФИЯ
Метод тонкослойной хроматографии (ТСХ), получивший в настоящее время широкое распространение, был разработан Н. А. Измайловым и М. С. Шрайбер еще в 1938 г.
В методе ТСХ неподвижная твердая фаза тонким слоем наносится на стеклянную, металлическую или пластмассовую пла стинку. В 2...3 см от края пластинки на стартовую линию вносят пробу анализируемой жидкости и край пластинки погружают в растворитель, который действует как подвижная фаза жидкостной адсорбционной хроматографии. Под действием капилляр
342
ных сил растворитель движется вдоль слоя сорбента и с разной скоростью переносит компоненты смеси, что приводит к их пространственному разделению. Диффузия в тонком слое происходит в продольном и поперечном направлениях, поэтому процесс следует рассматривать как двумерный.
17.9.1. Основные характеристики ТСХ
Схема ТСХ и экспериментально определяемые величины, которые характеризуют этот процесс, приведены на рис. 17.8.
Сорбционные свойства системы в ТСХ характеризуются п о-движностью Rf, которая рассчитывается из экспериментальных данных (рис. 17.8) по уравнению
Ri=
х.
Xf '
(17.17)
где Xi — расстояние от стартовой линии до центра зоны: X/—расстояние, пройденное за это же время растворителем.
Строго говоря, при определении подвижности вместо расстояний Л, и Xf следовало бы учитывать скорость движения соответствующих веществ в тонком слое, так как в теории ТСХ принято считать, что скорость движения центра зоны хроматографируемого вещества составляет строго определенную долю скорости движения растворителя. Однако измерение этих величин затруднительно, а значения Rf, рассчитанные по соотношению (17.17), также характеризуют подвижность. По смыслу определения Rf как свойство, характерное для данной системы, не должно зависеть от концентрации и других факторов. Опыт показывает, однако, что воспроизводимость и постоянство значений Rf не всегда достаточны, особенно при анализе неорганических ионов. На Rf влияет качество и активность сорбента, его влажность, толщина слоя, качество растворителя и другие факторы, не все
гда поддающиеся достаточному контролю. На практике часто пользуются относительной величиной
где /?f,CT также рассчитывается по уравнению (17.17).
Стандартное вещество (свидетель) в том же растворителе наносится на стартовую линию рядом с анализируемой пробой и, таким образом, хроматографируется в тех же условиях.
Число теоретических тарелок п в методе ТСХ рассчитывается по соотношению ,
Рис. 17.8. ТСХ-схема;
а — а — стартовая линия; b — b — граница фронта растворителя в конце опыта; заштрихованные пятна характеризуют положение компонентов в тонком слое в конце опыта
343
Значения /„ и /п определяются в соответствии с рис. 17.8.
Тогда высота И, эквивалентная теоретической тарелке, будет равна
П 16/н
Коэффициент разделения в тонком слое Kf связан с числом теоретических тарелок и подвижностями Rf уравнением
iz________ R> [ — «г ,/й-
где R[,xi> Rf,xi— подвижности соседних компонентов смеси.
Теоретический анализ показывает, что при небольших значениях Rf и уменьшении длительности анализа размывание зоны вещества на сорбенте бывает максимальным, следовательно, концентрация вещества будет максимальна и чувствительность анализа увеличится. Уменьшение диаметра зерна в тонком слое приводит к увеличению продолжительности анализа и усиливает диффузное размывание.
17.9.2. Основные элементы установок ТСХ
Подложки для сорбента (пластинки) обычно изготовляют из стекла, алюминиевой фольги или полиэфирной пленки. Одно из достоинств пленки состоит в том, что она прозрачна примерно до 320 нм и это позволяет проводить прямое фотометрирование многих веществ непосредственно в слое.
Сорбент может быть нанесен на подложку в виде пасты (закрепленный слой), тонкого порошка (незакрепленный слой) или быть приготовленным при обжиге силикагеля на стеклянной пластине.
В качестве сорбента, в ТСХ применяют силикагели, оксид алюминия, крахмал, целлюлозу и некоторые другие вещества с высокой адсорбционной способностью. Для характеристики сорбционной активности оксида алюминия часто используется шкала Брокмана, составленная по Rf стандартного набора красителей. Эффективно применяются в ТСХ в качестве сорбентов жидкие иониты и другие вещества, обладающие ионообменными свойствами и свойствами молекулярных сит (например, сефадексы)
Выбор растворителя зависит от природы сорбента и свойств анализируемых соединений. Часто применяют смеси растворителей из двух или трех компонентов. Например, при хроматографировании аминокислот используют смесь н-бутанола с уксусной кислотой и водой, при анализе неогранических ионов — водные буферные растворы, создающие постоянное значение pH.
В. восходящей хроматографии растворитель поднимается снизу вверх под действием капиллярных сил, в нисходящей — растворитель передвигается по слою вниз
344
под действием и капиллярных и гравитационных сил. Горизонтальная хроматография выполняется в виде круговой и со свободным испарением растворителя. В круговой хроматографии в центр горизонтально установленной пластинки вносят каплю анализируемой смеси и непрерывно подают растворитель, который под действием капиллярных сил движется в радиальном направлении от центра. Компоненты смеси располагаются в слое в виде концентрических колец.
По окончании хроматографирования непроточным методом зоны на хроматограмме проявляют химическим или физическим способом. При химическом способе пластинку опрыскивают раствором реактива, взаимодействующего с компонентами смеси. При обработке, например, парами иода четко проявляются непредельные соединения. В физических способах проявления используется способность некоторых веществ флуоресцировать под действием ультрафиолетового излучения, часто при добавлении флуоресцирующего индикатора, взаимодействующего с компонентами смеси. Вещества, обладающие радиоактивным излучением, обнаруживаются методом радиоавтографии с помощью различных фотоматериалов (бумаги или пленки). После проявления хроматограммы приступают к идентификации веществ и дальнейшему анализу.
17.9.3. Качественный анализ
Проще всего идентификация вещества может быть сделана, если пятно определяемого вещества имеет характерную окраску или определяемое вещество образует на хроматограмме окрашенное пятно под действием специального реактива. Однако число таких веществ, особенно органических, невелико, поэтому метод имеет ограниченное применение.
Наиболее общий подход к качественному анализу основан на значениях Rf. Хроматографическая подвижность является чувствительной характеристикой вещества, однако она существенно зависит от условий определения. Эта трудность преодолевается путем проведения опыта в строго фиксированных стандартных условиях, которые регламентируют размер пластин, толщину слоя сорбента, объем пробы, длину пути фронта растворителя и другие факторы. При соблюдении стандартных условий получаются воспроизводимые значения Rf, которые можно использовать в аналитических целях при сравнении с табличными, если они получены в тех же условиях опыта.
Самым надежным является метод свидетелей, когда на стартовую линию рядом с пробой наносятся индивидуальные вещества, соответствующие предполагаемым компонентам смеси. Влияние различных факторов на все вещества будет одинаковым, поэтому совпадение R; компонента пробы и одного из свидетелей дает основания для отождествления веществ с учетом возможных наложений. Несовпадение R; интерпретируется более одно
345
значно: оно указывает на отсутствие в пробе соответствующего компонента.
Интересные результаты получаются при сочетании ТСХ с другими методами. При сочетании ТСХ с газовой хроматографией пластинка становится своеобразным детектором. Выходящий из колонки газ направляется на стартовую линию пластинки и затем хроматографируется по методике ТСХ выбранным растворителем. Анализ тонкослойных хроматограмм позволяет независимым методом идентифицировать компоненты смеси, что увеличивает надежность анализа. Хроматографирование вещества методом ТСХ после прохождения газовой колонки может дать дополнительную информацию о составе смеси, в частности о компонентах, разделение которых методом газовой хроматографии было неполным. Сочетание ТСХ и газовой хроматографии позволяет также установить, все ли компоненты смеси вымываются из колонки, происходят ли химические изменения при хроматографировании, и решить некоторые другие вопросы.
Сочетание ТСХ с электрофорезом расширяет возможности разделения, в частности, неорганических ионов и значительно ускоряет процесс разделения. ТСХ комбинируется также с экстракцией и другими методами анализа.
17.9.4. Количественный анализ
Количественные определения в ТСХ могут быть сделаны или непосредственно на пластинке, или после удаления вещества с пластинки. При непосредственном определении на пластинке измеряют тем или иным методом площадь пятна (например, с помощью миллиметровой кальки) и по заранее построенному градуировочному графику находят количество вещества.
Применяют также прямое спектрофометрирование пластинки с помощью спектроденситометров. Для количественных расчетов также предварительно строят градуировочный график, используя оптическую плотность в центре пятна.
Наиболее точным считается метод, в котором вещество после разделения удаляется с пластинки и анализируется спектрофотометрическим или иным методом. Удаление вещества с пластинки обычно производят механическим путем, хотя иногда применяют вымывание подходящим растворителем.
В настоящее время ТСХ является одним из важных методов аналитической химии. Это непревзойденный метод анализа сложных смесей. Он прост по методике выполнения и аппаратуре, экспрессен, не требует для анализа больших количеств веществ.
17.10. ЖИДКОСТНО-ЖИДКОСТНАЯ РАСПРЕДЕЛИТЕЛЬНАЯ ХРОМАТОГРАФИЯ
Жидкостно-жидкостная хроматография по сути близка к газожидкостной хроматографии. На твердый носитель также наносится пленка жидкой фазы и через колонку, наполненную таким
346
сорбентом, пропускают жидкий раствор. Этот вид хроматографии называют жидкостно-жидкостной распределительной хроматографией или просто распределительной хроматографией. Жидкость, нанесенную на носитель, называют неподвижной жидкой фазой, а растворитель, передвигающийся через носитель, — подвижной жидкой фазой. Жидкостно-жидкостная хроматография может проводиться в колонке (колоночный вариант) и на бумаге (бумажная хроматография, или хроматография на бумаге).
17.10.1. Колоночный вариант
Разделение смеси веществ в жидкостно-жидкостной хроматографии основываются на различии коэффициентов распределения вещества между несмешивающимися растворителями. Коэффициент распределения вещества равен
К п. н- £ п/ С н,
где сп и ск — концентрация вещества в подвижной и неподвижной фазах.
Для членов одного гомологического ряда установлены некоторые закономерности в величинах Кпн. Известна, например, зависимость Кп и в данном гомологическом ряду от числа атомов углерода.
Поиск несмешивающихся фаз, обеспечивающих разделение, обычно производится эмпирически на основе экспериментальных данных. Широкое применение в жидкостно-жидкостной хроматографии получили тройные системы, состоящие из двух несмешивающихся растворителей и третьего, растворимого в обеих фазах. Такие системы позволяют получать набор несмешивающихся фаз различной селективности. В качестве примера можно привести систему из несмешивающихся между собой гептана и воды, в которую введен этанол, растворяющийся в обоих растворителях.
Хотя в качестве подвижной и неподвижной фаз выбираются растворители, не смешивающиеся между собой, все же во многих системах наблюдается некоторая взаимная растворимость. Чтобы предотвратить процессы взаимного растворения жидкостей в ходе хроматографирования, подвижную жидкую фазу предварительно насыщают неподвижной. Для сохранения неизменного состава фаз применяют также метод химического закрепления неподвижной фазы на сорбенте. При этом используют взаимодействие растворителя с группами ОН- на поверхности носителя. Адсорбенты с закрепленной на их поверхности жидкой фазой выпускаются промышленностью.
Эффективность колонки связана с вязкостью, коэффициентом диффузии и другими физическими свойствами жидкостей. С уменьшением вязкости подвижной фазы сокращается продолжительность анализа, но с увеличением вязкости несколько возрастает эффективность. В практике обычно используют маловяз
347
кие растворители, так как возрастание эффективности колонок при увеличении вязкости не очень велико.
Носитель неподвижной фазы должен обладать достаточно развитой поверхностью, быть химически инертным, прочно удерживать на своей поверхности жидкую фазу и не растворяться в применяемых растворителях. В качестве носителей используют вещества различной химической природы: гидрофильные носители — силикагель, целлюлоза и др. и гидрофобные — фторопласт, тефлон и другие полимеры. Успешно развивается применение в жидкостно-жидкостной распределительной хроматографии высокого давления.
17.10.2. Распределительная хроматография на бумаге
Кроме обычных носителей, используемых для заполнения колонок, в распределительной хроматографии применяют специфический носитель, позволяющий обходиться вообще без колонки. Таким носителем является специальная хроматографическая бумага, а методика, основанная на ее применении, получила название распределительной хроматографии на бумаге или распределительной бумажной хроматографии. Во многом она сходна с хроматографией в тонком слое (ТСХ).
Важной характеристикой в бумажной распределительной хроматографии, так же как и в ТСХ, является где х — сме-
щение зоны компонента; Xf — смещение фронта растворителя. Методика определения Rf в бумажной хроматографии не отличается от соответствующей методики в ТСХ, основанной на измерениях в соответствии с рис. 17.8. В начальный момент времени хроматографируемая проба наносится на начальную (стартовую) линию бумажной полоски и подвергается действию подвижной фазы (растворителя). Если компоненты окрашены, через некоторое время на хроматограмме можно будет видеть отдельные цветные пятна. Первый компонент будет иметь Rfi=xi/Xf, второй — Rf2=X2/Xf и т. д.
При идеальных условиях коэффициент Rf определяется только природой вещества, параметрами бумаги и свойствами растворителей, но не зависит от концентрации вещества и присутствия других компонентов. В действительности коэффициент R; в некоторой степени оказался зависящим от этих факторов и техники эксперимента. Тем не менее при достаточном постоянстве условий опыта и не слишком больших колебаниях в составе смеси эти коэффициенты остаются вполне постоянными и могут быть использованы для идентификации компонентов смеси.
Хроматографическая бумага должна быть химически чистой, нейтральной, инертной по отношению к компонентам раствора и подвижному растворителю и быть однородной по плотности. Имеют значение также такие свойства, как структура молекул целлюлозы в бумаге, набухаемость, ориентация волокна и дру
348
гие, влияющие на скорость движения растворителя и на иные характеристики процесса. В СССР выпускается четыре сорта специальной бумаги для хроматографирования, различающиеся по номерам (№ 1, № 2 и т. д.).
В атмосфере водяных паров бумага поглощает значительное количество влаги (до 20...25 % своей массы), поэтому когда неподвижной жидкой фазой является вода, никакого дополнительного увлажнения бумаги не делают. При выборе в качестве неподвижной фазы некоторых органических веществ, гидрофильную бумагу превращают в гидрофобную, пропитывая ее растворами различных гидрофобных веществ (парафина, растительного масла и др.).
В выбранных растворителях компоненты пробы должны иметь разную растворимость, иначе разделения вообще не произойдет. В растворителе, являющемся подвижной фазой, растворимость каждого компонента должна быть меньшей, чем в растворителе неподвижной фазы, но все же составлять вполне заметное значение. Это ограничение связано с тем, что если растворимость вещества будет очень велика, вещество будет двигаться вместе с фронтом растворителя, а если растворимость будет очень мала, вещество останется на начальной линии.
Для разделения водорастворимых веществ в качестве подвижной фазы обычно берут органический растворитель, а в качестве неподвижной — воду. Если вещество растворимо в орга-ническйх растворителях, вода используется уже в качестве подвижной фазы, а органический растворитель является неподвижной фазой. Это так называемый метод обращенных фаз.
К растворителям обычно предъявляются следующие требования: растворители подвижной и неподвижной фаз не должны смешиваться, состав растворителя в процессе хроматографирования не должен изменяться, растворители должны легко удаляться с бумаги, быть недефицитными и безвредными для человека.
Индивидуальные растворители в распределительной хроматографии используют относительно редко. Чаще для этой цели употребляют смеси веществ, например бутилового или амилового спирта с метиловым или этиловым, насыщенные водные растворы фенола, крезола и др., смеси бутилового спирта с уксусной кислотой, аммиаком и т. д. Применение различных смесей растворителей позволяет плавно изменять Rf и тем самым создавать наиболее благоприятные условия разделения.
По технике выполнения различают следующие виды бумажной хроматографии: одномерную, двумерную, круговую и электрофоретическую. Для получения двумерных хроматограмм хроматографирование производят дважды во взаимно противоположных направлениях: после обработки пробы одним растворителем хроматограмму поворачивают иа 90° и хроматографируют вторично уже другим растворителем.
349
Такая методика позволяет производить более тонкие разделения компонентов смеси.
Весьма эффективным оказалось сочетание хроматографии и электрофореза, т. е. получение электрофоретических хроматограмм на бумаге. Воздействие электрического поля можно использовать или одновременно с хроматографированием, или последовательно, т. е. сначала провести электрофорез, а затем хроматографирование.
Качественный состав пробы в методе бумажной распределительной хроматографии так же, как и в ТСХ, может быть установлен или по специфической окраске отдельных пятен на хроматограмме, или по числовому значению Rf каждого компонента.
Количественные определения в распределительной, так же как и в тонкослойной хроматографии, выполняются или по хроматографическим характеристикам (площади пятна на хроматограмме и интенсивности его окраски), или по методу вымывания. Нередко хроматограмму разрезают на отдельные части по числу пятен, каждое пятно обрабатывают соответствующим экстрагентом и определяют количество экстрагированного вещества любым подходящим методом: фотометрическим, полярографическим и т. д.
Методики, основанные на использовании хроматографических характеристик, довольно многообразны. Количество вещества можно определить методом шкалы, когда на одной и той же хроматограмме получают пятно раствора с неизвестной концентрацией и пятна нескольких стандартов. Концентрацию исследуемого раствора находят прямым сравнением площади пятна и интенсивности его окраски у исследуемого и у стандартного растворов.
Более точным является метод, основанный на применении градуировочного графика S — Igc, где S — площадь пятна, с — концентрация. В указанных координатах график линеен. Используется также измерение интенсивности окраски пятна, которая пропорциональна концентрации. Методами распределительной жидкостной хроматографии успешно анализируют смеси катионов в неорганическом качественном анализе, смеси аминокислот и других органических кислот, смеси красителей и т. д.
17.11. ГЕЛЬ-ХРОМАТОГРАФИЯ
Это совершенно своеобразный вид хроматографии, основанный на использовании различия в размерах молекул. Его называют также гель-фильтрацией или ситовой хроматографией. Неподвижной фазой в гель-хроматогпафии является растворитель, находящийся в порах геля, а подвижной — сам растворитель, т. е. и подвижную и неподвижную фазы составляет одно и то же вещество или одна и та же смесь вещества. Гель готовят на основе, например, декстрана, полиакриламида или других природных и синтетических соединений.
350
В процессе гель-хроматографирования могут быть отделены крупные молекулы, которые гелем не сорбируются, так как их размеры превышают размеры пор, от мелких, которые проникают в поры, а затем могут быть элюированы. Проводятся и более тонкие разделения, так как размеры пор можно регулировать, изменяя, например, состав растворителя и, как следствие, набу-хаемость геля. Гель-хроматография может быть выполнена в колоночном варианте и в тонкослойном.
Применяемые на практике гели обычно подразделяют на мягкие, полужесткие и жесткие. Мягкими гелями являются высокомолекулярные органические соединения с незначительным число поперечных связей. Фактор емкости, равный отношению объема растворителя внутри геля к его объему вне геля, у них равен 3. При набухании они значительно увеличивают собственный объем. Это сефадекс ы или декстрановые гели, агарочные гели, крахмал и др. Они применяются для разделения смесей низкомолекулярных веществ, часто в тонкослойном варианте. Хроматографирование на мягких гелях называют гель-фильтрацией.
Полужесткие гели получают путем полимеризации. Большое распространение получили стирогели — продукты сополимеризации стирола и дивинилбензола с большим числом поперечных связей. Фактор емкости полужестких гелей лежит в пределах 0,8...1,2, их объем при набухании увеличивается не очень значительно (в 1,2...1,8 раза). Хроматографирование на полужестких гелях называют гель-проникающей хроматографией.
К жестким гелям относят силикагели и часто пористые стекла, хотя они и не являются гелями. Жесткие гели имеют небольшой фактор емкости (0,8...1,1) и фиксированный размер пор. Эти материалы используют в гель-хроматографии при высоком давлении.
Растворители гель-хроматографии должны растворять все компоненты смеси, смачивать поверхность геля и не адсорбироваться на ней.
Практическое применение гель-хроматографии связано, главным образом, с разделением смеси высокомолекулярных соединений, хотя нередко они используются для разделения и низкомолекулярных, так как разделение этим методом возможно при комнатной температуре.
17.12. ИОНООБМЕННАЯ ХРОМАТОГРАФИЯ
Ионообменная хроматография основана на обратимом стехиометрическом обмене ионов, находящихся в растворе, на ионы, входящие в состав ионообменника. Хотя явление, известное в настоящее время как ионный обмен, фактически было известно с середины прошлого века, широкое применение ионообменных процессов в практике началось после создания с и н-
351
тетических ионообменников — так называемых ионообменных смол или ионитов. Используемые ранее естественные ионообменники (различные алюмосиликаты и другие соединения) не обладали достаточной воспроизводимостью свойств, не были химически устойчивыми и т. д. и поэтому существенного практического значения не имели.
Применяемые в настоящее время синтетические ионообменники лишены многих недостатков, присущих естественным ионо-обменникам, и обладают рядом важных достоинств- они имеют высокую обменную емкость и воспроизводимые ионообменные и другие свойства, устойчивы к действию кислот и оснований, не разрушаются в присутствии многих окислителей и восстановителей и т. д. Обычно синтетический ионообменник представляет собой высокополимер, например поперечно-сшитый полистирол, содержащий различные функциональные группы, которые и определяют наиболее характерные свойства смол. Известны также синтетические неорганические иониты, например различные пермутиты, активированный оксид алюминия, гели на основе соединений железа или соединений циркония и т. д. Однако органические ионообменные смолы имеют намного большее практические применение.
17.12.1. Типы ионообменных смол
В зависимости от знака разряда функциональных групп ионообменные смолы являются катионитами или анионитами. Катиониты содержат кислотные функциональные группы SO3; —СОО-; —РОз; —NfCHzCOz)], поэтому каркас катионита, несущий фиксированные отрицательные заряды, заряжен отрицательно. Отрицательные заряды каркаса компенсируются положительными зарядами противоионов, так что в целом катионит остается электронейтральным. Однако противоионы, в данном случае катионы, в отличие от функциональных групп каркаса обладают подвижностью и могут переходить в раствор в обмен на эквивалентное количество ионов из раствора. Этот обмен приводит к установлению подвижного равновесия между ионами, находящимися в фазе смолы, и ионами в растворителе. Наиболее распространенными катионитами являются сульфокислоты, образованные сульфированными продуктами сополимеризации стирола и дивинилбензола. Это отечественные смолы КУ-2, СДВ-3 и др., иностранные дауэкс-50, амберлит IR-120 и др. Сульфокатиониты характеризуются высокой химической стойкостью и механической прочностью, большой скоростью установления ионообменного равновесия.
Функциональными группами каркаса анионитов являются чет вертичные —NR£ третичные —NRzH+ или первичные — NH^ аммониевые, пиридиновые или другие основания, а в качестве подвижных противоионов выступают анионы. Анионообменные смолы получаются также путем проведения реакций полимери-
352
зации или поликонденсации с использованием различных аминосоединений (фенилендиамина, полиэтиленполиамина и т. п.), формальдегида и др. Так были получены аниониты АН-1, АН-2Ф, амберлит. Широкое распространение получил полифункциональ-ный анионит ЭДЭ-10П, содержащий амины различной степени замещения, включая четвертичные. Амфотерные иониты или амфолиты способны осуществлять одновременный обмен катионов и анионов. Биполярным, или амфотерным, ионитом является продукт поликонденсации диэтилентриамина, фенола и формальдегида, так как в его состав наряду с аминогруппами входят слабокислотные фенольные группы. Амфотерными и комплексообразующими свойствами обладают смолы, содержащие остатки комплексонов, например ЭДТА.
Важной характеристикой ионообменника является его о б-менная емкость, определяемая в первом приближении числом функциональных групп каркаса и степенью их ионизации при данном pH раствора. Обменную емкость ионита численно можно выразить количеством молей эквивалента противоиона на единицу массы или объема смолы. В аналитической химии емкость ионита обычно выражают количеством молей эквивалента обменивающегося иона на 1 г сухой смолы в Н+-форме для катионита и С1-- или ОН_-форме для анионита. Оговорка относительно сухой смолы необходима, так как в контакте с водой смола набухает в 1,5—2 раза, а некоторые виды смол — в 5 раз и более. Величина обменной емкости характеризуется несколькими (3...5 до 10) моль экв иона на 1 г смолы. Емкость, найденную в статических условиях, когда навеску смолы помещают в раствор насыщающего иона достаточной концентрации и выдерживают при встряхивании до полного насыщения, называют статической обменной емкостью (СОЕ). Величину емкости, полученную в динамических условиях при пропускании насыщающего раствора через колонку с ионитом, называют динамической обменной емкостью (ДОЕ). Эта емкость ионита, определяемая по первому появлению насыщающего иона в вытекающем растворе. Полная обменная емкость (ПДОЕ) находится по полному насыщению ионита данным ионом.
17.12.2. Ионообменное равновесие
Взаимодействие ионообменной смолы с раствором электролита включает несколько сложных процессов, наиболее важными из которых являются собственно ионный обмен, физическая абсорбция ионов и молекул на смоле и набухание смолы за счет поглощения растворителя и проникновения электролита внутрь смолы.
Процесс собственно ионного обмена протекает стехиометриче-ски. Если, например, катионит в водородной форме RH ввести в раствор, содержащий ионы Са2+, в системе установится равновесие:
12-1533
353
2HR-I-С а2+= Ca R2-I-2Н+
т. е. в растворе появятся ионы водорода, а эквивалентное количество ионов Са2+ будет поглощено катионитом.
Аналогичный процесс обмена имеет место при взаимодействии раствора, содержащего, например, хлорид, с анионитом ROli:
ROH + Cr = RCI4-OH
Распределение каждого иона между смолой и раствором можно охарактеризовать коэффициентом распределения Р:
D _ [CaR2] j D _ [RC1]
^Ca'+ [Ca2+] ИЛИ [Cl“] ’
Концентрация иона в растворе обычно выражается в моль/л, а в Лазе ионита — молярными долями.
Более строго это равновесие характеризуется константой ионного обмена:
дО Дц'ДСаКл
где а — активности частиц.
Уравнение ионного обмена нередко записывают так же, как
Н+ + */гСа2+ = 72Са2+ + Н+ где горизонтальная черта показывает принадлежность иона к фазе ионита.
По Никольскому константа ионного обмена имеет вид К________________________а'с^,
Активности ионов в фазе смолы выражаются в ммоль экв/г смолы.
Во многих случаях оказываются достаточными концентрационные константы равновесия:
к + _ [Н+1 [CaRs]
Лн /Са [Ca2+][HR]2’
Константы обмена связаны с коэффициентом распределения соотношением
TS _ Р C;i?'
ЛН /Са у,2+ •
Хотя количественный подход к ионообменным равновесиям на основании закона действующих масс, когда константы равновесия выражены через концентрации, а не через активности, является приближенным, все же он оказывается весьма полезным. На основе так называемых концентрационных констант ионного
354
обмена могут быть, например, построены ряды сродства катионов к данной смоле, позволяющие предвидеть возможности ионообменных разделений.
К настоящему времени установлено несколько эмпирических закономерностей, связывающих константы ионного обмена со свойствами ионов. Так, в частности, найдено, что с ростом заряда сродство ионов к смоле увеличивается и, например, в ряду Na+<Ca2+<Al3+<Th4+ оно возрастает.
С повышением температуры избирательность поглощения несколько уменьшается, хотя этот эффект не очень велик.
Введение в раствор веществ, способных образовывать комплексные соединения с присутствующими ионами, сдвигает равновесие ионного обмена, так как в результате комплексообразования уменьшается равновесная концентрация иона в растворе.
17.13. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Методы ионообменной хроматографии используются преимущественно для разделения ионов. Количественные определения компонентов после разделения могут быть выполнены любым подходящим методом.
Простейшая методика ионообменного разделения состоит в поглощении компонентов смеси ионитом и последовательном элюировании каждого компонента подходящим растворителем.
Например, катионы щелочных металлов легко элюируются разбавленным раствором соляной кислоты (0,1 М HCI). Выходная кривая на рис. 17.9 показывает эффективность этого разделения. Как видно, элюирование раствором 0,1 М НС1 позволяет легко разделить Na+ и К+.
В качестве другого примера можно указать на методику разделения ионов циркония (IV) и гафния (IV). Для разделения эти катионы сначала переводят в анионные сульфатные комплексы, которые поглощают анионитом. При последующем элюировании 1 М раствором H2SO4, содержащим сульфат натрия, происходит их полное разделение: сначала вымывается гафний, а затем цирконий.
Применение ионообменной хроматографии к анализу смеси лантаноидов с использованием в качестве элюентов растворов лактата, цитрата, ЭДТА и других привело к разработке эффективного способа разделения этих элементов. На основе полученных данных была предложена и успешно осуществлена на практике технологическая схема промышленной переработки руд лантаноидов.
V (НО,)
Рис 17 9 Элюирование поглощенных на катионите ионов Na+ и К4 раствором 0,1 М НС1
12
355
Известны ионообменные методики даже для разделения изотопов. Например l4N и l5N могут быть разделены на сульфосмоле в виде NHt так как оказалось, что l4NH^ сорбируется хуже и потом вымывается раньше, чем 15NH£
Ионообменные методы применяют для определения суммарного содержания катионов или анионов в растворе и для анализа растворов чистых солей. При пропускании через катионит в Н+-форме раствора, например, соли натрия в результате ионообменного процесса
HR4-Na+ = NaR4-H+ в растворе появляется эквивалентное количество Н+-ионов. Концентрация ионов Н+ в этом растворе может быть определена, например, титрованием и, таким образом, найдена концентрация Na* в исходном растворе.
Большое практическое значение имеет основанный на ионном обмене процесс деминерализации воды. Сущность его заключается в следующем. Солевой раствор или вода, предназначенная для деминерализации, обрабатывается одновременно катионитом в Н+-форме и анионитом в ОН“-форме. В результате обмена на катионите в растворе появятся Н"*-ионы:
HR4-M+=MR4-H+
а в результате обмена на анионите ОН_-ионы:
ROH4-X-= RX4-OH-
Но Н+ и ОН- в растворе взаимодействуют Н+4-ОН_ = Н2О, сдвигая равновесие ионного обмена до полного извлечения ионов из раствора и получения чистой деминерализованной воды. Такая вода используется в лабораториях вместо дистиллированной.
Ионообменные процессы применяются также для переведения в раствор малорастворимых соединений. Для этого взвесь малорастворимой соли MX следует обработать ионитом HR до наступления равновесия:
MX4-HR=MR + H+4-X"
и десорбировать М+ с ионита подходящим растворителем. Возможность растворения будет, очевидно, определяться сродством М к R и растворимостью MX. В настоящее время известны методики растворения с помощью ионного обмена таких осадков, как BaSO4, AgCl и др.
Интенсивно разрабатываются электрохимические аспекты применения ионообменных смол. Специально для электрохимических целей изготавливают так называемые ионообменные мембраны. Их получают в виде листов из ионообменной смолы, поэтому они обладают одновременно и свойствами ионообменника, способного к ионному обмену, и свойствами мембраны, как полупроницаемой перегородки. Однако ионная проницаемость ионооб-
356
менных мембран селективна — мембраны из катионита проницаемы только для катионов, а мембраны из анионита — для анионов. Это открывает широкие возможности их практического использования.
Так, например, с помощью ионообменных мембран можно приготовить растворы чистых NaOH и H2SO4 при электролизе раствора NagSO^ в ячейке,
Рис 17.10 Электролиз раствора
Na2SO4 в ячейке с ионообменными мембранами
разделенной двумя ионообменными мембранами (рис. 17.10).
Катодное пространство отделено от раствора Na2SO4 катионитом MR, пропускающие ионы Na+, но задерживающим ионы SO?-, а анодное пространство отделено мембраной из анионита RA, проницаемой для SO?- и непроницаемой для иона Na+.
При электролизе водного раствора в катодном пространстве
происходит восстановление воды:
2Н2О + 2е- = Н2 + 2ОН-
а в анодном пространстве идет окисление воды: Н2О = 72О2 + 2Н++ 26-
Таким образом в катодном пространстве образуется чистый NaOH, а анодном — чистая H2SO4, а в центральном отделении, в конце концов, остается деминерализованная вода. В принципе, такое устройство может быть использовано для водоочистки или обессоливания морской воды.
Рассмотренные способы применения ионообменных смол, конечно, не исчерпывают всего многообразия, однако они показывают широкие возможности, которые открывает использование ионитов в аналитической химии и технологии.
17.14. ИОННАЯ ХРОМАТОГРАФИЯ
Это один из вариантов разделения на ионитах, характеризующийся более высокоэффективной техникой, чем обычная ионообменная хроматография. В методе ионной хроматографии используются поверхностно-слойные сорбенты с небольшой емкостью (1О-2...1О-1 ммоль экв/г) и небольшим размером частиц (5...50 мкм), повышенное давление на входе в колонку (2.. 5 МПа) и высокочувствительные детекторы с автоматической записью сигнала. Для ионной хроматографии характерны экс-прессность, удобство работы и более высокая разделительная способность.
Детекторы ионообменных разделений должны регистрировать концентрацию анализируемых ионов в элюате в присутствии ионов элюента. Особенно широко в ионной хроматографии исполь
357
зуют кондуктометрический детектор, являющийся универсальным, так как он реагирует на все ионы в растворе. Применяют также селективные детекторы: электрохимические (полярографический, кулонометрический и т. д.) и спектрофотометрический. В кондуктометрическом детекторе должны быть предусмотрены какие-либо средства для компенсации фонового сигнала, который может на 2...3 порядка превышать аналитический сигнал. Фоновый сигнал может быть скомпенсирован с помощью электронной схемы. Элюат можно обработать также на второй колонке, чтобы ослабить электрическую проводимость элюента и усилить проводимость образца.
17.14.1. Методы ионной хроматографии
В ионной хроматографии используют два основных метода: одноколоночный и двухколоночный.
В одноколоночной анионной хроматографии применяют элюенты с низкой электрической проводимостью, например, разбавленные растворы (10“3...10“4 ммоль/л) бензоата или фталата щелочного металла или бензойной кислоты (Ас,н5ог = 32 См-см2/ /моль экв). Электрическая проводимость хлорида, нитрата и многих других анионов существенно превышает электрическую проводимость бензоата или фталата (Х^о3 = 71 См-см2/моль экв), чго вызывает при элюировании увеличение электрической проводимости и на хроматограмме регистрируется соответствующий пик. В качестве элюента используют также разбавленные растворы щелочных гидроксидов. В этом случае регистрируются так называемые отрицательные пики, которые появляются вследствие уменьшения электрической проводимости при замене гидро-ксид-иона в растворе на какой-либо анион.
В катионной хроматографии элюирование проводят разбавленным раствором азотной или другой минеральной кислоты. Здесь также получаются отрицательные пики как результат замены ионов Н+, имеющих высокую подвижность, на какой-либо катион.
В двухколоночном методе используются две колонки: разделительная и компенсационная. Для разделения применяют колонки с малой емкостью: в качестве элюентов при анализе анионов используют гидроксиды щелочных металлов и соли слабых кислот, а при анализе катионов — азотную или другую минеральную кислоту. В анионной хроматографии компенсационная колонка заполняется сильнокислотным катионитом, при пропускании через который щелочной раствор нейтрализуется. В катионной хроматографии компенсационную колонку с той же целью заполняют сильноосновным анионитом. Электрическая проводимость раствора на выходе из компенсационной колонки определяется главным образом ионами анализируемого образца.
358
17.15. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Методами ионной хроматографии определяют очень многие анионы в питьевой и технической воде, в продуктах технологической переработки в пищевой, фармацевтической и других отраслях промышленности. Известны методики определения галогенидов, нитрата, нитрита, сульфата, ацетата и т. д., всего свыше 70 анионов неорганических и органических кислот. Число катионов значительно меньше. Методами ионной хроматографии определяют главным образом катионы щелочных и щелочно-земельных металлов, а также органические катионы замещенных солей аммония. Определение многих других катионов оказывается ненадежным, так как они выпадают в осадок в компенсационной колонке с сильноосновной смолой. Ионная хроматография успешно применяется в анализе объектов окружающей среды (атмосферы, воды и т. д.), в клинических исследованиях и многих отраслях промышленности.
17.16. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА
Хроматография является эффективным методом разделения и анализа сложных по составу газообразных и жидких смесей. Твердые вещества могут быть проанализированы после перевода их в жидкое (растворенное) или газообразное состояние. Качественный и количественный анализ проводится по характеристикам удерживания. Универсальность газовой и газо-жидкостной .хроматографии значительно возрастает при сочетании хроматографического разделения и анализа компонентов масс-спектраль-ным или иным подходящим методом. Жидкостная распределительная хроматография особенно эффективна при разделении веществ, близких по химическим свойствам, например аминокислот.
В органическом и биохимическом анализе большое значение имеет бумажная хроматография — простейший вариант хроматографического метода, обладающий высокой чувствительностью. Более воспроизводимые рузультаты дает тонкослойная хроматография, широко применяемая в анализе лекарств, биохимических проб и различных природных объектов. Ионообменная хроматография ценна как метод разделения сложных смесей ионов и как метод концентрирования микропримесей. Успешно развиваются также новые хроматографические методы, например высокоэффективная жидкостная и ионная хроматография.
J Вопросы
1. В чем сущность методов хроматографии?
2. В чем сущность хроматографического разделения по методу: а) газо-адсорбционной хроматографии; б) газо-жидкостной хроматографии; в) распределительной жидкостной хроматографии; г) осадочной хроматографии; д) тонкослойной хроматогра
359
фии, е) ионообменной хроматографии; ж) молекулярно-ситовой хроматографии?
3. Каковы области применения, достоинства и недостатки методов адсорбционной хроматографии?
4. Какие требования предъявляются к адсорбентам и растворителям? Назовите наиболее распространенные растворители и адсорбенты в жидкостной хроматографии.
5. Какие способы применяют для определения эффективности хроматографических разделений?
6. Каковы области применения, достоинства и недостатки методов газовой хроматографии?
7. Какие устройства используются в качестве дозаторов?
8. Что представляют собой: а) дифференциальные детекторы; б) интегральные детекторы?
9. Каков принцип работы дифференциальных детекторов: а) катарометра; б) термохимического; в) ионизационного (или пламенно-ионизационного); г) селективного (термоионного) ?
10. Какими будут относительные значения сигнала пламенноионизационного детектора при введении эквимолярных количеств следующих соединений: н-бутана; нзо-бутана; н-гексана; н-дека-на; 2,2,5-триметилгептаьа, если для н-бутана сигнал равен 8? Ответ: 8; 8; 12; 20; 20.
И. Каковы области применения, достоинства и недостатки методов газо-жидкостной хроматографии?
12. Какие требования предъявляются к жидкой фазе в газожидкостной хроматографии? Какие вещества используются в качестве жидкой фазы, в качестве твердого носителя?
13. В чем сущность капиллярной газо-жидкостной хромато графин?
14. Дать определения понятий: а) высота хроматографического пика; б) ширина хроматографического пика: в) приведенный удерживаемый объем; г) общий удерживаемый объем.
15. Что такое коэффициент селективности работы колонки? Каково условие количественного разделения двух компонентов смеси3
16. Коэффициент распределения К для вещества А в данной хроматографической колонке больше, чем для вещества В. Какое вещество выйдет из колонки первым?
17. В чем сущность качественного хроматографического анализа по величине удерживаемого объема?
18. В чем сущность методов количественного анализа: а) абсолютной калибровки; б) внутренней нормализации (нормировки) ; в) внутреннего стандарта?
19. В чем сущность ионообменной хроматографии3
20. Как определяется: а) статическая обменная емкость ионита; б) динамическая емкость ионита?
21. Коэффициенты распределения ионов на катионите КУ-2 из 5 М СНзСООН составляют Zn2+ — 3,3; Со2+ — 4,5; Fe3+ — 3; 1п3+ — 3; Ga3+ — 3,3; Pb2+ — 5; Bi3+ — 2. Какие из ионов могут
360
быть выделены в отдельные фракции в результате пропускания через катионит следующих смесей: a) Bi , Fe3+, Pb2+; б) Zn2+ Ga3+, Co2+; в) Fe3+, In3+, Pb2+?
22. В чем сущность распределительной хроматографии на бумаге? Дать определение Rf.
23. Как выполняется качественный анализ методом распределительной жидкостной хроматографии на бумаге: а) смеси катионов; б) смеси органических соединений (аминокислот)?
24. Значения Rf при хроматографическом разделении ионов на бумаге в среде бутанола, насыщенного 2М НС1, составляют: Cd — 0,6; Zn — 0,6; Bi — 0,5; Al — 0,1, Co — 0,1; Ca — 0,0. Какие из ионов не могут быть четко идентифицированы из смеси: a) Zn, Al, Со; б) Cd, Zn, Со; в) Bi, Al, Са?
25. Как выполняется количественный анализ методом распределительной жидкостной хроматографии на бумаге?
26. Каковы области применения, достоинства и недостатки: а) тонкослойной хроматографии; б) осадочной хроматографии; в) ионообменной хроматографии; г) молекулярно-ситовой хроматографии?
ZA Задачи
1. Через колонку, содержащую 5,0 г катионита, пропустили 250,0 мл 0,050 М ZnSCV Вытекающий из колонки раствор собирали порциями по 50,0 мл, в каждой порции определяли содержание ионов цинка и получили следующие значения концентраций (моль/л): I—0,008; II — 0,029; III — 0,038; IV — 0,050; V — 0,050.
Определить полную динамическую емкость катионита (ммоль/г).
Вычисляем количество эквивалента Zn2+, поглощенное катионитом из каждой порции раствора, принимая молярную массу эквивалента равной М (72м2+у
(0,050-0,008)2-50-1000 л _п , 2+. ...
----------iOQO---------=4,20 ммоль (72Zn2+); (I)
(0’050"°100902'50'1000 = 2-Ю ммоль C/2Zn2+); <П)
(0,050^,038)2-50-1000 = j 20 мммь (Щ)
0 ммоль (’/2Zn2+) (IV, V)
Всего в пяти порциях раствора поглощено
4.20 + 2,10 + 1,20 = 7,50 ммоль (‘/2Zn2+). Отсюда динамическая емкость катионита для ионов цинка равна
k = = 1,50 ммоль (’/> Zn2+)/r.
361
2. Определить массовую долю (%) компонентов газовой сме-
си по следующим данным:
Компоненты смеси S, мм2 Пропан 175 Бутан 203 Пентан 182 Циклогексан 35
fe 0,68 0,68 0,69 0,85
Расчеты проводим по методу внутренней нормализации, согласно которому
2 SA
I
где wt — массовая доля i-го компонента в смеси, %, S, — площадь пика i-ro компонента; fe, — поправочный коэффициент, определяемый чувствительностью детектора хроматографа к i-му компоненту.
Найдем приведенную суммарную площадь пиков.
2 S,fe, = 175 • 0,68 + 203 - 0,68 +182 0,69 + 35 0,85 = 412,4. ।
Отсюда массовая доля (%) пропана равна
ю(пропана) = 100=28,6 %.
Аналогично находим массовые доли (%) остальных компонентов смеси: w (бутана) =33,46 %, w (пентана) = 30,46 %, w (циклогексана) = 7,22 %.
При выполнения анализа по методу внутреннего стандарта расчет проводят по формуле
S fe
100 %,
*-^СТ"'СТ
где SCT - площадь пика вещества, введенного в качестве внутреннего стандарта; kCi — его поправочный коэффициент; R — отношение массы внутреннего стандарта к массе анализируемой пробы.
3. К V мл 0,05 н. М(ЫО3)г (/экв = ‘/г) прибавили т г катионита в Н-форме. После установления равновесия концентрация уменьшилась до с'.
Определить статическую обменную емкость катионита (ммоль/г), принимая молярную массу эквивалента Л4(1/2М2+):
Вариант V, мл м m, г с’у моль/л
1 50 Cd 3 0,003
2 75 Ni 5 0,008
3 100 Zn 10 0,006
Ответ: 1) 0,78 ммоль/г; 2) 0,63 ммоль/г; 3) 0,44 ммоль/г.
4. Какая масса кобальта останется в растворе, если через колонку, заполненную m г катионита, пропустили V мл раствора CoSO4 с концентрацией снач. Полная динамическая емкость в условиях разделения равна 1,6 ммоль/г [молярная масса эквива лента равна Af(‘/2M2+)].
362
Вариант т, г V, мл Снач, МОЛь/л
1 5,0 200 0,050
2 10,0 250 0,050
3 10,0 500 0,100
Ответ: 1) 0,29 г/л; 2) 2,53 г/л; 3) 2,00 г/л.
5. Через колонку, заполненную катионитом массой 10 г, пропустили 250,0 мл 0,08 М C11SO4. Выходящие из колонки порции раствора по 50,0 мл титровали 0,1 н. раствором тиосульфата натрия (/экв= 1) и получили следующие результаты:
Порция раствора . . 1
Расход тиосульфата яа титрование, мл 0
2 3 4 5
12,00 25,00 39,20 39,20
Вычислить динамическую емкость катионита по меди если молярная масса эквивалента составляет Af(‘/2M 1,66 ммоль (*/2 Си2+)/г.
'ммоль/г), 2+). Ответ:
6. При определении фурфурола в смеси методом газовой хроматографии площадь его пика 5фУРфУр0ля сравнивали с площадью пика о-ксилола S ксилола, который вводили в качестве стандарта. Для стандартного образца, содержащего 25 % фурфурола, и исследуемого образца получили следующие результаты:
Вариант Стандартный образец Исследуемый образец
S фур фу рол а» мм2 И 2 1 м 6) фурфурола, мм2 5ксилола, мм2
1 11 25 18,5 22
2 15 28 19,5 24
3 21 35 25 32
Принять k равным единице для обоих компонентов. Определить массовую долю (%) фурфурола в исследуемом образце. Ответ: 1) 47,78 %; 2) 37,92 %; 3) 32,55 %.
7. Рассчитать массовую долю (%) компонентов газовой смеси по следующим данным, полученным методом газовой хроматографии:
Вариант 1 Вариант 2 Вариант 3
Газ S, мм2 k Газ S, мм2 k Газ S, мм2 k
Бензол 20 6 0,78 о-Ксилол 16,7 0,84 Бензол 85 1,0
Толуол 22,9 0,79 т-Ксилол 20,3 0,81 Гексан 27 1,1
Этилбензол 30,5 0.82 п-Ксилол 8,5 0,81 Пропилен 34 1,1
Кумол 16,7 0,84 Этилбензол 30,4 0,82 Этанол 11 1,8
Ответ: 1) 21,95; 24,72; 34,17; 19,16 %; 2) 22,52; 26,40; 11,05;
40,03%; 3) 49,45; 17,28; 21,75; 11,52%.
363
8. Реакционную массу после нитрования толуола проанализировали методом газо-жидкостной хроматографии с применением этилбензола в качестве внутреннего стандарта.
Определить массовую долю (%) непрореагировавшего толуола по следующим экспериментальным данным:
Вариант Взято •^толуола» мм2 k 5этилбензола» мм2 k
m(толуола), г m(этилбензола), г
1 12,75 1,25 307 1,01 352 1,02
2 15,26 1,09 108 0,79 158 0,82
3 25,16 1,28 80 0,79 109 0,82
Ответ: 1) 8,47%; 2) 4,70%; 3) 3,60%.
9. Цис-1,2-дихлорэтилен в винилиденхлориде определяли методом газовой хроматографии, используя толуол в качестве внутреннего стандарта, и получили следующие данные для градуировочного графика:
Sx/ST 0,72 0.90 1,08 1,28
w. % 0,5 1,0 1,5 2,0
Рассчитать массовую долю (%) ^ис-1,2-дихлорэтилена в исследуемом образце по следующим данным о пиках определяемого и стандартного вещества (принять k— 1):
Вариант Пик цис-1,2-дихлорэтилен Пик толуола
основание высота основание высота
1 18 35 15 52
2 14 42 18 45
3 12 60 15 50
Ответ: 1) 0,75%; 2) 0,55%; 3) 1,15 %.
10. При определении адипиновой кислоты в продукте гидрокарбоксилирования бутадиена методом бумажной хроматографии полученные пятна, проявленные метиловым красным, вырезали, высушили и взвесили. Для стандартных смесей с различным содержанием адипиновой кислоты получили данные:
Масса кислоты, мкг
Масса бумаги с пятном, мг
5 10 15 20
61 106 146 186
Навеску анализируемого образца m мг растворили в V мл воды и порции полученного раствора по 0,05 мл хроматографировали. Масса полученных пятен составила тъ мг.
Определить массовую долю (%) адипиновой кислоты в анализируемом продукте.
364
Вариант Ш\, мг V, мл та, мг
1 100 10 85
2 150 20 107
3 200 25 165
Ответ: 1) 1,60%; 2) 2,93 %; 3) 4,37 %.
11. Навеску природного продукта массой тг растворили в V мл спирта и 0,05 мл раствора нанесли на бумагу. После хроматографического разделения полученные пятна уридиловой кислоты, рибозо-1,5-дифосфата и адениловой кислоты вырезали, сплавили с КОН и K2S2O8; после растворения плава определили фосфор фотометрически, получив значения оптической плотности Лурнд, Лриб и А аденил соответственно; из стандартного раствора фосфата, содержащего 20 мкгР/мл, приготовили раствор с оптической плотностью Лсг.
Определить массовую долю (%) этих производных в природном продукте.
Вариант т, г V, мл ^урид ^риб ^аденил ^СТ
1 0,560 50 0,350 0,520 0,120 0,730
2 0,780 100 0,450 0,630 0,180 0,850
3 1,050 100 0.580 0,790 0,230 0,950
Ответ: 1) 1,71; 2,54; 0,59%; 2) 2,71; 3,80; 1,09%; 3) 2,33; 3,17; 0,92%.
12. Для определения диоксидифенилметана в пищевых продуктах использовали метод тонкослойной хроматографии. Для стандартных образцов получены следующие результаты:
Концентрация диоксидифенилметана, мкг/0,02 мл . . .
Площадь пятна, мм2 .
5,0 10,0 15,0 35,0
7,94 12,59 15,85 27,10
Для построения градуировочного графика использована зависимость IgS — Igc. Навеску овощей массой m г обработали V мл спирта, который затем упарили до 5,00 мл, Затем 0,02 мл его хроматографировали методом ТСХ и получили пятно площадью S мм2.
Определить концентрацию диоксидифенилметана в овощах (мг/кг)
Вариант т, г . . S, Мм2
1 2 3
250 100 38
26,55 20.42 14,79
Ответ: 1) 34,4 мг/кг; 2) 36,5 мг/кг; 3) 16,48 мг/кг.
365
Глава 18
МАТЕМАТИЧЕСКОЕ ПЛАНИРОВАНИЕ ЭКСПЕРИМЕНТА В АНАЛИТИЧЕСКОМ ХИМИИ
18.1. ОСНОВНЫЕ ПОНЯТИЯ И ОПРЕДЕЛЕНИЯ
Традиционный классический метод изучения сложных химических процессов и определения оптимальных условий проведения реакций состоит в том, что исследуется влияние одного параметра при постоянстве остальных. Например, при выявлении оптимальных условий фотометрического определения обычно изучают влияние концентрации реагентов, pH раствора, времени развития окраски и других факторов на оптическую плотность раствора в некотором спектральном интервале. Изучение влияния каждого фактора при постоянстве остальных требует больших затрат времени и средств, а порядок изучения факторов определяется, в основном, личным опытом и интуицией исследователя. Современные статистические методы пла
нирования эксперимента позволяют отыскивать опти
мальные условия проведения реакции при значительном сокра
щении числа опытов.
Метод анализа, являющийся в данном случае объектом иссле
дования, можно представить в виде «черного ящик а», как это обычно делается в кибернетике (рис. 18.1). Незави
симые величины xi, Х2 и т. д., влияющие на протекание реакции, называются факторами, параметрами или входа-м и. Их можно фиксировать с определенной точностью. Величину у, являющуюся характеристикой реакции, называют выходом, оптимизируемым параметром или ф у н к-ц и е-й отклика.
При разработке, например, фотометрического метода анализа факторами, или параметрами, являются концентрация раствора, его pH, концентрация реагента и др. Функцией отклика или оптимизируемым параметром будет оптическая плотность раствора. Координатное пространство, по осям кото-
рого отложены Xi, Х2 и т. д., называют факторным пространством. Функция отклика в этом пространстве может быть изображена в виде поверхности отклика. При
У
х„
Рис 18 1 Модель объекта исследования в виде «черного ящика»
Xi х2 Хз — входы, факторы или параметры, y = f(xi, х2 х3)— выход, функция отклика или оптимизируемый параметр
геометрической интерпретации поверх-ности отклика обычно пользуются двумерным сечением, т. е. оставляют постоянными все факторы, кроме двух, действие которых изучается в данном сечении.
Поверхность отклика может иметь вид «вершины», «хребта», «оврага», «седла» и т. д.; встречаются и более сложные конструкции.
366
\| Систему уравнений, связывающих г функцию отклика с факторами, назы-1 вают математическим описанием про-I цесса, или математической моделью.
I Математические методы планирования эксперимента дают возможность получить математическую модель процесса даже при отсутствии сведений о его механизме или о физически обоснованных математических зависимостях между факторами и функцией отклика.
Математическую модель процесса можно получить методом полного факторного эксперимента или м е-тодом дробных реплик.
Рис 18.2 Введение кодированных переменных
В методе полного факторного
эксперимента исследуется локальная область факторного пространства вблизи выбранной точки с координатами хоь *02, х->и (рис. 18.2), т. е зависимость функции отклика (параметра оптимизации) от всех факторов во всех возможных комбинациях. В методе дробных реплик проводится только некоторая часть полного факторного эксперимента.
Начало координат факторного пространства перенесем в выбранную точку, окрестности которой исследуются, и введем новую переменную X,:
х,—хо, Ах, ’
(18.1)
где х, — координата в старой системе, т. е. естественное, или натуральное, значение фактора I вблизи исследуемой точки; хо< — координата выбранной точки, в окрестности которой ведется исследование, в старой системе, т. е. натуральное значение фактора t, взятое за нуль отсчета в новой системе; Дх, — интервал варьирования или масштаб по оси л(.
Величину X обычно называют кодированной переменной. В ходе полного факторного эксперимента в аналитической химии все факторы варьируют на двух уровнях: верхнем и нижнем. Если, например, нужно исследовать влияние pH на оптическую плотность раствора в области pH от 5,0 до 10,0, то, очевидно, х, = рН, верхний уровень х,'=10,0. нижний хГ = 5,0, а интервал варьирования в данном случае составляет Дх,=2,5. Выбранная точка лежит в середине исследуемого интервала: xoi = 7,5. Кодированное значение X по уравнению (18.1) будет равно Х'= 1^~7,5-=-|-1 для верхнего предела и Х"= 5’°^57,5 = — 1 для нижнего. Очевидно, натуральное значение
367
фактора на верхнем уровне кодируется всегда как +1, а на нижу нем как —1. Таблицу кодированных значений факторов называ/ ют матрицей планирования эксперимента Вполне понятно, что она кодирует, в сущности, условия проведения опытов, предусматривающие в полном факторном эксперименте все возможные комбинации факторов. В качестве прр мера в табл. .18.1 приведены натуральные значения факторов, составленные для изучения влияния pH и концентрации растворов на оптическую плотность. В габл. 18.2 дана матрица планирования эксперимента, которая получена по натуральным значениям факторов из табл. 18.1.
Таблица 181 Натуральные значения факторов и интервал варьирования
Факторы Основной уровень Интервал варьиро вания Уровни
верхний ннжний
=в pH 7,5 2,5 10 5
х2 = концентрация раствора 0,6 0,2 0,8 0,4
Таблица 18 2. Матрица планирования при полном двухфакторном эксперименте
Опыт Факторы Функция отклика у
л, Х2
1 —1 — 1 1/1
2 +1 — 1 1/2
3 —1 + 1 'Л
4 +1 + 1
Общее число опытов N в матрице планирования равно
W=2n, (18.2)
где п — число факторов.
В табл 18.3 представлена матрица трехфакторного эксперимента
При составлении матрицы полного факторного эксперимента уровень варьирования первого фактора изменяется от опыта к опыту, уровень варьирования второго фактора изменяется с частотой, уже в 2 раза меньшей, чем первого. Как общее правило можно отметить, что уровень варьирования каждого последующего фактора в матрице изменяется с частотой вдвое меньшей, чем предыдущего. Это правило четко проявляется в матрицах в табл. 18.2 и 18.3. Например, при трехфакторном эксперименте (табл. 18.3) третий фактор Хз остается постоянным (поддерживается на нижнем уровне) во всех первых четырех опытах и опять
368
Таблица 18 3 Матрица планирования при полном трехфакторном эксперименте
Опыт 1 Факторы Функция отклика У
Xi Х2 Хз Лз = Л|Х2
1 —1 —1 —1 +1 У>
2 +1 —1 —1 —1 У*
3 1 +1 —1 -1 Уз
4 +1 4-1 —1 +1 1/4
5 —1 — 1 +1 Уз
6 +1 -1 +1 — Уб
7 —1 + 1 +1 — У1
8 +1 + 1 +1 — Уб
остается постоянным (на верхнем уровне) в последних четырех опытах.
Матрица планирования имеет следующие свойства:
2 Л,.=0;
/=1
2 X2=/V;
2 адт=о,
j=i
где TV — число опытов полного факторного эксперимента; / — номер опыта; i, I, т — номера факторов.
18.2. УРАВНЕНИЕ РЕГРЕССИИ И РЕГРЕССИОННЫЙ АНАЛИЗ
Математическое описание процесса по данным полного факторного эксперимента находят в виде уравнения регрессии:
y=bo-^-biX\ -)-bzXi +...+bnXn-}~b 12X1X2 +.b(n— i)nXn—iXn-}-
-)-bi1X1 ЬггХг-j-.-.-j-bnnXn-}-...,
где bo — константа; bi, Ьг, bn — коэффициенты, характеризующие линейные эффекты; bn, bz2, .... bnn — квадратичные эффекты; bi2,b(n-i)n — эффекты взаимодействия.
Коэффициенты этого уравнения называют коэффициентами регрессии.
Коэффициенты регрессии рассчитываются по следующим формулам:
b0=±iy>, (18-3)
369
1 N
Ъ,=^^Х,.у,-, (18.4)'
1 N
bim=-^-Ti X/iXi/ny,. при (18.5)
Для решения задач аналитической химии в уравнении регрессии можно ограничиться лишь линейными членами. Тогда расчетные формулы для коэффициентов регрессии, например, при полном двухфакторном эксперименте в развернутой форме будут иметь вид:
У|+«д+уз + {/4 .
(—l)j/l +(+1)*Д+(—^)*/3 + (4-1)*/4 .
4 ’
h = (-l)yi+(—1){/г+(+1){/з + (+1){/4 2 4
В соответствии с требованиями регрессионного анализа дисперсии строк матрицы должны быть однородными. Для неоднородных дисперсий математические методы планирования эксперимента неприменимы. Однородность дисперсий проверяется по критерию Кохрена G:
s2
(18.6)
5 s?
где S2 тах—наибольшая из дисперсий, рассчитанных как S = п
= если d — отклонение от среднего; k — число параллельных.
Дисперсия считается однородной, если рассчитанное по уравнению (18.6) значение критерия Кохрена удовлетворяет условию
G<G табл,
где GTa6jI— табличное значение критерия Кохрена при данном 7V и числе степеней свободы f, причем f=k— 1, если k, как и ранее, число параллельных опытов. Табличные значения GTa6jI, соответствующие доверительной вероятности 0,95, приведены в табл. 18.4.
Однородную дисперсию нескольких серий можно усреднить и найти дисперсию воспроизводимости Sf по соотношению
1 N
Sl—ZSl (18.7)
Число степеней свободы у S2 равно f=N(k—1).
370
Значимость коэффициентов регрессии устанавливается по данным о погрешностях их определения St и критерию Стьюдента tp. Если выполняется условие
b^Sbta или tP^b/Sb, (18.8)
то коэффициент регрессии значим, т. е. отличается от нуля. Погрешности в определении коэффициентов регрессии одинаковы и рассчитываются по формуле
s‘= <18-9> I
о
Числовые значения критерия Стьюдента даны в ®
табл. 18.5. Слагаемые с не- "
значащими коэффициента- »
ми из уравнения регрессии исключаются. Полученное об
после этого уравнение регрессии проверяется на а
адекватность, т. е. на s
точность описания поверх- ю
ности отклика. Проверка ®
производится с помощью критерия Фишера. Для этого сначала находят расчетное значение функции отклика по уравнению регрессии и определяют дисперсию адекватно с-Т И Saa ПО формуле
(18.10) /=1
где В — число коэффициентов в уравнении регрессии, включая свободный член; yf и у;р — экспериментальное и рассчитанное по уравнению регрессии значения
о о о о о о о о о о с О
II
СП Г- о 1Л LO тГ тг тГ СО СО СЧ сч о о о о о' о о о о о о o'
СЧ СО -Ф Ю ф
371
Таблица 18 5. Коэффициенты Стьюдента (/р /)
/ Р = = 0,75 Р = = 0,90 Р = = 0,95 6 = = 0,98 6 = = 0,99 / р = = 0.75 Р = = 0,90 6 = = 0,95 6 = = 0,98 Р = = 0,99
1 2,41 6,31 1271 31,82 63,66 13 1,20 1,77 2,16 2,65 3,01
2 1,60 2,92 4.30 6,97 9,92 14 1,20 1,76 2,14 2,62 2,98
3 1,42 2,35 3,18 4,54 5,84 15 1,20 1,75 2,13 2,60 2,95
4 1,34 2,13 2,78 3,75 4,60 16 1,19 1,75 2 12 2,58 2,92
5 1,30 2,01 2,57 3,37 4,03 17 1,19 1,74 2,11 2,57 2,90
6 1.27 1,94 2,45 3,14 3,71 18 1,19 1,73 2,10 2,55 2,88
7 1,25 1,89 2,36 3,00 3,50 19 1,19 1,73 2,09 2,54 2,86
8 1,24 1 86 2,31 2,90 3,36 20 1,18 1,73 2,09 2,53 2,85
9 1,23 1,83 2,26 2,82 3,25 30 1,17 1.70 2,04 2,46 2,75
10 1,22 1,81 2,23 2,76 3,17 40 1,17 1,68 2 02 2,42 2,70
11 1,21 1,80 2,20 2,72 3,11 60 1,16 1,67 2,00 2,39 2,66
12 1,21 1,78 2,18 2,68 3,05 120 1 16 1,66 1,98 2,36 2,62
оо 1,15 1,64 1,96 2,33 2,58
функции отклика в /-м опыте. Число степеней свободы у 5ад равно /ад = (V — В.
Расчетное значение критерия Фишера вычисляют по формуле F=-'S. (18.11)
Оу
Если полученное по этому соотношению значение критерия Фишера не превышает табличной величины (FTa6J1) при данном числе степеней Свободы, уравнение регрессии является адекватным и условие адекватности имеет вид F^FTa6a. Табличные значения критерия Фишера даны в табл. 18.6.
В качестве примера найдем уравнение регрессии, связывающее оптическую плотность раствора тиомочевинных комплексов олова (IV) (у) с временем развития окраски (xi), концентрацией тиомочевины (х2) и концентрацией хлорной кислоты (хз). Полный трехфакторный эксперимент проводился в окрестностях точки факторного пространства с координатами xoi = 4Omhh, хо2= 1,7 моль/л и хоз=2,5 моль/л. Условия проведения полного факторного эксперимента приведены в табл. 18.7.
Введем кодированные переменные:
v Х1 — Xoi v х2 —Х02 Хз —Хоз
Л,=-------; Л2=--т---; Лз=--;---
Ах, Дх2 Дх3
Таблица 18.6. 6-критерий при различной вероятности появления Р
6 6=1 6=2 6=3 6=4 6=5 6=6 6=8 6=ю 6 = 12 6=20
1 161 200 216 Р= 0 225 ,95 230 234 239 242 244 241
2 18,51 19,00 19,16 19,25 19,30 19,33 19,37 19,39 19,41 19,44
3 10,13 9,55 9,28 9,12 9,01 8,94 8,84 8,78 8,74 8,66
4 7,71 6,94 6,59 6,39 6,26 6,16 6,04 5,96 5,91 5,80
5 6,61 5,79 5,41 5,19 5,05 4,95 4,82 4,74 4,68 4,56
6 5,99 5,14 4,76 4,53 4,39 4,28 4,15 4,06 4,00 3,87
7 5,59 4,74 4,35 4,12 3,97 3,87 3,73 3,63 3,57 3,44
8 5,32 4,46 4,07 3,84 3,69 3,58 3,44 3,34 3,28 3,15
9 5,12 4,26 3.86 3,63 3,48 3,37 3,23 3,13 3,07 2,93
10 4,96 4,10 3,71 3,48 3,33 3,22 3,07 2 97 2,91 2,77
372
Таблица 18.7. Характеристики плана экспериментов
Характеристика X, МИИ Х2 ThiO, моль/л X3HC1O4, моль/л
Основной уровень 40 1,7 2,5
Интервал варьирования 10 0,2 0,5
Верхний уровень 50 1,9 3,0
Нижиий уровень 30 1,5 2,0
Матрица планирования и результаты эксперимента представлены в табл. 18.8.
Для проверки однородности дисперсий рассчитываем по уравнению (18.6) критерий Кохрена:
S2,., _ 8,82-10~4 * „2 2,384-10-3
Л
0,3700.
и сравниваем это значение с табличным. Из табл. 18.4 для f= 1, /’=0,95 и /V=8 находим Сгайл = 0,6798. Табличное значение превышает рассчитанные из экспериментальных данных (0,37), поэтому дисперсии следует квалифицировать как однородные.
Дисперсия воспроизводимости по уравнению (18.7) будет равна
S2d= 2’384'10 = 2,98 -10"4 О
и дисперсия среднего
Число степеней свободы дисперсии воспроизводимости составляет f=N(k—1)= = 8(2—1)=8. Теперь по формулам (18.3).. (18.5) рассчитываем коэффициенты уравнения регрессии:
Ьо = 1 /8(0,392 + 0,429 + 0,531 + 0,571 + 0,512 + 0,528 + 0,646 + 0,678) = = 0,5359 = 0,536;
b, = 1 /8(—0,392 + 0,429 — 0,531 + 0,571 - 0,512 + 0,528 — 0,646 + 0,678)= = 0,01563 = 0,0156;
Ь2 == 1 /8(—0,392 — 0,429 + 0,531 + 0.571 — 0,512 — 0,528 + 0,646 + 0,678)= =0,07063 = 0,0706;
Ь3= 1 /8(—0.392 - 0 429 — 0,531 — 0,571 + 0,512 + 0,528 + 0,646 + 0,678)= =0,05513=0,0551
Значимость полученных коэффициентов оцениваем по критерию Стьюдента Для этого сначала определим погрешность коэффициентов по формуле (18.9):
S„ =
Sb =
J 2,98-10-4 _
V 8-2 —
4,31-IO'3.
Si .
Nk ’
Затем составляем / отношение для всех коэффициентов уравнения регрессии-
to
bo
Sb
0,536 4,31-10-’
124,4;
fci 0,0156
SZ 4,31-10-’
3,62;
373
Таблица 18.8. Матрица планирования и результаты полного трехфакторного эксперимента (опыты дублирования)
7 (£5 40 LO ю m Ю 40 CZ) 111)1)01 — оооооооо^ о • Г"- союфс; —• СЧ —г О м
расч 1 —0,003 0,003 —0,005 0,004 0,007 —0,008 0,000 0,001
hoed у СО LO LO ю 1О О О оооооооо
X) <п ' 1 1 1 1 1 1 1 -Оооооооо • • оо °1СЧООООСМСЧСЧОСО СО О Tf ООО ^,00? — О со СО || и
1 3 IDin^-Tr-t-riOlO 1 1 1 1 1 1 1 1 оооооооо О —1 о О LQ — со со сч~ СО — тг
Оптическая плотность 'X СЧ О СЧ СО О СО О СЧ СО Is СЧ-^1^ СО ’Ф LO Ю lQ lO О С0 о o' о о о о о о
соосо-^ — ООСЧСО X CN ’С со О. — in Е-СО^гООг+гООО о о о~ о о о" о о
LOQOCOOOCOOOOC4 О СО — Ю СО СО хЕ ОО СО LO ГО LO ГО О СО о о о о о о o' о
Хз НС1О4, моль/л о о о о_о о о о_ СЧ СЧ СЧ СЧ со СО СО со
х2 Thio , моль/л LO го О О Ю 1-0 О О
„ а ОООООООО СО LO со ю со Ю СО го
1111++++
11++11++
>7 1 +1 + 1 + 1 +
Опыт — СЧ СО о 1-0 to Ь ос
62 0,0706
ST- 4,31 -10 "
bs 0,0551
Зь 4,31-10*
16,38;
12,78.
Для Р = 0,95 и f = 8(2—1) = 8 табличное значение критерия Стьюдента равно <0.95.8= 2,31 Это удовлетворяет условию (18.8), следовательно, все коэффициенты уравнения регрессии значимы и уравнение регрессии имеет вид
Ара„=0,536 +0,0156X1 + 0,0706%2 +
0,0551Хз- (18.12)
Найденные по этому уравнению значения оптической плотности Лрасч приведены в табл. 18.8.
Адекватность уравнения эксперименту проверяем по критерию Фишера. Дисперсию адекватности рассчитываем по соотношению (18.10) и данным табл 18.6:
S2
° ад'
2
f (А-Арасч)2=
=-1,73-10 4=8.65-10“5.
о—4
По формуле (18.11) находим F-otho-шение:
J98U0-
Л 8,65-10“5 ’ b
(в числитель /•'-отношения ставят большую из дисперсий). Из табл. 18.6 находим, что для Р = 0,95, Fi = 4 и Fi = 8 критерий Фишера равен Fо 95(4 в) = 3,8, т. е. > F. Следовательно, уравнение регрессии (18.12) адекватно эксперименту.
18.3. ДРОБНЫЙ ФАКТОРНЫЙ ЭКСПЕРИМЕНТ
Уравнение (18.2) показывает, что с увеличением числа факторов резко возрастает объем полного факторного эксперимента. Если в уравнении регрессии можно ограничиться только линейными членами, число опытов уменьшают с помощью дробного факторного эксперимен-т а или методом дробных реплик от полного факторного эксперимента. В качестве реплики берут
374
полный факторный эксперимент для меньшего числа факторов. При этом число опытов должно остаться превышающим или равным числу неизвестных в уравнении регрессии. Например, в полном трехфакторном эксперименте, как показывает табл. 18.3, должно быть проведено восемь опытов. Можно, однако, ограничиться четырьмя опытами, если в матрице планирования в качестве плана для Хз использовать произведение XiX? Такой сокращенный план называют полурепликой полного факторного эксперимента. Используют также четверть реплики и более дробные реплики. Если парные взаимодействия относятся к статистически значимым, то соответствующие коэффициенты регрессии не будут равны нулю и, следовательно, полученные в методе дробных реплик коэффициенты регрессии будут смешанные, отражающие эффект фактора и парного взаимодействия. Вид планирования эксперимента, когда некоторые факторы приравниваются к произведению факторов, называют планированием со смешиванием и обозначают как 2"-р, где п — общее число факторов, р — число факторов, которые приравниваются произведению. Соотношение типа
Х3=Х,Х2 (18.13)
называют генерирующим соотношением. Оно показывает, с каким эффектом смешан данный эффект.
При умножении соотношения (18.13) на Хз получаем Х|=Х1Х2Хз,
но X,2 = 1, поэтому
1=Х,Х2Х3. (18.14)
Произведение (18.14) называют определяющим контрастом. С его помощью определяют смешанные элементы. Для этого его умножают поочередно на Xi, Х2 и Х3:
Xi =Х|Х2Хз=Х2Хз; Х2=Х[Хз; Хз=Х1Х2.
Отсюда получаем систему смешанных оценок:
6i = Pi +₽2з; ^2 = Рг-1-Р1 з; Ьз=Рз-1-Р12,
где р — истинные коэффициенты ряда Тейлора.
Статистическую значимость имеют не все парные взаимодействия, поэтому не все коэффициенты регрессии являются смешанными. Составление уравнения регрессии и его анализ в методе дробных реплик не отличаются от соответствующих операций в полном факторном эксперименте.
18.4. ОПТИМИЗАЦИЯ ПО МЕТОДУ КРУТОГО ВОСХОЖДЕНИЯ
Положительное значение коэффициентов уравнения регрессии (18.12) показывает, что оптическая плотность раствора будет возрастать с увеличением всех факторов: времени развития окрас
375
ки, концентрации тиомочевины и концентрации хлорной кислоты. Поскольку количественной мерой влияния каждого из факторов на параметр оптимизации является коэффициент регрессии, наиболее существенное влияние на оптическую плотность раствора при выбранных интервалах варьирования будет оказывать концентрация тиомочевины. С увеличением интервалов варьирования абсолютное значение коэффициентов уравнения регрессии также возрастает, что может привести к изменению относительного вклада каждого из факторов. Это обстоятельство играет существенную роль при выборе интервалов варьирования.
Задача оптимизации в данном случае заключается в поиске условий или значений факторов, при которых оптическая плотность раствора заданной концентрации по олову будет максимальной. Вполне понятно, что при изменении независимых переменных пропорционально коэффициентам регрессии функция отклика будет эффективно отражать суммарное действие всех факторов. Для получения максимального значения функции отклика необходимо увеличивать те переменные, которые входят в уравнение регрессии с положительным знаком, и уменьшать те, которые входят с отрицательным. Бокс и Уилсон разработали прием достижения максимума в функции отклика, получивший название крутого восхождения. Техника расчета по этой методике заключается в следующем. Один из факторов, например Хг, выбирают как базовый и вычисляют для него произведения коэффициента регрессии bz на интервал варьирования АХг, т. е. Ь^КХ?, и определяют шаг движения по градиенту АХ*. Выбор АХ* является очень важным элементом расчета. Чрезмерно малый шаг потребует очень большого числа опытов, а при слишком большом шаге могут быть не замечены важные особенности системы или будет очень быстро превышен предел физически возможных значений фактора (например, концентрация раствора превысит растворимость соединения и т. д.). Величина шага должна, конечно, существенно превышать погрешность измерения фактора. Обычно принимают АХ*^ДХ. Затем вычисляют отношение
(18.15)
Величину шага для других факторов рассчитывают по формуле
АХ,*=Т6,ДХ,.
Движение начинают из центра плана путем прибавления АХ* к основному уровню и последующим значениям факторов. В данном примере выбираем шаг AXJ=O,1 и по уравнению (18 15) находим пересчетный множитель у:
У = 0,0706-0,2 = 7’08'
376
Тогда шаг по времени развития окраски будет равен
AXf=yfeiAXi = 7,08-0,0156-10=1,1 ~ 1
и шаг по концентрации хлорной кислоты АХ£ = yb3AX3 = 7,08 X X 0,0551-0,5 = 0,2.
Для удобства расчетов значение шага обычно округляют. Результаты расчетов по методу крутого восхождения приводятся в табл. 18.9. Значение Лрасч получено по уравнению регрессии (18.21). Это результат так называемых мысленных опытов. При адекватной модели в экспериментально изученной области влияющих факторов результаты мысленных опытов экспериментально обычно не проверяются. В рассматриваемом примере (табл. 18.9)
Таблица 18.9. Расчет крутого восхождения
Характеристика и номер опыта X, мин Hi ТЬЮ, моль/л Х3 НС1О4, моль/л Кодированные значения А л расч
X, Х2 Х3
Основной уровень (центр 40 1,7 2.5 0 0 0 0,536
плана) Интервал варьирования 10 0,2 0,5 1 1 1 —
Шаг движения 1 0,1 0,2 0,1 0,5 0,4 —
Опыт 1 41 1,8 2,7 0,1 0,5 0,4 0,595
» 2 42 1,9 2,9 0,2 1,0 0,8 0,654
» 3 43 2,0 3,1 0,3 1,5 1,2 0,713
» 4 44 2,1 3,3 0,4 2,0 1,6 0,772
» 5 45 2,2 3,5 0,5 2,5 2,0 0,831
» 6 46 2,3 3,7 0,6 3,0 2,4 0,889
это относится только к опытам с первого по третий. Опыт четвертый и следующие по концентрации тиомочевины уже выходят за пределы экспериментально изученной области, поэтому результаты мысленных опытов, начиная с четвертого, следует проверить экспериментально. Однако возможности экспериментальной проверки ограничиваются растворимостью тиомочевины в воде, составляющей при 25 °C примерно 2 моль/л. Можно зафиксировать АХг примерно на этом уровне и еще раз рассчитать крутое восхождение с учетом изменения только А/ и Х3. Однако более целесообразным представляется проведение полного факторного (двухфакторного) эксперимента, зафиксировав постоянную концентрацию тиомочевины в 2 моль/л и взяв в качестве центра плана оптимальные условия, например условия второго опыта
ЛИТЕРАТУРА
Айвазов Б. В. Введение в хроматографию.—М.: Высшая школа, 1983.
Булатов М. И., Калинкии И. П. Практическое руководство по фотоколори-метрическим методам анализа. 5-е изд , перераб. — Л : Химия, 1986.
Васильев В. П. Теоретические основы физико-химических методов анализа.— М.: Высшая школа, 1979.
Васильев В. П. Термодинамические свойства растворов электролитов. — М.: Высшая школа, 1982.
Вяхирев Д. А., Шушунова А. Ф. Руководство по газовой громатографии. 2-е изд., перераб. и доп. — М.: Высшая, школа, 1987.
Головина А. П., Левшин Л. В. Химический люминесцентный анализ неорганических веществ. — М.: Химия, 1978.
Золотов Ю. А., Кузьмин Н. М. Концентрирование микроэлементов — М : Химия, 1982.
Ляликов Ю. С. Физико-химические методы анализа.—М.- Химия, 1974.
Лурье Ю. Ю. Справочник по аналитической химии. 5-е изд., перераб. и доп. — М.: Химия, 1979.
Нефедов В. Д., Текстер Е. Н., Торопова М. А. Радиохимия. — М.: Высшая школа, 1987.
Петерс Д., Хайес Д., Хифтье Г. Химическое разделение и измерение. Теория и практика аналитической химии. — М.: Химия, 1978. Т. 1, 2.
Пешкова В. М., Громова М. И. Методы абсорбционной спектроскопии в аналитической химии. — М.: Высшая школа, 1976.
Практикум по физико-химическим методам анализа/Под ред. О. М. Петрухина. — М.: Химия, 1987.
Практическое руководство по физико-химическим методам анализа/Под ред И. П. Алимарина, В. М. Иванова. — М.: МГУ, 1987.
СкугД., УэстД. Основы аналитической химии. — М : Мир, 1979. Т. 1, 2.
Терек Т., Мика И., Гегуш Э. Эмиссионный спектральный анализ. —М.: Мир, 1982. Т. 1, 2.
Физико-химические методы анализа/Под ред. В. Б. Алесковского, К. Б. Яци-мирского. — Л.: Химия, 1971.
Чарыков А. К. Математическая обработка результатов химического анализа.— Л.’ Химия, 1984.
Яцимнрскнй к. Б. Кинетические методы анализа. — М.: Химия, 1967.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсорбционная спектроскопия 50
Адсорбция 316
Активационный анализ 268
Амперометрическое титрование 232
Анноннты 352
Атомно-абсорбционная спектроскопия 97
Болометр 64
Вольтамперометрия 220
Время удерживания 321
Высота, эквивалентная теоретической тарелке 323
Выход люминесценции 107
Гальванический элемент 189
— — стандартный 192
— — исследуемый 192
Гейгера — Мюллера счетчик 126
Генераторы
— дуги 20
— звуковой 173
— искры конденсированной 21
Гомологическая пара 34
Детектор (ы) 330
— пламенно ионизационный 331
— рефрактометрический 342
— спектрометрический 342
— по теплопроводности (катарометр) 330
— термоионный 331
— термохимический 330
Дисперсия 149
— адекватности 371
— воспроизводимости 370
— однородности 370
— оптического вращения 155
— спектральных приборов 22
Дифракционная решетка 22
Дихроизм круговой (циркулярный) 156
Дозатор 375
Дуга электрическая 20
Емкость обменная 352
Закон(ы)
— Больцмана 16
— Бугера — «Ламберта — Бера 51
- Вавилова 108
— Вульфа — Брегга 123
— Кольрауша 170
— Мозли 121
— преломления 147
— радиоактивного распада 265
— распределения 298
— Рэлея 159
Стокса — «Ломмеля 107
Фарадея 244
Зонд электронный 6, 123
Извлечения степень 300
Излучение рентгеновское 119 120
Изотерма адсорбции 317
Изотопного разбавления метод 271
Иммерсионный метод 150
Индекс удерживания 332
— — Ковача 332
Иониты типы 352
Ионометрия 203
Ионообменное равновесие 353
Искра высоковольтная 21
Катализатор 284
Катарометр 330
Катиониты 352
Квантовое число
— — главное 11
— — магнитное 12
— — побочное 11
— — спиновое 12
Квантометры 29
Кинетические методы анализа 283
— теория в хроматографии 324
Колонки хроматографические 327
Колтупа карта 66
Кондуктометрия 168
— аппаратура 172
— высокочастотная 171
— практика 174
Константа
— ионного обмена 354
— распределения 299
— скорости реакции 283
— экстракции 303
Коэффициент (ы)
— поглощения молярный 51
— преломления 147
— пропускания 51
— разделения 301
— распределения 298, 354
Критерий
— разделения 322
— Фишера 371
Крутое восхождение 375
Кулонометрия 251
— амперостатическая 253
— потенциометрическая 251
— практика 255
Кулонометры 252
Лазер 18, 136
Лампа с полым катодом 98
«Линия
— базовая 74
— аналитическая 29
— комбинационного рассеяния 135
Линии спектральные 15—17
Люминесцентный анализ 104
— — аппаратура 111
— — качественный 111
— _ количественный 112
— — теория 104
Люминесценция 104
— и интенсивность 108
— и спектры 104
Масс спектр 281
Масс-спектрометр 280
Масс-спектрометрический метод а нал и за 279
Матрица планирования эксперимента 368
Метод
— градуировочного графика 8
— дифференциальной фотометрии 71
— добавок 8
379
— молярного свойства 8
— постоянного графика 38
— твердого графика 39
— трех эталонов 37
— фотоэлектрический 40
Модель математическая 367
Монохроматизаторы 63
Мульти плетность 14
Нефелометрия 158
Определение pH 196
Орбитали молекулярные 58
Ослабитель ступенчатый 35
Осциллятор
— ангармонический 56
— гармонический 55
Отбора правила 14
Отклика поверхность 366
Пара гомологическая 34
Параметр оптимизируемый 366
Переменная кодированная 367
Переход
— запрещенный 14
— разрешенный 14
— резонансный 15
— — магнитный 139
Период полураспада 265
Пламена 19
Планирование эксперимента математическое 366
Плоскость поляризации 154
Плотность оптическая 51
Подвижность ионов 170
Показатель преломления 147
Полуволны потенциал 223
Поляризация молекулы 134, 171
Поляриметрический метод 153
— аппаратура 156
— теория 154
Полярограмма 223
Полярографический метод 220
— — анализа органических соединений 231
--- аппаратура 225
---количественный 227
--- практика 225
— — теория 220
Полярография
— инверсионная 231
— осциллографическая 230
Постоянная
— Планка 10
— распада радиактивного 265
— Ридберга 12
— силовая 56
— сосуда 294
— экранирования 120
Потенциал (ы)
— полуволны 223
— стандартный 188
Потенциометр 191
Потенциометрический метод 188
Потенциостаты 251
Почернение фотопластинки 23
Преломление света 146
Предел обнаружения 4, 31
Пространство координатное 366
Радиоспектроскопия 138
Радиометрические методы анализа 263
Рассеяние света
- комбинационное 134
— рэлеевское 134
Регрессии уравнение 369
Рентгеновская трубка 122
Рентгеноспектральный анализ 119
— — аппаратура 122, 128
— количественный 130
- — практика 129
Рефрактометр 151
Рефрактометрический метод 146
Рефракция 149
Ротатор жесткий 54
Самопоглошение 17
Светофильтры 64
Сдвиг химический 140
Сила осциллятора 61
Смолы ионообменные 352
Спектр
— —вращательный 54
— инфракрасный 66
— колебательный 55
— комбинационного рассеяния 133
— Мессбауэра 274
— поглощения 52
— полярографический 224
— рентгеновский 199
— электронный 58
— эмиссионный 11
— ЭПР 146
— ЯМР 140
Спектрометр ЯМР 141
Спктрофотометр 65
— атомно-абсорбционный 98
Спин
— электрона 11, 145
— ядра 138
Спиновое эхо 143
Степень разделения 322
Стилометры 28
Стилоскопы 27
Стьюдента критерий 371
Счетчики
Гейгера — Мюллера 126
— ионизационные 124 266
- кристаллические 127
самогасящиеся 126
сцинтилляционные 126, 267
черенковские 267
Температура удерживания 333
Теоретических тарелок метод 323
Терм
— рентгеновский 121
— спектральный 10, 14
Термометрическое титрование 292
— практика 296
— теория 292
Титраторы автоматические 77, 210
Титрование
— амперометрическое 232
высокочастотное 180
— каталиметрическое 287
— кондуктометрическое 175
— кулонометрическое 253
- потенциометрическое 206
— радиометрическое 272
380
- термометрическое 292
— турбидиметрическое 169
— фотометрическое 76
— хронокондуктометрическое 175
Ток диффузионный 222
Турбидиметрический метод 158
Удерживания
— время 321
— объем 321
— температура 333
Уитстона мостик 173
Уравнение
— Ван Деемтера 324
— Илькевича 225
— Ломакина 17
— Лорентца—Лоренца 149
— Лэнгмюра 317
— Нернста 188
— Онзагера 153
— полярографической волны 223
— Шредингера 11
Фактор
— натуральное значение 367
— извлечения 299
— контрастности пластинки 24
— обогащения 301
Флуоресценция
— атомная 110
— выход 107
— интенсивность 108
— спектры 181
Флуорометры 111
Фосфоресценция 106
Фотометрический анализ
— — аппаратура 62
— — методы 70
— — — экстрацнонно фотометрический 74
Фотометрия пламени 42
Фотопластинка 23
Фотоумножители 26
Фотоэлектроколорнметры 65
Фотоэлементы
— с внешним фотоэффектом 25
— с внутренним фотоэффектом 25
— с запирающим слоем 26
Фотоэффект 25
Функция отклика 366
Хроматограмма 319
Хроматографические меточы 318
— — качественного анализа 331
— — количественного ана 1иза 334
— — абсолютной калибровки 334
— — внутренней стандарiизацни 335
— — нормировки 334
— — нормировки с калибровочным коэф фициентом 334
Хроматографический пик 120
Хроматография 316
— аналитическая реакционная гаювая 337
— бумажная 348
— восходящая 344
— газовая 327
— влияние температуры 135
— газо-жидкостная 328
— гель 350
— жидкостная адсорбционная 338 жидкостно-жидкостная распределитель ная 346
— ионная 357
— ионообменная 351
— капиллярная 329
— круговая 345
— нисходящая 344
— теоретические представления 323 тонкослойная 342
— электрофоретическая 350
Хроматермография 336
Хроматомасс спектрометрия 333
Частота(ы)
— антнетокова 135
— резонансная 139
— нормальных колебаний 56
— характеристическая 57
— стокова 134
Число
— волновое 11
— квантовое 11
— степеней свободы 57
— теоретических тарелок 323
Шкала pH 197
Экстрационные системы
— галогенидные и тиоцианатные 307
— хелатные 305
Экстракция 298
Электрическая проводимость 168
--- предельная 170
--- удельная 168
— — эквивалентная 169
Электрогравиметрические разделения 246 Электрод(ы)
— амальгамные 193
— вращающийся 226
— второго рода 193
— индикаторный 192
— ионоселективный 200
— — жидкостной 202
--- твердый 201
— каломельный 194
— первого рода 193
— редокс 193
— ртутный капающий 225
- сравнения 193
— стеклянный 198
— хлорсеребряный 193
Электролиз
— аппаратура 246
— внутренний 250
— на ртутном катоде 249
Электромагнитный спектр 10
Электронный парамагнитный резонанс 145
Этюцнониые характеристики 320
Энергия
вращательная 54
колебательная 55
- электромагнитного излучения 10
— электронная 58
Эффект
Допплера 274
Зеемана 18
- Коттона 155
Мессбауэра 273
- Оже 120
Шпольского 107
— Штарка 18
№1
ОГЛАВЛЕНИЕ
Предисловие 3
Глава 1. Общая характеристика физико-химических методов анализа . 4
1.1. Особенности и области применения физико-химических методов анализа ....................................................... 4
1.2. Основные физико-химические методы анализа ..................... 6
1.3. Основные приемы, используемые в физико-химических методах анализа 7
Глава 2. Эмиссионный спектральный анализ . . . 9
2.1. Основные характеристики электромагнитного излучения 9
2.2. Теоретические основы эмиссионной спектроскопии . 10
2.3. Основные узлы спектральных приборов . 18
2.4. Конструкция спектральных приборов . 27
2.5. Качественный спектральный анализ . . 29
2.6. Количественный спектральный анализ............................ 31
2.7. Фотометрия пламени (пламенная эмиссионная спектроскопия) 42
2.8. Практическое применение . ...... 43
2.9. Общая характеристика метода . 43
Глава 3. Абсорбционная спектроскопия . 50
3.1. Основной закон светопоглощения (закон Бугера — Ламберта — Бера) . . . ... 50
3.2. Спектры поглощения............................................ 52
3.3. Основные узлы приборов абсорбционной спектроскопии . 62
3.4. Качественный анализ . 65
3.5. Количественный анализ 66
3.6. Практическое применение . . 78
3.7. Общая характеристика метода . 79
Глава 4. Атомно-абсорбционный спектральный анализ 97
4.1. Теоретические основы метода.................................... 97
4.2. Основные узлы приборов для атомно-абсорбционного анализа . 98
4.3. Количественные определения 99
4.4. Практическое применение 101
4.5. Общая характеристика метода . 101
Глава 5. Люминесцентный анализ . 104
5.1. Спектры люминесценции . . 104
5.2. Схема прибора для люминесцентного анализа 111
5.3. Качественный анализ . 111
5.4. Количественный анализ . 112
5.5. Практическое применение . . 112
5.6. Общая характеристика метода . . 113
Глава 6. Рентгеноспектральные методы анализа 119
6.1. Рентгеновские спектры......................................... 119
6.2. Поглощение рентгеновского излучения .... 122
6.3. Основные узлы рентгеноспектральных приборов................... 122
382
6.4. Конструкции рентгеновских спектральных приборов 128
6.5. Качественный рентгеноспектральный анализ . . 129
6.6. Количественный рентгеноспектральный анализ 130
6.7. Практическое применение . 131
6.8. Общая характеристика метода 132
Глава 7. Другие спектральные и оптические методы анализа 133
7.1. Анализ по спектрам комбинационного рассеяния . 133
7.2. Радиоспектроскопия . 13g
7.3. Рефрактометрические методы анализа 146
7.4. Поляриметрия .... 153
7.5. Нефелометрия и турбидиметрия 158
7.6. Практическое применение . . 161
7.7. Общая характеристика методов 161
Глава 8. Кондуктометрия (анализ по электрической проводимости) 168
8.1. Электрическая проводимость растворов .... 168
8.2. Схема установки для определения электрической проводимости 172
8.3. Прямая кондуктометрия ... . 174
8.4. Кондуктометрическое титрование 175
8.5. Высокочастотное титрование 180
8.6. Практическое применение . . 181
8.7. Общая характеристика метода 183
Глава 9. Потенциометрия . 188
9.1. Электродный потенциал......................................... 188
9.2. Схема установки для потенциометрических измерений . 189
9.3. Прямая потенциометрия ... . . 195
9.4. Потенциометрическое титрование................................ 206
9.5. Потенциометрическое определение физико-химических свойств веществ ............................. . .............. 211
9.6. Практическое применение . 213
9.7. Общая характеристика метода 213
220
Глава 10. Вольтамперометрия
10.1. Кривая ток—потенциал .... 220
10.2. Схема полярографической установки . 225
10.3. Прямая полярография ... 226
10.4. Амперометрическое титрование 232
10.5. Практическое применение . . 236
10.6. Общая характеристика метода 237
Глава 11. Электролиз и кулонометрия 244
11.1. Законы электролиза.......................................... 244
11.2. Потенциал разложения и перенапряжения 245
1L3. Электрогравиметрический анализ 246
11.4. Кулонометрия 251
11.5. Практическое применение . . 255
11.6. Общая характеристика метода 257
Глава 12. Радиометрические методы анализа . 263
12.1. Типы радиоактивного распада и радиоактивного излучения 263
12.2. Закон радиоактивного распада . . . 265
12.3. Взаимодействие радиоактивного излучения с веществом и счетчики излучения ........................................................ 266
12.4. Ядерная химия и искусственная радиоактивность............... 267
12.5. Методики анализа, основанные на измерении радиоактивности . . 268
12.6. Практическое применение . . . . . 274
12.7. Общая характеристика метода................................. 275
383