/































































































































































































































































































































































Текст
К. Г. ЗЫРИН и Д. С.
* h >
» : /« v. i
Ш1КО
М/ИИЧЕСКИЕ
Т 6 Д Ы %
[•КГ41ЩЦ
|<1ЫИ|1МНЩ|1ШГ11
Н. Г. ЗЫРИН, Д. С. ОРЛОВ
ФИЗИКО-ХИМИЧЕСКИЕ
МЕТОДЫ
ИССЛЕДОВАНИЯ ПОЧВ
ИЗДАТЕЛЬСТВО
МОСКОВСКОГО УНИВЕРСИТЕТА
1964
/
Введение
in
Решение современных проблем почвоведения базируется
на детальном изучении химических и физических свойств
почвы. Вопросы химизации сельского хозяйства и повышения
почвенного плодородия, рационального использования
земельных угодий и удобрений, природа и генезис отдельных
почвенных типов и обширных почвенно-географических зон
и провинций — все это требует предварительной
химико-аналитической характеристики почв в сочетании с глубоким
пониманием закономерностей почвообразования и
специфических особенностей химии почв.
В почвоведении с помощью приемов обычного анализа
накоплен огромный фактический материал, освещающий
химический состав почв и многие ее свойства. Это позволило
обосновать нуждаемость почв в удобрениях, описать историю
и закономерности образования типов почв. Классические
объемные и весовые методы анализа, которые часто
сокращенно называют просто химическими методами, останутся
и в дальнейшем как один из важнейших приемов изучения
состава почв. Существенным недостатком этих методов
является их громоздкость и большая трудоемкость; кроме того,
их применение сопровождается более или менее сильным
воздействием на почву различных реактивов и в силу
длительности определения они мало пригодны для изучения
динамики почвенных процессов.
Перспективными для изучения почвообразования и
плодородия почв являются физико-химические методы анализа.
Современные физико-химические методы не отвечают,
конечно, всем предъявляемым требованиям и дают ответ
далеко не на все вопросы, но их основные принципы и приемы
применения близко совпадают с потребностями науки о
почвах.
Физико-химические методы анализа — это большая группа
методов, в которую часто включают все приемы химических
исследований, базирующиеся на количественном измерении
физических свойств. Предварительно изученная зависимость
состав — свойства позволяет посредством простых
физических измерений анализировать любую систему. Если в
химическом анализе для определения количественного состава
измеряют количество вещества, вступающего в реакцию, или
весовое (объемное) количество продуктов реакции, то в
физико-химическом анализе непосредственного измерения
объема или веса не производят, а количественно определяю/
какое-либо физическое свойство вещества или системы.
Поэтому первым этапом разработки и применения любого
физико-химического метода является установление зависимости
между составом и свойствами, выражаемой математически
в виде формулы или графика.
Зависимости, используемые в физико-химических методах
анализа, опираются на общие законы физики и химии;
специфичность свойств веществ, характер реакций и
особенности изучаемых систем находят отражение в величинах
параметров уравнений. Это придает физико-химическим методам
универсальность, позволяющую применять одни и те же
приборы для исследования разнообразных соединений. В связи
с этим классификация методов и последовательность их
изучения основывается на общности используемых законов
(свойств) и применяемой аппаратуры. Вместе с тем
специфика состава, структуры и свойств почвы требует уточнения,
иногда разработки особых условий проведения исследования,
а подчас и новых приемов и деталей аппаратуры. Следует
подчеркнуть, что еще далеко не все физико-химические
методы исследования в полной мере проверены и приспособлены
к изучению состава, структуры и свойств почвы.
Другая особенность физико-химического анализа связана
с тем, что свойства вещества или системы не зависят от
взятого объема вещества. Любые свойства: окраска,
интенсивность излучения, показатель преломления, величина
потенциала— определяются только концентрацией, а не
абсолютным количеством изучаемого компонента. Это позволяет
значительно повысить чувствительность методов количественного
определения и вносит некоторые особенности в технику
работы по сравнению с обычными химическими методами.
Ряд физико-химических методов позволяет определять та-
4
кие свойства вещества, или компонента в смеси, которые
нельзя изучить обычными приемами:
окислительно-восстановительный потенциал, активности ионов, светопоглощение и
отражательная способность почвы и т. п.
Разнообразие физико-химических методов столь велико,
что сейчас уже трудно установить границы, в пределах
которых тот или иной метод следует считать
физико-химическим. При почвенных исследованиях наиболее
употребительными за последнее время оказались следующие:
1) потенциометрические методы, применяемые в
почвоведении для определения рН, окислительно-восстановительного
потенциала, активности ионов натрия, калия, хлора и др.;
2) кондуктометрические методы, используемые в
почвоведении для определения солесодержания в почвах и
почвенных растворах;
3) полярографические методы, нашедшие применение в
почвоведении для количественного определения многих
катионов и анионов, особенно присутствующих в
микроколичествах;
4) фотометрические и нефелометрические методы
анализа, позволяющие определять практически любые компоненты
почв и почвенных растворов;
5) спектрофотометрический анализ, используемый в
почвоведении как для количественных определений, так и для
изучения структуры гумусовых веществ и минералов
тонкодисперсной фракции;
6) методы пламенной фотометрии, используемые в
почвоведении преимущественно для определения содержания в
почвах катионов щелочных и щелочноземельных металлов;
7) методы термического анализа, применяемые в
почвоведении для изучения минералогического состава почв и
почвенных коллоидов.
Кроме перечисленных в почвоведении находят применение
рефрактометрия, поляриметрия, люминесценция; все шире
используют спектральный эмиссионный атомный анализ, рент-
геноструктурный, электронномикроскопический анализ и др.
В настоящем руководстве излагаются только те методы,
которые наиболее широко применяются в почвенно-химических
лабораториях.
Необходимо подчеркнуть, что в большинстве случаев
проведение анализа физико-химическими методами требует
•очень немного времени и, хотя используется часто
дорогостоящая аппаратура, все же достигается экономия средств
■благодаря быстроте определения и малому расходу реакти-
5
bob. Вместе с тем по чувствительности и точности определения
(особенно малых количеств) физико-химические методы
безусловно превосходят обычные объемные и весовые
методы анализа. С точки зрения почвоведения особенно важно,
что многие почвенные характеристики могут быть получены
этими методами без какого-либо нарушения естественного
состояния почвы. И, наконец, физико-химические методы
позволяют глубоко изучить принципы построения вещества, в том
числе таких важнейших компонентов почвы, как
тонкодисперсные минералы и органические гумусовые вещества.
Физико-химические приемы анализа осуществимы при
наличии специальной, часто дорогостоящей аппаратуры,
безотказно работающей только при умелом обращении с ней.
Теория самих методов довольно сложна и требует достаточно
высокой подготовки сотрудников и умелого толкования
получаемых данных. Успех и более широкое использование
физико-химических методов в почвоведении зависит прежде
всего от подготовки почвоведов в этой области.
Данное руководство должно служить учебным пособием
при обучении студентов по специальностям почвоведение и
агрохимия, а также для ознакомления сотрудников
лабораторий, обслуживающих сельское хозяйство, с
физико-химическими методами исследования почв.
Глава I
ОБЩИЕ СВОЙСТВА ДИСПЕРСНЫХ СИСТЕМ КАК
ПРЕДМЕТА АНАЛИЗА
(способы выражения концентрации, общие законы
и расчетные формулы)
При исследовании почв приходится иметь дело с
различными дисперсными системами, дисперсионной средой
которых, как правило, является вода. Наиболее простые из них —
■молекулярные растворы, более сложные — коллоидные
растворы (размеры частиц дисперсной фазы колеблются в
пределах от 1 до 100 ммк) и взвеси (величина частиц >
> 100 ммк). Последняя группа представлена золями и
суспензиями, хотя в редких случаях не исключена возможность
образования и эмульсий. Почвенная масса является
сложной полидисперсной и химически неоднородной
системой, поэтому при взаимодействии почвы с водой
одновременно образуется истинный раствор, коллоидный раствор и
взвесь Такую сложную систему обычно для краткости
называют просто суспензией.
В природе золи и суспензии встречаются очень часто
(пойменные воды, делювиальные потоки, потеки или суспензии
по трещинам в почвенном профиле). Различные вещества
в ландшафте мигрируют преимущественно в форме суспензий
и коллоидных растворов. В почвах коллоидные растворы
чрезвычайно лабильны и могут существовать при наличии
свободной влаги; периодическое просыхание и промораживание
яючв, наблюдающееся в большинстве почвенных зон, приводит
к свертыванию почвенных коллоидов и седиментации частиц;
7
основная масса коллоидных частиц в почве находится в
коагулированном состоянии, в виде гелей. Поэтому без особой
ошибки можно принимать почвенные растворы за истинные.
ИСТИННЫЕ РАСТВОРЫ
Частицы растворенного вещества в истинном растворе
представлены ■ молекулами и ионами. Это определение
несколько условно, но оно охватывает большинство истинных
растворов, представляющих собой гомогенные однофазные
системы.
Выбор способа выражения концентрации определяется
тем, какие свойства растворов исследуются, а также
удобством и простотой вычислений в зависимости от того, весовые
или объемные меры используются в аналитической работе.
Наиболее простая форма — весовые проценты, т. е.
количество граммов растворенного вещества в 100 г
раствора. В практике почвенного и гидрохимического анализа
количество граммов растворенного вещества часто относят не
к 100 г раствора, а к 100 мл (или чаще к 1 л) раствора. Для
разбавленных водных растворов концентрация вещества,
отнесенная к 100 г или к 100 мл раствора, близко совпадает
с весовыми процентами.
Физические и химические свойства растворов зависят от
соотношения числа молекул и ионов компонентов,
составляющих раствор. Поэтому принято также характеризовать
концентрацию числом молей в единице объема или в
единице веса, или же отношением молей компонентов
раствора, включая растворитель:
1. Молярная концентрация выражается числом
молей растворенного вещества в одном литре раствора.
2. Моляльная концентрация — число молей
растворенного вещества, приходящееся на 1000 г растворителя.
Моляльная концентрация не зависит от температуры, тогда
как значение молярной концентрации изменяется при
повышении или понижении температуры вместе с изменением
объема раствора.
3. Если число молей данного соединения относят к сумме
числа молей всех компонентов раствора (в том числе и
растворителя), то полученная величина носит название
молярной доли (N). Удобно пользоваться молярными
процентами (мол. %), которые показывают, сколько молой
данного вещества содержится в 100 молях веществ,
составляющих раствор.
8
S
I
1
I
I
g
к
c£
1
1
5
M
£
S;
J.
ычисли
X
71
5
о.
О
8
£
^
sf
о
о
+•
9э
5
£
о
о
?
^
2;
+
!?
5
W
^
1
II
о
1
^
eg"
5
i
1
<J
-t-
—
,->
!
О
О
О
+
S
i
^
^
%
и
<о
S?
О
1
8
^
5
II
Й;
о
о
о
5
9Э
5
1
О,
О
8
1
1
О
о
о
Й;
.*--.
w
2
"--<
1
g
о
г*
"s?
и
1
1
о
о
"^
50
5
II
см
£
1
о.
о
о
о
1
^
+
О
о.
о
о
о
Й;
?
?
1
ТЙ
+•
1
о.
О
о~
tf
5
11
5
4. Пользуясь величинами молярных долей, иногда
рассчитывают средний молекулярный вес раствора Mecv:
Мвср = N1Mel + WaMe2 + ... NnMen,
где Мви Мв2, ..., Мвп — молекулярные веса веществ,
составляющих раствор, и Nlt N2, ..., Nn — их молярные доли.
Соотношения между разными способами выражения
концентрации связаны простыми формулами. В табл. 1, где
приведены эти формулы, молярная концентрация обозначена
буквой М (моль/л), молярная доля — N, моляльность — т
(молей на 1000 г растворителя) и процентная
концентрация— С% (граммов вещества в 100 г раствора); величины,
отнесенные к растворителю, обозначены индексом 1, к
растворенному веществу — индексом 2, р—плотность раствора,
Мв — молекулярный вес.
Приведенные в таблице формулы пригодны для двухком-
понентных систем (растворитель и одно растворенное
вещество) . В случае многокомпонентных систем вычисления,
связанные с молярными долями, усложняются. Выбирать тот
или иной способ выражения концентрации надо так, чтобы
можно было легко производить расчеты, а числовые
значения должны правильно и наглядно подтверждать выводы.
Неудачно выбранный способ вычисления затрудняет
обобщение результатов и выявление закономерностей. Вот
несколько примеров. Ниже приведено содержание некоторых ионов
в водных вытяжках, выраженное четырьмя различными
способами.
Солончаковатый солонец. Саратовская обл., 60—65 см
(по В. А. Ковда)
Содержание
г/л ....
9о
мг-экв/л . .
ммол/л . .
Na+
0,17
0,017
7,4
7,4
Са2+
0,12
0,012
6,0
3,0
Иовы
Mg2+
0,12
0,012
9,8
4,9
ci—
0,60
0,060
16,9
16,9
9
SO4
0,27
0,027
5,6
2,8
Как видно из таблицы, для разбавленных растворов с
плотностью, близкой к плотности воды, безразлично, выражать ли
содержание в процентах или граммах на литр, меняется
10
только в 10 раз масштаб величин. Иное дело при переходе
к миллиграмм-эквивалентам или миллимолям. Если
процентное содержание натрия в солончаковатом солонце выше,
чем содержание кальция и магния, то по количеству
эквивалентов натрий значительно уступает магнию. При сравнении
магния и кальция оказывается, что при одинаковом весовом
количестве число атомов и эквивалентов магния в 1,6 раза
превышает те же величины для кальция. Таким образом,
вывод об одинаковом накоплении кальция и магния в этом
горизонте, как это следует из процентного содержания, был бы
неверен. Очевидно, что магния накопилось в 1,5 раза больше,
чем кальция (по числу атомов). В то же время если магния
накопилось меньше, чем натрия (соответственно 4,9 и
7,4 мол/л), то по числу анионных остатков, которые могут
быть ими связаны, Mg2+ значительно превалирует над Na+
(9,8 и 7,4 мг-экв). Иными словами, способ обработки
результатов анализов определяется поставленной целью
исследования. Однако во всех случаях наименее выразительными
следует признать процентные вычисления.
Рассмотрим еще один случай, относящийся уже не к
растворам, а к смесям и сложным многокомпонентным системам.
Потребление азота и зольных элементов дубовым древостоем
в возрасте 48 лет выражается (по Н. П. Ремезову)
следующими величинами в кг/га:
Элементы
кг/га . .
N
50
Са
76
К
22
S
6
Si
15
Р
8
Mg
10
При первом рассмотрении этих данных создается
впечатление, что в наибольших количествах растение потребляет
кальций, затем азот; прочие элементы располагаются в
порядке: K>Si>Mg>P>S, что явно не вяжется с особой ролью
азота в живых организмах. При пересчете на грамм-атомы
и атомные доли получим
Элементы
г-атом
ат. %
(доли)
N
3570
48,0
Са
1900
25,6
К
564
7,6
S
188
2,5
Si
535
7,2
р
266
3,6
Mg
412
5,5
11
Теперь уже очевидно, что половину всех потребляемых
растением атомов составляет азот, второе место занимает
кальций и ряд поглощения иной: N>Ca>K>Si>Mg>P>S.
Напомним, что при оценке валового состава почв
исследователи уже давно пользуются вычислением атомных
(молекулярных) отношений С : N и БЮг : R2O3.
При обработке аналитических результатов правильнее,
конечно, вычислять процентное содержание не окислов, а
элементов. Вычисление окислов часто оправдывают тем, что это
позволяет проверить точность анализа простым
суммированием. Однако почвы могут содержать окислы элементов с
переменной валентностью, тогда эта проверка в известной мере
обесценивается. Если учесть еще, что в большинстве случаев
производится сокращенный валовой анализ, то никаких
видимых преимуществ для вычисления процентного
содержания окислов не остается.
Выражение результатов анализов в эквивалентах, в
молярных долях или атомных процентах дает ясное и истинное
представление о составе образцов почв и об относительной
роли различных частиц (атомов, ионов, молекул) в
построении почвенной массы.
Приведенные способы выражения концентрации
недостаточны для характеристики растворов электролитов или
ионных и окислительно-восстановительных реакций. Часто
пользуются нормальными концентрациями,
показывающими количество грамм-эквивалентов ' вещества в литре
раствора (обозначается н.).
Для нахождения эквивалентного веса вещества
в окислительно-восстановительных реакциях следует
молекулярный вес его разделить на суммарное изменение
валентности атомов, составляющих ион или молекулу
реагирующего вещества.
В реакциях нейтрализации и в обменных реакциях
эквивалентный вес определяется делением молекулярного веса на
число единиц валентности ионов, участвующих в реакции.
Активная концентрация (активность). Все разобранные
формы выражения концентрации являются аналитически
определяемыми концентрациями и указывают на количество
1 Г р а м м-э к в и в а л е и т о м вещества называют количество
вещества в граммах, равноценное (эквивалентное) в реакции одному грамм-
атому водорода или половине грамм-атома кислорода.
Величина грамм-эквивалеита, а следовательно, и нормальность
концентрации зависят от характера реакции, в которой участвует
вещество.
12
вещества, которое содержится в единице объема (или веса)
раствора. Молярную, нормальную или иную концентрацию
раствора находят путем обычных прямых методов анализа —
весового, объемного.
Понятие активности впервые введено Льюисом.
Формально активную концентрацию можно характеризовать как
эффективную, проявляющую себя в действии. Между
аналитически определяемой С и активной а концентрациями имеется
соотношение
а == Су,
где y — средний коэффициент активности электролита.
Например, в 0,10 н. растворе НС1 средний коэффициент
активности равен при 15е — 0,80. Тогда активность 0,1 н.
раствора НС1 равняется:
а =- 0,10-0,80 -0,08.
Это значит, что вычисляя, например, потенциал электрода,
обратимого к водородным ионам, мы должны подставить в
уравнение Нернста величину 0,08 (а не 0,10), только в этом
случае вычисленные и измеренные значения потенциалов
совпадут.
Иными словами, 0,1 н. раствор НС1 проявляет себя в
действии не как 0,1 н., а только как 0,08 н. Следовательно, в
данном случае эффективная, действующая концентрация
раствора меньше, чем аналитически определяемая. В других
случаях активность может быть больше или равной
аналитической концентрации.
Величина коэффициента активности зависит от
концентрации раствора, заряда иона, диэлектрической постоянной
среды, степени гидратации ионов и др. Нам важно
установить следующие закономерности:
1. Величина коэффициента активности одного и того же
вещества изменяется при изменении концентрации раствора.
2. В очень разбавленных растворах (<0,001—0,0001 н.)
величина коэффициента активности для всех веществ близка
к единице; значения активности близки к аналитическим
концентрациям.
3. При повышении концентрации величина коэффициента
активности сначала уменьшается до некоторого предела, а
затем снова возрастает, вплоть до значений, больших
единицы.
4. В разбавленных растворах наибольшими значениями
коэффициентов активности характеризуются 1,1-валентные
13
электролиты (типа NaCl), наименьшими — сложные
электролиты, составленные многозарядными ионами.
Средние коэффициенты активности чистых растворов
некоторых солей (наиболее часто встречающихся в почвах)
приведены в табл. 2.
Таблица 2
Коэффициенты активности электролитов при 25°
Концентрация,
моль1л
0,01
0,05
0,10
0,20
0,30
0,40
0,50
0,70
1,00
2,00
2,50
3,00
3,50
4,00
Электролит
NaCl
0,903
0,822
0,778
0,732
0,679
0,656
0,670
0,691
0,719
0,752
0,791
КС1
0,904
0,819
0,769
0,719
0,688
0,651
0,628
0,606
0,576
0,572
0,571
0,574
0,579
KN03
0,916
0,733
0,659
0,607
0,542
0,494
0,441
0,327
0,293
0,266
0,244
СаС12
0,732
0,582
0,531
0,482
0,462
0,456
0,457
0,469
0,509
0,807
C.39)
MgCIj
0,565
0,520
0,507
0,508
0,514
0,542
0,613
1,143
A1CI3
0,409
0,360
0,308
0,323
0,354
0,415
0,578
1,81—
—1,95
K2S04
0,745
0,529
0,441
0,361
0,313
0,265
Na^Oi
0,721
0,529
0,445
0,365
0,322
0,268
0,234
0,204
Mgso,
0,262
0,195
0,142
0,088
0,072
0,068
0,057
0,049
0,042
0,044
0,0495
Коэффициенты активности отдельных ионов какой-либо
соли МаАу связаны со средним коэффициентом
активности y± уравнением
i_
y±-(yx+-yL)x+y-
Для 1,1-валентного электролита (NaCl)
Wi = (YNa+ ' Уа-У" =^Уш+~Т^г:'
Приближенное значение коэффициента активности иона
в сильно разбавленных растворах вычисляют по уравнению
Дебая и Гюккеля
где \j — коэффициент активности ионов /-того рода; z —
валентность, А — константа, приближенно равная для водных
14
растворов 0,5 (при 20°), и Г—ионная сила раствора,
рассчитываемая по формуле
„ CjZ{ + С2г2 ...+ Спгп
~~~ ~ 2
где С — концентрация иона, а индексы 1, 2, ...; п — разные
ионы.
Ионную силу растворов, содержащих только один какой-
либо электролит, легко вычислить умножением молярной
концентрации на соответствующий множитель, различный для
электролитов разного типа. Для солей типа NaCI множитель
равен 1, для солей типа СаС12, Na2S04 — 3, для солей типа
MgS04 — 4, для солей типа А1СЬ — 6 и для солей типа
A12(S04K—15.
Для растворов с концентрацией выше 0,01 М выведены
более сложные уравнения, пользуясь которыми можно
получить достаточно точные значения. Так, средний коэффициент
активности хлористого натрия в растворах с концентрацией
до 4,0 М можно точно вычислить по уравнению
, JU549KT___ + 00410Ci
1 + 0,9736/Г
где числовые параметры определяются свойствами
присутствующих в растворе ионов.
В практике почвенных исследований определение
активности ионов чаще всего проводят для водородных ионов, в
виде показателя активности рН — отрицательного
логарифма активности водородных ионов, — 1 gan.
За последние годы большое внимание уделяется изучению
активности ионов натрия и калия в почвах и почвенных
растворах. Особая роль теории активности в понимании
почвообразовательного процесса обусловлена специфическими
свойствами почвенной среды. Невысокая влажность,
сложный химический состав почвенных растворов и резкие
колебания в их концентрации, высокая удельная поверхность
почвенных коллоидов — все это резко влияет на свойства
веществ, входящих в состав почвенных растворов. Степень
участия тех или иных ионов в обменных реакциях или
других процессах может изменяться в несколько раз в
зависимости от величины коэффициента активности. Фактическое
влияние на почвохимические процессы даже одновалентных
ионов, как например ионов натрия, может быть снижено в
два раза по сравнению с влиянием, которого можно было
15
ожидать исходя из аналитически определяемых
концентраций. Примером могут служить результаты определения
активности ионов натрия в луговом солончаке при помощи
Na-стеклянного электрода, (табл. 3).
|Таблица 3
Активность и коэффициент активности ионов натрия в луговом солончаке
(по Д. С. Орлову и Н. Н. Цикуриной)
Название
почвы
Луговой
солончак, сред-
несуглини-
стый
Глубина взятия
образца, см
корка
0—40
65—75
115—160
160—200
Иональность
водной
ВЫТЯЖКИ
0,660
0,340
0,332
0,168
0,103
Коэффициент
активности
Na+
(измеренный)
0,56
0,66
0,63
0,73
0,84
Концентрация
г же л
0,174
0,132
0,091
0,048
0,035
Активность
Na+
0,097
0,087
0,057
0,035
0,029
Отсюда вытекает необходимость прямого
непосредственного определения активности всех ионов (молекул)
почвенного раствора.
Прямое определение активности водородных ионов (рН)
служит для диагностики нуждаемости кислых почв в
известковании. Аналогично для некоторых почв Прикаспийской
низменности было показано, что активность ионов натрия в
водных суспензиях может служить диагностическим
признаком степени солонцеватости. Если ввести обозначение
pNa =—'gaNa+> TO оказывается, что среди исследованных
почв несолонцеватые разности имели pNa> 3,0 слабо- и
среднесолонцеватые — 1,5—3,0 и в солонцах pNa<l,5
(Н. Г. Зырин и Д. С. Орлов).
Произведение растворимости. Растворение <и осаждение
труднорастворимых соединений в их насыщенных растворах
подсчитывают с помощью произведения растворимости. Если
растворенная часть электролита состава МтАп диссоциирует
нацело на ионы: МтЛ„ ^,mMz+ -|-пЛг_(где z+ и г~ —
валентности ионов), то произведение растворимости ПрмтА„'-
при этом [Mz+] и [А2-] — концентрации, а у+ и
у-—коэффициенты активности соответствующих ионов.
При постоянной температуре величина произведения
растворимости остается постоянной. Прибавление к
насыщенному раствору электролита соединения с одноименным ионом
16
ведет к выпадению в осадок части электролита,
насыщающего раствор, даже если он не относится к
труднорастворимым соединениям. Произведение растворимости позволяет
учитывать взаимодействие двух труднорастворимых
соединений, имеющих одноименные ионы. Если ввести в раствор
некоторое количество электролита, не имеющего общих
ионов с насыщающим, то уменьшение активности
насыщающей соли вызовет дополнительное растворение осадка; таким
образом, растворимость труднорастворимого электролита
повышается в присутствии солей. Но произведение
растворимости, выраженное через активности ионов, остается
постоянной величиной. Величина произведения растворимости
некоторых электролитов имеет решающее значение в процессах
соленакопления в почвах и образования засоленных почв и
солончаков. Почвенные минералы и гумусовые вещества
сильно влияют на активность солей в почвенном растворе.
Поскольку речь идет о двухфазной системе насыщенный
раствор соли — осадок, отметим еще одно обстоятельство.
В равновесной системе во всех фазах активность отдельного
компонента одна и та же, если только она определяется по
отношению к одному и тому же стандартному состоянию.
Это положение вытекает из равенства химических
потенциалов компонента во всех фазах равновесной системы.
К тем случаям, когда на границе раздела фаз существует
скачок потенциала (например, имеется некоторая разность
потенциалов между твердой фазой и раствором), последнее
положение неприменимо. В таких системах говорят о
равенстве электрохимических потенциалов.
Константа и степень диссоциации. Диссоциирующий в
водном растворе электролит можно характеризовать
степенью диссоциации «. Эта величина представляет
собой отношение числа распавшихся на ионы молекул к
общему числу находящихся в растворе молекул электролита.
Степень диссоциации не является постоянной величиной, а
изменяется с изменением концентрации раствора. При
разбавлении степень диссоциации электролита увеличивается.
Более общей характеристикой является константа
диссоциации К, определяемая для процесса диссоциации
электролита МА:
МАг±М+ + А"\
По уравнению закона действующих масс
к_ [М+][А-]
[МА]
17
Это уравнение справедливо только в тех случаях, когда
концентрация раствора и степень диссоциации электролита
очень малы, т. е. когда соответствующее отношение
коэффициентов активности близко к единице:
Точнее, константа диссоциации определяется путем
подстановки в уравнение не концентраций, а активностей, т. е.
„ [М+] [Л~] Y+Y- йм+яа-
[MA] Y±
JMA
Зависимость между величиной степени диссоциации и
константой диссоциации слабого электролита выражается
законом разбавления В. Оствальда
-,Са-- = к,
1 —а
где С — концентрация электролита.
Если степень диссоциации <0,02—0,01, то в расчетах
величиной 1—а можно пренебречь и пользоваться
приближенной упрощенной формулой
К ~ Со?.
Ионное произведение воды. Константа диссоциации воды,
как и любого электролита, равна
v йн+аон-
д =
ан2о
Величина К при обычной температуре очень мала
A,8-Ю-16 при 22°), поэтому без заметной ошибки можно
принять, что концентрация (активность) недиссоциированных
молекул воды равна общему количеству воды и,
следовательно, остается практически величиной постоянной. Тогда
йп+аон_ - ЛаН20 - 1,8-10-16-55,56 - 1 • 10~14 = КНг0,
где 55,56 — количество молей воды в литре воды.
Величина Кн2о носит название ионного
произведения воды и имеет константное значение при постоянной
температуре:
18
сс
0
18
25
50
100
Vo
1,139-Ю-15
5,702-Ю-15
1,008-Ю-14
5,474-Ю-14
5,9- Ю-13
аи+ = аОН-
3,38 -10~8
7,64 -Ю-8
1,004-Ю-7
2,339-Ю-7
7,7 -Ю-7
—lg°H-f = РН
7,972
7,117
6,999
6,631
6,12
Для ,воды при всех температурах pH = lgCoH_—lgKii2o-
Гидролиз. В процессе гидролиза происходит связывание
водородных или гидроксильных ионов (или тех и других) с
ионами растворенного вещества с образованием плохо
диссоциируемых соединений, например:
Na2COs + Н20 z± NaHC03 + NaOH.
В этой реакции водородный ион связывается в малодиссо-
циированную группу НСОГ и равновесие реакции
смещается вправо. Образующаяся NaOH придает раствору
щелочную реакцию. Реакция раствора зависит от типа гидролизуе-
мой соли, и концентрацию водородных ионов в нем можно
рассчитать по следующим формулам.
Если соль образована сильным основанием и слабой
кислотой, то
Сн+= у Ян,о-£-А-.
где КигО — ионное произведение воды, Кна — константа
диссоциации слабой кислоты и С — концентрация соли.
Если соль образована сильной кислотой и слабым
основанием, то
Сн+ = 1/ ^н*° ~к *
V лмон
где Кмон — константа диссоциации слабого основания.
Для соли слабой кислоты и слабого основания
С„+ —А/ /Сн.о -jr~ ~ (обозначения те же).
н У Кмон
Гидролиз солей многоосновных кислот и оснований может
проходить ступенчато, образуя ряд основных или кислых
19
солей. Так, гидролиз солей алюминия может проходить в
три стадии:
Al3+ -f НОН г± АЮН2+ + Н+,
А13+ + 2НОН ^ А1 (ОН)^ + 2Н+,
А13+ + ЗНОН ?± А1 (ОНK + ЗН+.
Изучая природу почвенной кислотности, В. А. Чернов
показал, что в KCl-вытяжке гидролиз протекает по первому
уравнению реакции при малых концентрациях солей
алюминия (Ю-2—10_4М). Пользуясь одним из приведенных выше
уравнений, можно рассчитать концентрацию водородных
ионов и рН раствора А1СЬ:
рН ,-: 0,5рЛ:гидр- 0.5 lg([AI^J - [H+D,
где /Сгидр — константа гидролиза.
Это уравнение, в частности, было использовано для
доказательства гипотезы, объясняющей почвенную кислотность
наличием обменного алюминия.
Приведенные выше формулы позволяют вычислить рНг
при котором начинается образование осадка гидроокиси или
основной соли, что имеет огромное значение для оценки
миграционной способности соединений таких элементов, как
Al, Fe, Zn, Ni, Со, Си и т. п. в профиле различных почв.
Величина рН образования гидроокисей вычисляется по
следующей формуле:
рН -= — lg Пр — lg/CH2o lg om2+,
г г '
где Mz+ ■— обозначение катиона, а г — его заряд; остальные
обозначения прежние.
Буферность растворов. Растворы солей слабых кислот if
сильных оснований в присутствии слабой кислоты, например
СНзСОСЖа + СНзСООН, и растворы солей слабых оснований
и сильных кислот в присутствии слабого основания,
например NH4CI + NH4OH, способны сохранять постоянство
активности водородных ионов после прибавления сильной кислоты
или щелочи. Незначительно меняется рН этих растворов при
разбавлении. Способность сглаживать, «буферить», реакцию
растворов получило название буферности, а сами
растворы — буферных растворов. Буферные растворы
разнообразны по составу и по диапазонам рН. Буферные
растворы служат эталонными в отношении рН. Когда нужно
20
создать в растворе определенную концентрацию водородных
ионов, прибавляют к нему буферный раствор.
Достаточно точные расчеты концентраций компонентов и
значений активности (рН) буферных растворов легко провести
по следующим приближенным формулам, принимая, что соль
нацело диссоциирована.
Для раствора слабой кислоты и соли сильного основания
и слабой кислоты
1Н+] ~- /Снл Снл
смл
где /Сна — константа диссоциации кислоты, Сна —
концентрация кислоты. Смл —■ концентрация соли.
рН - — lg [H+] ---= - lg/Снл - Ig Сна + lg СМА.
Для раствора сильной кислоты и соли сильной кислоты и
слабого основания:
гон-1 -^ Кьюн —н, [н+| - СлАо
смл ^мон^мон
рН --= — lgfH+J -- — lg/fH,o — lgCMA + Ig/Слюн + JgCftioH,
рН ■--- 14 — р/Слюн ~ lg Смл + lg Смон,
где р/Смон = —Ig^MOH, Смон — концентрация основания,.
Смл — концентрация соли, /Смон — константа диссоциации
основания, /Снго — ионное произведение воды.
Состав некоторых буферных смесей приведен в табл. 4.
Электропроводность электролитов. Удельная
электропроводность раствора электролита — это
электропроводность 1 см% раствора, помещенного между двумя
плоскими параллельными электродами, каждый площадью J см2,
при расстоянии между электродами 1 см, и равная 1/р, где
р — удельное сопротивление.
Для растворов бинарных электролитов, имеющих только
одновалентные ионы, удельная электропроводность
X = -L - -^- (U + V) ом-1-см-1,
р 1000
где и — степень диссоциации, С —концентрация электролита
вг-экв/л, F — число Фарадея=96 49б к, U и
V—абсолютные скорости движения соответственно катиона и аниона в
см/сек при напряженности поля в I в(см.
21
Таблица 4
Состав буферных смесей
а. Фосфатные смеси Зеренсена—Вальбума
рН
4,94
5,29
5,59
5,91
6,24
6,47
6,64
6,81
"i-, М
КН2Р01( мл
9,9
9,75
9,5
9,0
8,0
7,0
6,0
5,0
"« М
Na2HPO„ мл
0,1
0,25
0,5
1,0
2,0
3,0
4,0
5,0
РН
6,98
7,17
7,38
7,73
8,04
8,34
8,67
9,18
'« м
КН.ГО,. мл
4,0
3,0
2,0
1.0
0,5
0,25
0,1
0,0
V,» М
Na.HPO,, мл
6,0
7,0
8,0
9,0
9,5
9,75
9,9
10,0
б. Б о р а т н ы е смеси Зеренсен а—В а л ь б у м а
рН
7,62
7,94
8,14
8,29
8,51
8,68
8,80
8,91
9,01
9,09
9,17
0,05 М
NaaB407,
мл
5,25
5,5
5,75
6,0
6,5
7,0
7,5
8,0
8,5
9,0
9,5
0.1 н. НС1,
4,75
4,5
4,25
4,0
3,5
3,0
2,5
- 2,0
1,5
1,0
0,5
РН
9,24
9,36
9,50
9,68
9,97
11,07
12,37
0,05 М
NaB.O,,
МЛ
10,0
9,0
8,0
7,0
6,0
5,0
4,0
0,1 н.
NaOH,
мл
0,0
1,0
2,0
3,0
4,0
5,0
6,0
в. Ацетатные смеси (к 50 мл 1 н. СН3СОО№ добавляют х мл 1 н. НС1
и разбавляют водой до 250 мл)
РН
0,65
0,75
0,91
1,09
1,24
1,42
1,71
1,85
X
100
90
80
70
65
60
55
53,5
РН
1,99
2,32
2,64
2,72
3,06
3,29
3,49
3,61
X
52,5
51,0
50,0
49,75
48,5
47,5
46,25
45,00
рН
3,79
3,95
4,19
4,39
4,58
4,76
4,95
5,20
X
42,5
40,0
35,0
30,0
25,0
20,0
15,0
10,0
г Универсальные буферные смеси для области рН=1,81—11,98 при 18°
(к 100 мл смеси равных объемов 0,04 М Н3Р04, 0,04 н. СН3СООН
и 0,04 М НзВОз добавляют х мл 0,2 н. NaOH)
X
0
2,5
5,0
7,5
10,0
12,5
15,0
17,5
20,0
22,5
РН
1,81
1,89
1,98
2,09
2,21
2,36
2,56
2,87
3,29
3,78
X
25,0
27,5
30,0
32,5
35,0
37,5
40,0
42,5
45,0
47,5
рН
4,10
4,35
4,56
4,78
5,02
5,33
5,72
6,09
6,37
6,59
X
50,0
52,5
55,0
57,5
60,0
62,5
65,0
67,5
70,0
72,5
рН
6,80
7,00
7,24
7,54
7,96
8,36
8,69
8,95
9,15
9,37
X
! 1М
75,0
77,5
80,0
82,5
85,0
87,5
90,0
92,5
95,0
97,5
100,0
РН
9,62
9,91
10,38
10,88
11,20
11,40
11,58
11,70
11,82
11,92
11,98
е. Буферные смеси для области рН= 10,17—11,4
(смесь двух компонентов разбавляют водой до 100 мл)
рН
10,17
10 35
10,55
0.1 М № СОз,
МЛ
50
50
50
0,1 и.
на, ял
20
15
10
ри
10,86
11,04
0.1М
Na2C03, мл
50
50
0,1 н.
НС1, мл
5,0
3,0
ж. Буферные смеси с Н3ВОз и Na2C03
рн
8,0
8,2
8,4
8,6
8,8
9,0
9,2
9,4
Раствор 6,2 г
HsBO, и
7,45 г КС1
в литре, мл
88,8
85,0
80,7
75,7
69,5
63,0
56,4
49,7
0,1М растнор
Na_C03, мл
11,2
15,0
19,3
24,3
30,5
37,0
43,6
50,3
рН
9,6
9,8
10,0
10,2
10,4
10,6
10,8
11,0
Раствор 6,2 г
Н»ВО,
и 7,45 г КС1
в литре, мл
42,9
36,0
29,1
22,1
15,4
9,8
5,7
3,5
0,1М раствор
Na2COs, мл
57,1
64,0
70,9
77,9
84,6
90,2
94,3
96,5
Абсолютные скорости движения ионов изменяются при
разбавлении раствора, так как меняется сила взаимодействия
между ионами, что влечет за собой и изменение
электропроводности.
Эквивалентная электропроводность раствора Л
электролита равна электропроводности объема раствора,
содержащего 1 г • же электролита, помещенного между двумя
плоскими параллельными электродами с расстоянием между
ними в 1 см. Связь между удельной х и эквивалентной Л
электропроводностью выражается \ равнением
где С — число грамм-эквивалентов электролита в 1 л
раствора и
A-=aF(U+ V).
Методом электропроводности пользуются в почвоведении
для ориентировочного, быстрого, определения солесодержания
в почве, в анализе при титровании (кондуктометрическое
титрование) и в случаях, когда электропроводность служит
индикатором (модельные опыты промывки засоленных
почв и т. п.).
КОЛЛОИДНЫЕ РАСТВОРЫ
В почвенной литературе коллоиды часто рассматривают
как частицы определенного размера (<0,1—0,2 мк).
Коллоидальное состояние вещества, широко распространенное в
природе, характеризуется рядом специфических,
качественных особенностей, обусловленных как размерами частиц, так
и гетерогенностью, или многофазностью, системы.
Существование поверхности раздела между дисперсной фазой
и дисперсионной средой коренным образом отличает
коллоидные растворы от истинных. Вторая особенность
коллоидальных растворов и близких к ним суспензий — агрегат и в-
ная неустойчивость.
Классификация и методы изучения коллоидных систем
разработаны преимущественно для органических
высокомолекулярных соединений. К таким системам в почвах могут
быть отнесены гумусовые вещества: гуминовые и фульво-
кислоты, слабогумифицированные органические остатки,
продукты распада плазмы микроорганизмов. К лиофобным
* 24
суспензоидам среди почвенных компонентов относятся
гидроокиси железа, алюминия, марганца, кремневая кислота.
Большой и самостоятельный класс составляют почвенные
вторичные минералы со слоистой кристаллической
решеткой.
Одна из важнейших характеристик коллоидных систем и
суспензий — степень дисперсности, которая хорошо
описывается величиной удельной поверхности, т. е.
поверхности, приходящейся на единицу объема или веса
вещества.
Существует много методов определения поверхности
высокодисперсных систем. Они позволяют учитывать как внешнюю,
так и внутреннюю поверхность. Последняя может состоять
из поверхности пор разного диаметра, внутренней
поверхности в межпакетном пространстве глинных минералов.
Поверхность определяют сорбционными методами или
учетом теплоты смачивания. Если сорбированное соединение
покрывает частицы дисперсной системы мономолекулярным
слоем (или другой, известной толщины), то, зная площадь
одной молекулы, легко вычислить поверхность сорбента.
Многие соединения, имеющие большой объем, не проникают
в поры сорбента и не могут поглощаться внутренней
поверхностью, что позволяет определить порознь оба вида
поверхности.
По теплоте смачивания определяют степень гидрофильно-
сти поверхности путем нахождения этой теплоты для одного
и того же вещества при смачивании его водой и бензолом.
Уменьшение отношения идет пропорционально снижению
гидрофильности изучаемого вещества.
Концентрация коллоидных растворов. Поскольку
коллоидные частицы (мицеллы) неодинаковы по размеру и состоят
обычно из нескольких молекул, а реакции взаимодействия
частиц между собой и с ионами и молекулами происходят
на поверхности частиц, то понятие концентрации коллоидных
растворов и ее определение значительно усложняются.
При изучении монодисперсных систем используют ч а-
стичную концентрацию, т. е. определяют количество
частиц в единице объема. Для монодисперсных систем
частичная концентрация пропорциональна в то же время
величине суммарной поверхности частиц.
Полидисперсная система не может быть однозначно
охарактеризована числом частиц; разные по размерам частицы
играют неодинаковую роль в формировании общих свойств
изучаемого коллоидного раствора. Поэтому определяют
25
содержание фракций частиц различного размера и выражают
состав либо в процентах, либо в виде так называемой
кривой распределения, часто используемой для
характеристики механического состава почв.
Концентрация коллоидных растворов не всегда
одинаковая и постоянная во всем объеме раствора, как это имеет
место для истинных растворов; в последних не происходит
оседания ионов или молекул под действием силы тяжести,
поскольку ее влияние компенсируется хаотическим тепловым
движением молекул, приводящим к равномерному
диффузному распределению последних в дисперсионной среде.
По мере увеличения размеров частиц диффузия их
уменьшается, тогда как скорость оседания под действием силы
тяжести возрастает и в коллоидальных растворах изменение
концентрации по вертикали подчиняется гипсометрическому
закону:
п,
h = 2,3 Ъ—(—*—\
Nmg \ d — d0 J
где do — удельный вес дисперсионной среды, d — удельный
вес дисперсной фазы, т-—масса частицы и N — число Аво-
тадро; — — отношение числа частиц в равных объемах
системы, разность высот которых равна 1г. Величина h
является характеристикой кинетической устойчивости системы.
Если положить —— = 2, то изменение высоты, равное 1г,
п.
2
отвечает изменению концентрации в два раза.
Практически заметное изменение концентрации
наблюдается лишь для частиц более крупных, чем коллоидные, т. е.
в суспензиях. Собственно коллоиды показывают столь
высокое значение 1г, что часто в коллоидных растворах,
хранившихся несколько лет, не замечается какого-либо изменения
концентрации.
Скорость оседания суспензий более значительна, что
позволяет использовать для их дисперсионного анализа
формулу Стокса
ц_ 2 r*(dK-d)g
9 Ц
где т] — вязкость жидкости, g — ускорение силы тяжести,
dK — плотность дисперсной фазы, d — плотность дисперсион-
26
ной среды, а г — эффективный радиус шарообразной
частицы, оседающей со скоростью V.
Пользуясь формулой, найдем радиус частиц, подставив
значение g=981:
г = 0,06771/ Щ см.
V dK-d
Аналитическое определение размеров частиц по скорости
их оседания в значительной степени условно, что
объясняется, во-первых, неодинаковой плотностью различных
составных частей почвы и, во-вторых, тем, что реальные частицы
почвенных коллоидов и суспензий никогда не имеют
идеальной шарообразной формы. Иными словами, в анализе
учитывается не истинный, а эффективный радиус, или радиус таких
шарообразных частиц, которые способны оседать с
наблюдаемой скоростью. Радиус реальных частиц на самом деле
меньше эффективного. Кроме того, уравнение Стокса не
учитывает взаимодействия поверхности частиц с дисперсионной
средой.
Для минеральных частиц с d=2,7 время т, за которое
частица опускается в водной среде на 1 см, равно (по
А. В. Думанскому):
Радиус
частиц, см
t
Ю-3
31,03 сек
Ю—4
51,7 мин
ю—5
86,2 ч
ю—6
359 дней
ю-7
100 лет
В связи с малой скоростью оседания метод практически
применим только к грубым суспензиям, и поэтому в
механическом анализе почв он используется для фракционирования
частиц с эффективным диаметром >1 мк.
Анализ коллоидных систем, выделение дисперсной фазы,
определение размера частиц и молекулярных весов проводят
с помощью центрифуги, где сила тяжести заменяется
центробежной силой. В центрифуге при определенной скорости
вращения процессы диффузии и седиментации частиц
уравновешиваются после того, как достигается определенный
градиент концентрации в направлении, перпендикулярном
оси вращения. В этих условиях можно вычислить вес частиц
(или вес больших молекул), если измерить концентрации
вещества С{ и Cz на расстоянии Х\ и х2 от центра вращения:
27
Со
2RT In —-
С,
где со — угловая скорость вращения B я умноженное на число
■оборотов в секунду), v — парциальный удельный объем
макромолекулы (частиц) и р — плотность раствора.
Эти расчеты сделаны для анализа однородных по
размерам частиц. Для почв характерны полидисперсные и
гетерогенные системы, обычное понятие молекулярного веса для
которых теряет смысл. Так, гумусовые вещества неоднородны
по размерам частиц и молекулярному весу. Поэтому
определяемый молекулярный вес таких соединений является лишь
средней статистической величиной.
Способ усреднения может быть разный, в связи с чем различают три
вида средних молекулярных весов.
Сред нечисловой молекулярный вес Мп. Если весовая
доля вещества с молекулярным весом Mi равна Wt , то Wi =tiiMi,
где гц—число молей вещества с данным молекулярным весом. Средне-
числовой вес вычисляется путем деления суммы весовых долей всех
фракций на общее число молей:
Среднечисловое значение получают обычно при определении
молекулярного веса химическими методами.
Сред неве совой молекулярный вес Mw. В этом случае
весовая доля каждой фракции дополнительно умножается на
молекулярный вес. В результате каждая молекула учитывается пропорционально
се весовой доле:
м - ^ntM"
mw~ •
Средневесовое значение получают при определении молекулярных весов
методом светорассеивания.
Средний молекулярный вес Мг вычисляется по формуле
Им*?
Если при исследовании какого-либо вещества найдено, что
ТЛг ^MW=M„,
то можно говорить об его однородности по молекулярному весу.
м
w
28
Осмотическое давление р присуще и коллоидным
растворам, причем величина его зависит от частичной концентрации
раствора, что находится в соответствии с молекулярно-кине-
тической теорией, т. е.
р = — пти2,
3
где п — число частиц в единице объема, т — масса и
и — средняя скорость движения частиц.
Сравнивая два раствора с различной концентрацией и
учитывая, что при одной и той же температуре
произведение ти2 имеет постоянное значение, легко заметить, что
р1:рг--п1: пг,
т. е. осмотическое давление прямо пропорционально числу
частиц в единице объема.
При одинаковой плотности частиц
Pi "• Pz ^ (гг •• М3
величина осмотического давления обратно пропорциональна
кубу радиуса частиц. Два последних уравнения позволяют
определить радиус и количество частиц в растворе по
осмотическому давлению. Малые величины осмотического
давления и изменение степени дисперсности коллоидов во времени
ограничивают применение этого метода. Значительные
погрешности вносит также присутствие истинно
растворенных веществ, которые не всегда можно удалить, не изменяя
при этом некоторых свойств коллоидных растворов.
Рассеяние света. Количество света, рассеянного при
прохождении через коллоидный раствор, зависит от
концентрации, величины частиц и длины волны света. Суммарное
изменение интенсивности света, проходящего через систему, по
Теореллу определяется уравнением
„ nvH
где п — число частиц в золе, v — их объем, / — длина пути,
проходимого лучом в слое, и h — длина волны рассеянного
света.
Применимость уравнения Теорелла ограничивается
размерами частиц <20—30 ммк. Рассеивание света такими
частицами вызвано дифракцией света, возникающей, если
размеры частиц меньше длины световой волны. При умень-
29
шении степени дисперсности показатель степени у
величины X начинает убывать (вплоть до нуля). Этим явлением
можно воспользоваться для определения степени
дисперсности золя. Рассеивание света в грубодисперсных системах
(г>30—50 ммк) вызвано уже не дифракцией, а отражением
света от поверхности частиц.
Количество света, отраженного системой, размер и форма
частиц в которой не изменяется, пропорционально количеству
частиц (концентрации), что позволяет определять
концентрацию мутных сред, измеряя при помощи нефелометров
яркость рассеянного системой (отраженного) света.
Для определения молекулярных весов вычисляют отношение Рэ-
лея R$, Если /о — интенсивность светового пучка, претерпевающего
рассеяние, а /в — интенсивность рассеянного света на расстоянии г, то
/?.--=-4—
или, при измерении рассеяния под углом 90°:
R —^
io
Отношение Рэлея связано со свойствами рассеивающей среды
уравнением
2я2 2 • дпк \2 RTC _ KRTC
дС дС
где N0— число Авогадро, Я,0 — длина волны света в вакууме, п^—пока-
дя,
затель преломления среды, — — инкремент показателя преломления
дС
для растворенного вещества, р — осмотическое давление, С —
концентрация растворенного вещества. Решение этого выражения приводит к
формуле
КС 1
+ 2ВС,
*» Mw
где В — константа.
Измерив несколько значений /?эо для разных концентраций и построив,
график зависимости KC/Rw от С, находят, что отрезок, отсекаемый на
оси ординат, равен обратной величине молекулярного веса. Этот метод
дает средневесовое значение молекулярного веса Mw-
Электрофорез. В результате сорбции ионов на
поверхности коллоидной частицы, или диссоциации ее молекул,
частицы дисперсной системы несут электрический заряд
30
{+ или —). В электрическом поле частицы золя движутся
к положительному или отрицательному полюсу. Это явление
называется электрофорезом.
Электрофорез может быть использован для определения
знака заряда частиц и величины электрокинетического
потенциала. Зависимость между скоростью движения частиц
к электроду и величиной электрокинетического потенциала
определяется уравнением
ZED
и-= -^ ,
4т\1
где и — скорость в см/сек, £ — електрокинетический
потенциал, Е — разность потенциалов между электродами,
/—расстояние между ними, г\ — вязкость жидкости в динах,
D — диэлектрическая постоянная жидкости.
Если представить коллоидную частицу в виде
шарообразного конденсатора, то заряд е частицы равен
в = -ь—L_f
где г\— радиус частицы, г—радиус частицы вместе с
внешней поверхностью двойного электрического слоя.
Дисперсионная среда заряжена противоположно заряду
дисперсной фазы. В электрическом поле дисперсионная среда
движется к другому полюсу, нежели частицы вещества. Этим
пользуются при определении знака заряда и величины
электрокинетического потенциала. Движение жидкости при
наложении электрического поля на дисперсную систему получило
название электроосмоса. По скорости движения
жидкости при электроосмосе можно вычислить электрокинетический
потенциал
4лт)
где и — скорость передвижения жидкости при градиенте
падения напряжения поля 1 в/см
Поглотительная способность. Поглотительная и
ионообменная способность почв зависит от свойств и строения
.поверхности коллоидных частиц. Заметной поглотительной
способностью обладают не только коллоидные, но и
значительно более крупные по размерам частицы. Но поскольку
емкость поглощения для частиц, одинаковых по составу и
различных по размеру, определяется величиной удельной
31
поверхности, то крупные фракции, как правило, не
принимаются во внимание. О том, насколько быстро уменьшается
удельная поверхность крупных фракций, можно судить по
результатам исследований А. Д. Воронина (табл. 5).
Таблица 5
Удельная поверхность фракций механических элементов в м-
Почва
Светло-каштановая, тяжело-
суглинистая
Горизонт
и глубина,
см
А 0—26
В 30—40
С 70—90
250—270
Фракции, мм
0,1—0,05
1,9
2,3
0,05—0,01
2,9
2,8
3,2
0,01—0,005
15,0
5,0
5,0
4,8
0,005—0,001
65,0
31,0
17,2
16,6
< 0,001
187
226
218
230
Поглощение недиссоциированных (или м алодиссоцииро-
ванных) веществ различными адсорбентами происходит в
соответствии с изотермой адсорбции Фрейндлиха:
т
где С — вес адсорбированного вещества, т — вес адсорбента,
С — равновесная концентрация адсорбтива, а а и Ъ —
константы. Уравнение Фрейндлиха выведено для случая
адсорбции, т. е. когда на поверхности раздела твердое тело —
жидкость, или твердое тело — газ происходит увеличение
концентрации газа или растворенного вещества.
Для почвенных коллоидов значительно большее значение
имеет ионообменная способность. При обмене ионов не
происходит одностороннего поглощения вещества из раствора;
поглощаемые ионы обмениваются на ионы твердой фазы.
Явление поглощения сводится к распределению ионов между
твердой фазой и раствором
nNa + K+^nK + Na+,
где riNa и ПК — почвенный поглощающий комплекс,
содержащий ионы Na+ и К+. Концентрация поглощенных Na+ и К+
равна соответственно [ПЫа] и [ПК]. Количественная
характеристика реакции обмена сводится к нахождению функции
распределения ионов между обеими фазами. Этим
ионообменная способность отличается от адсорбции, например,
газа твердым телом. Поглощаемые из раствора ионы
удерживаются силами межионного притяжения, образуя мало-
32
Диссоциированные соединения с ионогенными группами на
поверхности коллоидных частиц.
Количественно реакция обмена, принимая, что
коэффициенты активности поглощенных ионов близки к единице,
выражается уравнением закона действующих масс
ак+
[ПК] oNa+ [ПК]
= Л, или
к
Если активные центры, способные к поглощению ионов, не
одинаковы на всей поверхности почвы и различны по
прочности связи соединений, образуемых с поглощаемыми
ионами, тэ величина К в уравнении не остается постоянной.
Изотермы ионного обмена характеризуются графиками,
выражающими распределение ионов между твердой фазой н раствором. Например
при обмене ионов Л},+ на Мг+ по оси ординат откладывают отношение
количеств поглощенных катионов (/И,//И2)„, а по оси абшиес -
отношение активностей те\ же ионов раство- "ткоше
ра. При идеальном подчинении систе- rju \
мы закону действующих масс иа 1'тг )
графике получается прямая линия z n
(рис. 1, /), причем тангенс угла
наклона этой прямой равен константе
обменной реакции. По Е. Н. Гапону.
для двух различных групп активных
центров, вступающих в реакцию
последовательно, изотерма выражается
ломаной линией (рис. !,//).
Константы обмена для каждой из групп
могут быть вычислены из
соответствующих углов наклона. При достаточно
большом числе разнородных
активных центров изотерма обмена
выражается выпуклой кривой с большей
или меньшей степенью крутизны
(рис. 1, ///). Вычислять в этом
случае какую-либо константу уже не
имеет смысла; расчет состава
поглощенных катионов в практических
целях при такой форме кривой лучше
вести графическим методом.
В некоторых случаях обмен ионов
в почвах значительно лучше
характеризуется логарифмической изотермой
адсорбции, выведенной для обмена
потенциалопределяющих ионов (уравиие Фрумкина-Гортикова-Гапона):
rk ^ а + Р рН — у рМе,
ГаДкетитю<Т„К^ЧеСТВ0 П0ГЛ0Ще"«°го катиона, отрицательный логарифм
V ™0И( ™ °Р0Г° В раСТ80ре раВоеН РМе; "• PY-константы, причем
Y - и,ьр для двухвалентных и Y= P Для одновалентных катионов.
2 Зак. 51
33
Рис. 1. Изотермы ионного обмена
(по Е. Н. Гапону), (-^~) -
отношение количества нонов
металла Mt и Мг в поглощенном
\ м9 L
состоянии, | • ,/ 1 —их
тнвности в растворе
ак-
Коагуляция коллоидов. Процесс коагуляции протекает по
крайней мере в две стадии. Первая стадия — агрегация
частиц (или скрытая коагуляция) не приводит к каким-либо
заметным внешним изменениям. Во второй стадии
(явной коагуляции) коллоидный раствор мутнеет,
изменяется окраска или начинается выпадение осадка —
седиментация.
В лиофобных золях, как правило, очень быстро
изменяется степень дисперсности; начавшись с изменения
цвета или помутнения, явная коагуляция лиофобных золен"
обязательно заканчивается выпадением в осадок. Этст
процесс необратим.
Условия коагуляции лиофобных коллоидов весьма
разнообразны; коагуляция может наступить при изменении
концентрации золя, реакции среды (рН), температуры, при
добавлении электролитов.
Лиофильные золи значительно более устойчивы, в
связи с этим начавшийся процесс скрытой коагуляции не
всегда доходит до стадии явной коагуляции. При хранении
лиофильных золей степень дисперсности самопроизвольно
изменяется во времени, но эти изменения невелики и
постепенны.
Коагуляция лиофобных золей электролитами происходит
под действием иона, знак заряда которого противоположен
заряду коллоидной частицы, и наступает только при
определенной для каждого золя концентрации электролита,
которая носит название порога коагуляции. Согласно
правилу Шульце коагулирующая способность электролита
резко возрастает с увеличением заряда иона. Для одно-,
двух- и трехвалентных катионов Шульцем было установлено
соотношение: I : 20 : 350 (по другим авторам это отношение
несколько иное).
Вызываемая электролитами коагуляция объясняется
влиянием ионов на электрокинетический потенциал частиц и
толщину двойного электрического слоя, с уменьшением которых
возрастают силы притяжения между частицами и
уменьшаются силы отталкивания. Степень воздействия
электролитов, как мы видели, неодинакова; общей характеристикой в
этом отношении (по Во. Оствальду) может служить
коэффициент активности. Оствальд нашел, что коагуляция
электролитами наступает при одинаковых значениях коэффициентов
активности или, другими словами, при одинаковой
интенсивности взаимодействующих в системе сил.
Установлено, что некоторые золи при освещении солнеч-
34
ним светом значительно легче коагулируют. Такое же
влияние оказывает воздействие ультрафиолетовыми лучами или
освещение мощной лампой накаливания. Облучение светом,
видимо, вызывает изменение заряда частиц или способствует
химическим реакциям, в частности окислению золей. В обоих
случаях изменяется исходное строение или заряд золя, что
и ускоряет процесс агрегации н коагуляции.
Лиофильные коллоидные растворы отличаются от лио-
фобных растворов энергичным взаимодействием дисперсной
фазы с дисперсионной средой, малой величиной свободной
поверхностной энергии на границе раздела частица — среда
и значительно большей агрегативной устойчивостью.
Коагуляция лиофильных коллоидов в большинстве
случаев обратима и часто не достигает стадии седиментации.
Небольшие примеси электролитов вызывают
незначительное изменение степени дисперсности. Явная коагуляция
наступает лишь в концентрированных и даже насыщенных
растворах солей. Поэтому часто употребляют термин
высаливание для характеристики коагуляции лиофильных
коллоидов. По Гофмейстер)^, высаливающая сила
электролитов зависит от аьионов, которые располагаются по силе
воздействия в лиотропный ряд: цитрат>сульфат> ацетат>
> хлорид > нитр ат > роданид.
Расположение ионов в лиотропном ряду (или ряду
Гофмейстера) связано не только с их высаливающим действием,
но и с рядом иных, общих свойств растворов.
Лиотропные ряды в некотором роде универсальны, так
как порядок расположения ионов отвечает их влиянию на
растворимость солей, на величину поверхностного
натяжения, на скорость реакций и т. д. Из общей теории
электролитов, кроме того, следует, что положение ионов в лиотропном
ряду зависит (или суммарно выражается) от коэффициента
активности соли, в состав которой входит данный ион.
2*
Глава II
АППАРАТУРА ДЛЯ ФИЗИКО-ХИМИЧЕСКИХ
ИССЛЕДОВАНИЙ
Приборы, используемые в практике почвенных и
агрохимических лабораторий, имеют самую разнообразною
конструкцию и принципы действия Наибольшее
распространение получили те приборы, в которых непосредственно
измеряемой величиной являются потенциал, сила тока или
электрическое сопротивление. Так, при определении рН
измеряется потенциал, возникающий на электроде, пламенно-
фотометрическое определение концентрации катионов
щелочных и щелочноземельных металлов сводится к измерению
силы тока, возникающего в фотоэлементе под действием
светового излечения и т. п. Развитие электрических методов
объясняется рядом преимуществ, таких, как быстрота и
простота измерения, высокая точность и чувствительность,
возможность непрерывной автоматической записи.
Почвоведа, агрохимика или агронома редко интересуют
собственно электрические свойства почвы; более подробно
изучают только немногие показатели — электропроводность,
фазовые потенциалы. Основная задача исследования
сводится обычно к измерениям неэлектрических параметров:
концентрации почвенных растворов; состава почв и изучения
строения слагающих почву органических и минеральных
компонентов
Переход от одних величин к другим осуществляется с
помощью приборов, в которых те элементы, которые
выполняют функцию преобразования неэлектрических величин в
электрические, называют преобразователями, или
датчиками (по А. М. Туричину). Такими преобразова-
36
телями в практике почвенных исследований служат
различные виды электродов, обратимых по отношению к
определяемым ионам, фотоэлементы, термопары, термометры
сопротивления и др.
Измерение электрических величин осуществляется с
помощью вольтметров, амперметров и гальванометров. Многие
приборы требуют дополнительной электрической энергии
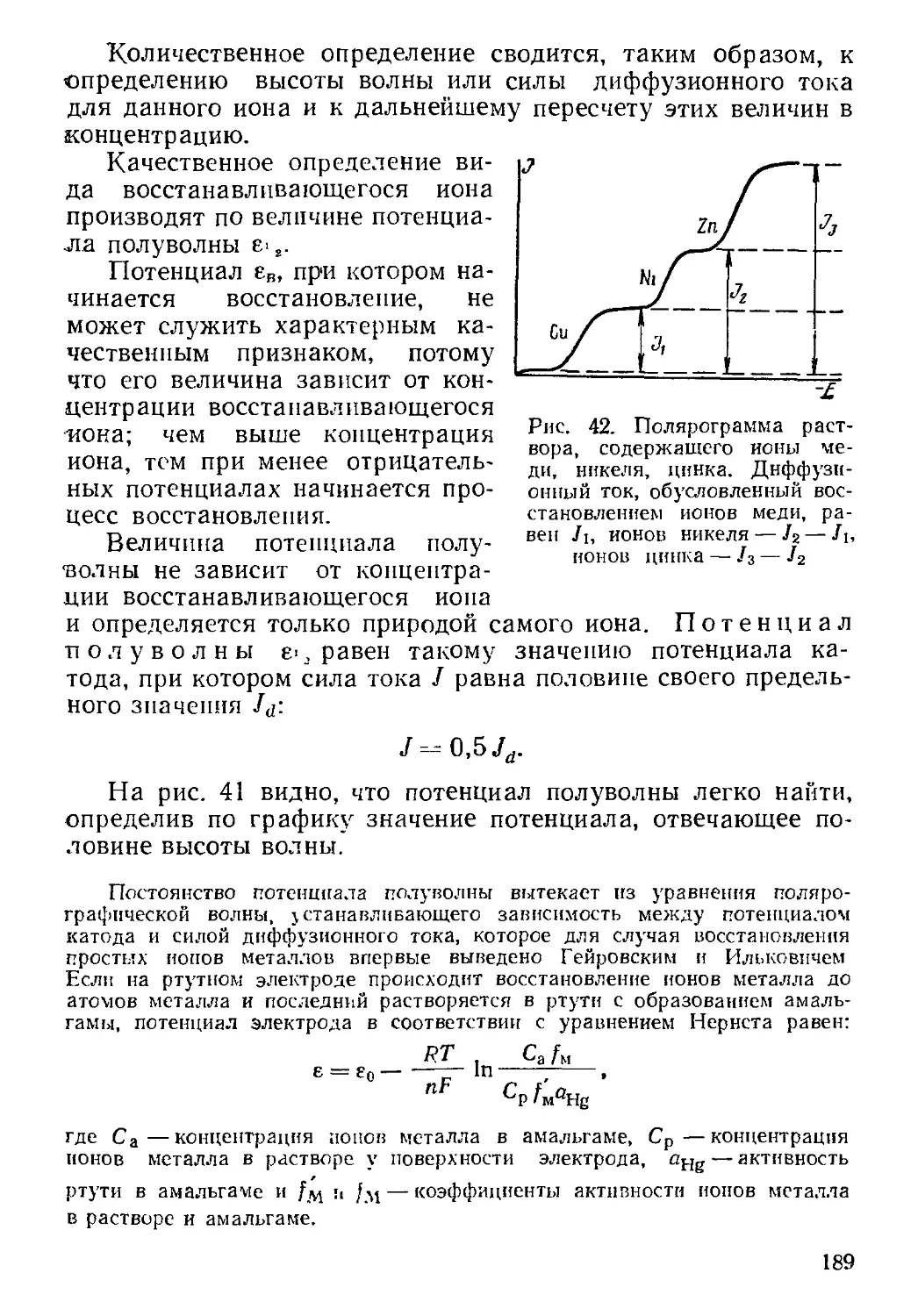
для возбуждения процесса или для питания усилителей,
двигателей и осветительных устройств. Наблюдение за
правильностью работы всех элементов аппаратуры проводится с
помощью контрольно-измерительных приборов.
ИСТОЧНИКИ ЭЛЕКТРОПИТАНИЯ
При физико-химических исследованиях используются
различные источники электроэнергии для питания приборов,
потребляющих в зависимости от конструкции и метода
исследования постоянный или переменный ток, разнообразный по
напряжению, силе и мощности. Неумелое или небрежное
обращение с источниками питания и неправильные
включения могут вызывать серьезные повреждения приборов и
источников питания.
Сетевые источники
В лаборатории подается 3 вида токов: переменный ток
напряжением 220 и 127 в, постоянный генераторный ток
напряжением в 110 и 220 в и постоянный аккумуляторный
ток. Переменное напряжение (обозначаемое ~ } подается на
лабораторные двух- и трехфазные щитки ЛЩ и в
штепсельную сеть ШС, как правило, для двухфазного тока. Имеются
штепсельные колодки и на три фазы
Для питания приборов, требующих напряжения, не
подаваемого в сеть, используются трансформаторы и реостаты.
Трансформаторы применяются в тех случаях, когда
напряжение, на которое рассчитан прибор, не соответствует
напряжению в сети. В этих условиях прибор подключают
через понижающий или повышающий трансформаторы
(РАТ-200 на 200 вт, АОС-3 на 300 вт и др.).
Трансформатор пригоден для питания прибора, если его мощность не
ниже мощности, потребляемой прибором.
Для специальных целей выпускаются ступенчатые
трансформаторы, например, типа ТП-50, рассчитанный на
вторичный ток в 4 с и позволяющий получать 2, 3, 4, 5, 6, 8, 9, 12 в
37
Сеть
-'О"ОО"О0ОШ6,|
Г
шш^
Нагрузка.
от сети 220 в, или Т-3 мошностью 50 вт на 4, 6. 8, 12, 14 и
16 в. Эти трансформаторы позволяют получать
фиксированное напряжение и быстро переходить от одного режима
питания к другому.
Регулировочные автотрансформаторы типа ЛАТР
(ЛАТР-1 на 9с. ЛАТР-2 на 2а) позволяют плавно изменять
напряжение в пределах
0—127—250 в независимо
от величины исходного
A27 или 220 в).
Автотрансформаторы ЛАТР-1
и ЛАТР-2 имеют шкалу,
показывающую
подаваемое на прибор
напряжение, однако регулировка
у них довольно грубая, и
ими не рекомендуется
пользоваться, если нужно
получить низкое
напряжение A0—15 в). Для более
точной регулировки напряжения к автотрансформатору
параллельно нагрузке подключается вольтметр.
Вместо трансформатора для понижения напряжения
можно включить дополнительную нагрузку (например, реостат,
электролампу или использовать реостат, включенный в
качестве потенциометра (рис. 2).
Использование реостатов в качестве потенциометров
ограничивается заданным сопротивлением прибора и
напряжением сети. Сила тока, на которую рассчитан реостат, должна
быть всегда несколько выше, чем ток, потребляемый
нагрузкой. Для цепи (рис. 2) легко вычислить общую силу тока /,
если известно сопротивление реостата R = r2 + r3, нагрузки гх и
подаваемое напряжение Е
Рис.
2. Схема
в качестве
Г\, /2, Г3-
включення реостата
потенциометра:
•сопротивления
Г&
П + Ъ
Сила тока J\, проходящего через нагрузку, определяется
по закону Кирхгофа:
А =
П +г2
Для приборов большей мощности (более 1000 вт)
используют рычажные реостаты, рассчитанные на ток от 10 до 30 а.
38
Расчет реостата, вводимого в качестве нагрузочного
сопротивления, производится по закону Ома. Так, если
требуется погасить избыточное напряжение в 60 в при силе тока
в'цепи = 5й, то сопротивление реостата должно быть равно
60: 5=12 ом.
Источники постоянного тока
Постоянный ток в лабораторных условиях получают от
аккумуляторов, сухих батарей или от выпрямителей.
Выпрямители делятся по своему устройству на вакуумные,
газовые, электролитические и твердые. Из них наиболее
употребительны в лабораторных работах вакуумные и твердые.
Вакуумные выпрямители, или кенотроны, состоят из двух
металлических электродов, находящихся в сосуде под
высоким вакуумом.
При нагревании катода (рис. 3) из металла в вакуум
начинают переходить электроны (термоэлектронная эмиссия),
/ ?
Рис 3. Схема однополупери- Рис. 4. Схема двухполуиериодного
одного выпрямители: выпрямителя на двух кенотронах:
/ — нить накала катода, Ui — напряжение переменного тока,
2—катод, 3 — акод, U\ — на- 1/2—напряжение постоянного тока
пряжение сети, U —
выпрямленное напряжение
образуя у поверхности электрода электронное облако. Если
к электродам приложить некоторую разность потенциалов
(к горячему — минус, а к холодному — плюс), то электроны
начнут двигаться от катода к аноду. Обратное направление
движения при перемене напряжения невозможно, так как у
холодного гнода не образуется электронного облака.
Кенотрон, включенный в цепь с переменным напряжением,
допускает движение электронов только в одном направлении —
ют катода к аноду, когда горячий электрод (катод) имеет
39
отрицательный заряд. Поэтому постоянный ток,
выпрямленный кенотроном, имеет пульсирующий характер.
Уменьшения пульсации можно достичь применением двух-
анодного выпрямителя или включением двух одноанодных
кенотронов по схеме, данной на рис. 4.
Максимальная сила выпрямленного тока большинства
кенотронов не превышает 0,3 а. Поэтому они мало пригодны
для питания приборов большой мощности, но очень удобны
для получения малых токов при высоком напряжении. В
качестве выпрямителей используют и двухэлектродные лампы,
заполненные газом, так называемые газотроны.
Баллоны ламп чаще всего заполняют парами ртути или
инертным газом (аргоном). Газотроны с инертным газом носят
название тунгаров.
В отличие от кенотронов при отрицательном напряжении
на аноде через газотрон проходит кратковременный
обратный ток.
Твердые выпрямители состоят из двух металлических
электродов, между которыми находится полупроводниковый
слой, проводящий ток только в одном направлении. В
селеновых выпрямителях таким слоем является слой селена,
который наносится на никелированный железный диск.
Вторым электродом служит сплав олова, висмута и кадмия.
Предельное напряжение, которое может выдержать такой
выпрямитель, не превышает 15—18 в. Для выпрямления
более высокого напряжения соединяют последовательно нужное
количество дисков. Сила тока, которую могут пропускать
селеновые столбики, зависит от их диаметра (при диаметре
50 мм до 500 ма). Параллельное соединение селеновых
столбиков позволяет получить ток большой силы. Кроме
селеновых широкое применение находят купроксные
выпрямители, в которых полупроводящим слоем служит СигО.
Удобный селеновый выпрямитель ВСА-10, выпускаемый
нашей промышленностью, предназначен для зарядки
кислотных аккумуляторных батарей. Питание выпрямителя от сети
127 или 220 в. Переключением перемычек на панели
выпрямителя можно получить постоянный ток 7 или 12 а.
Постоянный ток, снимаемый с выпрямителя ВСА-4, не
превышает 2 а, но выходное напряжение может быть
получено 120 и 240 в. С помощью других выпрямителей типа ВСА
и ВАК можно получить выпрямленный ток до 10—12 с при
различном напряжении.
Аккумуляторы применяются в схемах, когда требуется
строго постоянное напряжение. Важнейшими характеристика-
40
ми аккумуляторов служат электродвижущая сила Е (э. д. с),
напряжение V, внутреннее сопротивление RBU и емкость Q.
Напряжение аккумулятора при разряде всегда меньше
его э. д. с. и зависит от силы тока, протекающего в цепи
разряда:
Up " С, «р".ВН>
где L'p — напряжение разряда, а /р — разрядный ток.
Точно так же при заряде аккумулятора напряжение
должно превышать э. д. с. на ту же величину.
U3 — Е + J3Re„
(значок «з» указывает на зарядный ток).
При монтаже схемы немаловажное значение имеет выбор
аккумулятора с достаточной емкостью. Аккумуляторы с низкой
емкостью требуют частой подзарядки и пригодны только для
слаботочных цепей. Под емкостью аккумулятора
понимают количество электричества, которое может отдать
аккумулятор при разряде до предельно допустимого
напряжения на клеммах; вычисляют емкость в ампер-часах:
Q - Л.
где Q — емкость в ампер-часах; J — сила разрядного тока в
амперах, t — время в часах. В соответствии с этой
формулой для выбора аккумулятора необходимо знать время, в
течение которого предполагается без перезарядки
использовать аккумулятор при заданной силе тока в цепи.
Свинцовые, или кислотные, аккумуляторы имеют
один электрод из свинца, а второй из двуокиси свинца,
погруженные в раствор серной кислоты. Напряжение на
пластинах свинцового аккумулятора около 2 в. Для получения
большего напряжения аккумуляторы соединяют в батареи.
Например, стартерные батареи СТ состоят из 3
аккумуляторов, число которых указано перед маркировочными буквами;
емкость аккумулятора указывается после букв — 3 СТ-60 —
батарея емкостью 60 с/ч.
Для заливки аккумуляторов готовится «электролит», т. е.
раствор серной кислоты (квалификации «аккумуляторная
х. ч.») в дистиллированной воде. Плотность электролита
колеблется в пределах 1,1 —1,3 и обычно указывается в
паспорте аккумулятора. Уровень электролита должен превышать
на 5 мм кромку изолирующих сепараторных прокладок,
разделяющих положительно и отрицательно заряженные пластины
аккумулятора.
41
Перед зарядкой аккумуляторы тщательно вытирают, зачищают кок-
такты, проверяют плотность и уровень электролита и прочищают
вентиляционные отверстия в пробках. Подготовленный аккумулятор
соединяют с источником постоянного тока (от сети . через выпрямитель);
для 6- и 12-вольтовой батареи напряжение источника должно быть
соответственно 7,5 и 15 в. В процессе зарядки регулярно проверяется
плотность и температура электролита, которая не должна превышать 45°
Об окончании зарядки свидетельствует постоянство плотности
электролита н напряжения в течение 2 ч. Бурное выделение газов, кпавным
образом водорода, — «кипение» электролита, — может служить внешним
признаком окончания зарядки.
Заряжая аккумулятор, необходимо проявлять осторожность,
поскольку выделяющийся на пластинах газ с воздухом образует гремучую смесь
Длительность эксплуатации аккумуляторов обеспечивается
правильным уходом. Эксплуатируемые аккумуляторы необходимо обязательно
подзаряжать один раз в месяц, а аккумуляторы, находящиеся на
хранении, но залитые электролитом, — один раз в два месяца. Необходимо
также регулярно, раз в 10—15 дней, протирать батареи от пыли и грязи,
очищать контакты и вентиляционные отверстия. Одновременно
проверяются плотность и уровень электролита и напряжение каждого
аккумулятора. Нельзя допускать снижения напряжения на контактах менее
1,8 в, так как это ведет к порче пластин.
Все аккумуляторы подвергаются саморазрядке.
Нормальная саморазрядка кислотных аккумуляторов составляет
до 1 % в сутки от емкости при температурах 25—30° и
повышается при загрязнении электролита и утечке тока по
влажной поверхности аккумуляторного ящика.
Щелочные аккумуляторы имеют ряд преимуществ
по сравнению с кислотными. Они допускают значительные
перезарядки и разрядку до нулевого положения (даже
короткое замыкание), хранятся в незаряженном состоянии.
Щелочные аккумуляторы мало чувствительны к окружающей
температуре, просты и безопасны в обращении, в меньшей
степени подвержены саморазрядке. В то же время они менее
экономичны и имеют меньшее среднее рабочее напряжение
(свинцовые аккумуляторы 1,9 в, щелочные только 1,2 в).
Наиболее распространены к а д м и ево-ни к ел е в ы е
аккумуляторы (КН) и железно-никелевые (ЖН).
Электролитом для щелочных аккумуляторов служит раствор
КОН уд. в. 1,19—1,21. Для повышения емкости и увеличения
срока службы к электролиту часто добавляют соединения
лития. Так, калиево-литиевый обычный электролит (К.ЛО)
готовится путем добавления к раствору КОН (уд. в.=
= 1,19—1,21) едкого лития из расчета 20 г LiOH • НгО на 1 л
раствора. КОН очень активно поглощает из воздуха
углекислоту, что со временем приводит к серьезным нарушениям
в работе и даже порче аккумулятора. Поэтому при использо-
42
вании аккумуляторов даже в сравнительно благоприятных
лабораторных условиях обязательно проводят смену
электролита один раз в год.
Сухие батареи применяются благодаря их
портативности и малому весу, особенно удобны они в полевых
условиях. В то же время при длительной работе питание
приборов от сухих батарей не обеспечивает достаточного
постоянства тока. Емкость сухих батарей не велика, срок хранения
6—12 месяцев.
Стабилизаторы
Для устойчивой работы радио-, электроламп, усилителей
и т. п., применяемых в измерительных схемах, необходимо
осуществлять питание током, стабилизированным как по
напряжению, так и по величине. С этой целью используются
различные виды стабилизаторов напряжения и тока.
Дроссельн о-е м костные и реостатн о-е м к о с т-
ные фильтры позволяют значительно сглаживать пуль-
е TffllPS^
f/л It/rrpjiMV/ntjs ^-L. Нагрузка "- v£r Л Нагрузка
О м i. i i 1 e
Рис. 5. Схемы стабилизации напряжения:
а — реостатно-емкостной фильтр, б — газовый стабилизатор
напряжения
сации выпрямленного кенотроном тока. Реостатно-емкостной
фильтр состоит из сопротивления R, включенного
последовательно с выпрямителем, и конденсатора С, подключенного
параллельно нагрузке (рис. 5, о). При повышении
напряжения источника питания усиливается ток, проходящий через
сопротивление R. В результате падение напряжения на
сопротивлении /? увеличивается, а напряжение на
конденсаторе С повышается уже на меньшую величину, чем на входе
фильтра. Конденсатор С в свою очередь еще больше
сглаживает пульсацию, потому что в момент повышения
напряжения конденсатор заряжается и забирает на себя избыток
энергии. При уменьшении входного напряжения конденсатор
разряжается и поддерживает в цепи нагрузки требуемую
43
величину напряжения. Вместо сопротивления R может быть
включен дроссель с сердечником из трансформаторной стали.
Дроссель, обладая индуктивностью, препятствует всякому
изменению проходящего через него тока и вместе с
конденсатором стабилизирует питание нагрузки.
Неоновые лампы имеют постоянное падение
напряжения между электродами независимо от силы проходящего
через лампу тока и начинают пропускать ток только при
определенной разности потенциалов между электродами
(потенциал зажигания). Дальнейшее повышение напряжения
вызывает увеличение силы тока и уменьшение сопротивления
лампы вследствие дополнительной ионизации газа. Таким
образом, падение напряжения JR между электродами
остается постоянным (рис. 5,6).
Кроме указанных типов применяют и другие
стабилизаторы напряжения, например феррорезонансные,
электромеханические и др.
Бареттеры служат для поддержания постоянного
значения силы тока в цепи при меняющемся напряжении
Конструктивно бареттер представляет собой электролампу,
наполненную водородом. Рабочей частью бареттера является
тонкая стальная или вольфрамовая проволока. Увеличение
напряжения и силы тока, проходящего через проволоку,
вызывает нагрев и возрастание ее сопротивления, что
препятствует дальнейшему увеличению силы тока. Нормальная
работа бареттера начинается только через некоторое время
после включения тока; для большинства типов нормальный
ток бареттирования устанавливается через 5 мин.
Стабилизированные выпрямители.
Высоковольтное стабилизированное напряжение в пределах
500—1000 в позволяет получить прибор ВВС-1,
представляющий собой кенотронный двухполупериодный выпрямитель с
электронной стабилизацией выпрямленного напряжения.
Максимальная сила тока 100 ма. Если пределы колебаний
напряжения в питающей сети составляют от —15 до +5%,
то выходное напряжение меняется не более чем на ±0,5%.
Пульсация напряжения не превышает ±0,1%.
В пределах от 100 до 400 в удобно пользоваться
выпрямителем ВУС-1, стабилизирующие характеристики которого
такие же, как у ВВС-1, но сила постоянного тока достигает
уже 300 ма. ВУС-1 одновременно может быть использован
и как понижающий трансформатор с выходным переменным
напряжением в 2; 2,5; 4; 6,3; 12,6; 24 в.
44
КОНТРОЛЬНО-ИЗМЕРИТЕЛЬНЫЕ ПРИБОРЫ
Амперметры и вольтметры служат для контроля и
измерения напряжения и силы тока в цепи. Эти приборы
выпускаются разных типов и рассчитаны на измерение
постоянного или переменного тока в разных пределах Нельзя
включать прибор в цепи с большим напряжением или силой тока,
чем это указано в паспорте прибора Если паспорт
отсутствует, то необходимые
характеристики можно
прочитать на шкале
самого прибора.
По общепринятым
правилам на шкало прибора
(рис 6) указывается
порядковый номер, номер
государственного
стандарта, тип и система прибора.
Кроме того, на шкале
указано, в каких величинах
проградуирована шкала
(в, а, мв, ма, яка и т. д.),
вид тока, на который
рассчитан прибор (—
постоянный, ~ переменный,
^ постоянный и
переменный), класс точности
прибора. Обозначаемый COOT- р"с г' Шкала миллиамперметра
встствующей цифрой в
кружке, например, (м) и др (общий перечень условных
обозначений см в конце главы).
Класс точности указывает на величину погрешности
прибора в процентах от предельного значения измеряемой
величины Так, для вольтметра на 150 в класса точности 0,5
погрешность измерения составит 0,75 в. Очевидно, что
относительная ошибка в начале шкалы значительно выше, чем
в конце. Поэтому для точных измерений не рекомендуется
пользоваться градуировкой в начале шкалы.
По величине погрешности амперметры и вольтметры
делятся на семь классов точности: 0,1; 0,2; 0,5; 1,0; 1,5; 2,5; 4,0.
Пределы измерений можно прочитать по градуировке
шкалы. У многопредельных приборов пределы измерений
указаны на панели у соответствующих клемм. Приборы высокого
класса точности снабжаются кнопкой корректора, при помо-
45
щи которой стрелка устанавливается на нулевое деление
шкалы перед началом измерений.
Тестер ТТ-1 предназначен для контрольных измерений
напряжения и силы постоянного тока, напряжения
переменного тока и сопротивления постоянному току в следующих
пределах: 1) постоянное и переменное напряжение
измеряется на четырех шкалах: 0—10, 0—50, 0—200, 0—1000 в; 2)
сила постоянного тока 0—0,2; 0—1; 0—5; 0—20; 0—100;
0—500 ма; 3) сопротивления: 1—200 ом; 10 ом— 20 ком;
100 ом — 200 ком, 1 ком — 2 мгом. Погрешность измерения
сопротивлений ±10%.
Для измерения сопротивлений прибор снабжен сухой
батареей на 6 в.
Ампервольтметр АВО-5 имеет следующий диапазон
измерений:
1. Напряжение постоянного и переменного тока
ступенчато в пределах 0-3—12—30—300—600—1200—6000 в.
2. Сила постоянного и переменного тока в пределах: 0—■
60—300 мка; 0—3—30—120 ма; 0—1, 2—12 а для
постоянного тока и 0—3—30—120 ма; 1 —1,2—12 а для переменного
тока.
3. Сопротивление постоянному току: 0—3 ком; 0—300 ком;
0—30 мгом. Погрешность прибора ±3% при измерении
постоянного тока; ±5%—при измерении переменного тока и
±10% при измерении сопротивлений.
Аккумуляторный пробник типа АП для
измерения напряжения на клеммах аккумуляторных батарей
рассчитан на измерения до 3 в постоянного тока.' К прибору
приложены нагрузочные сопротивления, позволяющие измерить
напряжение под нагрузкой током пятичасового разряда. Это
позволяет судить о степени разряда аккумулятора.
Перечисленные переносные контрольные приборы служат
для поверочных измерений и позволяют быстро обнаружить
неисправности в электрической цепи.
Гальванометры используются как нуль-инструменты,
показывающие отсутствие тока в цепи в различных потенцио-
метрических схемах, и как приборы непосредственного
отсчета (пламенный фотометр, пирометры).
Стрелочные гальванометры ГМП и М-122
чувствительностью 10~5—Ю-7 а на 1 мм шкалы используются
в потенциометрических схемах Поггендорфа. Гальванометры
имеют арретир и корректор нуля. Оба типа приборов можно
использовать в цепях с невысоким сопротивлением
( — 1000—500 ож).
46
Зеркальные гальванометры по своему
устройству и правилам пользования отличаются от большинства
контрольно-измерительных приборов. В почвенно-химических
лабораториях наиболее часто применяются
магнитоэлектрические гальванометры с подвижной катушкой. В приборах
этого типа имеется
постоянный магнит, в поле которого
расположен железный
цилиндр (рис 7). В зазоре
между магнитом и цилиндром
находится легкая рамка с
обмоткой из тонкой
проволоки. Рамка подвешивается на
тонкой нити (подвесе) из
фосфористой бронзы или
кварца и может свободно
вращаться вокруг цилиндра.
На той же нити крепится
миниатюрное легкое
зеркальце. Принцип действия
гальванометра сводится к
следующему: измеряемый
ток протекает через обмотку
рамки и создает магнитное
поле, взаимодействующее с полем постоянного магнита; в
результате рамка поворачивается так, чтобы плоскость ее
заняла положение, перпендикулярное силовым линиям постоянного
магнита. Вращению рамки противодействует сила кручения
упругой нити, на которой она подвешена. При равновесии
моменты сил, действующие на рамку со стороны магнитного поля
и крутящего упругого момента нити подвеса, равны
Рис. 7. Устройство зеркального
магнитоэлектрического гальванометра:
/ — подвес; 2 — зеркальце; 3 — рамка
с обмоткой
HnSJ = Мка,
где И— напряженность магнитного поля, 5 — площадь
витка, п — число витков обмотки рамки, J — сила тока,
протекающего через обмотку, Мк — момент сил кручения и а —
угловой поворот рамки.
Следовательно, угол поворота рамки прямо
пропорционален силе протекающего через обмотку тока
а
HnS
47
Постоянная называется токовой
чувствительностью гальванометра и равна силе тока, отклоняющей
рамку на угол а=1. Обозначается токовая чувствительность
символом Si.
Рис 8 Зеркальный гальванометр-
а — внешний вид, б — его установка на вертикальной стене. / —
гальванометр, 2 — осветитель, 3 — зеркало, 4 — шкала, 5 — кронштеш.
Для измерения угла поворота рамки гальванометры
снабжаются осветителем и шкалой. Луч света от лампочки
осветителя падает на зеркальце гальванометра и отражается на
шкалу, по которой ведутся отсчеты (рис. 8).
48
Кроме обычных гальванометров, в которых отсчет
производится в момент установления равновесия, применяют
баллистические гальванометры, отличающиеся большим
моментом инерции подвижной части прибора и предназначенные
для измерения кратковременных импульсов тока. В этих
гальванометрах прохождение тока через обмотку
заканчивается еще до того, как рамка успеет заметно отклониться.
Благодаря этому первый отброс «зайчика» пропорционален
количеству электричества, прошедшего через рамку.
После включения или выключения тока рамка
гальванометра не сразу останавливается (успокаивается) в
положении равновесия или в нулевом положении, а совершает
некоторое время затухающие колебания. Время затухания
зависит от торможения рамки вследствие трения ее о воздух
и от электромагнитного торможения, возникающего в
результате того, что в обмотке пр-и ее движении индуцируется
электрический ток. Величина тока, а следовательно, и степень
торможения определяются сопротивлением внешней цепи
гальванометре и "внутренним сопротивлением самого
гальванометра. Оптимальный режим работы гальванометра
заключается в том, что рамка быстро асимптотически приближается
к.положению равновесия, не переходя его. Такого результата
можно достичь при строго определенной величине индчнируе-
мого в рамке тока. Индуцируемый в обмотке ток
определяется следующим равенством:
I — HnS da
~ Rr+R ' dt '
где RT — внутреннее сопротивление гальванометра и R —
сопротивление внешней цепи, / — время.
Если сопротивление в цепи гальванометра подобрано
правильно, то поворот рамки совершается апериодически, т. е.
она подходит к положению равновесия бистро, не переходя
его. Гальванометр с таким сопротивлением называется
критически успокоенным, а величина Rr+R называется
полным критическим сопротивлением RKvm.
Соответственно наибольшее сопротивление внешней цепи, при
котором рамка совершает апериодические колебания,
называется внешним критическим сопротивлением
пви крит-
Понятие внешнего критического сопротивления приводит
к важным практическим выводам. Наилучшие результаты при
использовании зеркального гальванометра могут быть полу-
49
чены лишь в том случае, если сопротивление внешней цепи,
в которую включен гальванометр, будет близким к его
критическому сопротивлению. Если сопротивление цепи резко
отличается от внешнего критического, то приходится
дополнительно включать балластные или шунтирующие
сопротивления.
Шунтирование гальванометров. Шкалы зеркальных
гальванометров имеют обычно не более пятисот делений, тогда
как диапазон измеряемых величин (по силе тока или напря-
-£Ъ
SJ/t 0,3R ff/Л 0,OSR O,03R O,0tR 0,№R ffJffS/T
1 -ИЙ-" " " ' ' '
л\
ь
в
Рис. 9 Шунтирование гальванометра. В данной схеме сопротивления слева
от подвижного контакта составляют балластное сопротивление /?,-,,
справа — шунтовое R ш
женню) может меняться в сотни и даже тысячи раз.
Шунтирование гальванометра позволяет пользоваться одним и тем
же прибором для измерения резко различных по,масштабу
величин. Схема шунтирования гальванометра показана на
рис. 9. Гальванометр замыкается на сопротивление, величина
которого близка к внешнему критическому сопротивлению.
Источник тока подключается к одному из концов
сопротивления В и к подвижному контакту А, положение которого
может изменяться от С до D. Гальванометр подбирается
с расчетом, чтобы его внешнее критическое сопротивление
было меньше сопротивления цепи источника тока; в этом
случае источник тока не будет заметно влиять на характер
колебаний рамки гальванометра, и последний будет работать
в критически успокоенном режиме независимо от положения
подвижного контакта А, поскольку суммарное сопротивление
R(s+Rm, на которое замкнут гальванометр, остается
практически постоянным (см. рис. 9).
50
Чувствительность гальванометра в такой схеме всецело
зависит от положения подвижного контакта. Чем больше
сопротивление R6 (слева от подвижного контакта А), тем
меньшая часть тока пройдет через гальванометр, а большая —
через шунтирующее сопротивление Rm, и относительная
чувствительность гальванометра снижается в определенное
число раз.
Такими ступенчатыми делителями, или редукторами
чувствительности, снабжаются все современные полярографы,
пламенные фотометры и многие другие приборы с
зеркальными гальванометрами.
При использовании редукторов вводят понятие
относительной чувствительности а гальванометра, которая равна
отношению силы тока, идущего через гальванометр, к
суммарной силе тока, протекающего через гальванометр и шунт:
Jr
а = ——,
Jc
где /г — сила тока, протекающего через гальванометр, и /с—
суммарная сила тока. Если к клеммам А и В приложено
напряжение, равное Е, то
L =-- Е- '
Кб + «г
1 1 \ г. °крит
\ Дб + #г RmJ
(Яб + RT) Ru
где RT— сопротивление гальванометра, /?б —
последовательное (балластное) сопротивление и Rm— шунтовое
сопротивление. Отсюда ясно, что относительная чувствительность
может быть вычислена как отношение
a R»
Акрит
или если сопротивление гальванометра достаточно малс по
сравнению с полным критическим
а = —^—.
Rm + Яб
Зеркальные гальванометры требуют в работе большого
внимания и осторожного обращения. Начинать измерения
всегда нужно при минимальной относительной
чувствительности гальванометра; убедившись, что измеряемый ток не
51
превышает допустимых для гальванометра величин, можно
увеличивать силу тока. Гальванометры очень чувствительны
к механическим сотрясениям, поэтому их устанавливают на
основаниях, не имеющих вибраций, на кронштейнах,
укрепленных в капитальной стене, используют специальные
амортизирующие подвесы или прокладки.
Промышленность выпускает целую серию зеркальных
гальванометров с различными характеристиками, из которых
широкое применение находят гальванометры типов М-21,
М-25, ГБЗС и некоторые другие. Характеристики
гальванометров М-21 и М-25 приведены в табл. 6.
Таблица б
Характеристики гальванометров типа М-21 к М-25
Марка гальванометра
М-21
М-21/1
М-21/4
М-21/5
М-21/6 двух-
обглоточ-
нь:й
М-25/3
М-25/5
М-25/7
М-25/11
М-25/13
1-я
обм
2-я
обм.
Постоянная
по току.
а мм м*
1,5-Ю-9
1,5-Ю-9
4-10-9
2-10~9
1,5-Ю-9
1,5- Ю-9
12-Ю-9
1,5-Ю-9
0,5-10-9
4,5-10~9
0,4-10-9
Постоянная
по
напряжению, в/ММ М
0,2-Ю-6
2-Ю-6
—
0,6-Ю'6
3,0-Ю-6
7,0-Ю-6
—
Внутреннее
сопротивление рамки, ом
500
1000
20
30
100
100
16
350
2500
35
3500
Критическое
сопротивление, ом
20000
100 000
50
1000
3 500
3 500
50
2 500
20 000
70
15 000
Период
колебаний
в секунду
не более
5
12
12
18»
12
10
10
10
15
15
* а/.им'м —размерность, показывающая, что ток силой 1 а вызывает
отклонение «зайчика» на 1 мм прн шкале, расположенной от зеркальца на 1 м.
ЭЛЕКТРОННЫЕ ЛАМПЫ И УСИЛИТЕЛИ
Двухэлектродные лампы, или диоды, применяющиеся
в качестве выпрямителей, описаны в предыдущем разделе.
В стабилизирующих, преобразовательных и усилительных
схемах используются также трехэлектродные лампы
(триоды) и многоэлектродные лампы. Многие современные модели
приборов для физико-химических исследований конструиру-
52
ютсл с применением электронных ламп, которые используют
также в регуляторах температуры (ЭРМ-47), выпрямителях
(ВВС-1), автоматических записывающих потенциометрах
(ЭПП-Э9).
Трехэлектродная лампа, или триод, устроена наиболее
просто. Кроме катода и анода в триоде имеется третий
электрод— сетка, расположенная между анодом и катодом.
Как и в диоде, в триоде накаленный катод излучает
электроны, которые направленно движутся под действием
положительного потенциала анода. Сила анодного тока зависит от
температуры катода и разности потенциалов, приложенной
к электродам.
Подавая на сетку положительный или отрицательный
потенциал, можно регулировать силу анодного тока. Если
потенциал сетки относительно анода будет положителен, то сила
анодного тока возрастает и продолжает увеличиваться с
ростом положительного потенциала. Увеличение отрицательного
потенциала на сетке препятствует движению электронов от
катода к анод> и приводит к уменьшению анодного тока.
Поэтому анодный ток триода является функцией трех
переменных величин: накала катода, напряжения на аноде и
напряжения на сетке.
Важнейшей характеристикой триода является
коэффициент усиления, который показывает степень влияния
потенциала сетки на анодный ток при одном и том же
напряжении в анодной цепи. При постоянном накале катода
анодный ток можно регулировать двумя способами:
изменением анодного напряжения или изменением напряжения на
сетке. Оказывается, что увеличение или уменьшение
напряжении на сетке гораздо сильнее воздействует на анодный
ток, чем анодное напряжение. Это объясняется близким
расположением сетки к катоду. Относительное влияние анода и
сетки выражается коэффициентом усиления, который
показывает, во сколько раз сильнее действует на электронный
поток изменение напряжения на сетке, чем такое же по
величине изменение напряжения на аноде, и численно показывает,
на сколько вольт изменяется напряжение на аноде лампы
при изменении потенциала управляющей сетки на 1 е:
Ди.,
Дис
где [I — коэффициент усиления; Д«а— изменение анодного
напряжения; Аис — изменение напряжения на сетке.
53
Вторая, важная для ламп величина — крутизна
характеристики, показывающая, как быстро
увеличивается анодный ток при увеличении напряжения на сетке.
Практически крутизна характеристики S триода выражается углом
наклона зависимости анодного тока от потенциала сетки и
может быть вычислена по формуле
где А/а — изменение анодного тока.
Коэффициент усиления и крутизна характеристики
связаны уравнением
Ц - SRt,
где Ri — внутреннее сопротивление лампы.
Параметры триодов, как и других электронных ламп,
даются в виде таблиц и графиков. Значительно более сложны
по устройству и работе многоэлектродные лампы.
Дополнительные сетки улучшают характеристики и расширяют
возможности применения электронных ламп.
Сильное влияние сетки на анодный ток позволило
использовать трехэлектродную лампу в качестве усилителя. Токи,
возникающие в фотоэлементах, термоэлементах, некоторых
электродах, часто настолько малы, что их не удается
измерить даже самыми чувствительными гальванометрами. Если
же включить маломощный источник тока Ux в цепь трехвлек-
тродной лампы между сеткой и катодом (рис. 10,/). то
потенциал, принимаемый сеткой, сразу же изменяет величину
анодного тока /а. Амперметр, включенный в цепь анода,
позволяет зафиксировать изменение анодного тока и измерить
"тем самым величину потенциала сетки. Собственно говоря,
в этом процессе ток и напряжение маломощного источника
не увеличиваются; изменяется лишь анодный ток лампы или
падение напряжения на нагрузочном сопротивлении в
анодной цепи, но эти изменения пропорциональны напряжению
(или колебаниям напряжения) источника тока. Роль лампы
сводится, таким образом, к регулированию анодного тока.
Существенный недостаток простых усилительных схем
заключается в том, что через измерительный прибор проходит
ток даже в том случае, когда величина измеряемого
напряжения равна нулю (нулевой анодный ток); это снижает
чувствительность прибора. Вместе с тем на величине анодного
тока сильно сказываются случайные колебания напряжения
54
накала Uu и анодного напряжения Ua. Первый недостаток
легко устранить при помощи дополнительного
потенциометра, который позволяет компенсировать нулевой анодный ток.
Значительно стабильнее мостовые, или балансные,
схемы усилителей. Балансные схемы собираются с двумя
одинаковыми электронными лампами, которые являются
плечами уравновешенного моста. В диагональ моста включается
//.
& ^ JU
М>1
i!iV
Ю
Рис. 10. Схема усилителей:
/ — по напряжению; 2 — по
току; 3 —- балансная схема
=1= Ua
гальванометр, показывающий наличие тока лишь тогда, когда
на сетку одной из ламп подано измеряемое напряжение
(рис. 10,5). В этой схеме на обе лампы Лх и Л2 подано
питающее напряжение от одного источника тока. Лампы и
сопротивления Ri и Я2 подбираются по возможности
идентичными для того, чтобы потенциалы в точках подключения
гальванометра были одинаковыми. Перед работой усилитель
настраивается до нулевого положения потенциометром У?4.
подаюшим на управляющую сетку лампы «/72 дополнительный
регулировочный потенциал. Изменение сеточного потенциала
уравнивает падение напряжения в лампах Л{ и /72, которое
могло быть неодинаковым вследствие небольших различий
в характеристиках ламп. После настройки на сетку и катод
55
лампы Л] подается измеряемое напряжение Ux, которое
нарушает равновесие, и в цепи гальванометра возникает ток
разбаланса. Показания гальванометра пропорциональны
напряжению Ux.
Измерение очень малых токов и напряжений
осуществляется многокаскадными усилителями, в которых падение
напряжения на нагрузочном сопротивлении анодной цепи
первой лампы последовательно подвергается усилению два,
три и более раз.
ФОТОЭЛЕМЕНТЫ
Преобразование лучистой энергии в электрическую
осуществляется фотоэлектронными индикаторами,
в которых используются три вида фотоэлектрического
эффекта: внешний, внутренний и в запирающем слое.
Внешний фотоэффект используется в вакуумных
фотоэлементах и фотоумножителях, внутренний — в фотосопротивлениях,
а эффект в запирающем слое — в вентильных фотоэлементах.
Наибольшее применение в аналитической практике нашли
фотоэлементы с внешним фотоэффектом и фотоэлементы
вентильного типа, применяемые в фотоэлектроколориметрах,
пламенных фотометрах, спектрофотометрах,
фотоэлектрических приставках к спектрографам, микрофотометрах, а
также в реле различных конструкций.
Фотоэлементы с внешним фотоэффектом. Устройство
простейшего вакуумного фотоэлемента с внешним фотоэффектом
показано на рис. 11. В стеклянном баллоне, из которого
выкачан воздух, расположены катод и анод. Поверхность
катода изготовлена из металла или сплава, чувствительного
к световому излучению; в этих целях используют цезий,
серебро, окись цезия, сурьму, висмут в виде тонкого слоя на
внутренней поверхности баллона. Например, фотокатоды,
чувствительные к ближней инфракрасной области спектра,
состоят из серебряной подкладки (зеркало), на .которой
располагается слой окиси цезия, покрытый с поверхности тонким
слоем металла цезия.
Под действием света фотокатод излучает электроны, т. е.
происходит фотоэлектронная эмиссия. На анод фотоэлемента
подается положительное напряжение, под действием которого
фотоэлектроны движутся от катода к аноду и в цепи
возникает электрический ток.
Фотоэлектронная эмиссия возникает только в том случае,
если энергия кванта света, падающего на фотокатод,
превышает работу выхода электрона. Энергия кванта света погло-
56
шается электроном и расходуется на преодоление
потенциального энергетического барьера на границе металла и на
сообщение электрону некоторой скорости. Если энергия
одного кванта падающего света равна e = /iv. а работа выхода
электрона
W -ец,
(е — заряд электрона, ф = е2—ei — разность потенциала в
точках, между которыми перемещается электрон),
Рис. 11 Схемы фотоэлементов
вакуумного фотоэлемента (/) и его
включения в цепь B); А — анод, К—катод, Б —
стеклянный баллон, В — межэлектродное пространство (вакуум
~10~7 мм pi. ст.), Ц — цоколь с выводами анода и катода,
ФЭ — фотоэлемент, U — анодное напряжение, R
—сопротивление; вентильных фотоэлементов с
тыловым C) и фронтальным D) фотоэффектами;
п — подложка, з — запирающий слой, пп —- полупроводники,
э — электрод
57"
-то пс закону сохранения энергии
hv = —— + ар,
mv2
где кинетическая энергия фотоэлектрона.
Из этого уравнения, в частности, следует, что при
некоторой минимальной частоте световых колебаний,
обозначаемой vo, энергия фотона будет ниже, чем работа выхода, и
электрон не сможет вылететь из металла. Эта минимальная
частота (или максимальная длина волны К0) излучения
называется красным порогом фотоэффекта, или
длинноволновой границей фотоэффекта.
Наименьшую работу выхода имеют щелочные и
щелочноземельные металлы, особенно соединения цезия с окисью
цезия, что и обусловило их выбор для изготовления
большинства фотокатодов.
Фотоэлементы различных конструкций отличаются по
своей общей чувствительности и по чувствительности к
различным участкам спектра. Чувствительность фотоэлемента
равна величине фототока, возникающего под действием
светового потока, интенсивность которого равна единице. Чем
выше чувствительность, тем большей силы ток течет в
фотоэлементе при освещении его светом, одинаковым по составу
и интенсивности. Различают интегральную и
спектральную чувствительности фотоэлементов.
Интегральная чувствительность К равна отношению силы
фототока J к энергии падающего светового потока,
состоящего из разных длин волн и соответствующего по
спектральному составу излучению нити вольфрамовой лампы при
температуре 2840° К. Интегральная чувствительность выражается
в 2!!1E_L (а/вг), а для видимой части спектра — обычно
ватт
микроампер , , ч
в — (мка/лм):
люмен
Ф
где Ф — интенсивность светового потока.
Спектральная чувствительность выражает зависимость
между силой фототока и интенсивностью монохроматического
излучения, или зависимость чувствительности от длины
волны. Спектральная характеристика фотоэлемента
изображается графически: по оси ординат откладывают силу фототока,
.58
а по оси абсцисс — длину волны излучения. Интегральная
чувствительность может быть вычислена как площадь,
ограничиваемая кривой спектральной чувствительности (табл. 7,
рис. ]2).
Таблица 7
Интегральная чувствительность некоторых фотоэлементов
Фотоэлемент
Интегральная
чувствительность, мка дм
Вакуумный кислородноцезиевый ЦВ-1 .
» > » ЦВ-2.
Газонаполненный кислородноцезиевый ЦГ-1 .
» » » ЦГ-2.
» » » ЦГ-3.
20
20
75
150
150
Интегральная и спектральная чувствительности даже
одного фотоэлемента неодинаковы в разных участках его
поверхности. Тем в большей степени это относится к разным
фотоэлементам даже одной партии.
I
|
t
/\
Д-Q
\ у
г\
Ш 800 /200
Длит болш, ммх
600
!
Ц200
Z
/1
2<t0
I
КО |
!*:
80 j=|
J
200 600 WOO
Освещенность, ля
S
Рис. 12. Характеристики фотоэлементов:
а — спектральная характеристика селенового (/) и
сернисто-серебряного B) фотоэлементов; б — зависимость фототока (/) и ф.э.д.с. B)
сернисто-серебряного фотоэлемента (ФЭСС) от освещенности
Важнейшим условием применимости фотоэлементов для
количественных измерений интенсивности световых потоков,
является пропорциональность фототока интенсивности
облучения или освещенности. Практически прямая пропорцио-
58,
дальность сохраняется в вакуумные фотоэлементах только
для световых потоков относительно малой интенсивности.
Для газонаполненных фотоэлементов зависимость между
фототоком и освещенностью нелинейная, только при очень
малых световых потоках (менее 0,1 .гм) н в этих фотоэлементах
наблюдается прямая пропорциональность.
Интегральная и спектральная чувствительности и
зависимость фототока от освещенности еще недостаточно полно
характеризуют фотоэлемент. Практически важными свойствами
являются утомляемость и инерционность фотоэлементов.
Утомлением называется понижение чувствительности при
длительном облучении фотоэлемента, причем с течением
времени может изменяться не только интегральная, но и
спектральная чувствительность. При освещении интенсивными
световыми потоками утомляемость проявляется значительно
сильнее, чем при более слабом освещении.
Вакуумные фотоэлементы практически безынерционны,
т. е. при быстрых изменениях освещенности сила фототока
мгновенно изменяется до нормального значения; некоторая
инерционность проявляется только при большой частоте
световых колебаний. Для газонаполненных фотоэлементов
характерно возникновение и увеличение инерционности с ростом
приложенного напряжения.
Фотоэлементы имеют определенные пределы
применимости. Повышенная интенсивность освещения приводит к
неустойчивости работы фотоэлемента и сокращению срока его
службы. Нижний предел освещенности фотоэлемента
определяется величиной темнового тока, который проходит
через фотоэлемент даже тогда, когда последний находится
в полной темноте. Темновой ток имеет небольшую величину
и возникает как вследствие утечек тока, идущего по
поверхности баллона, так и вследствие термоэлектронной эмиссии.
Кроме темнового тока чувствительность фотоэлементов
ограничивается помехами, создаваемыми в цепи самого
фотоэлемента и в цепи усилителя. Нижний предел освещенности
можно понизить, а чувствительность фотоэлементов повысить
путем замораживания катода.
Вентильные фотоэлементы, или элементы с, запирающим
слоем, не требуют в отличие от вакуумных и
газонаполненных фотоэлементов наложения дополнительной разности
потенциалов между катодом и анодом. Это значительно
упрощает приборы с применением вентильных фотоэлементов и
позволяет, в частности, широко использовать их в полевых
походных лабораториях. Действие фотоэлементов вентильно-
.60
го типа основано на явлении односторонней проводимости
запирающего слоя, образующегося на контакте между
проводником и полупроводником. В зависимости от устройства
и направления движения электронов вентильные
фотоэлементы делятся на фотоэлементы с тылов и м и л и ц е в ы м
(фронтальным) фотоэффектами (рис. 11,3 и 4).
Облучение поверхности фотоэлемента светом приводит
к высвобождению электронов, которые становятся
электронами проводимости;
движение фотоэлектронов
ограничивается запирающим слоем,
который пропускает
электроны в одном направлении.
В результате по одну
сторону запирающего слоя
скапливается избыток
электронов и возникает разность
потенциалов между
подложкой фотоэлемента и внешним
электродом. Разность
потенциалов препятствует
дальнейшей концентрации электронов по одну сторону
запирающего слоя, и в системе устанавливается динамическое
равновесие (постоянство величины фототока), зависящее от
интенсивности освещения. Для вентильных фотоэлементов используют
те же характеристики, что и для фотоэлементов с внешним
фотоэффектом.
Обе группы фотоэлементов весьма различны по
свойствам, что влияет на выбор и пригодность тех или других
элементов в приборах различного типа. Одно из преимуществ
вентильных фотоэлементов заключается в том, что они
работают без дополнительных источников питания. Спектральная
чувствительность этих фотоэлементов весьма разнообразна,
что позволяет выбрать фотоэлемент с хорошими
показателями для любого участка спектра (см. рис. 12). Другое
положительное качество — высокая интегральная
чувствительность; если для вакуумных фотоэлементов она редко
превышает 150 мка/лм, то чувствительность вентильных
фотоэлементов достигает тысяч и десятков тысяч микроампер на
люмен (табл. 8).
Существенными недостатками фотоэлементов с
запирающим слоем являются относительно высокая инерционность и
нелинейная зависимость между силой фототока и
освещенностью, в чем они значительно уступают фотоэлементам
Таблица 8
Интегральная чувствительность
вентильных фотоэлементов
Фотоэлемент
... ... _. .. .
Меднозакисный . . .
Сернисто-серебряный
Сернисто-талляевый
Германиевый ....
Интегральная
чувствительность, мка ям
100
600
8 000
11000
30000
61
с внешним фотоэффектом — вакуумным и газонаполненным.
Прямолинейная зависимость между фототоком и
освещенностью сохраняется при световых потоках не более 1 лм
только в цепях с малым сопротивлением.
МОНОХРОМАТОРЫ И СВЕТОФИЛЬТРЫ
В практике эмиссионного и абсорбционного спектрального
анализа, при колориметрических работах и некоторых других
измерениях (например, работах с интерферометрами)
возникает необходимость выделить из светового потока излучение
с определенной шириной спектрального интервала.
Электромагнитное излучение, характеризующееся
определенной частотой или длиной волны, называют
монохроматическим, а процесс его выделения из светового
потока— моно хромат и за ц и ей света. Идеальное
монохроматическое излучение невозможно получить ни от
источника света, ни выделить с помощью приборов. Всякое
излучение охватывает некоторый спектральный интервал
Ак=Кг—к\, и поэтому определение «монохроматизация»
употребляется всегда несколько условно и относительно и
понимается как выделение из сплошного или линейчатого спектра
участков электромагнитного излучения в более или менее
узком интервале длин волн. Степень монохроматизации,
требуемая экспериментальной практикой, весьма различна и
колеблется от выделения отдельных спектральных линий (очень
узких интервалов частот) до широких участков спектра в
несколько десятков и даже сотен А. Созданные в этих целях
приборы основаны преимущественно на явлениях дисперсии
света, интерференции и избирательного поглощения.
Монохроматоры и спектрографы построены «а принципе
дисперсии света, разложения сложного излучения на
монохроматические составляющие с помощью призм,
дифракционных решеток. На рис. 13 показана простая схема призменного
монохроматора УМ-2. Луч света от источника попадает на
входную щель, которая ограничивает входящий пучок.
Объектив собирает лучи в параллельный пучок, после чего
в призме происходит разложение пучка на
монохроматические составляющие. Второй объектив фокусирует
монохроматические пучки света в плоскости выходной щели, образуя
спектр. Нужный участок спектра вырезается выходной
щелью; ширина получаемого спектрального интервала
зависит от конструкции прибора и, в первую очередь, от
дисперсии призмы и ширины щели.
62
При помощи монохроматора можно выделить любой
участок спектра; это достигается или поворотом призмы, или
передвижением щели вдоль спектра. Монохроматор УМ-2
рассчитан на работы в области от 380 до 1000 ммк. Широкая
рабочая область спектра достигается применением сменных
призм.
Степень дисперсии призменного прибора характеризуется
da ,
величиной угловон дисперсии призмы —— , где da— разность
аХ
углов отклонения для двух лучей с разностью длин волн,
„ ,, ,. ., ., dl тт
равной аК, или линейной дисперсией . Линейная диспер-
Рис. 13. Схема монохроматора УМ-2.
а — ход лучей в приборе: 1 — призма, 2 — линза,
3 — щели, 4 ■— источник света; б — разрез щели
сия —— дает расстояние на спектре (в миллиметрах) между
длинами волн К и %+а\. Чаще пользуются обратной величи-
„ dk . , v
ной —— (интервал длин волн на 1 мм длины спектра).
Отдельные участки спектра диспергируются призмами,
изготовленными из различных материалов; для видимой
области спектра обычно используются стеклянные призмы, для
ультрафиолетовой области — кварцевые, а для
инфракрасной — призмы из кристаллов некоторых солей — NaCl, KC1,
KBr, LiF.
Другая ответственная часть монохроматора — щели.
63
Щель представляет собой механическое устройство, рабочей
частью которого служат два ножа; расстояние между ними
(ширину щели] можно менять с помощью микрометрического
винта (на рисунке не показан). Края ножей делаются
острыми и скошенными (рис. 13,6), причем скосы расположены
в сторону распространения лучей (у входной щели скосы
обращены внутрь прибора, у выходной — наружу). Для
правильной работы прибора края щели должны быть строго
параллельны, находиться в одной плоскости и не иметь
зазубрин. Фактически в плоскости выходной щели образуется
спектральное изображение входной щели, поэтому
предъявляемые к ней требования очень высоки. Малейшие
загрязнения щелей сильно сказываются на работе прибора.
Недостатками призменных монохроматоров являются
относительно небольшая мощность выходящего светового
пучка, сложное устройство, большой вес и размеры прибора.
Поэтому монохроматорами пользуются только тогда, когда
необходимо выделить возможно узкий (до 2 ммк)
спектральный интервал, или когда предполагается подробное изучение
спектрального состава излучения.
Светофильтры находят широкое применение почти во всех
оптических приборах. По спектральным характеристикам
светофильтры делятся на три класса. Первые два класса —
ограничивающие, или срезающие, фильтры,
которые хорошо пропускают лучи с длиной волны, большей или
меньшей заданной. Все остальные лучи фильтры этих классов
сильно поглощают. Третий класс-—это фильтры полосо-
в ы е, или монохроматические, которые сильно
поглощают весь спектр, за исключением одной или нескольких
узких полос пропускания. В физико-химической аппаратуре
обычно используют последние.
Оптические характеристики светофильтров выражаются
графиками зависимости пропускания от длины волны. На
рис. 14 по оси ординат отложена пропускающая способность
светофильтра, равная процентному отношению интенсивности
светового потока, прошедшего через фильтр, к интенсивности
исходного светового потока. По оси абсцисс отложена длина
волны излучения. Практическая пригодность светофильтра
для экспериментальных работ определяется следующими
характеристиками: пропусканием света в максимуме кривой
'■макс полушириной пропускания, или интервалом длин
волн К2—К\ между двумя точками кривой при Т—Л/2 * *.макс
и остаточной величиной пропускания Гост в остальной части
спектра. Лучшими светофильтрами считаются те, для кото-
64
•паке
^
^
■t
Ъ
V;
Vj
?*
t;
ъ
£"
/
/|
/ 1
У i
\ Полуширина
/*\
/Л
|\
I \
Л,
''man *?
ММУ
рых полуширина пропускания имеет минимальное значение,
а 7\макс — максимальное.
По принципу действия светофильтры делятся на
абсорбционные, отражающие (с селективным отражением),
фильтры с различными показателями
преломления и интерференционные.
Абсорбционные фильтры изготовляются из желатина,
окрашенного стекла или пластмасс и водных окрашенных
растворов. В желатиновых
фильтрах между двумя пло- 7^
скими стеклянными
пластинками помещается тонкая
@,1—0,5 мм) желатиновая
пленка, окрашенная
органическими красителями.
Желатиновые фильтры непрочны
и неустойчивы; они изменяют
спектральную
характеристику со временем, мутнеют,
чувствительны к влажности
и температуре. Значительно
более прочны, устойчивы и
воспроизводимы стеклянные
светофильтры. Стекло для
фильтров этого типа
окрашивается при варке молекулярными или коллоидными
красителями. В качестве красителей применяют окислы Со, Ni, Fe или
коллоидальные Ag, Au, Си, редкоземельные элементы и
некоторые другие.
Абсорбционные светофильтры обычно отличаются
невысоким коэффициентом пропускания в максимуме и широкой
полосой пропускания по спектру. Чаще всего их используют для
фильтрации излучения источника, в фотоработах и для
повышения чувствительности фотоколориметрических методов
анализа.
Высокая степень монохроматизации света, достаточная,
в частности, для пламеннофотометрического анализа,
достигается применением интерференционных светофильтров
(рис. 15).
Простейший интерференционный светофильтр состоит из
лвух полуотражающих металлических слоев, между которы-
рыми помещена тонкая пленка прозрачного диэлектрика
(рис. 16,А). Световой луч, проходящий через такой фильтр,
дважды претерпевает отражение от полуотражающих слоев
Рис. 14. Оптические характеристики
светофильтров (по Полуэк'гову)
3 Зак. 51
65
400
500
600
Рис
тика
ного
так, что в какой-либо точке К совмещаются два луча:
прямой в и отраженный а. В результате интерференции эти лучи
могут взаимно усилиться (суммироваться) или погаситься
Максимальное увеличение
амплитуды колебаний
произойдет при условии, что
разность хода двух лучей
равна четному числу
полуволн, т. е. 2 — =Х, 4- =2%;
2 2
%
6 — —2>% и т. д. Разность хо-
tJ 2
700мм* да практически равна
удвоенной толщине слоя
диэлектрика, поэтому
максимальное пропускание возможно в
том случае, если толщина б
диэлектрика равна целому
числу полуволн:
2
где N — целое число.
Практически полное
гашение лучей наблюдается,
если разность хода равна
нечетному числу полуволн, т. е.
— , о — , о— и т. д. или
2 2 2
BЛМ-1) j, где 2N+ 1—
нечетное число, а луч падает
перпендикулярно
поверхности фильтра. Пропускание
15. Сравнительная характерис-
пропускания интерференцион-
(/) и абсорбционного (f[)
светофильтров
300
500
гоос
ммк
Рис. 16. Устройство
интерференционного светофильтра (А) и его
пропускающая способность (£) при
толщине слоя диэлектрика, равной
1,16 мк;
I — падающий световой поток;
// — световой поток, прошедший
через фильтр;
/ — стеклянные пластины; 2 — слои
серебра, 3 —■ диэлектрик (сульфид
цинка)
66
простого интерференционного светофильтра зависит также от
свойств диэлектрика и полуотражающих слоев.
Коэффициент пропускания таких фильтров в максимуме
около 40%, а в минимумах менее 1%- В многослойных
фильтрах удается, не увеличивая ширины пропускания, достичь
увеличения пропускающей способности до 80%.
Любой интерференционный фильтр, как вытекает из
изложенного, имеет серию максимумов и минимумов
пропускания (рис. 16,Б). Совмещение интерференционного фильтра
с абсорбционным устраняет ненужные максимумы и дает
фильтры, пропускающие только один узкий спектральный
интервал шириной всего лишь в 3—4 ммк (а иногда и менее).
КОНТРОЛЬ И ИЗМЕРЕНИЕ ТЕМПЕРАТУРЫ
В лаборатории постоянно приходится контролировать и
измерять температуру. Поправки на температуру вводятся
при вычислениях потенциалов электродов, измеряемых
с целью определения рН и окислительно-восстановительных
потенциалов почвы. Влияние температуры на подвижность
ионов, объем различных тел, величину фототока и т. п.
заставляет термостатировать ячейки солемеров, полярографов,
фотоэлементы пламенных фотометров и др. Измерение
температуры является основным приемом термического анализа
тонкодисперсной фракции почв и служит приемом
определения интенсивности инфракрасного излучения при изучении
молекулярных спектров поглощения.
При обычных потенциометрических определениях
температура измеряется ртутными термометрами с
точностью, требуемой постановкой эксперимента. Для
регулирования температуры с точностью 0,01—0,001° в пределах от 0
до 150° находят применение водяные ультратермоста-
т ы, работающие от ртутных контактных термометров.
Контролирование и программное регулирование температуры в
электропечах осуществляется с помощью термопар и
электронных потенциометров. Термопары
используют также для прямого измерения температуры образцов
в процессе термического анализа. Высоко чувствительными
приборами являются термостолбики
(термоэлементы) и термосопротивления, необходимые для
измерения незначительных изменений температуры
(инфракрасные спектрометры).
Ртутные термометры пригодны как для измерения, так и
для регулирования температуры (контактные ртутные термо-
3*
67
метры). В среде с измеряемой температурой обычно
находится только нижняя часть столбика ртути, а верхняя часть
столбика и стеклянной трубки принимает некоторую
промежуточную температуру между температурами измеряемой и
окружающей среды. Это заставляет вводить в показания
термометра поправку на выступающий столбик ртути, особенно
при точных измерениях и при измерениях высоких
температур. Поправка на выступающий столбик ртути вычисляется
по формуле
Дг — кп (гТСрМ *в0зд),
где \t—поправка в °С; k—коэффициент линейного
расширения стеклянного капилляра, который зависит от сорта
стекла и конструкции термометра и обычно принимается
равным 1/6000; h — число градусных делений в выступающей
части ртутного столбика; ^теРм —температура, показываемая
термометром, и ^В03д — температура окружающего воздуха
(или, точнее, температура средней части выступающего
столбика, определяемая вспомогательным термометром).
В контактных термометрах с магнитной головкой
(рис. 17, а) в капилляр впаян проводник, который постоянно
погружен в ртутный столбик; второй проводник укреплен на
винте; он приводится во вращение при поворотах магнитной
головки. Поднимая или опуская штифт вращением магнита,
можно установить конец проводника на заданном делении
термометра. Когда температура поднимается, между обоими
проводниками возникает электрический контакт через
столбик ртути. Контактный термометр включается в схему
нагревателя, как показано на рис. 17,6. При замыкании контактов
реле срабатывает и отключает нагреватель от источника
питания. Если температура опускается ниже заданного уровня,
контакт между столбиком ртути и верхним проводником
нарушается и реле вновь включает нагреватель. Такой
терморегулятор позволяет поддерживать заданную температуру
длительное время с точностью, зависящей от конструкции
.термометра.
, Термопары — приборы для измерения температуры -—
состоят из двух проводников, концы которых сварены с одной
стороны, а с другой соединены с гальванометром или
милливольтметром (рис. 18). Возникновение тсрмоэлектродвижу-
[щей силы (т. э. д. с.) обусловлено разницей работы выхода
Электронов для металлов, образующих термопару. Величина
работы выхода меняется с температурой, и поэтому при на-
68
Контактный
термометр
Обмотка
нагревателя
Рис. 17. Ртутный контактный термометр.
а — общий вид: 1,2 — электрические контакты,
3 -магнитная головка, 4-—клеммы для
подключения к реле; б включение термометра в цепь
нагревателя; Эм — электромагнит; Пр — ртутный
прерыватель; Тр — трансформатор
Рис. 18. Устройст
во термопары
типа ПП:
1 — горячий спай,
2 ■— платиновая
проволока, 3 —
проволока из
сплава Pt+10%
Rh, 4 —
соединительные провода,
5 —
милливольтметр
) гревании спая т. э. д. с. возрастает. Согласно электронной
i теории величина т.©.д.с. определяется уравнением:
N.
т. э. д. с.
-In
N,
■(Tt-TJ,
в
где к — постоянная Больцмана, равная 1,38- 10~16 эрг/град;
е — заряд электрона; NA и N-ц— число свободных электронов
в 1 см3 материалов, из которых изготовлена термопара,
Т2 — температура горячего спая и Ti — температура
холодного спая в замкнутой термопаре.
Влияние температуры на потенциал выхода (и работу
выхода) очень мало, и 'поэтому величина т. э. д. с. невелика и ис-
\ числяется всего лишь
единицами или десятками милли-
/ вольт. Соединение холодных
концов термопары с
подвозящими проводами не
меняет т.э.д.с, если только
подводящие провода однородны
и температура в местах
соединения строго одинакова.
Помещая концы
холодных спаев в термостат,
можно поддерживать
постоянство их температуры
достаточно длительное время.
Практически в лабораторных
условиях прибегают к термо-
статированию с помощью
сосудов Дьюара,
заполненных смесью льда с водой (см. гл. VIII). Иногда применяют
компенсаторы, автоматически вводящие поправки на
изменение температуры холодных спаев. Принципиальная схема
такого компенсатора показана на рис. 19: термопара Тп и
милливольтметр V включаются в диагональ моста, питаемого
от аккумулятора Акк на 4 в. Сопротивление R, являющееся
одним из плеч моста, располагается в непосредственной
близости от свободных концов термопары и имеет одинаковую
с ними температуру; это сопротивление изготавливается из
медной или никелевой проволоки, а остальные плечи моста —
из манганитовой проволоки, сопротивление которой
практически не меняется с повышением температуры. Перед работой
мост уравновешивается с таким расчетом, чтобы напряжение
Рис. 19. Схема автоматической
компенсации температуры холодного
спая
70
на диагонали моста было равно нулю. При изменении
температуры холодного спая изменяется и величина
сопротивления R, в результате чего мост выходит из равновесия.
Возникшее дополнительное напряжение на диагонали моста
компенсирует уменьшение т.е. д. с. термопары. Точность ком-
ленсации такого рода устройств достигает 0,04 мв на 10°.
Выбор термопары зависит от измеряемой температуры,
т. э. д. с, развиваемой термопарой, и устойчивости материала,
Рис 20 Схема ЭРМ-47:
1 — клеммы для подключения прибора к сети, 2 — клеммы для
включения обмотки нагревателя, 3—ртутное реле, 4 — трансформатор,
5 — шкала и стрелка милливольтметра, 6 — алюминиевый флажок,
7— катушки индуктивности, 8 — клеммы для термопары
из которого изготовлена термопара, к той среде, в которой
предстоят измерения. Всегда желательно использовать
термопару с наибольшой т. э. д. с, что позволяет значительно
легче и с большей точностью провести измерения. Наиболее
i распространены нлатина-платинородиевые, хромель-алюмеле-
j вые, хромель-копелевые термопары и пары: константан—медь
i и константан—железо.
Термопары более удобны при измерениях температуры,
чем ртутные термометры, поскольку они пригодны для
измерения высоких (выше 1000°) температур и для
дистанционных измерений; с помощью термопар легко осуществляется
программное регулирование температуры. Простой регулятор
для электропечей состоит из платино-плагинородиевой
термопары и электронного регулятора типа ЭРМ-47 (или любого
71
автоматического регулирующего потенциометра). В приборе
ЭРМ-47 (рис. 20) имеется милливольтметр, к которому
подключается термопара; шкала 5 милливольтметра
отградуирована в градусах Цельсия, и по ней всегда можно узнать
температуру среды, в которую помещена термопара. Вторая
стрелка шкалы контрольная (задающая), предназначена для
ограничения нагрева. Контрольная стрелка несет на себе две
катушки индуктивности, включенные в сеточный контур
лампы 6ПЗ, а на стрелке вольтметра укреплен легкий
алюминиевый флажок 6, который при совпадении положения обеих
стрелок входит в зазор между катушками. В результате
индуктивность катушек изменяется, что приводит к резкому
снижению анодного тока в цепи лампы 6ПЗ. Уменьшение
анодного тока, питающего обмотку электромагнитного реле.3,
вызывает его выключение. При выходе флажка из зазора
катушек реле включается снова (устройство ртутного реле
см. на рис. 17).
При неподвижном положении контрольной стрелки прибор
поддерживает в печи постоянную заданную температуру; если
контрольную стрелку перемещать вдоль шкалы с
постоянной скоростью, то температура печи возрастает с той же
скоростью.
Термоэлементы высокой чувствительности, пригодные для
обнаружения теплового излучения очень небольшой
мощности (порядка Ю-10 вт), нашли применение в инфракрасных
спектрометрах. Термоэлементы изготавливают из очень
тонких проволочек или фольги, получаемой специальными
способами; по одному из них спаивают две проволоки,
образующие термопару, и прокатывают их. Из полученной фольги
вырезают нужной ширины ленту и собирают термобатареи или
термостолбики, содержащие часто до нескольких десятков
последовательно соединенных горячих спаев. Для нанесения
очень тонких слоев пользуются также распылением металла
в вакууме. Термобатареи или термостолбики обычно
помещают в вакуум, что повышает их чувствительность во
много раз.
Термометры сопротивления .построены на так называемых
термосопротивлениях, представляющих собой обычный
проводник (медь, платина) или полупроводник, нагреваемый
током и находящийся в теплообмене с окружающей средой.
Термосопротивление включают как одно из плеч моста,
питаемого от сухого элемента. В зависимости от диапазона
измеряемых температур мост уравновешивают так, чтобы
минимальная температура соответствовала нулевому деле-
72
нию шкалы. Вследствие нагрева сопротивления баланс моста
нарушается и стрелка измерительного прибора отклоняется.
Важным условием хорошей работы термометра
сопротивления является минимальная нагрузка током, чтобы
выделяемое при прохождении тока тепло было значительно меньше
тепла, получаемого от изучаемой среды. В качестве
чувствительных элементов применяют как металлические
проводники, так и полупроводниковые термосопротивления (терми-
сторы), у которых температурный коэффициент в несколько
раз больше, чем у металлов. Из чистых металлов используют
платину, медь и никель. Величина температурного
коэффициента для чистых металлов близка к 0,004, для
полупроводников порядка 0,03—0,04.
В широком диапазоне температур зависимость между
сопротивлением и температурой нелинейная, так, сопротивление
платиновой проволоки в пределах от —40° до +630°
выражается уравнением
R --- ■/?„(! + 3,940- Ю-? + 5,810- 10~7Г2),
где R0 — сопротивление при 0°.
Применение термосопротивлений из того или иного
металла определяется преимущественно устойчивостью металла
в изучаемой среде при заданных температурах. Вследствие
химической инертности часто применяется платина, верхним
пределом применения которой считают 1000—1200°. Никель
при условии хорошей изоляции работает до 250—300°, а медь—
до температуры 100—150°.
Широкое использование, в том числе для измерения
температуры почвы, находят термисторы, отличающиеся высоким
температурным коэффициентом и высоким собственным
сопротивлением, что дает определенный выигрыш при их
эксплуатации. Термисторы изготавливаются из соединений
окисей металлов Си, Мп, Ni, Co путем термической обработки
пастообразной смеси окислов, нанесенной тонким слоем на
■стеклянную пластинку. По внешнему виду они напоминают
обычные радиосопротивления.
Термисторы изготовляют различных типов и выбирают в
зависимости от назначения и испытуемой среды. Интервал
измеряемых температур относительно невелик (—70 (-150°),
но чувствительность термисторов благодаря высокому
температурному коэффициенту значительная: так, термисторы из
соединений окиси марганца и никеля могут реагировать на
изменение температуры в 0,0005е.
73
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ, ПРИМЕНЯЕМЫЕ В СХЕМАХ ПРИБОРОВ»
А ТАКЖЕ СЛУЖАЩИЕ ДЛЯ МАРКИРОВКИ ШКАЛ*
1. Обозначения приборов и их технические характеристики
Система приборов
Магнитоэлектрическая
Электрома пштная
Электродинамическая
Классы точности приборов:
Испытательное
Напряжение
изоляции
прибора (например
на 2 кв)
Магнитный экран
Постоянный ток
Электростатическая
Тепловая
Термоэлектрическая
Выпрямитель вообще
Гальванометр или
амперметр
2. Обозначения в схемах приборов
Ответвление проводов
©
па
С=Ь
V.
н#-
Переменный ток
Постоянный и
переменный ток
Провод
<"^ Пересекающиеся
(в схеме) провода
г^>
Провод экраниро- gs=
ванный
Здесь представлены только наиболее важные обозначения.
74
Сопротивление
достоянное
или
-Сопротивление
переменное
Потенциометр
Диоды прямего
накала
Диод с
подогревным катодом
Триод
Тетрод
Пентод
-VWW-
?L
о
W
Газотрон
Газоразрядный
стабилизатор
напряжения
Реостат
проволочный
или
Катушка
индуктивности
Конденсатор
постоянной емкости
Конденсатор
переменной емкости
Конденсатор
электролитический
Колебательный
контур
Дроссель
Трансформатор
•ЛЛЛЛЛ-
-/щяг
О -у-
Hi
-у^ШШЬ
75
Автотрансформатор
Регул ировочный
автотрансформатор
Фотоэлемент
вакуумный
Бареттер
to
Лампа осветительная
Неоновая ламна
Фотоэлемент
газонаполненный
Термистор
Лампа сигнальная
Важнейшие физические константы*
Ускорение силы тяжести на уровне моря
и широте 45°, g] . . •
Абсолютный нуль температуры
Температура таяния льда при давлении
1 атм
Нормальная атмосфера
980, 665 см -сек—2
—273, 16Х
273, 16 °К
1,03325 кг/см* -
= 1,01326- Ю-6 дин-см~2
22414 см*-моль~х
Объем грамм-молекулы идеального газа прн
нормальных условиях
Универсальная газовая постоянная, R. . . 8,ШЗ-№ эрг-град~1- моль~к
=0,08205 л-атм/град-мсь =
= 1,98645 кал/град-моль
6,0228-1023
2,687-1019 cm-
Число Авогадро (число молекул в одной
грамм-молекуле), Ne
Число Лошмидта (число молекул в 1 см3
при нормальных условиях), п0
Постоянная Больцмана (отношение
универсальной газовой постоянной к числу
Авогадро), К 1,380-Ю-16 эрг-град-1
* Значения многих констант подвергались изменениям по мере
повышения точности измерения. Приведенные в таблице значения обеспечивают
достаточную для практических целей точность вычислений.
76
Масса атома водорода 1,673 ■ 10 24 г
Масса протона' 1,672-Ю-24 г
Масса электрона 9,107-10 28 г
Заряд электрона 4,80- 10-10ои2/3-г1/2- сек =
-1,59-Ю-19 к
Масса кванта света с частотой \ 0,728-Ю-47 м г
Квантовая постоянная (постоянная
Планка), Л 6,625-Ю-27 зрг-сек
Скорость света в вакууме B,997928±0,000004)-10"> см Ксек.-*
Число Фарадея, F 96 491 к-г-экв .
Кратные и дольные производные физических единиц*
Наименование
Во
сколько раз
изменяется
основная
величина
Сокращенные
обозначения
Наименование
Во
сколько раз
изменяется
основная
величина
Сокращенные
обозначения
Пико
Нано
Микро
Милли
Санти
Деци
10
10~
10"
10~
10-
10
—12
,-1
п
н
мк
м
с
д
Дека
Гек то
Кило
Мега
Гига .
Тера .
10
10*
103
10«
Ю9
10»г
да
г
к
М
Г
т
* Наименования кратных и дольных производных всех единиц содержат
приставки, которые указывают во сколько раз увеличивается или уменьшается
числовое значение единицы.
Глава III
ПОТЕНЦИОМЕТРИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ
ПОЧВ
'Потенциометрические методы анализа широко
применяются для исследования почв; по распространенности в
научно-исследовательских и производственных почвенных и
агрохимических лабораториях они занимают одно из первых мест
среди физико-химических методов анализа. Всеобщее
признание получили потенциометрические методы
определения рН, окислительно-восстановительных потенциалов. В
последние годы разработаны новые типы электродов, пригодных
для определения в почвах активности ионов натрия и калия,
совершенствуются электроды для определения ионов
кальция. Потенциометрические методы определения активности
яонов позволяют определять в почвах содержание
водорастворимых солей, поглощенных оснований, буферность почв и
нуждаемость кислых почв в известковании, изучать свойства
и поведение органических и минеральных веществ почв.'
Бесспорными преимуществами этих методов являются
простота техники измерения и аппаратуры, доступной любой
почвенно-химической лаборатории, и малое время,
затрачиваемое на одно определение. В большинстве случаев
оказывается возможным изучать свойства почвы и почвенных
растворов, не изменяя их состава и концентрации, и получать
тем самым характеристики почв в естественном, природном
состоянии. Это позволяет применять некоторые электроды
в вегетационных опытах, непрерывно следить за состоянием
почвы по фазам развития растений.
Практически техника работы потенциометрическими
методами сводится к измерению э. д. с.
78
ПОТЕНЦИАЛ ЭЛЕКТРОДА
Потенциометрические методы основаны на измерении
потенциалов электродов, обратимых по отношению к каким-
либо ионам. Уравнение зависимости потенциала от
активности ионов можно вывести, если принять, что разбавленные
растворы во многом подчиняются газовым законам.
Известно, что бесконечно малая работа dA, совершаемая
газом при изотермическом расширении, пропорциональна
давлению газа р и бесконечно малому изменению объема dv:
dA = pdv.
n'RT
Поскольку р = , где п' — число молей газа, для изо-
V
термического процесса
dA = n'RT~.
V
Если объем изменяется при этом от Vi до v2, то
интегрируя последнее уравнение в тех же пределах, получим
конечную величину работы А:
щ
[dA=[ n'RT—, A=— n'RT In-^и
J J V V!
По аналогии:
n'RTIn -£-;
Pi
A — максимальная работа, которую можно получить при
расширении л' молей газа от объема Vi до v2 или при изменении
давления от р\ до р2 в изотермическом процессе.
В разбавленных растворах вместо давления ставят
осмотическое давление, объемы заменяют обратной им
величиной— концентрацией. Для раствора и соответствующих
концентраций уравнение будет таким:
А = n'RT In -£-.
Здесь А — максимальная работа, которая совершается при
изменении концентрации раствора от С\ до Сг. Если вся
работа заключается в перемещении зарядов, то ее можно
вычислить и как произведение разности потенциалов Е на
количество перемещенных зарядов. Если Е — скачок потенциала
79
между электродом и раствором, или сокращенно — потенциал
электрода, то
Аэл — EQ>
где Q — количество электричества, которое подсчитывают по
формулам:
Q =--= n'nF --- n'nNe и Лэл = En'nF;
F— число Фарадея, /V —число Авогадро, в — заряд
электрона и п — число зарядов, переносимых одной частицей
(ионом) вещества. При А =Аал
Аъл ■--- En'nF = п'КГ In
Р — ° \ Съ
Это уравнение называют уравнением Нернста. Оно
применяется как для одного электрода, погруженного в раствор,
так и для цепи, состоящей из двух одинаковых электродов,
погруженных в растворы с концентрациями
потенциалопределяющих ионов, равными Ci и С2. Когда речь идет об одном
электроде, то уравнение записывают так:
4 е=-е<Н — 1пС,
где С — концентрация потенциалопределяющих ионов в
растворе, а со — постоянная для данного электрода величина.
Очевидно, что е = е0, когда С—\. Такой электрод,
концентрация потенциалопределяющих ионов в котором равна
единице, называют нормальным электродом, а его
потенциал — потенциалом нормального
электрода. Величину е0 экспериментально находят для каждого
типа электродов и приводят в таблицах справочных изданий.
Э. д. с. элемента, состоящего из двух одинаковых электродов,
погруженных в растворы, различающиеся только
концентрацией (концентрационная цепь), не зависит от потенциала
нормального электрода. Для каждого из таких электродов
справедливо уравнение
RT
и э. д.
80
Для простоты изложения во всех предыдущих выводах в
уравнения подставлялась концентрация С. Реальные
растворы подчиняются законам, выведенным для идеальных систем,
только в том случае, когда вместо величин концентрации в
уравнения подставляются величины активности:
i8 — е„ 4-
RT
nF
\па-
RT
nF
In Су;
Y — коэффициент активности.
Разобранные уравнения потенциала электрода служат
основой для любых расчетов, принятых в потенциометриче-
ских методах анализа.
Для элементов из двух или нескольких различных
электродов обязательно учитывается стандартная э. д. с. Из
уравнения Нернста для двух разных электродов с потенциалами
е' и г":
-^-Ina'— (el + -$£- inаЛ =
nF { ° nF j
RT , e'_ r, , RT
э. д. с = e — e — e„
= Eo-eo-
nF
In
-= E0 -f
nF
In
если величина п одинакова для обоих электродов, а а' и а"-
активности ионов и £о=е0—е0.
Переходя от натуральных логарифмов к десятичным,
получим
RT
е-= еп
2,303
nF
Iga
или
)
' '*' ,j ■ v э. д. с. - Е0 + 2,303^- lg -4
-vstw . - nF
i Потенциал одного, отдельно взятого электрода измерить
практически невозможно. Цепь для измерений составляется
минимум из двух электродов, один из которых — электрод
сравнения — имеет заранее известный и постоянный скачок
потенциала. Электрод сравнения осуществляет контакт
между раствором и клеммой измерительного прибора; вторая
клемма прибора соединяется с индикаторным электродом.
Например, при измерении рН раствора составляют цепь из
водородного и каломельного полуэлементов:
Pt
НС1
н2 ;
2н+
КС1
насыщ.
HgaCl2
Hg
81
В этой схеме вертикальная черта означает границу
раздела двух фаз.
Э. д. с. элемента равна алгебраической сумме скачков
потенциалов в цепи; для написанной выше цепи она
складывается из потенциала водородного электрода еь
потенциала Е2 каломельного электрода и диффузионного
потенциала ез, возникающего в месте соприкосновения двух
электролитов
э. Д. с. = et + е2 + е3.
Величина диффузионного потенциала невелика, и ею
можно пренебречь. Чтобы свести до минимума возникающую при
этом ошибку, используют соединение электродов череа
электролитический ключ, содержащий концентрированный
раствор соли, подвижность обоих ионов которого практически
одинакова; чаще всего это растворы КС1, NH4NO3.
Постоянное значение имеет и потенциал каломельного-
электрода (электрод сравнения); суммируя все скачки
потенциалов в цепи, находим
э. д. с. = k + ех = е' + 2,303-^- Igfl,
" nF
где к — постоянная величина, включающая потенциал
каломельного электрода и диффузионный потенциал.
Следовательно, всякое изменение э. д. с. рассматриваемого-
элемента обусловлено только изменением потенциала
водородного электрода или активной концентрацией
определяемого иона. Поэтому при графических построениях и в тех
случаях, когда требуется знать не абсолютное значение
потенциала (по отношению к водородному электроду), а лишь
его изменения, можно пользоваться значениями э. д. с. такого
элемента в целом.
Изменение потенциала при уменьшении активности ионов
от ах до «2 равно:
или
, = е'0 + 2,303 -^-Igfli-
.^+2,303-^-lga^.
■в,- 2,303-^-Ig-^-
nF c2
82
тт л, и оол-э RT 0.0001983T
Численное значение коэффициента 2,6кN —
nF n
ОЛ10 0,0581 „ ,
а при 20 равно . Если п—\, тогда
п
ех — е2 = 0,0581 (lgox — lg a2).
При изменении активной концентрации в 10 раз разность
логарифмов активных концентраций равна единице, а э. д. с.
цепи изменяется на 0,0581 в или на 58,1 мв. В тех реакциях,
где п — 2, численное значение коэффициента уменьшится
вдвое @,029 в) и т. д.
До сих пор мы рассматривали возникновение скачка
потенциала как следствие перехода атомов металла в раствор
и обратно, иначе говоря, как процесс окисления и
восстановления с участием атомов элемента самого электрода или
сорбированного им газа (водорода).
В некоторых случаях обе формы: окисленная и
восстановленная, одновременно находятся в растворе, например в
системе
Fe3+ + ei±Fe2+.
Металлический электрод для таких систем служит лишь
источником или акцептором электронов, а
окислительно-восстановительный процесс не сопровождается переходом
вещества из одной фазы в другую. Потенциал^ индифферентного
электрода, например платинового, в растворе, содержащем
окисленную и восстановленную форму железа (Fe3+—Fe2+),
определяется по уравнению
s = e0 + 2,606—— lg — .
nF °Fe2+
B отличие от концентрационных элементов здесь в
формулу входит не логарифм активности, а логарифм отношения
активных концентраций окисленной и восстановленной форм.
Иными словами, потенциал такого электрода не зависит от
абсолютного содержания ионов, а только от их отношения.
Понятие нормального потенциала
окислительно-восстановительных систем имеет важное значение. Нормальный
окислительно-восстановительный потенциал служит мерой
окислительных свойств системы. Например,
нормальный окислительно-восстановительный потенциал системы
Cu2+/Cut-= +0.17 в, а потенциал системы Fe3+/Fe2+=H?75 в;
сравнивая эти системы, легко прийти к выводу, что Fe*+ яв-
83
ляется более сильным окислителем, чем Си2+, и поэтому при
взаимодействии этих систем первая из них окисляется. По
окончании реакции суммарное значение потенциала
установится между 0,17 и 0,75 в.
Фактически уравнение Нернста позволяет вычислить
только приращение потенциала вследствие изменения
концентрации. Суммарный скачок потенциала всегда вычисляется по
отношению к условно выбранному стандартному электроду.
В электрохимии принято нулевым значением потенциала
считать потенциал нормального водородного
электрода, т. е. водородного электрода, насыщенного при
давлении в одну атмосферу газообразным водородом и
находящегося в равновесии с раствором, активность водородных
ионов которого равна единице. Потенциал нормального
водородного электрода считают равным нулю при всех
температурах.
Величина и знак любого электрода определяется в
сравнении с водородным электродом. Считают, что электрод имеет
положительный знак, если при самопроизвольной работе
элемента электроны покидают водородный электрод. На таком
электроде происходит восстановление вещества. При
обратном процессе считают, что электрод имеет отрицательный
знак по отношению к нормальному водородному электроду.
То же правило сохраняется и для определения знака
электродов в цепи без водородного электрода. В этом случае
определяют знаки электродов относительно друг друга. По
отношению к водородному оба электрода могут иметь одинаковый
знак — положительный или отрицательный; при составлении
цепи из таких электродов положительным будет тот,
потенциал которого выше по отношению к водородному электроду
(или тот, который в процессе реакции приобретает
электроны).
Определение знака электрода необходимо при
вычислении электродных потенциалов по результатам измерения
э. д. с. Так как общая разность потенциалов в цепи равна
алгебраической сумме скачков потенциалов
э.д.с. =(ех) +(е2) +...,
то при расчетах необходимо знать знак каждого электрода
в данной цепи. Если, например, цепь составлена каломельным
электродом, потенциал которого равен +0,245 в, и хингид-
ронньш электродом со значением потенциала +0,703 в, то
э.д.с. = ( +0,703)+ ( — 0,245) = +458 мв.
84
Потенциал каломельного электрода берется со знаком'
минус, так как в данной цепи каломельный электрод более
отрицателен, чем хингидронный.
В экспериментальной практике знак электрода определяют
из опыта. При определении э. д. с. компенсационным методом
(см. ниже) сравнивают э. д. с. изучаемого элемента и
стандартного источника тока. По условиям определения
сравниваемые элементы должны соединяться одинаковыми
полюсами. В правильно собранной цепи положительным электродом
считается тот, который соединен с положительным полюсом
аккумулятора или элемента Вестона. Если же цепь собрана
неверно, то найти точку компенсации не удастся; поменяв
местами электроды, ошибку исправляют и определяют знаки
электродов.
ИЗМЕРЕНИЕ Э.Д.С.
Наиболее правильно и точно измеряют э. д. с.
компенсационным методом, по которому разность потенциалов между
полюсами элемента находят в тот момент, когда в цепи не
протекает электрический ток. Именно это условие и
позволяет определить разность потенциалов элемента, весьма
близкую к его э. д. с.
В замкнутой цепи э. д. с. элемента равна сумме падения
напряжения внутри элемента и напряжения на зажимах
элемента (или падения напряжения во внешней цепи).
Измеряемое вольтметром напряжение на зажимах элемента
меньше, чем его э. д. с, на величину внутреннего падения
напряжения. Кроме того, некоторая (небольшая) часть напряжения
гасится в проводах, соединяющих элемент с измеряющим
прибором. Таким образом, чем выше внутреннее
сопротивление элемента и чем выше сопротивление соединительных
проводов, тем больше при пользовании вольтметром будет
разница между измеренной величиной напряжения и э. д. с.
Если же тока в цепи нет (цепь разомкнута), то очевидно,
что ни внутреннее сопротивление элемента, ни сопротивление
проводов не повлияют на измерения. Замкнутую цепь, в
которой не протекает ток, можно приравнять к цепи
разомкнутой или к цепи с бесконечно большим сопротивлением. Когда
сила тока равна нулю, а сопротивление — бесконечности, то'
естественно, что никакое конечное изменение сопротивления
внешней или внутренней цепи не отразится на результатах.
При прохожднии тока в элементе электроды могут
поляризоваться. Поляризация электродов в результате электро-
8S
лиза раствора и последующих реакций сопровождается
выделением газов или изменением концентрации компонентов
раствора в слое около электродов. Изменение состояния или
концентрации у электродов вызывает возникновение
поляризационной э. д. с, направленной противоположно основной и
снижающей тем самым э. д. с. элемента. Влияние поляриза-
.ции можно снизить включением сопротивления в цепь, умень-
Рис. 21. Компенсационная схема Поггендорфа:
Акк— аккумулятор; Вк—выключатель источника тока; Я —реостат
для регулирования напряжения, АВ — реохорд (потенциометрическая
проволока), С — скользящий контакт, Г—гальванометр,
К—телеграфный ключ, Пк — переключатель; Ее—нормальный элемент Вестона;
Ех — исследуемый элемент
щением промежутка времени замыкания цепи при измерении
э. д. с. При компенсационном методе определения э.д. с,
поскольку сила тока в момент измерения и вблизи точки
компенсации практически равна нулю, можно считать, что в
элементе не будет побочных процессов, ведущих к поляризации
электродов.
На рис. 21 изображена простейшая схема, используемая
для измерения э.д. с. компенсационным методом (схема
Поггендорфа), состоящая из аккумулятора на 1,5—2 в, потен-
циометрической проволоки (реохорд) с сопротивлением
порядка 20 ом, гальванометра, нормального элемента Вестона,
ключей и переключателя.
Проволока либо растянута на линейке с делениями, либо
намотана на барабан. Вся проволока должна иметь строго
86
одинаковое сечение, однородный состав и малый
температурный коэффициент сопротивления, достаточную механическую
прочность. Часто используют для реохорда проволоку из
нержавеющей стали. Реохорд можно заменить двумя
штепсельными магазинами сопротивления. Напряжение с реохорда
снимается с помощью скользящего контакта.
Подводящие провода имеют очень малое сопротивление,
и поэтому все напряжение аккумуляторов падает практически
на потенциометрической проволоке, равномерно от точки А
к точке В. На барабане (линейке) реохорда нанесены
деления, разделяющие всю проволоку на 1000 равных частей; на
каждое деление приходится падение напряжения, равное '/юоо
напряжения аккумулятора. Если передвигать подвижный
контакт реохорда, то оказывается, что в некоторый момент
падение напряжения на участке АС равно э. д. с. нормального
элемента. Поскольку токи от аккумулятора и от нормального
элемента направлены навстречу друг другу, то суммарное
значение силы тока, протекающего в этот момент через
нормальный элемент и гальванометр, равно нулю.
Процесс измерения сводится к простым операциям:
включают аккумулятор и элемент Вестона, замыкают на короткое
время ключ К и передвигают подвижный контакт вдоль
проволоки до тех пор, пока гальванометр не покажет отсутствия
тока в малой цепи. Соотношение между величиной падения
напряжения £Лкк на всей потенциометрической проволоке и
э. д. с. нормального элемента определяются формулой:
- -—■= £в.
АВ
где АВ — число делений на всей потенциометрической
проволоке, АС — число делений от начала реохорда до подвижного!
контакта, отсюда
р _ ЕвАВ
tAKK~~AC •
Измерения наиболее точны, если величина АС равна
примерно половине Л В. Это условие соблюдается, когда э. д. с.
нормального элемента составляет около половины напряжения
аккумулятора. При иных соотношениях точность измерения
снижается; измерение практически становится невозможным,
если э. д. с. меньше, чем 0,01 ЕАкк (или э. д. с. больше, чем
U,99 ЕЛкь). Указанными соотношениями руководствуются при
8Z
выборе источника питания и нормального элемента;
используя элемент Вестона, э. д. с. которого около 1 в, получают
хорошие результаты, если общее падение напряжения на
реохорде составляет 1,6—2,0 в.
После измерения £Лкк измеряют э. д. с. неизвестного
элемента, подключив его переключателем Пк вместо
нормального элемента; из приведенной выше формулы получим
£rr АСХ с АС\
х -■ САкк —7— = ев — —,
* АВ АС
где Ех — э. д. с. неизвестного элемента, АС] — отсчет по
реохорду в момент, когда э. д. с. изучаемого элемента была
полностью компенсирована падением напряжения на этом
участке реохорда.
В качестве нормального элемента для определения £акк
обычно используют нормальный элемент Вестона, принятый
в качестве международного стандарта сравнения, э. д. с
которого (табл. 9) очень мало изменяется в пределах от 15
до 25° и может быть вычислена по уравнению
Ев = 1,01830 + 0,00004 B0° — *°) е.
Таблица 9
Э. д. с. элемента Вестона при различных температурах
°с
15
16
17
18
19
20
Э. д. с.
1,01850
1,01846
1,01842
1,01838
1,01834
1,01830
СС
21
22
23
24
25
а д. с.
1,01826
1,01822
1,01818
1,01814
1,01810
Для нормальной работы элемента Вестона включать его
'в цепь можно только на очень короткое время во избежание
поляризации. Короткое замыкание сильно изменяет э. д. с.
элемента и на длительное время выводит его из строя.
Э.д. с. элемента может быть правильно измерена, если за
время измерений напряжение аккумулятора остается строго
■постоянным, что проверяется по крайней мере двумя
контрольными отсчетами — до и после работы. Если показания
на реохорде при этом увеличиваются больше, чем на 1
деление, значит, аккумулятор разряжен и его необходимо заме-
88
нить новым. В некоторых случаях уменьшение показаний
может быть вызвано тем, что первое измерение было сделано
сразу после включения аккумулятора; устойчивое значение
Еаик, особенно свежезаряженного, получается только через
8—10 мин после включения аккумулятора в цепь.
Порядок определения напряжения
аккумулятора и э.д.с. гальванического
элемента
1. Собрать компенсационную схему, как показано на
рис. 21 (Пк в положении 1).
2. Замкнуть цепь аккумулятора и через 10—15 мин
приступить к измерениям напряжения аккумулятора.
3. Для измерения напряжения аккумулятора изменять
положение подвижного контакта С, замыкая после каждого ■>
передвижения ключ К на возможно короткое время. Наблю- ,
дая за показаниями гальванометра, добиться, чтобы стрелка
гальванометра перестала отклоняться при замыкании ключа.
4. Проверить правильность точки компенсации; для этого
слегка смещать подвижный контакт от точки равновесия
вправо и влево. При небольших смещениях (на 1—2 деления)
стрелка гальванометра должна заметно отклоняться;
направление тока при переходе через точку компенсации изменяется:
записать отсчет по реохорду и повторить компенсацию еще
два раза, каждый раз записывая отсчеты.
5. Включить в цепь измеряемый гальванический элемент,
для чего переключатель Пк поставить в положение 2, вновь,
найти точку компенсации и записать отсчеты.
6. Переключатель Пк перевести в положение 1 и
проверить напряжение аккумулятора.
7. Вычислить напряжение аккумулятора ЕМхК и э. д. с.
гальванического элемента.
Вычисление результатов вести до десятитысячных долей
вольта; при записи в журнал результаты округляются до
тысячных долей вольта, которые и являются достоверными для
данной схемы. Неправильно было бы записывать результаты
с большим числом значащих цифр, чем это обусловлено
точностью измерения.
Простая компенсационная схема очень надежна и
устойчива в работе; пользуясь ею, можно получать прекрасные
результаты, если подготовка к работе и монтаж проведены
достаточно аккуратно.
При ошибках, допущенных в монтаже, схема работает
89.
ттлохо или вообще не работает. Наиболее часто встречаются
следующие неполадки:
а. Стрелка гальванометра не отклоняется при замыкании
ключа; причины (в порядке проверки): арретирован
гальванометр, неверно подключен гальванометр к переключателю,
нет контакта в клеммах или имеется разрыв в
соединительных проводах малой цепи.
б. Стрелка гальванометра при всех положениях
подвижного контакта отклоняется в, одну сторону; причины: не
включен в цепь аккумулятор (или провода большой цепи
разорваны), неверно подключены полюса измеряемого
элемента, £дкк равно или меньше э. д. с. измеряемой цепи.
в. Стрелка гальванометра отклоняется очень слабо,
а вблизи точки компенсации вовсе не движется в пределах
5—10 делений; причины: большое сопротивление (нарушены
контакты) в измеряемой цепи (прежде всего в
электролитическом ключе), отросток электрода сравнения перекрыт
краном.
г. Повторные измерения дают все повышающиеся
отсчеты — разряжен аккумулятор.
ПРИГОТОВЛЕНИЕ ВОДОРОДНОГО И КАЛОМЕЛЬНОГО
ЭЛЕКТРОДОВ И ИЗМЕРЕНИЕ ПОТЕНЦИАЛА КАЛОМЕЛЬНОГО
ЭЛЕКТРОДА
Вспомогательные электроды сравнения необходимы при
всех потенциометрических работах, и к ним предъявляют
весьма высокие требования. Потенциал электрода сравнения
1,1 должен быть постоянным во времени и точно известен,
поскольку все расчеты ведутся по разности между э. д. с.
гальванического элемента и потенциалом электрода сравнения
Кроме того, электроды сравнения должны быть просты в
изготовлении, а потенциалы—-хорошо воспроизводимы.
Каломельные электроды отвечают всем этим требованиям и
получили широкое распространение в почвенных лабораториях
В качестве стандарта и нулевой точки шкалы потенциалов
принят нормальный водородный электрод. Водородный
электрод иногда применяется и для определения рН в почвах.
Водородный электрод. Возникновение скачка потенциала
в водородном электроде обусловлено реакцией
Н2^2Н+ + 2е.
Водород хорошо растворим в платине, как и в некоторых
других металлах (никеле, палладии). В связи с этим и бла-
90
годаря высокой химической инертности платина
употребляется для изготовления водородных электродов. Водородный,
электрод состоит из платиновой проволоки, покрытой
платиновой чернью, погруженной в раствор, насыщенный
газообразным водородом при атмосферном давлении (рис. 22).
ad к
Рис 22. Электроды сравнения:
а — колоколообразный водородный электрод, б — грушевидный
водородный электрод; в — каломельный электрод;
/ — платиновые проволочки, 2—раствор КО, 3—слой кристаллов
КО, 4 — слой каломели, 5 — ртуть
Адсорбция водорода на платине, растворение и установление-
равновесия между молекулярным и ионным водородом
облегчаются благодаря рыхлому, губчатому покрытию на
поверхности электрода. Процесс образования такого покрытия-
обычно называют платинированием. Первой стадией при
работе водородного электрода является процесс адсорбции:
H2pa3 + Pt-*PtlH2aTO.
Затем происходит реакция
Pt|H4|e*±Pt + 2e + 2H+,
которая активизируется благодаря большой удельной
поверхности губчатой платины. Суммарную реакцию можно
записать так:
Н2гя,^ 2Н+ + 2е.
9L
Ионы водорода в губчатой платине находятся в
равновесии с ионами водорода раствора; всякое изменение
активности водородных ионов в растворе сдвигает равновесие
электродной реакции. Из этого следует, что потенциал
водородного электрода (как и любого
окислительно-восстановительного электрода) определяется величиной отношения
активных концентраций окисленных и восстановленных
форм, т. е.
„ _ „ Л_ *? In (а»+J '
Концентрация молекулярного водорода пропорциональна его
парциальному давлению, тогда
. , RT, (ан+>2
0 ' nF Ph. '
где рн — парциальное давление водорода, а п = 2, так как
в реакции участвуют два электрона на каждый моль водорода.
Вводя численные значения и производя преобразования,
получим
0,0581 , г ан+ J , , „„,„,. ан+
е
lg(l^} =^+°да.1в-
Для электрода, в котором активность водородных ионов
в растворе и давление водорода равны единице, е = е0; такой
электрод называют нор м ал ьны м водородным
электродом, а величину его потенциала условно принимают
равной нулю. При любых температурах величины
потенциалов различных электродов даются по отношению к
нормальному водородному электроду. Следовательно, величина
любого электродного потенциала равна э.д. с. элемента,
состоящего из данного электрода и нормального водородного
электрода.
Обычно водородный электрод насыщают газообразным
водородом при атмосферном давлении и считают, что
давление водорода постоянно и равно 1 атм. Давление воздуха
не всегда равно 1 атм, газовая фаза электрода содержит
пары воды, поэтому парциальное давление водорода
несколько ниже. При особо точных измерениях расчеты ведут по
приведенной выше формуле, внося соответствующие
поправки, однако они очень невелики. Так, даже при
барометрическом давлении 730 мм рт. ст. и давлении водяных паров
32 мм ошибка вычисления едва достигает 1 мв, что не имеет
существенного значения в обычной аналитической практике.
Если считать, что
'92
, , 0,0581 , Г °Н+ I2 ' , n nRoi 1 ан+
я Ph., -- const = 1,
то уравнение можно упростить, записав
е =г. ер + 0,0581 lg а^ = г'0 — 0,0581 рН,
«о так как принято, что е^ = 0,
то е = —0,0581 рН.
Последнее уравнение используют в расчетах при
определении рН почвенных вытяжек и при использовании
водородного электрода в качестве стандартного полуэлемента.
Для приготовления водородного электрода берут гладкую
платину (в виде пластинки или проволоки, впаянной в
стеклянную трубочку) и тщательно очищают. Очищенную
поверхность промывают горячим раствором хромовой смеси и
дистиллированной водой; рекомендуют выдержать электрод
в дистиллированной воде не менее 12 ч.
Затем приступают к покрытию электрода губчатым слоем
платиновой черни (платинирование). Для этого готовят
раствор, содержащий 2—3 г PtCU и 0,02 г (СН3СООJРЬ в 100 мл
раствора; уксуснокислый свинец добавляют, чтобы получить
более прочное и равномерное покрытие. В этот раствор
опускают электрод и присоединяют его через реостат к
отрицательному полюсу аккумулятора на 2—4 в. Анодом служит
платиновая пластинка или проволока. Платинирование
проводят в течение 4—6 мин (в зависимости от напряжения
аккумулятора) до тех пор, пока электрод не покроется ровным
бархатисто-черным слоем губчатой платины. После
платинирования электрод промывают дистиллированной водой и
снова подвергают катодной поляризации A5—20 мин)
в 0,05 н. растворе H2SO4 для удаления с поверхности
адсорбированных ионов хлора. Готовый электрод промывают
дистиллированной водой (рекомендуется оставить в воде на
сутки) и хранят погруженным в воду.
Работа с водородным электродом при использовании его
в качестве электрода сравнения сводится к следующему.
Электродный сосуд наполняют буферным раствором с
известным рН и затем насыщают водородом из аппарата Киппа
в течение 20—30 мин Для этого пропускают водород с такой
скоростью, чтобы можно было считать пузырьки B—3 в
секунду). Ло окончании насыщения соединяют водородный
93
электрод с каломельным при помощи электролитического
ключа и измеряют е. д. с. цепи (см. стр. 89). Повторяют
насыщение водородом и измерения до тех пор, пока не
установится постоянное значение потенциала. По полученным
данным вычисляют потенциал каломельного электрода.
Измерения рН проводят аналогично с той лишь разницей,
что электродный сосуд заполняют исследуемым раствором,
рН которого требуется узнать, а в качестве электрода
сравнения берут каломельный полуэлемент с заранее
установленным потенциалом.
Водород для насыщения электрода получают из аппарата
Киппа путем растворения металлического цинка в 25%-ной
H2SO4. Полученный водород очищают, пропуская его через
промывные склянки, наполненные растворами в следующем
порядке: 5%-ный KMnCU в 5% -ной H2S04; 10%-ный
РЬ(СНзСОО)г; смесь 10%-ного КОН и 10%-ного раствора
пирогаллола; вода.
Для прямого определения рН раствора при почвенных
исследованиях водородный электрод редко применяют потому,
что он легко отравляется в присутствии многих веществ.
Особенно сильно влияют соли хромовой и азотной кислот,
соединения закиси железа, сульфиды, сулема, аммиак, амины,
поверхностно-активные вещества. Измерениям в почвенных
суспензиях препятствует еще и легкость загрязнения
платинированной поверхности адсорбирующимися гумусовыми
веществами и минеральными коллоидами, почти всегда
присутствующими в почвенных вытяжках и суспензиях.
Каломельные электроды сравнения. Скачок потенциала
в каломельном электроде обусловлен равновесием:
Hg|++2e^2Hg0,
которое устанавливается между металлической ртутью и
насыщенным раствором каломели HgaCb.
Для приготовления каломельного электрода используют
обычно сосуд Михаэлиса (рис. 22). На дно сосуда наливают
немного ртути, в которую погружают предварительно
промытую и амальгамированную платиновую проволоку, служащую
контактом.
Для амальгамирования платиновую проволоку, впаянную
в стеклянную трубочку (она одновременно служит пробкой
сосуда Михаэлиса), промывают в конц. H2SO4, воде и
прокаливают на пламени спиртовки. Промытую проволоку
опускают в раствор Hg2(N03b, подкисленный HN03, и соеди-
94
мяют с отрицательным полюсом аккумулятора. Анодом
служит платиновая пластинка. После прохождения тока в
течение I—2 мин проволока покрывается серым налетом ртути.
Амальгамированную проволоку промывают
дистиллированной водой и осторожно высушивают фильтровальной
бумагой. В сосуд Михаэлиса наливают немного ртути, в которую
■погружают амальгамированную проволоку. Стеклянную
пробку с контактом плотно вставляют в сосуд на шлифе и при
всех последующих операциях не вынимают. Количество ртути
должно быть достаточным, чтобы покрыть всю поверхность
платиновой проволоки и часть стеклянной трубки.
Недостаток ртути может привести к образованию поверхности
раздела между суспензией каломели и платиной, что нарушит
.нормальную работу электрода.
На поверхность ртути через длинный отросток сосуда
насасывается суспензия каломели в растворе КС1. Если
готовится насыщенный каломельный электрод, то несколько
граммов каломели отмывают насыщенным раствором КС1 (путем
растирания и декантации) и затем густую суспензию (пасту)
насасывают в сосудик. Слой пасты на поверхности рт}тн
должен быть в 1—2 мм. Затем насасывают горячий (при 50°)
насыщенный раствор КО. По охлаждении сосуда из раствора
выделяются кристаллы КС1; достаточно, если образуется слой
кристаллов KCI толщиной в 2—3 мм. На поверхности ртути
готового электрода расположены последовательно слой
каломели и слой кристаллов КО; над осадком находится
насыщенный раствор КО.
Нормальный и децинормальный каломельные электроды
готовят так же, но используя раствор КО соответствующей
концентрации. Естественно, что в этом случае нагревать
раствор КО перед засасыванием не требуется, а в готовых
электродах слой кристаллов КО не образуется. Каломельный
электрод необходимо приготовить не менее, чем за 24 ч до
иачала работы.
Каломельные электроды с различной концентрацией
хлористого калия отличаются по величинам потенциалов и
отношению к условиям окружающей среды. Скачок потенциала
в электроде равен
Активность ионов ртути в каломельном электроде всецело
зависит от активности ионов хлора (или КО) в силу ничтож-
95
ной растворимости каломели. Произведение растворимости
каломели очень мало:
и активность ионов закиси ртути в растворе может быть
вычислена по уравнению
v2+=-
2-10-
,-18
^СГ
Подставляя это значение в приведенное выше уравнение,
получим
е = е0 + --- in 2-1"~'8 = е0 + -^ In 2- КМ8 —
"СГ
2F
RT , р . RT .
In <к.._ — еп In aci—-
2f ci op w
При 20°
е=е; — 0,0581 Igacl_.
В результате оказывается, что потенциал каломельного
электрода обратим к ионам хлора. Такие электроды получили
название электродов второго рода.
Растворимость КС] довольно быстро возрастает с
температурой (примерно на 1,5—1,7 г на 100 г воды при
увеличении температуры на 10е). Этим объясняется, что потенциал
насыщенного каломельного электрода заметно уменьшается
с ростом температуры (табл. 10). В то же время потенциалы
нормального и особенно децинормального каломельного
электрода в диапазоне 10—30° от температуры практически не
зависят. Преимущество насыщенного каломельного электрода
проявляется в том, что концентрация КО в насыщенном
растворе с течением времени не меняется и электродом можно
очень долго пользоваться даже в полевых условиях. В
ненасыщенных электродах при длительном хранении всегда
возможно заметное увеличение концентрации вследствие
испарения воды, и поэтому потенциалы таких электродов менее
устойчивы.
Приготовленный каломельный электрод выдерживают в
течение суток, после чего измеряют его потенциал. Для
измерения собирают цепь, как показано на рис. 21, соединяют
каломельный электрод с водородным электролитическим
ключом и измеряют э.д. с. В качестве наполнителя для
электролитического ключа употребляют насыщенный раствор КО,
96
Таблица 10
Потенциалы каломельных электродов в вольтах
Температура,
1С
15
16
17
18
19
20
21
22
23
24
25
насыщенный
0,2503
0,2497
0,2490
0,2483
0,2477
0,2471
0,2464
0,2458
0.2451
0,2445
0,2438
Концентрация KCI
1,0 н.
0,2852
0,2850
0,2847
0,2845
0,2842
0,2840
0,2838
0,2835
0,2833
0,2830
0,2828
0,1 н.
0,3371
0,3370
0,3370
0,3369
0,3369
0,3368
0,3367
0,3367
0,3366
0,3366
0,3365
который предохраняют от выливания пробочками из
фильтровальной бумаги. Можно наполнять электролитический ключ
агар-агаровым или' желатиновым гелем, содержащим КО
или NH4N03; для этого набухший в воде агар-агар
(желатин) нагревают на кипящей водяной бане и к
образовавшемуся гелю добавляют такое количество КО, чтобы получился
40%-ный раствор соли C г агара, 100 мл воды, 40 г КО).
Вычисление потенциала каломельного
электрода
Э. д. с. элемента равна разности потенциалов между
водородным и каломельным электродами. Насыщенный
каломельный электрод имеет положительный знак по отношению к
водородному, тогда
Э. Д. С. = 8кал - ^водородн»
ИЛИ
^кад == Э- Д- С. -+- Бводородн-
Но ПОСКОЛЬКУ Еводороди = —0,0581 рН, ТО ЕКал = э-Д-с- —
-0,0581 рН (при 20°).
Возможные неисправности и их устранение.
Кроме общих неисправностей цепи, указанных в разделе
по измерению э. д. с, можно обнаружить, что при повторных
насыщениях водородного электрода газообразным водородом
результаты измерений плохо воспроизводимы. Причиной мо-
4 Зак. 51 97
жет быть загрязнение водорода и плохая очистка его
промывными жидкостями. Для улучшения результатов необходимо
сменить реактивы в промывных склянках. Иногда плохая
воспроизводимость наблюдается при использовании
свежезаряженного аппарата Киппа; это обусловлено тем, что
отбираемый от свежезаряженного аппарата газ содержит
значительное количество кислорода. В этом случае надо дать
поработать аппарату Киппа некоторое время вхолостую.
Плохая воспроизводимость или грубые расхождения
найденного потенциала каломельного электрода с табличными
значениями могут быть и по причине низкого качества ртути,
каломели, хлористого калия, а также неаккуратного
обращения с каломельным электродом, в частности при
перемешивании слоя ртути с суспензией каломели (каломельный
электрод нельзя сильно наклонять, встряхивать). В этом случае
необходимо заново приготовить каломельный электрод.
ПОТЕНЦИОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ АКТИВНОСТИ
ВОДОРОДНЫХ ИОНОВ В ПОЧВЕННЫХ РАСТВОРАХ, ВЫТЯЖКАХ
И СУСПЕНЗИЯХ
Ни одно почвенное исследование не обходится без
определения рН. Реакция почвенной среды является важнейшим
генетическим показателем, она используется в качестве
одного из классификационных признаков и характеризует
пригодность почв для возделывания различных
сельскохозяйственных культур.
Чувствительность растений к величине рН неодинакова и
зависит не только от вида растений, но и от состава почвы.
Вредное влияние повышенной кислотности или щелочности
обусловлено как прямыми, так и косвенными причинами; при
повышении кислотности изменяется химический состав
почвенного раствора, увеличивается содержание подвижного
алюминия, марганца, препятствующих нормальному ходу
обмена веществ.
Необходимость регулирования рН при окультуривании
почв послужила одной из причин, благодаря которым
определение рН — один из наиболее распространенных видов
почвенного анализа.
На территории СССР преобладают почвы
дерново-подзолистой зоны, отличающиеся в своем большинстве
повышенной кислотностью. Поэтому изучение природы почвенной
кислотности и разработка мер по борьбе с ней — одна из
важнейших задач химии почв.
98
Различают несколько видов почвенной кислотности,
которые количественно характеризуют или путем определения
общего количества свободных кислот, титруемых щелочью,
или путем определения реакции среды, выражаемой в
единицах рН. Окончательного решения вопроса о природе
почвенной кислотности нет.
Принято характеризовать: 1) реакцию почвенной среды
по величине рН водной вытяжки; 2) реакцию солевой
вытяжки из почв по величине рН вытяжки 1,0 н. раствором КО;
3) обменную кислотность по количеству щелочи,
затрачиваемой на титрование солевой A,0 н. КО) вытяжки; 4)
гидролитическую кислотность по количеству щелочи, затраченной
на титрование вытяжки, приготовленной на нейтральном рас*
творе гидролитически щелочной соли.
Активная реакция водной вытяжки зависит от присутствия
в почве легко растворимых кислот, солей и от содержания
углекислого газа. Активную реакцию определяют для всех
типов почв, причем на величину рН в солончаках влияют
преимущественно легкорастворимые соли, а в
солониах-—поглощенный натрий или карбонаты и бикарбонаты натрия; в
карбонатных почвах и там, где поглощающий комплекс насыщен
кальцием, реакция почвы определяется буферной системой
С02+Са(НС03J.
В водные вытяжки могут переходить такие простейшие
кислоты, как муравьиная, уксусная, щавелевая, молочная,
яблочная, винная и др. Вместе с тем растворяются и
некоторые специфические органические вещества почвы — фульво-
кислоты. В почвах, не насыщенных основаниями, эти
кислоты и углекислота преимущественно определяют величину рН
водной вытяжки. Прямой связи между общим содержанием
кислот и реакцией водных вытяжек установить не удается,
поскольку рН является функцией многих переменных и в том
числе ионной силы и степени диссоциации всех компонентов
раствора.
Реакция почв, содержащих карбонаты, в значительной
мере снижается в присутствии С02; под влиянием
углекислого газа карбонаты переходят в бикарбонаты, растворимость
которых выше, и рН снижается. Для равновесной системы
СаСОз—С02 влияние углекислоты на реакцию среды
выражается, по Е. Бэру, формулой
pH = ptf1-0,5/r+IgJ-^,
l^^l
где Д]=3' \0-7 — первая константа диссоциации угольной
кислоты, а Г — ионная сила раствора.
Легкорастворимые соли солончаков и засоленных почв
значительно меняют активную реакцию почвенного раствора.
Соли сильных оснований и слабых кислот обусловливают
щелочную реакцию, что имеет место в содовых солончаках. Хло-
ридные солончаки, особенно с высоким содержанием
кальциевых и магниевых солей, имеют кислую реакцию.
Щелочная реакция свойственна водным вытяжкам из
некоторых горизонтов солонцов, что может быть объяснено
гидролитическим отщеплением поглощенного натрия. Так,
если в почве присутствуют гуматы натрия, то при
взаимодействии почвы с водой проходит реакция
№Гум + Н20 -* NaOH -f НГум,
образуется гумпновая кислота; щелочной раствор быстро
поглощает углекислоту:
2NaOH + С02 -^ Na2COa + Н20.
В результате величина рН зависит как ог константы
гидролиза гумата натрии, так и от равновесия в системе
гидроокись— карбонат натрия. Аналогично гидролизуется
минеральный поглощающий комплекс, насыщенный ионами
натрия.
Активную реакцию солевой вытяжки, обменную
кислотность и гидролитическую кислотность определяют только для
почв, ненасыщенных основаниями. Дискуссия о природе
обменной кислотности продолжается уже на протяжении
полстолетия. Так как в солевой вытяжке обнаруживаются
одновременно ионы водорода и ионы алюминия, то обменную
кислотность можно объяснить при помощи двух реакций:
1. пн+ + ш = пк+ + на,
где П — почвенный поглощающий комплекс;
2. ПА13+ + ЗКС1 = ПКз+ + А1С13;
хлористый алюминий гидролизуется:
А1С1, + ЗН20 = А1 (ОНK + 3HC1
и подкисляет раствор.
Или, при титровании:
А1С13 + 3NaOH -у Al (OHK1 + 3NaCl.
Та и другая гипотеза позволяет удовлетворительно
объяснить большинство явлений, наблюдаемых в связи,.с почвен-
«00
ной кислотностью. Для образцов гумусированных почв было
экспериментально показано: кислотность солевых вытяжек
выше, чем это может быть вызвано реакцией гидролиза; это
прямо указывает на присутствие обменных ионов водорода.
Малогумусные горизонты отличаются, как правило,
эквивалентностью в содержании алюминия и титруемой
кислотности. В. А. Черному удалось вычислить величины рН,
пользуясь формулой гидролиза алюминиевых солей:
рИ = 2,635 — 0.523 lgCAi
при концентрациях ионов А13+ 10~2—10~~4 М. Проверка на
•большом числе почвенных образцов показала, что для
минеральных горизонтов величины рН, вычисленные по
содержанию алюминия и измеренные потенциометрическим методом,
удовлетворительно совпадают.
Гидролитическая кислотность также служила предметом
длительной дискуссии, не оконченной и в настоящее время.
Сущность явления заключается в значительном увеличении
поглощения оснований при обработке почвы растворами
гидролитически щелочных солей; аналогичное явление
наблюдается и при обработке почвы нейтральными солями, но при
повышенных значениях рН. Характер происходящей реакции
■был объяснен Д. Н. Прянишниковым, взгляды которого
развивал в дальнейшем Н. П. Ремезов. Прянишников
рассматривал почвенный поглощающий комплекс как многоосновный »
кислотный остаток (ацидоид), содержащий водородные ионы,
способные к диссоциации и обмену при различных
значениях рН. Величина рН, при которой водородный ион может
замещаться металлическим катионом, зависит от прочности
связи (константы диссоциации; водородного иона с
почвенным поглощающим комплексом. Чем выше прочность связи,
тем больше энергии затрачивается на обменную реакцию.
Очевидно, что при щелочной реакции среды водородные ионы,
связываются с образованием молекул воды и реакция
сдвинута в сторону наиболее полного вытеснении Н^-ионов. То же
происходит и при взаимодействии почвы с раствором
гидролитически щелочной соли. Анионный остаток слабой кислоты
прочно связывает вытесненные водородные ионы, что
способствует увеличению полноты замещения.
Аналогичное увеличение кислотности можно ожидать и
при вытеснении ионов алюминия, так как при более высоких
значениях рН, или при введении солей слабых кислот,
степень гидролиза образующихся алюминиевых солей
увеличивается, а следовательно, растет и полнота вытеснения.
101
Наглядная классификация видов почвенной кислотности
дана Н. П. Карпинским, который вводит понятия
количества кислотности (фактор емкости) и
интенсивности кислотности (фактор интенсивности).
Количество кислотности охватывает общее содержание компонентов,
способных реагировать но кислотному типу. Фактор
интенсивности указывает, в какой мере эти компоненты проявляют
себя в действии в конкретных условиях среды или
эксперимента.
Таблица 11
Виды кислотности (no H. П. Карпинскому)
Количество кислотности (потенциальная кислотность)
Интенсивность кислотности
Л Кислотность твердой фазы почвы
1. Обменная кислотность А. Степень кислотности
а) поглощенный алюминии, почв
б) собственно обменная кислотность:
поглощенные ионы водорода
2. Гидролитическая кислотность Б. Степень подвижности
поглощенного
алюминия
П. Кислотность почвенного раствора (и вытяжек)
Титруемая кислотность Активная кислотность
(рН)
Диапазон величин рН, наблюдаемый для почв, весьма
широк; большинство почв, важнейших в сельскохозяйственном
отношении, характеризуется умеренно кислой, нейтральной
или слабощелочной реакцией (рН от 4,5—5,0 до 7,5); однако
приходится встречаться как со значительно более
кислыми (рН = 3,0—3,2), так и с сильнощелочными почвами
(рН = 9,3—9,8).
ИЗМЕРЕНИЯ АКТИВНОСТИ ВОДОРОДНЫХ ИОНОВ
ХИНГИДРОННЫМ ЭЛЕКТРОДОМ В ВОДНЫХ И СОЛЕВЫХ
ВЫТЯЖКАХ ИЗ ПОЧВ
Благодаря простоте устройства и надежности в работе
большинство измерений рН в почвах проводятся с помощью
хингидронного электрода.
102
Потенциал хингидронного электрода зависит от
соотношения активных концентраций окислителя (хинона) и
восстановителя (аниона гидрохинона) в растворе, т. е. является
потенциалом окислительно-восстановительной системы (см.
стр. 83). Для изготовления электрода погружают
платиновую пластинку или проволоку в насыщенный раствор хин-
гидрона; диссоциация последнего и создает необходимую
•окислительно-восстановительную систему, потенциал которой
воспринимается платиновым электродом.
Хингидрон — труднорастворимое эквимолекулярное
соединение хинона и гидрохинона; при растворении хингидрона
в воде происходит реакция
с12н8о2 (онJ «± с6н4а. + с(;н4 (ОНJ.
Хингидрон Хинон Гидрохинон
"Гидрохинон диссоциирует, образуя анион гидрохинона:
ед (ОИJ г± сен4о1~ + 2И+,
или
НО-/ У~ОН ?± -О-^^^-—О- + 2И+.
Анион гидрохинона — это в то же время восстановленная
■форма хинона:
С6Н4о!~ ^ С6Н4Оа + 2е;
s результате в системе устанавливается
окислительно-восстановительное равновесие, а платиновый электрод приобретает
яютенциал, величина которого определяется формулой
е - е0 + -^ Ш J°± = £о + Ж In-I™™,
nF [Red] ' nF [CeH40|-]
если считать, что коэффициенты активности окислителя и
восстановителя равны единице.
При увеличении концентрации водородных ионов
диссоциация хингидрона подавляется в соответствии с законом
действующих масс, а количество анионов гидрохинона (или
восстановленной формы хинона) уменьшается, что влияет на
величину электродного потенциала. Если известны
концентрация гидрохинона и константы его диссоциации, то можно
вычислить концентрацию анионов гидрохинона при любом
значении рН. Это позволяет установить интересующую нас
103
зависимость между потенциалом электрода и величиной рН.
Для удобства вычислений введем обозначения:
Н2А— недиссоциироваиный гидрохинон, НО — <? ^> — ОН,
НА-—однозаряженныи анион гидрохинона, НО—/ у—О-,
А2"-—двухзаряженный анион гидрохинона, ~0—<^" у—0~,
Cs —общая концентрация гидрохинона, в растворе,
Съ = [Н2А] + [НА-] + [А*-].
К\ — первая константа диссоциации гидрохинона,
Кг — вторая константа диссоциации гидрохинона,
[Н2А], [НА-] и [А ] — концентрации соответствующих компонентов
в равновесном растворе.
Первая и вторая константы диссоциации гидрохинона
соответственно равны:
Ai — И Да = " •
[Н2А] [НА"]
Тогда
(НА-1 = В*Ш. и [#-} = [НАП/С.
[Н2А] КгКг
ан+ °н+ «н+
В то же время, по условию
СЕ = [Н2А1 + [НА-] + [AM-
Производя замену, можно записать:
С. = [Н2А] + [Нг-А]-^ + -^^
°н+ °н+
jh+ "н+
что позволяет вычислить концентрацию недиссоциированного
гидрохинона в зависимости от активности водородных ионов
в растворе
[И2А] = С, : fl + ~^~ +«£l)=Ci :
Ч °н+ йн+ /
а^+ + К,ан+ + KiK,
н+ °н+ J ан+
104
сн+
^4-+/CIflH+ + fi/fs
/Ci/C2
°н+
Но, как было определено выше,
[Д2-] = JH^LMl..
Подставив в это уравнение величину Н2А, вычисленную
как функцию активной концентрации водородных ионов,
получим
[Л*-] = С,
В этом уравнении [A2-]=[C6H4Oi2-] — концентрация
анионов гидрохинона или восстановленной формы хинона,
входящей в уравнение потенциала хингидронного электрода,
которое теперь можно записать так:
nF С, KiK2
Величина Сх выражает сумму концентраций
диссоциированного и недиссоциированного гидрохинона. Поскольку при
распаде хингидрона образуется одна молекула хинона и одна
молекула гидрохинона, впоследствии диссоциирующая, то
можно принять, что
С, -- [С6Н402],
а отношение
[С6Н402] : С. == I,
и упростить уравнение:
, RT , ан+ + ^ан+ + #1^2
е -- е0 + -1— In •
nF I^Ki
Но и в такой форме уравнение еще слишком сложно и
неудобно для практического применения. В целях дальнейшего
преобразования перепишем, объединив постоянную величину
RT
In /Cj/Ca с константой уравнения:
пГ
в "-= е0 + ~ In (cfa. + KtaH+ + КМ - ~ In КуКг =
nF " " nF
= e' + -ff In (dfa + K,aH+ + KxKJ.
105
Последнее слагаемое (А1А2) в сумме, стоящей под знаком
логарифма, при всех реально возможных условиях
значительно меньше, чем первые. Константы диссоциации
гидрохинона равны
Kt = 1-Ю-10 и tf2 = 3-Ю-12,
т. е. произведение A'iA'2=3- Ю-22.
Этой величиной можно пренебречь, не внося сколь-ни-
будь заметных ошибок. Тогда
RT
6 = Е'° + ~^F 1П (G"+ + ^lGH+)-
При невысоких значениях рН можно пренебречь и вторым
слагаемым, если удовлетворяется неравенство
G2J+>/C1aH+ или aH+~pKi,
т. е. при условии, что ан+>Ь1040.
Такому условию в практических целях вполне
удовлетворяет величина ан±=10~&, т. е. когда активная концентрация
водородных ионов по крайней мере в 100 раз превышает по
величине первую константу диссоциации.
Пренебрегая величиной Kiau+ и учитывая, что число
участвующих в реакции электронов равно двум, получил!
. , 'RT , „ , RT .
е ^ е°+ 17 н+''" 8° "" ~Т °н+ ^
~е0- 2,303 -^-рН.
р
RT
При температуре 20° произведение 2,303 —0,0581, что
позволяет переписать уравнение потенциала хингидроиного
электрода в следующем окончательном виде (для
температуры 20°):
е = 6; — 0,0581 рН.
На рис. 23 показана зависимость потенциала
хингидроиного электрода от величины рН, вычисленная по этому
уравнению и по полному уравнению, учитывающему величины
констант диссоциации гидрохинона В пределах до рН=9 обе
зависимости выражаются практически одной прямой линией.
106
Величина г'0 — потенциал нормального электрода,
равная потенциалу такого хингидрониого электрода,
активность водородных ионов в котором равна единице. Если
«11+=1, рН = 0 и 8 = 8о'. Величина ео определяется путем гра-
S, мб
-Ь. 1 1 I I I I I I I ♦ у I I
/ 2 3 4- S 6 7 8 9 10 II /Z\!3 /V рц
\
Рис 23. Потенциал хингидрониого электрода, рассчитанный по
формулам
/—е=е0— 0,0581 рН; // —е=е0+0,029 lg(o^+ +KiOH+ +
+/&*,), где /С, = 1 • Ю-10 и К2=3- 10-»
фической экстраполяции ряда измерений электродного
потенциала до значения рН=0.
Рассматриваемая зависимость выражается уравнением
прямой линии (t/ = a + Kx). Если на графике по оси абсцисс
отложены значения рН, а по оси ординат — величины в, то
отрезок, отсекаемый на оси ординат прямой линией, равен
107
ютенциалу нормального хингидронного электрода. Наклон
гоямой определяется величиной коэффициента
2,303-^ = —tg а.
F ъ
При аналитических определениях график зависимости г —
>Н обычно не строят, однако при подготовке прибора и про-
зерке электродов он весьма полезен. Путем построения гра-
гжка можно быстро и надежно проверить качество собранной
•хемы. Надежность электрода считается достаточной, если
гостроенный график удовлетворяет следующим признакам:
1. Зависимость между в и величинами рН буферных
растворов прямолинейна в пределах рН от 2 до 8.
2. Тангенс угла наклона прямой (Ле/ЛрН) равен
1,0001983 Т или 0,058 при 20°.
3. Величина ео', найденная по графику, отличается от
табличного значения не более чем на ±2 мв; если же
определенный таким способом потенциал нормального электрода
■-начительно отличается от теоретического, то причиной мо-
кет быть неправильно установленный потенциал электрода
•равнения (или для хингидрон-хингидронного элемента
незерно определенная величина рП буферного раствора в элек-
~ооде сравнения). Все последующие аналитические измере-
-шя, проводимые с тем же элементом, также включают
постоянную систематическую ошибку, равную разности между
теоретическим и экспериментальным значением е0'.
Величина потенциала нормального хингидронного элек-
-оода при различных температурах вычисляется с помощью
:мпирической формулы
е0 ■-=-- + 0,6990 — 7,4 • 10-" (t — 25),
-по / — температура (см. табл. 12). 1
Таблица 12
Потенциал нормального хингидронного электрода
при различных температурах
?
5
6
7
8
9
Ео
0,7064
0,7057
0,7049
0,7042
0,7034
<с
20
21
22
23
24
Ео
0,7027
0,7020
0,7012
0,7004
0,6997
t°
25
26
27
28
29
to
0,6990
0,6983
0,6975
0,6968
0,6960
При выводе уравнения потенциала хингидронного
электрода были сделаны некоторые допущения и показано, что до
значения рН = 8 упрощенное уравнение дает те же
результаты, что и полное. При дальнейшем возрастании рН оба
уравнения уже дают различные результаты, а зависимость
между потенциалом и величиной рН отличается в
действительности от прямолинейной (см. рис. 23). Это и является
одной из причин, которая не позволяет использовать хингид-
ронный электрод в средах с рН>8. Другая причина,
ограничивающая применение электрода указанным интервалом,
обусловлена заметным окислением гидрохинона в щелочной
среде кислородом воздуха.
Составление цепей с хингидронным электродом
и вычисление рН
Растворимость хингидрона в воде очень мала—1,1 г/л,
поэтому для насыщения раствора в хингидронном электроде
достаточно к 10 мл добавить 15—20 мг хингидрона (на
кончике ножа). Гладкий платиновый электрод воспринимает
возникающий в системе потенциал, величина которого
измеряется на компенсационной схеме.
Электродом сравнения служит каломельный электрод или
второй хингидронный электрод с буферным раствором,
имеющим точно определенное значение рН.
Схема цепи хингидрон-хингидронного элемента
выражается формулой
Pt
раствор -f-
хингидрон
КС1
насыщ.
раствор -f-
хингидрон
РН,
Pt
и, если электродом сравнения служит каломельный полуэлемент:
раствор +
хингидрон
Pt—Hg
Hg
Hg2Cl2
KCl
kci
иасыщ.
РН,
Pt
При составлении концентрационного элемента (хингидрон—
хингидрон) наиболее часто в качестве буферного раствора
ислользуют раствор Вайбеля с рН=2,04, для приготовления
которого смешивают 9 частей точно 0,1 н. раствора КС1 и
1 часть 0,1 н. раствора НС1. Величину рН буферного раствора
рекомендуется дополнительно проверить водородным элек-
109
тродом. В практике почвенного анализа хингидронный
электрод, приготовленный на растворе Вайбеля, всегда имеет
положительный знак по сравнению с электродом, погруженным
в почвенную вытяжку или суспензию. Это обусловлено тем,
что при анализе почв в вытяжках практически никогда не
встречаются величины рН ниже 2,5. Даже если почвенный
раствор имеет рН равный 2,5, то и в этом случае электрод
сравнения уже значительно более положителен (примерно
на 28 мв). Поэтому составляя цепь для измерения рН почв
хингидрон-хингидронным элементом, электрод сравнения
всегда соединяют с положительным полюсом аккумулятора.
Использование каломельного электрода сравнения требует
предварительного определения полюсности. Потенциал
насыщенного каломельного электрода при Г8° равен 0,248 в.
Такое же значение приобретает потенциал хингидронного
электрода в растворе с рН = 7,9 (из формулы: 0,248 = 0,704—
—0,0577 рН). Таким образом, при рН хингидронного электрода
ниже 7,9 каломельный электрод имеет более низкое значение
потенциала, чем хингидронный, и, следовательно,
отрицательный знак. При рН более 7,9 знаки в цепи меняются.
Последнее обстоятельство необходимо всегда учитывать и при
работе с каломельным электродом менять полюсность, если для
первоначальной собранной цепи не удается найти точку
компенсации. Одновременно это нужно отметить в рабочей
тетради.
Для 1,0 н. и 0,1 н. каломельных электродов перемена
знаков в цепи происходит соответственно при значениях рН,
равных 7,2 и 6,1. Насыщенный каломельный электрод позволяет
работать без переключения цепи в более широком
диапазоне рН, что значительно удобней. Если учесть, что
хингидронный электрод при рН = 8 дает уже неверные показания, то
можно окончательно рекомендовать насыщенный
каломельный электрод сравнения, принимая его отрицательным
электродом, а при отсутствии точки компенсации (рН>7,9)
переходить к определениям рН с помощью стеклянного
электрода.
Вычисление рН по результатам измерений
1. Хингидрон-хингидронный элемент.
Измеренная величина разности потенциалов равна
где ев — потенциал хингидронного электрода в растворе
Вайбеля и 8Х — потенциал хингидронного электрода в растворе
с неизвестным значением рН. Раскрывая формулу, получим
ПО
Е иэм = % — °.°581 Рнв — (её — 0,0581 рНх) =
= 0,0581 рНх — 0,0581 рНв.
(где рНв — величина рН в электроде сравнения) или
0,0581 рНх =-- Ент + 0,6581 рНв и рНх - -??£- + рНв,
если рНх>рНв.
При рНх<рНв изменяются полюса цепи в хингидрон-хин-
гидронном элементе, тогда
рНх = рНв-
0,0581
Если температура отличается от 20°, то вместо
коэффициента 0,0581 подставляют соответствующее табличное
значение. В этих расчетах величины потенциалов выражены
в вольтах.
При массовых определениях по схеме Поггендорфа в
целях экономии времени можно несколько изменить порядок
вычислений. Не рекомендуется отдельно вычислять
величину £ИЭм и Делить ее затем на 0,0581. Поскольку
£„зм = Ев ~- (см. стр. 88),
то
pHx--= ЕвАС> +рНв
х 0.058UC, '
Ев
тогда удобно сразу вычислить величину , обозначив
ее К, получим
рНх = КАСг + рНв.
Этой формулой, предварительно вычислив К, можно
пользоваться в течение целой серии измерений, если температура
и э. д. с. аккумулятора остаются постоянными. Естественно,
что при работе на потенциометрах, показания которых
даются непосредственно в вольтах, последняя формула
неприменима.
2. Хингидронный электрод в цепи с
насыщенным каломельным электродом
сравнения. В большинстве случаев каломельный электрод имеет
отрицательный знак в цепи (р!1<7,9).
Ш
Тогда
-'ИЗМ
ИЛИ
^изм — Ео 2,303 ——- рНх екал,
отсюда
RT
рнх =
2,303^рНх^е;-екал-£нзМ
е0 екал ^изм С|) ек<)Л £и
2,Ш-~- 2,-т~ 2,303 —
Если обозначить 2,303— = #,
то
рнх= Р"~
Объединив нормальный потенциал хингидронного
электрода и потенциал каломельного электрода в постоянную
уравнения, запишем
рНх- а— -^Ц где а
0 #
В щелочных растворах (рН > 7,9)
рНх = -_-. а л. _■
О
е0 екал
Величина а-- при разных температурах имеет сле-
дующие значения:
<°С 15 16 17 18 19 20 21
е0 еьал
7,99 7,96 7,93 7,90 7,87 7,83 7,81
t°C 22 23 24 25 26 27
g= e°""lE!^i- 7,78 7,74 7,72 7,70 7,67 7,65
&
112
При 18° и рН = 7,9 э. д. с. измеряемого элемента
становится равной нулю Последнее позволяет производить
настройку ламповых потенциометров (см. стр. 132) путем замыкания
накоротко входных клемм потенциометра.
Пределы применимости и ошибки при измерении рН
хингидронным электродом
Достоверность получаемых результатов зависит от
погрешностей двух видов. Прежде всего ето погрешности
измерения, зависящие от качества имеющейся аппаратуры;
погрешности второго рода возникают вследствие того, что
реальные, изучаемые, растворы оказывают влияние на
электрод и изменяют до некоторой степени характер зависимости
между величиной рН и потенциалом электрода, найденной
теоретически. Точность измерений ограничивается в первую
очередь ошибками приборов; эти ошибки легко поддаются
расчету. При работе с компенсационной схемой нужно знать
цену деления шкалы — падение напряжения на единицу
шкалы реохорда — и тогда можно рассчитать ошибку измерения.
В схеме (рис. 21) используется аккумулятор с э. д. с. = 2 в,
а число делений на линейке=1000. Следовательно, цена
одного деления равна 2 мв, и возможная ошибка измерения =
= ±2 мв. В соответствии с уравнением потенциала хингид-
ронного электрода изменение рН раствора на единицу
вызывает изменение потенциала на 58 мв (при 20°), тогда
58 же соответствуют 1
2 мв соответствуют х
2-1
х= — ~0,034,
58
т. е. погрешность определения — ±0,03 рН.
Точность измерений на различных промышленных
потенциометрах не превышает величины 0,05. Отсюда ясно, что при
обработке результатов измерений рН в почвах и почвенных
растворах можно принимать во внимание только такие
различия в величинах рН, которые превышают по крайней
мере 0,1.
Более высокой точности измерения можно достичь,
применяя аккумуляторы с меньшей э д. с. и используя линейки
с большим числом делений; однако это требует
одновременно и замены гальванометра на более чувствительный
(Ю~«—10-* а/мм).
113
Хингидронный электрод имеет ограниченную область
применения, и если не учитывать влияние, которое оказывают
почвенные растворы на электродную реакцию, то могут
возникнуть весьма значительные погрешности. Ошибки самого-
метода объясняются окислительно-восстановительным
характером скачка потенциала и свойствами хингидрона.
Присутствие окислителей или восстановителей смещает
равновесие в растворе хингидрона. Насколько сильно влияние
окислителей, можно судить уже по тому, что даже хлорное
железо легко окисляет гидрохинон до хинона. В щелочной
среде гидрохинон окисляется и кислородом воздуха.
Почвенные вытяжки и суспензии содержат ряд
окислительно-восстановительных систем, из которых наибольшее
значение имеет система Fe3+/Fe2+ с нормальным
окислительно-восстановительным потенциалом (ОВ-потенциалом) =
= +0,77 в. Обычно при низкой
окислительно-восстановительной буферное™ почв и почвенных суспензий потенциал хин-
гидронного электрода практически не смещается. Однако
в некоторых почвах содержание подвижных форм соединений
железа может быть достаточно велико, и тогда измеренные
величины потенциалов оказываются завышенными. Более
высокие значения потенциалов соответствуют более низким
значениям рН, т. е. результаты определения рН занижаются.
К таким почвам, где можно ожидать заниженных
результатов при определении рН хингидронным электродом, относятся
иллювиальные горизонты подзолов и дерново-подзолистых,
почв, некоторые пойменные почвы, красноземы и другие
кислые и богатые железом почвы и горизонты. Величина ошибки
зависит от состава образцов. Для исследования указанных
почв и горизонтов хингидронный электрод иногда все же
применяют, но если в вытяжки переходит очень много ионов
железа, или если результаты определений оказываются
неоправданно низкими, то следует провести проверку
измерений при помощи стеклянного электрода. Аналогичная
ошибка возникает и в солонцовых горизонтах, где она
усугубляется еще и щелочной реакцией. Поэтому для горизонтов В
солонцов и солонцеватых почв хингидронный электрод не
применяется.
В присутствии заметных количеств восстановителей в
почвах потенциал хингидронного электрода снижается, а
результаты определения рН оказываются завышенными. Это
наблюдается для некоторых почв, формирующихся в
условиях избыточного увлажнения. Ошибки измерений в сильно-
оглеенных почвах могут достигать нескольких единиц рН.
114
В щелочной среде гидрохинон легко окисляется
кислородом воздуха, а зависимость между потенциалом электрода
а величиной рН отклоняется от прямолинейной. Это не
позволяет использовать хингидронный электрод при рН>8.
Щелочная реакция характерна для содовых и боратных
солончаков и для некоторых горизонтов солонцов и
солонцеватых почв; по этой причине нельзя пользоваться хингидрон-
ным электродом для определения рН в перечисленных
почвах.
Если реакция почвенной вытяжки близка к нейтральной,
а буферность ее невелика, то в результате диссоциации
гидрохинона вытяжка подкисляется. Растворимость хингидрона
мала, но все же концентрация его насыщенного раствора
примерно 0,005 М. Если принять во внимание только первую
константу диссоциации, то оказывается, что хингидрон
подкисляет насыщенный им раствор до рН —6. Поэтому
применять хингидронный электрод в мало буферных растворах
'При рН более чем 5,5—6,0 не рекомендуется.
Кроме указанных отмечают для хингидронного электрода
еще солевую ошибку, возникающую при высоком содержании
солей в почвенном растворе. Однако ошибка невелика и для
1,0 н. растворов солги не превышает сотых долей единиц рН.
Такой ошибкой при региональных, агрохимических и
генетических исследованиях можно пренебречь, что позволяет
использовать хингидронный электрод для определения рН в
солончаках и в солевых вытяжках из почв, ненасыщенных
основаниями. Также невелика ошибка за счет адсорбции
гидрохинона почвенными частицами в разбавленных
•суспензиях.
Приготовление вытяжек и суспензий и измерение в них
концентрации водородных ионов
Величина активности водородных ионов, рН водных
вытяжек из почв характеризует кислотность
(щелочность)— реакцию почв; измеренные в водных вытяжках
значения рН, конечно, далеко не всегда идентичны
величинам рН почвенных растворов. Обменная кислотность
определяется в солевых вытяжках из почв.
При анализе водных вытяжек не безразлично, измеряется
ли рН истинного или коллоидного раствора (суспензии).
Коллоидные растворы ацидоидов и суспензии из кислых почв
амеют более низкие значения рН, чем фильтрат (центрифу-
115
гат), в силу так называемого суспензионного эффек-
т а. Поэтому наиболее целесообразным способом подготовки,
пробы к измерениям является центрифугирование почвенных
суспензий до полного осветления раствора. Фильтровать
суспензии не рекомендуют, так как при этом рН вытяжек
может измениться. Для фильтрования, если оно неизбежно,
нужно использовать фильтры, проверенные на чистоту или
предварительно промытые водой, высушенные и хранящиеся
в банках с притертой пробкой.
Водные и солевые вытяжки для измерения рН готовят
при соотношении почва: раствор = 1 : 2,5, как это принято по
международному соглашению. При анализе торфов, лесных
подстилок и т. п. это соотношение приходится изменять
до 1 : 10, а иногда и еще больше, так как органическая
масса впитывает большое количество раствора, который не
удается отделить простым фильтрованием или
центрифугированием.
Соотношение почва: раствор изменяют только в случае
особых задач исследований.
Для ускорения анализа при массовых определениях
можно воспользоваться объемным способом взятия
соответствующего количества почвы для приготовления вытяжки.
Водные вытяжки готовят на бидистиллированной воде,
предварительно прокипяченной и охлажденной без
доступа СОг. Такую воду хранят в бутылях с тубусом; бутыль
закрывается пробкой, в которую вставлена трубка с
поглотителем CCV
Можно сделать и так: через дистиллированную воду
просасывать воздух, лишенный СОг. Длительность просасывания
3—4 ч. Воздух предварительно пропускают через
поглотитель СОг (обычно раствор щелочи). Для поглощения NH3
ставят дополнительную поглотительную колонку с конц.
H2S04.
Для солевых вытяжек применяют 1 н. раствор КО.
Раствор КС1 должен иметь рН —6 E,6—6,0). В противном
случае нужно раствор КС1 с большой осторожностью довести
титрованием щелочью (обычно раствор кислее, чем следует)
по индикатору до нужного значения рН. Еще лучше дважды
перекристаллизовать КС1 из воды, а затем уже готовить
1 н. раствор.
Почву готовят так же, как для водных вытяжек: отбирают
корни, разминают комки; торф измельчают до однородной
порошистой массы. Как и в других случаях, берут для
вытяжек среднюю пробу из мелкозема.
116
Порядок работы
1. Взяв навески почвы и поместив их в плоскодонные
колбы на 100 мл, заливают требуемым количеством воды или
раствора КС1 и взбалтывают. Водные и солевые суспензии,
встряхивают на ротаторе в течение 5 мин; наклоняя колбы,
смывают раствором частицы почвы, приставшие к стенкам.
2. Дают отстояться суспензии 18—24 ч и сливают в сухую
пальчикообразную пробирку 2—3 мл жидкости для
определения р!1 водной суспензии; остаток переносят в
центрифужные пробирки, центрифугируют до полного осветления и
раствор сливают в пробирки. Солевым вытяжкам дают
отстояться и прозрачный раствор переносят в пробирки. Пробирки
располагаются в штативе в том же порядке, в котором
занесены в журнал исследуемые образцы.
3. В каждую пробирку вносят небольшое количество хин-
гидрона (на кончике ножа) и осторожно взбалтывают; хин-
гидрон образует насыщенный раствор и растворяется не
полностью. Через 3---5 мин можно приступить к измерениям.
Измерения проводят на компенсационной схеме или на
потенциометрах П-6, ЛП-4E) (см. стр. 85 и 128),
использовав в качестве электрода сравнения хингидронный электрод
с рН=2,04. Для его приготовления чистый и сухой
электродный сосуд заполняют буферным раствором Вайбеля с рН = 2,04
(предварительно ополоснув его тем же раствором). К
раствору добавляют немного хингидрона с таким расчетом,
чтобы после растворения в сосуде остался небольшой осадок
или взвесь хингидрона. Электрод сравнения соединяют с
почвенной суспензией электролитическим ключом, а в почвенную-
суспензию погружают платиновый электрод.
4. После каждого измерения платиновый электрод и
соединительный агар-агаровый сифон ополаскивают
дистиллированной водой без С02 и просушивают легким
прикосновением фильтровальной бумаги.
5. По результатам измерений рассчитывают величины рН
и активность водородных ионов в анализируемых растворах.
СТЕКЛЯННЫЙ ЭЛЕКТРОД И ЕГО ПРИМЕНЕНИЕ
В ПОЧВЕННЫХ ИССЛЕДОВАНИЯХ
Стеклянный электрод позволяет проводить определения рН
практически в любых почвах, он не отравляется как
водородный электрод, не боится присутствия окислителей и
восстановителей и пригоден в широком диапазоне величины рН.
11Т
Поэтому стеклянный электрод находит все более широкое
применение в почвоведении и постепенно вытесняет из
лабораторной практики другие типы электродов. Кроме того,
специальные стеклянные электроды пригодны для определения
активности ионов щелочных металлов, что открыло новые
перспективы их использования. С помощью стеклянного
электрода легко наладить прибор для непрерывного
дистанционного измерения рН.
Стекло имеет подвижные, гидролизуемые щелочные
катионы в узлах алюмосиликатного или силикатного остова.
Интересно отметить, что стекло — проводник второго рода:
в нем электрический ток проводят катионы, а сам алюмосн-
ликатный остов остается неподвижным. Пространство,
занимаемое катионами, имеет размеры, близкие к размеру
радиуса иона.
В этом электроде скачок потенциала возникает на
поверхности раздела между тонкой стеклянной мембраной и
раствором. Стеклянная пластинка взаимодействует с раствором,
в который она погружена, и вода постепенно проникает в
поверхностный слой — идет «набухание» стекла. При этом
одновременно протекает гидролиз силикатных соединений
щелочных и щелочноземельных металлов.
Ионы натрия и водорода могут диссоциировать и
обмениваться с ионами раствора, соприкасающегося со
стеклянной пластинкой. При вымачивании стеклянного электрода
з растворе с высокой активностью водородных ионов
поверхностный слой стекла теряет щелочные катионы и насыщается
водородным ионом. Это создает предпосылки для появления
у стекла водородной функции.
Переходящие в раствор ноны водорода уносят с собой
положительные заряды; избыток неподвижных силикатных
анионов придает стеклянной пластинке отрицательный заряд
Увеличение концентрации водородных ионов в растворе
подавляет диссоциацию силикатов и бисиликатов в стекле,
избыток Н+-ИОНОВ связывается с неподвижным силикатным
скелетом, в результате чего изменяется заряд стекла. Таким
образом, заряд стекла, и, следовательно, разность
потенциалов между стеклом и раствором зависят от активности
водородных ионов в растворе или от величины рН. В тоже время
влияние металлических катионов на потенциал стекла
ограничено рядом факторов. Одним из них является размер ионов-
крупные ионы неспособны легко проникать внутрь стекла и
не могут повлиять на величину потенциала. Другим
ограничивающим фактором служит прочность связи водородных и
«18
металлических катионов с анионами стекла. Чем выше
прочность связи металлического катиона, например натрия, со
стеклом, тем с большим трудом он замещается водородными
ионами и тем сильнее сказывается влияние натрия на
потенциал стеклянного электрода. Аналогичное влияние оказывает
и калий. Развивая эти взгляды, проф. Б. П. Никольский
предложил уравнение, выражающее потенциал стеклянного
электрода в зависимости от активности водородных и натриевых
ионов в растворе:
, RT 2,303 , , , „ .
6 = е„ 4- - Ig (aH+ + KaNa+),
где
Л = ;
°Na+°H+ f с.
и #к+ и с'н+— активности иона водорода соответственно в
растворе и в стекле, a aNa+ и а'^я+ — тоже для ионов натрия.
Из уравнения следует, что если а„+ ~^>Ка^а+, то
практически
2,303 ЯГ „
е-е0— рН
и потенциал электрода зависит только от величины рН. В
щелочной среде, когда концентрация водородных ионов очень
мала, ак+ может стать равным KciNa+ или даже меньше, и
гогда стеклянный электрод начинает проявлять себя как
натриевый.
Разница в относительной прочности связи ионов Na+ и Н+
со стеклам характеризуется константой К- Чем выше
значение К, тем при более низких величинах рН проявляется
натриевая функция стеклянного электрода. Увеличение
константы К и появление у стекла свойств натриевого электрода
достигаются введением в состав стекла окислов бора и
алюминия. По Никольскому, стекло, обычно употребляемое для
изготовления электродов с водородной функцией, имеет
величину К=Ю~12; стекло, содержащее 9% ВгСЬ, характеризуется
величиной К=Ю"9 и содержащее 7% А12СЬ, — величиной
Л'=10-ь.
Состав стекла, употребляемого для стеклянных
электродов, различен (табл. 13).
Потенциал стеклянных электродов, употребляемых для
определения рН, в кислой, нейтральной и слабощелочной сре-
119'
Таблица 13
Состав стекла для изготовления стеклянных электродов
Тип электрода
Электрод с
водородной функцией
Электрод с
водородной функцией малого
сопротивления
Электрод с
натриевой функцией
То же
Si02, %
60—75
64
62
75
А120,, %
--
И
3
СаО, %
8-10
—
3
-
MgO, %
_
8
—
■
Е_,0„, %
_
—
—
И
Nj^O, %
20-30
28
24
11
де зависит только от активности водородных ионов. При
повышении рН до 9—10 постепенно усиливается металлическая
-функция электрода и прямолинейная зависимость между
потенциалом и величиной рН нарушается (рис. 24).
Диапазон .значений рН, которые
могут быть измерены стеклянным
электродом, зависит от
состава стекла; некоторые стекла
сохраняют прямолинейную
зависимость между
потенциалом и рН в широком
интервале и позволяют измерять
величины рН от 1 до 10, а иногда
до 11 — 12.
Расхождение свойств
отдельных стеклянных
электродов заставляет проводить
обязательную градуировку
каждого вновь изготовленного
электрода, чтобы определить
пределы его 'применимости.
Устройство стеклянных
электродов показано на рис. 25.
Для изготовления берут стеклянную трубку диаметром от 3
до 5 мм, запаивают один конец в пламени газовой горелки
и осторожно выдувают шарик. Толщина стенок шарика
должна быть достаточно малой, чтобы обеспечить необходимую
электропроводность. Стекла с высокой электропроводностью
позволяют изготовлять толстостенные прочные электроды, ко-
Рис. 24. Зависимость потенциала
■стеклянного электрода от
активности водородных ионов;
2,303/?Г
tga=©= ; :
г
/ — теоретическая зависимость,
2 — экспериментальные данные
.120
торые не разбиваются даже при легком постукивании о
твердые предметы. Тонкостенные шарики очень неудобны в
обращении, так как легко бьются.
Для увеличения надежности в работе и предупреждения)
от механических повреждений электродам придают иногда
специальную вогнутую форму (рис. 25); в таких электродах.
Рис. 25. Виды стеклянных электродов:
А — стеклянный электрод Мак-Иннеса; Б — шариковый электрод; В —
электрод с защитной муфтой; /'—вогнутый электрод; 1 — стеклянная
мембрана, 2 — 0,1 и. раствор НС1, 3 — хлоросеребряный электрод, 4 — наружная'
(утолщенная) часть электрода, 5 — защитная муфта
тонкостенная рабочая часть электрода защищена от
механических повреждений. Снаружи стеклянные электроды часто
защищают специальным колпачком (рис. 25, в).
Внутрь стеклянного электрода наливают 0,1 н.
раствор НС1, в который погружают хлоросеребряный электрод.
В результате получается цепь, состоящая из:
Ag-AgCI
НС1
0,1 н.
РНХ
рНх,
где двойная черта обозначает стеклянную мембрану.
121.
Хлоросеребряный электрод можно приготовить в виде
серебряной проволоки, погруженной в раствор НО или в соля-
■ло-кислую суспензию AgCl. Поверхность серебряной
проволочки, находящейся в НС1, быстро покрывается слоем AgCl.
Для улучшения качества хлоросеребряное покрытие лучше
заносить электролитическим путем. Иногда в качестве
внутреннего электрода сравнения рекомендуют платиновую
проволоку в 0,1 н. НС1, но такой электрод не имеет устойчивого
скачка потенциала.
Измеряемый в опытах потенциал стеклянного электрода
равен разности между скачками потенциалов на внутренней
и внешней поверхности мембраны. Можно было бы ожидать,
как это следует из уравнения потенциала, что при равенстве
рН по обе стороны мембраны суммарный потенциал
окажется равным нулю, поскольку скачки потенциалов
одинаковы и имеют противоположные знаки. Однако в
действительности этого не наблюдается. То значение скачка
потенциала, которое имеет место при равенстве величины рН по
обе стороны стеклянной мембраны, носит название
потенциала асимметрии. Возникновение потенциала
асимметрии обусловлено неодинаковым воздействием температуры
пламени на внешнюю и внутреннюю поверхность стеклянного
шарика в процессе его выдувания. В результате состав и
физическое состояние поверхностных пленок оказывается
различным, что влияет на равновесие между стеклом и
раствором. При длительном вымачивании стеклянного электрода
в воде или слабом растворе кислоты поверхностные пленки
растворяются и в соприкосновение с раствором приходит
внутренняя, практически не измененная в процессе обработки,
часть стекла. Эта операция позволяет значительно снизить
потенциал асимметрии и довести его всего лишь до
нескольких единиц или даже долей милливольта; однако полностью
устранить потенциал асимметрии не удается.
Величина потенциала асимметрии не остается постоянной
при изменении рН раствора, именно поэтому с ней
приходится считаться при практическом использовании электродов.
Учесть влияние потенциала асимметрии на показания
прибора можно путем градуировки электродов.
В цепях со стеклянным электродом в качестве второго
полуэлемента, или внешнего электрода сравнения, служат
каломельный, хлоросеребряный, водородный или хингидрон-
ный электрод. Наиболее часто употребляется каломельный
электрод; цепь, составленная из стеклянного и каломельного
полуэлементов, имеет вид:
122
Ag-AgCl
HC1
PHX
0,1 н.
pHx
KCl
насыщ.
Hg2Cl2
Hg-Pt
В некоторых случаях удобно пользоваться хлоросеребря-
ным электродом сравнения, тогда цепь выглядит так:
Ag-AgCl
НС1
0,1 н.
рН,
рНх
КС1
насыщ.
НС1
pHi
0,1 н.
AgCl-Ag
Измеряемая величина э. д. с. первого элемента
алгебраически складывается из скачков потенциалов хлоросеребряно-
го ех с. и каломельного екал электродов, потенциала
асимметрии гас и скачков потенциалов на внутренней ест и
внешней ех поверхности стеклянного электрода:
э. д. с. = ех.с. + екал + еас -j- ест + ех.
Потенциалы каломельного и хлоросеребряного электродов
остаются постоянными в течение всей серии измерений; их
можно объединить в константу: ех с + £кал=А Раскрывая
величину ест. и ех по уравнению Никольского, получим
2,303 ЯГ
э. д. с. =-= А + еас + е0
■pHi + e0p —
2,303 RT
рнх
Если не учитывать потенциала асимметрии при изменении-
рН, то можно упростить выражение, объединяя все
постоянные величины под значком а: два значения е0 стек-
ст
лянного электрода в цепи имеют противоположные знаки,
неравное абсолютное значение, тогда
э. д. с.
-ФрН,
где
о, 2,3fiT . , „ 2,2,RT „„
* = — ~ и о. = ех.с. + екал + еас — рН,.
F F
Э. д. с. второй цепи может быть вычислена по тому же
уравнению, с той лишь разницей, что два хлоросеребряных
электрода в цепи имеют равные потенциалы, но
противоположные знаки. Таким образом, постоянная а для уравнения
123
'второй цепи включает только потенциал асимметрии и
некоторую величину, обусловленную значением рН внутреннего
раствора:
2,303 RT т, 2,303 ЯТ „
э. д. с = еас рН, рНх,
'ИЛИ
э. д. с. = а — ftpHx, где а -- еас — ftpHr
Такой характер зависимости позволяет определять рН, не
вычисляя отдельно скачок потенциала на внешней
поверхности стеклянного электрода, а пользуясь суммарной
величиной э. д. с. всего элемента в целом. Достаточно точное
значение рН получают по градуировочному графику. Как следует
из последнего уравнения, зависимость между э. д. с. всей цепи
и величиной рН прямолинейная.
Градуировочный график строится по результатам
измерений: стеклянный электрод последовательно помещают в
буферные растворы с точно известными значениями рН; для
каждого раствора измеряют э. д. с. цепи. Результаты
измерений наносят на график в координатах э. д. с. — рН. Затем
по графику находят величины а и ft и при исследовании
растворов с неизвестным значением рН производят вычисления
по формуле
а-з Д. с
Н &
Числовое значение коэффициентов не остается постоянным
>во времени. Градуировку электрода приходится проводить
каждый раз перед измерениями и контролировать ее в
процессе работы.
Расчет по этой формуле дает хорошие результаты только
при идеальной прямолинейной зависимости между э. д. с. и рН;
нарушения зависимости, вызываемые влиянием потенциала
асимметрии и появлением металлической функции у
электрода в щелочном интервале рН, наблюдаются довольно часто.
Практически в широком диапазоне рН график приобретает
вид S-образной кривой (рис. 24). Поэтому наиболее точные
результаты получают, когда искомые значения рН находят
прямо по графику. Таким образом, процесс измерения рН
с помощью стеклянного электрода сводится к измерениям
э. д. с. цепей, в которых участвуют буферные растворы, и к
составлению градуировочного графика, измерению э. д. с. цепи,
J24
составленной с участием исследуемого раствора, и
нахождению искомой величины по графику. Потенциометры и рН-мет-
ры позволяют автоматически учитывать градуировку
электродов и определять рН непосредственно по отсчетной шкале
потенциометра, не прибегая к построению графика.
ИЗМЕРЕНИЕ Э.Д.С. ЦЕПИ СО СТЕКЛЯННЫМ ЭЛЕКТРОДОМ
Особенностью и недостатком стеклянного электрода
является его высокое омическое сопротивление, которое
настолько велико, что компенсационная схема (см. стр. 85)
■оказывается совершенно непригодной. Измерительный
инструмент в потенциометре должен быть настолько чувствителен,
чтобы фиксировать отклонение подвижного контакта от
точки компенсации на 1 деление шкалы. В обычных схемах цена
одного деления порядка 1—2 мв. Если стеклянный электрод
имеет сопротивление ~ 107 ом, то сила тока, протекающего
через гальванометр, равна 10-3: 107=10-10 а, если подвижный
контакт отстоит от точки равновесия на одно деление шкалы.
Такого же порядка A0~10 а) должна быть и чувствительность
измерительного инструмента. Стрелочные гальванометры,
употребляемые при работе с хингидронным и водородным
электродами, .имеют чувствительность до Ю-7 а и могут быть
использованы только после предварительного усиления тока.
•Стандартные зеркальные гальванометры имеют
чувствительность порядка 10~8—Ю-9 а/мм, но и они оказываются
непригодными при прямом включении в обычную компенсационную
схему Поггендорфа. Поэтому на практике используют
зеркальные гальванометры в сочетании с компенсационной
конденсаторной схемой или работают по баллистическому
методу. Наиболее современным измерительным прибором является
потенциометр с ламповым усилением.
Компенсационная схема с конденсатором. Схема такой
цепи показана на рис. 26,/. При замыкании ключа
протекающий в цепи ток заряжает конденсатор С; после переключения
в положение 2 конденсатор разряжается на гальванометр.
Максимальная эффективность достигается при правильно
подобранных характеристиках гальванометра и конденсатора.
Правильно рассчитанная конденсаторная схема позволяет
■обнаружить ток даже в цепи с очень высоким сопротивлением.
Однако такие высокие сопротивления уже становятся
соизмеримыми с сопротивлениями изолирующих материалов:
лаков и красок, покрывающих поверхность деталей. Это
приводит к значительным утечкам тока. Утечка тока часто
125
происходит по тонкой пленке жидкости, обволакивающе»
соединительные трубки и стеклянные части электродов. Для
предупреждения утечек схемы со стеклянным электродом
монтируют на специальных парафинированных подставках;
все детали, соприкасающиеся с электрической цепью, должны
быть чистыми и насухо вытертыми; там, где возможно,
поверхность деталей парафинируют или покрывают
специальными лаками высокого сопротивления, или эти детали
изготавливают из негигроскопичных материалов с высоким
сопротивлением.
UI
ш
Рис. 26. Схемы для измерения рН со стеклянным электродом:
/ — конденсаторная схема; // — баллистическая схема; Ех—элемент со
стеклянным электродом; £в —элемент Всстона; К—ключ; С —
конденсатор; З.г. — зеркальный гальванометр; Акк — аккумулятор; 111 — шунт
гальванометра
Измерения и расчеты потенциалов осуществляются так
же, как и на обычной компенсационной схеме. Отличие лишь
в том, что «зайчик» гальванометра движется не после
замыкания ключа, а после его размыкания, когда конденсатор
соединяется с гальванометром. Время заряца конденсатора
подбирается экспериментально и должно быть постоянным в
максимальным вблизи точки компенсации. Во избежание
порчи гальванометра в начале работы его следует шунтировать, а
конденсатор заряжать в течение возможно короткого времени,
увеличивая время заряда и уменьшая шунтирование по мере
приближения к точке компенсации.
Схема для работы баллистическим методом. При работе
по баллистическому методу отпадает необходимость
вычислять потенциал электрода. Вся схема состоит из стеклянного
электрода, электрода сравнения, конденсатора, ключа и зер-
126
кального гальванометра (рис. 26,//). Для точного
определения времени заряда необходим секундомер.
Ход определения сводится к зарядке конденсатора в
течение определенного времени и последующему измерению
показаний гальванометра при разряде конденсатора на
гальванометр.
Если для компенсационной схемы с конденсатором
величина показаний гальванометра безразлична (отсчет берут,
когда сила тока в малой цепи равна нулю), то при работе
но баллистическому методу обязательно сохранение
пропорциональности между показаниями гальванометра и э. д. с.
элемента. Для определения рН гальванометр в собранной
цепи градуируют, откладывая на графике показания
гальванометра в зависимости от рН.
Время заряда при баллистическом методе имеет
решающее значение. Расчеты показывают, что если заряжать
конденсатор до разности потенциалов Е, близкой к э. д. с.
измеряемого элемента, то надо затратить много времени; чтобы
довести £^0,95 э.д. с. для конденсатора емкостью 10 мкф,
включенного в цепь с сопротивлением 108 ом, требуется
3000 сек, а для конденсатора 1,0 мкф— 300 сек. В связи
с этим оказывается выгодным заряжать конденсатор не
полностью. Так как разность потенциалов на обкладках
конденсатора £ пропорциональна э. д. с. элемента Е\\
*_
£-=£хA — е RC)
^ — сопротивление цепи и С — емкость конденсатора), то
при постоянном времени заряда t показания гальванометра
пропорциональны э.д. с. элемента. Последнее позволяет
в уравнении цепи со стеклянным электродом заменить э.д. с.
на пропорциональную ей величину показаний
гальванометра А:
э. д. с. = kA,
тогда э. д. с. = а — ФрН ^= kA.
Отсюда А = а' — ЬрН,
Практически заряжают конденсатор строго определенное
время (по секундомеру), разряжают его на гальванометр,
записывают показания гальванометра и строят график. Если
отсчеты малы, то увеличивают время зарядки или емкость
конденсатора, хотя последнее эффективно лишь в
ограниченных пределах.
127
Порядок определения рН баллистическим
методом
1. Собрать схему, как показано на рис. 26,/I, и установить
сопротивление шунта, равное внешнему критическому
сопротивлению гальванометра.
2. В сосуд со стеклянным электродом налить буферный
раствор с известным значением рН; стеклянный электрод
соединить с каломельным посредством электролитического
ключа.
3. Замкнуть ключ, нажав одновременно пуск
секундомера; ровно через 30 сек отпустить ключ и записать
максимальный отброс гальванометра. Повторить измерения еще
2 раза и вычислить среднее значение.
4. Определить таким способом показания гальванометра
для 5—7 буферных растворов и построить график
зависимости показаний гальванометра от величины рН.
5. В сосуд со стеклянным электродом налить исследуемый
раствор и определить показания гальванометра.
6. Найти по графику величину рН исследуемого раствора.
7. Если нужно с помощью баллистического потенциометра
измерить э. д. с, то гальванометр градуируют в
милливольтах, для чего в схему включают стандартный элемент,
например элемент Вестона, через делитель напряжения. Меняя
поданное напряжение, можно получить зависимость между
э. д. с. заряжения конденсатора и отбросами «зайчика»
гальванометра.
Ламповые потенциометры
Во всех ламповых приборах измеряется сила тока,
протекающего в анодной цепи электронной лампы (рис. 10, 27).
Потенциал стеклянного электрода подается на управляющую
сетку электронной лампы и изменяет тем самым силу
анодного тока (см. стр. 54).
Использование усилителей с электронными лампами
обеспечивает следующие важные преимущества: ламповые
усилители позволяют измерять очень слабые токи и малые
разности потенциалов; при работе с ламповыми усилителями
сила тока, протекающего через измеряемый элемент,
практически равна нулю; на измерение затрачивается мало
времени.
Электронные лампы могут быть применены для прямого
усиления, когда измеряемой величиной являются показания
128
Клип К I
Рис 27. Схема (А) и панель (Б) потенциометра ЛП-4E).
/ — потешшометрическая часть: Ri— реохорд; К—ключ; Кл и
Ст — клеммы для подключения каломельного и стеклянного электродов;
#ю — настройка потенциометра; /?2 — температурный компенсатор;
/7j — переключатель настройки; /72—переключатель диапазонов
измерений, Б — сухая батарея; Н. э.— нормальный элемент;
// — усилитель: /'—гальванометр; Л г и Л*—лампы моста,
RJS—настройка усилителя;
/// — блок питания: Тр — трансформатор; Л\— двухполупернодный
выпрямитель; Сг и Лг— конденсатор и газотрон, сглаживающие
пульсации тока; Л—сигнальная лампа (пунктиром обведена часть,
экранируемая от электромагнитных полей)
5 Зак. 51
гальванометра, включенного в анодную цепь; в другом
варианте гальванометр служит только как нуль-инструмент и
включается в диагональ моста, одно или два плеча которого
представлены электронными лампами (см. рис. 10). Прямое
усиление используется, в частности, в рН-метрах.
Недостатком однолампового усилителя является
неустойчивость его работы; всякие изменения питающего анодную
цепь напряжения, э. д. с. батарей и т. д. отражаются на
работе усилителя и искажают результаты. Усилитель, собранный
по мостовой схеме, лишен указанных недостатков.
Ламповый потенциометр ЛП-4E) предназначен для
измерения рН в цепях со стеклянным, с хингидронным
электродом, а также для измерения э. д. с. элементов с различным
внутренним сопротивлением. Прибор рассчитан на
измерения рН от 0 до 14 с точностью до 0,05 рН и на измерения
э. д. с. в пределах от —1400 до +1400 мв.
Прибор состоит из трех основных узлов (см. схему на
рис. 27). Потенциометрическая часть / включает реохорд R\,
нормальный элемент И. э„ сухой элемент Б, переключатели
и дополнительные сопротивления. Вторая часть //
(нуль-инструмент) представлена двухламповым усилителем Л3 и
Ль—6Ж7, собранным по мостовой схеме, в диагональ моста
которого включен стрелочный гальванометр Г,
показывающий разность анодных токов ламп Лъ и Л4. Для настройки
усилителя используется потенциометр Rl5. Третий узел ///
обеспечивает питание усилителя и состоит из
трансформатора Тр и системы, поддерживающей постоянство питающего
напряжения: двухполупериодного выпрямителя Ли
газотрона Л2 и емкостного стабилизатора С2, сглаживающего
пульсации выпрямленного тока. Эта схема очень мало
чувствительна к изменениям питающего напряжения; все помехи
одинаково влияют на обе лампы моста и не нарушают его
уравновешенности.
Порядок работы на потенциометре ЛП-4E)
и измерения
Соединяют прибор с источником переменного тока
127/220 в и, включая тумблер Вкл, расположенный на правой
боковой панели футляра, подают питающее напряжение на
лампы усилителя. При этом должна загореться индикаторная
-лампочка Л (рис. 27,Б). После прогрева ламп в течение
3—5 мин переводят переключатель Пх в положение Р
(рабочее) и настраивают усилитель. Для этого, вращая руч-
130
ку #15, изменяют сопротивление в цепи лампы Лз и тем
самым добиваются полного уравновешивания плеч моста.
Стрелка гальванометра должна остановиться в нулевом
положении. Затем настраивают прибор по сухому элементу.
Переключатель П\ переводят в положение Н.э. (нормальный
элемент), подавая один полюс Н. э. на сетку Л4. При втом
стрелка гальванометра сдвинется с нуля; сопротивлением R\o,
связанным с рукояткой на верхней панели прибора, гасят
избыток напряжения сухого элемента; после этой настройки
на каждое деление шкалы приходится ровно 10 мв. Когда-
это условие соблюдено, стрелка гальванометра возвращается
в нулевое положение. Снова переводят переключатель П^
в положение Р, и если стрелка гальванометра отклонилась,
от нуля, то повторяют настройку, начиная с первой операции.
В случае изменения положения переключателя П2 (переход,
от измерения в единицах рН к милливольтам и обратно),
настройка прибора проводится заново.
Затем стеклянный и каломельный электроды соединяют
с гнездами на панели потенциометра (обозначены «Ст» и
«Кл»). Нажимают кнопку К и вращают ручку реохорда Ri,
пока стрелка гальванометра не займет нулевого положения1.
Если вращением реохорда не удается добиться компенсации,,
то меняют полюсность, ставя переключатель П2 в положение
+мв (или —мв). Если кнопку К нажать и немного повернуть
вправо по часовой стрелке, то она остается в фиксированном
положении, а цепь оказывается замкнутой. Этим можно
пользоваться для быстрого отыскания точки компенсации. Когда
стрелка гальванометра займет нулевое положение, кнопку К
следует отпустить и убедиться, что при кратковременном
замыкании цепи стрелка гальванометра не отклоняется.
Отсчеты берут по шкале реохорда и записывают, учитывая
полюсность (+ или — по переключателю П2).
По измеренным значениям э. д. с. для ряда буферных
растворов строят график зависимости Е — рН, по которому
рассчитывают константы а и b уравнения стеклянного электрода
и находят величины рН анализируемых растворов.
В небольших интервалах прибор позволяет измерять рН
и без градуировочного графика. Для этого переключатель U%
ставят в положение «рН» и настраивают потенциометр.
Стеклянный электрод помещают в буферный раствор и на
реохорде устанавливают значение, равное величине рН
буферного раствора, а на температурном компенсаторе /?2 —
температуру раствора. Нажав кнопку К, рукояткой «установки
нуля» подводят стрелку гальванометра к нулевому делению.
5*
131
Затем заменяют буферный раствор испытуемым и, нажимая
кнопку К, добиваются компенсации. По шкале реохорда
считают величину рН.
Точно также определяют величину рН и в цепи с хингид-
ронным электродом с той разницей, что градуировка по
буферному раствору заменяется следующим простым способом.
Как было показано на стр. 112, нулевое значение э. д. с. хин-
гидрон-каломельного элемента достигается при рН = 7,8 B0°).
Поэтому в градуировке такой элемент заменяют коротким
замыканием входных клемм потенциометра. Замкнув входные
клеммы перемычкой и установив на шкале значение рН=7,8,
рукояткой установки нуля стрелку гальванометра подводят
к нулевому делению. Отсчеты берут непосредственно по
шкале рН.
Успех измерения величин рН на потенциометрах зависит
в значительной мере от аккуратности в обращении с
прибором, электродами и тщательности настройки. Здесь, как и в
других схемах, все детали должны быть насухо протерты,
электроды чисты и хорошо отградуированы. Перед сменой
раствора, рН которого измеряется, необходимо обязательно
выключить кнопку реохорда К.
ОПРЕДЕЛЕНИЕ рН ВОДНЫХ И СОЛЕВЫХ ВЫТЯЖЕК ИЗ ПОЧВ
НА ПОТЕНЦИОМЕТРАХ
Потенциометр П-6 позволяет измерять э. д. с. в пределах
от 0 до 1300 мв с погрешностью ±5 мв. Электрическая цепь
позволяет также брать отсчеты непосредственно в
величинах рН, работая с одним из приложенных к прибору
электродов: хингидронным или сурьмяным. Для измерения э. д. с.
в цепях со стеклянным электродом потенциометр соединяют
с ламповым усилителем.
Электрическая схема состоит из четырех самостоятельных
цепей, используемых для: 1) настройки прибора по
нормальному элементу; 2) нахождения рН хингидронным электродом;
3) измерения рН сурьмяным электродом; 4) определения
э.д. с. в милливольтах.
Переключение цепей осуществляется ключами (рис. 28) И2
(настройка по нормальному элементу, рабочее положение и
отключение сухого элемента) и И\ (переключения для работы
с хингидронным, сурьмяным электродом или для измерений
э.д. с. в милливольтах). Источником питания служит сухой
элемент ЗСЛ-30 на 1,5 в (Е). Настройка прибора по
нормальному элементу достигается вращением рукоятки гру-
132
+ х —
ШШ-
V 19
L^ww-Ji
■я. "'U LK
3 „в р
Рис. 28. Потенциометр П-6:
А — схема; Е — сухой элемент; Н.э. — элемент Вестона; х — клеммы
для подключения изучаемого элемента; Г — гальванометр; К — кнопка;
Mi и Mi—ключи; Ri—реостат грубой настройки; R2—реостат плавной
•настройки; Яз— температурный компенсатор; Rt— корректор рН;
Rs— реохорд; Б — панель
бой Ri и плавной R2 настройки. Температурная поправка
вводится реостатом /?з. Основная рабочая часть потенциометра —
реохорд R5 для компенсации разности потенциалов
измеряемого элемента и падения напряжения от сухого элемента.
Все детали прибора смонтированы в одном деревянном
футляре. Правая часть футляра занята отделением, в котором
помещается сухой элемент, футлярчик с электродами и
специальная подставка для электродов. На верхней панели
расположены все рукоятки управления и клеммы для
подключения электродов.
Подготовка прибора и измерения
Перед работой открывают крышку футляра и
устанавливают прибор в рабочее положение. Электроды и электродные
сосуды ополаскивают дистиллированной водой, поворотом
рукоятки арретира переводят стрелку гальванометра в
свободное положение и с помощью корректора устанавливают
ее на нулевое деление шкалы.
Измерение э.д.с. в милливольтах. Для
измерения э. д. с. поступают так.
1. Вставляют электроды в сосуд, заполненный
анализируемым раствором, и соединяют их шнуром с гнездами
прибора. Если при подключении нарушена полярность, то в
дальнейших операциях (п. 3) не удается найти точку
компенсации; тогда необходимо изменить последовательность
включения электродов в измерительную цепь.
2. Поставив ручку И2 в положение «Н. э.», а рукоятку И\
в положение «Мв», нажимают кнопку К и, вращая ручку
реостатов R\ и R2, добиваются, чтобы стрелка гальванометра
оставалась на нулевом делении шкалы. В этом положении
прибор скомпенсирован по нормальному элементу и на
каждое деление шкалы приходится точно 10 мв.
3. Поставив ручку И2 в рабочее положение «Я»,
нажимают кнопку К и вращают ручку реохорда R5 до тех пор»
пока при очередном замыкании кнопки К стрелка
гальванометра не перестанет отклоняться от нулевого деления.
4. Записывают показания (в мв) по шкале реохорда и
повторяют измерения.
Измерения в единицах рН. Последовательность
операций при определении рН с хингидронным или
сурьмяным электродами одинакова; отличается только порядок
градуировки шкалы.
1. Заполняют электродный сосудик испытуемым раство-
134
ром и погружают в него пару электродов (хингидронный —
каломельный или сурьмяный — каломельный). В паре
хингидронный— каломельный к гнезду « + » на панели
потенциометра подключают хингидронный электрод, а к
гнезду «—» -— каломельный. В паре сурьмяный — каломельный
к гнезду « + » подключают каломельный электрод, а к
гнезду «—» — сурьмяный.
2. Настраивают прибор по нормальному элементу. Для
этого ключ И2 ставят в положение «Н. э.», ключ Их — в
положение «Хинг. эл.» при работе с хингидронным электродом
или в положение «Сурьмян. эл.» — при работе с сурьмяным
электродом. Ручкой компенсатора Яз устанавливают
температуру исследуемого раствора. Нажав кнопку К, приводят
стрелку гальванометра к нулевому делению шкалы при
помощи рукояток настройки.
3. При работе с сурьмяным электродом градуируют
шкалу по буферным растворам. Для этого наливают в
электродный сосудик буферный раствор с известным значением рН,
ключ И2 переводят в положение «Я» и ручкой реохорда R$
устанавливают шкалу на деление, отвечающее величине рН
буферного раствора. Затем, нажав кнопку К, вращают
рукоятку корректора рН /?4 до тех пор, пока стрелка
гальванометра не установится на нуль.
Дальнейшие операции с хингидронным и сурьмяным
электродами одинаковы.
4. Заполняют сосуд испытуемым раствором,
предварительно ополоснув сосуд и электроды сначала водой, а затем
раствором. При работе с хингидронным электродом к
раствору добавляют несколько миллиграммов хингидрона (на
кончике ножа). Ключ И2 ставят в положение «Я», ключ И\ —
в положение, соответствующее выбранному электроду («Хинг.
эл.» или «Сурьмян. эл.»). Нажав кнопку К, вращают ручку
реохорда Rs, пока стрелка гальванометра не установится на
нулевом делении.
5. Записывают показатели на шкале реохорда, каждое
деление которого соответствует 0,1 рН.
Определения рН со стеклянным электродом
на потенциометрах П-5 (П-6)
Высокое сопротивление стеклянного электрода не
позволяет непосредственно измерять рН на потенциометрах
типа П-5 без дополнительного усиления. Специальные
усилительные приставки (типа ЛУ-2) предназначены для
135
измерения э. д. с. высокоомных элементов и определения рН
стеклянным электродом с помощью любого потенциометра.
Использование потенциометров в комбинации с усилительной
приставкой повышает точность измерения рН любыми
электродами, так как при этом устраняются явления поляризации.
Рис. 29. Панель усилителя ЛУ-2
Приставка ЛУ-2 (рис. 29) по своей принципиальной схеме
аналогична потенциометру ЛП-4 E).
Порядок измерения э.д.с. с приставкой ЛУ-2
1. Присоединить усилитель ЛУ-2 к потенциометру,
включить прибор в сеть, освободить (арретиром) стрелку
гальванометра и установить ее корректором в нулевое положение.
2. Поставить выключатель в положение «Вкл.» и дать
прогреться лампам усилителя 3—5 мин. При этом должна
загореться индикаторная лампочка. Настроить потенциометр
по нормальному элементу.
3. Ручкой усилителя с обозначением «настройка»
установить стрелку гальванометра в нулевое положение.
4. Налить в электродный сосуд испытуемый раствор, по-
136
грузить в него стеклянный и каломельный электроды,
соединив их с гнездами на панели усилителя.
5. Переключатель потенциометра перевести в
положение «Р» и нажать кнопку К на потенциометре; последняя
должна оставаться замкнутой в течение всех последующих
измерений. Нажать кнопку К\ усилителя и ручкой реохорда
потенциометра вернуть стрелку гальванометра в нулевое
положение. Записать показания (в мв) по шкале реохорда
потенциометра.
6. Отградуировать электрод по буферным растворам и по
графику найти величину рН испытуемого раствора.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ И АКТИВНОСТИ ИОНОВ НАТРИЯ
В ПОЧВЕННЫХ ВЫТЯЖКАХ И СУСПЕНЗИЯХ С ПОМОЩЬЮ
СТЕКЛЯННОГО ЭЛЕКТРОДА
Стеклянные электроды с металлической функцией
используются для прямого потенциометрического определения
активности ионов натрия и калия особенно в засоленных и
солонцеватых почвах. Для натриевых электродов уравнение
Никольского (см. стр. 119) выглядит так:
е - ее + 2,303 М- lg Ка^+ = г'0 + 2,303 -^ lgaNa+.
F г
Вместе с натрием на электродную функцию влияют ионы
калия и, частично, кальция. Избирательность, или
селективность, стеклянного электрода по отношению к катионам
одного какого-либо вида достигается изменением химического
состава стек-гцуВ результате работ Никольского и его
сотрудников в Советском Союзе и Айзенмана в США сейчас уже
изготавливают электроды, селективные по отношению к
ионам натрия и калия, даже если примесь мешающего катиона
превосходит концентрацию определяемого в 10 раз и более.
-, J Диапазон концентраций, измеряемых стеклянным
электродом с металлической функцией, значительно уже, чем для
электродов с водородной функцией^Прямолинейная
зависимость между потенциалом и отрицательным логарифмом
активности ионов металла, равным рМ, сохраняется в пределах
от 5-10~4 до 1,2—2,0 н., т. е. от 0 до 4 рМ, тогда как рН может
быть измерен от 1—2 до 10—11.Причиной относительно
невысокой чувствительности являются водородные ионы, которые
в разбавленных растворах заметно влияюпна потенциал любого
стеклянного электрода.
137
Первоначально стеклянные электроды применялись
почвоведами только для определения активности ионов натрия
в почвенных вытяжках и природных водах. За последние годы
разработаны методы прямого определения содержания ионов
натрия в засоленных почвах, водах и растворах, а также
методы определения поглощенного натрия со стеклянным
электродом.
Особая ценность прямого определения активности
металлических катионов обусловлена тем, что активная
концентрация (активность) вещества является количественной мерой
участия этого вещества в реальных химических процессах.
Определение активности ионов натрия в водных суспензиях
солончаков и солонцов
Активность ионов натрия в почвенных суспензиях
определяют путем измерения потенциала стеклянного электрода
с натриевой функцией. Для измерения потенциала
составляют цепь:
Ag-AgCl
NaCl
0,1 н.
Почвенная j Агар - агар +
суспензия I +КС1 насыщ.
КС1
насыщ.
X
X
Hg2Cl2
Hg-Pt,
где две вертикальные черты означают стеклянную мембрану.
Перед определением стеклянный электрод градуируют по
стандартным растворам. Для этого готовят путем
разбавления точно 5-10-4, 1 • 10-3, 5- Ю-3, 1 • Ю-2, 5- 1С)-2, 1 • 10 и
5«Ю-1 н. растворы NaCl и, вводя их последовательно в цепь,
измеряют э. д. с. Для каждого из растворов находят
табличный коэффициент активности и вычисляют активную
концентрацию. По результатам измерений строят градуировочный
график, откладывая по оси ординат э. д. с. цепи с Na-стеклян-
ным электродом, а по оси абсцисс — отрицательный логарифм
активности ионов натрия.
"~ Почву для анализа растирают в фарфоровой ступке,
просеивают через сито с отверстиями 1 мм и берут навески по
10 г, которые помещают в колбы на 100 мл. Навески почв
в колбах заливают при помешивании 50 мл
дистиллированной воды (лишенной С02), закрывают пробками и
встряхивают на ротаторе 5 мин.
138
После встряхивания суспензии дают отстояться; верхний
слой жидкости сливают в стаканчик, куда погружают
стеклянный электрод и электролитический ключ (агар-агаровый
сифон), соединенный с каломельным электродом. Через
2—3 мин измеряют э. д. с. цепи.
Отстаивание суспензии можно заменить
центрифугированием, что ускоряет определение и позволяет получить
прозрачные растворы. Навески почв в этом случае удобно брать
сразу в центрифужные пробирки на 100 мл, в пробирки с
почвой наливают воду и встряхивают на ротаторе, закрыв
пробками, а затем центрифугируют, вынув пробки.
По окончании измерений находят -а1*тивнееть ионов натрия
по градуировочному графику. г^лг-^-^к^^
"""'Измерение активности ионов натрия проводят в образцах
засоленных почв и солонцов; для контрольных измерений
берут несколько образцов незаселенных и несолонцеватых
разностей.
Определение содержания натрия в водных вытяжках
из засоленных почв
В почвах и почвенных растворах, содержащих среди
легкорастворимых солей преимущественно хлориды натрия, этим
же методом** можно определять не только активность, но и
концентрацию ионов натрия. Для этого достаточно сделать
допущение (Н. О. Авакян), что коэффициенты активности
в стандартных и анализируемых растворах одинаковы. Тогда
градуировочный график следует построить в координатах
Е — lg С, а весь ход определения остается прежним.
Результат получается в тех единицах концентрации, которые были
выбраны для построения графика (безразлично: %, г/л,
М,н.).
Почвы с более сложным солевым составом, особенно при
высоком содержании двухвалентных ионов, а также водные
вытяжки, в которых много органических Ееществ или
соединений железа и алюминия, этим методом анализировать
нельзя.
,- ' Для выравнивания коэффициентов активности в
стандартных и испытуемых растворах было предложено
пользоваться избытком индифферентной соли (Д. С. Орлов и
Н. Н. Цикурина), нивелирующей разницу в ионной силе
растворов. Так, например, коэффициенты активности ионов натрия
остаются практически постоянными, если раствор содержит
139
в концентрации от 0,3 до 0,5 М. Это позволило опре-
содержание натрия в засоленных почвах, пользуясь
следующим методом.
Готовят три исходных раствора: 0,01 М. NaCl, 1,0 М NaCl
и раствор СаС12, содержащий 180 г соли в литре раствора.
В колбы на 100 мл вносят последовательно 3, 10 и 30 мл
0,01 М раствора NaCl и далее— 1, 3, 10 и 30 мл 1,0 М
раствора NaCl. В каждую колбу добавляют по 25 мл раствора
СаС12 и доливают до метки дистиллированной водой,
лишенной С02. Получается серия стандартных растворов, каждый
из которых содержит СаС12 из расчета 45 г в литре и NaCl
в следующей последовательности: З-Ш-4, 1-10-3, 3-Ю-3,.
1 • Ю-2, 3-10-2, 1 • Ю-1 и 3-Ю-1 М. По этим растворам
градуируют электрод и строят градуировочный график в
координатах- Е — lg Cxai ■
Для приготовления водной вытяжки образцы засоленных
почв заливают водой в соотношении 1 : 5 и встряхивают
5 мин. Частицам почвы дают осесть, раствор над осадком
сливают в центрифужные пробирки и центрифугируют до
полного осветления при скорости 6—7 тыс. об/мин.
Отбирают 50 мл прозрачного раствора и переносят в
мерные колбы емкостью 100 мл, добавляют в каждую колбу по
25 мл исходного раствора СаС12 и доливают водой до метки.
После перемешивания раствор наливают в электродную'
ячейку и измеряют э. д. с. цепи. По графику находят
содержание ионов натрия в растворе.
Полученный результат выражает концентрацию ионоь
натрия в анализируемом растворе в грамм-молях на литр и.
численно совпадает с содержанием грамм-молей натрия
в 100 г почвы. Если вытяжку готовят при соотношении почвы
к раствору=1 : 5, то 100 г почвы отвечает 500 мл вытяжки;
последнюю разбавляют перед измерением активности в 2
раза, следовательно, 100 г почвы отвечает 1000 мл раствора.
Таким образом, если концентрация натрия в растворе
оказалась равной 2-10~2 М, то в 100 г почвы содержалось
2- Ю-2 г-моль натрия. Чтобы найти содержание натрия в
граммах на 100 г почвы, найденную величину надо умножить
на атомный вес натрия: 2 • 10~2 • 22,99 = 0,46 г/100 г почвы,
или 0,46%.
Чтобы ошибка определения не превышала 3—4%
(относительных), необходимо использовать потенциометр с
чувствительностью порядка 0,5 мв. Хорошие результаты
позволяет получить потенциометр ППТВ с ламповым усилителем
или баллистическая схема.
140
Определение воднорастворимого и поглощенного натрия
по Боуэру
Изложенный выше метод пригоден для почв с реакцией,
близкой к нейтральной. Если реакция сильно отличается от
нейтральной, то в качестве индифферентного электролита
применяют буферные растворы. В методе Боуэра роль
буфера и индифферентного электролита выполняет
Mg(CHsCOOJ. Измерения э. д. с. осуществляют на
ламповом потенциометре с чувствительностью до ± 1 мв.
Стеклянный электрод с натриевой функцией градуируют по
стандартам, содержащим 1,10 и 100 мг • же. Na+ в 1 л 1,0 н. раствора
Мё(СШСООJ.
Порядок определения натрия в водах
и водных вытяжках из почв
10 мл воды или водной вытяжки из почвы помещают в
стакан емкостью 50 мл. Добавляют 10 мл 2,0 н. раствора
Mg(CHsCOOJ и перемешивают. В раствор погружают
стеклянный и каломельный электроды и измеряют э. д. с. цепи.
Содержание ионов Na+ (в мг-экв/л) находят по градуиро-
вочному графику.
Порядок определения содержания
поглощенного натрия в солонцеватых почвах
5 г почвы помещают в круглодонную центрифужную
пробирку емкостью на 50 мл. Добавляют в пробирку 33 мл
1,0 н. раствора Mg(CHsCOOJ, закрывают пробкой и
встряхивают 5 мин. После встряхивания снимают пробку и
суспензию центрифугируют при скорости вращения
1500—2000 об/мин до полного осветления раствора.
Прозрачную жидкость сливают в мерную колбу на 100 мл, в
центрифужную пробирку снова доливают 33 мл 1,0 н. раствора
Mg(CHsCOOJ, встряхивают и центрифугируют; такую
обработку проводят 3 раза, сливая экстракты в мерную колбу.
По окончании экстракции объем раствора в колбе доводят
до 100 мл, добавляя 1,0 н. раствор М^(СНзСООJ.
Полученный раствор переносят в электродный сосуд и
измеряют э. д. с. цепи. Найденное по графику количество
ионов натрия равно сумме поглощенного натрия и воднора-
створимых форм его соединений. Вычитая из найденной
суммы количество натрия, входящего в состав воднораствори-
141
мых соединений, получают содержание поглощенного
натрия. Результат, как и при других потенциометрических
измерениях, выражен в единицах концентрации (мг- экв/л);
для пересчета на 100 г почвы необходимо знать соотношение
почвы и раствора. В данном случае 5 г почвы отвечает
100 мл раствора, следовательно, количество миллиграмм-
эквивалентов натрия в литре раствора соответствует
содержанию натрия в 50 г почвы. Для пересчета на 100 г почвы
полученный результат умножают на два.
Порядок определения емкости поглощения
(по натрию)
Взвешивают 5 г почвы и помещают в центрифужную
пробирку на 50 мл. Добавляют 33 мл 1,0 н. раствора
CH?,COONa с рН = 8,2, взбалтывают 5 мин и центрифугируют.
Раствор над осадком сливают и отбрасывают. Обработку
раствором СНзСОО№ повторяют 3—4 раза до полного
насыщения поглощающего комплекса ионами натрия. Почву,
насыщенную ионами натрия, отмывают в тех же пробирках
от избытка солей 95%-ным этиловым спиртом, обрабатывая
осадок порциями спирта по 33 мл и центрифугируя.
Отмывание продолжают до тех пор, пока сопротивление жидкости
над осадком не станет менее 40 мком/см (при 25°). Для
полного отмывания требуется 3—4 порции этанола по 33 мл.
Насыщенную натрием и отмытую от избытка CH3COONa
навеску почвы обрабатывают в той же центрифужной
пробирке 1,0 н. раствором Mg(CH3COOJ, как это принято при
определении поглощенного натрия. Собранные экстракты
доводят в мерной колбе до 100 мл, раствор переносят в
электродную ячейку и измеряют э. д. с. цепи с Ка-стеклянным
электродом. Умножив на два найденное количество
миллиграмм-эквивалентов натрия в литре раствора, получают
емкость поглощения (по натрию) почвы, выраженную
в мг -же на 100 г.
Бариевый метод определения поглощенного натрия
Простой и быстрый метод определения поглощенного
натрия предложен С. Н. Алешиным. Как и в предыдущих
вариантах, здесь использовано нивелирующее влияние
индифферентной соли на ионную силу раствора и величину
коэффициента активности.
По этому методу навеску почвы обрабатывают 1,0 н.
ранг
створом ВаСЬ, затем почву отфильтровывают и в фильтрате
определяют концентрацию ионов натрия при помощи
Na-стеклянного электрода. Градуировка электрода
осуществляется по растворам NaCl на фоне 1,0 н. раствора ВаС12.
В отличие от метода Боуэра в этом случае
индифферентный электролит не оказывает буферного действия, и поэтому
реакция почвенной вытяжки может влиять на результаты.
Другим недостатком метода следует считать применение
бариевых солей. Хотя ион бария отличается высокой сорб-
ционной способностью, что обеспечивает полноту вытеснения
поглощенных оснований, но в его присутствии образуются
труднорастворимые ВаСОз и BaS04 при наличии в почве
карбонатов или гипса. Пленки BaSC>4 обволакивают
почвенные частицы и препятствуют полному вытеснению ионов
натрия. Поэтому бариевый метод, видимо, наиболее пригоден
для бескарбонатных и безгипсовых почв.
Ход анализа
Навеску почвы в 5 г помещают в плоскодонную колбу
на 200 мл, приливают 100 мл 1,0 н. раствора ВаС12 и
встряхивают на ротаторе в течение 30 мин. Затем почву
отфильтровывают или отделяют от раствора на центрифуге, а
прозрачный раствор переносят в электродную ячейку и
измеряют э. д. с. цепи с Na-стеклянным электродом.
Содержание натрия находят по градуировочному графику.
Очевидно, что при расчетах необходимо ввести поправку
на то количество ионов натрия, которое находилось в почве
в форме легкорастворимых солей.
ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Потенциал электрода, обратимого к какому-либо виду
ионов, может служить индикатором при титровании ионов
данного вида. Методы потенциометрического титрования
имеют значительные преимущества в сравнении с обычным
титрованием, так как позволяют анализировать окрашенные
и мутные растворы, где невозможно применение цветных
индикаторов. Потенциалопределяющим может быть не только
определяемый ион, но и добавляемый при титровании или
один из продуктов реакции.
Кроме прямого аналитического назначения потенциомет-
рическое титрование часто используют для определения
свойств титруемых соединений, таких, как константы диссо-
143
циации, количество активных групп. Потенциометрическое
титрование слабых кислот сильным основанием и слабых
оснований сильными кислотами позволяет определить
концентрацию слабой кислоты (или слабого основания), что
нельзя сделать кондуктометрическим методом из-за
гидролиза соответствующих солей.
Используемая аппаратура та же, что и при любых потен-
циометрических измерениях, но она может быть значительно
упрощена. Для построения кривой потенциометрического
титрования достаточно знать, как изменяется потенциал
электрода в процессе титрования, но нет необходимости
точно определять абсолютные величины потенциалов.
Поэтому из схемы потенциометра можно исключить элемент
Вестона, а для построения графика ограничиться просто
показаниями приборов.
Результаты потенциометрического титрования обычно
выражают в координатах: потенциал электрода —
количество добавленного реактива. При титровании кислоты
щелочью по оси ординат откладывают величины рН;
перегиб на кривой титрования отвечает точке эквивалентности.
Значительно точнее точку эквивалентности находят по
дифференциальной кривой. Для построения последней обычный
график потенциометрического титрования делят по оси
абсцисс на небольшие отрезки Au = t/—v" (рис. 30, /). Затем
узнают, насколько изменилась величина рН (ДрН = рН| —
— рН2) при добавлении объема реактива, равного Af.
Результаты строят в виде нового графика (рис. 30, //), откладывая
по оси ординат приращение рН (ДрН), а по оси абсцисс объем
добавленного раствора, принимая, что для каждой точки
v' +V
v — -———.
2
Небольшие и постоянные изменения потенциала вдали от
точки эквивалентности находят выражение на графике в
виде прямой, практически параллельной горизонтальной оси
(ЛрН/Аи = const). Скачок потенциала в конце титрования
отражается на дифференциальной кривой в виде максимума.
Положение максимума точно совпадает с перегибом на
кривой титрования (рис. 30, /), поскольку до перегиба ДрН/Ди
увеличивается, а после перегиба — уменьшается.
Титруемый раствор помещают в стаканчик, закрытый
пробкой с отверстиями для бюретки, двух электродов и
мешалки (если в процессе титрования пропускается газ, то
мешалка заменяется стеклянной трубкой, через которую
144
в раствор поступает газ). Один из электродов является
индикаторным; это может быть стеклянный или хингидронный
электрод при кислотно-основном титровании, хлоросеребря-
ный электрод — при определении СГ-иона, платиновый
электрод— при титровании окислительно-восстановительных
систем. Второй полуэлемент — электрод сравнения; чаще всего
это каломельный электрод, хотя при
окислительно-восстановительном титровании иногда применяют и второй
металлический электрод (например, пара: платина — вольфрам). Элек-
Точка экВибалснт-
ности
Рис. 30. Построение дифференциальной кривой потенциометрического
титрования
троды соединены с входом потенциометра или через ключ и
гальванометр с реохордом, на который подано напряжение
от аккумулятора.
Техника титрования проста. После добавления
определенного объема реактива находят точку компенсации
(см. стр. 85) и записывают показания прибора (отсчет по
реохорду, или милливольты при работе с потенциометром).
Титрование проводят дважды; первый раз грубо, приливая
каждый раз большой объем реактива; второй раз титруют
вблизи точки эквивалентности осторожно, малыми порциями.
По графику находят объем реактива, пошедшего на
титрование.
Иногда используют упрощенные варианты схем, в
которых вместо съемки полных кривых ограничиваются
титрованием до какого-либо наперед заданного значения рН.
Если, например, при кислотно-основном титровании
используется хингидронный и каломельный электроды, то э. д. с. та-
145
кого элемента определяется уравнением
э. д. с.= е0хинг—0,0581 рН—екал,
или э. д. с. = 0,7027 в—0,0581 рН—0,2471 в = 0,4556 в—0,0581 рН,
т. е. направление тока в цепи меняется в зависимости от
величины рН. Если 0,4556>0,0581 рН, то хингидронный электрод
положителен и ток течет в одном направлении, а если
0,4556<0,0581 рН, —в другом. Когда 0,4556 = 0,0581 рН,
э. д. с. = 0. Иными словами, э. д. с. и ток в цепи равны нулю-
при условии, что величина рН раствора в хингидронном*
Рис. 31. Упрощенная схема для потенциометрнческого титрования:
1—гальванометр, 2 — ключ, 3 — хингидронный электрод, 4 —
каломельный электрод
электроде равна 0,4556:0,0581=7,84. Это позволяет
титровать до рН = 7,84 при помощи простой схемы (рис. 31),
включающей платиновый электрод в насыщенном растворе
хингидрона, каломельный электрод, ключ, гальванометр.
При замыкании ключа в этой цепи идет ток, обнаруживаемый
гальванометром. Титруя раствор, в который опущен
платиновый электрод, и замыкая ключ после каждого добавления
раствора щелочи, заметим, что сила тока постепенно
уменьшается, а когда рН станет равным 7,84, гальванометр
покажет нулевое значение. Дальнейшее прибавление щелочи
вызовет снова появление тока, но уже в обратном направлении.
Таким образом, изменение направления тока указывает на
точку эквивалентности для всех кислот, точка нейтрализации
146
которых лежит вблизи рН = 7,84. Этот метод приближенный,
так как точно отметить момент изменения направления тока
трудно. Кроме того, в области высоких значений рН сам хин-
гидронный электрод не дает устойчивых показаний.
Ту же схему легко применить в другом варианте,
использовав в качестве электрода сравнения вместо каломельного
электрода — хингидронный, приготовленный на буферном
растворе с точно известным и заранее определенным
значением рН. Перемена направления тока в этом случае укажет,
что рН титруемого раствора стал равным рН буферной смеси
электрода сравнения. Этим способом легко титровать
растворы до любого заранее заданного значения рН, располагая
только простейшим оборудованием — парой электродов и
гальванометром.
Определение рН почвенных суспензий методом
потенциометрического титрования «на нуль»
Упрощенная установка для потенциометрического
титрования может быть использована для нахождения рН
почвенных вытяжек и суспензий. Этот метод, предложенный
Л. И. Вигоровым, почти не уступает в точности обычному
потенциометрическому определению рН, но не требует
относительно сложного потенциометрического оборудования.
Сущность метода сводится к выравниванию потенциалов
двух хингидронных электродов, один из которых помещен в
испытуемый раствор. Второй электрод готовится на буферном
растворе. Если величины рН буферного и испытуемого
растворов совпадают, то сила тока в цепи становится равной
нулю.
Выравнивание потенциалов достигается титрованием
буферной смеси. Для этого в электродный сосуд наливают
определенный объем одного из компонентов буферной смеси
и добавляют хингидрон; в другой сосуд помещают
испытуемый раствор и также добавляют хингидрон. В оба раствора
опускают платиновые электроды и соединяют их через ключ
и гальванометр, растворы замыкают агар-агаровым сифоном.
При замыкании ключа стрелка гальванометра отклоняется.
Затем в первый сосуд добавляют из микробюретки второй
компонент буферной смеси до тех пор, пока потенциалы
электродов не сравняются и стрелка гальванометра не
перестанет отклоняться при замыкании ключа. Зная
соотношение компонентов буферной смеси, по табл. 14 находят
величину рН. Для титрования удобно пользоваться цитратно-
147
фосфатным буфером, позволяющим работать в интервале
рН от 2,4 до 7,8, перекрывающим реакцию большинства
почв В более узких интервалах рекомендуется ацетатный
буфер (рН 3,8—5,6) и фосфатный буфер (рН 5,5—7,7).
Ход анализа
В один из электродных сосудов наливают произвольный
объем почвенной суспензии или вытяжки, а во второй сосуд
отмеривают бюреткой точно 10 мл 0,1 М раствора лимонной
кислоты. В оба сосуда добавляют по 10—20 мг хингидрона,
погружают платиновые электроды и соединяют растворы
агар-агаровым сифоном. Замыкают ключ и наблюдают
отклонение стрелки гальванометра. Затем к лимонной кислоте
приливают фосфатный компонент буферной смеси @,1—0,2 мл
раствора Na2HP04), перемешивают и снова замыкают ключ.
Приливание раствора фосфата продолжают до тех пор, пока
сила тока в цепи не станет равной нулю В точке равенства
потенциалов записывают количество фосфата натрия,
добавленного к лимонной кислоте, и по табл 14 находят
величину рН.
Компонентами цитратно-фосфатного буфера служат 0,1 М
раствор лимонной кислоты B1,008 г лимонной кислоты в 1 л)
и 0,2 М раствор фосфата натрия C5,60 г Na2HP04-2H20
в 1 л). Для получения препарата с двумя молекулами воды
свежеперекристаллизованный двузамещенный
фосфорнокислый натрий сушат в термостате при 37° в течение двух
суток при периодическом перемешивании. После растирания
порошок хранят в склянке с притертой пробкой.
Таблица 14
Величины рН цитратно-фосфатного буфера. К Ю мл
0,1 М раствора лимонной кислоты добавлено п мл
раствора Na2HP04
п, мл
0,2
0,7
1,2
1,9
2,6
3,3
4,0
4,8
5,5
рН
2,2
2,4
2,6
2,8
3,0
3,2
3,4
3,6
3.8
п, мл
6,3
7,1
7,9
8,8
9,7
10,6
11,6
12,6
13,8
рН
4,0
4,2
4,4
4,6
4,8
5,0
5,2
5,4
5,6
п, мл
15,3
17,1
19,5
22,5
26,7
34,0
45,4
66,6
—
РН
5,8
6,0
6,2
6,4
6,6
6,8
7,0
7,2
—
148
Определение констант диссоциации слабых кислот
При титровании слабой кислоты происходит реакция:
НЛ + ОН- -* Н20 + А-,
где НА — органическая кислота, а А- — ее анион. В процессе
титрования гидроксильные ионы щелочи полностью
связываются, образуя воду, и в любой точке сохраняется
соотношение:
НА^А- + Н+
К _ -Ж+] [A_I
А [HAI '
где К— константа диссоциации кислоты.
Если принять во внимание гидролиз образующейся соли,
то можно считать, что концентрация соли равна
концентрации аниона кислоты [соль]=[А ], и тогда уравнение
константы диссоциации можно переписать в виде
к - -Ш+11соль1
А '"" [НА]
Логарифмируя, получим
lg^.-lg[H+)+lgrfCOTbJT
[кислота]
Если обозначить — lg К — рК,
то
[соль]
p^pH-lgr
[кислота]
Эта формула и служит для определения константы
диссоциации. В тот момент, когда концентрации соли и кислоты
одинаковы, т. е. [соль] = [кислота!, р^С=рН. Другими словами,
в точке полунейтрализации, когда оттитровано точно
половинное количество взятой кислоты, величина рН равна
отрицательному логарифму константы диссоциации кислоты.
Практически определение сводится к следующим
операциям. Раствор органической кислоты титруют щелочью и
строят график потенциометрического титрования. По графику
находят точку эквивалентности и узнают количество щелочи,
пошедшее на титрование. Затем по тому же графику
находят величину рН, соответствующую половинному количеству
149
щелочи, израсходованной на нейтрализацию кислоты.
Найденная величина равна отрицательному логарифму константы
диссоциации (рНу2 =р/().
При титровании очень слабых кислот возникает
трудность в определении точки эквивалентности. Поэтому для
уточнения берут среднее значение из 6—10 точек, лежащих
по обе стороны от предполагаемой точки эквивалентности.
При особо точных вычислениях необходимо учитывать
коэффициенты активности и константу гидролиза соли.
Потенциометрическое определение обменного А13+ в почвах
Определение алюминия методом потенциометрического
титрования описано В. М. Тараяном, а затем использовано
Черновым для анализа почв. Метод основан на титровании
раствора, содержащего ионы алюминия, фтористым натрием
в присутствии системы Fe3+/Fe2+. В результате реакции
образуется фторидный комплекс алюминия [Л1Рб]3-. В тот момент,
когда весь алюминий связан в комплекс, начинается
образование аналогичного фторидного комплекса железа (III),
вследствие чего резко изменяется ОВ-потенциал системы, что
легко фиксируется обычным потенциометром. Титрование
производят из микробюретки с ценой деления 0,02 мл,
изменения потенциала фиксируют посредством измерения э. д. с.
цепи, составленной из платинового и каломельного
электродов. Первый перегиб на кривой потенциометрического
титрования (рис. 32) отвечает концу титрования Л13+ и выражен
весьма резко.
Основным мешающим элементом является кальций.
Железо, обычно затрудняющее определение А13+ иными
методами, не только не мешает, но необходимо (и может быть
определено одновременно). В процессе титрования потенциал,
соответствующий равновесному значению, устанавливается
не сразу; после прибавления фтористого натрия наблюдается
резкое изменение потенциала вследствие связывания железа.
Спустя 0,5—1 мин в начале титрования и 5—10 мин вблизи
точки эквивалентности весь фтор связывается алюминием и
потенциал возвращается к прежнему устойчивому значению.
Титрование можно проводить в довольно широком диапазоне
рН, наилучшие результаты получаются при рН от 4 до 6,5.
Необходимым условием является удаление кислорода, что
достигается пропусканием из аппарата Киппа постоянного
тока СОг, который одновременно перемешивает раствор
(рис. 33).
150
Ход анализа
К раствору, содержащему от 0,2 до 20 мг алюминия,
добавляют 3,0 мл 0,1 н. раствора FeCl3 и 3—4 капли
насыщенного раствора FeS04 (при большом содержании в вытяжке
SOOr
Ш
300
ZOO
wo
К потенциометру
1,0
2,0 мл
Рис. 32 Кривая потенциометриче-
ского титрования ионов алюминия
в солевой вытяжке из
иллювиального горизонта
дерново-подзолистой почвы
ионов Fe3+ раствор хлорного
железа можно не добавлять).
Затем приливается равный
объем спирта и раствор
насыщают NaCl (или КО).
Пропускают СОг и через 5—8 мин
приступают к титрованию, анализ
ведут все время в токе СОг-
Титр фтористого натрия
предварительно устанавливают
при помощи стандартного раствора алюминия. Результаты*
титрования наносят на график и находят точку эквивалент*
ности.
Рис 33 Сосуд для потенцио-
метрического титрования в
токе СОг:
1 — платиновый электрод, 2 —
каломельный электрод, 3 —■
бюретка
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЙ ПОТЕНЦИАЛ ПОЧВ
И МЕТОДЫ ЕГО ИЗМЕРЕНИЯ
Окислительные процессы в почвах мало изучены, хотя им
принадлежит немалая роль в почвообразовании и плодородии-
почв. Окислительно-восстановительные реакции в почвах,
протекающие с участием микроорганизмов, происходят при
151
разложении органических остатков и синтезе гуминовых
кислот и фульвокислот, ими обусловлены превращения форм
соединений различных элементов: железа, марганца, азота в
почвах. При восстановительных условиях образуется ряд
легкоподвижных форм соединений марганца и железа, что
повышает их миграционную способность. Восстановительные
реакции могут способствовать также накоплению токсичных
для растений форм соединений. Известно, что большинство
культурных растений требует для нормального развития
оптимальных значений ОВ-потенциалов D00—600 же).
Измерение ОВ-потенциалов (обозначается ел) позволяет
суммарно определить напряженность
окислительно-восстановительных процессов в почвах и тем самым выявить общую
направленность окислительных процессов; ОВ-потенциал
может служить мерой потребности в мероприятиях,
регулирующих окислительный режим почв и газообмен.
Окислительно-восстановительная реакция происходит с
передачей электронов от восстановителя к окислителю:
Ox + e^Red,
где Ох — окислитель, Red — восстановитель и е — электрон.
Так, для системы ферри-ферро:
Fe3+ + e^!Fe2+.
Константа этой реакции, как и любой другой, может быть
выражена уравнением
(Red] '
Введение в систему дополнительного количества
электронов ведет к уменьшению "кониентратщи "окисленной и кТПтбвг-
шению содержания восстановленной фоЩТ^УШЕШяиёше кбн-
центрации^эле"к?р^нав~^ыз1^ает~ог)ратТГый эффект. Характер
и направление процесса в окислительно-восстановительной
реакции зависит, таким образом, от «равновесной
концентрации электронов», или, точнее, от того, насколько легко
восстановитель отдает свои электроны и насколько прочно эти
электроны связывает окислитель. Окислительные свойства
вещества или системы определяются, следовательно,
прочностью связи электронов с окисленной и восстановленной
формами. Это позволяет выражать
окислительно-восстановительные свойства любых систем при помощи одной и той
же меры — величины ОВ-потенциала.
152
ОВ-потенциал воспринимается электродом из
индифферентного металла (платины), погруженным в раствор,
содержащий одновременно окисленную и восстановленную
формы одного и того же вещества. Между платиновым
электродом и обратимой окислительно-восстановительной
системой устанавливается равновесие, определяемое относительной
прочностью связи электронов с восстановденной^фо^мой
вещества и с металлом (платина), из которого изготовлен
электрод. При увеличении положительного потенциала на
электроде восстановитель отдает часть электронов;
увеличение отрицательного заряда платинового электрода, наоборот,
вызывает переход электронов с электрода к окислителю.
Если платиновый электрод не связан с внешним
источником тока, то в растворе, содержащем обратимую
окислительно-восстановительную систему, он приобретает некоторый
потенциал, величина которого зависит от того, насколько
легко система отдает свои электроны. Тогда можно считать,
что потенциал электрода равен:
" ' nF [e'\ '
где [ем] — концентрация электронов (или упругость
электронного газа) в металле, [е'] — та же величина для окислительно-
восстановительной системы, которую можно вычислить, зная
константу окислительно-восстановительной реакции.
Если
Ox + e^Red,
то
К [Red]
W\ =
[Ох]
Подставляя это значение в уравнение электродного
потенциала, получим
eft = Eo + J^lni£JiM
h ° nF К [Red]
Вследствие большой емкости металлического электрода
величину [еы] можно считать постоянной. Объединяя ее и
постоянную К с константой уравнения, получим
- RT , [Ох]
nF [Red]
если коэффициенты активности равны единице.
153
Последнее уравнение и выражает величину ОВ-потенциа-
ла системы. Более высокий потенциал показывает, что
реакция
Red^Ox-fe
сдвинута в сторону образования окисленных форм.
Величина ОВ-потенциала зависит от соотношения
активных концентраций окислителя сох и восстановителя aRed,
поэтому сравнивать различные системы можно только при
одинаковых условиях. Основное условие вытекает из самого
уравнения. Когда ОТНОШеНИе GOx/^Red== 1j ТО И Е — Ео.
Потенциал системы, в которой активности окислителя и
восстановителя одинаковы и равны единице (е = ео),
называется нормальным потенциалом
окислительно-восстановительной системы (см. стр. 83).
Фактически свойства окислительно-восстановительной
системы, т. е. сродство восстановителя к электрону, определяются
величиной ео, объединяющей такие важнейшие показатели,
как К и [е]. Изменения в соотношении aox/«Red вызывают
только большие или меньшие отклонения потенциала от
величины е0. Знание величин нормальных потенциалов
позволяет сравнивать различные системы, предсказывать
направление реакций и подбирать необходимые окислители и
восстановители. Окислительно-восстановительная система с
более высоким (положительным) потенциалом является
окислителем по отношению к системе, нормальный
потенциал которой ниже (конечно, при прочих равных условиях).
Константу реакции взаимодействия двух
окислительно-восстановительных систем можно вычислить, пользуясь
величинами их нормальных потенциалов ко и во :
Б ч 0,0581
где К — константа окислительно-восстановительной реакций
при температуре 20°.
Кроме Е0 в уравнение потенциала входит другая постоян-
.ная величина — п, также зависящая от свойств системы;
п — это число электронов, участвующих в реакции, которое
в большинстве случаев находят как разность валентностей
окисленной и восстановленной форм. Когда п=1, то
численное значение коэффициента уравнения
2,303-^-^0,0581.
nF
154
При п = 2 коэффициент лменьшается вдвое и т. д.
Рассматриваемое уравнение выведено для простейшей
реакции, когда кроме окислителя и восстановителя в реакции
не участвуют другие компоненты (например, только
Fe3++el;Fe2+; Cu2+e^Cu+).
В наиболее общем случае под знаком логарифма
находятся равновесные концентрации всех участвующих в
реакции соединений. Часто, особенно при реакциях органических
соединений, окисление и восстановление происходит с
участием водородных ионов. Например, при окислении
двухвалентного железа кислым раствором перманганата реакция
идет по уравнению
5Fe2+ + MnOJ" + 8Н+ -> 5Fe3+ + Мп2+ + 4Н20.
Концентрация водородныл-лонов здесь оказывает решающее
влияние на характер—взаимодействия и величину ОВ-потен-
циала. Уравнение потенциала для системы
МпОГ + 8Н+ +5е = Мп2+ + 4Н20
записывается уже с учетом концентрации водородных
ионов
а -о"
RT , Мп04 Н+
ел = е0Н -in
Понятие нормального потенциала в этой реакции
несколько отличается от предыдущего; очевидно, что равенство
е = е0 справедливо, когда не только активные концентрации
окислителя и восстановителя равны единице, но и когда
активность водородных нонов также равна единице (точнее,
а _<*.
МпОл Н \
при 51—= 1 • В этом случае величина рН очень
амп2+ }
сильно влияет на ОВ-потенциал; в кислой среде при рН = 3
и ашпо—/амп2+= 1 получим
4
ей= 1,51+ °'°581 lgA0~3)8=l,23e и при рН - 5
5
ей= 1,04 в.
Поскольку активность водородных ионов в уравнении
потенциала входит в степени, равной числу ионов водорода,
участвующих в реакции (в данном случае=8), то очевидно,
155
что здесь изменение реакции среды значительно сильнее
влияет на окислительные свойства системы, чем концентрация
окислителя или восстановителя.
Для характеристики окислнтелыго-восстановнтельных систем кроме
величины ей часто используют и другую величину, обозначаемую гН2
(читается «эр аш», или «эр аш два»). Понятие гНг возникло в соответствии
с ранними представлениями об окислительно-восстановительных процессах,
как о реакциях присоединения или отдачи водорода. С этой точки
зрения напряженность окислительно-восстановительных процессов можно
характеризовать условным давлением газообразного водорода, которое
способно создавать такое же значение окислительно-восстановительного
потенциала, которое реально имеет место в изучаемой системе. Сводя
любой окислительно-восстановительный процесс к реакции
Н2 = 2Н+ + 2е,
можно записать, что е/, такой системы (при давлении водорода рн)
равен
2,3 RT , сн+
ел = е0+ Ig ,
nF Ph.
т. е. равен потенциалу водородного электрода, для которою е0=0.
Тогда
0,0581 °н+
ей = lg- (при 20°),
2 Рц,
0,0581 , 0,0581
eft = 2 !^ан+- % -IgpH,-
Давление водорода в такой системе равно
0,0581 0,0581 , „
lg РНа = еп — 9 !g аи+,
или
Но
2 &"н* " 2
— Ig Pu = — " lg aH-f-.
6 н* 0,029 н
f ' Eft
- lg aH+ = рН, тогда — lg %2 - -^-^ + 2рН.
Отрицательный логарифм давления водорода и
называют величиной гН2:
rH,= -lgPHj = ^+2PH.
156
Следует, конечно, помнить, что величина rll2 не выражает
зависимости между рП и ей системы, а лишь в некоторой степени отражает
суммарное влияние той и другой величины на напряженность
окислительных процессов. В ряде биологических объектов величина гН2 хорошо
коррелирует с интенсивностью окислительных процессов. В
действительности же эта величина лишена всякого физического смысла, что
нетрудно показать расчетом, и ее применение имеет только условное значение.
Можно вычислить, например, условное давление водорода гН2 в
нормальном хингидроином электроде, потенциал которого равен 0,690, а рН
в таком электроде равен нулю. Тогда
0,699
гН2 = —- ~24,
0,029
— lgp„ =24, а ри= 10"
-24
Теперь можно подсчитать, какое количество молекул водорода
создает такое давление в хиигидронном электроде объемом в 1 см3.
Каждая грамм-молекула газа при нормальных условиях занимает объем
22,4 л, и поскольку
PlVi
PlVl — P2.V2 И D2 = ,
Р-2
то при давлении в 10~~ одна грамм-молекула займет объем, равный
1-22,4
10_
«Ь= .п-24 =22,4-10» л.
В то же время каждая грамм-молекула вещества содержит
6,023- 10 23 молекул, следовательно, объем, приходящийся на одну
молекулу, составит 22,4 ■ 1024 : 6,023 • 1023=37 л.
Если приготовленный нами хингидронный электрод имеет объем
= 1 л«л=0,001 л, то в нем может находиться только 10— : 37 ~ 3-10~
части молекулы водорода. Этот абсурдный вывод показывает, что rll2
следует рассматривать как чисто расчетную величину, которая довольно
часто употребляется в биологии и почвоведении, но смысл которой
совершенно не соответствует первоначально данному определению.
Изучение окислительно-восстановительных процессов в почвах
должно базироваться на одновременном рассмотрении величин гн и рН, но
к вычислению гН2 следует подходить осторожно.
Роль водородных ионов во всех
окислительно-восстановительных процессах нельзя рассматривать одинаково, а
следует оценивать, исходя из конкретных условий реакции.
Здесь возможны три случая:
1) водородные ионы вовсе не участвуют в окислительно-
восстановительной реакции;
2) прямое участие водородных ионов в окислительных
процессах;
157
3) косвенное влияние водородных ионов.
Все три случая представлены в почвенных процессах.
Первый случай. Ионы водорода не участвуют, как.
правило, в окислительных реакциях простых ионов металлов-
и ряда анионов, как например:
Fe3+ + е ^ Fe2+, e0 = 0,77 в,
МпОГ + е ^ MnOTf е0 = 0,54 б,
Fe (CN)I- + е ^ Fe (CN)|_, e0 -= 0,36 в,
Cu2+ + e^?Cu+, е0 = 0,17в.
Во всех перечисленных случаях кн не зависит от рН, если
оба компонента смеси одинаково хорошо растворимы.
Второй случай. Реакции многих органических
соединений и некоторых анионов (кислородсодержащих) часто
сопровождаются образованием воды или слабодиссоцииро-
ванных кислот. В этих реакциях прямо участвуют водородные
ионы и величина ги зависит от рН. Степень влияния рН
определяется числом участвующих в реакции водородных ионов.
Примером таких окислительно-восстановительных систем
может служить хингидронный электрод (см. стр. 103) и
такие реакции неорганических веществ, как например:
Н202 + 2е + 2Н+ ^ 2Н20, е0 - 1,77 в,
МпОГ + 5е + 8Н+ ^ Мп2+ + 4Н20, е0 = 1,51 в,
N05" + 8е + 10Н+ ^ NHf + ЗН20, е0 = 0,87 в,
2Н+ + 2е ^ Н2 (газ), е0 = + 0,00 в.
Уравнение гн для таких систем записывается в общей
форме с участием т ионов водорода:
Ox + ne + mH+^Red и ел-е0 + —lg- °х п+
п aRed
, $ °Ох . ft , т . ft . «Ох .
п aRed n n aRed
+ —-lg%+.
158
По мере подкисления раствора вп таких систем
увеличивается. Если пит равны единице, то е& повышается на 58 мв
при уменьшении рН на единицу. В общем случае сдвиг
потенциала зависит от числа электронов и числа ионов
водорода, участвующих в реакции, и определяется
коэффициентом последнего уравнения:
■&т 2,303 RT __ т 2,303 ^Г _ т n nt-01
п nF n F n
При окислении перманганатом сдвиг потенциала равен
0,0581 - —; для реакции NOr+8e+10H+^NH| + 3H2O он
5
равен 0,0581 • — и т. п.
^ 8
Третий случай. Косвенное влияние водородных
ионов на величину гн наблюдается в результате изменения
растворимости компонентов системы при подкислении или
подщелачивании раствора. Характерным примером для почв
может служить равновесие в системе ферри-ферро. Допустим,
что одновременно имеется 0,1 н. раствор закиси и окиси
железа. Величина е& определяется уравнением (если
коэффициенты активности близки к единице)
е = е0 + 0,0581 lg l^-] .
[Fe2+]
Если рН раствора довести до 6,0, то количество ионов
железа в растворе можно вычислить из произведений
растворимости:
[Fe2 +] • [ОН-]2 = 4,8-10-16 и [Fe3+] • [ОН-]3 = 4-10-38.
При jqF[=j6,0 концентрация [ОН~] = Ю-8, тогда
гс 24-1 4,8 Ю-16 4,8-Ю-16 . с..
[Fe2+] = — = = 4,8М
[ОН-]2 КГ16
4. I Г)—38 а 1 г»—38
[Fe3+] = -^ = -^ = 4- Ю-14 М,
[ОН-]* Ю-24
т. е. ионы Fe2+ находятся в растворе, тогда как ионы Fes+ —
в осадке. Потенциал фактически определяется только [Fe2+1,
если в р_астворе нет органических rphtpptr, сохраняющих
подвижность Fe3+. При нодкислении до рН=ЛД) количество
159
ионов Fe2+jB_j>acjBope не изменится, а гидроокись железа
полностью растворится. В результате получим, что [Fe2+J=.
={Fe+3], а ел = Ео. "
Можно ожидать также значительных изменений
потенциалов в присутствии комплексообразователей, избирательно
связывающих окислитель или восстановитель. Влияние
комплексообразователей, как и рН, широко распространено
в почвах, богатых подвижными органическими соединениями.
Разберем простой случай образования комплексных ионов
Fe(CNN~ и Fe(CN)eT. Нормальный ОВ-потенциал системы
Fe(CNN~7Fe(CNN~ = +0,36 в, тогда как для системы
Fe34/Fe2+ — ео=0,77 в. Снижение нормального потенциала
вызвано неодинаковой прочностью комплексных соединений. Это
подтверждает следующий расчет.
Константы нестойкости двух комплексных ионов железа
соответственно равны:
^ [РеЗ+Исм-)в ^5-10^ и *,= J*?iH2nL=5.l<H*
[Fe(CN)|-] [Fe(CN)^-]
Ионы CN~ для обеих систем общие, поэтому можно
приравнять
Xi[Fe(CN)g-l ^ JC,[Fe(CN)g-l
(Fe3 + 1 ' lFe2+l
Поскольку константы нестойкости очень малы, то
концентрация i[Fe(CNN_], как и [Fe(CN)<5~], практически равна
общему содержанию вещества, а для нормальной системы
[Fe(CN)!~)=[Fe(CNJ-l.
При таких условиях
IFe3+L ^ Jk. = 5-ю~" = ю-7
[Fe2+] ^2 5-Ю-37
Найденное отношение концентраций простых ионов трех-
и двухвалентного железа можно подставить в обычное
уравнение потенциала для системы ферри-ферро.
8 ^ е0 + 0,0581 lg |Fe3+1 =- + 0,77 + 0,0581 lg 10~7 =
[Fe2+]
== +0,77—0,41 - +0,36 e.
В результате получили значение потенциала, характерное
для нормальной окислительно-восстановительной системы
цианидных комплексов железа. Ясно, что окислительно-вос-
160
становительная способность ионов железа в такой системе
не меняется, а нарушается только отношение концентраций
окисленных и восстановленных форм. Для данного случая
нормальный потенциал комплексной системы можно
вычислить по уравнению
Эти зависимости имеют большое значение в оценке
почвенных процессов. Зная константы нестойкости почвенных
комплексных органических соединений с ионами железа,
меди, марганца, можно предсказывать динамику ОВ-потен-
циала. В то же время этот метод позволяет определить
характер соединений, образуемых элементами переменной
валентности с органическими веществами почвы.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ РЕАКЦИИ
И ПОТЕНЦИАЛЫ В ПОЧВАХ
Особенностью почвенных условий является необратимость
большинства реакций окисления и восстановления.
Обратимые реакции, которые подчиняются разобранным выше
теоретическим положениям, свойственны только некоторым
почвенным системам при благоприятных условиях:
окислительно-восстановительному равновесию в почвенном растворе
ионов двух- и трехвалентного железа, разных форм
соединений марганца. Важнейшим и наиболее сильно действующим
агентом окисления в почвах служит кислород. С другой
стороны, восстановительные процессы обязаны преимущественно
микробиологической деятельности. Поэтому общая
направленность процессов, напряженность
окислительно-восстановительного взаимодействия, а следовательно, и величины
ОВ-потенциалов складываются преимущественно под
воздействием двух последних факторов и зависят от
интенсивности развития в почвах биологических процессов, с одной
стороны, и концентрации кислорода, определяемой степенью
аэрации почвы, с другой.
В нормально аэрируемых почвах ОВ-потенциал варьирует
в пределах 300—650 мв. Колебания е/, в течение года для одной
и той же почвы часто превышают различия в окислительных
потенциалах важнейших почвенных типов (их средние
значения). Поэтому трудно установить какие-либо характерные
показатели для почв, развивающихся в плакорных условиях.
Резко отличаются только почвы, образующиеся при избыточ-
6 Зак. 51
161
ном увлажнении. Заболачивание и оглеение снижают е^ до
величин, меньших 200 мв.
Направленность окислительно-восстановительных
процессов обусловливается рядом факторов и прежде всего
степенью аэрации, зависящей от порозности, влажности и
температуры почвы. Особенно сильно препятствует
поступлению кислорода почвенная корка, в результате образования
которой после дождя величины ел снижаются на 200—300 мв.
Развитие биологических процессов, как правило, также
снижает потенциал вследствие потребления кислорода
микроорганизмами и корнями высших растений.
В любой почве имеется большое количество
разнообразных окислительно-восстановительных систем. Заметное
влияние на величину ОВ-потенциала оказывают только те из них,
которые обладают большой емкостью, т. е. такие, содержание
которых достаточно велико (кислород, двух- и трехвалентное
железо, органические вещества). Многие системы сами не
могут значительно повлиять на е/,, но играют важную
физиологическую роль. Преобладание окислительных или
восстановительных форм этих соединений определяется ходом
окислительно-восстановительных процессов и величиной ей.
Можно указать на следующие, часто встречающиеся в
почвах, пары окисленных и восстановленных компонентов:
С02 —CH,; ШГ — N07 — NH,; SOt~H2S; PO4-—РН3;
Мп2+ — Мп3+ — Mn<+; Cu+ — Си2"*; Со2+ —Со3+.
В наибольшей степени от величины еп в почвах зависит
судьба соединений азота, железа, марганца и серы. Фиксация
азота азотобактером снижается при увеличении содержания
в почвенном растворе веществ с высоким окислительным
потенциалом. Процессы нитрификации, денитрификации и
аммонификации идут только при определенных
окислительных условиях. Сердобольским были намечены условия
развития процессов денитрификации и условия нахождения разных
форм соединений азота на черноземах, рендзинах и
подзолистых почвах:
ен
Преобладающие формы
соединений азота
> 480 же
Нитраты
480—340 мв
Нитраты—
нитриты
340—200 мв
Нитриты
< 200 мв
Окислы
азота,
молекулярный азот
162
Соединения серы в почвах обычно представлены
сульфатами. В растительных остатках, поступающих в почву, кроме
сульфатов содержатся органические соединения, в которые
сера входит в виде сульфгидрилыюй группы — SH-. При
разложении органических остатков в условиях высоких Eh
образуются сульфаты, в анаэробных условиях — сульфиды.
Превращения соединений серы полностью обусловлены
окислительно-восстановительными условиями. Так, сульфиды
могут окисляться серобактериями, а сульфаты
восстанавливаются сульфатредуцирующими бактериями по схеме:
so!- ^ s4c|~ 7? s2c|- ^ s ^ sh-.
Значительный материал накоплен по содержанию в
почвах соединений железа и марганца разных степеней
окисленное™. Железо в почвенных условиях легко образует как
двух-, так и трехвалентные соединения. Это обусловлено тем,
что нормальный потенциал системы Fe3+/Fe2+=0,77 в близок
к реальным почвенным условиям. Изменение рН и наличие
комплексообразователей в почвах, как было показано выше,
может резко изменить соотношение ферри-ферро даже при
высоких потенциалах. Снижение потенциалов до 200—250 мв
и ниже, что имеет место при заболачивании, ускоряет
переход железа в закисную форму. Усиление аэрации при
обсыхании почв морфологически проявляется в виде пленок,
конкреций, прослоек и линз окисных соединений железа.
Высокий окислительный потенциал может быть причиной
недостатка подвижного железа в карбонатных почвах и
хлороза растений.
Сердобольский предложил уравнение, связывающее
величину ед почвы и содержание закиси железа:
eft= 1,112 +0,1451/Г—0,174 рН —0,0581 lg[Fe2+],
где Г— ионная сила раствора.
Марганец в почвах присутствует только в низших
степенях окисленности. Соединения шести- и семивалентного
марганца очень сильные окислители с потенциалами порядка
1,2—1,6 вив почвенных условиях неустойчивы. Для почв
характерны соединения двухвалентного марганца и
четырехвалентного (Мп02 — пиролюзит); последний легко
образуется путем окисления неустойчивых соединений Мп3+.
Количество двухвалентного марганца, по Е. А. Яриловой, связано
с величиной ОВ-потенциала уравнением
е„ = 0,991 + 0,0581 у/Т — 0,116 рН — 0,029 lg [Mn»+].
6'=
163
Исходя из этих положений, Сердобольский определил
граничные условия нормального питания высших растений
марганцем, железом и нитратами в зависимости от величин
рН и 8/,. На графике (рис. 34) прямоугольник АВСД
охватывает условия, обычные для большинства культурных почв.
В пределах графика с помощью приведенных выше уравне-
70S
600
500
4-00
300
гоо
\л
в
- м —
А
\/г
cVVvv \
AXXXxV
VyvSAXXX
лЛЛфЛсххХ
i
Ш^\
с
N
п\ \
N£3"
N0,"
6
рН
8
Рис 34. Граничные условия нормального питания растений
(по Сердобольскому):
£^23 — область нормального снабжения железом и
марганцем,
gsgggj — область нормального снабжения желе-
628283 зом, марганцем и нитратами
ний можно нанести изоплеты (линии равных концентраций)
Мп и Fe. Прямая RS ограничивает область нормального
снабжения растений марганцем. Если сп и рН таковы, что
условия питания попадают в область, расположенную выше
и правее прямой RS, то растения могут испытывать
марганцевое голодание. Аналогично прямая KL ограничивает
область нормального обеспечения железом. Линия МЛ',
проведенная на уровне 0,34 в, отделяет область усиленной
денитрификации. Таким образом, наиболее благоприятные
для питания марганцем и железом условия ограничиваются
164
заштрихованной площадью, а с учетом денитрификационных
процессов — площадью, отмеченной двойной штриховкой.
Одной из причин резких изменений еь в почвах является
низкая буферная способность и емкость
окислительно-восстановительных систем в почве. Буферность (или обратное
ей понятие — податливость) в отношении ън можно оценить
по изменению гп при 'добавлении к
окислительно-восстановительной системе окислителя или восстановителя. Если В —
концентрация окисленных и восстановленных форм в
окислительно-восстановительной системе, а X— концентрация
окисленной формы, то потенциал системы равен
2,3 ЯГ. X
пг В— X
После добавления к системе исчезающе малого
количества dX эквивалентов окислителя потенциал увеличивается до
e/i + deh:
, , 2,3RT , X + dX ,
h nF B — X
Дифференцируя первое уравнение, получаем:
deh 2,3RT / 1 , 1 \ 2,3RT В
^ 2^3 RT (_
nF \
dX nF \X B — X ) nF X(B--X)
Здесь величина deu/dX— податливость
окислительно-восстановительной системы, a dA7de=p— мера ее буферное™-.
o___nF_ j* (В — Х)_
2,3 RT В
Максимальное значение буферность, или величина р,
приобретает, когда производная р по X равна нулю. Для этого
дифференцируем р по X:
сф nF ( В — X X
dX 2,3RT
если
или
/ В — X __ Х\
\~ в ' в Г
d^
dX
= 0, то
В
в — х х
в в '
X
в
о,
165
тогда
В — Х = Х и В = 2Х, а Х = —.
2
Расчет приводит к важному выводу, что максимальная
буферность системы наблюдается при равных активностях
окислителя и восстановителя в системе. В почве отношение
[Ox]/[Red] редко может быть равно единице, а концентрация
системы в целом низкая, поэтому почва чрезвычайно
податлива к изменению e/t и снижению его величины при
увлажнении (как говорят, к закисанию). Повышение содержания
гуминовых соединений повышает буферность почвы."Буфер-
ность-почв по отношению к ОВ-потенциалу мало изучена, но
она является важной характеристикой почвы.
ЭЛЕКТРОДЫ И СХЕМЫ ДЛЯ ОПРЕДЕЛЕНИЯ
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫХ ПОТЕНЦИАЛОВ
В ПОЧВАХ
ОВ-потенциал в почвах можно измерять любым
индифферентным металлическим электродом, т. е. электродом,
изготовленным из вещества, хорошо проводящего электрический
ток, но химически не реагирующего ни с одним из
компонентов почвы. В качестве такого электрода используют металлы
с достаточно высокими значениями нормальных электродных
потенциалов. Обычно берут платиновые электроды.
Поверхность электрода должна быть гладкой, желательно
отполированной; площадь электрода выбирается достаточно
большой (порядка 25 мм2), чтобы на показаниях не сказывалась
неоднородность состава и строения почвы. Платиновая
пластинка впаивается в стеклянную трубку и в таком виде
погружается в почвенный образец (рис. 35). Электродом
сравнения служит каломельный электрод.
При желании определить локальное значение потенциала
берут электрод малой площади или тонкую, достаточно
прочную платиновую проволоку. Значительное увеличение
поверхности платиновой пластинки повышает ее
электрическую емкость, что может неблагоприятно отразиться на
измерении гн почвы, так как большая емкость платинового
электрода ведет к переходу значительного количества
электронов из слоя почвы к электроду (или обратно) и
искажает истинную величину ън. Для восстановления
равновесия потребуется время, чтобы вблизи электрода снова
установился первоначальный потенциал.
166
При голевых определениях удобны платинированные
«стеклянные электроды (рис. 35,Б), на их изготовление
расходуется небольшое количество платины, что позволяет
значительно экономить дорогостоящий металл. Из одного грамма
хлорной платины можно
получить до 200 электродов.
Платинированные
электроды, как это описали
В. А. Рабинович и О. В. Ку-
ровская, изготавливают
следующим образом. Канифоль
растворяют в скипидаре (по
весу 1:1) при нагревании
на песчаной бане; канифоль
вносят небольшими
порциями при постоянном
помешивании. После охлаждения
полученную маслообразную
коричневую жидкость
смешивают с равным
количеством спирта. Затем 1 г
хлорной платины растворяют в
12 мл спирта, добавляют
равный объем насыщенного
■спиртового раствора борной
кислоты и полученный
раствор смешивают с 25 мл
раствора канифоли. Эту
-смесь используют для
платинирования.
Электроды изготовляют
■из стеклянных трубок,
диаметр и толщина стенок которых выбираются произвольно, но
так, чтобы трубки обладали достаточной механической
прочностью. Удобны толстостенные трубки внешнего диаметра
4 мм и диаметра отверстия 2—3 мм. Конец стеклянной
трубки погружают на I—2 см в жидкость для платинирования и
затем обжигают в коптящем пламени газовой горелки.
Трубку при этом держат в наклонном положении (платинируемым
концом вниз) и нагревание начинают несколько выше границы
жидкости, постепенно вводя в пламя всю платинируемую
поверхность. После того как растворитель испарится и на
внешней поверхности появится блестящий слой восстановленной
платины, продолжают обжиг в окислительном пламени горел-
Рис. 35. Электроды для определения
окислительно-восстановительного
потенциала почв:
А — платиновые; Б —<
платинированные: /—стеклянная трубка, 2 —
платиновое покрытие, 3 — ртуть,
4 — пробка из менделеевской
замазки, 5 — ватная пробка
167
ки, пока вся внутренняя поверхность не покроется
равномерным серым налетом. Трубке дают остыть, снова погружают
в раствор для платинирования и повторяют обработку, пока
вся нижняя поверхность не будет покрыта ровным слоем
металлической платины. Платинированная поверхность должна
быть ровной и блестящей. При слишком быстром, сильном и
неравномерном обжиге поверхность может получиться
матовой, пузырчатой; такие электроды бракуют.
Конец платинированного отверстия заливают
менделеевской замазкой или битумом. Для этого подогретый
платинированный конец погружают в расплавленную менделеевскую-
замазку и быстро вынимают. По охлаждении образуется
прочная пробка. Внутрь электрода наливают немного ртути,
в которую погружают медную проволоку для соединения с
потенциометром. Верхнее отверстие закрывают ватной
пробкой и также заливают менделеевской замазкой. В таком виде
электрод готов к употреблению (рис. 35).
Платинированный конец электрода можно не заливать
замазкой, а запаять. При осторожном вращении конца
трубки в окислительном пламени горелки стенки трубки
постепенно размягчаются, внутреннее отверстие сужается*
а затем исчезает. На месте отверстия остается узкая
прожилка металлической платины, осуществляющая электрический
контакт между внешней и внутренней платинированной
поверхностями. Эти операции требуют известного навыка в.
стеклодувном деле. Если неправильно выбран сорт стекла и
нарушен режим охлаждения готового электрода, то
платинированные головки лопаются и отваливаются от электрода..
В полевых условиях применимы портативные
каломельные электроды сравнения Крюкова. В этих электродах
контакт с почвой осуществляется через асбестовую нить,,
впаянную в конец стеклянной трубки. Электроды имеют
сравнительно небольшое омическое сопротивление и легко
погружаются в почву. Устройство и изготовление таких
каломельных электродов ясно из рис. 36.
Каломельный электрод перед измерениями погружают в
почву возможно ближе к платиновому. Это позволяет
уменьшить сопротивление элемента и повысить чувствительность.
Если электроды приходится опускать на большую глубину,,
то для соединения их с каломельными пользуются
удлиненными агаровыми сифонами.
Достоверные сведения о величинах ОВ-потенциалов в
почвах можно получить только при полевых измерениях со
стационарно расположенными электродами. Погружение
168
электродов в почву нарушает естественное сложение
последней: по скважине, в которой расположен электрод, проникает
атмосферный воздух, содержание кислорода и влажность
•около электрода меняются. После усадки почвы условия
постепенно возвращаются к нормальным.
В связи с этим однократное
измерение потенциала сразу после
погружения электрода в почву не
рекомендуется; оно не дает уверенности в точности
•измерения.
Полевые измерения е^ по
генетическим горизонтам проводят в двух
вариантах. В первом случае специальным
тонким буром («иглой») делают узкие
скважины до глубины, на которой
намечено измерение потенциалов. В
скважины плотно вставляют
электроды, стараясь погрузить платиновый
или платинированный электрод в дно
скважины. При этом нельзя прибегать
к значительным усилиям во избежание
механических повреждений
электродов. Стенки скважины с поверхности
слегка обжимают, предотвращая тем
самым поступление атмосферного
воздуха в глубь почвы. Если почва
слишком сухая, ее можно слегка смочить.
Через один-два дня приступают к
измерениям ОВ-потенциалов. Для этого
рядом с электродом погружают в
почву каломельный полуэлемент и
соединяют оба электрода с потенциометром.
При измерениях в слое 15—20 см и
глубже в почву погружают агаровый
сифон (через отдельную скважину),
одно из плеч которого увеличено до
требуемой глубины.
Во втором варианте проводят
разовые измерения е^, сопровождающие
крупномасштабную почвенную съемку. Каломельные и
платиновые (или платинированные) электроды устанавливают на
ступеньках, вырезанных -в стенке почвенного разреза по
генетическим горизонтом. Платиновые электроды погружают
з почву за 15—20 мин до начала измерений, чтобы в системе
Рис. 36. Каломельный
электрод Крюкова:
/ — каломель, 2 — ртуть,
3 — платиновый контакт,
4 — ватная пробка, 5 —
насыщенный раствор
КС1, 6 — пробка, 7 —
кристаллы КО, 8 —
асбестовый фильтр, 9 —
асбестовая нить
169
почва — электрод установилось равновесное состояние.
Измерения проводят несколько раз. За истинный отсчет принима -
ют тот, который повторился подряд три раза в течение часа
Желательно дополнительно проверить e/t через один-два часа.
Относительно быстро постоянство ОВ-потенциала
устанавливается во влажной почве. Сухая почва непригодна для
измерений.
Для лабораторных определений почвенные образцы берут-
в алюминиевые стаканчики или стеклянные широкие бюксы
с плотно закрывающимися крышками, которые
предотвращают газообмен и потерю влаги. С доставленных в
лабораторию образцов осторожно снимают верхний слой и
электроды погружают непосредственно в почву, оставшуюся в
стаканчике.
Лабораторные опыты по изучению влияния
ОВ-потенциала на развитие микроорганизмов, разложение органического-
вещества, переход различных элементов из одного
окислительного состояния в другое и т. п. ставят обычно так, что в
течение всего опыта электроды находятся в среде, потенциал
которой регулярно измеряется. Даже кратковременное
извлечение электродов серьезно нарушает равновесие,
установившееся на поверхности электрода, и вносит ошибки в
результаты наблюдений.
Окислительный режим в почвенной толще отличается
большой неоднородностью; часто на расстоянии всего
нескольких миллиметров величины потенциалов меняются
скачкообразно; внутри и на поверхности агрегата, как
показано Сердобольским, может быть иное значение е/4. Это*
заставляет все измерения осуществлять минимум в трех
повторностях, что удобно делать при помощи трех
электродов, смонтированных вместе.
Для измерения ОВ-потенциалов в буферных (по
отношению к ОВ-потенциалу) системах пригодна любая известная
потенциометрическая схема (см. стр 86) Почвы
отличаются, наоборот, очень невысокой буферностыо, в связи с чем
прохождение тока через почву, даже только в момент
измерения, сильно влияет на окислительно-восстановительную*
систему: если не предпринять специальных мер, то потенциал
«ползет» во времени Во избежание этого применяют схемы,
ограничивающие количества электричества, протекающего в
цепи. Таким прибором является любой ламповый
потенциометр (см. стр. 128) или видоизмененная схема Поггендорфа
с конденсатором. Для ограничения токов в схему
Поггендорфа вводится дополнительно конденсатор возможно малой/
170
«емкости (см. рис. 26). При замыкании цепи ключом К
проходящий ток заряжает конденсатор С; второе положение
ключа («разомкнуто») позволяет конденсатору разрядиться
на гальванометр. Таким образом, в отличие от обычной
схемы отклонение гальванометра наблюдается не при замыка-
«нии ключа, а при его размыкании. Время заряда
конденсатора выбирают наименьшим, но достаточным для того, чтобы
отклонения гальванометра были хорошо заметны. По мере
приближения к точке компенсации время заряда
увеличивают. В остальном техника работы на конденсаторной схеме
ничем не отличается от работы на обычной компенсационной
схеме. Такое же изменение схемы легко осуществимо в любом
потенциометре.
Вычисления ОВ-потенциалов
Э. д. с. цепи, составленной для измерения ОВ-потенциала
почвы, равна алгебраической сумме скачков потенциалов
платинового электрода гн и электрода сравнения екал:
Э. д. с. =(ей) + (екал).
В этой сумме знак плюс имеет потенциал с большим
абсолютным значением, а знак минус — потенциал меньшей
величины. Знаки соответствуют полюсности электродов в
компенсационной схеме: электрод, соединенный с
положительным полюсом аккумулятора, имеет большее абсолютное
значение и ему приписывается положительный знак. В реальных
цепях могут быть два случая. Первый—платиновый электрод
соединен с положительным полюсом аккумулятора и имеет
знак + , а каломельный— (минус).
Тогда
э. д. с. = eh — екал,
и
ел = екал + э. д. с.
Бторой случай — платиновый электрод ' соединен с
отрицательным полюсом аккумуляторов и имеет знак минус, тогда
э. д. с. = — Eh + екал и Eh = екал — э. Д. с.
1 Правильное положение полюсов определяется по наличию точки
•компенсации. Если на всем протяжении реохорда найти точку
компенсации не удается, то электроды необходимо поменять местами.
171
Потенциал каломельного электрода равен примерно
250 мв, поэтому перемена полюсов происходит, когда ОВ-по-
тенциал оказывается меньше этой величины, что характерно
для почв, в которых развивается глеевый процесс. При всех
измерениях ОВ-потенциалов обязательно записывают знак
платинового (окислительно-восстановительного)' электрода.
Глава IVi
ОПРЕДЕЛЕНИЕ СОЛЕСОДЕРЖАНИЯ В ПОЧВАХ
МЕТОДОМ КОНДУКТОМЕТР И И
Широкое распространение засоленных почв в южных
районах нашей страны ставит как одну из важнейших
практических задач определение солесодержания в почвах в связи
с их освоением и мелиорацией. Степень засоленности часто
является решающим фактором при отборе новых земель и
выборе наиболее рациональных методов орошения. На
культурных орошаемых массивах контроль за динамикой солей в
почвенном профиле необходим для проведения
профилактических мероприятий по предупреждению вторичного
засоления. ч
В зависимости от природных условий количество и состав
солей, накапливающихся в почвенном профиле, различны.
Общая схема соленакопления в почвах разработана Ковдой.
Его исследования показали, что состав и содержание солей
вполне закономерно меняются по мере увеличения
минерализации грунтовых и почвенных вод.
По химическому составу солончаки делят на хлорид-
ные, сульфат но-хлоридные, натриево-каль-
ц и е в ы е и т. п. Эквивалентное отношение С1_ : SCu- в
водной вытяжке хлоридных солончаков >4, сульфатно-хло-
ридных—1—4, хлоридно-сульфатных — 0,5—1, сульфатных —
<0,5. Разнообразие состава солей в солончаках и засоленных
почвах очень велико, что является основной причиной,
затрудняющей применение кондуктометрии для определения
степени засоленности почв.
Классический метод характеристики солесодержания по
173
величине плотного остатка имеет два существенных
недостатка. Прежде всего, при фильтровании водных вытяжек в
фильтраты часто переходят органические и минеральные
коллоиды, освободиться от которых простыми способами не
всегда возможно. Кроме того, фильтрование и выпаривание
требуют значительных затрат времени и энергии. Поэтому
был предложен ряд методов быстрого физико-химического
определения солесодержания, осуществимых в полевой
(экспедиционной) обстановке. Наибольшего внимания среди
них заслуживают приемы, основанные на измерении
электропроводности. Попытки разработать метод прямого
определения путем измерения электропроводности почвы не
увенчались пока успехом, поскольку электропроводность почвы
зависит не только от солесодержания, но и от влажности и
общих свойств почвы. Вполне приемлемым оказался другой
вариант — определение солесодержания по
электропроводности водной вытяжки из почвы, и на этой основе были
созданы приборы для быстрых полевых определений,
получившие название солемеров.
Кондуктометрический метод имеет существенные
недостатки в силу неодинаковой электропроводности различных солей
и весьма изменчивого солевого состава почв, однако его
простота и большая экономия времени дают такие
преимущества, что солемеры с успехом применяются во многих
(преимущественно полевых) исследованиях.
Кондуктометрическим методом измеряют удельную
электропроводность %, которая зависит от концентрации
электролита, степени его диссоциации и скорости движения
ионов в электрическом поле (см.гл.I):
%=--Ca(u + v)-F,
где С — концентрация электролита в г • экв/мл, а — степень
диссоциации, F — число Фарадея, и и v — скорости движения
катионов и анионов при напряжении электрического поля в
1 в/см. Обозначив эквивалентную электропроводность Л,
получим
х = сл.
Последнее уравнение используют для определения
солесодержания, но оно пригодно лишь при известных значениях
эквивалентной электропроводности. Поскольку водные
вытяжки из почв содержат различные соли и часто в
сложных сочетаниях, то прямое использование эквивалентных
элсктропроводностей становится невозможным. Если в разбав-
174
ленных растворах эквивалентные электропроводности солей,
встречающихся в почвах, относительно близки, то в более
концентрированных растворах различия достигают слишком
больших величин, чтобы ими можно было пренебречь
(табл. 15).
Поэтому М. А. Берлинер и Н. Н. Долгополов пришли к
правильному решению, что следует проводить эмпирическую
градуировку солемеров для работы в пределах одной
почвенной провинции (районе) с примерно одинаковым составом
солей. При градуировке солемера на почвах Кура-Араксин-
ской низменности ими были получены следующие
характеристики:
Интервал солесодержания, % . . . . . 0.1—1 1—3 3—10
Прирост электропроводности, ом. -10~4 • •-28 24 15
Таблица 15
Эквивалентная электропроводность водных растворов электролитов
различных концентраций при 25"
Электролит
NaCl
KCl
MgCl2
СаС12
Na2S04
CaS04
MgS04
NaHC03
Ca(HC03J
Na2C03
Электропроводность для концентраций, выраженных в г-экв/л
0
126,5
149,9
129,4
135,8
129,9
139,3
123,2
94,6
104,0
122,1
0,0005
124,5
147,8
125,6
131,9
125,7
127,2
123,2
94,4
102,3
0,001 0,005
123,7
147,0
124,1
130,4
124,2
121,4
117,6
93,5
99,3
120,7
143,6
118,3
124,3
117,2
100,0
98,8
90,3
91,7
110,7
0,010
118,5
141,3
114,6
120,4
112,4
90,0
88,9
88,1
87,0
108,0
0,50
111,1
133,4
103,1
108,5
97,8
66,5
80,6
93,6
0,100
106,7
129,0
97,1
102,5
90,0
57,8
76,1
85,3
0,500
93,3
84,0
40,7
63,4
1,00
73,5
77,5
33,6
53,4
Составление градуировочных кривых для различных типов
почв позволит широко внедрить солемеры в практику
почвенных исследований.
Значение кондуктометрического метода не ограничивается
только определением солесодержания в почвах. Значительно
большие возможности имеет этот метод в экспериментальном
почвоведении. Кондуктометрические измерения
целесообразно ставить для контроля в опытах по промывкам засоленных
почв, при изучении диффузии солен в почвенном профиле
и т. п.
175
Кондуктометрические работы выполняют в помещении
с постоянной температурой, или термостатируя сосуд, в
котором проводят 'измерения. Зависимость электропроводности
электролитов от температуры выражается уравнением
K = KH+a(t — t0) + b(t — t0)'l^KtH \-a{t-t0)\.)
Величина температурного коэффициента а, по Берлинеру,
мало зависит от концентрации раствора и для всего предела
измерений, обычных при почвенных анализах, остается
равной 0,022, что совпадает с литературными данными для
водных растворов солей.
ПРИБОРЫ ДЛЯ КОНДУКТОМЕТРИЧЕСКИХ ИССЛЕДОВАНИЙ
ПОЧВЕННЫХ ВЫТЯЖЕК И РАСТВОРОВ
Прибор для определения электропроводности состоит из
ячейки, или электродного сосуда, в который наливается
анализируемая почвенная вытяжка, и измерительного мостика
на переменном токе (мостик Кольрауша). Простейшая
ячейка представляет собой сосуд, в котором находятся два
платиновых электрода (рис. 37), впаянных в стенки сосуда или
постоянно фиксированных в пробке. Воспроизводимые
результаты получают, когда положение электродов не
меняется, т. е. не изменяется и постоянная сосуда. Поверхность
платиновых электродов платинируется для увеличения
рабочей поверхности электродов и предотвращения
поляризации. Кроме платинированных платиновых электродов
используют графитовые.
Измерительный мостик состоит из источника тока Е
(рис. 38), сосуда с исследуемым раствором Rx, постоянного
сопротивления Ri, индикатора тока Г, сопротивлений плеча
сравнения ^2, Rn и выключателя К- В качестве источника
тока применяют генераторы переменного тока или
пользуются сетевым током с частотой 50 гц. Индикатором может
служить телефон, гальванометр переменного тока или осцил-
лографический индикатор нуля. Чтобы найти сопротивление
исследуемого раствора с помощью моста, передвигают
подвижный контакт С, меняя соотношение сопротивлений R2 и Rs
и отыскивая такую точку, когда в диагонали моста ток не
обнаруживается. Очевидно, что при этом потенциалы в
точках С и d равны, что обеспечивается соблюдением равенства
Rx _. fli
Rs «2 '
176
откуда
it электропроводность ах — a1 .
R3
Если длина реохорда ab = R2+Rz= 1000 (реохорд разделен
на 1000 делений]^ то вычисление сводится к формуле
аС
1 1000—аС
где аС — число делений от точки а до точки С.
Лучше измерения электропроводности проводить при
частотах порядка 10 000 гц; применение переменного тока
необходимо для предупреждения поляризации электродов.
Рис 37. Ячейки для измерения электро- Рис. 38. Мостик для изме-
проводности: рения электропроводности:
/ — платинированные электроды Ru R2, Rs — сопротивления;
Rx—искомое
сопротивление; Е — источник тока;
К — выключатель; С —
скользящий контакт
Однако значительное увеличение частоты может вызвать и
резкие изменения электропроводности вследствие эффекта
релаксационного торможения, обусловленного отставанием
центра ионной атмосферы от самого иона при его движении.
Этот эффект особенно сильно проявляется при частотах от
п • 104 до п • 109 гц и в большой степени зависит от
концентрации электролита. Поэтому правильный выбор и постоянство
177
частоты колебаний являются одним из важнейших условий
кондуктометрического анализа.
Сопротивление раствора, измеренное в электродном
сосуде, не позволяет сразу рассчитать удельное сопротивление
раствора по той причине, что расстояние между электродами
в сосуде не всегда равно 1 см, а площадь их не точно 1 см2.
Измеренное сопротивление нужно помножить на
коэффициент я];, чтобы получить удельное сопротивление раствора.
Коэффициент-ф называют постоянной сосуда.
Определение if сводится к измерению в электродном
сосуде сопротивления раствора с известно» величиной
удельного сопротивления:
-J- = ф или р0 = Rx ф,
где Rx — измеренное сопротивление раствора, ро — его
удельное сопротивление (находят по таблицам).
Растворы 0,02 и 0,01 н. КО чаще других используют как
стандарты удельного сопротивления. Приготавливают их из
химически чистой соли, прокаленной при температуре
темно-красного каления и хранящейся без доступа водяных
паров, например, в эксикаторе над конц. H2SO4. Для
растворения соли используют дважды перегнанную воду без С02.
Сопротивление растворов измеряют, как всегда, не менее
трех раз, сменяя раствор и термостатируя сосуд.
Погрешность измерений не должна превышать точности прибора.
Для вычисления постоянной сосуда измеряют сопротивления
по крайней мере двух растворов КО разной концентрации.
Промышленность выпускает приборы, пригодные для
непосредственного использования в аналитических работах.
Одним из них является реохордный мост Р-38, рассчитанный
на работу с постоянным и переменным током промышленной
частоты E0 гц); подключив внешний индикатор тока, этот
прибор можно применять и для определения сопротивления
на иных частотах.
Реохордный мост Р-38 позволяет определить
сопротивление проводников I и II рода. Сопротивление электролитов
определяется на переменном токе, твердых проводников — на
постоянном. Диапазон сопротивлений, измеряемых мостом,
достаточно широк: от 0,3 до 30 000 ом. Погрешность зависит
от измеряемой величины и составляет ± 1 % при
сопротивлении более 30 ом и ±5% при сопротивлении до 30 ом.
Почвенный солемер — специальный прибор для
определения солесодержания в почвах — был разработан Берлине-
178
ром с сотрудниками. Принципиальная электрическая схема
почвенного солемера ПС-3 показана на рис. 39. В отличие от
мостика Кольрауша, в этом приборе использована
дифференциальная (разностная) схема, повышающая чувствительность
прибора.
В двух контурах I и II действуют одинаковые по величине
э. д. с В один контур включен электродный сосуд / с
измеряемым раствором, в другой — переменное сопротивление 3
После выпрямления токи поступают на гальванометр 5, кото-
Рис. 39. Принципиальная схема почвенного солемера ПС-3:
J—электродный сосуд, 2 — трансформатор питания,
3—переменное отсчетное сопротивление; 4—выпрямители, 5 —
нулевой гальванометр, 6 — потенциометр установки нуля
рый показывает разность токов. Изменяя переменное
сопротивление 3, можно добиться, чтобы гальванометр
показывал нуль.
В этом положении выпрямленные токи равны по величине
и противоположны по направлению, а сопротивление
электродного сосуда равно переменному сопротивлению.
В приборе предусмотрено питание от сети переменного
тока или от сухих батарей, вмонтированных в прибор.
Ламповый генератор, собранный на пентоде 1К1П, обеспечивает
питание от сухих батарей переменным током с частотой
50 гц. Для настройки солемера — установки гальванометра
на нуль перед началом работы — имеется специальный
потенциометр 6. Дополнительное шунтирование гальванометра
позволяет работать в широком диапазоне измерений.
179
Водные вытяжки для определения электропроводности с
помощью почвенного солемера можно готовить на обычной
(недистиллированной) воде, электропроводность которой
компенсируется перед началом измерений особым
компенсационным потенциометром.
Шкала прибора, связанная с отсчетным потенциометром,
позволяет по внутренней окружности отсчитывать показания
в процентах солесодержания от 0,1 до 1%, или в единицах
электропроводности — от 3 до 28* Ю-4 о/и-1; на внешней
окружности солесодержание — от 1 до 10% и
электропроводность от 28 до 200 • Ю-4 • ом~х. Переход от одного предела
измерений к другому осуществляется переключателем.
Подготовка солемера к работе и выполнение
измерений
Перед началом работы необходимо проверить,
соответствует ли положение контактного штырька напряжению сети, и
включить его в гнездо, соответствующее источнику питания.
Ручку компенсационного реостата поворачивают до упора
(положениеоо) и устанавливают стрелку гальванометра на
нуль при помощи корректора.
Электродный сосуд споласкивают исследуемым раствором
и, заполнив до краев, устанавливают в гнездах. Переводят
ручку переключателя в положение «контр» (контроль нуля),
и если стрелка гальванометра отклоняется, то добиваются
нулевого положения вращением ручки реостата установки
нуля. Поставив ручку переключателя в среднее положение
«измерение» и вращая рукоятку отсчетного потенциометра,
снова добиваются нулевого показания гальванометра. Если
точки компенсации найти не удается, то следует перейти на
другой предел измерений. Концентрированные растворы, для
которых нельзя найти нулевой точки даже на верхнем
пределе измерений, следует разбавить водой в 10 раз. По
достижении точки компенсации берут отсчет по шкале.
Температурная поправка вводится в соответствии с приведенной
ранее формулой.
Если водная вытяжка приготовлена на
недистиллированной воде, то перед началом работы ручку переключателя
переводят предварительно в положение «вода» и вращением
компенсационного реостата добиваются нулевого положения
гальванометра для ячейки, заполненной водой. Затем
проводят измерения, как описанр выше.
180
При пользовании солемером ПС-3 и аналогичными
приборами следует учитывать условность градуировки шкалы в
показателях солесодержания. Более точные результаты
можно получить, если измерения проводить непосредственно
в величинах электропроводности, составив предварительно-
градуировочную шкалу применительно к исследуемому типу
почв.
Глава V
ПОЛЯРОГРАФИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ
ПОЧВ
Возникновение и развитие полярографического метода
анализа связано с именем чешского профессора Я- Гейров-
ского, который впервые установил связь между силой тока
и концентрацией вещества, восстанавливающегося на
ртутном капельном электроде. Первая установка для получения
поляризационных кривых была создана Гейровским в 1922 г.,
а первая автоматическая конструкция полярографа
осуществлена в 1925 г.
Быстрое развитие метода и широкие возможности его
использования в почвенных исследованиях определяются в
основном двумя факторами: универсальностью метода и
высокой чувствительностью и точностью.
Универсальность метода основывается на следующих
особенностях.
! Полярографический метод применим как для целей
количественного анализа, так и для изучения структуры и
строения различных соединений.
2. Применение этого метода позволяет определить очень
большое число веществ как в ионной, так и в молекулярной
•форме. Принципиально возможно определение любых
соединений, способных электроокисляться или электровосстанав-
ливаться. Многие другие соединения могут быть определены
косвенными приемами.
3. Одновременно по одной полярограмме возможно
количественное определение сразу нескольких компонентов (до
пяти — шести), присутствующих в растворе, причем можно
182
осуществлять непрерывный контроль за содержанием
отдельных компонентов раствора.
При полярографическом определении различных
соединений используются единая аппаратура и технические приемы.
Значительным преимуществом полярографического
метода является высокая чувствительность его и большая
точность определения очень малых количеств веществ, что
позволяет определять содержание микроэлементов в почвах.
Количественный анализ возможен при содержании
определяемого иона порядка 10_5%; для анализа достаточно
Ю-4—Ю-3 мг вещества. Используя некоторые приемы
(увеличенные диффузионные токи, висячую каплю), можно еще
более повысить чувствительность. Точность анализа,
достигаемая при определении столь малых количеств, значительно
превышает точность анализа тех же образцов, проведенного
иными методами.
Полярографический метод в почвоведении наиболее часто
используется для определения содержания микроэлементов.
В последнее время предложены методики и для определения
ряда макроэлементов и некоторых почвенных характеристик
(емкость поглощения), разработанные преимущественно Ско-
бец с сотрудниками.
Полярографические методы анализа различных
промышленных и природных объектов (руд, минералов) известны
для большинства элементов периодической системы
Д. И. Менделеева. Перенести эти методы в почвенный анализ
далеко не всегда возможно, так как почвы резко отличаются
от руд и минералов свойствами и составом. Трудности,
возникающие при анализе почв, обусловлены следующими
причинами:
а) почвы содержат почти все элементы периодической
системы, причем преобладают соединения Si, Fe, Л1 и
катионы щелочных и щелочноземельных металлов; различные
микроэлементы содержатся в соизмеримых количествах, а
набор их весьма велик;
б) большинство важных в сельскохозяйственном
отношении почв содержат значительные количества органических
веществ A—8—10%);
в) почвоведу и агрохимику приходится определять не
только валовое содержание элементов, но и различные
формы их соединений, отличающиеся по подвижности.
В почвах и близких к ним объектах полярографически
определяют пока относительно небольшой набор элементов.
Из элементов I группы были сделаны попытки определения
183.
Na, Li, К, но эти методы не получили распространения в силу
низких отрицательных потенциалов восстановления катионов
щелочных металлов. Щелочноземельные элементы, в том
числе Са, определяются косвенными методами с применением
комплексонов (по Пршибилу). Хорошо разработаны методы
анализа элементов IV, V, VI, и VII групп; в почвах с
успехом определяют Pb, V, Bi, Mo, Co, Nj, Cu Zn, Mn.
Выполнены работы по определению в почвах соединений
фосфора, азота и молекулярного кислорода.
Варьирование условий позволяет в большинстве случаев
проводить одновременное полярографическое определение
такого набора элементов, который требуется для решения
поставленной задачи, что особенно удобно при анализе
микроэлементов. По этим показателям полярография
занимает второе место после эмиссионного спектрального
анализа, однако чувствительность последнего по отношению ко
многим элементам остается пока ниже чувствительности
полярографического анализа.
ОСНОВЫ ТЕОРИИ ПОЛЯРОГРАФИЧЕСКОГО МЕТОДА
В основе метода лежат процессы, происходящие при
электролизе анализируемого раствора. Для осуществления
электролиза в анализируемый раствор погружают два
электрода, к которым подводится напряжение от внешнего
источника тока (рис. 40). Один из электродов—-слой ртути на
дне сосуда или каломельный полуэлемент 6, роль катода
чаще всего выполняет ртутный капельный электрод 4,
состоящий из капилляра, соединенного с резервуаром 7, в
который налита ртуть. При подъеме резервуара под
влиянием создающегося давления ртуть начинает капать из
капилляра.
Контакт с раствором и процесс электролиза происходит во
время роста ртутной капли от ее появления до отрыва от
капилляра. Характер реакции зависит от свойств металла, из
которого изготовлен электрод (для капельного электрода —
это ртуть), состава анализируемого раствора и потенциала
электрода.
Зависимость между потенциалом ртутного
микроэлектрода, характером и направлением реакции при восстановлении
катионов удобней всего проследить графически, откладывая
по оси ординат силу тока, протекающего через раствор, а по
оси абсцисс — потенциал электрода (рис. 41). До тех пор
пока потенциал электрода остается более положительным,
184
чем потенциал восстановления присутствующих в растворе
ионов, никаких реакций на электроде не происходит и сила
тока близка к нулю, что выражается на графике
отрезком АВ. Как только потенциал электрода достигнет
величины, равной потенциалу восстановления ев данного иона, на
электроде начнется процесс восстановления и через раствор
проходит ток, сила которого увеличивается с возрастанием,
потенциала электрода (отрезок ВС на рис. 41).
Рис. 40. Схема простой установки для
полярографического анализа:
АВ— реохорд; 1 — аккумулятор ; 2—вольтметр, 3--
гальванометр, 4 — ртутный капельный электрод; 5 —
электролизер; 6 — каломельный электрод, 7 —
резервуар со ртутью
Однако возрастание силы тока, протекающего через,
раствор, происходит не беспредельно. Если раствор не
перемешивается в ходе электролиза, то при потенциале
электрода е<ев (интервал CD на рис. 41) сила тока достигает
предельной величины и не изменяется при увеличении
электродного потенциала. В этом интервале потенциалов катионы,
расположенные вблизи поверхности электрода,
восстанавливаются за очень короткий промежуток времени и
растворяются в ртути с образованием амальгамы. Концентрация
ионов вблизи электрода быстро уменьшается, т. е.
происходит концентрационная поляризация электрода,
185
я поступление новых порций ионов идет теперь только
вследствие движения ионов с постоянной скоростью из основной
массы раствора к поверхности электрода. Это движение
происходит под влиянием, во-первых, диффузионных сил и, во-
вторых, электростатического притяжения положительно
заряженных катионов отрицательным зарядом катода. Суммарная
сила тока, протекающего через раствор, обусловлена общим
количеством ионов,
разряжающихся на электроде в единицу
времени, и складывается из двух
составляющих:
диффузионного тока, сила которого зависит
от скорости диффузии, и
миграционного тока.
Миграционную составляющую,
обусловленную взаимодействием зарядов
Рис 41. Подпрограмма ио- иона и электрода, в полярографии
нов металла v ^ г г -г
сводят к нулю, добавляя в
раствор избыток индифферентного
электролита. Поэтому можно считать, что сила тока
обусловлена только скоростью диффузии и, исходя из этого
положения, определить зависимость между силой предельного тока
и концентрацией восстанавливающегося вещества в растворе.
По закону Фика скорость диффузии прямо
пропорциональна разности концентраций веществ в двух объемах раствора,
между которыми происходит диффузия. Если диффузия идет
по направлению к электроду с плоской поверхностью, то
, __ nFCS \/rT> •«
У nt
где Jd — сила тока, обусловленная скоростью диффузии ионов
к электроду (диффузионный ток); С — исходная
концентрация диффундирующего вещества в растворе; D —
коэффициент диффузии; t — время, в течение которого протекает
процесс диффузии; 5 — площадь поверхности электрода;
а — заряд иона. Сила диффузионного тока не зависит,
следовательно, от потенциала электрода, что выражается
горизонтальным участком кривой на рис. 41 (отрезок CD).
Кривая зависимости между силой тока и потенциалом
носит название полярограммы, а участок, на котором
сила тока возрастает, достигая предельного значения,
называют волной восстановления ионов данного вида.
Прямая пропорциональность между скоростью диффузии и
186
силой диффузионного тока, с одной стороны, и
концентрацией восстанавливающихся ионов раствора, с другой,
является основным соотношением количественного
полярографического анализа.
В случае ртутного капельного электрода характер
зависимости несколько меняется. Ртутная капля, образующаяся на
конце капилляра, все время растет, в связи с чем площадь
поверхности электрода также увеличивается во времени. Если,
принять, что масса ртути, вытекающей из капилляра за
1 сек, равна т, то очевидно, что масса капли, образующейся
за время t, будет
где г — радиус капли и d — плотность ртути.
Теперь уже легко подсчитать радиус, а следовательно, и
площадь поверхности ртутной капли в любой момент
времени:
V 4nd J \ 4jtd J
Подставляя найденное значение площади поверхности
электрода 5 в любой момент времени в уравнение
диффузионного тока, можно определить силу диффузионного тока на
капельном ртутном электроде. Объединяя все числовые
коэффициенты, входящие в это уравнение, получим
Jd = 0,732 nFCW> m*'> Z"/«.
Время, выраженное в секундах, характеризует любой
момент существования ртутной капли.
Подставляя в уравнение время, равное времени
существования одной капли, получим выражение для максимальной
силы тока в процессе роста и отрыва капель от капилляра.
Из уравнения следует, что изменение силы тока в период
существования одной капли выражается параболой шестого
порядка, т. е. по мере роста капли сила тока увеличивается
сначала быстро, а затем все медленнее; при отрыве капли от
капилляра сила тока падает почти до нуля. В обычной
лабораторной практике невозможно (да в этом и нет
необходимости) записать полностью кривую изменения силы тока во
времени. Наиболее точно измеряется среднее значение
диффузионного тока. Под средним током понимается
постоянный ток такой cHJjbi, который за время существования
187
■одной капли дает такое же количество электричества, какое
фактически связано с каждой каплей ртути. Расчет
показывает, что средний ток связан с максимальным значением тока
соотношением
^ср = Ь/7 «/макс.
Подставляя в уравнение диффузионного тока значение
-среднего тока и выражая силу тока в микроамперах,
концентрацию в миллимолях на литр и т в мг-секг1,
получим
/ср = 605яСЯ/.т*/./Й«..
Это уравнение Ильковича.
В растворах, близких по составу и концентрации,
величина коэффициента диффузии определяемого иона остается
практически постоянной. Если уровень ртути в резервуаре
поддерживать в процессе анализа на одной высоте, то при
работе с одним и тем же капилляром период капания ртути
и масса ртути, вытекающая из капилляра в единицу времени,
также остаются постоянными. Это позволяет объединить все
постоянные величины в правой части уравнения Ильковича
и обозначить суммарную постоянную символом К.
Тогда получим J = KC.
В такой форме уравнение наиболее удобно применять при
обычных количественных определениях.
В реальных условиях сила тока на отрезке АВ (рис. 41)
никогда не бывает равной нулю, а всегда имеет • некоторое,
хотя часто и очень малое, конечное значение; этот ток
называют остаточным током. Очевидно, для определения
истинного значения диффузионного тока нужно из
измеренной величины предельного тока вычесть величину
остаточного тока или найти разность графически.
Каждый из определяемых ионов дает свою волну на по-
лярограмме, и суммарная сила тока при этом равна сумме
диффузионных токов всех восстанавливающихся ионов. Если
для первого иона (в нашем примере для иона меди, рис. 42)
диффузионный ток находят простым измерением силы
тока /ь то для иона никеля он равен разности /г — /i и т. д.
Практически удобно определять сразу высоту волны,
т. е. разность высот между горизонтальными площадками,
ограничивающими волну данного иона.
Л 88
Количественное определение сводится, таким образом, к
определению высоты волны или силы диффузионного тока
для данного иона и к дальнейшему пересчету этих величин в
концентрацию.
Качественное определение
вида восстанавливающегося иона
производят по величине
потенциала ПОЛУВОЛНЫ Е' г.
Потенциал ев, при котором
начинается восстановление, не
может служить характерным
качественным признаком, потому
что его величина зависит от
концентрации восстанавливающегося
иона; чем выше концентрация
иона, тем при менее
отрицательных потенциалах начинается
процесс восстановления.
Величина потенциала
полуволны не зависит от
концентрации восстанавливающегося иона
и определяется только природой самого иона. Потенциал
полуволны е-_, равен такому значению потенциала
катода, при котором сила тока / равна половине своего
предельного значения 7^:
/-0,5/d.
На рис. 41 видно, что потенциал полуволны легко найти,
определив по графику значение потенциала, отвечающее
половине высоты волны.
Рис. 42. Подпрограмма
раствора, содержащего ионы
меди, никеля, цинка.
Диффузионный ток, обусловленный
восстановлением ионов меди,
равен /i, ионов никеля — /г — /ь
ионов цинка — /з — ^г
Постоянство потенциала полуволны вытекает из уравнения
полярографической волны, устанавливающего зависимость между потенциалом
катода и силой диффузионного тока, которое для случая восстановления
простых ионов металлов впервые выведено Гейровским и Ильковичем
Если на ртутном электроде происходит восстановление ионов металла до
атомов металла и последний растворяется в ртути с образованием
амальгамы, потенциал электрода в соответствии с уравнением Нернста равен:
е = 80 —
RT
nF
1п-
Са /м
£р ^MeHg
где Са—концентрация попов металла в амальгаме, Ср—концентрация
ионов металла в растворе у поверхности электрода, aHg — активность
ртути в амальгаме и /^ и /м — коэффициенты активности ионов металла
в растворе и амальгаме.
189
Поскольку образующиеся амальгамы очень разбавлены, активность
ртути в амальгаме можно считать постоянной и объединить
соответствующую величину с константой уравнения, обозначая ее е'с.
Концентрацию металла в амальгаме и концентрацию нонов у поверхности
электрода легко подсчитать, пользуясь уравнением диффузионного тока.
Величина предельного диффузионного тока, как было показано выше,
определяется
J = KC.
Если же сила тока не достигла предельного значения и концентрация
ионов у поверхности электрода равна Ср , то в этом случае сила тока
для любой точки волны может быть выражена соотношением
J'=K(C-Cp).
Отсюда следует, что концентрация ионов металла вблизи поверхности
электрода равна
ср =
Ц:
с
J
~ к
г
к
~"
J'
к
КС
к
J
—
J'
к'
— J'
к '
Пользуясь последним выражением, вычисляют концентрацию ионов
металла Ср вблизи поверхности электрода для любой точки волны, если
известна сила тока /' в той же точке волны и сила предельного
диффузионного тока J.
Концентрация амальгамы на поверхности ртутной капли
определяется аналогично:
Г
" К."
Подставляя эти соотношения в вышеприведенное уравнение
потенциала электрода, получим i
. RT , J'Kfu • RT Kfu
е = е0— —— In — = е0 — —— ш — —
nF (J~J')KafM nF Kafu
RT , J'
-In
nF J —J'
При условии /'==0,5 / последний член уравнения становится равным
нулю, а потенциал электрода равен
RT Kfu
In
nF
*а/м
Величины коэффициентов активности и констант К и Ка постоянны,
а следовательно, потенциал электрода при /'=0,5 / тоже величина
постоянная и не зависит от силы тока и концентрации ионов металла,
восстанавливающихся на электроде.
190
Это значение потенциала, как было определено выше, носит название
.потенциала полуволны и обозначается е» . Вводя потенциал полуволны
а качестве константы в уравнение полярографической волны, получим
в окончательном виде у"
RT , Г
-1п
'' nF J — J'
Величина потенциала полуволны' для ионов различных
металлов складывается из нескольких составляющих, но для
большинства металлов определяется в основном
нормальными электродными потенциалами. Потенциалы полуволн
некоторых элементов приведены в табл. 16.
Практически потенциал полуволны и вид восстанавлива-
.ющегося иона определяют путем измерений е», на поляро-
грамме и сравнения найденной величины с потенциалами
полуволн различных ионов, измеренных ранее при условии
постоянства состава растворов в отношении прочих солей.
Присутствие в растворе веществ, способных образовывать
комплексные соединения с определяемыми ионами,
вызывает смешение потенциала полуволны в сторону более
отрицательных значении потенциалов. При обратимой реакции
восстановления комплексного иона до металла величина £■/»
зависит также от концентрации комплексообразующего
вещества, координационного числа и константы диссоциации
комплексного иона металла:
nF fKK nF
где Сх — концентрация комплексообразующего вещества у
поверхности электрода, fK — соответствующий коэффициент
активности, р — координационное число, Кых — константа
.диссоциации комплексного иона металла и
/мх—коэффициент активности комплексного иона металла.
Изменение потенциала полуволны в присутствии ком-
плексообразователей имеет важное практическое значение.
В анализируемых растворах часто одновременно
присутствуют ионы двух (или более) различных металлов,
восстанавливающихся приблизительно при одном значении потенциала.
Одновременное определение таких ионов" оказывается
невозможным, так как не удается отличить волну одного иона от
волны другого. Если же добавить к анализируемому
раствору вещество, образующее с одним из ионов комплексное
■ соединение, то потенциал полуволны этого иона сдвигается в
191
Потенциалы полуволн
Таблица 16
Элемент
Алюминий, А13+
Аммоний, NHJ"
Барий, Ва2+
Ванадий, VO^~
»
Водород, Н30^"
Железо, Fe3+
»
»
» Fe2+
»
Калий, К-*"
Кальций, Са2+
Кислород, 02
»
Кобальт, Со2+
»
»
»
Марганец Мп2"*"
»
Медь, Си2+
»
»
Молибден, МоОТ"
»
Никель, Ni2+
»
Свинец, РЬ2+
»
»
Цинк, Zn2+
»
»
»
»
Фон
0,05 н. ВаС12
(CH3LNOH и (CH3LNC1
0,1 н. LiCl
0,1 М НС1
0,1 н. LiOH
0,1 М КС1
1 М (КН4JШ3
0,5 М цитрат натрия
1,0 М оксалат калня
Fe(OHJ в 1 н. NaOH
ВаС12
(СН3L NOH или (CH3LNC1
(СН3L NOH или (CH3LNCl
0,1 н. КС1, НС1
ОН- в 0,1 н. KN03
1,0 М KCNS
1,0 М раствор тартрата
0,1 н. КС1
1,0 н. NH4C1-H,0 н. NH4OH
1,0 и. КС1
1,0 н. КН4С1 + 1,0 и. NH4OH
1,0 М NH4OH
0,1 н. KCNS
1,0 н. NH4OH + 2,0 h.NH4C1
0,5 М H2S04
10 М H2S04
1,0 н. KCNS
0,2 н. NH4CI + 1 М NH4OH
1,0 н. KNOa
1,0 и. НС1
0,5 М тартрат калия-натрия
1,0 н. NH4OH + 2,0 н. NH4C1
0,1 н. KCNS
1,0 н. KNOs
1,0 н. КС1
1,0 н. NaOH
Е'/г
—0,77
—2,09
—1,90
0; —0,98
—1,7
—1,58
-0,44; —1,52
—0,426—0,108 пЬ
—0,24
—1,46
—1,3
—2,17
—2,26
—0,1; —ОД
«Н),03
—1,03
—1,66
—1,20
—1,51
—1,51
—1,45
—0,20; -0,4;
—0,02; —0,&
—0,2; — 0.1
—0,29; —0,8-
—0,23
—0,70
— 1,02
—0,45
-0,4
—0,50
— 1,4
—1,01
— 1,012
— 1,022
—1,50
отрицательную сторону и на полярограмме получают две
хорошо различимые волны.
Уравнение полярографической волны и уравнение Илько-
вича для диффузионных токов являются двумя основными
уравнениями, применяемыми в практике полярографического
анализа. Оба уравнения выведены с некоторыми
допущениями. Уравнение Ильковича (по Т. А. Крюковой) не учитывает
кривизны поверхности ртутной капли, изменения раствора в
процессе электролиза, экранирования части ртутной капли
поверхностью капилляра. При выводе обоих уравнений
предполагается, что коэффициенты диффузии ионов постоянны и
что коэффициенты активности ионов в растворах разной
концентрации имеют одно и то же значение. Все эти допущения
приводят часто к довольно значительным отклонениям
наблюдаемых величин диффузионных токов и потенциалов
полуволн от теоретических. Эти отклонения при количественных
определениях учитываются построением градуировочного
графика.
* *
*
Получение четких полярограмм и линейной зависимости
между силой тока и концентрацией возможно лишь при
устранении влияния миграционного и остаточного токов и
максимумов на полярографических кривых.
Остаточный ток. Остаточный ток возникает вследствие
того,,что анализируемые растворы почти всегда содержат
некоторое количество примесей веществ, восстанавливающихся
при потенциалах более положительных, чем потенциалы
полуволн определяемых элементов; в воде обычно имеются следы
растворенного кислорода, некоторые металлические катионы,
особенно цинка и меди, которые обнаруживаются даже в
дистиллированной воде.
Остаточный ток, однако, не может быть равен нулю, даже
если из раствора тщательно удалены все электровосстанавлн-
ваюшиеся вещества. Причиной этому служит ток заряжания,
или ток, который расходуется на заряжание каждой новой
капли ртути на капельном микрокатоде. В измеренные
величины предельных токов всегда вводится поправка иа
остаточный ток.
Миграционный ток. Движение ионов к электроду
происходит не только за счет диффузии, но также и .под влиянием
электрической силы, пропорциональной градиенту
электрического потенциала у поверхности электрода. Та часть пре-
7 Зак. 51
193
дельного тока, которая обязана своим происхождением
электрической миграции ионов, носит название миграционного
тока, и его величина возрастает с увеличением приложенной
к электродам э. д. с. при восстановлении катионов или
уменьшается при восстановлении анионов.
Уменьшение и даже практически полное устранение
миграционного тока достигается прибавлением индифферентного
электролита (фона) в концентрации, превышающей
концентрацию определяемых ионов в 50—100 раз. В качестве фона
чаще употребляются 1,1-валентные электролиты, потенциалы
полуволн катионов которых лежат в пределах от —1,8 до 2,2 в.
При избытке индифферентного электролита доля тока,
переносимого определяемыми ионами путем миграции, очень мала,
и поступление к электроду восстанавливающихся ионов
определяется только скоростью их диффузии.
Введение фона при снятии полярограмм обеспечивает
правильность уравнения Ильковича. Если избыток
индифферентного электролита отсутствует, то уравнение приобретает
значительно более сложный вид и для вычисления предельного
тока требуется знать величины чисел переноса определяемых
ионов и отношение удельной электропроводности основной
соли к сумме удельных электропроводностей всех
присутствующих в растворе солей.
Максимумы. Часто в начале полярографической волны
появляются максимумы, свидетельствующие о возникновении
тока большего, чем обычный предельный ток, и
затрудняющие измерение высоты волны. По теории А. Н. Фрумкина
максимумы возникают из-за перемешивания раствора,
которое происходит вследствие роста и движения поверхности
капель ртути.
Устранить максимумы можно путем добавления к
раствору малых количеств поверхностно-активных веществ, таких,
как желатин, агар-агар, некоторые красители и др. Чаше
всего добавляют несколько капель слабого, например
0,25%-ного, раствора желатина или агар-агара. Следует
учитывать при этом, что добавление желатина в больших
концентрациях может повлиять и на диффузионный ток. В
некоторых случаях максимумы используют в целях
количественного анализа.
ПРИБОРЫ ДЛЯ ПОЛЯРОГРАФИЧЕСКОГО АНАЛИЗА
В простейшей установке для полярографического анализа
имеются сосуд, в котором производится полярографирование,
два электрода, погружаемые в анализируемый раствор.
194
источник постоянного тока и приборы для регистрации
потенциала электрода и силы тока (см. рис. 40). Основной
рабочей частью является капельный ртутный электрод,
представляющий собой капилляр, соединенный с резервуаром, в
котором находится тщательно очищенная ртуть. Вторым
электродом служит большая поверхность ртути, налитой на дно
электролизерного сосуда, или каломельный электрод. Вместо
каломельного электрода можно воспользоваться
электродами других типов, имеющих устойчивое значение потенциалов,
не меняющееся при прохождении тока в несколько десятков
миллиампер (неполяризующиеся электроды). Наиболее часто
в полярографических работах применяется насыщенный
каломельный электрод; потенциалы полуволн обычно вычисляют
по отношению к потенциалу этого электрода.
Напряжение на электроды подается от аккумулятора
через потенциометр (реохорд) и измеряется вольтметром,
подключенным параллельно потенциометру. Сила тока,
протекающего через электролизер, измеряется зеркальным
гальванометром, включенным последовательно с
электролитической ячейкой. Эта общая схема лежит в основе большинства
промышленных полярографов.
Ртутный капельный электрод. Капельный ртутный
электрод обладает рядом преимуществ: его можно применять в
области низких отрицательных потенциалов, поверхность
ртутной капли весьма однородна, постоянное возобновление
капель обновляет поверхность электрода, что позволяет вести
электролиз в условиях, когда свойства поверхности
электрода практически не меняются.
Для изготовления капельного электрода (по И. М. Кольт-
гофу) берут капиллярные стеклянные трубки длиной
10—20 см, внешнего диаметра ~ 5—6 мм и внутреннего
0,5—1,0 мм; конец трубки оттягивается, пока внутренний
диаметр не станет равным 0,03—0,05 мм. Оттянутый кончик
капилляра должен быть однородным по всей длине.
Значительно лучше воспользоваться готовыми
термометрическими трубками, внутренний диаметр которых имеет
приблизительно те же размеры. Длина капилляра
подбирается так, чтобы период капания ртути был порядка 2—4 сек.
Наилучший период капания находят уже на готовой схеме
путем изменения высоты уровня ртути над кончиком
капилляра.
Бюретка, резиновые трубки и резервуар для ртути
(рис. 43) перед монтажом прибора должны быть тщательно
вымыты горячим раствором щелочи, пропарены и высушены.
7*
195
Загрязнившийся при работе кончик капилляра (о чем можно
судить по очень малому или большому периоду капания
ртути) промывают, опуская его в концентрированную азотную
кислоту и затем отмывая дистиллированной водой. Можно
также прочистить капилляр насосом, просасывая под
давлением ртуть. Если прочистить
капилляр не удается, то необходимо
отрезать его кончик или заменить
новым. При перерывах в работе
кончик капилляра погружают в
дистиллированную воду, в
противном случае капилляр
просыхает и часто загрязняется
настолько, что его не всегда
удается промыть.
В ряде случаев капельный
ртутный электрод с успехом
может быть заменен твердым
микроэлектродом.
Твердый микроэлектрод. Такой
электрод, изготовленный из
тонкой платиновой проволоки,
можно использовать при анодной
поляризации вплоть до
потенциала + 1,45 в, тогда как ртутный
электрод применим только до
потенциала 0,65 в (при более
высоких потенциалах начинается
заметное растворение ртути).
Были предложены различные
формы микроэлектродов. Для
изготовления электрода, по Скобец.
платиновую проволочку сечением
0,3—0,5 мм и длиной 35—40 мм
припаивают серебром к медной, а
затем платиновую проволоку
вплавляют в тонкую стеклянную
трубку. Оставшийся вне трубки
конец платиновой проволоки
длиной 25—30 мм заматывают в виде
Рис. 43. Приспособление для спирали. Медную проволочку, вы-
ртутного капельного электро- ХОдящую из незапаянного конца,
/-капилляр,ЛЙ2-платиновый плотно прижимают медной муф-
контакт той, надеваемой на конец стек-
196
ляшюй трубки. Изготовленный электрод крепится на оси
моторчика Уоррена, причем выходным контактом служит
звонковый провод, припаянный непосредственно к кожуху
моторчика Уоррена.
При работе с твердым электродом поверхность последнего
необходимо периодически обновлять, для чего перед каждым
■определением промывают электрод в тигелечке горячей
Рис 44. Виды электролизеров:
А —■ простой; Б — для работы
с донной ртутью; В — из
органического стекла (по
Большакову) ; / — платиновые
контакты, 2 — отверстия для
капилляра и соединительного
сифона, 3 — прокладка из мягкой
резины. 4 отводная трубка
H2SO4 A:1) и затем дистиллированной водой. Такой
электрод служит и как стационарный и как вращающийся;
хорошие результаты получаются при скорости вращения
■60 об/мин.
Электролизеры. Количественный анализ можно проводить
в обычных конических колбочках на 25 мл с пробками. Если
в ходе анализа требуется пропускать через раствор газ (азот,
водород), то электролизеры (рис. 44) изготавливают с
отростками, через которые поступает газ. В пробке, закрывающей
электролизер, делают два отверстия, одно из которых
предназначено для капилляра, а второе — для соединительного
сифона или каломельного электрода. Более сложно
устройство электролизеров, в которых анодом служит донная ртуть;
197
V,
Л
з-3
гг
V
в стенку такого сосудика должна быть впаяна платиновая
проволочка 1, через которую осуществляется контакт между
анодом и остальной частью цепи.
При массовых анализах очень удобен электролизер из
органического стекла, предложенный В. А. Большаковым для
небольших объемов растворов (рис. 44, В); во время
пропускания газа и полярографирования трубку 4 помещают в
пробирку с водой, которая предохраняет анализируемый
раствор от кислорода воздуха. Сосуд устанавливают в
специальной стойке и прижимают к резиновой прокладке 3,
заменяющей пробку. Капилляр и агар-агаровый сифон
постоянно закреплены в отверстиях 2 корпуса стойки и резиновой,
прокладки.
Источник тока. Источником тока, 'питающего электроды,
служит аккумулятор или сухая батарея достаточно большой
емкости (80—120 а-ч), позволяющая получить напряжение
4—6 в.
Напряжение, снимаемое с зажимов аккумулятора,
подается на электроды через делитель напряжения или
потенциометр; наиболее удобны барабаны с несколькими витками
потенциометрической проволоки. Сопротивление и длина по-
тенциометрической проволоки выбираются из расчета, чтобы
напряжение, подаваемое на электроды, можно было
контролировать с точностью до 15—30 мв. Параллельно
потенциометрической проволоке подключают вольтметр на 5—6 в
класса точности 1,0—1,5 для контроля величины падения
напряжения по всей длине потенциометрической проволоки.
Гальванометр для регистрации диффузионных токов
должен иметь чувствительность порядка Ю-8—10~19 а/л1м/м;
хорошими гальванометрами являются отечественные
зеркальные гальванометры типа М-21 или М-25 с чувствительностью
10~9—Ю-10 а\жж\ж. При выборе гальванометра следует
обратить внимание на период колебаний, который не должен
быть слишком мал, так как иначе осцилляции тока,
возникающие при росте и отрыве капель ртути от капилляра, будут
сильно отражаться на показаниях гальванометра. Для
уменьшения осцилляции подключают параллельно гальванометру
конденсатор большой емкости (до 5000 мкф).
Шунт, включенный параллельно гальванометру,
позволяет уменьшать чувствительность гальванометра; шунт
включается так (см. рис. 9), что суммарное сопротивление
внешней цепи гальванометра остается постоянной величиной; это
позволяет легко изменять чувствительность гальванометра в
определенное число раз, ие нарушая режима его работы.
198
Устройство полярографов
Отечественная и зарубежная промышленность выпускает
ряд полярографов, различных по конструкции; некоторые из
них получили массовое распространение. Широко
используются в почвенных и агрохимических лабораториях модели
чешских приборов типа полярографа Гейровского и ЛП-55.
За исключением электронных и осциллографических
полярографов все приборы имеют принципиально общую схему и
отличаются только деталями конструкции, поэтому ниже
дается описание только некоторых приборов с
автоматической записью'.
Полярограф Гейровского A951 г.) предназначен для
автоматической фотозаписи полярограмм и снабжен рядом
приспособлений, упрощающих работу и расширяющих
возможности применения прибора.
Кроме оборудования, входящего в комплект прибора, для
начала работы необходимо приобрести: аккумулятор на
4—б в, аппарат Киппа или электролизер для получения
водорода, промывные склянки, каломельный электрод, штативы
и эмалированные кюветы с высокими стенками,
необходимые для работы с ртутными приборами.
Существенны*! недостатком этой конструкции являются
трущиеся контакты, через которые подается напряжение
на вращающийся реохордный барабан. Контакты быстро
снашиваются, что приводит к незакономерным скачкам
напряжения на реохорде. В позднейших конструкциях чешских
полярографов (ЛП-55) этот недостаток устранен.
Электрическая схема прибора показана на рис. 45.
Полярограф Гейровского состоит из четырех основных
частей: собственно полярографа, зеркального гальванометра,
редуктора чувствительности и осветителя гальванометра.
Собственно полярограф смонтирован на одной панели", на
которой собраны: потенциометрический барабан,
электромотор, приводящий в движение потенциометрический барабан и
барабан фотокамеры, лампа абсцисс, вольтметр, рукоятки
управления и зажимные контакты (рис. 46).
Аккумулятор подключается к клеммам, обозначенным
«Акк»; после включения тумблера «Л/с/с» вольтметр на панели
показывает напряжение, поданное на потенциометрическую
лроволоку. Напряжение на проволоке можно менять в не-
1 Выпускавшиеся ранее полярографы для визуальной записи (типа
«Гинцветмет», ВП-1, ВП-5 и др.) в настоящее время почти полностью
вытеснены приборами с автоматической записью полярограмм.
199
больших пределах потенциометром, рукоятка которого
выведена на панель с надписью «Регуляция э. д. с». Изменять
напряжение можно также с помощью переключателя,
позволяющего вводить добавочное сопротивление в начале и конце
£в 4АГЛ
Рис. 45. Электрическая схема полярографа Гейровского:
Акк — аккумулятор; ав — потеициометрическая проволока; С —
скользящий контакт; V—вольтметр; /'-•- гальванометр; Эл—электролизер.
Клеммы для подключения: А— анода, К—катода; Ев — нормальный
элемент Вестона; Т — выключатель аккумулятора; Ri и
R2—сопротивления по 16 ом; Rz — переменное сопротивление, регулирующее
напряжение; #4 — переменное сопротивление для введения компенсации.
Переключатели: П\ — включение катодно-аноднон поляризации, Л2 —
катодной поляризации, П3 — нормального элемента; В, и В2 — выключатели
реохордной проволоки по 16 ом каждое. Если общее
напряжение, снимаемое с аккумулятора, равно 4 в, то при
положении переключателя «16 ом перед» на электродах действует
200
э. д. с. от 2 до 4 в, при положении переключателя «16 ом
за»— на электродах от 0 до 2 в, при положении «16 ом перед
и за» —от 1,3 до 2,7 в.
Переключатель поляризации позволяет производить
катодную поляризацию и осуществлять, если требуется,
плавный переход от анодной к катодной поляризации, а также
Рис. ЛИ. Внешний вид полярографа Гейровского:
/, 2 — зажимы для аккумулятора, 3, 4 — зажимы для элемента Вес-
тона, 5, 6 — зажимы для электролизера (анод, катод), 7, 8— зажимы
.редуктора чувствительности, 9 -заземление, 10-- переключатель по-
лярияации, // — переключатель напряжения, 12 регулировка
напряжения, 13 регулятор компенсационных токов.
Выключатели: М---аккумулятора, 15 — лампы абсцисс, 16 — лампы
•осветителя гальванометра, 17—мотора; 18 — электромотор; 19 — рычаг
скорости мотора; 20 — редуктор; 21— подвижный контакт; 22—потен-
шклиетрнческш! барабан; 2<У трущиеся контакты; 24—крепящая
гайка фотокамеры; 25—лампа абсцисс; 26—фотокамера; 27 — вольтметр;
28— колпачок подвижной оси стойки
подключать нормальный элемент Вестона для точного
измерения падения напряжения на каждом витке потенциометри-
ческой проволоки.
Для подключения электродов электролизера на панели
выведены две клеммы — «анод» и «катод». Напряжение,
поданное на электроды, определяют по положению подвижного
201
контакта на потенциометрическом барабане. Всего на
барабане 19 витков проволоки; если общее напряжение,
показываемое вольтметром, равно 3,8 в, то на каждый виток
проволоки приходится 200 мв. На барабане нанесены, кроме того,
деления, позволяющие определить и доли витка.
Потенциометрический барабан соединен с мотором
посредством редуктора, который изменяет скорость и
направление вращения барабана и разъединяет мотор и барабан.
Фотокамера снабжена крепящей гайкой, позволяющей
вращать фотокамеру независимо от потенциометрического
барабана и устанавливать барабан в любое положение,
соответствующее делениям на крышке фотокамеры, т. е. начинать
запись полярограмм с любого значения абсциссы. Обычно
съемку начинают с нулевой абсциссы и при наибольшей для
данного случая чувствительности гальванометра. Вторую
кривую снимают, передвинув начало записи на абсциссу «1»
или «2» и т. д. Это позволяет одновременно снять до 18
полярограмм, не перезаряжая фотокамеру.
После установки фотокамеры на нужное деление и
закрепления гайки фотокамера вращается вместе с потенцио-
метрическим барабаном с такой скоростью, что одному
полному обороту потенциометрического барабана соответствует
поворот фотокамеры на одно деление. При каждом обороте
замыкаются контакты лампы абсцисс («моргуна»),
засвечивающей вертикальные полосы на фотобумаге («абсциссы»).
Расстояние между «абсциссами» на фотобумаге в точности
соответствует падению напряжения в '/ш долю от общего
напряжения на всей проволоке.
Зеркальный гальванометр чувствительностью Ю-8 а/мм/м
через редуктор чувствительности подключается к зажимам
редуктора на панели полярографа. Редуктор
чувствительности позволяет уменьшать чувствительность гальванометра в
2, 3, 5... 5000 раз. Установленная чувствительность может
быть прочитана в окошке редуктора и обозначена
соответственно как Vi (максимальная), '/г, ••■ Vsooo.
Осветитель гальванометра имеет специальный
трансформатор (лампа осветителя рассчитана на 12в); на крышке
трансформатора при помощи шарнира крепится труба
проекционной лампы, внутри которой помещены [цель и
фокусирующая система. Осветитель устанавливается так, чтобы угол
падения луча на зеркальце гальванометра был возможно
меньшим, а расстояние от щели до зеркальца гальванометра
было порядка 25 см.
202
Установка и настройка полярографа Гей-
ровского. Полярограф устанавливают на прочном
основавши, не испытывающем механических сотрясений. Перед
включением в сеть проверяют положение переключателя
напряжения A27 или 220 в) в нижней части клеммной
сборки мотора и под нижним защитным листом трансформатора
осветителя. Зажимы гальванометра подсоединяют к
зажимам Г редуктора, а зажимы редуктора Р — к клеммам на па-
мели полярографа. Вилку трансформатора осветителя
включают в гнездо на передней стенке полярографа. Затем
подключают полярограф к сети переменного тока и включают
тумблеры мотора, осветители гальванометра и лампы
«абсцисс». При этом мотор вместе с барабаном и фотокамерой
должен вращаться, загорается лампа осветителя, а лампа
«абсцисс» дает кратковременные вспышки при каждом
обороте потенциометрического барабана. Выключив мотор и
лампу «абсцисс», фокусируют осветитель, добиваясь, чтобы
на щель фотокамеры падал узкий вертикальный, хорошо
сфокусированный, пучок света, полностью перекрывающий всю
ширину щели.
После соединения клемм аккумулятора с зажимами
полярографа « + » и «—» и при включении тумблера «Лк/с»
вольтметр показывает величину падения напряжения на потенцио-
метрической проволоке. Проверив исправность электрической
схемы, окончательно устанавливают гальванометр по
уровню, освобождают подвижную часть от арретира и выводят
«зайчик» гальванометра на нулевое деление шкалы
фотокамеры. Затем устанавливают полярографическую ячейку и
проводят проверку качества прибора, как это описано дальше.
Микрополярограф Гейровского выпускается в нескольких
вариантах и отличается малыми габаритами и
компактностью конструкции, что делает его весьма удобным для
работы в полустационарных (полевых) условиях. Все детали мик-
рополярографа, включая гальванометр, собраны в одном
кожухе (рис. 47); необходимое расстояние от зеркальца
гальванометра до барабана с фотобумагой обеспечивается
системой зеркал. Положение гальванометра относительно
осветителя и фотокамеры строго фиксировано, что исключает
неточности в настройке. В этом приборе, как и в
последующих конструкциях, имеется матовая стеклянная шкала с
делениями, позволяющая следить за положением «зайчика»
гальванометра. Осветитель при помощи специальной
рукоятки можно несколько поворачивать вокруг оси и
устанавливать «зайчик» гальванометра на любое деление шкалы.
203
В микрополярографе напряжение на реохордную
проволоку подается непосредственно от аккумулятора; имеется
только одно дополнительное сопротивление для точно»
регулировки напряжения Диапазоны поляризации ограничены, а
реохордная проволока имеет один виток, что снижает точ-
Рис 47 Внешний вид микрополярографа
/—гальванометр, 2—уровень, 3—вольтметр, 4—рукоятка
редуктора чувствительности, 5—реостат регулирования напряжения,
6 — регулятор компенсационных токов, 7—рукоятка регулятора
скорости записи, 8—регулировка осветителя, 9 — крышка освети
теля, 10- -шкала потенциометра, 11 — фотокамера, 12 - - персе.™
чатель записи (обычная, по схеме производных, «выключено»),.
13 — переключатель поляризации (катодная, анодно катодная),
14—переключатель гальванометра, 15 — барабан часового
механизма, 16— шкала гальванометра
ность измерения потенциалов полуволн в сравнении с поля-
рографом Гейровского.
Микрополярограф предусматривает также съемку
производных полярографических кривых, т. е запись в
координатах dJ/dE—Е, что обеспечивается введением в схему поляро-
204
графа дополнительных конденсаторов Все эти изменения
делают прибор удобным для производственных целей, но
снижают его пригодность для исследовательской работы
К миьрополярографам поздних конструкций прилагается
вмонтированный в общий кожух кислотный аккумулятор для
питания ячейки Потенциометрический барабан в этих
приборах полностью скрыт в кожухе, а контроль за напряжением
на ячейке осуществляется вольтметром, подключенным
параллельно электролизерной ячейки Запись по схеме
производных в этих приборах отсутствует
Полярограф ЛП-55 значительно усовершенствован по
сравнению с предыдущими конструкциями В нем
потенциометрический барабан сделан неподвижным, а контакт,
снимающий напряжение, вращается вокруг барабана
Снимаемое с проволоки напряжение регулируется переключателем
диапазонов поляризации (рис 48)
Для точной регулировки напряжения используется
реостат в сочетании с элементом Вестона При включении
последнего кнопкой 10 «зайчик» гальванометра не должен
отклоняться от нулевого положения, если падение
напряжения на потенциометрической проволоке равно точно 4 в При
отклонении «зайчика» падение напряжения регулируется
рукояткой 11
Питание потенциометра осуществляется от аккумулятора,
для зарядки которого в приборе предусмотрен выпрямитель
Для зарядки аккумулятора включается тумблер на боковой
панели прибора, а синяя лампочка 3 сш нализирует включе
ние выпрямителя в цепь (вольтметр при этом должен быть
отключен) Схема позволяет соединять выпрямитель через
добавочное сопротивление с потенциометрической проволокой
и питать электроды от выпрямителя, в этом случае
подключенный параллельно выпрямителю аккумулятор играет роль
стабилизатора напряжения Такой схемой включения можно
пользоваться, если небольшие колебания напряжения не
влияют на качество измерений
Напряжение от потенциометрической проволоки подается
на электроды через редуктор чувствительности, позволяющий
изменять силу тока, протекающего через гальванометр, в
определенное число раз Переключатель 17 соединяет
гальванометр со схемой, позволяющей получать запись производных
dJ/dE—Е Потенциометр 12 служит для компенсации оста
точного тока
205
На рис. 4& показана электрическая схема прибора.
Общее питание прибора осуществляется промышленным током
120/220 е, напряжение подается на первичную обмотку трансформатора;
со вторичной обмотки снимается напряжение' в 4, 6 и 10 в для питания
выпрямителя Се, ламп, засвечивающих «абсциссы» Л2 и обмотки пуска
электродвигателя «пуск». С первичной обмотки трансформатора напря-
Рис. 48. Внешний вид полярографа ЛМ-55:
1—рукоятка шкалы потенциометра, 2 — рукоятка
автоматического выключения Мотора, 3— контрольная лампа (синяя,
зарядка аккумулятора); -^--контрольная красная лампа (работа
мотора), 5--кнопка включения мотора, 6—вольтметр, 7 —
выключатель абсцисс, 8 — кнопка для засвечивания абсцисс
вручную, 9—переключатель интервалов поляризации,
10—регулировка напряжения, //— реостат регулятора напряжения, 12—
регулятор компенсационных токов, 13 — барабан фотокамеры,
14 — шкала, 15 — входная щель фотокамеры, 16 — редуктор
гальванометра, 17 — тумблер для включения дифференциальной
записи
жение подается на электродвигатель, осветитель гальванометра Трз и Л*
и красную лампу Л$, сигнализирующую работу электродвигателя
При замыкании переключателя /7| в положение / осветитель
гальванометра непосредственно соединен с трансформатором, и лампа горит
постоянно. Положение 2 обеспечивает синхронную работу электродвигателя и
лампы осветителя, автоматическое выключение мотора и осветителя
гальванометра при помощи блокирующего устройства Кг. Для пуска
мотора надо нажать кнопку Ki, при этом ток, прошедший через обмотку
206
пускового устройства, замыкает контакт Кь вследствие чего мотор
начинает вращаться, а на травсформатор осветителя подается питающее
напряжение. Ток, протекающий через обмотку двигателя, проходит и через
обмотку трансформатора 7р2, на выходных клеммах которого создается
напряжение 6 е. Вследствие этого загорается красная сигнальная
лампа J/s. Блокирующее устройство связано со шкалой потенциалов. Когда
показания на шкале достигнут заданной величины, устанавливаемой
рукояткой автоматического выключения, произойдет замыкание контакта Кг,
и через обмотку пускового устройства снова пройдет ток; контакт К*
будет разомкнут, электродвигатель остановится, сигнальная лампа и
лампа осветителя гальванометра /югаснут. Эти приспособления делают
работу на приборе весьма удобной.
Скорость изменения потенциала на реохорде
регулируется переключателем скоростей (редуктором
электродвигателя), имеющим следующие диапазоны: 10, 20, 100, 200 и
400 мв/мин.
Гальванометр на специальной консоли крепится к стене.
Положение «зайчика» регулируется системой зеркал, а
изображение щели фокусируется путем выдвижения тубуса
осветителя. Для установки прибора необходимо укрепить консоль
с гальванометром на стене, проверить правильность работы
всех выключателей и отфокусировать изображение щели на
фотокассете.
Полярографы других конструкций. Советский полярограф
СГМ-8 в отличие от ЛП-55 позволяет проводить как
фотографическую, так и визуальную запись, для чего он снабжен
полупрозрачной шкалой длиной 40 см. Чувствительность
гальванометра порядка 10~8 а/мм/м; редуктор позволяет
менять чувствительность от '/г до V2000 от максимальной через
15 ступеней. Многие зарубежные фирмы изготавливают
различные конструкции, приспособленные как для
автоматической, так и для визуальной записи. Большинство моделей
снабжено приспособлениями для подавления осцилляции,
кривые вычерчиваются пером на специальных бланках.
Выпущен ряд катодно-лучевых полярографов,
представляющих собой усовершенствованные модели осциллографи-
ческих приборов, которые позволяют наблюдать исследуемые
кривые (полярограммы) непосредственно на экране катодно-
лучевой трубки. В некоторых приборах вместо
потенциометра, автоматически изменяющего подаваемое на электроды
напряжение, используется конденсатор большой емкости,
который постепенно заряжается от вспомогательного
источника постоянного напряжения. Возрастающая разность
потенциалов на обкладках конденсатора и служит источником
207
XJ U
S CJ
га E
о. а
i- о
« 2 8 Я S.»
с о Ь5 -
со
«a.
К. га
s
H
8*
5 *
qj ra
в1 н
К E
S- О
га :*:
соя
г о
О I-
га с &
X в |
с I
щ к
■■' га *> о.
t>
га
BJ'S Cra
5
1Л
■ - к
~ о
-go
= ^ о
— S
а , .
) а к
: о га
СЗ f- SC
Sot
га
га m
-©■„ R
га ci, s
ug в:
H QJ
rai<
QJ
О ra
^5 щ ь-
^ О о С
oe-o u
<= о о
в ~
гага ™
2 о. га Е:
cj f- с: ^
2 л
J3 О ^
о£5
га о
н Е г-
U з< £
•X >, л
о. —
£ « с
В* с
- СЗ QJ
-ё
2*-
'» С
-^ QJ О
CM |J-
O-fc; О
О ^ й" В
I- £ О
га с ьй | ш г-
.-гага
ь: го и.
ft да
^?95<
о .,
о
= £?
н ё ™
MS
•Я
О
в
QJ
н
(_ ^ га
о •-
о *э х
Е S
О
в ^
■о Ч
ГО QJ
га f-
se s
P*i ra
н
QJ CJ
ft
QJ
[-
ra
о x ft
О QJ X
>, С О
o a
cl о ^" и ft о ^"
постепенного повышения напряжения на
электродах ячейки. Конструкции
советского электронного самопишущего иоля-
рографа разработаны Центральной
лабораторией автоматики.
Подготовка полярографов
к работе
Особое внимание следует уделить
подготовке и очистке ртути', которой
заполняются капилляр и резервуар. Ртуть
лучше брать перегнанную, но можно
применять и тщательно промытую
подкисленным раствором Hg2(NOaJ и затем
дистиллированной водой. Воду с поверхности
ртути снимают фильтровальной бумагой.
Полярографический анализ растворов
можно начинать после того, как прибор
установлен в рабочее положение,
капилляр и резервуар заполнены очищенной
ртутью, определена чувствительность
гальванометра и проградуирован
капельный ртутный электрод.
Когда резервуар и капилляр заполнены
ртутью, отверстие бюретки закрывают
ватной пробкой, а отверстие
резервуара— резиновой или корковой пробкой, в
которую вставлен платиновый электрод,
служащий для контакта с ртутью.
Тщательно собранный и правильно
установленный прибор нормально работает в
течение длительного времени.
Перед началом работы каждый раз
проводят осмотр прибора. При этом
необходимо проверить: 1) степень заряжен-
ности акумулятора; 2) качество
выносного анода и электролитического ключа;
3) имеет ли рамка гальванометра
свободный ход и правильно ли освещается
щель фотокамеры. Перед включением
1 См П. П. П > г а ч е в и ч Техника работы
со ртутью в лабораторных условиях. Госхимиз-
дат, М, 1961.
209
прибора все контакты и потенцнометрическую проволоку
промывают бензином и насухо вытирают (тряпочкой, но не ватой)
н устанавливают требуемую скорость капания ртути, поднимая?
или опуская резервуар с ртутью. После составления схемы и
включения прибора проверяют, есть ли ток в цепи электроли-
зерной ячейки.
Проверка качества схемы
Одновременную проверку всех основных элементов схемы
проводят следующим простым способом. Вместо
электролитической ячейки в цепь включают магазин сопротивлений,,
например типа Р-14, на котором устанавливают
сопротивление 20000-—40 000 ом с таким расчетом, чтобы «зайчик»
гальванометра отклонялся от нулевого до максимального
деления шкалы, при изменении напряжения на потенциомет-
рической проволоке от 0 до Зе.
Дальнейшие операции, как и при полярографированипг
сводятся к измерениям отклонений «зайчика» гальванометра
(силы тока) при различных значениях поданного на магазин
напряжения. Измерения силы тока проводят через каждые
40—60 мв, начиная с нулевого деления. Закончив первую
серию измерений, увеличивают чувствительность
гальванометра и проводят вторую серию измерений, и так несколько1
раз (на автоматических полярографах несколько раз
повторяют автоматическую запись кривой).
В этих условиях опыта сопротивление цепи в каждой1
серии измерений остается постоянным, а по закону Ома сила-
тока прямо пропорциональна величине поданного
напряжения. При графическом изображении полученных данных в
координатах сила тока — напряжение эта зависимость
выражается прямой линией. Семейство прямых, полученных
с разной чувствительностью гальванометра, свидетельствует
о пригодности электрической схемы для дальнейших работ.
Различные неисправности в схеме отражаются на графике
нарушением прямолинейной зависимости между силой тока
и напряжением.
Причины некоторых неисправностей можно найти,
сравнивая кривые, полученные при различной чувствительности
гальванометра.
1. Плохое качество контакта между потенциометрической
проволокой и скользящим контактом приводит к резким и
незакономерным изменениям силы тока при любых чувстви-
тельностях гальванометра; диапазон колебаний возрастает
210
•с увеличением чувствительности. Для устранения неисправно-
•сти надо промыть потенциометрическую проволоку или
усилить натяжение пружины, прижимающей скользящий контакт
к проволоке
2. Плохой контакт на щетках, подводящих напряжение к
потенциометрическому барабану, проявляется аналогично;
если нарушения происходят при одном и том же положении
барабана, то резкое падение силы тока можно наблюдать
через равные промежутки при каждом обороте барабана.
3. Отдельные витки проволоки на барабане уложены
неправильно, при вращении барабана подвижный контакт
замыкается на соседний виток проволоки; это вызывает
кратковременное изменение силы тока, происходящее всегда при
•одном и том же напряжении.
4. Рамка гальванометра при своем вращении задевает за
неподвижные части гальванометра; при этом наблюдаются
перегибы или резкие изломы на кривой сила тока —
напряжение, происходящие при одной и той же силе тока,
независимо от приложенного напряжения (это легко проверить,
•сравнивая кривые, полученные при разной чувствительности
гальванометра или при различном сопротивлении цепи).
Нсли при тщательной установке гальванометра по уровню
эти нарушения не устраняются, необходимо заменить
гальванометр.
5. Если источник напряжения (аккумулятор) разряжен н
•его напряжение во времени падает, то вместо прямых линий
на графике могут получиться плавные кривые, причем
степень отклонения от прямолинейной зависимости во времени
возрастает. Надо заменить аккумулятор на
свежезаряженный или перезарядить старый.
6. При одном или нескольких положениях редуктора
чувствительности наблюдаются незакономерные изменения силы
тока (как в случае 1) или же при некоторых положениях
«зайчик» гальванометра вовсе не отклоняется от нулевого
положения. Причиной неисправности в первом случае
является плохое качество контактов в переключателе редуктора чув-
-ствшельности, во-втором — обрыв соединительных проводов
в самом редукторе.
Определение чувствительности гальванометра
Чувствительность гальванометра необходимо определять
в тех случаях, когда вычисляют концентрации по уравнению
Ильковича. Простой способ определения предложен Кольт-
211
гофом Вместо электролизера в цепь включается
прецизионный магазин сопротивлений и затем измеряется величина
отклонения «зайчика» гальванометра при различной величине
поданного на магазин напряжения
Последовательность определения чувствительности
гальванометра следующая Устанавливают на реохорде некоторое
произвольное значение напряжения, подаваемого на магазин
A —1,5 в), и подбирают такое сопротивление, чтобы
получить отклонение «зайчика» гальванометра примерно на
половину шкалы При этом, согласно закону Ома
E = J(Rx-\-Ru),
где / — сила тока в цени, Е- напряжение, поданное на
магазин сопротивлении, Rn— сопротивление на магазине и Rx—
сопротивление остальной части цепи, величина которого
остается неизвестной
Для вычисления силы тока величину RK надо исключить,
что можно сделать путем составления второго равенства Для
этого увеличивают сопротивление магазина примерно вдвое
и повышают Е (изменяя положение подвижного контакта на
потенциометре) до тех пор, пока не будет достигнута
прежняя сила тока и «зайчик» гальванометра возвратится точно в
прежнее положение При этом
E1--J(RX + R'M).
Вычитая почленно из второго равенства первое, получим
р р
Ех — Е ■--■ J (RM — Rv) или J -■= -^ —
Теперь легко вычислить чувствительность гальванометра,
т е. силу тока, вызывающую отклонение гальванометра на
одно деление шкалы Для этого найденную величину / деляг
на число делений d, на которое отклонялся «зайчик»
гальванометра при измерениях Кроме того, чтобы получить
максимальное значение чувствительности (полную
чувствительность), эту величину нужно еще умножить на показатель
положения редуктора чувствительности f, при котором
производились измерения Нсли Е выражено в вольтах,
сопротивление— в омах, а одно деление шкалы равно 1 мм, то
общая формула для вычисления максимальной
чувствительности гальванометра приобретает вид
5- -(Е1~Е)± о/мм/м
,д^ IR'H-Ru)d
V)
212
ПОЛУЧЕНИЕ ПОЛЯРОГРАММ И ИХ РАСШИФРОВКА
Порядок работы практически одинаков и на визуальном
и на самопишущем (автоматическом) полярографе
Несколько миллилитров исследуемого раствора помещают в
электролизер, добавляют туда же равный объем фона и 5—6 капель
0,25%-ного раствора желатина Затем пропускают в течение
10 мин чистый водород для удаления растворенного
кислорода (или, в зависимости от метода, добавляют несколько
кристаллов NazSO-j).
Электролизер с анализируемым раствором закрывают
пробкой с двумя отверстиями для ртутного капельного и
каломельного электродов Не рекомендуется погружать
капельный электрод слишком глубоко в раствор, так как при этом
ртуть, накапливающаяся на дне сосуда, может соприкасаться
с капельным электродом, а это приводит к резким и
незакономерным изменениям силы тока
Резервуар с ртутью устанавливают на требуемую высоту,
включают осветитель гальванометра и, проверив нулевое
положение «зайчика» на шкале, замыкают цепь электролизера
на гальванометр Аккумулятор включается за 10—15 мин до
начала работы, чтобы падение напряжения на потенциометре
приобрело устойчивое значение
Перед съемкой просматривают всю полярограмму
непосредственно по показаниям гальванометра, начиная с
наименьшей чувствительности Предварительный просмотр
позволяет обнаружить волны восстановления ионов и определить
приближенные значения потенциалов начала и конца каждой
из волн.
Чувствительность гальванометра подбирают для каждой
волны в отдельности Для этого при помощи потенциометра
придают капельному электроду потенциал несколько
меньший, чем потенциал восстановления определяемого иона, и
«зайчик» гальванометра устанавливают на нулевое деление
шкалы при помощи механического корректора или
электрокорректора Затем придают капельному электроду потенциал,
соответствующий максимальному диффузионному току
данного иона; и постепенно увеличивают чувствительность
гальванометра, пока «зайчик» не сместится почти до конца
шкалы Проверяют положение нуля гальванометра (при
начальном потенциале) и в случае изменения последнего повторяют
настройку, но уже не с минимальной, а с подобранной в
первом опыте чувствительностью гальванометра
Затем устанавливают исходное значение потенциала, ба-
213
л/а
$>абан с фотобумагой ставят на нулевое деление и
одновременно включают мотор, лампу «абсцисс» и открывают щель
«фотокамеры. Когда сила тока достигнет в процессе записи
предельного значения, сначала закрывают щель фотокассеты,
выключают мотор и переводят подвижный контакт
потенциометра в нулевое положение. Запись полярограммы повторяют
те менее трех раз, последовательно уменьшая
чувствительность гальванометра.
Расшифровка полярограммы заключается в определении
потенциала полуволны и силы диффузионного тока (высоты
волны). Наиболее точное
определение высоты волны
может быть сделано по
методу Г. Хона. Для этого три
линии, составляющие волну,
продолжают до взаимного
попарного пересеченая
(рис. 50). Через точки лере-
сечения проводят прямые,
параллельные оси абсцисс.
Расстояние между этими
прямыми по вертикали
считают равным высоте волны.
Потенциал полуволны
находится из того же построения,
для чего проводят третью
линию параллельно оси
абсцисс на уровне, равном половинному значению диффузионного
тока; потенциал полуволны определяется как проекция точки
пересечения этой прямой и волны на ось абсцисс.
Определение концентрации по высоте волны.
Вычислением концентрации по уравнению Ильковича, как правило,
пользуются редко. Это вызвано сложностью определения
коэффициента диффузии, величина которого не остается
постоянной в растворах различного состава и концентрации, и
тем обстоятельством, что диффузионный ток не всегда прямо
пропорционален концентрации. Значительно более просто
(и более точно) концентрацию можно определить либо по
градуировочному графику, либо по методу добавления
стандартов.
Для построения градуировочного графика снимают
полярограммы серии стандартных растворов; концентрацию
стандартов выбирают так, чтобы содержание определяемого иона
в наиболее разбавленном растворе было несколько меньше,
Рис. 50 Определение высоты
волны при значительных осцилляци-
ях
214
а в наиболее концентрированном — несколько больше, чем в-
исследуемых образцах. Исходный раствор готовят из солей,
имеющих устойчивую весовую форму, или путем растворения
чистых металлов. Концентрация исходного раствора обычно
равна 1 • 10_2% или 0,1 мг определяемого вещества в 1 мл;
стандартные растворы получают разбавлением до нужной
концентрации исходного раствора.
По величинам измеренных диффузионных токов серии
стандартных растворов строят график, откладывая по оси
ординат значения диффузионных токов (или просто высоты
волн), а по оси абсцисс — концентрацию стандартных
растворов.
Удобно пользоваться не абсолютными значениями высот
волн, а их приведенными значениями, т. е. высотой волны в
миллиметрах, деленной на показатель чувствительности
гальванометра (положение редуктора чувствительности). Так,
если были найдены следующие высоты волн h: при '/з
максимальной чувствительности 7 мм, при '/ю — 11 мм и при
'/юо—15 мм, то вычисленные для построения графика
высоты h/f соответственно равны: 7:'/з = 21; 11:1/ю=110 и
15 : 7ioo= 1500. Необходимо помнить, что сравнимые величины
высот воли стандартных и исследуемых растворов
получаются только в том случае, когда условия съемки полярограмм
совершенно одинаковы (состав раствора, скорость капания
ртути из капилляра, температура и т. п.).
По методу Хона условия съемки сохраняются строго
постоянными, и тем самым обеспечивается достаточная
точность результатов измерений. Постоянство условий
достигается тем, что обе полярограммы (стандарта и образца)
снимаются в одном и том же растворе. Для этого сначала
снимают полярограмму определяемого иона в анализируемом
растворе, а затем в тот же электролизер добавляют из
пипетки известный объем стандартного раствора (часто 0,1 —
0,2 мл), содержащего ионы того же вида, но известной
концентрации, и получают вторую полярограмму.
Концентрацию исследуемого раствора рассчитывают
путем сравнения высот волн на обеих полярограммах. Если
высота волны определяемого иона, полученная в растворе
неизвестной концентрации, равна hi, объем его — v\, высота
волны после добавления стандартного раствора равна /г2,
объем добавленного раствора — v2 и концентрация
стандарта— С2,то при смешении растворов концентрация стандарта
вследствие разбавления уменьшится до Са- , а высо-
vt + v2
215
та первоначальной волны до Лх- —; увеличение волны в
vi + Щ
результате добавления стандартного раствора равно
АА-=А, —Аг —?!—.
Исходя из этого, при помощи простой пропорции легко
подсчитать, что первоначальная концентрация была равна
,-, CJi,v, ,-, C,h,v>
(Ag — hj) Vi + А,у> AAnt -f- Ajtia
Концентрацию добавляемого стандартного раствора
выбирают так, чтобы высота волны увеличилась примерно в два
раза.
Если состав исследуемого раствора резко отличен от
состава стандарта, а зависимость силы тока от концентрации не
подчиняется уравнению Ильковича, то ни метод стандартов,
ни метод добавок не могут дать хороших результатов. В этом
случае можно рекомендовать применение двух добавок или
разбавление раствора по следующем схеме.
Сила предельного тока сначала измеряется в исходном
анализируемом растворе с неизвестной концентрацией
определяемого иона х (высота волны равна h\); затем повторяют
измерение в том же растворе, но разбавленном в п раз
(высота волны равна /г2), и в исходном растворе с добавкой
стандарта (концентрация добавленного стандарта равна С\,
высота волны с добавкой равна h3). Если три измеренные
высоты волны отличаются не более чем в 1,5—2 раза и в этих
пределах высота волны прямолинейно зависит от
концентрации, то можно найти искомую концентрацию по
интерполяционной формуле
х .. (А, — h2) п с
(hs-~hv)(n-\) 1-
О точности и воспроизводимости определений
Общин анализ отдельных факторов, влияющих на силу
предельного тока, по уравнению Ильковича выполнен Кольт-
гофом и Крюковой. Возможные колебания температуры,
давления ртути, коэффициента диффузии и др приводят к
ошибкам порядка 1—2%. Эти ошибки по Кольтгофу могут быть
сведены к минимуму, если обобщенная константа К
уравнения Ильковича определяется экспериментально в тех же
216
точно условиях, которые применяются реально при анализе,
а всю установку термостатируют. Наибольшие погрешности
возникают при измерении силы предельного тока (высоты
волны); дополнительные ошибки, часто очень значительные,
вносятся при подготовке пробы почвы к полярографированию.
Практическое измерение силы предельного тока при
автоматической записи кривых производят путем промера высо-
Рис 51 Полярограммы никеля и цинка (/) и кобальта B) в
экстрактах из почвенных образцов при малом содержании указанных
элементов в почвах
ты волны. При содержании определяемого иона порядка
Ю~5% анализ приходится вести при высокой
чувствительности гальванометра. Это вызывает значительные осцилляции
в записи тока; кроме того, при анализе почв с малой
концентрацией элементов участки остаточного и предельного тока
имеют, как правило, значительный наклон (рис. 51), причем
угол наклона неодинаков в начале (остаточный ток) и конце
волны (предельный ток).
Измерение высоты волны в таких условиях не может
быть абсолютно точным. Различные способы измерения (по
минимальным, максимальным или средним точкам
осцилляции) дают одинаковые количественные результаты, как
показал П. Н. Терещенко, изменяется несколько только наклон
градуировочной кривой. Но при всех способах не удается
добиться промеров с абсолютной ошибкой, меньшей, чем 1 мм.
Снижение чувствительности гальванометра обычно значи-
217
тельно улучшает форму волны, но одновременно увеличивает
относительную ошибку измерения Поэтому наиболее
правильным является полярографирование в трехкратной
повторное™ при различных чувствительностях гальванометра.
Если при количественных измерениях точность промера
высоты волны ограничена I мм, то нетрудно подсчитать
возможную погрешность в зависимости от величины волны.
При высоте волны 50—100 мм (что встречается довольно
часто) погрешность измерения составляет 1—2%; при
меньшей концентрации, когда высота волны не превышает
10—20 мм, погрешность составляет 5—10%.
ПРИМЕНЕНИЕ ПОЛЯРОГРАФИЧЕСКОГО АНАЛИЗА К ИЗУЧЕНИЮ
СОСТАВА И СВОЙСТВ ПОЧВ
Определение меди, никеля, цинка и кобальта в почвах
При анализе почв и других природных объектов
микроэлементы Си, Ni, Zn и Со (или Zn, Ni, Cd и Pb) определяют
обычно одновременно, так как они дают хорошо выраженные
волны на одинаковом фоне. Подготовка образцов и обработка
пробы при определении каждого из перечисленных элементов
одна и та же.
Си, Ni, Zn, Cd, Pb, Bi, Co принадлежат к группе
элементов, определение которых возможно путем прямого поляро-
графирования с ртутным катодом. Для всех элементов при
содержании их в растворе около 10~4—10~3% сохраняется
прямолинейная зависимость силы диффузионного тока от
концентрации. Состав фона может быть различен, чаще
других применяют смеси NH4CI + NH3, K.CNS или оксалатные
растворы (табл.17).
Ход анализа почв и других природных объектов на
содержание микроэлементов складывается из ряда операций:
переведение анализируемой навески в раствор, обогащение
раствора определяемыми элементами, полярографирование.
При определении микроэлементов особое внимание
должно быть уделено химической посуде и чистоте реактивов.
Все операции, а особенно с применением кислот или
щелочей при нагревании (кипячение, выпаривание), проводят
только в кварцевой (иногда платиновой) посуде.
Низкотемпературные обработки (до 250—300°) можно вести в
фторопластовой (тефлоновой) посуде Обычную лабораторную
посуду, например марки «Дружная горка» № 23,
можно использовать для проведения кратковременных опе-
218 \
Таблица 17
Потенциалы полуволн Си, Pb, Cd, Ni, Zn и Со
Определяемый
элемент
Медь
»
Свинец
Кадмий
»
Никель
»
Цинк
»
Кобальт
»
Состав фона
1М NH3+1M NH4C1
0,Ш KCNS
0,Ш KCNS
1М NH4OH
0,1М KCNS
1М NH3-f 0,2M NH4C1
0.1М KCNS
1М NH3+0,2M NH4CI
0,5М KCNS
1М NH4C1+1M NH4OH
1М KCNS
Изменение
заряда иона
1-^0
2-»1
1—0
2-0
2-0
2-0
2—0
2-Л
2—0
2-0
2—0
2—0
е1/2
—0,54
—0,02
-0,39
—0,38
—0,74
—0,59
— 1,06
—0,69
—1,33
—1,02
—1,30
—1,03
раций при комнатной температуре (экстракция
микроэлементов дитизоном, иолярографирование).
Реактивы, употребляемые при анализе, обычно требуют
дополнительной очистки. Вода должна быть дважды
перегнана. Продажные реактивы, даже марки «х. ч.», содержат
примеси различных элементов в количествах, часто
соизмеримых с содержанием этих элементов в почвах и природных
водах. Наибольшие погрешности при определении
микроэлементов полярографическим методом обусловлены
загрязнением реактивов; постановка «холостых» определений, обычно-
позволяющая учесть влияние примесей определяемого иона в
реактивах, при определении микроэлементов оказывается
недостаточной. Содержание микроэлементов в «холостом»
опыте при использовании реактивов с маркой «х. ч.» может
быть одинаковым или даже значительно большим, чем в
исследуемом образце, поэтому введение поправок на «холостое»
определение не дает положительных результатов. Растворы
кислот и щелочей, очищенных для микроэлементного анализа,
хранятся в кварцевой или парафинированной стеклянной
посуде, в посуде из полиэтилена.
Переведение навески почвы в раствор достигается
сплавлением почвы с содой или обработкой ее плавиковой
кислотой в соответствии с обычными методами валового анализа.
Растительный материал подвергается озолению. Получаемый
таким образом раствор содержит ряд элементов, мешающих
или затрудняющих определение (Fe, Al, Si), в то время как
концентрация определяемых микроэлементов может быть
219
•слишком низкой. Поэтому определяемые элементы извлекают
из раствора и концентрируют в малом объеме растворителя.
Для отделения определяемых ионов от других,
присутствующих в образце, используют способность дитизона (дифе-
нилтиокарбазона)
s_c/NH-NH-C6H6
\n~n-ch,
образовывать очень прочные соединения со многими
тяжелыми металлами. При взаимодействии дитизона с металлами
происходит замещение водородных атомов NH-групп на
металл с образованием комплексных соединении.
Образующиеся соединения, дитизонаты различных металлов, ярко
окрашены, сравнительно трудно растворимы в воде и
значительно легче растворимы в хлороформе и четыреххлористом
углероде. Если к раствору, содержащему ионы тяжелых
металлов, добавить раствор дитизона в ССЦ и энергично
взболтать, то после отстаивания смесь разделяется на две
фазы; нижняя представляет раствор дитизонатов и избытка
дитизона в ССЦ. При этом изумрудно-зеленый цвет дитизона
переходит в красную (бордово- или фиолетово-красную)
окраску дитизонатов.
Экстракция ионов различных металлов происходит при
неодинаковых условиях. Наиболее сильно влияет на
образование дитизонатов и экстракцию ионов металлов присутствие
•комплексообразователей, способных связывать определяемые
ионы более прочно, чем дитизон; другим важным фактором
является рН раствора. Путем изменения рН и добавления
комплексообразователей (роданид-ион, тиосульфат) можно
подобрать условия, когда будут экстрагированы только те
ионы, содержание которых предполагается определять.
Области экстракции и окраска дитизонатов приведены в
табл. 18.
Определение валового содержания микроэлементов в почвах
по Н. Г. Зырину, Д. С. Орлову, Н. М. Гриндель
Навеску почвы 1—1,5 г сплавляют с 6-кратным
количеством соды и отделяют кремнекислоту обычным методом;
■осадок кремнекислоты на фильтре тщательно промывают,
чтобы избежать потерь микроэлементов вследствие сорбции.
Фильтрат после отделения кремнекислоты выпаривают
досуха, добиваясь возможно полного удаления НО. Сухой
220
Таблица 18
'Области 100 %-ной'экстракции дитизонатов СС14 в зависимости от рН
(по Г. Иваичеву)
Соединение
H.Dz
AgHDz
Ug (HDz)
HgDz
PdfHDz),
Pt(HDz),
Au(HD7.),
Po(HDz)'
CutHDz),
CuDz
BI{HDz),
In(HD),
Sn(HDz),
Zn(HDz),
Cd(HDzb
Co(HDz),
CoDz
Ni(HDz),
Pb(HD7)8
Fe(HDz)!
Mn(HDz),
TlHDz
pH
-1 1
9 11 13 15
Цвет
i.liliMllllliJi^
-j^jfianiKLL
~SP I in.! iiMiniiimg
-*дпашлшгр'
изумрудно зеленый
оранжено желтый
золотисто-желтый
оранжеио-желтый
фиолегоиый
бледно-зеленый
коричнево желтый
золотисто-желтый
ьрасно-коричневый
красно-фиолет оный
желто-коричнеи ый
красно-оранжевый
малиново-крлсный
красный
пурпурно-красный
розово-красный
красно-фислетовый
желто-коричневый
коричнево-фиолетовый
карминово-красный
фиолетово-краен ый
коричневый
(растворим в хлороформе)
малиново- красный*
Обозначения: HyDz
дитизон:
однозамещенные дитизонаты;
днузамещенные дитизонаты;
экстракция HDz~ в водном растворе.
В водной суспензии.
221
остаток растворяют в малом количестве 0,5 Л\ лимонной
кислоты; затем раствор подщелачивают аммиаком по-
фенолфталеину, переносят в делительную воронку и
добавляют 15—20 мл раствора дитизона в ССЦ.
Тщательно встряхивают полученную смесь в течение
5 мин; образующиеся дитизонаты переходят в фазу ССЦ,
которая приобретает ярко-красную, вишневую или буроватую,
окраску (в зависимости от содержания того или другого
элемента). Смеси дают отстояться до полного разделения на
две фазы и сливают раствор дитизонатов (фаза с CCU) в
кварцевую чашку на 100 мл. Повторяют экстракцию до тем
пор, пока зеленая окраска дитизона не перестанет изменяться
после взбалтывания с анализируемым раствором. Экстракты
собирают вместе и сгущают на водяной бане. Сгущенные
экстракты в кварцевых чашках обрабатывают на водяной
бане 5 мл конц. НЫОз, если при этом дитизонаты не будут
полностью разрушены, то снова добавляют 2 мл HNO3, 0,5 мл
НС104, 2—3 капли конц. H2SO4 и выпаривают до появления,
белых паров. Вновь повторяют операцию до полного
разрушения дитизонатов, о чем можно судить по обесцвечиванию
раствора и сухого остатка. Сухой остаток прокаливают при
температуре 450—500°, после остывания чашек 2—3 раза
обрабатывают несколькими каплями конц. НС1, которую затем
почти нацело удаляют нагреванием на песчаной бане, и
растворяют осадок в 2—3 мл воды.
Поскольку Zn и Со имеют одинаковые потенциалы
полуволн, Со отделяют от остальных элементов осаждением,
а-нитрозо-р-нафтолом, для чего добавляют 2 мл 80%-ной
СНзСООН и 0,5 мл 3%-ного раствора а-нитрозо- р-нафтола в
60%-ной СНзСООН. Чашки с осадком закрывают стеклом и
нагревают на водяной бане; после охлаждения добавляют
1 мл 24%-ной НС1 и отфильтровывают через маленький
фильтр, предварительно промытый 10%-ной НО. Осадок на
фильтре промывают 2 раза той же кислотой и затем
небольшой порцией горячей воды.
Фильтр с осадком кобальтовой соли озоляют и
прокаливают, а фильтрат, содержащий остальные определяемые
элементы, выпаривают досуха, остаток смачивают несколькими
каплями 10%-ной H2SO4 и тоже прокаливают. Прокаленные
остатки растворяют в нескольких каплях конц. НС1, которую
затем выпаривают на водяной бане, и остаток растворяют в
2—3 мл бидистиллированной воды (если осадок плохо
растворяется, можно добавить 1 каплю разбавленной НС1).
Приготовленные таким образом растворы переносят в
222
■мерные цилиндры на 10 мл с притертыми пробками, обмывая
чашки сначала 2—3 мл воды, а затем 5 мл раствора фона.
Общий объем должен быть 10 мл. Раствор в цилиндрах
перемешивают, переносят в электролизеры и после добавления
нескольких E—6) капель 0,25%-ного раствора желатина и
нескольких кристалликов Na2S03 полярографируют. Фоном
•служит смесь 200 мл 0,1 н. NH4C1, 50 мл 0,1 н. СаС12 и 100 мл
1,0 н. NH4OH в 500 мл воды.
Потенциалы полуволн определяемых элементов имеют
следующие значения: Си—0,36 в; Cd—0,65 в; Ni—0,90 е;
Zn—1,20 в; Со—1,20 в.
Тем же методом можно определить подвижные (водно-
растворимые и обменные) формы соединений
микроэлементов, причем для приготовления водной вытяжки необходимо
взять 50—100 г почвы, при анализе обменных форм — от 10
до 20 г. Поглощенные Zn, Си, Ni и Со вытесняют 1,0 н
CH3COONH4. Весь ход анализа сохраняется, отпадает только
необходимость отделения кремнекислоты.
Ускоренный полярографический метод определения железа
в почвах и растениях
Быстрый метод определения железа в продуктах
органического происхождения разработан Синяковой с
сотрудниками. Аналогичный вариант применили П. А. Власюк и
Е. С. Косматый для определения железа в почвах и
растениях. Метод основан на прямом полярографировании
Fe3+-HOHa в солянокислой среде. В 0,8—1,0 н. растворе НО
волна восстановления начинается от нуля приложенной
э. д. с; сила тока достигает предельного значения при
потенциалах от —0,2 до —0,3 е. В 1,0 н НС1 соблюдается линейная
зависимость между силой предельного тока и концентрацией
железа. Ионы Ni, Zn, Cd, Pb, Mn не мешают определению.
Ход анализа
К 10 г почвы прибавляют 250 мл 0,5 н. НС1, взбалтывают
на ротаторе в течение часа и фильтруют. Из приготовленной
вытяжки берут 10 мл, подкисляют 10%-ной НС1 с таким
расчетом, чтобы общая концентрация НС! была равна 1,0 н,
и доливают водой до 20 мл. Полученный раствор помещают в
электролизер, пропускают через него в течение 20 мин
водород и полярографируют в пределах от нуля до 0,6 в. Ошибка
223
определения по сопоставлению с химическими методами не
превышает J %.
Определение железа в растительном материале проводят
аналогично. Навеску растений озоляют, золу растворяют
в 2—3 мл 6,0 н. НС1 и раствор разбавляют водой до
концентрации НО = 1,0 н. Далее поступают как при анализе
вытяжек из почвы.
Определение кальция в почвах и растениях
Ион кальция восстанавливается на ртутном капельном
электроде при потенциалах от —2,0 до —2,2 в на фоне
(CHsLNOH или (CH^NCl. Близкие величины потенциалов
полуволн в тех же растворах имеют К, Li и Na. Это
осложняет прямое полярографическое определение кальция в
почвах, природных водах и образцах растений. Поэтому были
предложены косвенные методы определения кальция, в
частности, с применением трилона Б.
Методика полярографического определения кальция с
применением трилона Б описана Р. Пршибилом, а затем
применена Скобец для анализа почв. Метод основан на
способности ионов кальция количественно вытеснять ионы цинка
из соответствующего комплекса в сильно аммиачной среде.
Если обозначить двузарядный ион этилендиаминтетрауксус-
ной кислоты символом НгУ2~, то образование комплексоната
цинка можно представить но схеме
Н2У2~ + Zn2+ -^ Zny2~ -!- 2Н+.
Константа устойчивости комплексоната цинка (—\gKZny~ --
= 16,16) значительно выше константы устойчивости
комплексоната кальция (—-lgAcay*- =10,59), поэтому полное
количественное вытеснение цинка из комплекса происходит только
в среде с концентрацией NH4OH не менее, чем 4 М. При
введении ионов кальция в сильно аммиачный раствор
комплексоната цинка происходит реакция:
Zny2~ + Са2+ -» Zn2+ + СаУ2~.
Вытесненное из комплекса количество ионов цинка
эквивалентно содержанию кальция в анализируемом растворе и
легко может быть определено полярографически.
Определению мешает присутствие магния, поскольку последний
реагирует с комплексонатом цинка аналогично иону кальция.
Поэтому магний следует удалять из раствора осаждением в
виде MgNH4P04.
224
Перед определением готовят раствор комплексоната
цинка растворением 1,9170 г ZnS04 • 7Н20 и 2,4800 г
натриевой соли этилендиаминтетрауксусной кислоты в 1 л 8,0 М
NH4OH. Этот раствор комплексоната дает на полярограмме
хорошо выраженную волну цинка. При определении кальция
для построения градуировочного графика берут разность
высот волн исследуемого и исходного растворов
комплексоната.
Выполнение анализа сводится к следующему. В
электролизер помещают 10 мл раствора комплексоната цинка, 1,0 ял
раствора (NH4JHP04, добавляют 10 капель желатина и
после доведения объема раствора водой до 20 мл пропускают
в него водород в течение 10—15 мин. Затем снимают волну
цинка.
К тому же раствору добавляют 1—2 мл анализируемого
раствора, содержащего ионы кальция; по разности высот
волн находят приращение силы диффузионного тока,
пропорциональное содержанию кальция в растворе.
Содержание кальция во всем объеме раствора, взятого
для анализа, не должно превышать 2 мг. При более высокой
концентрации кальция приходится брать не 10, а 15—20 мл
исходного раствора комплексоната цинка. Калибровочная
кривая строится по растворам СаНР04 • 2Н20.
Этим методом удобно определять в почвах содержание
обменного и воднорастворимого кальция. Для вытеснения
обменного кальция можно воспользоваться как методом
К- К. Гедройца (обработка 10—20 г почвы 1,0 н. раствором
NH4C1 до полного замещения), так и методом настаивания.
В последнем длительный процесс отмывания почвы на
воронках заменяется 12-часовым настаиванием 1 г почвы с 100 мл
1,0 н. NH4C1.
Несомненное преимущество полярографического метода в
данном случае заключается в возможности определения очень
малых количеств кальция, а также в том, что можно брать
малые навески и значительно экономить время,
затрачиваемое на вытеснение поглощенных оснований, и уменьшить
расход реактивов.
Определение емкости поглощения почв
В основу ускоренного полярографического метода
определения емкости поглощения почв (Е. М. Скобец, И. Л. Абар-
барчук, К. П. Костицина, Н. И. Белинская), положено
определение емкости по Е. В. Бобко и Д. Л. Аскинази и по
А. Шмуку с полярографическим определением бария или
8 Зак. 51
225
никеля, применявшихся для насыщения почвенного
поглощающего комплекса (полярографическое окончание).
Определение емкости поглощения почв по барию. Навеска
почвы в 0,5—1,0 г помещается в стеклянную воронку
с плотным фильтром; к концу воронки посредством резиновой
трубки прикреплен кран Почву на фильтре обрабатывают
небольшим количеством 1,0 н. раствора ВаС12; через 3—5 мин
кран открывают и дают раствору BaCU полностью
профильтроваться. Для полного насыщения барием
поглощающего комплекса обработку повторяют 12—15 раз. После
отмывания почвы дистиллированной водой до исчезновения
реакции на С1-ион воронку помещают в горло мерной колбы
на 100 мл и обрабатывают 1 н. раствором НС1.
Для полного вытеснения поглощенного бария достаточно,
по данным авторов, обработать почву 60—70 мл 1 н. НС1.
Получаемую солянокислую вытяжку нейтрализуют СаО до
ясно щелочной реакции и по охлаждении разбавляют водой
до 100 мл.
В растворе полярографически определяют барий на фоне
СаСЬ, образовавшегося в процессе подготовки раствора.
Потенциал полуволны бария—-1,83 в (анодом служит
донная ртуть).
Определение емкости поглощения по никелю. Аналогично
'предыдущему методу почву обрабатывают на воронке 0,5 н.
раствором NiCl2 и отмывают затем водой до потери реакции
на Ni2+-noH. Поглощенный никель вытесняют 0,5 н. НС1, и
собранный фильтрат нейтрализуют 25%-ным NH4OH до
щелочной реакции. Потенциал полуволны никеля но
отношению к насыщенному каломельному электроду — 1 в. Перед
определением никеля из раствора удаляют кислород одним
из описанных выше методов п для подавления максимума
добавляют 1 мл 0,5%-ного раствора желатина.
Определение емкости поглощения по свинцу. Поскольку
полярографическое определение бария несколько
затруднительно, а соединения никеля стоят довольно дорого, Скобец
с сотрудниками предложили для насыщения почвы при
определении емкости поглощения пользоваться солями свинца.
При этом почву на воронке, как и ранее, обрабатывают
1,0 н. раствором Pb(NOjJ. После промывания
дистиллированной водой поглощенный почвой свинец вытесняют
60—70 мл 0,5 н. HNOs, порциями но 8—10 мл.
Добавив 1 мл 0,5%-ного раствора желатина и пропустив
водород, снимают полярограмму свинца. Потенциал
полуволны свинца в этих условиях 0,40 в.
226
Подавление полярографических максимумов гумусовыми
веществами почвы
/ \тл№
Рис 52 Максимум волны кислорода:
и — в 1,0 и. растворе NH4OH; б —
в присутствии гуминовой кислоты
Гуминовые и фульвокислоты не обладают способностью
восстанавливаться на ртутном капельном электроде.
Растворимая в спирте фракция гуминовой кислоты, называемая
гиматомелановой кислотой по данным Токуока и Ружички,
восстанавливается на
фоне LiOH при потенциале
1,4 е. В то же время все
фракции гуминовых и
фульвокислот активно
подавляют
полярографические максимумы. Эта
способность к подавлению
максимумов настолько
хорошо выражена, что
может быть использована
для качественной
характеристики органических
веществ почвы и
количественного определения
отдельных фракций.
Наиболее эффективно
гуминовые и
фульвокислоты подавляют
положительные максимумы и в
меньшей степени влияют
на отрицательные. На
рис. 52 показан максимум
ка волне кислорода в
1,0 М растворе NH4OH.
Чтобы получить эту поля-
рограмму, в ячейку
наливают несколько
миллилитров 1,0 М раствора NH^OH
и снимают волну
растворенного кислорода в
диапазоне от 0 до 0,5 в.
На полярограмме
хорошо очерчен максимум, высоту которого определяют по
разности между наибольшей силой тока и силой предельного
тока. Для изучения полярографической активности гуминовой
кислоты аммиачный раствор последней добавляют из микро-
Нонцентрация гума/па
Рис. 53 Зависимость высоты максимума
от концентрации гумата
8*
227
бюретки в ячейку небольшими порциями. Концентрация
добавляемого раствора—10—20 мг гуминовой кислоты в
100 мл 1 н. NH4OH. После добавления каждой порции
гуминовой кислоты раствор перемешивают и вновь снимают поля-
рограмму кислородного максимума. Высота максимума
постепенно уменьшается (рис. 53). Построив кривую
зависимости высоты максимума от концентрации гумата в
электролизере, получают характеристику полярографической
активности гуминовой кислоты, которую можно использовать
в качестве градуировочного графика для определения
концентрации гуминовых кислот в различных вытяжках из почв.
Качественная характеристика гумусовых веществ, по
Лгоцена-Конде, дается величиной коэффициента
полуподавления, который равен концентрации гумусовых веществ
(мг/л), необходимой для уменьшения высоты
полярографического максимума на половину (табл. 19).
Таблица 19
Коэффициенты полуподавления полярографических максимумов
гумусовыми веществами в мг/л
Вещества
Гиматомелановая кислота . .
Максимумы
положительные, на
волне
кислорода
0,40
4,00
85,0
отрицательные, на волне
N.2+
26,0
275,0
не подавляет
Zn2+
62,5
662,0
не подавляет
V
Глава VI
ИССЛЕДОВАНИЯ ПОЧВ МЕТОДАМИ АБСОРБЦИОННОЙ
СПЕКТРОФОТОМЕТРИИ
Методы спектрального анализа включают
спектрофотометр ию (абсорбционный спектральный анализ) и
эмиссионную спектроскоп и го, один из разделов
которой представлен методами анализа по фотометрии
пламени.
При помощи спектрофотометрии возможно изучение очень
широкого круга явлений и решение различных задач
почвоведения. Одним из методов спектрофотометрии является
колориметрический анализ, который давно уже
применяют в почвоведении и агрохимии при определении в
почвах, растениях и удобрениях содержания фосфора,
аммиака, нитратов, нитритов, двух- и трехвалентного железа,
титана, алюминия и др. Колориметрирование— наиболее
простой метод анализа. Введение фотометров, фотоэлектроколо-
риметров и спектрофотометров значительно повысило
чувствительность, точность и скорость определения. Вместе с тем
стало возможным одновременное определение нескольких
элементов по смешанным окраскам. В последние годы
разрабатываются спектрофотометр ическ ие методы
определения гумусовых" веществ почвы: гуминовых и фульво-*
кислот и неспецифических органических соединений, таких,
как аминокислоты, уроновые кислоты, гексозы, пуриновые
основания и др.
Не меньшее значение имеет качественная характеристика
гумусовых веществ но спектрам поглощения, расширение
диапазона исследований в ультрафиолетовую и
инфракрасную области спектра позволило решить ряд вопросов, связан-
229
ных со строением органических и минеральных веществ
почвы.
Кроме спектров поглощения в почвоведении находят
применение и спектры отражения, пользуясь которыми
можно проводить быстрые количественные и гюлуколичественные
определения ряда минеральных и органических компонентов
почвы. Спектральная отражательная способность почв имеет
самостоятельное значение для характеристики цвета почвы,
расчета теплового баланса и разработки методов
спектральной аэрофотосъемки.
Абсорбционная спектрофотометрия включает все методы,
основанные на законе Бугера— Бэра, устанавливающего
зависимость между ослаблением света растворами и
концентрацией поглощающего свет компонента. Определение
концентрации в колориметрах и фотометрах сводится к уравниванию
освещенности двух полей, находящихся в поле зрения
аналитика. Уравнивание освещенности в фотометрах
достигается изменением степени раскрытия диафрагмы, стоящей
на пути светового пучка, а в колориметрах — путем
регулирования высоты столба жидкости, поглощающей свет.
Фотоэлектроколориметры отличаются тем, что интенсивность
светового потока (освещенность поля) оценивается не
визуально, на глаз, а с помощью фотоэлектрического
устройства, состоящего из фотоэлементов и гальванометров.
К спектрофотометрам относят призменные приборы с
фотоэлектрическим отсчетным устройством; во многих
спектрофотометрах предусмотрена автоматическая регистрация
показаний прибора.
СПЕКТРЫ. ОСНОВНЫЕ ЕДИНИЦЫ
Наиболее важными характеристиками электромагнитного
излучения для аналитических целей являются частота
колебаний или длина волны и величина
энергии одного кванта (фотона).
Частота колебаний v не зависит от среды, в которой
распространяется излучение, и выражается числом световых
колебаний, совершаемых в одну секунду. Вместо частоты,
имеющей очень большие численные значения в видимой
части спектра (порядка п- 1014 сект1), используют волновые1
числа, равные количеству волн, укладывающихся на
протяжении одного сантиметра в вакууме. Обе величины часто
обозначают одним символом v (греческое «ню»), но волновое
число — без значка сверху. Длина волны Я зависит от скоро-
230
сти распространения излучения в данной среде, и если
излучение распространяется в вакууме, то длина волны связана с
частотой соотношением
, с 3 • 1010
Л — ^т- — -- ~— см.
v v
Частота имеет размерность сек ', волновое число см~1.
Волновое число v = — • 108 смГ1, если длина волны выра-
жена в ангстремах.
Для отдельных участков спектра применяют различные
единицы измерения длины волны; это связано с удобством
использования небольших чисел. Для ультрафиолетовой и
видимой части спектра вычисления делают в ангстремах
(А = 10~8 см) « миллимикронах A ммк = 10А=10~7 см).
В ближней инфракрасной области длину волны выражают а
микронах A мк= 10~4 см) и часто используют волновые
числа.
Световой луч состоит из непрерывного потока отдельных квантов
с частотой v и энсршей Е. Связь между обеими величинами строю
постоянная:
E = hv.
где Л — постоянная Планка, равная 6,625+0,008x107" эрг/сек, поэтому
энергию кванта можно выразить в единицах частоты
При встрече с атомом (или молекулой) луч отдает ему часть
энергии. Передача световой энергии веществу наиболее вероятна, когда
разность двух энергетических состояний, в которых может находиться атом
или молекула, точно равна (резонансна) величине кванта излучения
Д£ — £излуч т: "У'
Излучение электромагнитных колебаний происходит по тем же
правилам. Сначала атом (или молекула) под действием тепла илн света
переходит в возбужденное состояние, которое длится около 10~ сек.
Затем частица возвращается в нормальное (основное) состояние, выделяя
избыток энергии в виде электромагнитных колебаний, частота которых
зависит от разности энергий в возбужденном и стационарном состоянии.
hv — Е2 — Ei.
Последовательно освещая вещество монохроматическим
светом различной длины волны и определяя изменение
энергии луча после поглощения света веществом, можно
установить, что наиболее интенсивное поглощение света происходит
при некоторых значениях длин волн, характерных для
данного вещества. Чередование участков, в которых свет мало
231
или совсем не поглощается, с участками интенсивного
поглощения образует спектр поглощения. Такой спектр
удобней всего представить графически, откладывая по оси абсцисс
частоту колебаний (длину волны), а по оси ординат —
уменьшение энергии светового пучка. Следовательно, в этом случае
мы будем понимать под спектром поглощения
последовательное расположение по длинам волн
относительного количества энергии электромагнитных колебаний, по-
1Лощаемых веществом.
Спектром излучения называют последовательное
расположение энергии излучения по длинам волн. Оба
определения вытекают из общего понятия спектра как
совокупности интенсивностей (или амплитуд) гармонических
колебаний, на которые можно разложить данное сложное колебание.
При взаимодействии света и вещества происходит не
только уменьшение энергии колебаний в результате
поглощения, но и ряд других явлений. Так, например, широко
известны изменение направления падающего луча при
преломлении и отражении, рассеяние, дифракция, вращение
плоскости поляризации и др. Все эти явления используются,
как и поглощение света, в исследованиях состава структуры
и свойств вещества, а в абсорбционной спектрометрии
влияние их приходится учитывать, чтобы избежать возможных
ошибок измерений. Процесс поглощения света
сопровождается часто превращением энергии электромагнитных колебаний
в другие виды: тепловую, энергию химических превращений
и др.
СПЕКТРЫ ПОГЛОЩЕНИЯ МОЛЕКУЛ
Взаимодействие света и вещества сопровождается
разнообразными процессами, ведущими к изменению
энергетического состояния вещества. Наиболее простая форма
превращения лучистой энергии — это переход ее в теплоту, в
поступательное неквантуемое движение частиц вещества Еа.
В других случаях почти вся лучистая энергия идет на
образование квантуемых форм энергии: вращательное движение
всей частицы £вв и отдельных групп атомов внутри молекул
£п, изменение характера колебаний ядер атомов в пределах
молекулярных структур Еп и электронов в отношении ядра Ес,
изменение ориентации спинов ядер атомов Ес. Таким
образом, световая энергия Е, поглощаемая веществом,
расчленяется на ряд слагаемых:
Е = Еп + Евв + Ев + Ея + Ее — £с. ;-
232 *V<" ^ \
Формы превращения и диапазон поглощаемых частот зависят
от величины кванта света, структуры и состава вещества.
Вращательное движение молекул вызывается, как правило,
действием квантов малой величины, поэтому спектры,
обусловленные изменением энергии вращения, наблюдаются в
далекой инфракрасной области. По мере увеличения частоты
излучения энергия кванта достигает величины, достаточной
для изменения энергии колебания атомов в молекуле, и
соответствующие спектры обнаруживаются в ближней
инфракрасной области (Х= 1,0—25 ж/с). В видимой D00—750 ммк)
и ультрафиолетовой (<400 ммк) областях поглощение
обусловлено преимущественно электронными переходами.
Различные энергетические состояния молекул схематично
изображают в виде отдельных уровней (рис. 54).
Положения А и Б отвечают двум электронным уровням (состояниям)
молекулы. Изменение энергии колебаний атомов вызывает
переходы от низшего колебательного подуровня ■Оо к
высшему #ь #2 — или от #о к #ь #2— в пределах одного
электронного состояния или сопутствуя электронным переходам
А—> Б. Переходы от подуровня / = 1 к /=2 и т. д. связаны с
изменением энергии вращения молекулы в целом. Расстояния
между уровнями (горизонтальные линии на рис. 54) условно
без соблюдения масштаба показывают разность энергий; чем
больше расстояние, тем выше разность £2 — £\. Излучение
относительно высокой частоты может переводить молекулу из
основного состояния А в возбужденное, электронное
состояние Б так, что молекула оказывается на одном из
колебательных (вращательных) подуровней. Это показано на рисунке
вертикальными стрелками. Картина спектра в таком случае
становится весьма сложной. Вместо одной линии или полосы
поглощения возникает серия линий, близко расположенных
друг к другу.
Если обозначить энергию электронного перехода Еол,
колебательных переходов — £'Кол, E"K01U E'"K0:i... и т. д., то полосы
поглощения соответствуют частотам, при которых величины
квантов приобретают значения, равные ^эл + Е КОл, Еэп +
+ ЕЮл и т. д.; но поскольку Е-м > £кол, то Еал +Е'К0Л si
= Сад Т С КОЛ =^ СэлТЕ КОЛ» И Т. Д.
Поэтому поглощаемые частоты расположены близко друг
к другу, хотя величины Е1;0п сами по себе значительно
отличаются друг от друга. В результате колебательные и
вращательные переходы как бы накладываются на электронный
переход и образуют в области электронных спектров серию
233
близко расположенных полос, часто сливающихся настолько,
что в видимом и ультрафиолетовом спектре различить
полосы, вызванные колебательным и вращательным
движением, бывает почти невозможно. Те же колебания хорошо
выявляются в инфракрасной области спектра.
•5
■К
«*
*1
,
:
']
■ 1
;,"j
1 ,.
j_
ЧГ
-I
*ЦДч1^
f .
£,
Г'
Рис 54 Возможные энср1етические уровни молекулы
Общий вид спектра зависит не только от возможных
энергетических состояний отдельной молекулы, но так же от
взаимного влияния молекул и их взаимодействия с
молекулами растворителя. Сближение молекул, образование
ассоциаций из нескольких молекул и т. п. затрудняет,
например, их вращательное движение или ослабляет связи
между атомами. Все это сказывается на количестве и
вероятности возможных переходов.
Анализ спектров сложных молекул и особенно молекул
многих наиболее важных природных соединений (белки,
234
пигменты, гуминовые кислоты и др.) представляет большие
трудности. Спектры таких соединений содержат большое
число полос с трудно разрешаемой или даже неразрешаемой
современными приборами тонкой структурой. Тем не менее
спектральный анализ сложных природных органических и
минеральных соединений используется с большим успехом.
Длительность возбужденного состояния оценивается для
большинства соединений очень малыми отрезками времени
(порядка Ю-8 сек). Через определенный промежуток времени
молекула вновь возвращается в наиболее устойчивое
состояние, обладающее минимально возможным запасом энергии.
Этот переход сопровождается выделением энергии. Характер
обратных переходов может быть весьма различным.
Наиболее простой механизм перехода в основное
состояние состоит в излучении кванта света, что известно под
названием люминесценции. Отличительная особенность
этого процесса заключается в том, что излучаемый свет, как
правило, имеет значительно большую длину волны, чем
поглощенный. Это происходит потому, что колебательная энергия
довольно легко передается другим молекулам при
столкновениях. В процессе столкновений молекула отдает частично
колебательную энергию и переходит на более низкий уровень
возбужденного электронного состояния. Последующий
переход в основное электронное состояние происходит уже с
меньшим изменением энергетического состояния и в результате
величина излучаемого кванта оказывается меньше величины
кванта, поглощенного молекулой, а длина волны
люминесцентного света возрастет.
Значительно чаще переход в основное состояние
сопровождается превращением световой энергии в тепловую.
Безызлучательные переходы совершаются тогда, когда
молекула и в основном и в возбужденном состояниях обладает в
некоторых положениях равной потенциальной энергией при
одинаковом расположении ядер Такие случаи часто
наблюдаются для сложных многоатомных молекул.
Характерные области поглощения органических соединений
Частоты поглощаемых веществом колебаний зависят от
множества факторов: числа атомов в молекуле, их радиусов,
зарядов, массы, взаимного расположения атомов в
пространстве и т. д. Сочетание всех этих факторов для каждого
индивидуального химического соединения строго специфично
и неповторимо, а поэтому и спектры поглощения имеют свои
235
характерные особенности. В отдельных участках спектры
близких по строению веществ, конечно, могут частично или
полностью совпадать, тогда как в широком диапазоне частот,
включая далекие ультрафиолетовую и инфракрасную
области, легко обнаруживаются индивидуальные особенности.
Совпадение полос в отдельных участках спектра вызвано
поглощением света определенными атомами или
молекулярными группировками, общими для многих веществ. Это
свойство значительно облегчает задачи исследователя,
поскольку позволяет по характеру спектра быстро распознавать
структурные элементы в исследуемом веществе
Спектры поглощения в ультрафиолетовой и видимой
областях называют электронными спектрами
поглощения, поскольку их происхождение связано с
возбужденными электронными состояниями молекул Электронные
спектры состоят из одной или нескольких широких полос, в
которых при благоприятных условиях может быть различима
и тонкая колебательная структура. В наиболее
коротковолновой части наблюдается, как правило, значительное
поглощение простыми молекулами. Так, углеводороды ряда метана
поглощают излучение в области длин волн <2000А. Двойные
связи сдвигают максимум поглощения в длинноволновую
сторону. Аналогичным эффектом спровождается введение в
молекулу заместителей, увеличение числа двойных связей.
Около 2600 А наблюдается спектр «бензольного
поглощения», состоящий из ряда узких, хорошо очерченных полос.
Соединения с конденсированными бензольными кольцами
имеют максимум поглощения в более длинноволновой
области и значительно более высокую интенсивность. Предельным
случаем конденсированных бензольных колец является
графит, который равномерно поглощает излучение во всей
видимой области спектра.
Все насыщенные углеводороды, соединения с одной двой-%.
ной связью, эфиры не поглощают свет в области 2000—10 000 А
и потому бесцветны. Окраска появляется при введении в
бесцветные соединения групп, называемых хромофорами
(табл. 20), а также при увеличении степени
конденсированное™ и увеличении числа двойных связей. В сложных по
строению соединениях, содержащих несколько хромофорных
групп, полосы могут перекрываться, что значительно
затрудняет или даже делает невозможным идентификацию
отдельных полос.
Для почвенных исследований большой интерес
представляют спектры поглощения органических соединений, содер-
236
Таблица 20
Типичные простые хромофорные группы (по Гиллему
и Штерну)
Хромофорные группы
Карбонильная
»
Карбоксильная
Амидная
Этиленовая
Ацетиленовая
Нитроэо-
Нитро-
Нитратная
Нитрнткая
Система
RC=0
RMC=0
—СООН
—CONHa
RHC=-CHR
RC-CR
—N=0
1N\o
-ONO,
-ONO
Пример соединения
ацетон
ацетальдегид
уксусная кислота
ацетамид
этилен
ацетилен
нитрозобутан
нитрометан
этилнитрат
октилнитрит
%
макс»
ммк
270,6
293,4
204
<208
193
173
300; 665
271
270
230
жащих бензольные кольца и гетероциклы, особенно
включающие азот.
Для некоторых из этих соединений и природных пигментов
максимумы поглощения даны в табл. 21.
В инфракрасной области спектра специфичность
поглощения отдельных атомных групп и индивидуальных соединений
проявляется еще более резко. Ближняя инфракрасная область
(длина волны менее 8 ж/с) обычно используется для
обнаружения и количественного определения атомных группировок.
Положение полос поглощения в этой области лишь в малой
степени зависит от общего строения молекулы. В более
длинноволновой области обнаруживаются очень тонкие
различия в строении даже близких по структуре молекул. Для
исследования характерных групп в составе молекулы иногда
ограничиваются областью от 2 до 8 мк, а в некоторых
случаях и значительно более узкими отрезками. Такими легко
обнаруживаемыми группами могут быть:
> СН2> - СН3, — ОН, — СООН, R — О — R',
>С-С<,—NHa, — N03
и т. д. (табл. 22). Так, присутствие в спектре полосы около
1700 см~1 указывает на карбоксильную группу. Правильность
отнесения может быть доказана замещением водорода
карбоксильной группы на металлический катион, в результате
которого в спектре исчезает полоса около 1700 см~1 и появ-
237
Таблица 21
Максимумы поглощения излучения в видимой и ультрафиолетовой
частях спектра некоторыми органическими соединениями
Соединение
Главнейшие максимумы поглощения в ммк
Бензол .
Нафталин
Антрацен
Феиантрен
3, 4-Бензпирен
Фураи . . ,
Пиррол . . .
Пиримидин .
Адемин . . .
Гу<шин . . .
а-Каротин
Р-Каротин
Хлорофилл а
Хлорофилл b
В ультрафиолетовой части спектра
200
220, 275;
250,
210, 250;
275;
280;
293;
225, 265
284;
296
200;
210;
243,
261;
249;
В видимой
477,
450
428
458
509
485
662
255
314
380
316
323
330
338-
346,
252
340
273
части
520
640
-346
363
384
403
спектра
ляются полосы поглощения ионизированной карбоксильной
группы (в диапазоне 1610—1550 и 1420—1300 см,-1).
Положение отдельных полос в спектре зависит также от
агрегатного состояния, растворителя, величины рН и др.
Поэтому при расшифровке спектров приходится учитывать
'возможные смещения максимумов и в целях сравнения
пользоваться спектрами, полученными в одинаковых "условиях
(если предварительно не выявлена роль отдельных факторов).
КОЛИЧЕСТВЕННЫЕ ЗАКОНОМЕРНОСТИ ПОГЛОЩЕНИЯ СВЕТА
РАСТВОРАМИ
Интенсивность света, прошедшего через кювету с
раствором, ослабляется вследствие того, что одна часть световой
энергии поглощается, а другая рассеивается и отражается
гранями кюветы и границей раздела стекло — раствор. Коли-
238
чественные определения основаны на зависимости между
концентрацией раствора и количеством световой энергии,
поглощенной растворенным веществом Для измерения
количества энергии, поглощаемой веществом при прохождении
через него светового пучка пользуются относительными
величинами, выражающими изменение интенсивности света
в результате взаимодействия
Величина пропускания Т одр_еделяется формулой
'о
где — h интенсивность светового пучка до взаимодействия с
веществом и / — то же после взаимодействия.
Значительно чаще используют величину оптической
плотности D (иногда обозначают Е — экстинкция), определяемой
соотношением
D = -lg-f = -lgT.
Оптическая плотность, как это следует из закона Бугера—■
Бэра, прямо пропорциональна концентрации вещества, и
поэтому ею удобно пользоваться при количественных
определениях. Связь между концентрацией и оптической плотностью
вытекает из следующих рассуждений Если
монохроматический световой поток интенсивностью /() падает на
поглощающий слой (рис. 55), то выходящий пучок / оказывается
ослабленным. Измей©ш+е-+шч«и-си.ццасги. ayvuia_jiC..B
результате взаимодействия с одним из тонких cjum&-dir на которые
можно разделить всю толщину слоя /, пропорционально
толщине слоя и интенсивности светового п>чка
dl -■- — k/odl,
где k — коэффициент пропорциональности, минус указывает
на уменьшение энергии в результате поглощения.
Если бы световой пучок, падающий на каждый из слоев
dl, был одинаков, то формула была бы справедлива для
любой толщины. Однако интенсивность света, проходящего
через слои dl, последовательно уменьшается. Поэтому общее
решение находится путем интегрирования
дифференциального соотношения
— —kdl, или \ = — \kdl,
/о .1 Л> .3
/о О
239
откуда
In — — — kl, или / = /0e
-ы
Это соотношение выведено Бугером. Поскольку
поглощение света определяется количеством частиц вещества,
расположенных на пути светового пучка, то увеличение
концентрации вызывает такое же изменение интенсивности света, как
и изменение толщины
поглощающего слоя (закон Бэра), fi j " iT
Тогда
-ыс
где С
I = he'
концентрация.
J„
швш
___ с
Рис 55 Схема для вывода закона Рис 56. Графическое выражение за-
Бугера кона Бугера — Бэра
Выведенное положение строго справедливо для
монохроматического излучения. Вводя обозначение еА =2,303 k и
переходя к десятичным логарифмам, получим -—-—*•*
/ - I0\Q~e>tc
h
Так как по определению оптической плотности
или lg-
- е?,/С.
D=-lg-
ч
то получим D -- е%1С.^
В такой форме объединенный закон Бугера — Бэра
используется в количественном анализе.
Коэффициент пропорциональности е^ называется м о -
лярным коэффициентом погашения; его
численное значение равно оптической плотности при толщине слоя
240
1 см и концентрации раствора 1,0 М. Значок К указывает, что
оптическая плотность определена при данном (%) значении
длины волны. В тех случаях, когда величина молекулярного
веса исследуемого вещества неизвестна, Гиллем и Штерн
предложили пользоваться ^-величиной, выражающей
оптическую плотность 1%-ного раствора толщиной 1 см
(обозначается Еш< ).
Графически закон Бугера — Бэра выражается прямой
линией, проходящей через начало координат. Получение
Рис 57. Приложение закона Бугера — Бэра к растворам гумата
натрия
а — оптическая плотность раствора гумата и величины е^, б —
условия количественных определении в разных участках спектра
прямой зависимости между D и С и является собственно
целью логарифмирования уравнения Бугера — Бэра, тогда
как зависимость между Т и С (Т=— — 10 я ) не является
Л>
прямолинейной, что значительно усложняет расчеты, а для
построения графика по последнему уравнению (в
координатах Т—С) требуется большое число отдельных определений
В то же время зависимость D — С может быть построена
всего по трем точкам, а для расчетов пользуются уравнением
прямой пропорции Тангенс угла наклона прямой
зависимости D — С (рис. 56), как следует из уравнения, равен
величине молярного коэффициента погашения £^/ = tgcc или при
толщине слоя 1 см tga=ex.
Последнее соотношение указывает на необходимость
монохроматизации света при количественных определениях.
На рис. 57,а представлена кривая оптической плотности
241
раствора гумата натрия; если толщина слоя 1 см, а
концентрация — 1 г • моль/л, то вертикальные отрезки, опущенные из
любой точки кривой на ось абсцисс, равны молярному
коэффициенту погашения для данной длины волны. Измерив
оптическую плотность для двух длин волн Xi и }.2 и построив
график в координатах D — С (рис. 57,6), получим две
прямые линии, причем
tg<*i ^«х, и tga2--=£V
Очевидно, что прямая с большим углом наклона
значительно лучше отвечает требованиям количественного
определения. Действительно, для двух растворов с концентрациями
С\ и С2 разница в оптических плотностях тем более заметна,
чем больше тангенс угла наклона. Если разница в оптических
плотностях для двух концентраций при длине волны hi равна
ADt --- ея,ДС,
а при h2 AD2 -- е^ДС,
ADi с,
то —- = --^,
Д£)о е,
т е. различие в оптических плотностях возрастает во столько
раз, во сколько молярный коэффициент погашения при К\
больше того же коэффициента при ?.2- Тем самым достигается
значительное увеличение точности количественных
определений. Вместе с тем увеличивается и чувствительность
метода. Если под чувствительностью понимать
минимальное количество вещества, обнаруживаемое данным методом,
то очевидно, что чувствительность прямо пропорциональна
величине ел . Так как при фотометрировании нижний предел
измерений зависит от величины оптической плотности,
которую можно измерить без больших погрешностей, а для
большинства приборов эта величина равна 0,1, то, сравнивая
графики на рис. 57, легко обнаружить, что при одном и том
же значении D @,1) соблюдается равенство
Qe*,, - С2ея2,
или Ct: Сг = еяг: е*,,.
Другими словами, минимально обнаруживаемое
количество вещества обратно пропорционально величине ея .
242
Таблица 22
Полосы поглощения, характерные для различных групп
Группы
^С—Н
=с—н
-с—н
\с/Н
/С\н
-с/Н
~С\н
—с—н
X
X
*
X
\
=с—н*
—CsC—
N:=o
—с—о-
II
О
Полосы поглощения
мк
3,5
3,3
3,0
3,4—3,5
3,2—3,3
3,4—3,5
8,0
6,0—6,2
6,3—6,7
-3,3
4,4—4,8
5,7—6,1
6,2—6,5 и 7,1—7,7
см 1
2890
3010—3040
около 3300
2920 и 2850
3090—3080
2962 и 2872
1250
1680—1620
1600 и 1500
3030
2260—2100
1750—1650
1610—1550 и 1420—1300
^С-О—
8,7—9,5
1150—1060
Продолжение табл. 22
^С—N^=
=C--N=**
—C~_-N
V;=n—
^C—Si—
—O—II
Водородная связь
Nn—н
NH3
_ n- N—
R-NOa
-N«r
^P=0
=t-°-<
Полосы поглощения
ж
7,1—7,2
7,4—7,8
4,4—4,5
5,9—6,1
11,1—14,3
2,7—2,8
2,8—2,9
2,9—3,00
3,2—3,3
6,1—6,3
6,1—6,7 и 7,4—8,0
7,1—7,5 и 11,6—12,5
7,7—8,0
9,5—10,0
см 1
1420—1400
1360—1280
2260—2215
[690—1630
900—700
3650—3590
3600—3400
3500—3300
3130—3030
1630—1580
1650—1500; 1350—1250
1410—1340; 860—800
1300—1250
1050—1000
Продолжение табл 22
Полосы поглощения
Группы
1 1
_-,р_О— Р=
1 1
-Si(CH3K
-Si-QH5
мк
10,3-10,6
8,0
7,0 и 8,9—9,2
см~~'
970—940
1250
1430 и 1130—1090
^Si- O-Si^
)со3
/S04
—NH4
Силикаты
9,2--9,9
6,9—7,1 и 11,3—11,7
8,9—9,3 и 14,7—16,4
3,0—3,3 и 7,0—7,2
9,1-11,1
1090—1010
1450—1410 и 880—860
1130—1080 и 680—610
3300—3030 и 1430-1390
1100—900
* В ароматических соединениях.
н* В ароматических аминах.
Аналогично можно показать, что использование неразло-
женного (белого) света дает худшие результаты в сравнении
с монохроматическим пучком, для которого ел имеет высокое
значение. С другой стороны, применение монохроматического
света при малых значениях г% может значительно снизить
точность результатов измерения в сравнении с белым светом.
Отсюда вытекает важность монохроматизации света и,
главное, правильного выбора длины волны света при
количественном анализе, поскольку неправильный выбор может не
только не улучшить результаты, но даже снизить
чувствительность и точность метода.
Фактически применение светофильтров устраняет
«балластный» свет, который увеличивает освещенность фотомет-
245
рических полей, но не взаимодействует с определяемым
веществом. Правильный выбор светофильтров не
ограничивается в общем случае подбором максимальной величины е?.
Максимум поглощения может оказаться в той области
спектра, где понижена чувствительность глаза или
фотоэлектрических приемников, и тогда приходится применять
светофильтры, выделяющие область спектра, в которой e>
исследуемого вещества меньше максимального, но зато
чувствительность глаза или прибора достаточно высока. Кроме этого,
возможны и иные ограничения. По Е. Стирнсу, выбор длины
волны, при которой проводят количественные определения
на данном приборе, зависит по крайней мере от шести
факторов: 1) величины коэффициентов ослабления света, или £я;
2) ошибки измерения длины волны; 3) свойства
анализируемого раствора; 4) ошибки определения пропускания
(оптической плотности); 5) разрешающей способности прибора;
6) влияния примесей. Наивыгоднейшие условия подбираются
в результате внимательного сопоставления всех факторов,
зависящих как от свойств окрашенного раствора, так и от
характеристик приборов.
Независимо от степени монохроматизации прямая
пропорциональность между концентрацией поглощающего свет
вещества и оптической плотностью соблюдается далеко не
всегда. Отклонения от закона вызываются различными
причинами, и, прежде всего, диссоциацией окрашенного
соединения на бесцветные ионы, изменением степени окраски под
влиянием кислотности среды и коллоидальным состоянием
красителя. В таких случаях прибегают к особым приемам,
устраняющим влияние указанных факторов, или делающих
их стабильными: введение избытка реактива, применение
буферных растворов и т. п. Поэтому проверка применимости
закона Бугера ■—Бэра к исследуемым растворам перед
началом работы обязательна. Отклонения от закона могут быть
связаны и с техникой эксперимента.
Вывод основного уравнения Бугера — Бэра был дан без
учета того, что прошедший через кювету с раствором свет
ослабляется не только вследствие поглощения его
окрашенным компонентом, но и по другим причинам.
В общем случае световая энергия /, прошедшия через кювету с
раствором, равна
i-ij-(fa + h + Ъ^г + 2/,).
где Ij—световой поток, падающий на кювету, 1а и I ь—количество
энергии, поглощенной растворенным веществом и растворителем, 2 /г —
246
полная энергия, потерянная в результате отражения от граней кюветы,
и 2/.S — полная энергия, рассеянная на поверхности кюветы и в
поглощающем слое.
Аналогично для кюветы с растворителем, используемой для
определения /о, можно записать
Очевидно, что истинное пропускание равно:
т - !j ■(/" + /» + 2У' + 21М _ 1_
//-(/;+z/;+x/s) '<>'
Поглощение света, потерн на рассеивание и отражение
пропорциональны интенсивности исходного светового пучка (составляет
определенную долю Ij ). Если влияние отдельных факторов мысленно разделить
пространственно, то можно представить, что сначала происходит
отражение света, затем рассеивание, а окрашенное вещество взаимодействует
с оставшимся световым потоком. Тогда при условии, что
1ь-1'ь=ж - 2/; - 2/, - ж - о,
Следовательно, потерн света вследствие рассеивания и отражения не
влияют на результат измерения D или Т, если кювета с окрашенным
раствором и кювета с растворителем одинаково отражают и рассеивают
свет. Несоблюдение этих условий приводит к значительным ошибкам.
Практически принимают, что отражательная способность
различных кювет одинакова; нельзя только использовать для
сравнения пустые кюветы, поскольку отражение на границе
воздух — стекло значительно больше, чем на границе
раствор — стекло. Рассеивание в истинных растворах столь
невелико, что им пренебрегают без всяких последствий, однако
в растворах коллоидных и высокомолекулярных систем
рассеивание вызывает заметные отклонения от закона Бугера —
Бэра. Так, для растворов гуматов натрия влияние
рассеивания становится заметным уже при D > 0,5 и учесть его
можно только построением подробного градуировочного графика.
В количественном фотометрическом анализе часто
употребляют окрашенные реактивы, когда окрашенное вещество Л
реагирует с определяемым компонентом В, образуя новый
комплекс АВ, интенсивность окраски которого измеряется
при фотометрировании. В этом случае закон Бугера — Бэра
не нарушается, но чувствительность и точность определения
несколько снижаются вследствие изменения наклона градуи-
247
ровочного графика. Изменение наклона объясняется тем, что
оптическая плотность раствора, в котором находятся
определяемый окрашенный комплекс и избыток окрашенного
реактива, равна сумме оптических плотностей двух
компонентов:
D^DAB + DA,
или D -- ъАВСАВ1 + еАСА1.
В стандартном растворе, используемом для сравнения,
D' = еАВС'А1.
Концентрация реактива в первом растворе меньше, чем в
стандартном (Сл<С'л), так как часть реактива
израсходована на образование окрашенного комплекса. Если при этой
реакции одна часть реактива отвечает 11п части окрашенного
комплекса, то
СА --- СА — пСАв-
Отсюда
D -^ гАВСАВ1 + ел {CA~nCAB)l =
= влвСАВ1 -\- ъАСА1 — гАпСАв1.
Определяемая из опыта оптическая плотность D„3M равна
разности
DllJM--D~D'.
Тогда
Д|зм =^ ZaePabI + ъАСА1 — гАпСАВ1 — гАСА1 = EABCABt —
— еА пСАВ1 — (елв — пгА )САВ1.
Следовательно, оптическая плотность уменьшается, а гра-
дуировочный график имеет меньший угол наклона. Степень
отклонения зависит от относительного количества
связываемого реактива и молярного коэффициента погашения
реактива при данной длине волны. В предельном случае, когда
елс<«ел, D<0
и анализ становится невозможным.
ПОРЯДОК ИЗМЕРЕНИЙ И ОБРАБОТКА РЕЗУЛЬТАТОВ
Методы абсорбционной спектроскопии используют для
изучения строения вещества и для количественного определе-
248
ния содержания различных компонентов. В первом случае с
помощью фотометров со светофильтрами или
спектрофотометров измеряют поглощение света в зависимости от длины
волны. Достоверность исследования обеспечивается
применением двухлучевых приборов и подбором таких
растворителей, которые сами слабо поглощают свет в исследуемом
спектральном интервале (для видимой части спектра это
вода, спирты, растворы минеральных и органических кислот
и оснований). Результаты представляют в виде графика, на
котором по оси ординат откладывают величины оптической
плотности D, коэффициента пропускания
Т~-—( или — • 100%}
или коэффициента погашения 1 —Т= — .
По оси абсцисс наносят характеристику излучения: длину
волны, частоту колебаний, волновые числа. Выбор того или
иного способа построения графика зависит от свойств
вещества и цели исследования с т^ким расчетом, чтобы
получить наиболее выразительную и удобную для работы
спектрограмму.
В двухлучевых самопишущих приборах запись спектра
производится автоматически. Следует иметь в виду, что
качество получаемых спектров зависит от ширины
спектрального интервала, выделяемого прибором (ширины щели,
дисперсии, свойств светофильтров), а на регистрирующих
приборах и от скорости записи спектра. При уменьшении
ширины щели наблюдаемая интенсивность поглощения в
максимуме полосы обычно возрастает, а сама полоса
становится более резкой и четкой. Некоторые сложные полосы с
уменьшением ширины щели разделяются на 2—3 более
тонкие.
Скорость записи на регистрирующих приборах влияет не
только на интенсивность поглощения, но и на положение
полосы в спектре. Вследствие инерции прибора при слишком
большой скорости записи регистрирующее устройство
запаздывает и максимум поглощения оказывается сдвинутым;
одновременно близко расположенные небольшие максимумы
могут сливаться в общую полосу.
Количественный анализ не требует в большинстве случаев
полного изучения спектров поглощения. Светофильтры для
249
работы в видимой части спектра можно подобрать простым
сравнением (выбирается светофильтр, дающий
максимальное значение D). Для сложных смесей и особенно в
ультрафиолетовой и инфракрасной областях спектра длину волны
выбирают по полной кривой спектрального поглощения.
Общий порядок количественных измерений для впервые
изучаемого вещества следующий:
1) получить спектр поглощения;
2) выбрать оптимальный интервал длин волн
(светофильтр);
3) с выбранным светофильтром (а при работе на
спектрофотометре в выбранном спектральном интервале для
нескольких длин волн) проверить приложимость закона Буге-
ра — Бэра;
4) построить градуировочный график в координатах
D — С по растворам с известной концентрацией;
5) определить оптические плотности исследуемых
растворов и по графику найти их концентрацию.
В количественном анализе благодаря исключительной
простоте получил очень широкое распространение метод
колориметрирования, основанный на законе Бугера — Бэра,
в котором ход определения сводится к визуальному
уравниванию окрасок двух растворов путем изменения толщины
поглощающего слоя. После уравнивания
Dx — еСУх и D2 eC2/2>
но так как Di=ZJ, то Cll\ — C2h. Если известна толщина слоя
каждого из растворов и концентрация одного из них, то
концентрация второго определяется по формуле
С2- —.
Для уравнивания окрасок применяют колориметры типа
Дюбоска, КОЛ-1 и др. Очевидно, что этот способ
непригоден для анализа растворов, не подчиняющихся закону
Бугера — Бэра.
Хорошие результаты дает метод колориметрического
титрования. В этом случае готовят два раствора: первый
содержит определяемое вещество и все реактивы,
необходимые для возбуждения окраски; второй раствор содержит
только реактивы. Ко второму раствору добавляют по каплям
раствор, содержащий определяемое вещество в известной
концентрации, пока интенсивность окрасок обоих растворов не
250
сравняется. Титрование проводят в мерных цилиндрах и
интенсивность окраски сравнивают, наблюдая сверху
(просматривая всю толщу раствора). При равной интенсивности
окрасок количество окрашенных веществ (но не их
концентрации) в обоих цилиндрах одинаковы.
Весьма употребителен в почвоведении метод шкал, по
которому готовят серию стандартных растворов (в
пробирках или мерных колбах), содержащих точно известные
количества определяемого иона, и окрашивают их выбранным
реактивом, а затем сравнивают с окраской исследуемого
раствора. Искомая концентрация приравнивается к
концентрации стандартного раствора в той из пробирок, окраска
которой наиболее близка.
Фотометрические методы отличаются тем, что они
основаны на непосредственном измерении ослабления интенсивности
светового пучка, т. е. величин D или Т. Измерение оптической
плотности или пропускания — наиболее универсальный метод;
толщину поглощающего слоя и молярный коэффициент
поглощения можно легко измерить и тогда для определения
концентрации достаточно знать только величину D.
Измерениями D можно пользоваться, кроме того, и в сочетании со
шкалой стандартов, при колориметрическом титровании и др.
Определение концентрации проводят следующими способами.
1. Если раствор содержит только один окрашенный
компонент, то сначала строят градуировочный график по серии
стандартных растворов. Для построения графика по
вертикальной оси откладывают измеренные значения оптической
плотности для монохроматизированного света,- а по
горизонтальной оси — концентрацию стандартных растворов. При
правильно выбранных условиях измерения градуировочная
прямая обязательно проходит через точку начала координат.
Затем измеряют оптическую плотность исследуемого
раствора, окраска в котором возбуждена тем же способом,
что и в стандартах, и по графику находят искомую
концентрацию.
Устойчивость окраски и постоянство величины молярного
коэффициента поглощения во времени дает возможность
пользоваться градуировочным графиком длительное время.
2. Для веществ и растворов, строго подчиняющихся
закону Бугера — Бэра, нет необходимости в построении градуи-
ровочного графика. Достаточно приготовить только один
стандартный раствор и, измерив для него оптическую
плотность, рассчитать концентрацию, используя прямую
пропорциональность между D и С.
^ 251
3. Одновременное определение нескольких окрашенных
компонентов из одного раствора по смешанной окраске
вполне возможно, если известны коэффициенты поглощения света
этих соединений по крайней мере для двух различных длин
волн.
Оптическая плотность смеси из двух окрашенных
компонентов равна сумме оптических плотностей, обусловленных
каждым из этих компонентов в отдельности:
D1 = U + D", ' -
но D' = e'KC'l, ".
и D" - гкС,
тогда
D1 = eKlC,t + eKlCl.
Измерив оптическую плотность при другой длине волны
?>.2, ПОЛуЧИМ
Так как молярные коэффициенты поглощения е^, ъХг, е*.
и е^ известны, а толщина слоя определяется
размерами кюветы, то получаются два уравнения с двумя
неизвестными, которые легко решить. Если последнее уравнение
умножить на
Л-2
то «D2 = n&xfi'l + пъ%£,
где mKC'l = elfi'l
и /zDjj = elfi'l + n&ijC"l.
Почленное вычитание этих уравнений дает
D1 — nDi = ei,C7 — nelfi"{,
откуда
С" = Pi — «Pa
252
Измерение оптических плотностей при спектрофотометрн-
ческом анализе проводят на универсальных фотометрах
(типа ФМ), фотоэлектроколориметрах (ФЭК-М, ФЭК-52,
ФЭК-57), спектрофотометрах (СФ-2М, СФ-10, СФ-4). Для
измерений в инфракрасной области спектра промышленность
выпускает инфракрасные спектрометры типа ИКС-2, ИК.С-11,
ИКС-12, ИКС-14 и др.
ОПИСАНИЕ ПРИБОРОВ
В основе фотометрических измерений лежит сравнение
интенсивностей двух световых пучков, которое может быть
осуществлено визуально, «на глаз», когда сопоставляется
яркость двух половинок поля, освещаемых сравниваемыми
пучками, или путем объективных методов сравнения, когда
интенсивность световых пучков измеряется с помощью фото-
или термоэлементов. Фотометры (ФМ и др.) предназначены
для визуальных измерений. Приборы типа ФЭК-М, ФЭК-57
дают возможность объективно измерить относительную
интенсивность света. Приборы СФ-2М, СФ-10, ИКС-12, ИКС-14
обеспечивают автоматическую запись спектров поглощения.
Спектрофотометрические приборы бывают однолучевые и
двухлучевьге. В двухлучевых приборах световой поток
делится на два одинаковых по интенсивности луча, что позволяет
измерять поглощающую способность раствора по отношению
к поглощающей способности растворителя.
Эти приборы автоматически учитывают влияние кювет
и растворителей на результаты измерений. В однолучевых
приборах для учета влияния растворителя, кюветы и
спектральной характеристики источника излучения приходится
проводить дополнительные измерения интенсивности
светового потока, прошедшего через кювету с растворителем.
На современных приборах предусматривается
программированное управление, позволяющее изменять скорость записи
спектров во время съемки, а также интенсивность исходного
светового потока в соответствии с намечаемой целью
исследования.
Универсальный фотометр ФМ
Прибор предназначен для измерения оптических
плотностей или коэффициентов светопропускания жидкостей,
растворов и твердых веществ, а также для определения
коэффициентов отражения света поверхностями твердых тел. Прибор
253
позволяет получать простейшие спектральные
характеристики окрашенных веществ
Оптическая схема фотометра показана на рис 58
Источником служит низковольтная A2 в) лампа накаливания, свет
от которой при помощи зеркал 2 делится на два пучка и
через конденсоры 3 попадает
на зеркало б, направляющее оба
пучка света на кюветы 6 с
раствором и с растворителем
Прошедшие через раствор пучки света
ограничиваются диафрагмами 7,
а затем при помощи системы
призм попадают в окуляр, через
который наблюдатель видит
освещенный круг, разделенный на две
равные части Призмы изменяют
направление световых пучков так,
что левая половина освещенного
поля соответствует пучку света,
прошедшему через правую
кювету, и наоборот Линия раздела
полей фокусируется поворотом
кольца 6 (рис 59) Перед
линзами окуляра помещена
вращающаяся обойма со
светофильтрами 4, номер светофильтра,
перекрывающего световой поток,
можно прочесть в окошке на обойме
Характеристики светофильтров
приведены в табл 23 Рядом с
окуляром помещена свободно
вращающаяся линза 5, через которую
наблюдают изображение
диафрагм Пользуясь этой линзой,
проверяют равномерность осве
щения диафрагмы
Кюветы с раствором и
растворителем помещаются на столик 8,
поднимаемый или опускаемый
винтом 9
Осветител ь / питается от сете
вого тока через трансформатор
Необходимо следить, чтобы вилка
подводящего провода находилась
VIR*'
w/
фо-
Ри1_ 58 Оптическая схема
тометра ФМ
/ — лампа 2—зеркала 3—
конденсоры, 4 — рассеиватели,
5 — черкало 6 — кюветы 7 —
диафрагмы, 8 — барабаны 9—•
06 ьективы, 10 - - ромбические
призмы, // — бипризма 12 —
светофильтр, 13 — оку пир
254
в гнездах трансформатора, обозначение которых отвечает
напряжению накала лампы, указанному на ее цоколе.
Измерение оптической плотности производится с помощью
диафрагм. Закрывая или открывая одну из них, уравнивают
освещенности полей, наблюдаемых в окуляр. Если в- одну из
кювет налит окрашенный раствор, а в другую растворитель,
Рис. 59. Внешний вид фотометра ФМ-
/ — осветитель, 2, 3-—регулировочные винты осветителя,
4 — обойма со светофильтрами, 5 — линза, 6 —
фокусирующее кольцо, 7—барабаны, 8 — предметный столик, 9 —
подъемный винт, 10 — зеркало, II — конденсор с рассеивающими
стеклами
255
Таблица 23
Характеристики светофильтров
Светофильтр
М43
М47
М50
М53
М61
М66
М72
К-2
К-4
К-6
^эфф.
ммк
436
465
496
533
619
665
726
633
541
478
К максимума пропускания,
ммк
416+10
455 + 10
500 + 10
540 ±10
610 + 10
660 + 10
—
—
—
—
Ширина области
пропускания, ммк
41
43
40
36
41
64
64
85
51
77
то освещенность первого поля меньше, чем второго. Для
уравнивания их уменьшают раскрытие диафрагмы на пути
второго пучка. В результате степень раскрытия этой
диафрагмы служит мерой коэффициента пропускания. На
барабанах 7 (рис. 59), связанных с диафрагмами и управляющих
их раскрытием, нанесены две шкалы. Деления черной шкалы
выражают процентное отношение
площади S раскрытия диафрагмы
в момент измерения к площади
S0 максимального раскрытия. Это
отношение и есть величина пропу-
екания Т= ■— «100%. Вторая,
красная шкала получена простым
логарифмированием обратных
величин черной шкалы, она
позволяет сразу отсчитать оптическую
плотность раствора.
Рис. 60 Кюветы к фотометру Кюветы показаны на рис. 60.
ФМ ^ '
Свет проходит через кюветы с
раствором вертикально, толщина
поглощающего слоя равна расстоянию между крышкой и дном
кюветы и остается строго постоянной для одной и той же пары:
кювета-—крышка. На кювете и крышке указана их высота.
Заполняют кюветы так, чтобы под крышкой не было пузырьков
воздуха; слишком полно наливать кювету не следует, иначе
крышка вытеснит избыток раствора и снаружи кювета будет
загрязнена.
256
Прибор очень прост в обращении и устройстве, однако
хорошие результаты получают только при правильной
настройке и обращении. Длительная работа на фотометре
сильно утомляет глаз, что снижает качество определений. Для
удобства работы прибор снабжен парой наглазников, один из
которых предохраняет левый глаз наблюдателя от попадания
постороннего света. Наглазники позволяют держать оОа
глаза открытыми, что значительно уменьшает их утомляемость.
Настройка прибора
Перед началом работы устанавливают достаточно яркое
и равномерное освещение обоих полей, наблюдаемых в
окуляр. Все регулировки освещенности рекомендуют проводить
с зеленым светофильтром. Для регулировки вынимают
рассеивающие стекла, помещенные после конденсоров
осветителя, затем, вращая осветитель и поворачивая зеркало (см. 10
на рис. 59), добиваются, чтобы в поле зрения была видна
нить лампы накаливания, симметрично пересекающая оба
поля. Хорошей установке может помочь перемещение самой
лампы внутри кожуха осветителя, что достигается винтами 2
и перемещением патрона лампы, укрепленного на кожухе
осветителя винтом (см. 3 на рис. 59). Резкость изображения
нити достигается выдвижением конденсоров.
После того как получено резкое и равномерное
изображение нити, снова вставляют рассеивающие стекла в гнезда
конденсора и проверяют освещенность полей. Если поля
освещены неодинаково, добиваются равномерного освещения
расфокусировкой конденсоров или переменой местами
рассеивающих стекол.
Чтобы убедиться в одинаковой освещенности полей,
ставят один из барабанов на деление 50 по черной шкале и,
вращая другой барабан, уравнивают освещенность. При этом
отсчет на втором барабане также должен быть 50.
Наблюдение повторяют три раза и берут среднее значение.
Допустимые отклонения не должны превышать ±0,5%, т. с. одного
деления на шкале светопропускания. Если не удается
незначительной расфокусировкой конденсоров получить
одинаковую освещенность, то на пути одного из пучков можно
поместить нейтральный фильтр (в гнездо конденсора рядом с
рассеивающим стеклом), входящий в комплект прибора.
Иногда при уменьшении диафрагмы можно наблюдать,
что освещенное поле темнеет не равномерно, а начиная с
9 Зак 51
257
верхнего угла; причиной этого является неправильная
установка осветителя но отношению к отражающему зеркалу
(свет от осветителя частично не попадает на зеркало).
Порядок определения оптической плотности
или пропускания на ФМ
1. Одну кювету наполняют исследуемым раствором, а
другую — растворителем, содержащим те же компоненты, что и
исследуемый раствор, кроме определяемого соединения.
Когда все исходные вещества бесцветны, то вторую кювету
заполняют просто дистиллированной водой. Водные
растворы готовят на предварительно лишенной газа
(прокипяченной) дистиллированной воде, чтобы избежать появления на
стенках и пробках кювет воздушных пузырьков.
2. Кюветы помещают на столик прибора, совмещая центр
кюветы с центром светового пучка, вводят светофильтр.
3. Полностью раскрывают диафрагму, находящуюся на
пути пучка, прошедшего через раствор, для чего барабан
ставят на деление 0 по шкале оптической плотности.
4. Наблюдая в окуляр освещенность полей, вращают
второй барабан, пока освещенность полей не сравняется. Не
прекращая наблюдения, несколько изменяют положение
барабана и снова уравнивают освещенность. Повторяют эту
операцию два-три раза. Такой прием позволяет быстро и точно
найти правильное положение барабана, поскольку небольшие
смещения его в обе стороны от точки равновесия легко
фиксируются рукой и позволяют уверенно взять отсчет.
5. Записывают отсчет но барабану и снова повторяют
измерения. Для уточнения результатов кюветы меняют
местами и уравнивают освещенность, беря отсчет по другому
барабану. Это устраняет ошибки, возникающие вследствие
недостаточно равномерного освещения полей или неточной
градуировки барабанов.
6. Вычисляют средние значения из нескольких повторных
измерений D и Т.
Поскольку величина оптической плотности прямо
пропорциональна концентрации, то среднее арифметическое
оптических плотностей отвечает среднему значению концентрации,
т. е. Dcp = {D\ + D2) : 2. Пропускание связано с оптической
плотностью уравнением
Подставляя это выражение вместо величины D в
предыдущую формулу, получают среднее значение Т по формуле
тср юо ^ v loo/
или
•— lg —- = 1о — 4- 1е— ) — Щ ■ ■ —
100 2 \к100^ s100/ 2 1002
6 V 10№ /
Тогда
Тср __ (Tfj\h = VW,
100 " V 1002 ] ~ 100 '
или Гср -■ VTJ\.
Определение коэффициентов отражения производится
также с той лишь разницей, что предметный столик опускают в
нижнее положение, на пути пучков (вместо зеркала)
помещают стандартные отражатели и уравнивают освещенности
полей. Затем на пути одного из пучков вместо стандартного
отражателя, состоящего из порошка MgO, помещают объект,
отражательную способность которого требуется измерить.
Уравнивают освещенность полей и записывают показания,
выражая отражательную способность в процентах.
Спектры поглощения растворов и кривые спектральной
яркости твердых тел. Порядок работы при определении
спектральных характеристик растворов и твердых тел тот же,
что и при определении оптических плотностей и
коэффициентов отражения. Для построения кривой, выражающей спектр
поглощения, измеряется оптическая плотность при разных
длинах волн, для чего последовательно включаются
узкополосные светофильтры (от № 1 до № 8); по результатам
измерений строится кривая, характеризующая изменение
оптической плотности в зависимости от длины волны.
Найденные значения оптических плотностей откладывают на
графике против отметки «эффективная длина волны», указанной в
паспорте светофильтра (см. табл. 23). Так же строится
кривая спектральной яркости или спектраль-
9*
259
ной отражательной способности, но только
отражательную способность измеряют в процентах.
Спектральная характеристика служит мерой
качественного сравнения различных веществ; кроме того, она нужна для
правильного выбора светофильтра, используемого при
количественных определениях. Правильно выбранный светофильтр
отвечает следующим требованиям: оптическая плотность
раствора, измеренная с этим светофильтром, должна быть
наибольшей, т. е. чтобы область пропускания светофильтра
соответствовала максимуму на спектральной кривой
оптической плотности раствора; в то же время желательно, чтобы
область пропускания светофильтра была ограничена
горизонтальным (или близким к горизонтальному) участком на
кривой оптической плотности раствора. Если второе условие
выполнить невозможно (горизонтальный участок имеется
только в области малых оптических плотностей), то
ограничиваются при выборе первым условием.
Выбор кюветы для работы с растворами. Кювета
подбирается после того, как выбран светофильтр. Размер кюветы
должен обеспечить оптимальный диапазон измеряемых
оптических плотностей исследуемых растворов от 0,15 до 0,9 D
(в случае необходимости диапазон можно расширить до
£) = 1,2—1,4). Подбирают кювету по наиболее
концентрированному раствору так, чтобы оптическая плотность не
выходила за указанные пределы. Если плотность даже в самой
малой кювете оказывается слишком высокой, то приходится
разбавлять растворы или заменять светофильтр. После того
как кювета и светофильтр подобраны для
концентрированного раствора, дополнительно промеряют с той же кюветой
наиболее разбавленные, с тем чтобы минимальная оптическая
плотность в той же кювете была не ниже 0,15. Иногда для
разбавленных растворов берут кюветы большего размера, чем
для концентрированных, а при построении градуировочного
графика и расчетах все измеренные значения оптических
плотностей приводят к одной и той же толщине
поглощающего слоя (по формуле Бугера — Бэра). Однако наилучшие
результаты дает применение одинаковых кювет во всем
диапазоне концентраций.
Фотоэлектроколориметры
В фотоэлектроколориметрах интенсивность световых
потоков сравнивается при помощи фотоэлементов.
Принципиальная схема прибора ФЭК-М^показана на рис. 61. Световые
260
пучки, идущие от лампы / через систему линз и зеркал
2—4, 6, попадают на кюветы с раствором 7. Затем правый
пучок света ограничивается щелевой диафрагмой 8,
связанной с отсчетным барабаном 9, и попадает на
фотоэлемент 14. Левый пучок света также попадает на фотоэлемент,
пройдя сначала через
нейтральные оптические клинья 3
12, позволяющие
регулировать интенсивность светового
пучка при настройке
прибора. Фотоэлементы соединены
с гальванометром 15 по
компенсационной схеме и при
равенстве фототоков стрелка
гальванометра находится на
нулевом делении шкалы. Для
прибора подбирается пара
фотоэлементов с
одинаковыми свойствами, поэтому при
равной освещенности
фотоэлементов сила тока в цепи
гальванометра равна нулю.
В компенсационной
схеме важное значение
приобретает стабилизация
питания лампы накаливания и
возможность работы при
постоянной освещенности
фотоэлементов. Первое
достигается введением в цепь
питания стабилизатора, а
постоянство освещенности
фотоэлементов обеспечивается при работе
измерений (на правом барабане).
В приборе ФЭК-М имеется только три широкополосных
светофильтра 5, в фотоколориметрах других марок набор
светофильтров расширен до девяти. В отличие от ФЭК-М
приборы ФЭК-52, ФЭК-54, ФЭК-57 имеют электронный
усилитель тока, повышающий чувствительность прибора; селеновые
фотоэлементы в них заменены на сурьмяно-цезиевые, в связи
с чем используемый спектральный интервал расширен в
коротковолновую область до 360 ммк.
Рис. 61. Оптическая и электрическая
схемы прибора ФЭК-М:
1—лампа, 2—конденсоры, 3—
теплозащитные фильтры, 4 — зеркала,
5 —светофильтры, 6 — линзы, 7 —
кюветы, 8 — диафрагма, 9 — барабан,
10 — линзы, 11— призмы,
/2—/круговые оптические нейтральные
клинья; 13 — шторки, 14 —
фотоэлементы, 15 — гальванометр
по второму способу
261
Установка и настройка прибора
Прибор включают в сеть напряжением 120—220 в, в
зависимости от положения переключателя стабилизатора, подают
напряжение накала на лампу осветителя и через 10—15 мин
приступают к измерениям. Правильность установки лампы
проверяют по изображению нити лампы, которое можно ви-
- Рис. 62. Внешний вид фотоэлектроколориметра ФЭК-М:
1 — кожух лампы, 2 — корректор; 3 — шкала гальванометра,
4—барабаны, 5—шкала левою и 6 — правого барабанов, 7 — рукоятка
установки светофильтров, 8, 9—рукоятки нейтральных клиньев,
10— переключатель гальванометра
деть, поместив папиросную бумагу перед линзой 6.
Изображение нити должно быть четким, резко сфокусированным; в
правом и левом пучке света нить должна быть расположена
вертикально по центру линзы и симметрично в обоих пучках.
Для установки лампу накаливания перемещают с помощью
регулировочных винтов на патроне. Отъюстировав лампу,
перекрывают световые пучки шторками, освобождают
арретир гальванометра и устанавливают стрелку гальванометра
на нулевое деление при помощи корректора (рис. 62,2). Затем
открывают шторки, переключатель чувствительности 10 галь-
262
ванометра ставят в положение / и, вращая барабаны 4 и
нейтральные клинья 8, 9, убеждаются, что стрелка
гальванометра имеет свободный ход и отзывается на изменение
освещенности обоих фотоэлементов. После этого можно
приступать к измерениям, подобрав светофильтр и кювету.
Измерение оптической плотности на фотоэлектроколори-
метре. Все измерения и настройка .прибора проводятся в две
ступени. Первоначально делают грубую настройку, для чего
переключатель чувствительности 10 гальванометра переводят
из положения 0 в положение 1, а затем вращением рукоятки
нейтральных клиньев 8, 9 или измерительного барабана 4
стрелка гальванометра сначала грубо, а затем точно
подводится к нулю. Только после этого можно включать
гальванометр на полную чувствительность (положение 2) и
производить точную компенсацию. Перед сменой кювет обязательно
перевести переключатель гальванометра в положение 0.
В фотоэлектроколориметрах применяют четырехгранные
кюветы с плоскопараллельными стенками; кюветы
устанавливают в держатель так, чтобы грань, на которой надписана
длина кюветы, была параллельна проходящему через нее
световому пучку.
1-й способ измерений. В левый пучок света
помещают кювету с растворителем, в правый — кювету с
раствором. Щелевую диафрагму полностью открывают, для чего
ставят шкалу левого барабана в положение «100%
пропускания» (или «нуль оптической плотности»). На левом барабане
шкала круговая, и одно и то же деление показывает и 0 и
«100% пропускания»; правильное положение барабана
узнают, вращая барабан до упора и наблюдая, в какую сторону
изменяются величины Т (оба барабана жестко сидят на
одной оси, поэтому при работе удобно вращать барабан
правой рукой, тогда как левая рука занята переключением чув-
ствительностей гальванометра).
Установив барабан на деление «100% пропускания»,
переводят переключатель чувствительности в положение 1 и
подводят гальванометр к нулю при помощи поворота дисков 8,9
(рис. 62) оптических клиньев сначала грубо, а затем точно;
при этом освещенность левого фотоэлемента уравнивается
с освещенностью правого. Затем кювету с раствором,
освещаемую правым пучком света, заменяют на кювету с
растворителем (в пучке слева остается та же кювета с
растворителем) и вращением барабана возвращают гальванометр к
нулевому делению (последовательно меняя чувствительность
гальванометра). При этом диафрагма закрывается и умень
263
шает интенсивность левого пучка ровно настолько, насколько
ослабляет свет исследуемый раствор. По шкале левого
барабана 5 берут отсчет в величинах пропускания Т (черная
шкала) или оптической плотности D (красная шкала). Этот
способ позволяет определять любые коэффициенты пропускания
от 0 до 100%, поскольку первоначально полностью открытая
диафрагма в процессе измерения может быть полностью
закрыта, а деления на черной шкале, как и у фотометра,
выражают отношение площадей закрытой н полностью
открытой диафрагм.
При таком способе измерения освещенность
фотоэлементов все время меняется и зависит от оптической плотности
растворов. Это снижает качество измерений, так как
характеристики фотоэлементов могут* несколько различаться, а
зависимость между освещенностью и силой фототока не вполне
прямолинейна. Эти недостатки полностью устраняются при
втором способе измерений.
2-й способ. На пути левого пучка света, как и ранее,
ставят кювету с растворителем, и такую же кювету с
растворителем ставят на пути правого пучка. Диафрагму
закрывают на 70%, для чего на шкале правого барабана ставят
«нуль оптической плотности». Нейтральными клиньями
уравнивают световые потоки, подводя стрелку гальванометра к
нулевому делению. Заменяют справа кювету с
растворителем на кювету с раствором и открывают постепенно
диафрагму, подводя стрелку гальванометра к нулю. Отсчет берут по
шкале правого барабана 6. В этом случае при любых концен
трациях исследуемых растворов освещенность фотоэлементов
в момент измерения остается постоянной. Постоянство
освещенности обеспечивает независимость результатов от
характеристик фотоэлементов, поэтому второй способ точнее.
Однако измеряемые по второму способу величины ограничены
значениями оптической плотности от 0 до 0,52 и
коэффициентов пропускания от 100 до 30%, что вытекает из самого
принципа измерения, поскольку перед началом работы
приходится частично закрывать диафрагму.
Второй способ измерений не только точнее, но и
значительно проще, так как установка нейтральных клиньев
производится только один раз. Поэтому во всех случаях
рекомендуется работать по второму способу. Первым способом
можно пользоваться только, когда необходимо измерить
высокие оптические плотности (более 0,5), а по условиям
работы нет возможности или нецелесообразно разбавлять
исследуемые растворы.
264
Спектрофотометр СФ-2М
:ь''>'Р"Г'"кчн'тГЩИГ1 С11е«рофотомеТр СФ-2М предназ^чеи
жсния „ „п" ,, ш,т"ческИХ плотностей, коэффициентов отра-
от 400 10 7&ускаи,!2 Ра"воров и твердых тел в диапазоне
и wile cup*'-™ ЛЛЛ'- РезУльтаты автоматически записываются
полней -tm,ui)альиои кР"вой на специальном бланке. Время
iioMoiuiw, п, "велико и может непрерывно изменяться с
чк-т,',! РЛ>КТ°ра мот°Ра от Зю10*и. Весь прибор
vciniiTt-TM / °Хроматора с за""сываюишм устройством и
лш ании прибо^'Г К°ЖуХе ° ,юследн,,м смонтирован блок
■ ,.нш'ТтЧеСКаи СХема "Р"б°Ра "оказана иа рис. 63.
Изобрази J J" о1'""'" ,1акал,1Ва""" / фокусируется посредством
кон.и тора 2 через входную щель 3 в фокальной плоскости
°' hT,,Ba коллиматора. Затем световой пучок разлагается в
снекгр призмой 5. Спектральное изображение входной щели
совпадает с плоскостью средней щели 8, 9; требуемый
участок спектра вырезается „рн перемещении средней и1ели
„доль линии спектра. Пройдя через вторую диспергирующую
при.му 12, монохроматический пучок проектируется в
плоскость выходной щели 14. При помощи двояконреломляющен
призмы Pomona /6 свет поляризуется и, проходя призму 18,
делится н<1 два пучка, расходящихся симметрично онтнче'ской
пси. иоа пучка проходит через кюветы 22, заполненные ана-
лизщпемым раствором и растворителем, отражаются от
эталонных отражателен 25, суммируются в интегрирующей
сфере J.S и попадают на фотоэлемент 24.
В оптической системе прибора происходит сдвиг фазы
одною из лучей но отношению ко второму, поэтому при
равенстве световых пучков в интегрирующей сфере
'напряжение, снимаемое с фотоэлемента 6 (рис. 64) на усилитель 5,
равно нулю. Если же интенсивность одного из пучков ока-
жччея больше интенсивности другого, то в фотоэлементе
возни!.ает фототок, который через усилитель подается на обмот-
к\ электродвигатели 3, заставляя его вращаться.
Электродвигатель передвигает перо 2 записывающего механизма и
одновременно поворачивает первую призму Рошона 4. При
вращении призмы распределение энергии в обоих пучках
изменяется, пока их интенсивности не сравняются. По
достижении равенства напряжение фотоэлемента, подаваемое на
нчод усилителя, снова обращается в нуль, вращение
электродвигателя и призмы Рошона прекращается, а перо
останавливается на делении, отвечающем пропускающем снособно-
26Г>
Si—v у 1
Рис. 63. Оптическая схема
спектрофотометра СФ-2М:
1 — лампа накаливания; 2—конденсор; 3—
входная щель; 4, 13 — объективы; 5, 12 —
диспергирующие призмы; 6, 7, 10, И, IS,
20 — линзы; 8, 9—средняя щель; 14 —
выходная щель; 16, 18, 19 — поляризующие
призмы; 17 — диафрагма; 21 — полулинзы,
22 — кюветы; 23—интегрирующая сфера;
24 — фотоэлемент; 25 — стандартные
отражатели
сти раствора. Таким образом, мерой отражения или
пропускания образца по отношению ж эталону служягттаэте'нение
углового положения первой призмы Рошона. — —
Общий вид прибора показан на рис. 65. Источником света
служит кинопроекционная лампа К-30, расположенная под
кожухом /. Изображение нити лампы полностью и
равномерно заполняет объектив коллиматора, что можно
наблюдать через левое окошко в кожухе прибора. Рядом с окошком
Рис 64 Блок-схема записывающего устройства спектрофотометра
СФ-2М:
J — барабан с бланком, 2 — перо, 3 -~ мотор отработки, 4 — призма
Рошона, 5 — усилитель, б — фотоэлемент
расположена рукоятка, изменяющая ширину входной щели.
Юстировка лампы осуществляется рукоятками в нижней
части кожуха.
Кюветы с жидкщ№- образцами устанавливают на
параллельных тискаГ'в "кюветном отделении 6, закрытом крышкой.
В окна на задней «оверхностй'интегрирующей сферы
вставляют эталонные отражатели. При измерении отражающей
способности держатели с кюветами вынимают из кюветного
-отделения, а образец прижимают к левому окну
интегрирующей сферы.
— Изменение длины волны света, проходящего через
образец, достигается перемещением средней щели, что можно
сделать вручную с помощью рукоятки 2, предварительно завер-
267
нув винт 4 до упора. Установленная длина волны
определяется по шкале в окошке на кожухе прибора. Вращение
рукоятки 2 в сторону увеличения длины волны неограниченно^
вращение в обратную сторону ограничено диапазоном шкалы
длины волн от 750 до 400 ммк
Автоматическое изменение длины волны обеспечивается
мотором 14, перед включением мотора выключателем с
Рис 65 Внешний вид спектрофотометра СФ 2М
/—кожух лампы, 2 — рукоятка длин волн, 3 — рукоятка редуктора
мотора, 4—стопорный винт редуктора, 5 — переключатель способа
записи, 6 — кюветное отделение, 7 — интегрирующая сфера
Выключатели, 8 — мотор модулятора, 9- пуск мотора модулятора, 10—
Jiawiiui, 11— мотор длин волн, 12—-мотор отработки, 13 —
переключатель реверсивного двигателя, 14 — мотор длин волн, 15 — мотор
отработки, 16 — барабан
надписью «мотор длин волн» // необходимо отпустить винт 4,
расположенный на коробке скоростей Скорость записи
спектра регулируется рукояткой 3.
На лицевой стороне прибора расположены также
выключатель и кнопка мотора модулятора 8, 9, обеспечивающего
модуляцию световых пучков с частотой 50 гц, выключатель
мотора отработки 12, подающий напряжение с усилителя на
обмотку реверсивного двигателя, и переключатель 13,
изменяющий движение записывающего пера на обратное.
С правой стороны кожуха имеется окошко, открывающее
доступ к рукоятке установки ширины выходной щели и к вин-
268
ту, регулирующему равенство интенсивностей световых
пучков, попадающих в кюветное отделение.
Сверху на кожухе расположена рукоятка 5
переключателя способа записи. Если желательно записать оптическую
плотность, то поворачивают рукоятку так, чтобы в ее окне
появилась буква Д; при записи коэффициентов отражения и
пропускания в окне должна быть буква Т.
На передней стенке блока усилительного устройства
имеются выключатели сети и усилителя и рукоятка регулировки
усиления. Степень усиления определяется раскрытием
светящегося сектора оптического индикатора (лампа 6Е5С).
Включение и подготовка прибора к работе. Перед
началом работы устанавливают бланк на барабане
записывающего устройства. Для этого по двум нанесенным на бланке
рискам обрезают его (если риски не нанесены, то обрезают
по размеру барабана) и ставят на шкале длин волн отсчет
400 ммк Бланк навертывают на барабан так, чтобы край
бланка, вдоль которого нанесены величины длин волн, ровно
прилегал к правому бортику барабана, а перо стояло на
абсциссе 400 ммк, и прижимают бланк пружиной. К обоим
окошкам интегрирующей сферы поджимают эталонные
отражатели баритовые пластинки, пластинки из MgO или чистый лист
хорошее белой чертежной бумаги
Сетевой шланг включают в розетку электросети, а на
приборе устанавливают требуемую ширину щелей и скорость
записи. В зависимости от цели рукоятку 5 ставят в
положение «Д» или «Т». Включают последовательно выключатели
«сеть» и «усилитель» и дают прогреться 3—5 мин. Включив
лампу, юстируют ее, добиваясь полного заполнения
объектива коллиматора. Включают мотор-модулятор, на 2—-3 сек
нажимают кнопку «пуск модулятора» и включают мотор
отработки. Когда рукоятка 5 установлена в положение «Т»,
перо должно двигаться в сторону «100% пропускания», в
положении «Д» перо движется к «0 оптической плотности».
Если перо движется в противоположную сторону,
перебрасывают переключатель 13.
В зависимости от масштаба записи перо должно
остановиться на делении «100% пропускания» или «0 оптической
плотности»; при отклонении более чем на 0,5% перо
устанавливают на требуемое деление винтом в правом окошке
кожуха. Качество эталонных отражателей и работы прибора
проверяют записью по всему спектру 100%-ной пропускаемо-
сти. Для строгой проверки и юстировки прибора применяют
стандартные светофильтры, входящие в комплект прибора.
269
Порядок работы при измерении оптической
плотности и коэффициентов отражения
на СФ-2М
1 Установить бланк для записи, как указано выше
2 Включить выключатели «сеть» и «усилитель» и дать
прогреться лампам усилителя 3—5 мин Проверить при
включенном приборе положение пера
3 Заполнить две одинаковые кюветы исследуемым
раствором и растворителем и закрепить их в тисочках
держателей, для правильной установки держателей в кюветном
отделении имеются направляющие Кювету с раствором
помещают на пути левого светового пучка, растворитель — на пути
правого.
4 Закрыть крышку кюветного отделения и включить
лампу и мотор модулятора
5. Включить мотор отработки. После того как мотор
перестанет двигаться и перо займет положение, отвечающее
оптической плотности раствора при данной длине волны, включить
мотор длин волн, предварительно отпустив винт 4 на коробке
скоростей (I). В области 400—410 ммк чувствительность
прибора несколько понижена, поэтому при включении мотора
отработки перо довольно медленно достигает равновесного
положения Во избежание ошибок лучше выждать некоторое
время и только после этого включать мотор длин волн;
слишком раннее включение может привести к появлению ложного
максимума в области 405—415 ммк
6 Провести запись оптической плотности по всему
спектру
Для записи коэффициентов пропускания рукоятку 5
переводят в положение «Т» и, убрав кюветы из кюветного
отделения, устанавливают перо на «100% пропускания» Затем
снова помещают кюветы на пути световых пучков и проводят
запись по спектру.
Измерение коэффициентов отражения проводят так же,
как и измерение коэффициентов пропускания, но образец
прижимается к левому окошку интегрирующей сферы, а кю-
ветное отделение остается пустым.
Спектрофотометр СФ-4
Спектрофотометр СФ-4 представляет собой призменный
монохроматор, предназначенный для измерения
коэффициентов пропускания и оптической плотности растворов и твер-
270
дых тел в ультрафиолетовой и видимой частях спектра от
210 до 1100 ммк. При работе в ультрафиолетовой области
используют водородную лампу в качестве источника света и
сурьмяно-цезиевый фотоэлемент, в видимой области
спектра— лампу накаливания и кислородно-цезиевый
фотоэлемент (от 650 до 1100 ммк).
Для измерения интенсивности света в приборе
использован компенсационный метод. Фототок, возникающий в цепи
Рис. 66. Внешний вид спектрофотометра СФ-4:
/ — рукоятка длин волн, 2—шкала длин волн, S — рукоятка и шкала
отсчетного потенциометра, 4 — миллиамперметр, 5, 6 — рукоятка и шкала
установки ширины щели, 7—переключатель, 8 — потенциометр темпового
тока, 9—потенциометр чувствительности, 10 — кюветное отделение;
11— рукоятка передвижения кювет, 12—установка фотоэлементов;
13 — включение фотоэлементов
фотоэлемента, проходит через высокоомное сопротивление,
на котором создается падение напряжения,
пропорциональное освещенности фотоэлемента. Компенсация фототока
осуществляется током противоположного направления,
снимаемым с потенциометра. Если падение напряжения на
сопротивлении и напряжение, снимаемое с потенциометра, равны и
противоположно направлены, то стрелка миллиамперметра 4
(рис. 66), включенного в компенсационную схему, находится
на нулевом делении шкалы. В результате показания шкалы
потенциометра оказываются пропорциональными
освещенности фотоэлемента.
271
При измерениях в световой пучок поочередно вводят
кювету с растворителем и кювету с раствором. После установки
кюветы с растворителем стрелку миллиамперметра подводят
к нулю, раскрывая щель 6 и пользуясь рукояткой 9
потенциометра чувствительности. Затем в световой поток вводят
кювету с раствором и, вращая ручку отсчетного
потенциометра 3, добиваются полной компенсации. Результат получают
на шкале отсчетного потенциометра.
Порядок работы на приборе
Перед началом работы в приборе устанавливают источник
света: лампу накаливания или водородную лампу, в
зависимости от того, в какой области спектра предполагается
работать.
Включение водородной лампы осуществляется через
стабилизатор; для этого рукоятку потенциометра на
стабилизаторе (на рисунке не показан) переводят в крайнее левое
положение и включают ток накала. Постепенно увеличивают
ток накала, поворачивая ручку потенциометра вправо, пока
контрольный амперметр не покажет значение,
соответствующее току накала, указанному в паспорте лампы. Через
несколько минут после достижения паспортного значения силы
тока включают высокое напряжение и £разу уменьшают ток
накала в соответствии с паспортными данными (или
переводят потенциометр в крайнее левое положение). После
включения лампы приступают к измерениям.
Рукояткой 12 (рис. 66) устанавливают на пути светового
пучка один из фотоэлементов (для сурьмяно-цезиевого при
работе в области 210—650 ммк рукоятка полностью
вдвинута; для кислородно-цезиевого при работе от 650 до ПО ммк
рукоятка полностью выдвинута). Шторкой 13 перекрывают
световой пучок и компенсируют темновой ток
потенциометром 8.
Одну из кювет заполняют исследуемым раствором,
вторую — растворителем и помещают их в гнездо кюветного
отделения 10.
Затем устанавливают по шкале длин волн требуемую
длину волны, открывают шторку 13, в световой пучок вводят
кювету с растворителем и переключатель 7 устанавливают в
положение «включ.». Потенциометр чувствительности 9
переводят примерно в среднее положение и подбирают ширину
щели так, чтобы стрелку миллиамперметра 4 можно было
установить на нулевое деление при одном обороте
потенциометра чувствительности.
272
Установив стрелку миллиамперметра на нулевое деление,
вводят в световой пучок кювету с раствором;
переключатель 7 ставят в положение «XI» и подводят стрелку
миллиамперметра снова к нулевому делению, вращая ручку 3 отсчет-
ного потенциометра. По шкале потенциометра берут отсчет
процента пропускания или оптической плотности.
Количественные определения при постоянной длине
волны осуществляют, не меняя настройку прибора. В случае
неустойчивых показаний через 3—5 измерений проверяют
компенсацию темнового тока и настройку прибора по
растворителю. Для снятия спектров поглощения требуется
непрерывная настройка по растворителю для каждого значения длины
волны. Эту настройку проводят или при положении
переключателя 7 «включ.» — тогда положение отсчетного
потенциометра безразлично, или при положении «XI» — тогда отсчет -
ный потенциометр необходимо поставить на нуль оптической
плотности.
Инфракрасные спектрометры
Особенности длинноволнового излучения вносят
значительную специфику в устройство инфракрасных
спектрометров. Инфракрасное излучение с длиной волны >2,5 мк
сильно поглощается стеклом, поэтому призмы, защитные стекла
и окна кювет изготовляют из кристаллов различных солей:
NaCl, KC1, LiCl, КВг. Вместо линз устанаа/щшцшив©мтуты"е"
зеркала с покрытием из материалов, ".хорошо от«рджающих
инфракрасное излучение. Источниками излучения служат
силлитовые стержни — «глобары», или другие излучатели, ксР
торые разогреваются до высокой температуры под действием
электрического тока. Иногда используют для этой же цели и
значительно более простые излучатели, например ленту из
нихрома или платины.
Приемником излучения служат термоэлементы;
возникающие в-имх-очень слабые токи усиливаются фотоэлектроопти-
ческими или электронными усилителями.
Спектрометр ИКС-11. Оптическая схема однолучевого
спектрометра ИКС-11 показана на рис. 67. Свет от источника
(«глобар» /) при помощи системы зеркал подается на
входную щель 5 монохроматора. Затем через параболическое
зеркало он попадает на призму 10 и разлагается в спектр.
После отражения от плоского зеркала )/ свет претерпевает
вторичное разложение и, отразившись от зеркал 12, 7,
фокусируется сферическим зеркалом 8, на рабочую поверхность
273
термоэлемента 9. Возникающий в термоэлементе ток
измеряется зеркальным гальванометром. Изучаемый объект
помещают на пути светового пучка перед входной щелью.
На приборах ИК.С-11 предусмотрена также
автоматическая запись показаний гальванометра на фотобумаге; более
поздние конструкции инфракрасных спектрометров
снабжены электронными потенциометрами ЭПП-09, и запись спектра
ведется пером на бумаге.
X
7 -~i
'1 '
-P^a-f-r
12
Pise. 67. Оптическая схема инфракрасного спектрометра ИКС-11:
1 — источник излучения, 2, S — зеркала, 4 — кювета с образцом,
5 — входная щель, б—выходная щель, 7, 12—плоские зеркала,
8 — сферическое зеркало, 9 — термоэлемент, 10—призма, 11 — плоское
зеркало, IS—параболическое зеркало
Наиболее точные результаты на одиолучевом приборе
получают при визуальной записи показаний гальванометра по
точкам. Для этого вращают барабан, связанный с
устройством, изменяющим длину волны излучения, попадающего на
термоэлемент; для каждого положения барабана
записывают два показания гальванометра, помещая последовательно
в световой пучок кювету с раствором и кювету с
растворителем. Затем изменяют положение барабана и снова делают
два измерения и т. д., продолжая запись, пока измерениями
не будет охвачена вся интересующая область спектра.
Пропускающая способность образца для каждого значения
длины волны вычисляется просто как отношение двух показаний
гальванометра: для кюветы с образцом и контрольной.
Подробный спектр, на котором видны даже узкие полосы
274
поглощения, получают при работе с минимальным
раскрытием щели, когда вырезаемый спектральный участок достаточно
мал. Минимальная ширина щели зависит как от
чувствительности приемника излучения (термоэлемента), так и от
свойства источника излучения. Распределение энергии в спектре
источника неравномерное, и с увеличением длины волны
энергия излучения резко падает. Поэтому, работая в области
малых частот, приходится увеличивать размер щели, а при
переходе к меньшим длинам волн, ширину щели уменьшают.
Ширина щели подбирается таким образом, чтобы отклонения
«зайчика» гальванометра можно было измерить с
достаточной степенью точности.
Подготовка ИКС-11 к работе и порядок работы
1. В зависимости от требуемой спектральной области в
прибор устанавливают одну из призм, входящих в комплект.
2. Проверяют правильность освещения щели и, слегка
перемещая источник излучения, добиваются, чтобы щель
была освещена равномерно и полностью.
3. Проводят градуировку прибора с каждой призмой в
отдельности, используя вещества с точно известными
максимумами поглощения. По результатам градуировки строят
график перевода делений отсчетного барабана в длины волн.
4. За 30 мин до начала работы включают фотоэлектрооп-
тический усилитель и дают ему прогреться. Затем пускают
воду в холодильник источника излучения и включают ток,
разогревающий источник. Разогревать источник надо
постепенно, медленно увеличивая силу тока до значения,
указанного в паспорте излучателя. При помощи корректора
«зайчик» гальванометра устанавливают на нулевое деление
шкалы'. На барабане длин волн подбирают деление,
отвечающее максимальной длине волны исследуемого
спектрального участка, и такой размер щели, чтобы отсчет по
гальванометру был не менее 10—20 делений.
5. В держателе кювет закрепляют две кюветы: одну с
образцом, другую с растворителем. Перекрыв световой пучок
глухим окошком, проверяют нулевое положение
гальванометра. Затем, поместив на пути светового пучка кювету с
образцом и открыв окошко, записывают показания
гальванометра. Снова закрывают окошко, передвигают кюветы,
помещая на пути светового пучка кювету с растворителем, и,
проверив нулевое положение гальванометра, производят из-
275
мерение. Затем барабан переводят на одно деление в сторону
уменьшения длины волны и повторяют отсчеты. Таким
образом измеряется весь исследуемый спектральный участок.
Подготовка образцов
В инфракрасной области исследуют как жидкие, так и
твердые образцы. Почвенные вторичные минералы и гумино-
вые кислоты исследуют обычно в виде порошков, поскольку
они не растворимы в органических растворителях, пригодных
для работы в инфракрасной области спектра. Существенным
недостатком порошкообразных объектов является их высокая
рассеивающая способность. Для уменьшения влияния
рассеивания порошки минералов, коллоидов гуминовых кислот и
других аналогичных объектов или запрессовывают под
давлением в таблетки из КВг, или помещают между двумя
полированными солевыми пластинками суспензию,
приготовленную из парафинового масла и тонкорастертого исследуемого
образца. В качестве контроля применяется такая же кювета,
заполненная чистым парафиновым маслом. При работе с
суспензиями (пастой), поверхности солевых пластин должны
быть идеально отполированы.
Для заполнения кюветы одну солевую пластину помещают
в металлическую оправу разборной кюветы и сверху кладут
свинцовую или алюминиевую прокладку (рис. 68), в вырез
прокладки наносят каплю подготовленной суспензии,
закрывают сверху другой пластинкой и готовую кювету прочно
зажимают между кольцами металлической оправы. В
хорошо заправленной кювете нет пузырьков воздуха, а
дисперсная фаза равномерно распределена по всей площади окна
кюветы.
Наиболее часто суспензию готовят на парафиновом
масле, однако оно дает сильные полосы поглощения около
3 и 6.5 мк и для работы в этих областях непригодно.
В последние годы получает распространение помещение
различных биологических материалов в прозрачные пленки.
Этот метод значительно проще, чем приготовление солевых
пластин, и дает хорошие результаты. Способ приготовления
прозрачных агаровых пленок, по И. Андрашина и С. Крупа,
следующий.
На стеклянную пластинку наносят тонкий слой
силиконового масла, затем пластинку нагревают 60—100 мин в
термостате при 200—220°. Одновременно готовится 1%-ный водный,
раствор агар-агара на кипящей водяной бане. Раствору дают
276
отстояться, фильтруют через плотный фильтр и медленно
охлаждают до 35° К этому раствору прибавляют равный
объем ~ 1%-ного раствора (или суспензии) исследуемого
материала
На покрытую силиконовым маслом стеклянную пластинку
устанавливают стеклянный цилиндр (форму) без дна высотой
2 см и диаметром 3—4 см В цилиндр наливают 5 мл приго-
Рис 68 Разборная кювета к ИКС 11
1- корпус кюветы, 2 — солевая пластина, 3—прокладка и
алюминиевой фольги — кольцо оправы
товленной смеси агар-агара с исследуемым веществом
Раствор в форме охлаждают до комнатной температуры, поьа
он не станет достаточно твердым, и затем медленно сушат в
термостате при температуре 37° в течение 16—20 ч После
такой сушки получают очень хорошую прозрачную пленку,
которая свободно лежит на стеклянной пластинке Если
пленка не отстает, то ее можно отделить от стеклянной пластинки
тонким острым лезвием
Контрольную пленку для сравнения готовят точно так же,,
только вместо образца добавляют к агар-агару равный объем
дистиллированной воды
Вместо стеклянной пластинки для получения пленок
может быть использован тонкий слой целофана, который перед
нанесением пленки увлажняют и натягивают на кювету. Для
контроля толщш'ы пленок к раствору агар-агара подмешива-
277
ют внутренний стандарт с точно известной полосой
поглощения; так, при изучении белковых спектров пользуются
полосой поглощения KCNS при 2120 см~1.
СПЕКТРОФОТОМЕТРИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ ПОЧВ
В почвенном анализе спектрофотометрические методы
используют для:
1) количественного определения различных химических
элементов как при валовом анализе почв, так и при анализе
подвижных форм соединений;
2) качественной характеристики органических веществ
почвы;
3) количественного определения гуминовых и фульвокис-
лот и неспецифических органических соединений;
4) изучения структуры органических веществ почвы и
почвенных вторичных минералов;
5) изучения отражательной способности почв в целях
аэрофотосъемки, расчета теплового баланса и
количественных определений окрашенных компонентов почвенного
профиля.
Излагаемые ниже методы охватывают все указанные
разделы.
Определение обменного алюминия на фотометре ФМ
Алюминий образует ярко-красный лак при взаимодействии
с алюминоном — аммонийной солью ауринтрикарбоновой
кислоты C22H2»N». Эта реакция используется для
фотометрического определения алюминия.
Образующийся лак, по Сенделу, имеет следующее
строение:
COONH4
v/^Vo...
v_/—
но—^ ^/
\=/
I
COONH4
COO —
AI
278
Кроме алюминия окрашенные соединения с алюминоном
образует и железо, поэтому последнее отделяют
экстрагированием роданидного комплекса железа смесью амилового
спирта и эфира.
Сам алюминон также имеет сходную с цветом
образующегося соединения окраску. Интенсивность окраски
растворов алюминона и его солей зависит в значительной степени
от величины рН, поэтому необходимым условием
количественного определения является применение буферных
растворов. Образующийся лак алюминия с алюминоном имеет ярко
выраженный коллоидный характер, и его растворы
подчиняются закону Бугера —Бэра в узком диапазоне концентраций.
Отделение железа и возбуждение окраски. 20 г почвы
помещают в колбу, приливают 100 мл 1,0 н. раствора КО и
взбалтывают на ротаторе в течение часа. Суспензию
фильтруют, несколько миллилитров фильтрата (содержание
алюминия во взятом объеме должно быть порядка 0,03—0,12 мг)
переносят в небольшую фарфоровую чашку и обрабатывают
1—2 каплями конц. НО; затем добавляют 1 мл 10%-ного
раствора NH4CNS, перемешивают и, добавив 5 мл смеси
эфира и амилового спирта A :2), снова перемешивают
стеклянной палочкой. Смесь переносят в делительную воронку и
дают ей отстояться. Водную фазу сливают в чашку, экстракт
отбрасывают, снова добавляют эфиро-амиловую смесь и
повторяют экстракцию до обесцвечивания раствора.
Раствор, не содержащий железа, переносят в мерную
колбу на 100 мл, нейтрализуют до слабо-розовой окраски по
фенолфталеину и разбавляют водой до 20—25 мл. К
слаборозовому раствору приливают 1 мл 1,0 н. НО, 5 мл 3,0 н.
CH3COONH4 и 2,5 мл 0,1%-ного раствора алюминона. Через
20 мин добавляют 0,5 мл 5,0 н. NH4OH, 0,5 мл 5,0 н.
(NH4JCOa и доливают колбу до метки дистиллированной
водой.
Окрашенные растворы исследуют на ФМ или ФЭК- Для
выбора светофильтра предварительно снимается
спектральная характеристика раствора. Во вторую кювету наливают
раствор, содержащий все те же реактивы, кроме алюминия.
Стандартные растворы готовятся аналогично. Для
приготовления шкалы в колбы на 100 мл вносят различные
количества образцового раствора алюминия: 1, 2, 5, 8, 10, 15 и
20 мл. Измерив оптические плотности образцовых растворов,
строят график зависимости D—С, по которому определяют
концентрацию алюминия в почвенных вытяжках и затем
рассчитывают содержание алюминия в почвенном образце.
279
Определение железа в виде сульфосалицилата
Сульфосалициловая кислота образует с железом (III)
несколько комплексов, состав которых зависит от кислотности
среды. В кислой среде образуется моносульфосалицилатный
комплекс железа фиолетовой окраски, в среде, близкой к
нейтральной,— красный дисульфосалицилат ив щелочной
среде (рН 8—11)—желтый трисульфосалицилат, [Sal3Fe]3~,
Sal — остаток сульфосалициловой кислоты. Молярный
коэффициент погашения трисульфосалицилатного комплекса пр;>
>.=416 ммк равен 4000.
В анализе почв используют трисульфосалицнлатные
комплексы в аммиачной среде, отличающиеся большой
устойчивостью. Проведению анализа в щелочной среде может
помешать образование гидроокисей, выпадающих в осадок и
вызывающих помутнение раствора. Чтобы этого не
происходило, к исследуемому раствору добавляют избыток
сульфосалициловой кислоты, образующей с ионами алюминия,
кальция и магния бесцветные комплексы, хорошо растворимые в
аммиачной среде. Для удержания в растворе марганца
прибавляют также 0,5—1,0 г солянокислого гидроксиламина
(Е. В. Аринушкина). ч
Определение железа в солянокислой вытяжке из почв.
К 10 г почвы прибавляют 250 мл 0,5 н. НО, взбалтывают на
ротаторе в течение часа и фильтруют. Из приготовленной
вытяжки берут такой объем раствора, чтобы в нем
содержалось от 0,05 до 0,3 мг железа, и переносят его в мерную колбу
на 100 мл. Добавляют 10 мл 25%-ного раствора
сульфосалициловой кислоты и нейтрализуют конц. NH4OH до
появления слабого запаха. При помутнении раствора необходимо
вновь добавить сульфосалициловой кислоты до полного
растворения осадка. Количество добавленного аммиака
достаточно, если желтая окраска раствора не меняется от
прибавления нескольких капель аммиака. Одновременно
окрашивают и эталонные растворы, содержание железа в
которых находится в указанных выше пределах. Окрашенные
растворы доводят водой до метки и фотометрируют.
Светофильтр выбирают по предварительно снятой спектральной
характеристике раствора.
Определение подвижных соединений фосфора (по Кирсанову)
на фотоэлектроколориметре
В кислой среде легко образуется фосфорномолибденовая
гетерополикислота состава Hs[P(Mo3OiQ)i]-пН20. При
действии восстановителей молибден из гетерополианиона восста-
280
навливается и раствор окрашивается в интенсивно синий"
цвет. Состав образующегося соединения, по Дениже, может
быть выражен формулой (Мо02 -4Мо03J • Н3Р04 • 4Н20. Эта
реакция лежит в основе фотометрического определения
фосфорной кислоты. В качестве восстановителя обычно
используют SnCb; более мягким восстановителем, часто дающим
более воспроизводимые результаты, является аскорбиновая
кислота. Рекомендованы также методы, по которым вовсе не
используются восстановители, а в качестве реактива
применяют раствор, содержащий смесь пяти- и шестивалентного
молибдена.
Для получения хороших результатов необходимо строго
следить за кислотностью раствора; присутствие сильных
окислителей, например Fe (III), искажает результаты в силу
окисления Мо (V) до Мо (VI).
Ход определения
К 5 г почвы добавляют 25 мл 0,2 н. раствора НС1,
взбалтывают и через 15 мин отфильтровывают. 10 мл фильтрата
переносят в колбу на 100 мл, разбавляют водой и добавляют
4 мл 2,5%-ного сернокислого (ЫН4)бМо7024-4Н20.
Перемешивают, вносят 5 капель 2,5%-ного раствора SnCb> в 10%-ный
НС1 и доводят до метки дистиллированной водой. Через 10мин
начинают фотометрирование. Стандартные растворы
окрашивают обязательно одновременно с испытуемым.
Измерение степени окраски проводят на фотоэлектроколо-
риметре; для работы выбирают тот светофильтр, при
использовании которого получается наибольшее значение оптической
плотности.
ХАРАКТЕРИСТИКА ОРГАНИЧЕСКИХ ВЕЩЕСТВ ПОЧВЫ
Г^миновые и фульвокислоты интенсивно поглощают свет
в области 400—450 ммк; с увеличением длины волны
интенсивность поглощения быстро падает, относительно мало-
меняясь в диапазоне 600—700 ммк. Наиболее сильно
гумусовые вещества поглощают энергию ультрафиолетового
излучения. Кислоты, выделенные из образцов различных почв, мало
отличаются по общему характеру поглощения, однако весьма
заметные .различия наблюдаются в величинах молярных
коэффициентов погашения, что, по М. М. Кононовой,
характеризует степень конденсированное™ гуминовых кислот различ-
281
ного происхождения. Для численного сравнения
интенсивности поглощения света гумусовыми веществами удобно
пользоваться £-величинами; для концентрации в 0,001%
соответствующая величина обозначается как Щ'^1%.
Для всех изученных почв ^-величины фульвокислот
значительно меньше (в 5—10 раз), чем ^-величины гуминовых
кислот. Кроме того, окраска фульвокислот в значительной
степени зависит от рН раствора: в щелочных средах,
оптическая плотность фульвокислот увеличивается на 0,025 при
изменении рН на единицу (при длине волны 450 ммк).
Характер поглощения света растворами фульвокислот в видимой
части спектра также несколько отличается от растворов
гуминовых кислот. В коротковолновой части спектра
D00—500 ммк) кривые поглощения света гуминовыми
кислотами и фульвокислотами имеют разную степень крутизны,
что четко выражается отношением оптических плотностей,
измеренных при 420 и 650 ммк. Это отношение для гуматов
натрия в среднем 4,4, а для фульвокислот >7.
Сравнивая спектры поглощения гуминовых кислот и
фульвокислот как по ^-величинам, так и по отношению
оптических плотностей при разных длинах волн, можно получить
материал для сравнительной характеристики гумусовых
веществ различных типов почв.
Выделение гуминовых и фульвокислот из почвы. Из 50 г
почвы отбирают неразложившиеся растительные остатки,
почву растирают и просеивают через сито с диаметром
отверстий 1 мм. Подготовленную навеску декантируют 0,05 н.
H2SO4 до удаления кальция, промывают водой и переносят
в цилиндр емкостью 1 л. Почву заливают 800 мл 0,1 н.
раствора NaOH, суспензию взбалтывают, добавляют в качестве
коагулятора Na2S04 C0—40 г на 1 л раствора) н оставляют
стоять до полного оседания частиц твердой фазы. Затем
сливают отстоявшийся раствор при помощи сифона в десчти-
литровую бутыль. Обработку щелочью повторяют до тех
пор, пока раствор не станет только слабо окрашиваться.
Темноокрашенный щелочный раствор центрифугируют на
проточной центрифуге Мошева. Операцию повторяют, если в
волчке (роторе) центрифуги скапливается заметное
количество ила.
К отцентрифугированному раствору добавляют 2—3 н.
H2SO4 до начала выпадения хлопьевидного осадка гуминовой
кислоты, которому дают полностью осесть, жидкость над
осадком сливают, а избыток раствора, оставшийся в
гуминовой кислоте, отделяют центрифугированием. Осадок гумино-
.282
вой кислоты промывают на центрифуге 0,05 н. НС1,
добавляют 0,1 н. NaOH до полного растворения осадка и
повторяют осаждение. Отмытый осадок суспендируют водой (до
состояния густой пасты) и слегка подщелачивают 10%-ным
NaOH до рН 8,0—8,5. Полученную суспензию подвергают
электродиализу, после чего препарат высушивают при
60—70°.
Кислый фильтрат, оставшийся после первого и второго
осаждения гуминовой кислоты и содержащий фульвокислоты,
пропускают через активированный уголь, предварительно
промытый разбавленной 0,1 н. H2SO4. Фульвокислоты
сорбируются на угле. Для отделения неспецифических органических
соединений уголь последовательно промывают водой,
ацетоном и спиртом.
Фракцию собственно фульвокислот (по Форситу) снимают
с угля 0,1 н. NaOH. Щелочной раствор фульвокислот,
имеющий вишнево-красный цвет, диализуют в целофановых
мешочках, пока реакция среды не станет слабокислой. Затем
подкисляют раствор фульвокислот 1—2 каплями 10%-ной
HCI и продолжают диализ до отрицательной реакции на
Cl-ион. Водный раствор фульвокислот выпаривают в вакууме
при температуре 35° и получают сухой препарат в виде
темно-красных блестящих чешуек или пленок.
Полученные препараты гуминовых и фульвокислот
используют для изучения оптических свойств.
Получение спектров поглощения
фульвокислот и г у матов натрия
4—6 мг гуминовой кислоты растворяют в 100 мл 0,1 н.
раствора NaOH. Подбирают кювету, чтобы оптическая
плотность в области 420 ммк была порядка 0,9—1,2, и снимают
спектр поглощения на спектрофотометре СФ-2М. как это
описано на стр. 269. Для сравнения можно использовать
такую же кювету, заполненную дистиллированной водой.
Также поступают с фульвокислотами, с той лишь разницей,
что навески фульвокислот по 10—15 мг растворяют в 100 мл
дистиллированной воды.
Растворы гуматов натрия и фульвокислот хорошо
подчиняются закону Бугера-Бэра, поэтому по спектрам поглощения
находят оптические плотности образцов яри длинах волн 420
и 650 ммк и пересчитывают их на концентрацию гуминовых
кислот и фульвокислот, равную 0,001%, и толщину ^тоглощаю-
28»
щего слоя 1 см; т. е. вычисляют величину £,i£«I%- На
основании полученных величин располагают все изученные
образцы в ряд по степени конденсированное™.
Спектры поглощения в ультрафиолетовой области
получают на спектрофотометре СФ-4. В качестве эталонного
раствора (раствор сравнения) в этом случае берут
обязательно те компоненты, в которых растворен исследуемый
образец, т. е. для фульвокислот — воду, а для гуматов
натрия— 0,1 н. раствор NaOH. Раствор NaOH надо
предохранять от поглощения СОг из воздуха.
Спектрофотометрическое определение гуминовых
и фульвокислот (ускоренный метод)
Гуминовые и фульвокислоты подчиняются закону Буге-
ра — Бэра и имеют неодинаковые коэффициенты молярного
погашения в разных областях спектра. Это позволяет
одновременно определять гуминовые и фульвокислоты из одного
раствора, если известны их коэффициенты молярного
погашения или ^-величины. Оптические свойства гуминовых
кислот различных типов почв неодинаковы, поэтому
коэффициенты погашения должны быть предварительно измерены для
каждого типа почв в отдельности. Фульвокислоты
характеризуются близкими показателями и для них можно
использовать только одно — среднее значение. Метод сводится к
измерению оптической плотности смешанного раствора при
двух длинах волн и последующему вычислению
концентрации, как это описано на стр. 252.
Ход определения
1 г предварительно подготовленной почвы подвергают
декальцированию и после промывания водой обрабатывают
0,2 н. раствором NaOH. Обработку продолжают декантацией
малыми порциями раствора щелочи до тех пор, пока он не
станет окрашенным в слабо-желтый цвет. Щелочной раствор
сразу фильтруют через плотный складчатый фильтр в мерную
колбу на 100 мл (или в зависимости от содержания
органического вещества — на 250 мл). По окончании выделения
гуминовых и фульвокислот, о чем судят по почти полному
обесцвечиванию раствора щелочи, доводят жидкость в колбе
до метки дистиллированной водой и снимают спектр
поглощения. Пользуясь формулами, приведенными на стр. 252,
рассчитывают содержание гуминовых и фульвокислот во
взятой навеске почвы, учитывая фактор разбавления.
284
Исследование реакции взаимодействия гуминовых кислот
с катионами тяжелых металлов
Гуминовые кислоты легко образуют нерастворимые в
воде осадки при взаимодействии с катионами Ni2+, Co2+, Fe3+,
•Cu2+, Zn2+ и др. Характер взаимодействия различен в
зависимости от условий протекания реакции. В щелочной среде
образуются гидроокиси указанных металлов, вместе с
которыми осаждается гуминовая кислота. При взаимодействии
концентрированных растворов гуминовых кислот и солей
происходит коагуляция гуминовых кислот без образования
гуматов. Разбавленные растворы гуминовых кислот (или
гуматов натрия) взаимодействуют с медленно добавляемой
солью так, что образуются нерастворимые гуматы, в которых
Н-ион карбоксильных групп гуминовой кислоты замещен на
металлический катион.
Проследить ход реакции и обнаружить образование
гуматов можно при помощи инфракрасной спектрометрии.
Карбоксильная группа обнаруживается в спектре гуминовых
кислот по очень сильной полосе поглощения с максимумом
около 5,9 мк. При замещении водорода карбоксильной
группы на металлический катион образуется ионизированная
группа — СОО-, имеющая полосы поглощения в области
■6,48—6,50 и 7,10—7,15 мк, т. е. спектр гумата отличается от
спектра гуминовой кислоты отсутствием полосы около 5,9 мк
и появлением двух новых полос в вышеуказанной области
спектра.
Ход работы
Для получения спектров готовят два образца. Первый
образец — это сухой препарат гуминовой кислоты,
выделенной из почвы. Второй образец — гумат меди. Для получения
последнего разбавленный раствор гумата натрия A00—200 мг
в литре) диализируют до нейтральной реакции и добавляют
к нему по каплям 10%-ный раствор сульфата меди. После
добавления каждой капли раствор тщательно перемешивают
и наблюдают, не появился ли осадок. После появления
первых признаков коагуляции прекращают добавление медной
соли и дают раствору постоять ночь. На следующий день
добавляют 10 мл 10%-ного раствора CuS04, осадок
взмучивают и тщательно перемешивают. Через 2 ч препарат
отфильтровывают, промывают несколькими порциями
дистиллированной воды и высушивают при 60°.
Далее с сухим образцом гумата меди и гуминовой кисло-
285
ты поступают одинаково. Из обоих образцов берут
одинаковые навески (от 20 до 40 мг) тщательно растирают их в
агатовой ступке; к растертому до тонкой пудры образцу
добавляют 0,4 мл парафинового масла и снова тщательно
растирают в течение часа.
Заранее готовят отполированные солевые пластины для
кювет; две пластины для пасты гуминовой кислоты в масле
и две для чистого масла. Одну из пластин помещают на
оправу сборной кюветы и покрывают сверху прокладкой из*
алюминиевой фольги толщиной около 15 мк (см. рис. 68).
В вырез прокладки наносят одну каплю пасты из
парафинового масла и гуминовой кислоты, закрывают сверху второй
солевой пластиной, которую плотно поджимают винтами.
Давление, создаваемое винтами в кювете, должно быть
равномерным на всей площади пластин, тогда образец
распределяется ровным слоем и между пластинами не остается
пузырьков воздуха. Так же готовится вторая кювета,
заполненная чистым парафиновым маслом. Съемка спектра
проводится на инфракрасном спектрометре с призмой NaCl в
интервале длин волн от 5,5 до 8 мк.
После того как получен спектр гуминовой кислоты, кювету
разбирают, пластины протирают фланелью, смоченной в
парафиновом масле, и снова собирают, помещая между
пластинами каплю пасты, приготовленной с гуматом меди.
Для сравнения используют ту же кювету с чистым
парафиновым маслом, что и в первый раз.
По результатам измерений строят графики зависимости
пропускания от длины волны, на графиках находят
минимумы, отвечающие полосам поглощения групп — СООН и —
СОО~; по соотношению интенсивностей полос при 5,9 мк
устанавливают степень замещения водорода карбоксильных
групп катионом меди.
ХАРАКТЕРИСТИКА ПОЧВ ПО КРИВЫМ СПЕКТРАЛЬНОЙ ЯРКОСТИ
Спектральная отражательная способность почв
объективно характеризует окраску почв и может быть использована
как в целях спектрозональной аэрофотосъемки почв, так и
для быстрого полуколичественного определения содержания
в почвах гуминовых кислот и соединений железа. Типичные
кривые отражения света поверхностью почвы показаны на
рис. 69, где по оси ординат отложено отражение R света в
процентах. Повышенное содержание гумуса резко уменьшает
отражательную способность почв в области 700—750 ммк:
286
во
£0
30
20
10
в,
i i i
л,
, 1
перегиб на кривой около 600 ммк связан с присутствием
окисных соединений железа.
Из органических соединений на окраску почв наиболее
сильно влияют гуминовые кислоты, поэтому удается
установить почти прямолинейную зависимость между содержанием
гуминовых кислот и коэффициентом отражения излучения с
Я=720 ммк. Эта
зависимость хорошо проел ежи- #,%_
вается при сопоставлении
почв одного генетического
типа. То же можно
показать и для зависимости
коэффициента отражения
от содержания железа в
прокаленных образцах
почвы. Прокаливание при
800—900° приводит к
полному удалению
органических веществ,
маскирующих окраску,
обусловленную соединениями железа.
Кроме того, в результате
прокаливания минералы,
составляющие твердую фазу почв, распадаются на окислы и
интенсивность окраски прокаленного образца оказывается
пропорциональной валовому содержанию железа.
Получение кривых спектральной отражательной
способности почв. 3—5 г почвы растирают в агатовой ступке
(пестиком с резиновым наконечником) и просеивают через сито с
диаметром отверстий 0,1 мм. Просеянный образец помещают
в кювету спектрофотометра СФ-2М; вторая кювета
наполняется чистым порошком MgO. Кювету с почвой прижимают
к левому окошку интегрирующей сферы спектрофотометра
СФ-2М, кювету с MgO — к правому. Включают прибор и
записывают отражение в процентах, как описано на
стр. 259, 270.
W
SOO
600
700ммк
Рис. 69. Спектральная
отражательная способность горизонтов Ai и Bi
дерново-подзолистой почвы
ХАРАКТЕРИСТИКА ПОЧВЕННЫХ ПРОФИЛЕЙ ПО СОДЕРЖАНИЮ
ГУМУСА И СОЕДИНЕНИИ ЖЕЛЕЗА
Кривые спектральной яркости позволяют быстро
установить, как распределяются в почвенном профиле соединения
железа н органические вещества. Для большинства
почвенных типов поглощение света при 720—750 ммк пропорцио-
287
нально содержанию гумуса. Чтобы выявить характер
распределения гумуса по генетическим горизонтам, поступают
следующим образом: снимают кривые спектрального
отражения образцов, предварительно растертых и пропущенных
через сито в 0,1 мм; вычисляют коэффициенты поглощения
света в области 720—750 ммк для всех исследуемых
образцов (коэффициент поглощения равен разности 100 — R, где
R — отражение в процентах); строят график изменения
коэффициентов поглощения по почвенным генетическим
горизонтам (он характеризует изменение содержания гумуса с
глубиной).
Этот метод пока что можно применить только для
относительной характеристики генетических горизонтов. Делать
абсолютные количественные выводы не представляется
возможным. Для почв с ярко выраженным иллювиальным
горизонтом (дерново-подзолистых, солонцов) описываемый метод
пригоден только в пределах перегнойно-аккумулятивного и
элювиального горизонтов. Резкое увеличение поглощения
света иллювиальными горизонтами обусловлено накоплением
окислов железа и не связано с содержанием гумуса.
Значительно более четко можно выявить распределение
по профилю соединений железа. Для этого растертые
образцы почв прокаливают в муфельной печи при температуре
900—950°. После прокаливания образцы повторно растирают,
помещают в кюветы и записывают спектры отражения на
спектрофотометре СФ-2М.
Разность коэффициентов отражения при 620 и 520 ммк
(^620—Rs2a) пропорциональна содержанию железа. По
полученным данным строят график, характеризующий
распределение соединений железа по генетическим горизонтам.
Глава VII
ОПРЕДЕЛЕНИЕ В ПОЧВАХ КАЛИЯ И НАТРИЯ МЕТОДОМ
ПЛАМЕННОЙ ФОТОМЕТРИИ
Метод пламенной фотометрии является одним из
вариантов спектрального эмиссионного анализа, в котором
возбуждение спектров атомов и молекул происходит в сравнительно
низкотемпературных газовых пламенах. Под действием
температуры атомы элементов, находящихся в пламени,
переходят в возбужденное состояние и излучают
электромагнитные колебания, интенсивность которых измеряется
фотоэлектрическими приборами и служит мерой
количественного содержания элементов в анализируемом растворе.
В практике почвенных исследований методы пламенной
фотометрии поистине незамешшы. Почвенные и
агрохимические анализы, как правило, включают определение калия,
натрия и кальция, химические методы определения которых
трудоемки и часто неточны; применение пламенных
фотометров позволяет вести быстрые массовые определения калия и
натрия с требуемой степенью точности. Интересно отметить,
что один из первых пламенных фотометров конструкции
Шукнехта и Вайбеля использовался в основном для анализа
почв и удобрений.
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
Область применения пламенной фотометрии очень широка.
Набор элементов, определяемых этим методом прямыми или
косвенными способами, охватывает почти всю периодическую
систему Менделеева, а быстрота определения сочетается с
высокой чувствительностью (табл. 24).
10 Зак 51
289
Таблица 24
Чувствительность пламенно-фотометрического метода
Элемент
Литий . . .
Натрий . .
Калий . . .
Рубидий . .
Цезий . . .
Магний . .
Кальций . .
Стронций .
Спектральная
линия или
полоса, ммк
670,8
589,0—589,6
766,5—769,9
794,8
852,1
285,2
422,7
460,7
Чувствительность,
у/ли
0,01
0,001
0,01
0,1
0,1
5,0
0,06
од
1
I Элемент
1
Барий . .
Кобальт
Марганец .
Хром . . .
Железо . .
Медь . . .
Серебро . .
Никель . .
Спектральная
линия или
полоса, ммк
870
352,7
403.1; 403,3
425,4
373,5 и 373,7
324,8
338,3
352,5
Чувствительность,
У 1мл,
0,6
5,0
0,06
2,0
12,5
1,0
2,0
3,0
Возбуждение спектров
Поглощение и излучение световой энергии обусловлено
переходом атомов из нормального состояния в возбужденное
и обратно (см. стр. 230). В нормальном состоянии атом
обладает наименьшей энергией; поглощение энергии
сопровождается переходом атома (электронов) в возбужденное
состояние. Источником энергии для перевода атомов в
возбужденное состояние служит пламя газовой горелки.
Время существования атомов в возбужденном состоянии
невелико, порядка Ю-8 сек; переход в нормальное состояние
сопровождается излучением кванта света, величина которого
зависит от разности энергетических уровней возбужденного
и нормального состояний:
Е2— Ег = hv (обозначения прежние).
Таким образом, каждый единичный акт перехода
сопровождается излучением электромагнитного колебания с
частотой v. В любых, даже наиболее простых атомах
щелочных металлов число возможных энергетических состояний
достаточно велико. Каждый из переходов характеризуется
определенной разностью энергии, поэтому атом обладает
способностью излучать кванты различной величины.
Поскольку величина кванта связана с частотой излучаемых
колебаний по вышеприведенной формуле, то очевидно, что
излучение атомов любого элемента имеет сложный
спектральный состав и складывается из нескольких серий
колебаний с разными частотами, или из набора спектральных линий. (
290
Для количественных определений выбирается одна из
линий-спектра, которая легко обнаруживается уже при
минимальном количестве вещества. Измерив интенсивность этой
линии, устанавливают количество атомов данного элемента
во взятой пробе.
Интенсивность / излучения в обшей форме выражается
следующим уравнением:
т. е. интенсивность излучения линии }.тп пропорциональна:
Nm — числу атомов данного элемента, возбужденных до
уровня т, Атп — вероятности энергетического перехода
атомов из состояния m (верхний уровень) до п (нижний
уровень) и энергии излучаемых при этом квантов с частотой
vm„. Интенсивность по уравнению зависит от величины
кванта света. Вероятность А близка для всех разрешенных
электронных переходов.
Число атомов Nm, возбужденных до уровня т,
определяется из уравнения
gn
где N0 — число всех атомов данного рода, gm и gn —
статистические веса возбужденного и невозбужденного уровня
атомов; Ет — потенциал возбуждения уровня т;
к—постоянная Больцмана; Т—абсолютная температура пламени.
Объединяя оба уравнения, получим
1 — yv0 — е nmnnvmn.
gn
С увеличением температуры число возбужденных атомов
быстро растет, количество переходов, совершаемых в единицу
времени, увеличивается и интенсивность излучения данной
линии спектра также возрастает. Поэтому повышение
температуры значительно увеличивает чувствительность метода;
однако роль температурного фактора весьма ограничена, так
как с ростом температуры усложняется спектр, а при
использовании атомных линий чувствительность может даже
понизиться вследствие ионизации атомов. Очень большое влияние
на интенсивность излучения оказывает величина энергии
возбужденного состояния. Чем меньше энергия возбуждения
10* 291
атома до нужного уровня, тем легче совершаются
электронные переходы и тем выше интенсивность излучения.
В случае, когда анализ ведется при постоянной
температуре, для единичной линии спектра почти все величины
рассмотренного выше уравнения имеют постоянное значение.
Тогда )
I - a'N0,
Е tn
где константа а' = Amnh\mn ■ — • е кТ ,
8п
т. е. интенсивность излучения прямо пропорциональна числу
атомов излучающего элемента в пламени. Допуская, что
число атомов в пламени пропорционально их концентрации С
в веществе, и заменяя jVo на С, получим
Прямолинейная зависимость между интенсивностью
излучения и концентрацией соблюдается только в узком
диапазоне концентраций даже для растворов, содержащих один
компонент. В многокомпонентных смешанных растворах,
какими являются почвенные вытяжки, характер зависимости
осложняется. Особенно сильное влияние оказывают
самопоглощение и ионизация атомов при использовании атомных
линий.
Если степень ионизации а, то число атомов будет не N0,
а Л/0A—а). Ионизация резко возрастает с повышением
температуры, а для щелочных и щелочноземельных
элементов характерна большая величина а при относительно низких
потенциалах (температурах). Поэтому для анализа на
щелочные катионы не следует применять пламен с очень высокой
температурой, достаточна такая степень нагрева, которую
дают при горении светильный газ в смеси с воздухом или
ацетилен с воздухом. Для получения линий с высоким
потенциалом возбуждения нужны пламена с высокой
температурой.
Температура некоторых пламен приведена в табл. 25.
Очень высокую температуру порядка 5000° К можно получить
при горении дициана в кислороде, она близка к температуре
электрической дуги.
Повышение концентрации элемента влечет за собой
самопоглощение излучения, когда невозбужденные атомы
поглощают часть излученной энергии. В результате прямолинейная
292
Таблица 25
Температура некоторых пламен, используемых в анализе
(по Н. С. Полуэктову)
Горючая см(сь
Светильный газ--воздух .
Пропан -воздух
Водород- воздух
Ацетилен- -воздух . . . .
Водород—кислород . . .
Светильный газ- кислород
Пропан— кислород ....
Ацетилен—кислород . . .
Температура пламени, 'С
измеренная
1700—1840
1925
2000 - 2045
2125- 2397
2550- 2660
2730
3100—3137
вычисленная
1840
1925
2115
2250
2690—2780
2800
2850
3110
зависимость между интенсивностью излучения и
концентрацией элемента в пламени нарушается, а графическое
изображение этой зависимости в координатах / — С дает S-образную
кривую. Снижение интенсивности излучения за счет
самопоглощения можно до известной степени учитывать, вводя в
уравнение эмпирический коэффициент в<1; тогда уравнение
приобретает вид:
/ =.- аС.
Логарифмируя последнее выражение, приводят его к
уравнению прямой линии, что удобно для количественных
определений:
lg/-lga + MgC.
Откладывая по оси ординат lg/, а по оси абсцисс lgC, на
графике получают прямую линию, тангенс угла наклона
которой равен Ь, а отрезок, отсекаемый на оси ординат, равс- \
\ga (рис. 70,/). Однако и при таком способе построения
прямолинейная зависимость часто сохраняется только к
нешироком интервале концентраций (от 1 до 100—150 мг/л).
Кроме концентрации элемента в пламени и температуры
на интенсивность излучения влияет и ряд других факторов.
Процессы, происходящие в пламени, начинаются испарением
раствора и образованием аэрозоля; под влиянием
температуры молекулы солей диссоциируют и вступают в реакции с
атомами или радикалами, присутствующими в пламени. В
результате одновременно могут существовать небольшое
количество свободных электронов, ионы, атомы и молекулы.
293.
Равновесие между этими формами зависит от температуры
пламени, состава испаряемого раствора и
окислительно-восстановительных условий, которые по-разному складываются
в различных зонах пламени. Этим и объясняется очень
сильное влияние состава раствора на интенсивность
излучения. Точные количественные результаты получаются, когда
коэффициенты а и b уравнения 1=аСь одинаковы и для
анализируемого и для стандартного растворов. Поэтому,
приготавливая стандартные растворы, стремятся к
наибольшему совпадению их состава с
составом проб. В почвенных
исследованиях, где приняты
водные, солевые и кислотные
вытяжки, пробы отличаются
переменным составом и, конечно,
невозможно приготовить
стандарты, полностью идентичные,
например по содержанию
различных компонентов, солевой
или кислотной вытяжке из
почвы. В этих случаях наиболее
существенно, чтобы пробы и
стандарты были максимально
близки по содержанию
определяющего компонента.
Так, при анализе солевыл
вытяжек из почв стандарты готовят
на том же солевом растворе,
который был использован
для получения вытяжек из почвы. То же относится и к
кислотным вытяжкам. Большой избыток кислоты или соли
нивелирует условия возбуждения и позволяет получить
сравнимые результаты. Труднее анализировать водные вытяжки
и почвенные растворы, особенно из засоленных почв, ибо
состав солей и их содержание резко колеблется даже в
пределах одного почвенного профиля, что сильно влияет на
качество определения по методу обычного градуировочного
графика.
Состав растворов оказывает воздействие не только на
процессы, происходящие в пламени, но и на качество работы
распылителя. Оценивая в целом влияние состава на
результаты анализов, следует иметь в виду следующее.
1. Температура, вязкость и поверхностное натяжение
изменяют количество раствора, засасываемого распылителем
Рис. 70. Калибровочный график
для определений содержания
элементов методом пламенной
фотометрии:
/ — теоретический, 2 —
экспериментальный
294
и поступающего в пламя горелки. Тем самым яркость
свечения пламени меняется. Несоответствие свойств
анализируемых и стандартных растворов в этом отношении поведет к
ошибкам в определениях концентраций.
2. Большой избыток посторонних катионов сдвигает
равновесие в реакциях диссоциации и ионизации ионов.
Интенсивность излучения кальция и других щелочноземельных
металлов снижается в присутствии алюминия, образующего
(по Д. Н. Иванову) трудно диссоциирующие соединения
состава пСаО • А120з- С увеличением концентрации кальция
в растворе п повышается. Гасящая способность алюминия
возрастает в ряду:
A12(S04K>A1(N03K>A1C13
и понижается от кальция к магнию и затем к барию. В
высокотемпературном пламени эффект действия алюминия
выражен наиболее ярко. В меньшей степени А1 снижает
интенсивность излучения щелочных металлов.
Щелочные катионы усиливают излучение друг друга, при
этом влияние катиона тем больше, чем меньше величина
ионизационного потенциала атомов элемента. Влияние
щелочных катионов убывает в ряду:
Cs>Rb>K>Na>Li,
что связано с низким ионизационным потенциалом их атомов.
При ионизации, отщеплении электрона от атома, образуются
сразу две частицы — ион металла и электрон
где М° — нейтральный атом, М+ — однократно ионизованный
атом не — электрон.
Реакция ионизации носит обратимый характер и имеет
константу, уравнение которой выглядит аналогично
уравнению константы диссоциации слабого электролита:
[м+lUI _к
[М°]
где К — константа ионизации.
Присутствие посторонних, легко ионизирующихся атомов
дает дополнительное количество электронов, которые
подавляют ионизацию основного компонента, увеличивает
концентрацию его атомов и соответственно интенсивность их
излучения. Поскольку в пламенной фотометрии используют для
295
количественного определения элемента измерение
интенсивности излучения его атомов, то меняющиеся количества
посторонних щелочных катионов могут повести к ошибкам в
определении концентрации щелочного металла. При
почвенных исследованиях прежде всего нужно учитывать взаимное
влияние натрия и калия при анализе водных вытяжек из
засоленных почв и определениях калия (или натрия) в солевых
вытяжках. Существенные помехи возможны в анализах
лития, рубидия и цезия, содержащихся в почве в
микроколичествах.
Степень ионизации падает с увеличением общей
концентрации элементов в пламени, но она увеличивается с
возрастанием температуры. Щелочноземельные металлы не
оказывают влияния на ионизацию щелочных металлов; даже
при молярном соотношении кальций (магний) : натрий
(калий) = 100 интенсивность излучения щелочных элементов
не меняется.
3. Не все соли одинаково хорошо диссоциируют в
пламени, поэтому в присутствии некоторых анионов снижается
концентрация и интенсивность излучения атомов металла.
Азотнокислый анион меньше других оказывает влияние на
излучение щелочных металлов. Степень влияния растет в
ряду: сульфат<хлорид<фосфат.
Для кальциевых солен интенсивность излучения при
одной и той же концентрации располагается в следующий
ряд:
СаС12 > Ca(NO,)„ > CaS04 > Ca3(PO„J.
Максимум гашения наступает при соотношении
Ca2+:SC>4-=1 : 1. При необходимости снизить интенсивность
излучения кальция оказывается эффективным прибавление
к раствору фосфорной или серной кислоты.
4. Многие вещества, присутствующие в растворе, часто
сами дают излучение с длиной волны, близкой к
аналитической линии определяемого элемента. Наложение излучения
примеси на излучение определяемого элемента приводит к
завышенным результатам.
Полоса излучения СаО накладывается на аналитическую
линию натрия E89,0—589,6 А) и тем создает затруднение в
точном определении натрия. Очень близки по длине волны
линии калия, лития и рубидия, их совместное присутствие
может внести ошибки в определения каждого из этих
элементов при плохом разрешении спектра.
296
Несмотря на большое число взаимосвязанных явлений,
изменяющих излучение элементов в пламени, методом
пламенной фотометрии можно проводить количественные
определения с большой точностью. Снижение погрешностей
достигается как специальными аналитическими приемами
(например, метод добавок), так и совершенствованием конструкции
приборов.
АППАРАТУРА ДЛЯ ПЛАМЕННОЙ ФОТОМЕТРИИ
Любои пламенный фотометр обязательно состоит из
следующих основных узлов (рис. 71): распылитель,
засасывающий анализируемый раствор н переводящий его в состояние
аэрозоля; горелка, в пламени которой происходит
возбуждение атомов определяемого элемента и излучение спектраль-
Горслка.
Монохрвмаггшза
ция излучения
(свепофилб/пр.
монохроме тер )
Приемник
излучения
(дютеэлеиент)
ГальВанометр
баллон
с сжатым
Роздыхом
Ста.чанчик с
анализируемым
рсспСорсм
Рис. 71. Блок-схема пламенного фотометра
ных линий; монохроматизирующее устройство, выделяющее
излучение с длиной волны, соответствующей аналитической
линии определяемого элемента; приемник излучения,
позволяющий измерить интенсивность излучения; в наиболее
распространенных приборах приемник излучения представлен
фотоэлементами, фототок которых измеряется
чувствительным микроамперметром или зеркальным гальванометром.
Распылители
Простейший распылитель состоит из двух трубок,
расположенных под углом 90° (рис. 72,А). Одна из трубок
опущена в раствор, через другую подается сжатый воздух. Воздуш-
297
ная струя проходит с большой скоростью мимо отверстия
первой трубки и увлекает за собой_ молекулы воздуха,
находящиеся около отверстия капилляра, создавая тем самым в
зоне капилляра пониженное давление. В результате раствор
поднимается по трубке, захватывается струей воздуха и
Воздух /*
CZZ
Газ
Ж
J
А
Рис. 72. Распылители:
А— стеклянный; Б — металлический; В — из пластмассы; / —
корпус распылителя, 2 — трубка для подачи раствора, 3 — трубка для
сжатого воздуха, 4 — гайка, 5 — асбест, 6 — горелка, 7 — аэрозоль,
8 — шланг для слива избытка раствора
превращается в туман (аэрозоль). Качество распыления
зависит от скорости поступления раствора н воздуха, сечения
отверстий трубок и расположения двух трубок распылителя
относительно друг друга.
Распылитель впаивается в стеклянный баллон,
направляющий аэрозоль в пламя горелки. Баллон обычно имеет еще
два отверстия: одно для сливания избытка раствора и
второе— для промывания распылителя. Такой упрощенный
распылитель имеет ряд недостатков: трудно промыть
капилляры при засорении, а заменить их вовсе невозможно; нельзя
регулировать взаимное расположение капилляров и давление
воздуха в газовой смеси независимо от работы распылителя.
Поэтому часто используют разнообразные конструкции
распылителей, включающие каплеуловители и позволяющие
менять детали, регулировать положение, прочищать и менять
капилляры. Удобными в эксплуатации оказались также
распылители из металла и пластмасс (рис. 72, Б, В).
Количество раствора, подаваемого через распылитель в
пламя горелки, зависит от конструкции прибора, давления
воздуха и таких свойств раствора, как вязкость и поверхост-
ное натяжение. Последнее может быть использовано для
значительного повышения чувствительности метода. Так, для
повышения чувствительности определения кальция в водах
В. Дзиедзианович предложила добавлять пропиловый спирт
к анализируемому раствору. Такое добавление снижает
поверхностное натяжение и повышает температуру пламени, в
результате чего уже при введении 20—25% пропилового
спирта чувствительность определения повышается примерно
в 3 раза.
Горелки
Горелки должны обеспечить стабильное бесцветное пламя;
их характеристики зависят от состава горючей смеси. В
каждой лаборатории легко изготовить простую горелку из
тугоплавкого стекла в виде усеченного конуса (рис. 73,1). Такая
горелка дает устойчивое пламя с хорошо выраженным
внутренним конусом. Устойчивость пламени зависит от
соотношения между количеством поступающей в горелку горючей
смеси и диаметром выходного отверстия. Для смесей с малой
скоростью распространения пламени размер выходного
отверстия должен быть больше, например для природного газа
около 10 мм, для воздушно-ацетиленового пламени — меньше.
Хорошее устойчивое пламя получается в горелках Меккера
(рис. 73,2); диаметр отверстий насадки горелки Меккера
299
так же, как и обычной, изменяется в зависимости от скорости
распространения пламени.
Правильно подобранная горелка дает почти бесцветное
пламя с четко выраженным внутренним зеленоватым кону-
У=~
Гаэ из
J
JU-J
1' i i
I?
t
ч
Клри£сру
Рис 74.
Стабилизатор
газа
давления
сом. Регулировка давления
воздуха и газа, обеспечивающая
стабильность работы горелки,
осуществляется системой кранов или
редукторов, а постоянство
давления проверяют по водяным или
стрелочным манометрам.
Относительную стабилизацию давления
воздуха создают применением
больших емкостей (бутылей),
через которые проходит воздух.
Давление газа можно также
автоматически регулировать при помощи устройства, показанного
на рис. 74. Для предупреждения засорения распылителя
воздух и газ рекомендуется фильтровать через ватные и
стеклянные фильтры.
Рис. 73. Горелки для
пламенных фотометров:
/ — простая коническая
горелка, 2—горелка е
металлической сеткой ло Мек-
керу
Фотометрические ячейки
Излучение пламени воспринимается фотометрической
ячейкой (рис. 75), в которую входят диафрагма, светофильтр
и фотоэлемент. Между пламенем и фотометрической ячейкой
помещают теплозащитный фильтр.
300
Фотометрическая ячейка располагается так, чтобы на
фотоэлемент попадало преимущественно излучение наиболее
горячей, центральной части пламени. Для этого нижний срез
диафрагмы устанавливают несколько выше зеленоватого
внутреннего конуса пламени. Важнейшей частью
фотометрической ячейки являются светофильтр и фотоэлемент, от
которых зависит качество показаний прибора в целом. Поэтому
выбор светофильтра и
фотоэлемента имеет решающее значение
при монтаже прибора.
Светофильтры
Светофильтры (см. также
стр. 64—67) предназначены для
выделения из сложного спектра
излучения одной аналитической
линии определяемого элемента.
Характеристики светофильтров
показаны на рис. 14, 15, 16. По
этим характеристикам
определяют пригодность светофильтров
для анализа тех или иных
элементов. Ширина пропускания должна
быть возможно меньшей, чтобы
даже близко расположенные
линии или полосы посторонних
элементов были задержаны
светофильтром. Длина волны в
максимуме пропускания должна
совпадать с длиной волны аналитической линии определяемого
элемента, а пропускающая способность для излучения
определяемого элемента — наибольшей, что обеспечивает высокую
чувствительность метода. Всем этим требованиям в
наибольшей степени удовлетворяют интерференционные светофильтры
(см. стр. 65—66). По Р. Смиту, 15-слойные интерференционные
фильтры, содержащие в качестве диэлектриков криолит и
сернистый цинк, имеют коэффициент пропускания в
максимуме более 80%. Максимальная монохроматизация света
в широком диапазоне длин волн происходит при сочетании
интерференционного фильтра с абсорбционным.
Полуэктов предложил простые жидкостные светофильтры,
пригодные для пламенно-фотометрического определения
некоторых элементов. Эти светофильтры состоят из двух окрашен-
Рис. 75. Устройство
фотометрической ячейки:
/ — теплозащитный фильтр,
2 — ирисовая диафрагма; 3 —
интерференционный
светофильтр, 4 — фотоэлемент, 5 —
внутренний конус пламени
301
пых растворов, которые помещают один за другим в кюветах
толщиной 1 см (см. табл. 26).
Высокая степень монохроматизации излучения
достигается на призменных приборах, хотя монохроматоры, как
например универсальный монохроматор УМ-2 (см. стр. 62),
обычно громоздки и дороги и применение их должно быть
оправдано условиями эксперимента. Рекомендуется
использование монохроматоров в лабораторных условиях для
исследовательских целей, когда предполагается определять
методом пламенной фотометрии большое число элементов.
Кроме того, монохроматоры могут оказаться незаменимыми
в том случае, когда линии определяемого и мешающего
элементов очень близки друг к другу. Вместе с тем, прошедший
через монохроматор свет обычно сильно ослаблен, и поэтому
на выходе прибора ставят высокочувствительные приемники
(фотоэлементы с усилителями, фотоумножители).
Таблица 26
Состав жидкостных светофильтров для определения
натрия и калия (по Н. С. Полуэктову)
Определяемый
элемент
Na
К
Аналитические
линии, ммк
589,0—589,6
766,5—769,9
Растворы
50%-ный водный раствор
NaaCraO,-2H20
596 -ный раствор
СиС1а-2НаО в 8 н. НС1
2 % -ный раствор КаСг207
0,02%-ный раствор
анилиновой сини в
этиловом спирте
Т,
%
42
98
Полуширина,
ммк
40
Фотоэлементы
В пламенных фотометрах чаще применяют фотоэлементы
вентильного типа (см. стр. 56—62), обладающие достаточно
высокой чувствительностью; возникающий в них фототок
измеряется непосредственно микроамперметром или
гальванометром. Вакуумные фотоэлементы ставят тогда, когда фото-
ток предполагается усиливать. Важнейшими требованиями,
предъявляемыми к фотоэлементам в пламенной фотометрии,
надо считать высокую чувствительность в той спектральной
302
области, где расположена аналитическая линия
определяемого элемента, прямолинейную зависимость между
освещенностью и силой фототока, малую инерционность и
утомляемость.
При анализе почвенных образцов (за исключением
солончаков) концентрация катионов калия, кальция и натрия в
водных вытяжках колеблется от десятых долей до нескольких
десятков миллиграммов в литре раствора. Поэтому только
высокая чувствительность фотоэлемента в пламенном
фотометре обеспечивает достаточную точность анализа вытяжки.
Подбором фотоэлементов для различных участков спектра
добиваются высокой чувствительности метода. При
определении натрия и кальция применяют селеновые фотоэлементы,
для калия — сернисто-серебряные (см. рис. 12).
В схемах с вентильными фотоэлементами фототок
регистрируется зеркальными гальванометрами (М-21, М-17, М-25
и др.), или микроамперметрами. Шунтирование
гальванометра и ступенчатое изменение чувствительности осуществляется
по обычным схемам (см. гл. II).
Пламенные фотометры
Пламенный фотометр простой конструкции можно собрать
в любой лаборатории самостоятельно. Такой прибор состоит
из фотометрической ячейки, гальванометра и распылителя с
горелкой. Под действием сжатого воздуха в распылитель
засасывается и распыляется в нем раствор. Крупные брызги
оседают на стенках стеклянного баллона распылителя и
стекают по трубке в бутыль с жидкостным затвором. Аэрозоль
смешивается с газом и поступает в горелку 5 (см. рис. 75).
Поток излучения, возникшего в пламени, попадая в
фотометрическую ячейку, ограничивается диафрагмой 2, проходит
светофильтр 3 и попадает на фотоэлемент 4, в котором
возникает фототок, регистрируемый гальванометром.
Правильно собранный и хорошо отрегулированный
прибор дает устойчивые и точные показания. При регулировке
прибора добиваются, чтобы раствор быстро засасывался и
тонко распылялся (без крупных капель). Пламя должно
иметь четкий внутренний конус и быть стабильным; высота
пламени обычно не превышает 5—6 см. Если не удается
добиться стабильности и хорошей формы пламени подбором
давления воздуха и газа, то можно заменить распылитель,
изменить диаметр выходного отверстия горелки или
поставить сетку на горелку. Качество показаний гальванометра
303
сильно зависит от величины шунтового сопротивления
(см. гл. II).
В сборные схемы и промышленные конструкции
пламенных фотометров вводят разнообразные приспособления,
улучшающие работу приборов. Для увеличения освещенности
фотоэлементов используют сферические зеркала и
фокусирующие линзы, конденсирующие световой поток на рабочей
площади фотоэлемента. Линзы, располагаемые между
пламенем горелки и светофильтром, собирают свет в пучок
параллельных лучей, что необходимо для правильной работы
интерференционных светофильтров. Любой пламенный
фотометр можно дополнить приспособлением для воспламенения
газовой смеси, состоящим из электродов и конденсатора,
заряжаемого от источника постоянного тока. Электроды
вводят в пространство над горелкой, и при соударении их
конденсатор разряжается, а между электродами
проскакивает искра, воспламеняющая газовую смесь.
Порядок работы на сбор н о й схеме
1. Приготовить эталонные растворы для градуировки
прибора. Эталонные растворы готовятся с концентрацией
определяемого элемента от I до 150 мг/л, а состав их
подбирается максимально близким к составу проб.
2. Включить воздух и проверить, как засасывается и
распыляется раствор; шланг для слива избытка раствора должен
быть опущен в бутыль с водой; необходимо проверить, чтобы
при полном давлении газа и воздуха через воду не
проскакивали пузырьки газовой смеси. Если пузырьки проскакивают,
то в бутыль доливают воду.
3. Не выключая воздуха, открыть газовый кран и поджечь
газ у края горелки.
4. Отрегулировать давление воздуха и газа, добиваясь
устойчивости и хорошей формы пламени. При уменьшении
давления воздуха следить, чтобы скорость засасывания
раствора не стала слишком малой.
5. Открыть диафрагму фотоэлемента и включить
гальванометр на минимальную чувствительность. Через
распылитель ввести в пламя горелки эталонный раствор с
максимальной концентрацией определяемого элемента. Если показания
гальванометра при этом выходят за пределы шкалы,
уменьшить диафрагму. Продолжать вводить эталонный раствор в
пламя горелки в течение нескольких минут, пока показания
гальванометра не перестанут изменяться.
304
6. Промыть распылитель и горелку дистиллированной
водой (подавая воду в распылитель, как и раствор).
7. Ввести в пламя эталонный раствор с минимальной
концентрацией определяемого элемента и записать показания
гальванометра. Снова промыть распылитель небольшой
порцией воды и продолжать градуировку, последовательно
вводя в пламя эталонные растворы в порядке увеличения
концентрации. После каждого измерения распылитель
промывают водой.
8. Так же поступают с анализируемыми растворами,
записывая показания гальванометра в лабораторный журнал.
9. Через каждые 30 мин работы необходимо проверить
показания прибора с эталонными растворами (градуировку).
Если условия возбуждения меняются и показания прибора
неустойчивы, то пробы сравнивают с эталонными растворами
через 2—3 определения.
10. По полученным данным строят градуировочный график
в координатах: lg/1—igC (или А— С), где А — показания
гальванометра и С — концентрация, и по графику находят
содержание определяемого элемента в исследуемых
растворах. Градуировочный график пригоден только для одной
серии измерений, проведенных без перерыва.
11. По окончании работы сначала выключают газ,
закрывают диафрагму фотоэлемента и накоротко замыкают
гальванометр. Затем тщательно промывают распылитель и
горелку дистиллированной водой, а все коммуникации продувают
воздухом в течение 15 мин. После этого выключают воздух.
Портативный пламенный фотометр ППФ1. Портативный
пламенный фотометр, разработанный Украинским
научно-исследовательским институтом земледелия, предназначен для
определения К, Na и Са в почвенных вытяжках и растворах.
В качестве источника возбуждения используется пламя
различных горючих смесей: природный газ — воздух, пропан —
бутан — воздух, ацетилен — воздух, а также пропан — бутан в
смеси с 30—40% кислорода. Нижний предел измерения для
К, N а и С а 0,5 мг/л.
Схема прибора показана на рис. 76. Анализируемый
раствор из бюкса / засасывается в распылитель 2 током
воздуха, который поступает из баллона или нагнетается
компрессором через редуктор 3 и фильтр 4. Давление воздуха
измеряется манометром 5. Распыленный раствор проходит
1 Для лабораторных работ можно рекомендовать пламенный
фотометр завода «Геологоразведка» и импортные фотометры фирмы «Цейс»
н «Ланге».
Ц1;4 Зак 51
305
через смеситель 6 и каплеуловитель и подается в горелку 7„
Горючий газ также проходит через редуктор и фильтр и
смешивается с аэрозолем в смесителе 6.
Рис. 76. Схема портативного пламенного фотометра ППФ:
/ — бкже с анализируемым раствором, 2 — распылитель, 3 —
редуктор, 4 — воздушный фильтр, 5 — манометр, 6 — смеситель, 7 —
горелка, 8—переключатель фотометрических ячеек, 9 —
микроамперметр, 10 — баллон с газом, // — баллон со сжатым воздухом (или
компрессор), 12 — жидкостной манометр
При работе с природным газом и пропан — бутаном на
горелку надевается насадка с трехмиллиметровыми
отверстиями; для сжигания ацетилена и пропан — бутана в смеси
306
с кислородом используется насадка с одномиллиметровыми
отверстиями.
Симметрично вокруг горелки расположены три
фотометрические ячейки (головки). Фототоки через переключатель 8
попадают на микроамперметр М-95 9. В цепи измерения
интенсивности излучения Na и Са стоят селеновые фотоэлементы
(тип АФИ-5; чувствительность 460—500 мка/лм), излучение
калия измеряется сернисто-серебряными фотоэлементами
ФЭСС-УЗ (чувствительность 6000—9000 мка/лм).
Характеристики интерференционных светофильтров прибора показаны
а табл. 27.
Таблица 27
Характеристики интерференционных светофильтров
фотометра ППФ-УНИИЗ
Определяемый
элемент
з>1
такс
ммк
766 ±5
589 ±5
615±5
Ширина пропускания
в середине максимума, ммк
8—13
8—13
8—13
Коэффициент
upon ускаиня,
%
20—40
30—40
20—40
Порядок работы на пламенном
фотометре ППФ. Устанавливая прибор (рис. 77), входные штуцера
«а задней стенке соединяют резиновыми шлангами с
воздушной и газовой сетью. Давление воздуха не должно быть более
1,0—1,5 ат и газа-—40 мм вод. ст. К сливной трубке 3
распылителя присоединяют резиновый шланг, который
опускают в сосуд с водой; слой воды высотой 25—35 см играет
роль водяного затвора. Затем производят юстировку
положения капилляров в распылителе / и положения горелки. Для
этого пробку с капиллярными трубками 2 вынимают из
горловины распылителя, отвертывают стягивающий винт и к
«боковому капилляру присоединяют источник сжатого воздуха,
а нижний капилляр опускают в стаканчик с водой. Доводят
по манометру 9 давление воздуха до 0,4—0,8 ат и
наблюдают за распылением воды. Хороший распылитель быстро
засасывает воду и образует высокодисперсный аэрозоль без
крупных капель. Поворачивая всасывающую трубку и трубку
для воздуха, добиваются наилучшего распыления. Завернув
винт, снова помещают пробку-в горловину распылителя.
Перед включением газа заполняют манометр 4
подкрашенной водой; не прекращая тока воздуха, открывают доступ
горючего газа в смеситель и горелку; спустя 10—20 сек (дав-
I1V» Зак. 51
307
ление газа 8—10 мм вод. ст.) поджигают газ у края насадки
горелки. Регулируя давление воздуха рукояткой 5 и газа —-
рукояткой 6, добиваются наилучшей формы пламени, когда
четко ограниченные внутренние конуса имеют яркую
зеленоватую, а внешний конус — голубовато-синюю окраску. Регу-
Рис. 77. Внешний вид пламенного фотометра ППФ:
/ — распылитель, 2 — пробка с капиллярами, 3 — трубка и шланг для
слива избытка раствора, 4 — жидкостной манометр, 5 — регулятор давления
воздуха, 6—регулятор давления газа, 7—переключатель
фотометрических ячеек, 8 — микроамперметр, 9 — воздушный манометр, 10 —
фотометрические ячейки, // — ручка регулировки положения горелки
лируя пламя, нельзя слишком сильно уменьшать давление
воздуха, иначе распылитель перестанет засасывать раствор.
Положение горелки можно изменять с помощью ручки И.
Наилучшие результаты получаются, когда вершины
внутренних конусов пламени находятся на 5—6 мм ниже входного
30 8
-отверстия диафрагм фотометрических ячеек 10.
Окончательная установка давления газа и положения горелки
контролируется показаниями микроамперметра 8. При оптимальных
условиях показания микроамперметра оказываются
максимальными.
После того как прибор настроен и подготовлены растворы,
можно приступать к измерениям, проводя все операции в
■следующем порядке:
1. Включить воздух и рукояткой 5 отрегулировать его
давление.
2. Включить газ и через 10—20 сек зажечь пламя.
3. Отрегулировать давление воздуха и газа, как указано
выше.
4. Переключателем 7 соединить микроамперметр с нужным
•фотоэлементом и открыть диафрагму.
5. Поставить микроамперметр на минимальную
чувствительность и ввести в распылитель эталонный раствор,
содержащий 15—20 мг(.г определяемого элемента. В течение
20 мин засвечивать фотоэлементы излучением пламени, затем
распылитель хорошо промыть дистиллированной водой.
6. Вращая головку корректора, установить
микроамперметр на нулевое деление шкалы.
7. Ввести в распылитель эталонный раствор с
максимальной концентрацией определяемого элемента и подобрать
диафрагму и чувствительность так, чтобы получить на
микроамперметре максимально возможное показание. Далее
поступают, как указано на стр. 305 (пункт 6 и следующие).
Компенсационная схема соединения фотометрических
ячеек позволяет автоматически вводить поправки на влияние
■мешающих элементов. При определении Na в почвах
мешающим элементом обычно является Са. Для того чтобы
устранить его влияние, поступают следующим образом. К микро-
амперметру подключают фотометрическую ячейку,
воспринимающую излучение Са, и, открыв полностью диафрагму,
■отмечают максимальное отклонение, вызываемое
исследуемым раствором. Затем подбирают раствор СаСЬ такой
концентрации, которая вызывает такое же отклонение
микроамперметра, и включают по компенсационной схеме
обе фотометрические ячейки (Са — Na). Подавая в
распылитель подобранный предварительно раствор СаСЬ уменьшают
диафрагму кальциевой фотометрической ячейки, пока
микроамперметр не остановится на нулевом делении шкалы,
промывают распылитель водой и проводят измерения (см. выше),
сохраняя постоянной степень раскрытия обеих диафрагм.
II1/.*
зеэ
ОПРЕДЕЛЕНИЕ КАЛИЯ И НАТРИЯ В ПОЧВАХ
При определении Na и К в почвах выгодно использовать
низкотемпературные пламена, поскольку при этом
снижаются помехи, вызываемые щелочноземельными элементами, и в
первую очередь кальцием. В спектре
воздушно-ацетиленового пламени при температуре порядка 2200° К и Na дают по
несколько линий, отличающихся по чувствительности
(табл.28).
Таблица 28
Длины волн и чувствительность некоторых линий К и Na
в воздушно-ацетиленовом пламени
Элемент
К
Я,, ммк
766,5 \
769,9 /
404,4
404,7
344,7
Чувствительность,
у/мл
0,001
0,5*
0,5*
2
Элемент
Na
X, ммк
819,4
589,6 1
589,0 }
330,2
Чувствительность,
У/мл
10
0,0002
1
* В электрической дуге.
Для количественных определений натрия используют
только желтый дублет 589,0—589,6 ммк, для определения
калия — 766,5—769,9 ммк. Зависимость между концентрацией
и интенсивностью излучения для калия выражается S-образ-
ной кривой; для натрия (при малых концентрациях) она
прямолинейная, а при содержании натрия более 10—20 у J ли
интенсивность излучения прямо пропорциональна корню
квадратному из концентрации. Многие элементы, такие, как
Си, Zn, Mg, Fe, Ba, снижают интенсивность излучения калия
и натрия. Часто наблюдается сильное влияние на определение
обоих элементов Са, Mn, A1, которые преобладают среди
посторонних элементов, переходящих в различные почвенные
вытяжки. Количество мешающих элементов в составе пробы
зависит как от свойств почвы, так и от применяемых вытяжек.
Особенно много А1 и Са переходит в кислотные вытяжки и
значительно меньше при обработке почвы раствором:
CH3COONH4 с рН = 7. Почти всегда присутствующие в
вытяжках органические вещества могут значительно увеличивать
310
фон пламени. В связи с этим при разработке методик
определения К и Na в почвах прибегают к ряду приемов,
снижающих влияние и количество мешающих компонентов в
пробах. Так, при анализе природных вод рекомендуют
добавление буферов (к 25 мл воды добавляют 1 мл буфера,
полученного насыщением дистиллированной воды СаС12 и MgCb),
нивелирующих условия возбуждения в стандартных
растворах и пробах, а определение обменного Na и К проводят
путем выщелачивания почвы нейтральным раствором
CH3COONH4, избыток которого впоследствии удаляется при
выпаривании и прокаливании. Для освобождения от кальция
иногда пользуются его предварительным осаждением в виде
оксалата.
В почвах, не содержащих больших количеств
легкорастворимых соединений Са, Mn, A1, вполне допустимо также
прямое определение водорастворимых соединений К и Na в
водных вытяжках и обменных К и Na в 0,05 н. солянокислых
вытяжках (по К. К- Гедройцу).
Определение водорастворимых соединений калия и натрия
Методом пламенной фотометрии можно определить
количество К и Na в водной вытяжке без ее обогащения, когда
содержание определяемых соединений в почве не менее, чем
1 мг на 100 г почвы A0_3%). Для приготовления водной
вытяжки образец почвы доводят до воздушно-сухого
состояния, растирают пестиком с резиновым наконечником и
пропускают через сито с отверстиями 1 мм. Из приготовленной
таким образом почвы берут среднюю пробу в количестве Юг
(в расчете на абсолютно-сухой вес).
Навеску почвы помещают в колбу емкостью 100 мл,
приливают 50 мл дистиллированной воды, лишенной СОг, и
взбалтывают на ротаторе 5 мин. После взбалтывания дают
почвенным частицам осесть на дно колбы и осторожно
сливают раствор над осадком в центрифужные пробирки.
Пробирки с раствором уравновешивают попарно на весах и
размещают в гнездах центрифуги так,- чтобы в
противоположных гнездах находились пробирки со строго одинаковым
весом. В случае нечетного числа образцов одну из пробирок
заполняют водой. Растворы центрифугируют в течение
30 мин при 6000—8000 об/мин, после чего раствор над
осадком сливают в стаканчик. Если после центрифугирования в
растворе обнаруживаются какие-либо взвешенные частицы
(часто в верхней части пробирки собираются мелкие кореш-
311
кн), то необходимо дополнительное фильтрование, поскольку
яаже мелкие частицы могут засорить капилляр распылителя
и вывести прибор из строя. Первые порции фильтрата в этом
случае отбрасывают.
Прозрачную вытяжку используют для определения К и Na
на пламенном фотометре, как описано выше. Анализ ведут
последовательно, снимая сначала показания эталонных
растворов и проб для одного из элементов, например Na,
а затем переключают гальванометр и проводят определение
второго элемента.
Количество раствора, взятого для анализа и введенного в
пламя, не имеет значения. Важно только, чтобы скорость
поступления раствора оставалась постоянной.
Результаты определения получают в тех единицах,
которыми была выражена концентрация эталонных растворов.
Если результат выражен в миллиграммах на литр, то, зная
отношение почва : вода, взятое для приготовления вытяжки,
легко рассчитать содержание Na и К в почве. Для
приготовления вытяжки берут 50 мл воды на 10 г почвы, что
соответствует 1000 мл воды на 200 г почвы. Результат определения
выражен в миллиграммах на литр, или, следовательно, в
миллиграммах на 200 г почвы. Чтобы узнать содержание К
и Na в миллиграммах на 100 г почвы, полученный результат
необходимо уменьшить в два раза. Например, если найдено,
что водная вытяжка содержит 74 мг/л Na, то содержание Na
в почве равно 37 мг на 100 г почвы, или, что то же самое, —
0,037%.
Определение содержания обменных калия и натрия
в почвах
Для определения обменных калия и натрия пользуются
растворами солей или кислот, обеспечивающими полноту
вытеснения. Мало пригодны растворы солей, образующих
труднорастворимые соединения с компонентами почвы, как
например бариевые соли в почвах, содержащих гипс.
Используя разбавленные кислоты, необходимо учитывать их
растворяющее действие на твердую фазу почвы и возможность
перехода в раствор элементов, гасящих излучение щелочных
металлов (как алюминий).
Удобно для пламенной фотометрии вытеснение
поглощенных оснований 0,05 н. НС1 по К- К. Гедройцу, особенно в
почвах с невысоким содержанием подвижного А1, хотя в
солянокислую вытяжку всегда переходит заметное количест-
312
во Са, мешающего точному определению содержания Na.
Поэтому кислотную вытяжку целесообразно забуферивать
катионами щелочноземельных металлов или вводить в
стандарты такое количество Са, которое можно ожидать в
солянокислых вытяжках из образцов почв. Наилучшие результаты в
этом случает дает компенсационная схема Иванова
(см. стр. 309).
При анализе по компенсационной схеме 20 г почвы
промывают на воронке или декантируют 0,05 н. НС1 до
прекращения реакции на Са-ион. Фильтрат помещают в мерную
колбу, а при малом содержании Na и К фильтрат
предварительно концентрируют выпариванием, доводят до метки тем
же раствором кислоты и перемешивают. Затем определяют
концентрацию К- и Na-ионов, как описано выше.
Эталонные растворы для градуировки готовят на 0,05 н.
НС1.
Глава VIII
ТЕРМИЧЕСКИЙ АНАЛИЗ ПОЧВ
Почвенная масса состоит преимущественно из минералов,
находящихся в дисперсном состоянии. Свойства минеральной
почвенной массы определяются ее минералогическим
составом. В малогумусных почвах, особенно их илистой фракции,
от минералогического состава зависит способность почвенных
частиц к агрегированию, липкость, удельный вес,
ионообменная способность (а иногда и состав поглощенных оснований),
водоудерживающая способность почв и другие свойства.
Поэтому при оценке потенциального плодородия и
технологических свойств почв приходится обращать внимание на
минералогический состав. Огромное значение приобретает
минералогический состав в решении вопросов генезиса почв, так как
преобладание тех или иных минералов указывает на условия
почвообразования, историю и современное направление
почвообразовательного процесса.
Наибольшие трудности при изучении минералогического
состава представляет анализ высокодисперсных фракций, в
котором наиболее заинтересованы почвоведы. Фракции с
размером частиц <0,001 мм обладают высокой удельной
поверхностью (десятки и сотни квадратных метров на грамм)
и в силу этого весьма активны во всевозможных почвенных
процессах. Тонкодисперсная фракция наиболее легко
подвергается различным изменениям, к ней относятся и вновь
образованные почвенные вторичные минералы.
Для определения минералогического состава почв
применяют методы термического, рентгеноструктурно-
314
го и электронном и кроскоп и чес кого анализа,
используют и такие способы, как изучение окраски минералов,
изменение окраски красителей при сорбции их на
поверхности частиц и др.
Полное определение минералогического состава почв —
очень сложная и трудоемкая задача. Наилучшие результаты
дает сочетание ряда перечисленных методов, причем для
полном расшифровки результатов необходимо знание валового
химического состава (в первую очередь, содержания Si02,
AI2O3, Fe20,j, CaO. MgO, K2O и Na20), емкости поглощения,
присутствия свободных окислов и др
Термические методы анализа, пожалуй, наиболее простые,
для их использования не требуется слишком сложной
аппаратуры и специальных знании в области физики и
математики. Поэтому термография находит широкое применение в
почвенных исследованиях для общей характеристики
минералогического состава.
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДА
Методы термического анализа основаны на изучении
фазовых превращений, происходящих при
нагревании или охлаждении вещества. В почвенных исследованиях
используют только процессы нагревания, поскольку
большинство фазовых превращений, совершающихся в почвах,
протекают необратимо.
По определению Раковского, «ф^за есть совокупность
комплексов, обладающих одинаковым химическим свойством
и термодинамическими свойствами». Поэтому понятие
фазовых превращений очень широкое и включает переходы из
одного агрегатного состояния в другое (плавление,
затвердевание, испарение, конденсация), химические превращения
без изменения агрегатного состояния, а также сложные
процессы, при которых одновременно происходят как химические,
так и агрегатные изменения.
В большинстве случаев фазовые превращения
сопровождаются заметным поглощением (эндотермическая реакция)
или выделением тепла (экзотермическая реакция), что
возможно использовать для изучения происходящих процессов.
Нагревание почв или почвенных коллоидов вызывает
преимущественно такие необратимые процессы, как
разрушение органического вещества, распад минералов на
составляющие их окислы, кристаллизацию некоторых аморфных
315
™ степени обратимым процессом
соединений До «^с™°0Ипической и частично кристаллиза-
„вляется потеря гцнгР0™°"Ипочвенный образец ие нагревался
ционной воды, да и то еми окале„ные при более высо-
выше "О-'^^епяют способность сорбировать воду в
к„х температурах. ™РЛ™ с матриВаем только кривые
прежних "°™4e™f"Zl?* почвенных минералов,
(процессы) "агРе^гн"яП"°аЧнВНе почв ведется в пределах от
ТЖИЧеСПМ I2M» В этом диапазоне температур воз-
20-100" до 11М_ЫШ- ьп„" ашеиия. Наиболее
характерны самые Ра^ичнь* "р*°Р^ралов является потеря
„ым для почвенных ^'^ZeZZ^os на составляющие
Р™Ь2 to'lZx лочвГосушествляются процессы перехо-
ГиГаморфного состояния в кристаллическое, образование
шнераГв группы полуторных окислов, реакции окисления,
в том числе окисления органических соединении Некоторые
вещества испытывают полиморфные преврашения, например
кварц (а ^ Р) Для образцов засоленных почв,
исследуемых без предварительной обработки, могут быть
характерными процессы обезвоживания, полиморфных превращении н
плавления солей В карбонатных почвах наблюдается
разложение карбонатов на составляющие окислы и образование
газовой фазы.
CaC03(MgC03) = CaO(.MgO) + СОг.
Разнообразные процессы термических превращений
протекают у разных минералов при неодинаковых температурах,
что позволяет качественно определить минерал по
температуре фазового превращения.
Температурные особенности превращения минералов обусловлены как
ш составом, так и строением кристаллической решетки При Tcpviwec-
ми^,я™"™.1ГВе'ШЫ* К0ЛЛ°"Д°8 наибольший интерес представляют
минералы групп каолинита, монтмориллонита, иллита (гидрослюды) и
смешанно-слоистых минералов. Иоиы Si4+ , Al3+ Fe3+ Fe2+ Mb2*
0< .№ в глинистых минералах образуют параллельные слои.' причем
Ь.иОлаюткремнеК„слород„ыеслонсостава [Sia0sO, ноны А13-и ОН~-
пирартиллитовыйслой — (AI2(OHU„ Mo2* rf- „ m-
с»нсостава|Че3(ОН|,1„ .ilviJni п ' и 0H ~ гиббентовый
Дает гексагональную «tkv nS = Проекиия ноноввелое на плоскость
в«х и ткббситГыГсе^ок н«е^ /Г,^ ,кРем1кк»-'">родиых, гилрвр.иллито-
ственш* наложение «оевТТоиТтк? гТЧ'РЫ' ЧТ0 ^«гчает простран-
ведет к образованию электт.и.ь™ ' ПогеРИ ПРИ этом части ОН-групп
»ь через кислород ЧежТу „акетГ,^„""* "!С1Ш- слои в К01°РЫ* соедиие-
в слоях В кремне-ислоро, шУ „ " "Р°Ч,Ю0ТЬ связи значительно ниже, чеу
ньи замещения: Si4 + на Af4- «1^ ТзЭТй+ слс""' часты >«очорф-
3,6 ' на Fc • ^е+и др, ведущие прн re-
NPWHMmmMi ««мешимм^.ище^ WKtfiflU£utaaflwioctM пакетов r™™
ишючиозе-
MW* ЭПрМДМ ,
дольними шипнйми.
JMUAHllOTUMte минералы не имеют ичоморфимх замещений г слоя*
ттт т ^ктрои^шиты, еммиы между'собой 4;шми СдороХх
пмт-в которые прочно сишнйют нйксты н кристалле. В моотморилмии-
teX *ШШШДаити рп-ышчиыг ияоморфимс чамещения, и" поэтому"между
К|н-мигМ№ЛОролиыми слоями располагаются катионы 1-Й", Na+, M^+,
Ся** обршуи Са моншприллоиит и др Монтмориллониты, имеющие
иа*ДНЧМ1<№ жмлоиимшыс основания, различаются но своим термическим
xaj>«Ktrj»iu4tHKnM.
60 ©ОН © А1 #Я Э5«,И ©К* фМ,М|Г
Рис. 78. Схемах структуры пакета слоистых минералов:
1 — каолинит, 2 — галлуазит (гидратированный), 3 — слюда,
4 — монтмориллонит
Глинистые минералы, сходные по структуре с мусковитом,
называются иллитами или гидрослюдами, отличаясь от слюд большим
содержанием воды и меньшим — щелочей.
Из минералов группы гидроокислов А1 и Fe часто в почвах
встречаются гидраргиллит — А1(ОНK (иногда пишут — А1203-ЗН20—тригид-
рат окиси алюминия); полиморфные формы моногидрата алюминия: бе-
мит и диаспор — АЮ(ОН), или А1203 • НгО, а также безводный
глинозем в метастабильной форме ■у-глинозема и наиболее устойчивой форме —
а-глинозема, или корунда. Железистые соединения представлены гети-
том — a-Fe203 • Н20 и безводными окислами в виде гематита Fe2C>3 и
магнетита — Fe304. При повышенном содержании воды образуется
модификация, называемая лимонитом—2Fe203-3H20. Содержание воды в
лимоните непостоянно, и, видимо, связано с ярко выраженной
адсорбционной способностью минерала, находящегося в высокодисперсном состоянии.
ТЕРМИЧЕСКИЕ ХАРАКТЕРИСТИКИ НЕКОТОРЫХ МИНЕРАЛОВ
Группа каолинита. Для этой группы кроме эффекта
удаления гигроскопической воды, который у каолинитовых
минералов обычно очень невелик, обнаруживаются сильный эндо-
12 Зак 51
317
термический аффект при температурах 560-^0» ,
Первый эндотермический эффект (около юл* й
дегидратацией ••- потерей конституционной boW»06*****.
лящнх в решетку каолинита ОН-гру,," пД«* За ««гЭ
зависит от степени упорядоченностТс^^?^ ^Ф^
упорядоченные или слабо упорядоченные^ ctdvk^^S
воду при более низкой температуре.. Так у УЖ1*1 *р8г
,ффектонаблЮд,с1Ся в средне"- %hJ S^jV^SS^S
Идеальный состав каолинита (OH)sSi4AUO,0.
Изучение продуктов превращения показывает, что они не являются
простой смесью окислов Si02 и А12Оэ, а сохраняют некоторую
упорядоченность структуры Образующееся вещество с упорядриенн*>н--с1Диаурой
называют м е т а к а*"о л и И я т о м Цоследующие экзотермические
реакции объясняются полиморфными пренращениямй"*!!" Образованием—иттого
минерала—муллита — из возникших при распаде окйттптв—алюминия
и кремния Характер этих прекращений, по ХайТЛОпу,—может- -быть
выражен схемой.
При 550°:
А1,03 • 2Si02 ■ 2Н20 -* А1203 • 2Si02 + Н20 f.
Каолииозый минерал Метакаолин
От 850 до 1050°:
А1203 • 2Si02^Y"Al203-f-SiOi (n твердом растьоре с А12Оэ).
Выше 900°:
у - А12Оа + Si02 + А1203 -+ ЗА1203 ■ 2Si02.
Твердый раствор Муллит
Группа монтмориллонита включает большой набор
минералов со сходными структурами. Собственно
монтмориллонитом называют минерал состава Al2 [Si4Oi0] (OHJ • пН20, из
которого часть воды можно обратимо удалить. Из других
представителей наиболее важным являются бейделлит,
у которого отношение Si:Al = 3. Al3+ гидраргиллитового слоя
может замещаться на Mg2+, Zn2+, Fe3+, Сг3+ и др. Нонтронит
имеет состав (Fe, Al) [Si40(o] (ОН^-пНгО, в нем до половины
атомов А1 замещены на Fe.
Монтмориллонитовые минералы способны к набуханию и
поглощению большого количества воды,ч заключенной между
слоями. Потеря £е происходит в„ди-ааазоне от 120 до 200°, где
и наблюдается первый эндотермический эффект: "Процесс
318
высушивания до.200_°_ обкатам; при нагревании до 300°
происходит уменьшение расстоя'ни5Г~'М5Жду" "слоями, и~~по'теря
воды оказывается уже иедбр^х^шт^
При дальнейшем нагревании в области температур от 500
до 700° теряется консгитуцдщшдя, вода и образуется безвсаг—
н1ая медшфИкация" ми11с'р^ла^1рич£М_дегидратация начинается
р^ньш^'"у~1Яинералов с .шжьшхешЫм.. содержанием -~ж"еле5гагг---
Так, "но'нтрТЗнит тсрист конституционную воду (структурные
группы ОН~) при температуре ниже 500°. Полного
разрушения структуры при этом не происходит, поэтому около 900°
наблюдается новый эндотермический эффект, обусловленный
уже полным распадом. Около 950° происходит экзотедщш©-
екая реакция_2ек£истал.шзлц11и. Ог900 До'1300е."образ^£1ся
шгПОТельГ кристаллизуютсч окислы железа и кремнезема.
превращагощептстгТГн'ачала >; кварц, а затем в кристчэ-
б ал лит. Так"Же7*как и при нагревании каолинита,
появляется муллит (примерно при 1050°).
Слюдистые минералы характеризуются теми же
основными эффектами, что и предыдущие группы. Гидрослюды
теряют адсорбированную „воду, затем идет удаление
конституционной воды, которое начинается"" "при 600—650° и часто
имеет максимум в интервале 750—850°"." Последняя эндотер-"
мическая реакция около 850—900° сопровождается слабым
экзотермическим эффектом.
Минералы окиси алюминия. Разложение тригидрата —
гидраргиллита—происходит при температуре около 150°, и
при температуре около 200° на 1 моль А12Оя приходится уже
менее 0,5 моля Н2О. Последние молекулы воды из
моногидрата удаляются около 420\ В■ результате- -цбезвоживШия
образуется метастабильная форма — у-глинозем, который
при нагревании до 1000° перехоАЙ.ЖЗтабй^ную "форму —
u-г линоаем, или кор у н д.__
Гидраты окиси железа тоже легко разлагаются с
образованием безводных форм. Гетит начинает разлагаться при
250°, магнетит окисляется на воздухе при бледно-красном
калении. Образующийся при разложении некоторых
модификаций Y"Fc203 ПРИ 400° медленно переходит в гематит —
и-РсгОз- ' —
Температуры важнейших фазовых превращений,
происходящих с первичными и вторичными почвенными минералами
при их нагревании, приведены в табл. 30. Более подробные
сведения можно найти в указанной литературе.
Даже краткое изложение термических свойств минералов
тонкодисперсной фракции почв показывает, что расшифровка
12*
319
почвенных термограмм не является легким делом.
Тонкодисперсная фракция почв представляет собой сложную смесь
минералов с близкорасположенными термическими
эффектами. При взаимном наложении эффектов преобладающий
минерал маскирует минералы, содержащиеся в небольших
количествах. Второе осложняющее обстоятельство вызвано
относительно высоким содержанием органического вещества.
Окисление гумуса происходит медленно и в широком
диапазоне температур — до 700—800° (И. Д. Седлецкий), этот
длительный экзотермический эффект может значительно
видоизменить характер термограммы. Для устранения
влияния гумуса прибегают к предварительному разрушению
органических веществ почвы путем обработки образцов перекисью
водорода, которая, однако, не окисляет весь гумус.
ПРИБОРЫ ДЛЯ ТЕРМИЧЕСКОГО АНАЛИЗА ПОЧВ И ПОРЯДОК
РАБОТЫ
Для обнаружения термического эффекта в процессе
нагревания образца непрерывно измеряют температуру послед-
н£щ,7 Измерение температуры принципиально возможно
[любым известным методом, однако наиболее удобным
инструментом, особенно при измерении температуры почвенных
'(и близких к ним) образцов, является термопара.
Термопары
Термопара (см. также гл. II) состоит из двух различных
проводников. В месте соприкосновения проводников
возникает контактная разность потенциалов, которая обусловлена
неодинаковым энергетическим состоянием электронов в
металлах. Согласно электронной теории при выходе из
металла электрон должен преодолеть работу выхода,
которая зависит от свойств металла.
В ейду разных работ выхода при соприкосновении
металлов начинается переход электронов из одного металла в
другой, продолжающийся до тех пор, пока между металлами
не установится^Текоторая разность потенциалов,
препятствующая дальнейшему переходу электронов. Поскольку
электроны переходят из металла с меньшей работой выхода
в металл с большей работой выхода, последний заряжается
отрицательно. Возникшая разность потенциалов и является
причиной термотока. Кроме того, термоток может возникать
даже в однородном проводнике, если он нагрет неодинаково
320
по всей длине, но эта вторая составляющая термотока мала
по величине.
Работа выхода электронов, а следовательно, и
термоэлектродвижущая сила (т. э. д. с.) термопары зависит от
температуры, причем для многих пар металлов эта
зависимость прямолинейна. Это позволяет пользоваться термопарой
для измерения температур по величине возникающей т. э. д. с.
Для измерения составляется простая цепь, состоящая из двух
сваренных проводников и милливольтметра (см. рис. 18).
Спаянные концы нагреваются, а температура свободных
концов термопары, соединенных с измерительным прибором,
поддерживается постоянной. Шкала милливольтметра или
гальванометра градуируется непосредственно в градусах
стоградусной школы (°С).
В табл. 29 приведены величины т. э. д. с. термопар,
состоящих из платины в паре с одним из указанных в таблице
проводников, когда температура рабочего конца равна 100°,
а температура свободных концов — 0°.
Таблица 29
Т. э. д. с. термопар из платины в паре с одним
из указанных проводников
Материал
Кремний ....
.Медь
Родий
Т. э. д. с,
мв
+44,8
+ 2,2
+ 1.8
+ 0.8
+ 0,76
+ 0,75
+ 0,64
Материал
Сплав 90%Pt +
+ 10?ь Rh . .
Алюминий . . .
Графит ....
Алюмель ....
Константан . .
Копель ....
Т. э. д. с,
мв
+0,64
+0,40
+0,32
— 1,7
—3,4
—3.6
Наиболее пригодны для термического анализа почв
термопары из сплава 90% платины +10% родия, позволяющие
измерять температуру до 1600° и устойчивые к окислению.
Т. э. д. с. платина-платинородиевой термопары почти не
меняется со временем.
Для изготовления термопары берут две проволоки: одну
из чистой платины, другую из сплава 90%Pt+10%Rh;
проволоки должны быть однородными по всей длине. Для
проверки однородности подключают два конца отрезка проволоки
к чувствительному милливольтметру и последовательно по
321
всей длине прогревают на газовой горелке небольшие участки
проволоки, идя от одного конца к другому. Если где-либо
однородность нарушена, то при прогреве этого участка
милливольтметр покажет т. э. д. с, такую проволоку следует
заменить другой. Лучше использовать для изготовления
термопар платиновую проволоку, специально предназначенную
для этой цели. Перед изготовлением термопары проволоку
обжигают при светло-красном калении, после чего она
становится мягче. Толщина проволоки 0,3—0,5 мм, длина
каждого отрезка 100—120 см.
Т. э. д. с. платино-платинородиевой термопары в пределах
от 300 до 1200° может быть вычислена по уравнению
т. э. д. с. = —0,323 + 8,276 ■ Ю-3/— 1,638 • Ю-6/2,
где t — температура.
Последним слагаемым в уравнении можно пренебречь,
поскольку коэффициент 1,638- 10' G очень мал, и тогда
т. э. д. с. s~ 0,323+ 8,276- 10~3 t.
Существенным недостатком платиновых термопар
является способность платины образовывать хрупкие карбиды при
высоких температурах. Поэтому при работе с образцами почв,
содержащими значительные количества органических
веществ, термопара быстро изнашивается, а на термограммах*
появляются ложные эффекты. Для предохранения термопар
горячие спаи иногда помещают в защитные кварцевые
колпачки.
Установки для получения кривых нагревания почв
и почвенных коллоидов
Простейшая установка для получения термограмм состоит
из печи, в которой нагревается образец, термопары и
гальванометра (или милливольтметра). Записывая регулярно
показания гальванометра по мере нагревания образца и
откладывая затем на графике по оси ординат показания гальванометра
(пропорциональные температуре), а по оси абсцисс — время,
получают т е р м о г р а м м у, или кривую нагревания.
Такая термограмма имеет существенные недостатки,
связанные прежде всего с тем, что тепловые эффекты,
наблюдающиеся в образцах, обычно очень невелики. Поэтому заметить
изменение скорости нагревания, особенно при высоких
температурах, весьма трудно. Второй и очень важный недостаток
322
вызван непостоянством скорости нагрева самой печи.
Колебания напряжения в сети, питающей обмотку электропечи, и
конвекционные токи воздуха, отводящие тепло, вызывают
замедление или ускорение нагрева, которые могут быть
восприняты при расшифровке термограммы как эндо- или
экзотермический эффект.
Значительное снижение этих
погрешностей и повышение
чувствительности дает
применение дифференциальной (или,
вернее, разностной)
термопары.
Дифференциальная __тер_мо-
пара состоит in двух простые;
термопар, соедннснных_1аващ.
образом, что возникающие в
НИХ ТОКИ KOjMIKMK'np\IOX_J£yr
друга (рис. 79). По
компенсационной схеме обе термопары
соединяют одноименными
проводниками. Гальванометр.^
(милливольтметр),
"находящийся в цепи одной -из термопар,
показывает температуру
образца, а гальванометр 3
подключается к одноименным концам 1ЛЫ| ^i
двух термопар и показывает-
разность т. э. д. с. между двумя
горячими спаями."
Первый горячий спай погру-
жец.£~~йссЛёду"ё'мое вещество,
второй — в эта7юн~1::::=7М£Г©Ц!И-
ЛЬОз. Эталон подбирается
так, чтобы во всем диапазоне
используемых температур в
нем не происходило никаких
фазовых превращений. Скорость его нагревания зависит
только от скорости нагрева печи и теплоемкости и
теплопроводности самого эталона.
Откладывая на графике по оси ординат показания
дифференциального гальванометра, а по оси абсцисс — время,
получают дифференциальную термограмму. Если
теплоемкость и теплопроводность исследуемого образца
почвы и эталона одинаковы, то запись термограммы для мине-
Рис. 79 Схема устройства
дифференциальной термопары:
/ — образец, 2 — эталон, 3 —
дифференциальный гальванометр, 4 —
пирометрический гальванометр,
5 — коробка холодных спаев;
R6 и R6 — балластные
сопротивления, Яш и /?ш — шунтовые
сопротивления
323
рала с одним эндо- и одним экзотермическим эффектом
выглядит, как показано на рис. 80,6. В диапазоне температур
от точки А до точки В ни минерал, ни эталон не испытывают
никаких фазовых превращений, поэтому и скорости
нагревания обоих веществ, как и температуры, в любой момент
времени практически одинаковы. Это обусловливает равенство
т. э. д. с. двух горячих спаев, но поскольку термотоки
направлены навстречу друг другу, суммарная т. э. д. с. равна нулю,
и стрелка («зайчик») дифференциального гальванометра
остается на нулевом делении шкалы. Второй
(пирометрический) гальванометр в то же время отмечает увеличение
температуры образца.
$ начале эндотермического процесса скорость нагревания
образца~~за-медлнется, так как, ..часть энергии расходуется
на фазовое превращение. Температура эталона продолжает
увеличиваться с постоянной скоростью, и в некоторый
промежуток времени остается выше, чем температура образца.
Э. д. с. первой- термопары при этом оказывается меньше, чем
второй, и дифференциальный гальванометр регистрирует
разность т. э. д. е.; изменения т. э. д. с. при эндотермических
эффектах считаются отрицательными и откладываются на
графике вниз от нулевой линии. По окончании превращения
температуры образца и эталона постепенно вновь
выравниваются, и «зайчик» гальванометра в идеальном случае
возвращается к нулевому делению. В результате на
дифференциальной кривой возникает более или менее четко
выраженный минимум (участок BCD, на рис. 80,6), указывающий на
появление первого эффекта. На простой термограмме этот
эффект обнаруживается замедлением скорости нагрева
(ЛГЫ.
Также проявляется на термограмме и экзотермический
эффект с той лишь разницей, что вследствие выделения тепла
температура образца на некоторое время становится выше
температуры эталона; возникающий суммарный термоток
имеет направление, обратное тому, какое было при
эндотермическом эффекте. Соответствующие значения на графике
откладываются вверх от нулевой линии (участок EFH).
Термограмма на рис. 80 представляет идеальный случай.
Фактически на графике линии АВ и DE могут проходить
выше или ниже нулевой линии и даже идти под некоторым
углом к ней. Это объясняется влиянием неодинаковой
теплоемкости и теплопроводности образца и эталона на скорость
нагревания, а также изменением последних величин при росте
температуры или фазовых превращениях.
324
При дифференциальной записи все колебания в скорости
нагрева почти одинаково влияют и на образец и на эталон и
полностью компенсируются. Вместе с тем если применять в
качестве измерительного инструмента высокочувствительный
зеркальный гальванометр, то на дифференциальной кривой
легко зафиксировать даже малейшую разность температур
между образцом почвы и эталоном. В связи с этим при
монтаже прибора особое внимание уделяется качеству термо-
Рис. 80. Термограммы каолинита, полученные при помощи простои
(о) и дифференциальной (б) термопары:
С —эндотермический эффект; F — экзотермический эффект
пар. „Обе тчетви^ должны-йьиъ- абсддцщю одинаковы, поэтому
их nejflgioTliT^HHaKOBbix кусков однородной проволокиГДля
изготовления"" дпффер^нщ1ад1>но111 термопары оба конца..
йлатинородиевом 11роволоки_сваривают Ъ ^"ЧШШи._платиновоп
щюволрки* в пламени вольтовой „дуги (при этой операции
обязательно надевают темные защитные очки). Затем
платиновую проволоку режут пополам, а платинородиевую
сгибают точно посередине (рис. 81). Два спаянных конца
являются горячими спаями и помещаются в нагреваемый
образец и эталон. Разрезанные концы платиновой проволоки
и место перегиба платинородиевой проволоки служат
холодными спаями. Холодные спаи соединяют с медными
соединительными проводниками и помещают в термостат. Все плечи
термопар помещают в защитные фарфоровые одно- или двух-
канальные трубки, оставляя оголенными лишь горячие спаи.
Дифференциальный гальванометр или милливольтметр в
325
этой схеме имеет шкалу с нулем посередине, так как
измеряются и положительные и отрицательные т. э. д. с.
Термостатирование холодных спаев. На контакте между
платиновой или платинородиевой проволокой и
соединительными медными проводами также возникает т. э. д. с. Для
устранения погрешностей температура холодных „спаев
должна оставаться строго постоянной в течение всего опыта.
В простейшем случае холодные спаи помещают в пробирки
с маслом, предохраняющим их от коррозии, и вместе с про-
| *
1? Pt'/<7%Rh
Рис. 81. Изготовление дифференциальной термопары.
й — спаянные проволоки до разрезания; б —
готовая термопара: / — место разреза, 2 — место сгиба,
3 — места спаев
бирками погружают в сосуд Дьюара, заполненный смесью
воды со льдом (рис. 82). Сосуд Дьаюра позволяет
длительное время сохранять холодные спаи при температуре тающего
льда.
Гальванометры и их шунтирование. Регистрация
термотока простой термопары возможна довольно грубыми
приборами ■— стрелочными милливольтметрами (примерно 20 мв па
всю шкалу). Дифференциальная термопара требует
высокочувствительных гальванометров — зеркальных или стрелочных
с чувствительностью порядка 10-8—10~9 а/мм/м с малым R —
200—800 ом, Rmi— 150—300 ом. Практически оказывается
выгодным использовать два зеркальных гальванометра,
которые обеспечивают одновременную фотографическую запись
обеих кривых, что избавляет исследователя от визуальной
записи и дает очень полную картину нагревания. В то же время
зеркальный гальванометр в цепи простой термопары
позволяет легко уравновесить оба плеча. Па рис. 79 видно, что
гальванометр 4 шунтирует цепь дифференциальной
термопары. Включая дополнительные сопротивления в эту цепь,
уменьшают шунтирующее влияние. Очевидно, что и
необходимое здесь большое по величине балластное сопротивление
допустимо лишь при условии, что
гальванометр 4 имеет высокую
чувствительность.
Балластные R'q, R"z и шунтовые
сопротивления R'm и Я"ш включают
в обе цепи. Величины сопротивлений
подбирают с таким расчетом, чтобы
во всем диапазоне измерений
показания гальванометра не выходили
за пределы шкалы, учитывая,
конечно, и величину внешнего
критического сопротивления (гл. II,
стр. 49—52).
Печи. Почвенные образцы
исследуются на минералогический состав
при нагревании от 100 до 1200°.
Температуру до 1000° можно
получить в обычных тигельных печах
типа ТП-1, ТП-3. Более высокие
температуры достигаются
применением специальных нагревательных
элементов, например сплава № 2
и др. Очень удобна обмотка из
платиновой проволоки, дающая в печах
температуру до 1300°. Однако она
стоит дорого.
Обычные тигельные печи
обладают двумя существенными
недостатками, ограничивающими их
применение в термографии.
Во-первых, они неравномерно
нагреваются и, во-вторых, медленно остывают.
Характерная кривая скорости
нагревания печи имеет S-образную
форму. В начале нагрева в силу
инерционности печи скорость
нагревания мала, затем она возрастает, и кривая имеет участок,
близкий к прямолинейному. При высокой температуре резко
увеличивается теплоотдача стенками печи, и скорость
нагревания снова падает. Более плавный нагрев достигается раз-
Рис 82 Терчостатировачие
холодных спаев-
/ — сосуд Дьюара, 2 —
смесь воды со льдом, 3 —
пробирки с маслом, 4 —
спаи термопар, 5 — пробка
327
личным монтажом нагревательных обмоток; открытое
расположение обмотки внутри печного пространства снижает
инерцию печи, а влияние теплоотдачи может быть компенсировано
второй внутренней спиралью. Однако наиболее постоянную
скорость нагрева получают только при применении
программных регуляторов температуры печи.
Медленное остывание печи снижает производительность
работы. Пользуясь одной печью, в течение дня удается
получить всего две, в лучшем случае три термограммы. Поэтому
необходимо иметь в лаборатории несколько запасных печей
или применять специальные разборные печи с быстро
остывающими или сменными нагревательными блоками.
Программное регулирование температуры
Для обеспечения постоянной скорости нагрева предложено
несколько способов и конструкций приборов. Все они связаны
с программным изменением питающего напряжения или силы
тока, протекающего в обмотке электропечи. Поскольку
кривая нагрева имеет обычно S-образную форму, то задача
регулирования сводится к тому, чтобы в начале нагрева
быстрее увеличивать напряжение, затем скорость подъема
напряжения может быть снижена или оставлена постоянной,
а в конце нагрева вновь значительно увеличена. Наиболее
простая регулировка осуществляется вручную, когда обмотка
печи питается от сети через реостат или трансформатор.
Исследователь, наблюдающий за ходом нагрева, изменяет
положение подвижного контакта реостата (трансформатора),
добиваясь, чтобы «зайчик» пирометрического гальванометра
перемещался с постоянной скоростью. Такая регулировка
может быть очень надежной, но отнимает много времени,
требуя пребывания исследователя у прибора в течение всего
эксперимента.
Автоматические регуляторы делятся на два типа. К
первому типу относятся такие регуляторы, у которых изменение
питающего напряжения достигается при помощи мотора,
вращающего рукоятку автотрансформатора. Мотор связан с
автотрансформатором через редуктор и механическое
устройство, повышающее напряжение неравномерно, в соответствии
с заранее подобранным режимом. Заданный режим
регулирования здесь рассчитан только на постоянное напряжение
сети и на печи (обмотки) одного типа; при замене печей
режим регулирования приходится подбирать заново, поэтому
подобные способы не универсальны.
328
В наибольшей степени отвечают требованиям
термического анализа методы регулирования, основанные на
непрерывном контроле температуры печи Блок-схема такого
регулятора описывается на стр. 71—72. Температура печи измеряется
контрольной термопарой, которая связана с измерительно-
регулирующим устройством — электронным регулятором
температуры ЭРМ-471. Если сигнал, воспринимаемый с
термопары, превышает заданную величину, то регулятор отключает
питание нагревателя; при понижении температуры ниже
заданной обмотки нагревателя снова подключаются к
питающей цепи через реле. С помошью такого потенциометра
можно регулировать и скорость нагрева, если заставить
указатель перемещаться вдоль шкалы с постоянной скоростью.
Тогда в любой момент времени температура печи
соответствует положению указателя на шкале потенциометра.
Движение на указатель шкалы передается от синхронного
моторчика, число оборотов которого постоянно и зависит только от
частоты питающего напряжения.
Полное выключение и последующее включение тока в
обмотке нагревателя приводит к значительным колебаниям
температуры около заданной, одновременно в цепи
дифференциальной термопары могут возникать наводки тока. Чтобы
избежать этого, регулятор соединяют не с нагревательными
элементами печи, но с мотором автотрансформатора,
непрерывно повышающим питающее напряжение. Как только
температура превысит заданную, реле отключает мотор
автотрансформатора, и скорость нагревания замедляется. Если
правильно выбрано начальное напряжение и скорость
вращения мотора, то этого вполне достаточно для обеспечения
прямолинейного нагрева. Для уменьшения пульсаций в
записи т. э. д. с, вызванных наведенными токами,
рекомендуется заземлять нейтральную точку термопары (точка 2
на рис. 80).
Градуировка термопар
Каждую вновь изготовленную термопару обязательно
градуируют, причем в той цепи (вместе со всеми
сопротивлениями и измерительным прибором), которая будет в дальнейшем
использована в работе. Цель градуировки — установить
зависимость между показаниями пирометрического гальванометра
1 Используют также стрелочные и электронные потенциометры ЭПД,
ЭПП и др.
329
и температурой горячего спая (или определение цены одного
деления шкалы гальванометра в °С).
В практике принята градуировка по веществам с точно
известными температурами фазовых превращений. Набор
веществ для градуировки термопары в диапазоне от 0 до
1100° приведен в табл. 30.
Таблица 30
Стандартные вещества дли градуировки термопары
Вещество и превращение
Вещество и превращение
Плавление льда
Плавление Na2SO4-10H2O .
Кипение воды
Кипение Na-jSCVlOHaO . .
Плавление гидрохинона* . .
Кипение нафталина* . . . .
Полиморфное превращение
Na2S04
Плавление бихро\ата калия*
Кипение серы*
0,0
32,4
100
102
169
217,96
240
397,5
444,6
Плавление эвтектики
45%КС1-г-55% NasS04 . .
Полиморфное превращение
K2S04
Плавление эвтектики 30,5%
NaCl+69,5% NasS04 . .
Пла!!ленне КС!
Плавление NaCl
Плавление Na2S04 . . . .
Плавление K{S04
517,1
585
627
770,3
800,5
884
1069,1
Примечание. Работа с веществами, стоящими под звездочкой,
бует осторожности и особых условий.
тре-
Определение показаний термопары в тающем льде и
парах кипящей воды производят просто после помещения
термопары в соответствующую систему. Установить же
визуально плавление солей, особенно при высокой
температуре, довольно трудно. Поэтому при градуировке простой
термопары поступают следующим образом. Горячий спай
помещают в тигель, плотно набитый солью (или
эвтектической смесью), и начинают нагрев, непрерывно (через 1 мин)
записывая показания гальванометра. С начала фазового
превращения показания прибора остаются постоянными,
пока вся соль полностью не расплавится. После этого
показания прибора вновь начинают повышаться. По окончании
превращения выключают нагреватель и наблюдают за
охлаждением и затвердеванием и снова записывают показания
1 альванометра. Усредняя результаты, наносят их на график
в координатах показания гальванометра — время.
Горизонтальная площадка на графике отвечает показанию гальвано-
330
метра при температуре фазового превращения. Расхождение
значений, найденных при нагревании и охлаждении, не должно
превышать одного деления шкалы гальванометра.
По окончании опыта тигель снова нагревают до
расплавления соли, из расплава вынимают термопару, охлаждают
термопару и тигель с солью и моют их в кипящей воде.
Выливать расплавленную соль в раковину
или в воду нельзя, это может привести к
тяжелым ожогам вследствие взрыва и
разбрызгивания расплава.
Промытую термопару подсушивают, особенно тщательно
сушат фарфоровые изолирующие трубки. Если в следующем
опыте будет использована трубка, в порах которой осталась
вода, то при нагревании и испарении воды стенки
фарфоровой трубки разрушаются.
Вымыв и высушив термопару, приступают к опытам со
следующим стандартным веществом. Результаты опытов
оформляют в виде градуировочного графика, на котором но
оси абсцисс отложены температуры фазовых превращений, а
по оси ординат — соответствующие им показания
гальванометра. Результатами градуировки можно пользоваться как
по графику, так и по таблицам, в которых против каждого
значения шкалы гальванометра проставлены температуры.
Дифференциальная термопара градуируется в том же
режиме работы, который выбран для исследования. Один
тигель плотно набивают стандартным веществом, второй —
порошком Al2Os или MgO (последние предварительно нагре
вают до максимальной температуры, которой можно ожидать
в дальнейших опытах, и хранят без доступа влаги и С02).
Спай пирометрической термопары, соединенной с тепловым
гальванометром, помещают в стандартное вещество, стараясь
расположить место спая в центре образца. Второй спай
погружают в эталон (MgO или Al2Os). Оба малых тигля
(с веществом и эталоном) ставят в большой общий тигель
или в нагревательный блок внутри печи. «Зайчик»
дифференциального гальванометра устанавливают на середину шкалы,
а «зайчик» пирометрического — на нулевое деление. Затем
включают обмотки нагревателя и ведут запись показаний
гальванометров, делая отсчеты через 1 мин по секундомеру.
В результате опыта получают две термограммы,
совмещенные на одном бланке. Показания дифференциальной
термограммы пропорциональны разности т. э. д. с. или AT,
показания простой — температуре образца. Обе кривые
объединены одной общей осью времени (ось абсцисс на рис. 83,й).
331
По дифференциальной термограмме устанавливают в
какой момент времени произошло фазовое превращение Для
этого из точки перегиба минимума (для эндотермического
эффекта) или максимума (для экзотермического эффекта)
опускают перпендикуляр на ось времени Пересечение
перпендикуляра с простой (пирометрической) термограммой Т
(рис. 83,а) позволяет определить показания А
пирометрического гальванометра в тот же момент времени Для каждого
МП
h(T)
700 "С
Рис 83 Градуировка термопар
а — термограмма смеси 30,5% NaCl + 69,5% Na2S04 (плавление при 627°),
б — построение градуировочного графика, А — показания
пирометрического гальванометра в максимуме эндотермического эффекта при 630°
из эффектов показания пирометрической термопары
измеряют циркулем и переносят на график, где но оси абсцисс
отложены температуры соответствующих эффектов
(рис 83,6) Таким образом получают градуировочкый график,
по которому в дальнейшем определяют температуры
эффектов Исходной точкой при построении графика служит
отметка 100°; показания гальванометра при 100° находят, помещая
термопару в пары кипящей воды При визуальной записи
стоградусную отметку ставят на пирометрической кривой от
руки, а при автоматической записи на пирометре
выключают осветитель пирометрического гальванометра на
5—10 сек, когда температура образца достигла 100°.
Пирометр Курнакова
Первая модель фоторегистрирующего прибора для
термического анализа создана в 1903 г акад Н. С. Курнаковым.
Усовершенствованные конструкции были выпущены в
1947—1948, 1952 и 1955 г.
332
Модель пирометра 1955 г. (ПК-55) предназначена для
автоматической регистрации термических кривых при
нагревании или охлаждении образцов. Прибор работает на
дневном свету, и затемненное помещение необходимо только для
заряжания камеры и обработки фотобумаги.
Рис 84 Внешний вид пирометра Курнакова:
J — барабан с фотобумагой, 2 — редуктор мотора, 3 — моторчик
Уоррена, 4 — осветители гальванометра, 5 — шкала
гальванометров
Пирометрическая установка (см. рис. 84) состоит из
барабана / для фотозаписи, зеркальных гальванометров с
осветителями 4 и магазинами сопротивлений, трансформатора,
333
печи, термопары и термостата для холодных спаев. Кроме
того, может быть дополнительно подключен автоматический
регулятор скорости нагрева печи.
Гальванометры установлены внутри прибора и
изолированы от дневного света. Луч света, отраженный зеркальцем
гальванометра, попадает на входную щель, перед которой на
поверхности фотокамеры установлена цилиндрическая линза,
фокусирующая световой
пучок и обращающая его
в точку, проектируемую
на поверхность
фотобумаги. На передней стенке
прибора расположена
шкала 5, позволяющая
визуально наблюдать
положение «зайчика»
гальванометров.
Фотобумага
закрепляется на барабане /
двумя зажимами, а на
торцевой стороне
фотокамеры имеется лимб с
делениями и указательная
положением и скоростью
воемя
Рис. 85. Дифференциальная
термограмма карбонатного образца
стрелка, позволяющие следить за
вращения барабана с фотобумагой.
Вращение на барабан передается от синхронного
моторчика 3 (тина Уоррена). Время оборота барабана задается
редуктором 2. Эта модель пирометра является по существу
вариантом установки для автоматической записи любых
процессов на фотобумаге. В комбинации с дифференциальной
термопарой осуществляется термический анализ, но
возможно использование и любых иных схем, где наблюдение за
процессами осуществляется с помощью регистрации тока.
Кроме пирометров этого типа применяют сложные установки,
позволяющие получать одновременно кривые нагревания и
кривые обезвоживания, показывающие весовые потери воды
образцом при различных температурах. Кривые
обезвоживания помогают уточнить результаты термического анализа.
Подготовка образцов
Термическому анализу редко подвергают образцы почв
в целом, чаще исследуют фракции, отличающиеся степенью
дисперсности, уделяя особое внимание илистой фракции или
почвенным коллоидам.
334
Тонкодисперсную фракцию выделяют методом размучива-
ния и центрифугирования. После предварительного декаль-
цирования 0,05 н. раствором НС1 карбонатных почв или
растирания почв с водой (по Айдиняну) образцы суспендируют
в воде, добавляя для повышения устойчивости взвеси
несколько капель 10%-ного раствора едкой щелочи (до
слабо-розовой окраски по фенолфталеину). Суспензию тщательно
взбалтывают и оставляют на сутки. Через сутки сливают
верхний слой толщиной ~ 7 см, содержащий частицы размером
<С 1 мк (см. гл. I). Размучивание продолжают до полного
осветления верхнего семисантиметрового слоя после
суточного отстаивания. Суммарная фракция частиц<0,001 мм может
быть выделена из суспензии фильтрованием через плотные
фильтры на воронке Бюхнера под вакуумом или коагуляцией
(при р_Н = 3—4) небольшой порцией соляной кислоты. Так
как суспензия с частицами < 0,001 мм легко проходит через
фильтры, то, собирая фракцию первым способом, приходится
пропускать ее через фильтр несколько раз, пока поры
фильтра не будут забиты коллоидными частицами, а фильтрат не
станет совсем прозрачным. Влажный образец коллоидной
фракции высушивают при температуре 50—70°, растирают и
подвергают термическому анализу.
Коллоидную фракцию с помощью проточной центрифуги
разделяют на иодфракции с частицами от 1 до 0,2 мк и
с частицами < 0,2 мк. В суперцентрифугах, имеющих
50 тыс. об/мин, можно выделить фракцию с частицами
<0,06 мк.
Получение термограмм и их расшифровка
Термический анализ образцов почв или почвенных
коллоидов технически ничем не отличается от снятия термограмм
при градуировке дифференциальной термопары.
Предварительно подготовленный образец плотно набивают в один из
тиглей, в середину образца помещают горячий спай
пирометрической термопары и снова сверху уплотняют образец, чтобы
он хорошо прилегал к концам термонары. Второй тигель
набивают эталоном и в него помещают вторую ветвь термопары.
Тигли ставят в печь и включают нагреватель. Когда
температура в образце поднимается до 100°, начинают записывать
показания гальванометров (или открывают щель
фотобарабана на самописце).
По окончании записи выключают нагреватель и сразу, не
давая образцу остыть, извлекают обе ветви дифференциаль-
335
Температуры
Таблица 31
фазовых превращений некоторых минералов (по Бергу)
Минерал
Лллофан
Арагонит
Бейделит
(группа
.монтмориллонита)
Биотит
Боксит
Браунит
Брусит
Вермикулит
(группа слюд)
Галлуазит
Гстнт
Глдробиотит
(группа слюд)
Гидрогематит
Гидраргиллит
Гипс
Дикнт (грулла
каолинита)
Каолинит
Кварц
Лшюнит
Монтмориллонит
Формула
Al203Si02-3H20
СаС03
(KHJ(Mg,rcJ(Al,FeJSi3012
смесь каолинита с
диаспорой или гидраргиллитом
Мп203
Mg(OHJ
—
Al203-3Si02-3H20
FeO(OH)
---
Fe203aq
A1203-3H20
Ca2S04-2H20
—
Al203-2Si02-2H20
Si02
2Fe203-3H20
H2Al2Si40J2-«H20
Температура (ГС) и природа эффекта
170—дегидратация
960—1000 (экз.)—перестройка
решетки
450 (энд)—монотропное
превращение
900—диссоциация
320— дегидратация
580—дегидратация
1100—распад решетки
285—310—дегидратация гидрар-
гиллита
505—дегидратация диаспора
550— 600—дегидратация
каолинита
960 (экз.)—перестройка решетки
910—1000—превращение в Мп304
405—410—дегидратация
120—170— »
270—300— »
860 (энд )—необратимое
превращение
125—170—дегидратация
560—650— »
960 (экз.)—перестройка решетки
300—360—дегидратация
160-180— »
220—250— »
800—1000—необратимое
превращение
120—140—дегидратация
340 — »
285—300— »
120, 180— »
360 (экз.)—перестройка решетки
665— 670—дегидратация
970—990 (экз.)—перестройка ре-
щетки
550—600—дегидратация
960 (экз.)—перестройка решетки
575—полиморфное превращение
120— 140,
280—360—дегидратация
160—240—потери адсорбционной
воды
660—700—потеря
конституционной воды
Продолжение
Минерал
Формула
Температура ('С) и природа эффекта
Мусковит
Нонтронит
K20-3Al203-6Si02-2H20
(Fe, AlJ(S>4O10)(OHJ nH20
850— 900—экзотермический
эффект
125—потеря гигроскопической
воды
450—650—потеря
конституционной воды
850—900—разрушение решетки
300—дегидратация
515—560—дегидратация
752—(экз.)
940—(экз.)
Определитель минералов (по Бергу)
Таблица 32
Минерал
■Гипс, алюминит
Алюминит, вермикулит, галлуазит, гидрогематит, эмеевик,
лимонит, метагаллуазит, сепиолит
Аллофан (вермикулит), гидробиотит, гидромусковит, гипс,
глауконит, змеевик, монтмориллонит, алюминит
Боксит, вермикулит, гидробиотит, гндраргнллит, манганит,
нонтронит, сингенит
Бейделлит, вермикулит, гетит, гидромагнезит, гипс (экз.),
манганит (экз.), полигалит, сапонит, сидерит
Анальцим, аратнит, брусит, гидромагцезит, натролит,
сидерит
Алунит, мусковит, родохрозит
Боксит, гидромагнезит (экз.), глауберит, диаспор, манганит,
нонтронит
Боксит, бейделит, галлуазит, гидромагнезит, глауконит,
каолинит, кварц, магнезит, метагаллуазит, пиролюзит,
сапонит
Аноксит, галлуазит, гидромусковит, накрнт
Дикит, змеевик, манганит, монтмориллонит
Алуннт, анкерит, доломит, пирофиллит, родохрозит (экз.)
Алуннт, нонтронит (экз.), пирофиллит
Анкерит, алюминит, змеевик (экз.), сепиолит
Кальцит, анкерит, доломит, алюминит, арагонит,
монтмориллонит, мусковит, серицит, тальк, ксонтолит
Браунит, нонтронит (экз.), парагонит, лепидолит
Аноксит (экз.), боксит (экз.), галлуазит (экз.), дикит (экз.),
каолинит (экз), манганит, метагаллуазит (экз.),
монтмориллонит (экз.), накрит (экз.), пиролюзит, аллофан
(экз.)
Биотит, гаусманит, гидромусковит, манганит, парагонит,
флогопит, пиролюзит, серицит, стронцианит
337
ной термопары из образца и эталона. Спекшийся образец
после охлаждения вынимают из тигля и легким
постукиванием или осторожным раздавливанием спекшейся массы
типцами освобождают термопару, стараясь не повредить ее
при этом.
Расшифровка термограммы проводится в два этапа.
Сначала на кривой находят характерные максимумы и
минимумы, свидетельствующие о прошедших фазовых превращениях.
Для каждого эффекта определяют момент времени
превращения, опуская перпендикуляр из точки перегиба на ось
абсцисс. Измерив циркулем расстояние от оси абсцисс до
точки пересечения перпендикуляра с пирометрической кривой
(рис. 85), откладывают эту величину на градуировочном
графике, находя тем самым температуру эффекта. Найденные
величины температур надписывают на дифференциальной
термограмме около соответствующих эффектов.
Второй этап расшифровки заключается в сопоставлении
оформленной термограммы с таблицами (табл. 31, 32) и
термограммами стандартных минералов. В почвенных образцах
наиболее часто встречаются слоистые минералы с парой
характерных эффектов (эндотермическим при 400—700° и
экзотермическим при 800—1100°). Поэтому расшифровку удобно
начинать с нахождения этой пары (или — пар) эффектов, а
затем переходить к минералам группы гидроокислов железа
и алюминия.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
Общие руководства
Сб. «Агрохимические методы исследования почв». Изд-во АН СССР,
М., 1960.
Анализ следов элементов. Материалы симпозиума. Под ред. Дж. йо и
Г. Коха. ИЛ, М, 1961.
Л я л и к о в Ю. С. Физико-химические методы анализа. Госхимиздат,
М., 1960.
П л о х и н с к и й Н. А. Биометрия. Изд-во Сиб. отд. АН СССР,
Новосибирск, 1961.
Сб. «Современные методы исследования физико-химических свойств
почв», вып. 1. Изд-во АН СССР, М., 1945.
Сб. «Современные методы исследования физико-химических свойств
почв», вып. 2. Изд-во АН СССР, М., 1947.
Сб. «Современные методы исследования физико-химических свойств
почв», вып. 3. Изд-во АН СССР, М., 1948.
Ю и н г Г. В. Инструментальные методы химического анализа.
Госхимиздат, М., 1960.
Яковлев К. П. Математическая обработка результатов измерений.
Гостехиздат, М., 1953.
338
Справочники и руководства по технике лабораторных работ
Беклемишев А. В. Меры н единицы физических величин. Гостех-
нздат, М., 1963.
Воскресенский П. И. Техника лабораторных работ.
Гослитиздат, М., 1962.
Карякин Ю. В., Ангелов И. II. Чистые химические реактивы.
Госхимиздат, М., 1955.
Коростелев П. II. Приготовление растворов для
химико-аналитических работ. Изд-во АН СССР, М., 1962.
Лурье Ю. Ю. Справочник по аналитической химии. Химиздат,
М., 1962.
Н аде и некий Б. П. Теоретические обоснования и расчеты в
аналитической химии. «Сов. наука», М., 1959.
Справочник химика, т. 1. Под ред. Б. П. Никольского. Госхимиздат,
М.—Л., 1962.
Справочник химика, т. 2. Госхимиздат, М. — Л , 1963.
Справочник химика, т. 3. Госхимиздат, М.- -Л., 1952.
Строи г Д. Техника физического эксперимента. Лениздат, 1948.
Приборы и аппаратура для физико-химического анализа
Алексеев Н. Г., Прохоров В. А., Чмутов К. В.
Электронные приборы и схемы в физико-химическом исследовании. Госхимиздат,
М., 1961.
Арутюнов В. О. Электрические измерительные приборы и
измерения. Госэнергоиздат, М.- Л., 1958.
Куликов И. Г. Аккумуляторы. Воениздат, М., 1961.
Сом и некий М. С. Вентильные фотоэлементы. Изд-во АН СССР,
Л., 1957.
Тури ч и v A. M. Электрические измерения неэлектрических величин.
Госэнергоиздат, М.—Л., 1954.
Чмутов К. В. Техника физико-химического исследования.
Госхимиздат, М., 1954.
Чудновский И. Я- Электровакуумные приборы и усилители.
«Искусство», М., 1955.
Ш к у р и н Г. П. Справочник по электроизмерительным и
радиоизмерительным приборам. Воениздат, М., 1956.
Потенциометрические и кондуктометрические методы анализа
Б е р л и н е р М. А., Д о л г о п о л о в Н. Н. Электрометрическое
определение солесодержания почв, грунтов и грунтовых вод. Изд-во
АН СССР, М., 1954.
Виноградова Е. Н. Методы определения концентрации
водородных ионов. Изд-во МГУ, 1956.
Кольтгоф И. М., Л а й т и н е н Г. А. Определение концентрации
водородных ионов и электротитрование. ИЛ, М., 1947.
Командин А. В. Электродвижущие силы. Изд-во МГУ, 1951.
П ч е л и и В. А. Измерение активности водородных ионов (рН)
Гослегпром, М., 1955.
Швабе К. Основы техники измерения рН. ИЛ, М., 1962.
339
Полярография
Виноградова Е. Н., Галлай 3. А., Финогенова 3 М.
Методы полярографического и амперометрического анализа. Изд-во МГУ,
1963.
Г е й р о в с к и й Я. Техника полярографического исследования. ИЛ,
М., 1961.
Иванов И. Д. Полярография белков, энзимов и аминокислот.
Изд-во АН СССР, М., 1961.
Крюкова Т. А., С и н я к о в а С. II., Арефьева Т. В.
Полярографический анализ. Госхимиздат, М., 1959.
Орлов Д. С. Полярографические методы исследования почв.
Изд-во МГУ, 1960.
Терентьев А. П., Яновская Л. А. Полярографический метод
в органической химии (реакции и методы исследования органических
соединений. Книга пятая). Госхимиздат, М., 1957.
Цфасман С. Б. Электронные полярографы. Металлургиздат,
М., 1960.
Колориметрия и спектрофотометрия
Асатиани В. С. Биохимическая фотометрия. Изд-во АН СССР,
М, 1957.
Б а б к о А. К-, П и л и п е н к о А. Т. Колориметрический анализ.
Госхимиздат, М. — Л., 1951.
Бабушкин А. А„ Б а ж у л и и П. А., Королев Ф. А., Лев-
шин Л. В., П р о к о ф ь е в В. К-, С т р и г а н о в А. Р. Методы
спектрального анализа. Изд-во МГУ, 19G2.
Г и л л е м А , Штерн Е. Электронные спектры поглощения
органических соединений. ИЛ, М., 1957.
Сб. «Колориметрические (фотометрические) методы определения1
неметаллов». ИЛ, М., 1963.
Л е к о н т Ж- Инфракрасное излучение. Фнзматгиз, М., 1958.
Пешкова В. М., Громова М. И. Практическое руководство
по спектрофотометрии и колориметрии. Изд-во МГУ, 1961.
С е н д е л Е. Б. Колориметрическое определение следов металлов.
Госхимиздат, М., 1949.
Чулан овский В. М. Введение в молекулярный спектральный
анализ. Гостехнздат, М. —Л., 1951.
Методы анализа по фотометрии пламени
Б о р о в и к-Р оманова Т. Ф. Спектрально-аналитическое
определение щелочных и щелочноземельных элементов. Изд-во АН СССР, М., 1959.
Б у р р и е л ь-М а р т и Ф., Р а м и р е с-М у н ь о с X. Фотометрия
пламени ИЛ, М., 1962.
Полуэктов Н. С. Методы анализа по фотометрии пламени.
Госхимиздат, М., 1959.
Русанов А. К-, И л ь я с о в а Н. В. Атлас пламенных, дуговых и
искровых спектров элементов. Госгеолтехиздат, М., 1958.
340
Термический анализ почв
Берг Л. Г. Введение в термографию. Изд-во АН СССР, М., 1961.
Горбу иов Н. И., Цюрупа И. Г., Ш у р ы г и н а Е. А.
Рентгенограммы, термограммы и кривые обезвоживания минералов,
встречающихся в почвах и глинах. Изд-во АН СССР, М, 1952.
Горбунов Н. И. Высокодисперсные минералы и методы их
изучения. Изд-во АН СССР, М., 1963.
Грим Р. Е. Минералогия глин. ИЛ, М., 1959.
Попов М. М. Термометрия и калориметрия. Изд-во МГУ, 1954.
Ц у р и н о в Г. Г. Пирометр Н. С. Курнакова. Изд-во АН СССР,
М., 1953.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Аккумуляторы 40—43, 46, 86—89
— условия эксплуатации 41, 42, 89,
205
Активность (активная концентрация)
12—17, 33, 78, 81
-- водородных ионов 15, 98, 104, 106,
107, 115, 118, 119, 120
— водородных ионов, определение
в почвах водородным электродом
90-94
-— водородных ионов, определение в
почвах стеклянным электродом
117—128, 131, 135—137
— водородных попов, определение
в почвах хиигидронным электродом
102—1O, 132, 134, 135
— ионов натрия, определение в
почвах 5, 16, 78. 137—139
Активности коэффициент 13—16, 33,
35, 81, 103, 153
■— коэффициент ионов натрия в
почвах 15, 16, 139
■— коэффициент гредний электролита
13—15
Алюминия обменного определение по-
тенщюметрическое 150—151
— обменного определение
фотометрическое 278—279
Алюминон 278
Амальгамирование 94, 95
Баллистический метод измерения э.д.с.
125—128
Батареи сухие 39, 43
Бугера закон 240
Бугера— Бэра закон 238—241, 246,
250, 251, 260, 279, 283
—, влияние окрашенных
реактивов 247, 248
—, влияние рассеивания света
246, 247
, отклонения от закона 246,
247
Буферность кислотно-основных систем
20, 21
— окиелнтелыю-восстановительиых
систем 165, 166
Буферные растворы 22, 23, 109, 148
Бэра закон 240
Всстона элемент 85, 88
Вода дистиллированная без СОг 116
Водород, получение и очистка 94
Водородный электрод 81, 82, 84, 90 —
94, 97, 98, 156
— электрод нормальный 84, 92
— электрод, приготовление 93
Возбуждение атомов и молекул 231,
235, 290—293
Волна полярографическая 186, 188,
214—216
— полярографическая, определение
высоты 188, 189, 214
— полярографическая, уравнение
189—193
Волновое число 231
Выпрямители вакуумные 39
— твердые 40
Вытяжки водные из почв для
определения рН 115—117
— солевые из почв 116
Гальванометры зеркальные 47—50,
125, 126, 211, 212, 297, 326
, определение чувствительности
211, 212
• , полное критическое
сопротивление 49, 51
342
. , характеристики 52
шунтирование 50—52, 326
— стрелочные 46, 86, 125
Гидролиз 19, 20, 100, 101
Горелки для пламенной фотометрии
299, 300
Гуматов инфракрасные спектры 285
Гумнновые кислоты, выделение из
почвы 282, 283
, подавление полярографических
максимумов 227, 228
спектрофотометрическое
определение 229, 284
■ спектры поглощения 281, 282
Диссоциации константа 17, 18, 149,
150
— степень 17, 18
Дитизон 220
Дитизонаты, образование и условия
экстракции 220—222
Дифференциальные кривые
титрования 144, 145
Дифференциальные термограммы
323—326, 331, 332
Диффузионный ток (см. ток
диффузионный)
Диффузия ионов 186
Емкость поглощения,
полярографическое определение 225, 226
, потенциометрическое
определение 142
Железа определение
полярографическое 223
фотометрическое 280, 287, 288
Ильковича уравнение 188
Инфракрасная спектрометрия 231 —
234, 237
, подготовка образцов 276
Инфракрасные спектрометры 273—
278, 285, 286
Ионная сила 15, 99
— — водных вытяжек из почв 16
Ионное произведение воды 18, 19
Ионного обмена изотермы 33
Ионообменная способность почо 31—
33
Калий, определение по фотометрии
пламени 289 и ел., 310—313
Каломельный электрод 90—98
по Крюкову 169
■, потенциалы при различных
температурах 97
, приготовление 91, 94, 95, 96
Кальция полярографическое
определение 224
Каолинитопые минералы, строение
316
Каолинитовые минералы, термические
характеристики 317, 318
Квант излучения 231
Кенотроны (см. вакуумные
выпрямители)
Кислотность почв 20, 98—102, 115, 116
— ■— гидролитическая 99—102
обменная 99—102, 115
Ключ электролитический 82, 90, 97
Кобальта полярографическое
определение 218, 220—223
Коллоидные растворы 7, 24—31, 115
— —, коагуляция 34, 35
Коллоиды почвенные 7, 24, 25, 94,
314, 335
Колориметрическое титрование 250,
251
Кольрауша мостик 176, 177
Кондуктометрия 173 и ел.
Константы физические 76, 77
Концентрация моляльная 8—10
— молярная 8—11
— нормальная 12
-- процентная 8—11
— коллоидных растворов 25, 26
Кюветы фотометрические, подбор 256,
260
Лампы электронные 52—56
Люминесценция 235
Максимумы полярографические 194
, их подавление 194, 227, 228
Меди полярографическое определение
218, 220—223
Микроэлементы, определение в почвах
220
Минералогический состав почв,
определение 314, 315, 335—338
Минералы слоистой структуры 316
Молекулярный вес гредневесовой 28,
30
— — среднечисловой 28
средний 28
раствора 10
Моляльиая концентрация (см.
концентрация моляльная)
343
Молярная доля 8—10
Молярная концентрация (см.
концентрация молярная)
Молярный коэффициент погашения
240
трисульфосалицплата
железа 280
Монохроматизация света 62
Монохроматоры 62—64, 265, 270,
273, 302
Монтмориллопитоные минералы 316—
318
Натрия определение по фотометрии
пламени 310—313
— определение стеклянным
электродом 138—143
— обменного, определение по
фотометрии пламени 312
■— обменного, определение
стеклянным электродом 141—143
Нернста уравнение 80—84
Никеля полярографическое
определение 218—223
О кисл ител ьно- восста нонительны й
потенциал 83, 84, 151-155
, влияние комплексообразова-
телей 160, 161
рН 155, 157—160
Окислительно-восстановительный
потенциал нормальной системы 83,
154, 161
■ определение в почвах 166 и
ел.
почвы 161—172
Окислительно-восстановительные
реакции 83, 84, 151—156
• , вычисление гН2 156, 157
в почвах 151, 152, 161—166
, константа 152
Осмотическое давление 29, 79
Оствальда закон разбавления 18
Отражательная спектральная
способность почв 286, 287
Пирометр Курнакова 332—334
Платинирование водородных
электродов 93
— стеклянных электродов 167
Плотность оптическая 239—242, 250—
252, 258
раствора гумата 241, 247, 281—
284
344
Поггендорфа потенциометрическая
схема (см. потенциометры)
Полуподавления коэффициент 228
Поляризация электродов 85, 86
концентрационная 185
Полярограф Гейровского 199—203
— Л И-55 205-207
— - микро- 203—205
— электронный 209
Полярографы, проверка качества
схемы 209 -211
-•-, типичные неисправности 210, 211
Потенциал асимметрии стеклянного
электрода 122, 123
— восстановления 184, J85, 189
— диффузионный 82
— окислительно-восстановительный
(см.
окислительно-восстановительный потенциал)
— полуволны 189—192
— полуволны, влияние комплексооб-
разователей 191, 193
— полуволны, таблицы 192
— химический 17
— электрода 79—85
— электрокинетический 31
— электрохимический 17
Потепциометрическос титрование
143—151
«на нуль» 145—148
Потенциометры 85—90, 125—132
—, компенсационная схема
Поггендорфа 86—89, 125
—, конденсаторная схема 125
— ламповые 128—132
— П-6 132—137
Произведение растворимости 16, 17
— — каломели 96
Пропускание излучения 239
, коэффициент 249
, полуширина 65
Рассеяние света 29, 30, 247, 276
Распылители 297—299
Реакция почв 98—102
, влияние поглощенных ионов
100, 101
— солей 100
, определение методом
титрования «на нуль» 147, 148
Реостаты 38, 39
Реохорд 86, 87
Реохордный мост Р-38 178
Ртутный капельный электрод 184, 187,
195, 196
, градуировка 209, 214
, площадь поверхности 187
Рэлея отношение 30
Светофильтров подбор 245, 246, 259
Светофильтры абсорбционные 64 и ел.
— абсорбционные, спектральная
характеристика 64—66
Светофильтры абсорбционные,
желатиновые 65
— абсорбционные жидкостные 65, 302
— абсорбционные стеклянные 65
— интерференционные 65—67, 301
— интерференционные, спектральная
.характеристика 66, 67
—, оптические характеристики 64—66
Слюдистые минералы, термические
характеристики 319
Солесодержания определение 173—181
Солемер почвенный ПС-3 178—181
-•- —, градуировка 175, 181
Спектрофотометр СФ-2М 253, 265—
270, 287
— СФ-4 270—273
Спектрофотометрический анализ 229,
230—249—253, 272—288
, выбор светофильтров (длины
волны) 241, 246, 250
, поправка на кювету с
растворителем 246, 247
— — по смешанным окраскам 252
Спектры излучения 232, 289—292
— —, влияние температуры 291, 292
состава пробы 293, 294, 295—
297
— —, возбуждение в пламени 292—
294
Спектры поглощения 232 и ел., 248,
249
— — вращательные 233
гумусовых веществ 241, 276,
283—286
колебательные 232—234
— — молекул, характерные области
поглощения 235—238
электронные 233, 234, 236
Стеклянный электрод 117—125
, градуировка 124, 128, 138, 140,
141
— —, изготовление и устройство 120,
121
■ , недостатки 122, 125
, пределы применимости 120
Стокса формула 26, 27
Теорелла уравнение 29
Термисторы 73
Термограмма дифференциальная
322—325, 331, 332
— дифференциальная, расшифровка
332, 338
— простая 324, 325
Термометры ртутные 67 и ел.
— ртутные контактные 68, 69
— ртутные, поправка на
выступающий столбик ртути 68
— сопротивления 67, 72, 73
Термопары 67—71, 320-323, 325,
326
— дифференциальные 323, 325, 326
— дифференциальные, градуировка
331
— платина-платинородиевые 321, 322
—, холодного спая термостатирование
326
— т.э.дс. при 100° 321
Терморегуляторы 68, 69, 71, 72, 328
Терыоэлектродвижущая сила 68—71,
320, 321, 322
Термоэлементы 67, 72
Ток диффузионный 186, 188, 193
— миграционный 186, 193
— миграционный, устранение 193, 194
— остаточный 188, 193
— предельный 186
Трансформаторы 37, 38, 328
Уровни энергетические 233, 234
Усилители 52—56
Фазовые превращения 315
в почвах при термическом
анализе 316
Фои (см. электролит
индифферентный)
Фосфора фотометрическое
определение 280, 281
Фотометр пламенный 297, 303—309
ППФ 305—309
— универсальный 253—260
Фотометрия пламени 289 и ел.
— —, выбор светофильтров 301, 302,
307
' фотоэлементов 302, 303
Фотоэлектроколориметры 253, 260—
264
Фотоэлементы, инерционность 60
— с внешним фотоэффектом 56, 57
— с внутренним фотоэффектом 56, 57
— селеновые 59, 61
345
— сернистосеребряные 59, 61
— с запирающим слоем 56, 60—62
—, темновой ток 60
—, утомляемость 60
—, чувствительность интегральная 58,
59
спектральная 58, 59
Фотоэффект 56—58
—, порог 58
Фрейндлиха уравнение 32
Фрумкииа уравнение 33
Хиигидрон 103
Хингидронный электрод 102 и ел.
нормальный 106, 107
, определение рН 109—117, 135
, приготовление 103, 109
, проверка качества 108
Хлоросеребряный электрод 121—123
Хромофорные группы 236, 237
■ , таблица 237
Цинка полярографическое
определение 218, 220—223
Экзотермические реакции 315
— эффекты минералов 336—337
Экстннкция (см. плотность
оптическая)
Электродвижущая сила (э. д. с.) 78,
80, 81, 85
, вычисления 81, 82, 97, 110—112,
123, 124
, измерения 85—90, 125-128,
134—137
■ концентрационной цепи 80
Электроды второго рода 96
— для определения ОВ-потенциала
166 и ел.
— для определения
электропроводности 176
—, определение знака заряда 85, 171
— твердые для полярографии 196
Электролизеры полярографические 197
Электролит индифферентный (фон)
186, 192, 194, 219
Электронный регулятор ЭРМ-47, 71,
72, 329
Электропроводность, влияние
частоты 177, 178
—, зависимость от температуры 176
—, измерение 178
— удельная 21, 24, 174
— эквивалентная 21, 24, 174, 175
Электроосмое 31
Электрофорез 30, 31
Элементы со стеклянным электродом
121—123, 138
•— хингидрон-каломельные 109—112,
145, 146
— хингидрои-хингидроиные 109-—111»
147
Эндотермические реакции 315
— эффекты минералов 336, 337
Эталоны для градуировки термопар
330
Этилендиаминтетрауксусная кислота,
комплексонаты цинка и кальция 224
Ячейки для определения
электропроводности (см. электроды)
— полярографические (см.
электролизеры полярографические)
— фотометрические 300, 301
ОГЛАВЛЕНИЕ
Введение 3
Глава I. Общие свойства дисперсных систем как предмета анализа
(способы выражения концентрации, общие законы и расчетные
формулы) 7
Истинные растворы ... 8
Коллоидные растворы 24
Глава II. Аппаратура для физико-химических исследовании . . 36
Источники электропитания 37
Контрольно-измерительные приборы . . . .45
Электронные лампы и усилители 52
Фотоэлементы 56
Монохроматоры и светофильтры 62
Контроль и измерение температуры 67
Условные обозначения, применяемые в схемах приборов,
а также служащие для маркировки шкал 74,^
Важнейшие физические константы 7t>',
Глава III. Потенциометрические методы исследования почв . 78
Потенциал электрода 79
Измерение э.д.с 85
Приготовление водородного и каломельного электродов и
измерение потенциала каломельного электрода 90
Потенциометрическое определение активности водородных
ионов в почвенных растворах, вытяжках и суспензиях . . 98
Измерения активности водородных ионов хингидроиным
электродом в водных и солевых вытяжках из почв .... 102
Стеклянный электрод и его применение в почвенных
исследованиях 117
Намерение эд.с цепи со стеклянным электродом .... 125
Определение рИ водных и солевых вытяжек из почв на
потенциометрах 132
Определение содержания и активности ионов натрия в
почвенных вытяжках и суспензиях с помошью стеклянного электрода 137
Потенциометрическое титрование 143
Окислительно-восстановительный потенциал почв и методы его
измерения 151
Окислительно-восстановительные реакции и потенциалы в поч-
Электроды и схемы для определения
окислительно-восстановительных потенциалов в почвах 16*-
Глава IV. Определение солесодержания в почвах методом коидук-
тометрии 17.(
Приборы для кондуктометрических исследований почвенных
вытяжек и растворов 17'>
Глава V. Полярографические методы исследования почв . 18'
Основы теории полярографического метода . 18'
Приборы для полярографического анализа . ... 19*
Получение полярограмм и их расшифровка 21,
Применение полярографического анализа к изучению состава
и свойств почв 216
Глава VI. Исследование почв методами адсорбционной спектрофо-
томерии 229
Спектры. Основные единицы ... ... 230
Спектры поглощения молекул 232
Количественные закономерности поглощения света растворами 238
Порядок измерений и обработка результатов 248
Описание приборов 253
Спектрофотометрические методы исследования почв . . . 278
Характеристика органических веществ почвы 281
Характеристика почв по кривым спектральной яркости . . 286
Характеристика почвенных профилей по содержании гумуса
и соединений железа 287
Глава VII. Определение в почвах калия и натрия методом
пламенной фотометрии .... .... 289
Теоретические основы 289
Аппаратура для пламенной фотометрии 297
Определение калия и натрия в почвах . .310
Глава VIII. Термический анализ почв 314
Теоретические основы метода ... 315
Термические характеристики некоторых минералоо ... . 317
Приборы для термического анализа почв и порядок работы . 320
123, Рекомендуемая литература 338
Лредметный указатель 342
Николай Георгиевич Зырин
Дмитрий Сергеевич Орлов
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ ПОЧВ
Сводный тематический план сельскохозяйственной литературы 1964 г. № 6
Редактор Коробцова Н. А. Технический редактор Георгиева Г. И.
Переплет художш/ка Самариной Л. М.
Сдано в набор 11/11 1964 г. Подписано к печати 12/Х 1964 г.
Л-79834 Формат бОХЭО'/ш Печ. л. 21,75 Уч.-изд. л. 20,75 Изд. № 61 J
Заказ № 51 Тираж 5500 экз. Цена I р. 60 к.
Издательство Московского университета.
Москва, Ленинские горы. Административный корпус.
Типография Изд-ва Л1ГУ. Москва, Ленинские горы